/


























































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































Author: Шерякова А.А.
Tags: фармация аптечное дело фармакология токсикология фармакопея
ISBN: 978-985-6967-17-0
Year: 2012
Similar




Text
...
v
..........y .........
: ...
.............-J-...........
..-,..
....-..
=
-
.. ............
.._ .......""'
.....
....-J.. :...,...o"
""""" J \.../......J
\...
.--=.
.... .......
""""'.
........-..:.
K.....
..........
.
.Jr
""'........
.
.....,.
.......
...,
..././
-
,
.........
;::
..r
-..
-----'
'---'
.......
/
--------'
...............
..::.................J
.......:.
'-......./
.....
'""
......
J
\.
......J
...
.:t...
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РЕСПУБЛИКИ БЕЛАРУСЬ
Республиканское унитарное предприятие
«Центр экспертиз и испытаний в здравоохранении»
rОСУДАРСТВЕННАЯ
ФАРМАКОПЕЯ
РЕСПУБЛИКИ БЕЛАРУСЬ
Разработана на основе Европейской Фармакопеи
(rФ. РБ 11)
в двух томах
Том 1
Общие методы
контроля качества
лекарственных средств
Введено в действие с 1 января 2013 аода
приказом Министерства здравоохранения Республики Беларусь
от 25.04.2012 аода NQ 453
Молодечно
. Типоrрафия «Победа»
2012
УДК 615.11 (476)(083.7)
ББК 52.8(4Беи)
172
Под общей редакцией А. А. Шерякова
r 72 rосударственная фармакопея Республики Беларусь. (rФ. РБ 11): Разработана на
основе Европейской фармакопеи. В 2 т. Т. 1. Общие методы контроля лекарственных
средств 1 M вo здравоохр. Респ. Беларусь, УП «Центр экспертиз и испытаний в
здравоохранении" ; под общ. ред. А. А. Шерякова. Молодечно: Тип. «Победа". 2012.
1220 с.
ISBN 978 985 6967 17 o
Первый том BToporo издания содержит обязательные стандарты и положения. реrламентмрующие качество
лекарственных средств и субстанций для фармацевтическоrо использования: общие статьи на методы анализа
(физические, физико химические, биолоrические и фармакоrностические методы, определение подлинНOCТ1I, определение
примесей, количественное определение, фармацевтико технолоrические испытания), контейнеры, реакrивы, общие тексты
(по микробиолоrии, по биолоrическим продуктам, общие статьи и таблицы физических характеристик). ЭICCТeIIпоральные
лекарственные средства, общие статьи, дозированные лекарственные формы, rомеопатические лекарственные средства.
Книrа предназначена для специалистов, занимающихся разработкой, производством. контролем качества,
хранением и реализацией лекарственных средств.
УДК 615.11(476)(083.7)
ББК 52. (4Беи)
ISBN 978 985 б9б7 17-0 (Т.1.)
978 985 б9б7 19 4
@ УП «Центр экспертиз и испытаний в здравоохранении.», 2012
@Оформление УП ..Типоrpафия «Победа», 2012
3
СОДЕРЖАНИЕ
Введен ие ................. ....... ................................................ ............ ...................................... 15
1. Общие сведения ....... .... ................... ........................ .... ....... ........... ......... .............. 17
2. Методы анализа ............... ..... ..................... ............. ........... ... ............... ................. ЗЗ
2.1 Оборудование ... ....... ................................ .............. ....... ........ .. ............... ......... ................... ............. ... 33
2.1.1. Каплемер ................................................................................................................................ 33
2.1.2. Сравнительная таблица пористости стеклянных фИfiЬТрОВ ............................................... 33
2.1.3. Лампы с ультрафиолетовым излучением для аналитических целей................................. 34
2.1.4. Сита ............. ..... ........... ... .............. ...... ...... ........ ...... ...... ........ ........ ..... ...................... ....... ......... з4
2.1.5. Пробирки для сравнительных испытаний ............................................................................ 35
2.1.6. Индикаторные трубки...... ..... ............................................................... ................................... 35
2.2. Физические и физ-ико химические методы ....................................................................................... 36
2.2.1. Определение прозрачности и степени мутности жидкостей .............................................. 36
2.2.2. Определение степени окрашивания жидкостей .................................................................. 39
2.2.3. Потенциометрическое определение рН ............................................................................... 41
2.2.4. Зависимость между реакцией раствора, приблизительным значением рН
и цветом индикаторов....... ................ ......... ............................................................................ 43
2.2.5. Относительная плотность .. ....... .... ...... .... ... ................................... ............................. ........... 45
2.2.6. Показатель преломления (индекс рефракции).................................................................... 47
2.2.7. Оптическое вращение... ... ... ..... ...... ........ ... ................ ......... ....... ......... ............ ........ ....... .......... 47
2.2.8. Вязкость ....... ..... ................... ........ ..... .... ..... .......................... ........ ..... ...... ........ ........... ..... ........ 48
2.2.9. Метод капиллярной вискозиметрии........... ....................................................... .................... 49
2.2.10. Метод ротационной вискозиметрии ...................................................................................... 51
2.2.11. Температурные пределы переroнки ................................................................................ ..... 53
2.2.12. Температура кипения........... .................................................................................................. 54
2.2.13. Определение воды методом отrонки .................................................................................... 54
2.2.14. Температура плавления капиллярный метод ................................................................. 55
2.2.15. Температура плавления открытый капиллярный метод ................................................ 56
2.2.16. Температура плавления метод MrHoBeHHoro плавления ................................................ 56
2.2.17 . Температура каплепадения. ..................... ............................................................................. 57
2.2.18. Температура затвердевания .......................... ............................................ ..... ...................... 59
2.2.19. Амперометрическое титрование.............................. ............................................................. 60
2.2.20. Потенциометрическое титрование.................... ................................................................... 60
2.2.21. флуориметpI4я................................................................................................................... ...... 62
2.2 .22. Атомно эмиссионная спектрометрия................................. . ................................................ 62
2.2.23. Атомно абсорбционная спектрометрия.................................... ............................................ 64
2.2.24. Абсорбционная спектрофотометрия в инфракрасной области.......................................... 67
2.2.25. Абсорбционная спектрофотометр.ия в ультрафиолетовой
и видимой областях .................................................................................................. ... .......... 70
2.2.26. Бумажная хроматоrрафия ..................................................... ................................................ 76
2.2.27 . Тонкослойная хроматоrрафия.. ............. ................................................................................ 77
2.2.28. rазовая хроматоrрафия ................................... ........ .................... ......................... ................. 80
2.2.29. Жидкостная хроматоrрафия................:....................................................................... .......... 82
2.2.30. Эксклюзионная хроматоrрафия ........................ ..................... ....................... ..--.................... 84
2.2.31. Электрофорез.................................................................................................. --..................... 85
2.2.32. Потеря в массе при высушивании ........................................................................................ 92
2.2.33. Спектрометрия ядерноro маrнитноro резонанса ............................................ . .................. 93
2 .2.34. Термический анализ.................................. .......................................... ................... ................ 98
2.2.35. Осмоляльность ..... ... ..... ................................... .......................... .................. .. ..................... 101
2.2.36. Потенциометрическое определение концентрации ионов
с использованием ионоселективных электродов . ........................ .... ............................... 103
2.2.37. Рентrенофлуоресцентная спектрометрия..................... ......................................... .... ........ 104
2.2.38. Электропроводность................................................ ....... ........... ......................................... 105
2.2.39. Молекулярно массовое распределение дeKcтpaHOВ..... ........ ............................. ............ 106
2.2.40. Спектрофотометрия ближнеro инфракрасноrо диanaзoнa .................__........................... 109
2.2.41. Круroвой дихроизм ... .... ....._.. . .............................. ............................. ................. ...... .... ........ 115
4
rосударственная фармакопея Республики Беларусь
2.2.42. Плотность твердых тел ......................... ................................ ....... ............. ...................... ..... 116
2.2.43. Масс спеКТрометрия .. .......... ............................ ..... ............ ..... .... .............. ....... ........ ............. 117
2.2.44. Определение содержания общеro орrаническоro уrлерода
в воде для фармацевтическоro применения ..................................................................... 121
2.2.45. Сверхкритическая флюидная хроматоrрафия........ ....... .............................. ... ......... .......... 123
2.2.46. Хроматоrрафические методы разделения ......................................................................... 123
2.2.4 7. Капиллярный элекТрофорез........ ..... .................... ... ........ ..... .............................. ................. 132
2.2.48. Рамановская спектрометрия ..... .... ........... ............ ............. .... ......... ................................. .... 140
2.2.49. Измерение вязкости на вискозиметре с падающим шариком.......................................... 142
2.2.54. Изоэлектрическое фокусирование................................ ............. ............................. ........... 142
2.2.55. Пептидное картирование... .......................................................................... ........................ 145
2.2.56. Анализ аминокислот .............................................................................................. .......... .... 150
2.2.57. Атомно эмиссионная спектрометрия с использованием индуктивно
связанной плазмы ...... ................................. ... ........... ......... ....... ... ............. ........................... 161
2.2.58. Масс спектрометрия с использованием
индуктивно связанной плазмы .... ....................... ...... ........... ............. ............. .......... ............ 164
2.2.59. Анализ rликанов в rликопротеинах ......:... .................... ................................................... .... 166
2.2.60. Температура плавления инструментальный метод...................................................... 174
2.3. Подлинность (идентификация).. ... ...... ....... ......................... .... .... ........ ....... .............. ......... ............... 175
2.3.1. Реакции подлинности (идентификации) на ионы и функциональные rруппы................. 175
2.3.2. Идентификация жирных масел методом тонкослойной хроматоrрафии ........................ 181
2.3.3. Идентификация фенотиазинов методом тонкослойной хроматоrрафии ........................ 182
2.3.4. Определение запаха .............. ................ ..... .............. ................. ..... .... ..... ................. ........... 182
2.4. Испытания на предельное содержание примесей......................................................................... 183
2.4.1. Аммония соли ....... ............. ............. .............. ......... ................ ....... ................ ..... ...... ............. 183
2.4.2. Мышьяк..... ...... ..... ...... ............... .......... ......... ......... ...... ...... ........... .............. ......... .... .... .... ...... 183
2.4.3. Кальций.... ......... ............. .......... ....... ... ......... ............................. ................... .......................... 184
2.4.4. хлориды................................................................................................ ................................ 185
2.4.5. Фториды ................................... ............................................................................................. 185
2.4.6. Маrний......................................................................................................................... .......... 185
2.4.7. Маrний и щелочноземельные металлы.............................................................................. 186
2.4.8. Тяжелые металлы ................................................................................................................ 186
2.4.9. Железо .................. ........ ......................... ...... ........... ................ ..... .... ....... ... .... ................... .... 191
2.4.10. Свинец в сахарах ................................................................................................................. 191
2.4.11. Фосфаты ..... ....... ...... ....................... .... ..... ........... ........ ..................... ...... ............................... 191
2.4.12. Калий.... ........... ............ ....... .... ...... ...... ....... ............ .... ................... ....... ....... ............... ........ .... 191
2.4.13. Сульфаты.... ......... .......... ........ .... ......................... ..... ..................... ...... ................... ............... 192
2.4.14. Сульфатная зола..... ....... ........... .... ............ .... ............. ........ .... ..... ............................ ............. 192
2.4.15. Никель в полиолах ............................................................................................................... 192
2.4.16. Общая зола. ......... .... ........... ................ ......... ......... .......... ..... ..................... ....... ...... ............... 192
2.4.17. Алюминий.. .... ....... ...... ...... .... ....... ...... ....... ................ ............ ....... ................. ........................ 193
2.4.18. Свободный формальдеrид .................................................................................................. 193
2.4.19. Щелочные при меси в жирных маслах .. .............................................................................. 194
2.4.21. Посторонние масла в жирных маслах методом тонкослойной хроматоrрафии ............. 194
2.4.22. Посторонние жирные кислоты в маслах методом rазовой хроматоrрафии ..... .............. 195
2.4.23. Стерины в жирных маслах .................................................................................................. 197
2.4.24. Идентификация и контроль содержания остаточных растворителей ............................ 200
2.4.25. Остаточные количества этиленоксида идиоксана............................................................ 204
2.4.26. N,п диметиланилин............... ........... ........... ..... ........... ... ...... ........... ............................ ......... 207
2.4.27. Тяжелые металлы в лекарственном растительном сырье и жирных маслах ................. 208
2.4.28. 2 Этилrексановая кислота...................... ...... ................... ..... ................................... ............ 209
2.4.29. Состав жирных кислот в маслах, обоrащенных OMera 3 кислотами ................................ 210
2.4.30. Этиленrликоль и диэтиленrликоль в этоксилированных субстанциях ............................ 212
2.4.31. Никель в rидроrенизированных растительных маслах ..................................................... 213
в
5
2.4.З2. Общий холестерин в маслах, обоraщенных OMera 3 кислотами ...................................... 213
2.5. Методы количественноro определения ........... ....... ........ ........................... ... .... ......... ... .................. 215
2.5.1. Кислотное число........... ........ .... ...... ...... ... ...... .... ............................................ ...... ....... .... ...... 215
2.5.2. Эфирное число.............. ....... .... ..... ...... ....... ... ............................. ...:....... ............. ..... ............. 215
2.5.3. rидроксильное число.............. ....... ....... ....... ........................................ ................ .......... ...... 215
2.5.4. Йодное число....... ....... ....................... ... ..... ........ ................................. .... ............. ................. 216
2.5.5. Перекисное (пероксидное) число ....................................................................................... 217
2.5.6. Число омыления....................... ....... .... ......... ...... .......................... ......... .... ....... ........ ..... ....... 218
2.5.7. Неомыляемые вещества ...........................:......................................................................... 219
2.5.8. Определение аминноro азота в соединениях, которые содержат первичную
ароматическую аминоrруппу .... ........ ............. ........................................ ........... ......... .......... 219
2.5.9. Определение азота после минерализации серной кислотой ........................................... 220
2.5.10. Метод сжиrания в колбе с кислородом............................................................................... 220
2.5.11. Комплексометрическое титрование..... ............... .......................... ... .... ............ .... ............... 221
2.5.12. Вода: полумикрометод .. ...... ..... .... ........ ....... .... ......... ................ ............ ........... ......... .... ....... 222
2.5.1 З. Алюминий в адсорбированных вакцинах ........................................................................... 224
2.5.14. Кальций в адсорбированных вакцинах .............................................................................. 224
2.5.15. Фенол в иммуносыворотках и вакцинах ............................................................................. 224
2.5.16. Белок в полисахаридных вакцинах..................................................................................... 224
2.5.17. Нуклеиновые кислоты в полисахаридных вакцинах.......................................................... 225
2.5.18. Фосфор в полисахаридных вакцинах ................................................................................. 225
2.5.19. О ацетил в полисахаридных вакцинах ............................................................................... 226
2.5.20. rексозамины в полисахаридных вакцинах......................................................................... 226
2.5.21. Метилпентозы в полисахаридных вакцинах ...................................................................... 227
2.5.22. Уроновые кислоты в полисахаридных Вакцинах................................................................ 227
2.5.23. Сиаловая кислота в полисахаридных вакцинах ................................................................ 228
2.5.24. Уrлерода диоксид в rазах .................................................................................................... 228
2.5.25. Уrлерода монооксид в rазах ................................................................................................ 229
2.5.26. Азота монооксид и азота диоксид в rазах .......................................................................... 230
2.5.27. Кислород в rазах ..... ......... ................. ................ ............. .................... ........ ....... ................... 2З 1
2.5.28. Вода в rазах ....... ................... ......... .... ...... ... ....... ..... ..... ..... ... ..... ............ .... ...... ... ................... 231
2.5.29. Серы диоксид ............................................... ..... .......... ............... ...... ........................ ............ 231
2.5.30. Окисляющие вещества ................................................................................................ ... ..... 232
2.5.31. Рибоза в полисахаридных вакцинах................................................................................... 232
2.5.З2. Вода: микроопределение................ ...... ................... ............. ... ..................... ...... ..... ...... ...... 2ЗЗ
2.5.33. Общий белок... .... .............................. ... .... ... ....... ....... ....... ........ .... ........ ... ...... .... ....... ............. 23З
2.5.34. Уксусная кислота в синтетических пептидах...................................................................... 239
2.5.35. Азота закись в rазах ...................................................... . ...................................................... 2З9
2.5.36. Анизидиновое число ............................. ..... ................. ...... .......... ...... ... ................... ....... ...... 240
2.5.37. Метил , этил и изопропилметансульфонат в метансульфоновой кислоте..................... 240
2.5.38. Метил , этил и изопропилметансульфонат
в фармацевтических субстанциях .... ...... ...... ......... ........ ......... ....................... ......... ... ........ 241
2.5.39. Метансульфонилхлорид в метансульфоновой кислоте.................................................... 243
#2.5.50. Титрование в неводных растворителях............................................................................ 244
2.6. Биолоrические испытания ........ .......... ....... .... .......... ..... .......... ... ....... ... ...... ..................... ................. 252
2.6.1. Стерильность....... ................. ...... .......... .... ......................... ... .......... ................. ................. .... 252
2.6.2. Микобактерии .. ........ ...... ........... .......................... ......... .......... ................. ............ .................. 257
2.6.7. Микоплазмы... ...... ... .... .......... ....... ...... .... ....... ............... ... ....... ....................... ...... ........ ..... ..... 257
2.6.8. Пироrенность. ... ........ ......... .... ........ ........ ............ ........ .......... ............ ... .......... ..... ................... 265
2.6.9. Аномальная токсичность... _.. .................................... ..... ...... ...... ....................... ................... 266
2.6.10. rистамин .................. ................................................... ....... ..... ................ .............................. 267
2.6.11. Депрессорные вещества ......................................._............................................................. 268
2.6.12. Микробиолоrические испытания нестерильной продукции:
общее количество жизнеспособных аэробов .................................................................... 269
2. Зак 1712
6 rосударственная фармакопея Республики Беларусь
2.6.13. Микробиолоrические испытания нестерильной продукции:
испытания на наличие специф ';ческих микроорrаНизмов................................................ 276
2.6.14. Бактериальные эндотоксины .................. .... ...... ..................... ................................. ............. 288
2.6.15. Активатор прекалликреина................................................................ .................................. 294
2.6.16. Испытания на посторонние areHTbI в вирусных вакцинах для медицинскою
применения............................ ............................................................. ....... ........................... 295
2.6.17. Испытание на антикомплементарную активность иммуноrлобулина.............................. 298
2.6.18. Испытание живых вирусных вакцин на . нейровирулентность.......................................... 301
2.6.19. Испытание пероральной вакцины полиомиелита на нейровирулентность..................... 301
2.6.20. Анти А и анти В rемаrrлютинины (непрямой метод) ......................................................... 303
2.6.21. Методы амплификации нуклеиновых кислот ..................................................................... 303
2.6.22. Активированные факторы свертывания крови .................................................................. 311
2.6.26. Испытание на анти D антитела в иммуноrлобулине человека
для внутривенноro введения .................. ................. .... .............................. ............. ....... ...... 311
2.6.27. Микробиолоrический контроль клеточных продуктов ....................................................... 312
2.6.30. Испытание на активацию моноцитоВ.................................................................................. 314
2.6.31. Микробиолоrические испытания лекарственных средств растительною
происхождения для внутреннеro применения . .................................................................. 323
2.7. Биолоrические методы количественноro определения ........................................................ ......... 325
2.7.1. Иммунохимические методы ............................................ ...... .................................. ............. 325
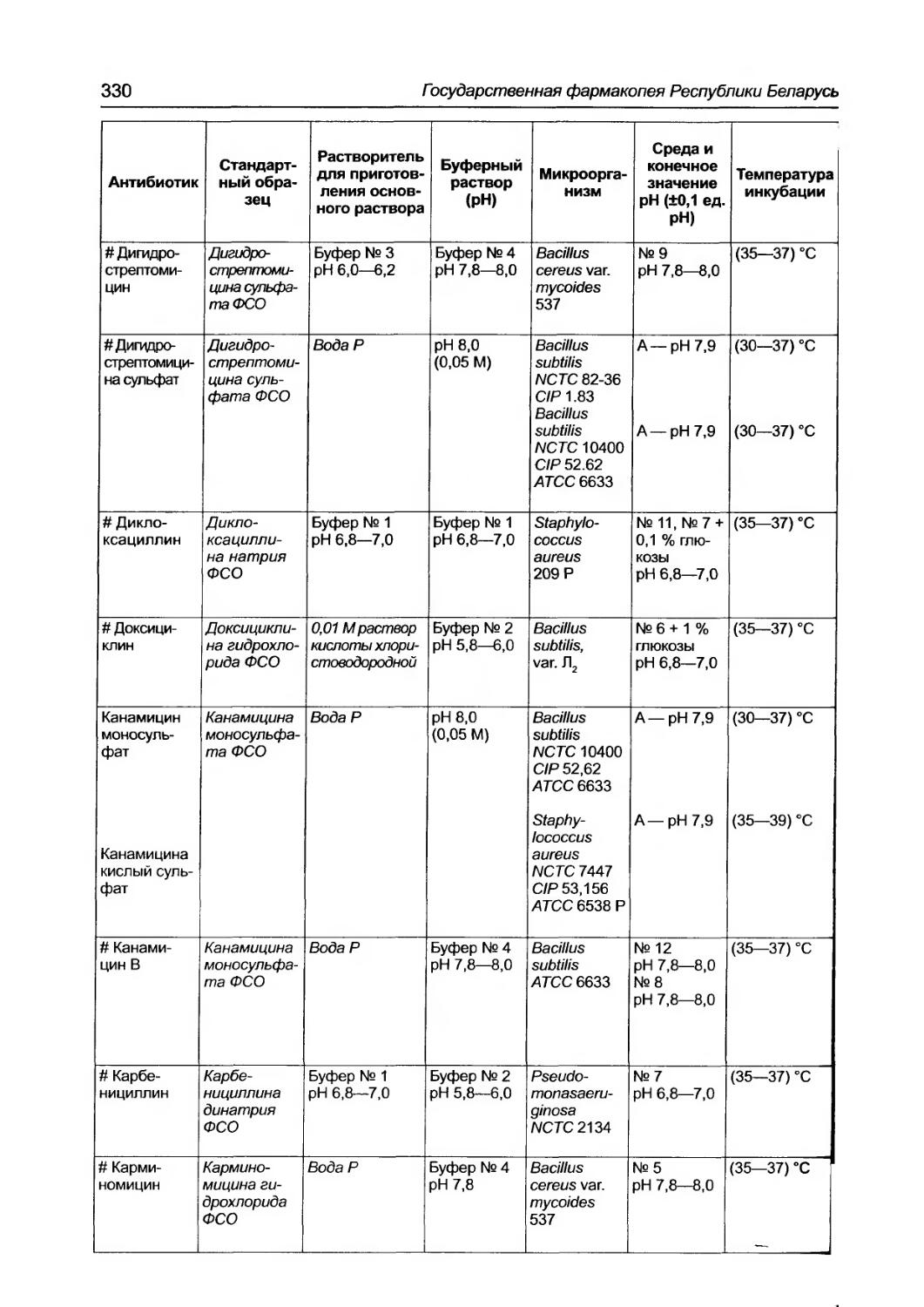
2.7.2. Количественное определение антибиотиков микробиолоrическим методом ................. 327
2.7.4. Количественное определение фактора свертывания крови V,l,...................................... 342
2.7.5. Количественное определение rепарина............................ .......... ..... ........................ .......... 344
2.7.6. Количестенное определение адсорбированной дифтерийной вакцины ......................... 346
2.7.7. Количественное определение вакцины . коклюша............................................................. 353
2.7.8. Количественное определение адсорбированной столбнячной вакцины......................... 354
2.7.9. Определение функциональною состояния Fс фраrмента иммуноrлобулина ................ 361
2.7.10. Количественное определение фактора свертывания крови человека Vll....................... 363
2.7.11. Количественное определение фактора свертывания крови человека IХ........................ 364
2.7.12. Количественное определение rепарина в концентратах факторов
свертывания крови........ .......... .................. ........ ................. ......................... .................... ..... З65
2.7.13. Количественное определение анти D иммуноrлобулина человека................................. 366
2.7.14. Количественное определение антиrенной (иммуноrенной)
активности вакцины rепатита А................ ............................................... ...... ............ .......... З69
2.7.15. Количественное определение вакцины rепатита В (рДНК) .............................................. 370
2.7.16. Количественное определение вакцины коклюша (бесклеточной).................................... 371
2.7.17. Количественное определение аНТИТР9мбина 111 человека................................................ 372
2.7.18. Количественное определение фактора свертывания крови 11 человека ......................... 373
2.7.19. Количественное определение фактора свертывания крови Х человека......................... 374
2.7.20. Количественное определение инактивированной вакцины полиомиелита iп vivo ....,.... 375
2.7.21. Количественное определение фактора ВИ{lебранда человека ........................................ 376
2.7.22. Количественное определение фактора свертывания крови ХI человека........................ 378
2.7.23. Подсчет клеток СО34/СО45+ в rемопоэтических продуктах ............................................. З79
2.7.24. Проточная цитометрия.................................... ................................................ .... .., .... .......... 381
2.7.25. Определение содержания инrибитора плазмина человека.............................................. 384
2.7.27. Значение флоккуляции (Lf) токсинов и анатоксинов дифтерии
и столбняка (проба рамона) ................................................................................................ 385
2.7.28. Определение количества rемопоэтических клеток предшественниц человека
по колониеобразующим клеткам.................................................................... ..................... 386
2.7.29. Определение количества и жизнеспособности
ядросодержащих клеток..................................................... ................................................. 388
2.7.30. Количественное определение человеческоro белка С..................................................... 391
2.7.31. Количественное определение человеческоro белка S...................................................... 392
2.7.32. Количественное определение инrибитора а 1 протеиназы человека ............................. 393
7
#2.7.50. Определение биолоrической активности инсулина......................................................... 394
2.8. Методы фармакоrнозии ................ ....... ............... ........................ .............. ......................... ............... 396
2.8.1. Зола, нерастворимая в хлористоводородной кислоте...................................................... 396
2.8.2. Примеси ........ ........ .................... .......................... ......... .............. ............... .... ........................ 396
2.8.3. Устьичный коэффициент........ ..... ............... ... ....... ................. ....................... ....................... 397
2.8.4. Коэффициент набухания ....... ................... ... ........... ................ ................ ....... ...................... 397
2.8.5. Определение воды в эфирных маслах............................................................................... 397
2.8.6. Посторонние эфиры в эфирных маслах ............................................................................ 398
2.8.7. Жирные и минеральные масла в эфирных маслах .......................................................... 398
2.8.8. Запах и вкус эфирных масел............................................................................................... 398
2.8.9. Остаток после выпаривания эфирною масла ................................................................... 398
2.8.10. Растворимость эфирных масел в спирте ......................................................................... . 398
2.8.11. Определение 1 ,8 цинеола в эфирных маслах................................................................... 399
2.8.12. Определение эфирноrо масла в лекарственном растительном сырье........................... 399
2.8.13. Остаточное количество пестицидов ................................................................................... 403
2.8.14. Определение дубильных веществ .:.................................................................................... 405
2.8.15. Определение показателя roречи......................................................................................... 405
2.8.16. Сухой остаток экстрактов .................................................................................................... 406
2.8.17. Потеря в массе при высушивании экстракта ..................................................................... 406
2.8.18. Определение афлатоксина В 1 в лекарственном растительном сырье............................ 407
2.8.20. Лекарственное растительное сырье: отбор проб.............................................................. 409
2.8.22. Определение охратоксина а в лекарственном растительном сырье............................... 410
2.8.23. #Макроскопический и# микроскопический анализ лекарственноro
растительноrо сырья.. ....... ..... ... ...... ................................ ...... ........ ........ ........... ........ ........ .... 412
2.9. фармацевтико Технолоrические испытания ........................... ........................................................ 417
2.9.1. Распадаемость таблеток и капсул ...................................................................................... 417
2.9.2. Распадаемость суппозиториев и пессариев...................................................................... 419
2.9.3. Тест «Растворение» для твердых дозированных форм.................................................... 420
2.9.4. Тест «Растворение» для трансдермальных пластырей .................. ................................ 428
2.9.5. Однородность массы для единицы дозированноro лекарственноrо средства ............... 430
2.9.6. Однородность содержания действующеro вещества
в единице дозированноro лекарственноro средства......................................................... 431
2.9.7. Прочность таблеток без оболочки на истирание ...............................................................432
2.9.8. Прочность таблеток на сжатие ............................................................................................ 433
2.9.9. Измерение консистенции методом пенетрометрии........................................................... 433
2.9.10. Содержание эта нола . ....... ...... ...... ......... .............. .......... ......... ......... ....... .......... ... ... .............. 435
2.9.11. Испытание на содержание метанола и 2 пропанола ........................................................ 438
2.9.12. Ситовой анализ...... ........ .................. ........................ ....... ......... ..... ....... .......................... ...... 440
2.9.14. Удельная площадь поверхности ....................................................................................... . 441
2.9.16. Сыпучесть. ............... ....... ........ ................. ...... .......... ..... ........... ....... ........... .... ......... .............. 443
2.9.17. Определение извлекаемоro объема парентеральных лекарственных средств ..... . 444
2.9.18. Лекарственные средства для инrаляций: аэродинамическое
испытание мелких частиц ................................................................................___............ ..... 445
2.9.19. Заrрязнение механическими включениями: невидимые частицы.................................... 459
2.9.20. Заrрязнение механическими включениями: видимые частицы..................__.................... 462
2.9.22. Определение времени размяrчения липофильных суппозиторИеВ................................. 469
2.9.23. Определение плотности твердых частиц при помощи rазовоro ПИlCНOМетра .................. 470
2.9.25. Высвобождение действующею вещества из лекарственных жевательных резинок ..... 471
2.9.26. Определение удельной площади поверхности методом raэoвoй адсорбции .................473
2.9.27. Однородность массы одной дозы, высвобожденной
из мноroдозовоro контейнера.. ...... ......... ............... .......................................................... .... 477
2.9.29. Характеристическое растворение ...... ........ ...... ...................................................... ............ 477
2.9.31. Определение размера частиц методом дифракции лазерною излучения ..................... 479
2.9.32. Пористость и определение размера пер с ИСПОЛЬЗОванием ртутной порозиметрии...... 484
8
rосударственная фармакопея Республики Беларусь
2.9.33. Определение параметров KPl-1сталлических и частично кристаллических
твердых веществ при помощи .L'.ифракции рентrеновскоro излучения на порошКе........ 487
2.9.34. Насыпная плотность и плотность после усадки ................................................................ 494
2.9.35. Степень измельчения порошков ...... .... .......... ................. ................ ...... ...................... ........ 497
2.9.36. Текучесть порошков ............ .......... ....... .......................... ........ .... ....... ........ ..... ...................... 497
2.9.37. Оптическая микроскопия.... ......... ...... ................ ....... ..................... ...................................... 501
2.9.38. Определение размера частиц методом аналитическоro просеивания............................ 504
2.9.40. Однородность дозированных единиц. ....... .............. .......................... .......................... ....... 508
2.9.41. Истираемость rранул и сфероидов .................................................................................... 512
2.9.42. Тест «Растворение» для липофильных твердых дозированных форм ........................... 513
2.9.43. Наблюдаемое растворение...... ........ ...... ....... ................. .... .... ............. ....... ............ ..... ........ 515
2.9.44. Лекарственные средства для распыления: характеристика ............................................ 515
2.9.45. Смачиваемость пористых твердых материалов, включая порошки ................................ 520
з. Контейнеры.. .... ................. .... ...... .... ....... ...... .......... .......................................... .... 525
3.1. Материалы, используемые для производства контейнеров......................................................... 525
3.1.1. Материалы, используемые для производства контейнеров для человеческой
крови и компонентов крови......... ................ .... ........ ..... ....... ........... .............. ................. ....... 525
3.1.1.1. Материалы на основе пластифицированноrо поливинилхлорида,
используемые для производства контейнеров для человеческой
крови и ее компонентов... ................................................ .................. ........... ......... 525
3.1.1.2. Материалы на основе пластифицированноro поливинилхлорида для
трубок, используемых в комплектах для переливания
крови и компонентов крови ................ ............. .... ............ ........ ............. ..... ............ 530
3.1.3. Полиолефины ........ ................... ....... ....... ............... ............ ...................................... ..... ........ 533
3.1.4. Полиэтилен без добавок для контейнеров для парентеральных
и офтальмолоrических лекарственных средств ................................................................ 538
3.1.5. Полиэтилен с добавками для контейнеров для парентеральных
и офтальмолоrических лекарственных средств ................................................................ 538
3.1.6. Полипропилен для контейнеров и укупорочных материалов
для парентеральных и офтальмолоrических лекарственных средств ............................ 545
3.1.7. Полиэтиленвинилацетат для контейнеров и трубок
для лекарственных средств для парентеральноro питания ............................................. 550
3.1.8. Силиконовое масло, используемое в качестве смазывающей
добавки......................................................................... ......................................................... 553
3.1.9. Силиконовый эластомер для укупорочных средств и трубок........................................... 554
3.1.10. Материалы на основе непластифицированноro поливинилхлорида
для контейнеров для неинъекционных водных растВоров............................................... 556
3.1.11. Материалы на основе непластифицированноrо поливинилхлорида
для контейнеров для твердых лекарственных форм для пероральноro применения... 559
3.1.13. Добавки к пластмассе .......................................................................................................... 561
3.1.14. Материалы на основе пластифицировэнноro поливинилхлорида
для контейнеров для водных растворов для внутривенноro применения ...................... 565
3.1.15. Полиэтилентерефталат для контейнеров для лекарственных средств
для непарентеральноro применения.................... ............. ................................................. 568
3.2. Контейнеры. ............................................................................................................................... ....... 571
3.2.1. Стеклянные контейнеры для фармацевтическоro использования .................................. 571
3.2.2. Пластмассовые контейнеры и укупорочные средства для фармацевтическоro
использования ........ .............................................................................................................. 579
3.2.2.1. Пластмассовые контейнеры для водных растворов для инфузий .................... 580
3.2.3. Стерильные пластмассовые контейнеры для человеческой крови и ее компонентов.. 581
3.2.4. Пустые стерильные контейнеры из пластифицированноro поливинилхлорида
для человеческой крови и ее компонентов ........................................................................ 584
3.2.5. Стерильные контейнеры из пластифицированноro поливинилхлорида
9
для человеческой крови, содержащие раствор антикоаryлянта ...................................... 585
3.2.6. Комплекты для переливания крови и компонентов крови ...........................................0.... 586
3.2.8. Стерильные одноразовые пластмассовые шприцы.......................................................... 588
3.2.9. Резиновые укупорочные средства для контейнеров,
предназначенных для водных лекарственных средств
для парентеральноro применения, порошков
и лиофилизированных порошков....... ........... .................. ............... ......... ....... .......... ........... 590
4. Реактивы .................... ..... ......................................................'.' ........ ..................... 593
4.1. Реактивы, эталонные растворы, буферные растворы ...............................................................0.. 593
4.1.1. Реактивы ...... ......... ............... ....... .................. ............ "о." ..................... .................. ........... .... 594
4.1.2. Эталонные растворы для испытаний на предельное содержание примесей ...............0. 737
.4.1.3. Буферные растворы....................................................................о........................................ 743
4.2. Реактивы, титрованные растворы для объемноro анализ8.........................................................0 750
4.2.1. Исходные стандартные вещества для титрованных растворов....................................... 750
4.2.2. Титрованные растворы........... .......0...:..0...................... ............... ..0.....00............................ о" 751
5. Общие тексты ....................................... ............................ ............................ ....... 761
5.1. Общие тексты по микробиолоrии................................................................00.....00.0......................... 761
5.1.1. Методы приroтовления стерильных продуктов......................................о.........о................. 761
5.1.2. Биолоrические индикаторы стерилизации .....................................0.....0..0.......................... 764
5.1.3. Эффективность антимикробных консервантов ............................0..0....0.........000................ 766
5.1.4. Микробиолоrическая чистота нестерильных лекарственных
средств и фармацевтических субстанций.......... ............ ....... ..........000...0..00........................ 768
5.1.5. Применение fo концепции при стерилизации паром водных
растворов...................... .......... "0" ....... ......................................... ........o......o.o............... ..."" 770
5.1.6. Альтернативные методы контроля микробиолоrической чистоты 000.0...0..00...................... 771
5.1.7. Вирусная безопасность..............................................................................0..0000.............. ..... 789
5.1.8. Микробиолоrическая чистота лекарственных средств растителыюro
происхождения для внутреннеro применения ...........................0.................00.................... 789
5.1.9. Руководство по применению испытания на стерильность ....0...................0...................0.. 791
5.1.10. Руководство по применению испытания на бактериальные ЭНДОТOI<сины ...................... 792
5.2. Общие тексты по биолоrическим продуктам.........................................о..........оооо......................... 796
5.2.1. Общепринятая терминолоrия в статьях на биолоrические продукты...о.......................... 796
5.2.2. Стаи кур, не имеющих конкретных патоrенов и используемых
для производства и контроля качества вакцин.....................................оо.оо........................ 798
5.2.3. Клетки продуценты для производства вакцин для медицинскoro применения .............. 802
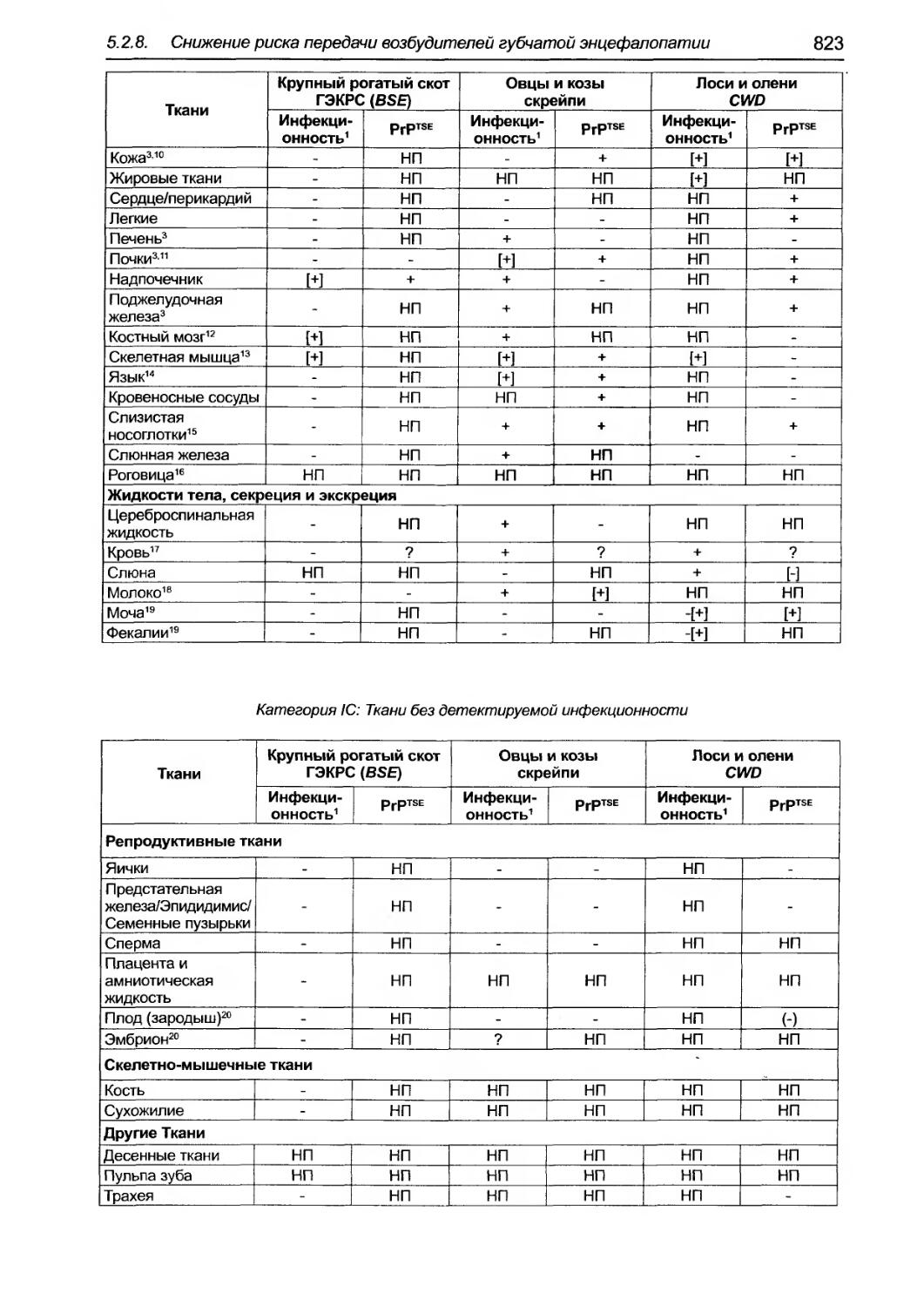
5.2.8. Снижение риска передачи возбудителей rубчатой энцефалопатии животных
при применении медицинских лекарственных средств .....00........0..0.............0................... 807
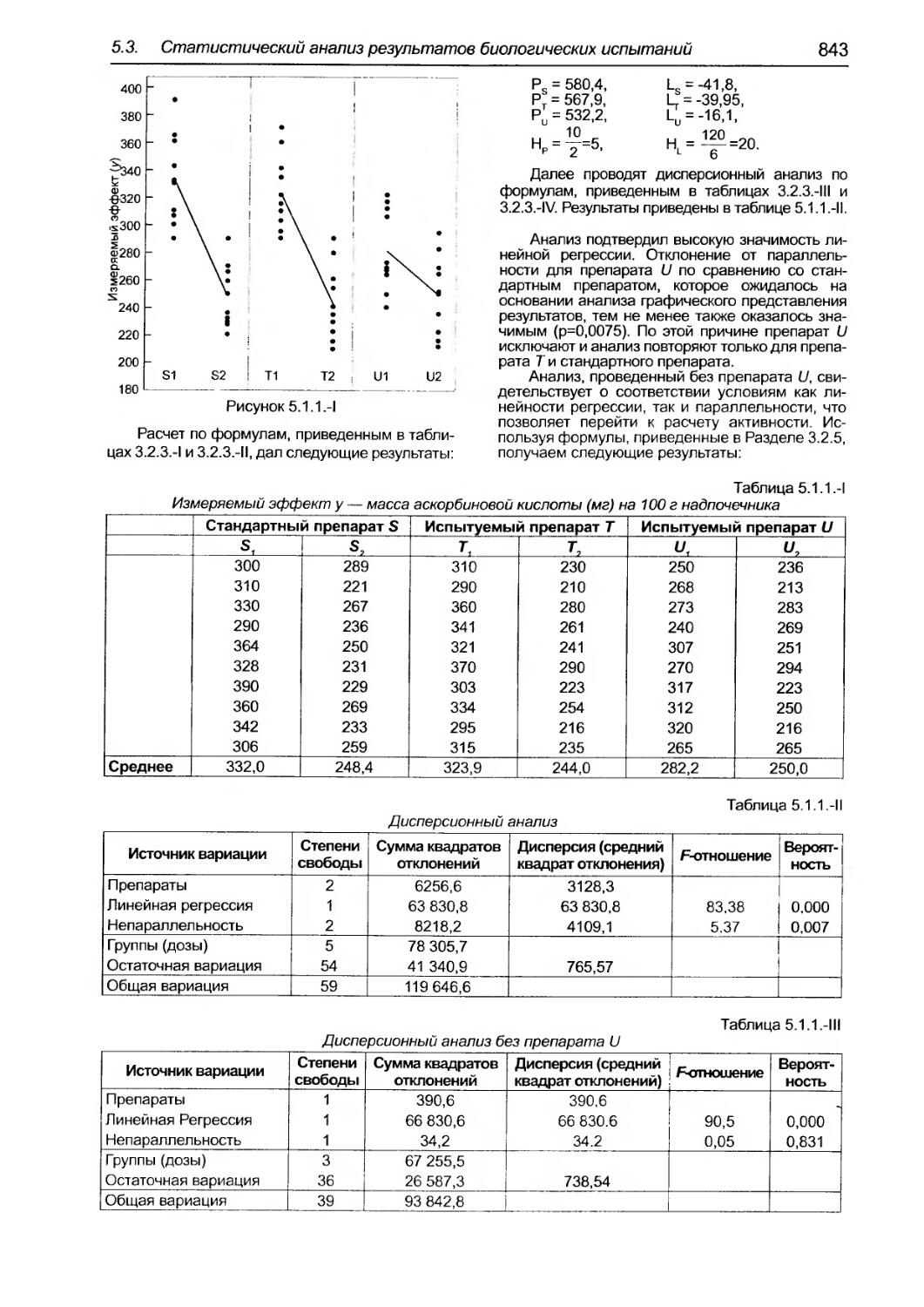
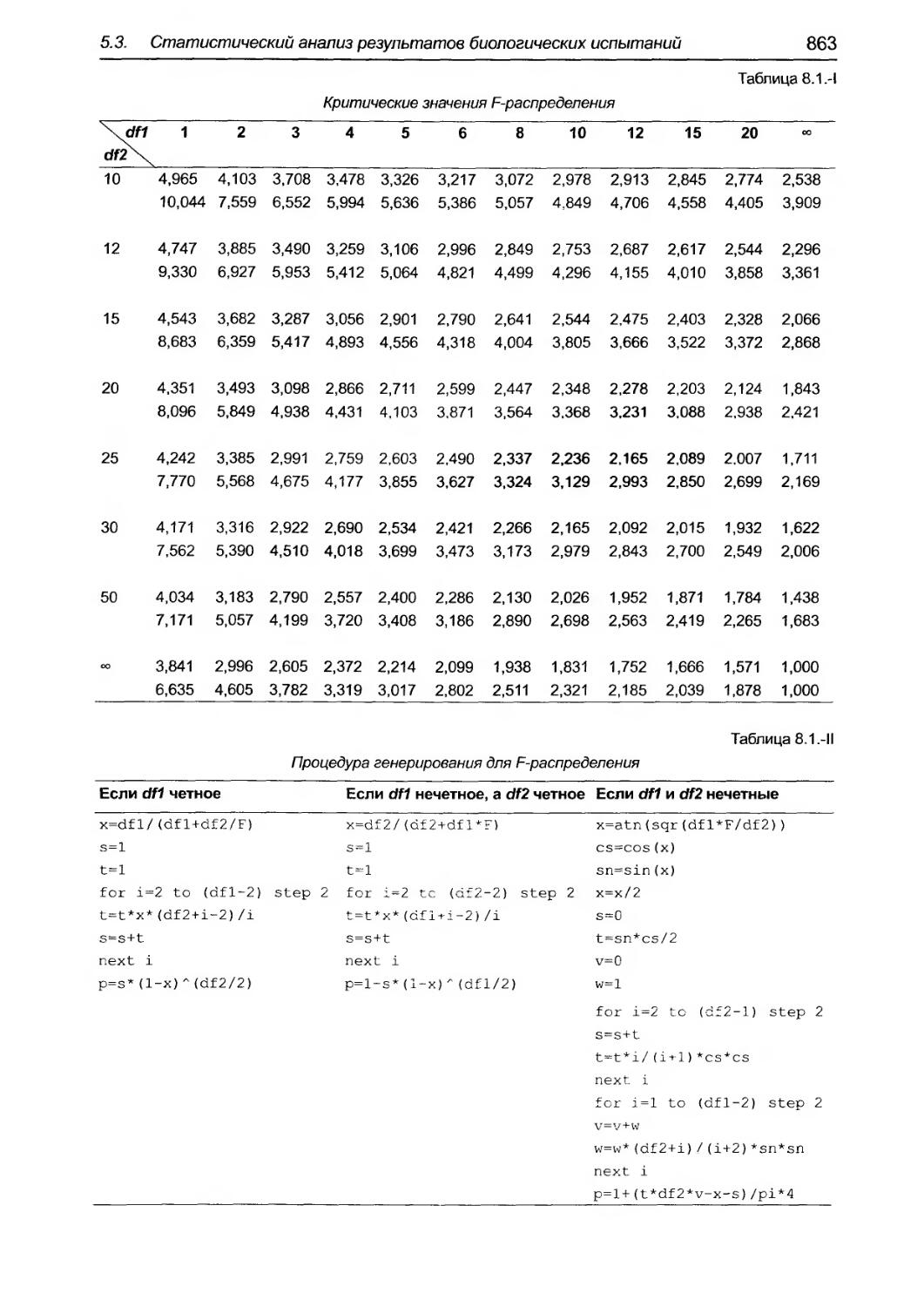
5.3. Статистический анализ результатов биолоrических испытаний и тестов....о............................... 825
#5.3.1. Статистический анализ результатов химическоro экспериМентаоо.о...о...о.о....................... 868
#5.3.2. Валидация аналитических методик и испытаний ..............0.0.0..........0................................ 895
5.4. Остаточные количества орrанических растворителей .........0.......0.000...........000.............................. 908
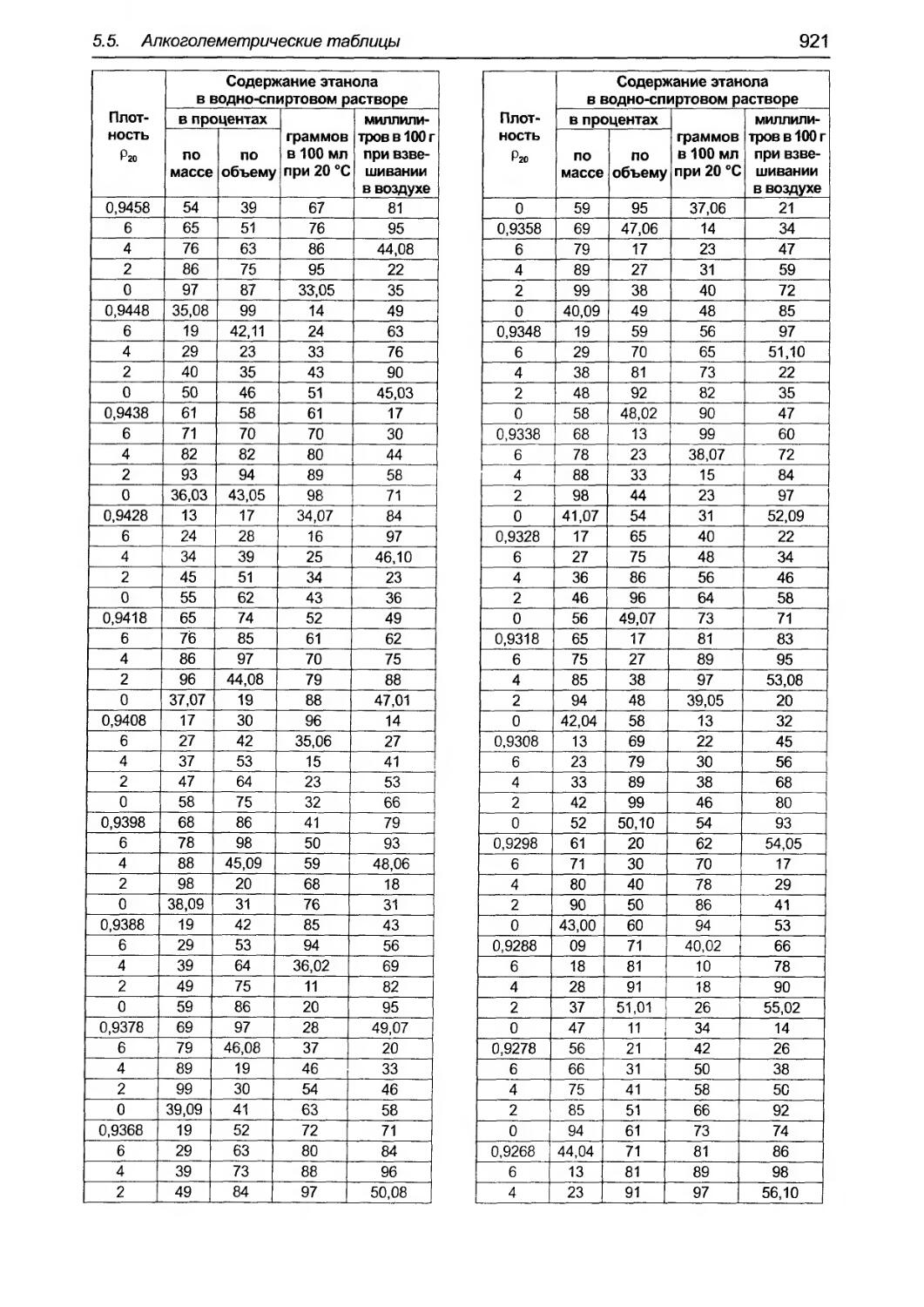
5.5 Алкоroлеметрические таблицы......... ........... .......................... ...... ...... ....... .0. .............................. ..... 918
5.6. Количественное определение интерферонов................... ........ .... ...... .................................. ..... .... 934
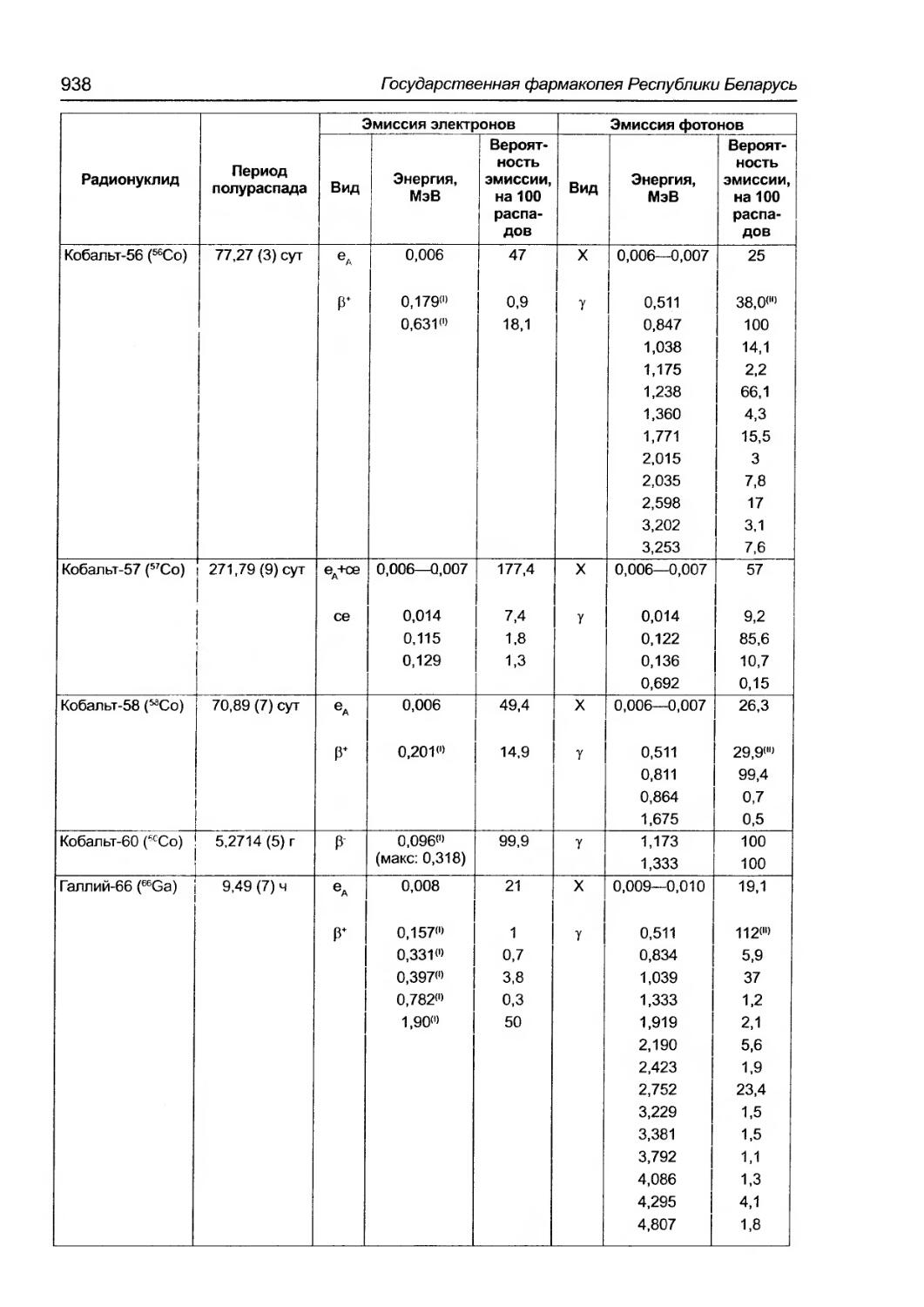
5.7. Таблица физических характеристик радионуклидов, упоминаемых в Фармакопее ................... 937
5.9. Полиморфизм .......... ........... .................. ...0........................ ........ ........0................................ ............... 943
5.10. Контроль примесей в субстанциях для фармацевтическоro использования.............................. 944
5.11. Раздел «Описание (свойства)>> в частных статьях ........................................................................ 949
5.12. Стандартные образllы.... .................................................. ......... ......... ..... ...... ........ ........................... 950
5.14. Лекарственные средства для rенной терапии для медицинскоro применения....................о...... 955
5.15. функционально обусловленные характеристики вспомоrательных веществ.............................. 976
З.3ак.I712
1 О rосударственная фармакопея Республики Беларусь
5.16. Кристалличность.... ............ ........... ..... ...... ...... ..................................... ... ...... ....... .......... ...... '" .......... 980
5.17. Рекомендации по проведению испыта--lИЙ дозированных лекарственных средств.................... 982
5.17.1. Рекомендации по проведению теста «растворение»......................................................... 982
#6. Экстемпоральные лекарственные средства ................................................. 987
#6.1. Приroтовление экстемпоральных лекарственных средств........................................................... 987
#6.1.1. Жидкие лекарственные средства........................................................................................ 987
#6.1.2. Твердые лекарственные средства .................................................................................... 1006
#6.1.3. Мяrкие лекарственные средства....................................................................................... 101 О
#6.1.4. Режимы стерилизации экстемпоральных лекарственных средств ................................ 1012
#6.2. Экспресс анализ экстемпоральных лекарственных средств...................................................... 1013
#6.3. Оценка качества экстемпоральных лекарственных средств...................................................... 1047
#6.3.1. Нормы отклонений. допустимых при изroтовлении лекарственных средств
(в том числе roмеопатических) в . аптеКах........................................................................ 1048
#6.3.2. Нормы отклонений, допустимых при фасовке в аптеках
лекарственных средств промышленноro производства ................................................. 1050
Общие статьи ............... .............................................................................................. 1053
Вакцины для медицинскоrо применения ......... ............................................................................ 1053
Иммунные сыворотки животноro происхождения для медицинскоro применения ..................-. 1058
Лекарственное растительное сырье...................... ....... .... .......... ... .......... ........................ ........ ..... 1061
#Лекарственное растительное сырье цельное
или измельченное фасованное ....... ..................................................... ......................................... 1063
Лекарственные средства на основе аллерrенов ......................................................................... 1064
Моноклональные антитела для медицинскою применения ....................................................... 1068
#Настои, отвары и чаи... .......................... ........... ..... ..... ............. .......................... ............ ........ ...... 1072
Продукты технолоrии рекомбинантной ДНК ................................................................................ 1074
Продукты ферментации......................... ....... ...... ..... ...... .................. ................................ .... .......... 1077
Продукты, которые MOryT быть переносчиками ryбчатой энцефалопатии животною
происхождения ... ...... ......... .................... .... ................. ........ .... ............................ ............... ........... 1079
Радиоактивные фармацевтические препараты ......................................... .................................. 1079
Растительные жирные масла. ...................... ............ ......... .................................................. .......... 1088
Растительные чаи.......... ............................... ...... ..... ........ .... ........................ ...... ..... .......... .......... ... 1090
#Сборы .......... ...... ......................... .... .................... ........ .... ...... .................... ... ........ ................. ......... 1091
#Реrистрационные требования и правила проведения
исследований биодоступности и биоэквивалентности
rенерических лекарственных средств .............. ....... .............................. ........... .................. .......... 1092
Субстанции для фармацевтическоro использования.................................................................. 1130
Экстракты........ ....... ...... ......... .......................... ........ ........ ....... ....... ... ............. .......... ...... ........ .......... 1133
Эфирные масла..... ... .... ............. ....... ............. ..... ...... .................. ......... ...... .............. ............. ..... ..... 1136
Дозированные лекарственные формы ................................................................. 1139
Основные термины и определения.. ..... .... .................... .... ................... ........ ..... .......... .................. 1139
rлазные лекарственные средства.............. ....... .... .............. ...... .............. ................. ......... .... ........ 1140
r ранулы ............................................................................................................................... ............ 1143
#Драже .............................................................................................................................. .............. 1145
Жевательные резинки лекарственные..... .................................. ............. .............. ..... .................. 1145
Жидкие лекарственные средства для внутреннеro применения ............................................... 1146
Жидкие лекарственные средства для наружноro применения................................................... 1150
Капсулы. ....... ............ ....... ...... .......... ... ...... ............. .................... ......... ....... ... ...... ...... ............. .... ... ... 1151
Лекарственные средства ДЛЯ ваrинальною применения ........................................................... 1153
Лекарственные средства для инrаляций ................. ................ .................... ..... ..... ............ ...... ..... 1156
11
Лекарственные средства для орошения...... ................ ................. ...... ......................................... 1162
Лекарственные средства для парентеральноrо применения ..................................................... 1163
Лекарственные средства для ректальноro применения ............................................................. 1166
Лекарственные средства для слизистой оболочки полости рта................................................. 1169
Лекарственные средства, находящиеся под давлением ............................................................ 1173
Мяrкие лекарственные средства для наружною применения.................................................... 1174
Назальные лекарственные средства .............. .......... ....... ................... ............. ..... ........................ 1178
Палочки.............. ...... ............. ............ ........... ......... ...... .... ............. .................................. ................. 1180
Пены медицинские ......... ...... ............ ...... ....... ............. ............................................. ........... ............ 1181
Порошки для внутреннеro применения ........................................................................................ 1182
Порошки для наружноro применения ........................................................................................... 1183
Таблетки..... ....... ... .... ......................... ....... ....... ...... ....... ......... ....... ......... ............. ................... ...... .... 1184
Тампоны медицинские ... .... ................................. ........... ........ ............. ..................... ............ .......... 1188
Трансдермальные пластыри ........... ............. ............. ............ ....... ..................................... .... .... .... 1188
Ушные лекарственные средства ................................................................................................... 1189
rомеопатические лекарственные средства ......................................................... 1193
rомеопатические лекарственные средства.. ............... ...... .... .................. ......... ....... ................. .... 1193
Методы приrотовления roмеопатических базисных препаратов и потенцирование................. 1197
Матричные настойки для roмеопатических лекарственных средств ......................................... 1211
Лекарственное растительное сырье для roмеопатических лекарственных средств................ 1213
12
/осударственная фармакопея Республики Беларусь
Редакционный совет
rосударственной фармакопеи Республики Беларусь
Шеряков А. А. председатель редакционноro совета
Марченко С. И. заместитель председателя, координатор
Члены редакционноrо совета
Общие сведения
Марченко С. И.
Стреха И. С.
Шеряков А А., к. ф. н.
Иванова Н. А.
Иванова Т. Д.
Ковальчук Н. В.
Ковшик А. В.
Кравец М. М.
Ларченко Е. И.
Лежава Т. И.
Лемешевская Т. А
Либерова С. Е.
Марченко С. И.
Мельникова r. r.
Милькевич В. В.
Назарова Е. Ф.
Никифорова Л. Н.
Осипова В. Н.
Плаксицкая Т. д.
Позняк И. А.
Покачайло Л. И., к. Ф Н.
Политова Е. В.
Посконная Л. Б
PorYTbKo И. В.
Рябкова Л. П.
Снарская r. С.
Стреха И. С.
Сыресина Н. В.
Тарасова Т. И.
Торбина Т. Д.
Филиппова r. Н.
Хвеженко О. В.
Хишова О. М., д. ф. н.
Чернявекая А. А., к. Х. н.
Шеряков А. А, к. ф. н.
Шерякова Ю. А.
Методы анализа
Александрова Н. В.
Алексеев Н. А, к. ф. н.
Бузук r. Н., д. ф. н.
Марченко С. И.
Покачайло Л. И., к. ф. н.
Потапнев М. П., д. М. н.
Стреха И. С.
Черношей С. И.
Шеряков А. А., к. ф. н.
Контейнеры
Марченко С. И.
Шеряков А А., к. ф. н.
Реактивы
Зенько Л. А
Коцур О. М.
Марченко С. И.
Стреха И. С.
Шеряков А. А, к. ф. н.
Общие тексты
Марченко С. И.
Рождественский Д. А, к. м. н.
Стреха И. С.
Чернявская А. А, к. Х. н.
Шеряков А. А, к. ф. н.
Общие статьи
Бузук r. Н., д. ф. н.
rолик r. И.
Марченко С. И.
Рождественский Д. А, к. М. н.
Стреха И. С.
Шеряков А. А., к. ф. н.
Экстемпоральные лекарственные
средства
Аrафонова И. В.
Артемьева О. И.
Будай А. И.
Валюкевич Я. М.
Власик Е. Л.
Войтюль Е. И.
rалаrанова т. r.
rолик r. И.
rрибанова Б. П.
Демидова О. Н.
Евдокимова Л. Н.
Закацура Л. Ф.
Залесская С. В.
Захарова Т. В.
Зейдина Т. В.
Дозированные лекарственные
формы
rолик r. И.
Марченко С. И.
Стреха И. С.
Шеряков А А., к. ф. н.
rомеопатические лекарственные
средства
Стреха И. С.
Шеряков А А., к. ф. Н.
13
Список авторов
Аrафонова И. В.
Александрова Н. В.
Алексеев Н. А., к. ф. н.
Артемьева О. И.
Будай А. И.
Бузук r. Н., д. ф. н.
Валюкевич Я. М.
Власик Е. Л.
Войтюль Е. И.
rалаrанова т. r.
repMaHeHKo Е. В.
rолик r. и.
rрибанова В. П.
Демидова О. Н.
Евдокимова Л. Н.
Закацура Л. Ф.
Залесская С. В.
Захарова Т. В.
Зейдина Т. В.
Зенько Л. А.
Иванова Н. А.
Иванова Т. Д.
Кенькова Н. Н.
Ковальчук Н. В.
Ковшик А. В.
Кравец М. М.
Ларченко Е. И.
Лежава Т. И.
Лемешевская Т. А.
Либерова С. Е.
Марченко С. И.
Мельникова r. r.
Милькевич В. В.
Назарова Е. Ф.
Никифорова Л. Н.
Осипова В. Н.
Плаксицкая Т. д.
Позняк И. А.
Покачайло Л. И., к. ф. н.
Политова Е. В.
Посконная Л. Б.
Потапнев М. П., д. м. н.
POryTbKO И. В.
Рождественский Д. А., к. м. н.
Рябкова Л. П.
Сеткина С. Б.
Снарская r. С.
Стреха и. С.
Сыресина Н. В.
Тарасова Т. И.
Торбина Т. д.
Филиппова r. Н.
Хвеженко О. В.
Хишова О. М., д. ф. н.
Черношей С. И.
Чернявская А. А., к. Х. н.
Шеряков А. А., к. ф. н.
Шерякова Ю. А.
4 3a1l 1712
14
rосударственная фармакопея Республики Беларусь
Список орrанизаций, учреждений и предприятий
Республики Беларусь, принимавших участие в разработке
rосударственной фармакопеи Республики Беларусь
РУП «Центр экспертиз и испытаний в здравоохранении»
Уа Витебский roсударственный медицинский университет
РУП «Белмедпрепараты»
ОАО «Борисовский завод медицинских препаратов»
000 «Фармтехнолоrия»
УП «Минскинтеркапс»
РУП «Несвижский завод медицинских препаратов»
АДА «Фармалэнд»
rY «Республиканский научно практический центр трансфузиолоrии и медицинских
биотехнолоrий»
rY «Республиканский научно практический центр эпидемиолоrии и микробиолоrии»
Отдел контроля качества аптечноro склада ТП РУП «БелФармация»
Контрольно аналитическая лаборатория ТП РУП «Минская Фармация»
Контрольно аналитическая лаборатория Брестскоro ТП РУП «Фармация»
Контрольно аналитическая лаборатория Витебскоrо ТП РУП «Фармация»
Контрольно аналитическая лаборатория rомельскоro ТП РУП «Фармация»
Контрольно аналитическая лаборатория rродненскоro ТП РУП «Фармация»
Контрольно аналитическая лаборатория Моrилевскоro ТП РУП «Фармация»
Введение
15
ВВЕДЕНИЕ
Во втором издании 1 20 тома rосудар
ственной фармакопеи Республики Беларусь в
новой редакции приведены все общие статьи
и тексты, опубликованные в первом издании
Фармакопеи (тома 1, 2, 3).
Введены новые общие статьи и тексты:
2.2.57. Атомно эмиссионная спектроме
трия с использованием индуктивно связанной
плазмы:
2.2.58. Масс спектрометрия с использова
нием индуктивно связанной плазмы;
2.2.59. Анализ 2ликанов в 2ликопротеинах;
2.2.60. Температура плавления иHcтpy
ментальный метод;
2.4.29. Состав жирных кислот в маслах,
об02ащенных OMe2a 3 кислотами;
2.4.31. Никель в 2идР02енизированных pac
тительных маслах;
2.4.32. Общий холестерин в маслах, об02а
щенных OMe2a 3 кислотами;
2.5.35. Азота закись в 2азах;
2.5.37. Метил , этил и изопропилметан
сульфонат в метансульфоновой кислоте;
2.5.38. Метил , этил и изопропилметан
сульфонат в фармацевтических субстанциях;
2.5.39. Метансульфонилхлорид в MeтaH
сульфоновой кислоте;
#2.5.50. Титрование в неводных pacтвo
рителях (изменение номера статьи, ранее
#2.5.35);
2.6.26. Испытание на aHти O антитела в
иММУН02лобулине человека для внутривеНН020
введения;
2.6.30. Испытание на активацию моноцитов;
2.6.31. МикробиОЛ02ические испыта
ния лекарственных средств растительно
20 происхождения для внутренне20 приме
нения;
2.7.22. Количественное определение фак
тора свертывания крови Х/ человека;
2.7.23. Подсчет клеток СО34/СD45+ в 2e
мопоэтических продуктах;
2.7.24. Проточная цитометрия;
2.7.25. Определение содержания иН2ибито
ра плазмина человека;
2.7.27. Значение флоккуляции (Lf) токсинов
и анатоксинов дифтерии и столбняка (проба
Рамона);
2.7.28. Определение количества 2емопоэ
тических клеток предшественников человека
по колониеобразующим клеткам;
2.7.29. Определение количества и жизне
способности ядросодержащих клеток;
2.7.30. Количественное определение чело
вечеСК020 белка с;
2.7.31. Количественное определение чело
вечеСК020 белка s;
2.7.32. Количественное олределение ИН2И
битора а 1 протеиназы человека;
#2.7.50. Определение биОЛ02ической aKтив
ности инсулина (изменение номера статьи,
ранее #2.7.22);
2.8.2. Примеси (изменение номера статьи,
ранее #2.8.2);
2.8.20. Лекарственное растительное
сырье: отбор проб (взамен статьи #2.8.19.
Правила приемки и методы отбора проб);
2.8.22. Определение охра токсина А в ле
карственном растительном сырье;
2.8.23. #Макроскопический и# микроскопи
ческий анализ лекарствеНН020 раститель
Н020 сырья (взамен статьи #2.8.3. Техника
макроскопичеСК020 и микроскопичеСК020 aHa
лиза);
2.9.29. Характеристическое растворение;
2.9.32. Пористость и определение разме
ра пор с использованием ртутной порозиме
трии;
2.9.34. Насыпная плотность и плотность
после усадки;
2.9.35. Степень измельчения порошков;
2.9.41. Истираемость 2ранул и сфероидов;
2.9.42. Тест «Растворение» для липофиль
ных твердых дозированных форм:
2.9.43. Наблюдаемое растворение:
2.9.44. Лекарственные средства для pac
пыления: характеристика:
2.9.45. Смачиваемость пористых твердых
материалов, включая порошки;
5.1.6. Альтернативные методы контроля
микробиОЛ02ической чистоты;
5.1.7. Вирусная безопасность;
5.1.8. МикробиОЛ02ическая чистота пе
карственных средств растuтеЛЬН020 про
исхождения для внутренне20 применения;
5. 1.9. Руководство по применению испыта
ния на стерильность;
5. 1. 10. Руководство по примененuю испы
тания на бактериальные эндотоксины:
5.3. Статистический анализ результатов
биОЛ02ических испытаний и тестов (измене
ние номера статьи, ранее 5.3.1):
#5.3.1. Статистический анализ резуль
татов химичеСК020 эксперимента (изменение
номера статьи, ранее #5.3.2);
#5.3.2. Валидация аналитических Meтo
дик и испытаний (изменение номера статьи,
ранее #5.3.3);
5.14. Лекарственные средства для 2енной
терапии для медициНСК020 применения;
5. 15. Функционально бусловленные xapaK
теристики вСПОМ02ательных веществ;
5.16. Кристалличность:
5.17.1. Рекомендации по проведению теста
«Растворение»;
#6.1.4. Режимы стерилизации экстемпо
ральных лекарственных средств;
Моноклональные антитела для MeдициH
СК020 применения (2031);
Продукты теХНОЛО2ии рекомбинантной
ДНК (0784);
16
rосударственная фармакопея Республики Беларусь
#Ре2истрационные требования и прави
ла проведения исследований биодоступн()стu
и биоэквивалентностu zенерических леКбр
ственных средств (РБОО05) (изменение номера
статьи, ранее #5.8);
Эфирные масла (2098);
Методы при20товления 20меопатических
базисных препаратов и потенцирование (2371).
Исключены следующие общие статьи:
#2.4.29. Цинк;
#2.4.30. Ле2кооБУ2ливающиеся вещества;
#2.5.35. Титрование в неводных pacтвopи
телях (изменение номера статьи на #2.5.50);
2.6.3. Испытание на посторонние вирусы
с использованием куриных эмбрионов (oтcyт
ствует в Европейской Фармакопее);
2.6.4. Испытание на вирусы лейкоза (oт
сутствует в Европейской Фармакопее);
2.6.5. Испытание на посторонние вирусы
с использованием клеточных культур (oтcyт
ствует в Европейской Фармакопее);
2.6.6. Испытание на посторонние а2енты
с использованием цыплят (отсутствует в Eв
ропейской Фармакопее);
2.7.3. Количественное определение Kopти
котропина (отсутствует в Европейской Фар
макопее);
#2.7.22. Определение биОЛО2ической aK
тивности инсулина (изменение номера статьи
на #2.7.50);
#2.8.2. Допустимые примеси (изменение
номера статьи на 2.8.2);
#2.8.3. Техника макроскопичеСК020 и MиKpO
скопичеСК020 анализа (новая редакция, статья
2.8.23);
#2.8.18. Определение содержания экстрак
тивных веществ;
#2.8.19. Правила прuемкu u методы отбора
проб (новая редакция, статья 2.8.20);
#2.8.20. Определение содержания токсиче
ских веществ методом атомно абсорбционной
спектроскопии (см. статью 2.4.27);
#2.8.22. Определение содержания paдиOHY
клидов;
2.9.13. Определение размера частиц Meтo
дом микроскопии (см. статью 2.9.37);
2.9.15. Насыпной объем (см. статью
2.9.34);
2.9.24. Устойчивость суппозиториев и
пессариев к разрушению (отсутствует в Eв
ропейской Фармакопее);
2.9.28. Определение массы или объема
содержиМ020 контейнера для жидких и МЯ2ких
лекарственных средств (см. общие статьи
на жидкие и МЯ2кие дозированные формы,
статья отсутствует в Европейской Фарма
копее) ;
5.2.6. Оценка безопасности вакцин (oтcyт
ствует в Европейской Фармакопее);
5.2.7. Оценка эффективности вакцин (oт
сутствует в Европейской Фармакопее);
5.3.1. Статистический анализ резуль
татов биОЛ02ических исследований и коли
чественнЬ/х определений (изменение номера
статьи на 5.3);
#5.3.2. Статистический анализ резуль
татов химичеСК020 эксперимента (изменение
номера статьи на #5.3.1);
#5.3.3. Валидация аналитических Meтo
дик испытаний (изменение номера статьи на
#5.3.2).
Общие сведения
17
1. ОБЩИЕ СВЕДЕНИЯ
1.1. ОБЩИЕ ПОЛОЖЕНИЯ
#Положения rлавы «Общие сведения» pac
пространяются на все общие статьи, частные
статьи и друrие материалы rосударственной
фармакопеи Республики Беларусь.
В материалах rосударственной фармако
пеи Республики Беларусь слово «Фармакопея»
без уточнений подразумевает rосударственную
фармакопею Республики Беларусь. Наряду с
этим для обозначения rосударственной фарма
копеи Республики Беларусь может быть исполь
зова но также официальное сокращение rФ РБ.
Все частные и общие статьи ФармакопеЙ
rармонизированы с Европейской Фармакопеей и
построены в следующем формате:
адаптированный перевод соответствую
щеro материала Европейской Фармакопеи;
национальные общие и частные статьи,
дополнительные испытания, информационные
и иные материалы отмечены значком «#» перед
названием rлавы, статьи, раздела, пункта на
которые он распространяется; текст, отличаю
щийся от текста Европейской Фармакопеи, OT
мечается между двумя значками «#», например,
#контрольный опыт#;
статьи и нормы на лекарственное расти
тельное сырье, соответствующие требованиям
Европейской Фармакопеи, обозначены значком
« ЕФ ».#
Ссылка в материалах Фармакопеи на какую
либо статью и/или ее раздел означает, что про
дукт соответствует требованиям этой статьи. Ha
звание статьи, на которую дается ссылка, и/или
ее номер выделены курсивом.
rOToBoe лекарственное средство должно co
ответствовать требованиям Фармакопеи на про
тяжении срока rодности. Компетентный упол
номоченный opraH может принять решение о
необходимости установления отдельноro срока
roдности и/или требования спецификации для
лекарственноro средства во вскрытом контейне
ре. Объект любой частной статьи должен COOT
ветствовать установленным требованиям в Te
чение всеro периода еro использования. Срок
roдности и дата, с которой он должен отсчиты
ваться, соrласовываются компетентным упол
номоченным opraHoM на основании эксперимен
тальных исследований по стабильности данноro
roтовоro продукта.
Требования частной статьи являются обя
зательными, если нет специальных указаний в
разделе «Общие сведения» или в данной част
ной статье. Общие статьи становятся обязатель
ными, Korдa на них при водится ссылка в той или
иной частной или общей статье, если только спе
циально не указано, что ссылка приводится ис
ключительно как информация или рекомендация.
Действующие вещества (фармацевтические
субстанции), вспомоrательные вещества, roто
вые лекарственные средства и друrие продукты,
описываемые в частных статьях Фармакопеи,
предназначены для использования в медицине.
Качество продукта является фармакопейным
только при ero соответствии всем требованиям
фармакопейной статьи. Это условие не подраз
умевает необходимость выполнения производи
телем всех испытаний, описанных в статье, при
оценке соответствия требованиям Фармакопеи
при выпуске в обращение. Для подтверждения
соответствия продукта требованиям Фармакопеи
производитель может использовать данные, по
лученные, например, в ходе валидации производ
cTBeHHoro процесса и внутрипроизводственноrо
контроля. Таким образом, требование COOTBeT
ствия продукта Фармакопее может выполняться
и при выходном контроле в виде выпуска по па
раметрам (parametric ,e/ease) при наличии разре
шения компетентноro уполномоченноro opraHa.
Субстанции, вспомoraтельные вещества и
друrие продукты, на которые распростраНЯIQТСЯ
требования Фармакопеи, мoryт иметь различные
параметры качества в зависимости от цели ис
пользования. Если на этот счет нет указаний в co
ответствующей частной стаТЬе, ее требования pac
пространяются на продукт независимо от целей еro
применения. В некоторых случаях, в частности, в
случае вспомоrательных веществ, для информа
ции частная статья может быть дополнена списком
функционально обусловленных характеристик, KO
торые являются важными для использования дaH
ноro вещества. Также в информационных целях
MOryr быть приведены методики контроля одной
или нескольких таких характеристик.
Системы качества. Стандарты качества.
установленные в фармакопейных статьях, при
менимы к продукту только при условии еro про
изводства в рамках соответствующей системы
обеспечения качества.
Общие статьи. Субстанции и лекарствен
ные средства, описанные в частных статьях,
также должны выдерживать требования соот
ветствующих подходящих общих статей. В част
ных статьях обычно не указывают перекрестные
ссылки на общие фармакопейные статьи.
Действие общих статей распространяется на
все субстанции и лекарственные средства, YKa
занные в разделе «Определение» общей статьи,
за исключением случаев, коrда вводная часть
оrраничивает ее применение, например, только
для субстанций и лекарственных средств, опи
санных в этой фармакопейной статье.
Требования общих статей на лекарственные
формы распространяются на все лекарствен
ные средства, изrотовленные в виде этой лекар
ственной формы. Для конкретноro лекарствен
ноro средства требования соответствующей
общей статьи не обязательно являются исчер
18
rосударственная фармакопея Республики Беларусь
пывающими, и компетентный уполномоченный
opraH может ввести дополнительные требова
ния, помимо указанных в статье.
Общие и частные статьи являются взаимо
дополняемыми. Если требования общей статьи
неприменимы к определенному лекарственному
средству, это однозначно указывается в частной
статье.
Валидация фармакопейных методик. Me
тодики, приведенные в частных и общих статьях,
были валидированы в соответствии с общепри
нятой научной практикой и современными peKO
мендациями по валидации аналитических MeTO
дик. Если иное не указано в частной или общей
статье, валидация методик испытания аналити
ком не требуется.
#Испытания и методики определения, приве
денные в Фармакопее, являются официальными
методиками, однако по соrласованию с компетент
ными уполномоченными орrанами MOryт использо
ваться и друrие методики, при условии. что они дают
результаты, соответствующие фармакопейным Me
тодикам. В случае сомнений или разноrласий реша
ющей является фармакопейная методика."
Общепринятые термины. #Термин «компе
тентный уполномоченный opraH» означает Ми
нистерство здравоохранения Республики Бела
русь и Республиканское унитарное предприятие
«Центр экспертиз и испытаний в здравоохране
нии» В соответствии с ero компетенцией.#
Выражение «если нет друrих указаний в
частной статье» #(под частной статьей под
разумевается частная статья Фармакопеи на
субстанцию или нормативный документ по
контролю качества (НД), утвеР '1енный уполно
fv10ченным opraHoM)# означает, что требования
общей статьи должны быть выполнены, за ис
ключением случаев, коrда компетентный упол
номоченный opraH соrласовал (утвердил) изме
нения или исключения для таких требований,
что указывается в частной статье.
Положения со словом «следует» носят peKO
мендательный или информационный характер.
В некоторых общих и частных статьях Фар
макопеи при описании реактива, микроорrаниз
ма, методики и т. д. используется термин «подхо
дящий». Если при этом критерии их приroдности
не сформулированы, то приroдность конкретных
реактивов, методик и т. д., используемых в част
ной статье, должна быть обоснована перед KOM
петентным уполномоченным opraHoM.
Лекарственное средство. #Вещество или
комбинация веществ природноrо, синтетическо
ro или биотехнолоrическоrо происхождения, ()б
ладающие фармаколоrической активностью и в
определенной лекарственной форме применяе
мые для профилактики и диаrностики заболева
ний, лечения и медицинской реабилитаuии па
циентов, предотвращения беременности путем
BHYTpeHHero или внешнеro применения.#
Лекарственное средство раститеЛЬН020
происхождения. Лекарственное средство, co
держащее в качестве действующеro веще
ства (действующих веществ) только лекар
ственное растительное сырье или продукты
из лекарственноro растительноro сырья или
лекарственное растительное сырье в комби
нации с продуктами из лекарственноro расти
тельноrо сырья.
#Продукт из лекарствеНН020 растительно
20 сырья однородный продукт, полученный из
лекарственноro растительноro сырья путем экс
тракции, переroнки, отжима, фраrментирова
ния, очистки, концентрирования или фермента
ции. Продукты из лекарственноro растительноro
сырья включают, например, экстракты, эфир
ные масла, соки, эксудаты и лекарственное pac
тительное сырье, измельченное для специфи
ческоro применения, например, измельченное
лекарственное растительное сырье для после
дующей фасовки, приrотовления чаев и сборов,
капсулирования или таблетирования. Продукт
из лекарственноro растительною сырья может
представлять собой фармацевтическую суб
станцию, промежуточный продукт или roтовое
лекарственное средство.
#Лекарственное растительное сырье. Ис
пользуемые для промышленноrо производства,
аптечноro изroтовления лекарственных средств
цельные лекарственные растения или части ле
карственных растений, на которые имеются co
ответствующие фармакопейные статьи.
Фармацевтическая субстанция (деЙству
ющее вещество). "Вещество или комбинация
нескольких веществ природноro, синтетическо
ro или биотехнолоrическоro происхождения, об
ладающие фармаколоrической активностью.
используемые для промышленноrо производ
ства, аптечною изroтовления лекарственных
средств.# Такие вещества предназначены для
оказания фармаколоrическоro или иноrо непо
средственноrо действия при диаrностике, лече
нии и профилактике заболеваний. или для воз
действия на opraHbJ и функции орrанизма.
ВСПОМ02ательное вещество. #Вещество
или комбинация нескольких веществ, не обла
дающих фармаколоrической активностью и ис
пользуемых в процессе промышленноrо произ
водства, аnтечноro изrотовления лекарственноro
средства для придания ему определенной ле
карственной формы.# Например, вспомоrатель
ными веществами являются стабилизаторы,
антимикробные консерванты, антиоксиданты,
растворители, адъюванты.
1.2. друrИЕ ПОЛОЖЕНИЯ, РАСПРОСТРА
НЯЮЩИЕСЯ НА ОБЩИЕ И ЧАСТНЫЕ ФАРМА
КОПЕЙНЫЕ СТАТЬИ
Количество вещества. При описании коли
чественноrо определения или испытания с чис
лен но заданными пределами, количество веще
ства, необходимое для проведения испытания.
может отклоняться в пределах :t1 О % от указан
Общие сведения
19
ноro количества. Необходимо взять точную Ha
веску анализируемою вещества (или отмерить
ero каким либо друrим способом) и все вычисле
ния производить для этоro точноrо количества.
Если пределы испытания заданы не числен
но, а определяются путем сравнения со CTaH
дартом при тех же условиях, для испытания
берут указанное количество вещества. Реакти
вы Bcerдa берут в указанных количествах.
Количества вещества взвешивают или OTMe
ривают с точностью. соответствующей указанной
степени прецизионности. Точность взвешивания
должна быть ж5 единиц после последней YKa
занной цифры (например, навеска 0.25 r может
находиться в пределах от 0,245 r до 0,255 r).
Объемы отмеривают следующим образом: если
после десятичной запятой стоит ноль или число,
заканчивающееся ноль (например, 10,0 мл или
0,50 мл), требуемый объем отмеривают с по
мощью пипетки, мерной колбы или бюретки. В
остальных случаях можно использовать rрадуи
рованный мерный цилиндр или rрадуированную
пипетку. Микролитры отмеривают с помощью
микропипетки или микрошприца.
Тем не менее, в некоторых случаях точ
ность. с которой указаны количества вещества,
не совпадает с количеством значащих цифр,
указанных в заданных числовых пределах. В
таких случаях взвешивания и отмеривания про
водят с существенно более высокой точностью.
Оборудование и аналитические опера-
ции. Стеклянная мерная посуда должна COOT
ветствовать требованиям класса А Международ
ноro стандарта, выпущенноro Международной
орrанизацией по стандартизации (/50). "или Ha
циональноrо стандарта Республики Беларусь".
Аналитические операции, если нет друrих
указаний, осуществляют при температуре от
15 ос до 25 ОС.
Сравнительные испытания, если нет друrих
указаний, проводят с использованием идентичных
пробирок из бесцветною прозрачноro нейтрально
ro стекла с плоским основанием и внутренним диа
метром 16 мм, так как указываемые объемы жидко
стей рассчитаны ДПЯ этоro диаметра; однако, при
условии корректировки объе мов, MOryт быть ис
пользованы также пробирки с большим внутренним
диаметром (2.1.5). Сравнивают равные объемы
жидкостей оль вертикальной оси пробирки на
белом (или, при необходимости, на черном) фоне.
Испытания проводят в рассеянном свете.
Если для проведения испытания или количе
ственноro определения требуется использовать
растворитель с растворенным в нем индикато
ром и при этом не предусмотрен контропьный
опыт, этот растворитель предварительно ней
трализуют по этому индикатору.
#Под контрольным опытом подразумева
ют определение, проводимое с теми же количе
ствами реактивов и в тех же условиях, но без ис
пытуемоro образца.#
Водяная баня. Если не указана вода с
друroй температурой, то подразумевается баня
с кипящей водой. Можно использовать и друrие
способы наrревания, если они rарантированно
обеспечивают температуру, близкую, но не пре
восходящую 100 ос (или друryю указанную TeM
пературу).
#Ледяная баня. Подразумевается баня с TeM
пературой О ОС. Если необходимо охлаждение до
более низкой температуры, применяют смесь льда
с некоторыми электролитами (соли, кислоты).
Высушивание и прокаливаниедопостоян-
ной массы. Результаты двух последних взвеши
ваний должны отличаться не более чем на 0,5 Mr;
интервал времени между двумя взвешиваниями
определяется свойствами и количеством BЫCY
шиваемоrо/прокаливаемоrо остатка.
В тех случаях. Korдa требуется высушивание
«в эксикаторе» или «в вакууме», оно осущест
вляется в соответствии с условиями, описанны
ми в статье 2.2.32. Потеря в массе при высуши
вании.
Реактивы. Надежность результатов, полу
чаемых с помошью описанных в фармакопей
ных статьях аналитических операций, зависит, в
частности, от качества используемых реактивов.
Реактивы описаны в rлаве 4. Реактивы. Под
разумеваемая степень чистоты не ниже KBa
лификации «аналитической чистоты» (aпa/ytica/
grade) Пили квалификации «чистый для анали
за» (ч. д. а.)#. длq некоторых реактивов включе
ны испытания для определения приrодности.
Растворители. Если для растворов не
Уf.;эзан растворитель, то подразумевают водные
оастворы.
Для проведения описанных в фармакопей
НЫХ статьях аналитических операций и для при
ютовления реактивов используют воду, COOTBeT
ствующую требованиям частной статьи «Вода
очищенная», за исключением случаев, Korдa
требования по содержанию бактериальных эн
дотоксинов (Вода очищенная ((iп bu/k») или по
микробиолоrической чистоте (Вода" очищенная
в контейнерах) не являются существенными.
Под термином «вода дистиллированная» пони
мают воду очищенную, полученную путем дис
тилляции.
Термин «этанол» без уточнений означает
абсолютный этиловый спирт. Термин «спирт»
без уточнений означает 96 % эта нол. Друrие
степени разбавления обозначаются термином
«этанол» или «спирт» С указанием содержания
этанола (С 2 Н 6 О) в объемных процентах.
#Термин «эфир» без уточнений означает ди
этиловый эфир.#
Способы выражения концентрации. Bыpa
жение «%» может иметь одно из трех значений:
20
rосударственная фармакопея Республики Беларусь
массовый процент (м/м) число rpaM
мов вещества в 100 rpaMMax конечноro про
дукта;
объемный процент (об/об) число мил
лилитров вещества в 100 миллилитрах конеч
ноro продукта;
#массо объемный про цент (м/об)
число rpaMMoB вещества в 100 миллилитрах
конечноro продукта#.
Если не указано иноro, обозначение
«ррт» (частей на миллион) подразумевает
массовое соотношение.
#Если указано, что при приroтовлении
смеси растворителей их берут в соотношении
(а:Ь), то имеется в виду соотношение объе
мов. Например, соотношение reKcaH бензол
(1 :З) означает, что смешивают 1 объем reKca
на с З объемами бензола.#
Температура. Кроме KOHKpeTHoro указа
ния температуры, при проведении испытаний
используют также следующие термины:
rлубокое охлаждение ниже 15 ос;
в холодильнике от 2 ос до 8 ОС;
в холодном или прохладном месте от
8 ос до 15 ОС;
при комнатной температуре от 15 ос до
25 ОС.
#Кроме терминов, приведенных выше,
MorYT использоваться также следующие Tep
мины:
теплый (от 40 ос до 50 ОС);
rорячий (от 80 ос до 90 ОС);
температура «водяной бани» от 98 ОС
до 100 ОС;
температура «ледяной бани» (О ОС).#
1.З. ОБЩИЕ ФАРМАКОПЕЙНЫЕ СТАТЬИ
Контейнеры. Материалы, используемые
для контейнеров, описаны в rлаве 3.1. Для
материалов, используемых для производ
ства контейнеров, особенно для полимерных
материалов, используют общие названия,
каждое из которых охватывает ряд материа
лов, отличающихся как свойствами OCHOBHOro
компонента, так и используемыми добавками.
Испытания и пределы нормирования зависят
от конкретноro состава материала и, таким
образом, применимы только при условии,
что материал соответствует вводной части
ero спецификации. По соrласованию с компе
тентным уполномоченным opraHoM MorYT ис
пользоваться материалы друrих составов, а
также испытания для них.
Спецификации на контейнеры, включен
ные в rлаву 3.2, разрабатывались для всех
контейнеров указанной катеrории. Однако,
учитывая большое разнообразие существу
ющих контейнеров и возможность появления
новых контейнеров, публикация специфика
ции не исключает возможности использова
ния контейнеров, соответствующих друroй
спецификации, если это обосновано и соrла
сова но с компетентным уполномоченным op
raHOM.
В статьях Фармакопеи MorYT даваться
ссылки на определения и спецификации KOH
тейнеров, приведенные в rлаве 3.2. Контей
неры. В разделах «Определение» или «Про
изводство» общих статей на лекарственные
формы может содержаться требование по ис
пользованию определенноro типа контейне
ра. В разделе «Хранение» некоторых статей
может указываться тип рекомендуемоro KOH
тейнера.
1.4. ЧАСТНЫЕ ФАРМАКОПЕЙНЫЕ СТАТЬИ
НА3ВАНИЯ
#Кроме названий на русском языке, при
водятся также анrлийское и латинское назва
ния.#
ОТНОСИТЕЛЬНЫЕ АТОМНЫЕ И
МОЛЕКУЛЯРНЫЕ МАССЫ
Относительную атомную массу (А. м.) или
относительную молекулярную массу (м. м.)
указывают, при необходимости, в начале
частной фармакопейной статьи. Относитель
ную атомную массу, относительную моле
кулярную массу, молекулярную формулу и
rрафическую формулу приводят как инфор
мационный материал.
РЕrИСТРАЦИОННЫЕ НОМЕРА CAS
Реrистрационные номера Химической pe
феративной службы CAS (Chemica/ Abstracts
Se,vice) MorYT включаться для информации в
частные фармакопейные статьи, rдe это при
менимо, с целью предоставления пользовате
лю удобноro доступа к полезной инфрмации.
Реrистрационный номер CA (CAS Regist,y
NubefFJ) является зареrистрированной торro
вой маркой Американскоro Химическоro Об
щества (Americaп Chemica/ Society).
ОПРЕДЕЛЕНИЕ
В разделе «Определение», идущей после
названия частной статьи, при водится офици
альное определение субстанции, roTOBOro ле
карственноro средства или иноro продукта"
являющеroся предметом частной статьи.
Пределы содержания. Если указаны
пределы содержания, то это пределы, 11OЛУ
ченные с использованием метода, указанно
ro в разделе «Количественное определение».
Лекарственное растительное сырье. В
частных статьях на лекарственное раститель
ное сырье в разделе «Определение» указыва
Общие сведения
21
ют на предмет частной статьи. Это может быть,
например, лекарственное растительное сырье в
исходном виде или лекарственное растительное
сырье, измельченное в порошок. Если частная
статья распространяется на несколько вариан
тов, например. на оба из указанных, то это оro
варивается в разделе «Определение».
ПРОИЗВОДСТВО
Информация в разделе «Производство»
призвана привлечь внимание к некоторым
важным аспектам процесса производства и не
обязательно является исчерпывающей. Coдep
жащиеся в ней инструкции адресованы произ
водителю. Они MOryT относиться, например, к
материалам, к процессу производства, к ею Ba
лидации и контролю, к постадийному контролю,
а также к испытаниям, которые производитель
должен проводить перед выпуском для каждой
серии продукта или для выбранных серий. Эти
положения не обязательно должны быть под
тверждены посредством анализа конечноrо про
дукта. Компетентным уполномоченным opra
ном может быть установлено, что приведенные
выше аспекты были выполнены. Такое заключе
ние может быть сделано на основании проверки
полученных от производителя данных, или при
инспектировании производства или при испыта
нии соответствующих образцов.
Отсутствие раздела «Производство» не оз
начает, что аспекты производственноro процес
са, отмеченные выше, не требуют внимания
Выбор вакцинноrо штамма, Выбор со-
става вакцины. В разделе «Производство»
частной фармакопейной статьи на вакцину
MOryT быть указаны характеристики вакцинноro
штамма или состав. Если иное не указано, опи
сываемые в этом разделе методики испытаний
для подтверждения этих параметров приводятся
для информации в качестве примера. В случае
разрешения компетентным уполномоченным op
raHoM, MOryT использоваться иные методики ис
пытаний без проведения их валидации в cpaBHe
нии с методиками, приведенными в статье.
В03МОЖНОСТЬ ФАЛЬСИФИКАЦИИ
В связи с увеличением мошеннической aK
тивности и случаев фальсификации, COOTBeT
ствующая осведомленность может помочь
пользователям Фармакопеи в обнаружении
фальсификатов (т. е. фармацевтических суб
станций, вспомоrательных веществ, промежу
точных продуктов, нерасфасованых продуктов и
rOToBbIx лекарственных средств).
С учетом этоro, в раздел «Возможность
фальсифицирования» частных статей на суб
станции, для которых встречались случаи фаль
сифицирования или у которых присутствует риск
наличия умышленно добавленных примесей.
может быть включена методика обнаружения
возможных фальсификатов и соответствующие
пределы вместе с напоминанием, что все стадии
производства и снабжения должны быть частью
надлежащей системы качества. Частота испыта
ний, проводимых производителем или потреби
телем (например, производителем промежуточ
ных продуктов, нерасфасованных продуктов и
roтовых лекарственных средств, rдe применимо)
зависит от оценки риска, с учетом национальных
требований и информированности о всей цепоч
ки поставок.
Данный раздел устанавливает требования
ко всей цепочке поставок в целом, от производи
телей до потребителей (например, производите
лей промежуточных продуктов, нерасфасован
ных продуктов и roтовых лекарственных средств.
rдe применимо). Отсутствие данноro раздела не
предполаrает тоro, что не должно уделяться вни
мание моментам, упомянутым выше.
ОПИСАНИЕ (СВОЙСТВА)
Информация. приведенная в этом разделе,
не должна рассматриваться как прямое указа
ние и не является требованием.
Растворимость. #Под «растворимостью»
подразумевают свойство вещества растворяться
в различных растворителях, принятых в rф РБ.
Показатели растворимости вещества в различ
ных растворителях приводятся в частных фар
макопейных статьях.#
Для обозначения растворимости в разделе
«Описание (Свойства)>> используют описатель
ные термины, которые в температурном интер
вале от 15 ос дО 25 ос имеют следующие зна
чения:
Примерное количество рас-
Термин творителя (мл), необходи-
мое для растворения 1 r
вещества
Очень леrко 1
менее
растворим
Леrкорастворим от 1 до 10
Растворим от 10 до 30
Умеренно pac от 30 до 100
творим
Малорастворим от 100 до 1000
Очень мало от 1000 до 1 О 000
растворим
Практически более 1 О 000
нерастворим
Термин «частично растворим» используют
для характеристики смесей, у которых раствори
мы толькО некоторые компоненты. Термин «CMe
шивается с ...» используют для характеристики
жидкостеЙ. смешивающихся с указанным paCT
ворителем во всех соотношениях.
=Прuмечание: Для веществ, образующих при
растворении мутные растворы, соответствую
щее указание должно быть приведено в частной
22
rосударственная фармакопея Республики Беларусь
фармакопейной статье. Если указано, что суб
станция растворима в жирных маслах, то име
ется в виду, что она растворима в любом масле,
относящемся к классу жирных масел.1'
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Область применения. Приводимые в этом
разделе испытания не рассчитаны на полное
подтверждение химической структуры или cocтa
ва продукта. Они предназначены для подтверж
дения с приемлемой степенью достоверности
тоro, что продукт соответствует информации,
приведенной на этикетке.
Первая и вторая идентификации. В HeKO
торых частных статьях есть подразделы «Первая
идентификация» и «Вторая идентификация».
Испытания, описанные в подразделе «Первая
идентификация», MOryT быть использованы во
всех случаях. Испытания, описанные в подраз
деле «Вторая идентификация», MOryT быть ис
пользованы в аптеках, если есть доказательства
тоro, что данная серия субстанции была серти
фицирована на соответствие всем друrим Tpe
бованиям частной фармакопейной статьи.
#Для лекарственноro растительноro сырья
Moryт использоваться испытания, описанные в
подразделе «Вторая идентификация», если есть
доказательства тоro, что данная серия сырья
была сертифицирована на соответствие всем
требованиям частной фармакопейной статьи.#
В некоторых частных статьях для первой
идентификации приводятся два или более вида
испытаний, которые являются эквивалентными и
MOryт использоваться независимо друr от друrа.
Один или более из этих рядов обычно coдep
жат пере крестные ссылки на испытания, приве
денные в разделе «Испытания» частной статьи.
Это может быть использовано для облеrчения
работы аналитика, проводящеrо идентифика
цию и предписанные испытания. Например,
один ряд испытаний для идентификации имеет
ссылку на определение энантиомерной чистоты,
в то время как в друroм ряде проводится опреде
ление удельноro вращения: оба ряда преследу
ют одну и ту же цель подтвердить присутствие
необходимоro энантиомера.
Порошкообразное лекарственное pac
тительное сырье. Частные статьи на лекар
ственное растительное сырье MOryT содержать
схематические рисунки порошкообразноro ле
карственноro средства. Эти рисунки дополняют
описание, приведенное в соответствующем ис
пытании на подлинность.
ИСПblТАНИЯ И КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
Область применения. Эти требования не
рассчитаны на охват всех возможных приме
сей. В частности, если примесь не определяет
ся с помощью описанных испытаний, а здравый
смысл и надлежащая производственная прак
тика не допускают ее присутствия, не следу
ет делать вывод, что она допустима. См. также
ниже раздел «Примеси».
#Если в испытаниях с использованием xpo
матоrрафических методов после указания BBO
димоro или наносимоro объема раствора в ми
кролитрах в скобках указывается количество
вещества в микроrраммах, то имеется в виду
приблизительное количество.
Если указано, что испытание проводят «в
защищенном от света месте», то это означает,
что следует принять меры для избеrания попа
дания прямоro солнечноro света, любоro друroro
яркоrо света, а также исключить попадание уль
трафиолетовоro света, например, путем исполь
зования посуды из специальноro стекла, работы
в затемненной комнате и т. д.#
Расчеты. Если при проведении вычислений
требуется выполнить пересчет на сухое веще
ство или безводное вещество или оroворено Ka
кое либо друrое условие, то потерю в массе при
высушивании, содержание воды или иной пока
затель определяют с помощью метода, описан
ноro в частной фармакопейной статье. Слова «в
пересчете на сухое вещество» или «в пересче
те на безводное вещество» и друrие указывают
после результата.
Пределы. Указываемые пределы OCHOBЫBa
ются на результатах, полученных в рамках обыч
ной аналитической практики; в них уже учтены
обычные аналитические поrрешности, допусти
мый разброс при производстве и приroтовлении,
а также ухудшение качества в процессе xpaHe
ния в пределах, которые считаются приемле
мыми. При определении соответствия продук
та требованиям частной фармакопейной статьи
к указанным пределам не должны добавляться
никакие дополнительные допуски.
Результат, полученный в испытании, OKPy
rляют до указанноro в пределе количества знача
щих цифр (если нет друrих указаний). Пределы,
независимо от тоro, выражены они в процентах
или в абсолютных значениях, считаются значи
мыми до последнеro указанноro знака (напри
мер, число 140 обозначает три значимых знака).
При этом последнюю цифру увеличивают на
единицу, если цифра, отбрасываемая при oKpyr
лении, больше или равна пяти. Если цифра, OT
брасываемая при окруrлении, меньше пяти, по
следнюю цифру оставляют неизменной.
Определение допустимоrо предела при-
месей. Для сравнительных испыта/-lИЙ пример
ное допустимое содержание при меси или суммы
примесей может быть указано в скобках только
для информации. Решение об одобрении или OT
клонении принимают на основании соответствия
либо несоответствия приведенному в статье ис
пытанию. Если для данной примеси не указано
Общие сведения
23
использование стандартноro образца, ее coдep
жание может быть выражено исходя из номи
нальной концентрации вещества, используемоrо
для приroтовления указанноro в частной фарма
копейной статье раствора сравнения (если нет
друrих указаний).
Лекарственное растительное сырье. Для
лекарственноro растительноro сырья сульфатную
золу, общую золу, растворимые в воде вещества,
растворимые в спирте вещества, содержание
воды, содержание эфирных масел и содержание
действующих веществ рассчитывают в пересче
те на лекарственное средство, которое не было
специально высушено (если нет друrих указаний
в частной фармакопейной статье).
Эквиваленты (титры). В тех случаях, коrДа
приводят эквивалент, еro дают с таким коли
чеством значащих цифр, которое требуется в
данной частной статьи.
Питательные среды. Питательные среды,
описанные в частных и общих статьях, являются
удовлетворительными для использования их по
предназначению. Однако из за непостоянноro
качества компонентов среды, особенно биоло
rическоrо происхождения, может по надобиться
коррекция концентраций некоторых инrредиен
тов, например:
пептонов, мясных или дрожжевых экстрак
тов с учетом их питательных свойств;
буферных веществ;
желчных солей, желчноro экстракта, дезок
сихолата и красящих веществ, в зависимости от
их селективных свойств;
антибиотиков, в зависимости от их актив
ности.
ХРАНЕНИЕ
Информация и рекомендации, при водимые
в разделе «Хранение», не являются исчерпы
вающими фармакопейными требованиями, и
компетентные уполномоченные opraHbI MoryT
указывать конкретные условия хранения, обяза
тельные для исполнения.
Описанные в Фармакопее продукты следу
ет хранить таким образом, чтобы предотвратить
их заrрязнение И, по возможности, разложение.
Если рекомендуются особые условия хранения,
включая тип контейнера (см. раздел 1.3. Общие
фармакопейные статьи) и температурные пре
делы, эти рекомендации при водятся в частной
статье.
Ниже разъясняются термины, используе
мые в частных фармакопейных статьях в разде
ле «Хранение».
«В воздухонепроницаемом конrnейнере»
обозначает, что лекарственное средство должно
храниться в воздухонепроницаемом контейнере
(3.2). При вскрытии контейнера во влажнои aT
мосфере принимают меры предосторожности.
При необходимости низкая влажность содержи
моro можеr поддерживаться с помощью осуша
ющеro вещества, помещаемоro в контейнер. при
условии отсутствия прямоrо контакта между ним
Таблица #1.4. 1
. I . 1 1 дополнительно l'
, ! Темпе ат ные n е елы
,Условия хранения лекарственноrо средства р ур р д , указание (при
указываемые на упаковке I б )
нео ходимости
Лека ственное средство Не требует особых # 1 Н ет I «Не охлаждать» и ли
: ловии хранения I . . « Не замо раживать'>..j
Лекарственное средство требует условий xpa ! «Хранить при температуре. е Не охла wдать» I-1ЛИ ) '
, нения при температуре не выше 30 ос (TeMlle I выше 30 ОС» или «Хранить I «Не замораживать»
; ратура хранения от +2 ос до +30 ОС) при температуре ниже 30 ОС» I
! Лекарственное средство требует условий xpa «Хранить при температуре не «Не охлаждать» или
: нения при температуре не выше 25 ос (темпе выше 25 ОС» или «Хранить «Не замораживать»
! ратура хранения от +2 ос до +25 ОС) при температуре ниже 25 ОС»
! Лекарственное средство требует хранения при «Хранить при температуре от «Не охлаждать» или
I комнатной температуре 15 ос до 25 ОС» «Не замораживать»
I Лекарственное средство требует хранения в «Хранить при температуре от «Не замораживать»
I холодном или прохладном месте 8 ос до 15 ОС»
I Лекарственное средство требует хранения в «Хранить при температуре от «Не замораживать»
i холодильнике 2 ос до 8 ОС»
I Лекарственное средство требует хранения при «Хранить В морозильной
I температуре ниже О ОС камере» или «Хранить И
i транспортировать в морозиль
ной камере»
Прuмечанuя:
r:ри дополнительном указании «Не охлаждать» не допускается подверrать лекарственное средство воздействию температуры
ниже 8 'С.
;юnускается кратковременное изменение условий хранения в процессе местной транспортировки (в пределах одноro населен
нoro пункта) для лекарственных средств. не имеющих дополнительное указание по хранению «Не охлаждать» или «Не замора
--ЗТЬ .
rосударственная фармакопея Республики Беларусь
ФУНКЦИОНАЛЬНО ОБУСЛОВЛЕННЬ
ХАРАКТЕРИСТИКИ ВСПОМОiАТЕЛЬНblХ
ВЕЩЕСТВ
24
и лекарственным средством. #При указании «за
щищать от вла2и», относительная влажность в
условиях хранения должна быть не более 60 %. #
«В защищенном от света месте» обозна
чае что лекарственное средство должно xpa
ниться в контейнере, изrотовленном из матери
ала, в достаточной степени поrлощающею свет,
способный вызвать фотохимические превраще
ния (актиничный свет); или контейнер должен
быть помещен во внешний контейнер, обеспечи
вающий такую защиту; или лекарственное cpeд
ство должно храниться в месте, исключающем
возможность попадания такою света.
#«Температура». Лекарственное средство
должно храниться в соответствии с требования
ми, указанными в таблице #1.4. 1.#
МАРКИРОВКА
Маркировка реrламентируется компетент
ным уполномоченным opraHoM с изданием co
ответствующеro нормативною правовою акта.
Таким образом, информация в разделе «Марки
ровка» не претендует на полноту. Она ориенти
рована прежде всеro на фармакопейные цели, и
обязательными являются только те положения,
которые необходимы для подтверждения co
ответствия продукта статье. Вся остальная ин
формация носит рекомендательный xapaKTep
В тех случаях, Korдa в Фармакопее употребля
ется термин «этикетка», соответствующая ин
формация может быть помещена на контейне
ре, на упаковке, во вкладыше или в сертификате
анализа, сопровождающем лекарственное cpeд
ство, в зависимости от решения компетентною
уполномоченноrо opraHa.
ПРЕДОСТЕРЕЖЕНИЯ
Описываемые в Фармакопее продукты и
реактивы MorYT оказаться опасными для здоро
вья, если не предпринять необходимые Mepы
Во всех случаях следует придерживаться прин
ципов Надлежащей лабораторной практики
(НЛП, GLP), а также соответствующих положе
ний техники безопасности. В некоторые статьи
включены специальные указания о необходи
мых мерах предосторожности, но отсутствие
таких указаний не следует трактовать как OTCYT
ствие всякоro риска.
ПРИМЕСИ
В частной фармакопейной статье может
быть приведен перечень всех известных и по
тенциальных примесей, для которых показано,
что они контролируются испытаниями. См. также
статью 5. 10. Контроль прuмесей в субстанциях
для фармацевтичеСК020 использования. При
меси обозначают буквой или буквами латинско
ro алфавита Если какая либо буква пропущена.
это означает, что примесь, обозначенная с по
мощью нее, исключена из списка примесей при
разработке частной статьи до публикации либо
при пересмотре этой статьи.
Частные статьи на вспомоrательные Be
щества MorYT содержать раздел с перечнем
функционально обусловленных характери
стик. Параметры, любые методики определе
ния и нормы, приведенные в этом разделе. не
являются обязательными требованиями; тем
не менее, они MorYT быть важны для исполь
зования вспомоrательных веществ и приве
дены для информации (см. также раздел 1.1.
Общие положения).
I
СТАНДАРТНЫЕ ОБРА3ЦbI
Некоторые частные статьи предусматри
вают использование стандартных образцов
(фармакопейных стандартных образцов, био
лоrических стандартных препаратов, эталон
ных спектров). См. также статью 5. 12. CтaH
дартные обраЗЦbl. #Стандартные образцы,
стандартные препараты и эталонные спектры
вводятся в действие компетентным уполно
моченным opraHoM. Полный перечень может
быть получен в соответствующей орrаниза
ции.# Эти стандартные образцы являются
официальными в случае арбитража.
1.5 СОКРАЩЕНИЯ И ОБОЗНАЧЕНИЯ
А Оптическая плотность
A1; Удельный показатель поrлощения
А. м. Относительная атомная масса
[а I C
БСП
Удельное оптическое вращение
Биолоrический стандартный пре
парат
Фармакопейный стандартный об
разец
ФСО
d 20
20
Относительная плотность
1"
МЕ
М
М. М.
Длина волны
Международные Единицы
Молярность
Относител ьная молекулярная
масса
п 2О
о
Показатель преломления
Единица действия Европейской
Фармакопеи
Частей на миллиард (микроrрам
мов на килоrрамм)
Частей на миллион (миллиrрам
мов на килоrрамм)
Вещество или раствор. указанный
в rлаве 4. Реактивы
Ph. Еш. U
ррЬ
ррт
Р
общие сведения
25
R. Фактор подвижности (статья
2.2.46)
R Используется в хроматоrрафии
для обозначения пути, пройденно
ro испытуемым веществом, к пути,
пройденному стандартным образ
цом
РО Исходное стандартное вещество
для установки титра титрованных
растворов в объемном анализе
(статья 4.2.1)
Аббревиатуры, используемые в частных
фармакопейных статьях на иммуноrлобули
ны, сыворотки и вакцины
LD 50 Медианная летальная доза. CTa
тистически определенное количе
ство вещества, которое при COOT
ветствующем применении может
вызвать смерть 50 % испытуемых
животных за исследуемый период
MLD Минимальная летальная доза
L +/1 О dose Наименьшее количество токсина,
которое в смеси с 0,1 МЕ антиток
си на при обычном пути введения
способно вызвать rибель лабора
торных животных в эксперимен
тальных условиях за исследуемый
период
L + dose Наименьшее количество токсина,
которое в смеси С 1 МЕ аНТИТОI<
сина при обычном пути введения
способно вызвать rибель лабора
торных животных в эксперимен
тальных условиях за исследуемый
период
Ir/100 dose Наименьшее количество токсина,
которое в смеси с 0,01 МЕ анти
токсина при внутрикожном BBeдe
нии способно вызвать xapaKTep
ные реакции в месте введения у
лабораторных животных в экспе
риментальных условиях за иссле
дуемый период
Lp/1 О dose Наименьшее количество токсина,
которое в смеси с 0,1 МЕ антитокси
на при внутрикожном введении спо
собно вызвать характерные реакции
в месте введения у лабораторных
животных в экспериментальных yc
лов иях за исследуемый период
Lo/1 О dose Наименьшее количество токсина,
которое в смеси с 0,1 МЕ антиток
сина и обычном пути введения He
способно вызвать токсическую pe
акцию у лабораторных животных
в экспериментальных условиях за
исследуемый период
Lf dose
ССI0 50
Е 1050
1050
РD 50
ЕD 50
PFU
8PF
Количество токсина или токсино
подобноro вещества, которое спо
собно связать 1 МЕ антитоксина
за кратчайшее время
Медианная инфицирующая доза
кулыурыклеток. Статистиче
ски определенная доза вирусных
частиц, которая при внесении в
клеточную культуру способна ин
фицировать 50 % клеток
Медианная инфицирующая доза
яиц. Статистически определенная
доза вирусных частиц, которая при
внесении в оплодотворенные яйца
способна инфицировать 50 % эм
брионов
Медианная инфицирующая доза.
Статистически определенная доза
вирусных частиц, которая при BBe
дении в орrанизм животных спо
собна инфицировать 50 % особей
Медианная защитная доза. Стати
стически определенная доза BaK
цины, которая в эксперименталь
ных условиях способна защитить
50 % животных от инфицирующей
дозы микроорrанизмов или токси
нов, В отношении которых эта BaK
цина активна
Статистически определенная доза
вакцины, которая в эксперимен
тальных условиях способна ин
дуцировать выработку антител к
соответствующим антиrенам BaK
цинЬ! у 50 % животных
Оспинообразующие единицы или
бляшкообразующее число
Свободный от специфических
патоrенов
Коллекции микроорrанизмов
АТСС
С/Р
/М/
Американская коллекция типовых
культур
Атеriсап Туре Cu/ture Collectioп .
10801 Uпivеrsitу Bou/eva,d
Мапаssаs, Virgiпiа 20110 2209,
USA
Коллекция Пастеровскоro институ
та (штаммы бактерий)
Collectioп de Bacteries /'/пstititиtе
Pasteur
В. р. 52,25 rue du Docteur Roux
75724 Pa,is Cedex 15, Fraпce
Международный институт
миколоrии
/пtеrпаtiопа/ Myc%gica//пstitute
Bakeham Lапе
Surrey TW20 9 ТУ, Great Britaiп
26
rосударственная фармакопея Республики Беларусь
'Р
NC/MB
NCPF
NCTC
NCYC
Национальная коллекция культур
микроорrаНИЗМ08
Соllесtiоп Nationa'e de Cufture de
Мiсrооrgапisтеs (CNCM)
/nstititute Pasteur
25, (ие du Docteur Roux
75724 Paris Cedex 15, France
Национальная коллекция промыш
ленных и морских бактерий
Nаtiопа' Соllесtiоп of fndustriaf апd
Marine Bacteria Ltd.
23 5t Machar Orive
Aberdeen АВ2 1 RY, Great Britaiп
Национальная коллекция
патоrенных rрибов
Nationa' Collection of Pathogeпig
Fungi
Lопdоп Schoo' of Hyg/ene and Tro
pica' Medicine
Керре/ 5treet
Lопdол WC1 Е 7 НТ, G,eat Вritаiп
Национальная коллекция типовых
культур
Nаtiопа' Collection of Туре Cu'tures
Сепtrа' РиМс Hea'th Laboratory
Соliпdа/е Avenue Lопdоп NVV9 5НТ,
Great Britain
Национальная коллекция
дрожжевых культур
Nationa' Соllесtiоп of Yeast Cultures
AFCR Food Research /nstitute
Со'пеу Lапе
Norwich NR4 7 ИА, Great Britain
N'TE
55/
Центр биолоrических ресурсов
Bio'ogica/ Resources Center
Oepartment of Biotechпofogy
Nаtiопа//пstitиtе of Techпo'ogy aпd
Еvа/иаtiоп
2 5 8 Kazusakamatary, Кisarazu shi,
Chiba, 292 0818
Japaп
rосударственный институт CЫBO
роток
5tateпs 5е,ит /пstitиt
80 Amager Bou'evard, Сорепhаgеп,
Deпmark
#Украинская коллекция микроор
rанизмов
Институт микробиолоrии
им. д. К. Заболотноro НАН Украины
01000, Киев,ул.Заболотноrо, 154,
Украина
#Коллекция штаммов Российско
ro музея патоrенных бактерий
rосударственный институт CTaH
дартизации и контроля медицин
ских биолоrических препаратов
им.Л.А. Тарасевича
121002, Москва, Сивцев Вражек,
41, Российская Федерация
#Белорусская коллекция непато
reHHbIx микроорrанизмов (научная
коллекция непатоrенных типовых
и промышленно ценных микроор
rанизмов института микробиоло
rии НАН Беларуси)
rHY «Институт микробиолоrии»
НАН Беларуси
220141, Минск, ул. Купревича. 2,
Республика Беларусь
1.6. ЕДИНИЦЫ МЕЖДУНАРОДНОЙ СИСТЕМЫ (СИ), ИСПОЛЬЗУЕМЫЕ В ФАРfv1АКОПЕЙНЫХ CTA
ТЬЯХ, И ИХ СООТВЕТСТВИЕ дРУrим ЕДИНИЦI\М
МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ (СИ)
Международная система единиц состоит из трех классов единиц физических величин, а именно:
основные единицы, производные единицы и вcnоМоraтельные единицы'. Основные единицы и их опре
деления приведены в таблице 1.б. 1.
основные единицы СИ
Таблица 1.6. 1
Величина ! Единица i
!
СИМВОЛ, i ' I I Определение
Наименование принятый в I #=азме : Наимено I СИМВОЛ I
Фармакопее ость : вание .
! I 1 Один метр представляет собой
Длина I L метр м I путб, который проходит свет в
I ; i вакууме за 1/299 792 458 долю
! i секунды
I I
I : килоrрамм I Один килоrрамм равен массе
Масса т М Kr международноrо прототипа
! I килоrрамма
1 Определения единиц, используемых 8 МеждународноЙ системе, даны в буклете "Le 5ysteme /пteтatioпa/ O'Uпites (5/»>, опу
бликованной Bиreaи /п/етаtiОПа/ des Poids et fvIesиres. PaVll10п de Bre/eui/. F 92310 5evres.
Общие сведения
27
Наименование
Величина
Символ,
i принятый в
; Фармакопее
Единица
#Разме р Наимено
Символ
НОСТЬ вание
Определение
Время
т
секунда
с
Одна секунда представля
ет собой продолжительность
9 192 631 770 периодов излу !
чения, соответствующих пе
реходу между двумя CBepx
тонкими уровнями OCHOBHoro
состояния атома цезия 133
I Щ r я т ка, к торыи, при
I прохождении по двум пря
I молинейным параллель
I ным проводникам бесконеч
I Электрический ток I ( ампер А I ной длины и пренебрежимо
малой площади круroвоro
I I поперечноro сечения, pac
I : положенным на расстоянии
I
I 1 метр один от друrоrо в Ba
кууме, вызывал бы силу вза
! I имодействия, равную 2'10 7
ньютона на один метр длины
I Один кельвин представляет
jтермодинамическая собой 1/273,16 часть TepMO
(=абсолютная#) I т е кельвин К
I температура i I I динамической температуры
I тройной точки воды I
I I '
I : ,
I I i I I Один моль представля
I ет собой количество веще
, ства системы, содержащей
I I
i Количество п 1'11 моль моль столько же структурных эле
I вещества I ментов, сколько атомов co j
I
I держится в 0,012 Kr уrлеро i
i , :
дa 12'
Кандела представляет собой I
силу света источника, испу
скающеrо в данном направ
лении монохроматическое ;
,
Сила света I J кандела кд излучение частотой 540 -1 О' z !
.
rерц, энерrетическая сила
KOToporo в этом направле
нии составляет 1/683 ватт на
один стерадиан
Один ампер представля
ет собой силу неизменяю
е ос о о
П Если использованы моль. то следует указывать, к чему они относятся, например, атомы, Мaлe«yJ1bI. ионы_ электроны, иные ча
стицы или определенные rpYnnbI таких объектов_
С) Размерность приведена в соответствии с техническим реrламентом «Единицы измерений. дonyweHHыe к применению на Tep
ритории Республики Беларусь)} (ТР 2007/О0З/ВУ).
Производные единицы MorYT быть образованы путем комбинирования основных единиц, соrлас
но алrебраическим взаимоотношениям, связывающим соответствующие количества. Некоторые из
таких производных величин имеют свои собственные названия и обозначения. Единицы СИ, исполь
зуемые в Фармакопее, приведены в таблице 1.6. 2.
28
rосударственная фармакопея Республики Беларусь
Величина Единица
Символ, #Раз- I Выраже- Выраже- Преобразование
Наименова- принятый Наименова- Сим- ние в ос- ние в иных иных единиц в
ние в Фарма- мер- ние вол новных единицах единицы СИ
.
копее НОСТЬ СИ
единицах
Волновое v единица на 1/м M '
число метр
Длина волны л микрометр мкм 10 6 м
нанометр нм 10 9 м
Площадь A,S метр квадрат м 2 м 2
ный
Объем v метр кубиче м 3 м 3 1 мл = 1 см 3 =
ский 1 O м 3
Частота v Т 1 rерц rц c 1
Плотность Р килоrрамм на Kr/M 3 Kr'M 3 1 r/мл = 1 r/CM 3 =
метр кубиче 103 Kr'M 3
ский
Скорость v метр в ceKYH м/с M'C '
ду
Сила F LMT 2 ньютон Н M'Kr'c 2 1 дин = 1 r'cM'c 2 =
1 0 5 Н
1 Krc = 9,80665 Н
Давление р L lMТZ паскаль Па M "Kr.c 2 H'M 2 1 дин/см 2 = 1 o , Па
= 10 ' H'M 2
1 атм = 101 325 Па
= 101,325 кПа
1 бар = 105 Па =
0,1 МПа
1 мм рт. ст. =
133,322387 Па
1 торр =
133,322368 Па
1 фунт на KBa
i дратный дюйм
! (psi) = 6,894757
кПа
Динамиче f] паскаль се Па'с M "Kr'c ' H'C.M 2 1 пз = 10 ' Па'с =
ская вязкость кунда 10 ' H.C'M 2
! 1 спз = 1 мПа'с
Кинематиче v метр квадрат м 2 /с M2.c , Па'с'м 3 .кr , 1 Ст = 1 см 2 .с, =
ская вязкость ный в ceKYH H.M'C-Kr ., 10-4 м2.с.,
ду
Энерrия W иMТZ джоуль Дж m2'kr,c- 2 Н'М 1 зрr = 1 cM 'r'c 2 =
1 дин см = 10 7 Дж
1 кал = 4,1868 Дж
Мощность р uмтз ватт Вт ? , H'M'C ' 1 эрr/с =
M 'Kr.c I
Поток излу Дж'с ' 1 дин'см'с ' =
чения I 10 7 Вт =
i I 1СУ Н-М'С' = 1СУ Дж'С'
Поrлощенная О иТ 2 rрей I rp I M2.c 2 Дж'кr' 1 рад = 10 2 rp
!
доза ионизи I
I I
рующеro из I
лучения ! !
Электриче И eMT3/ 1 вольт В ! M;;'Kr'c 3'A '1 Вт'А'
ский потен I
I I I
циал, элек i
тродвижущая !
сила I
Таблица 1.6. 2
Единицы СИ, используемые в Фармакопее, и их соответствие дРУ2им единицам
Общие сведения 29
Величина Единица
Символ, #Раз- Выраже- Выраже- Преобразование
Наименова- принятый Наименова- Сим- ние в ос- ние в иных иных единиц в
ние в Фарма- мер- ние вол новных единицах единицы СИ
.
копее ность СИ
единицах
Электриче R еМТЗ/ 2 ом Ом м2'кr'с З'А2 В-А 1
ское сопро
тивление
Количество Q ТI кулон кл Ас
электриче
ства
Радиоактив А Р беккерель Бк c 1 1 Ки = 37.109 Бк =
ность веще 37.109 c 1
ства
KOHцeHTpa с моль на метр молы моль,м з 1 молы/л = 1 М =
ция (коли кубический м з 1 моль/дм з =
чества Be 103 моль,м- з
щества), >
молярная
KOHцeHTpa
ция
Массовая р килоrрамм на кr/м З кr'м З 1 r/л = 1 r/дм з =
KOHцeHTpa метр кубиче 1 кr'м з
ция ский
(") Размерность приведена в соответствии с техническим реrламентом «Единицы измерений. допyщetll8ol8lr l1l 91e1lИlО на Tep
ритории Республики Беларусь» (ТР 2007ЮОЗ/ВУ).
Некоторые важные и широко используемые единицы, не входящие в МеЖДУНЩXQfYIOOICТему СИ,
приведены в таблице 1.6. 3.
Тaбnицa 1.6. 3
Единицы, используемые наряду с Международной системой единиц
Величина Единица Значение в eдмr r- си I
Наименование Символ
Время минута мин 1 мин = 60 с
час ч 1 ч = 60 мин = 3600 с
сутки сут 1 сут = 24 ч = 86 400 с
Плоский уroл rрадус о 10 = (п/180) рад
Объем литр л 1 л = 1 дм 3 = 1 0-3 м 3
Масса тонна т 1 т = 1 ОЗ Kr
Частота вращения оборот в минуту обlмин 1 об/мин = (1/60) с- 1
Множительные приставки для образования десятичных кратных и дольных единиц npмвeдeны в Ta
блице 1.6А.
Тaбnица 1.6А
Множители и приставки для образования десятичных кратных и дольных единиц
Множитель Приставка Обозначение Множитель Приставка Обозначение
10 1В экса Э 10-1 деци д
1015 пета П 1 0 2 санти С
1012 тера Т 10 З милли м
109 rиrа r 10 6 микро мк
106 Mera М 10 9 нана н
10 З кило к 10 12 ПИКО ; П
10: reKTO r 1 0 15 фемто I Ф
10' дека да 10-18 апо а
ПРИМЕЧАНИЯ
1. В Фармакопее используется температура по Цельсию (символ t). Температура по Цельсию опре
.::\еляется соrласно формуле:
30
rосударственная фармакопея Республики Беларусь
t == Т То '
rде:
То 273,15 К.
Температура по Цельсию выражается в rpa
дусах Цельсия (символ ОС). Один «rрадус Цель
сия» равен одному «кельвину».
2. Практические выражения для KOHцeHTpa
ций определены в Общих положениях.
3. Радиан представляет собой плоский уrол,
вырезающий на окружности дуry, равную по
длине радиусу.
4. Условия центрифуrирования определяют
ся центробежным ускорением по отношению к
ускорению свободноro падения (g), которое при
нимается равным
9 = 9,80665 M.c 2.
5. Некоторые величины используются без
размерности, как, например. относительная
плотность (2.2.5), оптическая плотность (2.2.25),
удельный показатель поrлощения (2.2.25) и по
казатель преломления (2.2.6).
6. Микрокатал определяется как энзимати
ческа я активность, которая при указанных yc
ловиях приводит К превращению (например, к
rидролизу) 1 микромоль субстрата за одну ce
кунду.
#1.7. ОТБОР ПРОБ
ОСНОВНЫЕ ПОНЯТИЯ
Выборка количество штучной продук
ции, отобранной из контролируемой серии
(партии).
,отовое лекарственное средство (про
дукт) лекарственное средство (продукт),
прошедший все стадии технолоrическоrо про
цесса, включая маркировку, упаковку, лабора
торный контроль.
Контейнер изделие, которое содержит
продукцию или предназначено для ее xpaHe
ния и находится или может находиться в He
посредственном контакте с ней. Укупорочное
средство является частью контейнера.
Контроль процедура оценки COOTBeT
ствия путем измерений, наблюдений, испыта
ний или калибрования соответствующих xa
рактеристик.
Контаминация процесс заrрязнения.
Нефасованный продукт (аН2РО или ,п bu/k
product) продукт, прошедший все стадии
технолоrическоro процесса, кроме упаковы
ван ия.
Отбор проб действия по изъятию проб
сырья, материалов, полупродуктов, промежу
точной и rотовой продукции для исследова
ния их качества.
Объем выборки число выборочных
единиц в выборке.
Объединенная проба .проба продукции,
состоящая из нескольких точечных проб, OTO
бранных из контролируемой партии.
Образец продукции единица конкретной
продукции, используемая в качестве представи
теля этой продукции при исследовании, KOHTpO
ле и оценке.
Промежуточная продукция частично
обработанное сырье или продукция, которые
должны пройти последующие технолоrические
операции прежде, чем стать нефасованной про
дукцией.
Проба количество нештучной продукции,
отобранное из контролируемой серии (партии)
.для принятия решения и состоящее из несколь
ких точечных проб.
Сырье субстанции для фармацевтиче
скоro использования, части лекарственных pac
тений или продукты их переработки, вспомоrа
тельные вещества собственноro производства
или получаемые от внешних постаВЩI4IЮВ, ис
пользуемые в производстве лекарственных
средств, за исключением маркировочных и упа
ковочных материалов.
Серия (партия) количество продукции
одноrо наименования, полученное в одном Tex
нолоrическом цикле или в течение определенно
ro интервала времени, в одних и тех же услови
ях и одновременно представленное на контроль.
Качество серии (партии) должно быть YДOCTOBe
рено одним документом.
Точечная проба количество продук
ции, взятое за один раз из одноro места серии
(партии) одномоментно.
Упаковка средство или комплект средств,
обеспечивающее защиту продукции от повреж
дений и потерь, окружающей среды от заrрязне
ния, а также процесс обращения продукции.
Упаковочная единица упаковка, содержа
щая установленное количество продукции.
Чистая зона зона. в которой окружающая
среда контролируется на наличие контаминиру
ющих частиц и микроорrанизмов, построенная и
эксплуатируемая таким образом, чтобы YMeHb
шить их проникновение и образование контами
нантов внутри зоны.
ОБЩИЕ ПРАВИЛА
Если в частных статьях не указано иное,
отбор проб для испытаний должен проводиться
в соответствии с процедурой отбора.
Процедура отбора проб включает:
план или схему отбора проб;
место и время отбора проб;
извлечение и подroтовку проб продукции
для испытаний;
объем и тип отбора проб;
параметры окружающей среды при отборе
и подrотовке проб для испытаний.
Все оборудование, используемое для
отбора проб, включая измерительное оборудо
вание для проведения испытаний, связанных
Общие сее)вJ..
31
с отбором проб. ДОЛЖНО удовлетворять требо
ванияu норматиsной документации или проце
дуре отбора проб и работать в соответствии с
данной npoцедурой или инструкциями (эксплу
тационной документацией) на оборудование.
Измерительное оборудование должно пройти
в установленном порядке поверку или апеста
цию
Персонал, выполняющий отбор проб,
должен владеть процедурой отбора. ДOKYMeH
тация по процедуре отбора проб должна Haxo
диться в местах отбора проб и быть доступной
для персонала.
В процоссе проведения отбора проб необ
ходи'Мо учитывать факторы, которые должны
контролироваться с тем, чтобы обеспечить
достоверность результатов испыта.ний. К ним
относятся: показатели, обеспечивающие co '
ответствие контролируемых показателей каче
ства и безопасности проб показателям объек
та испытаний и стабильность этих показателей
в течение периода проведения испытаний; па
раметры окружающей среды и друrих воздей
ствующих на пробы факторов.
Пробы, прошедшие отбор, должны COOT
ветствующим образом идентифицироваться с
использованием единой маркировки и оформ
ляться актом отбора или друrим документом,
включающим дату, время и место отбора, усло
вия окружающей среды при отборе, фамилию,
имя и отчество лица, проводившеro отбор, и
друrую необходимую информацию.
Если поставка сырья или roтовой продук
ции состоит из нескольких серий (партий), то
каждую серию (партию) необходимо paCCMa
тривать как отдельную в отношении отбора
проб и проведения испытаний.
Во избежание ошибок при отборе проб не
допускается отбор проб одновременно от двух
и более наименований, двух и более серий
(партий) сырья или roтовой продукции.
Отбор проб для нефасованной продукции
(aHrpo или bu/k product) должен осуществлять
ся в стерильные контейнеры. Упаковка должна
обеспечивать приroдность пробы для проведе
ния испытаний.
К отбору от следующей серии (партии)
поступившеro сырья или roтовой продукции
можно приступать только после выполнения
всей процедуры отбора от предыдущей серии
(партии).
Отбор проб следует производить только
из неповрежденных укупоренных и упакован
ных соrласно нормативной документации упа
ковочных единиц.
До liI после проведения испытаний пробы
должны храниться в отдельном помещении. Yc
ловия в помещении должны обеспечивать co
хранность проб в течение всеro срока хранения.
Перед отбором проб необходимо произве
сти внешний осмотр упаковочной тары (ящики,
короба, барабаны, бутыли и т. д.), определить
ее количество, целостность, наличме П11OIo1б.
правильность маркировки и оформленме
проводительной документации, а также COOT
ветствие тары и упаковки требованиям специ
фикации.
Количество упаковочных единиц, отобран
ных для отбора, рассчитывают по формуле:
0,4.JП ,
rдe:
п общее количество упаковочных единиц
одной серии (партии).
Количество вскрытых упаковочных единиц
должно быть не менее 3 и не более ЗО.
Отбор проб сырья необходимо производить
в отдельном помещении или в складском поме
щении таким образом, чтобы предотвратить KOH
таминацию.
Для проведения испытаний лекарственных
средств на соответствие требованиям норматив
ной документации проводят мноroступенчатый
отбор проб. При мноroступенчатом отборе пробу
образуют по ступеням и продукцию в каждой CTY
пени отбирают случайным образом в пропорци
ональных количествах из единиц, отобранных в
предыдущей ступени. Число ступеней определя
ется видом упаковки.
1 ступень: отбор единиц упаковочной тары
(ящиков, коробок, мешков и др.)
11 ступень: отбор упаковочных единиц, Ha
ходящихся в упаковочной таре (пакетов, флако
нов, банок, бутылок, рулонов и др.)
111 ступень: отбор продукции в первичной
упаковке (ампул, флаконов, туб, контурных упа
ковок и др.)
При отборе проб необходимо принимать
меры предосторожности и требования безопас
ности с учетом токсичности, взрывчатости, orHe
опасности, rиrроскопичности и друrих свойств
сырья, а также меры, направленные на предо
хранение проб (образцов) от повреждения и
заrрязнения во время работы с ними, к их упа
ковке, транспортировке, складированию и xpa
нению с учетом требований и методов последу
ющих испытаний.
При отборе проб лекарственных средств
списка «А», наркотических и психотропных ле
карственных средств следует PYKOBOДCTBOBaTb
ся правилами, инструкциями и положениями, YT
вержденными компетентным уполномоченным
opraHoM, и частными статьями на эти лекар
ственные средства.
При отборе проб запрещается принимать
пищу, пить, I{УРИТЬ, а также хранить еду, cpeд
ства для курения в специальной одежде или
месте отбора проб.
Персонan, занятый отбором проб, должен
строю соблюдать инструкции, реrламентирую
щие состояние здоровья и требования личной
rиrиены, носить специальную обувь.
32
Тосударственная фармакопея Республики Беларусь
ОТБОР ПРОБ ИЗ НЕФАСОВАННОЙ
ПРОДУКЦИИ (AHrpO ИЛИ /N BULK
PROOUCT)
Проба из нефасованной продукции должна
представлять собой объединенные точечные
пробы, взятые примерно в равных количествах.
Отбор точечных проб (объединенная проба)
необходимо производить из верхнеro, среднеro
и нижнеro слоев каждой упаковочной единицы,
убедиться в однородности сыпучих, вязких и re
TeporeHHbIx препаратов.
При отборе проб сыпучих и вязких лекар
ственных средств, фармацевтических субстан
ций и вспомоrательных веществ для предотвра
щения перекрестной контаминации точечные
пробы необходимо отбирать чистыми и стериль
ными пробоотборниками, изroтовленными из
материала, не реаrирующеro с данным сырьем.
Детали пробоотборников, контактирующие с
сырьем, должны быть обработаны специальны
ми антисептиками, разрешенными к примене
нию компетентным уполномоченным opraHoM.
В случае, если перемешивание жидкости
затруднено (большие емкости), точечные пробы
отбирают без перемешивания из разных слоев.
ОТБОР ПРОБ ,ОТОВЫХ
ЛЕКАРСТВЕННblХ СРЕДСТВ (rотовой
ПРОДУКЦИИ)
Отбор проб roтовых лекарственных средств
(продукции) должен производиться из HeHapy
шенных заводских упаковочных единиц.
Отбор проб roтовых лекарственных средств
для инъекций и rлазных лекарственных средств
на отсутствие в них механических включений
должен про изводиться соrласно соответствую
щей документации, утвержденной компетент
ным уполномоченным opraHoM.
Отбор проб аэрозолей проводят в соответ
ствии с требованиями частных статей.
2.1.2. Сравнительная таблица пористости стеКЛЯННblХ фильтров
зз
2.
МЕТОДЫ АНАЛИЗА
2.1. ОБОРУДОВАНИЕ
01/2013:20101
2.1.1. КдПЛЕМЕР
Термин «капли» обозначает стандартные
капли, вытекающие из стандартноro каплемера,
как описано ниже.
Стандартные каплемеры (рисунок 2.1.1 1 )
изrотавливают из бесцветноro стекла. Нижний
конец имеет круrлое отверстие, расположенное
в плоскости, перпендикулярной оси.
Друrие каплемеры #(пипетки)# MOryT быть
использованы, если они отвечают требованиям
следующеro теста.
20 капель воды Р при температуре (20:t1) ОС,
свободно вытекающих из каплемера #(пипетки)#,
удерживаемоrо в вертикальном положении, со
скоростью одна капля в секунду. должны иметь
массу (1000:t50) Mr.
Каплемер #(пипетки)# перед использовани
ем должен быть тщательно вымыт. Проводят три
определения для каждоro каплемера #(пипетки)#.
Ни один из результатов не должен отклонять
ся более чем на 5 % от среднеro значения трех
определений.
5
'1
о
I
о
с')
l
о
1,0
3,00 3,05
Рисунок 2.1.1. 1. Стандартный каплемер
(размеры указаны в миллиметрах)
01/2013:20102
2.1.2. СРАВНИТЕЛЬНАЯ ТАБЛИЦА ПОРИСТОСТИ СТЕКЛЯННЫХ ФИЛЬТРОВ
Пори Макси 1:1; 1:1; ,1:1;
:S: :S: 0:S:
стость мальный Ж ::r :о: Ж
со Ж :S:CO #
фильтра ::Е со I:;
диаметр о. о. ф:s:
(Ph. Eur.)(1) пор,мкм е то.
\D
1.6 менее 1,6 5f пор 1,6
1 2,5 5 5
1,6 З пор 3,0
4 1,6
4 5
3 10 ПОР 10
10 4 10 4f 4
! 16 10 16 4 4 пор 16
40 ! 16 0 3 3 3 пор 40
: 40 50 2
I
100 40 100 2 2 пор 100
5 3.а.. J "!2
Таблица 2.1.2. 1
Пори Макси 1:1; 1:1; , 1:1;
:S: :S: о :S:
СТОСТЬ мальный Ж ::r :.: :t
со Ж :S: со #
фильтра диаметр ::Е со к::: I
Q. Q. ф:s: I
(Ph. Eur.)(1) пор,мкм е т !
100 120 1 I
160 100 160 1 1 I !пор 160
150 200 О О
250 160 250 I I ПОР 250
200 500 '00
250 500 пор 500
." Европейская Фармакoneя приняла систему. предложенную
Международной по Стандартизации (150).
з4
rосударственная фармакопея Республики Беларусь
Возможно следующее применение филь
тров (диаметр в микрометрах), пределы KOl pЫX
являются приблизительными:
#<1 ,6 фильтрование коллоидных pac
творов;#
#1 ,6 16 фильтрование аморфных ocaд
ков;#
<2,5 бактериолоrическое фильтрова
ние;
4 10 ультратонкое фильтрование, OT
деление микроорrанизмов большоro диаметра;
10------40 аналитическое фильтрование,
очень тонкое фильтрование ртути, очень тонкое
дисперrирование rазов;
#16------40 фильтрование мелкокристалли
ческих осадков;#
40 1 00 тонкое фильтрование, филь
трование ртути, тонкое дисперrирование rазов;
100 160 rрубое фильтрование, дис
перrирование v. промывка rазов, использование
в качестве подложки для друrих фильтрующих
материалов; #фильтрование rрубозернистых и
студнеобразных осадков#;
160 500 очень rрубое фильтрование
частиц, дисперrирование и промывка rазов.
01/2013:20103
2.1.3. ЛАМПЫ
С УЛЬТРАФИОЛЕТОВЫМ
ИЗЛУЧЕНИЕМ ДЛЯ u
АНАЛИТИЧЕСКИХ ЦЕЛЕИ
В качестве источника улырафиолетово
ro излучения используются пары ртути в KBap
цевых лампах. Для устранения испускаемой
лампой видимой части спектра может ИСI10ЛЬ
зоваться подходящий фильтр. Если Фармакопея
предписывает использование в испытании уль
трафиолетовой лампы с длиной волны 254 нм
или З65 нм, используют прибор, состоящий из
лампы, содержащей пары ртути, и фильтра, дa
ющеro спектр излучения с максимальной интен
сивностью около 254 нм или 365 нм. Использу
емая лампа должна позволять без затруднений
обнаруживать стандартное пятно натрия сали
цилата диаметром около 5 мм на пластинке со
слоем силика2еля G Р, причем пятно должно ис
следоваться в положении, перпендикулярном к
излучению лампы.
Для этой цели используют 5 мкл раствора
0,4 r/л натрия салицилата Р в 96 % спирте Р
(спирт не должен флуоресцировать) для ламп с
максимальным излучением 254 нм и 5 мкл paCT
вора 2 r/л в. 96 % спирте Р для ламп с макси
мальным излучением 365 нм. Расстояние между
лампой и хроматоrрафической пластинкой, при
проведении испытаний, указанных в общих и
частных статьях, не должно превышать расстоя
ния при проведении вышеуказанноro испытания.
01/2013:20104
2.1.4. СИТА
Сита с квадратными отверстиями изroтав
ливают из соответствующих материалов. Для
неаналитических процедур MOryT быть исполь
зованы сита с круrлыми отверстиями, диаметр
которых в 1,25 раза превышает размер стороны
квадратноro отверстия сита соответствующею
номера. Не должно быть взаимодействия между
материалом, из которою изютовлено сито, и Be
ществом, которое просеивают. Степень измель
чения в частной статье указывают, используя
номер сита, соответствующий номинальному
размеру стороны отверстия в микрометрах, KO
торый приводится В скобках после названия Be
щества (таблица 2.1.4. 1).
'S
::a
:1: :е
.lI ::.::
Е:; :е
Ii
S S
:et
Оа.
:1: Ф
co
ra ...
"'0
sQ.
I i
i Q.
I
! 11 200
i 8000
5600
, 4000
2800
2000
1400
1000
710
500
355
250
180
125
90
63
45
38
Таблица 2.1.4. 1
Диаметр прово
ЛОКИ,мкм
I Допуск дЛЯ OT
rРСТИ М
'::.:: I I
u 1 :1:... U
>. MU >.
50; eg. l ' 5
qS ФСО q
... :1:..., S
'S U qo '
::аа. Ф ra ..а
:l: Ф аоа. :1:
.D U Ф б
r::; о I ...
qra
q ::.::аоl Ф i
::.:: uo;,:e
ra I о l '
:;: ОФ!
11:17 ,
. .+ . i
+Х ! :tY i +Z d d max
77 60 .1 2900
600 250 I 430 2000 2300
470 180 320 1600 1900
370 130 250 1400 1700
290 90 190 1120 1300
230 70 150 900 1040
180 50 110 710 820
140 30 90 560 640
112 25 69 450 520
89 18 54 315 360
72 13 43 224 260
58 9,9 34 160 190
47 7,6 27 125 150
38 5,8 22 90 104
32 4,6 18 63 72
26 3,7 15 45 52
22 3,1 13 32 37
30 35
Q.
...
'S Ф
:е
.lI ra
:l:s
q
co,s
&::а
:1: :1:
ф.lI ,
l '
(l) S
а.:е
I
!
r::;
Ф
q
Ф
Q.
Е:
'S
::а
:е
S
...
U
>.
Е:
о
Q
d. ; ,
mю
2100 i
1700 i
1300 I
1200
950
770
600
480
380
270
190
130
106
77
54
38
27
24
.2.1.6. Индикаторные трубки
35
Максимальный допуск (см. Международ
ttый Стандарт /SO 3310/1(1975» для разме
ра отверстия (+Х): не должно быть отверстий,
размер которых превышает номинальный
размер более чем на величину х:
Х = 2(т О . 75 ) +4(т О . 25 ).
3
rде:
т номинальный размер отверстия.
Допуск для среднеro значения размера
отверстия (:1: У): средний размер отверстия не
должен отклоняться от номинальноrо размера
более чем на величину :1: У:
т о . 9В
Y= +16.
27 '
Промежуточный допуск (+Z): не более 6 %
общеro числа отверстий MorYT иметь разме
ры между {(номинальный +Х» и «номинальный
+Z»:
z= x+y .
2
Диаметр проволоки d: диаметр проволо
ки, применяемой для плетения металлич
ской проволочной ткани, вставленной в рамку,
представлен в таблице 2_1.4. 1. Номинальный
диаметр проволоки может отличаться от зна
чения d внутри пределов d max и d miл . Пределы
установлены в допустимом диапазоне :t15 0/0
от рекомендуемых номинальных размеров.
Диаметр проволоки по всему ситу должен быть
одинаковым.
01/2013:20105
2.1.5. ПРОБИРКИ
ДЛЯ СРАВНIt1ТЕЛЬНЫХ
ИСПЫТАНИИ
Пробирки, используемые для сравнитель
ных испытаний, это специально подобран
ные пробирки из бесцветноro стекла с одина
ковым внутренним диаметром и прозрачным
плоским дном.
Слой жидкости исследуется сверху вниз по
вертикальной оси пробирки на белом или, при
необходимости, на черном фоне. Испытание
проводят при рассеянном свете.
Обычно используются пробирки с BHYTpeH
ним диаметром 16 мм. Пробирки с большим BHY
тренним диаметром MorYT быть использованы
вместо вышеупомянутых при условии увеличе-
ния объема испытуемой жидкости на столько,
чтобы высота жидкости в пробирках была не
ниже, чем при аналоrичном испытании с исполь
зованием жидкости в пробирках с внутренним
диаметром 16 мм.
01/2013:20106
2.1.6. ИНДИКАТОРНЫЕ ТРУБКИ
Индикаторные трубки это rерметизи
рованные цилиндрические трубки, состоящие
из инертноro прозрачноrо материала и CKOH
струированные с учетом возможности пропу
скания через них rаза. Они содержат peareHT,
подходящий для визуализации обнаруживае
моro вещества, адсорбированный на инерт
ном носителе, и, при необходимости, очищаю
щие слои и/или адсорбирующие фильтры для
удаления веществ, которые MorYT взаимодей
ствовать с обнаруживаемым веществом. Слой
индикатора содержит либо один peareHT, по
зволяющий обнаружить определенное веще
ство, либо насколько peareHToB для обнару
жения нескольких веществ (однослойные и
мноroспойные трубки).
Испытания проводят путем пропускания тре-
буемоro объема rаза через индикаторную трубку.
Длина окрашенноro слоя или интенсивность из
менения цвета на rрадуировочной шкале явля
ется функцией и мерой массовой концентрации
определяемоro компонента.
Проверка индикаторных трубок проводится
в соответствии с инструкциями изroтовителя.
Под20товка к измерению. Про водится со-
rласно инструкциям изroтовителя или следую
щим образом. Устройство для подачи rаза под
соединяют к реryлятору давления с иrольчатым
клапаном. Соединяют rибкий шланr трубки с
У частью клапана и продувают систему (рисунок
2.1.6. 1). Присоединяют открытый конец индика
торный трубки к короткому концу шланrа и pery
лируют насосом объем анализируемою rаза, про-
ходящеro через трубку. Записывают значения,
соответствующие длине окрашенною слоя или
интенсивности цвета на rрадуировочной шкале.
При отрицательном результате анализа индика
торная трубка должна быть откалибрована rазом,
содержащим соответствующую примесь.
Ввиду использования в воздухозаборном
устройстве масла необходимо проверить peaK
цию трубки на используемое масло. Информа
ция о реакционной способности для различных
масел приводится в сопроводительном листке,
прилаrаемом к трубке. Если используемое
масло не указано в сопроводительном листке,
изrотовитель трубки должен проверить реакци
онную способность и при необходимости обе
спечить при60р специальной трубкой для дан-
I-юю масла.
36
rосударственная фармакопея Республики Беларусь
2
з
4
5
6
1 подача rаза;
2 реryлятор давления;
3 иrольчатый клапан;
4 "у" конец;
5 индикаторная трубка;
б насос для индикаторной трубки;
7 открытый конец для выхода rаза в атмосферу.
Рисунок 2.1.6. 1. Прибор для индикаторных
трубок
Индикаторная трубка для диоксида уrле-
рода. rерметизированная стеклянная трубка,
содержащая адсорбирующие фильтры и подхо
дящие носители для индикаторов: rидразина и
кристаллическоro фиолетовоro. Минимальная
определяемая концентрация 100 ррт с OTHO
сительным стандартным отклонением :1:15 %.
Индикаторная трубка для диоксида серы.
rерметизированная стеклянная трубка, coдep
жащая адсорбирующие фильтры и подходящие
носители для индикаторов: йода и крахмала.
Минимальная определяемая концентрация
0,5 ррт с относительным стандартным отклоне
нием :1:15 %.
Индикаторная трубка для масел. rермети
зированная стеклянная трубка, содержащая aд
сорбирующие фильтры и подходящие носители
для индикатора кислоты серной. Минималь
ная определяемая концентрация 0,1 мr/м З
с относительным стандартным отклонением
:1:30%.
Индикаторная трубка для монооксида
азота и диоксида азота. rерметизированная
стеклянная трубка, содержащая адсорбирую
щие фильтры и подходящие носители для окис
ляющеro слоя (соль Cr (VJ» и индикатора ди
фенилбензидина. Минимальная определяемая
концентрация 0,5 ррm с относительным CTaH
дартным отклонением :1:15 %.
Индикаторная трубка для моноокси-
да уrлерода. rерметизированная стеклянная
трубка, содержащая адсорбирующие фильтры и
подходящие носители для индикаторов: йода (V)
оксида, селена диоксида и кислоты серной ды
мящей. Минимальная определяемая KOHцeHTpa
ция 5 ррm или менее с относительным CTaH
дартным отклонением :1:15 %.
7
Индикаторная трубка для сероводорода.
rерметизированная стеклянная трубка, coдep
жащая адсорбирующие фильтры и подходящие
носители для индикатора соли свинца. Мини
мальная определяемая концентрация 1 ррm
или менее с относительным стандартным откло
нением :1:10 %.
Индикаторная трубка для паров воды.
rерметизированная стеклянная трубка, coдep
жащая адсорбирующие фильтры и подходящие
носители для индикатора перхлората маrния.
Минимальная определяемая концентрация
67 ррт или менее с относительным CTaHдapT
ным отклонением :1:20 %.
2.2.
ФИЗИЧЕСКИЕ И ФИЗИКО-
ХИМИЧЕСКИЕ МЕТОДЫ
01/2013:20201
2.2.1. ОПРЕДЕЛЕНИЕ
ПРОЗРАЧНОСТИ И СТЕПЕНИ
МУТНОСТИ ЖИДКОСТЕЙ
ВИЗУАЛЬНЫЙ МЕТОД
Для определения прозрачности и степени
мутности жидкостей используют одинаковые про
бирки из бесцветноro, прозрачноro и нейтрально
ro стекла с плоским дном, которые имеют BHYTpeH
ний диаметр от 15 мм до 25 мм. Слой испытуемой
жидкости толщиной 40 мм сравнивают с 40 мил
лиметровым слоем свежеприrотовленноro, как
описано ниже, эталона. Сравнение растворов
проводят при рассеянном дневном освещении
через 5 минут после приroтовления эталона, про-
сматривая объекты вдоль вертикальной оси про
бирок на черном фоне. Рассеянный свет должен
быть таким, чтобы эталон I леrко отличался от
воды, а эталон 11 леrко отличался от эталона 1.
Прозрачными считаются жидкости, которые
по прозрачности не отличаются от воды Рили
раствора, который используют при приroтовле
нии жидкости в описанных выше условиях, или
которые не превышают по интенсивности мут
ность эталонной суспензии 1.
Раствор rидразина сульфата. 1,0 r 2идpa
зина сульфата Р растворяют в воде Р и доводят
водой Р до объема 100,0 мл. Раствор выдержи
вают в течение 4 6 ч.
Раствор rексаметилентетрамина. 2,5 r 2eK
саметилентетрамина р растворяют в 25,0 мл
2.2.1. Определение прозрачности и степени мутности жидкостей
37
воды Р в колбе вместимостью 100 мл со CTe
клянной притертой пробкой.
Исходная опалесцирующая суспензия
(суспензия формазина). 25,0 мл раствора rидра
_на сульфата прибавляют к приroтовленному
раствору rексаметилентетрамина, перемеши
вают и выдерживают в течение 24 ч. Суспензия
стабильна в течение 2 месяцев при хранении в
стеклянной посуде, которая не имеет дефектов
rюверхности. Суспензия не должна прилипать к
стеклу и перед применением должна быть тща
тельно перемешана.
Основная опалесцирующая суспензия.
15,0 мл исходной опалесцирующей суспензии
доводят водой Р до объема 1000,0 мл. Срок rод
ности основной опалесцирующей суспензии 24
часа.
Эталоны. Приroтовление эталонов прово
дят в соответствии с таблицей 2.2.1. 1. OCHOB
ную опалесцирующую суспензию и воду Р CMe
шивают и встряхивают перед применением.
Таблица 2.2.1. 1
1 11 111 IV
Основная опа
лесцирующая 5,0 10,0 30,0 50,0
суспензия, мл
Вода Р, мл 95,0 90,0 70,0 50,0
Эталон мутности. Суспензия формазина,
приroтовленная смешиванием равных объемов
раствора rидразина сульфата и раствора reKca
метилентетрамина, является исходной суспен
зией с 4000 NTU (Nephe/omet,ic Tu,bidity Uпits
нефелометрические единицы мутности). Эта
лоны 1, 11, 111 и IV имеют значения 3 NTU, 6 NTU,
18 NTU и 30 NTU соответственно. Стабилизиро
ванные суспензии формазина, приrодные для
приroтовления стабильных разведенных этало
нов мутности, являются коммерчески доступны
ми и MOryT быть использованы в случае, если
выдерживают сравнение с эталонами, приroтов
пенными, как указано выше.
Формазин обладает характеристиками, дe
пающими ero превосходным эталоном MYTHO
сти. Он может быть воспроизводимо приroтов
пен из реактивов. Физические характеристики
Qелают ero желательным эталоном для rрадуи
:ювки светорассеяния. Полимер формазин co
::тоит из цепей различной длины, которые при
имают случайные конфиrурации, что ПРИВОДИТ
[ получению частиц с широким спектром форм
1 размеров, аналитически соответствующих ча
:тицам С различными размерами и формами,
lаходящимся в испытуемых образцах. Блаroда
'я трассируемости и воспроизводимости CBeTO
,ассеивающих свойств формазина инструмен
альные калибровочные алroритмы и критерии
'. Зак 1712
выполнения по большей части основаны на этом
стандарте,
ИНСТРУМЕНТАЛЬНЫЕ МЕТОДЫ
ВВЕДЕНИЕ
Степень мутности раствора может быть
определена с помощью инструментальноro из
мерения поrлощения или рассеяния света за
счет субмикроскопических неоднородностей оп
тической плотности опалесцирующих растворов
и суспензий. На практике используются 2 метода:
нефелометрия и турбидиметрия. Для измерения
мутности окрашенных испытуемых образцов ис
пользуют методы относительной турбидиметрии
и нефелометрии с выбором отношения.
Эффект рассеяния света суспендированны
ми частицами может быть измерен с помощью
либо проходящеro света (турбидиметрия), либо
рассеянноro света (нефелометрия). Относитель
ная турбидиметрия сочетает принципы и нефе
лометрии, и турбидиметрии. Тубидиметрия и He
фелометрия MOryT использоваться в случае со
слеrка опалесцирующими суспензиями, Усло
вия приroтовления эталона должны быть четко
оroворены. Для количественных определений
существенным является построение калибро
вочноro rрафика, так как зависимость между оп
тическими свойствами суспензий и KOHцeHTpa
ции дисперrированной фазы является в лучшем
случае полуэмпирической. .
Определение опалесценции окрашенных
жидкостей проводят методом относительной
турбидиметрии или нефелометрии с выбором
отношения, так как окрашивание оказывает He
rативное воздействие, ослабляя как падающий,
так и рассеянный свет, и уменьшает значение
мутности. Эффект настолько велик, что даже в
случае умеренно окрашенных образцов обыч
ные нефелометры не MOryт быть использованы.
Инструментальная оценка прозрачности и
опалесценции является более селективным Me
тодом, который не зависит от остроты зрения
аналитика. Числовые результаты более наrляд
ны для мониторинrа качества и контроля про
цесса, особенно в проверке стабильности при
хранении. Например, предварительные число
вые данные по стабильности MOryT быть исполь
зованы для определения возможности тоro, что
данная партия лекарственноro средства или aK
тивноro фармацевтическоrо инrредиента превы
сит предельное значение для срока rодности до
окончания срока хранения.
НЕФЕЛОМЕТРИЯ
при просматривании суспензии под опреде
ленным уrлом к направлению падающею света
система кажется мутной вследствие отраже
ния света от частичек суспензии (эффект Тин
даля). Опредeneнная часть световоro луча, по
падающеro в мутную жидкость, проходит через
нее, друraя часть поrлощается, а оставшаяся
38
rосударственная фармакопея Республики Беларусь
часть рассеивается суспендированными части
цами. Если измерение проводят под уrЛОlv: 900
к падающему лучу света. рассеянный суспен,L'И
рованными частицами свет может быть исполь
зован для определения их концентрации, обу
словленной колlt1чеством и размерами частиц,
влияющих на константу остаточноro рассеяния.
Эталон должен иметь постоянную степень мут
ности, а испытуемый образец и эталон должны
быть приroтовлены в идентичных условиях.
Эффект Тиндаля зависит и от числа частиц, и
от их размеров. Нефелометрические данные
более достоверны при низкой мутности суспен
зий, Korдa наблюдается линейная зависимость
между значением нефелометрической единицы
мутности (NTU) и относительным сиrналом дe
тектора. При возрастании степени мутности па
дающий свет попадает не на все частицы, а pac
сеянное излучение друrих частиц поrлощается
на пути к детектору. Максимальное нефеломе
трическое значение, при котором может быть
проведено достоверное измерение, находится
в диапазоне 1750 2000 NTu. ДЛЯ подтвержде
ния линейности необходимо построение rрадуи
ровочноro rрафика как минимум по 4 M KOHцeH
трациям.
ТУРБИДИМЕТРИЯ
Мутность это оптическое свойство, воз
никающее при взаимодействии между светом и
суспендированными в жидкости частицами. Это
определение оптическою свойства, обусловли
вающеro то, что падающий свет в большеи CTe
пени рассеивается или поrлощается. чем про
ходит через испытуемый образеu по прямой.
Количество твердых частиu В суспензии r,'ожет
быть определено с помощью измерения ин
тенсивности прошедшеrо света. Линейная за
висимость между мутностью и концентрацией
наблюдается в случае однородности и roMoreH
ности размера частиц в суспензии. Это справед
ливо только для очень разведенных суспензий,
содержащих маленькие частицы. Для подтверж
дения линейности необходимо построение rpa
дуировочноro rрафика как минимум по 4 M KOH
центрациям.
ОТНОСИТЕЛЬНАЯ ТУРБИДИМЕТРИЯ
В относительной турбидиметрии находит
ся отношение интенсивности прошедшеro света
к интенсивности рассеянноrо света под уrлом
900. Проведение такоro испытания компенсиру
ет влияние окрашивания испытуемоro образца.
Этоro эффекта можно добиться также за счет
использования инфракрасноro светоиcnycкакr
щеro диода при длине волны 860 НМ. ФоТОI\И
одные детекторы прибора получают и измеря
ют интенсивность рассеянноro света под уrлом
900, а также интенсивность рассеяния в прямом
направлении (отраженный свет) перед испытуе
мым образцом и интенсивность света, прошед
шеro через испытуемый образец. Измеренные
результаты в единицах NTU (отношение) рассчи
тываются делением интенсивности рассеянно
ro света под уrлом 900 на сумму интенсивности
рассеяния в прямом направлении и интенсивно
сти прошедшеro света. В относительной rурби
диметрии влияние постороннею света становит
ся незначительным. Нефелометры используют
для измерения стеПЕ:НИ мутности бесцветных
жидкостей.
Определение степени мутности эталuнов I IV
с помощью относитеЛЫ-iОИ 1 урбидимеТРIi:И показа
ло, что зависимость между концентрациями и изме
ренными значениями NTU (таблица 2.2.1. 2) явля
ется линейной. Эталоны I IV trф РБ) Muryт быть
использованы для rpадуировки прибора.
Таблица 2.2.1. 2
I Сусп .нзия форма- Тз н а ч.;;и. о ';ап.сцен-I
зина I ции (NTU) !
I Э эталон "" j !
I талон ..
l ' 18 '
! Этаrюн 111
! Эталон IV r ' 301
i Основная I I
: опалесцирующая I 60 '
: суспензия 1 i
I Исходная i I
lопалесцирующая! 4000
l суспензия . . . .
ИНСТРУМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ
ОПАЛЕСЦЕНЦИИ
Требования, приведенные в частных фар
макопейных статьях, указаны в терминах для
визуальноro определения степени мутности
в сравнении с конкретным эталоном. Может
быть использован также и инструментальный
метод определения степени мутности при yc
ловии,чтоприБОРСGответствуеттр Бованиям,
указанным ниже, и если ПIJОБедена rрадуиров
ка с использованием эталонов I IV и воды Р
или используемоro растворителя.
Прибор. В качестве используемых источ
ников света в приборах для относительной TYP
бидиметрии или нефелометрах с Вl:?lбором OT
ношения используют вольфрамовую лампу со
спектральной чувствительностью около 550 нм,
работающую при температуре 2700 К, или ин
фракрасный светоиспускающий диод, имеющий
максимум испускания при 860 нм со спектраль
ной полосой пропускания 60 нм. Moryт исполь
зоваться И друrие подходящие источники света.
В качестве детекторов, записывающих измене
ния в рассеянии или пропускании света испыту
емым образцом, обычно используют кремние
вые фотодиоды и фотоумножители. Рассеянный
свет под уrлом (90:!:2,5)0 реrистрируется первич
ным детектором. Остальные детекторы реrи
стрируют интенсивность рассеяния в прямом и
В обратном направлениях. Используемые при
боры rрадуируют с использованием эталонов с
39
2.2.2. Определение степени окрашивания жидкостей
известной степенью мутности; кроме этою, при
боры должны быть способны к автоматическому
определению степени мутности. Результаты ис
пытания, выраженные в NTU, считываются непо
средственно с дисплея прибора и сравниваются
со спецификацие , приведенной в частной фар
макопейной статье.
Прибор считают приroдным, если он COOTBeT
ствует следующим техническим условиям.
Единицы измерения: NTU NTU основана на
мутности первичноro стандартноro образца фор
мазина. Т аюке MOryт использоваться FTU (Forтaziп
Turbidity Units единицы мутности по формазину)
или FNU (Fоrтаziп Nephe/ometry Uпits формази
новые нефелометрические единицы), которые эк
вивалентны NTU при низких значениях (до 40 NТИJ
Эти единицы используются во всех трех инстру
ментальных методах (нефелометрия, турбидиме
трия и относительная тур6идиметрия).
Диапазон измерениЙ от 0,01 NTU до
1100 NTU.
Разрешение: 0,01 NTU в диапазоне 10
NTU; 0,1 NTU в диапазоне 1 100 NTU; 1 NTU в
диапазоне более 100 NTU Прибор rpадуируют и
контролируют с использованием эталонов форма
зина.
Точность: в диапазоне 10 NTU: :1:(2 % от
показания прибора + 0,01) NTU; в диапазоне 1
1000 NTU::1:5 %.
Повторяемость: в диапазоне 1 10 NTU:
:1:0,01 NTU; в диапазоне 1 1000 NTU: :1:2 % от из
меренною значения.
rрадуировка: используют 4 эталона форма
зина в интересующем диапазоне. Moryт быть ис
пользованы стандартные образцы, описанные
выше, или подходящие стандартные образцы, от
калиброванные по исходным суспензиям опалес
ценции.
Посторонний свет: посторонний свет явля
ется существенным источником ошибок при низких
значениях турбидиметрических измерений; посто
ронний свет попадает на оптическую систему дe
тектора, но приходит не от испытуемоro образца:
менее 0,15 NTU в диапазоне 10 NTU, менее
0,5 NТUвдиапазоне 1 1000 NTU
Приборы, отвечающие требованиям, указан
ным выше, и поверенные с использованием эта
лонов, описанных в разделе «Визуальный метод»,
MOryт быть использованы для подтвер>!Щения co
ответствия требованиям частной фармакопейной
статьи вместо визуальноro определения.
Moryт быть использованы приборы с xapaK
теристиками (диапазоном измерения, разреше
нием, точностью, повторяемостью), отличными от
указанных выше, при условии, что они полностью
валидированы и подходят для предполаrаемоrо
использования. Для отдельных испытуемых фар
мацевтических субстанций/лекарственных средств
дпя подтвер>!Щения возможности использования
также должна быть валидирована методика испы
1ания. Прибор и методолоrия не должны противо
речить свойствам испытуемою образца.
01/2013:20202
2.2.2. ОПРЕДЕЛЕНИЕ СТЕПЕНИ
ОКРАШИВАНИЯ ЖИДКОСТЕИ
Определение степени окрашивания жид
костей в ряду коричневый желтый красный
проводят визуально путем сравнения с COOT
ветствующими эталонами (растворами cpaB
нения) одним из двух описанных ниже методов
настоящей статьи.
Раствор считается бесцветным, если он
выдерживает сравнение с водой Р или paCT
ворителем, или окрашен не более интенсивно,
чем эталон В(К)9'
=Степень окрашивания испытуемоro paCT
вора не должна превышать степени окраши
вания соответствующеro эталона, а цвет испы
туемоro раствора должен быть максимально
приближен к цвету соответствующеro эталона.#
МЕТОД I
2,0 мл испытуемой жидкости сравнива
ют с 2,0 мл воды р, растворителя или эталона
(см. таблицы эталонов), описанноro в HaCTa
ящей статье, используя одинаковые пробир
ки из бесцветноro, прозрачнorо, нейтральноro
стекла с внешним диаметром 12 мм. CpaBHe
ние окраски проводят при рассеянном ДHeB
ном освещении, просматривая объекты roри
зонтально (перпендикулярно оси пробирок) на
белом матовом фоне.
#По методу 1 обычно проводят сравнение
окрашивания жидкостей с эталонами [В(К),
ВУ(КЖ), У(Ж), GУ(ЗЖ), R(Кр)],з,#
МЕТОД 11
40 миллиметровыи слой испытуемой жид
кости сравнивают с 40 милпиметровым слоем
воды Р. растворителя или эталона (см. табли
цы эталонов), указанноro в частной статье, ис
пользуя одинаковые пробирки из бесцветноro,
прозрачноro, нейтральноro стекла с плоским
дном, которые имеют внутренний диаметр от
15 мм до 25 мм. Сравнение окраски проводят
при рассеянном дневном освещении, просма
тривая объекты вдоль вертикальной оси про
бирок на белом фоне.
#По методу 11 обычно проводят сравнение
окрашивания жидкостей с эталонами [В(К),
ВУ(КЖ), У(Ж), GУ(ЗЖ), R(Kp)]4 9'#
ЭТАЛОНЫ (РАСТВОРЫ СРАВНЕНИЯ)
Исходные растворы
Желтый раствор. 46 r железа (111) хлорида Р
растворяют в 900 мл смеси кислота хлористов(l
дородная Р вода Р (25:975, 06/06) и доводят до
объема 1000,0 мл этим же растворителем. Опре
деля ют концентрацию полученноro раствора и
разводят раствор этим же растворителем таким
образом, чтобы содержание FeCl 3 '6НР в 1 мл paB
нялось 45,0 Mr:
40
rосударственная фармакопея Республики Беларусь
Раствор хранят в защищенном от света месте.
Определение концентрации. 10,0 мл полу
ченноro раствора помещают в коническую кол5у
вместимостью 250 мл с притертой пробкой, при
бавляют 15 мл воды Р, 5 мл кислоты хлористово
дородной Р и 4 r калия йодида Р, колбу закрывают
и выдерживают в течение 15 мин в темном месте.
Прибавляют 100 мл воды Р и выделившийся йод
титруют 0,1 М раствором натрия тиосульфата,
прибавляя в конце титрования в качестве индика
тора 0,5 мл раствора крахмала Р.
1 мл 0,1 М раствора натрия тиосульфата
соответствует 27,03 Mr FеСl з '6Нр.
Красный раствор. 60 r кобальта хлорида Р
растворяют в 900 мл смеси кислота хлористово
дородная Р вода Р (25:975, об/о6) и доводят до
объема 1000,0 мл этим же растворителем. Опре
деля ют концентрацию полученноro раствора и
разводят раствор этим же растворителем таким
образом, чтобы содержание CoCI 2 '6H 2 0 в 1 мл paB
нялось 59,5 Mr:
Определение концентрации. 5,0 мл полученно
ro раствора помещают в коническую колбу вмести
мостью 250 мл с притертой пробкой, прибавляют
5 мл раствора водорода пероксида разведенно
20 Р И 10 мл раствора 300 r/л натрия 2идроксида
Р, осторожно кипятят в течение 1 О минут, охлаж
дают и прибавляют 60 мл кислоты серной разве
денной Р и 2 r калия йодида Р. Колбу закрывают
и осторожно встряхивают до полноro растворения
осадка. Выделившийся йод титруют 0,1 М pacтвo
ром натрия тиосульфата, прибавляя в конце ти
трования в качестве индикатора 0,5 мл раствора
крахмала р, и титруют до появления бледно розо
воro окрашивания.
1 мл 0,1 М раствора натрия тиосульфата
соответствует 23,79 Mr CoCI 2 '6Hp.
Синий раствор. 63 r меди сульфата Р paCT
воряют в 900 мл смеси кислота хлористоводо
родная Р вода Р (25:975, 06/06) и доводят до
объема 1000,0 мл этим же растворителем. Опре
деля ют концентрацию полученноro раствора и
разводят раствор этим же растворителем таким
образом, чтобы содержание Cu80 4 -5H 2 0 в 1 мл
равнялось 62,4 Mr:
Определение концентрации. 10,0 мл полу
ченноro раствора помещают в коническую колбу
вместимостью 250 мл с притертой пробкой, при
бавляют 50 мл воды Р, 12 мл кислоты уксусной
разведенной Р и 3 r калия йодида Р Выделивший
ся йод титруют 0,1 М раствором натрия тиосуль
фата, прибавляя в конце титрования в качестве
индикатора 0,5 мл раствора крахмала р, и титруют
до появления бледно коричневоro окрашивания.
1 мл 0,1 М раствора натрия тиосульфата
соответствует 24,97 Mr Cu80 4 '5H 2 0.
основные растворы
Пять основных растворов приroтавливают
с использованием трех исходных, как указано в
таблице 2.2.2. 1.
Основные растворы
Таблица 2.2.2. 1
Объем,мл
Основной раствор Желтый Красный Синий Раствор кислоты хлори
раствор раствор раствор стоводородной (10 r/л HCI)
В (Ьrowп) 3,0 3,0 2,4 1.6
К (коричневый)
ВУ (Ьrоwпish уеllоw) 2,4 1,0 0,4 6,2
кж (коричневато желтый)
У (yellow) 2,4 0,6 0,0 7,0
Ж (желтый)
GY (grеепish уеllоw) 9,6 0,2 0,2 0,0
ЗЖ (зеленовато желтый)
R (,ed) 1,0 2,0 0,0 7,0
Кр (красный)
Эталоны для методов 1 и 11.
Эталоны приroтавливают с использованием пяти основных растворов, как указано в таблицах
2.2.2. 2 2.2.2. 6.
Эталоны шкалы В(К)
Таблица 2.2.2. 2
Объем,мл
Эталон Основной Раствор кислоты
раствор хлористоводородной
В(К) (10 r/л HCI)
В(К)1 75,0 25,0
В(К)2 50,0 50,0
Объем,МЛ
Эталон Основной Раствор кислоты
раствор хлористоводородной
В(К) (10 r/л HCI)
В(К)з 37,5 62,5
В(К)4 25,0 75,0
2.2.3. Потенциометрическое определение рН
41
Объем,МЛ
Эталон Основной Раствор кислоты
раствор хлористоводородной
В(К) (10 r/л HCI)
В(К)5 12,5 87,5
В(К)в 5,0 ! 95,0
!
В(К)7 2,5 I 97,5
В(К)в 1,5 98,5
В(К)9 1,0 99,0
Таблица 2.2.2. 3
Эталоны шкалы ВУ(КЖ)
Объем,мл
Эталон Основной Раствор кислоты
раствор хлористоводородной
ВУ(КЖ) (10 r/л HCI)
ВУ(ЮК)1 100,0 0,0
ВУ(ЮК)2 75,0 25,0 ,
ВУ(ЮК)З 50,0 50.0
ВУ(ЮК)4 25,0 75,0
ВУ(ЮК)5 12,5 87,5
ВУ(ЮК)6 5,0 95,0
ВУ(ЮК)7 2,5 97,5
Таблица 2.2.2А
Эталоны шкалы У(Ж)
Объем,МЛ
Эталон Основной Раствор кислоты хло-
раствор ристоводородной
У(Ж) (10 r/л НС/)
У(Ж)1 100,0 0,0
У(Ж)2 75,0 25,0
У(Ж)З 50,0 50,0
У(Ж)4 25,0 75,0
У(Ж)5 12,5 87,5
У(Ж)в 5,0 95,0
У (Ж)7 2,5 97,5
Таблица 2.2.2. 5
Эталоны шкалы GУ(ЗЖ)
Объем,мл
Эталон Основной Раствор кислоты хло-
раствор ристоводородной
GУ(3Ж) (10 r/л HCI)
GУ(ЗЖ)1 25,0 75,0
GУ(ЗЖ)2 15,0 85,0
GУ(ЗЖ)з 8,5 91,5
GУ(ЗЖ) 5.0 95,0
GУ(ЗЖ): I 3.0 97,0
GY(ЗЖ)_ I 1.5 I 98,5
(ЗЖ ) 0,75 i 99,25
Таблица 2.2.2. 6
Эталоны шкалы R(Kp)
Объем,мл
Эталон Основной Раствор кислоты
раствор хлористоводородной
R(Kp) (1 О r/л HCI)
R(Kp )1 100,0 0,0
R(Kp )2 75,0 25,0
R(Kp )з 50,0 50,0
R(Kp )4 37,5 62,5
R(Kp )5 25,0 75,0
R(Kp )6 12,5 87,5
i R(Kp )7 5,0 95,0
Хранение
Эталоны для определения степени OKpa
шивания жидкостей по методу I хранят в защи
щенном от света месте в запаянных пробир
ках из бесцветноro, прозрачноrо, нейтральноro
стекла С внешним диаметром 12 мм. #Для при
roТ08ления эталонов для определения степени
окрашивания жидкостей по методу I используют
основные растворы, приroтовленные непосред
ственно перед применением.#
Эталоны для определения степени окраши-
вания жидкостей по методу 11 приroтавливают
из основных растворов непосредственно перед
применением.
#Срок roдности первичных и основных pac
творов при хранении в защищенном от света
месте в штанrлазах с притертой пробкой 1 roд.#
01/2013:20203
2.2.3. ПОТЕНЦИОМЕТРИЧЕСКОЕ
ОПРЕДЕЛЕНИЕ рН
Водородный показатель (рН) число, xapaK
теризующее концентрацию (активность) ионов
водорода в водных растворах. На практике рН
определяют экспериментально. Значение рН ис
пытуемоro раствора связано со значением рН стан-
дартноro раствора (pHs) следующим уравнением:
E E
pH==PHS k'
rдe:
Е потенциал электрода в испытуемом
растворе, В;
Е, потенциал Toro же электрода в paCTBO
ре с известным значением рН (pHJ, В;
k температурный коэффициент (изменение
потенциала при изменении значения рН на едини
цу, рассчитанное по уравнению Нернста) (таблица
2.2_3. 1), В.
42
rосударственная фармакопея Республики Беларусь
Таблица 2.2.3. 1
Значения k при различных температУРаХ
Температура, ос k,B
15 0,0572
20 0,0582
25 0,0592
30 0,0601
35 0,0611
Потенциометрическое определение рН
проводят путем измерения разности потенциа
лов между двумя электродами, поrруженными
в испытуемый раствор: один из электродов чув
ствителен к ионам водорода (обычно это CTe
клянный электрод), друroй электрод cpaB
нения (например, насыщенный каломельный
электрод).
Прибор. Измерительным прибором служит
вольтметр с входным сопротивлением, как ми
нимум в 100 раз превышающим сопротивление
используемых электродов. Прибор обычно rpa
дуирован в единицах рН и должен иметь такую
чувствительность, чтобы можно было провести
измерение с точностью не менее 0,05 единиц
рН или не менее 0,003 В.
Методика. Если иноro не указано в част
ной статье, все измерения проводят при одной
И той же температуре в интервале температур
от 20 ос до 25 ОС. В таблице 2.2.3. 2 приведена
зависимость значения рН от температуры для
различных стандартных буферных растворов,
используемых для калибровки. При необходи
мости используют температурные поправки 8
соответствии с инструкцией предприятия про
изводителя. Прибор калибруют с помощью бу
ферноro раствора калия rидрофталата (пер
вичный стандарт) и одноro из буферных
растворов с друrим значением рН (предпочти
тельно одноro из указанных в таблице 2.2.3. 2).
Показания прибора для третьеro буферноro
раствора с промежуточным значением рН не
должны отличаться более чем на 0,05 единиц
рН от табличноrо значения рН, соответствую
щеro этому раствору. Электроды поrружают в
исследуемый раствор и измеряют рН при тех же
условиях, что и для буферных растворов.
Если прибор используется ежедневно, ero
rрадуировку проводят реryлярно. В противном
случае rрадуировку прибора проводят перед
каждым измерением.
Все испытуемые растворы и стандартные
буферные растворы должны быть приroтовлены
с использованием воды, свободной от У2лерода
диоксида, Р.
приrОТОВЛЕНИЕ СТАНДАРТНЫХ
БУФЕРНЫХ РАСТВОРОВ
0,05Мрастворкалиятетраоксалата.12,61 r
С 4 Н з КО в '2Н 2 О растворяют в воде, свободной
от У2лерода диоксида, Р и доводят до объема
1000,0 мл этим же растворителем.
Насыщенный при 25 ос раствор калия rи
дротартрата. Избыток С 4 Н 5 КО 6 смешивают при
энерrичном встряхивании с водой, свободной от
У2лерода диоксида, Р при температуре 25 ос в
течение 30 мин. Фильтруют или сливают Haдo
садочную жидкость. Раствор roтовят непосред
ственно перед использованием.
0,05 М раствор калия диrидроцитрата.
11,41 r С 6 Н 7 КО 7 растворяют в воде, свободной
от У2лерода диоксида, Р и доводят до объема
1000,0 мл этим же растворителем. Раствор ro
товят непосредственно перед использованием.
0,05 М раствор калия rидрофталата. 1 О, 1 3 r
С в Н е КО 4 , предварительно высушенноro при TeM
пературе (110:t2) ос в течение 1 Ч, растворяют в
воде, свободной от уелерода диоксида, Р и ДOBO
дят до объема 1000,0 мл этим же растворителем.
0,025 М раствор калия диrидрофосфата
и 0,025 М раствор динатрия rидрофосфата.
3,39 r КНРО4 и 3,53 r Na2HP04' предварительно
высушенных при температуре (120:t2) ос в тече
ние 2 ч, растворяют в воде, свободной от У2ле
рода диоксида, Р и доводят до объема 1000,0 мл
этим же растворителем.
0.0087 М раствор калия диrидрофосфата
и 0,0303 М раствор динатрия rидрофосфата.
1,18 r кнро 4 и 4,30 r NэzНРО.,. Ilредваритель
но высушенных при температуре (120:t2) сс в
течение 2 часов, растворяют в воде. свободной
от уелерода диоксида, Р и доводят до объема
1000,0 мл этим же растворителем.
0,01 М раствор натрия тетрабората. 3,80 r
Na 2 B..0 7 '10H 2 0 растворяют 8 воде, свободной от
У2лерода диоксида, Р и доводят до объема 1000,0
мл этим же растворителем. Хранят в защищен
ном от атмосферноrо уrлерода диоксида месте.
0,025 М раствор натрия карбоната и 0,025
М раствор натрия rидрокарбоната. 2,64 r
Nа 2 СО з и 2,09 r NаНСО з растворяют в воде,
свободной от У2лерода диоксида, Р и доводят
до объема 1000,0 мл этим же растворителем.
Хранят в защищенном от атмосферноro уrлеро
да диоксида месте.
Насыщенный при 25 ос раствор каль-
ция rидроксида. Избыток кальция 2идроксuда
р встряхивают с водой, свободной от У2лерода
диоксида, Р и сливают надосадочную жидкость
при температуре 25 ОС. Хранят в защищенном от
атмосферноro уrлерода диоксида месте.
ХРАНЕНИЕ
Буферные растворы хранят в химически стой
ких плотноукупориваемых контейнерах, таких как
контейнеры из стекла класса I или пластмассо
вые контейнеры для водных растворов.
2.2.4. Зависимость между реакцией раствора, приблизительным значением рН 43
Таблица 2.2.3. 2
Значение рН стандартных буферных растворов при разных температурах
i I I s: I S..,., I
u I " с:: о:: Io::t; .u
о:: о о:: о:: S ro q ('1') i\j S I ro I rn '
L() I ro qa. 1 0::0 -& а. ...а. ro NO::
roro No:: ro ! ro'" roro o:: ro S u !ro ...' S
:.::'" ! ss'" I:'::ro :.::... ,SL()O::", CtS О ro Ж o;ro S:1ro
ro a.t:;ro а.а. a.ro I t:;NSro:.::o -& ж... а.L()sЖ a..aq
а. t:; I 1:: ro а. ! , ' о'" о t:; I q -& а. S а. о а. ro О N о.. О 1:: t:; S
о ro ,s :.:: !i r:a S r:a CtI I а. О CtI О ro g а. о а. r:a q, ... t!) 'S CtI U
t ! ]g- t g t€- gs -& Iot; t f; S
ctlro l ' Жr:aО ctla. ctlO I... qo -&CtI a. CtI 0..:':: 0 ЖОа.
а. а. Ж ... а. а. q а. о.. u CtI а. а. ё.. u а. 0::'" а. CtI r:a q ,
... I Q)uq S q ctI.dog О S a.t; ...Оа. Q)...S I
ф l 3'ctls :Е.... s 0..1fr:a , -& а. ... :Eror:aq 3'u....
Lt')'" ]а..... L()....::2:0 1......0 10 L()5 ]CtI I
О u ..... 1 0 L()-& &- Ж q INt!) "а.
О CtI О О 8. q .... s О 6f;
::I: ciq О :.::
u
о
CtI
о..
»
...
CtI
а.
Q)
1::
:!
Q)
.....
,
,
!
I
i
15
20
25
30
35
1 1 L\pH(1)
Ы
С 4 Н з КО в '
'2НР
1,67
1,68
1,68
1,68
1,69
Nа 2 СО з +
NаНСО з
10,12
10,06
10,01
9,97
9,93
Са(ОН)2
12,81
12,63
12,45
12,29
12.13
С Н КО КНР04+ КНРО4+
в 5 4 I Na;HP04 I Na 2 HP0 4
4,00 I 6,90 1 7,45
4,00 6,88 7,43
4,01 6,87 I 7,41 I
4,02 i 6,85 I 7,40 I
4,02 I 6,84 7,39
+0'00121 0,0028 1 1 O,0028
I
Na 2 B 4 0 7 '
'10НР
9,28
9,23
9,18
9,14
9,10
С 4 Н 5 К0 6 С 6 Н 7 КО 7
3,56
3,55
3,55
3,80
3,79
3,78
3,77
3,77
0,0082
0,0096
O,034
+0,001
0,0014
0,0022
. ... ... .,.. .
(1) дpH изменение рН на rрадус по Цельсию.
01/2013:20204
2.2.4. ЗАВИСИМОСТЬ МЕЖДУ РЕАКЦИЕЙ РАСТВОРА, ПРИБЛИЗИТЕЛЬНЫМ
ЗНАЧЕНИЕМ рН И ЦВЕТОМ ИНДИКАТОРОВ
к 10 мл испытуемоro раствора прибавляют 0,1 мл раствора индикатора, если иноrо не указано в
таблице 2.2.4. 1.
Таблица 2.2.4..1
Реакция раствора рН Индикатор Окраска J
.J
UЦелочная >8 Лакмусовая бума2а красная Р Синяя !
Раствор тимолов020 сине20 Р Серая или фиолеТ080 серая :
(0,05 мл)
Слабо щелочная 8,O 10,0 Раствор фенолфталениа Р Бесцветная или розовая :
(0,05 мл) .
I Раствор тимолов020 сине20 Р
(0,05 мл) Серая
Сильнощелочная >10 Фенолфталеиновая бума2а Р Красная
1 Нейтральная 6,O 8,O Раствор метиловО20 краСНО20 Р Желтая
I Раствор Фенолов020 краСН020 Р
! (0,05 мл)
I Нейтральная по 4,5 6,0 Оранжево-красная
i метиловому Kpac Раствор метилов020 краСНО20 Р
jHOMY
: Бесцветная; розовая или
I Нейтральная по <8,0 Раствор фенолфталеина Р красная после прибавления
I фенолфталеину I (0,05 мл) 0,05 мл 0,1 М раствора OCHO
I
I вания
44
rосударственная фармакопея Республики Беларусь
Реакция раствора рН Индикатор Окраска
Кислая <6 Раствор i:lJетилов020 краСН020 Р Оранжевая или красная
Раствор бромтимолов020 сине20 Р1 Желтая
Слабокислая 4,0 6,0 Раствор метиловоzо KpaCHOZO Р Оранжевая
Раствор бромкрезолов020 Зеленая или синяя
зеленоzо Р
Сильнокислая <4 Бума28 КОН20 краСН020 Р Зеленая или синяя
#Зависимость между интервалом рН и переходом окраски индикаторов представлена в таблице
#2.2.4. 2.#
Таблица #2.2.4. 2
Индикаторный раствор Интервал рН Переход окраски
перехода окраски
Метилов020 фиолетов020 раствор Р1 O,1 1,5 От желтой до зеленой
Малахитов020 зелеН020 раствор Р О, 1 2,0 От желтой до зеленовато roлубой
Крезоловоzо KpaCHozo раствор Р 0,2 1,8 От красной до желтой
Метанилов020 желтО20 раствор Р 1 ,2 2,3 От красной до оранжево желтой
м Крезолов020 пурпуровоzо раствор Р 1 ,2 2,8 От красной до желтой
Тимолов020 сине20 раствор Р 1 ,2 2,8 От красной до желтой
Тролеолина 00 раствор Р . 1 ,4 3,2 От красной до желтой
Хинальдинов020 краСНО20 раствор Р 1 ,4 3,2 От бесцветной до красной
Метиловоzо фиолетов020 раствор Р1 1 ,5 3.2 От зеленой до фиолетовой
Бромфенолов020 cUHezo раствор Р 2,8 ,4 От желтой до синевато фиолетовой
ДиметuловО20 желт020 раствор Р r 3,0 ,O ОТ красной до желтой
I
Метилов020 оранжев020 раствор Р 3.0 .4 I От красной ДО желтой
I ;
,
Метилов020 оранжевО20 смешанный ОТ оранжевой до желтовато зеле
раствор Р i 3.0 ,4 I ной
КОН20 краСНО20 раствор Р 3,()"""",5,О От синей до розовой
Бромкрезолов020 зелеНО20 раствор Р 3,6 5,2 От желтой до синей
Ализарина S раствор Р 3,7 5,2 От желтой до фиолетовой
Метилов020 краСН020 раствор Р 4,4 ,O От красной до желтой
Лакмус Р 5,0 8,0 От красной до синей
Метилов020 краСН020 смешанный 5,2 5,6 ОТ красно Фиолетовой до зеленой
раствор Р
Бромкрезолов020 ПУрПУРО8020 раствор Р 5,2 ,8 От желтой до синевато фиолетовой
Бромтимолов020 сине20 раствор Р1 5,8 7,4 От желтой до синей
НейтраЛЬН020 краСН020 раствор Р 6,8 8,O От красной до желтой
Фенолов020 краСН020 раствор Р 6,8 ,4 От желтой до красновато фиоле
товой
Крезолов020 краСН020 раствор Р 7,O 8,6 От желтой до красной
а Нафтолфталеина раствор Р 7,4 8,6 От желтовато розовой до зелено
вато синей
м КрезоловО20 ПУРПУРН020 раствор Р 7,4 9,0 От желтой до красно фиолетовой
Тимолов020 сине20 раствор Р 8.o 9,6 От оливково зеленой до синей
Фенолфталеина раствор Р 8,2 10,0 От бесцветной до ярко розовой
НиЛЬСК020 сине20 А раствор Р 9,0 13,O От синей до красной
Тимолфталеина раствор Р 9,3 10,5 ОТ бесцветной до синей
ПротравН020 желт020 ЗR раствор Р 10,2 12,0 От желтой до красной
Интервалы рН и переход окраски индикаторов
2.2.5. Относительная плотность
45
Индикаторный раствор Интервал рН Переход окраски
перехода окраски
Малахитов020 зелеН020 раствор Р 11, 13,O От зеленовато roлубой до бесцвет
ной
Инди20кармина индикаторный pacт 11,6 14,0 От синей до желтой
ворР
2.2.5. ОТНОСИТЕЛЬНАЯ ПЛОТНОСТЬ
Относительная плотность d:' представляет
собой отношение массы определенноro объема
вещества при температуре t, к массе paBHO
ro ему объема воды при температуре t 2 . Если в
частной статье не указано иноro, используют OT
носительную плотность d;g. Часто используется
также относительная плотность d: O . Может ис
пользоваться плотность Р20' определяемая как
масса единицы объема при температуре 20 ос
и выражаемая в килоrраммах на кубический
метр или в rpaMMax на кубический сантиметр
(1 Kr/M 3 = 0,001 r/CM 3 ). Эти величины связаны
между собой следующими уравнениями, в KOTO
рых плотность выражена в rpaMMax на кубиче
ский сантиметр:
Р20 = 0,998203. d;g или d;g = 1,00180. Р20 '
Р20 = 0,999972. d;o или d;o = 1,00003 . Р20 '
d;o = 0,998230 . d;g .
Относительную плотность или плотность
определяют с точностью до десятичных знаков,
обозначенных в частной статье, с помощью пик
нометра (твердые вещества и жидкости), rидро
статических весов (твердые вещества), apeo
метра (жидкости) или цифровою плотномера с .
осциллирующим датчиком (жидкости и rазы). AT
мосферное давление в определениях с исполь
зованием взвешивания не учитывают, так как
связанная с ним ошибка не превышает единицы
в третьем десятичном знаке. Давление воздуха
не оказывает влияние на определения с исполь
зованием плотномера.
Плотномер с осциллирующим датчиком.
Прибор состоит из следующих основных COCTaB
ляющих:
U образная трубка, обычно из боросили
катноro стекла, в кот()рую помещают испытуе
мую жидкость;
reHepaTop маrнитоэлектрическоrо или пье
зоэлектрическоro возбуждения, заставляющий
колебаться U образную трубку с характеристи
ческой частотой, зависящей от плотности испы
туемой жидкости;
8 За.. 1712.
01/2013:20205
измерительный аппарат, определяющий
период колебаний (7), который может быть пре
образован прибором непосредственно в плот
ность либо использован для расчета плотности
с использованием постоянных А и В, как указа
но ниже.
Резонансная частота (f) является функцией
динамической жесткости (с) и массы (т) систе
мы:
(2 . .
Т 2 т 4л- 2
Отсюда:
2 ( М p-V ) 2
т с + .4л- ,
rдe:
м масса трубки;
V внутренний объем трубки.
Введение двух постоянных А с / (4л- 2 . V) И
в М/ V приводит уравнение к классическому
виду для осциллирующеro датчика:
р = А- т 2 В .
Постоянные А и В определяют для KOHKpeT
ною прибора, заполняя U образную трубку двумя
различными образцами с известными плотно
стями, например, деrазированной водой Р и воз
духом. Проверку получаемых данных проводят
ежедневно с использованием деrазированной
воды Р. Результаты, полученные при провер
ке с использованием деrазированной воды Р,
не должны отличаться от стандартных значе
ний (Р20 = 0,998203 r/CM 3 , d;g = 1,000000) более
чем на специфицированную ошибку. Например,
прибор, специфицированчый до :1:0,0001 r/CM 3 ,
считается приюдным для дальнейших измере
ний, если выдает значение (0,9982:1:0,0001) r/CM 3 .
В противном случае необходима повторная Ha
стройка. Реrулярно должна проводиться rраду
ировка с использованием сертифицированных
стандартных образцов. Измерения должны пpo
46
rосударственная фармакопея Республики Беларусl:.
водиться при тех же условиях, что и rрадуиров
ка. Перед помещением в трубку ИСПЫТУL''V1УЮ
жидкость при необходимости термостатиру от
при 20 ос для предотвращения образования ПУ
зырьков rаза и для уменьшения времени, необ
ходимоro для измерения.
Факторы, влияющие на точность:
неоднородность температуры во всем
объеме трубки;
отсутствие линейности в диапазоне изме
ряемоrо значения плотности;
мешающие резонансные эффекты;
вязкость, вследствие чеro растворы с ВЯЗ
костью, большей, чем у раствора, по которому
проводилась rрадуировка, показывают плот
ность заметно более высокую, чем истинная.
Проблемы, связанные с эффектами OTCYT
ствия линейности и вязкости, MOryT быть решены
при использовании для rрадуировки веществ со
значениями плотности и вязкости, близкими к Ta
ковым для испытуемой жидкости (:!:5 % для плот
ности И :!:50 % для вязкости). Плотномер может
иметь функцию для автоматической коррекции
вязкости и для коррекции ошибок, связанных с
отсутствием линейности и с изменениями TeM
пературы .
Прецизионность является функцией воспро
изводимости и стабильности частоты ОСЦИЛJ1И
рующеrо датчика, которая зависит от стабиль
ности объема, массы и динамической жесткости
ячейки.
Плотномеры способны проводить измере
ния с точностью от 1.103 r/cM 3 до 1'105 r/CM 3 и
повторяемостью от 1 .104 r/CM 3 до 1.10-6 r/CM 3 .
#Определение плотности при помощи ПИК
нометра проводят, как указано в методах 1 и 2,
при помощи ареометра как указано в методе
3, если нет друrих указаний в частной статье.#
#Метод 1. Применяют в случае определения
плотности жидкостей с точностью до 0,001 r/см З .
Чистый сухой пикнометр взвешивают с ТОЧ
ностью до 0,0002 r, заполняют при помощи сухой
воронки водой Р чуть выше метки, закрывают
пробкой и термостатируют в течение 20 мин при
температуре (20:!:0,1) ос. При этой температуре
уровень воды в пикнометре доводят до метки,
быстро отбирая избыток воды при помощи пи
петки или завернутой в трубку полоски филь
тровальной бумаrи. Пикнометр снова закрывают
пробкой и термостатируют еще 1 О мин, прове
ряя положение мениска по отношению к метке.
Затем пикнометр вынимают из термостата,
фильтровальной бумаroй вытирают внутреннюю
поверхность шеЙКlA пикнометра, а также весь
пикнометр снаружи, выдерживают под стеклом
весов в течение 1 О мин и взвешивают с точно
стью, указанной выше.
Пикнометр освобождают от воды, высуши
вают, ополаскивая последовательно 96 % спир
том Р и эфиром Р (сушить пикнометр путем
наrревания не допускается), удаляют остаТОI
эфира продуванием воздуха, заполняют пикно.
метр испытуемой жидкостью и затем ПрОВОДЯl
те же операции, что и с водой Р.
Плотность Р20 (r/см З ) рассчитывают по фор.
муле:
(т 2 т).O.99703
... : + 0,0012.
т, П?
rдe:
т масса пустоro пикнометра. r;
т 1 . масса пикнометра с водой Р, r;
т 2 масса пикнометра с испытуемой жид.
костью, r;
0,99703 значение плотности воды прv
20 ос, r/см З (с учетом плотности воздуха);
0,0012 значение плотности ВОЗДУХе
при 20 ос, смЗ И барометрическом давлениv
101,3 кПа (760 мм рт. ст.).
#Метод 2. Применяют для определеНИf
плотности твердых жиров и воска.
Взвешивают пустой пикнометр, затем взве-
шивают тот же пикнометр, заполненный водой Р
После этоrо ВОДУ удаляют и пикнометр высуши-
вают. Все операции проводят. соблюдая усло-
вия. указанные в MeTO.ц 1.
В пикномеТ/J вливают при помощи пипет-
ки или небольшо воронки с тонкооттянутым
концом расплавленны.i жир или воск в таком ко-
пичестве, чтобы он занимал от 1/3 до 1/2 объема
пикнометра. Пикнометр выдерживают в течение
1 ч без пробки в roрячей воде, затем охлаждают
до температуры 20 ос, взвешивают, доводят до
метки водой Р при температуре 20 ос, вытирают
насухо и снова взвешивают. В обеих фазах и на
поверхности их раздела не должно быть пузырь
ков воздуха.
Плотность Р20 (r/cM 3 ) рассчитывают по фор
муле:
(т т).O.99703 +0,0012,
(щ +т2) (т+тз)
rдe:
т масса пустоro пикнометра, r;
т 1 масса пикнометра с водой р, r;
т 2 масса пикнометра с испытуемым об
разцом, r;
тз масса пикнометра с испытуемым об
разцом и водой Р, r.
#Метод 3. Применяют в случае определения
плотности жидкостей с точностью до 0,01 r/см З .
Испытуемую жидкость помещают в цилиндр,
и при температуре жидкости 20 ос осторожно
опускают в нее чистый сухой ареометр, шкала
котороro позволяет определи1'ь ожидаемую Be
личину плотности. Ареометр не выпускают из
рук, пока не станет очевидным, что он плава
ет; при этом необходимо следить, чтобы apeo
2.2.7. Оптическое вращение
47
метр не касался стенок и дна цилиндра. Отсчет
плотности проводят через 3------4 мин после по
rpужения ареометра по делению на шкале, co
ответствующему нижнему мениску жидкости (в
случае определения темноокрашенных жидко
стей отсчет ПрОИЗ80ДЯТ по верхнему мениску).
При отсчете rлаз должен быть на уровне мени
ска. Определение плотности сильнолетучих Be
ществ ареометром не допускается.
01/2013:20206
2.2.6. ПОКАЗАТЕЛЬ ПРЕЛОМЛЕНИЯ
(ИНДЕКС РЕФРАКЦИИ)
Показатель преломления среды относи
тельно воздуха равняется отношению синуса
уrла падения луча света в воздухе к синусу уrла
преломления луча света в данной среде.
#Абсолютным показателем преломления Ha
зывают отношение скорости распространения
света в вакууме к скорости распространения
света в испытуемом веществе. Относительный
показатель преломления это отношение CKOpO
сти распространения света в воздухе к скорости
распространения света в испытуемом веществе. #
Если в частной статье нет друrих указаний,
определение показателя преломления проводят
при температуре (20:tO,5) ос при длине волны
линии О спектра натрия (л.=589,3 нм); показатель
преломления, определенный в таких условиях,
обозначают символом п O.
#Показатель преломления зависит от темпе
ратуры и длины волны света, при которой про
водят определение. В растворах показатель
преломления зависит также от концентрации Be
щества и при роды растворителя.
Определение показателя преломления при
меняется для установления подлинности и чи
стоты вещества. Метод применяют также для
определения концентрации вещества в paCTBO
ре, которую находят по rрафику зависимости по
казателя преломления от концентрации. В таком
случае точность измерения показателя прелом
ления должна быть не ниже :t2'10 . На rрафи
ке выбирают интервал концентраций, в котором
соблюдается линейная зависимость между KO
эффициентом преломления и концентрацией. В
этом интервале концентрацию вещества в pac
творе в процентах рассчитывают по формуле:
п П'J
F
rдe:
п показатель преломления раствора:
по показатель преломления растворителя
при той же температуре;
F фактор, равный величине прироста по
казателя преломления при увеличении KOHцeH
трации на 1 % (устанавливается эксперимен
тально).#
Рефрактометры обычно определяют крити
ческий уroл. В таком приборе rлавной частью
является призма с известным показателем пре
ломления, которая контактирует с анализируе
мой жидкостью.
Для калибровки прибора используют серти
фицированные эталонные вещества.
#rрадуировка прибора проводится по эта
лонным жидкостям, прилаrаемым к прибору, или
по воде Р, полученной методом дистилляции,
для которой п O = 1,3330 (l1п/lJ.t = 0,000085).#
При использовании белоro света рефрак
тометры должны быть оборудованы компенса
ционной системой. Прибор должен давать по
казания с точностью как минимум до TpeTbero
десятичноro знака и обеспечивать возможность
проведения операций при заданной температу
ре. Цена деления термометра не должна превы
шать 0,5 ос.
01/2013:20207
2.2.7. ОПТИЧЕСКОЕ ВРАЩЕНИЕ
Оптическое вращение это свойство Be
щества вращать плоскость поляризации поляри
зованноro света.
Оптическое вращение считается положи
тельным (+) для правовращающих веществ (то
есть тех, которые вращают плоскость поляри
зации по часовой стрелке) и отрицательным ( )
для левовращающих веществ.
Удельное оптическое вращение [aт] ' BЫ
раженное в радианах (рад), представляет собой
вращение, вызванное слоем жидкости или paCT
вора толщиной 1 м, содержащим 1 Kr/M 3 оптиче
ски активноro вещества при прохождении через
неro поляризованноro света с длиной волны
.'" при температуре t. Для практических целей
удельное оптическое вращение обычно выража
ют в миллирадиан метрах квадратных на кило
rpaMM (мрад . м 2 . Kr .1).
В Фармакопее используются следующие об
щепринятые определения.
У20Л оптичеСК020 вращения жидких Be
ществ представляет собой уroл вращения а пло
скости поляризации, выраженный в rрадусах (О),
при длине волны IlИНИИ О спектра натрия
(1,=589,3 нм), измеренный при температуре 20 ос
в толщине слоя 1 дм. Для растворов способ при
roтовления указывают в частной статье.
Удельное оптическое вращение [а ] O жид
кости представляет собой уroл вращения а ПЛ
скости поляризации, выраженный в rpaдycax С).
48
rосударственная фармакопея Республики Беларусь
при длине волны линии D спектра натрия (л.=589,3
нм), измеренный при температуре 20 ОС, рас чи
танный для толщины слоя 1 дм испытуемоro Ee
щества и деленный на плотность, выраженную в
rpaMMax на кубический сантиметр.
Удельное оптическое вращение [a] O веще
ства в растворе представляет собой уrол Bpa
щения а плоскости поляризации, выраженный
в rрадусах (О), при длине волны линии О спек
тра натрия (л. = 589,3 нм), измеренный при TeM
пературе 20 ос в растворе испытуемоro веще
ства и рассчитанный для слоя 1 дм в пересчете
на содержание вещества 1 r/мл в растворе.
Для удельноro вращения вещества в растворе
Bcerдa указывают используемый растворитель и
концентрацию раствора.
В Фармакопее удельное оптическое враще
ние выражают без размерности; реальная раз
мерность выражается в rрадус миллилитрах на
дециметр rрамм [(О) . мл . ДM 1 . r 1].
Пересчет удельноro вращения в единицах
по Международной Системе в единицы, исполь
зуемые Фармакопеей, проводят по формуле:
[aт] := [a] , .0,1745.
В отдельных случаях, указанных в частной
статье, уroл вращения может быть измерен при
температурах, отличных от 20 ОС, и при друrих
длинах волн.
Используемый поляриметр должен обеспе
чивать измерения с точностью до 0,010. Шкалу
обычно проверяют при помощи сертифици
рованных кварцевых пластинок. Линейность
шкалы может быть проверена при помощи pac
творов сахарозы.
Методика. Определяют ноль поляриметра и
уroл вращения плоскости поляризации при длине
волны линии D спектра натрия (л. = 589,3 нм) при
температуре (20:tO,5) ОС. Измерения оптическо
ro вращения MOryT проводиться при друrих TeM
пературах только в тех случаях, если в частной
статье указан способ учета температуры. Опре
деляют ноль прибора с закрытой трубкой; для
жидкостей с пустой трубкой; для растворов
твердых веществ с трубкой, заполненной co
ответствующим растворителем.
#Проводят не менее 5 измерений и рассчи
тывают среднее значение.#
Удельное оптическое вращение рассчиты
вают по следующим формулам.
Для жидкостей:
[а];О = .
,. Р20
Для веществ в растворе:
[ ] 20 1000'а
а D ,
'.с
rдe:
с концентрация вещества в растворе, r/л.
Содержание с (r/л) или с' (% (м/м») paCTBO
peHHoro вещества рассчитывают по формулам:
1000'а
с= ,
1'[a] O
. 100'а
с = ,
1'[a] O' Р20
rдe:
a уroл вращения, измеренный при темпе
ратуре (20:t0,5) ОС, в rрадусах;
, длина поляриметрической трубки, в дe
циметрах;
Р20 плотность при температуре 20 ОС, в
rpaMMax на кубический сантиметр. В фармако
пейном анализе плотность заменяют относи
тельной плотностью (2.2.5).
#Если нет друrих указаний в частной статье,
оптическое вращение растворов должно быть
измерено в течение 30 мин после их приrо
товления. Если известно, что вещества под
вержены рацемизации или мутаротации, He
обходимо установить время (интервал) между
добавлениями вещества в растворитель и BBe
дением приrотовленноrо раствора в трубку по
ляриметра.#
01/2013:20208
2.2.8. ВЯЗКОСТЬ
#Вязкость (внутреннее трение) свойство
текучих тел оказывать сопротивление передви
жению одной их части относительно друroй.
Ньютоновскими жидкостями называют си
стемы, вязкость которых не зависит от напряже
ния сдвиrа и является постоянной величиной в
соответствии с законом Ньютона. Неньютонов
ские жидкости не попадают под действие закона
Ньютона, так как их вязкость зависит от напря
жения сдвиrа.
Для ньютоновских жидкостей различают ди
намическую, кинематическую, относительную,
удельную, приведенную и характеристическую
вязкости. Для неньютоновских жидкостей xapaK
терна, rлавным образом, структурная вязкость.#
Динамическая вязкость, или коэффици
ент вязкости 17 это танrенциальная сила,
приходящаяся на единицу поверхности, KOTO
рая также называется напряжением сдви2а т,
выраженная в паскалях (Па), которую необхо
димо приложить для Toro, чтобы переместить
слой жидкости площадью 1 м 2 со скоростью (v)
1 метр в секунду (м . c 1) на расстояние (х) 1 м
относительно друroro слоя, параллельно пло
щади скольжения.
Величина dvldx представляет собой rради
ент скорости и обусловливает скорость сдви2а
О, выраженную в обратных секундах (c 1). Таким
образом, rz:= r 1 D .
2.2.9. Метод капиллярной вискозuметрии
49
01/2013:20209
Единицей динамической вязкости явля
ется паскаль секунда (Па' с). Чаще всеro ис
пользуется дольная единица миллипаскаль
секунда (м Па . с).
Кинематическая вязкость и, выраженная
в метрах квадратных в секунду (м 2 . c 1), pac
сматривается как отношение величины дина
мической вязкости 1] к плотности жидкости р, BЫ
раженной в килоrраммах на метр кубический
(Kr . м З), измеренной при этой же температуре:
v == 1] / р. Кинематическую вязкость обычно
выражают в миллиметрах квадратных в ceKYH
ду (мм 2 . c 1).
#Структурная (эффективная, или кажу
щаяся) вязкость вязкость при данном Ha
пряжении сдвиrа. В ряде случаев необходимо
определить вязкость одной жидкости относи
тельно друroй относительную вязкость
1] отн'
Часто используют удельную вязкость '7 уд'
показывающую, какой вклад в вязкость paCT
вора вносит присутствие в нем pacTBOpeHfJOrO
вещества:
17 170 17
17 у д == '70 == 7i: 1 == 170тн 1,
rдe:
'7 вязкость раствора;
'70ТН вязкость растворителя.
Удельную вязкость, отнесенную к единице
концентрации раствора, называют приведенной
вязкостью 17 прив:
'7 прив
'7 у д
== '
С
rдe с концентрация раствора.
Для растворов полимеров вязкость явля
ется функцией молекулярных масс, формы,
размеров и rибкости макромолекул. Чтобы
определить структурные характеристики по
лимеров, приведенную вязкость экстраПОJ1И
руют к нулевой концентрации. В таком случае
вводится понятие характеристической
вязкости ['7]:
[ ] 1 . 1 . 17 уд
'7 == Im'7npue == ,т
C O C O С
Характеристическая вязкость выражается в
единицах, обратных единицам концентрации.#
Для определения вязкости ньютоновских
жидкостей можно использовать капиллярный
вискозиметр; для определения вязкости как
НЬЮТОНОВGКИХ, так и неньютоновских жидко
стей можно использовать ротационный виско
зиметр. Допускается использование и друrих
вискозиметров с учетом, что точность и пра
вильность измерений будут не худшими, чем
на при мере использования вискозиметров,
описанных ниже.
2.2.9. МЕТОД КАПИЛЛЯРНОЙ
ВИСКОЗИМЕТРИИ
Определение вязкости проводят. исполь
зуя подходящий капиллярный вискозиметр. при
температуре (20:1:0,1) ос, если в частной статье
не указана друrая температура. Время BЫTeKa
ния от одной отметки вискозиметра до друroй
измеряется секундомером с точностью до одной
пятой секунды. Полученные данные считаются
достоверными при условии, что результаты двух
последовательных измерений отличаются не
более чем на 1 %.
Время вытекания испытуемой жидкости опре
деля ют как среднее не менее трех измерений.
Динамическую вязкость 17 (2.2.8), Bыpa
женную в миллипаскаль секундах (мПа . с), pac
считывают по формуле:
'7 = kpt ,
rде:
k постоянная вискозиметра, в миллиме
трах квадратных на секунду в квадрате (мм 2 . c 2);
Р плотность испытуемой жидкости.
в миллиrраммах на миллиметр кубический
(Mr . мм З), полученная умножением относитель
ной плотности (d;g) на 0,9982;
t время вытекания испытуемой жидкости,
в секундах (с).
Постоянную k определяют с использовани
ем соответствующей жидкости для калибровки
вискозиметров.
Кинематическую вязкость, выраженную в
миллиметрах квадратных на секунду (мм 2 . c 1),
рассчитывают по формуле:
l} = kt.
Определение вязкости может проводиться
при помощи прибора (предложен Международ
ной орrанизацией по стандартизации), xapaK
теристики котороro представлены на рисунке
2.2.9. 1 и в таблице 2.2.9. 1.
Минимальное время вытекания должно
быть 350 с для размера 1 и 200 с для осталь
ных размеров
Методика. Испытуемую жидкость, имею
щую температуру 20 ос, если в частной статье
не указана иная, заливают в вискозиметр через
трубку (L) в таком количестве, чтобы заполнить
расширение (А), но при этом уровень жидкости
в расширении (В) должен остаться ниже выхода
к веНТИЛЯЦИОI-fНОЙ трубке (М). Вискозиметр в
вертикальном положении поrружают в водяную
баню при температуре (20:1:0:1) ос, если в част
ной статье не указана иная температура, yдep
живая ero в этом положении не менее 30 минут
для установления температурноrо равновесия.
Трубку (М) закрывают и повышают уровень жид
50
rосударственная фармакопея Республики Беларусь
м N
1
I
t:::1
I
I
Ni
С"?!
+
i
..
о
(J)
!
1
1
01
..;t,
+
1
L
(N) и открыв трубку (М). Затем открывают трубку
(N) и измеряют время, за которое уровень жидко
сти снизится от метки (Е) до метки (F), ceKYHДO
мером с точностью до одной пятой секунды.
"Работа с прибором, описанным выше, дo
пускает использование вискозиметров капил
лярных стеклянных с висячим уровнем (напри
мер, вискозиметры капиллярные стеклянные
типа ВПЖ 1), параметры которых аналоrичны
приведенному на рисунке 2.2.9. 1. Вязкость из
меряют в соответствии с инструкцией по приме
нению вискозиметра.
Для определения относительной вязкости
жидкости измеряют время t,'cп вытекания между
верхней и нижней отметкоЙ вискозиметра той
жидкости, относительно которой проводят из
мерение '70тн' Затем в том же чистом и сухом
вискозиметре при тех же условиях определяют
время вытекания t Co. испытуемой жидкости.
Одновременно измеряют плотность ЖИДКО
стей, которые изучаются, пикнометром (Ро ир)
при той же температуре, при которой определя
ют вязкость, и рассчитывают относительную вяз
кость по формуле:
Е
F
R
-:
t ep ' Р
, 1
Orr.H t
Оер . Ро
Для определения характеристической вязко
сти roтовят не менее пяти испытуемых растворов
разной концентрации. При этом должно выполнять
ся условие возможности линейной экстраполяции
приведенной вязкости к нулевой концентрации, то
есть нужно выбирать минимальные концентрации
раствора в пределах чувствительности и точности
метода измерения. Для каждой концентрации paCT
вора определяют t cc и рассчитывают приведенную
вязкость. Затем строят зависимость '7 прuв от KOH
центрации с и rpафически или линейным методом
наименьших квадратов экстраполируют приведен
ную вязкость к нулевой концентрации, то есть Ha
ходят характеристическую вязкость.#
Рисунок 2.2.9. 1. Вискозuметр
с висячим уровнем
(размеры указаны в миллиметрах)
кости в трубке (N) таким образом. чтобы она Haxo
дилась примерно на 8 мм выше метки (Е). Yдep
живают жидкость на этом уровне, закрыв трубку
Таблица 2.2.9. 1
I
Номиналь I Внутрен-
Диапазон I Объем pac Внутренний ди
Размер ная постоя н- кинематической ний диа- аметр трубки
ная вискози- метр трубки ширения
вязкости С N
метра R
мм 2 . с. 2 мм 2 . с" мм (:t2 %) мл (:t5 %) мм
1 0,01 От 3.5 до 10 0,64 5,6 От 2,8 до 3,2
1А 0,03 ОТ 6 до 30 0,84 5,6 От 2,8 до 3,2
2 0,1 От 20 до 100 , 1,15 5,6 I От 2,8 до 3,2
2А 0,3 От 60 до 300 1,51 5,6 I От 2,8 до 3,2
3 1,0 От 200 до 1000 2,06 5,6 I От 3,7 до 4,3
I
3А 3,0 От 600 до 3000 2,74 5,6 i От 4,6 до 5,4
4 10 От 2000 до 1 О 000 3,70 5.6 I От 4,6 до 5,4
I
4А 30 От 6000 до 30 000 4,07 5,6 От 5,6 до 6,4
5 100 I 6,76 5,6 От 6,8 до 7,5
От 20 000 до 100000 i
2.2.10. Метод ротационной вискозиметрии
01/2013:20210
51
2.2.10. МЕТОД РОТАЦИОННОЙ
ВИСКОЗИМЕТРИИ
Принцип метода заключается в измере
НИИ силы, действующей на ротор (вращаю
щий момент) во время ero вращения с посто
янной уrловой скоростью (скорость вращения)
в жидкости. Ротационные вискозиметры ис
пользуются для измерения вязкости ньюто
новских жидкостей (вязкость не заsи,:::и т от
напряжения сдвиrа) и неньютоновских жидко
стеи (вязкость зависит от напряжения сдвиrа,
кажущаяся вязкость). Ротационные вискози
метры MorYT быть разделены на две туппы.
а именно: абсолютные вискозиметры v. 01 HO
сительные вискозиметры. В абсолютнь,:< ви
скозиметрах время вытекания в ИСllопозvе
мой rеометрии хорошо известно. РеЗУ:1Ы аты
измерений представляются в единицзл aoco
лютнои вязкости, которые MorYT cpaBf-li'1БЭ Т ЬСЯ
с любыми друrими абсолютными знач ниями.
В относительных вискозиметрах время BЫTe
кания в используемой rеометрии не опреде
лено. Результаты измерений представляются
в единицах относительной вязкости, которые
не MorYT быть соотнесены с абсолютными ;jча
чениями вязкости или со значениями относи
тельной вязкости, полученными не на юм же
самом приборе.
Для данных диапазонов вязкости и для
различных скоростей вращения доступны раз
личные измерительные системы.
ПРИБОР
Наиболее обычными являются следующие
типы инструментов.
(J)
J
м
Ro
R,
r
h I
I
.
, "
; // _///" ,//"//,//// ///,,'
./', ,./',,// //,,, ,//.' ./,'. ,/,;///",/'
Рисунок 2.2.10. 1.
КОНЦЕНТРИЧЕСКИЕ ЦИЛИНДРИЧЕСКИЕ
ВИСКОЗИМЕТРbI (АБСОЛЮТНblЕ
ВИСКОЗИМЕТРbI)
В концентрических цилиндрических вискози
метрах (коаксиальный двойной цилиндрический
вискозиметр или просто коаксиальный вискози
Melp) вязкость определяют, помещая испытуе
мую жидкость в зазор между внутренним и внещ
ним цилиндрами. При проведении измерений
вращают -'lибо внутренний цилиндр (вискозиметр
типа Сирла (5e8r/e», либо внешний цилиндр (ви
скозиметр типа Куэтта (Couette», как показано
на рисунках 2.2.10. 1 111 2.2.10. 2 соответственно.
Для ламинарноrо потока вязкость (или кажущую
ся вязкость) 17. выраженную в паскаль секундах
(Па' с), рассчитывают по формуле:
1 . М \( 1 1 ') М
17 : '! i k ,
( ) 4лh Л R,L R;; J {jJ
,j:;, .
м . - вращаюLЦИЙ момент, ,:..J,ействующий на
. ИЛИf--щрическую поверхность. в HbfuToH MeTpax;
()) уrловая скорость, 8 радианах в секунду;
h rлу'БИН2 поrружения внутреннеro ЦИЛИf-l
др<з в жидкую среду, в метрах;
R, радиус внутреннею цилиндра. в
Fv1eTpax:
R, радиус внешнеro цилиндра, в метрах;
k' постоянная прибора, в раf1ианах на
метр кубический.
Для неньЮТОНОВСКИХ жидкостей необходимо
указывать напряжение при сдвиrе (т ) или CKO
рость сдвиrа (У), при которых измеряется вяз
кость. В узком интервале условий (подходящих
для абсолютных вискозиметров) наблюдается
пропорциональная зависимость между М и т , а
также между йJ и у:
/ j '/ / / / / / / j/ '/
/ I/ J ///, '///
h
/////,////" /
/ / / / / / / / / / ','
м
Ro
R, !
ro
, /
Рисунок 2.2.1 0. 2.
52
rосударственная фармакопея Республики Беларусь
r == АМ r == Вт ,
rдe А и В постоянные прибора, которые
рассчитывают по следующим формулам:
для концентрической поверхности:
А == 1R j 2 +R;
4лhR,2R;
в == R,2 + R
R R,2
для устройства конус плита:
A==
2nR З
1
B== ,
а
rде:
М вращающий момент, действующий на
коническую или цилиндрическую поверхность, в
ньютон метрах;
йJ уrловая скорость, в радианах в
секунду;
Rj радиус внутреннеro цилиндра, в
метрах;
Ro радиус внешнеro цилиндра, в метрах;
R радиус конуса, в метрах;
h rлубина поrружения внутреннеro цилин
дра в жидкую среду, в метрах;
а уroл между плоским диском и конусом,
в радианах;
r напряжение при сдвиrе, в паска
лях (Па);
r скорость сдвиrа, в обратных ceKYH
дах (c 1).
ВИСК03ИМЕТРbl КОНУС ПЛИТА
(АБСОЛЮТНblЕ ВИСКОЗИМЕТРbl)
В вискозиметрах типа конус плита жид
кость помещают в промежуток между пло
ским диском И конусом с определенным уrлом.
Определение вязкости проводят, вращая конус
или плоский диск, как показано на рисунках
2.2.1 0. 3 и 2.2.10.А соответственно. Вязкость
(или кажущуюся вязкость) 1], выраженную в
паскаль секундах (Па . с), для ламинарноro
потока рассчитывают по формуле:
[ M j( За ) М
'7 k ,
{iJ, 2лR З (iJ
rдe:
м вращающий момент, действующий на
коническую поверхность или поверхность пло
cKoro диска, в ньютон метрах;
йJ уrловая скорость, в радианах в
секунду;
а уroл между плоским диском и конусом,
в радианах;
R радиус конуса. в метрах;
k постоянная прибора, в радианах на
метр кубический.
Постоянные А и В прибора определяют aHa
лоrично, как для концентрических цилиндриче
ских вискозиметров.
о)
/
м
R
\
la
/j// ///// ////
м
R
a
Рисунок 2.2.10А.
ШПИНДЕЛЬНblЕ ВИСК03ИМЕТРbl
(ОТНОСИТЕЛЬНblЕ ВИСК03ИМЕТРbl)
В шпиндельном вискозиметре вязкость
определяют с помощью вращающеroся шпин
деля (например, цилиндрическоro или диско
образноro, как показано на рисунках 2.2.1 0. 5 и
2.2.1 0. 6 соответственно), помещенноro в жид
кость. Относительные значения вязкости (или
кажущейся вязкости) MOryT быть рассчитаны He
посредственно с использованием коэффициен
тов пересчета исходя из показаний прибора при
данной скорости вращения.
В общем случае постоянная k прибора может
быть определена для разных скоростей враще
ния с использованием жидкости, сертифициро
ванной для калибровки вискозиметра. После
этоro вязкость 1] рассчитывают по формуле:
м
'] == k '
{iJ
МЕТОДИКА
Измеряют вязкость (или кажущуюся вязкость)
в соответствии с инструкциями производителя po
тационноrо вискозиметра. Температуру, при KO
торой измеряют вязкость, указывают в частных
статьях. Для неньютоновских систем в частных
статьях указывают тип используемоro вискозиме
тра и, если используются абсолютные вискозиме
2.2.11. ТемператУРНblе пределы переzонки
53
/\
Рисунок 2.2.1 0. 5.
тры, уrловую скорость или скорость сдвиra, при
которых производится измерение. В случае, если
точно получить необходимую скорость сдвиrа He
возможно, используют значения скорости сдвиrа
выше и ниже необходимоro и интерполируют.
В случае относительных вискозиметров CKO
рость сдвиrа не является постоянной по всему
объему образца, и, таким образом, она не может
быть определена. В этих условиях вязкость He
ньютоновских жидкостей, определенная по BЫ
шеприведенной формуле, имеет относительный
характер, который зависит от типа шпинделя,
уrловой скорости, а также от размеров контейне
ра для образцов (@ = не менее 80 мм) и rлубины
поrружения шпинделя. Получаемые результа
ты сопоставимы только при соблюдении CTporo
одинаковых условий эксперимента.
100
L[).
о
со
r
I
I
.,
I
I
Рисунок 2.2.10. 6.
01/2013:20211
2.2.11. ТЕМПЕРАТУРНЫЕ ПРЕДЕЛЫ
ПЕРЕrонки
Температурные пределы переroнки пред
ставляют собой интервал температур, приве
денных к давлению 101,3 кПа (760 мм рт. ст.), В
пределах KOToporo переroняется жидкость или
некоторая ее фракция в следующих условиях.
Прибор. Прибор (рисунок 2.2.11. 1) состоит
из переroнной колбы (А), прямоro холодильни
ка (В), присоединенноro к отводной трубке пере
roнной колбы, и вставной трубки (алонжа) (С),
присоединенной к концу холодильника. В каче
стве альтернативы нижний конец холодильника
может быть изоrнут для тоro, чтобы исv.лючить
1
Ri
I
J
Рисунок 2.2.11. 1. Прибор для определения температурных пределов пере20НКU
(размеры указаны в миллиметрах)
54
rосударственная фармакопея Республики Беларусь
алонж. В rорловину колбы помещают термометр
таким образом, чтобы верхний конец PTYTHO
ro резервуара находился на 5 мм ниже нижнеro
края отводной трубки переroнной колбы. Исполь
зуют термометр с диапазоном шкалы, близким к
50 ОС, с ценой деления 0,2 ОС. Во время опреде
ления колбу, включая и rорловину, предохраня
ют от охлаждения подходящим экраном.
Методика. 50,0 мл испытуемой жидкости и
несколько кусочков пористоro материала поме
щают в колбу (А). ДЛЯ сбора отroна используют
цилиндр вместимостью 50 мл С ценой деления
1 мл. Для жидкостей, кипящих при температу
ре ниже 150 ОС. при меняют охлаждение цирку
лирующей водой. Колбу наrревают таким обра
зом, чтобы быстро достичь кипения, и отмечают
температур при которой в цилиндр поступа
ет первая капля отroна. Устанавливают Harpe
вание, которое обеспечивает переroнку от 2 до
3 мл В минуту, И отмечают температуру, при KO
торой вся жидкость или некоторая ее фракция,
объем которой измеряют при температуре 20 ОС,
OTorHaHbI.
Вносят поправку в наблюдаемую температу
ру для приведения к нормальному давлению по
формуле:
t 1 == t 2 + k(1 01,3 Ь) .
rдe:
t 1 исправленная температура;
t 2 наблюдаемая температура при бароме
трическом давлении Ь;
k поправочный коэффициент в соот
ветствии с таблицей 2.2.11. 1, если не указано
иноro;
Ь барометрическое давление во время
переrонки, в килопаскалях.
Таблица 2.2.11. 1
Коэффициент поправки для приведения
к нормальному давлению
1 , ' Поправочный
Температура переrонки коэффициент k
0,30
0.34
0.38
0.41
0.45
Д0100 0 С
Свыше 100 ос до 140 ос
Свыше 140 ос дО 190 ос
Свыше 190 ос до 240 ос
Свыше 240 ос
01/2013:20212
2.2.12. ТЕМПЕРАТУРА КИПЕНИЯ
Точкой кипения называется скорректирован
ная температура, при которой давление паров
жидкости равно 101,3 кПа.
Прибор. В данном случае используется тот же
прибор, что и для определения пределов переrонки
(2.2.11), за исключением тоro, что термометр BBO
дится в roрло колбы таким образом, чтобы нижний
конец ртутноro резервуара был на уровне нижнеrо
конца roрла переrонной колбы и чтобы сама колба
располаrалась на пластине из изолирующеro MaTe
риала со сквозным отверстием диаметром 35 мм.
Методика. 20 мл испытуемой жидкости и He
сколько кусочков пористоro материала помещают
в колбу (А). Колбу наrревают таким образом, чтобы
быстро достичь кипения, и отмечают температуру,
при которой жидкость начинает вытекать из OTBO
дной трубки в холодильник
Вносят поправку в наблюдаемую температуру
для приведения к нормальному давлению по фор
муле:
t, ==t 2 +k(101,3 b),
rдe:
t 1 исправленная температура;
t 2 наблюдаемая температура при бароме
трическом давлении Ь;
k поправочный коэффициент в COOTBeT
ствии с таблицей 2.2.11. 1;
Ь барометрическое давление во время
переroнки, в килопаскалях.
01/2013:20213
2.2.1 з. ОПРЕДЕЛЕНИЕ БОДЫ
МЕТОДОМ отrонки
Прибор (рисунок 2.2.1 3. 1) состоит из стеклян
ной круrлодонной колбы (А), соединенной труб
кой (О) с цилиндрической трубкой (В), снабжен
ной rрадуированным приемником (Е) и обратным
холодильником (С). Цена деления приемника (Е)
0,1 мл. Для соответствующеrо наrревания целесо
образно использовать электрический наrреватель
с реостатом или масляную #(или песчаную)" баню.
Верхняя часть колбы и соединительная трубка
MOryт быть покрыты теплоизоляцией.
Методика. Приемник и холодильник прибора
очищают, промывают водой и высушивают.
200 мл толуола Р и около 2 мл воды Р поме
щают в сухую колбу и переroняют в течение 2 ч.
Колбу охлаждают в течение 30 мин и записывают
объем воды с точностью до 0,05 мл. В колбу поме
щают количество вещества, отвешенноro с точно
стью ДО 1 %, которое содержит приблизительно от 2
до 3 мл воды. Если вещество имеет пастоподобную
консистенцию, еro взвешивают на кусочке металли
ческой фольrи. В колбу вносят несколько кусочков
пористоro материала и осторожно наrревают в Te
чение 15 мин. Korдa толуол начнет кипеть, отroня
ют со скоростью около двух капель в секунду, пока
2.2.14.
Температура плавления капиллярный метод
01/2013:20214
55
' k
( и=\j :
11' О!
I !
1, с 1\1:
п l
'V
u 1\
\
,
в
LO .
N
о
.
.4/
ж
ж- Е
!I ЮI
) :1. "; .
:1101-"
I
I.?t ,)
V r<J,
------- ---T
17
I
I '
I 500мл 'д
\ ' )
165
Pl1CYHOK 2.2.13. 1. Прuбор для определенu;l
воды методом отеонки
(размеры указаны в миллиметрах)
большая часть воды не отrонится, а затем увеличи
вают скорость отroнки до четырех капель в секунду.
'Кипячение прекращают, Korдa объем воды
8 приемнике перестанет увеличиваться и Bepx
ний слой растворителя в приемнике станет про
зрачным.
Korдa вода отroнится полностью, внутреннюю
трубку холодильника промывают толуолом Р. Ha
rревание продолжают еще 5 мин, затем HarpeBa
тель убирают, дают приемнику охладиться дО KOM
натной температуры и стряхивают все капли воды,
которые находятся на стенках приемника. После
полноro разделения воды и толуола записывают
объем воды и определяют ее содержание в мил
лилитрах на килоrрамм по формуле:
1 00О(П 2 П 1 )
т
rдe:
П 1 объем воды, определенный при первом
отroне, в миллилитрах;
П 2 общий объем отоrнанной воды, опре
деленный в обоих отroнах, в миллилитрах;
т масса испытуемоro образца, в rpaM
мах.
2.2.14. ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
КАПИЛЛЯРНЫЙ МЕТОД
Температураплавления,определеннаякапил
лярным методом, представляет собой темneрату
ру, при которой последняя твердая частичка ynлoт
ненноro столбика вещества в капиллярной трубке
переходит в жидкую фазу.
#Весь процесс плавления протекает в течение
определенноro промежутка времени и определен
ноro интервала температур: начало плавления
появление первой капли жидкости и конец плав
ления (температура плавления) полный переход
вещества в жидкое состояние. Этот интервал TeM
ператур не должен преВbJшать 2 ос, если в частной
статье нет друrих указаний.
Целый ряд орrанических соединений при плав
лении разлаrается (происходит резкое изменение
Rн€шнеro вида вещества, например. вспенивание).
Такую температуру называют температурой раз
ложения. Она в значительной мере зависит от cкo
j.JOсти HarpeBa, поэтому при определении темпе
ратуры разложения в частных статьях указывают
скорость HarpeBa.
Капиллярный метод при меняют для опреде
пения температуры плавления твердых веществ,
леrко превращаемых в порошок.#
Если есть указание в частной статье, тот же
прибор и методику применяют для определения
друrих показателей, таких, как образование мени
ска или диапазона плавления, характеризующих
поведение вещества при плавлении.
Прибор. Составными частями прибора яв
ляются:
подходящий стеклянный сосуд, содержа
щий жидкость (например, воду, вазелиновое или
силиконовое масло), используемый в качестве
бани и оснащенный подходящим устройством
ДЛЯ HarpeBa;
устройство для перемешивания. обеспечи
вающее однородную температуру внутри бани;
термометр с меткой поrружения и ценой де--
ления не более 0,5 ос. Разность между верхним и
нижним делениями термометра в области измеря
емой температуры не более 100 сс:
запаянные с одноro конца капиллярные
трубки из бесщелочноro прочноro стекла диаме
тром от 0,9 мм до 1,1 мм и толщиной стенок от
0,10 мм до 0,15 мм.
#Во время определения температуры плав
ления ни колба, ни пробирка не должны быть за
крыты rерметически.'
Методика. Если в частной статье нет друrих
указаний, тонкоизмел енное вещество сушат в
вакууме над силика2елем безводным Р в течение
24 ч. Достаточное количество вещества помеща
ют в капиллярную трубку до получения уплотнен
ноro столбика высотой от 4 мм до 6 мм. #Необ
ходи мое уплотнение вещества при заполнении
капиллярной трубки можно получить, если ее He
56
rосударственная фармакопея Республики Беларусь
сколько раз бросить запаянным концом вниз в
стеклянную трубку длиной не менее 1 м, постав
ленную вертикально на твердую поверхность."
Повышают температуру бани до темпе
ратуры приблизительно на 1 О ос ниже пред
полаrаемой температуры плавления и затем
продолжают наrревание со скоростью около
1 ОС/мин. Коrда температура достиrнет значе
ния на 5 ос ниже предполаrаемой температу
ры плавления, помещают капиллярную трубку
с веществом в прибор. Для описанноro выше
прибора капиллярную трубку помещают таким
образом, чтобы ее запаянный конец распола
rался около центра шарика термометра, метка
поrружения котороro находится на уровне по
верхности жидкости. Отмечают температуру,
при которой последняя твердая частичка пере
ходит в жидкую фазу.
#Если в частной статье нет друrих указа
ний, наrревание реryлируют так, чтобы к MO
менту плавления была достиrнута необходи
мая скорость подъема температуры:
от 0,5 до 1 ОС/мин при определении TeM
пературы плавления ниже 100 ос;
от 1 до 1,5 ОСIмин при определении TeM
пературы плавления от 100 ос до 150 ос;
от 1,5 до 2 ОС/мин при определении TeM
пературы плавления выше 150 ос;
от 2,5 до 3,5 ОСIмин для веществ,
неустойчивых при наrревании.
Проводят не менее двух определений. За
температуру плавления принимают среднее
значение. Расхождение между определениями
не должно превышать 1 ос.#
iрадуировка прибора. Для rрадуировки
при бора используют подходящие вещества,
приroдные для этих целей.
#Допускается применение друrих прибо
ров, использующих капиллярный метод, если
показано, что точность и правильность измере
ний будут не хуже, чем в случае применения
прибора, описанноro выше.#
01/2013:20215
2.2.15. ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
ОТКРЫТЫЙ КАПИЛЛЯРНЫЙ
МЕТОД
Данный метод используют для определения
температуры плавления (также известной как
температура сдвиrа или подъема, либо темпе
ратура разжижения) некоторых веществ.
#Открытый капиллярный метод применяют
для веществ, имеющих аморфную структуру, не
растирающихся в порошок и плавящихея ниже
температуры кипения воды, таких как жиры,
воск, парафин, вазелин. смолы.#
Используют стеклянную капиллярную
трубку, открытую с обоих концов, длиной около
80 мм, наружным диаметром от 1,4 мм до 1,5 мм
и внутренним диаметром от 1,0 мм до 1,2 мм.
Вещество, предварительно обработан
ное, как указано в частной статье, помещают в
каждую из пяти капиллярных трубок (капилляр
ную трубку, открытую с обоих сторон, поrружают
в вещество так, чтобы оно заполнило нижнюю
часть трубки)" в количестве, достаточном для
формирования в каждой трубке столбика BЫ
сотой около 1 О мм. Трубки выдерживают опре
деленное время при температуре, указанной в
частной статье.
Если не указано иноro, вещества с BOCKO
образной консистенцией осторожно полностью
расплавляют на водяной бане перед заполнени
ем капиллярных трубок. Трубки выдерживают в
течение 2 ч при температуре от 2 ос до 8 ос.
Прикрепляют одну из капиллярных трубок к
термометру с ценой деления 0,5 ос таким обра
зом, чтобы вещество находилось в непосредствен
ной близости к шарику термометра. Термометр с
прикрепленной капиллярной трубкой помещают в
стакан так, чтобы расстояние между дном CTaKa
на и нижней частью шарика термометра составля
ло 1 см. Стакан наполняют водой таким образом,
чтобы высота слоя составляла 5 см. Повышают
температуру воды со скоростью 1 ОС/мин.
За температуру плавления принимают TeM
пературу, при которой вещество начинает подни
маться по капиллярной трубке. #В тех случаях,
Korдa столбик вещества в капилляре не подни
мается, за температуру плавления принимают
температуру, при которой столбик вещества в
капилляре становится прозрачным.#
Повторяют эту операцию с четырьмя друrи
ми капиллярными трубками и рассчитывают pe
зультат как среднее из пяти показаний.
01/2013:20216
2.2.16. ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
МЕТОД MrHOBEHHOrO
ПЛАВЛЕНИЯ
Температуру плавления по методу MrHoBeH
ноro плавления рассчитывают по формуле:
(1 +(2
,
2
rдe:
(1 первая температура, определяемая в
условиях, приведенных ниже;
(2 вторая температура, определяемая в
условиях, приведенных ниже.
Прибор. Прибор состоит из металлическоro
блока, изroтовленноro из материала, обладаю
56
rосударственная фармакопея Республики Беларусь
сколько раз бросить запаянным концом вниз в
стеклянную трубку длиной не менее 1 м, постав
ленную вертикально на твердую поверхность."
Повышают температуру бани до темпе
ратуры приблизительно на 1 О ос ниже пред
полаrаемой температуры плавления и затем
продолжают наrревание со скоростью около
1 ОС/мин. Korдa температура достиrнет значе
ния на 5 ос ниже предполаrаемой температу
ры плавления, помещают капиллярную трубку
с веществом в прибор. Для описанноro выше
при бора капиллярную трубку помещают таким
образом, чтобы ее запаянный конец распола
rался около центра шарика термометра, метка
поrружения котороro находится на уровне по
верхности жидкости. Отмечают температуру,
при которой последняя твердая частичка пере
ходит в жидкую фазу.
#Если в частной статье нет друrих указа
ний, наrревание реryлируют так, чтобы к MO
менту плавления была достиrнута необходи
мая скорость подъема температуры:
от 0,5 до 1 ОСIмин при определении TeM
пературы плавления ниже 100 ос;
от 1 до 1,5 ОС/мин при определении TeM
пературы плавления от 100 сс до 150 ос;
от 1,5 до 2 ОС/мин при определении TeM
пературы плавления выше 150 ос:
от 2,5 до 3,5 ОС/мин для веществ,
неустойчивых при наrревании.
Проводят не менее двух определений. За
температуру плавления принимают среднее
значение. Расхождение между определениями
не должно превышать 1 ОС.-
'радуировка прибора. для rpадуировки
прибора используют подходящие вещества,
приroдные для этих целей.
"Допускается применение дрyrих прибо
ров, использующих капиллярный метод. если
показано, что точность и пpaвмl1bНOCТb измере
ний будут не хуже, чем в спучае npименения
прибора, описанноro ВЫUJe. 8
0112013:20215
2.2.15. ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
ОТКРЫТЫЙ КАПИЛЛЯРНЫЙ
МЕТОД
Данный метод используют для определения
температуры плавления (также известной как
температура сдвиrа или подъема, либо темпе
ратура разжижения) некоторых веществ.
#Открытый капиллярный метод при меняют
для веществ, имеющих аморфную структуру, не
растирающихся в порошок и плавящихся ниже
температуры кипения воды, таких как жиры,
воск, парафин, вазелин, смолы.#
Используют стеклянную капиллярную
трубку, открытую с обоих концов, длиной около
80 мм, наружным диаметром от 1,4 мм до 1,5 мм
и внутренним диаметром от 1,0 мм до 1,2 мм.
Вещество, предварительно обработан
ное, как указано в частной статье, помещают в
каЖдУЮ из пяти капиллярных трубок #(I<апилляр
ную трубку, открытую с обоих сторон, поrружают
в вещество так, чтобы оно заполнило нижнюю
часть трубки)# в количестве, достаточном для
формирования в каждой трубке столбика BЫ
сотой около 1 О мм. Трубки выдерживают опре
деленное время при температуре, указанной в
частной статье.
Если не указано иноrо, вещества с BOCKO
образной консистенцией осторожно полностью
расплавляют на водяной бане перед заполнени
ем капиллярных трубок. Трубки выдерживают в
течение 2 ч при температуре от 2 ос до 8 ос.
Прикрепляют одну из капиллярных трубок к
термометру с ценой деления 0,5 ос таким обра
зом, чтобы вещество находилось в непосредствен
ной близости к шарику термометра. Термометр с
при крепленной капиллярной трубкой помещают в
стакан так, чтобы расстояние между дном CTal<a
на и нижней частью шарика термометра составля
ло 1 см. Стакан наполняют водой таким образом,
чтобы высота слоя составляла 5 см. Повышают
температуру воды со скоростью 1 ОС/мин.
За температуру плавления принимают TeM
пературу, при которой вещество начинает подни
маться по капиллярной трубке. #В тех случаях,
Korдa столбик вещества в капилляре не подни
мается, за температуру плавления принимают
температуру, при которой столбик вещества в
капилляре становится прозрачным.#
Повторяют эту операцию с четырьмя друrи
ми капиллярными трубками и рассчитывают pe
зультат как среднее из пяти показаний.
01/2013:20216
2.2.16. ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
МЕТОД MrHOBEHHOrO
ПЛАВЛЕНИЯ
Температуру плавления по методу MrHoBeH
ноro плавления рассчитывают по формуле:
t 1 + t 2 ,
2
rде:
t 1 первая температура, определяемая в
условиях, приведенных ниже;
t 2 вторая температура, определяемая в
условиях, приведенных ниже.
Прибор. Прибор состоит из металлическоro
блока, изroтовленноrо из материала. облада
2.2.17. Температура каплепаденuя
57
щеro высокой теплопроводностью и не взаимо
действующеro с испытуемым веществом, Ha
пример, из латуни. Верхняя поверхность блока
должна быть плоской и тщательно отполирован
ной. Блок равномерно наrревают по всей массе
rазовой rорелкой с микрореryлировкой или элек
трическим наrревателем с тонкой реryлировкой.
Блок имеет достаточно широкую цилиндриче
скую полость для размещения термометра, стол
бик ртути котороro должен находиться в одном и
том же положении как при калибровке, так и при
определении температуры плавления испытуе
мою вещества. Цилиндрическая полость разме
шена параллельно отполированной верхней по
верхности блока и на расстоянии около 3 мм от
нее. Прибор rрадуируют, используя подходящие
вещества с известной температурой плавления.
Методика. Блок быстро наrревают до TeM
пературы на 1 О ос ниже предполаrаемой TeM
пературы плавления и затем устанавливают
скорость наrревания около 1 ОС/мин. Несколь
ко частиц тонко измельченною вещества, BЫ
сушенноro в вакууме над силика2елем безво
дHыM Р в течение 24 ч, бросают через равные
промежутки времени на поверхность блока в He
посредственной близости от шарика TepMOMe
тра, очищая поверхность после каждоro испы
тания. Записывают температуру t 1 , при которой
вещество плавится MrHoBeHHo при соприкосно
вении с металлом. Останавливают наrревание.
Во время охлаждения через равные временные
промежутки бросают несколько частичек веще
ства на поверхность блока, очищая ее после
каждоro испытания. Записывают температуру
t 2 , при которой вещество прекращает MrHoBeHHo
плавиться при соприкосновении с металлом.
,радуировка прибора. Для rрадуировки при
бора используют подходящие вещества, приroд
ные для этих целей.
01/2013:20217
2.2.17. ТЕМПЕРАТУРА
КдПЛЕПАДЕНИЯ
Температура каплепадения представляет
собой температуру, при которой в условиях, при
веденных ниже, первая капля расплавленноro
испытуемоro вещества падает из чашечки.
#Определение температуры каплепадения
проводят для веществ, не растирающихся в по
рошок И плавящихся ниже температуры кипения
воды, таких как жиры, воск, парафин, вазелин,
смолы. п
Если в частной статье нет друrих указаний,
определение проводят по методу А. Замена
метода А методом В должна быть валидиро
вана.
МЕТОД А
Прибор. Прибор (рисунок 2.2.17. 1) состо--
ит из двух металлических rильз (А) и (В), сое--
диненных одна с друroй посредством резьбы.
rильза (А) прикреплена к ртутному термометру. В
нижней части rильзы (В) с помощью двух уплот--
нителей (Е) свободно закреплена металлическая
чашечка (F). Точное положение чашечки опреде--
ляется фиксаторами (О) длиной 2 мм, которые
используются также для центровки термометра.
Отверстие (С) в стенке rильзы (В) предназначено
для выравнивания давления. Отводящая поверх
ность чашечки должна быть плоской, а края BЫ
ходноro отверстия под прямым уrлом к поверх
ности. Нижняя часть pTYTHoro термометра имеет
форму и размер, как показано на рисунке; рабо--
чий диапазон термометра от О ОС до 11 О ос и pac
стояние на шкале в 1 мм соответствует разности
температур в 1 ос. Ртутный шарик термометра
имеет диаметр (3,5:!::0,2) мм и высоту (6,0:!::0,3) мм.
Прибор устанавливают по оси пробирки
длиной около 200 мм и наружным диаметром
около 40 мм.
Прибор прикрепляют к пробирке с помо
ЩЬЮ пробки, В которую вставлен термометр и
которая имеет боковую прорезь. Отверстие ча
шечки должно находиться на расстоянии около
15 мм от дна пробирки. Все устройство поrружа
ют в стакан вместимостью около 1 л, заполнен
ный водой. Дно пробирки должно находиться на
расстоянии около 25 мм от дна стакана. Уровень
воды должен достиrать верхней части rильзы
(А). ДЛЯ равномерноro распределения темпера
туры в стакане используют мешалку.
10
1U
,,,1
,
с.
. .
_!
..
д
в
....
N
CjI
i
C Y I
. I
I
с
. "!I'
, .'
i 1 r j
о
Е
F
5
i
i
i
б 1: 0.05
31:0.05
Рисунок 2.2.17. 1. Прибор для определения
температуры каплепадения
(размеры указаны в миллиметрах)
58
rосударственная фармакопея Республики Беларусь
Методика. Испытуемое вещество подroтав
ливают. как указано в частной статье. Заполняю,.
чашечку до краев испытуемым веществом. Из
быток вещества удаляют с обеих сторон шпате
лем. После тоro, как rильзы (А) и (В) COeJJtAHe
ны, проталкивают чашечку B YTPb на ее месте,
в rильзе (8) до упора. Удаляют шпателем веще.
ство, выдавленное термометром. Прибор поме
щают в водяную баню, как описано выше. Водя
ную баню наrревают до температуры примерно
на 1 О ос ниже предполаrаемой температуры Ka
плепадения и устанавливают скорость HarpeBa
около 1 ОС/мин. Отмечают температуру п;:щения
первой капли. Проводят не менее трех опреде
лений, каждый раз с новым образцом вещества
Разность между показаниями не д()лжна пре
вышать 3 ОС. Среднее из полученных значениЙ
представляет собой температуру капл€пздения
МЕТОД В АВТОМАТИЧЕСКИЙ МЕТОД
Прибор. Прибор (рисунок 2.2.17. 2) cocтo
ит из сборноro картриджа, включающеro держа
тель чашечки, в котором свободно фиксируется
чашечка с испытуемым образцом. и приемноro
стаканчика с roризонтальной щелью для света,
фиксирующеroся ниже чашечки. Этот узел поме
щают в наrревательный блок. Блок это метал
лическии цилиндр с цилиндрическим отверсти
ем, расположенным вдоль вертикальной оси. в
который помещается сборный картридж. Кроме
тоro, присутствует второе более узкое верти
кальное отверстие, в которое помещается TeM
пературный датчик, располаraющийся на одном
уровне с чашечкой с испt.nyeМым образцом.
Снаружи наrревательноro бnoка располаrается
электрический наrревательный элемент Ниже
наrревательноro блока находится лампа, pac
положенная таким образом. что луч света про
ходит сквозь roризонталblfYlO щель приемноro
рукава и падает на фотоэлектрический датчик,
расположенный налротив. Наrpeвательный блок
с помощью наrревателbНOrO элемента может
быть точно выведен на заданную температуру
и, после начальноro И30терммческоrо периода,
способен медленно и с постоянной скоростью
наrреваться.
Методика. Испытуемый образец расплав
ляют, помещают в чашечку соrласно указаниям
частной статьи и проводят испытание, как указа-
но ниже или в соответствии с инструкциями про-
изводителя. Избыток вещества удаляют с обеих
сторон шпателем. Перед проведением испыа..
ния испытуемый образец выдерживают при тем-
пературе и в течение времени, указанных в част-
ной статье. Чашечку протаЛЮ-1вают в держатель
и наталкивают приемный рукав I-Ja чашечку. Co
бранный картридж помещают в наrревательный
блок. Устанавливают начальную температуру и
скорость последующеro наrревания, как указано
в частной статье, и начинают проведение испы-
тания. Автоматически реrистрируется темпера-
тура, при которой первая капля расплавленноro
Е
к
А
F
G
в
X.
С .'
Ш o (l) 11 J <
н
д держатель чашечки.
Б нarревательныЙ блок;
С . источник света:
D щель;
Е сборный картр.щж:
F наrревательный элемент:
G чашечка с испытуемым образцом;
Н фотоэлеКТРИLlес"ий даТЧI'К:
J приемный стаканчи ;
К температурныЙ датчик
Рисунок 2.2.17. 2. Пример прибора
автоматичеСК020 определения
температуры каплепадения
испытуемоro образца капает из отверстия в дне
чашечки. прерывая луч света. падающий на Фо
тоэлектрический датчик.
,радуuровка прибора. Прибор используют
в соответствии с инструкциями производителя
и реrулярно проводят предписанные rрадуиров-
ки и проверки приrодности системы в зависи
мости от использования прибора и испытуемых
образцов. В качестве сертифицированных стан-
дартных веществ обычно используют бензойную
кислоту и бензофенон. MoryT быть использова-
ны и друrие вещества при условии, что они не
обладают полиморфизмом. rрадуировку про во-
дят в соответствии с инструкциями производи
теля или следующим образом. Подroтавливают
по три чашечки каждым из двух сертифициро
ванных веществ. Помещают чашечки на чистую
поверхность. В каждую чашечку помещают H
большое количество испытуемоro образца и
прижимают ко дну с помощью стержня (диаметр
около 4,5 мм). Необходимо убедиться. что OT
верстие полностью заполнено. Далее наполня
ют чашечку примерно до ПОЛОВИНbI, уплотняют
испытуемый образец стержнем (диаметр около.
9 мм) И, уплотняя, заполняют чашечку до краев. 1
2.2.18.
Температура затвердевания
59
Температурная проrрамма для бензойной
кислоты: начальная температура 118,0 ос;
скорость HarpeBa 0,2 аС/мин; конечная TeM
пература 126,0 C. После установки чашечки
при температуре 118 ос выдерживают в течение
ЗО с и продолжают наrревание.
Температурная проrрамма для бензосрено
на: начальная температура 44,0 ОС; скорость
HarpeBa 0,2 ОС/мин; конечная температура
56,0 ос. После установки чашечки при темпера
туре 44 ос выдерживают в течение 30 с и про
должают наrревание.
Приroдность системы: 3 единичных резуль
тата испытания не должны отличаться более
чем на 0,3 ос от среднеro значения.
Рассчитывают скорректированную среднюю
температуру (Tz>:
T1 F ,
rдe:
Т 1 . средняя Te A'lepaTYf.'a каПJlAf' ",".., .,'1>",
рассчитанная по трем I;спытачилм. ос:
F . компенсирование различия MP')j( lY TeM
пературой испытуемою образца и температурой
в месте измерения температуры; это значение
сильно зависит от конструкции прибора aBTOMa
тическоro определения температуры каПj,еП;:jде
ния и представляется производителем.
Принимая во внимание температуру каriПе
падения (ТО) сертифицированноro вещества,
точность температурной шкалы считается IlрИ
емлемой, если IT7 Тоl не более 0,3 ос.
01/2013:20218
2.2.18. ТЕМПЕРАТУРА
ЗАТВЕРДЕВАНИЯ
Температура затвердевания представл ет
собой максимальную температуру, при которой
происходит затвердевание переохлажденной
жидкости.
Прибор. Прибор (рисунок 2.2.18. 1) состоит
из пробирки для проведения определения диа
метром около 25 мм и длиной около 150 мм,
помещенной вовнутрь друrой пробирки диаме
тром около 40 мм и длиной около 160 мм. BHY
тренняя пробирка закрыта пробкой, снабжен
ной термометром длиной около 175 мм с ценой
деления 0,2 О С, который закреплен таким обра
зом, чтобы ртутный шарик Н8ХОДИЛСЯ на уровне
около 15 мм от дна пробирки. В пробке имеется
отверстие, через которое проходит вал мешал
ки, изrотовленный из стеклянноrо стержня или
друrоro подходящеro материала, заrнутый на
конце под прямым уrлом в виде петли, внеш
ний диаметр которой около 18 мм. Внутреннюю
пробирку вместе с внешней пробиркой разме
щают в центре сосуда вместимостью 1 л, в KO
торый помещают подходящую охлаждающую
жидкость, уровень которой находится в преде
лах 20 мм от верхнею края сосуда. Охлаждаю
щая баня также должна быть снабжена TepMO
метром.
I
i
I
I
I
, !
l'
I '!
,i
Ш
о
;;
..
со
j.
ц) ,
i
. 'f
..
i
о
"
I
.... L
40
Рисунок 22 .18. 1. Прибор для определения
температуры затвердевания
(размерbl у,,<азаНbI в миллиметрах)
Методика. Во внутреннюю IlIJOбирку 110
мещают достаточное количество жидкости или
предварительно расплавленноro вещества,
чтобы покрыть ртутный шарик термометра
(ртутный шарик термометра должен находить
ся посередине слоя испытуемою вещества), и
при быстром охлаждении определяют прибли
зительную температуру затвердевания. BHY
треннюю пробирку помещают в водяную баню
с температурой на 5 ос выше приблизительно
определенной температуры до полноro расплав
ления кристаллов. Затем заполняют сосуд водой
или насыщенным раствором натрия хлорида с
температурой на 5 сс ниже ожидаемой темпе
ратуры затвердевания. Внутреннюю пробирку
вместе с внешней помещают в сосуд, убежда
ются в наличии зародышей кристаллов и тща
тельно перемешивают испытуемый образец в
течение затвердевания. Отмечают наиболее BЫ
сокую температуру, наблюдаемую во время за
твердевания.
60
rосударственная фармакопея Республики Беларусь
#Вначале ПРОИСХОДИТ постепенное пониже
ние температуры, затем, при появлении TBep
ДОЙ фазы, она остается некоторое время по
стоянной или повышается перед тем, как стать
ПОСТОЯННОЙ (в этот момент прекращают переме
. шивание), а затем снова падает. Если вещество
остается жидким при ожидаемой температуре
затвердевания, еro затвердевание вызывают по
тиранием о стенки внутренней пробирки TepMO
метром или внесением кристаллика испытуемо
ro вещества.#
01/2013:20219
2.2.19. АМПЕРОМЕТРИЧЕСКОЕ
ТИТРОВАНИЕ
При амперометрическом титровании конеч
ную точку титрования определяют по измене
нию тока между поrруженными в испытуемый
раствор электродами (один из них поляризущий
ся индикаторный, а друrой неполяризующийся
электрод сравнения, либо два поляризующихся
индикаторных электрода) как функции от коли
чества прибавленноro титранта при постоянной
и контролируемой разности потенциалов.
Потенциал индикаторноro электрода должен
обеспечивать предельный диффузионный ток
для электрохимически активноro соединения.
Прибор. Прибор состоит из источника по
стоянноro тока с реryлируемым напряжением и
чувствительноro микроамперметра. Детектиру
ющая система обычно состоит из индикаторно
ro электрода (например, платиновоrо, ртутноro
капельноro, вращающеroся дисковоro или rpa
фитовоro электрода) и электрода сравнения (Ha
пример, каломельноro или хлоридсеребряноro
электрода).
Иноrда используют трехэлектродную Си
стему, состоящую из индикаторноro электрода,
электрода сравнения и поляризованноrо вспо
моrательноro электрода.
Методика. Электроды помещают в испытуе
мый раствор, устанавливают постоянный потен
циал, указанный в частной статье. и прибавляют
титрант порциями. По значениям силы началь
ноro тока и значениям, полученным в процессе
титрования, строят rрафик зависимости силы
тока от количества прибавляемоrо титранта. Ти
трант прибавляют последовательно, не менее
чем тремя порциями, составляющими в сумме
около 80 % от теоретическоro объема, COOTBeT
ствующеro предполаrаемой точке эквивалентно
сти. Три полученных значения силы тока должны
укладываться на прямую. Продолжают последо
вательно прибавлять титрант после предполаrа
емой точки эквивалентности не менее трех раз.
Полученные значения должны укладываться на
прямую. Точка пересечения этих двух прямых
представляет конечную точку титрования.
#При амперометрическом титровании с
двумя индикаторными электродами (без элек
трода сравнения) оба электрода изroтовлены
из одноro и Toro же материала и имеют одина
ковую относительно небольшую поверхность. В
этом случае реrистрируют всю кривую титрова
ния и определяют конечную точку титрования по
минимальному значению силы тока. Наиболь
шая точность амперометрическоro титрования
достиrается при потенциале на индикаторном
электроде, соответствующем предельному диф
фузионному току.
При амперометрическом титровании, как пра
вило, концентрация титранта в 1O 20 раз превы
шает концентрацию определяемоro вещества.
В фармакопейном анализе амперометриче
ское титрование целесообразно применять в ни
тритометрии при определении воды полумикро
методом (по К. Фишеру) и в йодометрии.
В частных статьях необходимо указывать
параметры, необходимые для корректноro BЫ
полнения методики: типы электродов, задава
емый потенциал, массу навески вещества, KOH
центрацию титранта, температуру и т. д.#
01/2013:20220
2.2.20. ПОТЕНЦИОМЕТРИЧЕСКОЕ
ТИТРОВАНИЕ
При потенциометрическом титровании KO
нечную точку титрования находят. измеряя элек
тродвижущую силу (3ДС) электродной пары,
состоящей из индикаторноrо электрода и элек
трода сравнения или двух индикаторных элек
тродов, поrруженных в испытуемый раствор, как
функцию количества прибавленноro титранта.
ЗДС обычно измеряют при нулевом или
практически нулевом токе.
=Потенциометрическое титрование обычно
дает более точные результаты, чем индикатор
ное, особенно при анализе мутных и окрашен
ных растворов, позволяет автоматизировать
процесс титрования.
Как правило, электродную пару поrружа
ют в испытуемый раствор, кроме случаев, Korдa
ионы из электрода сравнения мешают титрова
нию. В этом случае электрод сравнения соеди
няют с испытуемым раствором электролитиче
ским МОСТОМ.#
Прибор. Используемый прибор (простой по
тенциометр, электронное устройство, #pH MeTp#,
#иономер#) включает вольтметр с разрешением
около милливольта.
Выбор индикаторноro электрода зависит
от природы определяемоro вещества #и типа
2.2.20. Потенциометрическое титрование
61
Таблица # 2.2.20. 1
Электродные системы для потенциометричеСК020 титрования
Тип аналитической Индикаторные 3лектродысравнения Применение !
реакции электроды ;
Кислотоно основной Стеклянный Хлоридсеребряный, Ka Титрование кислот, oc I
ломельный нований и солей
Осаждения Серебряный, сульфид Хлоридсеребряный, Ka Титрование rалоrени ,
серебряный ломельный, стеклянный ДОВ,роданидов, циани I
дов, сульфидов
Комплексонометриче Ртутный, ионоселектив Хлоридсеребряный, Ka Титрование катионов,
ский ный ломельный, стеклянный образующих прочные
комплексонаты
Окислительно восстано Платиновый Хлоридсеребряный, Ka Титрование восстанови
вительный ломельный, стеклянный телей различными окис
лителями, например,
броматом,дихроматом,
перманrанатом, йодом,
церием (IV). Титрование
I окислителей различны
ми восстановителями,
например,арсенитом,
тиосульфатом, нитри
том.
аналитической реакции#. Этот электрод может
быть стеклянным или металлическим (напри
мер, платиновым, золотым, серебряным или
ртутным). #Индикаторный электрод выбира
ют так, чтобы еro потенциал закономерно из
менялся при протекании химической реакции
между титруемыми ионами и ионами титранта
(таблица # 2.2.20. 1).#
Электродом сравнения обычно служит Ka
ломельный или хлоридсеребряный электрод.
Для кислотно основноrо титрования, если
нет друrих указаний в частной статье, исполь
зуют системы стеклянноro и каломельноrо или
стеклянноro и хлоридсеребряноro электродов.
Методика. При необходимости смесь раст
ворителейнейтрализуют перед растворением
испытуемоro образца. Строят rрафик зависимо
сти изменения ЭДС от количества прибавлен
ноro титранта, продолжая прибавлять титрант
сверх предполаrаемой точки эквивалентности.
Конечная точка титрования соответствует резко
му изменению ЭДС.
=При проведении анализа титрованный
раствор прибавляют из бюретки равными объе
мами при постоянном перемешивании. Вблизи
точки эквивалентности прибавляют по 0,1 или
0,05 мл и после каждоro прибавления измеря
ют ЭДС. Величина ЭДС особенно сильно изме
няется вблизи точки эквивалентности, абсолют
ное значение отношения изменения ЭДG (дЕ) к
приросту объема приБЭl3ляемоrо титранта (Д v) в
этой точке будет максимальным.
Объем титранта в точке эквивалентности
VЭ"В может быть определен описанными ниже
способами.
1. По rpафику кривой титрования в коорди
натах [V: Е], применяя метод касательных.
2. По rрафику:
[ V' Д Е ]
'ДV'
rдe:
дЕ изменение ЭДС;
/ V соответствующее приращение объема
титранта.
При этом конечной точке титрования соот
ветствует максимальное значение дЕ/д V;
3. Расчетным путем по максимальному зна
чению дЕ/д V и соответственно д(дЕ/д V), как YKa
зано в таблице # 2.2.20 2. Эквивалентный объем
титранта VЭ"В рассчитывают по формуле:
Ау
V экв =о V, + (V 2 V, ) ,
Ау, Ау.
Таблица #2.2.20. 2
V 1 ' дV Е, дЕ дЕ/д V Av= ( EHV)
мл мВ
5,00 250 I
I
0,1 13 130 ;
5,10 263 +150
0,1 28 280 i
5,20 291 i +720
0,1 100 i 1000
5,30 391 , 450
,
0,1 I 55 i 550
5,40 446 i ззо
0,1 I 22 ! 220
5,50 468 120
0,1 10 100
5,60 478
62
rосударственная фармакопея Республики Беларусь
rдe:
V 1 объем титранта, соответствующий по
следнему положительному (отрицательному)
значению величины Av;
V 2 объем титранта, соответствующий пер
вому отрицательному (положительному) значе
нию величины А.,;
Av = д(дЕ/д v) приращенная величина дЕ/д V.
При прохождении через точку эквивалентно
сти Av меняет знак на противоположный.
Пример:
720
V экв = 5,20 + (5,30 5,20) = 5,26 мл
720 ( 50) .
Потенциометрическое титрование может
быть автоматизировано путем применения при
боров двух типов: использующих математиче
ский анализ кривой титрования или прекращаю
щих прибавление титранта при достижении ЭДС
электронной пары, соответствующей точке экви
валентности.#
01/2013:20221
2.2.21. ФЛУОРИМЕТРИЯ
Флуориметрия метод анализа, OCHOBaH
ный на измерении интенсивности флуорес
ценции, излучаемой испытуемым образцом,
относительно флуоресценции, излучаемой CTaH
рартным образцом.
Методика. Испытуемый образец paCTBO
ряют в растворителе или смеси растворителей,
указанных в частной статье. Полученный pacт
вор помещают в кювету или камеру флуориме
тра и облучают возбуждающим светом. имею
щим как можно большую монохроматичность,
при длине волны. указанной в частной статье.
Измеряют интенсивность испускаемоro
света под уrлом 90 с относительно воз6уждаю
щеro света после прохождения сквозь фильтр,
пропускающий избирательно свет с длиной
волны максимальной флуоресценции. MorYT
быть использованы и друrие типы приборов при
условии получения аналоrичных результатов.
При проведении количественных опреде
лений в прибор помещают растворитель или
смесь растворителей, используемых для paCT
ворения испытуемоrо образца, и устанавли
вают реrистрирующее устройство прибора на
нулевое значение. Затем помещают раствор
стандартною образца и устанавливают чув
ствительность прибора таким образом. чтобы
отклик показаний был больше 50 %. Если было
проведено вторичное реrулирование с измене
нием ширины щели, повторно устанавливают
реrистрирующее устройство прибора на нуле
вое значение и снова измеряют интенсивность
флуоресценции раствора стандартноrо об
разца. Затем в прибор помещают испытуемый
раствор с неизвестной концентрацией и реrи
стрируют показания прибора. Концентрацию
(с) испытуемоro образца в растворе рассчиты
вают по формуле:
/xcs
с = .
х /,
rдe:
С х концентрация испытуемоro образца в
растворе;
С 5 концентрация стандартноro образца в
растворе;
/ интенсивность флуоресценции раст
вора Хиспытуемоrо образца;
15 интенсивность флуоресценции paCT
вора стандартною образца.
Если значения интенсивности флуорес
ценции не CTpOro пропорциональны значениям
концентрации растворов, измерения MOryт про
водиться с использованием rрадуировочноro
rрафика.
В некоторых случаях измерения MOryт про
водиться С использованием qpиксированно
ro стандарта (например. флуоресцирующеro
стекла или раствора флуоресцирующей жидко
сти). В таких случаях концентрация испытуемо
ro вещества может определяться с использова
нием rрадуировочноro rрафика, построенноrо в
тех же условиях.
01/2013:20222
2.2.22. АТОМ НО-ЭМИССИОННАЯ
СПЕКТРОМЕТРИЯ
ОСНОВНОЙ ПРИНЦИП
Атомная эмиссия это процесс испуска
ния электромаrнитноro излучения возбужденны
ми атомами или ионами. В атомно эмиссионной
спектрометрии испытуемый образец подверrа
ется воздействию достаточно высоких темпе
ратур, вызывающих как диссоциацию на атомы,
так и значительное количество соударений, при
водящих к возбуждению и ионизации атомов ис
пытуемоrо образца. Атомы и ионы, будучи в воз
бужденном состоянии, r.пособны возвращаться
в основное энерrетическое состояние с переда
чей тепловой или излучающей энерrии и испу
сканием (эмиссией) электромаrнитноro излуче
ния. Эмиссионный спектр элемента содержит
несколько больше линий, чем соответствующий
абсорбционный спектр.
2.2.22. Атомно эмuccuонная спектрометрия
63
Атомно эмиссионная спектрометрия это
метод определения содержания химическоro
элемента в испьпуемом образце посредством
измерения интенсивности одной из эмиссион
ных линий атомноro пара элемента. Определе
l1ие проводят при длине волны, соответствую
щей выбранной эмиссионной линии.
В данной статье описан только метод атоми
зации с использованием пламени. Метод aTOM
но--эмиссионной спектрометрии с индуктивно
связанной плазмой (АЭС ИСП) описан в друrой
общей статье.
ПРИ БОР
rлавными составляющими частями прибора
являются:
система введения и распыления образца;
пламенный reHepaTop атомноro пара,
монохроматор;
детектор;
блок сбора данных.
Для получения пламени MOryт быть исполь
зованы водород, ацетилен, пропан или бу",-ан в
сочетании с кислородом или воздухом. Выбор
reHepaTopa атомноro пара является критическим
моментом, так как он должен обеспечивать дo
статочную энерrию для возбуждения и распы
ления атомов. Атомные спектры, полученные с
использованием пламенною reHepaTopa aTOM
ноro пара, имеют преимущества, будучи более
простыми, по сравнению со спектрами. получен
ными с использованием reHepaTopoB атомноro
пара друrих типов, однако основным лимитиру
ющим фактором в ею использовании являет
ся недостаточная мощность для возбуждения
атомов мноrих элементов. Предпочтительным
растворителем для приroтовления испытуемых
растворов и растворов сравнения является под
кисленная вода, но MOryT быть использованы и
орrанические растворители, если доказано, что
они не влияют на стабильность пламеН I.
ИНТЕРФЕРЕНЦИЯ
Спектральную интерференцию уменьша
ют или исключают путем выбора для измере
ния подходящей линии спектра либо подбора
ширины щели. Физическую интерференцию Kap
ректируют разведением раствора испытуемо
ro образца, подбором матрицы или использова
нием метода стандартных добавок. Химическую
интерференцию уменьшают использованием
химических модификаторов или ионизационных
буферов.
ЭФФЕКТ ПАМЯТИ
Эффект памяти, вызванный остатками aHa
лита в приборе, может быть лимитирован тща
тельным промыванием прибора между испы
таниями, разведением, если это возможно,
измеряемых растворов, уменьшая таким обра
зом содержание соли, и как можно быстрейшим
впрыскиванием растворов.
МЕТОД
По возможности рекомендуется использо
вать пластмассовую лабораторную посуду.
Атомно эмиссионный спектрометр BЫBO
ДЯТ на режим в соответствии с инструкцией за
вода производителя и устанавливают необхо
димую длину волны. Устанавливают пара метры
эксперимента (температура пламени, настрой
ка roрелки, использование ионноro буфера, KOH
центрация растворов), необходимые для опре
деления анализируемою элемента, учитывая
матрицу образца. В reHepaтop атомноro пара
вводят холостой раствор и настраивают реrи
ст рирующее устройство на нулевое значение
или на значение холостою опыта. Вводят paCT
вор сравнения определяемоro элемента с наи
большей концентрацией и настраивают прибор
так, чтобы получить реrмстрируемый сиrнал в
оптимальном диапазоне измерений.
Предпочтительно, чтобы концентрации pac
ТЕОрОВ находились в линейной части rрадуиро
вочноro rрафика. Если это невозможно, MOryт
быть использованы КРИВОШ1нейные rрадуиро
BO'-lНbIе rрафики С применением подходящею
проrраммною обеспечения.
Определения ПРОВОДЯТ путем сравнения со
стандартными растворами с звестными KOH
цент рациями определяемою элемента методом
rрадуировочною rрафика (метод 1) .1Ш1 методом
стандартных добавок (метод 11).
МЕТОД i МЕТОД rрАДУИРОВОЧНО,О
,РАФИКА
Обычно для измерений rотовят и использу
ют три раствора сравнения определяемоro эле
мента и холостой раствор.
Раствор испытуемоrо образца (испытуемый
раствор) ютовят, как указано 6 'iастной статье.
Не менее трех растворов сравнени определя
GMQrO элемента roТОВЯl [ак. ';тсбы д..аr.азсн KOH
цьнтраций этих растворив вКJ. ал ожидаемое
зна ение концентраций ,-,пределяемurо элемен-
та в испытуемом растворе. Для количественно
ro анализа оптимальные значения калибровки
должны находиться в диапазоне от 0,7 до 1,3 от
ожидаемоro содержания определяемою элемен
та или от предела, указанноro в частной статье.
Для анализа на содержание примесей оптималь
ные значения калибровки должны находиться
в диапазоне от предела обнаружения до 1,2 от
предельноro значения для определяемоro эле
мента. Любые реактивы. используемые при при
ютовлении испытуемоro раствора, прибавляют в
холостой раствор и растворы сравнения в таких
же количествах, как и 13 испытуемый раствор.
Вводят растворы, используя одинаковое KO
личество повторов, для получения устойчивых
результатов.
Расчет. Строят rрадуировочный rрафик за
висимости средних значений эмиссий растворов
64
rосударственная фармакопея Республики Беларусt.
сравнения от концентрации, по которому опре
деляют концентрацию элемента в испытуемом
растворе.
МЕТОД 11 МЕТОД СТАНДАРТНЫХ
ДОБАВОК
Раствор испытуемоro образца (испытуемый
раствор) roтовят. как указано в частной статье.
Равные объемы испытуемоro раствора помеща
ют не менее, чем в три мерные колбы одинако
воro объема. Во все мерные колбы кроме одной
прибавляют пропорционально увеличивающие
ся объемы раствора сравнения с известной KOH
центрацией определяемоro элемента, получая
серию растворов, содержащих устойчиво воз
растающие концентрации элемента, значения
эмиссии котороro, если это возможно, находят
ся в линейной области rрадуировочноrо rрафи
ка. Доводят содержимое каждой колбы paCTBO
рителем до метки.
Вводят растворы, используя одинаковое KO
личество повторов. для получения устойчивых
результатов.
Расчет. Методом наименьших квадратов
рассчитывают линейное уравнение rрафика и по
нему концентрацию определяемоrо элемента
в испытуемом растворе.
ВАЛИДАЦИЯ МЕТОДА
Через определенные временные интервалы
верифицируют удовлетворительное исполнение
метода, описанноro в частной статье.
ЛИНЕЙНОСТЬ
rотовят и анализируют не менее четырех
растворов сравнения, концентрация которых Ha
ходится в пределах диапазона rрадуировки, и
холостой раствор. Проводят не менее пяти по
второв.
Используя все полученные данные, рассчи
тывают линейное уравнение rрафика методом
наименьших квадратов. Строят кривую pefpec
сии, отмечая средние значения, измеренные
значения и доверительный интервал rрадуиро
вочноro rрафика. Метод является приrодным при
условии соблюдения следующих требований:
коэффициент корреляции должен быть не
менее 0,99;
на rрадуировочном rрафике поrрешности
каждоro калибровочноro уровня должны быть
распределены случайным образом.
Рассчитывают среднее значение и относи
тельное стандартное отклонение для наимень
шеro и наибольшеro калибровочноro уровня.
В случае если отношение рассчитанноro
стандартноro отклонения наименьшеro и наи
большеrо калибровочноro уровня менее 0,5
или более 2,0, то более точная оценка rрадуи
ровочноrо rрафика может быть получена с ис
пользованием взвешенной линейной реrрессии.
Линейная и квадратичная весовая функции при
меняются к полученным данным для нахожде.
.ния наиболее ПОДХОДЯLЦей для использоваНИf
весовой qpункции. Если средние значения пр
сравнении с rрадуировочным rрафиком про.
являют отклонение от линейности, использую,
двухмерную линейную реrрессию.
ПРАВИЛЬНОСТЬ
Предпочтительно верифицировать правиль
ность с использованием сертифицированных
стандартных образцов. Если это невозможно,
проверяют открываемость.
Открываемость. В случае методик количе
cTBeHHoro определения открываемость должна
быть от 90 % до 110 %. Для друrих определе
ний, например для определения следовых коли
честв элемента, открываемость должна быть от
80 % до 120 % от теоретическоro значения. OT
крываемость может быть определена с исполь
зованием подходящеrо раствора сравнения (Ma
триксноrо раствора), содержащеrо известное
количество аналита (средняя концентрация rpa
дуировочноro rраqpика).
СХОДИМОСТЬ
Сходимость должна быть не более 3 % для
количественноrо определения и не более 5 %
для испытания на содержание примесей.
ПРЕДЕЛ КОЛИЧЕСТВЕННО/О
ОПРЕДЕЛЕНИЯ
Удостоверяются, что предел количественно
ro определения (например, определенный с ис
пользованием приближения 10а) ниже измеряе
моro значения.
01/2013:20223
2.2.23. АТОМНО АБСОРБЦИОННАЯ
СПЕКТРОМЕТРИЯ
ОСНОВНОЙ ПРИНЦИП
Атомная абсорбция это процесс поrло
щения электромаrнитноro излучения специфи
ческой длины волны атомом в основном cocтo
янии С переходом в возбужденное состояние.
Атомы в основном состоянии поrлощают энер
rию с резонансной частотой, и вследствие TaKoro
резонансноro поrлощения электромаrнитное из
лучение ослабляется. Поrлощенная энерrия
фактически прямо пропорциональна количеству
присутствующих атомов.
Данная rлава приводит общую информа
цию и определяет порядок действия при опре
делении элементов атомно абсорбционной
спектрометрией либо с помощью атомизации с
2. 2.23. Атомно абсорбцuонная спектрометрия
65
использованием пламени, электротермическоro
испарения в rрафитовой печи, получения rидри
дов либо методом холодноro пара для опреде
ления ртути.
Атомно абсорбционная спектрометрия
это метод определения концентрации элемента
в испытуемом образце путем измерения поrло
щения электромаrнитноro излучения атомным
паром элемента испытуемоro образца. Испыта
ние проводят при длине волны одной из линий
поrлощения (резонансных линий) определяемо
ro элемента. Количество поrлощенноrо излуче
ния, в соответствии с законом Буrера Ламберта
Бера, пропорционально концентрации элемента.
ПРИБОР
Основными составляющими частями прибо
ра являются:
источник излучения;
система введения и распыления образца:
атомизатор;
монохроматор или полихроматор;
детектор;
блок сбора данных.
Прибор обычно оснащается системой KOp
рекции фона. В качестве источника излуче
ния используют лампы с полым катодом (ЛПК,
HCL hollow cathode /атр) и безэлектродные
rазоразрядные лампы (Бэrл, EDL e/ectrode
/ess discha'ge /атр). Излучение таких ламп co
стоит из спектра, представляющеro собой очень
узкие линии с полушириной около 0,002 нм.
Существует три типа атомизаторов:
Пламенный метод
Пламенный атомизатор состоит из систе
мы распыления с пн евматическим приспосо
блением для получения аэрозоля, реrулятора
rаза и roрелки. Для получения температуры от
2000 К до 3000 К используют различные смеси
rорючеrо rаза (пропан, водород и ацетилен) и
окислителя (воздух и оксид азота). Конфиrу
рацию roрелки адаптируют под используемые
rазы, скорость подачи rаза реrулируется. Об .
разцы распыляют, используя подкисленную
воду как предпочтительный растворитель для
приroтовления испытуемых растворов и pac
творов сравнения. MOryT быть использованы
и орrанические растворители, если rарантиро
вано, что они не влияют на стабильность пла
мени.
Метод электротермической атомизации
Основными составляющими электротер
мическоro атомизатора являются rрафитовая
трубчатая пе% и источник электроэнерrии. При
использовании rрафитовой трубчатой печи про
исходит полная атомизация образца и атомный
пар удерживается на пути излучения в тече
ние длительноro времени, что улучшает предел
определения. Образцы (жидкости и твердые Be
щества) вводят непосредственно в rрафитовую
9 Заh 1712
трубчатую печь, которая наrревается постепен
но по заданной проrрамме, сначала высушивая
образец, затем удаляя основные компоненты
матрикса путем пиролиза, после чеrо атоми
зируя весь аналит. Очищают печь. наrревая ее
до температуры более высокой, чем темпера
тура атомизации. Продувание rрафитовой печи
инертным rазом во время пиролиза приводит к
более качественному процессу атомизации.
Метод холодН020 пара и 2идридный
метод
Атомный пар может быть получен и вне
спектрометра. Такой способ получения aTOMHO
ro пара используют в методе холодноro пара при
определении ртути или для определения таких
элементов, образующих rидриды, как мышьяк,
сурьма, висмут, селен и олово. В случае опре
деления ртути, атомы rенерируют химическим
восстановлением с помощью хлорида олова
или борrидрида натрия, после чеro атомный пар
быстро переносят с помощью инертноro rаза в
холодную кварцевую кювету, расположенную на
пути излучения, испускаемоro пампой. rидриды,
rенерированные таким образом, пере носятся с
помощью инертноrо rаза в roрячую кювету, rдe
они диссоциируют на атомы.
ИНТЕРФЕРЕНЦИЯ
При измерениях атомной абсорбции MOryT
иметь место химическая, физическая, иониза
ционная и спектральная интерференции. Хи
мическую интерференцию компенсируют ис
пользованием модификаторов матрикса или
высвобождающих areHToB либо использовани
ем высоких температур пламени оксид азота
ацетилен. Ионизационную интерференцию
компенсируют использованием специальных
ионизационных буферов (например, лантановых
или цезиевых). Физическую интерференцию,
обусловленную высоким содержанием соли или
вязкостью, компенсируют, используя разведе
ние образца, посредством метода стандартных
добавок или подбором матрикса. Спектральная
интерференция происходит при перекрывании
р зонансных линий, и для ее исключения ис
пользуют друryю резонансную линию. Исполь
зование коррекции фона Зеемана также компен
сирует спектральную интерференцию и помехи,
связанные с поrлощением излучения моле
кулами, особенно при использовании метода
электротермической атомизации. Также к спек
тральной интерференции может при водить ис
пользование мноroэлементных ламп С полым
катодом. Специфическое или неспецифическое
поrлощение ИЗМАРЯЮТ в спектральном диапа
зоне, определяемом выбранной шириной щели
монохроматора (0,2 2 нм).
КОРРЕКЦИЯ ФОНА
Рассеивание и фон увеличивают значе
ние измеряемоro поrлощения при атомизации
66
rосударственная фармакопея Республики Беларусь
пламенем или при электротермической атоми
зации. Поrлощение фоном охватывает Ш:lрО
кий диапазон длин волн, в то время как aTorv:bI
поrлощают в очень узких диапазонах порядка
O,005 ,02 нм. Поrлощение фоном может быть
скорректировано использованием контрольно
ro раствора, имеющеro точно такой же состав,
что и раствор образца, за исключением опреде
ляемоro элемента. что, однако, часто является
неосуществимым. При электротермической ато'
мизации температура пиролиза должна быть
подобрана так, чтобы исключить продукты раз
ложения матрикса, вызывающие фоновое по.
rлощение. Коррекция фона может быть прове
дена также с использованием двух различных
источников энерrии: лампы с полым катодом. с
помощью которой измеряют общее поrлощение
(элемент + фон), и дейтериевой лампы с непре
рыв ной эмиссией, с помощью которой измеря
ют фоновое поrлощение. Для коррекции фона
сиrнал, полученный от дейтериевой ламы, выи
тают из сиrнала, полученною от лампы с полым
катодом. Этот способ лимитирован спектраль
ным диапазоном дейтериевой лампы (от 190 нм
до 400 нм). Фоновое поrлощение также может
быть найдено путем измерения поrлощения при
двух длинах волн: резонансной линии и длине
волны, близкоЙ к резонансной, но при которой не
наблюдается поrпощения образцом, и последу
ющим вычитанием из поrлощения резочянсной
линии поrлощения второй длины волны.
Еще один способ коррекции фона эффект
3еемана (основан на расщеплении 3€er"aHa
линии поrлощения в маrнитном поле). Даннь:й
способ особенно полезен в случае, если фон
имеет тонкую структуру, а также позволяет зна
чительно скорректировать фон в диапазоне от
185 нм до 900 нм.
ВЫБОР УСЛОВИЙ ПРОВЕДЕНИЯ
ИСПЫТАНИЯ
После подбора подходящей длины волны и
ширины щели для определяемоro элемента pac
сматривают следующие моменты:
коррекцию неспецифическоro фоновоro
поrлощения;
необходимость прибавления химических
модификаторов или ионизационных буферов к
образцу, а также к контрольному раствору и pac
творам сравнения;
разведение образца (например, для мини
мизации физической интерференции);
детали температурной проrраммы, пред
варительный HarpeB, высушивание. пиролиз,
атомизацию, постатомизацию со временем ли
нейноrо нарастания и временем удерживания:
расход инертноro rаза;
модификаторы матрикса для электротер
мической атомизации (печь);
химические восстанавливающие peareH
ты для определения ртути или друrих rидри
добразующих элементов вместе с температурой
кюветы с холодным паром или температурой Ha
rреваемой кюветы;
технические требования по исполнению
печи (камера, платформа Львова и друrие).
МЕТОД
По ВОЗМОХНОСТИ рекомендуется использо
вать пластмассовую Л8бораторную посуду При
roтовление образцов может вкпючать paCTBope
ние, варку (преимущественно с использованием
микроволновоro излучения), стадию прокалива
ния или их сочетание для Toro, чтобы О<JИСТИТЬ Ma
трикс образца и/или удалить уrлеродсодержащие
вещества. Если процесс происходит в открытой
системе, если в частной статье не указано иноrо,
TeMf1epaTypa прокаJ"1ивания не должна превышать
600 ос по причине летучести некоторых металлов.
Атомно абсорбциончый спектрометр BЫ
водят на режим 8 соответствии с инструкци
ей завода произвrщителя и устанавливают He
обходимую длину волны. В reHepaToP aTOMHoro
пара вводят холостой раствор и настраивают
реrистрирующее устройство на максимум про
пускания. Значение холостоro опыта может
быть найдено при использовании растворителя
для выведения при60ра на нулевое значение.
Вводят раствор сравнения определяемоro эле
мента с наибольшей концентрацией и настраи
вают прибор так, чтобы получить максимальный
реrИСТРli1руемый сиrнал. Во избежание заrряз
нения и эффекта памяти тщательно промывают
прибор. После завершения анализа прибор про
r,lывают водой Рили ПО,'1кисленной водой.
В С,'lучае использс::,ан; ;'1 техники ввода T8ep
дых проб все УСJlОВИ пооведения испытания
описывают в частной статье.
Предпочтительно. чтобы концентрации pac
творов находились в линейной части rрадуиро
вочноro rрафика. Если это невозможно, MOryT
быть использованы криволинейные rрадуиро
вочные rрафики с применением подходящеro
проrраммноro обеспечения.
Определения проводят путем сравнивания
со стандартными растворами с известными co
держаниями определяемоro элемента или MeTO
дом rрадуировочноro rрафика (метод 1), или Me
тодом стандартных добавок (метод 11).
МЕТОД / МЕТОД rРАдУиРовочноrо
rРАФИКА
Обычно для измерений roтовят и использу
ют три раствора сравнения определяемоro эле
мента и холостой раствор.
Испытуемый раствор rотовят, как указано в
частной статье. Не менее трех растворов cpaBHe
ния определяемоro элемента rотовят так, чтобы
диапазон концентраций этих растворов включал
ожидаемое значение концентрации определя
емоro элемента в испытуемом растворе. ДЛЯ KO
личественноrо анализа оптимальные значения Ka
либровки должны находиться в диапазоне от 0,7
до 1,3 от ожидаемою содержания определяемо
2.2.24. Абсорбционная спектрофотометрuя в инфракрасной области
67
ю элемента или от предела, указанноro в част
ной статье. Для анализа на содержание примесей
оптимальные значения калибровки должны Haxo
диться в диапазоне от предела обнаружения до 1 ,2
от предельноro значения для определяемоro эле
мента. Любые реактивы, используемые при приro
товлении испытуемою раствора, прибавляют в xo
лостой раствор и растворы сравнения в таких же
количествах, как и в испытуемый раствор.
Вводят растворы, используя одинаковое KO
личество повторов, для получения устойчивых
результатов.
Расчет. Строят rрадуировочный rрафик за
висимости средних значений поrлощений pac
творов сравнения от концентрации, по которому
определяют концентрацию элемента в испыту
емом растворе.
МЕТОД 11 МЕТОД СТАНДАРТНЫХ
ДОБАВОК
Испытуемый раствор roтовят, как указа
но в частной статье. Равные объемы испытуе
моro раствора помещают не менее, чем в три
мерные колбы одинаковоro объема. Во все
мерные колбы кроме одной прибавляют пропор
ционально увеличивающиеся объемы раствора
сравнения с известной концентрацией опре
деляемоro элемента, получая серию paCTBO
ров, содержащих устойчиво возрастающие KOH
центрации определяемоro элемента, получая,
если это возможно, значения в линейной обла
сти кривой. Доводят содержимое каждой колбы
растворителем до метки.
Вводят растворы, используя одинаковое KO
личество повторов, для получения устойчивых
результатов.
Расчет. Методом наименьших квадратов
находят линейное уравнение rрафика и по нему
рассчитывают концентрацию определяемоro
элемента в испытуемом растворе.
ВАЛИДАЦИЯ МЕТОДА
Через определенные временные интервалы
верифицируют удовлетворительное исполнение
метода, описанною в частной статье.
ЛИНЕЙНОСТЬ
rотовят и анализируют не менее четырех pac
творов сравнения, концентрация которых Haxo
дится в пределах диапазона rрадуировки, и холо
стой раствор. Проводят не менее пяти повторов.
Используя все полученные данные, рассчи
тывают линейное уравнение rрафика методом
наимеНhШИХ квадратов. Строят кривую perpec
сии, отмечая средние значения, измеренные зна
чения и доверительный интервал rрадуировоч
ною rрафика. Метод является приroдным при
условии соблюдения следующих требований:
коэффициент корреляции должен быть не
менее 0,99;
на rрадуировочном rрафике поrрешности
каждоrо калибровочноro уровня должны быть
распределены случайным образом.
Рассчитывают среднее значение и относи
тельное стандартное отклонение для наимень
шеro и наибольшею калибровочноro уровня.
В случае если отношение рассчитанноro
стандартною отклонения наименьшею и наи
большею калибровочноro уровня менее 0,5 или
более 2,0. может быть получена более точная
оценка rрадуировочною rрафика с использова
нием взвешенной линейноЙ реrрессии. И пиней
ная, и квадратичная весовая функции приме
няются к полученным данным для нахождения
наиболее подходящей для использования Beco
вой функции. Если средние значения при cpaB
нении с rрадуировочным rрафиком проявляют
отклонение от линейности. используют ДBYXMep
ную линейную реrрессию.
ПРАВИЛЬНаСТЬ
Предпочтительно верифицировать правиль
ность с использованием сертифицированных
стандартных образцов. Если это невозможно,
проверяют открываем ость.
Открываемость. В случае методик количе
ственною определения открываемость должна
быть от 90 % до 110 %. Для друrих определе
ний, например для определения следовых коли
честв элемента, открываемость должна быть от
80 % до 120 % от теоретическою значения. OT
крываемость может быть определена с исполь
зованием подходящеro раствора сравнения (Ma
триксноro раствора), содержащею известное
количество аналита (средняя концентрация rpa
дуировочноro rрафика).
СХОДИМОСТЬ
Сходимость должна быть не более 3 % для
количественноro определения и не более 5 %
для испытания на содержание примесей.
ПРЕДЕЛ КОЛИЧЕСТВЕННО/О
ОПРЕДЕЛЕНИЯ
Удостоверяются,чтопределколичественно
ro определения (например, определенный с ис
пользованием приближения 100) ниже измеряе
моro значения.
01/2013:20224
2.2.24. АБСОРБЦИОННАЯ
СПЕКТРОФОТОМЕТРИЯ
В ИНФРАКРАСНОЙ ОБЛАСТИ
=Спектрофотометрию в инфракрасной об
ласти спектра обычно используют для исследо
68
rосударственная фармакопея Республики Беларусь
вания строения молекул, идентификации, коли
чественною определения, а также для контроля
примесей в субстанциях.#
Инфракрасные спектрофотометры приме
няют для записи спектров в области от 4000 CM 1
ДО 650 CM 1 (от 2,5 мкм до 15,4 мкм), а в HeKOТO
рых случаях до 200 CM 1 (до 50 мкм).
ПРИ БОР
Спектрофотометры для снятия спектров co
стоят из подходящеro источника излучения, MO
нохроматора или интерферометра и детектора.
В спектрофотометрах с Фурье преобразо
ванием используется полихроматическое из
лучение и рассчитывается спектр в заданной
области частот путем Фурье преобразования ис
ходных данных. Также MOryT быть использова
ны спектрофотометры, снабженные оптической
системой, способной выделять монохроматиче
ское излучение в измеряемой области. Обычно
спектр представляется как функция пропускания,
т. е. отношение интенсивности прошедшеrо к ин
тенсивности падающеro на образец излучения.
Но также может быть представлен как функция
поrлощения.
Оптическую плотность (А) определяют как
десятичный лоrарифм величины, обратной про
пусканию (7):
(1\ (/ )
А =lgl 1=lg l ,
,Т) /,
rде:
T= '
/0 '
/0 интенсивность излучения, падающеro
на вещество;
I интенсивность излучения, прошедшеro
через вещество.
#Каждый инфракрасный спектр характеризу
ется серией полос поrлощения, максимумы KO
торых определяются волновым числом v или
длиной волны Л и интенсивностью максимумов
поrлощения. Волновое число v, выраженное в
обратных сантиметрах (CM 1), определяется из
соотношения:
104
\' == ,
).
rдe:
л длина волны, мкм.#
подrОТОВКА ОБРАЗЦА
ДЛЯ ПОЛУЧЕНИЯ СПЕКТРОВ
ПРОПУСКАНИЯ ИЛИ поrЛОЩЕНИЯ
Жидкости. Жидкости исследуют в форме
пленки, находящейся между двумя пластинка
ми, прозрачными для инфракрасноrо излучения,
или в кювете с соответствующей толщиной слоя,
также прозрачной для инфракрасноrо излучения.
Жидкости или твердые вещества в рас-
творе. rотовят раствор испытуемоro образца
в подходящем растворителе. Выбирают KOH
центрацию вещества и толщину слоя кюветы,
позволяющие получить удовлетворительный
спектр. Обычно хорошие результаты получа
ют при концентрациях от 1 О r/л до 100 r/л при
толщине слоя от 0,5 мм до 0,1 мм. Поrлощение
растворителя компенсируют путем помещения в
канал сравнения аналоrичной кюветы, содержа
щей выбранный растворитель. При использова
нии прибора с Фурье преобразованием записы
вают поочередно спектр растворителя и спектр
образца и вычитают поrлощение растворителя с
учетом коэффициента компенсации.
Твердые вещества. Твердые вещества
исследуют дисперrированными в подходящей
жидкости в виде суспензии или в твердом co
стоянии (диски из rалоrенидов щелочных метал
лов). Если указано в частной статье, формиру
ют пленку из расплавленной массы между двумя
пластинами, прозрачными для инфракрасноro
излучения.
а) Суспензия. Небольшое количество ис
пытуемоrо образца растирают с минимальным
количеством вазелинов020 масла Рили друrой
подходящей жидкости; обычно от 5 Mr до 1 О Mr
испытуемоrо образца достаточно для получе
ния подходящей суспензии с использованием
одной капли вазелинов020 масла Р. Полученную
суспензию сжимают между двумя пластинками,
прозрачными для инфракрасною излучения.
б) Диски. От 1 Mr до 2 Mr испытуемоro об
разца растирают с ЗОО ОО Mr, если нет друrих
указаний, тщательно измельченною калия бро
мида Р или калия хлорида Р. Обычно этих KO
личеств достаточно для получения диска ди
аметром 1 o 15 мм и спектра соответствующей
интенсивности. Смесь тщательно растирают, по
мещают равномерно в пуансон и прессуют при
давлении около 800 МПа (8 т,см 2 ). Диски из He
стабильных при обычных атмосферных услови
ях или rиrроскопичных веществ прессуют в Ba
кууме. Причиной образования некачественных
дисков MOryT быть такие факторы, как HeДOCTa
точное или чрезмерное растирание, влажность
или иные примеси в дисперсионной среде и He
достаточное измельчение частиц. Диск неприro
ден для испытания, если он при визуальном oc
мотре неоднороден по прозрачности или если
пропускание 2000 CM 1 (5 мкм) составляет менее
60 % без компенсации при отсутствии специфи
ческой полосы поrлощения вещества.
rаЗi>I. rазы исследуют в кювете, прозрачной
для инфракрасноro излучения, с длиной оптиче
CKoro пути около 100 мм. Кювету откачивают и
заполняют через кран или при помощи иroльча
тоro клапана через подходящую rазовую линию
между кюветой и контейнером с субстанцией,
предназначенной для испытания.
2.2.24. Абсорбционная спектрофотометрия в инфракрасной области
69
Если необходимо, доводят давление в
кювете до атмосферноro, используя rаз, про
зрачный для инфракрасноro излучения (напри
мер, азот Рили ар20Н Р). Мешающее влияние
поrлощения воды, уrлерода диоксида или друrих
атмосферных rазов исключают путем помеще
ния в канал сравнения идентичной кюветы, KOTO
рая либо вакуумирована, либо заполнена rазом,
прозрачным для инфракрасноrо излучения.
ДЛЯ ПОЛУЧЕНИЯ СПЕКТРОВ
диФФУзноrо ОТРАЖЕНИЯ
Твердые вещества. Растирают в порошок
смесь из испытуемоro образца и мелко измель
ченноro и высушенноro калия бромида Рили
калия хлорида Р. Если иноro не указано в част
ной статье, roтовят смесь, содержащую около
5 % испытуемоro образца. Смесь растирают,
помещают в чашку для образца и записывают
спектр отражения.
После обработки записанноro спектра по
функции Кубелка Мунка (КиЬе/kа Мипk) может
быть получен спектр поrлощения испытуемоro
образца.
ДЛЯ ПОЛУЧЕНИЯ СПЕКТРОВ
НАРУШЕнноrо полноrо BHYTPEHHErO
ОТРАЖЕНИЯ
Нарушенное полное отражение (включая
MHoroKpaTHoe отражение) касается света, обычно
MHoroKpaTHo внутренне отраженноro пропуска
ющей средой. Существуют приборы, rдe проис
ходит только одно отражение. Испытуемый об
разец помещают на внутренний отражающий
элемент (ВОЭ, /RE), например, из алмаза, repMa
ния, цинка селенида, таллия бромида таллия
йодида (KRS 5) или из друrorо подходящеro Be
щества с высоким коэффициентом преломления,
и добиваются плотноro и однородноro контакта
со всей поверхностью кристалла внутреннеro OT
ражающеro элемента с помощью давления или
растворяя испытуемый образец в подходящем
растворителе и высушивая полученный раствор
на внутреннем отражающем элементе. Просма
тривают спектр нарушенноro полноro BHyтpeHHe
ro отражения (Н ПВО, ATR).
ИДЕНТИФИКАЦИЯ С ИСПОЛЬЗОВАНИЕМ
СТАНДАРТНЫХ ОБРАЗЦОВ
Образцы испытуемоro вещества и стандарт
HOro вещества roтовят по одной и той же методи
ке и записывают спектры в области от 4000 CM 1
ДО 650 CM 1 (от 2,5 мкм до 15,4 мкм) в одних И тех
же условиях. Минимумы пропускания (макси
мумы поrлощения) в спектрах испытуемой суб
стзнции должны соответствовать по положению
и относительной величине таковым в спектре
стандартноro образца.
Если спектры, полученные в твердом co
стоянии, отличаются по положению минимумов
пропускания (максимумов поrлощения), то ис
пытуемый образец и стандартный образец обра
JO.Зак.1712.
батывают одним и тем же способом так, чтобы
они кристаллизовались или получались в одной
и той же форме, или обрабатывают способом,
указанным в частной статье, а затем снимают
спектры.
ИДЕНТИФИКАЦИЯ С ИСПОЛЬЗОВАНИЕМ
ЭТАЛОННЫХ СПЕКТРОВ
Контроль разрешающей способности. В
случае приборов с монохроматором записыва
ют спектр пленки полистирола толщиной около
35 мкм. Разность х (см. рисунок 2.2.24. 1) между
процентом пропускания в максимуме пропуска
ния А при 2870 CM 1 (3,48 мкм) и минимуме про
пускания В при 2849,5 CM 1 (3,51 мкм) должна
быть более 18. Разность у между процентом
пропускания в максимуме пропускания С при
.1589 CM 1 (6,29 мкм) и минимуме пропускания О
при 1583 CM 1 (6,32 мкм) должна быть более 10.
В случае приборов с Фурье преобразова
нием устанавливают подходящее разрешение с
подходящей аподизацией, рекомендуемое про
изводителем. Для контроля разрешающей спо
собности используют подходящие методы, Ha
пример, записывают спектр пленки полистирола
толщиной около 35 мкм. Разность между поrло
щением в минимуме поrлощения при 2870 CM 1 и
максимуме поrлощения при 2849,5 CM 1 должна
быть более 0,33. Разность между поrлощени
ем в минимуме поrлощения при 1589 CM 1 и MaK
симуме поrлощения при 1583 CM 1 должна быть
более 0,08.
<F.
1\ " n
)
с
f
1
D
ф
:s;
:х:
ro
""
u
>.
с:
о
Q.
с:
80
80
60
60
40
40
20
А
20
6
о о
3200 3000 2800 2600 1800
1600
1400
волновое число, СМ '
Рисунок 2.2.24. 1. Типичный спектр полисти
рола, используеМ020 для проверки разрешаю
щей способности
70
rосударственная фармакопея Республики Беларусь
01/2013:20225
Проверка шкалы волновых чисел. Шкала
волновых чисел может быть проверена с ис;->оль
зованием пленки полистирола, которая имеет
минимум пропускания (максимум поrлощения)
при волновых числах (CM '), приведенных в Ta
блице 2.2.24. 1.
Таблица 2.2.24. 1
Максимумы ПРОПУСКдния и допустимые
пределы пленки полистирола
М Допустимые приделы (CM 1) !
.инимум
пропускания Прибор ! Прибор с Фу- I
(смо1) С монохрома-I рье-преОбра- 1
3060,0 I Т + З В 1
2849,5 :t2,0 :t1.0!
1942,9 :t1,5 :t1,0!
1601,2 :t1,0 I :t1,0 1
1583,0 :t1,O I - :t1 ,0 l
, ,
1154,5 :t1,O I :t1,0 !
1028,3 :t1,O :t1 ,О i
#Для проверки шкалы волновых чисел MOryT
использоваться методики, приведенные в ин
струкции К прибору. #
Методика. Испытуемый образец rOTo
вят к испытанию соrласно инструкции, при
ложенной к эталонному спектру/стандартно
му образцу. Использу УСJlСВ 'Я. при KOTOpblX
записывали эталонныЙ спектр обы'-iНО СО8па
дающие с условиями для проверки разреша
ющей способности, записывают спектр испы
TyeMoro образца.
Положения и относительные величины
полос на спектре испытуемоro образца и на
эталонном спектре должны соrласовываться
между собой.
Компенсация влияния паров воды и ат-
мосферноrо уrлерода диоксида. В случае
приборов с Фурье-преобразованием спектраль-
ная интерференция, вызываемая парами воды
идиоксидом уrлерода, компенсируется с ис
пользованием подходящих алroритмов в соот-
ветствии с инструкциями производителя. Кроме
тоro, спектры MorYT быть получены либо с ис
пользованием продутых приборов, либо при аб
солютно одинаковых условиях записи однолу
чевоro спектра фона и спектра образца.
ПРИМЕСИ В rАЗАХ
Для анализа примесей используют кювету,
прозрачную для инфракрасноrо излучения и
имеющую соответствующую длину оптическо
ro пути (например, от 1 м до 20 м). Кювету за
полняют так, как указано в разделе «rазы». Для
определения и количественной оценки приме
сей используют методики, указанные в частных
статьях.
2.2.25. АБСОРБЦИОННАЯ
СПЕКТРОФОТОМЕТРИЯ
В УЛЬТРАФИОЛЕТОВОЙ
И ВИДИМОЙ ОБЛАСТЯХ
Определение оптической плотности. Оп
тическая плотн сть (А) раствора представляет
собой десятичный ЛОi арифм обратной величи
ны пропускания (т) для монохроматическоro из-
лучения и выражается соотношением:
1 по '\
А = I g .. I = I g ,1 1,
\ Т J 1C , j
rдe:
T= :
'о
'о интенсивность падающеrо MOHoxpOMa
тическоro излучения;
/ интенсивность прошедшеro MOHoxpOMa '
тическоro излучения.
В отсутствие друrих физико химических
факторов измеренная оптическая плотность (А)
пропорциональна длине пути (Ь), через который
проходит излучение, и концентрации (с) веще
ства в растворе в соответствии с уравнением:
А = ;, с . Ь,
rдe:
[; молярный kо-эфсрициент ПОrJ IOщения;
Ь длина оптичеекоrо пути. в сантиметрах,
с концентрация вещества в растворе, в
моль на литр.
Величина Ас.. представляет собой удель-
ный показатель поrлошения. то есть оптическую
плотность раствора вещества с концентрацией
1 О r/л в кювете с толшиной слоя 1 см:
А ''''' = 10,[, .
,.,.,
Мм.
Если нет друrих указаний в частной статье,
измерение оптической плотности проводят
при указанной длине волны с использованием
кюветы 1 см. Если нет друrих указаний в част-
ной статье, измерение проводят по сравнению
с тем же растворителем или той же смесью
растворителей, в которой растворено веще
ство. Оптическая плотность растворителя, из
меренная против воздуха при указанной длине
волны, не должна превышать 0,4 и желатель
но, чтобы она была менее 0.2. Спектр поrло
щения представляют таким образом, чтобы оп
тическая плотность или ее некоторая функция
были приведены по оси ординат, а длина волны
или некоторая функция от длины волны по
оси абсцисс.
Если в частной статье приводят только одно
значение для положения максимума поrлоще
ния, то это означае что полученное значение
2.2.25. Абсорбционная спектрофотометрия в уф и видимой областях
71
максимума не должно отличаться от указанноro
более чем на :!::2 нм.
Прибор. Спектрофотометры, предназна
ченные для измерений в ультрафиолетовой и
видимой областях спектра, состоят из оптиче
ской системы, выделяющей монохроматиче
ское излучение в области от 200 нм ДО 800 нм.
И устройства для измерения оптической плот
ности.
Проверка шкалы длин волн. Для провер
ки шкалы длин волн используют линии BOДO
родной или дейтериевой разрядной лампы или
линии паров ртути, а также максимумы поrло
щения раствора 20льмия перхлората Р, KO
торые пред ставлены в таблице 2.2.25. 1. Дo
пустимое отклонение составляет :!::1 нм для
ультрафиолетовоrо и :!::3 нм для видимоro диа
пазонов.
Таблица 2.2.25. 1
Максимумы ПО2лощения (или испускания) для
проверки шкалы длин волн
241,15 нм (Но) 404,66 нм (Hg)
253,7 нм (Hg) 435,83 нм (Hg)
287,15 нм (Но) 486,0 нм (013)
302,25 нм (Hg) 486,1 нм (Н13)
313,16 нм (Hg) 536,3 нм (Но)
334,15 нм (Hg) 546,07 нм (Hg)
361,5 нм (Но) 576,96 нм (Hg)
365,48 нм (Hg) 579,07 нм (Hg)
Проверка шкалы оптической плотности.
Проверяют значения оптических плотностей,
используя подходящие фильтры или раствор
калия дихромата Р при длинах волн, указан
ных в таблице 2.2.25. 2. В таблице 2.2.25. 2
приведены точные значения удельноro пока
зателя поrлощения и ero допустимые пределы
для каждой длины волны. Данные таблицы oc
нованы на допустимой поrрешности при изме
рении поrлощения :!:О,01.
ДЛЯ проверки шкалы оптических плотно
стей используют растворы калия диxpOMa
та Р, который предварительно сушат при TeM
пературе 130 0 С ДО постоянной массы. Для
проверки шкалы оптических плотностей при
длинах волн 235 нм, 257 нм, 313 нм и 350 нм
растворяют 57 ,0 63,0 Mr калия дихромата Р
в 0,005 М растворе кислоты серной и доводят
до объема 1000,0 мл этим же растворителем.
Для проверки шкалы оптических плотностей
при длине волны 430 нм растворяют 57,0
63,0 Mr калия дихромата Р в 0,005 М pac
творе кислоты серной и доводят до объема
100,0 мл этим же растворителем. Также MorYT
быть использованы и друrие сертифицирован
ные стандартные вещества.
Таблица 2.2.25. 2
Длина Удельн ый I
показатель Допустимые
волны, поrлощения пределы А;;:
нм А ' % I
1см
235 124,5 от 122,9 до 126,2
257 144,5 от 142,8 до 146,2
313 48,6 от 47,0 до 50,3
350 107,3 от 105,6 до 109,0
430 15,9 от 15,7 до 16,1
Предельный уровень рассеянноrо
света. Рассеянный свет может быть опреде
лен при данной длине волны с использовани
ем соответствующих фильтров или растворов.
Например, оптическая плотность раствора
2 r/л калия хлорида Р в кювете с толщиной
слоя 1 см резко возрастает между 220 нм и
200 нм И имеет значение более 2,0 при 198 нм
при использовании воды в качестве компенса
ционноro раствора. Также MorYT быть исполь
зованы и друrие сертифицированные CTaH
дартные вещества.
Разрешающая способность (для каче
ственноro анализа). Если указано в частных
статьях, то определяют разрешающую способ
ность спектрофотометра следующим образом.
Записывают спектр 0,02 % (об/об) раствора
толуола Р в 2ексане Р. Минимаf1ЬНО допусти
мое значение отношения оптической плотно
сти в максимуме поrлощения при 269 нм к оп
тической плотности в минимуме поrлощения
при 266 нм указывают в частной статье. Также
MorYT быть использованы и друrие сертифици
рованные стандартные вещества.
Ширина спектральной щели (для коли
чественноro анализа). В случае использования
спектрофотометра с изменяемой шириной спек
тральной щели при выбранной длине волны
возможны поrрешности, связанные с шириной
этой щели. Для их исключения ширина спек
тральной щели должна быть малой по cpaB
нению с полушириной полосы поrлощения и в
то же время должна быть максимально велика
для получения высокоro уровня 'о' Таким обра
зом, ширина щели должна быть такой, чтобы
дальнейшее ее уменьшение не изменяло вели
чину измеряемой оптической плотности.
Кюветы. Допустимые вариации в толщи
не слоя используемых кювет должны быть не
более :!:О,005 см. Кюветы, предназначенные для
испытуемоro и компенсационноrо растворов,
должны иметь одинаковое пропускание (или
оптическую плотность) при заполнении одним и
тем же растворителем. В противном случае это
различие следует учитывать.
Чистка и обращение с кюветами должны
быть аккуратными.
72
rосударственная фармакопея Республики Беларусь
ПРОИЗВОДНАЯ СПЕКТРОФОТОМЕТРИЯ
В производной спектрофотометрии исr:()ль
зуется преобразование исходною спектра поrло
щения (нулевой порядок) в производные спек
тры первоro, второro и более высоких порядков.
ПроизводНblЙ спектр перв020 порядка
представляет собой rрафик зависимости rради
ента кривой поrлощения (скорость изменения
оптической плотности с длиной волны, dА/dл) от
длины волны.
ПроизводНblЙ спектр втОр020 порядка
представляет собой rрафик зависимости кри
визны спектра поrлощения от длины волны
(d 2 А/dл 2 ). Вторая производная при любой длине
волны л связана с концентрацией следующим
соотношением:
d 2 А d 2 А;с: с'Ь d 2 АЕ сЬ
== ' ' I
d/. 2 dл 2 1 О dл. 2 1 О
rдe:
с' концентрация поrлощающеro paCT
вора, в rpaMMax на литр.
Прибор. Используют спектрофотометр, OT
вечающий указанным выше требованиям и oc
нащенный аналоювым резистентноемкостным
дифференцирующим модулем, цифровым диф
ференциатором или друrими средствами полу
чения производных спектров. Некоторые методы
получения производных спектров второro поряд
ка сдвиrают их относительно спектра нулевою
порядка, что следует учитывать там, rдe это He
обходимо.
Разрешающая способность. Если указа
но в частных статьях, записывают производный
спектр второю порядка для раствора 0,2 r/л тo
луола Р в метаноле Р, используя метанол Р в
качестве компенсационноrо раствора. На спек
тре должен присутствовать небольшой отри
цательный экстремум, расположенный между
двумя большими отрицательными экстрему
мами при 261 нм и 268 нм соответственно, как
показано на рисунке 2.2.25. 1. Если нет друrих
указаний в частных статьях, отношение NB (см.
рисунок 2.2.25. 1) должно быть не менее 0,2.
Методика. rотовят раствор испытуемоro Be
щества, устанавливают различные инструмен
тальные характеристики в соответствии с ин
струкцией к прибору и рассчитывают количество
определяемою вещества, как указано в частной
статье.
#ИДЕНТИФИКАЦИЯ
Абсорбционную спектрофотометрию в уль
трафиолетовой и видимой областях спектра
обычно применяют для идентификации лекар
ственною средства в следующих вариантах.
1. Сравнение спектров поrлощения испыту
емоro раствора и раствора сравнения; в указан
d 2 А/dл 2
26З
+
+
А
265
в
261
268
240
260
280 290 нм
Рисунок 2.2.25. 1.
ной области спектра должно наблюдаться co
впадение положений максимумов, минимумов,
плеч и точек переrиба.
2. В указанной облаС1И спектра при YKa
занных длинах волн должны наблюдаться MaK
симумы, минимумы. плечи и точки переrиба;
возможно указание только некоторых из этих
характеристик. Расхождение между наблюда
емыми и указанными длинами волн не должно
обычно превышать :t2 нм.
3. В дополнение к варианту 2 при водят еще
и удельные показатели поrлощения при указан
ных длинах волн.
4. В дополнение к варианту 2 при водят OT
ношения оптических плотностей при указанных
длинах волн.
Возможны и друrие варианты применения,
оroворенные в частных статьях.
#КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Рекомендуется такая схема про ведения
спектрофотометрических измерений: «В изме
рительную кювету помещают иСПblтуеМblЙ
раствор и определяют ezo оптическую плот
ность по сравнению с компенсациОННblМ pac
твором. 3атем кювету вblнимаюm, удаляют
ее содержимое, снова помещают иСПblтуе
МblЙ раствор и снова определяют оптическую
плотность. Операцию повторяюm, получая не
менее трех значений оптической плотности
для иСПblтуеМ020 раствора. Таким же образом
получают не менее трех значений оптической
плотности для раствора сравнения. Следует
избе2ать попадания исследуеМ020 раствора
2.2.25. Абсорбционная спектрофотометрия в уф и видимой областях
73
на внешнюю стенку кюветы. Для расчетов иc
пользуют среднее значение полученных опти
ческих плотностей испытуеМ02О раствора и
раствора сравнения».
Однокомпонентный ОДНОВОЛНОВОЙ
анализ. Однокомпонентный одноволновой
анализ (или «обычная спектрофотометрия» )
это количественное определение одноro из KOM
понентов лекарственноro средства посредством
измерения оптической плотности раствора ис
пытуемою образца при одной аналитической
длине волны (АДВ).
Такой анализ может проводиться методом
показателя поrлощения (МПП) и методом CTaH
дарта (М С).
При использовании МПП количественное
определение проводят посредством измерения
оптической плотности (А) раствора испытуемо
ю образца при АДВ и расчете концентрации (с)
анализируемою компонента по формуле:
c А , (1)
А 1 %
1см
rде:
А;;: удельный показатель поrлощения
анализируемою компонента при АДВ;
с концентрация анализируемою веще
ства, % (м/об).
При использовании МС количественное
определение про водят посредством измерения
при АДВ оптических плотностей раствора испы
туемоro образца (А) и раствора сравнения (Ао)
с концентрацией со и расчета концентрации (с)
анализируемою компонента, исходя из форму
лы:
= . (2)
Gэ Ао
Измерение оптических плотностей испы
туемоro раствора и раствора сравнения следу
ет проводить в одних И тех же условиях с мини
мальным интервалом времени.
В общем случае более надежным является
МС. Возможность применения МПП необходи
мо в каждом конкретном случае обосновывать,
исходя из допусков количественною содержания
анализируемоrо компонента, метролоrических
характеристик методики и требований к спектро
фотометру. Обычно МПП применим при допу
сках содержания анализируемоro компонента не
менее :!:10 % от номинальноro содержания.
Во всех случаях применения одновслновоro
однокомпонентноro анализа необходимо, чтобы
остальные компоненты препарата не оказывали
существенною влияния на результаты. Обычно
доля их суммарноro поrлощения в оптическом
поrлощении образца при АДВ не должна превы
шать десятой части допусков содержания aHa
лизируемоro компонента.
11.30K.1712
Мноrокомпонентный спектрофотометри-
ческий анализ. Мноroкомпонентный спектро
фотометрический анализ применяют для oд
HOBpeMeHHoro количественноrо определения
компонентов лекарственных средств.
В обычной спектрофотометрии поrpешность
собственно спектрофотометрических измере
ний (<<спектрофотометрическая поrрешность»)
мало зависит от типа анализируемоro вещества и
выбора аналитической длины волны, а определя
ется классом спектрофотометра и не превышает
обычно 0,5 %. С учетом поrрешностей приroтовле
ния растворов это приводит к суммарной поrреш
ности анализа, не превышающей обычно 1 %.
В отличие от обычной спектрофотометрии,
спектрофотометрическая поrрешность мноro
компонентною анализа определяется не только
> классом прибора, но и сильно зависит от COCTa
ва анализируемоrо лекарственноrо средства и
особенно выбора аналитических длин волн. Эта
поrрешность может быть охарактеризована KO
эффициентом усиления К, который показывает,
во сколько раз спектрофотометрическая поrреш
ность определения данноro вещества в анали
зируемой смеси с помощью мноroкомпонентной
спектрофотометрии превышает спектрофотоме
трическую поrрешность определения этою же
вещества в чистом растворе (без друrих компо
нентов) методом обычной спектрофотометрии.
Способы расчета коэффициентов усиления для
каждоro компонента при использовании различ
ных методов представлены ниже.
Обычными являются величины К в
диапазоне 10, но возможны и значения К
равные 100 и более, что может приводить к
общей поrрешности анализа, составляющей дe
сятки и даже сотни процентов.
Для получения надежных результатов при
количественном определении лекарственных
средств коэффициенты усиления К не должны
обычно превышать 5.
Поэтому проrноз поrрешности определения
и сравнение ее с допусками содержания aHa
лизируемоro компонента является обязатель
ным условием при обосновании применимости
методик мноroкомпонентной спектрофотоме
трии. Если нет соответствующею обоснования,
то должно выдерживаться следующее COOTHO
шение между полной относительной поrрешно
стью количественноro определения k компонен
та анализируемоro образца (Д k r %) и' допусками
(:1:8) содержания этоro компонента в образце:
l\k.r:5: 0,32. В, (3)
l\k.r = 2. SCk,r' (4)
rдe:
SCkr относительное rенеральное CTaH
дартное отклонение полной поrрешности коли
чественноro определения k, компонента анали
зируемоro образца.
74
rосударственная фармакопея Республики Беларусь
Количественное определение в MHoroKOM
понентном спектрофотометрическом ана,lизе
основывается обычно на использовании ypaBl;e
ния:
т
А; == L,Eij 'С, j ==1...п, (5)
i=-1
rдe:
А, оптическая плотность испытуемоro
раствора при i ой длине волны;
Е показатели поrлощения (зависящие
q
от способа выражения концентрации) J oro KOM
понента образца при i ой аналитической длине
волны;
С ] концентрация j oro компонента об
разца.
Для решения данноro уравнения MorYT
при меняться различные подходы, среди KOТO
рых можно выделить три основных: метод наи
меньших квадратов (МНК), модифицирован
ный метод наименьших квадратов (ММНК) и
метод отношения рассчитанных концентраций
(МОРК).
Метод наименьших квадратов. Метод наи
меньших квадратов (МНК) является об06щени
ем метода показателя поrлощения одноволно
воro однокомпонентноro анализа. В рамках МНК
решение уравнения (3) имеет вид:
п
C k == Ia k , .А" 1< =1...п, (6)
1=1
Расчетные коэффициенты a MHk находят в
соответствии с матричным соотношением:
а МНК == (Е Т . E) 1 ЕТ, (7)
rдe:
а МНК матрица расчетных коэффициен
тов;
Е матрица показателей поrлощения;
т символ транспонирования.
Выбор аналитических длин волн (АДВ).
Выбор АДВ описан в разделе «Модифициро
ванный метод наименьших квадратов».
ПрО2НОЗ П02решности определения.
Полная поrрешность количественноro опреде
ления компонента с помощью МНК определя
ется из соотношения:
S;k., ==(к;нк )2 .(S ., +S ,)+S " (8)
(к;нк )2 == t( ak : ,51 J, (9)
rде:
SCk., относительное стандартное отклоне
ние полной поrрешности количественноro опре
деления k oro компонента образца;
А,51 оптическая плотность раствора MO
дельной смеси образца, содержащей номи
нальные концентрации всех компонентов;
c;st номинальная концентрация k oro
компонента образца в модельной смеси;
SA, относительное стандартное OT
клонение сходимости оптической плотности
на спектрофотометре (включая кюветную по
rрешность);
SfC' относительное стандартное откло
нение правильности оптической плотности на
спектрофотометре;
Sv,r относительное стандартное OT
клонение поrрешности приroтовления pac
творов.
Величины SA.r И SE.r извеСТНbI из паспорт
ных данных спектрофотометра. а Sv., оцени
вают, исходя из поrрешностей взятия навесок
и разбавлений.
При этом должны выполняться соотноше
ния (3 4).
Преимуществом МНК является то, что еro
применение не требует использования CTaH
дартных образцов. Однако из за значитель
ной поrрешности правильности оптической
плотности (из таблицы 2.2.25. 2 видно, что Be
личины Sv. MorYT достиrать нескольких про
центов) и ее неконтролируемости полная по
rрешность анализа посредством МНК может
достиrать десяти и более процентов, что
делает МНК ненадежным методом.
Ero применение предъявляет очень BЫ
сокие требования 1( спектрофотометрам и
уровню работы аналитическоro персонала,
поэтому он применим обычно только в Ha
учных исследованиях на стадии разработки
лекарственных средств при больших колеба
ниях в концентрации анализируемых компо
нентов.
Модифицированный метод наимень-
ших квадратов. Модифицированный метод
наименьших квадратов является одним из
вариантов обобщения метода стандарта на
случай мноroкомпонентной спектрофотоме
трии и основан на уравнениях:
А m С. т
d == = 'r . == 'r.X ( 10)
, A SI L.- 1) 51 IJ 1,
J=1 С ] J=1
Е 51
'! 'C j
r ==
" т
IEik .С: ,
k=1
, (11)
rде переменные имеют тот же смысл, что
и в уравнениях (5) и (9), величины 'q представ
ляют собой информационные коэффициенты,
а Х ; .100 представляет собой концентрацию
j oro компонента препарата в процентах к ero
номинальному содержанию.
Решение уравнения (10) имеет вид:
2.2.25. Абсорбционная спектрофотометрия в уф и видимой областях
75
п
Х '" ММНК d .
k==L,.a ki . ,I=1...п,
, 1
(12)
rдe расчетные коэффициенты а ММНК находят
по матричному уравнению
а ММНК =(r T .r(1r T , (13)
Выбор аналитических длин волн (АДВ).
АДВ находят исходя из критерия минимума KO
эффициента усиления КММНК, получаемоro из co
отношения:
т т п
(KMMHK)L == I-K: == I-I-(а;МНК)2, (14)
J 1 j 1 ; 1
rдe:
к J коэффициент усиления для j oro компо
нента образца.
В случае анализа двух соединений по двум
длинам волн АДВ можно находить из условия
максимума информационных коэффициентов
каждоro компонента при одной из двух длин
волн.
Схема проведения анализа. rотовят MO
дельную смесь, содержащую все компоненты
препарата точно в номинальных концентраци
ях (раствор сравнения). Проводят необходимые
разбавления, измеряют попеременно оптиче
ские плотности испытуемоro раствора и paCT
вора сравнения при АДВ и проводят расчет по
уравнениям (1 o 11 ).
ПрО2НОЗ П02решности определения.
Полную поrрешность количественноro опреде
ления компонента с помощью ММНК определя
ют из соотношения:
S2 = 2. [(кММНК ) 2 .S2 +s2] ( 15)
ck,r k А" V,r J
Должно выполняться соотношение (3).
Недостатком ММНК является необходи
мость приroтовления точной номинальной смеси
образца, однако он является самым надежным и
точным из всех мноroволновых методов количе
ственноro определения лекарственных средств.
Частный случай количественное опре
деление одН020 компонента смеси по одной
длине волны. Если информационный коэффици
ент ( J k компонента при i ой длине волны зна
чительно превосходит все остальные, то количе
ственное определение этоro компонента можно
проводить по упрощенной формуле:
X k =dj> (16)
Максимальная поrрешность такоro прибли
жения не превосходит 2B(1 k)' Данное прибли
жение является обоснованным при k 0,95.
Метод отношения рассчитанных KOH
центраций. Метод отношения рассчитанных
концентраций (МОРК) является одним из вари
антов обобщения метода стандарта на случай
мноroкомпонентной спектрофотометрии и oc
нован на допущении, что отношение KOHцeH
траций, рассчитанных для испытуемоro paCT
вора и раствора сравнения, является более
точным, чем сами рассчитанные KOHцeHTpa
ЦИИ, то есть:
п
Iakj.A j
Х = = j 1 , (17)
k 5t п
C k Ia k ; .A;st
1=1
rдe:
a kj коэффициенты расчетной матрицы,
полученные с помощью МНК (уравнение (7»
или друrими методами цифровой фильтрации,
например, методом производной спектрофото
метрии. В отсутствии фона поrлощения самым
точным является применение МНК.
Выбор аналитических длин волн (АДВ).
Смотрите выбор АДВ и ммнк.
Процедура проведения анализа. Такая же,
как для ММНК, но для изrотовления модель
ной смеси можно использовать концентрации
компонентов, близкие (а не точно равные) к HO
минальным. Расчет концентраций проводят по
уравнению (17).
Пр02НОЗ П02решности определения (в
случае использования МНК) проводят по COOT
ношению:
S;k" = 2. [(к;нк)2 .S;" +S .,]. (18)
Должно выполняться соотношение (3).
МОРК менее точен, чем ММНК, но он не
требует приroтовления точно номинальной
смеси и поэтому проще в применении.
#ПРОИЗВОДНАЯ СПЕКТРОФОТОМЕТРИЯ
Производную спектрофотометрию исполь
зуют для количественноro определения обычно
в тех случаях, Korдa имеется фоновое поrло
щение, вызванное присутствием веществ, co
держание которых не реrламентируется. Она
может применяться в двух вариантах.
В первом случае получение производной
проводит сам аналитик с помощью произво
дных полиномов, которые представляются в
уравнении (17) вместо величин a k ;. Проrноз по
rрешности концентрации должен учитывать в
этом случае поrрешность неполноro обраще
ния в нуль поrлощения друrих компонентов
смеси.
Во втором случае непосредственчо ис
пользуют значения производных, получаемых
на спектрофотометре. Процедура анализа при
этом аналоrична применению метода CTaHдap
та в обычной спектрофотометрии, но вместо
оптических плотностей используют произво
дные.
76
01/2013:20226
/осударственная фармакопея Республики Беларусь
2.2.26. БУМАЖНАЯ ХРОМАтоrРАФt'fЯ
#Хроматоrрафия на бумаrе представляет
собой хроматоrрафический метод разделения.
при котором подвижная жидкая фаза перемеща
ется по капиллярам и поверхности xpoMaTorpa
фической бумаrи либо веществ, предварительно
нанесенных на ее волокна. В бумажной xpOMaTO
rрафии используют хроматоrрафическую бумаry
нужной плотности квалификации «Для xpOMaTO
rрафии».#
ВОСХОДЯЩАЯ ХРОМАтоrРАФИЯ НА
БУМАrЕ
Оборудование. Оборудование представляет
собой стеклянную камеру с тщательно пришли
фованной крышкой, соответствующую по разме
рам используемой хроматоrрафической бумаrе.
Камера в верхней части должна быть снабжена
приспособлением для закрепления xpoMaTorpa
фической бумаrи, с помощью котороro ее можно
опускать, не открывая камеры. На дне камеры
помещена емкость (лодочка) для подвижной
фазы, в которую должен быть поrружен нижний
край бумаrи. Хроматоrрафическая бумаrа пред
ставляет собой соответствующую фИЛЫр08аль
ную бумаry, разрезанную на ПОЛОСКИ достаточ
ной длины и шириной не менее 2.5 см: бумаrа
должна быть разрезана так, чтобы подвижная
фаза двиrалась вдоль направления волокна
бумаrи.
#В некоторых случаях допускается использо
вание хроматоrрафических камер друrих типов с
описанием их в частной статье."
Методика. Обозначенную в частной СТЭlъе
подвижную фазу помещают в лодочку в количе
стве, достаточном для образования слоя 2,5 см.
Если указано в частных статьях. между CT€Hl\a
ми камеры и лодочки помещают хроматоrрафи
ческую бумаry. Для насыщения камеру закрыва
ют крышкой и выдерживают в течение 24 ч при
температуре от 20 ос до 25 СС. Подцерживают
заданную температуру камеры на протяжении
всеro испытания.
На расстоянии 3 см от одноro края по всей
ширине полоски бумаrи карандашом наносят
тонкую roризонтальную линию. Обозначенный в
частной статье объем раствора наносят на полу
ченную линию при помощи микропипетки. Если
весь обозначенный раствор образует пятно диа
метром более 10 мм, раствор наносят неболь
шими порциями, давая каждой из них высохнуть
перед нанесением следующей. Если на одной
полоске бумаrи должно быть получено больше
одной xpoMaTorpaMMbI, растворы наносят вдоль
линии так, чтобы расстояние между точками Ha
несения отдельных проб было не менее 3 см.
#Если условия насыщения хроматоrрафи
ческой камеры, нанесения пятен или xpOMaTO
rрафирования отличаются от условий, описан
ных выше, они должны быть описаны в частной
статье."
Бумаry помещают в камеру, закрывают
камеру крышкой и выдерживают в течение 1,5 ч.
Бумаry опускают в подвижну.о фазу и проводят
элюирование в соответствии с указанным Bpe
менем или на указанное расстояние, которое
должна пройти подвижная фаза. На протяжении
всеro процесса злюирсвзния бумаrу защищают
от воздействия яркоrо света. Полоску бумаrи BЫ
нимают из камеры и сушат на воздухе.
#После прохождения подвижной фазой He
обходимоrо расстояния, обозначенною в част
ной статье, бумаry вынимают, отмечают KapaH
дашом конец положения фронта подвижной
фазы, сушат и проявляют пятна обозначенным
в частной статье способом.
Визуальную оценку и количественные из
мерения проводят в соответствии со статьей
2.2.27. Тонкослойная хромат02рафия.#
НИСХОДЯЩАЯ ХРОМАтоrРАФИЯ НА
БУМАrЕ
Оборудование. Оборудование представляет
собой стеклянную камеру с тщательно пришли
фованной крышкой, соответствующую по разме
рам используемой хроматоrрафической бумаrе.
В центре КрЫШКИ должно быть отверстие с диа
метром около 1,5 см, закрытое тяжелой стеклян
ной пластиной или пробкой. В верхней части
камеры располаrается лодочка для подвижной
фазы, снабженная пр.1способлением для закре
пления хроматоrраф 1ческой бумаrи. От каждой
стороны лодочки параллельно ей инемною
выше верхнеro ее края размещены два прямых
стеклянных стержня, которые подцерживают
бумаry в таком положении, чтобы она не сопри
касалась со стенками "амеры. Хроматоrрафиче
екая бумаrа представляет собой соответствую
щую фильтровальную бумаry, разрезанную на
полоски достаточной длины шириной не менее
2,5 см и не более длины лодочки; бумаrа должна
быть разрезана таким образом, чтобы подвиж
ная фаза двиrалась вдоль направления волок
на бумаrи.
#В необходимых случаях допускается ис
пользование хроматоrрафических камер друrих
типов с описанием их в частной статье.#
Методика. Обозначенную в частной статье
подвижную фазу помещают на дно xpoMaтorpa
фической камеры в количестве, достаточном
для образования слоя 2,5 см. Для насыщения
камеру закрывают крышкой и выдерживают в Te
чение 24 ч при температуре от 20 ос дО 25 ос.
Подцерживают заданную температуру камеры
на протяжении всеro испытания.
По всей ширине полоски бумаrи KapaHдa
шом наносят тонкую rоризонтальную линию от
закрепленною в лодочке края на таком paCCTO
янии, чтобы линия была размещена параллель
но направляющему стержню и несколькими caH
тиметрами ниже еro. Обозначенный в частной
2.2.27.
Тонкослойная хромат02рафuя
77
статье объем раствора наносят на полученную
линию при помощи микропипетки. Если весь
обозначенный раствор образует пятно диаме
тром более 1 О мм, раствор наносят небольшими
порциями, давая каждой из них высохнуть перед
нанесением следующей. Если на одной поло
ске бумаrи должно быть получено больше одной
xpoMaTorpaMMbI, растворы наносят вдоль линии
так, чтобы расстояние между точками нанесения
отдельных проб было не менее 3 см.
#Если условия насыщения хроматоrрафиче
ской камеры, нанесения пятен или хроматоrрафи
рования отличаются от условий, описанных выше,
они должны быть описаны в частной статье.#
Бумаry помещают в камеру, закрывают
камеру крышкой и выдерживают в течение 1 ,5 ч.
Через отверстие в крышке в лодочку помещают
достаточное количество подвижной фазы и прd
водят элюирование в соответствии с указанным
временем или на указанное расстояние, которое
должна пройти подвижная фаза. На протяжении
всеro процесса элюирования бумаry защищают
от воздействия яркоro света.
#После прохождения подвижной фазой He
обходимоro расстояния, обозначенноrо в част
ной статье, бумаry вынимают, отмечают KapaH
дашом конец положения фронта подвижной
фазы, сушат и проявляют пятна обозначенным
в частной статье способом.
Визуальную оценку и количественные из
мерения проводят в соответствии со статьей
2.2.27. Тонкослойная хромато?рафия.#
01/2013:20227
2.2.27. ТОНКОСЛОЙНАЯ
ХРОМАтоrРАФИЯ
Тонкослойная хроматоrрафия (ТСХ) пред
ставляет собой метод разделения, в котором
используется неподвижная фаза, состоящая из
подходящеro материала, нанесенноro в виде
тонкоro слоя и зафиксированноro на подложке
(пластинке) из стекла, металла или пластмас
сы. Перед хроматоrрафированием растворы
анализируемых веществ наносят на пластин
ку. Разделение основано на процессах адсорб
ции, распределения, ионноro обмена или на их
комбинации и осуществляется посредством пе
ремещения в тонком слое (не подвижной фазе)
исследуемых веществ, растворенных в paCTBO
рителе или в соответствующей смеси раствори
телей (подвижной фазе).
ОБОРУДОВАНИЕ
Пластинки. Хроматоrрафирование прово
дят с использованием пластинок, полученных,
как указано в статье 4.1.1. Реактивы.
12. За.. 1712.
#Допускается использование пластинок,
приroтовленных в промышленных условиях,
если они отвечают требованиям статьи 4.1.1.
Реактивы, а также выдерживают испытание
«Проверка приroдности хроматоrрафической
системы», описанное в частной статье. PeKO
мендуется каждую партию получаемых пласти
но к, приroтовленных в промышленных условиях,
проверять на близость величин R F . #
#Близость величин R F . Испытания прово
дят не менее чем на трех пластинках испыту
емой партии. Для этоro используют методику
проверки хроматоrрафической разделяющей
способности, указанной в статье 4. 1. 1. PeaKти
вы (ТСХ пластинка со слоем силика2еля Р). На
линию старта каждой пластинки наносят по 5
пятен раствора для проверки при20дности ТСХ
пластинок Р, хроматоrрафируют и рассчитыва
ют величины R F красителей для каждоro HaHece
ния. В пределах каждой пластинки наибольшая
разница величин R F среди разных нанесений
для каждоro красителя не должна превышать
0,02. В противном случае такие пластинки не pe
комендуется использовать для фармакопейноro
анализа.
Предварительная под20товка пластинок.
В некоторых случаях может по надобиться про
мывка пластинок перед хроматоrрафировани
ем, которая может быть выполнена посредством
предварительноro элюирования чистых пла
стинок подходящим растворителем. Пластинки
MOryт быть также импреrнированны (пропитаны)
посредством таких процедур, как элюирование,
поrружение или опрыскивание. Перед использо
ванием пластинки активируют, если необходимо,
посредством наrревания в термостате при TeM
пературе от 100 ос до 105 ос в течение 1 ч.
#Допускаются друrие условия активации
пластинок, описанные в частных статьях.#
Хроматоrрафическая камера представля
ет собой емкость с плотно подоrнанной крыш
кой и С плоским дном или дном с двумя же
лобами из инертноro прозрачноrо материала,
соответствующими по размеру используемым
пластинкам. Для rоризонтальноro элюирова
ния хроматоrрафическая камера имеет желоб
для подвижной фазы и дополнительно coдep
жит устройство для подачи подвижной фазы к
неподвижной фазе.
#Допускается использование xpoMaTorpa
фических камер друrих типов С описанием их в
частных статьях.#
Микропипетки, микрошприцы, калибро
ванные капилляры или друrиЕ:' устройства,
приrодные для нанесения растворов.
Устройство для обнаружения флуорес
ценции для обнаружения непосредственной
флуоресценции или для обнаружения тушения
флуоресценции.
78
rосударственная фармакопея Республики Беларусь
Проявляющие устройства и реаК""ивы.
Для проявления разделенных веществ иcri. }пь
зуют подходящие устройства, приroдные для
нанесения реактивов на пластинку (посред
ством опрыскивания, поrружения или обработ
ки парами) и, если это необходимо, для Harpe
вания.
Документирование. Для обеспечения дo
кументирования проявленных xpoMaTorpaMM
MOryт быть использованы различные приемы,
например фотоrрафирование или создание KOM
пьютерноro файла.
МЕТОДИКА
#Если в частной статье нет друrих указаний,
хроматоrрафическое разделение выполняют
восходящим способом.
Предпочтительнее использовать такие под
вижные фазы, которые обеспечивают величи
ны R F испытуемых соединений в пределах от 0,3
до 0,7.
Слой жидкости в хроматоrрафической
камере должен быть таким, чтобы после поме
щения в неro пластинки пятна ипи полосы Haxo
дились над уровнем жидкости."
Нанесение. Указанные объемы растворов
наносят на подходящем расстоянии от нижнеro
края и от боковых краев пластинки на линию, па
раллельную нижнему краю пластинки: расстоя
ние между центрами круrлых пятен должно co
ставлять не менее 10 мм (5 мм для пластинок
для высокоэффективной тонкослойной XOOMa
тоrрафии (ВЭТСХ), расстояние между краями
полос не менее 5 мм (2 мм для пластинок ДЛfl
ВЭТСХ). Растворы наносят минимальными пор
циями для получения пятен диаметром 2 5 мм
(1 2 мм для пластинок для ВЭТСХ) или полос
длиной 10 20 мм ( 10 мм для пластинок дЛЯ
ВЭТСХ) и шириной 1 2 мм.
Если частная статья предусматривает ис
пользование наряду с обычными пластинками и
пластинок для ВЭТСХ. рабочие условия дЛЯ BЫ
сокоэффективной тонкослойной хроматоrрафии
указываются в скобках [ ] после указания усло
вий для обычных пластинок.
Вертикальное элюирование. Стенки xpo
матоrрафической камеры выстилают филь
тровальной бумаroй. Подвижную фазу нали
вают в камеру в количестве, достаточном для
тоro, чтобы после смачивания фильтровальной
бумаrи покрыть дно камеры слоем жидкости, He
обходимым для хроматоrрафирования. Для Ha
сыщения хроматоrрафическую камеру с подвиж
ной фазой закрывают крышкой и выдерживают в
течение 1 ч при температуре от 20 ос до 25 ос.
Если в частной статье нет друrих указаний, xpo
матоrрафирование проводят в насыщенной
камере. Наносят указанные объемы растворов,
как описано выше. После испарения раствори
телей из нанесенных проб пластинку помещают
в хроматоrрафическую камеру как можно более
вертикально, следя за тем, чтобы пятна или
полосы находились выше поверхности подвиж
ной фазы. Камеру закрывают, оставляют ее при
температуре от 20 ос дО 25 ос в защищенном
от попадания прямых солнечных лучей месте.
После тоro, как подвижная фаза пройдет paCCTO
яние, указанное в частной статье, пластинку BЫ
нимают, сушат и обнаруживают пятна способом,
указанным в частной статье.
В случае двухмерной хроматоrрафии после
первоro хроматоrрафирования пластинку сушат
и выполняют второе хроматоrрафирование в Ha
правлении, перпендикулярном первому.
rоризонтальное элюирование. HaHO
сят указанные объемы растворов, как описано
выше. После испарения растворителей из Ha
несенных проб в желоб хроматоrрафической
камеры вводят с помощью шприца или пипетки
достаточное количество подвижной фазы, поме
щают пластинку. убеждаясь, что она расположе
на roризонтально и подсоединена к устройству
для подачи подвижной фазы, в соответствии с
инструкциями производителя. Если указано в
частной статье. пластинку элюируют, начиная
одновременно с двух концов. Камеру закрыва
ют и проводят хроматоrрафирование при темпе
ратуре от 20 ос дО 25 'с. После тоro, как под
вижная фаза пройдет расстояние, указанное в
частной статье, пластинку вынимают, сушат и
обнаруживают пятна YKClzaHHbIM способом.
В случае двухмернои )(роматоrрафии после
первоro хроматоrрафирования пластинку СУШа1
и выполняют второе хроматоrрафирование в Ha
правлении, перпендикулярном первому.
ВИЗУАЛЬНАЯ ОЦЕНКА
Идентификация. Основное пятно на xpo
MaTorpaMMe испытуемоro раствора сравнивают
с соответствуюшим пятном на xpoMaTorpaMMe
раствора сравнения, сравнивая ЦBe размер и
фактор подвижности (R F ) "(или фактор удержи
вания (R,»" обоих пятен.
Фактор подвижности (R F ) определяют как OT
ношение расстояния от точки нанесения пятна
до центра пятна после хроматоrрафирования к
расстоянию, пройденному фронтом растворите
ля от точки нанесения.
Проверка разделяющей способности He
подвижной фазы для идентификации. Обычно
для оценки приroдности достаточно испытания
на приroдность неподвижной фазы, описанноro
в статье 4.1.1. Реактивы. В особых случаях дo
полнительные требования указывают в частных
статьях.
Сопутствующие примеси. Дополнитель
ное пятно (пятна) на xpoMaтorpaMMe испытуе
моro раствора сравнивают визуально с COOTBeT
ствующим пятном (пятнами) на xpoMaTorpaMMe
2.2.27. Тонкослойная хроматоzрафия
79
раствора сравнения. В качестве стандартноrо
образца для приrотовления раствора cpaBHe
ния используют как саму примесь (при меси), так
и различные разведения испытуемоro раствора.
Проверка разделяющей способности. Tpe
бования для проверки разделяющей способности
приводят в соответствующих частных статьях.
Проверка чувствительности. Чувстви
тельность считается удовлетворительной, если
на xpOMaтorpaMMe наиболее разведенноro paCT
вора сравнения четко обнаруживается пятно или
полоса.
#Способы оценки содержания примесей
методом ТСХ. При контроле примесей нежела
тельно включение в частную статью требования
отсутствия пятна контролируемой при меси на
xpoMaтorpaMMe испытуемоro раствора. Методом
ТСХ MOryT контролироваться как конкретно YKa
зываемые (специфицированные) при меси, так и
сумма примесей.
Контроль специфицированных примесей.
Контроль специфицированных примесей приме
няют в тех случаях, коrда содержание каких то
конкретных примесей, возникающих в процес
се производства лекарственноrо средства или
еro хранения, должно быть оrраничено ввиду их
токсичности или по друrим соображениям. При
контроле специфицированных примесей обычно
используют сравнение пятен реrламентирован
ных примесей на xpoMaTorpaMMax испытуемоro
раствора и раствора сравнения.
Контроль обще20 содержания примесей. В
тех случаях, Korдa нет оснований считать какие
то примеси особо токсичными, часто не так
важно знать их истинное содержание. Важно
знать, что это содержание не превосходит опре
деленноro уровня. В таких случаях использу
ют метод внутренней нормализации: в качестве
растворов сравнения обычно используют pac
творы самой испытуемой субстанции различной
концентрации, а содержание примесей находят
в пересчете на эту субстанцию. В зависимости
от количества различных растворов субстанции,
наносимых на xpoMaTorpaMMY в виде растворов
сравнения, контроль общеro содержания приме
сей может быть одноуровневым, двухуровневым
и трехуровневым. В ДBYX и трехуровневых вари
антах возможна реrламентация и общей суммы
примесей.
КОЛИЧЕСТВЕННЫЕ ИЗМЕРЕНИЯ
Требования к разрешению и разделению
при водят в частных статьях.
В том случае, KOrдa вещества, разделяемые
методом тонкослойной хrюматоrрафии, поrло
щают или флуоресцируют в ультрафиолетовом
или видимом свете, их можно количественно
определить непосредственно на пластинке, ис
пользуя подходящее оборудование. Для этоro
измеряют отражение или пропускание падающе
ro света, передвиrая пластинку или измеряющее
устройство. Аналоrичным образом, используя
подходящее оптическое оборудование, можно
измерять флуоресценцию. Вещества, содержа
щие радионуклиды, MOryт быть количественно
определены тремя способами:
непосредственно на пластинке передви
жением пластинки вдоль подходящеro счетчика
радиоактивности или счетчика радиоактивности
вдоль пластины (см. статью Радиофармацевти
ческие препараты);
разрезанием пластинки на полосы и из
мерением радиоактивности на каждой полосе, с
использованием подходящеro счетчика радиоак
тивности;
соскребанием неподвижной фазы, pac
творением ее в подходящем сцинтилляционном
коктейле и измерением радиоактивности с ис
пользованием жидкостноrо сцинтилляционноrо
счетчика.
Оборудование. Оборудование для измере
ний непосредственно на пластинке включает в
себя:
устройство для точноro нанесения в опре
деленном месте пластинки необходимоro коли
чества вещества;
механическое устройство для передвиже
ния пластинки или измерительноrо устройства
вдоль осей Х или У;
реrистрирующее устройство и интеrратор
или компьютер;
для веществ, П02лощающих или флуо
ресцирующих в ультрафиолетовом или види
мом свете: для измерения отражения или про
пускания используются фотометр с источником
света, оптическое устройство, rенерирующее
монохроматический свет, и фотоэлемент COOT
ветствующей чувствительности; в том случае,
Korдa измеряется флуоресценция, требуется дo
полнительно монохроматический фильтр для
выбора соответствующей спектральной области
излучаемоro света;
для веществ, содержащих радионуклиды:
подходящий счетчик радиоактивности; для неro
необходимо проверить линейность диапазона.
Методика. rотовят раствор испытуемоro об
разца (испытуемый раствор), как указано в част
ной статье, и, при необходимости, растворы
стандартных образцов анализируемых веществ
(растворы сравнения), используя тот же раствори
тель, что и для приroтовления испытуемоro paCT
вора. Одинаковые объемы каждоro из растворов
наносят на пластинку и хроматоrрафируют.
Вещества, П02лощающие или флуорес
цирующие в ультрафиолетовом или видимом
свете. rотовят и наносят не менее трех paCTBO
ров сравнения, концентрации которых OXBaTЫBa
ют ожидаемое значение концентрации в испыту
емом растворе (около 80 %, 100 % и 120 % от
этой концентрации). Опрыскивают, если необхо
димо, указанным реактивом и реrистрируют OT
80
rосударственная фармакопея Республики Беларусь
ражение, пропускание или флуоресценцию на
xpoMaTorpaMMax испытуемоro раствора и !)ac
творов сравнения. По полученным данным pc.c
считывают количество вещества в испытуемом
растворе.
Вещества, содержащие радионуклиды. ro
товят и наносят испытуемый раствор, содержа
щий около 100 % ожидаемоro значения KOHцeH
трации. Измеряют радиоактивность как функцию
длины пути и записывают радиоактивность каж
доro полученноro пика в процентах от CYMMap
ной радиоактивности.
Критерии приroдности системы описаны в
статье 2.2.46. Хромат02рафические методы
разделения. Допустимые величины изменений,
которые можно вносить с хроматоrрафическую
систему для удовлетворения критериям приroд
ности системы, также указаны в данном разделе.
01/2013:20228
2.2.28. rАЗОВАЯ ХРОМАтоrРАФИЯ
rазовая хроматоrрафия (rx) представля
ет собой метод разделения, основанный на раз
личном распределении образца между двумя
несмешивающимися фазами, в котором подвиж
ной фазой является rаз (rаз носитель), а непод
вижной фазой, помещенной в колонку, TBep
дое вещество или жидкость, нанесенные на
твердый инертный носитель или равномерно по
крывающие внутренние стенки колонки.
rазовая хроматоrрафия основана на механиз
мах адсорбции, распределения или эксклюзии.
ПРИБОР
Прибор состоит из блока ввода проб (ин
жектора), хроматоrрафической колонки, поме
щенной в термостат, детектора и реrистрирую
щеro устройства (интеrрирующее устройство
или самописец). rаз носитель проходит с задан
ной скоростью или давлением через колонку, а
затем через детектор.
Определение проводят при постоянной TeM
пературе колонки или в соответствии с заданной
температурной проrраммой.
БЛОК ВВОДА ПРОБ (ИНЖЕКТОР)
Прямой ввод растворов является обычным
способом ввода проб, если в частной статье нет
друrих указаний. В90Д пробы может осущест
вляться либо непосредственно в начало колон
ки с использованием шприца или инжекторноro
клапана, либо в испаритель, который может oc
нащаться делителем потока.
Ввод паровой фазы может осуществляться
с использованием статической или динамиче
ской парофазной системы ввода.
Динамическая парофазная (продувка и уло
витель ) система ввода включает барботажную
установку, при помощи которой летучие веще
ства в растворе продуваются в абсорбирующую
колонку при невысокой температуре. Задержан
ные на колонке вещества затем десорбируются
в подвижную фазу при быстром наrревании аб
сорбирующей колонки.
Статическая парофазная система ввода
включает термостатируемую наrревающую
камеру для образцов, в которую помещены за
крытые виалы с твердыми или жидкими образ
цами на фиксированный период времени, позво
ляющий летучим компонентам образца достичь
равновесия между неrазовой и паровой фазами.
После достижения равновесия заданное количе
ство паровой фазы из виал вводится в rазовый
хроматоrраф.
НЕПОДВИЖНАЯ ФАЗА
Колонки с помещенной в них неподвижной
фазой MOryT быть описаны следующим образом:
капиллярные кварцевые колонки с покры
тыми неподвижной фазой внутренними CTeHKa
ми;
колонки, заполненные инертными части
цами, импреrнированными неподвижной фазой;
колонки, заполненные твердой неподвиж
ной фазой.
Капиллярные колонки имеют внутренний
диаметр (0) от 0,1 мм до 0.53 мм и длину от 5 м
до 60 м. Жидкость или неподвижная фаза, KOTO
рая может быть химически связана с внутренней
поверхностью, представляет собой пленку с тол
ЩИ ной слоя от 0,1 мкм до 5.0 мкм.
Набивные колонки. изroтовленные из стекла
или металла, имеют длину обычно от 1 м до 3 м
и внутренний диаметр (0) от 2 мм до 4 мм. He
подвижная фаза обычно состоит из пористых
полимеров или твердых носителей, импреrниро
ванных жидкой фазой.
Носители для анализа полярных соеди
нений на колонках, набитых малоемкой мало
полярной неподвижной фазой. должны быть
инертными для предотвращения образования
пиков с размытым тылом. Реакционная способ
ность носителей может быть снижена путем си
ланизации, предшествующей нанесению жидкой
фазы. Обычно используют обработанный кис
лотой и прокаленный диатомит. Размер частиц
может быть разным, но наиболее часто исполь
зуются частицы размером от 150 мкм до 180 мкм
и от 125 мкм до 150 мкм.
ПОДВИЖНАЯ ФАЗА
Время удерживания и характеристики пика
зависят от скорости потока rаза носителя; время
удерживания прямо пропорционально длине KO
лонки, а разрешение пропорционально KBaдpaT
ному корню из длины колонки. Для набивных
колонок скорость потока rаза носителя обычно
выражают в миллилитрах в минуту при атмосфер
2.2.28. rазовая хромат02рафия
81
ном давлении и комнатной температуре. Скорость
потока измеряется при рабочей температуре KO
лонки на выходе из детектора как rрадуирован
ным механическим устройством, так и с помощью
пенноrо измерителя. Линейная скорость rаза но
сителя через набивную колонку обратно пропор
циональна квадратному корню из внутреннеro
диаметра колонки для данноro объема потока.
Скорости потока 60 мл/мин при внутреннем диа
метре колонки 4 мм и 15 мл/мин при внутреннем
диаметре 2 мм дают одинаковые линейные CKOpO
сти и близкие времена удерживания.
В качестве rаза носителя для набивных KO
лоно!< обычно используют rелий или азот, для Ka
пиллярных азот, rелий и водород.
ДЕТЕКТОРЬ!
Обычно используется пламенно ионизаци
онные детекторы; кроме этоro, в зависимости от
цели анализа моryтприменяться следующие типы
детекторов: электронноro захвата, азотно фос
форный, масс спектрометрический, TepMO KOH
дуктометрический, ИК спектрофотометрический
с преобразованием Фурье и друrие.
МЕТОДИКА
Колонку, устройство ввода пробы и детектор
термостатируют при указанной в частной статье
температуре и скорости rаза носителя до полу
чения стабильной базовой линии. rотовят испы
туемый раствор(ы) и раствор(ы) сравнения, как
указано в частной статье. Растворы не должны
содержать твердых частиц.
Критерии для проверки приroдности xpo
матоrрафической системы описаны в статье
2.2.46. Хромат02рафические методы разделе
ния, rдe кроме этоro приводятся интервалы, в
пределах которых MOryт изменяться различные
пара метры хроматоrрафических испытаний без
принципиальноro изменения методики, для тоro,
чтобы удовлетворять критериям приroдности
хроматоrрафической системы.
Парофазная rазовая
хроматоrрафия
Парофазная rазовая хроматоrрафия явля
ется методом, наиболее подходящим для раз
деления и определения летучих соединений,
которые присутствуют в твердых или жидких
образцах. Метод основан на анализе rазовой
фазы, находящейся в равновесии с твердой или
жидкой фазой.
ПРИБОР
Прибор состоит из rазовоrо xpoMaтorpa
фа, снабженноro уr.тройством для ввода испы
TyeMoro образца, которое может быть связано
с модулем автоматическоro контроля давления
и температуры. При необходимости используют
устройство для удаления растворителей.
Испытуемый образец вводят в контейнер,
снабженный подходящей пробкой и клапанной
системой, которая реryлирует прохождение ra
за носителя. Контейнер помещают в TepMOCTa
тируемую камеру с температурой, устанавлива
емой в соответствии со свойствами испытуемоro
образца.
Пробу выдерживают при заданной темпе
ратуре в течение времени, достаточноro для
установления равновесия меЖдУ твердой или
жидкой фазой и rазовой фазой.
В контейнер вводят rаз носитель и по исте
чении указанноro времени открывают клапан,
чтобы rаз поступал в хроматоrрафическую KO
лонку, перенося с собой перешедшие в rазовую
фазу компоненты.
Вместо специально оснащенноro устрой
ства для ввода образцов хроматоrрафа воз
можно использование rазовых шприцов и обыч
ноro хроматоrpафа. Уравновешивание в таком
случае проводится в отдельной камере, а па
ровая фаза вводится в колонку с соблюдением
необходимых мер предосторожности для пре
дотвращения любых изменений в равновесной
системе.
МЕТОДИКА
Настраивают прибор для получения необхо
димоro сиrнала, используя подroтовленные об
разцысравнения.
МЕТОД ПРЯМОЙ ,РАДУИРОВКИ
В одинаковые флаконы раздельно помеща.
ют испытуемый образец и каждый из образцов
сравнения, приroтовленные, как указано в част
ной статье, избеrая контакта между устройством
для ввода проб и образцами.
Флаконы rерметично закрывают и помеща
ют в термостатируемую камеру с температу
рой и давлением, указанными в частной статье.
После установления равновесия паровую фазу
хроматоrрафируют в указанных условиях.
МЕТОД СТАНДАРТНblХ ДОБАВОК
Равные объемы испытуемоro образца поме
щают в одинаковые указанные в частной статье
контейнеры. Во все контейнеры, кроме одноro,
прибавляют указанные количества раствора
сравнения, содержащеro известную KOHцeHTpa
цию анализируемоro вещества, для получения
ряда образцов с равномерно увеличивающими
ся концентрациями этоro вещества.
Контейнеры rерметично закрывают и поме
щают в термостатируемую камеру с температу
рой и давлением, указанными в частной статье.
После установления равновесия хроматоrрафи
руют rазовую фазу в указанных условиях.
Уравнение линейной зависимости рассчи
тывают методом наименьших квадратов. По по
лученному уравнению определяют KOHцeHTpa
цию анализируемоro вещества в испытуемом
образце.
Допускается определение концентрации с
использованием rрафическоro метода. Для этоro
82
rосударственная фармакопея Республики Беларусь
по оси ординат откладывают средние значения
полученных результатов, а по оси абсцисс
концентрации стандартных добавок анализиру
емоro вещества. Экстраполируют линию, прохо
дящую через полученные точки, до пересечения
с осью абсцисс. Расстояние между этой точкой и
началом координат представляет собой KOHцeH
трацию анализируемоro вещества в испытуемом
растворе.
МЕТОД ПОСЛЕДОВАТЕЛЬНЫХ ОТБОРОВ
(мноrОКРАТНАЯ ПАРОФА3НАЯ
ЭКСТРАКЦИЯj
Применение данноro метода описано в част
ной статье.
01/2013:20229
2.2.29. ЖИДКОСТНАЯ
ХРОМАтоrрдФия
Жидкостная хроматоrрафия (ЖХ) пред
ставляет собой метод разделения, основанный
на различном распределении образца между
двумя несмешивающимися фазами. rдe под
вижной фазой является жидкость, которая про
ходит сквозь неподвижную фазу, помещенную
в колонку.
Жидкостная хроматоrрафия основана на
механизмах адсорбции, распределения, ион
ноro обмена, разделения по размерам молекул
(эксклюзии) или стереохимическоrо взаимодей
ствия.
ПРИБОР
Прибор обычно состоит из системы подачи
подвижной фазы, блока ввода пробы (инжек
тора), хроматоrрафической колонки (может ис
пользоваться устройство контроля температуры
колонки), детектора и реrистрирующеro устрой
ства (интеrрирующее устройство или самопи
сец). Подвижная фаза из одной или нескольких
емкостей протекает обычно с постоянной CKOpO
стью через колонку, а затем через детектор.
СИСТЕМА ПОДАЧИ ПОДВИЖНОЙ ФАЗbI
Система подачи подвижной фазы (насосная
установка) необходима для подцержания посто
янной скорости подачи подвижной фазы. Коле
бания давления должны быть минимизированы,
например, путем пропускания подвижной фазы,
находящейся под давлением, через устройство,
сrлаживающее пульсацию давления (демпфер).
Пр080дящая система капилляров и соединитель
ные узлы должны выдерживать давление, которое
развивается при работе насосной установки. Ha
сосные установки, использующиеся в жидкостной
хроматоrрафии, MOryт оснащаться деrазаторами.
Система, контролируемая микропроцессо
ром, должна обеслечивать точную подачу под
вижной фазы постоянноro (изократическое
элюирование) или переменноro (rрадиентное
элюирование) состава в соответствии с опреде
ленной проrpаммой. В случае rpадиентноrо элю
ирования необходима насосная установка, по
дающая растворители из нескольких емкостей, а
смешивание растворителей должно проводить
ся на стороне либо низкоro либо высокоro дaB
ления насоса(ов).
БЛОК ВВОДА ПРОБЫ (ИНЖЕКТОР)
Раствор испытуемоro образца вводится в
поток подвижной фазы, поступающей в колон
ку, при помощи блока ввода, функционирующе
ro при высоком давлении. Для ввода пробы ис
пользуют петлевые дозаторы фиксированноrо
объема или устройства с реryлируемым объе
мом, которые MOryт управляться вручную либо
посредством автосамплера. Частичное запол
нение петлевоro дозатора, осуществляющееся
вручную, приводит к снижению точности вводи
моro объема пробы.
НЕПОДВИЖНАЯ ФАЗА
В жидкостной хроматоrрафии используется
большое количество типов неподвижных фаз:
силикаrель, оксид алюминия или пори
стый rрафит. используемый в нормально фазо
вой хроматоrрафии, в которой разделение ос-
новано на разнице в адсорбции и/или массовом
распределении;
смолы или полимеры, модифицированные
кислотными или основными r'руппами, использу
ющиеся в ионообменной хроматоrрафии, в KO
торой разделение основано на конкурирующем
взаимодействии между разделяемыми ионами и
ионами подвижной фазы;
пористый силикаrель или полимеры, ис
пользующиеся в эксклюзионной xpoMaTorpa
фии, в которой разделение основано на разнице
в размерах молекул, соответствующих стериче
ской эксклюзии;
большое количество химически модифи
цированных носителей, изroтовленных из по
лимеров, силикаrеля или пористоro rрафита,
использующихся в обращенно фазовой xpOMa
тоrрафии, в которой разделение основано на
разделении молекул между подвижной и непод
вижной фазами;
специальные химически модифицирован
ные неподвижные фазы, например, производны
ми целлюлозы или амилозы, белками и пептида
ми, циклодеКСТРll'нами и т. д., использующиеся
для разделения энаl-iтиомеров (хиральная xpo
матоrрафия).
Большая часть разделений основана на
механизме распределения между химически
модифицированным силикаrелем, использу
ющимся в качестве неподвижной фазы, и по
лярными растворителями, использующимися в
2.2.29. Жидкостная хромат02рафия
83
качестве ПОДВИЖНОЙ фазы. Поверхность носи
теля, например, силанольные rруппы силикаrе
ля, реаrирует с различными силановыми pea
rентами с образованием ковалентно связанных
силильных производных, покрывающих различ
ное число активных rрупп на поверхности носи
теля. При рода модифицированной фазы явля
ется важной характеристикой, определяющей
разделяющие свойства хроматоrраф ческой
системы.
Ниже представлены наиболее часто исполь
зуемые модифицированные фазы:
Октильная
Октадецильная
Фенильная
Цианопропильная
Аминопропильная
Диольная
Si [СН2]7 СНЗ СВ
SНСН2]17 СНЗ С 1В
Si [CH2Jп C6H5 C6H
Si [СН2)З CN CN
Si [CH2kNH2 NHz
SНСН 2 ]з O CH(OH ) CH2 OH
Если производитель не дает друroй инфор
мации, находящаяся в колонках обращенно фа
зовая неподвижная фаза на основе силикаrеля
считается стабильной в подвижных фазах при
значениях рН в диапазоне от 2,0 до 8,0. Непод
вижная фаза, представляющая собой пористый
rрафит или частицы полимерноro материала,
такою как сополимер стирол дивинилбензола,
стабильна в более широком диапазоне значе
ний рН.
В некоторых случаях для анализа приме
няют нормально фазовую хроматоrрафию, ис
пользуя в качестве неподвижной фазы HeMO
дифицированный силикаrель, пористый rрафит
или силикаrель, химически модифицированный
полярными rруппами (например, цианопропиль
ными или диольными).
Размер частиц для большинства неподвиж
ных фаз, использующихся для аналитическоro
разделения, составляет от 3 мкм ДО 1 О мкм. Ча
стицы MOryT иметь сферическую или неправиль
ную форму, различную пористость и удельную
площадь поверхности. Эти параметры опреде
ляют хроматоrрафические свойства конкретных
неподвижных фаз. В обращенно фазоврй xpo
матоrрафии дополнительными определяющи
ми факторами являются природа неподвижной
фазы, степень связывания, выраженная в co
держании уrлерода, а также эндкепирование
подвижной фазы (т. е. силилирование остаточ
ных силанольных rрупп). Остаточные силаноль
ные rруппы MOryт являться причиной размытия
тыла пика, особенно для веществ основною xa
рактера.
Если нет друrих указаний в частной статье,
в аналитической хроматоrрафии используют KO
лонки, изroтовленные из нержавеющей стали, с
различными длиной и внутренним диаметром
(0). Колонки с внутренним диаметром менее
2 мм часто относят к микроколонкам. Темпера
туры подвижной фазы и колонки в течение aHa
лиза должны сохраняться постоянными. Боль
шинство анализов проводится при комнатной
температуре, однако для повышения эффек
тивности допускается наrревание колонки. Pe
комендуется не HarpeBaTb хроматоrрафическую
колонку выше 60 ОС, так как это может привести
к разрушению неподвижной фазы или измене
нию состава подвижной фазы.
ПОДВИЖНАЯ ФАЗА
В нормально фазовой хроматоrрафии ис
пользуют малополярные растворители. Для по
лучения воспроизводимых результатов нормаль
н<rфазовой хроматоrpафии необходимо cтporo
контролировать содержание воды в подвижной
фазе. В обращенно--фазовой жх используют
водные подвижные фазы, содержащие либо не
содержащие орrанический модификатор. Компо
ненты подвижной фазы обычно фильтруют для
удаления частиц с размером более 0,45 мкм.
При приrотовлении мноroкомпонентных подвиж
ных фаз смешивают отмеренные объемы (или
массы) индивидуальных компонентов. Кроме
этоro, растворители MOryт подаваться инди
видуальными насосами и с помощью дозиру
ЮIЦI1Х клапанов смешиваться в необходимых
пропорциях. Для предотвращения образования
пузырьков rаза и попадания их на детектор pac
творители обычно деrазируют при помощи бар
ботирования rелием, обработкой ультразвуком
или с использованием мембранно вакуумных
модулей, работающих в режиме «оп liпе».
Растворители для приrотовления подвиж
ных фаз обычно не содержат стабилизаторов, а
при использовании ультрафиолетовых дeTeKTO
ров должны быть прозрачными при длине волны
детектирования. Растворители и друrие компо
ненты подвижной фазы должны быть соответ
ствующей степени чистоты. Корректировку рН
при необходимости проводят для водноro KOM
понента подвижной фазы, а не для смеси. Для
предотвращения кристаллизации солей после
проведения хроматоrрафирования с использо
ванием буферных растворов проводят промыв
ку системы с использованием смеси воды и op
rаническоro модификатора (5 % (об/об».
Подвижные фазы MOryT содержать друrие
компоненты, например, контр ионы для ион
парной хроматоrрафии или хиральные модифи
каторы для хроматоrрафии, использующей ахи
ральные неподвижные фазы.
ДЕТЕКТОРЫ
Наиболее часто используемыми дeTeKтopa
ми являются спектрофотометры, работающие в
ультрафиолетовой/видимой (UVNis) областях,
а также диодно матричные детекторы. Кроме
этоro MOryT использоваться специальные дe
текторы: флуоресцентные спектрофотометры,
дифференциальные рефрактометры, электро
84
rосударственная фармакопея Республики Беларусь
химические детекторы, масс спектрометры, дe
текторы радиоактивности и друrие.
МЕТОДИКА
Колонку уравновешивают при указанном
составе и скорости подвижной фазы, а также
при комнатной температуре или температуре,
указанной в частной статье, до получения CTa
бильной базовой линии. rотовят испытуемый
раствор(ы) и раствор(ы) сравнения, как указа
но в частной статье. Растворы не должны coдep
жать твердых частиц.
Критерии для проверки приroдности xpOMa
тоrрафической системы описаны в статье 2.2.46.
Хромат02рафuческие методы разделения, rде
также при водятся интервалы, в пределах KOTO
рых MOryт изменяться различные параметры
хроматоrрафических испытаний без принципи
альноro изменения методики для тою, чтобы
удовлетворять критериям приroдности xpOMaTO
rрафической системы.
01/2013:20230
2.2.30. ЭКСКЛЮЗИОННАЯ
ХРОМАтоrРАФИЯ
Эксклюзионная хроматоrрафия представля
ет собой хроматоrрафический метод, в котором
процесс разделения молекул в растворе проис
ходит в соответствии с их размерами. В случае
использования орrанической подвижной фазы
метод называют 2ель проникающей xpOMaтo
2рафией, а в случае использования водной
подвижной фазы 2ель-фuльтрационной
хромат02рафией. Проба вводится в колонку,
заполненную rелем или пористыми частичками
наполнителя, и переносится подвижной фазой
через колонку_ Разделение в соотвеТСТВИI4 с
размерами происходит в процессе движения и
мноrоразовых обменов молекул растворенною
вещества между молекулами растворителя под
вижной фазы и тем же самым растворителем,
находящимся в порах материала, которым за
полнена колонка (неподвижная фаза). Диапазон
размеров распределенных молекул определяет
ся диапазоном размеров пор наполнителя.
Достаточно маленькие молекулы, способ
ные проникать во все поры материала, элю
ируются в полном проникающем объеме колон
ки (V/, #объем пор#). Молекулы с размерами, пре
вышающиlV'И размер всех пор материала колон
ки, миrрируют лишь сквозь пространство между
частицами наполнителя, не препятствуя им, и
элюируются В свободном объеме колонки или
объеме эксклюзии (V O ' объем пустот). Разделе
ние молекул в соответствии с размерами про
исходит между объемом эксклюзии и полным
проникающим объемом колонки; наиболее эф
фективное разделение происходит в первые две
трети данноro диапазона.
Прибор. Специфичной частью оборудования
является хроматоrрафическая колонка с различ
ными длиной и внутренним диаметром (0), за
полненная материалом, который обеспечивает
разделение молекул соответствующих размеров
в необходимом диапазоне. При необходимости
колонку термостатируют. Через колонку с посто
янной скоростью пропускают элюент. К одному
концу колонки обычно присоединяют приспосо
бление для введения пробы, например, инжек
тор с прерывателем потока, шприцевой инжек
тор с мембраной или инжектор с клапаном. К
этому же концу колонки также может быть присо
единен соответствующий насос для подачи элю
ента с контролируемой скоростью. Проба может
также наноситься непосредственно на сухую по
верхность материала колонки или, если вязкость
пробы превышает вязкость элюента, может Ha
слаиваться на поверхность материала колонки
под элюент. Друroй конец колонки обычно при
соединяют к соответствующему детектору с при
способлением, реrистрирующим и обеспечива
ющим контроль соответствующих концентраций
распределенных компонентов пробы. Обычно
используют следующее детекторы: фотометри
ческий, рефрактометрический или люминес
центный. При необходимости может быть присо
единен автоматический коллектор фракций.
В качестве наполнителя может использо
ваться или мяrкий носитель, такой как набухший
rель, или твердый пористое стекло, силика
rель или подходящий для данноro растворителя
поперечносшитый орrанический полимер. При
использовании твердых носителей обычно при
меняют принудительную подачу подвижной
фазы под давлением, что ускоряет разделение.
Подвижную фазу выбирают исходя из природы
пробы, наполнителя и метода детектирования.
Указания по обработке материала для запол
нения колонки перед выполнением разделения
и наполнения колонки представлены в COOTBeT
ствующей частной статье или инструкции изro
товителя.
Критерии для проверки приroдности xpOMa
тоrрафической системы описаны в статье 2.2.46.
Хромат02рафические методы разделения, rдe
также приводятся интервалы, в пределах KOTO
рых MorYT изменяться различные параметры
хроматоrрафических испытаний без принципи
альноro изменения методики для тою, чтобы
удовлетворять критериям приroдности xpOMaTO
rрафической системы.
ОПРЕДЕЛЕНИЕ ОТНОСИТЕльноrо
КОМПОНЕНТНОiО СОСТАВА СМЕСЕЙ
Разделение проводят, как описано в частной
статье. По возможности постоянно контролиру
ют элюирование компонентов и измеряют пло
щади соответствующих пиков. Если контроль
2.2.31. Электрофорез
85
пробы проводится по физико химическим свой
ствам, по которым все интересующие компонен
ты обладают одинаковыми откликами (напри
мер, они имеют одинаковое удельное оптическое
поrлощение), относительное содержание каж
дою компонента рассчитывают как отношение
площади пика соответствующеro компонента к
сумме площадей пиков всех компонентов. Если
отклики компонентов по детектируемому свой
ству отличаются, содержание каждою компо
нента рассчитывают с помощью rрадуировоч
ных rрафиков, полученных с использованием
соответствующих стандартных образцов для
rрсщуировки, как указано в частной статье.
ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНblХ МАСС
Эксклюзионная хроматоrрафия может быть
использована для определения молеКУllЯРНЬ
масс веществ путем сравнения с соответсrвую
щими стандартными образцами для rрС1ДУИрОВ
ки, указанными в частной статье. Для молекуляр
ных масс стандартных образцов строят rрафик
зависимости объема удерживания от лоrариф
ма молекулярных масс. rрафик, выстроенный
в пределах, оrраниченных значениями объема
эксклюзии и полноrо проникающеro объема,
обычно для данной колонки в данных экспери
ментальных условиях приближается к ПрЯflliOИ
линии. На основании этоro rрафика MorYT быть
рассчитаны молекулярные массы. ИСПОЛЬЗ0ва
ние молекулярно массовой rрадуировки позво
ляет получать достоверные результаты лишь
для частных случаев систем высокомолекуляр
ное вещество/растворитель в описанных эк(.rlе
риментальных условиях.
ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНО
МАССОВО/О РАЗДЕЛЕНИЯ ПОЛИМЕРОВ
Эксклюзионная хроматоrрафия может быть
использована для определения молеКУIlЯРНО
массовоro распределения полимеров. Однако
сравнить между собой результаты можно лишь
в одинаковых экспериментальных условиях.
Стандартные образцы, используемые для rpa
дуировки, и методики анализа описывают в co
ответствующих частных статьях.
01/2013:20231
2.2.31. ЭЛЕКТРОФОРЕЗ
ОБЩИЙ ПРИНЦИП
Под действием электрическоro поля
заряженные частицы, растворенные или
дисперrированные в растворе электро
лита, передвиrаются в направлении элек
трода противоположной полярности. В rель
электрофорезе движение частиц замедляется
взаимодействием с окружающей матрицей
rеля, который действует как молекулярное
сито. Встречные взаимодействия электри
ческой силы и молекулярною сита приводят
к дифференциации скорости передвижения
частиц в зависимости от их размеров, форм
и зарядов. В ходе электрофореза из за раз
личия физико химических свойств разные Ma
кромолекулы смеси передвиrаются с разной
скоростью и, таким образом, разделяются
на дискретные фракции. #Скорость передви
жения прямо пропорциональна суммарному
заряду частицы и обратно пропорциональна
ее размеру либо молекулярной массе.#
#Электрофоретическая подвижность яв
ляется величиной, характерной для данною
вещества. Различают абсолютную и относи
тельную электрофоретическую подвижность.
Абсолютная электрофоретическая подвиж
насть под влиянием rрадиента потенциала 1 В
на 1 см измеряется в сантиметрах в секунду.
Относительная электрофоретическая подвиж
нооь это отношение подвижности испыту
eMoro вещества к подвижности друrorо веще
ства, принятоrо за стандарт.#
Электрофоретическое разделение может
бы f ь 11роведено 8 системах без неподвижных
фаз (например, свободное разделение paCT
вора в капиллярном электрофорезе) и в CTa
билизираванных средах, таких как тонкослой
ные пластинки, пленки или rели.
#Существует два различных метода элек
трофореза: фронтальный и зональный. Фрон
тальный электрофорез проводят в свободной
незакрепленной среде, и он является един
ственным способом определения абсолютной
электрофоретической подвижности. Зональ
i-IЫЙ электрофорез про водят в закрепленной
среде, цель которой состоит в стабилизации
электрофоретических зон.#
ЭЛЕКТРОФОРЕЗ СО СВОБОДНОЙ
ИЛИ ПОДВИЖНОЙ rРАНИЦЕЙ
Этот метод обычно используется для
определения электрофоретической подвиж
ности, экспериментальной характеристики Be
ществ, непосредственно измеренной и воспро
изведенной. Этот метод обычно применяется
для веществ с высокой относительной моле
кулярной массой, которые имеют малую спо
собность к диффузии. Перед началом опреде
ления определяют месторасположение rраниц
физическими методами, например, рефракто
метрией или. кондуктометрией. После нало
жения определенноrо электрическоrо поля в
течение точно измеренноrо времени опреде
ляют новые rраницы и их относительное по
ложение. Условия испытания подбирают так,
чтобы можно было определять столько rраниц,
сколько веществ присутствует в испытуемом
образце.
86
rосударственная фармакопея Республики Беларусь
ЗОНАЛЬНЫЙ ЭЛЕКТРОФОРЕЗ
С ИСПОЛЬЗОВАНИЕМ ФАЗЫ НОСИТЕЛЯ
Для проведения этою испытания требует
ся лишь небольшое количество испытуемоro об
разца.
Природа носителя (бумаrи, rеля arapa. aцe
тата целлюлозы, крахмала, аrарозы, полиакри
ламида, смешанноro rеля) служит причиной
ряда дополнительных факторов, влияющих на
подвижность:
а) вследствие наличия каналов в фазе носи
теля кажущееся пройденное расстояние меньше
реальноro лройденноrо расстояния;
б) некоторые фаЗbl носителя не являются
электрически нейтральными. Поскольку носи
тели являются неподвижной фазой, это иноrда
может приводить к значительно большему элек
троэндоосмотическому потоку;
в) любое наrревание в результате эффек
та Джоуля может вызвать некоторое испарение
жидкости с фаЗbl носителя, что вследствие Ka
пиллярности приводит к движению раствора в
направлении от краев к центру. Ионная сила в
этом случае возрастает.
Следовательно, скорость передвижения за
висит от четырех rлавных факторов: подвижно
сти заряженной частицы, скорости потока элек
троэндоосмоса, испарения жидкости С фазы
носителя, напряженности поля. Поэтому необхо
димо проводить испытание в строro определен
ных экспериментальных условиях и, по возмож
ности, использовать стандартные вещества.
Основными составляющими прибора для
электрофореза являются следующие.
Источник постОЯНН020 тока, напряжение
KOToporo можно контролировать и, желательно,
стабилизировать.
Электрофоретическая камера. Обычно
это камера из стекла или твердой пластмас
сы, состоящая из двух отдельных резервуа
ров анодною и катодною, содержащих раст
вор электролита. В каждый резервуар камеры
поrружается электрод, например, платиновый
или rрафитовый. Они присоединяются изолиро
ванной схемой к соответствующему выходу ис
точника питания и образуют анод и катод. Во
избежание сифонирования уровень жидкости в
обоих резервуарах камеры поддерживается оди
наковым.
Электрофоретическая камера снабжена
воздухонепроницаемой крышкой, которая под
держивает атмосферу насыщенной влажности
на протяжении испытания и уменьшает испаре
ние растворителя. При снятии крышки ток отклю
чается предохранителем. Если электродвижу
щClЯ сила измерена в интервале, превышающим
10 В, желательно охладить носитель.
Приспособление установки носителя.
Электрофорез на полосках. Полоски с HO
сителем, предварительно смоченные одним и
тем же электродным раствором, поrружают в
электродный резервуар, закрепляют и фикси
руют соответствующим держателем для преду
преждения диффузии электролита. Такими дep
жателями MOryт быть roризонтальная рамка,
перевернуто V образный штатив или OДHOpOД
ная поверхность с точками контакта через опре
деленные интервалы.
r ель электрофорез. Основной частью при
способления является стеклянная пластинка
(например, предметное стекло), на поверхность
которой нанесен слой хорошо закрепленноro
rеля одинаковой толщины. Соединение rеля с
электродным раствором осуществляется разны
ми способами в зависимости от типа использу
емоro прибора. Необходимо принять меры для
предупреждения конденсации воды или Bыcыxa
ния твердоro слоя.
Измерительное устройство или дeтeK
тор.
Методика. Раствор электролита помеща
ют в электродный резервуар. Соответствующим
образом импреrнированныи носитель С электро
литным раствором помещают в камеру в соот
ветствии с инструкцией для используемоro при
бора. Устанавливают линию старта и наносят
образец. Подают электрический ток в течение
определенноro времени. После отключения тока
носитель вынимают из камеры, высушивают и
проявляют.
ЭЛЕКТРОФОРЕЗ НА КОЛОНКАХ
С ПОЛИАКРИЛАМИДНЬifv1 rЕЛЕМ
При электрофорезе на колонках с полиакри
ламидным rелем стационарной фазой являет
ся rель, приroтовленный из смеси акриламида
и N,N' метиленбисакриламида. Колонку с rелем
roтовят с использованием трубок длиной 7,5 см
и внутренним диаметром 0.5 см; во всех трубках
должен быть использован один и тот же раствор.
Прибор. Прибор состоит из двух BCTaB
ленных вертикально один над одним резер
вуаров для буферноrо раствора, изroтовлен
ных из подходящею материала, например,
поли(метилметакрилата). КаЖдЫЙ резервуар
снабжен платиновым электродом. Электроды
присоединены к источнику тока, что позволяет
проводить испытание I'IЛИ при пос'юянном токе,
или при постоянном напряжении. В основании
BepxHero резервуара имеется ряд держателей,
равноудаленных от электрода.
Методика. Обычно растворы деrазируют до
начала полимеризации и rели используют сразу
после приroтовления. rотовят предписанную
смесь компонентов, заливают в соответствую
щие закрытые снизу стеклянные колонки до оди
наковоro уровня около 1 см от верхнеro края, из
беrая попадания пузырьков воздуха в колонки.
На смесь rеля наслаивают слой воды Р, чтобы
исключить попадание воздуха, и оставляют для
полимеризации. rелеобразование обычно зани
мает около 30 минут и завершается, Korдa об
разуется четкое разделение поверхности rеля
и водноro слоя. Водный слой удаляют. Нижний
2.2.31. Электрофорез
87
резервуар наполняют указанным буферным pac
твором. Колонки открывают И вставляют в дep
жатели верхнеro резервуара так, чтобы дНО KO
лонок было поrружено в буферный раствор
нижнеro резервуара. Колонки осторожно напол
няют указанным буферным раствором. rотовят
испытуемые образцы и образцы сравнения, co
держащие индикаторный краситель, и уплотня
ют путем растворения в них, например, caxapo
зы Р. Приroтовленные образцы наслаивают на
поверхность rеля, используя для каждоro об
разца отдельную колонку. Верхний резервуар
наполняют тем же буферным раствором. Элек
Tp ДЫ подключают к источнику тока и прово
дят процесс электрофореза при указанных yc
ловиях: температуре и постоянном напряжении
или токе. Источник тока отключают, Korдa инди
каторный краситель почти переходит в нижний
резервуар. Каждую колонку сразу вынимают из
прибора и извлекают rель. Определяют местопо
ложение полос на электрофореrрамме.
ЭЛЕКТРОФОРЕЗ
С НАТРИЯ ДОДЕЦИЛСУЛЬФАТОМ
В ПОЛИАКРИЛАМИДНОМ rЕЛЕ
(ДСН ПАr, SOS PAGE)
Область применения. Электрофорез в
полиакриламидном rеле применяется для Ka
чественной идентиqpикации белков в биолоrи
ческих препаратах, контроля их чистоты и коли
чественных определений.
Цель. Аналитический rель электрофорез
метод, позволяющий идентифицировать и oцe
нить roMoreHHocTb белков в лекарственных
средствах. Метод обычно используется для
определения молекулярных масс белковых
субьединиц и субьединичноro состава очищен
ных белков.
Имеется разнообразный ассортимент KOM
мерчески доступных roTOBbIX к использованию
rелей и реактивов, которые MOryT быть исполь
зованы вместо описанных в общей статье, если
получаются эквивалентные результаты и выпол
няются условия раздела «Валидация испыта
ния», приведенноrо ниже.
СВОЙСТВА ПОЛИАКРИЛАМИДНЫХ rЕЛЕЙ
Ситовые свойства полиакриламидных rелей
обусловлены трехмерной сеткой rеля, образо
ванной поперечными сшивками соседних акри
ламидных цепей бифункциональным бисакрила
мидом. Процесс полимеризации катализируется
системой rенерирующей свободные радикалы
состоящей из аммония персульфата и TeTpaMe
тилэтилендиамина.
При увеличении концентрации акрилами
да в rеле уменьшается эффективный размер
пор. Практически эффективный размер пор rеля
определяется еro ситовыми свойствами, то есть
сопротивлением миrрации макромолекул. Из
этоro следует, что акриламид может использо
ваться только В определенных концентрациях.
При высоких концентрациях акриламида reли
чаще ломаются и являются сложными 8 обра
ботке. Поскольку размер пор rеля уменьшает
ся, уменьшается скорость миrрации белка через
rель. Реryлируя размер пор rеля, изменяя KOH
центрацию акриламида, можно оптимизиро--
вать условия разделения испытуемоro белков<r
ro продукта. Следовательно, данный конкретный
rель характеризуется содержанием акриламида
ибисакриламида.
Кроме состава rеля, важным компонентом
электрофоретической подвижности является
структура белка. В случае определения белков
электрофоретическая подвижность зависит от
значения рК заряженных rрупп и размера M<r
лекулы. На это влияют тип, концентрация и рН
буфера, температура и сила электрическоro
поля, а также природа материала носителя.
ДЕНАТУРИРУЮЩИЙ ЭЛЕКТРОФОРЕЗ
В ПОЛИАКРИЛАМИДНОМ rЕЛЕ
Данный метод является примером анали
за мономерных полипептидов с молекулярной
массой от 14 000 дальтон до 100 000 дальтон.
Возможно расширение rраниц молекулярных
масс (например, путем использования rрадиент
ных rелей, особенностей буферной системы),
но такие методики анализа не обсуждаются в
данной статье.
Денатурирующий электрофорез в полиакри
ламидном rеле с использованием натрия дoдe
цилсульфата (ДСН ПАr; SOS PAGE) является
наиболее общепринятым методом электрофо
реза, который используется для оценки качества
белковых лекарственных средств и описан ниже
в качестве примера указанноro метода. Обычно
аналитический электрофорез белков ПРОВОДЯТ в
полиакриламидных rелях в условиях, обеспечи
вающих диссоциацию белков на отдельные по
липептидные субьединицы и минимизирующих
их аrреrацию. Чаще всеro перед нанесением на
rель белки подверrают диссоциации, наrревая
их с сильным анионным детерrентом натрия
додецилсульфатом (ДСН). Денатурированные
полипептиды связываются с ДСН, превращаясь
в отрицательно заряженные частички с посто
янным отношением массы к заряду независимо
от типа белка. Поскольку количество связанно
ro ДСН почти Bcerдa пропорционально молеку
лярной массе полипептида и не зависит от ero
последовательности, ДСН полипептидные KOM
плексы миrрируют в полиакриламидном rеле со
скоростью, зависящей от размера полипептида.
Электрофоретическая подвижность по
лучР.нных детерrент полипептидных комплек
сов находится в функциональной взаимосвязи
с их молекулярными массами. Миrрация ДCH
комплексов происходит в направлении к аноду
предсказуемым образом: комплексы с низкими
молекулярными массами движутся быстрее, чем
комплексы с большими молекулярными Macca
88
rосударственная фармакопея Республики Беларусь
ми. Следовательно, молекулярная масса белка
может быть определена калибровкой ДСН ПАr,
и наличие единичной полосы в rеле является
критерием чистоты белка.
Однако модификации полипептидной цепи.
например, N или О rликозилирование при
водят к значительному влиянию на кажущую
ся среднюю молекулярную массу белка, так как
ДСН не связывается с уrлеводным компонентом
так, как с полипептидным. В этом случае посто
янное отношение заряда к молекулярной массе
не сохраняется. Средняя молекулярная масса
белков, подверrнутых поспрансляционной MO
дификации, не является истинным отображени
ем массы полипептидной цепи.
Восстанавливающие условия. Полипеп..
тидные субъединицы и трехмерная структура
белков часто поддерживаются блаrодаря при
сутствию дисульфидных связей. Целью ДCH
пдr испытаний в восстановленных условиях
является разрушение структуры белка 80CCTa
новлением дисульфидных связей. Полная дe
натурация и диссоциация белков обработкой
2 меркаптоэтанолом или дитиотреитолом (ДТТ)
приводит к развертыванию полипептидной цепи
и дальнейшему комплексообразованию с ДСН.
В этих условиях молекулярную массу попипеп
тидных субъединиц можно рассчитать по линей
ной зависимости в присутствии соответствую
щих стандартов молекулярных масс.
Невосстанавливающие условия. Для
ряда испытаний полная диссоциация белка на
субъенидичные пептиды невозможна. Из за OT
сутствия восстанавливающих areHToB. таких как
2 меркаптоэтанол или ДТТ, дисульфидные KOBa
лентные связи становятся недоступными, coxpa
няя олиroмерную Форму белка. Олиroмерные
ДСН белковые комплексы миrрируют медлен
нее, чем их ДСН полипептидные субъединицы.
Кроме TOro, невосстановленные белки не MorYT
быть целиком насыщены ДСН и, COOTBeTCTBeH
но, поэтому не MOryт связывать детерrент с по
стоянным соотношением масс. Это делает MO
лекулярно массовое определение этих молекул
с помощью ДСН ПАr неадекватным в сравнении
с анализом полностью денатурированных поли
пептидов, так как необходимо наличие CTaHдap
тов, которые имеют идентичную с испытуемым
неизвестным белком конфиryрацию для aдeK
BaTHoro сравнения. Однако проявление одной
полосы в таком rеле является критерием чисто
ты белка.
СВОЙСТВА ЭЛЕКТРОФОРЕ3А В rЕЛЕ
С ПРЕРblВИСТОЙ БУФЕРНОЙ СИСТЕМОЙ
Наиболее широко используемый электро
форетический метод исследования сложных
белковых смесей включает использование пре
рывистой буферной системы, которая состоит
из двух rелей: разрешающеrо или разделяюще
ro (нижнеro) rеля и концентрирующеro (Bepx
неro) rеля. Два rеля отличаются пористостью,
рН и ионной силой. Кроме тоro, используются
разные по подвижности ионы в rеле и электро--
дных буферах. Блаroдаря буферной прерыви
стости происходит концентрация больших объе
мов образца, приводя к лучшему разрешению
После подключения источника тока перепад Ha
лряжения приводит к тому, что белки проника
ют в концентрирующий rель. rлицинат ионы из
электродноro буфера двиrаются вслед за белка
ми в концентрирующий rель. Быстро образуется
область подвижной rраницы с высокоподвижны
ми хлорид ионами на переднем краю и относи
тельно медленными rлицинат ионами на заднем
краю. Образуется локализованный высоковольт
ный rрадиент между фронтами лидирующих
и отстающих ионов, заставляя ДСН белковые
комплексы формировать тонкие зоны (полосы) и
миrрировать между фазами хлорида и rлицина..
та. В широких rраницах, независимо от высоты
HaHeceHHoro образца, все ДСН белки KOHцeH
трируются в очень узкую область и двиrаются к
разделяющему rелю в виде четко определенной
тонкой зоны с высокой плотностью белка. Круп
нопористый концентрирующий rель не задержи
вает миrрацию БОЛЫIJинства белков и, в OCHOB
ном, служит антиконвективной средой.
На rранице раздела концентрирующеro и
разделяющеro rелей происходит резкое возрас--
тание эффекта задерживания белков вслед
ствие оrраниченноrо размера пор разделяющеro
rеля. Одновременно движение белков в разде
ляющем rеле продолжает замедляться вслед
ствие ситовых свойств матрицы. rлицинат ионы
доroняют белки, которые далее передвиrаются в
пространстве с постоянным рН. образованным
трис(rидроксиметил)аминометаном и rлицином.
Молекулярно ситовые свойства фазы при водят
к разделу ДСН полилептидных комплексов в co
ответствии с их молекулярными массами.
приrОТОВЛЕНИЕ ВЕРТИКАЛЬНblХ
ПРЕРЫВИСТО БУФЕРНЫХ
ДСН ПОЛИАКРИЛАМИДНЫХ rЕЛЕЙ
Сборка кассет, формирующих rель. Две
стеклянные пластинки (например, размером
10 см х 8 см). политетрафторэтиленовую rpe
бенку. две прокладки и силиконовые трубки (на--
при мер, диаметром 0,6 мм и длиной 35 см) моют
с мяrким моющим средством и ополаскивают
водой. Все предметы сушат бумажным полотен
цем или тканью. Прокладки и трубки смазывают
несиликоновым маслом. Прокладки укладыва--
ют вдоль двух коротких сторон стеклянных пла
стин на расстоянии 2 мм от их краев и 2 мм от
длинной стороны, являющейся дном rеля. Ис
пользуя одну прокладку в качестве основы, Ha
чинают укладывать трубку. Осторожно просо
вывают трубку к низу прокладки и протяrивают
вдоль длинной стороны стеклянной пластинки.
Придерживая трубку вдоль длинной стороны
2.2.31. Электрофорез
89
пластинки, укладывают трубку по короткой CTO
роне стеклянной пластинки, используя проклад
ку в качестве направляющей. Накрывают друrой
стеклянной пластинкой, плотно прижимая Kacce
ту руками. Устанавливают по два зажима на две
короткие стороны кассеты. Осторожно YCTaHaB
ливают четыре зажима на длинную сторону Kac
сеты rеля, формируя дно кассеты. Необходимо
удостовериться, что трубка проходит вдоль края
стеклянной пластинки и ниrде не выдавливает
ся при размещении зажимов. Кассета rOTOBa для
заливания rелем.
Приrотовление rеля. В случае прерыви
стоro буферноro ДСН полиакриламидноro rеля
рекомендуется залить разделительный rель,
дождаться еro полимеризации и затем залить
концентрирующий rель, поскольку rели отлича
ются содержанием акриламида бисакриламида,
буфером и рН.
При20товление разделитеЛЬН020 2еля.
В конической колбе roтовят соответствующий
объем раствора с необходимой концентрацией
акриламида для формирования rеля, исполь
зуя указания, приведенные в таблице 2.2.31. 1.
Компоненты смешивают в указанной после
довательности. Если необходимо, перед при
бавлением раствора аммония персульфата и
тетраметилэтилендиамина (ТЕМЭД) раствор
фильтруют под вакуумом сквозь ацетатцел
люлозную мембрану (диаметр пор 0,45 мкм);
раствор выдерживают под вакуумом, взбал
тывая фильтрационное приспособление до
окончания образования в растворе пузырь
ков. Прибавляют соответствующее количество
раствора аммония персульфата и ТЕМЭД, как
указано в таблице 2.2.31. 1, взбалтывают и
сразу заливают в пространство между двумя
пластинками кассеты. Оставляют необходи
мое место для концентрирующеro rеля (к длине
зубца rребенки прибавляют 1 см). Используя
заостренную стеклянную пипетку, осторожно
наслаивают насыщенный водой раствор изо
бутанола. rель выдерживают в вертикальном
положении при комнатной температуре для по
лимеризации.
При20товленuе разделитеЛЬН020 2еля
Таблица 2.2.31. 1
Компоненты раствора Объем компонента (мл) на 1 кассету rеля вместимостью
5мл 10 мл 15 мл 20мл 25мл 30 мл 40мл 50мл
6 % акриламид
Вода Р 2,6 5,3 7,9 10,6 13,2 15,9 21,2 26,5
Раствор акриламида (1) 1,0 2,0 3,0 4,0 5,0 6,0 8,0 10,0
1,5 М Трис (рН 8,8)(2) 1,3 2,5 3,8 5,0 6,3 7,5 10,0 12,5
Раствор 100 r/л ДСН(З) 0,05 0,1 0,15 0,2 0,25 0,3 0,4 0,5
Раствор 100 r/л АПС(4) 0,05 0,1 0,15 0,2 0,25 0,3 0,4 0,5
ТЕМЭД(5) 0,004 0,008 0,012 0,016 0,02 0,024 0,032 0,04
8 % акриламид
Вода Р 2,3 4,6 6,9 9,3 11,5 13,9 18,5 23,2
Раствор акриламида (1) 1,3 2,7 4,0 5,3 6,7 8,0 10,7 13,3
1,5 М Трис (рН 8,8)(2) 1,3 2,5 3,8 5,0 6,3 7,5 10,0 12,5
Раствор 100 r/л ДСН(З) 0,05 0,1 0,15 0,2 0,25 0,3 0,4 0,5
Раствор 100 r/л АПС(4) 0,05 0,1 0,15 0,2 0,25 0,3 0,4 0,5
ТЕМЭД(5) 0,003 0,006 0,009 0,012 0,015 0,018 0,024 0,03
1 О % акриламид
Вода Р 1,9 4,0 5,9 7,9 9,9 11,9 15,9 19,8
Раствор акриламида (1) 1,7 3,3 5,0 6,7 8,3 10,0 13,3 16,7
1,5 М Трис (рН 8,8)(2) 1,3 2,5 3,8 5,0 6,3 7,5 10,0 12,5
Раствор 100 r/л ДСН(З) 0,05 0,1 0,15 0,2 0,25 0,3 0,4 0,5
Раствор 100 r/л АПG<4) 0,05 0,1 0,15 0,2 0,25 0,3 0,4 0,5
ТЕМЭД(5) 0,002 0,004 0,006 0,008 0,01 0,012 0,016 0,02
12 % акриламид
Вода Р 1,6 3,З 4,9 6,6 8,2 9,9 13,2 16,5
Раствор акриламида (1) 2,0 4,0 6,0 8,0 10,0 12,0 16,0 20,0
1,5 М Трис (рН 8,8)(2) 1,3 2,5 3,8 5,0 6,3 7,5 10,0 12,5
Раствор 100 r/л ДСН(З) 0,05 0,1 0,15 0,2 0,25 0,3 0,4 0,5
Раствор 100 r/л АПС(4) 0,05 0,1 0,15 0,2 0,25 0,3 0,4 0,5
ТЕМЭД(5) 0,002 0,004 0,006 0,008 0,01 0,012 0,016 0,02
14 % акриламид
Вода Р 1,4 2,7 3,9 5,3 6,6 8,0 10,6 13,8
90
rосударственная фармакопея Республики Беларусь
r ! Объем компонента (мл) на 1 кассету rеля вместимостью
I Компоненты раствора rs мл 10 мл I 15 мл 20 мл 25мл 30 мл 40мл 50 мл
I
i Раствор акриламида (11 : 2,3 4.6 9 93 11,6 13,9 18,6 23,2
11,5 М Трис (рН 8,8}12) J 1,2 2,5 3,6 ! 5.0 6,3 7,5 10,0 12,5
, I
Раствор 100 r/л ДСН(3) i 0,05 0,1 o, o,2 0,25 0,3 0,4 0,5
Раствор 100 r/л АПС,-1, I 0,05 0,1 0,15 0,2 0,25 0,3 0,4 I 0,5
ТЕМЭД(5 J i 0,002 0,004 0,006 0,008 0,01 0,012 0,016 0,02
15% акрил мид
Вода Р I 1,1 2.3 3,4 4,6 5,7 6,9 9,2 11,5
Раствор а р лам да (1) I 2,5 5,0 7,5 10,0 12,5 i 15,0 I 20,0 25,0
1,5 М Трис (рН 8,8)(2) =h 1 ,3 2,5 3,8 5,0 6,3 ! 7.5 I 10,0 12,5
0:31 -
Раствор 100 r/л ДСН(З1 0,05 0,1 I 0,15 0,2 0.25 I 0,4 0,5
I Раствор 100 r/л АПС(4) I 0,05 0,1 i 0,15 0,2 0,25 i 0,3 ! 0,4 0,5
: ТЕМЭД(5) I 0,002 0,004 ! 0,006 0,008 O, 0,012 0,016 0,02
,
(1) Раствор акриламида. 30 % раствор акриламид бuсакриламида (29.'1) Р.
(2) 1,5 М Трис (рН 8,8): 1,5 М буферный раствор тpиC 2идpoxr;пpидa РН 8.8 Р.
(3) Раствор 100 r!л ДСН: раствор 100 r/л натрия додецuлсульфата Р.
(4) Раствор 100 r/л АПС: раствор 100 r!л аммония персульфата Р Блаrодаря аммония персульфату появляются свободные pa
дикалы. ускоряющие полимеризацию акриламида и бисакриламида. Поскольку раствор аммония персульфата медленно разла
rается, свежие растворы следует roтовить еженедельно.
(5) ТЕМЭД: тетраметилэтилендиамuн Р
При20товление концентрирующе20 2еля.
После завершения полимеризации (около
30 мин) сливают изобутанол и промывают Bepx
нюю поверхность rеля несколько раз водой для
удаления нанесенноro изобутанола и любых
неполимеризованных излишков акриламида.
Удаляют как можно больше жидкости с поверх
ности rеля при помощи кончика бумажной сал
фетки.
В конической колбе roтовят COOTBeTCTBY
ющий объем раствора с необходимой KOHцeH
трацией акриламида для формирования rеля,
используя указания. приведенные в таблице
2.2.31. 2. Компоненты смешивают в указанной
последовательности. Если необходимо, перед
прибавлением раствора аммония персульфа
та и ТЕМЭД раствор фильтруют под вакуумом
сквозь ацетатцеллюлозную мембрану (диаметр
пор 0,45 мкм); раствор выдерживают под Ba
куумом, встряхивая фильтрационное приспо
собление до окончания образования в paCTBO
ре пузырьков. Прибавляют соответствующее
количество раствора аммония персульфата и
ТЕМЭД. как указано в таблице 2.2.31. 2, взбал
тывают и сразу заливают в пространство между
двумя пластинками кассеты прямо на поверх
ность полимеризованноro разделительноro
rеля. В раствор концентрирующеro rеля сразу
вкладывают чистую политетрафторэтиленовую
rребенку во избежание появления воздушных
пузырьков. Прибавляют еще раствор KOHцeH
трирующеrо rеля до полноro заполнения про
странства rребня. rель выдерживают в верти
кальном положении при комнатной температуре
для полимеризации.
Сборка электрофоретическоrо прибо
ра и проведение электрофоретическоrо раз-
деления. После завершения полимеризации
(около 30 мин) осторожно удаляют политетра
фторэтиленовую rребенку. Сразу ополаскива
ют стенки водой или буферным рабочим pac
При20товление концентрuрующе20 2еля
Таблица 2.2.31. 2
Компоненты раствора Объем компонента (мл) на 1 кассету rеля вместимостью
1 мл 2 мл Змл 4мл 5мл 6 мл 8 мл 10 мл
Вода Р I 0,68 1,4 2,1 2,7 3,4 4,1 5,5 6,8
Раствор акриламида (1, 0,17 0,33 0,5 0,67 0,83 1,0 1,3 1,7
1,0 М Трис (рН 6,8)(2) i 0,13 0,25 0.38 ! 0,5 0,63 0,75 1,0 1,25
Раствор 100 r/л ДСН(З) 0,01 I 0,02 0,03 I 0,04 0,05 0,06 0,08 0,1
i I
I Раствор 100 r/л АПС(4) 0,01 I 0,02 0.03 0,04 I 0,05 0,06 0,08 0,1
I !
I 0,001 ! 0,002 I 0,003 ! 0,004 0,005 0,006 I 0,008 0,01
ТЕМЭЦ5! i
(1) Раствор акриламида: 30 % раствор акриламидlбисакрuламuда (29'1) Р.
(2) 1.0 М Трис (рН 6,81: 1 М буферный раствор mpUC 2UapCK7cr;ura рН 6.8 Р
(3) Раствор 100 r/л ДСН: раствор 100 r/л натрия додецuлсульфата Р
(4) Раствор 100 r/л АПС: раствор 100 r/л аммония персульфата Р Блаroдаря аммония персульфату появпяются свободные pa
дикалы. ускоряющие полимеризацию акриламида и бисакриламида. Поскольку раствор аммония персульфата медленно разла
rается, свежие растворы следует roтовить еженедельно.
(5) ТЕМЭД: тетраметuлэтилендuамин Р.
2.2.31. Электрофорез
91
твором для электрофореза в системе натрия
додецилсульфат полиакриламидный 2ель
(SOS PAGE) Р для удаления излишков неполи
меризованноro акриламида. При необходимости
поправляют зубцы концентрирующеro rеля тупой
rиподермичной иrлой, надетой на шприц. Снима
ют зажимы с одной короткой стороны. осторож
но вытяrивают трубку и возвращают зажимы на
место. Повторяют эти операции с друroй корот
кой стороны. Затем удаляют трубку со дна rеля.
Помещают rель в прибор для электрофореза.
Верхний и нижний резервуары наполняют Ьуфе
ром для электрофореза. Уцаляют все пузырь
ки, образующиеся да дне rеля между двумя CTe
клянными пластинками. Для этих целей лучше
всеro использовать изоrнутую иrлу, надетую на
шприц. Не следует проводить первоначальный
электрофорез до нанесения образцов. так как
это приведет к нарушению прерывистостv. бу
ферной системы. Перед нанесением образцов
осторожно ополаскивают лунки буферным pa
бочим раствором для электрофореза в cиcтe
ме натрия додецилсульфат полиакрuламид
ный 2ель (SOS PAGE) Р rотовят испытуемые
растворы и растворы сравнения в peKOMeHДO
ванном буфере для образцов и обрабатывают,
как указано в частной статье. Наносят необхо
димое количество каждоro раствора в лунки KOH
центрирующеro rеля. Начинают электрофорез
в условиях, указанных в инструкции к прибору.
Изroтовители ДСН ПАr приборов MOryT постав
лять rели разных размеров и толщины. Для по
лучения оптимальноrо разделения необходимо
подбирать время проведения электрофореза,
а также силу тока/напряжения в соответствии с
указанием изroтовителя приборов.
Необходимо удостовериться, что окрашен
ный фронт доходит до разделительноro rеля.
Korдa краситель доходит до нижнею края rеля,
электрофорез заканчивают. Кассету rеля выни
мают из прибора и отделяют стеклянные пла
стинки. Вытаскивают прокладки, отрезают и OT
деляют концентрирующий rель и немедленно
окрашивают полученный rель.
ОБНАРУЖЕНИЕ БЕЛКОВ В iЕЛЯХ
Наиболее широко применяется метод OKpa
шивания белков красителем Кумасси, позволя
ющий определить от 1 MKr до 1 О MKr белков в
одной полосе. Окрашивание серебром наи
более чувствительный метод для окрашивания
белков в rелях, при этом определяются полосы,
содержащие от 1 О Hr до 100 Hr белка.
Все стадии окрашивания rеля проводят при
комнатной температуре, осторожно встряхивая
(например, на платформе встряхивателя с KPy
roвыми движениями) в подходящей посуде. При
окрашивании rелей необходимо надевать перчат
ки, так как на rеле остаются отпечатки пальцев.
Окрашивание Кумасси. rель поrружают в
большой объем красяще20 раствора Кумасси Р,
выдерживают в течение не менее 1 ч. Затем сли
вают раствор для окрашивания.
rель обесцвечивают большим объемом
обесцвечивающе20 раствора Р. Обесцвечива
ющий раствор меняют несколько раз до четко
ю проявления белковых полос на прозрачном
фоне. Чем тщательнее обесцвечивают rель, тем
меньшее количество белков можно найти этим
методом. Обесцвечивание можно ускорить ис
пользованием нескольких rpaMMoB анионооб
мен ной смолы или маленьким кусочком пори
стою материала, смоченным обесцвечивающим
раствором Р.
ПРИМЕЧАНИЕ. Кислотно спиртовые pac
творы, используемые в указанной методике, не
полностью фиксируют белки в 2еле. Это может
привести к потерям некоторых низкомолеку
лярных белков в процессе окрашивания и обесц
вечивания тонких 2елей. Стойкая фиксация
обеспечивается выдерживанием 2еля в смеси
трихпоруксусная кислота Р метанол Р
вода Р (1:4:5, об/об/об) в течение 1 ч перед по
2ружением 2еля в красящий раствор Кумасси Р
Окрашивание серебром. rель поrружают
в большой объем фиксирующе20 раствора Р и
выдерживают в течение 1 ч. Фиксирующий paCT
вор сливают, rель заливают свежим фиксиру
ющим раствором, выдерживают в течение не
менее 1 ч или, если возможно, в течение ночи.
Фиксирующий раствор сливают и rель промы
вают большим объемом воды Р в течение 1 ч.
Затем rель выдерживают в растворе 1 % (об/об)
2лутаров020 альде2uда Р в течение 15 мин,
дважды промывают большим объемом воды Р,
каждый раз в течение 15 мин, помещают в CBe
жеприrотовленный реактив серебра нитрата Р
и выдерживают в течение 15 мин в темном
месте. Затем трижды промывают большим объе
мом воды Р, каждый раз в течение 5 мин. rель
выдерживают в растворе проявителя Р в Te
чение около 1 мин до достаточноro окрашива
ния. Проявление останавливают, помещая rель
в блокирующий раствор Р на 15 мин. Ополаски
вают rель водой Р.
ВblСУШИВАНИЕ ОКРАШЕННblХ
ДСН ПОЛИАКРИЛАМИДНblХ ТЕЛЕЙ
rели высушивают в зависимости от исполь
зованноro метода окрашивания. Кумасси окра
шенные rели после стадии обесцвечивания BЫ
держивают в растворе 100 r/л 2лицерина Р в
течение 2 ч (возможно инкубирование в течение
ночи). При окрашивании серебром на конечной
стадии ополаскивания выдерживают rель в pac
творе 20 r/л 2лицерина Р в течение 5 мин.
Два листа пористой целлюлозной пленки
поrружают в воду Р и выдерживают в тече
ние 5 10 мин. Один из листов помещают на
рамку высушивания, осторожно наносят на
нею rель, удаляют воздушные пузырьки и Ha
носят несколько миллилитров воды Р по краям
92
rосударственная фармакопея Республики Беларусь
rеля, накрывают друrим листом, удаляют воз
душные пузырьки и завершают сборку рамки
высушивания. Рамку помещают в сушилку и
оставляют при комнатной температуре дО BЫ
сыхания rеля.
ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОЙ МАССЬ!
Молекулярные массы белков определя
ют, сравнивая их подвижность с подвижностью
маркерных белков с известной молекулярной
массой. Для rрадуирования rелей используют
доступные rOToBbIe смеси равномерно окраши
ваемых маркерных белков с точно известными
молекулярными массами. Такие смеси доступ
ны для различных диапазонов молекулярных
масс. Концентрированные растворы белков с
известной молекулярной массой разводят co
ответствующим буферным раствором для об
разцов и наносят на тот же rель, что и испыту
емый образец.
Сразу после проведения электрофореза
фиксируют электрофоретический фронт Kpa
сителя бромфеноловоro синеrо. Это можно
сделать нанесением насечки на край rеля или
введением иrлы, смоченной индиroкармином,
в rель на линии фронта красителя. После OKpa
шивания измеряют пройденное расстояние
каждой белковой полосы (известной и неиз
вестной) от верха разделительноrо rеля. Pac
стояние, пройденное каждым белком, делят
на расстояние, пройденное красителем. По
лученные результаты называются относи
тельной подвижностью белков (относительно
фронта красителя) и условно обозначаются Rr
Строят rрафик зависимости лоrарифма OTHO
сительных молекулярных масс (М м.) MapKep
ных белков от условно полученных значений
Rf' Обычно rрафик имеет слеrка сиrмовидную
форму Неизвестные молекулярные массы
определяют с помощью линейной зависимо
сти или интерполяцией rрафика зависимости
19 М. м. от Rf' если значения, полученные для
неизвестных образцов, располаrаются на ли
ней ной части rрафика.
ВАЛИДАЦИЯ ИСПblТАНИЯ
Результаты испытания признаются дocтo
верными, если белки, которые являются MapKe
рами молекулярных масс, расположены вдоль
80 % длины rеля и по всей области разделения
(то есть области, включающей образец и ero
димеры или образец и сопутствующие примеси),
разделение соответствующих белковых полос
проявляет линейную зависимость лоrарифма
молекулярной массы от Rr Дополнительные Tpe
бования к валидации по отношению к Io-'спытуе--
мым растворам указываются в частной статье.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
ПРИМЕСЕЙ
Если в частной статье указывают предел
содержания примесей, в испытании необхо
димо использовать раствор сравнения COOT
ветствующеrо разведения. Например, если
предел содержания примесей составляет
5 %. необходимо использовать раствор cpaB
нения. разведенный по отношению к испы
туемому раствору в соотношении 1 :20. На
электрофореrрамме испытуемоro раствора
ни одна примесь (ни одна полоса, кроме oc
новной ) не должна быть интенсивнее OCHOB
ной полосы на электрофореrрамме раствора
сравнения.
Для валидированной методики содержа
ние примесей может определяться методом
внутренней нормализации по отношению к
основной полосе с использованием интеrри
рующеro денситометра. При валидации Me
тодики необходимо подтвердить линейность
сиrнала.
01/2013:20232
2.2.32. ПОТЕРЯ В МАССЕ
ПРИ ВЫСУШИВАНИИ
Определение потери в массе при высушива
нии проводят одним из приведенных способов и
выражают в процентах (м/м).
Методика. Указанное в частной статье KO
личество испытуемоro образца помещают во
взвешенный бюкс, предварительно высушенный
в условиях, описанных для испытуемоro веще
ства. Вещество сушат до постоянной массы или
в течение времени, указанноrо в частной статье,
одним из следующих ниже способов. Если TeM
пературный интервал не указан, то высушива
ние проводят при указанной температуре :t2 ос.
а) «в эксикаторе»: высушивание проводят
над фосфора (V) оксидом Р при атмосферном
давлении и комнатной температуре;
Ь) «в вакууме»: высушивание проводят над
фосфора (V) оксидом Р при давлении от 1,5 кПа
до 2.5 кПа и комнатной температуре;
с) «в вакууме в пределах указанноro темпе
ратурноro интервала»: высушивание проводят
над фосфора (V) оксидом Р при давлении от 1.5
кПа до 2.5 кПа и температуре, указанной в част
ной статье;
d) «В пределах указанноro TeMnepaTYPHoro
интервала»: высушивание проводят в сушиль
ном шкафу в температурном интервале, указан
ном в частной статье;
е) «в rлубоком вакууме»: высушивание про-
водят над фосфора (V) оксидом Р при давле--
нии не более 0,1 кПа и температуре. указанной
в частной статье.
Если указаны иные условия, используемая
методика полностью описывается в чаcтнoil
статье.
01/2013:20233
2.2.33. Спектрометрия ядерН020 ма2нитН020 резонанса
93
2.2.33. СПЕКТРОМЕТРИЯ ЯДЕРноrо
МАrнитноrОРЕЗОНАНСА
ВВЕДЕНИЕ
Спектрометрия ядерноro маrнитноro pe
З0нанса (ямр) это аналитический метод,
позволяющий выяснить химическую CTPYКТY
ру орrанических молекул по интерпретации
их ЯМР спектров по маrнитным моментам 'Н
или изотопов '3С, '9F, '5N, 3'р. ямр спектры
MorYT использоваться для качественноrо и KO
личественноrо определения.
В соответствующих эксперименталь
ных условиях интеrрированная интенсив
ность ЯМР сиrналов прямо пропорциональ
на числу спинов ядер молекулярной rруппы,
ответственной за сиrнал. Эти интеrралы
MorYT использоваться для количественноrо
анализа.
ОБЩИЕ ПРИНЦИПЫ
При помещении rруппировки ядер с уrло
вым моментом и маrнитным моментом в CTa
тическое маrнитное поле (ВО) ядра занимают
по отношению к оси маrнитноrо поля опре
деленные ориентации, разрешенные в KBaH
товой механике. Эти уровни энерrетич€ски
различны. Колебательное высокочастотное
маrнитное поле (В,), перпендикулярное ВО'
вызывает переходы между этими уровня
ми С поrлощением энерrии. Соrласно усло
вию резонанса 0)0 = УВ О (у rиромаrнитное
отношение, 0)0 частота Лармора), для по
лучения спектра MorYT варьироваться Mar
нитное поле ВО или частота поля В, (метод
непрерывной волны (НВ; cw. coпtiпuous
wave)). В настоящее время излучение В1 по
лучают при помощи радиочастотноrо импуль
са (метод Фурье преобразования (ФП; FТ,
Fourier trапsfоrт». KorepeHTHoe излучение,
.1спускаемое при возвращении в первона
чальное состояние, реrистрируют в виде за
тухающей кривой, называемой спадом CBO
бодной индукции (ССИ; F/D, free iпductioп
decay). Последующее Фурье преобразование
дает спектр в области частоты, предоставляя
информацию о молекулярной структуре. Дo
полнительные радиочастотные области при
меняются во время реrистрации сиrнала ССИ
дЛЯ Toro. чтобы подавить скалярные взаимо
действия (прямая связь) между ядрами (Ha
зывается «распаривание»). Для качествен
ноro и количественноrо анализа жидких или
твердых образцов можно применить OДHO и
MHoroMepHble методы.
Важную информацию о структуре веще
ства получают из следующих спектроскопиче
ски"Х параметров:
резонансная частота
определяется ..
ядер
I число резонансных
сиrналов (синrлеты,
! мулыиплеты)
I химический сдвиr l)
i (ррт)
I
I
количество OTдenb-
ных химических
rрупп ядер
определяется хим
ческая природа и
окружение CTPYКТY
ных rрупп I
относительное число I
резонансных ядер в
отдельной химиче
ской rруппе
интенсивность резо-
I нансных сиrналов
I
t
мультиплетность со-
! пряженной структуры
!
количество ядер, KO
торые скалярно свя
заны с наблюдае
мым ядром
'константа спин- I количество взаимо I
' спиновоrо взаимодей- J действующих ядер и
! ствия nj (rц) rеометрия связей I
Корреляции различных спектральных па
раметров (например, химический сдвиr и KOH
станта спин спиновоro взаимодействия, или
химические сдвиrи различных ядер в преде
лах одной молекулярной системы) MorYT быть
получены rOMO и rетероядерными ДBYMep
ными и более мноroмерными методами. Ин
формацию о времени релаксации Т 1 и ТЗ' О
ядерных эффектах Оверхаузера (ЯЭО, NOE)
и кинетике процессов, зависящих от BpeMe
ни, также можно получить с помощью COOT
ветствующих экспериментов.
ПРИБОР
ЯМР спектрометр с высокой разреша
ющей способностью состоит из следующих
частеЙ:
маrнит для создания постоянноro Mar
нитноrо поля Во;
термостатируемый датчик с держате
лем образца, предназначенный для подачи
высокочастотноrо импульса и определения
излучения, испускаемоrо образцом;
электронное устройство для создания
мощных высокочастотных импульсов, реrи
страции и преобразования сиrнала ССИ в циф
ровую форму; это устройство также поддержи
вает стабильность электроники прибора;
устройства сбора и обработки данных
(компьютер);
и может также включать:
проточную кювету для проведения жид
костной ЯМР хроматоrрафии или проточно
инъекционноro анализа;
систему для создания импульсноrо rpa
диента маrнитноrо поля ЯМР
Сильное маrнитное поле r'енерируется Ka
тушкой сверхпроводимости в сосуде Дьюара,
94
rосударственная фармакопея Республики Беларусь
заполненноrо ЖИДКИМ I елием. Датчик, как пра
вило, содержит образец в пробирке с внеш
ним диаметром 5 мм ИЛИ в riРС,ОЧНОЙ кювете
и соединен радиочастотными кабелями с
электронным блоком, настроенным на часто
ту 1Н ядер и изотопов Х ядер. Необходимы
ми являются дополнительные устройства для
настройки и реrулировки электронных KOHTY
ров; кроме Toro, часто используют TepMOCTa
тирование образцов.
Следует проверять надлежащее функ
ционирование ЯМР спектрометра Для про
верки используют соответствующие испыта
ния, включающие. как правило, измерение
ширины спектральной линии на полувысо
те опредепенных пиков при определенных
условиях, отношение сиrнал/шум (8/N) для
стандартных смесей. интенсивносrь иr,,1ПУЛЬ
са (измеренная как 900 ширины импульса) и
воспроизводимость импульса. Все изroтови
тели при боров предоставляют спецификации
и протоколы измерения указанных параме
тров для определенных комбинаций приборl
датчик, и необходимо проводить проверку co
ответствия этим спецификациями.
ямр С ФУРЬЕ ПРЕОБРАЗОВАНИЕМ
(ЯМР ФП)
Как правило, в современных спектроме
трах используется принцип Фурье преобра
зования (ФП): после возбуждения образца
высокочастотным импульсом соответствую
щей частоты (u), амплитуды (В 1 ) и ПРОДОЛЖИ-
тельности (Т р ) И последующей коро. кой паузы
(td) (время восстановления чувствительности
приемника), усиленный аналоrовый сиrнал
ССИ реrистрируют в течение определенноro
Б емени (t ac ) и переводят 8 цифровую форму
аналоrово цифровым преобразователем
(АЦП), результаты сохраняют !3 памяти спек
трометра. Выходной сиf нал перед оцифров
кой усиливается, чтобы максимально повы
сить чувствительность, не переrружая АЦП.
В случае наблюдения за Х ядрами CTaHдapT
ный эксперимент включает, в случае необхо
димости, широкополосное 'Н распаривание,
то есть возбуждение всех протонов во время
эксперимента. Чтобы увеличить отноше
ние сиrнал/шум, MHoroKpaTHbIe сиrналы ССИ
MorYT быть KorepeHTHo накоплены и сумми
рованы. При Фурье преобразовании данных
этоrо BpeMeHHoro интервала получают спектр
области частот.
ПАРАМЕТРЫ
Следующие параметры I3лияют на резуль
тат эксперимента ФП и должны контролиро
ваться.
Ширина импульса (1). Импульс возбуж
дения направлен вдоль оси х, так называемой
рамки вращения. ее продолжительность (или
«ширина», Т р ) определяет уroл пере ворота (8:
и, таким образом, интенсивность (1) резонанс-
ноro си нала:
в = у' . В 1 . Т р, (1)
Му = МО .sinB, (2)
Наблюдаемая намаrниченность М мак-
у
симальна при В = 900. ПродолжитеЛЬНОСТI:
импульса должна быть короткой, чтобы ra-
рантировать, что все сиrналы возбуждения
в спектральной ширине (SW. spectra/ width;
имеют одинаковую интенсивность. Намаrни
ченность затухает вследствие процессов ре-
лаксации.
Длительность паузы (t d )- Пауза
время между окончаниеrll подачи импуль
са и началом сбора данных, необходимое
по техническим причинам, поскольку ее дли
тельность может влиять на интенсивность
сиrнала и фазу пика. Быстро затухающие сиr
налы (дающие начало широким спектраль
ным линиям) уменьшаются в интенсивности
больше, чем медленно затухающие сиrналы
(которые дают начало узким спектральным
линиям).
Время сбора данных (taJ. Время сбора
данных (t ac ) связано со спектральной шири
ной (то есть всей наблюдаемой областью)
и количеством точек цифровоrо разреше
ния (ОР), собранных 80 время обнаружения
сиrнала.
DР
t =- - . (3)
ar 2SW
Максимальная интенсивность сиrнала
и отношение сиrнала к шуму будут достиr
нуты, если (ас 1 ,2/(лт, 2),rдe V 1l2 полная
ширина на полувысоте, но для минимизации
искажения сиrнала эта величина должна быть
больше чем 5/(::т. J.
Время повторения (t.). Спин решеточная
релаксация (Т.) влияет на время, требуемое
для спиновой системы, чтобы прийти в paB
новесие после импульса. Релаксация может
быть уменьшена при помощи специальных
реактивов. В количественном анализе ис
пользуемое время повторения должно YCTa
навливаться относительно Т 1 и В для Toro.
цтобы избежать эффектов насыщения.
Усиление приемника. Аналоrовыи
сиrнал. детектируемый датчиком, усилива
ют до оцифровки и хранения. Амплификация
сиrнала должна быть отреrулирована aBTO
матически или вручную так, чтобы сиrнал не
2.2.33. Спектрометрия ядеРН020 ма2нитН020 резонанса
95
переrрузил АЦП, что может привести к иска
жению сиrнала, и позволит случайному шуму,
произведенному при исследовании, быть пе
реведенным в цифровую форму (то есн. быть
отличным от нуля).
ОПТИМИЗАЦИЯ ПАРАМЕТРОВ
СБОРА И ОБРАБОТКИ ДАННЫХ
ПРИ КОЛИЧЕСТВЕННОМ АНАЛИЗЕ
Помимо параметров сбора данных. ин
тенсивность сиrнала зависит еще от неr!(')ль
ких парамеТР08 После сбора ДOCTaTC'-!HO
ro количества циклов сканирования сводный
сиrнал ССИ преобразовывается с ПОfJ:ОЩЬЮ
Фурье преобразования. Для надежных коли
честв€нных определений должны быть опти
мизированы следующие параметры.
Цифровое разрешение. Цифров:'е раз
решение разнесение по частоте Mew,c:y из
меренными точками. У обработанноr'J r;иr
'ала должно быть не меньше 5 измереч ъ!х
точек выше полувысоты интеrрируеМIJ!Х сиr
налов. Чтобы улучшить цифровое разре!lJе
ние, перед преобразованием к концу экспе
риментальной ССИ MorYT быть прибавпены
ДОПОJlнительные точки нулевой интеНСИRНО
сти (<<нулевое заполнение»).
Отношение сиrнал/шум. Это отнсшсрие
между интенсивностью (высотой пика) опроде
ленноro сиrнала в ЯМР спектре и интенсиsно
стью случайных колебаний, которые оfычно
измеряются в области спектра, который H co
держит сиrналов от анализируемоro вещества.
Недостаточное отношение сиrнал/шум crpa
ничивает точность интеrрирования пика и KO
личественных определений. При отношении
сиrнал/шум равным или более 150:1 CTaHдapT
ное относительное отклонение при интеrриро
вании пика может быть менее 1 %. В проrрамм
ном обеспечении современных спектроме-тров
имеются алroритмы по определению отноше
ния сиrнал/шум соответствующих пиков. При
анализе разбавленных растворов получить дo
статочно высокое отношение сиrнал/шум дo
вольно трудно, особенно при детектировании
ядер, кроме 1Н. Способы увеличения отноше
ния сиrнал/шум включают в себя:
увеличение числа суммируемых изме
рений (п), поскольку отношение сиrнал/шум
увеличивается с увеличением Jп ;
использование экспоненциальноrо YM
ножения сиrнала ССИ перед Фурье преобра
зованием: экспонен иальный коэффициент
умножения должен быть в зоне полной пико
вой ширины на полувысоте;
использование спектрометров с более
высоким маrнитным полем Во' так как отноше
ние сиrнал/шум пропорционально ag/ 2 ;
использование цифровоrо фильтра для
уменьшения шума;
использование датчиков, которые MaK
симизируют фактор заполнения;
использование криодатчиков, которые
уменьшают тепловые помехи.
Область интеrрирования. Интенсив
ность ЯМР сиrналов получают интеrрирова
нием квази аналоrовоro сиrнала или с помо
щью ступенчатоro rрафика, или более точным
интеrрированием отдельных линий и цифро
вым представлением данных. При исследова
НИ!1 жидких образцов получают ЯМР сиrналы
в виде линий лоренцевой формы. Если в
частной статье не определено иначе или
Korpa происходит перекрывание пика, для ис
пытуемоrо пика и пика сравнения должен ис
ользоваться одинаковый диапазон интеrри
рования, выраженный в виде кратноro числа
rюлной ширине на полувысоте пика.
Динамический диапазон. Динамический
диапазон аналоrово цифР080rо преобразова
теля (АЦП) определяет минимальную линию
интенсиЕ'НОСТИ. которую можно наблюдать
или количественно определить при интеrри
рова 1ИИ двух сиrналов с той же самой ши
Р!. I-IОЙ спеv.трэпьной линии. 16 битовый АЦП
ПfJЗ:ЗОЛ9ет идентифицировать сиrнал интен
сивностью 0,003 % относительно сильноro
сиrнала, rЮrJНОСТЬЮ заполняющеro дин<'!миче
ский диапазон АЦП.
ЯМР СПЕКТРОСКОПИЯ ОБРАЗЦОВ
В РАСТВОРАХ
Большинство ЯМР исследований про
водят на разведенных растворах (приблизи
тельно 1 %) испытуемоro вещества 8 COOTBeT
ствующем растворителе, к которому может
быть прибавлен в известной концентрации
подходящий стандарт для калибровки хими
ческоro сдвиrа.
Растворители. Растворитель должен
растворять испытуемое вещество без даль
нейшеro взаимодействия, если не обозна
чено иначе. Для минимизации интенсивных
сиrналов растворителей следует ИСIJОЛЬЗО
вать полностью дейтерированные раствори
тели (дейтерия оксид Р, дейтерированный
хлороформ Р, дейтерированный диметил
сульфоксид Р, дейтерированный ацетон Р,
дейтерированный метанол Рит. д.). Атомы
растворителя дают сиrналы, которые леrко
идентифицируются по их химическому сдвиrу
и MorYT использоваться для калибровки оси
химическоro сдвиrа (ВТОрИЧl-iЫЙ стандарт).
Сравнение. Спектральная характери
стика, наиболее зависящая от химическо
ro окружения атома в молекуле, называется
химическим сдвиrом, обозначается буквой ь
и измеряется в миллионных долях. Химиче
96
rосударственная фармакопея Республики Беларусь
Ядра Вода 1 Друrие
.::. .::.
растворители
1Н 0882 1,00000000 ТМС 1,00000000
1ЗС 0882 0,25144953 ТМС 0,25145020
15N NН з 0,10132912 СН з N0 2 0,10136767
19F СFзСООН не указан ССlзF 0,94094011
З1р НРО4 (85 %) 0,40480742 (СНР)з РО 0,40480864
1 Химический сдвиr зависит от значения рН.
2 DSS натрия 2.2 диметил 2 силапентан 5 сульфонат-
ский сдвиr между резонансом для ЯМР aK
тивноrо ядра Х (ь х ' б ) измеряется в мил
о разец
лионных долях (ррт) как разность между
частотой резонанса этоro ядра (\' х ' б ) И
о разец
внутреннеro сдвиrа ядра стандартноro об
разца (V x ' ), вы р аженная в rе р цах, дe
сравнение
ленная на основную рабочую частоту ЯМР
спектрометра (\lX' сравнение)' в меrаrерцах, при
данном Во:
8 (v Х, образец }/ Х. сравнение)
Х,образец . (4)
\' Х. сравнение
Принято, что стандарт точноro химиче
cKoro сдвиrа устанавливается по 1Н резонан
су тетраметилсuлана Р (ТМС) и обознача
ется как Отмс = О ррт. Формально, установив
шкалу сдвиrов 1Н относительно ТМС, можно
вычислить точную частоту любоro друrоro Х
резонанса и откалибровать масштаб ero хи
мическоro сдвиrа. Частота (вторичноro) CTaH
дартноro образца при о х = о ррm (v x ' )
сравнение
рассчитывается по 1Н частоте ТМС (V H ' тмс) и
табличному значению отношения (:::: Х ' )
сравнение
определенной для изотопа частоты относи
тельно 'Н в ТМС:
\' Х. сравнение == V н тмс':=: Х, сравнение . (5)
Стандартные образцы при <'\ = о ррт и
соответствующие значения ::::Х' cpaChEHUE приве
дены в таблице.
На практике химические сдвиrи Х cpaB
нивают, непосредственно используя COOTBeT
ствующий стандарт. В 1Н и 1ЗС ямр rлавным
образом используется внутренний CTaHдap
который при исследовании непосредственно
прибавляют к испытуемому образцу. В 15N, 19F
и З1р ЯМР часто используется внешний CTaH
дарт, коrда образец и стандарт находятся в
отдельных коаксиальных цилиндрических
проб ирках.
Стабилизация. Чтобы предотвратить
дрейф спектра во времени, выполняется CTa
билизационная процедура, названная дей
териевой стабилизацией (дейтериевый лок).
Для этоro используется сиrнал 2Н (дейтерия),
вызываемый дейтерированными растворите
лями, если в частной статье не указано иначе.
КАЧЕСТВЕННЫЙ АНАЛИЗ
Качественные ямр спектры используют
ся как испытания на подлинность, при KOTO
рых 1Н или 1ЗС спектр испытуемоrо образца
сравнивают со спектром стандартноro образ
ца или, реже, с опубликованным стандартным
спектром. Спектры стандартных и испытуемых
образцов должны быть получены с использо
ванием тех же самых методик и условий. Пики
в этих 2 спектрах или характеристичных об
ластях спектров должны совпадать по поло
жению, интенсивности и мультиплетности. В
соответствующих случаях может быть прове
дено математическое сопоставление, напри
мер. вычисление коэффициента корреляции.
При отсутствии стандартноrо образца может
использоваться рабочий стандартный обра
зец, идентичность котороro была подтверж
дена альтернативными методами или путем
подтверждения Toro. что ямр спектр полно
стью совпадает с заявленной структурой Ma
териала.
КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
Интенсивность сиrнала в основном ис
пытании ЯМР представляет собой интеrри
рованную площадь под кривой измеренноro
сиrнала. Высота сиrнала может быть мерой
интенсивности только при одинаковом значе
нии полной ширины на полувысоте и одинако
вой мультиплетности двух сиrналов. В усло
виях практически полной релаксации между
реrистрируемыми сиrналами интенсивность
сиrнала (/J является истинной мерой количе
ства ядер (N A ), ответственных за COOTBeTCTBY
ющий сиrнал:
'A=Ks.N A .(6)
Константа Ks включает фундаменталь
ные постоянные, свойства образца и пара
метры приемника и может не учитываться в
2.2.33. Спектрометрия ядеРН020 ма2нитН020 резонанса
97
случаях, коrда интенсивности сиrналов сопо
ставимы и можно получить прямую зависи
мость между количеством ядер в двух cpaB
ниваемых структурных rруппах А и В:
!А == N А . (7)
'в N B
Количества (N,) ядер, принадлежащих
различным структурным rруппам одной моле
кулы, представляют собой небольшие целые
числа. Измеренные значения окруrляют до
целых чисел. Однако правильное функцио
нирование блока сбора и обработки данных
леrко проверяется путем сравнения точной
интенсивности в пределах спектра любоro
подходящеro орrаническоro соединения из
вестной структуры.
Помимо тоro, что интенсивность сиrна
лов, полученных от каждоro компонента в
смеси, связана друr с друrом небольшими
целыми числами, относительные молярные
количества этих компонентов MorYT быть из
мерены сравнением нормализованных интен
сивностей резонансов различных компонен
тов. Молярное отношение двух компонентов
смеси вычисляют с помощью следующеro
уравнения:
ПА == . N B . (8)
П в 'в N A
Результаты определения являются ДOCTO
верными только в случаях, коrда структуры
молекул, для которых определены ,д и I B , из
вестны (или, по крайней мере, известны зна
чения N для проверенных rрупп). Определе
ния проводя используя метод BHYTpeHHero
стандарта или метод нормализации пиков.
Метод BHYTpeHHero стандарта. Масса
(тА) аналита (А) может быть определена,
если к раствору в качестве стандарта интен
сивности прибавлена известная масса (тв)
вещества (В) с известным процентным coдep
жанием (Р В )' Уравнение (8) может быть пре
образовано в уравнение (9):
тА ==!А. N B . М А .т в . Р В ,(9)
/в N A Мв 100
rдe М ; молекулярные массы.
Необходимо тщательно выбирать CTaH
дарт интенсивности; он должен быть полно
стью растворим в растворителе, использу
емом для иr.пытуемоrо вещества, должен
производить лишь небольшое количество
сиrналов, и у «мониторной rруппы» должен
быть сиrнал в пустой спектральной области.
Для этих целей рекомендуется выбирать coe
динение с высокой чистотой и с относительно
высокой молекулярной массой.
11 __ J7IZ
Метод внутренней нормализации. OT
носительное содержание компонентов в
смеси, степень замещения в структурно из
мененном полимере или количество при меси
можно определить с помощью сравнения OT
носительных интенсивностей полученных pe
зонансов.
Методика должна быть валидирована в
отношении отсутствия наложения COOTBeT
ствующих сиrналов. Коrда в образце при
сутствуют вещества с плохо определенной
структурой или молекулярной массой (Ha
при мер, эмульrатор), прибавление извест
Horo количества этоro материала в пробирку
для ЯМР позволяет построить калибровоч
ную кривую.
МЕТОД
Подrотовка испытуемоrо образца. Ис
пытуемый образец растворяют в раствори
теле, к которому может быть прибавлен co
ответствующий стандартный образец для
калибровки химическоro сдвиrа, как указано в
частной статье. Для количественноro анализа
растворы не должны содержать HepaCTBopeH
ных частиц. При некоторых количественных
определениях может потребоваться прибав
ление внутреннеro стандарта для сравнения
интеrрированных резонансов испытуемоro
и стандартноro образцов. Соответствующие
стандартные образцы и их концентрации YKa
заны в частных статьях. В друrих количе
ственных исследованиях результат получают
путем сравнения относительной интенсивно
сти двух или всех резонансов, полученных
при исследовании образца. После помеще
ния образца в пробирку и укупорки, образец
вводят в маrнит ЯМР установки устанавлива
ют параметры испытания и проводят измере
ния. Основные параметры испытания приво
ДЯТСЯ в частных статьях.
Процедура измерения. Уравновешива
ют образец в датчике и реrулируют прибор
для получения наиболее оптимальных усло
вий резонанса и максимальноro отношения
сиrнал/шум путем подбора настройки датчи
ка для данноro объема образца для обеспе
чения максимальной однородности маrнитно
ro поля во всем объеме испытуемоro образца
(процедура называется «шиммирование»).
Параметры настройки записывают или co
храняют в компьютере. Условия испытания
MorYT включать выполнение мноroкратных по
следоватеЛЬНlJстей циклов «импульс сбор
данных пауза», и отдельные ССИ сиrналы
суммируются в памяти компьютера, с ycpeд
нением уровня шума. Коrда соответствующий
уровень отношения сиrнал/шум достиrнут,
ССИ сохраняют и спектр области частот по
лучают путем Фурье преобразования сумми
рованных ССИ сиrналов.
98
rосударственная фармакопея Республики Беларусь
ЯМР СПЕКТРОМЕТРИЯ ОБРАЗЦОВ
В ТВЕРДОМ СОСТОЯНИИ
Анализ образцов в твердом состоян и
может быть выполнен с помощью специально
оборудованных ЯМР спектрометров. Опреде
ленные технические операции обеспечивают
выделение отдельных линий для определен
ных положений атомов, а также позволяют ис
пользовать ЯМР для оценки неорrанических
материалов.
Одна из таких операций быстрое Bpa
щение (4 30 кrц) порошкообразноro образ
ца в роторе (внешний диаметр около 4 мм)
находящеrося под уrлом 54,70 (<<маrический
уroл») к оси маrнитноrо поля Во. Этот способ
называется вращением под маrическим
уrлом. Второй эффективный способ сило
вое распаривание, и третий способ пере
нос поляризации от леrковозбудимых ядер к
менее поляризуемым ядрам, то есть KpOCC
поляризация. Комбинация этих методов по
зволяет получить спектры с высоким разре
шением, содержащие большое количество
информации о химических и структурных xa
рактеристиках твердых стекловидных тел, Be
ществ в аморфном состоянии, кристаллов Ke
рамической, полимерной или минеральной
природы.
Если ЯМР используют для оценки образ
ца в твердом состоянии, полное описание
процедуры приводят в частной статье.
01/2013:20234
2.2.34. ТЕРМИЧЕСКИЙ АНАЛИЗ
Термический анализ объединяет rруппу
методов, при помощи которых определяется
зависимость различных физических свойств
веществ от температуры. Обычно в большин
стве используемых методов определяют из
менение энерrии или массы испытуемоro об
разца.
#Характерной особенностью термических
методов анализа является их универсаль
ность. Инструментальные методы термиче
скоro анализа дают информацию о кристал
лической структуре, температуре кипения,
способности к сублимации, деrидратации,
твердофазном взаимодействии. Эта инфор
мация может быть использована для иденти
фикациl.1, определения чистоты, влажности и
стабильности веществ, количественноro опре
деления термонестойких веществ, выявления
присутствия полиморфных форм, для xapaK
теристики совместимости субстанций и вспо
моrательных веществ в лекарственных cpeд
ствах, выбора упаковки и контроля качества.
Различают термоrравиметрию, TepMOMe
трическое титрование, дифференциальный
термический анализ (ДТА) , дифференциаль
ную сканирующую калориметрию (ДСК), дe
риватоrрафию, термомикроскопию.
Термические методы анализа. как прави
ло, не требуют больших количеств веществ,
непродолжительны по времени выполнения.
В частности, определение потери в массе
при высушивании методом тepM02paвиMe
трии включают в частные статьи на дороrие
субстанции. В частных статьях следует YKa
зывать пара метры, необходимые для KOp
ректноro выполнения методики, например,
скорость и температуру наrревания, инерт
ный rаз и скорость ero потока.#
ТЕРмоrРАВИМЕТРИЯ
Термоrравиметрия представляет собой
Me TOД, при помощи котороro реrистрируют
изменение массы испытуемоrо образца в за
висимости от проrраммированноrо измене
ния температуры.
Прибор. Основными составляющими ча
стями прибора являются: приспособление
для наrревания или охлаждения вещества в
соответствии с заданной температурной про
rраммой. камера для образца с контролиру
емой атмосферой, электронные весы и pe
rистрирующее устройство. К прибору может
быть присоединено приспособление для aHa
лиза летучих веществ.
Проверка температурной шкалы. Про
верку температурной шкалы проводят в COOT
ветствии с инструкцией изroтовителя.
rрадуировка электронных весов. Под
ходящее количество подходящеro сертифи
цированноrо стандартноro образца (напри
мер, ФСО кальция оксалата МОН02идрата)
помещают в держатель и реrистрируют еro
массу. Начинают наrревание образца по за
проrраммированному изменению темпера
туры соответствии инструкции изrотовителя.
Термоrравиметрическую кривую реrистриру
ют в виде rрафика: показания температуры
или времени откладываются по оси абсцисс
по возрастанию значений слева направо; по
казания массы откладываются по оси орди
нат по возрастанию значений снизу вверх.
При температуре около 230 ос повышение
температуры останавливают. На rрафике из
меряют расстояние между начальным и KO
нечным плато масса температура или плато
масса время, что соответствует изменению
массы образца. Полученную величину потери
в массе сертифицированноro испытуемоro
образца сопоставляют с величиной, указан
ной на маркировке.
2.2.34. Термический анализ
99
Методика. Изменение массы испытуемо
ro образца определяют в условиях, указанных
в частной статье. Потерю в массе испытуемо
ro образца определяют, измеряя расстояние
между начальным и конечным плато на rpa
виметрической кривой, и выражают в процен
тах (д.м/м).
При реrулярном использовании прибора
периодически проводят rрадуировку и про
верку температурной шкалы. В противном
случае эти операции проводят перед каждым
использованием.
Так как характеристики атмосферы при
проведении испытания очень важны, для каж
доro измерения отмечают следующие пара
метры: давление или скорость потока, состав
rаза.
ДИФФЕРЕНЦИАЛЬНАЯ СКАНИРУЮЩАЯ
КАЛОРИМЕТРИЯ
Дифференциальная сканирующая кало
риметрия (ДСК, OSC) является методом, KOTO
рый используется для демонстрации энерrети
ческих явлений, происходящих при наrревании
(или охлаждении) вещества (или смеси Be
ществ), и для определения изменений в энталь
пии и удельной теплоемкости, а так же темпе
ратур, при которых эти изменения происходят.
Этот метод используется для нахождения
различий в количестве теплоты, выделенной
или поrлощенной испытуемым образцом, по
сравнению с ячейкой сравнения в зависимо
сти от температуры. Существует два типа при
боров дЛЯ ДСК: приборы, использующие KOM
пенсацию энерrии для удерживания нулевой
разницы температуры между испытуемым об
разцом и образцом сравнения; приборы, под
держивающие постоянную скорость HarpeBa и
определяющие температурный дифференци
ал как разность в тепловом потоке между ис
пытуемым образцом и образцом сравнения.
Прибор. Прибор дЛЯ ДСК с компенсаци
ей энерrии состоит из печи, имеющей держа '
тель образцов с испытуемой ячейкой и ячей
кой сравнения. Прибор дЛЯ ДСК с тепловым
потоком имеет единственную ячейку с держа
телем образца для испытуемоro тиrля и тиrля
сравнения.
К прибору присоединяют устройство для
проrраммирования температуры, темпера
турный детектор (детекторы) и реrистрирую
щую систему, которая может быть подсоеди
нена к компьютеру. Измерения про водятся в
контролируемой атмосфере.
Калибровка прибора. Прибор калибру
ется на изменения температуры и энталь
пии с использованием индия высокой чисто
ты или любоro друroro сертифицированноro
стандартноrо образца в соответствии с ин
струкцией изroтовителя. Использование двух
металлов, например, индия и цинка, может
применяться для проверки линейности.
Проведение испытания. В подходящий
тиrель взвешивают подходящее количество
испытуемоro образца и помещают в держа
тель для образца. Устанавливают температу
ру начала и конца испытания и скорость Ha
rpeBa, указанные в частной статье.
Начинают испытание и реrистрируют
кривую дифференциальноrо термическоro
анализа, откладывая значения температуры
или времени по оси абсцисс (по возрастанию
значений слева направо) и изменения энер
rии по оси ординат (уточняют, процесс эндо
термичный или экзотермичный).
Температура, при которой происходит
явление (начальная температура), COOTBeT
ствует точке пересечения (А) продолжения
базовой линии и касательной к точке caMOro
большоro наклона (точке переrиба) кривой
(см. рисунок 2.2.34. 1). Конечная температура
тепловоro явления соответствует пику на
кривой.
А
ЭНДQтерма
... .__.__.__.__. _._____.__.__. .! ________._ .______ ._ . _._! пература
Рисунок 2.2.34. 1. ТеРМ02рамма
Значение энтальпии явления пропорцио
нально значению площади под кривой, orpa
ниченной базовой линей; коэффициент про
порциональности находят путем измерения
теплоты плавления известноro вещества (Ha
пример, индия) при тех же условиях проведе
ния испытания.
Каждая TepMorpaMMa может СОПрОВО
ждаться следующими данными: условия ис
пытания, запись последней калибровки,
размер и идентификация (включая термиче
скую историю) образца, контейнер, aTMOC
фера (состав, скорость потока, давление),
направление и скорость температурных из
мерений, чувствительность прибора и реrи
стрирующеrо устройства.
Область применения
Фазовые превращения. Определение
температуры, изменения теплоемкости и эн
тальпии при фазовых превращениях, проис
ходящих с веществом, как функции темпе
ратуры.
100
rосударственная фармакопея Республики Беларусь
переход твердое вещество твердое BeUleCTBf): аллотропия полиморфизм
прозрачность стекла
десольватация
аморфность кристалличность
exoд твердое вещество жидкость: плавление
переход твердое вещество rаз: , сублимация
переход жидкость твердое вещество: ! замерзание I
перекристаллизация
переход жидкость rаз: испарение
1 ос
Температура
..
.............._....._ . O... ;;- r
"'-- :' I I
"'-- .
.0.... ... ........ : I I
.. ......... ;
..... .... "': ,
....0.. .... ...... '\, ! I I
........ ......, \ I I
..... \ ; \ I
\ ,[ \ I
..... \; \ I I
.... I
99,10 % \. " \ I I
.. ........: \ \ I
99 3 5 0/ \ J \ I
, /О .. ".... \
99.50 % \./
............... .
дН
о
"tJ
С
Ш
99,80 %
Рисунок 2.2.34. 2. Термические диа2раммы вещества в зависимости от чистоты
Изменения химичеСК020 состава. Измеряя
теплоту и температуру реакции при данных yc
ловиях. можно определить, например, кинетику
разложения или десольватации.
Использование для фазовых диа2рамм. Соз
дание фазовых диаrрамм для твердых смесей.
Создание фазовой диаrраммы может быть
важным шаroм при предварительной постановке
и оптимизации процесса лиофилизации.
Определение чистоты. Измерение тепло
ты плавления и температуры плавления с по
мощью ДСК делает возможным определение
содержания примесей в веществе по одной
диаrрамме с использованием нескольких мил
лиrрамм образца и без необходимости прове
дения повторных точных измерений истинной
температуры.
Теоретически плавление полностью чисто
ro кристаллическоro вещества при постоянном
давлении характеризуется теплотой плавления
Ь.Н, с бесконечно узким диапазоном, COOTBeT
ствующим температуре плавления То. Расшире
ние этоrо диапазона является чувствительным
индикатором присутствия примесей. Таким об
разом, при испытании образцов одноro и тоro же
вещества с содержанием примесей, отличаю
щимся на насколько десятых процента, получа
ют визуально отличающиеся друr от друrа Tep
мические диаrраммы (см. рисунок 2.2.З4. 2).
Определение молярной чистоты с помощью
ДСК основано на использовании математиче
скоro приближения интеrральной формы ypaB
нения Вант rоффа применительно к KOHцeH
трациям (не к активностям) в бинарной системе
[ln(1 x2)== X2 и Т.То ==To2J:
RТo 2
т == т. !L.. х , (1)
о ""Н 2
,
rдe:
Х 2 молярная доля примеси, то есть число
молекул примеси, разделенное на общее число
молекул в жидкой фазе (или в расплавленной
фазе) при температуре Т (выраженной в Кель
винах);
ТО температура плавления химически чи
стоro вещества, в кельвинах;
R универсальная rазовая постоянная для
идеальных rазов, в джоуль'кельвин' 1 ,моль'\
""Н, молярная теплоемкость плавления
вещества, в джоулях.
Таким образом, определение чистоты
с помощью ДСК оrраничено обнаружением
примесей, образующих эвтектические смеси
с основным веществом и присутствующих
в испытуемом образце с молярной долей
менее 2 %.
2.2.35. Осмоляльность
101
Данный метод не применяют для:
аморфных веществ;
сольватов или полиморфных соединений,
которые нестабильны в пределах используемых
при испытании температур;
примесей, образующих твердые растворы
с основным веществом;
примесей, которые нерастворимы в жидкой
фазе или в расплаве основноro вещества.
Во время наrревания испытуемоro вещества
при меси полностью плавятся при температуре
эвтектической смеси. Выше этой температуры
твердая фаза содержит только чистое вещество.
Так как температура постепенно увеличивается
от температуры эвтектической смеси до темпе
ратуры плавления чистоro вещества, молярная
доля примеси в жидкой фазе постоянно падает,
так как количество расплавленноro чистоro Be
щества постоянно увеличивается. Для всех TeM
ператур выше эвтектической точки:
Х 2 = . х;, (2)
F
rдe:
F расплавленная доля анализируемоrо
образца;
Х; молярная доля примеси в анализируе
мом образце.
Если образец расплавлен полностью, то
F = 1 и Х 2 = Х;.
При объединении уравнений (1) и (2) полу
чается следующее выражение:
т = т. x; RT o2 . .
О дН F
,
Значение теплоты плавления получают
путем интеrрирования пика плавления.
Температура плавления То чистоro веще
ства экстраполируется из rрафика зависимости
lIF от температуры, выраженной в кельвинах.
Наклон (а) rрафика, при необходимости полу
ченноrо после линеаризации, соответствующий
х.
RTo2 позволяет определить значение х;.
дН,
Доля х;, умноженная на 100, дает молярную
долю всех эвтектических примесей в процентах.
ТЕРМОМИКРОСКОПИЯ
Фазовые изменения MOryT быть визуализи
рованы с помощью термомикроскопии метода,
который позволяет исследовать образец, KOТO
рый подверrается температурному воздейств ю
по заданной проrрамме, под микроскопом в по
ляризованном свете.
Наблюдения, сделанные с помощью Tep
момикроскопии, позволяют точно идентифи
амювать природу явления, обнаруженноro с
81С1ЮЛьзованием термоrравиметрии и диффе
ренциальной сканирующей калориметрии.
и _ 1712
Прибор. Прибор состоит из микроскопа,
снабженноro поляризатором света, наrреватель
ной пластинки, проrраммноro реryлятора TeM
пературы и скорости HarpeBa и/или остывания
и реrистрирующеro устройства для температур
фазовых переходов. Moryт дополнительно ис
пользоваться видеокамера и видеомаrнитофон.
01/2013:20235
2.2.35. ОСМОЛЯЛЬНОСТЬ
Осмоляльность это показатель, позволя
ющий оценить суммарный вклад различных pac
творенных веществ в осмотическое давление
раствора.
Приближенный расчет осмоляльности qm
водноro раствора проводят по формуле:
т ==итФ,
rде:
v суммарное число ионов, образующих
ся из одной молекулы растворенноro вещества
в результате диссоциации; в случае если pac
творенное вещество не диссоциирует на ионы,
и = 1;
т моляльность раствора, т. е. число
молей растворенноro вещества на килоrрамм
растворителя;
Ф моляльный осмотический коэффи
циент, учитывающий взаимодействие между
ионами противоположноro знака в растворе и
зависящий от величины т. По мере усложнения
состава раствора усложняется и определение
величины Ф.
Единицей осмоляльности является осмоль
на килоrрамм (осмоль/кr), но на практике обычно
используется единица миллиосмоль на кило
rpaMM (мосмоль/кr).
Если нет друrих указаний в частной статье,
осмоляльность определяют по понижению
температуры замерзания раствора. Зависи
мость между осмоляльностью и понижением
температуры замерзания !'J. Т выражают COOT
ношением:
дТ
т = .1 000 мосмоль/кr.
1,86
Прибор. Составными частями прибора (oc
мометра) являются:
приспособление для охлаждения сосуда с
измерительной кюветой;
система для измерения температуры, co
стоящая из чувствительноro к температуре co
противления (термистора) с соответствующим
устройством для измерения тока или разно
102
/осударственная фармакопея Республики Беларусь
Таблица 2.2.35. 1
Масса натрия
хлорида Р, в
rpaMMax на кило
rpaMM воды Р
3,087
6,260
9,463
12,684
15,916
19,147
22,380
Стандартные раСl., оры ля 2радуировки осмометра
Фактическая Те,)реР1' еская Моляльный
осмоляльность осмоляльность осмотический
(мосмоль/кr) i идеальноrо paCT коэффициент
вора (мосмоль/кr)
105,67 I 0,9463 0,186
! 2',4,20 F ,9 _ , 0.372
? З, З 0,9264 J. 0,558
4Э4.о7 0,9264 () 558
! 5; '<
'T ' t:t:;"
; БJ :,
765.
I
t
100
200
300
400
500
600
700
сти потенциалов; измерительное устройство
может быть rрадуировано в rрадусах пониже
ния температуры или непосредственно в едй
ницах осмоляльности;
как правило, приспособление для переме
шивания образца.
Методика. rотовят стандартные paCTBO
ры в соответствии с таблицей 2.2.35. 1. YCTa
навливают нулевое значение на шкале прибо
ра, используя воду Р. Проводят rрадуировку
прибора, используя стандартные растворы:
помещают от 50 мкл ДО 250 мкл CTaHдapTHO
ro раствора в измерительную кювету и начина
ют охлаждение системы. Чтобы предотвратить
переохлаждение, измерительное устройство,
как правило, проrраммируют на работу при
температурах более низ"их, чем ожидаt';МОt;
криоскопическое понижение температуры.
Подходящее устройство указывает на AOCl и
жение равновесия. Перед каждым измерением
ячейку ополаскивают соответствующим CTaH
дартным раствором.
Те же операции проводят с испытуемым
раствором. При этом перед каждым измерением
кювету ополаскивают испытуемым раствором.
Результаты либо непосредственно определяют
по шкале прибора, либо рассчитывают по изме
ренному понижению температуры замерзания.
Результаты считают достоверными, если полу
ченное значение осмоляльности испытуемою
раствора не выходит за пределы значений OCMO
ляльности двух стандартных растворов, исполь
зованных для rрадуировки.
#Наряду с понятием «осмоляльность» В
практике используется понятие «осмоляр
ность». Данные показатели близки и отличают
ся друr от друrа только различным способом
выражения концентрации растворов MO
ляльной И молярной:
осмоляльность количество осмоле.::! на
1 килоrрамм раствора;
осмолярность количество осмолей на
1 литр раствора. Единицей осмолярности явля
ется осмоль на литр (осмоль/л), но на практике
обычно используется единица миллиосмоль на
литр (мосмоль/л).
Криоскопическое
I понижение
I температуры
05 О 9180 I . О:9ЗО
24 0,9157 1, 16 I
.
86 0,9140 I 1,302 I
Для идеальных растворов масса осмоля в
rpaMMax представляет собой отношение rpaMM
молекулярной массы вещества к числу частиц
или ионов, образующихся при ею растворении.
Для разбавленных растворов, близких к иде
альным, осмоляльность и осмолярность MorYT
быть рассчитаны теоретически.
Осмолярность идеальных растворов может
быть рассчитана по формуле:
Осмолярность !<.онцентрацuя ве щ т. .!<.олu ч ест 'lCJ с;тULi ,
молекулярная масса
rдe:
конценrпрация вeLЦecrпвa количество
растворенною вещества на литр раствора, в
rpaMMax;
количество частиц число частиц или
ионов, образующихся ПрИ растворении одной
молекулы вещества.
При повышении концентрации раствора
взаимодействие между частицами вещества
возрастает, и фактическая осмолярность по
нижается по сравнению с осмолярностью иде
альноrо раствора. Теоретический расчет oc
молярности растворов веществ с большой
молекулярной массой (например, белковых
rидролизатов) и высококонцентрированных
растворов невозможен. В таких случаях опре
деля ют осмоляльность экспериментальным
путем по понижению температуры замерзания
раствора или по понижению давления пара
над раствором. Понижение температуры за
мерзания на 1,86 ос и понижение давления
пара на 0,3 мм рт. ст. при температуре 25 ос
соответствует 1 осмолю на килоrрамм воды.
Растворы, равные по осмоляльности (OCMO
лярности) 0,9 % раствору натрия хлорида, назы
вают изотоничными.
Наряду с прибором, описанным выше, для
определения осмоляльности MOryT использо
ваться также осмометры, основанные на изме
рении давления пара над раствором. Они Tpe
буют малоro объема вводимой пробы (около
5 мкл), при этом точность и правильность опре
деления сравнима с величинами, получаемыми
на приборах, основанных на измерении темпе
ратуры замерзания.
2.2.36. Потенциометрическое определение концентрации ионов
103
в осмометрах, основанных на измерении
понижения температуры замерзания, объем
вводимой пробы образца можно варьировать.
Значение осмоляльности (осмолярности)
необходимо указывать на этикетках растворов
для инфузий.#
01/2013:20236
2.2.36. ПОТЕНЦИОМЕТРИЧЕСКОЕ
ОПРЕДЕЛЕНИЕ
КОНЦЕНТРАЦИИ ИОНОВ
С ИСПОЛЬЗОВАНИЕМ
ИОНОСЕЛЕКТИВНЫХ
ЭЛЕКТРОДОВ
Теоретически потенциал Е ионоселективно
ro электрода изменяется линейно от лоrариф
ма активности а; данноro иона, что выражается
уравнением Нернста:
RT
Е -=Ео +2.303 lga;'
z;F
rде:
Ео стандартный электродный потенциал
используемоrо электрода;
R универсальная rазовая постоянная;
т абсолютная температура;
F число Фарадея;
z, величина заряда иона с обозначением
ero знака.
При постоянной ионной силе выполняется
уравнение:
k
Е -= Е О + Ig (С.,
z. '
,
rде:
С, молярная концентрация иона;
( коэффициент активности (а ; = ('С;);
k RT .
F
Если: Ео + Ig (-= E и S == , rдe
Zj z.
S наклон rрадуировочной кривой электрода
(наклон электродной функции), в таком случае
выполняется уравнение:
Е = E +Slg С, и для lgC; -= рС;: Е == E SpC;.
Потенциометрическое определени KOHцeH
трации иона проводят путем измерения разницы
noтeнциaпoв между двумя подходящими элек
И, поrpуженными в испытуемый раствор;
ЫЙ электрод является селективным
по orнowe нию к анализируемому иону, друroй
_1ЯeТCЯ электродом сравнения.
Прибор. Используют вольтметр, позволяю
щий выполнять измерения с точностью О, 1 мил
ливольта, с входным сопротивлением, в 100 раз
превышающим сопротивление используемых
электродов.
Ионоселективные электроды преимуще
ственно электроды с кристаллической или некри
сталлической мембраной или с твердой основой
(например, стеклянные электроды); или элек
троды с заряженными (положительно или отри
цательно) или с незаряженными подвижными
носителями; или сенсибилизированные электро
ды (ферментные электроды, rаз индикаторные
электроды). Электродом сравнения обычно яв
ляется хлорсеребряный или каломельный элек
трод с подходящими соединяющими жидкостя
ми, исключающими их перемешивание.
Методика. Измерения проводят при посто
янной температуре с точностью ::1:0,5 ОС, учиты
вая изменение наклона электродной <рункции
электрода в зависимости от температуры (см.
таблицу 2.2.36. 1 ).
Таблица 2.2.36. 1
3начения k при различных температурах
Температура, ос k
20 0,0582
25 0,0592
30 0,0602
Реryлируют ионную силу и, если необходи
мо, рН испытуемоro раствора, используя буфер
ные растворы, указанные в частной статье, и
уравновешивают электрод, поrружая еro в пере
мешиваемый с постоянной скоростью испытуе
мый раствор, до получения постоянноro значе
ния потенциала.
Если электродная система используется
часто, то реryлярно проверяют воспроизводи
мость и стабильность сиrнала, линейность rpa
дуировочной кривой или расчетноro алroритма в
пределах концентраций испытуемоro раствора;
если нет, то проводят проверку перед каждой
серией измерений.
Показания электрода можно считать линей
ными, если наклон S rрадуировочной кривой
приблизительно равен k/z, на единицу рС;.
МЕТОД I (МЕТОД ПРЯМОЙ rРАДУИРОВКИ)
Измеряют последовательно не менее трех
раз потенциалы не менее трех растворов cpaB
нения предположительно близких к испытуемо
му раствору концентраций. Рассчитывают ypaB
нение rрадуировочноro rрафика или строят
rрадуировочный rрафик в координатах cp ДHee
значение потенциала Е соответствующее ему
значение концентрации анализируемоrо иона,
выраженное как logC; или pCj'
rотовят испытуемый раствор, как указано в
частной статье. Измеряют потенциал три раза и
по среднему значению потенциала рассчитыва
104
Тосударственная фармакопея Республики Беларусь
ют концентрацию анализируемоro иона, исполь
зуя rрадуировQЧНЫЙ rрафик.
МЕТОД 11 (МЕТОД мноrОРАЗОВЫХ
СТАНДАРТНЫХ ДОБАВОК)
rотовят испытуемый раствор, как указано в
частной статье. Измеряют равновесный потен
циал Е т в объеме V T этоro раствора с неизвест
ной концентрацией С т анализируемою иона. Не
менее трех раз последовательно к объему V т ис
пытуемоro раствора прибавляют объем V s раст
вора сравнения, незначительный в сравнении с
V T (V s S 0,01 V 7 ), с концентрацией C s ' значения
которой находятся в пределах линейной части
rрадуировочноrо rрафика. После каждоro при
бавления измеряют потенциал и рассчитыва
ют разницу потенциалов ДЕ между измеряемым
потенциалом и Er ДЕ связано с концентрацией
анализируемоro иона уравнением:
или
( С V '1
E Slog 1+ )
CTV T
106: o=1+ CsVs ,
CTV T
rдe:
V T объем испытуемою раствора;
С т концентрация анализируемоrо иона в
испытуемом растворе;
V s объем прибавляемоro раствора cpaB
нения;
C s концентрация анализируемоro иона в
растворе сравнения;
S наклон электродной функции, YCTa
новленный экспериментально при постоянной
температуре путем измерения разницы между
потенциалами двух растворов сравнения, KOH
центрации которых отличаются в 1 О раз и pac
считаны в пределах линейной области rрадуиро
вочноro rрафика.
6Е
Строят rрафик зависимости 10 s (ось у) OT
носительно V s (ось х) и экстраполируют полу
ченную линию до пересечения с осью Х. В точке
пересечения концентрацию С т анализируемою
иона в испытуемом растворе рассчитывают по
формуле:
С CsV s .
т
V T
МЕТОД 111 (МЕТОД ОДНОЙ СТАНДАРТНОЙ
ДОБАВКИ)
К объему V T испытуемоro раствора, приro
товленною, как указано в частной статье, при
бавляют объем V s раствора сравчения, вмеща
ющеro такое количество анализируемоro иона,
чтобы сиrнал находился в линейной части rpa
дуировочноro rрафика. Параллельно в тех же
условиях ютовят контрольный раствор. Потен
циалы испытуемоro раствора и контрольно
ro раствора измеряют не менее трех раз перед
прибавлением и после прибавления раствора
сравнения. Рассчитывают концентрацию С т aHa
лизируемоrо иона, используя уравнение и учи
тывая контрольный раствор:
С т 6Е CsV s
10 S (V7 +VS) V7
rдe:
С т концентрация анализируемою иона в
испытуемом растворе;
C s концентрация анализируемоro иона в
растворе сравнения;
V s объем прибавляемоrо раствора cpaB
нения;
дЕ среднее значение разницы потен
циалов, измеренных до и после прибавления
объема V s ;
S наклон электродной функции, YCTa
новленный экспериментально при постоянной
температуре путем измерения разницы между
потенциалами двух растворов сравнения, KOH
центрации которых отличаются в 1 О раз и pac
считаны в пределах линейной области rрадуиро
вочноrо rрафика;
V T объем испытуемою раствора или KOH
трольноro раствора.
01/2013:20237
2.2.37. РЕнтrЕНОФЛУОРЕСЦЕНТНАЯ
СПЕКТРОМЕТРИЯ
Рентrенофлуоресцентная спектрометрия с
волновой дисперсией это метод анализа, в
котором измеряют интенсивность флуоресцент
ноro излучения, испускаемоro элементом, имею
щим атомный номер от 11 до 92, возбуждаемоro
непрерывным первичным рентrеновским излу
чением. Интенсивность флуоресценции эле
мента зависит от ею концентрации в образце, а
также от поrлощения матрицей возбуждающеro
и испускаемоrо излучения. При следовых коли
чествах определяемоro вещества в области ли
нейности rрадуировочною rрафика измеренная
при определенной длине волны интенсивность
флуоресценции элемента, находящеroся в Ma
трице определенноrо состава, прямо пропор
циональна концентрации данноrо элемента и
обратно пропорциональна маСС090МУ коэффи
циенту ослабления (поrлощения) МClтрицы при
этой длине волны.
Методика. Настройку и использование при
бора проводят в соответствии с инструкция
ми производителя. Жидкие образцы помещают
непосредственно в прибор; твердые образцы
предварительно прессуют в таблетки (rранулы),
2.2.38. Электропроводность
105
01/2013:20238
иноrда после смешивания с подходящим связы
вающим материалом.
Для определения концентрации элемента
в образце измеряют скорость сетевоro импуль
са, создаваемоro одним или несколькими стан-
дартными образцами, содержащими известные
количества данноrо элемента в матрицах опре
деленноro состава, и рассчитывают или измеря
ют массовый коэффициент ослабления матри
цы анализируемоro образца.
rрадуuровка. с помощью rpадуировочно
ro раствора или серии растворов, содержащих
разные концентрации определяемоro элемента в
различных матрицах, определяют уrловой коэф
фициент rрадуировочноro rpафика Ь о по формуле:
1 ,N
bo =....f:...
11м С
rде:
11м рассчитанный или измеренный массо-
вый коэффициент ослабления матрицы М;
, скорость ceTeBoro импульса;
С концентрация определяемоrо элемента
в стандартном образце.
Массовый коэффициент ослабления ма-
трицы образца. Если эмпирическая формула
анализируемоro образца известна, ero массовый
коэффициент ослабления рассчитывают исходя
из справочных значений массовых коэффициен
тов ослабления составляющих элементов. Если
элементный состав образца неизвестен, изме-
ряют интенсивность рассеянноrо peHTreHoBcKoro
излучения 'и (Комптоновское рассеяние) и рас-
считывают величину MaccoBoro коэффициента
ослабления матрицы образца по формуле:
1
=a+Ыи'
I1MP
rде:
I1 MP массовый коэффициент ослабления
образца;
'и рассеянное peHTreHoBcKoe излучение.
Определение скорости сетевоео импульса
для определяеМ020 элемента в образце. Pac
считывают скорость ceTeBoro импульса ' P дЛЯ
определяемоro элемента по измеренным интен-
сивностям флуоресцентной линии и фоновой
линии (линий), принимая во внимание все за
rрязняющие вещества пробирки.
Расчет содержания следов. Если KOHцeH
трация элемента входит в область линейности
rрадуировочноrо rрафика, ее можно рассчитать
по формуле:
с = ' P . "
1
Ь О H
J.J МР
rде:
f фактор разведения.
15 э-- 1712
2.2.38. ЭЛЕКТРОПРОВОДНОСТЬ
Сила тока' (в амперах) в проводнике пря-
мопропорциональна приложенной электродви-
жущей силе Е (в вольтах) и обратнопропорцио-
нальна сопротивлению R (в омах) проводника:
Е
'= '
R
Электропроводность (ранее удельная
электропроводность) раствора (к) величина,
обратная удельному сопротивлению (р). Удель-
ное сопротивление определяют как отношение
напряженности электрическоro поля к плотности
тока. Сопротивление R (Ом) проводника, име-
ющеrо площадь сечения $ (см 2 ) и длину L (см),
рассчитывают по формуле:
L
R=PS'
1 L 1 L
Таким образом: R = . или к = . ,
к $ R $
rде L/$ соответствует постоянной идеаль-
ной камеры.
Единица электропроводности в системе
СИ сименс на метр (См . M 1). На практике элек-
тропроводность растворов выражают в сименсах
на сантиметр (См . CM 1) или В микросименсах на
сантиметр (мкСм . CM 1). Единица удельноro со-
противления в системе СИ ом-метр (Ом . м).
На практике сопротивление раствора обычно вы-
ражают единицах ом-сантиметр (Ом' см). Если
нет друrих указаний в частной статье, значения
электропроводности или удельноro сопротивле-
ния обозначают для температуры 25 ос.
Описание прибора и порядок проведения
испытания, приведенные ниже, приrодны для
лабораторноrо измерения электропроводности
большей чем 1 О мкСм . см- 1 . Определение элек-
тропроводности воды проводят, как указано в co
ответствующих частных статьях.
ПРИБОР
Принцип работы ИСПОЛЬЗуемоro прибора (кон-
дуктометра или омметра) основан на измерении
удельноro сопротивления столба жидкости между
электродами иммерсионноro измерительноro при-
способления (электропроводной камеры). Во из
бежание поляризации электродов используют пе-
ременный ток. Прибор снабжают температурным
компенсатором или прецизионным термометром.
Измерительная камера вмещает два плати-
новых электрода, каждый с площадью поверх-
ности $, располаrающихся параллельно один к
друrому на расстоянии L и покрытых платиновой
чернью. Обычно оба электрода защищены сте-
клянной трубкой. Moryт использоваться и друrие
типы измерительных камер.
106
rосударственная фармакопея Республики Беларусь
ПОРЯДОК ПРОВЕДЕНИЯ ИСПЫТАНИЯ
Определение постоянной камеры
Выбирают измерительную камеру, COOT
ветствующую свойствам и электропроводно
сти испытуемоro раствора. Чем выше ожидае
мая электропроводность, тем большей должна
быть постоянная камеры (ниже р). Обычно ис
пользуемые измерительные камеры имеют
постоянную порядка О, 1 CM 1, 1 см. 1 И 10 см. 1 ,
Используют сертифицированный стандарт.
ный образец, например, раствор калия хло
рида, который приrоден для измерения.
Значение электропроводности сертифициро
BaHHoro стандартноro образца должно быть
близким к ожидаемому значению электропро
водности испытуемоro раствора. MorYT быть
использованы друrие сертифицированные
стандартные образцы, особенно для камер с
постоянной порядка О, 1 CM 1. Камеру опола
скивают несколько раз водой дистиллирован
ной Р и как минимум дважды используемым
для определения постоянной измерительной
камеры сертифицированным стандартным
образцом. Измеряют сопротивление измери
тельной камеры, используя сертифицирован
ный стандартный образец при температуре
(25:t1) ос. Постоянную К (CM 1) измерительной
камеры рассчитывают по формуле:
К=- Rcco . Косо'
rдe:
Rooo измеренное сопротивление, МОм:
Кооо электропроводность ИСПОЛЬ2уемо
ro сертифицированноro стандартноro образца.
мкСм . см. 1 .
Измеренное значение постоянной К
камеры не должно отличаться от указаннсrо
более чем на 5 %.
Если определение постоянной камеры
проводится при температуре, отличающейся
от указанной для сертифицированноro CTaH
дартноro образца, значение проводимости
рассчитывают по формуле:
КТ =- к тесе' [1 + а(Т Тссо)],
rдe:
кт значение электропроводности при
отличающейся температуре;
К теоо значение электропроводности cep
тифицированноro стандартноro образца;
т температура при проведении rраду
ировки;
Теео температура, указанная для серти
фицированноro стандартноro образца;
а температурный коэффициент для
значения электропроводности сертифициро
ванноro стандартноro образца; для калия хло
рида а=0,021.
Определение удельной электропровод
ности испытуемоrо раствора
После тоro, как прибор отrрадуирован с ис
пользованием одноro из стандартных растворов,
измерительную камеру промывают несколь
ко раз водой дистиллированной Р и как мини
мум дважды испытуемым водным раствором.
Измерения проводят так, как указано в частной
статье.
01/2013:20239
2.2.39. МОЛЕКУЛЯРНО-МАССОВОЕ
РАСПРЕДЕЛЕНИЕ
ДЕКСТРАНОВ
Определение проводят методом эксклюзи
он ной хроматоrрафии (2.2.30).
Испытуемый раствор. 0,200 r испытуемо
ro образца растворяют в подвижной фазе и дo
водят до объема 1 О мл этим же растворителем.
Маркерный раствор. 5 Mr 2ЛЮКОЗЫ Р и 2 Mr
ФСО декстрана V o растворяют в 1 мл подвиж
ной фазы.
iрадуировочные растворы. Навеску каж
доro из указанных стандартных образцов: 15 Mr
ФСО декстрана 4 для 2радуировки, 15 Mr ФСО
декстрана 10 для 2радуировки, 20 Mr ФСО дeK
страна 40 для 2радуировки, 20 Mr ФСО дeKcтpa
на 70 для 2радуировки и 20 Mr ФСО оекстрана
250 для 2радуировки по отдельности растворяют
в 1 мл подвижной фазы.
#Допускается использование стандартных
образцов декстранов для rрадуировки с друrими
паспортными данными, в этом случае для rpa
дуировки хроматоrрафической системы peKO
мендуется использовать дополнительно еще не
менее двух стандартных образцов декстранов. #
Раствор для проверки при20дности xpOMa
т02рафической системы. 20 Mr ФСО дeKcтpa
на 40 для проверки при20дности xpOMaт02pa
фической системы (для анализа субстанции
декстрана 40) или 20 Mr ФСО декстрана 60ПО
для проверки при20дности хромат02рафиче
ской системы (для анализа субстанций дeK
страна 60 и декстрана 70) растворяют в 1 мл
подвижной фазы.
Если нет друrих указаний в частной статье,
можно использовать следующие условия xpOMa
тоrрафирования,:
колонка длиной 0,3 м и внутренним диаме
тром 10 мм, заполненная а2арозой поперечно
сшитой для хромат02рафии Р, или серия коло
нок длиной 0,3 м и внутренним диаметром 1 О мм
каждая, заполненных полиэфирным 2идPOKcи
лированным 2елем для хромат02рафии Р;
подвижная фаза: раствор 7 r натрия сулъфа
та безводН020 Р и 1 r хлорбутанола Р в 1 л воды Р;
2.2.39. Молекулярно массовое распределение декстранов
107
скорость подвижной фазы: от 0,5 мл/мин
до 1 мл/мин, поддерживаемая постоянно с точ
ностью:t1 % в час;
детектор: дифференциальный рефрак
тометрический;
петлевой дозатор: объем от 100 мкл ДО
200 мкл;
температура системы: поддерживают
постоянной с точностью :1:0,1 ОС.
rРАДУИРОВКАХРОМАтоrРАФИЧЕСКОЙ
СИСТЕМЫ
Хроматоrрафируют несколько раз BЫ
бранный объем MapKepHoro раствора. На xpo
MaTorpaMMe должны присутствовать два пика:
первый пик соответствует ФСО декстрана V o '
второй пик соответствует 2люкозе Р. Исходя
из объема элюирования декстрана V Q , рассчи
тывают объем эксклюзии V Q ; полный объем
V/ рассчитывают по пику, соответствующему
rлюкозе.
Хроматоrрафируют определенный объем
каждоro из rрадуировочных растворов. Для
каждой полученной xpoMaTorpaMMbI проводят
базовую линию. Каждую xpoMaтorpaMMY делят
вертикальными равноудаленными линиями на
р секций (количеством не менее 60), что co
ответствует равным объемам элюирования.
Для каждой секции i, соответствующей объему
элюирования V i , измеряют высоту (у) от базо
вой линии ДО линии xpoMaTorpaMMbI и рассчи
тывают коэффициент распределения (К,) по
формуле:
(\I; Vo) ,(1)
(\1; V o )
rде:
V o объем эксклюзии, определенный по
пику, соответствующему ФСО декстрана V o на
xpoMaTorpaMMe MapKepHoro раствора;
V/ полный объем колонки, определенный
по пику, соответствующему rлюкозе на xpOMaTO
rpaMMe маркерноro раствора;
объем элюирования для секции i, полу <
ченный для xpoMaTorpaMMbI каждоro из rрадуи
ровочных растворов.
rрадуировку проводят, используя один из
следующих методов.
rрадуировка путем построения rpa-
дуировочноrо rрафика. По уравнению (1)
для каждоrо из декстранов для rрадуировки
рассчитывают коэффициент распределения
Kma.' соответствующий максимальной высоте
линии xpoMaTorpaMMbI. На полулоrарифми
ческой бумаrе строят зависимость значений
Krra. (ось абсцисс) от молекулярной массы для
максимальной высоты линии xpoMaTorpaMMbI
(AI....) каждоro из декстранов для rрадуировки
.. fПЮкозы (ось ординат). Через все получен
ные точки про водят первую rрадуировочную
линию, экстраполируя ее из точки Ктах' полу-
ченной для ФСО декстрана 250 для 2paдyи
ровки, до более низких значений К, получен
ных для тех же стандартных растворов (см.
рисунок 2.2.З9. 1).
I I I
I I
I I
, !
,
, I i
\1 I I
i
t . . ..,
,
' j
I , I
1 , 1 r
i , I
f ,
, , i
, ,; ,
'r -.. Декстран 250 I
5 ; ,1
"'1 _..
, .
." Де.стран 70
, '\. . ' I --_. .
: Декстран -IO
I I I ш
I I , Ш
I
! i \ I дJстран 10
!
i ш 1\ I
1 ;
I 1 ш\ шшr
ш ш шf Ш - I\. Де.'стра
I , ш\
i
3 ;
;
! ! I \
i i I
! I I
I ; i
! I
250 i
7 4
i 1 4
м
1()б
10
1()4
H-l
10
rлюкоза
к
Q1 Q2 QЗ Q4 Q5 Q6 Q, Q8 Q9 1Ю
Рисунок 2.2.39. 1. Пример 2радуировОЧН020
2рафика
Пунктиром обозначена часть 2радуировочно
20 2рафика, полученная экстраполированием.
tоризонтаЛЬНblми линиями в нижней части pи
сунка указаНbI ширина и положения xpOMaтo
2рафических линий, полученных для кажд020
декстрана для 2радуировки
Используя полученный первый rрадуиро
вочный rрафик, находят для всех значений К;
для всех xpoMaTorpaMM соответствующие зна
чения молекулярных масс Mi' получая таким об
разом rрафическую зависимость для расчета
молекулярно массовоro распределения. Для
всех декстранов для rрадуировки рассчитыва
ют среднюю молекулярную массу по уравне-
нию (3), приведенному ниже. rрадуировочный
rрафик может быть Иi;пользован для расчетов,
если рассчитанные значения для каждоro из
декстранов для rрадуировки не отличаются от
указанных в паспорте значений больше чем на
5 % и среднее отклонение для всех дeKCTpa
нов не превышает :1:3 %. Если данные условия
не выполняются, rрадуировочный rрафик CMe
108
rосударственная фармакопея Республики Беларусь
щают ВДОЛЬ оси ординат и повторяют вышео
писанные операции, пока рассчитанные и YKa
занные в паспорте значения будут отличаться
не более чем на 5 %.
rрадуировка при помощи расчетноrо
метода. При помощи уравнений (2) и (3), при
веденных ниже, и подходящеro метода (воз
можно использование итеративноro метода,
такоro как модифицированноro Хартли метода
rаусса Ньютона; также возможно построение
кривой при помощи компьютерных проrрамм,
которые используют принцип нелинейноro pe
rрессивноro анализа) рассчитывают значе
ния коэффициентов Ь 1 , Ь 2 , Ь З ' Ь 4 И Ь 5 , которые
MorYT отличаться не более чем на 5 % от co
ответствующих паспортных значений для каж
доro декстрана для rрадуировки и 180:t2 для
rлюкозы.
#При расчетах rлюкозы в уравнении (2) дo
пускают К;=1.#
М, = Ь 5 + е{ь. ь,К,+Ь2К:+ЬЗК ), (2)
р
I(y;M;)
М = ; , , (3)
w р
LY;
1==1
rAe:
р число секций, на которые поделена xpo
MaTorpaMMa;
У; высота хроматоrрафической линии над
базовой линией в i той секции;
М, молекулярная масса для секции i.
приrодность ХРОМАтоrРАФИЧЕСКОЙ
СИСТЕМЫ
Хроматоrрафируют подходящий объем co
ответствующеro раствора для проверки приroд
ноСТи хроматоrрафической системы.
Средняя молекулярная масса ФСО дек-
страна для проверки приrодности хро-
матоrрафической системы. Рассчитывают
значение М,." как указано в разделе «rраду
ировка хроматоrрафической системы», ис
пользуя rрадуировочный rрафик или значения
коэффициентов Ь" Ь 2 , Ь З ' Ь 4 И Ь 5 . Результаты
анализа считаются достоверными, если полу
ченное значение находится в пределах:
от 41 000 до 47 000 (для ФСО дeKcтpa
на 40 для проверки при20дности xpOMaтo
2рафической системы);
от 67 ООО до 75 000 (для ФСО дeKcтpa
на 60/70 для проверки при20дности xpOMa
m02рафической системы).
Средняя молекулярная масса 10% вы-
сокомолекулярной фракции декстрана.
Значение М,.) для 1 О % высокомолекулярной
фракции декстрана, которая элюируется в
секциях от 1 до п включительно, рассчитыва-
ют по формуле:
n
L (у;М; )
М = м , (4)
W n
LYi
1..::1
rдe п определяется неравенствами:
t y , so,{t y ,}, (5)
Yj > о,{ Y. ), (6)
rде:
р количество секций, на которые разделе
на xpoMaTorpaMMa;
У; высота хроматоrрафической линии над
базовой линией для секции i;
М; молекулярная масса для секции i.
Результаты анализа считаются ДOCTOBepHЫ
ми, если полученное значение для 1 О % BЫCO
комолекулярной срракции декстрана находится
в пределах:
от 11 О 000 до 130 000 (для ФСО декстра-
на 40 для проверки приеодности хроматоара-
фической системы);
от 190 000 до 230 000 (для ФСО дeKcтpa
на 60/70 для проверки приаодности xpOMaтo
2рафической системы).
Средняя молекулярная масса 1 О % низ-
комолекулярной фракции декстрана. Значе
ние для 1 О % низкокомолекулярной фракции
декстрана, которая элюируется в секции и после
секции xpoMaTorpaMM т, рассчитывают по фор-
муле:
р
м = t.;(Y;M') ,(7)
W р
LY;
i m
rAe m определяется неравенствами:
Y, so,{t y ;), (8)
,tr i > о,{ y;), (9)
rAe:
р количество секций, на которые разделе
на хроматоrраммэ;
У; высота хроматоrрафической линии над
базовой линией для секции i;
М; молекулярная масса для секции i.
Результаты анализа считаются достоверными,
если полученное значение для 1 О % низкомолеку-
лярной фракции декстрана находится в пределах:
2.2.40. Спектрофотометрия ближне20 инфракраСН020 диапазона
109
от 6000 до 8500 (для ФСО декстрана 40
для проверки при20дности хромат02рафиче
СКОЙ системы);
от 7000 до 11 000 (для ФСО декстрана
60ПО для проверки при20дности xpOMaт02pa
фической системы).
МОЛЕКУЛЯРНО МАССОВОЕ
РАСПРЕДЕЛЕНИЕ ИСПЫТУЕмоrо
ДЕКСТРАНА
Хроматоrрафируют выбранный объем ис
пытуемоro раствора и рассчитывают: значение
Мы для молекулярно массовоro распределения
декстрана, значение Мы для 10 % высокомоле
кулярной фракции декстрана и значение Мы для
1 О % низкомолекулярной фракции декстрана
рассчитывают, как описано в разделе «Приrод
насть хроматоrрафической системы».
01/2013:20240
2.2.40. СПЕКТРОФОТОМЕТРИЯ
БЛИЖНЕrо ИНФРАКРАсноrо
ДИАПАЗОНА
Спектрофотометрия ближнеro инфракрас
ноro (БИК) диапазона метод с широкими
м разнообразными способами применения в
фармацевтическом анализе. Ближний инфра
красный спектральный диапазон охватывает
oбnасть от около 780 нм до около 2500 нм (от
около 12 800 CM 1 ДО около 4000 CM 1). В HeKO
торых случаях наиболее полезная информация
находится в спектральном диапазоне от около
1700 нм ДО около 2500 нм (от около 6000 CM 1
ДО около 4000 CM 1). В спектрах БИК диапазона
представлены rлавным образом обертона коле
баний C H, N H, O H и S H и комбинации oc
новных типов колебаний; они являются очень
информативными при условии, если информа
ция обрабатывается с помощью COOTBeTCTBY
ющих хемометрических алrоритмов. Полосы в
БИК области значительно более слабые, чем
полосы основных колебаний в средней ИК
области, от которых они происходят. Так как
значения молярноro поrлощения в БИК области
малы, излучение обычно проникает в материал
(включая твердые вещества) на несколько мил
лиметров. Кроме тоro, мноrие вещества, такие
как стекло, являются прозрачными в этом диа
пазоне длин волн.
Измерения MOryT быть проведены как на об
разцах iп situ, так и с использованием CTaHдapT
ноro отбора образцов и методов испытаний. Из
БИК спектра доступна как физическая инфор
мация, так и химическая, а также качественная
и количественная информация. Однако непо
средственное сравнение полученноrо спектра
16 30. 1712
со спектром химическоro стандартноro образца,
как это происходит в абсорбционной спектро
фотометрии в среднем ИК диапазоне, в данном
случае не приемлемо. Требуется проведение
соответствующей валидированной математиче
ской обработки полученных данных.
БИК спектрофотометрия широко использу
ется как для химическоro анализа, так и для фи
зическоrо.
Химический анализ:
идентификация активных субстанций,
вспомоrательных веществ, rOToBbIx лекарствен
ных форм, промежуточных продуктов производ
ства, химическоro сырья и упаковочных матери
алов;
количественное определение содержа
ния активных субстанций и вспомоrательных Be
ществ, определение химических чисел, таких как
rидроксильное число, йодное число, кислотное
число, определение содержания воды, опреде
ление степени rидроксилирования, контроль co
держания растворителей;
контроль процесса производства.
Физический анализ:
кристаллическая форма и кристаллич
ность, полиморфизм, псевдополиморфизм,
размер частиц;
процесс растворения, схема распада,
твердость;
исследование свойств пленок;
контроль процесса производства, напри
мер, контроль смешивания и rрануляции.
На измерения в БИК диапазоне влияют
мноrие химические и физические факторы, опи
санные ниже; воспроизводимость и релевант
ность результатов зависят от контроля этих фак
торов; измерения справедливы обычно только
для конкретной модели rрадуировки.
ПРИБОР
БИК спектрофотометры используются для
получения спектров в области от около 780 нм
< ДО около 2500 нм (от около 12 800 CM 1 ДО около
4000 CM 1). Все измерения в БИК диапазоне oc
нованы на прохождении луча света через об
разец или в неro и измерении ослабления
выходящеro (прошедшеro, рассеянноro или OT
раженноro) излучения. Спектрофотометры для
измерений в БИК диапазоне включают подходя
щий источник света и монохроматор или интер
ферометр. Обычно монохроматорами являются
акустико оптические перестраиваемые фильтры
(AOTF), дифракционные решетки или призмы.
Обычно используются источники света высокой
интенсивности, такие как кварцевые или воль
фрамовые лампы или аналоrичные. Вольфра
мовые лампы в качестве источника излучения
MOryт быть высоко стабилизированы. Поэтому
мноrие БИК спектрофотометры имеют однолу
чевую конструкцию. В качестве материала дe
тектора обычно используются кремния диоксид,
110
rосударственная фармакопея Республики Беларусь
свинца сульфид, индия арсениД, индия rаллия
арсен ид, ртути кадмия теллурид (МС7) и Аей
терированный триrлицинсульфат. CTaHдapTHЫ
ми приспособлениями для образцов являются
обычные кюветные держатели, оптико волокон
ные зонды, трансмиссионная кювета для по
rружения, вращающиеся или траверсные дep
жатели образцов. Выбор производят исходя из
предполаrаемоro применения, уделяя особое
внимание приrодности приспособления отбора
проб для типа анализируемых образцов. Подхо
дящие блоки по обработке данных и их оценке
обычно являются частью системы.
МЕТОДЫ ИЗМЕРЕНИЙ
Режим пропускания. Пропускание (7) явля
ется мерой ослабления интенсивности излуче
ния при данной длине волны при прохождении
излучения сквозь образец. Образец помещают
в оптический пучок между источником и дeTeK
тором. Расположение аналоrично таковому во
мноrих традиционных спектрофотометрах_ Pe
зультат может быть представлен непосредствен
но в единицах пропускания (7) и/или поrлоще
ния (А).
1
T '
10
rдe:
1 интенсивность падающеro излучения;
/0 интенсивность прошедшеro излучения.
A== lo910 Т == 10910 (;) == 10910 ( 1; )-
Режим диффузноrо отражения. Режим
диффузноro отражения основан на измере
нии отражения (R) отношения интенсивности
света, отраженноro от образца (1) к интенсивно
сти света, отраженноro от фона или стандартной
отражающей поверхности (/,). БИК излучение
может проникать в образец на существенную
rлубину, rде может быть поrлощено колебатель
ными комбинациями и обертонами аналитов,
присутствующих в образце. Непоrлощенное из
лучение отражается от образца на детектор.
БИК спектр отражения обычно получают расче
том и построением зависимости Ig(1/R) от длины
волны или волновоro числа.
1
R '
1,
rдe:
1 интенсивность излучения, диффузноо
траженноrо от образца;
10 интенсивность излучения, диффузно
отраженноro от фона или отраженноro от по
верхности сравнения.
A R 10910( ) 10910( 1; )-
Режим пропускания-отражения. Этот
режим является комбинацией пропускания и OT
ражения. При измерении пропускания отраже
ния (Т*) используется зеркало или диффузная
отражающая поверхность для отражения излу
чения, прошедшеro сквозь образец второй раз,
удваивая, таким образом, оптический путь. He
поrлощенное излучение отражается от образца
на детектор.
T* '
17
rде:
/т интенсивность прошедшеro и отражен
ноro излучения без образца;
/ интенсивность прошедшеro и отражен
ною излучения, измеренная с образцом.
, 1 I
A*== I0 91Ol т * )"
приrОТОВЛЕНИЕ/ПОДАЧА ОБРАЗЦА
Режим пропускания. Измерение' и расчет
пропускания (7) зависят от фоновоro спектра
пропускания. В качестве фона MOryT выступать
воздух, пустая кювета, используемый раствори
тель или, в специальных случаях, стандартный
образец. Метод в основном применяется для
исследования разведенных инеразведенных
жидкостей, дисперсионных систем, растворов
и твердых веществ. Для измерения пропуска
ния твердых веществ необходимо использовать
подходящие приспособления для образцов. Об
разцы исследуются либо в кюветах подходящей
длины (обычно от 0,5 мм до 4 мм), прозрачных в
БИК диапазоне, либо путем поrружения оптико
волоконноro зонда подходящей конфиryрации,
позволяющеro получать спектр, находящийся в
зоне, отвечающей требованиям спецификации
прибора и подходящей для намеченных целей.
Режим диффузноrо отражения. Данный
метод обычно при меняется для исследования
твердых веществ. Образец исследуется в под
ходящем приспособлении. Должны быть приня
ты меры по соблюдению как можно более BOC
производимых условий измерений от образца к
образцу. При использовании оптико волоконноro
зонда внимание должно быть уделено позицио
нированию зонда, чтобы убедиться, что он OCTa
ется неподвижным во время получения спектра
и что условия испытания воспроизводимы от об
разца к образцу. Излучение, отраженное от CTaH
дартной отражающей поверхности (фона), CKa
нируется для получения базовой линии, и затем
измеряется отражение одноro или более анали
тическоro образца. Распространенными CTaH
дартными отражающими веществами являются
керамическая плитка, перфторированные по
лимеры и золото. MorYT быть использованы и
друrие подходящие вещества. Непосредствен
2.2.40. Спектрофотометрия ближне20 инфракраСНО20 диапазона
111
ное сравнение возможно только лишь между
спектрами, полученными при использовании
фона с одинаковыми оптическими свойствами.
Должны учитываться влияния размера частиц,
rидратационной воды и степени сольватации.
Режим пропускания-отражения. Отража
тель размещается позади образца таким обра
зом, чтобы удваивать оптический путь. Такая
конфиryрация может быть предназначена для
разделения одной и той же rеометрии прибора
на систему с отражателем и систему с оптико
волоконным зондом, Korдa источник излучения
и детектор располаrаются по одну сторону об
разца. Образец исследуется в кювете с зерка
лом или подходящим диффузным отражателем,
сделанным либо из металла, либо из инертноro
вещества (например, титана оксида), которое не
поrлощает в БИК диапазоне.
ФАКТОРЫ, ВЛИЯЮЩИЕ
НА СПЕКТРАЛЬНЫЙ ОТКЛИК
Температура образца. Данный параметр
важен при исследовании водных растворов и
мноrих жидкостей, Korдa различие на несколько
rрадусов может приводить к значительным отли
чиям спектров. Кроме тоro, температура иrрает
важную роль при исследовании твердых Be
ществ и порошков, содержащих воду.
Влаrа и остаточные растворители. Влаrа
и остаточные растворители, присутствующие в
образце, будут приводить к значительным поло
сам поrлощения в БИК диапаЗ0не.
Толщина образца. Толщина образца яв
ляется известным источником спектральной
вариабельности и должна учитываться и/или
контролироваться. Например, при измерении
отражения образцы должны быть «бесконечно»
толстыми, а более тонкие образцы постоянной
толщины должны иметь стабильный диффузи
онно отражающий материал подложки с посто
янной и, желательно, высокой отражающей спо
собностью.
Оптические свойства образца. Для TBep
дых веществ должны учитываться как поверх
ностные свойства, так и объемные свойства
образца. Для записи спектров физически, хи
мически или оптически неоднородных образцов
может понадобиться усреднение путем увели
чения пучка излучения или путем проведения
большоro числа измерений или путем враще
ния зонда. Определенные факторы, такие как
различltlЯ в степени уплотнения или в размерах
частиц порошков, а также в качестве поверхно
сти, MOryT вызывать значительные спектраль
ные изменения.
Полиморфизм. Вариации в кристалличе
ской структуре (полиморфизм) влияют на спек
тры. По этой причине различные кристалличе
ские формы, так же как и аморфные формы,
твердых веществ MOryт быть отличены друr от
друrа на основании БИК спектров. В случае
присутствия мноroчисленных кристаллических
форм необходимо обращать особое внимание
на то, чтобы rрадуировочные стандартные об
разцы имели распределение кристаллических
форм, подходящее для решения поставленной
задачи.
Возраст образца. С течением времени в
образце MOryT происходить изменения физиче
ских, химических или оптических свойств. He
обходимо убедиться, что образцы, предназна
ченные для БИК спектрофотометрии, являются
однотипными с образцами, взятыми для rраду
ировки. В случае анализа образцов различноro
возраста необходимо принимать в расчет потен
циальные отличия в их свойствах.
КОНТРОЛЬ ПАРАМЕТРОВ ПРИБОРА
Прибор используют в соответствии с ин
струкциями производителя. Реryлярно проводят
необходимые проверки в соответствии с исполь
зованием прибора и испытуемыми веществами.
Проверка шкалы длин волн (за исклю-
чением прибора с фильтром). Проверяют ис
пользуемую шкалу длин волн обычно в диапазо
не от около 780 нм до около 2500 нм (от около
12800 см. 1 ДО около 4000 см. 1 ) или В планируемом
для анализа диапазоне, используя один или He
сколько более подходящих стандартных образцов
для проверки длин волн, имеющих характеристи
ческие максимумы или минимумы в данном диа
пазоне. Подходящими стандартными образцами
являются, например, метиленхлорид или смесь
оксидов редкоземельных металлов. Записывают
спектр с таким же спектральным разрешением,
как и при получении сертифицированноro значе
ния, и определяют расположение не менее трех
пиков, расположенных в используемом диапазо
не. Допустимыми отклонениями являются :!::1 нм
при 1200 нм, :!::1 нм при 1600 нм и :!::1,5 нм при
2000 нм (:!::8 см. 1 при 8300 см. 1 , :!::4 см. 1 при 6250 см. 1
и :!::4 см. 1 при 5000 см. 1 ). В зависимости ОТ'исполь
зуемоro стандартноro образца проверку допусти
моro отклонения для каждоro используемоro пика
осуществляют, применяя требования для ближай
шей длины волны (волновоro числа) из описанных
выше. Для приборов с преобразованием Фурье
rpадуировка шкалы длин волн может быть прове
дена с использованием узкой линии паров воды
при 7299,86 см. 1 или узкой линии сертифициро
ванноro образца. Из оксидов редкоземельных ме---
таллов наиболее подходящим стандартным об
разцом является NI8T 1920 (а).
Измерения в режиме пропускания. Может
быть использован метиленхлорид Р с длиной
оптическоro пути 1,0 мм. Метиленхлорид имеет
характеристические резкие полосы при 1155 нм,
112
/осударственная фармакопея Республики Беларусь
1366 нм, 1417 нм, 1690 нм, 18З8 нм, 1894 нм,
2068 нм и 2245 нм. Полосы при 1155 нм, 1417 нм,
1690 нм и 2245 нм используются для rрадуиров
ки. Moryт быть использованы и друrие подходя
щие стандартные образцы.
Измерения в режиме диффУЗН020 отраже
ния. Может быть использована смесь оксидов
диспрозия, roльмия и эрбия (1 + 1 + 1 по массе) или
друroй подходящий стандартный образец. Указан
ный стандартный образец имеет характеристиче
ские пики при 1261 нм, 1681 нм и 1935 нм. Если
использование внешних твердых стандартных об
разцов является невозможным и если измерения
диффузною рассеивания проводят в кюветах или
с использованием оnтоволоконноro зонда, то He
посредственно в кювету или зонд помещают тща
тельно перемешанную суспензию из 1,2 r титана
диоксида Р в около 4 мл метиленхлорида Р.
Спектр записывают через 2 мин. Титана оксид не
обладает поrлощением в БИК диапазоне. Спек
тры записывают с максимальной номинальной ин
струментальной шириной щели 1 О нм при 2500 нм
(16 CM 1 при 4000 CM 1). Измерения осуществля
ют на не менее трех пиках, расположенных в ис
пользуемом диапазоне. Допустимые отклонения
такие же, как указаны выше (при проверке шкалы
длин волн). В зависимости от используемоro CTaH
дартноro образца, проверку допустимою отклоне
ния для каждою используемоro пика осуществля
ют, применяя требования для ближайшей длины
волны (волновою числа) из описанных выше.
Проверка СХОДИМОСТИ длин волн (за ис-
ключением прибора с фильтром). Проверяют
сходимость длин волн с использованием подхо--
дящих стандартных образцов. Стандартное OT
клонение длины волны должно соответствовать
спецификации производителя прибора.
Проверка фотометрической линейности
и стабильности отклика. Проверку фотометри
ческой линейности проводят с использованием
стандартных образцов пропускания или oтpa
жения и известными значениями пропускания
и отражения, выраженными в процентах. При
измерениях отражения используют уrлеродсо
держащие стандартные образцы. В диапазоне
1 0 90 % используют не менее 4 стандартных
образцов: 10 %, 20 %, 40 % и 80 % со значени
ями оптическою поrлощения 1,0, 0,7, 0,4 и 0,1
соответственно. Если система используется для
аналитов с оптической плотностью более 1,0,
дополнительно используют 2 % и/или 5 % CTaH
дартные образцы. Строят rрафик зависимости
наблюдаемой оптической плотности стандарт
ны)( образцов от их стандартных оптических
плотностей и рассчитывают линейную perpec
сию. Допустимые отклонения: для уrла накло
на (1,00:1:0,05); для точки пересечения с осью
абсцисс (0,00:1:0,05).
Спектры, полученные с использованием
стандартных образцов отражения, проявляют
вариабельность по причине отличий условий
испытаний, при которых они калибровались
в заводских условиях, от условий испытаний,
при которых они далее используются. По этой
причине процентные значения отражения CTaH
дартных образцов, используемых для rрадуи
ровки, MOryT быть не достаточны для «абсолют
ной» rрадуировки данноro прибора. Однако, до
тех пор, пока стандартные образцы не изменя
ются химически или физически, и до тех пор,
пока используется та же стандартная отражаю
щая поверхность (фон), что использовалась и
при получении сертифицированноro значения,
последующие измерения тех же стандартных
образцов при идентичных условиях, включая
точное расположение образца, дают информа
цию о долroсрочной стабильности фотометри
ческою отклика. Для долroсрочной стабиль
ности допустимыми отклонениями являются
:1:2 %; это необходимо в случае, только если
спектры записывают без предварительной об
работки образца.
Проверка фотометрическоrо шума. Ha
ходят фотометрический шум при помощи под
ходящеrо стандартноro отражающеrо образца,
например, белых керамических плиток или OT
ражающих термопластических смол (например,
ПТФЭ). Сканируют отражение стандартноro об
разца в необходимом диапазоне длин волн или
волновых чисел с соблюдением рекомендаций
производителя прибора и рассчитывают фото
метрический шум как пик/пик шум. Фотометри
ческий шум должен соответствовать специфика
ции спектрофотометра.
ИДЕНТИФИКАЦИЯ И СПЕКТРАЛЬНЫЕ
ХАРАКТЕРИСТИКИ (КАЧЕСТВЕННЫЙ
АНАЛИЗ)
Создание библиотеки спектров сравне-
ния. Записывают спектры подходящею количе
ства серий субстанции, которые были провере
ны на соответствие требованиям спецификации
и которые проявляют типичную для данной суб
станции вариабельность (обусловленные. Ha
пример, различными производителями, физиче
ской формой, размером частиц). Набор спектров
представляет информацию для идентификации
и характеристики, которая определяет rрани
цы сходства для данной субстанции; этот набор
вносят в библиотек используемую для иден
тификации данной субстанции. Количество Be
ществ в библиотеке зависит от специфики ее ис
пользования, но слишком большие библиотеки
способны вызывать некоторые сложности в BЫ
явлеНИL1 отличий между различными вещества
ми и при валидации. Все спектры используемой
библиотеки должны иметь одинаковые:
спектральный диапазон и количество
точек получения данных;
методику измерения;
предварительную обработку данных.
2.2.40. Спектрофотометрuя ближне20 инфракраСНО20 диапазона
113
Если создаются подrруппы (библиотеки), то
указанные выше параметры применяются неза
висимо для каждой rруппы. Коллекция спектров
библиотеки может быть представлена различны
ми способами в соответствии с математически
ми методами, используемыми для идентифика
ции. Коллекции MOryT включать:
все индивидуальные спектры субстанции;
средний спектр каждой серии субстанции;
при необходимости, описание вариабель
ности спектра субстанции.
Электронные рабочие данные, использо
ванные для создания библиотеки, должны быть
сохранены.
Предварительная обработка данных.
Во мноrих случаях, особенно для спектров,
снятых в режиме диффузноro отражения, MOryT
применяться различные виды предваритель
ной математической обработки спектров перед
разработкой классификации или проведени
ем rрадуировки. Целью этоro может быть, Ha
при мер, уменьшение вариаций базовой линии,
уменьшение влияния известных изменений, Me
шающих дальнейшему использованию MaTeMa
тических моделей, или сжатие данных перед
использованием. Типичными методами являют
ся коррекция мультипликативноrо рассеивания
(MSC), преобразование Кубелка Мунка, спосо
бы сжатия спектральных данных, которые MOryт
включать использование весовой функции и по
давление шумов, а также числовой расчет про
изводных спектра первоro или второro порядка.
Производные более высокою порядка не peKO
мендуются. В некоторых случаях спектры также
MOryт быть нормализованы, например, по MaK
симуму поrлощения, среднему поrлощению или
интеrрированной площади под спектром поrло
щения.
При проведении любых математических
преобразований должна соблюдаться осторож
ность во избежание появления помех или воз
можной потери важной информации (важных
для квалификации методик). Необходимо пони
мание алroритма обработки, и во всех случаях
лоrическое обоснование использования преоб
разования должно документироваться.
Оценка данных. Спектры, полученные в
испытании, сравнивают напрямую с отдельны
ми или усредненными спектрами сравнения
всех субстанций, находящихся в базе данных,
на основании их математической корреляции
или друrих подходящих алюритмов. Набор из
вестных усредненных спектров сравнения и
ОТJ<лонений от них может быть использован в
соответствии с алюритмом классификации. Раз
личные алroритмы, основанные на анализе oc
новных компонентов в комбинации с кластер
ным анализом, S/MCA (soft iпdерепdепt тоdе'iпg
Ьу c'ass апа'оgу проrраммное независимое
моделирование на основе сходства классов),
COMPARE функциях, использующих фильтры,
UNEQ (ипеqиа/ dispe,sed c/ass HepaBHOMep
но рассеянный класс) и друrие, используются в
проrраммном обеспечении приборов для БИК
спектрофотометрии, а также в стороннем про
rpaMMHoM обеспечении. Достоверность выбран
ноro алroритма должна валидироваться для
конкретноro использования. Например, коэффи
циент корреляции, сумма квадратов разниц или
расстояний при кластерном анализе, должны co
ответствовать критериям приемлемости, опре
деленным в процессе валидации.
Валидация базы данных
Специфичность. В процессе валидации
должна быть установлена селективность клас
сификации, использующей базу данных спектра
для положительной идентификации данноro
вещества и достоверноro отличия от спектров
друrих веществ. Должны быть установлены
пределы приемлемости. Строrие критерии при
емлемости обладают большей способностью к
различению спектров, но MOryT вызывать HeKO
торые ошибки, связанные с присущей данному
веществу вариабельностью. Мяrкие критерии
приемлемости решают эти проблемы, но MorYT
приводить к появлению неоднозначных pe
зультатов. Исследование веществ, близких по
внешним признакам, химической структуре или
наименованию к веществам, входящим в базу
данных спектров, должно дать отрицательные
результаты при идентификации. Образцы Be
ществ, входящих в базу данных, но не исполь
зованных для ее создания (т. е. друrие серии,
смеси) при идентификации должны давать по
ложительные результаты.
Робастность. Робастность качествен
ноro анализа должна быть оценена для YCTa
новления влияния незначительных изменений
стандартных условий на проведение испыта
ний. Не допускаются изменения параметров
предварительной обработки и алroритма rpa
дуировки. Факторами, влияющими на робаст
ность, являются:
эффект различных условий окружающей
среды (например, температуры и влажности в
лаборатории );
эффект температуры образца, размеще
ния образца в оптическом окне, rлубины зонда
и уплотнения/наполнения вещества;
замена частей прибора или устройств
подачи образца.
КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
Создание библиотеки стандартных спек
тров для rрадуировки. rрадуировка это
процесс получения математической модели co
отношения отклика, полученною от прибора,
и свойств испытуемоro образца. Используют
любой алroритм rрадуировки, который может
описываться точной математической форму
лой и давать удовлетворительные результаты.
114
rосударственная фармакопея Республики Беларусь
Записывают спектры необходимоro количества
образцов с известными значениями определя
емой величины во всем диапазоне rрадуировки
(например, содержание воды). При rрадуировке
проверяют правильности выбора длин волн для
испытуемоro образца путем сравнения полос ис
лытуемоrо образца и матрицы. Создают модель
rрадуировки с использованием около 213 из
меренных образцов. Оставшуюся 1/3 часть из
меренных образцов сравнивают с полученной
базой данных. Все образцы должны иметь зна
чения количественноro содержания в точно ycтa
новленных пределах в соответствии с постав
ленной задачей. Корректность количественноro
определения должна быть продемонстрирова
на изменениями матрицы в пределах заданно
ro интервала. Обычно используют множествен
ную линейную реrрессию (MLR, тиltiр/е liпеаr
,eg,essioп), метод частных наименьших KBaдpa
тов (PLS, partia/ Jeast squa,es) и реrpeссию oc
новноro компонента (PCR, priпcip/e сотропепt
,eg,еssiоп). В случае rpадуировок PLS и PCR на
rрафик наносят коэффициенты или наrрузки, и
области высоких коэффициентов сравнивают
ся со спектрами анализируемоro образца. Рабо
чие данные, использованные для rрадуировки,
должны быть сохранены без предварительной
обработки.
Предварительная обработка данных.
Предварительная обработка данных может быть
представлена как математическое превращение
БИК спектральных данных для улучшения Ka
чества спектров и/или для удаления или YMeHb
шения нежелательных источников отклонений
перед проведением rрадуировки. Существует
мноro подходящих алroритмов для предвари
тельной обработки данных и калибровки. Выбор
алroритма основывается на приroдности для
решения поставленной задачи. Выбор длины
волны может повысить эффективность Moдe
лей rрадуировки, таких как MLR (например, для
определения размера частиц). В некоторых слу
чаях весьма полезным является удаление части
шкалы длин волн, например, в случае опреде
ления rидратной воды. Кроме этоro используют
прием компрессии шкалы длин волн.
Параметры валидации. При валидации
методик в БИК области спектра используются
такие же аналитические характеристики, как и
для друrих аналитических методик. Специфиче
ские критерии приемлемости для каждоro пара
метра валидации должны учитывать предпола
raeMoe использование метода.
Специфичность. Относительная различи
тельная способность и селективность дЛЯ KO
личественноro анализа должны быть анало
rичны таковым для «Качественноro анализа».
Объем испытаний на специфичность определя
ется предполаrаемым использованием метода и
контролируемыми рисками. Отличия KOHцeHTpa
ций матрицы в пределах рабочеro диапазона не
должны значительно влиять на количественное
измерение.
Линейность. Валидация по показателю ли
нейность включает в себя корреляцию БИК
результатов, рассчитанных из БИК откликов,
полученных по используемым алroритмам, от
результатов, полученных стандартным методом
и распределенным в пределах определенноro
диапазона модели rрадуировки. Также MOryт Ba
лидироваться БИК отклики, находящиеся в He
линейном диапазоне.
Ди пазон примененuя. Диапазон CTaHдapT
ных значений испытуемоro образца определяе-т
диапазон БИК методики и ее пределов количе
cTBeHHoro определения. Необходимо контроли
ровать, чтобы результаты определения не BЫ
ходили за пределы валидированноro диапазона.
Правильность. Правильность может быть
определена путем сравнения результатов aHa
лиза с результатами, полученными по валиди
рованной методике, или с результатами анализа
извес-тных образцов (холостые образцы и образ
цы, к которым добавлено известное количес-тво
испытуемой субстанции). Правильность может
быть выражена как с-тандартная ошибка про
rнозирования (SEP, stапdаrd e,ror о' рrеdiсtiоп)
БИК методики, которая должна соrласовывать
ся С данными валидированной методики. CTaH
дартная ошибка проrнозирования представляет
собой стандартное отклонение разниц, получен
ных при сравнении данных, полученных по ис
пользуемой БИК методике, с аналитическими
данными метода сравнения. Путем сравнения
стандартной ошибки проrнозирования с MeTO
дом сравнения, использованным для валида
ции, демонстрируется корреляция результатов
БИК методики и аналитических данных метода
сравнения. Для сравнения результатов БИК
методики со стандартными результатами MOryт
при меняться альтернативные статистические
методы (двухсторонний Tec оценка система
тической поrрешности).
Точность (прецизионность). Точность Bыpa
жает степень близости результатов для серии из
мерений при заданных условиях. Она устанавли
вается на не менее 6 измерениях, выполненных в
соответствии с разработанной аналитической Me
тодикой. Точность рассматривается на двух ypOB
нях: сходимость (повторяемость), которая опре
деляется повторными измерениями одноrо и тoro
же образца с изменениями еro положения или
без них, и внутрилабораторная точность (прове
дение измерений различными аналитиками, BЫ
полнение измерений в различные дни).
Робастность. Робастность включает влия
ние изменений температур, влажности, обработки
образца, а также инструментальных изменений.
Выбросы (outliers). Выбросы данных БИК
измерений образца за пределы диапазона rpa
дуировки указывают на необходимость дальней
ших испытаний. Если дальнейшие испытания
2.2.41. Круаовой дихроизм
115
образца соответствующей аналитической MeTO
ДИКОЙ дают значения, укладывающиеся в нормы
спецификации, полученный результат paccмa
тривается как соответствующий спецификации.
Таким образом, выбросы данных БИК измерений
образца MOryт рассматриваться как COOTвeTCTBY
ющие спецификации для испытуемоro образца.
ОЦЕНКА ДЕЙСТВУЮЩЕЙ МОДЕЛИ
Валидированные для использования БИК
модели подверrаются непрерывной оценке
производительности и контролю за валидаци
онными параметрами. При обнаружении He
соответствий необходимо применять KoppeK
тирующие действия. Степень необходимой
ревалидации зависит от при роды изменений.
Ревалидация модели качественноrо анализа
необходима в случае внесения HOBOro веще
ства в библиотеку стандартов и может быть He
обходима, если наблюдаются изменения фи
зических свойств вещества и если изменяется
поставщик материалов. Ревалидация модели
количественноro анализа необходима при из
менении состава roTOBOro продукта, производ
cTBeHHoro процесса и поставщиков/квалифи
кации сырья.
ПЕРЕНОС БАЗЫ ДАННЫХ
При переносе базы данных на друroй прибор
должны быть учтены спектральный диапазон,
количество экспериментальных точек, спек
тральное разрешение и друrие параметры . Для
демонстрации TOro, что модель остается приroд
ной для новой базы данных или новоro прибора,
должны быть проведены последующие валида
ционные процедуры и установлены COOTBeTCTBY
ющие критерии.
ХРАНЕНИЕ ДАННЫХ
Электронные версии БИК спектров, библио
теки и данные сохраняют в соответствии с дей
ствующими правилами.
БИК спектры сохраняют с предварительной
обработкой данных для конкретноro использо
вания (например, для идентификации, анализа
размера частиц, определения содержания воды
и др.) в соответствии с действующими специфи
кациями.
01/2013:20241
2.2.41. круrовой ДИХРОИЗМ
Круroвым дихроизмом называют разность
оптических плотностей в пределах полосы по
rлощения для света с левой и правой круrовой
поляризацией, присущую оптически активным
веществам.
Прямое измерение дает величину:
A == A L AR,
rде:
М оптическая плотность круroвоro дих
роизма;
A L оптическая плотность для света с
левой круroвой поляризацией;
A R оптическая плотность для света с
правой круroвой поляризацией.
Круroвой дихроизм рассчитывают по фор
муле:
дА
Дr:= Е E == ,
l. R с./
rде:
ДЕ молярный круroвой дихроизм или
молярный дифференциальный дихроич
ный коэффициент поmощения, выраженный
л . моль. 1 . см. 1 ;
E L молярный коэффициент поmощения
(2.2.25) для света с левой круrовой поляриза
цией;
E R молярный коэффициент поmощения
для света с правой круrовой поляризацией;
с концентрация вещества в испытуемом
растворе, моль. л. 1 ;
f длина оптическоro пути, см.
Для характеристики KpyroBoro дихроизма
MOryт быть также использованы следующие еди
ницы:
Фактор диссимметрии:
ДЕ
g== .
Е
rдe:
Е молярный коэффициент поrлощения
(2.2.25).
Молярная эллиптичность:
Некоторые типы приборов непосредствен
но показывают величину эллиптичности 0, BЫ
раженную в rрадусах. При использовании таких
приборов молярная эллиптичность [0] может
быть рассчитана по формуле:
] 0.М
L0 == ,
c.f.10
rде:
[0] молярная эллиптичность,
rрадус' см 2 . дмоль. 1 ;
o величина эллиптичности, показывае
мая прибором;
М относительная молекулярная масса ис
пытуемоro вещества;
с концентрация испытуемоro вещества в
растворе, r/мл;
f длина оптическоro пути, см.
116
rосударственная фармакопея Республики Беларусь
Молярная эллиптичность также связана с
молярным круroвым дихроизмом следующим
уравнением:
4500
е == 2,30ЗДЕ ;:; З300ДЕ.
П
Молярная эллиптичность часто использует
ся при анализе белков и нуклеиновых кислот. В
этом случае молярная концентрация, выражен
ная в единицах мономерных остатков, рассчиты
вается, исходя из выражения:
молекулярная масса
число аминокислот
Для белков среднее значение относитель
ной молекулярной массы мономерноro остатка
составляет от 100 до 120 (в среднем 115), для
нуклеиновых кислот (в виде натриевой соли)
около 330.
Прибор. Источником света (S) является Kce
ноновая лампа (рисунок 2.2.41. 1); свет прох
дит через двойной монохроматор (М), снабжен
ный кварцевыми призмами (Р1, Р2).
so
/' '}м
" '" Q , "" < ' ]]
m т ,: c :?: ]:
М4[ ,' ,::: ' --: ::=," СС', , .,g...-"", [}{I-,,: : o"' f""" r Ф
С. Р.М
Рисунок 2.2.41. 1. Оптическая схема
дихроарафа
Линейный луч из первоro монохроматора
расщепляется на два компонента, поляризуе
мые под правым уrлом вторым MOHoxpOMaTO
ром. Дополнительный луч устраняется выходной
щелью монохроматора.
Поляризованный и монохроматический свет
проходит через двоякопреломляющий модуля
тор (Cr); в результате образуется свет с круrовой
поляризацией.
Затем луч проходит через исследуемый
образец (С) и попадает на qpотоумножитель
(Р.М.), за которым следует усилитель, произ
водящий два электрических сиrнала: один по
стоянноro тока V c ' а друroй переменноro тока
с частотой модуляции V ac ' характерной для ис
следуемоro образца. Фаза является показа
телем круroвоro дихроизма. Отношение VjV c
пропорционально дифференциальной оптиче
ской плотности М, которая создается сиrна
лом. Дихроrраф обычно позволяет проводить
измерения в диапазоне длин волн от 170 нм
до 800 нм.
rрадуировка прибора
Точность шкалы оптической плотно
сти. 10,0 Mr изоандростерона Р растворяют в
диоксане Р и доводят до объема 10,0 мл этим
же растворителем. Снимают спектр KpyrOBoro
дихроизма раствора в диапазоне от 280 нм до
360 нм. Величина ДЕ, измеренная в максимуме
при 304 нм, равна +3,3.
Также может быть использован раствор
(1S) (+) 10 камфорсульфоновой кислоты Р.
Линейность модуляции. 10.0 Mr 1(S) (+)
1 О камфорсульфоновой кислоты Р растворяют
в воде Р и доводят до объема 10,0 мл этим же
растворителем. Определяют точную KOHцeHTpa
цию камфорсульфоновой кислоты в растворе Me
тодом спектрофотометрии в ультрафиолетовой
области (2.2.25), используя величину удельноro
показателя поrлощения при 285 нм, равную 1,49.
Снимают спектр KpyroBoro дихроизма paCT
вора в диапазоне от 185 нм до 340 нм. Величина
ДЕ, измеренная в максимуме при 290,5 нм, co
ставляет от +2,2 до +2,5, а измеренная в макси
муме при 192,5 нм ОТ -4,3 до 5.
Может быть использован (1 S) (+) или еro оп
тический изомер (1R) (+) 10 камфорсульфонат
аммония р
01/2013:20242
2.2.42. ПЛОТНОСТЬ ТВЕРДЫХ ТЕЛ
Плотность твердых тел соответствует их
средней массе единицы объема и обычно Bыpa
жается в rpaMMax на сантиметр кубический (r/см З ),
хотя международной единицей является кило
rpaMM на метр кубический (1 r/cM 3 = 1000 кr/м З ).
В отличие от rазов и жидкостей, плот
ность которых определятся только температу
рой и давлением, плотность твердых тел зави
сит также от их структуры и поэтому изменяется
в зависимости от кристаллической структуры и
степени кристалличности.
В том случае, коrда твердые частицы явля
ются аморфными или частично аморфными, их
плотность зависит от способа получения, обра
ботки и хранения.
Поэтому, в отличие от жидкостей, плотно
сти двух химически эквивалентных твердых Be
ществ MOryT быть различными, и это отличие OT
ражает соответствующие различия в строении
твердых тел. Плотность частиц важная физи
ческая характеристика лекарственных средств в
форме порошка.
Плотность твердой частицы может при
нимать различные значения в зависимости
от метода, используемоro для измерения ее
объема. Следует различать три уровня выраже
ния плотности:
2.2.43. Масс спектрометрuя
117
истинная плотность, которая использу-
ется только для твердой фракции материала;
истинную плотность таюке называют кристал-
лической плотностью;
плотность частиц, которая характеризу-
ет таюке объем пор внутри твердых частиц;
объемная ппотность, которая включает
объемы пустот между частицами, образованные
в слое порошка; 'объемную плотность называ-
ют таюке кажущейся плотностью или насыпной
массой" .
ИСТИННАЯ ПЛОТНОСТЬ
Истинная плотность вещества представля-
ет собой отношение массы к объему элементар-
ной ячейки кристалла за исключением объема
пор, которые не являются неотъемлемой частью
кристаллической решетки вещества. Истинная
плотность является свойством, присущим инди-
видуальной кристаллической структуре веще-
ства, и поэтому ее величина не должна зависеть
от способа определения. Истинная плотность
определяется расчетным путем.
Истинную мотность рассчитывают на осно-
вании кристаллоrpaфических данных (размер и
состав элементарной ячейки кристалла), напри
мер, из данных дифракции рентreновскоro излу-
чения монокристалла или путем уточнения кри-
сталлической структуры на основании данных
дифракции рентreновскоro излучения на порошке.
ПЛОТНОСТЬ ЧАСТИЦ
Плотность частиц вещества учитывает как
истинную плотность, так и пористость самой ча-
стицы вещества (закрытые и/или недоступные в
условиях эксперимента открытые поры). Таким
образом, плотность частиц зависит от величины
установленноro объема, который, в свою оче-
редь, зависит от метода измерения. Плотность
частицы вещества может быть установлена
QQНИМ из двух следующих методов.
Пикнометрическая плотность определя-
ется путем измерения объема, занимаемоro из-
вестной массой порошкообразноro вещества, ко- <
торый эквивалентен объему raза, BbITecHeHHoro
веществом, используя пикнометр raзовоro вы-
теснения (2.9.23). Объем вещества, установлен-
ный таким способом, не включает объем, зани-
маемый открытыми порами, однако он включает
объем закрытых пор или пор, недоступных для
raза. Блаroдаря высокой диффузионной актив-
ности reлия, который является наиболее пред-
почтительным для определения raзом, большин-
ство открытых пор доступны для raза. Поэтому
пикнометрическая плотность тонко измельчен-
HOro порошка, в общем, не сильно отличается от
значений истинной плотности. Таким образом,
с помощью пикнометрической плотности можно
оценить истинную плотность аморфных или ча-
стично кристаллических образцов, и поэтому
данный способ широко применим для образцов
лекарственных средств в форме порошков.
Плотность, устанавливаемая ртутным
порозиметром, таюке называемая аранулярной
плотностью. Объем, определяемый этим мето-
дом, включает объем, занимаемый закрытыми
пора ми или порами, недоступными для ртути,
однако объем открытых пор включается только в
случае, если их размер меньше HeKoтoporo пре-
дела. Этот предел размера пор или минимально
доступный диаметр зависит от давления, nQQ ко-
торым подается ртуть во время измерения; при
обычных условиях измерения ртуть не прони-
кает в мельчайшие поры, доступные для reлия.
Для OДHoro образца MOryт быть получены раз-
личные значения rpанулярной мотности, так
как возможно определение мотности для каж-
доro используемоro рабочеro давления, что со-
ответствует различным значениям предельных
размеров пор при различных давлениях.
ОБЪЕМНАЯ ПЛОТНОСТЬ И ПЛОТНОСТЬ
ПОСЛЕ УСАДКИ
Плотность при свободной засыпке порош
ка учитывает вклад свободноro объема между
частицами порошка. Следовательно, плотность
при свободной засыпке зависит как от мотности
частиц порошка, так и от их npocTpaHcTBeHHoro
расположения в слое порошка.
Плотность при свободной засыпке зачастую
очень сложно измерить с хорошей воспроизво-
димостью, так как самые незначительные нару-
шения в слое MOryт приводить к новому значению
плотности. Таким образом, важным требовани-
ем при сообщении значений насыпной плотно-
сти является указание метода ее определения.
Объемную плотность и плотность после
усадки определяют, как указано в статье
2.9.34. Объемная плотность и плотность
после усадки.
01/2013:20243
2.2.43. МАСС-СПЕКТРОМЕТРИЯ
Масс-спектрометрия основана на прямом
измерении отношения массы к числу элемен-
тарных положительных или отрицательных за
рядов ионов (т/z), полученных из анализируе-
MOro вещества и находящихся в raзовой фазе.
Данное отношение выражается в атомных еди-
ницах массы (1 а. е. м. равна одной двенадцатой
массы нуклида 12С) или в даль тонах (1 Да равен
массе атома водорода).
Ионы, образовавшиеся в ионизаторе прибо-
ра, ускоряются и перед попаданием в детектор
разделяются с помощью масс-внвлизатора. Все
эти действия происходят в камере, в которой на-
сосная система поддерживает вакуум от 10-3 Па
до 10.6 Па.
118
rосударственная фармакопея Республики Беларусь
Результирующий масс--спектр является rpa
фиком зависимости относительноro количества
различных ионов от отношения m/z. Сиrнал,
отвечающий иону, представлен несколькими
пиками, соответствующими статистическому
распределению различных изотопов этою иона.
Такая диаrрамма называется изотопным про
филем, а пик (по крайней мере, для небольших
молекул), представляющий наиболее распро
страненные изотопы для каждою атома, MO
ноизотопным пиком.
Масс спектрометрический анализ дает
важную качественную (определение молекуляр
ных масс; информация, касающаяся структуры
определяемых фраrментов) и количественную
(с использованием внешнеro или внутреннеro
стандартов) информацию с пределом обнаруже
ния от пикомоля до фемтомоля.
ВВОД ОБРАЗЦА
Первой стадией анализа является ввод об
разца в прибор без значительноro нарушения
вакуума. В широко распространенном методе,
называемом прямым вводом жидкости, об
разец помещается на конце цилиндрическоro
штока (в кварцевом тиrле, на проволоке или на
металлической поверхности). Этот шток через
вакуумный шлюз, в котором поддерживается
первичный промежуточный вакуум между aTMOC
ферным давлением и вторичным вакуумом при
бора, вводится в спектрометр.
Друrие системы ввода позволяют проанали
зировать компоненты смеси, которые разделя
ются с помощью соответствующеro прибора, co
единенноrо с масс спектрометром.
rазовая хроматоrрафия/масс спектромет-
рия. При использовании подходящих колонок
(капиллярных или полукапиллярных) возможно
непосредственное введение конца колонки в ио
низатор прибора без применения сепаратора.
Жидкостная хроматоrрафия/масс-спектро-
метрия. Такая комбинация особенно полезна
для анализа полярных соединений, которые яв
ляются недостаточно летучими либо слишком
термолабильными для тoro, чтобы их можно
было проанализировать методом rаЗQ.80Й xpOMa
тоrрафии в сочетании с масс спектрометрией.
Данный метод осложняется трудностью получе
ния ионов в rазовой фазе из жидкой фазы, что
требует применения специальных интерфейсов,
таких как:
прямой ввод жидкости: подвижная фаза
распыляется, и растворитель испаряется перед
ионизатором прибора;
интерфейс с пучком частиц: подвиж
ная фаза, скорость которой может достиrать
0,6 мл/мин, распыляется в десольватационной
камере, в результате в ионизатор прибора по
падают лишь анализируемые вещества в ней
тральной форме; данный способ может быть
использован для соединений с относительно
низкой полярностью и молекулярными массами
меньше 1000 Да;
интерфейс С движущейся лентой: под
вижная фаза, скорость которой может достиraть
1 мл/мин, наносится на поверхность движущей
ся ленты; после выпаривания растворителя aHa
лизируемые вещества переносятся к ионизатору
прибора. rдe подверrаются ионизации; данный
способ мало продуктивен для очень полярных
или термолабильных соединений.
Друrие виды интерфейсов (электрораспы
ление. термораспыление, химическая иони
зация при атмосферном давлении) MOryT pac
сматриваться как самостоятельные методы
ионизации и описаны в разделе, посвященном
способам ионизации.
Сверхкритическая флюидная xpOMaTO
rрафия/масс-спектрометрия. Подвижная
фаза, обычно состоящая из находящеrося в
сверхкритическом состоянии диоксида уrлеро
да, переходит в rазообразное состояние после
прохождения через наrpeтую заслонку, находя
щуюся между колонкой и ионизатором.
Капиллярный электpoфopeзlмасс-спектро--
метрия. Элюент вводится в ионизатор, в HeKOTO
рых случаях после добавления дополнительноro
растворителя для тoro, чтобы достичь CKOpO
сти потока порядка нескольких миллилитров в
минуту. Оrраничениями данноrо метода являют
ся малые количества вводимоro образца и He
обходимость использования летучих буферных
растворов.
СПОСОБЫ ИОНИЗАЦИИ
Электронный удар. Образец, находящий
ся в rазообразном состоянии, ионизируется по
током электронов, энерrия которых (обычно
70 эВ) больше энерrии ионизации образца. При
этом кроме молекулярноro иона М+ образуются
осколочные ионы, характерные для данной MO
лекулярной структуры. rлавным оrраничением
данноro способа является необходимость ис
парения образца, что делает невозможным еro
применение для полярных, термолабильных
или высокомолекулярных соединений. Иониза
ция электронным ударом может быть использо
вана в rазовой хроматоrрафии, соединенной с
масс спектрометрией, а в некоторых случаях и в
жидкостной хроматоrрафии.
Химическая ионизация. Этот тип иониза
ции предполаrает использование peareHTH'Jro
rаза, TaKoro как метан, аммиак, монооксид азота,
диоксид азота или кислород. Спектр характери
зуется ионами (М + Н)+ или (М НУ типов или
ионами аддуктами, образованными из анали
та и используемоro rаза. Осколочные ионы об
разуются реже, чем при ионизации электрон
ным ударом. Для термолабильных веществ
2.2.43. Масс пектрометрuя
119
используется разновидность данною метода
ионизации, при которой образец, находящийся
на проволоке, очень быстро испаряется вслед
ствие эффекта Джоуля Томсона (десорбцион
ная химическая ионизация).
Бомбардировка быстрыми aTOMa
ми (FAB) или ионизация бомбардиров
кой быстрыми ионами (жидкостная Macc
спектрометрия вторичных ионов LSIMS).
Образец, растворенный в вязкой матрице (Ha
пример, в rлицерине), наносится на металличе
скую поверхность и ионизируется потоком ней
тральных атомов, например, aproHa, ксенона
или обладающими большой кинетической энер
rией ионами цезия. Образуются ионы (М + НУ
или (М H) типов либо ионы аддукты, сформи
рованные матрицей или образцом. Данный тип
ионизации хорошо подходит для полярных, Tep
молабильных соединений, имеющих молекуляр
ную массу до 1 О 000 Да. При прибавлении к под
вижной фазе 1 2 % rлицерина он может быть
использован для жидкостной хроматоrрафии,
однако скорость подвижной фазы должна быть
очень низкой (несколько микролитров в минуту).
При нанесении тонкоro слоя матрицы на по
верхность хроматоrрафических пластинок такой
способ ионизации может быть использован и в
тонкослойной хроматоrрафии.
Полевая десорбция или полевая иони-
зация. Образец испаряется около вольфрамо
вой проволоки, покрытой микроиrлами (полевая
ионизация) или помещается на эту проволо
КУ (полевая десорбция). Сильное электриче
ское поле (напряжение около 10 кВ), возника
ющее между проволокой и противоэлектродом,
ионизирует образец. При данных способах ио
низации образуются преимущественно молеку
лярные ионы М+ и (М + Н)+ ионы. Эти способы
используются для малополярных и/или TepMO
лабильных соединений.
Матрично-активированная лазерная дe <
сорбция/ионизация (MALDI). Образец, CMe
шанный с подходящей матрицей и помещенный
на металлическую подложк ионизируется им
пульсным лазерным излучением с длиной волны
от Уф до ИК диапазона (продолжительность
импульсов может составлять от пикосекунды
до нескольких наносекунд). Данный способ ио
низации иrрает важную роль при анализе co
единений с очень большой молекулярной массой
(более 100000 Да), но оrраничен использовани
ем времяпролетноro анализатора (см. ниже).
Электрораспылительная ионизация.
Данный способ ионизации проводится при ат
мосферном давлении. Образец, находящийся в
растворе, вводится в источник через капилляр,
конец котороro имеет потенциал порядка 5 кВ.
Для облеrчения распыления может использо
ваться raз. Испарение молекул растворителя из
образующихся микрокапель приводит к образо
ванию в rазовой фазе однозарядных или мноro
зарядных ионов. Скорости подвижной фазы при
данном виде ионизации MOryт изменяться от
нескольких микролитров в минуту до 1 мл/мин.
Такая ионизация подходит для полярных соеди
нений, а также для исследования биомолекул
с молекулярными массами до 100 000 Да. Она
может сочетаться с жидкостной хроматоrрафией
или капиллярным электрофорезом.
Химическая ионизация при атмосфер-
ном давлении (APCI). Ионизация проводит
ся при атмосферном давлении под действи
ем электрода, имеющеro потенциал несколько
киловольт и помещенноro на пути подвижной
фазы, которая распыляется как вследствие T
пловых эффектов, так и блаroдаря использова
нию потока азота. Образующиеся ионы являют
ся однозарядными и относятся к (М + ну типу В
случае положительноro заряда и к (М Н)- типу
в случае отрицательноro. Возможность исполь
зования высоких скоростей подвижной фазы (до
2 мл/мин) делает этот способ ионизации идеаль
ным для сочетания с жидкостной хроматоrpафией.
Термораспылительная ионизация. Обра
зец, находящийся в подвижной фазе, состоящей
из воды и орrанических модификаторов и coдep
жащей летучий электролит (обычно ацетат aM
мония), вводится В распыленной форме после
прохождения через металлический капилляр,
температура котороro контролируется. Допу
скаются скорости подвижной фазы порядка
1 мл/мин 2 мл/мин. Ионы электролита ионизи
руют анализируемое соединение. Такой процесс
ионизации может быть заменен или усилен элек
трическим разрядом величиной около 800 вольт,
особенно при использовании только орrаниче
скоro растворителя. Данный способ ионизации
может быть использован в методе жидкостной
хроматоrрафии с масс спектрометрией.
АНАЛИЗАТОРЫ
Различия в работе анализаторов зависят,
rлавным образом, от двух пара метров:
диапазона, в котором MOryт быть измере
ны отношения m/z (массовый диапазон);
разрешающей способности, характеризу
ющейся возможностью разделить два иона оди
наковой интенсивности с отношениями m/z, OT
личающимися на f1M и перекрывающиеся при
определенном процентном превышении базо
вой линии; например, разрешающая способ
ность (M/f1M), равная 1000 с 10 % превышени м
базовой линии, позволяет провести разделение
отношений m/z 1000 и 1001 с интенсивностями,
на 10 % превышающими базовую линию. В He
которых случаях (времяпролетные анализато
ры, квадрупольные анализаторы, ионные ло
вушки) разрешающая способность может быть
120
rосударственная фармакопея Республики Беларусь
определена как отношение молекулярной массы
к ширине пика на половине высоты (50 % превы
шение базовой линии).
Маrнитные и электростатические анали-
эаторы. Ионы, образовавшиеся в ионизаторе,
ускоряются потенциалом V и сосредотачивают
СЯ, в зависимости от устройства прибора, между
маrнитным (маrнитное поле В) или электроста
тическим анализаторами (электростатическое
поле Е). В соответствии с законом Лапласа они
перемещаются по траектории с радиусом r.
т В 2 ,2
............= .
z 2V
Для накопления и измерения различных
ионов, образовавшихся в ионизаторе, MOryT быть
использованы два типа сканирования: измене-
ние В при постоянной величине V или измене
ние V при постоянной величине В. за маrнитным
анализатором обычно следует электрический
сектор, который действует как фильтр кинетиче--
ской энерrии и позволяет заметно увеличить раз
решающую способность прибора. Максимальная
разрешающая способность тaKOro прибора (двой
ной сектор) изменяется от 1 О 000 до 150 000 и
в большинстве случаев позволяет достаточно
точно оценить величину отношений mIZ для опре
деления элементноro состава соответствующих
ионов. Для однозарядных ионов массовый диа
пазон составляет от 2000 Да до 15 000 Да. Не-
которые ионы MOryт разрушаться спонтанно (Me
тастабильные переходы) или при столкновении
с raзом (диссоциация, активированная столкно-.
вением (CAD» в oбnастях между ионизатором и
детектором, rде отсутствует поле. Исследование
этих процессов разрушения очень полезно для
определения структуры и характеристики опреде--
ленноro соединения, входящеro в состав смеси,
и делает возможным использование тандемной
маcc-cnектрометрии. В зависимости от места, в
котором протекают процессы разрушения, из
вестно MHOro методик ero проведения:
режим дочернеао иона (определение co
става ионов, образовавшихся при разрушении
определенноro родительскоro иона): ВlE = KOH
станта, MIKES (Mass4lпalysed 10п Kiпetic Eпergy
Spectroscopy);
режим родительскоао иона (определе
ние всех ионов, образовавшихся при разруше
нии иона с определенным отношением m/z):
ВЦЕ = константа;
режим нейтральных частиц (определе--
ние всех ионов, которые теряют идентичные
фраrменты): ВlE(1 - Е/Е о )"2 = константа, rде Ео
базовый потенциал электрическоro сектора.
Кввдрупольные анализаторы. Анализа
тор состоит из четырех параллельных металли-
ческих стержней, имеющих цилиндрическое или
rиперболическое поперечное сечение. Они рас-
положены симметрично по отношению к TpaeK
тории движения ионов; пары противоположных
стержней присоединены к электрическому ис
точнику. Потенциалы двух пар стержней противо
положны. Они состоят из постоянноro и изменя
ющеroся компонентов. Ионы, образовавшиеся в
ионизаторе, пропускаются и разделяются вслед
ствие изменения потенциалов, приложенных к
стержням, причем отношение постоянноro по
тенциала к переменному должно OCTaBaTb
ся неизменным. Квадрупольные анализаторы
обычно имеют массовый диапазон от 1 а. е. м.
до 2000 а. е. м., хотя в некоторых случаях он
может достиraть 4000 а. е. м. Несмотря на то,
что такие анализаторы имеют меньшее разре
шение, чем маrнитные, они позволяют получать
моноизотопный профиль однозарядных ионов
во всем массовом диапазоне. Для получения
спектров можно использовать три квадруполя,
соединенных друr Сдруroм, О,, 02' Оз (02 служит
ионизационной камерой и в действительности
не является анализатором; чаще всеro в каче-
стве ионизирующеro raза применяют aproH).
Наиболее часто используемыми режимами
сканирования являются:
режим дочернеао иона: 01 выбирает mlz
ион, фраrменты котороro, полученные при иони-
зации в квадруполе 02' анализируются квадру-
полем оз;
режим родительскоао иона: Оз пропуска
ет ионы только с определенным соотношени
ем m/z, в то время как О, сканирует определен-
ный массовый диапазон. Детектируются только
ионы, при разрушении которых образуется ион,
отбираемый квадруполем ОЗ;
режим нейтральных частиц: 01 и Оз CKa
нируют определенный массовый диапазон, но в
интервале, соответствующем фраrментам, KOTO
рые теряет определенное вещество или rруппа
веществ.
Для получения спектров возможно сочета
ние квадрупольных анализаторов с маrнитными
или электростатическими. Такие приборы назы
вают аибридными масс пектрометрами.
Ионные ловушки. Данные анализаторы
имеют такой же принцип работы, что и квадру-
польные, но в них используются трехмерные
электрические поля. Анализаторы TaKoro типа
позволяют получать спектры ионов нескольких
reнераций (MS").
Циклотронно-резонансные масс-анали-
заторы. Ионы, образовавшиеся в ячейке и под
Beprнyтыe действию интенсивноro маrнитноro
поля, движутся по KpyroBbIM траекториям с ча
стотами, которые MOryт быть непосредствен
но связаны с величинами отношений m/z для
этих ионов при помощи преобразования Фурье.
Данное явление называется ионно-циклотрон-
ным резонансом. Анализаторы TaKoro типа со-
стоят из сверхпроводящих маrнитов и обладают
2.2.44. Определение содержания общеzо орzаническоzо уzлерода
121
очень высокой разрешающей способностью (до
1 000 000 и выше), а также позволяют получать
MSn спектры. Их недостатком является необхо
димость использования очень rлубокоrо вакуума
(порядка 10 7 Па).
Времяпролетные анализаторы. Ионы, об
разовавшиеся в ионизаторе, ускоряются потен
циалом величиной от 10 кВ до 20 кВ. Они прохо
дят через анализатор, состоящий из бесполевой
трубы длиной от 25 см до 1,5 м, обычно называ
емой пролетной трубой. Время (t), за которое
ион достиrает детектора, пропорционально KBa
дратному корню из отношения m/z. Теоретиче
ски массовый диапазон для такоro анализатора
бесконечен. Практически он определяется MeTO
дом ионизации или десорбции. Времяпролетные
анализаторы используются, rлавным образом,
для высокомолекулярных соединений (вплоть
до нескольких сотен тысяч дальтон). Данный
анализатор обладает очень высокой чувстви
тельностью (достаточно несколько пикомолей
вещества). Точность измерений и разрешающая
способность таких приборов MOryт быть значи
тельно улучшены при использовании электро
статическоro зеркала (рефлектрона).
ПОЛУЧЕНИЕ сиrНАЛА
Существует три возможных режима получе
ния сиrнала.
Режим полноrо спектра. Записывает
ся полный сиrнал, полученный для выбранно
ro массовою диапазона. Спектр представляет
собой зависимость относительной интенсивно
сти различных ионов от величины m/z. Получа
емые результаты являются rлавным образом
качественными. Для более быстрой идентифи
кации возможно использование библиотек спек
тров сравнения.
Фраrментометрический режим (селек-
тивное сканирование ионов). Получаемый
сиrнал оrраничен одним (сканирование одноrо
иона, sing/e ion топitоriпg (S/M» или нескольки
ми (множественное ионное сканирование, mu/ti
ple jon топitоriпg (М/М» ионами, характерными
для анализируемоro вещества. В этом режиме
предел обнаружения значительно ниже. При ис
пользовании внутреннеro или внешнеrо CTaH
дартов (например, дейтерированные CTaHдap
ты) MOryT быть проведены количественные или
полуколичественные измерения. При использо
вании времяпролетных анализаторов такие из
мерения невозможны.
Двойной фраrментометрический масс-
спектрометрический режим (множественный
мониторинr реакций, multiple reactioп топi
toring (МRМ». Включает специфическую MOHO
молекулярную или бимолекулярную реакцию
разрушения выбранною иона предшествен
ника, характерноro для анализируемоro веще
ства. Селективность и высокая специфичность
данноrо режима получения сиrнала обеспечи
вают превосходную чувствительность и делают
ero наиболее подходящим для количествен
ных определений с использованием подходя
щих внутренних стандартов (например, дейте
рированных). Данный вид анализа может быть
проведен только при использовании приборов,
снабженных тремя соединенными квадруполя
ми, ионными ловушками или циклотронно резо
нансными анализаторами.
rРАДУИРОВКА
rрадуировка позволяет установить COOT
ветствие между величиной m/z и детектируе
мым сиrналом. Как правило, она проводится с
использованием вещества сравнения. rрадуи
ровка может быть внешней (данные находятся
в отдельном файле) или внутренней (вещество
(вещества) сравнения смешивается с исследуе
мым веществом и результаты вносятся в файл с
данными анализа). Число ионов или точек, He
обходи мое для надежной rрадуировки, зависит
от типа анализатора и требуемой точности из
мерений, например, в случае маrнитноrо анали
затора, rдe отношение m/z экспоненциально из
меняется при изменении величины маrнитноrо
поля, необходимо брать настолько мноro точек,
насколько это возможно.
ОБНАРУЖЕНИЕ сиrНАЛА И ОБРАБОТКА
ДАННЫХ
Ионы, разделенные анализатором, преоб
разуются в электрические сиrналы такими дe
тектирующими системами, как фотоумножитель
или электронный умножитель. Затем данные
сиrналы усиливаются и превращаются в циф
ровые сиrналы, которые используются при об
работке данных и позволяют проводить различ
ные операции: rрадуировку, получение спектров,
автоматические количественные расчеты, архи
вирование данных, создание или использование
библиотек масс спектров. Различные физиче
ские параметры, требуемые для работы прибо
ра, в целом контролируются компьютером.
01/2013:20244
2.2.44. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ
ОБЩЕrо оРrАНИЧЕскоrо
уrЛЕРОДА В ВОДЕ ДЛЯ
ФАРМАЦЕВТИЧЕскоrо
ПРИМЕНЕНИЯ
Определение содержания общеro op
rаническоro уrлерода (ООУ, ТОС, tota/ o'ga
nic carbon) является непрямым методом опре
122
rосударственная фармакопея Республики Беларусь
деления содержания орrанических веществ
в воде для фармацевтическоro применения.
Определение содержания ООУ может также
использоваться при контроле выполнения раз
личных операций в производстве лекарствен
ных средств.
Поскольку для определения содержания
ООУ MorYT применяться различные методи
ки, в данной статье приведены не описания
методик, а их квалификация и интерпретация
результатов в предельных испытаниях. Ис
пытания раствора сравнения проводят через
определенные интервалы времени в зависи
мости от частоты измерений; раствор rотовят с
использованием леrкоокисляющейся субстан
ции (например, сахарозы) с такой KOHцeHTpa
цией, чтобы отклик прибора соответствовал
измеряемому пределу содержания ООУ При
roдность системы проверяют с использовани
ем трудноокисляющейся субстанции (напри
мер, 1 ,4 бензохинона).
Разные типы приборов для определения
ООУ в воде для фармацевтическоro примене
ния, как правило, предназначены для полно
ro окисления орrанических молекул в образ
це воды до уrлерода диоксида с последующим
измерением ero количества, используемоro
затем для расчета концентрации уrлерода в
воде.
Используемый прибор в процессе работы
может определять орrанический и неорrани
ческий уrлерод (неорrанический уrлерод при
сутствует в виде карбонатов). Это может обе
спечиваться путем определения количества
неорrаническоrо уrлерода и вычитанием еro из
количества общеro уrлерода или удалением He
орrаническоro уrлерода из образца при помощи
продувания перед окислением. В процессе про
дувания из испытуемоro образца MorYT yдa
ляться и орrанические молекулы, но часть
связанноro с ними уrлерода в воде для фарма
цевтическоro применения незначительна.
Прибор. Используют отrрадуированный
прибор, установленный в режим «oп liпe» или
«автономно». Приrодность системы прове
ряют, как описано ниже, через определенный
промежуток времени. Прибор должен иметь
предел определения уrлерода 0,05 мr/л или
менее, в соответствии с паспортом изroтови
теля прибора.
Вода для определения содержания ООУ.
Используют воду высокоочищенную, COOTBeT
ствующую следующим требованиям:
электропроводность: не более 1 ,О MкCM'CM 1
при температуре 25 ос;
содержание общеro орrаническоro уrле
рода: не более 0,1 мr/л.
В зависимости от типа используемоro при
бора критическим параметром может быть
также содержание в воде тяжелых металлов
или меди, что должно быть указано в инструк
ции изroтовителя прибора.
Под20товка посуды. Используют посуду,
тщательно вымытую с помощью метода, позво
ляющеrо удалить орrанические вещества. Для
последнеro промывания используют воду для
определения содержания ООУ.
Раствор сравнения. Сахарозу Р, предва
рительно высушенную при температуре 105 ос
в течение 3 ч, растворяют в воде для определе
ния содержания ООУ, получая раствор, coдep
жащий 1,19 мr/л сахарозы (0,50 мr/л уrлерода).
Испытуемый раствор. Испытуемую воду
собирают. исключая минимальное воздушное
пространство. в воздухонепроницаемый KOH
тейнер, используя все возможные меры для
предотвращения заrрязнения. Испытание про
водят с минимальной задержкой по времени
с целью уменьшения возможноro заrрязнения
воды от контейнера и еro укупорочноro MaTe
риала.
Раствор для проверки при20дности cи
стемы. rотовят раствор 0,75 мr/л 1.4 бензо
хинона Р в воде для определения содержания
ООУ (0,50 мr/л уrлерода).
Контрольная вода для определения coдep
жания ООУ. Используют воду для определения
содержания ООУ, полученную одновременно с
водой для приroтовления раствора сравнения
и раствора для про верки приroдности системы.
Контрольные растворы. Кроме контроль-
ной воды для определения содержания ООУ
roтовят подходящие холостые растворы или
друrие растворы, необходимые для BOCCTa
новления базовой линии или корректирования
rрадуировки в соответствии с инструкцией из
rотовителя прибора; используя контрольные
растворы, устанавливают ноль прибора.
Проверка при20дности системы. Прово
дят испытания указанных растворов и записы
вают отклики лрибора: вода для определения
содержания ООУ (rJ, раствор сравнения ('s)'
раствор для проверки при20дности cиcтe
мы ('s). Эффективность откликов, в процентах,
рассчитывают по формуле:
rss rw .100.
rs rw
Система считается приroдной, если эф
срективность отклика прибора составляет не
менее 85 % и не более 115 % от теоретическо
ro отклика.
Методика. Записывают отклик (r) для ис
пытуемоrо раствора. Испытуемый раствор BЫ
держивает испытание, если значение ru не пре
вышает значение r r .
s w
Данная методика может быть выполнена
в режиме «оп-liпе» на приборе, который COOT
ветствующим образом отrрадуирован и СООТ-
BeTcTByeT требованиям приroдности системы.
Прибор должен быть установлен таким образом,
чтобы обеспечить показания откликов, COOTBeT
ствующих испытуемой воде.
01/2013:20245
2.2.46. Хроматоzрафuческие методы разделения
123
2.2.45. СВЕРХКРИТИЧЕСКАЯ
ФЛЮИДНАЯ
ХРОМАТОrPАФИЯ
Сверхкритическая флюидная xpoMaTorpa
фия (СФХ, SFC) это метод хроматоrрафиче
скоro разделения, в котором подвижная фаза
является флюидом, находящимся в сверхкрити
ческом или субкритическом состоянии. Непод
вижная фаза, содержащаяся в колонке, cocтo
ит либо из тонко измельченных твердых частиц,
таких как силикаrель или пористый rpафит; хи
мически модифицированная неподвижная фаза
такая же, как и в жидкостной хроматоrpафии,
или, в случае капиллярных колонок, пленка
жидкости, равномерно нанесенная на стенки KO
лонки.
СФХ основана на механизмах адсорбции
или распределения.
ПРИ БОР
Прибор обычно состоит из охлаждаемой
насосной системы, инжектора, хроматоrрафи
ческой колонки, находящейся в термостате,
детектора, реryлятора давления и реrистри
рующеro устройства (интеrратор или самопи
сец).
Насосная система
Насосная система необходима для подцер
жания постоянной скорости подвижной фазы.
Колебания давления должны быть сведены к ми
нимуму, например, путем пропускания сжатоro
растворителя через устройство, подавляющее
пульсацию. Насосно компрессорная система и
соединительные линии должны быть устойчивы
к давлению, создаваемому насосом.
Точная доставка подвижной фазы при по
стоянных либо изменяющихся по определен
ной проrрамме условиях осуществляется систе
мами, контролируемыми микропроцессором. В
случае rрадиентноrо элюирования MOryT быть
использованы насосные системы, доставляю
щие растворитель (растворители) из нескольких
резервуаров; смешивание растворителей может
быть проведено как со стороны насоса (Haco
сов) низкоro давления, так и со стороны BЫCOKO
ro давления.
Инжекторы .
Ввод пробы может быть осуществлен непо
средственно в колонку с помощью клапана.
Неподвижные фазы
Неподвижные фазы находятся в колонках,
которые описаны в статьях 2.2.29. Жидкостная
хромат02рафия (набивные колонки) и 2.2.28.
rазовая хромат02рафия (капиллярные колон
ки). Максимальный внутренний диаметр (0) Ka
пиллярной колонки 100 мкм.
Подвижные фазы
Обычно подвижной фазой является диоксид
уrлерода, который может содержать полярный
модификатор, такой как метанол, 2 пропанол
или ацетонитрил. Состав, давление (плотность),
температура и скорость используемой подвиж
ной фазы MOryт быть как постоянными в теч
ние всеro хроматоrрафическоro процесса (из
кратическое, изотермическое элюирование,
элюирование при постоянной плотности), так и
изменяться по определенной проrрамме (rpади
ентное элюирование модификатора, давления
(плотности), температуры или скорости потока).
Детекторы
Наиболее часто используемыми дeTeктopa
ми являются спектрофотометры, работающие в
ультрафиолетовой и видимой областях (UVN;s)
и пламенно ионизационные детекторы. Кроме
тоro, MOryт при меняться детекторы, работа KO
торых основана на измерении рассеяния света,
ИК спектрофотометры, катарометры или друrие
специальные детекторы.
МЕТОДИКА
Испытуемый раствор (растворы) и раствор
(растворы) сравнения roтовят так, как описано
выше. Растворы не должны содержать твердых
частиц.
Критерии для оценки приrодности систе
мы были описаны в статье 2.2.46. XpOMaтo
2рафические методы разделения. В данной
rлаве были также приведены уровни, до которых
можно реryлировать пара метры хроматоrрафи
ческой системы для тоro, чтобы она оставалась
приroдной.
01/2013:20246
2.2.46. ХРОМАтоrРАФИЧЕСКИЕ
МЕТОДЫ РАЗДЕЛЕНИЯ
Хроматоrрафическими называют мнС?roста
дийные методы разделения, в которых компо
ненты образца распределяются между двумя
фазами неподвижной и подвижной. Непод
вижная фаза может быть твердым веществом,
жидкостью, нанесенной на твердый носитель,
либо rелем; она помещается в колонку, распре
деляется в виде тонкоro слоя, пленки и т. д. Под
вижная фаза может быть rазсм, жидкостью или
сверхкритическим rазом (флюидом). Разделение
основано на адсорбции, распределении, ионном
обмене и т. д. либо на различиях в физико хи
мических свойствах молекул, таких как размер,
масса, объем и т. д.
Данная rлава содержит определения и pac
четы общих параметров и применимых ко всем
124 rосударственная фармакопея Республики Беларусь
ПИК 1 Пик2
ro
Q.
О
1;:
Q)
....
Q)
q
:.:
:s:
....
о
V M
( м
V R1
t R1
V R2
( ю
N
wh :t:
Объем (v)
Время (t)
Рисунок 2.2.46. 1.
хроматоrрафическим методам требований для
приrодности системы. Принципы разделения,
аппаратура и методики приводятся в следующих
общих статьях:
Бумажная хромат02рафия (2.2.26);
Тонкослойная хромат02рафия (2.2.27);
rазовая хромат02рафия (2.2.28);
Жидкостная хромат02рафия (2.2.29);
Эксклюзионная хромат02рафия (2.2.30);
Сверхкритическая флюидная xpOMaтo
2рафия (2.2.45).
ОПРЕДЕЛЕНИЯ
Для установления при20дности xpOMaтo
2рафической системы и расчета пределов в
частных статьях использованы приведенные
ниже определения. Ряд параметров (например,
отношение си2нал/шум и разрешение) может
быть рассчитан с помощью Пр02раММН020 обе
спечения, поставляеМ020 производителем иc
пользуеМ020 оборудования. На пользователе
лежит ответственность за соответствие
способов расчета, используемых в Пр02рамм
ном обеспечении, требованиям Фармакопеи. В
противном случае должны быть сделаны co
ответствующие корректировки.
XpoMaTorpaMMa
XpOMaTorpaMMa представляет собой rpa
фическое или иное представление отклика дe
тектора на концентрацию веществ в элюате или
друroй количественной величины, используемой
для измерения концентр=щии веществ в элюате,
относительно времени или объема. Идеализи
рованные xpoMaTorpaMMbI представлены соче
танием rауссовских пиков и базовой линии.
Пик
Участок xpoMaTorpaMMbI с записанным от
кликом детектора при элюировании из колонки
одноro компонента (или двух и более неразде
ленных компонентов).
Пик может быть охарактеризован площа
дью пика, или высотой пика (h) и шириной
пика на половине высоты (w h ), или высотой
пика (h) и шириной пика между точками пе
реrиба (wJ Для rауссовских пиков (рисунок
2.2.46. 1) справедливо соотношение:
W h o=1,18w j .
Время удерживания (t R )
Время, необходимое для элюирования KOM
понента (рисунок 2.2.46. 1, rдe базовая линия
время).
Объем удерживания (V R )
Объем подвижной фазы, необходимый для
элюирования компонента. Он может быть pac
считан из времени удерживания (в минутах) и
скорости подвижной фазы (F) (в миллилитрах в
минуту) по формуле:
V R o=tR.F.
Мертвое время (t M )
Время, необходимое для элюирова
ния неудерживаемоrо компонента (рисунок
2.2.46. 1, rдe базовая линия время). В
эксклюзионной хроматоrрафии используют
символ ( о (см. ниже).
Мертвый объем (V M )
Объем подвижной фазы, необходимый для
элюирования неудерживаемоro компонента. Он
может быть рассчитан из «MepTBoro» времени (в
минутах) и скорости подвижной фазы (F) (в мил
лилитрах в минуту) по формуле:
VMo=tM.F.
2.2.46. Хроматоzрафические методы разделения
125
в эксклюзионной хроматоrрафии использу
ют символ V o (см. ниже).
Фактор удерживания (k)
Фактор удерживания (также известный как
концентрационный коэффициент распределе
ния (От) или коэффициент емкости (k) пред
ставляет собой соотношение:
k = количество вещества в неподвижной фазе К ,
количество вещества в подвижной фазе с V..
rдe:
Кс коэффициент распределения;
V s объем неподвижной фазы;
V м объем подвижной фазы.
Значение фактора удерживания компонен
та может быть определено из xpoMaTorpaMMbI по
формуле:
k== tR tM ,
t M
Общее время подвижной фазы (t,)
В эксклюзионной хроматоrрафии время
удерживания компонента, молекулы котороro
меньше, чем размер мельчайшей поры rеля (ри
сунок 2.2.46. 2).
Iю'Vт
(\)
Q.
О
Х
L. I!!
;s;
х
;;Е
о
tR1/\I R1
Io/V"
u: u:
t1:I :s:
:1:-&
:1: t1:I
00.
:s: L..
м О
Q
r:;t1:I
'"
UO
"'о.
(1)х
Время (t)
Объем (v)
107
1O t '8
rn
"
Ю !Е
lO
'"
C::
Ю
Ic/'VQ"""
............... 1,'11,
Рисунок 2.2.46. 2.
Общий объем подвижной фазы (V,)
в эксклюзионной хромаТ(lrрафии объем
удерживания компонента, молекулы котороro
меньше, чем размер мельчайшей поры rеля.
ОН может быть рассчитан из общею времени
подвижной фазы (в минутах) и скорости под
вижной фазы (F) (в миллилитрах в минуту) по
формуле:
v, == t, . F .
Время удерживания неудерживаемоrо
компонента (t o )
В эксклюзионной хроматоrрафии время
удерживания компонента, молекулы котороro
больше, чем размер наибольшей поры rеля (ри
сунок 2.2.46. 2).
Объем удерживания неудерживаемоro
компонента (V o )
в эксклюзионной хроматоrрафии объем
удерживания компонента, молекулы KOTOpO
ro больше, чем размер наибольшей поры rеля.
Он может быть рассчитан из времени удержива
ния неудерживаемоro компонента (в минутах) и
скорости подвижной фазы (F) (в миллилитрах в
минуту) по формуле:
V o == t o . F _
Коэффициент распределения (КО)
В эксклюзионной хроматоrрафии элюент
ные свойства компонента в определенной KO
лонке MOryт быть представлены коэффициен
том распределения, который рассчитывают по
формуле:
К tR to
о .
t, to
Фактор подвижности (R F >
Фактор подвижности (также известный как
относительная скорость или фактор удержива
ния (R,», используемый в плоскостной xpOMaTO
rрафии, представляет собой отношение paCCTO
яния от точки нанесения пробы до центра пятна
к расстоянию. пройденному фронтом растворите
ля от точки нанесения пробы (рисунок 22.46. 3).
ь
R F = ,
а
O B
а
Ь
A .
C
Рисунок 2.2.46. З.
А фронт подвижной фазы;
В пятно;
С линия нанесения пробы ("линия старта").
126
rосударственная фармакопея Республики Беларусь
rдe:
Ь расстояние, пройденное компонентом;
а расстояние, пройденное фронтом pac
творителя.
Число тарелок (н)
Эффективность колонки в зависимости от
метода может быть paCC'-lитана как число таре---
лок (также известное как число теоретических Ta
релок) из данных полученных в зависимости от
методики как при изотермическом либо изокра
тическом режиме, так и при режиме постоянной
плотности. по формуле, в которой величины t R И W h
должны быть выражены в одинаковых единицах:
N 5,54 ( ;J 2,
rдe:
t R время удерживания пика, COOTвeTCTBY
ющеro компоненту;
W h ширина пика на половине высоты.
Число теоретических тарелок зависит как от
компонента, так и от используемой колонки и ее
температуры, а также от подвижной фазы и Bpe
мени удерживания.
Объемrрадиентнойзадержки(D)
Объем rрадиентной задержки #(объем
формирования rрадиента в хроматоrрафиче
ской системе)# это объем между точками,
при которых элюенты достиrнут конца колон
ки. Он может быть определен с использовани
ем следующей методики.
Условия хроматоrрафирования:
колонка: хроматоrрафическую колонку
заменяют подходящей капиллярной трубкой
(например, длиной 1 м и внутренним диаме
тром 0,12 мм);
подвижная фаза:
подвижная фаза А: вода Р;
. подвижная фаза В: 0,1 % (об/об) paCT
вор ацетона Р;
,
Подвижная Подвижная
Время (мин) фаза А фаза В
(%, об/об) (%, об/об)
0 20 1 00 о о 100
20 30 О 100
скорость подвижной фазы: устанавлива
ют до получения достаточноro давления (напри
мер, 2 мл/мин);
детектор: спектрофотометрический,
длина волны 265 нм.
Определяют время (t 05 ) (в минутах), при KO
тором оптическая плотность увеличится на 50 %
(рисунок 2.2.46,А).
о t o . F ,
rдe:
t o представляет собой разницу t O . 5 0,5t G
(в минутах);
t G время rрадиента (20 мин);
F скорость ПОд.ва1жной фазы.
100% ш_шш_ш___ш_; __
.D
r-
u
О
I
r-
О
1:;
с
о::
ro
::.::
U
Ф
:т
:s:
r-
с
.0
50%-ш_ш-
t O . 5
Время
Рисунок 2.2.46.-4.
Фактор асимметрии (А)
Фактор асимметрии пика (также извест
ный как коэффициент или фактор симметрии)
(рисунок 2.2.46. 5) рассчитывают по формуле:
w
A ,
s 2d
rдe:
W O . 05 ширина пика на одной двадцатой еro
высоты;
d расстояние между перпендикуляром,
опущенным из максимума пика, и передним
краем пика на одной двадцатой высоты.
Если фактор ассиметрии равен 1,0, это
означает полную (идеальную) симметрию.
Если As > 1,0, это означает, что пик с размы
ты м тылом; если As < 1,0 пик с размытым
фронтом.
.....d
WO,05
Рисунок 2.2.46. 5.
2.2.46. Хроматосрафuческие методы разделения
1.27
Разрешение (Rs)
Разрешение между пиками Дf3YX компонентов
(рисунок 2.2.46. 1) рассчитывают по формуле:
R =о 1,18 (t R2 tR1) ,
s w h1 +W h2
rдe:
t R2 > t R1
t R1 И t ю времена удерживания пиков;
W h1 и W h2 ширины пиков на половине
высоты.
В количественной плоскостной xpoMaTorpa
фии с использованием денситометрии вместо
времен удерживания используются пройденные
расстояния, и разрешение между пиками двух
комrюнентов рассчитывают по формуле:
R =о 1,18а (R F2 RF1) .
s
W h1 +W h2
rдe:
R F1 и R F2 факторы подвижности пиков;
W h1 и W h2 ширина пиков на половине
высоты;
а расстояние, пройденное фронтом pac
творителя.
Коэффициент разделения пиков (p/v)
Коэффициент разделения не полностью
разделенных пиков (или p/v napaMeTp) может
быть использован в качестве критерия приroд
ности хроматоrрафической системы при выпол
нении испытания на сопутствующие примеси в
случае неполноrо разделения (не до базовой
линии) двух пиков (рисунок 2.2.46. 6).
Нр
p/v == ,
Hv
Нр
Hv
Рисунок 2.2.46. 6.
rде:
Н высота меньшеro пика CYrНOCIofТепьНQ
р
экстраполированной базовой линии;
Hv расстояни между экстраnoлироваlТ
ной базовой линией. и ни ей точкой кривой,
разделяющей меньш й и больший пики.
Ohносительноеу рживание(ry
Относительное удерживание (,) рассчиты
вают по формуле: !
, t Ri tM
[== ,
,t Rst tM
rде:
( А; время удерживания интересующеro
пика;
t RsI время удерживания пика сравнения
(обычно пик, соответствующий испытуемому Be
ществу);
t M мертвое время.
Неоткорректированное относительное yдep
живание ('G) рассчитывают по формуле:
t R
r G == .
t Rst
Относительное удерживание, указанное
в частных статьях, соответствует HeOTKoppeK
тированному относительному удерживанию,
если нет друrих указаний.
В плоскостной хроматоrрафии вместо t Rs /
и t Ri используются факторы подвижности R Fst
И R Fi .
Отношение сиrнал/шум (5/Н)
Отношение сиrнал/шум влияет на точность
(прецизионность) количественноro определения
и рассчитывается по формуле:
S/N=o 2Н ,
h
rде:
Н высота пика (рисунок 2.2.46. 7), COOT
ветствующая рассматриваемому компоненту,
на xpoMaTorpaMMe, полученной при использо
вании рекомендованноro раствора сравнения,
измеренная от максимума пика до экстрапо
лированной базовой линии для сиrнала, Be
личина KOToporo эквивалентна пятикратному
превышению величины ширины пика на поло
вине высоты;
h размах для фоновоro шума (уровень
шума) на xpoMaTorpaMMe, полученной при BBe
дении или нанесении холостой пробы, величи
на KOTOpOro эквивалентна пятикратному пре
вышению ширины пика на половине высоты
для пика на xpoMaTorpaMMe, полученной при
использовании рекомендованноro раствора
сравнения и, если возможно, расположенноro
на одном и том же расстоянии от места воз
можноrо обнаружения пика.
128
rосударственная фармакопея Республики Беларусь
н
Рисунок 2.2.46. 7.
Сходимость (повторяемость)
Сходимость (повторяемость) отклика
выражается в виде рассчитанною процент
ною относительноro стандартною отклоне
ния (s"%» последовательных серий измере
ний не менее чем 3 введений или нанесений
раствора сравнения и рассчитывается по
формуле:
S,(%)= 100 L(Y; у)2
У п 1
rдe:
У; индивидуальные значения площади
пика, высоты пика или отношения площадей в
методе внутреннеro стандарта;
у среднее индивидуальных значений;
п число индивидуальных значений.
приrодность ХРОМАтоrРАФИЧЕСКОЙ
СИСТЕМЫ
Используемое оборудование (в том числе
отдельные компоненты) должно быть при
roдным и способным выдерживать требова
ния к приrодности системы, необходимые для
проведения испытания или количественноro
определения.
Испытания на определение приюдности
системы являются неотъемлемой частью Me
тодики и используются для тою, чтобы yдo
стовериться в адекватном функциониро
вании хроматоrрафической системы. Для
оценки работы колонки обычно используют
ся следующие параметры: эффективность,
фактор удерживания (концентрационный KO
эффициент распределения), разрешение,
относительное удерживание и фактор асим
метрии.
На хроматоrрафическое поведение MorYT
влиять следующие факторы:
состав, ионная сила, температура и рН
подвижной фазы;
скорость потока, размеры колонки,
температура колонки и давление;
характеристика неподвижной фазы:
тип хроматоrрафической основы (может
представлять собой частицы либо быть
монолитной), размер частиц или макропор,
пористость, удельная площадь поверхно
сти;
химическая модификация поверхно
сти неподвижной фазы (обращенно фазовая
и друrие модификации), степень химиче
ской модификации (блокирование концевых
rрупп (эндкепирование), число атомов уrле
рода и т. д.).
При отсутствии друrих указаний должны
быть соблюдены следующие требования и
любые друrие дополнительные требования,
описанные в частной статье.
В испытании на сопутствующие приме
си и при количественном определении вели
чина фактора асимметрии пика, полученною
на xpoMaTorpaMMe раствора сравнения, ис
пользуемою для количественных расчетов,
должна находиться в пределах от 0,8 до 1,5,
если в частной статье нет иных указаний.
При количественном определении
фармацевтической субстанции, при количе
ственном содержании 100 % для чистой суб
станции, максимально допустимое относи
тельное стандартное отклонение (S,(%» дЛЯ
установленных пределов рассчитывают из
серии введений раствора сравнения по фор
муле:
о KB.Jп
s,(Yo)max = ,
(90%Л 1
rдe:
к константа (0,349), полученная из ypaB
06 ( 9О О; 5 06
нения К = . , в котором требуемое
12 -J6 -J2
значение относительноro стандартноro отклоне
ния (в процентах) при 6 повторных вводах пробы
для В = 1,0;
В верхний предел количественноro co
держания, приведенный в определении част
ной статьи минус 100 %;
п число повторных вводов пробы для
раствора сравнения (3 :::; п :::; 6);
(90 %.п 1 коэффициент Стьюдента ( для
90 % ной доверительной вероятности (ДBY
сторонняя критическая область) с числом
степеней свободы п 1.
Если иноrо не указано в частной статье,
максимально допустимое относительное
стандартное отклонение не должно пре
вышать величин, приведенных в таблице
2.2.46. 1. Данное требование не распростра
няеТСfl на испытания на сопутствующие при
меси.
2.2.46. ХроматО2рафические методы разделения
129
в испытании на сопутствующие приме
си предел определения пика (соответствующий
отношению сиrнал/шум равному 10) равен или
меньше, чем неучитываемый предел.
Таблица 2.2.46. 1.
Требования к сходимости (повторяемости)
I Число отдельных вводов пробы
j 3 4 5 6
В(%) Максимально допустимое относи-
тельное стандартное отклонение
2,0 0,41 0,59 0,73 0,85
2,5 0,52 0,74 0,92 1,06
3,0 0,62 0,89 1,10 1,27
Соответствие критериям приroдности систе
мы является необходимым в течение всеro про
цесса хроматоrрафирования. В зависимости от
различных факторов, таких как частота исполь
зования методики, опыта и знания xpoMaTorpa
фической системы, аналитик подбирает подхо
дящую схему верификации для их контроля.
РЕrYЛИРОВАНИЕ
ХРОМАтоrРАФИЧЕСКИХ УСЛОВИЙ
Ниже приведены интервалы, в пределах KO
торых MOryT изменяться различные параметры
хроматоrрафических испытаний без принципи
альноro изменения методики, для тоro, чтобы
удовлетворять критериям приroдности xpOMaTO
rрафической системы. Реryлирование условий
хроматоrрафирования при rрадиентном элюи
ровании более критичны, чем при изократиче
ском элюировании, и MOryт привести к смеще
нию пиков на различных уровнях rрадиента, что
в свою очередь приведет к неверному YCTaHOB
лению пиков, маскированию пиков или к такому
смещению, при котором элюирование будет про
исходить вне предписанноro времени элюиро
вания. Друrие, не описанные ниже изменения,
требуют ревалидации методики. Описанные
хроматоrрафические условия были валидирова
ны в процессе подrотовки этой статьи.
Проверка приrодности хроматоrрафической
системы включена для тоro, чтобы rарантировать
требуемое разделение для удовлетворительно
ro проведения испытания или количественно
ro определения. Неподвижные фазы описаны
только в общем виде, так как существует orpoM
ное количество их доступных коммерческих раз
новидностей, отличающихся по хроматоrрафи
ческому поведению, и для тоro, чтобы достиrнуть
предписанных требований приrодности систе
мы. в ряде случаев приходится вносить HeKOTO
рые изменения в хроматоrрафические условия.
В частности, в меТQдиках обращенно фазовой
хроматоrpафии реryлирование условий xpOMa
тоrрафирования не Bcerдa приводит к удовлет
ворительному разделению. В этом случае может
возникнуть необходимость заменить одну колон
ку друrой однотипной колонкой (например, сили
каrель октадецилсилильный), обеспечивающей
J7 30. 1712
желаемое хроматоrрафическое поведение. Ин
формацию о хроматоrрафических колонках, ис
пользованных для разработки частных статей,
описанных в Европейской Фармакопее, можно
найти на сайте ЕООМ (www.edqm.eu) в разделе
«Kпow/edge database».
Реryлирование критических параметров для
rарантии приrодности системы четко указывает
ся в частной статье.
Тонкослойная и бумажная xpoMaTorpa-
фия
Состав подвижной фазы: количество pac
творителя, содержание котороro в смеси мини
мально, может изменяться в пределах :t30 %
(относительное содержание) или :t2 % (абсо
лютное содержание), 8 зависимости от тоro, что
больше. Например, для растворителя, содержа
ние котороro в смеси минимально и составляет
10 %, относительное изменение в 30 % допуска
ет предельные значения 7 13 %. а абсолютное
изменение в 2 % допускает предельные значе
ния 12 %, т. е. изменение количества paCTBO
рителя по относительному содержанию больше;
для растворителя, содержание котороro в смеси
минимально и составляет 5 %, относительное
изменение в 30 % допускает предельные значе
ния 3,5 ,5 %, а абсолютное изменение в 2 %
допускает предельные значения 3 7 %, т. е.
изменение количества растворителя по абсо
лютному содержанию больше. Абсолютное co
держание друrих компонентов не может быть из
менено более чем на 1 О %.
рН водН020 компонента подвижной фазы:
:t0,2 рН при отсутствии друrих указаний в част
ной статье, или :t1,0 рН в случае исследования
неионизированных веществ.
Концентрация солей в буферном компо
ненте подвижной фазы: :t10 %.
Наносимый объем пробы: 1 0 20 % от Tpe
буемоro объема при использовании пластинок с
мелким размером частиц (2 10 мкм).
Жидкостная хроматоrрафия: изократиче-
ское элюирование
Состав подвижной фазы: количество pac
творителя, содержание котороro в смеси мини
мально, может изменяться в пределах :t30'% (OT
носительное содержание) или:t2 % (абсолютное
содержание), в зависимости от тоro, что больше
(см. пример выше). Абсолютное содержание
друrих компонентов не может быть изменено
более чем на 10 %.
рН водН020 компонента подвижной фазы:
:tO,2 рН при отсутствии друrих указаний в част
ной статье, или :t1,0 рН в случае исследования
неионизированных веществ.
Концентрация солей в буферном компо
ненте подвижной фазы: :t10 %.
Скорость подвижной фазы: :t50 %; боль
шие отклонения допустимы в случае изменения
размеров колонки (см. формулу ниже).
130
rосударственная фармакопея Республики Беларусь
Характеристики колонки:
Неподвижная фаза:
не допускаются изменения в назван.1И
химическоro модификатора неподвижной
фазы (т. е. не допускается замена С'8 на Са);
размер частиц: допускается YMeHb
шение размера на 50 %. увеличение не дo
пускается.
Размер колонки:
длина: :1:70 %;
внутренний диаметр: :1:25 %.
Если изменяется размер колонки. то
требуемая скорость подвижной фазы может
быть определена по формуле:
F F 1 2 d;
2 " d 2 .
1 1
rдe:
F , скорость подвижнои фазы, YKa
занная в частной статье, в миллилитрах в
минуту;
F 2 измененная скорость, в миллили
трах в минуту;
/1 длина колонки, указанная в частной
статье, в миллиметрах;
'2 длина используемой колонки, в
миллиметрах;
d, внутренний диаметр колонки, YKa
занный в частной статье. в миллиметрах:
d 2 внутренний диаметр используемой
колонки, в миллиметрах.
Температура: :t10 ос при отсутствии друrих
указаний в частной статье.
Длина волны детектора: изменения не дo
пускаются.
Объем вводимой пробы: может быть
уменьшен при условии, что используемое дe
тектирование и сходимость пика (пиков) OCTa
ются удовлетворительными; увеличение не дo
пускается.
Жидкостная хроматоrрафия: rрадиент-
ное элюирование
Реryлирование хроматоrрафических yc
ловий при rрадиентном элюировании требует
большей осторожности, чем при изократическом
элюировании.
Состав подвижной фаЗЫ/2радиентН020
элюирования: минимальные изменения состава
подвижной фазы и rрадиента допустимы при BЫ
полнении следующих условий:
соответствие требованиям приroдности
хроматоrрафической системы;
отклонение от указанноro в частной статье
времени удерживания OCHOBHOro пика (или
пиков) не должно превышать :t15 %;
элюирующая сила конечноro состава под
вижной фазы должна быть не ниже, чем у пред
писанноro состава.
В случае, если не удается достичь критери
ев приrодности хроматоrрафической системы,
зачастую рассчитывают объем задержки или
меняют хроматоrрафическую колонку.
Объем задержки. Конфиryрация используе
Moro оборудования может значительно изменить
разрешение, время удерживания или относитель
ные удерживания, описанные в методике. К таким
эффектам может привести избыточный объем за
держки. В частных статьях обычно указывается
изократическая стадия, предшествующая началу
rрадиентноro элюирования, поэтому различие
в объеме задержки для системы, используемой
при разработке частной статьи, и для фактически
используемой системы можно откорректировать
изменением временных точек проrраммы rради
ента. Таким образом. должна проводиться адап
тация продолжительности изократической стадии
в зависимости от используемоro аналитическо
ro оборудования. Если 8 частной статье приве
ден объем задержки, установленный при ее раз
работке, то временные точки (t. мин), указанные
в таблице rрадиента, MOryт быть заменены на
адаптированные временные точки (t c ' мин), pac
считанные по формуле:
(O Oo)
t c = t F
rдe:
D объем задержки, в миллилитрах;
00 объем задержки, использованный при
разработке частной статьи, в миллилитрах;
F скорость подвижной фазы, в миллили
трах в минуту.
Изократическая стадия, введенная для этих
целей, может быть удалена при наличии данных
по валидации методики без указанной стадии.
рН водН020 компонента подвижной фазы:
изменения не допускаются.
Концентрация солей в буферном компо
ненте подвижной фазы: изменения не допуска
ются.
Скорость подвижной фазы: отклонения дo
пустимы в случае изменения размеров колонки
(см. формулу ниже).
Характеристики колонки:
Неподвижная фаза:
не допускаются изменения в названии
химическоro модификатора неподвижной
фазы (т. е. не допускается замена С ,в на СВ);
размер частиц: изменения не допу
скаются.
Размер колонки:
длина: :t70 %;
внутренний диаметр: :1:25 %.
Если изменяется размер колонки, то
требуемая скорость подвижной фазы может
быть определена по формуле:
F =F I2d ;
2 1 I d 2 '
1 1
rдe:
2.2.46. Хроматоzрафическuе методы разделения
131
F 1 скорость подвижной фазы, YKa
занная в частной статье, в миллилитрах в
минуту;
F 2 измененная скорость, в миллили
трах в минуту;
/1 длина колонки, указанная в частной
статье, в миллиметрах;
'2 длина используемой колонки, в
миллиметрах;
d, внутренний диаметр колонки, YKa
занный в частной статье, в миллиметрах;
d, внутренний диаметр используемой
колонки, в миллиметрах.
Температура: :t5 ос при отсутствии друrих
указаний в частной статье.
Длина волны детектора: изменения не дo
пускаются.
Объем вводимой пробы: может быть YMeHb
шен при условии, что используемое детектиро
вание и сходимость пика (пиков) остаются YДOB
летворительными; увеличение не допускается.
rазовая хроматоrрафия
Характеристики колонки:
Неподвижная фаза:
размер частиц: допускается YMeHb
шение размера на 50 %, увеличение не дo
пускается (набивные колонки);
толщина слоя: от 50 % до +100 %
(капиллярные колонки).
Размер колонки:
длина: :t70 %;
внутренний диаметр: :t50 %.
Скорость потока: :t50 %.
Температура: :t10 %.
Вводимый объем пробы: может быть изме
нен при условии, что используемое детектиро
вание и повторяемость остаются удовлетвори
тельными.
Сверхкритическая флюидная xpOMaTO
rрафия
Состав подвижной фазы: для набивных KO
лонок количество растворителя, содержание KO
торою в смеси минимально, может изменяться
в пределах :t30 % (относительное содержание)
или:t2 % (абсолютное содержание), в зависимо
сти от Toro, что больше. Для капиллярных коло
нок изменения не допускаются.
Длина волны детектора: изменения не дo
пускаются.
Характеристики колонки:
Неподвижная фаза:
размер частиц: допускается YMeHb
шение размера на 50 %, увеличение не дo
пускает (набивные колонки);
Размер колонки:
длина: :t70 %;
внутренний диаметр:
:t25 % (набивные колонки);
:t50 % (капиллярные колонки).
Скорость потока: :t50 %.
Температура::t5 0 С.
Объем вводимый пробы: может быть YMeHb
шен при условии, что используемое детектиро
вание и повторяемость остаются удовлетвори
тельными; увеличение не допускается.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
При количественном определении не учиты
вают пики растворителей и реактивов, а также
пики подвижной фазы и матрикса испытуемоrо
образца.
Чувствительность детектора. Чув
ствительность детектора определяется ero BЫ
ходным сиrналом на концентрацию или едини
цу массы вещества, попадающею на детектор.
Относительный фактор отклика детектора, или
коэффициент отклика, выражает чувствитель
ность детектора к данному веществу относи
тельно стандартноro вещества. Поправочный
коэффициент обратно пропорционален коэф
фициенту отклика.
Метод внешне20 стандарта. KOHцeHTpa
цию анализируемоro компонента (компонентов)
определяют путем сравнения отклика (откликов)
детектора (пика (пиков)), полученноro для испы
TyeMoro раствора, и отклика (откликов) дeTeK
тора (пика (пиков)), полученноro для раствора
сравнения.
Метод внутренне20 стандарта. В ис
пытуемый раствор и раствор сравнения вводят
одинаковые количества компонента (BHYTpeH
нею стандарта), который разделяется с иссле
дуемым веществом. Внутренний стандарт не
должен взаимодействовать с исследуемым Be
ществом, должен быть стабильным и не co
держать примесей со временем удерживания,
соответствующим исследуемому веществу. KOH
центрацию исследуемоrо вещества определя
ют путем сравнения отношения площадей или
высот пиков, соответствующих исследуемому
веществу и внутреннему стандарту для испы
туемоro раствора и отношения площадей или
высот пиков, соответствующих исследуемому
веществу и внутреннему стандарту для paCT
вора сравнения.
Методика нормализации. Процентное
содержание компонента исследуемоr9 веще
ства рассчитывают путем определения площа
ди соответствующеro пика в процентах от общей
площади всех пиков, исключая пики, COOTBeT
ствующие растворителям, реактивам, пикам
подвижной фазы и матрикса испытуемоro образ
ца, а также пики, содержание которых равно или
ниже неучитываемоro предела.
Методика 2радуировки. Определяют связь
между измеренным или рассчитанным сиrна
лом (у) и количеством (концентрацией, массой
и т. д.) исследуемою вещества (х) и рассчиты
вают уравнение rрадуировочной функции. AHa
ли:rические результаты рассчитывают из изме
ренноro или рассчитанною сиrнала с помощью
обратной функции.
132
rосударственная фармакопея Республики Беларусь
в испытаниях на сопутствующие примеси
с применением, как метода внешнеro cTa 'f1ap
та с использованием разведенною испытуемс ro
раствора, так и методики нормализации, исполь
зуют поправочные коэффициенты, приведенные
в частной статье (т. е. коrда коэффициент откли
ка выходит за рамки 0,8 1 ,2).
Korдa в испытании на сопутствующие при
меси указывается сумма примесей или прово
дится количественное определение отдельной
примеси. очень важно выбрать COOTBeTCTBY
ющие настройки для предела определения и
подходяtЦие условия для интеrрирования пло
щадей пиков. В таких испытаниях неучитывае
мый предел, т. е. предел, при котором и ниже KO
торою пики не учитываются, обычно составляет
0,05 %. Таким образом, предел определения в
системе обработки данных должен COOTBeTCTBO
вать не менее чем половине значения неучиты
ваемоro предела.
Интеrрирование площади пика любой при
меси, которая не полностью разделяется с oc
новным пиком, лучше всею проводить экстрапо
ляцией по нижним точкам пика (по касательной).
01/2013:20247
2.2.47. КАПИЛЛЯРНЫЙ
ЭЛЕКТРОФОРЕЗ
ОБЩИЕ ПРИНЦИПЫ
Капиллярный электрофорез это физиче
ский метод анализа, основанный на миrрации
внутри капилляра заряженных аналитов, pac
творенных в растворе электролита, под влияни
ем постоянною электрическою поля.
Скорость миrрации аналита под влиянием
электрическоrо поля с напряженностью Е опре
деляется электрофоретической подвижностью
аналита и электроосмотической подвижностью
буферноro раствора внутри капилляра. Элек
трофоретическая подвижность вещества (jJ ер)
зависит от ero свойств (электрический заряд,
размер и форма молекул) и от свойств буфер
ноro раствора, в котором происходит процесс
миrрации (тип и ионная сила электролита, зна
чение рН, вязкость и наличие добавок). Электро
форетическая скорость (v e ) вещества, частицы
котороro принимаются за сферические, описы
вается уравнением:
V =р 'E= ( ) ' ( V ) ,
ер ер 6ттr fJ L
rдe:
q эффективный заряд вещества;
r Стоксовский радиус частиц вещества;
fJ вязкость раствора электролита;
V приложенное напряжение;
L общая длина капилляра.
При помещении капилляра, заполненно
ro буферным раствором, в электрическое поле
внутри капилляра начинается перемещение pac
творителя, называемое электроосмотическим
потоком. Скорость электроосмотическоro потока
зависит от электроосмотической подвижности
(jJeo)' которая, в свою очередь, зависит от плот
ности заряда на внутренней стенке капилляра и
свойств буферноro раствора. Электроосмотиче
ская скорость (V e ) описывается уравнением:
V eo = Рео. Е =( &/} ( }
rдe:
Е; диэлектрическая постоянная буферно
юраствора;
дзета потенциал поверхности капилляра.
Скорость вещества (v) определяется как:
v == v ер + v ей'
в зависимости от заряда вещества элек
трофоретическая подвижность аналита и элек
троосмотическая подвижность MoryT быть Ha
правлены одинаково или противоположно. В
условиях нормальною капиллярноro электро
фореза анионы перемещаются в направлении,
противоположном направлению электроосмо
тическоrо потока, а их скорости меньше элек
троосмотической скорости. Катионы миrрируют
в направлении, совпадающем с направлени
ем электроосмотическоro потока, а их скорости
превышают электроосмотическую скорость. В
условиях, коrда электроосмотическая скорость
превышает электрофоретическую, катионы и
анионы MOryT быть разделены в течение одноro
анализа.
Время (t), необходимое веществу для ми
rрации на расстояние (1) от конца капилляра, в
который вводится вещество, до точки детекции
(эффективная длина капилляра), определяется
уравнением:
t=
Vep+V eo
/.L
( Рер + р ео ) . V
в общем, при значении рН более 3 капил
ляры с немодифицированной поверхностью, из
roтовленные из плавленоro кварца, имеют отри
цательный заряд, обусловленный ионизацией
силанольных rрупп, расположенных на BHYTpeH
ней стенке капилляра. Соответственно, электро
осмотический поток направлен от анода к катоду.
Если необходимо получить хорошую воспроиз
водимость скорости миrрации веществ, электро
осмотический поток должен оставаться постоян
ным. В некоторых случаях требуется уменьшить
или устранить электроосмотический поток путем
2.2.47. Капиллярный электрофорез
1ЗЗ
модификации внутренней стенки капилляра или
изменения концентрации, состава и/или рН бу
ферноro раствора.
После введения испытуемоro образца
в капилляр каждый ион аналита, входящеro
в состав пробы, миrрирует.в среде фоново
ro электролита соrласно своей электрофоре
тической подвижности как независимая зона.
Дисперсия зоны, представляющая собой pac
ширение полосы каждоro вещества, являет
ся следствием различных явлений. При иде
альных условиях вклад в процесс расширения
зоны вещества вносит только молекулярная
диффузия вещества вдоль капилляра (про ,
дольная диффузия). В этом идеальном случае
эффективность зоны, выражаемая как число
теоретических тарелок (N), определяется как:
( ep + eJv./
N== ,
2.O.L
rдe:
D коэффициент молекулярной диффузии
вещества в буферном растворе.
На практике значительный вклад в дис
персию полосы вносят процессы тепловой
конвекции, адсорбции образца на стенках Ka
пилляра, а также неодинаковая проводимость
между образцом и буферным раствором,
длина устройства для ввода пробы, размер
ячейки детектора и расположение емкостей с
буферными растворами на разных уровнях.
Разделение двух полос (выражаемое как
разрешение, Rs) может быть достиrнуто при
изменении электрофоретической подвижно
сти аналитов либо электроосмотической под
вижности, а также при увеличении эффектив
ности полосы для каждоro аналита, соrласно
уравнению:
R == .JN(J1ePb Рера) ,
s 4 (д ер + Р ео )
rдe:
Р ера и Jl epb электрофоретические подвиж
ности двух разделяемых аналитов;
Рер средняя электрофоретическая под
вижность двух аналитов Рер == (р ерЬ + Jl ера)-
2
ПРИБОР
Прибор для капиллярноro электрофореза
состоит из:
управляемоro высоковольтноro источника
постоянноro тока;
двух рl3зервуаров с буферными paCTBopa
ми, расположенных на одном и том же уровне и
содержащих предписанные анодный и катодный
растворы;
двух электродов (катода и анода), поrру
женных в резервуары с буферными растворами
и соединенных с источником тока;
1В.3ак. 1712.
капилляра, в котором проводится раз
деление (обычно изroтовленноrо из плавлено
ro кварца); при использовании некоторых типов
детекторов капилляр имеет оптическое окошко,
расположенное на уровне детектора; концы Ka
пилляра помещены в резервуары с буферными
растворами; капилляр заполнен раствором, YKa
занным в частной статье;
подходящей системы ввода пробы;
детектора, способноro контролировать KO
личество определяемых веществ, проходящих
через разделяющий cerMeHT капилляра в течение
определенноro времени; детектирование обычно
основано на абсорбционной спектрофотометрии
(в ультрафиолетовой и видимой областях) или
флуориметрии, в ряде случаев может быть ис
пользовано также кондуктометрическое, ампе
рометрическое или масс спектрометрическое
детектирование; альтернативным способом дe
тектирования веществ, которые не поrлощают
УФ излучение и не флуоресцируют, является He
прямое детектирование;
для получения хорошо воспроизводимых
результатов разделения рекомендуется исполь
зовать термостат, способный поддерживать по
стоянную температуру внутри капилляра;
устройства записи и походящеro интеrра
тора или компьютера.
Описание процесса ввода пробы и еro aB
томатизация являются критическими фактора
ми для точноro количественноro анализа. Ввод
пробы может быть основан на использовании
силы тяжести, давления, вакуума и электроки
нетических сил. Количество каждоro компонен
та образца, введенноro электрокинетически, за
висит от еro электрокинетической подвижности,
что приводит К возможным неравным условиям
для разных веществ при использовании данноro
режима ввода пробы.
Используют капилляр, буферные paCTBO
ры, способ пробоподrотовки и создания условий
анализа, раствор пробы и условия миrрации,
указанные в частной статье для испытуемо
ro образца. Применяемый раствор электроли
та фильтруют для удаления твердых частиц и
деrазируют для предотвращения образования
пузырьков, мешающих работе детек.тора и соз
данию электрическоro контакта в капилляре во
время процесса разделения. Для обеспечения
воспроизводимых значений времени миrрации
веществ для каждой аналитической методики
должна быть разработана методика тщательной
промывки системы.
КАПИЛЛЯРНЫЙ ЗОННЫЙ ЭЛЕКТРОФОРЕЗ
ПРИНЦИП
В капиллярном зонном электрофорезе aHa
литы разделяются в капилляре, содержащем
только буферный раствор без антиконвектив
ной среды. В данном методе разделение проис
ходит вследствие TOro, что различные компонен
134
rосударственная фармакопея Республики Беларусь
ты образца миrрируют с различной скоростью в
виде отдельных полос. Скорость перемеu,ения
каждой полосы зависит от электрофоретичеu< )Й
подвижности вещества и электроосмотическоrо
потока в капилляре (см. Общие принципы). Для
улучшения разделения веществ, адсорбирую
щихся на кварцевой поверхности, MOryT быть
использованы капилляры с модифицированной
поверхностью.
При использовании данной разновидности
капиллярною электрофореза может быть ocy
ществлен анализ как малых (М, < 2000), так и
больших молекул (2000 < М, < 100 000). Блаroда
ря высокой эффективности зонноro капиллярно
ro электрофореза в свободном растворе может
быть проведено разделение молекул, имеющих
очень незначительные различия в величинах OT
ношений заряда к массе. При добавлении к раз
деляющему буферному раствору хиральных
селекторов можно разделять хиральные соеди
нения.
ОПТИМИ3АЦИЯ
Оптимизация разделения является ком-
плексным процессом, в котором основную роль
MOryT иrрать несколько параметров разделения.
Основными факторами, которые следует прини-
мать во внимание при разработке методик раз
деления, являются инструментальные параме-
тры, а также параметры раствора электролита.
Инструментальные параметры
Напряжение. При оптимизации при клады-
ваемоro напряжения и температуры капилляра
важно учитывать HarpeB, возникающий при воз
действии электрическоro тока. Время разделе
ния обратно пропорционально приложенному
напряжению. Однако увеличение напряжения
может вызвать избыточное выделение тепла, по
вышение температуры и, как результат, образо-
вание rрадиентов вязкости в буферном paCT80
ре, находящемся в капилляре. Данный эффект
при водит к расширению полосы и уменьшению
разрешения.
Полярность. Полярность электродов
может быть нормальной (анод на входе и катод
на выходе), электроосмотический поток при
этом перемещается по направлению к катоду.
В случае обращенной полярности электро
дов электроосмотический поток направлен от
выхода из капилляра, и только заряженные aHa
литы с электрофоретическими подвижностями,
превышающими величину электрофоретическо
ro потока, попадают к выходу.
Температура. Температура влияет, rлав
ным образом, на вязкость и электрическую
проводимость и, как следствие, на скорость
миrрации. В некоторых случаях увеличение
температуры капилляра может вызвать KOH
формационные изменения в молекулах белков,
что изменяет их времена миrрации и эффек
тивность разделения.
Капилляр. Размеры капилляра (длина и BHY
тренний диаметр) влияют на время анализа, эф
фективность разделения и заrрузочную емкость.
Увеличение как эффективной, так и общей
длины капилляра может с:,пабить электриче
ское поле (в случае работы пр.1 постоянном Ha
пряжении), что приводит к увеr,ичению времени
миrрации. Для определенноro буферноro paCT
вора и электрическою поля тепловая KOHBeK
ция и, следовательно. расширение полос образ
ца заВИС jТ от величины внутреннеro ;::.иаметра
капилляра. Кроме этоro, величина внутреннеro
диаметра капилляра влияет на преД6Л обнару
жения, зависящий от объема введенной пробы и
используемоro детектора.
Поскольку адсорбция компонентов образ
ца на стенке капилляра влияет на эффектив
ность, при разработке методики разделения
должны быть предложены способы предотвра
щения таких взаимодействиЙ. В некоторых слу
чаях, касающихся белков, были предложены
способы, позволяющие избежать адсорбции на
стенке капилляра. Некоторые из таких спосо
бов (использование экстремальных значений рН
и адсорбция положительно заряженных буфер
ных добавок) для предотвращения адсорбции
белков требуют лишь изменения состава буфер
ной смеси. В друrих способах внутренняя стенка
капилляра покрывается полимером, ковалентно
связанным с поверхностью, что предотвращает
взаимодействия между белками и отрицатель
но заряженной ilОБерХI--!ОСТЬЮ кварца. Для этих
целей выпускаются roтавые к использованию
капилляры с покрытиями, состоящими из ней
тральных rидрофильных, К3ТИШiНЫХ или анион
ных полимеров.
Параметры раствора электролита
Тип и концентрация буфеРН020 раствора.
Буферные растворы, подходящие для капилляр
ноro электрофореза, имеют соответствующую
буферную емкость в выбранном диапазоне рН
и низкую подвижность для уменьшения образо
вания тока.
Подбор соответствующей подвижности бу
ферноro иона по отношению к подвижности ис
пытуемоro вещества важен для уменьшения
возможноro изменения полосы. Тип раствори
теля, использованноro для растворения опреде
ляемоro вещества, также важен для достижения
фокусирования вещества на колонке, что увели
чивает эффективность разделения и улучшает
детектирование.
Увеличение концентрации буферноro раст
вора (для данноro значения рН) уменьшает элек
троосмотический поток и скорость вещества.
Значение рН буферН020 раствора. Значе
ние рН буферноro раствора может влиять на
разделение вследствие изменения заряда aHa
лита или добавок, а также электроосмотическоro
потока. При разделении белков и пептидов изме
нение рН буферноro раствора от значения, пре
2.2.47. Капиллярный электрофорез
135
вышающеrо изоэлектрическую точку (pl), до зна
чения, которое меньше ее, изменяет суммарный
отрицательный заряд вещества на положитель
ный. Увеличение рН буферноro раствора обычно
увеличивает электроосмотический поток.
ОР2анические растворители. К водным бу
ферным растворам для увеличения растворимо
сти веществ, а также (или) для изменения степе
ни ионизации компонентов образца MOryт быть
добавлены орrанические модификаторы (MeTa
нол, ацетонитрил и друrие). Добавление таких
орrанических модификаторов к буферному pac
твору обы'-lНО вызывает уменьшение электроос
мотическоro потока.
Добавки для хиральных разделений. Для
разделения оптических изомеров к разделяю
щему буферному раствору добавляют хираль
ный селектор. Наиболее часто используемыми
хиральными селекторами являются циклодек
стрины, но кроме этоro MOryт быть использованы
краун эфиры, полисахариды и белки. Поскольку
хиральное распознавание управляется различ
ными взаимодействиями между хиральным ce
лектором и каждым из энантиомеров, разреше
ние, достиrаемое для хиральных соединений,
зависит, rлавным образом, от типа использован
ноro хиральноro селектора. В этом отношении
при разработке методик определенных разделе
ний может оказаться полезным использование
циклодекстринов с различным размером поло
стей (a , или у циклодекстрин) либо модифи
цированных циклодекстринов с нейтральны
ми (метил , этил , rидроксиалкил и друrие) или
способными к ионизации (аминометил . карбок
симетил , сульфобутиловые эфиры и друrие)
rруппами. При использовании модифицирован
ных циклодекстринов следует принимать во вни
мание различия в степени замещения между
разными партиями данных веществ, поскольку
это может оказывать влияние на селективность.
Друrими факторами, контролирующими разре
шение при хиральных разделениях, являются
концентрация хиральноro селектора, состав и
значение рН буферноro раствора, а также TeM
пература. Достиrнутую величину разрешения
также может изменять использование орrаниче
ских добавок, таких как метанол или мочевина.
КАПИЛЛЯРНЫЙ rЕЛЬ ЭЛЕКТРОФОРЕЗ
ПРИНЦИП
8 капиллярном rель электрофорезе раз
деление происходит внутри капилляра, запол
ненноro rелем, который действует как молеку
ЛЯРНlJе сито. Поскольку более малые молекулы
леrче пrюникают в структуру rеля и миrриру
ют быстрее, чем большие, разделение моле
кул с близкими величинами отношения заряда
к массе происходит в соответствии с их разме
рами. Таким образом, методом капиллярноro
rель электрофореза соrласно величинам моле
кулярных масс MOryT быть разделены различ
ные биолоrические макромолекулы (например,
белки и фраrменты ДНК), часто имеющие близ
кие величины отношения заряда к массе.
ХАРАКТЕРИСТИКИ rЕЛЕЙ
В капиллярном электрофорезе используют
rели двух типов: химически модифицированные
и динамически модифицированные. Химически
модифицированные rели, такие как поперечно
сшитый полиакриламид, получают полимериза
цией мономеров внутри капилляра. Они обычно
химически связаны с поверхностью плавленоro
кварца и не MOryт быть удалены без разрушения
капилляра. Если rели используются для анализа
белков, разделяющий буфер обычно содержит
натрия додецилсульфат и пробы перед вводом
денатурируют наrреванием в смеси натрия дo
децилсульфата и 2 меркаптоэтанола или дити
отреитола. При использовании не восстанавли
вающих условий (в случае анализа интактноro
антитела) 2 меркаптоэтанол и дитиотреитол не
применяют. Разделение с помощью поперечно
сшитых rелей может быть улучшено при изме
нении буферноro раствора, используемоro при
разделении (как указано в разделе, посвящен
ном капиллярному зонному электрофорезу), и
контролировании пористости rеля во время ero
получения. Для поперечно сшитых полиакрила
мидных rелей пористость может быть модифи
цирована путем изменения концентрации акри
ламида и/или пропорции сшивающеro peareHTa.
Как правило, уменьшение пористости rеля при
водит к уменьшению подвижности разделяемых
веществ. Вследствие жесткости подобных rелей
может быть использован только электрокинети
ческий ввод пробы.
Динамически модифицированные rели пред
ставляют собой rидрофильные полимеры, такие
как линейный полиакриламид, производные цел
люлозы, декстран и друrие, которые MOryт pac
творяться в водных буферных растворах, исполь
зуемых при разделении, образуя разделяющую
среду, которая также действует как молекулярное
сито. Такие разделяющие среды получить леrче,
чем поперечно сшитые полимеры. Они MOryT
быть приroтовлены в пробирке и помещены под
давлением в капилляр с модифицированной по
верхностью (без электроосмотическоrо потока).
Удаление rеля после каждоro ввода пробы
обычно улучшает воспроизводимость разделе
ния. Пористость rелей может быть увеличена при
использовании полимеров с большей молекуляр
ной массой (при определенной концентрации по
лимера) или путем уменьшения концентрации по
ли мера (при определенной молекулярной массе
полимера). Уменьшение пористости rеля приво
дит к уменьшению подвижности вещества для
тоro же самоro буферноro раствора. Поскольку
растворение таких полимеров в буферном pac
творе дает растворы с низкой вязкостью, может
быть использован как rидродинамический, так и
электрокинетический ввод пробы.
136
rосударственная фармакопея Республики Беларусь
КАПИЛЛЯРНОЕИЗОЭЛЕКТРИЧЕСКОЕ
ФОКУСИРОВАНИЕ
ПРИНЦИП
При изоэлектрическом фокусировании MO
лекулы миrрируют под влиянием электрическоro
поля до тех пор, пока они остаются заряженны
ми в rрадиенте рН, создаваемом амфолитами,
имеющими широкий диапазон значений рl (по
лиаминокарбоновые кислоты), растворенными в
буферном растворе.
Тремя основными стадиями изоэлектриче
cKoro фокусирования являются заrрузка, фоку
сирование и мобилизация.
Стадия заrрузки. MoryT быть использованы
два способа:
одноступенчатая заrрузка: проба смеши
вается с амфолитами и вводится в капилляр под
давлением или с помощью вакуума;
последовательная заrрузка: в капилляр
вводят вначале ведущий буферный раствор,
затем амфолиты, затем пробу, смешанную с aM
фолитами, после чеrо снова амфолиты и в конце
замыкающий буферный раствор. Объем пробы
должен быть таким, чтобы не повлиять на вели
чину rрадиента рН.
Стадия фокусирования. При наложении
напряжения амфолиты, в зависимости от своеro
заряда, миrрируют по направлению к катоду или
аноду, тем самым создавая rрадиент от анода
(более низкие рН) к катоду (более высокие рН).
Во время данной стадии разделяемые компо
ненты миrрируют до тех пор, пока не достиrнут
значения рН, соответствующеro своей изоэлек
трической точке и ток не упадет до очень низких
значений.
Стадия мобилизации. Если для детекти
рования необходима мобилизация, используют
один из следующих способов:
в первом способе мобилизация проводит
ся в течение стадии фокусирования вследствие
действия электроосмотическоro потока; элек
троосмотический поток должен быть достаточно
малым, чтобы позволить провести фокусирова
ние компонентов;
во втором способе мобилизация проводит
ся после стадии фокусирования путем использо
вания положительноro давления;
в третьем способе мобилизация проводит
ся после стадии фокусирования путем добав
ления солей в катодный или анодный резерву
ар (в зависимости от направления, выбранноro
для мобилизации) для изменения рН в капилля
ре при наложении напряжения. Поскольку рН из
меняется, белки и электролиты перемещаются
по направлению к резервуару, который содержит
добавленные соли, и проходят через детектор.
Достиrаемое разделение, выраженное как
дрl, зависит от rрадиента рН (dpH/dx), числа aM
фолитов, имеющих различные величины pl, KO
эффициента молекулярной диффузии (О), Ha
пряженности электрическоro поля (Е) и тоro, как
изменяется электрофоретическая подвижность
аналита при изменении рН ( dJ.1/dpH):
!lpl == 3. D dpH/dx) .
Е( ф/dрН)
ОПТИМИЗАЦИЯ
Основными параметрами, которые следует
учитывать при разработке методик разделения,
являются следующие.
Напряжение. В капиллярном изоэлектри
ческом фокусировании на стадии фокусирова
ния используются электрические поля с очень
высокой напряженностью от 300 BlcM ДО
1000 В/см.
Капилляр. В зависимости от способа моби
лизации (см. выше) электроосмотический поток
должен быть уменьшен или подавлен. Капилля
ры с модифицированной поверхностью склонны
уменьшать электроосмотический поток.
Растворы. Резервуар с анодным буферным
раствором заполняют раствором, значение рН
KOToporo меньше, чем значение рl большинства
кислотных амфолитов, а катодный резервуар
раствором, значение рН котороro больше, чем
значение р' большинства основных амфолитов.
Для анода часто используют фосфорную кисло
ту, а для катода натрия rидроксид.
Добавление полимера, такоro как метилцел
люлоза, в раствор амфолита приводит к пода
влению конвективных сил (если такие имеются)
и электроосмотическоro потока, что обусловле
но увеличением вязкости. Имеющиеся в прода
же амфолиты покрывают множество диапазонов
рН, а при необходимости получить более широ
кий диапазон рН их можно смешивать. Широ
кие диапазоны рН используются для оценки Be
личины изоэлектрической точки, в то время как
более узкие применяются для улучшения точно
сти анализа. Для rрадуировки можно использо
вать зависимость между временем миrрации и
изоэлектрической точкой для серии белковых
маркеров.
Если необходимо, осаждение белков в изо
электрической точке во время стадии фокусиро
вания может быть предотвращено с помощью
добавления к буферному раствору rлицерина,
поверхностно активных веществ, мочевины или
цвиттер ионных буферных растворов. Следу
ет иметь в виду, что мочевина в зависимости от
концентрации денатурирует белки.
МИЦЕЛЛЯРНАЯ ЭЛЕКТРОКИНЕТИЧЕСКАЯ
ХРОМАтоrРАФИЯ
ПРИНЦИП
В мицеллярной электрокинетической xpOMa
тоrрафии (МЭКХ, МЕКС) разделение происхо
2.2.47. Капиллярный электрофорез
137
дит В растворе электролита, который содержит
поверхностно активное BeuцecTBo в KOHцeHTpa
ции, превышающей критическую KOHцeHTpa
цию мицеллообразования (ккм). Молекулы pac
творенноro вещества распределяются между
водным буферным раствором и псевдонепод
вижной фазой, образованной мицеллами, в co
ответствии со своим коэффициентом распре
деления. Таким образом, данный метод можно
рассматривать как rибрид электрофореза и xpo
матоrрафии. Он может быть использован для
разделения как заряженных, так и нейтральных
веществ и сохраняет эффективность, скорость
и оборудование, присущие капиллярному элек
трофорезу. Одним из поверхностно активных Be
ществ, широко используемым в МЭКХ, являет
ся анионное поверхностно активное вещество
натрия додецилсульфат. Применяются также и
друrие поверхностно активные вещества, Ha
пример, катионные поверхностно активные Be
щества, такие как соли цетилтриметиламмония.
Механизм разделения заключается в следу
ющем. В нейтральной или щелочной среде воз
никает сильный электроосмотический поток, KO
торый перемещает ионы буферноro раствора,
используемоro при разделении, по направлению
к катоду. Если в качестве поверхностно активно
ro вещества используется натрия додецилсуль
фат, электрофоретическая миrрация анионных
мицелл происходит в противоположном направ
лении, т. е. к аноду. В результате общая скорость
миrрации мицелл уменьшается по сравнению
со скоростью потока раствора электролита. В
случае нейтральных разделяемых веществ CKO
рость миrрации аналита будет зависеть только
от величины еro коэффициента распределения
между мицеллой и водным буферным paCTBO
ром, поскольку аналит способен распределяться
между мицеллой и водным буферным раствором
и не имеет электрофоретической подвижности.
На электрофореrрамме пики, соответствующие
каждому из разделяемых незаряженных Be
ществ, Bcerдa находятся между пиком маркера
электроосмотическоro потока и пиком мицеплы
(время между этими двумя пиками называется
окном разделения). Для заряженных разделяе
мых веществ скорость миrрации зависит как от
коэффициента распределения вещества между
мицеллой и водным буферным раствором, так и
от электрофоретической подвижности вещества
в отсутствии мицеллы.
Поскольку механизм разделения в МЭКХ
для нейтральных и слабоионизированных Be
ществ rлавным образом хроматоrрафический,
миrрация вещества и разрешение MOryT быть
оха!)актеризованы с помощью фактора удержи
вания вещества (k), также известноro как KOH
центрационный коэффициент распределения
(От)' который представляет собой отношение
количества разделяемоro вещества, находяще
rося в мицелле, к количеству данноro вещества
в подвижной фазе.
19 За" 1712
Для нейтральных соединений k определяет
ся как:
k= tR to K. Vs ,
t o . ( 1 tfi.. ) VM
t me
rдe:
t R время миrрации вещества;
t o время анализа неудерживаемоrо веще
ства (определяемое с помощью маркера элек
троосмотическоro тока, не входящеro в мицеллу,
например, метанола);
t me время миrрации мицеллы (измеря
ется с помощью маркера мицеллы, такоro как
Судан 111, который миrрирует, полностью Haxo
дясь в мицеллах);
К коэффициент распределения веще
ства;
V S объем мицеллярной фазы;
V M объем подвижной фазы.
Аналоrично, разрешение пиков двух близко
миrрирующих веществ (R s ) определяется как:
1 ( o )
R JN а 1 kb t me ,
s 4'a' kb +1 '1+ka . ( t )
t me
rдe:
N число теоретических тарелок для
одноro из веществ;
а селективность;
ka и kb факторы удерживания для обоих
веществ соответственно (k b > kJ
Подобные, но не идентичные уравнения для
значений k и Rs используются и в случае разде
ления заряженных веществ.
ОПТИМИЗАЦИЯ
Основными параметрами, которые следует
принимать во внимание при разработке методик
разделения в МЭКХ, являются инструменталь
ные, а также пара метры раствора электролита.
Инструментальные параметры
Напряжение. Время разделения обратно
пропорционально приложенному напряжению.
Однако увеличение напряжения может вызвать
избыточное выделение тепла, повышение TeM
пературы и, как результат, образование rрадиен
тов температуры и вязкости в буферном paCTBO
ре, находящемся в капилляре. Данный эффект
особен!-ю характерен для буферных растворов
с высокой электропроводностью и содержащих
мицеллы. Неудовлетворительная тепловая KOH
векция при водит к расширению полосы и YMeHb
шает разрешение.
Температура. Изменения температуры Ka
пилляра влияют на коэффициент распределе
138
rосударственная фармакопея Республики Беларусь
ния вещества между буферным pacTBorOM и
мицеллой, на критическую концентрацию ми. \f'л
лообразования и вязкость буферноro pactbor-а.
Данные параметры вносят вклад в величину
времени удерживания вещества. ИСПОЛЬЗ0ва
ние хорошей охлаждающей системы улучшает
воспроизводимость времен миrрации веществ.
Капилляр. Как и в случае капиллярноrо элек
трофореза со свободным раствором, размеры
капилляра (длина и внутренний диаметр) влияют
на время анализа и эффективность разделения.
Увеличение как эффективной, так и общей длины
капилляра может ослабить электрическое поле (в
случае работы при постоянном напряжении), YBe
личить время миrрации и улучшить эффектив
ность разделения. Внутренний диаметр KOHTpO
лирует тепловую конвекцию (для определенноro
буферноro раствора и электрическоro поля) и, co
ответственно, расширение полосы вещества.
Параметры раствора электролита
Тип и концентрация поверхностно актив
Н020 вещества. Тип поверхностно активноro Be
щества таким же образом, как инеподвижная
фаза в хроматоrрафии, влияет на разрешение.
поскольку изменяет селективность разделения.
Величина 19 k нейтральноrо соединения ЛИl1ей
но увеличивается при увеличении КОНllf.:нтраЦИi/l
поверхностно активноrо вещества в подвижной
фазе. Поскольку разрешение в М:ЭКХ п.остиrа
ет мсш симума при приближении величины k i{
Jt:c / t o , изменение концентрации поверхностно
активноro вещества в подвижной фазе изменяеl
величину разрешения.
3начение рН буферН020 раствора. HeCMO
тря на то, что значение рН не изменяет коэф
фициент распределения неионизированных Be
ществ, оно может изменять электроосмотический
поток в капиллярах с немодифицированной по
верхностью. Уменьшение значения рН буферноro
раствора уменьшает электроосмотический ПОТОI<
и тем самым увеличивает разрешение нейтраль
ных веществ в МЭКХ вследствие увеличения Bpe
мени анализа.
ОР2анические растворители. Для улучше
ния мэкх. разделения rидрофобных соединений
к раствору электролита MOryт быть добавлены
орrанические модификаторы (метанол, пропа
нол, ацетонитрил и друrие). Добавление этих MO
дификаторов обычно уменьшает время миrрации
и селективность разделения. Поскольку добав
ление орrанических модификаторов влияет на
величину критической концентрации мицелло
образования, определенная концентрация по
верхностно активноro вещества может быть
IIIспользована только в пределах некотороro про
центноro содержания орrаническоrо модифика
тора до прекраLЦения мицеллообразования или
отрицательноro влияния на этот процесс, приво
дящеro к исчезновению мицелл и прекращению
разделения. Диссоциация мицелл в присутствии
большоro количества орrаническоro растворите
ля не Bcerдa означает не возможность дальней
шеro разделения; в некоторых случаях rидрофоб
ное взаимодействие между мономером ионноro
поверхностно активноro вешества и нейтраль
ным разделяемым веществом ПDИВОДИТ к образо--
ванию сольвофобных комплексов, которые MOryт
быть разделены элеКТDофоретически.
Добавки для хираЛhНЫХ разделений. Для раз
деления энантиомеРОR с использованием мэкх.
хиnальный селектор ВКJlючается в мицеллярную
систему, либо ковалентно связывается с поверх
ностно активным веществом либо добавляется
1( раствору электролита, в котором происходит
разделение мицелл. Мицеллы. способные к хи
ральным разделениям, обычно включают в себя
соли N додеканоил L аминокислот. соли желчных
кислот и т. д. Хиральное разделение может быть
также проведено с помощью циклодекстринов,
добавленных к растворам электролитов, coдep
жащих мицеллы ахиральнЬ!х поверхностно ак
тивных веществ.
Друсие добавки. Существует ряд подходов,
предполаrающих добавление различных peareH
тов к буферному раствору для изменения селек
тивности. Добавление некоторых типов циклодек
стринов к буферному раствору также может быть
использовано для уменьшения взаимодействия
rидрофобных веществ с мицеллой, тем самым
увеличивая селективность разделения для такоro
типа соединений.
Добавление вешеств. которые способны из
менять взаимодействие между разделяемым Be
ществом и мицеллой вследствие адсорбции на
последней, используется для улучшения селек
тивности разделения в мэкх.. Данные добавки
MOryT представлять собой второе поверхностно
активное вещество (ионное или неионное), KOТO
рое увеличивает количество смешанных мицелл
или катионов металлов, которые растворяются в
мицелле и образуют координационные комплек
сы с разделяемыми веществами.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Пики должны быть разделены COOTBeTCTBY
ющими временами миrрации, чтобы в итоrе по
лучить исправленную площадь, позволяющую
компенсировать:
смещения времен удерживания от анали
за к анализу и тем самым уменьшить различия в
величине отклика;
различные отклики компонентов пробы с
раЗЛИЧНblМИ временами миrрации.
При использовании внутреннеro стаНдарта сле
дует убедиться в том, что пик испытуемою вещества
не перекрывается пиком внутренноro стаНдарта.
РАСЧЕТbI
По полученным данным рассчитывают co
держание испытуемоro компонента или KOM
понентов. Если требуется, рассчитывают про
центное содержание одноro или нескольких
компонентов образца путем определения ис
2.2.47. Капиллярный электрофорез
139
правленной площади пика или пиков как про
центной части исправленных площадей всех
пиков, исключая пики, соответствующие paCTBO
рителям или любым добавленным peareHTaM
(метод нормализации). Рекомендуется использо
вание автоматической системы интеrрирования
(интеrpатора или системы получения и обработ
ки данных).
приrодность СИСТЕМЫ
Параметры приrодности системы использу
кwся для проверки поведения системы капил
лярноro электрофореза. Выбор этих параметров
зависит от используемой разновидности капил
лярноro электрофореза. К ним относятся: фактор
удерживания (k) (только для мицеллярной элек
трокинетической хроматоrрафии), число Teope
тических тарелок (N), фактор симметрии (As) и
разрешение (RJ В предыдущих разделах были
описаны уравнения для расчетов N и Rs' ниже
приведены уравнения, позволяющие рассчитать
эти параметры из электрофореrрамм.
ЧИСЛО ТЕОРЕТИЧЕСКИХ ТАРЕЛОК
Число теоретических тарелок (N) может
быть рассчитано по формуле:
N 5'54-( ;J 2,
rдe:
t R время миrрации или расстояние по ба
зовой линии от точки ввода пробы до перпенди
куляра, опущенною из максимума пика, COOTBeT
ствующеro компоненту;
W h ширина пика на половине высоты.
РАЗРЕШЕНИЕ
Разрешение (R s ) близких по высоте пиков
двух компонентов может быть рассчитано по
формуле:
R 1,1B.(t R2 tR1)
s W h1 +W h2
t R2 > t R1 ,
rдe:
t R1 и t R2 времена миrрации или расстояния
по базовой линии от точки ввода пробы до пер
пендикуляров, опущенных из максимумов двух
соседних пиков;
W hr и W h2 ширины пиков на половине
высоты.
При необходимости может быть измерена
высота седловины (Н) между двумя частично
разделенными пиками в стандартном образце, а
также высота меньшеro пика (Н ) и рассчитана
р
величина коэффициента разделения не полно
стью разделенных пиков:
Нр
p/v== .
Hv
ФАКТОР АСИММЕТРИИ
Фактор асимметрии пика (As) рассчитыва
ют по формуле:
W
A .
s 2d
rдe:
W O ,05 ширина пика на одной двадцатой
высоты;
d расстояние между перпендикуля
ром, опущенным из максимума пика, и пе
редним краем пика на одной двадцатой
высоты.
Испытания на повторяемость площадей
(стандартное отклонение площадей или OTHO
шений площады/ремяя миrрации) и времен ми
rрации (стандартное отклонение времени ми
rрации) используются в качестве параметров
приrодности. Повторяемость времени миrра
ции обеспечивает испытание по определению
приroдности методики промывки капилляра.
Альтернативным способом предупреждения
ухудшения повторяемости времени миrрации
является использование времени миrрации
относительно времени миrрация внутреннею
стандарта.
Для определения родственных соеди
нений может быть полезно испытание на
подтверждение отношения сиrнал/шум для
стандартною образца (или определение
предела количественною определения).
ОТНОШЕНИЕ сиrНАЛ/ШУМ
Пределы обнаружения и количествен
ною определения отвечают отношению
сиrнал/шум 3 и 1 О соответственно. OTHO
шение сиrнал/шум (S/N) рассчитывают по
формуле:
SIN 2H ,
h
rдe:
Н высота пика, соответствующеro ею
рассматриваемому компоненту, на электро
фореrрамме, полученной при использова
нии рекомендованноrо раствора сравнения,
и измеренная от максимума пика до экстра
полированной базовой линии для сиrнала,
величина котороro эквивалентна двадцати
кратному превышению ширины пика на поло
вине высоты;
h размах для фоновою шума (уровень
шума) на электрофореrрамме, полученной
при введении контрольной пробы. Величина
Эi{вивалентна двадцатикратному превыше
нию ширины пика на половине высоты для
пика на электрофореrрамме, полученной при
использовании рекомендованноro раствора
сравнения и, если возможно, расположенною
на одном и том же расстоянии от места воз
можноrо обнаружения пика.
140
01/2013:20248
rосударственная фармакопея Республики Беларусь
2.2.48. РАМАНОВСКАЯ
СПЕКТРОМЕТРИЯ
Рамановская спектрометрия (неупруrое
рассеяние света) "(или спектрометрия комби
национноro рассеяния)" это процесс pacce
яния света, при котором испытуемый образец
в условиях испытаний облучается интенсивным
монохроматическим светом (обычно излучение
лазера), и для света, рассеиваемоro образцом,
определяется сдвиr частоты.
Рамановская спектрометрия является
дополнительным методом по отношению к
ИК спектрометрии в том смысле, что оба этих
метода основаны на молекулярных колеба
ниях частиц испытуемоro вещества. Однако
рамаНОВСl<ая и ИК спеl<трометрия отличают
ся по относительной чувствительности для
различных функциональных rрупп. PaMaHOB
ская спектрометрия особенно чувствительна
1< неполярным связям (например, одинарные
или кратные связи C C), Torдa как вода, для
которой характерно сильное поrлощение в
ИК области, обладает слабым рамановским
рассеянием и поэтому хорошо подходит на
роль растворителя в рамановской спектро
метрии.
Прибор. Спектрометры для получения pa
мановских спектров обычно состоят из следу
ющих компонентов:
источник монохроматическоrо света,
чаще всеro лазер С длиной волны в ультрафи
олетовой, видимой или ближней инфракрас
ной области;
подходящая оптика (линзы, зеркала,
оптико волоконные устройства). которые Ha
правляют возбуждающее излучение на обра
зец и собирают свет, рассеиваемый образцом:
оптическое устройство (монохроматор
или фильтр), которое пропускает сдвинутое
по частоте рамановское рассеяние и препят
ствует попаданию на детектор интенсивноro
света, частота KOToporo равна частоте возбуж
дающеro света (рэлеевское рассеяние);
дисriерrирующее устройство (дифрак
ционная решетка или призма), соединенное
со щелью, реrулирующей длину волны, и дe
тектором (обычно фотоумножитель);
или:
дисперrирующее устройство (дифрак
ционная решетка или призма), соединенное с
мноroканальным детектором (обычно прибор
с зарядовой связью (ПЗС, ССD));
или:
интерферометр, реrистрирующий ин
тенсивность рассеянноrо света во BpeMe
ни, и устройство обработки данных, которое
с помощью Фурье преобразования перево
дит данные в диапазон частот или волновых
чисел.
подrотовКА ОБРАЗЦА
Рамановские спектры MorYT быть получе
ны для твердых веществ, жидкостей и rазов.
причем как помещенных в стеклянные контей
неры или трубки, как правило, без предвари
тельной подrотовки образца или растворения,
так и напрямую.
Основным оrраничением рамановской
спектрометрии является флуоресценция при
месей, которая мешает детектору улавли
вать значительно более слабый рамановский
сиrнал. Флуоресценции можно избежать при
использовании в качестве возбуждающеro
света лазерноrо излучения С большой длиной
волны, например, находящейся в ближней ин
фракрасной области. Интенсивность HeKOTO
рых рамановских линий может быть усилена
несколькими способами. например, путем ис
пользования резонансной рамановской спек
трометрии (RR) или усиленной поверхностью
рамановской спектрометрии (SERS).
Из за узости световоro потока падающеro
лазерноrо излучения для получения спектра
необходимо Bcero лишь несколько микроли
тров образца. Тем не менее следует учиты
вать неоднородность образца, если только
ero объем не увеличен, например, путем Bpa
щения.
ИДЕНТИФИКАЦИЯ И КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ С ИСПОЛЬЗОВАНИЕМ
СТАНДАРТНЫХ ОБРАЗЦОВ
Испытуемый образец и стандартный обра
зец подверrают одинаковой обработке и затем
при одинаковых условиях получают их спек
тры. Максимумы в спектре испытуемоrо образ
ца должны совпадать по расположению и интен
сивности с соответствующими максимумами в
спектре стандартноrо образца (ФСО).
Если спектры. полученные для твердых Be
ществ, имеют различия в положениях максиму
мов, испытуемый образец и стандартный обра
зец подверrают одинаковой обработке для тоro,
чтобы они выкристаллизовались или образова
лись в одинаковой форме, либо поступают так,
как описано в частной статье, и затем снимают
спектры.
Поскольку закон Бера Ламберта для paMa
новской спектроскопии не выполняется, интен
сивность рамановскоro излучения прямо пропор
циональна концентрации рассеивающих частиц.
Как и в случае друrих спектроскопических Meтo
дов, количественное определение может быть
проведено с использованием известных коли
честв или концентраций стандартных образцов.
Вследствие небольшоro пространственноro раз
решения метода следует обращать внимание на
качество испытуемых образцов и стандартных
образцов, например, необходимо быть YBepeH
ным в том, что они находятся в одном и том же
физическом состоянии либо использовать BHY
тренний стандарт для жидких образцов.
2.2.48. Рамановская спектрометрия
141
ИДЕНТИФИКАЦИЯ И КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ С ИСПОЛЬЗОВАНИЕМ
СПЕКТРАЛЬНЫХ БИБЛИОТЕК
И СТАТИСТИЧЕСКИХ МЕТОДОВ
КЛАССИФИКАЦИИ И rрАДУИРОВКИ
Контроль за работой прибора. При ис
пользовании прибора следуют инструкциям
производителя; реryлярно, в зависимости от
применения прибора и испытуемых образцов,
проводят предписанные rрадуировки и испы
тания работы системы. При использовании pa
мановской спектрометрии для количественных
определений либо при создании спектральных
библиотек сравнения для (хемометрической)
классификации или rрадуировки необходимо
обращать особое внимание на то, чтобы были
сделаны все поправки к измерениям либо пред
приняты меры для контроля за изменяемостью
величин волновых чисел и интенсивности сиr
нала прибора.
Проверка шкалы волновых чисел. Шкалу
волновых чисел рамановских сдвиrов (обычно
выражаемую в обратных сантиметрах) проверя
ют с помощью подходящих стандартов, которые
имеют характерные максимумы при исследуе
мых величинах волновых чисел (например, opra
ническое вещество, неоновая лампа или линии
Ar плазмы арroн ионноro лазера).
rрадуировка прибора должна COOTBeTCTBO
вать типу образца, т. е. для твердых испытуе
мых образцов следует использовать твердые
образцы сравнения, а для жидких жидкие.
Выбирают подходящее вещество (например,
инден, циклоrексан или нафталин), для котороro
установлены точные значения сдвиrов волновых
чисел. Образец индена для предотвращения еro
разрушения помещают в пробирку для ЯМр, Ba
куумируют и затем хранят в атмосфере инертно
ro rаза в прохладном и темном месте.
Таблица 2.2.48. 1
Сдви2и волновых чисел (и допустимые откло
нения) для циКЛ02ексана, индена и нафталина
Циклоrексан А Инден В НафталинА
3056,4 (:1:1,5)
2938,3 (:1:1,5)
2923,8 (:1:1,5)
2852,9 (:1:1,5)
1609,7 (:1:1,0) 1576,6 (:1:1,0)
1444,4 (:1:1,0) 1552,6 (:1:1,0) 1464,5 (:1:1,0)
1266,4 (:1:1,0) 1205,2 (:1:1,0) 1382,2 (:1:1,0)
1157,6 (:1:1,0) 1147,2 (:1:1,0)
1028,3 (:1:1,0) 1018,6 (:1:1,0) 1021,6 (:1:1,0)
801,3 (:1:1,0) 730,5 (:1:1,0) 763,8 (:1:1,0)
533,9 (:1:1,0) 513,8 (:1:1,0)
А Standard gu;de for Raтan shift standards for sресtютеtеrсаliЬratiоп
(Атепсап Soc;ety for Test;ng aпd Materia/s ASTM Е 1840).
в D. А. Carter. W R. Thoтpsoп, С. Е Tay/or and J. Е РетЬепоп.
Applied Spectroscopy, 1995,49 (11). 1561 1576.
20 3а.. 1712.
Проверка шкалы интенсивностей. На аб
солютную и относительную интенсивность pa
мановских полос MOryт оказывать влияние сле
дующие факторы:
состояние поляризации падающеro света;
состояние поляризации собирающей
оптики;
интенсивность падающеro света;
различия в отклике прибора;
различия в фокусе и rеометрии образца;
различия в насыпной плотности для TBep
дых образцов.
Подходящие критерии приемлемости MOryт
изменяться в зависимости от применения, но
в большинстве случаев допустимы колебания
(day to day vаriаtiопs) в относительных интен
сивностях полос :1:1 О %.
Создание спектральных библиотек cpaв
нения. Снимают спектры подходящею числа
полностью исследованных (например, так, как
описано в частной статье) веществ, имеющих
отличия в производителе, серии, кристалличе
ской модификации, размерах частиц и т. д., ти
пичные для анализируемоro материала. Набор
спектров дает информацию, определяющую
rраницы подобия или количественноro опреде
ления, которые MOryT быть использованы для
тоro, чтобы, например, идентифицировать Be
щество или проконтролировать ею количество,
образовавшееся в процессе производства. Ko
личество веществ в базе данных зависит от oco
бенностей ее применения. Коллекция спектров в
базе данных может быть представлена различ
ными способами, определяемыми математиче
ской методикой, используемой для классифика
ции или количественноro определения.
Селективность базы данных, которая позво
ляет положительно идентифицировать данный
материал либо достоверно отличить еro от
друrих материалов базы данных, должна быть
установлена во время процедуры валидации.
Для уверенности в приroдности базы данных
эта селективность должна реryлярно проверять
ся; в особенности это необходимо делать после
любых значительных изменений, произошедших
с веществом (например, изменения поставщика
или производственноro процесса), либо при Ha
стройке прибора для рамановской спектроме
трии (например, при проверке шкалы волновых
чисел или повторяемости отклика спектрометра).
База данных приroдна только для исходною
прибора либо для подобною прибора при усло
вии, что перенесенная на прибор база данных
остается приroдной.
Методика. Подroтовку и исследование об
разца проводят таким же образом, как и при соз
дании базы данных. Для облеrчения сравнения
спектров и количественных расчетов MOryT быть
использованы подходящие математические пре
образования рамановских спектров.
Сравнение или преобразования спектров,
количественный проrноз свойств веществ, Haxo
142
rосударственная фармакопея Республики Беларусь
ДЯЩИХСЯ в испытуемом материале, MOryт ПDОВО
ДИТЬСЯ С использованием подходящих меl<)ДОВ
хемометрики, статистической классификаЦИlrl и
rрадуировки.
01/2013:20249
2.2.49. ИЗМЕРЕНИЕ ВЯЗКОСТИ
НА ВИСКОЗИМЕТРЕ
С ПАДАЮЩИМ ШАРИКОМ
Определение динамической вязкости нью
тоновских жидкостей проводят, используя подхо
дящий вискозиметр с падающим шариком, Пр/li
температуре (20:t0,1) ОС, если в частной статье
не указана друrая температура. Измерение вяз
кости основано на определении времени па
дения шарика в жидкости от одной отметки ви
скозиметра до друrой. Если для используемоrо
оборудования не установлены точные пределы,
то полученные результаты считаются приемле
мыми при условии, что результаты двух после
довательных измерений отличаются не более,
чем на 1,5 %.
Оборудование. Вискозиметр с падающим
шариком состоит из стеклянной трубки, заклю
ченной в КОЖУХ, позволяющий контролировать
температуру; шести шариков различной плот
ности и диаметра, изrотовленных из стекла, же
лезно никелевоro сплава или стали. Трубку фик
сируют таким образом, чтобы отклонение от
вертикальной оси составляло (1 O:t 1 )0. На трубке
имеются две отметки, которые определяют pac
стояние падения шарика. К промышленным ви
скозиметрам прилаrаются таблицы со значения
ми постоянных, плотности шариков и данные по
приrодности определенноro шарика для исполь
зования в предполаrаемом диапазоне вязкости.
Методика. Чистую, сухую трубку виско
зиметра, предварительно выдержанную при
температуре (20:t0,1) ОС, заполняют испы
туемой жидкостью, избеrая образования пу
зырьков. Помещают подходящий для предпо
лаrаемоro диапазона вязкости шарик, чтобы
получить время падения не менее 30 с. За
крывают трубку и термостатируют раствор при
(20:t0,1) ос не менее 15 мин. Пропускают шарик
между двумя метками без измерения BpeMe
ни падения. Шарик ставят в исходное положе
ние, включают секундомер, Korдa нижняя часть
шарика коснется верхней метки, и останавлива
ют, коrда шарик достиrнет нижней метки. Время
движения шарика измеряют не менее 3 раз с
точностью до 1/5 секунды.
Рассчитывают динамическую вязкость
('1, мПа'с) по формуле:
'1 =о k(P1 Р2)' t,
rде:
k постоянная вискозиметра, в ммЧс 2 ;
Р 1 плотность шарика, в r/cM 3 ;
Р 2 плотность испытуемой жидкости,
в r/см З , полученная умножением относительной
плотности (d;g) на 0,9982;
t среднее время падения шарика между
крайними метками, в секундах.
01/2013:20254
2.2.54. ИЗОЭЛЕКТРИЧЕСКОЕ
ФОКУСИРОВАНИЕ
ОБЩИЕ ПРИНЦИПЫ
Изоэлектрическое фокусирование (ИЭФ,
/EF) это вариант электрофореза, в котором
разделение белков происходит в соответствии с
их изоэлектрическими точками. Разделение про
водится на пластине из полиакриламидноro или
аrарозноrо rеля, содержащеro смесь амфотер
ных электролитов (амфолитов). При действии
электрическоro поля амфолиты миrрируют в
rеле, создавая rрадиент рН. В некоторых слу
чаях используют rели. имеющие фиксирован
ный rрадиент рН, полученные путем включения
слабых кислот или оснований в определенные
места rеля во время ею приroтовления. Korдa
нанесенные белки достиrают фракции rеля, KO
торая имеет такое же значение рН. что и их изо
электрическая точка (pl), заряд данных белков
нейтрализуется и миrрация прекращается. rpa
диенты MOryT быть созданы в различных диапа
зонах рН в зависимости от выбранной смеси aM
фолитов.
ТЕОРЕТИЧЕСКИЕ АСПЕКТЫ
В изоэлектрической точке молекула белка
не заряжена и не может передвиrаться в Ma
трице rеля под действием электрическою поля.
Ее перемещение, однако, может происходить
в результате диффузии. rрадиент рН вынуж
дает белок оставаться в изоэлектрическом co
стоянии, концентрируя данное вещество. Такое
концентрирование называется «фокусировани
ем». Увеличение приложенноro напряжения или
уменьшение количества нанесенноro вещества
улучшает разделение полос. Величина прило
женноrо напряжения оrраничена выделяющей
ся теплотой, которая должна быть рассеяна. Ис
пользование тонких слоев rеля, а также пластин
с эффективным охлаждением, контролируемых
термостатирующим устройством, предотвраща
ет возroрание rеля и в тоже время обеспечивает
точное фокусирование. Разделение оценивают
по минимальной разности р' (др/), которая He
обходима для разделения двух соседних полос:
2.2.54. Изоэлектрическое фокусирование
143
D(dpH/ dx)
pl=3. E( d,u/dpH)'
rде:
D коэффициент диффузии белка;
dpH rрадиент рН;
dx
Е напряженность электрическоro поля, в
вольтах на сантиметр;
dp
изменение подвижности вещества
dpH
при изменении рН в диапазоне, близком к pl.
Поскольку О и d,u для определенноro
dpH
белка не MOryT быть изменены, улучшить раз
деление можно при использовании более узкоro
диапазона рН либо при увеличении напряженно
сти электрическоro поля.
Разрешение между полосами белков в
случае использования ИЭФ rеля, содержащеro
ведущие амфолиты, достаточно хорошее. Улуч
шить разрешение можно при использовании
фиксированных rрадиентов рН, для создания
которых применяются буферные вещества, aHa
лоrичные ведущим электролитам, сополимери
зованные с матрицей rеля. При использовании
rеля, содержащеro ведущие электролиты, MOryт
быть разделены белки, значения pl которых OT
личаются на 0,02 единицы рН, в то время как
фиксированные rрадиенты рН позволяют разде
лить белки, изоэлектрические точки которых раз
личаются приблизительно на 0,001 рН.
ПРАКТИЧЕСКИЕ АСПЕКТЫ
Следует уделять особое внимание xapaK
теристикам и/или подrотовке образца. Наличие
солей в образце может вызвать ряд проблем,
поэтому лучше всеro при еro приroтовлении ис
пользовать деионизированную воду или 2 %
раствор амфолитов, используя при необходимо
сти диализ или rель фильтрацию.
Время, необходимое для завершения фо
кусирования в тонком слое полиакриламидных <
rелей, определяют путем нанесения окрашенно
ro белка (например, rемоrлобина) в различные
области поверхности rеля и применения элек
трическоro поля: стабильное состояние достиrа
ется Torдa, Korдa все нанесения дают идентич
ный образец полос. В некоторых протоколах о
завершении фокусирования судят по времени,
прошедшему после нанесения образца.
ИЭФ может быть использовано в качестве
испытания на подлинность. В этом случае ис
пытуемый образец, миrрирующий на reJ1e, cpaB
нивается с подходящим стандартным образцом
и ИЭФ rрадуировочными белками. ИЭФ может
быть использовано в качестве испытания на
предельное содержание. При этом плотность
полосы на ИЭФ сравнивается, соответственно,
с полосой на ИЭФ, появляющейся при исполь
зовании стандартноro образца, если измере
ние производится с помощью денситометра или
аналоrичноro устройства, позволяющеro опре
делить относительную концентрацию белка в
полосе. ИЭФ может быть также использовано
при испытании на количественное содержание
белков.
Прибор. Прибор для ИЭФ состоит из:
управляемоrо reHepaTopa для создания
постоянноro потенциала, тока и мощности; ис
пользуются потенциалы величиной 2500 В: они
считаются оптимальными для используемых yc
ловий работы; рекомендуется источник тока,
имеющий постоянную мощность до 30 Вт;
жесткой пластиковой ИЭФ камеры, в KOTO
рой находится охлаждаемая пластинка или под
ходящий материал, служащий основой для Ha
несения rеля;
пластиковой крышки с платиновыми элек
тродами. которые соединены с rелем с помощью
бумажных фитилей подходящей ширины, длины
и толщины, пропитанных растворами анодных и
катодных электролитов.
ИЗО ЭЛЕКТРИЧЕСКОЕ ФОКУСИРОВАНИЕ
В ПОЛИАКРИЛАМИДНЫХ rЕЛЯХ:
ПОДРОБНАЯ МЕТОДИКА
Следующая методика представляет
собой подробное описание процедуры ИЭФ в
пластинах полиакриламидН020 2еля, использу
емой, если в частной статье не указано иН020.
ПРИ,ОТОВЛЕНИЕ ,ЕЛЕЙ
Матрица формования. Матрица формо
вания (см. рисунок 2.2.54. 1) состоит из CTe
клянной пластинки (А) на которую для облеr
D
Рисунок 2.2.54. 1. Матрица формования
144
rосударственная фармакопея Республики Беларусь
чения работы с rелем помещена полиэфирная
пленка (В), одной или нескольких раСПО :--IЫХ
деталей (С), второй стеклянной пластинки (.J)
и зажимов, скрепляющих всю конструкцию.
7,5 %-ный полиакриламидный rель.
29,1 r акриламида Р и 0,9 r метиленбисакри
ламида Р растворяют в 100 мл воды Р. К 2,5
объемам полученноrо раствора прибавляют
смесь амфолитов. указанных в частной статье.
и доводят водой Р до объема 10 мл. Раствор
аккуратно перемешивают и деrазируют.
Приrотовление формы. Полиэфирную
пленку помещают на нижнюю стеклянную
пластинку, вставляют распорную деталь, по
мещают вторую стеклянную пластинку и CKpe
пляют конструкцию зажимами. Перед исполь
зованием раствор помещают на маrнитную
мешалку и прибавляют к нему 0,25 объема
100 r/л раствора аммония персульфата Р
и 0,25 объема тетраметuлэтилендиами
на Р. Немедленно заполняют раствором про
странство между стеклянными пластинками
формы.
МЕТОДИКА
Форму разбирают на составные части и, ис
пользуя полиэфирную пленку, переносят rель
на охлажденную подложку, увлажненную He
сколькими миллилитрами подходящей жидко
сти, избеrая образования воздушных пузырь
ков. rотовят испытуемые растворы и растворы
сравнения, как указано в частной статье. Для
нанесения образца помещают полоски бумаrи
размером приблизительно 1 О мм х 5 мм на по
верхность rеля и пропитывают каждую пред
писанным количеством испытуемоro раствора
и раствора сравнения. Также наносят предпи
санное количество раствора белков с извест
ными величинами изоэлектрических точек.
служащих в качестве маркеров рН для rраду
ировки rеля. В некоторых протоколах вместо
импреrнированных бумажных полосок исполь
зуют rель, который имеет желобки, предна
значенные для помещения раствора образца.
Отрезают 2 полоски бумаrи длиной, COOTBeT
ствующей длине rеля, и пропитывают их pac
творами электролитов: кислотой для анода и
щелочью для катода (составы анодноro и Ka
тодноrо растворов при водятся в частных CTa
тьях). Эти бумажные фитильки помещают на
каждую сторону rеля на расстоянии несколь
ких миллиметров от ero rраницы. Устанавли
вают крышку так. чтобы электроды контакти
ровали с полосками бумаrи (соответственно,
анодным и катодным полями). Выполняют изо
электрическое фокусирование при исполь
зова нии электрических параметров, описан
ных в частной статье. Коrда миrрация смеси
стандартных белков стабилизируется, элек
трический ток выключают. С помощью пин
цета удаляют полоски, предназначенные для
нанесения образца, и 2 электродных фитиль
ка. Поrружают rель в фиксирующий раствор
для uзоэлекrпричеСК020 фокусирования в по
лиакриламидном 2еле Р. Выдерживают при
аккуратном встряхивании при комнатной TeM
пературе в течение 30 мин. Удаляют раствор
высушиванием и прибавляют 200 мл обесц
вечивающе20 pacrпвopa Р. Выдерживают при
перемешивании в течение 1 часа. Высушива
ют rель, прибавляют кума сси красящий pacт
вор Р. Выдерживают в течение 30 мин. Обесц
вечивают rель путем пассивной диффузии
обесцвечивающе20 раствора Р до тех пор,
пока на чистом фоне не станут хорошо видны
полосы. Отмечают положение и интенсив
ность полос на электрофореrрамме, как указа
но в частной статье.
ИЗМЕНЕНИЯ ПОДРОБНОЙ МЕТОДИКИ
(ПРЕДМЕТ ВАЛИДАЦИИ)
Там, rдe делается ссылка на общую MeTO
дику изоэлектрическоro фокусирования, изме
нения в методолоrии или методике изоэлектри
ческоro фокусирования MorYT быть предметом
валидации. Они включают в себя:
использование имеющихся в продаже
rелей заводскоrо производства, а также OKpa
шивающих и обесцвечивающих наборов;
использование фиксированных rрадиен
тов рН;
использование стержневых rелей;
использование кассет rеля различных
размеров, включая ультратонкие (0,2 мм) rели;
изменения в методике нанесения образ
ца, включающие различные объемы образца
или использование шаблонов или фитильков,
изrотовленных не из бумаrи;
использование альтернативных условий
перемещения, включающих изменения электри
ческоro поля в зависимости от размеров rеля и
оборудования, а также использование фиксиро
ванных времен миrрации вместо субъективной
интерпретации стабильности полосы;
включение стадии предварительноro фо
кусирования;
использование автоматизированноro обо
рудования;
использование rелей аrарозы.
ВАЛИДАЦИЯ МЕТОДИК
ИЗОЭЛЕКТРИЧЕскоrо ФОКУСИРОВАНИЯ
Если используются альтернативные спо
собы по отношению к описанной методике, они
должны быть валидированы. Для валидации
разделения MorYT быть использованы следую
щие критерии:
образование устойчивоrо rрадиента рН
с желаемыми характеристиками, оцениваемо
ro, например, с помощью окрашенных MapKe
ров рН с известными величинами изоэлектри
ческих точек;
2.2.55. Пептuдное картирование
145
01/2013:20255
сравнение электрофореrраммы, прила
rаемой к стандартному образцу, с электрофо
реrраммой, полученной при про ведении испы
тания;
любые друrие критерии валидации, YKa
занные в частной статье.
СПЕЦИФИЦИРОВАННЫЕ ИЗМЕНЕНИЯ
ОБЩЕЙ МЕТОДИКИ
Изменения общей методики, необходимые
при анализе конкретных субстанций, MOryT быть
подробно описаны в частных статьях. Они вклю
чают:
добавление в rель мочевины (обычно
3 М концентрация достаточна для поддержива
ния белка в растворенном состоянии, но может
быть использована концентрация до 8 М): HeKO
торые белки в изоэлектрической точке выпада
ют в осадок, в этом случае для тоro, чтобы белок
оставался в растворе, в rель вводится мочеви
на; при использовании мочевины следует при
менять только ее свежие растворы, чтобы не дo
пустить карбамоилирования белка;
использование альтернативных способов
окрашивания;
использование добавок для rеля, таких как
неионные детерrенты (например, октилrлюко
зид) или цвиттер ионные детерrенты (например,
CHAPS или CHAPSO), и добавление амфолита
к образцу для предотвращения процессов arpe
rации и преципитации белков.
ПОЛОЖЕНИЯ, КОТОРЫЕ СЛЕДУЕТ
ПРИНИМАТЬ ВО ВНИМАНИЕ ПРИ
ИСПОЛЬЗОВАНИИ ДАнноrо МЕТОДА
Образцы MorYT наноситься на любой уча
сток поверхности rеля, но для TOro, чтобы за
щитить белки от экстремальных значений рН,
образцы не должны наносится в непосред
ственной близости от электродов. В ходе BЫ
полнения анализа аналитик может нанести
белок на rель в трех местах (посередине и
на оба конца). Образцы белка, наносимые на
противоположные концы rеля, MorYT быть раз <
личными.
Известно явление, называемое катодным
дрейфом, при котором rрадиент рН с течени
ем времени разрушается. Это может происхо
дить, если фокусирование в rеле происходит
слишком долro. Причины данноro явления не
совсем ясны, но факторами, вызывающими Ka
тодный дрейф, MorYT быть электроэндоосмос и
абсорбция диоксида уrлерода. Для исследова
ния этой проблемы MorYT быть использованы
фиксированные rрадиенты рН.
В течение фокусирования важно прово
дить эффективное охлаждение (до около 4 СС)
емкости, 8 которую помещен rель. Высокие Ha
пряженности электрическоro поля, используе
lIые во время изоэлектрическоro фокусирова
ИМЯ, MOryт ПрИ80ДИТЬ К переrреванию и влиять
на качество фокусируемоrо rеля.
2.2.55. ПЕПТИДНОЕ КАРТИРОВАНИЕ
Пептидное картирование (или составление
пептидных карт) является методом идентифика
ции белков, в особенности получаемых методом
рекомбинантных ДНк. Метод включает в себя
этапы химическоro или энзиматическоro расще
пления белков до образования пептидных фраr
ментов с последующим разделением и иденти
фикацией этих фраrментов в воспроизводимых
условиях. Это надежный метод испытания, по
зволяющий произвести идентификацию измене
ния практически каждой отдельной аминокисло
ты, произошедшеro в результате таких явлений,
как ошибка в считывании последовательности
комплементарной ДНК (кДНК), либо являюще
roся результатом мутации. Составление пептид
ных карт является сравнительным испытанием,
поскольку получаемая информация сравнива
ется со стандартным образцом, подверrающим
ся аналоrичному воздействию. Данное испыта
ние позволяет подтвердить первичную структуру
белка, определить возможные изменения пер
вичной структуры в случае наличия таковых,
подтвердить соответствие процесса и rенетиче
скую стабильность. Для каждоro из белков xa
рактерна совокупность уникальных характери
стик, которые должны быть описаны Научные
и аналитические методики позволяют осущест
влять развитие метода пептидноrо картирова
ния, который, в свою очередь, характеризуется
достаточной специфичностью.
Данная статья содержит детальное опи
сание аспектов использования и валидации
метода пептидноro картирования С целью по
лучения характеристики анализируемоro белка,
оценки стабильности клеточных систем экспрес
сии, используемых для получения продуктов pe
комбинантных ДНК и оценки соrласованности
процесса в целом в отношении как оценки
стабильности продукта, так и обеспечения иден
тичности белка, либо определения содержания
протеинов с изменениями в структуре.
Пептидное картирование не является
общим методом, а заключается в составлении
индивидуальных карт для каждоro отдельно
ro белка. Хотя технолоrии продолжают стреми
тельно развиваться, существует ряд методов,
которые являются общепризнанными. При нали
чии определенных расхождений с данными Me
тодами они будут описываться в соответствую
щих частных статьях.
Составление пептидных карт может pac
сматриваться как метод получения «отпечат
ков пальцев» белка и представляет собой
получаемую в результате ряда химических про
цессов полную расшифровку анализируемоro
белка. Метод включает четыре принципиальные
стадии: выделение и очистка белка в случае,
если белок входит в состав какой либо смеси;
146
rосударственная фармакопея Республики Беларусь
избирательное расщепление пептидных связей;
хроматоrрафическое разделение образующихся
пептидных фраrментов; анализ и идентифика
ция пептидов. Анализируемый образец подвер
rается расщеплению и анализу параллельно со
стандартным образцом. Полное расщепление
пептидных связей более вероятно, коrда вместо
химических rидролизующих peareHToB использу
ются такие ферменты, как эндопептидазы (Ha
пример, трипсин). Для тоro, чтобы карта была
информативной. она должна содержать дocтa
точное количество пептидов. С друroй стороны, в
случае, если пептидных фраrментов будет слиш
ком мноro, карта может утратить свою специ
фичность, поскольку у мноrих белков может OKa
заться аналоrичный профиль.
ВЫДЕЛЕНИЕ И ОЧИСТКА
Выделение и очистка являются обязатель
ным этапом при проведении анализа нерасфа
сованных или дозированных форм лекарствен
ных средств, содержащих мешающие анализу
вспомоrательные вещества или белковые носи
тели и, если это требуется, указываются в част
ной статье. Количественное извлечение пептида
из дозированной формы должно быть валидиро
вано.
ИЗБИРАТЕЛЬНОЕ РАСЩЕПЛЕНИЕ
ПЕПТИДНЫХ СВЯЗЕЙ
Выбор метода расщепления пептидных
связей зависит от анализируемоro белка. Про
цедура выбора включает определение требу
ющеroся типа расщепления (химическоro или
ферментативноro), а также типа протеолитиче
скоro peareHTa в рамках выбранной катеroрии.
В таблице 2.2.55. 1 приводится ряд протеоли
тических ферментов и параметры их специфич
ности. Данный список не является исчерпываю
щим И будет дополняться по мере определения
друrих peareHToB, способных расщеплять пеп
тидные связи.
Подrотовка образца. В зависимости от
размера или конфиryрации белка используют
ся различные подходы к подroтовке образца. В
случае если используется трипсин для расще
пления пептидных связей белка с молекулярной
массой более 100 000 Да, лизиновые остатки
должны быть защищены путем получения про
изводных с лимонной или малеиновой кислотой,
так как в противном случае может образоваться
слишком мноro пептидных фраrментов.
Подrотовка peareHTa для расщепления
пептидных связей. Подroтовка peareHTa для
расщепления пептидных связей, в особенности
ферментов, может быть необходимым этапом
для обеспечения требуемой чистоты и воспро
изводимости карты. Например, трипсин, исполь
зуемый в качестве peareHTa для расщепления
пептидных связей, следует обработать тозил L
фенилаланинхлорметилкетоном с целью инак
тиваuии химотрипсина. Хорошо зарекомендо
вали себя. в особенности при оrраниченном
количестве имеющеrося белка, друrие методы
очистки, такие как высокоэффективная жидкост
ная хроматоrрафия (В3ЖХ) при очистке трипси
на или иммобилизация фермента на rелевом HO
сителе.
Подrотовка белка. При определенных усло
виях может быть необходимо концентрирование
образца либо отделение белка от используемых
в roтовой лекарственной форме сопутствую
щих веществ и стабилизаторов, которые MOryT
искажать результаты картирования. Физиче
ские методы подroтовки MOryT включать ультра
Примеры реа2ентов для расщепления пептидных связей
Таблица 2.2.55. 1
Тип PeareHT Специфичность
Ферментативные Трипсин (ЕС 3.4.21.4) C KOHцeBыe арrинин илизин
. Химотрипсин (ЕС 3.4.21.1 ) C KOHцeBыe rидрофильные остатки (Ha
I пример,лейцин, меТИОНИН,алаНИН,аро
матические аминокислоты)
i Пепсин (ЕС 3.4.23.1 и 2) Неспецифический peareHT
Лизил эндопептидаза (Lys C эндо С концевой лизин
пептидаза) (ЕС 3.4.21.50)
rлутамил эндопептидаза (из C KOHцeBыe rлутамин и аспараrин
S. aureus штамм V8) (ЕС 3.4.21.19)
Пептидил Аsр металло эндопепти N концевой аспараrин
да за (эндопротеаза Asp N)
Клострипаин (ЕС 3.4.22.8) С концевой арrинин
Химические Цианобромид С концевой метионин
2 Нитро 5 тио цианобензойная кис N концевой цистеин
лота
О Йодозобензойная кислота C KOHцeBыe триптофан и тирозин
Разведенная кислота Аспараrин и пролин
ВNРS скатол Триптофан
2.2.55. Пептидное картирование
147
фильтрацию, колоночную хроматоrрафию и ли
офилизацию. С целью про ведения денатурации
белка до процедуры пептидноrо картирования
возможно использование друrих методов подю
товки, таких как добавление хаотропных areHToB
(например, мочевины). Для обеспечения полно
ro доступа фермента к местам разрыва связей и
частичной денатурации белка во мноrих случа
ях до этапа протеолитическоro расщепления He
обходимым представляется сокращение числа
дисульфидных связей путем их восстановле
ния и алкилирования. Расщепление пептидных
связей с использованием трипсина может сопро
вождаться появлением в пептидной карте ряда
неточностей ввиду протекающих параллель
но с реакцией протеолитическоrо расщепления
побочных реакций, таких как неспецифическое
расщепление,дезаминирование,дисульфидная
изомеризация, окисление остатков метионина,
образование пироrлутаминовых rрупп при деза
минировании rлутамина и N концевых участков
пептида. Более тоro, при автоrидролизе трип
сина также MOryт появляться дополнительные
пики. Интенсивность их образования зависит
от соотношения содержания трипсина к белку.
Для предотвращения rидролиза растворы про
теаз MOryT быть приroтовлены при рН, отличном
от оптимальною (например, при рН 5 для трип
сина), блаroдаря чему фермент не перейдет в
активное состояние до момента разведения с
буферным раствором при проведении расще
пления.
Установление оптимальных условий для
расщепления пептидных связей. Факторы,
которые влияют на полноту и эффективность
расщепления протеинов, являются факторами,
оказывающими влияние на химические и фер
ментативные реакции.
рН реакционной средЬ/. Значение рН смеси,
в которой осуществляется расщепление пеп
тида, определяется эмпирически и направлено
на оптимизацию состояния расщепляющеro pe
areHTa. Например, при использовании циано
бромида в качестве расщепляющеro peareHTa
необходимым является создание сильнокислой
среды (например, рН 2, муравьиная кислота);
однако при использовании трипсина в качестве
расщепляющею areHTa оптимальной является
слабощелочная среда (рН 8). В целом значение
рН реакционной среды не должно изменять хи
мическую структуру белка в ходе реакции pac
щепления и не должно изменяться в ходе прове
дения реакции фраrментации.
Температура. Для большинства реакций
расщепления наиболее ПОДХОДЯLl..(ей является
температура в интервале от 25 ос до 37 ос. Соз
даваемая температура должна способствовать
минимизации побочных химических реакций. Тип
тестируемоro протеина определяет выбор TeM
пературы реакционной среды, поскольку HeKOTO
рые белки с повышением температуры склонны к
денатурации. Например, расщепление рекомби
нантноro бычьеro соматропина проводится при
температуре 4 ос, поскольку при более высокой
температуре он осаждается в ходе проведения
реакции расщепления.
Время. В случае если имеется достаточное
количество тестируемоro образца, следует про
вести испытание по определению времени, яв
ляющеroся оптимальным для получения воспро
изводимой карты и достаточным для проведения
полноro расщепления. Время расщепления
варьируется от 2 ч до 30 ч. Реакция прекращает
ся либо прибавлением кислоты, которая не OKa
зывает влияния на полученную пептидную карту,
либо замораживанием.
Количество используеМ020 расщепляюще
20 реа2ента. Для обеспечения достаточноro бы
строю расщепления (от 6 ч до 20 ч) используется
избыточное количество расщепляющеro peareH
та; тем не менее, еro количество должно быть
минимизировано, чтобы предотвратить вклад
peareHTa в структуру пептидной карты. Обычно
анализируемый протеин и peareHT берут в COOT
ношении 20: 1 или 200: 1. Для оптимизации про
цедуры расщепления рекомендуется добавлять
расщепляющий areHT в два или более этапов.
При этом конечный объем реаrирующей смеси
должен оставаться достаточно небольшим для
обеспечения следующеro этапа пептидноro Kap
тирования разделения. Для тоro, чтобы ис
ключить возможное влияние искажающих про
дуктов расщепления (артефактов) на результаты
последующеrо анализа, проводится контроль
ное определение со всеми реаrентами за исклю
чением анализируемоro белка.
ХРОМАтоrРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ
Для разделения пептидных фраrментов ис
пользуются различные методы. Выбор метода
зависит от белка, для котороro составляется
пептидная карта. В таблице 2.2.55. 2 перечис
лены методы, которые на данном этапе успеш
но используются для разделения пептидных
фраrментов при составлении пептидных карт. В
данном разделе описывается один из методов
хром поrрафическоro разделения наиболее
часто используемый метод обращенно фазовой
вэжх. .
Критическими факторами при вэжх раз
делении являются чистота растворителей и
подвижной фазы. Для проведения обращен
но фазовой вэжх рекомендуется использо
вание растворителей и воды степени чистоты
«для вэжх». Растворенные rазы являются про
блемой для rрадиентных систем в тех случаях,
коrда растворимость rаза в растворителе может
быть меньше в смеси, чем в самом растворите
ле. В качестве эффективноro способа деrаза
ции может быть использована вакуумная деrаза
ция и воздействие ультразвуком. В случае если
твердые частицы в растворителе попадут в xpo
матоrрафическую систему, они MOryT нарушить
148
rосударственная фармакопея Республики Беларусь
rерметичность клапанов насоса или засорить
верхнюю часть хроматоrрафической колонки. В
связи с этим рекомендуется проведение филь
трации до и после насоса.
Таблица 2.2.55. 2
Методы, используемые для разделения
пептuдных фра2ментов
Обращенно фазовая высокоэффективная
жидкостная хроматоrрафия
Ионообменная хроматоrрафия
Хроматоrрафия rидрофобною взаимодей
ствия
Полиакриламидный rель электрофорез, Heдe
натурирующий
rель электрофорез в полиакриламидных
слоях с использованием мицеллярных систем
(натрия додецилсульфат)
Капиллярный электрофорез
Бумажная электро хроматоrрафия
Электрофорез на бумаrе при высокой разно
сти потенциалов
Хроматоrрафическая колонка. Для каж
доrо белка выбор колонки осуществляется эм
пирически. Колонки с размером пор от 1 О нм ДО
30 нм, заполненные сорбентом на основе сили
каrеля, MOryT обеспечить оптимальное разделе
ние. Для более мелких пептидных фраrментов
силика2ель октилсилильный для xpOMaт02pa
фии р (З 10 мкм) и силика2ель октадецилси
лильный для хромат02рафии Р (3 10 мкм) в
качестве наполнителей колонки являются более
эффективными, чем силика2ель бутилсилиль
ный для хроматО2рафии Р (5 10 мкм).
Растворитель. Наиболее часто в качестве
растворителя используется вода и ацетони
трил в качестве орrаническою модификатора с
добавлением не более 0,1 % кислоты трифто
руксусной. При необходимости для повышения
растворимости компонентов смеси прибавляют
пропанол или 2 пропанол. Прибавление спиртов
не должно приводить к чрезмерному повыше
нию вязкости компонентов.
Подвижная фаза. Подвижная фаза, co
держащая фосфатный буфер, используется
для обеспечения возможности выбора рН, по
скольку изменение рН в интервале от 3,0 до 5,0
улучшает разделение пептидов. содержащих
кислотные остатки (например. rлутаминовая и
аспараrиновая кислоты). Натрия или калия фос
фаты, ацетат аммония, фосфорная кислота при
значении рН между 2 и 7 (или выше при исполь
зовании полимерноrо наполнителя) используют
ся с ацетонитрильным rрадиентом. Достаточно
часто используется ацетонитрил с трифторук
сусной кислотой.
rрадиент. rрадиент может быть линейным,
нелинейным или включать ступенчатое измене
ние состава. Для разделения сложных смесей
рекомендуется низкая скорость изменения co
става. rрадиент оптимизируют, добиваясь чет
коro разрешения с одним или двумя пиками, KO
торые становятся «маркерными» пиками для
теста.
Изократическое элюирование. Изокра
тическая ВЭЖХ с подвижной фазой постоян
Horo состава используется ввиду ее удобства и
улучшенноrо отклика детектора. В ряде случаев
сложно подобрать оптимальный состав подвиж
ной фазы, позволяющий добиться хорошею раз
решения для каждоro пика. Для проведения изо
кратической ВЭЖХ не должны использоваться
подвижные фазы, для которых незначительное
изменение в соотношении компонентов или в
значении рН существенно влияет на время yдep
живания пиков на пептидной карте.
Друrие параметры. Для достижения xo
рошей воспроизводимости, как правило, требу
ется контроль температуры колонки. Скорость
подвижной фазы варьируется от 0,1 мл/мин до
2,0 млlмин, детектирование пептидов осущест
вляется спектрофотометрически детектором при
длине волны от 200 нм ДО 230 нм. Используют
ся И друrие методы детектирования (например,
постколоночная дериватизация), но они не столь
надежны и универсальны, как детектирование в
ультрафиолетовой области.
Валидация. В данном разделе при водятся
экспериментальные средства для оценки общих
характеристик используемоro метода. Критерии
оценки приroдности системы зависят от опреде
ления критических параметров испытания, вли
яющих на интерпретацию данных и их приемле
мость. Критические пара метры являются также
критериями, по которым производится контроль
расщепления и анализа пептидов. Для контроля
полноты требуемою расщепления используется
сравнение со стандартным образцом, который
был подверrнут такому же воздействию, как и ис
пытуемый белок. Использование стандартноro
образца параллельно С испытуемым белком яв
ляется необходимым для разработки и YCTaHOB
ления значений критериев оценки приrодности
системы. Кроме этою, с целью дополнительно
ro сравнения включается образец xpoMaтorpaM
мы стандартноro образца. Друrие показатели
MorYT включать визуальный контроль раствори
мости белка или пептидов, отсутствие интактно
ro белка. измерение откликов труднорасщепля
емых пептидов. Критические параметры оценки
приroдности системы для пептидноro анали
за будут зависеть от KOHKpeTHoro используемо
ro метода разделения и детектирования, а также
требований к получаемым в результате анализа
данным.
В случае, Korдa пептидное картирование ис
пользуется в качестве испытания по идентифи
2.2.55. Пептидное картирование
149
кации, требования к приrодности системы для
идентифицируемых лептидов включают избира
тельность и воспроизводимость. В этом случае,
как и в случае идентификации отличающихся по
составу белков, определение первичной CTPYK
туры пептидных фраrментов при картировании
обеспечивает как верификацию известной пер
вичной структуры, так и идентификацию CTPYK
туры отличающихся белков путем сравнения
с пептидной картой стандартноrо образца для
белка установленной структуры. Использование
подверrнутою расщеплению стандартною об
разца при определении разделения пептидов и
аминокислот является референтным методом.
Для анализа отличающихся по составу белков
может быть использована охарактеризованная
по составу смесь отличающеюся белка и CTaH
дартноro образца, в особенности если отличные
по составу белки находятся в зоне худшеro раз
решения на пептидной карте. Критерием выбора
системы определения структуры белка может
являться число основных определяемых пепти
дов. Критерии выбора системы для определения
структуры белка лучшим образом определяются
разрешением пиков пептидов. Хроматоrрафиче
ские параметры, такие как разрешение между
пиками, ширина пика у основания, площадь
пика, степень размывания заднеro фронта лика
и эффективность колонки, MOryT быть использо
ваны для определения разрешения пептидов. В
зависимости от испытуемоro белка и используе
мою метода разделения MOryт устанавливаться
требования к разрешению одноro или несколь
ких пелтидов.
Повторный анализ продуктов расщепления
стандартноro образца для испытуемоro белка
позволяет установить количественные xapaKTe
ристики прецизионности и открываемости. OT
крываемость определяемых пептидов в целом
устанавливается при помощи внутренних и
внешних пептидных стандартов. Прецизион
ность выражается как относительное стандарт
ное отклонение. Следует ожидать различий в
извлечении и воспроизводимости идентиqpици:
руемоro протеина; следовательно, критерии дo
пуска для оценки приroдности системы должны
быть установлены как для извлечения, так и для
воспроизводимости идентифицируемых пепти
дов. Установленные критерии допуска являются
специфическими для данноro протеина и указы
ваются в частных статьях.
Визуальное сравнение относительных
времен удерживания, параметров пиков (площа
ди пика или высоты пика), числа пиков и в целом
метода элюирования производится изначально.
Оно дополняется и подтверждается математи
чески м анализом соотношений анализируемых
параметров пиков и хроматоrрафическоrо про
филя смеси 1:1 (об/об) продуктов расщепле
ния испытуемоro образца и образца сравнения.
Идентичность анализируемою образца под
тверждается, если все пики продуктов расще
пления анализируемою белка и стандартноro
образца имеют аналоrичные времена удержи
вания и соотношения оцениваемых параметров
пиков.
Если пики, которые изначально характери
зовались значительно различающимися Bpe
менами удерживания, затем в смеси 1:1 об
наруживались единым пиком, изначально
выявленное различие является показателем
нестабильности системы. Однако если в смеси
1:1 наблюдаются разделенные пики, это явля
ется доказательством наличия различных пеп
тидов в каждом из пиков. Если пик в смеси 1: 1
существенно шире, чем соответствующий пик в
анализируемом белке и стандартном образце,
это может означать наличие различных пепти
дов. Было предложено использование компью
терной проrраммы распознавания для анали
за данных пептидноro картирования, однако
аспекты валидации компьютерной проrрам
мы исключают ее использование в качестве
справочноro теста в ближайшем будущем. Ис
пользовались также иные автоматизирован
ные подходы с использованием математиче
ских формул, моделей и систем распознавания.
Одним из таких подходов, например, является
автоматическое определение соединений Me
тодом ИК спектроскопии и применение диод
но матричноro УФ спектральноro анализа для
идентиqpикации пептидов. Данные методы
имеют оrраничения, обусловленные HeДOCTa
точным разрешением, одновременным элюиро
ванием фраrментов. различиями в абсолютных
значениях лараметров ликов между продукта
ми расщепления анализируемоro и CTaHдapT
ных образцов.
Можно про извести множественное cpaB
нение времен удерживания и площадей или
высоты пиков для выбранной rруппы соответ
ствующих пиков, которые правильно иденти
фицируются на пептидной карте. Площади
ликов MOryT быть рассчитаны с использова
нием одноro лика, демонстрирующеrо относи
тельно небольшую вариабельность в качестве
внутреннеro стандарта, с учетом тоro, что ин
теrрирование площади пика является чувстви
тельным к базовой вариабельности. и может
служить причиной ошибки анализа. Как аль
тернативный вариант для испытуемоro образ
ца может быть рассчитан процент высоты пика
каждоro пептида относительно суммы высоты
всех пиков. Затем полученный процент сравни
вается со значением аналоrичноrо пара метра
соответствующею пика стандартноro образца.
Возможность автоrидролиза трипсина KOHTpO
лируется при помощи создания пептидной Kap
ты плацебо, получаемой при обработке трипси
ном раствора без определяемоro белка.
Минимальным требованием для оценки Ka
чества пептидноrо картирования является при
нятая процедура тестирования, которая включа
ет приrодность системы в качестве контроля. В
150
rосударственная фармакопея Республики Беларусь
целом на начальном этапе реryляторноro про---
цесса качественный анализ пептидной карты
анализируемоro белка является достаточным. С
усовершенствованием процесса реryляторноro
одобрения белков дополнительные испытания по
оценке качества MOryт включать частичную вали
дацию аналитической процедуры для обеспече
ния rарантии тоro. что метод будет произведен в
соответствии с запланированным при разработке
пептидной карты определяемоrо белка.
АНАЛИЗ И ИДЕНТИФИКАЦИЯ ПЕПТИДОВ
В данном разделе приводится PYKoвoд
ство по использованию метода составле
ния пептидных карт лри разработке част
ных фармакопейных статей и нормативных
документов по контролю качества (нД) при
рееистрации лекарственных средств.
Использование метода составления пеп
тидных карт в качестве метода качественно
ro определения не требует подробной xapaK
теристики индивидуальных пептидных пиков.
Однако валидация метода составления пеп
тидных карт при разработке частных фармако
пейных статей и НД требует исчерпывающей
характеристики каждоrо из индивидуальных
пиков пептидной карты. Для характеристики
индивидуальных пиков MorYT быть использо
ваны как метод N концевоro секвенирования
с последующим анализом аминокислот, так и
метод с использованием масс спектроскопии.
В случаях, коrда для составления xapaK
теристики используется метод N концевоrо
секвенирования с последующим анализом
аминокислот. аналитическое разделение Mac
штабируют для целей препаративноro разде
ления. НеоЬходимо убедиться на основании
эмпирических данных, что ухудшения разре
шения между пиками вследствие проведения
масштабирования не происходит. Элюаты,
соответствующие специфическим пептидным
пикам, собирают, концентрируют в вакууме
и, при необходимости. повторно xpoMaTorpa
фируют. Аминокислотный анализ фраrмен
тов может быть оrраничен размером пепти
дов. В случае если N концевая аминокислота
заблокирована, может потребоваться ее BЫ
свобождение до секвенирования. С целью по
лучения характеристики может также исполь
зоваться C KOHцeBoe секвенирование белков
в комбинации с карбоксипептидазой и MeTO
дом матрично активированной лазерной дe
сорбции/ионизации с времяпролетным Macc
анализатором (MALD/ TOF).
Использование маr.с спектроскопии для
характеристики пептидных фраrментов про
изводится либо путем непосредственноrо
введения выделенных пептидов, либо с ис
пользованием оп /iпе системы ВЭЖХ с Macc
спектрометром для структурною анализа.
В целом он включает электрораспыление
и MALO/ TOF масс спектрометрию, а также
бомбардировку быстрыми атомами. TaHдeM
ная масс спектроскопия также использует
ся для определения последовательности MO
дифицированных белков и для определения
типа произошедшей аминокислотной модифи
кации. Определение расположения дисуль
фидных связей в различных сульфrидрил--со
держащих пептидах может быть произведено
методом сравнения масс спектров продук
та расщепления до и после восстановления.
Если некоторые части первичной CTPYКТY
ры не MorYT быть достаточно четко отражены
пептидной картой, может потребоваться co
ставление вторичной пептидной карты. Целью
валидированноro метода характеристики про
теина методом пептидноrо картирования яв
ляется определение как минимум 95 % Teo
ретическоrо состава структуры белка.
01/2013:20256
2.2.56. АНАЛИЗ АМИНОКИСЛОТ
Анализ аминокислот это комплекс Me
тодов, используемых для определения амино
кислотноrо состава или количественноrо co
держания аминокислот в белках, пептидах и
пекарственных средствах. Белки и пептиды
являются макромолекулзми, состоящими из
ковалентно связанных остатков аминокислот,
которые образуют линейные полимеры. После
довательность аминокислот в белках или пеп
тидах определяет свойства молекулы. Белки
представляют собой крупные молекулы, KOTO
рые обычно существуют в виде трехмерных
структур со специфической конформацией, в
то время как пептиды имеют меньший размер
и MorYT состоять только из нескольких амино
кислот. Анализ аминокислот может быть ис
пользован для количественноrо определения
белков и пептидов, определения подлинности
белков или пептидов на основе их аминокис
лотноrо состава, структурноro анализа белков
и пептидов, для оценки стратеrий расщепле
ния для пептидных остатков и для обнаруже
ния атипичных аминокислот, которые MorYT
присутствовать в белках или пептидах. Перед
анализом аминокислот необходимо rидролизо
вать белки/пептиды до получения отдельных
аминокислотных составляющих. После rидро
лиза белков/пептидов ПрОI \есс анализа ами
нокислот может быть таким же, как и дЛЯ CBO
бодных аминокислот в друrих лекарственных
средствах. Аминокислотные составляющие ис
пытуемоrо образца обычно подверrаются хи
мической модификации (или дериватизации)
для анализа.
2.2.56. Анализ аминокислот
151
ОБОРУДОВАНИЕ
Методы, используемые для анализа ами
нокислот, обычно основаны на хроматоrрафи
ческом разделении аминокислот, присутству
ющих в испытуемом оБРf.!зце. Современные
методики анализа основаны на использова
нии автоматизированных хроматоrрафиче
ских приборов для решения аналитических
задач. В качестве прибора для анализа ами
НОf<ИСЛОТ обычно используют жидкостный xpo
матоrраФ низкоrо или BbIcoKoro давления, сп<r
собный создавать rрадиентное элюирование
подвижной фазой, что обеспечивает разделе--
ние анализируемых аминокислот на xpOMaTO
rрафической колонке. Если анализ образца не
осуществляется с использованием предколо
ночной дериватизациИ', прибор должен иметь
устройство для постюапоночной дериватиза
ЦИIA. В качестве детектора обычно исполь
зуется спектрофотометрический детектор в
улырафиолетовой/видИ'моЙ области или флу
оресцентный детектор в зависимости от ис
пользуемоro метода дериватизации. Реrистри
рующее устройство (например, интеrратор)
используется для преобразования аналоrово
ro сиrнала из детектора и для проведения KO
личественных вычислений. Предпочтительно,
чтобы прибор IIIспользовался исключительно
для анализа аминокислот.
ОСНОВНЫЕ МЕРЫ
ПРЕДОСТОРОЖНОСТИ
При выполнении анализа аминокислот aHa
питик Bcerдa должен учитывать возможность
фоновоro заrрязнения. Необходима высокая
степень чистоты реактивов (например, низкая
чистота кислоты хлористоводородной может
способствовать заrрязнению rлицина). Анали
тические реактивы должны меняться реryлярно
каждые несколько недель, при этом должны ис
пользоваться только растворители класса «для
высокоэффективной жидкостной xpoMaTorpa
фии (ВЭЖХ)>>. Возможное заrрязнение микроор
rанизмами и посторонними частицами, которые.
MOryт присутствовать в растворителях, устраня
ется путем фильтрования этих растворителей
перед применением, хранением растворителей
в закрытых емкостях и проведением анализа с
защитой от прямоro солнечноro света.
Качество анализа аминокислот определя
ется лабораторной практикой. Приборы раз
мещают в местах наименьшеro передвижения
персонала. Лабораторию содержат в чистоте.
Очищают и калибруют пипетки соrласно rрафи
ку. Наконечники ПИrJеток хранят в закрытой KO
робке; аналитикам не позволяется TporaTb их
руками. Аналитик должен использовать непри
пудренные латексные или аналоrичные перчат
ки. Оrраничивают число открытий и закрытий
виалы с испытуемым образцом, так как пыль
может приводить к завышению результатов по
содержанию rлицина, серина и алан ина.
Для получения удовлетворительных ре--
зультатов анализа аминокислот прибор необхо
димо обслуживать надлежащим образом. Если
прибор используется по стандартной проrрам
ме, он должен ежедневно проверяrься на OT
сутствие течи, на стабильную работу детектора
и ламп и на способность колонки поддерживать
необходимую степень разделения индивидуаль
ных аминокислот. Соrласно rрафику проводят
очистку или замену всех фильтров прибора и
друroе техническое обслуживание
СТАНДАРТНЫЕ ОБРАЗЦЫ
Необходимые стандартные образцы ами
нокислот для проведения анализа аминокислот
коммерчески доступны и обычно представляют
собой водный раствор смеси аминокислот. При
определении аминокислотноro состава наряду
с испытуемым образцом анализируются cтaH
дартные образцы белков или пелтидов, KOTO
рые lAСПОЛЬЗУЮТСЯ в качестве контроля для под
тверждения полноты Bcero процесса. Для этой
цели в качестве стандартноro образца белка
используют высокоочищенный бычий CЫBOpO
точный альбумин.
rРАДУИРОВКА ОБОРУДОВАНИЯ
rрадуировка оборудования для анализа
аминокислот обычно включает анализ CTaHдapT
ноro образца аминокислот, который состоит из
смеси аминокислот, при разных концентрациях
для определения величины отклика и рабоче
ro диапазона концентраций для каждой амино
кислоты. Концентрация каждой аминокислоты в
стандартном образце известна. В процессе rpa
дуировки при выполнении анализа аминокис
лот стандартный образец разводят до различ
ных концентраций анализируемоro вещества в
пределах ожидаемой области линейности co
rласно используемой аналитической методике.
Затем анализируют растворы с различными KOH
центрациями анализируемоro вещества. По оси
ординат наносят площади пиков каждой амино
кислоты, а по оси абсцисс соответствующие
известные концентрации каждой аминокислоты
с учетом разведений. Эти результаты позволяют
определить область концентраций аминокислот,
в которой зависимость площади пиКа данной
аминокислоты от ее конц нтрации является ли
нейной функцией. Важно, чтобы образцы для
анализа аминокислот были приroтовлены таким
образом, чтобы концентрации аминокислот в
этих образцах находились в пределах аналити
ческих rраниц (т. е. в рабочей линейной обла
сти) используемой меидики с целью получения
точных и воспроизводимых результатов.
Для определения коэффициента отклика
каждой аминокислоты анализируют от 4 до 6
концентраций стандартноro образца. Коэффи
циент отклика рассчитывается как средняя пло
щадь или высота пика в расчете на наномоль
(10-9 моль) аминокислоты, представленной в
152
rосударственная фармакопея Республики Беларусь
стандартном образце. Коэффициенты отклика
каждой аминокислоты используют для расчета
концентрации каждой аминокислоты, представ
ленной в испытуемом образце. Количество Be
щества (наномоль) анализируемой аминокисло
ты рассчитывают путем деления площади пика.
соответствующею данной аминокислоте, на KO
эффициент отклика этой аминокислоты. Для py
тинною анализа может быть достаточно rрадуи
ровки по одной точке; при этом rрадуировка по
коэффициентам отклика каждой аминокисло
ты должна часто обновляться и проверятся для
контроля достоверности.
ПОВТОРЯЕМОСТЬ
Полный анализ аминокислот требует от aHa
литической лаборатории контроля повторяе
мости результатов количественноro определе
ния. Во время хроматоrрафическою разделения
аминокислот или их производных на xpOMaTO
rpaMMe наблюдается множество пиков, относя
щихся К аминокислотам. Большое количество
пиков делает необходимым наличие проrрамм
ною обеспечения, способноro неоднократно
идентифицировать пики, основываясь на Bpe
мени удерживания, и интеrрировать площади
пиков для количественных расчетов. Типичная
оценка повторяемости включает приroтовление
стандартною раствора аминокислот и анализ
мноroкратных повторных вводов пробы (напри
мер, 6 или более повторных вводов) одноro и
тоro же стандартноro раствора. Относительное
стандартное отклонение (RSD) определяют для
времени удерживания и интеrрированной пло
щади пика каждой аминокислоты. Оценку по
вторяемости дополняют включением мноroкрат
ных количественных определений, проводимых
в течение нескольких дней разными аналитика
ми. Мноroкратные количественные определения
включают приrотовление стандартных разведе
ний из исходных материалов для определения
различий результатов, возникающих из за мани
пуляций с образцом. При оценке повторяемости
часто проводят анализ аминокислотноrо состава
стандартноro образца белка (например, бычий
сывороточный альбумин). На основе оценки от
носительных стандартных отклонений (RSO) ла
боратория рассчитывает аналитические пре
делы для подтверждения тою. что анализы в
данной лаборатории проведены надлежащим
образом. Для rарантирования наилучших pe
зультатов желательно установить наименьшие
практические пределы отклонений. Факторы, на
которые следует обратить внимание для YMeHb
шения отклонений в результатах анализа ами
нокислот, включают приroтовление образца,
высокие фоновые спектральные помехи из за
качества реактивов и/или из за лабораторных
манипуляций, работу и обслуживание приборов,
анализ и интерпретацию данных, профессио
нальные качества аналитика и еro состояние.
Все параметры, о которых идет речь, должны
всесторонне исследоваться в рамках проведе
ние валидации.
приrОТОВЛЕНИЕ ОБРАЗЦА
Точные результаты анализа аминокислот
требуют использования очищенных образцов
белков и пептидов. Компоненты буфера (Ha
пример, соли, мочевина. детерrенты) MorYT по
влиять на анализ аминокислот и должны yдa
ляться из образца перед анализом. Методики,
которые используют постколоночную дерива
тизацию аминокислот, обычно зависят от вли
яния компонентов буфера в меньшей степени,
чем методики с предколоночной дериватизаци
ей. Для уменьшения потенциально возможно
ro фоновоro заrрязнения, улучшения OTKpЫBae
мости анализируемою вещества и уменьшения
трудоемкости число операций с образцом жела
тельно оrраничить. Общие технические приемы,
которые используются для удаления компонен
тов буфера из образцов белка, включают сле
дующие методы: (1) введение образца белка в
обращенно фазовую систему В3ЖХ. удаление
белка с помощью летучеro растворителя, coдep
жащею достаточное количество орrаническоro
компонента, и высушивание образца в BaKYYM
ной центрифуrе; (2) диализ с летучим буфер
ным раствором или водой; (3) ультрафильтраци
онное центрифуrирование для замены буфера
летучим буферным раствором или водой;
(4) осаждение белка из буферноrо раствора с
использованием орrаническоrо растворителя
(например, ацетона); (5) rель фильтрация.
ВНУТРЕННИЕ СТАНДАРТЫ
В ходе аминокислотноrо анализа peKOMeH
дуется использовать внутренний стандарт для
контроля физических и химических потерь и KO
лебаний результатов. Перед rидролизом к pac
твору белка может быть прибавлено точно из
вестное количество BHYTpeHHero стандарта.
Открываемость внутреннею стандарта указыва
ет на общую открываемость аминокислот белко
вою раствора. Однако свободные аминокисло
ты ведут себя иначе. чем аминокислоты белка
во время rидролиза. скорость высвобождения
которых всеrда разная. Поэтому использование
BHYTpeHHero стандарта для введения поправки
на потери в ходе rидролиза может давать HeHa
дежные результаты, что необходимо учитывать
при их интерпретации. Внутренние стандарты
также MorYT быть добавлены к смеси аминокис
лот после rидролиза для введения поправки на
различия во введениях пробы образца, на изме
няющуюся стабильность реактивов и на откло
нения скорости подвижной фазы. В идеальном
случае в качестве BHYTpeHHero стандарта ис
пользуют коммерчески доступную аминокисло
ту, которая отличается от природных аминокис
лот. Кроме тоro, внутренний стандарт должен
быть стабильным в ходе rидролиза, еro коэффи
циент отклика должен иметь линейную зависи
2.2.56. Анализ аминокислот
153
мость от концентрации и выходить из xpOMaтo
rрафической колонки со свойственным только
этому стандарту временем удерживания без пе
рекрывания пиков друrих аминокислот. Наибо
лее часто используемые стандартные образцы
аминокислот включают норлейцин, нитротиро
зин и а аминомасляную кислоту.
rидролиз БЕЛКОВ
ДЛЯ анализа аминокислот белков и пеnти
дов необходимо проводить rидролиз. Лабора
торная посуда, используемая для проведения
rидролиза, должна быть очень чистой. Оnyдри
вающий порошок перчаток, а также отпечатки
пальцев на посуде, в которой проводится rидP<r
лиз, MOryт привести к заrрязнению. Для очист
ки стеклянных пробирок для проведения rидро
лиза используют кипячение в течение 1 ч в 1 М
растворе кислоты хлористоводородной или за
мачивание в концентрированной азотной кисло
те или в смеси из равных объемов концентриро
ванных хлористоводородной и азотной кислот.
Чистые пробирки, используемые для rидролиза,
промывают водой высокоочищенной, затем опо
ласкивают метанолом для хроматоrрафии, BЫ
сушивают в течение ночи в сушильном шкафу
и хранят в закрытом состоянии вплоть до ис
пользования. Для удаления заrрязнения из про
бирок, которые используются для rидролиза,
также можно использовать прокаливание чистой
стеклянной посуды при температуре 500 сс в
течение 4 ч. Допускается использование COOT
ветствующих одноразовых лабораторных при
надлежностей.
Кислотный rидролиз является наиболее
часто используемым методом для расщепления
белка в образце перед анализом аминокисло
Метод кислотною rидролиза может повлиять на
отклонение результатов анализа из за полно
ro или частичноro разрушения некоторых ами
нокислот: триптофан разрушается, серин и Tpe
онин частично разрушаются, метионин может
подверrаться окислению, а цистеин обычно
определяется как цистин (но открываемость ци:
стина обычно низкая вследствие частичноro
разрушения или восстановления до цистеина).
Применение соответствующеro вакуума (менее
200 мкм ртутною столба, или 26,7 Па) или BBe
дение инертноro rаза (арroна) в пространство
реакционноro сосуда может уменьшить степень
окислительной деструкции. Пептидные связи
иэолейцин изолейцин, валин валин, изолейцин
валИН и валин изолейцин расщепляются частич
но; аcnараrин и rлутамин дезамидируются с об
разованием соответственно аспараrиновой и
mутаминовой КИСJlОТ. Потеря триптофана, аспа
раrина И rлутамина в ходе кислотноro rидролиза
оrpаничивает количественное определение до
17 аминокислот. Некоторые из описанных ниже
методов кислотноro rидролиза используются
для решения этих задач. Некоторые методы rи
дролиза (например. методы 4 11) MOryт приво
дить К превращениям в друrие аминокислоты.
Поэтому преимущества использования KOHKpeT
ной методики оцениваются относительно задач,
решаемых с ее помощью, и всесторонне изуча
ются перед выбором методики, отличающейся
от обычноro кислотноro rидролиза.
Для определения исходной концентрации
аминокислот, которые частично разрушаются
или медленно отщепляются, часто используют
исследование rидролиза во времени (анализ
аминокислот при кислотном rидролизе в тече
+lие 24 ч, 48 ч и 72 ч). Путем построения rрафика
зависимости найденной концентрации частично
разрушающихся (лабильных) аминокислот (Ha
пример, серина и треонина) от времени rидроли
за и экстраполяции полученной кривой к началу
координат определяют исходную концентрацию
этих аминокислот. Концентрацию остатков ами
нокислот, которые медленно отщепляются (Ha
пример, изолейцин ивалин), принимают равной
высоте плато на полученном rрафике зависи
мости концентрации остатков от времени rи
дролиза. Если rидролиз будет протекать слиш
ком долro, концентрация аминокислот в образце
начнет уменьшаться, что укажет на их разруше
ние при данных условиях rидролиза.
Приемлемая альтернатива исследованию
rидролиза во времени подверrнуть стандарт
аминокислот с известными концентрациями
таким же условиям rидролиза, как и в случае с
испытуемым образцом. Однако аминокислоты в
свободном состоянии MOryT не в полной мере OT
ражать превращения, происходящие в ходе rи
дролиза: степень разрушения лабильных ами
нокислот, находящихся в составе пептидов или
белков, и, в особенности, степень расщепле
ния медленно расщепляемых связей (напри
мер, связи изолейцин валин). Эта методика по
зволяет аналитику учесть лишь некоторую часть
разрушающихся аминокислот. Возможно приме
нение кислотноro rидролиза с использованием
микроволновоro излучения, который отличается
быстротой, но требует специальноro оборудова
ния, а также специальных мер предосторожно
сти. Оптимальные условия rидролиза с ИСПОЛЬ
зованием микроволновоro излучения должны
быть установлены для каждоro отдельноro бел
ковоrо/протеиновоro образца. Микроволновый
rидролиз обычно протекает в течение несколь
ких мину но отклонение даже в одну минуту
может привести к неудовлетворительным pe
зультатам (например, неполный rидролиз или
разрушение лабильных аминокислот). Полный
протеолиз с применением смеси протеаз может
быть слишком СЛО)l{НЫМ, он требует надлежаще
ю контроля и обычно fiолее подходит для пепти
дов, чем для белков.
Для определения оптимальных условий в
ходе первоначальноro анализа белка неизвест
ноro состава проводятся эксперименты с раз
личными временами rидролиза и при разных
температурных режимах.
154
rосударственная фармакопея Республики Беларусь
МЕТОД 1
Для rидролиза используется кислота хло
ристоводородная, содержащая фенол, это
наиболее общая процедура, используемая для
rидролиза белка/пептида при анализе аминокис
лот. Добавление фенола предупреждает peaK
цию rалоrенирования тирозина.
rидролизный раствор. 6 М кислота хлори
стоводородная, содержащая от 0,1 % до 1,0 %
фенола.
Методика.
Жидкофазный rидролиз Образец белка
или пептида помещают в rидролизную ампулу
и высушивают (образец высушивают для тоro,
чтобы вода в образце не разводила кислоту, ис
пользуемую для rидролиза). Прибавляют rидро
лизный раствор из расчета 200 мкл на 500 MKr
лиофилизированноrо белка. Образец в ампуле
замораживают в бане с сухим льдом и aцeTO
ном и запаивают в вакууме при помощи пла
мени. Образцы обычно rидролизуют при TeM
пературе 110 ос в течение 24 ч в вакууме или
в атмосфере инертноro rаза для предотвраще
ния окисления. Если есть подозрения, что ис
пытуемый белок полностью не rидролизуется,
рассматривают возможность проведения rи
дролиза в течение более длительноro времени
(например, 48 ч и 72 ч).
Парофазный 2идролиз. Это один из наибо
лее общих методов кислотною rидролиза. Он
предпочтителен для микроанализа, Korдa дo
ступны лишь небольшие количества 06раз
ца. При использовании парофазноrо rидролиза
контаминация образца кислотными реактива
N И минимальна. Контейнеры с сухими образца
ми помещают в сосуд, содержащиЙ подходящее
количество rидролизноro раствора. rидролиз
ный раствор не должен контактировать с испы
туемым образцом. В свободном пространстве
сосуда используют атмосферу инертноro rаза
или вакуум (менее 200 мкм ртутноro столба, или
26,7 Па) и для проведения rидролиза HarpeBa
ют при температуре около 110 ос в течение 24 ч.
Пары кислоты rидролизуют сухой образец. Воз
можность конденсации кислоты в контейнере
с образцом должна быть сведена к минимуму.
После rидролиза испытуемый образец высуши
вают в вакууме до удаления остатков кислоты.
МЕТОД 2
Использование меркаптоэтансульфоновой
кислоты в качестве восстанавливающей кисло
ты уменьшает окисление триптофана в процес
се rИДрО.llиза.
rидролизный раствор. 2,5 М раствор Mep
каптоэтансульфоновой кислоты.
Парофазный rидролиз. От 1 MKr до
100MKr испытуемою белка/пептида высушива
ют в rидролизной ампуле. rидролизную ампулу
помещают в большую ампулу, содержащую
окопо 200 мкл rидролизноrо раствора. repMe
тично запаивают большую ампулу в вакууме
(около 50 мкм ртутноro столба, или 6,7 Па).
Наrревают rидролизную ампулу до температу
ры от 170 ос до 185 ос в течение 12,5 мин.
После rидролиза rидролизную ампулу BЫCY
шивают в вакууме в течение 15 мин до удале
ния остатков кислоты.
МЕТОД 3
Использование тиоrлиv.олевой кислоты
(TrK) в качестве восстанавливающей кислоты
предотвращает окисление триптофана при rи
дролизе.
rидролизный раствор_ 7 М раствор кис
лоты хлористоводородной, содержащий 1 %
фенола, 1 О % кислоты трифторуксусной И 20 %
кислоты тиоrликолевой.
Парофазный rидролиз. От 1 О MKr до
50 MKr испытуемою белка/пептида высушива
ют в rидролизной ампуле. rидролизную ампулу
помещают в большую ампулу, содержащую
около 200 мкл rидролизноrо раствора. repMe
тично запаивают большую ампулу в вакууме
(около 50 мкм ртутною столба, или 6,7 Па). Ha
rревают rидролизную ампулу до температуры
166 ос в течение 15 30 мин. После rидроли
за rидролизную ампулу высушивают в вакууме
в течение 5 мин до удаления остатков кислоты
Окрываемость триптофана в этом метод€' за-
висит от количества испытуемоrо образца.
МЕТОД 4
Перед rидролизом белка проводят окисле
ние цистеина/цистина и метионина кислотой
надмуравьиной.
Окисляющий раствор. Смешивают 1
объем раствора пероксида водорода (30 %) и
9 объемов кислоты муравьиной безводной и BЫ
держивают при комнатной температуре в тече
ние 1 ч. Полученную кислоту надмуравьиную ис
пользуют свежеприrотовленной.
Методика. Образец белка/пептида paCT
воряют в 20 мкл кислоты муравьиной безво
дной, наrревают при температуре 50 ос в
течение 5 мин и прибавляют 100 мкл окисля
ющеrо раствора. Процесс окисления проводят
в течение 1 О ЗО мин. В этой реакции цистеин
превращается в цистеиновую кислоту, а мети
онин в МР'тионин сульфон. Избыток реакти
ва удаляют из образца при помощи BaKYYM
ноro центрифуrирования. Окисленный белок
может быть затем rидролизован по методу 1
или методу 2. При наличии rалоrенидов эта
методика может при водить к модификации
остатков тирозина.
2.2.56. Анализ аминокислот
155
МЕТОД 5
Окисление цистеина/цистина происходит во
время жидкофазноro rидролиза с натрия азидом.
rидролизный раствор. К 6 М раствору кис
лоты хлористоводородной, содержащей 0,2 %
фенола, прибавляют натрия азид до получе
ния конечной концентрации 2 r/л. Прибавление
фенола предотвращает rалоrенирование тиро
зина.
Жидкофазный rидролиз. rидролиз белка/
пептида проводят при температуре околе 11 О ос
в течение 24 ч. Во время rидролиза присутству
ющий в образце цистеин/цистин превращается в
кислоту цистеиновую под воздействием натрия
азида, содержащеrося в rидролизном растворе.
Эта методика приводит к лучшей открываемости
тирозина, чем метод 4, но она не подходит для
количественноro определения метионина. Мети
онин превращается в смесь из метионина и двух
продуктов ero окисления метионинсульфокси
да и метионинсульфона.
МЕТОД 6
Окисление цистеина/цистина происходит
под воздействием диметилсульфоксида (ДМСО).
rидролизный раствор. К раствору 6 М
кислоты хлористоводородной, содержащей от
0,1 % до 1,0 % фенола, прибавляют ДМСО до
получения раствора с конечной концентрацией
2 % (об/об).
Парофазный rидролиз. rидролиз белка/
пептида проводят при температуре около 11 О ос
в течение 24 ч. Во время rидролиза присутству
ющий в образце цистеин/цистин превращается в
кислоту цистеиновую под воздействием ДМСО,
содержащеrося в rидролизном растворе. С
целью оrраничения разброса данных и компен
сации частичноro разрушения рекомендуется
оценивать открываемость цистеиновой кислоты
при окислительном rидролизе стандартноro об
разца белка, содержащеro 1.........а остатков цисте
ина. Коэффициент отклика rидролизата белка/
пептида обычно на 30 % ниже, чем для неrидро
лизованноro стандартноro образца цистеиновой
кислоты. Так как rистидин, метионин, тирозин
и триптофан также модифицируются, полный
анализ состава при использовании этой методи
ки невозможен.
МЕТОД 7
Восстановление и алкилирование цистеина/
цистина происходит при пиридилэтилировании в
паровой фазе.
Восстанавливающий раствор. 83,3 мкл
пиридина, 16,7 мкл 4 винилпиридина, 16,7 мкл
трибутилфосфина и 83,3 мкл воды помещают в
подходящий контейнер и перемешивают.
Методика. rидролизную ампулу с образ
цом белка/пептида (от 1 MKr до 100 MKr) по
мещают в большую ампулу. Переносят BOC
стана вливающий раствор в большую ампулу,
rерметично запаивают в вакууме (около
50 мкм ртутноro столба, или 6,7 Па) и Harpe
вают при температуре около 100 ос в течение
5 мин. Затем внутреннюю rидролизную ампулу
помещают в вакуумный эксикатор и высуши
вают в течение 15 мин для удаления OCTaT
ков реактивов. Пиридилэтилированный обра
зец затем rидролизуют соrласно описанной
ранее методике. Для оценки открываемости
пиридилэтилцистеина параллельно проводят
реакцию пиридилэтилирования стандартноrо
образца белка, содержащею 1 8 остатков ци
стеина. Длительное время инкубации при pe
акции пиридилэтилирования может быть при
чиной модификации конечных а аминоrрупп и
Е аминоrрупп лизина в белке.
МЕТОД 8
Восстановление и алкилирование цистеина/
цистина происходит при пиридилэтилировании в
жидкой фазе.
Исходные растворы. rотовят и фильтру
ЮТ 3 раствора: 1 М раствор трис rидрохлорида
с рН 8,5, содержащий 0,004 М динатрия эдетата
(исходный раствор А); 8 М раствор ryанидина rи
дрохлорида (исходный раствор В); и 10 % paCT
вор 2 меркаптоэтанола (исходный раствор С).
Восстанавливающий раствор. Для полу
чения буферноro раствора 6 М раствора rya
нидина rидрохлорида в 0,25 М растворе трис
rидрохлорида смешивают 1 объем исходноro
раствора А и 3 объема исходноro раствора В.
Методика. Около 1 О MKr испытуемоro об
разца растворяют в 50 мкл восстанавливающеro
раствора и прибавляют около 2,5 мкл исходноro
раствора С. Выдерживают при комнатной темпе
ратуре в защищенном от света месте в aTMOC
фере азота или aproHa в течение 2 ч. Для прове
дения реакции пиридинэтилирования к раствору
белка прибавляют около 2 мкл 4 винилпиридина
и выдерживают дополнительно 2 ч при KOMHaT
ной температуре в защищенном от света месте.
Обессоливают белокlпептид методом обращен
но фазовой ВЭЖХ, собирая белковую/пептид
ную фракцию. Перед кислотным rидролизом
собранный образец может быть высушен при
помощи вакуумноro центрифуrирования.
МЕТОД 9
Восстановление и алкилирование цистеина/
цистина происходит при карбоксиметилирова
нии в жидкой фазе.
Исходные растворы. rотовят, как указано
в методе 8.
156
rосударственная фармакопея Республики Беларусь
Карбоксиметилирующий раствор. rотовят
раствор 100 r/л йодацетамида в 96 % спирте.
Буферный раствор. Используют BOCCTa
навливающий раствор, приютовленный, как YKa
зано в Методе 8.
Методика. Испытуемый образец paCTBO
ряют в 50 мкл буферноro раствора и прибавляют
около 2,5 мкл исходною раствора С. Выдержива
ют при комнатной температуре в защищенном от
света месте в атмосфере азота или арюна в Te
чение 2 ч. Прибавляют карбоксиметилирующий
раствор в 1 ,5 KpaTHOM количестве от общеrо Te
оретическоro содержания тиолов и выдержива
ют дополнительно в течение 30 мин при комнат
ной температуре в защищенном от света месте.
Если содержание тиолов в белке неизвестно,
прибавляют 5 мкл 0,1 М раствора йодацетами
да на каждые 20 наномоль испытуемоro образца
белка. Для прекращения реакции прибавляют из
быток 2 меркаптоэтанола. Обессоливают белок/
пептид методом обращенно фазовой ВЭЖХ, co
бирая белковую/пептидную фракцию. Перед кис
лотным rидролизом собранный образец может
быть высушен при помощи вакуумноro центри
фуrирования. В процессе кислотноro rидроли
за полученный S карбоксиамидометилцистеин
будет превращаться в S карбоксиметилцистеин.
МЕТОД 10
Цистеин/цистин. прореаrировавший с ди
тиоrликолевой кислотой или дитиодипропионо
вой кислотой, образует смешанный дисульфид.
Выбор дитиоrликолевой кислоты или дитиоди
пропионовой кислоты зависит от требуемою
разделения при анализе аминокислот.
Восстанавливающий раствор. Раствор
1 О r/л дитиоrликолевой кислоты (или дитиоди
пропионовой кислоты) В 0,2 М растворе натрия
rидроксида.
Методика. Около 20 MKr испытуемоro об
разца помещают в rидролизную ампулу и при
бавляют 5 мкл восстанавливающеro раствора.
Прибавляют 1 О мкл изопропиловоro спирта и
удаляют всю жидкость из образца при помощи
вакуумноro центрифуrирования. Образец rи
дролизуют, используя метод 1. Преимущество
данноro метода заключается в том, что друrие
остатки аминокислот не участвуют в побочных
реакциях, а образец не нуждается в обессолива
нии перед rидролизом.
МЕТОД 11
Аспараrин и rлутамин в процессе кислот
ною rидролиза превращаются в аспараrино
вую кислоту и rлутаминовую кислоту соответ
ственно. Остатки аспараrина и аспараrиновой
кислоты обозначают как Asx, а остатки rлу
тамина и rлутаминовой кислоты обозначают
как G/x. Белки/пептиды MOryT реаrировать с
бис( 1, 1 трифторацетокси )йодбензолом (В71),
превращая при rидролизе остатки аспараrина и
rлутамина в остатки диаминопропионовой кис
лоты и диаминомасляной кислоты COOTвeTCTBeH
но. Эти превращения позволяют определять в
белке/пептиде содержание аспараrина и rлута
мина в присутствии остатков аспараrиновой кис
лоты и rлутаминовой кислоты.
Восстанавливающие растворы. rотовят
и фильтруют 3 раствора: 0,01 М раствор три
фторуксусной кислоты (раствор А); 5 М раствор
ryанидина rидрохлорида, содержащий 0,01 М
трифторуксусной кислоты (раствор В); свеже
приroтовленный раствор диметилформамида,
содержащий 36 мr/мл BТI (раствор С).
Методика. Около 200 MKr испытуемоro об
разца помещают в чистую rидролизную ампулу
и прибавляют 2 мл раствора А или раствора В и
2 мл раствора С. rерметично запаивают rидро
лизную ампулу в вакууме. Испытуемый обра
зец наrревают при температуре 60 ос в течение
4 ч в защищенном от света месте. Затем обра
зец диализируют водой для удаления избытка
реактивов. Диализированный образец трижды
экстраrируют равными объемами бутил ацетата
и лиофилизируют. Белок может быть затем rи
дролизован соrласно описанной ранее методи
ке. Остатки а,l3 диаминопропионовой кислоты и
а, у диаминомасляной кислоты обычно не OTдe
ляются от остатков лизина ионообменной xpo
матоrрафией, используемой при анализе ами
нокислот. Таким образом, при использовании
ионноro обмена в качестве метода разделения
при анализе аминокислот содержание аспара
rина и rлутамина представляет собой разни
цу между количественным содержанием аспа
раrиновой кислоты и rлутаминовой кислоты,
полученным при кислотном rидролизе без дe
риватизации и полученным при кислотном rи
дролизе с ВТI дериватизацией. Количественное
содержание треонина, метионина, цистеина, ти
розина и rистидина может быть изменено при
ВТI дериватизации; при необходимости опре
деления этих аминокислотных остатков белка/
пептида следует проводить rидролиз без BТI
дериватизации.
МЕтодолоrия АНАЛИЗА
АМИНОКИСЛОТ: ОБЩИЕ ПРИНЦИПЫ
Существует множество способов анализа
аминокислот. Выбор конкретною способа зача
стую определяется требуемой чувствительно
стью количественноro определения. В целом
около половины применяемых способов анали
за аминокислот основаны на разделении свобод
ных аминокислот посредством ионообменной
хроматоrрафии с последующей постколоночной
дериватизацией (например, с нинrидрином или
о фталевым альдеrидом). Метод постколоноч
2.2.56. Анализ аминокислот
157
ной дериватизации может быть использован для
образцов, содержащих небольшое количество
буферных компонентов (таких как соли и моче
вина), и обычно требует от 5 MKr до 10 MKr испы
туемоro образца белка для проведения одноro
анализа. Остальные способы анализа амино
кислот обычно включают предколоночную дери
ватизацию свободных аминокислот (например,
с фенилизотиоцианатом; с 6 аминохинолил N
rидроксисукцинимидилкарбаматом или о--фта
левым альдеrидом; с (диметиламино)азобензол
сульфонилхлоридом; с 9 фторенилметилхлор
формиатом; с 7 фтор 4 нитробензо 2 окса 1 ,з
диазолом) с последующим обращенно фазовым
ВЭЖХ анализом. Метод предколоночной дe
риватизации отличается высокой чувствитель
ностью (обычно требуется от 0,5 MKr до 1,0 MKr
испытуемоro образца белка для проведения
одноro анализа), однако на неro MOryт оказывать
влияние компоненты солевоro буфера, coдep
жащиеся в испытуемом образце. В результате
предколоночной дериватизации MOryт образо
вываться множественные производные одной
аминокислоты, что приводит к усложнению ин
терпретации результатов.
Для количественноro анализа аминокислот
MOryT быть использованы приведенные ниже
методы. Приборы и реактивы для проведения
этих испытаний коммерчески доступны. Кроме
тоro, мноrие из этих методов имеют модифи
кации с различным приroтовлением реактивов,
проведением химических реакций, различными
хроматоrрафическими системами и так далее.
Специфические параметры MOryT отличаться в
зависимости от конкретноro оборудования и Me
тодики выполнения анализа. Мноrие лаборато
рии используют более одной методики анализа
аминокислот, так как каждая из них имеет свои
преимущества. В каждой из этих методик анало
roвый сиrнал обнаруживается с помощью дeTeK
тора, а площади пиков используются для коли
чественноro определения.
МЕТОД1.ПОСТКОЛОНОЧНАЯ
ДЕРИВАТИ3АЦИЯ С нинrидрином
Ионообменная хроматоrрафия с постколо
ночной дериватизацией с нинrидрином является
одним из наиболее распространенных методов,
предназначенных для количественноro анали
за аминокислот. Как правило, для анализа боль
шинства сложных физиолоrических образцов
ПРИnQДной является катионообменная система
на основе ионов лития, а катионообменная си
стема на основе ионов натрия используется для
более простых смесей аминокислот, полученных
при rидролизе белков (обычно 17 аминокислот).
Разделение аминокислот на ионообменной KO
лонке достиraется подбором рН и ионной силы.
Для улучшения разделения часто используют
температурный rрадиент.
При взаимодействии аминокислоты с нин
rидрином образуется продукт с характерным
фиолетовым или желтым цветом. Аминокисло
ты, за исключением иминокислот, образуют про
дукт фиолетовоro цвета, имеющий максимум по--
rлощения при 570 нм. Иминокислоты, такие как
пролин, образуют продукт желтоro цвета, имею
щий максимум поrлощения при 440 нм.
Продукты постколоночной реакции между
нинrидрином и элюируемыми из колонки ами
нокислотами детектируются при длинах волн
440 нм и 570 нм, а полученная xpoMaTorpaMMa
используется для Оr:!ределения аминокислотно
ro состава.
Предел обнаружения для большинства
производных аминокислот обычно составля
ет 1 О пикомоль, а для производных пролина
50 пикомоль. Линейность отклика наблюдается
в области 20 500 пикомоль с коэффициентом
корреляции более 0,999. Для получения YДOB
летворительных данных рекомендуется перед
rидролизом использовать образцы массой
более 1 MKr.
МЕТОД2.ПОСТКОЛОНОЧНАЯ
ДЕРИВАТИЗАЦИЯ С О ФТАЛЕВЫМ
АЛЬДЕrидом
о Фталевый альдеrид (ОФА) реаrирует с
первичными аминами в присутствии тиольных
соединений, образуя производные изоиндола с
сильной флуоресценцией. Эта реакция исполь
зуется для постколоночной дериватизации при
анализе аминокислот методом ионообменной
хроматоrрафии. Условия разделения такие же,
как указано в методе 1.
Для образования флуоресцирующих произ
водных с ОФА вторичные амины (иминокисло
ты, такие как пролин) предварительно окисляют
натрия rипохлоритом или хлорамином Т. В Me
тодике используются сильнокислотные катионо
обменные колонки для разделения свободных
аминокислот с последующим постколоночным
окислением натрия rипохлоритом или хлора
мином Т и постколоночной дериватизацией с
использованием ОФА и тиольных соединений,
таких как N ацетил L цистеин или 2 меркаптоэ
танол. Длительное воздействие натрия rипохло
рита или хлорамина Т не оказывает заметноrо
влияния на дериватизацию первичных амино
кислот.
Разделение аминокислот на ионообмен
ной колонке достиrается подбором рН и ионной
силы. После постколоночной дериватизации
элюируемых аминокислот с ОФА продукты peaK
ции проходят через флуориметрический дeTeK
тор. Интенсивность флуоресценции ОФА про
изводных аминокислот измеряется при длине
волны возбуждающеro света 348 нм и длине
волны излучаемоro света 450 нм.
Предел обнаружения для большинства
ОФА производных аминокислот обычно COCTaB
ляет несколько десятков пикомоль. Линейность
отклика наблюдается в области от нескольких
пикомоль до нескольких десятков наномоль. Для
158
rосударственная фармакопея Республики Беларусь
получения удовлетворительных данных peKO
мендуется перед rидролизом использовать об
разцы массой более 500 Hr.
МЕТОД 3. ПРЕДКОЛОНОЧНАЯ
ДЕРИВАТИЗАЦИЯ С ФЕНИЛИЗО
ТИОЦИАНАТОМ
Фенилизотиоцианат (ФИТЦ) реаrирует с
аминокислотами с образованием фенилтиокар
бам ильных производных (ФТК производных),
которые MOryT быть обнаружены с высокой
чувствительностью при длине волны 254 нм.
Предколоночная дериватизация аминокислот
с ФИТЦ проводится перед разделением смеси
аминокислот методом обращенно фазовой
ВЭЖХ с УФ спектрофотометрическим детекти
рованием.
После удаления реактива под вакуумом
сухие замороженные производные аминокис
лот MOryT храниться без заметной деструкции в
течение нескольких недель. Раствор пробы для
введения может храниться в холодном месте в
течение 3 дней без заметных изменений xpOMa
тоrрафическоrо отклика.
Разделение ФТК производных аминокислот
методом обращенно фазовой ВЭЖХ на колон
ках, заполненных октадецилсилилсиликаrелем
(С. В ) достиrается путем подбора концентрации
ацетонитрила и ионной силы буферноro раст
Bopa Элюируемые из колонки ФТК производные
аминокислот определяют при длине волны
254 нм.
Предел обнаружения для большинства ФТК
производных аминокислот обычно составляет
1 пикомоль. Линейность отклика наблюдается
в области 20 500 пикомоль с коэффициентом
корреляции более 0,999. Для получения YДOB
летворительных данных рекомендуется перед
rидролизом использовать образцы массой
более 500 Hr
МЕТОД 4. ПРЕДКОЛОНОЧНАЯ
ДЕРИВАТИЗАЦИЯ С 6 АМИНОХИНОЛИЛ
N rидроксисУкцини
МИДИЛКАРБАМАТОМ
Предколоночная дериватизация амино
кислот с 6 аминохинолил N rидроксисукцини
мидилкарбаматом (АХК) проводится перед раз
делением смеси аминокислот методом об
ращенно фазовой ВЭЖХ. АХК реаrирует с
аминокислотами с образованием стабильных
флуоресцирующих несимметричных произво
дных мочевины (АХК аминокислот), которые
леrко поддаются анализу методом обращенно
фазовой ВЭЖХ с флуориметрическим детекти
рованием.
Разделение АХК производных аминокислот
методом обращенно фазовой ВЭЖХ на колон
ке С'8 достиrается подбором концентрации aцe
тонитрила и ионной силы буферною раствора.
Селективное флуоресцентное детектирование
производных при длине волны возбуждающе
ro света 250 нм и при длине волны излучаемоro
света 395 нм допускает прямой ввод реакцион
ной смеси без каких либо существенных попра
вок на основной флуоресцирующий побочный
продукт 6 аминохинолин. Избыток реактива
быстро rидролизуется (t 1l2 < 15 с) с образовани
ем 6 аминохинолина, N rидроксисукцинимида и
диоксида уrлерода, а через 1 мин дальнейшее
образование производных не происходит.
Раствор пробы АХК аминокислот для BBe
дения может храниться при комнатной темпе
ратуре в течение одной недели без заметных
изменений хроматоrрафическоro отклика. Сле
довательно, АХК аминокислоты обладают более
чем достаточной стабильностью для проведе
ния круrлосуточноro автоматическоro xpOMaTO
rрафическоro анализа. Предел обнаружения
для каждой аминокислоты, кроме цистеина, Ha
ходится в области от 40 фемтомоль до 320 фем
томоль.
Предел обнаружения для цистеина COCTaB
ляет около 800 фемтомоль. Линейность откли
ка наблюдается в области 2,5 500 микромоль
с коэффициентом корреляции более 0,999. Для
получения удовлетворительных данных peKO
мендуется перед rидролизом использовать об
разцы массой более 30 Hr.
МЕТОД 5. ПРЕДКОЛОНОЧНАЯ
ДЕРИВАТИ3АЦИЯ С О ФТАЛЕВЫМ
АЛЬДЕrидом
Предколоночная дериватизация аминокис
лот с о фталевым альдеrидом (ОФА) прово
дится перед разделением смеси аминокислот
методом обращенно фазовой ВЭЖХ с флуори
метрическим детектированием. Метод не позво
ляет определять аминокислоты, являющиеся
вторичными аминами (например, пролин).
ОФА в присутствии тиольных соедине
ний реаrирует с первичной аминоrруппой, об
разуя производные изоиндола с сильной флуо
ресценцией. В качестве тиольных соединений
MOryT быть использованы 2 меркаптоэтанол и
3 меркаптопропионовая кислота. ОФА не обла
дает флуоресценцией и, следовательно, не об
разует мешающих пиков. Кроме тоro, еro pac
творим ость и стабильность в водном растворе,
наряду с высокой скоростью реакции, позволя
ют проводить автоматическую дериватизацию с
использованием автодозатора для смешивания
испытуемоro образца и peareHTa. Основным He
достатком ОФА является отсутствие у неro спо
собности реаrировать со вторичными аминокис
лотами (например, пролином). Компенсировать
данный недостаток можно сочетанием с методи
кClМИ, описанными в методах 7 или 8.
Предколоночная дериватизация аминокис
лот с ОФА используется для пробоподroтовки
перед обращенно фазовой ВЭЖХ. Так как ОФА
производные аминокислот неустойчивы, анализ
и разделение методом ВЭЖХ проводят HeMeд
ленно после дериватизации. Хроматоrраф для
2.2.56. Анализ аминокислот
159
жидкостной хроматоrрафии должен быть обо
рудован флуометрическим детектором для об
наружения производных аминокислот. Интен
сивность флуоресценции ОФА производных
аминокислот измеряют при длине волны возбуж
дающеro света 348 нм и длине волны излучае
моro света 450 нм.
Заявленный предел обнаружения при флуо
риметрическом детектировании не ниже 50 фем
томоль, однако на практике предел обнаружения
составляет 1 пикомоль.
МЕТОД 6. ПРЕДКОЛОНОЧНАЯ
ДЕРИВАТИЗАЦИЯ С (ДИМЕТИЛАМИНО)
АЗОБЕН30ЛСУЛЬФОНИЛХЛОРИДОМ
Предколоночная дериватизация аминокис
лот с (диметиламино )азобензолсульфонилхло
ридом (ДАБС) проводится перед разделением
смеси аминокислот методом обращенно фазо
вой ВЭЖХ со спектрофотометрическим детекти
рованием в видимой области спектра.
ДАБС является хромофорным реактивом
и используется для маркировки аминокислот.
Аминокислоты, маркированные ДАБС (ДАБС
аминокислоты), обладают высокой стабильно
стью и проявляют максимум поrлощения при
436 нм.
ДАБС аминокислоты всех встречающихся в
природе производных аминокислот MOryT быть
разделены на колонке С,в методом обращенно
фазовой ВЭЖХ с использованием rрадиентноro
элюирования и подвижной фазы, состоящей из
ацетонитрила и водной буферной смеси. Элюи
руемые из колонки ДАБС аминокислоты опреде
ляют при длине волны 436 нм.
Метод позволяет проводить с одинаковой
чувствительностью анализ как аминокислот, так
и иминокислот (таких как пролин). Метод дерива
тизации с ДАБС позволяет количественно опре
делять остатки триптофана, полученные при
rидролизе белка/пептида с сульфоновыми кис
лотами (такими как кислота меркаптоэтансуль
фоновая, кислота п толуолсульфоновая или кис
лота метансульфоновая), как указано в методе 2, <
описанном в разделе «rидролиз белков». Друrие
лабильные остатки аминокислот, такие как аспа
раrин и rлутамин, можно анализировать, как и
предыдущие, после превращения их в кислоту
диаминопропионовую и кислоту диаминомас
ляную соответственно, как указано в методе 11,
описанном в разделе «rидролиз белков».
В качестве внутреннеro стандарта для этоro
метода не может быть использован норлейцин,
так как он элюируется в области xpoMaTorpaMMbI
со скоплением пиков первичных аминокислот. В
качестве внутреннеro стандарта может быть ис
пользован нитротирозин, который элюируется в
свободной от пиков области xpoMaTorpaMMbI.
Предел обнаружения ДАБС аминокислот
обычно составляет 1 пикомоль. Предел опре
деления ДАБС аминокислот 2 5 пикомоль.
Для получения удовлетворительных данных pe
комендуется для одноro анализа ИСПОЛЬЗОВать
1 о зо Hr ДАБС производных белковоro rидро--
лизата.
МЕТОД 7. ПРЕДКОЛОНОЧНАЯ
ДЕРИВАТИ3АЦИЯ С 9 ФТОРЕНИЛМЕТИЛ
ХЛОРФОРМИАТОМ
Пред колоночная дериватизация ами
нокислот с 9 фторенилметилхлорформиа
том (ФМФ) проводится перед разделением
смеси аминокислот методом обращенно фа
зовой ВЭЖХ с флуориметрическим детекти
рованием.
ФМФ взаимодействует с первичными и
вторичными аминокислотами с образованием
производных с сильной флуоресценцией. Pe
акция проходит при мяrких условиях в водном
растворе в течение 30 с. Полученные произ
водные стабильны. за исключением подвер
женных деструкции производных rистидина.
Избыток флуоресцирующею ФМФ и флуо
ресцирующие побочные продукты MOryT быть
удалены без потерь ФМФ аминокислот.
ФМФ аминокислоты MorYT быть разде
лены на колонке С,в методом обращенно
фазовой ВЭЖХ. Разделение проводят с ис
пользованием rрадиентноrо элюирования с
использованием линейно изменяющеroся co
става подвижной фазы со смеси из 1 О объе
мов ацетонитрила, 40 объемов метанола и 50
объемов ацетатноro буфера до смеси из 50
объемов ацетонитрила и 50 объемов aцeTaT
ною буфера. В результате такоro элюирова
ния 20 производных аминокислот разделяют
ся в течение 20 мин. Элюируемые из колонки
производные аминокислот определяют при
длине волны возбуждающею света 260 нм и
длине волны излучаемоro света 313 нм.
Предел обнаружения находится в нижнем
фемтомолярном диапазоне. Линейность OT
клика для большинства аминокислот наблю
дается в области O,1 50 микромоль.
МЕТОД 8. ПРЕДКОЛОНОЧНАЯ
ДЕРИВАТИ3АЦИЯ С 7 ФТОР
4 НИТРОБЕН30 2 ОКСА 1,3 ДИА30ЛОМ
Предколоночная дериватизация аминокис
лот с 7 фтор 4 нитробензо 2 окса 1 :3 диазо
лом (НБД) про водится перед разделением
смеси аминокислот методом обращенно фазо
вой ВЭЖХ с флуориметрическим детектирова
нием.
НБД взаимодействует с первичными и BTO
ричными аминокислотами с образованием про
изводных с сильной флуоресценцией/Аминокис
лоты дериватизируются с НБД при наrревании
до 60 ос в течение 5 мин.
НБД производные аминокислот MorYT быть
разделены на колонке С'8 методом обращенно
фазовой ВЭЖХ с использованием rрадиентно
ro элюирования и подвижной фазы, состоящей
из ацетонитрила и водной буферной смеси. В
160
rосударственная фармакопея Республики Беларусь
резулыате такою элюирования 17 производных
аминокислот разделяются в течение 35 мин. В
качестве внутреннею стандарта может быть ис
пользована Е аминокапроновая кислота. кото.
рая элюируется в свободной от пиков области
xpoMaTorpaMMbI. Элюируемые из колонки про
изводные аминокислот определяют при длине
волны возбуждающеro света 480 нм и длине
волны излучаемоro света 530 нм.
Чувствительность этоrо метода почти
такая же. как в предколоночной дериватиза
ции с ОФА (метод 5), за исключением про
лина, который не реаrирует с ОФА (преиму
щественное отличие метода с НБД). Предел
обнаружения для каждой аминокислоты co
ставляет около 1 О фемтомоль. Для получения
удовлетворительных данных рекомендуется
для одноrо анализа использовать 10 30 Hr
ДАБС производных белковоrо rИДРОflизата.
Анализ проводят с использованием 1,5 Mr
белковою rидролизата в предколоночной pe
акционной смеси.
РАСЧЕТ И АНАЛИЗ ПОЛУЧЕННЫХ
ДАННЫХ
При определении содержания аминокис
лот в белковом/пептидном rидролизате сле
дует учитывать, что при кислотном rидроли
зе разрушаются триптофан и цистеин, сери н
и треонин разрушаются частично, в то время
как связи по остаткам изолейцина ивалина
расщепляются не полностью. Метионин во
время кислотною rидролиза может подвер
rаться окислению, в результате чеrо появля
ются свободные аминокислоты (например,
rлицин и серин). заrрязняющие образец. Ис
пользование вакуума (менее 200 мкм PTYTHO
ro столба, или 26.7 Па) или введение инертно
ro rаза (aproH) в пространство реакционноro
сосуда может уменьшить степень окислитель
ной деструкции. Поэтому количественные pe
зультаты, полученные для цистеина, трип
тофана, треонина, изолейцина, валина,
метионина, rлицина и серина из белковоrо/
пептидноrо rидролизата. MorYT быть непосто
янными и служить основанием для дальней
шеro исследования и анализа.
Мольный процент аминокислот
это количество конкретных аминокислотных
остатков на 100 остатков белка. Эта величи
на может быть полезной при оценке данных,
полученных при анализе аминокислот, если
молекулярная масса исследуемоrо белка He
известна. Информация также может быть It'с
пользована для подтверждения подлинности
белка/пептида или для друrих целей. Моль
ный процент каждой аминокислоты в испыту
ем ом образце рассчитывают по формуле:
100.'и ,
,
rдe:
ru отклик пика аминокислоты в испыту
емом образце, проинтеrрированный в HaHO
моль;
r сумма откликов пиков всех аминокислот
в испытуемом образце, проинтеrрированных в
наномоль.
Сравнение полученноro в испытании моль
ноro процента аминокислот с данными извест
ных белков может помочь установить или под
твердить подлинность испытуемою образца
белка.
Образцы неизвестноrо белка. Этот метод
расчета может быть использован для оценки KOH
центрации белка в образце неизвестноro белка с
использованием данных, полученных при анали
зе аминокислот. Массу каждой высвобождаемой
аминокислоты рассчитывают в микроrраммах по
формуле:
т.М, .
1000
rдe:
т количество аминокислоты, определен
ное в испытании, в наномоль;
М , средняя молекулярная масса данной
аминокислоты, скорректированная с учетом
массы молекулы воды, удаленной при образова
нии пептидной связи.
Суммируя массы высвобожденных амино
кислот, получают общую массу анализируемо
ro белка после соответствующей корректировки
с учетом частично или полностью разрушен
ных аминокислот. Если известна молекуляр
ная масса неизвестноro белка (например, опре
деленная методом rель электрофореза в
полиакриламидном rеле С использованием
натрия додецилсульфата или методом Macc
спектрометрии), можно установить амино
кислотный состав неизвестноro белка. Число
остатков каждой аминокислоты рассчитывают
по формуле:
т
(1000. М ) '
l. м"
rде:
т количество аминокислоты, определен
ное в испытании, в наномоль;
М общая масса белка, в микроrраммах;
М" молекулярная масса неизвестноro
белка.
Образцы известноrо белка. Этот метод
расчета может быть использован для YCTaHOB
ления аминокислотноro состава и KOHцeHTpa
ции белка в образце белка с известной моле
кулярной массой и аминокислотным составом
с использованием данных, полученных при
2.2.57. Атомно эмuссионная спектрометрия
161
01/2013:20257
анализе аминокислот. При анализе состава
известноro белка учитывают, что некоторые
аминокислоты высвобождаются хорошо, в то
время как открываемость друrих аминокис
лот может быть затруднена вследствие пол
ноro или частичноro разрушения (например,
триптофан, цистеин, треонин, серин, метио
нин), неполноro расщепления связей (напри
мер, изолейцин и валин), контаминации CBO
бодными аминокислотами (например, rлицин
и серин).
Для определения количественноrо coдep
жания белка используют аминокислоты, OTKpЫ
ваемость которых наилучшая. Хорошо OTKpЫ
ваемыми аминокислотами обычно являются:
аспартат аспараrин, rлутамат rлутамин, аланин,
лейцин, фенилаланин, лизин и арrинин. Этот
перечень может быть модифицирован на oc
новании наработанноro опыта по проведению
анализа аминокислот. Для определения KO
личественноrо содержания белка по каждой
хорошо открываемой аминокислоте количество
каждой хорошо открываемой аминокислоты,
в наномоль, делят на ожидаемое число остат
ков этой аминокислоты в белке. Рассчитывают
среднее содержание белка. Содержание белка,
установленное по каждой хорошо открываемой
аминокислоте, должно быть равномерно pac
пределено около средней величины. Значения
содержания белка, определенные по этим ами
нокислотам, имеющие неприемлемые отклоне
ния (обычно более 5 %) от средней величины,
отбрасывают. С учетом оставшихся значений пе
ресчитывают среднее содержание белка в испы
туемом образце. Для определения аминокислот
ноro состава испытуемоrо образца содержание
каждой аминокислоты делят на рассчитанное
среднее содержание белка. Относительную
ошибку определения аминокислотноro состава
рассчитывают в процентах по формуле:
100.т
,
m s
rде:
т экспериментально установленное KO
личество аминокислот, в наномоль на аминокис
лотный остаток;
m s известное значение остатков для этой
аминокислоты.
Средняя величина относительной ошибки
определения аминокислотноro состава является
средним значением абсолютных величин OTHO
сительных ошибок определения каждой амино
кислоты, обычно кроме триптофана и цистеина.
Средняя величина относительной ошибки опре
деления аминокислотноro состава может дать
важную информацию об устойчивости анализа
во времени. Соответствие найденноro аминокис
лотноro состава образца белка и известноro co
става может служить подтверждением подлин
ности и чистоты белка в испытуемом образце.
21. Зак. 1712.
2.2.57. АТОМНО-ЭМИССИОННАЯ
СПЕКТРОМЕТРИЯ
С ИСПОЛЬЗОВАНИЕМ
ИНДУКТИВНО СВЯЗАННОЙ
ПЛАЗМЫ
ОБЩИЙ ПРИНЦИП
Атомно эмиссионная спектрометрия с ис
пользованием индуктивно связанной плазмы
(ИСП АЭС, /CP AES iпductive/y coup/ed p/as
ma atomic етissiоп spect,ometry) представляет
собой метод атомно эмиссионной спектроме
трии, в котором В качестве источника возбужде
ния атомов используется индуктивно связанная
плазма (ИСП).
Индуктивно связанная плазма представля
ет собой ионизированный инертный rаз (обычно
aproH) с одинаковым числом электронов и ионов,
поддерживаемых радиочастотным (РЧ) полем.
Высокая температура плазмы последователь
но десольватирует, испаряет, возбуждает (дeTeK
тирование методом атомно эмиссионной спек
трометрии (АЭС» и ионизирует (детектирование
методом масс спектрометрии (МС» атомы испы
туемоro образца. Пределы обнаружения обычно
находятся в диапазоне от менее HaHorpaMMa
(ИСП МС) до менее микроrрамма (ИСП АЭС) на
литр.
Плазма образуется при прохождении TaH
rенциальноro потока rаза носителя через «ro
релку», т. е. систему, состоящую из трех KOH
центрических кварцевых трубок. Металлическая
катушка (индуктор электронаrревателя) OKPy
жает верхний конец rорелки и подсоединена к
радиочастотному (РЧ) reHepaTopy. На катуш
ку подается мощность (обычно 700 1500 Вт),
в результате чеrо образуется осциллирующее
маrнитное поле с частотой, соответствующей
частоте reHepaTopa (в большинстве случаев
27 мrц, 40 мrц). Плазма образуется, коrда rаз
носитель становится проводящим под воздей
ствием электрическоro разряда, который вносит
затравочные электроны и ионы. В индуцирован
ном маrнитном поле заряженные частицы (ионы
и электроны) движутся по замкнутой кольцевой
траектории. Из за наличия сопротивления дви
жению происходит разоrревание, в результа
те котороro появляется дальнейшая ионизация.
Процесс происходит почти MrHoBeHHo, и плазма
развивается до своих полных размеров и мощ
ности. Радиочастотная осцилляция мощности,
подающаяся на катушку, вызывает образование
около верха roрелки радиочастотных электри
ческоro и маrнитноro полей. Korдa искра (проду
цируемая либо трубкой Тесла, либо иным при
способлением) воздействует на rаз носитель,
протекающий через roрелку, из rаза носителя
выбиваются некоторые электроны. Эти электро
ны подхватываются маrнитным полем и ускоря
162
rосударственная фармакопея Республики Беларусь
ются. Придание энерrии электронам с помощью
катушки называется индуктивным связываНt:ем.
Эти высокоэнерrетические электроны сталкив,]
ются с друrими атомами rаза носителя, выбивая
все больше электронов. Ионизация rаза носите
ля при столкновениях, происходящая в режиме
цепной реакции, приводит к превращению rаза
в физическую плазму, состоящую из атомов ra
за носителя, электронов и ионов rаза носителя.
Плазма затем поддерживается ме>IЩУ rорелкой
и катушкой постоянной подачей энерrии с помо
щью процесса индуктивноro связывания.
ИСП представляет собой интенсивную,
очень яркую перовидную плазму. Основание
плазмы тороидальное и оно является местом
индукции, то есть местом, в котором индуктив
ная энерrия передается от индуктора электро
наrревателя плазме. Образец вводится через
место индукции в центр плазмы.
ПРИБОР
rлавными составляющими частями при бора
являются:
система ввода образца, состоящая из пе
ристальтическоro насоса, подающеro раствор с
постоянной скоростью в распылитель;
радиочастотный (РЧ) reHepaTop;
плазменная юрелка;
оптика, фокусирующая изображение
плазмы на входной щели спектрометра; ради
альное наблюдение больше подходит для слож
ных матриц (щелочи, орrанические вещества), в
то время как аксиальное наблюдение дает боль
шую интенсивность и лучший предел определе
ния в случае с простыми матрицами;
приспособление для разложения света на
спектр, включающее в себя дифракционные pe
шетки, призмы, фильтры или интерферометры;
детектор, превращающий энерrию излуче
ния в электрическую энерrию;
блок сбора данных.
ИНТЕРФЕРЕНЦИЯ
Интерференцией называется явление, BЫ
зывающее несоответствие аналитическоrо сиr
нала аналита в образце сиrналу аналита в rpa
дуировочном растворе такой же концентрации.
Хорошо известная химическая интерференция,
которая встречается в пламенной атомно аб
сорбционной спектрометрии, обычно слабо про
является в ИСП АЭС. В редких случаях, Korдa
имеет место интерференция, может помочь YBe
личение мощности радиочастот или уменьшение
потока BHyтpeHHero rаза носителя. Интерферен
ция в ИСП АЭС может быть спектральчоrо про
исхождения или может быть результатом BЫC
ких концентраций определенных элементов или
компонентов матрицы. Физическая интерферен
ция (возникающая из за различий в вязкости и
поверхностном натяжении раствора образца и
rрадуировочноro раствора) может быть YMeHb
шена путем разведения, подбора матрицы, ис
пользования внутренних стандартов или исполь
зованием метода стандартных добавок.
Друrим типом интерференции, иноrда BCTpe
чающимся в ИСП АЭС, является так называе
мый «эффект леrко ионизируемых элементов».
Леrко ионизируемыми элементами называются
элементы, которые ионизируются значительно
леrче, например, щелочные и щелочноземель
ные металлы. В образцах, которые содержат
высокие концентрации таких элементов (более
0,1 %) может происходить подавление либо YBe
личение эмиссии.
Спектральная интерференция. Она может
происходить из за присутствия друrих линий
или сдвиroв в интенсивности базовой линии.
Эти линии MOryт соответствовать арюну (Ha
5людаются после 300 нм); ОН линии, появляю
щиеся из за разложения воды (около 300 нм);
NO линии, появляющиеся из за взаимодействия
азота из окружающей среды с плазмой (между
200 нм И 300 нм); MOryT присутствовать линии
друrих элементов, особенно тех, которые Haxo
ДЯТСЯ в высокой концентрации в образце. Ин
терференция распадается на четыре различные
катеrории: простой сдвиr базовой линии, наклон
ный сдвиr базовой линии, прямое спектральное
наложение, сложный сдвиr базовой линии.
Абсорбционная интерференция. Данный
вид интерференции возникает Torдa, Korдa часть
сиrнала аналита поrлощается до достижения
детектора. В частности, этот эффект обнаружи
вается, Korдa концентрация элемента с сильной
эмиссией настолько высока, что атомы или ионы
этою элемента с более низкой энерrией абсор
бируют значительное количество излучения, ис
пускаемоro соответствующими возбужденными
атомами или ионами. Этот эффект, называемый
самопоrлощением, предопределяет верхнюю
rраницу линейноro участка рабочею диапазона
для данной длины волны эмиссии.
Мноrокомпонентный спектральный
подбор. Во избежание проблем со спектральной
интерференцией обычно проводят определение с
использованием нескольких эмиссионных линий.
Для более точной коррекции спектральной интер
ференции получают информацию с использова
нием усовершенствованной системы детекторов
при помощи мноroкомпонентноro спектрально
ro подбора. Этот способ учитывает не только ин
терференцию, но также фоновый вклад матри
цы, создавая, таким образом, формулу поправки.
Мноrокомпонентный спектральный подбор ис
пользует модель множественных линейных K a
дратов, основанную на анализе чистою аналита,
матрицы и холостоro раствора, создавая MaTeMa
тическую модель с учетом интерференции. Это
позволяет определять эмиссию аналита в слож
ной матрице с более низкими пределами дeTeK
тирования и более высокой точностью.
2.2.57. Атомно эмиссионная спектрометрия
163
МЕТОД
ПРОБоподrОТОВКА И ВВОД ОБРА3ЦА
ОСНОВНОЙ целью пробоподrотовки является
raрантирование попадания концентрации aHa
лита после использования разведения или KOH
центрирования в рабочий диапазон прибора и
получения раствора испытуемоro образца, спо
собноrо воспроизводимо распыляться.
Некоторые распылительные системы устой
чивы к действию высоких концентраций кислот,
но использование серной и ФосФорной кислот
может вносить вклад в базовую линию ИСП
спектров. Более предпочтительным является
использование азотной и хлористоводородной
кислот. Фтористоводородную кислоту можно ис
пользовать при наличии устойчивых к ней систем
BBqдa образца (например, из перфторалкокси
полимера) и roрелок. При выборе метода ввода
образца учитывают требования по чувствителtr
насти, стабильности, скорости, размеру образ
ца, устойчивости к коррозии и устойчивости к
закупорке. В большинстве случаев подходит ис
пользование распылителя с поперечным пото
ком вместе с распылительной камерой и roрел
кой. Перистальтический насос, использующийся
в ИСП АЭС, подает испытуемый раствор и paCT
вор сравнения со скоростью 1 мл/мин или менее.
В случае использования орrанических раст
ворителей необходимо обеспечить разделение
кислорода и орrанических слоев.
ВЫБОР УСЛОВИЙ ПРОВЕДЕНИЯ
АНАЛИ3А
При выборе условий проведения анализа
следуют рекомендациям производителя прибо
ра. Анализ водных растворов и орrанических
растворителей обычно происходит при различ
ных условиях. Надлежащим образом должны
быть выбраны следующие аналитические пара
метры:
длины волн;
скорости потоков rаза носителя (внешняя,
промежуточная и внутренняя трубки rорелки); <
мощность радиочастотноro излучения;
положение для наблюдения (радиальное
или аксиальное);
скорость насоса;
условия для детектора (усиление/напря
жение для детектора с фотоумножающей
трубкой, друrие для матричноro детектора);
время интеrрирования (установленное
время для измерения интенсивности эмиссии на
каждой длине волны).
КОНТРОЛЬ РАБОТОСПОСОБНОСТИ
ПРИБОРА
Приrодность системы
Для подтверждения надлежащей работы
ИСП АЭС при бора с использованием мноroэле
ментноro раствора MOryт быть проведены следу
ющие испытания:
передача энерrии (reHepaTop, rорелка,
плазма); может быть использовано соотношение
Mg 11 (280,270 HM)/Mg 1(285,213 нм);
подача образца, путем проверки эффек
тивности и стабильности распылителя;
разрешение (оптическая система), путем
измерения ширины пика на половине высоты,
например, для As (189,042 нм), Мп (257,610 нм),
Cu (З24,745 нм) или Ва (455,403 нм);
аналитические характеристики, путем pac
чета пределов обнаружения выбранных элемен
тов в данном диапазоне длин волн.
ВАЛИДАЦИЯ МЕТОДА
Через определенные временные интервалы
верифицируют удовлетворительное исполнение
метода, описанноro в частной статье.
ЛИНЕЙНОСТЬ
rотовят и анализируют не менее четырех
растворов сравнения, концентрация которых Ha
ходится в пределах диапазона rрадуировки, и
холостой раствор. Проводят не менее пяти по
второв.
Используя все полученные данные, рассчи
тывают линейное уравнение rрафика методом
наименьших квадратов. Строят кривую perpec
сии, отмечая средние значения, измеренные
значения и доверительный интервал rрадуиро
вочноrо rрафика. Метод является приroдным при
условии соблюдения следующих требований:
коэффициент корреляции должен быть не
менее 0,99;
на rрадуировочном rрафике поrрешности
каждоro калибровочноro уровня должны быть
распределены случайным образом.
Рассчитывают среднее значение и относи
тельное стандартное отклонение для наимень
шеro и наибольшеro калибровочноro уровня.
В случае если отношение рассчитанноro
стандартноro отклонения наименьшеro и наи
большеro калибровочноro уровня менее 0,5
или более 2,0, то более точная оценка rрадуи
ровочноro rрафика может быть получена с ис
пользованием взвешенной линейной реrрессии.
Линейная и квадратичная весовая функции при
меняются к полученным данным для 'нахожде
ния наиболее подходящей для использования
весовой функции. Если средние значения при
сравнении с rрадуировочным rрафиком про
являют отклонение от линейности, используют
двухмерную линейную реrрессию.
ПРАВИЛЬНОСТЬ
Предпочтительно верифицировать правиль
ность с использованием сертифицированных
стандартных образцов. Если это невозможно,
проверяют открываемость.
Открываемость. В случае методик количе
cTBeHHoro определения открываемость должна
быть от 90 % до 110 %. Для друrих определе
164
rосударственная фармакопея Республики Беларусь
ний, например, для определения следовых KO
личеств элемента, открываемость должна быть
от 80 % до 120 % от теоретическоro значенv.я.
Открываемость может быть определена с ис
пользованием подходящеro раствора сравнения
(матриксноrо раствора), содержащеro известное
количество аналита (средняя концентрация rpa
дуировочноro rрафика).
СХОДИМОСТЬ
Сходимость должна быть не более 3 % для
количественноro определения и не более 5 %
для испытания на содержание примесей.
ПРЕДЕЛ КОЛИЧЕСТВЕННО/О
ОПРЕДЕЛЕНИЯ
Удостоверяются, что предел количественно
ro определения (например, определенный с ис
пользованием приближения 10а) ниже измеряе
моro значения.
01/2013:20258
2.2.58. МАСС СПЕКТРОМЕТРИЯ
С ИСПОЛЬЗОВАНИЕМ
ИНДУКТИВНО СВЯЗАННОЙ
ПЛАЗМЫ
Масс спектрометрия с использовани
ем индуктивно связанной плазмы (ИСП МС,
/CP MS iпdисtivе/у coup/ed p/asma mass spec
trometry) представляет собой метод, использу
ющий индуктивно связанную плазму в качестве
ионизирующеro источника. Основные принципы
ИСП описаны в статье 2.2.57. Атомно эмисси
онная спектрометрия с использованием иHдYK
тивно связанной плазмы (ИСП АЭС).
В ИСП МС используется способность ИСП
rенерировать заряженные ионы элементов, BXO
дящих в состав образца. Полученные ионы Ha
правляют в масс спектрометр, который разде
ляет их в соответствии с отношением массаl
заряд (m/z). Большинство масс спектрометров
имеют квадрупольные или маrнитные анализа
торы. Ионы переносятся из плазмы в ионную
оптику через два сопла (сопло ввода пробы и
сепараторное сопло, образующие область ин
терфейса). Ионная оптика состоит из электро
статических линз, которые переносят ионы
из пространства с атмосферным давлением к
масс фильтру с давлением 10 B Па или менее,
поддерживаемым с помощью турбшюлекуляр
ноro насоса. После фильтрации ионы с выбран
ным соотношением массаlзаряд направляют
ся на детектор (канальный электроумножитель,
чашка Фарадея, диноды), rдe поток ионов KOH
вертируется в электрические сиrналы. Количе
ственно определяют число появляющихся и re
нерирующих электрические импульсы ионов в
единицу времени.
Система ввода образца и способы обработ
ки данных аналоrичны используемым в ИСП
АЭС.
ПРИБОР
rлавными составляющими частями прибора
являются:
система ввода образца, состоящая из пе
ристалыическоrо насоса, подающеro раствор с
постоянной скоростью в распылитель;
радиочастотный (РЧ) reHepaтop;
плазменная rорелка;
область интерфейса, включающая в себя
сопла для переноса ионов в ионную оптику;
масс спектрометр;
детектор;
блок сбора данных.
ИНТЕРФЕРЕНЦИЯ
Наибольшей проблемой является Macco
вая интерференция. Например, изобариче
ские виды значительно перекрывают массовый
сиrнал искомых ионов, особенно 8 централь
ной части диапазона масс (например, 40 80
а.е.м.). Комбинация атомных ионов приводит
к полиатомной или молекулярной интерферен
uиям (то есть 4°Ar 16 0 с 56Fe или 4°Ar"°Ar с ВО8е).
Матрица, в случае с некоторыми аналитами,
также может вызывать интерференцию. HeKO
торые образцы оказывают влияние на образо
вание капель или на температуру ионизации.
Эти явления MorYT приводить к подавлению
аналитическоro сиrнала. Обойти физическую
интерференцию можно, используя метод BHY
треннеro стандарта или метод стандартных
добавок. Выбор элемента, используемоro в Ka
честве внутреннеro стандарта, зависит от ис
следуемоro элемента: например, в качестве
внутренних стандартов MorYT быть использо
ваны 59Со и 1151п.
Основной характеристикой приборов
ИСП МС является разрешение, то есть способ
ность к разделению двух близких масс. С этой
точки зрения приборы с квадрупольными анали
заторами проиrрывают приборам с маrнитными
анализаторами.
МЕТОД
ПРОБОПОД,ОТОВКА И ВВОД ОБРАЗЦА
Пробоподroтовка обычно включает в себя
стадию разложения матрицы с помощью под
ходящеro метода, например, в микроволновой
печи. Кроме тоro, важно попадание KOHl\eHTpa
ции аналита после использования разведения
или концентрирования в рабочий диапазон при
бора и получение раствора испытуемоro образ
ца, способноro воспроизводимо распыляться.
Некоторые распылительные системы устой
чивы к действию высоких концентраций кислот,
2.2.58. Масс спектрометрия с использованием индуктивно связанной плазмbt 165
но использование серной и фосфорной кислот
может вносить вклад в базовую линию ИСП
спектров. Более предпочтительным является
использование азотной и хлористоводородной
кислот. Фтористоводородную кислоту можно ис
пользовать при наличии устойчивых к ней (Ha
при мер, из перфторалкоксиполимера) систем
ввода образца и roрелок. При выборе метода
ввода образца учитывают требования по чув
ствительности, стабильности, скорости, размеру
образца, устойчивости к коррозии и устойчиво
сти К закупорке. В большинстве случаев подхо
дит использование распылителя с поперечным
потоком вместе с распылительной камерой и ro
релкой. Перистальтический насос подает испы
туемый раствор и раствор сравнения со CKOpO
стью 20 1000 мкл/мин.
В случае использования орrанических раст
ворителей необходимо обеспечить разделение
кислорода и орrанических слоев.
ВЫБОР УСЛОВИЙ ПРОВЕДЕНИЯ
АНАЛИЗА
При выборе условий проведения анализа
следуют рекомендациям производителя прибо
ра. Анализ водных растворов и орrанических
растворителей обычно происходит при различ
ных условиях. Надлежащим образом должны
быть выбраны следующие аналитические пара
метры:
сопла (материал сопла ввода проб и ce
параторноro сопла);
скорости потоков rаза носителя (внешняя,
промежуточная и внутренняя трубки rорелки);
мощность радиочастотноro излучения;
скорость насоса;
должен быть осуществлен выбор одноro
или более изотопов измеряемоrо элемента
(масса).
ВЫБОР ИЗОТОПОВ
Выбор изотопов осуществляют по несколь
ким критериям. Для получения максимальной
чувствительности выбирают наиболее распро '
страненный изотоп данноro элемента. Кроме
тоro, следует выбирать изотоп с минимальной
интерференцией, вызываемой друrими видами
изотопов, присутствующими в матрице и в rазе
носителе. В проrраммном обеспечении произво
дителей приборов ИСП МС обычно присутствует
информация об изобарической интерференции
и интерференции, вызываемой полиатомны
ми ионами, например, rидридными, оксидными,
хлоридными и т. д.
КОНТРОЛЬ РАБОТОСПОСОБНОСТИ
ПРИБОРА
Приroдность системы
Настройка прибора позволяет отследить
и настроить измерение перед введением об
разца. Точность определения масс прибором
ИСП МС проверяют с помощью раствора для
22 3n 1712
настройки, содержащеro несколько изотопов, ox
ватывающих весь диапазон, например, 9Ве, 59Со,
В9У. 1151п, 140Се и 209Bi.
Записывают чувствительность и KpaTKO
временную и долroвременную стабильность.
Параметры прибора (условия плазмы, параме
тры ионных линз И квадруполей) настраивают на
получение максимально возможноro числа об
наружений ионов.
Для подтверждения наблюдения прием
лемоro отклика в широком диапазоне масс раз
решение и ось масс настраивают с использова
нием раствора, содержащеro Li, У и TI.
Должна про водиться про верка эффек
тивности разложения плазмой оксидов. Xopo
шими индикаторами являются отношения Се/
СеО и/или BaIBaO, требуемый уровень при этом
должен составлять менее З %.
Подавление образования двухзарядных
ионов осуществляют с использованием Ва и Се.
Отношение сиrнала двухзарядных ионов к иско
мому элементу должно составлять менее 2 %.
Долroвременную стабильность проверя
ют, измеряя стандарт в начале и в конце испыта
ния образца, при этом проверяется влияние OT
ложения солей в соплах на уменьшение сиrнала
во время испытания.
ВАЛИДАЦИЯ МЕТОДА
Через определенные временные интервалы
верифицируют удовлетворительное исполнение
метода, описанноro в частной статье.
ЛИНЕЙНОСТЬ
rотовят и анализируют не менее четырех
растворов сравнения, концентрация которых Ha
ходится в пределах диапазона rрадуировки, и
холостой раствор. Проводят не менее пяти по
второв.
Используя все полученные данные, рассчи
тывают линейное уравнение rрафика методом
наименьших квадратов. Строят кривую perpec
сии, отмечая средние значения, измеренные
значения и доверительный интервал rрадуиро
вочноrо rрафика. Метод является приrодным при
условии соблюдения следующих требований:
коэффициент корреляции должен быть
не менее 0,99;
на rрадуировочном rрафике поrрешности
каждоro калибровочноro уровня должны быть
распределены случайным образом.
Рассчитывают среднее значение и относи
тельное стандартное отклонение для наимень
шеrо и наибольшеro калибровочноro уровня.
В случае если отношение рассчитанноro
стандартноro отклонения наименьшР.ro и наи
большеrо калибровочноro уровня менее 0,5
или более 2,0, то более точная оценка rрадуи
ровочноro rрафика может быть получена с ис
пользованием взвешенной линейной реrрессии.
Линейная и квадратичная весовая функции при
меняются к полученным данным для нахожде
166
rосударственная фармакопея Республики Беларусь
ния наиболее подходящей для использования
весовой срункции. Если средние значения ри
сравнении с rрадуировочным rрафиком пр,)
являют отклонение от линейности, используют
двухмерную линейную реrрессию.
ПРА ВИЛЬНОС ТЬ
Предпочтительно верифицировать правиль
ность с использованием сертифицированных
стандартных образцов. Если это невозможно,
проверяют открываемость.
Открываемость. В случае методик количе
ственноro определения открываемость должна
быть от 90 % до 110 %. Для друrих определе
ний, например для определения следовых коли
честв элемента, открываемость должна быть от
80 % до 120 % от теоретическоro значения. OT
крываемость может быть определена с исполь
зованием подходящеro раствора сравнения (Ma
триксноro раствора), содержащеro известное
количество аналита (средняя концентрация rpa
дуировочноrо rрафика).
СХОДИМОСТЬ
Сходимость должна быть не более 3 % для
количественноro определения и не более 5 %
для испытания на содержание примесей.
ПРЕДЕЛ КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ
Удостоверяются, что предел количественно
ro определения (например, определенный с ис
пользованием приближения 10а) ниже измеряе
моro значения.
01/2013:20259
2.2.59. АНАЛИЗ rлиКАНОВ
в rЛИКОПРОТЕИНАХ
1. ВВЕДЕНИЕ
Анализ rликанов это испытание, исполь
зуемое для исследования rликановых фраrмен
тов rликопротеинов. Он может включать в себя:
анализ целоro rликопротеина;
разделение и обнаружение rликоформ
белка;
анализ rликопептидов, полученных при
ферментативной обработке rликопротеина;
анализ высвобожденных rликанов, полу
ченных при химической ИЛL1 ферментативной
обработке rликопротеина.
Анализ моносахаридов может служить дo
полнением к информации, полученной при aHa
лизе rликанов.
rликозилирование может иrрать важную
роль в проявлении лекарственным средством
биолоrическоrо происхождения ero функцио
нальных, фармакокинетических, фармакодина
мических и иммуноrенных свойств, а также еro
устойчивости. rликозилирование, в отличие от
транскрипции, является ферментативным моди
фицирующим процессом, не имеющим матрич
ноro управления, результатом чеro является re
TeporeHHocTb rликанов. На reTeporeHHocTb также
влияет процесс производства. Поэтому анализ
rликанов rликопротеинов является важным ис
пытанием для идентификации изменений в
схеме rликозилирования rликопротеинов иlили
для наблюдения за стабильностью схемы rлико
зилирования при производстве.
Анализ rликанов может быть сравнитель
ным методом, потому что полученная инфор
мация, при сравнении с аналоrично анализи
руемым стандартным образцом, подтверждает
соответствие испытуемоro образца.
В приведенном разделе представлены под
ходы для анализа rликанов rликопротеинов и
требования к использованию и валидации MeTO
дов.
Анализ rликанов не является единым общим
методом, а включает в себя применение опре
деленных процедур и получение специфиче
ских карт rликанов для каждоro rликопротеина.
Особые процедуры указаны в соответствующих
частных статьях.
1 1. rЛИК03ИЛИРОВАНИЕ БЕЛКОВ
Существует 3 вида ферментативноro rлико
зилирования белков:
N rликозилирование, под которым понима
ют присоединение олиroсахаридов к азоту в Tep
минальной амидной rруппе аспараrина;
О rликозилирование, под которым пони
мают присоединение олиroсахаридов к rидрок
сильным rруппам серина, треонина и/или rи
дроксипролина;
С rликозилирование, под которым по
нимают присоединение о маннопиранозы к
С2 уrлероду индольноro цикла триптофана.
При инкубировании белков с восстанавли
вающими сахарами может происходить нефер
ментативное присоединение, известное как rли
кирование.
Приведенный раздел описывает аналитиче
ские методики для N и О связанных rликозили
рований, обнаруживающихся в основном в rли
копротеиновых лекарственных средствах.
1 2. rETEPOrEHHOCTb
rЛИКОЗИЛИРОВАНИЯ БЕЛКОВ
При производстве rликопротеинов может
проявляться различная степень rетероrенности
rликанов. Эта reTeporeHHocTb может быть след
ствием следующих изменений:
степени заполненности (полная, частич
ная, свободная);
типа rликозилирования (N или О связан
ная);
2.2.59. Анализ 2ликанов в 2ликопротеинах
167
АНАЛИЗ ИНТ АКТНЫХ rЛИКОПРОТЕИНОВ
МС
ИЭФ
КЭ
Ионо06менная хроматоарафия
Эксклюзионная хроматоерафия
дсн пАr электрофорез
I rЛИКОПРОТЕин l
+
АНАЛизrЛИКОПЕПТИДОВ
Прямой анализ
МС
ПРЕДВАРИТЕЛЬНАЯ
ОБРАБОТКА
(выделение и очистка)
при необходимости
Протеолиз
Разделение перед прямым анализом
ЖХ/КЭ
АНАЛИЗ ВЫСВОБОЖДЕННЫХ rлиКАНОВ
Анализ немеченых rликанов
Десиалирование
HPAEC PAD
пrx MC
МС
Высвобождение rликанов
o и/или N связаные
2ликаны:
ферментативное или
химическое
Анализ меченых rликанов
Мечение
rликанов
Обработка
экзоrликозидазами
!
..... Разделение
ЖХ/КЭ
АНАЛИЗ МОНОСАХАРИДОВ
HPAEC PAD
пrx MC
Кислотный
rидролиз
Мечение
флуорофором
.- ЖХ
КЭ
I Колориметрические методы
Рисунок 2.2.59. 1. Общая схема методик анализа 2ликанов
Леrенда:
КЭ капиллярный электрофорез;
HPAEC PAD анионно обменная хроматоrрафия при высоких значениях рН с импульсным амперометрическим детектировани
ем;
ИЭФ изоэлектрическое фокусирование;
ЖХ жидкостная хроматоrрафия;
МС масс спектрометрия;
пrх пористо rрафитная хроматоrрафия;
ДСН ПАr электрофорез электрофорез с натрия додецилсульфатом в полиакриламидном rеле.
168
rосударственная фармакопея Республики Беларусь
олиroсахаридных структур (удлинение,
разветвление и присоединение ).
Результатом такой rетероrенности rли
козилирования является набор rликоформ
для одноro определенноro rликопротеина.
Такие изменения появляются по причине
тоro, что, несмотря на транскрипцию и TpaHC
ляцию, rликозилирование является пост
трансляционным модификационным процес
сом, не имеющим матричноrо управления.
Схема rликозилирования в определенном по
ложении зависит от мноrих факторов, вклю
чая специфичное для клетки и/или раз
мер зависимое наличие rликотрансфераз и
экзоrликозидаз, обнаруженных в комплексе
rольджи и эндоплазматическом ретикулуме.
На rликозилирование также влияют структура
белка, процесс производства, хозяин вектор
ная ДНК система экспрессии и условия кле
точных культур.
2. МЕТОДИКИ ПРОВЕДЕНИЯ АНАЛИЗА
rлиКАНОВ
reTeporeHHocTb в rликозилировании может
быть оценена четырьмя отдельными и взаимо
дополняющими методами:
анализ интактноro rликопротеина;
анализ rликопептидов;
анализ высвобожденных rликанов;
анализ моносахаридов.
В настоящей статье представлены методы
и общие требования, применяемые для анали
за rликанов в rликопротеинах, содержащих N
и О связанные rликаны.
Анализ rликанов обычно представляет
собой мноroступенчатый процесс. Существу
ет большое количество методик анализа rлика
нов, обусловленное разнообразием и сложно
стью rликановых структур, числом доступных
методик и систем детектирования, а также ши
роким выбором подходов, в зависимости от
уровня необходимой информации.
На рисунке 2.2.59. 1 представлена схема
аналитических методик анализа rликанов, KO
торая может быть использована для выбранно
ro подхода (подходов). Вследствие разнообра
зия и сложности структуры и происхождения
rликанов, доступны различные вариации одних
и тех же методик и условий.
Выделение и очистка. Выделение и
очистка MorYT быть необходимы для анализа
фармацевтических субстанций или для aHa
лиза дозированных лекарственных форм, co
держащих мешающие вспомоrательные веще
ства, и описываются в частных статьях.
2 1. АНАЛИЗ ИНТАКТНЫХ
,ЛИКОПРОТЕИНОВ
Анализ интактноrо rликопротеина предо
ставляет информацию о rликозилировании rли
копротеина в целом.
В том случае, если молекула крупная и co
держит MHoro положений rликозилирования,
этот подход дает оrраниченное количество ин
формации.
Moryт использоваться такие методы, как
капиллярный электрофорез (КЭ, СЕ) (2.2.47) и
масс--спектрометрия (МС, MS) (2.2.43). Размер
зависимые методы, такие как эксклюзионная
хроматоrрафия (2.2.ЗО) и электрофорез с натрия
додецилсульфатом в полиакриламидном rеле
(ДСН ПАr электрофорез, SOS PAGE) (2.2.31),
MOryT предоставлять информацию о состоянии
rликозилирования белка. Если степень сиалиро
вания значительно влияет на биолоrическую aK
тивность rликопротеина, то для ее мониторинrа
MOryт быть использованы ионообменная xpOMa
тоrрафия (2.2.46), изоэлектрическое фокусиро
вание (ИЭФ, /EF) (2.2.54) или КЭ (2.2.47). Должна
быть выбрана методика, обеспечивающая дo
стоверную корреляцию между степенью сиали
рования и биолоrической активностью продукта.
2 2. АНАЛИЗ ,ЛИКОПЕПТИДОВ
Анализ rликопептидов предоставляет ин
формацию о свойствах rликозилирования в за
висимости от местоположения, о степени запол
ненности и об олиroсахаридных структурах. Он
включает протеолитическое расщепление rли
копротеинов. Подходы к расщеплению белко
воro остова в зависимости от местоположения
представлены в общей статье 2.2.55. Пептид
ное картирование.
После протеолиза rликопротеина MOryT
быть выбраны следующие подходы.
Прямой анализ методом МС (2.2.43). Необ
ходимо тщательно следить, чтобы сиrнал rли
копептида не подавлялся присутствием друrих
пептидов, так как rликопептиды составляют
малую долю в общей смеси пептидов и интен
сивность сиrнала у них меньше, чем у неrлико
зилированных пептидов.
Разделение, предшествующее анализу МС.
Эта дополнительная стадия помоraет преодолеть
проблемы, упомянутые выше. Приемы насыще
ния или фракционирования MOryт быть исполь
зованы как параллельно с прямым анализом, так
и после неro. Такие методы разделения как жид
костная хроматоrрафия (ЖХ, LC) (2.2.29) и КЭ
(2.2.47) также приroдны. Эти методы MOryт быть
совмещены с МС, позволяя получать результаты
МС в режиме реальноro времени (оп liпе).
Де2лuкозилuрование 2лuкопептидов. При
сравнении пептидных карт, полученных при проте
олитическом расщеплении интактноrо rликопроте
ина и деrликозилированчоro перед протеолитиче
ским расщеплением или поr.ле протеолитическоro
расщепления, возможно определение различных
положений rликозилирования rликопротеина. Пеп
тидная масса предоставляет информацию о rли
козилированных участках, и с помощью подсчета
разницы масс между интактным и деrликозилиро
ванным rликопептидом можно получить информа
2.2.59. Анализ 2ликанов в 2ликопротеинах
169
цию о составе и reтероrенности присоединенных
rликанов. Способы деrликозилирования белково
ro остова представлены в разделе 2 З 1. Стадия
разделения может быть проведена до или после
деrликозилирования.
2 3. АНАЛИЗ ВblСВОБОЖДЕННblХ
rЛИКАНОВ
Анализ высвобожденных rликанов являет
ся удобным способом получения информации
о различных видах rликанов, представленных
в белке (профилирование по характеру BeT
вления: двойное, тройное или более BЫCOKO
ю порядка). На этой стадии определяют также
степень сиалирования. В зависимости от BЫ
бранноro метода, для детектирования rликанов
может быть необходима предварительная дери
ватизация/мечение.
Анализ высвобожденных rликанов обычно
включает в себя высвобождение и очистку rлика
нов из реакционной смеси, сопровождающиеся
при необходимости мечением/дериватизацией
rликанов; полученные rликаны затем профили
руют (фракционирование или разделение).
2 З 1. Высвобождение rликанов
Выбор метода, который используется для
высвобождения rликанов, зависит от испытуе
моro rликопротеина. Применяемый areHT pac
щепления выбирается в зависимости от типа
требуемоro расщепления и уровня необходи
мой информации. Может быть использовано
ферментативное или химическое расщепление.
В таблице 2.2.59. 1 представлен неполный
список areHToB ферментативною расщепления
и их особенности.
Обычно эффективность расщепления за
висит от доступности rликанов в rликопротеине.
Следовательно, для увеличения доступности
мест rликозилирования белок может быть дeHa
турирован, за исключением случаев, коrда He
обходимо различить поверхностные и поrружен
ные rликаны.
Также MOryT быть использованы areHTbI
химическоro расщепления, например, rидра
зин или борrидриды щелочных металлов для
l3 элиминирования.
2 3 2. Анализ rликанов
Высвобожденные rликаны MOryт быть про
анализированы или профилированы xpOMa
тоrрафическими, электрофоретическими и
масс спектрометричекими методами, а также co
четанием этих всех методов. Выбор метода про
изводят в зависимости от природы rликанов и
уровня необходимой информации.
Анализ rликанов предоставляет информа
цию о различных видах rликанов, присутствую
щих в белке (с высоким содержанием маннозы,
комплексных, rибридных). Информация об OTHO
сительных количествах разветвленных структур
может быть получена с помощью анализа деси
алированных rликанов.
Может потребоваться стадия разделения.
Она подразумевает использование ЖХ (2.2.29)
и КЭ (2.2.47) в качестве промежуточных MeTO
Примеры а2ентов ферментативН020 расщепления
Таблица 2.2.59. 1
AreHTbI Специфичность
Высвобождение N--связанных rликанов
Пептид-N4-(N-ацетил-l3-rлюкозаминил)аспара- rидролиз N4-(ацетил-Р-D-rлюкозаминил)аспа-
rинамидаза (3.5.1.52) раrиновых остатков, в которых остаток rлю-
козамина может быть далее rликозилирован,
с получением (замещенноrо) N-ацетил-Р-D-
< rлюкозаминиламина и пептида, содержащеrо
остатокаспартата
Пептид N rликозидаза F (PNGase F) Высвобождение N rликановой цепи, кроме
N rликановой цепи, содержащей (а.1 3) связанное
ядро фукозы
Пептид N rликозидаза А (PNGase А) Высвобождение N rликановой цепи, содержащей
(0.1 з ) связанное ядро фукозы
Маннозил-rликопротеинэндо-Р-N- Эндоrидролиз N,N'-диацетилхитобиозиловых
ацетилrлюкозаминидаза (3.2.1.96) единиц в rликопептидах/rликопротеинах с вы-
соким содержанием маннозы, содержащих
структуру -[Мап(GlсNАс),]Аsп
Эндо Р N ацетилrлюкозаминидаза F (эндо F) Высвобождение rибридных и комплексных олиro
сахаридов с высоким содержанием маннозы
Эндо Р N ацетилrлюкозаминидаза Н (эндо Н) Высвобождение rибридных олиrосахаридов с BЫ
соким содержанием маннозы
Высвобождение О-связанных rликанов
rликопептид а.-N-ацетилrалактозаминидаза rидролиз терминальных D-rалактозил-N-
(3.2.1.97)* ацетил-а.-D-rалактозаминидиновых остатков
.Данный фермент используется редко из за своей высокой специфичности к субстрату.
23. З,к 1712.
170
rосударственная фармакопея Республики Беларусь
дов. ЖХ (2.2.29) может при меняться как препа
ративная, со сбором индивидуальных фракций
(обычно требуется мечение), так может быть и
совмещена непосредственно с МС (2.2.43).
2 З 2 1. Анализ немеченых 2ликанов
Нативные rликаны MOryт быть проанализи
рованы с помощью анионообменной xpOMaтo
rрафии при высоких значениях рН с импульсным
амперометрическим детектированием (HPAEC
РАО), пористо rрафитной хроматоrрафией (пrх,
PGC) и МС (2.2.43).
Метод HPAEC PAO обладает высокой чув
ствительностью и способностью к разделению
некоторых изомеров. Коэффициенты откли
ков разных сиrналов не одинаковы из за разли
чий олиюсахаридных структур. Полный количе
ственный анализ rликанов невозможен, кроме
случаев, KorAa доступна информация о CTaHдap
тах олиroсахаридов. Количественное определе
ние может быть проведено путем сравнения ис
пытуемоrо образца с соответствующим хорошо
изученным стандартным образцом или путем
соотнесения площади пика каждою rликана с
общей площадью пиков всех rликанов на карте.
Для разделения при родных rликанов также
может при меняться пrх, обладающая более BЫ
сокой селективностью по сравнению с общепри
нятой хроматоrрафией на неполярных колонках.
Для прямою анализа rликанов может приме
няться пrх мс с электрораспылительной иони
зацией.
2 З 2 2. Анализ маркированных 2ликанов
Л1аркированиеzликанов
Тип производимоro химическоro изменения
(дериватизации) зависит от метода, используе
мою для детектирования rликанов: уф или флу
оресцентный.
Дериватизация с флуоресцентными MeTKa
ми наиболее используемый метод для марки
рования rликанов на восстанавливающем остат
ке путем восстановительноro аминирования. Один
маркер может прикрепляться к каждому MOH и
олиrосахариду, позволяя определить молярное K
личество. В таблице 2.2.59. 2 приведен неполный
список обычно используемых флуоресцентных
маркеров и подходящих аналитических методов.
Для детектирования с использованием
только МС (2.2.43) также может использоваться
предельное метилирование rликанов. ОНО OCHO
вано на метилировании олиroсахаридов.
Анализ маркированных 2ликанов
Маркированные rликаны MOryT быть проана
лизированы с помощью таких аналитических Me
тодов, как ЖХ (2.2.29), КЭ (2.2.47) и МС (2.2.43).
В соответствии со свойствами разделяю
щихся rликанов, они MOryT быть профилиро
ваны и определены количественно с исполь
зованием подходящеro маркера с помощью
нескольких систем ЖХ (2.2.29): обращенно фаз
ной (разделение по rидрофобности), нормаль
но фазной (разделение по размеру), и анионо
обменной (разделение по заряду) ЖХ.
2 4. АНАЛИ3 МОНОСАХАРИДОВ
Анализ моносахаридов дает информа
цию о моносахаридной структуре rликопро
теина. Анализ может быть выполнен как KO
лори метрическими, так и разделительными
методами.
2--4 1. Колориметрические методы
Колориметрические методы, основанные
на химическом окрашивании, дают информа
цию о количестве специфических классов caxa
ров, таких как сиаловые кислоты, нейтральные
сахара и rексозамины.
2 4 2. Методы разделения
Разделительные методы предоставля
ют информацию о полном моносахаридном
составе. Перед использованием методов
требуется предварительный кислотный rи
дролиз олиrосахаридных цепей интактных
rликопротеинов или высвобожденных rли
канов. Для высвобождения сиаловых кислот
при меняется умеренный кислотный rидро
лиз или ферментативная обработка. Стадия
rидролиза это значимый источник вариа
бельности, и она требует продукт специфич
ной валидации.
Методы для разделения и количественноro
определения моносахаридов включают:
использование HPAEC PAO и пrх мс, KO
торые позволяют определить молярные количе
ства нативных моносахаридов (сиаловые кисло
ты, нейтральные сахара и альдиты);
флуорофорное мечение моносахаридов с
последующим использованием методов разде
ления, таких как обращенно фазная или ионооб
менная хроматоrрафия или капиллярный элек
трофорез.
Таблица 2.2.59. 2
Примеры флуоресцентных меток и подходящих аналитических методов
Название Аббревиатура Аналитические методы
2 Аминобензойная кислота 2 AA ЖХ (2.2.29), МС (2.2.43)
2 Аминобензамид 2 AB ЖХ (2.2.29), МС (2.2.43)
2 Аминопиридин 2 AP ЖХ (2.2.29), МС (2.2.43)
2 Амино 9( 1 ОН) акридинон АМАС rель электрофорез (2.2.31)
Три натрия 8 аминопирин APTS КЭ (2.2.47)
1 ,З,6 трисульфонат
2.2.59. Анализ 2ликанов в 2ликопротеинах
171
3. ОЦЕНКА И АНАЛИЗ РЕЗУЛЬТАТОВ
Исследование и оценка результатов, полу
ченных в ходе анализа rликанов, производится
с 3 различными целями:
подтверждение подлинности индивиду
альных структур или rрупп структур;
подтверждение соответствия испытуемо
ro образца при качественном анализе;
подтверждение соответствия испытуемо
ro образца при количественном анализе.
Особые соображения касательно CTaHдapT
ных образцов и методов проведения каждой
стадии анализа приведены в разделах 4 и 5 co
ответственно.
3--1. ПОДТВЕРЖДЕНИЕ ПОДЛИННОСТИ
ИНДИВИДУАЛЬНblХ СТРУКТУР
ИЛИ rрупп СТРУКТУР
Аналитической мишенью метода анализа
rликанов может быть отдельный моносахарид
(например, сиаловая кислота, фукоза), опреде
ленная олиroсахаридная структура (например.
тетра сиалиловый, тетра антеннальный rликан)
или rpynna структур, имеющая общую анали
тическую особенность (например, тетра--сиа
лиловые rликаны, три антеннальные rликаны,
rликопротеиновые изоформы с одинаковым за
рядом). Подтверждение подлинности аналити
ческой мишени это важнейший шаr при aHa
лизе и оценке результатов, который может быть
достиrнут абсолютно (путем подтверждения MO
лекулярной структуры) или сравнительно (путем
сравнения с соответствующим стандартным об
разцом).
З 1 1. Абсолютное подтверждение под
линности
Абсолютное подтверждение подлинности
rликановых структур выполняется обычно во
время разработки лекарственноro средства, и
оно не обязательно является целью рутинноro
анализа. Подлинность аналитической мишени
устанавливают с помощью известноro молеку:
лярноro свойства молекулы. Такая абсолютная
идентификация индивидуальных структур Tpe
бует мноroступенчатых подходов, включающих
в себя ферментативные и химические реакции,
разделительные методики и оп liпе или оfМiпе
методы детектирования, и обычно использует
в качестве основополаrающеro принципа для
ПQДтверждения структуры отношение заряда к
массе молекулярноro иона, которое определяет
ся rюдxодящим масс спектрометрическим MeTO
дом.
1 2. Сравнительное определение под-
линности
При рутинном применении аналитическо
ro метода подлинность аналитической мишени
может быть подтверждена путем сравнения со
стандартными образцами процесса или приroд
насти системы. Они MOryт быть получены из из
вестных хорошо изученных rликопротеинов, KO
торые MOryт принадлежать тому же классу, что
и испытуемый образец (например, фетуин для
N связанных rликопротеинов), или MOryт быть
произведены из хорошо изученной серии испы
туемоro продукта, которая была определена как
стандартный образец. Следующие утверждения
применяются к сравнительному определению
структурной подлинности:
в случае подтвержденной высокой BOC
производимости времени удерживания, для KOp
ректной интерпретации может быть использова
но абсолютное время удерживания;
в качестве альтернативы, в начале и в
конце испытания может быть введен rликановый
маркер для проверки любых отклонений BpeMe
ни удерживания; на основе этих эталонных xpo
MaTorpaMM MOryт быть интерпретированы rлика
ны испытуемых образцов;
в случаях, Korдa стандарт для определе
ния всех пиков rликанов в испытуемом образце
отсутствует, для контролирования и мечения He
известных пиков rликанов может использовать
ся абсолютное или нормализованное время
удерживания.
3 2. ПОДТВЕРЖДЕНИЕ СООТВЕТСТВИЯ
ИСПblТУЕмоrо ОБРАЗЦА ТРЕБОВАНИЯМ
КАЧЕСТВА
На этом этапе определения аналитические
результаты, полученные для испытуемоro об
разца, оцениваются на предмет соответствия
спецификации. Обычно это достиrается путем
сравнения с данными, полученными при парал
лельном испытании соответствующеro стандарт
ноro образца. При оценке данных необходимо:
для проверки приroдности системы YCTa
новить, что аналитический результа получен
ный при использовании стандартноro образца,
соизмерим с ожидаемым результатом; напри
мер, при картировании rликанов это будет дo
стиrнуто путем сравнения карты для стандарт
ноro образца с представленной картой образца,
полученной при создании этоro стандартноro об
разца, и с помощью подтверждения приroдности
всех критериев системы;
продемонстрировать сходство apT CTaH
дартноro образца и испытуемоro образца, ис
пользуя любые специфические критерии приroд
ности, указанные в частной статье.
3 3. ПОДТВЕРЖДЕНИЕ СООТВЕТСТВИЯ
ИСПblТУЕмоrо ОБРА3ЦА ТРЕБОВАНИЯМ
КОЛИЧЕСТВЕнноrо АНАЛИ3А
3 3 1. Количественное определение со-
держания анализируемоrо вещества и пред-
ставление результатов
В некоторых случаях, например, при опре
делении сиаловой кислоты или друrих MOHO
сахаридов, данные предоставляют в виде
молярноrо соотношения сиаловой кисло
172
rосударственная фармакопея Республики Беларусь
ты к rликопротеину. Данные рассчитывают с
учетом стандартноro образца сиаловой кис
лоты и валидированноrо метода опред€rе
ния белка. Может быть использован как метод
внутреннею стандарта, так и метод внешне
ro стандарта (2.2.46. Хроматоарафические
методы разделения).
3 3 2. Количественное выражение про-
филя разделения
Профили или структуры распределения
MorYT быть представлены в цифровом виде
несколькими путями, включая процесс HOp
мализации; процент содержания каждой aHa
литической мишени, например, части rлика
на, рассчитывают путем определения отклика
части rликана как процентную составляющую
общею отклика всех частей, не учитывая OT
клики растворителей, друrих добавленных pe
активов и сиrналов ниже неучитываемоrо пре
дела. Кроме тоro, MorYT использоваться такие
численные выражения, как Z число, которые
специфичны методу и продукту и описаны в
частных статьях.
4. СТАНДАРТНЫЕ ОБРАЗЦЫ
Стандартные образцы для анализа rлика
нов выполняют две функции: проверка приrод
ности системы и подтверждение соответствия
испытуемоrо образца специфицированным Tpe
бованиям.
Стандартными образцами, использующи
мися для проверки приroдности системы, MOryT
быть:
стандартный образец испытуемоro веще
ства;
rликановая часть, высвобожденная из
полностью изученноro стандартною образца ис
пытуемоro вещества;
хорошо изученные rликановые части, BЫ
свобожденные из rликопротеинов (например,
фетуин, IgG);
маркеры rликановых частей, изученные
по показателям идентификации и чистоты.
Стандартный образец, используемый для
получения заключения о соответствии испыту
eMoro rликопротеина, является препаратом ис
пытуемоrо вещества. Ранее отмечено, что Me
тодики анализа rликанов, описанные в частных
статьях, предписывают использование CTaH
дартных образцов для испытуемою вещества и
валидированной методики анализа rликанов.
5. ПУНКТЫ, КОТОРЫЕ УЧИТЫВАЮТСЯ
ПРИ РАЗРАБОТКЕ МЕТОДА
Этот раздел представляет способы оценки
исполнения в целом метода при еro разработке.
Степень проработки метода и аналитическая Ba
лидация выбираются на основе их приroдности
для определенноro продукта. В зависимости от
выбранноro метода, для анализа rликанов необ
ходимы определенные действия, например, как:
выделение и очистка (или деминерализа
ция) rликопротеина;
ферментативная (или химическая) обра
ботка rликопротеина для селективноro BЫCBO
бождения как N , так и О СВfJзанных rликанов от
белковою остова;
выделение и очистка высвобожденных
rликанов;
контроль высвобожденной сиаловой кис
лоты и моносахаридных остатков;
хромофорное мечение высвобожденных
rликанов;
разделение rликанов, нативных или флу
оресцентно меченных;
идентификация и количественное опреде
ление rликанов (например, вычисление Zчисла);
определение положений связывания на
основании относительных количеств rликозили
рованных и неrликозилированных пептидов.
Выделение и очистка белков. Для удале
ния мешающих веществ (например, BcnoMora
тельные вещества, соли) MOryT понадобиться
выделение и очистка rликопротеинов из матри
цы, что при необходимости описывают в частной
статье. Эти процедуры необходимо проводить
воспроизводимо для rарантии количественной
открываемости белка.
Высвобождение и выделение олиrоса-
харидов. Подход, выбранный для высвобожде
ния rликанов, зависит от анализируемых белков
и основан на типе rликозилирования, т. е. N или
О связанное rликозилирование. Не описанные
в Фармакопее подходы, применяемые дЛЯ BЫ
свобождения rликанов, должны быть оптими
зированы для подтверждения количественно
ю профилирования rликановых составляющих.
Факторы, влияющие на эффективность расще
пления, такие как соотношение концентраций
фермент белок, температура, время протекания
реакции и предварительная денатурация белка,
должны быть оптимизированы.
Следует отметить, что ферментативная/
химическая реакция не должна изменять CTpO
ение rликана, например, не должна разрушать
остатки сиаловой кислоты. Там, rдe присутствует
более одноro положения rликозилирования, при
ферментативной обработке должны пропорцио
нально высвобождаться все фраrменты олиro
сахаридов, присоединенные к белку, независи
мо от их структуры и своею конкретною места в
белке. Должна быть подтверждена воспроизво
димость открываемости всех rликановых COCTaB
ляющих из реакционной смеси.
Дериватизация высвобожденных rли-
канов. Дериватизацию обычно выполняют в
соответствии с методиками, неописанными в
фармакопее. По этой причине повторная дe
риватизация всех rликановых rрупп должна
верифицироваться, что может быть достиrну
2.2.59. Анализ 2ликанов в zликопротеинах
173
Необходим анализ rликана rликопротеина
Создание метода анализа rликана
Небольшой белок,
оrpаниченное rликозилирование
и/или простое строение?
Нет
Да Анализ интактноrо rликопротеина
(например, масс спектрометрический
анализ)
Большие молекулы, сложные разветвленные
rликаны и/или мноroчисленные места rликозилирования
Требуется определение Да
положений rликозилирования?
Нет
Определение отрицательно
заряженноrо содержимоrо Нет
Определение сиаловой кислоты
и/или моносахаридов
Требуется подробный
анализ rликанов?
Протеолитическое расщепление
и разделение rликопептидов
Да
Создание методов для анализа расщепленных
rликанов (например, антенальный профиль,
определение индивидуальной структуры)
Рисунок 2.2.59. 2. Руководство по выбору методики для проведения анализа 2ликанов
то при оптимизации условий реакций, таких
как количество areHToB дериватизации, TeM
пература и время реакции. Реакция деривати
зации не должна изменять строение rликана,
например, не должна разрушать остаток сиа
ловой кислоты.
Разделение, идентификация и проверка
приrодности системы. Методы, используемые
для анализа rликанов, должны позволять обна
ружение и разделение разных фраrментов rли
канов для достоверности идентификации и KO
личественноrо анализа.
Критерии приемлемости для проверки при
rодности системы, а также расщепления, OTKpЫ
ваемости и анализа rликанов, зависят от крити
ческих параметров испытания, которые влияют
на конечный результат.
Сравнение rликановых карт испытуемоrо
образца и стандартноro образца, полученных
в одинаковых условиях, является пара метром
для оценки правильности выполнения аналити
ческой процедуры. Для дальнейшеrо подтверж
дения полученных результатов анализы MOryт
быть повторно проведены с помощью независи
моro метода. Использование стандартноro об
разца (например, стандартный образец испыту
eMoro вещества, маркер rликана для про верки
приrодности системы) является необходимым
24 За.. 1712
для установления пара метров приroдности си
стемы и валидации аналитической процедуры.
Воспроизводимость результатов количе
cTBeHHoro определения (например, расчет Z
числа) rликана должна быть подтверждена.
Определение положений связывания на
основании относительных количеств rлико
зилированных и неrликозилированных пеп-
тидов. При установлении положений связы
вания путем сравнения rликозилированных и
неrликозилированных пептидов с ферментатив
но расщепленными rликопротеинами должна
быть продемонстрирована воспроизводимость
расщепления обеих форм пептидов.
6. СХЕМА ПРИНЯТИЯ РЕШЕНИЙ
ПРИ АНАЛИЗЕ rлиКАНОВ
Эта система принятия решений приводит
ся для информации и не является обязательной
частью Фармакопеи.
Выбор метода для анализа rликанов про
изводят в зависимости от уровня информации,
необходимой для подтверждения качества rли
копротеина, и устанавливают при разработке
продукта.
Руководство по выбору методики для прове
дения rликановоrо анализа, при необходимости,
представлено на рисунке 2.2.59. 2.
174
01/2013:20260
rосударственная фармакопея Республики Беларусь
2.2.60. ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
ИНСТРУМЕНТАЛЬНЫЙ МЕТОД
В разделе описано измерение температуры
плавления капиллярным методом с использова
нием инструментальноro метода определения.
ПРИ БОР
Существуют 2 варианта автоматической pe
rистрации:
вариант А: по пропусканию света через Ka
пилляр с испытуемым образцом;
вариант В: по свету, отраженному образ
цом в капилляре.
В обоих вариантах капиллярную трубку по
мещают в отверстие электронаrреваемоro Me
таллическоro блока, снабженноro температур
ным датчиком, размещенным в друroм отверстии
блока. Наrревательный блок должен при Harpe
вании выдерживать заданную температуру с
точностью до :t0, 1 ОС, а также обеспечивать Meд
ленную и устойчивую скорость HarpeBa 1 ОС/мин
после начальной изотермической стадии.
В варианте А луч света проходит через ro
ризонтальное отверстие и вертикально YCTaHOB
ленную капиллярную трубку и попадает на фо
тодатчик.
В варианте В луч света освещает капилляр
ную трубку снаружи, а датчик записывает изо
бражение.
Некоторые модели приборов позволяют
проводить визуальное определение точки плав
ления.
Температура, при которой сиrнал датчика из
менит начальное показание, называется началом
плавления, а температура, при которой сиrнал
достиrнет окончательноro значения концом
плавления, или температурой плавления.
А
А
F
Е Е)
+
+
+
+
+
+
D 8 +
F
Е:Э в о с
с
о
Е
Рисунок 2.2.60. 1. Вариант А пропускание
А стеклянная капиллярная трубка;
В испытуемый образец;
С фотодатчик;
D температурный датчик;
Е наrревательный блок;
F источник излучения.
...G
Е
Рисунок 2.2.60. 2. Вариант В отражение
А стеклянная капиллярная трубка;
В испытуемый образец;
С даТ'lИК;
D температурный датчик;
Е наrревательный блок;
F источник излучения;
G прозрачнаяпластинка.
2.3.1. Реакции подлинности на ионы и функциональные zруппы
175
Используют стеклянные капиллярные
трубки с запаянным концом длиной 100 мм, Ha
ружным диаметром 1 ,3 1,5 мм и внутренним
диаметром 0,8 1,3 мм. Толщина стенки капил
лярной трубки О, 1 0,3 мм.
Некоторые модели приборов позволяют
определять температуру плавления более чем
на одной капиллярной трубке.
МЕТОДИКА
Испытуемый образец, предварительно об
работанный, как указано в частной статье, по
мещают в капиллярную трубку в количестве, дo
статочном для формирования в трубке столбика
высотой около 4 мм. Трубку выдерживают в Te
чение определенноro времени при температуре,
указанной в частной статье.
Далее поступают, как указано в инструкции
изrотовителя по эксплуатации прибора или сле
дующим образом. Наrревают наrревательный
блок до температуры на 5 0 С ниже ожидаемой
температуры плавления. Помещают капилляр
ную трубку в наrревательный блок запаянным
концом вниз. Включают температурную проrрам
му. Korдa субстанция начнет плавиться, изме
нится ее внешний вид, в результате чеro после
изменения сиrнала фотодатчика, обусловлен
ноro пропусканием света (рисунок 2.2.60. 1) или
изменением изображения (2.2.60. 2), автомати
чески будет записана температура наrреватель
ноro блока.
Повторяют испытание еще на 2 x образцах
и рассчитывают среднее значение из З х резуль
татов.
КАЛИБРОВКА
Температурная шкала прибора периоди
чески должна проверяться путем измерения
температуры плавления сертифицированных
веществ сравнения. При этом используют Ka
пиллярные трубки такоro же размера, что и при
обычном измерении температуры плавления
(см. «Прибор» ). <
rотовят по 3 капиллярные трубки на каждое
сертифицированное вещество сравнения (не
менее 2 x). Проводят испытание и рассчитыва
ют среднее значение из 3 x результатов для каж
доro вещества.
приrодность СИСТЕМЫ
В дополнение к калибровке перед проведени
ем испытания осуществляют верификацию с ис
пользованием подходящеro сертифицированноro
вещества сравнения с температурой плавления,
близкой к таковой для испытуемоro образца.
rотовят по 3 капиллярные трубки. Проводят
испытание и рассчитывают среднее значение из
3 x результатов.
Среднее значение должно укладываться в
пределы, установленные в сертификате, прила
raeMoM к сертифицированному веществу cpaB
нения.
2.3.
ПОДЛИННОСТЬ
(ИДЕНТИФИКАЦИЯ)
01/2013:20301
2.3.1. РЕАКЦИИ ПОДЛИННОСТИ
(ИДЕНТИФИКАЦИИ)
НА ИОНЫ
И ФУНКЦИОНАЛЬНЫЕ rруппы
АЛКАЛОИДЫ
Несколько миллиrраммов или указанное в
частной статье количество испытуемоro образца
растворяют в 5 мл воды Р, прибавляют кислоту
хлористоводородную разведенную Р до кислой
реакции раствора (2.2.4), затем 1 мл раствора
калия йодовисмутата Р; тотчас образуется
оранжевый или оранжево красный осадок.
АЛЮМИНИЙ
Около 15 Mr испытуемоro образца paCTBO
ряют в 2 мл воды Р. К полученному раствору или
к 2 мл раствора, указанноro в частной статье,
прибавляют 0,5 мл кислоты хлористоводород
ной разведенной Р и около 0,5 мл реактива ти
оацетамида Р; осадок не образуется. Затем
прибавляют по каплям раствор натрия 2идpOK
сида разведенный Р; образуется rелеобразный
белый осадок, растворяющийся при последую
щем прибавлении раствора натрия 2идроксида
разведеНН020 Р. К полученному раствору посте
пенно прибавляют раствор аммония хлорида Р;
вновь образуется rелеобразный белый осадок.
АМИНЫ АРОМАТИЧЕСКИЕ ПЕРВИЧНЫЕ
Испытуемый раствор, указанный в частной
статье, подкисляют кислотой хлористоводо
родной разведенной Р #(или 0,05 r испытуемо
ro образца растворяют в кислоте клористово
дородной разведенной Р)# и прибавляют 0,2 мл
раствора натрия нитрита Р. Через 1 2 мин
прибавляют 1 мл раствора fЗ нафтола Р; по
является интенсивное оранжевое или красное
окрашивание и, как правило, образуется осадок
такоro же цвета.
АММОНИЯ СОЛИ
К испытуемому раствору, указанному в част
HO< статье, прибавляют 0,2 r ма2ния оксида Р.
Через жидкость пропускают воздух и направля
ют выходящий rаз в смесь из 1 мл 0,1 М pacт
вора кислоты хлористоводородной и 0,05 мл
раствора метилов020 краСН020 Р; окраска ИН
дикатора переходит в желтую. Затем прибав
ляют 1 мл свежеприroтовленноrо раствора
176
rосударственная фармакопея Республики Беларусь
100 r/л натрия кобальтинитрита Р; образует
ся желтый осадок.
АММОНИЯ СОЛИ И СОЛИ ЛЕТУЧИХ
ОСНОВАНИЙ
Около 20 Mr испытуемоro образца paCTBO
ряют в 2 мл воды Р. К полученному раствору или
к 2 мл раствора, указанноro в частной статье,
прибавляют 2 мл раствора натрия 2идроксида
разведеНН020 Р. При наrревании раствора Bыдe
ляются пары аммиака, которые обнаруживаются
по запаху и щелочной реакции (2.2.4).
АЦЕТАТЫ
а) Испытуемый образец наrревают с равным
количеством кислоты щавелевой Р; выделяется
кислота уксусная, обнаруживаемая по запаху и
кислой реакции (2.2.4).
Ь) Около 30 Mr испытуемоro образца раст
воряют в 3 мл воды Р. К полученному paCTBO
ру или к 3 мл раствора, указанноro в частной
статье, последовательно прибавляют 0,25 мл
раствора лантана (111) нитрата Р, 0,1 мл
0,05 М раствора йода и 0,05 мл раствора aM
миака разведеНН020 Р2. Смесь осторожно Ha
rревают до кипения; в течение нескольких
минут образуется синий осадок или появляется
синее окрашивание.
#с) К 2 мл нейтральноro раствора испыту
емоro образца, содержащеro 20 0 Mr aцeTaT
иона (СНзСОО ), прибавляют 0,2 мл раствора
30 r/л железа (111) хлорида Р; появляется KpaCHO
коричневое окрашивание, исчезающее при при
бавлении минеральных разведенных кислот.
#d) 2 мл раствора испытуемоro образца, co
держащеro 20 0 Mr ацетат иона (СНзСОО ), Ha
rревают с равным количеством кислоты серной
концентрированной Р и 0,5 мл 96 % спирта Р;
образуется этилацетат, обнаруживаемый по
запаху.
АЦЕТИЛ
Около 15 Mr или указанное в частной статье
количество испытуемоro образца помещают в
пробирку длиной около 180 мм и наружным ди
аметром 18 мм и прибавляют 0,15 мл кислоты
фосфорной Р. Пробирку закрывают пробкой,
через которую пропущена небольшая пробир
ка длиной около 100 мм и наружным диаметром
10 мм, содержащая воду Р и выполняющая роль
холодильника. На внешнюю поверхность MeHb
шей пробирки помещают 1 каплю раствора
лантана (111) нитрата Р. Если субстанция OTHO
сительно леrко rидролизуется, устройство поме
щают на 5 мин в водяную баню, затем вынимают
меньшую пробирку. Для трудно 2идролизуемых
субстанций смесь медленно наrpевают на OT
крытом пламени до кипения. Каплю снимают,
смешивают на фарфоровой пластинке с 0,05 мл
0,01 М раствора йода. На край капли наносят
0,05 мл раствора аммиака разведеНН020 Р2;
через 1 2 мин в месте соединения двух капель
появляется синее окрашивание, которое усили
вается и сохраняется в течение короткоro про
межутка времени.
БАРБИТУРАТЫ (3А ИСКЛЮЧЕНИЕМ
N ЗАМЕЩЕННЫХ)
Около 5 Mr испытуемою образца растворяют в
3 мл метанола Р, прибавляют 0,1 мл раствора, со-
держащеro 100 r/л кобальта нитрата Р и 100 r/л
кальция хлорида Р, перемешивают и прибавля
ют при встряхивании 0,1 мл раствора натрия 2и
дроксида разведенною р. появляется фиолетово
синее окрашивание и образуется осадок.
БЕНЗОАТЫ
а) К 1 мл раствора, указанноro в частной
статье, прибавляют 0,5 мл раствора железа (111)
хлорида Р1; образуется розовато желтый oca
док, растворимый в эфире Р.
Ь) 0,2 r испытуемоro образца, при необхо
димости подrотовленноro, как указано в частной
статье, помещают в пробирку, смачивают 0,2 мл
или 0,3 мл кислоты серной Р и осторожно Ha
rpевают дно пробирки; на внутренних стенках
пробирки появляется белый налет.
с) 0,5 r испытуемоro образца растворяют
в 1 О мл воды Р. К полученному раствору или к
10 мл раствора, указанноro в частной статье,
прибавляют 0,5 мл кислоты хлористоводород
ной р. образуется осадок, который после пере
кристаллизации из теплой воды Р и высушива
ния под вакуумом (2.2.32) имеет температуру
плавления (2.2.14) от 120 ос до 124 ОС.
БРОМИДЫ
а) Навеску испытуемоro образца, содержа
щую около 3 Mr бромид иона (Br ), растворяют в
2 мл воды Р. Полученный раствор или 2 мл раст
вора, указанноro в частной статье, подкисляют
кислотой азотной разведенной Р, прибавляют
0,4 мл раствора серебра нитрата Р1, переме
шивают и отстаивают; образуется светло жел
тый творожистый осадок. Осадок отделяют цeH
трифуrированием и промывают тремя порциями
воды Р по 1 мл каждая. Эти операции проводят
быстро в защищенном от яркоro света месте,
при этом допускается, чтобы жидкость над ocaд
ком не была полностью прозрачной. Получен
ный осадок суспендируют в 2 мл воды Р и при
бавляют 1,5 мл раствора аммиака Р; осадок
трудно растворяется.
Ь) Навеску испытуемоro образца, эквива
лентную около 5 Mr бромид иона (Br ), или KO
личество образца, указанное в частной статье,
помещают внебольшую пробирку, прибавляют
0,25 мл воды Р, около 75 Mr свинца (/V) оксида Р,
0,25 мл кислоты уксусной Р и осторожно встря
хивают. Верхнюю внутреннюю часть пробирки
высушивают с помощью фильтровальной бумаrи
и выдерживают в течение 5 мин. Полоску филь
тровальной бумаrи необходимоro размера про
питывают, помещая ее край в каплю раствора
2.3.1. Реакции подлинности на ионы и функциональные аруппы
177
фуксина обесцвечеНН020 Р и тотчас помещают
пропитанную часть в пробирку. В течение 1 О с У
нижнеro края фильтровальной бумаrи появляет
ся фиолетовое окрашивание, которое четко OT
личается от красной окраски фуксина, наблю
даем ой в верхней пропитанной части полоски
бумаrи.
#с) К 1 мл раствора испытуемоrо образца,
содержащеro 2 30 Mr бромид иона (Br ), при
бавляют 1 мл кислоты хлористоводородной
разведенной Р, 0,5 мл свежеприrотовленноrо
раствора 50 r/л хлорамина Р, 1 мл хлорофор
ма Р и встряхивают; хлороформный слой приоб
ретает желто коричневую окраску.
ВИСМУТ
а) 0,5 r испытуемоro образца растворяют в
10 мл кислоты хлористоводородной разведен
ной Р. Полученный раствор или 1 О мл раствора,
указанноro в частной статье, кипятят в течение
1 мин, охлаждают и при необходимости филь
труют. К 1 мл полученноro раствора прибавляют
20 мл воды Р; образуется белый или светло жел
тый осадок, цвет котороro после прибавления от
0,05 мл до 0,1 мл раствора натрия сульфида Р
изменяется на коричневый.
Ь) Около 45 Mr испытуемоro образца paCT
воряют в 1 О мл кислоты азотной разведен
ной Р. Полученный раствор или 1 О мл раствора,
указанноro в частной статье, кипятят в течение
1 мин, охлаждают и при необходимости филь
труют. К 5 мл полученноro раствора прибавляют
2 мл раствора 100 r/л тиомочевины Р; появля
ется желтовато оранжевое окрашивание или об
разуется оранжевый осадок. Затем прибавляют
4 мл раствора 25 r/л натрия фторида Р; раст
вор не обесцвечивается в течение ЗО мин.
#с) Навеску испытуемоro образца, coдep
жащую около 50 Mr висмут иона (ВР+), встряхи
вают с 5 мл кислоты серной разведенной Р и
фильтруют. К фильтрату прибавляют 2 капли
раствора калия йодида Р1; образуется черный
осадок, растворимый в избытке реактива с<об
разованием раствора желтовато оранжевоrо
цвета.
ЖЕЛЕЗО
а) Навеску испытуемоro образца, содержа
щую около 10 Mr железо иона (Fe 2 +), растворяют
в 1 мл воды Р. К полученному раствору или к
1 мл раствора, указанноro в частной статье, при
бавляют 1 мл раствора калия феррицианида Р;
образуется синий осадок, не растворяющийся
при прибавлении 5 мл кислоты хлористоводо
родной разведенной Р.
Ь) Навеску испытуемоro образца, содержа
щую около 1 Mr железо иона (Fе З +), растворяют в
30 мл воды Р. К 3 мл полученноro раствора или
к 3 мл раствора, указанноro в частной статье,
прибавляют 1 мл кислоты хлористоводородной
разведенной Р и 1 мл раствора калия тиoциa
ната Р; появляется красное окрашивание. Отби
рают две порции полученноro раствора по 1 мл
каждая. К одной порции прибавляют 5 мл спирта
изоамиловО20 Рили 5 мл эфира Р, встряхивают
и оставляют до расслоения; орrанический слой
окрашивается в розовый цвет. К друrой порции
прибавляют 2 мл раствора ртути (11) хлори
да Р; красное окрашивание раствора исчезает.
с) Навеску испытуемоro образца, содержа
щую не менее 1 Mr железо иона (Fе З +), paCTBO
ряют в 1 мл воды Р. К полученному раствору или
к 1 мл раствора, указанноro в частной статье,
прибавляют 1 мл раствора калия ферроциа
нида Р; образуется синий осадок, не растворя
ющийся при прибавлении 5 мл кислоты хлори
стоводородной разведенной Р.
ЙОДИДЫ
а) Навеску испытуемоro образца, coдep
жащую около 4 Mr йодид иона (I ), растворяют
в 2 мл воды Р. Полученный раствор или 2 мл
раствора, указанноro в частной статье, под
кисляют кислотой азотной разведенной Р,
прибавляют 0,4 мл раствора серебра Hитpa
та Р1, перемешивают и отстаивают до обра
зования светло желтоro творожистоro осадка.
Осадок отделяют центрифуrированием и про
мывают 3 порциями воды Р по 1 мл каждая.
Эту операцию проводят быстро в защищенном
от яркоro света месте; при этом допускается,
чтобы жидкость над осадком не была полно
стью прозрачной. Осадок суспендируют в 2 мл
воды Р и прибавляют 1,5 мл раствора aMMиa
ка Р; осадок не растворяется.
Ь) К 0,2 мл раствора испытуемоro образ
ца, содержащеro около 5 Mr йодид иона (I ) в
1 мл, или К 0,2 мл раствора, указанною в част
ной статье, прибавляют 0,5 мл кислоты серной
разведенной Р, 0,1 мл раствора калия диxpOMa
та Р, 2 мл воды Р, 2 мл хлороформа Р, встряхи
вают в течение нескольких секунд и оставляют
до расслоения; хлороформный слой приобрета
ет фиолетовую или фиолетово красную окраску.
#с) При наrревании 0,1 r испытуемоro образ
ца с 1 мл кислоты серной Р выделяются фиоле
товые пары йода.
КАЛИЙ
а) 0,1 r испытуемоro образца растворяют в
2 мл воды Р. К полученному раствору или к 2 мл
раствора, указанною в частной статье, прибав
ляют 1 мл раствора натрия карбоната Р и Ha
rревают; осадок не образуется. К roрячему pac
твору прибавляют 0,05 мл раствора натрия
сульфида Р; осадок не образуется. Раствор ox
лаждают в ледяной воде, прибавляют 2 мл paCT
вора 150 r/л кислоты винной Р и отстаивают; об
разуется белый кристаллический осадок.
Ь) Около 40 Mr испытуемоrо образца paCTBO
ряют в 1 мл воды Р. К полученному раствору или
к 1 мл раствора, указанноro в частной статье,
прибавляют 1 мл кислоты уксусной разведен
ной Р и 1 мл свежеприroтовленноro раствора
178
rосударственная фармакопея Республики Белаpj
100 r/л натрия кобальтинитрита Р; тотчас об
разуется желтый или оранжево желтый осадок.
#с) Соль калия, внесенная в бесцветное
пламя, окрашивает еro в фиолетовый цвет или,
при рассматривании через синее стекло, в пур
пурно красный.
КАЛЬЦИЙ
а) К 0,2 мл нейтральноro раствора испыту
емоro образца, содержащеro около 0,2 Mr каль
ций иона (Са 2 +) в 1 мл, или К 0,2 мл раствора,
указанноrо в частной статье, прибавляют 0,5 мл
раствора 2 r/л 2лиоксаЛЬ2идроксианила Р в 96 %
спирте Р, 0.2 мл раствора натрия 2идроксида
разведеНН020 Р и 0,2 мл раствора натрия Kap
боната Р Смесь встряхивают с 1 мл или 2 мл
хлороформа Р и прибавляют от 1 мл ДО 2 мл
воды Р; хлороформный слой приобретает Kpac
ную окраску.
Ь) Около 20 Mr или указанное в частной
статье количество испытуемоro образца paCTBO
ряют в 5 мл кислоты уксусной Р К полученно
му раствору прибавляют 0,5 мл раствора калия
ферроцианида Р; раствор остается прозрачным.
К раствору прибавляют около 50 Mr аммония
хлорида Р; образуется белый кристаллический
осадок.
#с) К 1 мл раствора испытуемоro образ
ца, содержащеrо около 2 20 Mr кальций иона
(Са 2 +), прибавляют 1 мл раствора 40 r/л аммония
оксалата Р; образуется белый осадок, Hepac
творимый в кислоте уксусной разведенной Р,
растворе аммиака Р и растворимый в разведен
ных минеральных кислотах.
#d) Соль кальция, смоченная кислотой хло
ристоводородной Р и внесенная в бесuветное
пламя, окрашивает еro в оранжево красный цвет.
КАРБОНАТЫ И rИДРОКАРБОНАТЫ
а) 0,1 r испытуемоro образца помещают в
пробирку и суспендируют в 2 мл воды Р К полу
ченной суспензии или к 2 мл суспензии, указан
ной в частной статье, прибавляют 3 мл кислоты
уксусной разведенной Р Пробирку тотчас закры
вают притертой пробкой со стеклянной трубкой,
дважды изоrнутой под прямым уrлом; наблюда
ется бурное выделение пузырьков rаза без цвета
и запаха. Пробирку осторожно наrревают и про
пускают выделяющийся rаз через 5 мл раствора
бария 2идроксида Р; образуется белый осадок,
растворяющийся при прибавлении избытка Kиc
лоты хлористоводородной Р1.
#Ь) 0,2 r испытуемоro образца растворяют в
2 мл воды Р К полученному раствору прибавля
ют 0,5 мл насыщеНН020 раствора ма2ния суль
фата Р; образуется белый осадок (в отличие от
rидрокарбонатов, растворы которых образуют
осадок только при кипячении смеси).
#с) 0,2 r испытуемоro образца растворяют в
2 мл воды Р. К полученному раствору прибав
ляют 0,05 мл раствора фенолфталеина Р; по
является красное окрашивание (в отличие от
rидрокар60натов, растворы которых остаю
бесцветными ).
КСАНТИНЫ
К нескольким миллиrраммам или к указе
ному в частной статье количеству испытуеМ(
образца прибавляют 0,1 мл раствора водоро
пероксида концентрироваНН020 Р, 0,3 мл кис/.
ты хлористоводородной разведенной Р и в
паривают на водяной бане до получения сухо
желтовато красноro остатка. К остатку прибавл
ют 0,1 мл аммиака разведеННО20 Р2; цвет оста
ка изменяется на фиолетово красный.
ЛАКТАТЫ
Навеску испытуемоrо образца, эквивалек
ную около 5 Mr кислоты молочной, растворяк
в 5 мл воды Р. К полученному раствору или
5 мл раствора, указанноro в частной статье, пр'
бавляют 1 мл бромной воды Р. 0,5 мл кислота
серной разведенной Р и наrревают на водянOI
бане, периодически перемешивая стекляннOI
палочкой, до обесцвечивания раствора. К рас
твору прибавляют 4 r аммония сульфата Р .
перемешивают. Прибавляют по каплям, не пе
ремешивая, 0,2 мл раствора 100 r/л натрш
нитропруссида Р в кислоте серной разведен.
ной Р, осторожно прибавляют. также не переме-
шивая, 1 мл раствора аммиака концентриро-
ваНН020 Р и выдерживают в течение 30 мин; на
rранице двух жидкостей образуется темно зеле
ное кольцо.
МАrний
Около 15 Mr испытуемоro образца paCTBO
ряют в 2 мл воды Р. К полученному раствору или
к 2 мл раствора, указанноro в частной статье,
прибавляют 1 мл раствора аммиака разведен
носо Р1; образуется белый осадок, растворя
ющийся при прибавлении 1 мл раствора aM
мония хлорида Р К полученному раствору при
бавляют 1 мл раствора динатрия 2идрофосфа
та Р; образуется белый кристаллический осадок.
МЫШЬЯК
а) 5 мл испытуемоro раствора наrревают на
водяной бане с равным объемом реактива 2и
пофосфuта Р; образуется коричневый осадок.
#Ь) Мышьяк (1/1) (арсениты). К 0,3 мл paCT
вора испытуемоro образца, содержащеro около
30 Mr арсенит иона (АsОзЗ-), прибавляют 0,5 мл
кислоты хлористоводородной разведенной Р
и 0,1 мл раствора натрия сульфида Р; образу
ется желтый осадок, нерастворимый в кислоте
хлористоводородной Р и растворимый в pac
творе аммиака Р.
#с) Мышьяк (V) (арсенаты). К 0,3 мл pacт
вора испытуемоrо образца, содержащеro около
1 Mr арсенат иона (АsО 4 З-), прибавляют по 1 мл
раствора 100 r/л аммония хлорида Р, pacт
вора аммиака Р и раствора 100 r/л ма2ния суль
фата Р; образуется белый кристаллический
2.3.1. Реакции подлинности на ионы и функциональные 2руппы
179
осадок, растворимый в кислоте хлористоводо
родной разведенной Р (отличие от арсенитов).
НАТРИЙ
а) 0,1 r испытуемоro образца растворяют в
2 мл воды Р. К полученному раствору или к 2 мл
раствора, указанноro в частной статье, прибав
ляют 2 мл раствора 150 r/л калия карбоната Р
и наrревают до кипения; осадок не образуется. К
раствору прибавляют 4 мл раствора калия пи
роантимоната Р и наrревают до кипения, затем
охлаждают в ледяной воде и при необходимо
сти потирают внутренние стенки пробирки CTe
клянной палочкой; образуется плотный осадок
белоro цвета.
Ь) Навеску испытуемоro образца, эквива
лентную около 2 Mr натрий иона (Na+), paCTBO
ряют в 0,5 мл воды Р. К полученному paCTBO
ру или к 0,5 мл раствора, указанноro в частной
статье, прибавляют 1,5 мл реактива MeтOKcи
фенилуксусной кислоты Р, охлаждают в ледя
ной воде в течение 30 мин; образуется объемный
белый кристаллический осадок. Смесь помеща
ют в воду при температуре 20 ос и перемешива
ют в течение 5 мин; осадок не исчезает. К смеси
прибавляют 1 мл раствора аммиака разведен
Н020 Р1; осадок полностью растворяется. К по
лученному раствору прибавляют 1 мл раствора
аммония карбоната Р; осадок не образуется.
#с) Соль натрия, смоченная кислотой хло
ристоводородной Р и внесенная в бесцветное
пламя, окрашивает ero в желтый цвет.
НИТРАТЫ
а) Навеску испытуемоro образца, содержа
щую около 1 Mr нитрат иона (NОЗ о ), или количе
ство, указанное в частной статье, прибавляют к
смеси из 0,1 мл нитробензола Р и 0,2 мл Kиc
лоты серной Р и через 5 мин охлаждают в ле
дяной воде. Продолжая охлаждение, медленно
при перемешивании прибавляют 5 мл воды Р,
5 мл раствора натрия 2идроксида KOHцeHтpи
роваНН020 Р, 5 мл ацетона Р, взбалтывают и OT <
стаивают; верхний слой приобретает темно фи
олетовую окраску.
#Ь) Раствор испытуемоrо образца, coдep
жащий около 2 Mr нитрат иона (NОЗ"), не обесц
вечивает раствор 1 r/л калия пермаН2аната Р,
подкисленный кислотой серной разведенной Р
(в отличие от нитритов).
#НИТРИТЫ
а) Несколько кристаллов феназона Р paCT
воряют в фарфоровой чашке в 0,1 мл кислоты
хлористоводородной разведенной Р, прибавля
ют 0,1 мл испытуемоro раствора, содержащеro
около 1 Mr нитрит иона (N02 o ); появляется зеле
ное окрашивание (в отличие от нитратов).
Ь) К навеске испытуемоro образца, эквива
лентной ЗО Mr нитрит иона (N02-), прибавляют
1 мл кислоты серной разведенной Р; выделяются
желто коричневые пары (в отличие от нитратов).
РТУТЬ
а) Около 0,1 мл раствора испытуемоro об
разца помещают на тщательно очищенную по
верхность медной фольrи; появляется TeMHO
серое пятно, которое при натирании становится
блестящим. Фольry высушивают и наrревают в
лробирке; пятно исчезает.
Ь) К раствору, указанному в частной статье,
прибавляют раствор натрия 2идроксида раз
веденный Р до сильнощелочной среды (2.2.4);
образуется плотный осадок желтоro цвета
(ртути (11) соединения).
#с) К 1 мл раствора испытуемоrо образца,
содержащеro 10 30 Mr ртуть иона (H g 2+), при
бавляют осторожно по каплям раствор калия
йодида Р; образуется красный осадок, раствори
мый в избытке этоro реактива.
САЛИЦИЛАТЫ
а) К 1 мл раствора, указанноro в частной
статье, прибавляют 0,5 мл раствора железа
(111) хлорида Р1; появляется фиолетовое OKpa
шивание, которое не исчезает после прибавле
ния 0,1 мл кислоты уксусной Р, #но исчезает при
прибавлении кислоты хлористоводородной
разведенной Р; при этом образуется белый кри
сталлический осадок салициловой кислоты#.
Ь) 0,5 r испытуемоro образца растворяют
в 1 О мл воды Р. К полученному раствору или к
1 О мл раствора, указанноro в частной статье,
прибавляют 0,5 мл кислоты хлористоводород
ной Р. Полученный осадок после перекристал
лизации из roрячей воды Р и высушивания под
вакуумом (2.2.32) имеет температуру плавления
(2.2.14) от 156 ос до 161 ОС.
СВИНЕЦ
а) 0,1 r испытуемоro образца растворяют в
1 мл кислоты уксусной Р. К полученному pac
твору или к 1 мл раствора, указанноro в частной
статье, прибавляют 2 мл раствора калия xpo
мата Р; образуется желтый осадок, растворяю
щийся при прибавлении 2 мл раствора натрия
сидроксида концентрироваНН020 Р.
Ь) 50 Mr испытуемоro образца растворяют
в 1 мл кислоты уксусной Р. К полученному pac
твору или к 1 мл раствора, указанноro в част
ной статье, прибавляют 1 О мл воды Р и 0,2 мл
раствора калия йодида Р, образуется желтый
осадок. Смесь кипятят в течение 1 2 мин;
осадок растворяется. Раствору дают остыть;
вновь образуется осадок в виде блестящих
желтых пластинок.
СЕРЕБРО
а) Около 10 Mr испытуемоro образца paCT
воряют в 1 О мл воды Р. К полученному paCTBO
ру или к 1 О мл раствора, указанноro в частной
статье, прибавляют 0,3 мл кислоты хлористо
водородной Р1, образуется белый творожистый
осадок, растворяющийся при прибавлении 3 мл
раствора аммиака разведеНН020 Р1.
180
rосударственная фармакопея Республики Беларусl.
#Ь) К 1 мл раствора испытуемоro образ
ца, содержащеrо около 5 Mr серебро иона,
прибавляют раствор аммиака разведенно
20 Р1 до растворения образующеrося BHa
чале осадка, затем прибавляют 2 3 капли
раствора формальде2ида Р и наrревают;
на стенках пробирки образуется блестящий
налет металлическоro серебра.
СИЛИКАТЫ
Количество испытуемоro образца, YKa
занное в частной статье, смешивают в свин
цовом или платиновом тиrле с помощью
медной проволоки с примерно 1 О Mr натрия
фторида Р и несколькими каплями кислоты
серной Р до образования суспензии. Тиrель
накрывают тонкой прозрачной пластиковой
пластинкой с висящей каплей воды Р и OCTO
рожно наrревают; через короткий промежуток
времени BOKpyr капли воды появляется белое
кольцо_
СУЛЬФАТЫ
а) Около 45 Mr испытуемоro образца
растворяют в 5 мл воды Р. К полученному
раствору или к 5 мл раствора, указанноro в
частной статье, прибавляют 1 мл кислоты
хлористоводородной разведенной Р и 1 мл
раствора бария хлорида Р1; образуется
белый осадок.
Ь) К суспензии, полученной в резулыа
те реакции (а), прибавляют 0,1 мл 0,05 М
раствора йода; желтая окраска йода не ис
чезает (в отличие от сульфитов и дитиони
тов), но обесцвечивается при прибавлении
по каплям раствора олова хлорида Р (в OT
личие от йодатов). Смесь кипятят; осадок
не окрашивается (в отличие от селенатов и
вольфраматов).
#СУЛЬФИТЫ
а) К 2 мл раствора испытуемоro образца,
содержащеro 10 30 Mr сульфит иона (80/-),
прибавляют 2 мл кислоты хлористоводород
ной разведенной Р и встряхивают; постепенно
выделяется сернистый rаз, обнаруживаемый
по характерному резкому запаху.
Ь) К указанному 8 частной статье paCTBO
ру, содержащему сульфит ион (80з2-). при
бавляют 0.1 мл 0,5 М раствора йода; реактив
обесцвечивается.
СУРЬМА
Около 1 О Mr испытуемоrо образца paCTBO
ряют при наrревании в растворе 0,5 r калия
натрия тартрата Р в 1 О мл воды Р и охлаж
дают. К 2 мл полученноrо раствора или к 2 мп
раствора, указанноro в частной статье, при
бавляют по каплям раствор натрия сульфи
да Р; образуется оранжево красный осадок,
растворяющийся при прибавлении раствора
натрия 2идроксида разведеНН020 Р.
ТАРТ РАТ Ы
а) Около 15 Mr испытуемоro образца раст-
воряют в 5 мл воды Р. К полученному раство-
ру или к 5 мл раствора, указанноro в частной
статье, прибавляют 0,05 мл раствора 1 О r/л
железа (11) сульфата Р и 0,05 мл раствора
пероксида водорода разведеНН020 Р; появ
ляется неустойчивое желтое окрашивание.
После обесцвечивания раствора к нему при
бавпяют по каплям раствор натрия 2идpOK
сида разведенный Р; появляется интенсивное
синее окрашивание.
Ь) К 0,1 мл раствора испытуемоro образца,
содержащеrо около 15 мr/мл кислоты винной,
или к 0,1 мл раствора, указанноro в частной
статье, прибавляют 0,1 мл раствора 100 r/л
калия бромида Р, 0,1 мл раствора 20 r/л pe
зорцина Р, 3 мл кислоты серной Р и Harpe
вают на водяной бане от 5 мин до 10 мин; по
является темно синее окрашивание. Раствор
охлаждают и вливают в воду Р; окраска paCT
вора изменяется на красную.
ФОСФАТЫ (ОРТОФОСФАТЫ)
а) К 5 мл раствора, указанноro в частной
статье, при необходимости нейтрализован
HOro, прибавляют 5 мл раствора серебра Hи
трата Р1; образуется желтый осадок, цвет
KOTOpOro не изменяется при кипячении и KOTO
рый растворяется при прибавлении раствора
аммиака Р.
Ь) 1 мл раствора, указанноro в частной
статье, смешивают с 2 мл молибденованади
ев020 реактива Р; появляется желтое OKpa
шивание.
#с) К 1 мл раствора испытуемоrо образца,
содержащеro 10 ЗО Mr фосфат иона (РО/-) в
кислоте азотной разведенной Р, прибавля
ют 2 мл раствора аммония молибдата Р и
наrревают; образуется желтый кристалличе
ский осадок, растворимый в растворе aMMи
ака разведенном Р1.
#d) К 1 мл раствора испытуемоro образца,
содержащеro 10 30 Mr фосфат иона (Р0 4 3-),
прибавляют 1 мл раствора аммония хлори
да Р, 1 мл раствора аммиака разведенно
20 Р1 и 0.5 мл раствора 100r/л ма2ния суль
фата Р: образуется белый кристаллический
осадок. растворимый в разведенных мине
ральных кислотах.
ХЛОРИДЫ
а) Навеску испытуемоro образца, coдep
жащую около 2 Mr хлорид иона (CI-), paCTBO
ряют в 2 мл воды Р. Полученный раствор или
2 мл раствора, указанноrо в частной статье,
подкисляют кислотой азотной разведен
ной Р, прибавляют 0,4 мл раствора серебра
нитрата Р1, перемешивают и отстаивают; об
разуется белый творожистый осадок, который
центрифуrируют и промывают тремя порция
ми воды Р по 1 мл каждая. Эту операцию про
2.3.2. Идентификация жирных масел методом тонкослойной хроматоарафии 181
водят быстро в защищенном от яркоro света
месте, при этом допускается, чтобы жидкость
над осадком не была полностью прозрачной.
Осадок суспендируют в 2 мл воды Р и при
бавляют 1,5 мл раствора аммиака Р; осадок
быстро растворяется; допускается наличие
нескольких крупных частиц, растворяющихся
медленно.
Ь) Навеску испытуемоro образца, coдep
жащую около 15 Mr хлорид иона (CI ), или
количество, указанное в частной статье, по
мещают в пробирку, прибавляют 0,2 r калия
дихромата Р и 1 мл кислоты серной Р. У
входноro отверстия пробирки помещают
фильтровальную бумаrу, пропитанную 0,1 мл
раствора дифенилкарбазида Р (при этом
пропитанная бумаrа не должна соприкасать
ся с калия дихроматом); бумаrа окрашивается
в фиолетово красный цвет.
ЦИНК
а) 0,1 r испытуемоro образца растворяют
в 5 мл воды Р. К полученному раствору или к
5 мл раствора, указанноro в частной статье,
прибавляют 0,2 мл раствора натрия 2и
дроксида концентрироваНН020 Р; образует
ся белый осадок. Затем прибавляют еще 2мл
раствора натрия 2идроксида KOHцeHтpиpo
ваНН020 Р; осадок растворяется. К получен
ному раствору прибавляют 1 О мл раствора
аммония хлорида Р; раствор остается про
зрачным. К раствору прибавляют 0,1 мл pacт
вора натрия сульфида Р; образуется белый
хлопьевидный осадок.
#Ь) К 2 мл раствора испытуемоro образ
ца, содержащеro 5 20 Mr цинк иона (Zn 2 +),
прибавляют 0,5 мл раствора калия ферроци
анида Р; образуется белый осадок, HepaCTBO
римый в кислоте хлористоводородной раз
веденной Р.
ЦИТРАТЫ
а) Навеску испытуемоro образца, coдep
жащую около 50 Mr кислоты лимонной, paCT
воряют в 5 мл воды Р. К полученному paCTBO
ру или к 5 мл раствора, указанноro в частной
статье, прибавляют 0,5 мл кислоты серной Р
и 1 мл раствора калия пермаН2аната Р.
Раствор наrревают до обесцвечивания, при
бавляют 0,5 мл раствора 100 r/л натрия Hи
тропруссида Р в кислоте серной разведен
ной Р, 4 r кислоты сульфаминовой Р. К смеси
прибавляют раствор аммиака KOHцeHтpиpo
ванный Р до щелочной реакции среды, при
бавляя еro по каплям до полноro растворения
кислоты сульфаминовой. ПрибаВJlение из
бытка раствора аммиака KOHцeHтpиpoвaHHO
20 Р приводит К появлению фиолетовоro OKpa
шивания, переходящеro в фиолетово синее.
#Ь) К 1 мл нейтральноro раствора испыту
емоro образца, содержащеro 2 10 Mr цитрат
иона, прибавляют 1 мл раствора 200 r/л каль
ция хлорида Р; раствор остается прозрачным;
при кипячении раствора образуется белый
осадок, растворимый в кислоте хлористово
дородной разведенной Р.
#с) К навеске испытуемоro образца, co
держащей 1 2 Mr цитрат иона, прибавляют
0,5 мл аН2идрида УКСУСН020 Р и наrревают;
через 20 0 с появляется красное окрашива
ние.
ЭФИРЫ СЛОЖНЫЕ
К 30 Mr или к указанному в частной статье
количеству испытуемоro образца прибавля
ют 0,5 мл раствора 70 r/л 2идроксиламина 2и
дрохлорида Р в метаноле Р, 0,5 мл раствора
100 r/л калия 2идроксида Р в 96 % спирте Р,
наrревают до кипения и охлаждают. Полу
ченный раствор подкисляют кислотой хло
ристоводородной разведенной Р, прибавля
ют 0,2 мл раствора железа (111) хлорида Р1,
разбавленноro в 1 О раз; появляется синевато
красное или красное окрашивание.
01/2013:20302
2.3.2. ИДЕНТИФИКАЦИЯ
ЖИРНЫХ M CEJ] МЕТОДОМ
ТОНКОСЛОИНОИ
ХРОМАтоrРАФИИ
Определение проводят методом TOHKOC
лойной хроматоrрафии (2.2.27), используя в Ka
честве тонкоro слоя силикаrель октадецилси
лильный для высокоэффективной тонкослойной
хроматоrрафии.
Испытуемый раствор. Если нет друrих YKa
заний в частной статье, около 20 Mr (1 каплю) жир
ноro масла растворяют в 3 мл метиленхлорида Р.
Раствор сравнения. Около 20 Mr (1 каплю)
масла КУКУРУЗН020 Р растворяют в 3 мл Meти
ленхлорида Р.
На линию старта хроматоr'pафической
пластинки отдельно наносят по 1 мкл испы
туемоro раствора и раствора сравнения. Пла
стинку дважды хроматоrрафируют на paCCTO
яние 0,5 см, используя в качестве подвижной
фазы эфир Р, и дважды хроматоrрафируют на
расстояние 8 см, используя в качестве под
вижной фазы смесь растворителей: метилен
хлорид Р кислота уксусная ледяная Р
ацетон Р (20:40:50, об/об/об). Пластинку
сушат на воздухе, опрыскивают раствором
100 r/л кислоты фосфорномолибденовой Р в
96 % спирте Р, наrревают при температуре
120 ос в течение 3 мин и просматривают при
дневном свете.
Типичная xpoMaTorpaMMa для идентифика
ции жирных масел приведена на рисунке 2.3.2. 1.
182
rосударственная фармакопея Республики Беларусь
:
t -:. <-;::li
. .;I t
"" "" "'" ..
G ... "'" ....
F ....
с
Ь
2
з
7
4
5
..6
8
9
10
11
12
13
14
15
Рисунок 2_З.2. 1. Типичная хромато2рамма для идентификации жирных масел
1 арахисовое масло;
2 кунжутное масло;
3 кукурузное масло;
4 рапсовое масло;
5 соевое масло;
б рапсовое масло (не содержащее эруковой ислоты);
7 льняное масло;
8 оливковое масло;
01/2013:20303
2.3.3. ИДЕНТИФИКАЦИЯ
ФЕНОТИАЗИНОВ
МЕТОДОМ ТОНКОСЛОЙНОЙ
ХРОМАтоrРАФИИ
Определение проводят методом ТOHKO
слойной хроматоrрафии (2.2.27), используя
в качестве тонкоro слоя кизеЛЬ2УР G Р. Пла
стинку помещают в закрытую камеру с необ
ходи мы м количеством пропитывающей смеси,
содержащей 10 % (об/об) феноксиэтанола Р
в растворе 50 r/л маКР020ла 300 Р в aцeтo
не Р, таким образом, чтобы она была поrруже
на в пропитывающую смесь примерно на 5 мм.
Коrда фронт растворителей пройдет 17 см
от нижнеro края пластинки, ее вынимают из
камеры и тотчас используют для xpoMaTorpa
фирования. Хроматоrрафирование проводят в
том же направлении, что и пропитывание.
Испытуемый раствор. 20 Mr испытуемо
ro образца растворяют в хлороформе Р и дo
водят до объема 1 О мл этим же растворите
лем.
Раствор сравнения. 20 Mr соответствую
щеro ФСО растворяют в хлороформе Р и дo
водят до объема 1 О мл эти . же растворите
лем.
На линию старта хроматоrрафической пла
стинки отдельно наносят по 2 мкл испытуемо
ro раствора и раствора сравнения. Пластинку
помещают в камеру со смесью петролейный
эфир Р диэтиламuн Р (50:1, об/об), Hacы
щенной феноксиэтанолом Р (т. е. от 3 мл до
9 подсолнечное масло;
10 миндальное масло;
11 масло зародышей пшеНI4ЦЫ;
12 масло бурачника лекарственнoro;
1 3 энотеровое масло;
14 сафлоровое масло (тип 1);
15 сафлоровое масло (тип 11).
4 мл феноксиэтанола Р прибавляют к вышеу
казанной смеси растворителей, взбалтывают
до образования устойчивой мутности, дeKaH
тируют и используют верхний слой, который
может быть мутным). Хроматоrрафирование
про водят в темном месте. Коrда фронт paCT
ворителей пройдет 15 см от линии старта, пла
стинку вынимают из камеры. помещают в уль
трафиолетовый свет с длиной волны 365 нм и
через несколько минут Пр080ДЯТ оценку xpo
MaTorpaMMbI.
На xpoMaTorpaMMe испытуемоro раствора
обнаруживается пятно, соответствующее
по расположению, флуоресценции и разме
ру пятну на xpoMaTorpaMMe раствора cpaB
нения. Затем пластинку опрыскивают 10 %
(об/об) раствором кислоты серной Р в 96 %
спирте р. На xpoMaTorpaMMe испытуемоro
раствора обнаруживается пятно, COOTBeTCTBY
ющее по цвету, а также стабильности окраски
в течение не менее 20 мин, пятну на xpOMaTO
rpaMMe раствора сравнения.
01/2013:20304
2.3.4. ОПРЕДЕЛЕНИЕ ЗАПАХА
От 0,5 r до 2,0 r испытуемоro образца pac
пределяют тонким слоем на часовом стекле ди
аметром от 6 см до 8 см. Через 15 мин опреде
ляют запах или делают вывод о еro отсутствии.
2.4.2. A4bIU1bJ1K
183
2.4. ИСПЫТАНИЯ
НА ПРЕДЕЛЬНОЕ
СОДЕРЖАНИЕПРИМЕСЕЙ
01/2013:20401
2.4.1. АММОНИЯ СОЛИ
Если нет друаих указаний в частной
статье, используют метод А
МЕТОД А
Количество испытуемоro образца, указан
ное в частной статье, помещают в пробирку,
растворяют в 14 мл воды Р, при необходимости
подщелачивают раствором натрия 2идpOKcи
да разведенным Р и доводят водой Р до объема
15 мл. Прибавляют 0,3 мл раствора калия тe
трайодмеркурата щеЛОЧН020 Р. Пробирку за
крывают.
В качестве эталона используют раствор, по
лученный прибавлением к 1 О мл этаЛОНН020
раствора аммония (1 ррт NH) Р 5 мл воды Р
и 0,3 мл раствора калия тетрайодмеркурата
щеЛОЧН020 Р. Пробирку закрывают.
Через 5 мин желтая окраска испытуемоro
раствора должна быть не интенсивнее окраски
эталона.
МЕТОД В
Количество мелкоизмельченноro испытуе
моro образца, указанное в частной статье, по
мещают в сосуд вместимостью 25 мл, снаб
женный полиэтиленовой пробкой, и растворяют
или суспендируют в 1 мл воды Р. Прибавляют
0,30 r ма2ния оксида тяжеЛ020 Р, помещают в
сосуд под пробку полоску серебряно маР2анце
вой бума2и Р, смоченную несколькими каплями
воды Р, таким образом, чтобы отрезок бумаrи
размером 5 х 5 мм находился ниже нижнеro края
пробки, после чеrо сосуд немедленно закрыва
ют пробкой. Перемешивают содержимое сосуда
круroвыми движениями, не допуская попадания
брызr на бумаry, и выдерживают при температу
ре 40 ос в течение 30 мин.
Параллельно с теми же количествами peaK
тивов и в этих же условиях rотовят эталон. К YKa
занному в частной статье количеству эталонно
20 раствора аммония (1 ррт NH) Р прибавляют
1 мл воды Р, 0,30 r ма2ния оусида тяжеЛ020 Р и
далее поступают, как с испытуемым раствором.
Серая окраска серебряно маР2анцевой
бума2и Р, полученная в опыте с испытуемым
раствором, должна быть не интенсивнее OKpa
ски серебряно маР2анцевой бума2и Р, получен
ной в опыте с эталоном.
#МЕТОД С
Применяют для образцов, содержащих
щелочноземельные и тяжелые металлы.
Количество испытуемоro образца, указан
ное в частной статье, помещают в пробирку,
растворяют в минимальном объеме воды Р,
прибавляют при охлаждении 2 мл раствора
натрия 2идроксида разведеНН020 Р и 2 мл
раствора натрия карбоната Р. Раствор раз
водят водой Р до указанной в частной статье
концентрации, встряхивают и фильтруют.
1 О мл полученноro фильтрата помещают в
пробирку, доводят водой Р до объема 15 мл
и прибавляют 0,3 мл раствора калия тeтpa
йодмеркурата щеЛОЧН020 Р.
В качестве эталона используют смесь
10 мл этаЛОНН020 раствора аммония (1 ррт
NH) Р, 5 мл воды Р и 0,3 мл раствора калия
тетрайодмеркурата щеЛОЧН020 Р. Пробирки
закрывают.
Через 5 мин желтая окраска испытуемоro
раствора должна быть не интенсивнее OKpa
ски эталона.
#МЕТОД О
Применяют для образцов, содержащих
более 300 ррт примеси железа.
Количество испытуемоro образца, YKa
занное в частной статье, помещают в про
бирку, растворяют в 1 О мл воды Р, прибав
ляют 2 капли раствора натрия 2идроксида
разведеНН020 Р и 3 мл раствора 200 л Ka
лия натрия тартрата Р. Тщательно переме
шивают, доводят водой Р до объема 15 мл и
прибавляют 0,3 мл раствора калия тeтpa
йодмеркурата щеЛОЧН020 Р. Пробирку закры
вают.
Параллельно с теми же количествами pe
активов и в тех же условиях roтовят эталон,
используя вместо 1 О мл испытуемоro paCT
вора 1 О мл этаЛОНН020 раствора аммония
(1 ррт NH) Р.
Через 5 мин желтая окраска испытуемоro
раствора должна быть не интенсивнее OKpa
ски эталона.
01/2013:20402
2.4.2. МЫШЬЯК
МЕТОД А
Прибор (см. рисунок 2.4.2. 1) состоит из KO
нической колбы вместимостью 100 мл, закры
той стеклянной притертой пробкой, через KOTO
рую проходит стеклянная трубка длиной около
200 мм с внутренним диаметром 5 мм. Нижняя
часть трубки вытянута до внутреннеro диаметра
184
rосударственная фармакопея Республики Беларусь
1,0 мм; на расстоянии 15 мм от кончика трубки
расположено боковое отверстие диаметром от
2 мм до 3 мм. Трубка помещена таким обра
зом, чтобы боковое отверстие находилось мини
мум на 3 мм ниже нижней поверхности пробки.
Верхний конец трубки должен иметь совершен
но плоскую притертую поверхность, располо
женную под прямым уrлом к оси трубки. Друrая
стеклянная трубка длиной 30 мм с таким же BHY
тренним диаметром и такой же плоской притер
той поверхностью помещается наверху первой и
плотно прикрепляется к ней двумя пружинами.
Нижнюю трубку неплотно заполняют
50 0 Mr свинцово ацетатной ваты Р или по
мещают небольшой ватный тампон и скручен
ную в трубочку полоску свинцово ацетатной
бума2и Р массой 50 0 Mr. Между плоскими по
верхностями трубок помещают кусочек pтyтHO
бромидной бума2и Р такоro размера, чтобы за
крыть отверстие трубки (15 х 15 мм).
Указанное количество испытуемоro образ
ца помещают в коническую колбу и растворяют
в 25 мл воды Р или указанный объем испытуе
моro раствора помещают в коническую колбу,
доводят водой Р до объема 25 мл. Прибавляют
15 мл кислоты хлористоводородной Р, 0,1 мл
раствора олова (11) хлорида Р, 5 мл раствора
калия йодида Р, выдерживают в течение 15 мин
и затем прибавляют 5 r цинка aKтuвиpoвaHHO
20 Р. Немедленно соединяют две части прибора,
колбу помещают в водяную баню, температура
которой поддерживается так, чтобы обеспечить
равномерное выделение rаза #(температура BO
дяной бани не должна превышать 40 ОС)#.
Параллельно с теми же количествами peaK
тивов и в тех же условиях проводят опыт С эта
лоном, состоящим из 1 мл этаЛОНН020 pacт
вора мышьяка (1 ррт As) Р и 24 мл воды Р.
По истечении не менее 2 ч окраска PTYТHO
бромидной бумаrи, полученная в опыте с испы
туемым раствором, должна быть не интенсивнее
окраски ртутно бромидной бумаrи, полученной в
опыте с эталоном.
МЕТОД В
Количество испытуемоro образца, указан
ное в частной статье, помещают в пробирку, co
держащую 4 мл кислоты хлористоводород
ной Р и около 5 Mr калия йодида Р, и прибавляют
3 мл реактива 2ипофосфита Р. Смесь HarpeBa
ют на водяной бане в течение 15 мин, время от
времени встряхивая.
Параллельно с теми же количествами pe
активов и в тех же условиях roтовят эталон, ис
пользуя вместо испытуемоro образца 0,5 мл
этаЛОНН020 раствора мышьяка (10 ррт As) Р.
После наrревания на водяной бане окраска
испытуемою раствора должна быть не интен
сивнее окраски эталона.
#Метод В применяют в случае определения
наряду с мышьяком селена и теллура, а также
при определении мышьяка в образцах, coдep
1 1 1 . 5
.I"'
I i
oi
С")!
..
-+
i
,
о:
r---I
.......1
I
I
I
I
Рисунок 2.4.2. 1. Прибор для испытания
на мышьяк (метод А) (размеры указаны
в миллиметрах)
жащих сурьму, висмут, ртуть и серебро, а также
сульфиды и сульфиты, и в друrих случаях, YKa
занных в частных статьях.#
01/2013:20403
2.4.3. КАЛЬЦИЙ
При при20товлении всех растворов, при
меняемых в данном испытании, должна исполь
зоваться вода дистиллированная Р.
К 0,2 мл этаЛОНН020 раствора кальция
спиртов020 (100 ррт Са) Р прибавляют 1 мл
раствора аммония оксалата Р. Через 1 мин
прибавляют смесь из 1 мл кислоты уксусной
разведенной Р и 15 мл раствора, содержащею
указанное в частной статье количество испытуе
моro образца, и встряхивают.
Параллельно с теми же количествами pe
активов и в тех же условиях rотовят эталон, ис
пользуя смесь из 1 мл кислоты уксусной разве
денной Р, 1 О мл этаЛОНН020 раствора кальция
водН020 (10 ррт Са) Р и 5 мл воды дистиллиро
ванной Р.
2.4.6. Маzний
185
Через 15 мин опалесценция испытуемоro
раствора не должна превышать опалесценцию
эталона.
01/2013:20404
2.4.4. ХЛОРИДЫ
К 15 мл раствора, указанноro в частной
статье, прибавляют 1 мл кислоты азотной раз
веденной Р и выливают смесь в один прием в
пробирку, содержащую 1 мл раствора серебра
нитрата Р2.
Параллельно с теми же количествами peaK
тивов и в тех же условиях roтовят эталон, исполь
зуя вместо 15 мл испытуемоro раствора 1 О мл
этаЛОНН020 раствора хлорида (5 ррт С/) Р и
5 мл воды Р. Пробирки помещают в защищенное
от света место.
Через 5 мин пробирки просматривают на
черном фоне roризонтально (перпендикулярно оси
пробирок). Опалесценция испытуемоro раствора
не должна превышать опалесценцию эталона.
01/2013:20405
2.4.5. ФТОРИДЫ
Количество испытуемоro образца, указан
ное в частной статье, 0,1 r песка Р, промыто
ro кислотой, и 20 мл смеси из равных объемов
кислоты серной Р и воды Р помещают во BHY
треннюю пробирку прибора, изображенноro
на рисунке 2.4.5. 1. Рубашку, заполненную тe
трахлорэтаном Р, наrревают до температуры
кипения тетрахлорэтана (146 ОС). Подсоединяют
reHepaтop водяноro пара и отroняют содержимое
пробирки с переrретым водяным паром, собирая
OTroH в мерную колбу вместимостью 100 мл, co
держащую 0,3 мл 0,1 М раствора натрия 2и
дроксида и 0,1 мл раствора фенолфталеина Р.
Объем раствора в пробирке во время отroнки
должен быть постоянным (20 мл). Подцержи
вают щелочную реакцию содержимоro мерной
колбы, при необходимости прибавляя по каплям
0,1 М раствор натрия 2идроксида. Доводят
объем дистиллята водой Р до 100 мл И переме
шивают (испытуемый раствор).
Параллельно с теми же количествами pe
активов и в тех же условиях rотовят эталон, ис
пользуя вместо испытуемоro образца 5 мл эта
ЛОНН020 раствора фторида (10 ррт F) Р.
В цилиндр со стеклянной притертой проб
кой помещают 20 мл испытуемоro раствора, в
друrой такой же цилиндр 20 мл эталона, затем
в каждый цилиндр прибавляют по 5 мл peaKти
ва аминометилализариндиуксусной кислоты Р.
Через 20 мин синяя окраска испытуемоro paCT
вора, появившаяся вместо первоначальной Kpac
ной, должна быть не интенсивнее окраски эталона.
180
I
t
I
78
30
о
о
v
I
I
Рисунок 2.4.5. 1. Прибор
для испытания на фториды
(размеры указаны в миллиметрах)
01/2013:20406
2.4.6. МАrний
К 1 О мл раствора испытуемоro образца,
приrотовленноrо, как указано в частной статье,
прибавляют 0,1 r динатрия тетрабората Р.
Определяют значение рН раствора и при необ
ходимости доводят до рН 8, 9,2, используя
кислоту хлористоводородную разведенную Р
или раствор натрия 2идроксида разведенный Р.
186
rосударственная фармакопея Республики Беларусь
Раствор помещают в делительную воронку,
встряхивают в течение 1 мин с двумя порциями,
по 5 мл каждая, раствора 1 r/л 2идроксихиноли
на р в хлороформе Р, оставляют до расслоения
и отбрасывают орrанический слой. К водному
слою прибавляют 0,4 мл бутиламина Р и 0,1 мл
трuэтаноламина Р Определяют значение рН
раствора и при необходимости доводят до рН
10,5 11,5. Прибавляют 4 мл раствора 1 r/л 2и
дроксихинолина Р в хлороформе Р, встряхивают
в течение 1 мин и оставляют до расслоения. Для
испытания используют нижний слой.
Параллельно с теми же количествами pe
активов и в тех же условиях ютовят эталон, ис
пользуя вместо 10 мл раствора испытуемоro
вещества смесь из 1 мл этаЛОНН020 раствора
ма2ния (10 ррт Mg) Р и 9 мл воды Р
Окраска испытуемоrо раствора должна быть
не интенсивнее окраски эталона.
01/2013:20407
2.4.7. мдrний
и ЩЕЛОЧНОЗЕМЕЛЬНЫЕ
МЕТАЛЛЫ
к 200 мл воды Р прибавляют 0,1 r 2идpo
ксиламина 2идрохлорида Р, 1 О мл аммиачно
20 буферН020 раствора рН 10. О Р, 1 мл 0,1 М
раствора цинка сульфата и около 15 Mr иHди
каторной смеси протравН020 черНО20 11 Р Ha
rревают до температуры около 40 ОС. Получен
ный раствор титруют 0,01 М раствором натрия
эдетата до перехода окраски раствора от фи
олетовой к синей. К полученному раствору при
бавляют указанное в частной статье количество
испытуемоro образца, растворенное в 100 мл
воды Р, или используют указанный в частной
статье раствор. Если окраска раствора CTaHO
вится фиолетовой, снова титруют до перехода
окраски раствора к синей.
Объем 0,01 М раствора натрия эдета
та, израсходованный на второе титрование, не
должен превышать объем титранта. указанный в
частной статье.
01/2013:20408
2.4.8. ТЯЖЕЛЫЕ МЕТАЛЛЫ
Все описанные ниже методы требуют ис
пользования тиоацетамидН020 peaKти
ва Р. Допускается использование раствора
натрия сульфида Р1 (0,1 мл) вместо тиoaцe
тамидН020 реактива Р. Если предписанная в
частной статье методика испытания разрабо
тана с использованием тиоацетамидН020 pe
актива Р, но вместо Hero используется pacт
вор натрия сульфида Р1, то для методов А, В
и Н необходимо введение проверочноro paCT
вора, приroтовленноro из такоro же количе
ства испытуемоro образца, что используется
при приrотовлении испытуемоro раствора, к
которому прибавляют такой же объем эталон
ноro раствора свинца. что и при приrотовле
нии раствора сравнения. Испытание считают
недействительным, если окраска проверочно
ro раствора менее интенсивная, чем окраска
раствора сравнения.
МЕТОД А
Испытуемый раствор. 12 мл указанною в
частной статье водноro раствора испытуемоro
образца.
Раствор сравнения (эталон). Смесь из
1 О мл этаЛОНН020 раствора свинца (1 ррт РЬ) Р
или эталОНН020 раствора свинца (2 ррт РЬ) Р,
указанноro в частной статье, и 2 мл указанноro
в частной статье водною раствора испытуемо
ю образца.
Контрольный раствор. Смесь из 1 О мл
воды Р и 2 мл указанною в частной статье BO
дноro раствора испытуемоro образца.
К каждому раствору прибавляют по 2 мл бу
ферНО20 раствора рН 3,5 Р и перемешивают.
Полученные растворы прибавляют к порциям
по 1,2 мл тиоацетамидН020 реактива Р и He
медленно перемешивают. Сравнение растворов
про водят через 2 мин.
При20дность системы: раствор сравнения
имеет светло коричневую окраску по сравнению
с контрольным раствором.
Результат: окраска испытуемою раствора
должна быть не интенсивнее окраски раствора
сравнения.
Если невозможно дать однозначную оценку
результату испытания, растворы фильтруют
через подходящий мембранный фильтр (HO
минальный размер пор 0,45 мкм). Давление
поршня шприца должно быть умеренным и по
стоянным для обеспечения медленноrо и paB
HOMepHoro фильтрования. Сравнивают окраски
мембранных фильтров, полученные в опытах с
разными растворами.
МЕТОД В
Испытуемый раствор. 12 мл указанноro в
частной статье раствора, приroтовленноro с ис
пользованием орrаническою растворителя, co
держащеro минимальное количество воды (Ha
пример, диоксан или ацетон, содержащие по
15 % воды).
Раствор сравнения (эталон). Смесь из
1 О мл эталонноro раствора свинца (1 ррт или
2 ррт РЬ), указанною в частной статье, и 2 мл
указанноro в частной статье раствора испыту
2.4.8. Тяжелые металлы
187
емоro образца в орrаническом растворителе.
Эталонный раствор свинца (1 ррm или 2 ррт
РЬ) roтовят путем разведения этаЛОНН020 pacт
вора свинца (100 ррт РЬ) орrаническим paCTBO
рителем, применяемым для растворения испы
туемоro образца.
Контрольный раствор. Смесь из 1 О мл pac
творителя, применяемоro для растворения ис
пытуемоrо образца, и 2 мл указанноro в частной
статье раствора испытуемоro образца в орrани
ческом растворителе.
К каждому раствору прибавляют по 2 мл бу
феРН020 раствора рН 3,5 Р и перемешивают.
Полученные растворы прибавляют к порциям
по 1,2 мл тиоацетамидН020 реактива Р и He
медленно перемешивают. Сравнение растворов
проводят через 2 мин.
При20дность системы: раствор сравнения
имеет светло коричневую окраску по сравнению
с контрольным раствором.
Результат: окраска испытуемоro раствора
должна быть не интенсивнее окраски раствора
сравнения.
Если невозможно дать однозначную оценку
результату испытания, растворы фильтруют
через подходящий мембранный фильтр (HO
минальный размер пор 0,45 мкм). Давление
поршня шприца должно быть умеренным и по
стоянным для обеспечения медленноro и paB
номерноro фильтрования. Сравнивают окраски
мембранных фильтров, полученные в опытах с
разными растворами.
МЕТОД С
Испытуемый раствор. В кварцевый тиrель
помещают 4 мл раствора 250 r/л ма2ния суль
фата Р в кислоте серной разведенной Р и
прибавляют количество испытуемоro образ
ца, указанное в частной статье (не более 2 r).
Перемешивают тонкой стеклянной палочкой и
осторожно наrревают. Если смесь жидкая, OCTO
рожно выпаривают на водяной бане до сухоro
остатка. Затем постепенно наrревают до обуrли
вания и продолжают наrревание до получения
почти белоro или, в крайнем случае, cepOBaTO
ro остатка. Сжиrание проводят при температу
ре не более 800 ОС. Оставляют до охлаждения,
затем остаток в тиrле смачивают несколькими
каплями кислоты серной разведенной Р. Bы
паривают до сухоro остатка, вновь сжиrают и
оставляют до охлаждения. Общее время сжиrа
ния не должно превышать 2 ч. Остаток из тиrля
количественно пере носят в пробирку двумя
порциями кислоты хлористоводородной раз
веденной Р по 5 мл каждая. Прибавляют 0,1 мл
раствора фенолфталеина Р, затем нейтра
лизуют раствором аммиака KOHцeHтpиpoвaH
ным Р до появления розовой окраски. Охлаж
дают, прибавляют кислоту уксусную ледяную Р
до обесцвечивания раствора и прибавляют еще
0,5 мл кислоты уксусной ледяной Р. При He
обходимости фильтруют и промывают фильтр
водой Р. Доводят объем раствора водой Р до
20 мл И перемешивают.
Раствор сравнения (эталон). rотовят aHa
лоrично испытуемому раствору, используя YKa
занное в частной статье количество этаЛОНН020
раствора свинца (10 ррт РЬ) Р вместо испыту
емоro образца. К 1 О мл полученноro раствора
прибавляют 2 мл испытуемоro раствора.
Проверочный раствор. rотовят аналоrично
испытуемому раствору, прибавляя к испытуемо
МУ образцу указанный в частной статье объем
этаЛОНН020 раствора свинца (10 ррт РЬ) Р для
приroтовления раствора сравнения. К 10 мл по
лученноrо раствора прибавляют 2 мл испытуе
моro раствора.
Контрольный раствор. Смесь из 10 мл
воды Р и 2 мл испытуемоro раствора.
К 12 мл каждоro раствора прибавляют по
2 мл буфеРН020 раствора рН 3,5 Р и перемеши
вают. Полученные растворы прибавляют к пор
циям по 1,2 мл тиоацетамидН020 реактива Р и
немедленно перемешивают. Сравнение paCTBO
ров проводят через 2 мин.
При20дность системы:
раствор сравнения имеет светло коричне
вую окраску по сравнению с контрольным pac
твором;
окраска проверочноro раствора не менее
интенсивная, чем окраска раствора сравнения.
Результат: окраска испытуемоro раствора
должна быть не интенсивнее окраски раствора
сравнения.
Если невозможно дать однозначную оценку
результату испытания, растворы фильтруют
через подходящий мембранный фильтр (HO
минальный размер пор 0,45 мкм). Давление
поршня шприца должно быть умеренным и по
стоянным для обеспечения медленноro и paB
номерноro фильтрования. Сравнивают окраски
мембранных фильтров, полученные в опытах с
разными растворами.
МЕТОД О
Испытуемый раствор. Количество испыту
eMoro образца, указанное в частной статье, по
мещают в кварцевый тиrель и тщательно CMe
шивают с 0,5 r ма2ния оксида Р1. .Сжиrают при
слабом красном калении #(около 600 ОС)# до об
разования однородноro остатка белоro или cepo
вато белоro цвета. Если после ЗО мин сжиrания
смесь остается окрашенной, тиrель оставля
ют до охлаждения, содержимое перемешивают
тонкой стеклянной палочкой и повторяют сжиrа
ние. При необходимости операцию повторяют.
Наrревают при температуре 800 ос около 1 ч.
Остаток из тиrля количественно переносят в про
бирку двумя порциями по 5 мл смеси из равных
объемов раствора кислоты хлористоводо
родной Р1 и воды Р. Прибавляют 0,1 мл pacт
вора фенолфталеина Р, затем подщелачива
ют раствором аммиака концентрированным Р
до появления розовой окраски. Охлаждают, под
188
rосударственная фармакопея Республики Беларусь
кисляют кислотой уксусной ледяной Р до обесц
вечивания раствора и прибавляют еще 0,5 мл
кислоты уксусной ледяной Р При необходимо
сти фильтруют и промывают фильтр водой Р Дo
водят объем раствора водой Р до 20 мл И пере
мешивают.
Раствор сравнения (эталон). rотовят aHa
лоrично испытуемому раствору, используя YKa
занное в частной статье количество этаЛОНН020
раствора свинца (10 ррт РЬ) Р вместо испыту
емоro образца и наrревая при температуре от
100 ос до 105 ОС. К 1 О мл полученноro раствора
прибавляют 2 мл испытуемоro раствора.
Проверочный раствор. rотовят аналоrично
испытуемому раствору, прибавляя к испытуемо
му образцу указанный в частной статье объем
этаЛОНН020 раствора свинца (10 ррт РЬ) Р для
приrотовления раствора сравнения и наrревая
при температуре от 100 ос до 105 ОС. К 10 мл
полученноro раствора прибавляют 2 мл испыту
емоro раствора.
Контрольный раствор. Смесь из 1 О мл
воды Р и 2 мл испытуемоrо раствора.
К 12 мл каждоro раствора прибавляют по
2 мл буфеРН020 раствора рН 3,5 Р и перемеши
вают. Полученные растворы прибавляют к пор
циям по 1,2 мл тиоацетамидН020 реактива Р и
немедленно перемешивают. Сравнение paCTBO
ров про водят через 2 мин.
При20дность системы:
раствор сравнения имеет светло коричне
вую окраску по сравнению с контрольным pac
Т80рОМ;
('1")
сх:>
т--
со
со
т"""
.А
с 13 ..
с 15,7 .
с 17,4 .
Соединение
окраска проверочноro раствора не менее
интенсивная, чем окраска раствора сравнения.
Результат: окраска испытуемоro раствора
должна быть не интенсивнее окраски раствора
сравнения.
Если невозможно дать однозначную оценку
результату испытания, растворы фильтруют
через подходящий мембранный фильтр (HO
минальный размер пор 0,45 мкм). Давление
поршня шприца должно быть умеренным и по
стоянным для обеспечения медленноro и paB
номерноro фильтрования. Сравнивают окраски
мембранных фильтров, полученные в опытах с
разными растворами.
МЕТОД Е
Испытуемый раствор. Количество испыту
емоro образца, указанное в частной статье, paCT
воряют в 30 мл или В указанном объеме воды Р.
Раствор сравнения (эталон). Количество
этаЛОНН020 раствора свинца (1 ррт РЬ) Р, YKa
занное в частной статье, разводят водой Р до
объема испытуемоro раствора.
В держатель устройства для фильтрования,
на подложку котороro помещен мембранный
фильтр (номинальный размер пор 3 мкм), а над
ним предфильтр (рисунок 2.4.8. 1), устанавли
вают шприц вместимостью 50 мл без поршня.
Испытуемый раствор помещают в шприц
и вводят поршень, создавая такое давление,
чтобы профильтровался весь раствор. OTKpЫ
вают держатель, вынимают предФильтР и про
веряют отсутствие примесей на мембранном
Предфиль тр
1 : . .' .. ':.., .. ' : ., . 1
...". .. ".'" .. ....
Мембранный
фильтр
Мембранный
фильтр
r " .. :, . . .'.'" . . ...... I
. . - .. .. - .... . - .
Предфильтр
Предфиль трация
раствора
(метод Е)
Фильтрация раствора
после прибавления
peareHToB
(метод Е)
Рисунок 2.4.8. 1. Прибор для испытания на соли тяжелых металлов
(размеры указаны в миллиметрах)
2.4.8. Тяжелые метаЛЛbl
189
фильтре. В случае обнаружения примесей ero
заменяют друrим мембранным фильтром и
операцию повторяют в тех же условиях.
К фильтрату или к указанному в частной
статье объему фильтрата прибавляют 2 мл
буфеРН020 раствора рН 3,5 Р и перемешива
ют. Полученные растворы прибавляют к пор
циям по 1,2 мл реактива тиоацетамида Р и
перемешивают, оставляют на 1 О мин и вновь
фильтруют, как описано выше, но располо
жение фильтров изменяют таким образом,
чтобы жидкость проходила вначале через
мембранный фильтр, а затем через пред
фильтр (рисунок 2.4.8. 1). Давление поршня
шприца должно быть умеренным и постоян
ным для обеспечения медленною и paBHO
MepHoro фильтрования. После окончания
фильтрования держатель открывают, MeM
бранный фильтр вынимают и высушивают при
помощи фильтровальной бумаrи.
Параллельно проводят аналоrичное испы
тание с раствором сравнения.
Результат: окраска мембранноro фильтра,
полученная в опыте с испытуемым раствором,
должна быть не интенсивнее окраски мембран
ноro фильтра, полученной в опыте с раствором
сравнения.
МЕТОД F
Испытуемый раствор. Количество испы
туемоro образца, указанное в частной статье,
помещают в чистую сухую колбу Кьельдаля
вместимостью 100 мл (в случае интенсивноro
пенообразования следует использовать колбу
вместимостью 300 мл). Колбу закрепляют под
уrлом 450 и, если испытуемый образец пред
ставляет собой твердое вещество, прибавля
ют смесь из 8 мл кислоты серной Р и 1 О мл
кислоты азотной Р в количестве, достаточ
ном для полноro смачивания испытуемоro об
разца; если испытуемый образец представляет
собой жидкость, прибавляют несколько мил
лилитров смеси из 8 мл кислоты серной Р< И
1 О мл кислоты азотной Р. Осторожно Harpe
вают до начала реакции. После прекращения
реакции прибавляют дополнительные порции
этой же смеси кислот, наrревая после каждоro
прибавления. Операцию повторяют до тех пор,
пока объем прибавленной смеси кислот не дo
стиrнет 18 мл. Увеличивают температуру Ha
rревания и осторожно кипятят до потемнения
раствора. Охлаждают, прибавляют 2 мл кисло
ты азотной Р и вновь наrревают до потемне
ния раствора. Продолжают прибавление кисло
ты азотной Р с последующим наrреванием до
тех пор, пока раствор не перестанет темнеть,
затем сильно наrревают до появления плотных
белых паров. Охлаждают, осторожно прибавля
ют 5 мл воды Р, кипятят с предосторожностями
до появления плотных белых паров и продол
жают наrревание до получения остатка объ
ем ом от 2 мл ДО 3 мл. Охлаждают, осторожно
прибавляют 5 мл воды Р и определяют окраску
раствора. Если раствор имеет желтую окраску,
прибавляют по каплям 1 мл раствора вoдopo
да пероксида концентрироваНН020 Р, вновь Ha
rревают до появления плотных белых паров и
продолжают наrревание до получения остатка
объемом от 2 мл ДО 3 мл. Если окраска paCT
вора все еще остается желтой, повторно при
бавляют 5 мл воды Р и 1 мл раствора водорода
пероксида концентрироваНН020 Р до обесц
вечивания раствора. Охлаждают и осторожно
разводят водой Р. Ополаскивая колбу водой Р,
полученный раствор пере носят в пробирку BMe
стимостью 50 мл И доводят до объема 25 мл
этим же растворителем.
Доводят рН раствора до 3,0.......-4,0 pacтвo
ром аммиака концентрированным Р1 (при при
ближении к указанному значению рН можно
применять раствор аммиака разведенный Р1),
используя в качестве внешнеro индикатора ин
дикаторную бумаry, действующую в узком интер
вале рН, затем доводят объем раствора водой Р
до 40 мл И перемешивают. Прибавляют 2 мл бу
феРН020 раствора рН 3,5 Р и перемешивают.
Полученный раствор прибавляют к 1,2 мл тиo
ацетамидН020 реактива Р и немедленно пере
мешивают. Доводят водой Р до объема 50 мл и
перемешивают.
Раствор сравнения (эталон). rотовят па
раллельно с теми же количествами реактивов
и в тех же условиях, используя вместо испытуе
моro образца указанный в частной статье объем
этаЛОНН020 раствора свинца (10 ррт РЬ) Р.
Проверочный раствор. rотовят аналоrично
испытуемому раствору, прибавляя к испытуемо
му образцу указанный в частной статье объем
этаЛОНН020 раствора свинца (10 ррт РЬ) Р для
приrотовления раствора сравнения.
Контрольный раствор. rотовят аналоrично
испытуемому раствору, но без прибавления ис
пытуемоrо образца.
Просматривают растворы через 2 мин после
приrотовления вертикально напротив белоro
фона.
При20дность системы:
раствор сравнения имеет коричневую
окраску по сравнению с контрольным paCTBO
ром;
окраска проверочноro раствора не менее
интенсивная, чем окраска раствора сравнения.
Результат: коричневая окраска испыту
емоro раствора должна быть не интенсивнее
окраски раствора сравнения.
Если невозможно дать однозначную оценку
результату испытания, растворы фильтруют
через подходящий мембранный фильтр (HO
минальный размер пор 0,45 мкм). Давление
поршня шприца должно быть умеренным и по
стоянным для обеспечения медленноro и paB
номерноro фильтрования. Сравнивают окраски
мембранных фильтров, полученные в опытах с
разными растворами.
190
rосударственная фармакопея Республики Беларусь
МЕТОД G
ПРЕДОСТЕРЕЖЕНИЕ при использовании
сосудов для сжи2ания при высоком давлении
необходимо соблюдать требования по тex
нuк-е безопасности, указанные производите
лем оборудования. Циклы сжи2ания должны
соответствовать типу используемой Mи
кроволновой печи (например, микроволновые
печи с контролем мощности, печи с KOHтpo
лем температуры или печи с контролем дaв
ленuя). Кроме тоео, циклы сжиzaния должны
соответствовать инструкции по эксплуата
ции используеМ020 оборудования. Циклы сжи
2ания применяют в случае образования про
зрачных растворов.
Испытуемый pacтвop Количество испы
туемоro образца, указанное в частной статье
(не более 0,5 r), помещают в подходящий
чистый лабораторный стакан с маrнитной Me
шапкой, прибавляют последовательно 2.7 мл
кислоты серной Р, 3,3 мл кислоты азотной Р
и 2,0 мл раствора водорода пероксида KOH
центрированноео Р, прибавляя каждый peaK
тив после завершения реакции между испы
туемым образцом и предыдущим реактивом.
Полученную смесь количественно переносят в
сухой, устойчивый к высокому давлению сосуд
для сжиrания (фторсодержащий полимер или
кварцевое стекло).
Раствор сравнения. rотовят аналоrич
но испытуемому раствору, используя вместо
испытуемоro образца указанный в частной
статье объем этаЛОНН020 раствора свинца
(10 ррт РЬ) Р.
Проверочный раствор. rотовят аналоrично
испытуемому раствору, прибавляя к испытуемо
му образцу указанный в частной статье объем
этаЛОНН020 раствора свинца (10 ррт РЬ) Р
для приroтовления раствора сравнения.
Контрольный раствор. rотовят аналоrич
но испытуемому раствору. но без прибавления
испытуемоrо образца.
Сосуды укупоривают и помещают в лабора
торную микроволновую печь. Процедуру сжиrа
ния проводят, последовательно используя две
подходящие проrраммы. В зависимости от типа
используемой микроволновой печи (следят за
давлением, температурой или мощностью)
разрабатывают мноroступенчатые проrраммы
сжиrания для адекватноrо контроля над про
теканием реакции. После завершения первой
проrраммы оставляют сосуды до охлаждения
перед их вскрытием. Затем в каждый сосуд
прибавляют по 2,0 мл раствора водорода пе
роксида концентрироваНН020 Р и сжиrают, ис
пользуя вторую проrрамму. После завершения
второй проrраммы оставляют сосуды до охлаж
дения перед их вскрытием. При необходимости
для получения прозрачноrо раствора повтор
но прибавляют раствор водорода перокси
да концентрироваНН020 Р и проводят вторую
проrрамму сжиrания. Охлаждают и осторожно
разводят водой Р. Ополаскивая сосуд водой Р,
полученный раствор переносят в колбу и ДOBO
дят до объема 25 мл этим же растворителем.
Доводят рН растворов до З, ,О pacтвo
ром аммиака концентрированным Р1 (при пр*
ближении к указанному значению рН можно
применять раствор аммиака разведенный Р1),
используя в качестве внешнеro индикатора ин
дикаторную бумаry, действующую в узком ин
тервале рН. Для предотвращения наrревания
растворов используют ледяную баню и Mar
нитную мешалку. Доводят объем растворов до
40 мл водой Р и перемешивают. Прибавляют
2 мл буфернО2О раствора рН 3,5 Р и переме--
шивают. Полученные растворы прибавляют к
порциям по 1,2 мл тиоацетамидНО20 реактива
р и немедленно перемешивают. Доводят объем
растворов водой Р до 50 мл, перемешивают и
выдерживают в течение 2 мин.
Растворы фильтруют через подходящий
мембранный фильтр (номинальный размер пор
0,45 МКМ). Давление поршня шприца должно быть
умеренным и постоянным для обеcnечения Meд
ленноro и paвHoMepHoro фильтрования. Сравни
вают окраски мембранных фильтров, полученные
в опытах с разными растворами
При20дность системы:
окраска мембранноrо фильтра, получен
ная в опыте с раствором сравнения, должна
быть коричневая по сравнению с окраской MeM
бранноro фильтра, полученной в опыте с KOH
трольным раствором;
окраска мембранноro фильтра, получен
ная в опыте с проверочным раствором, не менее
интенсивна, чем окраска мембранноro фильтра,
полученная в опыте с раствором сравнения.
Результат: коричневая окраска мембран
ноro фильтра, полученная в опыте с испытуе
мым раствором, должна быть не интенсивнее
окраски мембранноro фильтра, полученной в
опыте с раствором сравнения.
МЕТОД Н
Испытуемый раствор. Количество испы
TyeMoro образца, указанное в частной статье,
растворяют в 20 мл указанноro в частной статье
растворителя или смеси растворителей.
Раствор сравнения. Указанный в частной
статье объем этаЛОНН020 раствора свинца
(10 ррт РЬ) Р разводят указанным в частной
статье растворителем или смесью растворителей.
Контрольный раствор. 20 мл указанноro в
частной статье растворителя или смеси paCTBO
рителей.
К каждому раствору прибавляют по 2 мл бу
фе,ОН020 раствора рН 3,5 Р и перемешивают.
(В некоторых случаях образуется осадок, поэ
mofv!y в частной статье необходимо описать
повторное растворение в определенном коли
честве даНН020 растворителя). Полученные
растворы прибавляют к порциям по 1 ,2 мл тиo
ацетамидН020 реактива Р, немедленно пе
2.4.12. Калий
191
ремешивают и выдерживают в течение 2 мин.
Полученные растворы фильтруют через под
ходящие мембранные фильтры (номинальный
размер пор 0,45 мкм). Сравнивают окраски MeM
бранных фильтров, полученные в опытах с раз
ными растворами.
При20дность системы: окраска мембран
ною фильтра, полученная в опыте с раствором
сравнения, должна быть коричневато черной
по сравнению с окраской мембранноrо филь
тра, полученной в опыте с контрольным pac
твором.
Результат: коричневато-черная окраска
мембранноro фильтра, полученная в опыте с ис
пытуемым раствором, должна быть не интенсив
нее окраски мембранноro фильтра, полученной
в опыте с раствором сравнения.
01/2013:20409
2.4.9. ЖЕЛЕЗО
Количество испытуемоro образца, указан
ное в частной статье, растворяют в воде Р, дo
водят до объема 1 О мл этим же растворителем
и перемешивают; или используют 10 мл paCT
вора, указанноro в частной статье. Прибавля
ют 2 мл раствора 200 r/л кислоты лимонной Р и
0,1 мл кислоты ти02ликолевой Р. Перемешива
ют, подщелачивают раствором аммиака Р и дo
водят водой Р до объема 20 мл.
Параллельно с теми же количествами pe
активов и в тех же условиях roтовят эталон, ис
пользуя 1 О мл этаЛОНН020 раствора железа (/1/)
(1 ррт Fe) Р.
Через 5 мин розовая окраска испытуемоro
раствора должна быть не интенсивнее окраски
эталона.
01/2013:20410
2.4.10. СВИНЕЦ В САХАРАХ
Определение свинца проводят методом
атомно абсорбционной спектрометрии (2.2.23,
метод 11).
Испытуемый раствор. 20,0 r испытуемоro
образца растворяют в смеси из равных объемов
кислоты уксусной разведенной Р и воды Р, дo
водят до объема 100 мл этой же смесью раст
ворителей и перемешивают. Прибавляют 2,0 мл
прозрачноro насыщенноro (около 10 r/л) paCT
вора аммония пирролидиндитиокарбамата Р и
10,0 мл метилизобутилкетона Р и встряхива
ют в течение 30 с в заr.ц.ищенном от яркою света
месте. Оставляют до расслоения. Для испыта
ния используют слой метилизобутилкетона.
Растворы сравнения. rотовят три раствора
сравнения аналorично испытуемому раствору,
но с добавлением к 20.0 r испытуемоro образца
соответственно 0,5 мл, 1,0 мл И 1,5 мл эталон
Н020 раствора свинца (10 ррт РЬ) Р.
Устанавливают нулевую точку на приборе,
используя метилизобутилкетон Р, обработан
ный аналоrично испытуемому раствору, но без
добавления испытуемою образца. Измеряют по
rлощение при длине волны 283,З нм, используя
в качестве источника излучения лампу с полым
свинцовым катодом и воздушно-ацетиленовое
пламя.
Испытуемый образец должен содержать не
более 0,5 ррm свинца, если нет дрyrих указаний
в частной статье.
01/2013:20411
2.4.11. ФОСФАТЫ
К 100 мл приroтовленноro и при необходи
мости нейтрализованноro, как указано в частной
статье, раствора прибавляют 4 мл сульфомо
либденов020 реактива Р3. Встряхивают и при
бавляют 0,1 мл раствора олова (11) хлорида Р1.
Параллельно с теми же количествами pe
активов и в тех же условиях rотовят эталон, ис
пользуя вместо 100 мл раствора испытуемоro
образца смесь из 2 мл этаЛОНН020 раствора
фосфата (5 ррт Р0 4 ) Р и 98 мл воды Р.
Через 1 О мин сравнивают окраску 20 мл каж
доro раствора. Окраска испытуемоro раствора
должна быть не интенсивнее окраски эталона.
01/2013:20412
2.4.12. КАЛИЙ
К 1 О мл раствора, приroтовленноrо, как YKa
зано в частной статье, прибавляют 2 мл свеже
приroтовленноro раствора 1 О r/л натрия тeтpa
фенилбората Р.
Параллельно с теми же количествами pe
активов и в тех же условиях roтовят эталон, ис
пользуя вместо 1 О мл раствора испытуемоro
образца смесь из 5 мл этаЛОНН020 раствора
калия (20 ррт К) Р и 5 мл воды Р.
Через 5 мин опалесценция испытуемоro
раствора не должна превышать опалесценцию
эталона.
01/2013:20413
rосударственная фармакопея Республики Беларусь
192
2.4.13. СУЛЬФАТЫ
При при20товлении всех растворов, при
меняемых в данном испытании, должна исполь
зоваться вода дистиллированная Р.
К 4,5 мл этаЛОНН020 раствора сульфа
та (10 ррт SO) Р1 прибавляют 3 мл раствора
250 r/л бария хлорида Р. Встряхивают и Bыдep
живают в течение 1 мин. К 2,5 мл полученноro
раствора прибавляют 15 мл испытуемоro pacт
вора, приrотовленноrо, как указано в частной
статье, и 0,5 мл кислоты уксусной Р.
Параллельно с теми же количествами peaK
тивов и в тех же условиях roтовят эталон, исполь
зуя вместо испытуемоro раствора 15 мл эталон
Н020 раствора сульфата (10 ррт S04) Р.
Через 5 мин опалесценция испытуемою
раствора не должна превышать опалесценцию
эталона.
01/2013:20414
2.4.14. СУЛЬФАТНАЯ ЗОЛА
Подходящий тиrель (платиновый, фарфо
ровый или кварцевый) прокаливают при темпе
ратуре (600:t50) ОС, выдерживают до остывания
в эксикаторе над силикаrелем или друrим под
ходящим влаroпоrлотителем и взвешивают. В
тиrель помещают указанное в частной статье KO
личество испытуемоro образца и взвешивают.
Испытуемый образец увлажняют небольшим KO
личеством кислоты серной Р (обычно 1 мл) и
осторожно наrревают при насколько возможно
невысокой температуре до полноro обуrливания
образца. После охлаждения остаток увлажня
ют небольшим количеством кислоты серной Р
(обычно 1 мл), осторожно наrревают до прекра
щения выделения белых паров и прокаливают
при температуре (600:t50) ос до полноrо озо
ления остатка. Во время проведения анализа в
тиrле не должно появляться пламя.
Тиrель с остатком выдерживают до OCTЫBa
ния в эксикаторе над силикаrелем или друrим
подходящим влаюпоrлотителем, взвешивают и
рассчитывают процентное содержание остатка.
Если количество полученноro остатка пре
вышает предписанный предел, ero повторно
увлажняют кислотой серной Р и сжиrают, ка!'
указано выше, в течение 30 мин. Сжиrание по
вторяют до тех пор, пока разница между двумя
последующими взвешиваниями будет отличать
ся не более чем на 0,5 Mr, или пока полученное
количество не уложится в предписанный предел.
Количество испытуемоro образца, исполь
зуемое для анализа (обычно 1 2 r), выбирают
таким образом, чтобы при данном предписан
ном пределе масса остатка (обычно около 1 Mr)
моrла быть измерена с достаточной точностью.
01/2013:20415
2.4.15. НИКЕЛЬ ВПОЛИОЛАХ
Определение никеля проводят методом
атомно абсорбционной спектрометрии (2.2.23,
метод 11).
Испытуемый раствор. 20,0 r испытуемоro
образца растворяют в смеси из равных объемов
кислоты уксусной разведенной Р и воды Р, дo
водят до объема 100 мл этой же смесью раст
ворителей и перемешивают. Прибавляют 2,0 мл
насыщенноro (около 10 r/л) раствора аммония
пирролидиндитиокарбамата Р и 10,0 мл Me
тилизобутилкетона Р и встряхивают в тече
ние 30 с в защищенном от яркою света месте.
Оставляют до расслоения. Для испытания ис
пользуют слой метилизобутилкетона.
Растворы сравнения. rотовят три раствора
сравнения аналоrично испытуемому раствору,
но с добавлением к 20,0 r испытуемоro образца
соответственно 0,5 мл, 1,0 мл И 1,5 мл эталон
Н020 раствора никеля (10 ррт Ni) Р.
Устанавливают нулевую точку на приборе,
используя метилизобутилкетон Р, обработан
ный аналоrично испытуемому раствору, но без
добавления испытуемою образца. Измеряют по
rлощение при длине волны 232,0 нм, используя
в качестве источника излучения лампу с полым
никелевым катодом и воздушно ацетиленовое
пламя.
Испытуемый образец должен содержать не
более 1,0 ррm никеля, если в частной статье нет
друrих указаний.
01/2013:20416
2.4.16. ОБЩАЯ ЗОЛА
Кварцевый или платиновый тиrель HarpeBa
ют при красном калении (около 500 ОС) в тече
ние 30 мин, выдерживают до остывания в экси
каторе и взвешивают. Если нет друrих указаний в
частной статье, 1,00 r испытуемою образца или
измельченноro в порошок лекарственноrо расти
тельноro сырья помещают в тиrель и paBHOMep
но распределяют по дну тиrля. Высушивают при
температуре от 100 ос до 105 ос в течение 1 ч и
затем сжиrают до постоянной массы в муфель
ной печи при температуре (600:t25) ОС, выдержи
2.4.18. Свободный формальде ид
193
вая тиrель до остывания в эксикаторе после каж
доro сжиrания. Во время проведения анализа в
тиrле не должно появляться пламя. Если после
длительноro сжиrания зола все еще содержит
темные частицы, содержимое тиrля количе
ственно переносят roрячей водой на беззольный
фильтр и сжиrают остаток на фильтре вместе с
фильтровальной бумаroй. Фильтрат объединяют
с золой, осторожно выпаривают до сухоro OCTaT
ка и сжиrают до постоянной массы.
01/2013:20417
2.4.17. АЛЮМИНИЙ
Раствор испытуемоro образца, приroтов
ленный, как указано в частной статье, поме
щают в делительную воронку, встряхивают с
двумя порциями, по 20 мл каждая, 10 мл paCT
вора 5 r/л 2идроксихинолина Р в хлороформе Р,
затем с 1 О мл этоro же раствора. Хлороформные
слои объединяют и доводят хлороформом Р до
объема 50,0 мл (испытуемый раствор).
Эталон rотовят аналоrично, используя YKa
занный в частной статье раствор сравнения.
Контрольный раствор rотовят аналоrично,
используя указанный в частной статье контроль
ный раствор.
Измеряют интенсивность флуоресценции
(2.2.21) испытуемоro раствора (/1)' эталона (/)
и холостоro раствора ('3)' используя возбуждаю
щее излучение при длине волны З92 нм и BТO
ричный фильтр С полосой пропускания, име
ющей максимум при длине волны 518 нм, или
монохроматор, установленный на пропускание
этой длины волны.
Флуоресценция (', '3) испытуемоro paCT
вора не должна превышать флуоресценцию
('2 /3) эталона.
01/2013:20418
2.4.18. СВОБОДНЫЙ
ФОРМАЛЬДЕrид
Если в частной статье нет друrих указаний,
используют метод А. Метод В является подхо
дящим для вакцин, в которых для нейтрализа
ции избыточноro формальдеrида использован
натрия метабисульфит.
МЕТОД А
Вакцины, используемые в медицине, разво
дят в 10 раз.
25 Зак 1712.
1 мл испытуемой вакцины, разведенной, как
указано выше, помещают в пробирку и прибав
ляют 4 мл воды Р и 5 мл реактива ацетилаце
тона 'Р1. Пробирку помещают в водяную баню с
температурой 40 ос на 40 мин.
Параллельно в этих же условиях roтовят
эталон, используя вместо разведенной вакцины
1 мл раствора формальде2ида Р, разведенноro
до содержания формальдеrида (снр) 20 мкr/мл.
Пробирки просматривают вдоль их верти
кальных осей. Окраска испытуемоro раствора
должна быть не интенсивнее окраски эталона.
МЕТОД В
Испытуемый раствор. Испытуемую вакци
ну разводят 1 :200 водой Р. Если вакцина пред
ставляет собой эмульсию, такое разведение
проводят с водной фазой, отделенной подходя
щим способом (см. ниже). Если для отделения
используют один из перечисленных ниже спосо
бов, то В дальнейшем водную фазу используют
в разведении 1 :20.
Раствор сравнения. rотовят растворы 0,25 r/л,
0,50 r/л, 1,00 r/л и 2,00 r/л снр путем разведе
ния раствора формальде2ида Р в воде Р. Полу
ченные растворы разводят 1 :200 водой Р.
В пробирки помещают по 0,5 мл испытуе
моro раствора и каждоro из растворов cpaBHe
ния. В каждую пробирку прибавляют по 5,0 мл
раствора 0,5 r/л метилбензотиаЗОЛОН2идразо
на 2идрохлорида Р. Пробирки закрывают, встря
хивают и выдерживают в течение 60 мин. Затем
прибавляют по 1 мл реактива железа (111) хлори
да и сульфаминовой кислоты Р и выдерживают
в течение 15 мин.
Измеряют оптическую плотность (2.2.25) по
лученных растворов при длине волны 628 нм.
Содержание формальдеrида в испытуемой
вакцине рассчитывают по rрадуировочному rpa
фику, построенному с использованием paCTBO
ров сравнения. Испытание считается недей
ствительным, если коэффициент корреляции (,)
rрадуировочноro rрафика менее 0,97.
Эмульсии. Если испытуемая вакцина пред
ставляет собой эмульсию, водную фазу oтдe
ляют подходящим способом и используют для
приroтовления испытуемоro раствора. Перечис
ленные ниже способы являются подходящими
для отделения водной фазы.
(а) К 1,0 мл изопропилмиристата Р прибав
ляют 1,0 мл испытуемой вакцины и перемеши
вают. Прибавляют 1,3 мл 1 М раствора кисло
ты хлористоводородной, 2,0 мл хлороформа Р
и 2,7 мл раствора 9 r/л натрия хлорида Р. Тща
тельно перемешивают и центрифуrируют с YCKO
рением 15 000 9 в течение 60 мин. Водный слой
помещают в мерную колбу вместимостью 1 О мл
И доводят водой Р до метки. Если описанная про
цедура не позволяет достичь разделения слоев,
к раствору натрия хлорида прибавляют раствор
100 r/л полисорбата 20 Р и повторяют процеду
ру, центрифуrируя с ускорением 22 500 g.
194
rосударственная фармакопея Республики Беларусь
(Ь) к 1,0 мл раствора 100 r/л натрия хпори
да Р прибавляют 1,0 мл испытуемой BaKЦi1HЫ,
перемешивают и центрифуrируют с ускореhИ
ем 1000 9 в течение 15 мин. Водный слой по
мещают в мерную колбу вместимостью 1 О мл И
доводят водой Р до метки.
(с) К 2,0 мл раствора 100 r/л натрия хло
рида Р и 3,0 мл хлороформа Р прибавляют
1,0 мл испытуемой вакцины, перемешивают и
центрифуrируют с ускорением 1000 9 в тече
ние 5 мин. Водный слой помещают в мерную
колбу вместимостью 1 О мл И доводят водой Р
до метки.
01/2013:20419
2.4.19. ЩЕЛОЧНЫЕ ПРИ МЕСИ
В ЖИРНЫХ МАСЛАХ
В пробирку помещают 10 мл свежепереr
нанноro ацетона Р и 0,3 мл воды Р, прибавля
ют 0,05 мл раствора 0,4 r/л бромфенолов020
сине20 Р в 96 % спирте Р. При необходимости
раствор нейтрализуют 0,01 М раствором кисло
ты хлористоводородной или 0,01 М раствором
натрия 2идроксида. Затем прибавляют 1 О мл
испытуемоro масла, встряхивают и оставляют
до разделения слоев.
Для изменения окраски верхнею слоя на
желтую должно быть израсходовано не более
0,1 мл 0,01 М раствора кислоты хлористово
дородноЙ
01/2013:20421
2.4.21. ПОСТОРОННИЕ МАСЛА
В ЖИРНЫХ МАСЛАХ
МЕТОДОМ ТОНКОСЛОЙНОЙ
ХРОМАтоrРАФИИ
Испытание проводят методом тонкослой
ной хроматоrрафии (2.2.27) с использованием в
качестве ТOHKoro слоя кизеЛЬ2ура G Р Пластин
ку пропитывают, поместив в камеру, содержа
щую смесь вазелиновое масло Р петролей
ный эфир Р (10:90, об/об) в таком количестве,
чтобы нижний край пластинки поrрузился на
5 мм ниже поверхности жидкости. Korдa смесь
для пропитывания поднимется не менее чем на
12 см от нижнеro края пластинки, ее вынима
ют из камеры и дают растворителю испариться
в течение 5 мин. Последующее хроматоrрафи
рование проводят в том же направлении, что и
пропитывание.
При20товление смеси жирных кислот. К
2 r масла прибавляют 30 мл 0,5 М раствора
калия 2идроксида спиртов020 и наrревают с
обратным холодильником в течение 45 мин.
Прибавляют 50 мл воды Р, выдерживают до ox
лаждения, переносят в делительную воронку
и встряхивают с тремя порциями эфира Р, по
50 мл каждая. Эфирные слои отделяют, водный
слой подкисляют кислотой хлористоводород
ной Р и вновь встряхивают с тремя порциями
эфира Р, по 50 мл каждая. Все эфирные слои
объединяют и встряхивают с тремя порциями
воды Р, по 10 мл каждая, отбрасывая промыв
ные воды. Объединенные эфирные слои BЫCY
шивают с помощью натрия сульфата безво
дН020 Р и фильтруют. Затем эфир выпаривают
на водяной бане, а из остатка rотовят испытуе
мый раствор. Жирные кислоты также можно из
влечь из раствора, полученноro при определе
нии неомыляемых веществ.
Испытуемый раствор. 40 Mr смеси жирных
кислот, полученной из испытуемою образца,
растворяют в 4 мл хлороформа Р.
Раствор сравнения. 40 Mr смеси жирных
кислот, полученной из смеси 19 объемов KYKY
РУЗН020 масла Р и 1 объема рапсов020 масла Р,
растворяют в 4 мл хлороформа Р
На линию старта хроматоrрафической пла
стинки наносят по 3 мкл каждоro раствора. По
мещают пластинку в камеру со смесью paCTBO
рителей: вода Р кислота уксусная ледяная Р
(10:90, об/об). Коrда фронт растворителей
пройдет 8 см от линии старта, пластинку выни
мают из камеры, высушивают при температуре
11 О ос в течение 1 О мин и оставляют до охлаж
дения. Затем, если нет друrих указаний в част
ной статье, пластинку помещают в закрытую
плотно подоrнанной крышкой хроматоrрафи
ческую камеру, предварительно насыщенную
парами йода (на дно камеры в выпарительной
чашке помещен йод Р). Через некоторое время
проявляются коричневые или желтовато корич
невые пятна. Пластинку вынимают из камеры
и оставляют на несколько минут. Коrда корич
невый фон исчезнет, пластинку опрыскивают
раствором крахмала Р. Появляющиеся синие
пятна MorYT приобретать коричневую окраску
при высушивании и вновь становятся синими
после опрыскивания водой Р
На xpoMaTorpaMMe испытуемоrо раст
вора всеrда должны обнаруживаться пятно с
R F около 0,5 (олеиновая кислота) и пятно с R F
около 0,65 (линолевая кислота), соответствую
щие пятнам на xpoMaTorpaMMe раствора cpaB
нения. На xpoMaTorpaMMax некоторых масел
может обнаруживаться пятно с R F около 0,75
(линоленовая кислота). Путем сравнения пятен
на xpoMaTorpaMMe испытуемоro раствора с
пятнами на xpoMaTorpaMMe раствора cpaBHe
ния убеждаются в отсутствии на xpoMaTorpaM
ме испытуемоro раствора пятна с R F около 0,25
(эруковая кислота).
2.4.22. Посторонние жирные кислоты в маслах
01/2013:20422
195
2.4.22. ПОСТОРОННИЕ ЖИРНЫЕ
КИСЛОТЫ В МАСЛАХ
МЕТОДОМ rАЗОВОЙ
ХРОМАтоrРАФИИ
Испытание на посторонние жирные кисло
ты проводят путем перевода жирных кислот, co
держащихся в испытуемом масле, в метиловые
эфиры, которые определяют методом rазовой
хроматоrрафии (2.2.28).
МЕТОД А
Этот метод неприменим для масел, coдe,r
жащuх 2Пицерuды жирных кислот с эпокси , 2и
дроэпокси , 2идроперокси , циклопропиловыми
или циклопропениловblми 2руппами, а также
для масел, в составе которых большая часть
жиРНblХ кислот имеет длину цепи менее восьми
атомов У2лерода, и для масел с кислотным
числом более 2,0.
ИСПblтуеМblЙ раствор. Испытуемое масло
высушивают перед метилированием, если это
указано в частной статье. 1,0 r масла помеща
ют в круrлодонную колбу вместимостью 25 мл
со шлифом, снабженную обратным холодильни
ком и rазоотводной трубкой. В колбу прибавля
ют 10 мл метанола безводН020 Р, 0,2 мл paCT
вора 60 r/л калия 2идроксида Р в метаноле р,
присоединяют обратный холодильник и, пропу
ская через смесь азот Р со скоростью около
50 мл/мин, встряхивают и наrревают до кипения.
Коrда раствор станет прозрачным (обычно через
10 мин), продолжают наrpевание в течение по
следующих 5 мин. Затем колбу охлаждают под
проточной водой и содержимое переносят в дe
лительную воронку. Колбу промывают 5 мл 2еп
тана Р, переносят промывные воды в ту же
делительную воронку и встряхивают. Прибавля
ют 10 мл раствора 200 r/л натрия хлорида Р и
энерrично встряхивают. Оставляют до расслое
ния, затем переносят орrанический слой в сосуд,
содержащий натрия сульфат безводНblЙ Р, и
через некоторое время фильтруют.
Раствор сравнения (а). rотовят 0,50 r смеси
веществ, при меняемых для rрадуировки (rpa
дуировочной смеси), состава, приведенноro в
одной из таблиц 2.4.22, как указано в частной
статье (если в частной статье не указан опреде
ленный раствор, roтовят смесь, состав которой
приведен в таблице 2.4.22. 1). Смесь paCTBO
ряют в 2ептане Р и доводят до объема 50,0 мл
этим же растворителем.
Раствор сравнения (Ь). 1,0 мл раствора
сравнения (а) доводят 2ептаном Р до объема
10,0 мл.
Раствор сравнения (с). rотовят 0,50 r смеси
метиловых эфиров жирных кислот, COOTBeTCTBY
ющих составу смеси жирных кислот, указанной в
частной статье для данноro испытуемоro образ
ца. Смесь растворяют в 2ептане Р и доводят до
объема 50,0 мл этим же растворителем. Допу
скается использование roтовых смесей метило
вых эфиров жирных кислот.
Таблица 2.4.22. 1
rрадуировочная смесь (для 2азовой xpOMaтo
2рафии с применением капиллярной колонки и
делением потока рекомендуется прибавлять
к 2радуировочной смеси компонентbl иСПblтуе
мой смеси с большей длиной цепи, К02да каче
ственный анализ проведен с использованием
2радуировОЧН020 2рафика)
Смесь следующих Состав ( %, м/м)
веществ
Метиллаурат Р 5
Метилмиристат Р 5
Метилпальмитат Р 10
Метилстеарат Р 20
Метиларахидат Р 40
Метилолеат Р 20
Таблица 2.4.22. 2
rрадуировочная смесь (для 2азовой xpOMaтo
2рафии с применением капиллярной колонки и
делением потока рекомендуется прибавлять
к 2радуировочной смеси компоненты иСПblтуе
мой смеси с большей длиной цепи, К02да каче
ствеННblЙ анализ проведен с использованием
2радуировОЧН020 2рафика)
Смесь следующих Состав ( %, м/м)
веществ
Метилкапроат Р 10
Метилкаприлат Р 10
Метилкапрат Р 20
Метиллаурат Р 20
Метuлмиристат Р 40 i
Таблица 2.4.22. 3
rрадуировочная смесь (для 2азовой xpOMaтo
2рафии с применением капиллярной колонки и
делением потока рекомендуется прибавлять
к 2радуировочной смеси компонентbl иСПblтуе
мой смеси с большей длиной цепи, К02да каче
ственный анализ проведен с использованием
2радуировОЧН020 zрафика)
Смесь следующих Состав ( %, м/м)
веществ
Метилмиристат Р 5
Метилпальмитат Р 10
Метилстеарат Р 15
Метиларахидат Р 20
Метилолеат Р 20
Метилэйкозеноат Р 10
Метилбе2енат Р 10
Метилли2ноцерат Р 10
Условия хроматоrрафирования:
колонка: капиллярная кварцевая или
стеклянная длиной от 1 м до 1 О м и BHYTpeH
ним диаметром от 0,2 мм до 0,8 мм, покрытая
196
rосударственная фармакопея Республики Беларусь
слоем маКРО20ла 20 000 Р (толщина слоя О, 1
0,5 мкм) или друrой подходящей неподви,,<ной
фазой;
2аз носитель: 2елий для xpOMaт02pa
фии р или водород для хромат02рафии Р;
скорость ааза носителя: 1,3 мл/мин (для
колонки С внутренним диаметром 0,32 мкм);
деление потока: 1:100 или меньше,
в зависимости от внутреннеro диаметра KO
лонки (для колонки С внутренним диаметром
0,32 мкм 1 :50);
температура:
колонка: при изотермических услови
ях 160 200 ос в зависимости от длины
и типа колонки (200 ос для колонки длиной
30 м, покрытой слоем маКР020ла 20 000 Р);
при необходимости использования про
rpaMMbI линейноro rрадиента температу
ры, например, повышают температуру от
170 ос до 230 ос со скоростью 3 ОС/мин;
блок ввода проб: 250 ос;
детектор: 250 ос;
детектор: пламенно ионизационный;
объем вводимой пробы: 1 мкл.
При20дность хромат02рафической cи
стемы при использовании rрадуировочной
смеси в соответствии с таблицей 2.4.22. 1 или
таблицей 2.4.22. 3:
разрешение: не менее 1,8 между пиками
метилолеата и метилстеарата на xpoMaTorpaM
ме раствора сравнения (а);
отношение си2нал/шум: не менее 5 для
пика метилмиристата на xpoMaTorpaMMe paCT
вора сравнения (Ь);
число теоретических тарелок: чуть
более ЗО 000, рассчитанных по пику метилстеа
рата на xpoMaTorpaMMe раствора сравнения (а).
Приаодность хроматоzрафической cи
стемы при использовании rрадуировочной
смеси в соответствии с таблицей 2.4.22. 2:
разрешение: не менее 4,0 между пиками
метилкаприлата и метилкапрата на xpOMaTO
rpaMMe раствора сравнения (а);
отношение си2Нaлlwум: не менее 5 для
пика метилкапроата на xpoMaTorpaMMe paCT
вора сравнения (Ь);
число теоретических тарелок: не менее
15 000, рассчитанных по пику метилкапрата на
xpoMaTorpaMMe раствора сравнения (а).
ОЦЕНКА ХРОМАТО,РАММ
Следует избеrать условий хроматоrрафи
рования, которые MorYT дать неразделенные
пики (наличие компонентов с небольшим раз
личием между временами удерживания, напри
мер, линоленовая и арахидоновая I{ИСЛОТЫ).
Качественный анализ. Идентифицируют
пики, полученные при хроматоrрафировании
раствора сравнения (с) (изотермические усло
вия или условия линейноro rрадиента темпера
туры).
При использовании изотермических yc
ловий, пики MorYT быть идентифицированы
при построении rрадуировочных rрафиков,
используя xpoMaTorpaMMY раствора cpaBHe
ния (а) и данные таблиц 2.4.22. 1, 2.4.22. 2,
2.4.22. 3.
Измеряют приведенное время удержива
ния (t'R) для каждоro пика на xpoMaTorpaMMe
раствора сравнения (а). Приведенное время
удерживания представляет собой время yдep
живания, измеренное от времени выхода пика
растворителя, а не от времени ввода пробы.
Строят rрадуировочный rрафик, который
представляет собой прямую линию:
/9 (t',J = f(эквивалент длины цепи)
Лоrарифмы приведенных времен удержи
вания ненасыщенных кислот расположены на
этой линии как точки, соответствующие Heцe
лым значениям «эквивалента длины цепи»;
эквивалент длины цепи это длина теорети
чески насыщенной цепи, которая будет иметь
такое же приведенное время удерживания, как
идентифицируемая жирная кислота. Напри
мер, линолевая кислота имеет такое же приве
денное время удерживания, как и теоретиче
ски насыщенная жирная кислота, содержащая
18,8 атомов уrлерода.
Пики на xpoMaTorpaMMe испытуемоro раст
вора идентифицируют по rрадуировочному rpa
фику и приведенным временам удерживания.
Эквиваленты длины цепи приведены в таблице
2.4.22А.
Таблица 2.4.22. 4
Эквиваленты длины цепи (эти значения, pac
считанные по 2радуировочному 2рафику, даны
в качестве примера для колонки, покрытой
слоем маКР020ла 20 000 Р)
Жирная кислота Эквивалент
длины цепи
Капроевая кислота 6,0
Каприловая кислота 8,0
Капровая кислота 10,0
Лауриновая кислота 12,0
Миристиновая кислота 14,0
Пальмитиновая кислота 16,0
Пальмитолеиновая кислота 16,3
Марrариновая кислота 17,0
Стеариновая кислота 18,0
Олеиновая кислота 18,3
Линолеваякислота 18,8
rамма линоленовая кислота 19,0
Альфа линоленовая кислота 19,2
Арахидиновая кислота 20,0
Эйкозеноевая кислота 20,2
Арахидоноваякислота 21,2
Беrеновая кислота 22,0
2.4.23. Стер ины в жирных маслах
197
Жирная кислота Эквивалент
длины цепи
Эруковая кислота 22,2
12 Оксостеариновая кислота 22,7
Рицинолеиновая кислота 23,9
12 rидроксистеариновая 23,9
кислота
Лиrноцериновая кислота 24,0
Нервоновая кислота 24,2
Количественный анализ. Обычно исполь
зуют метод внутренней нормализации; при этом
сумму площадей всех пиков на xpoMaTorpaM
ме, кроме пиков, относящихся к растворителю,
принимают за 100 %. Содержание каждоro KOM
понента рассчитывают как отношение площа
ди соответствующеro пика к сумме площадей
всех пиков. Пики, площадь которых составляет
менее 0,05 % от суммы площадей всех пиков,
не учитывают.
В определенных случаях, т. е. при наличии
жирных кислот с 12 или менее атомами уrлеро
да, в частной статье должен быть указан попра
вочный коэффициент для преобразования пло
щадей пиков в проценты (м/м).
МЕТОД В
Этот метод неприменим для масел, co
держащих 2Лицериды жирных кислот с эпокси ,
2идроэпокси , 2идроперокси , циклопропило
выми и циклопропениловыми 2руппами и для
масел с кислотным числом более 2, О.
Испытуемый раствор. 0,100 r испыту
емоro образца помещают в центрифужную
пробирку с завинчивающейся крышкой, paCT
воряют в 1 мл 2ептана Р и 1 мл диметилкар
боната Р и энерrично перемешивают при YMe
ренном наrревании (от 50 ос до 60 ОС). К еще
теплому раствору прибавляют 1 мл раствора
12 r/л натрия Р в метаноле безводном Р, при
roтовленноro с необходимыми предосторожно
стями, и энерrично перемешивают в течение
5 мин. Прибавляют 3 мл воды дистиллироваt-r--
ной Р и энерrично перемешивают в течение
30 с. Центрифуrируют в течение 15 мин с YCKO
рением 1500 g. Хроматоrрафируют 1 мкл op
rаническоrо слоя.
Растворы сравнения и оценка xpOMaтo
2рамм. Если в частной статье нет друrих указа
ний, поступают, как указано в методе А.
Условия хроматоrрафирования:
колонка: капиллярная кварцевая длиной
30 м и внутренним диаметром 0,25 мм, покры
тая слоем маКР020ла 20 000 Р (толщ&.'lна слоя
О, 1 0,5 мкм) или друrой пор,ходящей непод
вижной фазой;
2аз носитель: 2елий для xpOMaт02pa
фииР;
скорость 2аза носителя: 0,9 мл/мин;
деление потока: 1 :100;
температура:
26.32К 1712
Время (мин) Температура
( ОС)
Колонка 0 15 100
15 36 100 -----+ 225
36 1 225
Блок ввода 250
проб
Детектор 250
детектор: пламенно ионизационный;
объем вводимой пробы: 1 мкл.
МЕТОД С
Этот метод неприменим для масел,
содержащих 2лицериды жирных кислот с
эпокси , 2идроэпокси , 2идроперокси , альде
2идными, кетоновыми, циклопропиловыми и
циклопропениловыми 2руппами и сопряжен
ными полиненасыщенными и ацетиленовыми
компонентами из за частиЧН020 или ПОЛН020
разрушения этих 2РУПп.
Испытуемый раствор. 0,10 r испытуе
моro образца помещают в коническую колбу
вместимостью 25 мл, растворяют в 2 мл paCT
вора 20 r/л натрия 2идроксида Р в MeтaHO
ле Р и кипятят с обратным холодильником в
течение 30 мин. Затем через холодильник при
бавляют 2,0 мл раствора бора фторида в Me
та ноле Р и кипятят еще 30 мин, после чеrо
прибавляют через холодильник 4,0 мл 2епта
на Р и кипятят 5 мин. Охлаждают, прибавляют
10,0 мл раствора натрия хлорида насыщен
Н020 р, встряхивают в течение 15 с и прибав
ляют такой объем раствора натрия хлорида
насыщеНН020 Р, чтобы верхний слой поднялся
к roрлу колбы. Отбирают 2,0 мл верхнеro слоя,
помещают в делительную воронку, промыва
ют тремя порциями воды Р, по 2 мл каждая,
и высушивают над натрия сульфатом
безводным Р
Растворы сравнения, хромат02рафиче
ская методика и оценка хромат02рамм. Если
в частной статье нет друrих указаний, посту
пают, как указано в методе А.
01/2013:20423
2.4.23. СТЕРИНЫ В ЖИРНЫХ
МАСЛАХ
ОТДЕЛЕНИЕ СТЕРИНОВОЙ ФРАКЦИИ
Получают неомыляемые вещества жирно
ro масла и затем отделяют стериновую фракцию
методом тонкослойной хроматоrрафии (2.2.27),
используя ТСХ пластинки со слоем силика2е
ля Р толщиной от 0,2 мм до 0,5 мм.
198
rосударственная фармакопея Республики Беларусь
Испытуемый раствор (а). В колбу ВМР'СТИ
мостью 150 мл, снабженную обратным х(,ло
дильником, помещают раствор 2 r/л бетулина Р
вметиленхлориде Р в таком объеме, чтобы KO
личество бетулина соответствовало около 1 О %
от содержания стеринов в образце, взятом для
определения (т. е. в случае оливковоro масла
прибавляют 500 мкл, в случае друrих раститель
ных масел 1500 мкл раствора бетулина). Если
в частной статье определяется содержание ин
дивидуальных стеринов в процентах к стерино
вой фракции, раствор бетулина можно не при
бавлять. Выпаривают до сухоro остатка в токе
азота Р. В колбу помещают 5,00 r (т) испытуе
моro образца. Прибавляют 50 мл 2 М раствора
калия 2идроксида спиртов020 Р и наrревают на
водяной бане в течение 1 ч, часто перемешивая
содержимое колбы круroвыми движениями. Ox
лаждают до температуры ниже 25 ос и количе
ственно переносят содержимое колбы в дели
тельную воронку, содержащую 100 мл воды Р.
Осторожно встряхивают с тремя порциями
эфира, свободН020 от пероксидов Р, по 100 мл
каждая. Эфирные слои собирают в друryю дe
лительную воронку, содержащую 40 мл воды Р,
осторожно встряхивают в течение нескольких
минут, оставляют до расслоения и отбрасывают
водный слой. Эфирный слой встряхивают с He
сколькими порциями воды Р, по 40 мл каждая, до
тех пор, пока водный слой не перестанет давать
щелочную реакцию по фенолфталеину. Эфир
ный слой количественно переносят в колбу, BЫ
сушенную до постоянной массы, промывая дe
лительную воронку эфиром, свободным от
пероксидов Р Эфир отroняют с необходимыми
предосторожностями, к остатку прибавляют 6 мл
ацетона Р Осторожно удаляют растворитель в
токе азота Р Высушивают до постоянной массы
при температуре от 100 ос до 105 ОС, охлажда
ют в эксикаторе и взвешивают. Остаток перено
сят с помощью метиленхлорида Р в маленькую
пробирку и упаривают в токе азота Р до объема
1 мл. В зависимости от содержания неомыляе
мых веществ в масле. доводят конечную KOHцeH
трацию раствора до 25 50 мr/мл.
Испытуемый раствор (Ь). В колбу вмести
мостью 150 мл, снабженную обратным холо
дильником, помещают 5,00 r рапсов020 масла Р
и далее поступают, как указано в приrотовле
нии испытуемоro раствора (а), начиная со слов
«Прибавляют 50 мл 2 М раствора калия 2идpOK
сида спиртов020 Р».
Испытуемый раствор (с). В колбу BMe
стимостью 150 мл, снабженную обратным xo
лодильником, помещают 5,00 r подсолнеЧН020
масла Р и далее поступают, как указано в при
roтовлении испытуемоro раствора (а), начиная
со слов «Прибавляют 50 мл 2 М раствора калия
2идроксида спиртов020 Р».
Раствор сравнения. 25 Mr холестерина Р и
1 О Mr бету лина Р растворяют в 1 мл метилен
хлорида Р.
Для каждоro испытуемоro раствора ис
пользуют отдельную пластинку. Наносят в виде
полосы длиной 1 О мм (на расстоянии 20 мм
от нижнеro края пластинки и 1 О мм от левою
края пластинки) 10 мкл раствора сравнения и
в виде полос длиной 150 мм (на расстоянии
20 мм от нижнеro края пластинки) по 0,5 мл ис
пытуемых растворов (а), (Ь) или (с). Пластин
ки помещают в камеру со смесью растворите
лей эфир Р 2ексан Р (35:65, об/об). Коrда
фронт растворителей пройдет 17 см от линии
старта, пластинки вынимают из камеры, BЫCY
шивают в токе азота Р, опрыскивают paCTBO
ром 2 r/л дихлорфлуоресцеина Р в этаноле Р
и просматривают в ультрафиолетовом свете
при длине волны 254 нм. На xpoMaTorpaMMe
раствора сравнения обнаруживаются полосы,
соответствующие холестерину и бетулину. На
xpoMaTorpaMMax испытуемых растворов обна
руживаются полосы с близкими значениями RF'
соответствующие стеринам. С xpoMaTorpaM
мы каждоro испытуемоro раствора снимают
область покрытия, на которой расположены
полосы стеринов, а также дополнительно зоны
на 2 З мм выше и ниже видимых зон paCT
вора сравнения, и помещают раздельно в три
колбы вместимостью 50 мл каждая. В каждую
колбу прибавляют по 15 мл метиленхлори
да Р и наrревают с обратным холодильником
в течение 15 мин при перемешивании. Каждый
раствор фильтруют через стеклянный фильтр
(ПОР 40) (2.1.2) или подходящую фильтроваль
ную бумаrу и промывают каждый фильтр тремя
порциями метиленхлорuда Р, по 15 мл каждая.
Объединенные фильтраты и смывы помещают
в три колбы, упаривают до объема 5 10 мл в
токе азота Р, переносят в маленькие пробирки
и выпаривают досуха в токе азота Р.
ОПРЕДЕЛЕНИЕ СТЕРИНОВ
rазовая хроматоrрафия (2.2.28). Испы
тание проводят с защитой от попадания
вла2и, и растворы 20товят непосредствен
но перед использованием.
Испытуемый раствор. К выделенным из
испытуемою образца методом тонкослойной
хроматоrрафии стеринам прибавляют CBe
жеприrотовленную смесь из 0,04 мл хлор
триметилсилана Р, 0,1 мл 2ексаметилди
силазана Р и 0,5 мл пиридина безводН020 Р.
Выдерживают в течение не менее 5 мин и ис
пользуют надосадочную жидкость.
Раствор сравнения (а). К 9 частям CTe
ринов, выделенных из рапсов020 масла Р
методом тонкослойной хроматоrрафии, при
бавляют 1 часть холестерина Р К получен
ной смеси прибавляют свежеприroтовленную
смесь из 0,04 мл хлортриметилсилана Р,
0,1 мл 2ексаметилдисилазана Р и 0,5 мл пи
ридина безводН020 Р. Выдерживают в тече
ние не менее 5 мин и используют надосадоч
ную жидкость.
2.4.23. Стерины в жиРНblХ маслах
199
Раствор сравнения (Ь). К стеринам, BЫ
деленным из подсолнечН020 масла Р методом
тонкослойной хроматоrрафии, прибавляют CBe
жеприrотовленную смесь из 0,04 мл хпортриме
тилсилана Р, 0,1 мл 2ексаметилдисилазана Р
и 0,5 мл пиридина безводН020 Р. Выдерживают
в течение не менее 5 мин и используют Haдoca
дачную жидкость.
Условия хроматоrрафирования:
колонка: кварцевая, длиной от 20 м до 30 м
и внутренним диаметром от 0,25 мм до 0,32 мм,
покрытая поли{метил(95)фенил(5)]силоксаном Р
или поли(цианопропил)(7)(фенил)(7)(метил)(86)
силоксаном Р с толщиной слоя 0,25 мкм;
2аз носитель: водород для xpOMaт02pa
фии Рили 2елий для хромат02рафии Р;
линейная скорость: 30 50 см/с (водород)
или 20 35 см/с (rелий);
деление потока: 1 :50 или 1: 100;
температура: колонка 260 ос, блок
ввода проб 280 ос, детектор 290 ос;
детектор: пламенно ионизационный;
объем вводимой пробы: 1 мкл.
Результаты: на xpoMaTorpaMMe paCT
вора сравнения (а) обнаруживаются 4 OCHOB
ных пика, соответствующих холестерину, брас
сикастерину, кампестерину и fЗ ситостерину; на
xpoMaтorpaMMe раствора сравнения (Ь) обна
руживаются 4 основных пика, соответствующих
кампестерину, стиrмастерину, fЗ ситостерину и
Д7 стиrмастенолу. Времена удерживания стери
нов относительно fЗ ситостерина представлены
в таблице 2.4.23. 1.
Пик внутреннеro стандарта (бетулин)
должен быть четко отделен от пиков определя
емых стеринов.
Идентифицируют пики, обнаруженные на
xpoMaTorpaMMe испытуемоro раствора, и pac
считывают процентное содержание каждоro CTe
рина в стериновой фракции испытуемоro веще
ства по формуле:
А
.100,
S
rдe:
А площадь пика, соответствующеrо опре
деляемому стерину;
S сумма площадей всех пиков, соответ
ствующих компонентам, указанным в таблице
2.4.23. 1.
Если это указано в частной статье, рассчи
тывают количество каждоro стерина в милли
rpaMMax, содержащееся в 100 r испытуемоro об
разца, по формуле:
А.т' .100
А/.т
rдe:
А площадь пика, соответствующеro опре
деляемому компоненту;
А' площадь пика, соответствующею бетулину;
т масса навески испытуемоro образца, r;
т' масса навески бету лина Р, Mr.
Таблица 2.4.23. 1
Времена удерживания стеринов по отношению ко времени удерживания fЗ ситостерина
для двух различных колонок
Поли(цианопропил)(7)( фенил) Поли[метил(95) фенил(5)]
(7)(метил)(86)силоксан силоксан
Холестерин 0,64 0,63
Брассикастерин 0,70 0,71
24 Метиленхолестерин . 0,79
0,80
Кампестерин 0,82 0,81
Кампестанол 0,83 0,82
Стиrмастерин 0,87 0,87
Д7 Кампестерин 0,93 0,92
Д5,23 Стиrмастадиенол 0,95 0,95
Клеростерин 0,96 0,96
fЗ Ситостерин 1 1
Ситостанол 1,01 1,02
Д5 Аванастерин 1,03 1,03
Д5,24 Стиrмастадиенол 1,09 1,08
Д7 Стиrмастенол1 1,13 1,12
Д 7 Авенастерин 1,18 1,16
Бетулин 1,4 1,4
1 L':.7 Стиrмастенол в литературе может упоминаться какL':.7 стиrмастерин.
200
01/2013:20424
rосударственная фармакопея Республики Беларусь
2.4.24. ИДЕНТИФИКАЦИЯ
И КОНТРОЛЬ СОДЕРЖАНИЯ
ОСТАТОЧНЫХ
РАСТВОРИТЕЛЕЙ
Методики испытаний, приведенные в этой
общей статье, MorYT быть использованы:
1) для идентификации большинства OCTa
точных растворителей классов 1 и 2 в фарма
цевтических субстанциях, вспомоrательных
веществах и rOToBbIx лекарственных cpeд
ствах, если остаточные растворители неиз
вестны;
2) для определения предельноrо coдep
жания растворителей классов 1 и 2, если они
присутствуют в фармацевтических субстанци
ях, вспомоrательных веществах и rOToBbIx ле
карственных средствах;
3) для количественноro определения
растворителей класса 2, если их предельное
содержание превышает 1000 ррm (0,1 %),
или, если необходимо, для количественноrо
определения растворителей класса 3.
Остаточные растворители классов 1. 2 и 3
перечислены в статье 5.4. Остаточные коли
чества ОР2анических растворителей.
Для приrотовления испытуемых растворов
ниже предложены три растворителя, а также
условия введения rазовых проб при парофаз
ной rазовой хроматоrрафии. Хотя описаны
две хроматоrрафические системы, предпо
чтительнее использовать систему А; система
В обычно используется для идентификации.
Выбор методики приroтовления испытуемых
растворов зависит от растворимости испыту
емоro образца и, в некоторых случаях, от KOH
тролируемых остаточных растворителей.
Формамид, 2 этоксиэтанол, 2 метокси
этанол, этиленrликоль, N метилпирролидон
и сульфолан плохо определяются с помощью
парофазной rазовой хроматоrрафии, и поэто
му их следует определять друrими валидиро
ванными методиками.
Если приведенную ниже методику ис
пытаний при меняют для количественно
ro определения остаточных растворителей в
конкретном веществе, она должна быть вали
дирована.
МЕТОДИКА
Испытания проводят методом парофазной
rазовой хроматоrрафии (2.2.28).
ПробоподrОТQвка 1. Применяют при опре
делении остаточных растворителей в веще
ствах, растворимых в воде.
Исходный раствор испытуеМ020 образ
ца (1). 0,200 r испытуемоro образца растворяют
в воде Р и доводят до объема 20,0 мл этим же
растворителем.
Пробоподrотовка 2. Применяют при опре
делении остаточных растворителей в веще
ствах, нерастворимых в воде.
Исходный раствор испытуеМ020 образ
ца (2). 0,200 r испытуемоro образца растворяют
в N,N диметилформамиде Р (ДМФ) и доводят
до объема 20,0 мл этим же растворителем.
Пробоподrотовка 3. Применяют при
контроле N,N диметилацетамида иlили N,N
диметилформамида, если известно или допу
скается, что один или оба растворителя при
сутствуют в испытуемом образце.
Исходный раствор испытуеМО20 образ
ца (3). 0,200 r испытуемоro образца paCTBO
ряют в 1,3 диметил 2 имидазолидиноне Р
(ДМИ) и доводят до объема 20,0 мл этим же
растворителем.
Если ни одна из вышеприведенных MeTO
дик пробоподroтовки не подходит, возможно
использование друrorо растворителя и друrих
подходящих условий парофазноro анализа.
Раствор остатОЧН020 pacтвopитe
ля (а). К 1,0 мл ФСО раствора остаточно
20 растворителя класса 1 прибавляют 9 мл
диметилсульфоксида Р и доводят водой Р до
объема 100,0 мл. 1,0 мл полученноro paCT
вора доводят водой Р до объема 100 мл.
1,0 мл полученноro раствора доводят водой
р до объема 10,0 мл.
Растворы сравнения должны выдержи
вать следующие пределы:
бензол: 2 ррт (0.0002 %);
четыреххлористый уrлерод: 4 ррт
(0,0004 %);
1 ,2 дихлорэтан: 5 ррт (0,0005 %);
1,1 дихлорэтен: 8 ррт (0,0008 %);
1,1,1 трихлорэтан: 10 ррт (0,001 %).
Раствор остаточных pacтвopитe
лей (Ь). Соответствующие количества OCTa
точных растворителей класса 2 растворяют в
диметилсульФоксиде Р и доводят водой Р до
объема 100,0 мл. Полученный раствор разво
дят водой Р до концентрации, соответствую
щей 1/20 значений пределов концентраций,
указанных в таблице 2 статьи 5.4. Остаточ
ные количества ОР2анических pacтвopитe
лей.
Раствор остаточных pacтвopитe
лей (с). 1,00 r растворителя или растворите
лей, присутствующих в испытуемом образце,
растворяют в диметилсульфоксиде Р или в
воде Р и разводят водой Р до концентрации,
соответствующей 1/20 значений пределов
концентраций, указанных в таблице 1 или Ta
блице 2 статьи 5.4. Остаточные количества
ОР2анических растворителей.
Холостой раствор (контроль). rотовят
аналоrично раствору остаточных растворите
лей (с), но без добавления остаточных paCT
ворителей (используют для проверки OTCYT
ствия мешающих пиков).
2.4.24. Идентификация и контроль содержания остатОЧНblХ растворителей 201
Испытуемый раствор. 5,0 мл исходноro
раствора испытуемоro образца и 1,0 мл холо
стоro раствора помещают во флакон.
Раствор сравнения (а) (класс 1). 1,0 мл
раствора остаточноro растворителя (а) и
5,0 мл подходящеro растворителя #(исполь
зуемоrо для растворения испытуемоro образ
ца)# помещают во флакон.
Раствор сравнения (а 1 ) (класс 1). 5,0 мл
исходноro раствора испытуемоrо образца и
1,0 мл раствора остаточноro растворителя (а)
помещают во флакон.
Раствор сравнения (Ь) (класс 2). 1,0 мл
раствора остаточноro растворителя (Ь) и
5,0 мл подходящеro растворителя помещают
во флакон.
Раствор сравнения (с). 5,0 мл исходноro
раствора испытуемоro образца и 1,0 мл paCT
вора остаточноro растворителя (с) помещают
во флакон.
Раствор сравнения (d). 1,0 мл холостоro
раствора и 5,0 мл подходящеro растворителя
#(используемоro для растворения испытуемо
ro образца)# помещают во флакон.
Флаконы плотно закрывают резиновыми
пробками, покрытыми политетрафторэти
леном, и обжимают алюминиевыми колпач
ками. Встряхивают до получения oдHopoд
Н020 раствора.
Для проведения парофазноro анализа
MorYT быть использованы следующие усло
вия:
Параметры Методика
хроматоrрафической пробоподrотовки
системы
1 I 2 3
Равновесная температу 80 105 80
ра (ОС)
Время достижения 60 45 45
равновесия (мин)
Температура линии 85 110 105
подачи rазовой пробы <
(ОС)
rаз носитель: азот для хромат02рафии Рили
2елий для хромат02рафии Р под COOTBeTCTBY
ющим давлением
Время нахождения под 30 30 30
давлением (с)
Объем вводимой пробы 1 1 1
(мл)
Для проведения хроматоrрафическоrо
анализа MorYT быть использованы следую
щие системы.
СИСТЕМА А
колонка кварцевая капиллярная длиной
30 м и внутренним диаметром 0,32 мм или
0,53 мм, покрытая слоем поперечносшитоro
"!.7 Зак.1712
полимера с 6 % полицианопропилфенилси
локсана и 94 % полидиметилсилоксана тол
щиной 1,8 мкм или 3 мкм;
2аз носитель: азот для xpOMaтo
арафии Рили 2елий для хромат02рафии Р
с делением потока 1:5 и линейной скоростью
около 35 см/с;
пламенно ионизационный детектор
(для хлорсодержащих остаточных раствори
телей класса 1 может быть также использо
ван масс спектрометрический детектор или
детектор электронноro захвата);
температура колонки: изотермиче
ский режим при 40 ОС в течение 20 мин с по
следующим проrраммированным повышени
ем температуры со скоростью 10 ОСIмин ДО
240 ОС, затем изотермический режим при
температуре 240 ос в течение 20 мин;
температура блока ввода проб:
140 ОС;
температура детектора: 250 ОС.
Если возможно мешающее влияние Ma
трицы, используют систему В.
СИСТЕМА В
колонка кварцевая капиллярная дли
ной 30 м и внутренним диаметром 0,32 мм или
0,53 мм, покрытая слоем макроаола 20 000 Р
толщиной 0,25 мкм;
2аз носитель: азот для xpOMaт02pa
фии Рили 2елий для хромат02рафии Р с
делением потока 1:5 и линейной скоростью
около 35 cM/c;
пламенно ионизационный детектор
(для хлорсодержащих остаточных раствори
телей класса 1 может быть также использо
ван масс спектрометрический детектор или
детектор электронноro захвата);
температура колонки: изотермиче
ский режим при 50 ОС в течение 20 мин с по
следующим проrраммированным повышени
ем температуры со скоростью 6 ОС/мин до
165 ОС, затем изотермический режим при TeM
пературе 165 ос в течение 20 мин;
температура блока ввода проб:
140 ОС;
температура детектора: 2.50 ОС.
Испытания в системе А
Вводят 1 мл равновесной rазовой фазы
раствора сравнения (а) и записывают xpo
MaTorpaMMY в таких условиях, чтобы можно
было измерить отношение сиrнал/шум для
1,1, 1 трихлорэтана; отношение сиrнал/шум
должно быть не менее 5. Типичная xpOMaTO
rpaMMa предстаВЛf!на на рисунке 2.4.24. 1.
Вводят 1 мл равновесной rазовой фазы
раствора сравнения (а 1 ). Пики, COOTBeTCTBY
ющие остаточным растворителям класса 1,
должны обнаруживаться.
Вводят 1 мл равновесной rазовой фазы
202
rосударственная фармакопея Республики Беларусь
j
I
I
I
i
I
1
j
I
1
2
4/5
3
T 2 4 5 6
7
8 9 H 12 13 M
Рисунок 2.4.24. 1. Типичная хромат02рамма остаточных растворителей класса 1 в условиях,
описаннЬ1Х для системы А и пробопод20товки 1. Пламенно ионизационнЬ1Й детектор
1 1, 1 дихлорэтен; 2 1.1,1 трихлорэтан; 3 четыреххлористый уrлерод; 4 бензол; 5 1,2 дихлорэтан.
345/68
11
14
1617
18
10
15
7
2
9
12
13
о
5
10
15
20
25
30
мин
Рисунок 2.4.24. 2. Типичная хромат02рамма остаточнЬ1Х растворителей класса 2 в условиях,
описаннЬ1Х для системЬ1 А и пробопод20товки 1. Пламенно ионизационнЬ1Й детектор
1 метанол; 6 нитрометан; 11 метилциклоrексан; 16 хлорбензол;
2 ацетонитрил; 7 хлороформ; 12 1.4 диоксан; 17 opтo-, мета и пapa
3 дихлорметан; 8 циклоrекС8Н; 1 3 пиридин; ксилол;
4 reKcaH; 9 1.2 диметоксиметан; 14 толуол; 18 тетралин.
5 циc 1 ,2 дихлорэтен; 1 О 1.1.2 трихлорэтен; 15 2 reKcaHoH;
2.4.24. Идентификация и контроль содержания остатОЧНblХ растворителей 203
раствора сравнения (Ь) и записывают xpo
MaTorpaMMY в таких условия, чтобы можно
было определить разрешение между пиками
ацетонитрила и метиленхлорида. XpOMaTO
rрафическая система считается приrодной,
если полученная xpoMaTorpaMMa имеет вид
xpoMaTorpaMMbI, представленной на рисунке
2.4.24. 2, а разрешение между пиками aцeTO
нитрила и метиленхлорида не менее 1,0.
Вводят 1 мл равновесной rазовой фазы
испытуемоro раствора. Если на полученной
xpoMaTorpaMMe не обнаруживаются пики, co
ответствующие пикам остаточных раствори
телей на xpoMaTorpaMMax, полученных для
растворов сравнения (а) или (Ь), то испыту
емый образец выдерживает испытания. Если
какой либо пик на полученной для испытуе
моro раствора xpoMaTorpaMMe соответствует
одному из пиков остаточных растворителей
на xpoMaTorpaMMax, полученных для paCTBO
ров сравнения (а) или (Ь), то должна быть ис
пользована система В.
Испытания в системе В
Вводят 1 мл равновесной rазовой фазы
раствора сравнения (а) и записывают xpo
MaTorpaMMY в таких условиях, чтобы можно
было измерить отношение сиrнал/шум для
2/3
4
о
бензола; отношение сиrнал/шум должно быть
не менее 5. Типичная xpoMaTorpaMMa пред
ставлена на рисунке 2.4.24. 3.
Вводят 1 мл равновесной rазовой фазы
раствора сравнения (а 1 ); пики, COOTBeTCTBY
ющие остаточным растворителям класса 1,
должны обнаруживаться.
Вводят 1 мл равновесной rазовой фазы
раствора сравнения (Ь) и записывают xpo
MaTorpaMMY в таких условия, чтобы можно
было определить разрешение между пиками
ацетонитрила и трихлорэтена. XpOMaTO
rрафическая система считается приrодной,
если полученная xpoMaTorpaMMa имеет вид
xpoMaTorpaMMbI, представленной на рисунке
2.4.24. 4, а разрешение между пиками aцe
тонитрила и трихлорэтена не менее 1,0.
Вводят 1 мл равновесной rазовой фазы ис
пытуемоrо раствора. Если на полученной xpo
MaтorpaMMe не обнаруживаются пики, COOTBeT
ствующие пикам остаточных растворителей на
xpoMaтorpaMMax, полученных для растворов
сравнения (а) или (Ь), то испытуемый образец
выдерживает испытания. Если какой либо пик
на полученной для испытуемоro раствора xpo
MaTorpaMMe соответствует одному из пиков OCTa
точных растворителей на xpoMaTorpaMMax, по
лученных для растворов сравнения (а) или (Ь),
и подтверждает соответствие, полученное при
5
2
3
мин
Рисунок 2.4.24. 3. Типичная хромат02рамма остаточных растворителей класса 1 в условиях,
описаННblХ для системы В и пробопод20товки 1. Пламенно ионизациОННblЙ детектор
1 1,1 дихлорэтен; 2 1,1,1 трихлорэтан; 3 четыреххлористый уrлерод; 4 бензол; 5 1,2 дихлорэтан.
204
rосударственная фармакопея Республики Беларусь
использовании системы А, то далее поступают
следующим образом.
Вводят 1 мл равновесной rазовой фа::ы
раствора сравнения (с) в условиях, указан
ных для системы А или системы В. При He
обходимости реrулируют чувствительность
системы таким образом, чтобы высота пика,
соответствующеrо устанавливаемому OCTa
точному растворителю, составляла не менее
50 % шкалы реrистрирующеrо устройства.
Вводят 1 мл равновесной rазовой фазы
раствора сравнения (d). Не должно наблю
даться никаких мешающих пиков.
Вводят 1 мл равновесной rазовой фазы
испытуемоro раствора и 1 мл rазовой фазы
раствора сравнения (с). Повторяют введение
еще дважды.
Средняя площадь пика остаточноro pac
творителяlрастворителей на xpoMaTorpaMMe,
полученной для испытуемоro раствора, не
должна превышать половину площади пика
остаточноrо растворителя/растворителей на
xpoMaTorpaMMe, полученной для раствора
сравнения (с). Хроматоrрафическая систе
ма считается приrодной, если относительное
стандартное отклонение разностей площадей
4811
5/10 14
17
3/9
17
пиков анализируемоro вещества, полученное
из трех повторных парных введений paBHO
весной rазовой фазы раствора сравнения (а)
и испытуемою раствора, не превышает 15 %.
Схема проведения испытаний представ
лена на рисунке 2.4.24. 5.
Если уровень содержания остаточноro
растворителя (классов 2 или 3) составляет
0,1 % и более, для ero количественноrо опре
деления может быть использован метод CTaH
дартных добавок.
01/2013:20425
2.4.25. ОСТАТОЧНЫЕ КОЛИЧЕСТВА
ЭТИЛЕНОКСИДА ИДИОКСАНА
Испытание предназначено для определения
содержания остаточных количеств этиленокси
да и диоксана в веществах, растворимых в воде
или диметилацетамиде. Для веществ, HepaCТBO
римых или недостаточно растворимых в этих
16
7 15
6 13
О 2 3 4 5 6 7 8 9 мин
Рисунок 2.4.24. 4. Типичная хромат02рамма остаточных растворителей класса 1 в условиях,
описанных для системы В и пробопод20товки 1. Пламенно ионизационный детектор
1 метанол;
2 ацетонитрил;
3 дихлорметан;
4 reKcaH;
5 цuc 1.2 дихлорэтен;
6 нитрометан;
7 хлороформ;
8 циклоrексан;
9 1 ,2 диметоксиэтан;
10 1.1,2 трихлорэтен;
11 меТИЛЦИl<Лоrексан;
12 1 ,4 диоксан;
13 пиридин;
14 толуол;
15 2 reKcaHoH;
16 хлорбензол;
17 opт , Meтa и пара ксилол;
18 тетралин (t R =28 мин).
2.4.25. Остаточные количества этиленоксида идиоксана
205
ИСПЫТУЕМЫЙ РАСТВОР
I СИ7МА I
приrОТОВЛЕНИЕ ИСПЫТУЕмоrо РАСТВОРА
И РАСТВОРА СРАВНЕНИЯ
НЕТ
I ВЫДЕРЖИВАЕТ ИСПЫТАНИЕ I
НЕТ
I ВЫДЕРЖИВАЕТ ИСПЫТАНИЕ I
МЕНЬШЕ ПОЛОВИНЫ ПЛОЩАДИ ПИКА,
СООТВЕТСТВУЮЩЕro РАСТВОРУ СРАВНЕНИЯ
I ВЫДЕРЖИВАЕТ ИСПЫТАНИЕ I
БОЛЬШЕПОЛОВИНЫ ПЛОЩАДИ ПИКА,
СООТВЕТСТВУЮЩЕrо РАСТВОРУ СРАВНЕНИЯ
I HE ВЫДЕРЖИВАЕТ ИСПЫТАНИЕ I
Рисунок 2.4.24. 5. Ход испытаний при идентификации и определении пределов
растворителях, приroтовление раствора испыту
емоro образца и условия определения методом
парофазной rазовой хроматоrрафии указывают
в частной статье.
Испытание проводят методом парофазной
rазовой хроматоrрафии (2.2.28).
А. ДлCl веществ, растворимых или смеши
вающихся с водой, при меняют следующую Me
тодику.
Испытуемый раствор. 1,00 r (М т) испы
TyeMoro образца помещают во флакон BMe
стимостью 1 О мл (в зависимости от условий
проведения испытания MorYT при меняться
28 Зак. 1712.
флаконы друrorо объема) и прибавляют 1 мл
воды Р. Флакон закрывают, содержимое пере
мешивают до получения однородноro paCT
вора и выдерживают в течение 45 мин при
температуре 70 ос.
Растеор сравнения (а). 1,00 r (M R ) испы
туемоro обра1ца помещают в такой же флакон
вместимостью 10 мл, прибавляют 0,50 мл
раствора этиленоксида Р3 и 0,50 мл pacт
вора диоксана Р1. Флакон закрывают, содержи
мое перемешивают до получения однородноro
раствора и выдерживают в течение 45 мин при
температуре 70 ос.
206
rосударственная фармакопея Республики Беларусь
Раствор сравнения (Ь). 0,50 мл расrrвора
этиленоксида Р3 помещают во флакон BMe
стимостью 10 мл, прибавляют 0,1 мл свеж.э
приroтовленноro раствора 1 О мr/л ацеталь
де2ида Р и 0,1 мл раствора диоксана Р1.
Флакон закрывают, содержимое перемешива
ют до получения однородноro раствора и BЫ
держивают в течение 45 мин при температу
ре 70 ос.
В. ДЛЯ веществ, растворимых или смешива
ющихся с диметилацетамидом, применяют сле
дующую методику.
Испытуемый раствор. 1,00 r (М Т ) испы
туемоro образца помещают во флакон BMe
стимостью 10 мл (в зависимости от условий
проведения испытания MOryT при меняться фла
коны друroro объема), прибавляют 1,0 мл диMe
тилацетамида Р и 0,20 мл воды Р. Флакон за
крывают, содержимое перемешивают до получе
ния однородноro раствора и выдерживают в Te
чение 45 мин при температуре 90 ос.
Раствор сравнения (а). 1,00 r (M R ) испыту
eMoro образца помещают во флакон вместимо
стью 10 мл И прибавляют 1,0 мл диметилацета
мида Р, 0,10 мл раствора диоксана Р и 0,10 мл
раствора этиленоксида Р2. Флакон закрывают,
содержимое перемешивают до получения oд
нородноro раствора и выдерживают в течение
45 мин при температуре 90 ос.
Раствор сравнения (Ь). 0,10 мл раствора
этиленоксида Р2 помещают во флакон вмести
мостью 10 мл, прибавляют 0,1 мл свежеприro
товленноro раствора 10 мr/л ацетальде2ида Р
и 0,1 мл раствора диоксана Р Флакон закры
вают, содержимое перемешивают до получения
однородноro раствора и выдерживают в течение
45 мин при температуре 90 ос.
MoryT быть использованы следующие yc
ловия подrотовки и ввода равновесной паровой
фазы:
температура уравновешивания: 70 ос
(90 ос для растворов в диметилацетамиде);
время уравновешивания: 45 мин;
температура линии подачи пробы: 75 ос
(150 ос для растворов в диметилацетамиде);
rаз носитель: еелий для xpOMaт02pa
фииР;
время подачи rаза носителя в контейнер с
анализируемой пробой: 1 мин;
время ввода rазовой пробы: 12 с.
Условия хроматоrрафирования:
колонка капиллярная стеклянная или
кварцевая длиной 30 м и внутренним диаметром
0,32 мм, внутренняя поверхность которой по
крыта слоем полидиметилсилоксана Р толщи
ной 1,0 мкм;
2аз носитель: 2елий для xpOMaт02pa
фии Р или азот для хромат02рафии Р;
линейная скорость 2аза носителя: около
20 см/с;
деление потока: 1 :20;
детектор: пламенно ионизационный;
температура: колонка 50 ос в течение
5 мин, затем повышают до 180 ос со скоростью
5 ОС/мин, затем до температуры 230 ос со CKOpO
стью 30 ОС/мин и поддерживают такую темпера
туру в течение 5 мин; блок ввода проб 150 ос;
детектор 250 ос.
Хроматоrрафируют подходящий объем (Ha
пример. 1,0 мл) пробы rазовой фазы раствора
сравнения (Ь). Реryлируют чувствительность си
стемы таким образом, чтобы высоты пиков эти
леноксида и ацетальдеrида на полученной xpo
MaтorpaMMe составляли не менее 15 % шкалы
реrистрирующеrо устройства. Хроматоrрафиче
ская система считается приroдной, если разре
шение для пиков ацетальдеrида и этиленоксида
не менее 2,0, а отношение сиrнал/шум для пика
диоксана не менее 5.
Хроматоrрафируют поочередно подходя
щие объемы, например, по 1,0 мл (или такой
же объем, как для раствора сравнения (Ь) проб
rазовых фаз испытуемоro раствора и раствора
сравнения (а). Повторяют введение еще дважды.
ПРОВЕРКА ТОЧНОСТИ
ДЛЯ каждой пары введений вычисляют раз
ность площадей пиков этилен оксида и диокса
на на xpoMaTorpaMMe раствора сравнения (а)
и xpoMaTorpaMMe испытуемоro раствора. Xpo
матоrрафическая система считается приroд
ной, если относительное стандартное отклоне
ние, рассчитанное для трех значений разности
площадей пиков этиленоксида, составляет не
более 15 %, а относительное стандартное OT
клонение, рассчитанное для трех значений раз
ности площадей пиков диоксана, составляет не
более 10 %.
Если навески испытуемоro образца, взятые
для приroтовления испытуемоrо раствора и
раствора сравнения (а), отличались от 1,00 r
более чем на 0,5 %, необходимо внести COOTBeT
ствующие поправки.
Содержание этиленоксида в миллионных
частях (ррт) рассчитывают по формуле:
Ar'C
(A R .MT) (Ar .M R )
rдe:
А т площадь пика этиленоксида на xpOMa
TorpaMMe испытуемоro раствора;
A R площадь пика этиленоксида на xpOMa
TorpaMMe раствора сравнения (а);
М7 масса навески испытуемоro образца,
взятая для приroтовления испытуемоro раст
вора, r;
M R масса навески испытуемоrо образ
ца, ВЗЯТ2Я дЛЯ приrотовления раствора cpaB
нения, r;
С количество этиленоксида, прибавлен
ноro к раствору сравнения (а), MKr.
Содержание диоксана в миллионных частях
(ррm) рассчитывают по формуле:
2.4.26. N,N диметиланилин
207
От'С
(D R .MT) (DT .M R )
rдe:
О т площадь пика диоксана на xpOMaTO
rpaMMe испытуемоro раствора;
O R площадь пика диоксана на xpOMaTO
rpaMMe раствора сравнения (а);
С количество диоксана, прибавленноro к
раствору сравнения (а), MKr:
01/2013:20426
2.4.26. N,N-ДИМЕТИЛАНИЛИН
МЕТОД А
Испытание проводят методом rазовой xpo
матоrрафии (2.2.28), используя в качестве BHY
треннеro стандарта N,N диэтиланилин Р
Раствор внутренне20 стандарта. 50 Mr
N,N диэтиланилина Р растворяют в 4 мл 0,1 М
раствора кислоты хлористоводородной и дo
водят водой Р до объема 50 мл. 1 мл полученно
ro раствора разводят водой Р до объема 100 мл.
Испытуемый раствор. В колбу с притер
той стеклянной пробкой помещают 0,50 r испы
туемоro образца и растворяют в 30,0 мл воды Р,
затем прибавляют 1,0 мл раствора внутреннеro
стандарта и термостатируют раствор при TeM
пературе от 26 ос до 28 ос. Прибавляют 1,0 мл
раствора натрия 2идроксида KOHцeHтpиpoвaH
Н020 Р и перемешивают до полноro paCTBope
ния. Прибавляют 2,0 мл триметилпентана Р,
встряхивают в течение 2 мин и оставляют до
расслоения. Используют верхний слой.
Раствор сравнения. 50,0 Mr N,N диме
тиланилина Р растворяют в 4,0 мл 0,1 М pacт
вора кислоты хлористоводородной и доводят
водой Р до объема 50,0 мл. 1,0 мл полученноro
раствора разводят водой Р до объема 100,0 мл.
1,0 мл полученноro раствора разводят водой Р
до объема 30,0 мл. Прибавляют 1,0 мл paCT
вора внутреннеro стандарта и 1,0 мл раствора
натрия 2идроксида концентрироваНН020 Р При
бавляют 2,0 мл триметилпентана Р, встряхи
вают в течение 2 мин и оставляют до расслое
ния. Используют верхний слой.
Испытание может быть проведено при ис
пользовании следующих условий xpoMaTorpa
фирования:
колонка кварцевая капиллярная длиной
2ry м и внутренним диаметром 0,32 мм, покры
тая слоем поперечносшитоro полиметилфенил
силоксана Р толщиной 0,52 мкм;
еаз носитель: 2елий для xpOMaт02pa
фии Р с делением потока 1 :20, давлением на
входе в колонку 50 кПа и объемной скоростью
сбрасываемоro rаза носителя 20 мл/мин;
детектор: пламенно ионизационный;
кварцевая вставка испаритель длиной
1 см, заполненная слоем диатомита для
2азовой хромат02рафии Р с нанесенным
поли(диметил)силоксаном Р в количестве
1 О % (м/м);
температура: колонка 150 ос в теч
ние 5 мин, затем повышают до 275 ос со CKOp(r
стью 20 ОС/мин и поддерживают такую темпера
туру в течение 3 мин; блок ввода проб 220 ос;
детектор 300 ос.
Времена удерживания: N,N диметилани
лин около 3,6 мин, N,N диэтиланилин
около 5,0 мин.
Хроматоrрафируют 1 мкл испытуемоrо
раствора и 1 мкл раствора сравнения.
МЕТОД В
Испытание проводят методом rазовой xpo
матоrрафии (2.2.28), используя в качестве BHY
треннеro стандарта нафталин Р
Раствор внутренне20 стандарта. 50 Mr
нафталина р растворяют в циКЛО2ексане Р и
доводят до объема 50 мл этим же растворите
лем. 5 мл полученноro раствора разводят цикло
2ексаном Р до объема 100 мл.
Испытуемый раствор. В пробирку с при
тертой стеклянной пробкой помещают 1,00 r
испытуемоro вещества, прибавляют 5 мл 1 М
раствора натрия 2идроксида и 1, О мл раствора
внутреннею стандарта. Пробирку закрывают
и энерrично встряхивают в течение 1 мин. При
необходимости центрифуrируют и используют
верхний слой.
Раствор сравнения. К 50,0 Mr N.N
диметиланилина Р прибавляют 2 мл кислоты
хлористоводородной Р и 20 мл воды Р. Встряхи
вают до растворения и доводят объем раствора
водой Р до объема 50,0 мл. 5,0 мл полученноro
раствора разводят водой Р до объема 250,0 мл.
1,0 мл полученноro раствора помещают в про
бирку с притертой стеклянной пробкой, прибав
ляют 5 мл 1 М раствора натрия 2идроксида и
1,0 мл раствора внутреннеro стандарта. Пробир
ку закрывают и энерrично встряхивают в тече
ние 1 мин. При необходимости центрифуrируют
и используют верхний слой.
Испытание может быть проведено при ис
пользовании следующих условий xpoMaTorpa
фирования:
колонка стеклянная длиной 2 м и BHyтpeH
ним диаметром 2 мм, заполненная диатомитом
силанизированным для 2азовой xpOMaт02pa
фии р, импреrнированным 3 % (М/М) полиметил
фенилсилоксана Р:
2аз носитель: азот для хромат02рафии Р;
скорость 2аза носителя: 30 мл/мин;
детектор: пламенно ионизационный;
температура: колонка 120 ос; блок
ввода проб 150 ос; детектор 150 ос.
Хроматоrрафируют 1 мкл испытуемоro
раствора и 1 мкл раствора сравнения.
208
01/2013:20427
rосударственная фармакопея Республики Беларусь
2.4.27. ТЯЖЕЛЫЕ МЕТАЛЛЫ
В ЛЕКАРСТВЕННОМ
РАСТИТЕЛЬНОМ СЫРЬЕ
И ЖИРНЫХ МАСЛАХ
Определение проводят методом aTOMHO
абсорбционной спектрометрии (2.2.23).
При использовании закрытых peaKциOH
ных сосудов вЫСОК020 давления и микровол
нов020 лаборатОРН020 оборудования необхо
димо стР020 придерживаться инструкций по
технике безопасности и эксплуатации обо
рудования, прилаеаемых производителем.
ОБОРУДОВАНИЕ
колбы для минерализации rерметич
но укупориваемые политетрафторэтилено
вые колбы вместимостью около 120 мл, име
ющие клапан для реrулирования давления
внутри колбы, и политетрафторэтиленовые
трубки для удаления rазов;
система для rерметичноrо укупорива
ния колб, использующая одинаковую силу за
крутки для каждой из них;
микроволновая печь с частотой MarHe
трона 2450 мrц, с возможностью реrулирова
ния мощности от О Вт до (6ЗО:t70) Вт с шаroм
в 1 %, с проrраммируемым цифровым блоком
управления, покрытая изнутри политетрафто
рэтиленом, с реrулируемой скоростью венти
ляции, с вращающейся подставкой;
атомно абсорбционный спектрометр,
снабженный лампами с полым катодом в Ka
честве источника излучения и .дейтериевой
лампой в качестве корректора фона, а также:
(а) rрафитовой печью в качестве reHepa
тора атомноro пара при определении кадмия,
свинца, меди, железа, никеля и цинка;
(Ь) автоматизированным reHepaTopOM rи
дридноro пара для определения мышьяка и
ртути.
МЕТОД
В случае использования альтернатив
Н020 оборудования может быть необходима
дополнительная настройка.
Перед использованием вся стеклянная
посуда и лабораторное оборудование должны
быть обработаны раствором 1 О r/л кислоты
азотной Р.
Испытуемый раствор. Указанное в част
ной статье количество испытуемоro образ
ца (около 0,50 r измельченноrо сырья (1400)
(2.9.12) или 0,50 r жирноro масла) помещают
в колбу для минерализации, прибавляют 6 мл
кислоты азотной, свободной от тяжелых
металлов, Р и 4 мл кислоты хлористоводо
родной, свободной от тяжелых металлов, Р.
rерметично укупоривают.
Помещают колбы для минерализации в
микроволновую печь и проводят минерализа
цию в соответствии со следующей проrрам
мой в три стадии, используя семь колб, coдep
жащих испытуемый раствор: 80 % мощности
в течение 15 мин; 100 % мощности в течение
5 мин; 80 % мощности в течение 20 мин.
По окончании процесса колбы охлаж
дают на воздухе и прибавляют в каждую по
4 мл серной кислоты, свободной от тяжелых
металлов, Р. Укупоривают и повторяют про
rpaMMY минерализации, как описано выше.
После охлаждения на воздухе колбы OTKpЫ
вают, полученные прозрачные бесцветные
растворы помещают в мерные колбы вмести
мостью 50 мл. Каждую колбу для минерали
зации дважды ополаскивают водой Р порци
ями по 15 мл и переносят промывные воды в
те же мерные колбы. В каждую мерную колбу
прибавляют 1,0 мл раствора 1 О r/л ма2ния
нитрата Р и 1,0 мл раствора 100 r/л aMMO
ния ди2идрофосфата Р и доводят водой Р до
объема 50,0 мл.
Холостой раствор. 6 мл кислоты азот
ной, свободной от тяжелых металлов, Р и
4 мл кислоты хлористоводородной, свобод
ной от тяжелых металлов, Р помещают в
колбу для минерализации и обрабатывают
аналоrично испытуемому раствору.
Таблица 2.4.27. 1
Cd Cu Fe Ni РЬ Zn
Длина волны нм 228,8 324,8 248,3 232 283,5 213,9
Ширина щели нм 0,5 0,5 0,2 0,2 0,5 0,5
Сила тока в мА 6 7 5 10 5 7
лампе
Температура ос 800 800 800 800 800 800
сжиrания
Температура ос 1800 2300 2300 2500 2200 2000
атомизации
Корректор фона вкл. Выкл. Выкл. Выкл. Выкл. Выкл.
Ток азота л/мин 3 3 3 3 3 3
2.4.28. 2 ЭтиЛ2ексановая кислота
209
КАДМИЙ, МЕДЬ, ЖЕЛЕ30, СВИНЕЦ
НИКЕЛЬ И ЦИНК
Содержание кадмия, меди, железа,
свинца, никеля и цинка определяют методом
стандартных добавок (2.2.23, метод 11), ис
пользуя растворы сравнения каждоro тяжело
ro металла и пара метры, описанные в табли
це 2.4.27. 1.
Значение абсорбции холостоro раствора
отнимается от значения абсорбции испытуе
моro раствора.
МЫШЬЯК И РТУТЬ
Содержание мышьяка и ртути определя
ют методом rрадуировочноro rрафика (2.2.23,
метод /).
Значение абсорбции холостоro раствора
отнимается от значения абсорбции испытуе
моro раствора.
Мышьяк
Раствор образца. К 19,0 мл испытуемоro
раствора или холостоro раствора, приroтов
ленных, как описано выше, прибавляют 1 мл
раствора 200 r/л калия йодида Р. Выдержива
ют испытуемый раствор при комнатной TeM
пературе в течение около 50 мин или при TeM
пературе 70 ос в течение около 4 мин.
Кислотный реактив. Кислота хлористо
водородная, свободная от тяжелых метал
лов, Р.
Восстанавливающий реактив. Раствор
6 r/л натрия тетра2идробората Р в paCTBO
ре 5 r/л натрия 2идроксида Р.
Используют параметры, описанные в Ta
блице 2.4.27. 2.
Ртуть
Раствор образца. К 19,0 мл испытуемоro
раствора или холостоro раствора, приroтов
ленных как описано выше, прибавляют 1 мл
раствора 200 r/л калия йодида Р. Выдержива
ют испытуемый раствор при комнатной TeM
пературе в течение около 50 мин или при TeM
пературе 70 ос в течение около 4 мин.
Кислотный реактив. Раствор 515 r/л Kиc
лоты хлористоводородной, свободной от
тяжелых металлов, Р.
Восстанавливающий реактив. Раствор
10 r/л олова (11) хлорида Р в кислоте хлори
стоводородной разведенной, свободной от
тяжелых металлов, Р.
Используют параметры, описанные в Ta
блице 2.4.27. 2.
01/2013:20428
2.4.28. 2 этилrЕКСАНОВАЯ
КИСЛОТА
Испытание проводят методом rазовой xpo
матоrрафии (2.2.28), используя в качестве BHY
TpeHHero стандарта кислоту 3 циКЛ02ексилпро
пионовую р
Раствор внутренне20 стандарта. 100 Mr
кислоты 3 циКЛ02ексилпропионовой Р раст
воряют в циКЛ02ексане Р и доводят до объема
100 мл этим же растворителем.
Испытуемый раствор. 0,300 r испытуемоro
образца растворяют в 4,0 мл 33 % (об/об) paCT
вора кислоты хлористоводородной Р. Энерrич
но встряхивают в течение 1 мин с 1,0 мл paCT
вора внутреннеro стандарта. Выдерживают до
расслоения, при необходимости центрифуrиру
ют. Для испытания используют верхний слой.
Раствор сравнения. 75,0 Mr кислоты
2 этиЛ2ексановой Р растворяют в растворе
BHYTpeHHero стандарта и доводят до объема
50,0 мл этим же растворителем. К 1,0 мл по
лученноro раствора прибавляют 4,0 мл 33 %
(об/об) раствора кислоты хлористоводо
родной Р и энерrично встряхивают в течение
1 мин. Выдерживают до расслоения, при He
обходимости центрифуrируют. Для испытания
используют верхний слой.
Хроматоrрафируют по 1 мкл испытуемоro
раствора и раствора сравнения в следующих yc
ловиях:
колонка капиллярная кварцевая длиной
1 О м и внутренним диаметром 0,53 мм, покры
тая слоем маКР020ла 20 000 2 нитротерефта
лата Ртолщиной 1,0 мкм;
2аз носитель: 2елий для xpOMaт02pa
фииР;
Таблица 2.4.27. 2
As Hg
Длина волны нм 193,7 253,7
Ширина щели нм 0,2 0,5
Сила тока в лампе мА 10 4
Кислотный реактив мл/мин 1,0 1,0
Восстанавливающий мл/мин 1,0 1,0
реактив
Раствор образца мл/мин 7,0 7,0
Кювета Кварцевая (подоrретая) Кварцевая (не подоrретая)
Корректор фона Выкл. Выкл.
Ток азота лlмин 0,1 0,1
210
rосударственная фармакопея Республики Беларусь
Время (МИН) Температура (ОС) Скорость подъема Примечание
температуры (ОС/мин)
Колонка 0 2 40 I Изотермический режим
2 7,3 40 200 30 Линейный rрадиент
температуры
7,3 10,3 200 Изотермический режим
Блок ввода 200
проб
Детектор 300
скорость 2аза носителя: 10 мл/мин;
детектор: пламенно ионизационный;
температура колонки и скорость подъ
ема температуры: в соответствии со следую
щей проrраммой:
Хроматоrрафическая система считает
ся приrодной, если разрешение между пиком
2 этилrексановой кислоты (первый пик) и пиком
внутреннеro стандарта составляет не менее 2,0.
Содержание 2 этилrексановой кислоты в
про центах вычисляют по формуле:
LI ./ .т .2
< 'Т R R
А р .I T .т т
rдe:
А т площадь пика 2 этилrексановой кисло
ты на xpoMaтorpaMMe испытуемоro раствора;
A R площадь пика 2 этилrексановой кисло
ты на xpoMaTorpaMMe раствора сравнения;
/т площадь пика внутреннеro стандарта
на xpoMaTorpaMMe испытуемоro раствора;
/R площадь пика внутреннеro стандарта
на xpoMaTorpaMMe раствора сравнения;
т т масса навески испытуемоrо образца,
взятая для приrотовления испытуемоrо paCT
вора, r;
m R масса навески 2 этилrексановой кис
лоты, взятая для приrотовления раствора cpaB
нения, r.
01/2013:20429
2.4.29. СОСТАВ ЖИРНЫХ КИСЛОТ
В МАСЛАХ, ОБоrАЩЕННЫХ
OMErA-З КИСЛОТАМИ
Испытание применяется для количе
ствеНН020 определения содержания эйкозапен
таеновой кислоты (ЭПК) и докоза2ексаеновой
кислоты (Д/К). содержащихся в рыбьем жире,
об02ащенном OMe2a 3 кислотами в раЗНblХ KOH
центрациях. Метод применяется для трисли
церидов или сложных этиловых эфиров, и pe
зультатbl выражаются как три2лицериды или
сложные этиловые эфиры соответственно.
ЭПК И дrк
rазовая хроматоrрафия (2.2.28). Испыта
ние выполняют по возможности быстро, избе
2ая воздействия актиниЧН020 света, окисляю
щих а2ентов, окислительных катализаторов
(например, медь и железо) и воздуха.
Метод применяется для определения Me
тиловых и этиловых сложных эфиров (al/ Z)
эйкоза 5,8, 11,14, 17 пентаеновой кислоты (ЭПК
(ЕРА); 20:5 п З) и (аll Z) докоза 4,7, 1 О, 1 3, 16, 19
rексаеновой кислоты (дrк (ОНА); 22:6 п 3) в ис
пытуемом образце.
Внутренний стандарт. Метилтрикозано
атР
Испытуемый раствор (а)
А. Количество испытуемоro образца в co
ответствии с таблицей 2.4.29. 1 и около 70,0 Mr
BHYTpeHHero стандарта растворяют в растворе
50 мr/л бутиЛ2идрокситолуола Р в трLJметил
пентане р и доводят до объема 10,0 мл этим же
растворителем. При необходимости, осторожно
наrревают (до температуры 60 ОС) до полноro
растворения внутреннеro стандарта.
Таблица 2.4.29. 1
Приблизительная Количество
сумма ЭПК и дrк испытуемоrо образца
(%) (r)
30 50 0,4 0,5
50 70 0,3
70 90 0,25
Сложные этиловые эфиры подroтовлены к
анализу. Стадия В необходима для подroтовки к
анализу триrлицеридов.
В. 2,0 мл раствора, полученноro на стадии
А, помещают в кварцевую пробирку и выпа
ривают растворитель в слабом токе азота Р.
Прибавляют 1,5 мл раствора 20 r/л натрия 2и
дроксида Р в метаноле Р, плотно укупорива
ют пробкой, покрытой политетрафторэтиле
ном, в атмосфере азота Р, перемешивают и
наrревают на водяной бане 8 течение 7 мин.
Охлаждают. Прибавляют 2 мл раствора бора
хлорида в метаноле Р, плотно укупоривают в
атмосфере азота Р, перемешивают и HarpeBa
ют на водяной бане в течение 30 мин. Охлаж
дают до температуры (40 50) ОС, прибавляют
1 мл триметилпентана Р, укупоривают и ин
2.4.29. Состав жирных кислот в маслах
211
тенсивно встряхивают не менее 30 с. HeMeд
ленно прибавляют 5 мл насыщеНН020 pacт
вора натрия хлорида Р, плотно укупоривают
в атмосфере азота Р и тщательно встряхива
ют не менее 15 с. Переносят верхний слой в
отдельную пробирку. Встряхивают еще раз Me
танольный слой с 1 мл триметилпантана Р.
Объединенные триметилпентановые извлече
ния дважды промывают водой Р nорциями по
1 мл И высушивают с помощью натрия суль
фата безводН020 Р. rотовят по 3 раствора для
каждоro образца.
Испытуемый раствор (Ь). 0,300 r испытуе
моro образца растворяют в растворе 50 мr/л бу
тиЛ2идрокситолуола Р в триметилпентане Р
и доводят до объема 10,0 мл этим же раствори
телем. Далее поступают, как указано для испы
туемоro раствора (а).
Раствор сравнения (aJ 70,0 Mr BHYTpeHHe
ro стандарта и 90,0 Mr ФСО этилов020 эфира
эйкозапентаеновой кислоты растворяют в pac
творе 50 мr/л бутиЛ2идрокситолуола Р в тpи
метилпентане Р и доводят до объема 10,0 мл
этим же растворителем. При необходимости,
осторожно наrревают (до температуры 60 ОС) до
полноro растворения внутреннеro стандарта.
Раствор сравнения (а 2 ). 70,0 Mr BHYTpeHHe
ro стандарта и 60,0 Mr ФСО этилов020 эфира
докоза2ексаеновой кислоты растворяют в pac
творе 50 мr/л бутиЛ2идрокситолуола Р в тpи
метилпентане Р и доводят до объема 10,0 мл
этим же растворителем. При необходимости.
осторожно наrревают (до температуры 60 ОС) до
полноro растворения внутреннею стандарта.
При анализе этиловых эфиров раствор
сравнения (а 1 ) и раствор сравнения (а 2 ) обраба
тывают аналоrично испытуемому раствору (а),
стадия А. Для анализа триrлицеридов посту
па ют, как указано в стадии В для испытуемоro
раствора (а), и roтовят по 3 раствора для каждо
ro образца.
Раствор сравнения (Ь). 0,3 r метиларахи
дата Р, 0,3 r метилбе2ената Р, 0,3 r метил <
пальмитата Р и 0,3 r метилстеарата Р по
мещают в мерную колбу вместимостью 1 О мл
И растворяют в растворе 50 мr/л бутиЛ2идрок
ситолуола Р в триметилпентане Р и ДOBO
дят до объема 10,0 мл этим же растворителем.
Раствор сравнения (с). Образец, coдep
жащий около 55,0 Mr метилов020 эфира дo
коза2ексаеновой кислоты Р и около 5,0 Mr
метилов020 эфира тетракоз 15 еновой Kиc
лоты р, помещают в мерную колбу 8местимо
стью 1 О мл, растворяют в растворе 50 мr/л бу
тиЛ2идрокситолуола Р в триметилпентане
Р и доводят до объема 10,0 мл этим же paCT
ворителем.
Условия хроматоrрафирования:
колонка: кварцевая капиллярная длиной
не менее 25 м и внутренним диаметром
0,25 мм, покрытая слоем маКР020ла 20 000 Р
(толщина слоя 0,2 мкм);
2аз носитель: водород для xpOMaт02pa
фии Рили zелий для хромат02рафии Р;
скорость 2аза носителя: 1 мл/мин;
деление потока: 1 :200; в качестве альте
нативы без деления потока с температурным
контролем (перед введением растворы образца
должны быть разведены 1/200 раствором 50 мr/л
бутиЛ2идрокситолуола Р в триметилпен
тане Р);
температура:
Время Температура
(мин) (ОС)
0 2 170
Колонка 2 25,7 170-----+240
25,7 28 240
Блок ввода 250
проб
Детектор 270
детектор: пламенно ионизационный;
объем вводимой пробы: 1 мкл, дважды.
При20дность хромат02рафической cи
стемы:
на xpoMaTorpaMMe раствора сравнения (Ь)
процент площади пиков компонентов возрас
тает в следующем порядке: метилпальмитат,
метилстеарат, метиларахидат, метилбеrенат;
разница между площадями пиков метилпаль
митата и метилбеrената (в %) должна быть
менее 2,0 %;
разрешение: не менее 1 ,2 между пиками
метиловоro эфира докозаrексаеновой кислоты и
метиловоro эфира тетракоз 15 еновой кислоты
на xpoMaTorpaMMe сравнения (с);
на xpoMaTorpaMMe испытуемоro раствора
(а) пики, соответствующие метилтрикозаноату
и метиловому или этиловому эфирам rенэйко
запентаноеновой кислоты (С21 :5), обнаружива
емые при сравнении с хроматоrраммой испы
туемоro раствора (Ь), должны быть полностью
разделены (если нет, используют поправочный
коэффициент).
Принимая 80 внимание указанную чистоту
стандартных образцов, процентное содержание
ЭПК и дrк рассчитывают по формуле: .
А хз т 1 т х. 1
А . . . . .C.100,
х т х . з А Ах" т 2
rде:
т 1 масса навески BHyтpeHHero CTaHдap
та, взятая для приroтовления испытуемоro paCT
вора (а), Mr;
т 2 масса навески испытуемоro образца,
взятая для приrотовления испытуемоro paCT
вора (а), Mr;
т х . з масса навески внутреннеro CTaHдap
та, взятая для приroтовления раствора cpaBHe
ния (а 1 ) (определение ЭПК) или раствора cpaB
нения (а 2 ) (определение дrк), Mr;
212
rосударственная фармакопея Республики Беларусь
т масса навески ФСО этилов020
Х.'
эфира эйкозапентаеновой кислоты, взятая
для приrотовления раствора сравнения (а,)
или масса ФСО этиловоro эфира докозаrек
саеновой кислоты, взятая для приrотовления
раствора сравнения (а 2 ), Mr;
Ах площадь пика эфира эйкозапента
еновой кислоты или докозаrексаеновой кис
лоты на xpoMaTorpaMMe испытуемоro paCT
вора (а);
А площадь пика эфира эйкозапента
Х.'
еновой кислоты на xpoMaTorpaMMe раствора
сравнения (а,) или эфира докозаrексаеновой
кислоты на xpoMaTorpaMMe раствора cpaBHe
ния (а 2 );
А, площадь пика внутреннеro CTaH
дарта на xpoMaTorpaMMe испытуемоro paCT
вора (а);
Ах.з площадь пика BHYTpeHHero CTaH
дарта на xpoMaTorpaMMe раствора сравнения
(а,) (определение ЭПК) или на xpoMaTorpaMMe
раствора сравнения (а 2 ) (определение дrк);
с коэффициент пересчета между эти
ловым эфиром И триrлицеридами:
С=1 ,00 для этиловых эфиров,
С=0,954 для ЭПк.
С=0,957 для дrк.
СУММА OMErA 3 КИСЛОТ
Процентное содержание суммы OMera 3
кислот рассчитывают по формуле, идентифи
цируя соответствующие пики на xpoMaTorpaM
мах:
А ( ЭПК + дrк )
ЭПК + дrк + п 3 ,
А эпк + А ДП<
rдe:
ЭПК процентное содержание ЭПК;
дrк процентное содержание дrк;
Ап з сумма площадей пиков, COOTBeTCTBY
ющих эфирам С18:3 п 3, С18:4 п з, С20:4 п 3,
С21:5 n 3 и С22:5 n 3, на xpoMaTorpaMMe испы
туемоro раствора (Ь);
А эпк площадь пика эфира ЭПК на xpOMa
TorpaMMe испытуемоrо раствора (Ь);
А дrк площадь пика сложноro эфира дrк
на xpoMaтorpaMMe испытуемоro раствора (Ь).
01/2013:20430
2.4.30. ЭТИЛЕнrликоль
И ДИЭТИЛЕнrликоль
В ЭТОКСИЛИРОВАННЫХ
СУБСТАНЦИЯХ
Этоксилированные субстанции MorYT co
держать различные количества этиленrли
коля и диэтиленrликоля, образующихся при
производственном процессе. Метод, описан
ный ниже, может использоваться для коли
чественноro определения ЭТИХ веществ, в
частности в случае следующих поверхност
но активных веществ: рицинолеата MaKpo
rолrлицерина, rидроксистеарата макроroл
rлицерина, rидроксистеарата макроrола 15,
цетостеариловоro эфира наноксинола 9 и Ma
кроroла.
rДЗОВАЯ ХРОМАтоrРАФИЯ (2.2.28).
Раствор внутренне20 стандарта. 30,0 Mr
1,2 пентандиола Р растворяют в ацетоне Р
и доводят до объема 30,0 мл этим же paCTBO
рителем. 1,0 мл полученноro раствора ДOBO
щп ацетоном Р до объема 20,0 мл.
Испытуемый раствор. 0,500 r испытуе
моro образца растворяют в растворе BHYTpeH
неro стандарта и доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (а). 30,0 Mr этuлен
2ликоля Р растворяют в ацетоне Р и ДOBO
дят до объема 100,0 мл этим же раствори
телем. 1,0 мл полученноro раствора доводят
раствором внутреннеro стандарта до объема
10,0 мл.
Раствор сравнения (Ь). rотовят раствор
диэтилеН2ликоля Р с концентрацией, COOT
ветствующей заданному пределу, используя
те же растворители, что и для приrотовления
раствора сравнения (а).
Условия хроматоrрафирования:
колонка: кварцевая капиллярная длиной
30 м и внутренним диаметром 0,53 мм, покры
тая слоем маКР020ла 20 000 Р (толщина слоя
1 мкм);
2аз носитель: 2елий для xpOMaт02pa
фииР;
скорость 2аза носителя: 10 мл/мин;
деление потока: 1 :3;
температура:
Время Температура
(МИН) (ОС)
0-------40 80........200
Колонка 40-------45 200........230
45 5 230
Блок ввода проб 250
Детектор 250
детектор: пламенно ионизационный;
объем вводимой пробы: 2 мкл.
Относительное удерживание (по отноше
нию к 1 ,2 пентандиолу; время удерживания
около 19 мин): этиленrликоль около 0,7;
диэтиленrликоль около 1,3.
01/2013:20431
2.4.32. Общий холестерин в маслах, обоzащенных омеzа З кислотами
213
2.4.31. НИКЕЛЬ
В rидРоrЕНИЗИРОВАННЫХ
РАСТИТЕЛЬНЫХ МАСЛАХ
Атомно абсорбционная спектрометрия
(2.2.23, метод 1).
ВНИМАНИЕ при использовании сосудов
для разложения под высоким давлением и Mи
кроволновых лабораторных печей тщательно
ознакомьтесь с инструкциями производителя
по безопасности и применению.
Реактивы ма2ния нитрат Р и аммония ди
2идрофосфат Р перед использованием должны
быть проверены на содержание никеля. При
расчете содержания никеля в испытуемом об
разце учитывают фактическое содержание
никеля.
Испытуемый раствор. 0,250 r (т) испыту
емоro образца помещают в сосуд для разложе
ния под высоким давлением (из фторированных
полимеров или кварцевоro стекла), прибавляют
6,0 мл азотной кислоты, свободной от никеля, Р
и 2,0 мл концентрироваНН020 раствора вoдopo
да пероксида Р. Аналоrично rотовят контрольный
раствор. Закрытые сосуды помещают в лабора
торную микроволновую печь и обрабатывают по
подходящей проrрамме, например, при 1000 Вт
в течение 40 мин. Перед открытием сосуды ox
лаждают. Прибавляют 2,0 мл KOHцeHтpиpoвaHHO
20 раствора водорода пероксида Р и повторяют
процедуру разложения. Перед открытием сосуды
охлаждают. Содержимое сосуда количественно
переносят в мерную колбу вместимостью 25 мл,
прибавляют 0,5 мл раствора 10 r/л ма2ния Hи
трата Р и 0,5 мл раствора 100 r/л аммония ди2и
дрофосфата Р, доводят водой для xpOMaт02pa
фии Р до объема 25,0 мл и перемешивают.
Раствор сравнения. По 25 мкл, 50 мкл,
75 мкл и 100 мкл этаЛОНН020 раствора никеля
(5 ррт Ni) Р помещают в 4 мерные колбы вмести
мостью 25 мл. В каждую колбу прибавляют 0,5 мл
раствора 10 r/л маения нитрата Р, 0,5 мл paCT
вора 100 r/л аммониядиеидрофосфата Ри 6,0 мл
азотной кислоты, свободной от никеля, Р,
доводят водой для хромат02рафии Р до объема
25,0 мл и перемешивают. Полученные растворы
содержат соответственно 5 нr/мл, 1 О нr/мл, 15 Hrl
мл и 20 нr/мл (ррЬ) никеля.
Нулевой раствор. В мерную колбу вмести
мостью 50 мл помещают 1,0 мл раствора 1 О r/л
ма2ния нитрата Р, 1,0 мл раствора 100 r/л
аммония ди2идрофосфата Р, 12,0 мл азот
ной кислоты, свободной от никеля, Р, д()водят
водой для хромат02рафии Р до объема 50,0 мл
И перемешивают.
Методика. Измеряют поrлощение каждо
ro раствора при 232,0 нм, используя подхо
дящий атомно абсорбционный спектрометр с
rрафитовой печью, оборудованный системой
коррекции фона, пиролитически покрытой кю
ветой и никелевой лампой с полым катодом.
Высушивание образца проводят при темпе
ратуре 120 ос в течение 35 с после подъема
температуры в течение 5 с; озоление прово
дят при температуре 1100 ос в течение 1 О с
после подъема температуры в течение 30 с;
охлаждение проводят при температуре 800 ос
в течение 5 с после уменьшения температу
ры в течение 5 с; атомизацию проводят при
температуре 2600 ос в течение 7 с. Нуле
вой раствор используют для настройки при
бора на ноль. Используя наклон полученных
кривых, определяют концентрации в испытуе
мом и контрольном растворах. При необходи
мости разводят нулевым раствором для полу
чения показаний в калиброванном диапазоне
поrлощения.
Содержание никеля в микроrраммах на
rpaMM (ррm) рассчитывают по формуле:
с.,
,
т.40
rде:
с измеренная концентрация никеля, нr/мл;
, коэффициент разведения испытуемоro
раствора;
т масса навески испытуемоro образца, r.
01/2013:20432
2.4.32. ОБЩИЙ ХОЛЕСТЕРИН
В МАСЛАХ, ОБоrАЩЕННЫХ
OMErA-3 КИСЛОТАМИ
Данный метод может быть использован для
количественноro определения суммы свободно
. ro и этерифицированноrо холестерина в рыбьем
жире, обоrащенном омеrа З кислотами (в виде
сложных этиловых эфиров или триrлицеридов).
rазовая хроматоrрафия (2.2.28).
Исходный раствор внутренне20 cтaHдap
та. 0,15 r (5а) холестана Р растворяют в 2еп
тане Р и доводят до объема 50,0 мл этим же
растворителем.
Рабочий раствор внутренне20 cтaHдap
та. rотовят непосредственно перед исполь
зованием. 1,0 мл исходноro раствора BHyтpeH
нею стандарта доводят 2ептаном Р до объема
10,0 мл.
Исходный раствор холестерина. 30,0 Mr xo
лестерина Р растворяют в 2ептане Р и доводят
до объема 10,0 мл этим же растворителем. По
лученный раствор помещают в сосуды для rазо
вой хроматоrрафии, он может храниться в замо
роженном состоянии до 6 месяцев.
214
Тосударственная фармакопея Республики Беларусь
Таблица 2.4.З2. 1
При20товление испытуемых растворов и растворов сравнения
Испытуемый раствор Раствор сравнения (а) Раствор
менее более либо менее более либо сравнения (Ь)
3 Mr/r равно 3 Mr/r 3 Mr/r равно 3 urfr
ИСХОДНЫЙ раствор + + +
внутреннеro стандарта
Рабочий раствор + +
внутреннеro стандарта
Исходный раствор + +
холестерина
Рабочий раствор +
холестерина
Раствор a токоферола +
Рабочий раствор холестерина. rотовят
непосредственно перед использованием. 1,0 мл
исходноro раствора холестерина доводят zеп
таном Р до объема 10,0 мл.
Раствор а токоферола. 15,0 Mr ФСО
а токоферола доводят 2ептаном Р до объема
10,0 мл.
Испытуемый раствор. 0.100 r испытуемою
образца помещают в кварцевую пробирку BMe
стимостью 15 мл и прибавляют 1,0 мл исходно
rо/рабочеrо раствора BHyтpeHHero стандарта в
зависимости от ожидаемоro содержания холе
стерина в масле (таблица 2.4.З2. 1).
Полученный раствор выпаривают досуха
при температуре 50 ос в слабом токе азота при
перемешивании. Прибавляют 0,5 мл раствора
50 % калия 2идроксида Р и 3,0 мл 96 % спирта Р.
Пробирку укупоривают в атмосфере азота Р и
наrревают при температуре 100 ос в течение 1 ч
при перемешивании. Охлаждают около 1 О мин и
прибавляют 6 мл воды дистиллированной Р. Экс
траrируют 4 раза эфиром Р порциями по 2,5 мл,
перемешивая каждый раз в течение 1 мин с
использованием вихревой мешалки. Объе
диненные эфирные слои помещают в большую
центрифужную пробирку или делительную BO
ронку и промывают 5 мл воды дистиллирован
ной Р, аккуратно перемешивая определенное
количество раз, например, 60 раз. Водную фазу
отбрасывают, к эфирной фазе прибавляют 5 мл
3 % раствора калия 2идроксида Р и аккуратно
перемешивают 20 раз. Водную фазу отбрасыва
ют, к эфирной фазе прибавляют 5 мл воды диc
тиллированной Р и аккуратно перемешивают 20
раз. Эфирную фазе переносят в маленькую цeH
трифужную пробирку, избеrая попадания воды.
В случае образования эмульсии прибавляют He
большое количество натрия хлорида Р для раз
деления фаз.
Полученный раствор выпаривают в слабом
токе азота при аккуратном подоrреве. Остаток
растворяют в 600 мкл этилацетата Р.
В зависимости от ожидаемоro содержания
холестерина в масле, далее растворы ютовят,
как указано ниже:
содержание менее 3 Mrlr: 200 мкл полу
ченноro раствора доводят этилацетатом Р до
объема 2,0 мл;
содержание более либо равно 3 Mr/r: 20 мкл
полученноro раствора доводят этилацета
том Р до объема 2,0 мл.
Раствор сравнения (а). 1,0 мл исходноro/ра
бочеrо раствора внутреннею стандарта и 1,0 мл
исходноro/рабочеro раствора холестерина, в за
висимости от ожидаемоro содержания холесте
рина в масле (таблица 2.4.З2. 1), помещают в
кварцевую пробирку вместимостью 15 мл и об
рабатывают аналоrично испытуемому раствору,
начиная со слов «Выпаривают досуха при TeM
пературе . . .».
Раствор сравнения (Ь). Смешивают 1 ,Омлис
ходноro раствора внутреннеro стандарта, 1,0 мл
исходноro раствора холестерина и 2,0 мл paCT
вора a токоферола. Выпаривают досуха в
слабом токе азота при аккуратном подоrреве.
Полученный остаток растворяют в этилаце
тате Р и доводят до объема 50,0 мл этим же
растворителем. 1,0 мл полученною раствора
доводят этилацетатом Р до объема 10,0 мл.
Полученный раствор в замороженном состоянии
может храниться до 6 месяцев.
Условия хроматоrрафирования:
колонка кварцевая капиллярная длиной
30 м и внутренним диаметром 0,25 мм, покры
тая слоем поли(диметuл)(дифенил)силоксана Р
(толщина слоя 0,25 мкм);
2аз носитель: 2елий для xpOMaт02pa
фииР;
скорость 2аза носителя: 1,3 мл/мин;
температура:
Время Температура
(мин) (ОС)
0 1 170
Колонка 1 38 170 320
38------40 320
Блок ввода 320
проб
Детектор 300
2.5.3. rидроксильное число
215
детектор: пламенно ионизационный;
объем вводимой пробы: 1 мкл.
Приzодность хромат02рафической cиcтe
мы: раствор сравнения (Ь):
разрешение: не менее 1,2 между пиками
холестерина и a токоферола.
Содержание общеro холестерина, Bыpa
женное в милиrpаммах холестерина на rpaMM
масла, рассчитывают по формуле:
A.m 2 .F
A.z.щ.R
rдe:
А 1 площадь пика холестерина на XpoMa
TorpaMMe испытуемоro раствора;
А 2 площадь пика (5а) холестана на xpo
MaTorpaMMe испытуемоro раствора;
т 1 масса навески испытуемоro образца, r;
т 2 масса навески (5а) холестана, взятоro
для приrотовления исходноro раствора BHyтpeH
неro стандарта, r;
F коэффициент разведения: 20 для
масел с ожидаемым количеством холестерина
более либо равным З Mr/r, 2 для масел с ожи
даемым содержанием холестерина менее 3 Mr/r;
R коэффициент отклика.
Коэффициент отклика рассчитывают по
формуле:
.т2
А4 .т з .5
rде:
Аз площадь пика холестерина на xpOMa
TorpaMMe раствора сравнения (а);
А4 площадь пика (5а) холестана на xpo
MaTorpaMMe раствора сравнения (а);
тз масса навески холестерина, взятоro
для приrотовления исходноro раствора холесте
рина.
2.5.
МЕТОДЫ
КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ
01/2013:20501
2.5.1. КИСЛОТНОЕ ЧИСЛО
Кислотным числом /А называют массу (Mr)
калия rидроксида, необходимую для нейтрали
зации свободных кислот, содержащихся в 1 r ис
пытуемоro образца.
10,00 r или указанное в частной статье KO
личество испытуемоro образца (т, r) paCTBO
ряют в 50 мл смеси из равных объемов 96 %
этанола Р и петролейноzо эфира РЗ, пред
варительно нейтрализованной 0,1 М pacтвo
ром калия zидроксида или О, 1 М раствором
натрия zидроксида, если в частной статье нет
друrих указаний, используя в качестве инди
катара 0,5 мл раствора фенолфталеина Р1.
При необходимости используют наrpевание
до температуры около 90 ос для растворения
испытуемоro образца. После растворения ти
труют 0,1 М раствором калия zидроксuда или
0,1 М раствором натрия zидроксида до п<r
явления розоваro окрашивания, не исчезаю
щеro в течение 15 с (п мл титранта). В случае
использования наrревания для достижения
растворения при титровании поддерживают
температуру около 90 ОС.
'А 5,610.п .
т
01/2013:20502
2.5.2. ЭФИРНОЕ ЧИСЛО
Эфирным числом /Е называют массу (Mr)
калия rидроксида, необходимую для омыле
ния эфиров, содержащихся в 1 r испытуемо
ro образца. Эфирное число рассчитывают
исходя из значений числа омыления /s и кис
лотноro числа /А:
'Е 's 'A'
01/2013:20503
2.5.3. rИДРОКСИЛЬНОЕ ЧИСЛО
rидроксильным числом 'ОН называют массу
(Mr) калия rидроксида, эквивалентную массе
кислоты, связывающейся при ацилировании 1 r
испытуемоro образца.
МЕТОД А
Навеску испытуемоro образца Et COOTBeT
ствии с таблицей 2.5.3. 1 (т, r), если иное
количество не указано в частной статье, по
мещают в круrлодонную колбу со шлифом
вместимостью 150 мл. Прибавляют объем
раствора УКСУСН020 аН2идрида Р1, указанный
в таблице 2.5.З. 1.
216
rосударственная фармакопея Республики Беларусь
Таблица 2.5.З. .1
Предполаrаемое значение 'ОН Навеска испытуемоrо образца, r Объем раствора УНСУСН020
аН2идрида Р1, мл
10 100 2,0 5,0
100 150 1,5 5,0
150 200 1,0 5,0
200 250 0,75 5,0
250 300 0,60 или 1,20 5,0 или 10,0
300 350 1,0 10,0
350 700 0,75 15,0
700 950 0,5 15,0
к колбе присоединяют воздушный холо
дильник, помещают в водяную баню, поддержи
вая уровень воды в бане на 2,5 см выше уровня
жидкости в колбе, и наrревают в течение 1 ч.
Колбу извлекают из водяной бани, охлаждают
и через верхний конец воздушноro холодильни
ка прибавляют 5 мл воды Р. При помутнении к
раствору прибавляют пиридин Р до полноro ис
чезновения мути, отмечая израсходованный
объем. Колбу встряхивают, возвращают в водя
ную баню и наrревают в течение 10 мин. Затем
колбу извлекают из водяной бани и охлаждают.
Воздушный холодильник и стенки колбы промы
вают 5 мл спирта Р, предварительно нейтра
лизованноro по раствору фенолфталеина Р1.
Полученный раствор титруют 0,5 М раствором
калия 2идроксида спиртовым, используя в каче
стве индикатора 0,2 мл раствора фенолфтале
ина Р1 (п, мл 0,5 М раствора калия 2идроксида
спиртов020).
Параллельно проводят контрольный опыт
(П 2 мл 0,5 М раствора калия 2идроксида спир
товОiЮ).
/ 28,05. (П 2 п,) /
он + Д'
т
МЕТОД В
Указанную навеску испытуемоro образца
(т, r) помещают в хорошо высушенную кони
ческую колбу вместимостью 5 мл С притертой
пробкой или подходящей пластмассовой проб
кой и прибавляют 2,0 мл реактива пропионо
в020 аН2идрида Р Колбу закрывают, осторожно
встряхивают до растворения вещества и BЫ
держивают в течение 2 ч, если нет друrих указа
ний в частной статье. Вынимают пробку и колбу
вместе с содержимым помещают в коническую
колбу вместимостью 500 мл С широким rорлом,
содержащую 25,0 мл раствора 9 r/л анилина Р в
циКЛ02ексане Р и 30 мл кислоты уксусной ле
дяной Р. Содержимое колбы взбалтывают и BЫ
держивают в течение 5 мин, прибавляют 0,05 мл
раствора кристалличеСК020 фиолетово ю Р и
титруют 0,1 М раствором хлорной кислоты до
появления изумрудно зеленоro окрашивания (п,
мл 0,1 М раствора хлорной кислоты).
Параллельно проводят контрольный опыт
(П 2 мл 0,1 М раствора хлорной кислоты).
/ 5,610'(П' П2)
ОН
т
Количество воды (у, %), содержащейся в ис
пытуемом образце. определяют полумикромето--
дом (2.5.12).
Пересчет rидроксильноrо числа /ОН осущест
вляют по формуле:
I он = (полученное значение аидроксильноао числа) 3 1,1у.
01/2013:20504
2.5.4. ЙОДНОЕ ЧИСЛО
Йодным числом называют массу (r) rалоrе
на в пересчете на йод, которая может связаться
<;:0 100 r испытуемою образца в описанных yc
ловиях.
Если в частной статье нет дРУ2их указа
ний, используют метод А. 3амена метода А
методом В требует проведения валидации.
МЕТОД А
Если в частной статье нет друrих указаний,
навеску испытуемоro образца в соответствии с
таблицей 2.5.4. 1 (т. r), помещают в предвари
тельно высушенную или промытую кислотой
уксусной ледяной Р колбу вместимостью 250 мл
с притертой проб кой и растворяют в 15 мл хло
роформа Р. К полученному раствору очень
медленно прибавляют 25,0 мл раствора йода
бромида Р. Колбу закрывают пробкой и Bыдep
живают в темном месте при част(\м перемешива
нии в течение 30 мин, если нет друrих указаний
в частной статье. Прибавляют 1 О мл раствора
100 r/л калия йодида Р, 100 мл воды Р и титру
ют О, 1 М раствором натрия тиосульфата при
интенсивном перемешивании до светло желтоro
окрашивания, затем прибавляют 5 мл раствора
2.5.5. Перекuсное (пероксидное) число
217
крахмала Р и титруют 0,1 М раствором натрия
тиосульфата по каплям до обесцвечивания
(П 1 мл 0,1 М раствора натрия тиосульфата).
Параллельно проводят контрольный опыт
(п 2 мл 0,1 М раствора натрия тиосульфата).
'/ == 1,269'(П 2 П1) .
т
Таблица 2.5.4. 1
Предполаrаемое Масса навески
значение 1 испытуемоrо образца, r
менее 20 1,0
20 60 0, 0,25
60 100 0,2 0,15
более 100 0,1 ,10
МЕТОД В
Масса испытуемоro образца должна быть
такая, чтобы избыток добавленноro раствора
йода хлорида Р составлял 50 0 % от общеro
количества, Т.е. 100 150 % от связанноro коли
чества.
Если в частной статье нет друrих указа
ний, навеску испытуемоro образца в COOTBeT
ствии с таблицей 2.5.4. 2 (т, r) помещают в
предварительно высушенную или промытую
кислотой уксусной ледяной Р колбу вмести
мостью 250 мл с притертой пробкой и paCT
воряют в 15 мл смеси из равных объемов ци
КЛ02ексана Р и кислоты уксусной ледяной Р.
Если необходимо, образец расплавляют перед
растворением (температура плавления более
50 ОС). Очень медленно прибавляют указан
ный в таблице 2.5.4. 2 объем раствора йода
хлорида Р. Колбу закрывают пробкой и Bыдep
живают в темном месте при частом перемеши
вании в течение 30 мин, если в частной статье
нет друrих указаний. Прибавляют 1 О мл paCT
вора 100 r/л калия йодида Р, 100 мл воды Р и
титруют 0,1 М раствором натрия тиосульфа
та при постоянном интенсивном перемешива
нии до светло желтоro окрашивания. Прибав
ляют 5 мл раствора крахмала Р и продолжают
титрование, прибавляя по каплям О, 1 М pacт
вор натрия тиосульфата до полноro обес
цвечивания (П 1 мл О, 1 М раствора натрия тиo
сульфата ).
Параллельно проводят контрольный опыт
(П 2 мл 0,1 М раствора натрия тиосульфата).
, == 1,269'(П 2 П1) .
/ т
01/2013:20505
2.5.5. ПЕРЕКИСНОЕ
(ПЕРОКСИДНОЕ) ЧИСЛО
Перекисным (пероксидным) числом 'р назы
вают количество миллиэквивалентов активноro
кислорода, соответствующее количеству перок
сидов, содержащемуся в 1000 r испытуемоro об
разца и определяемому методами, приведенны
ми ниже.
Если в частной статье нет дРУ2их указа
ний, используют метод А. 3амена метода А
методом В требует проведения валидации.
МЕТОД А
5,00 r испытуемоro образца (т, r) помещают
в коническую колбу вместимостью 250 мл с при
тертой стеклянной пробкой, прибавляют 30 мл
смеси хлороформ Р кислота уксусная ледя
ная Р (2:3, об/об). Колбу встряхивают до paCTBO
рения испытуемоro образца, прибавляют 0,5 мл
Таблица 2.5.4. 2
Масса (r) навески Масса (r) навески
Предполаrаемое испытуемоrо образца испытуемоrо образца Раствор йода
значение 1. (соответствует избытку (соответствует хлорида (мл)
150 % ICI) избытку 100 % ICI)
<З 10 10 25
3 8,4613 10,5760 25
5 5,0770 6,3460 25
10 2,5384 3,1730 20
20 0,8461 1,5865 20
40 0,6346 0,7935 20
60 0,4321 0,5288 20
80 0,3173 0,3966 20
100 0,2538 0,3173 20
120 0,2115 0,2644 20
140 0,1813 0,2266 20
160 0,1587 0,1983 20
180 0,1410 0,1762 20
200 0,1269 0,1586 20
218
rосударственная фармакопея Республики Беларусь
насыщеНН020 раствора калия йодида Р, встря
хивают в течение ровно 1 мин и прибавляют 30 мл
воды Р. Полученный раствор титруют 0,01 М
раствором натрия тиосульфата, медленно
добавляя титрованный раствор при непрерыв
ном перемешивании почти до полноro исчезно
вения желтоro окрашивания. Затем прибавляют
5 мл раствора крахмала Р и продолжают титро
вать при интенсивном перемешивании до обес
цвечивания раствора (п, мл 0,01 М раствора
натрия тиосульфата).
Параллельно проводят контрольный опыт
(П 2 мл 0,01 М раствора натрия тиосульфата).
Объем 0,01 М раствора натрия тиосульфата Р,
израсходованный на титрование в контрольном
опыте, не должен превышать 0,1 мл.
' р 10.(п 1 П2) .
т
МЕТОД В
Испытания проводят с защитой от света,
спосоБН020 вызывать фотохимические пре
вращения.
Навеску испытуемоro образца (т, r) в COOT
ветствии с таблицей 2.5.5. 1 помещают в кони
ческую колбу с притертой стеклянной пробкой,
прибавляют 50 мл смеси триметилпентан Р
кислота уксусная ледяная Р (2:3, об/об). Колбу
закрывают пробкой и взбалтывают до paCTBope
ния испытуемоro образца. К полученному pac
твору подходящей мерной пипеткой прибавляют
0,5 мл насыщеНН020 раствора калия йодида Р.
Колбу закрывают пробкой, выдерживают в тече
ние 60:t1 с при постоянном тщательном встряхи
вании и прибавляют 30 мл воды Р. Титруют 0,01
М раствором натрия тиосульфата (V 1 , мл),
прибавляя титрованный раствор постепенно
при постоянном и интенсивном перемешивании
почти до полноrо исчезновения желтоro OKpa
шивания йода. Затем прибавляют около 0,5 мл
раствора крахмала Р1 и продолжают титрова
ние при постоянном интенсивном перемешива
нии, особенно вблизи точки эквивалентности, с
целью полноro высвобождения йода из слоя op
rаническоro растворителя. Раствор натрия тио
сульфата прибавляют по каплям до исчезнове
ния синеro окрашивания.
При необходимости, вместо 0,01 М pacт
вора натрия тиосульфата может быть исполь
зован 0,1 М раствор натрия тиосульфата.
Таблица 2.5.5. 1
Предполаrаемое Масса навески испы-
значение ' D TyeMoro образца, r
0 12 2,00 5,OO
12 20 1 ,20 2,00
20 30 0,80 1,20
30 50 0,500 ,800
50 90 0,300 0,500
ПРИМЕЧАНИЕ: для перекисных чисел ЛJ
и более наблюдается задержка от 15 с до 30 с
в обесцвечивании крахмальноro индикатора,
что обусловлено свойством триметилпента
на всплывать на поверхность водной фазы.
Поэтому необходимо время для перемеши
вания растворителя с титрованным paCTBO
ром для полноro высвобождения йода. Для
перекисных чисел более 150 peKOMeHдyeT
ся использовать О, 1 М раствор натрия ти
осульфата. С целью предотвращения pac
слоения фаз и быстроro высвобождения
йода к реакционной смеси допускается при
бавление небольшоro количества (от 0,5 %
до 1,0 % (м/м» эмульrатора с высоким rидро
фильно липофильным балансом (rЛБ) (Ha
.при мер, полисорбат 60).
Параллельно проводят контрольный опыт
(V O ' мл). Если объем титрованноro раствора, из
расходованный на титрование в контрольном
опыте, превышает 0,1 мл, испытание повторяют
со свежеприroтовленными реактивами.
Пероксидное число рассчитывают по фор
муле:
1000'(V 1 Vo).c
' р ,
т
rдe:
с концентрация раствора натрия тиосуль
фата, моль/л.
01/2013:20506
2.5.6. ЧИСЛО ОМЫЛЕНИЯ
Числом омыления /5 называют массу (Mr)
калия rидроксида, необходимую для нейтрали
зации свободных кислот и омыления сложных
эфиров, содержащихся в 1 r испытуемоro об
разца.
Если в частной статье нет друrих указаний,
навеску испытуемоro образца (т, r) в COOTBeT
ствии с таблицей 2.5.6. 1 помещают в колбу из
боросиликатноro стекла вместимостью 250 мл,
снабженную обратным холодильником.
#Если испытуемый образец в целях KOHcep
вации был насыщен уrлерода диоксидом, перед
взвешиванием еro выдерживают в выпаритель
ной чашке в течение 24 ч в вакуум эксикаторе.#
Прибавляют 25,0 мл 0,5 М раствора калия
2идроксида спиртов020 и несколько стеклянных
шариков, присоединяют холодильник и Harpe
вают в течение 30 мин, если иноro не указано в
частной статье. Прибавляют 1 мл раствора фе
нолфталеина Р1 и немедленно титруют (пока
раствор roрячий) 0,5 М раствором хлористово
дородной кислоты (П 1 мл 0,5 М раствора хлори
стоводородной кислоты).
2.5.8. Определение амиННО20 азота в соединениях
219
Параллельно проводят контрольный опыт
(п 2 мл 0,5 М раствора хлористоводородной Kиc
лоты).
18 == 28,05.(п 2 П1) .
т
Таблица 2.5.6. 1
Предполаrаемое Масса навески
значение , испытуемоrо образца, r
<3 20
3 10 12 15
10 0 8 12
40 0 5------8
60 100 3 5
100 200 2,5---------З
200 ЗОО 1 2
ЗОО ОО 0,5---------1
01/2013:20507
2.5.7. НЕОМЫЛЯЕМЫЕ ВЕЩЕСТВА
Термин «неомыляемые вещества» применя
ется к веществам, нелетучим при температуре от
100 ос до 105 ОС, которые экстраrируются opra
ническим растворителем из испытуемою образ
ца после еro омыления. Содержание неомыля
емых веществ рассчитывают в процентах (м/м).
Следует использовать стеклянную посуду
со шлифами без смазки.
Навеску испытуемоro образца (т, r), YKa
занную в частной статье, помещают в колбу
вместимостью 250 мл, снабженную обратным
холодильником. Прибавляют 50 мл 2 М pacт
вора калия 2идроксида спиртов020 Р и Ha
rревают на водяной бане в течение 1 ч, часто
перемешивая круroвыми движениями. Затем
охлаждают до температуры ниже 25 ос и ко'nи
чественно переносят содержимое колбы в дe
лительную воронку с помощью 100 мл воды Р.
Полученный раствор осторожно встряхивают с
тремя порциями эфира, свободН020 от перок
сидов, Р по 100 мл каждая. Все эфирные извле
чения собирают в отдельную делительную BO
ронку, в которую предварительно помещают 40
мл воды Р, осторожно встряхивают в течение
нескольких минут, выдерживают до полноro pac
слоения, после чеrо отбрасывают водный слой.
Эфирный слой дважды промывают водой Р
порциями по 40 мл каждая. Затем тщательно
промывают поочередно 40 мл раствора 30 r/л
калия 2идроксида Р и 40 мл воды Р; повторя
ют данную процедуру три раза. Затем эфирный
слой несколько раз промывают 40 мл воды Р до
отсутствия щелочной реакции по фенолфтале
ину в водном слое. Эфирный слой количествен
но переносят в предварительно взвешенную
колбу с помощью эфира, свободН020 от перок
сидов, Р.
Эфир отrоняют С соответствующими предо
сторожностями и к остатку прибавляют 6 мл aцe
тона Р. Затем аккуратно удаляют растворитель
в потоке воздуха. Остаток в колбе сушат до по
стоянной массы при температуре от 100 ос до
105 ОС, охлаждают в эксикаторе и взвешивают
(а, r).
100'8
Неомыляемые вещества == %.
т
Остаток растворяют в 20 мл 96 % спирта Р,
предварительно нейтрализованноro по pacтвo
ру фенолфталеина Р, и титруют 0,1 М pacтвo
ром натрия 2идроксида спиртовым. Если объем
0,1 М раствора натрия 2идроксида спиртово
20, пошедшеro на титрование, более 0,2 мл, раз
деление слоев прошло не полностью; при этом
взвешенный остаток не может рассматриваться
как «неомыляемые вещества». В этом случае
испытания необходимо повторить.
01/2013:20508
2.5.8. ОПРЕДЕЛЕНИЕ
Аминноrо АЗОТА
В СОЕДИНЕНИЯХ, КОТОРЫЕ
СОДЕРЖАТ ПЕРВИЧНУЮ
АРОМАТИЧЕСКУЮ
Аминоrруппу
Указанное количество испытуемоro образ
ца растворяют в 50 мл кислоты хлористоводо
родной разведенной Р или в друroм указанном
растворителе и прибавляют З r калия бромида
Р. Охлаждают в воде со льдом и затем медлен
но титруют, при постоянном перемешивании,
0,1 М раствором натрия нитрита, поддержи
вая температуру раствора около 15 ОС, если нет
друrих указаний в частной статье.
Конечную точку титрования определяют
электрометрическими методами или с помощью
индикатора, указанноro в частной статье.
#Рекомендуемая скорость прибавления ти
транта в начале титрования около 2 млlмин, В
конце титрования около 0,05 мл/мин.
При потенциометрическом титровании в
качестве индикаторноro электрода применяют
платиновый электрод, электродом сравнения
служит хлоридсеребряный или насыщенный Ka
ломельный электрод.
В качестве внешнею индикатора использу
ют йодкрахмальную бума2У Р. Титрование про
водят, пока через 1 мин, если нет друrих указа
ний в частной статье, после прибавления О, 1 М
220
/осударственная фармакопея Республики Беларусь
раствора натрия нитрита капля титруемо
ro раствора, взятая с помощью стеклянной па
лочки, не будет вызывать синее окрашивание
бумаrи. Параллельно проводят контрольный
опыт.
В качестве внутренних индикаторов исполь
зуют тропеолин 00 (0,2 мл раствора тропеолина
00 Р), тропеолин в смеси с метиленовым синим
(0,2 мл раствора тропеолuна 00 Р и 0,1 мл
раствора метиленов020 сине20 Р) или раствор
нейтральноro красноro 5 r/л (0,1 мл раствора в
начале титрования и 0,1 мл В конце титрования).
Титрование с тропеолином 00 проводят до пе
рехода окраски от красной к желтой, со смесью
тропеолина 00 с метиленовым синим от Kpac
но фиолетовой к roлубой, с нейтральным Kpac
ным от красно фиолетовой к синей. Выдержку
в конце титрования с нейтральным красным YBe
личивают до 2 мин.
Нитритометрическое титрование применя
ют для количественноro определения соеди
нений, содержащих первичную ароматическую
аминоrруппу, для определения rидразидов, а
также ароматических нитросоединений после
предварительноro восстановления нитроrруппы
до аминоrруппы.#
01/2013:20509
2.5.9. ОПРЕДЕЛЕНИЕ АЗОТА ПО ЛЕ
МИНЕРАЛ ЗАЦИИ СЕРНОИ
КИСЛОТОИ
ПОЛУМИКРОМЕТОД
Указанную массу испытуемоro образца (т, r),
содержащеro около 2 Mr азота, помещают в колбу
для сжиrания, прибавляют 4 r измельченной
смеси, состоящей из 100 r калия сульфата Р,
5 r меди сульфата Р и 2,5 r селена Р, и три CTe
клянных шарика. Смывают прилипшие к roрлу
колбы частицы 5 мл кислоты серной Р таким об
разом, чтобы кислота стекала по стенкам колбы.
Содержимое колбы перемешивают круroвыми
движениями. Во избежание чрезмерных потерь
серной кислоты roрло колбы неплотно закрыва
ют пробкой (например, подходящей rрушевидной
пробкой с коротким отростком). Колбу наrревают,
постепенно доводя до кипения с конденсацией
серной кислоты в rорле колбы; при этом необхо
димо следить за тем, чтобы верхняя часть колбы
не переrреВ<'Iлась. Наrревание продолжают в Te
чение 30 мин, если нет друrих указаний в частной
статье. Охлаждают, растворяют твердый OCTa
ток, осторожно прибавляя к смеси 25 мл воды Р,
снова охлаждают и подсоединяют к прибору для
переroнки с водяным паром. Прибавляют 30 мл
раствора натрия 2идроксида KOHцeHтpиpo
ваНН020 Р и немедленно переroняют, пропу
екая пар через смесь. Собирают около 40 мл
отroна в приемник, содержащий 20,0 мл 0,01 М
раствора кислоты хлористоводородной и дo
статочное количества воды Р для тоro, чтобы
конец холодильника был поrружен в жидкость.
В конце переroнки приемник опускают таким об
разом, чтобы конец холодильника находился над
поверхностью жидкости. Необходимо исключить
попадание жидкости на внешнюю поверхность
холодильника из содержимоro приемника.
#По окончании отrонки конец холодильни
ка промывают снаружи небольшим количеством
воды Р, собирая промывную воду в тот же при
емник. #
OтroH титруют 0,01 М раствором натрия 2и
дроксида, используя в качестве индикатора CMe
wанный раствор метuлов020 красно;ю Р до пе
рехода окраски из красно фиолетовой в зеленую
(П I мл 0,01 М раствора натрия 2идроксида).
Испытания повторяют, используя вместо ис
пытуемоro образца около 50 Mr 2ЛЮКОЗЫ Р (П 2 мл
0,01 М раствора натрия 2идроксида).
001401.(п п )
Содержание азота == ' 2 1 %.
т
01/2013:2051 О
2.5.10. МЕТОД сжиrАНИЯ в КОЛБЕ
С КИСЛОРОДОМ
Если нет друrих указаний, колба для сжиrа
ния это коническая колба вместимостью не
менее 500 мл из боросиликатноro стекла с при
тертой пробкой, снабженной держателем для
образца, который может быть, например, плати
новым или платино иридиевым.
Указанноевчастнойстатьеколичествомелко
измельченноro испытуемоro образца помещают
в центр фильтровальной бумаrи размером 30 мм
на 40 мм с небольшой полосой шириной 1 О мм
и длиной 30 мм.
#Фильтровальную бумаry заворачивают в
виде пакетика, оставляя узкую полоску. В случае
испытания жидкостей их помещают в капилляр,
запаянный парафином, или в капсулу из поли
этилена, нитропленки или метилцеллюлозы.
Для труднолетучих жидкостей возможно при
менение двойноro бумажноro пакетика. При ис
пытании мазеподобных образцов применяют
капсулу из нитропленки или пакет из вощеной
бумаrи. Капсулы и капилляры заворачивают в
пакетик из фильтровальной бумаrи, оставляя
узкую полоску. При анализе твердых и мазепо
добных образцов, сroрающих со вспышкой, к Ha
веске прибавляют 3 5 Mr парафина. Приroтов
ленную пробу помещают в держатель.#
2.5.11. Комплексометрическое титрование
221
Если в частной статье указана бумаrа,
пропитанная лития карбонатом, перед исполь
зованием центр бумаrи увлажняют насыщен
ным раствором лития карбоната Р и сушат
в сушильном шкафу. Испытуемый образец за
ворачивают в бумаrу и помещают в держатель
образца. Помещают в колбу воду Р или YKa
занный раствор, предназначенный для поrло
щения продуктов roрения. С помощью трубки,
конец которой находится выше уровня жид
кости, вытесняют воздух из колбы, пропуская
поток кислорода. Смачивают roрловину колбы
водой Р, поджиrают узкий конец фильтроваль
ной бумаrи с соблюдением мер предосторож
ности и закрывают колбу пробкой.
#Для соблюдения мер предосторожности
необходимо: надеть защитные очки, колбу по
местить в предохранительный чехол, YCTaHO
вить защитный экран.#
Во время сжиrания необходимо придер
живать пробку колбы рукой. После сжиrания
колбу энерrично встряхивают до полноro paCT
ворения продуктов roрения. Колбу охлаждают
в течение 5 мин, если нет друrих указаний, и
осторожно открывают. Обмывают дно, стенки
колбы и держатель образца водой Р. Объеди
няют продукты rорения и жидкость после про
мывки. Проводят определение методом, YKa
занным в частной статье.
01/2013:20511
2.5.11. КОМПЛЕКСОМЕТРИЧЕСКОЕ
ТИТРОВАНИЕ
#Комлексометрическое титрование это
rруппа методов титрования, основанных на
реакциях образования растворимых в воде
комплексных соединений.
Комплексонометрическое титрование как
частный случай комплексометрическоro ти
трования основано на реакции комплексо
образования катионов металлов с комплексо
нами аминополикарбоновыми кислотами и
их солями. Образующиеся комплексные coe
динения называют комплексонатами.
Для комплексонометрическоro титрова
ния в качестве титранта обычно применя
ют динатриевую соль этилендиаминтетраук
сусной кислоты, известную под названиями:
натрия эдетат, трилон Б, комплексон 111, хела
тон 111 и др.
Натрия эдетат образует с катионами MHO
roвалентных металлов устойчивые и хорошо
растворимые в воде комплексонаты в стехи
ометрическом отношении 1: 1 и использует
ся для количественноrо определения алюми
ния, висмута, кальция, свинца, маrния и цинка
в лекарственных средствах и фармацевтиче
ских субстанциях.
Индикаторы, применяемые для визуальноro
обнаружения конечной точки титрования, назы
ваются металлоиндикаторами. Большинство из
них (металлохромные индикаторы) являются op
rаническими красителями и обладают свойством
изменять окраску при образовании комплексных
соединений с катионами металлов. Металлоин
дикаторы для комплексонометрии подбирают
таким образом, чтобы их взаимодействие с Ka
тионами определяемых металлов было обрати
мым и устойчивость их комплексов была значи
. тельно меньше устойчивости ком плексонатов ,
образующихся в процессе титрования.
Прямое титрование растворами натрия эде
тата проводят следующим образом: к испытуе
МОМУ раствору, при необходимости нейтрали
зованному, прибавляют буферный раствор для
создания требуемоro значения рН, затем при
бавляют металлоиндикатор. В процессе титро
вания раствором натрия эдетата в конечной
точке титрования окраска раствора изменяется
от окраски комплекса металлоиндикатора с ти
труемым катионом металла до окраски свобод
ноro металлоиндикатора.
При обратном титровании к испытуемому
раствору прибавляют натрия эдетат и ero из
быток оттитровывают при определенном значе
нии рН в присутствии соответствующеro метал
лоиндикатора растворами солей цинка, маrния,
свинца и др.
Допускается использование в качестве ти
транта 0,05 М раствор натрия здетата, что
должно быть указано в частной статье.#
АЛЮМИНИЙ
20,0 мл раствора, указанноro в частной
статье, помещают в коническую колбу вмести
мостью 500 мл, прибавляют 25,0 мл 0,1 М pacт
вора натрия эдетата и 1 О мл смеси из равных
объемов раствора 155 r/л аммония ацетата Р
и кислоты уксусной разведенной Р, кипятят в
течение 2 мин и охлаждают. Прибавляют 50 мл
этанола Р и 3 мл свежеприrотовленноrо раст
вора 0,25 r/л дитизона Р в этаноле Р. Титруют
избыток натрия эдетата О, 1 М раствором цинка
сульфата до перехода зеленовато синеro OKpa
шивания раствора в красно фиолетовое.
1 мл 0,1 М раствора натрия здетата COOT
ветствует 2,698 Mr AI.
#Допускается проводить определение алю
миния по следующей методике.
Навеску испытуемоro образца, эквивалент
ную 0,02 0,03 r алюминия, растворяют в 2 мл
1 М раствора кислоты хлористоводородной и
50 мл воды Р. Прибавляют 50 мл 0,05 М pacт
вора натрия эдетата и нейтрализуют 1 М pac
твором натрия 2идроксида, используя в каче
стве индикатора метиловый красный Р. Раствор
наrревают до кипения и выдерживают на кипя
222
rосударственная фармакопея Республики Беларусь
щей водяной бане в течение 1 О мин, охлаждают,
прибавляют 50 Mr индикаторной смеси ксилено
Л08О20 оранжев020 Р, 5 r 2ексаметилентетра
мина Р и титруют избыток натрия эдетата 0,05 М
раствором свинца нитрата до появления розо
вато--фиолетовоro окрашивания.
1 мл 0,05 М раствора натрия эдетата co
ответствует 1,349 Mr AI.#
ВИСМУТ
Раствор, указанный в частной статье, по
мещают в коническую колбу вместимостью
500 мл, доводят водой Р до объема 250 мл
и, если нет друrих указаний в частной статье,
прибавляют по каплям при перемешивании
раствор аммиака концентрированный Р до
помутнения смеси. Затем прибавляют 0,5 мл
кислоты азотной Р, наrревают при темпе
ратуре около 70 ос до полноro исчезновения
помутнения. Прибавляют около 50 Mr иHдиKa
торной смеси ксиленолов020 оранжев020 Р и
титруют 0,1 М раствором натрия эдетата до
перехода розовато фиолетовоro окрашивания
в желтое.
1 мл 0,1 М раствора натрия эдетата COOT
ветствует 20,90 Mr Bi.
КАЛЬЦИЙ
Раствор, указанный в частной статье, поме
щают в коническую колбу вместимостью 500 мл
И доводят водой Р до объема 300 мл. Прибавля
ют 6,0 мл раствора натрия 2идроксида KOHцeH
трироваНН020 Р и около 15 Mr индикаторной
смеси кальконкарбоновой кислоты Р. Титруют
0,1 М раствором натрия эдетата до перехода
фиолетовоro окрашивания в насыщенно синее.
1 мл 0,1 М раствора натрия эдетата COOT
ветствует 4,008 Mr Са.
#Допускается проводить определение каль
ция по следующей методике.
Навеску испытуемоro образца, эквивалент
ную 0,04 0,05 r кальция, растворяют, как указа
но в частной статье, в воде Р или кислоте хлори
стоводородной разведенной Р, доводят водой Р
до объема 50 мл, прибавляют 10 мл аммиачно
20 буферН020 раствора рН 10,0 Р, 0,1 r иHди
каторной смеси хромовО20 тeMHo cиHe20 Рили
7 капель раствора хромов020 тeMHo cиHe20 Р и
титруют 0,05 М раствором натрия эдетата до
появления сине фиолетовоro окрашивания.
1 мл 0,05 М раствора натрия эдетата co
ответствует 2,004 Mr Са.#
МАrний
Р8СТВОр, указанный в частной статье, поме
щают в коническую колбу вместимостью 500 мл
и доводят водой Р до объема 300 мл. Прибавля
ют 1 О мл аммиаЧН020 буферН020 раствора рН
1 О, О Р и около 50 Mr индикаторной смеси про
травН020 черН020 11 Р (эриохром черный Т).
Раствор наrревают до температуры около 40 ос
и титруют при этой температуре 0,1 М pacтвo
ром натрия эдетата до перехода фиолетовоro
окрашивания в синее.
1 мл 0,1 М раствора натрия эдетата COOT
ветствует 2,431 Mr Mg.
#Допускается проводить определение
маrния по следующей методике:
Навеску испытуемоro образца, эквивалент
ную 0,02 О,ОЗ r маrния, растворяют, как указано
в частной статье. Прибавляют 50 мл воды Р, 1 О мл
аммиаЧН020 буферН020 раствора рН 10,0 Р;
0,1 r индикаторной смеси протравН020 черН020
11 Р (эриохром черный Т) или 7 капель раствора
протравН020 черН020 11 Р (эриохром черный Т)
и титруют 0,05 М раствором натрия эдетата
до появления синеro окрашивания.
1 мл 0,05 М раствора натрия эдетата co
ответствует 1,215 Mr Mg.#
СВИНЕЦ
Раствор, указанный в частной статье, поме
щают в коническую колбу вместимостью 500 мл
и доводят водой Р до объема 200 мл. Прибав
ляют около 50 Mr индикаторной смеси ксилено
лов020 оранжев020 Р, а затем 2ексаметиленте
трамина Р до появления фиолетово розовоro
окрашивания. Титруют 0,1 М раствором натрия
эдетата до перехода фиолетов розовоro OKpa
шивания в желтое.
1 мл 0,1 М раствора натрия эдетата соот
ветствует 20,72 Mr РЬ.
ЦИНК
Раствор, указанный в частной статье, поме
щают в коническую колбу вместимостью 500 мл и
доводят водой Р до объема 200 мл. Прибавляют
около 50 Mr индикаторной смеси ксиленолово
20 оранжев020 Р, а затем 2ексаметилентетра
мина Р до появления фиолетово розовоro OKpa
шивания раствора. После этоro дополнительно
прибавляют 2 r 2ексаметилентетрамина Р и
титруют 0,1 М раствором натрия эдетата до
перехода фиолетово розовоro окрашивания в
желтое.
1 мл 0,1 М раствора натрия эдетата соот
ветствует 6,54 Mr Zп.
01/2013:20512
2.5.12. ВОДА: ПОЛУМИКРОМЕТОД
#(МЕТОД К. ФИШЕРА)#
Определение воды полумикрометодом oc
новано на использовании количественной peaK
ции воды с диоксидом серы и йодом в подходя
щей безводной среде в присутствии основания с
достаточной буферной емкостью.
2.5.12. Вода: полумикрометод
ПРИБОР
Прибор состоит из сосуда для титрования
снабженноro:
двумя одинаковыми платиновыми элек
тродами;
rерметичными входными отверстиями
для ввода растворителя и титранта;
входным отверстием для доступа возду
ха через осушитель;
входным отверстием для испытуемоro
образца с плотно прилеrающей пробкой или,
в случае жидких образцов, с переroродкой.
Также MorYT присутствовать системы для
подачи сухоro азота или для удаления (OTca
сывания) растворителей.
Титрование проводят в соответствии с
инструкцией производителя, прилаrаемой к
при бору. При проведении испытания необхо
димо исключить воздействие на реактивы и
растворители атмосферной влаrи. Конечная
точка титрования определяется с помощью
двух одинаковых индикаторных электродов,
подсоединенных к электрическому источни
ку, поддерживающему между электродами
либо постоянную силу тока, либо постоянное
напряжение. В случае применения прямоrо
метода титрования (метод А), прибавление
титранта вызывает либо уменьшение напря
жения (в случае, если поддерживается посто
янная сила тока), либо увеличение силы тока
(в случае, если поддерживается постоянное
напряжение) до тех пор. пока не будет достиr
нута точка эквивалентности.
Установка титра. В сосуд для титрова
ния помещают высушенный при необходимо
сти метанол Рили друrой растворитель, pe
комендованный производителем титранта. В
зависимости от характеристики прибора yдa
ляют остатки воды из измерительной ячейки
или проводят предварительное титрование.
Прибавляют подходящее количество воды в
подходящем виде (вода Р или сертифициро
ванный стандартный образец) и титруют при
перемешивании в течение необходимоro Bpe
мени. Титр используемоro титранта должен
быть не менее 80 % от указанноro производи
телем. Титр устанавливают перед первым ис
пользованием и затем через подходящие ин
тервалы времени.
Если нет друrих указаний в частной статье,
используют метод А.
Метод А. В сосуд для титрования поме
щают метанол Р или растворитель, KOTO
рый указан в частной статье или peKOMeHДO
ван производителем титранта. В зависимости
от характеристики прибора удаляют остатки
воды из измерительной ячейки или проводят
предварительное титрование. Быстро вносят
испытуемый образец и титруют при переме
223
шивании в течение необходимоro для извле
чения времени.
Метод В. В сосуд для титрования поме
щают метанол Р или растворитель, который
указан в частной статье или рекомендован про
изводителем титранта. В зависимости от xa
рактеристики прибора удаляют остатки воды из
измерительной ячейки или проводят предвари
тельное титрование. Быстро вносят измельчен
ный до необходимой степени испытуемый об
разец. Прибавляют точно отмеренный объем
титранта, взятоro в избытке приблизительно на
1 мл, или объем, указанный в частной статье.
Выдерживают в защищенном от света месте
в течение 1 мин или в течение времени, YKa
занноro в частной статье, при перемешивании.
Избыток реактива титруют метанолом Рили
указанным в частной статье растворителем, co
держащим точно известное количество воды.
Приrодность. Правильность использо
вания выбранноro титранта для определения
содержания воды должна быть верифициро
вана для каждоro испытуемоro образца. Ha
пример, в случае образцов, содержащих воду
в количестве от 2,5 Mr до 25 Mr, применима
следующая методика.
Проводят определение содержания воды
с использованием выбранной системы pe
актив/растворитель. После этоro последо
вательно прибавляют в подходящей форме
известные количества воды Р (не менее 5
прибавлений) и после каждоrо прибавления
определяют общее содержание воды. Рассчи
тывают процентное значение открываемости
(,) в каждой точке по формуле:
W 2
r =100. ,
W 1
rдe:
W 1 масса прибавленной воды, Mr;
W 2 масса найденной воды, Mr.
Рассчитывают линейную зависимость
общеro количества найденной воды от коли
чества прибавленной воды. Находят значение
наклона прямой (Ь), точку пересечения с осью
ординат (а) и точку пересечения экстраполи
рованной калибровочной прямой с осью аб
сцисс (d).
Рассчитывают среднее процентное зна
чение открываемости (' ). Рассчитывают про
цеl-!тные значения ошибок (е 1 и е 2 ) по форму
лам:
a M
е 1 =100. ,
М
Idl M
е 2 =100. ,
М
224
rосударственная фармакопея Республики Беларусь
01/2013:20514
rде:
а величина отрезка, отсекаемоro кали
бровочной прямой на оси ординат, в миллиrрам
мах воды;
d величина отрезка, oTceKaeMoro кали
бровочной прямой на оси абсцисс, в милли
rpaMMax воды;
М содержание воды в испытуемом об
разце, в миллиrраммах воды.
Используемая система реактив/раствори
тель считается приrодной, если:
le11 и le21 не более 2,5 %;
значение Ь находится в диапазоне от
0,975 до 1,025 (отклонение :t2,5 %);
значение r находится в диапазоне от
97,Б % до 102,5 %.
01/2013:20513
2.5.13. АЛЮМИНИЙ В
АДСОРБИРОВАННЫХ
ВАКЦИНАХ
Испытуемый образец roмоrенизируют и KO
личество с предполаrаемым содержанием алю
миния от 5 Mr до 6 Mr переносят в колбу для сжи
rания вместимостью 50 мл. Прибавляют 1 мл
серной кислоты Р, 0,1 мл азотной кислоты Р
и несколько стеклянных шариков. Получен
ный раствор наrревают до выделения плот
ноro белоro дыма. Если на данной стадии Ha
блюдается обуrливание, прибавляют несколько
капель азотной кислоты Р и продолжают ки
пячение до исчезновение окраски. Смесь ox
лаждают несколько минут, с осторожностью
прибавляют 1 О мл воды Р и кипятят до полу
чения прозрачноrо раствора. Охлаждают, при
бавляют 0,05 мл раствора метиловО20 opaH
жевоzо Р и нейтрализуют раствором натрия
2идроксида концентрированным Р (от 6,5 мл
до 7 мл). Если образуется осадок, еro paCTBO
ряют, прибавляя по каплям достаточный объем
серной кислоты разведенной Р. Переносят
раствор в коническую колбу вместимостью
250 мл, промывая колбу для сжиrания 25 мл
воды Р. Прибавляют 25,0 мл 0.02 М раствора
натрия эдетата, 1 О мл ацетатН020 буфер
Н020 раствора рН 4,4 Р, несколько стеклянных
шариков и осторожно кипятят в течение 3 мин,
прибавляют 0,1 мл раствора пиридилазона
фтола Р и титруют roрячий раствор 0,02 М pac
твором меди сульфата до изменения цвета
раствора на пурпурно коричневый.
Проводят контрольный опыт без использо
вания вакцины.
1 мл 0,02 М раствора натрия эдетата co
ответствует 0,5396 Mr AI.
2.5.14. КАЛЬЦИЙ
В АДСОРБИРОВАННЫХ
ВАКЦИНАХ
Все растворы, используемые для даНН020
исследования, должны 20товится с использо
ванием воды Р.
Кальций определяют методом атомно эмис
сионной спектрометрии (2.2.22, метод 1).
Испытуемыйобразецrомоrенизируют. К 1 ,Омл
прибавляют 0.2 мл кислоты хлористоводород
ной разведенной Р и разводят до 3,0 мл водой Р.
Измеряют интенсивность излучения при длине
волны 620 нм.
01/2013:20515
2.5.15. ФЕНОЛ
В ИММУНОСЫВОРОТКАХ
И ВАКЦИНАХ
Испытуемый образец rомоrенизируют и
разводят до необходимоro объема водой Р,
чтобы получить раствор с предполаrаемым
содержанием фенола 15 мкr/мл. rотовят ряд
растворов сравнения с фенолом Р, содержа
щих 5 мкr/мл, 10 мкr/мл, 15 мкr/мл, 20 мкr/мл и
30 мкr/мл фенола соответственно. К 5 мл ис
пытуемоro раствора и к 5 мл каждоro из pac
творов сравнения прибавляют 5 мл буфеРН020
раствора рН 9,0 Р, 5 мл раствора 1 r/л aMи
нопиразолона Р и 5 мл раствора калия фер
рицианида Р. Выдерживают в течение 1 О мин
и измеряют интенсивность окрашивания при
длине волны 546 нм.
Строят rрадуировочный rрафик и рассчи
тывают содержание фенола в испытуемом
образце.
01/2013:20516
2.5.16. БЕЛОК В ПОЛИСАХАРИДНЫХ
ВАКЦИНАХ
Испытуемый раствор. Используют
меrную колбу необходимоro объема для при
rотовления раствора, содержащеro около
5 мr/мл cyxoro полисахарида. Количественно
переносят содержимое контейнера в мерную
колбу и доводят водой Р до метки. 1 мл paCT
вора помещают в стеклянную пробирку, при
бавляют 0,15 мл раствора 400 r/л кислоты
224
rосударственная фармакопея Республики Беларусь
01/2013:20514
rде:
а величина отрезка, отсекаемоro кали
бровочной прямой на оси ординат, в миллиrрам
мах воды;
d величина отрезка, отсекаемоro кали
бровочной прямой на оси абсцисс, в милли
rpaMMax воды;
М содержание воды в испытуемом об
разце, в миллиrраммах воды.
Используемая система реактив/раствори
тель считается приrодной, если:
le 1 1 и 'е21 не более 2,5 %;
значение Ь находится в диапазоне от
0,975 до 1,025 (отклонение :!:2,5 %);
значение r находится в диапазоне от
97,5 % до 102,5 %.
01/2013:20513
2.5.13. АЛЮМИНИЙ В
АДСОРБИРОВАННЫХ
ВАКЦИНАХ
Испытуемый образец rомоrенизируют и KO
личество с предполаrаемым содержанием алю
миния от 5 Mr до 6 Mr переносят в колбу для сжи
rания вместимостью 50 мл. Прибавляют 1 мл
серной кислоты Р, 0,1 мл азотной кислоты Р
и несколько стеклянных шариков. Получен
ный раствор наrревают до выделения плот
ноro белоro дыма. Если на данной стадии Ha
блюдается обуrливание, прибавляют несколько
капель азотной кислоты Р и продолжают ки
пячение до исчезновение окраски. Смесь ox
лаждают несколько минут, с осторожностью
прибавляют 1 О мл воды Р и кипятят до полу
чения прозрачноrо раствора. Охлаждают, при
бавляют 0,05 мл раствора метилов020 opaH
жевО20 Р и нейтрализуют раствором натрия
2идроксида концентрированным Р (от 6,5 мл
до 7 мл). Если образуется осадок, ero paCTBO
ряют, прибавляя по каплям достаточный объем
серной кислоты разведенной Р. Переносят
раствор в коническую колбу вместимостью
250 мл, промывая колбу для сжиrания 25 мл
воды Р. Прибавляют 25,0 мл 0.02 М раствора
натрия эдетата, 10 мл ацетатН020 буфер
Н020 раствора рН 4,4 Р, несколько стеклянных
шариков и осторожно кипятят в течение 3 мин,
прибавляют 0,1 мл раствора пиридилазона
фтола Р и титруют roрячий раствор 0,02 М pac
.rneopoM меди сульфата до изменения цвета
раствора на пурпурно коричневый.
Проводят контрольный опыт без использо
вания вакцины.
1 мл 0,02 М раствора натрия эдетата co
ответствует 0,5396 Mr AI.
2.5.14. КАЛЬЦИЙ
В АДСОРБИРОВАННЫХ
ВАКЦИНАХ
Все растворы, используемые для даНН020
исследования, должны 20товится с использо
ванием воды Р.
Кальций определяют методом атомно эмис
сионной спектрометрии (2.2.22, метод /).
Испытуемыйобразецroмоrенизируют. К 1 ,Омл
прибавляют 0,2 мл кислоты хлористоводород
ной разведенной Р и разводят до 3,0 мл водой Р.
Измеряют интенсивность излучения при длине
волны 620 нм.
01/2013:20515
2.5.15. ФЕНОЛ
В ИММУНОСЫВОРОТКАХ
И ВАКЦИНАХ
Испытуемый образец rомоrенизируют и
разводят до необходимоro объема водой Р,
чтобы получить раствор с предполаrаемым
содержанием фенола 15 мкr/мл. rотовят ряд
растворов сравнения с фенолом Р, содержа
щих 5 мкr/мл, 10 мкr/мл, 15 мкr/мл, 20 мкr/мл и
30 мкr/мл фенола соответственно. К 5 мл ис
пытуемоro раствора и к 5 мл каждоrо из pac
творов сравнения прибавляют 5 мл буфеРН020
раствора рН 9,0 Р, 5 мл раствора 1 r/л aMи
нопиразолона Р и 5 мл раствора калия фер
рицианида Р. Выдерживают в течение 1 О мин
и измеряют интенсивность окрашивания при
длине волны 546 нм.
Строят rрадуировочный rрафик и рассчи
тывают содержание фенола в испытуемом
образце.
01/2013:2051 б
2.5.16. БЕЛОК В ПОЛИСАХАРИДНЫХ
ВАКЦИНАХ
Испытуемый раствор. Используют
мерную колбу необходимоro объема для при
rотовления раствора, содержащеro около
5 мr/мл сухоro полисахарида. Количественно
переносят содержимое контейнера в мерную
колбу и доводят водой Р до метки. 1 мл paCT
вора помещают в стеклянную пробирку, при
бавляют 0,15 мл раствора 400 r/л кислоты
2.5.18. Фосфор в полисахаридных вакцинах
225
трихлоруксусной Р, встряхивают, выдержива
ют в течение 15 мин, центрифуrируют в тече
ние 1 О мин при скорости вращения 5000 об/мин
и отбрасывают надосадочную жидкость. К по
лученному осадку прибавляют 0,4 мл 0,1 М
раствора натрия 2идроксида.
Растворы сравнения. 0,100 r альбумина
бычье20 Р растворяют в 100 мл 0,1 М натрия
2идроксида (основной раствор, содержащий
1 r/л белка). 1 мл OCHOBHOro раствора ДOBO
дят 0,1 М раствором натрия 2идроксида до
объема 20 мл (рабочий раствор 1 с KOHцeHTpa
цией белка 50 мr/л). 1 мл основноro раствора
доводят 0,1 М раствором натрия 2идpOKcи
да до объема 4 мл (рабочий раствор 2 с KOH
центрацией протеина 250 мr/л). В 6 стеклян
ных пробирок помещают 0,10 мл, 0,20 мл и
0,40 мл рабочеro раствора 1 и 0,15 мл, 0,20 мл
и 0,25 мл рабочеro раствора 2. Доводят
объем в каждой пробирке до 0,40 мл, ИСПОЛЬ
зуя О, 1 М раствор натрия 2идроксида.
rотовят КОНТРОЛЬНЫЙ раствор, используя
0,40 мл 0,1 М раствор натрия 2идроксида.
В каждую пробирку прибавляют 2 мл
раствора MeдHo тapтpaтH020 Р3, встря
хивают и выдерживают в течение 10 мин.
В каждую пробирку прибавляют по 0,2 мл
смеси из равных объемов фосфомолибде
новольфрамов020 реактива Р и воды Р,
приrотовленной непосредственно перед ис
пользованием. Закрывают пробирки, переме
шивают, переворачивая их, и дают отстоять
ся в темном месте в течение 30 мин. rолубой
цвет остается стабильным в течение 60 мин.
При необходимости центрифуrируют для по
лучения прозрачных растворов.
Измеряют оптическую плотность (2.2.25)
каждоro раствора при длине волны 760 нм,
используя контрольный раствор в качестве
компенсационноro раствора. Строят rрадуи
ровочный rрафик на основе оптической плот
ности 6 растворов сравнения и COOTBeTCTBY
ющей концентрации белка и с ero помощью
находят содержание белка в испытуемом
растворе.
01/2013:20517
2.5.17. НУКЛЕИНОВЫЕ КИСЛОТЫ
В ПОЛИСАХдРИДНЫХ
ВАКЦИНАХ
Испытуемый раствор. Используют мерную
1C0лбу необходимоro объема для приroтовления
раствора, содержащеro около 5 мr/мл сухоro по
лисахарида. Количественно переносят содержи
мое контейнера в мерную колбу и доводят водой
р до метки.
3.J1( 1712
При необходимости разводят испытуемый
раствор до достижения оптической плотности,
подходящей для используемоro прибора. Изме
ряют оптическую плотность (2.2.25) при длине
волны 260 нм, используя воду Р в качестве KOM
пенсационноro раствора.
Оптическая плотность раствора 1 r/л HY
клеиновой кислоты при длине волны 260 нм
равна 20.
01/2013:20518
2.5.18. ФОСФОР
В ПОЛИСАХдРИДНЫХ
ВАКЦИНАХ
Испытуемый раствор. Используют мерную
колбу необходимоro объема для приroтовления
раствора, содержащеro около 5 мr/мл сухоro
полисахарида. Количественно переносят co
держи мое контейнера в мерную колбу и ДOBO
дят водой Р до метки. Разводят раствор так,
чтобы объем, используемый в опыте (1 мл), co
держал около 6 MKr фосфора. Переносят 1,0 мл
раствора в пробирку из термостойкоro стекла
вместимостью 1 О мл.
Растворы сравнения. 0,2194 r калия ди
2идрофосфата Р растворяют в 500 мл воды Р
для получения раствора с содержанием <poc
фора 0,1 мr/мл. 5,0 мл полученноro раствора
доводят водой Р до объема 100,0 мл. Перено
сят 0,5 мл, 1,0 мл И 2,0 мл разведенноro paCT
вора в три пробирки из термостойкоro стекла.
rотовят контрольный раствор, используя
2,0 мл воды Р в пробирке из термостойкоro
стекла.
Во все пробирки прибавляют по 0,2 мл Kиc
лоты серной Р и подоrревают на масляной
бане при температуре 120 ос в течение 1 ч, а
затем при температуре 160 ос до появления
белоro дыма (около 1 ч). Прибавляют 0,1 мл
кислоты хлорной Р и наrревают при темпера
туре 160 ос до обесцвечивания раствора (около
90 мин). Охлаждают и прибавляют в каждую
пробирку 4 мл воды Р и 4 мл реактива aMMO
ния молибдата Р. Наrревают на водяной бане
при температуре 37 ос течение 90 мин, охлаж
дают и доводят водой Р до объема 10,0 мл. ro
лубой цвет остается стабильным в течение He
скольких часов.
Измеряют оптическую плотность (2.2.25)
I<аждоrо раствора при длине волны 820 нм, ис
пользуя контрольный раствор в качестве KOM
пенсационноrо раствора. Строят rрадуировоч
ный rрафик на основе оптической плотности
6 растворов сравнения и соответствующей KOH
центрации фосфора и с ero помощью находят
содержание фосфора в испытуемом растворе.
226
01/2013:20519
rосударственная фармакопея Республики Беларусь
2.5.19. О-АЦЕТИЛ
В ПОЛИСАХАРИДНЫХ
ВАКЦИНАХ
Испытуемый раствор. Используют мерную
колбу необходимоro объема для приroтовления
раствора, содержащеrо около 5 мr/мл сухоro по
лисахарида. Количественно переносят содержи
мое контейнера в мерную колбу и доводят водой Р
до метки. Разводят раствор так, чтобы объемы,
используемые в опыте, содержали от 30 MKr до
600 MKr ацетилхолина хлорида (О ацетил). По
мещают по О,З мл, 0,5 мл и 1,0 мл полученно
ro раствора в двух повторностях в 6 пробирок
(3 реаrирующих раствора и З корректирующих
раствора).
Растворы сравнения. 0,150 r ацетилхоли
на хлорида Р растворяют в 1 О мл воды Р (OCHOB
ной раствор, содержащий 15 r/л ацетилхолина
хлорида). Непосредственно перед использова
нием 1 мл OCHoBHoro раствора доводят водой
Р до объема 50 мл (рабочая концентрация
1: 300 мкr/мл ацетилхолина хлорида). Непосред
ственно перед использованием 1 мл основноro
раствора доводят водой Р до объема 25 мл (pa
бочая концентрация 2: 600 мкr/мл ацетилхолина
хлорида). Помещают по 0,1 мл И 0.4 мл рабочей
концентрации 1 в двух повторностях (реаrирую
щий и корректирующий растворы) в 4 пробирки
И по 0,6 мл и 1,0 мл рабочей концентрации 2 в
двух повторностях (реаrирующий и корректиру
ющий растворы) в друrие 4 пробирки.
rотовятконтрольный раствор, используя 1 мл
воды Р.
Объем в каждой пробирке доводят водой Р
до 1 мл. Прибавляют 1,0 мл 4 М раствора Kиc
лоты хлористоводородной в каждую из KoppeK
тирующих пробирок и В контрольный раствор.
Прибавляют 2,0 мл раствора 2идроксиламина
щеЛОЧН020 Р в каждую пробирку. Выдерживают
в течение ровно 2 мин для протекания реакции
и прибавляют в каждую из пробирок с реаrирую
щим раствором 1,0 мл 4 М раствора кислоты
хлористоводородной. В каждую пробирку при
бавляют 1,0 мл раствора 100 r/л железа хлори
да Р в 0,1 М растворе кислоты хлористоводо
родной, закрывают пробирки пробками и сильно
встряхивают для удаления пузырьков.
Измеряют оптическую плотность (2.2.25)
каждоro раствора при длине волны 540 нм, ис
пользуя В качестве компенсационноro раствора
контрольный раствор. Для каждоro реаrирующе
ro раствора вычитают оптическую плотность co
ответствующеro корректирующею раствора.
Строят rрадуировочный rрафик на OCHOBa
нии скорректированных оптических плотностей
для 4 растворов сравнения и соответствующе
ro содержания ацетилхолин хлорида и с ero по
мощью определяют содержание ацетилхолина
хлорида в испытуемом растворе для каждою
исследуемоro объема. Рассчитывают среднее
по трем значениям.
1 моль ацетилхолина хлорида (181,7 r) COOT
ветствует 1 моль О ацетила (43,05 r).
01/2013:20520
2.5.20. rЕКСОЗАМИНЫ
В ПОЛИСАХдРИДНЫХ
ВАКЦИНАХ
Испытуемый раствор. Используют мерную
> колбу необходимоro объема для приroтовления
раствора, содержащею около 5 мr/мл сухоro по
лисахарида. Количественно переносят coдep
жимое контейнера в мерную колбу и доводят
водой Р до метки. Разводят раствор так, чтобы
объемы, используемые в опыте. содержали от
125 MKr до 500 MKr rлюкозамина (rексозамина).
Помещают 1,0 мл полученноro раствора в rраду
ированную пробирку.
Растворы сравнения. 60 Mr 2люкозамина
2идрохлорида Р растворяют в 100 мл воды Р (oc
новной раствор. содержащий 0,500 r/л rлюкоза
мина). Помещают по 0,25 мл, 0,50 мл, 0.75 мл. и
1,0 мл рабочей концентрации в 4 rрадуирован
ные пробирки.
rотовят контрольный раствор, используя 1 мл
воды Р.
Объем в каждой пробирке доводят водой Р до
1 мл. В каждую пробирку прибавляют 1 мл paCT
вора (292 r/л) кислоты хлористоводородной Р.
Закрывают пробирки пробками, выдерживают в
водяной бане в течение 1 ч и охлаждают дО KOM
натной температуры. В каждую пробирку прибав
ляют 0,05 мл раствора 5 r/л тимолфталеина Р
в спирте Р. Прибавляют раствор 200 r/л натрия
2идроксида Р до появления roлубоro окрашива
ния, а затем раствор 1 М кислоты хлористово
дородной до обесцвечивания раствора. Объем
в каждой пробирке доводят водой Р до 1 О мл
(нейтрализованные rидролизаты).
Во вторую серию rрадуированных пробирок
объемом 1 О мл помещают по 1 мл каждоro ней
трализованноro rидролизата. В каждую пробирку
прибавляют по 1 мл ацетилацетоновоro реактива
(приroтовленная непосредственно перед исполь
зованием смесь из 1 объема ацетилацетона Р
и 50 объемов раствора 53 r/л натрия карбоната
безводН020 Р). Закрывают пробирки пробками и
выдерживают в водяной бане при температуре
90 ос в течение 45 мин. Охлаждают до KOMHaT
ной температуры. В каждую пробирку прибав
ляют по 2,5 мл спирта Р и по 1,0 мл раствора
диметиламинобензальдеrида (непосредствен
но перед использованием растворяют 0,8 r ди
метиламинобензальде2ида Р в 15 мл спирта Р
и прибавляют 15 мл кислоты хлористоводород
2.5.22. Уроновые кислоты в полисахаридных вакцинах
227
ной Р) и доводят объем в каждой пробирке спир
том Р до 10 мл. Пробирки закрывают, содержи
мое перемешивают, переворачивая пробирки, и
выдерживают в темном месте в течение 90 мин.
Измеряют оптическую плотность (2.2.25) каждо
ro раствора при длине волны 530 нм, используя
контрольный раствор в качестве компенсацион
ноro раствора.
Строят rрадуировочный rрафик на OCHO
вании оптических плотностей для 4 растворов
сравнения и соответствующеro содержания reK
созамина и с еro помощью определяют содержа
ние rексозамина в испытуемом растворе.
01/2013:20521
2.5.21. МЕТИЛПЕНТОЗЫ
В ПОЛИСАХАРИДНЫХ
ВАКЦИНАХ
Испытуемый раствор. Используют
мерную колбу необходимоro объема для при
rотовления раствора, содержащеro около
5 мr/мл сухоro полисахарида. Количественно
переносят содержимое контейнера в мерную
колбу и доводят водой Р до метки. Разводят
раствор так, чтобы объемы, используемые
8 опыте, содержали от 2 MKr до 20 MKr paM
нозы (метилпентозы). Помещают по 0,25 мл,
0,50 мл и 1,0 мл полученноro раствора в 3
пробирки.
Растворы сравнения. 0,100 r рамнозы Р
растворяют в 100 мл воды Р (основной paCT
вор, содержащий 1 r/л метилпентозы). Непо
средственно перед использованием 1 мл oc
HOBHOro раствора доводят водой Р до объема
50 мл (рабочая концентрация: 20 мr/л метил
пентозы). В 5 пробирок помещают по 0,10 мл,
0,25 мл, 0,50 мл, 0,75 мл и 1,0 мл рабочей
концентрации.
rотовят контрольный раствор с использо
ванием 1 мл воды Р.
Объем в каждой пробирке доводят водой Р
до 1 мл. Пробирки помещают в ледяную
воду, прибавляют по каплям при постоян
ном помешивании по 4,5 мл охлажденной
смеси, состоящей из 1 объема воды Р и 6
объемов кислоты серной Р. Пробирки Ha
rревают до комнатной температуры и BЫ
держивают на водяной бане в течение He
скольких минут. Охлаждают до комнатной
температуры. В каждую пробирку прибав
ля ют по 0,10 мл приroтовленноro непосред
ственно перед применением раствора 30 пл
цистеина 2идрохлорида Р. Встряхивают и BЫ
держивают в течение 2 ч.
Измеряют оптическую плотность (2.2.25)
каждоro раствора при длинах волн 396 нм и
430 нм, используя контрольный раствор в Ka
честве компенсационноro раствора. Для каж
доro раствора рассчитывают разницу между
оптической плотностью, измеренной при
длине волны 396 нм, и оптической плотно
стью, измеренной при длине волны 430 нм.
Строят rрадуировочный rрафик на OCHO
вании разницы оптических плотностей для 5
стандартных растворов и соответствующей
концентрации метилпентозы и с ero помощью
определяют содержание метилпентозы в ис
пытуемом растворе для каждоro исследуе
моro объема. Рассчитывают среднее по трем
значениям.
01/2013:20522
2.5.22. УРОНОВЫЕ КИСЛОТЫ
В ПОЛИСАХдРИДНЫХ
ВАКЦИНАХ
Испытуемый раствор. Используют
мерную колбу необходимоro объема для приrо
товления раствора, содержащеro около 5 мr/мл
cyxoro полисахарида. Количественно перено
сят содержимое контейнера в мерную колбу и
доводят водой Р до метки. Разводят раствор
так, чтобы объемы, используемые в опыте, co
держали от 4 MKr до 40 MKr rлюкуроновой кис
лоты (уроновых кислот). Помещают по 0,25 мл,
0.50 мл и 1,0 мл полученноrо раствора в 3 про
бирки.
Растворы сравнения. 50 Mr натрия 2ЛЮКУ
роната р растворяют в 100 мл воды Р (основной
раствор: содержит 0,4 r/л rлюкуроновой кисло
ты). Непосредственно перед использованием
5 мл основноro раствора разводят водой Р до
объема 50 мл (рабочая концентрация: 40 мr/л
rлюкуроновой кислоты). В 5 пробирок помеща
ют по 0,1 О мл, 0,25 мл, 0,50 мл, 0,75 мл, и 1,0 мл
рабочей концентрации.
rотовят контрольный раствор с использо
ванием 1 мл воды Р
Доводят водой Р объем в каждой пробирке
до 1 мл. Помещают пробирки в ледяную воду и
при постоянном перемешивании по каплям при
бавляют в каждую пробирку по 5,0 мл боратно
20 раствора Р. Закрывают пробирки пробками и
выдерживают в водяной бане в течение 15 мин.
Охлаждают до комнатной температуры. В
каждую пробирку добавляют по 0,20 мл paCT
вора 1,25 r/л карбазола Р в этаноле Р. Закры
вают пробирки пробками и выдерживают в BO
дяной бане в течение 15 мин. Охлаждают до
комнатной температуры. Измеряют оптическую
плотность (2.2.25) каждоro раствора при длине
волны 530 нм, используя контрольный раствор
в качестве компенсационноro раствора.
228
rосударственная фармакопея Республики Беларусь
Строят rрадуировочный rрафик оптиче
ских плотностей для 5 стандартных раствсров
и соответствующих концентраций rлюкурон
вой кислоты и определяют содержание rлюку
роновой кислоты в испытуемом растворе для
каждоro исследуемоro объема. Рассчитывают
среднее по трем значениям.
01/2013:20523
2.5.23. СИДЛОВАЯ КИСЛОТА
В ПОЛИСАХАРИДНЫХ
ВАКЦИНАХ
Испытуемый раствор. Используют мерную
колбу необходимоro объема для приroтовления
раствора, содержащеro около 250 мкr/мл сухоro
полисахарида. Количественно переносят содержи
мое одною или нескольких контейнеров в мерную
колбу и доводят водой Р до метки. 4,0 мл получен
ноro раствора переносят с помощью шприца в уль-
трафильтрационную кювету вместимостью 10 мл,
пропускающую молекулы с относительной молеку
лярной массой менее 50 000. Дважды промывают
шприц водой Р и промывную жидкость тоже пере
носят в ультрафильтрационную кювету. Проводят
ультрафильтрацию при постоянном перемешива
нии в присутствии азота Р под давлением около
150 кПа. Наполняют кювету водой Р каждый раз,
коrда объем жидкости в ней уменьшается до 1 мл,
до тех пор, пока не профильтруется 200 мл, а
оставшийся в кювете объем не будет составлять
около 2 мл. ИсполЬЗуЯ шприц, переносят оставшу
юся жидкость в мерную колбу вместимостью 1 О мл.
Кювету трижды промывают водой Р порциями по
2 мл. Смывную жидкость neреносят в ту же мерную
колбу и доводят водой Р до объема 10,0 мл (ис
пытуемый раствор). В две пробирки помещают по
2,0 мл испытуемою раствора.
Растворы сравнения. Используют растворы
сравнения, указанные в частной статье.
r отовят две серии из 3 пробирок. В пробир--
ки каждой серии помещают 0.5 мл, 1,0 мл И 1.5 мл
раствора сравнения, соответствующею типу BaK
цины, предназначенной для исследования, и
объем в каждой пробирке доводят водой Р до 2 мл.
rотовят контрольный раствор с использовани
ем воды Р по 2,0 мл В двух пробирках.
В каждую пробирку прибавляют по 5,0 мл pe
зорцина реактива Р. Наrревают при температу
ре 105 ос в течение 15 мин, охлаждают в холод
ной воде и помещают в баню с ледяной водой. 130
все пробирки прибавляют 5 мл спирта изоамило
в020 Р, тщательно перемешивают и выдерживают
в ледяной воде в течение 15 мин. Центрифуrиру
ют пробирки и держат их в ледяной воде до про
ведения исследования методом абсорбционной
спектрофотометрии. Измеряют оптическую плот
ность (2.2.25) надосадочной жидкости при 580 нм
и 450 нм, используя спирт изоамиловый Р в Ka
честве компенсационноro раствора. Для каждой
длины волны рассчитывают оптическую плотность
как среднее значений, полученных в двух идентич--
ных растворах. Вычитают среднее значение для
контрольною раствора из средних значений, полу
ченных для друrих растворов.
Строят rрафик зависимости разницы
между оптическими плотностями при 580 нм и
450 нм стандартных растворов от содержания
N ацетилнейраминовой кислоты и с еro помощью
определяютсодержаниеN ацетилнейраминовой
кислоты (сиаловой кислоты) в испытуемом pac
творе.
01/2013:20524
2.5.24. уrЛЕРОДА ДИОКСИД В rАЗАХ
rазы поrлощают свет при единственных
характерных длинах волн. Это широко ис
пользующееся свойство rазов позволяет про
водить высокоселективное измерение их KOH
центрации.
Описание и принцип измерения. KOHцeH
трация уrлерода диоксида в друrих rазах опреде
ляется с помощью инфракрасноro анализатора.
Инфракрасный анализатор rлавным обра
зом состоит из источника широкополосноro ин
фракрасноro излучения. оптическою устройства,
ячейки для испытуемоro образца и детектора. Оп
тическое устройство. размещаемое либо перед
ячейкой для испытуемоro образца. либо за ней,
состоит из одноro или нескольких оптических
фильтров, через которые проходит широкополос
ное инфракрасное излучение. В данном случае
используют оптическое устройство для опреде
ления уrлерода диоксида. Измеряемый световой
пучок проходит через ячейку с испытуемым об
разцом и ячейку сравнения. если анализатор Ta
ковой оборудован (в некоторых приборах вместо
ячейки сравнения используется электронная си
стема).
Если в ячейке с испытуемым образцом при
сутствует уrлерода диоксид, происходит поrлоще
ние энерrии измеряемоro световоro пучка в co
ответствии с законом Буrера Ламберта Бера, что
при водит к изменению сиrнала детектора. Изме
ренный сиrнал сравнивается с сиrналом cpaBHe
ния и на выходе образуется сиrнал, отражающий
содержание уrлерода диоксида. Выходной сиrнал
линеаризируется таким образом, чтобы получить
концентрацию уrлерода диоксида. Для предот
вращения попадания на датчики твердых частиц,
приводящих к явлению светорассеяния, прибор
снабжается подходящим фильтром.
2.5.25. Уzлерода монооксид в Z8зах
229
Требования к техническим характеристи-
кам. Для определения предельных содержаний
инфракрасный анализатор должен выдерживать
следующие требования:
предел обнаружения: (обычно определя
емый по отношению сиrнал/шум более 2) не
более 20 % от максимально допустимой KOH
центрации;
повторяемость: относительное CTaндapT
ное отклонение для максимально допустимой
концентрации не более 1 О %, определенное на
шести повторных определениях;
линейность: не более 1 О % от максимально
допустимой концентрации.
Требования к техническим характеристикам
должны выдерживаться и в присутствии приме
сей друrих rазов в испытуемом образце.
01/2013:20525
2.5.25. уrЛЕРОДА МОНООКСИД
в rАЗАХ
МЕТОД I
Прибор. Прибор (рисунок 2.5.25. 1) состоит
из следующих частей, соединенных последова
тельно:
U образная трубка (И 1 ), содержащая без
водный силика2ель Р, пропитанный хрома (V/)
оксидом Р;
промывочная емкость (F I ), содержащая
100 мл раствора 400 r/л калия 2идроксида Р;
U1
F1
U2
11
11
II
1 ,...
"'1 '_
-= I 1
100мл
-:1 i
... I 1
I
11
:,I
. ':"
U образная трубка (И 2 ), содержащая ша
рики калия 2идроксида Р;
U образная трубка (Из), содержащая фос
фора (V) оксид Р, распределенный на предвари
тельно rранулированную плавленную пемзу;
U образная трубка (И 4 ), содержащая 30 r
йода (V) оксида перекристаллизоваНН020 Р в
rранулах, предварительно высушенноro при TeM
пературе 200 ос и поддерживаемоro при темпе
ратуре 120 ос (1) во время испытания. Йода (V)
оксид упакован в трубке в односантиметровые
столбики, разделенные односантиметровыми
столбиками стекловаты для достижения эффек
тивной длины 5 см;
реакционная трубка (F 2 ), содержащая 2,0 мл
раствора калия йодида Р и 0,15 мл раствора
крахмала Р.
Методика. 5,0 л ар20на Р пропускают
через аппарат и при необходимости обесц
вечивают roлубой цвет йодноro раствора,
прибавляя минимально необходимое коли
чество свежеприroтовленноro 0,002 М pacт
вора натрия тиосульфата. rаз продолжают
пропускать до тех пор, пока после прохожде
ния 5,0 л ар20на Р для обесцвечивания йод
Horo раствора будет требоваться не более
0,045 мл 0,002 М раствора натрия тиосуль
фата. Испытуемый rаз пропускают из ци
линдра через аппарат, учитывая указанные
в частной статье объем и скорость потока.
Остатки выделившеroся йода в реакционной
пробирке извлекают, пропустив через аппа
рат 1,0 л ар20на Р. Выделившийся йод титру
ют 0,002 М раствором натрия тиосульфа
та. Проводят контрольный опыт, используя
одинаковый объем ар20на Р. Разница между
объемами 0,002 М раствора натрия тиo
UЗ
U4
F2
. ....
_"" _
== .I"'
.. ...
8
Рисунок 2.5.25. 1. Прибор для определения У2лерода монооксида
(размеры указаны в миллиметрах)
1(1 За... 1712
230
rосударственная фармакопея Республики Беларусь
сульфата, использованноro при ТИТр09ани
ях, не должна превышать указанное в ч зст
ной статье значение.
МЕТОД 11
rазы поrлощают свет при единственных xa
рактерных длинах волн. Это широко использу
ющееся свойство rазов позволяет проводить
высокоселективное измерение их KOHцeHTpa
ции.
Описание и принцип измерения. KOH
центрация уrлерода монооксида в друrих rазах
определяется с помощью инфракрасною анали
затора.
Инфракрасный анализатор rлавным об
разом состоит из источника широкополос
ноro инфракрасноro излучения, оптическою
устройства, ячейки для испытуемоro образца
и детектора. Оптическое устройство, размеща
емое либо перед ячейкой для испытуемоro об
разца, либо за ней, состоит из одноro или He
скольких оптических фильтров, через которые
проходит широкополосное инфракрасное излу
чение. В данном случае используют оптическое
устройство для определения уrлерода MOHO
оксида. Измеряемый световой пучок проходит
через ячейку с испытуемым образцом и ячейку
сравнения, если анализатор таковой оборудо
ван (в некоторых приборах вместо ячейки cpaB
нения используется электронная система).
Если в ячейке с испытуемым образцом при
сутствует уrлерода монооксид, происходит по
rлощение энерrии измеряемоro световоro пучка
в соответствии с законом Буrера Ламберта Бе
ра, что при водит к изменению сиrнала дeTeKTO
ра. Измеренный сиrнал сравнивается с сиrна
лом сравнения и на выходе образуется сиrнал,
Фильтр для
ввода пробы
Преобразователь
N02"""';:' NO
Насос
-< Q- ..
Фильтр для
удаления озона
reHepaTop
озона
отражающий содержание уrлерода моноокси
да. Выходной сиrнал линеаризируется таким об
разом, чтобы получить концентрацию уrлерода
монооксида. Для предотвращения попадания на
датчики твердых частиц, приводящих к явлению
светорассеяния, прибор снабжается подходя
щим фильтром.
Требования к техническим характеристи
кам. Для определения предельных содержаний
инфракрасный анализатор должен выдерживать
следующие требования:
предел обнаружения: (обычно опреде
ляемый по отношению сиrнал/шум более 2) не
более 20 % от максимально допустимой KOHцeH
трации;
повторяемость: относительное CTaHдapT
ное отклонение для максимально допустимой
концентрации не более 10 %. определенное на
шести повторных определениях;
линейность: не более 1 О % от максималь
но допустимой концентрации.
Требования к техническим характеристикам
должны выдерживаться и в присутствии приме
сей друrих rазов в испытуемом образце.
01/2013:20526
2.5.26. АЗОТА МОНООКСИД И АЗОТА
ДИОКСИД В rАЗАХ
Азота монооксид и азота диоксид в rазах
определяется хемилюминесцентным анализато
ром (рисунок 2.5.26. 1).
Реrулировка скорости пробы
Фотоумно
жительная
трубка
Контрольная панель
NO (NO+N02)
NO
(NO+N02)
N02
Рисунок 2.5.26. 1. Хемилюминесцентный анализатор
2.5.29. Серы диоксид
231
Конструкция аппарата включает в себя:
устройство для фильтрования, проверки и
контроля скорости потока испытуемоro rаза;
преобразователь, восстанавливающий
азота диоксид в азота МОНООКGИд для определе
ния суммарноro содержания азота монооксида и
азота диоксида; перед использованием необхо
димо проверить эффективность преобразовате
ля;
reHepaTop озона с контролем скорости
потока; озон производится высоковольтными
электрическими разрядами между двумя элек
тродами; в reHepaTop озона подается чистый
кислород или безводный воздух окружающей
среды, а концентрация озона должна HaMHO
ro превышать максимальное содержание всех
определяемых оксидов азота;
камеру, в которой взаимодействуют азота
монооксид с озоном;
систему определения слабоro излучения
при длине волны 1,2 мкм, состоящую из селек
тивноro оптическоro фильтра и фотоумножи
тельной трубки.
01/2013:20527
2.5.27. КИСЛОРОД В rдзАХ
Кислород в rазах определяется с помощью
парамаrнитноro анализатора.
Принцип метода основан на высокой па
рамаrнитной чувствительности молекулы кис
лорода. Кислород проявляет сильное вза
имодействие с маrнитными полями. Это
взаимодействие измеряется количеством элек
тричества, зависящим от величины содержа
ния кислорода. Измерения концентрации КИС
лорода зависят от давления и температуры,
и, если анализатор автоматически не реаrиру
ет на изменения температуры и давления, He
обходимо произвести rрадуировку прибора He
посредственно перед самым использованием.
Поскольку парамаrнитный эффект кислорода
имеет линейный характер, прибор должен по
зволять проводить измерения в подходящем
диапазоне с возможностью считывания показа
ний до 0,1 % и ниже.
,радуировка прибора. rрадуировка произво
дится следующим образом:
устанавливают нулевой уровень, пропу
ская через прибор азот Р1 до получения посто
янных показателей;
устанавливают шкалу на 100 % пропуская
через прибор кислород Р со скоростью потока,
аналоrичной используемой для азота Р1, до по
лучения постоянных показателей.
Количественное определение. Пропускают
испытуемый rаз через прибор с постоянной CKO
ростью потока до получения стабильных показа
телей. Записывают концентрацию кислорода в
испытуемом rазе.
01/2013:20528
2.5.28. ВОДА В rАЗАХ
Вода в rазах определяется с помощью элек
тролитическоro rиrрометра, описанноro ниже.
Измерительная камера состоит из тонкой
пленки фосфора (V) оксида, расположенной
между 2 платиновыми Пр080дами в качестве
электродов. Водный пар в испытуемом rазе по
rлощается фосфора (V) оксидом, КОТОРЫЙ пре
образуется в фосфорную кислоту, являющуюся
электрическим проводником. Постоянное напря
жение BOKpyr электродов вызывает электролиз
воды и реrенерацию фосфора (V) оксида. Затем
измеряется возникающий электрический ток, KO
торый пропорционален содержанию воды в ис
пытуемом rазе. Данная система является caMO
калибрующейся, поскольку она соответствует
закону Фарадея.
Берут образец испытуемоrо rаза и оставляют
еro для уравновешивания при комнатной темпе
ратуре. Постоянно пропускают rаз через камеру,
пока не получат стабильные показатели. Изме
ряют содержание воды в испытуемом rазе. Необ
ходимо следить, чтобы в течение всеro периода
работы прибора температура была постоянной.
01/2013:20529
2.5.29. СЕРЫ ДИОКСИД
В колбу (А) вливают 150 мл воды Р (рисунок
2.5.29. 1) и через всю систему пропускают У2ле
рода диоксид Р в течение 15 мин со скоростью
100 мл/мин. К 1 О мл раствора водорода перокси
да разведеНН020 Р прибавляют 0,15 мл раствора
1 r/л бромофеНОЛО8020 сине20 Р в спирте Р (20 %,
об/об). Затем прибавляют 0,1 М раствор натрия
2идроксида Р до появления фиолетово сине
ro оттенка. Раствор помещают в пробирку (О).
Не прерывая поток диоксида уrлерода, снимают
воронку (В) и через образовавшееся отверстие
вводят в колбу (А) 25,0 r испытуемою образца
при помощи 100 мл воды Р. Через воронку при
бавляют 80 мл кислоты хлористоводородной
разведенной Р и кипятят в течение 1 ч. OTKpЫBa
ют пробку воронки И останавливают поток диок
сида уrлерода, а также наrревание и охлаждение
воды. Переносят содержимое пробирки с неболь
232
rосударственная фармакопея Республики Беларусь
шим количеством воды Р в коническую колбу
вместимостью 200 мл с широким roрлом. К:->лбу
наrревают на водяной бане в течение 15 MIIH
и выдерживают до охлаждения. Прибавляют
0,1 мл раствора 1 r/л бромфенолов020 сине20 Р
в спирте Р (20 %, об/об) и титруют О, 1 М pac
твором натрия 2идроксuда до изменения цвета
с желтоro на фиолетово синий (V 1 , мл).
Параллельно проводят контрольный опыт
(V 2 , мл).
Содержание серы диоксида в частях на мил
лион рассчитывают по формуле:
320ЗО. ( V v ) . ,
1 2 т
rдe:
п молярность раствора натрия rидрокси
да, использованноro в качестве титранта.
Е
о
i
I
I
:;1
I
с
C
Рисунок 2.5.29. 1. Прибор для определения
серы диоксuда
01/2013:20530
2.5.30. ОКИСЛЯЮЩИЕ ВЕЩЕСТВА
В коническую колбу с притертой пробкой
вместимостью 125 мл вносят 4,0 r испытуемою
образца и прибавляют 50,0 мл воды Р. Закрыва
ют пробкой и взбалтывают в течение 5 мин. Пе
реносят содержимое в центрифужную пробирку
вместимостью 50 мл с притертой пробкой и цeH
трифуrируют. 30 мл прозрачной надосадочной
жидкости переносят в коническую колбу вмести
мостью 125 мл с притертой пробкой, прибавляют
1 мл кислоты уксусной ледяной Р и от 0,5 r до
1,0 r калия йодида Р. Закрывают пробкой, взбал
тывают и выдерживают в течение 25 ЗО мин в
темном месте. Прибавляют 1 мл раствора Kpax
мала р и титруют 0,002 М раствором натрия
тиосульфата до исчезновения окраски.
Параллельно проводятконтрольный опыт. На
титрование расходуется не более 1,4 мл 0,002 М
раствора натрия тиосульфата (0,002 % в пе
ресчете на H 2 0J
1 мл 0,002 М раствора натрия тиосульфа
та соответствует 34 MKr окисляющих веществ в
пересчете на водорода пероксид.
01/2013:20531
2.5.31. РИБОЗА
В ПОЛИСАХдРИДНЫХ
ВАКЦИНАХ
Испытуемый раствор. Для приroтовле
ния раствора, содержащеro около 5 мr/мл
сухоro полисахарида, используют мерную
колбу необходимоro объема. Количественно
переносят содержимое контейнера в мерную
колбу и доводят водой Р до метки. Разводят
раствор так, чтобы объемы, используемые в
опыте, содержали от 2,5 MKr до 25 MKr рибозы.
Помещают по 0,20 мл и по 0,40 мл полученно
ro раствора в пробирки в трех повторностях.
Растворы сравнения. 25 Mr рибозы Р
растворяют в воде Р и доводят до объема
100,0 мл этим же растворителем (основной
раствор: 0,25 r/л рибозы). Непосредственно
перед использованием 1 мл основноro paCT
вора доводят водой Р до объема 10,0 мл (рабо
чая концентрация: 25 мr/л рибозы). Помеща
ют 0,10 мл, 0,20 мл, 0,40 мл, 0,60 мл, 0,80 мл
и 1,0 мл рабочей концентрации в шесть про
бирок.
rотовят контрольный раствор с использова
нием 2 мл воды Р.
Доводят объем в каждой пробирке водой Р
до 2 мл. Встряхивают. Прибавляют в каждую про
бирку 2 мл раствора 0,5 r/л железа хлорида Р в
кислоте хлористоводородной Р и встряхивают.
Прибавляют 0,2 мл раствора 100 r/л орцина Р
в этаноле Р. Пробирки выдерживают в водяной
бане в течение 20 мин. Охлаждают в ледяной
воде. Измеряют оптическую плотность (2.2.25)
каждоro раствора при длине волны 670 нм,
используя контрольный раствор в качестве KOM
пенсационноro раствора. На основании показа
2.5.33. Общий белок
2ЗЗ
телей 6 растворов сравнения и соответствующе
ro содержания рибозы строят rрадуировочный
rрафик и с ero помощью определяют содержа
ние рибозы в испытуемом растворе для каждо
ro объема. Рассчитывают среднее по трем зна
чениям.
01/2013:20532
2.5.32. ВОДА: МИКРООПРЕДЕЛЕНИЕ
ПРИНЦИП
Кулонометрическое титрование воды oc
нова но на количественной реакции воды с
сернистым rазом и йодом в безводной среде в
присутствии основания с большой буферной
емкостью. В отличие от объемноro метода
(2.5.12), йод получается электрохимическим
способом в реакторной камере. Йод, получае
мый на аноде, сразу взаимодействует с водой
и диоксидом серы, содержащимися в peaK
торной камере. Количество воды в веществе
прямо пропорционально количеству электри
чества, выделенноro до конечноro результата
титрования. Конечная точка титрования опре
деляется по появлению избытка йода после
тою, как расходуется вся вода, присутствую
щая в реакторной камере. Один моль йода co
ответствует одному моль воды, а количество
электричества 10,71 Кл соответствует 1 Mr
воды.
Вода удаляется из системы предвари
тельным электролизом. Индивидуальные
определения MorYT выполняться последова
тельно в том же растворе peareHTa при BЫ
полнении следующих условий:
каждый компонент испытуемой смеси
совместим с друrими компонентами;
друrих реакций не происходит;
объем и емкость воды электролитиче
скою peareHTa достаточны.
Кулонометрическое титрование оrрани
чено количественным определением малых
объемов воды (рекомендуется от 1 О MKr
до 10 Mr).
Точность метода предопределяется CTe
пенью, до которой атмосферная влажность
исключается из системы. Система должна
контролироваться измерением количества
базовоro отклонения.
ПРИБОР
Прибор состоит из реакторной камеры, элек
тродов и маrнитной мешалки. Реакторная камера
состоит из большоro анодноro отдела и меньше
ю катодною отдела. В зависимости от KOHCTPYK
цИИ электрода оба отделения MOryт быть разде
31 За. 1712.
лены диафраrмой. Каждое отделение содержит
платиновый электрод. Жидкость или paCTBopeH
ные образцы вводятся через септу при помощью
шприца. В качестве альтернативы может ис
пользоваться техника выпаривания, при которой
проба наrревается в трубке (печке) и вода выпа
ривается и переносится в реакторную камеру по
током сухоro инертноro rаза. Следует избеrать
попадания в камеру твердых образцов. Однако,
если существует такая необходимость, исполь
зуют rерметично закрывающееся отверстие; при
этом для предотвращения попадания атмосфер
ной влаrи должны быть приняты меры предосто
рожности, такие как, например, работа в защит
ной камере с перчатками в атмосфере сухоro
инертною rаза. Процесс анализа должен KOHTpO
лироваться с помощью электронноro приспосо
бления, которое также показывает и результаты.
МЕТОД
Отделение реакторной камеры заполняют
электролитическим реактивом для микроопре
деления воды Р в соответствии с инструкциями
производителя и выполняют кулонометрическое
титрование до получения стабильноro результа
та. В реакторную камеру вносят соответствую
щее количество испытуемоro образца, переме
шивают в течение 30 с, если в частной статье не
указано иное, и снова титруют до получения CTa
бильноrо результата. В случае использования
печи необходимое количество образца вносят в
трубку и наrревают. После выпаривания воды из
образца в титрующую камеру проводят титрова
ние. Записывают показания прибора и, при He
обходимости, рассчитывают процентный состав
или количество воды, присутствующее в образ
це. При необходимости, Korдa этоro требует тип
образца или еro пробоподroтовка, проводят KOH
трольный опыт.
ПОДТВЕРЖДЕНИЕ ТОЧНОСТИ
Между двумя последовательными титрова
ниями образца вносят тщательно взвешенное
количество воды Рили стандартН020 pacт
вора для микроопределения воды Р тою же по
рядка, что и в образце, и производят кулономе
трическое титрование. Степень открываемости
должна быть в диапазоне от 97,5 % до 102,5 %
для навески 1000 MKr Н 2 О и в диапазоне от
90,0 % до 110,0 % для навески 100 MKr нр.
01/2013:20533
2.5.33. ОБЩИЙ БЕЛОК
Мноrие из методов анализа, которые описа
ны в данном разделе, MOryT выполняться с ис
пользованием коммерчески доступных наборов.
234
rосударственная фармакопея Республики Беларусь
МЕТОД 1
Белок в растворе поrлощает ультрафи')ле
товый свет при длине волны 280 нм блаrо.ца
ря присутствию в ею структуре ароматических
аминокислот, rлавным образом тирозина и трип
тофана. Это свойство белков может использо
ваться в целях их количественною определе
ния. Если буферный раствор, используемый
для разведения белка, имеет большую оптиче
скую плотность относительно плотности воды,
в нем присутствует мешающее вещество. Этот
мешающий эффект можно устранить, исполь
зуя буферный раствор в качестве компенсаци
онноro раствора, однако, если мешающее Be
щество имеет высокую оптическую плотность,
результаты MOryт быть подверrнуты сомнению.
При низких концентрациях белок может сорби
роваться на кювете, что приводит к существен
ному занижению концентрации еro в растворе.
Этоro можно избежать, roтовя образцы с BЫCO
кой концентрацией или используя детерrенты
неионноro характера.
Испытуемый раствор. Необходимое коли
чество испытуемою образца растворяют в YKa
занном в частной статье буферном растворе
для получения раствора с концентрацией белка
в диапазоне от 0.2 мr/мл до 2 мr/мл.
Раствор сравнения. rотовят оаствор CTaH
дартноro образца. предназначенноrо для опре
деления белка, в том же буферном растворе и
той же концентрации, что и у испытуемою paCT
вора.
Методика. Во время выполнения дaHHO
ro исследования испытуемый раствор, раствор
сравнения и компенсационный раствор хранят
при одинаковой температуре. Определяют опти
ческие плотности (2.2.25) испытуемоro раствора
и раствора сравнения в кварцевой кювете при
длине волны 280 нм. используя указанны!/' бу
ферный раствор в качестве компенсационною
раствора. Отклик должен быть линейным в ди
апазоне концентраций определяемоro белка.
Рассеяние света. Точность определения
белка может быть снижена из за светорассеяния
раствором испытуемоro образца. Если белок в
растворе присутствует в виде частиц, сопостави
мых по размерам с длиной волны используемоro
света (от 250 нм до 300 нм), рассеяние световоro
потока при водит к значительному увеличению оп
тической плотности испытуемою образца. Чтобы
рассчитать оптическую плотность при длине
волны 280 нм с учетом светорассеяния, опреде
ляют оптическую rJЛОТНОСТЬ испытуемоro раст
вора при 320 нм, 325 нм, 330 нм, 335 нм, 340 нм,
345 нм и 350 нм. Строят rрафик зависимости ло
rарифма оптической плотности от лоrарифма
длины волны и с помощью линейной реrрессии
находят кривую, наилучшим образом совпадаю
щую с нанесенными точками. Для определения
лоrарифма оптической плотности при 280 нм
экстраполируют полученную кривую. Антило
rарифмом данноro значения является оптиче
ская плотность, характеризующая CBeTopacce
яние. Для получения оптической плотности
белка в растворе вычитают оптическую плот
ность, характеризующую светорассеяние, из
общей оптической плотности, измеренной при
длине волны 280 нм. Для снижения эффекта
светорассеяния MorYT применяться фильтрова
ние с использованием не поrлощающеro белок
фильтра с размером пор 0,2 мкм или осветление
центрифуrированием, особенно в случае явной
мутности раствора.
Расчеты. Для расчетов используют OTKOp
ректированные значения оптической плотности.
Рассчитывают концентрацию белка в испытуе
мом растворе (С И > по уравнению:
С,) C s (Аи / Ао ),
rдe:
C s концентрация белка в растворе cpaB
нения;
Al! и As откорректированные значения оп
тических плотностей испытуемоro раствора и
раствора сравнения соответственно.
МЕТОД 2
Данный метод (обычно называемый MeTO
дом Лоури) основан на восстановлении белком
xpoMoreHa (смешанной кислоты фосфорномо
либденово вольфрамовой) в фосфорномолиб
деново вольфрамовом реактиве, что при водит
к появлению максимума оптической плотно
сти при длине волны 750 нм. Фософорномо
либденово вольфрамовый реактив реаrирует
преимущественно с остатками тирозина. Раз
витие окраски достиrает своеro максимума
через 20 30 мин при комнатной температуре,
после чеro происходит постепенное обесцвечи
вание. Поскольку данный метод является чув
ствительным к мешающим веществам, можно
использовать процедуру для осаждения белка
из испытуемоro образца. Большинство меша
ющих веществ вызывают ослабление окраши
вания, хотя применение некоторых детерrен
тов может вызывать усиление цвета. Высокая
концентрация солей может приводить к обра
зованию осадка. Поскольку различные белки
MorYT давать цветные реакции различной ин
тенсивности, стандартный образец и испыту
емый белок должны быть одинаковыми. В тех
случаях, Korдa необходимо отделение мешаю
щих веществ от белкз в испытуемом образце,
перед тем как приroтовить испытуемый раствор
используют процедуру удаления мешающих Be
ществ, описанную ниже. Эффект мешающих
веществ может быть минимизирован с помо
щью разведения, обеспечивающеro KOHцeHTpa
цию испытуемоro белка на уровне, достаточном
для проведения точных измерений.
2.5.33. Общий белок
235
Для приrотовления всех реактивов и буфер
ных растворов, применяемых в данном методе,
используют воду дистиллированную Р.
Испытуемый раствор. Достаточное коли
чество испытуемоro образца растворяют в YKa
занном в частной статье буферном растворе для
получения раствора с концентрацией в пределах
интервала стандартною rрафика. При использо
вании указанною буферноro раствора приroтов
ленный раствор имеет рН от 10,0 до 10,5.
Растворы сравнения. Стандартный обра
зец, предназначенный для определения белка,
растворяют Б указанном в частной статье буфер
ном растворе. Полученный раствор разводят
тем же буферным раствором, чтобы получить не
менее пяти растворов сравнения с концентраци
ями белка, равномерно распределенными в He
обходимом диапазоне в интервале от 5 мкr/мл
до 100 мкr/мл.
Контрольный раствор. Используют буфер
ный раствор, используемый для приroтовления
испытуемоro раствора и растворов сравнения.
Реактив меди сульфата. 100 Mr меди (11)
сульфата р и 0,2 r натрия тартрата Р paCT
воряют в воде дистиллированной Р и доводят
до объема 50 мл этим же растворителем. 1 О r
натрия карбоната безводН020 Р растворяют в
воде дистиллированной Р и доводят до объема
50 мл этим же растворителем. Медленно при пе
ремешивании вливают раствор натрия карбона
та в раствор меди сульфата. Раствор использу
ют в течение 24 ч.
Реактив щелочной меди. Смешивают
1 объем реактива меди сульфата, 2 объема
раствора 50 r/л натрия додецилсульфата Р и
1 объем раствора 32 r/л натрия 2идроксида Р.
Хранят при комнатной температуре и использу
ют В течение 2 x недель.
разведенный фосфорномолибденово оль
фрамовый реактив. Смешивают 5 мл фосфорно
молибденово вольфрамово20 реактива Р и 55 мл
воды дистиллированной Р Хранят в контейнере
из темноro стекла при комнатной температуре.
Методика. К 1,0 мл каждою раствора cpaB
нения, испытуемою раствора и контрольноro
раствора прибавляют по 1,0 мл реактива ще
лочной меди и перемешивают. Выдерживают
в течение 10 мин. Прибавляют 0,5 мл разве
денною фосфорномолибденово вольфрамо
воro реактива, перемешивают и выдержива
ют при комнатной температуре в течение 30
мин. Измеряют оптические плотности (2.2.25)
растворов при длине волны 750 нм, используя
контрольный раствор в качестве компенсаци
онноro раствора
Расчеты. Зависимость оптической плотно
сти от концентрации белка не является линей
ной; тем не менее, если диапазон концентраций,
используемый для построения стандартноro rpa
фика, невелик, он близок к линейности. Строят
rрафическую зависимость оптических плот
ностей растворов сравнения от концентраций
белка и используют линейную реrрессию для
построения стандартною rрафика. На OCHOBa
нии стандартноro rрафика и оптической плотно
сти определяют концентрацию белка в испытуе
мом растворе.
Мешающие вещества. В представленной
методике перед началом определения для yдa
ления мешающих веществ с помощью осаж
дения белков к испытуемому образцу прибав
ляют дезоксихолево трихлоруксусную кислоту.
Данная методика также может быть применена
для концентрирования белков из разведенноro
раствора.
0,1 мл раствора 1,5 r/л натрия дезоксихо
лата Р прибавляют к 1 мл раствора испыту
емоro образца. Используя вихревой встряхи
ватель (вортекс) перемешивают полученный
раствор и выдерживают при комнатной темпе
рату ре в течение 1 О мин. Прибавляют 0,1 мл
раствора 720 r/л трихлоруксусной кислоты Р
и перемешивают, используя вихревой встряхи
ватель. Центрифуrируют при ускорении 3000 9
в течение 30 мин, декантируют и удаляют
оставшуюся жидкость пипеткой. Повторно раз
водят полученный осадок в 1 мл реактива ще
лочной меди.
МЕТОД 3
Данный метод (известный как метод Бред
форда) основан на абсорбционном сдвиrе с
длины волны 470 нм до 595 нм, наблюдаемом
при связывании белков с красителем кислотным
синим 90. Краситель кислотный синий 90 наибо
лее активно связывает в белке остатки арrини
на и лизина, что может приводить к поrрешно
сти при исследовании различных белков. Белок.
используемый в качестве стандартною образца,
должен быть такими же, как и белок, взятый для
испытания. Мешающих веществ относительно
немноro, однако следует избеrать присутствия в
испытуемом образце детерrентов и амфолитов.
Высокощелочные образцы MOryT взаимодей
ствовать с кислотным peareHToM.
Для приroтовления всех реактивов и буфер
ных растворов, применяемых в данном методе,
используют воду дистиллированную Р.
Испытуемый раствор. Для получения раст
вора с концентрацией в пределах интервала
стандартноro rрафика растворяют достаточное
количество испытуемоro образца в указанном в
частной статье буферном растворе.
Растворы срзвнения. Стандартный образец,
предназначенный для определения белка, paCTBO
ряют в указанном в частной статье буферном pac
Т80ре. Полученный раствор разводят тем же бу
ферным раствором, чтобы получить не менее пяти
растворов сравнения с концентрациями белка,
равномерно распределенными в необходимом ди
апазоне в интервале от 0,1 мr/мл до 1 мr/мл.
236
rосударственная фармакопея Республики Беларусь
Контрольный раствор. Используют бу
ферный раствор, применяемый для приro
товления испытуемоro раствора и растворов
сравнения.
Реактив кислотН020 сине20 90. 0,1 О r
кислотН020 сине20 90 Р растворяют в 50 мл
96 % спирта Р. Прибавляют 100 мл кислоты
фосфорной Р, доводят дистиллированной
водой Р до объема 1000 мл И перемешива
ют. Фильтруют раствор и хранят в контейнере
из темноro стекла при комнатной температу
ре. При хранении выпадает осадок красите
ля, поэтому перед использованием реактив
необходимо фильтровать.
Методика. По 5 мл реактива кислотно
ro синеro 90 прибавляют к 0,100 мл каждоrо
раствора сравнения, испытуемоro раствора и
контрольноro раствора. Смешивают, при этом
избеrают образования пены, приводящей
к ухудшению воспроизводимости. Измеря
ют оптические плотности (2.2.25) растворов
сравнения и испытуемоro раствора при длине
волны 595 нм, используя контрольный paCT
вор в качестве компенсационноro раствора.
Не следует использовать кварцевые (кремни
евые) спектрофотометрические кюветы, по
скольку с этим материалом связывается Kpa
ситель.
Расчеты. Зависимость оптической плот
ности от концентрации белка не является ли
нейной; тем не менее, если диапазон KOH
центраций, используемый для построения
стандартноrо rрафика, невелик, он близок
к линейности. Строят rрафическую зави
симость оптических плотностей растворов
сравнения от концентраций белка и исполь
зуют линейную реrрессию для построения
стандартноro rрафика. На основании CTaH
дартноro rрафика и оптической плотности
определяют концентрацию белка в испытуе
мом растворе.
МЕТОД 4
Данный метод (известный как метод бицин
хонинической кислоты или БХК, ВСА) основан
на восстановлении белком иона двухвалентной
меди (Cu 2 +) до иона одновалентной меди (Cu 1 +).
PeareHT бицинхонинической кислоты приме
няется для определения иона одновалентной
меди. Реакции мешают некоторые вещества.
Их влияние может быть минимизировано с по
мощью разведения, обеспечивающеro KOHцeH
трацию исследуемоro белка на уровне, ДOCTa
точном для проведения точных измерений.
Для удаления мешающих веществ также может
быть использовано осаждение белка, описан
ное в методе 2. Поскольку различные белки
MorYT давать цветные реакции различной ин
тенсивности, стандартный образец и испытуе
мый белок должны быть одинаковыми.
Для приrотовления всех реактивов и бу
ферных растворов, при меняемых в данном
методе, используют воду дистиллирован
нуюР.
Испытуемый раствор. Для получения
раствора с концентрацией в пределах интер
вала стандартноro rрафика растворяют дo
статочное количество испытуемоro образца
в указанном в частной статье буферном pac
творе.
Растворы сравнения. Стандартный об
разец, предназначенный для определе
ния белка, растворяют в указанном в част
ной статье буферном растворе. Полученный
раствор разводят тем же буферным pac
твором, чтобы получить не менее пяти pac
творов сравнения с концентрациями белка,
равномерно распределенными в необходи
мом диапазоне в интервале от 1 О мкr/мл до
1200 мкr/мл.
КонтРОЛЬНblй раствор. Используют бу
ферный раствор, применяемый для приrо
товления испытуемоro раствора и растворов
сравнения.
БХК реактив. 10 r динатрия бицинхони
ната Р, 20 r натрия карбоната MOH02идpa
та Р, 1,6 r натрия тартрата Р, 4 r натрия
2идроксида Р, и 9,5 r натрия 2идрокарбона
та Р растворяют в воде дистиллированной Р.
При необходимости доводят значение рН
раствора до 11,25 раствором натрия 2идpOK
сида Р или раствором натрия 2идрокарбона
та Р, доводят водой дистиллированной Р до
объема 1000 мл И перемешивают.
Медно бицинхонинатовblЙ реактив.
Смешивают 1 мл раствора 40 r/л меди СУЛЬ
фата Р и 50 мл БХК реактива.
Методика. 0,1 мл каждоro раствора cpaB
нения, испытуемоro раствора и контрольноro
раствора смешивают с 2 мл медно бицинхо
нинатовоrо реактива и перемешивают. Инку
бируют полученные растворы при температу
ре З7 ос в течение 30 мин, отмечают время
и выдерживают до охлаждения до комнатной
температуры. В течение 60 мин после оконча
ния инкубации в кварцевых кюветах измеря
ют оптическую плотность (2.2.25) растворов
сравнения и испытуемоro раствора при длине
волны 562 нм, используя контрольный paCT
вор в качестве компенсационноro раствора.
Необходимо учитывать, что после остывания
растворов до комнатной температуры, интен
сивность цвета постепенно возрастает.
Расчеты. З висимость оптической плот
ности от концентрации белка не является ли
нейной; тем не менее, если диапазон KOH
центраций, используемый для построения
стандартноro rрафика, невелик, он близок
к линейности. Строят rрафическую зави
симость оптических плотностей растворов
2.5.33. Общий белок
237
сравнения от концентраций белка и исполь
зуют линейную реrрессию для построения
стандартноrо rрафика. На основании CTaH
дартноro rрафика и оптической плотности
определяют концентрацию белка в испытуе
мом растворе.
МЕТОД 5
Данный метод (известный как биурето
вый метод) основан на взаимодействии ДBYX
валентноro иона меди (Cu 2 +) С белком в ще
лочной среде и появлении окрашивания
раствора, оптическая плотность котороro из
меряется при длине волны 545 нм. Метод
проявляет минимальное различие между
равными количествами иммуноrлобулино
вых и альбуминовых образцов. Прибавление
натрия rидроксида и биуретовоrо реактива в
качестве комбинированноro peareHTa, Heдo
статочное перемешивание после прибавле
ния натрия rидроксида или продолжительный
период времени между прибавлением натрия
rидроксида и прибавлением биуретовоrо pe
актива завышают значения для иммуноrлобу
ли новых образцов по сравнению с альбуми
новыми образцами. Метод трихлоруксусной
кислоты, используемый для минимизации
влияния мешающих веществ, также может ис
пользоваться для определения содержания
белка в испытуемых образцах при KOHцeHTpa
циях ниже 500 мкr/мл.
Для приroтовления всех реактивов и бу
ферных растворов, при меняемых в данном
методе, используют воду дистиллированную Р.
Испытуемый раствор. Для получения
раствора с концентрацией в пределах интер
вала стандартноro rрафика растворяют дo
статочное количество испытуемоrо образца в
растворе 9 r/л натрия хлорида Р.
Растворы сравнения. Стандартный об
разец, предназначенный для определения
белка, растворяют в растворе 9 r/л натрия
хлорида Р. Полученный раствор разво.qят
раствором 9 r/л натрия хлорида Р, чтобы по
лучить не менее трех растворов сравнения с
концентрациями белка, равномерно распре
деленными в необходимом диапазоне в ин
тервале от 0,5 мr/мл до 10 мr/мл.
Контрольный раствор. Используют paCT
вор 9 r/л натрия хлорида Р.
Биуретовый реактив. 3,46 r меди (11) СУЛЬ
фата Р растворяют в 1 О мл roрячей воды диc
тиллированной Р и охлаждают (раствор А).
34,6 r натрия цитрата Р и 20,0 r натрия
карбоната безводН020 Р растворяют в 80 мл
roрячей 130ды дистиллированной Р и охлаж
дают (раствор В). Смешивают растворы А и
В и доводят водой дистиллированной Р до
объема 200 мл. Используют полученный paCT
вор в течение 6 месяцев. В случае помутне
ния или выпадения осадка реактив не подле
жит использованию.
12 За.. 1712.
Методика. К одному объему испытуемо
ro раствора прибавляют равный объем paCT
вора 60 r/л натрия 2идроксида Р и переме
шивают. Немедленно прибавляют биуретовый
реактив в количестве, равном 0,4 объема ис
пытуемоrо раствора, и быстро перемешива
ют. Выдерживают при температуре от 15 ос
до 25 ос в течение не менее 15 мин. В Te
чение 90 мин после прибавления биуретово
ro реактива измеряют оптическую плотность
(2.2.25) растворов сравнения и испытуемоro
раствора при длине волны 545 нм, используя
контрольный раствор в качестве компенсаци
онноro раствора. Мутные растворы или pac
творы с осадком неприroдны для определе
ния концентрации белка.
Расчеты. Зависимость оптической плот
ности от концентрации белка имеет почти
линейный характер в рамках указанноro ин
тервала концентрации белка для растворов
сравнения. Строят rрафическую зависимость
оптических плотностей растворов сравнения
от концентраций белка и используют линей
ную реrрессию для построения CTaHдapTHO
ro rрафика. Рассчитывают коэффициент KOp
реляции для стандартноro rрафика. Система
считается приrодной, если линейная зави
симость имеет коэффициент корреляции не
менее 0,99. На основании стандартноro rpa
фика и оптической плотности определяют
концентрацию белка в испытуемом растворе.
Мешающие вещества. Для минимизации
эффекта мешающих веществ белок может
быть осажден из испытуемоro образца следу
ющим образом: прибавляют 0,1 объема paCT
вора 500 r/л кислоты трихлоруксусной Р к
1 объему раствора испытуемоro образца,
удаляют надосадочную жидкость и осадок
растворяют в небольшом объеме 0,5 М pacт
вора натрия 2идроксида. Полученный paCT
вор используют для приrотовления испытуе
моro раствора.
МЕТОД 6
Данный флуориметрический. метод oc
нован на дериватизации белка о фталевым
альдеrидом, который взаимодействует с пер
вичными аминами белка (N концевая амино
кислота и Е аминоrруппа остатков лизина).
Чувствительность анализа может быть по
вышена rидролизом белка перед прибав
лением о фталевоro альдеrида. rидролиз
делает С! аминоrруппы аминокислот, входя
щих в струпуру белка, доступными для pe
акции с реактивом фталевоrо альдеrида.
Данный метод требует очень малых коли
честв белка. Первичные амины, например,
трис(rидроксиметил)аминометан и аминокис
лотные буферные растворы. взаимодейству
ют с фталевым альдеrидом, вследствие чеro
238
rосударственная фармакопея Республики Беларусь
их следует избеrать или удалять. Аммиак при
высоких концентрациях взаимодействует с
фталевым альдеrидом. Флуоресценция, полу
ченная при взаимодействии амина с фтале
вым альдеrидом. может быть нестабильной.
Использование автоматизированных про
цедур для стандартизации данноro метода
может повысить ero качество и точность.
Для приroтовления всех реактивов и бу
ферных растворов, при меняемых в данном
методе, используют воду дистиллированную Р.
Испытуемый раствор. Растворяют дo
статочное количество испытуемоro образца в
растворе 9 r/л натрия хлорида Р для полу
чения раствора с концентрацией в пределах
концентраций растворов сравнения. Перед
прибавлением реактива фталевоrо альдеrи
да устанавливают рН раствора от 8 до 10.5.
Растворы сравнения. Стандартный об
разец, предназначенный для определения
белка, растворяют в растворе 9 r/л натрия
хлорида Р. Полученный раствор разводят
раствором 9 r/л натрия хлорида Р, чтобы по
лучить не менее пяти растворов сравнения с
концентрациями белка, равномерно распре
деленными в необходимом диапазоне в ин
тервале от 1 О мкr/мл до 200 мкr/мл. Перед
прибавлением реактива фталевоrо альдеrи
да устанавливают значение рН растворов от
8 до 10,5.
Контрольный раствор. Используют paCT
вор 9 r/л натрия хлорида Р.
Боратный буфер%/й раствор. 61,83 r
кислоты борной Р растворяют в воде диc
тиллированной Р, доводят раствором калия
2идроксида Р до рН 10,4, разводят водой диc
тиллированной Р до объема 1000 мл И пере
мешивают.
Основной раствор фталев020 альде2и
да. 1,20 r фталев020 альде2ида Р paCTBO
ряют в 1,5 мл метанола Р, прибавляют 100 мл
боратноro буферноro раствора и перемеши
вают. Прибавляют 0.6 мл раствора 300 r/л Ma
КР020Л 23 лаурилов020 эфира Р и перемеши
вают. Хранят при комнатной температуре и
используют в течение 3 x недель.
Реактив фталев020 альде2ида. К 5 мл
основноro раствора фталевоrо альдеrида при
бавляют 15 мкл 2 меркаптоэтанола Р. PaCT
вор roтовят не менее чем за 30 мин до начала
использования. Используют в течение 24 ч.
Методика. К 10 мкл испытуемоro paCT
вора и каждоro из растворов сравнения при
бавляют по 0,1 мл реактива фталевоrо аль
деrидэ, перемешивают и выдерживают при
комнатной температуре в течение 15 мин.
Прибавляют 3 мл 0,5 М раствора натрия 2и
дроксида и перемешивают. Определяют флу
оресценцию (2.2.21) растворов сравнения и
испытуемоro раствора при длине волны воз
буждающеro света 340 нм и длине волны ис
.......
пускаемоro света от 440 нм до 455 нм. Изме
ряют флуоресценцию полученных растворов
только один раз, поскольку облучение снижа
ет интенсивность флуоресценции.
Расчеты. Зависимость Флуоресценции
от концентрации белка является линейной.
Строят rрафическую зависимость флуорес
ценции растворов сравнения от KOHueHTpa
ций белка и используют линейную реrрессию
для построения стандартноro rрафика. На oc
I-Iовании стандартноro rрафика и интенсивно
сти флуоресценции определяют KOHцeHTpa
цию беJlка в испытуемом растворе.
МЕТОД 7
Данный метоц основан на определении
соцержания азота как способе определения
белка. ПРИСУТС19ие друrих азотсодержащих
веществ в испытуемом образце может OKa
зывать мешающее влияние на определение
rеЛl{а при помощи данноro метода Техника
азотноro анализа основана на разрушении
испытуемоro образца, но не зависит от co
держания белка в водной среде.
Методика А. 8ыпспняется в соответствии
с методом определения азота после мине
рализации серной кислотой (2.5.9) или при
помощи коммерчески ,L:;оступноrо прибора
ДJ:Я оr:ределения азов O Кьельдалю.
Методика В. Анализ может выполнять
ся при помощи коммерчески доступных при
боров. Большинство приборов, применяемых
для азотноro анализа, используют пиро
лиз (т. е. сжиrание образца в присутствии
кислорода при температуре, достиrающей
1000 ОС). При этом из азота, присутствующе
ro в испытуемых образцах, образуется оксид
азота (NO) и друrие оксиды азота (NO). В He
которых приборах оксиды азота преобразует
ся в азот, который обнаруживается теплопро
водным детектором. В друrих приборах оксид
азота (NO) смешивается с озоном (0з), обра
зуя при этом активированный диоксид азота
(NO/), который испускает свет при распаде и
может быть определен с помощью хемилюми
несцентноro детектора. Стандартный обра
зец белка, который относительно чист и схож
по своему составу с исследуемыми белками,
используется для оптимизации параметров
пиролиза и оценки стабильности анализа.
Расчеты. Содержание белка рассчиты
вают путем деления содержания азота в ис
пытуемом образце на известное содержание
азота в белке. Известное содержание азота в
белке можно определить на основе химиче
скоro состава белка или сравнивая с подхcr
дящим стандартным образцом.
2.5.35. Азота закись в 2азах
01/2013:20534
239
2.5.34. УКСУСНАЯ КИСЛОТА
В СИНТЕТИЧЕСКИХ ПЕПТИДАХ.
Жидкостная хромат02рафия (2.2.29).
Испытуемый раствор. rотовят испыту
емый раствор, как указано в частной статье.
Концентрация пептида в растворе может быть
адаптирована в зависимости от ожидаемоro
содержания уксусной кислоты в образце.
Раствор сравнения . rотовятраствор О, 1 О r/л
кислоты уксусной ледяной Р в смеси из 5
объемов подвижной фазы В и 95 объемов
подвижной фазы А.
Условия хроматоrрафрования:
колонка из нержавеющей стали длиной
0,25 м и внутренним диаметром 4,6 мм, запол
ненная силика2елем октадецилсилильным
для хромаm02рафии Р с размером частиц
5 мкм;
скорость подвижной фазы: 1,2 мл/мин;
подвижная фаза:
подвижная фаза А: 0,7 мл кислоты фос
форной Р разводят водой Р до объема 1000 мл
И доводят раствором натрия 2идроксида
концентрированным р до рН 3,0;
подвижная фаза В: метанол Р2;
Подвижная Подвиж ная I
Время (мин) фаза А фаза В
(%, 06/06) (%, 06/06) I
0 5 95 5
10 95 50 5 50
10 20 50 50
20 22 50 95 50 5
22 30 95 5
спектрофотометрический детектор,
длина волны 210 нм;
объем вводимой пробы: 1 О мкл.
ВЫХОД raза
t
ВХОД rаза
t
Детектор
На полученных xpoMaTorpaMMax пик, co
ответствующий уксусной кислоте, имеет
время удерживания 3 4 мин. После начала
rрадиента базовая линия круто поднимается,
что соответствует элюированию из колонки
пептида.
Определяют содержание уксусной кисло
ты В пептиде.
01/2013:20535
2.5.35. АЗОТА ЗАКИСЬ В rАЗАХ
Азота закись (N 2 0) в rазах определяют с ис
пользованием инфракрасноro анализатора (ри
сунок 2.5.35. 1).
Инфракрасный анализатор включает в
себя 2 reHepaTopa идентичных инфракрас
ных лучей, которые оснащены зеркалами и
катушками, электрически наrреваемыми до
слабоro красноro каления. Один луч проходит
через испытуемую ячейку, в которую посту
пает струя испытуемоrо rаза, друrой через
ячейку сравнения, содержащую азот Р1. Две
камеры детектора, заполненные азота за
кисью Р автоматически селективно реrистри
руют излучение. Проходя через испытуемый
rаз, излучение поrлощается закисью азота и
по разному HarpeBaeT rаз в камерах дeTeKTO
ра, приводя к различию в расширении. Раз
личие в давлении между камерами детектора
приводит к растяжению металлической диа
фраrмы, которая их разделяет. Диафраrма яв
ляется частью конденсатора, чья емкость Ba
рьирует в зависимости от разности давления,
которое в свою очередь зависит от содержа
ния закиси азота в испытуемом rазе. Так как
инфракрасные лучи периодически прерыва
ются вращающимися лопастями, электриче
ский сиrнал является частотно модулирован
ным.
reHepaTop
ИК излучения
а
reHepaTop
ИК излучения
G
Конденсатор
Лопасти
Мотор
Рисунок 2.5.35. 1. Инфракрасный анализатор
240
01/2013:20536
rосударственная фармакопея Республики Беларусь
2.5.36. АНИЗИДИНОВОЕ ЧИСЛО
Анизидиновое число представляет собой
1 ОО кратную измеренную в кювете с толщиной
слоя 1 см оптическую плотность раствора, co
держащеro 1 r испытуемоro образца в 100 мл
смеси из растворителей и реактивов.
Все при20товления и измерения необходu
мо проводить насколько это возможно быстро,
избе2ая воздействия света, спосоБН020 вызы
вать фотохимические превращения.
Испытуемый раствор (а). 0,500 r испытуе
моro образца растворяют в триметuлпентане Р
и доводят до объема 25,0 мл этим же раствори
телем.
Испытуемый раствор (Ь). К 5.0 мл испыту
eMoro раствора (а) прибавляют 1.0 мл раствора
2,5 r/л п анизидина Р в кислоте уксусной ледя
ной Р, встряхивают и хранят в защищенном от
света месте.
Раствор сравнения. К 5,0 мл триметил
пентана Р прибавляют 1,0 мл раствора 2,5 r/л
п низидина Рв кислоте уксусной ледяной Р, встря
хивают И хранят в защищенном от света месте.
Измеряют оптическую плотность (2.2.25) ис
пытуемоrо раствора (а) при 350 нм, используя
триметилпентан Р в качестве компенсацион
ноro раствора. Измеряют оптическую плотность
испытуемоro раствора (Ь) при 350 нм ровно
через 10 мин после еro приrотовления, исполь
зуя раствор сравнения в качестве компенсаци
онноro раствора.
Анизидиновое число рассчитывают по фор
муле:
25.(1,2А A2) ,
т
rде:
А 1 оптическая плотность испытуемоro
раствора (Ь) при 350 нм;
А 2 оптическая плотность испытуемоro
раствора (а) при 350 нм;
т масса навески испытуемоro образца, r.
01/2013:20537
2.5.37. МЕТИЛ-, ЭТИЛ-
И ИЗОПРОПИЛМErАНСУЛЬФОНАТ
В МЕТАНСУЛЬФОНОВОЙ
КИСЛОТЕ
Следующий метод был валидирован для
определения метиловых, этиловых и изопропи
ловых эфиров метансульфоновой кислоты в ди
апазоне концентраций от 0,5 ррm до 100 ррт.
Если данный метод предполаrается ис
пользовать для определения концентраций
эфиров метансульфоновой кислоты, выходя
щих за rраницы валидированноrо диапазона
концентраций, например, на ранних стадиях
синтеза до их удаления, концентрация испы
TyeMoro раствора должна быть соответствую
щим образом скорректирована.
МЕТОДИКА
rазовая хроматоrрафия (2.2.28) спарен
ная с масс спектрометрией (2.2.43).
Раствор внутренне20 стандарта. 7 мкл
ФСО бутилметансульфоната (БМС) ДOBO
дят метиленхлоридом Р до объема 10,0 мл.
1 О мкл полученноro раствора доводят Meти
ленхлоридом Р до объема 100,0 мл.
Испытуемый раствор. К 0,74 r испыту
емоro образцаприбавляют 10,0 мл воды Р и
проводятэкстракциюс использованием 10,0 мл
раствора внутреннеro стандарта. Выдержи
вают до расслоения фаз, переносят орrани
ческий слой во флакон, содержащий натрия
сульфат безводный Р. Встряхивают и филь
труют.
Раствор сравнения (а). По 50 Mr метил
метансульфоната Р (ММС), этилметан
сульфоната Р (ЭМС) и изопропилметан
сульфоната Р (ИМС) растворяют в растворе
внутреннеro стандарта и доводят до объема
50,0 мл этим же раствором. 74 мкл получен
HOro раствора доводят раствором BHYTpeH
Hero стандарта до объема 10,0 мл. 100 мкл
полученноrо раствора доводят раствором
внутреннеro стандарта до объема 10,0 мл.
Раствор сравнения (Ь). 3,0 мл раствора
сравнения (а) доводят раствором BHYTpeHHe
ro стандарта до объема 10,0 мл.
Условия хроматоrрафирования:
колонка: кварцевая капиллярная,
длиной 15 м и внутренним диаметром 0,25 мм,
заполненная полuдиметипсилоксаном Р
(толщина слоя 1 мкм);
2аз носитель: 2епий дпя xpOMaт02pa
фииР;
скорость потока: 1 мл/мин;
импульсный ввод без деления потока:
250 кПа, 0,25 мин;
температура:
Время Температура
(мин) (ОС)
0 1 55
Колонка
1 9 55------+1З5
Блок ввода проб 240
автоматиче 280
ская линия
Детектор: источник 230
анализатор 150
2.5.38. Метил , этuл и изопропилметансульфонат
241
детектор: масс спектрометр, описан
ный ниже; корректируют настройку детектора
для соответствия критериям приrодности си
стемы:
квадрупольный масс спектрометр,
снабженный системой ионизации электрон
ным ударом (70 эВ);
для режима фраrментометрии (OДHO
ионный мониторинr (S/M» параметры Macc
спектрометра устанавливают следующим об
разом:
Вещество m/z Длительность
мониторинrа
Бутилметансульфонат 56 t R между 7,0 мин
(БМС) и 9,0 мин
Метилметансульфонат 80 t R между 2,0 мин
(ММС) и З,5 мин
Этилметансульфонат 79 t R между 4,0 мин
(ЭМ С) и 4,7 мин
Изопропилметансуль 123 t R между 4,7 мин
фонат (ИМС) и 5,5 мин
объем вводимой пробы: 2 мкл.
Относительное удерживание (по OTHO
шению к внутреннему стандарту (БМС) (время
удерживания около 7,6 мин»: ММС около
0,3; ЭМС около 0,5; ИМС около 0,6.
При20дность хромат02рафической cи
стемы:
разрешение: не менее 3,0 между пиками
ЭМС и ИМС на xpoMaTorpaMMe раствора cpaB
нения (а);
отношение сиzнал/шум: не менее 1 О для
пиков ММС, ЭМС и ИМС на xpoMaтorpaMMe paCT
вора сравнения (Ь).
Содержание ММС, ЭМС или ИМС (в частях
на миллион, ррm) рассчитывают по формуле:
.lI ./ .Иt: .С. О 148
1'2 1 1 '
.lI./ .Иt:
2 2
rдe:
А 1 площадь пика ММС, ЭМС или ИМС на
xpoMaтorpaMMe раствора сравнения (а);
А 2 площадь пика ММС, ЭМС или ИМС на
xpoMaTorpaMMe испытуемоro раствора;
С концентрация ММС, ЭМС или ИМС, в
процентах;
/1 площадь пика BHYTpeHHero стандарта
на xpoMaтorpaMMe раствора сравнения (а);
/2 площадь пика внутреннеro стандарта
на xpoMaTorpaMMe испытуемоro раствора;
W 1 масса навески ММС, ЭМС или ИМС,
взятой ДЛЯ приrотовления раствора сравнения
(а), в миллиrраммах;
W 2 масса навески испытуемоro образца,
взятой для приrотовления испытуемоro раст
вора, в миллиrраммах;
0,148 коэффициент разведения.
01/2013:20538
2.5.38. МЕТИЛ , ЭТИЛ
ИИЗОПРОПИЛМЕТАНСУЛЬФОНАТ
В ФАРМАЦЕВТИЧЕСКИХ
СУБСТАНЦИЯХ
Следующий метод был валидирован для
определения метиловых, ЭТИЛОВЫХ и изопро
пиловых эфиров метансульфоновой кислоты
(в диапазоне концентраций от 0,2 ррmдо 5 ррт)
в бетаrистина мезилате.
Если данный метод предполаrается ис
пользовать для определения эфиров Me
тансульфоновой кислоты в друrих фар
мацевтических субстанциях, содержащих
отличающиеся концентрации эфиров Me
тансульфоновой кислоты, концентрация ис
пытуемоro раствора и раствора сравнения
должна быть соответствующим образом
скорректирована и проведена подходящая
валидация.
МЕТОДИКА
Парофазная rазовая хроматоrрафия
(2.2.28), спаренная с масс спектрометрией
(2.2.43). Испытуемый раствор и раствор
сравнения 20товят непосредственно перед
использованием.
Смесь растворителей. Вода Р aцe
тонитрил Р (20:80, об/об).
Раствор А. 30 Mr натрия тиосульфата
безводН020 Р и 60,0 r натрия йодида Р paCT
воряют в воде Р и доводят до объема 50,0
мл этим же растворителем.
Растворвнутренне20стандарта. 1 О мкл
ФСО бутилметансульфоната (БМС) ДOBO
дят смесью растворителей до объема 10,0 мл.
20 мкл полученноrо раствора доводят
смесью растворителей до объема 10,0 мл.
10,0 мл полученноrо раствора доводят
смесью растворителей до объема 100,0 мл.
Испытуемый раствор. 25,0 Mr испытуе
MOro образца помещают во флакон для паро
фазноro пробоотборника вместимостью 20 мл,
прибавляют 0,50 мл раствора А и 0,50 мл
раствора BHYTpeHHero стандарта, немедлен
но закрывают флакон силиконовой мембра
ной с политетрафторэтиленовым покрыти
ем и обкатывают алюминиевым колпачком.
В ходе реакции дериватизации может об
разоваться осадок, который не влияет на
достоверность количествеННО20 опреде
ления.
Раствор сравнения (а). По 25,0 Mr Me
тилметансульфоната р (ММС), этил
метансульфоната Р (ЭМС) и изопропил
метансульфоната Р (ИМС) растворяют в
толуоле Р и доводят до объема 5,0 мл этим
же растворителем. 50 мкл полученноro paCT
вора доводят раствором BHYTpeHHero CTaH
дарта до объема 25,0 мл.
242
rосударственная фармакопея Республики Беларусь
Раствор сравнения (Ь). 20 мкл раствора
сравнения (а) доводят раствором BHYTpeH
неro стандарта до объема 20,0 мл. 0,50 мл
полученноro раствора и 0,50 мл раствора А
помещают во флакон для парофазноro про
боотборника вместимостью 20 мл, немедлен
но закрывают флакон силиконовой мембра
ной с политетрафторэтиленовым покрытием
и обкатывают алюминиевым колпачком.
Раствор сравнения (с). 500 мкл раствора
сравнения (а) доводят раствором BHYTpeH
неro стандарта до объема 20,0 мл. 0,50 мл
полученноrо раствора и 0,50 мл раствора А
помещают во флакон для парофазноro про
боотборника вместимостью 20 мл, немедлен
но закрывают флакон силиконовой мембра
ной с политетрафторэтиленовым покрытием
и обкатывают алюминиевым колпачком.
Условия хроматоrрафирования:
колонка: кварцевая капиллярная,
длиной 30 м и внутренним диаметром
0,25 мм, заполненная полярно дeaKтивиpo
ванным полиэтилеН2ликолем Р (толщина
слоя 1 мкм);
2аз носитель: 2елий для xpOMaт02pa
фииР;
скорость потока: 0,5 мл/мин;
деление потока: 1 :20;
условия для парофазноro пробоотбор
ника:
температура уравновешивания: 60 ОС;
время уравновешивания: 30 мин;
температура автоматической линии:
120 ОС;
температура:
т Время I Температура
i (мин) (ОС)
Колонка* ! 0 1 40
I 1 10 40 130
Блок ввода проб 220
aBTOMa
тическая 280
Детектор: линия
источник 250
анализатор 200
. в конце анализа поднимают температуру колонки до 240 ос
и поддерживают ее в течение 7 мин.
детектор: масс спектрометр, описан
ный ниже; корректируют настройку детектора
для соответствия критериям приroдности си
стемы; альтернативно может быть использо
ван подходящий детектор с электронной ло
вушкой:
квадрупольный масс спектромеч:1, снаб
женный системой ионизации электронным
ударом (70 эВ);
для режима фраrментометрии (OДHO
ионный мониторинr (S/M) параметры Macc
спектрометра устанавливают следующим об
разом:
ИОН для ИОН ДЛЯ
количе-- квалифика
Вещество cтвeHHoro
определения ции
(m/z) (m/z)
Бутилйодид 184 127
(Ви/)**
Метилйодид 142 127
(Ме/)**
Этилйодид (EtI)** 156 127
--;
Изопропилйодид I
(iP,/)** 170 127 I
.. Получены из БМС. ММС. ЭМС ИМС в результате реак-
ции дериватизации.
объем вводимой пробы: по 1 мл paBHO
весной rазовой фазы испытуемоro раствора,
раствора сравнения (Ь) и раствора cpaBHe
ния (с).
Относительное удерживание (по OTHO
шению к внутреннему стандарту (бутилйо
дид) (время удерживания около 8,5 мин)):
метилйодид около 0,51; этилйодид
около 0,63; изопропилйодид около 0,68.
При20дность хромат02рафической cи
стемы:
разрешение: не менее 1,5 между
пиками этилйодида и изопропилйодида на
xpoMaTorpaMMe раствора сравнения (с);
отношение сиzнал/шум: не менее 1 О
для пиков каждоro алкилйодида на xpOMaTO
rpaMMe раствора сравнения (Ь).
Содержание каждоrо алкилметансуль
фоната (в частях на миллион, ррт) рассчи
тывают по формуле:
Az '/1 . . с . 0,05
Lt ./ .и':
'1 2 2
rде:
А 1 площадь пика каждоro алкилйоди
да на xpoMaTorpaMMe раствора сравнения (с);
А 2 площадь пика каждоro алкилйоди
да на xpoMaTorpaMMe испытуемоro раствора;
С концентрация каждоro эфира, в про
центах;
/, площадь пика BHYTpeHHero CTaHдap
та на xpoMaTorpaMMe раствора сравнения (с);
/2 площадь пика внутреннеro CTaHдap
та на xpoMaTorpaMMe испытуемоro раствора;
W 1 масса навески каждоro эфира.
взятой для приrотовления раствора cpaBHe
ния (а), в миллиrраммах;
W 2 масса навески испытуемоro образ
ца, взятой для приroтовления испытуемоro
раствора, в миллиrраммах;
0,05 коэффициент разведения.
01/2013:20539
2.5.39. Метансулъфонuлхлорuд в метансульфоновой кислоте
243
2.5.39. МЕТАНСУЛЬФОНИЛХЛОРИД
В МЕТАНСУЛЬФОНОВОИ
КИСЛОТЕ
СледУЮЩИЙ метод был валидирован для опр&-
деления метансульфонилхлорида в метансулbф<r
новой кислоте в диапазоне концентраций от 0,05 ррm
до 50 ррm.
МЕТОДИКА
rазовая хроматоrpафия (2.2.28), спаренная с
масс спектрометрией (2.2.43).
Раствор внymреннеzo стандарта. 68 мкл
ФСО бутилметансульфоната (БМС) доводят Me
тиленхлоридом Р до объема 10,0 мл. 0.5 мл полу
ченноro раствора доводят метиленхлоридом Р до
объема 50,0 мл.
Испытуемый раствор. К 5 мл воды Р цобав
ляют 7,4 r испытуемоro образца и медленНО пере--
мешивают. После охлаждения прибавляют 5.0 мл
метиленхлорида Р, 100 мкл раствора внутреннею
стандарта и встряхивают. Выдерживают до расслcr
ения фаз, переносят орraнический слой во флакон,
содержащий 1 r натрия сульФата безводНО20 Р.
Экстракцию повторяют дважды, каждый раз ис
пользуя 5,0 мл метиленхлорида Р. Орraнические
спои смешивают и фильтруют.
Раствор сравнения (а). 50 Mr метансулЬфОнuлх
лорида Ррастворяют вметиленхлориде Р и доводят
до объема 10,0 мл этим же растворитепем. 1 0 мл
полученною раствора доводят метuлeнxлopuдoм Р
до объема 10,0 мл. 300 мкл полученною раствора дcr
водят метuленхлоридом Р до объема 10,0 мл.
Раствор сравнения (Ь). 500 мкл раствора cpaB
нения (а) и 100 мкл раствора внутреннеro cтaндap
та доводят метиленхлоридом Р до объема 15,0 мл.
Раствор сравнения (с). 25 мкл раствора cpaB
нения (а) и 100 мкл раствора внутреннею стандар.-
та доводят метиленхлоридом Р до объема 15,0 мл.
Условия хроматоrрафирования:
колонка: кварцевая капиллярная, длиной 15 м
и внутренним диаметром 0,25 мм, заполненная<по
лидиметuлсилоксаном Р (толщина слоя 1 МКМ);
2аз носитель: zелий для хроматО2рафии р;
скорость потока: 1 млlмин;
импульсный ввод без деления потока: 60 кПа,
0,1 мин;
температура:
Время Температура
(мин) (ОС)
Колонка* O 40
40 200
Блок ввода проб 240
автоматиче 280
ская линия
Детектор: источник I I 230
анализатор i 150
. в конце анализа поднимают температуру колонки до 270 ос
и подцерживают ее в течение 8 мин.
детектор: масс спектрометр, описан
ный ниже; корректируют настройку детектора
для соответствия критериям приrодности си
стемы:
квадрупольный масс спектрометр, снаб
женный системой ионизации электронным
ударом (70 эВ);
для режима фраrментометрии (OДHO
ионный мониторинr (S/M» параметры Macc
спектрометра устанавливают следующим об
разом:
' Вещество m/z Длительность
I мониторинrа
I МетансулЬфонилхлорид 79 t R между З,З
мин и 6,0 мин
I утилметэнсульфонат 56 t R между 6,0
(БМС) мин и 8,0 мин
объем вводимой пробы: по 5 мкл испы
TyeMoro раствора, раствора сравнения (Ь),
раствора сравнения (с), раствора внутреннеro
стандарта и метиленхлорида Р.
Относительное удерживание (по OTHO
шению к внутреннему стандарту (БМС) (время
удерживания около 7,2 мин)): метансульфо
нилхлорид около 0,68.
При20дность хромат02рафической cи
стемы:
на xpOMaTorpaMMe раствора BHYTpeHHe
ro стандарта не должно обнаруживаться пика
со временем удерживания, идентичным Me
тансульфонилхлориду;
разрешение: не менее 5,0 между пиками
метансульфонилхлорида и БМС на xpOMaTO
rpaMMe раствора сравнения (Ь);
отношение си2нал/шум: не менее 1 О
для пика метансульфонилхлорида на xpOMa
TorpaMMe раствора сравнения (с).
Содержание метансульфонилхлорида (в
частях на миллион, ррm) рассчитывают по
формуле:
11 ./.W .С.15
r1;2 1 1 J
11./ .W
Т""'I 2 2
rдe:
А 1 площадь пика метансульфонилхлори
да на xpoMaTorpaMMe раствора сравнения (Ь);
А 2 площадь пика метансульфонилхлори
да на xpoMaTorpaMMe испытуемоro раствора;
С концентрация метансульфонилхлори
да, в процентах;
/1 площадь пика внутреннеro CTaHдap
та (БМС) на xpoMaтorpaMMe раствора c:paBHe
ния (Ь);
/2 площадь пика внутреннеro стандарта
(БМС) на xpoMaTorpaMMe испытуемоro раствора;
W 1 масса навески метансульфонилхлори
да, взятой для приroтовления раствора cpaBHe
ния (а), в миллиrраммах;
244
rосударственная фармакопея Республики Беларусь
W 2 масса навески испытуемоro образца,
взятой для приroтовления испытуемоro paCT
вора, в миллиrраммах;
1 ,5 коэффициент разведения.
01/201З:РБ20550
#2.5.50. ТИТРОВАНИЕ ВНЕВОДНЫХ
РАСТВОРИТЕЛЯХ
Метод кислотно основноro титрования в He
водных растворителях применяется для коли
чественноro определения веществ, представ
ляющих собой кислоты, основания или соли,
титрование которых в воде затруднено или He
возможно из за слабых кислотно основных
свойств или малой растворимости.
В неводных растворителях резко изменяются
кислотно основные свойства различных веществ.
В зависимости от растворителя одно и то же вещ&
ство может быть кислотой, основанием или вообще
не проявлять кислотнcroсновных свойств. Приме
нение различных растворителей позволяет управ
лять кислотнcroсновными свойствами веществ.
Выбор растворителя осуществляется на oc
новании величин констант титрования КТ или
их отрицательных лоrарифмов РК Т ' Величина
РК Т является критерием возможности и точно
сти титрования. Чем больше эта величина, тем
лучше условия титрования. Константа титрова
ния определяется двумя основными величина
ми: константой кислотности титруемой кислоты
(Ка) или константой кислотности кислоты, сопря
женной с титруемым основанием (Квн'), и KOH
стантой автопротолиза растворителя (K SH )'
При титровании кислот:
К K SH или Р К p K p K.
т ,SH а
К.
При титровании оснований:
КТ Квн или рК т РК вн . .
При титровании смеси двух кислот:
КТ К а (2) или РК т РК.(2) РК.(1) .
К.(1)
При титровании смеси двух оснований:
К Квн' (1) или Р К Р К + Р К .
т т BH (1) BH (2)
KBH (2)
Индекс 1 относится к более сильной
кислоте (основанию), а индекс 2 к более
слабой кислоте (основанию).
Значения величин pK SH для ряда paCT
ворителей и РКа кислот и РК вн . оснований в
воде и различных неводных растворителях
приведены в таблицах #2.5.50. 2, #2.5.50. 3,
#2.5.50. 4. Эти таблицы позволяют быстро
определять РКТ- Задачу выбора растворите
ля помоrает решить также линейная зависи
мость РК Т кислот И оснований, принадлежа
щих к одной rруппе химических соединений,
в воде и неводных растворителях. Это позво
ляет определять РК Т кислот и оснований в He
водных растворителях, если известны их РК Т
в воде.
Как кислоты можно титровать: карбоно
вые кислоты, фенолы, барбитураты, сульфа
миды, аминокислоты и друrие.
Как основания можно титровать: амины,
азотсодержащие rетероциклические соедине
ния, амиды, четвертичные аммониевые OCHO
вания и друrие.
Оптимальные условия титрования для
слабых кислот достиrаются в основных HeBO
дных растворителях, таких как пиридин, ди
метилформамид; для слабых оснований в
кислотных неводных растворителях, таких как
уксусная кислота и уксусный анrидрид.
Соли некоторых орrанических и мине
ральных кислот MorYT быть опитрованы как
основания в кислотных растворителях.
В случае титрования солей rалоrеново
дородных кислот перед титрованием прибав
ляют обычно раствор ртути (11) ацетата для
связывания rалоrенид ионов в малодиссоци
ирующие соединения. При использовании YK
cycHoro анrидрида в качестве растворителя
возможно титрование солей rалоrенводород
ных кислот, преимущественно хлоридов, без
прибавления ртути (11) ацетата.
Для раздельноrо титрования смесей
кислот или смесей оснований используют
дифференцирующие растворители, т. е. pac
творители с величиной pK SH ' обычно превы
шающей 15, не обладающие выраженными
кислотно основными свойствами, такие как
кетоны, нитрилы, нитрометан.
В ряде случаев для титрования применя
ют смеси неводных растворителей, один из
которых является апротонным (бензол, хло
роформ и дp ). Присутствие апротонноrо pac
творителя уменьшает константу автопрото
лиза среды (К), что может способствовать
улучшению условий титрования.
Метод неводноrо титрования применя
ют также для количественноrо определения
веществ, содержащих карбонильную rруппу,
обладающих слабовыраженными кислотно
основными свойствами и не поддающихся пря
мому титрованию в неводных растворителях.
В этом случае проводят реакцию оксимирова
ния в неводных растворителях и осуществля
ют количественное определение карбонилсо
держащих веществ путем титрования избытка
#2.5.50. Титрование в неводных растворителях
245
peareHTa. Метод при меняют для количествен
ноro определения различных классов орrани
ческих соединений: альдеrидов, кетонов, xpo
монов, стероидных roрмонов, rликозидов и др.
Титрование в неводных средах может
быть проведено как с индикаторами, так и по
тенциометрически с использованием в каче
стве индикаторноro стеклянноro или друrих
обратимых к протону электродов. В качестве
электрода сравнения обычно используют хло
ридсеребряный электрод.
При проведении потенциометрическоro
титрования в неводных средах электролити
ческий мост или электрод сравнения запол
няют растворами калия хлорида или лития
хлорида в соответствующих неводных pac
творителях.
При титровании в основных растворите
лях следует принимать меры для защиты ис
пытуемоro раствора и титрованноro раствора
от уrлерода диоксида, содержащеrося в воз
духе. Титрование лучше проводить в aTMOC
фере инертноro rаза (азота, rелия).
В таблице #2.5.50. 1 приведены наиболее
часто применяемые неводные растворители,
индикаторы и титранты.
Таблица #2.5.50. 1
Растворители, индикаторы и титранты, наиболее часто применяеМblе в неводном титровании
Растворители Индикаторы Титранты
Кислотные Кристаллический фиолетовый,
Уксусная и муравьиная кислоты, судан 111, тропеолин 00, метило Раствор хлорной кислоты в YK
уксусный анrидрид и их смеси с вый фиолетовый, нейтральный сусной кислоте или нитрометане
красный, малахитовый зеленый,
друrими растворителями диметиламиноазобензол
Растворы натрия rидроксида,
Основные Тимоловый синий, бромтимоло калия rидроксида, натрия мети
Диметилформамид, пиридин, вый синий, rз нафтолбензеин, лата, лития метилата, TeTpa
этилендиамин о нитроанилин этиламмония в метаноле или
смеси метанола и бензола
Растворыхлористоводородной
Дифференцирующие кислоты в метаноле или rлико
Метиловый оранжевый, тимоло левых смесях; растворы хлор
Ацетон, диоксан, нитрометан, вый синий, нейтральный Kpac ной кислоты в нитрометане,
метилэтилкетон, метанол, изо ный, метиловый красный, бром в метаноле или в rликолевых
пропанол,третбутанол,диме
тилсульфоксид тимоловый синий смесях; растворы, при меняемые
при титровании в основных pac
творителях
Таблица #2.5.50. 2
Величины pK SH различных растворителей (pK SH = /gKsJ при температуре от 20 ос до 25 ос
Растворитель pK j Растворитель pK i
1. Серная кислота 3,62 17. Нитрометан 24,0
2. Муравьиная кислота 6,10 18. N Метилпирролидон 24,2
3. Уксусная кислота 14,4 19. Пиридин 24,2
4. Уксусный анrидрид 14,5 20. Диметилсульфоксид 97% (*) 24,5
5. Этилендиамин 15,3 21. Диметилформамид 25;3
6. Этиленrликоль 15,6 22. Метилбутилкетон 25,3
7. Формамид 16,7 23. Сульфолан 25,5
8. Метанол 16,7 24. Метилэтилкетон 25,7
9. Пропиленrликоль 16,8 25. Ацетон 25,9
10. Диэтиленrликоль 17,5 26. Ацетон 90% (*) 20,3
11. Этанол 19,1 27. трет Бутанол 26,8
12. Масляная кислота 19,2 28. Пропиленкарбонат 29,2
13. н Бутанол 20,1 29. Аммиак жидкий 29,8
14. Метилцеллозольв 20,7 ЗО. Ацетонитрил 32,2
15. Изопропанол 22,0 31. Диметилсульфоксид 33,3
16. Диметилацетамид 23,9 (*) Друrой компонент вода.
246
rосударственная фармакопея Республики Беларусь
Таблица #2.5.50. 3
:1:
JI aI О
JI с; JI ...
I Ф
";!. с; с; с; о а :.:
I о о о :.: о с;
с; It) с; :1: :1: :.: s с') :1: с) S
Кислота <11 о Ф о <11 <11 S Е о о ф ...
E:t :1: :1: 1: ... Е с; >-
<11 с; <11 О >- :1: с; ... :1:
О Ф \о
m ... о ... а. LO :1: Ф Ф :::r о о
ф ж >- 1: . Ф с; :::r <t ... с')
ra LO о ... с; s с; ф s
... ф :::r
м а. s 1: S с;
s ... ... о ... <с s
а. ф ... I
I t: ф
I I
!
Хлорная 3,94 2,90
Серная 1,44 3,42 I
Азотная 0,2 3,17 З.75 >
Хлористоводородная 0,8 1,05 1,95 3,10 5,50 8,90 2,47
Бромистоводородная 0,2 2,0 5,0
п ТолуолсулЬфоновая 3,82
Уксусная 4,75 9,70 10,41 10,З5 11,З5 14,27 8,32 9,10 11,10 12,55 10,27
Монохлоруксусная 2,86 7,80 8,51 8,50 9,23 12,24 6,05 9,10 11,20 7,60
Дихлоруксусная 1,31 6,30 7,14 7,30 7,80 10.27 4,50 10,20
Трихлоруксусная 0,70 4,90 5,70 6,30 I 5,90 8,20
Фенилуксусная 4,31 8,06 8,78
Муравьиная 3,75 9,15 8,82 9,70 J
Бензойная 4,20 9,52 10,13 10.24 15,10 8,16 8,8З 10,70 11,95 9,70 !
п Нитробензойная 3,40 8,40 8,87 9,10 9,60 12,04 10,59 8,09 !
м Нитробензойная 3,46 , 8,30 9,0 9,15 I 9,20 10,66 8,03 +
п Аминобензойная 4,92
3,5 Динитробензойная 2,80 18,31 10,60 9,63 18,80;
Салициловая 2,89 7,90 8,60 7,73 9,53 8,90 9,53 7,22 I
Ацетилсалициловая 3,50 I
Никотиновая 4,73 16,60 !
Барбитуровая 4,01 I
Винная 3,03 7,40
i I
Лимонная 3,10 I i
i
Фенол 9,89 14,2 19,08 i
I !
Резорцин 9,20
о Нитрофенол 7,17 13,82 10,75 I
п Нитрофенол 17,15 11,0 11,0 11,19 14,48 13,52 10,93
м Нитрофенол ! 8,30 12,6 I
2,4 Динитрофенол 4,02 7,85 8,21 8,36 10,68 8,76 6,45
2,5 Динитрофенол i 5,22 8,98 8,10
2,6 Динитрофенол I 3,71 7,63 6,77 17,60
3,5 Динитрофенол ' 6,70 I 10,83 13,40
Пикриновая 0,80 4,80 3,93 4,50 3,70 3,65 3,17 2,18 11,0
Барбитал 7,43 12,69 16,8
Фенобарбитал 7,21 19,20
Сульфадимезин 7,51 19,60
Сульфадиметоксин 5,90
Сульфамеразин 7,10
Норсульфазол 6,80 I
I
Сульrин 13,00
Метилурацил 9,70
Фторурацил 7,98
Тиоурацил 7,82
Величина РКа кислот в разных растворителях (РКа = gK;
#2.5.50. Титрование в неводных растворителях
247
ь
астворитель
I I I
q q q ::1: CG q
:s; :s: q о с
::1: :s: u u :s: CG q ':S: C'G о 15 :s:
о ::Е :.:: :.:: ж t:; Q.
i C'G о о ::Е о r:; ж :s: :s: о u r:; q
q са :s: r:; :.:: r:;
Ф ::Е €- .е- 1.0 са ::1: q :s: u :s:
:.:: :s: Q. а. о :s: :.:: :s: ...
r:; ::Е а. I ..Q ..Q о Ф CtI Ф а. :s: :s: u :.:: ж
:s: са о I r:;C;' ::r :.:: :s: ::Е а. q :Е :.:: а:: CG
::Е .е- >-...... са :1: Ж О :s: :s :.:: а:: са а::
i (') r:; I u иО) r:; ф о а. 1:: а. CG CtI Ж са ':s
I r:; а. r:; r:; :s I r:; :s ж :s ж ::n
о :s i , ф r:; :s; ..Q а:
I :s: е :s: :s s s :s t: ::Е u ж
ф ф 1:: ::r ::J: >- ш r:; (J
I ф ::Е I ф ф ::Е о с;( ф ::Е u са u >-
I :!Е :s: I ::Е ::Е :s Q. i ::Е <с :.:: а. са CJ
I:I s s I:I t: . >- >-
I:I I:I <: :!Е
2,20 5.34 1,90 2,23 3,2З 2,27 2,70 0,28 12,1 0,90
5,48 3,10 4,60 5,10 4,25 0,58 4,90
4,66 8,80 4,30 2,37 5,10 14,2 8,20
. 8,30 7,77 6,20 8,10 4,08 5,40 2,89 5,30 0,89 13,3 8,30
1,80 5,51 4,36 4,90
1,55 5,30 2,68 0,34 12,90
16,6 6,91 13,50 12,60 12,60 18.81 22,30! 20,50 13,30 11,44 4,11
. .. . . .. ,
15 4,50 10,10 8,90 8,75 16.55' 18,8() Ш,О 10,90
. 10,26 15,80 14,10 8,30
8,86 1,46 10,60 11,30 !
T _-Т__ ' 6,57 12,90 11,60 20,10
16,70 5,74 11,55 17,09, ! 112.0 8,84
I ;
I
16,6 6,36 12,20. 11.10 11,0 19.67 20.70119,60 12,30 9,80
. .
5,88 10,60 9,0 18,70 17,60 10,50 7,94
I
I 5,40 10,82 9,20 19,29 i 17,60 10,70 8,16
.L
.. 10,20
9,78 8,95 7,40 16,90 i
16,701
! 13 О 4,73 8,30 i 6,80 8,34 6,90 15,23
I : .j-.--
! 16,30 . . 11,30 t i
..
. i 15,0 10,80 9,60 9,60
12,32 , 6,67
...
- -, 8,90
. t 10,1 10,60
,22,3 18,0 16,40 15,62 26,60 25,70 17,60 16,20
- .
I 14,73
,._ 11,0 ,
I 12,14 22,0 21,40 12,60 10,03
I 8,53 11,83 11,0 19,71 2ЩЮ 20,10 12,50 9,60
21,24
9,31 4,52 6,36 5,20 14,87 16,0 15,90 6,80 4,38
5,94 8,78 16,45 17,50 17,9 5,76
4,17 6,18 4,90 16,8 16,0
11,42 10,60 20,50 19,40
3,70 1,33 3,65 1,0 9,90 11,0 10,50 3,65
19,0 1З,0 11,15 23,4
13,30 13,40 10,98 10,9
18,70 13,0 11.0
7,47
7,43
11,22
18,05
15,30 13,0
12,40 10,1
12,20 10,8
248
rосударственная фармакопея Республики Беларусь
ж
.Q 111 О
.Q 1:; .Q ...
Ф
1:; 1:; 1:; о 1:;
о О О О О о 1:;
1:; It) 1:; ж ж s м ж Q S
Кислота со о с» о со со s Е о о с» ...
I:t ж ж 1: ... Е 1:; >-
со 1:; со о >- ж 1:; ... ж
о ф \о
m ... о ... Q. Lй ж ф ф :r о о
ф ж >- 1: . Ф 1:; :r ...
со Lй ... ос( ф м
... О Ф 1:; S 1:; :r s
м Q. S 1: S 1:;
('1) :s: ... ... о ... ос( s
('1) Q. Ф ...
t:: :Е ф
Цинхофен 6,22
Рутин 8,96 10,34 13,15
Кверцетин 8,21 9,18 14,41
Ликуразид 9,46 12,30 14,34
Изосалипурпозид 7,67 11,66 12,90
Неодикумарин 4,37 5,71 7,37
300кумарин 4,70 9,63 7,81
Таблица #2.5.50А
Величина РК вн . оснований в разных растворителях (рК вн . = pK SH рК в = рК т )
ж со
О ...
.Q 111 I:t о
1:;
1:; .Q S U
.Q О 1:; 1:; s
1:; о о
Основание о s м Q.
Е о Q. о::
о 1:; со
s s
It) 1:; ж 1:; 1: Ж
1:; (7) ... ф ф 1:; s
о 1:; ж 1:; :r s .Q
Ж О Ф S 1:; ... 111
со со ж 1:; 1: S Ф со
I:t ... со s о ... Q.
О Ф ... ... Q. Ф . >-
m ('1) ('1) t::
т етраметилryанидин 13,60
rуанозин 12,40 15,0
Пиперидин 11,20 11,0 12,51 12,52 11,71 10,40
Диэтиламин 10,90 12,20 9,20 5,19
Бензиламин 9,62 11,30
Цитидин 9,80 13,20
Аденин 9,90 13,70
Трибутиламин 9,85
Триэтиламин i 10,70 11,15 10,87 8,70
Диметиламин , 10,60
н Бутиламин 10.60
Аденозин 10,55 14,90
Дифенилryанидин 10.10 13,60 15,26 8,90
Аммиак 9,30 11,70
Анилин 4,58 6,10 5,70 6,12 6,12 5,49
Диметиланилин 5,10 4,50 4,40
Диэтиланилин 6,52
п Хлоранилин 4,00 4,66
м Хлоранилин 3,52 4,35
о Нитроанилин 0,29
м Нитроанилин 2,50 4,25
п Нитроанилин 1,02 1,29 1,01
Пиридин 5,15 5,54 4,30 5,92 5,69 5,50 I
п Толуидин 5,07 6,60 6,30 I
#2.5.50. Титрование в неводных растворителях
249
Растворитель
J:l J:l J:l 1- :Е: cu J:l
:s: :s: J:l о 1- cu
:Е: :s: u u :s: cu J:l ':s: cu о 1- :s:
о :Е :.: :.: :Е: Ь 1:; о а.
1- cu О О :Е о 1:; :Е: :s: :s: u 1:; J:l
J:l cu :s: 1:; :.: 1:;
CI) :Е -& -& \о cu :Е: J:l :s: u :s:
:.: :s: а. 1- а. а. 1- о :s: :s: :.: :s: ...
1:; :Е .13 .13 о CI) cu 1- CI) а. :s: u :.: :Е:
:s: cu О 1:; 1:;;0:::: :r :.: :s: :Е а. J:l :Е :.: о:: cu
:;; :Е -& >. >.,... cu :Е: :Е: О :s: :s: :.: о:: cu о::
а. 1:; u Ф 1:; CI) о а. Е: а. cu cu :Е: cu ':s:
1:; о :s: 1:; :s: 1:; 1- 1- 1:; :s: :s: :Е: :s: :Е: :ii
:s: е 1- :s: :s: 1- :s: CI) :s: :s: Е:: :Е u .13 о:: :Е:
CI) 1- 1- CI) Е: :r :I: 1- >. ш t; u
:Е CI) CI) :Е о <{ CI) :Е u cu u >.
:Е :s: :Е :Е :s: а. :Е <{ :.: а. cu u
1:[ :s: :s: 1:[ t: . >- >. :Е :.:
1:[ 1:[ :Е >-
11,28
11,12 12,1
11,22 11,63
13,15 1З,28
11,83 12,20
5,75 6,2З
8,45 9,90
астворитель
:Е: J:l
J:l о J:l :s: 1-
:s: 1- :s: u cu
cu а. CI) :Е :.: :Е:
1- :Е: :.: О
О J:l о t; cu -& о
1:; :s: 1- :s: :Е \о
U ... CI) 1- а. .13 а. t;
:Е:
:s: cu :.: >. о t; cu :Е: :s:
:.: t; \о J:l -& >. :.: cu а.
о:: ':s: :s: о :s: u :Е: 1- 1-
:ii I- М :Е t; t; CI) CI) :s:
cu :Е: со) :s: :s: :s: t; :Е :Е:
:Е: :Е: О 1:; t; cu 1- 1- :s: о о
u u 1- :s: :s: :Е CI) CI) Е: а. 1-
>. >. CI) 1- 1- а. :Е :Е о 1- CI)
U U
:.: :.: :r CI) CI) о :s: :s: а. :s: :r
>- >- <{ :Е :Е е 1:[ 1:[ Е:: :I: <{
20,30 13,65 13,20 23,3
10,1 12,24 1 З,48 11,08 10,40 18,53 18,22 18,92
10,10 13,44 10,10 10,50 18,69 17,95 18,70
10,10 17,77 18,10
10,20 11,50 11,62 12,40 9,99 9,25 9,0 17,91 18,З5 18,46
10,0 10,40 19,96 . 18,73
10,50 11,10 16,86 18,26
10,0 15,90 8,90 17,00 17,20 17,90
10,10 9,45 10,50 15,90 15,70 16,46
8,60 5,92 9,63 4,10 4,36 3,60 10,56 9,07 10,56
9,93 4,91 6,20 2,51 11,15 11,04
10,20 10,60 6,26 7,20 12,08
9,27 5,34 3,26 7,99
9,25 4,85 8,40 2,75 7,74
6,95
6,70 3,97 7,40 6,62
3,52 5,18
10,0 9,90 5,77 6,94 4,4З 3,30 3,40 11,92 12,16 19,33
6,57 6,81 5,81 10,90 9,80 11,25
250
rосударственная фармакопея Республики Беларусь
ж CII
О
.D ID q О
1:;
1:; .D :s u
.D О 1:; 1:; :s
1:; х о о х
Основание о :s м а.
х Е о а. а:
о 1:; CII
an :s ж 1:; :s ж
1:; cn Е ф ф с :s
о 1:; ж 1:; ::r 1:; .D
:s
ж О ф :s 1:; ID
CII CII Ж 1:; С :s ф CII
q CII :s О :Е а.
о ф а. ф . >.
m :!: (') (') t: :!: :!:
м Толуидин 4,71 3,20 5,90
Q ПИКОЛИН 5,95 16,24 5,54 6,38 6,09
а Нафтиламин 3,92 5,66 5,10 5,05
Дифениламин 0,90 : 3,18
Ацетамид 0,48 :
Ацетоксим 1,81 i
Тиомочевина 0,96 !
Эфедрин 9,70 11,29
Атропин 9,60
Цитозин ;9,40 12,50
Новокаин i8,80
Промедол 8,40 !
Морфолин 18,70 10,14 9,19
Тебаин 18,30
Димедрол 8.20 I
Платифилин 18,10
Кодеин :8,00 8,60 11 ,40 9,38 5,11
Морфин i7,80 8,60 9,51 5,08
rидразин 18,11 11,10
Хинин 8,00 5,23
Бруцин 7,96
Этилморфин 7,90
Спазмолитин 7,70
Пилокарпин 6,80
Резерпин 6,60
Наркотин 6,20 7,20
rидроксиламин 5,97 9,00
Папаверин 5,90 6,92 7,25
Тифен 16,30
Уротропин 4,90
Хинолин 4,80 4,58 5,39 5,27
Амидопирин 14,80
Дибазол 4,20
Акридин 14,11
Теофилин 2,60
Фенацетин 2,20
Антипирин 1,51 .
Кофеин 0,60 5,17 ;
Теобромин 0,10 5,26
Мочевина 0,20 4,67
Ацетанилид 0,40
#2.5.50. Титрование в неводных растворителях
251
Растворитель
х tI
tI о tI S
S S U CII
CII а. ф ::Е :.: х
х :.: О
О tI о t; CII -е- о
t; S S ::Е -.о
u ... ф а. .lJ а. t;
S Х :.: >- О t; CII Х S
CII t; -.о tI -е- >- :.: CII а.
>S S о S u х ....
о:: Ji м ::Е t; t; ф ф s
CII х (') S S S t; ::Е х
х х о t; t; CII .... s о о
u u .... S S ::Е ф ф 1: а. ....
>- >- ф .... а. ::Е ::Е о .... CIJ
U U :r ф ф о s S а. S :r
:.: :.:
>- >- <с :iE :iE е Е::{ Е::{ t: :I: <с
6,15 9,70
6,64
9,60 5,42 6,38 9,71
7,45 З,87 5,24
6,75 8,60 9,50
8,42
7,75
8,90
11,30
11,30 8,60
11,30 14,40 8,20
16,61 15,18 16,61 15,80
11,01
7,70
11,50
10,80 9,62 11,81 8,30
9,57 8,30
11,50 8,75
12,90
11,10
11,60 13,60 7,00
10,90
9,54
8,10 8,30
10,80 8,03 6,60
11,30 1З,40
9,70 7,00 9,80 <
9,86 5,41 11,40
9,13 6,20
9,00 6,40
6,60
6,00 3,20
8,35 9,60 6,04
6,30
6,10
7,65 9,36
6,80 7,ЗО
252
rосударственная фармакопея Республики Беларусь
2.6.
БиолоrИЧЕСКИЕ
ИСПЫТАНИЯ
01/2013:20601
2.6.1. СТЕРИЛЬНОСТЬ
Испытание применяется для субстанций,
лекарственных средств или изделий, которые,
в соответствии с Фармакопеей, должны быть
стерильнbI. Однако удовлетворительный pe
зультат показывает лишь то, что в условиях
испытания в испытуемом образце не обнару
жено миКРООР2анизмов.
МЕРЫ ПРЕДОСТОРОЖНОСТИ ПРОТИВ
МИКРОБНОЙ КОНТАМИНАЦИИ
Испытание на стерильность выполняют в
асептических условиях. Для достижения таких
условий обстановка, в которой выполняется ис
пытание, должна быть адаптирована к способу
ero проведения. Меры предосторожности, при
нимаемые против контаминации, не должны
влиять ни на один из микроорrанизмов, обнару
живаемых в ходе испытания. Рабочие условия, в
которых выполняется испытание, реryлярно KOH
тролируются путем отбора проб Bo yxa и по
верхностей рабочей зоны, а также проведением
соответствующих контрольных испытаний.
ПИТАТЕЛЬНЫЕ СРЕДЫ И ТЕМПЕРАТУРЫ
ИНКУБАЦИИ
Среды для проведения испытаний МО2ут
быть при20товлены в соответствии с при
веденными ниже указаниями; также М02ут иc
пользоваться эквивалентные коммерчески
доступные среды при условии, что они вы
держивают испытания на питательные свой
ства.
Показано, что следующие питательные
среды являются подходящими для испытания
на стерильность. Жидкая тиоrликолевая среда
предназначена в первую очередь для культи
вирования анаэробных бактерий, но также при
roдна для обнаружения и аэробных бактерий.
Среда на основе rидролизатов соевых бобов и
казеина #(и среда Сабуро)# подходит для культи
вирования как rрибов, так и аэробных бактерий.
MorYT быть использованы также и друrие
среды при УСf1QВИИ их соответствия требовани
ям испытаний на приroдность.
ЖИДКАЯ ТИОiЛИКОЛЕВАЯ СРЕДА
L Цистин 0,5 r
Arap 0,75 r
Натрия хлорид
rлюкоза моноrидрат/rлюкоза
безводная
Дрожжевой экстракт
(водорастворимый)
Панкреатический rидролизат
казеина
Натрия тиоrликолят или
тиоrликолевая кислота
Раствор 1 r/л резазурина натрия
свежеприroтовленный
Вода Р 1000 мл
рН среды после стерилизации (7,НО,2)
2,5 r
5,5 r 1 5,0 r
5,0 r
15,0 r
0,5 r
О,З мл
1,0 мл
Смешивают L цистин, arap, натрия хлорид,
rлюкозу, водорастворимый дрожжевой экс
тракт и панкреатический r ролизат казеина с
1000 мл воды Р и наrpевают до получения paCT
вора. Натрия тиоrликолят или тиоrликолевую
кислоту растворяют в полученной смеси и, при
необходимости, прибавляют 1 М раствор 2и
дроксида натрия так, чтобы после стерилиза
ции значение рН раствора составляло 7,НО,2.
Если необходима фильтрация, раствор снова
наrревают, не доводя до кипения, и фИЛЬТf'уют в
roрячем виде через увлажненную фильтроваль
ную бум а ry. Прибавляют раствор резазурина
натрия, перемешивают и помещают среду в под
ходящие сосуды, которые обеспечивают такое
отношение поверхности к rлубине среды, чтобы
изменению окраски, указывающему на поrлоще
ние кислорода в конце периода инкубирования,
был подвержен только верхний слой, составля
ющий не более трети от общеro объема среды.
Стерилизуют с использованием валидированно
ro процесса. Среду хранят при температуре от
2 ос до 25 ос в стерильном воздухонепроница
емом контейнере. Если верхний слой, составля
ющий не более трети от общеro объема среды,
приобретает розовую окраску, среду можно oд
нократно реrенерировать путем HarpeBa KOH
тейнеров на водяной бане или в струе пара до
исчезновения розовой окраски с последующим
быстрым охлаждением; при этом должны быть
приняты меры предосторожности против по
падания в контейнер нестерильноro воздуха.
Среду не используют по истечении валидиро
ванных сроков хранения.
Жидкую тиоrликолевую среду инкубируют
при температуре от 30 0 С до 35 ОС.
ДЛЯ продуктов, содержащих ртутьсодержа
щие консерванты, которые не представляется
возможным контролировать методом мембран
ной фильтрации, может использоваться жидкая
тиоrликолевая ср"О!да, инкубированная при TeM
пературе от 20 ос до 25 ОС, вместо среды на
основе rидролизатов соевых бобов и казеина
при условии ее приrодности на основании pe
зультатов испытаний на ростовые свойства.
В тех случаях, Korдa это описано или оправ
дано и обосновано, может быть использова
2.6.1. Стерильность
253
на следующая альтернативная тиоrликолевая
среда. rотовят смесь, имеющую тот же состав
что и жидкая тиоrликолевая среда, но не вклю
чающую arap и раствор резазурина натрия; CTe
рилизуют, как указано выше. Значение рН среды
после стерилизации (7,1хО,2). Наrревают на
водяной бане перед использованием и инкуби
руют при температуре от 30 ос до 35 ос в анаэ
робных условиях.
Среда на основе rидролизатов соевых
бобов и казеина
Панкреатический rидролизат Ka 17,0 r
зеина
Папаиновый rидролизат сои
Натрия хлорид
Дикалия rидрофосфат
rлюкоза моноrидрат/rлюкоза без
2,5 r / 2,3 r
водная
Вода Р 1000 мл
рН среды после стерилизации (7,З:tО,2)
3,0 r
5,0 r
2,5 r
Твердые компоненты растворяют в воде Р,
слеrка наrревая для получения раствора. PaCT
вор охлаждают до комнатной температуры. При
необходимости прибавляют 1 М раствор 2и
дроксида натрия так, чтобы после стерилиза
ции значение рН раствора составляло (7,3хО,2).
Раствор фильтруют (при необходимости YCTpa
нения мутности), разливают по подходящим co
судам и стерилизуют с использованием валиди
pOBaHHoro процесса. Если среда не подлежит
немедленному использованию, ее хранят при
температуре от 2 ос до 25 ос в стерильном плот
ноукупоренном контейнере. Среду не использу
ют по истечении валидированных сроков xpaHe
ния.
Среду на основе rидролизатов соевых бобов
и казеина инкубируют при температуре от 20 ос
до 25 ОС.
#Допускается использование среды Сабуро:
Пептон ферментативный 10,0 r
rлюкоза моноrидрат/rлюкоза без 40,0 r / 36,4 r
водная
Вода Р до 1000 мл
рН среды после стерилизации 5,6хО,2
Среду Сабуро инкубируют при температуре
от 20 ос до 25 ОС.#
Используемые среды должны выдерживать
следующие испытания, проводимые до испыта
ния продукта или параллельно с ним.
Стерильность. Порции среды инкубируют в
течение 14дней. Роста микроорrанизмов наблю
даться не должно.
Ростовые свойства сред в отношении
аэробов, анаэробов и rрибов. Проверке
подверrают каждую партию roтовой среды
и каждую партию среды, приroтовленной из
обезвоженных составов или инrредиентов.
Подходящие штаммы микроорrанизмов при
ведены в таблице 2.6.1. 1.
Порции жидкой тиоrликолевой среды ино
кулируют небольшим количеством (не более
100 КОЕ) микроорrанизмов, используя отдель
ную порцию среды для каждоro из следующих
штаммов: C/ost,idium sроrogепеs, Pseudomo
паs ае,иgiпоsа, Staphy/ococcus au,eus. Порции
среды на основе rидролизатов соевых бобов
и казеина #(или среды Сабуро)# засевают He
большим количеством (не более 100 КОЕ)
микроорrанизмов, используя отдельную
порцию среды для каждоro из следующих
штаммов: Aspe,gillus Ь,аsi/iепsis, Bacillus sub
ti/is, Caпdida a/bicaпs. Инкубируют в течение
не более 3 дней #(при температуре от ЗО ОС до
35 ОС)# в случае бактерий и не более 5 дней
#(при температуре от 20 ос до 25 ОС)# в случае
rрибов.
При получении рабочей культуры, исполь
зуемой при проведении испытания, число
пересевов, произведенных от исходноro
штамма, не должно превышать пяти.
Среды являются приrодными при усло
вии наличия четкоro визуально наблюдаемоro
роста микроорrанизмов.
Таблица 2.6.1. 1
Штаммы тест миКрООр2анизмов, при20дные
для испытаний питательных сред
Аэробные бактерии
Staphy/ococcus АТСС 6538, С/Р 4.83,
au,eus NCTC 10788, NC/MB 9518,
NBRC 13276, #АТСС 6538 P#
Bacillus subtilis АТСС 6633, С/Р 52.62,
NC/MB 8054, NBRC 3134
Рsеиdотопаs АТСС 9027, NC/MB 8626,
аеrиgiпоsа CtP82.118, NBRC 13275
Анаэробные бактерии
Ctost,idium АТСС 19404, С/Р 79.3,
sроrogепеs NCTC 532, АТСС 114З7,
NBRC 14293, #rИСК 272#
rрибы
Caпdida АТСС 10231, /Р48.72,
а/Ысапs NCPF 3179, NBRC 1594,
#АТСС 885 65З#
Aspergillus АТСС 16404, 'Р 1431.83,
Ьrаsiliепsis /М/149007, NBRC 9455
ПРОВЕРКА приrодности
Проверку приrодности методики выполня
ют в соответствии с нижеприведенным опи
санием испытания на стерильность испыту
eMoro образца с использованием TaKoro же
метода, но со следующими модификациями.
254
rосударственная фармакопея Республики Беларусь
Мембранная фильтрация. После переноса
содержимоro испытуемоro контейнера или KOH
тейнеров на мембрану заключительную порцию
стерильною растворителя, используемоro для
промывки фильтра, инокулируют небольшим KO
личеством жизнеспособных микроорrанизмов
(не более 100 КОЕ).
Прямая инокуляция. После внесения в пи
тательную среду содержимоro испытуемоro KOH
тейнера или контейнеров ту же среду инокули
руют небольшим количеством жизнеспособных
микроорrанизмов (не более 100 КОЕ).
В обоих случаях используют штаммы микро
орrанизмов, приведенные выше в описании ис
пытания ростовых свойств в отношении аэро
бов, анаэробов и rрибов. За положительный
контроль принимают результаты испытания po
стовых свойств иcnользуемых сред. Все контей
неры, содержащие питательную среду. инкуби
руют не более 5 дней.
Если после периода инкубации отчетливо
наблюдается рост микроорrанизмов, визуаль
но сравнимый с ростом в контрольном сосуде,
не содержащем испытуемоro образца, делают
вывод, что либо продукт в условиях испытания
не обладает антимикробной активностью, либо
такая активность была в достаточной степени
исключена. В таком случае испытание на CTe
рильность может проводиться без дальнейших
модификаций.
Если после периода инкубации не наблюда
ется роста микроорrанизмов, визуально сравни
моro с ростом в контрольном сосуде, не coдep
жащем испытуемою образца, делают вывод, что
продукт обладает антимикробной активностью,
которая в условиях испытания не была в ДOCTa
ючной степени исключена. Условия подверrа
ют модификациям С целью исключения антими
кробной активности и повторяют испытание на
приюдность.
Испытание на проверку приюдности следу
ет проводить в следующих случаях:
а) Korдa про водится испытание на стериль
ность новой продукции;
б) при внесении любых изменений в экспе
риментальные условия испытания.
Проверка приrодности может выполняться
одновременно с контролем стерильности испы
туемой продукции.
#Частные статьи должны содержать CBeдe
ния о наличии антимикробноrо действия продук
та и рекомендации по еro устранению.1'
ИСПЫТАНИЕ НА СТЕРИЛЬНОСТЬ
ИСПЫТУЕмоrо ОБРАЗЦА
Испытание может быть выполнено с исполь
зованием метода мембранной фильтрации или
путем прямой инокуляции питательной среды
испытуемым образцом. Должны про водиться
соответствующие отрицательные контрольные
опыты. В тех случаях, Korдa позволяет природа
продукта (фильтруемые водные, спиртовые или
масляные образцы, а также образцы, смешива
емые или растворимые в водных или масляных
растворителях, не обладающих антимикробным
эффектом в условиях испытания), следует пред
почесть метод мембранной фильтрации.
Мембранная фильтрация. Используют
мембранные фильтры с номинальным разме
ром пор, не превышающим 0,45 мкм, с YCTaHOB
ленной способностью к удерживанию бактерий.
Например, фильтры на основе нитрата целлю
лозы используют для водных, масляных и раз
бавленных спиртовых растворов, а фильтры на
основе ацетата целлюлозы в частности, для
растворов с высоким содержанием спирта. Для
некоторых видов продукции, например, для aH
тибиотиков, MOryт потребоваться специально
подютовленные фильтры.
В нижеописанном методе предполаrается
использование мембран диаметром около 50 мм.
При использовании фильтров друrих диаметров
объемы разведений и промывок должны быть
изменены соответствующим образом. Аппарат
для фильтрации и мембрану стерилизуют лодхо
дящим способом. Конструкция аппарата должна
обеспечивать введение и фильтрацию испытуе
моro раствора в асептических условиях, извле
чение мембраны для ее переноса в питательную
среду или проведение инкубации после помеще
ния питательной среды в аппарат.
Водные растворы. Небольшое количество
подходящеro стерильноro растворителя, не по
давляющеro рост микроорrанизмов, например,
нейтральноro раствора 1 r/л мясною или казе
иновоro пептона (рН (7,1:1:0,2», помещают на
мембрану аппарата и фильтруют. Растворитель
может содержать подходящие нейтрализующие
и/или инактивирующие вещества, например, в
случае антибиотиков.
Содержимое контейнера или контейнеров,
подлежащих испытанию, переносят на мембра
ну (мембраны), при необходимости после разве
дения до объема, используемоrо в испытании на
приюдность, выбранным стерильным раствори
телем; в любом случае, используют количество
испытуемоro образца не менее указанноrо в Ta
блице 2.6.1. 2. Немедленно фильтруют. Если ис
пытуемый раствор обладает антимикробным
действием, мембрану не менее трех раз про
мывают путем пропускания через нее каждый
раз объема выбранноro стерильною раствори
теля, указанноro в испытании на приroдность.
Число циклов промывки должно быть не более 5
из расчета по 100 мл на фильтр, даже если при
проверке приroдности было показано, что такая
промывка не полностью исключает антимикроб
ное действие. Мембрану целиком переносят в
питательную среду или в асептических услови
ях делят ее на две равные части, каждую из KO
торых помещают в две подходящие среды. Ис
пользуют такие же объемы питательных сред,
2.6.1. Стерильность
255
Таблица 2.6.1. 2
Минимальные количества испытуеМ020 продукта, используемые для каждой
питательной среды
Минимальное количество, которое следует ис
Количество в контейнере пользовать для посева на каждую среду, если
не обосновано и не разрешено использование
друrих количеств
Для жидкостей
менее 1 мл I Все содержимое каждоro контейнера
1-........40 мл I Половина содержимоro каждоrо контейнера, но
не менее 1 мл
более 40 мл, но не более 100 мл 120 мл
более 100 мл 11 О % содержимоro контейнера, но не менее 20 мл
Для жидкостей, содержащих антибиотики 1 мл
Нерастворимые лекарственные средства, Все содержимое каждоro контейнера, составляю
кремы и мази, подлежащие суспендированию щее не менее 200 Mr
или ЭМУЛЬ2ированию
Для твердых веществ
Менее 50 Mr I Все содержимое каждоro контейнера
50 Mr и более, но менее 300 Mr I Половина содержимоro каждоro контейнера, но
не менее 50 Mr
От 300 Mr до 5 r ! 150 Mr
Более 5 r i 500 Mr
как и в испытании на приrодность. Кроме тоro,
среда может быть нанесена на мембрану в ап
парате. Среды инкубируют в течение не менее
14 дней.
Твердые растворимые вещества. Для
каждой среды используют количество продукта,
не менее указанноro в таблице 2.6.1. 2, paCTBO
peHHoro в подходящем растворителе, например,
растворителе, прилаrаемом к лекарственному
cpeДCTB изотоническом растворе натрия хло
рида или нейтральном растворе 1 r/л мясноro
или казеиновою пептона, и продолжают испыта
ние по методике, описанной выше, для водных
растворов, с использованием мембраны, COOT
ветствующей выбранному растворителю.
Масла и масляные растворы. Для каждой
среды используют количество продукта не менее
указанноro в таблице 2.6.1. 2. Масла и масляные
растворы, обладающие достаточно низкой вяз
костью, MOryT быть подверrнуты фильтрованию
через сухую мембрану без разбавления. Вязкие
масла MOryT быть при необходимости разбавле
ны подходящим стерильным растворителем, для
котороro показано отсутствие антимикробной aK
тивности в условиях испытания, например, изо
пропилмиристатом. Дают маслу проникнуть в
мембрану под действием собственной тяжести,
затем фильтруют, постепенно увеличивая дaB
ление или вакуум. Мембрану промывают путем
фильтрования не менее трех порций по 100 мл
подходящею стерильною растворителя, напри
мер, нейтральноro раствора 1 r/л мясноro или
казеиновоro пептона, содержащеro подходящий
эмульrатор в концентрации, приемлемость KO
торой была подтверждена в ходе испытания на
приroдность, например, 1 О r/л полисорбата 80.
Мембрану или мембраны переносят в пита
тельную среду или среды (или наоборот) в co
ответствии с указаниями, приведенными выше
для водных растворов, и инкубируют при таких
же температурах в течение тех же промежутков
времени.
Мази и кремы. Для каждой среды использу
ют количество продукта не менее указанноro в
таблице 2.6.1. 2. Мази на жирной основе и BO
дно масляные эмульсии MOryT быть разбавлены
до содержания 1% изопропилмиристатом, как
описано выше, при необходимости при Harpe
вании до температуры, не превышающей 40 ОС.
В исключительных случаях может оказаться He
обходимым HarpeB до температуры, не превы
шающей 44 ОС. Фильтруют насколько возможно
быстро и продолжают испытание в соответствии
с указаниями, приведенными выше для масел и
масляных растворов.
Прямая инокуляция питательной среды.
Количество испытуемою продукта, предписан
ное в таблице 2.6.1. 2, помещают непосред
ственно в питательную среду. При этом объем
продукта должен составлять не более 1 О %
объема среды, если нет друrих указаний.
Если испытуемый продукт обладает анти
микробной активностью, испытания выполняют
после нейтрализации подходящим нейтрализу
ющим areHToM ИJ1И путем разведения в ДOCTa
точном количестве питательной среды. При He
обходимости использования большоro объема
продукта предпочтительнее использовать KOH
центрированную питательную среду, приroтов
ленную с учетом последующеro разбавления.
Если это удобно, концентрированная среда
256
rосударственная фармакопея Республики Беларусь
может быть добавлена непосредственно к про
дукту в контейнере.
Масляные жидкости. Используют среды с
добавкой подходящеro эмульrатора в KOHцeH
трации, приемлемость которой была подтверж
дена в ходе испытания на приrодность, напри
мер, 1 О r/л полисорбата 80.
Мази и кремы. rотовят разбавлением в co
отношении около 1: 1 О путем эмульrирования с
использованием избранною эмульrатора в под
ходящем стерильном растворителе, например,
растворе мясноro или казеиновоro пептона, KOH
центрацией 1 r/л. Разбавленный продукт пере
носят в среду, не содержащую эмульrатора.
Инокулированную среду инкубируют в Te
чение не менее 14 дней. Состояние культур
оценивают несколько раз в течение инкубаци
онноro периода. Инокулированные среды, co
держащие масляные продукты, ежедневно OCTO
рожно встряхивают. Однако при использовании
тиоrликолевой или друюй аналоrичной среды
для определения анаэробных микроорrанизмов
встряхивание или перемешивание сводят к ми
нимуму для поддержания анаэробных условий.
НАБЛЮДЕНИЕ И ИНТЕРПРЕТАЦИЯ
РЕЗУЛЬТАТОВ
Время от времени в течение периода инку
бации, а также по еro завершении оценивают
наличие макроскопических признаков микроб
ноro роста в средах. Если внесение испытуе
моro материала приводит к помутнению пита
тельной среды, вследствие чею наличие или
отсутствие микробноro роста не может быть
леrко определено визуально, следует через 14
дней после начала инкубации перенести порции
среды (каждая объемом не менее 1 мл) в друrие
сосуды, содержащие свежую аналоrичную среду,
и инкубировать исходные сосуды и сосуды с пе
ренесенными порциями в течение 4 дней.
Если признаков микробноrо роста не об
наруживается, то продукт признают выдержи
вающим испытание на стерильность. При Ha
личии признаков микробноro роста продукт
Таблица 2.6.1_ 3
Минимальное количество единиц продукции для испытания
Минимальное количество единиц продукции для
каждой среды, если не обосновано и не разрешено
иное**
Количество единиц в партии*
Лекарственные средства для
парентераЛЬН020 применения
Не более 100 контейнеров
Более 100, но не более 500 контейне
ров
Более 500 контейнеров
ОфтаЛЬМОЛ02ические и дРУ2ие HeиHъeK
ционные лекарственные средства
Не более 200 контейнеров
Более 200 контейнеров
В случаях, коrда продукция представ
лена в форме однодозовых контейне
ров, применяют указанную выше схему
для лекарственных средств для парен
теральноro применения
Твердые продукты «in bu/k»
До 4 контейнеров
Более 4 контейнеров, но не более 50
контейнеров
Более 50 контейнеров
1 О % или 4 контейнера, в зависимости от тоro, что больше
1 О контейнеров
2 % или 20 контейнеров (10 контейнеров для лекарствен
ных средств для парентеральноro применения большою
объема), в зависимости от Toro, что меньше
5 % или 2 контейнера, в зависимости от тоro, что больше
1 О контейнеров
Каждый контейнер
20 % или 4 контейнера, в зависимости от тоro, что больше
2 % или 1 О контейнеров, в зависимости от тоro, что больше
*Если размер серии неизвестен, используют максимальное количество единиц для испытания.
**Если содержимоrо одноro контейнера достаточно для инокуляции двух сред, то в данной колонке
дано количество контейнеров, необходимое для обеих сред.
2.6.7. Микоплазмы
не выдерживает испытание на стерильность,
если не может быть показано путем повтор
ноro испытания или друrим способом, что pe
зультаты испытания не являются ДOCTOBepHЫ
ми по причинам, не связанным с испытуемым
продуктом. Результаты испытания MorYT быть
признаны недостоверными лишь при выполне
нии не менее одноrо из перечисленных ниже
критериев:
а) данные микробиолоrическоro мониторин
ra зоны проверки стерильности указывают на
произошедшие сбои;
Ь) проверка методики, используемой в дaH
ном испытании, привела к обнаружению недоче
тов;
с) в отрицательном контроле был обнару
жен микробный рост;
d) после установления типа микроорrаниз
мов, выделенных в ходе испытания, рост этоro
штамма (этих штаммов) может быть однознач
но приписан ошибкам, вносимым материалами
и/или методиками, используемыми при проведе
нии испытания.
Если результаты испытания признаны Heдo
стоверными, испытание повторяют, используя то
же количество образцов, что и в первоначаль
ном испытании.
Если признаков микробноro роста в ходе по
вторноro испытания не обнаруживается, то про
дукт признают выдерживающим испытание на
стерильность. Если в ходе повторноro испыта
ния обнаруживается рост, то продукт не Bыдep
живает испытание на стерильность.
ПРИМЕНЕНИЕ ИСПЫТАНИЯ
К ЛЕКАРСТВЕННЫМ СРЕДСТВАМ ДЛЯ
ПАРЕНТЕРАльноrо ПРИМЕНЕНИЯ,
ОФТАльмолоrИЧЕСКИМ И дРУrим
НЕИНЪЕКЦИОННЫМ ЛЕКАРСТВЕННЫМ
СРЕДСТВАМ, ДЛЯ КОТОРЫХ ТРЕБУЕТСЯ
ВЫПОЛНЕНИЕ ИСПЫТАНИЯ
НА СТЕРИЛЬНОСТЬ
При использовании метода мембранной
фильтрации используют, rдe это возможно, все
содержимое контейнера, но не менее чем в коли
честве, указанном в таблице 2.6.1. 2, при необ
ходимости разбавляя приблизительно до 100 мл
подходящим стерильным раствором, например.
нейтральным раствором 1 r/л мясноro или казе
иновоro пептона.
При использовании метода прямой инокуля
ции питательной среды используют количества,
указанные в таблице 2.6.1. 2, если не оправдано
и не разрешено применение друrих количеств.
Испытания на стерильность в отношении бакте
рий и rриfiов выполняют на одном и том же об
разце испытуемоro продукта. В случаях, Korдa
объем или количество, содержащиеся в контей
нере, являются недостаточными для проведе
ния испытаний, для инокуляции различных сред
используют содержимое двух или более контей
неров.
ЗЗ Зак. 1712
257
МИНИМАЛЬНОЕ КОЛИЧЕСТВО ЕДИНИЦ
ПРОДУКЦИИ ДЛЯ ИСПЫТАНИЯ
Минимальное количество единиц продукции
для испытания в зависимости от размера серии
указано в таблице 2.6.1. 3.
Руководства по применению испытания на
стерильность приведены в 2лаве 5.1.9.
01/2013:20602
2.6.2. МИКОБАКТЕРИИ
Если испытуемый образец может coдep
жать микроорrанизмы, отличные от микобакте
рий, еro обрабатывают подходящим дeKOHTa
минирующим раствором, например, раствором
ацетилцистеина и rидроксида натрия или pac
твором натрия лаурилсульфата.
На каждую из двух подходящих твердых пи
тательных сред (например, среда Левенштей
на Йенсена или среда Миддлбрук 7Н10) HaHO
сят по 0,2 мл образца. Эксперимент выполняют
в трех повторностях. В подходящую жидкую
питательную среду также в трех повторностях
вносят по 0,5 мл образца. Все среды инкуби
руют при температуре 37 ос в течение 56 дней.
Устанавливают способность среды к
обеспечению роста в присутствии испыту
емоro образца путем инокуляции подходящеro
штамма семейства Mycobacte,ium, например,
BCG, и, при необходимости, используют подхо
дящий нейтрализующий areHT.
Если в течение первых восьми дней ин
кубации наблюдается развитие посторонних
микроорrанизмов, испытание повторяют с oд
новременным проведением испытания на бак
териолоrическую стерильность.
Испытуемый образец выдерживает испы
тание, если в конце инкубационноro периода ни
в одной из пробирок не наблюдается роста ми
кобактерий.
01/2013:20607
2.6.7. МИКОПЛАЗМЫ
Если испытание на микоплазмы пред
писано для rлавноrо или рабочеro банка
клеток, для посевной партии вирусов или для
контрольных клеток, применяют как метод
культивирования, так и метод индикаторной
клеточной культуры. Если испытание пред
писано для вирусноro сбора, нефасованной
вакцины (iп bu/k) или roтовой партии (серии)
258
rосударственная фармакопея Республики Беларусь
продукта, при меняют метод культивирова
ния. Метод индикаторной клеточной культу
ры может при необходимости также исполь
зоваться для скрининrа сред.
При условии надлежащей ваЛИД8ЦИИ
вместо обоих этих методов MorYT использо
ваться методы амплификации нуклеиновых
кислот (АНК, NAT пuc'eic acid атр'ifiсаtiоп
tесhпiqие).
МЕТОД КУЛЬТИВИРОВАНИЯ
ВЫБОР ПИТАТЕЛЬНОЙ СРЕДЫ
Испытание выполняют с использованием дo
статочноrо количества как твердых, так и жидких
питательных сред для TOro, чтобы в выбранных
условиях инкубации был обеспечен рост He
большоro количества микоплазм, которые MOryт
присутствовать в испытуемом образце. Жидкие
среды должны содержать феноловый красный.
Для ряда сред показано наличие удовлетвори
тельных питательных свойств, по крайней мере
для ниже перечисленных орrанизмов. Для каждой
новой партии среды должны быть подтвержде
ны питательные свойства в отношении соответ
ствующих орrанизмов из списка. При проведении
испытания на содержание микоплазм в испыту
емом образце по крайней мере один штамм из
перечисленных ниже должен быть включен в Ka
честве положительноro контроля:
Acho/ep/asma 'aid'awii (вакцины для Me
дицинскоro и ветеринарноro применения, в
процессе производства которых используют
ся антибиотики);
Mycop'asma gal1isepticum (в случаях,
Korдa для производства вакцин используют
ся материалы, имеющие птичье происхожде
ние, или для вакцин, предназначенных для
применения в птицеводстве);
Mycop'asma hyorhiпis (ветеринарные
вакцины, кроме птичьих);
Mycop'asma ora'e (вакцины для меди
цинскоro и ветеринарноro применения);
Mycop'asтa pпeumoпiae (вакцины для
медицинскоrо применения) или друrие под
ходящие виды, связанные с ферментацией
D rлюкозы, например. Micop'asma fe,meп
taпs;
Mycop/asma syпoviae (в случаях, коrда
для производства вакцин используются MaTe
риалы, имеющие птичье происхождение, или
для вакцин, предназначенных для примене
ния в птицеводстве).
Тест штаммы представляют собой изо
ляты, ПС1дверrнутые оrраниченному числу
пассажей (не более чем 15) и хранящиеся в
замороженном или лиофилизированном co
стоянии. После клонирования принадлеж
ность штамма к требуемому виду определя
ется подходящим методом путем сравнения с
типовыми культурами, например:
А. /aid/awii NCTC С/Р АТСС
10116 75.25 23206
М. gallisepticum NCTC С/Р АТСС
10115 104967 19610
М. fermeпtas NCTC С'Р АТСС
10117 105680 19989
М. hуо,hiпis NCTC С/Р АТСС
10130 104968 17981
М. ora'e NCTC С'Р АТСС
10112 104969 23714
М. pпeumoпiae NCTC С/Р АТСС
10119 103766 15531
М. sупоviае NCTC С/Р АТСС
10124 104970 25204
БСП Acho/ep'asma /aid/awii, БСП Micop/asma
fermeпtaпs, БСП Mycop/asma hуоrhiпis, БСП
Mycop'asma ora'e и БСП Mycop/asma sупоviае
MOryт быть использованы в качестве CTaHдapT
ных штаммов с малым количеством пассажей.
УСЛОВИЯ ИНКУБАЦИИ
Жидкие питательные среды помещают в
плотно укупоренные контейнеры и инкубирубт
при температуре от 35 ос до 38 ОС. Твердые пи
тательные среды инкубируют в микроаэрофиль
ной атмосфере (азот, содержащий от 5 % до
1 О % уrлерода диоксида и с достаточной влаж
ностью, чтобы не высыхала поверхность arapa)
при температуре от 35 ос до 38 ОС.
ПИТАТЕЛЬНЫЕ СВОЙСТВА
Питательные свойства испытывают
для каждой новой партии среды. Выбран
ные среды засевают соответствующими TeCT
микроорrанизмами. На чашку диаметром 60 мм,
содержащую 9 мл твердой среды, или в KOH
тейнер вместимостью 100 мл, содержащий
жидкую среду, вносят не более 1 00 колоние
образующих единиц (КОЕ); для каждоro вида
орrанизмов используют отдельные чашки и
контейнеры. Среды инкубируют и через YKa
занные интервалы делают пересев 0,2 мл
жидкой среды на твердую среду (см. Испыта
ние анализируемоro продукта на микоплаз
мы). Твердая среда выдерживает испытание,
если наблюдается адекватный рост каждоro
тест микроорrанизма (наблюдаемый рост OT
личается не более чем в 5 раз по отношению к
первичному материалу). Жидкая среда Bыдep
живает испытание, если обнаруживается рост
хотя бы одноro пересева для каждоro испыту
емоro тест микроорrанизма на чашке с arapoM,
на которую был совершен пересев из жидкой
среды.
инrИБИРУЮЩИЕ ВЕЩЕСТВА
Испытание данноro продукта на инrибирукr
щие вещества проводят только один раз и повт<r
ряют в случае изменений в способе производ
ства, которые MorYT повлиять на определение
микоплазм.
2.6.7. Микоплазмы
259
Для демонстрации отсутствия инrибиру
ющих веществ проводят испытание на лита
тельные свойства в присутствии испытуемоro
образца и в еro отсутствии. Если в отсутствии
испытуемоro образца рост хотя бы одною пе
ресева тест микроорrанизмов происходит бы
стрее, чем в присутствии испытуемоro образ
ца, или если чашки, на которые напрямую был
нанесен испытуемый образец, имеют менее
1/5 количества колоний, которое обнаружива
ется на чашках без испытуемоro образца, это
указывает на присутствие инrибирующих Be
ществ, и их влияние должно быть нейтрализо
вано или учтено каким либо иным способом,
например, посевом на субстраты, не содержа
щие инrибиторов, или разведением в большем
объеме среды перед испытанием. Если ис
пользуется разведение, MorYT быть использо
ваны или большие объемы среды, или объем
первичноrо материала может быть разделен
между несколькими колбами вместимостью
100 мл. Эффективность нейтрализации или
иноro способа проверяется повторением испы
тания на инrибирующие вещества после ней
трализации.
ИСПblТАНИЕ ИСПblТУЕМО/О ПРОДУКТА
НА МИКОПЛА3МbI
В каждую жидкую среду вносят испытуе
мый образец в количестве 1 О мл на 100 мл
среды. Если при прибавлении испытуемоro
образца происходит существенное изменение
показателя рН, исходное значение BOCCTaHaB
ливают прибавлением раствора кислоты хло
ристоводородной или натрия rидроксида. На
каждую чашку наносят по 0,2 мл испытуемо
ro образца. Жидкие среды инкубируют в Te
чение 20 21 дня. Твердые среды инкубируют
в течение не менее 14 дней, за исключени
ем соответствующих 20 21 дневным пере
севам, которые инкубируют в течение 7 дней.
Одновременно инкубируют лорции по 100 мл
каждой жидкой среды и чашки с arapoM без
испытуемоro образца в качестве отрицатель
ною контроля. На 2 4 день после инокуля
ции выполняют пересев по 0,2 мл каждой из
жидких культур по крайней мере на 1 чашку
каждой твердой среды. Данную процедуру по
вторяют на 6 8 день, на 13 15 день и на
19 21 день испытания. Состояние жидких
сред контролируют каждые два или три дня
и, в случае изменения окраски, выполняют
пересев. Если в жидкой среде обнаружива
ются бактериальные или rрибковые KOHTa
минации, испытание считают недействитель
ным. ИС'lытание считают действительным,
если может быть проведена оценка по край
ней мере одной чашки с каждой средой и
каждым выполненным посевом. В испыта
ние включают положительный контроль, при
rотовленный инокуляцией не более 100 КОЕ
по крайней мере одноro тест микроорrанизма
на среду с arapoM или в бульонную среду. В
случае реrулярноro лроведения испытания
на микоплазмы рекомендуется по возможно
сти чередовать тест микроорrанизмы. TeCT
микроорrанизмы перечислены в разделе
«Выбор питательной среды».
ИНТЕРПРЕТАЦИЯ РЕ3УЛЬТАТОВ
В конце периодов инкубации все инокули
рованные твердые среды изучают под микро
скопом для установления наличия колоний
микоплазм. Образец выдерживает испыта
ние, если ни на одной инокулированной среде
не обнаружено роста микоплазм. Образец не
выдерживает испытание, если на любой из
твердых сред обнаруживается рост типичных
колоний микоплазм. Испытание является He
действительным, если в одном или более по
ложительном контроле не обнаруживается
рост микоплазм хотя бы на одной чашке пе
ресева. Испытание является недействитель
ным, если в одном или более отрицательном
контроле обнаруживается рост микоплазм.
При обнаружении подозрительных колоний
может быть использован валидированный
метод определения их принадлежности к ми
коплазмам.
Следующий раздел приводится для инфор
мации
СРЕДЫ, РЕКОМЕНДУЕМЫЕ ДЛЯ МЕТОДА
КУЛЬТИВИРОВАНИЯ
Рекомендуются нижеследующие среды.
Moryт использоваться и друrие среды при yc
ловии, что на каждой партии в присутствии и
отсутствии испытуемоro продукта продемон
стрирована способность к поддержанию роста
микоплазм.
СРЕДЫ ХЕЙФЛИКА (НАVFLIСК)
(РЕКОМЕНДОВАНЫ ДЛЯ ОБЩЕ/О
ОПРЕДЕЛЕНИЯ МИКОПЛАЗМ)
Жидкая среда
Бульон из экстракта
roвяжьеro сердца (1)
Лошадиная сыворотка
(не подверrнутая HarpeBY)
Дрожжевой экстракт (250 r/л)
Феноловый красный (раствор 0,6 r/л)
Пенициллин (20 000 МЕ/мл)
Дезоксирибонуклеиновая кислота
(раствор 2 r/л)
90,0 мл
20,0 мл
10,0 мл
5,0 мл
0,25 мл
1,2 мл
Значение рН доводят до 7,8.
Твердая среда
rотовят, как указано выше, заменяя бульон
из экстракта roвяжьеro сердца на arap из экстрак
та roвяжьеro сердца, содержащий 15 r/л arapa.
260
/осударственная фармакопея Республики Беларусь
СРЕДЫ ФРЕЯ (FREY) (РЕКОМЕНДОВАНЫ
ДЛЯ ОПРЕДЕЛЕНИЯ М. SYNOVJAE)
Жидкая среда
Бульон из экстракта 90,0 мл
roвяжьеro сердца (1)
Смесь незаменимых 0,025 мл
витаминов (2)
rлюкоза моноrидрат 2,0 мл
(раствор 500 r/л)
Свиная сыворотка 12,0 мл
(инактивированная при температуре
56 ос в течение 30 мин)
(3 Никотинамидадениндинуклеотид 1,0 мл
(раствор 10 r/л)
Цистеина rидрохлорид 1,0 мл
(раствор 10 r/л)
Феноловый красный 5,0 мл
(раствор 0,6 r/л)
Пенициллин (20 000 МЕ/мл) 0,25 мл
Смешивают растворы J3 никотинамида
дениндинуклеотида и цистеина rидрохлорида
и через 1 О мин прибавляют к остальным компо
нентам. Значение рН доводят до 7,8.
Твердая среда
Бульон из экстракта
roвяжьеro сердца (1)
Arap очищенный (3)
90,0 мл
1,4 r
Значение рН доводят до 7,8, стерилизуют
автоклавированием, после чеro прибавляют:
Смесь незаменимых витаминов (2) 0,025 мл
rлюкоза моноrидрат 2,0 мл
(раствор 500 r/л)
Свиная сыворотка 12,0 мл
(не подверrнутая HarpeBY)
(3 Никотинамидадениндинуклеотид 1,0 мл
(раствор 10 r/л)
Цистеина rидрохлорид 1,0 мл
(раствор 10 r/л)
Феноловый красный 5,0 мл
(раствор 0.6 r/л)
Пенициллин (20 000 МЕ/мл) 0,25 мл
СРЕДЫ ФРИИСА (FRIIS)
(РЕКОМЕНДОВАНЫ ДЛЯ ОПРЕДЕЛЕНИЯ
МИКОПЛА3М, ИМЕЮЩИХ ОТЛИЧНОЕ
ОТ ПТИЧЬЕrо ПРОИСХОЖДЕНИЕ)
Жидкая среда
Сбалансированный солевой раствор
Хэнкса (модифицированный) (4)
Вода дистиллированная
Экстракт из сердца и мозroв (5)
Бульон PPLO (6)
Дрожжевой экстракт (170 r/л)
Бацитрацин
Метициллин
Феноловый красный (5 r/л)
800 мл
67 мл
135 мл
248 мл
60 мл
250 Mr
250 Mr
4,5мл
Лошадиная сыворотка 165 мл
Свиная сыворотка 165 мл
Значение рН доводят до 7,40 7,45.
Твердая среда
Сбалансированный солевой раствор 200 мл
Хэнкса (модифицированный) (4)
OEAE дeKCTpaH 200 Mr
#(диэтиламиноэтилдекстран )#
Arap очищенный (3) 15,65 r
Хорошо перемешивают и стерилизуют aB
токлавированием. Охлаждают до температуры
100 ОС. Прибавляют к 1740 мл вышеописанной
жидкой среды.
(1) Бульон из экстракта 20вяжье20
сердца
rовяжье сердце
(для приroтовления экстракта)
Пептон
Натрия хлорид
Вода дистиллированная
Стерилизуют автоклавированием.
500 r
10 r
5r
до 1000 мл
(2) Смесь незаменимых витаминов
Биотин
Кальция пантотенат
Холина хлорид
Фолиевая кислота
i Инозит
Никотинамид
Пиридоксаляrидрохлорид
Рибофлавин
Тиаминаrидрохлорид
Вода дистиллированная
100 Mr
100 Mr
100 Mr
100 Mr
200 Mr
100 Mr
100 Mr
10 Mr
100 Mr
до 1000 мл
(3) А2ар очищенный (иона2ар)
Высокочистый arap для использования в
микробиолоrии и иммунолоrии roтовят ионо
обменным методом с получением продук
та исключительной чистоты, прозрачности
и прочности образующеrося rеля. Содержит
приблизительно:
Вода
Зола
Зола, нерастворимая в кислоте
Хлор
Фосфаты (в пересчете на Р205)
Общий азот
Медь
Железо
Кальций
Маrний
12,2 %
1,5 %
0,2%
О
0,3%
0,3%
8ррт
170 ррт
0,28%
0,32 %
2.6.7. Микоплазмы
261
(4) Сбалансированный солевой раствор
Хэнкса (модифицированный)
Натрия хлорид
Калия хлорид
Маrния сульфат rептаrидрат
Маrния хлорид rексаrидрат
Кальция хлорид безводный
Натрия rидрофосфат диrидрат
Калия диrидрофосфат безводный
Вода дистиллированная
(5) Экстракт из сердца и МОЗ20в
Экстракт мозrа теленка
Экстракт rовяжьеro сердца
Протеозный пептон
rлюкоза моноrидрат
Натрия хлорид
Натрия rидрофосфат безводный
Вода дистиллированная
(6) Бульон PPLO
Экстракт ювяжьеrо сердца
Пептон
Натрия хлорид
Вода дистиллированная
6,4 r
0,32 r
0,08 r
0,08 r
0,112 r
0,0596 r
0,048 r
до 800 мл
200 r
250 r
10 r
2 r
5r
2,5 r
до 1000 мл
50 r
10 r
5r
до 1000 мл
МЕТОД ИНДИКАТОРНОЙ КЛЕТОЧНОЙ
КУЛЬТУРЫ
Клеточные культуры окрашиваются флу
оресцентным красителем, обладающим спо
собностью связываться с ДНк. Микоплазмы дe
тектируются по их характерной точечной или
волокнистой флуоресценции на клеточной по
верхности и, при высоком уровне заrрязнения, в
прилеrающем пространстве. Митохондрии в ци
топлазме также MOryT окрашиваться, однако они
леrко распознаются.
Если в случае вирусных суспензий затрудне
на интерпретация результатов из за значитель
ных цитопатических эффектов, вирусы MOryт быть
нейтрализованы с помощью специфичной им
мунной сыворотки, которая бы не влияла на ми
коплазмы, или с помощью субстрата клеточной
КУЛЬТУРЫ, препятствующей росту вирусов. Для дo
казательства отсутствия инrибирующеro эффек
та сыворотки проводят положительные контроли
в присутствии И отсутствии иммунной сыворотки.
ПРОВЕРКА СУБСТРАТА
ДЛЯ обнаружения микоплазм используют
клеточную культуру Веро или иную эквивалент
ную по эффективности клеточную культуру (Ha
пример, производственную линию клеток). Про
веряют эффективность используемых клеток,
используя описанную ниже методику и иноку
лируя не более 100 КОЕ или КОЕ подобных ми
н ЗоН, 1712
кроорrанизмов подходящих штаммов сравнения
М. hуоrhiпis и М. о,а/е. MOryT быть использованы
следующие штаммы:
м. hуоrhiпis
АТСС
29052
АТСС
23714
м. ora/e NCTC
10112
С/Р
104969
Клеточная культура является подходящей,
если обнаруживаются оба штамма сравнения.
Индикаторные клетки должны быть пересе
яны без антибиотика перед использованием для
испытания.
МЕТОД ИСПЫТАНИЯ
1. Среду однородно засевают культурой с
подходящей плотностью (например, от 2'104 до
2.105 клеток в миллилитре; от 4.103 до 2,5'104
клеток на см 2 ), которая через 3 дня роста приве
ла бы к слиянию. 1 мл испытуемою образца ино
кулируют в сосуд С клеточной культурой и инку
бируют при температуре от 35 ос до 38 ОС.
2. Не менее чем через 3 дня инкубирования,
Korдa клеточный рост привел к слиянию, прово
дят пересев на покровные стекла в подходящих
контейнерах или на друryю подходящую поверх
ность (например, разделенные на камеры пред
метные стекла), приroдную для методики испы
тания. Клетки высевают с низкой плотностью
так, чтобы через 3 5 дней инкубирования было
достиrнуто 50 % слияние. Необходимо избеrать
полною слияния, так как оно ослабляет визуали
зацию микоплазм после окрашивания.
3. Удаляют среду и ополаскивают индика
торные клетки фосфатным забуференным фи
зиОЛ02ическим раствором рН 7.4 Р, затем при
бавляют подходящий фиксирующий раствор
(при использовании бисбензимида Р для OKpa
шивания, может быть использована свежеприro
товленная смесь из 1 объема уксусной кислоты
ледяной Р и 3 объемов метанола Р).
4. Удаляют фиксирующий раствор и промы
вают клетки стерильной водой Р. Если стекла
должны будут окрашиваться более чем через
1 ч, их полностью высушивают (при окрашива
нии высушенных стекол должны быть приняты
особые меры предосторожности во избежание
возникновения артефактов).
5. Прибавляют подходящий краситель ДНК и
выдерживают в течение необходимою времени
(для рабоче20 раствора бисбензимида Ртакое
время составляет 1 О мин).
6. Удаляют flраситель и промывают MOHO
слой водой Р.
7. При необходимости подroтавливают по
кровные стекла для исследования (для подro
товки может быть использована смесь из равных
объемов 2лицерuна Р и фосфатно цитратН020
буфеРН020 раствора рН 5,5 Р). Просматривают
с использованием люминесцентною микроскопа
262
rосударственная фармакопея Республики Беларусь
(при применении бисбензимида в качестве Kpa
сителя MOryT быть использованы фильтр B03
буждения 330 нм/380 нм и барьерный фильтр LP
440 нм) при увеличении 400х или более.
8. Сравнивают микроскопическую карти
ну тест культур с микроскопическими картина
ми отрицательноro и положительных контролей ,
изучая внеядерную флуоресценцию. Микоплаз
мы проявляют себя в виде точек или волокон,
расположенных над цитоплазмой и иноrда в
межклеточном пространстве. Количество иссле
дуемых микроскопических полей указывается в
протоколе испытаний, установленном в процес
се валидации.
ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ
Испытуемый образец выдерживает испыта
ние, если в тест культурах, на которые он был
нанесен, нет признаков присутствия микоплазм.
Результаты испытания недостоверны, если в по
ложительных контролях не обнаруживается при
сутствие микоплазм. Результаты испытания He
достоверны, если в отрицательных контролях
обнаруживается присутствие микоплазм.
МЕТОД АМПЛИФИКАЦИИ НУКЛЕИНОВЫХ
КИСЛОТ
Методы амплификации нуклеиновых кислот
(2.6.21) MOryт быть использованы для обнаруже
ния микоплазм с помощью амплификации нукле
иновых кислот, экстраrированных из испытуемо
ro образца, и с использованием специфических
праймеров, которые позволяют выявить при
сутствие целевых нуклеиновых кислот. Данные
методы указывают на присутствие отдельных
последовательностей нуклеиновых кислот и не
обязательно на присутствие жизнеспособных
микоплазм. Для проведения определения дo
ступны несколько различных методик. В данном
общем разделе не описываются конкретные Me
тодики проведения испытаний. Используемая
методика должна быть соответствующим обра
зом валидирована с учетом рекомендаций, при
веденных в конце данноro раздела. В случае ис
пользования коммерчески доступных наборов
некоторые элементы валидации MOryT быть про
ведены производителем, и данная информация
может быть представлена пользователю, однако
необходимо помнить, что полная информация
относительно праймеров может быть недоступ
на, а производство наборов может быть измене
но или прервано.
Методы амплификации нуклеиновых кислот
используются, если это указано в частной фар
макопейнС'й статье. Также, после надлежащей
валидации, С'НИ MOryT использоваться вместо
метода культивирования и метода индикаторной
клеточной культуры.
Прямой метод амплификации нуклеиновых
кислот может быть использован в присутствии
цитотоксическоrо материала и в случае необхо
димости быстроro проведения испытания.
Об02ащение клеточной культуры с по
следующей амплификацией нуклеиновых
кислот: испытуемый образец и подходящий
клеточный субстрат (указанный при описании
метода индикаторной клеточной культуры)
культивируют вместе в течение определенно
ro периода времени; после этоro экстраrиро
ванные из клеток и плавающие над осадком
нуклеиновые кислоты используют для опре
деления с помощью методов амплификации
нуклеиновых кислот.
ВАЛИДАЦИЯ
Стандартные образцы необходимы на раз
личных стадиях валидации, а также дЛЯ KOH
троля при проведении рутинных испытаний. В
, качестве стандартных образцов MOryT использо
ваться микоплазмы или нуклеиновые кислоты.
Для валидации предела обнаружения сле
дующие штаммы представляют собой наилуч
ший выбор блаroдаря частоте встречаемости в
качестве контаминантов и блаroдаря филоrене
тическому родству:
А. /aid/awii;
М. fеrтепtаs;
М. hуоrhiпis (при обоrащении клеточной
культуры включают штамм, требовательный к
питательным средам, такой как АТСС 29052);
М. о,а/е;
М. рпеитопiае или М. gаШsерtiсит;
М. syпoviae (в случае использования или
обработки материалами nтичьеro происхожде
ния в процессе производства);
Mycop/asma аrgiпiпi;
Spirop/asma citri (в случае использования
или обработки материалами насекомых или pac
тений в процессе производства).
Демонстрация специфичности требует ис
пользования подходящеro ряда штаммов бакте
рий, отличных от микоплазм. Наиболее подходя
щими для валидации являются рода бактерий с
близким к микоплазмам филоrенетическим poд
ством; в их число входят C/ostridiuт, Lactobacil
/us и Streptococcus.
Сравнительные исследования для исполь
зования методов амплификации нуклеиновых
кислот в качестве альтернативН020 метода.
Для каждоro штамма испытуемых микоплазм:
в качестве альтернативы методу культи
вирования: должна быть показана возможность
обнаруживать методами амплификации нукле
иновых кислот 10 КОЕ/мл;
В качестве альтернативы методу инди
каторной клеточной культуры: должна быть
показана возможность обнаруживать MeTO
дами амплификации нуклеиновых кислот
100 КОЕ/мл;
или эквивалентный предел обнаружения,
выраженный в единицах числа копий нуклеино
вой кислоты микоплазмы в испытуемом образце
(с использованием подходящих стандартных об
разцов нуклеиновой кислоты микоплазм).
2.6.7. Микоплазмы
263
КОНТРОЛИ
Внутренний контроль. Внутренние KOH
троли необходимы при рутинных анализах для
верификации отсутствия инrибирования. BHY
тренний контроль может содержать место свя
зывания праймера, или может быть использо
вана друrая подходящая последовательность.
Внутренний контроль, как правило, добавляет
ся в исследуемый материал перед изолирова
нием нуклеиновой кислоты и вследствие этоro
действует как общий контроль (экстракция, об
ратная транскрипция, амплификация и детекти
рование ).
Внешние контроли. Внешний положитель
ный контроль содержит определенное число
копий целевых последовательностей или КОЕ из
одноro или более видов микоплазм, выбранных
из числа используемых для валидации усло
вий испытания. Один положительный контроль
должен располаrаться близко около положи
тельной точки отсечения для подтверждения
достижения ожидаемой чувствительности. OT
рицательный внешний контроль не содержит
целевой последовательности, однако не обяза
тельно воспроизводит тот же матрикс, что и ис
пытуемый образец.
ИНТЕРПРЕТАЦИЯ РЕ3УЛЬТАТОВ
Используемые праймеры MOryT также aM
плифицировать немикоплазменные бактери
альные нуклеиновые кислоты, при водя к оши
бочному положительному результату. При
необходимости во время валидации устанавли
ваются процедуры обращения с положительны
ми результатами.
Следующий раздел приводится для инфор
мации
Валидация методов амплификации
нуклеиновых кислот для обнаружен.ия
микоплазм:руководство
1. ОБЛАСТЬ ПРИМЕНЕНИЯ
Методы амплификации нуклеиновых кислот
являются качественными или количественны
ми методами определения присутствия нукле
иновой кислоты. Для обнаружения заrрязнения
микоплазмами различных образцов, таких как
вакцины и клеточные субстраты, подходят коли
чественные методы, которые должны быть pac
смотрены на предмет возможности быть пре
дельным испытанием.
ДаНf-Iое руководство описывает методы вали
дации количественных аналитических методик aM
плификации нуклеиновых кислот для оценки зара
женности микоплазмами. Эти подходы также MOryт
быть приroдны для методики амплификации нукле
иновых кислот в реальном времени в качестве пре
дельноro испытания по контролю за контаминацией.
К наиболее важным характеристикам для
валидации аналитической методики относят
специфичность и предел обнаружения. Кроме
тоro, должна быть оценена робастность анали
тической методики.
В данном документе под аналитической Me
тодикой понимают методику целиком, начиная
от выделения нуклеиновой кислоты и заканчи
вая обнаружением амплифицированных продук
тов.
В случае использования коммерчески дo
ступных наборов для выполнения методики в
целом или ее части, документы по валидации от
производителя этих наборов MOryT заменить Ba
лидацию, проводимую пользователем. Однако
соответствие набора для предполаrаемоrо aHa
лиза должно быть оценено пользователем (Ha
при мер, предел обнаружения. робастность, вза
имное обнаружение классов бактерий).
Амплификация нуклеиновых кислот может
использоваться:
в качестве дополнительноro испытания
(например, для цитотоксичных вирусных суспен
зий) или в целях контроля процесса;
в качестве альтернативноro метода для
замены общепринятой методики (метода инди
каторной клеточной культуры или метода кулыи
вирования).
Данное руководство, таким образом, будет
разделяться на руководство по валидации MeTO
дики амплификации нуклеиновых кислот и на py
ководство по изучению сопоставимости метода
амплификации нуклеиновых кислот и общепри
нятых методов.
2. РУКОВОДСТВО ПО ВАЛИДАЦИИ
МЕТОДИКИ АМПЛИФИКАЦИИ
НУКЛЕИНОВblХ КИСЛОТ МИКОПЛАЗМ
Следует оценить три параметра: специфич
ность, предел обнаружения и робастность.
2 1. Специфичность. Специфичность
это способность методики однозначно опреде
лять специфические нуклеиновые кислоты в
присутствии друrих компонентов, которые MOryT
присутствовать в образце.
Специфичность амплификации зависит от
выбора праймеров, проб (для анализа roтово
ro продукта) и строroсти условий испытания (как
для стадии амплификации. так и для стадии дe
тектирования ).
Возможность метода амплификации нукле
иновых кислот обнаруживать большой список
видов микоплазм зависит от выбора праймеров
и параметров пробы и метода. Эта возможность
должна быть продемонстрирована с использо
ванием охарактеризованных стандартных па
нелей (например, штаммы сравнения, предла
raeMbIe ЕООМ). Так как методы амплификации
нуклеиновых кислот зависят от смеси прайме
ров, теоретический анализ праймеров и проб,
основанный на сравнении с базами данных, не
264
rосударственная фармакопея Республики Беларусь
рекомендуется, так как интерпретация результа
тов может быть довольно сложной и не отражать
экспериментальных результатов.
Более тоro, так как есть вероятность, что
праймеры будут обнаруживать друrие виды бак
терий, должно быть задокументировано и OTBa
лидировано потенциальное перекрестное обна
ружение. Наиболее подходящими для данной
валидации являются такие виды бактерий, как
rрам положительные бактерии с близким фило
rенетическим родством к микоплазмам; это C/os
tridiuт, Lactobacillus и Streptococcus. Однако этот
перечень не является исчерпывающим, и выбор
видов, необходимых для исследования, зависит
от теоретической возможности (основанной на
последовательности праймеров/проб) методов
амплификации нуклеиновых кислот обнаружи
вать такие виды.
На основании результатов, полученных при
валидации специфичности, при обнаружении
проблемных мест в специфичности методики
(таких как немикоплазматические бактериаль
ные нуклеиновые кислоты), при изучении Ba
лидации должна быть предложена стратеrия,
позволяющая интерпретировать положитель
ные результаты рутинных испытаний. Напри
мер. повторное испытание может выполняться
с использованием альтернативноro метода без
данных проблемных мест в специфичности либо
с использованием общепринятоro метода.
2 2. Предел обнаружения. Предел обнару
жения отдельной аналитической методики это
наименьшее количество специфической нукле
иновой кислоты в образце, которое может быть
обнаружено. но необязательно должно быть
определено количественно.
Для установления предела обнаружения
должна быть определена крайняя положитель
ная точка для аналитической методики ампли
фикации нуклеиновой кислоты. Крайняя положи
тельная точка (как определено в общем разделе
2.6.21) это минимальное число копий специ
фической последовательности в объеме об
разца, которое может быть обнаружено в 95 %
случаев проведения испытания. На крайнюю
положительную точку влияет распределение ми
коплазматическоrо reHoMa в отдельно взятых
испытуемых образцах и друrие факторы (напри
мер, эффективность фермента), что может при
водить к различным значениям 95 % пороroвоro
значения в индивидуальных аналитических ис
пытаниях.
Для определения крайней положительной
течки должны быть исследованы серии разве
дени"" охарактеризованных и калиброванных
(либо в КОЕ, либо в копиях нуклеиновой кисло
ты) внутренних рабочих штаммов или CTaHдap
тов EDQM в различные дни для определения Ba
риабельности рабочих циклов.
Для валидации предела обнаружения MeTO
дики рекомендуется использовать следующие
виды с учетом распространенности и филоrене
тических связей:
А. 'aid/awii;
М. fеrтепtаs;
М. hyorhiпis;
М. о,а/е;
М. рпеитопiае или М. gallisepticuт;
М. syпoviae (в случае использования или
обработки материалами птичьеro происхожде
ния в процессе производства);
М. аrgiпiпi;
S. citri (в случае использования или обра
ботки материалами насекомых или растений в
процессе производства).
Для статистическоrо анализа полученных
результатов следует исследовать не менее 3
независимых 1 O KpaTHЫX разведений каждо
ro штамма с достаточным числом повторов для
каждоrо разведения для тoro, чтобы получить в
конечном итоrе 24 результата испытания каждо
ro разведения.
Например, лаборатория может исследо
вать 3 серии разведений в разные дни с KO
личеством повторов для каждоrо разведения
равным 8, или 4 серии разведений в разные
дни с количеством повторов для каждоrо раз
ведения равным 6, или 6 серий разведений в
разные дни с количеством повторов для каж
доrо разведения равным 4. Чтобы удержать KO
личество разведен ий в приемлемых рамках,
следует провести предварительные испыта
ния для получения предварительноro значения
крайней положительной точки. Диапазон раз
ведений может выбираться в области предва
рительно определенноro пороroвоro значения.
Концентрацию микоплазм (в КОЕ или в количе
стве копиях), которая может быть обнаружена
в 95 % случаев проведения испытаний, рассчи
тывают с помощью подходящеro статистиче
cKoro подхода.
Полученные результаты также MorYT слу
жить для оценки вариабильности аналитической
методики.
2 3. Робастность (устойчивость). Робаст
ность (устойчивость) аналитической методики
это мера устойчивости к небольшим HaMepeH
ным изменениям параметров методики, она
дает представление о надежности методики при
стандартном использовании.
Оценку робастности следует проводить на
стадии разработки. Следует показать надеж
ность аналитической методики по отношению к
намеренным изменениям параметров методи
ки. В случае амплификации нуклеиновых кислот
небольшие изменения условий эксперимента
MOryT быть критическими. Однако, робастность
методики может быть продемонстрирована при
ее разработке в случае изучения малых измене
ний концентраций реактивов (например, MgCI 2 ,
праймеров или дезоксирибонуклеотидов). Также
MorYT быть оценены модификации наборов для
2.6.8. Пuроzенность
265
экстракции или методик экстракции и различные
типы термоциклера (амплификатора).
В конечном итоrе, робастность методики
может быть оценена с помощью межлаборатор
ных испытаний.
3. РУКОВОДСТВО ДЛЯ ИЗУЧЕНИЯ
СОПОСТАВИМОСТИ МЕТОДА
АМПЛИФИКАЦИИ НУКЛЕИНОВЫХ
КИСЛОТ И ОБЩЕПРИНЯТЫХ МЕТОДОВ
Метод амплификации нуклеиновых кислот
может быть использован вместо общепринятых
методов (метода культивирования и метода ин
дикаторной клеточной культуры). В этом случае
следует провести изучение сопоставимости Me
тодов. В изучение следует включать в основном
сравнение соответствующих пределов обнару
жения альтернативной и общепринятой методи
ками. При этом должна оцениваться также специ
фичность (rруппа определяемых микоплазм,
предполаrаемые ложноположительные резуль
таты ).
Для предела обнаружения определяют кри
терии приемлемости, как указано далее:
если альтернативный метод использует
ся вместо метода культивирования, то должно
быть показано, что он способен обнаруживать
1 О КОЕ/мл каждоrо испытуемоro вида мико
плазм, описанных в разделе 2 2;
если альтернативный метод используется
вместо метода индикаторной клеточной культу
ры, то должно быть показано, что он способен
обнаруживать 100 КОЕIмл каждоro испытуемою
вида микоплазм, описанных в разделе 2 2.
В обоих случаях для установления факта
достижения критериев приемлемости MOryT ис
пользоваться подходящие стандарты, калибро
ванные по количеству копий нуклеиновой кисло
ты или по количеству КОЕ. Зависимость между
КОЕ и числом копий нуклеиновой кислоты
должна быть предварительно установлена для
возможности сравнения характеристик альтер
нативных методов амплификации нуклеиновых
кислот с характеристиками общепринятых MeTO
дов.
Для проведения сравнительноro исследова
ния может быть использована одна из следую
щих методик:
альтернативный метод проводят парал
лельно с общепринятым методом (методами)
для одновременноro установления предела об
наружения обеих методик с использованием
одинаковых образцов калиброванных штаммов;
сравнивают характеристики методов aM
плификации нуклеиновых кислот с данными,
полученными ранее при валидации общепри
нятоro метода. В этом случае должна быть тща
тельно задокументирована калибровка исполь
зуемых стандартов, а также их стабильность.
В отчетах о сравнительных исследовани
ях (проверке сопоставимости методов) должны
быть отмечены все элементы валидации, упомя
35 Зак. 1712.
нутые в разделе 2 (специфичность, предел об
наружения, вариабельность и робастность), для
тоro, чтобы оценить все преимущества и/или He
достатки альтернативноrо метода по сравнению
с общепринятыми методами.
01/2013:20608
2.6.8. пироrЕННОСТЬ
Испытание состоит в измерении pO Ta TeM
пературы тела, вызванною у кроликов внутри
венным введением стерильноro раствора испы
туемоro образца.
Выбор животных. Используют здоровых
взрослых кроликов обоеrо пола массой не менее
1 ,5 к!; получавших полноценное сбалансиро
ванное питание, не включающее антибиотиков,
масса тела которых не снижалась в течение
недели, предшествующей испытанию. Кролика
не следует использовать в испытании на пиро
reHHocTb, если:
а) он использовался в аналоrичном испыта
нии, давшем отрицательный результат, в тече
ние предшествующих 3 дней;
Ь) он использовался в аналоrичном испы
тании, в котором испытуемая субстанция была
признана несоответствующей требованиям, в
течение предшествующих 3 недель.
Помещение для животных. Кроликов co
держат индивидуально в тихом помещении при
однородной подходящей температуре. Живот
ных не кормят в течение ночи и до окончания
испытания, в течение испытания не дают воду.
Испытание проводят в тихом помещении, rдe OT
сутствует риск возникновения помех, которые
MOryT возбудить животных, и в котором темпера
тура поддерживается на уровне, не более чем
на 3 ос отличающемся от температуры, поддер
живаемой в месте постоянноro содержания KpO
ликов или В помещении, в котором кролики co
держались в течение не менее 18 ч до начала
испытания.
Материалы. Стеклянная посуда, шприцы и
иrлы. Тщательно моют всю стеклянную посуду,
шприцы и иrлы водой для инъекций и наrревают
в сухожаровом шкафу при температуре 250 ос
в течение 30 мин или при температуре 200 ос в
течение 1 ч.
Клетки (станки). Клетки для кроликов, TeM
пературу которых измеряют с помощью элек
трическою прибора, устроены таким образом,
чтобы животные фиксировались лишь с по
мощью свободно закрепленных на шее XOMY
тов; остальная часть тела остается относитель
но свободной так, чтобы кролики моrли сидеть
в обычном положении. Они не удерживаются с
помощью ремней или друrими подобными Meтo
266
rосударственная фармакопея Республики Беларусь
дами, которые MorYT причинить вред животному.
Животные помещаются в клетки не менее чем за
1 ч до первоro измерения температуры и оста ют
ся там в течение всеro испытания.
Термометры. Используют термометр или
электрическое устройство, показывающее TeM
пературу с точностью до 0,1 ос, вводя ero в
прямую кишку кролика на rлубину около 5 см.
rлубина введения постоянна для каждоro из
кроликов в течение каждоro из испытаний. Если
используется электрическое устройство, оно
может оставаться на месте в течение Bcero ис
пытания.
Предварительное испытание. После
отбора животных за 1 3 дня до проведения ис
пытания продукции тем из них, которые не ис
пользовались для опытов в течение последних
двух недель, вводят внутривенно апироrенный
раствор 9 r/л натрия хлорида Р, наrретый до
температуры около 38,5 ос, в количестве 1 О мл
на килоrрамм массы тела. Записывают темпера
туру животных, начиная не менее чем за 90 мин
до введения и продолжая в течение 3 ч после
введения раствора. Животные, температура KO
торых колеблется в пределах более 0,6 ос, не
используются в основном испытании.
Основное испытание. Испытание проводят
С использованием rруппы из трех кроликов.
Подrотовка и введение продукта. Испы
туемую жидкость перед введением HarpeBa
ют до температуры около 38,5 ос. Испытуемый
продукт может быть растворен или разведен в
апироrенном растворе 9 r/л хлорида натрия Р
или друroм растворителе, указанном в частной
статье. Раствор медленно вводят в крайнюю
вену уха каждоro из кроликов в течение не более
4 мин, если в частной статье нет друrих указа
ний. Количество ВВОДИМОro продукта варьиру
ется в зависимости от испытуемоro продукта и
указывается в частной статье. Вводимый объем
должен составлять не менее 0.5 мл и не более
1 О мл на 1 Kr массы тела.
Определение исходной и максимальной
температуры. За «исходную температуру» каж
доrо из кроликов принимают среднее из двух
значение температуры, записанных для данною
кролика с интервалом 30 мин в течение 40 мин,
предшествующих введению испытуемоrо продук
та. «Максимальная температура» каждоro из KpO
ликов самая высокая температура, записанная
для данною кролика в течение 3 ч после BBeдe
ния. Температуру каждоro из кроликов записы
вают с интервалами, не превышающими 30 мин,
начиная не менее чем за 90 мин до введения ис
пытуемоro продукта и продолжая в течение 3 ч
после введения. За результат испытания при
нимают разность между максимальной и исход
ной температурами каждоrо из кроликов. При
отрицательной разности результат принимают
равным нулю.
При определении исходной температуры
кролики, у которых два последовательных зна
чения температуры варьируются в пределах,
превышающих 0,2 ос, изымаются из испытания.
В каждом из испытаний используют животных,
исходная температура которых отличается друr
от друrа не более чем на 1 ос. Кролики, исход
ная температура которых выше 39,8 ос или ниже
38,0 ос, изымаются из испытания.
Интерпретация результатов. После про
ведения испытания на rруппе из трех кроликов
еro при необходимости повторяют на друrих
rруппах из трех кроликов (суммарно до четырех
rрупп, в зависимости от полученных результа
тов). Если суммированный результат, получен
ный в первой rруппе, не превышает значение,
данное во второй колонке таблицы 2.6.8. 1, счи
тают, что продукт выдерживает испытание. Если
суммированный результат превышает значение,
данное во второй колонке таблицы, но не пре
вышает значение, данное в третьей колонке Ta
блицы, испытание повторяют, как указано выше.
Если суммированный результат превышает зна
чение, данное в третьей колонке таблицы, счи
тают, что продукт не выдерживает испытание.
Коли-
чество
кроли
ков
Продукт вы-
держивает ис
пытание, если
суммирован-
ный результат,
не превышает:
1 ,15 ос
2,80 ос
4,45 ос
6,60 ос
Таблица 2.6.8. 1
Продукт не вы-
держивает ис
пытание, если
суммированный
результат, превы-
шает:
2,65 ос
4,30 ос
5,95 ос
6,60 ос
3
6
9
12
Кролики, которые использовались в про
ведении испытаний на пироrенность, в случае
если повышение их температуры составило
1,2 ос, исключаются из дальнейших испытаний.
01/2013:20609
2.6.9. АНОМАЛЬНАЯ ТОКСИЧНОСТЬ
ОБЩЕЕ ИСПЫТАНИЕ
Каждой из пяти здоровых мышей массой от
17 r до 24 r вводят внутривенно указанное в част
HOLl1 статье количество испытуемоro образца,
растворенное в 0,5 мл воды для инъекций Рили
стерильноrо раствора 9 r/л хлорида натрия Р.
Если нет друrих указаний в частной статье, paCT
вор вводят в течение 15 30 с.
Считается, что испытуемый образец Bыдep
живает испытание, если ни одна из мышей не
2.6.10. rucmaMuH
267
поrибает в течение 24 ч или времени, указанно
ro в частной статье. Если поrибает более одною
животною, считают, что испытуемый образец не
выдерживает испытание. Если поrибает одно
животное, испытание повторяют. Считается, что
испытуемый образец выдерживает испытание,
если ни одна из мышей второй rруппы не поrи
бает в течение указанноro промежутка времени.
ИММУННЫЕ СЫВОРОТКИ И ВАКЦИНЫ
ДЛЯ МЕдицинскоrо ПРИМЕНЕНИЯ
Если в частной статье нет друrих указаний,
каждой из пяти здоровых мышей массой от 17 r
до 24 r внутрибрюшинно вводят одну челове
ческую дозу, но не более 1,0 мл. Человеческая
доза указана на этикетке или листке вкладыше
испытуемоro образца. Животных наблюдают в
течение 7 дней.
Испытуемый образец выдерживает испыта
ние, если ни одно из животных не обнаружива
ет признаков недомоrания. Если поrибает более
одноro животноro, испытуемый образец не BЫ
держивает испытание. Если одно из животных
поrибает или обнаруживает признаки недомоrа
ния, испытание повторяют. Испытуемый образец
выдерживает испытание, если ни одно из живот
ных второй rруппы не поrибаэт и не обнаружива
ет признаков недомоrания в течение указанноro
промежутка времени.
Испытание также должно быть проведено
на двух здоровых морских свинках массой от
250 r до 400 r. Внутрибрюшинно вводят каждому
животному одну человеческую дозу, но не более
5,0 мл. Человеческая доза указана на этикет
ке или листке вкладыше испытуемоro образца.
Животных наблюдают в течение 7 дней.
Испытуемый образец выдерживает испыта
ние, если ни одно из животных не обнаружива
ет признаков недомоrания. Если поrибает более
одноro животноro, испытуемый образец не BЫ
держивает испытание. Если одно из животных
поrибает или обнаруживает признаки недомоrа
ния, испытание повторяют. Испытуемый образец
выдерживает испытание, если ни одно из жйвот
ных второй rруппы не поrибает и не обнаружива
ет признаков недомоrания в течение указанноro
промежутка времени.
01/2013:2061 О
2.6.10. rИСТАМИН
Убивают морскую свинку массой от 250 r
до 350 r, которая была лишена пищи в течение
предшествовавших 24 ч. Извлекают участок пе
риферийной тонкой кишки длиной 2 см и опо
рожняют отделенную часть путем тщательной
промывки раствором В, описанным ниже, с помо
щью шприца. К каждому из концов присоединя
ют тонкую нить и делают маленький поперечный
разрез в центре фраrмента кишки. Помещают ее
в ванночку для opraHoB вместимостью от 1 О мл
ДО 20 мл, содержащую раствор В, поддержива
емый при постоянной температуре (от 34 ос до
36 ОС), и пропускают через раствор смесь из 95
частей кислорода и 5 частей диоксида уrлерода.
Укрепляют одну из нитей рядом с дном ванночки
для opraHoB. Друryю нить присоединяют к изото
ническому миоrрафу и реrистрируют сокращения
opraHa с помощью кимоrрафа или друrим под
ходящим способом непрерывной реrистрации.
Если используется рычar, ею длина должна быть
такой, чтобы движение opraHa усиливалось при
близительно в 20 раз. Натяжение кишки должно
составлять около 9,8 мН (1 r) и быть отреryлиро
вано в соответствии с чувствительностью opraHa.
Промывают ванночку для opraHoB раствором В.
Выдерживают в течение 1 О мин. Промывку pac
твором В повторяют еще два или три раза. Сти
мулируют серию сокращений путем добавле
ния измеренных объемов от 0,2 мл до 0,5 мл
раствора еистамина ди2идрохлорида Р, имею
щеro концентрацию, которая вызывает воспро
изводимые субмаксимальные реакции. Эту дозу
обозначают как «высокую дозу». Перед каждым
добавлением rистамина ванночку для opraHoB
трижды промывают (предпочтительно путем пе
реполнения без опорожнения ванночки) paCTBO
ром В. Последовательные добавления следует
выполнять, соблюдая равные интервалы BpeMe
ни, достаточные для полной релаксации между
добавлениями (около 2 мин). Добавляют равные
объемы более разбавленноro раствора 2иcтa
мина ди2идрохлорида Р, приводящею к воспро
изводимым реакциям, составляющим пример
но половину от реакций, вызываемых «высокой
дозой». Эту дозу обозначают как «низкую дозу».
Продолжают периодическое добавление «BЫCO
ких» и «низких» доз раствора rистамина, следуя
вышеуказанному, и чередуют каждое добавле
ние с равным объемом разведения испытуемоrо
раствора, реryлируя степень разбавления таким
образом, чтобы сокращение кишки, в случае ero
наличия, было меньшим, чем сокращение, BЫ
зываемое «высокой дозой» rистамина. Опреде
ля ют, является ли сокращение, в случае ею Ha
личия, воспроизводимым и остаются ли реакции
на «высокую» И «низкую» дозы rистамина неиз
менными. Вычисляют активность испытуемоrо
образца, выраженную в ее эквиваленте на ми
KporpaMM основания rистамина, основываясь на
определенном выше разведении.
Определенное таким образом количество
не должно превышать количество, указанное в
частной статье.
Если испытуемый раствор не вызыва
ет сокращений, roтовят свежий раствор, дo
бавляя количество rистамина, соответствую
щее максимально допустимому в соответствии
с требованиями частной статьи, и отмечают.
268
rосударственная фармакопея Республики Беларусь
соответствуют ли сокращения, вызываемые ис
пытуемым раствором с добавкой rистамина, KO
личеству добавленноro rистамина. Если такоro
соответствия не наблюдается, сокращение, BЫ
зываемое испытуемым образцом, невоспроиз
водимо или последующие реакции на «высокие»
И «низкие» дозы rистамина уменьшаются, то pe
зультаты испытания считают недостоверными и
выполняют испытание на депрессорные веще
ства (2.6.11).
Раствор А
Натрия хлорид
Калия хлорид
Кальция хлорид, безводный
Маrния хлорид, безводный
Натрия rидрофосфат дoдeKa
rидрат
Вода для инъекций Р
160,0 r
4,0 r
2,0 r
1,0 r
0,10 r
до 1000 мл
Раствор В
Раствор А
Атропина сульфат
Натрия rидрокарбонат
rлюкоза моноrидрат
Вода для инъекций Р
50,0 мл
0,5 Mr
1,0 r
0,5 r
до 1000 мл
Раствор В должен быть свежеприroтовлен
ным, и еro используют в течение 24 ч.
01/2013:20611
2.6.11. ДЕПРЕССОРНЫЕ ВЕЩЕСТВА
Испытание проводят на кошке массой не
менее 2 Kr, анестезированной с использованием
хлоралозы или барбитурата, что позволяет под
держивать постоянное кровяное давление. Жи
вотное защищают от потери температуры тела и
поддерживают ее таким образом, чтобы ректаль
ная температура оставалась в физиолоrических
пределах. В трахею вводят дыхательную трубку.
В общую сонную артерию вводят канюлю, Ha
полненную rепаринизированным раствором 9 r/л
натрия хлорида, и присоединяют ее к устрой
ству, обеспечивающему постоянную реrистра
цию кровяноro давления. В бедренную вену
вводят друrую канюлю, наполненную rепарини
зированным раствором 9 r/л натрия хлорида,
через которую может быть введен раствор rиста
мина и испытуемоro образца. Определяют чув
ствительность животноro к rистамину путем BHY
тривенноro введения через равные промежутки
времени раствора 2истамина Р в дозах, соот
ветствующих 0,1 MKr и 0,15 MKr основания rиста
мина на килоrрамм массы тела. Введение MeHb
шей дозы повторяют не менее трех раз. Вторую
и последующие инъекции вводят не ранее, чем
через одну минуту после тоro, как кровяное дaB
ление возвращается к уровню, наблюдавшему
ся непосредственно перед предыдущей инъек
цией. Животное используют в испытании только
в случае, если получено четко реrистрируемое
снижение кровяноro давления, являющееся по
стоянным для меньшей дозы, а большая доза
вызывает более высокую реакцию. Испытуе
мый образец растворяют в растворе 9 r/л натрия
хлорида или в друroм растворителе, указанном
в частной статье и взятом в количестве, ДOCTa
точном для получения предписанной KOHцeHTpa
ции. Вводят внутривенно на килоrрамм массы
тела 1,0 мл раствора zucmaMUHa Р, затем две
последовательные инъекции указанноro в част
ной статье количества испытуемоro раствора и,
наконец, 1,0 мл раствора 2истамина Р. Вторую,
третью и четвертую инъекции вводят не ранее,
чем через одну минуту после тоro, как кровяное
давление возвращается к уровню, наблюдавше
муся непосредственно перед предыдущей инъе
кцией. Эти серии инъекций повторяют дважды и
завершают испытание введением 1,5 мл pacт
вора 2истамина Р на килоrрамм массы тела.
Если реакция на 1,5 мл раствора 2иcтa
мина р на килоrрамм массы тела не превышает
реакцию на 1,0 мл тоro же раствора, результа
ты испытания признают недостоверными. Ис
пытуемый образец не выдерживает испытание,
если среднее значение реакции на ero BBeдe
ние превышает среднее значение реакции на
1,0 мл раствора 2истамина Р на килоrрамм
массы тела или если любая из доз образца BЫ
звала более высокую депрессорную реакцию,
чем завершающая доза раствора rистамина. Ла
бораторное животное не следует использовать
в друroм испытании на депрессорные вещества,
если любая из испытанных доз вызывает более
выраженную депрессорную реакцию, чем завер
шающая доза раствора rистамина, или если pe
акция на высокую дозу rистамина, введенную
после испытуемоro образца, менее среднеro
значения реакции на введенные ранее меньшие
дозы rистамина.
#ИСПblтание на содержание веществ 2и
стамuноподоБН020 действия может бblть про
ведено также в соответствии с нижеизложен
ным методом.
Испытание проводят на кошках обоеrо пола
массой не менее 2 к!; анестезированных с ис
пользованием хлоралозы, зооксилазина, pOMe
тара или ба рбитурата , что позволяет поддержи
вать постоянное кровяное давление.
Для реrистрации артериальноro давления
в отпрепарированную сонную артерию вставля
ют канюлю, заполненную раствором, предупреж
дающим свертывание крови (насыщенный paCT
вор натрия rидрокарбоната, 25 % раствор маrния
сульфата, раствор rепарина, содержащий 50 ЕД в
1 мл И др.), И соединяют ее с ртутным манометром.
Запись давления производят на ленте кимоrрафа.
2.6. 12. Общее количество жизнеспособных аэробов
269
Испытуемые растворы вводят внутривенно.
Вначале проверяют чувствительность животною
к rистамину. Для приroтовления основноro раст
вора rистамина, содержащеro 1 Mr rистамина
основания в 1 мл воды, около 138,1 Mr 2иcтa
мина фосфата Р или около 82,8 Mr 2истамина
диеидрохлорида Р растворяют в 50 мл воды диc
тиллированной Р.
Раствор хранят не более 1 месяца в склянке
из темноro стекла с притертой пробкой в защи
щенном от света месте при температуре от 4 ос
до 10 ос.
В качестве основною раствора rистамина
можно использовать также 0,1 % раствор rиста
мина диrидрохлорида в ампулах, в 1 мл которою
содержится 0,6038 Mr rистамина основания.
Из основноro раствора rистамина непосред
ственно перед опытом путем последовательных
разведений в воде или в растворе 9 r/л натрия
хлорида в зависимости от растворителя для ис
пытуемоro образца, указанною в COOTBeTCTBY
ющей статье, ютовят стандартные растворы,
содержащие 0,5 мкr/мл и 1 мкr/мл rистамина ос
нования.
Для проверки реакции животноro на rиста
мин в вену последовательно вводят указан
ные стандартные растворы с промежутками не
менее 5 мин в дозах 0,1 MKr и 0,2 MKr rистами
на основания в 0,2 мл на 1 Kr массы. Скорость
введения раствора 0,1 миллилитр в секунду.
Животных, у которых при введении rистамина в
дозе 0,2 MKr на 1 Kr массы артериальное давле
ние снизится менее чем на 20 мм рт. ст., из опыта
исключают.
После проверки чувствительности животно
ro к rистамину уточняют величину rипотензив
ной реакции посредством повторноro введения
стандартноro раствора rистамина в дозе 0,1 MKr
(0,2 мл раствора) на 1 Kr массы со скоростью
0,1 миллилитр в секунду. Введение rистамина
с интервалом 5 мин повторяют несколько раз,
пока при двух последовательных введениях не
будет получено одинаковое снижение артери
альноro давления, которое принимается за CTaH
дартное в данных условиях опыта.
Затем с интервалом не менее 5 мин живот
ному вводят испытуемый раствор лекарственно
ю средства с той же скоростью, с которой вводи
ли раствор rистамина.
Растворитель и концентрация испытуемо
ro раствора (тест доза) указаны в соответствую
щей фармакопейной статье.
Испытуемый образец считают выдержав
шим испытание, если снижение артериальноro
давления после введения тест дозы не превы
шает реакции на введение 0,1 MKr/Kr rистаминi'l в
стандартном растворе.
При испытании в одном опыте разных ле
карственных средств необходимо проводить дo
полнительное определение реакции животноrо
на введение стандартноro раствора rистамина
в дозе 0,1 м Kr/Kr. Отмеченная при этом величи
Зб З", 1712.
на реакции принимается за стандартную для по
следующеro испытания образца.
В случае значительноro уменьшения вели
чины реакции по сравнению с наблюдаемой в
начале опыта необходимо вновь проверить чув
ствительность животноro к действию rистамина
в дозе 0,2 MKr/Kr. Если снижение артериальноro
давления при этом будет не менее 20 мм рт. ст.,
испытание образцов продолжают в соответствии
с указанными выше требованиями.#
01/2013:20612
2.6.12. МИКРОБиолоrИЧЕСКИЕ
ИСПЫТАНИЯ НЕСТЕРИЛЬНОЙ
ПРОДУКЦИИ: ОБЩЕЕ
КОЛИЧЕСТВО
ЖИЗНЕСПОСОБНЫХ
АЭРОБОВ
1. ВВЕДЕНИЕ
Испытания, описанные ниже, позволяют
осуществлять количественное определение Me
зофильных бактерий и rрибов, способных расти
в аэробных условиях.
Испытания предназначены в первую оче
редь для определения соответствия требова
ниям спецификации по микробиолоrической
чистоте как субстанций, так и лекарственных
средств. В случае использования методик для
таких целей следуют инструкциям, приведенным
ниже, включая количество образцов, которое
следует взять, и интерпретацию результатов, в
соответствии с тем, как изложено далее.
Эти методы не применимы к продуктам, co
держащим живые микроорrанизмы в качестве
активных инrредиентов.
Альтернативные микробиолоrические MeTO
дики, включая автоматические методы, MOryT ис
пользоваться в том случае, если доказана их эк
вивалентность фармакопейным методикам.
2. ОБЩИЕ ОПЕРАЦИИ
Выполняют испытания в условиях, позво
ляющих избежать случайную контаминацию ис
пытуемоro продукта. Меры предосторожности,
предпринимаемые для предотвращения KOHTa
минации, не должны влиять ни на один из ми
кроорrанизмов, обнаруживаемых в ходе испы
тания.
Если испытуемый продукт обладает анти
микробным действием, оно должно быть подхо
дящим образом нейтрализовано. Если для этой
цели используют инактиваторы, должна быть
продемонстрирована их эффективность и HeТOK
сичность в отношении микроорrанизмов.
270
rосударственная фармакопея Республики Беларусь
Если для приroтовления образца использу
ются поверхностно активные вещества, должна
быть продемонстрирована их нетоксичность и
их совместимость с применяемыми инактивато
рами.
3. МЕТОДЫ ПОДСЧЕТА
Суммарное количество жизнеспособных
аэробов определяют методом мембранной
фильтрации или методом чашечноro подсче
та в соответствии с указаниями частной статьи.
Метод наиболее вероятноrо числа (НВЧ) обычно
является наименее точным методом микробио
лоrических подсчетов, однако он может оказать
ся наиболее подходящим для некоторых rрупп
продуктов с очень низкой бионаrрузкой.
Выбор метода основывается на таких фак
торах, как природа продукта и требуемый
предел содержания микроорrанизмов. Выбран
ный метод должен позволять проводить испыта
ния образца подходящеro размера для возмож
ности вынесения заключения о соответствии
спецификации. Приroдность выбранноro метода
должна быть доказана.
4. РОСТОВЫЕ СВОЙСТВА СРЕД,
приrодность МЕТОДА ПОДСЧЕТА
И ОТРИЦАТЕЛЬНЫЕ КОНТРОЛИ
4 1. ОБЩИЕ ПОЛОЖЕНИЯ
Возможность обнаружения микроорrаниз
мов в испытании в присутствии испытуемоrо
продукта должна быть доказана.
При внесении в процедуру испытания или в
продукт изменений, способных повлиять на про
ведение испытания, должна быть подтверждена
приrодность.
4 2. приrОТОВЛЕНИЕ ТЕСТ ШТАММОВ
Используют стабильные стандартизован
ные суспензии тест штаммов или roтовят их, как
указано ниже. Используется техника coxpaHe
ния эталонноrо банка культур микроорrанизмов
таким образом, чтобы живые микроорrанизмы,
использованные для инокуляции, были удале
ны не более чем на 5 пересевов от ориrиналь
ноro контрольноro банка культур микроорrаниз
мов. Выращивают каждый бактериальный или
rрибковый тест штамм отдельно, как указано в
таблице 2.6.12. 1.
Для приroтовления испытуемой суспензии
используют забуференный раствор натрия хло
рида и пептона рН 7,0 или фосфатный буфер
ный раствор рН 7,2; для суспендирования спор
А. brasilieпsis в буфер может быть приба9лен по
лисорбат 80 в количестве 0,05 %. СуспензиVl ис
пользуют в течение 2 ч или, при условии xpaHe
ния при температуре от 2 ос до 8 ОС, в течение
24 ч. В качестве альтернативы приroтовлению и
последующему разведению свежей суспензии
веrетативных клеток А. Ьrаsi/iепsis или В. subti/is
roтовят стабильную суспензию спор и затем He
обходимый объем приroтовленной суспензии ис
пользуют для инокуляции. Стабильная суспен
зия спор может храниться при температуре от
2 ос до 8 ос в течение валидированноro периода
времени.
4 3. ОТРИЦАТЕЛЬНЫЙ КОНТРОЛЬ
ДЛЯ проверки условий испытания проводят
отрицательный контроль с использованием BЫ
бранноro разбавителя вместо испытуемоro об
разца. Не должно наблюдаться роста микроор
rанизмов. Отрицательный контроль проводят
также при испытании продуктов, как указано в
разделе 5. Неудавшийся отрицательный KOH
троль требует проведения расследования.
4 4. РОСТОВЫЕ СВОЙСТВА СРЕД
Испытания проводят на каждой серии roто
вой среды и на каждой серии среды, приroтов
ленной из деrидратированной среды или из YKa
занных инrредиентов.
Инокулируют порции/чашки с бульоном на
основе rидролизата казеина и соевых бобов и с
аrаризованной средой на основе rидролизата Ka
зеина и соевых бобов небольшим количеством
(не более 100 КОЕ) микроорrанизмов, указанных
в таблице 2.6.12. 1, используя отдельные порции/
чашки среды для каждоro вида. Инокулируют
чашки с декстрозным arapoM Сабуро небольшим
количеством (не более 100 КОЕ) микроорrаниз
мов, указанных в таблице 2.6.12. 1, используя OT
дельные чашки со средой для каждоro вида. Инку
бируют в условиях, указанных в таблице 2.6.12. 1.
Рост, обнаруживаемый на каждой твердой
среде, не должен отличаться от значения, най
денноro для стандартизованноro инокулята,
более чем в 2 раза. Для свежеприroтовленноro
инокулята рост микроорrанизмов сопоставим с
ростом, который был ранее получен с исполь
зованием ранее испытанной и утвержденной
серией среды. Жидкая среда приroдна, если об
наруживается отчетливо заметный рост микро
орrанизмов в сравнении с ростом, который был
ранее получен с использованием ранее испы
танной и утвержденной серией среды.
#Допускается использование альтернатив
ных питательных сред (2.6.13), например, жидкой
среды NQ 1 (мясо пептонный бульон) и аrаризо
ванной среды NQ 1 для приroтовления TeCT
штаммов и определения общеro количества аэро
бов (ОКА); жидкой среды NQ 2 и аrаризованной
среды NQ 2 для приroтовления тест штаммов
и определения общеro количества rрибов (ОКП.#
4 5. приrодность МЕТОДА ПОДСЧЕТА В
ПРИСУТСТВИИ ПРОДУКТА
4 5 1. Приrотовление образца. Способ
приroтовления образца зависит от физических
свойств испытуемоro продукта. В случае если ни
один из описанных способов не является YДOB
летворительным, должен быть разработан аль
тернативный способ.
2.6.12. Общее количество жизнеспосоБНblХ аэробов
271
Таблица 2.6.12. 1
Активация роста ПриroдностьмеТQДапqд а
в присутствии продукта
Микроорraнизм Приrотовление Общее Общее коли- Общее Общее коли-
тест-штамма количество чество rpибов количество чество rpибов
аэробов и плесени аэробов и плесени
Аraризован
Аraризованная ная среда на
Аrаризованная среда на основе основе rидро-
среда на основе rидролизата Ka лизата казе
Staphy/ococcus rидролизата Ka зеина и соевых ина и соевых
aureus: зеина и соевых бобов и бульон бобов/НВЧ
А ТСС 6538 бобов или бульон на основе rидро бульон на
NC/MB 9518 на основе rидро лизата казеина и основе rидро
CIP 4.83 лизата казеина и соевых бобов лизата казеина
NBRC 13276 соевых бобов $100 КОЕ и соевых бобов
3 35 ос 3 35 ос $100 КОЕ
1в-........24ч $ 3 дней 3 35 ос
$ 3 дней
Аrаризован
Araризованная ная среда на
Аrаризованная среда на основе основе rидро
среда на основе rидролизата Ka лизата каз&
Рsеиdотопаs rидролизата Ka зеина и соевых ина и соевых
aerugiпosa: зеина и соевых бобов и бульон бобов/НВЧ
АТСС 9027 бобов или бульон на основе rидро бульон на
NC/MB 8626 на основе rидро лизата казеина и основе rидро
С/Р82.118 лизата казеина и соевых бобов лизата казеина
NBRC 13275 соевых бобов $100 КОЕ и соевых бобов
3 35 ос 3 З5 ос $100 КОЕ
18 24 ч $ 3 дней 3 35 ос
< 3 дней
Аrаризован
Аrаризованная ная среда на
Аrаризованная среда на основе основе rидро
среда на основе rидролизата Ka лизата казе
Bacillus sиЫШs: rидролизата Ka зеина и соевых ина и соевых
зеина и соевых бобов и бульон бобов/НВЧ
А ТСС 6633 бобов или бульон бульон на
NC/MB 8054 на основе rидро
CIP 52.62 на основе rидро лизата казеина и основе rидро
NBRC 3134 лизата казеина и соевых бобов лизата казеина
соевых бобов $100 КОЕ и соевых бобов
3 35 ос 3 35 ос $100 КОЕ
1в-........24ч $ 3 ДHe 3 35 ос
< 3 дней
Аraризован
Аraризованная ная среда на
Декстрозный arap среда на основе Декстрозный основе rидро Декстрозный
Сапdidа a/bicaпs: Сабуро или дeK rидролизата Ka arap Сабуро лизата казеина arap Сабуро
АТСС 10231 строзный бульон зеина и соевых $100 КОЕ и соевых бобов $100 КОЕ
NC/MB 3179 Сабуро бобов 2 25 ос $100 КОЕ 2 25 ос
С/Р 48.72 20 25 ос $100 КОЕ $ 5 дней 3 35 ос $ 5 дней
NBRC 1594 2 3 дня 30 35 ос $ 5 дней
$ 5 дней НВЧ: не приме
няется
Декстрозный аrэр Аrаризованная Аraризованная
среда на основе
Aspergillus Сабуро или Kap среда на основе Декстрозный rидролизата Ka Декстрозный
ЬrаsШепsis: тофельн(}-дек rидролизата Ka аrэр Сабуро зеина и соевых arap Сабуро
АТСС 16404 строзный бульон зеина и соевых $100 КОЕ бобов $100 КОЕ
$100 КОЕ
NC/MB 149007 2 25 ос бобов 2 25 ос 3 35 ос 2 25 ос
С/Р 1431.83 7 дней или до $100 КОЕ $ 5 дней $ 5 дней $ 5 дней
NBRC 9455 получения xopo 3 35 ос НВЧ: не приме
шей споруляции $ 5 дней няется
При20товление и использование тест миКРООР2анизмов.
272
rосударственная фармакопея Республики Беларусь
Водорастворимые продукты. Растворяют
или разводят (обычно в соотношении 1: 1 О) ис
пытуемый продукт в забуференном растворе
натрия хлорида и пептона рН 7,0, в фосфат
ном буферном растворе рН 7,2 или в бульоне
на основе rидролизата казеина и соевых бобов.
При необходимости доводят до рН 6 . Даль
нейшие разведения, при необходимости, roтовят
с помощью этоro же разбавителя.
Нерастворимые в воде продукты, не oт
носящиеся к жирам. Суспендируют (обычно в
соотношении 1: 1 О) испытуемый продукт в забу
ференном растворе натрия хлорида и пептона
рН 7,0. в фосфатном буферном растворе рН 7,2
или в бульоне на основе rидролизата казеина и
соевых бобов. Для получения суспензии плохо
смачиваемых веществ может быть добавлено по
верхностно активное вещество, например 1 r/л
полисорбата 80. При необходимости доводят до
рН 6 8. Дальнейшие разведения, при необхо
димости, roтовят с помощью этоro же разбави
теля.
Жиры. Испытуемый продукт растворяют в
изопропилмиристате, стерилизованном с помо
щью фильтрации, или смешивают с минималь
ным необходимым количеством стерильною
полисорбата 80 или иноro не инrибирующеro
стерильноro поверхностно активноro вещества,
HarpeToro при необходимости до температуры
не выше 40 ос или, в исключительных случаях,
не выше 45 ос. Аккуратно перемешивают и при
необходимости поддерживают температуру в BO
дяной бане. Прибавляют предварительно подо
rретый до нужной температуры выбранный раз
бавитель до получения разведения испытуемою
продукта 1: 1 О. Аккуратно перемешивают, под
держивая температуру в течение минимальноrо
времени, необходимою для образования эмуль
сии. Дальнейшие серийные десятикратные раз
ведения MOryT быть приютовлены с использо
ванием выбранною разбавителя, содержащеro
подходящую концентрацию полисорбата 80 или
иноro не инrибирующеrо стерильною поверх
ностно активною вещества.
Жидкости или твердые вещества в форме
аэрозоля. Испытуемый продукт асептически пе
реносят на мембранный фильтр или помещают в
стерильный контейнер для дальнейшеro отбора
проб. Используют либо полный объем аэрозоля,
либо определенное количество доз из каждою
испытуемоrо контейнера.
Трансдермальные пластыри. С TpaHcдep
мальных пластырей удаляют защитное покрытие
и помещают их в стерильные пластмассовые или
стеклянные лотки клейкой поверхностью вверх.
Клейкую поверхность накрывают перильным
пористым материалом, например, марлей, для
предотвращения слипания пластырей, и пере
носят их в подходящий объем выбранною раз
бавителя, содержащеrо инактиваторы, такие как
полисорбат 80 и/или лецитин. Энерrично встря
хивают в течение не менее 30 мин.
4 5 2. Инокуляция и разведение. К приro
товленному как описано выше (4 5 1) образцу и
к контролю (не содержащему испытуемою про
дукта) прибавляют достаточный объем микроб
ной суспензии до получения инокулята, содер.-
жащеro не более 100 КОЕ. Объем суспензии
инокулята не должен превышать 1 % объема
разведенною продукта.
Для демонстрации приемлемой микробной
открываемости испытуемоro продукта для ис
пытания должен быть использован минималь
но возможный фактор разведения. Если это He
возможно из за антимикробною действия или
плохой растворимости, должны быть разрабо
таны иные подходящие методики. Если инrиби
рование роста образцом невозможно предот
вратить, аликвота микробной суспензии может
, прибавляться после нейтрализации, разведения
или фильтрации.
4 5 З. Нейтрализация/устранение анти-
микр06ноrо действия. Количество микроор
rанизмов, обнаруженных в приrотовленном
образце, разведенном, как указано в 4 5 2,
и инкубированном в соответствии с процеду
рой, описанной в 4 5A, сравнивают с количе
ством микроорrанизмов, обнаруженных в KOH
троле.
Если рост инrибируется (уменьшение на KO
эффициент более 2), изменяют процедуру точно
ro подсчета для подтверждения достоверности
полученных результатов. Изменения процеду
ры MOryT включать, например, (1) увеличение
объема разбавителя или питательной среды,
(2) включение специфическою или общею ней
трализующеro areHTa в разбавитель, (3) MeM
бранную фильтрацию или (4) комбинацию пере
численных способов.
Нейтрализующие а2енты. Нейтрализую
щие areHTbI MorYT быть использованы для ней
трализации активности антимикробных areHToB
(таблица 2.6.12. 2). Они MorYT прибавляться в
выбранный разбавитель или в среду, желатель
но перед стерилизацией. Если они используют
ся, их эффективность и отсутствие токсичною
действия на микроорrанизмы должны подтверж
даться контрольным испытанием, проводимым
с нейтрализующим areHToM и без испытуемою
продукта.
Таблица 2.6.12. 2
Общие нейтрализующие а2енты
для мешающих веществ
Мешающее вещество Возможный метод I
нейтрализации
rлутаровый альдеrид, Натрия
ртутьсодержащие rидросульфит
вещества (натрия бисульфит)
Фенольные вещества, Разведение
спирт, альдеrиды, сорбат
Альдеrиды rлицин
2.6.12. Общее количество жизнеспосоБНblХ аэробов
273
Мешающее вещество Возможный метод
нейтрализации
Четвертичные аммоние
вые соединения (ЧАС),
параrидроксибензо Лецитин
аты (парабены), бис
биryаниды
ЧАС, йод, парабены Полисорбат
Ртутьсодержащие Тиоrликолят
вещества
Ртутьсодержащие
вещества, rалоrены, Тиосульфат
альдеrиды
ЭДТА (эдетат) Ионы M g 2+ и Са 2 +
Если невозможно подобрать подходящий
метод нейтрализации, можно сделать заклю
чение, что невозможность изолирования ино
кулированноro микроорrанизма обуславливает
ся бактерицидным действием продукта. Такая
информация указывает на то, что продукт не
может быть контаминирован данным видом ми
кроорrанизма. Однако, возможно, что продукт
инrибирует только некоторые специфицирован
ные для неro микроорrанизмы, но не инrибирует
иные, не включенные в этот список штаммы или
для которых эти штаммы не являются xapaKTep
ными. Поэтому проводят испытание с большим
коэффициентом разведения, допускающим ми
кробный рост И специфические критерии при
емлемости.
4 5A. Обнаружение микроорrанизмов в
присутствии испытуемоrо продукта. Прово
дят отдельные испытания для каждоro перечис
ленноro микроорrанизма. Подсчитывают только
микроорrанизмы прибавленноro тест штамма.
4 5 4 1. Мембранная фильтрация. Исполь
зуют мембранные фильтры с номинальным
размером пор, не превышающим 0,45 мкм. Тип
фильтрующеro материала выбирают таким об
разом, чтобы компоненты испытуемоro образц
не влияли на эффективность удерживания бак
терий. Для каждоro перечисленноro микроорrа
низма используют один мембранный фильтр.
Подходящее количество испытуемоroобраз
ца, приroтовленноro, как описано в 4 5 1 5 3
(желательно, содержащеro 1 r испытуемоro
продукта или менее, если ожидают большое
количество КОЕ), переносят на мембранный
фильтр, немедленно фильтруют и промывают
мембранный фильтр подходящим количеством
разбавителя.
Для определения общеro количества аэро
бов (ОКА, ТАМС tota/ aerobic microbia/ соипt)
мембранный фильтр переносят на поверхность
аrаризованной среды на основе rидролизата Ka
зеина и соевых бобов. Для определения общеro
количества rрибов (OKr, ТУМС tota/ сотЫпеd
yeasts/mou/ds соипt) мембранный фильтр пере
носят на декстрозный arap Сабуро. Чашки инку
бируют, как указано в таблице 2.6.12. 1. Прово
дят подсчет.
4 5 4 2. Методы чашеЧН020 подсчета.
Проводят метод чашечноro подсчета не менее
чем в двух повторностях на каждой среде и ис
пользуют среднее значение.
4 5A 2 1. Метод rлубинноro посева.
На чашку Петри диаметром 9 см прибавля
ют 1 мл испытуемоro образца, приroтовленноro,
как описано в 4 5 1 5 3, и 15 20 мл аrари
зованной среды на основе rидролизата казеина
и соевых бобов или декстрозноro arapa Са буро
с температурой не более 45 ОС. Если исполь
зуются большие чашки Петри, соответственно
увеличивают количество аrаризованной среды.
Для каждоro микроорrанизма, перечисленноro в
таблице 2.6.12. 1, используют не менее 2 чашек
Петри. Инкубируют чашки, как указано в табли
це 2.6.12. 1. Рассчитывают среднее арифмети
ческое значение числа колоний и определяют
КОЕ в первоначальном инокуляте.
4 5 4 2 2. Метод поверхностноrо посева.
На чашку Петри диаметром 9 см прибавля
ют 15 20 мл аrаризованной среды на основе
rидролизата казеина и соевых бобов или дeK
строзноro arapa Сабуро с температурой около
45 ос и выдерживают до затвердевания. Если
используются большие чашки Петри, COOTBeT
ственно увеличивают количество аrаризован
ной среды. Чашки высушивают, например, в
ламинаре или в инкубаторе. Для каждоro микро
орrанизма, перечисленноro в таблице 2.6.12. 1,
используют не менее 2 чашек Петри. Наносят
измеренный объем (не менее 0,1 мл) испыту
емоro образца, приroтовленноrо, как описано в
4 5 1 5 3, на поверхность среды. Инкубиру
ют и проводят расчет, как описано в 4 5 4 2 1.
4 5A 3. Метод наиболее вероятН020 числа
(НВЧ). Точность и правильность метода НВЧ
меньше, чем у метода мембранной фильтра
ции и метода чашечноro подсчета. Ненадежные
результаты получаются rлавным образом при
подсчете rрибов. По этой причине метод НВЧ
используют только для расчета общеro количе
ства аэробов (ОКА) в случае, если иные методы
не доступны. Если данный метод указан, еro
проводят следующим образом.
rотовят серии из не менее 3 серийных дe
сятикратных разведений испытуемоro про
дукта, как описано в 4 5 1 5 3. Из каждоro
уровня разведений используют 3 аликвоты по
1 r или 1 мл для инокулирования 3 пробирок с
9 1 О мл бульона на основе rидролизата казеи
на и соевых бобов. При необходимости в среду
может быть добавлено поверхноr;тно активное
вещество, например, полисорбат 80 или инак
тиватор антимикробноro действия. Таким обра
зом, если приroтовлено 3 уровня разведений,
инокулируют 9 пробирок.
Пробирки инкубируют при температуре
30 35 ос в течение не более 3 дней. Если по
лучение результатов осложнено или они неточ
274
rосударственная фармакопея Республики Беларусь
Таблица 2.6.12. З
3начения наиболее вероятН020 числа миКРООР2анизмов
Наблюдаемые комбинации количества пробирок, в кото- НВЧ В rpaMMe
рых наблюдается рост в каждой серии пробирок Пределы
Количество rpaMMoB или миллилитров испытуемоrо или миллилитре при уровне
продукта в пробирке испытуемоrо значимости 95 %
0,1 0,01 0,001 продукта
О О О <3 0 9,4
О О 1 3 О, 1 9,5
О 1 О 3 0,1 10
О 1 1 6,1 1 ,2 17
О 2 О 6,2 1 ,2 17
О 3 О 9,4 3,5 35
1 О О 3,6 0,2 17
1 О 1 7,2 1 ,2 17
1 О 2 11 4 35
1 1 О 7,4 1 ,3 20
1 1 1 11 4 35
1 2 О 11 4 З5
1 2 1 15 5 З8
1 3 О 16 5 38
2 О О 9,2 1 ,5 35
2 О 1 14 4 35
2 О 2 20 5 38
2 1 О 15 4 38
2 1 1 20 5 38
2 1 2 27 9 94
2 2 О 21 5 0
2 2 1 28 9 94
2 2 2 35 9 94
2 3 О 29 9 94
2 3 1 36 9 94
3 О О 23 5 94
3 О 1 38 9 104
3 О 2 64 16 181
3 1 О 43 9 181
3 1 1 75 17 199
3 1 2 120 30 360
3 1 3 160 30 380
3 2 О 93 18 360
3 2 1 150 30 380
3 2 2 210 ЗО ОО
3 2 3 290 90 990
3 3 О 240 40 990
3 3 1 460 90 1980
3 3 2 1100 200 000
3 3 3 > 1100
ные вследствие природы испытуемоro продук
та, пересевают в аналоrичный бульон или на
аrаризованную среду на основе rидролизата
казеина и соевых бобов, инкубируют при той же
температуре в течение 1 2 дней и используют
полученные результаты. Определяют наиболее
вероятное число микроорrанизмов в rpaMMe
или миллилитре испытуемою продукта по Ta
блице 2.6.12. 3.
4 6. РЕЗУЛЬТАТbf И ИХ ИНТЕРПРЕТАЦИЯ
При проверке приroдности метода мембран
ной фильтрации или метода чашечною под
счета должно быть получено среднее значение
любоro из тест орrанизмов, не отличающееся на
коэффициент более чем 2 от значения контроля,
описанноro в 4 5 2 при отсутствии испытуемо
ю продукта. При проверке приroдности метода
НВЧ рассчитанное значение инокулята должно
2.6.12. Общее количество жизнеспосоБНblХ аэробов
275
укладываться в пределы при уровне значимости
95 % от результата, полученною в контроле.
Если указанные выше критерии не MOryт
быть достиrнуты для одноro или нескольких op
rанизмов, испытываемых любым из описанных
методов, для испытуемоro образца применяют
метод и условия испытания, наиболее близкие к
требуемым критериям.
5. ИСПЫТАНИЕ ПРОДУКТОВ
5 1. КОЛИЧЕСТВА, ИСПОЛЬЗУЕМЫЕ ДЛЯ
ИСПЫТАНИЙ
Если иноro не указано в частной статье, ис
пользуют 1 О r или 1 О мл испытуемою продукта,
отобранноro с указанными выше предосторож
ностями. Для жидкостей или твердых веществ
в форме аэрозоля используют 1 О контейнеров.
Для трансдермальных пластырей используют
1 О пластырей.
Количество, необходимое для испытания,
может быть уменьшено для фармацевтиче
ских субстанций, содержание которых в roтовой
форме будет составлять: количество в дозиро
ванной единице (например, таблетка, капсула,
форма для инъекции) менее или равно 1 Mr или
количество в rpaMMe или миллилитре (для Heдo
зированных лекарственных средств) менее 1 Mr.
В таких случаях количество испытуемоro образ
ца должно быть не менее чем количество, co
держащееся в 1 О дозированных единицах или
10 r или 10 мл продукта.
Для веществ, используемых в качестве фар
мацевтических субстанций, количество которых
оrраничено или объем серии очень мал (напри
мер, менее 1000 мл или 1000 r), количество, He
обходи мое для испытания, должно составлять
1 % от размера серии, если не подтверждена
возможность использования меньшеro количе
ства.
Для продуктов, общее количество в серии
которых менее 200 единиц (например, образ
цы, используемые для клинических исследова
ний) размер образца может быть уменьшен до
2 единиц, или 1 единицы, если объем серии
менее 100 единиц.
Образец (образцы) отбирают случайным
образом из iп bu/k продукта или из доступных
контейнеров лекарственною средства. Для по
лучения требуемоro количества смешивают co
держимое достаточноro числа контейнеров для
формирования образца для анализа.
5 2. ИСПЫТАНИЕ ПРОДУКТА
5 2 1. Мембранная фильтрация.
Используют аппарат для фильтрования,
позволяющий переносить фильтр на среду.
Для приrотовления образца используют
метод, который был установлен приroдным,
как указано в разделе 4, переносят подхо
дящее количество на каждый из двух MeM
бранных фильтров и немедленно фильтруют.
Допускается промывка фильтра после филь
трования.
Для определения общеro количества аэро
бов (ОКА) один из мембранных фильтров пере
носят на поверхность аrаризованной среды на
основе rидролизата казеина и соевых бобов.
Для определения общеro количества rрибов
(OKr) переносят друroй мембранный фильтр на
поверхность декстрозноro arapa Сабуро. Чашки
с аrаризованной средой на основе rидролизата
казеина и соевых бобов инкубируют при темпе
ратуре 30 35 ос в течение 3 5 дней, а чашки
с декстрозным arapoM Сабуро при температуре
20 25 ос в течение 5 7 дней. Рассчитывают
содержание КОЕ в rpaMMe или миллилитре ис
пытуемоro продукта.
При испытании трансдермальных пласты
рей 1 О % объема раствора, приroтовленноro
как указано в 4 5 1, фильтруют по отдельности
через каждый из двух стерильных мембранных
фильтров. Для определения общеro количества
аэробов (ОКА) один из мембранных фильтров
пере носят на поверхность аrаризованной среды
на основе rидролизата казеина и соевых бобов.
Для определения общею количества rрибов
(OKr) переносят друroй мембранный фильтр на
поверхность декстрозноro arapa Сабуро.
5 2 2. Методы чашечноrо подсчета.
5 2 2 1. Метод rлубинноro посева.
Для приroтовления образца используют
метод, который был найден приrодным, как YKa
зано в разделе 4. Для каждой среды roтовят не
менее двух чашек Петри для каждоro уровня
разведений. Чашки с аrаризованной средой на
основе rидролизата казеина и соевых бобов ин
кубируют при температуре 30 35 ос в тече
ние 3 5 дней, а чашки с декстрозным arapoM
Сабуро при температуре 20 25 ос в течение
5 7 дней. Отбирают чашки, соответствующие
данному уровню разведения, с максимальным
количеством колоний менее 250 для определе
ния общеro количества аэробов (ОКА) и менее
50 для определения общеrо количества rрибов
(OKr). Рассчитывают среднее арифметическое
значение числа колоний и определяют КОЕ в
rpaMMe или миллилитре испытуемою продукта.
5 2 2 2. Метод поверхностноrо посева.
Для приrотовления образца используют
метод, который был найден приroдным, как YKa
зано в разделе 4. Для каждой среды roтовят не
менее двух чашек Петри для каждоro уровня раз
ведений. Инкубацию и расчет числа КОЕ прово
дят, как указано для метода rлубинноro посева.
5 2 3. Метод наиболее вероятноrо числа.
Для приroтовления образца используют
метод, который был найден приrодным, как YKa
зано в разделе 4. Все пробирки инкубируют при
температуре 30 35 ос в течение 3 5 дней. При
необходимости пересевают, используя подходя
щий способ. Для каждоrо уровня разведения за
276
rосударственная фармакопея Республики Беларусь
писывают число пробирок, в которых обнаружи
вается микробный рост. Определяют наиболее
вероятное число микроорrанизмов в rpaMMe или
миллилитре испытуемоro продукта в COOTBeT
ствии с таблицей 2.6.12. 3.
#Частные статьи должны содержать CBeдe
ния о наличии антимикробноro действия продук
та и рекомендации по еro устранению.#
5 3. ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ
Общее количество аэробов (ОКА) считает
ся равным числу КОЕ, найденному с использо
ванием аrаризованной среды на основе rидро
лизата казеина и соевых бобов; если на этой
среде обнаруживаются колонии rрибов, они pac
считываются как часть общеro количества аэро
бов (ОКА). Общее количество rрибов (ОКП счи
тается равным числу КОЕ, найденному с ис
пользованием декстрозноro arapa Сабуро; если
на этой среде обнаруживаются колонии бакте
рий, они рассчитываются как часть общеro KO
личества rрибов (ОКП. Если из за микробноro
роста ожидается превышение предельных зна
чений общеro количества rрибов (OKr). допуска
ется использование декстрозноro arapa Сабуро,
содержащеrо антибиотики. Если расчеты произ
водят с использованием метода НВЧ, получен
ное значение рассматривается как общее коли
чество аэробов (ОКА).
Допустимые критерии приемлемости микро
биолоrической чистоты интерпретируют следую
щим образом:
101 КОЕ: максимально допустимое
число = 20;
102 КОЕ: максимально допустимое
число = 200;
103 КОЕ: максимально допустимое
число = 2000 и так далее.
Рекомендуемые растворы и питательные
среды описаны в общей статье 2.6.13.
01/2013:20613
2.6.13. МИКРОБиолоrИЧЕСКИЕ
ИСПЫТАНИЯ НЕСТЕРИЛЬНОЙ
ПРОДУКЦИИ: ИСПЫТАНИЯ НА
НАЛИЧИЕ СПЕЦИФИЧЕСКИХ
микроорrАНИЗМОВ
1. ВВЕДЕНИЕ
Испытания, описаННI:>,е далее в статье, по
зволяют определять отсутствие или предельное
содержание специфических микроорrанизмов,
которые MorYT быть обнаружены в приведенных
условиях.
Испытания предназначены в первую оче
редь для определения соответствия требова
ниям спецификации по микробиолоrической
чистоте, как субстанций, так и лекарственных
средств. В случае использования методик для
таких целей следуют инструкциям, приведенным
ниже, включая количество образцов, которое
следует взять, и интерпретацию результатов, в
соответствии с тем, как изложено далее.
Альтернативные микробиолоrические MeTO
дики, включая автоматические методы, MorYT ис
пользоваться в том случае, если доказана их эк
вивалентность фармакопейным методикам.
2. ОСНОВНЫЕ ОПЕРАЦИИ
Подroтовка образцов проводится так, как
описано в общей статье 2.6.12.
В случае если продукт обладает антими
кробным действием, то еro подавляют до такой
степени, насколько это возможно, или нейтрали
зуют, как описано в общей статье 2.6.12.
В случае если поверхностно активные Be
щества используются при подroтовке образцов
должно быть продемонстрировано, как это YKa
зано в общей статье 2.6.12, отсутствие их TOK
сичности для микроорrанизмов и их совмести
мость с используемыми инактиваторами.
3. РОСТОВЫЕ И инrИБИРУЮЩИЕ
РОСТ СВОЙСТВА СРЕД, приrодных
ДЛЯ ПРОВЕДЕНИЯ ИСПЫТАНИЙ
И ОТРИЦАТЕльноrо КОНТРОЛЯ
Способность методики определять микро
орrанизмы в присутствии испытуемоro образца
должна быть установлена предварительно. При
roдность методики должна быть подтверждена,
в случае если в выполнение испытаний или в
образец вводятся изменения, которые MOryT по
влиять на результат испытаний.
3 1. ПОЛУЧЕНИЕ ТЕСТ ШТАММОВ
Используют стабильные стандартизован
ные суспензии тест штаммов или roтовят их,
как указано ниже. Используется техника coxpa
нения эталонноro банка культур микроорrаниз
мов таким образом, чтобы живые микроорrа
низмы, использованные для инокуляции, были
удалены не более чем на 5 пересевов от ори
rинальноro контрольноro банка культур микро
орrанизмов.
3 1 1. Аэробные микроорrанизмы.
Бактериальные тест штаммы выращива
ют отдельно на бульоне или arape на основе
rидролизата казеина и соевых бобов при TeM
пературе 30 35 ос в течение 18 24 ч. TeCT
штаммы Caпdida a/bicaпs выращивают OT
дельно на декстрозном бупьоне Сабуро или
декстрозном arape Са буро при температуре
20 25 ос в течение 2 3 дней.
Staphy/ococcus aureus АТСС 6538, NC/MB
9518, С/Р 4.83 или NBRC 13276;
Рsеиdотопаs аеrиgiпоsа АТСС 9027,
NC/MB 8626, С/Р 82.118 или NBRC 13275;
2.6.13. Испытания на наличие специфических миКРООР2анизмов
277
Esche,ichia co/i АТСС 8739, NC/MB 8545,
С/Р 53.126 или NBRC З972;
Sa/moпella епtеriса подвид eпteri
са serovar Typhimu,ium АТСС 14028 или, как
альтернативный, Sa/moпella епtе,iса подвид eп
te,ica serova, АЬопу NBRC 100797, NCTC 6017
или С/Р 80.39;
Caпdida а/Ысапs АТСС 10231, NCPF 3179,
/Р48.72 или NBRC 1594.
Для приroтовления суспензий используют
забуференный раствор натрия хлорида и пеп
тона рН 7,0 или фосфатный буферный раствор
рН 7,2. Используют суспензию в течение 2 24 ч
при хранении при температуре 2 8 ОС.
3 1 2. Клостридии. Используют штаммы
C/ost,idium sро,оgепеs АТСС 11437 (NBRC
14293, NC/MB 12343, С/Р 100651) или АТСС
19404 (NCTC 532 или С/Р 79.03) или NBRC
14293. Тест штаммы клостридий выращивают
в анаэробных условиях на обоrащенной среде
для клостридий при температуре 30 35 ос в Te
чение 24 8 ч. В качестве альтернативы приro
товлению и последующему разведению свежей
суспензии веrетативных клеток С/. sporogeпes
для инокуляции используют стабильную суспен
зию спор. Стабильная суспензия спор может co
храняться при температуре 2 8 ос в течение
установленноro периода.
3 2. ОТРИЦАТЕЛЬНЫЙ КОНТРОЛЬ
ДЛЯ проверки условий испытания проводят
отрицательный контроль с использованием BЫ
бранноro разбавителя вместо испытуемоro об
разца. Не должно наблюдаться роста микроор
rанизмов. Отрицательный контроль проводят
также при испытании продуктов, как указано в
разделе 4. Неудавшийся отрицательный KOH
троль требует проведения расследования.
3 3. РОСТОВЫЕ И ИН,ИБИРУЮЩИЕ РОСТ
СВОЙСТВА СРЕД
Испытания проводят на каждой серии roто
вой среды и на каждой серии среды, приroтов
ленной из деrидратированной среды или из YKa
занных инrредиентов.
Таблица 2.б.13. 1
Ростовые, иН2ибирующие рост и индикаторные свойства питательных сред
Среда Свойство Тест-штамм
Обоrатительный питатель Ростовое Е. co/i
ный бульон Мосселя для эн Р aerugiпosa
Испытания на rрамотри теробактерий Инrибирующее S. aureus
цательные бактерии, тo Аrаризованная среда с rлю
лерантные к желчи козой, кристаллическим фи Ростовое и Е. coli
олетовым, нейтральным индикаторное Р aerugiпosa
красным и желчью
Бульон Макконки Ростовое Е. coli
Испытания на Инrибирующее S. aureus
Escherichia саП Ростовое и Е. co/i
Arap Макконки индикаторное
Sа/топеllа eпterica
Обоrатительный питатель подвид enterica se,ova,
ный бульон Раппапорта Ростовое Typhimurium или Sa/mo
Вассилиадиса для Sa/mo пеНа enterica подвид еп
пеllа . terica serova, АЬопу
Испытания на Инrибирующее S. aureus
Sa/moneHa Sa/moпella eпterica
Arap на основе ксилозы, Ростовое и подвид eпterica serova,
Typhimurium или Salmo
лизина,дезоксихолата индикаторное пеНа enterica подвид eп
terica serovar АЬопу
Испытания на Pseudoтo Arap на основе цетримида Ростовое Р ae,ugiпosa
пas аеrugiпоsа Инrибирующее Е. саП
Ростовое и S. au,eus
Испытания на Аrэр на основе маннита и индикаторное
Staphy/ococcus aureus соли Инrибирующее Е. coli
Испытания на Обоrащенная среда для Ростовое С/. sporogenes
клостридий
клостридии Колумбийский arap Ростовое С/. sроrogепеs
Испытания на Декстрозный бульон Са буро Ростовое С. a/bicaпs
Candida a/bicans Декстрозный arap Сабуро Ростовое и С. a/bicaпs
индикаторное
278
rосударственная фармакопея Республики Беларусь
Подтверждают приемлемость свойств подхо
дящих сред, как это указано в таблице 2.6.1 3. 1.
Испытания ростовых свойств жидких
сред: инокулируют порцию соответствующей
среды небольшим количеством подходящих
микроорrанизмов (не более 100 КОЕ). Инкуби
руют при определенной температуре в течение
периода времени, не большеro, чем самый KO
роткий период, указанный в испытании. Ha
блюдается отчетливый рост микроорrанизмов.
сравнимый с тем, что наблюдался для ранее
протестированной и одобренной серии среды.
Испытания ростовых свойств твердых
сред: выполняют методом поверхностноrо
посева, инокулируя каждую чашку небольшим
количеством (не более 100 КОЕ) COOTBeT
ствующих микроорrанизмов. Инкубируют при
определенной температуре в течение пери
ода времени, не большеrо. чем самый KOpOT
кий период, указанный в испытании. Наблю
дается отчетливый рост микроорrанизмов,
сравнимый с тем, что наблюдался для ранее
протестированной и одобренной серии среды.
Испытания на иН2ибирующие рост cвoй
ства жидких и твердых сред: инокулируют
порцию соответствующей среды не менее
чем 100 КОЕ подходящих микроорrанизмов.
Инкубируют при определенной температуре в
течение периода времени. не меньшerо, чем
самый длинный период. указанный в испыта
нии. Не должен наблюдаться рост микроорrа
низмов.
Испытания на индикаторные свойства:
выполняют методом поверхностноro посева,
инокулируя каждую чашку небольшим коли
чеством (не более 100 КОЕ) соответствующих
микроорrанизмов. Инкубируют при опреде
ленной температуре в течение периода Bpe
мени. находящеroся в специфицированных
для испытания рамках. Колонии сравнимы
по проявлению и индикаторным реакциям с
теми, что наблюдались для ранее протести
рованной и одобренной серии среды.
3 4. ИСПblТАНИЯ НА приrодность
МЕТОДА
Для каждоro испытуемоrо продукта BЫ
полняют подrотовку образцов, как описано в
соответствующем подразделе раздела 4. При
бавляют тест штаммы при перемешивании в
указанную питательную среду. Инокулируют
тест штаммы по отдельности. Используют коли
чество микроорrанизмов не более чем 100 КОЕ
при проведении испытания. Выполняют испы
тание, как описано в соответствующем под
разделе раздела 4, используя наименьший
период времени из предписанных.
Специфицированные микроорrанизмы
должны быть определены реакциями индика
ции, как описано в разделе 4.
Проявление любой антимикробной актив
ности продукта неизбежно влечет за собой
модификации методики проведения испыта
ний (см. 4 5 3 в общей статье 2.6.12). В случае
если для данноro продукта антимикробная aK
тивность не может быть нейтрализована без
влияния на тестируемые микроорrанизмы, то
предполаrается, что инrибированные микроор
rанизмы не будут присутствовать в продукте.
4. ИСПЫТАНИЕ ПРОДУКТОВ
4 1. rРАМОТРИЦАТЕЛЬНblЕ БАКТЕРИИ,
ТОЛЕРАНТНblЕ К ЖЕЛЧИ
4 1 1. Приrотовление образца и пред-
варительное инкубирование. rотовят обра
зец разведением 1 к 1 О не менее чем из 1 r
испытуемоrо продукта, как описано в общей
статье 2.6.12, но используя бульон на основе
rидролизата казеина и соевых бобов в каче
стве разбавителя, перемешивают и инкубиру
ют при температуре 20 25 ос в течение Bpe
мени, достаточноro для оживления бактерий,
но недостаточноro для увеличения числа op
rанизмов (обычно 2 ч, но не более 5 ч).
4 1 2. Испытание на отсутствие бакте
рий. Если иное не указано в частной статье,
используют объем, соответствующий 1 r про
дукта приrотовленноro, как указано в пункте
4 1 1, для инокуляции обоrатительноro пита
тельноrо бульона Мосселя для энтеробакте
рий. Инкубируют при температуре 30 35 ос
в течение 24 48 ч. Пересевают на чашку с
аrаризованной средой с rлюкозой, кристал
лическим фиолетовым, нейтральным Kpac
ным и желчью. Инкубируют при температуре
30 35 ос в течение 18 24 ч. Продукт Bыдep
живает испытание, если рост колоний не Ha
блюдается.
4 1 3. Количественная оценка
4 1 3 1. Селекция и пересев. Инокулиру
ют подходящие количества обоrатительноrо
питательноro бульона Мосселя для энтеро
бактерий образцом, приrотовленным, как YKa
зано в пункте 4 1 1, и/или ero разбавлениями.
содержащими, соответственно, 0,1 r, 0,01 r и
0,001 r (или 0,1 мл, 0,01 мл и 0,001 мл) испы
туемоro продукта. Инкубируют при температу
ре 30 35 ос в течение 24 48 ч. Каждую из
культур пересевают на чашку с аrаризованной
средой с rлюкозой, кристаллическим фиоле
товым, нейтральным красным и желчью. Ин
кубируют при температуре 30 35 ос в тече
ние 18 24 ч.
4 1 3 2. Интерпретация результатов.
Рост колоний свидетельствует о положитель
ном результате. Отмечают наименьшее коли
чество продукта, которое дает положительный
результат и наибольшее количество, которое
дает отрицательный результат. Вероятное
количество бактерий определяют по таблице
2.6.1 3. 2.
2.6.13. Испытания на наличие специфических микроорсанизмов
279
Таблица 2.6.1З. 2
Интерпретация результатов
Результаты для каждоrо Вероятное ко-
количества продукта личество бак-
0,1 r 0,01 0,001 r терий в rpaMMe
или rили или или миллили-
0,1 мл 0,01 мл 0,001 мл тре продукта
+ + + Более 103
+ + Менее 103, но
более 102
+ Менее 102, но
более 1 О
Менее 1 О
#В качестве альтернативноro испытания дo
пускается определение бактерий семейства Еп
terobacte,iaceae и друrих микроорrанизмов (Ha
пример, Аеroтопаs, Рsеиdотопаs) , как указано
ниже.
1 О мл испытуемоro образца, подroтовлен
ноro, как описано в общей статье 2.6.12, но с
использованием среды NQ 11 вместо буферноro
раствора с натрия хлоридом и пептоном рН 7,0,
инкубируют в течение 2 5 ч при температуре
30 35 ОС, вносят в 100 мл питательной среды
NQ 3, перемешивают и инкубируют при темпе
рату ре 30 35 ос в течение 24 8 ч. При Ha
личии роста делают пересев на среду NQ 4 в
чашке Петри. Посевы .инкубируют при темпе
рату ре 30 35 ос в течение 24 8 ч. Наличие
роста малиновых с металлическим блеском
или без неro; розовых, бесцветных, блестя
щих, выпуклых колоний rрамотрицательных
палочек указывает на возможную контамина
цию лекарственноro средства бактериями сем.
Eпterobacteriaceae и некоторыми друrими rpa
мотрицательными бактериями. Выросшие KO
лонии пересевают каждую отдельно со среды
NQ 4 на скошенную в пробирках среду NQ 1 и BЫ
ращивают при температуре 30 35 ос течение
18 20 ч. Из каждой пробирки с чистой культу
рой делают пересевы на среду NQ 6 (в двух по
вторностях) И среду NQ 7. <
После посева в половину пробирок со
средой NQ 6 вносят по 0,5 мл стерильноro Ba
зелиновоro масла. Все посевы инкубируют при
температуре 30 35 ос в течение 18 20 ч. Фер
ментацию rлюкозы устанавливают по измене
нию цвета среды NQ 6 из красноro в желтый в
пробирках с маслом и без неro. О наличии ни
тритов в среде NQ 7 судят по появлению KpaCHO
ro окрашивания при внесении в среду реактива
rрисса. Параллельно исследуют чистые куль
туры на наличие фермента цитохромоксидазы.
Если в образце обнаружены rрамотрицательные
неспорообразующие палочки, которые дают OT
рицательную оксидазную реакцию, ферментиру
ют rлюкозу С образованием кислоты (или кисло
ты И rаза) и восстанавливают нитраты в нитриты,
испытуемый продукт контаминирован бактерия
ми семейства ЕпtеroЬасtеriасеае.
Тест на цитохромоксидазу. Полоску филь
тровальной бумаrи смачивают реактивом и HaHO
сят стеклянной палочкой суточную чистую куль
туру исследуемых бактерий со среды NQ 1. Синее
окрашивание, появляющееся через 2 5 мин,
свидетельствует о положительной оксидазной
реакции.
Количественная оценка. Инокулируют под
ходящие количества среды NQ 3 roMoreHaTOM
и/или еro разбавления ми, содержащими, co
ответственно, 0,1 r, 0,01 r и 0,001 r (или 0,1 мл,
0,01 мл и 0,001 мл) испытуемоro продукта. Ин
кубируют при температуре 30 35 ос в течение
24 8 ч. При наличии роста делают пересев
на плотную среду NQ 4 (arap Эндо) и инкубиру
ют при той же температуре в течение 18 24 ч.
В случае появления характерных для ЕпtеroЬас
teriaceae колоний rрамотрицательных палочек,
определяют количество энтеробактерий в 1 r
или в 1 мл образца по таблице 2.6.1 3. 2.#
4 2. ESCHER/CH/A сои
4 2 1. Приrотовление образца и предва-
рительное инкубирование. rотовят образец
разведением 1 к 1 О не менее чем из 1 r испытуе
Moro продукта, как описано в общей статье 2.6.12
и используют 1 О мл или количество, COOTBeTCTBY
ющее 1 r или 1 мл, для инокуляции подходящеrо
количества (определенноro, как указано в пункте
3А) бульона на основе rидролизата казеина и
соевых бобов, перемешивают и инкубируют при
температуре 30 35 ос в течение 18 24 ч.
4 2 2. Селекция и пересев. Встряхивают
контейнер, переносят 1 мл бульона на основе
rидролизата казеина и соевых бобов в 100 мл
бульона Макконки и инкубируют при температу
ре 42 4 ос в течение 24 8 ч. Пересевают на
чашки с arapoM Макконки и инкубируют при TeM
пературе 30 35 ос в течение 18 72 ч.
4 2 3. Интерпретация результатов. Рост
колоний свидетельствует о возможном присут
ствии Е coli. Результат подтверждается иденти
фикационными испытаниями.
Продукт выдерживает испытание, если по
добные колонии не обнаруживаются' или испы
тания идентификации дают отрицательный pe
зультат.
#В качестве альтернативноro испытания дo
пускается определение Escherichia coli, как YKa
зано ниже.
10 мл образца в питательной среде NQ 11
или количество, представляющее 1 r или 1 мл
продукта, подroтовленное и инкубированное,
как описано для определения бактерий семей
ства Eпterobacteriaceae, помещают в 100 мл пи
тательной среды NQ 3 и инкубируют при темпера
туре 30 35 ос в течение 18 24 ч. Пересевают
на среду NQ 4 и инкубируют при температуре
30 35 ос в течение 1 &-------24 ч. На среде NQ 4 Esch
280
rосударственная фармакопея Республики Беларусь
erichia соН образуют, как правило, характерные
малиновые колонии с металлическим блеском
или без неro диаметром 2 мм. Подозритель
ные на принадлежность к Esche,ichia соН коло
нии микроскопируют. При обнаружении rpaMo
трицательных палочек отсевают на скошенную в
пробирках плотную среду NQ 1 и инкубируют при
температуре 30 35 ос в течение 18 24 ч. Из
каждой пробирки с чистой культурой делают пе
ресевы на среды NQ 14 и NQ 15, а также используют
для теста на цитохромоксидазу. Через 18 24 ч
инкубации при температуре 30 35 ос отмеча
ют бактериальный рост или ею отсутствие на
средах NQ 14 и NQ 15. Утилизацию цитрата YCTa
навливают по изменению цвета среды NQ 14 из
зеленоro в синий. Наличие индола определяют
по появлению красноro кольца на поверхности
среды NQ 15 при добавлении реактива Ковача
или Эрлиха.
Если в образце обнаружены rрамотрица
тельные неспорообразующие палочки, не об
ладающие ферментом цитохромоксидазой, не
утилизирующие цитрат натрия и образующие
и ндол , считают. что испытуемый продукт KOHTa
минирован Escherichia co/i.#
4 З. SALMONELLA
4 3 1. Приrотовление образца и пред.
варительное инкубирование. rотовят обра
зец испытуемоro продукта, как описано в общей
статье 2.6.12. и используют количество, COOTBeT
ствующее не менее 1 О r или 1 О мл для иноку
ляции подходящеro количества (определенноro,
как указано в пункте 3А) бульона на основе rи
дролизата казеина и соевых бобов, перемеши
вают и инкубируют при температуре ЗО 35 ос в
течение 1 24 ч.
4 3 2. Селекция и пересев. Переносят
0,1 мл бульона на основе rидролизата казеи
на и соевых бобов в 1 О мл обоrатительноro пи
тательноrо бульона Раппапорта Вассилиадиса
для Sa/тoпe//a и инкубируют при температу
ре 30 35 ос в течение 18 24 ч. Пересевают
на чашки с arapoM на основе ксилозы, лизина,
дезоксихолата. Инкубируют при температуре
30 35 ос в течение 18-------48 ч.
4 3 3. Интерпретация результатов. На воз
можное присутствие Sa/moпe//a указывает рост
хорошо развитых красных колоний или красных
колоний с черным центром. Присутствие Sa/mo
пе//а подтверждается в идентификационных ис
пытаниях.
Продукт выдерживает испытание, если KO
лонии опиr;анноrо типа не обнаруживаются или
испытания идентификации дают отрицательный
результат.
#В качестве альтернативноrо испытания дo
пускается определение Sa/moпella, как указано
ниже.
1 мл обоrащенной культуры на среде NQ 3
вносят в пробирку С 1 О мл среды NQ 12 и инку
бируют при температуре 30 35 ос в течение
16 18 ч. Делают пересев петлей на среду NQ 5
и инкубируют при температуре 30 35 ос в Te
чение 24 8 ч. На среде NQ 5 Sа/топе//а обра
зует, как правило, типичные черные колонии с
характерным металлическим блеском, при этом
участок среды под колонией прокрашивается в
черный цвет. Подозрительные на принадлеж
ность к Sа/топе//а колонии микроскопируют и
при обнаружении в мазках rрамотрицательных
палочек отсевают на среду NQ 13 (Tpexcaxap
ный arap с солями железа), нанося небольшое
количество культуры петлей сначала на скошен
ную часть arapa, а потом уколом в столбик. Па
I?аллельно ставят тест на цитохромоксидазу, ис
пользуя чистую культуру с плотной среды NQ 1.
Через 18 24 ч инкубации при 30 35 ос отмеча
ют изменение цвета среды из красноro в желтый
только в столбике питательной среды. Почерне
ние среды свидетельствует об образовании ce
роводорода.
Если в образце обнаружены rрамотрица
тельные неспорообразующие палочки, не обла
дающие ферментом цитохромоксидаза, не фер
ментирующие сахарозу и лактозу и выделяющие
сероводород, считают, что испытуемый продукт
контаминирован Sа/топе//а. #
4 4. PSEUOOMONAS AERUG/NOSA
4A 1. Приrотовление образца и предвари-
тельное инкубирование. rотовят образец раз
ведением 1 к 1 О не менее чем из 1 r испытуемо
ro продукта, как описано в общей статье 2.6.12, и
используют 1 О мл или количество, COOTBeTCTBY
ющее 1 r или 1 мл, для инокуляции подходящею
количества (определенноro, как указано в пункте
3А) бульона на основе rидролизата казеина и
соевых бобов, перемешивают. В случае если ис
пытанию подверrаются трансдермальные пла
стыри. фильтруют через стерильный мембран
ный фильтр объем образца, соответствующий 1
пластырю, обработанному, как указано в разделе
4 5 1 общей статьи 2.6.12, и переносят фильтр
в 100 мл бульона на основе rидролизата казеи
на и соевых бобов. Инкубируют при температуре
30 35 ос в течение 18 24 ч.
4 4 2. Селекция и пересев. Пересевают на
чашки с аrаризованной средой на основе цетри
мида и инкубируют при температуре 30 35 ос в
течение 18 72 ч.
4A 3. Интерпретация результатов. На воз
можное присутствие Р aerugiпosa указывает
рост колоний. Результат подтверждается иден
тификационными испытаниями.
Продукт выдерживает испытание, если по
добные колонии не обнаруживаются или испы
тания идентификации дают отрицательный pe
зультат.
2.6.13. Испытания на наличие специфических микроорzанизмов
281
#В качестве альтернативноrо испытания дo
пускается определение Pseudomoпas aerugiпo
sa, как указано ниже.
Образец в количестве 10 r (мл) вносят в
100 мл питательной среды NQ 8, перемешива
ют и инкубируют при температуре 30 35 ос
в течение 24 8 ч. Делают пересев на чашку
с плотной питательной средой NQ 9 и инку
бируют при температуре 30 35 ос в тече
ние 18 48 ч. Рост зеленоватых, как правило,
флюоресцирующих колоний, rолубых в уль
трафиолетовом свете (что свидетельствует
о наличии пиrмента пиоцианина), указывает
на возможность заrрязнения лекарственно
ro средства Pseudomoпas aerugiпosa. В этом
случае подозрительные колонии пересева
ют на скошенную плотную среду NQ 1, инку
бируют при температуре 30 35 ос в течение
18 24 ч. Выросшие колонии микроскопируют
и, при выявлении rрамотрицательных пало
чек, проводят тест на наличие фермента ци
тохромоксидаза. Если в образце обнаружены
rрамотрицательные неспорообразующие па
лочки, которые дают положительную оксидаз
ную реакцию и образуют сине зеленый пиr
мент, испытуемый продукт контаминирован
Рsеиdотопаs аеrиgiпоsа.#
4 5. STAPHYLOCOCCUS AUREUS
4 5 1. Приrотовление образцов и пред
варительное инкубирование. rотовят об
разец разведением 1 к 1 О не менее чем из 1 r
испытуемоrо продукта, как описано в общей
статье 2.6.12, и используют 1 О мл или количе
ство, соответствующее 1 r или 1 мл, для иноку
ляции подходящеrо количества (определенно
ro, как указано в пункте 3А) бульона на основе
rидролизата казеина и соевых бобов, переме
шивают. В случае если испытанию подверrа
ются трансдермальные пластыри, фильтруют
через стерильный мембранный фильтр объем
образца, соответствующий 1 пластырю, обра
ботанному, как указано в разделе 4 5 1 общей
статьи 2.6.12, и переносят фильтр в 100 мл бу
льона на основе rидролизата казеина и соевых
бобов. Инкубируют при температуре 30 35 ос
в течение 18 24 ч.
4 5 2. Селекция и пересев. Пересева
ЮТ на чашки с аrаризованной средой на основе
маннита и соли и инкубируют при температуре
30 35 ос в течение 18 72 ч.
4 5 3. Интерпретация результатов. На воз
можное присутствие S. aureus указывает рост
желтых/белых колоний, окруженных желтыми
зонами. Результат подтверждается идентифика
ционными испытаниями.
Продукт выдерживает испытание, если по
добные колонии не обнаруживаются или испы
тания идентификации дают отрицательный pe
зультат.
#В качестве альтернативноro испытания дo
пускается определение Staphy/ococcus aureus,
как указано ниже.
Образец в количестве 10 r (мл) вносят в
100 мл питательной среды NQ 8, перемешивают
и инкубируют при температуре 30 35 ос в тече
ние 24-------48 ч. Делают пересев на чашку с плотной
питательной средой NQ 1 О и инкубируют при TeM
пературе 30 35 ос в течение 18 8 ч. Рост зо
лотисто желтых колоний, окруженных желтыми
зонами (что свидетельствует о ферментации MaH
нита), указывает на возможность заrрязнения ле
карственною средства Staphy/ococcus aureus. В
этом случае колонии пересевают на скошенную
плотную среду NQ 1, инкубируют при температуре
30 35 ос в течение 1 24 ч. Выросшие колонии
микроскопируют и, при выявлении rрамоположи
тельных кокков, расположенных rроздьями, прово
дят тест на наличие фермента плазмокоаryлаза.
Тест на наличие плазмокоа2улазы (реакция
плазмокоа2уляции). Используют сухую кроличью
цитратную плазму промышленноro производства
в соответствии с инструкцией по ее применению.
Если в образце обнаружены rрамположи
тельные кокки, которые ферментируют маннит и
дают положительную реакцию плазмокоаryляции,
испытуемый продукт контаминирован Staphy
/ococcus aureus. #
4 6. CLOSTRID/A
4 6 1. Приrотовление образцов и тепло-
вая обработка. rотовят образец разведением
1 к 1 О (минимальный общий объем 20 мл) не
менее чем из 2 r или 2 мл испытуемоro продук
та, как описано в общей статье 2.6.12. Делят об
разец на две части, каждая из которых не менее
1 О мл. Наrревают одну порцию при температу
ре 80 ос в течение 1 О мин и быстро охлаждают.
Вторую порцию не наrревают.
4 6 2. Селекция и пересев. Используют
1 О мл или количество, соответствующее 1 r или
1 мл испытуемоrо продукта из обеих порций об
разца для инокуляции подходящеro количества
(определенноro, как указано в 3А) обоrащенной
среды для клостридий. Инкубируют в анаэроб
ных условиях при температуре 30 35 ос в Te
чение 48 ч. После инкубации делают пересев из
каждою контейнера на колумбийский arap и ин
кубируют В анаэробных условиях при темпера
туре 30 35 ос в течение 48 72 ч.
4 6 3. Интерпретация результатов. AHa
эробный рост палочек (с эндоспорами и без
них), дающих отрицательную реакцию на KaTa
лазу, свидетельствует о присутствии клостри
дий. Результат подтверждается идентификаци
онными испытаниями.
Продукт выдерживает испытание, если по
добные колонии не обнаруживаются или испы
тания идентификации дают отрицательный pe
зультат.
282
rосударственная фармакопея Республики Беларусь
4 7. CANOIDA ALB/CANS
4 7 1. Приrотовление образцов и пред
варительное инкубирование. rотовят образец
разведением 1 к 1 О не менее чем из 1 r испы
туемоro продукта, как описано в общей статье
2.6. 12, и используют 1 О мл или количество, co
ответствующее 1 r или 1 мл, для инокуляции
100 мл декстрозноro бульона Сабуро, переме
шивают. Инкубируют при температуре 30 35 ос
в течение 3 5 дней.
4 5 2. Селекция и пересев. Пересевают на
чашки с декстрозным arapoM Са буро и инкубиру
ют при температуре 30 35 ос в течение 24 8 ч.
4 7 3. Интерпретация результатов. Рост
белых колоний может свидетельствовать о при
сутствии С. а/Ысапs. Результат подтверждается
идентификационными испытаниями.
Продукт выдерживает испытание, если по
добные колонии не обнаруживаются или испы
тания идентификации дают отрицательный pe
зулыат.
Следующий раздел приводится для инфор
мации.
5. РЕКОМЕНДУЕМЫЕ РАСТВОРЫ
И ПИТАТЕЛЬНЫЕ СРЕДЫ
Нижеописанные растворы и питательные
среды удовлетворяют целям, для которых они
предназначены в фармакопейном испытании на
микробную контаминацию. MoryT быть исполь
зованы и друrие среды при условии, что их при
rодность может быть доказана.
Основной буферный раствор. 34 r калия
диrидрофосфата помещают в мерную колбу
вместимостью 1000 мл, растворяют в 500 мл
очищенной воды, доводят значение рН раствора
до (7,2:tO,2) натрия rидроксидом, доводят водой
очищенной до объема 1000,0 мл И перемеши
вают. Разливают в контейнеры и стерилизуют.
Хранят при температуре 2 8 ОС.
Фосфатный буферный раствор рН 7,2.
Смешивают основной буферный раствор и очи
щенную воду (1 :800, об/об) и стерилизуют.
Забуференный раствор натрия хлорида
и пептона рН 7,0
Калия диrидрофосфат
Динатрия rидрофосфат
диrидрат
Натрия хлорид
Пептон (мясной или
казеиновый)
Вода очищенная
3,6 r
7,2 r. что эквива
лентно 0.067 М
фосфата
4.3 r
1,0 r
1000 мл
Стерилизуют в автоклаве в соответствии с
валидированной процедурой.
Бульон на основе rидролизата казеина и
соевых бобов
Панкреатический rидролизат казеина
Папаиновый rидролизат соевых
бобов
Натрия хлорид
Дикалия rидрофосфат
rлюкоза моноrидрат
Вода очищенная
17,0 r
3,0 r
5,0 r
2,5 r
2,5 r
1000 мл
Значение рН доводят таким образом,
чтобы после стерилизации оно составляло
(7,3:tO,2) при температуре 25 ОС. Стерилизуют
в автоклаве в соответствии с валидированной
процедурой.
Аrаризованная среда на основе rидроли
зата казеина и соевых бобов
Панкреатический rидролизат казеина 15,0 r
Папаиновый rидролизат соевых 5,0 r
бобов
Натрия хлорид
Arap
Вода очищенная
5,0 r
15,0 r
1000 мл
Значение рН доводят таким образом,
чтобы после стерилизации оно составляло
(7,3:tO,2) при температуре 25 ОС. Стерилизуют
в автоклаве в соответствии с валидированной
процедурой.
Декстрозный arap Са буро
Декстроза
Смесь пепсиновоro rидролизата жи
вотных тканей и панкреатическоro
rидролизата казеина (1:1)
Arap
Вода очищенная
40,0 r
10,0 r
15,0 r
1000 мл
Значение рН доводят таким образом,
чтобы после стерилизации оно составляло
(5,6:tO,2) при температуре 25 ОС. Стерилизуют
в автоклаве в соответствии с валидированной
процедурой.
Аrаризованная среда
фельной декстрозы
Вытяжка из картофеля
Декстроза
Arap
Вода очищенная
на основе KapTO
200 r
20,0 r
15,0 r
1000 мл
Значение рН доводят таким образом.
чтобы после стерилизации оно составляло
(5,6:t0,2) при температуре 25 ОС. Стерилизуют
в автоклаве в соответствии с валидированнoii
процедурой.
2.6.13. Испытания на наличие специфических МИКрООр2анизмов
283
Декстрозный бульон Сабуро
Декстроза
Смесь пепсиновоro rидролизата
животных тканей и панкреатическоro
rидролизата казеина (1: 1 )
Вода очищенная
20,0 r
10,0 r
1000 мл
Значение рН доводят таким образом, чтобы
после стерилизации оно составляло (5,6:tO,2) при
температуре 25 ОС. Стерилизуют в автоклаве в
соответствии с валидированной процедурой.
Обоrатительный бульон Мосселя для
энтеробактерий
Панкреатическийrидролизат
желатина
rлюкоза моноrидрат
Деrидратированная бычья желчь
Калия диrидрофосфат
Динатрия rидрофосфата диrидрат
Бриллиантовый зеленый
Вода очищенная
10,0 r
5,0 r
20.0 r
3,0 r
8,0 r
15 Mr
1000 мл
Значение рН доводят таким образом, чтобы
после стерилизации оно составляло (7,2:t0,2)
при температуре 25 ОС. Наrревают при темпера
туре 100 ос в течение 30 мин и немедленно ox
лаждают.
Аrаризованная среда с rл, юкозой кри
сталлическим фиолетовым, нейтральным
красным и желчью
Дрожжевой экстракт
Панкреатический rидролизат
желатина
Соли желчных кислот
Натрия хлорид
rлюкоза моноrидрат
Arap
Нейтральный красный
Кристаллический фиолетовый
Вода очищенная
3,0 r
7,0 r
1,5 r
5,0 r
10,0 r
15,0 r
30 Mr
2 Mr
1000 мл
Значение рН доводят таким образом, чтобы
после стерилизации оно составляло (7,4:t0,2)
при температуре 25 ОС. Наrревают до кипения.
Не автоклавируют.
Бульон Макконки
Панкреатический rидролизат
желатина
Лактоза моноrидрат
Деrидратированная бычья желчь
Бромкрезоловый пурпурный
Вода очищенная
20,0 r
10,0 r
5,0 r
10 Mr
1000 мл
Значение рН доводят таким образом, чтобы
после стерилизации оно составляло (7,3:t0,2) при
температуре 25 ОС. Стерилизуют в автоклаве в
соответствии с валидированной процедурой.
Аrаризованная среда Макконки
Панкреатический rидролизат жела
тина
Пептоны (мясной и казеиновый)
Лактоза моноrидрат
Натрия хлорид
Соли желчных кислот
Arap
Нейтральный красный
Кристаллический фиолетовый
Вода очищенная
17,0 r
3,0 r
10,0 r
5,0 r
1,5 r
13,5 r
30,0 Mr
1 Mr
1000 мл
Значение рН доводят таким образом, чтобы
после стерилизации оно составляло (7,3:t0,2)
при температуре 25 ОС. Кипятят в течение одной
минуты при постоянном встряхивании, затем
стерилизуют в автоклаве в соответствии с вали
дированной процедурой.
Обоrатительный питательный бульон
Раппапорта Вассилиадиса для Sa/moпella
Соевый пептон 4,5 r
Маrния хлорид rексаrидрат 29,0 r
Дикалия rидрофосфат 0,4 r
Калия диrидрофосфат 0,6 r
Малахитовый зеленый 0,036 r
Вода очищенная 1000 мл
Растворяют при осторожном наrревании.
Стерилизуют в автоклаве в соответствии с вали
дированной процедурой при температуре, не пре
вышающей 115 ос. Значение рН доводят таким
образом, чтобы после наrревания и стерилизации
оно составляло (5,2:t0,2) при температуре 25 ОС.
Аrаризованная среда на основе ксилозы,
лизина,дезоксихолата
Ксилоза
L Лизин
Лактоза моноrидрат
Сахароза
Натрия хлорид
Дрожжевой экстракт
Феноловый красный
Arap
Натрия дезоксихолат
Натрия тиосульфат
Железа (111) аммония цитрат
Вода очищенная
3,5 r
5,0 r
7,5 r
.7,5 r
5,0 r
3,0 r
80 Mr
13,5 r
2,5 r
6,8 r
0,8 r
1000 мл
Значение рН доводят таким образом, чтобы
после наrревания оно составляло (5,2:t0,2) при
температуре 25 ОС. Наrревают до кипения, ox
лаждают до температуры 50 ос и разливают по
чашкам Петри. Не автоклавируют.
284
rосударственная фармакопея Республики Беларусь
Аrаризованная среда на основе цетри-
мида
Панкреатический rидролизат жела 20,0 r
тина
Маrния хлорид
Калия сульфат
Цетримид
Arap
Вода очищенная
rлицерин
1,4 r
10,0 r
0,3 r
13,6 r
1000 мл
10,0 мл
Наrревают до кипения и кипятят в течение
1 мин при встряхивании. Значение рН доводят
таким образом, чтобы после стерилизации оно
составляло (7,2:1:0,2) при температуре 25 ОС.
Стерилизуют в автоклаве в соответствии с вали
дированной процедурой.
Аrаризованная среда на основе маннита
и соли
Панкреатический rидролизат казеина 5,0 r
Пепсиновый rидролизат животных 5,0 r
тканей
r овяжий экстракт
D Маннит
Натрия хлорид
Arap
Феноловый красный
Вода очищенная
1,0 r
10,0 r
75,0 r
15,0 r
0,025 r
1000 мл
Наrревают до кипения и кипятят в течение
1 мин при встряхивании. Значение рН доводят
таким образом, чтобы после стерилизации оно
составляло (7,4:1:0,2) при температуре 25 СС.
Стерилизуют в автоклаве в соответствии С вали
дированной процедурой.
Обоrащенная питательная
клостридий
rовяжий экстракт
Пептон
Дрожжевой экстракт
Растворимый крахмал
rлюкоза моноrидрат
Цистеина rидрохлорид
Натрия хлорид
Натрия ацетат
Arap
Вода очищенная
среда для
10.0 r
10.0 r
3.0 r
1,0 r
5.0 r
0.5 r
5,0 r
3,0 r
0,5 r
1000 мл
Arap замачивают, растворяют при Harpe
вании до кипения при постоянном переме
шивании. При необходимости значение рН
доводят таким образом, чтобы после стери
лизации оно составляло около (6,8:1:0,2) при
температуре 25 ОС. Стерилизуют в автокла
ве в соответствии с валидированной проце
дурой.
Колумбийский arap
Панкреатический rидролизат казеина
Пепсиновый rидролизат мяса
Панкреатический rидролизат сердца
Дрожжевой экстракт
Кукурузный крахмал
Натрия хлорид
Arap, в зависимости от rелеобразую
щей способности
Вода очищенная
10,0 r
5,0 r
3,0 r
5,0 r
1,0 r
5,0 r
10,O
15,0 r
1000 мл
Arap замачивают, растворяют при HarpeBa
нии до кипения при постоянном перемешивании.
При необходимости значение рН доводят таким
образом, чтобы после стерилизации оно COCTaB
л ло (7,3:1:0,2) при 25 ОС. Стерилизуют в автокла
ве в соответствии с валидированной процедурой.
Охлаждают до температуры 45 50 ОС, и при He
обходимости прибавляют rентамицина сульфат
в количестве, соответствующем 20 Mr основания
rентамицина; разливают по чашкам Петри.
"Кроме описанных выше сред в некоторых
испытаниях М02ут использоваться следующие
альтернативные питательные среды и peaK
тивы:
Фосфатный буферный раствор с натрия
хлоридом и пептоном (1/15 моль)
Калия диrидрофосфат
Динатрия rидрофосфат диrидрат
Натрия хлорид
Пептон ферментативный сухой
Вода очищенная
рН
3,56 r
7,23 r
4,3 r
1,0 r
1000 мл
(7,0:1:0,2)
Компоненты растворяют в воде очищенной
при наrревании, фильтруют через бумажный
фильтр. Стерилизуют в паровом стерилизаторе
при температуре 121 ос в течение 15 мин.
Нейтрализующая жидкость
Полисорбат 80
Лецитин (яичный)
rистидина rидрохлорид
Пептон (мясной или казеиновый)
Натрия хлорид
Калия диrидрофосфат
Динатрия rидрофосфат диrидрат
Вода очищенная
30 r
3 r
1 r
1 r
4,3 r
3,6 r
7,2 r
1000 мл
Раствор стерилизуют в паровом стерилиза
торе при температуре 121 ос в течение 15 мин.
Если нейтрализующая способность paCT
вора недостаточна, концентрация полисор
ба та 80 или лецитина может быть увеличена.
Кроме тоro, MorYT быть добавлены нейтрализа
торы, перечисленные в статье 2.6.12 в таблице
2.6.12. 2.
2.6.13. Испытания на наличие специфических микроорzанизмов
285
Среда N!! 1 (для выращивания бактерий)
Пептон ферментативный сухой 10,0 r
Натрия хлорид 5,0 r
rлюкоза моноrидрат 1,0 r
Arap 13,0 r
Мясная вода 1000 мл
Все компоненты растворяют в мясной воде при
наrревании, вносят rлюкозу моноrидрат, значение
рН доводят таким образом, чтобы после стерили
зации оно составляло (7,3:tO,2). Кипятят в течение
1 мин, прибавляют arap, наrревают до полною еro
расплавления, фильтруют через ватно--марлевый
фильтр. Стерилизуют в паровом стерилизаторе при
температуре 121 ос в течение 15 мин.
Жидкая среда N!! 1 (мясо пептонный бульон)
Пептон ферментативный сухой 10,0 r
Натрия хлорид 5,0 r
rлюкоза моноrидрат 1,0 r
Мясная вода 1000 мл
к мясной воде прибавляют пептон и натрия
хлорид, растворяют при наrpевании, вносят rлю
козу моноrидрат, значение рН доводят таким об
разом, чтобы после стерилизации оно составляло
(7,3:t0,2). Кипятят в течение 1 мин, фильтруют через
бумажный фильтр. Стерилизуют в паровом стери
лизаторе при температуре 121 ос в течение 15 мин.
Среда N!! 2 (для выращивания rрибов, Ca
буро аrар )
Пептон ферментативный сухой
rлюкоза моноrидрат
Arap
Вода очищенная
10,0 r
40,0 r
13,0 r
1000 мл
Все компоненты растворяют в воде очи
щенной при наrревании, значение рН доводят
таким образом, чтобы после стерилизации оно
составляло (5,6:t0,2). Прибавляют arap, Harpe <
вают до полноro еro расплавления, фильтруют
через ватно марлевый фильтр. Для подавле
ния роста бактерий после стерилизации при
бавляют 100 Mr бензилпенициллина натрия и
100 Mr тетрациклина (либо перед стерилизаци
ей 50 Mr хлорамфеникола) на 100 мл среды.
Стерилизуют в паровом стерилизаторе при
температуре 121 ос в течение 15 мин.
Жидкая среда N!! 2 (среда Сабуро)
Пептон ферментативный сухой 1О.О r
rлюкоза моноrидрат 40,0 r
Вода очищенная 1000 мл
Все компоненты растворяют в воде очищен
tЮЙ при наrревании, значение рН доводят таким
образом, чтобы после стерилизации оно co
ставляло (5,6:tO,2). Фильтруют через бумажный
фильтр. Стерилизуют в паровом стерилизаторе
при температуре 121 ос в течение 15 мин.
Среда N!! 3 (среда обоrащения для бакте
рий семейства Eпterobacte,iaceae)
Пептон ферментативный сухой
Калия диrидрофосфат
Динатрия rидрофосфат диrидрат
rлюкоза моноrидрат
Феноловый красный
Малахитовый зеленый
Мясная вода
10,0 r
2,5 r
7,5 r
10,0 r
0,08 r
0,015 r
1000 мл
Пептон и соли растворяют в мясной воде при
наrревании, вносят rлюкозу моноrидрат, прибав
ляют 8 мл 1 % раствора феноловоro красноro и
3 мл 0,5 % раствора малахитовоro зеленоro, зна
чение рН доводят таким образом, чтобы после
стерилизации оно составляло (7,3:t0,2). Кипя
тят В течение 1 мин, фильтруют через бумажный
фильтр. Стерилизуют в паровом стерилизаторе
при температуре 121 ос в течение 15 мин.
Среда N!! 4 (для выделения бактерий ce
мейства ЕпtеroЬасtе,iасеае, arap Эндо)
Панкреатический rидролизат 12,0 r
рыбной муки
Экстракт пекарных дрожжей
Натрия хлорид
Натрия сульфит безводный
Динатрия rидрофосфат диrидрат
Лактоза моноrидрат
Фуксин основной
Arap
Вода очищенная
1,0 r
3,4 r
0,8 r
0,5 r
10,0 r
0,2 r
15,0 r
1000 мл
Все компоненты размешивают в воде очи
щенной, значение рН доводят таким обра
зом, чтобы после стерилизации оно составляло
(7,4:t0,2). Наrревают до полноro расплавления
arapa и кипятят в течение 2 3 мин. Фильтруют
через ватно марлевый фильтр, снова наrревают
до момента закипания. Охлаждают до температу
ры от 45 ос до 50 ос и разливают в чашки Петри.
Среда N!! 5 (для выделения бактерий семей
ства Eпterobacteriaceae, висмутсульфит arap)
Панкреатический rидролизат мяса 1 0,1 r
Динатрия rидрофосфат диrидрат 3,68 r
Натрия хлорид 2,6 r
Натрия карбонат 0,65 r
Висмута цитрат 2,38 r
Железа (11) аммония сульфат 0,97 r
rлюкоза моноrидрат 3,9 r
Бриллиантовый зеленый 0,028 r
Arap 15,0 r
Вода очищенная 1000 мл
286
rосударственная фармакопея Республики Беларусь
Все компоненты размешивают в воде очи
щенной, значение рН доводят таким образом,
чтобы после стерилизации оно составляло
(7,6:t0.2). Наrревают до полноrо расплавления
arapa и кипятят в течение 3 5 мин. Охлаждают
до температуры от 45 ос до 50 ос и разливают в
чашки Петри.
Среда N!! б (для определения ферментации
rлюкозы)
Пептон ферментативный сухой
Натрия хлорид
rлюкоза моноrидрат
Феноловый красный
Мясная вода
10,0 r
5,0 r
40,0 r
0,08 r
1000 мл
Пептон и натрия хлорид растворяют в
мясной воде при наrревании, вносят rлюкозу
моноrидрат, прибавляют 8 мл 1 % раствора фе
ноловоrо красноro, значение рН доводят таким
образом, чтобы после стерилизации оно COCTaB
ляло (7,2:t0,2). Кипятят в течение 1 мин, филь
труют через бумажный фильтр и разливают в
пробирки с поплавками по 4 5 мл. Стерилизу
ют в паровом стерилизаторе при температуре
121 ос в течение 15 мин. По окончании стерили
зации быстро охлаждают.
Среда N!! 7 (для определения восстановле
ния нитратов в нитриты)
Пептон ферментативный сухой
Натрия хлорид
Калия нитрат
Вода очищенная
5,0 r
5,0 r
1,5 r
1000 мл
Все компоненты растворяют в воде очи
щенной при наrревании. значение рН доводят
таким образом. чтобы после стерилизаuии оно
составляло (7.2:t0.2). Кипятят в течение 1 мин.
фильтруют через бумажный фильтр, разливают
в пробирки по 4 5 мл. Стерилизуют в паровом
стерилизаторе при температуре 121 ос в тече
ние 15 мин.
Среда N!! 8 (среда обоrащения для Pseudo
moпas aerugiпosa и Staphy/ococcus aureus)
Пептон ферментативный сухой 10,0 r
Натрия хлорид 5,0 r
Дикалия rидрофосфат 2,5 r
rлюкоза моноrидрат 2,5 r
Вода очищенная 1000 мл
Пептон и соли растворяют в воде ПрlA HarpeBa
нии, вносят rлюкозу моноrидрат, значение рН дo
водят таким образом, чтобы после стерилизации
оно составляло (7,3:tO,2). Кипятятвтечение 1 мин,
фильтруют через бумажный фильтр. Стерили
зуют в паровом стерилизаторе при температуре
121 ос в течение 15 мин.
Среда N!! 9 (для выявления
оцианина)
Пептон ферментативный сухой
Маrния хлорид безводный
Калия сульфат безводный
rлицерин
Arap
Вода очищенная
пиrмента пи
20,0 r
1,4 r
10,0 r
10,0 мл
15,0 r
1000 мл
Пептон и соли растворяют в воде очищен
ной. Затем вносят rлицерин, растворяют при
наrревании, значение рН доводят таким обра
зом, чтобы после стерилизации оно составляло
(7,2:t0,2). Кипятят в течение 1 мин, прибавляют
arap, наrревают до полноro еro расплавления,
фильтруют через ватно марлевый фильтр. CTe
рилизуют в паровом стерилизаторе при темпе
ратуре 121 ос в течение 15 мин.
Среда N!! 1 О (для идентификации Staphy/o
coccus aureus)
Пептон ферментативный сухой
Натрия хлорид
D Маннит
Феноловый красный
Arap
Вода очищенная
10,0 r
75,0 r
10,0 r
0,025 r
15,0 r
1000 мл
Все компоненты растворяют в воде очищен
ной, прибавляют 2,5 мл 1 % раствора феноло
воro красноro, значение рН доводят таким обра
зом, чтобы после стерилизации оно составляло
(7,4:t0,2). Кипятят в течение 1 мин, прибавляют
arap, наrревают до полноrо еro расплавления,
фильтруют через ватно марлевый фильтр. CTe
рилизуют в паровом стерилизаторе при темпе
ратуре 121 ос в течение 15 мин.
Среда N!! 11 (для предварительноro обоrа
щения бактерий семейства ЕпtеroЬасtеriасеае)
Пептон ферментативный сухой 8,0 r
Лактоза моноrидрат 5,0 r
Вода очищенная 1000 мл
Все компоненты растворяют в воде очищен
ной при наrревании, значение рН доводят таким
образом, чтобы после стерилизации оно co
ставляло (6,9:t0,1). Фильтруют через бумажный
фильтр. Стерилизуют в паровом стерилизаторе
при температуре 121 ос в течение 15 мин.
Среда N!! 12 (для накопления и вь'деления
бактерий рода Sa/moпella, селенитовая среда)
Панкреатический rидролизат казеина 5,26 r
Лактоза моноrидрат 4,21 r
Динатрия rидрофосфат диrидрат 6,23 r
Натрия rидроселенит (без теллура) 4,21 r
Вода очищенная 1000 мл
2.6.13. Испытания на наличие специфических миКрООр2анизмов
287
Все компоненты тщательно размешивают
в 1000 мл воды очищенной. Колбу закрывают
ватной пробкой, быстро доводят до кипения
(до образования пузырьков) и сразу прекра
щают HarpeB. Значение рН после стерилиза
ции (7,5:t0,2).
Среда Ng 13 (для выявления сероводорода
и определения ферментации лактозы, rлюкозы и
сахарозы; трехсахарный arap с солями железа)
Мясной экстракт
Дрожжевой экстракт
Пептон
Лактоза моноrидрат
Сахароза
rлюкоза моноrидрат
Железа (11) сульфат
Натрия хлорид
Натрия тиосульфат
Феноловый красный
Arap
Вода очищенная
3,0 r
3,0 r
20,0 r
10,0 r
10,0 r
1,0 r
0,2 r
5,С! r
0,3 r
0,024 r
15,0 r
1000 мл
Все компоненты растворяют в воде очи
щенной при наrревании, значение рН ДOBO
дят таким образом, чтобы после стерили
зации оно составляло (7,3:t0,2). Фильтруют
через ватно марлевый фильтр. Среду разли
вают в пробирки по 7 мл. Стерилизуют в паро
вом стерилизаторе при температуре 121 ос в
течение 15 мин. При охлаждении скашивают,
оставляя столбик высотой 2,5 3,0 см.
Среда Ng 14 (для определения утилизации
натрия цитрата, цитратный arap Симмонса)
Натрия хлорид 5,0 r
Маrния сульфат 0,2 r
Аммония диrидрофосфат 1,0 r
Дикалия rидрофосфат 1,0 r
Натрия цитрат 3,0 r
Бромтимоловый синий 0,08 r
Arap 20,0 r
Вода очищенная 1000 мл
Соли растворяют в воде очищенной,
вносят arap и наrревают до полноro ero pac
плавления, значение рН доводят таким обра
зам, чтобы после стерилизации оно составля
ло (7,2:tO, 1). Фильтруют через ватно марлевый
фильтр, прибавляют40 мл 0,2 % водноro paCT
вора бромтимоловоrо синеro. Среду разлива
IOТ в пробирку по 5 7 мл. Стерилизуют в па
ровом стерилизаторе при температуре 121 ос
. течение 15 мин. При охлаждении скашива
IOТ. оставляя столбик высотой 2,0 2.5 см.
Среда Ng 15 (для определения индола,
бульон Хоттинrера)
Натрия хлорид
Дикалия rидрофосфат
Раствор rидролизата мяса по
Хоттинrеру с содержанием
аминноro азота 1200 1400 мr/л
5,0 r
1,0 r
1000 мл
rидролизат Хоттинrера разводят водой до
содержания в среде 1200 1400 мr/л аминноro
азота, прибавляют соли, значение рН доводят
таким образом, чтобы после стерилизации оно
составляло (7,5:t0,1). Кипятят в течение 15 мин,
фильтруют через бумажный фильтр. Стерилизу
ют в паровом стерилизаторе при температуре
121 ос в течение 15 мин.
0,9 % раствор натрия хлорида
9,0 rнатрияхлорида Ррастворяютв 1 ООО,Омл
воды очищенной Р. Раствор фильтруют через
бумажный фильтр. Стерилизуют в паровом CTe
рилизаторе при температуре 121 ос в течение
15 мин.
Раствор Ng 1 (для промывания мембраны)
Пептон ферментативный 1,0 r
Вода очищенная 1000 мл
рН после стерилизации (7,1:1:0,2)
Раствор Ng 2 (для промывания мембраны)
Пептон ферментативный 5,0 r
Полисорбат 80 10,0 r
Вода очищенная 1000 мл
рН после стерилизации (7,1:1:0,2)
Растворы NQ 1 и NQ 2 стерилизуют в паровом
стерилизаторе при температуре 121 ос в тече
ние 15 мин.
1 % раствор феноловоrо KpacHoro
1,0 r фенолов020 краСН020 Р растирают в
ступке, прибавляя небольшими порциями 0,1 М
раствор натрия 2идроксида Р. Полученный
раствор переносят в мерную колбу вместимо
стью 100 мл И доводят объем раствора до метки
водой очищенной Р. Хранят во флаконе ней
тральноro стекла при температуре от 4 ос до
1 О ос в защищенном от света месте.
0,5 % раствор малахитовоro зеленоro
0,5 r малахитов020 зелеН020 Р растворяют
в roрячей стерильной воде очищенной Р и ДOBO
дят до объема 100,0 мл этим же раслюрителем.
Раствор выдерживают в термостате в течение
1 сут, периодически перемешивая.
Реактив rрисса
PacmeopNQ 1: 0,5 rкислотысульфаниловой Р
растворяют в 30 мл кислоты уксусной ледя
288
rосударственная фармакопея Республики Беларусь
ной Р, прибавляют 100 мл воды очищенной Р,
фильтруют.
Раствор NQ 2: 0,1 r нафтиламина Р paCTBO
ряют в 100 мл кипящей воды очищенной Р, ox
лаждают, прибавляют 30 мл кислоты уксусной
ледяной Р, фильтруют.
Перед употреблением смешивают равные
объемы растворов NQ 1 и NQ 2.
Реактив на цитохромоксидазу
Раствор NQ 1: 1 % раствор а нафтола Р в
96 % спирте Р.
Раствор NQ 2: 1 % раствор N,N диметил п
фенилендиамина диrидрохлорида.
Перед употреблением смешивают раствор
NQ 1 и NQ 2 в соотношении 2:3.
Реактив Ковача
5,0 r п диметиламинобензальде2ида Р
растворяют в 75,0 r пентанола Р или изоами
лов020 спирта Р при леrком наrревании до
50 55 ос (на водяной бане), охлаждают и Meд
лен но прибавляют 20,0 мл кислоты хлористо
водородной Р.
Раствор должен быть желтоro цвета.
Реактив Эрлиха
1 ,О r п иметиламинобензальде2ида Р
растворяют в 95 мл 96 % спирта Р и медленно
прибавляют 20 мл кислоты хлористоводород
ной Р.С
01/2013:20614
2.6.14. БАКТЕРИАЛЬНЫЕ
ЭНДОТОКСИНЫ
Испытание на бактериальные эндотокси
ны (ИБЭ) проводят для определения наличия
или количества эндотоксинов, источником KO
торых являются rрамотрицательные бакте
рии, с использованием лизата амебоцитов Me
чехвоста (Limu/us po/yphemus или Tachyp/eus
tridелtаtиs). Существует три способа проведе
ния данноrо испытания: способ rель тромба,
основанный на образовании rеля; турбидиме
трический способ, основанный на помутне
нии в результате расщепления эндоrенноro
субстрата; хромоrенный способ, основанный
на появлении окраски после расщепления
синтетическоrо пеПТИДНI) хромоrенноrо KOM
плекса.
В настоящем разделе описаны следующие
шесть методов.
Метод А. rель тромб метод: предельное ис
пытание.
Метод В. rель тромб метод: количественное
испытание.
Метод С. Турбидиметрический кинетиче
ский метод.
Метод D. Хромоrенный кинетический
метод.
Метод Е. Хромоrенный метод конечной
точки.
Метод F. Турбидиметрический метод KO
нечной точки.
Испытание выполняют любым из этих
шести методов. В сомнительных и спорных
случаях окончательное решение принимают,
основываясь на методе А, если иное не пред
писано в частной статье.
Испытание выполняют в условиях, не дo
пускающих заrрязнения посторонними эндо
токсинами.
1. ОБОРУДОВАНИЕ
ВСЮ стеклянную посуду и друrую TepMO
устойчивую аппаратуру депироrенизируют в
сухожаровом шкафу с использованием про
цесса с подтвержденной эффективностью.
Общеприняты минимальные значения BpeMe
ни и температуры обработки, составляющие
30 мин и 250 ос соответственно. При исполь
зовании пластиковой аппаратуры, например,
микротитрационных планшетов и наконеч
ников для автоматических пипеток, следует
продемонстрировать отсутствие на ней под
дающихся определению эндотоксинов и Me
шающих факторов.
Примечание: в данном разделе термин
«пробирка» включает в себя все типы сосудов,
например, микротитрационные планшеты.
2. РЕАКТИВЫ, ИСПЫТУЕМЫЕ
РАСТВОРЫ
(1) Лизат амебоцитов
Лизат амебоцитов это лиофилизиро
ванный продукт, полученный из лизата aMe
боцитов мечехвоста (Limu/us po/yphemus или
Tachyp/eus trideпtatus). Данный реактив произ
водят только в соответствии с инструкциями
компетентноrо opraHa.
Примечание: лизат амебоцитов pea2и
рует, кроме эндотоксинов, с некоторыми
{3 2люканами. Доступны также препараты
лизата амебоцитов, которые не реа2ируют
с 2люканами; их 20товят, удаляя из лизата
амебоцитов фактор G, реа2ирующий с 2ЛЮ
канами, или иН2ибируя реа2ирующий KOM
плекс фактора G лиза та амебоцитов. Эти
препараты М02ут быть использованы в при
сутствии 2люканов.
(2) Раствор лизата
Растворяют лизат амебоцитов в воде для
ИБЭ или в буфере, в зависимости от peKOMeH
даций производителя лизата, при аккуратном
перемешивании. Восстановленный раствор
хранят, в зависимости от рекомендаций произ
водителя, охлажденным или замороженным.
2.6.14. Бактериальные эндотоксuны
289
(3) Вода для ИБЭ (вода для испытания на
бактериальные эндотоксины)
Вода для инъекций Р или вода, полученная
иным способом и не реаrирующая с лизатом,
использованным на пределе обнаружения pe
areHTa.
З.пРиrОТОВЛЕНИЕисходноrо
СТАНДАРтноrо РАСТВОРА
ЭНДОТОКСИНА
Исходный стандартный раствор эндо
токсина rотовят из стандартноro образца эн
дотоксина, калиброванноro по отношению к
Международному стандартному образцу, Ha
пример, БСП стандартН020 образца эндо
токсина.
Количественное содержание эндотоксина BЫ
ражают в Международных Единицах (МЕ). Эквива
лентность Международноro стандартноro образ
ца в Международных Единицах устанавливается
Всемирной орrанизацией здравоохранения.
Примечание: одна Международная Единица
(МЕ) эндотоксина эквивалентна одной EдиHи
це Эндотоксина (ЕЭ).
Исходный стандартный раствор эндотокси
на roтовят и хранят, следуя условиям, приведен
ным на листке вкладыше и этикетке.
4. приrОТОВЛЕНИЕ СТАНДАРТНЫХ
РАСТВОРОВ ЭНДОТОКСИНА
Необходимые последовательные разведе
ния предварительно интенсивно перемешан
ноro исходноrо стандартноro раствора эндоток
сина roтовят с использованием воды для ИБЭ.
Приroтовленные растворы используют по
возможности быстро, чтобы избежать потери aK
тивности вследствие адсорбции.
5. приrОТОВЛЕНИЕ ИСПЫТУЕМЫХ
РАСТВОРОВ
Испытуемые растворы roтовят путем
растворения или разбавления фармацевти
ческих субстанций или медицинской ПрОДу'к
ции с использованием воды для ИБЭ. ДЛЯ He
которых субстанций и лекарственных средств
более подходящим является растворение или
разбавление с использованием друrих водных
растворов. При необходимости доводят рН
испытуемоro раствора (или ero разведения)
так, чтобы рН ero смеси с лизатом находи
лось в интервале, предписанном производи
телем лизата, обычно от 6,0 до 8,0. Значение
рН может быть доведено с использованием
кислоты, основания или подходящеrо буфер
HOro раствора в соответствии с рекомендаци
ями производителя лизата. Кислоты и OCHOBa
ния MorYT быть приroтовлены из концентратов
или порошков с использованием воды для
ИБЭ в контейнерах, не содержащих опреде
ляемых количеств эндотоксина. Должно быть
подтверждено отсутствие эндотоксинов и Me
шающих факторов в буферных растворах.
37 3., 1712
6. ОПРЕДЕЛЕНИЕ МАКСИМАЛЬНО
допУстимоrо РАЗВЕДЕНИЯ
Максимальнодопустимоеразведение(МДР)
это максимальное разведение образца, при KO
тором может быть определено предельное co
держание эндотоксина. МДР рассчитывают по
формуле:
пред ельное содержанuе зндотоксинов . концентрация испытуемоео рас твора
мдp= - A '
Предельное содержание эндотоксинов: для
фармацевтических субстанций, предназначен
ных для парентеральноro введения, равно:
к
,
м
rде:
К допустимая пироrенная доза эндоток
сина, на килоrрамм массы тела;
М максимальная рекомендованная доза
продукта, на килоrрамм массы тела.
Если продукт вводится через малые проме
жутки времени или если используется в течение
продолжительноro периода времени, в качестве
М используют максимальную общую дозу, BBO
димую В течение одноro часа.
Предельное содержание эндотоксинов в
фармацевтических субстанциях, предназна
ченных для парентеральноro введения, указы
вается в частных статьях и выражается в таких
единицах, как МЕ/мл, МЕ/м!; МЕ/Единица биоло
rической активности и т. д.
Концентрация испытуеМО20 раствора:
в мr/мл, если предельное содержание эн
дотоксинов выражено в массовых единицах
(ME/Mr);
в Ед/мл, если предельное содержание эн
дотоксинов выражено в пересчете на единицу
биолоrической активности (МЕ/Ед);
в мл/мл, если предельное содержание
эндотоксинов выражено в объемных единицах
(МЕ/мл).
Л чувствительность лизата (МЕ/мл) в
rель тромб методе, указанная на этикетке, или
низшая концентрация на стандартной кривой в
турбидиметрических или xpoMoreHHbIx методах.
7. СПОСОБ rЕЛЬ ТРОМБА (МЕТОДЫ А И В)
Способ rель тромба позволяет определять
наличие и количество эндотоксинов и OCHOBЫBa
ется на свертывании лизата в присутствии эндо
токсинов. Минимальная концентрация эндоток
синов, требующаяся для свертывания лизата в
стандартных условиях, представляет собой YKa
занную на этикетке чувствительность лизата.
Для обеспечения точности и достоверности ис
пытания указанную чувствительность лизата
следует подтвердить, а также провести испыта
ние на мешающие факторы, как описано в под
разделе 1. Предварительные испытания.
290
rосударственная фармакопея Республики Беларусь
1. ПРЕДВАРИТЕЛЬНЫЕ ИСПЫТАНИЯ
(1) Подтверждение заявленной чувстви-
тельности лизата
Перед использованием лизата в испытаниях
указанную на этикетке чувствительность Л, BЫ
раженную в МElмл, следует подтвердить в че
тырех повторностях. Подтверждение чувстви
тельности лизата выполняют при использовании
новой партии лизата или при любом изменении
экспериментальных условий, способном повли
ять на проведение испытания.
rотовят стандартные растворы не менее
чем четырех концентраций, эквивалентных 21\, Л,
0,5Л и 0,25Л, путем разбавления исходноro CTaH
дартноro раствора эндотоксина водой дЛЯ ИБЭ.
В каждой из пробирок смешивают раствор
лизата с равным объемом одноro из стандарт
ных растворов (например, по 0,1 мл каждоrо).
Если используются одноразовые флаконы или
ампулы с лиофилизированным лизатом, pac
творы добавляют непосредственно в ампулу
или флакон. Реакционную смесь инкубиру
ют в течение определенноro периода в COOT
ветствии с рекомендациями производителя
лизата (обычно при температуре (З7:t1) ос в
течение (60:t2) мин), избеrая вибрации. Ис
следуют целостность rеля: при использовании
пробирок каждую из них по очереди извле
кают из инкубатора и переворачивают одним
плавным движением приблизительно на 1800.
Если образуется прочный rель, остающийся
на своем месте после переворачивания, pe
зультат записывают как положительный. Pe
зультат отрицательный, если неповрежденно
ro rеля не образуется.
Результаты испытания считают ДOCTOBepHЫ
ми, если низшая концентрация стандартных pac
творов во всех повторностях дает отрицатель
ный результат.
За конечную точку принимают минималь
ную концентрацию в нисходящем ряду KOHцeH
траций эндотоксина, приводящую к CBepTЫBa
нию лизата. Находят среднее rеометрическое
значение концентрации в конечной точке, pac
считывая значение лоrарифмов концентраций в
конечных точках 4 серий разведений; берут aH
тилоrарифм этоro значения по следующей фор
муле:
Среднее аеометрuческое концентрации е конечной точке == antilog L е ,
f
rде:
Le сумма лоrарифмов концентраций в KO
нечных точках в используемом ряду разведений;
f число повторностей.
Среднее rеометрическое концентрации в
конечной точке представляет собой измерен
ную чувствительность раствора лизата (МЕ/мл).
Если это значение составляет не менее 0,5Л и
не более 2Л, чувствительность, указанную на
этикетке, считают подтвержденной и исполь-
зуют в испытаниях, выполняемых с этим ли
затом.
(11) Определение мешающих факторов
rотовят растворы А, В, С и О в соответствии
с таблицей 2.6.14. 1 и используют не содержа
щие определяемых эндотоксинов испытуемые
растворы в разведении, меньшем МДР в COOT
ветствии с указаниями подраздела 1. Предвари
тельные испытания, (1) Подтверждение заявлен
ной чувствительности лизата.
Средние rеометрические концентрации в KO
нечной точке для растворов В и С рассчитывают
по формуле, приведенной в подразделе 1. Преk
варительные испытания, (1) Подтверждение за
явленной чувствительности лизата.
Испытание на мешающие факторы повторя
ют в случае любых изменений эксперименталь
ных условий, которые MOryт повлиять на резуль
тат.
Результаты испытания считают ДOCTO
верными, если растворы А и О во всех no
вторностях дают отрицательный результат, а
результат, полученный для раствора С, под
тверждает чувствительность лизата, указан
ную на этикетке.
Если чувствительность лизата, определен
ная для раствора В, не менее 0,5Л и не более
2Л, считают, что испытуемый раствор в условиях
испытания не содержит мешающих факторов. В
иных случаях раствор оказывает влияние на pe
зультат испытания.
Если испытуемый образец оказывает вли
яние на результат испытания в разведен иях
менее МДР, испытание на мешающие факторы
повторяют с использованием больших разведе
ний, но не превышающих МДР. Использование
более чувствительноro лизата позволяет приме
нять большие разведения испытуемоro образца,
и это может содействовать исключению мешаю-
щих факторов.
Мешающие факторы MorYT быть удале
ны соответствующей обработкой, например,
путем фильтрации, нейтрализации, диализа
или тепловоro воздействия. Для подтвержде-
ния, что выбранный метод эффективно исклю-
чает мешающие факторы, не удаляя эндоток
сины, испытания повторяют с использованием
испытуемоro образца. к которому был добав-
лен стандартный образец эндотоксина и про-
ведена выбранная процедура удаления меша-
ющих факторов.
2. ПРЕДЕЛЬНОЕ ИСПЫТАНИЕ (МЕТОД А)
(1) Методика
rотовят растворы А, В, С и О в соответствии
с таблицей 2.6.14. 2 и выполняют испытание
для этих растворов в соответствии с указания.-
ми подраздела 1. Предварительные испытания,
(1) Подтверждение заявленной чувствитеЛЬН<r
сти лизата.
2.6.14. Бактериальные эндотоксины
291
Таблица 2.6.14. 1
Концентрация эндоток
Раствор си нов/Раствор, к KOTO Разбавитель Фактор Концентрация Количество
рому прибавляют эндо разведения эндотоксинов повторностей
токсины
А О/Испытуемый раствор 4
1 2Л 4
В 2I\/Испытуемый раствор Испытуемый 2 Л 4
раствор 4 0,5Л 4
8 0,25Л 4
1 2Л 2
С 2Л1Вода дЛЯ ИБЭ Вода дЛЯ ИБЭ 2 Л 2
4 0,5Л 2
8 0,25Л 2
D О/Вода дЛЯ ИБЭ 2
Раствор А раствор испытуемоro образца. не содержащий определяемых эндотоксинов.
Раствор В тест на мешающие вещества.
Раствор С контроль чувствительности, указанной на этикетке.
Раствор О отрицательный контроль (вода для ИБЭ).
Таблица 2.6.14. 2
Концентрация
эндотоксинов/ Количество
Раствор Раствор, к KOTO повторностей
рому прибавля
ют эндотоксины
А О/Испытуемый 2
раствор
В 2Л1Испытуемый 2
раствор
С 2Л1Вода дЛЯ ИБЭ 2
D О/Вода дЛЯ ИБЭ 2
Растворы А и В (положительный контроль с
образцом) roтовят в разведениях не более МДР
и обрабатывают в соответствии с указаниями
подраздела 1. Предварительные испытания, (11)
Определение мешающих факторов. Растворы В
и С (положительные контроли) содержат CTaH
дартный образец эндотоксина в концентрации,
соответствующей двукратной заявленной ч}!в
ствительности. Раствор D (отрицательный KOH
троль) представляет собой воду для ИБЭ.
(11) Интерпретация
Результаты испытания считают ДOCTOBepHЫ
ми, если растворы В и С в обеих повторностях
дают положительный результат, а раствор D
отрицательный результат.
Испытуемый образец выдерживает испыта
ние, если для раствора А в обеих повторностях
получен отрицательный результат.
Испытуемый образец не выдерживает испы
тание, если для раствора А в обеих повторно
стях получен положительный результат.
В случае, если для одной из повторностей
раствора А получен положительный результат,
а для друroй отрицательный, испытание по
вторя ют. Испытуемый образец выдерживает ис
пытание, если в повторном эксперименте для
раствора А в обеих повторностях получен отри
цательный результат. Испытуемый образец не
выдерживает испытание, если для раствора А в
одной или обеих повторностях получен положи
тельный результат.
Однако, если испытуемый образец не BЫ
держивает испытание при разведении, MeHb
шем, чем МДР, то испытание может быть
повторено при большем разведении, но не пре
вышающем МДР.
3. КОЛИЧЕСТВЕННОЕ ИСПЫТАНИЕ
(МЕТОД В)
(1) Методика
С помощью данноro испытания определя
ют количество бактериальных эндотоксинов
путем титрования до конечной точки. rOTO
вят растворы А, В, С и D в соответствии с Ta
блицей 2.6.14. 3 и выполняют испытание для
этих растворов в соответствии с указаниями
подраздела 1. Предварительные испытания,
(1) Подтверждение заявленной чувствительно
сти лизата.
(11) Вычисления и интерпретация
Результаты испытания считают ДOCТOBepHЫ
ми при выполнении следующих трех условий:
(а) раствор D (отрицательный контроль) в
обеих повторностях дает отрицательный резуль
тат;
(Ь) раствор В (положительный контроль с
образцом) в обеих повторностях дает положи
тельный результат;
(с) среднее rеШ. етрическое концентрации в
конечной точке для раствора С находится в пре
делах от О,5Л до 21\.
Для определения концентрации эндотокси
нов в растворе А находят концентрацию в KO
нечной точке для каждой из повторностей путем
умножения каждоro фактора разведения в KO
нечной точке на Л.
292
rосударственная фармакопея Республики Беларусь
Таблица 2.6.14. 3
Концентрация эндоток
Раствор си нов/Раствор, к KOTO Разбавитель Фактор Концентрация Количество
рому прибавляют эндо разведения эндотоксинов повторностей
токсины
1 2
А О/Испытуемый раствор Вода для ИБЭ 2 2
4 2
8 2
В 2Л1Испытуемый раствор 1 2Л 2
1 2Л 2
С 2NВода для ИБЭ Вода для ИБЭ 2 11 2
4 0,511 2
8 0,2511 2
D О/Вода для ИБЭ 2
Раствор А испытуемый раствор в разведении, не превышающем МДР, с которым проводилось определение мешающих фак
торов. Последующие разведения испытуемоrо раствора не должны превышать МДР. Используют ВОДУ для ИБЭ для получения
двух рядов разведений в отношениях 1. 1/2, 1/4, и 1/8 к разведению, с которым проводилось определение мешающих факторов.
При необходимости MorYT быть выполнены и друrие разведения.
Раствор В раствор А. содержащий стандартный образец эндотоксина в концентрации 2Л (положительный контроль с образ
цом).
Раствор С серия разведений из 4 пробирок воды для ИБЭ, содержащей стандартный образец эндотоксина в концентрациях
2Л, Л, О,5Л, О,25Л.
Раствор О вода дЛЯ ИБЭ (отрицательный контроль).
За концентрацию эндотоксина в испытуе
мом растворе принимают концентрации в KO
нечных точках в повторностях. Если испыта
ние проводят с разбавленным испытуемым
раствором, концентрацию эндотоксина в ис
ходном растворе получают путем умножения
на фактор разбавления.
Если ни одно из разведений испытуемо
ro раствора в ходе испытания с подтвержден
ной достоверностью не дает положительноrо
результата. концентрацию эндотоксинов реrи
стрируют как «менее Л» (или, В случае исполь
зования разбавленноro образца, как «менее
Л х наименьший фактор разведения образ
ца»). Если для всех разведений получен по
ложительный результат, то концентрация эн
дотоксинов реrистрируется как «равная или
превышающая максимальный фактор раз
ведения, умноженный на 11» (например, в Ta
блице 2.6.14. 3 это значение равно исходному
фактору разведения х 8 х Л).
Продукт соответствует требованиям ис
пытания, если концентрация эндотоксина в
обеих повторностях менее указанной в част
ной статье.
8. ФОТОМЕТРИЧЕСКИЕ
КОЛИЧЕСТВЕННЫЕ СПОСОБЫ (МЕТОДЫ
С, О, Е И F)
1. ТУРБИДИМЕТРИЧЕСКИЙ СПОСОБ
(МЕТОДЬ! С И F)
Данный способ представляет собой фото
метрическое испытание, основанное на измере
нии увеличения степени мутности. В зависимо
сти от используемоro подхода с использованием
данноro метода MorYT быть проведены турби
диметрическое испытание методом конечной
точки и кинетическое турбидиметрическое ис
пытание.
Турбидиметрический метод конечной точки
(метод F) основан на количественном COOTHO
шении между концентрацией эндотоксина и CTe
пенью мутности (выражающейся в пропускании
или поrлощении света) реакционной смеси в
конце инкубационноro периода.
Кинетическое турбидиметрическое испыта
ние (метод С) заключается либо в измерении
времени, требующеroся для достижения задан
ноro значения поrлощения в реакционной смеси,
либо в определении скорости увеличения степе
ни мутности.
Испытание проводят при температуре ин
кубации, рекомендованной производителем
лизата (обычно (37:1:1) ОС).
2. хРомоrЕННblЙ СПОСОБ
(МЕТОДЬ! D И Е)
Данный способ основан на измерении KO
личества хромофора, высвобождаемоro из co
ответствующеro xpoMoreHHoro пептида в pe
зультате реакции эндотоксинов с лизатом. В
зависимости от используемоro подхода с исполь
зованием данноrо способа MOryT быть проведе
ны xpoMoreHHoe испытание методом конечной
точки и кинетическое xpoMoreHHoe испытание.
Хромоrенный метод конечной точки (метод
Е) основан на количественном соотношении
между концентрацией эндотоксина и количе
ством хромофора, высвобождаемоro к концу ин
кубационноro периода.
Кинетическое xpoMoreHHoe испытание (метод
О) заключается либо в измерении времени, требу
ющеroся для достижения заданноro значения п
rлощения в реакционной смеси, либо в определ
нии скорости появления окраски.
2.6.14. БактериаЛЬНblе эндотОКСUНbI
293
Испытание проводят при
кубации, рекомендованной
лизата (обычно (37:t1) ОС).
3. ПРЕДВАРИТЕЛЬНЫЕ ИСПЫТАНИЯ
Предварительные испытания Пр080ДЯТ
для обеспечения точности и достоверности
результатов турбидиметрических и XpOMO
reHHbIx испытаний. В ходе предварительных
испытаний подтверждают выполнение кри
териев стандартной кривой и отсутствие Me
шающеrо влияния испытуемоro раствора на
результаты испытания.
При внесении любых изменений в экспе
риментальные условия, которые MorYT повли
ять на результаты, требуется подтверждение
приrодности метода испытания.
температуре ин
производителем
(1) Подтверждение выполнения критери
ев стандартной кривой
Данное испытание должно быть проведе
но для каждой серии peareHTa лизата.
Для получения стандартной кривой roто
вят не менее трех концентраций эндотоксина
в диапазоне, рекомендованном производите
лем лизата, с использованием стандартноro
раствора эндотоксина. Проводят испытание
с каждым стандартным раствором эндотокси
на не менее чем 8 трех повторностях в co
ответствии с рекомендациями производителя
лизата (объемные отношения, время инкуба
ции, температура, рН и т. д.).
Если при использовании кинетических
методов желаемый интервал 8 лоrарифми
ческой шкале превышает 2, в испытание
должны быть включены дополнительные
стандарты для включения каждоro увеличен
ноro лоrарифмическоro значения в диапазон
стандартной кривой.
В диапазоне концентраций эндотокси
на, указанном ПРОИЗ80дителем лизата, аб
солютное значение коэффициента корреля
ции I r I должно превышать или быть равным
0,980.
(11) Определение мешающих факторов
Выбирают концентрацию эндотоксина в центре
или недалеко от центра стандартной кривой.
rотовят растворы А, В, С и D в COOTBeT
ствии с таблицей 2.6.14.4. Проводят испытание
с каждым из этих растворов не менее чем в двух
повторностях в соответствии с рекомендациями
производителя лизата (объем испытуемоro paCT
вора и раствора лизата, объемное соотношение
испытуемоro раствора и раствора лизата, время
инкубации и т. д.).
Результаты испытания считают ДOCTOBepHЫ
ми при выполнении следующих трех условий:
абсолютное значение коэффициента KOp
реляции стандартной кривой, полученной при
помощи раствора С, более или равно 0,980;
результат, полученный с раствором О, не
превышает предельноro значения для контроля,
указанноro в описании используемоro реактива
лизата, или он меньше, чем предел обнаруже
ния используемоro реактива лизата.
Рассчитывают среднее значение OTKpЫBaeMO
сти прибавленноro эндотоксина вычитанием cpeд
ней концентрации эндотоксина в растворе (при ее
наличии) (раствор А, таблица 2.6.14.4) от KOHцeH
трации в растворе, содержащем прибавленный
эндотоксин (раствор В, таблица 2.6.14.--4).
Считают, что испытуемый раствор не coдep
жит мешающих факторов, если в условиях ис
пытания рассчитанная концентрация эндоток
сина, прибавленноro к испытуемому раствору,
находится в пределах 50 200 % от известной
концентрации прибавленноro эндотоксина после
вычитания концентрации эндотоксина, обна
ружен ной в растворе, не содержащем добавок
стандарта.
Если открываемость эндотоксина выходит за
специфицированные рамки, считают, что испыту
емый раствор обладает мешающими факторами.
Повторяют испытание, используя большее разве
дение, не превышающее МДР. Кроме тоro, меша
ющее влияние испытуемоro раствора или испыту
емоro раствора в разведении, не превышающем
Таблица 2.6.14.--4
! Концентрация Раствор, к КОТОРОМУ Количество
! Раствор эндотоксинов прибавляют эндотоксины повторностей
IA О Испытуемый раствор Не менее 2
В Средняя концентрация Испытуемый раствор Не менее 2
стандартной кривой
Не менее трех KOHцeH Каждая концентрация
С траций (низшая опре Вода для ИБЭ не менее 2
деляется Л)
D О Вода для ИБЭ Не менее 2
Раствор А испытуемый раствор, который может быть разведен, не превышая МДР.
Раствор В испытуемый образец в таком же разведении, что и раствор А, содержащий прибавленный эндотоксин в KOHцeHTpa
uии в центре или недалеко от центра стандартной кривой.
Раствор С стандартный раствор эндотоксина в концентрациях, использовавшихся при валидации метода в соответствии с YKa
занием подраздела З. Предварительные испытания, (1) Подтверждение выполнения критериев стандартной кривой (положитель
ные контроли)
Раствор О вода для ИБЭ (отрицательный контроль).
.>.... 3.а" 1 ,12
294
rосударственная фармакопея Республики Беларусь
МДР, может быть исключено подходящим под
твержденным способом, например, фильтраци
ей, нейтрализацией, диализом или термической
обработкой. Для подтверждения, что выбранный
способ эффективно удаляет мешающее влия
ние без потери эндотоксинов, повторяют испыта
ние на мешающие факторы с использованием ис
пытуемоro препарата, к которому был прибавлен
стандартный раствор эндотоксинов, и который
затем обработали выбранным способом.
4. ИСПЫТАНИЕ
(1) Метод
Следуют указаниям, приведенным в подраз
деле 3. Предварительные испытания, (11) Опре
деление мешающих факторов.
(11) Вычисления
Для раствора А в каждой повторности опре
деляют концентрацию эндотоксина с использо
ванием стандартной кривой, полученной с помо
щью раствора положительноro контроля С.
Результаты испытания считают ДOCTOBepHЫ
ми при выполнении следующих трех условий:
(1) результаты, полученные для раствора С,
соответствуют требованиям валидации, приве
денным в подразделе 1. Предварительные ис
пытания. (1) Подтверждение выполнения крите
риев стандартной кривой;
(2) открываемость эндотоксина, рассчитан
ная из концентрации эндотоксина, определен
ной в растворе В. после вычитания KOHцeHTpa
ции эндотоксина, определенной в растворе А,
находится в пределах 50 200 %.
(3) резулыат, полученный с раствором D (отри
цательный контроль), не превышает предельною
значения для контроля, указанною в описании ис
пользуемою реаreнта лизата, или он меньше, чем
предел обнаружения ИСПОЛЬЗуемою peareHTa лизата.
(111) Интерпретация
Испытуемый образец соответствует требова
ниям испытания, если средняя концентрация эн
дотоксинов в повторностях раствора А, с учетом
разведений и концентрации, меньше предельною
значения эндотоксина для испытуемоro образца.
Руководства по применению испытания
на бактериальные эндотоксины приведены в
2лаве 5.1.10.
01/2013:20615
2.6.15. АКТИВАТОР
ПРЕКАЛЛИКРЕИНА
Активатор прекалликреина (АПК) активиру
ет прекалликреин, переводя еro в калликреин, и
может быть определен по еro способности к от
щеплению хромофора от синтетическоro пептид
ноro субстрата в условиях, позволяющих опред
лять скорость распада спектрофотометрическим
методом; концентрация АПК вычисляется путем
сравнения со стандартным образцом, калибро
ванным в Международных Единицах.
За Международную Единицу принята актив
ность установленноro количества Международ
Horo стандартноro образца, представляющеro
собой лиофилизированный активатор прекалли
креина. Эквивалентность Международноro CTaH
дартноro образца в Международных Единицах
устанавливается Всемирной орrанизацией здра
воохранения.
РЕАКТИВЫ
БСП активатора прекалликреина в альбу
мине калибруется в Международных Единицах в
сравнении с Международным стандартным об
разцом.
Буфер А. 6,055 r триС(2идроксиметил)ами
нометана Р, 1,17 r натрия хлорида Р, 50 Mr
2ексадиметрина бромида Р и 100 Mr натрия
азида Р растворяют в воде Р. Доводят 2 М pac
твором кислоты хлористоводородной до
рН 8,0 и разводят водой Р до объема 1000 мл.
Буфер В. 6,055 r триС(2идроксиметил)ами
нометана Р, и 8,17 r натрия хлорида Р paCTBO
ряют в воде Р. Доводят 2 М раствором кисло
ты хлористоводородной до рН 8,0 и разводят
водой Р до объема 1000 мл.
приrОТОВЛЕНИЕ СУБСТРАТА
ПРЕКАЛЛИКРЕИНА
Для предотвращения активации коа2УЛЯ
ции кровь или плазма, используемая для при20
товленuя прекалликреина, должна KOHтaKти
ровать только с пластиком или стеклянными
поверхностями, обработанными силиконом.
9 объемов человеческой крови приливают к
1 объему антикоаryлянтноro раствора (АСО, СРD
или раствора 38 r/л цитрата натрия Р), к KOТOpO
му прибавлен 2ексадиметрина бромид Р в коли
честве 1 мr/мл. Смесь центрифуrируют при 3600 9
в течение 5 мин. Отделяют плазму и снова центри
фуrируют при 6000 9 в течение 20 мин для осаж
дения тромбоцитов. Плазму с уменьшенным CO
держанием тромбоцитов отделяют и подверrают
диализу против 1 О объемов буфера А в течение
20 ч. Диализированную плазму наносят на xpOMa
тоrрафическую колонку, заполненную ааарозой
ДЭАЭ для ионообменной хроматоzрафии Р, KOT
рая взята в количестве, равном двойному объему
плазмы, и которая уравновешена с помощью
буфера А. Колонку элюируют буфером А со CKOP<r
стью 20 мл/см 2 /ч. Собирают фракции элюата, pe
стрируя оптическую плотность при 280 нм (2.2.25).
Фракции, содержащие первый пик белка. объеди--
няют таким образом, чтобы объем объединенных
фракций составлял 120 % объема плазмы с пони--
женным содержанием тромбоцитов.
2.6.16. Испытания на посторонние azeHmbl в вирусных вакцинах
295
Проверяют отсутствие калликреиновой aK
тивности в объединенном растворе субстрата,
смешивая еro в соотношении 1 :20 с предвари
тельно подоrретым раствором xpoMoreHHoro суб
страта, который будет использоваться в опреде
лении, и инкубируют при 37 ос в течение 2 мин.
Субстрат может быть использован в испытании,
если увеличение оптической плотности COCTaB
ляет менее 0,001 единицы в минуту. К собранно
МУ раствору прибавляют натрия хлорид Р в KO
личестве 7 r/л и фильтруют через мембранный
фильтр (пористость 0,45 мкм). Порции филь
трата замораживают и хранят при температуре
25 ОС; субстрат перед хранением может быть
подверrнут лиофилизации.
Все процедуры от начала хроматоrрафии до
заморозки порций выполняют в течение одноro
рабочеro дня.
МЕТОД
Определение может быть выполнено с ис
пользованием автоматическоro анализатора
ферментов или подходящей системы с титра
ционным микропланшетом, позволяющей про
водить кинетические измерения, с подходящим
проrраммным обеспечением для обсчета pe
зультатов. Разведения стандартов, образцов и
субстрата прекалликреина MOryT быть приroтов
лены с использованием буфера В.
Разбавленные стандартные или испыту
емые образцы инкубируют с субстратом пре
калликреина в течение 1 О мин так, чтобы
объем неразбавленноro образца не превы
шал 1/10 общеro объема инкубируемой смеси,
во избежание ошибок, связанных с изменени
ем ионной силы и показателя рН инкубируе
мой смеси. Смесь или ее часть инкубируют с
не менее чем равным объемом раствора под
ходящеro синтетическоro xpoMoreHHoro суб
страта с установленной специфичностью к
калликреину (например, N бензоил L пролил
L фенилаланил L ар2инина 4 нитроанилида
ацетат Р или О пролил L фенилаланил
L ар2инина 4 нитроанилида ди2идрохлорид Р),
растворенноro в буфере В. Реrистрируют CKO
рость изменения оптической плотности в минуту
в течение промежутка времени от 2 до 1 О минут
при длине волны, специфичной для использу
eMoro субстрата. Для каждой смеси образца или
стандарта roтовят контроль, в котором вместо
субстрата прекалликреина содержится буфер В.
В зависимости от используемоro метода
вносят поправки в значение М/мин, вычитая pe
зультат, полученный для соответствующеro KOH
троля. Результат может быть рассчитан по rpa
дуиров()чной rрафику или с использованием
иноro метода статистической обработки. Строят
rрадуировочный rрафик, используя значения,
полученные таким образом для стандартноro
образца в соответствующих концентрациях; ис
пользуют полученный rрафик для определения
активности АПК в испытуемом образце.
01/2013:20616
2.6.16. ИСПЫТАНИЯ
НА ПОСТОРОННИЕ ArEHTbI
В ВИРУСНЫХ ВАКЦИНАХ
для МЕдицинскоrо
ПРИМЕНЕНИЯ
Если для проведения испытания требуется
предварительная нейтрализация вируса, исполь
зуют специфические антитела, не происходящие
от человека или обезьян; если выращивание
вируса производилось на тканях птичьеro проис
хождения, то антитела не должны, кроме тоro,
происходить от птиц. Для приroтовления антисы
воротки используют иммунизирующий антиrен,
произведенный клеточной культурой, имеющей
происхождение, отличное от происхождения
вакцины, и не содержащей посторонних areHTOB.
Если предписано использование яиц SPF (speci
fied pathogeпs (,ее, не зараженные конкретными
патоrенами (свободные от специфических пато
reHoB», яйца отбирают из rрупп, не содержащих
указанные патоrены (5.2.2).
ВИРУСНЫЙ ПОСЕВНОЙ МАТЕРИАЛ
Образцы вирусноro посевноro матери
ала отбирают во время еro сбора и, если их не
подверrают испытанию немедленно, хранят при
температуре ниже --40 ОС.
Половозрелые мыши. Каждой из не
менее 1 О половозрелых мышей массой от 15 r
до 20 r вводят интрацеребрально О,ОЗ мл и
внутрибрюшинно 0,5 мл вирусноro посевно
ro материала. Мышей наблюдают в течение
не менее 21 дня. Производят вскрытие всех
мышей, поrибших или имеющих симптомы за
болевания по окончании первых 24 ч испы
тания, и исследуют их на наличие признаков
вирусной инфекции как путем прямоrо Ma
кроскопическоro осмотра, так и перепривив
кой подходящих суспензий тканей интраце
ребральным и внутрибрюшинным методами
дополнительно не менее чем пяти мышам, KO
торых наблюдают в течение 21 дня. Вирусный
посевной материал выдерживает .испытание,
если ни одна мышь не обнаруживает симпто
мов инфекции, которая может быть вызвана
посевным материалом. Результаты испытания
считают достоверными, если не менее 80 %
привитых мышей выживают в течение пери
ода наблюдения.
Новорожденные мыши. Каждой из
не менее 20 мышей возрастом менее 24 ч
вводят интрацеребрально 0,01 мл и внутри
брюшин но не менее 0,1 мл вирусною посев
ною материала. Мышей наблюдают ежеднев
но в течение не менее 14 дней. Производят
вскрытие всех мышей, поrибших или име
ющих симптомы заболевания по окончании
296
rосударственная фармакопея Республики Беларусь
первых 24 ч испытания, и исследуют их на
наличие признаков вирусной инфекции как
путем прямоrо макроскопическоro осмотра,
так и перепрививкой подходящих суспензий
тканей интрацеребральным и внутрибрюшин
ным методами дополнительно не менее чем
пяти новорожденным мышам, которых наблю
дают ежедневно в течение 14 дней. Вирусный
посевной материал выдерживает испытание,
если ни одна мышь не обнаруживает симпто
мов инфекции, которая может быть вызва
на посевным материалом. Результаты испы
тания считают достоверными, если не менее
80 % при витых мышей выживают в течение
периода наблюдения.
Морские свинки. Каждой из не менее 5
морских свинок массой от 350 r до 450 r вводят
внутрибрюшинно 5,0 мл вирусноro посевноro
материала. Животных наблюдают в течение не
менее 42 дней, отмечая наличие симптомов за
болевания. Производят вскрытие всех морских
свинок, поrибших или имеющих симптомы забо
левания по окончании первых 24 ч испытания,
и исследуют их макроскопически; ткани иссле
дуют как микроскопически, так и культурально,
отмечая наличие признаков инфекции. Живот
ных, выживших В течение периода наблюдения,
умерщвляют и исследуют аналоrичным обра
зом. Вирусный посевной материал выдержи
вает испытание. если ни одна морская свинка
не обнаруживает признаков инфекции, которая
может быть вызвана посевным материалом.
Результаты испытания считают достоверными,
если не менее 80 % морских свинок выживают
в течение периода наблюдения.
Спироплазмы. Вирусный посевной Ma
териал, получаемый из клеток насекомых, не
должен содержать спироплазм, что подтверж
дается валидированным методом, одобрен
ным компетентным opraHoM.
ВИРУСНЫЙ ПОСЕВНОЙ МАТЕРИАЛ
И ВИРУСНЫЕ СБОРЫ
Образцы отбирают во время сбора и, если
их не подверrают испытанию немедленно,
хранят при температуре ниже АО ОС.
Бактериальная и rрибковая стериль-
ность. 1 О мл испытуемоro образца должны BЫ
держивать испытание на стерильность (2.6.1).
Микоплазмы. 1 О мл испытуемоrо образца
должны выдерживать испытание на микоплаз
мы (2.6.7).
Микобактерии (2.6.2). 5 мл испытуемо
ro образца подверrают испытанию на наличие
Mycobacterium spp. методами культивирования,
обладающими известной чувствительностью в
отношении определения этих орrанизмов.
Испытание клеточной культуры на
друrие посторонние areHTbI. Нейтрализо
ванные образцы, эквивалентные 500 челове
ческим дозам, но не менее 50 мл, если иные
количества не предписаны в частной статье,
испытывают на присутствие посторонних areH
тов путем введения в непрерывные почечные
обезьяньи и человеческие клеточные культуры.
Если вирус выращен на обезьяньих или чело
веческих клетках, нейтрализованный вирусный
сбор также испытывают на отдельной культуре
этих клеток. Если вирус вакцины выращен на
системе клеток млекопитающих или птиц, OT
личающихся от обезьяньих или человеческих
клеток, то производят инокуляцию клеток этих
видов из отдельной партии (серии). Клетки ин
кубируют при температуре (36:t1) ос и наблю
, дают в течение 14 дней. Вирусный посевной
материал или сбор выдерживает испытание,
если ни в одной из клеточных культур не об
наруживается признаков любых посторонних
areHToB, не внесенных в результате случайной
контаминации. Результаты испытания считают
достоверными, если не менее 80 % клеточных
культур остаются жизнеспособными.
Птичьи вирусы (требуется только для
вирусноrо noceBHoro материала, выра-
щенноrо на тканях птичьеrо происхожде-
ния, и вирусноrо сбора, выращенноrо на
первичных тканях птичьеrо происхожде-
ния). Нейтрализуют образец, эквивалентный
100 человеческим дозам вакцины или 1 О мл В
зависимости от Toro, что из них больше. Ис
пользуя по 0,5 мл на каждое яйцо, инокули
руют rруппу оплодотворенных яиц SPF воз
растом от 9 дней до 11 дней аллантоисным
путем, а друrую rруппу, возрастом от 5 до 7
дней, в желточный мешок. Инкубируют в
течение 7 дней. Вирусный посевной матери
ал или сбор выдерживает испытание, если
в аллантоисных жидкостях и жидкостях жел
точных мешков не обнаруживаются rемаrrлю
тинирующие areHTbI и если ни один из эм
брионов И хориоаллантоисных мембран не
обнаруживает макроскопической патолоrии.
Результаты испытания считают ДOCTOBepHЫ
ми, если не менее 80 % привитых яиц выжи
вают в течение 7 дней.
Вирусы насекомых (требуется только
для вирусов, выращенных на клетках на-
секомых). Если нет друrих указаний, ней
трализованные образцы, эквивалентные 500
человеческим дозам вакцины или 50 мл, В за
висимпсти от тоro, что из них больше, испы
тывают на наличие посторонних areHToB с по
мощью инокуляции на одну или более кле
точную культуру, отличающуюся от использо
вавшихся при получении и допустимую для
вируса насекомых, которая позволяет обна
руживать арбовирусы человека. Выбор клеток
2.6.16. ИСПblтания на посторонние azeHmbl в вирусных вакцинах
297
должен быть одобрен компетентным opraHoM,
учитывается также происхождение производя
щих клеток и вероятные контаминанты, KOTO
рые можно обнаружить в этих клетках. Клетки
инкубируют при температуре (27:t1) ос и Ha
блюдают в течение 14 сут. Посевной вирусный
материал или сбор выдерживает испытание,
если ни в одной из клеточных культур не об
наруживается присутствие посторонних areH
тов. Результаты испытания считаются дей
ствительными, если не менее 80 % клеточных
культур остаются жизнеспособными.
ПРОИЗВОДСТВЕННАЯ КЛЕТОЧНАЯ
КУЛЬТУРА: КОНТРОЛЬНЫЕ КЛЕТКИ
Контрольные клетки изучают под микро
скопом на отсутствие любой цитопатической
деrенерации вирусноro происхождения. Изу
чение проводят в течение периода инкубации,
но не менее чем в течение 14 дней после ино
куляции. Результаты испытания считают дo
стоверными, если не менее 80 % контрольных
клеточных культур выживают в течение пери
ода наблюдения.
В момент последнеro сбора вирусов, но не
ранее, чем на 14 й день, выполняют нижеопи
санные испытания.
Испытание на rемадсорбирующие
вирусы. Не менее 25 % контрольных культур
испытывают на наличие rемадсорбирующих
вирусов путем добавления эритроцитов MOp
ских свинок. Если испытание на rемадсорби
рующие вирусы невыполнимо, проводят ис
пытание на вирусы rемаrrлютинации. При
необходимости эритроциты морских свинок
можно хранить при температуре (5:t3) ос в Te
чение не более 7 дней. Состояние половины
культур оценивают после инкубации при TeM
пературе (5:t3) ос в течение 30 мин, а cocтo
яние друrой половины после инкубации
при температуре от 20 ос до 25 ос в течение
30 мин. Признаков присутствия rемадсорби
рующих areHToB не должно обнаруживаться.
Испытание клеточных культур на друrие
посторонние areHTbI. Собирают супернатан
ты контрольных клеток и испытывают на при
сутствие посторонних areHToB путем введения
в почечные обезьяньи и человеческие клеточ
ные культуры. Если вирус вакцины выращен в
системе клеток млекопитающих, то произво
дят также инокуляцию клеток данноro вида, но
из отдельной партии (серии). На каждой кле
точной системе испытывают не менее 5 мл.
Инокулированные культуры инкубируют при
температуре (36:t1) ос и наблюдают в течение
14 дней. Признаков присутствия посторонних
areHToB не должно обнаруживаться.
Если производственная клеточная культу
ра поддерживается при температуре, отлич
ной от (36:t1) ОС, выполняют дополнительное
39 Зак.1712.
испытание на посторонние areHTbI при этой
температуре с использованием клеток тоro же
типа, который используется для выращивания
вируса.
Если вирус выращен в клетках HaceKO
мых, то накопленную надосадочную жидкость
инокулируют на не менее чем одну клеточную
культуру, отличающуюся от использовавших
ся при получении и допускаемую для вируса
насекомых, которая позволяет обнаруживать
арбовирусы человека. Клетки инкубируют при
температуре (2Н1) ос в течение 14 сут. При
знаков присутствия посторонних areHToB не
должно обнаруживаться.
Вирусы птичьеrо лейкоза (требуется
только для вирусов, выращенных на пер-
вичных тканях птичьеrо происхождения).
Проводят испытания на вирусы причьеro лей
коза с использованием 5 мл надосадочной
жидкости от контрольных клеток.
КОНТРОЛЬНЫЕ ЯЙЦА
rемаrrлютинирующие areHTbI. По
0,25 мл аллантоисной жидкости каждоro из
яиц испытывают на rемаrrлютинирующие
areHTbI путем непосредственноro смешива
ния с куриными красными кровяными тель
цами и после засева яиц SPF, выполняемоro
следующим образом: инокулируют объеди
ненный образец амниотических жидкостей
общим объемом 5 мл в аллантоисную и амни
отическую полости яиц SPF. Объемы BBeдe
ния по 0,5 мл. Контрольные яйца выдержи
вают испытание, если ни в одном случае не
обнаруживаются признаки присутствия reMar
rлютинирующих areHToB.
Вирусы птичьеrо лейкоза. Используют
образец объединенных амниотических жидко
стей контрольных яиц общим объемом 10 мл.
Проводят усиление путем пяти пассажей на
клеточных культурах куриных эмбрионов, не
содержащих вирусов лейкоза; испытание на
птичий лейкоз выполняют с использовани
ем клеток, полученных в пятом пассаже. KOH
трольные яйца выдерживают исп'ытание, если
не обнаруживается признаков присутствия ви
русов лейкоза.
Друrие посторонние areHTbI. Образцы
объединенных амниотических жидкостей KOH
трольных яиц по 5 мл прививают на человече
ские и обезьяньи клеточные культуры. Клеточ
ные культуры наблюдают в течение 14 дней.
Контрольные яйца выдерживают испытание.
если не обнаруживается признаков присут
ствия посторонних areHToB. Результаты испы
тания считают достоверными, если не менее
80 % при витых культур выживают до оконча
ния периода наблюдения.
298
01/2013:20617
rосударственная фармакопея Республики Беларусь
2.6.17. ИСПЫТАНИЕ
НА АНТИКОМПЛЕМЕНТАРНУЮ
АКТИВНОСТЬ
иммУноrЛОБУЛИНА
Для определения антикомплементарной aK
тивности (АКА) иммуноrлобулина определенное
количество испытуемоro материала (10 Mr им
муноrлобулина) подверrают инкубации с опре
деленным количеством комплемента морских
свинок (20 СН 50 ), после чеrо титруют остаток
комплемента; антикомплементарная активность
выражается как поrлощение комплемента в про
центах к контрольному образцу комплемента,
принимаемому за 100 %.
За rемолитическую единицу активности KOM
племента (СН 50 ) принимают такое еro количе
ство, которое в данных реакционных условиях
приводит к лизису 2,5'10 В из общеrо количества
5.108 оптимальным образом сенсибилизирован
ных эритроцитов.
Основной раствор ма2НUЯ и кальция.
1,10З r кальция хлорида Р и 5,083 r ма2ния хло
рида Р растворяют в ваде Р и доводят до объема
25 мл этим же растворителем.
Основной буферный раствор барбитала.
207,5 r натрия хлорида Р и 25,48 r барбитал на
трия Ррастворяют в 4000 мл воды Ри доводят 1 М
раствором кислоты хлористоводородной до
значения рН 7.3. Прибавляют 12,5 мл OCHOBHO
ro раствора кальция и маrния и доводят водой Р
до объема 5000 мл. Фильтруют через мембран
ный фильтр (размер пор 0,22 мкм). Хранят при
температуре 4 ос в стеклянных контейнерах.
Раствор желатина. 12,5 r желатина Р
растворяют приблизительно в 800 мл воды Р и
наrревают на водяной бане до кипения. Охлаж
дают до 20 ос и доводят водой Р до объема 1 О л.
Фильтруют через мембранный фильтр (размер
пор 0,22 мкм). Хранят при температуре 4 ОС. Ис
пользованию подлежат только прозрачные pac
творы.
Раствор цитрата. 8.0 r натрия цитpa
та Р, 4,2 r натрия хлорида Р и 20,5 r 2ЛЮКОЗЫ Р
растворяют в 750 мл воды Р. Доводят раствором
100 r/л лимонной кислоты Р до рН 6,1 и разво
дят водой Р до объема 1000 мл.
Буферный раствор желатина и барбита
ла. 4 объема раствора желатина прибавляют к
1 объему основноro буферною раствора барби
тала и перемешивают. При необходимости ДOBO
дят 1 М раствором натрия 2идроксида или 1 М
раствором кислоты хлористоводородной до
рН 7,3. Хранят при температуре 4 ос. Ежеднев
но roтовят свежие растворы.
Стабилизированная овечья кровь. Смеши
вают равные объемы овечьей крови и раствора
цитрата и перемешивают. Хранят при темпера
туре 4 ос не менее 7 и не более 28 дней. (Стаби
лизированная овечья кровь и эритроциты овцы
доступны в различных коммерческих источн
ках).
,емолизин. Антисыворотку против красных
кровяных телец овцы roтовят с использованием
кроликов. (Такие антисыворотки доступны в раз
личных коммерческих источниках).
Комплемент морских свинок. rотовят пул
сыворотки из крови не менее 1 О морских свинок.
Сыворотку отделяют от свернувшейся крови
центрифуrированием при температуре около
4 ос. Сыворотку хранят в небольших количе
ствах при температуре ниже 70 ОС.
МЕТОД
Приrотовление стандартизированной
5 % суспензии эритроцитов овцы. Эритроци
ты овцы отделяют центрифуrированием COOTBeT
ствующеro объема стабилизированной овечьей
крови, промывают не менее трех раз буферным
раствором желатина и барбитала и rотовят в том
же растворе суспензию с содержанием клеток
5 % (об/об). Плотность клеток в суспензии опре
деляют следующим образом: 0,2 мл прибавляют
к 2,8 мл воды Р и центрифуrируют лизированный
раствор в течение 5 мин при 1000 g; плотность
клеток соответствует требованиям, если оптиче
ская плотность (2.2.25) супернатанта при 541 нм
составляет (0,62::1:0,01). Плотность клеток KoppeK
тируют прибавлением буферноro раствора жела
тина и барбитала в соответствии с формулой:
V== V;.A ,
, 0,62
rдe:
V, окончательный объем после KoppeK
ции;
v: исходный объем;
А оптическая плотность исходной суспен
зии при 541 нм.
Суспензия после коррекции содержит около
1.109 клеток в миллилитре.
Титрование rемолизина. rотовят разве
дения rемолизина в соответствии с таблицей
2.6.17. 1.
К каждой пробирке из серии разведении re
молизина, начиная с разведения 1 :75, прибавля
ют по 1,0 мл 5 % суспензии эритроцитов овцы
и перемешивают. Инкубируют при температуре
37 ос в течение 30 мин.
По 0,2 мл каждой из этих инкубированных
смесей переносят в новые пробирки и прибав
ляют по 1,1 О мл буферноro раствора желатина
и барбитала и по 0,2 мл разведенноro компле
eHTa морских свинок (например, в разведении
1:150). Эти операции выполняют дважды.
В качестве контрольноro образца неrемоли--
зированных клеток ютовят три пробирки, coдe
жащие по 1,4 мл буферноro раствора желатина
и барбитала и 0,1 мл 5 % суспензии эритроц
тов овцы.
2.6.17. Испытание на антикомплементарнуюактивность иММУНОёлобулина
299
Таблица 2.6.17. 1
Требуемое разведение rотовят с использованием
Буферный раствор желатина и барбитала rемолизин
rемолизина
Объем (мл) Разведение (1 : ...) Объем (мл)
7,5 0,65 неразведенный 0,1
10 0,90 неразведенный 0,1
75 1,80 7,5 0,2
100 1,80 10 0,2
150 1,00 75 1,0
200 1,00 100 1,0
300 1,00 150 1,0
400 1,00 200 1,0
600 1,00 300 1,0
800 1,00 400 1,0
1200 1,00 600 1,0
1600 1,00 800 1,0
2400 1,00 1200 1,0
3200* 1,00 1600 1,0
4800* 1,00 2400 1,0
.1,0 мл смеси отбрасывают.
В качестве полностью rемолизированноro
контрольноro образца rотовят три пробирки, co
держащие по 1,4 мл воды Р и 0,1 мл 5 % суспен
зии эритроцитов овцы.
Все пробирки инкубируют при температуре
37 ос в течение 60 мин и центрифуrируют при
1000 9 в течение 5 мин. Измеряют оптическую
плотность (2.2.25) супернатантов при 541 нм и
рассчитывают для каждой из пробирок степень
rемолиза в процентах по формуле:
Ав A1 .100
Ab A1 '
rде:
Ав оптическая плотность в пробирках с
разведениями rемолизина;
Аь средняя оптическая плотность в трех
пробирках с полным rемолизом;
А 1 средняя оптическая плотность в трех
пробирках без rемолиза. .
На миллиметровой бумаrе строят rрафик
зависимости степени rемолиза в процентах (по
оси ординат) от обратноro значения COOTBeT
ствующеro разведения rемолизина (по оси аб
сцисс). Определяют из rрафика оптимальное
разведение rемолизина. Выбирают разведение
таким образом, чтобы дальнейшее увеличение
количества rемолизина не приводило бы к cy
щественному изменению степени rемолиза. Это
разведение принимают за одну минимальную
rемолитическую единицу (1 MrE) в 1,0 мл. Оп
тимальное rемолитическое разведение rемоли
зина для приroтовления сенсибилизированных
эритроцитов овцы составляет 2 мrЕ/мл.
Результат титрования rемолизина считают
достоверным при условии, что максимальная
степень rемолиза составляет от 50 % до 70 %.
Если максимальная степень rемолиза не Haxo
дится в этих пределах, титрование повторяют с
использованием более или менее разведенноro
раствора комплемента.
Приrотовление оптимизированных ceH
сибилизированных эритроцитов (rемолити
ческой системы). rотовят подходящий объем
разведенноro rемолизина с содержанием
2 мrЕ/мл и равный объем стандартизированной
5 % суспензии эритроцитов овцы. Прибавляют
разведение rемолизина к стандартизированной
суспензии клеток и перемешивают. Инкубируют
при температуре 37 ос в течение 15 мин, после
чеro хранят при температуре от 2 ос до 8 ос и ис
пользуют в течение 6 ч.
Титрование комплемента. rотовят под
ходящее разведение комплемента (например,
1 :250) с использованием буферноro раствора
желатина и барбитала и выполняют титрование
в двух повторностях в соответствии с таблицей
2.6.17. 2.
К содержимому каждой пробирки прибавля
ют по 0,2 мл сенсибилизированных эритроцитов
овцы, хорошо перемешивают и инкубируют при
температуре 37 ос в течение 60 мин. Пробирки
охлаждают на ледяной бане и центрифуrируют
при 1000 9 в течение 5 мин. Измеряют оптиче
скую плотность супернатанта при 541 нм и pac
считывают степень rемолиза (У) по формуле
Ac A1
Ab A1
rдe:
Ас оптическая плотность в пробирках
1 12;
300
rосударственная фармакопея Республики Беларусь
Таблица 2.6.17. 2
Номер пробирки Объем разведения компле Объем буферноrо раствора
мента (например, 1 :250), мл желатина и барбитала
1 0,1 1,2
2 0,2 1,1
3 0,3 1,0
4 0,4 0,9
5 0,5 0,8
6 0,6 0,7
7 0,7 0,6
8 0,8 0,5
9 0,9 0,4
10 1,0 0,3
11 1,1 0,2
12 1,2 0,1
Три пробирки в качестве KOHTpO 1,3
ля rемолиз О %
Три пробирки со 100 % 1,3 мл воды
rемолизом
Аь средняя оптическая плотность в про
бирках со 100 % rемолизом;
А 1 средняя оптическая плотность в KOH
тролях С 0% rемолизом.
На миллиметровой бумаrе с лоrарифмиче
ским масштабом по обеим осям строят rрафик
со значениями У/( 1 у) на оси абсцисс и коли
чеством разведенною комплемента на оси op
динат. Строят наилучшую прямую, COOTBeT
ствующую нанесенным точкам, и определяют
ординату ДЛЯ дозы комплемента, приводя
щей к 50 % rемолизу, т. е. YI(1 У)=1,О. Рассчи
тывают активность в rемолитических единицах
(СН 5 /мл) по формуле:
Cd ,
Са .5
rдe:
C d обратное значение разведения KOM
племента;
Са объем (мл) разведенноro комплемен
та, при водящий к 50 % rемолизу;
5 масштабный множитель для учета коли
чества эритроцитов.
Результат испытания считают достоверным
при условии, что для диапазона rемодиализа
от 15 % до 85 % выполняется линейная зависи
мость, а TaHreHc уrла наклона прямой составляет
от 0,15 до 0,40, предпочтительно от 0,18 до 0,30.
Испытание на антикомплементарную
активность. rотовят разведение комплемен
та, содержащее 100 СН5iмл, путем разбав
ления титрованноro комплемента морских
свинок буферным раствором желатина и бар
битала. В зависимости от испытуемою им
муноrлобулина и основываясь на данные по
валидации, может быть необходимым ДOBeдe
ние значения рН до 7. Для иммуноrлобулина
с содержанием 50 мr/мл rотовят следующие
смеси для инкубации:
Таблица 2.6.17. 3
Испытуемый Контроль
образец комплемента
иммуноrлобулина (в повторе)
Иммуноrло
булин 0,2 мл
(50 мr/мл)
Буферный
раствор 0,6 мл 0,8 мл
желатина и
барбитала
Комплемент 0,2 мл 0,2 мл
Проводят испытание образца иммуноrлобу
лина и roтовят положительный и отрицательный
контроли АКд с использованием БСП иММУН02ЛО
булина человечеСК020 в соответствии с указания
ми на прилаrающемся к нему листке вкладыше.
Если концентрация иммуноrлобулина отличает
ся от 50 мrfмл, используют большие либо MeHb
шие объемы образца и буферноrо раствора
желатина и барбитала. Например, при KOHцeH
трации иммуноrлобулина 30 мr/мл к 0,33 мл
ею раствора прибавляют 0,47 мл буферною
раствора желатина и барбитала с получением
0,8 мл. Пробирки укупоривают и инкубируют при
37 ос в течение 60 мин. По 0,2 мл каждой из инку
бированных смесей прибавляют к 9,8 мл буфер
ноro раствора желатина и барбитала для разве
дения комплемента. Описанную выше операцию
титрования комплемента проводят для каждой
из пробирок, определяя остаточную активность
комплемента (таблица 2.6.17. 2). Антикомпле--
ментарную активность испытуемоro. Qбразца pac
считывают относительно контроля комплемента.
принимаемою за 100 %, по формуле:
2.6.19. Испытание пероральной вакцины полиомиелита на нейровuрулентность 301
a b
.100,
а
rдe:
а средняя активность комплемента
(СН 5 /мл) в контролях комплемента;
Ь активность комплемента (СНs/мл) в ис
пытуемом образце.
Результаты испытания считают ДOCТOBepHЫ
ми, если:
антикомплементарные активности,
определенные для АКА отрицательноro и
АКА положительноro контролей находятся в
пределах, указанных в листке вкладыше, при
лаrающемуся к стандартному биолоrическому
препарату;
активность комплемента в контроле KOM
племента (а) находится в пределах от 80 СН 5 /мл
ДО 120 СН 5 /мл.
01/2013:20618
2.6.18. ИСПЫТАНИЕ ЖИВЫХ
ВИРУС НЫХ ВАКЦИН
НА НЕИРОВИРУЛЕНТНОСТЬ
в каждом испытании используют не менее
десяти серонеrативных по отношению к испыту
емому вирусу обезьян. Каждой из обезьян в Ta
ламический отдел каждоrо полушария вводят
не менее 0,5 мл испытуемоro материала, если
нет друrих указаний в частной статье. Общее KO
личество привитоro каждой обезьяне вируса не
должно быть менее количества, содержащеro
ся в рекомендованной единичной человеческой
дозе вакцины. Для проверки наличия диких ней
ровирулентных вирусов контрольную rруппу, co
стоящую не менее чем из четырех обезьян, co
держат в тех же клетках, что и rруппу привитых
обезьян, или в непосредственной близости от
них. При витых обезьян наблюдают в течение
17 21 дней, отмечая возникновение паралича
и друrих невролоrических симптомов; обезьян
из контрольной rруппы наблюдают в течение
тоro же периода и дополнительно 1 О дней. Жи
вотные, поrибшие в течение первых 48 ч после
инъекции, считаются поrибшими по причинам
неспецифическоro характера и MorYT быть за
менены друrими. Результаты испытания призна
ют недостоверными, если более 20 % при витых
обезьян поrибают по неспецисрическим пrичи
нам иlили образцы сыворотки, отобранные у
обезьян контрольной rруппы, в момент привив
ки испытуемых животных и через 1 О дней после
умерщвления последних, имеют признаки ин
фекции, вызванной диким вирусом испытуемо
ro типа или вирусом кори. По окончании периода
40 Зак 1712
наблюдения выполняют вскрытие и rистопато
лоrическое изучение соответствующих отделов
мозrа на предмет наличия воздействия на цeH
тральную нервную систему. Материал Bыдep
живает испытание, если не обнаруживается He
ожиданных клинических и rистопатолоrических
признаков воздействия на центральную HepB
ную систему, которые MOryT быть вызваны BBe
денным вирусом.
01/2013:20619
2.6.19. ИСПЫТАНИЕ ПЕРОРАЛЬНОЙ
ВАКЦИ ЫПОЛИОМИЕЛИТА
НА НЕИРОВИРУЛЕНТНОСТЬ
Обезьяны, используемые в испытании на
нейровирулентность, должны соответствовать
требованиям, приведенным в частной статье
Вакцина полиомиелита, пероральная, и иметь
массу тела не менее 1,5 Kr:. Патоrенность для
обезьян семейств Масаса или Cercopithecus
топkеуs испытывается в сравнении с патоrен
ностью стандартноro вирусноro препарата для
испытания на нейровирулентность путем BBeдe
ния в поясничный отдел центральной нервной
системы после седации с использованием под
ходящеro areHTa, например, кетамина rидрох
лорида. Для образца сыворотки, отобранноro
до инъекции, должно быть показано отсутствие
нейтрализующих антител в разведении 1:4 при
испытании против не более чем 1000 ССID 50
каждоro из трех типов полиовируса.
Количество обезьян. Вакцину и подходя
щий roмотипический вирус сравнения испыты
вают одновременно в одной и той же rруппе
обезьян. Вакцину и препарат сравнения (CTaH
дартный образец) вводят равному количе
ству животных. Животных С различающимися
типами введенноro препарата размещают слу
чайным образом по клеткам, а их принадлеж
ность кодируется таким образом, ч.тобы тип
полученной ими прививки был скрыт от наблю
дателей. Количество привитых обезьян должно
быть таково, чтобы при оценке как вакцины,
так и препарата сравнения было включено
не менее одиннадцати обезьян, положитель
ных по отношению к вирусу типов 1 и 2, и не
менее восемнадцати обезьян, положительных
по отношению к вирусу типа 3 (положи'!'ель
ными считают обезьян, имеющих специфичр,
ские нейронные повреждения в центральной
нервной системе, вызванные полиовирусом).
С одним И тем же roмотипическим препара
том сравнения MorYT испытываться несколько
партий вакцины. По возможности, используют
обезьян из одной и той же карантинной rруппы.
302
rосударственная фармакопея Республики Беларусь
в друrих случаях используют обезьян из двух
rрупп, причем вакцина и препарат сравнения
прививаются равному количеству обезьян из
каждой rруппы. Если испытание проводится в
течение двух рабочих дней, в каждый из дней
вакциной и rомотипическим препаратом cpaB
нения прививают равное количество обезьян.
Содержание вируса. Содержание вируса в
вакцине и roмотипическом препарате сравнения
реryлируют таким образом, чтобы они были Ha
сколько возможно близки и находились в преде
лах от 105,5 до 106,5 ССlD 50 В 0,1 миллилитре.
Наблюдение. Всех обезьян наблюдают в Te
чение 17 22 дней, отмечая признаки полиоми
елита или друrих вирусных инфекций. Обезьян,
выживших в течение первых 24 ч, но поrибших
до 11 ro дня после прививки, подверrают BCKpЫ
тию для определения тоro, являлся ли причиной
rибели полиомиелит. Животные, поrибшие по
причинам, не связанным с полиоми елитом, не
учитываются при оценке результатов. Умираю
щих и в тяжелой степени парализованных живот
ных умерщвляют и вскрывают. Также вскрывают
всех животных, выживших в течение периода Ha
блюдения. Результаты испытания считают Heдo
стоверными, если более чем у 20 % животных в
течение периода наблюдения была обнаружена
интеркуррентная инфекция.
Количество срезов, подлежащих испы
танию. rистолоrическому исследованию подле
жат как минимум поясничный и шейный отделы,
верхний и нижний продолroватый мозr, средний
мозr, зрительный буroр и двиrательная область
коры rоловноro мозrа каждой обезьяны. Отбира
ют препараты толщиной 15 мкм и окрашивают их
rаллоцианином. Минимальные количества под
лежащих испытанию срезов составляют:
(а) 12 срезов, репрезентативных для пояс
ничноro утолщения;
(Ь) 1 О срезов, репрезентативных для шейно
ro утолщения;
(с) 2 среза продолrоватоro мозrа;
(d) 1 срез моста и мозжечка;
(е) 1 срез среднеro мозrа;
(f) 1 срез с левой и правой части зрительно
ro буrра;
(9) 1 срез с левой и правой части двиrатель
ной области коры roловноro мозrа.
Подсчет вирусной активности. Для оценки
вирусной активности в полусекциях поясничноro
корда и ствола мозrа используют систему под
счета тяжести повреждений, по котоrой следу
ющим образом дифференцируют клеточную ин
фильтрацию и разрушение нейронов:
1) наличие лишь клеточной инфильтрации
(обезьяна не считается положительной);
2) клеточная инфильтрация с минимальным
повреждением нейронов;
3) клеточная инфильтрация со значитель
ным повреждением нейронов;
4) массовое повреждение нейронов в соче
тании с клеточной инфильтрацией или без нее.
Полученные значения записывают на CTaH
дартном бланке (пример подходящей формы
имеется в требованиях к биолоrическим субстан
циям «Rеqиirетепts for Bio/ogica/ sиЬstапсеs» ,
NQ 7, Всемирной орrанизации здравоохранения
в разделе «Требования к вакцине полиомиели
та (пероральной)>> ). Обезьяна, на срезах тканей
которой имеются поврежденные нейроны, но
не имеется иroльчатых трактов, считается по
ложительной. Обезьяна, на срезах тканей KOTO
рой имеются иroльчатые тракты, но не имеется
поврежденных нейронов, не считается положи
тельной. Срезы, на которых имеются поврежде
, ния травматическоro происхождения, но не име
ется специфических вирусных повреждений, при
подсчете не учитываются.
Уровни тяжести повреждений основаны на
данных, полученных при изучении полусекций
поясничных (L), шейных (С) и мозrовых (В) rи
столоrических срезов. Для каждой положитель
ной обезьяны рассчитывают уровень поврежде
ний (LS) по формуле:
[ C :::H:e I + [ С :;ла: е , ] + [ С :;ла:;е ]
Число Число Число
LS полусекций полусекций полусекций .
З
Для каждой rруппы положительных обезьян
рассчитывают среднее значение уровня повреж
дений.
Оценка. Сравнение вирусной активно
сти в вакцине и в препарате сравнения OCHO
вано на активности в поясничном расширении
корда и степени распространения активности из
этоro реrиона в шейное расширение и rоловной
мозr. Положительное или отрицательное реше
ние основано на общем уровне для всех живот
ных, подверrнутых испытанию. Необычно BЫ
сокая активность как в поясничном отделе, так
и в результате распространения из этоro реrи
она, обнаруженная у отдельных животных, также
должна приниматься во внимание при BЫHece
нии окончательноro решения. Моновалентный
материал выдерживает испытание, если име
ется требуемое количество положительных жи
вотных и если ни в одном из клинических и rи
стопатолоrических исследований не выявлено
существенных различий в патorенности между
вирусом вакцины и препаратом сравнениq. Кри
терии положительноro решения приведены
ниже.
Критерии. Для каждой из вакцин сравнения
(типов 1, 2 и 3) проводят подходящее количе
ство (например, четыре) квалификационных ис
2.6.21. Методы амплификации нуклеиновых кислот
303
пытаний на вирулентность с целью получения
данных об их активности как основы критериев
для оценки испытуемых вакцин. Для повторных
испытаний каждоrо из вирусов сравнения pac
считывают общий средний уровень поврежде
ний (М). а также суммарную оценку дисперсии
(s2) и отклонения (s) результатов испытания.
Критерии достоверности результатов испы
тания препарата сравнения устанавливаются
на основе данных, накопленных в квалифика
ЦИОННЫХ испытаниях. Общепринятых критериев
не имеется; для лабораторий с оrраниченным
опытом работы может быть полезен следующий
эмпирический метод установления допустимых
пределов среднеro уровня повреждений для
препарата сравнения (Xef):
Типы 1 и 2
Тип 3
Нижний предел
M s
М s/2
Таблица 2.6.19. 1
Верхний предел
M+s
M+s
Если средний уровень повреждений испыту
емой вакцины равен X test ' а С " С 2 и С з KOHCTaH
ты, определенные ниже, то:
вакцина не выдерживает испытание при:
X tes \ X ref > С"
вакцина может быть однократно подверrну
та повторному испытанию при:
С, < X test X ref < С
Если вакцина испытывается повторно, cpeд
ние значения уровней повреждений для испы
туемой вакцины и препарата сравнения pac
считываются заново. Вакцина не выдерживает
испытание при:
X(test1+lest2) X(ref' ref2) > С .t
2 з
Константы С" С 2 И С з рассчитывают по фор
мулам:
с = 2з 2s2 ,
, , N,
с = 26 2S2 ,
2 , N,
1* S 2
С =16 ,
з ' N,
rде:
N, количество положительных обезьян
при испытании вакцины;
N 2 количество положительных обезьян в
двух испытаниях;
2,3 нормальное отклонение для 1 %
уровня;
2,6 нормальное отклонение для 0,5 %
уровня;
1,6 нормальное отклонение для 5 %
уровня.
Испытание на нейровирулентность, в ко-
тором получен средний уровень повреждений
препарата сравнения (X ref ), несовместимый с
предшествующим опытом, не используется для
оценки испытуемой вакцины. Если результаты
испытания достоверны, рассчитывают средний
уровень повреждений для испытуемой вакцины
() es\) И сравнивают ero с соответствующим зна
чением для roмотипическоro препарата cpaBHe
ния.
01/2013:20620
2.6.20. АНТИ-А И АНТИ-В
rЕМАrrлютинины
(НЕПРЯМОЙ МЕТОД)
rотовят два серийных разведения испытуемоro
образца в растворе 9 r/л натрия хлорида Р. К каж
дому разведению одною ряда прибавляют равный
объем 5 % (06106) суспензии эритроцитов rpуппы А"
предварительно трижды промытых раствором
натрия хлорида. К каждому разведению друroro
ряда прибавляют равный объем 5 % (06106) суспен
зии эритроцитов rруппы В, предварительно трижды
промытых раствором натрия хлорида. Суспензии
инкубируют при температуре 37 ос в течение 30 мин,
затем клетки трижды промывают раствором натрия
ХЛОрИда. Клетки оставляют в контакте с полива
лентным антиреаrентом человеческоro иммуноrло-
булина в течение ЗО мин. Каждую из суспензий. не
подверraя центрифуrированrию, исследуют под ми
кроскопом на аrrлютинацию.
01/2013:20621
2.6.21. МЕТОДЫ АМПЛИФИКАЦИИ
НУКЛЕИНОВЫХ КИСЛОТ
1. ВВЕДЕНИЕ
Методы амплификации нуклеиновых кислот
основаны на двух различных подходах:
1) амплификация последовательности ми
шени нуклеиновой кислоты с использованием,
например, полимеразной цепной реакции (ПЦР,
PCR), лиrазной цепной реакции (ЛЦР, LCR) или
изотермической амплификации рибонуклеино
вой кислоты (РНК, RNA);
304
rосударственная фармакопея Республики Беларусь
2) амплификация сиrнала rибридизации с
использованием, например, для дезоксирибо
нуклеиновой кислоты (ДНК, ONA) метода раз
ветвленной ДНК (bDNA); в этом случае ампли
фикация сиrнала достиrается без подверrания
нуклеиновой кислоты повторяющимся циклам
амплификации.
В данной общей статье в качестве стандарт
ноro метода описывается метод ПЦР. Moryт при
меняться и альтернативные методы, если они
выдерживают требования к качеству, перечис
ленные ниже.
2. ОБЛАСТЬ ПРИМЕНЕНИЯ
В данной статье устанавливаются требова
ния к подroтовке образца, к амплификации iп vitro
последовательностей ДНК и к детектированию
специфическоro продукта ПЦР. С помощью ПЦР
MOryT быть детектированы определенные после
довательности ДНк. Последовательности РНК
MOryT также быть детектированы путем обрат
ной транскрипции РНК на комплементарную ДНК
(cONA) с последующей амплификацией.
З. ПРИНЦИП МЕТОДА
ПЦР представляет собой проuедуру, позво
ляющую производить В условиях in vitro ампли
фикацию cerMeHTOB ДНК или РНК (после обрат
ной транскрипции на cONA).
После денатурации двунитевой ДНК в oд
нонитевую два синтетических олиroнуклеотид
ных праймера противоположной полярности
спариваются с их соответствующими компле
ментарными последовательностями в подлежа
щей амплификации ДНК. Короткие двунитевые
участки, формирующиеся в результате специ
фическоrо спаривания оснований праймеров
и комплементарной ДНК последовательности,
образуют rраницы амплифицируемоrо cerMeH
та ДНК и служат начальными точками для син
теза ДНК in vitro с участием термостабильной
ДНК полимеразы.
Процесс амплификация ДНК состоит из
циклов, включающих:
тепловую денатурацию нуклеиновой кис
лоты (последовательности мишени) на две OT
дельные нити;
специфический отжиr последовательно
сти мишени с праймерами в подходящих peaK
ционных условиях;
наращивание праймеров, присоединен
ных к обеим отдельным нитям, под воздействи
ем ДНК полимеразы при подходящей темпера
туре (синтез ДНК).
Повторяющиеся циклы тепловой дeHaTypa
ции, отжиrа с праймерами и синтеза ДНК приво
дят к экспоненциальной амплификации cerMeH
та ДНК, оrраниченноro праймерами.
Специфический продукт ПЦР, называе
мый ампликоном, может быть определен разно
образными методами соответствующей специ
фичности и чувствительности.
4. ИСПЫТУЕМЫЙ МАТЕРИАЛ
В связи с высокой чувствительностью ПЦР
образцы должны быть оптимальным образом за
щищены от попадания извне последовательнcr
стей мишеней. Отбор проб, хранение и TpaHC
портировку испытуемоro материала производят
в условиях, минимизирующих разрушение пcr
следовательности мишени. В случае проведения
испытания на РНК последовательности необхо
димо принять особые меры предосторожности
ввиду высокой чувствительности РНК к дeCTpYK
тивному воздействию рибонуклеаз. Следует учи
тывать, что некоторые добавляемые peareHTbI,
например, антикоаryлянты или консерванты.
MOryт оказывать влияние на ход определения.
5. МЕТОДИКА ИСПЫТАНИЯ
5.1. Предотвращение заrрязнения
В связи с риском заrрязнения следует cTporo
разrраничивать рабочие зоны в зависимости от
используемых материалов и применяемой Tex
нолоrии. Учитываемые факторы включают пере
движение персонала, использование спецодеж
ды, перемещение материалов, подачу воздуха и
процедуры деконтаминации.
Систему следует разделить на такие отсеки,
как:
зона основной смеси (зона, в которой
работы осуществляются только с материалами,
не содержащими шаблонных последовательно
стей, например, с праймерами, буферными pac
творами и т. д.);
пре ПЦР (зона, в которой ведется работа с
реаrентами, образцами и контролями);
ПЦР амплификация (амплифицируемый
материал содержится в закрытой системе);
пост ПЦР детектирование (единственная
зона, в которой операции с амплифицируемым
материалом производятся в открытой системе).
5.2. Подrотовка образцов
При подroтовке образцов подлежащая aM
плификации последовательность мишень
должна быть эффективным и воспроизводимым
образом экстраrирована или высвобождена из
испытуемоro материала так, чтобы амплифика
ция в избранных реакционных условиях была
осуществима. MoryT быть использованы различ
ные физико химические процедуры экстракции
и/или обоrащения.
Добавки, присутствующие в испытуемом Ma
териале, MoryT оказывать влияние на ход ПЦР.
Для контроля присутствия инrибиторов в испы
туемом материале следует провести процедуры,
описанные в пункте 7.3.2.
В случае РНК матриц следует уделять вни
мание предотвращению проявления рибонукле
азной активности.
5.3. Амплификация
Амплификация последовательности ми
шени методом ПЦР проводится в оnтимизирcr
2.6.21. Методы амплификации нуклеиновых кислот
305
ванных циклически изменяющихся условиях
(температурный профиль для денатурации ДBY
нитевой ДНК, отжиrа и наращивания праймеров;
периоды инкубации при выбранных температу
рах; скорость наращивания). Эти условия зави
сят от различных параметров, например:
длины и состава праймера и последова
тельности мишени;
типа ДНК полимеразы, буферной смеси и
реакционноro объема, используемых при ампли
фикации;
типа используемоro устройства для TepMO
циклирования и скорости передачи тепла между
аппаратом, реакционным сосудом и реакцион
ной жидкостью.
5.4. Детектирование
Ампликон, образовавшийся в результате
ПЦР, может быть идентифицирован по раз
меру, последовательности химической MO
дификации или комбинации этих параме
тров. Характеризация по размеру может быть
проведена методами rель электрофореза (с
использованием слоев аrарозноro или по
лиакриламидноro rелей или капиллярно
ro электрофореза) или колоночной xpOMaTO
rрафии (например, ВЭЖХ). Характеризация
по составу последовательности может быть
проведена путем специфической rибриди
зации зондов, содержащих последователь
ность, комплементарную мишени, или pac
щеплением амплифицированноro материала,
отражающим наличие мишень специфиче
ских сайтов ферментативной рестрикции. Xa
рактеризация по химической модификации
может быть проведена, например, введением
вампликоны флуорофора с дальнейшей дe
текцией флуоресценции, возникающей в pe
зультате возбуждения.
Детектирование ампликонов может быть дo
стиrнуто с помощью проб, помеченных для по
следующеro радиоизотопноro или иммунофер
MeHTHoro сопряженноro детектирования.
6. ОЦЕНКА И ИНТЕРПРЕТАЦИЯ
РЕЗУЛЬТАТОВ
Результат испытания является достоверным
лишь при условии получения однозначно поло
жительных результатов для положительноro KOH
троля (контролей) и однозначно отрицательных
результатов для отрицательноro контроля (KOH
тролей). В связи с исключительно высокой чув
ствительностью метода ПЦР и с риском попа
дания заrрязняющих примесей, положительные
результаты должны подтверждаться двукратным
полным повторением процедуры испытания по
возможности с новой аликвотой образца. Счи
тают, что образец дает положительную реакцию,
если не менее одноro из повторных испытаний
дает положительный результат. Как только опре
делен измеряемый предел мишени, необходима
система количественноro определения.
7. ОБЕСПЕЧЕНИЕ КАЧЕСТВА
7.1. Валидация системы для количе
CTBeHHoro определения методом ПЦР
Проrрамма валидации должна включать Ba
лидацию оборудования и используемоro метода
ПЦР. Следует учитывать положения, изложен
ные в руководстве /СН (тема О28) «Валидация
аналитическою метода: методолоrия».
Для валидации ПЦР испытания обязатель
ным является применение официальных pa
. бочих стандартных образцов или стандартных
образцов внутреннею применения, калиброван
ных относительно Международных стандартных
образцов, для последовательностей мишеней,
при определении которых будет использоваться
система.
7.1.1. Определение положитеЛЬН020 2pa
ниЧН020 значения
При валидации испытания должно быть
определено положительное rраничное значе
ние. Положительное rраничное значение опре
деляется как минимальное количество после
довательностей мишеней в объеме образца,
которые MOryт быть определены в 95 % испы
таний. Положительное rраничное значение за
висит от таких взаимосвязанных факторов, как
объем экстраrированноro образца и эффектив
ность методолоrии экстракции, транскрипция
РНК мишени в cONA, процесс амплификации и
детектирования.
При определении предела обнаружения си
стемы следует учитывать положительные rpa
ничные значения для каждой последовательно
сти мишени и результаты проведения испытания
выше и ниже этих значений.
7. 1.2. Система количествеННО20 определе
ния
При валидации количественноro определе
ния проверяются следующие параметры: пра
вильность, точность, специqpичность, предел
количественноro определения, линейность, ди
апазон и робастность.
7.2. Контроль качества peareHToB
Все peareHTbI, имеющие критическое зна
чение для используемой методолоrии,. должны
проходить контроль перед использованием в py
тинных испытаниях. Их приroдность или непри
roдность основывается на заранее определен
ных критериях качества.
Праймеры являются важнейшими компо
нентами ПЦР анализа, и поэтому следует yдe
лять особое внимание их строению, чисто
те и валидации для ПЦР анализов. Праймеры
MorYT быть подверrнуты модификации (напри
мер, связыванием с флуорофором или антиrе
ном) для проведения детектирования амплико
на специфическими методами при условии, что
такие модификации не приведут к инrибирова
нию правильноro и эффективноro протекания
процесса амплификации последовательности
мишени.
306
rосударственная фармакопея Республики Беларусь
7.3. Контроль хода испытания
7.3.1. Внешние контроли
Для минимизации риска заrрязнения и обе
спечения адекватной чувствительности в состав
каждоro ПЦР испытания вводят следующие
внешние контроли:
положительный контроль: в нем coдep
жится определенное количество копий последо
вательности мишени; количество копий близко
к положительному rраничному значению, опре
деляется индивидуально для каждой системы и
кратно положительному rраничному значению
данной системы;
отрицательный контроль: образец анало
rичноro основноro состава, для котороro доказа
но отсутствие последовательностей мишеней.
7.3.2. Внутренний контроль
Внутренние контроли представляют собой
определенные последовательности нуклеино
вых кислот, содержащие сайты связывания с
праймерами, если нет друrих указаний в част
ной статье. Внутренние контроли должны aM
плифицироваться так же эффективно, как и
испытуемая последовательность мишень, но
ампликоны должны иметь четкие отличия. Тип
нуклеиновой кислоты (ДНК/РНК), используе
мой в качестве внутреннеro контроля, должен
быть тем же, что и в испытуемом материале.
Внутренний контроль предпочтительно добав
лять к испытуемому материалу до выделения
нуклеиновой кислоты; при этом он может ис
пользоваться для общеro контроля процесса
(экстракция, обратная транскрипция, ампли
фикация, детектирование).
7.3.3. Контроль ПОр020вых значений
Контроль пороrовых значений для коли
чественноro определения это испытуемый
образец с таким содержанием аналита, KO
торое не должно превышаться. Он содержит
аналит, калиброванный желательно в Между
народных единицах, и анализируется парал
лельно при каждом количественном опреде
лен ии.
7.4. Внешняя оценка качества
Участие во внешних проrраммах оценки Ka
чества представляет собой важную процеду
ру обеспечения качества ПЦР анализов для
каждой лаборатории и каждоro оператора.
Следующий раздел приводится для инфор
мации.
Валидация метода амплификации
нуклеиновых кислот (МАНК, NАТ)
в определении РНК вируса rепатита
с (HCV) в пулах плазмы: руководство
1. ОБЛАСТЬ ПРИМЕНЕНИЯ
Большинство аналитических методик, OCHO
ванных на амплификации нуклеиновых кислот,
представляют собой качественные испытания
на наличие .нуклеиновых кислот. Существует
также некоторое количество методик количе
cTBeHHoro определения (либо внутренних, либо
коммерчески доступных). Для определения за
rрязнения пулов плазмы РНК HCV aдeKBaTHЫ
ми являются качественные тесты, которые MOryT
быть рекомендованы как предельные испыта
ния при контроле содержания примесей в COOT
ветствии с указаниями в журнале «Pharmeu,o
ра», декабрь 1999, Техническое руководство по
разработке моноrрафий, rлава 111 «Валидация
аналитических методию>. В этом руководстве
описаны лишь методы валидации качествен
ных аналитических методик оценки заrрязнения
пулов плазмы РНК HCV на основе амплифика
ции нуклеиновых кислот. Поэтому двумя наибо
лее важными характеристиками для валидации
аналитическоro метода являются еro специ
фичность и предел определения. Кроме тоro,
должна быть оценена робастность аналитиче
скоro метода.
Однако этот документ также может быть ис
пользован в качестве основы для валидации aM
плификации нуклеиновых кислот в общем.
В соответствии с данным документом за
аналитическую процедуру принимают всю про
цедуру от экстракции нуклеиновой кислоты до
детектирования амплифицированноro продукта.
При использовании в аналитическом методе
или еro части коммерческоro набора, валида
цию пользователя может заменить документиро
ванное подтверждение валидации вышеуказан
ных положений, произведенной изroтовителем
набора. Тем не менее, пользователем должна
быть продемонстрирована производительность
набора в отношении ero предполаrаемоro ис
пользования (т. е. предел определения, робаст
ность, перекрестная контаминация).
2. СПЕЦИФИЧНОСТЬ
Специфичность представляет собой спо
собность к однозначной оценке присутствия HY
клеи новой кислоты в смеси с компонентами,
присутствие которых можно ожидать.
Специфичность аналитических методов,
основанных на амплификации нуклеиновых
кислот, зависит от выбора праймеров, выбора
зонда (для анализа конечноro продукта) и обо
снованности условий проведения испытания
(как для стадии амплификации, так и для стадии
детектирования).
При разработке праймеров и зондов должна
быть исследована их специфичность в отноше
нии детектирования только РНК HCV путем cpaB
нения избранной последовательности с после
довательностями, опубликованными в банках
данных. В случае HCV праймеры (и зонды)
обычно выбирают из областей 5' некодируемоro
участка reHoMa HCV, которые хорошо сохраня
ются для всех rенотипов.
Амплифицированный продукт должен OДHO
значно идентифицироваться одним из мноrих
2.6.21. Методы амплификации нуклеиновых кислот
307
методов, например, проведением амплифика
ции с вложенными праймерами, анализом с ис
пользованием ферментов рестрикции, секвени
рованием или rибридизацией со специфическим
зондом.
Для валидации специфичности аналити
ческоro метода испытание должно быть про
ведено в отношении не менее 100 РНК HCV
отрицательных пулов плазмы, для которых
должно быть показано отсутствие реакции. Под
ходящие образцы пулов, не дающих реакции,
MOryт быть получены в Европейском управле
нии по качеству лекарственных средств и здра
воохранению при Совете Европы (Еи,ореап O
,ecto,ate for the Quality of Mediciпes & Hea/thca,e
(Соипсil of Еи,оре».
Способность аналитическоro метода к обна
ружению всех rенотипов HCV также зависит от
выбора праймеров, зондов и параметров метода.
Эта способность должна быть продеw.онстри
рована с использованием характеризованных
эталонных панелей. Однако, ввиду сложности
получения некоторых rенотипов (например, re
нотипа 6), должны быть на подходящем уровне
детектированы rенотипы преобладающих типов
(например, в Европе rенотипы 1 и 3).
3. ПРЕДЕЛ ОБНАРУЖЕНИЯ
За предел обнаружения данной аналитиче
ской процедуры принимают наименьшее коли
чество нуклеиновой кислоты в образце, которое
может быть детектировано, но не обязательно
определено как точное значение.
Аналитический метод с использованием aM
плификации нуклеиновых кислот, используемый
для детекции РНК HCV в пулах плазмы, обычно
дает качественные результаты. Количество воз
можных результатов оrраничено двумя: либо
положительным, либо отрицательным. Хотя
рекомендуется определение предела обнаруже
ния, в практических целях для аналитическоro
метода на основе амплификации нуклеиновых
кислот должно быть определено положитель
ное rраничное значение. Положительное rpa <
ничное значение (в соответствии с определени
ем в общем разделе 2.6.21) представляет собой
минимальное количество последовательностей
мишеней в объеме образца, которые MOryT быть
определены в 95 % испытаний. На это положи
тельное rраничное значение оказывают влияние
распределение вирусных reHoMoB в индивиду
альных испытуемых образцах и такие факторы,
как эффективность фермента. Это может приве
сти к получению различных 95 % rраничных зна
чений для отдельных испытаний.
Для определения положительноro rранич
ноro значения ряд разведен ий рабочею peareH
та или БСП вируса 2епатита С, калиброванно
ro в сравнении с Международным стандартным
образцом HCV ВОЗ 96/790, должен быть под
BeprHYT испытанию в разные дни для провер
ки изменчивости результатов анализа. Для про
ведения статистическоro анализа результатов
должно быть испытано не менее трех незави
симых рядов разведений в достаточном количе
стве повторностей для получения 24 результа
тов испытания для каждоro разведения.
Например, в лаборатории в разные дни
MOryT быть проведены испытания трех рядов
разведен ий в восьми повторностях для каждоro
из них, четырех рядов разведен ий в шести по
вторностях для каждоro из них или шести рядов
разведен ий в четырех повторностях для каждо
ro из них. Для тою, чтобы число разведений не
было слишком большим, следует провести пред
варительное испытание (например, с использо
ванием лоrарифмических разведений образца
пула плазмы) с получением предварительноro
положительною rраничноro значения (т. е. наи
большею значения, дающеro положительный
результат). После этоro диапазон разведен ий
может быть выбран в области полученною пред
варительноro значения (с использованием, Ha
пример, лоrарифмическоro фактора разведения
0,5 или менее и отрицательноro пула плазмы
для матрицы разведения). Концентрация РНК
HCV, которая может быть детектирована в 95 %
испытаний, может быть вычислена с использо
ванием соответствующих методов статистиче
ской оценки.
Эти результаты также MorYT служить для
демонстрации изменчивости результатов, по
лучаемых данным аналитическим методом, в
ходе испытания и в разные дни.
4. РОБАСТНОСТЬ
Робастность аналитическою метода
мера ею способности выдерживать воздействие
малых, но определенных изменений параметров
метода, являющаяся показателем ero надежно
сти при обычном использовании.
Оценка надежности должна проводиться в
фазе разработки. Она должна показать надеж
ность аналитической процедуры с учетом опре
деленных вариаций параметров метода. Для
случая МАНК небольшие изменения параме
тров метода MOryT иметь решающее значение.
Однако в ходе разработки метода ero устой
чивость может быть продемонстрирована He
большим изменением концентраций peareHToB
(например, MgCI 2 , праймеров или dNTP). Для дe
монстрации устойчивости не менее 20 РНК HCV
отрицательных пулов плазмы, отобранных слу
чайным образом и инфицированных РНК HCV с
получением конечной концентрации, в три раза
превышающей заранее определенное 95 % rpa
ничное значение, должны быть испытаны и при
знаны положитеЛЬН IМИ.
Проблемы с робастностью MorYT воз
никнуть также в связи с методами, в KOTO
рых стадия улырацентрифуrирования пред
шествует экстракции вирусной РНК. Поэтому
для испытания робастности таких методов
не менее 20 пулов плазмы, содержащих раз
З08
rосударственная фармакопея Республики Беларусь
личные уровни РНК HCV, но не содержащих
специфических антител к HCV, должны быть
испытаны и признаны положительными.
Предотвращение перекрестной KOHTa
минации должно быть продемонстрирова
но путем точноrо детектирования панели из
не менее чем 20 образцов, попеременно за
полненной образцами отрицательных пулов
плазмы и отрицательных пулов плазмы, ин
фицированных высокими концентрациями
HCV (не менее 1 OO KpaTHoro 95 % rраничноrо
значения или не менее 104 МЕ/мл).
5. ОБЕСПЕЧЕНИЕ КАЧЕСТВА
ДЛЯ таких биолоrических испытаний, как
МАНК, имеется вероятность возникновения
специфических проблем, которые MorYT по
влиять как на валидацию, так и на интер
претацию результатов. Методики испытаний
должны быть четко описаны в форме CTaH
дартных операционных процедур (СОП). В
них должно указываться:
способ отбора проб (тип контейнеров и
т. д.);
приroтовление мини пулов (по потреб
ности);
условия хранения до анализа;
четкое описание условий испытания,
включая меры предосторожности против пе
рекрестной контаминации и деструкции ви
русной РНК, используемых peareHToB и CTaH
дартных препаратов;
четкое описание используемой аппара
туры;
детальные формулы для вычисления
результатов, включая статистическую обра
ботку.
В качестве удовлетворительной провер
ки соответствия системы и надежности aHa
литической процедуры, коrда бы она ни ис
пользовалась, может быть рекомендован
подходящий контроль процесса (например,
подходящее разведение БСП вируса 2епати
та С или образец плазмы, инфицированной
HCV, калиброванный в сравнении с Между
народным стандартным образцом HCV ВОЗ
96/790).
Техническая квалификация. Каждой кри
тической части используемоro оборудования
должна соответствовать проrрамма квали
фикации по установке и эксплуатации. После
замены критически важноrо оборудования
(например, термореrуляторов) должна быть
документально подтверждена функциональ
ность аналитической процедуры путем прове
дения параплельноro испытания в отношении
8 повторных образцов пула плазмы, инфици
;)ованноro РНК HCV с получением конечной
концентрации, соответствующей ранее опре
деленному трехкратному 95 % rраничному
значению. Все результаты должны быть поло
жительными.
Квалификация оператора. В отношении
каждоro оператора, принимающеro участие
в испытании, должна действовать COOTBeT
ствующая квалификационная проrрамма. Для
подтверждения соответствующеro уровня
подrотовки каждый оператор должен прове
сти испытание не менее 8 повторяющихся об
разцов пула плазмы, инфицированноro РНК
HCV с получением конечной концентрации,
соответствующей предварительно опреде
ленному трехкратному 95 % rраничному зна
чению. Это испытание (8 повторяющихся об
разцов) должно быть повторено дважды, в
два различных дня. Таким образом, должно
быть проведено 24 испытания на протяжении
трех дней. Все результаты должны быть по
ложительными.
8алидация метода амплификации
нуклеиновых кислот (МАНК)
для количественною определения ДНК
вируса 819 (8 19V) в пулах плазмы:
руководство
1. ОБЛАСТЬ ПРИМЕНЕНИЯ
Фармакопея требует, чтобы пулы плазмы,
использующиеся при производстве некоторых
продуктов, были проверены на содержание
ДНК вируса В 19 (В 19V), допустимая KOHцeH
трация которой оrраничена. Для выполнения
этоrо требования выделены количественные
МАНК. Наиболее важными характеристика
ми для валидации количественноro МАНК яв
ляются: правильность, точность, специфич
ность, предел количественноro определения,
линейность и диапазон. Кроме тоro, должна
быть оценена робастность аналитическоro
метода.
В этом руководстве описаны способы Ba
лидации МАНК для определения контамина
ции пулов плазмы ДНК B19V, базирующиеся
на руководствах /СН. Однако этот документ
также может быть использован в качестве
основы для валидации амплификации нукле
иновых кислот в общем.
В соответствии с данным документом за
аналитическую процедуру принимают всю
процедуру от экстракции нуклеиновой кисло
ты до детектирования амплифицированноrо
продукта.
При использовании в аналитическом
методе или еro части коммерческоrо набора,
валидацию пользователя может заменить
документированное подтверждение вали
дации вышеук:ззанных положений, произве
денной изrотовителем набора. Тем не менее.
пользователем должна быть продемонстри
рована производительность набора в OTHO
шении ero предполаrаемоro использования
(т. е. точность, правильность, диапазон, po
бастность).
2.6.21. Методы амплификации нуклеиновых кислот
309
2. ПРАВИЛЬНОСТЬ
Правильность представляет собой бли
зость совпадения между найденным значе
нием и либо значением, которое определе
но как условно правильное, либо принятым
значением сравнения. Правильность количе
cTBeHHoro определения зависит от rрадуиро
вочноrо rрафика и от отклонений на различ
ных стадиях количественноro определения.
Хотя рекомендовано определять точность во
всем специфицированном диапазоне, наибо
лее важно подтвердить точность в диапазо
не около предельноrо значения KOHцeHTpa
ции. В случае количественноro определения
ДНК B19V в пулах плазмы рекомендуется
оценивать правильность метода путем коли
чественноro определения не менее 5 KOHцeH
траций (с лоrарифмическим коэффициентом
разведения 0,5) БСП вирусной днк 819 для
метода амплификации нуклеиновых кислот
или иноro препарата, калиброванноro в Меж
дународных единицах в сравнении с Между
народным стандартным образцом ВОЗ ДНК
В 19, в диапазоне рекомендованноro на данный
момент пороroвоro значения 10,0 МЕ/мкл ДНК
B19V (т. е. 105 МЕ/мл, 104.5 МЕ/мл, 104 МЕ/мл,
103,5 МЕ/мл И 103 МЕ/мл) не менее чем в трех
повторностях для каждоrо разведения. Пра
вильность должна быть документирована для
различных концентраций в процентных еди
ницах, определенных в сравнении с извест
ным количеством ДНК B19V. Необходимо
отражать уровень технолоrии соответствую
щеro метода количественноro определения,
который следует также определять, напри
мер, с помощью совместных испытаний.
3. ТОЧНОСТЬ
Точность представляет собой близость
совпадения между сериями измерений, по
лученных в мноroчисленных испытаниях раз
личных проб одноro и тоro же roMoreHHo
ro образца. Точность определяется на трех
уровнях: <
сходимость выражает точность при
одних и тех же условиях испытания в течение
короткоro промежутка времени (iпtrа аssау
рrесisiоп); она оценивается с использовани
ем одноro метода количественноrо определе
ния, с помощью KOToporo трижды анализиру
ют подходящие разведения положительных
образцов с ДНК B19V, калиброванных в Меж
дународных единицах и охватывающих весь
диапазон этоro метода количественноro опре
деления; коэффициент вариации рассчиты
вают для индивидуальных образцов (iпt'а аs
say variability);
внутрилабораторная точность выражает
внутрилабораторную вариабельность (iпte,
assay рrесisiоп); она устанавливается oцeH
1C0й повторов (как рутинный количественный
анализ) подходящих разведений положитель
ных образцов с дНК B19V, калиброванных в
Международных единицах и охватывающих
весь диапазон этоrо метода количественно
ro определения, в различной обстановке (т. е.
различные дни, различные аналитики, раз
личное оборудование, различные реактивы);
коэффициент вариации рассчитывают для
индивидуальных образцов;
воспроизводимость выражает точность
между различными лабораториями (межла
бораторная точность); она оценивается путем
участия в совместных количественных изу
чениях содержания дНК B19V, например, по
Проrрамме профессиональноro тестирова
ния (ППТ, PTS Proficieпcy Testiпg Scheme).
4. СПЕЦИФИЧНОСТЬ
Специфичность представляет собой спо
собность к однозначной оценке присутствия
нуклеиновой кислоты в смеси с компонен
тами, присутствие которых можно ожидать.
Специфичность аналитических методов, oc
нованных на амплификации нуклеиновых
кислот, зависит от выбора праймеров, выбора
зонда (для анализа конечноro продукта) и
обоснованности условий проведения испыта
ния (как для стадии амплификации, так и для
стадии детектирования).
При разработке праймеров и зондов
должна быть исследована их специфичность
в отношении детектирования только дНК
B19V путем сравнения избранной последо
вательности с последовательностями, опу
бликованными в банках данных. Не должно
обнаруживаться значительное сходство с
последовательностями, не относящимися к
B19V.
Амплифицированный продукт должен
однозначно идентифицироваться одним из
мноrих методов, например, проведением aM
плификации с вложенными праймерами, aHa
лизом С использованием ферментов рестрик
ции, секвенированием или rибридизацией со
специфическим зондом.
Для проверки специфичности аналити
ческоro метода испытание должно быть про
ведено в отношении не менее 20 ДНК B19V
отрицательных пулов плазмы, для которых
должно быть показано отсутствие реакции.
,енотипы парвовируса 819. Междуна
родная комиссия по таксономии вирусов
(/CTV) классифицирует представителей 3
rенотипов как штаммы человеческоro пар
вовируса В19. rенотип 1 представляет про
тотип В 19V, rенотип 2 представляет вирус
ные последовательности типа А6, и rенотип
3 представляет V9 подобные последователь
ности. При проведении удлинения последо
вательности с использованием COOTBeTCTBY
ющей последовательности rенотипа В 19V,
доступной из баз данных последовательно
стей нуклеиновых кислот, праймеры и зонды
310
/осударственная фармакопея Республики Беларусь
ДОЛЖНЫ быть выполнены с таким расчетом,
чтобы была возможность обнаружения и KO
личественноrо определения в равной степе
ни различных rенотипов парвовируса В19.
ДЛЯ проверки выбранноro подхода следует
использовать стандартные вещества. Так как
получение биолоrических препаратов cpaB
нения может быть затруднено, COOTBeTCTBY
ющие препараты плазмид или синтетических
нуклеиновых кислот также MorYT служить в Ka
честве источника характеризованных после
довательностей целей. Однако они не MorYT
быть использованы для валидации процеду
ры экстракции.
5. ПРЕДЕЛ КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ
Предел количественноrо определения
представляет собой минимальное количе
ство нуклеиновой кислоты в образце, которое
может быть количественно определено с Tpe
буемой правильностью и точностью. Предел
количественноrо определения метода aM
плификации нуклеиновых кислот B19V oцe
нивают при изучении сходимости и межла
бораторых сличен иях путем оrраничения
разведений. Определяют наименьшую KOH
центрацию нуклеиновой кислоты мишени, KO
торая может быть определена количественно
с достаточной правильностью и точностью.
6. ЛИНЕЙНОСТЬ
Линейность метода количественноro
определения это возможность получения
результатов, прямо пропорциональных KOH
центрации нуклеиновой кислоты. Линейность
метода амплификации нуклеиновых кислот
B19V оценивают при изучении сходимости и
межлабораторных сличениях путем испыта
ния повторностей разведенных образцов с
концентрациями, охватывающими весь ди
апазон количественноro определения. Опре
деляют интервал, расположенный между
верхней и нижней концентрациями нуклеино
вой кислоты мишени, при которых получае
мые результаты прямо пропорциональны ее
содержанию.
7. ДИАПАЗОН
Диапазон количественноro определе
ния это интервал, расположенный между
верхней и нижней концентрациями нуклеино
вой кислоты в образце, для котороro было по
казано, что процесс имеет приемлемый ypo
вень правильности, точности и линейности.
ДиапClЗОН метода амплификации нуклеиновых
кислот B19V оценивают при изучении сходи
мости и межлабораторных сличениях путем
испытаний повторностей разведенных образ
цов. Определяют интервал, расположенный
между верхней и нижней концентрациями HY
клеиновой кислоты мишени, который может
быть выражен с приемлемой степенью точно
сти и правильности.
8. РОБАСТНОСТЬ
Робастность аналитическоro метода
мера ero способности выдерживать воздей
ствие малых, но определенных изменений
пара метров метода, являющаяся показате
лем ero надежности при обычном использова
нии. Оценка надежности должна проводиться
в фазе разработки. Она должна показать Ha
дежность аналитической процедуры с учетом
определенных вариаций пара метров метода.
Для случая МАНК небольшие изменения па
раметров метода MorYT иметь решающее
значение. Однако в ходе разработки метода
ero устойчивость может быть продемонстри
рована небольшим изменением KOHцeHTpa
ций peareHToB. например, MgCI 2 , праймеров
или dNTP. Для демонстрации устойчивости
не менее 20 ДНК В19V отрицательных пулов
плазмы, инфицированных ДНК B19V с полу
чением конечной концентрации около rpa
ничноrо значения, должны быть испытаны
с получением приемлемых количественных
значений.
Предотвращение перекрестной контамина
ции должно быть продемонстрировано путем
точноrо детектирования панели из не менее
чем 20 образцов, попеременно заполненной
образцами пулов плазмы без ДНК B19V или
с концентрацией ниже пороroвоro значения
(10 образцов) и пулов плазмы, инфицирован
ных высокими концентрациями ДНК B19V (не
менее 100 кратноro rраничноro значения, 10
образцов ).
9. ОБЕСПЕЧЕНИЕ КАЧЕСТВА
ДЛЯ таких биолоrических испытаний, как
МАНК, имеется вероятность возникновения
специфических проблем, которые MOryT повли
ять как на валидацию, так и на интерпретацию
результатов. Методики испытаний должны быть
четко описаны в форме стандартных операцион
ных процедур (СОП). В них должно указываться:
способ отбора проб (тип контейнеров и т. д.);
приroтовление мини пулов производите
лем (по потребности);
условия хранения до анализа;
четкое описание условий испытания,
включая меры предосторожности против пере
крестной контаминации и деструкции вирусных
нуклеиновых кислот, используемых peareHToB и
стандартных препаратов;
четкое описание используемой аппаратуры;
детальные формулы для вычисления pe
зультатов, включая статистическую обработку.
В качестве удовлетворительной проверки
соответствия системы и надежности аналити
ческой процедуры, коrда бы она ни использcr
валась, может быть рекомендован подходящий
контроль процесса (например, плазма, инфици
2.6.26. Испытание на aHти D антитела в в иммуноzлобулине человека
311
рованная образцом ДНК вируса В 19V, калибро
ванным в Международных единицах в сравнении
с БСП вирусной дНК В19 для метода амплифи
кации нуклеиновых кислот).
Техническая квалификация. Каждой кри
тической части используемоro оборудования
должна соответствовать проrрамма квали
фикации по установке и эксплуатации. После
замены критически важноro оборудования (Ha
пример, термореrуляторов) должна быть ДOKY
ментально подтверждена функциональность
путем проведения параллельноro испытания в
отношении 8 повторных образцов пула плазмы,
инфицированноro ДНК B19V с получением KO
нечной концентрации около rраничноrо зна
чения. Все результаты должны быть приемле
мыми и должны отражать возможности метода
количественноro определения, как было опре
делено при валидации.
Квалификация оператора. В отн::>шении
каждоro оператора, принимающеrо участие в ис
пытании, должна действовать соответствующая
квалификационная проrрамма. Для подтвержде
ния соответствующеro уровня подroтовки каждый
оператор должен провести испытания не менее
8 повторяющихся образцов пула плазмы, инфи
цированноrо ДНК В 19V с получением конечной
концентрации около rраничноro значения, еже
дневно в течение трех дней (т. е. всеro 24 образ
ца). Все результаты должны быть приемлемыми
и должны отражать возможности метода коли
чественноro определения, как было определено
при валидации.
01/2013:20622
2.6.22. АКТИВИРОВАННЫЕ
ФАКТОРЫ СВЕРТЫВАНИЯ
КРОВИ
При необходимости определяют коли
чество присутствующеrо rепарина (2.7.12) и
нейтрализуют ero добавлением, например,
протамина сульфата Р (10 MKr протамина
сульфата нейтрализуют 1 МЕ rепарина). ro
товят разведения испытуемоro образца в co
отношениях 1: 1 О и 1: 100 с использованием
буферН020 раствора триС(2идроксиметил)
аминометана рН 7,5 Р. Ряд пробирок из по
листирола помещают в водяную баню с TeM
пературой 37 ос и прибавляют в каждую из
ПрQбирок по 0,1 мл плазмы с пониженным co
держанием тромбоцитов Р и по 0,1 мл под
ходящеrо разведения образца фосфолипида,
действующеro как заменитель тромбоцитов.
Выдерживают в течение 60 с. В каждую про
бирку прибавляют либо 0,1 мл одноro из разве
дений, либо 0,1 мл буферноro раствора (KOH
трольная пробирка). К содержимому каждой
пробирки немедленно прибавляют 0,1 мл
предварительно HarpeToro до 37 ос раствора
3,7 r/л кальция хлорида Р и в течение 30 мин
от момента первоначальноro разведения из
меряют время между прибавлением раствора
хлорида кальция и формированием crycT
ка. Результаты считают достоверными, если
время коаrуляции в контрольной пробирке co
ставило от 200 с до 350 с.
01/2013:20626
2.6.26. ИСПЫТАНИЕ
НА АНТИ-D АНТИТЕЛА
В иммУноrЛОБУЛИНЕ
ЧЕЛОВЕКА ДЛЯ
ВНУТРИВЕнноrо ВВЕДЕНИЯ
МАТЕРИАЛЫ
3абуференный солевой раствор (3СР). 8,0 r
натрия хлорида Р, 0,76 r натрия 2идрофосфа
та безводН020 Р, 0,2 r калия хлорида Р и 0,2 r
калия 2идрофосфата Р растворяют в воде Р и
доводят до объема 1000 мл этим же раствори
телем. Если полученный раствор необходимо
хранить в течение нескольких дней, прибавляют
0,2 r натрия азида Р с целью предотвращения
микробной контаминации.
Раствор папаина. Папаин серолоrической
степени чистоты из коммерческоro источника,
активность котороro подтверждена.
Эритроциты. Используют пул D поло
жительных эритроцитов от не менее 3 дo
норов, предпочтительно от rрупп OR 2 R 2 .
D положительные эритроциты MoryT также быть
получены от OR 1 R 1 или ОR 1 R 2 доноров. Смеше
ние различных эритроцитов не оценивалось, по
ЭТОМУ не рекомендуется.
Используют пул D отрицательные эритро
циты, предпочтительно от 3 доноров rруппы Orr.
Если доступен только 1 донор rруппы Orr, допу
стимо использовать D отрицательные эритроци
ты только 1 донора.
Клетки промывают ЗСР 4 раза или до тех
пор, пока надосадочная жидкость не станет про
зрачной. Клетки центрифуrируют при 1800 9 в
течение 5 мин с целью уплотнения. Уплотнен
ные клетки обрабатывают раствором папаина в
соответствии с инструкциями производителя.
Эrитроциты хранят не более 1 недели в KOH
сервирующем растворе. Состав приrодноro KOH
сервирующеro раствора:
Натрия цитрат
D rлюкоза
Кислота лимонная
Натрия хлорид
8 r/л
20 r/л
0,5 r/л
4,2 r/л
312
rосударственная фармакопея Республики Беларусь
Инозин
Аденозинтрифосфат (АТФ)
Хлорамфеникол
Неомицина сульфат
0,938 r/л
0,4 r/л
0,34 r/л
0,1 r/л
Микропланшеты. Используют жесткие ми
кропланшеты с V образным дном.
Стандартные образцы. БСП иММУН02ЛО
булина (испытание на aHти O антитела) и
БСП иММУН02лобулина (испытание на aHти O
антитела, отрицательный контроль) опти
мальны для использования в качестве CTaH
дартноrо образца и отрицательноrо контроля
соответственно.
МЕТОДИКА
Испытание, описанное в данном общем
разделе. выполняется при комнатной темпе
ратуре с использованием стандартных pac
творов, растворов отрицательноro контроля и
испытуемых растворов в одно и то же время
и в одних и тех же условиях.
Стандартные растворы и растворы
отрицатеЛЬН020 контроля. Стандартный
образец и отрицательный контроль BOCCTa
навливают в соответствии с инструкциями.
Концентрация иммуноrлобулина G (lgG) co
ставляет 50 r/л в каждом из восстановленных
препаратов. Выполняют 2 KpaTHoe разведе
ние каждоro восстановленноro препарата с
помощью забуференноrо солевоro раствора,
содержащеro альбумин бычий Р KOHцeHTpa
цией 2 r/л, получая раствор, содержащий IgG
в концентрации 25 r/л. rотовят по 7 последо
вательных 2 KpaTHЫX разведений для каждоrо
препарата с помощью забуференноro солево
ro раствора. содержащеrо альбумин бычий Р
концентрацией 2 r/л, для получения разведе
ний до 1/256 (0,195 r/л IgG). Помещают 20 мкл
каждоro разведения в микропланшет.
Испытуемые растворы. Испытуемый обра
зец разводят с помощью забуференноro соле
воro раствора. содержащеro альбумин бычий Р
концентрацией 2 л для получения начальной
концентрации IgG 25 r/л. Для препаратов с KOH
центрацией 50 r/л используют 2 KpaTHoe разве
дение; корректируют степень разведения для об
разцов, концентрация Ig в которых отличается от
50 r/л, таким образом, чтобы начальная KOHцeH
трация иммуноrлобулина в испытуемом образ
це составляла 25 r/л. Такому исходному 25 r/л
раствору присваивается номинальный 2 KpaT
i-IЫЙ коэффициент разведения для сравнения со
стандартными образцами, даже если это не OT
ражает истинную кратность разведения, исполь
зованную для получения концентрации 25 л.
rотовят по 7 последовательных 2 KpaTHЫX разве
дений для каждою образца с помощью забуфе
ренноro солевоro раствора, содержащеro альбу
мин бычий Р концентрацией 2 r/л, для получения
разведен ий до 1/256 (0,195 r/л IgG) для cpaBHe
ния со стандартными образцами в одинаковом
диапазоне концентраций JgG. rотовят 2 незави
симых ряда разведений. Помещают по 20 мкл
каждоro разведения в микропланшет.
rотовят 3 % (об/об) суспензию D положи
тельных (предпочтительно OR 2 R 2 , но OR 1 R 2
или OR 1 R 2 MorYT быть использованы) и
D отрицательных (Orr) эритроцитов, обрабо
танных папаином, в забуференном солевом
растворе, содержащем альбумин бычий Р
концентрацией 2 r/л. Прибавляют по 20 мкл
D положительных эритроцитов к 1 ряду раз
ведений каждоro испытуемоro образца, CTaH
дартноrо образца и отрицательноro KOHTpO
ля и по 20 мкл D отрицательных эритроцитов
к друroму ряду разведений каждоro испытуе
моro образца, стандартноro образца, отрица
тельноro контроля. Содержимое планшетов
перемешивают путем встряхивания вшейкере
в течение 1 О с.
Центрифуrируют при 80 9 в течение 1 мин
для уплотнения клеток. Планшет располаrа
ют под уrлом приблизительно 700. Снимают
показания не менее чем через 3 мин и более
после тоro, как эритроциты были добавле
ны в ячейки, содержащие отрицательный KOH
троль, и ячейки, в которые были добавлены
D отрицательные эритроциты. Аrrлютинация в
виде «кнопки» на дне ячейки свидетельствует о
положительном результате. Свободные клетки
свидетельствуют об отрицательном результате.
Фиксируют конечную точку титрования в
виде обратной дроби фактора наибольшеro
разведения, при котором отмечается положи
тельный результат. Отрицательный контроль
должен иметь титр не выше 2, в противном
случае необходимо провести проверку каче
ства реактивов и условий испытания. Титр ис
пытуемоrо образца не должен превышать титр
стандартноro образца при условии, что все
препараты титруются в концентрации 25 r/л.
01/2013:20627
2.6.27. МИКРОБиолоrИЧЕСКИЙ
КОНТРОЛЬ КЛЕТОЧНЫХ
ПРОДУКТОВ
Для данноro испытания была доказана еro
предпочтительность использования для опред
ленных клеточных продуктов вместо испытания
на стерильность (2.6.1), так как данное испыта
ние обладает лучшей чувствительностью, имеет
более широкий диапазон применения и пров
дится быстрее. Данное испытание применяется
вместо испытания на стерильность (2.6.1) в слу
чаях, Korдa это предусмотрено частной статьей.
Испытание выполняется ручным способом или с
использованием автоматической системы.
2.6.27. Мuкробuолоzическuй контроль клеточнЬ/х продуктов
313
ОБЩИЕ МЕРЫ ПРЕДОСТОРОЖНОСТИ
Испытание про водится в асептических усло
виях в соответствии с действующими правилами
обращения с потенциально инфекционным Ma
териалом.
Предпринимаемые меры предосторожности
по предупреждению контаминации не должны
оказывать влияния на любые микроорrанизмы,
которые должны быть обнаружены при данном
испытании. Испытание выполняется в рабо
чих условиях, которые реryлярно проверяются
путем отбора проб в рабочей зоне и проведения
соответствующих контролей.
ИСПЫТАНИЕ НА РОСТОВЫЕ СВОЙСТВА
Используют по крайней мере 2 подходящие
питательные среды обоrащения (например, KpO
вяные питательные среды), предназначенные
для обнаружения rрибов и аэробных и анаэроб
ных бактерий.
Стерильность каждой серии среды под
тверждают инкубированием репрезентативных
контейнеров при температуре от З5 ос до 37 ос
в течение не менее 7 дней. Каждая серия среды
испытывается производителем и/или пользо
вателем на наличие ростовых свойств путем
засева контейнеров с каждой питательной
средой 1 o 100 жизнеспособными микроорrа
низмами штамма каждоro вида, приведенными
в таблице 2.6.27. 1, в двукратной повторности.
Контейнеры инкубируют при температуре от
35 ос до 37 ос или в течение 7 дней при исполь
зовании автоматических систем обнаружения
микроорrанизмов, или в течение 14 дней при
визуальной оценке роста микроорrанизмов. Ис
пытуемые среды отвечают требованиям, если
во всех засеянных контейнерах наблюдается
четкий рост микроорrанизмов в течение указан
ноro периода.
Таблица 2.6.27. 1
МиКрООр2анизмы, используемые в испытании
на ростовые свойства
Среда для аэробных бактерий
Staphy/ococcus Например, АТСС 6538,
aureus С/Р 4.83, NCTC 10788,
NC/MB 9518
! Bacillus subtilis Например, АТСС 6633,
I С/Р 52.62, NC/MB 8054
Pseudomoпas Например, АТСС 9027,
aerugiпosa NC/MB 8626, С/Р 82.118
Сапdidа a/bicaпs Например, А ТСС 10231,
I /Р48.72, NCPF3179
I Aspergi1lus Например, АТСС 16404,
brasifieпtis /Р 1431.83, /М/149007
! Среда для анаэробных бактерий
i Cfostridium Например, АТСС 19404,
I sporogeпes С/Р 79.3, NCTC 532 или
I АТСС 11437
i
,
i Bacteroides Например, АТСС 25285,
i fragifis С/Р 77 .16, NCTC 9343
ВАЛИДАЦИЯ МЕТОДИКИ
Необходимость валидации методики в при
сутствии типа испытуемоro образца должна
определяться с учетом типа испытуемоro об
разца, метода ero получения, используемоro
объема инокулята и типа используемой в испы
тании системы. Если не обосновано и YCTaHOB
лено иначе, используемая система валидиру
ется в отношении специфичности (отсутствие
ложноположительных результатов), чувстви
тельности (предел обнаружения) и воспроиз
водимости. В процессе валидации, особенно
при определении предела обнаружения, испы
тание проводится с использованием образца,
в который специально добавлены в различном
количестве следующие микроорrанизмы, BЫ
бранные на основании вероятной контамина
ции и условий роста:
Aspergillus brasilieпtis, например, АТСС
16404, /Р 1431.83, /М/149007;
Baci1lus subtilis, например, АТСС 6633, С/Р
52.62, NC/MB 8054;
Сапdidа a/bicaпs, например, АТСС 10231, /Р
48.72, NCPF 3179;
C/ost,idium sро,оgепеs, например, АТСС
19404, С/Р 79.3, NCTC 532 или АТСС 11437;
Propioпibacte,ium acпes, например, А ТСС
11827;
Рsеиdотопаs аеrиgiпоsа, например, АТСС
9027, NC/MB 8626, С/Р 82.118;
Staphy/ococcus aureus, например, АТСС
6538, С/Р 4.83, NCTC 10788, NC/MB 9518
Streptococcus руоgепеs, например, АТСС
19615, С/Р 1042.26, NC/MB 1З285;
Yersiпia eпteroco/itica, например, АТСС 961 О,
CIP 80.27, NCTC 12982.
Перечень микроорrанизмов может быть из
менен в зависимости от происхождения клеток
и предварительно обнаруженных микроорrаниз
мов или микроорrанизмов, связанных со специ
фическим типом клеток.
MoryT быть также использованы друrие под
ходы к валидации методики, например, проведе
ние межлабораторноro сравнительноro испыта
ния.
ИССЛЕДОВАНИЕ ИСПЫТУЕмоrо
ОБРА3ЦА
Образец. Испытание про водится на pe
презентативном образце, включая клетки и/
или среду. Образец добавляется к питатель
ной среде сразу после ero отбора. Если обра
зец не используется сразу после ero отбора,
он хранится при температуре (5:t3) ос для
предупреждения фаrоцитоза микроорrаниз
мов клетками, присутствующими в опреде
ленных типах продуктов (например, ней
трофилы). Для rемопоэтических продуктов
минимальное количество, которое должно ис
пользоваться в испытании, в зависимости от
общеro объема продукта (V, мл) приведено
ниже.
314
rосударственная фармакопея Республики Беларусь
Общий объем
продукта Объем инокулята
(миллилитры)
V 10 1 % от общеro объема
1sV<10 100 мкл
V<1 Не применимо
Для rемопоэтических продуктов, дЛЯ KOТO
рых требуется разведение до замораживания,
объем инокулята должен увеличиваться с по
мощью фактора разведения. Для друrих клеточ
ных продуктов подходящие минимальные коли
чества определяются в зависимости от объема
или количества доз.
Анализ. Образцы вносятся в контейнеры с пи
тательной средой сразу после отбора и инкубиру
ются при температуре от 35 ос до 37 ос в течение
не менее 7 или 14 дней в зависимости от исполь
зуемой системы оценки роста микроорrанизмов.
Соответствующая порция инокулята добавляется
к среде для инкубирования в аэробных условиях,
а остаток инокулята добавляется к среде для инку
бирования В анаэробных условиях.
НАБЛЮДЕНИЕ И ОЦЕНКА РЕЗУЛЬТАТОВ
Посеянные среды исследуют на наличие
роста микроорrанизмов ежедневно, визуально
или с использованием автоматической систе
мы, и в конце периода наблюдения. Если в Te
чение и к концу периода наблюдения рост ми
кроорrанизмов отсутствует, продукт считается
«микробиолоrически чистым» на данном преде
ле обнаружения (<<неrативная культура»). Если в
испытании. признанном достоверным, наблюда
ется рост микроорrанизмов, продукт считается
«микробиолоrически контаминированным» (<<по
зитивная культура»); обнаруженный микроорrа
низм идентифицируется до соответствующеro
таксономическоro уровня (вид, род), и составля
ется антибиоrрамма.
01/2013:20630
2.6.30. ИСПЫТАНИЕ НА АКТИВАЦИЮ
МОНОЦИТОВ
1. ВВЕДЕНИЕ
Испытание на активацию моноцитов (ИАМ)
используется для обнаружения или количе
ственноro определения веществ, активиру
ющих моноциты человека или моноцитоподоб
ные клетки, и как результат активирующих BЫCBO
бождение эндоrенных медиаторов: например,
провоспалительных цитокинов, например, фак
тора некроза опухолей а (TNFa), интерлейкина
1 р (/L 1 Р) и интерлейкина 6 (/L 6). Данные цитоки
ны принимают участие в патоrенезе лихорадки.
Поэтому ИАМ будет выявлять наличие пироrе
нов в испытуемом образце. ИАМ подходит для
замены испытания на пироrенность на кроликах
после валидации методики для конкретноro про
дукта.
у лекарственных средств, содержащих пи
poreHHbIe вещества, отличные от эндотокси
нов, или провоспалительные вещества, часто
получают очень крутые кривые зависимости
доза эффект по сравнению с эндотоксиновыми
кривыми зависимости доза эффект. Часто наи
большую реакцию при испытании таких про
дуктов получают от неразведенных растворов
испытуемых препаратов или небольших разве
дений испытуемых препаратов. Поэтому пре
.параты, содержащие или которые MorYT co
держать пироrенные вещества, отличные от
эндотоксинов, должны испытываться при диа
пазоне концентраций, которые включают мини
мальное разведение.
В данном разделе описаны три метода.
Метод А. Количественное испытание.
Метод В. Полуколичественное испытание.
Метод С. Сравнительное испытание со CTaH
дартной партией.
Испытание про водится таким образом,
чтобы избежать контаминации пироrенными Be
ществами.
2. ОПРЕДЕЛЕНИЯ
Максимальное допустимое разведение
(МДР) это максимальное разведение образ
ца, при котором может быть определено пре
дельно допустимое пироrенное вещество. МДР
рассчитывают по формуле:
CLC.C
LOD
rдe:
CLC предельная концентрация пироrен
ноro вещества;
С концентрация испытуемою раствора;
LOO предел обнаружения.
Критерием приемлемости для принятия по
ложительноro или отрицательноro решения яв
ляется предельная концентрация пироrенноrо
вещества (CLC), которая выражается в эквива
лентах эндотоксина на миллиrрамм или милли
литр или В единицах биолоrической активности
испытуемоro образца.
CLC рассчитывают по формуле:
к
,
м
rдe:
к пороrовая пироrенная доза эндотокси
на на килоrрамм массы тела;
М максимальная рекомендованная разcr
вая доза лекарственноro средства на килоrрамм
массы тела.
2.6.30. Испытание на активацию моноцитов
315
В случае введения продукта через частые
интервалы или постоянно инфузионно, М
это максимальная общая доза, введенная в
течение одноro часовоro периода. В случае,
коrда для продукта установлена предельная
концентрация эндотоксина (ELC), CLC будет
такой же, что и ELC, если не указано иначе. В
этом случае концентрация испытуемоro раст
вора выражается в мr/мл, если предельное
содержание эндотоксина установлено в Mac
совых единицах (ME/Mr); в Ед/мл, если пре
дельное содержание эндотоксина выражено в
пересчете на единицу биолоrической активно
сти (МЕ/Ед); в мл/мл, если предельное coдep
жание эндотоксина выражено в объемных еди
ницах (МЕ/мл).
Эквиваленты эндотоксина это значе
ния концентрации пироrенноro вещества, BЫ
численные по кривой доза эффект стандарта
эндотоксина (метод А) или оцененные путем
сравнения реакций со стандартными paCTBopa
ми эндотоксина (метод В). Исходный CTaHдapT
ный раствор эндотоксина roтовят из CTaHдapT
ноro образца эндотоксина, калиброванноro
относительно Международноro стандартноro
образца, например, БСП стандартН020 образ
ца эндотоксина.
Пороroвое значение рассчитывают по фор
муле:
х+Зs,
ще:
х среднее значение 4 повторов для peaK
ций контрольноro опыта (Ro);
s стандартное отклонение 4 повторов для
реакций контрольноro опыта (Ro)'
Пороroвое значение выражается в COOTBeT
ствующих единицах полученных данных.
Предел обнаружения (ПО) определяется с
использованием кривой стандарта эндотоксина.
ПО это концентрация эндотоксина, COOTBeT
ствующая пороroвому значению. Для целей дaH
ноro испытания ПО выражается как эквиваленты
эндотоксина на миллилитр.
3. ОБЩАЯ ПРОЦЕДУРА
Испытуемый раствор инкубируют с источ
ником моноцитов человека, или с клетками MO
ноцитов человека, например, с rепаринизи
рованной периферической кровью человека,
отобранной предпочтительно не более чем за 4 ч
до испытания, или моноцит содержащей фрак
цией данной крови, например, мононуклеарные
mетки периферической крови человека {РВМС),
выделенные, например, путем центрифуrиро
вания в rрадиенте плотности, или с линией MO
ноцитоподобных клеток (моноцитов) человека.
rепаринизированная периферическая кровь че
lЮВека обычно разводится питательной средой
-ли физиолоrическим раствором, например, до
IOНцентрации от 2 % до 50 % (об/об) (конечная
концентрация). Обычно в испытании использу
ют РВМС или линии моноцитоподобных клеток
в питательной среде с добавлением или плазмы
донора, или АВ сыворотки с окончательной плот
ностью клеток 0,1 1,0'10б клеток/ячейка, про
бирка или друroй контейнер. При использовании
линий моноцитоподобных клеток АВ сыворотку
можно заменить на бычью эмбриональную cы
воротку, инактивированную наrреванием. Куль
тура клеток roтовится при температуре (37:1:1) ос
в подходящей атмосфере, например, увлаж
ненном воздухе с 5 % С0 2 . Устойчивость куль
туры должна быть достаточной для накопления
выбранных маркеров присутствия пироrенно
ro вещества. Реакцию выбранноro маркера, Ha
при мер, провоспалительных или пироrенных
цитокинов, сравнивают с реакциями на CTaH
дартный эндотоксин или на стандартную партию
испытуемоro лекарственноro средства. Выбран
ная реакция калибруется с использованием co
ответствующеro стандарта.
4. ОБОРУДОВАНИЕ
ВСЮ стеклянную посуду и друryю аппарату
ру, устойчивую к наrреванию, депироrенизируют
в стерилизационном воздушном шкафу с исполь
зованием валидированной процедуры. Широко
используемые минимальное время и температу
ра составляют 30 мин и 250 ос соответственно.
Если используется пластиковое оборудование,
в частности микропланшеты и наконечники для
автоматических пипеток. необходимо убедиться,
что данное оборудование не содержит обнару
живаемых пироrенов и не влияет на испытание.
5. ИСТОЧНИКИ КЛЕТОК
И КВАЛИФИКАЦИЯ
5 1. ЦЕЛЬНАЯ КРОВЬ
Цельную кровь получают от доноров или от
пулов цельной крови, которые квалифицируют
ся в соответствии с требованиями, описанными
в разделах 5 3 и 5 4 соответственно.
5 2. МОНОНУКЛЕАРНЫЕ КЛЕТКИ
ПЕРИФЕРИЧЕСКОЙ КРОВИ (РВМС)
РВМС выделяют из крови, полученной от
доноров или пулов цельной крови, которые
квалифицируются в соответствии с требовани
ями, описанными в разделах 5 3 и 5А COOTBeT
ственно.
5 3. КВАЛИФИКАЦИЯ ДОНОРОВ
Доноры, У которых берут кровь, должны co
ответствовать ниже приведенным квалифика
ционным критериям наряду с друrими действу
ющими требованиями в отношении соrласия
на процедуру, здоровья, безопасности и этиче
скими принципами. Доноры должны описывать
себя как «здоровые», «не страдающие любыми
бактериальными и вирусными инфекциями» и
«не имеющие симптомов любой инфекции в Te
316
rосударственная фармакопея Республики Беларусь
чение периода не менее 1 недели перед сдачей
крови». Доноры не должны принимать HeCTepo
идные противовоспалительные лекарственные
средства в течение 48 ч перед сдачей крови и
стероидные противовоспалительные лекар
ственные средства в течение 7 дней перед
сдачей крови. Физические лица не MOryт быть
донорами, если им были назначены иммуноде
прессанты или друrие лекарственные средства,
которые влияют на продукцию выбранных пока
зателей. Донорская кровь должна быть обследо
вана на инфекционные маркеры в соответствии
с национальными требованиями к трансфузион
ной медицине.
5 4. КВАЛИФИКАЦИЯ КЛЕТОК,
ОБЪЕДИНЕННblХ ОТ РЯДА ДОНОРОВ
Пулы (цельной крови или компонентов
крови, например, РВМС) должны состоять из
порций крови от минимум 4 индивидуальных дo
норов, но предпочтительно от 8 и более ДOHO
ров; пулы формируются путем отбора от каж
доrо пакета донорской крови приблизительно
одинаковых объемов крови или клеток от при
близительно одинаковоro объема крови. Для
квалификации объединенных в пул клеток дей
ствуют следующим образом: в течение 4 ч с MO
мента сбора крови строят кривые доза ответ
для пула с использованием стандартных paCTBO
ров эндотоксина в не менее чем в 4 x разведе
ниях, например, в диапазоне от 0,01 МЕ/мл до
4 МЕ/мл. Кривые доза ответ должны отвечать 2
критериям для стандартной кривой, описанным
в разделе 6 1.
5 5. КВАЛИФИКАЦИЯ
КРИОНОСЕРВИРОВАННblХ КЛЕТОК
Источник клеток, предназначенных для ис
пользования в ИАМ, например, цельная кровь че
ловека, компоненты крови, такие как РВМС или
линии моноцитоподобных клеток. может быть
криоконсервированным. Пулы криоконсервиро
ванных клеток получают объединением перед
замораживанием, или объединением единичных
криоконсервированных донорских порций сразу
же после оттаивания. Пулы должны состоять из
порций крови от минимум 4 индивидуальных дo
норов, но предпочтительно от 8 или более дo
норов; пулы формируются путем отбора прибли
зительно одинаковых объемов крови или клеток
от приблизительно одинаковоrо объема крови.
Квалификация криоконсервированной крови
или клеток выполняется сразу же после оттаива
ния (и объединения, в случае необходимости):
кривые доза ответ для криоконсервированных
клеток и крови должны отвечать 2 критеf)ИЯМ для
стандартной кривой, описанным в разделе 6 1.
5 6. НЕПРЕРblВНАЯ ЛИНИЯ
МОНОЦИТОПОДОБНblХ КЛЕТОК
Непрерывная клеточная линия моноцитов
(моноцитоподобных клеток) человека постоян
но культивируется для тоro, чтобы rарантиро
вать наличие соответствующеro субстрата для
ИАМ. ДЛЯ оптимизации метода MOryT использcr
ваться клоны, полученные от клеточной линии.
Клетки должны храниться в асептических услcr
виях и реryлярно испытываться на заражение
микоплазмой. Дополнительно клетки должны pe
ryлярно проверяться на подлинность (например,
время удвоения, морфолоrию и функцию) и CTa
бильность.
Клетки должны содержаться в асептиче
ских условиях и реryлярно контролироваться
на наличие микоплазм. Дополнительно клетки
должны реryлярно проверяться на идентичность
(т. е. время удвоения, морфолоrия и функции)
и стабильность. Функциональная стабильность
клеточной линии оценивается путем наблюде
ния за ее состоянием с учетом количества пас
сажей в процессе повседневных испытаний. Кри
терии функциональной стабильности должны
быть установлены и MorYT включать критерии
роста, максимальную реакцию, полученную в
испытании, фоновый шум и экспрессию рецеп
тора. Экспрессия рецептора может проверяться
с помощью специфических лиrандов, например.
липополисахарида (ЛПС) для tоJ/ подобноro pe
цептора 4 (ТLR4), липотейхоевой кислоты (LTA)
ТоJ/ подобноro рецептора 2 (ТLR2), синтетиче
скоro бактериальноro липопротеина для TLR2
TLR1 или синтетическоro бактериальноro липо
протеина для ТLR2 ТLR6.
6. ПРЕДВАРИТЕЛЬНЫЕ ИСПЫТАНИЯ
ДЛЯ обеспечения точности и ДOCTOBepHO
сти испытания проводят предварительные ис
пытания, в которых проверяют выполнение кри
териев для стандартной кривой, отсутствие
взаимодействия растворов в ходе испытания,
способность методики обнаруживать эндоток
сины и пироrенные вещества, отличные от эн
дотоксинов, отсутствие влияния растворов на
детектирующую систему.
6 1. КРИТЕРИИ ДОСТОВЕРНОСТИ
СТАНДАРТНОЙ КРИВОЙ
ИЗ стандартноro раствора эндотоксина ro
товят не менее 4 концентраций эндотоксина
для построения стандартной кривой. Прово
дят испытание, используя не менее 4 повторов
каждой концентрации стандартноrо раствора
эндотоксина.
Базовое высвобождение выбранноro Map
кера (контрольный уровень) в отсутствие дo
бавленноro стандартноro раствора эндотоксина
должно быть оптимизировано до наиболее низ
коro возможноro уровня.
Для стандартной кривой должны выполнять
ся 2 критерия приемлемости:
реrрессионная зависимость ответов (COOT
ветственно преобразованных при необходимcr
сти) от лоrарифма дозы должна быть статист
чески значима (р<О,01);
2.6.30. Испытание на активацию моноцитов
317
реrрессионная зависимость ответов от
лоrарифма дозы не должна существенно OT
личаться от линейности (р>0,05). При aHa
лизе 4 факторной лоrистической кривой Ha
блюдаемая кривая не должна существенно
отличаться от теоретической кривой, рассчи
танной с помощью стандартных статистиче
ских методов (см. статью 5.3. Cтaтиcти
ческий анализ результатов биОЛ02ических
испытаний и тестов).
6 2. ИСПЫТАНИЕ НА МЕШАЮЩИЕ
ФАКТОРЫ
ДЛЯ обеспечения достоверности испыта
ния проводятся предварительные испытания,
подтверждающие отсутствие влияния испы
туемоro раствора на результаты испытания.
Валидация аналитической методики требует
ся при внесении любых изменений в экспери
ментальные условия, которые способны по
влиять на результаты испытания. Используя
подходящий растворитель, roтовят разведе
ния испытуемоro лекарственноro средства с
коэффициентом rеометрической проrрессии
(1 :2, 1:4 и т. д.), не превышающие МДР. rOTO
вят такие же разведения испытуемоro лекар
ственноro средства и прибавляют эндоток
син, в концентрации, равной или близкой к
середине стандартной кривой (Метод А) или
равной удвоенному значению ПО (Метод В),
или используют растворитель, содержащий
добавленный эндотоксин в концентрации,
равной или близкой к середине стандартной
кривой (МетодА) или равной удвоенному зна
чению ПО (Метод В). Проводят испытания
полученных рядов разведений параллель
но в одном эксперименте. Для вычисления
концентрации эквивалентов эндотоксина в
каждом растворе используют стандартную
кривую. Вычисляют среднее значение полу
ченной концентрации раствора эндотокси
на, содержащеrо добавленный эндотоксин,
вычитанием средней концентрации эквива .
лентов эндотоксина в растворе (при ero Ha
личии) из средней концентрации раствора
эндотоксина, содержащеro добавленный эн
дотоксин.
Считают, что испытуемый раствор не co
держит мешающих факторов, если в усло
виях испытания вычисленная концентрация
эквивалентов эндотоксина, добавленноro к
испытуемому раствору, находится в преде
лах 50 200 % от известной концентрации
добавленноrо эндотоксина после вычитания
концентрации эндотоксина, обнаруженной в
растворе, не содержащем добавлечноro CTaH
дартноrо эндотоксина. Если испытание не OT
вечает этому критерию, следует провести ис
пытание Методом С. В методе С испытания
проводят на 3 разведениях раствора испыту
емоro лекарственноro средства: наибольшая
концентрация (наименьший фактор разведе
ния), при котором наблюдается наибольший
выброс выбранноrо маркера, и двукратные
разведения непосредственно больше или
меньше выбранной концентрации. Так как
концентрация испытуемоro лекарственноro
средства, при которой наблюдается наиболь
ший выброс выбранноro маркера, может за
висеть от особенностей донора клеток/крови,
а также серии клеток, должна быть проведе
на валидация методики в отношении исполь
зуемоro субстрата на трех независимых ис
пытаниях с использованием в каждом из них
клеток от различных доноров. Наибольшая
концентрация (наименьший фактор разведе
ния), при которой реrистрируется высвобож
дение наибольшеro количества выбранноro
маркера у большинства доноров и 2 KpaTHыe
разведения непосредственно ниже и выше
выбранноrо разведения, считается валиди
рованной для дальнейшеrо выполнения ис
пытания. Если наибольшее высвобождение
выбранноro маркера происходит при испыта
нии исходноro испытуемоro раствора, после
дующее испытание должно выполняться с ис
пользованием неразведенноro испытуемоro
раствора, а также испытуемоro раствора, раз
веденноrо в отношениях 1:2 и 1:4 перед ero
добавлением к РВМС. 3 разведения, исполь
зуемые в последующем исследовании, не
должны превышать МДР; коэффициенты раз
ведения для этих 3 растворов обозначаются
как f l' f 2 И f З' После валидации в отношении
субстрата выполняется обычное испытание с
клетками от 4 индивидуальных доноров, или
с единичным пулом или с клетками от 1 пас
сажа линии моноцитоподобных клеток чело
века.
6 3. ВАЛИДАЦИИ МЕТОДИКИ
В ОТНОШЕНИИ ВЕЩЕСТВ АКТИВАТОРОВ
МОНОЦИТОВ, ОТЛИЧНЫХ
ОТ ЭНДОТОКСИНОВ
В предварительных испытаниях также
подтверждают способность выбранной aHa
литической системы обнаруживать помимо
бактериальных эндотоксинов провоспали
тельные или пироrенные вещества, отличные
от эндотоксинов. Для этоro можно использо
вать ранее исследованные архивные серии,
наличие в которых пироrенных веществ, OT
личных от эндотоксинов, было определено на
основании положительных реакций в испы
тании «Пироrенность» на кроликах или раз
вития побочных эффектов у человека. При
отсутствии таких серий лекарственноrо cpeд
ства в предварительные испытания должна
быть включена валидация используемой aHa
литической системы при помощи специфиче
ских лиrандов для tоl1 подобных рецепторов,
например, пептидоrликанов, липотейхоевых
кислот или синтетических бактериальных ли
попротеинов.
318
rосударственная фармакопея Республики Беларусь
6 4. ВЛИЯНИЕ НА СИСТЕМУ
ДЕТЕКТИРОВАНИЯ
После подбора оптимальноro разведения
раствора испытуемоro лекарственноro cpeд
ства для дальнейшеro исследования данное
разведение испытывается на наличие влия
ния на систему детектирования (например,
ИФА, ELlSA) для выбранноro маркера. Раз
личие в определяемых концентрациях серий
разведений стандарта выбранноro маркера в
присутствии и отсутствии испытуемоro лекар
ственноro средства должно находиться в диа
пазоне :t20 %.
7. МЕТОДЫ
7 1. МЕТОД А: КОЛИЧЕСТВЕННОЕ
ИСПЫТАНИЕ
В методе А проводится сравнение peaK
ции испытуемоro лекарственноro средства
с кривой доза ответ стандартноrо раствора
эндотоксина. Концентрация пироrенных Be
ществ в испытуемом лекарственном средстве
не должна превышать CLC.
7-1-1. Процедура испытания
Используя валидированную методику ис
пытания, roтовят растворы, приведенные в
таблице 2.6.ЗО. 1, и культивируют 4 повтора
каждоro раствора с клетками от каждоro из 4
индивидуальных доноров или с единичным
пулом или с клетками от 1 пассажа линии MO
ноцитоподобных клеток человека.
7 1-2. Вычисления и оценка результатов
Все результаты, включаемые в данные
анализа, должны быть связаны с клетками,
для которых выполнены 2 критерия CTaHдapT
ной кривой. Определяемая концентрация эк
вивалентов эндотоксина, рассчитанная путем
вычитания концентрации эквивалентов эндо
токсина в растворе А из концентрации в pac
творе О находится в пределах 50 200 %. Для
каждоro различноro источника клеток, напри
мер, индивидуальноro донора, пула доноров,
клеточной линии, для вычисления KOHцeH
трации эквивалентов эндотоксина в каждом
из повторов растворов А, В и С используется
кривая стандарта эндотоксина R , R4' Ис
пытуемое лекарственное средство отвечает
требованиям испытания для определенноro
источника клеток, если средние KOHцeHTpa
ции эквивалентов эндотоксина, измеренные
в повторах растворов А, В и С с учетом раз
ведений и концентрации, меньше CLC, YCTa
новленноro для испытуемоro J1eKapCTBeHHoro
средства.
7 1 З. Критерии соответствия/несоответ-
ствия лекарственноrо средства
При использовании клеток от индиви
дуальных доноров испытуемое лекарствен
ное средство должно выдерживать испыта
ние, проведенное с клетками от каждоrо из 4
различных доноров. Если испытуемое лекар
ственное средство выдерживает испытание с
клетками от 3 из 4 доноров (клетки 1 донора
исключены, поскольку не отвечают критери
ям проведения испытания или имеют положи
тельную реакцию), испытание продолжают с
клетками от друrих 4 доноров, клетки от KOTO
рых не использовались в первом испытании;
испытуемое лекарственное средство должно
выдерживать испытание с клетками от 7 из
8 различных доноров (то есть допускается
только 1 положительная реакция при исполь
зовании клеток от 8 доноров). Если источник
Таблица 2.6.30. 1
PaCT Добавленный Число
вор Раствор эндотоксин повторов
(МЕ/мл)
А Испытуемый Нет 4
раствор/!
В Испытуемый Нет 4
раствор/2}
С Испытуемый Нет 4
раствор/4}
О Испытуемый Середина 4
раствор/! стандартной
кривой эндо
токсина (R з )
Ro Апироrенный Нет 4
физиолоrич (контроль)
ский раствор
или испыту
мый раство--
ритель
R Апироreнный 4 4 каждой
1
R4 физиолоrич концентрации KOHцeHTpa
ский раствор стандартноro ции
или испыту раствора
мый раство-- эндотоксина
ритель
Раствор А раствор испытуемоro лекарственноro
средства в разведении. обозначенном в таблице как J, при
котором было проведено испытание на мешающие факто
ры, то есть наибольшая концентрация (наименьшее разве
дение), для которой определяемая концентрация эндотокси
на находится в пределах от 50 200 %.
Раствор В 2 KpaTHoe разведение раствора А, не пре
вышающее мдР.
Раствор С 2 KpaTHoe разведение раствора В, не пре
вышающее МДР.
Раствор D раствор А, содержащий стандарт эн
дотоксина в концентрации около середины стандартной
кривой (R з ).
Раствор Ro контроль.
Растворы R, R. растворы стандарта эндотоксина
в концентрациях, использованных в испытании на мешаю
щие факторы.
2.6.30. Испытание на активацию моноцитов
319
моноцитов представляет собой клетки, объе
диненные от ряда индивидуальных доноров,
испытуемое лекарственное средство должно
выдерживать испытание с 1 пулом клеток.
При использовании линии моноцитоподобных
клеток человека испытуемое лекарственное
средство должно выдерживать испытание с 1
пассажем линии клеток.
2.МЕТОДВ.ПОЛУКОЛИЧЕСТВЕННОЕ
ИСПЫТАНИЕ
В методе В проводится сравнение испы
туемоro лекарственною средства со CTaHдap
том эндотоксина. Концентрация пироrенноrо
вещества в испытуемом лекарственном cpeд
стве должна быть не менее CLC. Раствор А
должен быть выбран для принятия решения
о соответствии, если не обоснованно и раз
решено друrое.
7 2 1. Процедура испытания
Используя валидированный метод испы
тания, roтовят растворы, приведенные в Ta
блице 2.6.30. 2. и культивируют 4 повтора
каждоro раствора с клетками от каждоro из 4
индивидуальных доноров, или с единичным
пулом или с клетками от 1 пассажа линии MO
ноцитоподобных клеток человека.
Таблица 2.6.30. 2
PaCT Добавленный Число
вор Раствор эндотоксин повторов
(МЕ/мл)
А Испытуемый Нет 4
раствор/ f
в Испытуемый Нет 4
раствор/ f 1
С Испытуемый Нет 4
раствор/ fo
о Испытуемый Стандарт эн 4
раствоpl f дотоксина
2ЛО дЛЯ ис
пытуемой си
стемы
Е Испытуемый Стандарт эн 4
pacтвop/f, дотоксина
2ЛО для ис
пытуемой си
стемы
F Испытуемый Стандарт эн 4
pacтвop/f 2 дотоксина
2ЛО для ис
пытуемой си
стемы
Ro Апироreнный Нет 4
физиолоrиче-- (контроль)
ский раствор
или испытуе--
мый раство--
ритепь
PaCT Добавленный Число
вор Раствор эндотоксин повторов
(МЕ/мл)
R, Апироrен Стандарт эн 4
ный физио дотоксина
лоrический 0,5ЛО для
раствор или испытуемой
испытуемый системы
раствори
тель
R 2 Апироrен Стандарт эн 4
ный физио дотоксина
лоrический 1.ПО дЛЯ ис
раствор или пытуемой си
испытуемый стемы
раствори
тель
R з Апироrен Стандарт эн 4
ный физио дотоксина
лоrический 2ЛО дЛЯ ис
раствор или пытуемой си
испытуемый стемы
раствори
тель
R4 Апироrен Стандарт эн 4
ный физио дотоксина
лоrический 4ЛО для ис
раствор или пытуемой си
испытуемый стемы
раствори
тель
Раствор А раствор испытуемоro лекарственноro
средства в разведении, обозначенном в таблице как /, при
котором было проведено испытание на мешающие факторы.
Раствор В раствор испытуемоro лекарственно
ro средства в разведении, обозначенном в таблице как /,.
не превышающем МДР, выбранном после рассмотрения
данных, полученных при валидации методики в отношении
субстрата, например 1:2'МДР (то есть 2 KpaTHoe разведение
выше МДР).
Раствор С раствор испытуемоro лекарственно
ro средства в разведении, обозначенном в таблице как /2'
не превышающем МДР, выбранном после рассмотрения
данных, полученных при валидации методики в отношении
субстрата, то есть МДР.
Раствор D раствор А. в который добавлен стандарт
эндотоксина в концентрации 2ЛО для испытуемой системы
(как определено в предварительном испытании).
Раствор Е раствор В, в который добавлен стандарт
эндотоксина в концентрации 2ЛО для испытуемой системы.
Раствор F раствор С, в который добавлен стандарт
эндотоксина в концентрации 2ЛО для испытуемой системы.
Раствор Ro контроль.
Раствор R, стандарт эндотоксина в количестве
О,SЛО для испытуемой системы.
Раствор R, стандарт эндотоксина в количестве 1. ПО
для испытуемой системы.
Раствор R з стандарт эндотоксина в количестве 2ЛО
для испытуемой системы.
Раствор R. стандарт эндотоксина в количестве 4ЛО
LlЛя испытvемой системы.
320
rосударственная фармакопея Республики Беларусь
7-2-2. Вычисление и оценка результатов
Все результаты, которые должны быть
включены в анализ данных, должны быть
связаны с клетками, для которых средние pe
акции на растворы Ro R4 увеличиваются
в проrрессии. Средняя реакция на Ro может
быть равной средней реакции на R 1 . ДЛЯ каж
доro различноro источника клеток, например,
индивидуальноro донора. пула доноров, кле
точной линии, средняя реакция на раствор R 2
должна быть больше положительноro поро
rOBoro значения. Результаты ниже этоro по
ложительноro пороrовоrо значения paCCMa
триваются как отрицательная реакция. Если
средний ответ на R 1 или R 2 превышает по
роroвое значение, реакция на раствор, BЫ
бранный для оценки соответствия/несоответ
ствия neKapcTBeHHoro средства требованиям,
должна быть отрицательной (лекарственное
средство выдерживает испытание).
Для каждоrо отрицательноro раствора
испытуемоrо лекарственноrо средства (А, В
и С) средняя реакция на соответствующий
раствор с добавленным стандартом эндоток
си на (О. Е или F соответственно) сравнива
ется со средней реакцией на R з для опреде
ления определяемой концентрации в % от
ожидаемой. Концентрация пироrенноro веще
ства в испытуемом лекарственном средстве
менее CLC дЛЯ данноro источника клеток,
если раствор испытуемоrо лекарственно
ro средства. выбранный для оценки COOTBeT
ствия/несоответствия, и разведения ниже ero
дают отрицательные результаты и определя
емая концентрация добавленноro в растворы
стандарта эндотоксина находится в диапазо
не от 50 200 %.
7 2 3. Критерии соответствия/несоответ-
ствия лекарственноrо средства
Критерии те же, что и для метода А (см. п.
7 1 3).
7 3. МЕТОД С: СРАВНИТЕЛЬНОЕ
ИСПЫТАНИЕ СО СТАНДАРТНОЙ
ПАРТИЕЙ
В методе С проводится сравнение ис
пытуемоro лекарственноrо средства с вали
дированной стандартной партией этоrо ле
KapcTBeHHoro средства. Стандартная партия
выбирается в соответствии с обоснованны
ми и утвержденными критериями. Данное ис
пытание предназначено для случаев, коrда
испытуемое лекарственное средство оказы
вает заметное мешающее действие, но не
может быть разведен() в диапазоне МДР дЛЯ
устранения мешающеro действия, поскопь
ку данное лекарственное средство содержит
(или предполаrается, что содержит) пироrен
ные вещества, отличные от эндотоксинов. Pe
акции на пироrенные вещества, отличные от
эндотоксинов, MorYT быть более быстрыми и
выраженными, чем ответы на эндотоксин, что
делает необходимым выполнение испытания
в диапазоне разведений, который включает
минимальное разведение. Процедура испы
тания описана ниже и включает пример cpaB
нения испытуемой партии со стандартной
партией.
7-3 1. Процедура испытания
Используя валидированную методику ис
пытания, rотовят растворы, указанные в Ta
блице 2.6.30. 3 и культивируют 4 повтора
каждоro раствора с клетками от каждоro из
4 индивидуальных доноров, или единичноro
пула или с клетками от 1 пассажа линии MO
ноцитарных лейкоцитов человека.
Таблица 2.6.30. 3
Раствор Раствор/фактор Число
разведения повторов
А Раствор стандартной 4
паРТИИ/!1
В Раствор стандартной 4
паРТИИ/!2
С Раствор стандартной 4
партии/!з
О Раствор стандартной 4
паРТИИ/!1
Е Раствор стандартной 4
паРТИИ/!2
F Раствор стандартной 4
партии/! з
G Положительный 4
контроль (стандарт
эндотоксина)
Ro Растворитель 4
(контроль)
Растворы А. В и С это разведения стандартной
партии в степенях разведения /1' /2 и /З' определенных в ис
пытании на мешающие факторы.
Растворы О, Е и F это разведения испытуемоro ле
KapcTBeHHoro средства в степенях разведения /1' /2 и /3'
определенных для стандартной партии в испытании на Me
шающие факторы.
Раствор G это положительный испытуемый контроль
жизнеспособности клеток, он содержит концентрацию стан.
дарта эндотоксина, при которой наблюдается четкий поло.
жительный ответ.
Раствор Ro это растворитель, используемый для раз
ведения испытуемоrо лекарственноro средства; отрицатель
ный контроль.
7 -3-2. Вычисления и оценка результатов
Все результаты, включаемые в анализ
данных, должны быть связаны с клетками, у KO
торых раствор G и хотя бы один из растворов А.
В и С вызывают ответ, превышающий базовый
уровень высвобождения выбранною маркера
2.6.30. Испытание на активацию моноцитов
321
(раствор Ro)' Для каждоro различноrо источни
ка клеток, например, индивидуальною донора,
пула доноров, клеточной линии, используют
стандартную кривую для маркера (калибровоч
ная кривая в двух повторах с отрицательным
контролем и не менее 4 разведен ий стандарта
маркера в rеометрической проrрессии) и вычис
ляют среднюю величину ответов на растворы
A F. Суммируют средние ответы на растворы
А, В и С и средние ответы на растворы О, Е и
F. Делят сумму средних ответов на растворы О,
Е и F на сумму средних ответов на растворы А,
В и С. Испытуемое лекарственное средство OT
вечает требованиям испытания для данноro ис
точника клеток, если результирующее значение
соответствует определенному критерию прием
лемости, не превышающему 2,5.
7 3-3. Критерии соответствия/несоответ-
ствия лекарственноrо средства
Критерии те же, что и для метода А
(см. 7 1 3).
Для более точноro количественноro опреде
ления уровня содержания пироrенноro вещества
методы А, В и С MOryт выполняться С использо
ванием друrих разведений раствора испытуемо
ro лекарственноro средства, не превышающих
МДР.
Следующий раздел приводится для инфор
мации.
Рекомендации
1. ВВЕДЕНИЕ
Испытание на активацию моноцитов (ИАМ)
в первую очередь предназначено для исполь
зования в качестве альтернативноro метода
испытанию на пироrенность на кроликах. ИАМ
позволяет обнаруживать пироrенные и провос
палительные вещества, включая эндотоксины
rрамотрицательных бактерий и пироrенные Be
щества, отличные от эндотоксинов, включая
патоrенно ассоциированные характерные MO <
лекулярные паттерны rрамположительных и
rрамотрицательных бактерий, вирусов и rрибов,
пироrенные биолоrические или химические Be
щества, характерные для продукта и/или обра
зующиеся в процессе производства.
Так как пироrенные вещества, отличные
от эндотоксинов, представляют собой молеку
лы с разнообразными физико химическими xa
рактеристиками, и обычно при рода пироrенно
ю вещества, присутствующеro в испытуемом
лекарственном средстве, неизвестна, уровень
содержания этих веществ выражается или в
единицах эндотоксин эквивалента, полученных
путем сравнения с реакциями на стандарт эн
дотоксина, или путем сравнения со стандартной
партией испытуемою лекарственноro средства.
В ИАМ концентрации маркеров, образую
IЦИхся в результате реакции на стандарт эндо
4. 3>, I I
токсина, обычно разводят приблизительно в 1 О
раз (на 1 лоrарифм), и при испытании продук
тов, содержащих пироrенные вещества, отлич
ные от эндотоксинов (одних или В комбинации
с эндотоксинами), часто получают очень крутые
кривые доза ответ, обычно только в диапазоне
1 или 2 степеней разведений при определении
их способности стимулировать моноциты.
Часто наибольший ответ на такие продук
ты получают при использовании неразведенных
растворов испытуемых лекарственных средств
или небольших разведений растворов испыту
емых лекарственных средств. По этой причине
испытуемые растворы лекарственных средств,
которые содержат или MOryT содержать пиро
reHHbIe вещества, отличные от эндотоксинов,
должны испытываться в диапазоне разведений,
который включает минимальное разведение.
2. МЕТОДЫ
2 1. ИНФОРМАЦИЯ ОТНОСИТЕЛЬНО
ВЫБОРА МЕТОДОВ
Методы А, В и С обычно не применяются в
случае, Korдa само испытуемое лекарственное
средство может активировать высвобождение
из клеток выбранноro маркера или Korдa одной
из примесей в испытуемом лекарственном cpeд
стве является выбранный маркер. В обоих слу
чаях эти обстоятельства должны привести к MO
дификации и валидации выбранноro метода.
Предполаrается, что при валидации выбранноro
метода в отношении субстрата (маркера) будет
определена частота отсутствия реакции на соот
ветствующую комбинацию продукт/пироrенные
при меси и будет обеспечен выбор мер по обеспе
чению достоверности результатов испытаний,
например, скрининr доноров, увеличение числа
доноров, У которых получают клетки для каждо
ro испытания, установление достаточно CTpO
rих критериев соответствия/несоответствия для
максимальноro увеличения вероятности обнару
жения серий продукта, содержащих существен
ное количество пироrенных веществ. Методы А
и В MOryT использоваться, коrда ответы на раз
ведения испытуемоro лекарственною средства
приблизительно параллельны ответам на раз
ведения стандарта эндотоксина. Метод В явля
ется полуколичественным испытанием, которое
может также применяться, Korдa ответы на раз
ведения испытуемоrо лекарственноrо средства
не параллельны ответам на разведения CTaH
дарта эндотоксина.
Метод С, сравнительное испытание со CTaH
дартной партией испытуемоro лекарственною
средства, был разработан для случаев значи
тельной индивидуальной вариабельности pe
акции донорских клеток на определенные KOM
бинации продукт/пироrенные примеси. Следует
отметить, что в то время, как реакция моноци
тов большинства доноров на бактериальный
эндотоксин приблизительно одинакова, ответы
322
rосударственная фармакопея Республики Беларусь
моноцитов отдельных доноров на пироrенные
вещества, отличные от эндотоксинов, MOryт cy
щественно различаться, поэтому возможно
идентифицировать клетки, у которых ответ на
определенные комбинации продукт/пироrенные
примеси отсутствует.
2 2. ВЫЧИСЛЕНИЕ ПРЕДЕЛЬНОЙ
КОНЦЕНТРАЦИИ пироrЕнноrо
ВЕЩЕСТВА
Критерием приемлемости для принятия pe
шения о соответствии/несоответствии являет
ся предельная концентрация пироrенноrо веще
ства (CLC), которая выражается в эквивалентах
эндотоксина на миллиrрамм или миллилитр или
в единицах биолоrической активности испытуе
моro лекарственноro средства. Если предельная
концентрация эндотоксина (ELC) для продукта
установлена, то CLC будет равен ELC, если не
указано иное. CLC выражается в эквивалентах
эндотоксина.
CLC рассчитывают по формуле:
к
,
М
rде:
К пороroвая пироrенная доза эндотокси
на на килоrрамм массы тела;
М максимальная рекомендованная разо
вая доза препарата на килоrрамм массы тела.
в случае введения продукта через частые
интервалы или постоянно инфузионно, М это
максимальная общая доза, введенная в течение
одноrо часовоro периода. Предельная KOHцeH
трация эндотоксина зависит от продукта и пути
ero введения и устанавливается в фармацевти
ческих статьях.
Значения для К представлены в Таблице
2.6.20,4.
Таблица 2.6.30,4
Путь введения К (МЕ эндотоксина на
килоrрамм массы тела)
Внутривенно 5,0
Внутривенно,для 2,5
радиофарма
цевтических ЛС
Интратекальный (в 0,2
полость позвоноч
ноro канала)
Для продуктов с друrими путями введения
предельная концентрация бактериальных эн
дотоксинов обычно определяется на основе pe
зулыатов, полуuенных в процессе разработки
лекарственноro средства.
2 3. ИНФОРМАЦИЯ ОТНОСИТЕЛЬНО
КРИОКОНСЕРВАНТОВ
Влияние криоконсервантов, например, ди
метилсульфоксида (ДМСО), и их остаточных
количеств в размороженных клетках должны
быть оценены: ДМСО оказывает токсичное
действие на клеточные культуры, и, даже если
клетки были тщательно промыты, криоконсер
вирование может изменять свойства клеток,
например, проницаемость клеточных MeM
бран.
2 4. ИСПЫТАНИЕ НА МЕШАЮЩИЕ
ФАКТОРЫ
Если требуется, проводится испытание на
мешающие факторы по крайней мере 3 различ
ных партий испытуемоro лекарственноro cpeд
ства. Испытуемые лекарственные средства, у
которых отмечается значительная вариабель
ность реакций от серии к серии, должны под
верrаться испытанию на мешающие факторы в
рамках каждоro индивидуальноro испытания, то
есть сопутствующей валидации.
Испытание на мешающие факторы в боль
шинстве случаев выполняется на сериях ис
пытуемоro лекарственноro средства, которые
не содержат эндотоксинов и друrих пироrен
ных/провоспалительных веществ, а если это
невозможно, на сериях с минимальным coдep
жанием пироrенных веществ. Если имеется
только 1 серия испытуемоro лекарственноro
средства, валидация должна выполняться на
этой серии в трех независимых испытаниях.
Должны выполняться пара метры точности для
воспроизводимости, например, х50о/0.
3. ЗАМЕНА ИСПЫТАНИЯ
НА пироrЕННОСТЬ НА КРОЛИКАХ
НА АКТИВАЦИЮ МОНОЦИТОВ
Как отмечалось ранее, испытание на акти
вацию моноцитов (ИАМ) прежде всеro предна
значено для использования в качестве альтер
нативы испытанию на пироrенность на кроликах.
В фармакопейных статьях на лекарственные
средства, предназначенные для парентеральнЬ
ro введения, которые MoryT содержать пироrен
ные вещества, содержатся требования проведе
ния испытания на бактериальные эндотоксины
или испытания на активацию моноцитов.
Общие принципы.
В любой фармакопейной статье при Ha
личии требований к содержанию пироrенных Be
ществ указывается только 1 испытание: или ис
пытание на бактериальные эндотоксины, или
ИАМ. Перед включением ИАМ в фармакопей
ную статью требуется подтверждение приroдно
сти одноro из трех методов, описанных в общем
разделе «испытания на активацию моноцитов»
для оценки продукта, являющеroся предметом
данной фармакопейчой статьи.
Необходимая информация предоставлsr
ется производителями. Заинтересованным про-
изводителям предлаrается предоставить любые
данные по валидации, относящиеся к применм-
мости ИАМ для контроля субстанций и лекар-
ственных форм. Такие данные включают ПQA-
2.6.31. МикробиОЛО2ические испытания лекарственных средств
323
робное описание приroтовления образцов, а
также любые процедуры по удалению мешаю
щих факторов. В дополнение требуются любые
доступные данные по параллельному испыта
нию продукта на пироrенность на кроликах, спо
собствующие доказательству тоro, что замена
испытания на пироrенность на кроликах на ИАМ
применима.
4. ВАЛИДАЦИЯ АЛЬТЕРНАТИВНЫХ
МЕТОДОВ
Замена испытания на пироrенность на KpO
ликах на ИАМ или замена метода определения
противовоспалительныxlпироrенных веществ на
друroй метод должна рассматриваться соrласно
положений раздела «Общие сведения» о замене
фармакопейных методик на альтернативные:
#«Испытания И методики определения, приве
денные в Фармакопее, являются официальны
ми методиками, однако по соrласованию с KOM
петентными уполномоченными орrанами MOryт
использоваться и друrие методики при условии,
что они дают результаты, соответствующие фар
макопейным методикам. В случае сомнений или
разноrласий решающей является фармакопей
ная методика»#.
Для валидации методики для ИАМ, отличной
от описанной в фармакопейной статье, предпо
лаrается выполнение следующих работ:
процедура, материалы и реактивы, ис
пользуемые в испытании, должны быть валиди
рованы в соответствии с рекомендациями для
данноro испытания;
присутствие мешающих факторов (и,
если необходимо, процедура по их устранению)
должно испытываться на образцах не менее 3
производственных серий.
ИАМ должен при меняться ко всем новым
продуктам, предназначенным для парентераль
ною введения, которые должны испытываться
на присутствие веществ, активирующих моноци
ты, в соответствии с требованиями Фармакопеи.
01/2013:20631
2.6.31. МИКРОБиолоrИЧЕСКИЕ
ИСПЫТАНИЯ
ЛЕКАРСТВЕННЫХ
СРЕДСТВ РАСТИТЕльноrо
ПРОИСХОЖДЕНИЯ
ДЛЯ BHYTPEHHErO
ПРИМЕНЕНИЯ
Общее количество аэробов (ОКА). Прово
ДЯТ подсчет, как описано в общей статье 2.6.12.
Общее количество rрибов (OKr). Про
водят подсче как описано в общей статье
2.6.12. В отношении продуктов, описанных в
общей статье 5.1.8, с высоким природным co
держанием бактерий может быть использован
декстрозный arap Сабуро, содержащий анти
биотики.
СПЕЦИФИЦИРОВАННЫЕ
микРооРrАНИЗМЫ
ESCHER/CH/A сои
Испытание на отсутствие
При20товление образцов и предвари
тельная инкубация. rотовят образец с ис
пользованием разведения 1:10 из не менее
1 r испытуемоro продукта, как указано в
общей статье 2.6.12, и используют 10 мл или
количество, соответствующее 1 r или 1 мл, для
инокуляции подходящеro количества (опреде
ленноro, как указано в подразделе 3 4 общей
статьи 2.6.13) бульона на основе rидролиза
тов казеина и соевых бобов, перемешивают и
инкубируют при температуре 30 35 ос в Te
чение 18 24 ч.
Селекция и пересев. Контейнер встряхи
вают, переносят 1 мл бульона на основе rи
дролизотов казеина и соевых бобов в 100 мл
бульона Макконки и инкубируют при темпера
туре 42 44 ос в течение 24 8 ч. Пересева
ют на чашку с аrаризованной средой MaKKOH
ки и инкубируют при температуре 30 35 ос в
течение 18 72 ч.
Интерпретация. Рост колоний указывает
на возможное присутствие Е co/i. Результат
подтверждается идентификационными испы
таниями.
Продукт выдерживает испытание, если
подобные колонии не обнаруживаются или
испытания идентификации дают отрицатель
ный результат.
Метод подсчета. Полуколичественный
метод (метод наиболее вероятноro числа).
При20товление образцов и предваритель
ная инкубация. rотовят образец с использовани
ем разведения 1:10 из не менее 1 r испытуемо
ro продукта, как указано в общей статье 2.6.12, и
используют количество, соответствующее 0,1 r,
0,01 r и 0,001 r (или 0,1 мл, 0,01 мл и 0,001 мл),
для инокуляции подходящеro количества (опре
деленноro, как указано в подразделе 3 4 общей
статьи 2.6. 13) бульона на основе rидролизатов
казеина и соевых бобов, перемешивают и ин
кубируют при температуре 30 35 ос в течение
18 24 ч.
Селекция и пересев. Контейнер встряхи
вают, переноr,ят 1 мл бульона на основе rи
дролизатов казеина и соевых бобов в 100 мл
бульона Макконки и инкубируют при темпера
туре 42 4 ос в течение 24 8 ч. Пересева
ют на чашку с аrаризованной средой MaKKOH
ки и инкубируют при температуре 30 35 ос в
течение 18 72 ч.
324
rосударственная фармакопея Республики Беларусь
Интерпретация. Рост колоний указывает
на возможное присутствие Е. соН. Результат под
тверждается идентификационными испытаниями.
Отмечают минимальное количество, KO
торое дает положительный результат, и MaK
симальное количество, которое дает отрица
тельный результат.
Определяют по следующей таблице наи
более вероятное число бактерий.
Результаты для
каждоrо взятоrо ко-
личества продукта НВЧв
s s I L rpaMMe или милли-
литре испытуемоrо
s :t ......s:t
s :t
L...... L...... ! О ...... продукта
.......0 ...... о sg
о . о -
о .0 о
о
+ + + Более 103
+ + Менее 103 И более 102
+ I Менее 102 И более 1 О
i
! Менее 1 О
iРАМОТРИЦАТЕЛЬНЬ1Е БАКТЕРИИ,
ТОЛЕРАНТНЬ1Е К ЖЕЛЧИ
Метод подсчета. Полуколичественный
метод (метод наиболее вероятною числа).
При20товление образцов и предваритель
ная инкубация. rотовят образец с использовани
ем разведения 1: 1 О из не менее 1 r испытуемо
ro продукта. как указано в общей статье 2.6.12,
но используя бульон на основе rидролизатов
казеина и соевых бобов в качестве разбавите
ля. перемешивают и инкубируют при температу
ре 20 25 сс в течение времени, достаточноro
для восстановления бактерий, но недостаточно
ro для значительною увеличения числа микро
орrанизмов (от 2 ч до 3 ч).
Селекция и пересев. Инокулируют подхо
дящие количества обоrатительноrо бульона
Мосселя для энтеробактерий раствором, при
rотовленным, как описано выше, и/или, в зави
симости от предельноrо содержания, предус
MOTpeHHoro для испытуемоro продукта, тремя
из четырех разведений, содержащих COOTBeT
ственно 0,1 r, 0,01 r, 0.001 r и 0,0001 r (или
0,1 мл, 0,01 мл, 0,001 мл и 0,0001 мл) испы
туемоro продукта. Инкубируют при температу
ре 30 35 ос в течение 24 8 ч. Пересевают
на чашку с аrаризованной средой с rлюкозой,
кристаллическим фиолетовым, нейтральным
красным и желчью. Инкубируют при температу
ре 30 35 ос в течение 18 24 ч.
Интерпретация. Рост колоний является
положительным результатом. Отмечают мини
мальное количество, которое дает положитель
ный результат, и максимальное количество, KO
торое дает отрицательный результат.
Определяют по следующей таблице наибо
лее вероятное число бактерий.
Результаты
ДЛЯ каждоrо взятоrо
количества продукта
s s нвч в rpaMMe или
s s миллилитре испыту
s:t s :t
s :t L...... L...... eMoro продукта
L...... L...... ...... О ...... О
...... О 00
...... . о . 00
.0 .0 о . 00
о о .0 о .
о .0
о
+ + + + Более 104
+ + + - Менее 104 И более 103
+ + Менее 103 И более 102
+ Менее 102 И более 1 О
Менее 1 О
SALMONELLA
Испытание на отсутствие
При20товление образцов и предваритель
ная инкубация. rотовят испытуемый продукт, как
указано в общей статье 2.6.12, и используют KO
личество, соответствующее не менее 25 r или не
менее 25 мл для инокуляции 225 мл буферизо
ванной среды на основе пептона и перемеши
вают (например, roмоrенизируют с использова
нием блендера) и инкубируют при температуре
30 35 ос в течение 18 24 ч.
Буферизованная среда на основе
пептона
Калия диrидрофосфат
Динатрия rидрофосфат
додекаrидрат
Натрия хлорид
Пептон
Вода очищенная
1,5 r
9,0 r
5,0 r
10,0 r
1000 мл
Значение рН доводят таким образом, чтобы
после стерилизации оно составляло 7,0:1:0,2 при
температуре 25 ОС. Стерилизуют в автоклаве в
соответствии с валидированной процедурой.
Селекция и пересев. Переносят 0,1 мл бу
феризованной среды на основе пептона в 1 О мл
обоrатительноro питательною бульона Раппапор
та Вассилиадиса для Sа/топе//а и инкубируют
при температуре 30 З5 ос в течение 1 24 ч.
Пересевают на чашку с аrаризованной средой на
основе ксилозы, лизина, дезоксихолата. Инкубиру-
ют при температуре 3 35 ос в течение 18 8 ч.
Интерпретация. На возможное присут
ствие Sа/топе//а указывает рост хорошо раз
витых красных колоний или красных колоний с
черным центром. Присутствие Sа/топе//а под
тверждается в идентификационных испытаниях.
Продукт выдерживает испытание, если KO
лонии описанною типа не обнаруживаются или
испытания идентификации дают отрицательный
результат.
Рекомендуемые растворы и среды описаны
в общей статье 2.6. 13.
2.7.1. Иммунохимuческие методы
325
2.7. БиолоrИЧЕСКИЕ
МЕТОДЫ
КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ
01/2013:20701
2.7.1. ИММУНОХИМИЧЕСКИЕ
МЕТОДЫ
Иммунохимические методы основаны на
селективном обратимом нековалентном связы
вании антиrенов антителами. Эти методы ис
пользуются для обнаружения и количественноro
определения как антиrенов, так и антител. Факт
образования комплекса антиrена с антителом и
количество этоro комплекса MOryт быть опреде
лены различными путями. Положения данноro
общеro метода относятся к иммунохимическим
методам с использованием меченых или HeMe
ченых peareHToB.
Результаты, получаемые иммунохимически
ми методами, зависят от реакционных условий,
а также природы и качества используемых pe
areHTOB. Важно стандартизировать компоненты,
используемые в ходе иммуноанализа и, rдe это
возможно, использовать для количественных
определений Международные стандартные об
разцы.
PeareHTbI, необходимые для мноrих имму
нохимических методов, содержатся в составе
коммерчески доступных наборов, т. е. наборов,
включающих peareHTbI (антиrены или антитела)
и материалы, предназначенные для оценки in
vitro указанной субстанции, а также инструкции
по их надлежащему применению. Наборы ис
пользуются в соответствии с инструкциями про
изводителя; важно убедиться в том, что наборы
являются подходящими для анализа испытуе
мой субстанции с особым учетом их селективно
сти и чувствительности. Всемирной орrанизаци
ей здравоохранения представлено руководство
по использованию иммунохимических наборов
(Тесhпiса/ Report Series 658 (1981».
МЕТОДЫ С ИСПОЛЬЗОВАНИЕМ
МЕЧЕноrо АнтиrЕНА ИЛИ АНТИТЕЛА
В методах с использованием меченых Be
ществ MorYT применяться подходящие метки,
например, ферменты, флуорофоры, люмино
форы И радиоизотопы. Если меткой является
радиоизотоп, метод называют «радиоимму
нолоrическим анализом». Рекомендации по
измерению радиоактивности, приведенные в
статье Радиоактивные фармацевтические
42 За. 1712
препараты, подходят также и для иммуноло
rических анализов с использованием радио
изотопов. Работа с радиоактивными матери
алами должна выполняться в соответствии с
национальным законодательством и между
народными документами по защите от ради
ационной опасности.
МЕТОДЫ С ИСПОЛЬЗОВАНИЕМ
НЕМЕЧЕноrо АнтиrЕНА ИЛИ АНТИТЕЛА
Методыиммунопреципитации
Методы иммунопреципитации включа
ют процессы флокуляции и осаждения. При
смешивании растворов антиrена с соответ
ствующим антителом в подходящих услови
ях реактанты образуют флокулирующие или
преципитационные arperaTbI. Отношение pe
актантов, при котором достиrается мини
мальное время флокуляции или наиболее за
метное осаждение, называют оптимальным
отношением. Такое отношение обычно co
ставляют эквивалентные количества антиrе
нов и антител. Иммунопреципитация может
оцениваться визуально или по CBeTopac
сеянию (нефелометрическое и турбидиме
трическое определение). Увеличение чув
ствительности может быть достиrнуто ис
пользованием в качестве реактантов частиц
(например, из латекса), ПО крытых антиrенами
или антителами.
В методах флокуляции обычно исполь
зуют ступенчатые разведения одноro из pe
актантов, в то время как в иммунодиффузи
онных (ИД) методах разведение достиrается
путем диффузии в rелеобразную среду: дo
стиrаются концентрационные rрадиенты
одноro из реактантов или обоих. Таким обра
зом, в rеле образуются зоны, в которых OTHO
шение реактантов способствует преципита
ции. В то время как флокуляционные методы
выполняются в пробирках, в иммунодиффузи
онных методах используются различные HO
сители: пробирки, пластины, стекла, ячейки
или камеры.
Если система иммунопреципитации co
стоит из одноro антиrена в комбинации с co
ответствующим антителом, система опреде
ляется как простая; если в ней участвуют
родственные, но серолоrически неидентич
ные реактанты, система является сложной, а
если в ней участвуют несколько серолоrиче
ски не связанных друr с друroм реактантов. ее
называют комплексной.
В методах простой диффузии KOHцeH
трационный rрадиент устанавливается только
для одноro из реактантов, диффундирующеro
из внешнеro источника в rелевую среду. co
держащую соответствующий реактант в OTHO
сительно низкой концентрации.
Простая радиальная uммунодиффузия
(ПРИД) представляет собой простой количе
ственный иммунодиффузионный метод. При
326
rосударственная фармакопея Республики Беларусь
достижении равновесия между внешним и
внутренним реактантами круroвая область
преципитации, начинающаяся от места лока
лизации внешнеro реактанта, прямо пропор
циональна количеству HaHeceHHoro антиrе
на и обратно пропорциональна концентрации
антитела в rеле.
В двойных диффузионных методах KOH
центрационные rрадиенты устанавливаются
для обоих реактантов. И антиrен, и антитело
диффундируют из различных участков в rель,
который первоначально был иммунолоrиче
ски нейтральным.
Сравнительные методы двойной диф
фузии используются для количественноro
сравнения различных антиrенов против под
ходящих антител или наоборот. Сравнение
основано на наличии или отсутствии вза
имодействия между образцами преципитации.
MorYT быть выделены реакции идентичности,
неидентичности и частичной идентичности
антиrенов или антител.
Иммуноэлектрофоретические методы
Иммуноэлектрофорез (ИЭ) качествен
ный метод, сочетающий две процедуры: rель
электрофорез и последующая иммунодиффу
зия.
Перекрестный иммуноэлектрофорез
представляет собой модификацию метода ИЭ
и является подходящим как для качественно
ro, так и для количественноro анализа. Первая
часть процедуры представляет собой обыч
ный rель электрофорез. По ее окончании про
тяженную полосу rеля, содержащую разде
ленные фракции, подлежащие определению,
вырезают и переносят на друrую пластину.
Электрофорез во втором направлении про
водят перпендикулярно предыдущему элек
трофоретическому процессу в rеле, содержа
щем относительно небольшую концентрацию
антитела, соответствующеro используемому
антиrену. Зависимость площади COOTBeTCTBY
ющих пиков преципитации от количества co
ответствующеrо антиrена является линейной
для данной концентрации антитела и толщи
ны rеля.
Электроиммунный анализ, часто назы
ваемый ракетным uммуноэлектрофорезом,
представляет собой быстрый метод коли
чественноrо определения антиrенов с заря
дом, отличным от заряда антител, и наоборот.
Выполняют электрофорез антиrена, подле
жащеro определению, в rеле, содержащем
относительно низкую концентрацию COOTBeT
ствующих антител. Испытуемый материал и
разведения стандартноro антиrена, использу
емые для калибровки, вносят в разные лунки
в rеле. В процессе электрофореза появляют
ся миrрирующие пикообразные зоны преци
питации, начинающиеся от лунок. Фронт пре
ципитации становится неподвижным, коrда
количество антиrена перестает быть избы
точным. Зависимость расстояния, пройденно
ro фронтом преципитации, от количества Ha
HeceHHoro антиrена является линейной для
данной концентрации антител.
Обратный иммуноэлектрофорез пред
ставляет собой быстрый метод количествен
ноro определения, позволяющий устанавли
вать концентрационные rрадиенты внешних
антител и антиrенов в электрическом поле в
зависимости от разницы зарядов. Разведения
стандарта для rрадуировки и разведения ис
пытуемоro образца вносят в ряд лунок в rеле,
а в противоположный ряд лунок вносят фик
сированное количество соответствующеro
реактанта. Титр испытуемоro образца может
. быть определен как максимальное разведе
ние, для котороro наблюдается линия преци
питации.
Существует ряд модификаций методов
перекрестноro иммуноэлектрофореза и элек
троиммунноro анализа.
В друrих методах сочетается разделение
антиrенов по размеру молекул и серолоrиче
ским свойствам.
Визуализация и характеризация линий
иммунопреципитации
Эти операции MorYT быть выполнены с ис
пользованием селективных или неселектив
ных красителей, по флуоресценции, введением
ферментных или изотопных меток или друrими
подходящими методами. Для характеризации
в преципитатах веществ небелковой природы
обычно используют методы селективноro OKpa
шивания.
В полупрозрачных rелях, например, на
основе arapa или аrарозы, линии осаждения
четко различимы визуально при условии нали
чия достаточной концентрации каждоro из peaK
тантов.
ВАЛИДАЦИЯ МЕТОДА
Критерии валидации
Количественный иммунохимический метод
является достоверным при выполнении следую
щих условий.
1. Антитела и антиrены не имеют суще
ственных отличий в стандартном и испытуе
мом образце. В случае меченых реактантов
различия меченоrо и немеченоro компонента
соответствующеro реактанта должны быть He
значительными.
2. На результат не оказывает влияние Ma
трица, т. е. любой компонент испытуемоro об
разца или используемые вспомоrательные
вещества, которые в разных образцах MOryT OT
личаться. В образцах MOryT содержаться BЫCO
кие концентрации посторонних белков, солей,
консервантов или веществ, обладающих проте
олитической активностью.
2.7.2. Количественное определение антибиотиков микробиОЛО2ическим методом 327
3. Предел количественноro определе
ния ниже критерия, установленноro в частной
статье.
4. Точность определения достаточна для
выполнения требований, предъявляемых в
частных статьях.
5. Порядок, в котором проводится опреде
ление, не ПрИ80ДИТ к увеличению системных
ошибок.
Методы валидации
Для проверки выполнения этих критериев
разрабатывается процесс валидации. вклю
чающий следующие элементы.
1. Испытание выполняют не менее чем в
трех повторностях.
2. Испытание проводят не менее чем для
трех различных разведений стандартноrо об
разца и трех разведений испытуемых образ
цов, предполаrаемая активность которых aHa
лоrична активности стандартноro образца.
3. Используют рандомизированную схему
количественноro определения.
4. Если испытуемый образец находится в
сыворотке или в смеси с иными компонента
ми, стандартный образец roтовят аналоrич
ным образом.
5. Испытание включает измерение He
специфическоro связывания меченоro peaK
танта.
6. В случае заместительноro иммунноro
анализа:
а) определяют максимальное связывание
(нулевое замещение);
б) разведения должны охватывать весь
диапазон откликов от значений, близких к He
специфическому связыванию, до максималь
Horo связывания, предпочтительно как для
стандартноro, так и для испытуемоrо образца.
СТАТИСТИЧЕСКИЕ РАСЧЕТЫ
Кривые откликов, полученные для испытуе
моro и стандартноro образцов, MorYT быть про
анализированы с использованием методов, опи
санных в разделе 5.3. Статистический анализ
результатов биОЛ02ических испытаний и
тестов.
Существенная степень непараллельности
указывает на то, что между антителом и антиrе
ном в испытуемом и стандартном образцах име
ются различия и результаты испытания являют
ся недостоверными.
В заместительных иммунолоrических коли
чественных определениях не должно быть cy
щественных различий между значениями He
специфическоro связывания и максимальноrо
замещения при высокой концентрации испыту
eMoro и стандартною образцов. Различия MOryT
указывать на наличие эффектов, связанных с
матрицей: инrибирование связывания либо раз
ложение метки.
01/2013:20702
2.7.2. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
АНТИБИОТИКОВ
МИКРОБиолоrИЧЕСКИМ
МЕТОДОМ
Активность антибиотика определяют
путем сравнения степени уrнетения роста чув
ствительных микроорrанизмов под действием
испытуемоro антибиотика и стандартноro об
разца в известных концентрациях.
Стандартные образцы, используемые для
количественноro определения, представляют
собой субстанции, активность которых точно
установлена по отношению к Международно
му стандартному образцу или Международно
му стандартному препарату.
Определение должно выполняться спосо
бом, позволяющим производить проверку дo
стоверности математической модели, на KOTO
рой основан расчет активности. Если выбрана
модель параллельных линий, линии лоrариф
мической зависимости активности от дозы для
испытуемоro и стандартною образцов должны
быть параллельны; они должны быть линейны
в области доз, использовавшихся при вычис
лении. Эти условия должны быть проверены
тестами соответствия для определенной Be
роятности, обычно Р = 0,05.
#Расчеты целесообразно проводить по
схеме латинских квадратов (если при про
ведении анализа методом диффузии в arap
были использованы прямоуrольные кюветы)
или по схеме рандомизированных блоков
(если при проведении анализа методом диф
фузии в arap были использованы чашки Петри
или анализ проводился турбидиметрическим
методом).#
MorYT быть использованы и друrие MaTe
матические модели, например, модель уrло
воro коэффициента, при условии доказатель
ства их достоверности.
Если в частной статье нет друrих указаний.
доверительные интервалы ошибки количе
ственноro определения активности (Р = 0.95)
должны составлять от 95 % до 105 % от oцe
ниваемой активности.
Определение выполняют методом А или
методом В.
А. МЕТОД ДИФФУЗИИ
Питательную среду определенноro соста--
ва расплавляют, засевают суспензией микроор.-
rанизмов, чувствительных к испытуемому aнт
биотику, при подходящей температуре, например.
(4&--------50) ос для веrетативных форм n и (6 70) ос
при использовании суспензии спор#. Количество
суспензии микроорrанизмов выбирают таким об.-
разом, чтобы образовывались четко определе
ные зоны инrибирования требуемоro диаметра при
328
rосударственная фармакопея Республики БеЛ81
концентрациях антибиотика, используемых в опре
делении. Инокулированная среда немедленно раз
ливается по чашкам Петри или по прямоуroльным
кюветам на строro roризонтальной поверхности в
количестве, достаточном для формирования oд
нородноro слоя толщиной от 2 мм до 5 мм. Среда
может также состоять из двух слоев, из которых
только верхний подверrался инокуляции.
Чашки хранят таким образом, чтобы не про
исходило заметноro роста или rибели микроор
rанизмов до их использования, а поверхность
среды была сухой к моменту использования.
Используя растворитель и буферный раст
вор, указанный в таблице 2.7.2. 1, roтовят pac
творы стандартноro образца и испытуемоro aH
тибиотика, имеющие известные концентрации и
предположительно равные активности. PaCTBO
ры наносят на поверхность среды, например, в
стерильных цилиндрах из фарфора, нержаве
ющей стали или друroro подходящеro матери
ала, или в лунки, сделанные в arape. В каждый
цилиндр или лунку должны быть помещены
равные объемы раствора. Кроме тоro можно ис
пользовать стерильные диски абсорбирующей
бумаrи подходящеro качества; диски пропитыва
ют растворами стандартноro образца и paCTBO
рами испытуемоro антибиотика и помещают на
поверхность arapa.
Для оценки достоверности количественно
ro определения используют не менее трех доз
стандартноro образца и трех доз испытуемо
ro антибиотика, имеющих равные предполаrа
емые активности. Предпочтительно использо
вать ряды доз, меняющихся в rеометрической
проrрессии #(например, в соотношении 1:2:4)#. В
рутинных анализах, Korдa линейность системы
продемонстрирована в адекватном количестве
экспериментов с определением по трем значе
ниям, по соrласованию с компетентными opra
нами может считаться достаточным определе
ние по двум значениям. Однако во всех спорных
случаях должно применяться вышеописанное
определение по трем значениям.
Растворы в каждой чашке Петри или пря
моуroльной кювете располаrают статистически
предпочтительным образом, кроме чашек Петри
малоro размера, на которых невозможно раз
местить больше шести растворов; растворы ис
пытуемоro антибиотика и стандартноro образца
чередуют таким образом, чтобы исключить вза
имное влияние более концентрированных pac
творов.
Количественное определение антибиотиков методом диффузии
Таблица 2.7.2. 1
Растворитель Среда и
Стандарт- Буферный конечное
Антибиотик ный обра- ДЛЯ приrотов раствор Микроорrа- значение Температура
ления основ- (рН) низм рН (:!:О,1 ед. инкубации
зец Horo раствора
рН)
# Азитроми Азитроми Буфер NQ 6. рН Буфер NQ 6, Bacil/us NQ9 (35 37) ос
цин цина ФСО 6,0 рН 6,0 subtilis рН 7, 8,0
АТСС 6633
(30 37) ос
Bacillus
subtilis
var. Л 2
1 мл спирта Р Буфер NQ 4, Bacillus NQ9 (35 37) ос
на 10 Mr Ha рН 7, ,0 cereus var. рН 7,8 8,0
вески, затем mycoides
буфер NQ 4 ДО НВ
1000 ЕДlмл
# Ампицил Ампицилли Буфер NQ 1, Буфер NQ 1, Staphy/o NQ11,NQ7+ (35 39) ос
лин на тpи2и рН 6,8 7,0 рН 6,8 7,0 coccus 0,1 % paCT
драта ФСО aureus вор rлюкозы
209 Р рН 6,8 7,0
Амфотери Амфотери Диметил рН 10,5 Saccha F рН 6,1 (35 37) ос
цин В цина В ФСО сульфоксид Р (0,2 М) romyces
cerevisiae
А ТСС 9763
/р 1432 83
# Буфер # Сапdidа # NQ 16 + # (29 31) ос
NQ 1 utilis 0,1 % rлю
рН 6,8 7,0 ЛИА 01 козы
рН 5, 6,O
2.7.2. Количественное определение антибиотиков микробиОЛО2ическим методом 329
Растворитель Среда и
Стандарт- Буферный Микроорrа- конечное Температура
Антибиотик ный обра- для приrотов- раствор значение
зец ления основ- (рН) низм рН (:!:О,1 ед. инкубации
Horo раствора рН)
Бацитрацин Бацитра 0,01 М pacт рН 7,0 Micrococcus A рН 7,0 (35 З9) ос
цинка цина цинка вор кислоты (0,05 М) /uteus
ФСО хлористо NCTC 7743
водородной С/Р 5З, 160
АТСС 10240
# Бензил Бензил Буфер NQ 1 Буфер NQ 1 Staphy/o NQ11,N27+ (35 37) ос
пенициллин пенициллин рН 6,8 7,0 рН 6,8 7,0 coccus 0,1 % rлю
натрия ФСО aureus козы
209Р рН 6,8 7,0
Блеомицина Блеомици Вода Р рН 6,8 Mycoba G pH7,0 (3 37) ос
сульфат на сульфата (0,1 М) cte,ium
ФСО smegmatis
АТСС 607
Ванкомици BaHKOMици Вода Р рН8,О Bacillus A pH8,0 (37 39) ос
на rидрохло на 2идрохло subtilis
рид рида ФСО NCTC 8236
С/Р 52,62
АТС С 6633
# rелиоми rелиомицина 0,1 М раствор 0,1 М pacт Bacillus NQ9 (35 37) ос
цин ФСО натрия 2идpOK вор натрия subtilis рН 7,8 8,0
сида 2идроксида А ТСС 6633
# rентами (eHтaMици Буфер NQ 4 Буфер N2 4 Bacillus NQ 9 (3 37) ос
цИН на сульфата рН 7,8 8,0 рН 7,8 8,0 pumilus рН 7,8 8,0
ФСО NCTC 8241
rентамицина rентамици Вода Р рН8,0 Baci/lus A pH 7,9 (35 39) ос
сульфат на сульфата (0,05 М) pumilus
ФСО NCTC 8241
С/Р76,18
Staphy/o- A pH7,9 (35 39) ос
coccus
epiderтidis
NC/B 8853
С/Р 68,21
АТСС 12228
# rрамици (рамицидина Спирт Вода Р Bacillus NQ 13 (З5 37) ос
дин С СФСО (95 %. об/об) Р cereus var. рН 7,0 7,2
mycoides
537
# Дактиноми ДaKтиHOMи Вода Р Буфер NQ 4 BacilJus N214 (35 37) ос
цин цина ФСО рН 7,8 8,O cereus var. рН 7,8 8,0
mycoides
НВ
Джозамицин Джозамици Метанол Р рН 5,6 BacilJus A рН 6,6 (35 37) ос
на ФСО (см. частную subtilis
статью) С/Р 52,62
А ТСС 6633
NCTC 10400
Джозамици Джозамици Метанол Р рН 5,6 BacilJus A pH 6,6 (35 37) ос
на пропио на пропиона (см. частную subtilis
нат та ФСО статью) С/Р 52,62
А ТСС 6633
NCTC 10400
43 Зак. 1712
ззо
rосударственная фармакопея Республики Беларусь
Растворитель Среда и
CTaHдapT Буферный конечное
Антибиотик ный обра ДЛЯ приrотов- раствор Микроорrа значение Температура
зец ления основ- (рН) низм рН (:tO,1 ед. инкубации
Horo раствора рН)
# Диrидро Ди2uдpcr Буфер NQ 3 Буфер NQ 4 Bacillus NQ 9 (З5 З7) ос
стрептоми стрепrrюмtr рН6,(}........6,2 рН 7.8 ,0 ce,eus var. рН 7,8 ,0
цин цuнa сулЬфа mycoides
та ФСО 537
#Диrидро-- Ди2идpo Вода Р рН 8,0 Bacillus A pH 7,9 (30 37) ос
стреnтомици-- стрептоми (0,05 М) subtilis
на сулЬФат цина суль NCTC 82 З6
фата ФСО С/Р 1.83
Bacillus
subtilis A pH7,9 (30 37) ос
NCTC 10400
С/Р 52.62
А ТСС 6633
# Дикло Дикло Буфер NQ 1 Буфер NQ 1 Staphy/o NQ11,NQ7+ (3 З7) ос
ксациллин ксацилли рН 6.8 7,0 рН 6. 7,0 coccus 0,1 % rлю
на натрия au,eus козы
ФСО 209Р рН 6.8 7,O
# Доксици Доксицикли 0,01 М раствор Буфер NQ 2 Bacillus NQ6+1% (3 37) ос
клин на 2идрохло кислоты хлори рН 5,8.........б,0 subtilis, rлюкозы
рида ФСО стоводородной var. Л 2 рН 6,8 7,0
Канамицин Канамицина Вода Р рН 8,0 Bacillus A рН 7,9 (ЗО 37) ос
моносуль моносульфа (0,05 М) subtilis
фат та ФСО NCTC 10400
С/Р 52,62
А ТСС 6633
Staphy A pH7,9 (35 39) ос
/ococcus
Канамицина aureus
кислый суль NCTC7447
фат С/Р 53,156
АТСС 6538 Р
# Канами Канамицина Вода Р Буфер NQ 4 Bacillus NQ 12 (35 37) ос
цин В моносульфа рН 7,8 ,0 subtilis рН 7,8 8,0
та ФСО А ТСС 6633 NQ 8
рН 7,8 8,0
# Карбе Карбе Буфер NQ 1 Буфер NQ 2 Pseudo NQ 7 (35 37) ос
нициллин нициллина рН 6,8 7,0 рН 5,8.........б,0 moпasaeru рН 6,8 7,O
динатрия giпоsа
ФСО NCTC 2134
# Карми KapMиHO Вода Р Буфер NQ 4 Bacillus NQ5 (35 37) ос
номицин мицина 2и рН 7,8 cereus var. рН 7,8 ,0
дрохлорида mycoides
ФСО 537
2.7.2. Количественное определение антибиотиков мuкробиОЛО2uческим методом ЗЗ1
Растворитель Среда и
CTaHдapT Буферный конечное
для приrотов Микроорrа- Температура
Антибиотик ный обра раствор значение инкубации
ления OCHOB (рН) низм рН (::!:0,1 ед.
зец Horo раствора
рН)
Колистиме Колистu-- Вода Р рН6,0 Bordetella
тат натрия метата (0,05 М) broпchi
натрия ФСО septica
NCTC 8344
СIР 53,157
АТСС4617
Escherichia B pH7,3 (35--------39) ос
co/i
NCIB 8879
СIР 54,127
АТСС 10536
# Леворин Ле80рина Диметил Диметил Caпdida utilis
ФСО сульфоксид Р сульфок ЛИА 01
сид Р до
KOHцeHTpa
ции
100 мкr/мл,
а затем в
буфере NQ 1,
рН 6, 7,0 NQ 16 + 1 %
(Срок rод rлюкозы (29 31) ос
ности pac рН 5'8.....-...6'0
Т80рОВ при
комнатной
температуре
не более
30 мин)
# Линкоми Линкомицu Вода Р Буфер N24 Bacillus N28 (35 37) ос
цин на 2идрохло рН 7,8 ,O subtilis рН 7, ,0
рида ФСО АТСС 6633
# Метаци Метацикли 0,01 М pacт ' Буфер N2 2 Bacillus N26+1% (30 37) ос
клин на 2идрохло вор кислоты рН 5,8.....-...6,0 subtilis, rлюкозы
рида ФСО хлористо var. Л 2 рН 6, 7 ,О
водородной
# Метицил Метицилли Буфер N2 1 Буфер N2 1 Staphylo N211,N27+ (35 37) ос
лин на натрия рН 6,8 7,0 рН 6,8 7,0 coccus 0,1 % rлю
ФСО au,eus козы
209 P рН 6,8 7,0
# Мидека MидeKaMи Метанол Р до Метанол Р Bacillus N29 (35 37) ос
мицин цина концентрации ДО KOH subtilis рН 7,8 8,0
ацетата 500 мкr/мл, а центрации А ТСС 66З3
ФСО затем в буфере 500 мкr/мл,
N27 а затем в
рН4,5 буфере NQ 7
рН4,5
ЗЗ2
rосударственная фармакопея Республики Беларусь
Растворитель Среда и
CTaHдapT Буферный конечное
Антибиотик ный обра ДЛЯ приrотов- раствор Микроорrа- значение Температура
ления OCHOB низм инкубации
зец (рН) рН (:1:0,1 ед.
Horo раствора рН)
# Микоrеп МиК02епти Диметил Диметил Сапdidа utilis NQ 16 + 1 % (29 31) ос
тин на ФСО сульфоксид Р сульфок ЛИА 01 rлюкозы
сид Р до рН 5, 6,0
KOHцeHTpa
ции
50 мкr/мл,
а затем в
буфере NQ 5,
рН 7,8 8,0
(Срок rод
ности pac
творов при
комнатной
температуре
не более
30 мин)
# Мономи Мономицина Вода Р 3 % раствор Bacillus NQ 9 (3 37) ос
цин ФСО калия хлори ce,eus var. рН 7,8 8,0
даР mycoides
537
# Неомицин Неомицина Буфер NQ 4 Буфер NQ 4 Bacillus NQ 9 (35 37) ос
сульфата рН 7 ,8 8,0 рН 7 , 8,0 cereus var. рН 7,8---------8,0
ФСО mycoides
537
Неомицина Неомицина Вода Р рН 8,0 Bacillus Е рН 7,9 (30 37) ос
сульфат сульфата (0,05 М) pumilus
для MиKpO NCTC 8241
биОЛ02иче С/Р76,18
СК020 опре
деления Bacillus Е рН 7,9 (30 37) ос
ФСО subtilis
NCTC 10400
С/Р 52,62
А ТСС 6633
Нетилмицин Нетuлмuцu Вода Р рН 8,02:0,1 Staphy/o A pH7,9 (32 35) ос
сульфат на сульфата coccus
ФСО aureus
А ТСС 6538Р
С/Р 53,156
Нистатин Нистатина Диметил рН 6,0 Caпdida F рН 6,0 (30 37) ос
ФСО формамид Р (0,05 М), co tropicalis
держащий С/Р 1433 83
5 % (об/об) NCYC 1393
дuметил
формами Saccha F рН 6,0 (30 32) ос
даР romyces
cerevisiae
NCYC 87
С/Р 1432 83
АТСС 9763
# Буфер # Сапdidа # NQ 16 + # (29 31) ос
NQ3 uti/is 0,1 % rлю
рН 6,0 ,2 ЛИА 01 козы
рН 5,8 6,0
2.7.2. Количественное определение антибиотиков микробиОЛО2uческим методом ЗЗЗ
Растворитель Среда и
CTaндapT Буферный Микроорrа- конечное Температура
Антибиотик ный обра для приrотов раствор значение
ления основ- (рН) низм рН (:tO,1 ед. инкубации
зец Horo раствора
рН)
# Оксаци Оксацuллuна Буфер N2 1 Буфер N!! 1 Staphy/o NQ11,NQ7+ (3 37) ос
лин натрия ФСО рН 6, 7.0 pH6, 7,0 coccus 0,1 % rлю
au,eus козы
209 P рН 6, 7,O
# Окситетра OKcитeтpa 0,01 М pacт Буфер N!! 2 Baci/lus NQ6+1% (30 37) ос
циклин циклuна 2и вор кuслоты рН5,8.........б,0 subti/is var. rлюкозы
дрохлорuда хлорист<rвo Л 2 рН 6,8 7,0
ФСО дородной
. # Олеандо Олеандо Буфер N2 З Буфер NQ 4 Baci/lus NQ9 (3 37) ос
мицин мицина фос рН 6, .2 рН 7,8 ,0 ce,eus var. рН 7,8 ,O
фата ФСО mycoides
НВ
# Оливоми Олuвомu Стандартный Буфер N9 1 Baci/lus NQ 11, NQ 18 (3 37) ос
цин цин кuслоты образец в 96 % рН 6. 7,0 subti/is рН 6,8 7,O
ФСО спирте Р из АТСС 663З
расчета 5 Mr
в 1 мл, затем
буфер NQ 1,
рН 6, 7,O
# Полимик Полимиксина Буфер NQ З Буфер NQ 5 Bordetella NQ 15 (35 37) ос
синВ В сульфата рН 6,0-------6,2 рН 6,0-------6,2 Ьroпсhi рН 7,o 7,2
ФСО septica
АТСС 4617
# Полимик Полимик Буфер NQ З Буфер NQ 5 Bo,detella NQ 15 (3 37) ос
синМ сина М суль рН 6,0-------6,2 рН 6,0-------6,2 b,oпchi рН 7,0 7,2
фата ФСО septica
АТСС 4617
# Рифампи Рифампи 1 мл диметил Буфер NQ 3 Bacillus NQ 10 + (35 37) ос
цИН цина формамида Р рН 6,0-------6,2 subtilis 0,1 % rлю
ФСО на 1 О Mr HaBe АТСС 6633 козы
ски, затем вода рН 6,0-------6,2
дистиллиро
ванная Р
Рифамицин Рифамицина Метанол Р рН 7,0 Mic,ococcus A рН 6,6 (3 39) ос
натрия натрия ФСО (0,05 М) /uteus
NCTC 8340
С/Р 53,45
АТСС 9341
# Рубомицин Рубомицина Вода Р Буфер NQ 4 Baci/lus NQ 5 (3 37) ос
2идрохлори рН 6,8 7,0 cereus var. рН 7,8 8,O
да ФСО mycoides
537
# Сизомицин Сизомицина Буфер NQ 4 Буфер NQ 4 Bacillus NQ 12, NQ 8 (3 37) ос
ФСО рН 6, 7,0 рН 6,8 7,0 pumilus рН 7,8 8,O
NCTC 8241
Спирамицин Спирамици Метанол Р рН 8,0 Baci/lus A рН 7,9 (30 32) ос
на ФСО (0,05 М) subtilis
NCTC 10400
С/Р 52,62
АТСС 6633
# Стрепто Стрепто Буфер NQ 3 Буфер NQ З Baci/lus NQ 9 (3 37) ос
мицин мицина ФСО рН 6,0-------6,2 рН 6,0-------6,2 ce,eus var. рН 7,8 ,0
mycoides
5З7
ЗЗ4
rосударственная фармакопея Республики Беларусь
I Среда и I
Растворитель I Б Ф
Стандарт- I у ерныи конечное I
Антибиотик ный обра- ДЛЯ приrотов- Микроорrа- значение Температура I
I раствор инкубации !
зец ления основ- Н низм рН (:1:0,1 ед.
i I I Horo раствора (р ) I
I рН) I
I I
Стрепто Стрепто Вода Р рН 8,0 Bacil1us A рН 7,9 (30 37) ос ,
I
мицина суль мицина суль (0,05 М) subtifis r
I , i
фат фата ФСО I NCTC 8236
I С/Р 1,83 I
I
I I
I
Bacillus A рН 7,9 (30 37) ос I
I subtiJis I
NCTC 10400 I
I С/Р 52,62
А ТСС 6633 i
Тейкопланин Тейкоплани рН 6,0 (0,05 М) рН 6,0 Вac;l/us I н (35 37) ос
на ФСО (0,05 М) subti/is рН 7,8 8,0
NCTC 10400
I С/Р 5262 i
А ТСС 6633
# Т етраци Тетрацикли 0,01 М pacт Буфер NQ 2 Bacil1us Ng6+1% (З5 37) ос
клин на 2идрохло вор кислоты рН 5,8 6,0 subti/is var. rлюкозы
рида ФСО хлористоводо- Л 2 рН 6,8 7,0
родной
# Тобрами Тобрамици Вода Р рН 8,0 Bacil1us A рН 7,9 (ЗО 37) ос
цИН ! на ФСО (0,05 М) subtilis
NCTC 10400
С/Р 5262
I А ТСС 6633
# Фенокси Феноксиме Буфер NQ 1 Буфер Ng 1 Staphy/o I NQ 11 + (З 37) ос
метил тил рН 6,8 7,O рН 6,8 7,0 coccus 0,1 % rлю
пенициллин пенициллина aureus козы
ФСО 209 P рН 6,8 7,0
# Флорими Флоримици Вода Р Буфер NQ 4 Bacil1us NQ 8 (35 З7) ос
цИН на сульфата рН 6,в.........7,0 subti/is рН 7,в.........в,0
ФСО А ТСС 66ЗЗ
# Фузидин Фузидие- Стандартный Буфер NQ 3 Bacil1us NQ 10 + (З5 37) ос I
вой кислоты образец в 96 % рН 6.o 6.2 ce,eus var. 0,1 % rлю I
ФСО спирте Риз mycoides козы I
i
расчета НВ рН 6,0 ,2
1 О мr/мл, затем
буфер NQ 4; ис
пытуемый об
разец в буфере
NQ4
рН 7,8 ,0
Фрамицети Фрамицети Вода Р рН8,0 Bacil1us E pH 7,9 (30 37) ос
на сульфат на сульфата (0,05 М) subtilis
ФСО NCTC 10400
С/Р 52,62
А ТСС 663З
Bacil1us Е рН 7,9 (ЗО 37) ос
pumilus
NCTC 8241
С/Р 76,18
# Цефалек Цефалекси Буфер NQ 1 Буфер NQ 1 Baci//us N211,NQ7+ (35 37) ос
син на ФСО рН 6,8 7,0 рН 6,8 7,0 subtilis 0,1 % rлю
А ТСС 6633 козы
рН 6,8 7,0
2.7.2. Количественное определение антибиотиков микробиоло;шческим методом ЗЗ5
Растворитель Среда и
Стандарт- Буферный Микроорrа- конечное Температура
Антибиотик ный обра- для приrотов- рзствор значение
ления основ- (рН) низм рН (:!:О,1 ед. инкубации
зец i Horo раствора
рН)
# Цефалотин Цефалоти . Буфер NQ 1 Буфер NQ 1 Bacillus NQ11,NQ7+ (зs...--..37) ос
I на натрия рН 6,8 7,0 рН 6.8 7,0 subtilis 0,1 % rлю
ФСО АТСС 6633 козы
рН 6,8 7,0
# Хлортетра ! Хлор 0,01 М pacт Буфер NQ 2 Bacillus NQ6+1% (З5 37) ос
циклин тeтpaциK вор кислоты рН 5,8 6,0 subtilis var. rлюкозы
лина 2идpox хлористо во Л 2 рН 6,8 7,O
лорида ФСО дородной .<
# Эритроми Эритроми 1 мл 96 % Буфер NQ 4 Bacillus NQ9 I (35 37) ос
I цин цина ФСО спирта Р на рН 6,8 7,O cereus var. рН 7,8 8,O
10 Mr навески, mycoides
затем буфер НВ
NQ 4 ДО
1000 ЕД/мл
# Эритроми Эритроми Метанол Р рН 8,0 Bacillus A рН 7,9 (ЗО 37) ос
цина эстолат цина ФСО (см. частные (0,05 М) pumilus
Эритромици статьи) NCTC 8241
на этилсук С/Р76.18
цинат Bacillus A рН 7,9 (30 37) ос
Эритромици subtilis
на стеарат NCTC 10400
С/Р 5262
А ТСС 6633
#Рекомендуется следующая последователь
ность внесения стандартноro (5) и испытуемо
ю (И) образцов в цилиндры или лунки каждой
чашки: первыми вносятся растворы CTaHдapTHO
ro и испытуемоro образца с малой концентраци
ей (51' И 1 ), затем со средней концентрацией (52'
И), последними вносят растворы с большими
концентрациями (5 з , Из).
Все растворы стандартноro и испытуемоro
образцов вносят в цилиндры или лунки одной
чашки Петри таким образом, чтобы растворы с
большими концентрациями не соприкасались
между собой. Предлаrаемый вариант закапыва
ния 5, Из 52 U 1 5 з U 2 .# .
Чашки инкубируют при подходящей темпе
рату ре около 18 ч. Чашки MOryT выдерживать
ся до инкубации при комнатной температуре
или, если необходимо, при температуре 4 ос
в течение определенною промежутка BpeMe
НИ, обычно от 1 до 4 ч для протекания диффу
ЗИИ, что уменьшает эффекты вариаций BpeMeH
ных интервалов между нанесением растворов и
улучшает качество кривой реrрессии.
Измеряют диаметры (с точностью не ниже
0,1 мм) или площади кольцевых зон инrибиро
вания (с соответствующей точностью) и вычис
l1Яют активность с использованием подходящих
статистических методов.
В каждом определении используют ДOCTa
1ОЧное для получения требуемой точности KO
-чество повторов. Может быть проведено He
oonько определений, результаты которых
статистически обработаны для получения требу
емой точности и для подтверждения тою, что aK
тивность испытуемоro антибиотика не ниже ми
нимальных требований.
В. МЕТОД ТУРБИДИМЕТРИИ
Подходящую питательную среду с суспен
зией выбранноrо микроорrанизма, обладаю
щею чувствительностью к тестируемому анти
биотику, инокулируют таким образом, чтобы в
условиях определения происходило достаточно
значительное инrибирование микробноro роста.
Количество суспензии выбирают таким обра
зом, чтобы получить после 4 ч инкубации помут
нение, леrко подверrающееся количественной
оценке.
Инокулированную среду используют HeMeд
ленно после ее приroтовления.
Используя растворитель и буферный
раствор, ук?занные в таблице 2.7.2. 2, roто
вят растворы стандартноro образца и испы
туемоro антибиотика, имеющие известные
концентрации и предположительно равные
активности.
Для оценки достоверности количествен
чоrо определения используют не менее трех
доз стандартноro образца и трех доз испыту
емоro антибиотика. Предпочтительно исполь
зовать ряды доз, меняющихся в rеометриче
ской проrрессии. Для получения необходимой
линейности может оказаться необходимым
осуществить выбор из большоro количества
ЗЗ6
rосударственная фармакопея Республики Беларусь
трех последовательных доз, используя COOT
ветствующие дозы для стандартноro обр зца
и испытуемоro антибиотика.
Помещают равные объемы каждоro из pac
творов в идентичные пробирки и прибавляют
в каждую пробирку равные объемы инокули
рованной среды (например, 1 мл раствора и
9 мл среды). Для количественноro определе
ния тиротрицина прибавляют 0,1 мл раствора
и 9,9 мл инокулированной среды.
Одновременно rотовят две контрольные
пробирки без антибиотика, содержащие иноку
лированную среду. В одну из них немедленно
прибавляют 0,5 мл формальде2ида Р. Эти про
бирки используют для настройки оптическоrо
прибора, используемоro для измерения роста.
Все пробирки, распределенные случай
ным образом, методом латинскоro квадрата
или рандомизированноrо блочноrо распреде
ления, помещают в водяную баню или в друroй
подходящий аппарат, позволяющий быстро дo
вести температуру всех пробирок до соответ
ствующей температуры инкубации и поддержи
вать пробирки при этой температуре в течение
3 ч, обеспечивая однородность темпера
TypHoro режима и равное время инкубации.
По окончании инкубации рост микроорrа
низмов останавливают прибавлением 0,5 мл
фОРМ8Льде2ида Р в каждую пробирку или Te
пловой обработкой и измеряют степень MYT
ности, используя подходящий оптический
при бор. Можно использовать метод, позволяю
щий измерять степень мутности в каждой про
бирке по окончании одинаковых периодов ин
кубации.
Рассчитывают активность с использовани
ем подходящих статистических методов.
Линейность преобразованной или непре
образованной зависимости реакции от дозы
часто получается лишь в оrраниченном интер
вале. Именно в этом промежутке необходимо
производить расчет активности. Для подтверж
дения линейности определение проводят на
трех последовательных дозах из этоro интер
вала. В рутинных анализах по соrласованию с
компетентными орrанами определение может
проводиться по двум значениям при условии,
что линейность подтверждена адекватным KO
личеством экспериментов с использованием
трех значений. Однако во всех спорных случа
ях должно применяться вышеописанное опре
деление по трем значениям.
В каждом определении используют ДOCTa
точное количество повторов для получения
требуемой точности. Определение может быть
проведено повторно, и результаты статистиче
ски объединены для получения требуемой точ
ности и подтверждения тоro, что активность
испытуемоro антибиотика не менее минималь
ных требований.
Таблица 2.7.2. 2
Количественное определение антибиотиков турбидиметрическим методом
Раствори- Среда и
тель для Буфер-
Стандартный приrотов- ный Микроорrа- конечное Температу-
Антибиотик образец ления ос- раствор низм значение ра инкуба-
рН (:tO,1 ед. ции
HOBHoro (рН) рН)
раствора
Ванкомицина Staphy/ococ
Ванкомицина 2uдрохлорида Вода Р рН8,0 cus au,eus C pH7,0 (37 39) ос
rидрохлорид ФСО С/Р 53,156
АТСС 65З8 Р
Staphy/ococ
rентамицина rентамицина cus aureus
Вода Р рН 7,0 NCTC7447 C pH7,O (35 37) ос
сульфат сульфата ФСО CIP 53,156
АТСС 6538 Р
Eпterococcu
shi,ae
rрамицuдина CIP 58,55
Метанол Р рН 7,0* АТСС 10541 C pH7,0 (З5 37) ос
ФСО Staphylococ
rрамицидин cus au,eus
А ТСС 65З8 Р
* может оказаться необходимым добавление детерrента для предотвращения
адсорбции материала в ходе ;эазведения, например, полисорбата 80 Р в KOHцeH
трации 0,1 мr/мл
Staphy/ococ
Джозамицина Метанол Р cus aureus
Джозамицин (см. част рН 5,6 С/Р 53,156 C pH8,0 (35 37) ос
ФСО ную статью) АТСС 6538 Р
NCTC 7447
2.7.2. Количественное определение антибиотtJКов мuкр06иолоаическuм методам 337
Раствори Среда и
тель для Б
Стандартный приrотов иый Микроорrа конечное Температу
Антибиотик ра инкуба-
образец ления oc раствор низм значение
HOBHoro (рН) рН (tO,1 ед. ции
раствора рН)
Staphylococcus
Джозамицина Джозамицина Метанол Р aиreus
пропионата (см. частную рН5,6 СIР5З,156 C pH8,0 (35--------37) ос
пропионат ФСО статью) АТСС 6538 Р
NCTC 7447
Канамицина MO Staphy/ococcиs
носульфат Канамицина aureus
моносульфата Вода Р рН8,О NCTC7447 C pH7,0 (35--------37) "С
Канамицина ФСО CIP 53,156
кислый сульфат АТСС 6538 Р
Колисти Escherichia coli
Колистиметат Вода Р рН7,0 NC/B 8666 C pН7,0 (35--------37) ос
метата CIP 2,8З
натрия натрия ФСО
АТСС 9637
Неомицина Staphy/ococcus
Неомицина сульфата для au,eus
сульфат микробиОЛ02и Вода Р рН8,О NCTC 7447 C pH7,O (35--------З7) "С
чеСК020 опреде- С/Р 53,156
ления ФСО АТСС 65З8 Р
Escherichia co/i
Рифамицин Рифамицина Метанол Р рН7,0 NC/B 8879 C pH7,O (З5--------37) ос
натрия натрия ФСО CIP54,127
АТСС 105З6
Staphy/ococcus
Спирамицина au,eus
Спирамицин Метанол Р рН8,0 NCTC7447 C pH7,0 (35--------37) ос
ФСО С/Р 53,156
А ТСС 6538 Р
К/еЬsiе//а рпеи-
Стрептомицина Стрепто- moпiae
мицина Вода Р рН8,0 NCTC 7427 C pH7,0 (35--------37) ос
сульфат сульфата ФСО С/Р53,153
АТСС 10031
rрамицидина 96% 96% Епtе,ососси-
Тиротрицин shi,ae C pH 7,0 З7 ос
ФСО спирт Р спирт Р АТСС 10541
Staphy/ococcus
Фрамицетина Фрамицетина au,eus
Вода Р < рН8,О NCTC7447 C pH 7,0 (35--------37) ос
сульфат сульфата ФСО CIP 53,156
АТСС6538 р
Следующий раздел приводится для инфор
мации.
Рекомендуемые микроорrанизмы
В нижеследующем тексте приводятся peKO
мендуемые микроорrанизмы и условия их при
менения. Moryт также использоваться и друrие
.,икроорrанизмы при условии, что доказана их чув
ствительность к испытуемому антибиотику и они
мoryт использоваться в подходящей среде и при
ПQD,Xодящих значениях температуры и рН. Кон-
центрации используемых растворов должны быть
..юраны таким образом, чтобы в условиях испы
'l8Ния обеспечивал ось наличие линейной зависи-
мости между лоrарифмом дозы и реакцией.
Приrотовление noceBHoro материала. Ба-
cillus cereus var. mycoides; Басi//иs subtilis; Baci//us
pumi/us. Суспензия спор микроорraнизмов, исполь-
зуемая в качестве посевноro материала, roтовится
следующим образом.
Микроорraнизм выращивается при температу-
ре (35--------37) ос в течение 7 дней на поверхности под
ходящей среды, к которой добавлен мараанца (11)
сульфат Р в концентрации 0,001 r/л. Выращенную
культуру, состоящую в основном из спор, смывают
стерильной вадой Р. Суспензию наrpевают при тем-
пературе 70 ос в течение 30 мин и разводят для по
лучения подходящей концентрации спор, обычно
от 10.106 до 100.106 В миллилитре. Суспензия спор
ЗЗ8
rосударственная фармакопея Республики Беларусь
может храниться в течение продолжительноro
времени при температуре, не превышающей 4 ОС.
Суспензия спор также может быть приroтовл&
на путем культивирования орraнизмов в среде С при
температуре 26 ос в течение дней с последу
ющим добавлением в асептических условиях Map
28нца (/Q сульфата Р в количестве, достаточном
для получения концентрации 0,001 r/л, и инкубиро
ванием в течение 48 ч. Суспензию микроскопиру
ют для подтверждения спорообразования (около
80 %) и центрифуrируют. Осадок ресуспендируют в
стерильной ваде Р, получая концентрацию спор от
10.106 до 100.106 в миллилитре, после чеro наrpэва
ют при температуре 70 ос в течение 30 мин. Суспен
зию хранят при температуре, не превышающей 4 ОС.
Вordetella broпchiseptica. Микрорrанизм Bыpa
щивают на среде В при температуре (3 З7) ос в
течение 1 18 ч. Выращенную культуру смывают
стерильной водой Р и разводят до требуемой cтe
пени мутности.
Staphylococcus aureus; К/еЬsiеllа рпеитопiае;
Escherichia coIi, Мicrococcus /uteus; Staphy/ococcus
epideпnidis. Посевной материал roтовят по методу,
описанному выше для В. Ьroпсhisерtiса, но с исполь
зованием среды А и доведением степени мутности
до значения, дающеro, в зависимости от метода,
удовлетворительную зависимость реакции от дозы
в турбидиметрическом определении или четкие
зоны инrибирования подходящеro диаметра.
Saccharomyces cerevisiae; Сапdidа tropica/is.
Микроорraнизм выращивают на среде F при TeM
пературе ( 37) ос в течение 24 ч. Смывают BЫ
ращенную культуру стерильным раствором 9 r/л
натрия хлорида Р. Разводят до требуемой степени
мутности этим же раствором.
#Допустимо использование следующих мето--
дов приroтовления посевноro материала.
Caпdida utilis ЛИА 01. Выращивают в пробир--
ках со скошенной средой NQ 3 в течение 48 ч при
температуре (3Ot1) ОС.
Staphylococcus aureus. Выращивают в пробир
ках со скошенной средой NQ 1 в течение 24 ч при
температуре (36I1) ОС.
Вo,detella broпchiseptica. Выращивают в '1ро--
бирках со скошенной средой NQ 1 в течение
3 36 ч при температуре (З6I1) ОС.
Рsеиdотопаs аеrugiпоsа NCTC 21 34. Выращи
вают на мясо--пептонном бульоне (МПБ) рН 7,2 7,4
в течение 1 20 ч при температуре (36I1) ОС.
Bacillus pumilus, NCTC 8241; Bacillus subtilis
А ТСС 6633; Вacillus cereus var. mycoides НВ; Bacillus
cereus var. mycoides 537.
Для получения суспензии спор тест
микроорraнизмы выращивают на скошенной среде
в пробирках (среда NQ 1 ДЛЯ В. subtilis var. L2' В. pит
i/us NCTC 8241, В. subtilis АТСС 6633, В. се,еиБ var.
mycoides НВ; среда NQ 2 для В. cereus var. mycoides
537), при температуре (36I1) ос В течение 18 20 ч.
Культуру, выращенную в пробирках, смывают
1 О мл стерильноro раствора 9 r/л натрия хло
рида Р засевают ею несколько матрацев с 300 мл
питательной среды со скошенной поверхностью,
(среда NQ 4 для В. pumi/us, В. cereus var. mycoides
537; среда NQ 5 дЛЯ В. subti/is АТСС 6633, В. cereиs
Таблица #2.7.2. 0
Характеристики культуральных свойств тест миКРООР2анизмов
I При росте на плотной При росте на жидкой ' При микроскопи :
Название : питательной среде питательной среде ческом изучении !
тест микро I условия описание условия описание мазка аrаровой :
орrанизма выращи- культуры, окра- ;
вания культуры выращивания культуры шенной по rpaMY i
Среда NQ 1, Однородные по I
(36I1) ос rладкие с pOBHЫ Мясо пептон Равномерное по величине и pac i
Staphy/ococcus 118 20 ч, ми краями коло ный бульон мутнение бу положению в виде I
au,eus ! затем при нии с paBHOMep рН 7,2 7.4 льона без плен rроздьев кокки с I
209 Р I комнатной ной золотистой 18 20 ч ки и слизистоro хо ошо выражен i
i температу пиrментацией осадка на дне нои rрамположи :
I ре 24 ч тельной окраской i
Круrлые KpeMOBO Овльные, иноrда с I
Среда NQ 3. , ro цвета с матовой МПБ, Рост в виде paB отростками, клет
Caпdida utilis (30I1) ос I поверхностью KO рН 7,2 7,4 номерной ки, располо жены i
ЛИА 01 с 1 % rлюкозы мути отдельно, цепоч I
48ч лонии с ровными 18 20 ч и осадка на дне ками или rруппа I
краями ми j
Колонии желто Палочки с закруr I
Среда NQ 1, BaToro цвета, KPy ленными краями, I
(36I1) ос rлой формы влаж
18 20 ч, но блестящие с МПБ, Рост в виде располаrающие .
Bacillus subtilis ся отдельно и цe I
var. Л 2 затем при шаrреневой по рН 7,2 7,4 пленки с ocaд
комнатной верхностью, слеr 18 20 ч ком на дне почками, с хоро: I
температу ка зазубренными шо выраженнои
ре 24 ч краями и припод rр мполож тель-I
нои окраскои
нятым центром j
2.7.2. Количественное определение антибиотиков микробиОЛО2ичеСIШМ методом ЗЗ9
Название
тест-микро-
орrанизма
При росте на плотной
питательной среде
условия
выращи-
вания
I
I
I
I
' 1 ' Bacillus subtilis
I АТСС 6633
I
I
I
l ' Bacillus ce,eus Среда Ng 2.
var. mycoides (36f:1) ос
1537 1 20ч
I
I
описание
культуры
При росте на жидкой
питательной среде
условия
, выращивания
Среда NQ 1, Мелкие cepOBa I МПБ,
I (36f:1) ос тые колонии с зуб I рН 6, 7,0
I 18 20 ч чатым краем 118 20 ч
I
Baci/1us ce,eus Среда NQ 1,
var. mycoides (З6f:1) ос
НВ 1 20ч
Baci/1us pumilus
NCTC 8241
Среда NQ 1.
(36f:1) ос
18 20 ч
Шероховатые ко--
лонии с краем, МПБ,
сформированным I рН 7,в-.......-в,0
из переплетаю 1 20 ч
щихся волокон I
rладкие колонии с
ровными краями и
сферической по
верхностью
Мелкие cepOBaTO
rолубоrо цвета KO
лонии с зазубрен
ными краями и
приподнятым цeH
тром
МПБ,
рН 7,2 7,4
1 20 ч
МПБ,
рН 7,2 7,4
18 20 ч
Мясо
пептон
Ь ный arap Мелкие мутные
roп
АТСС (М ПА), белые слеrка BЫ МПБ,
рН 7,2 пуклые фарфоро рН 7,2 7,4
7,4 видные колон и
(36f:1) ос
18 20 ч
Bo,detella
chiseptica
4617
Pseudoтoпas
ae,ugiпosa
NCTC 2134
МПА,
рН 7,2 7,4
(36f:1) ос
18 20 ч
Широкие расплыв
чатые прозрачные
с неровным краем
колонии
var. mycoides НВ). На матрацах посевы инкубиру
ют при температуре (313f:1) ос в течение 7 сут.
В процессе выращивания проводят микроскопи
ческий контроль культуры. Культуру, содержащую
80 90 % спор, смывают стерильной водой Р.
Полученную взвесь спор проrревают при
температуре от 60 ос до 70 ос в течение 30 мин.
Затем промывают стерильной водой Р при цeH
МПБ,
рН 7,2 7,4
описание
культуры
Рост в виде
пленки с ocaд
ком на дне
Морщинистая
пленка, вся cpe
да прозрачная
без осадков
Равномерное по-
мутнение бу
льона без обра
зования пленки
и осадка
Равномерное по-
мутнение буль
она без образо
вания пленки и
осадка
При микроскопи-
ческом изучении
мазка аrаровой
культуры, окра- ,
шенной по rpaMY i
Тонкие палочки, I
располаrающиеся i
отдельно или цe !
почками, с xopo !
шо выраженной!
rрамположитель
ной окраской
Палочки с закруr
ленными краями,
располаrающие
ся отдельно и цe
почками, с хоро--
шо выраженной
rpa мположитель
ной окраской
Тонкие с закруr
ленными концами
палочки, располо
rающиеся отдель<-
но или короткими
цепочками, с хо--
рошо выраженной
rрамположитель
ной окраской
Тонкие, мелкие
палочки с закру
rленными KOHцa
ми, располаrаю--
щиеся отдельно
или короткими цe
почками, с xopo
шо выраженной
rрамположитель
ной окраской
Одиночные корот
Заметное по кие тонкие палоч
мутнение, c ки с хорошо Bыpa
рая пленка, тя ' женной rрамотри
ryчий осадок цательной OKpa
ской
Заметное помут
нение, толстая
пленка, cpe
да может при
обретать жел
товато зеленую
окраску
Одиночные па
лочки либо корот
кие цепочки с xo
рошо выраженной
rрамотрицатель
ной окраской
трифуrировании до полной прозрачности Haдo
садочной жидкости. Промt-IТУЮ взвесь спор вновь
проrревают в течение 30 мин при температуре от
60 ос до 70 ОС. Взвесь спор хранят при температу
ре от 4 ос до 1 О ос и используют до тех пор, пока
интенсивность роста и четкость зон при опреде
лении антимикробной активности удовлетворяют
предъявляемым требованиям.#
340
rосударственная фармакопея Республики Беларусь
Буферные растворы. Буферные paCTBO
ры с noказатепем рН в интервале от 5,8 до
8,0 roтовят смешиванием 50,0 мл 0,2 М pacт
вора калия диzидрофосфата Р с указанным
в табпице 2.1.2. З объемом 0,2 М растоора
натрия 2идроксида. Разбавляют свежепрVl
roтовленной водой дистиллированной Р до
объема 200,0 мл.
Таблица 2.7 2.. з
рН 0,2 М раствор наmрия zидpoкcидa
5,8 3,72
6,0 5,70
6,2 8,60
6,4 12,60
6,6 17,80
6,8 23,65
7,0 29,63
7,2 35,00
7,4 З9,50
7,6 42,80
7,8 45,20
8,0 46,80
эти буФерные растворы ИСПОЛЬЗуЮтся для
всех микробиолоrических определений, указанных
в табnице 2.7.2. 1, за исключением бnеомицина
сульфата и амфотерицина В.
Буферный раствор рН 6,8 для блеомицина
сульфата roтовят следующим образом: 6,4 r калия
ди2идрофосфата Р и 18,9 r динатрия 2идрофос
фата р растворяют в ваде Р и доводят до объема
1000 мл этим же растворителем.
"Кроме описанных выше буферных растваров
МО2ут исrюльзооаться растворы следующею со--
става:
Буфер NQ 1
Калия ди2идрофосфат Р
Динатрия 2идрофосфат Р
Вода дистиллированная Р
рН раствора
3,6З r
7,13 r
до 1000 мл
(6,9:tO,1 )
Буфер NQ 2
Натрия цитрат Р 20,6 r
Кислота хлористоводородная Р 1,81 r
Вода дистиллированная Р до 1000 мл
рН раствора (5,9:t0,1)
Буфер NQ 3
Калия ди2идрофосфат Р
Динатрия 2идрофосфат Р
Вода дистиллированная Р
рН раствора
7,72 r
1,78 r
до 1000 мл
(6,1:t0,1)
Буфер NQ 4
Калия ди2идрофосфат Р
Динатрия 2идрофосфат Р
Вода дистиллированная Р
рН раствора
0,68 r
10,99 r
до 1000 мп
(7,9:tO,1 )
Буфер NQ 5
Калия ди2,uдpoфосфат Р
Динатрuя 2Uдpофосфаm Р
Вода дистиллированная Р
рН раСТ80ра
Буфер NQ 6
Дuнаmpия 2,идрофосфат Р
Вода дистиллированная Р
рН раствора
Буфер NQ 7
Натрия диzидрофосфат Р
Динатpuя 2Uдрофосфаm Р
Вода дистиллированная Р
рН раствора
З2,Оr
8,0 r
ДО 1000 мл
(6,1IO,1 )
35,8r
до 1 000 мл
(6,0:t0,1 )
O,64r
0,195 r
ДО 1000 мn
(4,51:0,1)
Буферные растворы стерилизуют Hacы
щенным паром под давлением при температуре
121 ос в течение 15 мин.#
Питательные среды. Moryт быть иcnользо
ваны следующие или эквивалентные питатель
ные среды.
Среда А
Пептон
Панкреатический rидролизат казеина
r овяжий экстракт
Дрожжевой экстракт
r люкоза моноrидрат
Arap
Вода
6r
4r
1,5 r
Зr
1 r
15 r
до 1000 мл
Среда В
Панкреатический rидролизат казеина 17 r
Папаиновый rидролизат соевых бобов 3 r
Натрия хлорид 5 r
Дикалия rидрофосфат 2,5 r
rлюкоза моноrидрат 2,5 r
Аrnр 15r
Полисорбат 80 1 О r
Вода до 1000 мл
Полисорбат 80 добавляется к roрячему
раствору остальных инrредиентов после кипя
чения непосредственно перед доведением до
объема.
Среда С
Пептон
r овяжий экстракт
Дрожжевой экстракт
Натрия хлорид
rлюкоза моноrидрат
Дикалия rидрофосфат
Калия диrИДрОфQсфат
Вода
Среда D
Экстракт сердца
Дрожжевой экстракт
Пептон казеин
6r
1,5 r
3r
3,5 r
1 r
3,68r
1,32r
до 1000 мл
1.5r
1.5.
5
2.7.2. Количественное определение антибиотиков микробиолоzическим методом 341
rлюкоза моноrидрат
Натрия хлорид
Дикалия rидрофосфат
Калия диrидрофосфат
Калия нитрат
Вода
1 r
3,5 r
3,68 r
1,32 r
2r
до 1000 мл
Среда Е
Пептон 5 r
Мясной экстракт 3 r
Динатрия rидрофосфат, 12НР 26,9 r
Arnp 10r
Вода до 1000 мл
Динатрия rидрофосфат добавляется в виде
стерильноrо раствора после стерилизации
среды.
Среда F
Пептон
Дрожжевой экстракт
rовяжий экстракт
Натрия хлорид
rлюкоза моноrидрат
Arap
Вода
9,4 r
4,7 r
2,4 r
10,0 r
10,0 r
23,5 r
до 1000 мл
Среда G
rлицерин 10 r
Пептон 1 О r
Мясной экстракт 1 О r
Натрия хлорид 3 r
Arnp 15r
Вода до 1000 мл
После стерилизации рН (7 ,0:t0, 1).
Среда Н
Пептон 5,0 r
Arnp 1 Or
rовяжий экстракт, порошок 3,0 r
Вода до 1000 мл
Доводят 0,1 М раствором натрия 2идpOKcи
да до рН (7,9:t0,1).
#Кроме описанных выше сред М02ут иc
пользоваться следующие питательные среды:
Среда N!! 1
Мясо пептонный бульон
Arap arap
После стерилизации рН 7, НО, 1.
1000 мл
20 r
Среда N!! 2
Мясо пептонный бульон
Arap arap
После стерилизации рН (7 ,9:t0, 1 ).
1000 мл
20 r
Среда N!! 3
Мясо пептонный бульон
Arap arap
rлюкоза моноrидрат
Натрия хлорид
После стерилизации рН (7, НО, 1).
1000 мл
17r
10 r
5r
Среда N!! 4
Раствор rидролизата мяса
по Хопинrеру с содержанием
аминноro азота З50-........400 мr/л
(35-........40 Mr %) 1000 мл
Arap arap 25 r
После стерилизации рН (6, НО, 1).
Среда N!! 5
Раствор rидролизата мяса
по Хопинrеру с содержанием
аминноro азота 350-........400 мr/л
(35-........40 Mr %) 1000 мл
Arap arap 25 r
После стерилизации рН (7,9:t0,1).
Среда N!! 6
Раствор rидролизата мяса
по Хопинrеру с содержанием
аминноro азота 1300 1400 мr/л
(130 140 Mr %) 1000 мл
Arap arap 10 15 r
После стерилизации рН (6,9:t0,1).
Среда N!! 7
Раствор rидролизата мяса
по Хопинrеру с содержанием
аминноro азота 1300 1400 мr/л
(130 140 Mr %) 1000 мл
Arap arap 10 12 r
Динатрия rидрофосфат, 12НР 3r
После стерилизации рН (6,9:t0,1).
Среда N!! 8
Раствор rидролизата мяса
по Хопинrеру с содержанием
аминноro азота 1300 1400 мr/л
(130 140 Mr %) 1000 мл
Arap arap 10 12 r
Динатрия rидрофосфат, 12НР 3r
После стерилизации рН (7,9:t0,1).
Среда N!! 9
Раствор rидролизата мяса
по Хопинrеру с содержанием
аминноro азота 350-........400 мr/л
(35-........40 Mr %) 1000 мл
Arap arap 15 r
Динатрия rидрофосфат, 12НР 3r
После стерилизации рН (7,9:t0,1).
Среда N!! 1 О
Раствор rидролизата мяса
по Хопинrеру с содержанием
аминноro азота 1 зоо 1400 мr/л
(130 140 Mr %) 1000 мл
Arap arap 1 15 r
Калия диrидрофосфат 5r
После стерилизации рН (6, НО, 1).
Среда N!! 11
Arap arap 1 15 r
342
rосударственная фармакопея Республики Беларусь
Динатрия rидрофосфат, 12НР З r
Вода дистиллированная до 1000 мл
После стерилизации рН (6,9:t0,1).
Среда N!! 12
Arap arap 15 r
Динатрия rидрофосфат, 12НР 3 r
Вода дистиллированная до 1000 мл
После стерилизации рН (7,9:t0,1).
Среда N!! 13
Раствор rидролизата мяса
по Хоттинrеру с содержанием
аминноro азота 350-------400 мr/л
(35-------40 Mr %)
Arap arap
Калия хлорид
После стерилизации рН (7,1 :t0, 1 ).
Среда N!! 14
Раствор rидролизата мяса
по Хоттинrеру с содержанием
аминноro азота 350-------400 мr/л
(З5-------40 Mr %)
Arap arap
После стерилизации рН (7 ,9:tO,1).
Среда N2 15
Раствор rидролизата мяса
по Хоттинrеру с содержанием
аминноro азота 350-------400 мr/л
(35-------40 Mr %)
Arap arap
Калия диrидрофосфат
После стерилизации рН (7, НО, 1).
1000 мл
20 r
20 r
1000 мл
15 r
1000 мл
15 r
25 r
Среда N!! 16
Arap arap 18 20 r
Динатрия rидрофосфат, 12НР 5 r
Натрия хлорид 20 r
Аммония цитрат 5 r
Калия хлорид 20 r
Вода дистиллированная до 1000 мл
После стерилизации рН (5,9:t0,1 ).
Среда N2 17
Arap arap 18 r
rлюкоза моноrидрат 5 r
Динатрия rидрофосфат 15 r
Мочевина 2 r
Аммония хлорид 2 r
Вода дистиллированная до 1000 мл
После стерилизации рН (7,9:t0,1).
Среда N2 18
Раствор rидролизата мяса
по Хоттинrеру с содержанием
аминноro азота 1300 1400 мr/л
(130 140 Mr %)
Arap arap
rлюкоза моноrидрат
Натрия хлорид
1000 мл
15 20 r
5r
30 r
После стерилизации рН (6,9:t0,1).
Среды стерилизуют насыщенным паром под
давлением при 121 ос в течение 15 мин.
При приrотовлении питательных сред
вместо раствора rидролизата мяса по Хоттин
repy можно использовать питательные среды на
основе рыбноro или мясо пептонноro бульона с
таким же содержанием аминною азота. 1I
01/2013:20704
2.7.4. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ ФАКТОРА
СВЕРТЫВАНИЯ КРОВИ VIII
Количественное определение срактора
свертывания крови VIII производят по ero био
лоrической активности в качестве кофактора aK
тивации фактора Х активированным фактором
'Х (фактором 'Ха) в присутствии ионов кальция
и фосфолипидов. Активность препарата факто
ра VIII оценивают путем сравнения количества,
необходимоro для достижения определенной
скорости образования фактора Ха в испытуе
мой смеси, содержащей вещества, принима
ющие участие в активации фактора Х, и коли
чества Международноro стандартноro образца
или стандартноro образца, калиброванноro в
Международных Единицах, которое требуется
для получения такой же скорости образования
фактора Ха.
За Международную Единицу принимают aK
тивность фактора VIII в установленном количе
стве Международноro стандартноro образца,
состоящеro из лиофильно высушенноro KOH
центрата человеческою фактора свертывания
крови VIII. Эквивалентность Международноro
стандартноro образца в Международных Едини
цах устанавливается Всемирной орrанизацией
здравоохранения.
БСП человечеСК020 фактора свертывания
крови V/II калиброван в Международных Едини
цах в сравнении с Международным CTaHдapT
ным образцом.
Хромоrенный метод количественноro
определения включает две последователь
ные стадии: зависящая от фактора V/II актива
ция фактора Х в peareHTe факторов CBepTЫBa
ния, состоящем из очищенных компонентов, и
ферментативное расщепление хромorенноro
субстрата фактором Ха с образованием xpo
мофора, который определяют спектрофотоме
трически. В соответствующих условиях испыта
ния существует линейная зависимость между
скоростью образования фактора Ха и KOHцeH
трацией фактора VIII. Содержание метода
количественноro определения описывается
следующей схемой:
1-я СТ ИЯ
2.7.4. Количественное определение фактора свертывания крови V/II
343
(активированный) фактор VIII
фактор Х
. фактор Ха
фактор (Ха, фосфолипид. Са 2 +
2-я СТ ИЯ
фактор Ха
хромоrенный суБстрат
пептид + хромофор
В обеих стадиях используются peareHTbI, дo
ступные из ряда коммерческих источников. Хотя
в состав peareHToB MOryт вноситься некоторые
изменения, их основные свойства должны COOT
ветствовать нижеприведенным спецификациям.
Отклонения от этоro описания допустимы при
условии, что с использованием Международно-
ro стандарта образца концентрата человеческо
ro фактора свертывания крови VIII в качестве
стандартноrо образца показано отсутствие су-
щественных различий в результатах.
Необходимо подтвердить приrодность ис-
пользуемоro набора путем проверки времени
образования фактора Ха. При этом определя-
ют время, необходимое для достижения 50 %
от максимальноro количества образовавшеrося
фактора Ха.
PEArEHTbI
PeareHT факторов коаryляции состоит
из очищенных белков человеческоro или ro
вяжьеro происхождения, включающих фактор Х,
фактор IXa и активатор фактора VIII, обычно
тромбин. Эти белки частично очищены, обычно
не менее чем до 50 %, и не содержат примесей,
мешающих активации фактора VIII или факто-
ра Х. Тромбин может присутствовать в форме
еro предшественника протромбина при yc
ловии, что еro активация в peareHTe протекает
с достаточной скоростью для практически MrHo
венной и полной активации в эксперименте фак
тора VIII. Фосфолипиды MOryT быть получены
из природных источников или синтетическим
путем и должны состоять в существенной сте-
пени из фосфатидилсеринов. Компоненты ре-
areHTa обычно разделяют не менее чем на 'Два
отдельных peareHTa, каждый из которых не об
ладает способностью самостоятельно rенери
ровать фактор Ха. Один из peareHТOB содержит
ионы кальция. После восстановления peareH
ты MOryT быть объединены при условии, что в
отсутствии фактора VIII не происходит образо
вания существенных количеств фактора Ха. В
окончательной инкубационной смеси фактор VIII
должен быть единственным компонентом, лими-
тирующим скорость.
Вторая стадия включает количественное
определение образовавшеroся фактора Ха с
использованием специфичноro в отношении
фактора Ха xpoMoreHHoro субстрата. Обычно
он представляет собой производное KOpOTKO
ro пептида, состоящеrо из 3 5 аминокислот
ных остатков, соединенное с хромофорной
rруппой. При отщеплении этой rруппы от пеп
тидноrо субстрата ее хромофорные свойства
смещаются к длине волны, при которой CTa
новится возможным произвести спектрофо
тометрическое определение. Субстрат также
может содержать подходящие инrибиторы для
прекращения дальнейшеrо образования фак
тора Ха (такие как хелатообразующие areHTbI)
и супрессирования активности тромбина.
МЕТОДИКА КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ
Восстанавливают полное содержимое
одной ампулы стандартноro образца и ис-
пытуемоro образца; используют немедлен
но. К восстановленным образцам прибавляют
прерастворитель в количестве, достаточном
для получения растворов с содержанием
0,5 2,O МЕ/мл.
Прерастворитель представляет собой
плазму, собранную у пациента, страдающеro
острой rемофилией А, или искусственно приro
товленный peareHT, содержащий достаточное
количество фактора фон Виллебранда. кото-
рый приводит к получению результатов, суще
ственно не отличающихся от результатов, по-
лучаемых при использовании rемофилической
плазмы. Полученные материалы предвари
тельноro растворения должны быть стабильны
в течение времени, необходимоro для прове
дения количественноro определения.
rотовят дальнейшие разведения испыту-
eMoro и стандартноro образцов с использова-
нием нехелатирующеro и буферизированноro
необходимым образом раствора (например,
трис(rидроксиметил)аминометан или ими
дазол), содержащеro 1 % человеческоro или
бычьеro альбумина. rотовят не менее трех
отдельных, независимых разведений каждо
ro препарата в не менее чем двух повторах.
Разведения roтовят таким образом, чтобы
окончательная концентрация фактора VIII на
стадии образования фактора Ха была ниже
0,01 МЕ/мл.
rотовят контрольный раствор, включаю
щий все компоненты, кроме фактора VIII.
Все разведения roтовят в пластиковых
пробирках и используют незамедлительно.
1-я стадия. Предварительно HarpeTbIe
разведения стандартноro образца фактора
VIII и испытуемоro образца смешивают с co
ответствующим объемом предварительно на-
rpeToro peareHTa факторов свертывания крови
или со смесью компонентов, ero составляю
щих, и инкубируют смесь в пластиковых про-
бирках или ячейках планшета при темпера
туре З7 ос. Активацию фактора Х про водят
в течение подходящеro промежутка BpeMe
ни, предпочтительно прерывая реакцию (2-ая
стадия) до достижения концентрацией фак-
тора Ха 50 % от ее максимальноro уровня
(плато) с целью получения удовлетворитель
344
rосударственная фармакопея Республики Беларусь
ной линейной зависимости отклика от дозы.
Время активации выбирается также таким об
разом, чтобы достичь линейноro во времени
образования фактора Ха. Подходящие перио
ды активации обычно находятся в пределах от
2 мин до 5 мин.
2--я стадия. Активацию прерывают добав
лением предварительно наrретою peareHTa, co
держащею хромоrенный субстрат. Проводят KO
личественную оценку скорости расщепления
субстрата, которая должна находиться в линейной
зависимости от концентрации образовавшеюся
фактора Ха, измеряя изменение оптической плот
ности при соответствующей длине волны с помо
щью спектрофотометра. Оптическую плотность
отслеживают постоянно, получая таким образом
возможность вычислить начальную скорость pac
щепления субстрата, либо прерывают реакцию rи
дролиза по окончании подходящеro промежутка
времени путем понижения рН прибавлением под
ходящею реактива, например, 50 % (06/06) paCT
вора уксусной кислоты или 1 М цитратноro буфер
ною раствора с рН 3. Время rидролиза реryлируют
с целью достижения линейноro характера обра
зования хромофора во времени. Подходящими
обычно являются периоды rидролиза от 3 мин до
15 мин, но допускаются и друrие значения при yc
лови и получения таким образом лучшей линейно
сти зависимости отклика от дозы.
Активность испытуемоro образца рассчиты
вают с использованием обычных статистических
методов (например, 5.3).
01/2013:20705
2.7.5. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕrЕПАРИНА
Антикоаryлянтную активность rепарина
определяют iп vitro путем сравнения еro способ
ности в заданных условиях замедлять CBepTЫBa
ние обоrащенной кальцием и цитратом овечьей
плазмы с аналоrичной способностью стандарт
ноro образца rепарина, калиброванноro в Меж
дународных Единицах.
Международная Единица (МЕ) актив
ность, проявляемая установленным количе
ством Международноro стандартноrо образца,
состоящею из лиофильно высушенноro rепари
на натрия, полученноro из слизистой оболочки
кишечника свиньи. Эквивалент Международно
ro стандартноro образца в МЕ устанавливается
Всемирной орrанизацией здравоохранения.
БСП 2епарина натрия откалиброван в МЕ
по сравнению с Международным стандартным
образцом с использованием изложенной ниже
методики количественноro определения.
Количественное определение выполня
ют одним из следующих методов установления
начала свертывания с использованием проби
рок и друroro оборудования, соответствующеro
выбранному методу:
а) прямое визуальное наблюдение, предпо
чтительно в отраженном свете на черном MaTO
вом фоне;
Ь) спектрофотометрическая реrистрация из
менения оптической плотности при длине волны
около 600 нм;
с) визуальная фиксация изменения текуче
сти путем опрокидывания пробирок;
d) механическая запись изменения текуче
сти, определяемоro по перемешиванию, с co
блюдением мер предосторожности, направлен
ных на минимизацию возмущений раствора в
начальной стадии свертывания.
МЕТОДИКА КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ
Приведенные в тексте 06ъемы являют
ся примерными и М02ут быть адаптированы к
используемой аппаратуре при условии соблю
дения соотношений между различными объе
мами.
БСП 2епарина натрия разводят paCTBO
ром 9 пл натрия хлорида Р до получения
точно известноro количества МЕ в миллилитре
и rотовят аналоrичный раствор испытуемо
ro образца, для котороro ожидается такая же
активность. Используя раствор 9 пл натрия
хлорида Р, из каждоro раствора rотовят ряд
разведений в rеометрической проrрессии так,
чтобы время свертывания, получаемое для
низшей концентрации, не менее чем в 1,5
раза превышало время в контрольном опыте
#(время рекальцификации)#, а время CBepTЫ
вания, получаемое для высшей концентрации,
находилось в пределах удовлетворительной
лоrарифмической кривой зависимости доза/
эффект, построенной в ходе предварительно
ro испытания.
Помещают двенадцать пробирок в ледяную
баню, маркируя их в дубликатах следующим об
разом: Т" Т 2 и Тз для разведений испытуемоro
образца и S1' S2 И 8 з для разведений CTaHдapTHO
ro образца. В каждую из пробирок помещают по
1,0 мл размороженноro субстрата плазмы Р1
и 1,0 мл соответствующеro разведения испыту
емоro образца или стандартноro образца. После
каждоro из добавлений про изводят перемеши
вание, не допуская образования пузырьков.
Пробирки обрабатывают в порядке: 81' 82'
8 з , Т1' Т2' ТЗ' при этом каждую из пробирок п<r
мещают в водяную баню, выдерживают при TeM
пературе 37 ос в течение около 15 мин и пр
бавляют к каждой из них по 1 мл подходящеro
АЧТВ (активированное частичное тромбоплcr
стиновое время, Activated Partia/ Th,ombop/asliп
Тiтe АРТТ) peareHTa, содержащеro фосфол.
2.7.5. Количественное определение 2епарина
345
пидный И контактный активаторы, в таком разве
дении, чтобы время контрольноro опыта #(время
рекальцификации)# было подходящим и не пре
вышало 60 с.
Ровно через 2 мин прибавляют 1 мл paCT
вора 3,7 r/л кальция хлорида Р, предварительно
HarpeToro до температуры 37 ОС, и реrистриру
ют в качестве времени свертывания интервал в
секундах междУ. последним прибавлением и Ha
чалом свертывания, определяемым с помощью
выбранноro метода. В начале и в конце проце
дуры аналоrичным образом определяют время
контрольноro опыта, используя вместо одноro
из разведений rепарина 1 мл раствора 9 пл
натрия хлорида Р; два полученных контроль
ных значения не должны существенно разли
чаться.
Время свертывания пере водят в лоrариф
мы, используя средние значения для дублика
тов. Процедуру повторяют со свежими разведе
ниями, проводя обработку пробирок в порядке:
Т1' Т2' ТЗ' S1' 82' Sз. Результаты рассчитывают с
использованием обычных статистических MeTO
дов (5.3).
Проводят не менее трех независимых опре
делений. Для каждоro из таких определений
roтовят свежие растворы стандартноro и ис
пытуемоro образцов и используют новую свеже
размороженную порцию субстрата плазмы.
Активность испытуемоrо образца рассчиты
вают путем комбинирования результатов этих
определений с использованием обычных стати
стических методов (5.3). Если отклонение, BЫ
званное различиями между результатами опре
делений, является достоверным при Р = 0,01,
комбинированная оценка активности может
быть получена путем расчета простоro среднеro
значения определенной активности.
#МЕТОДИКА КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ ПО РЕАКЦИИ
АНТИКОАrYЛЯЦИИ
Количественное определение 2епарина в
фармацевтических субстанциях и 20товых ле
карственных средствах может быть выполне
но с использованием реакции антиКО02уляцuи
следующим методом.
АнтиКО02улянтная активность. Опреде
ление активности rепарина проводят биолоrи
ческим методом по способности еro задержи
вать свертывание бараньей или бычьей плазмы
путем сопоставления активности испытуемоro
образца с активностью стандартноro образца re
парина.
В два ряда тщательно вымытых и BЫCY
шенных пробирок вносят возрастающее коли
чество рабочих растворов стандартноro образ
ца rепарина, испытуемоro образца и раствора
9 r/л натрия хлорида Р в количествах, указан
ных в таблице #2.7.5. 1. Затем в каждую про
бирку прибавляют по 1 мл плазмы, 0,2 мл paCT
вора 1 О r/л кальция хлорида безводН020 Р
(попеременно в пробирки из ряда с раствором
стандартноro образца rепарина и испытуемоro
образца), осторожно перемешивают содержи
мое поворачиванием и леrким встряхиванием,
не допуская образования пузырей. Отмечают
время и оставляют при комнатной температуре
(или в термостате при температуре (3НО,5) ОС)
на 1 ч. По истечении указанноro времени опре
деляют состояние плазмы в каждой пробирке
и отмечают в каждом ряду крайнюю слева про
бирку, в которой не произошло полное CBepTЫ
вание плазмы. Эти пробирки содержат наимень
шие объемы растворов стандартноro образца
rепарина и испытуемоro образца, которые за
держивают свертывание плазмы.
Степень свертывания (0,2; 0,4; 0,6; 0,8)
между нулем и полным свертыванием (1 ,О) опре
деляется визуально.
Таблица #2.7.5. 1
Наиме- Порядок внесения раствора
нование ..". Q ....
.... N М It) со r-- CIO 0'1
раствора .... ....
Раствор о L{) о L{) о L{) о L{) о L{) о
rепарина, м с") "<t "<t L{) L{) ф r-- r-- со
о о о о о о о о о о о
мл
PaCT
вор 9 r/л о L{) о L{) о L{) о L{) о L{)
натрия L{) "<t с") с") ('\j "!- ...... ...... о I
о о о о о о о о о о
хлори
да Р, мл
Антикоаryлянтную активность испытуемоro
образца, рассчитывают по формуле:
v, (v,' V,)'a1
.А
V 2 (v; V2),a2 '
rдe:
V 1 объем раствора стандартноro образца
rепарина, содержащийся в крайней левой про
бирке, в которой не произошло полное CBepTЫ
вание плазмы, в миллилитрах;
V 2 объем раствора испытуемоro образца
rепарина, содержащийся в крайней левой про
бирке, в которой не произошло полное CBepTЫ
вание плазмы, в миллилитрах;
V; объем раствора стандартноro образца
rепарина, содержащийся в крайней левой про
бирке, в которой произошло полное CBepTЫBa
ние плазмы, в миллилитрах;
v; объем раствора ИСflытуемоro образца
rепарина, содержащийся в крайней левой про
бирке, в которой произошло полное CBepTЫBa
ние плазмы, в миллилитрах;
а 1 степень свертываемости CTaHдapTHO
ro образца;
а 2 степень свертываемости испытуемоro
образца;
А предполаrаемая активность rепарина в
1 r испытуемоro образца.
346
rосударственная фармакопея Республики Беларусь
Определение проводят не менее 3 раз, ис
пользуя среднее значение из трех определений.
Приrотовление раствора 80 r/л натрия
цитрата. 8,0 r натрия цитрата Р растворяют в
70 мл воды Ри доводят до объема 100,0 мл этим
же растворителем. Раствор используют свеже
приroтовленным.
Приrотовление раствора 1 О r/л кальция
хлорида. 1,0 r кальция хлорида безводН020 Р
растворяют в 70 мл воды Р и доводят до объема
100,0 мл этим же растворителем. Хранят при
температуре от О ОС дО 4 ос в течение 1 мес.
Приrотовление плазмы. Кровь, вытекаю
щую из сосудов убойноro животноro (крупноro
poraтoro скота или баранов), собирают в банки
из полиэтилена вместимостью 1 л, содержа
щие 25-------30 мл раствора 80 r/л натрия цитpa
та Р. Кровь осторожно смешивают с paCTBO
ром 80 r/л натрия цитрата Р, поворачивая
банку, и как можно быстрее центрифуrируют при
2500 об/мин в течение 30 мин, после чеro отби
рают плазму.
Перед использованием плазму разморажи
вают на водяной бане или термостате при TeM
пературе (37:tO,5) ОС. Затем фильтруют через
марлю, проверяют время ее свертывания и
определяют оптимальное количество кальция
хлорида, необходимое для определения актив
ности, для чеro: в 5 пробирок С 1 мл плазмы в
каждой прибавляют возрастающие количества
раствора 10 r/л кальция хлорида Р (0,02; 0,04;
0,06; 0,08; 0,10 мл) и осторожно смешивают. Для
опыта используют плазму, время свертывания
которой не превышает 10 мин и то оптимальное
количество кальция хлорида, которое вызывает
полное свертывание плазмы в указанное время.
Найденное оптимальное количество кальция
хлорида должно содержаться в 0,2 мл рабоче
ro раствора (этот раствор roтовят разведением
раствора 1 О r/л кальция хлорида Р). Хранят при
температуре от 1 О ос до 20 ос в течение 1 roда.
Допускается использование лиофилизиро
ванной плазмы, разведенной в соответствии с
инструкцией производителя.
Приrотовление раствора рабочеrо CTaH
AapTHoro образца rепарина. Навеску рабоче
ro стандартноro образца rепарина, откалибро
ванноro по БСП 2епарина натрия, растворяют в
растворе 9 r/л натрия хлорида Р с таким расче
том, чтобы в 1 мл раствора содержал ось 50 ЕД
rепарина натрия. Хранят ПрllJ температуре от
О ОС до 4 ос в течение 6 мес. ДJ1Я определения
активности rепарина в фармацевтической суб
станции в качестве стандартноrо образца ис
пользуют БСП 2епарина натрия.
Из полученноrо раствора roтовят paCTBO
ры, содержащие в 1 мл 1,0; 1,5; 2,0; 2,5; 3,0 ЕД
rепарина натрия. По 0,5 мл и 0,72 мл получен
ных растворов помещают в пробирки, ДOBO
дят объем в пробирке до 0,8 мл раствором 9 r/л
натрия хлорида Р.
Прибавляют в каждый раствор по 1 мл
плазмы и 0,2 мл раствора кальция хлорида с оп
тимальной концентрацией и перемешивают.
Через 1 ч определяют состояние плазмы в
каждой пробирке и устанавливают активность
рабочеro стандартноro образца rепарина, при
которой в пробирке, содержащей 0,5 мл paCT
вора rепарина, происходит полное CBepTЫBa
ние плазмы, а в друroй пробирке, содержащей
0,72 мл этоro раствора rепарина, плазма не
свернулась. Выбранная активность обеспечи
вает наблюдение за влиянием испытуемоro pa
бочеrо стандартноro образца rепарина на CBep
тывание плазмы по истечении часа. Обычно эта
. активность находится в пределах 1 1,5 ЕД дЛЯ
плазмы крупноro poraтoro скота и 1 3,5 ЕД дЛЯ
плазмы барана.
Приrотовление раствора испытуемоrо
образца rепарина. Навеску испытуемоro образ
ца растворяют в растворе 9 r/л натрия хлори
да Р, исходя из еro предполаrаемой активности
с таким расчетом, чтобы 1 мл полученноro paCT
вора содержал столько же ЕД rепарина, сколько
содержит 1 мл рабочеro стандартноro образца
rепарина натрия. Раствор используют свежепри
roтовленным.
При необходимости допускается сдвиrать
ряд пробирок, указанный в таблице #2.7.5. 1, в
сторону уменьшения концентрации раствора re
парина.
01/2013:20706
2.7.6. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
АДСОРБИрОВ ННОИ
ДИФТЕРИИНОИ ВАКЦИНЫ
Активность дифтерийной вакцины опреде
ляется путем назначения вакцины морским свин
кам с последующей провокацией дифтерийным
токсином (методы А или В) или путем определе
ния титра антител против дифтерийноro токси
на или анатоксина в сыворотке крови морских
свинок (метод С). В обоих случаях активность
вакцины рассчитывается по сравнению со CTaH
дартным образцом, калиброванным в Междуна
родных Единицах.
За Международную Единицу принимают aK
тивность в установленном количестве Между
народноro стандартноro образца, состоящеro
из дифтерийноro анатоксина, адсорбированно
ro на rидроксиде алюминия. Эквивалентность
Международноro стандартноrо образца в Меж
2.7.6. Количественное определение адсорбированной дифтерийной вакцины
347
дународных Единицах устанавливается Всемир
ной орrанизацией здравоохранения (ВОЗ). БСП
адсорбированной дифтерийной вакцины может
использоваться в качестве стандартноro образ
ца (образца сравнения).
Выбор метода исследования адсорбирован
ной дифтерийной вакцины зависит от предпола
rаемой цели.
Методы А или В применяются:
1) в процессе разработки вакцины при KO
личественном определении на сериях продукта,
произведенных для валидации производствен
ноro процесса;
2) в любом случае необходимости ревали
дации (при значительных изменениях производ
ственноro процесса).
Метод А или В может также использоваться
для рутинной проверки серий вакцины, но в ин
тересах защиты животных, по возможности, сле
дует использовать метод С.
Метод С может использоваться за ис
ключением случаев, описанных в пунктах 1 и
2, после валидации ero для конкретноro про
дукта. Для этой цели подходящее количество
серий (обычно 3) испытываются методом С и
методом А или В.
Если различные вакцины (моновалентные
или комбинированные) приroтовлены из дифте
рийною анатоксина одною и тоro же происхожде
ния и содержат сопоставимые уровни анатокси
на (выраженные Lflмл), валидация, проведенная
для лекарственноro средства с максимальным
количеством компонентов, распространяется
на комбинированные лекарственные средства
с меньшим числом компонентов и на MOHOBaK
цины. Любые комбинированные лекарственные
средства, содержащие цельноклеточную KO
клюшную вакцину или конъюrированную вакци
ну против Haemophilus В и дифтерийный aHaТOK
син или дифтерийный белок CRM 197 в одном
флаконе, должны исследоваться отдельно.
Для комбинированных лекарственных
средств, содержащих дифтерийный и столбняч
ный анатоксины, серолоrический тест (метод С)
может быть проведен на rруппе животных, ис
пользовавшихея для количественною опреде
ления адсорбированной столбнячной вакци
ны (2.7.8), если общие условия иммунизации
для дифтерийноro и столбнячноro компонентов
(доза, продолжительность) валидированы для
комбинированноro лекарственноro средства.
Дизайн описанных ниже исследований ис
пользует множественные серийные разведения
испытуемых и стандартных образцов. При HaKO
плении определенноro опыта работы с методом
определения на конкретной вакцине возможен
переход на упрощенную схему исследования
одноro разведения испытуемоro и CTaHдapTHO
ro образцов. Такая схема позволяет установить
факт существенноro превышения активности
испытуемою образца минимально допустимоro
уровня, но не дает информации о линейности,
параллелизме и друrих параметрах зависимо
сти доза/эффект. Упрощенная схема позволяет
значительно сократить потребность в экспери
ментальных животных и должна приниматься
во внимание любой лабораторией в COOTBeT
ствии с положениями Европейской Конвенции
по защите позвоночных животных, используе
мых для экспериментальных и друrих научных
целей.
В случае перехода на упрощенную схему ис
пытаний соответствие метода тестирования сле
дует постоянно мониторировать по подходящим
индикаторам, а также путем периодической по
становки теста с множественными серийными
разведениями (каждые 2 roда). Для серолоrиче
скоro теста подходящими индикаторами дЛЯ MO
ниторинrа являются:
среднее значение и стандартное отклоне
ние относительных титров антитоксина или ин
дексов сывороток, полученных после назначе
ния фиксированных доз стандартноro образца
вакцины;
титры антитоксинов или индексы рабочих
контролей (положительные или отрицательные
образцы сыворотки);
соотношение титров антитоксина или ин
декса положительной контрольной сыворотки
и образцов сыворотки, соответствующих CTaH
дартному образцу вакцины.
МЕТОД А: ВНУТРИКОЖНЫЙ
ПРОВОКАЦИОННЫЙ ТЕСТ НА МОРСКИХ
СВИНКАХ
ОТБОР И РАСПРЕДЕЛЕНИЕ
ЭКСПЕРИМЕНТАЛЬНЫХ ЖИВОТНЫХ
Используют здоровых белых морских свинок
из одной партии; их размер должен быть ДOCTa
точным для проведения назначенною количе
ства тестов; разница в массе между наиболее
тяжелым и леrким животным не должна пре
вышать 100 r. Следует использовать животных
одноro пола; в противном случае самцов и самок
необходимо равномерно распределить между
rруппами. Животных распределяют не менее
чем на 6 равных rрупп. Число животных в rруппе
должно быть достаточным для получения дocтo
верных результатов в ходе описанных ниже ис
пытаний. Если токсин, используемый для про
вокации, предварительно не валидирован на
стабильность или не стандартизирован, следует
создать rруппу из 5 животных в качестве HeBaK
цинированноro контроля.
ВЫБОР ПРОВОКАЦИОННО,О ТОКСИНА
Следует выбрать стандартный образец TOK
сина, содержащий от 67 до 133 /r/100 в 1 U и от
25 000 до 50 000 минимальных доз, вызываю
щих реакцию на коже морских свинок в 1 и. Если
стабильность токсина подтверждена предвари
тельно, нет необходимости определять еro aK
тивность для каждою испытания.
348
rосударственная фармакопея Республики Беларусь
приrОТОВЛЕНИЕ РАСТВОРА ТОКСИНА
Непосредственно перед использованием
разводят токсин подходящим растворителем до
концентрации 0,0512 и в 0,2 мл. Далее roтовят
серию пятикратных разведений (0,0128, 0,0032,
0,0008,0,0002 и 0,00005 ив 0,2 мл).
РАЗВЕДЕНИЕ ИСПblТУЕМblХ
И СТАНДАРТНЫХ ОБРА3ЦОВ
С помощью раствора 9 r/л натрия хлори
да Р roтовят разведения испытуемой вакцины и
стандартноro образца таким образом, чтобы шаr
разведения в серии был не более 2,5 KpaTHЫM,
а внутрикожная инъекция промежуточных разве
дений морской свинке в дозе 1,0 мл приводила
бы к индексу 3 при постановке провокационно
ro теста.
ИММУНИ3АЦИЯ И ПРОВОКАЦИЯ
Назначают по одному разведению одной
rруппе морских свинок и вводят подкожно по
1,0 мл разведения каждой морской свинке в
rруппе. Спустя 28 дней каждой свинке выбри
вают оба бока и вводят внутрикожно по 0,2 мл
каждоro из 6 разведений токсина в 6 различных
мест на коже животноrо так, чтобы минимизиро
вать влияние соседних инъекций.
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ
ПРОВОКАционноrо ТОКСИНА
При необходимости вводят невакциниро
ванным контрольным животным разведения TOK
сина, содержащие 80, 40, 20, 10 и 5 . 10 6 Lf.
ПОЛУЧЕНИЕ
И ИНТЕРПРЕТАЦИЯ РЕ3УЛЬТАТОВ
Все места инъекций обследуют через
48 часов и записывают наличие специфической
дифтерийной эритемы. Записывают также число
мест инъекций без реакции (внутри кожный про
вокационный индекс, ВПИ). Сводят в таблицу
внутри кожные провокационные индексы всех
животных, получивших одинаковое разведение
вакцины, и производят преобразование данных:
(ВПИ)2 или arcsiп «ВПИ/6)2) для расчета относи
тельной активности каждоro испытуемоro образ
ца с применением параллельноro количествен
ноro определения.
ТРЕБОВАНИЯ К ДОСТОВЕРНОСТИ
ИСПblТАНИЯ
Тест является достоверным. если:
испытуемый и стандартный образцы
дают при введении минимальной дозы средний
индекс менее 3, а при введении максимальной
дозы более 3;
разведение токсина 40.1 О б Lf вызыва
ет дифтерийную эритему не менее чем у 80 %
контрольных животных, а разведение токсина
20.1 О б и менее чем у 80 % (если это требова
ние не выполняется, следует применить друroй
провокационный токсин);
rраницы доверительноro интервала
(Р = 0,95) составляют не менее 50 % и не более
200 % установленной активности;
статистический анализ подтверждает OT
сутствие отклонений в линейности и паралле
лизме.
Тест можно повторить, однако в случае про
ведения более одноro испытания результаты
всех достоверных испытаний должны учиты
ваться для определения активности.
МЕТОД В: ЛЕТАЛЬНЫЙ
ПРОВОКАЦИОННЫЙ ТЕСТ НА МОРСКИХ
СВИНКАХ
ОТБОР И РАСПРЕДЕЛЕНИЕ
ЭКСПЕРИМЕНТАЛЬНblХ ЖИВОТНblХ
Используют здоровых морских свинок из
одной партии весом 250 350 r. Следует ис
пользовать животных одноro пола; в противном
случае самцов и самок необходимо равномерно
распределить между rруппами. Животных pac
пределяют в 6 равных rрупп. Число животных в
rруппе должно быть достаточным для получе
ния достоверных результатов в ходе описанных
ниже испытаний. Если токсин, используемый
для провокации предварительно не валидиро
ван на стабильность или не стандартизирован,
следует создать 4 дополнительные rруппы по 5
животных в качестве невакцинированноrо KOH
троля.
ВblБОР ПРОВОКАционноrо ТОКСИНА
Выбирают стандарт дифтерийноro токсина,
содержащий не менее 100 LD5iмл. Если CTa
бильность токсина подтверждена предваритель
но. нет необходимости определять летальную
дозу для каждоro исследования.
приrОТОВЛЕНИЕ РА3ВЕДЕНИЙ
ПРОВОКАционноrо ТОКСИНА
Непосредственно перед использованием
разводят токсин подходящим растворителем
до концентрации 100 LD5iмл. При необходимо
сти выполняют разведения токсина 1:32, 1:100
и 1 :320.
РАЗВЕДЕНИЕ ИСПblТУЕМblХ
И СТАНДАРТНЫХ ОБРА3ЦОВ
С помощью раствора 9 r/л натрия хлори
да Р rотовят разведения испытуемой вакцины и
стандартноro образца таким образом, чтобы шаr
разведения в серии был не более 2,5 кратноro,
а подкожная инъекция промежуточных разведе-
ний морской свинке в дозе 1 мл защищала бы
50 % животных от летальноro действия опреде-
ленноro количества дифтерийноro токсина, YCT(r
новленноro для данноro теста.
ИММУНИ3АЦИЯ И ПРОВОКАЦИЯ
Назначают по одному разведению одной
rруппе морских свинок и вводят подкожно по 1 мn
2.7.6. Количественное определение адсорбированной дифтерийной вакцины
349
разведения каждой морской свинке в rруппе.
Спустя 28 дней каждой свинке вводят подкожно
по 1 мл раствора токсина (100 L0 50 )'
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ
ПРОВОКАЦИОННО,О ТОКСИНА
При необходимости назначают рабочее раз
ведение провокационноro токсина и три разве
дения, приroтовленных из неro, одной из 4 rрупп
(по 5 животных) невакцинированных животных
и вводят по 1 мл соответствующеro разведения
каждой свинке в rруппе.
УЧЕТ И ИНТЕРПРЕТАЦИЯ РЕ3УЛЬТАТОВ
Считают число выживших животных спустя
4 дня после инъекции токсина. Рассчитывают aK
тивность испытуемой вакцины по отношению к
активности стандартною образца на основании
пропорции выживших животных в каждой rруппе
вакцинированных морских свинок с применени
ем обычных статистических методов (5.3).
ТРЕБОВАНИЯ К ДОСТОВЕРНОСТИ ТЕСТА
Тест является достоверным, если:
для испытуемоro и стандартноro образцов
50 % протективная (защищающая) доза опре
деляется между минимальной и максимальной
дозами образцов, введенных морским свинкам;
количество поrибших животных в 4 HeBaK
цинированных rруппах после введения рабоче
ю раствор токсина и 3 ero разведений указыва
ет на то, что введенная доза токсина составляет
около 100 L0 50 ;
rраницы доверительноro интервала
(Р = 0,95) составляют не менее 50 % и не более
200 % установленной активности;
статистический анализ подтверждает OTCYТ
ствие отклонений в линейности и параллелизме.
Тест можно повторить, однако в случае про
ведения более одноro теста результаты всех
достоверных тестов должны учитываться для
определения активности.
МЕТОД С: ОПРЕДЕЛЕНИЕ АНТИТЕЛ
У МОРСКИХ СВИНОК
ОТБОР И РАСПРЕДЕЛЕНИЕ
ЭКСПЕРИМЕНТАЛЬНЫХ ЖИВОТНЫХ
Используют здоровых морских свинок из
одной партии с массой тела 250 350 r. Следует
использовать животных одною пола; в против
ном случае самцов и самок необходимо paBHO
мерно распределить между rруппами. Животных
распределяют не менее чем на 6 равных rрупп.
Число животных в rруппе должно быть достаточ
ным для получения достоверных результатов в
ходе описанных ниже испытаний. Для получе
ния неrативной контрольной сыворотки исполь
зуют дополнительную rруппу животных тоro же
происхождения. При условии валидации может
быть применена референсная отрицательная
контрольная сыворотка.
СТАНДАРТНЫЙ ОБРА3ЕЦ
Используют подходящий стандартный об
разец, например, БСП адсорбированной диф
терийной вакцины, серию вакцины с доказан
ной клинической эффективностью или друrую
репрезентативную серию, активность которой
измерена в Международных Единицах по cpaB
нению с БСП адсорбированной дифтерийной
вакцины или Международным стандартным об
разцом адсорбированноro дифтерийноrо aHa
токсина.
РАЗВЕДЕНИЕ ИСПЫТУЕМЫХ
И СТАНДАРТНЫХ ОБРАЗЦОВ
rотовят серийные разведения испытуемой
вакцины и стандартною образца на растворе
9 r/л натрия хлорида Р; разведения выполняют
с 2, 5 KpaTHЫM шаroм. Следует приroтовить не
менее трех разведений, например 0, 16 МЕ/мл
для стандартноro образца вакцины и 1 :2 1: 125
для испытуемой вакцины. Следует использо
вать разведения для иммунизации не позже
1 часа с момента приroтовления. Одно разведе
ние вводят одной rруппе свинок.
ИММУНИЗАЦИЯ
Каждой свинке вводят подкожно 1,0 мл раз
ведения, предназначенноro для данной rруппы
животных.
ЗАБОР КРОВИ
Через 35 2 дня после иммунизации под
ходящим методом берут образцы крови у каждо
ю вакцинированноrо и контрольною животною.
ПРИ,ОТОВЛЕНИЕ ОБРАЗЦОВ
СЫВОРОТКИ
Следует избеrать повторноro заморажива
ния/оттаивания образцов сыворотки. Для пре
дотвращения микробной контаминации об
разцов все манипуляции лучше производить в
ламинарном боксе.
ОПРЕДЕЛЕНИЕ ТИТРА АНТИТЕЛ
Определяют титры или индексы для каждо
ro образца сыворотки с помощью подходящею
иммунохимическою метода (2.7.1). Методы, при
веденные ниже, (иммуноферментный анализ
тИФА (eпzyme liпked immuпoso,beпt assay
ELlSA) и Vero TecT) признаны подходящими.
РАСЧЕТ АКТИВНОСТИ
Активность испытуемой вакцины в МЕ OTHO
сительно стандартною образца рассчитывают,
используя обычные методы статистики (напри
мер, 5.3).
ТРЕБОВАНИЯ К ДОСТОВЕРНОСТИ ТЕСТА
Тест является достоверным, если:
rраницы доверительноrо интервала
(Р = 0,95) составляют не менее 50 % и не более
200 % установленной активности;
350
rосударственная фармакопея Республики Беларусь
статистический анализ показывает значи
тельный наклон и отсутствие отклонений в ли
нейности и параллелизме rрафика зависимости
доза ответ (в rлаве 5.3 описаны возможные аль
тернативы при получении значительных откло
нений в линейности).
Тест может быть проведен повторно, при
этом при проведении более одноro теста для
расчета активности используют объединенные
результаты всех достоверных испытаний.
Следующий раздел прuводится для uнфор
мации.
Руководство по исследованию адсорби
рованной дифтерийной вакцины
МЕТОД С: ОПРЕДЕЛЕНИЕ АНТИТЕЛ
У МОРСКИХ СВИНОК
приrОТОВЛЕНИЕ ОБРА3ЦОВ
СblВОРОТКИ
Следующая методика приroдна для приrо
товления образца сыворотки. Переворачивают
пробирку с образцом крови 6 раз и выдержива
ют при температуре 37 ос в течение 2 ч, затем
при температуре 4 ос в течение 2 ч. Центрифуrи
руют при комнатной температуре с ускорением
800 9 в течение 20 мин. Сыворотку переносят в
стерильные пробирки и хранят при температуре
20 ос. Данная методика позволяет получить на
выходе не менее 40 % сыворотки.
ОПРЕДЕЛЕНИЕ ТИТРА АНТИТЕЛ
тИФА (ELlSA) и Vero TecT, приведенные
ниже, являются примером иммунохимических
методов, признанных подходящими для опреде
лени я титра антител.
Определение титра антител в сыворотке
морской свинки с помощью иммунофермент
Horo анализа (тИФА (ELlSA». в лунки планшета
для тИФА с адсорбированным дифтерийным
анатоксином вносят разведения испытуемой
и референсной сывороток. Положительную и
отрицательную контрольные сыворотки морской
свинки включают в каждую постановку для
контроля работы системы. Далее добавляют
конъюrированные с пероксидазой антитела
кролика или козы против IgG морской свинки,
после чеrо вносятсубстрат. Измеряют оптическую
плотность и рассчитывают относительный титр
антител, используя обычные методы статистики
(например, 5.3).
Реактивы и оборудование:
планшеты для тИФА: 96 лунок, колонки
1 12, строки A H;
противодифтерийная антисыворотка
морской свинки (положительная контрольная
сыворотка), полученная при иммунизации
морских свинок БСП дифтерийной адсорби
рованной вакцины;
КОНЪЮ2ат пероксидазы: конъюrированные
с пероксидазой хрена кроличьи или козьи
антитела против IgG морской свинки;
дифтерийный анатоксин;
карбонатный буферный раствор
для сорбции анти2ена рН 9,6. 1,59 r натрия
карбоната безводН020 Р и 2,93 r натрия
2идрокарбоната Р растворяют в 1000 мл воды Р.
Разливают в бутыли по 150 мл и стерилизуют
автоклавированием при температуре 121 ос в
течение 15 мин;
забуференный солевой раствор рН 7,4
(3СР). 80,0 r натрия хлорида Р, 2,0 r калия ди2и
дрофосфата Р, 14,3 r динатрия 2идрофосфа
(Па ди2идрата Р и 2,0 r калия хлорида Р paCT
воряют в 1000 мл воды Р. Хранят при комнатной
температуре для предотвращения кристаллиза
ции. Перед использованием разводят в 10 раз
водой Р;
раствор кислоты лимонной 10,51 r
кислоты лимонной Р растворяют в 1000 мл
воды Р и доводят до значения рН 4,0 раствором
400 r/л натрия 2идроксида Р:
промывочный буферный раствор. ЗСр,
содержащий 0,5 r/л полисорбата 20 Р;
блокирующий буферный раствор. ЗСр,
содержащий 0,5 r/л полисорбата 20 Р и 25 r/л
высушенноro обезжиренноro молока;
субстратный раствор. Незадолrо до
использования 10 Mr диаммоний 2,2 азинобис(3
этилбензотиазолин 6 сульфоната) Р (ABTS)
растворяют в 20 мл раствора кислоты лимонной.
Немедленно перед использованием прибавляют
5 мкл концентрироваНН020 раствора водорода
пероксида Р.
Методика
Ниже приведен пример подходящей pac
кладки образцов и контролей. Лунки ряда 1 A H
используют для отрицательной контрольной cы
воротки, лунки рядов 2A H и 12A H для по
ложительной контрольной сыворотки для мони
торинrа количественноro определения. Лунки
рядов 3 11 A H используют для испытуемых
образцов.
Покрывают лунки планшета для тИФА
100 мкл раствора дифтерийноro анатоксина
(0,5 Lf/мл в карбонатном буферном растворе для
сорбции антиrена рН 9,6). Планшет оставляют на
ночь при температуре 4 ос во влажной камере.
Чтобы избежать эффекта температурноro
rрадиента, не следует складывать в стопку
больше 4 пластин.
На следующий день ПJlастины тщательно
отмывают промывочным буферным раствором.
Планшеты блокируют прибавлением 100 мкл
блокирующеro буферноro раствора в каждую
лунку. Инкубируют во влажной камере при
температуре 37 ос в течение 1 ч. Отмывают
планшеты промывочным буферным раствором.
2.7.6. Количественное определение адсорбированной дифтерийной вакцины
351
Вносят по 100 мкл блокирующеro буферно
ro раствора во все лунки за исключением ряда А.
rотовят подходящие разведения отрицательной
и положительной контрольных сывороток (при
близительно от 0,01 МЕ/мл) и испытуемых cы
вороток. Располаrают сыворотки в соответствии
с раскладкой теста: отрицательную контроль
ную сыворотку для ряда 1, положительную KOH
трольную сыворотку для рядов 2 и 12 и испыту
eMbJe сыворотки для рядов 11 и прибавляют
по 100 мкл из каждой сыворотки в первые две
лунки соответствующих рядов. Используя MHO
roканальную микропипетку, делают двойные по
следовательные разведения от ряда В к ряду Н.
Удаляют 100 мкл из лунок последнеro ряда так,
чтобы все лунки содержали по 100 мл. Инкуби
руют при температуре 37 ос в течение 2 ч. OTMЫ
вают промывочным буферным раствором.
rотовят подходящее разведение (например,
2000 KpaTHoe) конъюrата пероксид(]зы в
блокирующем буферном растворе и прибавляют
по 100 мкл В каждую лунку. Инкубируют при
температуре 37 ос во влажной камере в течение
1 ч. Отмывают планшет промывочным буферным
раствором.
Прибавляют по 100 мкл субстратноro
раствора в каждую лунку. Инкубируют при
комнатной температуре в темноте в течение
30 мин.
Учитывают реакцию на ридере при длине
волны 405 нм в том же порядке, как прибавляли
субстрат.
Определение титра антител в сыворотке
морской свинки С помощью Vero-теста. Метод
основан на реrистрации уrнетения метаболиз
ма (1) или цитотоксичности (2) в качестве конеч
ноro эффекта. Реrистрация может быть выпол
нена путем микроскопии клеток (определение
клеточной морфолоrии) или макроскопически
(по изменению цвета).
Предел определения специфичен для каж
дою антитоксина и обычно составляет 0,015 МЕ/мл
для метода 1 и 0,05 МЕ/мл для метода 2.
Конечная точка метода может быть опреде
лена как максимальное разведение сыворотки,
которое защищает клетки от дифтерийноro TOK
сина. Активность антитоксина рассчитывается
по сравнению с референсной сывороткой MOp
ской свинки или стандартным образцом ВОЗ и
выражается в МЕ/мл.
Реактивы и оборудование:
плоскодонные планшеты для культур
клеток: 96 лунок, колонки 1 12, строки A H;
флаконы для KYf1bmyp клеток 75 см 2 ;
дифтерийный токсин;
противодифтерийная антисыворотка
морской свинки (положительная контрольная
сыворотка), полученная при иммунизации MOp
ских свинок БСП дифтерийной адсорбирован
ной вакцины;
культура клеток Ve,o (клетки почки аф
риканской зеленой мартышки). Для постановки
теста приroдны пассажи П2 П15.
Метод 1
Дифтерийный токсин оказывает цитопа
тическое действие (ЦПД) на клетки Vero и их
лизис. Антитела против токсина MOryт нейтра
лизовать указанное ЦПД. Следовательно, актив
ность дифтерийной вакцины можно определить
с помощью описанной культуральной клеточной
системы, если добавлять в культуру раЗЛИЧНbJе
разведения сыворотки от иммунизированных
животных и фиксированное количество токсина.
При проведении Vero TecTa желтый цвет среды
указывает, что клетки живые; красный что
клетки поrибли. В случае rибели части клеток
цвет среды может быть оранжевым.
Реактивы и оборудование:
модифицированная среда МОС (МЕМ): ми
нимальная основная среда (МОС, Мiпiтит Es
seпtia/ Medium МЕМ) на растворе Эрла (Еа,/е)
без rлутамина и бикарбоната натрия;
модифицированная среда 199: среда 199
на растворе Хэнкса (Напks) с rлутамином и без
бикарбоната натрия;
эмбриональная телячья сыворотка;
7,5 % раствор натрия бикарбоната;
раствор трипсина: 2,5 % раствор трип
сина;
раствор ЭДТА: 0,02 % раствор ЭДТА
(раствор Версена (Ve,seпe) 1 :5000);
модифицированный Д ФБСР (O PBS):
фосфатный буферный солевой раствор Дюль
бекко (Д ФБСР, Ou/becco's phosphate buffered
sаliпе O PBS) без кальция или маrния;
200 мМ раствор L 2лутамина;
раствор пенициллин/стрептомицина;
среда для первичной культуры: к 50 мл
модифицированной среды МОС (МЕМ) при
бавляют 440 мл воды Р, 5 мл 200 мМ раствора
L rлутамина и 10 мл 7,5 % раствора натрия би
карбоната. К 25 мл полученной среды прибавля
ют 1 ,25 мл эмбриональной телячьей сыворотки
(ЭТС);
поддерживающая среда: roтовится, как
среда для первичной культуры, но к 20 мл обо
rащенной среды МОС (МЕМ) прибавляют 0,5 мл
ЭТС вместо 1,25 мл;
среда А: к 50 мл модифицированной среды
199 прибавляют 440 мл воды Р, 5 мл 200 мМ
раствора L rлутамина и 10 мл 7,5 % раствора
натрия бикарбоната;
среда В: к 150 мл среды А прибавля
ют 3 мл эмбриональной телячьей сыворотки и
0,3 мл раствора пенициллин/стрептомицина;
среда С: к 22 мл среды А прибавляют
0,44 мл эмбриональной телячьей сыворотки и
0,44 мл раствора пенициллин/стрептомицина.
Клетки Vero культивируют в культуральных
флаконах (75 смЦ250 мл) в термостате при TeM
352
rосударственная фармакопея Республики Беларусь
пературе (36::1:1) ос в атмосфере с 5 % содержа
нием СО 2 при относительной влажности 90 %.
Сначала клетки культивируют в среде для пер
вичной культуры. После 2 3 дней роста среду
для первичной культуры заменяют на поддержи
вающую. После получения монослоя питатель
ную среду удаляют и аккуратно отмывают клетки
модифицированным Д ФБСР (D PBS). Прибав
ляют смесь из 1 объема раствора трипсина и
1 объема раствора ЭДТА во флакон. Флакон
слеrка встряхивают и инкубируют около 3 мин в
термостате (до отлипания клеток от субстрата).
Сильно встряхивают флакон для открепления
клеток. Ресуспендируют клетки в 5 6 мл свежей
среды С до получения roмоrенной суспензии. ro
товят суспензию клеток в среде С с KOHцeHTpa
цией 1 .105 клетокlмл.
Вносят по 25 мкл среды В в каждую лунку
планшета кроме колонки 1. Вносят по 25 мкл
антисыворотки морской свинки (положитель
ная контрольная сыворотка, рабочее разведе
ние в среде В 0.40 МЕ/мл) в лунки А1, А2 и
А 11. Вносят по 25 мкл испытуемых сывороток в
лунки B C колонок 1, 2 и 11. Вносят по 25 мкл
отрицательной контрольной сыворотки в лунки
ряда Н колонок 1, 2 и 11. Мноroканальной микро
пипеткой делают двукратные серийные разведе
ния колонок 2 1 О для рядов A C и 2 8 для
ряда Н. Удаляют по 25 мкл из лунок колонки 1 О
для рядов Д C и лунки колонки 8 ряд Н.
Дифтерийный токсин разводят физиолоrи
чески м раствором до получения концентрации
50 МЕiмл. rотовят 50 KpaTHoe разведение TOK
сина в среде В для получения рабочеro paCT
вора 1,0 МElмл. Прибавляют по 25 мкл pa
бочеrо раствора в лунки А12 и В12 (контроль
токсина). Делают двукратные серийные раз
ведения путем переноса 25 мкл жидкости
из одной лунки в друrую в лунках B12 H12,
каждый раз меняя наконечник. Удаляют 25 мкл
из лунки Н12. Прибавляют по 25 мкл среды В
в лунки B12 H12. Вносят по 25 мкл рабочеro
раствора токсина (1,0 МЕ/мл) в каждую лунку
рядов A H. колонки с 1 по 10, за исключением
лунок Н9 и Н10 (контроль клеток без токсина и
антисыворотки).
Планшет накрывают крышкой или пленкой и
осторожно встряхивают Инкубируют планшеты
не менее 2 ч при температуре 37 ос во влажной
камере в атмосфере 5 % СО..
Прибавляют по 200 MK суспензии клеток
Vero 1.105 клетокlмл в каждую лунку. Заклеивают
планшеты и инкубируют при температуре 37 ос
в течение 5 дней. Проверяют микробиолоrиче
скую чистоту МИl{роскопически.
Желтые лунки учитываются как неrативные,
красные лунки свидетельствуют о rибели клеток
и учитываются как положительные. Промежуточ
ные цвета (между желтым и красным) указыва
ют на наличие в лунке живых и мертвых клеток
и учитываются как положительные/отрицатель
ные. Результаты, полученные на основе учета
цвета лунок, MOryT быть подтверждены путем
микроскопическоro изучения живых и мертвых
клеток.
Активность антисыворотки морской свинки
рассчитывают, сравнивая последнюю лунку
с разведением стандартноro образца, полно-
стью нейтрализующим токсин, с последней
лункой разведения испытуемой сыворотки, де-
монстрирующим такой же эффект. При расче-
те активности следует помнить, что конечная
точка эксперимента может находиться между
отрицательной и положительной/отрицатеЛtr
ной лунками.
Метод 2
Тиазолил синий Л4ТТ восстанавливается 8
сине черный формазан митохондриальными де-
rидроrеназами живых клеток и, следовательно,
может быть использован для количественной
оценки присутствия живых клеток, указывая на
степень нейтрализации токсина антитоксином.
Белые или бесцветные лунки указывают на OT
сутствие живых клеток вследствие недостато'r!
ной нейтрализации токсина.
Реактивы и оборудование:
Л40С (Л4ЕЛ4) (минимальная основная
среда);
эмбриональная телячья сыворотка
(ЭТС);
раствор антибиотиков (содержит
10 000 единиц пенициллина, 10 Mr стрептом
цина и 25 Mr амфотерицина В в 1 мл);
200 мЛ4 раствор L 2лутамина;
трипсин ЭДТА.
тиазолил синий МТТ [3 (4,5 димеТИIr
тиазолил ) 2,5 дифенилтетразолий бромид];
1 л4 буферный (HEPES) раствор рН 8.1.
18,75 r 2 [4 (2 2идроксиэтил)пиперазин 1 илJ
этансульфоновой кислоты Р (HEPES) раство-
ряют в 82,5 мл воды Р и 30,0 мл 2 М раствора
2идроксида натрия Р;
1 О % раствор 2ЛЮКОЗЫ;
полная культуральная среда: смешивают
200 мл МОС (Л4ЕЛ4) с 1 О мл ЭТС, 3,0 мл 1 М бу
ферноro (HEPES) раствора рН 8,1, 2,0 мл 1 О ч6.
раствора rлюкозы, 2,0 мл раствора антибиот.
ков и 2,0 мл 200 мМ раствора L rлутамина:
забуференный солевой раствор рН 7,4
(ЗСР). 10,0 r натрия хлорида Р, 0,125 r калU1f
ди2идрофосфата Р, 1,44 r динатрия 2идрофсю-
фата ди2идрата Р и 0,75 r калия хлорида Р
растворяют в 1000 мл воды Р, при необходимо-
сти доводят ДО рН 7,4 (2.2.3), стерилизуют авто-
клавированием при температуре 120 ос в тече-
._ . ние 15 мин;
раствор тиазолил сине20 Л4ТТ: 0,1 r тиазо-
лил синеro МТТ растворяют в 20 мл ЗСР. Стери-
лизуют фильтрацией (0,2 мкм) и хранят во фла-
конах из TeMHoro стекла;
раствор для доведения рН: 40 мл Kиc
ты уксусной Р смешивают с 1 ,25 мл 1 л4 раство-
2.7.7. Количественное определение вакцины коклюша
353
ром кислоты хлористоводородной и 8,75 мл
воды Р;
буферный раствор для экстракции
рН 4,7: 1 О r натрия лаурилсульфата Р paCTBO
ряют в воде Р и прибавляют 50 мл диметилфор
мамида Р, доводят до 100 мл водой Р, доводят
рН с помощью раствора для доведения рН.
Клетки Vero культивируют в культуральных
флаконах (75 см 2 /250 мл) в термостате при TeM
пературе (36:t1 ) ос в атмосфере с 5 % содержа
нием С0 2 при относительной влажности 90 %.
Для выращивания используют полную пита
тельную среду. После 6 7 дней роста получают
монослой, после чеro среду удаляют, а клетки
трижды отмывают смесью трипсин ЭДТА: среду
осторожно удаляют пипеткой, прибавляют 1 мл
смеси трипсин ЭДТ А, распределяют смесь по
монослою и удаляют пипеткой. Повторяют про
цедуру дважды, а на третий раз помещают
флакон в термостат на 5 минут, пока клетки не
начнут отделяться от пластика. Сильно посту
кивают по боковой стенке флакона для отделе
ния клеток. Ресуспендируют клетки в 6 25 мл
свежей полной культуральной среды до получе
ния roмоrенной суспензии. rотовят суспензию
клеток в полной культуральной среде с KOHцeH
трацией 4.105 клетокlмл.
Вносят по 50 мкл полной культуральной
среды во все лунки планшета кроме колон
ки 1. Вносят 100 мкл антитоксической CЫBO
ротки морской свинки (положительная контроль
ная сыворотка; рабочее разведение на полной
культуральной среде 0,12 МЕ/мл) в лунку А1 и
50 мкл В лунку А 11. Вносят по 100 мкл испы
туемых сывороток (в случае необходимости их
разводят) в лунки B1 1. Добавляют по 50 мкл
этих же сывороток в лунки B11 11 COOTBeT
ствующеrо ряда. Вносят 100 мкл неrативной
контрольной сыворотки в лунку Н1 и 50 мкл
В лунку Н11. С помощью мноroканальноro доза
тора делают серийные двукратные разведения,
перенося по 50 мкл из лунки В лунку (колонки
1 10 для рядов A G и 1 8 для ряда Н).
Дифтерийный токсин с известной актив
ностью и Lf разводят полной культуральной
средой до удобной рабочей концентрации, co
держащей не менее 4 минимальных цитопа
тических доз. Вносят по 50 мкл разведенноro
токсина в каждую лунку, кроме Н9 и Н10 (KOH
троль клеток), A11 H11 (контроль сывороток) и
Д12 H12 (контроль токсина). Вносят 100 мкл
разведенною токсина в лунку А12 и выполня
ют двукратные серийные разведения, перенося
по 50 мкл из лунки В лунку (A12 H12). Удаля
ют 50 мкл из лунки Н12. Прибавляют по 50 мкл
полной среды в лунки Н9 и Н10.
Закрывают планшет крышкой или клейкой
лентой и инкубируют 1 час при комнатной TeM
nepатуре.
Прибавляют по 50 мкл суспензии 5.105
клеток/мл В каждую лунку. Планшет заклеива
.. 30. 1.12
ют и инкубируют в течение 6 дней при TeM
пературе 37 ос. Проверяют микробиолоrиче
скую чистоту микроскопически.
1 О мкл раствора тиазолил синеro МТТ при
бавляют в каждую лунку. Планшеты инкубиру
ют при температуре 37 ос еще в течение 2------4 ч.
Далее удаляют питательную среду и прибавля
ют в каждую лунку по 100 мкл буферноro paCT
вора для экстракции рН 4,7. Планшеты инкуби
руют при температуре 37 ос в течение ночи для
завершения процесса экстракции.
После завершения экстракции и paCTBO
рения формазана планшеты оценивают ви
зуально или на ридере при длине волны
570 нм. Сине черные лунки учитываются как
отрицательные (все клетки живы, токсин ней
трализован антитоксином); белые или бес
цветные лунки учитываются как положитель
ные (клетки поrибли, нейтрализация была не
эффективной ).
Активность антисыворотки морской
свинки рассчитывают, сравнивая последнюю
лунку с разведением стандартноro образ
ца, полностью нейтрализующим токсин, с по
следней лункой разведения испытуемой cы
воротки, демонстрирующим такой же эффект.
Титр нейтрализующих антител может быть
получен путем умножения фактора разведе
ния на число МЕ/мл стандартноro образца в
конечной точке. Тест считают достоверным,
если все клетки в лунках контроля токсина
поrибли, а референсный антитоксин полно
стью нейтрализует токсин, по крайней мере, в
первых двух разведен иях.
01/2013:20707
2.7.7. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ ВАКЦИНЫ
КОКЛЮША
Активность вакцины коклюша определяют
путем сравнения дозы вакцины, необходимой
для защиты мышей от эффектов летальной
дозы Bo,detella pertussis, вводимой интрацере
брально, с количеством стандартноro образца,
калиброванноro в Международных Единицах,
требующимся для получения такой же защиты.
За Международную Единицу принимают aK
тивность в установленном количестве Между
народною стандартноro образца, состоящеro
из высушенной вакцины коклюша. Эквивалент
ность Международноro стандартноro образца
в Международных Единицах устанавливается
Всемирной орrанизацией здравоохранения.
Выбор и распределение испытуемых
животных. В испытании используют здоровых
354
rосударственная фармакопея Республики Беларусь
мышей подходящей линии из одной парт. и воз
растом менее 5 недель. Разница в массе f\..СЖДУ
самым тяжелым и самым леrким животным не
должна превышать 5 r. Мышей делят на 6 rрупп
не менее чем по 16 особей и 4 rруппы по 1 С
особей. Используют мышей одноrо пола; в иных
случаях самцы и самки должны быть paBHOMep
но распределены по rруппам.
Выбор Пр080кационноrо штамма и при-
rотовление суспензии для провокации. Bы
бирают подходящий штамм В. pertussis, спо
собный привести к rибели мышей в течение
14 дней после интрацеребральной инъекции.
Если в течение 48 ч после инъекции поr.f,ба
ет более 20 % мышей, штамм является непод
ходящим. Выполняют один пересев штамма и
суспендируют собранные В. pertussis в paCTBO
ре, содержащем 1 О r/л 2uдролизата казеина Р
и 6 r/л натрия хлорида Р и имеющем показа
тель рН от 7,0 до 7,2, либо в друroм подходя
щем растворе. Определяют степень мутности
суспензии. rотовят ряд разведений с исполь
зованием Toro же раствора и назначают их
по одному каждой из rрупп по десять мышей.
Каждой мыши вводят интрацеребрально дозу
(0,02 мл или 0,03 мл) разведения, назначен
ноro rруппе, к которой она принадлежит. Через
14 дней подсчитывают в каждой rруппе коли
чество выживших мышей. Из этих результатов
вычисляют ожидаемую степень мутности cy
спензии, содержащей 100 LD 50 в каждой про
вокационной дозе. Для оценки активности
испытуемой вакцины производят свежий пе
ресев тоro же штамма В. pertussis и roтовят
из собранных орrанизмов суспензию со CTe
пенью мутности, соответствующей прибли
зительно 100 LD 50 В каждой провокационной
дозе. rотовят Зразведения провокационной
суспензии.
Определение активности. rотовят 3 серий
ных разведения испытуемой вакцины и З анало
rичных разведения стандартноro образца так,
чтобы для каждоro из промежуточных разведе
ний ожидаемая степень защиты мышей от ле
тальных эффектов провокационной дозы В. pe'
tussis составляла 50 %. Предлаrаемые дозы
составляют 1/8, 1/40 и 1/200 доли человеческой
дозы испытуемой вакцины и 0,5 МЕ, 0,1 МЕ и
0,02 МЕ препарата сравнения. Каждая из доз co
держится в объеме, не превышающем 0,5 мл.
6 разведен ий назначают по одному каждой из
rрупп, состоящих не менее чем из 16 мышей, и
вводят каждой из мышей внутрибрюшинно одну
дозу развед ния, назначенноro rруппе, к которой
относится данное животное. Через 14 17 дней
каждому животному из этих rрупп вводят интра
церебрально одну дозу суспензии для провока
ции. Суспензию для провокации и три ее разве
дения назначают по одному каждой из rрупп по
1 О мышей и вводят каждой из мышей интраце
ребрально одну дозу каждой суспензии, назна
ченной rруппе, к которой относится данное жи
вотное. Мышей, поrибших в течение 48 ч после
провокации, не учитывают при оценке резулыа
тов. В каждой из rрупп подсчитывают количество
мышей, выживающих после 14 дней. Активность
испытуемой вакцины рассчитывают относитель
но активности стандартноro образца на основе
количества животных, выживших в каждой и
rрупп из не менее чем 16 особей.
Результаты испытания являются ДOCTOBep
ными, если:
как для испытуемой вакцины, так и для
стандартноrо образца 50 % защитная доза за.
ключена в интервале между наибольшей и наи
меньшей дозами, введенными морским свинкам;
количество поrибших животных в 4 rруппах
по 1 О мышей, которым была введена провокаци
онная суспензия и ее разве;э,ения, указывает на
то, что провокационная доза приблизительно co
ответствует 100 LD 50;
при статистическом анализе не выявляет
ся отклонений от линейности и параллельности.
Испытание может быть выполнено повтор
но, но если было проведене более одноro опре
деления, то при оценке активности должны
учитываться результаты всех достоверных экс
периментов.
01/2013:20708
2.7.8. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
АДСОРБИРОВ ННОИ
СТОЛБНЯЧНОИ ВАКЦИНЫ
Активность столбнячной вакцины опред€
ляется путем назначения вакцины животным
(морским свинкам или мышам) с последующей
провокацией столбнячным токсином (методы А
или В) или путем определения титра антител
против столбнячноro анатоксина в сыворотке
крови морских свинок (метод С). В обоих случа
ях активность вакцины рассчитывается по cpaB
нению со стандартным образцом, калиброван
ным в Международных Единицах.
В странах, rде метод паралича не являет
ся обязательным, вместо методов А и В может
использоваться метод LD50' ДЛЯ метода LD 50 KO
личество животных и процедура исследования
идентичны описанным для метода паралича, но
конечной точкой является rибель животноro, а не
паралич.
3а Международную Единицу принимают aK
тивность в установленном количестве Междуна
родноro стаl"щартноro образца адсорбированно
ro столбнячноro анатоксина. Эквивалентность
Международноro стандартноro образца в Меж
2.7.8. Количественное определение адсорбированной столбнячной вакцины
355
дуна родных Единицах устанавливается Всемир
ной орrанизацией здравоохранения (ВОЗ).
БСП адсорбированная столбнячной вaK
цины откалибрована в МЕ по сравнению с
Международным стандартным образцом.
Выбор метода исследования адсорбиро
ванной столбнячной вакцины зависит от пред
полаrаемой цели.
Методы А или В применяются:
1) в процессе разработки вакцины при KO
личественном определении на сериях продук
та, произведенных для валидации производ
ственноro процесса;
2) в любом случае необходимости peBa
лидации (при значительных изменениях про
изводственноro процесса).
Метод А или В может также использовать
ся для рутинной проверки серий вакцины, но
в интересах защиты животных, по возможно
сти, следует использовать метод С.
Метод С может использоваться, за ис
ключением случаев, описанных в пунктах 1 и
2, после валидации ero для конкретноro про
дукта. Для этой цели подходящее количество
серий (обычно 3) испытываются методом С и
методом А или В.
Если различные вакцины (моновалент
ные или комбинированные) приroтовлены из
столбнячноro анатоксина одноro и Toro же
происхождения и содержат сопоставимые
уровни анатоксина (выраженные Lf/мл), вали
дация, проведенная для лекарственноro cpeд
ства с максимальным количеством компонен
тов, распространяется на комбинированные
лекарственные средства с меньшим числом
компонентов и на моновакцины. Любые KOM
бинированные лекарственные средства, co
держащие цельноклеточную коклюшную BaK
цину или конъюrированную вакцину против
Haemophilus В и столбнячный анатоксин в
одном флаконе, должны исследоваться OT
дельно.
Для комбинированных лекарственных
средств, содержащих дифтерийный и l;толб
нячный анатоксины, серолоrический тест
(метод С) может быть проведен на rруппе жи
вотных, использовавшихся для тестирова
ния адсорбированной дифтерийной вакци
ны (2.7.6), если общие условия иммунизации
для столбнячноrо и дифтерийноro компонен
тов (доза. продолжительность) валидированы
для комбинированноro лекарственноro cpeд
ства.
Дизайн описанных ниже исследований
использует множественные серийные разве
дения испытуемых и стандартных образцов.
Основываясь на данных активности, получен
ных при исследовании серийных разведений,
можно уменьшить количество животных для
получения статистически значимых результа
тов путем применения упрощенной модели,
включающей исследование одноro разведе
ния испытуемоro и стандартноro образцов.
Такая схема позволяет установить факт cy
щественноrо превышения активности испы
туемоro образца минимально допустимоro
уровня, но не дает информации о линейно
сти, параллелизме и друrих пара метрах зави
симости доза/эффект. Упрощенная схема по
зволяет значительно сократить потребность в
экспериментальных животных и должна при
ниматься во внимание любой лабораторией
в соответствии с положениями Европейской
Конвенции по защите позвоночных живот
ных, используемых для экспериментальных и
друrих научных целей.
В случае перехода на упрощенную схему
испытаний соответствие метода тестирова
ния следует постоянно мониторировать по
подходящим индикаторам, а также путем пе
риодической постановки теста с множествен
ными серийными разведениями (каждые 2
roда). Для серолоrическоrо теста подходящи
ми индикаторами для мониторинrа являются:
среднее значение и стандартное откло
нение относительных титров антитоксина или
индексов сывороток, полученных после Ha
значения фиксированных доз стандартноrо
образца вакцины;
титры антитоксинов или индексы рабо
чих контролей (положительные или отрица
тельные образцы сыворотки);
соотношение титров антитоксина или
индекса положительной контрольной CЫBO
ротки И образцов сыворотки, COOTBeTCTBY
ющих стандартному образцу вакцины.
МЕТОД А: ПРОВОКАЦИОННЫЙ ТЕСТ
НА МОРСКИХ СВИНКАХ
ОТБОР И РАСПРЕДЕЛЕНИЕ
ЭКСПЕРИМЕНТАЛЬНblХ ЖИВОТНblХ
Используют здоровых морских свинок из
одной партии с массой тела от 250 350 r. Сле
дует использовать животных одноro пола; в
противном случае самцов и самок необходимо
равномерно распределить между rруппами. Жи
вотных распределяют не менее чем на 6 равных
rрупп. Число животных в rруппе должно быть дo
статочным для получения достоверных резуль
татов в ходе описанных ниже испытаний. Если
необходимо определить активность используе
Moro токсина, следует создать еще 3 rруппы из
5 животных в качестве невакцинированноrо KOH
троля.
ВblБОР ПРОВОКАционноrо ТОКСИНА
Следует выбрать стандартный образец
столбнячноro токсина, содержащий не менее пя
тидесятикратной 50 % ной паралитической дозы
в миллилитре. Если стабильность токсина под
тверждена предварительно, нет необходимости
определять еro паралитическую дозу для каждо--
ro испытания.
356
rосударственная фармакопея Республики Беларусь
приrОТОВЛЕНИЕ РАСТВОРА ТОКСИНА
Непосредственно перед использоваНi1ем
разводят токсин подходящим растворител м
(например, забуференным солевым paCTBO
ром пептона рН 7,4) с получением раствора
провокационноro токсина, содержащеro около
пятидесятикратной 50 % ной паралитической
дозы в миллилитре. При необходимости для
определения активности токсина используют
порции раствора провакационноro токсина,
разведенные аналоrичным растворителем в
соотношениях 1:16, 1:50 и 1:160.
РА3ВЕДЕНИЕ ИСПblТУЕМblХ
И СТАНДАРТНЫХ ОБРАЗЦОВ
С помощью раствора 9 r/л натрия хло
рида Р rотовят разведения испытуемой BaK
цины и стандартною образца таким образом,
чтобы шаr разведения в серии был не более
2,5 KpaTHЫM, в котором промежуточные раз
ведения при подкожном введении морской
свинке в дозе 1,0 мл защищали около 50 %
животных от паралитических эффектов, BЫ
зываемых подкожным введением количества
столбнячною токсина, предписанноrо для
этоro испытания.
ИММУНИ3АЦИЯ И ПРОВОКАЦИЯ
Назначают по одному разведению одной
rруппе морских свинок и вводят подкожно по
1,0 мл разведения каждой морской свинке в
rруппе. Спустя 28 дней каждой свинке вводят
подкожно 1,0 мл раствора провокационно
ro токсина (содержащеrо пятидесятикратную
50 % ную паралитическую дозу).
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ
ПРОВОКАционноrо ТОКСИНА
При необходимости раствор провокаци
oHHoro токсина и три приютовленных из неro
разведения назначают по одному каждой из 3
rрупп по 5 морских свинок и вводят подкож
но по 1,0 мл каждою разведения каждой из
морских свинок rруппы, которой это разведе
ние было назначено. Активность и стабиль
ность провокационноrо токсина устанавли
вают проведением подходящею количества
исследований, подтверждающих 50 % ную па
ралитическую дозу. В дальнейшем нет необхо
димости в повторном установлении дозы для
каждою исследования.
ПОЛУЧЕНИЕ И
ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ
Состояние морских свинок оценивают
дважды в день. Животных с четкими призна
ками столбнячноro паралича изымают и ry
манным образом умерщвляют. Через 5 дней
после инъекции провокационноrо токсина
подсчитывают число непарализованных MOp
ских свинок. Активность испытуемой вакцины
рассчитывают относительно активности CTaH
дартноrо образца вакцины на основе пропор
ции непарализованных животных в каждой из
rрупп вакцинированных морских свинок. Pac
счеты проводят, используя обычные статисти
ческие методы (например, 6.3).
ТРЕБОВАНИЯ К ДОСТОВЕРНОСТИ
ИСПblТАНИЯ
Тест является достоверным, если:
для испытуемою и стандартною об
разца 50 % защитная доза лежит в интервале
между наибольшей и наименьшей дозами об
разцов, введенными морским свинкам;
rдe применимо, количество парализо
ванных животных в 3 rруппах по 5 морских
свинок, которым были введены разведения
раствора провокационною токсина указывает
на то, что провокация составляла около пя
тидесятикратной 50 % ной паралитической
дозы;
rраницы доверительноro интервала
(Р = 0,95) составляют не менее 50 % и не
более 200 % установленной активности;
статистический анализ показывает зна
чительный наклон и отсутствие отклонений
в линейности и параллелизме rрафика зави
симости доза ответ (в rлаве 5.3 описаны воз
можные альтернативы при получении значи
тельных отклонений в линейности).
Тест может быть проведен повторно, при
этом при проведении более одноro теста для
расчета активности используют объединен
ные результаты всех достоверных испытаний.
МЕТОД В: ПРОВОКАЦИОННЫЙ ТЕСТ
НА МЫШАХ
ОТБОР И РАСПРЕДЕЛЕНИЕ
ЭКСПЕРИМЕНТАЛЬНblХ ЖИВОТНblХ
Используют здоровых мышей из одной
партии в возрасте около 5 недель подходя
щей породы (линии) с массой тела 14 20 r.
Следует использовать мышей одною пола; в
противном случае самцов и самок необходи
мо равномерно распределить между rруппа
ми. Животных распределяют не менее чем
на 6 равных rpynn. Число животных в rруппе
должно быть достаточным для получения дo
стоверных результатов в ходе описанных ниже
испытаний. Если необходимо определить aK
тивность используемою токсина, следует соз
дать еще 3 rруппы из 5 мышей в качестве H
вакцинированноro контроля.
ВblБОР ПРОВОКАционноrо ТОКСИНА
Сле,nует выбрать стандартный образец
столбнячноro токсина, содержащий не менее
стократной 50 % ной паралитической дозы в
миллилитре. Если стабильность токсина п
тверждена предварительно, нет необходимости
определять ero паралитическую дозу для каждо-
ro испытания.
2.7.8. Количественное определение адсорбированной столбнячной вакцины
357
приrОТОВЛЕНИЕ РАСТВОРА ТОКСИНА
Непосредственно перед использованием раз
водят токсин подходящим растворителем (напри
мер, забуференным солевым раствором пептона
рН 7,4) с получением раствора провокационноro
токсина, содержащеro около пятидесятикратной
50 % ной паралитической дозы в 0,5 мл. При He
обходимости для определения активности токсина,
используют порции раствора провакационноro TOK
сина разведенные аналоrичным растворителем в
соотношениях 1:16, 1:50 и 1:160.
РА3ВЕДЕНИЕ ИСПЫТУЕМЫХ
И СТАНДАРТНЫХ ОБРА3ЦОВ
С помощью раствора 9 r/л натрия хлори
да Р rотовят разведения испытуемой вакцины и
стандартноro образца таким образом, чтобы шаr
разведения в серии был не более 2,5 KpaTHЫM,
в котором промежуточные разведения при под
кожном введении в дозе 0,5 мл на мышь защи
щали около 50 % животных от паралитических
эффектов, вызываемых подкожным введением
количества столбнячноro токсина, предписанно
ro для этоro испытания.
ИММУНИ3АЦИЯ И ПРОВОКАЦИЯ
Назначают по одному разведению одной
rруппе мышей и вводят подкожно по 0,5 мл раз
ведения каждому животному в rруппе. Спустя
28 дней каждому животному вводят подкожно
0,5 мл раствора провокационноro токсина (co
держащеro пятидесятикратную 50 % ную пара
литическую дозу).
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ
ПРОВОКАционноrо ТОКСИНА
При необходимости раствор провокационно
ro токсина и три приroтовленных из неro разве
дения назначают по одному каждой из 3 rрупп по
не менее 5 мышей и вводят подкожно по 0,5 мл
каждоro разведения каждому животному rруппы,
которому это разведение было назначено.
ПОЛУЧЕНИЕ И
ИНТЕРПРЕТАЦИЯ РЕ3УЛЬТАТОВ
Состояние мышей оценивают дважды в
день. Животных с четкими признаками столб
нячноrо паралича изымают и ryMaHHbIM образом
умерщвляют. Через 4 дня после инъекции прово
кационноro токсина подсчитывают число непа
рализованных мышей. Активность испытуемой
вакцины рассчитывают относительно активности
стандартноro образца с использованием обыч
ных статистических методов (например, 5.3) на
основе пропорции непарализованных животных
в каждой rруппе вакцинированных мышей.
Тест является достоверным, если:
для испытуемоro и стандартноro образца
50 % защитная доза лежит в интервале между
наибольшей и наименьшей дозами образцов,
введенными мышам;
46 За.. 1712.
rдe применимо, количество парализован
ных животных в 3 rруппах по 5 мышей, которым
были введены разведения раствора провокаци
онноro токсина, указывает на то, что провокация
составляла около пятидесятикратной 50 % ной
паралитической дозы;
rраницы доверительноro интервала
(Р = 0,95) составляют не менее 50 % и не более
200 % установленной активности;
статистический анализ показывает значи
тельный наклон и отсутствие отклонений в ли
нейности и параллелизме rрафика зависимости
доза ответ (в rлаве 5.3 описаны возможные аль
тернативы при получении значительных откло
нений в линейности).
Тест может быть проведен повторно, при
этом при проведении более одноro теста для
расчета активности используют объединенные
результаты всех достоверных испытаний.
МЕТОД С: ОПРЕДЕЛЕНИЕ АНТИТЕЛ
у МОРСКИХ СВИНОК
ОТБОР И РАСПРЕДЕЛЕНИЕ
ЭКСПЕРИМЕНТАЛЬНЫХ ЖИВОТНЫХ
Используют здоровых морских свинок из
одной партии с массой тела 250 350 r. Следует
использовать животных одноrо пола; в против
ном случае самцов и самок необходимо paBHO
мерно распределить между rруппами. Животных
распределяют не менее чем на 6 равных rрупп.
Число животных в rруппе должно быть достаточ
ным для получения достоверных результатов в
ходе описанных ниже испытаний. Для получе
ния неrативной контрольной сыворотки исполь
зуют дополнительную rруппу животных тою же
происхождения. При условии валидации может
быть применена референсная отрицательная
контрольная сыворотка.
СТАНДАРТНЫЙ ОБРА3ЕЦ
Используют подходящий стандартный об
разец, например БСП адсорбированной столб
нячной вакцины, серию вакцины с доказанной
клинической эффективностью или друryю репре
зентативную серию, активность которой измере
на в Международных Единицах по сравнению с
БСП адсорбированной столбнячной вакцины
или Международным стандартным образцом aд
сорбированноro дифтерийноro анатоксина.
РАЗВЕДЕНИЕ ИСПЫТУЕМЫХ
И СТАНДАРТНЫХ ОБРАЗЦОВ
rотовят серийные разведения испытуе
мой вакцины и стандартноro образца на pac
творе 9 r/л натрия хлорида Р; разведения
выполняютс2,5 5 кратным шаroм. Следует при
roтовить не менее трех разведений, например,
0, 16 МЕ/мл для каждой серии. Следует ис
пользовать разведения для иммунизации не
позже 1 часа с момента приroтовления. Одно
разведение вводят одной rруппе свинок.
358
rосударственная фармакопея Республики Беларусь
ИММУНИЗАЦИЯ
Каждой свинке вводят подкожно 1,0 м,. раз
ведения, предназначенноro для данной rpy,':-jbl
животных.
ЗАБОР КРОВИ
Через 35..-....42 дня после иммунизации ПОD
ходящим методом берут образцы крови у каждо
ro вакцинированною и контрольною животною.
приrОТОВЛЕНИЕ ОБРА3ЦОВ
СblВОРОТКИ
Следует избеrать повторноro заморажива
ния/оттаивания образцов сыворотки. Для пре
дотвращения микробной контаминации об
разцов все манипуляции лучше производить в
ламинарном боксе.
ОПРЕДЕЛЕНИЕ ТИТРА АНТИТЕЛ
Определяют титры или индексы для каждо
ro образца сыворотки с помощью подходящеrо
ИММУНОХИМИ'-l8СКОro метода (2.7.1). Методы. при
веденные ниже, (иммуноферментный анализ
тИФА (епzуте liпkеd immuпosorbeпt assay
ELlSA) и токсинсвязывающее инrибирование
(tохiп Ыпdiпg iпhiЫtiоп ToВl» признаны подхо
дящими.
РАСЧЕТ АКТИВНОСТИ
Активность испытуемой вакцины в МЕ OTHO
сительно стандартноro образца рассчитывают,
используя обычные методы статистики (напри
мер, 5.3).
ТРЕБОВАНИЯ К ДОСТОВЕРНОСТИ ТЕСТА
Тест является достоверным, если:
rраницы доверительноro интервала
(Р = 0,95) составляют не менее 50 % и не более
200 % установленной активности;
статистический анализ показывает значи
тельный наклон и отсутствие отклонений в ли
нейности и параллелизме rрафика зависимости
доза ответ (в rлаве 5.3 описаны возможные аль
тернативы при получении значительных откло
нений в линейности).
Тест может быть проведен повторно, при
этом при проведении более одноrо теста для
расчета активности используют объединенные
результаты всех достоверных испытаний.
Следующий раздел приводится для инфор
мации.
Руководство по исследованию адсорби
рованной столбнячной вакцины
МЕТОД А: ПРОВОКАЦИОННЫЙ ТЕСТ
НА МОРСКИХ СВИНКАХ
ПОЛУЧЕНИЕ
И ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ
С целью минимизации страданий испытуе
мых животных рекомендуется учитывать пара
литический ответ соrласно шкале, приведенной
ниже. Шкала учитывает типичные признаки,
Korдa подкожные инъекции провокационно
ro токсина проводятся в средостение прямо
позади rрудины острой ипюй по направлению
к шее морской свинки. Степень Т3 принимает
ся за конечную точку, но, при наличии опреде
ленноro опыта, может быть использована CTe
пень Т2.
Токсин столбняка вызывает rJаралич по
крайней мере одной из передних конечностей,
который может быть выявлен на ранчей стадии.
Шкала воздействия столбнячноro токсина на
морски)( C6I'1HKOf! может быть охарактеризована
следующим образом:
Т1: небольшая туroподвижность одной пе..
. редней конечности, идентификация паралича
затруднена;
Т2: парез одной передней конечности, KO
торая все еще может функционировать;
Т3: паралич одной передней конечности;
животное двиrается неохотно, тело часто HeMHO
ro искривлено (в форме банана) вследствие CKO
лиоза;
Т4: передняя конечность полностью Ha
пряжена, пальцы лапы неподвижны; мускульное
сокращение передней конечности очень Bыpa
жено, обычно наблюдается сколиоз;
Т5: стопбнячные судороrи, продолжитель
Hble тонические судороrи;
о: смерть.
МЕТОД В: ПРОВОКАЦИОННЫЙ ТЕСТ
НА МЫШАХ
ПОЛУЧЕНИЕ
И ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ
Чтобы минимизировать страдания испыту
емь!х животных, рекомендуется оценивать CTe
пень паралича по шкале, приведенной ниже.
Шкала учитывает типичные признаки, если инъ
екция токсина выполнена со спины, вблизи от
одной из задних HOr. Степень ТЗ является ко-
нечной точкой, но при приобретении HeKOTOpo
ro опыта вместо нее может быть использована
стадия Т2. Токсин столбняка вызывает парез и
последующий паралич задней ноrи, что может
быть выявлено на ранней стадии. Шкала пора
жения токсином на мышах учитывает следую
щие признаки:
Т 1: небольшая туroподвижность задней
ноrи, которая наблюдается только Korдa мышь
поднимают за хвост;
Т2: парез задней ноrи, которая все еще
может функционировать;
Т3: паралич задней ноrи, функция невоз
можна;
Т4: задняя Hora полностью риrидна.
пальцы неподвижны;
Т5: непрерывная тоническая судороrз
мышц ноrи;
о: смерть.
2.7.8. Количественное определение адсорбированной столбнячной вакцины
359
МЕТОД С: ОПРЕДЕЛЕНИЕ АНТИТЕЛ
У МОРСКИХ СВИНОК
подrОТОВКА ОБРА3ЦОВ СЫВОРОТКИ
Следующая методика приrодна для приrо
товления образца сыворотки. Переворачивают
пробирку с образцом крови 6 раз и выдержива
ют при температуре 37 ос в течение 2 Ч, затем
при температуре 4 ос в течение 2 ч. Центрифуrи
руют при комнатной температуре с ускорением
800 9 в течение 20 мин. Сыворотку переносят в
стерильные пробирки и хранят при температуре
20 ос. Данная методика позволяет получить на
выходе не менее 40 % сыворотки.
ОПРЕДЕЛЕНИЕ ТИТРА АНТИТЕЛ
ТИФА и ToВl, приведенные ниже, являются
примером иммунохимических методов, признан
ных подходящими для определения титра анти
тел.
Определение титра антител в сыворотке
морской свинки с помощью иммунофермент
Horo анализа (тИФА (ELlSA». в лунки планшета
для тИФА с адсорбированным столбнячным
анатоксином вносят разведения испытуемой
и референсной сывороток. Положительную и
отрицательную контрольные сыворотки морской
свинки включают в каждую постановку для
контроля работы системы. Далее добавляют
конъюrированные с пероксидазой антитела
кролика или козы против IgG морской свинки,
после чеrо вносят субстрат. Измеряютоптическую
плотность и рассчитывают относительный титр
антител, используя обычные методы статистики
(например, 5.3).
Реактивы и оборудование:
планшеты для тИФА: 96 лунок, колонки
1 12, строки A H;
БСП противостолбнячной aHтиcывo
ротки морской свинки (положительная KOH
трольная сыворотка);
КОНЪЮ2ат пероксидазы: конъюrирован
ные с пероксидазой хрена кроличьи или козьи
антитела против IgG морской свинки;
столбнячный анатоксин;
карбонатный буферный раствор для
сорбции анти2ена рН 9,6: 1,59 r натрия карбо
ната безводН020 Р и 2,93 r натрия 2идрокарбо
ната Р растворяют в 1000 мл воды Р Разливают
в бутыли по 150 мл и стерилизуют автоклави
рованием при температуре 121 ос в течение
15 мин;
забуференный солевой раствор рН 7.4
(ЗСР): 80,0 r натрия хлорида Р, 2,0 r калия ди2и
дрофосфата Р, 14,3 r динатрия 2идрофосфа
та ди2идрата Р и 2,0 r калия хлорида Р paCT
воряют в 1000 мл воды Р; хранят при комнатной
температуре для предотвращения кристаллиза
uми. перед использованием разводят в 1 О раз
8одой р;
раствор кислоты лимонной: 10,51 r Kиc
лоты лимонной Р растворяют в 1000 мл воды Р
и доводят до значения рН 4,0 раствором 400 r/л
натрия 2идроксида Р;
промывочный буферный раствор: ЗСр,
содержащий 0,5 r/л полисорбата 20 Р;
блокирующий буферный раствор: зср,
содержащий 0,5 r/л полисорбата 20 Р и 25 r/л
высушенноro обезжиренноro молока;
субстратный раствор: незадолro до ис
пользования 1 О Mr диаммоний 2, 2 азинобис(3
этилбензотиазолин 6 сульфоната) Р (ABTS)
растворяют в 20 мл раствора кислоты лимонной;
немедленно перед использованием прибавляют
5 мкл концентрироваННО20 раствора водорода
пероксида Р
Методика
Ниже приведен при мер подходящей pac
кладки образцов и контролей. Лунки ряда 1A H
используют для отрицательной контрольной cы
воротки, лунки рядов 2A H и 12A H для по
ложительной контрольной сыворотки для мони
торинrа количественноro определения. Лунки
рядов 3 11A H используют для испытуемых
образцов.
Покрывают лунки планшета для тИФА
100 мкл раствора столбнячноro анатоксина
(0,5 Lf/мл в карбонатном буферном растворе для
сорбции антиrена рН 9,6). Планшет оставляют на
ночь при температуре 4 ос во влажной камере.
Чтобы избежать эффекта температурноro
rрадиента, не следует складывать в стопку
больше 4 пластин.
На следующий день пластины тщательно
отмывают отмывочным буфером. Планшеты
блокируют прибавлением 100 мкл блокиру
ющеro буферноro раствора в каждую лунку.
Инкубируют во влажной камере при температуре
37 ос в течение 1 ч. Отмывают планшеты
промывочным буферным раствором.
Вносят по 100 мкл блокирующеro буферно
ro раствора во все лунки за исключением ряда А.
rотовят подходящие разведения отрицательной
и положительной контрольных сывороток (при
близительно от 0,01 МЕ/мл) и испытуеМ!=>IХ cы
вороток Располаrают сыворотки в соответствии
с раскладкой теста: отрицательную контроль
ную сыворотку для ряда 1, положительную KOH
трольную сыворотку для рядов 2 и 12 и испытуе
мые сыворотки для рядов 3 11 и прибавляют по
100 мкл из каждой сыворотки в первые две лунки
соответствующих рядов. Используя мноroканаль
ную микропипетку, делают двойные последова
тельные разведения от ряда В к ряду Н. Удаляют
100 мкл из лунок последнеro ряда так, чтобы все
лунки содержали по 100 мл. Инкубируют при TeM
пературе 37 ос в течение 2 ч. Отмывают промы
вечным буферным раствором.
rотовят подходящее разведение (например,
2000 KpaTHoe) конъюrата пероксидазы в
блокирующем буферном растворе и прибавляют
360
rосударственная фармакопея Республики Беларусь
по 100 мкл В каждую лунку. Инкубируют при
температуре З7 ос во влажной камере в те ение
1 Ч. Отмывают планшет промывочным буферным
раствором.
Прибавляют по 100 мкл субстратноro
раствора в каждую лунку. Инкубируют при
комнатной температуре в темноте в течение
30 мин.
Учитывают реакцию на ридере при длине
волны 405 нм в том же порядке, как прибавляли
субстрат.
Определение титра антител в CЫBO
ротке морской свинки методом токсин
связывающеrо инrбирования (ToBI). Токсин
или анатоксин столбняка прибавляют к
последовательным разведения м испытуемых
или референсных сывороток; смеси сыворотки/
антиrена инкубируют в течение ночи. Для
определения несвязанноrо токсина или
анатоксина смеси вносят в лунки планшета
для тИФА с сорбированными антителами.
Далее прибавляют коньюrат пероксидазы с
лошадиными противостолбнячными антито
ксическими антителами и субстратом. Измеряют
оптическую плотность и рассчитывают титр
антител, используя обычные статистические
методы (например, 5.3). Положительную и OT
рицательную контрольные сыворотки включают
в каждую постановку, чтобы контролировать
работу системы.
Реактивы и оборудование:
КРУ2лодонные планшеты из полисти
роола для при20товления разведений;
плоскодонные пластины для тИФА
(ELlSA);
столбнячный токсин или анатоксин;
БСП противостолбнячной сыворотки
морской свинки (положительная контрольная
сыворотка);
лошадиные противостолбнячные aHти
тела (/gG);
КОНЪЮ2ат пероксидазы хрена с лоша
диными противостолбнячными антителами
(lgG);
карбонатный буферный раствор рН 9,6:
1,5 r натрия карбоната безводН020 Р, 2,39 r
натрия 2идрокарбоната Р и 0,2 r натрия
азида Р растворяют в 1000 мл воды Р, доводят
до рН 9,6 и автоклавируют при температуре
121 ос в течение 20 мин;
ацетатный буферный раствор рН 5,5:
90,2 r натрия ацетата безводН020 Ррастворяют
в 900 мл воды Р, доводят до рН 5,5 насыщен
ным раствором кислоты лимонной Р и разводят
водой Р до объема 1000 мл.
забуференный солевой раствор рН 7,2
(3СР): 135,0 r натрия хлорида Р, 20,55 r
динатрия 2идрофосфата диаидрата Р и 4,80 r
натрия ди2идрофосфата МОН02идрата Р
растворяют в воде Р и доводят до объема 15 л
этим же растворителем; автоклавируют при
температуре 100 0 С в течение 60 мин;
буферный раствор для разведения: ЗСр,
содержащий 5 r/л бычье20 альбумина Р и 0,5 r/л
полисорбата 80 Р;
блокирующий буферный раствор: ЗСР,
содержащий 5 r/л бычье20 альбумина Р;
раствор тетраметилбензидина: paCT
вор 6 r/л тетраметилбензидина Р в 96 %
спирте Р; вещество растворяется в течение
ЗО О мин при комнатной температуре;
субстратный раствор: смешивают
90 мл воды Р, 10 мл ацетатноro буферноro paCT
вора рН 5,5, 1,67 мл раствора тетраметилбензи
дина и 20 мкл концентрироваНН020 раствора
водорода пероксида Р;
промывочный раствор: вода из под
крана, содержащая 0,5 r/л полисорбата 80 Р.
Методика
Блокируют планшеты для приrотовления
разведений, помещая в каждую лунку 150 мкл
блокирующеro буферноro раствора. Закрывают
планшеты крышкой или пленкой. Инкубируют
во влажной камере при температуре 37 ос в Te
чение 1 ч. Отмывают планшет промывочным
раствором.
Вносят по 100 мкл ЗСР В каждую лунку.
Вносят 100 мкл референсноro противо
столбнячноro антитоксина морской свинки в
первую лунку ряда А. Вносят по 100 мкл He
разведенных испытуемых сывороток в первые
лунки необходимоro числа рядов. Используя
мноroканальную микропипетку, делают двойные
последовательные разведения (до колонки 10),
пере нося по 100 мкл от одной лунки к следующей.
Удаляют 100 мкл из последней колонки так,
чтобы все лунки содержали по 100 мкл.
rотовят разведение токсина или анатоксина
столбняка 0,1 Lf/мл В ЗСР. Вносят по 40 мкл
этоro разведения во все лунки, за исключением
колонки 12. Колонка 11 содержит положительный
контроль. Вносят по 40 мкл ЗСР В лунки колонки
12 (отрицательный контроль). Осторожно
встряхивают планшеты и закрывают крышками.
Покрывают лунки планшета для тИФА:
немедленно перед использованием делают
подходящее разведение лошадиных протиВ<r
столбнячных антител (lgG) в карбонатном
буферном растворе рН 9,6 и прибавляют по
100 мкл полученных разведений в каждую лунку.
Инкубируют 2 серии планшетов во влажной камере
при температуре 37 ос в течение ночи. Чтобы
избежать эффекта температурноro rрадиента, не
следует складывать больше 4 пластин в стопку.
Закрывают планшеты крышками.
На следующий день планшеты отмывают
промывочным раствором. Блокируют планшеты.
прибавляя по 125 мкл блокирующеro буфернoro
раствора в каждую лунку. Инкубируют во влажнoi
камере при температуре 37 ос в течение 1 ч.
Отмывают планшеты промывочным раствором.
2. 7.9. Определениефункциональноzосостоянияfс--фраzментаиммуноzлобулuна
361
Переносят по 100 мкл преинкубирован
ных смесей из планшетов для разведения в
соответствующие лунки планшетов для тИФА,
начиная с колонки 12 и продолжая с 1 по 11.
Закрывают планшеты крышками. Инкубируют
во влажной камере при температуре 37 ос в
течение 2 ч. Отмывают планшеты промывоч
ным раствором.
rотовят подходящее разведение (напри
мер, 4000 KpaTHoe) конъюrата пероксидазы с
лошадиными противостолбнячными антите
лами (lgG) в буферном растворе для разве
дения. Вносят по 100 мкл В каждую лунку и
закрывают планшеты. Инкубируют во влаж
ной камере при температуре 37 ос в тече
ние 1,5 ч. Отмывают планшеты промывочным
раствором.
Вносят по 100 мкл раствора субстра
та в каждую лунку. Инкубируют при KOMHaT
ной температуре. Останавливают реакцию в
установленный срок (в пределах 10 мин) при
бавлением 100 мкл 2 М раствора кислоты
серной в каждую лунку в том же порядке, в
каком вносили субстрат.
Измеряют оптическую плотность при
длине волны 450 нм немедленно после при
бавления серной кислоты или хранят планше
ты в темноте до снятия результатов.
01/2013:20709
2.7.9. ОПРЕДЕЛЕНИЕ
ФУНКЦИОНАльноrо
СОСТОЯНИЯ Fc-ФРАrМЕНТА
иммУноrЛОБУЛИНА
Определение функционаЛЬН020 состояния
Fс фра2мента иММУН02лобулина выполняется
методом А или В. Метод В является адапта
цией метода А для микропланшетов при изме
рении комплемент зависиМ020 лизиса. Разли
чия в проведении теста между методами А и В
указаны ниже.
РЕАКТИВЫ
Стабилизированная человеческая кровь.
Человеческую кровь rруппы О собирают в aH
тикоаryлянтный раствор АСО. Стабилизирован
ную кровь хранят при температуре 4 ос не более
3 недель.
3абуференный солевой раствор рН 7,2.
1,022 r динатрия 2идрофосфата безводН020 Р,
0,336 r натрия ди2идрофосфата безводН020 Р
и 8,766 r натрия хлорида Р растворяют в 800 мл
воды Р и доводят до объема 1000 мл этим же
растворителем.
Раствор ма2ния и кальция. 1,103 r кальция
хлорида Р и 5,083 r ма2ния хлорида Р paCTBO
47 Зак 1712
ряют В воде Р и доводят до объема 25 мл этим
же растворителем.
Основной буферный раствор барбитала.
207,5 r натрия хлорида Р и 25,48 r барбитал
натрия Р растворяют в 4000 мл воды Р и дo
водят до рН 7,3, используя 1 М раствор кисло
ты хлористоводородной. Прибавляют 12,5 мл
раствора маrния и кальция и доводят водой Р до
объема 5000 мл. Фильтруют через мембранный
фильтр (размер пор 0,22 мкм). Хранят при TeM
пературе 4 ос в прозрачных контейнерах.
Альбумин барбиталовый буферный pacт
вор. 0,150 r бычье20 альбумина Р растворяют в
20 мл основноro буферноro раствора барбитала
и доводят водой Р до объема 100 мл. r отовят He
посредственно перед использованием.
Раствор таниновой кислоты. 1 О Mr тaHи
новой кислоты Р растворяют в 100 мл забуфе
ренноro солевоro раствора рН 7,2. rотовят непо
средственно перед.использованием.
Комплемент морской свинки. rотовят пул
сыворотки из крови не менее 1 О морских свинок.
Сыворотку отделяют от свернувшейся крови цeH
трифуrированием при температуре около 4 ОС.
Сыворотку хранят в небольших количествах при
температуре ниже:-70 ос. Непосредственно перед
использованием разводят альбумин барбитало
вым буферным раствором до 125-------200 СН 5d мл и
хранят в течение испытания в ледяной бане.
#СН 50 (rемолитическая единица активности
комплемента) концентрация комплемента в
контрольной сыворотке, вызывающая в данных
реакционных условиях теста аrrлютинации
лизис 50 % эритроцитов.#
Анти2ен краснухи. Подходит антиrен Kpac
нухи для титрования по инrибированию reMar
rлютинации (НIТ). Титр должен составлять более
256 НА единиц.
Приrотовление танизированных чело-
веческих эритроцитов. Эритроциты отделяют
центрифуrированием соответствующеro объема
стабилизированной человеческой крови. Про
мывают клетки не менее трех раз забуференным
солевым раствором рН 7,2 и ресуспендируют в
этом же растворе в концентрации 2 % (об/об).
0,2 мл раствора таниновой кислоты прибавляют
к 14,8 мл забуференноro солевоro раствора рН
7,2. Смешивают 1 объем свежеприroтовленноro
разведения с 1 объемом суспензии эритроцитов
человека и инкубируют при температуре 37 ос
в течение 10 мин. Клетки отделяют центрифуrи
рованием (800 9 в течение 10 мин), супернатант
удаляют, а клетки однократно промывают забу
ференным солевым раствором рН 7,2. Танизи
рованные клетки ресуспендируют в забуферен
ном солевым растворе рН 7,2 в концентрации
1 % (об/об).
Наrрузка танизированных человече-
ских эритроцитов антиrеном. К COOTBeTCTBY
ющему объему (V.) танизированных клеток дo
362
rосударственная фармакопея Республики Беларусь
бавляют антиrен краснухи (0,2 мл на 1,0 мл
клеток) и инкубируют при температуре 37 ос в
течение ЗО мин. Клетки отделяют центрифуrи
рованием (800 9 в течение 10 мин) и удаля
ют супернатант. Прибавляют альбумин барби
таловый буферный раствор в объеме, равном
объему удаленноro супернатанта, клетки pecy
спендируют и отделяют, как описано выше, и
повторяют операцию промывки. Клетки pecy
спендируют альбумин барбиталовым буфер
ным раствором, используя объем, эквивалент
ный 3/4 V s ' получая таким образом начальный
объем (V,).
900 мкл альбумин барбиталовоrо буфер
ною раствора смешивают с 100 МКЛ V i , получая
таким образом остаточный объем (V,), и опреде
ляют исходную оптическую плотность при длине
волны 541 нм (А). V, разводят альбумин бар
биталовым буферным раствором с коэффици
ентом разведения, эквивалентным А, получая
таким образом конечный скорректированный
объем = V, . А сенсибилизированных челове
ческих эритроцитов и устанавливая значение А
(1,0:10,1) для десятикратноro разведения.
Связывание антителами сенсибилизиро
ванных эритроцитов. rотовят один за друrим
в дубликатах перечисленные растворы, исполь
зуя для каждою из них отдельную одноразовую
кювету или пробирку.
(1) Испытуемые растворы. При необходи
мости доводят значение рН испытуемоro имму
ноrлобулина до 7,0.
При выполнении метода А объемы испытуе
моro образца разводят альбумин барбиталовым
буферным раствором до получения ЗО Mr и 40 Mr
иммуноrлобулина и доводят до объема 900 мкл
альбумин барбиталовым буферным раствором.
При выполнении метода В объемы испытуе
мою образца разводят альбумин барбиталовым
буферным раствором до получения 15 Mr и 30 Mr
иммуноrлобулина и доводят до объема 1200 мкл
альбумин барбиталовым буферным раствором.
(2) Растворы сравнения. rотовят так же, как
и испытуемые растворы с использованием БСП
человечеСК020 иММУН02лобулина.
(3) Контроль комплемента. 900 мкл альбу
мин барбиталовоro буферноro раствора.
При выполнении метода А к содержимо
му каждой кюветы или пробирки прибавляют по
100 мкл сенсибилизированных человеческих
эритроцитов и хорошо перемешивают.
Инкубируют при комнатной температуре в
течение 15 мин, прибавляют 1000 мкл альбу
мин барбиталовоro буферною раствора, oтдe
ляют клетки центрифуrированием (1001) 9 в тече
ние 10 мин) и удаляют 1900 мкл супернатанта.
Прибавляют 1900 мкл альбумин барбиталово
ro буферноro раствора повторяют операцию OT
мывки, оставляя объем 200 мкл. Испытуемые
образцы MOryT храниться в укупоренных кюветах
(пробирках) при температуре 4 ос не более 24 ч.
При выполнении метода В к содержи
му каждой кюветы или пробирки прибавляют по
300 мкл сенсибилизированных человеческих
эритроцитов и хорошо перемешивают (конеч
ная концентрация иммуноrлобулина колеблет
ся в пределах 1O 20 мr/мл). Инкубируют при
комнатной температуре в течение 15 мин, при
бавляют 1500 мкл альбумин барбиталовоrо бу
ферноrо раствора, отделяют клетки центрифуrи
рованием (1000 9 в течение 10 мин) и удаляют
супернатан Прибавляют 3000 мкл альбумин
барбиталовоro буферноro раствора и повторя
ют всю операцию промывки, оставляя объем
300 мкл. Испытуемые образцы MOryт храниться
в укупоренных пробирках при температуре 4 ос
не более 24 ч.
Комплемент-зависимый rемолиз. При
выполнении метода А для измерения rемоли
за к испытуемому образцу прибавляют 600 мкл
предварительно HarpeToro до температуры
37 ос альбумин барбиталовоro буферноro paCT
вора, тщательно ресуспендируют клетки путем
пипетирования (не менее пяти раз) и помеща
ют кювету в термостатированный держатель
кювет спектрофотометра. Через 2 мин прибав
ляют 200 мкл разведенноro комплемента MOp
ских свинок (125-------200 СН5iмл), тщательно пе
ремешивают путем двукратноro пипетирования
и тотчас после второro пипетирования начинают
реrистрацию оптической плотности при длине
волны 541 нм с использованием альбумин бар
биталовоro буферноro раствора в качестве KOM
пенсационною раствора. Измерение прекраща
ют при очевидном прохождении точки переrиба
rрафика зависимости оптической плотности от
времени.
При выполнении метода В для измерения
степени rемолиза к испытуемому образцу при
бавляют 900 мкл предварительно Harpeтoro до
температуры 37 ос альбумин барбиталовоro бу
ферноro раствора, осторожно ресуспендиру
ют клетки путем пипетирования (не менее пяти
раз). Микропланшеты должны быть предвари
тельно HarpeTbI до температуры 37 ос. Переносят
по 240 мкл каждоro раствора в 4 лунки микро
планшета и инкубируют при температуре 37 ос
в течение 6 мин, осторожно встряхивая каждые
1 О с. В каждую лунку микропланшета прибавля
ют по 60 мкл разведенноro комплемента морских
свинок (150 СН5iмл), тщательно перемешивают в
течение 1 О с и тотчас начинают реrистрацию оп
тической плотности при длине волны 541 нм при
температуре 37 ос; измерение проводят каждые
20 с. Измерение прекращают при очевидном
прохождении точки переrиба rрафика зависимо
сти оптической плотности от времени.
Оценка. Для каждой кюветы, пробирки.
лунки определяют уroл наклона (S) кривой reMo
лиза в районе точки переrиба путем cerMeHTa
ции участка с наибольшим уклоном на подходя
2.7.10. Количественное определение фактора свертывания крови V/I
363
щие временные отрезки (например, l1t = 1 мин)
и вычисляют значения 8 между соседними точ
ками пересечений, выраженные в М/мин. Наи
большее значение 8 принимают за (8 ). Кроме
ехр
тоro определяют оптическую плотность в начале
измерения (As) путем экстраполяции кривой,
практически линейной и параллельной оси Bpe
мени в течение первых нескольких минут. Kop
ректируют значение 8 ехр по формуле:
8
8'== .
As
Рассчитывают среднее арифметическое
значений S' для каждоro образца (испытуемых
растворов и растворов сравнения).
Рассчитывают индекс функции Fc (/FC) по
формуле:
100.( 8' 8' c )
/ == ,
Fc 8's 8'с
rдe:
8' среднее арифметическое корректи
рованноro уrла наклона для испытуемоro paCT
вора;
8' s среднее арифметическое корректиро
ванноro уrла наклона для раствора сравнения;
8' с среднее арифметическое корректиро
ванноro уклона для контроля комплемента.
Рассчитывают индекс функции Fc для испы
туемоro образца: значение должно быть не ниже
указанноro в листке вкладыше, сопровождаю
щем стандартный образец.
01/2013:20711
2.7.10. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ ФАКТОРА
СВЕРТЫВАНИЯ КРОВИ
ЧЕЛОВЕКА VII
Количественное определение фактора
свертывания крови VII производят по еro биоло
rмческой активности в виде комплекса фактора
Vlla с тканевым фактором при активации факто
ра Х в присутствии ионов кальция и фосфолипи
1JP8. Активность препарата фактора VII оценива
IOТ путем сравнения количества, необходимою
ДЛЯ достижения определенной скорости образо
вания фактора Ха в испытуемой смеси, coдep
_щей вещества, принимающие участие в акти
вации фактора Х, и количества Международноro
стандартноro образца или стандартноro образ
.... калиброванноro в Международных Едини
Q8X. которое требуется для достижения такой же
аюрости обр,азования фактора Ха.
За Международную Единицу принимают aK
тивность фактора VII в установленном количе
стве Международноro стандартноro образца,
состоящеro из лиофилизированной плазмы. Эк
вивалентность Международною стандартноro
образца в Международных Единицах устанавли
вается Всемирной орrанизацией здравоохране
ния.
БСП фактора свертывания крови VI1 Ka
либрван в МЕ в сравнении с Международным
стандартным образцом.
Хромоrенный метод количественноro опре
деления включает две последовательные
стадии. Первая стадия состоит в зависящей от
фактора VII активации фактора Х смесью pe
areHToB, включающей тканевый фактор, фос
фолипиды и ионы кальция. На второй стадии
происходит ферментативное расщепление xpo
MoreHHoro субстрата фактором Ха с образова
нием хромофора, который определяют спектро
фотометрически. В соответствующих условиях
испытания существует линейная зависимость
между скоростью образования фактора Ха и
концентрацией фактора. Содержание метода
количественноro определения описывается
следующей схемой.
1--я стадия
тканевый фактор, Са 2 +
а) фактор VII
.. фактор Vlla
фактор VII а, Са 2 +
Ь) фактор Х . фактор Ха
тканевый фактор/фосфолипид
2--я стадия
фактор Ха
хромоrенный субстрат .. пептид + хромофор
в обеих стадиях используются peareHTbI, дo
ступные из ряда коммерческих источников. Хотя
в состав peareHToB MOryT вноситься некоторые
изменения, их основные свойства должны COOT
ветствовать нижеприведенным спецификациям.
PEArEHTbI
PeareHT факторов коаryляции состоит из
очищенных белков человеческоro или ювяжьеro
происхождения, включающих фактор Х и TpOM
бопластиновый тканевый фактор/фосфолипид
в качестве активатора фактора VII. Эти белки
подверrнуты частичной очистке и не должны co
держать примесей, мешающих активации фак
тора VII или фактора Х. Фактор Х содержится
в количествах, приводящих к конечной KOHцeH
трации на первой стадии определения, находя
щейся в пределах 10 З50 нмолы/,' предпочти
тельно 14 70 нмоль/л. В качестве тканевоro
фактора/фосфолипида может использоваться
тромбопластин, полученный из природных ис
точников, например, из мозrа крупноro poraToro
скота или кроликов или синтетический препарат.
Тромбопластин, подходящий для использования
364
rосударственная фармакопея Республики Беларусь
в определении протромбиновою периода, раз
бавляют буфером в соотношении от 1:5 до 1 :50
так, чтобы окончательная концентрация ионов
кальция составляла 15 25 м моль/л. Образо
вание конечною фактора Ха проводят в paCTBO
ре, содержащем человеческий или бычий аль
бумин в концентрации, не допускающей потери
адсорбции, и соответствующим образом буфе
ризированноro до рН 7,3 8,o. В окончательной
инкубационной смеси фактор VII должен быть
единственным компонентом, лимитирующим
скорость.
Вторая стадия включает количественное
определение образовавшеrося фактора Ха с ис
пользованием специфичноro в отношении фак
тора Ха хромоrенною субстрата. Обычно он
представляет собой короткий пептид, состоя
щий из 3 5 аминокислотных остатков, присо
единенный к хромофорной rруппе. При отще
плении этой rруппы от пептидноro субстрата
ее хромофорные свойства смещаются к длине
волны, при которой становится возможным про
извести спектрофотометрическое определение.
Субстрат обычно растворяют в воде Р и исполь
зуют В конечных концентрациях 0,2 2 ммоль/л.
Субстрат также может содержать подходящие
инrибиторы для прекращения дальнейшеro об
разования фактора Ха (прибавляют эдетат).
МЕТОДИКА КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ
Восстанавливают полное содержимое одной
ампулы стандартноro образца и испытуемоrо об
разца путем прибавления подходящею количества
воды Р. используют в течение 1 ч. Восстановлен
ные образцы дополнительно разводят прерас
творителем. получая растворы с содержанием от
0,5 МЕ/мл до 2,0 МЕ/мл фактора VII.
rотовят дальнейшие разведения испытуемою
образца и стандартноro образца с использованием
изотоническоrо нехелатирующеro буфера, coдep
жащеrо 1 % человеческоrо или бычьеro альбуми
на и предпочтительно буферизированною при рН
от 7,3 до 8,0. rотовят не менее трех отдельных, He
зависимых разведений каждоrо образца. Предпо
чтительно каждое из разведен ий rотовить в двух
повторностях. Разведения roтовят таким образом,
чтобы окончательная концентрация фактора VII
была ниже 0,005 МЕ/мл.
rотовят контрольный раствор. включающий
все компоненты, кроме фактора VII.
Все разведения 20товят в пластиковых про
бирках и используют в течение 1 часа.
1--я стадия. Разведения стандартноrо образ
ца фактора VII и испытуемою образца смешива
ют с подходящим объемом предварительно Harpe
тою peareHTa факторов свертывания крови или со
смесью peareHTOВ, ею составляющих, и инкуби
руют смесь в пластиковых пробирках или ячейках
планшета при температуре 37 ос. Концентрации
компонентов в процессе образования фактора Ха
должны соответствовать спецификациям, прив
денным выше при описании peareHТOB.
Активацию фактора Х проводят в течение под
ходящеro промежутка времени, предпочтительно
прерывая реакцию до достижения концентрацией
фактора Ха ее максимальною уровня с целью по--
лучения удовлетворительной линейной зависимо--
сти отклика от дозы. Время активации выбирается
также таким образом, чтобы достичь линейною во
времени образования фактора Ха. Подходящие пе
риоды активации обычно находятся в пределах от
2 мин до 5 мин, но допускаются и друrие значения
при условии получения приемлемой линейности
зависимости отклика от дозы.
2-я стадия. Активацию прерывают добав
лением предварительно наrретою peareHTa, co
держащеro хромоrенный субстрат. Проводят KO
личественную оценку скорости расщепления
субстрата, которая должна находиться в линейной
зависимости от концентрации образовавшеюся
фактора Ха, измеряя изменение оптической nлот
ности при соответствующей длине волны с помо
щью спектрофотометра. Оптическую плотность
отслеживают постоянно, получая таким образом
возможность вычислить начальную скорость pac
щепления субстрата, либо прерывают реакцию rи
дролиза по окончании подходящеro промежутка
времени путем понижения рН добавлением под
ходящеrо peareHTa, например, уксусной кислоты
(500 r/л С 2 Н 4 О 2 ) или раствора цитрата (1 моль/л)
с рН 3. Время rидролиза реryлируют с целью до--
стижения линейною характера образования xpo
мофора во времени. Подходящими обычно явля
ются периоды rидролиза от 3 мин до 15 мин, но
допускаются и друrие значения при условии полу
чения таким образом лучшей линейности зависи
мости отклика от дозы.
Проверяют достоверность определения и pac
считывают активность испытуемоrо образца с ис
пользованием обычных статистических методов
(например, 5.3).
01/2013:20711
2.7.11. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ ФАКТОРА
СВЕРТЫВАНИЯ КРОВИ
ЧЕЛОВЕКА IX
Принцип исследования состоит в измере-
нии способности препарата фактора IX сокра-
щать время свертывания плазмы, дефицитнoii
по фактору IX. Реакция ускоряется при добавле-
нии peareHTa, содержащею фосфолипиды и KOtt-
тактные активаторы: каолин, кремнезем или эла-
rиевую кислоту. Активность оценивается путем
2.7.12. Количественное определение zепарина
365
сравнения кривой доза/эффект испытуемоro об
разца и стандартноro образца, калиброванноro в
Международных Единицах.
За Международную Единицу (МЕ) принима
ют активность фактора 'Х, проявляемую YCTa
новленным количеством Международною CTaH
дартноrо образца, состоящеro из лиофильно
высушенноro концентрата фактора свертывания
крови человека IX. Эквивалентность Междуна
родноro стандартноro образца в Международ
ных Единицах устанавливается Всемирной opra
низацией здравоохранения (ВОЗ).
БСП концентрата фактора свертывания
человека /Х калибруется в МЕ путем сравнения
с Международным стандартным образцом.
Восстанавливают по отдельности испыту
емый и стандартный образцы в соответствии с
указаниями на этикетке и используют немедлен
но. В случае необходимости определяют коли
чество присутствующею rепарина (2.7.12) и ней
трализуют rепарин прибавлением протамина
сульфата Р (1 О MKr протамина сульфата нейтра
лизуют 1 МЕ rепарина). Испытуемый и CTaHдapT
ный образцы разводят плазмой, дефицитной
по фактору IX (например, субстратом плазмы
R2) до получения растворов с содержанием от
0,5 МЕ/мл до 2,0 МЕ/мл. rотовят не менее трех
разведен ий всех образцов, предпочтительно в
дубликатах, с помощью подходящеro буфера
(например, имидаЗОЛЬН020 буферН020 pacт
вора рН 7,3 Р), содержащеro 10 r/л бычьею или
человеческою альбумина. Разведения исполь
зуют немедленно.
При определении при меняют аппарату
ру, позволяющую мноroкратно измерять время
свертывания, либо проводят исследование при
постоянной инкубации пробирок в водяной бане
при температуре 37 ОС.
В каждую пробирку помещают по 0,1 мл
плазмы, дефицитной по фактору IX (например,
субстрат плазмы Р2) и по 0,1 мл одноro из
разведений испытуемоro или стандартноro об
разцов. К содержимому каждой пробирки при
бавляют по 0,1 мл подходящеro peareHTa акти
вированноro частичноro тромбопластиновоro
времени (АЧТВ, Activated Partia/ Thrombop/astiп
Тiтe АРТТ), содержащеro фосфолипиды или
контактный активатор, и инкубируют смесь в Te
чение рекомендуемоro времени при темпера
туре 37 ос. К содержимому каждой пробирки
прибавляют по 0,1 мл раствора 3,7 r/л кальция
хлорида Р, предварительно подоrретою до TeM
пературы 37 ОС.
С помощью секундомера измеряют время
коаrуляции, т. е. интервал времени между при
бавлением кальция хлорида и первыми призна
ками образования фибрина. Объемы реактивов,
приведенные выше, MOryт быть адаптированы
для используемых peareHToB АЧТВ или аппара
туры. Рассчитывают активность с использова
нием обычных статистических методов (напри
мер, 5.3).
48 За.. 1712
Для подтверждения отсутствия в субстрате
плазмы Р2 существенною количества фактора
IX выполняют контрольный опыт с использова
нием вместо испытуемоro образца COOTBeTCTBY
ющеrо количества смеси из 1 объема раствора
38 r/л натрия цитрата Р и 5 объемов иMидa
ЗОЛЬН020 буферН020 раствора рН 7,3 Р. Резуль
таты испытания считают достоверными, если
время коаryляции в контрольном опыте COCTaB
ляет от 100 с до 200 с.
01/2013:20712
2.7.12. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕrЕПАРИНА
В КОНЦЕНТРАТАХ ФАКТОРОВ
СВЕРТЫВАНИЯ КРОВИ
rепарин определяют в виде комплекса с aH
титромбином 111 (АТ) по еro способности к ин
rибированию фактора свертывания крови Ха
(анти Ха активность). В реакционной смеси под
держивают избыток АТ для обеспечения посто
янной концентрации комплекса rепарина с Ат.
Фактор Ха нейтрализуется комплексом rепарина
с АТ, а ero избыток rидролизует специфический
хромофорный пептидный субстрат с высвобож
дением хромофора. Количество хромофора об
ратно пропорционально активности rепарина.
ХРОМ02енный субстрат фактора Ха.
Специфический хромоrенный субстрат для фак
тора Ха, например, N бензоил L изолейцил
L rлутамил rлицил L арrининА нитроанилида
rидрохлорид. Субстрат восстанавливают в COOT
ветствии с указаниями изroтовителя.
Буфер для разведениu. Раствор 6,05 r/л
триС(2идроксиметил)аминометана Р. При He
обходимости доводят кислотой хлористоводо
родной Р до рН 8,4.
Испытуемый раствор. Испытуемый обра
зец разводят буфером для разведений, получая
раствор с ожидаемым содержанием rепарина
0,1 МЕ/мл.
Раствор сравнения. Стандартный обра
зец разводят буфером для разведений, получая
раствор с ожидаемым содержанием rепарина
0,1 МЕ/мл.
Нижеописанные условия определения OT
носятся к титрационным микропланшетам. Если
определение производят в пробирках, объемы
MOryT изменяться при условии неизменности co
отношений компонентов в смесях.
Перед началом испытания все растворы
быстро наrревают на водяной бане до 37 ос.
В ряд ячеек распределяют по 20 мкл HOp
мальной человеческой плазмы и по 20 мкл pacт
вора антитромбина 111 Р1. Прибавляют в ячейки
ряда по 20,60,100 и 140 мкл испытуемоro paCT
366
rосударственная фармакопея Республики Беларусь
вора или раствора сравнения и доводят объемы
в каждой из ячеек до 200 мкл С использованием
буфера для разведений (содержание rепарина в
конечной смеси O,02 O.O8 МЕ/мл).
Метод конечной точки. По 40 мкл coдep
жимоro каждой из ячеек переносят во второй
ряд ячеек, прибавляют по 20 мкл раствора бы
чье20 фактора Ха Р и инкубируют при темпе
ратуре 37 ос в течение 30 с. Прибавляют по
40 мкл раствора 1 ммоль/л xpoMoreHHoro суб
страта фактора Ха и инкубируют при 37 ос в
течение 3 мин. Реакцию прерывают путем сни
жения показателя рН прибавлением походяще
ro peareHTa, например, 20 % (об/об) раствора
кислоты уксусной ледяной Р, и определяют оп
тическую плотность при длине волны 405 нм
(2.2.25). Обычно подходящей является продол
жительность реакции от 3 мин до 15 мин, но дo
пускаются отклонения от этоro интервала при
условии получения лучшей линейности зависи
мости отклика от дозы.
Кинетический метод. По 40 мкл содержи
моro каждой из ячеек переносят во второй ряд
ячеек, прибавляют по 20 мкл раствора бы
чье20 фактора Ха Р и инкубируют при темпе
ратуре З7 ос в течение 30 с. Прибавляют по
40 мкл раствора 2 ммоль/л xpoMoreHHoro суб
страта фактора Ха, инкубируют при температу
ре 37 ос и реrистрируют скорость расщепления
субстрата путем постоянноro измерения оптиче
ской плотности при длине волны 405 нм (2.2.25),
определяя таким образом начальную скорость
расщепления субстрата. Это значение должно
находиться в линейной зависимости от остаточ
ной концентрации фактора Ха.
Проверяют достоверность определения и
рассчитывают активность rепарина в испыту
емом образце с использованием обычных CTa
тистических методов, подходящих для анали
за результатов измерения уrлов наклона (Ha
пример, 5.3).
01/2013:20713
2.7.13. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ АНТИ-
D-иммУноrЛОБУЛИНА
ЧЕЛОВЕКА
МЕТОД А
Активность чеповеческоrо анти D иммуно
rлобулина определяют путем сравнения количе
ства образца, необходимоro для аrrлютинации
D положительных эритроцитов, с количеством
стандартноro образца, калиброванноro в Меж
дуна родных Единицах, необходимым для полу
чения тоro же эффекта.
За Международную Единицу принимают aK
тивность установленноro количества Между
народноro стандартноro образца. Эквивалент
ность МеЖдународноro стандартноro образца
в Международных Единицах устанавливается
Всемирной орrанизацией здравоохранения.
БСП человечеСК020 анти D иММУН02ЛО
булина калиброван в Международных Единицах
в сравнении с Международным стандартным
образцом и предназначен для использования в
количественных определениях человеческоro
анти D иммуноrлобулина.
Используют пул D положительных эритро
цитов, собранных не более чем за 7 дней до
испытания, хранившихся соответствующим об
разом и полученных не менее чем от четырех
rрупп доноров с rруппой О R 1 R 1 . К подходящему
объему клеток, предварительно трижды промы
тых раствором 9 r/л натрия хлорида Р, прибав
ляют равный объем раствора бромелайнов Р,
выдерживают при температуре 37 ос в течение
10 мин, центрифуrируют, отделяют супернатант
и промывают трижды раствором 9 r/л натрия
хлорида Р. 20 объемов эритроцитов суспендиру
ют в смеси из 15 объемов инертной сыворотки,
20 объемов раствора 300 r/л бычьесю альбумu
на Р и 45 объемов раствора 9 r/л натрия хлори
да Р. Полученную суспензию охлаждают ледя
ной водой при постоянном перемешивании.
С использованием калиброванноro aBTOMa
тическоrо прибора для разведения и раствора
для разведений, содержащеro 5 r/л бычье20
альбумина Р и 9 r/л натрия хлорида Р, roтовят
подходящие разведения испытуемоro образца и
стандартноro образца.
Определение обычно проводят по изложен--
ной ниже методике, используя соответствующий
прибор для непрерывноro автоматическоro re
молитическоro анализа. Температуру в трубо
проводе, за исключением инкубационных ячеек,
поддерживают на уровне 15,0 ос. Вводят с помо
щью насоса в трубопровод аппарата суспензию
эритроцитов со скоростью 0,1 млlмин И раствор
3 r/л метилцеллюлозы 450 Р со скоростью
0,05 мл/мин. В течение 2 мин со скоростью
0,1 мл/мин вводят разведения испытуемоro 06--
разца и стандартною препарата. Перед введени
ем очередноro разведения вводят в течение 4 мин
со скоростью 0,1 млlмин раствор для разведений.
Вводят воздух со скоростью 0,6 мл/мин. Инку
бируют при температуре 37 ос в течение 18 мин,
после чеro рассеивают столбики эритроцитов
путем введения со скоростью 1,6 мл/мин paCT
вора 9 r/л натрия хлорида Р, содержащеro подхо--
дящий увлажняющий areHT (например, полисор
бат 20 Р в окончательной концентрации 0,2 r/л)
для предотвращения разрушения аппликаций.
После оседания аrrлютинатов дваЖдЫ декантиру
ют: вначале со скоростью 0,4 млlмин, затем со
скоростью 0,6 мл/мин. Эритроциты, не подверr
шиеся аrrлютинации, лизируют раствором, содер.-
жащим 5 r/л окmoксинола 10 Р, 0,2 r/л калия фе{r
2.7.13. Количественное определение анти D иММУНО2лобулина человека
367
рицианида Р, 1 r/л натрия 2идрокарбоната Р и
0,05 r/л калия цианида Р и подаваемым со cкo
ростью 2,5 мл/мин. Система включает петлю для
получения десяти минутной задержки, необхо
димой для конверсии rемоrлобина. Оптическую
плотность (2.2.25) rемолизата реrистрируют He
прерывно при длине волны, выбранной в интер
вале от 540 нм ДО 550 нм. Определяют диапазон
концентраций антител, на протяжении котороro
существует линейная зависимость между KOHцeH
трацией и полученным изменением оптической
плотности (&4). Исходя из полученных результа
тов строят rрадуировочную кривую, участок ли
нейности которой используют для определения
активности испытуемоro образца.
Активность испытуемоro образца рассчиты
вают с использованием обычных статистических
методов (5.3).
МЕТОД В
Активность человеческоrо анти D
иммуноrлобулина определяют методом KOHKY
peHTHOro твердофазноro иммуноферментноrо
анализа, производимоro на титрационных ми
кропланшетах, по крытых эритроцитами. Метод
основан на конкурентном связывании между
поликлональным препаратом анти D
иммуноrлобулина и биотинированными MOHO
клональными анти D антителами, направленны
ми против специфическоro эпитопа D антиrена.
Активность испытуемоro образца сравнивают со
стандартным образцом, калиброванным в Меж
дуна родных Единицах.
За Международную Единицу принимают aK
тивность установленноro количества Между
народноro стандартноro образца. Эквивалент
ность Международноro стандартноro образца
в Международных Единицах устанавливается
Всемирной орrанизацией здравоохранения.
БСП человечеСК020 анти О иММУН02ЛО
булина калиброван в Международных Единицах
в сравнении с Международным стандартным
образцом и предназначен для использования в
количественных определениях человеческоro
анти D иммуноrлобулина.
МАТЕРИАЛЫ
PeareHTbI, для которых нет особых указаний,
должны быть аналитической степени чистоты.
СФБ (раствор натрия хлорида в фосфат
ном буферном растворе, PBS). 8,0 r натрия
хлорида Р, 0,76 r динатрия 2идрофосфата без
еодН020 Р, 0,2 r калия хлорида Р, 0,2 r калия ди
zидрофосфата Р и 0,2 r натрия азида Р paCTBO
ряют в воде Р и доводят до объема 1000 мл этим
асе растворителем.
СТБ (раствор натрия хлорида в тpиc
tiyфeрном растворе, TBS). 8,0 r натрия хло
рида Р и 0,6 r триС(2идроксиметил)амино
метана Р растворяют в воде Р, доводят 1 М
рествором хлористоводородной кислоты до рН
72 (2.2.3) и разводят водой Р до объема 1000 мл.
Раствор папаина. Раствор roтовят переме
шиванием при 37 ос в течение 30 мин 1 r па
паина Р в 10 мл 0,067 М фосфатН020 буфер
Н020 раствора рН 5,4 Р, центрифуrированием
при 1 О 000 9 в течение 5 мин и фильтровани
ем через мембранный фильтр или фильтр с
размером пор 0,22 мкм. Для активации смеши
вают 1 мл фильтрата, 1 мл раствора 48,44 r/л
L цистеина Р и 1 мл раствора 3,72 r/л натрия
эдетата Р и доводят 0,067 М фосфатным бу
ферным раствором рН 5,4 Р до объема 10 мл.
Аликвоты замораживают при температуре
200C или ниже.
Эритроциты. Используют пул D положи
тельных эритроцитов, полученных не менее чем
из трех rрупп доноров с rруппой О R 2 R 2 . Клетки
четырежды промывают СФБ. Центрифуrируют
при 1800 9 в течение 5 мин, смешивают подходя
щий объем предварительно HarpeTbIx упакован
ных клеток с подходящим объемом предвари
тельно HarpeToro раствора папаина (объемное
соотношение 2:1 является подходящим) и инку
бируют при температуре 37 ос в течение 1 О мин.
Клетки четырежды промывают СФБ. Хранят при
температуре 4 ос с подходящим стабилизато
ром не более недели.
Биотинированный B,ad 5. Используют в co
ответствии с инструкциями.
Щелочной авидин стрептавидиновый pe
а2ент, связанный с фосфатазой. Предпочти
тельно использовать модифицированный pe
areHT, сочетающий высокую специфическую
активность с низким уровнем неспецифическо
ro связывания. Используют в соответствии с ин
струкциями.
Раствор субстрата. Используют пара
нитрофенилфосфат в соответствии с инструк
циями.
Буфер для фиксации клеток. 18,02 r 2ЛЮ
козы Р, 4,09 r натрия хлорида Р, 1,24 r борной
кислоты Р, 10,29 r натрия цитрата Р и 0,74 r
натрия эдетата Р растворяют в воде Р. ДOBO
дЯТ 1 М раствором натрия 2идроксида или 1 М
раствором хлористоводородной кислоты до
рН 7,2 7,3 (2.2.3) и разводят водой Р до объема
1000 мл. Хранят при температуре 4 ОС. Исполь
зуют непосредственно из места хранения.
Раствор 2лутаров020 альде2ида. Непо
средственно перед применением 90 мкл paCT
вора 250 r/л 2лутаров020 альде2ида Р прибав
ляют к 24 мл холодноro СФБ.
Титрационные микропланшеты. Планше
ты, предназначенные для покрытия эритроцита
ми, представляют собой плоскодонные планше
ты из полистирола со свойствами поверхности,
оптимизироваННЫ 4И для иммуноферментно
ro анализа, и с большой емкостью в отношении
связывания с белками. Планшеты, использу
емые для приroтовления разведений иммуно
rлобулина, представляют собой планшеты из
полистирола или поливинилхлорида с U или
V образными формами ячеек.
368
rосударственная фармакопея Республики Беларусь
МЕТОДИКА
rотовят 0,1 % (об/об) суспензию обработан
ных папаином эритроцитов в холодном буфере
для фиксации клеток. В каждую ячейку плоско
донноro титрационноrо микропланшета помеща
ют по 50 мкл суспензии.
Планшет центрифуrируют при 350 9 в Te
чение 3 мин предпочтительно при температу
ре 4 ОС. Не удаляя супернатан к содержи
мому каждой из ячеек осторожно прибавляют
по 100 мкл раствора rлутаровоrо альдеrида и
выдерживают в течение 1 О мин. Содержимое
ячеек сливают путем быстроro переворачивания
планшета и трижды промывают каждую ячейку
250 300 мкл СФБ. Эта операция может произ
водиться либо вручную, либо с использовани
ем автоматическоro устройства для промывания
планшетов. Планшет либо используют в ниже
описанном испытании, либо после сливания
СФБ, прибавления в каждую ячейку по 100 мкл
буфера для фиксации клеток и запечатывания
пластиковой пленкой хранят при температуре
4 ос не более 1 месяца.
Испытуемые растворы. В случае лиофиль
но высушенных препаратов их восстанавливают
в соответствии с указаниями на этикетке. rOTO
вят в четырех независимых повторностях 5 ce
рийных двукратных разведен ий , начиная с KOH
центрации 30 МЕ/мл с использованием СФБ,
содержащеro 1 О r/л бычье20 альбумина Р. При
необходимости исходное разведение корректи
руют с целью получения откликов, попадающих
в область линейности rрафика зависимости OT
клика от дозы.
Растворы сравнения. Восстанавливают
стандартный образец в соответствии с указани
ями на этикетке. rотовят в четырех независимых
повторностях 5 серийных двукратных разведе
ний, начиная с концентрации 30 МЕ/мл с исполь
зованием СФБ, содержащеro 1 О r/л бычье20 аль
бумина Р.
В ячейки каждоro из рядов титрационноro
микропланшета с U или V образными формами
ячеек вносят по 35 мкл каждоro из разведений
испытуемоro раствора или раствора сравнения.
К содержимому каждой из ячеек прибавляют по
35 мкл биотинированноro Brad 5 с содержанием
250 нr/мл.
Освобождают ячейки планшета, покрытоro
эритроцитами, путем переворачивания и BЫCY
шивания бумажным полотенцем. Прибавляют по
250 мкл СФБ, содержащеro 20 r/л бычье20 аль
бумина Р и выдерживают при комнатной темпе
ратуре в течение 30 мин.
Освобождают ячейки планшета, покрыто
ro ЭРИТРОЦlIIтами, путем переворачивания и BЫ
сушивания бумажным полотенцем и помещают
в каждую из ячеек по 50 мкл каждоro из разве
дений испытуемоro раствора и раствора cpaB
нения, содержащих биотинированный B,ad 5.
50 мкл СФБ, содержащеro 1 О r/л бычье20 альбу
мина Р, используют в качестве отрицательноro
контроля. Планшет запечатывают пластиковой
пленкой и инкубируют при комнатной темпера
туре в течение 1 ч.
Удаляют жидкость из ячеек планшета, по
KpbIтoro эритроцитами, и трижды промывают
каждую ячейку 250 300 мкл СТБ.
Щелочной авидин стрептавидиновый pe
areHT, связанный с фосфатазой, разбавляют
СТБ, содержащим 1 О r/л бычье20 альбумина р, и
прибавляют в количестве по 50 мкл В каждую из
ячеек. Инкубируют в течение 30 мин при KOMHaT
ной температуре.
Удаляют жидкость из ячеек планшета, по
KpbIтoro эритроцитами, и трижды промывают
каждую ячейку 250 300 мкл СТБ.
В каждую из ячеек прибавляют по 100 мкл
раствора субстрата и инкубируют в темноте при
. комнатной температуре в течение 1 О мин. Для
остановки реакции в каждую из ячеек прибавля
ют по 50 мкл 3 М раствора натрия 2идроксида.
Измеряют значения оптической плотно
сти при длине волны 405 нм и вычитают зна
чение, полученное для отрицательноrо KOHTpO
ля. Значения оптической плотности в области
линейности кривой титрования используют для
оценки активности испытуемоro образца с ис
пользованием обычных статистических MeTO
дов (5.3).
МЕТОД С
Активность человеческоrо анти D иммуно
rлобулина определяют методом поточной ци
тометрии в формате титрационноro микро
планшета. Метод основан на специфическом
связывании между анти D иммуноrлобулином и
D положительными эритроцитами. Активность
испытуемоro образца сравнивают со CTaHдapT
ным образцом, калиброванным в Международ
ных Единицах.
За Международную Единицу принимают aK
тивность установленноro количества Между
народноro стандартноro образца. Эквивалент
ность Международноro стандартноro образца
в Международных Единицах устанавливается
Всемирной орrанизацией здравоохранения.
БСП человечеСК020 анти О иММУН02ЛО
булина калиброван в Международных Единицах
в сравнении с Международным стандартным
образцом и предназначен для использования в
количественных определениях человеческоro
анти D иммуноrлобулина.
МАТЕРИАЛЫ
PeareHTbI, для которых нет особых указаний,
должны быть аналитической степени чистоты.
СФБ. 8,0 r натрия хлорида Р, 0,76 r диHa
трия 2идрофосфата безводН020 Р, 0,2 r калия
хлорида Р, 0,2 r калия ди2идрофосфата Р и 0,2 r
натрия азида Р растворяют в воде Р и доводят
до объема 1000 мл этим же растворителем.
Раствор СФБ БСА. СФБ, содержащий
10,0 r/л БЫЧЬе20 альбумина Р.
2.7. 14. Копичественноеопределениеанти2енной(иММУНО2енной) актив насти
369
Эритроциты. ИспользуютD положительные
эритроциты, полученные из rруппы доноров с
rруппой О R 1 R 1 не более чем за две недели до
определения. При необходимости хранят с COOT
ветствующим стабилизатором при температуре
4 ОС. Клетки промывают не менее двух раз pac
твором СФБ БСА и rотовят суспензию, содержа
щую 1.104, но не более 5.104 клеток в микролитре
с использованием раствора СФБ БСА.
Используют D отрицательные эритроциты,
полученные из rруппы доноров с rруппой О rr и
подroтовленные аналоrичным образом.
Вторичные антитела. Используют подхо
дящий фраrмент анти lgG антитела, связанный
с флуоресцентным красителем, обладающий
специфичностью в отношении человеческоro
IgG или еro частей. Хранят и используют в соот
ветствии с инструкциями изroтовителя.
Титрационные микропланшеты. Использу
ют плоскодонные планшеты без поверхностной
обработки для иммуноферментноro анализа.
МЕТОДИКА
Испытуемые растворы. В случае лиофиль
но высушенных препаратов их восстанавливают
в соответствии с указаниями на этикетке. rOTO
вят не менее чем в трех независимых повтор
ностях не менее 3 серийных полутора или ДBY
кратных разведений, начиная с концентрации в
диапазоне 1 ,2 0, 15 МЕ/мл, с использованием
раствора СФБ БСА. При необходимости исход
ное разведение корректируют с целью получе
ния откликов, попадающих в область линейно
сти rрафика зависимости отклика от дозы.
Растворы сравнения. Восстанавливают
стандартный образец в соответствии с указания
ми на этикетке. rотовят не менее чем в трех неза
висимых повторностях не менее трех серийных
полутора или двукратных разведений, начиная
с концентрации в диапазоне 1.2 O,15 МЕ/мл, с
использованием раствора СФБ БСА. При необ
ходимости исходное разведение корректируют
с целью получения откликов, попадающих в б
ласть линейности rрафика зависимости отклика
от дозы.
В каждую ячейку титрационноro микроплан
шета распределяют по 50 мкл D положительных
эритроцитов. К содержимому ячеек каждоro
ряда прибавляют по 50 мкл каждоro из разведе
ний испытуемоro раствора или раствора cpaB
нения. Используют 50 мкл раствора СФБ БСА
в качестве отрицательноro контроля. По 50 мкл
D отрицательных эритроцитов распределяют в
четыре ячейки тоro же микропланшета и прибав
ляют по 50 мкл наименьшеro разведения испы
туемоro образца. Для отслеживания ложных pe
акций распределяют по 50 мкл D положительных
эритроцитов в четыре ячейки тоro же микро
планшета и прибавляют по 50 мкл раствора
СФБ БСА. Запечатывают пластиковой пленкой
и инкубируют при температуре 37 ос в течение
40 мин.
Планшеты центрифуrируют при 50 9 в тече
ние 3 мин, отбрасывают супернатант и промы
вают ячейки 200 250 мкл раствора СФБ БСА.
Повторяют промывку еще не менее одноro раза.
Планшеты центрифуrируют при 50 9 в тече
ние 3 мин, отбрасывают супернатант и прибавля
ют по 50 мкл вторичноro антитела, разведенноro
раствором СФБ БСА до подходящей KOHцeHTpa
ции белка. Запечатывают пластиковой пленкой и
инкубируют при комнатной температуре в защи
щенном от света месте в течение 20 мин.
Планшеты центрифуrируют при 50 9 в тече
ние 3 мин, отбрасывают супернатант и промы
вают ячейки 200 250 мкл раствора СФБ БСА.
Повторяют промывку еще не менее одноro раза.
Планшеты центрифуrируют при 50 9 в Te
чение 3 мин, клетки ресуспендируют в 200
250 мкл раствора СФБ БСА.
Суспензию клеток переносят в трубку, co
вместимую с имеющимся оборудованием для
поточной цитометрии, и проводят дальнейшее
разбавление прибавлением СФБ для обеспече
ния подходящей скорости потока.
Немедленно приступают к измерению Me
диан ной интенсивности флуоресценции в поточ
ном цитометре. Реrистрируют не менее 1 О 000
событий без rейтинrа, но с исключением шумов.
С использованием медианной интенсив
ности флуоресценции в области линейности
кривой зависимости отклика от дозы оценивают
активность испытуемоro образца обычными CTa
тистическими методами (5.3).
01/2013:20714
2.7.14. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ А,нтиrЕННОЙ
(иммУноrЕННОИ)
АКТИВНОСТИ ВАКЦИНЫ
rЕПАТИТА А
Количественное определение вакцины rепа
тита А производят либо iп vivo, путем сравнения
ее способности приводить в данных условиях к
образованию специфических антител у мышей
с такой же способностью стандартноrо образца,
либо iп vitro, путем иммунохимическоro опреде
ления содержания антиrена.
ОПРЕДЕЛЕНИЕ /N V/VO
Испытание на мышах, описанное ниже, при
водится в к<зчестве при мера подходящеro для
данной вакцины метода; MOryт использоваться и
друrие валидированные методы.
Выбор и распределение испытуемых
животных. В испытании используют здоровых
мышей подходящей линии из одной партии воз
370
tосударственная фармакопея Республики Беларусь
растом около 5 недель, одною пола. Животных
делят не менее чем на семь равных rрупп; KO
личество мышей в rруппе должно быть подходя
щим для выполнения требований испытания.
Определение антиrенной активности ис
пытуемой вакцины. rотовят не менее трех
серийных разведений испытуемой вакцины
и соответствующие разведения CTaHдapTHO
ю препарата с использованием раствора 9 r/л
натрия хлорида Р, содержащеro алюминийсо
держащий адъювант, используемый в вакцине.
Каждое разведение назначают одной из rрупп
мышей, каждой из которых вводят подкожно не
более 1,0 мл этою разведения. Контрольной He
вакцинированной rруппе вводят подкожно такой
же объем раствора 9 r/л натрия хлорида Р, co
держащею адъювант, используемый в вакцине.
Через 28 32 дня всех животных анестезируют
и производят забор крови. Индивидуальные cы
воротки хранят отдельно друr от друrа. Выполня
ют количественное определение специфических
антител к вирусу rепатита А в индивидуальных
сыворотках, используя подходящий иммунохи
мический метод (2.7.1).
Расчеты. Расчеты проводят обычными CTa
тистическими методами, используемыми дЛЯ KO
личественных определений с конечным числом
возможных результатов (5.3).
Из распределения уровней реакции, изме
ренных для всех сывороток rруппы невакцини
рованных животных определяют максимальный
ожидаемый уровень реакции невакцинирован
ноro животноro в данном количественном опре
делении. При получении отклика, превышающе
ro этот уровень, у вакцинированноro животноro,
еro по определению считают результатом cepo
конверсии.
Производят подходящее преобразование
(например, пробит преобразование) процент
ной численности животных с обнаруженной
сероконверсией в каждой из rрупп и анализи
руют данные в соответствии с моделью парал
лельных линий лоrарифмической зависимости
дозы от отклика. Активность испытуемою об
разца определяют относительно стандартноro
образца.
Условия достоверности. Результаты испы
тания являются достоверными. если:
как для испытуемой вакцины, так и для
стандартноro образца ЕD 50 заключена в интер
вале между наибольшей и наименьшей дозами,
введенными животным;
статистический анализ подтверждает от
сутствие отклонений в линейности и паралле
лизме;
rраницы доверительноrо интервала
(Р = 0,95) относительной активности не
менее З3 % и не более 300 % установленной
активности.
Требования к активности. Верхняя rрани
ца доверительною интервала (Р = 0,95) оцени
ваемой относительной активности должна быть
не менее 1,0.
ОПРЕДЕЛЕНИЕ /N V/TRO
Выполняют иммунохимическое определе
ние (2.7.1) содержания антиrена с критериями
приемлемости, валидированными в сравнении
с испытанием in vivo. Критерии приемлемости
для данною стандартноro образца подлежат ут
верждению компетентными орrанами с учетом
данных валидации.
БСП вакцина 2епатита А (иHaKтивиpoвaH
ная, не адсорбированная) приroдна для исполь
зования в качестве стандартноro образца.
01/2013:20715
2.7.15. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ ВАКЦИНЫ
rЕПАТИТА В (рДНК)
Количественное определение вакцины re
патита В (рДНК, rONA) производят либо iп vivo,
путем сравнения ее способности приводить в
данных условиях к образованию специфических
антител против поверхностноrо антиrена rепа
тита В (HbsAg) у мышей или морских свинок с
такой же способностью стандартноro образца,
либо in vitro, путем иммунохимическоro опреде
ления содержания антиrена.
ОПРЕДЕЛЕНИЕ /N V/VO
Выбор и распределение испытуемых
животных. В испытании используют здоро
вых мышей из одной партии возрастом около
5 недель. Используемая в данном испытании
линия мышей должна обеспечивать получение
существенною уклона кривой зависимости от
клика от дозы в отношении антиrена; подходя
щими являются мыши rаплотипов H 2q и H 2d.
Также MOryT использоваться здоровые морские
свинки из одной партии с массой тела от 300 r
до 350 r и возрастом около 7 недель. Использу
ют животных одною пола. Животных делят не
менее чем на семь равных rрупп; количество в
rруппе должно быть подходящим для выполне
ния требований испытания.
Оl'1ределение активности испытуемой
вакцины. rотовят не менее трех серийных
разведений испытуемой вакцины и COOTBeT
ствующие разведения стандартноro образца
с использованием раствора 9 r/л натрия хлcr
рида Р с добавлением алюминийсодержаще
ю адъюванта, используемою в вакцине, или
2.7.16. Количественное определение вакцины коклюша (бесклеточной)
371
друroro подходящею растворителя. Каждое из
полученных разведений назначают одной из
rрупп животных, каждому из которых вводят
внутрибрюшинно не более 1,0 мл этоro раз
ведения. Одну rруппу животных не вакциниру
ют, а вводят внутрибрюшинно такой же объем
растворителя. Через подходящий промежу
ток времени (например, от 4 до 6 недель) всех
животных анестезируют и производят забор
крови. Индивидуальные сыворотки хранят OT
дельно друr от друrа. Выполняют количествен
ное определение специфических антител
против HBsAg в индивидуальных сыворотках,
используя подходящий иммунохимический
метод (2.7.1).
Расчеты. Расчеты проводят обычными CTa
тистическими методами, используемыми дЛЯ KO
личественных определений с конечным числом
возможных результатов (5.3).
Из распределения уровней реакции, изме
ренных для всех сывороток rруппы невакцини
рованных животных, определяют максимальный
ожидаемый уровень реакции невакцинирован
ноro животноro в данном количественном опре
делении. При получении отклика, превышающе
ro этот уровень, у вакцинированноro животноro,
ero по определению считают результатом cepo
конверсии.
Производят подходящее преобразование
(например, пробит преобразование) процент
ной численности животных с обнаруженной ce
роконверсией в каждой из rрупп и анализируют
данные в соответствии с моделью параллель
ных линий лоrарифмической зависимости дозы
от отклика. Активность препарата определяют
относительно активности стандартноro образца.
Условия достоверности. Результаты испы
тания являются достоверными, если:
как для испытуемой вакцины, так и для
стандартноro образца ЕD 50 заключена в интер
вале между наибольшей и наименьшей дозами,
введенными животным;
статистический анализ подтверждает от
сутствие отклонений в линейности и паралле
лизме;
rраницы доверительноro интервала
(Р = 0,95) относительной активности не менее
З3 % и не более 300 % установленной актив
ности.
Требования к активности. Верхняя rрани
ца доверительноro интервала (Р = 0,95) оцени
ваемой относительной активности должна быть
не менее 1,0.
ОПРЕДЕЛЕНИЕ /N VIТRO
Выполняют иммунохимическое определе
ние (2.7.1) содержания антиrена с критериями
приемлемости, валидированными в сравнении с
испытанием iп vivo.
Показано, что методы твердофазноro имму
ноферментноrо анализа (тИФА, ELlSA) и радио
иммунолоrическоro анализа (РИА) с использcr
ванием моноклональных антител, специфичных
к индуцирующим защитную реакцию эпитопам
HBsAg, являются подходящими. Используют co
ответствующее число разведений испытуемой
вакцины и стандартноrо препарата. Полученные
данные, которые MOryт быть подверrнуты пре
образованию, анализируют методом параллель
ных линий. Наборы для оценки HBsAg iп vitro яв
ляются коммерчески доступными, и возможна
адаптация методов их использования для опре
деления активности iп vitro.
Критерии приемлемости для данноro CTaH
дартноro образца подлежат утверждению компе
тентными орrанами с учетом данных валидации.
01/2013:20716
2.7.16. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ ВАКЦИНЫ
КОКЛЮША (БЕСКЛЕТОЧНОИ)
Способность вакцины вызывать образова
ние специфических антител сравнивают с анало
rичным свойством параллельно испытываемоro
стандартноro препарата; антитела определяют
с использованием подходящеro иммунохимиче
скоro метода (2.7.1), например, твердофазно
ro иммуноферментноrо анализа (тИФА, ELlSA).
В приведенном ниже описании испытания на
мышах используется трехточечная модель,
однако, после валидации для рутинных испыта
ний может быть использован метод OДHOKpaTHO
ro разведения.
Стандартная вакцина. В качестве CTaH
дартной вакцины используют партию вакци
ны, эффективность которой доказана клини
ческими испытаниями, или репрезентативную
партию этой вакцины. Для приroтовления pe
презентативной партии необходимо cTporoe
соблюдение технолоrическоrо процесса при
производстве партии, подверrающейся кли
ническим испытаниям. Стабильность вакцины
сравнения должна быть документально под
тверждена.
Стандартная антисыворотка. В качестве
стандартной антисыворотки возможно использо
вать БСП мышиной антисыворотки Bordetefla
pertussis.
Требования. Способность вакцины вызы
вать образование антител не должна быть суще
ственно ниже (Р = 0,95) по сравнению с аналcr
rичным свойством стандартноro образца.
Следующая модель испытания приводится
в качестве примера метода, при20дность KOmcr
р020 для даНН020 испытания подтверждена.
372
rосударственная фармакопея Республики Беларусь
Отбор и распределение испытуемых
животных. В испытании используют здоро
вых мышей (например, линии СD1) из одной
партии возрастом от 4 до 8 недель. Животных
распределяют по шести rруппам, состоящим
из количества особей, достаточноro дЛЯ BЫ
полнения требований испытания. Использу
ют три разведения испытуемой вакцины и три
разведения стандартноrо образца и назнача
ют каждой из rрупп одно из разведений. Каж
дому животному внутрибрюшинно или под
кожно вводят по 0,5 мл разведения, которое
было назначено rруппе, к которой данное жи
вотное относится.
Отбор образцов сыворотки. Через 4 5
недель после вакцинации проводят забор крови
у мышей под анестезией. Сыворотки до опреде
лени я содержания антител хранят при темпера
туре 20 ОС.
Определение антител. Выполняют коли
чественное определение содержания специфи
ческих в отношении каждоro из компонентов aH
тител в сыворотках с использованием метода
с подтвержденной достоверностью, например,
ELlSA. методика котороro приведена ниже.
Испытание ELlSA. Титрационные ми
кропланшеты, выполненные из поливинил
хлорида или полистирола, в соответствии с
требованиями специфическоro антиrена по
крывают очищенным антиrеном в KOHцeHTpa
ции 100 Hr на одну ячейку. После промыв
ки непрореаrировавшие участки блокируют
путем инкубации с раствором бычьеro cы
вороточноro альбумина и последующей про
мывки. Производят на планшетах двукратные
разведения сывороток мышей, иммунизиро
ванных испытуемой вакциной и стандартной
вакциной. После инкубации при температуре
22 25 ос в течение 1 ч планшеты промыва
ют. После промывки добавляют хромоrенный
субстрат, высвобождающий под воздействи
ем присоединенноro ферментноro конъюrата
хромофор, который может быть количествен
но определен путем измерения оптической
плотности (2.2.25). Выбирают такие условия
испытания, чтобы в рабочем диапазоне из
мерений имелась линейная зависимость оп
тической плотности от содержания антител, а
значения оптической плотности находились в
пределах от 0,1 до 2,0.
В испытании используется контрольная aH
тисыворотка установленной активности, служа
щая основанием для вычисления содержания
антител в испытуемой сыворотке. Также в испы
тании используется стандартизированная KOH
трольная сыворотка.
Результаты испытания являются HeДOCTO
верными, если:
значение, определенное для контрольной
сыворотки, отличается от при писанной величи
ны более чем на удвоенное значение CTaHдapT
ноro отклонения;
rраницы доверительноro интервала
(Р = 0,95) относительной активности менее
50 % и более 200 % установленной активности.
Расчеты Рассчитывают титры антител в
сыворотках мышей, иммунизированных испыту
ем ой вакциной и стандартной вакциной, и из по
лученных значений с использованием обычных
статистических методов определяют активность
испытуемой вакцины относительно стандартной
вакцины (5.3).
01/2013:20717
2.7.17. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
АНТИТРОМБИНА 111
ЧЕЛОВЕКА
Содержание антитромбина 111 в испытуе
мом образце определяют путем сравнения еro
способности к инактивации тромбина в присут
ствии избыточноro количества rепарина с такой
же способностью стандартноro образца анти
тромбина 111 человека, калиброванноro в Между
народных Единицах. Различные количества ис
пытуемоro образца смешивают с определенным
количеством тромбина и определяют активность
остаточноrо количества тромбина с использова
нием подходящеro xpoMoreHHoro субстрата.
За Международную Единицу принимают aK
тивность установленноro количества Между
народноro стандартноro образца KOHцeHTpa
та антитромбина 111 человека. Эквивалентность
Международноro стандартноro образца в Меж
дуна родных Единицах устанавливается Всемир
ной орrанизацией здравоохранения.
Методика. rотовят два независимых ряда
из трех или четырех разведен ий испытуемоro
образца и стандартною образца в диапазоне от
1/75 до 1/200 исходя из концентрации 1 МЕ/мл
с использованием буферНО20 раствора тpиc
EOTA BSA рН 8,4 Р, содержащею 15 МЕ rепари
на в миллилитре.
По 200 мкл каждою из разведений выдержи
вают при температуре 37 0 С в течение 1 2 мин.
К каждому разведению прибавляют по 200 мкл
раствора 2 МЕ/мл БЫЧЬе20 тромбина Р в бу
ферном растворе mpuc EOTA BSA рН 8,4 Р, п
ремешивают и выдерживают при температуре
37 ос в течение ровно 1 мин. Прибавляют 500 мкл
подходящеro xpoMoreHHoro субстрата (напр
r-.,1ep, D фенилаланил L пипеколил L арrинин-
4 нитроанилида, растворенною в воде р, с по-
лучением раствора концентрацией 4 ммолtJn
и дополнительно разведенноrо до KOHцeH
ции, подходящей для проведения количествен-
ною определения с использованием буферноао
раствора mpuc EOТA BSA рН 8,4 Р без доб-,
2.7.18. Количественное определение фактора свертывания крови 11 человека 373
ления альбумина). Тотчас же начинают фикси
ровать изменение оптической плотности при
длине волны 405 нм (2.2.25) и продолжают из
мерение в течение не менее 30 с. Рассчитыва
ют скорость изменения оптической плотности
(М/мин). (Определение также может произво
диться методом конечной точки путем преры
вания реакции прибавлением уксусной кислоты
и измерением оптической плотности при длине
волны 405 нм).
Скорость изменения оптической плотности
(М/мин) обратно пропорциональна активности
антитромбина 111.
Проверяют достоверность определения и
рассчитывают активность испытуемоro образца
с использованием обычных статистических Me
тодов (5.3).
01/2013:20718
2.7.18. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ ФАКТОРА
СВЕРТЫВАНИЯ КРОВИ 11
ЧЕЛОВЕКА
Количественное определение фактора CBep
тывания крови 11 производят по еro специфиче
ской активации с образованием фактора Ila.
Фактор Ila оценивают на основании сопостав
ления еro активности по расщеплению специ
фическоro xpoMoreHHoro пептидноro субстрата
с такой же активностью Международноro CTaH
дартноro образца или стандартноro образца, Ka
либрованноrо в Международных Единицах.
За Международную Единицу принимают aK
тивность фактора 11 в установленном количестве
Международноro стандартноro образца, состоя
щеrо из лиофилизированноrо концентрата че
ловеческоro фактора свертывания крови 11. Эк
вивалентность Международноro стандартноrо
образца в Международных Единицах устанавли
вается Всемирной орrанизацией здравоохране
ния.
Хромоrенный метод количественноro опре
деления включает две последовательные
стадии: индуцируемая змеиным ядом актива
ция фактора 11 и ферментативное расщепление
xpoMoreHHoro субстрата фактором Ila с образо
ванием хромофора, который определяют спек
трофотометрически. В соответствующих услови
ях испытания существует линейная зависимость
между активностью фактора Ila и расщеплением
xpoMoreHHoro субстрата.
PEArEHTbI
Специфический активатор фактора 1/ из
яда эфы (Экарин). Белок, получаемый из яда
эфы (Echis cariпatus), специфически активиру
ющий фактор 11, восстанавливают в COOTBeT
ствии с инструкциями производителя. BOCCTa
новленный препарат хранят при температуре
4 ос и используют в течение 1 месяца.
ХрОМ02енный субстрат фактора I/а. Спе
цифический хромоrенный субстрат для фактора
Ila, например: Н D фенилаланил L пипеколил
L арrинин 4 нитроанилида диrидрохлорид,
4 толуолсульфонил rлицил пролил L арrин ин
4 нитроанилид, Н D циклоrексилrлицил а
а м и нобути рил L а рrи н и H 4 H итроа н ил ид,
D ци кл оrексил rл ицил L ала н ил L арrи н и H 4
нитроанилида диацетат. Восстанавливают в co
ответствии с инструкциями изrотовителя.
Буфер для разведений. Раствор, содержа
щий 6,06 r/л триС(2идроксиметил)аминомета
на Р, 17,53 r/л натрия хлорида Р, 2,79 r/л (эти
лендинитрил)тетрауксусной кислоты Р и 1 r/л
бычье20 альбумина Рили человечеСК020 альбу
мина Р. При необходимости доводят до рН 8,4
кислотой хлористоводородной Р.
МЕТОДИКА ОПРЕДЕЛЕНИЯ
Испытуемый раствор. Испытуемый обра
зец разбавляют буфером для разведений, по
лучая раствор с содержанием 0,015 МЕ фак
тора" в миллилитре. rотовят не менее трех
дальнейших разведений с использованием тоro
же буфера.
Раствор сравнения. Стандартный образец
разбавляют буфером для разведений, получая
раствор с содержанием 0,015 МЕ фактора 11 в
миллилитре. rотовят не менее трех дальнейших
разведений с использованием тоro же буфера.
Все растворы перед испытанием наrревают
на водяной бане до 37 ос.
Нижеописанные условия определения OT
носятся к титрационным микропланшетам. Если
определение производят в пробирках, объемы
MorYT изменяться при условии неизменности co
отношений компонентов в смесях.
Определение выполняют с использовани
ем титрационноro микропланшета, поддержи
BaeMoro при температуре 37 ОС. По 25 мкл каж
доro из разведен ий испытуемоrо раствора или
раствора сравнения вносят в каждый из рядов
ячеек. К содержимому каждой ячейки'прибавля
ют по 125 мкл буфера для разведений, затем
по 25 мкл эка рина и инкубируют ровно 2 мин.
К содержимому каждой ячейки прибавляют по
25 мкл xpoMoreHHoro субстрата фактора Ila.
Реrистрируют скорость изменения опти
ческой плотности (2.2.25) при длине волны
405 нм непрерывно в течение 3 мин и полу
чают среднюю скорость изменения оптиче
ской плотности (t.А/мин). При невозможности
непрерывной реrистрации оптическую плот
ность при 405 нм реrистрируют с подходя
щими последовательными интервалами, Ha
при мер, продолжительностью 40 с, строят на
миллиметровой бумаrе rрафик зависимости
оптической плотности от времени и вычис
374
rосударственная фармакопея Республики Беларусь
ляют из уклона полученной прямой значение
М/мин. Из значений дА/мин для каждоro раз
ведения испытуемою и стандартною образ
цов рассчитывают активность испытуемою
образца и проверяют достоверность опреде
ления с использованием обычных статистиче
ских методов (5.3).
01/2013:20719
2.7.19. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ ФАКТОРА
СВЕРТЫВАНИЯ КРОВИ Х
ЧЕЛОВЕКА
Количественное определение фактора
свертывания крови Х производят по еro специфи
ческой активации с образованием фактора Ха.
Фактор Ха оценивают на основании еro актив
ности по расщеплению специфическоro XpOMO
reHHoro пептидноro субстрата с такой же актив
ностью Международноro стандартноro образца
или стандартноro образца, калиброванною в
Международных Единицах.
За Международную Единицу принимают aK
тивность фактора Х в установленном количе
стве Международноro стандартною образца,
состоящеro из лиофилизированноrо KOHцeHTpa
та человеческоro фактора свертывания крови Х.
Эквивалентность Международноro стандартноro
образца в Международных Единицах устанавли
вается Всемирной орrанизацией здравоохране.-
ния.
Хромоrенный метод количественноro опре
деления включает две последовательные
стадии: индуцируемая змеиным ядом актива
ция фактора Х и ферментативное расщепление
xpoMoreHHoro субстрата фактором Ха с образо
ванием хромофора, который определяют спек
трофотометрически. В соответствующих услови
ях испытания существует линейная зависимость
между активностью фактора Ха и расщеплени
ем xpoMoreHHoro субстрата.
PEArEHTbI
Специфический активатор фактора Х из
яда цепочной 2адюки (RVV). Белок, полученный
из яда цепочной rадюки (Vipe,a ,usseJII), который
специфически активирует фактор Х. BOCCTaHaB
ливают в соответствии с инструкциями произ
водителя. Восстановленный препарат хранят
при температуре 4 ос и используют в течение
1 месяца.
ХрОМ02енный субстрат фактора Ха.
Специфический хромоrенный субстрат для фак
тора Ха, например: N а бензилоксикарбонил
D арrинил L rлицил L арrинин 4 нитроанилида
диrидрохлорид, N бензоил L изолейцил
L rлутамил rлицил L арrинин 4 нитроанилида
rидрохлорид, метансульфонил D лейцил rлицил
L арrинин 4 нитроанилид, метоксикарбонил
D ци клоrе ксилал а н ил rл и цил L a р rи н и н 4
нитроанилида ацетат. Восстанавливают в соот
ветствии с инструкциями изroтовителя.
Буфер для разведений. Раствор, содержа
щий 3,7 r/л триС(2идроксиметил)аминомета
на Р, 2,1 r/л имидазола Р, 0,02 r/л 2eKcaдиMe
трина бромида Р и 1 r/л бычье20 альбумина Р
или человечеСК020 альбумина Р. При необхо
димости доводят кислотой хлористоводород
ной Р до рН 8,4.
МЕТОДИКА ОПРЕДЕЛЕНИЯ
Испытуемый раствор. Испытуемый об
р.азец разбавляют буфером для разведений,
получая раствор с содержанием 0,18 МЕ фак
тора Х в миллилитре. rотовят не менее трех
дальнейших разведений с использованием
тою же буфера.
Раствор сравнения. Стандартный образец
разбавляют буфером для разведений, получая
раствор с содержанием 0,18 МЕ фактора Х в
миллилитре. rотовят не менее трех дальнейших
разведений с использованием тоro же буфера.
Все растворы перед испытанием быстро Ha
rревают на водяной бане до 37 ОС.
Нижеописанные условия определения от
носятся к титрационным микропланшетам. Если
определение производят в пробирках, объемы
MOryT изменяться при условии неизменности co
отношений компонентов в смесях.
Определение выполняют с использовани
ем титрационноro микропланшета, поддержива
емоro при температуре 37 ОС. По 12,5 мкл каж
доro из разведений испытуемоro раствора или
раствора сравнения вносят в каждый из рядов
ячеек. К содержимому каждой ячейки прибав
ляют по 25 мкл RVV и инкубируют ровно 90 с.
К содержимому каждой ячейки прибавляют по
150 мкл xpoMoreHHoro субстрата фактора Ха,
разбавленноro в соотношении 1:6 буфером для
разведен ий.
Реrистрируют скорость изменения опти
ческой плотности (2.2.25) при длине волны
405 нм непрерывно в течение 3 мин и полу
чают среднюю скорость изменения оптиче
ской плотности (дА/мин). При невозможности
непрерывной реrистрации оптическую плот
ность при 405 нм реrистрируют с подходя
щими последовательными интервалами, Ha
пример, продолжительностью 40 с, строят на
миллиметровой бумаrе rрафик зависимости
оптической плотности от времени и вычис
ляют из уклона полученной прямой значение
М/мин. Из значений МIмин для каждоro раз
ведения испытуемоro и стандартноro образ
цов рассчитывают активность испытуемою
образца и проверяют достоверность опреде
ления с использованием обычных статистиче
ских методов (5.3).
01/2013:20720
2.7.20. Количественное определение инактивированной вакцины полиомиелита 375
2.7.20. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
ИНАКТИВИРОВАННОИ
ВАКЦИНЫ ПОЛИОМИЕЛИТА
/Н V/VO
Способность вакцины вызывать образова
ние нейтрализующих антител определяют iп vivo
по одному из следующих методов.
ОПРЕДЕЛЕНИЕ С ИСПОЛЬЗОВАНИЕМ
ЦЫПЛЯТ ИЛИ МОРСКИХ СВИНОК
rотовят подходящий ряд, состоящий не
менее чем из трех разведен ий испытуемой
вакцины с использованием подходящеro бу
феризированноro раствора натрия хлорида.
Морских свинок с массой тела 250 З50 r или
цыплят возрастом 3 недели распределяют по
rруппам, состоящим из 10 особей, и назначают
каждой из rрупп одно из разведений вакцины.
Каждому животному внутримышечно вводят
по 0,5 мл разведения, которое было назначено
rруппе, к которой данное животное относится.
Через 5 6 дней про водят забор крови и oтдe
ляют сыворотки. Определяют наличие в разве
дениях сывороток (1:4) нейтрализующих анти
тел к каждому из человеческих полиовирусов:
1, 2 и 3. 100 ССID 50 вируса смешивают с разве
дением сыворотки и инкубируют при темпера
туре 37 ос в течение 4,5 ч. Хранят при TeM
пературе (5:1:3) ос в течение 12 18 ч. Смеси
прививают клеточным культурам для опреде
ления присутствия вирусов, не подверrшихся
нейтрализации, и ведут реrистрацию результа
тов в течение периода до 7 дней после иноку
ляции. Для каждой rруппы животных отмечают
количество сывороток, в которых обнаружено
наличие нейтрализующих антител, и вычисля
ют разведение вакцины, дающее такой отклик
у 50 % животных. Параллельно выполняют KOH
трольный опыт С использованием подходящеro
стандартноro образца. Вакцина выдерживает
испытание, если обнаруживается наличие ней
трализующих антител к каждому из трех типов
вируса у 50 % животных в разведении вакцины
в соотношении 1:100 или более.
ИСПЫТАНИЕ С ИСПОЛЬЗОВАНИЕМ
КРЫС
Метод количественноrо определения in
vivo состоит из внутримышечноro введения
в задние конечности не менее трех разведе
ний испытуемой вакцины и вакцины CpgBHe
ния с использованием для каждоro разведе
ния rруппы из 1 О крыс подходящей линии не
содержащих патоrенов. Для получения ДOCTO
верных результатов для всех серотипов часто
бывает необходимо использовать четыре
разведения. Количество животных в rруппе
должно быть достаточным для получения pe
зультатов, отвечающих критериям ДOCТOBep
ности; десяти крыс в rруппе обычно достаточ
но, хотя достоверные результаты MorYT быть
получены и с меньшим числом животных в
rруппе. Масса тела животноro не должна OT
личаться более чем на 1 О % от средней по
rруппе. Каждой крысе вводят по 0,5 мл MaTe
риала. Диапазон доз выбирают таким обра
зом, чтобы получить дозовый отклик для всех
трех типов полиовируса. Забор крови у живот
ных производят через 20 22 дня. Определяют
по отдельности нейтрализующие титры против
всех трех типов полиовируса с использовани
ем в качестве провокационных вирусов 100
ССID 50 штаммов SаЫп, в качестве индикатор
ных клеток Ve,o или Нер2 в условиях нейтра
лизации, состоящих в инкубации в течение 3 ч
при температуре 35 З7 ос и 18 ч при темпера
туре 2 8 ОС. ДЛЯ получения результатов про
изводят фиксацию и окрашивание после ce
мидневной инкубации при температуре З5 ОС.
ДЛЯ признания определения достоверным He
обходимо показать, что титр провокационных
вирусов находится в пределах от 10 ССID 50 ДО
1000 ССID 50 , а титр нейтрализующих антител
в контрольной сыворотке должен находиться
в пределах двух двукратных разведений cpeд
неro rеометрическоro титра сыворотки. Актив
ность вакцины рассчитывают путем сопостав
ления количества респондеров на испытуемую
вакцину и вакцину сравнения. Анализ выпол
няют с использованием пробит метода или,
после про верки достоверности, метода парал
лельных линий. В случае пробитметода для
определения респондера необходимо YCTaHO
вить rраничный титр нейтрализующих анти
тел для каждоro из типов полиовируса. В связи
с изменчивостью результатов в разных лабо
раториях определить общепринятые rранич
ные значения не представляется возможным.
Вместо этоro rраничные значения определяют
в каждой лаборатории, основываясь не менее
чем на трех результатах испытаний вакцины
сравнения. За rраничное значение принима
ют среднюю точку в шкале log2 минимально
ro и максимальноro значений среднеro reoMe
трическоro титров, вычисленных для ряда из
трех или более испытаний. Для каждоro из
трех типов полиовируса активность вакцины
не должна быть существенно ниже такой же
активности стандартноro образца. Результаты
испытания являются достоверными, если:
как для испытуемой вакцины, так и для
стандартноro образца ЕО 50 заключена в интер
вале между наибольшей и наименьшей дозами
препарата, введенными животным;
при статистическом анализе не выявляет
ся отклонений от линейности и параллельности;
rраницы доверительноro интервала
(Р = 0,95) относительной активности находятся
между 25 % и 400 % от оцененноro значения aK
тивности.
376
rосударственная фармакопея Республики Беларусь
Следующий раздел приводится для информации.
Руководство по отказу от проведения
исследования in vivo вакцины против
полиомиелита (инактивированной)
и ее комбинированных препаратов
Эта рекомендация относится к вакцинам,
полученным на основе «диких» штаммов полиови
русов. Описанную процедуру валидации следует
проводить для каждО20 продукта перед тем, как
отказаться от анализа in vivo. и необходимо по
вторять в случае существеНН020 изменения про
цесса производства, которое может повпиять
на результаты исследования iп vitro или iп vivo.
Европейская конвенция по защите позво
ночных животных, используемых для экспе
риментальных и иных научных целей требует,
чтобы тесты на животных не проводились, если
имеются удовлетворительные альтернативные
научно обоснованные и практически доступные
методы анализа.
Целью настоящеro руководства является
стимулирование отказа от исследований с ис
пользованием животных любоro данноro продук
та, для котороro существует метод iп vitro анали
за (определение D антиrена), обеспечивающий
удовлетворительные результаты в ходе рутинно
ro серийноro контроля.
Для in vivo анализа испытание на крысах
считается методом выбора.
Для вакцин, которые анализируют с исполь
зованием цыплят или морских свинок и дЛЯ KO
торых существуют утвержденные протоколы и
устоявшаяся история производства, от проведе
ния анализа in vivo можно отказаться, если ис
следования на крысах также применимы для их
анализа и включены в валидационные исследо
вания, описанные ниже.
Для еще не утвержденных вакцин, результа
ты анализа на крысах для всей roтовой партии
должны быть включены во все данные, пред
ставленные для демонстрации качества продук
ции до отказа от анализа iп vivo.
С момента отказа от анализа iп vivo партии
вакцины должны выпускаться на основе aHa
лиза in vitro, и анализ in vivo не должен ис
пользоваться как альтернатива для выпуска
партии, которая не соответствует требовани
ям при исследовании in vitro. Повторение aHa
лиза in vitro может быть выполнено в COOTBeT
ствии с утвержденной процедурой.
ИСПЫТАНИЯ
Следующие условия должны быть соблюде
ны перед выполнением валидационноro иссле
дования:
соответствующий опыт в использовании
анализа на крысах;
полная валидация исследования D анти
reHa (линейность, повторяемость, промежуточ
ная точность, поrрешность и пределы количе
ственноro определения);
установление критериев приемлемости
для D антиrен теста, основанных на определен
ном числе последовательных исследований;
установление соответствия продукции Tpe
бованиям на основе исследования последних
серий с применением действующей в настоящее
время процедуры iп vivo анализа; взятые в иссле
дование серии должны соответствовать лотам,
использованным для установления критериев
приемлемости для теста на D антиrен, и должны
представлять различные инактивированные куль
туры каждоro из 3 типов вируса полиомиелита.
Валидационные исследования должны быть
выполнены на:
roтовой серии, полученной в соответствии
с действующим реrламентом производства;
двух несоответствующих требованиям
сериях, полученных, например, путем проrрева
нормальной вакцины или смешивания нормаль
ной вакцины с термоинактивированной; титры
при исследовании несоответствующих требова
ниям вакцин должны составлять примерно поло
вину от нормальных.
Эти серии исследуются с применением в Ka
честве стандарта roмолоrичных серий продукта:
с применением утвержденноro в HaCTO
ящее время in vivo теста;
с применением исследования на крысах в
случае отсутствия утвержденноro iп vivo теста;
с применением теста на D антиrен.
Отказ от анализа с использованием лабора
торных животных является приемлемым, если
репрезентативная серия rотовой продукции co
ответствует требованиям при испытании in vivo
и in vitro, а заведомо несоответствующие требо
ваниям серии не проходят тесты. В случае, если
заведомо несоответствующие серии не проходят
тест на D антиrен, но удовлетворяют требовани
ям теста in vivo, последний может быть повторен.
01/2013:20721
2.7.21. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ ФАКТОРА
ВИЛЛЕБРАНДА ЧЕЛОВЕКА
Биолоrические функции фактора Вилле
бранда (von WillеЬ,апd) мноroчисленны. В Ha
стоящее время для количественноrо определе
ния MOryT использоваться еro ристоцетиновая
коферментная активность и коллаrен связыва
ющая активность. Активность фактора Вилле
бранда человека определяется путем cpaBH
ния еro активности в данных условиях с той же
активностью стандартноro образца, откалибрcr
BaHHoro относительно Международноro CTaH
дартноro образца, при необходимости в Между
народных Единицах.
2.7.21. Количественное определение фактора Виллебранда человека
377
За Международную Единицу принимают aK
тивность установленноro количества Между
народноro стандартноrо образца, состоящеro
из лиофилизированноrо концентрата фактора
Виллебранда че.ловека. Эквивалентность Меж
дународноro стандартноro образца в Междуна
родных Единицах устанавливается Всемирной
орrанизацией здравоохранения (ВОЗ).
РИСТОЦЕТИНОВАЯ КОФЕРМЕНТНАЯ
АКТИВНОСТЬ
Ристоцетиновая коqpерментная активность
фактора Виллебранда определяется путем из
мерения а лютинации суспензии тромбоцитов
в присутствии ристоцетина А. Испытание может
проводиться количественными методами с ис
пользованием автоматизированноro оборудова
ния или полуколичественным методом путем ви
зуальной оценки конечной точки аrrлютинации в
:;ерии разведений. Количественные методы яв
пяются предпочтительными.
РЕАКТИВЫ
Суспензия тромбоцитов. Используют CTaH
артизованные или фиксированные, например,
:I':юрмальдеrидом или параформальдеrидом,
lрепараты свежеполученных и отмытых TpOM
50ЦИТОВ человека. Суспензия может быть также
nиофилизированной. При необходимости добав
nяется соответствующее количество ристоцети
-ia А. Некоторые реактивы тромбоцитов MOryт
::одержать ристоцетин А.
Стандартный образец. Стандартный обра
lец фактора Виллебранда представляет собой
V1еждународный стандартный образец ВОЗ KOH
,teHTpaTa фактора Виллебранда.
МЕТОДИКА
Полуколичественный метод. rотовят под
садящие разведения испытуемоro и CTaHдapT
-ioro образцов с использованием в качестве pac
rворителя раствора, содержащеro 9 r/л натрия
иlОрида Р и 1 50 r/л альбумина человеческо
ю Р. К каждому разведению прибавляют co
)тветствующее количество суспензии TpOM
5оцитов и, при необходимости, ристоцетин А.
:мешивают на предметном стекле путем aKKY
)8ТНОro вращения в течение 1 мин. Выдержива
от в течение еще 1 мин и учитывают результаты
ia темном фоне при боковом освещении. По
:.neAHee разведение, при котором четко наблю
::аается видимая аrrлютинация, представляет
::о50Й ристоцетиновый коферментный титр об
)8зца. В качестве отрицательноro контроля ис
1Ользуют растворитель.
Количественный метод. Восстанаrзливают
х>держимое 1 ампулы со стандартным и испы
ryeMbIM образцами путем прибавления COOTBeT
:rвующеro рекомендуемоro растворителя (Ha
IpИмер, воды Р); используют незамедлительно.
( восстановленным образцам прибавляют дo
:raточное количество предварительноro paCTBO
рителя до получения растворов с активностью
0,5--------2,0 МЕ/мл. Предварительный раствори
тель состоит из изотоническоro нехелатирующе
ro буферноro раствора, содержащеro, например,
1 5 % человеческоro или бычЬеro альбумина и
трис(rидроксиметил)аминометан или имидазол,
забуференноrо соответствующим образом.
Испытание выполняется в соответствии с
инструкциями производителя не менее, чем на 2
сериях разведений, необходимых для получения
не менее 3 различных концентраций в линейном
диапазоне количественною определения.
Проверяют достоверность испытания и pac
считывают активность испытуемоro образца с
помощью обычных статистических методов (Ha
пример, 5.3).
КОЛЛАrEН СВЯЗЫВАЮЩАЯ
АКТИВНОСТЬ
Связывание коллаrена определяется с по
мощью иммуноферментноro анализа на ми
кропланшетах с сорбированным коллаrеном.
Метод основан на специфическом связыва
нии фактора Виллебранда с фибриллами кол
лаrена и последующим связыванием поли
клональных антител к фактору Виллебранда,
конъюrированных с ферментом, который при
добавлении к xpoMoreHHoMY субстрату об
разует продукт, который может быть количе
ственно определен спектрофотометрически.
В соответствующих условиях существует ли
нейная взаимосвязь между связыванием кол
лаrена с фактом Виллебранда и оптической
плотностью.
РЕАКТИВЫ
Колла2ен. Используют природные лоша
диные или человеческие фибриллы коллаrена
типа I или 111. Для удобства обращения MOryт ис
пользоваться растворы коллаrена.
Растворитель колла2ена. 50 r 2ЛЮКОЗЫ Р
растворяют в воде Р, доводят значение рН по
лученноro раствора до 2,7 2,9 с помощью 1 М
раствора кислоты хлорuстоводородной и дo
водят водой Р до объема 1000 мл.
СФБ (раствор натрия хлорида в фосфат
ном буферном растворе, PBS). 8,0. r натрия
хлорида Р, 1,05 r динатрия 2идрофосфата ди
2идрата Р, 0,2 r натрия ди2идрофосфата Р и
0,2 r калия хлорида Р растворяют в воде Р, ДOBO
дят до значения рН полученноro раствора 7,2 с
помощью 1 М раствора натрия 2идроксида или
1 М раствора кислоты хлористоводородной и
доводят водой Р до объема 1000 мл.
Промывочный буферный раствор. СФБ, co
держащий 1 r/л полисорбата Р.
Блокирующий реактив. СФБ, содержащий
1 r/л полисорбата Р и 10 r/л сыворотки альбу
мина бычье20 Р.
Буферный раствор для разведения. СФБ,
содержащий 1 r/л полисорбата Р и 50 r/л CЫBO
ротки альбумина бычье20 Р.
378
rосударственная фармакопея Республики Беларусь
КОНЪЮ2ат. Кроличья антисыворотка против
фактора Виллебранда человека. конъюrирован
ная с пероксидазой хрена. Используют в соот
ветствии с инструкциями производителя.
Раствор субстрата. Непосредственно
перед использованием растворяют таблетку
о фенилендиамина диrидрохлорида и таблет
ку rидроперита в 20 мл воды Р или используют
соответствующий объем водорода пероксида.
Хранят в защищенном от света месте.
Микропланшеты. Используют плоскодон
ные полистироловые планшеты со свойствами
поверхности. оптимизированными для иммуно
ферментною анализа и с высокой способностью
связывать белки.
МЕТОДИКА
Испытуемые растворы. Испытуемый обра
зец восстанавливают в соответствии с указаниями
на этикетке. Растворяют в буферном растворе для
разведения до получения раствора, содержащеro
приблизительно 1 МЕ Фактора Виллебрандэ. rOTo--
вят 2 серии из не менее 3 последовательных разве
дений с иаюльзованием буферноro раствора для
разведения.
Стандартные растворы. Стандартный обра
зец восстанавливают в соответствии с указаниями и
разводят буферным раствором для разведения до
получения раствора, содержащею приблизительно
1 МЕ Фактора Виллебранда. rотовят 2 серии из не
менее 3 последовательных разведений с использо
ванием буферноro раствора для разведения. PaCT
вор коллаreна доводят до комнатной температуры
и разводят растворителем коллаrена до получения
раствора содержащею З(}.......75 мкr/мл коллаrена,
осторожно смешивают до получения однородной
суспензии фибрилл коллаrена. С помощью микро--
пипетки в каждую лунку микропланшета вносят по
100 мкл полученной суспензии. Планшет HaKpЫBa
ют пластиковой пленкой и оставляют на ночь при
температуре 37 ОС. Лунки планшета с сорбирован
ным коллаrеном осушают, переворачивая и BЫ
сушивая бумажным полотенцем. В каждую лунку
планшета прибавляют по 250 мкл промывочною
буферноro раствора. Лунки планшета осушают, пе
реворачивая и высушивая бумажным полотенцем.
Повторяют данную процедуру 3 раза. В каждую
лунку планшета прибавпяют по 250 мкл блокиру
ющеro реактива, накрывают планшет пластиковой
пленкой и инкубируют при температуре 37 ос в Te
чение 1 ч. Лунки планшета осушают, переворачи
вая и высушивая бумажным полотенцем. В каждую
лунку планшета прибавляют по 250 мкл промывоч
ною буферною раствора. Лунки планшета осуша
ют, переворачивая и высушивая бумажным поло--
тенцем. Повторяют данную процеДУР 1 3 раза.
В лунки планшета при6авляют по 100 мкл каж
доro испытуемою и стандартною растворов. При
бавляют 100 мкл промывочною буферною paCT
вора в серию лунок, сохраняемых в качестве
отрицательноrо контроля. Планшет накрывают
пластиковой пленкой и инкубируют при температу
ре 37 ос в течение 2 ч. Удаляют из планшета жид
кость, переворачивая и высушивая бумажным по--
лотенцем. Прибавляют по 250 мкл промывочною
буферноro раствора. Лунки планшета осушают, пе--
реворачивая и высушивая бумажным полотенцем.
Повторяют данную процедуру 3 раза.
rотовят соответствующее разведение конъю
rэта (например, фактор разведения 1:4000) в СФБ.
содержащею 5 r/л альбумина бычьею Р, и прибав
ляют по 100 мкл В каждую лунку. Планшет HaKpы
вают пластиковой пленкой и инкубируют при TeM
пературе 37 ос в течение 2 ч. Удаляют из планшета
жидкость, переворачивая и высушивая бумажным
полотенцем. Прибавпяют по 250 мкл промывочноrо
буферноro раствора. Лунки планшета осушают, пе
реворачивая и высушивая бумажным полотенцем.
Ловторяют данную процедуру 3 раза.
В каждую лунку планшета прибавпяют по
100 мкл раствора субстрата и инкубируют при KOM
натной температуре в темном месте в течение
20 мин. В каждую лунку при6авляют по 100 мкл 1 М
раствора кислоты хлористоводородной.
Измеряют оптическую плотность при длине
волны 492 нм. Используют значения оптической
плотности для оценки активности испытуемоro об
разца с использованием обычных статистических
методов (5.3).
Результаты испытаний считают HeДOCToвepHЫ
ми, если оптическая плотность отрицательных KOH
тролей выше 0,05.
01/2013:20722
2.7.22. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ ФАКТОРА
СВЕРТЫВАНИЯ КРОВИ XI
ЧЕЛОВЕКА
Принцип исследования состоит в измерении
способности препарата фактора XI сокращать
время свертывания плазмы дефицитной по фак
тору XI. Реакция может быть ускорена путем дo
бавления peareHТOB, содержащих фосфолипиды
и контактные активаторы, каолин, кремнезем или
эллаrиевую кислоту.
Активность фактора XI в испытуемом лекар
ственном средстве оценивается путем сравнения
кривой дозаlэффект испытуемоro образца с Ta
ковой для референсноro образца, состоящеro из
нормальной человеческой плазмы.
1 единица фактора ХI равна активности 1 мл
нормальной человеческой плазмы. Ноrмальную
человеческую плазму получают путем объедине
ния в пул единиц плазмы, полученных не менее
чем от 30 доноров, и хранят при температуре
зо ос и ниже.
Восстанавливают по отдельности испыту
емый образец и стандартный образец в COOTBeT
2.7.23. Подсчет клеток СD34/СD45+ в zемопоэтuческuх продуктах
379
ствии С указаниями на этикетке и используют He
медленно. В случае необходимости определяют
количество присутствующеro rепарина (2.7.12) и
нейтрализуют rепарин добавлением протамина
сульфата Р (10 MKr протамина сульфата нейтра
лизуют 1 МЕ rепарина).
Испытуемый и стандартный образцы раз
водят человеческой плазмой, дефицитной по
фактору XI (например, субстрата плазмы Р3),
дЛЯ получения растворов с содержанием от
0,5 ЕД/мл до 2,0 ЕДiмл. rотовят не менее трех
разведений каждою образца, предпочтительно
в дубликатах, используя подходящий буферный
раствор (например, имидаЗОЛЬНblЙ буферныи
раствор рН 7,3 Р, содержащий 10 r/л бычьеro
или человеческоro альбумина. Разведения ис
пользуют немедленно.
При определении применяют аппаратуру, по
зволяющую мноroкратно измерять время CBepTЫ
вания, либо проводят исследование при посто
янной инкубации пробирок в водяной бане при
температуре З7 ос.
В каждую пробирку помещают по 0,1 мл
плазмы, дефицитной по фактору XI (например,
субстрата плаЗМbI Р3) и по 0,1 мл одноro из
разведений испытуемоro или стандартною об
разца. К содержимому каждой пробирки прибав
ляют по 0,1 мл peareHTa активированноro ча
стичноro тромбопластиновоrо времени (АЧТВ,
Activated Partial ТhroтЬорlаstiп Тiтe АРТ1),
содержащеro фосфолипиды или контактный aK
тиватор, и инкубируют при температуре 37 ос в
течение рекомендованноro времени. В каждую
пробирку прибавляют по 0,1 мл раствора З,7 r/л
кальция хлорида Р, предварительно подоrрето
ro до температуры 37 ос.
С помощью секундомера измеряют время
коаrуляции, т. е. интервал времени между при
бавлением кальция хлорида и первыми при
знаками образования фибрина. Объемы pe
активов, приведенные выше, MorYT быть
адаптированы для используемых peareHТOB
АЧТВ или аппаратуры. Рассчитывают актив
ность с использованием обычных статистиче
ских методов (например, 5.3).
01/2013:20723
2.7.23. ПОДСЧЕТ КЛЕТОК СDЗ4/СD45+
вrЕМОПОЭТИЧЕСКИХ
ПРОДУКТАХ
Эта rлава описывает методолоrию опреде
ления числа СО34/СО45+ клеток, содержашихся
в rемопоэтических биопрепаратах проведения
иммунною мечения и анализа методом проточ
ной цитометрии (2.7.24). Определение выпол
няют на основе одноплатформенноro метода с
использованием калиброванных флуоросфер
после лизиса эритроцитов в образце крови при
необходимости.
Этот метод применим ко всем типам био
препаратов и цельной крови. Однако уровень
точности делает ero особенно подходящим для
продуктов с очень низким содержанием СDЗ4/
СО45+ клеток.
Оценка качества трансплантата по под-
счету СО34/СО45+ клеток
С помощью разнообразных исследований
установлено, что 1 З % клеток в костном мозrе,
выделяющие антиrен поверхности клеток СDЗ4,
способны к восстановлению долrосрочноro,
мноroростковоro кроветворения после миело
аблативной терапии. СDЗ4/СD45+ клетки также
найдены в периферической крови нормальных
людей, но они чрезвычайно редки (от 0,01 % до
0,1 %). Однако СО34/СО45+ клетки MOryт также
быть мобилизованы из костноro мозrа в перифе
рическую кровь в значительных количествах под
действием rемопоэтических цитокинов, таких
как rранулоцитарный колониестимулирующий
фактор и/или химиотерапия. Техника, использу
емая для подсчета СDЗ4/СО45+ клеток, должна
соответствовать следующим требованиям:
высокая чувствительность, так как rемопо
этические стволовые клетки встречаются редко;
точность, чтобы обеспечить клинически
значимые результаты;
воспроизводимость, чтобы обеспечить
клинически надежные результаты;
скорость, чтобы обеспечить анализ в pe
альном времени.
Выбор параметров
Метод проточной цитометрии использует
коммерчески доступные, конъюrированные флу
орохром меченные моноклональные антитела,
стандартные процедуры окрашивания и лизиса
цельной крови, а также стратеrию установки си
стемы пропуска данных с использованием CBe
торассеивания и иммунофлуоресценцентно
ro анализа с применением комбинации всех
СО45/СО34 моноклональных антител.
Возможно определение жизнеспособности
СО34/СО45+ клеток с помощью соответствую
щей окраски нуклеиновых кислот красителями,
не окрашивающими мембраны неповрежденных
клеток (например, 7 аминоактиномицин О).
Выбор моноклональных антител
Антитела СО34. ДЛЯ обнаружения всех
rликозилированных вариантов молекулы (Ha
пример, клона 8G12 или 581) ИСПОJ1ЬЗУЮТ класс
111 антител СО34. ДЛЯ обнаружения редких про
исшествий используют антитела, конъюrирован
ные с флуорохромами (например, фикоэритри
ном (РЕ»), имеющими максимальное свечение
при использовании цитометра на основе арroно
воro лазера.
380
rосударственная фармакопея Республики Беларусь
Антитела СО45. Необходимы антитела, KO
торые способны обнаруживать все изоформы и
все rликоформы со структурой С045. Обычно
используют антитела С045, конъюrированные
с флуоресцеинизотиоцианатом (ФИТЦ, F/TC) в
роли флуорохрома (например, JЗ3, HLe1, 201).
Контроли изотипов и изоклонов. Для об
наружения неспецифичных сиrналов в области
использования РЕ анализируют неrативный KOH
троль. При применении контроля изотипов (MO
ноклональные антитела к нерелевантным анти
reHaM тоro же типа, что и используемые антитела
С045) РЕ конъюrированный изотип объединяют
с C045--FJТC (PerCP). При применении контроля
изоклонов неконъюrированные (в избытке) и PE
конъюrированные моноклональные тела, иден
тичные СDЗ4, объединяют с конъюrированными
СD45. Moryт быть использованы альтернатив
ные комбинации.
Абсолютное количество СDЗ4/СD45+
Калиброванные флуоросферы. В зависи
мости от при меняемой технолоrии, внутренний
стандарт или состоит из калиброванных сфер,
находящихся в суспензии, или помещен непо
средственно в присоединяемые пробирки про
изводителем.
Абсолютное количество клеток СО34/СО45+
в миллилитре рассчитывают по формуле:
А
.C'
в
rдe:
А количество подсчитанных клеток СОЗ41
СО45+;
В количество подсчитанных флуоросфер;
С известная концентрация флуоросфер.
Стратеrии селекции
Целью последовательной селекции являет
ся выбор популяции, представляющей интерес,
и одновременная минимизация интерференции,
вызываемой осколками и зрелыми клетками, с
которыми MOryT неспецифически связываться
антиrены. При использовании коммерческих Ha
боров применяют селекцию, рекомендованную
производителем набора. При использовании
собственной методики желательно применять
существующие рекомендуемые критерии селек
ции. Стратеrия селекции, учитывающая параме
тры рассеяния света и флуоресценцию СО34/
С045, будет способствовать правильной иден
тификации и подсчету клеток СО34/С045+.
Количество анализируемых событий
Для достижения приемлеl\llОЙ точности aHa
лизируют достаточное количество событий, Ha
пример, не менее 100 событий С034+ и не
менее 60 000 событий СО45+; общее количество
клеток может быть выше при условии, если про
центное содержание СО34 составляет 0,1 % или
менее.
Сбор образцов
Антикоаryлянтом при процедуре афереза
является формула А кислой цитратной дeKCTpo
зы (АСО). Данный антикоаryлянт позволяет про
водить на одном и том же образце как aBTOMa
тический подсчет лейкоцитов, так и испытание с
помощью проточной цитометрии. Антикоаryлян
том выбора при отборе проб периферической
крови является этилендиаминтетрауксусная кис
лота (ЭДТА, ЕОТА).
Транспортирование образцов
Условия транспортирования образцов
должны rарантировать физическую и темпера
турную безопасность образцов.
Целостность и хранение образцов
MorYT быть исследованы свежие (хранив
шиеся не более 24 ч) образцы продуктов афе
реза, цельной крови, пуповинной крови или
костноro мозrа. Старые образцы (хранившие
ся более 24 ч) и образцы, которые претерпели
заморозку и размораживание, должны OKpa
шиваться красителями, позволяющими oцe
нить количество живых клеток. При получении
образцов проверяется температура внутри
упаковки.
МЕТОДИКА
Приrотовление образца
Перед окрашиванием с помощью MOHO
клональных антител необходимо убедиться,
что концентрация лейкоцитов является подхо
дящей. При необходимости образец разводят
средой, совместимой с испытуемым образцом
и системой для лизиса. Отмечают коэффициент
разведения. Рекомендуется проводить испыта
ние с использованием отрицательноro контроля.
Анализ с помощью проточной цитоме-
трии
Автоматическая стандартизация
Для анализа клеток, меченных с примене
нием коммерчески доступных наборов, произ
водители разработали некоторые критерии для
настройки проточноro цитометра. Данные Ha
стройки затем автоматически переносятся в
протокол анализа образцов. Для настройки фcr
тоумножительной трубки на заданные значения
используют специальные флуоросферы, YCTa
навливают поправку и проверяют систему с ncr
мощью контрольных образцов.
Настройки системы
ДискриминатОР/ПОР02: во избежание
учета осколков (низкое прямое рассеяние), но
не мелких форм лимфоцитов, в rрафике pac
сеяния света устанавливают пороr прямоro
рассеяния.
Высоковольтные настройки фотоу
ножительной трубки: они должны быть со-
вместимы с анализом маркеров поверхности
клеток и должны быть установлены в каждoi
2.7.24. Проточная цитометрuя
381
лаборатории таким образом, чтобы была воз
можность различить неrативные и позитивные
популяции клеток с умеренной плотностью
антиrенов; напряжение фотоумножительной
трубки необходимо периодически проверять
и настраивать в соответствии с установлен
ными в лаборатории процедурами.
Поправка: она должна быть применима
в случае перекрываний в видимом диапазо
не спектра (например, F/TC/PE), случающих
ся при анализе маркеров поверхности клеток;
поправку на цвет анализируют и настраивают
в соответствии с установленными в лаборато
рии процедурами.
Скорость потока: должна подходить
для рутинноrо анализа маркеров поверхности
клеток.
Диапазоны селекции (ворота): диапазо
ны, установленные для анализа образцов СОЗ4/
СО45 оставляют неизменными для анализа от
рицательноrо диапазона.
Расчет абсолютноrо количества клеток
CD34/CD45+
Абсолютное количество клеток СО34/
СО45+ рассчитывают по формуле:
п.О.V,
rде:
п общее количество клеток СDЗ4/СD45+
в микролитре;
О фактор разведения;
V объем испытуемоro образца, мкл.
Результаты испытания выражаются как в
процентном содержании клеток СО34/СD45+,
так и в абсолютном числе клеток на микро
литр. rде это возможно, результаты MorYT
быть выражены в абсолютном числе на кило
rpaMM массы тела реципиента.
01/2013:20724
2.7.24. ПРОТОЧНАЯ ЦИТОМЕТРИЯ
Суть проточной цитометрии состоит в MHO
roпараметрическом анализе оптических свойств
отдельных частиц в жидкой системе.
Клетки или частицы в суспензии должны
быть индивидуально распределены в линейном
потоке, который протекает через измерительное
устройство. Плотные ткани для анализа должны
быть превращены в суспензию клеток
Спектр параметров, определяемых про
точной цитометрией, включает объем и MOp
фолоrическую структуру клеток или клеточ
ных частиц, клеточные пиrменты, содержание
ДНК, содержание РНК, белки, маркеры по
верхности клеток, внутриклеточные маркеры,
ферментативная активность, значение рН,
состояние мембраны, текучесть.
Существует возможность определять 2
морфолоrических пара метра и 1 или более
сиrналов флуоресценции на каждую отдель
ную клетку. Мноroпараметрический анализ
позволяет определять клеточные популяции
по их фенотипу.
ПРИ БОР
Фокусировка, умножение сиrнала и выбор
источника света должны бwь оптимизирова
ны для автоматическоro обнаружения и измере
ния морфолоrических разпичий и особенностей
окрашивания. Анализ методом проточной цито
метрии обязательно предусматривает:
выбор источника света в зависимости от
параметров, которые будут проанализированы;
настройку цитометра в зависимости от
типа анализируемых клеток (например, клеточ
ные культуры, лейкоциты, тромбоциты, бакте
рии, сперматозоиды, дрожжи) и вида анализа,
который должен быть выполнен (например, фе
нотипирование, клеточный цикл, апоптоз, цито
кины, мембранная текучесть, флуоресцентный
белок).
Проточная цитометрия характеризуется aB
томатизированным количественным определе
нием ряда установленных параметров для боль
шоrо числа единичных клеток во время каждоro
сеанса анализа. Например, 100 000 частиц или
более (фактически неоrраниченное количество)
анализируются одна за друroй, как правило, при
близительно в течение 1 минуты. Предел обна
ружения достаточно низок и составляет 1 00 флу
оресцентных молекул на клетку.
Прибор для проточной цитометрии (проточ
ный цитометр) имеет 5 основных блоков:
система подачи растворов и проточная
ячейка;
источник света;
система обнаружения и система аналоro
цифровоro преобразования (АЦП);
система умножения (амплификации);
компьютер с проrраммным обеспечением
для анализа сиrналов.
СИСТЕМА ПОДАЧИ РАСТВОРОВ
И ПРОТОЧНАЯ ЯЧЕЙКА
Единичные клетки освещаются лучом света
и детектируются в проточной ячейке. Систе
ма подачи растворов несет отдельные клетки
от пробирки с образцом клеточной взвеси до
точки детектирования, для чеrо поток клеточ
ной взвеси сужается до еДL1НИЧНЫХ клеток в
проточной ячейке (rидродинамическое фокуси
рование). Луч света фокусирован в эллипсоид
ном пространстве 2 конфокальными линзами в
канале проточной ячейки, через который прохо
дят клетки. Скорость потока во время анализа
маркеров поверхности клеток должна быть по
382
rосударственная фармакопея Республики Беларусь
стоянной И должна rарантировать подходящее
расстояние между клетками для возможности их
подсчета.
ИСТОЧНИКИ СВЕТА
Обычно используют следующие источники
света:
лампы (ртутная, ксеноновая);
охлаждаемые водой лазеры большой
мощности (арroновый, криптоновый и лазер на
красителях);
охлаждаемые воздухом лазеры низкой
мощности (арroновый (488 нм), красный rелий
неоновый (633 нм). зеленый rелий неоновый, re
лий кадмиевый, (ультрафиолетовый»);
полупроводниковые (диодные) лазеры
(синий, зеленый, красный, фиолетовый).
ОБНАРУЖЕНИЕ сиrНАЛА
Korдa частица проходит через пучок света,
она рассеивает часть света во всех направлени
ях. Флуоресцентные красители, добавленные к
частице, испускают свой собственный свет (флу
оресценцию), которая также излучается во всех
направлениях. Таким образом, MOryт rенериро
ваться 2 типа сиrналов:
рассеяние света;
эмиссия флуоресценции.
Световые датчики прибора собирают часть
этоro рассеянноro света и флуоресценции и пре
образуют в электрические сиrналы пропорцио
нально количеству собранноro света.
Рассеяние света. Измеряются 2 параметра
рассеяния света:
прямое рассеяние (FS, forwa,d scatter);
боковое рассеяние, под уrлом 900 к Ha
правлению пучка света (SS, side scatter).
Прямое рассеяние коррелирует с объемом
клетки, в то время как боковое рассеяние, свя
занное с такими параметрами, как форма ядра,
количество и тип цитоплазматических rранул
или шероховатость мембран, коррелирует с
морфолоrической сложностью клетки: чем выше
интенсивность боковоro рассеяния, тем выше
сложность клетки.
При анализе потока Bcerдa будут rенери
роваться сиrналы светорассеяния, являющи
еся функцией морфолоrических характеристик
клеток; они определяются как внутренние пара
метры.
Флуоресценция. В зависимости от типа и
количества источников света, Korдa клетка про
ходит через область освещения, она будет ис
пускать свет флуоресценции. Флуоресцентные
сиrналы образуются флуоресцентными краси
телями естественно находящимися в клетках
(например, коферменты, хлорофилл, пиrмен
ты морской водоросли) иlили от флуоресцент
ных проб, захваченных клетками в процессе
маркирования для анализа определенных oco
бенностей (например, флуоресцирующие aH
титела, красители нуклеиновых кислот, пробы
для исследования рН, пробы на кальций, флу
оресцирующие белки). В настоящее время дo
ступно большое количество флуоресцентных
меток различных типов. Оптические фильтры
должны быть приспособлены к используемым
флуорохромам. Каждая флуоресцентная метка
характеризуется своим спектром возбуждения
и спектром эмиссии. Они выбираются в зависи
мости от природы источника возбуждения, си
стемы обнаружения и соrласно специфической
цели анализа.
УПРАВЛЕНИЕ СИiНАЛОМ И АНАЛОiО
ЦИФРОВОЕ ПРЕОБРАЗОВАНИЕ
Сиrналы светорассеивания и флуоресцен
ции, испускаемые клетками при прохождении
через лазерный луч, разделяются и направля
ются к детекторам за счет использования опти
ческих фильтров. Детекторы являются преобра
зователями (фотоумножительные трубки (PMTs,
photomu/tip/ie, tubes», которые преобразуют CBe
товые сиrналы, испускаемые клетками, в им
пульсы напряжения.
Процесс подсчета каждоro импульса в co
ответствующем канале известен как анало
rо цифровое преобразование (АЦП). Процесс
отображается rрафически в виде частотной rи
cTorpaMMbI.
Умножение (амплификация). Для опти
мальной визуализации импульсы напряжеНИI1
должны быть усилены. Процесс умножения под
черкивает различия между сиrналами клетки И,
следовательно, увеличивает разрешение между
популяциями клеток с различными характери
стиками (например, отличие жизнеспособных
от нежизнеспособных клеток, или неспецифиче
ской флуоресценции от антиrен специфической
флуоресценции после окраски мечеными MOHO
клональными антителами).
Есть 2 метода умножения сиrнала: линей
ный или лоrарифмический. Выбор между двумя
типами делается для каждоro отдельноro сиr
нала в соответствии с морфолоrическими xa
рактеристиками клеток и использованными Kpa
сителями (например, флуоресцентно меченые
моноклональные антитела, красители нуклеино
вой кислоты).
Линейное умножение, которое усиливает
различия среди сильных импульсов, использует
ся с теми параметрами, которые производят сиr
налы высокой интенсивности, например:
параметры рассеяния клетки;
флуоресценция кр:зсителей нуклеиновых
кислот при изучении клеточноro цикла.
Л02арифмuческое умножение, напротив,
предназначено для слабых импульсов и параме
тров или аналитических условий, которые MOryT
произвести и слабый, и сильный импульсы, Ha
пример:
2.7.24. Проточная цитометрuя
383
антиrены клетки
светорассеивание от тромбоцитов, бакте
рий, дрожжей;
флуоресценция от красителей нуклеино
вых кислот при исследовании апоптоза.
Компенсация сиrналов флуоресцен-
ции. У каждоro флуоресцирующею красителя
есть определенный спектр поrлощения и спектр
эмиссии. При использовании 2 или более флуо
ресцентных проб одновременно для тоro, чтобы
окрасить клетки (например, иммунофенотипиро
вание по 4 антиrенам), спектры эмиссии флуо
рохромов MOryт перекрыаться.. Как следствие,
каждый детектор флуоресценции зареrистри
рует определенную люминесценцию «своей»
пробы и KaKoe TO количество света, излучаемоro
друrими флуоресцентными проба ми. Это может
привести к переоценке сиrнала и плохому разде
лению популяций клеток.
Решением является использование элек
тронной матрицы, которая позволяет селектив
но разделять перекрестные сиrналы после их
улавливания детектором (компенсация флуо
ресценции).
Компенсация флуоресценции требует ис
пользования калибраторов флуоресценции.
предпочтительно образцов позитивных клеток.
меченных необходимыми флуорохромами в по
рядке, соответствующем меченым антителам,
использованным для анализа.
ОТОБРАЖЕНИЕ сиrнАЛОВ
После усиления и компенсации сиrналы OTO
бражаются в виде ДBYX или трехмерных rисто
rpaMM. rистоrраммы показывают интенсивность
сиrнала и количество клеток для данноro па
раметра. Цитоrраммы, в которых каждая точка
представляет клетку, основаны на комбинации
2 сиrналов (двухсиrнальные точечные диаrрам
мы). Тип и число распределений и комбинаций
сиrналов выбирают в зависимости от образцов
клеток и использованных меток. KorAa анализи
руются полученные данные, проrраммное обе
спечение проточной цитометрии может также
rенерировать и друrие виды rрафических изо
бражений (такие как накладывания, поверхност
ные rистоrраммы, TOMorpaMMbI (срезовые rисто
rpaMMbI), контурные rистоrраммы, rистоrраммы
плотности/интенсивности, накладывающиеся
rистоrраммы). MoryT также использоваться CTa
тистические данные, такие как средние интен
сивности флуоресценции (и их изменения по
времени или в зависимости от функции клетки).
АНАЛИЗ ДАННЫХ
Клеточная взвесь для анализа может co
держать различные виды объектов, некоторые
из которых нежелательны (такие как мертвые
клетки, обломки клеток или MaKpoarperaTbI) или
просто не подходят для анализа (например, rpa
нулоциты при изучении лимфоцитов). Это за
висит от типа образца (цельная кровь, костный
мозr, клеточные культуры, биолоrические жид
кости, клеточные взвеси из плотных тканей) и
процедуры обработки (например, методы OKpa
шивая, лизиса (эритроцитов), фиксации, крио
консервирования, размораживания, депарафи
нирования ).
Как следствие, не все сиrналы, rенериро
ванные во время анализа, принадлежат изуча
емым клеткам. Чтобы исключить нежелатель
ные инесоответствующие сиrналы примеНЯЮ1
два подхода.
Первый используется при получении и HaKO
плени и данных. Это шумовой nopor, относящий
ся к одному (или более) существенному параме
тру (параметрам), устанавливающий включение
в анализ только клеток с интенсивностью сиrна
ла выше, чем предопределенное дискримина
ционное значение для данноro параметра. Бла
roдаря характеристике параметра как мощноro
сиrнала с низким уровнем перекреста, прямое
рассеивание является параметром, чаще всеro
используемым в качестве дискриминатора (фак
тора разделения).
Второй подход применяется при анаЛlAзе
данных и состоит в использовании систем пропу
ска данных, оrpаничивающих анализ только сиr
налами от тех популяций клеток, которые YДOB
летворяют определенным морфолоrическим tA
экспрессионным (по флуоресценции) профи
лям. Обычно используются 2 типа селекции сиr
нала (лоrических ворот). Первый тип основан на
морфолоrии клеток. Клеточные популяции иден
тифицируются по их морфолоrическим xapaктe
ристикам (FS и SS). Диапазон селекции сиrнала
определяет пределы представляющей интерес
популяции (например, лимфоциты, жизнеспо
собные клетки), затем rрафики флуоресцен
ции вводятся через выбранную область. Второй
тип селекции сиrнала основан на флуоресцен
ции. Интересующая нас клеточная популяция
идентифицируется на основании интенсивно
сти экспрессии антиrена или красителя, затем
определяются rраницы диапазона селекции.
Впоследствии rрафики флуоресценции строят
ся в рамках выбранной области. Аналитическое
проrраммное обеспечение позволяет создавать
множественные диапазоны селекции, используя
последовательный лоrический запрос. Это oco
бенно информативно при изучении редких попу
ляций клеток или с целью сортировки клеток.
КОНТРОЛИ
Внутренний контроль. Перед анализом
должна быть проведена настройка оптической
системы с использованием подходящих флуо
росфер, а также должна быть проверена стабиль
ность проточной системы. Полученные данные
сводятся в отчет и используются при проведе
нии периодическоro анализа показателей лабо--
раторной эффективности метода. Наличие по
ложительноro контроля весьма желательно для
384
rосударственная фармакопея Республики Беларусь
доказательства тоro, что используемые для Te
стирования антитела функциональны и позволя
ют надлежащим образом использовать проточ
ный цитометр. Положительный контроль должен
включать образцы, которые известны как noло
жительные для интересующеro нас маркера.
Внешний контроль. Чтобы raрантировать
надежность полученных данных или проверить
межлабораторную воспроизводимость, peкo
мендуется участвовать в межлабораторных сли--
чениях (проrраммах профессиональною тести
рования).
01/2013:20725
2.7.25. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ
инrИБИТОРАПЛАЗМИНА
ЧЕЛОВЕКА
Инrибитор плазмина человека, также Ha
званный человеческим Q2 антиплазмином, яв
ляется плазменным белком, который подавляет
связанный с плазмином (сериновой протеазы)
путь фибринолиза путем быстроro формирова
ния комплекса со свободным плазмином. Более
тоro, при свертывании крови инrибитор плазми
на человека поперечно связывается с нитями
фибрина посредством фактора XIII, и интерфе
рирует со связыванием профермента плазмино
reHa с фибрином.
Активность инrибитора плазмина челове
ка оценивают путем сравнения способности ис
пытуемых образцов подавлять расщепление
специфическоro xpoMoreHHoro субстрата плаз
мином с той же самой способностью CTaHдapT
Horo образца инrибитора плазмина человека.
Расщепление плазмином xpOMoreHHoro суб
страта приводит к образованию хромофора, KO
торый может быть определен количественно
спектрофотометрически.
Реактивы для испытания MOryт быть полу
чены отдельно или в составе коммерческих Ha
боров. При меняется как метод определения KO
нечной точки, так и кинетический метод. При
использовании различных наборов методики и
реактивы MOryT отличаться в соответствии с ин
струкциями производителя. Существенные oco
бенности методики описаны в следующем при
мере микропланшетноro кинетическоro метода.
РЕАКТИВЫ
Буферный раствор для разведения рН 7,5.
Используют подходящий буферный раствор co
rласно инструкциям производителя. При необхо
димости корректируют значение рН.
Плазмин. Предпочтительно использовать
препарат человеческоro плазмина, который не
содержит существенноro количества друrих про
теаз. rотовят раствор и хранят соrласно инструк
циям производителя.
ХРОМ02енный субстрат плазмина. Исполь--
зуется подходящий специфический хромоrенный
субстрат для плазмина: Н D циклоrексилаланил
норвалил лизил п нитроанилина rидрохлорид
(H D CHA Nva Lys pNA.HCI) или L пироrлутамил
L фенилаланил L лизил п нитроанилина rидро
хлорид (Glp Phe Lys pNA'HCI). rотовят раствор
в воде Р, чтобы получить подходящую KOHцeH
трацию соrласно инструкциям производителя.
МЕТОДИКА
f>азличные количества испытуемою образ
ца смешивают с определенным количеством
плазм ина и определяют остаточную активность
плазм и на с использованием подходящеro xpo
MoreHHoro субстрата.
rотовят или размораживают испытуемый
образец соrласно инструкциям производителя.
Разводят буферным раствором для разведения
рН 7,5 и rотовят по крайней мере по 2 независи
мые серии из 3 или 4 разведен ий для испыту
емоro образца и для стандартноro образца.
Смешивают по 0,020 мл каждоro разведе
ния с 0,020 мл буферноro раствора для раз
ведения рН 7,5 и наrревают до температуры
З7 ОС. Прибавляют по 0,04 мл раствора плазми
на (концентрации для испытания в диапазоне от
0,2 нкатIмл до 1,6 нкат/мл), ранее HarpeToro до
температуры 37 ОС, и выдерживают при темпе
ратуре 37 ос в течение 1 мин. К каждой смеси
прибавляют по 0,02 мл раствора xpoMoreHHoro
субстрата, предварительно HarpeToro до темпе
ратуры 37 ОС. Немедленно начинают измере
ние изменения оптической плотности при длине
волны 405 нм (2.2.25), используя микропланше
точный ридер (спектрофотометр). Рассчитыва
ют скорость изменения оптической плотности
(М/мин). Альтернативно может быть использо
ван метод определения конечной точки OCTa
новка реакции уксусной кислотой и измерение
оптической плотности при длине волны 405 нм.
В обоих случаях продолжительность pac
щепления xpoMoreHHoro субстрата должна быть
выбрана таким образом, чтобы наблюдалось ли
ней ное увеличение оптической плотности при
длине волны 405 нм прежде, чем существенное
влияние начнет оказывать недостаток субстра
та. Если испытание выполняют в пробирках или
кюветах с использованием спектрофотометри
ческоro метода, объемы растворов реактива из
меняют пропорционально.
Отнимают значение оптической плотно
сти контрольноro ()пыта (приrотовленноrо с бу
ферным раствором для разведения рН 7,5) от
оптической плотности испытуемых образцов.
Проверяют правильность выполнения испыта
ния и рассчитывают активность испытуемоro
образца, используя обычные статистические
методы (5.3).
01/2013:20727
2.7.27. Значение флоккуляции (Lf) токсинов и анатоксинов дифтерии и столбняка 385
2.7.27. ЗНАЧЕНИЕ ФЛОККУЛЯЦИИ
(Lf) ТОКСИНОВ
И АНАТОКСИНОВ ДИФТЕРИИ
И СТОЛБНЯКА (ПРОБА РАМОНА)
Содержание токсина или анатоксина (TOKCO
ида) в образце может быть выражено в виде зна
чения флоккуляции (Lf), определенноro пробой
Рамона. В этой пробе антитоксин добавляется
в повышающихся концентрациях к серии про
бирок, содержащих постоянное количество ТOK
сина или анатоксина. В точке эквивалентности
токсина/анатоксина и антитоксина в 1 или более
пробирках происходит образование <рлоккуля
тов (комочков). Первая пробирка, в которой про
исходит образование комочков. используется
для определения значения Lf образца.
Значение Lf для токсина или анатоксина
определяется количеством единиц антитокси
на, которое при смешивании с образцом обра
зует оптимально выпадающую хлопьями смесь
(проба Рамона).
Практический опыт показал, что результа
ты калибровки антитоксинов в Международных
Единицах (МЕ), например, в сравнении с Между
народными стандартными образцами антиток
сина, зависят от использованноro иммунохими
ческоro метода. По этой причине антитоксины,
используемые для пробы Рамона, должны быть
напрямую калиброваны с использованием меж
дународных биОЛ02ических стандартных pe
а2ентов для дифтерии или столБНЯЧН020
анатоксина для теста флоккуляции, исполь
зуя принципы, описанные ниже. KOHцeHTpa
ция, определенная таким образом, может быть
обозначена в Lf эквивалентах на миллилитр
(Lf экв.lмл ).
По определению 1 Lf это количество ТOK
сина или анатоксина, которое вызывает флок
куляцию в течение минимальноro времени с
1 Lf экв. специфическоro антитоксина.
Диапазон объемов стандартноro образ
ца антитоксина, доведенноrо до концентрации
1 00 Lf экв.lмл, распределяется в серию пробирок
(например, размером 7 см х 1 см) для реакции
на флоккуляцию. К каждой пробирке для полу
чения постоянноro суммарноro объема, напри
мер, 1 мл, прибавляют достаточное количество
раствора 9 r/л натрия хлорида Р. Испытуемый
образец разводят таким образом, чтобы полу
чить ожидаемую концентрацию приблизительно
50 Lf/мл, и, например, по 1 мл этоro раствора пе
реносят в каждую из пробирок, содержащих aH
титоксин. Пrобирки тщатепьно перемешиваются
путем встряхивания, затем помещаются в водя
ную баню с постоянной температурой в диапа
зоне от 30 ос до 52 ОС, затем наблюдают через
реryлярные интервалы времени до первоro по
явления флоккулятов. Это может потребовать
использования лупы и яркоro освещения.
4!; з.. I 12
Первая и вторая пробирки со смесями, rдe
наблюдалась флоккуляция, отмечаются, как и
время, потраченное для первоrо образования
визуально определяемых комочков. В 2 пробир
ках хлопья MOryT выпасть одновременно.
Первая пробирка, в которой выпадут хлопья,
является той, которая содержит количество aH
титоксина, наиболее эквивалентноro количеству
антиrена в образце. Содержание антитоксина
в этой пробирке может быть использовано для
расчета значения Lf образца. Если в 2 пробирках
выпадают хлопья в одно и то же время, как KO
нечный результат используют среднее значение
обеих пробирок.
Время флоккуляции в первой пробирке (Kf)
является полезным индикатором качества aH
тиrена. Если при используемых температуре и
концентрации анатоксина и антитоксина значе
ние Kf увеличено по сравнению с нормальным,
это указывает на то, что антиrен был поврежден.
Значение Kf может также измениться в связи с
качеством используемоro антитоксина.
Пробирка
Добавленный
антитоксин
(Lf экв.)
Добавленный
антитоксин
(мл)
Добавленный
физиолоrи
ческий раст
вор (мл)
Добавленный
разведенный
образец
Пример
А В С
40 45 50
D Е F
55 60 65
0,40 0,45 0,50 0,55 0,60 0,65
0,60 0,55 0,50 0,45 0,40 0,35
1,0 1,0 1,0 1,0 1,0 1,0
Если в этом примере первой пробиркой,
в которой выпали хлопья, является пробир
ка С, Torдa значение Lf разведенноro образ
ца равняется 50 Lf/мл. Однако, если первой
пробиркой, в которой выпали хлопья, являет
ся пробирка А или пробирка F, это указывает
на отсутствие эквивалентности на том уровне.
В этом случае необходимо выполнить повтор
ное испытание, используя или иное разведе
ние испытуемоro образца, или выбирая иной
диапазон доз стандартноro образца антиток
сина.
Большая точность может быть получена
при учете поправки на последовательность об
разования хлопьев после первой пробирки. Так
в указанном примере, если бы вторая пробирка
после той, rдe '3ыпали хлопья, была пробиркой
О, конечное значение для разведенноro образца
было бы 52, тorдa как, если бы вторая пробирка
за той, rдe выпали хлопья, была пробиркой В, KO
нечное значение было бы 48. Испытание может
быть выполнено повторно с немноro отличающи
мися разведения ми испытуемоro образца.
386
rосударственная фармакопея Республики Беларусь
Если нет никаких указаний на предпола
raeMoe значение Lf у доступноro образца, же
лательно получить rрубую оценку (значения),
используя более широкий диапазон содержа
ния антитоксина в пробирках перед проведе
нием завершающеro теста.
Пробирка
Содержание
антитоксина
(Lf экв.)
Пример
А В С
20 30 45
D Б F
70 100 150
Уровень токсина или анатоксина и KOH
центрация антитоксина в испытании MorYT
варьировать, что будет заметно влиять на
время флоккуляции. Таким образом, на очень
низких уровнях испытание будет длиться
слишком долro, в то время как при высокой
концентрации начало образования хлопьев
может быть настолько быстрым, что затруд
нит различие первой и второй пробирки, В KO
торых выпадают хлопья.
Определение низких концентраций Me
тодом совместной флоккуляции
Для очень низких концентраций жела
тельно измерять токсин или анатоксин Me
тодом совместной флоккуляции. Это пред
полаrает сравнение значения Lf известноrо
токсина или анатоксина и значения смеси об
разца с таким же токсином или анатоксином.
Коrда токсин или анатоксин с извест
ным значением Lf и токсин или анатоксин с
неизвестным значением Lf образуют хлопья
вместе, смесь сформирует хлопья как pe
зультат суммы их значений, если они были
rомоrенными. Если HerOMoreHHbIe токсины
или анатоксины будут смешаны, то они вызо
вут абберантную (отклонюящуюся от нормы)
флоккуляцию С 2 максимумами образования
хлопьев (флоккуляции).
01/2013:20728
2.7.28. ОПРЕДЕЛЕНИЕ
КОЛИЧЕСТВА
rЕМОПОЭТИЧЕСКИХ КЛЕТОК-
ПРЕДШЕСТВЕННИЦ
ЧЕЛОВЕКА
ПО КОЛОНИЕОБРАЗУЮЩИМ
КЛЕТКАМ
Система кроветворения представляет собой
совокупность клеток, фенотип которых и свой
ства изменяются по мере их развития от CTBO
ловых клеток до дифференцированных клеток.
rемопоэтические клетки предшествен
ницы (rCK, HPCs) способны к формирова
нию колоний или «клеточных кластеров» в
культурах, выращенных в плотных питатель
ных средах, и называются «колониеобра
зующими». Определение числа колониео
бразующих клеток (КОЕ, CFCs) в клеточном
продукте является индикатором функцио
нальной способности клеток предшествен
ниц и является показателем восстановле
ния кроветворения. Измеренное число КОЕ
коррелирует с минимальным числом клеток
предшественниц кроветворения, присутству
ющих в образце.
МАРКЕРЫ ПОВЕРХНОСТИ КЛЕТОК
Способность колониеобразующих клеток
давать начало кроветворным колониям iп vitro
и/или восстанавливать систему KpOBeTBope
ния коррелирует с экспрессией определен
ных антиrенов поверхности клеток. Экспрес
сия мембранноro антиrена СО34 является
приняты м маркером для большинства клеток
предшественниц кроветворения и стволовых
клеток.
СПЕЦИФИЧНОСТЬ ИСПЫТАНИЯ
Колониеобразующие клетки распозна
ются в соответствии с номенклатурой, OCHO
ванной на линейной принадлежности зрелых
клеток, присутствующих в колониях (напри
мер, КОЕ смешанные, КОЕ rранулоцитов,
эритроцитов, моноцитов, макрофаrов (KOE
rэмм), КОЕ rранулоцитов и макрофаroв
(KOE rM), КОЕ rранулоцитов (КОЕ П, КОЕ
макрофаrов (KOE M), бурст образующие еди
ницы эритроцитов (БОЕ Э), КОЕ эритроцитов
(КОЕ Э), КОЕ меrакариоцитов (KOE Mer», и
являются популяциями клеток предшествен
ниц, способных образовать колонии, coдep
жащие клетки одной или более линий (диф
ференцировки) кроветворных клеток. Для
этой популяции rCK по сравнению с самыми
незрелыми стволовыми клетками были опи
саны отсутствие или низкая способность к ca
мовосстановлению.
Количество и тип факторов роста, BHe
сенных во время культивирования, изменяют
тип и размер колоний, которые будут сфор
мированы.
Более высокая специфичность по общему
классу rCK и по их относительному пролифе
ративному потенциалу обеспечивается Bpe
менем, требуемым для дифференцировки in
vitro в зрелые клетки. Время, необходимое
для формирования колоний зрелых клеток
iп vitro у послеродовых колониеобразующих
клеток (взрослоro типа), составляет 10 14
дней.
2.7.28. Определение количества 2емопоэтических клеток предшественниц человека 387
ОБЕСПЕЧЕНИЕ КАЧЕСТВА
ПРИ ПРОВЕДЕНИИ ИСПЫТАНИЯ
Важнейшим условием для обеспечения
общеro качества испытания является исполь
зование CTPOro стандартизованных процедур.
Поэтому рекомендуется выполнить внутри и
межлабораторные валидации. Источник Ma
териалов, включая реактивы, факторы роста
и расходный материал, должен быть иденти
фицирован.
Основными факторами, влияющими на
вариабельность испытания, являются число
внесенных в лунки клеток и идентификация
колоний. Для одноro и TOro же испытания
может наблюдаться внутрилабораторная Ba
риабельность до 15 0/0. При необходимости
оценки числа колониеобразующих клеток в
очищенной популяции клеток возможно ис
пользование метода лимитирующих разведе
ний с подсчетом числа лунок, положительных
по клеточной пролиферации, с помощью aB
томатизированной системы.
Друrой важной причиной вариабельности
является использование в испытании Heo
пределенных материалов (например, эмбри
ональная бычья сыворотка или бычий CЫBO
роточный альбумин). Эти продукты проис
ходят из пулов исходных материалов и обе
спечивают неспецифическую стимуляцию
клеточной пролиферации. Однако довольно
часто встречаются серии/партии материалов
с особыми характеристиками, которые BЫ
борочно стимулируют пролиферацию клеток
определенных линий кроветворения.
Для клоноrенноrо количественноro aHa
лиза рекомендуется использовать материа
лы только с низким уровнем содержания эн
дотоксинов (менее 0,01 МЕ/мл или менее
0,01 ME/Mr), поскольку более высокие уровни
приводят сначала к проrрессивному искаже
нию линейной принадлежности rCK в культу
рах, а впоследствии к более общему подавле
нию клеточной пролиферации и образованию
клонов rCK.
клоноrЕННОЕ КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
Клоноrенное количественное определе
ние основано на способности клеток предше
ственниц формировать колонию при BHece
нии в плотную питательную среду или в rель в
присутствии специфических факторов роста.
В зависимости от желаемоro метода учета
MorYT использоваться различные типы плот
ных сред (например, метилцеллюлоза, колла
reH, arap и плазменный crycToK). Коммерчески
доступные среды обычно дают более воспро
изводимые результаты.
МАТЕРИАЛbI
Валидация выполняется, по крайней мере,
для следующих критических материалов.
Факторы роста. Для тоro, чтобы полу
чать наибольшее количество колоний от кле
точной взвеси, содержащей смешанную по
пуляцию rCK, требуются как мультилинейные
(такие как kit лиrанд или фактор стволовой
клетки (ФСК), интерлейкин 3, фактор сти
муляции колоний макрофаroв и rранулоци
тов (rм ксФ», так и линейно специфические
(эритропоэтин, фактор стимулирования коло
нии rранулоцитов (r ксФ» факторы роста.
Друrие компоненты питательной
среды. В среды MorYT быть добавлены CЫBO
ротки (особенно эмбриональная бычья CЫBO
ротка) и/или альбумин.
КЛЕТОЧНАЯ КУЛЬТУРА
Клетки. Образец, помещенный в культу
ру, должен быть представительным для Bcero
введенноro клеточноro продукта. Для этоrо
теста требуются суспензии клеток. В случае
использования пунктата KocTHoro мозrа такие
суспензии MorYT быть получены, продавливая
костный мозr через решетчатые фильтры или
через иrлы проrрессивно уменьшающеro Ka
либра. Повторные продавливания через иrлу
21 размера обычно достаточны, чтобы раз
бить клеточные кластеры до клеточной cy
спензии.
ПОМЕЩЕНИЕ В ЧАШКУ И ПОДСЧЕТ
Клетки, взвешенные в питательной среде,
смешивают с плотной питательной средой.
Обычно делают посев 1 мл смеси в необра
ботанную стерильную чашку Петри (диаме
тром 35 мм).
Из за вязкости среды раствор не может
быть помещен в пипетки со сменяемыми Ha
садками, и требуется использование шприцов
с иrлами большоro размера (:5 номер 18).
Число клеток, которые будут помещены в
чашку, зависит от концентрации rCK в испыту
емом образце. Так как колонии не будут полу
чены из 2 различных rCK, число помещенных
в чашку клеток должно позволить подсчитать
от 40 до 80 колоний на чашку (диаметром 35
мм). Конечное число колоний на <,iашку может
быть рассчитано из процентноro содержания
СО34+ (или из концентрации СО34+ клеток
в миллилитре), определенноro методом про
точной цитометрии (2.7.24) или от различных
разведений клеточной суспензии (обычно не
менее 2 концентраций для проверки).
Чашки инкубируются в аэробных услови
ях с концентрацией уrлекислоrо rаза 5 % при
температуре 37 ос во влажной (насыщенной)
атмосфере в течение 10 14 дней, число KO
лоний затем подсчитывают под инвертиро
ванным микроскопом. При манипуляциях с
чашками (платами), содержащими колонии,
должна быть проявлена осторожность, по
скольку среда, содержащая метил целлюлозу,
388
rосударственная фармакопея Республики Беларусь
ЧИСЛО КЛЕТОК
является вязкой, но не желеобразной. Куль
тивирование на наклоненной чашке может
привести к смещению и формированию KOMe
топодобных колоний, делающих подсчет He
корректным.
ИДЕНТИФИКАЦИЯ КОЛОНИЙ
Размер и структура колоний зависят от
типа зрелых клеток и их составляющих. Ko
личество в 50 клеток на колонию обычно
рассматривается как минимальное. Присут
ствие rемоrлобинизированных клеток указы
вает на наличие клеток предшественников
эритробластноrо ряда. Поскольку количество
зрелых клеток для каждой линии происхожде
ния в значительной степени зависит от фак
торов роста, добавленных к культурам, BЫ
полнение дифференцированноro подсчета не
рекомендуется, если иное не указано в част
ной статье.
ПРЕДСТАВЛЕНИЕ РЕЗУЛЬТАТОВ
Результаты испытания обычно выражают
как среднее арифметическое число колоний,
посчитанных не менее чем в 3 чашках. Cpeд
нее число колоний затем соотносится к 104
или 105 жизнеспособным ядросодержащим
клеткам, помещенным в культуру.
0112013:20729
2.7.29. ОПРЕДЕЛЕНИЕ
КОЛИЧЕСТВА И
ЖИЗНЕСПОСОБНОСТИ
ЯДРО СОДЕРЖАЩИХ КЛЕТОК
Определение качества клеточных суспен
зий требует точноrо измерения как количе
cTBeHHoro содержания, так и процента жиз
неспособных клеток. Эти данные важны для
процесса принятия решений в отношении
подrотовленных клеточных продуктов и под
держания оптимальных условий культивиро
вания. Количество клеток может быть пред
ставлено числом клеток на объем клеточной
суспензии, а жизнеспособность клеток
как число жизнеспособных клеток на объем
клеточной суспензии. Процедура подсчета
клеток может быть выполнена вручную (счет
чик клеток крови) или с использованием aB
томатическоrо аппарата (например, счет
чик частиц, проточная цитометрия). Кроме
описанных ниже MorYT быть использованы и
друrие методы.
РУЧНОЙ ПОДСЧЕТ
Описание прибора и принцuпа иСПblта
ния. Требуются следующие материалы.
Счетчик клеток крови: специализирован
ная камера для микроскопическоrо подсчета
доступна в различном исполнении. Камера
состоит из толстоro предметноro стекла и
покровноro шлифованноro стекла, YCTaHOB
ленноro так, чтобы разrраничить камеры с
определенным объемом для каждоro испол
нения. Толстое предметное стекло различных
счетчиков клеток крови состоит из счетной
камеры, разделенной rлубокими бороздка
ми, чтобы избежать перекрестноro заполне
ния. Счетная камера выrравирована в стекле
и содержит сетку, которая является специфи
ческой для каждой модели.
Оптический микроскоп слабое увели
чение от 1 ОХ до 40х.
Пипетки подходящеro объема.
Счетчик клеток крови используется для
определения количества клеток в данном pac
творе путем подсчета концентрации клеток в
миллилитре (С), используя следующее Bыpa
жение:
а .1 оп . d,
rде:
а число подсчитанных клеток;
d фактор разведения (если необходи
мо);
п фактор, меняющийся в зависимости
от объема камеры счетчика клеток крови.
Клетки в смешанной популяции различа
ют на основании их отличий по размеру или
пиrментации (например, лейкоциты и эритро
циты).
Под20товка камеры для подсчета и
анализ. Устанавливают покров ное стекло
(слеrка увлажненное по краям) на предмет
ное стекло. Двиrают покровное стекло взад
и вперед по предметному стеклу, нажимая
на края покровно r-o стекла. rотовят подходя
щее разведение клеточной суспензии в изо
тоническом или в rемолизирующем буферном
растворе.
Подходящий объем разведения вносят
в камеру подсчета. Жидкость помещают 8
щель, образующуюся между покровным CTe
клом и счетной камерой, и она заполняет
камеру подсчета блаrодаря капиллярности.
Осторожно помещают камеру для подсчета
клеюк крови под микроскоп и фокусируют.
Подсчитывают клетки в зоне сетки. Рассчит
вают концентрацию клеток в разведенных 11'
неразведенных образцах.
Чтобы увеличить точность измерения.
важно соблюсти следующие основные мер..'
предосторожности:
2.7.29. Определение количества и жизнеспособности ядросодержащих клеток 389
РУЧНОЙ МЕТОД ИСКЛЮЧЕНИЯ
КРАСИТЕЛЯ
используют покровное стекло только
соответственной толщины;
везде, rдe возможно, подсчитывают
более чем 100 клеток (в случае необходи
мости, считают большее количество KBaдpa
тов);
при обнаружении кластеров/rрупп
клеток (то есть взвесь клеток не является MO
ноклеточной), клетки ресуспендируют, отби
рают образец и снова подсчитывают:
необходимо избеrать неполноro запол
нения или переполнения счетной камеры,
иначе объем для подсчета не будет ДOCTOBep
ным.
АВТОМАТИЗИРОВАННЫЕ МЕТОДЫ
ПОДСЧЕТА
Счетчики частиц, основанные на изме
нении проводимости. Электронные счетные
устройства измеряют размер и число частиц
в растворе.
Перед использованием счетчики частиц
калибруют раствором частиц с известной
концентрацией и размером. Чтобы проводить
подсчет частиц большеro размера, применя
ют пробирки, оснащенные друrими калибро
вочными насадками. Эти приборы не позво
ля ют различать мертвые и живые клетки.
Поскольку осколки клеток MorYT также произ
водить импульсы, которые вызывают ошибки,
счетные устройства также оснащают пороro
ВЫМ контролем, позволяющим подсчитывать
только большие частицы.
Прибор должен быть приrоден для под
счета клеточных продуктов (с точки зрения
линейности, точности, и т. д.).
Счетчики частиц, основанные на про
точной цитометрии (2.7.24). Проточный ци
тометр калибруют по стандартным частицам
известной концентрации и размера, чтобы
подсчитать абсолютное количество клеток
в объеме. При этом калибровочный раствор
больше не является необходимым при щ:
пользовании приборов с двумя электродами,
поrруженными в счетную камеру для образ
ца, в которых постоянный размер камеры и
расстояние между двумя электродами позво
ляет про водить измерение в фиксированном
объеме. Этот тип оборудования редко требу
ет калибровки после первоначальной YCTa
новки.
ЖИЗНЕСПОСОБНОСТЬ
Приведенные методы определения про
цента жизнеспособных клеток основаны на
окрашивании жизнеспособных клеток краси
телями и ручном или автоматизированном
анализе с использованием оптическоro ми
кроскопа либо путем проточной цитометрии
суспензий клеток.
В зависимости от типа клеток и использу
eмoro метода результаты MorYT отличаться.
. з- т2
Принцип испытания. Испытание OCHO
вано на невключении красителя в жизнеспо
собные клетки, Torдa как мертвые или по
врежденные клетки абсорбируют краситель
и окрашиваются. Это дает информацию о цe
лостности цитоплазматической мембраны,
но ее результаты не обязательно отражают
функциональную активность клетки. HeдaB
но трипсинизированные или размороженные
живые клетки MorYT иметь липкие мембраны
способные абсорбировать краситель.
Краситель. Наиболее часто используе
мым красителем для тоro, чтобы различить
жизнеспособные и нежизнеспособные клетки,
является трипановый синий, но также MorYT
использоваться и друrие подходящие краси
тели, такие как эритрозин В или ниrрозин.
Трипановый синий кислотный краси
тель (М м. 961), анион с 4 сульфонатными
rруппами, которые MorYT леrко связаться с
белками; поэтому концентрация белка в ис
пытуемом образце должна быть настолько
низкой, насколько возможно.
Условия проведенuя испытания. Фикса
ция окраски сильно зависит от значения рН,
поэтому оно должно быть в пределах от 6,6 до
7,6. Фиксация оптимальна при рН 7,5. Друrие
условия, такие как концентрация красителя и
время окраски, должны быть валидированы.
Условия хранения красителя. Обычно ис
пользуют 0,4 % или 0,5 % раствор трипаново
ro синеro в стерильном фосфатном забуфе
ренном физиолоrическом растворе. Хранят в
защищенном от воздействия света и воздуха
месте.
Под20товка к испытанию и анализ. Cy
спензию клеток в требуемом разведении
(обычно в фосфатном забуференном физи
олоrическом растворе) окрашивают, напри
мер, с использованием раствора трипаново
ro синеro с конечной концентрацией от 0,1 %
до 0,2 %. Аккуратно перемешивают. Инкуби
руют в течение не более 2 4 мин при KOM
натной температуре. Аккуратно перемешива
ют и помещают подходящий объем в счетную
камеру. Без промедления проводят подсчет.
Определяют процент жизнеспособных
клеток как отношение числа неокрашенных
клеток к общему количеству клеток под опти
ческим микроскопом, считая все окрашенные
клетки как мертвые. Жизнеспособность (V) в
процентах рассчитывают по формуле:
п
.100,
N
rдe:
п число неокрашенных (жизнеспособ
ных) клеток;
N общее количество клеток (окрашен
ных инеокрашенных).
390
rосударственная фармакопея Республики Беларусь
Важно, чтобы время инкубации не пре
вышало 4 мин, поскольку впоследствии
число окрашенных клеток может значитель
но увеличиться. Поэтому для повторноro
определения необходимо подroтовить новый
образец.
АВТОМАТИЗИРОВАННblЕ МЕТОДЬ!
Проточная цитометрия
Принцип испытания. Испытание OCHOBa
но на способности определенных красителей
проникать через поврежденные мембраны
и связываться с ДНК таким образом, чтобы
мертвые клетки моrли флуоресцировать и pe
rистрироваться с помощью проточной цито
метрии (2.7.24). Нежизнеспособные клетки
оценивают по различиям, выявляемым по
положительному окрашиванию клеток, тоща
как жизнеспособные клетки остаются HeOKpa
шенными. Этот анализ обычно выполняют с
7 аминоактиномицином О (7 AAД, 7 AAO) или
пропидий йодидом (ПИ, Р/), но также MOryT
использоваться и друrие подходящие краси
тели.
Красители. 7 AAД и ПИ взяты как приме
ры мембранопроникающих веществ, которые
MorYT быть использованы в качестве красите
лей для определения жизнеспособности.
7 AAД является аналоroм актиномици
на О, у котороro в положении 7 хромофора
заменена аминокислотная rруппа. Он BCTaB
ляется между цитозиновыми и rуаниновыми
основаниями ДНК. Спектральные свойства
7 AAД делают эту молекулу наиболее подхо
дящей для анализа методом проточной ци
тометрии. Максимум поrлощения комплекса
7 AAД/ДHK расположен в зеленом спектраль
ном диапазоне и, таким образом, подходит
для определения с помощью цитометра, oc
нащенноro aproHoBbIM лазером (длина волны
возбуждения 488 нм). Сильная красная флуо
ресцентная эмиссия 7 AAД дЛЯ оценки жизне
способности (от 635 нм до 675 нм) облеrчает
использование пробы в комбинации с флуо
ресцеинизотиоцианат (ФИТЦ, F/TC) и фико
эритрин (ФЭ, РЕ) конъюrированными антите
лами, потому что в отличие от ПИ, комплекс
7 AAДIДHK имеет минимальное наложение с
ФИТЦ и ФЭ.
ПИ связывается с двунитевой ДНК, BCTpa
иваясь между пар оснований снебольшими
предпочтениями или безкаких либо предпо
чтений и со стехиометрией 1 молекула Kpac...,
теля на 4 5 пар оснований ДНК. Как только
краситель связывается с нуклеиновыми кис
лотами, их флуоресценция увеличивается
в 20 ЗО раз, максимум возбуждения флуо
ресценции перемещается приблизительно на
ЗО О нм к красному (пику флуЬресценции)
и максимум эмиссии флуоресценции (615 нм)
перемещается приблизительно на 15 нм к
синему (пику флуоресценции). Хотя способ
ность ПИ к оптическому поrлощению доволь
но низка, он проявляет достаточно большой
Стоксовский сдви что позволяет OДHOBpe
мен ное обнаружение нуклеиновых кислот и
флуоресцеин меченых антител при условии
использования подходящих оптических филь
тров.
Условия хранения растворов Kpacитe
лей нуклеиновой кислоты. При температуре
(5:1:3) ОС.
Под20товка к испытанию и анализ. В
случае rемопоэтических клеток краситель
может быть добавлен после маркирования
СD45 на клетках, чтобы получить лучшее раз
деление клеток от клеточных осколков и TpOM
боцитов на основе установления области
соответствия объектов по боковому CBeTO
рассеиванию (SS) и мечения СО45+. Условия
инкубации клеточной популяции с красителем
должны быть валидированы.
Инкубация выполняется при комнатной
температуре, в защищенных от света усло
виях. При необходимости лизиса эритроци
тов ero выполняют, используя, например, aM
мония хлорид. Если в проведении лизиса нет
необходимости, прибавляют только буфер
ный раствор.
Процентное содержание жизнеспособных
клеток определяется непосредственно с по
мощью проточноro цитометра и анализа поло
жительных клеток (мертвые клетки) на SS/7
ААД или SS/ПИ цитоrраммах (rрафиках).
Положительные контроли MorYT COCTO
ять из стабилизированных клеток (мертвые
клетки), смешанных со свежими жизнеспо
собными клетками в необходимом соотноше
нии.
Цифровое изображение окрашенных
клеток. Цифровое изображение позволяет
автоматизировать методы, основанные на
исключении красителя. Суспензия клеток и
раствор красителя смешиваются непосред
ственно прибором. Система, которая позво
ляет проводить аспирацию образца, работу
с реактивами и последующую очистку ин
струмента, полностью автоматизирована.
Как только клеточная суспензия была OTO
брана и смешана с раствором красителя,
она закачивается в проточную ячейку для
анализа. Окрашенная клеточная суспензия
прокачивается через камеру, rде стробоско
пический свет позволяет фотокамере фото
rрафировать протекающие через счетную
камеру клетки. Изображение оцифровывает
ся и число мертвых или живых клеток под
считывается с использованием проrраммно
ro обеспечения.
01/2013:20730
2.7.30. Количественное определение человечеСКО20 белка С
391
2.7.30. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
ЧЕЛОВЕЧЕскоrо БЕЛКА С
1. XPOMOrEHHOE КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
Человеческий белок С является витамин
К зависимым плазменным белком, который
после активации (активированный белок С
(АРС» может подавлять свертывание крови
путем протеолитическоrо расщепления фак
торов Va и Vllla. Активность человеческоrо
белка С определяют двухэтапным методом:
на первом этапе человеческий белок С
в образце активируют специфическим актива
тором, выделенным из яда змеи;
на втором этапе АРС расщепляет специ
фический хромоrенный субстрат с образова
нием окрашенноro продукта, который может
быть измерен количественно спектрофотоме
трически.
Стадия 1
человеческий белок С активатор человеческою белка С .. АРС
Стадия 2
хромоrенный субстрат
АРС
.. пептид + хромофор
Активность человеческоro белка С oцe
нивают путем сравнения способности испы
туемоro образца расщеплять хромоrенный
субстрат с активностью стандартноrо образ
ца человеческоro белка С, калиброванноro в
Международных Единицах. За Международ
ную Единицу принимают активность YCTaHOB
ленноro количества Международноrо CTaH
дартноrо образца человеческоro белка С.
Эквивалентность Международноro CTaHдapT
ноro образца в Международных Единицах
устанавливается Всемирной орrанизацией
здравоохранения.
Реактивы для испытания MorYT быть по
пучены отдельно или в составе коммерче
С/(ИХ наборов. Применяется как метод опре
деления конечной точки, так и кинетический
метод. При использовании различных набо
ров методики и реактивы MorYT отличаться в
соответствии с инструкциями производителя.
Существенные особенности методики опv.са
ны в следующем примере микропланшетноrс
метода определения конечной точки.
РЕАКТИВЫ
Буферный раствор для разведения
РН 8.4. 6.055 r тРИС(2идроксиметил)амино
метана Р и 16.84 r цезия хлорида Р paCTBO
ряют В воде Р, при необходимости доводят
значение рН (2.2.3) и разводят водой Р до
объема 1000,0 мл.
Активатор человечеСК020 белка С.
Белок, который специфично активирует че
ловеческий белок С, выделяют из яда rадюки
Аgkistrоdоп сопtоrtr;х сопtоrtriх. Восстанавли
вают и хранят в соответствии с инструкциями
производителя. Перед использованием в ис
пытании разводят водой Р до концентрации
0,25 ЕД/мл.
ХрОМ02енный субстрат aKтивиpoвaHHO
20 белка С. Специфический хромоrенный суб
страт для АРС, например, L пироrлутамил L
пролил L арrинин паранитроанилина
rидро хлорид (пироGlu Рro Аrg рNА' HCI).
Восстанавливают водой Р для получения KOH
центрации 4,5 ммоль/л. Перед использовани
ем разводят буферным раствором для разве
дения рН 8,4 до концентрации 1,1 ммоль/л.
МЕТОДИКА
Восстанавливают или размораживают
испытуемый образец соrласно инструкци
ям производителя. Разбавляя водой Р, для
каждоrо образца rотовят не менее 3 отдель
ных разведений с концентрацией в диапазо
не 0,050 0,200 МЕ/мл, желательно с повто
рами.
Этап 1. Смешивают по 0,025 мл каждоro
разведения с 0,050 мл активатора человече
скоro белка С (предварительно наrревают все
растворы до температуры 37 ОС), и инкуби
руют при температуре 37 ос в течение ровно
1 О мин. Для каждоrо разведения проводят xo
лостой опыт, используя воду Р вместо актива
тора человеческоro белка С.
Этап 2. К каждой смеси прибавляют по
0,150 мл разведенноro субстрата, предва
рительно HarpeToro до температуры 37 ОС, и
инкубируют ровно 10 мин при температуре
37 ОС. При необходимости время инкубации
должно быть уточнено для получения линей
ной зависимости развития окрашивания от
времени. Останавливают реакцию добавле
ни ем 0,050 мл раствора 50 % (об/об) кисло
ты уксусной ледяной Р.
Расщепление xpoMoreHHoro субстрата
под действием АРС вызывает образование
окрашенноrо ПJJодукта пропорционально KOH
центрации человеческоrо белка С в образце.
Измеряют оптическую плотность при длине
волны 405 нм. Оптическую плотность холосто
ro опыта вычитают из оптической плотности
испытуемоrо образца. Проверяют ДOCTOBep
ность испытания и рассчитывают активность
испытуемоro образца с использованием CTaH
дартных статистических методов (5.3).
392
rосударственная фармакопея Республики Беларусь
2. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
ПО ВРЕМЕНИ СВЕРТЫВАНИЯ КРОВИ
Активность человеческоrо белка С YCTa
навливают после расщепления ero до АРС
специфическим активатором, выделенным
из яда rадюки Аgkistrodоп coпtorfrix coпto,
trix. Полученный АРС инактивирует факто
ры Va и Vllla и таким образом удлиняет АЧТВ
(активированное частичное тромбопластино
вое время, АРТТ) системы, в которой присут
ствуют все факторы свертывания, на посто
янном уровне и в избытке, за исключением
человеческоro белка С, который находится
в испытуемом образце. Удлинение времени
свертывания пропорционально концентрации
человеческоro белка С в образце.
Активность человеческоro белка С oцe
нивают путем сравнения способности испы
ту емоro образца продлевать время CBepTЫ
вания с активностью стандартноrо образца
человеческоro белка С, калиброванноro в
Международных Единицах. За Международ
ную Единицу принимают активность YCTaHOB
ленноro количества Международноro CTaH
дартноrо образца человеческоro белка С.
Эквивалентность Международноro CTaHдapT
ноro образца в Международных Единицах
устанавливается Всемирной орrанизацией
здравоохранения.
Реактивы для испытания MorYT быть по
лучены отдельно или в составе коммерческих
наборов. При использовании различных Ha
боров методики и реактивы MorYT отличаться
в соответствии с инструкциями производите
ля. Ниже описаны существенные особенно
сти методики.
РЕАКТИВЫ
Буферный раствор для разведения рН 7,4.
Изотонический буфер, не содержащий хела
тирующих соединений.
Человеческая плазма, дефицитная по
белку С. Цитратная плазма человека без
определяемоro количества человеческоro
белка С. Восстанавливают и хранят в COOT
ветствии с инструкциями производителя.
Активатор человечеСКО20 белка С.
Белок, выделенный из яда rадюки Agkistrodoп
сопtоrfriх coпtortrix, который специфически
активирует человеческий белок С. BOCCTaHaB
ливают и хранят в соответствии с инструкци
ями производителя.
Активатор свертывания. Может быть
использован подходящий реактив для АЧТВ,
содержащий фосфолипиды и контактный aK
тиватор. Он может быть комбинирован с aK
тиватором человеческоro белка С.
МЕТОДИКА
Восстанавливают или размораживают ис
пытуемый образец соrласно инструкциям про
изводителя. Разбавляя буферным раствором
для разведения рН 7,4. для каждоro образца ro
товят не менее 3 отдельных разведений с KOH
центрацией в диапазоне 0,01 o o, 150 МЕ/мл,
желательно с повторами.
Смешивают 1 объем каждоro разведения
с 1 объемом человеческой плазмы, дефицит
ной по человеческому белку С, и 1 объемом
активатора человеческоrо белка С (объеди
ненноro с peareHToM для АЧТВ). Все paCTBO
ры должны быть предварительно HarpeTbI
до температуры 37 ОС. Прибавляют 1 объем
раствора кальция хлорида Р, предваритель
но HarpeToro до температуры 37 ОС, и реrи
стрируют время свертывания.
Время свертывания пропорциональ
но концентрации человеческоro белка С в
каждом разведении. Проверяют ДOCTOBep
ность испытания и рассчитывают активность
испытуемоro образца с использованием CTaH
дартных статистических методов (5.3).
0112013:20731
2.7.31. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
ЧЕЛОВЕЧЕскоrо БЕЛКА s
Человеческий белок 8 является витамин
К зависимым белком плазмы, который дей
ствует как кофактор активированноro белка
С (АРС). Активность человеческоro белка 8
может быть определена в испытании CBep
тывания, описанноro ниже, которое чувстви
тельно к способности человеческоro белка 8
ускорять инактивацию фактора Va под дей
ствием АРС. Постановка испытания включает
прибавление человеческоro белка 8 к смеси
реактивов, содержащей АРС, фактор Va и че
ловеческую плазму, дефицитную по белку S.
Увеличение времени свертывания пропорци
онально концентрации человеческоrо белка
8 в образце. Методы, в которых АРС добав
ляется непосредственно как peareHT, предпcr
чтительнее по сравнению с теми, в которых
АРС rенерируется во время проведения теста
путем добавления специфическоrо активатcr
ра человеческоrо белка С, выделенноro из
яда змеи. Активация свертывания иници иру
ется добавлением активирующеro peare
та, такоro как тромбопластин или активиро-
ванный фактор Х, вместе с фосфолипидам"
и хлоридом кальция. Во время теста фактор
Va rенерируется из фактора V в человеческoi
плазме, дефицитной по белку 8, с последу-
ющей активацией свертывания. Процедура
теста должна rарантировать, что человече-
ский белок 8 является единственным факт
ром оrраничения реакции свертывания.
2.7.32. Количественное определение UН2uбитора a 1 протеuназы человека
393
Активность человеческоro белка S оцени
вают путем сравнения способности испыту
eMoro образца удлинять время свертывания
с активностью стандартноro образца челове
ческоro белка 8, калиброванноrо в Междуна
родных Единицах. За Международную Еди
ницу принимают активность установленноrо
количества Международноro стандартноro
образца человеческоro белка S. Эквивалент
ность Международноro стандартноro образца
в Международных Единицах устанавливается
Всемирной орrанизацией здравоохранения.
Реактивы для испытания MorYT быть по
лучены отдельно или в составе коммерческих
наборов. При использовании различных Ha
боров методики и реактивы MorYT отличать
ся в соответствии с инструкциями производи
теля. Существенные особенности методики
описаны в следующем примере.
РЕАКТИВЫ
Буферный раствор для разведения
рН 7,4. Изотонический буфер, не coдep
жащий хелатирующих соединений, при
roтовленный следующим образом: 6,08 r
триС(2идроксиметил)аминометана Р и
8,77 r натрия хлорида Р растворяют в воде
Р и при необходимости доводят значение рН
(2.2.3); прибавляют 10 r бычье20 альбумина
Рили человечеСК020 альбумина Р и доводят
водой Р до объема 1000,0 мл.
Человеческая плазма, дефицитная по
белку S. Цитратная человеческая плазма
без определяемоro количества человеческо
ro белка S и, желательно, также свободная от
С4Ь связывающеro белка.
Активатор коа2уляции. Этот реактив
используется для тоro, чтобы инициировать
свертывание человеческой плазмы, дефи
цитной по белку, 8 и, следовательно, также
обеспечивать появление активированноrо
фактора V. Активатор может состоять из TKa
HeBoro фактора, активированноro фактора Х
..ли areHTa, способноro прямо активировать
фактор Х. Такой areHT может быть выделен из
--да rадюки Рассела (Vipera russe//i). Реактив
также содержит АРС, фосфолипиды и каль
quя хлорид Р, или, альтернативно, кальция
хлорид может быть добавлен отдельно после
определенноro периода активации.
МЕТОДИКА
Восстанавливают или размораживают ис
пытуемый образец соrласно инструкциям про
"зводителя. Разбавляя буферным раствором
для разведения рН 7,4, для каждоro образца ro
товят не менее 3 отдельных разведений с KOH
центрацией в диапазоне 0,020 0, 100 МЕ/мл,
.елательно с повторами.
SI з... 1 12
Смешивают по 1 объему каждоro разве
дения с 1 объемом человеческой плазмы, дe
фицитной по белку 8 (предварительно Harpe
вают все растворы до температуры 37 ОС).
Прибавляют 2 объема активатора CBepTЫBa
ния, HarpeToro до температуры 37 ОС, и OTMe
чают время свертывания.
Альтернативная процедура может преду
сматривать использование активатора Koary
ляции без кальция хлорида и требовать точ
Horo измерения периода активации перед
добавлением кальция хлорида и времени
свертывания.
Время образования crycTKa пропорци
онально концентрации человеческоrо белка
S в каждом разведении. Проверяют ДOCTOBep
ность испытания и рассчитывают активность
испытуемоro образца с использованием CTaH
дартных статистических методов (5.3).
01/2013:20732
2.7.32. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
инrИБИТОРА а-1-
ПРОТЕИНАЗЫЧЕЛОВЕКА
Содержание инrибитора а 1 протеиназы
человека (также известноro как а 1 анти
трипсин или а 1 антипротеиназа) опреде
ляют путем сравнения способности испыту
емоro образца инактивировать сериновую
протеазу эластазу (свиную панкреатиче
скую эластазу или человеческую эластазу
нейтрофилов) с такой же способностью CTaH
дартноrо образца инrибитора a 1 протеиназы
человека, калиброванноro в миллиrраммах
активноro (функциональноrо) инrибитора
а 1 протеиназы. Различные количества ис
пытуемоrо образца, смешивают с опреде
ленным количеством эластазы, остающуюся
активность эластазы измеряют С использова
нием подходящеro xpoMoreHHoro субстрата.
Метод. описанный ниже, приводится в каче
стве примера.
РЕАКТИВЫ
Трис альбуминовblЙ буфеРНblЙ раствор.
24,23 r трометамола Р растворяют в воде Р,
доводят кислотой хлористоводородной Р1
до значения рН (8,0:1:0,3) и разводят водой Р
до объема 1000 мл. К 100 мл полученноro
раствора прибавляют 0,5 мл 20 % раствора
человечеСК020 альбумина Рили бblчье20 аль
бумина Р.
Буферный раствор, содержащий чело
веческий или бычий альбумин, должен быть
приroтовлен в день ero использования; иначе
394
rосударственная фармакопея Республики Беларусь
01/2013:РБ20750
раствор стерилизуют фильтрацией (0,2 мкм)
и хранят при температуре от 2 ос до 8 ос до
2 недель.
МЕТОДИКА
Используя трис альбуминовый буфер
ный раствор, rотовят по 2 серии из 4 или 5
разведений с подходящим диапазоном KOH
центраций инrибитора a 1 протеиназы чело
века как для испытуемоro образца, так и для
стандартноro образца.
По 50 мкл разведений стандартноro об
разца помещают в лунки микропланшета
и в каждую лунку прибавляют по 150 мкл
раствора свиной панкреатической эла
стазы, разведенноro до необходимой KOH
центрации трис альбуминовым буферным
раствором. Инкубируют в течение опреде
ленноro промежутка времени, 3 10 мин,
при комнатной температуре. Так как актив
ность растворов различных свиных пан
креатических эластаз может варьировать,
концентрация эластазы может быть уточ
нена путем оценки активности в контроль
ных лунках, содержащих эластазу без ин
rибитора а 1 протеиназы человека, для
получения подходящеro изменения опти
ческой плотности при длине волны 405 нм
в данных конкретных условиях проведения
испытания.
В каждую лунку прибавляют по 100 мкл
рабочеro раствора xpoMoreHHoro субстрата
N сукци н ил три L аланил 4 п н итроанилида
(8uc Ala Ala Ala pNA), растворенноro в ди
метил сульфоксиде Р (концентрация 4,5 мr/мл),
затем разведенноro трис альбуминовым бу
ферным раствором до рабочей KOHцeHTpa
ции 0,45 мr/мл. Немедленно начинают из
мерение изменения оптической плотности
(2.2.25) при длине волны 405 нм, используя
миропланшетный ридер (спектрофотометр ),
снимая показания в течение не менее 5 мин.
Рассчитывают скорость изменения оптиче
ской плотности (М/мин). Альтернативно для
испытания может быть использован метод
определения конечной точки: реакцию OCTa
навливают уксусной кислотой и измеряют
поrлощение при длине волны 405 нм. Если
испытание выполняют в пробирках, исполь
зуют спектрофотометр для измерения опти
ческой плотности, объемы растворов peaK
тивов изменяют пропорционально.
Скорость изменения оптической плот
ности (М/мин) обратно пропорциональна
активности человеческоro инrибитора a 1
протеиназы.
Проверяют достоверность количествен
ноro определения и рассчитывают актив
ность испытуемоro образца обычными CTa
тистическими методами (5.3).
#12.7.50. ОПРЕДЕЛЕНИЕ
БиолоrИЧЕСКОЙ
АКТИВНОСТИ ИНСУЛИНА
Активность испытуемоro образца инсу
лина определят путем сопоставления ero
rипоrликемическоro (сахаропонижающеro)
действия с rипоrликемическим действием
стандартноro образца инсулина.
Стандартный образец инсулина пред
ставляет собой препарат высокоочищенно
ro инсулина, активность KOToporo опреде
лена путем мноroкратноro сопоставления с
активностью Международноro стандартноro
, образца и составляет не менее 25 ЕД в 1 Mr.
За единицу действия (ЕД) принимают спец
ифическую активность количества массы
стандартноro образца, эквивалентноro по
биолоrическому действию 1 Международ
ной Единице инсулина.
Примечание: приrотовление растворов
стандартноro образца и испытуемоro образ
ца инсулина. Навеску стандартноro образ
ца инсулина растворяют в растворе кисло
ты хлористоводородной Р (0,003 молы/)) с
рН 2,7 3,0, содержащем консервант в такой
же концентрации, как испытуемый образец.
Срок roдности основноro раствора CTaH
дартноrо образца инсулина 4 месяца при
температуре 4 ОС.
Перед опытом основной раствор CTaH
дартноrо образца инсулина разводят pac
твором хлористоводородной кислоты
(О,ООЗ моль/л) до концентрации 1 ЕД в
1 мл. Испытуемый образец инсулина раз
водят этим же раствором хлористоводород
ной кислоты до концентрации 1 ЕД в 1 мл,
исходя из ero предполаrаемой активности.
МЕТОДИКА ОПРЕДЕЛЕНИЯ
БиолоrИЧЕСКОЙ АКТИВНОСТИ
Определение биолоrической активности
инсулина проводят на здоровых кроликах
массой 2,5 З,5 Kr. Животных массой 1 ,8
2,3 Kr предварительно не менее 14 суток
содержат в условиях вивария, rдe они по
лучают овес, сено или свежую траву, KOp
неплоды, комбинированный корм и воду. По
истечении этоro срока определяют чувстви
тельность кроликов к инсулину. Для этоro
кроликам дважды с интервалом 7 8 суток
после 18 ч roлодания вводят подкожно paCT
вор стандартноrо образца инсулина из pac
чета 0,5 ЕД на 1 Kr массы тела животноrо
и определяют величину снижения KOHцeH
трации сахара в крови в процентах. Кровь
для исследования берут из краевой вены
уха животных до введения инсулина и через
1,5 и 2,5 часа после ero введения (для pac
#2.7.50. Определение биолоzической активности инсулина
395
чета берут среднюю концентрацию сахара в
крови из двух определений после введения
инсулина). Кровь берут в условиях, не дo
пускающих чрезмерноro волнения кроликов.
Из опытов исключают животных с исход
ной концентрацией сахара в крови ниже и
выше пределов, установленных для исполь
зуемоrо метода определения содержания
сахара в крови (ниже 75 Mr % и выше 120
Mr % для феррицианидноrо метода, ниже 52
Mr % и выше 94 Mr % для rлюкозооксидазно
ro метода), а также тех животных, которые
реаrируют на введение инсулина судороrа
ми или у которых снижение концентрации
сахара в крови составляет менее 15 % по
отношению к исходной концентрации. Опре
деление биолоrической активности инсули
на проводят не ранее чем через 7 8 суток
после испытания на чувствительность.
Отобранных для опытов кроликов
лишают корма (но не воды) на 18 ч, пред
шествующих введению инсулина. Животных
распределяют на две rpYnnbI способом слу
чайноro выбора (в каждой rpynne должно
быть не менее 9 кроликов). Непосредствен
но перед введением инсулина кроликов
взвешивают и определяют исходную KOH
центрацию сахара в крови. Кроликам одной
rруппы вводят подкожно по 0,5 ЕД CTaHдapT
HOro образца инсулина (S) на 1 Kr массы
тела, кроликам друrой rруппы такую же
дозу испытуемоro образца инсулина (Т).
Через 1,5 и 2,5 ч после введения инсулина у
животных снова определяют концентрацию
сахара в крови.
Для каждоro кролика рассчитывают сни
жение концентрации сахара в крови в про
центах по формуле:
a b
.100,
а
rдe:
а исходная концентрация сахара в
крови в миллиrрамм процентах;
Ь средняя концентрация сахара в
крови в миллиrрамм процентах из двух
определений, проведенных через 1,5 ч и
2,5 ч после введения инсулина.
Как и при испытании кроликов на чув
ствительность к инсулину, при расчетах не
учитывают животных с исходной KOHцeHTpa
цией сахара в крови ниже и выше пределов,
установленных для используемоrо метода, а
также реаrирующих на указанную дозу инсу
лина судороrами или дающих снижение KOH
центрации сахара в крови менее 15 % по OT
ношению к исходной концентрации.
Через 3 5 суток на этих же кроликах
проводят повторное испытание с той разни
цей, что животным из rруппы, получавшей
стандартный образец инсулина (S), вводят
испытуемый образец инсулина (Т), а живот
ным rpYnnbI, получавшей (Т), вводят (S).
Рассчитывают среднюю величину сни
жения концентрации сахара в крови на CTaH
дартный образец (%S) и на испытуемый
образец (% т) по данным Bcero опыта. OTHO
шение % Т / %S выражает относительную aK
тивность (R) испытуемоro образца (Т). Для
выражения активности испытуемоro образ
ца (т) в ЕД ero предполаrаемую активность
(А) умножают на относительную активность
(Т = R . А).
Пример: при определении биолоrической
активности инсулина для инъекций с предпо
лаrаемой активностью 40 ЕД в 1 мл получи
ли следующие величины снижения KOHцeH
трации сахара в крови в процентах (У).
Первая часть Вторая часть
испытания испытания
Y s У Т У Т Y s
49 48 50 50
61 50 58 51
35 48,5 53 41
55 65 55 55
39 46 39 45
28 22,5 68 60
53 15 38 4З
38 48 44 60
50 6З 44 56
LYs =869; %8 =869/18 =48,28,
R = % Т/ %8 = 45,83/48,28 = 0,949 ,
LY 7 =825; %Т=825/18=45,83
Активность Т в ЕД, по данным этоro опыта,
равна:
Т=0949.40=З7,96 ЕДв 1 мл.
Определение проводят не менее 2 раз
с каждым испытуемым образцом (Т). Cpeд
нюю величину при этом получают, объединяя
индивидуальные цифры снижения KOHцeH
трации сахара в крови в процентах в обоих
396
rосударственная фармакопея Республики Беларусь
опытах. При расчете окончательноro резуль
тата должны быть использованы данные,
полученные не менее чем на зо кроликах.
Если средняя величина R по данным двух
или большеro числа опытов будет меньше
0,85 или больше 1,15, испытание проводят
заново, исходя из новой величины предпола
rаемой активности испытуемоro препарата.
Кролики MorYT быть использованы по
вторно для определения биолоrической aK
тивности не ранее чем через 2 недели после
окончания предшествующеro опыта. Про
должительность использования кроликов в
опытах по определению биолоrической aK
тивности инсулина не должна превышать 5
месяцев.
ИСПЫТАНИЕ
НА пРолонrИРОВАННОЕ
(УДЛИНЕННОЕ) ДЕЙСТВИЕ
ПРЕПАРАТОВ ИНСУЛИНА
Испытание на пролонrированное дей
ствие препаратов инсулина основано на co
поставлении длительности rипоrликемиче
CKOro эффекта испытуемоro препарата и
стандартноro образца инсулина.
18 здоровых кроликов массой 3 4 Kr
распределяют на две rруппы по 9 животных
способом случайноrо выбора. Животных co
держат в отдельных клетках, за 18 ч до BBe
дения инсулина лишают корма (но не воды).
В начале опыта у кроликов определяют ис
ходное содержание сахара в крови и pac
считывают среднюю концентрацию сахара в
крови для каждой rруппы. Животным первой
rруппы вводят стандартный образец инсули
на, а животным второй rруппы испытуемый
препарат инсулина. Навеску стандартноro
образца инсулина растворяют в растворе
хлористо водородной кислоты (0,003 молы/))
рН 2,7 3,O, содержащем консервант в такой
же концентрации, как и препарат. Доза испы
TyeMoro препарата и стандартноrо образца
составляет 0,8 ЕД на 1 Kr массы тела живот
HOro. Испытуемый препарат вводят в нераз
веденном виде. Концентрация (количество
ЕД в 1 мл) стандартноro образца должна
быть равна концентрации неразведенноro
испытуемоro препарата. Через 1,5; 3,0; 4,5
и 6,0 ч после введения препарата инсули
на и стандартноrо образца определяют KOH
центрацию сахара в крови у кроликов и под
считывают среднее содержание сахара для
каждой rруппы.
Требования в отношении пролонrиро
BaHHoro действия отдельных препаратов ин
сулина изложены в соответствующих част
ных статьях.
2.8. МЕТОДЫ
ФАРМАкоrнозии
01/2013:20801
2.8.1. ЗОЛА, НЕРАСТВОРИМАЯ
ВХЛОРИСТОВОДОРОДНОИ
КИСЛОТЕ
Зола, нерастворимая в хлористоводород
ной кислоте, это остаток, полученный после
обработки сульфатной или общей золы хло
,ристоводородной кислотой И рассчитанный
на 100 r испытуемоro образца.
В тиrель, содержащий остаток после
определения общей или сульфатной золы,
прибавляют 15 мл воды Р и 1 О мл кислоты
хлористоводородной Р, раствор покрывают
часовым стеклом, аккуратно кипятят в тече
ние 1 О мин и охлаждают. Фильтруют через
обеззоленный фильтр, промывают фильтр
rорячей водой Р до тех пор, пока фильтрат
не станет нейтральным, затем фильтр сушат
и сжиrают при температуре слабоro KpaCHO
ro каления, после чеro охлаждают в эксика
торе и взвешивают. Процесс сжиrания необ
ходимо повторять до тех пор, пока разница
между двумя последовательными взвеши
ваниями будет не более 1 Mr.
01/2013:20802
2.8.2. ПРИМЕСИ
Допустимыми при меся ми в лекарствен
ном растительном сырье являются примеси,
включающие в себя:
1) несырьевые части растения: части
данноro лекарственноro растения, не яв
ляющиеся лекарственным растительным
сырьем;
2) посторонние примеси: примеси, не OT
носящиеся к данному лекарственному paCTe
нию, и имеющие растительное #(орrанические
примеси неядовитые части друrих paCTe
ний, солома, сено и Т. п.)# или минеральное
#(минеральные примеси земля, песок, Ka
мешки)# происхождение.
#Лекарственные средства на основе ле
KapcTBeHHoro растительноro сырья (лекар
ственное растительное сырье цельное или
измельченное фасованное, сборы, расти
тельные чаи) не должны обладать устойчи
вым посторонним запахом, содержать такие
2.8.5. Определение воды в эфирных маслах
397
при меси, как стекло, помет rрызунов и птиц,
ядовитые растения и т. п.; кроме этоro, не
должны обнаруживаться признаки rниения,
плесени, амбарных вредителей и друrих
примесей животноro происхождения.#
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ
ПРИМЕСЕЙ
ОТ 100 r до 500 r испытуемоro образца
лекарственноro растительноro средства (ле
карственноro растительноro сырья) или ми
нимальное количество, указанное в частной
статье, рассыпают тонким слоем. Осматри
вают невооруженным rлазом или с помощью
лупы (6 х ). Каждую rруппу примесей отделя
ют, взвешивают и рассчитывают их процент
ное содержание.
01/2013:20803
2.8.3. УСТЬИЧНЫЙ КОЭФФИЦИЕНТ
УСТЬИЦА
Различают несколько типов устьиц (ри
сунок 2.8.З. 1):
(1) аномоцитный тип: устьица окружены
неопределенным числом клеток, не отлича
ющихся по форме и размерам от остальных
клеток эпидермиса;
(2) анизоцитный тип: устьица окружены
тремя околоустьичными клетками, из KOTO
рых одна значительно меньше двух друrих;
(3) диацитный тип: устьица окружены
двумя околоустьичными клетками, смежные
стенки которых перпендикулярны устьичной
щели;
(4) парацитный тип: с каждой стороны
устьица вдоль ею продольной оси расположе
ны по одной или более околоустьичных клеток.
3
Рисунок 2.8.3. 1.
52. За". 1712.
УСТЬИЧНЫЙ КОЭФФИЦИЕНТ
100.S
Устьuчныи коэффициент = ,
E+S
rдe:
S количество устьиц на определенной
площади листа;
Е число клеток эпидермиса (включая
трихомы) на этой же площади листа.
Для каждоro испытуемоro образца листа
проводят не менее 1 О определений и рассчи
тывают среднее значение.
01/2013:20804
2.8.4. КОЭФФИЦИЕНТ НАБУХАНИЯ
2
Коэффициент набухания это объем в
миллилитрах, занимаемый 1 r лекарствен
HOro растительноro средства (лекарственно
ro растительноro сырья), включая налипшую
слизь, после набухания в водном растворе в
течение 4 ч.
В rрадуированный цилиндр с притер
той пробкой вместимостью 25 мл и BЫCO
той шкалы (125:t5) мм с делениями по 0,5
мл помещают 1,0 r испытуемоro сырья,
цельноro или измельченноro, как указано
в частной статье. Если иноro не указано в
частной статье, образец смачивают 1,0 мл
96 % спирта Р, прибавляют 25 мл воды Р и
закрывают цилиндр, интенсивно встряхивая
каждые 10 мин в течение 1 ч. Выдерживают
в течение 3 ч. Через 90 мин после начала
испытания сливают максимально возмож
ное количество жидкости и любые частички
сырья, плавающие на поверхности жидко
сти путем вращения цилиндра BOKpyr Bep
тикальной оси. Через 3 ч измеряют объем,
занимаемый образцом вместе с прилипшей
слизью. Параллельно проводят З испыта
ния.
Коэффициент набухания в.ыражается
средним значением, рассчитанным по pe
зулыатам трех параллельных испытаний.
01/2013:20805
4
2.8.5. ОПРЕДЕЛЕНИЕ ВОДЫ
В ЭФИРНЫХ МАСЛАХ
Смешивают 1 О капель эфирноro масла с 1 мл
У2лерода дисульфида Р. При стоянии раствор
должен оставаться прозрачным.
398
01/201 3: 0806
rосударственная фармакопея Республики Беларусь
2.8.6. ПОСТОРОННИЕ ЭФИРЫ
В ЭФИРНЫХ МАСЛАХ
Наrревают 1 мл эФирноro масла в течение
2 мин на ВОДЯНОЙ бане с 3 мл свежеприroтовлен
ноro раствора 100 r/л калия 2идроксида Р в 96 %
спирте Р. В течение 30 мин не должны образо
вываться кристаллы. даже при охлаждении.
01/2013:20807
2.8.7. ЖИРНЫЕ И МИНЕРАЛЬНЫЕ
МАСЛА В ЭФИРНЫХ МАСЛАХ
Капают 1 каплю эфирноro масла на филь
тровальную бумаry. Капля должна полностью ис
париться в течение 24 ч, не оставляя каких либо
просвечивающихся или жирных пятен.
01/2013:20808
2.8.8. ЗАПАХ И ВКУС ЭФИРНЫХ
МАСЕЛ
Перемешивают 3 капли эфирноro масла, 5 мл
спирта (90 %. об/об) Р и 1 О r порошкообразной
сахарозы Р. Запах и вкус должны быть идентич
ными запаху и вкусу растения или частей paCTe
ния, из которых получено эфирное масло.
01/2013:20809
2.8.9. ОСТАТОК ПОСЛЕ
ВЫПАРИВАНИЯ эФиРноrо
МАСЛА
Остаток эфирною масла после выпарива
ния это процентное (м/м) количество OCTaT
ка, образующеroся при выпаривании эфирноro
масла на водяной бане в условиях, описанных
ниже.
Прибор. Прибор (рисунок 2.8.9. 1) состоит
из следующих частей:
водяная баня с крышкой с отверстием в
ней диаметром 70 мм;
чашка для выпаривания из инертною к co
держимому материала;
эксикатор.
Методика. Взвешивают в предварительно Ha
rретую на водяной бане в течение 1 ч и охлажден
ную в эксикаторе чашку для выпаривания 5,00 r
эфирною масла, если иное ме указано в частной
статье. Наrревают масло на интенсивно кипящей
бане в вытяжном шкафу в течение указанноrо
времени. Охлаждают в эксикаторе и взвешивают.
Во время испытания уровень воды 8 бане
должен поддерживаться на расстоянии около
50 мм ниже уровня крышки.
86
о
u)
r
J
o;r!
70
i '
2.8.9. 1. Прибор для определения остатка
после выпаривания эфиРН020 масла
(размеры указаны в миллиметрах)
01/2013:20810
2.8.10. РАСТВОРИМОСТЬ ЭФИРНЫХ
МАСЕЛ В СПИРТЕ
1,0 мл эфирноro масла помещают в цилиндр
вместимостью 25 мл или 30 мл С притертой проб-
кой. Помещают в устройство, способное поддер-
живать температуру (20:t2) ОС. Используя бюретку
вместимостью не менее 20 мл, прибавляют спирт
указанной в частной статье концентрации при
частом и интенсивном встряхивании, приливая еro
сначала по 0,1 мл до образования раствора, затем
по 0,5 мл до истечения общею объема 20 мл. Фик
сируют объем использованною спирта в момент,
Korдa образуется прозрачный раствор или, если
раствор становится мутным или опалесцирующим
до тощ как прибавлено 20 мл спирта, фиксируют
объем, при котором появляется мутность или опа
лесценция и, rдe это применимо, объем, коrда MYТ
ность или опалесценция исчезает.
Если при прибавлении 20 мл спирта KOHцeH
трации, указанной в частноi;1 статье, прозрачный
раствор не образовался, повторяют испытание, ис
пользуя более высокую концентрацию спирта.
Считают, что эфирное масло «растворимо в П
или более объемах спирта данной концентрации Ь>,
Korдa прозрачный раствор в п объемах OCTaeT
ся прозрачным при сравнении снеразбавленным
2.8.12. Определение эфиРНО20 масла в лекарственном растительном сырье 399
маслом после дальнейшеro прибавления спирта
той же концентрации до общеro истечения 20 объе
мов спирта.
Считают, что эфирное масло «растворимо В П
объемах спирта данной концентрации t с появлени
ем помутнения при разбавлении», Korдa прозрач
ный раствор в n объемах становится мутным в п,
объемах (П 1 меньше 20) и остается таким же после
дальнейшею постепенноro прибавления спирта
той же концентрации до общеro истечения 20 объе--
мов спирта.
Считают, что эфирное масло «растворимо В П
объемах спирта данной концентрации t с появлени
ем помутнения в промежутке между объемами П 1
и П 2 », Korдa прозрачный в п объемах спирта paCT
вор становится мутным в П 1 объемах (П 1 меньше 20)
и остается таким же при последующем добавлении
спирта той же концентрации дО П 2 объемов спирта,
а затем становится прозрачным (П 2 меньше 20).
Считают, что эфирное масло «растворимо С по-
явлением опалесценции», Korдa спиртовой раствор
имеет rолубоватый оттенок, подобный оттенку CTaH
дарта опалесценции, приютовленною по следую.-
щей методике: смешивают 0,5 мл раствора серебра
нитрата Р2 и 0,05 мл кислоты азотной Р, при
бавляют 50 мл раствора 12 мr/л натрия хлорида Р,
перемешивают и выдерживают в течение 5 мин
в защищенном от света месте.
01/2013:20811
2.8.11. ОПРЕДЕЛЕНИЕ 1,8-ЦИНЕОЛА
В ЭФИРНЫХ МАСЛАХ
Взвешивают 3,00 r масла, предварительно
высушенноro с помощью натрия сульфата без
водН020 Р, в сухой пробирке и прибавляют 2,1 О r
расплавленноro крезола Р. Помещают пробир
ку в аппарат для определения точки замерзания
(2.2.18) и охлаждают при постоянном переме
шивании. При кристаллизации наблюдается He
большое повышение температуры. Фиксируют
точку наивысшей достиrнутой температуры (t 1 ).
Расплавляют смесь на водяной бане при
температуре, которая не превышает темпера
туру t 1 более чем на 5 ОС, и помещают пробир
ку в аппарат, в котором поддерживается TeM
пература на 5 ос ниже, чем t 1 . После тоro, как
прошла кристаллизация или после тоro, как TeM
пература смеси упала на 3 ос ниже температу
ры t 1 , перемешивают постоянно. Отмечают наи
высшую температуру J"lрИ кристаллизации смеси
(t 2 ). Повторяют операцию до тех пор, пока зна
чения двух максимумов температур t 2 не будут
отличаться не более чем на 0,2 ОС. Если про
исходит переохлаждение, инициируют кристал
лизацию прибавлением маленькою кристал
ла комплекса, состоящеro из 3,00 r цинеола Р и
2,10 r расплавленноro крезола Р. Если темпера
тура t 2 ниже 27,4 ОС, повторяют испытание после
добавления 5,10 r комплекса.
Содержание цинеола, соответствующее
наивысшей температуре t 2 , представлено в Ta
блице 2.8.11. 1. Если был прибавлен комплекс
в количестве 5,1 О r, рассчитывают процентное
(м/м) содержание цинеола по формуле:
2(А 50),
rде:
А значение, найденное по таблице
2.8.11. 1.
Содержание цинеола, соответствующее
наивысшей наблюдаемой температуре (t2)'
может быть определено, при необходимости,
путем интерполяции.
Таблица 2.8.11. 1
I u a u a u g- u a
о ::e о ::e о ::e о ::e
....о. s ....о. s ....<. S ....<. S
O O O O
24 45,5 32 56,0 40 67,0 48 82,0
25 47,0 33 57,0 41 68,5 49 84,0
26 48,5 34 58,5 42 70,0 50 86,0
27 49,5 35 60,0 43 72,5 51 88,5
28 50,5 36 61,0 44 74,0 52 91,0
29 52,0 37 62,5 45 76,0 53 93,5
I 30 53,5 38 63,5 46 78,0 54 96,0
31 54,5 39 65,0 47 80,0 55 99,0
01/2013:20812
2.8.12. ОПРЕДЕЛЕНИЕ эФиРноrо
МАСЛА В ЛЕКАРСТВЕННОМ
РАСТИТЕЛЬНОМ СЫРЬЕ
Определение эфирноro масла в лекар
ственных растительных средствах (лекарствен
ном растительном сырье) осущеСТВJ}яется путем
ero переroнки с паром в специальном приборе
в условиях, описанных ниже. Дистиллят собира
ют в rрадуированную трубку, используя ксилол
для поrлощения эфирноro масла; водная фаза
автоматически возвращается в дистилляцион
ную колбу.
Прибор. Прибор состоит из следующих
частей:
(а) круrлодонная колба подходящеro объема
с коротким притертым roрлышком, имеющим
внутренний диаметр в широком конце 29 мм;
(Ь) конденсирующая система (см. рисунок
2.8.12. 1), которая точно подоrнана к колбе и раз
личные части которой сплавлены в одно целое;
400
rосударственная фармакопея Республики Беларуа. !
используемое стекло должно иметь малый коэф-
фициент расширения, при этом система СОДбрЖИТ
следующие элементы:
пробка К' имеет отверстие, совпадающее с
отверстием диаметром 1 мм в трубке К; широкий
конец трубки К притерт и имеет внутренний ди
аметр 1 О мм;
rpуше06разное расширение J вместимостью
3мл;
трубка JL с делениями по 0,01 мл;
вместимость шарообразноro расширения L
около 2 мл;
трехходовой кран М;
переход (спай) В, находящийся на 20 мм
выше самой высокой риски rрадуировки;
(с) подходящий наrpевательный прибор с
точной реryлировкой температуры;
(d) штатив с кольцом, покрытым изолирующим
материалом.
о
:Е
с t
B
Омл
с>
v
N
О
15
с>
с>
А
1мл
C::J
Рисунок 2.8.12. 1. Прибор для определения
эфиРН020 масла в лекарственном раститель
но м сырье
(размеры указаны в миллиметрах)
.Конденсирующая система может отличаться по строению от
nриведенной на рисунке 2.8.12. 1."
МЕТОД А 1
Используют тщательно вымытый прибор. j
Испытание проводят в соответствии с особett-
ностями испытуемоro лекарственноro раст. :
тельноro средства (лекарственноro раститель-
ноro сырья). В колбу наливают указанный объем!
жидкости для переroнки, добавляют несколь-,
ко кусочков пористоro фарфора и присоедин.
ют к конденсирующей системе. Наливают воду Р
в трубку через воронку N до тех пор, пока она
не достиrнет уровня спая В. Извлекают пробку к- .
И наливают обозначенное количество ксилс;
ла Р, используя пипетку таким образом, чтобы
ее кончик находился в нижнем конце трубки К.
Возвращают на место пробку К' и убеждаются,
что отверстие в трубке совпадает с OTBepCT
ем в пробке. Наrревают жидкость и реryлируют
скорость переroнки, чтобы она cooTBeTcTBoвcr
ла 2 3 мл/мин, если иное не указано в частной
статье.
Для определения скорости переroнки He
ходимо во время переrонки понизить уровень
воды с помощью трехходовоro крана так, чтобы
мениск остановился на уровне нижней отметки (а)
(см. рисунок 2.8.12. 2). Затем закрывают кран и
засекают время, за которое уровень жидкости
достиrнет верхней отметки (Ь). Открывают кран и
продолжают переroнку, реryлируя подоrрев так,
чтобы достичь оптимальной скорости переroн
ки. Переrонку проводят в течение 30 мин. После
этоro прекращают HarpeB и по прошествии МИ
нимум 1 О мин измеряют объем ксилола в rраду
ированной трубке.
:3мл J
l' .
Отметкэ э
Рисунок 2.8.12Л.
Помещают в колбу указанное в частной
статье количество образца и продолжают пP<r
цесс переrонки, как описано выше, в течение
предписанноro времени и с обозначенной скорс;
стью. По истечении необходимоro времени п
реroнки прекращают наrревание, через 10 мин
измеряют объем жидкости, собранной в rраду
ированной трубке, и вычитают из полученнс;
ro значения ранее найденный объем ксилола.
Полученная разница соответствует количеству
эфирноro масла в массе взятоro образца. Pac
считывают результат в миллилитрах на килс;
rpaMM лекарственноro растительноro средства
(лекарственноro растительноrо сырья).
Если эфирное масло собираются ИСПОЛIr
зовать далее в аналитических исследованиях,
то безводная смесь ксилола и эфирноro масла
может быть получена следующим образом: дс;
2.8.12. Определение эфиРНО20 масла в лекарственном растительном сырье 401
стают пробку К'и прибавляютО, 1 мл раствора 1 r/л
натрия флуоресцеината Р и 0,5 мл воды Р.
Сливают смесь ксилола и эфирноro масла таким
образом, чтобы она оказалась в шарообразном
расширении L с использованием трехходовоro
крана, оставляют на 5 минут и спускают аккуратно
смесь до уровня крана М. Открывают кран против
хода часовой стрелки и дают воде вытечь из co
единяющей трубки вм. Промывают трубку aцe
тоном Ринебольшим количеством толуола р,
добавляя их через воронку N. Поворачивая кран
против часовой стрелки, сливают смесь ксилола
и эфирноro масла в подходящую колбу.
#МЕТОД В
Используют прибор. изображенный на рисун
ке #2.8.12. 3. Навеску измельченноro испытуемоro
образца помещают в широкоroрлую круrлодонную
или плоскодонную колбу А вместимостью 1000 мл,
приливают 300 мл воды Р (или иное количество,
указанное в частной статье) и закрывают резин
вой пробкой В С обратным шариковым холодиль
ником С. в пробке снизу укрепляют металлические
крючки, на которые при помощи тонкой провол
ки подвешивают rрадуированный приемник О так,
чтобы конец холодильника находился над ВOpOH
кообразным расширением приемника, не Kaca
ясь еro. Приемник должен свободно помещать
ся в roрле колбы, не касаясь стенок, и отстоять от
уровня воды не менее чем на 50 мм. Цена деления
rpaдуированной части приемника 0,025 мл.
D
v
r '
Рисунок #2.8.12. 3. Прибор для определения
содержания эфиРН020 масла методом В
Колбу с содержимым наrревают и кипя
тят В течение времени, указанноro в частной
статье. Объем масла в rрадуированной части
приемника О замеряют после окончания пе
реroнки и охлаждения прибора до комнатной
температуры.
После 6 8 определений холодильник и
rрадуированный приемник необходимо про
мыть последовательно ацетоном Р и водой Р.
Рассчитывают результат в миллилитрах на
килоrрамм лекарственноro растительноro cpeд
ства (лекарственноro растительноro сырья).
#МЕТОД С
Используется прибор, изображенный на
рисунке #2.8.12. 4. Прибор состоит из круrло
донной колбы А вместимостью 1000 мл, паро
проводной изоrнутой трубки в, холодильника
С, rрадуированной трубки приемника О, OKaH
чивающейся внизу спускным краном Е и слив
ной трубкой F. В верхней части приемника
имеется расширение G с боковой трубкой Н,
которая служит для внесения растворителя
эфирноro масла в дистиллят и сообщения BHY
тренней части прибора с атмосферой. Колба и
паропроводная трубка соединяются через HOp
мальный шлиф. rрадуированная трубка имеет
цену деления 0,02 мл. Для заполнения при
бора водой используется резиновая трубка I
с внутренним диаметром 4,5 5 мм, длиной
450 мм и воронка J диаметром ЗО О мм.
J
Рисунок #2.8.12.-4. Прибор для определения
содержания эфиРН020 масла методами С и О
402
rосударственная фармакопея Республики Беларусь
Перед каждым определением через
прибор пропускают пар в течение 15 20 мин.
После 6 8 определений прибор необходи
мо промыть последовательно ацетоном Р и
водой Р.
Навеску измельченноro сырья помещают
в колбу, приливают зоа мл воды Р, колбу co
единяют с паропроводнои трубкой и заполня
ют водой Р rрадуированную и сливную трубки
через кран при помощи трубки, оканчива
ющейся воронкой. Колбу с содержимым Ha
rревают и кипятят с интенсивностью, при
которой скорост':> стекания дистиллята co
ставляет 60 65 капель в 1 мин, в течение
времени, указанноrо в частной статье. Через
5 мин поспе окончания переrонки OTKpЫBa
ют кран, постепенно спуская дистиллят так,
чтобы эфирное масло заняло rрадуирован
ную часть трубки приемника, и еще через 5
мин измеряют объем эфирноro масла.
Рассчитывают результат в миллилитрах
на килоrрамм лекарственноro растительно
ro средства (лекарственноro растительноro
сырья).
#МЕТОД О
Используют прибор, изображенный на
рисунке #2.8.12. 4. Навеску измельченноro
сырья помещают в колбу, приливают 300 мл
воды Р, колбу соединяют с паропроводной
трубкой и заполняют водой Р rрадуированную
и сливную трубки через кран при помощи pe
зиновой трубки, оканчивающейся воронкой.
Затем через боковую трубку при помощи пи
петки вливают в приемник около 0,5 мл дeKa
лина Р и точно измеряют еro объем, опуская
для этоro уровень жидкости в rрадуирован
ную часть трубки. Далее поступают, как опи
сано в методе С.
По истечении необходимоro времени пе
реroнки прекращают наrревание. Через 5 мин
после окончания перerонки открывают кран,
постепенно спуская дистиллят так, чтобы
эфирное масло с декалином заняло rраду
ированную часть трубки приемника, и еще
через 5 мин измеряют объем жидкости и BЫ
читают из полученноrо значения ранее най
денный объем декалина. Полученная разница
соответствует количеству эфирноro масла в
массе взятоrо образца. Рассчитывают резуль
тат в миллилитрах на килоrрамм лекарствен
ноro растительноro средства (лекарственноro
растительноro сырья).
#МЕТОД Е
Испопьзуется прибор, изображенный на
рисунке #2.8.12. 5. Прибор состоит из круrло
донной колбы вместимостью 1000 мл с KOpOT
ким roрлом А, паропроводной трубки В, холо
дильника С, отстойника О с термометром до
100 ос Е, ртутный шарик котороro находится
на уровне отверстия холодильника, rрадуи
рованной трубки F с ценой деления 0,001 мл.
спускноrо крана G и сливной трубки Н. Дл
заполнения прибора водой используеlСЯ pe
зиновая трубка / с внутренним диаметром
4,5 5 мм, длиной 450 мм и воронка J диаме
тром 30 40 мм.
Перед каждым определением через прибор
пропускают пар в течение 15 20 мин. После
6 8 определений прибор последовательно
промывают ацетоном Р и водой Р.
Навеску измельченноro сырья помещают
в колбу, прибавляют необходимое количество
воды Р. Колбу соединяют с паропроводной
трубкой и заполняют водой Р rрадуированную
и сливную трубки через кран при помощи pe
зиновой трубки, оканчивающейся воронкой.
до тех пор, пока в нижней воронкообразной
части отстойника не наберется слой воды BЫ
сотой 8 12 мм. ВО время переrонки этот yp<r
вень воды должен оставаться без изменения.
Колбу с содержимым наrревают и кипятят в T
чение времени, указанноro в частной статье.
Во время переrонки температура в отстойнике
не должна превышать 25 ос. Через 5 мин после
окончания переroнки открывают кран. пост
пенно спускают дистиллят так, чтобы эфирное
масло заняло rрадуированную часть трубки.
Еще через 5 мин измеряют объем эфирноrо
масла.
Рассчитывают результат в миллилитрах
на килоrрамм лекарственноro раститеЛЬН<r
ro средства (лекарственноrо растительноrо
сырья).
Е
J
Рисунок #2.8.12. 5. Прибор для определенUII
содержания эфиРН020 масла методом Е
2.8.13. Остаточное количество пестицидов
01/2013:20813
403
2.8.13. ОСТАТОЧНОЕ КОЛИЧЕСТВО
ПЕСТИЦИДОВ
Определение. В Фармакопее под пестици
::;,ами понимают любые вещества или смесь Be
ществ, предназначенных для предотвращения
появления, уничтожения или контроля числен
ности любых вредителей, нежелательных видов
растений или животных, вызывающих повреж
дение или каким либо друrим образом оказы
вающих неrативное влияние на производство,
обработку, хранение, транспортировку или pe
ализацию лекарственноro растительноro сырья.
Также это понятие включает в себя вещества,
предназначенные для использования в каче.-
стве реryляторов роста, дефолиантов или ocy
шителей, и любые вещества, используемые
для обработки растений перед или после сбора
урожая с целью защиты от ухудшения качества
80 время хранения или транспортировки. Воз
можное наличие остаточных количеств пести
цидов в лекарственном растительном сырье и
лекарственных средствах на еro основе должно
контролироваться.
Предельное содержание. Если иное не
указано в частной статье, испытуемое лекар
ственное растительное сырье должно быть ис
следовано на наличие следов пестицидов в пре
делах как минимум списка, представленноro в
таблице 2.8.1З. 1. Предельное содержание пе
стицидов, не обозначенных в таблице, наличие
которых предполаrается по каким либо причи
нам, должно соответствовать требованиям дей
ствующеro законодательства. Предельное co
держание пестицидов, которые не указаны ни в
таблице 2.8.13. 1, ни в действующем законода
тельстве, рассчитывают по формуле:
АD/.М
MDD HD .100
rдe:
АО/ допустимая дневная доза, по предло
жению Орrанизации по продовольствию и сель
скому хозяйству ВОЗ (FAO WHO), в миллиrрам
мах на килоrрамм массы тела;
М масса тела, в килоrраммах (60 Kr);
MDO HD дневная доза лекарственноro pac
тительноro сырья, в килоrраммах.
Предельное содержание пестицидов в ле
карственных средствах на основе лекарственно
ro растительною сырья рассчитывают по фор
муле:
если OERs10:
MRL HD .DER ,
если DER>10:
AD1.M
МDD нр .100'
rдe:
MRL HD максимальное предельное coдep
жание пестицидов в лекарственном раститель
ном сырье в соответствии с таблицей 2.8.1 З. 1,
или в соответствии с действующим законода
тельством, или рассчитанное с использованием
формулы, указанной выше;
DER соотношение лекарственное pac
тительное сырье/экстракт, т. е. соотношение
между количеством лекарственноrо раститель
ноro сырья, используемоro при производстве ле
карственноro средства, и количеством получен
ною лекарственноro средства;
МОО нР дневная доза лекарственноro
средства на основе лекарственноro раститель
ноro сырья, в килоrpаммах.
Таблица 2.8.1З. 1
Предельное
Вещество содержание,
Mr/Kr
S421 0,02
Азинфос метил 1
Азинфос этил 0,1 i
Алахлор 0,05
Алдрин и диэлдрин (сумма) 0,05
Ацефат 0,1
Бромид неорrанический (в пе 50
ресчете на ион бромида)
Бромофос метил 0,05
Бромофос этил 0,05
Бромпропилат З
Винклозолин 0,4
rексахлорбензол 0,1
rептахлор (сумма rептахло
ра, цис rептахлорэпоксида и 0,05
транс rептахлорэпоксида)
rексахлорциклоrексан (сумма 0,3
изомеров a , JЗ . б и E )
ДЦТ (сумма о,п' ДЦЕ, п,п' ДЦЕ,
о,п' ДЦТ, п,п' ДЦТ, о,п' ТДЕ, П,п' 1
ТДЕ)
Дельтаметрин 0,5
Диазинон 0,5
Дикофол .0,5
Диметоат и ометоат (сумма) 0,1
Дитиокарбаматы (в пересчете 2
на CS,)
Дихлорфос 1
Дихлофлуанид 0,1
Квиналфос 0,05
Квинтоцен (сумма квинтоцена,
пентахлоранилина и метилпен 1
тахлорфенилсульфида)
Линдан 0,6
(v rексахлорциклоrексан )
Малатион и малаоксон (сумма) 1
Мекарбам 0,05
Метакрифос 0,05
404
rосударственная фармакопея Республики Беларусь
Предельное
Вещество содержание,
Mr/Kr
Метамидофос 0,05
Метидатион 0,2
Метоксихлор 0,05
Мирекс 0,01
Монокротофос 0,1
Паратион метил и параоксон 0,2
метил (сумма)
Паратион этил и параоксон 0,5
этил (сумма)
Пендиметалин 0,1
Пентахлоранизол 0,01
Перметрин и изомеры (сумма) 1
Пиперонилбутоксид 3
Пиретрум (сумма цинерина 1,
цинерина 11, джасмолина 1, 3
джасмолина 11, пиретрина I и
пиретрина 11)
Пиримифос метил (сумма пи
римифос метила и N дезэтил 4
пиримифос метила )
Пиримифос этил 0,05
Протиофос 0,05
Профенофос 0,1
Процимидон 0,1
Текназол 0,05
Тетрадифон 0,03
Фенвалерат 1,5
Фенитротион 0,5
Фенпропатрин 0,03
Фенсульфотион (сумма
фенсульфотиона,
фенсульфотион оксона, 0,05
фенсульфотион оксон
сульфона и фенсульфотион
сульфона)
Фентион (сумма фентиона,
фентион оксона. фентион
оксон сульфона, фентион 0,05
оксон сульфоксида,
фентион сульфона и фентион
сульфоксида)
Фенхлорофос (сумма фенхло
рофоса и фенхлорофос оксо 0,1
на)
т Флувалинат 0,05
Флуцитринат 0,05
Фонофос 0,05
Фозалон 0,1
Фосмет 0,05
Хлордан (сумма циc , тpaHC и 0,5
оксихлордана)
Хлорпириd:юс метил 0,1
ХлорпириФос этил 0,2
Хлортал диметил 0,01
Хлорфенвинфос 0,5
Циперметрин и изомеры 1
(сумма)
Предельное
Вещество содержание,
Mr/Kr
Цифлутрин (сумма) 0,1
Л Цихалотрин 1
Эндосульфан (сумма изомеров 3
и эндосульфана сульфата)
Эндрин 0,05
Этион 2
Этримфос 0,05
Компетентный уполномоченный opraH
может частично или полностью освободить
от проведения испытаний на содержание пе
стицидов при условии, что известна и может
,быть проверена полная история (природа
и количество использованных пестицидов,
дата каждой обработки во время выращива
ния и после сбора урожая) данной партии в
соответствии с Надлежащей практикой BЫ
ращивания, сбора, хранения растительноro
сырья (GACP).
#Поставщик культивируемоro лекарствен
ноro растительноro сырья должен предоста
вить протокол исследований на поставляемую
партию лекарственноro растительноro сырья, в
котором указываются использованные пестици
ды и их остаточное количество.
Для дикорастущих лекарственных растений
исследования на остаточное количество пести
цидов не требуется.#
Отбор проб лекарственноrо раститель-
Horo сырья. Отбор проб лекарственноro pac
тительноro сырья проводят в соответствии с
общей статьей 2.8.20. Лекарственное pacти
тельное сырье: отбор проб.
Качественный и количественный анализ
остаточных количеств пестицидов. Исполь
зуемые аналитические методики должны быть
валидированы (например, в соответствии с
Документом NQ SANCO/10232/2006). В част
ности они должны удовлетворять следующим
критериям:
выбранный метод, особенно стадии очист
ки, должен подходить для комбинации «OCTa
точное количество пестицидовlиспытуемый o
разец» и быть нечувствительным к влиянию
соэкстрактивных веществ;
при интерпретации результатов учитыва
ют природное происхождение некоторых компо-
нентов (например, дисульфид из сем. C,ucife,a-
севе);
концентрация испытуемоro раствора и
раствора сравнения, а также настройка пр
бора, должны быть такими, чтобы отклики, ис-
пользуемые для количественноro определения
остаточных количеств пестицидов, находились в
пределах динамическоro диапазона детектора.
2.8.15. Определение показателя 20речи
405
Испытуемые растворы, содержащие остаточные
количества пестицидов на уровнях, лежащих
вне динамическоro диапазона, MOryт быть раз
ведены в пределах диапазона калибровки при
условии, что концентрация матрицы в растворе
соответствует калибровочным растворам;
открываемость каждоro пестицида должна
находиться в пределах от 70 % до 11 О %;
сходимость метода: RSO не должно пре
вышать значения, указанные в таблице 2.8.1 3. 2;
воспроизводимость метода: RSO не долж
но превышать значения, указанные в таблице
2.8.1 3. 2.
Таблица 2.8.1З. 2
Диапазон
KOHцeHTpa Сходимость Воспроизводимость
ций пести
цидов, Mr/ (RSD), % (RSD), %
Kr
0,001 0,01 30 60
>0,01 O,1 20 40
>0,1 1 15 30
>1 10 20
01/2013:20814
2.8.14. ОПРЕДЕЛЕНИЕ ДУБИЛЬНЫХ
ВЕЩЕСТВ
Все операции экстракции и разведения
проводят с защитой от света.
В случае лекарственноrо растительноrо
сырья или сухоro экстракта в круrлодонную колбу
вместимостью 250 мл помещают указанное в
частной статье количество измельченноro сырья
(180) (2.9.12) или экстракта и прибавляют 150 мл
воды Р. Наrревают на водяной бане в течение
30 мин. Охлаждают под проточной водой и коли
чественно переносят в мерную колбу вместимо
стью 250 мл. Ополаскивают круrлодонную колбу и
сливают промывные воды в мерную колбу, после
чеro доводят водой Р до объема 250,0 мл. Дают
осесть твердым частичкам и фильтруют жид
кость через фильтровальную бумаry диаметром
125 мм. Первые 50 мл фильтрата отбрасывают.
В случае жидкоro экстракта или настойки
указанное количество жидкоro экстракта или Ha
стойки разводят водой Р до объема 250,0 мл.
Раствор фильтруют через фильтровальную
бумаry диаметром 125 мм. Первые 50 мл филь
TraTa отбрасывают.
Общее количество полифенолов. 5,0 мл
фильтрата доводят водой Р до объема 25,0 мл.
2,0 мл полученноro раствора смешивают с 1,0 мл
фосфорномолибденово вольфрамов020 peaK
тива Р и 10,0 мл воды Р и доводят раствором
290 r/л натрия карбоната Р до объема 25,0 мл.
Через 30 мин измеряют оптическую плотность
(2.2.25) при длине волны 760 нм (А 1 ), используя
воду Р в качестве компенсационноro раствора.
Полифенолы, не адсорбируемые кожным по
рошком. К 10,0 мл фильтрата прибавляют 0,10 r
ФСО КОЖН020 порошка и интенсивно встряхивают
в течение 60 мин. Фильтруют и 5,0 мл полученно
ro фильтрата доводят водой Р до объема 25,0 мл.
2,0 мл полученноro раствора смешивают с 1,0 мл
фосфорномолибденово вольфрамов020 peaKти
ва Р и 10,0 мл воды Р и доводят раствором 290 r/л
натрия карбоната Р до объема 25,0 мл. Через
30 мин измеряют оптическую плотность (2.2.25)
при длине волны 760 нм (А 2 ), используя воду Р в
качестве компенсационноro раствора.
Стандарт. Непосредственно перед ис
пользованием растворяют 50,0 Mr пир02алло
ла Р в воде Р и доводят до объема 100,0 мл
этим же растворителем. 5,0 мл получен
Horo раствора доводят водой Р до объема
100,0 мл. 2,0 мл полученноro раствора CMe
шивают с 1,0 мл фосфорномолибденово воль
фрамов020 реактива Р и 10,0 мл воды И дo
водят раствором 290 r/л натрия карбоната
Р до объема 25,0 мл. Через 30 мин измеря
ют оптическую плотность при длине волны
760 нм (Аз), используя воду Р в качестве KOM
пенсационноro раствора.
Процентное содержание дубильных Be
ществ в пересчете на пироrаллол рассчитывают
по формуле:
62,5'(А ).т2
.щ
rде:
т 1 масса испытуемоro образца, r;
т 2 масса пироrаллола, r.
#Допускается проводить определение дy
бильных веществ по методике, указанной в част
ной статье.#
О1/2013:20815
2.8.15. ОПРЕДЕЛЕНИЕ ПОКАЗАТЕЛЯ
rОРЕЧИ
Показатель roречи обратно пропорциона
лен разведению соединения, жидкости или экс
тракта, при котором все еще ощущается roрький
вкус. Он определяется по отношению к показате
лю roречи хинина rидрохлорида, значение KOТO
роro принято за 200 000.
Определение поправОЧН020 коэффициента
Деryстационная комиссия должна состоять,
по крайней мере из 6 человек. Перед деryстаци
ей рот необходимо ополаскивать водой Р.
406
rосударственная фармакопея Республики Беларусь
Для тоro, чтобы скорректировать индивиду
альные различия среди членов деryстационной
комиссии при определении показателя rоречи,
необходимо определять поправочный коэффи
циент для каждоro члена комиссии.
Основной раствор. 0,100 r хинина 2идpox
лорида Р растворяют в небольшом количестве
воды Р и доводят до объема 100,0 мл этим же
растворителем. 1,0 мл полученноro раствора дo
водят водой Р до объема 100,0 мл.
Раствор сравнения. rотовят серию разведе
ний, поместив в первую пробирку 3,6 мл OCHOB
ноro раствора, а в последующих пробирках YBe
личивают объем основноro раствора на 0,2 мл
до объема 5,8 мл. Объем каждой пробирки ДOBO
дят ДО 10,0 мл водой Р.
Находят наименее концентрированный roрь
кий раствор. Для этоro берут 10,0 мл наиболее
разведенноro раствора в рот и перекатывают еro
со стороны в сторону у корня языка в течение
30 с. Если в растворе не чувствуется roрько
ro вкуса, ero выплевывают и ждут 1 мин. После
этоro ополаскивают рот водой Р. Через 1 О мин
повторяют испытание со следующей по возрас
танию концентрацией хинина rидрохлорида.
Рассчитывают поправочный коэффициент k
для каждоro члена деryстационной комиссии по
формуле:
п
k=o ,
5,00
rдe:
п количество миллилитров основноro раст
вора в калибровочном растворе минимальной KOH
центрации, в котором был почувствован roрький
вкус. Лица, не почувствовавшие roречь в paCTBO
ре, приroтовленном из 5,8 мл основноro раствора,
должны быть исключены из состава комиссии.
Пробопод20товка
Если необходимо, измельчают образец в по
рошок (710) (2.9.12). К 1,0 r испытуемоro образца
прибавляют 100 мл кипящей воды Р и HarpeBa
ют на водяной бане в течение 30 мин, постоян
но взбалтывая. Выдерживают до остывания и
доводят водой Р до объема 100 мл. Тщательно
перемешивают и отфильтровывают, отбрасывая
первые 2 мл фильтрата. Этот раствор обозна
чается как C 1 и имеет фактор разведения (DF)
равный 100.
Если определяется roречь жидкости, 1 мл
ее разводят подходящим растворителем до
объема 100 мл И полученный раствор обознача
ют как C 1 (K 1).
Определение степени 20речи
Испытуемые растворы:
10,0 мл раствора C 1, разведенных водой Р
до 100 мл: C 2 (OF=1000);
10,0 мл раствора C 2, разведенных водой Р
до 100 мл: C 3 (OF=10 000);
20,0 мл раствора C 3, разведенных водой Р
до 100 мл: C 3A (OF=50 000);
10,0 мл раствора C 3, разведенных водой Р
до 100 мл: C 4 (OF=100 000).
Начиная с раствора C 4, каждый член KO
миссии определяет раствор с минимальной KOH
центрацией, который имеет rорький вкус. Этот
раствор обозначается О и имеет фактор разве
дения, который обозначается У.
Исходя из раствора О roтовят следующий
ряд разведений:
Раствор 1,2 1,5 2,0 3,0 6,0 8,0
D,мл
Вода Р, 8,8 8,5 8,0 7,0 4,0 2,0
мл
Определяют объем (в миллилитрах) paCT
вора О, который, будучи разведенным до 10 мл
водой Р, все еще имеет roрький вкус (Х).
Рассчитывают показатель roречи для каждо
ro члена деryстационной комиссии по формуле:
( Y.k )
Х.0,1
Показатель roречи образца считается, как
среднее арифметическое всех полученных pe
зультатов.
01/2013:20816
2.8.16. СУХОЙ ОСТАТОК ЭКСТРАКТОВ
в плоскодонную чашку диаметром около 50 мм
и высотой около ЗО мм быстро вносят 2,00 r или 2,0
мл испытуемоro экстракта. Выпаривают досуха на
водяной бане и сушат при температуре от 100 ос
до 105 ос в течение 3 ч. Охлаждают в эксикаторе
над фосфора (V) оксидом Рили силика2елем без
водным Р и взвешивают. Результат рассчитывают
как массовый процент или в rpaMMax на литр.
01/2013:20817
2.8.17. ПОТЕРЯ В МАССЕ ПРИ
ВЫСУШИВАНИИ ЭКСТРАКТА
в плоскодонную чашку диаметром 50 мм и
высотой 30 мм быстро взвешивают 0,50 r мелко
измельченноro испытуемоro экстракта. Сушат
при температуре от 100 ос до 105 ос в течение
3 ч. Охлаждают в эксикаторе над фосфора (V)
2.8.18. Определение афлатоксина 81 в лекарственном растительном сырье 407
оксидом Рили силика2елем безводным Р и
взвешивают. Результат рассчитывают в Mac
совых процентах.
01/2013:20818
2.8.18. ОПРЕДЕЛЕНИЕ
АФЛАТОКСИНА ВА
В ЛЕКАРСТВЕННuМ
РАСТИТЕЛЬНОМ СЫРЬЕ
ПРЕДОСТЕРЕЖЕНИЕ: афлатоксины яв
ляются высокотоксичными и KaHцep02eHHЫ
ми соединениями. Все манипуляции по возмож
ности проводят в вытяжном шкафу. Особые
меры предосторожности, такие как uсполь
зование закрыт020 бокса с перчатками, при
меняют при работе с токсинами в сухой
форме из за их электростатических свойств
и склонности к распространению по всей pa
бочей поверхности. Процедуры дeKOHтaMи
нации лабораторных отходов, содержащих
афлатоксины, были разработаны Междуна
родным а2ентством по изучению рака (/ARC).
Афлатоксины являются микотоксинами
природноro происхождения, продуцируемы
ми в основном Aspe,gillus f/avus и Aspergillus
pa,asiticus. Эти rрибы широко распростране
ны в природе и наиболее часто встречаются
при росте определенных зерен в стрессовых
условиях, таких как засуха. Плесень встреча
ется в почве, rниющей растительности, сене и
зернах, претерпевающих микробное rниение,
и поражает все типы орrанических субстратов
при любых условиях, блаrоприятствующих ее
росту. Блаroприятные условия включают BЫ
сокую влажность и высокую температуру. В
природе продуцируется как минимум 1 З раз
личных типов афлатоксинов, большинство из <
которых является высокотоксичными и KaHцe
роrенными. Афлатоксин В 1 считается наибо
лее токсичным. Лекарственное растительное
сырье, которое склонно к контаминации афла
токсинами, испытывают валидированным Me
тодом.
Если в частной статье нет друrих указаний,
лекарственное растительное сырье должно co
держать не более чем 2 MKr/Kr афлатоксина В 1 .
Уполномоченный opraH может установить пре
дельное содержание суммы афлатоксинов В1'
В2' G 1 И G 2 не более 4 MKr/Kr.
Метод, описанный ниже, приведен в каче
стве примера метода, Подходящеro для анали
за корневищ имбиря, плодов сенны и корней
дьявольскоro коrтя. Приroдность метода для
анализа друroro лекарственноro растительно
ro сырья должна быть доказана или должен ис
пользоваться друroй валидированный метод.
МЕТОД
Жидкостная хроматоrрафия (2.2.29).
Афлатоксины подвержены деструкции при
воздействии света. Определение проводят с
защитой от дневН020 света с помощью УФ
абсорбирующей пленки на окнах в комбинации
с при2лушенным светом или занавесок, жалю
зей, ставней в комбинации с обычным светом
(М02ут применяться флуоресцентные лампы).
Растворы, содержащие афлатоксины, защи
щают от воздействия дневН020 света.
Стеклянную посуду перед использованием
ополаскивают 10 % (об/об) раствором кислоты
серной Р и затем тщательно промывают водой
дистиллированной Р до полноro отсутствия кис
лоты.
Испытуемый раствор. Используют иммун
но аффинную колонку, содержащую антитела к
афлатоксину В 1 с емкостью не менее 100 Hr афла
токсина В 1 и открываемостью не менее 80 % при
прохождении через нее раствора 5 Hr афлаток
сина В 1 в смеси из 12,5 мл метанола Ри 87,5 мл
воды Р. Температуру иммунно аффинной колон
ки доводят до комнатной. К 5,00 r измельченноro
сырья (500) (2.9.12) прибавляют 100 мл смеси из
30 объемов воды Р и 70 объемов метанола Р и
экстраrируют при воздействии ультразвука в Te
чение 30 мин. Фильтруют через складчатый бу
мажный фильтр. 10,0 мл прозрачноro фильтра
та переносят с помощью пипетки в коническую
колбу вместимостью 150 мл, прибавляют 70 мл
воды Р. 40 мл полученноro раствора пропуска
ют через иммунно аффинную колонку со CKO
ростью 3 мл/мин (не допускается превышение
скорости до 5 мл/мин). Колонку дважды промы
вают водой Р порциями по 1 О мл со скоростью,
не превышающей 5 мл/мин, и высушивают с ис
пользованием леrкоro вакуума в течение 1 О с
или пропусканием через колонку воздуха с ис
пользованием шприца в течение 1 О с. В колон
ку вводят 0,5 мл метанола Р и дают пройти
под действием силы тяжести. Элюат собира
ют в мерную колбу вместимостью 5 мл. Через
1 мин наносят вторую порцию 0,5 мл .MeтaHO
ла Р. Через последующую 1 мин наносят третью
порцию 0,5 мл метанола Р. Собирают большую
часть элюата при помощи пропускания воздуха
через колонку или при помощи вакуума. Доводят
водой Р до объема 5 мл и тщательно перемеши
вают. Прозрачный раствор может быть исполь
зован для анализа. В ином случае полученный
раствор пропускают через одноразовый фильтр.
Используют одноразовый фильтр (например, по
литетрафторэтиленовый фильтр с размером пор
0,45 мкм), который не вызывает потери (не yдep
живает) афлатоксинов.
Первичный основной раствор афлаток
сина В 1 . Афлатоксин В 1 Р растворяют в смеси
из 2 объемов ацетонитрила Р и 98 объемов
толуола Р для получения раствора с KOHцeH
408
rосударственная фармакопея Республики Беларусь
трацией 10 мкпмл. Для определения точной
концентрации афлатоксина В 1 в основном pac
творе записывают спектр поrлощения (2.2.25) в
диапазоне длин волн 330 370 нм с использо
ванием кварцевых кювет. Массовую KOHцeHTpa
цию афлатоксина В 1 в мкr/мл рассчитывают по
формуле:
k М .100
" .{
rдe:
А оптическая плотность в максимуме по
лученноro спектра;
М молярная масса афлатоксина В 1 (312 r/мопь);
Е молярный коэффициент поrлощения
афлатоксина В, в смеси толуол ацетонитрил
(1930 м 2 /моль);
{ длина оптическоro пути (1 см).
Вторичный основной раствор афлаток
сина В,. Вторичный основной раствор, coдep
жащий 100 нr/мл афлатоксина В1' roтовят раз
ведением первичноro основноro раствора
афлатоксина В 1 смесью ацетонитрил толуол
(2:98, об/об). Колбу тщательно обертывают алю
миниевой фольroй и хранят при температуре
ниже 4 =с. Перед использованием алюминие
вую фольry не удаляют, пока содержимое не дo
стиrнет комнатной температуры. Если раствор
хранится в течение длительноrо периода Bpe
мени (например, 1 месяц), взвешивают колбу
и записывают массу перед и после каждоro ис
пользования раствора.
Стандартные растворы афлатоксина Bj'
В мерные колбы вместимостью 250 мл поме
щают объемы вторичноro основноro раствора
афлатоксина В.. указанные в таблице 2.8.18. 1.
Пропускают поток азота при комнатной темпе
ратуре до полноrо испарения растворителя. В
каждую колбу прибавляют 75 мл метанола Р,
выдерживают до растворения афлатоксина В 1 и
доводят водой Р до объема 250 мл.
Таблица 2.8.18. 1
Стандартные растворы афлатоксина В!
Объем I Конечная
Стандартный вторичноrо ! концентрация
OCHoBHoro I cTaHAapTHoro
раствор раствора, ! раствора,
мкл i нr/мл
1 125 0,05
2 250 0.1
3 500 0,2
4 750 0,3
5 1000 0.4
rрадуировочный 2рафик. Строят rрадуиро
вочный rрафик с использованием стандартных
растворов афлатоксина В 1 от 1 до 5, который ox
ватывает диапазон, эквивалентный 1 8 MKr/Kr
афлатоксина В 1 в лекарственном растительном
сырье. rрафик проверяют на линейность. Если
содержание афлатоксина В 1 в испытуемом об
разце выходит за пределы калибровки, испыту
емый раствор может быть разведен до получе
ния подходящей концентрации афлатоксина.
Условия хроматоrрафирования:
колонка длиной 0,25 м и внутренним ди
аметром 4,6 мм, заполненная силика2елем OK
тадецилсилильным для хроматО2рафии Р с
размером частиц 5 мкм;
подвижная фаза:
подвижная фаза А (для постколоноч
ной дериватизации с фотохимическим peaKTO
ром или пиридиния бромидом): ацетонитрил
Р метанол Р вода Р (2:3:6, об/об/об);
подвижная фаза В (для постколоноч
ной дериватизации с бромом, получаемым элек
трохимически): 0,12 r калия бромида Р и 350 мкл
кислоты азотной разведенной Р1 прибавляют к
1 л подвижной фазы А;
скорость подвижной фазы: 1 мл/мин;
флуоресцентный детектор, фильтр воз
буждения 360 нм, фильтр эмиссии 420 нм или
эквивалентные. Рекомендуемые настройки для
подходящих детекторов 365 нм (длина волны
возбуждения) и 435 нм (длина волны эмиссии);
объем вводимой пробы: 500 мкл.
Постколоночная дериватизация с пириди
ния 2идробромидом пербромидом (РВРВ):
безимпульсный насос;
Т отрезок с нулевым мертвым объемом;
политетрафторэтиленовая реакционная
трубка, длиной 0,45 м и внутренним диаметром
0,5 мм;
подвижная фаза А;
постколоночный дериватизирующий peaK
тив: 50 Mr пиридиниЯ2идробромида пербромида Р
растворяют в 1000 мл воды Р (хранят с защитой
от света и используют в течение 4 дней);
скорость потока дериватизирующеro peaK
тива: 0,4 мл/мин.
Постколоночная дериватизация с фотохи
мическим реактором (PHREO):
реактор с одной УФ лампой с максималь
ной интенсивностью при длине волны 254 нм с
низким давлением ртути (минимум 8 Вт);
полированная пластина держатель;
витой реактор: политетрафторэтиленовая
трубка, плотно намотанная BOKpyr УФ лампы,
длиной 25 м, внутренним диаметром 0,25 мм и
номинальным свободным объемом 1,25 мл;
время экспозиции: 2 мин;
подвижная фаза А.
Постколоночная дериватизация с бромом,
получаемым электрохимически (KOBRA):
КОВRА ячейка: электрохимическая
ячейка, rенерирующая реактивную форму брома
для дериватизации афлатоксинов, выражающу
2.8.20. Лекарственное растительное сырье: отбор проб
409
юся в усилении флуоресценции; доступен из
разных коммерческих источников;
источник постоянноro тока, соединенный с
КОВRA ячейкой, дающий постоянный ток в 100 мкА;
политетрафторэтиленовая реакторная
трубка длиной 0,12 м, внутренним диаметром
0,25 мм;
подвижная фаза В.
Порядок выхода пиков: афлатоксин G 2 , аф
латоксин G 1 , афлатоксин В2' афлатоксин В 1 .
Расчеты: находят уравнение прямой у=ах+Ь
по rрадуировочному rрафику, на котором по оси
абсцисс откладывают концентрацию афлатокси
на В 1 (нr/мл), а по оси ординат сиrнал (8). KOH
центрация афлатоксина В 1 в испытуемом pac
творе равна 8 Ь .
а
Содержание афлатоксина В 1 в лекарствен
ном растительном сырье (Hr/r) рассчитывают по
формуле:
.V2'C
m.V;
rде:
т масса лекарственноro растительноro
сырья, взятая для анализа, r;
V 1 объем растворителя, использованный
для экстракции, мл;
v; аликвота, взятая для иммунно аффин
ной очистки, мл;
V 2 конечный объем раствора после элю
ирования с иммунно аффинной колонки и разве
дения, мл;
С измеренная концентрация афлатокси
на В 1 в испытуемом растворе, нr/мл.
Присутствие афлатоксина В 1 может быть
подтверждено хроматоrрафированием без пост
колоночной дериватизации, при этом отклик аф
латоксина В 1 значительно меньше (более чем.. в
10 раз).
01/2013:20820
2.8.20. ЛЕКАРСТВЕННОЕ
РАСТИТЕЛЬНОЕ СЫРЬЕ:
ОТБОР ПРОБ
. Для уменьшения влияния способа отбfJра
. проб на качественный и количественный анализ
необходимо, чтобы состав испытуемою образ
ца репрезентативно характеризовал всю испы
туемую серию (партию). Для отбора проб ле
карственноro растительною сырья MOryT быть
использованы приведенные ниже процедуры.
ПРИМЕЧАНИЕ: М02ут быть использованы
дРУ2ие процедуры при условии, что в результа
те их будет получен репрезентативный обра
зец серии (партии).
#ОТБОР ПРОБ ЛЕКАРСТВЕнноrо
РАСТИТЕльноrо СЫРЬЯ
Каждую единицу продукции подверrают
внешнему осмотру для установления COOTBeT
ствия упаковки и маркировки требованиям HOp
мативной документации. Обращают внимание
на правильность упаковки, состояние тары (OT
сутствие подмочки, подтеков и друrих поврежде
ний, отрицательно влияющих на качество и co
хранность сырья).
Для проверки соответствия качества сырья
требованиям нормативной документации отби
рают выборку из неповрежденных единиц про
дукции, взятых случайным образом из разных
мест серии (партии) в количестве, указанном в
таблице 2.8.20. 1. Проверку качества сырья в
поврежденных единицах продукции производят
отдельно от неповрежденных, вскрывая каждую
единицу продукции.
Таблица 2.8.20. 1
Количество
контейнеров в Количество отбираемых
партии лекар- контейнеров
CTBeHHoro расти-
тельноrо сырья
1 5 Все контейнеры
6 50 5
>50 1 О % контейнеров от
серии (партии)*
. при получении дробною числа, окруrляют до следу юще
ю целою числа.
Попавшие в выборку единицы продукции
вскрывают и путем внешнеro осмотра опреде
ляют: однородность сырья по способу подrо
товки (цельное, измельченное, прессованное и
т. д.), цвету, запаху, засоренности; наличие при
месей (плесени, rнили, устойчивоro посторонне
ro запаха, не исчезающеro при проветривании;
засоренность ядовитыми растениями и такими
примесями, как камни, стекло, помет rрызунов
и птиц и т. п.). Одновременно невооруженным
rлазом и с помощью лупы (5 10x) определя
ют наличие амбарных вредителей, как указано в
статье «Лекарственное растительное сырье».
При обнаружении признаков rниения, пле
сени, устойчивоro постороннеro запаха, не ис
чезающеro при проветривании; засоренности
ядовитыми растениями и такими примесями,
как стекло, помет rрызунов и птиц и т. п. партия
сырья не подлежит приемке.
Из каждой единицы продукции, отобранной
для вскрытия, берут, избеrая измельчения, 3 TO
чечные пробы: сверху, снизу и из середины. Из
мешков, тюков и кип точечные пробы отбирают
на rлубине не менее 1 О см сверху, затем, после
410
rосударственная фармакопея Республики Беларусь
распарывания по шву, из середины и снизу; TO
чечные пробы семян и сухих плодов отбира
ют зерновым щупом. Из сырья, упакованноro в
ящик, первую точечную пробу отбирают из Bepx
неro слоя, вторую после удаления сырья при
мерно до половины ящика, и третью со дна
ящика. Точечные пробы должны быть примерно
одинаковыми по массе. Из всех точечных проб,
осторожно перемешивая. составляют объе
диненную пробу. Масса пробы, отбираемой из
каждоro контейнера. должна быть такой, чтобы
масса объединенной пробы была достаточна
для проведения всех испытаний; в противном
случае отбор точечных проб повторяют.
Из объединенной пробы методом KBapТOBa
ния (см. примечание ниже) выделяют:
при обнаружении присутствия амбарных
вредителей и при необходимости определения
степени зараженности амбарными вредителями
отбирают аналитическую пробу массой 500 r для
мелких видов сырья и массой 1000 r для круп
ных видов сырья:
аналитическую пробу для определения
микробиолоrической чистоты (50 r);
аналитическую пробу для определения
радионуклидов (размер пробы определяют в за
висимости от используемоro метода определе
ния радионуклидов ):
среднюю пробу в соответствии с табли
цей 2.8.20. 2 для проведения остальных видов
анализа.
Таблица 2.8.20. 2
Тип лекарственноrо Минимальный вес
растительноrо сырья средней пробы
Корни. корневища. кора,
травы (цельное и из 500 r
мельченное сырье)
Листья. uветки. семена.
плоды (цельное и из 250 r
мельченное сырье)
ПРИМЕЧАНИЕ: при квартовании лекар
ственное растительное сырье разравнивают
на 2ладкой, чистой. ровной поверхности в виде
квадрата по возможности тонким paвHOMep
ным по толщине слоем и по диа20нали делят
на четыре треУ20льника. Два противополож
ных треУ20льника удаляют. а два оставшихся
соединяют вместе и перемешивают. Эту про
цедуру повторяют до тех пор. пока не ocтa
нется количество сырья в двух противополож
ных треУ20льниках, соответствующее массе
заданных проб. Допустимые отклонения в
массе каждой из проб не должны превышать
:110%.
Оставшуюся после макроскопическоrо
анализа (2.8.23), определения степени из
мельчения (2.9.12) и содержания примесей
(2.8.2) часть средней пробы измельчают нож
ницами или секатором до размеров частиц
около 1 см, если в частной статье нет друrих
указаний, перемешивают и выделяют MeTO
дом квартования аналитические пробы для
определения влажности, золы и действу
ющих веществ, пестицидов и токсических
элементов.
#ОТБОР ПРОБ ФАСОВАННОЙ
ПРОДУКЦИИ
Лекарственное растительное сырье и
сборы расфасовываются в пачки, пакеты,
филыр пакеты (в цельном, резаном, дробле
ном, порошкованном виде), а также выпуска
ются в форме брикетов (в резано прессован
ном виде).
Единицы продукции в выборку необходи
мо отбирать из разных мест контролируемой
серии (партии).
Объем выборки зависит от размера серии
(партии) и определяется в соответствии с Tpe
бованиями раздела #1.7. Отбор проб.
В случае недостаточноrо количества по
требительских упаковок для проведения ис
пытания, их повторно отбирают, как указано
выше.
Отобранные потребительские упаковки
составляют объединенную пробу.
Из объединенной пробы выделяются
пробы:
в соответствии с таблицей 2.8.20. 2 (но
не менее 1 О невскрытых пачек или пакетов;
10 невскрытых контурных ячейковых упако
во к, брикетов; 20 невскрытых филыр пакетов
для определения допустимых отклонений на
промышленное фасование);
для определения микробиолоrической
чистоты 3 не вскрытых потребительских
упаковок общей массой не менее 50 r;
для определения радионуклидов в
соответствии с используемым методом опре
деления радионуклидов.
0112013:20822
2.8.22. ОПРЕДЕЛЕНИЕ
ОХРАТОКСИНА А
В ЛЕКАРСТВЕННОМ
РАСТИТЕЛЬНОМ СЫРЬЕ
ПРЕДОСТЕРЕЖЕНИЕ: охра токсин А яв
ляется нефротоксичным и нефроканцеро
2енным. Все манипуляции по возможности
проводят в вытяжном шкафу. Особые меры
предосторожности, такие как использова
ние закрыт020 бокса с перчатками, применя
ют при работе с токсинами в сухой форме
из за их электростатических свойств и
склонности к распространению по всей pa
бочей поверхности. Необходимо осущест
2.8.22. Определение охратоксина А в лекарственном растительном СЬ1рье
411
влять процедуры обеззараживания cтe
клянной лабораторной посуды, содержащей
охратоксин А (см. приложение).
Лекарственное растительное сырье, KOTO
рое склонно к контаминации охратоксином А,
испытывают валидированным методом.
Метод, описанный Ниже, приведен в Ka
честве при мера метода, ПОДХодящеro для
анализа корней солодки и экстракта солод
ки. Приrодность метода для анализа друroro
лекарственноro растительноro сырья должна
быть доказана или должен использоваться
друroй валидированный метод.
МЕТОД
Жидкостная хроматоrрафия (2.2.29).
Используют посуду из коричневоrо стекла,
не содержащую остатков детерrентов. При
необходимости стеклянную посуду перед ис
пользованием ополаскивают 10 % (об/об) pac
твором кислоты серной Р и затем тщатель
но промывают водой дистиллированной Р до
полноro отсутствия кислоты.
РастворА Смешивают 80 объемов воды Р,
предварительно доведенной кислотой Mypa
вьиной безводной Р до рН 2,3, и 20 объемов
ацетонитрила Р.
Испытуемый раствор. Используют им
мунно аффинную колонку, содержащую анти
тела кохратоксину А с емкостью не менее 100 Hr
охратоксина А и открываемостью не менее
70 %. Температуру ИМмунно аффинной KO
лонки доводят до комнатной.
К 2,00 r измельченноrо сырья (250) (2.9.12)
прибавляют 80 мл раствора ЗО r/л натрия 2и
дрокарбоната Р и экстраrируют при воздей
ствии ультразвука в течение 30 мин (через 15
мин сменяют воду в ультразвуковой бане). Ox
лаждают до комнатной температуры, доводят
до объема 100,0 мл (V 1 ) этим же растворите
лем и центрифуrируют. 5,0 мл (V:) прозрачной
надосадочной жидкости тщательно переме
шивают с 30 мл буфеРН020 раствора рН 7;4 Р
и весь полученный объем пропускают через
ИМмунно аффинную колонку со скоростью
3 млlмин (не допускается превышение CKOpO
сти ДО 5 мл/мин). Колонку сначала ПРоМЫва
ют 1 О мл буфеРН020 раствора рН 7,4 Р, затем
дважды промывают водой Р порциями по 1 О мл
со Скоростью, не превышающей 5 млlмин,
и высушивают с использованием леrкоro Ba
куума в течение 5 1 О с или Пропускани
ем через колонку воздуха с использованием
шприца в течение 1 О с. В колонку вводят 0,5 мл
метанола Р и дают пройти под действием
силы тяжести.
Элюат собирают в контейнер вместимо
стью 4 мл. Через 30 с наносят вторую порцию
0,5 мл метанола Р, дают пройти через KO
лонку под действием силы тяжести и собира
ют в этот же контейнер. Через последующие
30 с наносят третью порцию 0,5 мл MeтaHO
ла Р. Собирают весь удерживаемый в колон
ке объем при помощи пропускания воздуха
через колонку или при помощи вакуума. Объ
единенные элюаты выпаривают досуха с ис
пользованием наrревательноrо блока в среде
азота (40 ОС). Сухой остаток растворяют в
0,5 мл (V 2 ) раствора А. Прозрачный paCT
вор может быть использован для анализа. В
ином случае полученный раствор пропускают
через одноразовый фильтр. Используют OДHO
разовый фильтр (например, политетрафтор
этиленовый фильтр С размером пор 0,45 мкм),
который не вызывает потери (не удерживает)
охратоксина А.
Первичный основной раствор oxpaтOK
сина А 1, О мл раствора охратоксина А р
доводят метанолом Р до объема 100,0 мл и
тщательно перемешивают.
Вторичный основной раствор oxpaтOK
сина А 10,0 мл первичноrо основноro paCT
вора охратоксина А доводят метанолом Р
до объема 100,0 мл И тщательно перемеши
вают.
Стандартные растворы охратоксина А
Помещают объемы первичноrо OCHoBHoro
раствора охратокисна А или ВТОричноrо oc
новноro раствора охратоксина А, указанные в
таблице 2.8.22. 1, в отдельные колбы и ДOBO
дят раствором А до объема 50,0 мл.
Таблица 2.8.22. 1
Стандартные растворы охратоксина А
Объем Конечная
первично- концентра-
Стандартный ro OCHoBHoro ция охра-
токсина А в
раствор раствора стандартном
охратоксина А, растворе,
мкл нr/мл
1 5000 50
2 2500 25
3 1000 10
4 500 5
5 250 2,5
Объем Конечная
вторичноrо концентра-
Стандартный OCHoBHoro ция охра-
токсина А в
раствор раствора стандартном
охратоксина А, растворе,
мкл нr/мл
6 500 0,5
7 100 0,1
tрадуировочный 2рафик. Строят rрадуиро
вочный rрафик с использованием стандартных
растворов охратоксина А от 1 до 7, который OXBa
тывает диапазон, эквивалентный 0,5-..--250 MKr/Kr
охратоксина А в лекарственном растительном
412
rосударственная фармакопея Республики Беларусь
сырье. rрафик проверяют на линейность. Если
содержание охратоксина А в испытуемом образ
це выходит за пределы калибровки, испытуемый
раствор может быть разведен до получения под
ходящей концентрации охратоксина А.
Условия хроматоrрафирования:
колонка длиной 0,15 м и внутренним ди
аметром 4,6 мм, заполненная силика2елем OK
тадецилсилильным для хромат02рафии Р с
размером частиц 5 мкм;
температура: 45 ос;
подвижная фаза:
подвижная фаза А: вода Р, доведенная
кислотой фосфорной Р до рН 2,3;
подвижная фаза В: ацетонитрил Р;
Время Подвижная Подвижная фаза В
фаза А
(мин) (%, об/о6) (%, 06/06)
о зо 80 40 20 60
30 З5 40 20 60 80
35 37 20 80
37 0 20 80 80 20
скорость подвижной фазы: 0,8 мл/мин;
флуоресцентный детектор, peKOMeHДY
емые настройки 330 нм (длина волны возбуж
дения) и 460 нм (длина волны эмиссии);
объем вводимой пробы: 20 мкл.
Расчеты: находят уравнение прямой у=ах+Ь
по rрадуировочному rрафику, на котором по оси
абсцисс откладывают концентрацию охратокси
на А (нr/мл). а по оси ординат сиrнал (5). KOH
центрация охратоксина А в испытуемом paCTBO
ре равна 5 Ь .
а
Содержание охратоксина А в лекарствен
ном растительном сырье (Hr/r) рассчитывают по
формуле:
v..V 2 .C
т.\!;
rдe:
т масса лекарственноro растительноro
сырья, взятая для анализа. r;
V 1 объем разведения. мл;
v: аликвота, взятая для иммунно аффин
ной очистки, мл;
V 2 объем раствора. в котором был pac
творен сухой остаток, мл;
С найденная концентрация охратоксина
А в испытуемом растворе, нr/мл.
Приложение:процедурыобеззараживания
лабораторной посуды
Стеклянную посуду ополаскивают MeтaHO
лом Р и обеззараживают, выдерживая ее в pac
творенатрия 2идроксида концентрированном Р
в течение не менее 2 ч. Тщательно промывают
водой.
01/2013:20823
2.8.23. #МАКРОСКОПИЧЕСКИЙ
И#МИКРОСКОПИЧЕСКИЙ
АНАЛИЗЛЕКАРСТВЕнноrо
РАСТИТЕльноrо СЫРЬЯ
#МАКРОСКОПИЧЕСКИЙ АНАЛИЗ
Макроскопический анализ сводится к изуче
нию внешнеro вида лекарственноro раститель
ноro сырья, определению размеров отдельных
частей, орrанолептических показателей (цвета,
запаха), морфолоrических диаrностических при
знаков.
Размеры сырья определяются с помощью
измерительной линейки: для крупных объектов
(более 3 см) 3 5 измерений, для мелких
1 0 20 измерений. Мелкие семена и плоды из
меряют на миллиметровой бумаrе и рассчитыва
ют среднее значение.
Определяют цвет сырья поверхности на
изломе или в разрезе при дневном освещении.
Запах определяют при растирании между
пальцами на изломе или при растирании в
ступке.
Морфолоrические диаrностические при
знаки определяют для всех видов сырья. Bыcy
шенные и смятые части сырья предварительно
размяrчают во влажной камере или путем поrру
жения на несколько минут в roрячую воду, после
чеro раскладывают на стеклянной пластинке,
тщательно расправляя. Рассматривают HeBO
оруженным rлазом или с помощью лупы (10 х ).
Листья (Folia) лекарственное сырье,
представляющее собой высушенные или свежие
листья или отдельные листочки сложноro листа.
Диаrностическими признаками являются: тип ли
стьев (простые или сложные), форма и размеры
листовой пластинки и черешка, характер края,
жилкование, опушенность.
Цветки (F/ores) лекарственное сырье,
представляющее собой высушенные отдельные
цветки или соцветия, а также их части. Диаrно
стическими признакам и являются: тип соцветия,
опушенность, форма и размеры цветка, CTpO
ение околоцветника (число, форма и характер
срастания чашелистиков и лепестков), число и
строение тычинок и пестиков, характер завязи и
цветоложа.
Травы (Не,Ьае) лекарственное сырье,
представляющее собой высушенные или свежие
надземные части травянистых растений (стебли
с листьями и цветками, отчасти с бутонами и He
зрелыми плодами). Диаrностические признаки
для листьев и цветков указаны выше. Для CTe
блей: тип ветвления, форма поперечноro сече
ния, размеры (длина и диаметр у основания),
характер поверхности, опушенность, листора
сположение.
Плоды (Fructus) высушенные или свежие
простые или сложные плоды (соплодия) и их
2.8.23. Макроскопический и микроскопический анализ
413
части. Диаrностическими признаками являют
ся: консистенция околоплодника (перикарпия),
характер поверхности, размеры (длина, толщи
на, поперечник плода), расположение остатков
частей цветка и др.
Семена (Sетiпа) цельные семена или
отдельные семядоли. Исследуются сухими.
Диаrностические признаки: форма, размеры
(длина, толщина, поперечник), характер поверх
ности, цвет, запах, форма, размеры и располо
жение зародыша, наличие и форма рубчика или
семяшва.
Кора (Cortices) лекарственное сырье,
представляющее собой наружную часть CTBO
лов, ветвей и корней деревьев и кустарников,
расположенную к периферии от камбия. Диаrно
стические признаки: размеры и форма кусков,
особенности наружной и внутренней поверхно
сти и излома.
Корни, корневища, луковицы, клубни, клуб
нелуковицы (Radices, Rhizomata, Ви/Ы, Tubera,
Bu/botube,a) высушенные или свежие подзем
ные opraHbI мноюлетних растений, очищенные
или отмытые от земли, освобожденные от OCTaT
ков стеблей и листьев. Диаrностические призна
ки: форма, особенности наружной поверхности
и излома, размер, цвет поверхности и на свежем
изломе, запах.
Сборы (Species) смесь нескольких видов
измельченноro (реже цельноro) лекарственно
ro растительноrо сырья, иноrда с добавлением
солей, эфирных масел. Сырье, используемое
для приroтовления сборов, должно COOTBeTCTBO
вать требованиям нормативной документации
на каждый вид сырья.
Анализ зависит от морфолоrической rруппы
исследуемоro объекта, а также от состояния
сырья цельноro или измельченноro. Размер
лекарственноro растительною сырья указывают
в частных статьях.
Общие требования к измельченному лекар
ственному растительному сырью, в том числе
значения и допустимые нормы при ситовом aHa
лизе, описаны в общей статье Сборы, если не
указано иное в частных фармакопейных статьях.
Степень измельчения указывают в скобках, Ha
пример, измельченное сырье с частицами, про
ходящими сквозь сито с размером стороны OT
верстия 5600 мкм, обозначают как измельченное
сырье (5600).
МИКРОСКОПИЧЕСКИЙ АНАЛИЗ
#Микроскопический анализ зависит от MOp
фолоrической rруппы испытуемоro сырья, а
также от состояния сырья цельноro или из
мельченноro.#
#ЛИСТЬЯ, ТРАВЬ/, ЦВЕТКИ
Цельное и измельченное сырье. При ис
следовании цельною сырья берут кусочки пла
стинки листа С краем и жилкой; у трав берут лист,
иноrда также кусочек стебля и цветок, у цветков
чашечку и венчик. При исследовании резаноro
сырья берут несколько различных кусочков.
Просветление можно проводить двумя спо
собами.
1. Несколько кусочков сырья помещают в
колбу или пробирку, прибавляют раствор 25 r/л'
натрия 2идроксида Р и кипятят в течение
1 2 мин. Затем содержимое выливают в чашку
Петри (или фарфоровую), жидкость сливают и
сырье тщательно промывают водой Р. Из воды
кусочки сырья вынимают скальпелем или ло
паточкой и помещают на предметное стекло в
каплю раствора хлораЛ2идрата Р1 или pacт
вора 2лицерина Р.
11. Кусочки кипятят в растворе хлораЛ2и
драта Р1, разведенною водой Р (1:1, об/об), в
течение 5 10 мин (до просветления). Просвет
яенный кусочек сырья помещают на предметное
стекло в каплю раствора хлораЛ2идрата Р1
или 2лицерина Р, разделяют скальпелем или
препаровальной иrлой на две части, одну из них
осторожно переворачивают. Объект накрывают
покровным стеклом, слеrка подоrревают до yдa
ления пузырьков воздуха и, после охлаждения,
рассматривают лист с обеих сторон под микро
скопом сначала при малом, затем при большом
увеличении. При приroтовлении микропрепара
тов из толстых листьев их предварительно раз
давливают скальпелем.
Для исследования стеблей их отрезки кипя
тят в растворе 50 r/л натрия 2идроксида Р, тща
тельно промывают водой Р, снимают эпидер
мис скальпелем или препаровальными иrлами и
рассматривают еro с поверхности; из остальных
тканей roтовят препарат, раздавливая объект
скальпелем на предметном стекле в растворе
хлораЛ2идрата Р1 или растворе 2лицерина Р.
При необходимости приroтовления попереч
ных срезов листьев и стеблей их кипятят в pac
творе хлораЛ2идрата Р1 в течение 1 О мин и
делают срезы, зажимая кусочки листа в пробку
или сердцевину бузины. rOToBbIe срезы помеща
ют в воду Р и далее используют для приrотов
ления микропрепаратов, рассматривая их в pac
творе хлораЛ2идрата Р1.#
Исследование при дополнительном из-
мельчении. Для микроскопическою испыта
ния лекарственное растительное сырье дo
полнительно измельчают (355) (2.9.12), если в
частной статье не указано иное.
Наиболее широко используемой средой
для заключения в нее дополнительно измель
ченноrо сырья является раствор хлораЛ2и
драта Р. Однако в этом реактиве не всеrда
хорошо видны H€KOTOpыe элементы, в таком
случае MorYT быть использованы иные среды,
например, 50 % (об/об) раствор 2лицерина Р,
позволяющий обнаружить зерна крахмала.
При необходимости в частной статье может
быть отмечено использование специфических
реактивов, например, реактива молочной
414
rосударственная фармакопея Республики Беларусь
кислоты Р (использование котороro позволя
ет обнаруживать различные диаrностические
признаки), раствора рутения краСН020 Р (по
зволяющий увидеть присутствие слизи в клет
ках) или 2лицерина Р (в котором видны Kpax
мал и инулин) и друrих.
Использование раствора хлораЛ2идра
та. 2 3 капли раствора хлораЛ2идрата Р
помещают на предметное стекло. Небольшое
количество порошка суспендируют в paCTBO
ре и накрывают покровным стеклом. Препа
рат очень аккуратно наrревают на плитке или
на rазовой микроrорелке до кипения и кипятят
в течение непродолжительноrо времени, при
необходимости с помощью пипетки добавля
ют реактив. охлаждают и просматривают под
микроскопом Наrревание повторяют до тех
пор, пока крахмальные зерна и BoдopaCTBO
римое содержимое клеток не станет невиди
мым. Просматривают полученный препарат
под микроскопом. Так как хлоралrидрат скло
нен к кристаллизации в виде длинных нитей,
для предотвращения этоro после наrревания
удаляют покровное стекло, к препарату при
бавляют 1 каплю 10 % (об/об) смеси раствора
хлораЛ2uдрата Р и 2лицерина Р, накрывают
чистым покровным стеклом и просматривают
под микроскопом.
Использование 50 % (об/об) раствора
2лицерича. 2 капли 50 % (об/об) раствора 2ЛИ
церина р помещают на предметное стекло.
Небольшое количество порошка суспендиру
ют в растворе и накрывают покровным CTe
клом. Просматривают под микроскопом.
#ПЛОДЫ СЕМЕНА
Цельное сырье. rотовят препараты
кожуры семени и околоплодника с поверхно
сти или поперечные срезы.
Препараты поверхности кожуры и OKO
лоплодника. 2 3 семени или плода кипятят в
пробирке в рас--;-воре 50 r/л натрия 2идpOKcи
да Р в течение 2 З мин и тщательно промыва
ют водой Р. Объект помещают на предметное
стекло. препаровапьными иrлами отделяют
кожуру семени или ткани околоплодника и
рассматривают их в растворе хлораЛ2идра
та Р1 или растворе 2лицерина Р.
Срезы. Для приrотовления срезов сухие
плоды и семена предварительно размяrчают,
поместив их на сутки во влажную камеру (влаж
ной камерой служит эксикатор с водой, в KOTO
рую добавлено несколько капель хлорофор
ма) или водяным паром в течение 15 30 мин
или более в зависимости от твердости
объекта.
Мелкие плоды и семена запаива
ют в парафиновый блок размером около
0.5 см х 0,5 см х 1.5 см. Кончиком наrретой
препаровальной иrлы расплавляют парафин
и в образовавшуюся ямку быстро поrружа
ют объект. Поверхность объекта должна быть
сухой. Срезы объекта делают вместе с пара
фином; срезы выбирают из парафина препаро
вальной иrлой, смоченной жидкостью, и rOTo
вят микропрепараты в растворе 2лицерина Р
или растворе хлораЛ2идрата Р1.
Диаrностические признаки: форма и CTpO
ениеклетокэкзокарпия(эпидермиса),наличие
и строение трихом, расположение и форма
механических элементов в мезокарпии, число
и расположение эфиро масличных каналов,
проводящих пучков, наличие кристаллических
включений.
Исследование при дополнительном из
мельчении. Препарат roтовят так же, как YKa
зано в разделе «Листья, травы, цветки».
#КОРА
Цельное сырье и измельченное
сырье. rотовят поперечные или продольные
срезы коры. Кусочки коры размером около
2 3 см х 0,5 1 см кипятят в колбе или про
бирке с водой Р в течение 5 мин. Размяrчен
ные куски выравнивают скальпелем так, чтобы
они имели строro поперечное или продольное
сечение. Делают срезы и roтовят микропрепа
раты в растворе хлораЛ2идрата Р1 или pac
творе 2лицерина Р. При необходимости roто
вят препараты в соответствующих реактивах
для выявления различных структур или Be
ществ.
Диаrностические признаки: толщина,
окраска и характер пробки, наличие коллен
химы, толщина первичной и вторичной коры,
ширина сердцевинных лучей, особенности
расположения и количество лубяных волокон,
каменистых клеток, клеток с эфирным маслом,
включения кристаллов оксалата кальция,
млечники.
Соскоб коры или мелкие кусочки кипятят
в течение 3 5 мин в растворе 50 r/л натрия
2идроксида Р, промывают водой Р и rотовят
микропрепараты, раздавливая объект скаль
пелем в растворе 2лицерина Р или растворе
хлораЛ2идрата Р1.
Исследование при дополнительном из-
мельчении. Препарат rотовят так же, как указа
но в разделе «Листья, травы, цветки».
#КОРНИ, КОРНЕВИЩА, КЛУБНИ,
ЛУКОВИЦЫ, КЛУБНЕЛУКОВИЦЫ
Цельное сырье. rотовят поперечные и
продольные срезы. Небольшие куски подзем
ных opraHoB помещают в холодную воду Р и
выдерживают около суток, затем помещают
в смесь спирт Р 2лицерин Р (1: 1, об/об)
на 3 сут. Размоченные объекты выравнивают
скальпелем так, чтобы они имели строro по
перечное или продольное сечение. Делают
срезы и rотовят микропрепараты в растворе
хлораЛ2идрата Р1 или растворе 2лицерина р,
2.8.23. Макроскопический и микроскопический анализ лекарствеННО20
415
рассматривают диаrностические признаки
сначала при малом, затем при большом YBe
личении.
Диаrностические признаки: особенности
строения эпидермы или перидермы, распо
ложение и строение механических, проводя
щих, секреторных тканей, кристаллы оксалата
кальция, запасные вещества.
Измельченное сырье. Кусочки подзем
ных opraHoB кипятят в течение 3 5 мин в pac
творе 50 r/л натрия 2идроксида Р, тщательно
промывают водой Р и rотовят микропрепара
ты, раздавливая кусочки в растворе 2лицери
на Р или растворе хлораЛ2идрата Р1.
Исследование при дополнительном из-
мельчении. Препарат rотовят так же, как yкa
зано в разделе «Листья, травы, цветки».
#МИКРОХИМИЧЕСКИЙ АНАЛИЗ
Микрохимический анализ может прово
диться одновременно с микроскопическим
анализом.
Крахмал. rотовят два препарата в pac
творе ЛЮ20ЛЯ Р и В воде Р; от йода крахмаль
ные зерна окрашиваются в синий цвет. В воде
определяют их форму, строение. Также для
идентификации зерен крахмала проводят ис
пытание в поляризованном свете (между пе
рекрестными призмами Николя) на зернах
обнаруживается черный крестик.
Жирное и эфирное масло. Для обнаруже
ния жирноro и эфирноro масла rотовят пре
парат в растворе судана 111 Р и подоrревают;
капли жирноro или эфирноro масла окраши
ваются в оранжево розовый цвет.
Слизь. Для обнаружения слизи rотовят
препарат порошка в растворе черной туши Р
и тотчас рассматривают под микроскопом
(малое увеличение); слизь заметна в виде
бесцветных масс на черном фоне.
Либо помещают 2 капли раствора pyтe
ния краСН020 Р на предметное стекло, He
большое количество порошка суспендируют
в жидкости и накрывают покровным стеклом,
через 1 мин дают впитаться капле воды диc
тиллированной Р между предметным и по
кровным стеклами; слизь окрашивается в фи
олетово красный цвет.
Одревесневшие (ли2нифицированные)
элементы. Небольшое количество порошка
помещают на предметное стекло, прибавляют
1 2 капли 10 % (об/об) спиртовоro раствора
фЛОР02люцина Р, перемешивают и выдержи
вают до почти полноrо испарения раствори
теля, прибавляют 1 2 капли кислоты хло
ристоводородной Р, накрывают препарат
покровным стеклом и немедленно просма
тривают под микроскопом; красное окраши
вание указывает на присутствие лиrнина. Для
окраски одревесневших элементов можно ис
пользовать также раствор 1 О r/л сафрани
на Р. Срезы помещают в раствор 1 О r/л саф
ранина Р в спирте (50 % об/об) Р на 30 мин
(в закрытом бюксе или на часовом стекле),
промывают сначала спиртом (50 % об/об) Р,
затем подкисленным спиртом (на 100 мл 96 %
спирта Р прибавляют 2 капли хлористоводо
родной кислоты Р) и заключают на предмет
ном стекле в 2лицерин Р; одревесневшие обо
лочки окрашиваются в красный цвет.
Дубильные вещества. Наличие дубиль
ных веществ устанавливают, нанося 1 каплю
раствора 1 О r/л железа (111) аммония сульфа
та Р или раствора 30 r/л железа (111) хлорu
да Р на внутреннюю поверхность сухой коры;
появляется черно синее или черно зеленое
окрашивание.
Производные антрацена. Наличие про
изводных антрацена определяют, нанося 1 2
капли раствора 50 r/л натрия 2идроксида Р
на внутреннюю поверхность коры (красное
окрашивание) или проводя микросублима
цию описанным ниже способом. На предмет
ное стекло ставят трубку диаметром 1,5 см и
высотой 2 см. Внутрь стеклянной трубки по
мещают небольшое количество порошка (или
соскоба) испытуемоro образца, сверху HaKpы
вают друrим предметным стеклом, ставят на
асбестовую сетку, закрепленную в штативе, и
подоrревают. Пламя rорелки следует держать
от предметноro стекла на расстоянии 5 7 см.
На поверхность стекла, которое служит для
улавливания сублимата, помещают кусочки
фильтровальной бумаrи и смачивают время
от времени холодной водой. Через некоторое
время на нижней стороне стекла появляется
налет. Под микроскопом в сублимате видны
тонкие желтые иrолочки, которые в ультрафи
олетовом свете (люминесцентный микроскоп)
имеют яркое желтое или оранжево красное
свечение. В растворе 50 r/л калия 2идроксида Р
в 96 % спирте Р сублимат растворяется с по
явлением красноro окрашивания.
Инулин. Для обнаружения инулина на
предметное стекло помещают около 0.1 r
порошка образца, 1 2 капли раствора
{3 нафтола Р1 (или раствора резорцина Р.
или раствора тимола Р) и 1 каплю кислоты
серной Р; появляется красновато фиолето
вое окрашивание (от резорцина оранжево
красное). О наличии инулина можно делать
выводы только при отсутствии крахмала.
Использование реактива молочной Kиc
лоты. Помещают 2 3 капли реактива MO
лочной кислоты Р на предметное стекло,
небольшое количество порошка суспенди
руют в жидкости, накрывают покровным CTe
клом, препарат очень аккуратно наrревают
на плитке или на rазовой микроroрелке до ки
пения и кипятят в течение непродолжитель
ноro времени, при необходимости с помо
щью пипетки добавляют реактив, охлаждают
416
rосударственная фармакопея Республики Беларусь
и просматривают под микроскопом. Лиrнифи
цированные структуры окрашиваются в ярко
желтый цвет; структуры, содержащие целлю
лозу, остаются бесцветными. Зерна крахмала
окрашиваются в более или менее насыщен
ный фиолетовый цвет; некоторые секреты
(например, эфирные масла, смолы) окраши
ваются в оранжевый цвет; пробка окрашива
ется в красный цвет.
#ЛЮМИНЕСЦЕНТНАЯ МИКРОСКОПИЯ
Метод люминесцентной микроскопии при
меняется (rде это целесообразно) для опре
деления подлинности лекарственноro pac
тительноro сырья. Преимуществом метода
является возможность еro применения для
изучения cyxoro растительноro материала, из
котороro roтовят толстые срезы или препара
ты порошка и рассматривают их в падающем
свете при освещении препарата сверху через
опак иллюминатор или объектив.
Люминесцентная микроскопия выполня
ется с помощью люминесцентных микроско
пов, снабженных специальными люминес
центными осветителями.
Приrотовление микропрепаратов
Для приrотовления микропрепаратов ис
пользуют сухое лекарственное растительное
сырье или ero порошок. Предварительное
размачивание сырья исключается, так как это
при водит к вымыванию веществ из клеток; дo
пускается лишь непродолжительное размяr
чение во влажной камере.
Листья. rотовят обычно препараты из по
рошка листьев, которые рассматривают без
включающей жидкости. Наиболее яркая лю
минесценция характерна для одревесневших
элементов сосудов жилки, механических
волокон. а также кутикулы и кутинизирован
ных оболочек различных эпидермальных об
разований (волосков, железок и др.). В эпи
дермальных клетках часто содержатся
флавоноиды, обусловливающие коричневую.
желтую или зеленовато желтую люминесцен
цию. Клетки меЗОфИf'ла содержат различные
включения желтые. rолубые. зеленовато
желтые, коричневые в зависимости от их
химическоro состава. Хлорофилл в высушен
ном растительном материале не люминесци
рует. Кристаллы оксалата кальция также не
обладают люминесценцией. При необходимо
сти приrотовления среза лист предваритель
но размяrчают во влажной камере и с помо
щью бритвы делают толстый срез (2 3 мм),
который закрепляют на предметном стекле
пластилином. Более тонкие срезы помеща
ют во включающую жидкость и накрывают по
кровным стеклом.
В качестве включающей жидкости исполь
зуют воду Р, 2лицерин Р, раствор 50 r/л поли
винилов020 спирта Р, нефлуоресцирующее
вазелиновое масло Р. Включающая жидкость
не должна растворять содержащиеся в препа
рате люминесцирующие вещества.
Травы. При анализе трав rотовят микро
препараты листьев. При необходимости при
rотовления препарата стебля еro размяrчают
во влажной камере и roтовят срезы. Толстые
срезы (2 3 мм) закрепляют на предметном
стекле с помощью пластилина и рассматри
вают без включающей жидкости, тонкие по
мещают в подходящую жидкость и накрывают
покровным стеклом. Наиболее яркую люми
несценцию имеют одревесневшие элеменп
проводящих пучков сосуды и механически.
волокна, склеренхимные клетки, встреча
ющиеся в коре и сердцевине стебля. В клетках
эпидермиса и коры часто встречаются флавu
ноиды: у некоторых видов сырья в клетках об
кладки BOKpyr проводящих пучков содержатся
алкалоиды, которые обладают разнообраз
ным свечением: синим, rолубым, зеленым, зе
леновато желтым, золотисто желтым, opaH
жево красным в зависимости от состава.
Цветки. Чаще rотовят препараты из по
рошка цветков или отдельных частей цветка
(соцветия), которые рассматривают обычно
без включающей жидкости. В цветках часто
содержатся флавоноиды, каротиноиды и
друrие вещества, обладающие флюоресцен
цией. Отчетливо видны пыльцевые зерна.
имеющие желтое, зеленовато желтое или ro
лубоватое свечение.
Плоды. rотовят обычно поперечные
срезы плода после предварительноrо раз
мяrчения во влажной камере и рассматрива
ют во включающей жидкости или без нее в
зависимости от толщины среза. Для плодов
характерна люминесценция тканей около
плодника (экзокарпия, механических клеток
мезокарпия, проводящих пучков). Отчетли
во видны секреторные каналы: ярко светится
их содержимое; клетки выстилающеro слоя
обычно имеют желтовато коричневую люми
несценцию. В содержимом каналов нередко
видны ярко люминесцирующие кристалличе
ские включения, чаще всеro желтоrо или жел
то зеленоrо цвета.
Семена. rотовят обычно поперечные
срезы семени после предварительноro раз
мяrчения во влажной камере и рассматрива
ют их во включающей жидкости или без нее
в зависимости от толщины среза. Обращают
внимание на характер люминесценции ceMeH
ной кожуры, в которой отчетливо выделяются
склеренхимные слои. Клетки эпидермиса, co
держащие слизь, обычно имеют сине rолубое
свечение. Эндосперм и ткани зародыша, бо
raTbIe жирным маслом, характеризуются rолу
бой люминесценцией.
Кора. Кору предварительно размяrчают
во влажной камере. roтовят толстые попе
речные срезы (до 3 5 мм), которые закре
2.9.1. Распадаемость таблеток и капсул
417
пляют на предметном стекле пластилином,
и рассматривают без включающей жидко
сти; тонкие срезы заключают в жидкость.
Для некоторых видов сырья характерна лю
минесценция пробковоrо слоя коры: оболоч
ки клеток пробки светятся интенсивно синим,
их содержимое TeMHO KpaCHЫM (антоци
аны). Яркое и разнообразное свечение имеют
механические элементы (лубяные волокна и
каменистые клетки): rолубое, зеленовато
rолубое, желтовато зеленое. Люминесцен
ция паренхимы коры зависит от химическоro
t<:ocTaBa. Антрацен производные обуслов
flивают яркое оранжевое или KpaCHOBaTO
оранжевое свечение. Дубильные вещества
обладают свойством «тушить» люминесцен
!.{Ию, поэтому ткани, содержащие дубильные
вещества, темно коричневоrо, почти черно
ro, цвета.
Препарат, приroтовленный из порошка
коры или соскоба, рассматривают без включа
ющей жидкости. В нем наиболее ярко видны
механические элементы.
Корни, корневища, луковицы, клубни,
клубнелуковицы. rотовят поперечные срезы,
распилы, препараты порошка или соскоба.
Срезы rотовят из материала, предваритель
но размяrченноro во влажной камере, pac
пилы (из толстых корней и корневищ) из
сухоro материала с помощью тонкой пилы
или фрезы. С помощью бритвы с поверхности
распила снимают тонкий слой для удаления
слоя клеток, покрытых пылью. Толстые срезы
и распилы (до 3 5 мм) закрепляют на пред
метном стекле пластилином и рассматрива
ют без включающей жидкости. Слой пробки
У подземных opraHoB обычно тусклый, почти
черный. Ярко люминесцируют древесины (у
корней и корневищ) и проводящие пучки, а
также склеренхимные элементы. Их свече
ние весьма разнообразно: от буровато зе
леноro, желто зеленоro до светло roлубоro
и интенсивно синеro в зависимости от вида
сырья. Еще более разнообразна люминес:'
ценция паренхимных тканей и различных ce
креторных образований (вместилищ, каналов,
ходов, млечников, различных идиобластов),
что определяется их химическим составом. В
секреторных образованиях встречаются кри
сталлические включения кумаринов, алкало
идов, флавоноидов, обладающие яркой лю
минесценцией.
В препаратах порошка видны отдельные
сосуды, rруппы механических волокон, KaMe
нистые клетки, отдельные секреторные об
разования и их обрывки, ярко люминесциру
ющие клетки паренхимы, содержащие те или
иные вещества.
Препараты в люминесцентном микроско
пе рассматривают в ультрафиолетовом свете,
наблюдая первичную (собственную) люми
несценцию.
53. За.. 1712.
2.9. ФАРМАЦЕВТИКО..
ТЕхнолоrИЧЕСКИЕ
ИСПЫТАНИЯ
01/2013:20901
2.9.1. РАСПАДАЕМОСТЬ ТАБЛЕТОК
И КАПСУЛ
Испытание на распадаемость позволяет
определить, распадаются ли таблетки или кап
сулы в пределах установленноro времени, если
они помещены в жидкую среду в эксперимен
тальных условиях, указанных ниже.
В данном испытании под распадаемостью
не подразумевают полное растворение образ
ца или даже ее активноro компонента. Считают,
что образцы распались, если любой остаток об
разца, за исключением фраrментов нераствори
мой оболочки, прилипших к сетке или к нижней
поверхности диска, если они были использова
ны, состоит из мяrкой массы без явноro плотно
ro ядра.
В случае, если длина таблеток и капсул не
превышает 18 мм, используют прибор А, для Ta
блеток и капсул большеro размера используют
прибор В.
ТЕСТ А ТАБЛЕТКИ И КАПСУЛЫ
НОРМАЛьноrо РАЗМЕРА
Прибор. Прибор включает в себя корзинку;
низкий стакан вместимостью 1 литр, высотой
(149:t11) мм и внутренним диаметром (106:t9)
мм для жидкости для распадаемости; TepMOCTa
тическое устройство для HarpeBa жидкости до
температуры от 35 0 С до З9 0 С; приспособление
для поднимания и опускания корзинки в жид
кости для распадаемости с постоянной часто
той, лежащей в диапазоне от 29 до З2 циклов
в минуту на расстояние (55:t2) мм. Объем жид
кости в сосуде должен быть таким, qтобы при
нахождении корзинки в крайнем верхнем поло
жении край проволочной сетки был поrружен не
менее чем на 15 мм ниже поверхности жидко
сти; при нахождении корзинки в самом нижнем
положении расстояние от сетки до дна хими
ческоro стакана должно составлять не менее
25 мм. Верх корзинок не должен поrружаться
в жидкость. Время движения корзинки вверх
должно быть равным времени движР,ния KOp
зинки вниз, а изменение направления движе
ния должно происходить плавно, без резких
возвратных движений. Корзинка должна дви
rаться вертикально вдоль своей оси. Не должно
быть ощутимых rоризонтальных движений или
смещения оси от вертикальной.
418
rосударственная фармакопея Республики Беларусь
Корзинка. Корзинка состоит из шести ПI.:Jзрач
ных трубок, каждая длиной (77,5:t2,5) мм, BHY"TeH
ним диаметром (21,85:t1,15) мм и толщиной стенок
(1,9:tO,9) мм; трубки удерживаются в вертикальном
положении с помощью двух пластин, каждая ди
аметром (90:t2) мм и толщиной (6,75:t1,75) мм, с 6
отверстиями, каждое диаметром (24:t2) мм, paBHO
удаленными от центра пластины и на одинаковом
расстоянии одно от друrorо. К нижней поверхностlt1
нижней пластины прикреплена плетеная сетка с
размером квадратных отверстий (2,O:tO,2) мм и дv.
аметром проволоки (0,615:t0,045) мм. Части корзи
нок собираются и жестко удерживаются с помощью
трех болтов, проходящих сквозь две пластинки. Для
предотвращения поднимания и опускания корзинки
предусмотрено подходящее приспособление pac
полаrающееся на ее оси.
Схема корзинок может несколько отличаться
с учетом спецификаций для стеклянных трубок и
плетеной сетки. Размеры корзинки приведены на
рисунке 2.9.1. 1.
Диски. Использование дисков допускается
только в случаях, rде это указано или разрешено.
Каждая трубка снабжена цилиндрическим диском
толщиной (9,5:tO,15) мм и диаметром (20, 7:t0, 15) мм.
Диск изroтовлен из подходящеro прозрачноro пла
стика с удельной массой 1, 1 1,20. Между краями
цилиндра расположены пять параллельных отвер.-
стий (2:t0,1) мм. Одно из отверстий центрировано
по оси цилиндра. Остальные отверстия располо
жены на расстоянии (6:tO.2) мм от оси на вообра
жаемых линиях. перпенпикулярных оси и парал
лельных друr ЩJуry. На боковой поверхности диска
вырезаны 4 трапецеидальных уrлубления, почти
перпендикулярных краям цилиндра. Трапецеи
дальные формы симметричны; параллельные сто--
роны совпадают С концами цилиндра и параллель
ны воображаемой линии, соединяющей центры
двух распопоженных рядом отверстий, отстоящих
на 6 мм от оси цилиндра. Параллельная сторона
трапеции на нижней поверхности цилиндра имеет
длину (1 ,6:!:О, 1) мм и ее нижняя часть лежит на pac
стоянии (1 ,6:t0, 1) мм от края окружности. Парал
лельная сторона трапеции на верхней стороне ци
линдра имеет длину (9.4:tO,2) мм и ее центр лежит
на расстоянии (2,6:tO.1) мм от края окружности. Все
поверхности диска rладкие.
Если есть указание в частной статье, диски по--
мещают в каждую трубку и проводят испытание, как
указано в разделе Методика. Размеры диска приве
дены на рисунке 2.9.1. 1.
Использование автоматическоro определе
ния с использованием модифицированных дисков
допускается в случае, если использование дисков
указано или разрешено. Такие диски должны co
ответствовать требованиям по плотности и разме
рам, приведенным в данном разделе.
Методика. В каждую из 6 трубок помещают
одну дозированную единицу и, если указано, диск.
Проводят испытание, используя указанную среду,
поддерживаемую при температуре (37:t2) ОС, в Ka
честве жидкости для распадаемости. По истечении
указанноro времени вынимают корзинку из жидко
сти И исследуют состояние дозированных единиц:
все дозированные единицы должны полностью pac
пасться. Если одна или две дозированные единицы
не распались, повторяют испытание с использова
нием 12 дополнительных дозированных единиц. 16
из 18 дозированных единиц должны распасться.
Y.O;:'3i'!1l'f.;!
:]
11.б i
.t 1.15
L ... /
I
I
I
I
I
1 I
///п/// ///-
1.Ij1.U.9
Ol
6
2.6:10.' ....o
о>
_4,.()_2 I
ф
B.'J::...D<..
90!:.2
llJblШ3: В>щ "fiu.,
'"
о
"
,<'
,,:.' " () < о ;
о . ЬvщО:.....1"'
f О О .
15-1.6'
Рисунок2.9.1. 1. ПриборА
(размеры указаны в миллиметрах)
ТЕСТ В ТАБЛЕТКИ И КАПСУЛЫ
Большоrо РАЗМЕРА
Прибор. Основная часть прибора (рисунок
2.9.1. 2) представляет собой жесткую корзинку, ко--
торая поддерживает 3 прозрачных цилиндрических
трубки длиной (77,5:t2,5) мм, внутренним диаме
тром (33,0:t0,5) мм и толщиной стенки (2,5:t0,5) мм.
Каждая трубка имеет цилиндрический диск диа
метром (31,4:tO,13) мм и толщиной (15,3:t0,15) мм,
изroтовленный из прозрачноro материала с от
носительной плотностью 1,1 1,20. В каждом
диске просверлено по 7 отверстий, каждое диаме.-
тром (3, 15:t0, 1) мм, одно из которых расположено в
центре, а остальные шесть равномерно распреде.-
лены по KPYry диаметром 4,2 мм от центра диска.
Трубки удерживаются в вертикальном положении
сверху и снизу двумя жесткими пластиковыми пла
стинами диаметром 97 мм и толщиной 9 мм с тремя
отверстиями. Отверстия paBI-.юудалены от центра и
находятся на одинаковом расстоянии друr от друrэ.
К нижней поверхности нижней пластины прикре--
плена плетеная сетка, изroтовленная из проволоки
из нержавеющей стали диаметром (0,63:tO,03) мм.
с отверстиями размером (2.0:tO,2) мм. Пластины
жестко удерживаются на расстоянии 77,5 мм ЩJyr
2.9.2. Распадаемость суппозиториев и пессариев
419
от друra с помощью вертикальных металлических
стержней, расположенных по краям. Еще один ме--
таллический стержень прикреплен к центру Bepx
ней пластины, что позволяет прикрепить корзинку к
механическому устройству, способному поднимать
и опускать ее плавно с постоянной частотой 2g.........32
цикла в минуту на расстояние (55:1:2) мм.
Корзинку поrpужают в жидкость, указанную в
частной статье, в подходящем сосуде, предпочти
тельно в химический стакан вместимостью 1 литр.
Объем жидкости в сосуде должен быть таким,
чтобы при нахождении корзинки в крайнем вepx
нем положении, проволочная сетка была поrpуже
на не менее чем на 15 мм ниже уровня жидкости;
при нахождении корзинки в самом нижнем положе--
нии, расстояние от сетки до дна химическоro cтa
кана должно составлять не менее 25 мм, а Bepx
ние открытые концы трубок должны оставаться над
rюверхностью жидкости. Используют подходящее
приспособление для поддержания жидкости при
температуре от 35 ос до З9 ОС.
Схема корзинок может несколько отличаться
с учетом спецификаций для стеклянных трубок и
плетеной сетки.
Методика. Испытание проводят на 6 таблетках
и капсулах либо с одновременным использовани
ем 2 корзинок, либо повторяя испытание. В каждую
трубку помещают по 1 таблетке или капсуле и, если
указано, диск; корзинки помещают в стакан с YKa
занной жидкостью. По истечении указанноro вре.-
мени вынимают корзинку из жидкости и исследуют
состояние таблеток или капсул. Все 6 таблеток или
капсул должны распасться.
!
I
I
I
:1
::,
со
с
G
"
O,'
I &J
I 1 1
"1
'
°1
';;1
'"
е'
С,
;
Рисунок 2.9.1. 2. Прибор В
(размеры указаны в миллиметрах)
01/2013:20902
2.9.2. РАСПАДдЕМОСТЬ
СУППОЗИТОРИЕВ
И ПЕССАРИЕВ
Испытание на распадаемость позволяет
определить, размяrчаются или распадаются
суппозитории или пессарии в пределах YCTa
новленноro времени, если они помещены в
жидкую среду в экспериментальных услови
ях, указанных ниже.
Считают, что образцы распались, если:
а) наблюдается полное растворение;
Ь) компоненты суппозитория или песса
рия разделились: расплавленные жировые
вещества собрались на поверхности жидко
сти, нерастворимые вещества осели на дно,
и растворимые компоненты растворились; в
зависимости от состава и способа приroтов
ления компоненты лекарственноro средства
MorYT быть распределены по одному или He
скольким из вышеуказанных путей;
с) размяrчение образца сопровождает
ся заметным изменением формы без полно
ro разделения компонентов; размяrчением
считается также отсутствие у суппозитория
или пессария твердоro ядра, оказывающе
ro сопротивление давлению стеклянной па
лочки;
d) наблюдается разрыв желатиновой обо
лочки ректальной или ваrинальной капсулы,
позволяющий высвобождаться ее содержимо
му;
е) на перфорированном диске не OCTa
лось осадка или осадок, который остался, co
стоит из мяrкой или пенообразной массы, не
имеющей твердоro ядра, оказывающеro co
противление давлению стеклянной палочки
(ваrинальные таблетки).
Прибор. Прибор (рисунок 2.9.2. 1) COCTO
ит из прозрачноrо стеклянноro или пластмассо
воro полоro цилиндра с подходящей толщиной
стенок, внутри котороro с помощью трех держа
телей закреплено металлическое устройство,
представляющее собой два перфорированных
диска из нержавеющей стали, каждый с 39 от
верстиями диаметром 4 мм; диаметр дисков
близок внутреннему диаметру цилиндра; диски
закреплены на расстоянии 30 мм друr от друrа.
Испытание проводят, используя три таких прибо
ра, помещая в каждый по единственному образ
цу. Каждый прибор помещают в стакан вмести
мостью не менее 4 литров, наполненный водой
с температурой от 36 ос дО 37 ОС, если иноro не
указано в частной статье. Все приборы также
MOryT быть помещены в один сосуд емкостью не
менее 12 литров. Стакан оборудован медлен
ной мешалкой и устройством, удерживающим
цилиндры в вертикальном положении не менее
чем на 90 мм ниже поверхности воды и позво
420
Тосударственная фармакопея Республики Беларусь
ляющим их переворачивать, не вынимая из под
воды.
Методика. Испытывают три суппозито ия
или пессария. Каждый образец помещают на
нижний диск устройства, устанавливают устрой
ство в цилиндр прибора и закрепляют ею. Поме
щают прибор в сосуд с водой и начинают испыта
ние. Приборы переворачивают КЭ>lЩые 1 О мин. По
истечении времени, указанноro в частной статье,
исследуют образцы. Лекарственное средство BЫ
держивает испытание, если все образы распались.
'"
I (,/ ,', ,
./; 11'
r-;; , , ..',
;
) r/ \ f;
\
о
'"
"1/
О /-
'"
/ z
-'l';.
,
I :)
i
5Ь
. I .
. 5? .
!
....
t
24
Зб
.
Рисунок 2.9.2. 1. Прибор для определения
распадаемости суппозиториев и пессариев
(размеры указаны в миллиметрах)
МЕТОД ИСПЫТАНИЯ ВАrинАЛЬНЫХ
ТАБЛЕТОК
Применяют описанный выше прибор, YCTa
новленный на держателях (см. рисунок 2.9.2. 2).
Прибор помещают в химический стакан подхо
дящеro диаметра, который содержит воду с TeM
пературой от 36 ос до 37 ОС. Поверхность воды
должна быть немною ниже верхнеro перфориро
ванноro диска. Затем с помощью пипетки прибав
ляют воду с температурой от 36 ос до 37 ос до тех
пор, пока перфорацию диска не будет покрывать
однородная пленка воды. Испытывают три ваrи
нальные таблетки. Каждую в отдельности помеща
ют на верхний диск устройства, накрывают прибор
стеклянной пластинкой, чтобы поддерживать co
ответствующие условия влажности. По истечении
времени, указанноro в частной статье, исследуют
образцы. Лекарственное средство выдерживает
испытание, если все образцы распались.
iA
,
/
С
,
/ i
i
/ i
; / О
i
/ " I
I /
/.-
/ i .-
......U...... , )
'- '---'
'Е
i
Рисунок 2.9.2. 2.
А стеклянная пластина:
В ваrинальная таблетка;
С поверхность воды;
О вода;
Е стакан.
01/2013:20903
2.9.3. ТЕСТ «РАСТВОРЕНИЕ» для
ТВЕРДЫХ ДОЗИРОВАННЫХ
ФОРМ
Данное испытание используют для определе
ния соответствия требованиям по растворению для
оральных твердых дозированных форм. В данной
rлаве под дозированной единицей понимают 1 Ta
блетку, или 1 капсулу или указанное количество.
#Если нет друrих указаний в частной статье,
проведение теста «Растворение» не является
обязательным для жевательных таблеток, поли
витаминных лекарственных средств и в друrих
случаях, для которых обоснована неинформа
тивность данноro теста.#
ПРИ БОР
Прибор 1 (Прибор с корзинкой). Прибор
состоит из: выполненною из стекла или друroro
инертною прозрачноro материала(2) сосуда, KOТO
рый может быть закрыт крышкой; мотора; вала;
цилиндрической корзинки (вращающийся эле
мент). Сосуд частично поrружен в водяную баню
подходящеro размера или наrревается с помощью
, Материал не должен сорбировать испытуемый образец. pe
аrировать или взаимодействовать с ним
2.9.3. Тест «Растворение» для твердых дозированных форм
421
специальною приспособления, например HarpeBa
тельной рубашки. Водяная баня или приспособле
ние для Harpeвa должны позволять поддерживать
в течение испытания температуру внутри сосуда
(37::1:0,5) ос и постоянное леrкое движение среды
растворения. Никакие части прибора, а также OKPy
жение, в котором arperaT расположен, не должны
создавать чувствительных движений, тряски или
вибраций за исключением плавноro вращения пе
ремешивающеro элемента. Желательно, чтобы
прибор позволял наблюдать за раствором и за пе
ремешиванием во время испытания. Сосуд BMe
стимостью 1 литр имеет цилиндрическую форму
и полусферическое дно; высота от 160 мм до
210 мм, внутренний диаметр от 98 мм до 106 мм.
Стенки наверху сосуда заrнуты. Для предупрежде
ния испарения может быть использована плотно
подоrнанная крышка(З). Вал расположен таким об
разом, чтобы еro ось не отклонялась от верти
кальной оси сосуда более чем на 2 мм в любой ее
точке, и вращение было плавным, без заметною
качания, способноro повлиять на результаты. Ис
пользуют устройство для реryлирования скорости
вращения, способное устанавливать ее и подде
живать в заданном диапазоне ::1:4 %.
Компоненты вала и корзинки изroтавливают
из нержавеющей стали (тип З16 или эквивалент
ный) соrласно спецификации, приведенной на
рисунке 2.9.З. 1.
Moryт использоваться корзинки, покры
тые слоем золота толщиной около 2,5 мкм
(0,0001 дюйма). Дозированную единицу перед
началом каждоro испытания помещают в сухую
корзинку. Расстояние между внутренней частью
дна сосуда и дном корзинки во время испытания
поддерживают равным (25::1:2) мм.
Прибор 2 (Прибор с лопастью ешалкой).
Используют тот же прибор, что описан выше, за ис
ключением тою, что в качестве перемешивающеm
элемента используют лопасть и вал. Вал располо-
жен таким образом, чтобы еro ось не отклонялась
от вертикальной оси сосуда более чем на 2 мЬ1
В любой ее точке, и вращение было плавным, без
заметною качания, способною повлиять на резуль
таты. Вертикальная осевая линия лопасти про-
ходит сквозь ось вала таким образом, что нижняя
часть лопасти совпадает с нижней частью вала.
Спецификации для лопасти приведены на рисунке
2.9.З. 2. Расстояние между внутренней частью дна
сосуда и нижней частью лопасти во время испыта
ния поддерживают равным (25::!:2) мм. Металлич&
ские или подходящие инертные жесткие лопасть и
вал должны составлять единое целое. Может быть
использована модель, состоящая из двух частей,
при условии, что во время испытания они остают
ся надежно соединенными. Лопасть и вал мешал
ки MOryт быть покрыты подходящим инертным Ma
териалом. Раствор начинают перемешивать после
тоro, как дозированная единица достиrнет дна. Для
3 Если используется крышка, она должна иметь отверстия,
позволяющие быстро поместить туда термометр и отбирать
пробы.
54 Зак 1712
от 6,3 до 6,5
или от 9.4 до 10,1
Отверстие
2.0 :1:0.5
Удерживающая пружина
с З зажимами
ПОД уrлом 120 о
I I
Свободное сечение
20.2:1: 1.0
Наружный
диаметр сетки
22.2 10 1.0
со со
<')
+. +1
со со
..... .....
C'I <')
А
222+10
.
со
( ) ('")
+1
со
L()
" '/ N
Рисунок 2.9.3. 1.
Прибор 1. Вращающаяся корзинка
(размеры указаны в миллиметрах)
1. Сетка со спаянным швом: диаметр проволоки 0,22---....о,З 1 мм,
размер ячейки 0,Зб.......(),44 мм. После сплавления сетка может
быть слеrка видоизменена.
2. Максимально допустимое отклонение для «А» не должно
превышать 1,0 мм при вращении по центральной оси с ycтa
новленной корзинкой.
предотвращения всплывания дозированной еди
ницы К ней может быть прикреплен небольшой кy
сочек инертною материала, например, несколько
витков проволочной спирали. Одно из возможных
приспособлений для поrpуж ния приведено на ри
сунке 2.9.3. 3. Moryт быть использованы и друrие
валидированные приспособления для поrpужения
дозированной единицы.
Прибор 3 (Прибор с поршневым цилин
дром). Прибор состоит из набора цилиндрических
плоскодонных стеклянных сосудов; набора CTe
клянных поршневых цилиндров; инертной raрниту
ры (из нержавеющей стали или иноro подходяще
ю материала) и сит, выполненных из подходящеro
не сорбирующею и не реаrирующеro материала,
422
rосударственная фармакопея Республики Беларусь
I
'
! :1
" . i
. /1
!:: :'j
i
i
.,''''',' i
!
N
.
:..... . "-!
" r1
;с!
' .
",
"
.
.;-.(\
14.5 (\5
t " ,,"" ........ '\. ',SS;., ....:.... '\....... '..' " '"... I
Рисунок 2.9.3. 2.
Прибор 2. Вращающаяся лопасть
(размеры указаны в миллиметрах)
Расхождение в размерахА и Внедолжно превышатьО.5 мм при
Rрашении по центральной оси, Допуски составляют :!:1 ,О ММ.
если нет друrих указаний,
и соз.данных таким образом, чтобы подходить к
верхним и нижним отверстиям поршневых цилин
дров; мотора и передаточноro механизма для co
вершения возвратно поступательных движений
цилиндров вертикально внутри сосудов и, при He
обходимости, для roризонтальноro перемещения
поршневых цили! ;дров к различным рядам cocy
дов. Сосуды чаСl ично поrружены в водяную баню
подходящеro размера. поддерживающую в течение
испытания температуру (37:tO,5) ОС. Никакие части
arperaTa, а также окружение, в котором arperaT pac
35.4 О
А
'\
\
I
положен, не должны соз.давать чувствительных
движений, тряски или вибраций за искпючением
плавноro вертикальноro возвратно поступателtr
Horo движения цилиндра. Используют устройство
для реryлирования частоты движений, способное
устанавливать ее и поддерживать в заданном ди
апазоне :t5 %. Желательно, чтобы прибор позво
лял наблюдать во время испытания за образцами
и за двиrающимися цилиндрами. Сосуды снабжа
ют колпачками, предотвращающими испарение во
время испытания. Если не указано иною, COCTaB
ляющие прибора должны соответствовать специ
фикации, приведенной на рисунке 2.9.3.-4.
Прибор 4 (Прибор с проточной кюветой).
Прибор состоит из резервуара и насоса для среды
растворения; проточной кюветы; водяной бани, под
держивающей температуру среды при (37:tO,5) ОС.
Используют кюветы специфицированноro размера.
Насос прокачивает среду растворения через
проточную кювету. Насос имеет диапазон измене
ния объема подачи растворителя от 240 мл/ч до
960 мл/ч, стандартные скорости потока 4 мл/мин,
8 мл/мин И 16 мл/мин. Насос должен обеспечивать
стабильный поток (:t5 % от номинальной скорости
потока); профиль потока синусоидальный с пуль
сацией (120:t10) пульсов в минуту. Так же можеl
быть использован насос без пульсации. Процеду
ры проведения испытаний с использованием при
бора с проточной кюветой должны быть описань:
С учетом скорости потока и возможной пульсации.
Проточная кювета (см. рисунки 2.9.3. 5 и
2.9.3. 6) изroтавливается из прозрачною инертно
ro материала, устанавливается в вертикальном
положении и имеет фильтрующую систему, пре
дотвращающую просачивание нерастворившихся
частиц из верхней части кюветы. Стандартные ди
аметры кювет 12 мм и 22,6 мм. Конусовидное дно
обычно заполнено мелкими стекпянными шари
ками диаметром около 1 мм с одним шариком ди
аметром 5 мм, расположенным в вершине конуса
для предотвращения попадания жидкости в трубку.
Для позиционирования некоторых дозированных
форм возможно наличие держателя для таблетки
(см. рисунки 2.9.3. 5 и 2.9.3. 6). Кювету помещают в
водяную баню с температурой (37:t0,5) ОС.
3.0,3,5
" ". j
,н,
_ I
I
1
,
т 11
\ 1Л\I1
!/ \\
I
З,5 .0
F
1 ..
А ;
25.26
12.0 :t 0.2
Рисунок 2.9.3. 3. Альтернативное прuспособление для П02ружения (синкер)
(размеры указаны в миллиметрах)
А КИСЛОТОУСТОЙЧИВЫЙ ПРОВОЛОЧНЫЙ зажим;
В кислотоустойчивая проволочная основа,
2.9.3.
Тест ((Растворение» для твердых дозированных форм
423
Для фиксации кюветноro отделения исполь
зуют зажим и два уплотнительных кольца. Насос
находится отдельно от ячейки растворения для
предотвращения ее вибрации, вызываемой Ha
сосом. Насос не должен располаrаться на более
высоком уровне, чем колбы резервуары. Соеди
нения трубками должны быть ПО возможности
короткими. Используют ПОДХОДящие инертные
трубки, такие как политетрафторэтиленовые, с
внутренним диаметром 1.6 мм и инертные флан
цевые соединения.
+t
со
ф
w
I 50.8:!: 1 I
i . 38,1 :t 1 I Отверстия для воздуха
0з.9:tо.1
't'---l
I
I
I
I
Колпачок,
предотвращающий
испарение
Отверстия
0 8 для воздуха
03.9:1:0.1
Сито
I 2з126
+J
О
О
Стеклянный
nopшневой
цилиндр
Сито
I I I
I I
I I
I 47:!: 1.4 I
I . I
I I
I I I
I I
I I
I I
I I
I I I
I I I
I I
I I
I I
I I
I I I
I I I I
I
I
I
I I
I
I I
I
I
I
I
I I
I I
I "
I
I I
I I ,
I I
IL
Стеклянныи сосуд
+
О
со
t
Рисунок 2.9.3. 4. Прибор 3. Стеклянный сосуд
и поршневой цилиндр
(размеры указаны в миллиметрах)
При20дность прибора. Определе
ние приrодности при бора для растворения
должно включать соответствие требованиям
по размерам и допускам, приведенным выше.
Кроме TOro, критические параметры испыта
ния, которые должны проверяться периоди
чески при использовании, включают в себя
объем и температуру среды растворения,
скорость вращения (Прибор 1 и 2), скорость
поrружения (Прибор 3) и скорость потока
среды (Прибор 4).
Приrодность характеристик прибора для
растворения определяют периодически.
'"
Фильтровальная
камера
Сито
d =0.2 w=0.45
020 0.2
'"
с5
+<
LI
022.6 :!: 0.2
Бороздка
для держателя
ез
t2Ю.В :!: 0.05
_
'
L
t '"
N
с5
+1
'"
'"
I
/
24.0 . 8 :5
Рисунок 2.9.3. 5. Прибор 4. Большая кювета
для таблеток и капсул (сверху) и большой
держатель таблеток (снизу)
(размеры указаны в миллиметрах)
424
МЕТОДИКА
rосударственная фармакопея Республики Беларусь
ПРИБОРЫ1 И 2
Твердые дозированные формы с обыч
ным высвобождением
Методика. Указанный объем среды раст
ворения (:1:1 %) помещают в сосуд указанноro
прибора. Собирают прибор, уравновешивают
среду растворения при температуре (З7:1:0,5) ос
и вынимают термометр. Испытание также может
быть проведено с термометром, помещенным в
среду растворения при условии, что доказана
эквивалентность получаемых результатов с ис
пытанием без термометра в среде растворения.
Одну дозированную единицу помещают
в прибор, стараясь избежать появления воз
душных пузырьков на поверхности дозирован
ной единицы. Испытание проводят с указанной
скоростью вращения. По истечении указанноro
временноro интервала или через каждые YCTa
новленные периоды времени отбирают пробу
из пространства, расположенноro посереди
не между поверхностью среды растворения и
r
О
r.
r.,
ФильтровальнэS1
"амера
. ' СитоCl = С.2 w = С.oQ5
r.
с
с
,"
й 2 0.1
I
борО3Д,I<Э
ДЛЯ держателя
!
1 1
\.:),.
\ 11
03
еО.8:!:О,О5
,
", r
ciiV
"'
<.
Рисунок 2.9.3. 6. Прибор 4. Малая кювета для
таблеток и капсул (сверху) и малый
держатель таблеток (снизу)
(размеры указаны в миллиметрах)
верхом вращающейся корзинки или лопасти и
расположенною не менее чем 1 см от стенки
сосуда. Если происходит неоднократный отбор
проб, вместо отбираемой аликвоты в сосуд по
мещают равный ей объем среды растворения с
температурой 37 ос или, если доказано, что BOC
полнение не является необходимым, учитыва
ют изменение объема при проведении расчетов.
Сосуды закрывают крышками и периодически
проверяют температуру среды. Проводят анализ
с помощью подходящеro метода количественно
ю определения(4). Повторяют испытание со сле
дующими дозированными единицами.
Если для отбора проб используется aBTO
матизированное оборудование или прибор MO
дифицирован каким либо иным способом, He
обходимо подтверждение тоro, что получаемые
результаты эквивалентны результатам, получа
емым с использованием прибора, описанноro в
данном разделе.
Среда растворения. Используют подходящую
среду растворения. Указанный объем COOTBeTCТBY
ет объему, отмерянному при температуре от 20 ос
до 25 ОС. Если средой растворения является буфер,
доводят еro значение рН таким образом, чтобы оно
не отличалось более чем на 0,05 от указанною зна
чения рН. Растворенные raзы MOryт вызывать обра
зование пузырьков, способных изменять результа
ты испытания. В таких случаях перед проведением
испытания они должны быть удалены(S).
Время. Если в спецификации указано только
одно время растворения, испытание может быть
проведено в более короткий промежуток BpeMe
ни при условии, что выполняются требования
по минимальному растворенному количеству.
Пробы должны отбираться только в указанное
время с точностью :1:2 %.
Твердые дозированные формы с про
лонrированным высвобождением
Методика. Как указано для твердых дозиро
ванных форм с обычным высвобождением.
Среда растворения. Как указано для твердых
дозированных форм с обычным высвоБО>IЩением.
Время. Точки отбора проб (обычно 3) указы
вают в часах.
4 Если не указано, что фильтрование не является необхо-
димым, образцы фильтруют немедленно после отбора. Ис
пользую инертный фильтр, который не вызывает абсорбции
активною вещества или не содержит экстраrируемых B
ществ, способных повлиять на анализ.
5 Может быть использован следующий способ деаэрации:
среду наrревают при аккуратном перемешивании до темпе--
ратуры около 41 ос. немедленно фильтруют под вакуумом
через фильтр с размером пор 0,45 мкм или менее при интен-
сивном перемешивании и продолжают перемешивать ПQQ,
вакуумом в течение 5 мин. Также может быть использован
иной валидированный метод удаления растворенных rазов
2.9.3. Тест «Растворение» для твердых дозированных форм
425
Твердые дозированные формы с замед
ленным высвобождением
Методика. Используют Метод А или Метод В.
МЕТОД А
Кислотная стадия. 750 мл 0,1 М pacт
вора кислоты хлористоводородной помеща
ют в сосуд и собирают прибор. Выдерживают
среду для уравновешивания при температу
ре (37:1::0,5) ОС. Помещают одну дозированную
единицу в прибор, закрывают сосуд крышкой
и проводят испытание при указанной скорости
вращения. Через 2 часа растворения в среде
0,1 М раствора кислоты хлористоводород
ной отбирают аликвоту жидкости и немедлен
но продолжают испытание, как указано в Бу
ферной стадии. Анализ аликвот проводят с
помощью подходящеro метода количествен
ною определения.
Буферная стадия. Завершают опера
ции по прибавлению буфера и доведению рН
в течение 5 мин. Проводя испытание при YKa
занной скорости вращения, в сосуд прибавля
ют 250 мл 0,20 М раствора натрия фосфата
додека2идрата Р, предварительно подоrрето
ro до температуры (37:1::0,5) ОС. При необходи
мости доводят 2 М раствором кислоты хлори
стоводородной Рили 2 М раствором натрия
2идроксида до значения рН (6,8:1::0,05). Про
должают испытание в течение 45 мин или в Te
чение указанноro времени. По истечении YKa
занноro периода времени отбирают аликвоту
жидкости и проводят анализ с помощью подхо
дящеro метода количественноro определения.
МЕТОД В
Кислотная стадия. 1000 мл 0,1 М pacт
вора кислоты хлористоводородной помеща
ют в сосуд и собирают прибор. Выдерживают
среду для уравновешивания при температуре
(37:1::0,5) ОС. Помещают одну дозированную еди
ницу в прибор, закрывают сосуд крышкой и пр
водят испытание при указанной скорости Bpa
щения. Через 2 часа растворения в среде 0,1 М
раствора кислоты хлористоводородной отби
рают аликвоту жидкости и немедленно продол
жают испытание, как указано в Буферной стадии.
Анализ аликвот проводят с помощью ПОДходяще
ro метода количественноro определения.
Буферная стадия. Используют заранее
приroтовленный и уравновешенный при темпе
ратуре (37:1::0,5) ос буфер. Из сосуда выливают
кислоту и прибавляют 1000 мл буферноro paCT
вора со значением рН 6,8, приrlJтовленноrо CMe
шиванием 3 объемов 0,1 М раствора кисло
ты хлористоводородной с 1 объемом 0,20 М
раствора натрия фосфата додека2идрата Р,
доведенноro при необходимости 2 М pacтвo
ром кислоты хлористоводородной Рили 2 М
раствором натрия 2идроксида до значения
рН (6,8:1::0,05). Этот же результат может быть
55 За.. 1712.
достиrнут путем замены сосуда с кислотой на
друroй сосуд, содержащий буферный раствор,
и перенесением дозированной единицы в этот
сосуд. Продолжают испытание в течение 45 мин
или в течение указанноro времени. По истечении
указанноro периода времени отбирают аликвоту
жидкости и проводят анализ с помощью подхо
дящеrо метода количественноro определения.
Время. Если не указано иноro, все BpeMeH
ные периоды должны быть соблюдены с точно
стью :1::2 %.
ПРИБОР 3
Твердые дозированные формы с обыч
ным высвобождением
Методика. Указанный объем среды paCT
ворения (:1::1 %) помещают в каждый сосуд при
бора. Собирают прибор, уравновешивают среду
растворения при температуре (З7:1::0,5) ос и BЫ
нимают термометр. Помещают по одной дозиро
ванной единице в каждый поршневой цилиндр,
стараясь избежать образования пузырьков воз
духа на поверхности дозированных единиц, и
немедленно включают прибор. При поднятии и
поrружении поршневоrо цилиндра общее pac
стояние, на которое он дв И rается , должно co
ставлять 9,9 10, 1 см. По истечении указанноro
временноro интервала или через каждые YCTa
новленные периоды времени отбирают пробу
из пространства, расположенноrо между по
верхностью среды растворения и дном каждоro
сосуда. Анализ проводят, как указано. При He
обходимости повторяют испытание на дополни
тельных дозированных единицах.
Вместо отбираемой аликвоты в сосуд помеща
ют равный ей объем среды растворения с темпе
ратурой 37 ос или, если доказано, что восполнение
не является необходимым, учитывают измене
ние объема при проведении расчетов. Сосуды на
время проведения испытания закрывают колпачка
ми и периодически проверяют температуру среды.
Среда растворения. Как указано для TBep
дых дозированных форм с обычным высвобож
дением для Прибора 1 и 2. .
Время. Как указано для твердых дозирован
ных форм с обычным высвобождением для При
боров 1 и 2.
Твердые дозированные формы с про-
лонrированным высвобождением
Методика. Как указано для тверДhlХ дозиро
ванных форм с обычным высвобождением для
Приборов 3.
Среда растворения. Как указано для TBep
дых дозированных форм с пролонrированным
высвобождением для Приборов 1 и 2.
426
rосударственная фармакопея Республики Беларусь
Время. Как указано для твердых дозироранных
форм с пролонrированным высвобо>мениеrv; для
Приборов 1 и 2.
Твердые дозированные формы с замед-
ленным высвобождением
Методика. Как указано для твердых дози
рованных форм с замедленным высвобо>мени
ем для Приборов 1 и 2. метод В, используя один
ряд сосудов для среды кислотной стадии, а друroй
ряд для буферной стадии. Используют указан
ный объем среды (обычно 300 мл).
Время. Как указано для твердых дозирован
ных форм с замедленным высвобождением для
Приборов 1 и 2.
ПРИБОР 4
Твердые дозированные формы с обычным
высвобождением
В указанную кювету помещают стеклянные
шарики. Сверху стеклянных шариков или, если YKa
зано, в проволочный держатель помещают одну ДО--
зированную единицу. Присоединяют фильтрующий
элемент и соединяют части с помощью подходящеro
фиксирующеro приспособления. С помощью насоса
через дно кюветы подают среду растворения при
температуре (37:tO.5) ос со скоростью, измеренной
с точностью 5 %. Через каждые установленные пе.-
риоды времени собирают фракции элюата. Анализ
проводят. как указано. Повторяют испытание на ДО--
полнительных дозированных единицах.
Среда растворения. Как указано для твердых
дозированных ф:>рм с обычным высвобо>мением
для Приборов 1 И 2.
Время. Как указано для твердых дозирован
ных форм с обычным высвобождением для При
боров 1 и 2.
Твердые дозированные формы с пролон
rированным высвобождением
Методика. Как указано для твердых дозиро
ванных форм с обычным высвобо>мением для
Прибора 4.
Среда растворения. Как указано для твердых
дозированных форм с обычным высвобо>мением
для Прибора 4.
Время. Как указано для твердых дозированных
форм с обычным высвобождением для Прибора 4.
Твердые дозированные формы с замед.
ленным высвобождением
Методика. Как указано для твердых дозирован
ных форм с замедленным высвобо>мением для При
боров 1 и 2, метод В, используя указанные среды.
Время. Как указано для твердых дозированных
форм с замедленным высвобо>мением для Прибо--
ров 1 и 2.
ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ
Твердые дозированные лекарственные
формы с обычным высвобождением
Если нет друrих указаний в частной статье,
испытуемое лекарственное средство выдержива
ет требования теста «Растворение», Korдa коли
чество растворившеroся действующеro вещества
соответствует таблице 2.9.3. 1. Количество Q
нормируемое количество растворившеroся дей
ствующеro вещества, выраженное в процентах от
номинальноro содержания (содержания, указанно
ro в разделе «Состав»); кроме этоrо, значения 5 %,
15 % и 25 % выражены в процентах от номинально--
ro содержания и имеют ту же размерность, что и О.
Если результат не соответствует уровню 81' то про
должают испытание в соответствии с уровнем 82'
если результат не соответствует уровню 82' то про
должают испытание в соответствии с уровнем 8 з .
Таблица 2.9.3. 1
Количество Критерий
Уровень испытуемых
единиц приемлемости
8, 6 Каждая единица не
I менее О+5 %
82 I 6 Среднее значение
I из 12 единиц (8,+82)
равно или более О,
и ни одной единицы
менее O 15 %
8 з 12 Среднее значение из
24 единиц (8,+82+8)
равно или более О,
не более 2 единиц
менее O 15 % и ни
одной единицы менее
a 25 %
Твердые дозированные лекарственные
формы с пролонrированным высвобождением
Если нет друrих указаний в частной статье,
испытуемое лекарственное средство выдержива
ет требования теста «Растворение», Korдa коли
чество растворившеrося действующеro вещества
соответствует таблице 2.9.3. 2. Если результат не
соответствует уровню L1' то продолжают испыта
ние в соответствии с уровнем L2' если результат
не соответствует уровню L2' то продолжают ис
пытание в соответствии с уровнем L з . Предель
ные значения количества растворившеroся дей
ствующеro вещества выражают в процентах от
номинальноro содержания. ПреД13льные значе
ния включают ка>мое значение о; нормиру-
емое количество растворившеroся действующеro
вещества в ка>мый заданный момент времени. В
случае если нормируется более чем 1 интервал
значений, критерии приемлемости применяются
отдельно к каждому интервалу.
2.9.3. Тест «Растворение» для твердых дозироваННblХ форм
427
Количество
а! ИCllьпувмых
единиц
L 1 6
L 2 6
L3
Таблица 2.9.3. 2
Критерий приемлемости
Ни одно индивидуальное
значение не лежит за преде
лам и каждоro заданною ин
тервала значений и ни одно
ИНдивидуальное значе
ние не должно быть менее
нормируемоro значения на
момент завершения испы
тания
Среднее значение из 12
единиц (L 1 +Lz) не лежит
за пределами каждоro за
данноro интервала значе
ний и среднее значение не
должно быть менее норми
pyeMoro значения на момент
завершения испытания; ни
одно Индивидуальное зна
чение не выходит за пре
делы заданноrо интервала
значений более чем на 1 О %
от номинальноro содержа
ния; и ни одно индивиду
альное значение не должно
быть менее нормируемоro
значения на момент завер
шения испытания более чем
на 1 О % от номинальноro co
держания
12
Среднее значение из 24
единиц (L 1 +L 2 +L з ) не лежит
за пределами каждоrо за
данноro интервала значе
ний и среднее значение не
должно быть менее норми
pyeMoro значения на момент
завершения испытания; не
более 2 из 24 единиц MOryT
выходить за пределы зqдан
Horo интервала значений
более чем на 1 О % от HO
минальноrо содержания; не
более 2 из 24 единиц MOryT
выходить за пределы задан
Horo интервала значений
более чем на 1 О % от HO
минальноrо содержания на
момент завершения испыта
ния; ни одно ИНДивидуаль
ное значение не выходит за
пределы заданноro интерва
ла значений более чем на
20 % от номинальноro co
держания или не должно
быть менее нормируемоro
значения на момент завер
шения испытания более чем
на 20 % от номинальноro co
держания
Твердые дозированные лекарственные
формы с замедленным высвобождением
Кислотная стадия. Если нет друrих YKa
заний в частной статье, испытуемое лекар
ственное средство выдерживает требования
этой части теста «Растворение», Korдa коли
чество высвободившеroся/растворившеrося
действующеrо вещества в процентах от HO
минальною содержания соответствует табли
це 2.9.3. 3. Если результат обеих стадий (кис
лотной и буферной) не соответствует первым
двум уровням (А 1 , А 2 И В 1 , В), то продолжают
испытание в соответствии с третьим уровнем
(А з и В).
Таблица 2.9.3. 3
.а Количество
ж
ф Критерий приемлемости
ID испытуемых
о
а. единиц
>-
А 1 6 Ни одно ИНдивидуальное
значение степени paCTBope
ния не превышает 1 О %
А 2 6 Среднее значение степени
растворения из 12 единиц
I (A1+Az) не превышает 10 %,
и НИ одно индивидуальное,
значение степени paCTBope
ния не превышает 25 % I
I
Аз 12 Среднее значение степени
растворения из 24 единиц
(А 1 +Az +А з ) не превышает
1 О % и НИ одно индивиду
альное значение степени
растворения не превышает
25 %
Буферная стадия. Если нет друrих YKa
заний в частной статье, испытуемое лекар
ственное средство выдерживает требова
ния теста «Растворение», Korдa количество
высвободившеrося/растворившеrося дей
ствующеro вещества соответствует таблице
2.9.3. 4, rде Q нормируемое общее коли
чество растворившеroся действующеro Be
щества в ходе обеих стадий (кислотной и
буферной), выраженное в про центах от HO
минальноro содержания (кроме этоro, значе
ния 5 %, 15 % и 25 % выражены в про центах
от номинальноro содержания и имеют ту же
размерность, что и О). Если нет друrих YKa
заний в частной статье, то значение Q при
нимают равным 75 %. Если результат обеих
стадий (кислотной и буферной) не COOTBeT
ствует первым двум уровням (А 1 , А 2 И В1' BJ,
то продолжают испытание в соответствии с
третьим уровнем (Аз и в з )'
428
Таблица 2.9.3-А
rосударственная фармакопея Республики Беларусь
.о
:z: Количество
CII
ш испытуемых Критерий приемлемости
о
Q. единиц
>-
В 1 6 Каждая единица не менее
О+5%
В 2 6 Среднее значение из 12
единиц (В, +В 2 ) равно или
более О, и ни одной едини
цы менее O 15 %
В з 12 Среднее значение из 24
единиц (В, +В 2 +В з ) равно или
I более О, не более 2 единиц
менее o 15 %, и ни одной
! единицы менее O 25 %
Рекомендации по испытанию «Pacтвope
ние» приведеНbI в общей статье 5. 17. 1.
01/2013:20904
2.9.4. ТЕСТ «РАСТВОРЕНИЕ»
ДЛЯ ТРАНСIJЕРМАЛЬНЫХ
ПЛАСТЫРЕИ
Данный тест используется для определе
ния степени растворения действующих веществ
трансдермальных пластырей.
1. МЕТОД СБоРноrо ДИСКА
Оборудование. Используют мешалку и KOH
струкцию сосуда прибора с лопастью мешал
Сетка из нержаееюшеи стали
с размером отверстии 125 мкм
4.5 мм
1'-'1
'.
'-J
LJ 1 З,З мм
... 41,2 мм
Рисунок 2.9.4. 1. Сборный диск
кой, описанноro в тесте «Растворение» TBep
дых дозированных форм (2.9.3) с добавлением
сборноro диска из нержавеющей стали (ССД
сборный стальной диск) в виде сетки с разме
ром отверстий 125 мкм (рисунок 2.9.4. 1).
ССД удерживает систему на дне сосуда и
спроектирован таким образом, чтобы миними
зировать мертвый объем между ССД и дном
сосуда. На ССД параллельно низу лопасти Me
шалки крепится пластырь поверхностью BЫ
свобождения кверху. Во время испытания
расстояние между низом лопасти мешалки и по
верхностью пластыря в ССД должно составлять
(25:1:2) мм (см. рисунок 2.9.4. 2). Испытание BЫ
полняют при температуре (32:1:0,5) ОС. Во время
испытания сосуд следует закрывать, чтобы избе
>t.<aTb испарения.
Методика. Помещают указанный в част
ной статье объем среды растворения в сосуд
и доводят температуру до (32:1:0,5) ОС. Пла
стырь помещают на ССД, убедившись в том,
чтобы поверхность высвобождения пласты
ря была максимально распластана на диске.
Пластырь может крепиться к ССД с помощью
клея или двусторонней клейкой ленты. Пред
варительно следует проверить, не влияют ли
клей или лента на результаты испытания и не
адсорбируются ли на них действующие веще
ства. Прижимают пластырь поверхностью BЫ
свобождения кверху на адrезивную сторону
ССД таким образом, чтобы он не выходил за
края диска. Для этоro, а также для тоro, чтобы
убедиться в однородности лекарственноro
средства и равномерности еro распределе
ния на поверхности, для испытания можно OT
резать точно отмерянную часть пластыря. Эти
же действия MorYT использоваться для дости
жения необходимых условий поrружения. Но
эти действия не MorYT быть применены к пла
стырям мембранноro типа. Пластырь, прикре
пленный к ССД, помещают на дно сосуда по
верхностью высвобождения пластыря вверх.
Немедленно включают мешалку, например, со
скоростью 100 об/мин. Через определенные
промежутки времени отбирают пробу из участ
ка между поверхностью среды растворения и
верхушкой лопасти мешалки не менее чем в
1 см от стенки сосуда.
Анализируют пробу, при необходи
мости учитывают поправку на изменение
объема. Повторяют испытание с несколькими
пластырями.
2. МЕТОД ЯЧЕЙКИ
Оборудование. Используют мешалку и KOH
струкцию сосуда прибора с лопастью мешал
кой, описанноro в тесте «Растворение» твердых
дозированных форм (2.9.3) с добавлением экс
тракционной ячейки (ячейка).
Ячейка выполнена из химически инертноrо
материала и состоит из основания, Крblшки И.
2.9.4. Тест «Растворение» для трансдермальных пластырей
429
Сосуд для
растворения
Лопасть мешалка
д
Сборный диск
Рисунок 2.9.4_ 2. Лопасть мешалка и диск
при необходимости, мембраны, которая поме
щается на пластырь для ero изоляции от среды,
которая может повлиять на физико химические
свойства пластыря (рисунок 2.9.4. З).
.. 20 мм
З2ММ
.. 40 мм
.
.. rайl<.И
50 мм
.. Крышка
: 8.5 мм
c:
.. Мембрана
2.6 мм у
: 9,6 мм
... боЛТЫ
.. Уплотнительное
кольuо
.. Основание
52 мм
21.23 см'
70.5 мм
.27 ММ
.З8мм
...45 мм
..52 мм
Рисунок 2.9.4. З. Экстракционная ячейка
Основание. Центральная часть OCHOBa
ния имеет полость, в которую помещается пла
стырь. rлубина полости 2,6 мм, диаметр
должен соответствовать размеру испытуемоro
пластыря. Можно использовать следующие ди
аметры: 27 мм, 38 мм, 45 мм и 52 мм, что co
ответствует объемам 1,48 мл, 2,94 мл, 4,13 мл
и 5,52 мл.
56. З_к. 1712.
Крышка. Крышка имеет отверстие в центре,
диаметром в соответствии с размером испытуемо
ro пластыря. Таким образом, пластырь будет распо--
лаraться точно в центре, а поверхность высвобож
дения будет оrpаничена. Используются следующие
диаметры: 20 мм, З2 мм, 40 мм и 50 мм, что COOT
ветствует площадям 3,14 см 2 , 8,ОЗ см 2 , 12,56 см 2 и
19,63 см 2 . Крышка удерживается с помощью raeK,
накрученных на болты, вставленные в основание.
Ме>tЩу крышкой и основанием помещается уплот
нительное кольцо, которое надевается прямо на
сосуд.
Экстракционная ячейка. Ячейка позволяет
удерживать пластырь поверхностью высвобо>tЩе
ния кверху в roризонтальном положении параллел
но нижнему краю лопасти--мешалки. Расстояние
ме>tЩу лопастью и поверхностью пластыря должно
составлять (25:1:2) мм (см. рисунок 2.9.4.-4). Испы
тание выполняют при температуре (32:1::0,5) ос. Во
время испытания сосуд следует закрывать, чтобы
избежать испарения.
Методика. Помещают указанный в частной
статье объем среды растворения в сосуд и доводят
до необходимой температуры. Пластырь помеща
ют поверхностью высвоБО>tЩения вверх точно по
центру ячейки. Закрывают ячейку, если необходи
мо, используют rидрофобное вещество (например,
вазелин) для смазывания плоских поверхностей
для более плотноro соединения. Помещают ячейку
на дно резервуара. Немедленно включают мешал
ку, например, со скоростью 100 об/мин. Через опре
деленные промежутки времени отбирают пробу из
участка ме>tЩу поверхностью среды растворения и
верхушкой лопасти мешалки не менее чем в 1 см
от стенки сосуда.
Анализируют пробу, при необходимости учи
тывают поправку на изменение объема. Повторяют
испытание с несколькими пластырями.
Лопасть мешалка
Сосуд
для растворения
25:1:2 мм
Экстакционная
ячейка
Рисунок 2.9.4А.
Мешалка над экстракционной ячейкой
(размеры указаны в миллиметрах)
430
/осударственная фармакопея Республики Беларусь
3. МЕТОД ВРАЩАЮЩЕrося ЦИЛИh,1РА
Оборудование. Используют прибор ..:; ло
пастью мешалкой, описанный в тесте «PaCTBO
рение» твердых дозированных форм (2.9.3).
Мешалку и вал заменяют на цилиндрический
вращающийся элемент из нержавеющей стали
(цилиндр) (рисунок 2.9.4. 5). Перед началом
испытания пластырь помещают в цилиндр
Расстояние между внутренней поверхностью
дна сосуда и цилиндром в процессе испыта
ния составляет (25f:2) мм. Испытание выпол
няют при температуре (32:tO,5) ос. Во время
испытания сосуд следует закрывать, чтобы из
бежать испарения
Методика. Указанное в частной статье
количество среды растворения помещают в
сосуд и доводят до температуры (32:t:O,5) ос.
Удаляют защитную ленту и помещают пла
стырь клейкой стороной на кусочек инерт
ной пористой мембраны. Размер мембраны
со всех сторон до.r:жен быть на 1 см больше
пластыря. Пластырь помещают на чистую по
верхность таким образом, чтобы с ней KOHTaK
тировала мембрана. Можно использовать два
способа прикрепления пластыря к цилиндру:
наносят подходящий клей на мембрану
и при необходимости на заднюю поверхность
пластыря:
используют двустороннюю клейкую
ленту. которую крепят к внешней стенке ци
линдра.
Несильно надавливая, тщательно прикре
пляют пластырь нелипкой стороной к цилин
дру так. чтобы поверхность высвобождения
находилась в контакте со средой paCTBope
ния и длинная ось пластыря соответствовала
окружности цилиндра.
Предварительно следует проверить, не
влияют ли клей или лента на результаты испы
таний и e адсорбируются ли на них действую
щие вещества.
Цилиндр помещают в прибор и немедлен
но включают мешалку, например, со скоростью
100 об/мин. Через определенные промежутки
времени отбирают пробу из участка между по
верхностью среды растворения и верхушкой
вращающеrося цилиндра не менее чем в 1 см
от стенки сосуда.
Анализируют пробу, при необходимости
учитывают поправку на изменение объема.
Повторяют испытание с несколькими пласты
рями.
Оценка результатов. Лекарственное
средство выдР.рживает испытание, если коли
чество действующеro вещества, высвобожден
ноro из пластыря, во временных точках отбора
проб, выраженное в количестве на единицу
площади в единицу времени, соответствует
предписанным пределам, указанным в част
ных статьях.
A ....Q: А
. <ЭОd ) . :
. .. 1.270
Цe'ыo€ О ft:'(фСfИ .циамеI:::ЮМ ,.... &7 .........:.....
11' pP.Hr-IС''''f''РНО :JаUЮf:f,f:Леt1ы '1
ПО О РУi!<I..юст.1 !1 'r1MeTpo .! , 2222...1 ' '-Oll-; 5- ?;iJ.lfC1il'<'t
254(1p').,:\;,rr;;M63... =O - C.94 .C1
1\ повеРХ'-ЮСН1 6.j 4<> т О < JiШi
. .
.
tJ.;Зt-:сиr.Аап",,-.Ы.1 t: ;:.., ,c а 300
.. :-:::tl3.967
--.r "2 '..
'2.1З.j .:
l......
j :il . 3.670
2 0 40
nOf РЕШНОСТЬ :10,0127 ...... :
3.AI-' лючпr'lЬt---+АЯ .
05P.4[. .Tf\t.. Об . ЖИРИЕаl.('?Т ." .--" 9.3С3
.:t ; ' ' .::: : ::,p f 5.7 , ' . 12 .
саЛt>
. 2.222 .
3Т8 ceК!..;,,1
<=V1,2т-e;.:;,
1r;юль у€тс
Д!"(; БОf1ЬШ !)(
СИ l('М
Рисунок 2.9.4. 5. Вращающийся цилиндр
(размеры указаны в сантиметрах)
01/2013:20905
2.9.5. ОДНОРОДНОСТЬ МАССЫ ДЛЯ
Единицыдозировднноrо
ЛЕКАРСТВЕнноrо СРЕДСТВА
20 единиц дозированноrо лекарственно
ro средства или содержимое каждоro из 20
контейнеров, в случае однодозовых лекар
ственных средств в индивидуальных контей
нерах, отбирают по статистически обоснован
ной схеме, взвешивают каждую в отдельности
и рассчитывают среднюю массу. Лекарствен
ное средство считают выдержавшим испы
тание, если не более двух индивидуальных
масс отклоняются от средней массы на вели
чину, превышающую значение, указанное в
таблице 2.9.5. 1. При этом ни одна индивиду
альная масса не должна отклоняться от cpeд
ней массы на величину, в два раза превыша
ющую значение, указанное в таблице 2.9.5. 1.
Таблица 2.9.5. 1
I Лекарственная Допусти
форма Средняя масса мое отклоне
ние, %
!
I 80 Mr и менее 10
Таблетки (без I
оболочки и по I Более 80 Mr, но 7,5
крытые пленоч менее 250 Mr
ной оболочкой) I I
250 Mr или 5
; более !
;
,
. .. . . .. ..... . . L .
2.9.6. Однородность содержания действующеzо вещества в
431
I Лекарственная Допусти-
Средняя масса мое отклоне-
I форма ние, %
I Капсулы, rpaHY Менее ЗОО Mr 10
I лы (без оболоч
I ки, однодозо
вые) и порошки 300 Mr и более 7,5
i (однодозовые)
,
, Порошки для
приroтовления
лекарственных
средств для па Более 40 Mr 10
рентерально
ro применения*
(однодозовые)
Суппозитории и Для всех 5
пессарии случаев
Порошки для Менее 300 Mr 10
приroтовления
rлазных капель
и при мочек 300 Mr и более 7,5
. Если средняя масса равна 40 Mr и менее, лекарственное
средство подлежит испытанию на однородность содержания
действующею вещества в единице дозированною средства
(2.9.6).
Для капсул и порошков для приroтовления
лекарственных средств для парентеральноro
применения испытание про водят, как описано
ниже.
#Данное испытание не проводится для поли
витаминных лекарственных средств и для лекар
ственных средств, содержащих микроэлементы,
если нет друrих указаний в частной статье.#
КАПСУЛЫ
Взвешивают невскрытую капсулу. Затем
вскрывают капсулу таким образом, чтобы не
была потеряна какая либо часть оболочки, и
удаляют как можно полнее ее содержимое. В
случае капсул с мяrкой оболочкой промывают
оболочку подходящим растворителем и OCTaB
ляют на воздухе до удаления запаха раствори
теля. Затем взвешивают оболочку. По разности
взвешиваний рассчитывают массу содержимоro
капсулы. Повторяют процедуру с друrими 19 кап
сулами.
ПОРОШКИ ДЛЯ приrОТОВЛЕНИЯ
ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ
ПАРЕНТЕРАльноrо ПРИМЕНЕНИЯ
Удаляют бумажную этикетку с поверхности
контейнера. Контейнер моют снаружи и сушат.
Затем контейнер вскрывают и тотчас взвеши
вают. Осторожно постукиванием освобождают
контейнер от содержимоro как можно полнее,
ополаскивают еro, если необходимо, водой Р и
затем 96 % спиртом Р и сушат при температу
ре от 100 ос до 105 ос в течение 1 ч или, если
природа контейнера не позволяет использо
вать наrревание при этой температуре, сушат
при более низкой температуре до постоянной
массы. Охлаждают в эксикаторе и взвешивают.
По разности взвешиваний рассчитывают массу
содержимоro контейнера. Повторяют процедуру
с остальными 19 контейнерами.
01/2013:20906
2.9.6. ОДНОРОДНОСТЬ
со,gЕРЖАНИЯ
ДЕИСТВУЮЩЕrо ВЕЩЕСТВА
В ЕДИНИЦЕ ДОЗИРОВАнноrо
ЛЕКАРСТВЕнноrо СРЕДСТВА
Испытание на однородность содержания
действующею вещества в единице дозирован
ною лекарственноro средства основывается
на количественном определении содержания
в индивидуальных однодозовых единицах ле
карственноro средства с целью выяснения, Ha
ходится ли это содержание внутри пределов,
установленных по отношению к среднему coдep
жанию в испытуемом образце.
Данное испытание не про водится для поли
витаминных лекарственных средств и для лекар
ственных средств, содержащих микроэлементы,
если нет друrих указаний в частных статьях.
Метод. Используя аналитическую методи
ку. указанную в частной статье, определяют co
держание действующеro вещества в каждой из
1 О дозированных единиц лекарственноrо cpeд
ства, отобранных по статистически обоснован
ной схеме.
Применяют критерии тестов А. В или С, как
указано в статье для испытуемой дозированной
формы.
ТЕСТ А
Таблетки, порошки для при20товления
лекарствеННblХ средств для парентерально
20 применения, 2лаЗНblе вставки, суспензии
для инъекций. Лекарственное средство Bыдep
живает испытание, если содержание в каждой
еro однодозовой единице находится в пределах
85 115 % от среднеro содержания. Лекарствен
ное средство не выдерживает испытание, если
содержание более чем в одной единице выходит
за вышеуказанные пределы или если содержа
ние хотя бы в одной единице выходит за преде
лы 75 125 % от среднеro содержания.
Если содержание в одной единице лекарствен
ною средства выходит за пределы 8 115 %, но
находится в пределах 7 125 %, определяют co
держание в каждой из 20 дополнительных OДHO
дозовых единиц лекарственною средства, oтo
бранных по статистически обоснованной схеме.
Лекарственное средство выдерживает испытание,
если содержание не более чем в одной из проа
нализированных 30 единиц выходит за пределы
432
rосударственная фармакопея Республики Беларусь
' 10'0.:!:0.1 мм
+ I
,1<1 287.0 4.0 мм :-..
(внутренний) I \
I -\
. \\
. / '\\
1/1 , / \\;
'1: 11\
* t
111 N ;11
: !' J I . 1'1
, ' 11. ", ,!!
", 80.5.:!: 5.0 мм ! :i'.... /jI
\\:- (внутренний /11". /1/
'\ \\ :ади у : "",. JI
' '' 1/ .O!o ""
7
С З02.5 :1: 4.0 мм
Высота I
падения
1
(внутренний)
{ "
" ,
Рисунок 2.9.7. 1. Прибор для определения прочности таблеток на истирание
38.0.:!: 2.0 мм
8 115 % и ни в одной единице не выходит за пре
делы 7 125 % от среднеro содержания.
ТЕСТ В
Капсулы, порошки для при20товления ле
карственных средств не для парентерально
20 применения, 2ранулы, суппозитории, пес
сарии. Лекарственное средство выдерживает
испытание. если содержание не более чем в
одной единице выходит за пределы 85 115 %
и ни в одной единице не выходит за пределы
75 125 % от среднеro содержания в лекар
ственном средстве. Лекарственное средство
не выдерживает испытание, если содержание
более чем в трех единицах выходит за пре
делы 85 115 % от среднеro содержания или
если хотя бы в одной единице выходит за пре
делы 75 125 % от среднеro содержания.
Если содержание в двух или трех единицах
лекарственноrо средства выходит за пределы
85 115 %, но находится в пределах 75 125 %,
определяют содержание в каждой из 20 допол
нительных однодозовых единиц лекарственноro
средства, отобранных по статистически обосно
ванной схеме. Лекарственное средство Bыдep
живает испытание, если содержание не более
чем в трех из проанализированных 30 единиц
выходит за пределы 85 115 % и ни в одной
единице не выходит за пределы 75 125 % от
среднеro содержания.
ТЕСТ С
Т рансдермальные пластыри. Леl{арствен
ное средство выдерживает испытание, если
среднее содержание в 1 О однодозовых едини
цах находится в пределах 90 11 О % от coдep
жания, указанноro в разделе «Состав», И если
содержание в каждой из 1 О единиц находится в
пределах 75 125 % от среднеro содержания.
01/2013:20907
2.9.7. ПРОЧНОСТЬ ТАБЛЕТОК БЕЗ
ОБОЛОЧКИ НА ИСТИРАНИЕ
В данной статье приведено руководство для
определения истираемости таблеток без обо
лочки. Описанное испытание обычно приroдно
для большинства таблеток. Определение проч
ности таблеток на истирание дополняет друrие
данные о прочности, например, прочность Ta
блеток на излом.
Используют барабан с внутренним диаме
тром от 283 мм до 291 мм и rлубиной от 36 мм до
40 мм, изroтовленный из прозрачноro синтетиче
скоro полимера; внутренние поверхности бараба
на должны быть отполированы и не должны элек
тризоваться (см. рисунок 2.9.7. 1). Одна сторона
барабана съемная. При ка>tЩом обороте барабана
таблетки приводятся в движение посредством изо--
rнутой лопасти с внутренним диаметром от 75,5 мм
до 85,5 мм, расположенной ме>tЩу центром бара
бана и еro наружной стенкой. Внешний диаметр
центральноro кольца от 24,5 мм до 25,5 мм. Ба
рабан крепится к roризонтальной оси устройства,
обеспечивающеrо скорость вращения около (25:t1)
об/мин. Таким образом, при ка>tЩОМ обороте ба
рабана таблетки падают, переворачиваясь или
скользя, на стенку барабана или друr на друrа.
При массе одной таблетки 650 Mr или менее
для испытания берут такое количество целых
таблеток, чтобы их общая масса находилась как
можно ближе к 6,5 r. При массе одной таf)летки
более 650 Mr для анализа используют 1 О целых
таблеток. Перед испытанием таблетки тщатель
но обеспыливают. Затем таблетки точно взвеши
вают и помещают в барабан. После 100 оборотов
барабана таблетки извлекают, снова тщательно
обеспыливают и точно взвешивают.
2.9.9. Измерение консистенции методом пенетрометрии
4ЗЗ
Обычно испытание проводят QЦИн раз. Если
по окончании испьrrания обнаруживается явное
присутствие треснувших, расколотых или поло--
манных таблеток, считают, что лекарственное
средство не выдержало испытание. Если результа
ты испьrrания тяжело интерпретировать или если
потеря в массе получилась большей, чем необхо--
димо, испытание повторяют еще два раза и нахо--
дят среднее из трех полученных результатов. В кa
честве максимальной потери в массе (полученной
в одном испытании или средней, рассчитанной по
трем испытаниям) для большинства лекарствен--
ных средств обычно принимают значение 1,0 %.
Если размер таблеток или их форма приво--
дит к получению невоспроизводимых результа
тов, устанавливают основание барабана таким
образом, чтобы оно находилось под уrлом 100 к
rоризонтали для тоro, чтобы таблетки не образо
вывали rруппы, Korдa они лежат вплотную друr к
друry, и падали свободно.
Шипучие таблетки и таблетки для разже
вывания MOryт иметь иные требования к исти
раемости. В случае rиrроскопичных таблеток
испытание необходимо проводить в условиях
контролируемой влажности окружающей среды.
Допускается использование приборов с ба
рабаном с двойной лопастью или с более чем с
одним барабаном для проведения OДHOBpeMeH
ноro испытания нескольких образцов.
01/2013:20908
2.9.8. ПРОЧНОСТЬ ТАБЛЕТОК
НА СЖАТИЕ
Испытание позволяет определить прочность
таблеток на сжатие при определенных условиях
путем измерения .силы, необходимой для разру
шения таблеток.
ПРИБОР
Прибор представляет собой два располо
женные друr против друrа зажима, один из KO
торых может перемещаться по направлению к
друroму. Плоскости поверхностей зажимов пер
пендикулярны направлению движения. Сдавли
вающие поверхности зажимов должны быть пло
скими И превосходить по размеру зоны контакта
с таблеткой. Прибор калибруют с использовани
ем системы, обеспечивающей точность 1 Н.
МЕТОДИКА
Таблетку помещают между зажимами, при
нимая во внимание ее форму, а также раздели
тельную линию и надпись, если они есть. Для
всех измерений таблетка должна быть ориенти
рована одинаково по отношению к направлению
прилаrаемой силы. Измерения проводят для 10
таблеток. Перед каждым измерением тщательно
удаляют все фраrменты предыдущей таблетки.
Эта методuка непpuменима при использова
нии ПОЛНOCl77ЬЮ авmoматизироваНН020 прибора.
ПРЕДСТАВЛЕНИЕ РЕЗУЛЬТАТОВ
Необходимо указывать среднее, минималь
ное и максимальное значения измеренной силы
в ньютонах. Также указывают тип использован
нoro прибора и, если необходимо, ориентацию
таблеток.
01/2013:20909
2.9.9. ИЗМЕРЕНИЕ КОНСИСТЕНЦИИ
МЕТОДОМ ПЕНЕТРОМЕТРИИ
Испытание позволяет измерить в установлен
ных и валидированных условиях проникновение
объекта в испытуемый образец, находящийся в
контейнере определенной формы и размера.
в
с
D
Е
F
G
н
Рисунок 2.9.9. 1. Пенетрометр
А шкала, показывающая rлубину проникновения, проrра
дуированная в десятичных долях миллиметра;
В вертикальный стержень, который ПОДQерживает и
вводит проникающий объект;
С устройство для автоматическоrо введения проникаю
щеrо объекта в течение постоянноro времени;
О устройство, обеспечивающее вертикальное положе
ние проникающеro объекта и rоризонтальное положение
основания;
Е проникающий объект (см. рисунки 2.9.9Л и 2.9.9 З);
F контейнер;
G roризонтальное основание;
Н контроль rоризонтальноrо положения основания.
434
rосударственная фармакопея Республики Беларусь
'"
1"
А I
I
I
I ,
I //
//--
65:100.25 "
t
!
I
I
/ 1 i
!
/ С 0d
:% /
h
/
/
/ I
ф I
'"
/, "
... -: < ,/ ./, , .' / ' ,
3.141: С.О4"
.д
90 1: .
. -
t1
m
".;
"
,-"
"'-.....,.
с. l'
"':"'A
с'
...
Рисунок 2.9.9. 2. Воронка (т=102.5 2), контейнер (d=102 мм uпи 75 мм, h?62 мм) и стержень (/=162 мм, т=47,52)
(размеры указаны в миллиметрах)
ПРИ БОР
Прибор состоит из пенетрометра, представ
ляющеro собоЙ штатив и проникающий объект.
Образец пенетрометра показан на рисунке
2.9.9. 1,
Штатив состоит из следующих частей:
вертикальный стержень, который под
держивает и направляет проникающий объект;
rоризонталоный стержень;
устройство, которое обеспечивает Bep
тикальное положен;t18 проникающеrо объекта;
.з"1
· 8., ЪJ'
\ ,"" !
"-, V') t
\\ . /. j
I
со
т... ...
,'\ t
' 012.65 i
1( .j
! I ' i
11 tf
i! !
t I !
!
i
I
I
I
i
А !
/
"--
,/
/ '
//
/"//
/
'/
, '/
.<': /
,./
//
/
..... "'"
'25"
устройство, которое поддерживает ropv.
зонтальное положение основания;
устройство для ввода и извлечения про
никающеrс объекта;
шкала. проrрадуированная в десятич
НЫХ долях миллиметра, показывающая rлуби
ну проникновения.
Проникающий объект изroтовлен из COOT
ветствующеrо материала, имеет rладкую по
верхность и определенную форму, размер и
массу.
09.5
t 1
I !
I
"-- I
i.l) i
I I
I
I ф
I
I
. ,
!
.,
тI
ШI
I
i... 0 28.5 !
Рисунок 2.9.9. 3. Микроворонка (т=7,О 2), контейнер и стержень (/=116 мм, т=16,8 2)
(размеры указаны в миллиметрах)
2.9. 10. Содержание этанола
435
Пример проникающеrо объекта приведен на
рисунках 2.9.9. 2 и 2.9.9. З.
МЕТОДИКА
Перед испытанием образец подroтавливают
одним из методов:
А. Аккуратно наполняют доверху три KOH
тейнера, исключая образование пузырьков воз
духа. Если необходимо, разrлаживают поверх
t-<ость и выдерживают образцы при температуре
(25f:O,5) ос в течение 24 ч, если нет друrих YKa
заний в частной статье.
В. Три испытуемых образца выдерживают
при температуре (25f:0,5) ос в течение 24 ч. В
течение 5 мин подходящим способом нарезают
образцы и аккуратно наполняют три контейнера
доверху, исключая образование пузырьков воз
духа. Если необходимо, разrлаживают поверх
ность.
С. Расплавляют три испытуемых образ
ца и аккуратно наполняют три контейнера дo
верху, исключая образование пузырьков воз
духа. Образцы выдерживают при температуре
(25f:0,5) ос в течение 24 ч, если нет друrих YKa
заний в частной статье.
Определение проникновения. На OCHOBa
ние пенетрометра помещают испытуемый об
разец. Проверяют, чтобы поверхность образца
бы па перпендикулярна вертикальной оси прони
кающеro объекта. Доводят температуру прони
кающеro объекта до (25f:0,5) ос и устанавлива
ют в таком положении, чтобы наконечник слеrка
касался поверхности образца. Проникающий
объект разблокируют и выдерживают в таком
состоянии 5 с. Затем проникающий объект фик
сируют и измеряют rлубину проникновения. Ис
пытание повторяют с двумя оставшимися KOH
тейнерами.
ПРЕДСТАВЛЕНИЕ РЕЗУЛЬТАТОВ
Проникновение, выражают в десятых долях
миллиметра как среднее значение трех измере
ний. Если один из индивидуальных результатов
отличается от среднеro значения больше чем на
3 %, испытание повторяют и рассчитывают cpeд
нее значение и относительное среднеквадратич
ное отклонение шести испытаний.
01/2013:20910
2.9.10. СОДЕРЖАНИЕ ЭТАНОЛА
Эти методы предназначены для испытания
жидких лекарственных средств и их инrредиен
тов. которые содержат эта нол.
Содержание эта нола в жидкости Bыpa
жают количеством объемов эта нола, coдep
жащихся в 100 объемах жидкости при TeM
пературе (20f:0,1) ОС. Эта характеристика
называется «процентное содержание этано
ла по объему» (об/об). Содержание этано
ла можно также выражать в rpaMMax эта нола
в 100 r жидкости. Эта характеристика назы
вается «процентное содержание этанола по
массе» (м/м).
МЕТОД А
В случае лекарственных средств, coдep
жащих растворенные вещества, эти вещества
должны быть отделены от спирта дистилля
цией (отrонкой). Если при дистилляции кроме
спирта и воды MorYT отroняться друrие лету
чие вещества, в частной статье указывают
способ предотвращения этоro.
Соотношение плотности при температуре
(20f:0,1) ос относительной плотности (в BaI{Y
уме) и содержания эта нола в смеси воды и
спирта представлено в таблицах Междуна
родной орrанизации официальной метроло
rии (1972), Международная рекомендация
NQ 22.
Прибор. Прибор (рисунок 2.9.1 O. 1) пред
ставляет собой колбу с круrлым дном (А)
A
..........
"1
180
с>
с>
N
t
Е
Рисунок 2.9.1 O. 1. Прибор для определения
содержания этанола
(размеры указаны в миллиметрах)
436
rосударственная фармакопея Республики Беларусь
имеющую переходник (В) с улавливателем
водяноro пара, соединенную с вертикальным
холодильником (С). Нижняя часть холодиль
ника соединена с трубкой (0), через которую
дистиллят поступает в нижнюю часть мерной
колбы вместимостью 100 или 250 мл. Во
время дистилляции мерная колба поrружена
в смесь льда и воды (Е). ДЛЯ предотвраще
ния обуrливания растворенных веществ под
колбой (А) помещают диск, который имеет
круrлое отверстие диаметром 6 см.
Методика
Пикнометрический метод / метод с иc
пользованием плотномера с осциллиру
ющим датчиком. 25,0 мл испытуемоrо образ
ца, измеренных при температуре (20:1:0,1) ос,
помещают в Дистилляционную колбу, разво
дят водой дистиллированной Р до объема
100 мл или 150 мл и прибавляют несколько
кусочков пемзы. Присоединяют переходник и
холодильник. Отrоняют и собирают в мерную
колбу вместимостью 1 00 мл не менее 90 мл
дистиллята (oтroHa). Температуру oтrOHa дo
водят до температуры (20:1:0,1) ос и доводят
водой дистиллированной Р с температурой
(20:1:0,1) ос до объема 100 мл. Определяют
относительную плотность oтroHa при темпе
ратуре (20:1:0,1) ос с помощью пикнометра
или с помощью плотномера с осциллиру
ющим датчиком.
По таблице 2.9.10. 1 (колонка 3) находят
содержание этанола в oтroHe и рассчиты
вают содержание этанола в лекарственном
средстве (об/об) путем умножения найден
ноro табличноrо значения на четыре. Полу
ченный результат окруrляют до десятичноro
знака.
rидрометрический метод. 50,0 мл испы
TyeMoro образца, измеренных при температу
ре (20:1:0,1) ос, помещают в дистилляционную
колбу, прибавляют от 200 мл ДО 300 мл воды
дистиллированной Р и выполняют дистил
ЛЯЦИЮ, как описано выше, собирая в мерную
колбу вместимостью 250 мл не менее 180 мл
дистиллята. Температуру отroна доводят до
(20:1:0,1) ос и разводят до объема 250,0 мл
водой дистиллированной Р с температурой
(20:1:0,1) ос.
Помещают отroн в цилиндр, диаметр KO
тороro должен быть на 6 мм шире утолщения
ареометра. Если объем дистиллята HeДOCTa
точен, удваивают количество испытуемоro
лекарственноro средства и дистиллят разво
дят до объема 500,0 мл дистиллированной
водоЙ Р с температурой (20:1:0,1) ос.
Вносят поправку на разведение путем
умножения найденноro значения на пять. По
таблице 2.9.10. 1 рассчитывают процентное
содержание эта нола в лекарственном cpeд
стве (об/об), и результат окруrляют до деся
тичноrо знака.
Таблица 2.9.10. 1
Соотношение между плотностью, oтHocи
тельной плотностью и содержанием этанола
Относительная Содержание
Р20 плотность
(к2!м3) дистиллята на этанола в процентах
Bo yxe d g (06/06) при 20 ос
968,0 0,9697 25,09
968,5 0,9702 24,64
969,0 0,9707 24,19
969,5 0,9712 23,74
970,0 0,9717 23,29
970,5 0,9722 22,83
971,0 0,9727 22,З7
971,5 О,97З3 21,91
972,0 0,97З8 21,45
972,5 0,9743 20,98
973,0 0,9748 20,52
973,5 0,9753 20,05
974,0 0,9758 19,59
974,5 0,9763 19,12
975,0 0,9768 18,66
975,5 0,9773 18,19
976,0 0,9778 17,73
976,5 0,9783 17,25
977,0 0,9788 16,80
977,5 0,9793 16,34
978,0 0,9798 15,88
978,5 0,9803 15,43
979,0 0,9808 14,97
979,5 0,981З 14,52
980,0 0,9818 14,07
980,5 0,982З 13,6З
981,0 0,9828 16,18
981,5 0,9833 12,74
982,0 0,9838 12,31
982,5 0,9843 11,87
983,0 0,9848 11,44
983,5 0,9853 11,02
984,0 0,9858 10,60
984,5 0,9863 10,18
985,0 0,9868 9,76
985,5 0,9873 9,35
986,0 0,9878 8,94
986,5 0,9883 8,53
987,0 0,9888 8,13
987,5 О,989З 7,73
988,0 0,9898 7,34
988,5 0,9903 6,95
989,0 0,9908 6,56
989,5 0,9913 6,17
990,0 0,9918 5,79
990,5 0,9923 5,42
991,0 0,9928 5,04
991,5 0,993З 4,67
992,0 0,9938 4,30
992,5 0,9943 3,94
2.9.10. Содержание эта нола
437
Относительная Содержание
Р20 плотность
(кz/мЗ) дистиллята на этанола в процентах
воздухе d g (06/06) при 20 ос
993,0 0,9948 3,58
993,5 0,9953 3,22
994,0 0,9958 2,86
994,5 0,9963 2,51
995,0 0,9968 2,16
995,5 0,9973 1,82
996,0 0,9978 1,47
996,5 0,9983 1,13
997,0 0,9988 0,80
997,5 0,9993 0,46
998,0 0,9998 0,1З
#Если испытуемое лекарственное средство
содержит летучие вещества эфир, эфирные
масла, хлороформ, камфору, летучие кисло
ты или основания, свободный йод и др., ero
предварительно обрабатывают. Испытуемое
лекарственное средство, содержащее эфир,
эфирные масла, хлороформ или камфору, по
мещают в делительную воронку, прибавляют
равный объем наСblщеНН020 раствора натрия
хлорида Р и такой же объем петролеЙН020
эфира Р. Смесь взбалтывают в течение 3 мин.
После разделения слоев водно спиртовой
слой сливают в друrую делительную BOpOH
ку и обрабатывают таким же способом поло
винным количеством петролеЙН020 эфира Р.
Водно спиртовой слой сливают в дистилляци
онную колбу. Эфирные извлечения объединя
ют и взбалтывают с половинным количеством
наСblщеНН020 раствора натрия хлорида Р.
После разделения слоев водно спиртовой
слой присоединяют к жидкости, находящейся
в дистилляционной колбе.
Если жидкость при дистилляции сильно пе
нится, прибавляют 2 3 мл кислотbl серной Р
или кислоты фосфорной Р, 2 3 r кальция
хлорида Рили парафина Р
Если испытуемое лекарственное cpeд
ство содержит менее ЗО % спирта, то высали
вание проводят не наСblщенным раствором
натрия хлорида Р, а 1 О r натрия хлорида Р.
При содержании в испытуемом лекар
ственном средстве летучих веществ их ней
трализуют раствором щелочи, при coдep
жании летучих оснований кислотой
фосфорной Р или кислотой серной Р.
Испытуемые лекарственные средства,
содержащие свободный йод, перед дистил
ляцией обрабатывают порошком цинка Рили
рассчитанным количеством натрия тиосуль
фата Р до обесцвечивания. Для связывания
летучих сернистых соединений прибавляют
несколько капель раствора натрия 2идPOK
сида Р.#
МЕТОД В
Парофазная rазовая хроматоrрафия
(2.2.28).
Раствор внутренне20 стандарта. 1,0 мл
пропанола Р доводят водой Р до объема 100,0 мл.
1,0 мл полученноro раствора доводят водой Р
до объема 20,0 мл.
Испытуемый раствор. Количество испыту
емоro образца, содержащее 1 r этанола, доводят
водой Р до объема 50,0 мл. 1,0 мл полученноro
раствора доводят водой Р до объема 20,0 мл.
К 2,0 мл полученноro раствора прибавляют
1,0 мл раствора внутреннеro стандарта и дово--
дят водой Р до объема 20,0 мл.
Раствор сравнения (а). 5,0 мл зтанола Р1
доводят водой Р до объема 100,0 мл. 25,0 мл по
лученноro раствора доводят водой Р до объема
100,0 мл. 1,0 мл полученноro раствора доводят
водой Р до объема 20,0 мл.
Раствор сравнения (Ь). 0.5 мл раствора
сравнения (а) и 1,0 мл раствора внутреннеro
стандарта доводят водой Р до объема 20,0 мл.
Раствор сравнения (с). 1,0 мл раствора
сравнения (а) и 1,0 мл раствора внутреннею
стандарта доводят водой Р до объема 20,0 мл.
Раствор сравнения (d). 1,5 мл раствора
сравнения (а) и 1,0 мл раствора внутреннеro
стандарта доводят водой Р до объема 20,0 мл.
Раствор сравнения (е). 1 ,О мл метанола без
водН020 Р доводят водой Р до объема 100,0 мл.
1,0 мл полученноro раствора доводят водой Р до
объема 20,0 мл.
Раствор сравнения (f). Смешивают 1,0 мл
раствора внутреннеro стандарта, 2,0 мл paCT
вора сравнения (а), 2,0 мл раствора сравнения (е)
и доводят водой Р до объема 20,0 мл.
Условия хроматоrрафирования:
колонка кварцевая капиллярная длиной
30 м и диаметром 0,53 мм, покрытая слоем
поли[(цианопропил)(фенил)][диметил]силокса
на Р (толщина слоя О,З мкм);
2аз носитель: 2елий для хромат02рафии р,
скорость 2аза носителя: 1 мл/мин;
деление потока: 1 :50;
параметРbl парофаЗН020 пробоотборника:
равновесная температура: 85 C;
время достижения равновесия: 20 мин;
температура:
Время (мин) Температура (ОС)
0 1,6 40
Колонка 1 ,6 9,9 40 65
9,9 13,6 65 175
1 3,6 20,0 175
Блок
ввода 200
проб
Детектор 200
детектор: пламенно ионизационный;
объем вводимой пробы: по 1 мл rазовой
фазы испытуемоro раствора и растворов cpaB
нения (Ь), (с), (d) и (f), не менее 3 раз каждый.
438
rосударственная фармакопея Республики Беларусь
Порядок выхода пиков: метанол, эта нол ,
1 пропанол.
Относительное удерживание (по отношению
к этанолу, время удерживания около 5,3 мин):
метанол около 0,8; 1 пропанол около 1,6.
При20дность хромат02рафической cиcтe
мы: раствор сравнения (f):
разрешение: не менее 5 между пиками Me
танола и эта нола.
Строят rрадуировочный rрафик, откладывая
по оси абсцисс концентрации эта нола в paCTBO
рах сравнения (Ь), (с), (d) и (f), а по оси орди
нат среднее значение отношения площадей
пиков этанола и внутреннеro стандарта на xpo
MaTorpaMMax соответствующих растворов.
Рассчитывают процентное содержание эта
нола в испытуемом образце.
МЕТОД С
rазовая хроматоrpафия (2.2.28).
Раствор внутренне20 стандарта. 1,0 мл
пропанола Р доводят водой Р до объема
100,0 мл.
Испытуемый раствор. Количество испыту
емоro образца, содержащее 1 r этанола, ДOBO
дят водой Р до объема 50,0 мл. К 1,0 мл полу
ченноro раствора прибавляют 1,0 мл раствора
BHYTpeHHero стандарта и доводят водой Р до
объема 20,0 мл.
Раствор сравнения (а). 1,0 мл этанола Р1
доводят водой Р до объема 50,0 мл.
Раствор сравнения (Ь). 1, О мл метанола без
водН020 Р доводят водой Р до объема 100,0 мл.
1,0 мл полученноro раствора доводят водой Р до
объема 20,0 мл.
Раствор сравнения (с). Смешивают 1,0 мл
раствора BHYTpeHHero стандарта, 1,0 мл paCT
вора сравнения (а), 2.0 мл раствора cpaBHe
ния (Ь) и доводят водой Р до объема 20,0 мл.
Условия хроматоrрафирования:
колонка кварцевая капиллярная длиной
30 м и диаметром 0,53 мм, покрытая слоем
поли[( цианоп ропил) (фенил) ][диметил )силок
сана Р (толщина слоя 0,3 мкм);
2аз носитель: 2елий для xpOMaт02pa
фииР;
скорость 2аза носителя: 1 мл/мин;
деление потока: 1 :50;
температура:
Время (мин) I Температура (ОС)
0 1,6 I 40
1 ,6 9,9 40 65
Колонка
9,9 13,6 I 65 175
1 3,6 20,0 175
Блок ввода
проб 200
Детектор 200
детектор: пламенно--ионизационный:
объем вводимой пробы: по 1 мкл испы
туемоro раствора и раствора сравнения (с), не
менее 3 раз каждый.
Порядок выхода пиков: метанол, эта нол ,
1 пропанол.
Относительное удерживание (по отноше
ниюкэтанолу. времяудерживания окол05,3мин):
метанол около 0,8; 1 пропанол около 1,6.
При20дность хромат02рафической систе-
мы: раствор сравнения (с):
разрешение: не менее 5 между пиками Me
танола и этанола.
Содержание эта нола (в процентах (об/об»
рассчитывают по формуле:
А '/2'Р
./ . v.
"2 1 1
rдe:
А, площадь пика эта нола на хромато-
rpaMMe испытуемоro раствора;
А 2 площадь пика зтанола на xpOMaTO
rpaMMe раствора сравнения (с);
/1 площадь пика внутреннеro стандарта
на xpoMaTorpaMMe испытуемоro раствора;
/2 площадь пика внутреннеro стандарта
на xpoMaтorpaMMe раствора сравнения (с);
V 1 объем испытуемоrо образца в испыту
емом растворе, в миллилитрах;
р процентное содержание эта нола в эта-
ноле Р1.
01/2013:20911
2.9.11. ИСПЫТАНИЕ
НА СОДЕРЖАНИЕ МЕТАНОЛА
И 2 ПРОПАНОЛА
МЕТОД А
Парофазная rазовая хроматоrрафия (2.2.28).
Раствор внутренне20 стандарта. 1,0 мл
пропанола Р доводят водой Р до объема 100,0 мл
воды Р. 1,0 мл полученноrо раствора доводят
водой Р до объема 20,0 мл.
Испытуемый раствор. К 4,0 мл испыту
емоro образца прибавляют 1,0 мл раствора
BHYTpeHHero стандарта и доводят водой Р до
объема 20,0 мл.
Раствор сравнения (а). К 1,0 мл метанола
безводН020 Р прибавляют 1,0 мл 2 пропанола Р
и доводят водой Р до объема 100,0 мл. 1,0 мл по
лученноro раствора доводят водой Р до объема
20,0 мл.
Раствор сравнения (Ь). 5,0 мл этанола Р1
доводят водой Р до объема 100,0 мл. 25,0 мл по
лученноro раствора доводят водой Р до объема
2.9.11. Испытание на содержание метанола и 2 пропанола
439
100,0 мл. 1,0 мл полученноro раствора доводят
водой Р до объема 20,0 МЛ.
Раствор сравнения (с). Смешивают 1,0 мл
раствора внутреннеro стандарта, 2,0 мл paCT
вора сравнения (а), 2,0 мл раствора cpaBHe
ния (Ь) и доводят водой Р до объема 20,0 мл.
Условия хроматоrрафирования:
колонка кварцевая капиллярная длиной
30 м и диаметром 0,53 мм, покрытая слоем
поли[(цианопропил) ( фенuл)][диметилJсилокса
на Р (толщина слоя 0,3 мкм);
2аз носитель: 2елий для xpOMaт02pa
фииР;
скорость 2аз носителя: 1 мл/мин;
деление потока: 1 :50;
параметры парофазН020 пробоотборника:
равновесная температура: 85 ОС;
время достижения равновесия: 20 мин;
температура:
Время (мин) Температура (ОС)
0 1,6 40
1 ,6 9,9 40.........65
Колонка
9,9 13,6 65.........175
13,6 20,0 175
Блок
ввода 200
проб
Детектор 200
детектор: пламенно ионизационный;
объем вводимой пробы: по 1 мл rазовой
фазы испытуемоro раствора и раствора cpaBHe
ния (с). не менее 3 раз каждый.
Порядок выхода пиков: метанол, этанол,
2 пропанол, 1 пропанол.
Относительное удерживание (по OTHO
шению к этанолу, время удерживания около
5,3 мин): метанол около 0,8; 2 пропанол
около 1,2; 1 пропанол около 1,6.
При20дность 1fромат02рафической cиcтe
мы: раствор сравнения (с):
разрешение: не менее 5 между пиками Me
та нола и эта нола.
Содержание метанола (в процентах (об/об»
рассчитывают по формуле:
А "2 .р
А 2 ", .40
rдe:
А, площадь пика метанола на xpOMaTO
rpaMMe испытуемоro раствора;
А 2 площадь пика метанола на xpOMaTO
rpaMMe раствора сравнения (с);
" площадь пика внутреннеro стандарта
на xpoMaтorpaMMe испытуемоro раствора;
'2 площадь пика внутреннеro стандарта
на xpoMaTorpaMMe раствора сравнения (с);
р процентное содержание метанола в Me
maноле безводном Р.
Содержание 2 пропанола (в процентах (об/об»
рассчитывают по формуле:
Аз '/2 . Р
А4 "1 .40
rдe:
Аз площадь пика 2 пропанола на XpoMa
TorpaMMe испытуемоro раствора;
А4 площадь пика 2 пропанола на xpOMa
тorpaMMe раствора сравнения (с);
/, площадь пика BHYTpeHHero стандарта
на xpoMaTorpaMMe испытуемоro раствора;
'2 площадь пика внутреннеro стандарта
на xpoMaтorpaMMe раствора сравнения (с);
р процентное содержание 2 пропанола в
2 пропаноле Р.
МЕТОД В
rазовая хроматоrрафия (2.2.28).
Раствор внутренне20 стандарта. 1,0 мл
пропанола Р доводят водой Р до объема 100,0 мл.
Испытуемый раствор. К 4,0 мл испытуемою
образца прибавляют 1,0 мл раствора внутреннеro
стандарта и доводят водой Р до объема 20,0 мл.
Раствор сравнения (а). К 1,0 мл метанола без
воднО2О Р прибавляют 1,0 мл 2 пропанола Р и дово-
дят водой Р до объема 100,0 мл. 1,0 мл полученноro
раствора доводят водой Р до объема 20,0 мл.
Раствор сравнения (Ь). 1,0 мл этанола Р1
доводят водой Р до объема 50,0 мл.
Раствор сравнения (с). Смешивают 1,0 мл
раствора внутреннеro стандарта, 2,0 мл paCT
вора сравнения (а), 1,0 мл раствора сравнения (Ь)
и доводят водой Р до объема 20,0 мл.
Условия хроматоrрафирования:
колонка кварцевая капиллярная длиной
30 м и диаметром 0,53 мм, покрытая слоем
поли{(цианопропил) ( фенил)][диметил Jсилокса
на Р (толщина слоя 0,3 мкм);
2аз носитель: 2епий для хромат02рафии Р;
скорость 2аза носителя: 1 мл/мин;
деление потока: 1 :50;
температура:
Время (мин) Температура (ОС)
0 1,6 40
1 ,6 9,9 40.........65
Колонка
9,9 13,6 65.........175
1 3,6 20,0 175
Блок
ввода 200
проб
Детектор 200
детектор: пламенно ионизационный;
объем вводимой пробы: по 1 мкл испы
туемоro раствора и раствора сравнения (с), не
менее 3 раз каждый.
Порядок выхода пиков: метанол, этанол,
2 пропанол, 1 пропанол.
440
rосударственная фармакопея Республики Беларусь
Относительное удерживание (по отноше
нию к эта нолу, время удерживания около 5,3 мин):
метанол около 0,8; 2 пропанол около 1,2;
1 пропанол около 1,6.
При20дность хроматО2рафической cиcтe
МЫ: раствор сравнения (с):
разрешение: не менее 5 между пиками Me
танола и эта нола.
Содержание метанола (в процентах (об/об)
рассчитывают по формуле:
А "2 .р
А "1.40
rдe:
А 1 площадь пика метанола на xpOMaTO
rpaMMe испытуемою раствора;
А площадь пика метанола на xpOMaTO
rpaMMLe раствора сравнения (с);
'1 площадь пика внутреннеro стандарта
на xpoMaтorpaMMe испытуемоro раствора;
/2 площадь пика внутреннеro стандарта
на xpoMaтorpaMMe раствора сравнения (с);
р процентное содержание метанола в Me
таноле безводном Р.
Содержание 2 пропанола (в процентах (oБIо6))
рассчитывают по формуле:
Аз '/2 . Р
А4 "1.40
rдe:
Аз площадь пика 2 пропанола на xpOMa
тorpaMMe испытуемою раствора;
А4 площадь пика 2 пропанола на xpOMa
TorpaMMe раствора сравнения (с);
/1 площадь пика внутреннеro стандарта
на xpoMaTorpaMMe испытуемоro раствора;
'2 площадь пика внутреннеro стандарта
на xpoMaтorpaMMe раствора сравнения (с);
р процентное содержание 2 пропанола в
2 пропаноле Р.
01'2013:20912
2.9.12. СИТОВОЙ АНАЛИЗ
Степень измельчения образца может быть
выражена размерами отверстий сит, COOTBeTCTBY
ющих требованиям для неаналитических сит (2. 1.4).
Степень измельчения порошков в случае
определения ее с помощью просеивания Bыpa
жают, учитывая номер использованною сита,
либо с применением нижеприведенных терми
нов, либо, если такие термины не MorYT быть ис
пользованы, степень измельчения выражают в
процентах вещества (М/М), проходящеro через
сито определенноro размера.
При описании порошков используют следу
ющую терминолоrию:
rрубый порошок. Не менее 95 % массы
порошка проходит через сито (1400) и не более
40 % массы порошка проходит через сито (355).
Среднемелкий порошок. Не менее 95 %
массы порошка проходит через сито (355) и не
более 40 % массы порошка проходит через сито
(180).
Мелкий порошок. Не менее 95 % массы
порошка проходит через сито (180) и не более
40 % массы порошка проходит через сито (125).
, Очень мелкий порошок. Не менее 95 %
массы порошка проходит через сито (125) и
не более 40 % массы порошка проходит через
сито (90).
Если указано сито одноro номера, не менее
97 % массы порошка должно проходить через
указанное сито, если нет друrих указаний в част
ной статье.
#В случае измельченною лекарственноro
растительноro сырья должны выдерживаться
требования к степени измельчения, указанные в
статье «#Лекарственное растительное сырье
цельное или измельченное фасованное». если
нет друrих указаний в частной статье.#
Для определения степени измельчения по
рошка собирают сита, порошок полностью про
сеивают и взвешивают каждую фракцию.
#Степень измельчения указывают в скобках;
например, измельченный образец с частицами,
проходящими сквозь сито С размером стороны
отверстия 5600 мкм, обозначают как измельчен
ный образец (5600).#
#МЕТОДИКА ОПРЕДЕЛЕНИЯ СТЕПЕНИ
ИЗМЕЛЬЧЕНИЯ ПОРОШКОВ
Если нет друrих указаний в частной статье,
rрубые, среднемелкие порошки в количестве
2 100 r помещают на соответствующее сито,
встряхивают в течение 10 мин, периодически по
стукивая по ситу. Для мелких и очень мелких по
рошков навеска образца не должна превышать
25 r, сито встряхивают в течение 20 мин. Если
порошки закупоривают отверстия во время про
сеивания, допускается осторожная прочист
ка нижней поверхности сита. Навеску порошка,
время и условия просеивания указывают в част
ной статье.
#МЕТОДИКА ОПРЕДЕЛЕНИЯ СТЕПЕНИ
ИЗМЕЛЬЧЕНИЯ ЛЕКАРСТВЕнноrо
РАСТИТЕльноrо СЫРЬЯ
Если нет друrих указаний в частной статье,
лекарственное растительное сырье в количе
стве 25 100 r помещают на соответствующее
сито, снабженное плотно приrнанным приемным
лотком и крышкой, и просеивают, не допуская
2.9.14. Удельная площадь поверхности
441
016
дополнительноro измельчения. Просеивание
измельченных частиц считается законченным,
если количество сырья, прошедшеro сквозь сито
при дополнительном просеве в течение 1 мин,
составляет менее 1 % от оставшеrося на сите.
01/2013:20914
2.9.14. УДЕЛЬНАЯ ПЛОЩАДЬ
ПОВЕРХНОСТИ
Испытание предназначено для измере
ния удельной площади поверхности сухих
порошков, прошедших сквозь сито, и Bыpa
жается в метрах квадратных на rpaMM. Mo
лекулярный поток, который может повлиять
на результаты испытания порошков, размеры
частиц которых составляют несколько микро
метров, не учитывают в уравнении, которое
используется для расчета удельной площади
поверхности.
ПРИ БОР
Прибор состоит из следующих частей:
(а) пропускная камера (см. рисунок
2.9.14. 1), состоящая из цилиндра, имеюще
ro внутренний диаметр (12,6::1:0,1) мм (А), из
roтовленноro из стекла или нержавеющеro
металла. Дно камеры rерметично соедине
но (например, посредством адаптера) с Ma
нометром (рисунок 2.9.14. 2). На расстоя
нии (50::1:15) мм от верхушки ячейки имеется
выступ шириной 0,5 1 мм. На выступе фик
сируется перфорированный металлический
диск (В), выполненный из нержавеющеro Me
талла толщиной (0,9::1:0,1) мм. Диск перфори
рован ЗО О отверстиями с диаметром 1 мм,
равномерно распределенными по всей по
верхности.
Поршень (С) изroтовлен из нержавеюще
ro металла и подоrнан к камере с зазором не
более 0,1 мм. Дно поршня имеет острые края
перпендикулярно по отношению к централь
ной оси. На одной стороне поршня имеется
воздушный дренаж длиной З мм и О,З мм rлу
биной. Верхушка поршня имеет воротник, KO
торый при поrружении поршня в камеру co
при касается с верхушкой камеры. Расстояние
между дном поршня и верхушкой перфориро
ванноro диска (В) составляет (15::1:1) мм.
Диски из фильтровальной бумаrи (О)
имеют ровные края и диаметр, совпадающич
с внутренним диаметром камеры.
(Ь) U образный манометр (Е) (рисунок
2.9.14. 2) изroтовлен из стеклянной трубки с
внешним диаметром 9 мм и внутренним ди
8метром 7 мм. Верхушка одноro из плеч Ma
нометра rерметично соединена с пропускной
I
" 12.6:!: 0.1
I
'
/
(А)
ll'J
'"
,
ll'J
'"
(8)
..;:::.
..;. .
о
+t
<»
о
Рисунок 2.9.14. 1. Пропускная камера
(размеры указаны в миллиметрах)
камерой (F). На плечо манометра, соединен
ное с пропускной камерой, нанесена линия на
125 145 мм ниже верхушки выпускноro OTBep
стия, расположенноro сбоку. Еще три линии
расположены на 15 мм, 70 мм и 110 мм выше
этой линии (G). Выпускное отверстие, распо
ложенное на 250 305 мм выше дна MaHOMe
тра, используется для вакуумирования плеча
манометра, соединенноro с пропускной KaMe
рой. Кран выпускноro отверстия располаrает
ся не далее чем на 50 мм от плеча манометра.
Манометр установлен таким образом,
чтобы еro плечи были расположены верти
кально. До нижней метки он заполнен дибу
тилфталатом Р, содержащи липофильную
краску.
МЕТОДИКА
Испытуемый порошок следует предвари
тельно высушить и пропустить через сито (Ha
пример, N2 125). Массу порошка, необходимую
для выполнения испытания, рассчитывают по
формуле:
м = V. р.(1 Б),(1)
rдe:
V насыпной объем уплотненноro слоя п(r
рошка;
р плотность испытуемоro вещества, r/мл;
G пористость уплотненноrо слоя.
442
(F)
rосударственная фармакопея Республики Беларусь
(Е)
09
1.0
v о
'v
1.0 i
+
N 1
I --}(G)
I
I!) i
1D
"
I!)
у .......
А "
о
(JJ
1.0
N
у
Рисунок 2.9.14. 2. Манометр
(размеры указаны в миллиметрах)
Изначально пористость принимают равной
0,5 и учитывают при расчете массы (М) испыту
eMoro порошка по формуле (1).
Фильтровальный диск помещают на верхуш
ку металлическоro диска (В). Взвешивают He
обходимое количество испытуемоrо порошка с
точностью до 1 Mr. Помещают порошок в чистую
взвешенную пропускную камеру, осторожно за
полняют камеру таким образом, чтобы полу
чилась ровная поверхность, и накрывают слой
порошка вторым фильтровальным диском. Meд
ленно сдавливают порошок поршнем, стараясь
избеrать вращения. Надавливают на псршень
до тех пор, пока он полностью не поrрузится в
пропускную камеру. Если это невозможно, сле
дует уменьшить количество порошка, взятоro
для испытания. Если напротив, сопротивление
недостаточно, следует увеличить количество по
рошка. В этом случае пористость нужно рассчи
тать еще раз. Через 1 О с поршень удаляют.
Пропускную камеру rерметично соединяют с
манометром. Отсасывают из манометра воздух с
помощью резиновой rруши до тех пор, пока ypo
вень окрашенной жидкости не установится на
уровне верхней метки. Перекрывают кран и про
веряют rерметичность прибора, закрыв верхний
конец камеры, например, резиновой пробкой.
Удаляют пробку и с помощью таймера замеря
ют время, необходимое для опускания столбика
жидкости со второй до третьей метки.
Полученное значение используют при pac
чете удельной площади поверхности (S), KOТO
рую выражают в метрах квадратных на rpaMM, с
помощью следующей формулы:
s к...;;з. ,(2)
p.(1 E)'..fi
rдe:
t время, необходимое для опускания
столбика жидкости со второй до третей метки, с;
17 динамическая вязкость воздуха, мПalс
(см. таблицу 2.9.14. 1);
К константа прибора, рассчитанная по
формуле (4);
Р плотность испытуемоro вещества, r/мл;
E пористость уплотненноro слоя порошка.
rРАДУИРОВКА ПРИБОРА
Насыпной объем уплотненноrо слоя по
рошка определяют методом pTYТHoro смещения,
как описано ниже.
Два диска из фильтровальной бумаrи по
мещают в пропускную камеру, прижимают вниз
с помощью стержня, диаметр которою HeMHO
ro меньше диаметра камеры, до тех пор, пока
фильтровальные диски не будут rоризонтально
лежать на перфорированном металлическом
диске. Камеру заполняют ртутью, удаляя пу
зырьки воздуха со стенок, и отсасывают ее из
быток таким образом, чтобы на верху камеры
поучилась ровная поверхность. Если клетка из
roтовлена из материала, способноro вступать
в реакцию с ртутью, предварительно смазыва
ют камеру и металлический диск тонким слоем
вазелиновоro масла. Сливают ртуть в стакан и
определяют ее массу (М д ) и температуру.
Создают уплотненный слой, используя об
разец сравнения порошка, и снова заполняют
клетку ртутью до образования плоской поверх
ности на верху клетки. Затем ртуть сливают в
стакан и снова определяют ее массу (МВ) и TeM
пературу. Рассчитывают насыпной объем (v)
уплотненноro порошка по формуле:
v == М А MB ,(3)
Рну
rдe:
М д MB разница количества ртути, r;
P Hg плотность ртути при данной темпера
туре, r/мл.
2.9.16. Сыпучесть
443
01/2013:20916
Повторяют измерения дважды, используя
каждый раз новую порцию порошка. Колебания
рассчитанною объема (V) не должны превышать
0,01 мл. Используют среднее значение, рассчи
танное по трем испытаниям.
Константу прибора К определяют, исполь
зуя стандартный порошок с известной удельной
площадью поверхности, по методике, приведен
ной ниже:
Рассчитывают необходимое количество по
рошка по формуле (1), используя указанную
плотность и найденный по формуле (3) объем
уплотненноro порошка.
rомоrенизируют и разрыхляют порошок,
встряхивая ero в течение 2 мин в бутылке BMe
стимостью 100 мл. Создают уплотненный слой
порошка и измеряют время истечения, как YKa
зано ранее. Рассчитывают константу при бора
(К) по формуле:
Ssp . p.(1 E)'.Jq
К = ,(4)
J?x.Ji
ще:
SSP известная удельная площадь поверх
ности стандартноro порошка;
р плотность испытуемоro вещества, r/мл;
E: пористость компактноro слоя порошка;
t время, необходимое для опуска
ния столбика жидкости со второй до третьей
метки, с;
17 динамическая вязкость воздуха, мПа/с
(см. таблицу 2.9.14. 1).
Плотность ртути и вязкость воздуха при
различных температурах приведены в таблице
2.9.14. 1.
Таблица 2.9.14. 1
<
Плотность Вязкость
Температура, .....
ртути, BO y- -J1J
ос r/мл ха ('1),
мПа/с
16 13,56 0,01800 0,1342
17 13,56 0,01805 0,1344
18 13,55 0,01810 0,1345
19 13,55 0,01815 0,1347
20 13,55 0,01819 0,1349
21 13,54 0,01824 0,1351
22 13,54 0,01829 0,1353
23 13,54 0,01834 0,1354
24 13,54 0,01839 0,1356
2.9.16. СЫПУЧЕСТЬ
Испытание на сыпучесть предназначено
для определения способности материала, co
стоящеro из твердых частиц (например, порош
ков и rранул) течь в вертикальном направлении
при заданных условиях.
ПРИБОР
В зависимости от сыпучести испытуемоro
образца используют воронки с выходным ство.
лом или без выходною ствола, с разными уrлами
и диаметрами выходных отверстий. Типовые BO
ронки показаны на рисунках 2.9.16. 1 и 2.9.16. 2.
Воронка поддерживается в вертикальном полcr
0110
'
400 :t 5'
---...-;
/
Отпрлированная
повеохность
Воронка
Связующая
. муфта
Насадка 1. 2 иnи 3
030.2 .ъ.,
3.
Насадка 1, 2 ИЛИ 3
в увели"i€ННОМ масштабе
t 9.
i ·
°1
м,
.
,
.
,
!.. .
d
Рисунок 2.9.16. 1. Воронка и насадка.
Насадку иЗ20тавливают из нержавеющей
кислотоупорной стали (V4A, CrNi)
(размеры указаны в миллиметрах)
125
+1
1.25
Рисунок 2.9.16. 2.
(Размеры указаны в миллиметрах)
444
rосударственная фармакопея Республики Беларусь
жении при помощи специальноro приспособле
ния. Вся конструкция должна быть защищена от
вибрации #(метод неподвижной воронки)#.
Насадка Диаметр (d) BbIxoAHoro отверстия
(мм)
1 (1 0:tO,01)
2 (15:t0,01)
3 (25:t0,01 )
МЕТОДИКА
В сухую воронку, выходное отверстие .roтo
рой закрыто подходящим способом, помеща
ют без уплотнения навеску испытуемоrо образ
ца, взвешенную с точностью 0,5 %. Количество
испытуемоro образца зависит от ero насыпноro
объема и от типа использованноrо прибора. OT
крывают выходное отверстие воронки и опре.-
деляют время, необходимое для истечения ис
пытуемоro образца из воронки. Проводят три
измерения.
ПРЕДСТАВЛЕНИЕ РЕЗУЛЬТАТОВ
Сыпучесть выражают в секундах и десятых
долях секунд, отнесенных к 100 r образца.
Результаты зависят от условий хранения ис
пытуемоro образца.
Результаты MOryт быть представлены следу
ющим образом:
а) как среднее значение полученных резуль
татов при условии, что ни один из трех результа
тов не отклоняется от среднеro значения более
чем на 10 %;
Ь) в виде диапазона значений, если отдель
ные результаты отклоняются от среднеro значе
ния более чем на 1 О %;
с) в виде rрафика зависимости массы от
времени истечения;
d) указывают бесконечное время, если об
разец полностью не вытек через воронку.
01/2013:20917
2.9.17. ОПРЕДЕЛЕНИЕ
ИЗВЛЕКАЕмоrо ОБЪЕМА
ПАРЕНТЕРАЛЬНЫХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
Суспензии и эмульсии необходимо встря
хивать перед извлечением содержимоro и
перед определением еro плотности. Масляные
и вязкие лекарственные средства при необхо
димости предварительно наrреваю соrласно
указаниям на этикетке, и встряхивают перед
извлечением содержимоro. Перед измерением
объема содержимое охлаждают до температу
ры 20 25 ОС.
ОДНОДОЗОВЫЕ КОНТЕЙНЕРЫ
Если номинальный объем составляет 1 О мл
И более, испытывают один контейнер, если HO
минальный объем более 3 мл, но менее 1 О мл,
испытывают три контейнера, если номиналь
ный объем составляет 3 мл и менее, испыты
вают пять контейнеров. Содержимое каждоro
контейнера по отдельности извлекают с по
мощью сухих шприцев для подкожноro BBeдe
ния вместимостью не более чем тройной из
меряемый объем, имеющих иrлу 21 калибра
длиной не менее 2,5 см.
Вытесняют пузырьки воздуха из шприца и
иrлы, содержимое переливают в специальный
сухой rрадуированный цилиндр, избеrая опо
рожнения иrлы. Вместимость цилиндра должна
быть достаточной для тоro, чтобы измеряемый
объем составлял не менее 40 % rрадуирован
ной части цилиндра. Друrим способом объем
содержимоro в миллилитрах можно рассчи
тать путем деления массы испытуемоro лекар
ственноro средства на плотность.
Содержимое подходящеro числа контей
неров, имеющих номинальный объем 2 мл
и менее, может быть объединено при BЫ
полнении измерений. При этом для извлече
ния содержимоro каждоro контейнера сле
дует использовать отдельный сухой шприц.
Для контейнеров, содержимое которых имеет
объем 1 О мл И более, допускается перелива
ние лекарственноro средства непосредствен
но в цилиндр или предварительно взвешен
ный стакан.
Полученный объем не должен быть
меньше номинальноro объема. Для контей
неров с объемом 2 мл и менее полученный
объем должен быть не менее суммы объеди
ненных номинальных объемов.
мноrОДОЗОВЫЕ КОНТЕЙНЕРЫ
Для лекарственных средств, предназначен
ных для инъекционноro введения, на этикетке
обязательно указывают количество доз опре
деленноro объема. Выбирают один контейнер и
проводят испытание, как для однодозовых KOH
тейнеров, используя столько шприцев, сколько
доз лекарственноro средства указано на контей
нере.
Объем должен быть таковым, чтобы каждый
шприц высвободждал дозу, не менее указанной
на этикетке.
КАРТРИДЖИ И ПРЕДВАРИТЕЛЬНО
НАПОЛНЕННЫЕ ШПРИЦЫ
Если номинальный объем составляет 1 О мл
И более, испытывают один контейнер, если HO
минальный объем составляет более 3 мл, но
менее 10 мл, испытывают три контейнера, если
номинальный объем составляет 3 мл и менее,
испытывают пять контейнеров. При необходимо
сти к контейнеру присоединяют принадлежности
(иrлу, поршень, шприц) и переносят все coдep
2.9.18. Аэродинамическое испытание мелких частиц
445
жимое, избеrая опорожнения иrлы, в сухую Ta
рированную мензурку путем медленноro и по
стоянноro давления на поршень. Извлекаемый
объем рассчитывают путем деления массы co
держимоro каждоro контейнера на плотность ле
карственноro средства.
Измеренный объем для каждоro контей
нера должен быть не меньше номинальноro
объема.
ИНФУЗИИ ДЛЯ ПАРЕНТЕРАЛьноrо
ВВЕДЕНИЯ
Отбирают один контейнер. Содержимое пе
реносят в сухой мерный цилиндр такой вмести
мости, чтобы определяемый объем заполнил не
менее 40 % номинальноro объема цилиндра. Из
меряют извлекаемый объем.
Полученный объем должен быть не
меньше номинальноro объема, указанноrо на
контейнере.
01/2013:20918
2.9.18. ЛЕКАРСТВЕННЫЕ
СРЕДСТВА для инrАЛЯЦИЙ:
АЭРОДИНАМИЧЕСКОЕ
ИСПЫТАНИЕ МЕЛКИХ
ЧАСТИЦ
Испытание предназначено для определе
ния характеристик мелких частиц облаков аэро
золей, образующихся при использовании ле
карственных средств для инrаляций.
Для испытания используют один из опи
санных ниже приборов по указанной методике,
если нет друrих указаний в частных статьях.
Измерение размеров ступени проводят
периодически вместе с проверкой друrих раз
меров, являющихся критическими для эффек
тивной работы импактора.
Обратный захват (для приборов О и Е).
ДЛЯ тою чтобы убедиться в эффективно
сти захвата частиц, каждую пластинку по
крывают rлицерином, силиконовым маслом
или подобной жидкостью с высокой вязко
стью, обычно наносимой с помощью летуче
ro растворителя. Процесс покрытия пластин
ки должен быть частью валидации методики
и может отсутствовать, если это обосновано
и утверждено.
Баланс масс. Общая массз активною Be
щества должна быть не менее 75 % и не более
125 % от средней высвобождаемой дозы,
определенной в испытании на однородность
высвобождаемой дозы. Это не является ис
пытанием для инrалятора, однако, он может
служить для подтверждения правильности pe
зулыатов.
ПРИБОР А СТЕКЛЯННЫЙ ИМПИНДЖЕР
Прибор изображен на рисунке 2.9.18. 1 (см.
также таблицу 2.9.18. 1).
95
зз r
+
107
45'
t 40
Е
! 63
t 75
G
! f)5.з
G'
I
f) 1.85:t 0.125
Рисунок 2.9.18. 1.
Прибор А: стеклянный импинджер
(размеры указаны в миллиметрах; допустимые
отклонения :!1 мм, если нет дРУ2их указаний)
Методика ДЛЯ небулайзера
Помещают 7 мл и 30 мл ПОДХОДящеro
растворителя в верхнюю и нижнюю ударные
камеры соответственно.
Соединяют все части, расположив прибор
вертикально. Форсунка в нижней части прибо
ра должна только слеrка касаться дна нижней
ударной камеры. Присоединяют насос с филь
тром (с подходящим размером пор) к выпуск
ному отверстию прибора. Скорость возду
ха на входе в roрло устанавливают на уровне
(60:!5) л/мин.
Помещают жидкое лекарственное средство
для инrаляций в резервуар распылителя (небу
лайзера). Надевают мундштук и соединяют с
помощью адаптера с прибором.
Включают насос прибора, и через 10 с
включают распылитель.
Если иноro не указано в частной статье,
через 60 с выключают распылитель, через 5 с
после чеro выключают насос прибора. Демонти
руют прибор И промывают внутреннюю поверх
ность верхней ударной камеры растворителем,
указанным в частной статье. Промывную жид
кость собирают в мерную колбу. Затем промы
вают внутреннюю поверхность нижней yдap
446
rосударственная фармакопея Республики Беларусь
Таблица 2.9.18. 1
Спецификации компонентов для рисунка 2.9.18. 1
Код Компонент Описание Размеры.
! Адаптер Формованный резиновый адаптер для мундштука ;
IA мундштука i
I Видоизмененная круrлодонная колба: 50 мл j
В r орло входное отверстие из притерт020 стекла 29/32 I
HKa вЫПУСКН020 отверстия из притерт020 стекла 24/29
Видоизмененный стеклянный адаптер:
I входное отверстие из притерт020 стекла ,
24/29 ,
I
I воронка вЫПУСКН020 отверстия из притерт020 стекла 24/29 I
I
С Шея I Нижняя часть выпускною отверстия в виде трубки определен
ноro диаметра:
диаметр трубки 14
Отдельная тонкостенная трубка определенною диаметра:
I наружный диаметр 17 ;
I Верхняя ударная Видоизмененная круrлодонная колба: 100 мл
D входное отверстие из притерт020 стекла 24/29
камера I
воронка вЫПУСКН020 отверстия из притерт020 стекла 24/29 !
Стеклянная трубка со стенками средней толщины: !
воронка из притерт020 стекла 14/23 I
Е Соединительная Соrнутая часть и верхняя вертикальная часть: I
трубка наружный диаметр 13
Нижняя вертикальная часть: I
I
; наружный диаметр I
I 8
! Пластмассовый наконечник винта 28/13 I
I Силиконовая резиновая прокладка 28/11 I
I ;
Резьба винта бо PTFE шайба 28/11 i
F ;
ковоro адаптера Стеклянная резьба винта, размер 28 i
I Выпускное отверстие для присоединения вакуумноro насоса: !
I I
I
минимальный диаметр 5
I Модифицированный полипропиленовый держатель для филь
i см.при60Р
I тра, соединенный с нижней вертикальной частью соединитель
I ной трубки с помощью PTFE шайбы 2.9.18. 1
G Нижняя форсунка Ацетальный циркулярный диск, имеющий в центре четыре фор 10 I
сунки, расположенные в диаметре 5,3 мм от общей форсунки: ;
диаметр форсунки 2
выступ форсунки 2
Н Нижняя ударная Коническая колба: 250 мл
камера входное отверстие из притерт020 стекла 24/29
. Размеры указаны в миллиметрах, если нет друrих указаний в частных статьях.
ной камеры. Промывную жидкость собирают
в друrую мерную колбу. Промывают фильтр
насоса и еro соединение с нижней ударной Ka
мерой, промывную жидкость объединяют с
промывной жидкостью, полученной при мытье
нижней ударной камеры. Определяют количе
ственное содержание активноro вещества в
каждой из двух колб аналитическим методом,
описанным в частной статье. Результаты Bыpa
жают для каждой части прибора в процентах по
отношению к общему количеству активноro Be
щества.
Методика для инrаляторов под давлением
Адаптер мундштука помещают в конец
rорла таким образом, чтобы наконечник MYHД
штука при надевании ero на rлубину около 1 О мм
располаrался вдоль roризонтальной оси
roрла, а открытый конец мундштука, который
соединяется с контейнером под давлением,
располаrался выше остальных частей прибо
ра и находился в одной вертикальной плоско
сти с остальными частями прибора.
Помещают 7 мл и 30 мл подходящеrо
растворителя в верхнюю и нижнюю ударные
камеры соответственно.
Соединяют все части, расположив прибор
вертикально. Форсунка в нижней чаt;ти аппара
та должна только слеrка касаться дна нижней
ударной камеры. Присоединяют насос с филь
тром (с подходящим размером пор) к выпуск
ному отверстию прибора. Скорость воздуха
на входе в rорло устанавливают на уровне
60::!:5 л/мин.
2.9.18. Аэродинамическое испытание мелких частиц
447
Подютавливают ДОЗИрУЮЩИЙ клапан,
встряхивая инrалятор в течение 5 с и выпу
ская одну дозу в воздух; не менее чем через
5 с снова встряхивают и выпускают одну дозу
в воздух. Повторяют процедуру еще 3 раза.
Встряхивают инrалятор в течение 5 с,
подключают насос к прибору, соединяют Ha
конечник мундштука с адаптером и быстро
нажимают на дозирующий клапан, опорож
няя дозу лекарственноro средства. Отсоеди
няют инrалятор от адаптера, встряхивают в
течение не менее 5 с, соединяют наконечник
мундштука с адаптером и снова нажимают на
дозирующий клапан. Повторяют описанную
выше процедуру. Число нажатий может быть
минимизировано и обычно не превышает 1 О
раз. После последнею нажатия ожидают 5 с
и отсоединяют насос. Демонтируют прибор.
Промывают внутреннюю поверхность
и внешнюю поверхность (которая выступа
ет внутрь камеры) входной трубки нижней
ударной камеры растворителем, указанным
в частной статье. Промывную жидкость соби
рают в нижней ударной камере. Определяют
количественное содержание активною веще
ства в полученном растворе аналитическим
методом, описанным в частной статье. Pac
считывают количество активною вещества,
собранною в нижней камере, в пересчете
на каждое нажатие, и результаты выражают
в процентах по отношению к дозе, указанной
на этикетке.
Методика для порошковых инrаляторов
Помещают 7 мл и ЗО мл подходящеro
растворителя в верхнюю и нижнюю ударные
камеры соответственно.
Соединяют все части, расположив прибор
вертикально. Форсунка в нижней части прибо
ра должна только слеrка касаться дна нижней
ударной камеры. Присоединяют насос с
фильтром (с подходящим размером пор) к BЫ
пускному отверстию прибора. Скорость воз
духа на входе в rорловину устанавливают на
уровне 60:!::5 л/мин.
rотовят инrалятор к использованию и
вставляют через подходящий адаптер MYHД
штук инrалятора в прибор. Включают насос
на 5 с. Выключают насос и достают инrаля
тор. Повторяют описанную выше процедуру.
Число повторов может быть минимизировано
и обычно не превышает 1 О раз. Демонтируют
прибор.
Промывают внутреннюю поверхность
и внешнюю поверхность (которая выступа
ет внутрь камеры) входной трубки нижней
ударной камеры растворителем, указанным
в частной статье. Промывную жидкость соби
рают в нижней ударной камере. Определяют
количественное содержание активноro веще
ства в полученном растворе аналитическим
методом, описанным в частной статье. Pac
считывают количество активноrо вещества,
собранноro в нижней камере, в пересчете на
одну выпущенную дозу. и результаты выража
ют в процентах по отношению к дозе, указан
ной на этикетке.
Дозирование мелких частиц и распре
деление частиц по размеру
ПРИБОР С мноrОСТУПЕНЧАТЫЙ
ЖИДКОСТНЫЙ ИМПИНДЖЕР
Мноroступенчатый жидкостный импин
джер состоит из ступеней столкновения 1 (пре
сепаратор), 2, 3 и 4 и составной ступени фильтра
(5) (см. рисунки 2.9.18.-4 2.9.18. 6). Ступень
столкновения состоит из верхней roризонталь
ной металлической переroродки (В). через KO
торую выступает входное отверстие трубки (А)
с пластинкой столкновения (О), стеклянною
цилиндра (Е) с отверстием для отбора проб (F),
образующеro вертикальную стенку ступени. и
нижней roризонтальной металлической пере
roродки (G), через которую трубка (Н) соеди
няется со следующей ступенью. Трубка CTY
пени 4 (u) завершается мноroфорсуночно
конструкцией. Пластинка столкновения (О)
крепится на металлическом каркасе (J), KO
торый присоединен двумя проволоками (К) 1<
втулке (L), соединенной с распылителем. 'o
ризонтальная поверхность собирающей пла
стинки перпендикулярна оси распылителя и
расположена по центру. Верхняя поверхность
пластинки столкновения немноro приподня
та над краем металлическоro каркаса. Паз по
периметру rоризонтальной стенки предназна
чен для стеклянноro цилиндра. Стеклянные
цилиндры и rоризонтальные стенки скрепле
ны между собой шестью болтами (N), в месте
их контакта расположена уплотнительная про
кладка (М). Отверстия для отбора проб за
крыты пробками. Дно нижней стенки 4 ступе
ни имеет концентрический выступ, соединение
котороro с фильтром. который вставлен в спе
циальный держатель, уплотнено резиновым
кольцом (Р). Держатель фильтра (R) выполнен
в виде резервуара с концентрическим уrлубле
нием, в котором смонтирована перфориро
ванная подставка для фильтра (S). Держатель
фильтра имеет размер, позволяющий BCTaB
лять фильтры диаметром 76 мм. KOHCTPYK
ция, включающая все ступени, зафиксирована
на держателе фильтра двумя защелками (Т).
Порт индукции (рисунок 2.9.18. 7) соединяют
с распылителем первой ступени импинджера.
Резиновое уплотнительное кольцо распылите
ля обеспечивает rерметичность соединения с
портом индукции. Для rерметичноrо соедине
ния инrалятора и порта индукции используется
специальный адаптер мундштука. Передняя
поверхность мундштука инrалятора должна
совпадать с передней поверхностью входноrо
отверстия порта индукции.
448
rосударственная фармакопея Республики БеларуCh
Q
А
в
l'
I
С М
F/ L
IC .
G. i: 1', /
N
Е
К
Ступень 1
(пре сепаратор )
Н.
Ступень 2
Ступень 3
Ступень 4
Ступень 5
(фильтр)
т
ВЫХОД
R
Рисунок 2.9.18.4. Прибор с: МН020ступенча
тый жидкостный импинджер
центр ступени
{б:х}
i
I
I
.j
s
5
м
, r I
i
.II! I i
I
I н
', 9 j, !
, ' I 1 1
, 1
i С! <о
I I!
, !
I Mj
I
i
i
k
':: C<:
," О ;- ...
Q};:j" , " ,О :
'- @ '
i.j(7X)
I. h (7х)
структура распылителя
N
!
м
4
к
U распыnитель
о
, I
' '
I d !
,
Рисунок 2.9.18. 5. Прибор с: подробная схема
распылителя и пластинки столкновения. На
вставках изображены концы U распылителя
на ступени 4
(номера и строчные буквы соответствуют
обозначениям таблицы2.9.18. 3; прописные
буквы соответствуют обозначениям рисунка
2.9.18. 4)
1381
1360
р
O
10
5
010
R
Рисунок 2.9.18. 6. Прибор с: подробная схема
ступени фильтра (5 ступень).
Цифры обозначают размеры ((ZJ=диаметр).
Прописным буквам соответствуют обозначе
ния в таблице 2.9.18. 2
(размеры указаны в миллиметрах, если нет
дРУ2их указаний в частной статье)
Высверлить расточное отверстие
и соединеНI1е для винта М4,
Примечание: использовать минимапьный
зазор для винта в нижней части
дпя точноro совмещения
19.0
31.8
lза 1 i
! : j , j . с;
" :T : , . :n j ':;
1 , : I I :: ",,,1
. о.....,
I '! 145 rpa!1)'coe " : I . ...... .....i
U1 ", фаска .":i I
. r: о ',о I 1;5 <f' П j
" i .... "" 45". . З1.75+ С . 5 ' I
. :" " ,. ,,- -.00 :@5.4 '
'\: " Ш
",.Q) не повреждать край,
10 C ::t"1 C
"
q
1 .3
, 28-б
Уrловое соединение должно быть неплотным
'.r---....
в rнездо вставлен ..e\ _ "-
винт С roлов ой М4
/
Изометрическии вид порта индукции
Примечание:
(1) Материалом может служить алюминий, нержавеющая
сталь, а также друrое подходящее вещество.
(2) Провести обработку из стержневой заrотовки З8 мм.
(З) Просверлить в стержне канал диаметром 19 мм.
(4) Разрез трубки точно под уrлом 450, как показано.
(5) Внутренние отверстия и конусы должны быть rладкими
шлифовка приблизительно 0,40 мкм,
(6) Соединения выполняются таким образом, чтобы не было
протекания.
(7) Установить зажимное приспособление для выравнивания
канала диаметром 19 мм, а также для сверления и нареза
ния резьбы М4 х О,7. Не должно быть несовпадений внутрен-
них каналов в уrловых соединениях.
Рисунок 2.9.18. 7. Порт индукции
(размеры указаны в миллиметрах, если нет
дРУ2их указаний в частной статье)
2.9.18. Аэродинамическое испытание мелких частиц
449
Таблица 2.9.18. 2
Код* Компонент Описание Размеры**
Металлическая трубка с rладкой внутренней по см. рисунок
А,Н Трубка форсунки верхностью, прикрученная к разделительной 2.9.18. 5
стенке, с уплотнительной прокладкой (С)
Круrлая металлическая пластинка
B,G Разделительная диаметр 120
стенка толщина см. рисунок
2.9.18. 5
С Уплотнительная Например,РТFЕ для уплотнения
прокладка трубки форсунки
Пластинка Стеклянный диск с пористостью О
О диаметр см. рисунок
столкновения 2.9.18. 5
Плоская полированная разрезанная стеклянная
труба
высота, включая уплотнительные прокладки 46
Е Стеклянный внешний диаметр 100
цилиндр толщина стенок 3,5
диаметр отверстия для отбора проб 18
пробка для отверстия для отбора проб /80 24/25
L образный круrлый каркас с пазом
внутренний диаметр подходящий к
пластинке стол
J Металлический кновения
каркас высота 4
толщина 20ризонтальной секции 0,5
толщина вертикальной секции 2
Стальная проволока, связывающая металлический
К Проволока каркас и втулки (по 2 на каждый каркас)
диаметр 1
Металлическая втулка, закрепленная на трубке
форсунки резьбой
L Втулка внутренний диаметр подходящий к
трубке форсунки
высота 6
толщина 5
Уплотнительная Например, силиконовая подходящий к
М стеклянному
прокладка цилиндру
МеталлическиЙ винт в rайкой (6 пар)
N Винт длина 205
диаметр 4
Р Уплотнительное Резиновое уплотнительное кольцо
кольцо диаметр х толщина 66,З4 х 2,62
Q Уплотнительное Резиновое уплотнительное кольцо
кольцо диаметр х толщина 29,1 х 1,6
R Держатель фильтра Металлический корпус со станиной и выходным OT см. рисунок
верстием 2.9.18. 6
Перфорированный металлический лист
S Подставка для диаметр 65
фильтра диаметр отверстия 3
расстояние между центрами отверстий 4
т Защелка
U Распылитель конец распылителя (Н) в мноroфорсуночной KOH см. рисунок
струкции 2.9.18. 5
Спецификации компонентов для рисунков 2.9.18 4-------2.9.18 6
. Ссылки на рисунок 2.9.18.-4.
..Размеры в миллиметрах с допустимым отклонением в соответствии с /SO 2768 щ если нет друrих указаний в частной статье.
57 Зак. 1712.
450
iосударственная фармакопея Республики Беларусь
Таблица 2.9.18. 3
РазмерЫ(1) трубки фОРС}i'ки и f1ластинки столкновения прибора С
j Фильтр I
Тип Ко д ( 2) Ступень 1 Ступень 2 Ступень 3 Ступень 4
(Ступень 5) I
I Расстояние 1 ; 9,5 ( ,0+,5) 5,5 ( ,O+,5) 4,0 ( ,0+,5) 6,0 ( ,0+,5) I
н. о.
" Расстояние 2 26 31 33 30,5 О I
Расстояние 3 8 5 5 5 5 !
Расстояние 4 3 3 3 н. о. I
'-'
Расстояние 5 О 3 3 I 3 3 1
, I
Расстояние 6(3) 20 25 25 25 25
Расстояние 7 н. О. Н. О. н. о. 8,5 н. о.
Диаметр с 25 14 8,0 (х,1) 21 I 14
Диаметр d 50 30 20 30 н. о. i
Диаметр е 27,9 16,5 10,5 23,9 н. о. I
Диаметр f 31,75 ( ,0+,5) 22 14 31 22 I
Диаметр 9 25,4 21 13 30 21
Диаметр h н. О. н. О. н. о. 2,70 (х,5) н. о. I
.
Диаметр J н. О. н. О. н. о. 6,3 н. о. I
Диаметр k н. О. н. О. н. о. 12,6 н. о.
Радиус(4) r 16 22 27 28.5 О
Радиус s 46 46 46 46 н. о.
Радиус t н. о. 50 50 50 50
Уrол w 100 530 530 530 , 530
I Уroл u н. О. н. О. н. о. i 450 н. о.
Уrол , v н. О. н. О. н. о. 600 н. о.
(1) Размеры в миллиметрах с допустимым отклонением в соответствии с {50 2768 т. если нет друrих указаний в частной СТ2тье
(2) Относится 1( рисунку 2.9.18. 5.
(3) Включая уплотнительную ПрОКЛ8Дку.
(4) Относительно центра переrородки ступени.
н. о. не относится.
Методика для инrаляторов под
давлением
20 мл растворителя, указанноro в частной
статье, предназначенноrо для растворения aK
тивноrо вещества, распределяют по ступеням
1 и закрывают пробками. Наклоняют прибор,
чтобы смочить пробки для нейтрализации элек
тростатическоro заряда. Вставляют фильтр для
сборки активноro вещества на 5 ступени и соби
рают прибор. Соответствующий адаптер MYHД
штука присоединяют к концу входноro отверстия
порта индукции таким образом, чтобы наконеч
ник мундштука находился на одной линии с ro
ризонтальной осью входноro отверстия порта,
а инrалятор был ориентирован в рабочую пози
цию. К выходному отверстию прибора присоеди
няют вакуумный насос и реryлируют поток возду
ха через прибор со скоростью на входе 30 л/мин
(:1:5 %). Выключают насос.
Если в инструкции по применению нет друrих
указаний, встряхивают инrалятор в течение 5 с
и опорожняют одну дозу в воздух. Присоединя
ют насос к прибору, вставляют мундштук в адап
тер и высвобождают дозу инrалятора в прибор.
Нажимают дозирующий клапан до полноro BЫ
свобождения дозы. Через 5 с отсоединяют ин
rалятор от адаптера. Повторяют процедуру. Ko
личество выпусков должно быть минимальное,
как правило, не более десяти. Через 5 с после
последнеro высвобождения отключают насос.
Демонтируют ступень фильтра прибора. AK
куратно извлекают фильтр и смывают активное
вещество аликвотой растворителя, указанноrо
в частной статье. Отсоединяют порт индукции
и адаптер мундштука от прибора и смывают aK
тивное вещество аликвотой растворителя. При
необходимости ополаскивают внутреннюю по
верхность распылителя ступени 1 раствори
телем, позволяя ему свободно течь. Смывают
активное вещество с внутренних стенок и co
бирающей пластинки всех четырех верхних CTY
пеней прибора в раствор соответствующих CTY
пеней, осторожно поворачивая прибор. Следят,
чтобы жидкость не проникала из одной ступени
в друryю.
Используя аналитический метод, указанный
в частной статье, определяют количество актив
Horo вещества в каждой аликвоте растворите
лей.
Рассчитывают концентрацию мелких частиц
(см. раздел Расчеты).
Методика для порошковых инrаляторов
Вставляют низкорезистентный фильтр для
сбора актиноrо вещества на ступени 5 и соби
рают прибор. Присоединяют прибор к системе
реryлирования скорости потока в соответствии
со схемой, представленной на рисунке 2.9.18.--8
2.9.18. Аэродинамическое испытание мелких частиц
451
и в таблице 2.9.18. 4. Если нет друrих указаний,
выполняют испытание при скорости потока 0out'
как в испытании на определение однородности
дозы, пропуская через прибор с мундштуком ин
rалятора 4 литра воздуха.
К порту индукции присоединяют объем
ный счетчик, откалиброванный для измерения
объема выходноro потока, либо рассчитывают
выходной поток, используя закон для идеально
ro rаза (Оои"- Для измерительных приборов, Ka
либрованных на входной поток (чп>, используют
следующее уравнение:
о О;п . РО ,
ои! р. дP
о
rде:
РО атмосферное давление;
I::1P падение давления на измерительном
приборе.
Клапан реryлирования расхода настраива
ют на постоянную скорость потока 0ои/ (::1:5 %)
через систему. Отключают насос.
Удостоверяются, что критический поток KOH
тролируется в клапане реryлирования потока во
время проведения процедуры. Устанавливают
инrалятор и при установленной испытательной
скорости потока измеряют абсолютное давле
ние с обеих сторон контрольноro клапана (дaB
ление в точках Р2 и Р3 на рисунке 2.9.18. 8).
Соотношение Р3/Р2 равное 0,5 и меньше сви
детельствует о критическом потоке. Если крити
ческоro потока не достиrнуто, переключаются на
более мощный насос и измеряют испытатель
ную скорость потока.
Е
D
РЗ Р2
Вакуумный
насос
Импактор
А
Двухсторонний
электромаzнuтный
клапан
Клапан
реzулирования
расхода
с F
Вакуумная трубка
Соединитель
А
в
Рисунок 2.9.18. 8. Экспериментальный прибор
для испытания порошковых иН2аляторов
20 мл растворителя, указанноro в част
ной статье, предназначенноro для paCTBope
ния активноro вещества, распределяют по че
тырем верхним ступеням прибора и закрывают
пробками. Наклоняют прибор, чтобы смочить
пробки для нейтрализации электростатическоro
заряда. К входному отверстию порта индукции
присоединяют подходящий адаптер мундштука.
rотовят порошковый инrалятор в COOTBeT
ствии с инструкцией по применению. При pa
ботающем насосе и закрытом двустороннем
электромаrнитном клапане вставляют MYHД
штук инrалятора в адаптер. Высвобождают по
Спецификации компонентов для рисунка 2.9.18 8
Таблица 2.9.18. 4
КОД Компонент Описание
А Соединитель Внутренний диаметр 8 мм, например, короткое металлическое cцe
пление с отводом малоro диаметра к Р3
В Вакуумная трубка Отрезок подходящей трубки с внутренним диаметром 8 мм и BHY
тренним объ<емом (25::1:5) мл
Двусторонний Двусторонний электромаrнитный клапан с двумя выходами, с мини
С электромаrнитный мальной сопротивляемостью воздуха отверстия при внутреннем ди
аметре 8 мм и времени открытия s100 мс (например, тип 256 A08,
клапан Bu,kert GmbH, O 74653 /пgеlfiпgеп, или аналоrичный)
Насос должен обеспечивать необходимый поток воздуха через co
бранный прибор с порошковым инrалятором (например, тип 1023,
1423 или 2565, Gast Мапиfасtи,iпg /пс., Вепtоп На,Ьо" М/, 49022 или
D Вакуумный насос аналоrичный). Соединение насоса и электромаrнитноrо клапана
осуществляется с помощью короткоro и/или широкоro (внутренний
диаметр 10 мм) вакуумноro шланrа и соединителей, для уменьше
ния требований к емкости насоса
Таймер, способный управлять электромаrнитным клапаном на за
Е Таймер данном протяжении времени (например, тип G814 RS Сотропепts
/пtеrпаtiопа/, Со,Ьу, NN17 9RS, Великобритания, или аналоrичный)
Р2 Р3 Измерения давлений Измерения осуществляются в условиях постоянноro тока с TpaHCДYK
тором абсолютноro давления
Клапан реryлирования Реryлируемый клапан, имеющий максимальное значение Cv 1, (Ha
F при мер, тип 8FV12LNSS, Pa,ker Наппifiп р/с., Barпstap/e, ЕХ31 1NP,
расхода Великобритания, или аналоrичный)
452
rосударственная фармакопея Республики Беларусь
рошок В прибор, открывая клапан на необхо
димое время Т (:t5 %). Повторяют проце.,уру.
Количество высвобождений должно быть ми....и
мальным, как правило, не более десяти. Число
высвобождений должно быть достаточным для
BepHoro и точноrо определения доз мелких
частиц.
Демонтируют ступень фильтра прибора. AK
куратно извлекают фильтр и смывают активное
вещество аликвотой растворителя. Отсоединя
ют порт индукции и адаптер мундштука от при
бора и смывают активное вещество аликвотой
растворителя. При необходимости ополаски
вают растворителем внутреннюю поверхность
распылителя ступени 1, позволяя растворите
лю течь. Смывают активное вещество с BHY
тренних стенок и собирающей пластинки всех
четырех верхних ступеней прибора в раствор
соответствующих ступеней, осторожно повора
чивая прибор. Следят, чтобы жидкость не про
никала из одной ступени в друryю.
Используя аналитический метод, указанный
в частной статье, определяют количество aK
тивноro вещества в каждом собранном объеме
растворителей.
Рассчитывают концентрацию мелких частиц
(см. раздел Расчеты).
АППАРАТ О КАСКАДНЫЙ ИМПАКТОР
АНДЕРСЕНА
Каскадный импактор Андерсена (Апdеr
seп) 1 ACFM состоит из 8 ступеней и конеч
ноro фильтра. Прибор может быть выполнен
из алюминия, нержавеющей стали или иноro
подходящеrо материала. Ступени соедине
ны уплотнительными кольцами. Основные
критические размеры, закладываемые изro
товителями прибора О, приведены в табли
це 2.9.18. 5. При использовании прибора воз
можно появление закупорок и изнашивания
отверстий. Допуски размеров при использо
вании при бора необходимо обосновывать. В
конфиrурациях, используемых в инrаляторах
под давлением (рисунок 2.9.18. 9), входная
часть импактора соединена с портом индук
ции (рисунок 2.9.18. 7). Для обеспечения rep
метичности соединения инrалятора и порта
используют подходящий адаптер мундштука.
Передняя поверхность мундштука инrалятора
должна совпадать с передней поверхностью
входноro отверстия.
В конфиrурациях, используемых в по
рошковых инrаляторах, пре сепаратор pac
полаrается над верхней ступенью для сбора
порошка, не попавшеro в дыхательные пути.
Он соединяется с портом индукции (РИСУ'-IOк
2.9.18. 10). Чтобы обеспечить быстрое про
хождение потока через импактор, выступа
ющий патрубок, который используется для co
единения импактора с вакуумной системой,
увеличивают, чтобы ero внутренний диаметр
был не менее 8 мм.
Таблица 2.9.18. 5
Основные критические размеры для прибора О
Описание Число Размеры (мм)
Ступень о: диаметр 96 2,55:t0,025
распылителя
Ступень 1: диаметр 96 1,89:t0,025
распылителя
Ступень 2: диаметр 400 0,914:t0,0127
распылителя
Ступень 3: диаметр 400 0,711 :t0,0127
распылителя
Ступень 4: диаметр 400 0,533:t0,0127
распылителя
Ступень 5: диаметр 400 0,343:t0,0127
распылителя
Ступень 6: диаметр 400 0,254:t0,0127
распылителя
Ступень 7: диаметр 201 0,254:t0,0127
распылителя
/
/1
i
i
i
i
i
i
i
i
j
i
i
I
i
//
j у
{ \
r 1 ili Iirili . ..
t 11 III
: 1 l-1
!в . '. '1'.
;8'
I ,?: :
' O ']"
f L :1
.. . . ' I
. . ш 1ir iJJili ili
: !! I ! 'i. I
! . j- . . /i
1.. 1"
.. ,)
r.r;
:"=i
k 1
i
; I I
i
i
I
'т'
i I ,1=<
:... '";: . ' '
;f!
.II: ,
.' , :""dr" :,, . i
Ifh ' . . . LE": .
I
Рисунок 2.9.18. 9. Каскадный импактор
Андерсена для иН2аляторов под давлением
2.9.18. Аэродинамическое испытание мелких частиц
453
/107
//106
/ 102
"
'<"'100
'-. 94 Материал: алюминий. нержавеющая сталь
или дРУ20Й подходящий материал
Кроме ozoeopeHHblx случаее все края должны
быть отломаны и удалены заусенцы
Поверхность должна быть чисто отполирована
механическим способом
Шероховатость внутренней поверхности
отверстия Ra ОКОЛО 0.4 МКМ
о Уплотнительное КОЛЬЦО: размеры: внутренний
диаметр 29 мм. наружный диаметр 32 мм.
толщина 1,8 мм
44 Ji'?1
38 з+0.2 .
19" :..о I
15
14.4
13 .
8
Вид сверху
;/
/
Rб.7(h().ОЗ '
Не повреждать край
4 5 0+ 3 0 /
".,/
1О 0 :!: 10
...--....t.. .......
.......I... .
Вид сбоку
I
i
i
i
Не повреждать край
Поперечный разрез
R12.7...........J
/А12.7
А15
A15.87 ?o5
А14.4
.. Паз для уплотнительною
кольца
А19
А44
кольца
......А15.87 5
А8 А38.3 .2
А6.70 :t 0.03
27
/
:::"""22
14
Рисунок 2.9.18. 10. Присоединение порта индукции
к пре сепаратору каскадН020 импактора Андерсена
(размеры указаНbI в миллиметрах, если нет дРУ2их указаний в частной статье)
Методика для инrаляторов под давлением
Собирают импактор с соответствующим
фильтром и проверяют систему на rерметич
ность в соответствии с рекомендациями про
изводителя прибора. Адаптер мундштука pac
полаrают в KOHЦ входноro отверстия порта
индукции таким образом, чтобы конец наконеч
ника мундштука находился на одном уровне с
rоризонтальной осью входною отверстия, а ин
rалятор был ориентирован в рабочую позицию.
Насос соединяют с выходным отверстием при
бора и реryлируют поток воздуха через прибор
со скоростью на входе 28,3 л/мин (:1:5 %). OT
ключают насос.
Если нет друrих указаний в инструкции по
применению, встряхивают инrалятор в течение
5 с и высвобождают одну дозу в воздух. Присо
единяют насос к прибору, вставляют мундштук
в адаптер и высвобождают дозу инrалятора, пе
ревернув еro вверх дном, в прибор. На)l(имают
дозирующий клапан до полноro высвобождения
дозы. Через 5 с отсоединяют инrалятор от адап
тера. Повторяют процедуру. Количество выпу
сков должно быть минимальное, как правило, не
более десяти. Через 5 с после последнеro BЫ
свобождения отключают насос.
58. За.. 1712.
Демонтируют ступень фильтра прибора.
Аккуратно извлекают фильтр и смывают актив
ное вещество аликвотой растворителя, указан
ноro в частной статье. Отсоединяют порт индук
ции И адаптер мундштука от прибора и смывают
активное вещество растворителем. При необ
ходимости смачивают распылитель снаружи
растворителем. Смывают активное вещество
с внутренних стенок и собирающей пластинки
всех ступеней прибора али квота ми . растворите
ля, указанноro в частной статье.
Используя аналитический метод, указанный
в частной статье, определяют количество актив
ноro вещества в каждом собранном объеме pac
творителя.
Рассчитывают концентрацию мелких частиц
(см. раздел Расчеты).
Методика для порошковых инrаляторов
rраниЦbl аэродинамических диаметров для
каждой ступени прибора в настоящее время
являются недостаточно изученными при CKO
рости потока, отличающейся от 28,З л/мин.
Использование прибора при скорости потока,
отличающейся от 28,3 л/мин, должно быть вали
дировано.
454
rосударственная фармакопея Республики Беларусь
Собирают импактор с пре сепаратором и
соответствующим фильтром и проверяю" си
стему на rерметичность. В зависимости от
характеристик испытуемоro образца и если
это обосновано и утверждено, пре сепаратор
может отсутствовать. При соответствующем
обосновании ступени 6 и 7 также MorYT OTCYT
ствовать. Пре сепаратор может быть покрыт
таким же образом, как и пластины, или может
содержать 10 мл подходящеro растворителя.
Присоединяют прибор к системе реryлирова
ния потока в соответствии со схемой, пред
ставленной на рисунке 2.9.18. 8 и в таблице
2.9.18. 4. Если нет друrих указаний, выполняют
испытание при скорости потока 0ои!' как в ис
пытании на определение однородности дозы,
пропуская через прибор с мундштуком инrаля
тора 4 литра воздуха.
К порту индукции присоединяют объем
ный счетчик. откалиброванный для измерения
объема выходноro потока, либо рассчитывают
выходной поток, используя закон для идеаль
ноro rаза (Ос). Для измерительных приборов,
калиброванных на входной поток (Oi)' исполь
зу ют следующее уравнение:
О = От . Ро
ои! Ро др
rдe:
Ре атмосферное давление;
.'-.Р падение давления на измерительном
приборе.
Клапан реrулирования расхода HaCTpa
ивают на постоянную скорость потока Оои!
(:t5 %) через систему. Удостоверяются, что
критический поток контролируется в клапане
реrулирования потока в соответствии с про
цедурой, описанной для прибора С. Отключа
ют насос.
rотовят порошковый инrалятор в COOTBeT
ствии с инструкцией по применению. При pa
ботающем насосе и закрытом двустороннем
электромаrнитном клапане вставляют MYHД
штук инrалятора в адаптер. Высвобождают
порошок в прибор, открывая клапан на необ
ходимое время Т (:t5 %). Повторяют процеду
ру. Количество высвобождений должно быть
минимальным, как правило, не более десяти.
Число высвобождений должно быть достаточ
ным для верноro и точноrо определения доз
мелких частиц.
Демонтируют ступень фильтра прибора.
Аккуратно извлекают фильтр и смывают aK
тивное вещество аликвотой растворителя.
Отсоединяют пре сепаратор, порт индукции
и адаптер мундштука от прибора и смывают
активное вещество аликвотой растворителя.
Смывают активное вещество с внутренних
стенок и собирающей пластинки всех ступе
ней прибора аликвотами растворителя, YKa
занноrо в частной статье.
Используя аналитический метод, указан
ный в частной статье, определяют количе
ство активноro вещества в каждом собранном
объеме растворителя.
Рассчитывают концентрацию мелких
частиц (см. раздел Расчеты).
ПРИБОР Е
Прибор Е представляет собой каскадный
импактор с 7 ступенями и коллектором с ми
кроотверстиями (micro orifice collector, МОС).
В диапазоне скорости потока от 30 л/мин до
100 л/мин диапазон предельных диаметров,
определяемых с 50 % эффективностью (зна
чение 050)' составляет от 0,24 мкм до 11,7 мкм.
Кривая эффективности сбора для каждой CTY
лени имеет малый радиус кривизны и сводит к
минимуму наложения между ступенями.
Прибор может быть изroтовлен из алюми
ния, нержавеющей стали или иноro подходя
щеrо материала.
Конфиrурация импактора включает в себя
съемные импакторные (ударные) чашки, KO
торые располаrаются в одной плоскости (ри
сунки 2.9.18. 11 2.9.18. 14). При бор COCTO
ит из трех основных частей: нижней рамы, в
которой закрепляются импакторные чашки;
уплотнительноro устройства, включающеro в
себя распылители (форсунки); крышки, в KO
торой находятся межступеневые каналы (ри
сунки 2.9.18. 11 2.9.18. 12). Составные pac
пылители находятся на всех ступенях, кроме
первой (рисунок 2.9.18. 13). Поток протекает
через пилообразный импактор.
Основные критические размеры приведе
ны в таблице 2.9.18. 6.
Порт индукции
.),
Пре enаратор
Корпус и,,"пактора
. '\
r J?
. q . . /!
)y, I
J" "
т? / Выходное отверстие
потока воздуха
Зажимной механизм
Рисунок 2.9.18. 11. Прибор Е (изображено
с установленным пре сепаратором)
2.9.18. Аэродинамическое испытание мелких частиц
455
Форсунка ступени 1
Межступеневые пропускные каналы
Коллектор
с микроотверстиями
Крышка
с уплотняющим
устройством
Направляющий штифт
Выемка для
направляющеrо штифта
Нижняя рама
с установленными
импакторными чашами
Рисунок 2.9.18. 12. Прибор Е с указанием деталей
Стулень 2
6 отверстий
Ступень 4
52 отверстия
Ступень 6
396 отверстий
мое
4032 отверстий
Ступень 1
1 отверстие
Ступень 3 <
24 отверстия
Ступень 5
152 отверстия
Ступень 7
630 отверстий
Поддон
для чашек
Межступеневый пропускной
канал до следующей ступени
Межступеневый пропускной
канал с предыдущей ступени
!b
с
Крышка
Уплотняющее
устройство
Поддон для чашек
Нижняя рама
Коллекторная чаша J Мноrофорсуночная
ступень
Рисунок 2.9.18. 14. Прибор Е: конфи2урация межступеневых ПРОПУСКНЫХ каналов
456
rосударственная фармакопея Республики Беларусь
/ Корпус пре сепаратора
/
(
u
t
L.........J ] .......
u
t
[f
1J
D OWO " pт";
Диаметр распылителя,
размер а
Центральная чаша
'" Вставка
пре сепаратора
Рисунок 2.9.18. 15. Прибор Е: конфи2урация пре сепаратора
Таблица 2.9.18. 6
Основные критические размеры для прибора Е
Описание Размер (мм)
Пре сепаратор (размер а см. рисунок 2.9.18. 15) (12,8+0,05)
Ступень 1 * Диаметр распылителя (14,3:t0,05)
Ступень 2* Диаметр распылителя (4,88+0,04 )
Ступень 3* Диаметр распылителя (2,185:t0,02)
Ступень 4* Диаметр распылителя (1 ,207:t0,01)
Ступень 5* Диаметр распылителя (O,608:t0,01 )
Ступень 6* Диаметр распылителя (0,З2З:t0,01 )
Ступень 7* Диаметр распылителя (0,206:t0,01 )
МОС* около 0,070
rлубина чашки (размер Ь см. рисунок 2.9.18. 14) (14,625+0,10)
Шероховатость поверхности чашки (Ra) 0, 2 мкм
Ступень 1 расстояние от распылителя до уплотняющеro устройства** размер с (0+1,18)
Ступень 2 расстояние от распылителя до уплотняющеro устройства** размер с (5,236:t0,736)
Ступень 3 расстояние от распылителя до уплотняющеro устройства** размер с (8,445+0,41 О)
Ступень 4 расстояние от распылителя до уплотняющею устройства** размер с (11,379:t0,237)
Ступень 5 расстояние от распылителя до уплотняющею устройства** размер с (13,176+0,341 )
Ступень 6 расстояние от распылителя до уплотняющею устройства** размер с (1 3,999:t0,071)
Ступень 7 расстояние от распылителя до уплотняющеro устройства** размер с (14,000+0,071 )
МОС расстояние от распылителя до уплотняющеro устройства** размер с (14,429 14,571)
. Смотри рисунок 2.9.18. 1З.
.. Смотри рисунок 2.9.18. 14.
При рутинных операциях крышка и уплотни
тельное устройство скреплены вместе в единое
целое. После окончания испытания при подня
той верхней части импакторные чашки становят
ся доступными для обслуживания. Все чашки
расположены на одном поддоне таким образом,
что они одновременно все MOryT быть извлече
ны путем поднятия этоro поддона.
Порт индукции с внутренним диаметром
(иrрающим большую роль в скорости потока
воздуха), указанным на рисунке 2.9.18. 7, co
единяется с входным отверстием импактора.
При необходимости, обычно при испытании по
рошковых инrаляторов, может быть использо
ван пре сепаратор, располаrающийся между
портом индукции и импактором. Для обеспече
ния rерметичности соединения инrалятора и
порта индукции используют подходящий адап
тер мундштука.
Прибор Е имеет терминальный коллектор
с микроотверстиями (МОС), который, в случае
большинства лекарственных средств, при co
ответствующей валидации позволяет избежать
последней стадии фильтрования. МОС являет
ся пластинкой импактора с номинальным коли
чеством отверстий 4032, каждое с диаметром
2.9.18. Аэродинамическое испытание мелких частиц
457
около 70 мкм. Большинство частиц, не захва
ченных на седьмой ступени, будут захвачены
поверхностью чашки под МОС. В случае им
пактора, используемоrо при скорости потока
60 л/мин, МОС обеспечивает поимку 80 %
частиц с диаметром 0,14 мкм. Для лекарствен
ных средств, содержащих большую фракцию
частиц, не захватываемых МОС, может быть
использован держатель фильтра, который
может быть установлен вместо МОС либо pac
полаrаться после нею (подходящим является
фильтр из стекловолокна).
Методика для инrаляторов под давлением
Чашки устанавливают в поддон, который
затем вставляют в нижнюю раму и опускают на
место. Закрывают импактор крышкой с присо
единенным уплотнительным устройством и rep
метично закрывают с помощью ручки.
Присоединяют порт индукции с внутренними
диаметрами, указанными на рисунке 2.9.18. 7,
к входу импактора. Адаптер мундштука распо
лаrают в конце порта индукции таким образом,
чтобы конец мундштука находился на одном
уровне с rоризонтальной осью порта, а инrа
лятор был ориентирован в рабочую позицию.
Передняя поверхность мундштука инrалято
ра должна совпадать с передней поверхностью
порта индукции. Насос соединяют с выходным
отверстием прибора и реryлируют поток возду
ха через прибор со скоростью на входе 30 л/мин
(:t5 %). Отключают насос.
Если нет друrих указаний в инструкции по
применению, встряхивают инrалятор в течение
5 с и высвобождают одну дозу в воздух. Присо
единя ют насос к прибору, вставляют мундштук
в адаптер и высвобождают дозу инrалятора, пе
ревернув ero вверх дном, в прибор. Нажимают
дозирующий клапан до полною высвобождения
дозы. Через 5 с отсоединяют инrалятор от адап
тера. Повторяют процедуру. Количество выпу
сков должно быть минимальное, как правило, не
более десяти. Через S с после последнеro BЫ
свобождения отключают насос.
Демонтируют прибор и собирают активное
вещество: отсоединяют порт индукции и адап
тер мундштука от прибора и смывают отложения
активноro вещества аликвотой растворителя. С
помощью ручки открывают импактор и ПОДнима
ют крышку. Достают поддон для чашек вместе с
чашками и аликватами растворителя смывают
активное вещество из каждой чашки.
Используя аналитический метод, указанный
в частной статье, определяют количество актив
ноro вещества в каждом собранном объеме pac
творителя.
Рассчитывают концентрацию мелких частиц
(см. раздел Расчеты).
Методика для ПОрОШковых инrаляторов
Собирают импактор с пре сепаратором
(рисунок 2.9.18. 15). В зависимости от xapaK
59. Зак. 1712.
теристик испытуемоro образца и если это обо
сновано и утверждено, пре сепаратор может OT
сутствовать.
Чашки устанавливают в поддон, который
затем вставляют в нижнюю раму и опускают на
место. Закрывают импактор крышкой с присо
единенным уплотнительным устройством и rep
метично закрывают с помощью ручки.
При использовании пре сепаратора присо
единяют вставку пре сепаратора к основанию
пре сепаратора. Присоединяют основание пре
сепаратора к входному отверстию импактора.
Прибавляют 15 мл растворителя, использующе
roся для смыва активноrо образца, в централь
ную чашку вкладыша пре сепаратора. На верх
собранной конструкции помещают корпус пре
сепаратора и закрепляют с помощью двух зажи
мов.
Присоединяют порт индукции с внутренними
диаметрами, указанными на рисунке 2.9.18. 7,
к входу импактора или входу пре сепаратора.
Адаптер мундштука располаrают в конце порта
индукции таким образом, чтобы конец мундшту
ка находился на одном уровне с rоризонтальной
осью порта, а инrалятор был ориентирован в pa
бочую позицию. Передняя поверхность MYHД
штука инrалятора должна совпадать с передней
поверхностью порта индукции. Присоединяют
прибор к системе реryлирования потока в соот
ветствии со схемой, пред ставлен ной на рисунке
2.9.18. 8 и в таблице 2.9.18А.
Если нет друrих указаний, выполняют испы
тание при скорости потока Qou/' как в испытании
на определение однородности дозы, пропуская
через прибор с мундштуком инrалятора 4 литра
воздуха.
К порту индукции присоединяют объем
ный счетчик, откалиброванный для измерения
объема выходноro потока, либо рассчитывают
выходной поток, используя закон для идеально
ro rаза (QQ()' Для измерительных приборов, Ka
либрованных на входной поток (Q,п), используют
следующее уравнение:
Q О;п . РО
ои/ р. дp'
о
rдe:
РО атмосферное давление;
др падение давления на измерительном
при боре.
Клапан реryлирования расхода настраива
ют на постоянную скорость потока QOu! (:t5 %)
через систему. Отключают поток. Удостоверя
ются, что критический поток контролируется в
клапане реryлирования потока в соответствии с
процедурой, ('писанной для прибора С. Отклю
чают поток Воздуха.
rотовят порошковый инrалятор в COOTBeT
ствии с инструкцией по применению. При pa
ботающем насосе и закрытом двустороннем
электромаrнитном клапане вставляют MYHД
штук инrалятора в адаптер. Высвобождают по
rосударственная фармакопея Республики Беларусь
Таблица 2.9.18. 7
Расчеты для прuбора С. ИСПОЛЬЗУЮ.l1 q == .J (60 / О), 2де О скорость потока во время
испытания, л/мин (От для порошковых иН2аляторов)
458
I Масса отложившеrося Кумулятивная масса Кумулятивная !
Предельный I активноrо вещества, на отложившеrося I
фракция активноrо !
диаметр (мкм) активноrо вещества, на
одно высвобождение одно высвобождение вещества (%) :
d 4 =1,7'q масса на ступени 5, т 5 * с 4 =m 5 f 4 =(c/c)'100 I
d 3 =3,1'q масса на ступени 4, т 4 с з =с 4 +т 4 f з =(С/С)'100 I
d 2 =6.8.q масса на ступени 3, т з С 2 =с з +т з f 2 =(ci c )'100 !
масса на ступени 2, т 2 С=С 2 +т 2 100 I
* Ступень 5 является ступенью фильтра.
Таблица 2.9.18. 8
Расчеты для прибора О, использующе20СЯ при скорости потока 28,3 л/мин
Масса отложившеrося Ку'мулятивная масса Кумулятивная I
Предельный действующеrо веще- отложившеrося актив- фракция активноrо
диаметр (мкм) ства, на одно высво- Horo вещества, на одно
бождение высвобождение вещества (%)
d 7 =0,4 масса на ступени 8, тв С 7 =m 8 f 7 =(c/c)'100
d 6 =0.7 масса на ступени 7, m 7 С 6 =С 7 +т 7 f 6 =(сб'с)'100
d 5 =1.1 масса на ступени 6, т 6 с 5 =с 6 +т 6 f s =(ci c )'100
d 4 =2.1 масса на ступени 5, m 5 с 4 =с 5 +т 5 f 4 =(c/c)'100
d з =3,3 масса на ступени 4, m 4 с з =с 4 +m 4 f з =(С/С)'100
d 2 =4.7 масса на ступени 3, т з с 2 =с з +т з f 2 =(ci c )'100
d.=5.8 масса на ступени 2. т 2 с 1 =с 2 +т 2 f 1 =(c/c)'100
d=9,0 масса на ступени 1, то с о =с 1 +т 1 f o =(ci c )'100
масса на ступени О, т 1 с=с о +т о 100
Таблица 2.9.18_ 9
Расчеты для прибора Е Используют q == (60/ оу, 2де О скорость потока во время
испытания, л/мин, а х указанное в таблице значение
, Масса отложившеrо- Кумулятивная масса Кумулятивная
Предельный I ся активноrо веще- отложившеrося ак- фракция
диаметр (мкм) I х ства, на одно высво- тивноrо вещества, на
активноrо
бож.аение опно высвобожпение вешества (%)
d 7 =0,34'q 0,67 масса из МОС или с с 7 =т в f 7 =(c/c)'100
фильтра, m в
d 6 =0,55'q 0,60 масса на ступени 7, m 7 С 6 =С 7 +m 7 f 6 =(сб'с)'100
d 5 =0,94'q I 0,53 масса на ступени 6, m 6 С 5 =С 6 +m 6 f 5 =(ci c )'100
d 4 =1,66'q ! 0,47 масса на ступени 5, m 5 с 4 =с 5 +т 5 f 4 =(C/C)'100
d 3 =2,82'q 0,50 масса на ступени 4, m 4 С З =С 4 +m 4 f з =(с/с)'100
d 2 =4,46'q 0,52 масса на ступени 3, m з С 2 =с з +т з f 2 =(ci C )'100
d 1 =8,06'q 0.54 масса на ступени 2, m 2 С 1 =С 2 +т! f 1 =(c/c)'100
масса на ступени 1, mо С=С 1 +т 1 100
рошок В прибор, открывая клапан на необхо
димое время Т (:t5 %). Повторяют процедуру.
Количество высвобождений должно быть мини
мал ным, как правило, не более десяти. Число
высвобождений должно (5ыть достаточным для
верноro и точноro определения доз мелких
частиц.
Демонтируют прибор и собирают активное
вещество: отсоединяют порт индукции и адап
тер мундштука от пре сепаратора, если он был
использован, и смывают отложения активно
ro вещества аликвотой растворителя. Если ис
пользовался пре сепаратор, еro аккуратно OT
соединяют от импактора, избеrая попадания
жидкости в импактор. Смывают активное веще
ство с пре сепаратора.
С помощью ручки открывают импактор и
поднимают крышку. Достают поддон для чашек
вместе с чашками и аликвотами растворителя
смывают активное вещество из каждой чашки.
Используя аналитический метод, указанный
в частной статье, определяют количество актив-
2.9.19. Заzрязнение механическими включениями: невидимые частицы
459
ною вещества в каждом собранном объеме pac
творителя.
Рассчитывают концентрацию мелких частиц
(см. раздел Расчеты).
РАСЧЕТЫ
При испытании растворов рассчитывают
массу активноro вещества, отложившуюся на
каждой ступени в расчете на одно высвобожде
ние, и массу активною вещества в расчете на
одно высвобождение в порте индукции, адап
тере мундштука и, если используется, в пре
сепараторе.
Начиная с конечноro места сбора (фильтр
или МОС), определяют кумулятивную массу на
предельный диаметр соответствующей ступе
ни (таблица 2.9.18. 7 для прибора С, таблица
2.9.18. 8 для прибора О, таблица 2.9.18. 9 для
прибора Е). Методом интерполяции рассчитыва
ют массу активноro вещества менее 5 мкм. Это
концентрация мелких частиц (КМЧ KOHцeHTpa
ция мелких частиц).
Если необходимо, и rдe это применимо (Ha
пример, в случае, если наблюдается нормаль
ное лоrарифмическое распределение), строят
rрафик зависимости кумулятивной фракции
активноro вещества от предельноro диаме
тра (таблицы 2.9.18. 7 2.9.18. 9) на лоrариф
мической бумаrе. Этот rрафик используют для
определения значений средней массы аэроди
намическоro диаметра (СМАД средняя масса
аэродинамическоro диаметра) и стандартноro
rеометрическоrо отклонения (CrO CTaHдapT
ное rеометрическое отклонение). Для расчетов
можно использовать соответствующие компью
терные проrраммы.
01/2013:20919
2.9.19. ЗАrРЯЗНЕНИЕ
МЕХАНИЧЕСКИМИ
ВКЛЮЧЕНИЯМИ:
НЕВИДИМЫЕ ЧАСТИЦЫ
Механическими включениями в инъекцион
ных и инфузионных растворах являются ино
родные подвижные нерастворимые частицы, за
исключением пузырьков rаза, случайно оказав
шиеся в растворе.
Два метода определения заrрязнения Mexa
чическими включениями описаны ниже: метод 1
метод подсчета количества частиц в режиме CBe
тотени и метод 2 метод подсчета частиц при
помощи микроскопа. При испытании инъекцион
ных и инфузионных растворов на наличие Mexa
нических включений, невидимых невооруженным
rлазом, использование метода 1 является пред
почтительным. Однако чтобы сделать заклю
чение о соответствии требованиям, предъяв
ляемым к лекарственному средству, после про
ведения подсчета частиц в режиме светотени
для некоторых лекарственных средств может
возникнуть необходимость подсчета частиц Me
тодом 2.
Не все парентеральные лекарственные
средства MOryT быть испытаны на наличие Me
ханических включений, невидимых HeBOOpy
женным rлазом, с использованием одноro или
обоих этих методов. Korдa метод 1 неприменим
(например, если лекарственное средство Heдo
статочно прозрачно или обладает повышенной
вязкостью (например, эмульсии, коллоидные и
липосомальные лекарственные средства)), ис
пытание проводят по методу 2. Аналоrично для
испытания лекарственных средств, в которых
происходит выделение воздуха или пузырьков
rаза при их помещении в измерительное устрой
ство, может потребоваться проведение подсче
та количества частиц при помощи микроско
па. Если вязкость испытуемоro лекарственноro
средства слишком высока для тоro, чтобы про
водить испытание при помощи одною из описан
ных методов, для понижения вязкости проводят
количественное разбавление путем добавления
соответствующею количества разбавителя.
Результаты, полученные при испытании
каждоro отдельноro образца или rруппы образ
ЦОВ на заrрязнение механическими включени
ями, не MOryT быть с уверенностью экстрапо
лированы на друrие, неиспытанные, образ
цы. Таким образом, должны быть разработаны
статистически достоверные схемы отбора об
разцов, при условии, что выводы об уровне за
rрязнения частицами делаются на основании
анализа большой rруппы образцов.
МЕТОД 1. ПОДСЧЕТ КОЛИЧЕСТВА
ЧАСТИЦ В РЕЖИМЕ СВЕТОТЕНИ
Используют соответствующий прибор, прин
цип действия KOToporo основан на блокировке
подачи света, который позволяет автоматически
определять размеры и количество частиц в co
ответствии с их размером.
Прибор калибруют с испол зованием соот
ветствующеrо сертифицированною CTaHдapTHO
ro материала, представляющеro собой диспер
сионную взвесь сферических частиц с размером
от 1 О мкм ДО 25 мкм. Стандартные частицы дис
перrируют в воде, свободной от частиц, Р. Сле
дует избеrать аrреrации частиц во время дис
перrирования.
Общие указания
ИСllытание выполняют в условиях, оrрани
чивающих попадание механических включений,
предпочтительно в камере с ламинарным пото
ком воздуха.
Тщательно моют стеклянную посуду и ис
пользуемое оборудование для фильтрации, за
исключением мембранных фильтров, теплым
460
rосударственная фармакопея Республики Беларусь
раствором моющеro средства с последующим
ополаскиванием большим количеством !'ЮДЫ
для удаления следов моющеro средства. Неrю
средственно перед испытанием прибор опола
скивают сверху донизу, снаружи и затем внутри
водой, свободной от частиц, Р.
Необходимо исключать попадание пузырь
ков воздуха в испытуемый образец, особенно
при переносе пробы в емкость, в которой будет
проводиться испытание.
Необходимо убедиться в соответствии
среды требованиям к проведению испыта
ния: стеклянная посуда должна быть очище
на, в используемой воде должны OTCYTCTBO
вать механические включения. Для этоro
проводят следующий тест: определяют нали
чие механических включений в пяти пробах
воды, свободной от частиц, Р, каждая объе
мом по 5 мл, соrласно методике, описанной
ниже. Если в 25 мл для пяти объединенных
проб число частиц размером 1 О мкм И более
превышает 25, предпринятые меры предо
сторожности недостаточны. Обработку по
вторяют до тех пор, пока среда, стеклянная
посуда и вода не будут приrодными для про
ведения испытания.
Методика
Перемешивают содержимое образца, Meд
ленно и непрерывно переворачивая контейнер
20 раз. Если необходимо, осторожно удаляют
колпачки. Внешнюю поверхность вскрываемоro
контейнера очищают струей воды, свободной от
частиц. Ри вскрывают контейнер, избеrая какоro
либо заrрязнения содержимоro. Для удаления пу
зырьков воздуха дают отстояться в течение 2 мин
или обрабатывают ультразвуком.
При испытаниях парентеральных лекар
ственных средств большоro объема испыты
вают одиночные образцы. При испытаниях па
рентеральных лекарственных средств малоro
объема объемом менее 25 мл содержимое 1 О
или более образцов смешивают в чистой eM
кости до получения объема не менее 25 мл;
если указано в частных статьях, испытуемый
раствор может быть приroтовлен путем CMe
шивания содержимоro соответствующеro KO
личества контейнеров и разбавления водоц
свободной от частиц, Р или растворителем,
указанным в частной статье, не содержащим
механических включений, если вода, свобод
ная от частиц, Р является неподходящей. Па
рентеральные лекарственные средства малоro
объема объемом 25 мл и более MorYT испыты
ваться индивидуально.
Порошки для приroтовления лекарственных
средств для парентеральноro применения BOC
станавливают в воде, свободной от частиц, Р
или растворителе, указанном в частной статье,
не содержащем механических включений, если
вода, свободная от частиц, Р является непод
ходящей.
Количество испытуемых образцов должно
обеспечивать статистически достоверную
оценку данных. Для испытания больших объе.-
мов парентеральных лекарственных средств или
малых объемов парентеральных лекарственных
средств, имеющих объем 25 мл и более, можно
проводить испытание менее 1 О образцов при со-
ответствующей схеме отбора образцов.
Отбирают четыре пробы, каждая объемом
не менее 5 мл, и определяют количество частиц
размером равным или более 1 О мкм И 25 мкм.
Исключают результат, полученный для первой
пробы, и рассчитывают среднее количество
частиц в испытуемом образце.
Оценка результатов
, К лекарственным средствам, выпускаемым в
контейнере номинальным объемом более 100 мл,
применяют требования теста 1.А
К лекарственным средствам, выпускаемым в
контейнере номинальным объемом менее 1 00 мл,
применяют требования теста 1.В.
К лекарственным средствам, выпускаемым
в контейнере номинальным объемом 100 мл,
применяют требования теста 1.В.
Если среднее количество частиц превыша
ет допустимые нормы, испытание лекарственно
ro средства проводят, используя метод подсчета
количества частиц при помощи микроскопа.
Тест 1.А Растворы для инфузий или
инъекционные растворы, выпускаемые в KOH
тейнере номинальным объемом более 100 мл.
Лекарственное средство выдерживают ис
пытание, если в испытуемых образцах среднее
количество частиц размером 1 О мкм И более не
превышает 25 в одном миллилитре, а размером
25 мкм и более не превышает 3 в одном милли
литре.
Тест 1.8 Растворы для инфузий или
инъекционные растворы, выпускаемые в KOH
тейнере номинальным объемом менее 100 мл.
Лекарственное средство выдерживает ис
пытание, если в испытуемых образцах среднее
количество частиц размером 1 О мкм И более не
превышает 6000 в одном контейнере, а разме
ром 25 мкм и более не превышает 600 в одном
контейнере.
МЕТОД 2. ПОДСЧЕТ КОЛИЧЕСТВА
ЧАСТИЦ ПРИ ПОМОЩИ МИКРОСКОПА
Используют подходящий бинокулярный ми
кроскоп, комплект фильтров для сбора механи
ческих включений и мембранный фильтр для
проведения испытания.
Микроскоп должен быть оснащен окулярным
микrюметром, калиброванным по объект микро-
метру, предметным столиком для удерживания
и перемещения образца, двумя подходящими
осветительными приборами для обеспечения
эпископическоro освещения в дополнение к OT
раженному освещению, а также должен быть Ha
строен на увеличение в 100:1:10 раз.
462
rосударственная фармакопея Республики Беларусь
увлажняют несколькими миллилитрами еодt-I.
свободной от частиц, Р. В фильтрационну-,{; в<>
ронку помещают весь раствор или объем одною
образца и подключают вакуум. При необходимо
сти раствор добавляют постепенно, порциями,
пока не профильтруется весь объем лекарствен
ноro средства. После добавления последней
порции раствора промывают внутренние стенки
держателя фильтра струей воды, свободной orr,
частиц, Р. Не выключают вакуум до тех пор,
пока с поверхности мембранноro фильтра пол
ностью не исчезнет вода. Фильтр помещают 8
чашку Петри и сушат ero на воздухе со слеrка
приоткрытой крышкой. После тоro, как фильтр
высохнет, чашку Петри помещают на предмет
ный столик микроскопа, рассматривают MeM
бранный фильтр в отраженном свете и подсчи
тывают количество частиц размером 1 О 'мкм И
более и количество частиц размером 25 мкм и
более. В качестве альтернативы допускается
проведение частичноro либо полноro подсче
та количества частиц на фильтре. Вычисляют
среднее количество частиц в испытуемом ле
карственном средстве.
Размер частиц определяют при помощи раз
деленной по диаметру окружности путем вообра
жаемоro наложения изображения каждой части
цы на окружность с последующим сравнением
со стандартно размеченными окружностями раз
мером 10 мкм И 25 мкм. Таким образом, части
цы не перемещают с места их первоначальноro
расположения в поле зрения и не накладыва
ют на стандартные окружности для сравнения.
Внутренний диаметр прозрачных стандартных
окружностей используют для определения раз
мера белых и прозрачных частиц, в то время как
размер темных частиц определяют, используя
наружный диаметр черных непрозрачных CTaH
дартных окружностей.
При выполнении испытания методом под
счета количества частиц при помощи микроско
па не следует пытаться определить размер или
количество аморфных, полужидких или друrих
морфолоrически нечетких материалов, имею
щих вид пятна или обесцвечивающихся на MeM
бранном фильтре. Эти частицы MOryT иметь
небольшой поверхностный рельеф, быть желе
образными или иметь пленочную поверхность.
В таких случаях проводится испытание образца
раствора методом подсчета количества частиц в
режиме светотени.
Оценка результатов
к лекарственным средствам, выпускаемым в
контейнере номинальным объемом более 100 мл,
применяют требования теста 2.А.
К лекарственным средствам, выпускаемым в
контейнере номинальным объемом менее 1 00 мл,
применяют требования теста 2.В.
К лекарственным средствам, выпускаемым
в контейнере номинальным объемом 100 мл,
применяют требования теста 2.В.
Тест 2.А Растворы для инфузий или
инъекционные растворы, выпускаемые в KOH
тейнере номинальным объемом более 100 мл.
Лекарственное средс; ",о выдерживает ис
пытание, если в испытуемых образцах среднее
количество частиц с размером 1 О мкм И более
не превышает 12 в одном миллилитре, а раз
мером 25 мкм и более не превышает 2 в одном
МИЛлилитре.
Тест 2.В Растворы для инфу:юй ипи
инъекционные растворы, выпускаемые в
контейнере номинальным объемом менее
100 мл.
Лекарственное средство выдерживает ис
пытание, если в испытуемых образцах среднее
количество частиц размером 1 О мкм И более не
превышает 3000 в одном контейнере, а разме
ром 25 мкм и более не превышает 300 в одном
контейнере.
01/2013:20920
2.9.20. ЗАrРЯЗНЕНИЕ
МЕХАНИЧЕСКИМИ
ВКЛЮЧЕНИЯМИ: ВИДИМЫЕ
ЧАСТИЦЫ
Механическими включениями в инъекцион
ных и инфузионных растворах являются посто
ронние подвижные нерастворимые частицы, за
исключением пузырьков rаза, случайно оказав
шихся в растворе.
Испытание предназначено для визуальной
оценки качества парентеральных растворов.
#порошков для приroтовления инъекционных и
инфузионных лекарственных средств, rлазных
капель и примочек, порошков для приrотовления
rлазных капель и примочекfl на наличие видимых
частиц. Moryт использоваться друrие валидиро
ванные методы.
#Испытание распространяется как на лекар
ственные средства, находящиеся в процессе про
изводства, так и на roтовый продукт, находящий
ся на разных стадиях контроля (в отделе контроля
качества или орrанизации roсударственноro KOH
троля), а также на вышеуказанные лекарственные
средства, изroтовленные в аптеках по индивиду
альным прописям и внутриаптечной заroтовке.
Условия проведения испытаний лекарствен
ных средств в контейнерах из непрозрачных Ma
териалов указываются в частных фармакопей
ных статьях.#
ПРИ БОР
Прибор (см. рисунок 2.9.20. 1) состоит из
устройства для просмотра, которое включает в себя:
черный матовый экран подходящеro раз
мера, находящийся в вертикальном положении:
2.9.19. 3аzрязнение механическими включениями: невидимые частицы
461
Окулярный микрометр представляет
собой окружность, разделенную по диаметру
(см. рисунок 2.9.19.1 1). и состоит из большо
ro Kpyra, разделенноro перекрестиями на KBa
дранты, прозрачных и черных стандартных
окружностей диаметром 10 мкм И 25 мкм при
стократном увеличении, и линейной шкалы
с делениями по 10 мкм. Калибровка выпол
нена при помощи стандартноro микрометра,
соответствующеro международным или Ha
циональным стандартам. Допустимая OT
носительная поrрешность линейной шкалы
при делении на квадранты составляет ::!:2 %.
Большой Kpyr служит для определения поля
зрения.
Требуется два источника света: эпископи
ческий, с широким полем охвата, направленный
внутрь микроскопа, и внешний, дополнитель
ный, под уrлом 10 200 для обеспечения OTpa
женноro освещения.
Комплект фильтров для сбора механиче
ских включений состоит из держателя фильтра,
изrотовленноrо из стекла или друrоro подходя
щеrо материала, вакуумноro насоса и подходя
щеrо мембранноro фильтра.
Мембранный фильтр должен иметь подхо
дящий размер и быть черноro или темно сероro
цвета, с нанесенной разметкой или без размет
ки и номинальным диаметром пор 1,0 мкм или
менее.
;8
1/1
/ J
I
I
.' j .
.........,
.....
,
"
\.
\.
боЛЬШОЙ КРУ'"
I
.
Стандартная
окружность
\
\ ,
' J
/
/'
ЛИНИИ Визира
'1 ! 11 'Н' 1 :1: " 1: 11' 1 I Щ! 1I :' 1: !! '1 : 1: ! .
Линейная шкала
Рисунок 2.9.19. 1. Окружность, разделенная
по диаметру
Общие указания
Испытание выполняют в условиях, оrрани
чивающих попадание механических включений,
предпочтительно в камере с ламинарным пото
ком воздуха.
Тщательно моют стеклянную посуду и
используемое оборудование для фильтра
ции, за исключением мембранных фильтров,
теплым раствором моющеrо средства с по
следующим ополаскиванием большим коли
чеством воды для удаления следов моющеro
средства. Непосредственно перед испытани
ем обе стороны мембранноro фильтра и обо
60 Зак. 1712,
рудование сверху дониз снаружи и затем
внутри ополаскивают водой, свободной от
частиц, Р.
Необходимо убедиться в соответствии среды
требованиям к проведению испытания: стеклян
ная посуда должна быть очищена, в исполь
зуемой воде должны отсутствовать механиче
ские включения. Для этоro проводят следующий
тест: определяют наличие механических вклю--
чений в 50 мл воды, свободной от частиц, Р,
как описано ниже. Если на используемом MeM
бранном фильтре содержание частиц размером
1 О мкм И более превышает 20 или частиц разме
ром 25 мкм и более превышает 5, предпринятые
меры предосторожности недостаточны. Обра
ботку повторяют до тех пор, пока среда, стеклян
ная посуда, вода и мембранный фильтр не будут
приroдными для проведения испытания.
Методика
Перемешивают содержимое образца, Meд
ленно и непрерывно переворачивая контейнер
20 раз. Если необходимо, осторожно удаляют
колпачки. Внешнюю поверхность вскрываемоro
контейнера очищают струей воды, свободной от
частиц, Р и вскрывают контейнер, избеrая како-
ro либо заrрязнения содержимоro.
При испытаниях парентеральных лекаJr
ственных средств большоro объема испытыва
ются одиночные образцы. При испытаниях па
рентеральных лекарственных средств малоro
объема объемом менее 25 мл содержимое 1 О
или более образцов смешивают в чистой eM
кости; если указано в частных статьях, испыту
емый раствор может быть приroтовлен путем
смешивания содержимоro соответствующеro
количества контейнеров и разбавления водоц
свободной от частиц, Р или растворителем,
указанным в частной статье, не содержащим M
ханических включений, если вода, свободная от
частиц, Р является неподходящей, до получе
ния объема не менее 25 мл. Парентеральные
лекарственные средства малоro объема объе
мом 25 мл и более MOryт испытываться индиви
дуально.
Порошки для приroтовлени лекарственных
средств для парентеральноro применения BOC
станавливают водой, свободной от частиц, Р
или растворителем, указанным в частной статье,
не содержащим механических включений, если
вода, свободная от частиц, Р является непод
ходящей.
Количество испытуемых образцов должно
обеспечивать статистически достоверную
оценку данных. Для испытания больших объ
мов парентеральных лекарственных средств или
малых объемов парентеральных лекарственных
средств, имеющих объем 25 мл и более, можно
провести испытание менее 1 О образцов при со-
ответствующей схеме отбора образцов.
Внутреннюю поверхность держателя филь
тра, соединенноro с мембранным фильтром,
2.9.20. 3аzрязнение механическими включениями: видимые частицы
463
белый матовый экран ПОДХодящеro раз
мера, находящийся в вертикальном положении,
рядом с черный экраном;
реryлируемый плафон, снабженный под
ходящим затемненным источником дневноro
света с подходящим отражателем (осветитель
может состоять из 2 флуоресцентных ламп по
1З Вт каждая длиной 525 мм). Интенсивность oc
вещения в точке контроля должна быть от 2000
люкс до З750 люкс; для цветных стеклянных и
пластмассовых контейнеров предпочтительно
использовать более высокие мощности света.
плафон осветителя
/
черный
матовый
экран
белый
матовый
экран
Рисунок 2.9.20. 1. Прибор для обнаружения
видимых частиц
#Допускается использование реryлируемоro
плафона, снабженноro подходящим затемнен
ным источником дневноro света или электриче
ской лампой накаливания с подходящим OTpa
жателем. При мерная освещенность приведена в
таблице #2.9.20. 1.
Таблица #2.9.20. 1
Таблица мощности источника света
I Электрическая Лампа
Окраска лампа AHeBHoro
накаливания, Вт света, Вт
Бесцветные 60 20
Окрашенные
(или опалесци 100 30 <
рующие)
Примечания:
1. В rруппу «Окрашенные» включают бес
цветные растворы в контейнерах из светозащит
ноro стекла и окрашенные растворы в контейне
рах из бесцветноro стекла, а также растворы в
контейнерах из прозрачных полимерных MaTe
риалов и опалесцирующие растворы.
2. При просмотре на установках типа
KVLC 1 О допускается использование лампы Ha
каливания мощностью 40 Вт. #
#МЕТОДИКА
Плавно вращают или переворачивают контей
нер, избеrая образования воздушных пузырьков,
и просматривают в течение примерно 5 с перед
белым экраном. Повторяют эту процедуру перед
черным экраном. Отмечают наличие любых частиц.
Помещение для проведения контроля (ис
пытаний) на механические включения защища
ют от прямоro попадания солнечных лучей.
Рабочее место специалиста оснащают
столом и источником освещения.
Контроль (испытание) лекарственных
средств на механические включения должен про
водиться специалистом невооруженным rлазом
на черном и белом фоне (рисунок 2.9.20 1).
Зона испытания при просмотре должна быть oc
вещена электрической лампой накаливания или
лампой дневноro света соответствующей мощ
ности, указанной в таблице #2.9.20 1 в зависи
мости от степени окраски растворов или матери
алов упаковки.
При испытании допускается механизирован
ная подача емкостей в зону контроля с после
дующей их транспортировкой на дальнейшие
стадии операции, а также использование раз
личных типов специальных установок для про
смотра, обеспечивающих качество контроля.
Для проведения испытания специалист
должен иметь зрение «единицу». При необходи
мости производится очковая коррекция зрения.
Состояние зрения специалиста должно прове
ряться врачом окулистом не реже одноro раза в
6 месяцев, о чем должна быть отметка в меди
цинской карточке (книжке). Для снятия усталости
rлаз при просмотре через каждые 1, 2 ч работы
устанавливают десятиминутный перерыв.
Расстояние от rлаз специалиста до объек
та контроля должно быть в пределах 25 30 см.
Уroл между оптической осью просмотра и направ
лением лучей света соответствует примерно 900_
rлаза специалиста должны быть защищены
от попадания света непосредственно от источ
ника освещения, линия зрения должна быть Ha
правлена несколько вниз при вертикальном по
ложении rоловы.
#ПОРЯДОК КОНТРОЛЯ ИНЪЕКЦИОННЫХ
И ИНФУЗИОННЫХ РАСТВОРОВ
Порядок контроля на предприятиях
На предприятиях осуществляется трехкрат
ный контроль чистоты лекарственных средств
парентеральноrо введения больших и малых
объемов: первичный внутрицеховой сплош
ной, вторичный внутрицеховой выборочный
и третий выборочный контроль, осуществля
емый специалистом отдела контроля качества.
Первичному контролю подлежат 100 %
ампул, флаконов, бутылок, шприц тюбиков и
друrих полимерных контейнеров с лекарствен
ными средствами, прошедшими стадию стери
лизации или приroтовленными в асептических
условиях, перед маркировкой и упаковкой.
Первичный и вторичный контроль осущест
вляют специалисты (просмотрщики) цеха, участ
ка. Специалисты (просмотрщики) должны иметь
свои номера. Номер специалиста (просмотрщи
ка) вкладывают в упаковку продукции или штам
пуют на колпачке флакона.
rосударственная фармакопея Республики Беларусь
Таблица #2.9.20. 2
Нормативы объемов выборок для контроля растворов маЛ020 объема на механические включения
и параметры их оценки
464
Количество емкостей с растворами
Ступень Объем выборки малоrо объема, имеющими включе-
Объем серии, шт. визуальноrо ДЛЯ визуальноrо ния, шт.
контроля контроля, шт. Приемочное Браковочное
1 2 З 4 5
первая 80 2 5
1201 3200 суммарно (по 2 M 60 6 7
ступеням)
З201 10 000 200 6 10
тоже 400 15 16
Свыше 1 О 000 315 9 14
тоже 630 2З 24
Таблица #2.9.20. З
Нормативы объемов выборок для контроля растворов БОЛЬШ020 объема на механические
включения и параметры их оценки
Ступень Объем выборки Количество емкостей с растворами
малоrо объема, имеющими включе
Объем серии, шт. визуальноrо ДЛЯ визуальноrо ния, шт.
контроля контроля, шт. Приемочное Браковочное
1 2 3 4 5
первая 20 О 2
151 280 суммарно (по 2 M 40 1 2
ступеням)
281 500 32 О 2
тоже 64 1 2
501 1200 50 О 2
тоже 100 2 З
1201 З200 80 О 3
тоже 160 3 4
Свыше 3200 125 1 4
тоже 250 5 6
Для проведения вторичноro контроля от
каждой партии, прошедшей первичный KOH
троль, отбирают среднюю пробу 5 % от партии
до 2000 ампул, флаконов, бутылок, шприц
тюбиков и 250 штук от всех друrих партий. При
обнаружении более 2 % ампул, флаконов, буты
лок, шприц тюбиков С механическими включени
ями всю партию, от которой отобрана средняя
проба, возвращают для повторноro первичноrо
контроля.
Требования вториЧН020 выБОрОЧН020
контроля не распространяются на учреж
дения службы крови (станция переливания
крови).
Порядок контроля специалистами
отдела контроля качества
Третий выборочный контроль осущест
вляется специалистами отдела контроля каче
ства. Для контроля отбирают среднюю пробу от
каждой серии изrотовленной продукции перед
маркировкой и упаковкой ампул, флаконов, бу
тылок, шприц тюбиков и друrих полимерных
контейнеров.
Отбор образцов
Количество образцов, отбираемых от каждой
серии инъекционноro лекарственноro средства,
зависит от объема (малый или большой в COOTBeT
ствии с таблицами #2.9.20. 2 и #2.9.20. 3), колич&
ства единиц лекарственноro средства в серии.
Для проведения визуальноro контроля инъек
ционных лекарственных средств, не требующих
вскрытия и растворения (неразрушающий K
троль), производят отбор выборок продукции ..
оценку результатов контроля с помощью усилен-
ноro двухступенчатоro контроля.
Растворы маЛ020 объема (неразруи--
ющий контроль)
ОТ каждой серии произвольно отбирают Bbr
борку в два этапа 1 и 2 ступени в соответствии
с таблицей #2.9.20. 2.
Растворы БОЛЬШ020 объема (неразру
шающий контроль)
От каждой серии произвольно отбирают BЫ
борку в два этапа 1 и 2 ступени в соответ
ствии с таблицей #2.9.20. 3.
2.9.20. За2рязнение механическими включениями: видимые частицы
465
Таблица #2.9.20.--4
Нормативы количества одновременно контролируемых емкостей, времени и скорости контроля
Количество емкостей, одновременно взятых Время контроля OДH<r Скорость контроля,
для контроля временно взятых eMKO шт.lч
стей, с
Ампулы Флаконы, Шприц побики :.1: :.1:
бутылки о о
J:; J:;
jj ш jj ш
ф ф ф о о
.D ф ф .D ф :.1: :.1:
J:; .а J:; J:; >. s >. s
u о u о u о
о о о ri 2 ri 2
:Е ф :Е ф :Е ф о ';" о 1;-
s х s х s х х ::r х :r
х х х о s J:; о s
u u u :.1: >. :.1:
ф >. ф >. ф >. с ns а. с ns Q,
:Ес; :ЕЕ:; :Ес; :Е i с :Е J:; с
Ш:Е 3 Ш:Е 3 Ш:Е 3 « 3 с( е 3
1 2 3 4 5 6 7 8 9 10 11 12
Малоro объема
1,0 15 5,0 1,0 7 15 8-------1 О 15 до до до 870
2000 1700
2,O 13 ЗО, о------ 2 15 8 до до 600
З,О 50,0 1750
5,0 10 50, о------ 2 15 10 до до 400
100,0 1600
10,0 9 15 до 20 до до 300
1400
20,0 8 15 до
30,0 1250
Большоro объема
Свыше 1 до 20 до 300
100,0
На станциях переливания крови для просмо
тра инъекционных лекарственных средств отдел
контроля качества производит выборку 1 О %
контейнеров от серии, но не менее 10 бутылок.
При обнаружении хотя бы одноro контейнера с
механическими включениями всю серию возвра
щают для повторноro первичноrо контроля.
Подrотовка образцов
Для проведения визуальною контроля инъек
ционных лекарственных средств большоro и малою
объемов, не требующих вскрьrrия и растворения, по-
верхность контейнеров (ампул, Флаконов, бутылок,
шприц тюбиков и дрyrих емкостей из прозрачных по-
лимерных материалов) должна БЬrrb чистой и сухой.
Оценка результатов
Для просмотра растворов малоrо и боль
шоro объемов время контроля и количество oд
новременно взятых емкостей соответствуют
данным таблицы #2.9.20.--4.
Примечания:
1. Количество контейнеров (ампул), OДHOBp
менно взятых для контроля, должно быть не более
указанноro в таблице, но обычно не менее 50 %.
2. В случае необходимости (например, обу
чение учеников) количество контейнеров (ампул,
флаконов, бутылок, шприц тюбиков), OДHOBpe
мен но взятых в руки, уменьшают в 2 3 раза.
3. Время контроля определяется пери
одом, в течение котороro контролеры только
просматривают инъекционные лекарствен
ные средства в емкостях. Сюда не включает
ся время вспомоrательных операций, Korдa
специалист берет емкости, вносит их в зону
контроля, укладывает в тару. Время контроля
составляет от 40 % до 60 % от общеro BpeMe
ни, затраченноro на просмотр, с учетом вспо
моrательных операций, а при механизирован
ной подаче емкостей в зону контроля 70 %.
Время контроля инъекционных лекарственных
средств в сосудах из светозащитноrо стекла
и прозрачных полимерных материалов, OKpa
шенных растворов и неводных растворов YBe
личивается на 20 %, соответственно уменьша
ется и скорость контроля.
4. В rрафу «Скорость контроля» включе
ны емкости как с чистыми инъекционными ле
карственными средствами, так и с содержащи
ми механические включения, и учтено время на
вспомоrательные операции. Скорость контроля
при механизированной подаче инъекционных
лекарственных средств в зону контроля увели
чивается на 20 50 %.
Для просмотра инъекционных лекарствен
ных средств ампулы берут в руки за капилля
ры, флаконы и бутылки за rорловины, шприц
тюбики за колпачки, вносят их в зону контроля
в положении «вверх донышками» и просматри
вают на черном и белом фоне. Затем плавным
движением, без встряхивания, переводят их в
466
rосударственная фармакопея Республики Беларусь
ПОЛО>I<енv.е «<3НИЗ донышками» и вторично про..
:;матривают 143 черном и белом фоне. Для ле
'\арственных средств, требующих вскрытия и
растворения, КОНТРОЛt, '3 положении «вверх дo
нышками» 1\10ЖН ) ,1.:" : , "!ИТL,
Емкости с иньекt,,:' он":ЫМИ лекаРСТ8енны
ми средстваМI', !з t' 'U;Jt:iX оi5наружены видимые
механические "Н\f1'О ч ения, считают забракован
ными и укРаДЬН1ЗЮ'7' ,; отдельную таоу с оп..,ет
кой «Брак».
Если на пеоесй ступени контроля количе
ство емкостеЙ с ;;:НЪЕ'IЩИОННЫМИ лекарствен
ными среДСТ8d .j. Jдержащими механические
включения (таБЛIIЦЫ #2,9.20. 2 и #2.9.20. 3),
равно или I1ревышает указанное в rрафе 5, то
всю серию ПрО,1уции бракуют; если количество
таких емкостей меньше числа. указанноro в
rрафе 5, но f5C'J;L' 'Je числа в rрафе 4, то прово
дят вторую c:)''leHb контроля на таком же коли
честве емкостей аnдлизируемой продукции
rрафа 2.
Заключение о качестве анализируемой
серии Иl-1ъещионноro лекарственною средства
после второй с- тупени контроля делают на OCHO
вании количесва единиц продукции, имеющих
механические Вl{лючения, в суммарном (общем)
объеме первой и второй выборок в соответствии
с rрафами 4 и 5 таблиц #2.9.20. 2 и #2.9.20. З.
Всю серию бракуют, если количество единиц
ПР(о УКUИИ иr.1f:-r.JЩИХ механические включения.
превышает или равночислу, указанномувrрафе 5,
для cYMMapHofu объема первой и второй выбо
рок.
В случае бр:;> я
отдел I{ОНТРОЛЯ качества возвращает всю
:1РОЦУКЦИЮ в цех (на участок);
roсударственные контролирующие opraHbI
на'lравляют протокоп испытаний в установлен
ном порядке предприятию производителю и в
Министерство здравоохранения Республики Бе
ларусь
#ПОРЯДОК КОНТРОЛЯ ПОРОШКОВ
ДЛЯ приrОТОВЛЕНИЯ ИНЪЕКЦИОННЫХ
И ИНФУ3ИОННЫХ ЛЕКАРСТВЕННЫХ
СРЕДСТВ, П1А3НЫХ КАПЕЛЬ
И ПРИМОЧЕf<
Условия проведения контроля
Условия проведения визуаrlьноrо контроля
(разрушающий контроль) в порошках для приro
товления инъекционных и инфузионных лекар
ственных средств, rлазных капель и при мочек:
подroтовку образцов проводят в помеще
ниях чистоты класса В;
вскрытие флаконов или ампул. paCTBO
рение лекарственноro средства, контроль pac
творителя и лекарственноro средства про
водят на рабочем месте, соответствующем
классу чистоты А (в ламинарном потоке CTe
рильноro воздуха);
специалист должен работать в стериль
ном халате и шапочке из безворсовой ткани и
реЗИl-10ВЫХ перчатках, обработанных раСПЗ0рОМ
силиконовой эмульсии КЭ 10 16 (массовая доля
0,1 %) или др.;
оборудование, химическую посуду и Пр 1
надлежности для работы обрабатывают paCTB
ром разрешенноrо моющею средства (массовая
доля 0,1 %), несколько раз промывают rОРЯL;ей
водой и ополаскивают водой очищенной, не co
держащей механических 8кпючений.
Подrотовка образцов
Для визуальноro контроля порошков для
приrотовления инъекционных и инфузУ\ом
ных лекарственных средств, rлазных капель и
примочек отобранные образцы перед BCKPЫ
тием 3 раза промывают водой, не содержа
щей механических включений. При этом с
флаконов предварительно удаляют этикетки
и алюминиевые колпачки. Промытые образцы
подсушивают в ламинарном потоке стериль
ною воздуха.
Для растворения лекарственноrо cpeд
ства используют воду или друrой раствори
тель, указанный в частной фармакопейной
статье или в инструкции по применению ле
KapcTBeHHoro средства, предварительно f1pC
фильтрованный через мембрану с диаметром
пор не более 1,2 мкм,
Визуальный контроль растворителя OCj
ществляют по следующей методике: берут 1 О
тщательно отмытых контейнеров (флакOI-ЮВ)
вместимостью 1 О мл И С помощью промытсrа
медицинскоrо шприца или фильтрующеro rри
способления типа «Пистолел> В каждый контей
нер (флакон) вливают около 5 мл растворитепq,
Затем контейнеры (флаконы) закрывают про5ка
ми, свободными от механических включеНИl1, и
просматривают, как указано выше. РастворитеЛ6
приroден для испытания. если в 9 контейнерах
(флаконах) из 10 не обнаружено механических
включений, видимых невооруженным rлазом.
Вскрытие ампул производят следующим
образом: на поверхности капилляра HaHO
сят насечку с помощью победитовою ножа,
затем к краю насечки прикасаются раскален
ной докрасна молибденовой или вольфрамо
вой проволокой. После охлаждения капилляр
осторожно снимают. Возможен любой друrой
способ вскрытия, исключающий попадание
стекла в содержимое ампул.
Введение растворителя в контейнеры
(флаконы, ампулы) проводят через roрлови
ну с помощью фильтрующеrо приспособле
ния типа «Пистолет» И друrих или с помощью
предварительно промытоro шприца; допуска
ется введение растворителя через пробку с
помощью шприца с иrлой NQ 0840, предвари
тельно промытой внутри и снаружи водой, не
содержащей механических включений.
Растворитель вводят в количестве, ДOCTa
точном для полноro растворения лекарствен
ною средства около половины объема KOH
2.9.20. За2рязнение механическими включениями: видимые частицы
467
Таблица #2.9.20. 5
Нормативы объема выборок для контроля на механические включения
сухих лекарственных средств
I
I rpynna лекарственных
i средств
i
; 1. Лекарственные cpeд
! ства, предназначенные
I для внутривенноro BBe
. дения, а также с указани
I ем H этикетке «для инъ
екции»
I
' дo 1 r включительно
" более 1 r (до 5 r вклю
чительно)
12. Лекарственные cpeд
I ства, предназначенные
для внутримышечноro
I введения, rлазные капли
, и примочки
! до 1 r включительно
I более 1 r (до 5 r вклю
i чительно)
Количество флаконов (ампул) в серии
до 35 000 I до 70 000
I
включительно i включительно
I I
число кол-во i число кол во
выборок i образцов i выборок образцов
I
i
1
1
1
1
тейнера (флакона или ампулы), или в объеме,
указанном в частной фармакопейной статье или
в инструкции по применению. Затем контейне
ры вновь закрывают пробками. Лекарственное
средство должно быть полностью растворено
при встряхивании.
Примечания:
1. Леrко rидролизующиеся лекарственные
средства растворяют непосредственно перед
контролем.
2. Для высокомолекулярных соединений
(белки, полисахариды, rликопротеиды и др.) в
нормативной документации на лекарственное
средство или в инструкции по применению YKa
зывают растворители, рН, время и условия paCT
ворения, а также друrие факторы, влияющие на
процесс растворения.
При визуальном методе контроля просма
тривают образцы суммарной выборки в зависи
мости от числа флаконов (ампул) в серии (табли
ца #2.9.20. 5) и подсчитывают в каждом образце
число механических включений. При обнаруже
нии в одном флаконе (ампуле) свыше 5 механи
ческих включений дальнейший подсчет не про
изводят. За результат просмотра в этом случае
принимают цифру 7. Суммируют число механи
ческих включений, обнаруженных во всех образ
цах первой полной выборки, и делят на число
выборок.
Примечания:
1. Для лекарственных средств с дозировкой
более 5 r число выборок, количество образцов
в выборке и норму содержания механических
включений указывают в частных фармакопей
ных статьях.
до 105000
включительно
. I
число I кол во .
выборок I образцов I
I ;
8
5
!
I
I
I
i
I
!
!
,
I
I
!
i
i
i
i
!
i
I
!
j
2
2
16
10
3
3
24
15
5
3
I
15
9
2
2
10
б
3
3
2. Для проведения контроля в roсударствен
ных КОНТРОЛИРУЮЩИХ opraHax отбирают YДBO
енное количество образцов одной выборки вне
зависимости от rруппы, к которой отнесено ле
карственное средство (таблица #2.9.20. 5). Пр
необходимости roсударственные контролирую
щие opraHbI MOryт запросить дополнительное ко.
личество образцов, превышающее вышеуказан
ное удвоенное количество.
При обнаружении в лекарственных сред-
ствах, предназначенных для внутривенноro BBe
дения, и с указанием на этикетке «для инъекций»:
15 механических включений и менее
серию принимают с первой выборки;
20 механических включений и более
серию бракуют с первой выборки;
от 16 до 19 механических включений от
бирают вторую выборку в том же количестве (Ta
блица #2.9.20. 5) и просматривают по той же Me
тодике.
При обнаружении в лекарственных cpeд
ствах, предназначенных для внутримышечноro
введения:
23 механических включений и менее
серию принимают с первой выборки;
29 механических включений и более
серию бракуют с первой выборки;
от 24 до 28 механических включений OT
бирают вторую выборку в том же количестве (Ta
блица #2.9.20. 5) и просматривают по той же Me
тодике.
В случае контроля удвоенной выборки pe
зультаты первой и второй выборок суммируют.
При обнаружении в лекарственных сред-
ствах, предназначенных для внутривенно
468
rосударственная фармакопея Республики Беларусь
ro введения, и с указанием на этикетке «для
инъекций»:
34 механических включений и менее
серию принимают;
35 механических включений и более
серию бракуют;
При обнаружении в лекарственных средствах,
предназначенных для внутримышечноro введения:
52 механических включений и менее
серию принимают:
53 механических включений и более
серию бракуют.
При обнаружении в выборке хотя бы одной ча
стицы стекла отбирают дополнительную выборку в
том же количестве.
Серию считают rодной, если ни в одном из
флаконов (ампул) дополнительной выборки не об
наружено ни одной частицы стеклэ.
#поРядок КОНТРОЛЯ rЛАЗНЫХ КАПЕЛЬ
Порядок контроля на предприятиях
На предприятии осуществляется ДBYKpaT
ный контроль rлазных капель на механические
включения:
первичный контроль внутрицеховой, ocy
ществляемый специалистом цеха (участка). Спе--
циалист должен вложить талон со своим номером
в упаковку или кассету с доброкачественной про--
дукцией;
вторичный выборочный контроль, ocy
ществляемый специалистом отдела контроля Ka
чества.
Контейнеры с растворами rлазных капель,
в которых обнаружены видимые механические
включения, считаются забракованными. 3абрако---
ванную продукцию вкладывают в отдельную тару
с отметкой «Брак».
r лазные капли с опалесценцией не бракуются
в случае, если опалесценция допускается действу
ющей нормативной документацией.
Первичному контролю подлежат 100 % KOH
тейнеров, прошедших стадию стерилизации или
изroтовленных в асептических условиях, перед
маркировкой и упаковкой.
Вторичный выборочный контроль осущест
вляется контролером ОКК в процессе испытаний
лекарственноro средства на соответствие требо
ваниям частной фармакопейной статьи перед еro
подачей на склад roтовой продукции. Для этоro из
серии отбирают 1 % единиц продукции, но не менее
50 контейнеров с раствором rлазных капель.
Вторичный контроль rлазных капель осущест
вляют в соответствии с пунктом #Методика, опи
санным выше. В случае обнаружения ворсинок
фиксируется их число в кажцой единице контроли
руемой продукции. В случае обнаружения в едини
це продукции хотя бы одной твердой частицы или
более 5 ворсинок производят повторные испыта
ния на удвоенном количестве контейнеров, coдep
жащих rлазные капли. В случае обнаружения в
единице продукции хотя бы одной твердой части
цы или более 5 ворсинок при испытании на YДBO
енном количестве серия бракуется. Качество П
дукции в контейнерах (флаконах) определяется по
коэффициенту дефектности, который рассчитыва-
ется как среднее арифметическое число ворсинсх
из взятых на вторичный просмотр единиц продук
ции. Серия rлазных капель в контейнерах (флак
нах) считается rодной, если коэффициент деФект
ности не превышает 1,5.
Во взятом на просмотр количестве контейн
ров (тюбиков капельниц) допускается не более
4 % единиц продукции, содержащих механические
включения, причем в каждом контейнере (тюби
капельнице) не должно быть более 5 ворсинок
Порядок контроля 20сударственными
контролирующими Ор2анами
Отбор образцов rлазных капель и их испыта-
ния на механические включения осуществляют
ся орraнами roсударственноro контроля качества
лекарственных средств. Для этоro из серии o
рают 1 % единиц продукции, но не менее 50 КОН--
тейнеров с раствором rлазных капель. Испыта-
ния rлазных капель осуществляют в соответствии
с пунктом #Методика, описанным выше. В случае
обнаружения ворсинок фиксируется их число в
кажцой единице контролируемой продукции. В
случае обнаружения в единице продукции хотя бы
одной твердой частицы или более 5 ворсинок про--
изводят повторные испытания на удвоенном коли
честве контейнеров, содержащих rлазные капли. В
случае обнаружения в единице продукции хотя бы
одной твердой частицы или более 5 ворсинок при
испытании на удвоенном количестве серия браку
ется. Качество продукции в контейнерах (флако---
нах) определяется по коэффициенту дефектности,
который рассчитывается как среднее арифметич
ское число ворсинок из взятых на вторичный про---
смотр единиц продукции. Серия rлазных капель в
контейнерах (флаконах) считается roдной, если KO
эффициент дефектности не превышает 1,5.
Во взятом на просмотр количестве контейне--
ров (тюбиков капельниц) допускается не более
4 % единиц продукции, содержащих механические
включения, причем в кажцом контейнере (тюбике
капельнице) не должно быть более 5 ворсинок.
При необходимости opraHbI roсударственноro
контроля MOryт запросить дополнительное коли
чество образцов. В случае невыполнения условий,
описанных выше, серию бракуют.
На арбитражный контроль направляют образ
цы roтовой продукции в количестве, равном YДBO
енному количеству контейнеров, отобранных при
вторичном контроле, с соответствующим протоко---
лом испытаний. При необходимости opraHbI roсу
дарственноro контроля MOryт запросить дополни
тельное количество образцов.
#ПОРЯДОК КОНТРОЛЯ РАСТВОРОВ ДЛЯ
ПАРЕНТЕРАльноrо ПРИМЕНЕНИЯ,
rЛАЗНЫХ КАПЕЛЬ И ПРИМОЧЕК
В АПТЕКЕ
В аптеке осуществляют первичный KOH
троль на механические включения всех изroтов
2.9.22. Определение времени размяzченuя лuпофильных суппозиторuев
469
ленных контейнеров с растворами для парен
теральноro применения, rлазными каплями и
примочками в соответствии с пунктом «#Методи
ка», описанным выше. Лекарственные средства
подверraются контролю дважды: до стерилиза
ции после приroтовления и укупорки и после
стерилизации до этикетирования. При этом в
каждом контейнере должны отсутствовать Mexa
нические включения.
#АРБИТРАЖНЫЙ КОНТРОЛЬ
На арбитражный контроль направляют
образцы лекарственноro средства в количе
стве, равном суммарному объему выборки
(для двух ступеней контроля), с COOTBeTCTBY
ющим протоколом испытаний. При необходи
мости roсударственная контролирующая op
rанизация может запросить дополнительное
количество образцов.
01/2013:20922
2.9.22. ОПРЕДЕЛЕНИЕ
ВРЕМЕНИ РАзмяrЧЕНИЯ
ЛИПОФИЛЬНЫХ
СУППОЗИТОРИЕВ
Данное испытание позволяет определить
при заданных условиях время, необходимое для
размяrчения суппозитория с момента ero поме
щения в воду до момента, коrда он больше не
окажет сопротивления определенному прило
женному весу.
ПРИ БОР А
Прибор (см. рисунок 2.9.22. 1) состоит из
стеклянной трубки с плоским дном с BHYTpeH
ним диаметром 15,5 мм и длиной около 140 мм.
Трубка закрывается съемной пластмассо <
вой крышкой с отверстием диаметром 5,2 мм.
Прибор включает в себя также стержень диа
метром 5,0 мм, который расширяется книзу до
диаметра 12 мм. К нижней плоской стороне
стержня крепится металлическая иrла длиной
2 мм и диаметром 1 мм.
Стержень состоит из 2 x частей: нижней
части, изroтовленной из пластмассы, и верхней
части, изrотовленной из пластмассы или Me
талла, с диском определенной массы. Верхняя
и нижняя части либо соединены друr с друrом
(ручной вариант), либо отделяются (автоматизи
рованная версия). Вес всеro СТ'3ржня (ЗО:tО,4) r.
На верхней части стержня имеется свобод
но скользящее маркировочное кольцо. Korдa
стержень, введенный в стеклянную трубку, дo
стиrает дна, маркировочное кольцо Поднима
ется на уровень верхнеro края пластмассовой
крышки.
I i.
i 11
I
l J
I
0ЗО
I
t
I
I
, I
О" "'1
. '. I
i
.
..
.. s/
I
со ." 5.0 I
"!
.
I
I
I
i
I
I
I
-- I'
I
l ' 01В
I
! .5
I NI
i-n l t
il
Рисунок 2.9.22. 1. Прибор А для измерения вpe
мени раЗМЯ2чения липофильнbtх суппозиториев
(размерbt указаны в миллиметрах)
Методика. Стеклянную трубку, содержа
щую 10 мл воды, помещают в водяную баню
с температурой (36,5:tO,5) ОС. Стеклянную
трубку устанавливают вертикально и поrружа
ют на rлубину не менее 7 см ниже поверхно
сти, следя за тем, чтобы она не касалась дна
водяной бани. В трубку заостренным концом
вниз помещают суппозиторий, затем вводят
стержень со свободно скользящей пластико
вой крышкой до тех пор, пока металлическая
иrла не коснется плоскоrо конца суппозито
рия. Трубку закрывают пластиковой крышкой
и начинают отсчет времени. Реrистрируют
время, необходимое для достижения стерж
нем дна стеклянной трубки, и время Подъе
ма маркировочноro кольца до верхнеro края
пластмассовой крышки.
ПРИБОР В
Прибор (см. рисунок 2.9.22. 2) состоит из BO
дяной бани (В), в которую вставлена внутренняя
трубка (А), зафиксированная пробкой. Дно трубки
должно быть закрыто пробкой. Прибор снабжен
термометром. Имеется два вида вставок:
стеклянный стержень (С1) в форме трубки,
запаянной с обоих концов, имеющей ободок на
нижней части из свинцовой дроби; вес стержня
(ЗО:tО,4) r;
проникающая вставка (С2), состоящая
из стержня весом (7,5:tO,1) r в трубке, которая
имеет уrлубление для суппозитория; обе части
изroтовлены из нержавеющей стали.
470
rосударственная фармакопея Республики Беларусь
.f)
t
I
1 i
-" i
I
I
'
! -]
!
!
I
I
!
i
I
I
I
I
1
i
I
I
I
I I
1 .
I
I
I
1 +
II
! i
, i
i;
i8 1
i"1
II
! i
i I
!...!.
А
С1
С2
в
i r
,. ..
I.H
3;
пg
50
Рисунок 2.9.22. 1. Прибор В для измерения вpe
мени раЗМЯ2чения липофильных суппозиториев
(размеры указаны в миллиметрах)
Методика. Во внутреннюю трубку (А) OTMe
ривают 5 мл воды С температурой (36,5:tO,5) ОС,
помещают суппозиторий заостренным концом
вниз и вводят вставку (С1 или С2). Отсчет Bpe
мени начинают С этоro момента. Полное размяr
чение или растворение суппозитория считает
ся законченным. Korдa нижний край стеклянноro
стержня с ободком (С1) или стальноro стержня
(С2) достиrнет суженной части внутренней CTe
клянной трубки_
01/2013:20923
2.9.23. ОПРЕДЕЛЕНИЕ ПЛОТНОСТИ
ТВЕРДЫХ ЧАСТИЦ
ПРИ ПОМОЩИ rАзовоrо
ПИКНОМЕТРА
Испытание на определение плотности TBep
дых частиц при помощи rазовоro пикнометра
предназначено для определения объема, за
нятоro определенным количеством порошка,
путем измерения объема rаза, вытесненноro
при определенных условиях В объем, получен
ный в данном испытании, не входит объем, за
нимаемый открытыми порами, но учитывается
объем закрытых пор и пор, недоступных для
rаза.
Обычно в качестве rаза для испытания ис
пользуется rелий, способный проникать в Ma
:
ленькие открытые поры. При использовании
друrих rазов полученные значения MOryT отл
чаться от значений, полученных при исполЬ30-
вании rелия, так как проникновение rаза зависит
как от размера пор, так и от площади поперечнcr
ro сечения молекулы rаза.
Измеренная плотность является объемнcr
массовой средней плотностью индивидуальных
частиц порошка. Она называется плотностью
частиц, в отличие от действительной плотности
твердых веществ или насыпной плотности пcr
рошка. Плотность твердых веществ выражается
в rpaMMax на кубический сантиметр (r/см З ), хотя
единицей системы СИ является килоrрамм на
кубический метр (кr/м З ).
ПРИБОР
Прибор (см. рисунок 2.9.23. 1) состоит из
следующих частей:
rерметичная испытательная ячейка, с объе
мом пустой ячейки V c ' которая соединена по
средством клапана со стандартной ячейкой
объема V r ;
система, способная заполнять ячейки
rазом до достижения определенноro давления
(Р), отображаемоro на манометре;
система, которая соединена с источни
ком rаза, предпочтительно rелия, если не указан
друroй rаз, для выполнения измерений.
Температура в rазовом пикнометре должна
быть от 15 ос до 30 ос и не должна изменяться
в течение всею процесса измерения более чем
на 2 ос.
Прибор rрадуирован: объемы V c и V r опре
деляются при помощи подходящих rрадуиро
вочных стандартов, объем которых известен с
точностью до 0,001 см З . Процедура, описанная
1{
Рисунок 2.9.23. 1. Схема 2азов020 пикнометра
А клапан;
V r объем стандартной ячейки, см З ;
V объем испытательной ячейки, см З ;
V c объем образца, см З ;
s
М манометр.
2.9.25. Высвобождение действующеzо вещества
471
ниже, выполняется в два этапа: с использовани
ем пустой ячейки для проведения испытания и
с использованием стандарта, помещенноro в ис
пытательную ячейку. Объемы V c и V, рассчиты
вают при помощи уравнения для определения
объема образца (V s )' принимая, что на первом
этапе V s равен нулю.
МЕТОДИКА
Предварительно удаляют из порошка ле
тучие примеси, деrазируя порошок в токе rаза;
в некоторых случаях порошки деrазируют Ba
куумом. Так как при проведении измерения
также MOryT выделяться летучие компоненты,
взвешивание образца проводят после прове
дения пикнометрическоrо измерения объема.
Взвешивают ячейку для проведения испы
тания и реrистрируют ее массу. Наполняют ис
пытательную ячейку указанным количеством
порошка испытуемоro образца. rерметично
закрывают испытательную ячейку в пикноме
тре. Реrистрируют указанное на манометре
давление сравнения (Р,) в системе при OTKpЫ
том клапане, который соединяет cтaHдapT
ную ячейку с испытательной ячейкой. Закры
вают клапан на отделении стандартной ячейки
от испытательной ячейки. Заполняют испыта
тельную ячейку rазом под давлением и опре
деляют исходное давление (Р), реrистрируют
полученное значение. Открывают клапан для
соединения стандартной ячейки с испытатель
ной. Реrистрируют конечное давление (Р,). По
вторя ют испытания образцов Toro же порошка
до тех пор, пока показатель последовательных
измерений объема образца (V s ) не окажется в
пределах 0,2 %. Разrружают ячейку для прове
дения испытания и измеряют конечную массу
порошка (т), выраженную в rpaMMax. В случае
если пикнометр отличается по требуемым опе
рациям или по конструкции от описанноro на
рисунке 2.9.2З_ 1, то поступают соrласно ин
струкции произво,gителя пикнометра.
ОЦЕНКА РЕЗУЛЬТАТОВ
Объем образца (V) рассчитывают по фор
муле:
V s =V c
V,
P -= 1'
Р, P,
Плотность (р) рассчитывают по формуле:
т
p= .
V s
Результаты указывают вместе с описанием
условий подroтовки образца. Например, указы
вают, испытывался ли образец как есть или был
высушен в условиях, которые указаны для опре
деления потери в массе при высушивании.
01/2013:20925
2.9.25. ВЫ ВОБОЖДЕНИЕ
ДЕИСТВУЮЩЕrо ВЕЩЕСТВА
ИЗ ЛЕКАРСТВЕННЫХ
ЖЕВАТЕЛЬНЫХ РЕЗИНОК
ПРИНЦИП
Высвобождение действующеro вещества из
лекарственной жевательной резинки определя
ют путем механическоrо смешивания пластины
жевательной резинки, помещенной в неболь
шую камеру, моделирующую процесс жевания,
с определенным объемом буферноro раствора.
ПРИБОР
Прибор (рисунок 2.9.25. 1) состоит из следу
ющих частей:
камера, моделирующая процесс жевания;
вертикальный поршень;
2 roризонтальных поршня с уплотнитель
ными кольцами и манжетами.
Камера, моделирующая процесс жевания,
состоит из четырех отдельных частей:
центральная камера;
воронка (рисунок 2.9.25. 2);
2 направляющие с втулками (рисунок
2.9.25. 3).
Воронка и направляющие смонтированы на
центральной камере. Уплотнительные кольца
находятся в уrлублениях на поршнях и окруже
ны манжетами; манжеты предназначены для дo
полнительноro предохранения камеры от проте
кания у направляющих.
Имитацию жевания резинки обеспечивают
roризонтальные поршни, а вертикальный пор
шень предназначен для обеспечения правиль
ноro нахождения резинки между roризонтальны
ми поршнями.
Скорость машины контролируют, обеспе
чивая постоянство циклов. Один цикл вклю
чает в себя следующие стадии: roризонталь
ные поршни начинают свое движение от самых
дальних положений и двиrаются до своих самых
ближних положений, и затем возвращаются в
дальние положения. Один раз в течение цикла
вертикальный поршень движется из своею ниж
неro положения до верхнею положения и обрат
но до нижнеro.
Ход каждоro rоризонтальноro поршня co
ставляет 25,0 мм. Максимальное расстояние
между двумя поршнями составляет 50 мм. Ми
нимальное расстояние между поршнями COCTaB
ляет от 0,1 мм до 1,0 мм. Ход вертикальноro
поршня составляет 22,0 мм.
Движение roризонтальных поршней проис
ходит таким образом, что оба поршня находят
ся в своих ближних положениях в один и тот же
момент времени. Вертикальный поршень Ha
страивают так, чтобы он не мешал работе roри
зонтальных поршней.
472
rосударственная фармакопея Республики Бела
СЕКЦИЯ Н 1: '-- o...l I
,,50
0
040 G 112
ю=нt =: t ::=
,.". I .Ш."Ш..I "; . .т..
А в С
,,16 М6х16
-: I
I I i
i
,,70
,,68
СЕКЦИЯ G G
D
Рисунок 2.9.25. 1. Камера, моделирующая процесс жевания, и поршни
(размеры указаны в миллиметрах)
А roРИ30нтальный поршень;
В направляющая;
С камера, моделирующая процесс жевания;
D воронка:
Е вертикальный поршень.
При необходимости roризонтальные поршни
MOryT быть настроены так, чтобы они вращались
BOKpyr своей оси в противоположных друr друry
направлениях в конце каждоro движения, имити
рующеro жевание. обеспечивая, таким образом,
максимально эффективное жевание.
Все части прибора, вступающие в контакт с
лекарственным средством или средой paCTBO
рения, должны быть химически инертными и не
должны адсорбировать образец, реаrировать и
взаимодействовать с ним.
МЕТОДИКА
Для проведения испытания необходима
следующая информация:
состав, объем и температура среды paCT
ворения;
количество движений, имитирующих жева
ние, в минуту;
время и способ отбора образцов;
проводят ли анализ остатка жевательной
резинки или анализируют среду растворения;
описание аналитической методики.
Указанный объем среды растворения,
обычно это 20 мл фосфатН020 буфеРН020
раствора рН 6, О Р2, помещают в камеру, MO
делирующую процесс жевания. Температуру
среды поддерживают при (37:tO,5) ос с помо
щью электрическоro приспособления с внеш
ним контролем. Выставляют скорость движе
ния поршней с указанной частотой движений,
имитирующих жевание, в минуту (обычно 60).
В камеру, моделирующую процесс жевания,
помещают точно взвешенную часть жеватель
Рисунок 2.9.25. 2. Воронка
(размеры указаны в миллиметрах)
2.9.26. Определение удельной площади поверхности
473
0З6
024.95
и'1 'l'З.'\
м
и'1
1'--
1. 1.
32
50
..
6З
Рисунок 2.9.25. З. Направляющие (секция G---G)
(размеры указаны в миллиметрах)
ной резинки или целую резинку и запускают
прибор.
ОТБОР ПРОБ И ПРОВЕДЕНИЕ АНАЛИЗА
Через указанное время прибор останавли
вают. Удаляют остатки жевательной резинки и
отбирают образец среды растворения. Подходя
щим методом определяют содержание действу
ющеro вещества. После каждоro отбора образца
может проводиться замена среды растворения,
при этом при расчетах необходимо учитывать
изменение объема или разведение среды paCT
ворения. В качестве альтернативы может опре
деляться содержание действующеro вещества
в остатке жевательной резинки. Испытание про
водят на 6 лекарственных жевательных резин
ках. Все 6 лекарственных жевательных резинок
должны выдерживать испытание.
Количество деi1ствующеrо вещества, pac
творившееся за определенное время, выражают.
в процентах от указанноro на этикетке значения.
01/2013:20926
2.9.26. ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ
ПЛОЩАДИ ПОВЕР)tНОСТИ
МЕТОДОМ rАЗОВОИ
АДСОРБЦИИ
ВВЕДЕНИЕ
Удельная поверхность порошка определя
ется измерением количества физически aдcop
бированноrо поверхностью твердоrо вещества
rаза, соответствующеro мономолекулярному
СЕlO/О на поверхности. Физическая адсорбция
является результатом действия относительно
cnа6ых сил (силы Ван дер аальса), действу
ЮЩЮ( между молекулами адсорбируемою rаза
и адсорбирующей поверхностью испытуемо
ro rюpoшка. Испытание обычно проводят при
температуре жидкоro азота. Количество aдcop
бированноro rаза может быть измерено rрави
метрическим методом, методом объемноro aHa
лиза и методом HenpepbIBHoro потока.
ТЕОРИЯ БРУНАУЭРА ЭММЕТТА ТЕЛЛЕРА
(БЭТ) И ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ
ПЛОЩАДИ ПОВЕРХНОСТИ
мноrОТОЧЕЧНОЕ ИЗМЕРЕНИЕ
Данные обрабатываются соrласно ypaBHe
нию изотермы адсорбции Брунауэра Эммепа
Теллера (БЭТ) (8ruпаие" Emmett, Telle, (ВЕТ):
1 == C 1 . + , (1)
[ ( РО ) Vm,C РО V m .С
V. 1
а Р
rде:
Р парциальное давление пара адсорби
руемоro rаза в равновесии с поверхностью при
температуре 77,4 К (температура кипения жид
коro азота), Па;
РО давление насыщенноro пара адсорби
руемоro rаза, Па;
V a объем rаза, адсорбируемоro при CTaH
дартных температуре и давлении (СТД CTaH
дартные температура и давление) [273,15 К и
атмосферном давлении (1 ,013 х 10 5 Па)], мл:
V m объем rаза, адсорбируемоro при СТД
дЛЯ создания Видимоro мономолекулярноro
слоя на поверхности образца, мл;
С безразмерная константа, характери
зующая энтальпию адсорбции адсорбируемоro
rаза на поверхности образца.
Значение V a измеряется для каждоro из не
менее чем трех значений Р/Р О '
После этоro строят rрафическую зависи
мость значения БЭТ
1
[V a -( 1)]
от значения Р/Р О в соответствии с уравнени
ем (1). Этот rрафик имеет, как правило, вид
прямой линии в диапазоне относительноro
давления приблизительно от 0,05 до О,З. По
J1ученные данные считаются приемлемыми,
если коэффициент корреляции линейной pe
rрессии , составляет не менее 0,9975; то есть
f2 составляет не менее 0,995. На полученном
линейном rрафике при помощи анализа ли
ней ной реrрессии находят наклон кривой, KO
торый равен (C 1)/VmC' и отсекаемый OTpe
474
rосударственная фармакопея Республики Беларусь
зок, равный 1/V m C. Из полученных значений
рассчитывают V m как 1/(наклон+отрезок). в
то время как С рассчитывют как (наклон/от
резок)+1. Ис)(одя из полученноro значения V m
рассчитывают удельную площадь повер)(но--
сти S (M 2 /r) по уравнению:
s =о V т . N . а ,(2)
т . 22400
rде:
N число Авоrадро (6,022 х 10 23 моль 1);
а полезная площадь поперечноro сечения
одной молекулы адсорбированноro вещества, м 2
(0,162 нм 2 для азота и 0,195 нм 2 для криптона);
т масса испытуемою порошка, r;
22400 объем, занимаемый одним молем
адсорбируемоro raза при СТД, учитывающий не--
значительные отклонения от идеальною rаза, мл.
Для испытания необходимы как минимум
три точки. Дополнительные измерения MorYT
быть проведены особенно при получении He
линейной кривой при значениях PIP o около 0,3.
Из за частой нелинейности кривой при зна
чениях Р/Р О ниже 0,05, значения в этом диа
пазоне использовать не рекомендуется. Ис
пытание на линейность, обработка данных и
расчет удельной площади поверхности описа
ны выше.
ОДНОТОЧЕЧНОЕ И3МЕРЕНИЕ
Как правило, для определения удельной
площади поверхности методом динамическо
ro потока адсорбции rаза (метод 1) или методом
объемноro анализа rазовой адсорбции (метод 11)
требуются как минимум три измерения V a ,
каждое при различных значениях P1Po' Однако
при определенных описанных ниже условиях
приемлемо определять удельную площадь по
верхности порошка на основании единственно
ro значения V a . измеренною при единственном
значении Р/Р О ' таком как 0,300 (что COOTBeTCTBY
ет 0,300 моль азота или 0,001038 моль фракции
криптона), используя следующее уравнение для
расчета V m :
, Р \
V =oV 'i1 J '(3)
т а l РО
Удельную площадь поверхности рассчиты
вают на основании значения \1.", соrласно ypaB
нению (2), приведенному выше.
Одноточечный метод может применяться
непосредственно на сериях образцов порош
ка определенноro материала, для которою KOH
станта материала С нам НОЮ выше, чем едини
ца. Эти условия верифицируют путем сравнения
значений удельной площади поверхности, опре
деляемых одноточечным методом, со значени
ями, определяемыми мноюточечным методом,
для серии образцов порошка. Большое сходство
между значениями, определенными при помощи
одноточечною и мноroточечноro методов, по--
зволяет предположить, что 1/С приближается 1(
нулю.
Одноточечный метод может применяться
косвенно для ряда сходных образцов порошка,
для которых константа материала С не является
бесконечной, но может считаться инвариантной.
При этих обстоятельствах поrрешность, свя
занная с применением одноточечноro метода,
может быть уменьшена или устранена путем ис
пользования мноюточечноro метода для оценки
С одноro из образцов серии в соответствии с
БЭТ, сотасно которому С рассчитывают как
1+ (наклон/отрезок). Torдa V m рассчитывают на
основании единственноro значения V a , измеря
емоro при единственном значении Р/Р О соrласно
уравнению:
( РО ) [ 1 С 1 ( Р JJ
V m Va' p 1 . с+с' РО .(4)
Удельную площадь ловерхности рассчиты
вают на основании значения V m соrласно ypaB
нению (2), приведенному выше.
ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДИКИ
В данном разделе описываются методы, ис
пользуемые для подroтовки образца: метод ди
намическоro потока адсорбции rаза (метод 1)
и метод объемноro анализа адсорбции rаза
(метод 11).
подrотовКА ОБРА3ЦА
Деrазация
Перед определением удельной площа
ди поверхности образца необходимо удалить
rазы и пары, которые моrли адсорбироваться
на поверхности после производства и при об
ращении с ним при ею использовании и xpa
нении. Если удаление rазов не проведено,
удельная площадь поверхности может быть
меньше или меняться, так как промежуточ
ная площадь поверхности покрывается моле
кулами ранее адсорбировавшихся rазов или
паров. Крайне важно соблюдать условия дe
rазации для обеспечения требуемой точности
и правильности измерений удельной площади
поверхности фармацевтических лекарствен
ных средств по причине чувствительности по
верхности материалов.
Условия. Подтверждением тою, что усло
вия деrазации соблюдены. является воспро
изведение rрафика БЭТ, постоянная масса
испытуемоrо порошка и отсутствие физиче
ских или химических изменений в испыту
емом порошке.
Условия деrазации, определяемые темпе
ратурой, давлением и временем, должны BЫ
бираться таким образом, чтобы изначальная
поверхность твердоrо вещества воспроизво
дилась максимально точно. Деrазация боль
шинства веществ достиrается при помощи
вакуума, путем очистки образца в токе инерт
2.9.26. Определение удельной площади поверхности методом
475
Крутые
пробки
Клапан контроля
ТОtl;З rаза
Испытаtельная
ячейка
g-
ф
::!;
о
::!;
а.
QJ
Прибор контроля
дифференциальноro
тока rаза
Выключатель
Входное
отверстие
для rаза
А
Детектор
Цифровой
дисплей
Закрытый цикл
измерите
потока !
Клапан
выбора пути
Замедлитель
диффузии
При бор
для деrазации
Короткий путь
балласта
Длинный путь
балласта
в
Выходное
отверстие
для rаза
Детектор
Рисунок 2.9.26. 1. Схема прибора при использовании метода динамичеСК020 потока
HOro сухоro rаза или чередованием циклов
десорбции адсорбции. Иноrда применяют
ся повышенные температуры для повышения
скорости очистки поверхности от заrрязня
ющих веществ. Деrазация путем наrревания
образца порошка может изменить природу
поверхности и целостность образца, поэто
му данную процедуру не следует выполнять,
если только нет особых указаний.
Если наrревание применяется, peKOMeH
дованная температура и время деrазации
должны быть малы, насколько это возмож
но, чтобы достиrалась воспроизводимость из
мерений удельной площади поверхности за
приемлемый интервал времени. Для деrаза
ции чувствительных образцов применяются
друrие метqды деrазации, такие как циклич
ный метод десорбции и адсорбции.
Адсорбат
Стандартным методом является адсорбция
азота аналитическоro качества при температуре
жидкоro азота.
Для порошков с низкой удельной площа
дью поверхности «0,2 M2fr) пропорция aд
сорбции является низкой, и в таких случа
ях предпочтительно использовать криптон
при температуре жидкоro азота, поскольку
низкое давление пара этоro rаза значитель
но уменьшает поrрешность. Использование
более крупных образцов, rдe это может быть
осуществимо (эквивалент 1 м 2 или более при
использовании азота), может компенсировать
ошибки определения малой площади поверх
ности.
Все используемые rазы должны быть
сухими.
Количество образца
Точно взвешенное количество испытуемоro
образца порошка, соответствующеro общей по
верхности образца не менее 1 м 2 , если в каче
стве адсорбата используется азот, и 0,5 м 2 , если
в качестве адсорбата используется криптон.
Использование меньших количеств испыту
емоro образца должно быть валидировано.
И3МЕРЕНИЯ
Так как количество адсорбируемоro rаза при
данном давлении имеет тенденцию к увеличе
нию при понижении температуры, измерение aд
сорбции, как правило, производятся при низкой
температуре.
Измерения производят при температуре
77,4 К, точке кипения жидкоro азота.
Метод 1: метод динамическоrо потока
Характеристика метода
В методе динамическоro потока (см. рисунок
2.9.26. 1) рекомендуемым rазом адсорбатом яв
ляется сухой азот или криптон; rелий же исполь
зуется в качестве rаза разбавителя, который не
адсорбируется при рекомендуемых условиях.
В диапазоне Р/Р О от 0,05 до 0,30 необходи
мо использовать не менее 3 смесей rаза адСОJr
бата и rелия.
Детектор интеrратор должен обеспечивать
сиrнал, приблизительно пропорциональный
объему проходящеro через не.ro rаза при задан
ных условиях температуры и давления. Для этой
цели одним из мноrих подходящих видов прибо-
ров является детектор теплопроводности с элек
тронным интеrратором. Должны определяться
минимум три значения в рекомендуемом диапа
зоне Р/Р О от 0,05 до 0,30.
476
rосударственная фармакопея Республики Беларусь
Методика
Известную смесь rазов, как правило, азота и
rелия, пропускают через ячейку теплопроводно--
сти, потом через образец и снова через ячейку
теплопроводности, а затем через записыва
ющий потенциометр.
Поrружают ячейку с образцом в жидкий азот,
образец адсорбирует жидкий азот из подвижной
фазы. Изменение теплопроводности ячейки и
импульс реrистрируются записывающим прибо
ром.
Изымают образец из охладителя; опреде
ляют равный по площади пик десорбции и про
тивоположный ему пик адсорбции. Так как пик
десорбции определяется леrче, чем пик aдoo
ции, именно он и используется для расчетов.
Для проведения калибровки вводят дocтa
точное количество адсорбата в систему для
получения величины пика сходной с ликом де--
сорбции и рассчитывают пропорцию raза, aдcop
бируемоro на единицу пика площади.
Используют смесь азота и rелия для одно--
точечноro метода и несколько таких смесей или
предварительно смешанные два потока rаза для
мноrоточечноro метода.
Расчеты производят так же, как и при методе
объемноro анализа.
Метод 11: метод объемноrо анализа
Характеристика метода
При методе объемноro анализа (рисунок
2.9.2б. 2) рекомендуемым rазом адсорбатом яв
ляется азот, который помещается в разряженное
пространство над предварительно деrазирован
ным образцом порошка, чтобы обеспечить опре
деленное равновесное давление Р rаза. Исполь
зование raза збавителя, например, rелия, не
Инд .атор
ва"уу...а
7
8
10
11
Резервуар
ДЛ rел"
Манометр
является обязательным, хотя reлий может прм-
меняться для друrих целей, таких как измерение
MepTBoro объема.
Так как применяется только чистый
адсорбат вместо смеси rазов, при этом методе
удается избежать интерферирующих эффектов.
возникающих при термальной диффузии.
Методика
Помещают небольшое количество cyxoro
азота в пробирку с образцом, чтобы предотврcr
тить заrрязнение чистой поверхности, достcr
ют пробирку, закрывают пробкой и взвешивают.
Рассчитывают массу образца. Присоединяют
пробирку к прибору для проведения объемно--
ro анализа. Аккуратно вакуумируют образец до
уt<азанноro давления (например, до давления от
2 Па до 10 Па). Кроме этоro может быть исполь-
зован прибор, способный к созданию вакуума с
известной скоростью изменения давления (Ha
пример, менее 1З Па/30 с), которую поддерж
вают необходимое количество времени перед
началом следующей стадии.
Если принцип действия прибора требует из
мерения мертвоro объема пробирки, например,
путем подачи неадсорбированноrо rаза (напри
мер, rелия) данная процедура выполняется в
этот момент, после чеro образец вакуумируется.
Измерения мертвоro объема пробирки можно из
бежать, произведя измерения отличий с исполь
зованием стандартной пробирки и испытуемой
пробирки, соединенных дифференциальным
преобразователем. Затем измеряется aдcop
ция азота, как указано ниже.
Поднимают сосуд Дьюара, содержащий
жидкий азот при температуре 77,4 К, до опреде
ленной точки испытуемой ячейки. Добавляют He
9
4
Переключател" кулеров
" вакуумных насосов
5
Разр женныи
воздух
Рисунок 2.9.26. 2. Схема прибора при использовании метода объеМН020 анализа
2.9.29. Характеристическое растворение
477
обходимое количество адсорбируемоro rаза до
обеспечения минимально допустимой величины
относительноro давления. Измеряют адсорбиро
ванный объем V a . При мультиточечном измере
нии повторяют измерения при последовательно
увеличивающихся значениях Р/Р О ' При исполь
зовании азота в качестве адсорбата часто под
ходящими значениями являются значения Р/Р О
0,10,0,20 и 0,30.
СТАНДАРТНЫЕ ВЕЩЕСТВА
Периодически необходимо проверять
работу прибора, используя соответствующие
стандартные вещества с известной площадью
поверхности, такие как алЬфа оксид алюминия
(а АI20з)' которые должны иметь удельную пло
щадь поверхности, сходную с площадью поверх
ности испытуемоro образца.
01/2013:20927
2.9.27. ОДНОРОДНОСТЬ
МАССЫ ОДНОИ ДO Ы,
ВЫСВОБОЖДЕННОИ
ИЗ МНQrодозовоrо
КОНТЕИНЕРА
Данное испытание предназначено для
оральных лекарственных форм, таких как 2pa
нулы, порошки и жидкости для внутренне20
применения, выпускаемые в МН020дозовых KOH
тейнерах, снабженных дозирующим ycтpoй
ством.
Взвешивают индивидуально 20 доз, изъя
тых произвольно из одноro или более контей
неров при помощи дозирующеro устройства.
Определяют массу каждой индивидуальной
дозы и рассчитывают среднюю массу. Массы
не более двух индивидуальных доз Moryr
иметь отклонения более чем на 1 О % от cpeд
ней массы, и ни одна масса индивидуальной
дозы не может иметь отклонение более чем
на 20 %.
01/2013:20929
2.9.29. ХАРАКТЕРИСТИЧЕСКОЕ
РАСТВОРЕНИЕ
Данное испытание предназначено для
определения скорости характеристическоro
растворения чистых твердых веществ после
прессования (уплотнения). Испытание прово
дят в определенных экспериментальных yc
лов иях, которые позволяют непосредственно
измерять скорость характеристическою paCT
ворения.
Скорость характеристическоrо paCTBope
ния это теоретическое значение, относящее
ся к чистому твердому веществу, имеющему HY
левую пористость; однако практически скорость
собственноro растворения определяется на об
разцах, имеющих минимальную пористость.
ПРИНЦИП
Скорость характеристическоrо paCТBope
ния является скоростью растворения чистых
веществ после прессования при условии по
стоянной площади поверхности. Определение
скорости характеристическоro растворения дает
полезную информацию при описании фарма
цевтических субстанций и вспомоrательных Be
ществ.
Скорость растворения чистых веществ за
висит от всех свойств, характерных твердым
телам, таких как кристаллическая форма, CTe
пень кристалличности, степень аморфности,
полиморфизм, псевдополиморфизм, размер
частиц и удельная площадь поверхности. Кроме
тоro, на нее MOryт влиять и внешние факторы
(условия проведения испытания), такие как rи
дродинамика, температура, вязкость, рН, бу
ферная сила и ионная сила среды растворения.
Оценка скорости характеристическою paCT
ворения твердоro вещества включает стадию
приroтовления спрессованноro (уплотненноro)
образца. Перед проведением испытания необхо
димо убедиться в соблюдении соответствующих
условий прессования испытуемоro образца.
Скорость характеристическоro paCTBope
ния определяют путем обработки постоянной
площади спрессованноro вещества подходя
щей средой растворения при постоянном пере
мешивании, температуре, ионной силе и значе
нии рН.
Скорость собственноro растворения выража
ется в единицах растворенной массы вещества
в единицу времени на единицу площади, под
верrнутой обработке, обычно в миллиrраммах в
минуту на сантиметр квадратный (мr,мин 1,см 2).
ПРИ БОР
Обычно прибор включает в себя пуан
сон и штамп, изroтовленные из закаленной
стали. Основание штампа имеет три резьбо
вых отверстия для присоединения к внешней
пластинке, изroтовленной из полированной
стали, являющейся зеркально rладким OCHO
ванием для спрессованноro образца. Штамп
имеет уrлубление, диаметр которою COCTaB
ляет от 0,1 см до 1,0 см, в которое помещают
измеренное количество порошка испытуемо
ro образца. После этоro пуансон помещает
ся в уrлубление в штампе, и испытуемый об
разец, обычно с использованием настольною
пресса, сжимается. Сквозное отверстие в ro
478
/осударственная фармакопея Республики Беларусь
ЛОБке пуансона позволяет использовать Me
таллический стержень для ускорения удале
ния пуансона из штампа после испытания.
Спрессованный испытуемый образец обра
зуется в уrлублении, и имеет одну сторону с
известной площадью. которая находится на
нижней стороне штампа (см. рисунок2.9.29. 1).
Дно уrлубления штампа имеет такую резьбу,
что не менее 50 75 % спрессованноro об
разца может растворяться, не выпадая из
штампа. Верх штампа имеет резьбу, позволя
ющую присоединять держатель. Держатель
монтируют на лабораторном перемешиваю
щем устройстве, а штамп с находящимся в
нем образцом поrружают в среду paCTBope
ния и вращают.
МЕТОДИКА
Вещество взвешивают на бумаrе для взве
шивания. С помощью трех болтов к нижней CTO
роне штампа присоединяют внешнюю пластин
ку. В уrлубление штампа помещают взвешенный
образец, вставляют пуансон и закрепляют
сверху системы металлическую пластинку.
Сжимают порошок с использованием rидра.
лическоro пресса и выдерживают в течение Д€;
статочноro времени, ЧТ Ы получился стабилЬ"-
ный спрессованный оЬразец с минимальноМ
пористостью; как можно тщательнее защищают
спрессоваНtlый образец от разрушения, при к€;
тором происходит увеличение площади поверх
ности, что приведет 1( возрастанию скорости
растворения. Отсоединяют внешнюю пластин
ку, прикручивают штамп вместе с пуансоном 1(
держатеnю и плотно зажимают. Весь лишний
порошок убирают с поверхности штампа путем
продувания сжатым воздухом или азотом.
Плавно вставляют конструкцию «штамп--
держатель» в зажимное устройство испыт
тельноro прибора и закрепляют ero в нем.
> Вставляют вал в шпиндель таким образом, что
после опускания насадки внешняя поверхность
спрессованноro образца будет располаrаться
на расстоянии 3,В см от дна сосуда. Сборный
диск располаrают таким образом, чтобы ми
нимизировать ero раскачивание; нельзя допу
скать образования пузырьков воздуха, так как
они способны уменьшать площадь спрессован
. .75 :t 0.35 F
CL1 110A
1218.0
F
....
;;1 А
1- 101 -,
С'.
r.
I D
с
CI 54.С :!: С.-3
в
I
I
'" 7.985 0.005
I
= 1
;1
I А
/ o \
( I
,
с;
/ o \
I I
,
/ o \
I I
,
Рисунок 2.9.29. 1. Типичный прибор, используемый для получения спрессоваННО20 испытуеМ020
образца для определения характеристичеСК020 растворения
(размеры указаны в миллиметрах)
А внешняя пластина;
В штамп;
С неопреновая прокладка;
О пуансон;
Е держатель с валом.
F нижняя сторона штампа.
2.9.31. Определение размера частиц методом дифракции
479
ноro образца, контактирующую со средой раст
ворения. По возможности, условия поrружения
поддерживают постоянными в течение всеro
времени испытания. Однако, в целях получе
Н Я раствора определяемоro образца с KOHцeH
трацией выше предела детектирования вслед
ствие оrраниченной поверхности, доступной
для растворения, может быть необходимым
использование относительно малых объемов
среды растворения.
Среду растворения наrpевают до Bы6paH
ной для испытания температуры. Насадку опу
екают в рабочее положение перед началом
вращения. Необходимо убедиться, что на по
верхности спрессованноro испытуемоro образ
ца отсутствуют пузырьки воздуха, так как их
присутствие уменьшит площадь спрессованно
ro образца, находящеrocя в контакте со средой
растворения. Немедленно включают прибор с
выбранной для испытания скоростью враще
ния. Отбор проб осуществляют через опреде
ленные интервалы времени и проводят коли
чественное определение с помощью метода,
обладающеro достаточной чувствительностью
\11 точностью.
ОЦЕНКА РЕЗУЛЬТАТОВ
Данные о суммарном количестве paCTBO
рившеroся вещества в каждую единицу времени
корректируют в зависимости от уменьшения раз
мера испытуемоro образца. Для тоro, чтобы pac
считать собственное растворение, на rрафике
а, мечают зависимость суммарноro количества
растворившеrося вещества с единицы площа
ди спрессованноrо образца от времени. CYM
марное количество растворившеroся вещества
с единицы площади спрессованноro образца по
лучают делением суммарноro количества pac
творившеroся в определенное время вещества
на экспонированную площадь. Получают линей
ную зависимость для нормализованных данных
от времени от начала испытания до возможно
ro разрушения спрессованноro образца. Исходя
из наклона полученной прямой, находят CKO
рость характеристическоro растворения испыту
емоro образца, выраженную в миллиrраммах в
минуту на сантиметр квадратный. Результат CKO
рости характеристическоro растворения должен
сопровождаться точным описанием приroтовле
ния спрессованноro образца и метода испыта
ния(среда растворения, объем использованной
среды, скорость перемешивания, температура и
т. д.).
Примечание: при необходимости и по co
rласованию с уполномоченным opraHoM, может
быть использован прибор с иной конфиryра
цией, например, с держателем штампа, закре
пленным вместе со спрессованным образцом
в вертикальной позиции, и с лопастями, распо
ложенными на определенном расстоянии от по
верхности спрессованноro образца, обеспечива
ющими необходимое перемешивание.
01/2013:20931
2.9.31. ОПРЕДЕЛЕНИЕ РАЗМЕРА
ЧАСТИЦ МЕТОДОМ
ДИФРАКЦИИЛАЗЕРноrо
ИЗЛУЧЕНИЯ
Метод основывается на стандартах /80
13З20 1(1999) и 9276 1(1998).
ВВЕДЕНИЕ
Метод дифракции лазерноro излучения, при
меняющийся для определения распределения
частиц по размерам, основывается на анализе
дифрактоrpаммы, полученной при воздействии
на частицы монохроматическоro излучения.
Исторически ранее выпущенные приборы ла
зерной дифракции использовали рассеивание
только под маленькими уrлами. С тех пор метод
был расширен с помощью использования pacce
ивания лазерноro излучения в более широком
уrловом диапазоне и применения теории Мие
(Mie) в дополнение к аппроксимации Фраунrофе
ра (Frаипhоfе,) и аномальной дифракции.
Метод не позволяет отличить рассеивание,
вызываемое отдельными частицами и rруппа
ми отдельных частиц (то есть аrломератами или
аrреrатами). Поскольку большинство испыту
емых образцов содержат аrломераты или arpe
raTbI и основной интерес представляет опре
деление распределения отдельных частиц по
размерам, перед измерением rруппы обычно
дисперrируют на отдельные частицы.
Для несферических частиц получают экви
валентное распределение ссрерических разме
ров, так как техника воспринимает их как опти
ческую модель сферических частиц. Результат
определения распределения частиц по разме
рам будет отличаться от таковоro результата,
полученноro методами, основанными на друrих
физических свойствах (например, седимента
ция, просеивание).
Эта rлава служит руководством для измере
ния распределения частиц по размерам в раз
личных дисперсионных системах, .например,
в порошках, спреях, аэрозолях, суспензиях,
эмульсиях и в жидкостях с rазовыми пузырями,
посредством анализа их модели уrловоro pacce
яния. rлава не несет специальных требований
по измерению размера частиц конкретных про
дуктов.
ПРИНЦИП
Через репрезентативный образец, дис
перrированный в определеl-lНОЙ концентрации
в подходящей жидкости или rазе, пропуска
ют монохроматическое излучение (обычно ла
зерное). Рассеянное частицами под разными
уrлами излучение измеряется при помощи муль
тиэлементноro детектора. Числовые значения,
представляющие собой модель рассеяния, pe
480
Тосударственная фармакопея Республики БеЛд
rистрируются для последующеro анализа. Эти
значения затем преобразуются с использовани
ем соответствующей оптической модели и Ma
тематической обработки в отношение общею
объема к дискретным количествам размерных
классов, представляющее собой объемное pac
пределение частиц по размерам.
ОБОРУДОВАНИЕ
Оборудование размещают таким образом,
чтобы защитить ею от воздействия электри
ческих помех, механических вибраций, темпе
ратурных флуктуаций, влаrи и прямоro яркою
света.
Пример прибора для дифракции лазерно
ю излучения приводится на рисунке 2.9.31. 1.
Может использоваться и друroе оборудование.
Прибор состоит из источника лазерною из
лучения, оптики, формирующей луч. измери
тельною блока (или кюветы) для образца, линз
Фурье и мультиэлементноro детектора для изме
рения рассеянноro излучения. Систематизация
данных необходима для преобразования чис
ловых значений модели рассеяния в объемное
распределение по размерам, а также для анали
за сопряженных данных и отчетов.
Частицы MOryт подверrаться воздействию
лазерноro излучения в 2 положениях. В обыч
ном случае частицы находятся перед собира
ющей линзой в параллельном пучке излучения
#(дифракция Фраунroфера)/I. В так называемой
перевернутой оптике Фурье частицы находятся
после собирающей линзы в сходящемся (KOH
BepreHTHoM) пучке #(дифракция Френеля)/I. Пре
имущество обычной установки состоит в том,
что рабочее расстояние линзы обеспечивает
приемлемую длину измерительной кюветы для
образца. Вторая установка предполаrает Ma
ленькую длину кюветы, однако позволяет изме
рять рассеянный свет под большими уrлами, что
является полезным в случае присутствия субми
кронных частиц.
Результат взаимодействия падающеro луча
света и множества дисперсных частиц представ
ляется в виде модели рассеяния с разной ин
7
.
.
/
6 5
8
8
(/)
тенсивностью под различными уrлами. Полное
уrловое распределение интенсивности,
ящее из прямоro и из рассеянноro света, фоку.
сируется с помощью линз на мультиэлемett'F-
ном детекторе. Фокусирующие линзы создЗlOl'
модель рассеяния, которая в определенных
ницах не зависит от местоположения частиц .
луче света. Следовательно, непрерывное y
вое распределение интенсивности преобр
вывается в дискретное пространственное рас>
пределение интенсивности с отображением ..
элементах детектора.
Предполаraется, что измеренная модв..
рассеяния множеством частиц равна сумме мо-
делей всех отдельных рассеивающих частИIL
представленных в случайных взаимных по
жениях. Следует обратить внимание на то, Ч1О
линзы и, соответственно, детектор собираюr
только оrраниченный уrловой диапазон раСС&
янноro излучения.
РАЗРАБОТКА МЕТОДИКИ
С использованием дифракции лазерноro
излучения можно проводить измерения даже 8
субмикронном диапазоне при условии, что ис
пользуемый прибор и испытуемый образец тща.-
тельно контролируются с целью уменьшения
непостоянства условий испытания, таких как, Ha
пример, среды дисперrирования, способа приro-
товления дисперсии образца.
Традиционно измерение размера частицы
с использованием лазерной дифракции проВ<r
дилось в диапазоне приблизительно от 0,1 мкм
ДО 3 мм. Современные приборы способны pac
ширить этот диапазон блаroдаря достижениям
в разработке линз и оборудования. В отчете по
валидации указывают применимость метода дня
осуществления намеченной цели.
Отбор проб. Способ проведения отбора проб
должен быть подходящим для получения репр&
зентативноro образца подходящею объема для
измерения размера частицы. Moryт использоваТkr
ся такие методы, как метод вращающеroся разд
лителя или метод конусования и квартования.
9
11
10
.......
i I (8) /
3
1
2
4
Рисунок 2.9.3 1. 1. Схема дифракциОНН020 прибора с лазерным излучением
1 детектор; 2 рассеянный луч; 3 падающий луч; 4 линзы Фурье; 5 рассеянное излучение, не собранное линзами (4);
6 множество частиц; 7 источник лазерноro излучения; 8 устройство, образующее луч; 9 рабочее расстояние линз (4):
10 мультиэлементый детектор; 11 фокусное расстояние линз (4).
2.9.31. Определение размера частиц методом дифракции
481
Выбор метода дисперrирования. Ис
пытуемый образец исследуют визуально или
с помощью микроскопа для установления ди
апазона размеров частиц и их формы. Метод
дисперrирования должен соответствовать цели
измерения (отдельная частица или rруппа
частиц (кластер») Поэтому используют либо дe
аrломерацию кластеров на отдельные частицы в
максимально возмежной степени, либо C:::JxpaHe
ние неповрежденных кластеров, насколько это
возможно
Для проведения определения целесо
образно убедиться, что не происходит дробле
НИfl частиц и что частицы или кластеры YДOB
летворительно дисперrированы. Обычно это
производится путем изменения дисперrиру
ющей силы и контроля над вызванным изме
нением распределения частиц по размерам.
Коrда образец хорошо дисперrирован, а части
цы не хрупкие и не растворимые, то распре
деление по размерам не должно значительно
изменяться. Кроме тоro, интересующие части
цы можно контролировать визуально или при
помощи микроскопа. Если изменяются усло
вия производственноrо процесса (например,
;<ристаллизация, измельчение) испытуемоrо
:Jбъекта, то применимость метода необходимо
проверить (например, путем микроскопическо
,о сравнения).
Спреи, аэрозоли и жидкости с пузырька
ми rаза должны отмериваться точно при усло
В I 1И приroдности их концентрации, так как отбор
пробы или разведение обычно изменяют pac
пределение частиц по размерам.
В друrих случаях (речь идет об эмуль
СИSlх, пастах и порошках) репрезентативные
образцы MorYT быть дисперrированы в под
ходящих жидкостях. Для деаrломерации или
деаrреrации кластеров и стабилизации дис
персии используют механическое диспер
rирование (например, взбалтывание, обра
ботка ультразвуком) и/или вспомоrательные
вещества (увлажняющие средства, стабилt1
заторы). Для жидких дисперсий обычно ис
пользуют систему с рециркуляцией, которая
состоит из оптической кюветы, дисперси
онной бани, обычно оснащенной мешалкой
и ультразвуком, насоса и соединительных
трубок. Не способные к рециркуляции систе
мы, оснащенные мешалкой, используются
только в случае малоro количества образца
или при использовании специальных диспер
сионных жидкостей.
Сухие порошки MOryт также преобразовы
ваться в аэрозоли с помощью подходящих по
рошковых дисперrаторов, которые используют
механическое воздействие для деаrломерации
или дезаrреrации. Дисперrаторы обычно исполь
зуют энерrию сжатоro rаза или перепад давле
ния в вакууме для дисперrирования частиц до
аэрозоля, который уносится через измеритель
ную зону 80 входное отверстие rзакуумной YCTa
новки, собирающей частиц!::!. Однако, дЛЯ CBO
бодно текучих крупных частиц или rранул для
достаточной дисперсности может быть ДOCTa
точно силы тяжести.
Если частицы испытуемою образца, имею
щие наибольший размер, выпадают из диапа.
зона измерений данноrо прибора, материал, KO
торый является спИШКQМ rрубым, может быТ!-
удален с помощью просеивания с указанием ко
личества, в процентах, удаленною веществ
Однако необходимо учесть, что образец, OCTaB
шийся после просеивания, уже не является pe
презентативным, если не доказано иное.
Оптимизация жидкостной дисперсии
Жидкости, поверхностно активные вещества
(сурфактанты) и дисперrирующие areHTbI, I"С
пользующиеся для дисперrирозания порошков
должны отвечать следующим требованиям:
должны быть прозрачными при длине
волны лазерноro излучения и практически нЕ'
содержать пузырьков воздуха или посторонних
частиц;
иметь показатель преломления, отличны!<
от испытуемою образца,
не растворять испытуемый образец (чиста
жидкость или предварительно профильтрованный
насыщенный раствор);
не изменять размер испытуемоrо образца
(например, из за растворения, повышения рас
творимости, или эфФектов рекристаллизации);
способствовать формированию и стабиль
ности дисперсии;
не оказывать влияние на материалы, ис
пользующиеся в приборе (наrример прокладки,
соединительные трубки, и т. д.);
обладать подходящей вязкостью, способ
ствующей рециркуляции, перемешиванию и филь.
трации.
Сурфактанты и/или дисперrирующие areHTbi
часто используются для смачивания частиц и
стабилизации дисперсии. Для слабых кислот и
слабых оснований, буферирование дисперсион
ных сред при низких и высоких значениях рН, co
ответственно, может помочь в идентификации
подходящеro дисперrирующеrо areHTa.
Предварительная проверка качества дис
персии может быть выполнена визуально или
при помощи микроскопа. Можно пользовать
ся фракционными образцами, которые roтовят
разведением ПОРЦИИ хорошо перемешанной ба
зовой дисперсии. Базовые дисперсии получа
ют путем прибавления жидкости !( образцу при
постоянном перемешивании, например, CTe
клянной палочкой, шпателем или на вихревой
мешалке. Особое внимание уделяется репре
зентативности порции, отобранной из базовой
дисперсии; также следят за тем, чтобы не про
изошло оседание больших частиц. Таким обра
зом, приroтовление пасты из испытуемоro об
разца или отбор проб производят быстро из
перемешивэющеЙС Q суспечзии.
rосударственная фармакопея Республики Беларус.
482
Оптимизация rазовой дисперсии. Цля
аэрозолей и сухих порош"овых дисперсий может
использоваться сжатый rаз. не содержащий
масел, воды и друrих частиц. Чтобы удалить эти
материалы из сжатоrо rаза, можно использовать
сушилку с фильтром. Любое вакуумное приспо
собление должно быть расположено на takol-
расстоянии от измерительноro прибора, чтобы
не оказывать физи-.;ескоro воздействия на про
цесс измерения.
Определение диапазона концентра-
ций. Чтобы установить приемлемое отношение
сиrнал/шум в детекторе, концентрация чаСl'ИЦ
в дисперсии должна превышать минимальный
уровень. В то же время, чтобы избежать мноro
кратноro рассеяния. она должна быть ниже MaK
симальноrо УDС8НЯ. На диапазон концентраций
влияет ширина лазерноro луча, длина измери
тельноro участка, оптические свойства частиц и
чувствительность элементов детектора.
Ввиду вышеизложенноro, для определе
ния соответствующеrо диапазона концентраций
любоro типицноro образца необходимо выпол
нить измерения при различных концентраци
ях частиц. (Примечание: в различных прибо
рах концентрация частиц обычно выражается в
разных размерностях и единицах, например, за
темнение. сптическая концентрация, пропорци
ональное значение общей массы).
Определение времени измерения. Время
измерения. время и частота сбора данных дe
тектором определяют экспериментально таким
образом, чтобы достичь желаемой точности.
Обычно за время измерения происходит боль
шое количество сканирований за короткий про
межуток времени.
Выбор подходящей оптической модели.
Для расчета матрицы рассеяния в большинстве
приборов чаще используются теории Фраунro
фера или Мие. Выбор теоретической модели
зависит от предполаrаемоro применения и раз
личных допущений (размер, абсорбция, показа
тель преломления, шероховатости, ориентация
кристалла, смесь и т. д.) относительно испыту
емоro образца. Если значения показателя пре
ломления (реальные и мнимые части для ис
пользуемой длины волны) точно не известны,
то может использоваться аппроксимация Фра
унrофера или теория Мие с реалистической
оценкой показателя преломления. Аппроксима
ция Фраунroфера имеет преимущества, так как
она проста, не требует использования показа
теля преломления; теория Мие обычно предо
ставляет менее смещенное распределение по
размерам для маленьких частиц. Чтобы полу
чить прослеживаемые результаты, необходимо
записывать используемые значения показате
ля преломления, так как небольшие различия
в значениях, принятых для реальной и мнимой
части комплексноro показателя преломлен....
MorYT обуславливать существенные различия 8
измеренных распределен ях частиц по разме-
рам. Малые величины МНИМ:JЙ части показате-
ля преломления (приблизительно 0,01 , 1 i)
часто применяются для коррекции абсорбцим
с учетом поверхностной шероховатости частиц.
Необходимо отметить, что в общем спучае на
конечный результат оказывают ВЛИЯЫ!EJ оптиче-
ские свойства испытуемоro вещества, а так же
еro структура (например, форма, неровност-
поверхности и пористость).
Валидация. Обычно приroдность метода
может быть определена путем оценки ею специ--
фичности, линейности, диапазона, точност
I:Jравильности и робастности. При определении
размера частиц методом дифракции лазерно-
ro излучения специфичность, как она опреде-
лена Международным комитетом по rармонизcr
ции (/СН) , не используется, так как невозможно
обнаружить отличия между различными KO
понентами образца. а также невозможно отли--
чить аrломераты от ДИСllерrированных частиц.
если только данный метод не дополнен 8 полной
мере микроскопическими исследованиями. Для
данноro метода не проводят изучение линейно-
сти зависимости между концентрацией и откли
ком или математической модели для интерпре-
тации результатов. Вместо оценки линейности
для данноro метода необходимо определить д*
апазон концентраций, в котором результаты не
отличаются друr от дрУI'а в значительной степе-
ни. Концентрации ниже определенноro диапа
зона приводят к ошибкам, связанным с НИЗКИ'А
отношением сиrналiшум, в то время как KOHцe
трации. расположенные выше этоro диапазона,
приводят к ошибкам, вызванным множествен
ным рас еИБанием. Этот диапазон rлавным o
разом зависит от используемоrо оборудова
ния. Правильность должна быть подтверждена
путем подходящей поверки прибора и сравнения
с данными, полученными с помощью МИКрОСКС
пии, Torдa как точность может быть оценена при
определении сходимости.
Досяrаемая сходимость метода rлавным об.
разом зависит от свойств материала (молотый/
немолотый, прочный/хрупкий, степень распре-
деления еro частиц по размерам и т. д.), а тре-
бу ющаяся сходимость от цели измерения. В
этой rлаве не устанавливаются жесткие пределы,
поскольку сходимости (различные приroтовле-
ния образцов) MorYT существенно отличаться ДЛSI
разных веществ. Однако, по правилам надлежа--
щей практики приемлемым критерием сходи
сти для любоro центральноro значения расп
деления (например, дЛЯ Х 5О ) является S,el 1 О %
[п = 6]. Для значений по краям распределениl
(например, Х 1О и Xgo) применимы менее стp0rи8
приемлемые критерии S,el 15 % [п = 6]. Если ча-
стицы менее 10 мкм, то значения должны удва.
И13аТЬСG. РобаСТrЮСТЬ может бt,р ь оценена п..
2.9.31. Определение размера частиц методом дифракции
483
выборе и оптимизации дисперсионной среды и
силы дисперrирования. Изменение энерrии дис
перrирования может быть прослежено по измене
нию распределения частиц по размеру.
ПРОВЕДЕНИЕ ИЗМЕРЕНИЙ
Предосторожности. Руководство по экс
плуатации используемоro прибора включает
следующие требования:
никоrда не направлять лазерный луч или
еro отражение в rлаза;
все оборудование должно быть заземлено
для предотвращения воспламенения раствори
телей или взрыва пыли;
необходимо следить за установкой при
бора (например, проrрев, требуемый диапазон
измерений, линзы, соответствующее рабочее
расстояние, положение детектора, отсутствие
попадания прямых лучей дневноro света);
в случае влажной дисперсии, предупреж
дать появление воздушных пузырей, испаре
ния жидкости, неоднородностей в дисперсии; в
случае сухой дисперсии необходимо избеrать
неправильноro потока массы от дисперrатора
и турбулентноro потока воздуха; такие эффек
ты MOryт привести к ложному распределению
частиц по размерам.
Измерение рассеяния излучения диспер
rированным образцом (образцами). После
надлежащей настройки оптической части при
бора выполняется контрольное измерение дис
персионной среды, не содержащей частиц, с
использованием той же методики, которая ис
пользуется ДЛЯ измерения испытуемоro образ
ца. Фоновый сиrнал должен быть ниже пороrа
различимости сиrнала. Полученные данные co
храняют для тою, чтобы затем вычесть их из
данных, полученных при измерении испытуемо
ro образца. Измерение образца проводят по раз
работанной методике.
Для каждоro элемента детектора рассчи
тывается средний' сиrнал, иноrда вместе с ero
стандартным отклонением. Величина сиrнала из
каждоro элемента детектора зависит от области
обнаружения, интенсивности излучения и KBaH
товой эффективности. Координаты (размер и по
ложение) элементов детектора вместе с фокаль
ным расстоянием линзы определяют диапазон
уrлов рассеяния для каждоro элемента. Боль
шинство приборов также измеряет интенсив
ность центральноro (нерассеянноro) лазерно
ro луча. Отношение интенсивности, полученой
с использованием дисперrированноro образца,
к интенсивности контрольноro измерения опре
деляет долю рассеянною излучения и, следова
тельно, концентрацию частиц.
Преобразование модели рассеяния в
распределение частиц по размерам. Это пре
образование является инверсией вычисления
модели рассеяния для данноro распределения
частиц по размерам. Особенно важным являет
ся допущение о сферической форме частиц, по
скольку в большинстве алroритмов используется
математическое решение для рассеяния сфери
ческих частиц. Кроме тоro, измеренные данные
Bcerдa содержат некоторые случайные и систе
матические ошибки, которые MOryT исказить pac
пределение по размерам. Разработаны разные
математические приемы для использования на
доступных при борах. Они содержат отклонения
между измеренными и рассчитанными моделя
ми рассеяния (например, среднеквадратиче
ские), некоторые оrраничения (например, коли
чество частиц не может быть отрицательным) и/
или некоторое «сrлаживание» кривой распреде
ления частиц по размерам.
Используемые алroритмы являются специ
фическими и запатентованными для каждоro
вида и модели оборудования. Отличия в алro
ритмах для различных приборов MOryт приво
дить К росту различий в статистике рассчитан
ных размеров частиц.
Количество повторов. Рекомендуется,
чтобы число повторных измерений (с отдельны
ми приroтовлениями образцов), проводимое для
KOHKpeTHoro образца, определялось по специ
альному для данной субстанции методу.
ПРЕДСТАВЛЕНИЕ РЕЗУЛЬТАТОВ
Данные определения размера частиц обычно
представляют в виде интеrральной кривой CYM
марноro подразмерноro распределения и/или в
виде распределения плотности в определенном
объеме. Символ х используется для обозначе
ния размера частицы, который, в свою очередь,
определяется как диаметр сферы с эквивалент
ным объемом. аз(х) обозначает объемную долю
фракции с указанием подразмера при х. В rpa
фическом представлении по оси абсцисс от
кладывается х, а по оси ординат зависимая
переменная аз. Большинство общих xapaKTe
ристических значений рассчитываются из pac
пределения частиц по размерам путем интерпо
ляции. Часто используют размеры частиц с их
подразмерными величинами 10 %, 50 % и 90 %
(обозначают как Х 1О ' Х 50 и Х 9О соответотвенно). Х 5О
обозначает срединный (медианный) размер ча
стицы. Символ х может быть заменен символом d,
который также широко используется для обозна
чения размера частиц.
Кроме тоro, отчет должен содержать дo
статочную информацию об образце, ero при
roтовлении, условиях дисперrирования и типе
кюветы, а также о модели используемоro при
бора и еro оптической системt3, и проrрамме
анализа данных.
ПРОВЕРКА ЭКСПЛУАТАЦИОННЫХ
ХАРАКТЕРИСТИК ОБОРУДОВАНИЯ
Прибор должен использоваться в соответ
ствии с инструкциями изroтовителя, а ero повер
484
rосударственная фармакопея Республики Беларусь
ка (калибровка) должна r'рОИЗ80ДИТЬСЯ с peKO
мендованной периодичностью в зависимости от
частоты еro использования.
rрадуировка. Системы nазерной дифрак
ции учитывают идеаЛИ3v l ;х)ванные свойства
частиц и базируются нз тзвных принципах pac
сеяния лазерноrо излучения. Таким образом,
rрадуировки в бую?аJlЬНОМ смысле этоro слова
не требуется. Однако необходимо удостоверить
ся, что прибор работает правильно. Это можно
осуществить с использованием любоro прием
лемоro аттеСТСl3зчнпrо или эталонноro образца
сравнеCJИЯ. ПРОВЕ;ряется весь процесс измере
ния, включая сбор и дисперrирование образца,
ею перl';НОС чере"3 3CJHY измерения, само изме
рение и ПОСf1едующее преобразование. Весь
операционный процесс должен быть подробно
описан.
Предпочтите!lLНО использовать aттeCTO
ванные или эталонные образцы сравнения,
состоящие из сферических частиц с извест
ным распределением. Они аттестуются по
массо процентно;v1У распределению по разме
рам с помощью эталонноrо метода, если это
возможно, и испопьзуются с учетом соrласо
аанноro. детализированноrо порядка опера
ЦИЙ. Если для обработки данных используется
теория "'1ие. то необходимо указывать реаль
ные и мнимые части комплексноro показателя
,реломлеН.1Я вещества. При условии, что плот
ч')сть частиц одинакова для всех размерных
фракций. 06ъе:>1ное распределение частиц по
размерам буде"' соответствовать их массовому
расr.ределениfC' .
СUVlтается. ч ie показатели прибора лазер
ной дифра'.L!ИИ отвечают требованиям, если
среднее знаL.ение Х:50 как минимум трех неза
висимых ИЗi\.lЕ: ений ОТКЛОН}jется не более чем
на 3 % ОТ ус;тановленноro диапазона значений
(среднее ЗI' uение вместе с еro стандартным OT
КJlонением i дr:я aHeCT:JBaHHOro или эталонно
со образца. Сре;:1чие значения дЛЯ Х,О и Х 9О не
должны отклоl-'ЯТЬСЯ более чем на 5 % от YCTa
новленноrо диапазона значений. Если размер
частиц менее 10 мкм, то эти значения должны
удваиваться.
Кроме образцов, состоящих из сферических
частиц, допускается использование образцов
с несферическими частицами. Такие частицы
должны иметь аттестованные или характерные
значения, полученные методом лазерной диф
ракции в соответствии С учетом соrласованноro,
детализированноro порядка операций. Исполь
зование сравнительных значений, полученных
с использованием друrих методов, может при
вести к отклонению результата измерения от
истинноro значения. Причина этоro отклонения
заключается в том, что из за разных принципов
ряда методов для одной и той же несфериче
ской частицы MorYT быть установлены различ
ные диаметры эквивалентной сферы.
В дополнение к аттестованным образцам
сравнения, упомянутым выше, MOryT ИСПОЛ::.З<r
ваться образцы продуктов с характерным для
данноrо класса составом и распредепением
частиц по размерам при условии, что это р<1спре
деление будет устойчивым с течением BpeMe
ни. Результаты должны соответствовать рачее
определенным данным с тоЙ же точностью v1 OT
клонениями, как и для аттестооанных обрс.ЗЦ08
сравнения.
Поверка (квалификация) системы. В дo
полнение к rрадуировке рабочие характерис-r.1КИ
прибора должны проверяться через постоян."ые
промежутки времени или с подходящей ча::;то
той. Это может быть осуществлено с ИСПОЛьзо
ванием подходящеro стандартноro образца, ка",
описано выше.
Поверка системы основывается на предпо
ложении, что оборудование, электроника, про
rpaMMHoe обеспечение и аналитические опе
рации составляют общую систему, которая
рассматривается как единое целое. Таким обра
зом, поверяется весь процесс измерения, вклю
чая сбор и дисперrирование образца, еro пере
нос через зону измерения, само измерение и
последующее преобразование. Весь операцион
ный процесс должен быть подробно описан.
Обычно, если нет указаний в частных CTa
тьях, то отклик прибора лазерной дифракции
считается удовлетворительным, если значение
Х 5О отклоняется не более чем на 1 О % от диа
пазона значений образца сравнения (то есть от
среднеro значения вместе с еro стандартным
отклонением). Значения по краям распределе
ния (например, Х,О и Х 9О ) должны ОТКЛОНЯТЬС5-. <Ш
более чем на 15 % от аттесованноro диапазона
значений. Если размер частиц менее 1 О мкм, ТО
эти значения должны удваИ9аться.
ПРИМЕЧАНИЕ: при 2радуировке прибора,
как описано в разделе «/радуировка», применя
ются более жесткие требования.
01/2013:20932
2.9.32. ПОРИСТОСТЬ
И ОПРЕДЕЛЕНИЕ РАЗМЕРА
ПОР С ИСПОЛЬЗОВАНИЕМ
РТУТНОЙ ПОРОЗИМЕТРИИ
ВВЕДЕНИЕ
В общем случае поры MorYT быть изобра
жены как отверстия, каналы или полости внутри
твердоro тела или как пространство (щели или
пустоты) между твердыми частицами (спресC<r
ванными, аrреrированными или в слое). Пори--
стость это термин, часто используемый для
2.9.32. Пористость и определение размера пор
485
обозначения пористой природы твердоrо MaTe
риала, и более точно она определяется как OTHO
шение объема доступных пор и пустот к общему
объему, занимаемому данным количеством Ma
териала. Кроме доступных пор, твердый матери
ал может иметь закрытые поры, которые изоли
рованы от внешней поверхности и в которые не
способна проникать жидкость. Закрытые поры,
например, полости без выхода на поверхность,
в данном разделе не рассматриваются.
Пористые материалы MOryт иметь форму
мелких или rрубых порошков, спрессованных
образцов, экструдатов, листов или монолитов.
Их описание обычно включает в себя определе
ние общеro объема пор, а также размера пор.
Хорошо известно, что характеристики пори
стоro твердоro материала (например, прочность,
реакционная способность, проницаемость или
адсорбирующая способность) зависят от CTPYK
туры пор. Для ее описания было разработано
большое количество различных методов. Ввиду
сложности большинства пористых материалов,
не удивительно, что получаемые результаты не
Bcerдa совпадают и не существует метода, KOТO
рый cMor бы позволить получить полную картину
структуры пор. Выбор используемоro метода за
висит от применения пористоro материала, еro
химической и физической природы и диапазона
размеров пор.
Данная rлава приводит руководство по из
мерению пористости и размера пор с использо
ванием ртутной порозиметрии. Это сравнитель
ное испытание, обычно деструктивное, в котором
объем ртути, проникающей в поры или полости,
определяется как функция приложенноro rидро
статическоro давления, которое может быть COOT
несено с диаметром пор. Иная информация, как
форма пор и их взаимосвязь, площадь BHyтpeH
ней и наружной поверхностей, rранулометрия по
рошка, насыпная плотность и плотность после
усадки также MOryт зависеть от кривой зависи
мости объема 01" давления, однако, эти аспекты
метода выходят за рамки данноro раздела.
На данный момент по практическим сообра
жениям максимальное прилаrаемое абсолютное
давление, достиrаемое некоторыми приборами,
оrраничено 400 МПа, что соответствует мини
мальному диаметру пор около 0,003 мкм. Макси
мальный диаметр оrраничен rидростатическим
напором ртути из за значительной rлубины об
разцов. В большинстве случаев этот предел co
ставляет 400 мкм.
Может быть определена пористость внутри
частицы и пористость между частицами, однако,
при наличии обоих видов пористости данный
метод не разделяет их.
Метод подходит для испытаний большин
ства пористых материалов. Образцы, образую
щие амальrаму, такие как некоторые металлы,
должны быть предварительно пассивированы, в
ином случае они не MOryт быть Подверrнуты ис
пытанию данным методом. Некоторые матери
62 Зак.I712
алы MoryT деформироваться или сдавливаться
под действием прилаrаемоro давления, тorдa в
некоторых случаях применяют коррекцию, учи
тывающую сдавливание образца, для получе
ния сопоставимых данных.
Ртутная порозиметрия считается сравни
тельным методом, так как для большинства по
ристых сред отсутствуют теории, позволяющие
получить абсолютные данные о распределении
в соответствии с размером пор. Поэтому данный
метод рекомендован rлавным образом для ис
следований при разработке.
Так как ртуть токсична, должны быть приня
ты соответствующие меры предосторожности по
охране здоровья оператора и друrих работни
ков, находящихся в этом же помещении. Отходы
должны храниться в соответствии с требовани
ями реryлирующих opraHoB.
ПРИНЦИП
Методика основана на измерении объема
ртути, проникнувшей в поры твердоro матери
ала, при различном прилаrаемом давлении. Из
мерение охватывает только те поры, в которые
ртуть способна проникнуть при заданном давле
нии.
Несмачивающая жидкость проникает в поры
только под давлением. Необходимое для про
никновения давление находится в обратной за
висимости от внутреннеro диаметра отверстия
поры. В случае цилиндрических пор, зависи
мость между диаметром пор и давлением опи
сывается уравнением Уошборна (Washbuт):
4'а
d р = cose,
р
rдe:
d p диаметр пор, м;
cr поверхностное натяжение, H/M;
е краевой уrол ртути на образце, о.
р прилаrаемое давление, Па.
ПРИ БОР
Держатель образца, называемый пенетро
метр или дилатометр, оснащен rрадуирован
ной капиллярной трубкой, через которую обра
зец может быть вакуумирован и через которую
может подаваться ртуть. Капилляр присоеди
нен к более широкой трубке, в которую помеща
ют образец. Изменение объема проникнувшей в
образец ртути обычно отслеживают по измене
нию объема между столбом ртути в капилляре
и металлической втулкой BOKpyr капилляра. Для
обеспечения точности измерений необходимо,
чтобы предполаrаемый объем пустот и пор об
разца составлял от 20 % до 90 % от BHYTpeHHe
ro объема капиллярной трубки. Так как различ
ные материалы обладают широким диапазоном
пористости, может понадобиться большое коли
чество пенетрометров с различными диаметра
ми капиллярной трубки. Стандартная CTPYКТY
486
rосударственная фармакопея Республики Беларусь
ра прибора представлена на рисунке 2.9.32. 1.
Прибор может иметь различные входные O 8ep
стия для измерений при высоком и низком дaB
лении, либо для этоro MOryт использоваться раз
личные приборы.
Диапазон для измерений при низком давле
нии обычно составляет 4 300 кПа, для измере
ний при высоком давлении свыше 300 кПа, в
зависимости от используемоro прибора и целей
исследования.
II
11
III,..... , 11 1
I 11
[В
LG
I C;::J
I
I
I
I
I
I
I
I
I
I
I
GJ
Е
н
к
...
м
1111 ;$
N "'r Y H
т tt
I..Н".,..л ",
. .:...-rt .....«
с
Рисунок 2_9.З2. 1. Пример устройства прибора
для ртутной порозиметрии
А резервуар с низким давлением для rидравлической
жидкости:
В rидравлический насос;
С умножитель давления;
О даТ'!ик давления,
Е резервуар с ВbICOКJ.1M давлением дпя IV\DJJCIвлической жидкости;
F вакууJ.lНЫЙ насос с измерительным прибором;
G резервуар для ртути;
Н масло:
J индикатор объема проникновения;
К капиллярная трубка;
L камера с высоким давлением;
М ртуть;
N образец.
МЕТОДИКА
Приrотовление образца
Для удаления адсорбированноro вещества,
которое может скрыть доступные поры, образец
предварительно обрабатывают с помощью Ha
rревания и/или вакуумирования либо продувая
инертным rазом. Может понадобиться пассиви
рование поверхности смачиваемых или образу
ющих амальrаму образцов, например, формируя
тонкую пленку оксида или покрывая стеаратом.
Обработанный образец взвешивают и по
мещают в пенетрометр. Деrазируют систему пор
образца в вакууме при давлении, не превыша
ющем 7 Па.
Наполнение пенетрометра ртутью
Используют ртуть аналитической чисто
ты. Образец под вакуумом покрывают ртутью.
Вакуум необходим для тоro, чтобы переместить
ртуть из резервуара в пенетрометр. В Hanon-
ненном пенетрометре наполняющее давление
состоит из прилаrаемоro давления и давле-
ния, создаваемоro контактирующей с образцом
ртутью. Обычно наполнение проводят при да.
лении около 4 кПа. rидростатическое давление
ртути над образцом может быть минимизиро
но путем наполнения пенетрометра в roризо
тальном положении.
Измерения при низком давлении
Для увеличения давления наrнетают азот
или воздух в соответствии с определенным pa
мером пор или постоянно с малой скоростью.
Записывают изменения длины pTYTHoro столба
в капилляре. После тоro, как достиrнуто макси
альное требуемое давление, возвращаются к
атмосферному давлению.
Измерения при высоком давлении
После измерений при низком давлении, Ha
полненный ртутью пенетрометр присоединяют к
приспособлению для создания высокоro давл
ния. Ртуть вводят в поры с помощью rидравли
ческой жидкости. Увеличивают давление в сист
ме до максимальноro значения, достиrнутоro при
измерении при низком давлении, и записывают
показания проникнувшеro в поры объема, так как
последующие значения объема будут отсчиты
ваться от этою значения. Давление увеличивают
либо ступенчато в соответствии с интересующи
ми размерами пор, либо плавно с малой CKOpO
стью. Падение ртутноro столба измеряют вплоть
до максимальноro используемоro давления. При
необходимости для определения кривой BЫTec
нения ртути давление понижают либо ступенча
то, либо плавно с малой скоростью.
Коррекцию про водят для тоro, чтобы учесть
изменения объема ртути, пенетрометра и иных
компонентов системы определения объема
при данном давлении. Степень коррекции
может быть установлена с помощью проведе
ния холостоro испытания при тех же условиях.
Экспериментально полученная кривая объем
давление приведена на рисунке 2.9.32. 2.
300
240
,JJ
\о
О
':5:
180
u
ф
:5:
120
=r
ф
t:
, 60
:;;
:х:
Q.
ro
[ О ю'
u
10' 10'
Давление (МПа)
10'
Рисунок 2.9.32. 2. Кривая объем давление
в виде ПОЛУЛ02арифмичеСК020 2рафика
2.9.33. Дифракция peHmzeHOBCKOZO излучения на порошке
487
ОЦЕНКА РЕЗУЛЬТАТОВ
Полученные значения давления MOryт быть
переведены в диаметр пор с использованием
уравнения Уошборна либо с помощью друroй
модели.
Поверхностное натяжение ртути (о-) зависит
не только от температуры, но также, в случае
значительно искривленных поверхностей, от pa
диуса искривления. Обычно при комнатной TeM
пературе значения составляют от 0,41 HIM до
0,52 Н/м. Если точное значение неизвестно, при
нимают о- = 0,48 H/M.
Краевой уroл ртути в большинстве случа
ев составляет более 900. Он может быть опре
делен с использованием ПОДХодящеro прибора.
Если точное значение неизвестно, принимают
е = 1300. При отражении результатов указывают
используемые значения краевоro уrла и поверх
ностноro натяжения.
Визуализация данных может быть осущест
влена с помощью различных типов rрафиков.
Часто rрафическое представление диаметра
пор откладывают по оси абсцисс, а объем про
никнувший в поры ртути на единицу массы
по оси ординат, в результате чеro получают pac
пределение пор по диаметру. Для оси абсцисс
желательно использовать лоrарифмическую
шкалу (рисунок 2.9.32. 3). При обсчете резуль
татов пространство между частицами в образце
рассматривается как поры. Если поры отличают
ся по размерам от пустот, последние MOryт быть
отделены путем выбора конкретноro диапазона
давления, отвечающеro определенному диаме
тру пор.
, 300
::Е
:и 240
\о
о
."
180
'"
,.
"
120
::r
'"
>::
()
. 60
з:
с.
'"
::Е
О
u 10"
'"
....
::'
()
'"
:r
10' 10'
Диаметр пор (НМ)
10'
Рисунок 2.9.З2. 3. Распределение пор по ди
аметру в виде ПОЛУЛ02арифмичеСК020 2рафика
распределения суммарной и нормализованной
плотности
Кривые вытеснения ртути не Bcerдa MOryт
использоваться для расчета размера пор (для
rистерезиса, рисунок 2.9.32. 2), так как часть
проникнувшей ртути всеrда остается в порах. Ko
личество оставшейся ртути, однако, может быть
полезным для качественноro описания пор, име
ющих узкие отверстия.
Наиболее общие значения, такие, как общий
объем проникнувшей ртути и средний и средин
ный диаметры пор, рассчитывают исходя из pac
пределения по размеру пор. Кроме этоro должна
быть документирована информация об образце,
способе приroтовления образца, условиях экс
перимента и используемом приборе.
приrодность СИСТЕМЫ
Так как метод ртутной порозиметрии счита
ется сравнительным испытанием, конкретных
требований в данном разделе нет. Однако peKO
мендуется периодически использовать стабиль
ный образец сравнения для отслеживания кали
бровки прибора и еro эффективности.
01/2013:20933
2.9.33. ОПРЕДЕЛЕНИЕ ПАРАМЕТРОВ
КРИСТАЛЛИЧЕСКИХ
И ЧАСТИЧНО
КРИСТАЛЛИЧЕСКИХ
ТВЕРДЫХ ВЕЩЕСТВ ПРИ
ПОМОЩИ ДИФРАКЦИИ
PEHTrEHOBCKOrO
ИЗЛУЧЕНИЯ НА ПОРОШКЕ
Каждая кристаллическая фаза испытуемоrо
образца дает характерную картину дисрракции
peHтreHoBcKoro излучения.
Картину дифракции #(при использовании
дифрактометра дифрактоrрамму, при исполь
зовании рентrеновской пленки peHTreHorpaM
му)# можно получить из случайно ориентиро
ванноro кристаллическоro порошка, состоящеro
из кристаллитов и кристаллических фраrмен
тов оrраниченноro размера. На основе карти
ны дифракции на порошке можно вычислить три
важнейших типа информации: уrловое распо
ложение дифракционных линий (зависит от re
ометрии и размера кристаллической ячейки),
интенсивность дифракционных линий (зависит,
rлавным образом, от типа и структуры атома, а
также ориентации частиц внутри образца), про
филь дифракционных линий (зависит от разре
шения прибора, размера кристаллита, типа и
толщины образца).
Определение уrловоro расположения и ин
тенсивности линий может быть использовано
для качественноro фазовоro анализа (например,
идентификациЬ кристаллической фазы) и коли
чественноro фазовоrо анализа кристаллических
веществ. Так же можно рассчитать содержание
аморфной и кристаллической фракций.
Примечание: существуют и дРУ2ие, не опи
санные в данной 2лаве, способы использования
метода дифракции рент2еновСК020 излуче
ния на порошке (ДРИП или XRPO): определе
ние крисrnаллической crnPYKrnYPb уrnочнение
кристаллической структуры, определение
488
rосударственная фармакопея Республики Беларусь
кристаЛЛО2рафической чистоты кристалли
ческой фазы, характеристика кристалл вра
фической структуры и дРУ2ие.
Метод ДРИП имеет преимущество перед
друrими способами анализа, так как он явля
ется неразрушающим (приrотовление образца
обычно оrраничивается измельчением для полу
чения порошка со случайно ориентированными
составляющими). Исследования методом ДРИП
можно проводить при условиях iп situ на образ
цах, уязвимых к таким условиям как. например,
низкая и высокая температура или влажность.
ПРИНЦИП
Дифракция рентrеновских лучей происхо
дит в результате взаимодействия между peHтre
новским излучением и электронными облаками
атомов. Рассеянные рентrеновские лучи интер
ферируют в зависимости от положения атомов.
Такие интерференции возникают, Korдa разность
хода между двумя дифраrированными peHTre
новскими волнами отличается на целое число,
кратное длинам волн. Это избирательное COCTO
яние описывается с помощью уравнения Брэrrа
(Bragg), так же называемоro законом Брэrrа (ри
сунок 2.9.ЗЗ. 1):
2d"k!sine hkF ПА.
Длина волны peHTreHoBcKoro излучения А
имеет тот же порядок величины, что и paCCTO
яние О...., между последовательными отражаю
щими плоскостями кристаллической решетки
(также называемоro «d расстоянием» ). 8hkl это
уroл между падающим лучом и системой плоско
стей решетки. а sin8 hkl обратно пропорционален
расстоянию между соседними плоскостями кри
сталлической решетки или d расстоянию.
Направление и протяженность плоскостei
по отношению к осям одиночной кристалличе-
ской ячейки определяются при помощи индet(
сов Миллера {hkf}. Эти индексы являются обрат
ными величинами, приведенными к ближайшему
более низкому целому числу пересечений, кото-
рые совершает плоскость с осями единичной
кристаллической ячейки. Размеры единичной
кристаллической ячейки задаются через pac
стояния а, Ь и с, а также уrлами между ними:
а, 13, и у.
Межплоскостное расстояние для заданноro
набора параллельных hk/ плоскостей обознача
ется, как d hk ,. Каждая такая система плоскостей
может проявлять более высокие порядки диф-
ракции, rдe значения величины d, относящие--
с;я к связанным системам плоскостей пh, пk, пl.
уменьшаются на множитель 1/п (rдe n целое
число: 2, 3,4 и т. д.).
Каждый набор плоскостей по всему кри
сталлу имеет соответствующий уrол дифракции
Брэrrа 8hkl (для определенной длины волны А).
Предполаrается, что образец порошка явля
ется поликристаллическим, поэтому под любым
уrлом 8hkl всеrда есть кристаллиты, ориентация
которых вызывает дифракцию соrласно закону
Брэrrа.
Примечание: (<идеальный» порошок для
изучения состоит из БОЛЬШ020 количества
маленьких, случайно ориентированных сфе--
рических кристаллитов (К02ерентно дифра2и
рующие зоны кристалла). Если их количество
достаточно велико, то будет достаточно и
кристаллитов любой дифра2ирующей opиeH
тации для получения воспроизводимой Kapти
ны дифракции.
Для заданной длины волны peHTreHoBcKO-
ro излучения положения дифракционных пиков
dh
Рисунок 2.9.3З. 1. Дифракция рентееновских лучей в кристалле С02ласно закону БрЭ22а
2.9.33. Дифракция рентаеновскоао излучения на порошке
489
(также называемых «линии», «отражения» или
«Брэrroвские отражения») характеризуют кри
сталлическую решетку (d расстояния). Teope
тические интенсивности этих пиков зависят
от атомов единичной кристаллоrрафической
ячейки (от их природы и расположения). Про
фили дифракционных пиков зависят от COBep
шенства и величины кристаллической решетки.
При этих условиях дифракционный пик имеет
конечную интенсивность, которая обусловлена
атомным расположением, типом атомов, TeM
пературным колебательным движением, CTPYK
турными дефектами, а также характеристика
ми прибора.
Интенсивность зависит от мноrих факторов,
таких как структурный фактор, температурный
фактор, кристалличность, поляризационный
фактор, кратность и фактор Лоренца (Lо,епtz).
Основными характеристиками профилей
дифракционных линий являются: 28 положение,
высота пика, площадь пика и форма пика (xapaK
теризуемая, например, шириной пика и асимме
трией, аналитической функцией, эмпирическим
отображением). Примеры типовых картин диф
ракции на порошке для 5 различных твердых
фаз испытуемоro образца приведены на рисун
ке 2.9.33. 2.
В дополнение к дифракционным пикам,
при дифракции peHTreHoBcKoro излучения об
разуется более или менее однородное фо
новое излучение, на которое накладываются
пики. Кроме приrотовления образца, в фо
новое излучение вносит вклад, например,
устройство для удерживания пробы, диффуз
ное рассеяние от воздуха и оборудования,
шум детектора, общая радиация от peHтre
новской трубки и друrие. Отношение пика к
фоновому излучению можно увеличить путем
минимизации фоновоro излучения или ис
пользованием длительных времен экспози
ции.
ОБОРУДОВАНИЕ
Установка оборудования. Опыты с peHT
rеновской дифракцией обычно проводятся с ис
пользованием порошковых дифрактометров или
порошковых камер.
Порошковый дифрактометр обычно состоит
из пяти основных частей:
источник peHTreHoBcKoro излучения;
оптика, образующая падающий луч (обе.-
спечивает монохроматизацию, фильтрацию.
коллимацию и/или фокусировку луча);
roниометр;
Форма С
Форма В
Форма А
Аморфная фаза
.. & ...
...п... ... "" .."."...............
Io.
I J I! I I I J J II I t J I J I I I II ! ' I I I II I 1 I! II I J II III I 1 I I I J ! II I I
5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31
20(),Cu)
Рисунок 2.9.33. 2. Дифракт02раммы порошка, полученные для 5 различных твердых фаз
испытуеМ020 образца (интенсивности нормализованы)
63.3а.. 1712.
490
rосударственная фармакопея Республики Беларуа.
дифракционная оптика (обеспечивает моно--
хроматизацию, фильтрацию, коллимацию и фоку
сировку или обеспечивает параллельность Лучtй);
детектор.
Также требуются блоки сбора и обработ
ки данных, которые, как правило, входят в KOM
плект современноro оборудования для измере
ния дифракции.
В зависимости от проводимоro типа aHa
лиза (фазовая идентификация, количествен
ный анализ, определение пара метров решетки
и др.) используются ДРИП приборы различной
комплектации. Самыми простыми приборами
для определения картины дифракции на по
рошке являются порошковые камеры. Замена
фотоrрафической пленки как способа детекти
рования на фотонный детектор привела к раз
работке дифрактометров, в которых rеометри
ческая расстановка оптики является скорее не
фокусирующей, а парафокусирующей, как в re
ометрии Брэrrа Брентано (В,аgg Вrепtапо). Так
как в настоящее время наиболее широко pac
пространена пара фокусирующая комплектация
Брэrrа Брентано. ей уделено больше внимания
в данной статье.
Прибор в комплектации Брэrrа Брентано
может обеспечивать roризонтальную или Bep
тикальную 8/28 rеометрию или вертикальную
8/8 rеометрию. Для обеих rеометрий падающий
рентrеновский луч образует уroл 8 с плоскостью
образца. а дифраrированный (отраженный) луч
образует уrол 28 с направлением падающеro
рентrеновскоro луча (уroл 8 с плоскостью образ
ца). Основная rеометрическа модель представ
лена на рисунке 2.9.33. 3. Расходящийся луч из
рентrеновской трубки (так называемый «исход
ный луч») проходит через коллиматоры, через
рассеивающую щель и облучает плоскую по
верхность образца. Все лучи дифраrируют от
ориентированных соответствующим образом
в образце кристаллитов под уrлом 28 и соби
раются в линию у принимающей щели. Второй
набор из коллиматоров и рассеивающей щели
может быть помещен до или после принимаю
щей щели. Оси линейноro фокуса и принимаю
щей щели находятся на равном расстоянии от
оси roниометра. Кванты peHTreHoBcKoro излу
чения подсчитываются при помощи детектора
радиоактивности (сцинтилляционный счетчик,
пропорциональный счетчик. пространственно
чувствительный или полупроводниковый дeTeK
тор). Принимающая щель и детектор образуют
единый блок и движутся по касательной к фоку
сирующей окружности. Для 8/28 сканирования
roниометр вращает образец BOKpyr той же оси,
что ось детектора, но с половинной ск()ростью
вращения, в 8/28 движении. Таким образом, по
верхность образца находится по касательной к
фокусирующей окружности. Коллиматор orpa
ничивает осевое расхождение луча и таким об
разом частично контролирует форму профиля
дифраrированной линии.
/
В
\
/
!
\
\
1<
)
/
F
Рисунок 2.9.33. 3. Пространственное
представление парафокусирующей 2еометрии
БрЭ22а Брентано
А рентrеНОВСl<ая трубка;
В рассеивающая щель;
С образец;
D антирассеивающая щель;
Е при ни мающая щель;
F монохроматор;
G принимающая щель детектора;
Н детектор;
J Kpyr дифрактометра;
К фокусирующий Kpyr.
Дифрактометр Брэrrа Брентано также
можно использовать в режиме работы в прохо
дящем свете. Преимущество такоro способа в
том, что он уменьшает эффекты, связанные с
преимущественной ориентацией. Для малень
ких количеств образца можно использовать Ka
пилляры толщиной около 0,5 2 мм.
PeHTreHOBCKoe излучение. В лаборато
рии peHTreHoBcKoe излучение получают, бом
бардируя металлический анод электронами,
испускаемыми катодом вследствие TepMO
электронноro эффекта и ускоряемыми силь
ным электрическим полем (при использовании
reHepaTopa высокоro напряжения). Большая
часть кинетической энерrии электронов пе
реходит в тепло, что оrраничивает мощность
рентrеновских трубок и требует значитель
Horo охлаждения анода. При использовании
вращающихся анодов и рентrеновской оптики
можно увеличить интенсивность излучения в
20 30 раз. В качестве альтернативы, можно
получать peHTreHoBcKoe излучение большой
интенсивности (синхротрон).
Спектр, излучаемый рентrеновской труб
кой, работающей под высоким напряжением.
состоит из постоянноrо фона полихроматиче
скоro излучения и дополнительноro xapaKTe
ристическоrо излучения, которое зависит от
типа анода. В опытах с рентrеновской диф
ракцией используется только характеристиче
2.9.33. Дифракция рентаеновскоао излучения на порошке
491
ское излучение. Основными источниками из
лучения, используемыми для рентrеновской
дифракции, являются вакуумные трубки, co
держащие в качестве анода медь, молибден,
железо, кобальт и хром. PeHTreHoBcKoe из
лучение, полученное на основе медных, MO
либденовых и кобальтовых анодов, исполь
зуется в основном для орrанических веществ
(использование кобальтовых анодов особен
но предпочтительно для четкоrо разделения
рентrеновских линий). Выбор используемоrо
излучения зависит от абсорбционных свойств
образца и от возможной флуоресценции, об
условленной присутствующими в образце
атомами. Длины волн, используемые в диф
раКЦИ1ll на порошке, обычно соответствуют
Ка излучению, испускаемому анодом. PeHT
rеновский луч полезно сделать «MOHoxpOMa
тичным» путем удаления всех друrих компо
нентов испускаемоrо спектра. Частично это
достиrается при использовании K фильтров,
то есть металлических фильтров, подобран
ных таким образом, чтобы они имели rраницы
поrлощения между Ка и длинами волн, испу
скаемых рентrеновской трубкой.
Такие фильтры обычно вставляют между
рентrеновской трубкой и образцом. Друrой, наи
более часто используемый способ получения
монохроматическоro peHTreHoBcKoro луча ис
пользование большоro монохроматическоro кри
сталла (обычно называемый «монохроматор» ).
Этот кристалл помещается перед или после об
разца и преломляет различные характеристиче
ские полосы peHTreHoBcKoro луча ( то есть к К )
uи р
под различными уrлами, для тоro чтобы только
один из них Mor быть выбран для попадания на
детектор. При использовании специализирован
ных монохроматоров возможно разделение К а1 и
Кш излучений. К сожалению, выrода от получе
ния монохроматическоrо луча при использова
нии фильтра или монохроматора нивелируется
потерей интенсивности. Друrим способом разде
ления К К" длин волн является использование
n И ,... '"
изоrнутых рентrеновских зеркал, которые OДHO
временно монохромируют и фокусируют или
делают параллельными рентrеновские лучи.
ЗАЩИТА ОТ ИЗЛУЧЕНИя. Воздействие на
любую часть тела рент2еновСК020 излучения
может повредить здоровью. Поэтому важно
во время работы рент2еновСК020 оборудова
ния принять соответствующие меры предо
сторожности для защиты оператора и всех,
кто находится поблизости. Рекомендованный
установленный порядок защиты, также как и
пределы уровней воздействия peHт2eHoвCKO
20 излучения, устанавливаютGЯ национальным
законодательством в каждой стране. Если в
данной стране нет никаких официальных HOp
мативов и рекомендаций, то следует исполь
зовать рекомендации Международной KOMиc
сии по РадиОЛ02ической Защите в последней
редакции.
подrотовКА и РАЗМЕЩЕНИЕ ОБРАЗЦА
Подroтовка материала в виде порошка и раз
мещение образца в подходящем держателе яв
ляются важными этапами мноrих аналитических
методов и особенно в ходе анализа peHTreHoB
ской дифракции на порошке, так как они Moryт в
большой степени повлиять на качество получен
ных данных. Также в ходе сбора данных MOryT
происходить изменения в образце, вследствие
еro неравновесноro состояния (температура,
влажность). Далее будут приведены основные
источники ошибок, обусловленных приroтовле
нием и размещением образца, применительно
к приборам с парафокусирующей rеометрией
Брэrrа Брентана.
подrОТОВКА ОБРАЗЦА
Морфолоrия большинства кристаллических
частиц образца проявляет некоторую степень
предпочтительной ориентации в устройстве
прободержателя. Это особенно характерно для
иroльчатых или пластинчатых кристаллов, Korдa
измельчение такоro образца приводит к образо
ванию более мелких иrл и пластинок. Преиму
щественная ориентация в образце влияет на ин
тенсивность еro отражений (некоторые более
интенсивные, друrие менее), в отличие от co
вершенно неориентированною образца. Приме
няют несколько методов для улучшения случай
ной составляющей в ориентации кристаллитов
(и, следовательно, для минимизации преиму
щественной ориентации), но зачастую самым
лучшим и простым способом является дальней
шее уменьшение размера частиц. Оптимальное
количество кристаллитов зависит от rеометрии
дифрактометра, от требуемоrо разрешения и от
ослабления (аттенюации) рентrеновскоro луча в
образце. В некоторых случаях размер частиц по
рядка 50 мкм обеспечивает удовлетворительные
результаты фазовою анализа. Однако, чрезмер
ное измельчение (размер кристаллитов менее
0,5 мкм) может привести к уширению полос
(спектра) и вызвать значительные изменения в
самом образце, такие как:
заrрязнение образца частицами из обо
рудования для измельчения (ступка, пестик.
шарики и др.);
уменьшение степени кристалличности;
твердофазный переход к друrой поли
морфной модификации;
химическое разложение;
образование внутреннеro напряжения;
твердофазные реакции.
Поэтому целесообразно сравнить дифрак
ционную картину необработачноro образца с
дифракционной картиной, соответствующей об
разцу с более мелким размером частиц (напри
мер, измельченный образец). Если полученная
рентrеновская дифракционная картина, учиты
вая ее планируемое использование, имеет COOT
ветствующее качество, то измельчение не Tpe
буется.
492
rосударственная фармакопея Республики Беларусь
Следует отметить, что если проба coдep
жит более одной фазы и если для раздеJ 1 ения
частиц с заданным размером применяется IlpO
сеивание, исходный состав может изменяться.
РА3МЕЩЕНИЕ ОБРА3ЦА
Влияние смещения образца. Поверх
ность образца, которая смещена на pacco
яние О по отношению к оси вращения диф
рактометра, вызывает трудноисключаемые
систематические ошибки. происходящие
из за абсолютных О. cose сдвиroв в 28 поло
жен иях (обычно порядка 0,01 о в 28 при малых
уrлах (cos8 :::: 1) при смещении О = 15 мкм) и
из за асимметричноrо уширения профиля по
отношению к малым величинам 28. Исполь
зование подходящеrо внутреннеro стандарта
позволяет вести детектирование с коррекци
ей этоro эффекта одновременно с эффектом,
возникающим из за прозрачности образца.
Эффект смещения является самым большим
источником ошибок при получении данных на
хорошо настроенных дифрактометрах.
Влияние толщины и прозрачности об-
разца. Коrда метод ДРИП используется в
режиме отражения, то зачастую предпочти
тельнее работать с образцами «бесконечной
толщины». Для минимизации эффекта про
зрачности желательно использовать недиф
раrирующую подложку (нулевой фоновый
держатель), например, пластину из монокри
сталла кремния диоксида, расположенную
параллельно плоскости решетки 510. Пре
имущество режима передачи в том, что про
блемы с высотой пробы и прозрачностью об
разца менее значимы.
Использование подходящеro внутреннеro
стандарта позволяет вести детектирование с
коррекцией этоro эффекта одновременно с эф
фектом, возникающим из за смещения образца.
КОНТРОЛЬ приrодности ПРИБОРА
rониометр и соответствующая оптика для
падающеro и дифраrированоrо рентrеновских
лучей состоят из механических деталей, требу
ющих настройки. Степень настройки напрямую
влияет на качество результатов исследования
методом ДРИП. Поэтому все узлы дифрактоме
тра должны быть точно отреryлированы (оптиче
ская и механическая системы и друrие) для ми
нимизации в достаточной мере систематических
ошибок, оптимизируя при этом интенсивности
лучей, попадающих на детектор. При настрой
ке дифрактометра следует учитывать обратную
зависимость разрешения от интенсивности. По
этому при настройке прибора должно быть най
дено оптимальное компромиссное решение.
Существует большое количество различных KOH
фиryраций, и каждый прибор, в зависимости от
производителя, требует специфической проце
дуры настройки.
Периодически должна проводиться провер-
ка настройки Bcero прибора с использован
подходящих аттестованных стандартных обраэ-
цов. В зависимости от типа анализа MOryT быть
использованы и иные стандартные образцы.
хотя использование сертифицированных стан-
дартных образцов является более предпочти-
тельным.
КАЧЕСТВЕННЫЙ ФАЗОВЫЙ АНАЛИЗ
(ИДЕНТИФИКАЦИЯ ФАЗ)
Идентификация фазовоro состава неиз
вестноro образца методом ДРИП обычно oc
нована на визуальном или компьютерном
сравнении части еro дифракционной карти-
ны с экспериментальной или рассчитанной
к ртиной дифракции вещества сравнения.
Для получения наилучших результатов диф
ракционные картины сравнения получают на
хорошо изученных однофазных образцах.
Этот подход в большинстве случаев делает
возможным идентификацию кристаллическо-
ro вещества по ero 28 уrлам дифракции или
d-расстояниям, а также по ero относитель-
ным интенсивностям. Компьютеризированное
сличение дифракционной картины неизвест-
ноro образца со сравнительными данными
может основываться либо на расширенном
28 диапазоне полной дифракционной карти-
ны, либо на уменьшенном наборе данных,
взятых из дифракционной картины. Напри
мер, перечень d расстояний и нормализо-
ванных интенсивностей 'norm' так называемый
(d, /поrm) СПИСОК, взятый из дифракционной
картины, является кристаллоrрафическим OT
печатком материала, который можно cpaB
нить с (d, /пorm)-списками однофазных образ
цов, собранных в базе данных.
Для большинства орrанических кристал
лов при использовании Cu Ка излучения удобно
реrистрировать дифракционную картину в
28-диапазоне от значения, насколько возможно
близкоro к 00, до как минимум 400. Сходимость
28 уrлов дифракции между образцом и веще-
ством сравнения для одной и той же кристал
лической формы будет лежать в пределах 0,20,
Torдa как их относительные интенсивности Moryт
значительно отличаться из-за эффектов преи-
мущественной ориентации. В случае различных
rидратов и сольватов из-за их непостоянной при-
роды общепризнана изменчивость размеров их
элементарных ячеек, что приводит к сдвиrам на
дифрактоrраммах этих веществ. В таких случа
ях отличия для 28 уrлов более чем на 0,20 не
являются неожиданными. Для друrих типов об
разцов (таких как неорrанические соли) может
быть необходимым расширение 28 диапазона
сканирования значительно выше 400. Обычно
достаточно просканировать последние 1 О самых
сильных отражений (линий), идентифицирован
ных в файлах однофазной дифракции peHTr
новских лучей из базы данных.
2.9.33. Дифракция рентtЗеновскоtЗо излучения на порошке
493
Трудно или даже невозможно идентифици
ровать фазы в следующих случаях:
при определении некристаллических или
аморфных субстанций;
если компоненты, которые нужно иден
тифицировать, присутствуют в малых Macco
вых долях от анализируемых количеств (обычно
менее чем 1 О % м/м);
при резко выраженных эффектах преиму
щественной ориентации;
при отсутствии фазы в используемой базе
данных;
при образовании твердых растворов;
при наличии беспорядочных структур, KO
торые вносят изменение в элементарную ячейку;
если образец включает в себя слишком
мноro фаз;
при наличии деформации кристалличе
ской решетки;
при структурном подобии различных фаз.
КОЛИЧЕСТВЕННЫЙ ФАЗОВЫЙ АНАЛИЗ
Если исследуемый образец является смесью
двух или более известных фаз, а аморфная
только одна, то во мноrих случаях можно опре
делить их процентное содержание (по объему
или по массе). Количественный фазовый анализ
может основываться на интеrрированных ин
тенсивностях, на высоте пиков нескольких от
дельных дифракционных линий или на полной
картине дифракции. Эти интеrрированные ин
тенсивности, высоты пиков или результаты об
работки данных полной дифракционной картины
сравниваются с соответствующими значениями,
полученными для веществ сравнения. Эти Be
щества сравнения должны быть однофазными
или смесью известных фаз. Трудности, встреча
ющиеся в ходе количественноro анализа, свя
заны с приroтовлением образца (для ДOCТOBep
ности и точности результатов требуется особая
roMoreHHocTb всех фаз и подходящее распреде
ление частиц по размерам в каждой фазе) и с
эффектами матрицы. При блаroприятных усло
виях минимальное обнаруживаемое количеСтво
кристаллической фазы в твердой матрице может
составлять 1 О %.
ПОЛИМОРФНbJЕ ОБРАЗЦbJ
Для образца, состоящеro из двух полиморф
ных фаз а и Ь, можно вывести следующее соот
ношение для фракции Fa фазы а:
F == 1
а 1 + K(/b/IJ
Фракцию определяют путем измерения OT
ношения интенсивностей между двумя фазами
при известном значении константы К. KOHCTaH
та К представляет собой отношение абсолют
ных интенсивностей двух чистых полиморфных
фаз 108/1 оЬ' Эти значения можно определить при
помощи измерения стандартных образцов.
64. За.. 1712
МЕТОДЫ С ИСПОЛЬЗОВАНИЕМ
СТАНДАРТА
Наиболее часто используемые методы KO
личественноro анализа:
«метод внешнеro стандарта»;
«метод внутреннеro стандарта»;
«метод добавок» (часто также называется
«метод стандартных добавок»).
«Метод внешнеro стандарта» является наи
более общепринятым методом и состоит в cpaB
нении дифракционной картины peHTreHoBcKoro
излучения смеси или в сравнении COOTBeTCTBY
ющих линий интенсивности с этими же параме
трами, измеренными в сравнительной смеси,
или же в сравнении с теоретическими интенсив
ностями структурной модели, если она полно
стью известна.
Для оrраничения ошибок, возникающих
из за эффектов матрицы, можно использо
вать внутреннее вещество сравнения с разме
ром кристаллитов и коэффициентом peHTreHoB
ской абсорбции, сопоставимыми с таковыми
для компонентов образца, а также с картиной
дифракции, которая не перекрывается с карти
ной дифракции анализируемоro образца. Из
вестное количество вещества сравнения при
бавляют к анализируемому образцу и к каждой
смеси сравнения. При этом зависимость между
интенсивностью линий и концентрацией являет
ся линейной. Этот метод, называемый «метод
внутреннеro стандарта», требует точноro изме
рения интенсивностей дифракции.
В «методе добавою) (или «методе CTaHдapT
ных добавок» ) к смеси с неизвестной KOHцeHTpa
цией фазы а прибавляют известное количество
чистой фазы а. Измеряют интенсивности образ
цов, содержащих пропорционально увеличива
ющиеся концентрации добавок, и строят rрафик
зависимости интенсивности от концентрации,
на котором отрицательный отрезок х на оси аб
сцисс, отсекаемый построенной прямой, пред
ставляет собой концентрацию фазы а в исход
ном образце.
ОЦЕНКА АМОРФНОЙ
И КРИСТАЛЛИЧЕСКОЙ ФРАКЦИЙ
В ОБРАЗЦЕ
Кристаллическую и аморфную фракции в
смеси аморфной и кристаллической фаз можно
оценить несколькими способами. Выбор исполь
зуемоrо метода зависит от природы образца:
если образец состоит из кристаллических
фракций и одной аморфной фракции различ
ноro химическоro состава, количества каждых
отдельных кристаллических фаз можно pac
считать, используя подходящие стандартные Be
щества, как описано выше; аморфная фракция
определяется косвенно путем вычитания;
если образец состоит из одной аморф
ной и одной кристаллической фракций или пред
ставляет собой однофазную или двухфазную
смесь с одинаковым элементным составом, KO
494
rосударственная фармакопея Республики БеЛ817'1'2-
личество кристаллической фазы (<<степе....ь кри
сталличности» ) рассчитывают путем измеречия
трех площадей дифрактоrраммы:
А общая площадь пиков, возникающих в
результате дифракции кристаллической фрак
ции образца;
В общая площадь за исключением пло
щади А;
С площадь фоновоrо излучения (обуслов
лено рассеиванием в воздухе, флуоресценцией,
оборудованием и так далее).
Степень кристалличности рассчитывают по
формуле:
%кристаличностu =100A/(A+B C).
Этот метод не дает абсолютных величин
степени кристалличности и поэтому обычно ис
пользуется только для сравнительных целей.
Существуют так же усовершенствованные
методы, например, метод Рулэнда (Rи/апd).
СТРУКТУРА МОНОКРИСТАЛЛА
Обычно определение кристаллических
структур осуществляют по данным дифракции
peHTreHoвacoro излучения, полученным при ис
следовании монокристаллов. Однако анализ
кристаллической структуры орrанических кри
сталлов является сложной задачей, так как па
раметры решетки сравнительно велики, симме
трия недостаточна, а рассеивающие свойства
обычно очень слабы.
Для любой данной кристаллической формы
вещества знание кристаллической структуры
позволяет провести расчет соответствующей
картины дифракции, получая сравнительную
картину дифракции «без предпочтительной ори
ентации», которую можно использовать для ce
рийных сравнений.
01/2013:20934
2.9.34. НАСЫПНАЯ ПЛОТНОСТЬ
И ПЛОТНОСТЬ ПОСЛЕ УСАДКИ
Насыпная плотность
Насыпная плотность порошка это OTHO
шение массы неуплотненноrо образца порош
ка к ero объему, включая вклад свободноro
пространства между частицами. Следователь
но, насыпная плотность зависит и от плотно
сти частиц порошка, и от пространственно
ro размещения частиц в слое порошка. Так
как измерения проводят с использованием
цилиндра, насыпная плотность выражает
ся в rpaMMax на миллилитр, хотя единицей
СИ является килоrрамм на метр кубический
(1 r/мл = 1000 кr/м З ). Кроме тоro, она мо*...
быть выражена в rpaMMax на сантиметр куб.
ческий.
Насыпные свойства rюрошка зависят of
способа приroтовления, методов обработlCII
и условий хранения образца. Частицы MOryl'
скапливаться различными способами, приво-
дя, таким образом, t< целому ряду значений
насыпной плотности и, кроме тоro, мале
шее нарушение слоя порошка может приве-
сти к изменению насыпной плотности. По этой
причине определение насыпной плотности с
хорошей воспроизводимостью зачастую за
труднено, а итоrовые результаты должны со-
провождаться описанием TOro, каким образом
проводилось испытание.
> Насыпная плотность порошка определ
ется либо путем измерения объема предвари
тельно просеянноro через сито образца порош
ка известной массы в rрадуированном цилиндре
(Метод 1), либо измерением массы известноrо
объема порошка, прошедшеro через волюметр в
чашку (Метод 2) или который был помещен в из
мерительный сосуд (Метод 3).
Предпочтительнее использовать Методы
1 и3.
МЕТОД 1: ИЗМЕРЕНИЕ
В rРАДУИРОВАННОМ ЦИЛИНДРЕ
Методика. При необходимости для раз
рушения аrломератов, которые моrли обра
зоваться при хранении, количество порош
ка, используемое для проведения испытания,
пропускают через сито с отверстиями paBHЫ
ми или более 1,0 мм; эту процедуру проводят
аккуратно, избеrая изменения свойств веще
ства. В сухой rрадуированный цилиндр BMe
стимостью 250 мл (с делениями 2 мл) aKKY
ратно помещают, без уплотнения, около 100 r
(т) испытуемоro образца, взвешенноro с точ
ностью 0,1 %. При необходимости осторожно
без уплотнения разравнивают порошок и из
меряют наблюдаемый объем (V o )' Рассчиты
вают насыпную плотность в rpaMMax на мил
лилитр с использованием формулы т 1 V o .
При определении данноro свойства желатель
но провести повторные измерения.
Если плотность порошка слишком велика
или слишком мала, так что испытуемый образец
занимает объем более 250 мл или менее 150 мл,
использование образца массой 100 r невозмож
но. В таком случае используют образец иной
массы с таким расчетом, чтобы занимаемый им
объем составлял от 150 мл до 250 мл (наблюда
емый объем равен или более 60 % общеro
объема цилиндра); масса испытуемоro образца
отмечается при указании результатов испытания.
Для испытуемых образцов, имеющих наблю
даемый объем от 50 мл ДО 100 мл, может быть
использован цилиндр вместимостью 100 мл
С делениями 1 мл; объем цилиндра отмечают
при указании результатов испытания.
2.9.34. Насыпная плотность и плотность после усадки
495
МЕТОД 2: ИЗМЕРЕНИЕ ВВОЛЮМЕТРЕ
Прибор. Прибор (рисунок 2.9.34. 1) cocтo
ит из верхней воронки, снабженной ситом с раз
мером отверстий 1,0 мм, которая установлена
над экранной коробкой, имеющей 4 стеклянных
экрана, по которым скользит и отскакивает поро
шок при прохождении. Внизу экранной коробки
находится воронка, которая собирает порошок
и направляет еro в чашку, расположенную He
посредственно под ней. Чашка может быть ци
линдрической (вместимостью (25,00:!:0,05) мл и
внутренним диаметром (30,00:!:2,00) мм) или KY
бической (вместимостью (16,39:!:0,20) мл и BHY
тренними размерами (25,4:!:0,076) мм).
Методика. Избыток порошка пропускают
через прибор в приемную чашку до тех пор, пока
она не переполнится, используя не менее 25 СМ З
порошка для квадратной чашки и 35 см З для ци
линдрической чашки. Аккуратно снимают избы
ток порошка с верха чашки с помощью плавных
движений остроro края шпателя, располаrая еro
перпендикулярно и удерживая еro постоянно в
контакте с верхом чашки, обращая внимание
на то, чтобы шпатель был постоянно перпен
дикулярен верху чашки во избежание уплотне
ния порошка или удаления избыточноro количе
ства порошка из чашки. Удаляют все вещества с
внешних стенок чашки и определяют массу (М)
А
в
с
D
G
Е
F
Рисунок 2.9.34. 1. Волюметр
А сито с размером отверстий 1,0 мм;
В верхняя воронка;
С наrрузочная воронка;
О экранная коробка;
Е стеклянный экран;
F чашка;
G подставка.
порошка с точностью 0,1 %. Рассчитывают Ha
сыпную плотность в rpaMMax на миллилитр с ис
пользованием формулы М/ V o (rдe V o объем
чашки) и записывают среднее из трех измере.-
ний, проведенных с использованием каждый раз
новоro образца порошка.
МЕТОД 3: ИЗМЕРЕНИЕ В СОСУДЕ
Прибор. Прибор состоит из цилиндрическcr
ro сосуда вместимостью 100 мл, изroтовленноro
из нержавеющей стали, с размерами, указанны--
ми на рисунке 2.9.34. 2.
54.0 57.0
0 50'5 i [ 50'5 . i
52.0 . 50.0
· 1 10 . 0 . '
2.0 t 54.0
t
Рисунок 2.9.34. 2. Измерительный сосуд
(слева) и колпачок (справа)
(размеры указаны в миллиметрах)
Методика. При необходимости для
рушения аrломератов, которые моrли обpcr
зоваться при хранении, количество порошка,
используемое при проведении испытания, пpcr
пускают через сито с размером отверстий 1,0 мм,
и поток полученноro образца направляют в
мерительный сосуд, пока он не переполнится.
Осторожно удаляют избыток порошка с верха
сосуда, как описано в Методе 2. Определяют
массу (МО) порошка с точностью 0,1 %, вычитая
предварительно определенную массу пустоro
мерительноro сосуда. Рассчитывают насыпную
плотность в rpaMMax на миллилитр с испольэо-
ванием формулы Mi100 и записывают среднее
из трех измерений, проведенных с испол
нием каждый раз новоro образца порошка.
Плотность после усадки
Плотность после усадки это насыпная
плотность, полученная после механическoro
встряхивания резервуара, содержащеro обpcr
зец порошка.
Плотность после усадки получают с пoмc:r
щью механическоro встряхивания rрадуирова
ноro измерительноro цилиндра или сосуда, со-
держащеro образец порошка. После измерения
начальноro объема или массы порошка изме
тельный цилиндр или сосуд механически встря.-
хивают до получения постоянной массы или
объема. Механическое встряхивание создают,
поднимая цилиндр или сосуд на определенную
высоту и отпуская ero, позволяя упасть под дей.-
ствием ero собственноro веса, как это указано в
трех методах, описанных ниже. MOryT быть ис
пользованы различные приспособления, Bpcr
щающие цилиндр или сосуд при встряхивании
в целях минимизирования возможноro разд
ния массы при падении.
496
rосударственная фармакопея Республики Беларусь
МЕТОД 1
Прибор. Прибор (рисунок 2.9.34. 3) СQ( ТОИТ
из следующих частей:
rрадуированный цилиндр вместимостью
250 мл (с делениями 2 мл) массой (220I44) r;
приспособление, способное обеспечи
вать либо (250I15) падений в минуту с высоты
(3IO,2) мм, либо (300:1:15) падений в минуту с
высоты (14I2) мм. Подставка для rрадуиро
ванноro цилиндра вместе с держателем имеют
массу (450:1:10) r.
Методика. Проводят испытание, как YKa
зано при определении насыпноro объема (V o )'
Закрепляют цилиндр в подставке. Проводят
10, 500 и 1250 встряхиваний с использованием
одноro и Toro же образца порошка и отмечают
соответствующие объемы V,o' V 500 и V'250' Если
разница между V 500 и V'250 составляет менее 2
мл, значение V'250 является плотностью после
усадки. Если разница между V 500 и V'250 превы
шает 2 мл, испытание повторяют с инкрементом,
например, 1250 встряхиваний, до тех пор, пока
разница не станет менее 2 мл. Для некоторых
порошков может быть валидировано использо
вание меньшеro количества встряхиваний. Pac
считывают плотность после усадки с использо
ванием формулы mlV, (rдe V, конечный объем
уплотненноro образца). При определении дaH
Horo свойства желательно провести повторные
измерения. Вместе с результатами указывают
высоту падения.
rрад)'и
UИ 11д&
Ш 1
I . :>
" :
-. CIJ
; ::: r:. А
fIl '.:. : !
I ('\,1 ь!
'::=:: ! I
. =.. I со I
>. 0:./
1 ::: I CIJ
..... Q з: 10
-=- IL О
, i" ...
'1
Держатель
ЦИЛИt-lДра
--
. :>
:>
. N
ici
....
1:>
':>
'"'
Наковальня
. Данный рамер Должен
обеспечить высоту падения 1 .
удовлетворяюwую
. требованиям спеuификаЦИИ.
! и при ICOтором В На.1жней точке
Эl<сuеНТРИka держатель
U И Лl-1tiДра будет свобоДНО
стоять на верхней ....асТи
нзt<08апьни
Рисунок 2.9.34. 3. Встряхивающий прибор
для образцов порошков
(размеры указаны в миллиметрах)
МЕТОД 2
Методика. Проводят испытание, как указа
но в Методе 1, за исключением тоro, что прибор
обеспечивает фиксированную высоту падения
(3IO,2) мм с номинальной скоростью 250 раз в
минуту.
МЕТОД 3
Методика. Проводят испытание, как YK
за но в Методе 3 для определения насыпном
плотности, используя измерительный сосуд.
снабженный колпачком (см. рисунок2.9.34. 2)_
Измерительный сосуд вместе с колпачко"
поднимают 50 60 раз в минуту с использова
нием ПОДХОДящеro прибора для определения
плотности после усадки. Проводят 200 встря
хиваний, удаляют колпачок и аккуратно удаля
ют избыток порошка с верха измерительноro
сосуда, как описано в Методе 3 для опреде
ления насыпной плотности. Повторяют ИСПЫ
тание с 400 встряхиваниями. Если разница
между измерениями при 200 и 400 встряхива
ниях превышает 2 %, испытание повторяют,
дополнительно встряхивая до тех пор, пока
разница не станет менее 2 %. Рассчитывают
плотность в rpaMMax на миллилитр с исполь
зованием формулы М/100 (rдe M, масса по
рошка в измерительном сосуде) и записыва
ют среднее из трех измерений, проведенных
с использованием каждый раз HOBoro образ
ца порошка. Вместе с результатами указыва
ют высоту при встряхивании.
Меры сжимаемости порошка
По причине TOro, что взаимодействия
между частицами, влияющие на насыпные
свойства Порошка, также влияют на TeKY
честь Порошка, сравнение насыпной плотно
сти и плотности после усадки может позво
лить оценить относительную важность этих
взаимодействий для данноro Порошка. Такие
сравнения MorYT указывать на способность
порошков течь, например, коэффициент сжи
маемости или отношение Хауснера (Haus
пе,).
Коэффициент сжимаемости и отношение
Хауснера являются мерами склонности по
рошка к сжатию описанными выше методами.
По существу, они указывают на способность
порошка оседать и позволяют оценить OTHO
сительную важность взаимодействий между
частицами. В свободно текущем порошке
такие взаимодействия менее существенны
и значения насыпной плотности и плотности
после усадки близки. Для веществ, текущих
хуже, взаимодействия между частицами за
частую сильнее, и наблюдается большая раз
ница между значениями насыпной плотности
и плотности после усадки. Эти отличия OTpa
жаются коэффициентом сжимаемости и OTHO
шением Хауснера.
Коэффициент сжимаемости:
100(V o V, )
V o '
rдe:
V o насыпной объем до усадки;
V, конечный объем после усадки.
2.9.36. Текучесть порошков
497
Отношение Хауснера:
V o
V,
в зависимости от вещества коэффициент
сжимаемости может определяться с использ
ванием V 10 вместо V o ' Если использовалось зна
чение V 1o ' это должно быть указано вместе с по
лученным результатом.
01/2013:20935
2.9.35. СТЕПЕНЬ ИЗМЕЛЬЧЕНИЯ
ПОРОШКОВ
Размер частиц определяют с помощью
аналитическоro просеивания (2.9.38) или
иноro подходящеro метода. В данном раз
деле приведена простая описательная клас
сификация степени измельчения порошков.
Блаroдаря своему удобству, просеивание яв
ляется наиболее часто используемым MeTO
дом определения размера частиц. Просеива
ние является наиболее подходящим в случае
размера частиц более 75 мкм, хотя может
быть использован валидированный метод
просеивания и для порошков С меньшим раз
мером частиц. Кроме TOro, дифракция света
также является широко используемым спосо
бом измерения широкоro диапазона разме
ров частиц.
Если с помощью аналитическоro просе
ивания либо с использованием друrоrо
метода находят суммарное распределение
частиц по размерам, то размер частиц может
быть охарактеризован следующим образом:
Xgo размер частиц, соответствующий
суммарному распределению частиц по< раз
мерам с подразмерным значением 90 %;
X средний размер частиц (то есть
50 % частиц имеют размер менее номиналь
ноro и 50 % более номинальноro);
Х 10 размер частиц, соответствующий
суммарному распределению частиц по раз
мерам с подразмерным значением 10 %.
Для обозначения этих величин часто ис
пользуют символ d, и таким образом MorYT
быть использованы символы d go ' d 50 И d 10 ,
Основываясь на суммарном распределе
нии частиц по размерам, MorYT быть опреде
J1eHbI следующие параметры.
r Тип распределения
О Число
1 Длина
2 Площадь
3 Объем
О,(Х) суммарное распределение частиц
с размером менее или равным х, rдe индекс r
отображает тип распределения.
Таким образом, по определению:
О,(Х) = 0,90, если Х = Xgo;
О,(Х) = 0,50, если Х = Х 50 ;
О,(Х) = 0,10, если Х = Х 10 .
Альтернативным, но менее информатив
ным способом классификации порошков по CTe
пени измельчения является использование опи
сательных терминов, приведенных в таблице
2.9.35. 1.
Таблица 2.9.35. 1
Классификация порошков по степени
измельчения
Описательный Суммарное pac
термин Х 50 (мкм) пределение по
объему, Q,(x)
rрубый порошок >355 0,(355)<0,50
Среднемелкий 180 355 0з(180)<0,50 и
порошок O,(355) 0,50
Мелкий порошок 125 180 0з(125)<0,50 и
0,(180) 0,50
Очень мелкий S125 Оз(125) 0,50
порошок
01/2013:20936
2.9.36. ТЕКУЧЕСТЬ ПОРОШКОВ
Широкое использование порошков в фар
мацевтической промышленности привело к
возникновению множества методов для xapaктe
ристики текучести порошков. Цель этоro раздела
состоит в том, чтобы рассмотреть наиболее часто
встречающиеся в фармацевтической литерату
ре методы оценки текучести порошков, которые
MOryт быть стандартизированы и впоследствии
быть весьма полезными в процессе развития
фармацевтической промышленности. Так как не
существует одноro простоro способа адекватно
описать текучесть порошков, данная статья пред
лаrает стандартизацию методов испытаний, KOT
рые MOryт быть полезны при фармацевтической
разработке.
Для оценки текучести порошка используют 4
основных метода:
уrол eCTecTBeHHoro откоса;
коэффициент сжимаемости или отноше
ние Хауснера (Наиsпе,);
скорость вытекания через насадку;
метод сдвиroвой ячейки.
Кроме тоro, встречаются мноrочисленные
разновидности каждоro из этих основных MeTO
дов. Учитывая большое количество методов и
их разновидностей, необходима стандартизация
методики испытания.
498
rосударственная фармакопея Республики Беларусь
По этой причине наиболее часто исполь
зуемые методы описаны ниже. Для описан
ных ниже методов даются важнейшие условия
проведения испытаний, а также рекомендации
по их стандартизации. Любой метод измере
t-Iия текучести порошков ролжен давать знача
щие результаты, а также быть применимым на
практике, удобным, воспроизводимым и чув
ствительным. Однако ни один из методов опре
деления текучести порошков не может полно
стью охарактеризовать все свойства текучести
порошковой массы. которые являются важны
ми для фармацевтической промышленности.
Подходящей стратеrией может быть использо
вание нескольких стандартизованных методик
испытаний ДЛЯ описания различных аспектов
текучести порошков, необходимых для фарма
цевтической разработки.
уrол ECTECTBEHHOro ОТКОСА
Уrол ecтecтвeHHoro откоса используется в
различных отраслях науки для описания свойств
текучести rютока твердых тел. Уroл eCTeCTBeHHO
ro откоса это характеристика, учитывающая
трение частиц между собой, а также сопротив
ление между частицами при их движении. Pe
зультат определения уrла естественноro откоса
очень сильно зависит от выбранноro метода.
Трудности при испытании возникают из за cerpe
rации (расслаивания) материала и уплотнения
или разрыхления порошка при формировании
конуса. Hecмoт на эти трудности, метод про
должает использоваться в фармацевтической
промышленностм. а в литературе описано MHO
жество примеров. демонстрирующих еro значе
ние при решении производственных проблем.
Уrол ecтecтвeннoro откоса это величина
трехмерноro yma (относительно roризонталь
ной поверхности), образующеroся при насыпке
материала roркой в виде конуса, определенная
одним из кратко описанных ниже методов.
Основные методики определения уrла
eCTeCTBeHHoro откоса
В литературе описывается множество Me
тодик определения уrла естественноro откоса.
Большинство применяемых методик определе
ния статическоro уrла естественноro откоса за
висит от двух важных экспериментальных пере
менных:
высота трубы, через которую проходит
порошок, может фиксироваться относительно
основы или изменяться при образовании roрки;
основа, на которой образуется roрка,
может иметь определенный диаметр. или диа
метр основы конуса roрки из порошка может из
меняться при образовании roрки.
Разновидности методик определения
уrла eCTeCTBeHHoro откоса
В фармацевтической литературе описаны
разновидности вышеупомянутых методик:
стекающий У20Л естествеННО20 откоса
определяется после «стекания» из контейнера
дополнительноro количества материала, BЫXO
дящеro за пределы основы определенноro ди
аметра. Образование конуса порошка с основой
определенноro диаметра позволяет определить
стекающий уroл естественноro откоса;
динамический У20Л естествеНН020 откоса
определяется путем заполнения цилиндра (с rлад
ким, плоским дном) и вращения еro с определен
ной скоростью. Динамический уroл естественноro
откоса это уroл (относительно roризонтальной
поверхности), сформированный «текучим» по
рошком. Внутренний уroл кинетическоro трения
определяется плоскостью, разделяющей частицы,
которые скатываются с верхнеro слоя порошка, и
ЧqСТИЦЫ, которые вращаются вместе с цилиндром
(с шероховатой поверхностью).
Общая шкала текучести и уrла естествен-
Horo откоса
В то время как существуют некоторые раз
личия в качественном описании текучести по
рошка, основывающиеся на определении уrла
естественноro потока, большая часть описаний
в фармацевтической литературе соrласуется с
классификацией Карра [таблица 2.9.36. 1 (Carr
RL. Еvа/иаtiпg f/ow p,operties о( solids. Chem.
Eпg. 1965; 72:163 168)]. Для производственных
целей чаще подходят уrлы естественноro откоса
в диапазоне 40 50 rрадусов, и roраздо реже
более 50 rрадусов.
Таблица 2.9.36. 1
Текучесть и соответствующий У20Л
естествеННО20 откоса
Текучесть Уrол eCTeCTseHHoro
откоса (в rpaAycax)
Отличная 25-------ЗО
Хорошая 31 35
Умеренная (без З О
помощи)
Удовлетворительная 41 5
(может застревать)
Слабая (необходимо 46 55
встряхивание,
вибрация)
Плохая 56 5
Очень плохая > 66
Условия экспериментальноrо определе-
ния уrла eCTeCTBeHHoro откоса
Уroл естественноro откоса не является xa
рактерным свойством порошка, так как он изме
няется в зависимости от методики, используемой
для формирования конуса порошка. Поэтому He
обходимо соблюдать следующие условия:
вершина конуса порошка может деформи
роваться под воздействием порошка сверху, по
этому при построении порошковоro конуса, He
обходимо соблюдать аккуратность;
2.9.36. Текучесть порошков
499
природа основы, на которой строится
конус из порошка, влияет на уroл eCTeCTBeH
ноro откоса; рекомендуется, чтобы конус из
порошка формировался на «общей основе»
(то есть образование конуса на слое по
рошка); это достиrается путем использова
ния основания с определенным диаметром,
внешние края котороro имеют бортик для co
хранения слоя порошка, на котором форми
руется конус.
Рекомендуемая методика определения
уrла eCTeCTBeHHoro откоса
Формируют уroл естественноro откоса
на выбранной основе с блокировочным BЫ
ступом, удерживающим слой порошка на
основе. Основа не должна подверrаться ви
брации. Высоту подающей трубы изменяют
таким образом, чтобы порошок образовывал
симметричный конус. Необходимо следить за
вибрацией, возникающей I}РИ перемещении
трубы. Для уменьшения воздействия пада
ющеrо порошка на формирующуюся верши
ну конуса, край трубы должен находиться на
расстоянии 2 см от вершины roрки из по
рошка. Если не удается создать симметрич
ный или воспроизводимый конус из порошка,
то этот метод не может быть использован. Из
меряют высоту полученноro конуса из порош
ка и рассчитывают уroл естественноro откоса
а по формуле:
tg( а) = высота
0,5 . диаметр
КОЭФФИЦИЕНТ СЖИМАЕМОСТИ
И ОТНОШЕНИЕ ХАУСНЕРА
В последние roды определение коэффи
циента сжимаемости и близко связанноrо с
ним отношения Хауснера считается простым,
быстрым и популярным методом получения
характеристик текучести порошков. Коэффи
циент сжимаемости представляет собой KOC
венный показатель насыпной массы, размера
и формы, площади поверхности, коrезионной
способности и содержания влаrи в материа
ле. Коэффициент сжимаемости и отношение
Хауснера определяются путем измерения
объемов порошка при свободной засыпке и
при уплотнении порошка.
Основные методики определения KO
эффициента сжимаемости и отношения
Хауснера
Хотя существует несколько методик опре
деления коэффициента сжимаеМI)СТИ и OTHO
шения Хауснера, основной способ заключа
ется в измерении объема порошка до усадки
(V o ) и конечноro объема порошка после
усадки (VJ. Коэффициент сжимаемости и OT
ношение Хауснера рассчитывают по следую
щим формулам:
v v
Коэффициент сжимаемости = 100. ,
V o
Отношение Хауснера
110
V K
Как альтернатива, коэффициент сжимае
мости и отношение Хауснера MorYT быть pac
считаны с использованием измеренных зна
чений насыпной плотности (Рев) и плотности
после усадки (Ру) следующим образом:
Р p
Коэффициент сжимаемости = 100. ус св ,
Рус
Отношение Хауснера = Рус .
Рее
Иноrда в некоторых методиках определя
ют степень уплотнения как отдельный либо
дополнительный параметр изменения объема
при усадке. Для коэффициента сжимаемости
и отношения Хауснера используется обще
принятая шкала текучести [таблица 2.9.З6. 2
(Ca,r RL. Еvа/иаtiпg f/ow properties о( solids.
Chem. Eng. 1965; 72:163 168)].
Таблица 2.9.36. 2
Шкала текучести
Коэффициент Текучесть Отношение i
сжимаемости Хауснера ,
(%) !
1 10 Отличная 1,OO 1,11 i
11 15 Хорошая 1, 12 1, 18
16 20 Умеренная 1,19 1,25 !
21 25 Удовлетвори 1,26 1,34
тельная
2 31 Слабая 1,3 1,45
32 37 Плохая 1 ,46 1,59
>38 Очень плохая > 1,60
Условия экспериментальноrо опреде-
ления коэффициента сжимаемости и отно-
шения Хауснера
Коэффициент сжимаемости и отноше
ние Хауснера не являются характерными
свойствами порошка, так как они зависят от
используемой методики. На определение
объема порошка до усадки (V o )' конечноro
объема после усадки (VJ, насыпной плотно
сти (Рев) и плотности после усадки (Рус) влияют
следующие условия:
диаметр используемоro цилиндра;
количество ударов до достижения Ha
сыпной плотности после усадки;
масса материала, используемоrо в ис
пытании;
вращение образца во время ero уплот
нения.
500
rосударственная фармакопея Республики Бела
Рекомендуемая методика определения
коэффициента сжимаемости и отношения Xa
уснера
Используют мерный цилиндр объемом 250 мл
и массу испытуемою образца 100 r. Moryт ис
пользоваться меньшие количества и объемы, но
описания всех изменений в методиках должны
прикладываться к полученным результатам. PeKO
мендуется проводить в среднем три определения.
СКОРОСТЬ ВЫТЕКАНИЯ
Степень текучести материала зависит от
мноrих факторов. некоторые из которых связаны
с самими частицами, а некоторые зависят от
испытания. Контроль скорости вытекания MaTe
риала через отверстие является одним из лучших
способов измерения текучести порошков. Очень
важно контролировать прохождение непрерывно
потока, так как вытекающие с пульсацией потока
образцы встречаются даже среди свободно TeKY
чих материалов. Может наблюдаться также изме
нение скорости потока по мере тоro, как пустеет
контейнер. Выводятся эмпирические уравнения,
связывающие скорость потока с диаметром от
верстия, размером и плотностью частиц. Однако
определение скорости вытекания через oтвep
стие ИСПОЛЬЗуется только в случае со свободно
текучими материалами.
Скорость вытекания представляет собой
отношение массы, вытекающей из какоro ли
боконтейнера(цилиндр, воронка, заrрузочная
воронка), ко времени. Измерение скорости
вытекания может происходить в дискретных
приращениях или непрерывно.
Основные методики определения CKOpO
сти вытекания
В литературе при водится множество раз
нообразных методик определения скорости
вытекания через отверстие. Их можно класси
фицировать по 3 основным эксперименталь
ным переменным
Тип контейнера для порошка. Обычно ис
пользуются контейнеры в виде цилиндров, труб
и воронок из производственною оборудования.
Размер и форма используемоro OTBep
стия. Диаметр и форма отверстия являются
основными факторами при определении CKO
рости вытекания порошка.
Метод измерения скорости вытекания
порошка. Скорость потока может измеряться
непрерывно с использованием электронных
весов, оснащенных каким либо записываю
щим устройством (ленточный самописец, KOM
пьютер). Она может также измеряться в дис
кретных величинах (напримеr, время, которое
требуется для прохождения через отверстие
100 r порошка, измеренное с точностью до
ближайшей десятой секунды, или количество
порошка, проходящее через отверстие в Te
чение 1 О секунд, измеренное с точностью до
ближайшей десятой rpaMMa).
Различия в методиках определения с-.
расти вытекания
Можно определять массовую либо объе.
ную скорость вытекания. Определение Maccoвaii
скорости вытекания является более леrкой M
дикой, но В данном случае имеет место иск
ние результатов в случае летучих и высокопЛOJ
ных материалов. Так как наполнение матрицы
осуществляется по объему, предпочтительнее ис-
пользовать определение объемной скорости BЫТ
кания. Иноrда для облеrчения вытекания порошка
из контейнера используется виброусройство, Ч1О
усложняет интерпретацию результатов. YCT
ство подвижноro отверстия используется для tVO-
делирования условий роторноro пресса. Сможет
быть определен минимальный диаметр OTBe
ст.ия, через которое проходит поток порошка.
Общая шкала текучести и скорость BЫT
кания
Так как скорость вытекания зависит от ис
пользуемоro метода измерения, общей шкалы
для определения текучести не существует, в
связи с чем сравнение опубликованных резулtr
татов является затруднительным.
Условия экспериментальноrо определ
ния скорости вытекания
Скорость вытекания через отверстие не явля-
ется характерным свойством порошка, так как она в
значительной мере зависит от используемой мето-
дики. Литературные данные указывают на несколЬ.-
ко важных условий, влияющих на эти методики:
диаметр и форма отверстия;
тип материала, из котороro сделан контей
нер (металл, стекло, пластмасса);
диаметр и высота слоя порошка.
Рекомендуемая методика определения
скорости вытекания
Определение скорости вытекания через OT
верстие используется для материалов, име
ющих некоторую способность к течению. Эта
методика не при меняется для коrезионных Ma
териалов. Если высота порошковоro слоя зна
чительно выше, чем диаметр отверстия, то CTe
пень текучести фактически не зависит от высоты
слоя. Целесообразно использовать цилиндр в
качестве контейнера, чтобы стенки контейнера
не оказывали значительноro влияния на поток
порошка. Таким образом, скорость потока опре
деляется подвижностью порошка и в MeHb
шей степени движением порошка по стенке
контейнера. В большинстве случаев скорость
вытекания порошка увеличивается, если высота
столба порошка меньше, чем ero двойной диа
метр. Отверстие должно быть круrлым, а ци
линдр не должен вибрировать.
Основные требования к размерам цилиндра
следующие:
диаметр отверстия больше, чем шест
кратный диаметр частиц;
2.9.37. Оптическая микроскопия
501
диаметр цилиндра больше, чем двойной
диаметр отверстия.
Заrрузочная воронка в качестве контей
нера может использоваться для COOTBeTCTBY
ющей и репрезентативной оценки текучести
в производственных условиях. Не peKOMeHДY
ется использовать воронки с выходным CTBO
лом, так как скорость потока будет зависеть не
только от размера и высоты выходноro ствола,
но и от силы трения между порошком и CTBO
лом воронки. Можно использовать воронку в
виде усеченноro конуса, но в этом случае на
скорость потока будет влиять коэффициент
трения между стенками и порошком, поэтому
очень важно подобрать материал, из котороro
сделана воронка.
На плоском дне цилиндра делают отверстие
с изменяемым диаметром для обеспечения MaK
симальной подвижности и оптимальной CTPYКТY
ры потока (<<порошок по порошку»). Измерение
скорости вытекания может быть или дискрет
ным, или непрерывным. Непрерывное измере
ние с использованием электронных весов может
наиболее эффективно обнаруживать KpaTKOBpe
менные изменения скорости вытекания.
МЕТОДЫ сдвиrовой ЯЧЕЙКИ
Чтобы изучение текучести порошков и про
ектирование заrрузочных воронок поставить на
более фундаментальную основу, для разных
видов порошков были разработаны специаль
ные приборы и методы сдвиra, которые позво
ляют дать более полную и точную характеристи
ку текучести порошков. Методолоrия сдвиroвой
ячейки широко используется в изучении фарма
цевтических материалов. Используя эти методы,
можно получить множество различных пара
метров, включая зависимость текучести от Ha
личия деформации сдвиrа, уroл внутреннеro
трения, неоrраниченное напряжение текучести,
прочность при растяжении, а также производные
параметры, lакие как фактор потока и друrие KO
эффициенты текучести. Блаroдаря возможности
управлять экспериментальными пара метрами
более точно, свойства текучести можно опре
делять как функцию консолидационной наrруз
ки (то есть наrрузки при уплотнении), времени,
и друrих условий внешней среды. Эти методы
успешно используются для определения OCHOB
ных параметров заrрузочной воронки и друrих
емкостей.
Основные методики сдвиrовой ячейки
Одним из типов сдвиroвой ячейки является
цилиндрическая ячейка, которая в rоризонталь
ном разрезе формирует плоскость сдвиrа между
нижним неподвижным основанием и верхней
подвижной частью кольца сдвиroвой ячейки.
После уплотнения слоя порошка на сдвиrовой
ячейке (используется определенная методи
ка) определяют силу, необходимую для сдвиrа
слоя порошка движением верхнеro кольца.
Модель кольцевой сдвиroвой ячейки имеет He
сколько преимуществ по сравнению с моделью
цилиндрической сдвиroвой ячейки, включая по
требность в менЬшем количестве материала.
Недостатком, обусловленным конфиryрацией
ячейки, является то, что слой порошка сдвиrает
ся неоднородно, так как частицы материала на
внешней части кольца сдвиrаются больше, чем
частицы внутренней области. Третий тип сдвиrо
вой ячейки (тип пластины), представляет собой
тонкий слой порошка между нижней неподвиж
ной шероховатой поверхностью и верхней под
вижной шероховатой поверхностью.
Все методики сдвиrовой ячейки имеют свои
преимущества и недостатки, однако их деталь
ный обзор в рамках этоro раздела невозможен.
Как и в случае с друrими способами описания
текучести порошков, по данным методам суще
ствует большое количество информации в ли
тературе. Существенное преимущество метода
сдвиroвой ячейки заключается в возможности в
значительной степени контролировать экспери
ментальный процесс. В общем, этот метод яв
ляется достаточно трудоемким, требует значи
тельных количеств материала и наличия хорошо
обученноrо оператора.
Рекомендации к методу сдвиrовой ячейки
Мноrие существующие конфиryрации сдви
roвых ячеек и методы испытаний предоставля
ют большой объем данных и очень эффективно
MOryт использоваться для характеристики Teкy
чести порошков. Они также полезны при разра
ботке оборудования типа заrрузочных воронок
и бункеров. Из за разнообразия используемоro
оборудования и экспериментальных процедур
никакие определенные рекомендации ОТНОСИ
тельно методики в этом разделе не приводятся.
Рекомендуется, чтобы результаты определения
характеристик текучести порошков, получен
ные с использованием метода сдвиroвых ячеек,
включали полное описание оборудования и ис
пользуемой методики.
01/2013:20937
2.9.37. ОПТИЧЕСКАЯ МИКРОСКОПИЯ
Оптическая микроскопия используется
ДЛЯ характеристики частиц размером от 1 мим
и более. Нижний предеn определяется разр&
шающей способностью микроскопа. Верхний
предел менее оrраничен и обусловлен возраста.-
ющей сложностью, связанной с характеристикой
частиц больших размеров. Для характеристики
частиц вне диапазона оптической микроскопии
используются альтернативные методы. Оптич
ская микроскопия особенно подходит для xapaK
502
rосударственная фармакопея Республики Беларусь
теристики несферических частиц. Этот метод
может также служить основой для калибровки
разрабатываемых ускоренных и более рутинных
методов.
Оборудование. Используют устойчиво
установленный микроскоп, защищенный от ви
брации. Увеличение микроскопа (увеличение
объектива и окуляра, дополнительные компо
ненты увеличения) должно быть достаточное
для соответствующей идентификации наимень
ших частиц в испытуемом образце. Выбирают
самую большую числовую апертуру объектива
для каждою диапазона увеличеttия. Moryт ис
пользоваться поляризующие фильтры с под
ходящими анализаторами и замедляющими
пластинками. Светофильтры с узкополосной
спектральной пропускаемDCТЬЮ используются
с ахроматическими или, более предпочтитель
но, с апохроматическими объективами; они He
обходимы для соответствующей цветопередачи
в микрофотоrрафии. Конденсоры, откорректиро
ванные с учетом сферической аберрации, Haxo
дятся под предметным столиком и имеют BCT
енный осветитель. В рабочих условиях числовая
апертура конденсора соответствует апертуре
объектива; это достиrается с помощью апертур
ной диаqpраrмы конденсора и использованием
иммерсионных масел.
Реryлирование. Очень важна точная юсти
ровка всех элементов оптической системы и
соответствующая фокусировка. Фокусировка
элементов осуществляется в соответствии с pe
комендациями изroтовителя микроскопа. PeKO
мендуется проводить критическое осевое BЫ
равнивание.
Освещение. Требованием к освещению яв
ляется однородная и реryлируемая интенсив
ность света во всем поле обозрения; предпо
чтительно использовать настройку освещения
по Кёлеру. При наличии окрашенных объектов
выбирают цвет светофильтра таким образом,
чтобы следить за контрастом и деталями изо
бражения.
Визуальная характеристика. Увеличение
и числовая апертура должны быть достаточно
высокими для обеспечения необходимоrо раз
решения деталей исследуемых объектов. Фак
тическое увеличение устанавливают с исполь
зованием микрометра для проверки окулярноro
микрометра. Для уменьшения поrрешности BЫ
бирают такое увеличение, чтобы размер частицы
занимал не менее 10 делений окуляра. Каждый
объектив должен rрадуироваться отдельно.
Чтобы отrрадуировать шкалу окуляра, необходи
мо совместить шкалу микрометра и шкалу OKY
ляра. Таким способом можно точно определить
расстояния между делениями окуляра. Может
понадобиться использование нескольких раз
личных увеличений для характеристики объв.
тов, имеющих широкое распределение частиq
по размерам.
Фотоrрафическая характеристика. E
размер частицы должен определяться фотоrpa-
фическими методами, то следует предпринять
меры для тоro, чтобы объект был хорошо сфо
сирован на плоскости фотоrрафической эмуль-
сии. Определяют фактическое увеличение.
фотоrрафируя микрометр с .откалиброваннoi
шкалой, используя фотопленку с достаточнoi
разрешающей способностью и контрастностью.
Экспозиция и обработка фотоrрафии испыту
емоro образца и фотоrрафии, полученной при
определении увеличения, должны быть иде
т чными. Кажущийся размер фотоrрафическc:r
ro изображения зависит от экспозиции, проявкм
и печати фотоrрафии, а также Щ разрешающei
способности микроскопа.
ПРИl"отовление образца для микроско-
пии. Среду, в которую поrpyжaeтся или KOTOpoil
покрывается образец, выбирают в зависимости
от физических свойств иcnытуемоro образца
Для обеспечения резкоro изображения краев
образца необходимо наличие достаточноro, но
не чрезмерноro контраста между образцом и
средой. Образец должен располаrаться в одной
плоскости и быть распределен cooTBeTcTBYкr
щим образом для возможности выделения OT
дельных интересующих фраrментов. Кроме
тоro, частицы должны реально отражать rpaHY
лометрический состав образца и не изменять-
ся во время ero приroтовления. Этому важному
требованию должно быть уделено значитель
ное внимание. При выборе среды для приroтов
ления образца необходимо учитывать еro pac
творимость.
Характеристика кристаллическоrо COCTO
яния. Кристалличность испытуемоro образца
определяют для установления соответствия с
требованиями по кристалличности, указанными
в частной статье для данной фармацевтической
субстанции. Если нет указаний в частных CTa
тьях, несколько частиц образца в минеральном
масле помещают на чистое предметное стекло.
Смесь исследуют с использованием поляриза
ционноro микроскопа: частицы проявляют двой
ное лучепреломление (интерференционная
окраска) и зоны поrлощения при вращении пред
метноro столика микроскопа.
Определение размера частиц посред
ством микроскопии. ОТl3ешивают необходимое
количество испытуемоro порошка (например,
1 o 100 Mr), суспендируют в 1 О мл подходя
щей среды, в которой он не растворяется; при
необходимости прибавляют смачивающее Be
щество. rOMoreHHoe состояние частиц в cy
спензии поддерживают путем суспендирования
2.9.37.
Оптическая микроскопия
503
частиц в среде с подобной или схожей плотно
стью и путем обеспечения необходимоro пере
мешивания. Часть юмоreнной суспензии поме
щают в подходящую счетную ячейку и изучают
под микроскопом на площади, которую занимает
не менее 10 MKr испытуемою порошка. Подсчи
тывают все частицы, у которых размер больше,
чем нормированный в частной статье. Предель
ный размер и допустимое количество частиц,
превышающих этот размер, определяют для
каждоro вещества.
Характеристика размера частиц. Измере
ние размеров частицы отличается разной CT
пенью сложности в зависимости от их формы.
Количество исследуемых частиц должно быть
достаточным для обеспечения приемлемоro
уровня неопределенности измеренных пара
метров. Дополнительную информацию об из
мерении размеров частиц, размере образца и
анализе данных можно получить, например, в
/SO 9276. Для сферических частиц размер опре
деляется их диаметром. Для частиц неопре
деленной формы существуют различные спо
собы определения размера. Для таких частиц
характеристика их размеров должна включать
не только информацию о форме частицы, но и о
типе измеренноro диаметра (рисунок 2.9.37. 1):
диаметр по Ферету: расстояние между
воображаемыми параллельными касательными
линиями к произвольно ориентированной части
це, перпендикулярными шкале окуляра;
диаметр по Мартину: длина линии, па
раллельной тому же направлению, разделяю
щей профиль частицы на две равные по площа
ди части;
диаметр проекции: диаметр Kpyra, KOТO
рый имеет ту же площадь, что и проекция ча
стица;
длина: наибольшее расстояние между
краями частицы, ориентированной параллельно
шкале окуляра;
/
//
.
\
\
\ 1 диаметр по Ферету
.1
,.
I
диаметр по Мартину
Рисунок 2.9.37. 1. Наиболее используемые
способы измерения размеров частиц
ширина: наибольшее расстояние между
краями частицы, ориентированной перпендику
лярно шкале окуляра и проведенное под прямым
уrлом относительно длины.
Характеристика формы частиц. Xapaктe
ристика размера частиц неопределенной формы
должна содержать сведения и об их форме. Oд
нородность порошка проверяют при подходя
щем увеличении микроскопа. Наиболее часто
используют следующие описания формы частиц
(рисунок 2.9.37 _ 2):
и2Ловидная: узкая, иrлоподобная частица
с близкими значениями ширины и толщины;
палочковидная: длинная тонкая частица,
ширина и толщина которой больше аналоrичных
у иrлообразной частицы;
хлопьевидная: тонкая плоская частица с
близкими значениями длины и ширины;
пластинчатая: плоская частица с близки
ми значениями длины и ширины, но толще, чем
хлопьевидная частица;
в форме ПРЯМОУ20ЛЬНОЙ пластинки: длин
ная тонкая частица схожая по форме с лезвием;
кубовидная: частица с близкими значени
ями длины, ширины И толщины; сюда включают
кубические и сферические частицы.
ШJ
l<убоВI1ДН.ая
хлоriьеsv,дная
tf@
r;Ластинчатая
П2JlОЧ!ЮБЩJ.ная
,
в форме прямоуrолы-юи ллаеТVlI1Ь;
Рисунок 2.9.37. 2. Наиболее часто использу
емые описания формы частиц
Общие замечания. Считается, что части
ца является наименьшей дискретной единицей.
Частица может быть жидкой или полутвердой
капелькой; отдельным кристаллом или поли
кристаллом; в аморфном виде или в виде аrло
мерата. Частицы MOryT образовывать ассоци
аты. Степень их ассоциации может быть описа
на следующим образом:
МН020СЛОЙНЫЙ ассоциат: расположенные
друr над друroм пластины;
а2ре2ат: масса из слипшихся частиц;
а2ломерат: сплавившиеся или сцеплен
ные между собой частицы;
КОН2ломерат: ассоциат из 2 или более
типов частиц;
сферолит: радиальное скопление;
друза: частица, покрытая друrими крошеч
ными частицами.
Состояние частицы может описываться по
следующим параметрам:
504
rосударственная фармакопея Республики Беларусь
характер края: уrловатые, окруrлые, rлад
кие, острые, изломленные;
оптические параметры: цвет (использу
ют соответствующие компенсирующие CBeтo
фильтры), а также прозрачность, просвечива
емость и непрозрачность;
дефекты: окклюзия, инклюзия.
Характеристика поверхности может COCTO
ять из следующих описаний:
растрескавшаяся: наличие расщелин,
расколов или трещин;
2ладкая: без неровностей, шероховато
стей или проецирования;
пористая: наличие отверстий или пор;
неровная: буrристая, неоднородная, He
rладкая;
изрытая (или ямчатая): наличие малень
ких вмятин.
01/2013:20938
2.9.38. ОПРЕДЕЛЕНИЕ РАЗМЕРА
ЧАСТИЦ МЕТОДОМ
АНАЛИТИЧЕскоrо
ПРОСЕИВАНИЯ
Просеивание один из наиболее старых
методов, используемый для классификации
порошков и rранул в зависимости от rpaHY
лометрическоro состава. При использовании
плетеной ткани сита, просеивание сортирует
частицы в зависимости от их среднеro разме
ра (то есть ширины и толщины). Механическое
просеивание является наиболее применимым
методом в том случае, Korдa большая часть
частиц имеет размер более 75 мкм. Леrкий
вес более мелких частиц обуславливает Heдo
статочную силу для преодоления поверхност
ных сил коrезии и адrезии во время просеива
ния, что является причиной слипания частиц
между собой, а также с ситом, а это, в свою
очередь, является причиной задерживания
на сите частиц, которые по своему размеру
должны были бы пройти через отверстия. Для
таких образцов более подходящим является
воздушно струйное и ультразвуковое просе
ивания. Тем не менее, просеивание может
быть использовано для некоторых порошков
или rранул С размером частиц менее 75 мкм
в том случае, если метод может быть валиди
рован. В фармацевтической практике просеи
ВClние, как правило, является методом выбора
для классификации на rрубом уровне OДHO
компонентных порошков или rранул. Это наи
более подходящий метод для порошков или
rранул, классифицируемых только по размеру
частиц; в большинстве случаев анализ выпол
няют в сухом состоянии.
Среди оrраничений метода просеиванИII
наиболее существенными являются налич..
определенноro количества испытуемоro образ-
ца (в среднем не менее 25 r; в зависимости от
плотности порошка или rранул и от диаметра oт
верстий сита), а также сложность просеиванИII
маслянистых или друrих склеивающихся пороw-
ков или rранул, которые склонны к забиваниlO
отверстий сита. Метод в основном использует
ся для двухмерной оценки размера частиц, так
как прохождение частиц через отверстия сита
больше зависит от их ширины и толщины, чем
от длины.
Метод предназначен для оценки общеro rpa-
нулометрическоro состава однокомпонентноro
образца. Он не предназначен для определения
количественноro соотношения частиц, прош
ших или задержанных на 1 или 2 ситах.
Если нет указаний в частных статьях, то
определение rранулометрическоrо состава
проводят методом механическоro. встряхива-
ния (метод сухоro просеивания). В случае если
трудно определить конечную точку процесса
просеивания (т. е. частицы образца плохо про--
ходят через отверстия сита) или при необход
мости использовать сито с более мелкими OT
верстиями в конце определения (менее 75 мкм),
должна быть дана ссылка на использование ал
тернативноro метода.
Просеивание должно проводиться в услов
ях, исключающих увеличение или уменьшение
влаrи в испытуемом образце. Для предотвра-
щения поrлощения или потери влаrи испыту
емым образцом необходимо постоянно контро--
лировать относительную влажность окружаl(}-
щей среды, в которой проводят просеивание.
При отсутствии признаков несоответствия усло--
вий просеивание обычно проводят в условиях
относительной влажности окружающей среды.
Специальные условия, предусмотренные для
определенноro образца, должны быть указаны в
частных статьях.
Принципы аналитическоrо просеивания.
Аналитические сита изrотавливают из тканной
проволочной сетки, плетение которой обеспечи
вает квадратную форму отверстий и которая за
паяна в основание открытоro цилиндрическоro
контейнера. Сита устанавливают одно на друroе
по увеличению размера отверстий, и помеща
ют испытуемый порошок на верхнее сито. Набор
сит, установленных друr на друrа, встряхивают в
течение определенноro периода времени, затем
с необходимой точностью определяют количе
ство материала, оставшеroся на каждом сите.
В результате получают процентное содержание
фракций порошка в соответствии с диапазоном
размера отверстий используемых сит.
Такой способ просеивания в основном ис
пользуется для оценки rранулометрическоro со--
става однокомпонентных фармацевтических по--
рошков, не менее 80 % частиц которых имеют
2.9.38. Определение размера частиц методом аналитuчеСКО20 просеuванuя
505
размер более 75 мкм. Пара метр размера, ис
пользуемый при определении rранулометри
ческоro состава методом аналитическоrо про
сеивания, представляет собой длину стороны
минимальноro квадратноro отверстия, через KO
торое может пройти частица.
АНАЛИТИЧЕСКИЕ СИТА
Аналитические сита, используемые для
фармакопейноro анализа должны COOTBeTCTBO
вать текущему изданию /SO 33 1 o 1: Анали
тические сита Технические требования и
проверка Часть 1: Аналитические сита из
металлической сетки (см. таблицу 2.9.38. 1).
Если нет указаний в частных статьях, необхо
димо использовать /SO сита с размером OTBep
стий, указанным в таблице 2.9.З8. 1, которые pe
комендованы в определенной области.
Выбранные сита должны вкпючать весь ди
апазон размеров частиц, присутствующий в ис
пытуемом образце. Рекомендуется использовать
набор сит, имеющих .J2 проrрессию площадей от
верстий. Набор сит должен быть собран так, чтобы
сито с самым крупным размером отверстий было
верхним, а с самым мелким размером отверстий
нижним. Размер отверстий аналитических сИТ BЫ
ражается в микрометрах или миллиметрах.
Для изroтовления аналитических сит ис
пользуют проволоку из нержавеющей стали, или
(менее предпочтительно) из латуни или друroro
подходящеro химически неактивноro металла.
Калибровка и повторная калибровка анали
тических сит производится в соответствии с Te
кущим изданием /SO 33 1 o 1. Перед использо
ванием сита тщательно проверяются на rрубые
искажения и изломы, в особенности соединения
ситовоro каркаса. Сита MorYT быть откалиброва
ны оптически для определения среднеro разме
ра отверстия и различий в размерах отверстий.
В качестве альтернативноrо метода для оценки
эффективности просеивания аналитических сит
в диапазоне размеров 212 50 мкм Moryт быть
использованы доступные стандартные стекпян
ные сферы (шарики, rранулы). Если нет указа
ний в частных статьях, определение проводят
при контролируемой комнатной температуре и
относительной влажности окружающей среды.
Очистка аналитических сит. В идеале для
очистки аналитических сит используют только
воздушную струю под низким давлением или
струю жидкости. Если некоторые отверстия все
же блокированы частицами испытуемоro Be
щества, то можно применить мяrкую щеточную
очистку.
Таблица 2.9.38. 1
150 номинальный размер отверстия Рекомендованные
Основной США и5Р сита Европейские Японские
Дополнительный размер сито N!! сита N!! сита N!!
размер (мкм)
R 20/3 R20 R 40/3
11,20 мм 11,20 мм 11,20 мм 11 200
10,00 мм
9,50 мм
9,00 мм
8,00 мм 8,00 мм 8,00 мм
7,10 мм
6,70 мм
6,30 мм <
5,60 мм 5,60 мм 5600 3,5
5,00 мм
4,75 мм 4
4,50 мм
4,00 мм 4,00 мм 4,00 мм 5 4000 4000 4,7
З,55 мм
3,35 мм 6 5,5
3,15 мм
2,8 мм 2,80 мм 2,80 мм 7 2800 2800 6,5
2,50 мм
2,36 мм 8 7,5
2,24 мм
2,00 мм 2,00 мм 2,00 мм 10 2000 2000 8,6
1,80 мм
1,70 мм 12 10
506
rосударственная фармакопея Республики Беларусь
ISO номинальный размер отверстия Рекомендованные
Основной США иSP сита Европейские Японские
Дополнительный размер сито N!! сита N!! сита N!!
размер (мкм)
R 20/3 R20 R 40/3
1,60 мм
1,40 мм 1 ,40 мм 1,40 мм 14 1400 1400 12
1,25 мм
1,18 мм 16 14
1,12 мм
1,00 мм 1,00 мм 1,00 мм 18 1000 1000 16
900 мкм
850 мкм 20 18
800 мкм
710 мкм 710 мкм 71 О мкм 25 710 710 22
630 мкм
600 мкм 30 > 26
560 мкм
500 мкм 500 мкм 500 мкм 35 500 500 30
450 мкм
425 мкм 40 36
400 мкм
355 мкм 355 мкм 355 мкм 45 355 355 42
315 мкм
300 мкм 50 50
280 мкм
250 мкм 250 мкм 250 мкм 60 250 250 60
224 мкм
212 мкм 70 70
200 мкм
180 мкм 180 мкм 180 мкм 80 180 180 83
160 мкм
150 мкм 100 100
140 мкм
125 мкм 125 мкм 125 мкм 120 125 125 119
112 мкм
106 мкм 140 140
100 мкм
90 мкм 90 мкм 90 мкм 170 90 90 166
80 мкм
75 мкм 200 200
71 мкм
63 мкм 63 мкм 63 мкм 230 63 63 235
56 мкм
53 мкм 270 282
50 мкм
45 мкм 45 мкм 45 мкм 325 45 45 330
40 мкм
38 мкм 38 391
Испытуемый образец. Если в частной
статье нет указаний относительно массы H3
вески для конкретноro образца, то для испыта
ния на аналитических ситах диаметром 200 мм
берут навеску 25 100 r испытуемоro образца,
в зависимости от ero насыпной плотности. Для
сит диаметром 76 мм количество испытуемоro
образца должно составлять около 1/7 от коли
чества, рекомендованною для данною вида об
разца при определении на аналитических ситах
диаметром 200 мм. Массу навески для KOHKpeT
ноro образца определяют просеиванием в тече
ние определенноro периода времени, используя
механический встряхиватель, точных навесок
разной массы, например, 25 r, 50 r и 100 r (при
мечание: если результаты испытания сходны
для навесок образца 25 r и 50 r; но для навески
100 r получен более низкий процент просеива
2.9.38. Определение размера частиц методом аналитичеСКО20 просеивания
507
ния через самое мелкое сито, то навеска 100 r
слишком велика для данноro образца). Если в
наличии только 1 0 25 r испытуемою образца,
то MoryT быть использованы аналитические сита
с соответствующим номером меш, но меньшеro
диаметра, однако конечная точка просеивания
должна быть определена заново. Допускает
ся определение с навеской меньшей массы (то
есть менее 5 r). Для образцов с низкой насып
ной плотностью, а также для образцов, COCTO
ящих большей частью из частиц высокой изоди
аметрической формы, массы навески менее 5 r
для сит диметром 200 мм MOryт быть необходи
мы для предотвращения чрезмерноro прилипа
ния образца к ситу. При проведении валидации
определенноro метода аналитическоro просе
ивания необходимо учитывать возможность за
купоривания отверстий сита частицами испыту
емоro образца.
Если испытуемый образец имеет склон
ность к поrлощению или потере значительных
количеств воды при изменении влажности, то
испытание нужно проводить в условиях COOT
ветствующеro контроля окружающей среды.
Если известно, что испытуемый образец может
быть источником электростатическоro заряда,
то необходимо тщательное наблюдение за тем,
чтобы это не повлияло на результат анализа.
Для минимизации этоro эффекта к испытуемому
образцу может быть добавлен антистатический
peareHT, такой как кремния диоксид коллоидный
и/или алюминия оксид в количестве 0,5 % (м/м).
Если вышеуказанные эффекты не Moryт быть
устранены, то необходимо выбрать алыерна
тивную методику определения размера частиц.
Способы встряхивания. Для проведения
ситовою анализа MOryт быть использованы раз
личные виды коммерчески доступных сит и при
способлений для встряхивания порошков. Однако
различные способы встряхивания MOryт давать
разные результаты ситовоro анализа и YCTaHO.B
ления конечных точек, так как на каждую частицу
действуют силы различноro типа и величины. Ис
пользуют методы, включающие механическое или
электромаrнитное встряхивание, которые MOryт
вызвать вертикальное колебание или roризон
тальное круювое движение частиц, постукивание
или комбинацию постукивания и roризонтальноro
круroвою движения частиц. Может использовать
ся метод встряхивания с применением воздушной
струи. Так как изменение условий встряхивания
может привести к отличающимся инекорректным
результатам при ситовом анализе и установле
нии конечных точек просеивания, в результатах
должен быть указан используемый метод встряхи
вания, а также параметры встряхивания (в случае
их возможноro варьирования).
Определение конечной точки просеива-
ния. Ситовой анализ считается завершенным,
1C0rдa масса остатка на любом аналитическом
сите не отличается более чем на 5 % или на
0,1 r (10% при просеивании на сите диаметром
76 мм) от массы остатка на этом сите при пре
дыдущем взвешивании. Если масса остатка на
сите составляет менее чем 5 % от общей массы
навески, то конечная точка увеличивается таким
образом, чтобы разность масс на одном и том
же сите при двух повторных взвешиваниях co
ставляла не более 20 %.
Если масса остатка на одном сите COCTaB
ляет более 50 % от общей массы навески, при
отсутствии указаний в частной статье, испы
тание необходимо повторить, но с добавлени
ем к набору сит дополнительною сита, проме
жуточноrо по размеру отверстий между ситом
с избыточной массой и следующим (с большим
размером отверстий) ситом 150 серии из ориrи
нальноro набора.
МЕТОДЫПРОСЕИВАНИЯ
Механическое встряхивание (метод
cyxoro просеивания). Взвешивают каждое aHa
литическое сито с точностью до 0,1 r. Помещают
предварительно взвешенный с необходимой точ
ностью испытуемый образец на верхнее сито (с
наибольшим размером отверстий), накрывают
крышкой. Встряхивают набор сит в течение 5 мин,
затем аккуратно, без потери образца, вынимают
каждое сито из набора. Взвешивают каждое сито
и определяют массу остатка на сите после просе
ивания. Аналоrичным образом определяют массу
остатка в собирательном сосуде. Еще раз соби
рают набор сит и встряхивают в течение 5 мин.
Снимают каждое сито и взвешивают, как описа
но ранее. Повторяют процедуру до достижения
конечной точки просеивания (см. Определение
конечной точки просеивания в разделе Аналити
ческие сита). После завершения анализа сравни
вают массы фракций испытуемоro образца. CYM
марная потеря массы не должна превышать 5 %
от массы навески испытуемоro образца.
Повторяют просеивание с новым образцом,
проводя однократное просеивание в течение
времени, равною суммарному времени просе
ивания при предыдущем определении. Под
тверждают, что это время просеивания COOTBeT
ствует требованиям по достижению конечной
точки просеивания. После валидации конечной
точки просеивания для определенноrо образ
ца в последующих испытаниях может быть ис
пользовано однократное просеивание в течение
фиксированноro периода времени, обеспечива
ющеro равномерное распределение частиц в за
висимости от их размера.
При наличии признаков задержки на KaKOrJI
либо из сит arperaТOB частиц в большей степе
ни, чем отдельных частиц, получение хорошей
воспроизводимости результатов при использо
вании метода механическоro сухоro просеива
ния маловероятно. Анализ должен проводиться
с использованием друrorо метода определения
размера частиц.
508
rосударственная фармакопея Республики Беларусь
Методы с использованием воздуха (воз
душно-струйное просеивание и улыразвуко
вое просеивание). Для просеивания MOryт быть
использованы различные типы оборудования с
применением потока воздуха. Воздушно струй
ное просеивание представляет собой систему с
использованием однократноro просеивания. При
определении используется методика, аналоrич
ная указанной в методе cyxoro просеивания, но
вместо обычноro механизма встряхивания ис
пользуется стандартизованная воздушная струя.
Метод отличается проведением последователь
ных анализов на индивидуальных ситах, начи
ная с сита с наименьшим размером отверстий,
для распределения частиц по размеру. Воздуш
но струйное просеивание, как правило, включа
ет в себя использование сит с меньшим разме
ром отверстий, чем в ситах, используемых при
обычном сухом просеивании. Эта методика наи
более применима, если необходимо выделить
только фракции частиц наибольшеro или наи
меньшеro размера.
В методе ультразвуковоrо просеивания
также используется набор сит. Испытуемый об
разец вносится в вертикальную воздушную KO
лебательную колонку, которая поднимает обра
зец, затем в течение определенноro количества
импульсов в минуту возвращает еro через сито
вые отверстия. При использовании ультразвуко
BOro просеивания масса навески образца может
быть уменьшена до 5 r.
Методы воздушно струйноro и ультразву
KOBoro просеивания используются для порош
ков или rранул в тех случаях, Korдa механиче
ское просеивание не может дать достоверноrо
результата.
Эти методы в наибольшей степени зависят
от специфическоro распределения частиц по
рошка в воздушном потоке, поэтому основные
трудности методов MOryT возникнуть при исполь
зовании сит с малым размером отверстий (т. е.
менее 75 мкм), если частицы имеют тенденцию к
слипанию, а также, если образец может быть ис
точником электростатическоro заряда. В выше
указанных случаях особенно важно определение
конечной точки просеивания. а также подтверж
дение тоro, что наиболее крупные частицы явля
ются отдельными частицами, а не аrреrатами.
ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ
Необработанные данные должны включать
массу навески испытуемоro образца, общее
время просеивания, точную методику просеива
ния, заданные величины всех изменяемых пара
метров, кроме тoro, указывают массы остатков,
полученных на отдельных ситах и в собираТf!ЛЬ
ном сосуде.
Для удобства можно перевести получен
ные данные в суммарное распределение масс,
а при желании выразить распределение функци
ей прохождения суммарной массы, диапазон ис
пользуемых сит должен включать сито, через KO
торое проходит весь испытуемый образец. Если
есть признаки тоro, что остаток образца, задеJr
жавшийся на сите, представляет собой arperaTbl
частиц, образовавшиеся в процессе просеива
ния, то испытание считается недействительным.
01/2013:20940
2.9.40. ОДНОРОДНОСТЬ
ДОЗИРОВАННЫХ ЕДИНИЦ
Для обеспечения однородности дозирован
I-!bIх единиц (ОДЕ) содержание действующе
ro вещества в каждой дозированной единице в
серии должно находиться в пределах узкоro ди
апазона от номинальноro содержания, указанно
ro в разделе «Состав». Дозированными едини
цами называют дозированные формы, содержа
щие единицу дозы или часть дозы действующею
вещества в каждой единице дозированноro ле
KapcTBeHHoro средства. Если нет друrих указа
ний, требование ОДЕ не относится к суспензиям,
эмульсиям или rелям в однодозовых контейне
рах, предназначенных для наружноro примене
ния. Проведение данноrо испытания не требует
ся в случае мультивитаминных лекарственных
средств и лекарственных средств, содержащих
элементы в следовых количествах.
Термин «Однородность дозированных
единиц» определяется как степень OДHOpOДHO
сти распределения действующеro вещества в
дозированных единицах. Однако если нет друrих
указаний в Фармакопее, требования данной
статьи распространяются на каждое действу
ющее вещество, входящее в состав дозирован
ных единиц лекарственноro средства, содержа
щих одно или несколько активных веществ.
Для определения ОДЕ следует при менять
один из двух методов (таблица 2.9.40. 1):
метод прямоro определения;
расчетно весовой метод.
Метод прямоro определения OДHOpOДHO
сти дозированных единиц основывается на KO
личественном определении индивидуальноro
содержания действующеro вещества (или дей
ствующих веществ) в испытуемых дозирован
ных единицах лекарственноro средства с целью
выяснения, находится ли это содержание внутри
установленных пределов. Метод прямоro опре
деления может применяться во всех случаях.
Расчетно весовой метод определения при
меним для следующих дозированных лекар
ственных форм:
1) растворы в однодозовых контейнерах и
мяrких капсулах;
2) твердые лекарственные формы (включая
порошки, rранулы и стерильные твердые лекар
ственные формы) в однодозовых контейнерах,
510
rосударственная фармакопея Республики Беларуа.
I Перемен ные I - пределение
k I константа приемлемости I п = 1 О
i п=зо
! s стандартное отклонение в выбор I
ке I
I
I
м=х
(AV = ks)
М = 98,5 %
(AV=98,5 X +ks)
М=101,5%
(AV=X 101 ,5+ks)
М= Х
(AV=ks)
М = 98,5 %
(AV=98,5 X +ks)
М=Т
(AV=X T+ks)
общая формула:
IM x l+kS
максимальное допустимое при L 1 :: 15,0, если нет
емлемое значение друrих указаний
максимальный допустимый Результат нижнею значе L2 = 25,0, если нет'
предел отклонения для каждой ния дозированной еди друrих указаний
испытанной дозированной едини ! ницы не может быть
цы от рассчитанноro значения М меньше 0,75 м, а pe
зультат верхнеro значе
ния дозированной едини I
цы не должен превышать
1,25 М (основано на зна
чении L2=25,0)
IRSD
!
! м (случай 1)
i применяется,
I KOrдa
I TS 101,5
i
I
I
I
I
I м (случай 2)
I применяется,
I Korдa
, Т >101,5
относительное стандартное от
клонение
контрольное значение
I контрольное значение
IПриемлемое
I значение (А V)
IL1
I
!L2
I
i
I
I
I
I
[.
'т
Условия
98,5 % S Х s 101,5 %
Х < 98,5 %
х > 101,5 %
98,5 % s Х s Т
х < 98,5 %
Х>Т
искомое содержание определя
емоro компонента в дозирован
ной единице при выпуске, Bыpa
I женное в процентах от номиналь
,ноro значения. Если нет друrих
! указаний, Т= 100 % или Т иско I
J Moe содержание, установленное i
производителем
МЕТОД пРямоrо ОПРЕДЕЛЕНИЯ
Отбирают не менее 30 единиц лекарственно
ro средства и проводят определение, как указано
для данной дозированной формы. В случае если
используются разные методики для количествен
ноro определения и испытания однородности co
держимоro, то вводят корректирующий КQЭффИЦИ
ент, который применяют к конечным результатам.
Твердые дозированные формы. В
каждой 1-13 1 О отобранных единиц определяют
количественное содержание действующеro Be
щества, используя подходящий аналитический
!
2,4
2,0
Значение
r t,(x. х ' 1'"
l п 1 J
K ..
1005
Х
-
-
метод. Рассчитывают приемлемое значение
(см. таблицу 2.9.40. 2).
Жидкие или мяrкие дозированные
формы. В каждой из 1 О отобранных единиц
определяют количественное содержание дей
ствующеro вещества, используя подходящий
аналитический метод. Количественное опреде
ление про водят из хорошо перемешанноro MaTe
риала, взятоro из индивидуальноro контейнера
в условиях обычноro использования. Результаты
выражают на выходную дозу. Рассчитывают при
емлемое значение (см. таблицу 2.9.40. 2).
2.9.40. Однородность дозироваННblХ единиц
511
Расчет приемлемоrо значения
Приемлемое значение (AV) рассчитывают
по формуле:
I 'I
!M ,XI ks.
Расшифровка значений приведена в табли
це 2.9.40. 2.
РАСЧЕТНО ВЕСОВОЙ МЕТОД
Количественное определение действующе
ro вещества (или действующих веществ) про
водят на репрезентативном образце серии. ис
пользуя подходящий аналитический метод.
Получают значение А, выраженное в процен
тах от номинальноro содержания (см. «Расчет
приемлемоro значения»). Допускают. что KOH
центрация (масса действующеro вещества на
массу дозированной единицы) одинаковая ДЛЯ
всех дозированных единиц. Отбирают не менее
30 дозированных единиц и проводят определе
ние, как указано для каждой дозированноЙ л€
карственной формы.
Таблетки без оболочки или таблетки, f10-
крытые пленочной оболочкой. Точно взвеши-
вают каждую из 1 О отобранных таблеток. Pac
считывают содержание действующеro вещества
в каждой таблетке в процентах от номинальноro
содержания, исходя из индивидуальной массы
таблетки и результата количественноro опреде
ления. Рассчитывают приемлемое значение.
Твердые капсулы. Точно взвешивают
каждую из 10 отобранных капсул, тщательно
следя за их целостностью. Извлекают подхо
дящим способом содержимое каждой капсулы.
Точно взвешивают каждую пустую капсулу и pac
считывают для каждой капсулы чистую массу
содержимоro путем вычитания массы оболоч
ки из соответствующей массы капсупы. Рассчи
тывают содержание действующеro вещества в
каждой капсуле, исходя из извлеченной из к п
сулы индивидуальной массы и результата коли
чественноro определения. Рассчитывают прием
лемое значение.
Мяrкие капсулы. Точно взвешивают каждую
из 1 О отобранных неповрежденных капсул, тща
тельно следя за их целостностью. Разрезают
капсулы при помощи подходящеro сухоro и чи
стоro режущеro инструмента, например, ножниц
или скальпеля, и извлекают содержимое из кап
сулы. Промывают пустую капсулу подходящим
растворителем. Дают возможность раствори
телю испариться при комнатной температуре с
поверхности оболочек в течение около 30 мин,
предприняв меры по предотвращению поrлоще
ния или потери влаrи. По отдельности взвеши
вают каждую оболочку и рассчитывают массу
содержимоro в каждой капсуле. Рассчитывают
содержаниl': действующеro вещества в каЖj.iQЙ
капсуле, исходя из извлеченной из капсулы ин
дивидуальной массы и результата !(оличествен
ною определения. Рассчитывают приемлемо(-,
значение.
Друrие твердые дозированные формы,
кроме таблеток и капсул. Определение прове
дят так же, как и для твердых капсул, об рабаты..
вая каждую единицу, как указано в данном раз
деле. Рассчитывают приемлемое значение.
Жидкие или мяrкие дозированные
формы. Точно взвешивают количество жидко-
сти или мяrкоro содержи моro. извлеченное И,'
1 О отобранных индивидуальных контейнеров с
условиях оБЫЧI.,Оro v1СПОЛЬ' С8ания. Если необ..
ходимо, рассчитывают э..твиваJ:i?НТНЫЙ обье л
после определения плотности. Рассчитывают
содержание дейтсвующеro вещества в каж.цом
контейнере, исходя из изъятой из контейнера
индивидуальной массы и результа ов КО;'lиче.
ственноro определения. Рассчитывают прием.
лемое значение.
Расчет приемлемоrо значения. Рассчип:"!
вают приемлемое значение (AV) та!', как и ДЛ,'
метода прямоrо определения, заменяя ИНДИ8 '
дуальное содержание в единицах на рас,::;читэр
ное содержание, полученное, кш< f .3ЗС;Н(J ни,,;е.
Х 1 ' Х 2 '...) Х п
отдельные значения coдep
жания в испытанных дозиро
ванных единицах.
rдe
А
Х. =о W. '==00
I I W
W " W 2 "'" W,.
отдельные массы испытан
НЫХ дозированных единиц;
содержание деиствующею
вещества (процент от указан
HOro на этикетке), полученное
с использованием подходяще
ro аналитическоro метода (pe
зулыат количественноro опре
деления);
среднее значение отдель
ных масс (W" W 2 ' ..., v.... n ).
А
w
КРИТЕРИИ
Руководствуются следующими критериями,
если нет друrих указаний в частных статьях.
Твердые, мяrкие и жидкие дозирован-
ные формы. Требования для OДHOpOДHO
сти дозирования удовлетворяются, если при
емлемое значение первых 1 О дозированных
единиц является меньшим или равным L 1 %.
Если приемлемое значение выше, чем L 1 %,
то исследуют следующие 20 дозированных
е,с,ИНI1Ц и Е\ЫЧИСJ'ЯК-Т flриемлеr"ое :ачение.
512
rосударственная фармакопея Республики Беларусь
Требования удовлетворяются, если t,'1неч.
ное приемлемое значение 30 дозированIы,
единиц является меньшим или равным L 1 %
причем индивидуальное содержание дозиро
ванной единицы должно быть не менее чем
(1 L2'0,01)M и не более чем (1+L2'0,01)M
при расчете приемлемоrо значения по методу
прямоro определения или по расчетно весо
вому методу. Если нет друrих указаний в цaCT
ных статьях, то L 1 равно 15,0, а L2 равно 25,0
01/2013:20941
2.9.41. ИСТИРАЕМОСТЬ rРАНУЛ
И СФЕРОИДОВ
в данном разделе описаны два метода
определения истираемости 2ранул и сферо
идов, которые М02ут быть использованы на
стадии фармацевтической разработки. Кроме
эт020 М02ут быть использованы и мновие
дРУ2ие методы с эквивалентной при20дно
стью.
Данное испытание проводят для опреде
ления истираемости rранул и сфероидов в
определенных условиях. Истираемость опре
деляется как уменьшение массы rранул или
сфероидов или как образование фраrментов
rранул или сфероидов, появляющихся после
их механической деформации при обращении
(удары, вибрация, псевдоожижение и т. д.).
Примерами изменений являются истирание,
разламывание или деформация rранул или
сфероидов.
! 5:1:"; I
Н
I
I
r
"'
5. cт
,
.1
Е: S1
ш.....1
,
i
4.5.:fП
5,"
ё
С!
....1
Рисунок 2.9.41. 1. Прибор с псевдоожиженным
слоем
МЕТОД А
Прибор (прибор с псевдоожиженным
слоем). Прибор (рисунок 2.9.41 1) состоит из
стекпянною цилиндра (А) L конической нижней
частью. Цилиндр оборудован крышкой (В), изro
товленной из сетки с размером отверстий 500 мкм
или иноro подходящеro сита. Конический конец
присоединен к U образной стекпянной трубке (CI,
которая может быть отссединена от ЦИЛ'!Iндра для
очистки от rранул или сфероидов. L.: образная
трубка примыкает к Т 06разному соединению.
Одно входное отверстие Т образноro соединения
присоединяется с помощью силиконовой трубки к
манометру для реryлирования потока сжатою воз
духа (используют сжатый воздух выдерживающий
испытание на воду, как указано в статье Воздух Me
дицинский), второе входное отверстие соединяеl
tя с помощью силиконовой трубки с проточным
расходометром (Е) (0,10 1.00 мЗ/час).
Методика. Обычно используют описанную
ниже методику. Удаляют мелкие частицы с помо
щью просеивания (используют сито с размером Ol
верстия 71 О мкм или иное подходящее сито). По
мещают около 8,0 r (Щ) rранул или сфероидов в
цилиндр (А). Закрывают прибор крышкой (В), изro
товленной >'1з сита. Устанавливают скорость потока
сжатою воздуха 0,45 мЗ/час. Через 15 мин удаля.
ют rранулы или сфероиды из прибора путем OTCO
единеЧ;1Я U образной трубки и снова взвешивают
(т) r.рО80ДЯТ ИСПblтание на трех образцах и pac
считывают среднее значение. Для предотвраще
ния образования электрическою заряда рекомен,
дуется обрабатывать внутренние стенки прибора
антистатиком после каждых трех определений.
Потеря в массе при высушивании. Если 8
частной статье нет друrих указаний, сушат при
температуре 105 ос. В качестве альтернативы
MorYT быть использованы иные условия BЫCY
шивания, как указано в разделе 2.2.32.
Расчеты
F 0= ! (100 T1) т 2 (100 TJ .100,
т 1
rдe:
F истираемость;
Т 1 потеря в массе при высушивании, опре
деленная перед испытанием (среднее значение
из двух испытаний), %;
Т 2 потеря в массе при высушивании,
определенная после испытания (среднее значе
ние из двух испытаний), %;
т 1 масса rранул или сфероидов перед
проведением испытания, r;
т 2 масса rранул или сфероидов после
проведения испытания, r.
МЕТОД В
При бор (осциллирующий при бор).
Прибор (рисунок 2.9.41. 2) состоит из стеклянно
:--0 контейнера вместимостью 105 мл. содержа
2.9.42. Тест «Растворение» для лuпофuльных твердых дозированнЬ1Х форм
513
СlеkJl ННЫЙ
ttОНfейнер
'- BpeMSI
. ЧаСI01!!
i' . . ;'''.
- .......,
"
.! т '
LJ../
- '_:---_ $
.
/
/
/
\
СleкJl ННЫИ 'ЮНlейнер
8Месl....МОСllbю 105 ....11
с вну'ренним диамеlроМ 49.0 мм.
8ЫСОI<Ж 85.0 мм,
с 011ф 'Чl4вающеЙСSll<pblWкоИ
Рисунок 2.9.41. 2. Осциллирующий прибор
щеrо испытуемые rранулы или сфероиды, KOTO
рые подверrаются roризонтальным колебаниям.
Частота и продолжительность колебаний может
быть плавно отреryлирована: частота может
быть установлена в диапазоне O OO колеба
ний/мин, продолжительность может быть YCTa
новлена в диапазоне 0 9999 с.
Методика. Обычно используют описанную
ниже методику. Удаляют мелкие частицы с помо
щью просеивания (используют сито с размером
отверстия З55 мкм или иное подходящее сито).
В стеклянный контейнер помещают около 10,00 r
(т 1 ) rранул или сфероидов и устанавливают еro
в прибор. Встряхивают в течение 240 с при MaK
симальной частоте в случае твердых rранул или
сфероидов или в течение 120 с при более низкой
частоте (например, при 140 колебаний/мин) для
мяrких rранул и сфероидов. Просеивают (исполь
зуют сито С размером отверстия 355 мкм или
иное сито, использованное в начале испытания)
и взвешивают rранулы или сфероиды (mJ Испы
тание проводят с использованием трех образцов
и рассчитывают среднее значение.
Потеря в массе при высушивании. Если в
частной статье нет друrих указаний, сушат при
температуре 105 ос. В качестве альтернативы
MOryT быть использованы иные условия BЫCY
шивания, как указано в разделе 2.2.32.
Расчеты
F = щ(100 T1) т 2 (100 Т 2 ) .100,
т 1
rде:
F истираемость;
Т 1 потеря в массе при высушивании, опре
деленная перед испытанием (среднее значение
из двух испытаний), %;
Т 2 потеря в массе при высушивании,
определенная после испытания (среднее значе
ние из двух испытаний), %;
65 Зак. 1712.
т 1 масса rранул или сфероидов перед
проведением испытания, r;
т 2 масса rранул или сфероидов после
про ведения испытания, r.
01/2013:20942
2.9.42. ТЕСТ «РАСТВОРЕНИЕ» ДЛЯ
ЛИПОФИЛЬНЫХ ТВЕРДЫХ
ДОЗИРОВАННЫХ ФОРМ
ПРИБОР
Прибор (рисунок 2.9.42. 1) состоит из следу
ющих частей:
резервуар для среды растворения;
насос, направляющий среду растворения
вверх через проточную ячейку;
проточная ячейка (рисунок 2.9.42. 2), специ
ально предназначенная для липофильных твер.-
дых дозированных форм, таких как суппозитории
и мяrкие капсулы; она состоит из трех частей, из
roтовленных из прозрачноrо материала, подоrнан
ных друr к друry; нижняя часть (1) выполнена в виде
двух смежных камер, присоединенных к устрой
ству подачи среды растворения; среда pacтвope
ния проходит через камеру А, двиraясь снизу вверх;
в камере В среда растворения движется сверху
вниз к небольшому выходному отверстию, которое
выводит вверх к ситовому узлу; средняя часть (2)
ячейки имеет полость, предназначенную для соби
рания липофильных вспо .юraтельных веществ, KO
торые плавают на поверхности среды растворения;
металлическая сетка служит как rрубый фильтр;
верхняя часть (3) содержит фильтрующий элемент
для крепления бумажною, стеклянною или целлю
лозноro фильтра;
водяная баня, поддерживающая темпера
туру среды растворения при (37:tO,5) ос.
514
rосударственная фармакопея Республики Беларусь
1
.... .... .
' .' =. ....:. ;.
; ::;.ДM:
А
в
Рисунок 2.9.42. 1. Проточный прибор
А резервуар для среды растворения;
В насос;
С термостатируемая проточная ячейка и фильтр;
О собирающие сосуды.
СРЕДА РАСТВОРЕНИЯ
Если среда растворения представляет собой
буферный раствор, доводят еro рН до указанно
ro значения с точностью j:0,05. Из среды paCTBO
рения удаляют растворенные rазы, так как они
MOryт вызывать образование пузырьков, которые
значительно влияют на результат испытания.
МЕТОДИКА
Помещают одну единицу испытуемоro ле
KapcTBeHHoro средства в камеру А и закры
15';
(2)
....
(З)
Капиллярная трубка
I!)
I!) C"J
I!)
00
C"J
,/\
(1)
о ;;;;;;;;;;
о ====
)!!!!!!!!!!!!
t
1====
.....
.....
) ====
'\
) .....
==
')====
');;;;;;;;;;;;;
..
ЗО""
Сетка с иrnон
Рисунок 2.9.42. 2. Проточная ячейка
(размеры указаны в миллиметрах)
. '( :::-::/ bdd :
...... ..
. ;::.;::.:: :::.... :. :....::
.". . ....... .............
:::.: ::.;.......:; ..;.....:: .: :.\:
с
о
вают ячейку приroтовленным фильтрующим
элементом. В начале испытания из камеры
А необходимо удалить воздух через неболь
шое отверстие, соединенное с фильтрующим
элементом. Наrревают среду растворения до
необходимой температуры, принимая во вни
мание температуру плавления лекарственно
ro средства. С помощью подходящеro насоса
наrретую среду растворения вводят через
дно ячейки с заданной скоростью потока
(j:5 %) по открытому или закрытому циклу.
После тоro, как среда растворения достиrнет
края камеры А, она начнет заполнять камеру
В, а воздух будет выталкиваться через капил
ляр. Лекарственное средство распределяется
в среде растворения в соответствии с ero фи
зико химическими свойствами.
В некоторых случаях, при соответствующем
обосновании и соrласовании с уполномоченным
ОрfЭном, испытание может проводиться на репре.-
зентативной фракции суппозитория большоro раз--
мера.
ОТБОР ПРОБ И ОЦЕНКА РЕЗУЛЬТАТОВ
Отбор образцов BcerAa осуществляется на
выходном отверстии ячейки, независимо от тою.
замкнут ли цикл или открыт.
Фильтруют извлеченную жидкость через
фильтр из инертноro материала с подходящим
размером пор, который не вызывает значи
тельной адсорбции действующею вещества из
раствора и не содержит веществ, вымываемых
средой растворения и ВЛl.1яющих на результаты,
получаемые указанным методом. Полученный
фильтрат анализируют.
Количество активноro вещества, растворив
шеюся за указанное время, выражают в процен
тах от количества, указанноro на этикетке лекар
ственноro средства.
2.9.43. Наблюдаемое растворение
01/2013:20943
515
2.9.43. НАБЛЮДАЕМОЕ
РАСТВОРЕНИЕ
Данный метод используется для определе
ния скорости наблюдаемоro растворения чистых
твердых веществ. Также он может быть исполь
зован для определения скорости наблюда
емоro растворения действующих веществ в ле
карственных средствах в форме порошков или
rpанул.
ПРИБОР
Все части прибора, которые MOryт KOHTaK
тировать с образцом или средой растворения,
должны быть химически инертными и не должны
адсорбировать, реаrировать или влиять каким
либо друrим способом на образец. Не должно
быть никаких заметных движений, колебаний
или вибраций, происходящих от частей прибора
или окружающей среды, кроме тех, которые соз
даются проточной системой.
Предпочтительно использовать прибор, по
зволяющий наблюдать за образцом.
Прибор (рисунок 2.9.4З. 1) состоит из следу
ющих частей:
резервуар для среды растворения;
насос, направляющий среду растворения
вверх через проточную ячейку;
проточная ячейка, желательно из прозрач
ноro материала, установленная вертикально
с фильтрующей системой, предотвращающей
удаление нерастворившихся частиц;
водяная баня, поддерживающая выбран
ную температуру среды растворения (обычно
(з7:tО,5)ОС).
Проточная ячейка (рисунок 2.9.43. 2) co
стоит из трех частей, подоrнанных друr к друry.
Нижняя часть поддерживает систему сеток и
фильтров, на которые помещают порошок. Cpeд
I
1
. ;. . :' . :. ;
{. иiN:{
.......,...,....
А
в
Рисунок 2.9.4З. 1. Проточный прибор
А резервуар для среды растворения:
В насос;
С термостатируемая проточная ячейка и фильтр;
D собирающие сосуды.
няя часть, которая устанавливается на нижнюю
часть, содержит вкладыш, который просеивает
образец во время прохождения среды paCTBope
ния через ячейку. Этот вкладыш состоит из двух
частей: коническоro сита, которое размещает
ся на образце, и зажима, расположенноro на по
лови не расстояния до средней части, для yдep
живания сита на месте при прохождении среды
растворения. Второй фильтрующий элемент
(сетка и фильтр) расположен на вершине cpeд
ней части перед соединением с верхней частью,
через которую среда растворения выливается из
ячейки.
СРЕДА РАСТВОРЕНИЯ
Если среда растворения представляет
собой буферный раствор, доводят еro рН до YKa
занноro значения с точностью :t0,05. Из среды
растворения удаляют растворенные rазы, так
как они MoryT вызывать образование пузырьков,
которые значительно влияют на результат испы
тания.
МЕТОДИКА
На дно конической нижней части помеща
ют шарик диаметром (5:t0,5) мм, затем стеклян
ные шарики подходящеro размера, желательно
диаметром (1:t0,1) мм. Помещают сито (с раз
мером отверстий 0,2 мм), подходящий фильтр
и второе сито наверх нижней части. Собран
ный элемент прибора взвешивают. К нижней
части присоединяют среднюю часть прибора.
Испытуемый образец помещают на фильтру
ющий элемент и взвешивают образец в ячейке.
На испытуемый образец помещают сито вкла
дыша коническим концом вверх и закрепляют
ниже середины средней части. Наверх средней
части помещают сито (с размером отверстий
0,2 мм) и подходящий фильтр. Присоединяют
верхнюю часть. Наrревают среду растворения
до выбранной температуры. Наrретую среду
. ::':_::?': {: :
. :.:: '. :.': ;::' '::".::
. - ". .", ...... ","
-.:.<:.;... . :..._ ....:..<:-;
"0 о.".. . "0 о'"
с
о
516
rосударственная фармакопея Республики Беларусь
01/2013:20944
1"-
Ф
....
F
cil
Е
t:IJ
I!)
с
I"J
М
N
B
,
;]
0.8
Рисунок 2.9.43. 2. Проточная ячейка
(размеры указаны в миллиметрах)
А нижняя часть;
В сито;
С зажим;
D вкладыш;
Е средняя часть;
F верхняя часть.
растворения при помощи подходящеro насоса
направляют через дно ячейки с заданной CKO
ростью потока (:!:5 %) по открытому или закры
тому циклу.
ОТБОР ПРОБ
Отбор образцов всеrда осуществляется на
выходном отверстии ячейки, независимо от тоro,
замкнут ли цикл или открыт.
Немедленно фильтруют извлеченную
жидкость через фильтр из инертноro матери
ала с подходящим размером пор, который не
вызывает значительной адсорбции действу
ющеrо вещества из раствора и не содержит
веществ, вымываемых средой растворения
и влияющих на результаты, получаемые YKa
занным методом. Полученный фильтрат aHa
лизируют.
OO
00 =====
о !!!!!!!!
""
I =====
1'- ......
О ......
11'\
О ......
Z
сп ====
сп ;;;;;;;;;;;;
.....
ОЦЕНКА РЕЗУЛЬТАТОВ
При проведении испытания при выпуске
серии проводят достаточное количество повто
ров.
Результаты выражают следующим образом:
количество растворившеrося вещества в
единицу времени (если растворение линейно);
время растворения Bcero образца и на
подходящих промежуточных стадиях.
2.9.44. ЛЕКАРСТВЕННЫЕ СРЕДСТВА
ДЛЯ РАСПЫЛЕНИЯ:
ХАРАКТЕРИСТИКА
Характеристики лекарственных средств
для распыления, предназначенных для до--
ставки их в леrкие, определяются следующи
ми испытаниями:
скорость высвобождения действующеro
вещества и общее количество высвобожденнcr
ro вещества;
аэродинамическая оценка распыленных
аэрозолей.
Эти испытания стандартизируют под
ходы по оценке доз, которые будут получ
ны пациентом, но они не предназначены для
оценки caMoro приспособления для распыле
ния, которое описано в европейском стандарте
EN 1З544 1:2007+А1:2009, Оборудование для
респираторной терапии Часть 1: Системы для
распыления и их системы.
Массовое распределение более предпочти
тельно для оценки характеристик лекарствен
ноro средства, чем числовое распределение.
Кроме Toro, масса действующеro вещества как
функция аэродинамическоro диаметра лучше
описывает терапевтический эффект в дыхатель
ных путях.
СКОРОСТЬ ВЫСВОБОЖДЕНИЯ
ДЕЙСТВУЮЩЕrо ВЕЩЕСТВА И ОБЩЕЕ
КОЛИЧЕСТВО ВЫСВОБОЖДЕнноrо
ВЕЩЕСТВА
Испытания проводят для оценки скорости, с
которой пациент будет получать дозу, и общеro
количества действующеro вещества, получен
ноro пациентом, используя стандартные усло
вия для объемной скорости потока. Распылит
ли, инициализируемые дыханием и зависящие
от неro, должны проверяться с использованием
симулятора дыхания, так как выпуск дозы сильно
зависит от скорости вдоха. Приведенная ниже
методолоrия описывает использование CTaHдapT
ной модели дыхания взрослоro человека. Если
для конкретноro лекарственноro средства YKa
зано, что оно предназначено только для исполь
зования в педиатрии (т. е. для новорожденных,
младенцев или детей), должна быть использо
вана модель дыхания ребенка. Для проведения
соответствующих измерений массы действующе
ro вещества, получаемой пациентом, предпочти
тельнее использовать модель дыхания, чем ис
пытания при пос-юянной скорости потока.
Скорость высвобождения действующеro Be
щества и общее количество высвобожденно
ro вещества являются подходящими характери-
стиками, позволяющими описать в стандартных
условиях вне зависимости от используемоro
распылителя высвобождаемую массу. Таким 06--
разом, методолоrия испытания, приведенная
2.9.44. Лекарственные средства для раСПblления: характеристика
517
Таблица 2.9.44. 1
Спецификации сuмулятора дыхания
Элемент Спецификация
Взрослый Новорожденный Младенец Ребенок
Дыхательный 500 мл 25мл 50мл 155 мл
объем
Частота 15 циклов/мин 40 циклов/мин 30 цикловlмин 25 циклов/мин
Форма волны синусоидальная синусоидальная синусоидальная синусоидальная
Соотношение 1:1 1:З 1:3 1:2
вдох/выдох
ниже, позволяет измерить массу действующеro
вещества, высвобождаемою в первый период
времени (обычно за 1 мин) (оценка скорости 8Ы
свобождения действующеro вещества), а также
позволяет найти общую массу высвобождаемо
ro действующеro вещества.
ПРИБОР
Симулятор дыхания. Для испытания ис
пользуют коммерчески доступный симулятор
дыхания, приведенный на рисунке 2.9.44. 1, спо
собный воспроизводить модели дыхания. Ис
пользуют модель дыхания взрослоro челове
ка за исключением случаев, коrда указано, что
лекарственное средство предназначено для ис
пользования в педиатрии, в этом случае исполь
зуют альтернативные модели дыхания, как YKa
зано в таблице 2.9.44. 1.
Фильтрующая система. Для испытания ис
пользуют подходящий валидированный фильтр
с низким сопротивлением, способный количе
ственно удерживать аэрозоль и позволяющий
смывать подходящим растворителем действу
ющее вещество. Мертвый объем корпуса филь
тра не должен превышать 1 О % дыхательноro
объема симулятора дыхания.
МЕТОДИКА
Присоединяют фильтр (расположенный
в держателе фильтра) (А) к симулятору дыxa
ния (В) в соответствии с рисунком 2.9.44. 1. Рас"'"
пылитель (С) заполняют указанным в инструк
ции по применению объемом лекарственноro
средства. При необходимости, используя адап
тер мундштука, к фильтру вдоха rерметично при
соединяют мундштук распылителя. Необходимо
удостовериться, что распылитель ориентирован
с [J${J!
F
А В
Рисунок 2.9.44. 1. Экспериментальная ycтa
новка для испытания с симулятором дыхания
А фильтр вдоха и держатель фильтра;
В симулятор дыхания;
С распылитель.
66. Зак. 1712.
надлежащим для использования образом; это
может потребовать наклона симулятора дыxa
ния и держателя фильтра. Симулятор дыхания
настраивают на со ание специсрицированной
модели дыхания.
Запускают симулятор дыхания, затем в
начале дыхательноro цикла запускают распы
литель и позволяют ему работать в течение
определенноrо начальноro времени. Выбран
ное время, обычно (60:1:1) с, должно обеспечить
попадание на фильтр вдоха достаточноro для
определения содержания количества действу
ющеro вещества. Если количество вещества, по
павшеro на фильтр за 60 с, недостаточно для
проведения анализа, продолжительность Bpe
менноro интервала для сбора аэрозоля может
быть увеличена. Если фильтр забивается ле
карственным средством, это время может быть
уменьшено. В конце этоro начальноro времени
останавливают инrалятор.
Меняют фильтр и держатель фильтра на
свежие и продолжают распыление до полноro
ero прекращения. Во избежание перенасыщения
фильтра, при необходимости, распыление пре
рывают и меняют фильтр.
РЕЗУЛЬТАТЫ
С помощью подходящеro метода анали
за определяют массу действующеro вещества,
собранноro в каждый промежуток времени на
фильтре и держателе фильтра. Находят CKO
рость высвобождения действующею вещества
путем деления массы действующеro вещества,
собранноro на первый фильтр вдоха, на время,
в течение котороro осуществлялся еro сбор. Ha
ходят общую массу полученноro действующе
ro вещества суммируя массу действующею Be
щества, собранноro на фильтрах и держателях
фильтра.
АЭРОДИНАМИЧЕСКАЯ ОЦЕНКА
РАСПЫЛЕННЫХ АЭРОЗОЛЕЙ
Размер чаr.тиц (капель) лекарственных
средств для распыления должен быть опре
делен при более низких скоростях потока, чем
обычно используемых для порошковых инrа
ляторов и дозированных инrаляторов. EBpO
пейским стандартом рекомендована скорость
15 л/мин, так как это значение представляет xo
518
rосударственная фармакопея Республики БеларуCJj,
..;
рошую аппроксимацию средней скорости дыxa
ния здоровоro взрослою человека (дыхатель
ный объем 500 мл).
Несмотря на то, что приборы лазерной
дифракции с малым уrлом (лазерные дифрак
тометры) MOryT обеспечить быстрое измерение
распределения по размеру частиц аэрозолей,
образуемых распылителем, эти способы не по
зволяют определить содержание действующеro
вещества; они измеряют распределение по раз
меру капель вне зависимости от их состава. Это
не является проблемой в случае roMoreHHbIx
растворов, но может приводить к значитель
ным ошибкам, если лекарственное средство,
предназначенное для распыления, представ
ляет собой суспензию, или если происходит, в
случае некоторых распылителей, сильное ис
парение капель. Каскадные импакторы позво
ляют однозначно охарактеризовать аэрозоль
по массе действующеro вещества как функции
аэродинамическоro диаметра. Лазерная диф
ракция может быть использована в случае Ba
лидации по сравнению с методом, использую
щим каскадный импактор.
Для проведения данноro испытания
прибор Е (см. ниже в разделе Прибор), пред
ставляющий собой каскадный импактор, спе
циально калибруют на скорость 15 л/мин чтобы
удовлетворять требованиям европейскоro CTaH
дарта. Определение баланса масс таким же об
разом, как для порошковых инrаляторов и дози
рованных инrаляторов, не является прямым по
причине Toro, что доза захватывается в течение
продолжительноro выпуска, и поэтому не вклю
чено. При разработке метода для еro валида
ции должны быть проведены исследования по
оценке открываемости.
Во избежание ошибок в процессе измерения
размера капли важно контролировать испаре
ние образуемых распылителем капель. Испаре
ние может быть минимизировано путем охлаж
дения импактора до температуры около 5 ОС,
обычно достиrаемой при охлаждении импактора
в холодильнике в течение 90 мин. Из за BЫCO
коro риска коррозии, вызываемой конденсаци
ей/аккумулированием солесодержащих капель
на межкаскадных металлических конструкци
ях и охлаждением импактора, прибор должен
быть полностью очищен, включая межкаскад
ные каналы, обычно не реже чем после каждо
ro дня использования. После каждоro испытания
все поверхности прибора рекомендуется BЫCY
шивать, например, сжатым воздухом. Примеча
ние: сжатым воздухом нельзя сушить коллектор
с микроотверстиями (МОС).
ПРИБОР
Детальное описание Прибора Е и порта
индукции содержится в общей статье 2.9.18 и
включает подробные критические размеры и
процесс квалификации импактора (замеры CTY
пеней).
Для обеспечения полной открваемости дей-!
ствующеrо вещества из распыленноrо аэро;ю...
ля при специфицированной скорости потока.!
равной 15 л/мин, в добавление к коллектору ci
микроотверстиями (МОС) должен использоваn.-;
ся вспомоrательный фильтр. Он может распола- I
rаться под МОС (опция внутреннеro фильтра)'
или во внешнем держателе фильтра и служит,
для захвата мелких капель, вышедших за по-
Cflеднюю фракционирующую ступень.
В испытании аэрозолей, полученных с по-
мощью распылителя, пре сепаратор не иСПОЛir
зуют.
ВАЛИДАЦИЯ МЕТОДА
Переrрузка ступени импактора. При pa
работке и валидации метода важно доказат
что объем распыляемой ЖИДКОСТИ не переrру
жает импактор. При визуальном обследов
нии собирающих поверхностей ступеней, на ко-
торых скапливается наибольшее количество
капель, можно обнаружить образование поло-
сок, что свидетельствует о переrрузке. Это яв
ление также обычно связано с увеличением
массы действующеro вещества, собранноro на
последней ступени и на вспомоrательном фИЛir
тре. Наиболее эффективным способом изб
жать переrрузки для любой системы является
уменьшение времени распыления образца (ТО),
соrласовывая переrрузку и аналитическую чув
ствительность.
Обратный захват. Отскакивание и обрат
ный захват в случае капель, образующихся при
распылении, менее вероятны, чем в случае с
твердыми частицами из инrаляторов, поэтому
покрытие обычно не требуется.
МЕТОДИКА
Собранный импактор и порт индукции ox
лаждают в холодильнике (установленном на
температуру около 5 ОС) в течение не менее
90 мин и начинают определение в течение 5 мин
после извлечения импактора из холодильника.
Moryт использоваться друrие валидированные
способы поддержания постоянной температуры
импактора (например, холодовая комната).
Устанавливают распылитель с подведен
ным rазом (обычно воздух или кислород) или ис
пользуют компрессор, устанавливают указанные
производителем распылителя давление и CKO
рость потока. Следят за тем, чтобы линия подво
да rаза не отсоединялась во время нахождения
распылителя под давлением. В распылитель по
мещают указанное в инструкции по применению
количество лекарственною средства.
Достают импактор из холодильника, присо-
единяют порт индукции, к выходу импактора/внеw-
неro фильтра присоединяют источник вакуума,
способный поддерживать поток raза через импак
тор со скоростью 15 л/мин, как указано на рисунке
2.9.44. 2. Включают поток через импактор.
2.9.44. Лекарственные средства для распыления: характеристика
519
!
А
,
с
,
D
,
Е
Значение F представляют относительно
т,сотр
расположения в измерительном оборудовании,
начиная с порта индукции и заканчивая вспо
моrательным фильтром импактора (см. рисунок
2.9.44. З). При необходимости показать массу
фракции, собранной на rруппе стадий, как одно
значение, значения F для соседних ст у пе
т,сотр
ней MOryт быть объединены.
Рисунок 2.9.44. 2. Прибор Е для определения 04
распределения по размеру частиц лекарствен
ных средств для распыления
А Распылитель;
В Порт индукции;
С Импактор (прибор Е);
О Контрольный клапан;
Е Источник вакуума
к порту индукции подсоединяют объем
ный счетчик, откалиброванный для измерения
выходноro потока. Реryли руют контрольный
клапан, расположенный между импактором и ис
точником вакуума таким образом, чтобы обеспе
чить стабильный поток со скоростью 15 л/мин
(:t5 %). Отсоединяют объемный счетчик.
Убеждаются в том, что распылитель располо
жен необходимым для использования образом, и
rерметично присоединяют мундштук распылите
ля к порту индукции, используя адаптер мундшту
ка при необходимости. Включают подачу raза или
компрессор для распылителя и проводят распы
ление в течение предварительно установленноro
времени (ТО), Будучи установленным, это время
(ТО) должно быть оroворено и использоваться в
методике на данное лекарственное средство для
тоro, чтобы была возможность сравнивать массы
фракций. После распыления выключают подачу
rаза или компрессор для распылителя, отсоеди
няют распылитель от порта индукции и отключа
ют вакуум от импактора.
Разбирают импактор и, используя подходя
щий метод анализа, определяют массу действу
ющеro вещества, собранную в порте индукции,
на каждой из ступеней и на вспомоrательном/
внешнем фильтре, как указано для Прибора Е
в общей статье 2.9.18. Массу действующеro Be
щества, собранную в МОС, суммируют с массой,
собранной на вспомоrательном/внешнем филь
тре, и рассматривают это как единый образец
для последующих расчетов.
Рассчитывают массу фракции ( F ) дей
т,соmр
ствующеro вещества, собранноro на каждом
компоненте импактора, начиная с порта индук
ции и далее для всеro прибора, используя сле
дующее выражение:
F = теотр ,
щеотр М
rдe:
теотр масса, относящаяся к рассматрива
емому компоненту;
М общая масса, собранная системой.
03
tO.2
ц-
:
,.
"
) ,;
'';!
01
п
«) (D 1'-- U
.l) .l) .tI О
:х: :х: :х: :2;
Q) ф Q)
с: с: с: Q.
>- >- >- о--
о-- о-- о-- .tI
() U U с:
'"
е
00
'"
,.
::r
><
>-
<:[
:х:
'"
о--
Q.
О
t:
.l)
:r
Ф
с:
>-
о--
U
Рисунок 2.9.44. З. Пример масс фракций
капель, представленных в виде распределения
в приборе
Определяют кумулятивное массо оценоч
ное распределение по размеру частиц фрак
ционированноro с помощью импактора аэрозо
ля в соответствии с методикой, приведенной в
общей статье 2.9.18. Начиная с фильтра, диф
ференцируют кумулятивную массу по эффек
тивному предельному диаметру COOTBeTCTBY
ющей ступени (см. таблицу 2.9.44. 2 подходя
щих диаметров при скорости 15 л/мин). Строят
зависимость кумулятивной фракции от пре
дельноro диаметра в подходящем формате,
например, лоrарифмическом или лоrарифми
чески вероятносном. При необходимости с по
мощью интерполяции определяют или фрак
цию размером менее данноro размера; или
фракцию с размером между верхним и нижним
пределами.
Таблица 2.9.44. 2
Предельные размеры для Прибора Е
при скорости потока 15 л/мин
Ступень Предельный диаметр (мкм)
1 14,1
2 8,61
3 5,39
4 3,30
5 2,08
6 1,36
7 0,98
520
rосударственная фармакопея Республики Беларуа.
При необходимости и rде это применимо
определяют значения средней массы аэродина
мическоrо диаметра (СМАД) и стандартноro reo
метрическоro отклонения (CrO).
01/2013:20945
2.9.45. СМАЧИВАЕМОСТЬ ПОРИСТЫХ
ТВЕРДЫХ МАТЕРИАЛОВ,
ВКЛЮЧАЯ ПОРОШКИ
ВВЕДЕНИЕ
Смачиваемость твердых поверхностей в
общем случае характеризуется с помощью пря
моro или непрямоro определения краевоro уrла.
Краевым уrлом (8) между жидкостью и твердым
телом называется уroл, образующийся при поме
щении капли жидкости на твердую поверхность
(рисунок 2.9.45. 1). При рассмотрении KOHKpeT
ной жидкости в случае смачиваемых твердых
материалов краевой уroл мал, а в случае HeCMa
чиваемых твердых материалов краевой уroл co
ставляет 900 и более.
Рисунок 2.9.45. 1. Краевой У20Л (е) неподвиж
ной капли, наблюдаемой на непористой
поверхности
Ниже описаны два метода определения
смачиваемости. Эти методы приrодны при испы
тании пористых твердых тел, таких как порошки
или rранулы. Оба метода отображают смачива
емость с помощью KpaeBoro уrла между пори
стым материалом и выбранной жидкостью.
Метод неподвижной капли основан на
прямом измерении краевоro уrла неподвижной
капли на порошке, спрессованном в диск.
В методе Уошборна (Washburп) проводят
непрямое измерение краевоro уrла. Метод oc
нован на капиллярном эффекте пор порош
ка. Эффект (увеличение массы) записывается
специальными электронными весами, начиная
с момента касания испытуемым образцом по
верхности жидкости, которая не растворяет
или практически не растворяет ero. Измерение
не влияет или очень мало влияет на структуру
порошка.
Любая предварительная обработка испы-
TyeMoro образца нежелательна, так как Пpll
этом ero свойства MorYT быть в значитель-
ной степени изменены. Например, прессовcr
ние порошка в виде диска может уменьшать
свободную энерrию поверхности при измен
нии кристаллическоro состояния порошка (Ha
пример, метастабильные формы) или увел
чивать свободную энерrию поверхности из за
образования кристаллических дефектов (H
достаток метода неподвижной капли, так как
испытание про водится на порошке, спрессо--
ванном в виде диска).
Данные методы обычно применяются для
исследования следующих параметров:
постоянство образцов относительно CMcr
чиваемости от серии к серии;
эффект влияния вязкости жидкости на
смачиваемость;
эффект влияния поверхностноro натяж&
ния жидкости на смачиваемость;
изменение поверхностных свойств образ
цов.
МЕТОД НЕПОДВИЖНОЙ КАПЛИ
Этот метод может быть использован для
непосредственноrо определения смачива
емости покрытий и плотных лекарственных
форм, таких как таблетки. Кроме тоro, метод
неподвижной капли иноrда используется для
динамических измерений (динамическое из
мерение KpaeBoro уrла, рисунок 2.9.45. 2) в си
стеме пористый твердый материалlжидкость,
коrда происходит уменьшение краевоro уrла.
Скорость процесса растекания капли, сопро
вождающеrося проникновением в малопори
сто е твердое тело, может быть изучена путем
проведения нескольких измерений изменения
краевоro уrла во времени.
rаз
Жидкость
rS
YSl
Твердое вещество
Рисунок 2.9.45. 2. Метод неподвижной капли
с визуальным наблюдением за каплей
2.9.45. Смачиваемость пористых твердых материалов, включая
521
в равновесных условиях краевой уrол He
подвижной капли зависит от трех взаимосвязан
ных поверхностных натяжений и определяется
по уравнению Юнrа (Young) (рисунок 2.9.45. 2,
первая часть):
Ys =YSL +YL .cose,
rдe:
Y s поверхностное натяжение твердоro Be
щества на rранице с воздухом;
YSL натяжение твердоro вещества на rpa
нице с жидкостью;
Y L поверхностное натяжение жидкости на
rранице с воздухом.
МЕТОДИКА
Так как порошки не MOryт сформировать аб >
солютно плоскую поверхность, их обычно прес
суют, получая диски с более rладкой поверх
ностью. Каплю жидкости заданноro объема
помещают на поверхность (рисунок 2.9.45. 2),
измеряя краевой уroл с помощью rониометра,
оборудованноro окуляром с уrломером, или с
помощью rеометрическоrо построения на микро
фотоrрафии. Moryт быть использованы и иные
физические или математические способы об
работки данных. На результат может влиять ис
пользуемый объем капли. Обычно проводят He
сколько определений краевоro уrла (8) (п = 6) и
рассчитывают среднее значение.
МЕТОД УОШБОРНА
С помощью метода Уошборна возможно из
мерять краевые уrлы пористых твердых тел, ле
жащих в диапазоне от 00 до 900.
Испытанию подверrается комбинация из об
разца, держателя и фильтрующей системы. По
этой причине установление или определение
истинною значения невозможно определя
ют только кажущийся краевой уюл. Однако Kpa
евой уroл образца является функциональным
свойством, от которою в значительной степени
зависят получаемые результаты. В результате
испытания получают ранжирование различных
веществ или их смесей по смачиваемости, KOTO
рая характеризуется кажущимся краевым уrлом.
ПРИНЦИП
Если пористый твердый материал приводят
в контакт с жидкостью таким образом, что сам
материал не поrружается в жидкость, а только
касается ее поверхности, Torдa подъем жидко
сти по порам вследствие капиллярности будет
описываться следующим уравнением:
т2= ,(1)
А
rдe:
т масса жидкости, всосавшейся в TBep
дый материал;
67 За.. 1712
t время, прошедшее с момента контакта
твердоro материала и жидкости;
А константа, зависящая от свойств жид
кости и испытуемоro твердоro материала, pac
считанная по следующему уравнению:
А = 1] , (2)
c.p2.y.cose
rде:
fJ вязкость жидкости;
р плотность жидкости;
у поверхностное натяжение жидкости;
е краевой уrол между твердым материа
лом и жидкостью;
с постоянная вещества, зависящая от
структуры пор твердоro материала.
Из уравнений (1) и (2) следует уравнение (3):
т 2
cose= . 1]2 . (3)
t с'р'у
в методе Уошборна используют жидкость
с известными плотностью (р), вязкостью (7])
и поверхностным натяжением (у). При усло
вии, что масса жидкости, поднимающейся
по порам твердоro тела, отслеживается во
времени (таким образом, скорость проник
новения (m}fJ является экспериментальной
величиной), в уравнении (3) остается две He
известных: краевой уroл (О) жидкости на TBep
дом материале и постоянная вещества TBep
доro тела (с).
Определение постоянной вещества (с).
При условии цилиндричности пор, постоянную
вещества для пористоro твердоro материала
определяют по следующему уравнению:
2 2 N 2
П . (. , (4)
2
rде:
r средний радиус капилляра твердоro Ma
териала;
N число капилляров в единице объема.
При проведении метода Уошборна с ис
пользованием жидкости, краевой уrол KOTO
рой на твердом теле равен 00 (cosO° = 1), в
уравнении (3) остается только одна неизвест
ная постоянная вещества (с), и она может
быть рассчитана. Жидкостью выбора при
определении постоянной вещества является
н rептан, блаrодаря низкому поверхностно
му натяжению (20,14 мН/м при 25 ОС). Также
может исrользоваться H reKcaH (18,43 мН/м
при 25 ОС), но он более летучий. Если поро
шок растворяется в этих жидкостях слишком
быстро, может быть использован rексаметил
дисилоксан (15,9 мН/м при 25 ОС). Проводят
несколько определений (п = 6) и рассчитыва
ют среднее значение.
522 rосударственная фармакопея Республики Беларусь
После тоro, как постоянная вещества (с) Таблица 2.9.45. 1
для испытуемоrо твердою тела была YCTaHOB Технические параметры электронных весов.
лена, образец твердоro тела может быть ис Подъем Измерение
следован с использованием иной жидкости. По массы
стоянная вещества, определенная с помощью Диапазон > 110 мм 0 210 r ;
I
н rептана, используется в уравнении Уошборна Разрешение 0,1 мкм 1 О MKr
вместе со значением скорости проникновения Скорость 0,09 500 мм/
(т7(), найденным с использованием указанной мин
жидкости. Это позволяет рассчитать значение
краевоro уrла.
ПРИМЕЧАНИЕ: если в испытании даНН020
тверд020 тела используется несколько жидко
стей (не менее двух в дополнение к жидкости,
используемой для определения постоянной вe
щесmва), полученные в результате данные по
краевому У2ЛУ М02ут быть использованы для
расчета поверхностной энеР2ии порист020
тверд020 тела.
ПРИБОР
На рисунке 2.9.45. 3 представлены oc
новные компоненты прибора. Основным при
способлением являются электронные весы с
подходящим процессором, обеспечивающим
подходящее разрешение при измерении силы и
подходящее разрешение при поднятии иммер
сионной жидкости К образцу.
В таблице 2.9.45. 1 приведены параметры
подходящих электронных весов.
А
в
tJ
E
F
Рисунок 2.9.45. 3. Прибор для измерения
краев020 У2ла методом Уошборна
А электронные весы;
В компьютер;
С держатель образцов;
О фильтр;
Е иммерсионная жидкость;
F подъемник.
Держатель образцов. В качестве держа
теля образцов может быть использован He
большой цилиндр со стеклянным фильтром с
одноro конца.
Держатель образцов (рисунок 2.9.45А)
может быть изrотовлен из алюминия; он
менее хрупок, чем стеклянный, и имеет Ma
ленькие отверстия на дне, что позволяет чи
стить ero леrче, чем стеклянный фильтр. На
крышке ячейки присутствуют две винтовые
резьбы. Одна соединяет крышку с камерой
для образца, в то время как вторая позволяет
оператору двиrать поршень вниз, сжимая об
разец. За исключением держателя образца,
весь прибор похож на автоматический тензи
ометр.
A
с
D
Е
Вид поршня снизу
Вид прибора снизу
Рисунок 2.9.45А. Пример держателя образцов
с поршнем для прессования .
А крепление;
В крышка;
С резьба;
О поршень;
Е капиллярные отверстия;
F капиллярные отверстия
2.9.45. Смачиваемость порuстых твердых материалов
523
МЕТОДИКА
Наполнение держателя образцов. Для
предотвращения высыпания порошка со дна
ячейки на дно алюминиевоro или стеклянною
держателя помещают диск из фильтровальной
бумаrи. Фильтр не обязательно должен быть из
roтовлен из бумаrи, однако, он должен быть из
материала, который леrко смачивается жидко
стью, используемой в испытании. PeKOMeHДY
ется использовать фильтр с черной лентой (ис
пользуемый для обратноro осмоса) из за еro
высокой пористости и минимальноro сопротив
ления течению.
В ячейку помещают известное количество
порошка. Воспроизводимость при определении
постоянной вещества и краевоro уrла будет за
висеть от возможности взвешивать одинаковые
количества порошка для каждою определения,
чтобы порошок прессовался в идентичных yc
ловиях.
Для большинства порошков подходящим KO
личеством являются несколько rpaMMoB, обычно
заполняющих 2/3 объема держателя. Для пре
дотвращения поднятия порошка наверх при
прессовании и проведении испытания сквозь OT
верстия в поршне, на образец в ячейку помеща
ют второй кусочек ФИЛЬТРОВ6ЛЬНОЙ бумаrи.
Уплотнение/прессование порошка.
Обычно слой порошка очень порист и поэто
му чувствителен к небольшим воздействиям,
способным леrко изменить пористость и, COOT
ветственно, постоянную вещества (с). По этой
причине уплотненный порошок будет иметь пре
имущество и показывать более воспроизводи
мые результаты. Вначале устанавливают He
обходи мое количество встряхиваний: обычно
достаточно 50 100 встряхиваний.
При использовании алюминиевою держа
теля образцов, он может быть установлен в ци
линдр волюметра, который способен произвести
необходимое количество встряхиваний.
Если встряхивание не может быть исполь
эовано, слой порошка прессуют с помощью
rюршня в алюминиевом держателе, прилаrая
установленное давление.
Кроме этоro, можно использовать центрифу
rирование при определенных условиях. Иноrда
спрессованный диск из образца порошка может
быть установлен на весах без использования
держателя.
После присоединения весов, держатель
вместе с образцом помещают непосредственно
над поверхностью жидкости (рисунок 2.9.45. 3),
используя подъемник.
Затем жидкость поднимают до тех пор,
пока она не коснется дна пористоrо образца.
После этоro, по мере тоro, как жидкость подни
мается по образцу, отмечают изменение массы
с течением времени. Данные MorYT быть пред
ставлены в виде rрафика или таблицы. Прибор
способен проводить все определение aBTOMa
тически.
ВАЖНЫЕ УСЛОВИЯ
Должны быть рассмотрены следующие MO
менты.
Свойства образца:
содержание воды в образце;
кристаллические свойства или свойства
твердой фазы образца (полиморфные формы,
тип сольвата).
При20товление образца:
roMoreHHocTb всех испытуемых смесей по
рошков;
распределение по размеру частиц; перед
испытанием иноrда желательно просеять об
разец (например, через сито с отверстиями
250 мкм);
оптимальные условия прессования (коли
чество образца, число встряхиваний или масса
поршня);
спрессованное состояние различных об
разцов порошка должно быть одинаковым;
держатель образца или, если использует
ся, стеклянный фильтр должен быть тщательно
очищен;
однородность результатов улучшается при
использовании алюминиевоro держателя.
Иммерсионная жидкость:
должна быть приведена спецификация на
используемую иммерсионную жидкость.
3.1.1.1. Материалы на основе пластифuцироваННО20 поливинилхлорида
525
з.
КОНТЕЙНЕРЫ
01/2013:30100
3.1.
МАТЕРИАЛЫ,
ИСПОЛЬЗУЕМЫЕ
ДЛЯ ПРОИЗВОДСТВА
КОНТЕЙНЕРОВ
Материалы, описанные ниже, применяются
для производства контейнеров для фармацев
тическоrо использования. Кроме этоro, такие Ma
териалы MOryт при меняться при производстве
отдельных частей либо целых предметов для
медико хирурrическоrо использования.
Материалы и полимеры, не описанные в
Фармакопее, MOryт использоваться только после
их утверждения в каждом конкретном случае
компетентным уполномоченным opraHoM.
Инфекционные ryбчатые энцефалопатии
(5.2.8). Необходимо проводить оценку рисков
в отношении инфекционных ryбчатых энцефа
лопатий для продукта, а также предпринимать
меры по минимизации такоro риска.
01/2013:30101
3.1.1. МАТЕРИАЛЫ,
ИСПОЛЬЗУЕМЫЕ
ДЛЯ П ОИЗВОДСТВА
КОНТЕИНЕРОВ
ДЛЯ ЧЕЛОВЕЧЕСКОЙ КРОВИ
И КОМПОНЕНТОВ КРОВИ
ПРИМЕЧАНИЕ: о материалах на основе
пластифицироваНН020 поливинилхлорида, иc
пользуемых при производстве контейнеров
для водных растворов для внутривеНН020 ввe
дения, см. статью 3.1.14.
Пластиковые контейнеры, используемые
для сбора, хранения, обработки, введения крови
и ее компонентов, MOryт быть изroтовлены из
одноro или нескольких полимероl3 с использова
нием добавок, если это необходимо.
Если контейнер или часть еro изroтовлена
из материалов, описанных в статье Фармакопеи,
то качество этих материалов контролируется Me
тодами, описанными в соответствующих статьях
(см. статью 3.1.1.1. Материалы на основе пла
БS За" 1712
стифицироваНН020 поливинилхлорида, исполь
зуемые для производства контейнеров для че
ловеческой крови и ее компонентов).
В обычных условиях разрешается использо
вание материалов и контейнеров из этих MaTe
риалов, не выделяющих мономеров или друrих
веществ, вредно воздействующих или вызыва
ющих аномальные изменения крови или компо
нентов крови.
01/2013:90001
3.1.1.1. МАТЕРИАЛЫ НА ОСНОВЕ
ПЛАСТИФИЦИРОВАнноrо
ПОЛИВИНИЛХЛОРИДА,
ИСПОЛЬЗУЕМЫЕ
ДЛЯ П ОИЗВОДСТВА
КОНТЕИНЕРОВ
ДЛЯ ЧЕЛОВЕЧЕСКОЙ КРОВИ
И ЕЕ КОМПОНЕНТОВ
ОПРЕДЕЛЕНИЕ
Материалы на основе пластифицированно
ro поливинилхлорида представляют собой BЫCO
комолекулярные полимеры, полученные поли
меризацией винилхлорида, и содержат не менее
55 % поливинилхлорида и различные добавки.
Свойства материалов на основе пластифи
цированноrо поливинилхлорида, используемых
для производства контейнеров для человече
ской крови и ее компонентов, определяются при
родой и соотношением веществ, из которых они
получены.
ПРОИЗВОДСТВО
Материалы на основе пластифицированно
ro поливинилхлорида получают методами по
лимеризации, rарантирующими остаточное co
держание винилхлорида менее 1 ррт. Данный
метод должен быть валидирован для получения
продукта выдерживающеro испытание на ви
, нилхлорид.
Винилхлорид. Не более 1 ррт. Парофаз
ная rазовая хроматоrрафия (2.2.28).
Раствор внутренне20 стандарта. К
20,0 мл диметилацетамида Р с помощью ми
крошприца прибавляют 1 О мкл эфира Р, поrру
жая конец иrлы в растворитель. Непосредствен
но перед использованием разводят раствор
диметилацетамидом Р в 1000 раз.
Испытуемый раствор. 1,000 r испытуемоro
материала помещают во флакон вместимостью
50 мл и прибавляют 10,0 мл раствора BHYTpeH
неro стандарта. rерметично закрывают флакон
пробкой. Встряхивают, избеrая контакта жид
кости с пробкой. Помещают флакон в водяную
526
rосударственная фармакопея Республики Беларусь
баню при температуре (60:t1) ос и выдерживают
в течение 2 ч.
Основной раствор винилхлорида. rотовят
в вытяжном шкафу. 50,0 мл диметилацетами
да Р помещают во флакон вместимостью 50 мл,
rерметично закрывают пробкой и взвешивают с
точностью до 0,1 Mr. Наполняют полиэтиленовый
или полипропиленовый шприц вместимостью
50 мл rазообразным винилхлоридом Р, оставля
ют шприц в контакте с rазом в течение 3 мин,
затем rаз удаляют и снова наполняют шприц
50 мл rазообразноro винилхлорида Р. Присое
диняют к шприцу rиподермальную иrлу и YMeHb
шают объем rаза в шприце до 25 мл. Медлен
но вводят оставшиеся 25 мл винилхлорида во
флакон, осторожно встряхивая и избеrая KOHTaK
та жидкости с иrлой. Снова взвешивают флакон.
Увеличение массы должно составлять около
60 Mr (1 мкл раствора содержит около 1,2 MKr ви
нилхлорида). Выдерживают в течение 2 ч. Затем
хранят основной раствор в холодильнике.
Стандартный раствор винилхлорида. Oc
новной раствор винилхлорида диметилаце
тамид Р (1 :3, об/об).
Растворы сравнения. По 10,0 мл раствора
внутреннеro стандарта помещают в 6 одинаковых
флаконов вместимостью 50 мл. Флаконы repMe
тично закрывают пробками. В 5 колб с помощью
микрошприца прибавляют соответственно 1 мкл,
2 мкл, 3 мкл, 5 мкл И 10 мкл стандартноro paCT
вора винилхлорида. Полученные таким образом
6 растворов содержат соответственно О MKr, около
0,3 MKr, 0.6 MKr, 0,9 MKr, 1,5 MKr иЗ MKr винилхлорида.
Встряхивают, избеraя контакта жидкости с пробкой.
Помещают колбы в водяную баню при температу
ре (60:t1) ос и выдерживают в течение 2 ч.
Условия хроматоrрафирования:
колонка из нержавеющей стали длиной
3 м и внутренним диаметром 3 мм, заполнен
ная диатомитом силанизированным для 2азо
вой хромат02рафии Р, импреrнированным 5 %
(М/М) диметилстеариламида Р и 5 % (м/м) Ma
Кр020ла 400 Р;
2аз носитель: азот для хромат02рафии Р;
скорость 2аза носителя: 30 мл/мин;
детектор: пламенно ионизационный;
температура: колонка 45 ос, блок ввода
проб 100 ос, детектор 150 ос;
объем вводимой пробы: 1 мл пар080Й
фазы.
Рассчитывают содержание винилхлорида.
Добавки
В полимер вводят определенное количество
добавок для оптимизации ero химических, фи
зических и механических свойств с целью даль
нейшеro использования по назначению. Все дo
бавки выбирают из приведенноro ниже перечня,
который определяет для каждоro вещества MaK
симально допустимое еro содержание:
ди(2 этилrексил )фталат (добавка к пласт
массе 01): не более 40 %;
цинка октаноат (цинка 2 этилrексаноат)
(добавка к пластмассе 02): не более 1 %;
кальция стеарат или цинка стеарат: не
более 1 %, либо не более 1 % смеси этих ве-
ществ;
N,N' диацилэтилендиамины (добавка 1[
пластмассе 03): не более 1 %;
одно из следующих эпоксидированных
масел: не более 10 %, либо не более 10 % их
смеси:
эпоксидированное соевое масло (добаEr
ка к пластмассе 04) с содержанием кислорода
в эпоксидной rруппе от 6 % до 8 % и йодным
числом не более 6;
эпоксидированное льняное масло (добав
ка к пластмассе 05) с содержанием кислорода
в эпоксидной rруппе не более 1 О % и йодным
числом не более 7.
В полимере MOryT быть определены очень
малые количества антиоксидантов, добавле
ные к винилхлориду мономеру. В полимер анти
оксиданты не вводят.
В качестве красителя может быть использо-
ван только ультрамариновый синий.
Поставщик материала несет OTBeTCTBeH
ность за то, чтобы качественный и количествен
ный составы типовоro образца COOTBeTCTBOBa
ли качественному и количественному составам
каждой произведенной серии.
ОПИСАНИЕ
Бесцветный или бледно желтый порошок,
шарики, rранулы или после трансформа
ции полупрозрачные пластинки разной тол
щины со слабым запахом.
При сжиrании выделяется ryстой, черный
дым.
ИДЕНТИФИКАЦИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
к 2,0 r испытуемоro материала прибав
ляют 200 мл эфира, свободН020 от перок
сидов Р, и наrревают с обратным холодиль
ником в течение 8 ч. Отделяют осадок В от
раствора А фильтрованием. Выпаривают
раствор А до сухоro остатка под вакуумом в
водяной бане при температуре 30 ОС. OCTa
ток растворяют в 1 О мл толуола Р (раствор
А 1). Осадок В растворяют в 60 мл этилен
хлорида Р, наrревая на водяной бане с об
ратным холодильником. Фильтруют. Получен
ный раствор по каплям и при интенсивном
встряхивании прибавляют к 600 мл 2епта
на Р, HarpeToro почти до кипения. Коаrулят 81
и орrанический раствор разделяют rорячиМ
фильтрованием. Охлаждают последний, OTдe
ляют образующийся осадок В2 и фильтруют
через стеклянный фильтр (40) (2.1.2).
3.1.1.1. МатериаЛbl на основе пластифицироваННО20 поливинилхлорида
527
А. Абсорбционная спекторофотометрия в
инфракрасной области (2.2.24).
Коаrулят В 1 растворяют в 30 мл тeтpa
zидрофурана Р и прибавляют небольшими
порциями при встряхивании 40 мл этано
па безводН020 Р. Отделяют осадок В3 филь
трованием и сушат в вакууме над фосфора
(V) оксидом Р при температуре, не превыша
ющей 50 ОС. Несколько миллиrраммов осадка
В3 растворяют в 1 мл тетраzидрофурана Р,
помещают несколько капель полученноro
раствора на диск натрия хлорида и выпарива
ют досуха в сушильном шкафу при температу
ре 100 105 ОС.
Сравнение: ФСО поливинилхлрида.
В. Абсорбционная спекторофотометрия в
инфракрасной области (2.2.24).
Снимают спектр осадка С, полученноro в ис
пытании на добавки к пластмассе 01, 04 и 05.
Сравнение: ФСО добавки к пластмассе 01.
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
Раствор 51. 5.0 r испытуемоro материала
помещают в колбу для сжиrания, прибавляют
зо мл кислоты серной Р и наrревают до получе
ния черной, сиропообразной массы. Охлаждают
и прибавляют 10 мл раствора водорода перок
сида концентрированноzо Р. Осторожно Harpe
вают. Охлаждают и прибавляют 1 мл раствора
водорода пероксида концентрироваНН020 Р; по
вторяют поочередно выпаривание и прибавле
ние раствора водорода пероксида до получения
бесцветной жидкости. Уменьшают объем paCT
вора до 10 мл. Охлаждают и доводят водой Р до
объема 50,0 мл.
Раствор 52. 25 r испытуемоro материала
помещают в колбу из боросиликатноro стекла,
прибавляют 500 мл воды для инъекций Р и за
lCpывают roрло колбы стаканом из боросиликат
нoro стекла. Наrревают в автоклаве при TeM
пературе (121:t2) ос в течение 20 мин. Раствор
охлаждают, декантируют и доводят водой для
инъекций Р до объема 500 мл.
Внешний вид раствора 52. Раствор 82
должен быть прозрачным (2.2.1) и бесцветным
(2.2.2, метод 1/).
Кислотность или щелочность. К 100 мл
раствора S2 прибавл ют 0,15 мл раствора
индикатора BRP Р. При прибавлении не
более 1,5 мл 0,01 М раствора натрия 2идpOK
сида окраска раствора должна измениться
на синюю. К 100 мл раствора S2 прибавляют
0.2 мл раствора метилов020 оранжев020 Р.
При прибавлении не более 1,0 мл 0,01 М
раствора кислоты хлористоводородной
окраска раствора должна начать изменяться
от желтой до оранжевой.
Оптическая плотность (2.2.25). 100,0 мл
раствора S2 выпаривают досуха. Остаток paCT
воряют в 5,0 мл 2ексана Р. Оптическая плот
ность раствора в диапазоне длин волн от 250 нм
до 31 О нм не должна превышать 0,25.
Восстанавливающие вещества. Испыта
ние проводят в течение 4 ч после при20тов
ления раствора 82. К 20,0 мл раствора 82 при
бавляют 1 мл кислоты серной разведенной Р и
20,0 мл 0,002 М раствора калия пермаН2ана
та. Кипятят с обратным холодильником в Te
чение 3 мин и немедленно охлаждают. Прибав
ляют 1 r калия йодида Р и немедленно титруют
раствор 0,01 М раствором натрия тиосульФа
та, используя в качестве индикатора 0,25 мл
раствора крахмала Р. Параллельно проводят
контрольный опыт, используя 20 мл воды для
инъекций Р. Разность между объемами титран
та не должна превышать 2,0 мл.
Первичные ароматические амины. Не
более 20 ррm. К 2,5 мл раствора А 1, получен
ноro при проведении идентификации, прибавля
ют 6 мл воды Р и 4 мл 0,1 М раствора кислоты
хлористоводородной. Интенсивно встряхивают
и удаляют верхний слой. К водному слою при
бавляют 0,4 мл свежеприrотовленноrо раствора
10 r/л натрия нитрита Р. Перемешивают и BЫ
держивают в течение 1 мин. Прибавляют 0,8 мл
раствора 25 r/л аммония сульфамата Р, BЫ
держивают в течение 1 мин, затем прибавля
ют 2 мл раствора 5 r/л нафтилэтилендиамина
ди2идрохпорида Р. Через 30 мин окраска по
лученноro раствора должна быть не интенсив
нее окраски раствора сравнения, приroтовлен
ноro параллельно аналоrичным образом, но с
использованием вместо водноro слоя смеси из
1 мл раствора 0,01 r/л нафтиламина Р в 0,1 М
растворе кислоты хлористоводородной, 5 мл
воды Р и 4 мл 0,1 М раствора кислоты хлори
стоводородной.
Добавки к пластмассе 01, 04 и 05. TOHKOC
лойная хроматоrрафия (2.2.27).
Растворы сравнения. rотовят растворы
0,1 мr/мл ФСО добавки к пластмассе 01, ФСО
добавки к пластмассе 04 и ФСО добавки к
пластмассе 05, соответственно, в толуоле Р.
Пластинка: ТСХ пластинка со слоем сили
ка2еля GF 254 Р.
Подвижная фаза: толуол Р.
Наносимый объем пробы: 0,5 мл paCT
вора А 1, полученноro при проведении иденти
фикации, в виде полосы ЗО мм х 3 мм и по 5 мкл
каждоro из растворов сравнения.
Фронт подвижной фазы: 15 см.
Высушивание: на воздухе.
528
rосударственная фармакопея Республики Беларусь
Проявление А: просматривают в ультрафио
летовом свете при длине волны 254 нм.
Определяют положение зоны, COOTBeTCTBY
ющей добавке к пластмассе 01 (R, около 0,4).
Собирают силикаrель с этой зоны и встряхива
ют с 40 мл эфира Р в течение 1 мин. Фильтру
ют, промывают фильтр двумя порциями, каждая
по 1 О мл эфира Р, прибавляют их к фильтрату и
выпаривают досуха. Масса остатка С не должна
превышать 40 мс
Проявление В: обрабатывают парами йода
в течение 5 мин.
Просматривают xpoMaTorpaMMY, определяя
положение зоны, соответствующей добавкам к
пластмассе 04 и 05 (R,= О). Собирают силикаrель
с данной зоны. Аналоrично собирают с идентич
ной по размеру зоны силикаrель для контрольно
ro опыта. Раздельно встряхивают оба образца в
течение 15 мин с 40 мл метанола Р. Фильтруют,
промывают фильтр двумя порциями, каждая по
10 мл, метанола Р, прибавляя их к фильтрату,
и выпаривают досуха. Разность между массами
сухих остатков не должна превышать 1 О мс
Добавка к пластмассе 03. Осадок В2, по
лученный при проведении идентификации и
находящийся на предварительно взвешенном
стеклянном фильтре (40) (2.1.2), промывают
этанолом Р. Фильтр высушивают до постоянной
массы над фосфора (V) оксидом Р и взвешива
ют. Масса остатка не должна превышать 20 мс
Инфракрасный спектр (2.2.24) остатка
должен соответствовать спектру ФСО добавки к
пластмассе 03.
Барий. Не более 5 ррт.
Атомно эмиссионная спектрометрия
(2.2.57).
Испытуемый раствор. 1,0 r испытуемо
ro материала прокаливают в кварцевом тиrле.
Остаток растворяют в 1 О мл кислоты хлористо
водородной Р и выпаривают досуха на водяной
бане. Полученный остаток растворяют в 20 мл
0,1 М раствора кислоты хлористоводородной.
Раствор сравнения. Раствор, содержащий
0,25 ррт бария, приroтовленный разведением
этаЛОНН020 раствора бария (50 ррт Ва) Р О, 1 М
раствором кислоты хлористоводородной.
Длина волны: длина волны светоиспуска
ния бария 455,40 нм, спектральный фон
455,30 нм.
Проверяют отсутствие бария в использу
емой кислоте хлористоводородной.
Кадмий. Не более 0,6 ррт.
Атомно абсорбционная спектрометрия
(2.2.23, метод 1).
Испытуемый раствор. 1 О мл раствора 81
выпаривают досуха. Остаток растворяют в 5 мл
1 % (об/об) раствора кислоты хлористоводо
родной Р, фильтруют и доводят объем фильтра
та этим же растворителем до 10,0 мл.
Растворы сравнения. Растворы сравне-
ния rотовят разведением этаЛОНН020 раствора
кадмия (0,1 % Cd) Р 1 % (об/об) раствором Kиc
лоты хлористоводородной Р.
Источник излучения: лампа с полым кадми-
евым катодом.
Длина волны: 228,8 нм.
Атомизатор: воздушно ацетиленовое пламя.
Проверяют отсутствие кадмия в использу
емой кислоте хлористоводородной.
Кальций. Не более 0,07 %.
Атомно эмиссионная спектрометрия
(2.2.57).
Испытуемый раствор. Используют испы
туемый раствор, приroтовленный для определ
н,ия бария.
Раствор сравнения. Раствор, содержащий
50,0 ррт кальция, приroтовленный разбавлением
этаЛОНН020 раствора кальция (400 ррт Са) Р
0,1 М раствором кислоты хлористоводородной.
Длина волны: длина волны светоиспуска
ния кальция 315,89 нм, спектральный фон
315,60 нм.
Проверяют отсутствие кальция в использу
емой кислоте хлористоводородной.
Олово. Не более 20 ррт.
Атомно эмиссионная спектрометрия
(2.2.57).
Испытуемый раствор. Непосредственно
перед использованием раствор 81 разводят в 10
раз водой Р.
Раствор сравнения. Непосредственно
перед использованием 2 мл этаЛОНН020 pacт
вора олова (5 ррт Sn) Р помещают в мерную
колбу вместимостью 50 мл, содержащую 5 мл
20 % (об/об) раствора кислоты серной р, и дo
водят водой Р до объема 50 мл.
Длина волны: длина волны светоиспуска
ния олова 189,99 нм, спектральный фон
190,10 нм.
Проверяют отсутствие олова в использу
емой кислоте хлористо водородной.
Цинк. Не более 0,2 %.
Атомно абсорбционная спектрометрия
(2.2.23, метод 1).
Испытуемый раствор. Раствор 8 1 разво
дят в 100 раз О, 1 М раствором кислоты хлори
стоводородной.
Растворы сравнения. Растворы cpaBHe
ния roтовят разведением этаЛОНН020 раствора
цинка (100 ррт Zп) Р О, 1 М раствором кислоты
хлористоводородной.
Источник излучения: лампа с полым цинко
вым катодом.
Длина волны: 213,9 нм.
Атомизатор: воздушно ацетиленовое
пламя.
Проверяют отсутствие цинка в использу
емой кислоте хлористоводородной.
3. 1. 1. 1. Материалы на основе пластифицироваННО20 поливинилхлорида
529
Тяжелые металлы (2.4.8, метод А). Не
более 50 ррт. К 10,0 мл раствора S1 прибавля
ют 0,5 мл раствора фенолфталеина Р и затем
концентрироваНН020 раствора натрия 2идpOK
сида Р до получения бледно розовоro окрашива
ния. Доводят водой Р до объема 25 мл. 12 мл
полученноro раствора должны выдерживать ис
пытание на тяжелые металлы. Эталон rотовят с
использованием этаЛОНН020 раствора свинца
(2 ррт РЬ) Р.
Вещества, экстраrируемые водой. Не
более 0,3 %. 50,0 мл раствора S2 выпарива
ют на водяной бане досуха и высушивают в cy
шильном шкафу при температуре 100 105 ос
до постоянной массы. Параллельно проводят
контрольный опыт, используя 50,0 мл воды для
инъекций Р. Масса сухоro остатка не должна
превышать 7,5 Mr с учетом контрольноro опыта.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение проводят методом сжиrания в
колбе с кислородом (2.5.10), используя 50,0 Mr
испытуемоro материала. Продукты сжиrания
растворяют в 20 мл 1 М раствора натрия 2и
дроксида. К полученному раствору прибавля
ют 1 мл дибутилфталата Р, 2,5 мл кислоты
азотной Р, 5 мл раствора железа (111) аммония
сульфата Р2 и 10,0 мл 0,1 М раствора серебра
нитрата. Титруют раствор 0,05 М раствором
аммония тиоцианата до получения KpaCHOBa
то желтоro окрашивания. Параллельно прово
дят контрольный опыт.
1 мл 0,1 М раствора серебра нитрата co
ответствует 6,25 Mr поливинилхлорида.
Кроме эт020, для стерильных и пустых
контейнеров дополнительно проводят следу
ющие испытания.
Раствор 53. Если испытуемый контейнер
содержит раствор антикоаryлянта, то контейнер
опустошают, промывают внутри 250 мл воды для
инъекций Р при температуре (20:1:1) ос и перед
, приroтовлением раствора S3 удаляют промыв
ную жидкость. Помещают в контейнер воду для
инъекций Р в объеме, соответствующем объему
раствора антикоаryлянта. Закрывают контейнер
и наrревают в автоклаве таким образом, чтобы
температура жидкости составляла 110 ОС, в Te
чение 30 мин. После охлаждения доводят объем
раствора водой для инъекций Р до номинально
ro объема и юмоrенизируют.
Раствор сравнения. Воду для инъекций Р
,lI8rpeвают в КОJlбе из боросиликатноro стекла
. автоклаве при температуре 11 О ос в течение
!30 мин.
Восстанавливающие вещества. Непо
CTBeHHO после приroтовления раствор S3
. объеме, соответствующем 8 % номиналь
ною объема контейнера, помещают в колбу
из боросиликатноrо стекла. Параллельно в
колбе из боросиликатною стекла roтовят KOH
трольный раствор, используя равный объем
свежеприroтовленноro раствора сравнения.
К каждому раствору прибавляют по 20,0 мл
0,002 М раствора калия пермаН2аната и
1 мл кислоты серной разведенной Р. Bыдep
живают в защищенном от света месте в Te
чение 15 мин. Затем прибавляют к каждому
раствору по 0,1 r калия йод ида Р. Выдержи
вают в защищенном от света месте в течение
5 мин и немедленно титруют 0,01 М pacтвo
ром натрия тиосульфата, используя в каче
стве индикатора 0,25 мл раствора KpaXMa
ла Р. Разность между объемами титранта не
должна превышать 2,0 мл.
Кислотность или щелочность. К объему
раствора S3, соответствующему 4 % номи
нальноro объема контейнера, прибавляют
0,1 мл раствора фенолфталеина Р. Раствор
должен оставаться бесцветным. Прибавляют
0,4 мл 0,01 М раствора натрия 2идроксида.
Раствор должен приобрести розовое окраши
вание. Прибавляют 0,8 мл 0,01 М раствора
кислоты хлористоводородной и 0,1 мл pacт
вора метилов020 краСН020 Р. Раствор должен
приобрести оранжево красное или красное
окрашивание.
Хлориды (2.4.4). Не более 0,4 ррm, опреде
ленное с использованием раствора 83. Эталон
rотовят с использованием этаЛОНН020 pacт
вора хлорида (5 ррт С/) Р и 13,8 мл воды Р.
Аммоний (2.4.1, метод А). Не более 2 ррm.
5 мл раствора 83 доводят водой Р до объема
14 мл.
Вещества, экстраrируемые водой.
100 мл раствора S3 выпаривают досуха на BO
дяной бане. Высушивают в сушильном шкафу
при температуре 1 oo 105 ос до постоянной
массы. Параллельно проводят контрольный
опыт, используя 100 мл раствора сравнения.
Масса сухоro остатка, полученноro из paCT
вора S3, не должна превышать 3 Mr с учетом
контрольноro опыта.
Оптическая плотность (2.2.25). Измеря
ют оптическую плотность раствора 83. В обла
сти длин волн от 230 нм до 250 нм оптическая
плотность не должна превышать 0,30. В области
длин волн от 251 нм до 360 нм оптическая плот
ность не должна пrевышать 0,10. В качестве
компенсационноro раствора используют paCT
вор сравнения.
Экстраrируемая добавка к пластмассе 01.
В качестве экстраrента используют 96 %
спирт Р, разведенный водой Р, до получения
530
rосударственная фармакопея Республики Беларусь
01/2013:90002
относительной плотности (2.2.5) от 0,9389 до
0,9395, измеренной плотномером.
Основной раствор. 0,100 r ФСО добавки к
пластмассе 01 растворяют в экстраrенте и дo
водят до объема 100,0 мл этим же растворите
лем.
Стандартные растворы. В 5 мерных колб
вместимостью 100 мл вносят по 1,0 мл, 2,0 мл,
5,0 мл, 10,0 мл И 20,0 мл основноro раствора co
ответственно.
Измеряют оптическую плотность (2.2.25)
стандартных растворов в максимуме при
длине волны 272 нм, используя экстраrент
в кач.естве компенсационноro раствора, и
строят кривую зависимости оптической плот
ности от концентрации добавки к пластмас
се 01.
Методика экстракции. Используя ДOHOp
скую систему с иrлой или адаптер, помеща
ют в пустой контейнер экстраrент, предвари
тельно наrретый в хорошо укупоренной таре
до температуры 37 ОС, в объеме, COOTBeTCTBY
ющем половине номинальноrо объема KOH
тейнера. Полностью удаляют из контейнера
воздух и rерметизируют донорскую систему.
Поrружают наполненный контейнер в rори
зонтальном положении в водяную баню, TeM
пература которой поддерживается на уровне
(37::t1) ОС, на время (60::t1) мин без встряхи
вания. Вынимают контейнер из водяной бани,
аккуратно переворачивают 1 О раз и перено
сят содержимое в стеклянную колбу. Непо
средственно после этоro измеряют оптиче
скую плотность раствора в максимуме при
длине волны 272 нм, используя экстраrент в
качестве компенсационноro раствора.
По rрадуировочному rрафику определяют
концентрацию добавки к пластмассе 01 в мил
лиrраммах на 100 мл экстракта. Концентрация
не должна превышать:
10 Mr на 100 мл для контейнеров с номи
нальным объемом от 300 до 500 мл;
13 Mr на 100 мл для контейнеров с номи
нальным объемом от 150 до 300 мл;
14 Mr на 100 мл для контейнеров с номи
нальным объемом до 150 мл.
Если контейнер содержит раствор aH
тикоа2улянта, этот раствор должен cooт
ветствовать требованиям статьи AHтиKO
а2улянты и консервирующие растворы для
крови человека, а также требованиям следу
юще20 испытания.
Опти"!еская плотность (2.2.25). Не
более 0,5 в максимуме при 280 нм. Измеря
ют оптическую плотность раствора антико
аrулянта в диапазоне длин волн от 250 нм до
350 нм, используя в качестве компенсацион
ноro раствора идентичный по составу paCT
вор антикоаrулянта, не контактировавший с
пластмассой.
3.1.1.2. МАТЕРИАЛЫ НА ОСНОВЕ
ПЛАСТИФИЦИРОВАнноrо
ПОЛИВИНИЛХЛОРИДА
для ТРУБОК, ИСПОЛЬЗУЕМЫХ
В КОМПЛЕКТАХ
ДЛЯ ПЕРЕЛИВАНИЯ КРОВИ
И КОМПОНЕНТОВ КРОВИ
ОПРЕДЕЛЕНИЕ
Содержание: не менее 55 % поливинилхлcr
рида.
В качестве пластификатора использован
ди(2 этилrексил)фталат (добавка к пластмас
се 01).
ПРОИЗВОДСТВО
Материалы на основе пластифицированно
ro поливинилхлорида получают методами по
лимеризации, rарантирующими остаточное co
держание винилхлорида менее 1 ррт. Данный
метод должен быть валидирован для получ.ения
продукта, выдерживающеro испытание на ви
нилхлорид.
Винилхлорид. Не более 1 ррm. Парофаз
ная rазовая хроматоrрафия (2.2.28).
Раствор внутренне20 стандарта. К
20,0 мл диметилацетамида Р с помощью ми
крошприца прибавляют 1 О мкл эфира Р, поrру
жая конец иrлы в растворитель. Непосредствен
но перед использованием разводят раствор
диметилацетамидом Р в 1000 раз.
Испытуемый раствор. 1,000 r испытуемоro
материала помещают во флакон вместимостью
50 мл и прибавляют 10,0 мл раствора BHYTpeH
Hero стандарта. rерметично закрывают флакон
пробкой. Встряхивают, избеrая контакта жид
кости с пробкой. Помещают флакон в водяную
баню при температуре (60::t1) ос и выдерживают
в течение 2 ч.
Основной раствор винилхлорида. rотовят
в вытяжном шкафу. 50,0 мл диметилацетами
да Р помещают во флакон вместимостью 50 мл,
rерметично закрывают пробкой и взвешивают с
точностью до 0,1 Mr. Наполняют полиэтиленовый
или полипропиленовый шприц вместимостью
50 мл rазообразным винилхлоридом Р, оставля
ют шприц в контакте с rазом в течение 3 мин,
затем rаз удаляют и снова наполняют шприц
50 мл rазообразноro винилхлорида Р. Присое
диняют к шприцу rиподермальную иrлу и YMeHb
шают объем rаза в шприце до 25 мл. Медлен
но вводят оставшиеся 25 мл винилхлорида во
флакон, осторожно встряхивая и избеrая KOHTaK
та жидкости с иrлой. Снова взвешивают флакон.
Увеличение массы должно составлять около 60
Mr (1 мкл раствора содержит около 1,2 MKr ви
нилхлорида). Выдерживают в течение 2 ч. Затем
хранят основной раствор в холодильнике.
3.1.1.2. Материалы на основе пластифицироваННО20 поливинuлхлорuда
531
Стандартный раствор винилхлорида. Oc
новной раствор винилхлорида диметилаце
тамид Р (1 :3, об/об).
Растворы сравнения. По 10,0 мл раствора
внутреннеro стандарта помещают в 6 одинако--
вых флаконов вместимостью 50 мл. Флаконы
rерметично закрывают пробками. В 5 колб с по
мощью микрошприца прибавляют COOTBeTCTBeH
но 1 мкл, 2 мкл, 3 мкл, 5 мкл И 10 мкл CTaHдapT
ноro раствора винилхлорида. Полученные таким
образом 6 растворов содержат соответственно
О MKr, около О,З MKr, 0,6 MKr, 0,9 MKr, 1,5 MKr и 3 MKr
винилхлорида. Встряхивают, избеrая контакта
жидкости с пробкой. Помещают колбы в водя
ную баню при температуре (60:t1) ос и выдержи
вают в течение 2 ч.
Условия хромат02рафирования:
колонка из нержавеющей стали длиной
3 м и внутренним диаметром 3 мм, заполнен
ная диатомитом силанизированным для 2азо
вой хромат02рафии Р, импреrнированным 5 %
(м/м) диметилстеариламида Р и 5 % (м/м) Ma
Кр020ла 400 Р;
zаз носитель: азот для хромат02рафии р.
скорость 2аза носителя: ЗО мл/мин;
детектор: пламенно ионизационный;
температура: колонка 45 ос, блок ввода
проб 100 ос, детектор 150 ос;
объем вводимой пробы: 1 мл паровой фазы.
Рассчитывают содержание винилхлорида.
Поставщик материала несет OTBeTCTBeH
ность за то, чтобы качественный и количествен
ный составы типовоro образца COOTBeTCTBOBa
ли качественному и количественному составам
каждой произведенной серии.
ОПИСАНИЕ
Почти бесцветный или бледно желтый по
рошок, шарики, rранулы или после трансфор
мации полупрозрачные пластинки разной тол
щины со слабым запахом.
При сжиrании выделяется ryстой черный
дым.
ИДЕНТИФИКАЦИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
А. К 0,5 r испытуемоro материала прибав
ляют 30 мл тетра2идрофурана Р. Наrревают
при перемешивании на водяной бане в вытяж
tЮм шкафу в течение 1 О мин. Материал полно
стью растворяется. Прибавляют метанол Р по
каплям при перемешивании. Образуется rpaHY
лированный осадок Осадок отфильтровывают и
сушат при температуре 60 ос. Исследуют осадок
OДOM инqpракрасной абсорбционной спектро
метрии (2.2.24). Растворяют 50 Mr осадка в 2 мл
тетра2идрофурана Р и наносят на предметное
стекло. Сушат в сушильном шкафу при темпера
туре 80 ос. Достают предметное стекло и помеща
ют на подставку. Снимают спектр осадка методом
инфракрасной абсорбционной спектрометрии
(2.2.24) и сравнивают со спектром поливинилхло
рида ФСО.
В. Абсорбционная спекторофотометрия в
инфракрасной области (2.2.24).
Снимают спектр осадка, полученноro в ис
пытании на добавки к пластмассе 01.
Сравнение: ФСО добавки к пластмассе 01.
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
Раствор 51. 5,0 r испытуемоro материала
помещают в колбу для сжиrания, прибавляют
30 мл кислоты серной Р и наrревают до получе
ния черной, сиропообразной массы. Охлаждают
и прибавляют 1 О мл раствора водорода перок
сида концентрироваНН020 Р. Осторожно Harpe
вают. Охлаждают и прибавляют 1 мл раствора
водорода пероксида концентрироваНН020 Р; по
вторяют поочередно выпаривание и прибавле
ние раствора водорода пероксида до получения
бесцветной жидкости. Упаривают до уменьше
ния объема раствора до 10 мл. Охлаждают и дo
водят водой Р до объема 50,0 мл.
Раствор 52. 25 r испытуемоro материала
помещают в колбу из боросиликатноro стекла,
прибавляют 500 мл воды для инъекций Р и за
крывают rорло колбы стаканом из боросиликат
ноro стекла. Наrревают в автоклаве при TeM
пературе (121:t2) ос в течение 20 мин. Раствор
охлаждаю декантируют и доводят водой для
инъекций Р до объема 500 мл.
Внешний вид раствора 52. Раствор S2
должен быть прозрачным (2.2.1) и бесцветным
(2.2.2, метод 11).
Добавка к пластмассе 01. Тонкослойная
хроматоrрафия (2.2.27).
Испытуемый раствор. К 2,0 r испытуемоro
материала прибавляют 200 мл эфира, свобод
Н020 от пероксидов, Р и наrревают с обратным
холодильником в течение 8 ч. Разделяют осадок
и раствор фильтрованием. Раствор выпаривают
досуха в водяной бане при температуре 30 ос.
Осадок растворяют в 1 О мл толуола Р.
Раствор сравнения. 0,8 r ФСО добавки к
пластмаСGе 01 растворяют в толуоле Р и дo
водят до объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем сили
ка2еля G Р.
Подвижная фаза: толуол Р.
Наносимый объем пробы: 0,5 мл испыту
eMoro раствора и 5 мкл раствора сравнения в
виде полос 30 мм х 3 мм.
532
rосударственная фармакопея Республики Беларусь
Фронт подвижной фазы: 15 см.
Высушивание: на воздухе.
Проявление: просмэтривают в улырафио
летовом свете при длине волны 254 нм.
Определяют положение зоны, соответствую
щей добавке к пластмассе 01. Собирают силика
rель с этой зоны и встряхивают с 40 мл эфира Р.
Фильтруют без потерь и выпаривают досуха.
Масса остатка не должна превышать 40 Mr.
Барий. Не более 5 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. 1,0 r испытуемо
ro материала прокаливают в кварцевом тиrле.
Остаток растворяют в 1 О мл кислоты хлори
стоводородной Р и выпаривают досуха на BO
дяной бане. Полученный остаток растворяют в
20 мл 0,1 М раствора кислоты хлористоводо
родной.
Раствор сравнения. Раствор, содержащий
0,25 pplТ) :бария, приroтовленный разведением
этаЛQНН020 раствора бария (50 ррт Ва) Р О, 1 М
раствором кислоты хлористоводородной.
Длина волны: длина волны светоиспуска
ния бария 455,40 нм, спектральный фон
455,30 нм.
Проверяют отсутствие бария в использу
емой кислоте хлористоводородной.
Кадмий. Не более 0,6 ррт.
Атомно абсорбционная спектрометрия
(2.2.23, метод /).
Испытуемый раствор. 1 О мл раствора 81
выпаривают досуха. Остаток растворяют в 5 мл
1 % (об/об) раствора кислоты хлористоводо
родной Р, фильтруют и доводят объем фильтра
та этим же растворителем до 10,0 мл.
Растворы сравнения. Растворы cpaBHe
ния roтовят разведением этаЛОНН020 раствора
кадмия (0,1 % Cd) Р 1 % (об/об) раствором Kиc
лоты хлористоводородной Р.
Источник излучения: лампа с полым кадми
евым катодом.
Длина волны: 228,8 нм.
Атомизатор: воздушно ацетиленовое пламя.
Проверяют отсутствие кадмия в использу
емой кислоте хлористоводородной.
Олово. Не более 20 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Непосредственно
перед использованием раствор S 1 разводят в 1 О
раз водой Р.
Раствор сравнения. Непосредственно
перед использованием 2 мл этаЛОНН020 pacт
вора плова (5 ррт Sn) Р помещают в мерную
колбу вместимостью 50 мл, содержащую 5 мл
20 % (об/об) раствора кислоты серной Р и ДOBO
дят водой Р до объема 50 мл.
Длина волны: длина волны светоиспуска
ния олова 189,99 нм, спектральный фон
190,10 нм.
Проверяют отсутствие олова в использу
ем ой кислоте хлористоводородной.
Тяжелые металлы (2.4.8, метод А). Не
более 50 ррm. К 10,0 мл раствора S1 прибавля
ют 0,5 мл раствора фенолфталеина Р и затем
концентрироваНН020 раствора натрия 2идpOK
сида Р до получения бледно розовоro окрашива
ния. Доводят водой Р до объема 25 мл. 12 мл
полученноro раствора должны выдерживать ис
пытание на тяжелые металлы. Эталон rотовят с
использованием этаЛОНН020 раствора свинца
(2 ррт РЬ) Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,500 r испытуемоro материала прибав
ляют 30 мл тетра2идрофурана Р и наrревают
> при перемешивании на водяной бане в вытяж
ном шкафу в течение 10 мин. Материал полно
стью растворяется. Прибавляют 60 мл MeтaHO
ла Р по каплям при перемешивании. Образуется
rранулированный осадок поливинилхлорида.
Выдерживают в течение нескольких минут. Про
должают прибавление метанола Р до прекра
щения образования осадка. Осадок переносят
на стеклянный фильтр (40) (2.1.2), используя
три небольшие порции метанола р, для коли
чественноro перенесения и промывания осадка.
Высушивают фильтр и осадок при температуре
60 ос ДО постоянной массы и взвешивают.
Для стерильных комплектов дополни
тельно проводят следующие испытания.
Раствор 53. Делают замкнутую систему
циркуляции из трех комплектов и сосуда из боро
силикатноro стекла вместимостью 300 мл. При
соединяют к сосуду термостат для поддержания
температуры жидкости в сосуде (37:t1) ОС. Про
качивают по системе 250 мл воды для иHъeK
ций Р в направлении, которое используется для
переливания, в течение 2 ч со скоростью 1 л/час
(например, используя перистальтический насос,
присоединенный короткой частью подходящей
силиконовой трубки). Собирают весь раствор и
охлаждают.
Внешний вид раствора. Раствор 83
должен быть прозрачным (2.2.1) и бесцветным
(2.2.2, метод 11).
Кислотность или щелочность. К 25 мл
раствора 83 прибавляют 0,15 мл раствора
индикатора BRP Р. При прибавлении не
более 0,5 мл 0,01 М раствора натрия 2идpOK
сида окраска раствора должна измениться
на синюю. К 25 мл раствора 83 прибавляют
0,2 мл раствора метилов020 оранжев020 Р.
При прибавлении не более 0,5 мл 0,01 М pacт
вора кислоты хлористоводородной окраска
раствора должна измениться на желтую или
оранжевую.
.! 1.3. Полиолефuны
Оптическая плотность (2.2.25). Оптиче
аая плотность раствора S3 в области от 230 нм
до 250 нм не должна превышать 0,30. Оптиче
екая плотность раствора 83 в области от 251 нм
до 360 нм не должна превышать 0,15.
Восстанавливающие вещества. Испыта
ние проводят в течение 4 ч после при20тов
пения раствора S3. К 20,0 мл раствора S3 при
6авляют 1 мл кислоты серной разведенной Р и
20,0 мл 0,002 М раствора калия пермаН2ана
та. Кипятят в течение 3 мин и сразу охлаждают.
Прибавляют 1 r калия йодида Р и титруют 0,01 М
раствором натрия тиосульфата, используя в
качестве индикатора 0,25 мл раствора KpaXMa
ла Р. Параллельно проводят контрольный опыт,
используя 20 мл воды для инъекций Р. Разность
между объемами титранта не должна превы
шать 2,0 мл.
Вещества, экстраrируемые водой. 50,0 мл
раствора S3 выпаривают досуха на водяной бане
и высушивают в сушильном шкафу при темпе
ратуре 100 105 ос до постоянной массы. Па
раллельно проводят контрольный опыт, исполь
зуя 50,0 мл воды для инъекций Р. Масса сухою
остатка не должна превышать 1 ,5 Mr с учетом
контрольноro опыта.
01/2013:30103
3.1.3. ПОЛИОЛЕФИНЫ
ОПРЕДЕЛЕНИЕ
Полиолефины получают полимеризацией
этилена или пропилена или сополимеризаци
ей этих веществ с содержанием не более 25 %
высших rомолоrов (C4 C10)' карбоновых кислот
или эфиров. Некоторые материалы MOryT пред
ставлять собой смесь полиолефинов.
ПРОИЗВОДСТВО
В полимер вводят некоторое количество
добавок для оптимизации их химических, фи
зических и механических свойств с целью
обеспечения использования полимера по Ha
значению. Добавки выбирают из нижепере
численноro списка, в котором для каждой дo
бавки обозначен максимально возможный
состав.
Полимеры MOryT содержать не более трех
антиоксидантов, один или несколько смазыва
ющих или антиадrезивных areHToB, а также
титана диоксид как средство, придающее непро
зрачность для защиты материала от света:
бутилrидрокситолуол (добавка к пластмас
се 07): не более 0,125 %;
5ЗЗ
пентаэритритилтетракис[3 (3,5 ди трет
бутил 4 rидроксифенил)пропионат] (добавка к
пластмассе 09): не более 0,3 %;
1 ,3,5 трис(3,5 ди трет бутил-4 rидрокси
бензил) S триазин 2,4,6(1 Н,3Н,5Н) трион (дo
бавка к пластмассе 13): не более 0,3 %;
октадецил 3 (3,5 ди трет бутил 4 rидро
ксифенил )пропионат (добавка к пластмассе 11):
не более 0,3 %;
этиленбис[3,3 бис[3 (1,1 диметилэтил)-4
rидроксифенил]бутаноат] (добавка к пластмассе
08): не более 0,3 %;
диоктадецилдисульфид (добавка к пласт
массе 15): не более О,З %;
4,4',4" (2,4,6 триметилбензен 1 ,3,5 триил
трисметилен)трис[2,6 бис(1, 1 диметилэтил)
фенол] (добавка к пластмассе 10): не более
0,3 %;
2,2' бис(октадецилокси ) 5,5' спироби
[1 ,3,2 диоксафосфинан] (добавка к пластмассе
14): не более 0,3 %;
дидодецил 3,3' тиодипропионат (добавка к
пластмассе 16): не более 0,3 %;
диоктадецил 3,3' тиодипропионат (добав
ка к пластмассе 17): не более 0,3 %;
трис[2,4 бис( 1, 1 диметилэтил )фенил ]фос
фит (добавка к пластмассе 12): не более 0,3 %;
добавка к пластмассе 18: не более 0,1 %;
сополимер диметилсукцината и (4 rид
рокси 2 ,2,6, 6 тетраметилпиперидин 1 ил)
этанол (добавка к пластмассе 22): не более
0,3 %.
Сумма перечисленных выше антиоксидант
ных добавок не должна превышать 0,3 %.
rидротальцит: не более 0,5 %;
алканамиды: не более 0,5 %;
алкенамиды: не более 0,5 %;
натрия алюмосиликат: не более 0,5 %;
кремния диоксид: не более 0,5 %;
натрия бензоат: не более 0,5 %;
эфиры или соли жирных кислот: не
более 0,5 %;
натрия фосфат: не более 0,5 %;
вазелиновое масло (парафин жидкий): не
более 0,5 %;
цинка оксид: не более 0,5 %;
тальк: не более 0,5 %;
маrния оксид: не более 0,2 %;
кальция стеарат или цинка стеарат или их
сумма: не более 0,5 %;
титана диоксид: не более 4 %.
Поставщик материала несет OTBeTCTBeH
насть за то, чтобы качественный и количествен
ный составы типовоro образца COOTBeTCTBOBa
ли каl..lественному и количественному составам
каждой произведенной серии.
ОПИСАНИЕ
Порошок, шарики, rранулы или после
трансформации полупрозрачные пластинки
разной толщины или контейнеры.
534
rосударственная фармакопея Республики Беларусь
Практически нерастворимы в воде, в эта
ноле, в reKcaHe и в метаноле, растворимы в ro
рячих ароматических уrлеводородах. Размяrча
ются при температуре от 65 ос до 165 ОС. При
сжиrании пламя окрашивается в синий цвет.
ИДЕНТИФИКАЦИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
А. Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
При20товление: к 0,25 r испытуемоro MaTe
риала прибавляют 1 О мл толуола Р и кипятят с
обратным холодильником в течение 15 мин. He
сколько капель полученноro раствора помещают
на диск натрия хлорида и выпаривают раствори
тель в сушильном шкафу при температуре 80 ОС.
Максимумы П02лощения: при 2920 CM 1,
2850CM 1, 1475CM 1, 1465CM 1, 1380CM 1, 1170CM 1,
735 CM 1, 720 CM 1.
Полученный спектр должен соответствовать
спектру типовоro образца. Если материал имеет
форму пластин, идентификацию можно прове
сти непосредственно на вырезанном фраrменте
пластинки соответствующеro размера.
В. Испытуемый материал должен выдержи
вать дополнительные испытания в зависимости
от тех добавок, которые входят в еro состав.
с. 20 Mr испытуемоro материала смешива
ют в платиновом тиrле с 1 r калия 2идросульфа
та Р, наrревают до полноro расплавления и ox
лаждают. Прибавляют 20 мл кислоты серной
разведенной Р, аккуратно наrревают и фильтру
ют. К полученному фильтрату прибавляют 1 мл
кислоты фосфорной Р и 1 мл KOHцeHтpиpoвaH
Н020 раствора водорода пероксида Р. Если ис
пытуемый материал содержит титана диоксид,
появляется оранжево желтое окрашивание.
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
Раствор 51. Раствор S1 используют в тe
чение 4 ч после при20товления. 25 r испытуемо
ro материала помещают в колбу из боросиликат
Horo стекла с притертой пробкой. Прибавляют
500 мл воды для инъекций Р и кипятят с обрат
ным холодильником в течение 5 ч. Охлаждают и
декантируют. Часть раствора 81 сохраняют для
проведения испытаний на прозрачность и ЦBeT
ность. Остаток раствора фильтруют через CTe
клянный фильтр (16) (2.1.2).
Раствор 52. 2,0 r испытуемоro материала
помещают в коническую колбу из боросиликат
Horo стекла с притертой пробкой. Прибавляют
80 мл толуола Р и кипятят с обратным холо-
дильником при постоянном перемешивании в те-
чение 90 мин. Охлаждают до температуры 60 ос
и прибавляют 120 мл толуола Р при постоян-
ном перемешивании. Раствор фильтруют через
стеклянный фильтр (16) (2.1.2). Колбу и фильтр
промывают 25 мл смеси толуол Р метанол Р
(40:60, об/об), присоединяют ее к фильтрату и
доводят до объема 250 мл этой же смесью paCT
ворителей. rотовят контрольный раствор.
Раствор 53. 100 r испытуемоro материала
помещают в коническую колбу из боросиликат
ноro стекла с притертой пробкой и прибавляют
250 мл 0,1 М раствора кислоты хлористоводо
родной. Кипятят с обратным холодильником при
ПQ.СТОЯННОМ перемешивании в течение 1 ч. Ox
лаждают и декантируют раствор.
Внешний вид раствора 51. Раствор 81
должен быть прозрачным (2.2.1) и бесцветным
(2.2.2, метод 11).
Кислотность или щелочность. К 100 мл
раствора 51 прибавляют 0,15 мл раствора иH
дикатора BRP Р. При прибавлении не более
1,5 мл 0,01 М раствора натрия 2идроксида
окраска раствора должна измениться на синюю.
К 100 мл раствора 81 прибавляют 0,2 мл pacт
вора метилов020 оранжев020 Р. При прибавле
нии не более 1,0 мл 0,01 М раствора кислоты
хлористоводородной окраска раствора должна
измениться на желтую или оранжевую.
Оптическая плотность (2.2.25). Оптиче
екая плотность раствора 51 в области от 220 нм
до 340 нм не должна превышать 0,2.
Восстанавливающие вещества. К 20 мл
раствора 81 прибавляют 1 мл кислоты серной
разведенной Р и 20 мл 0,002 М раствора калия'
пермаН2аната. Кипятят с обратным холодильни
ком в течение 3 мин и сразу охлаждают. Прибав 1
ляют 1 r калия йодида Р и сразу титруют 0,01 М
раствором натрия тиосульфата, используя в
качестве индикатора 0,25 мл раствора KpaXMa
ла Р. Параллельно проводят контрольный опыт.
Разность между объемами титранта не должна
превышать 3,0 мл.
Вещества, растворимые в reKcaHe. 10 r
испытуемоro материала помещают в кониче
скую колбу из боросиликатноrо стекла с при
тертой пробкой вместимостью 250 мл. Прибав
ляют 100 мл 2ексана Р и кипятят с обратным
ХОJlОДИЛЬНИКОМ в течение 4 ч при постоянном
перемешивании. Охлаждают в ледяной бане и
быстро фильтруют через стеклянный фильтр
(16) (2.1.2), поддерживая температуру paCT
вора около О ос (время фильтрования должно
быть менее 5 мин; для ускорения процесса,
если необходимо, фильтрование про водят под
3.1.3. Полиолефины
535
давлением). 20 мл фильтрата помещают в BЫ
сушенный до постоянной массы стакан из бо
росиликатноro стекла и выпаривают на водя
ной бане. Остаток сушат в сушильном шкафу
при температуре 100 105 ос в течение 1 ч.
Масса полученноro остатка должна быть в
пределах 1 О % от массы остатка, полученно
ro для типовоro образца, и не должна превы
шать 5 %.
Экстраrируемый алюминий. Не более
1 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют paCT
вор 83.
Раствор сравнения. rотовят путем раз
ведения этаЛОНН020 раствора алюминия
(200 ррт А/) Р О, 1 М раствором кислоты хлори
стоводородной.
Длина волны: длина волны светоиспускания
алюминия 396,15 нм, спектральный фон
396,25 нм.
Проверяют отсутствие алюминия в исполь
зуемой кислоте хлористоводородной.
Экстраrируемый титан. Не более 1 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют раст
вор S3.
Раствор сравнения. rотовят путем раз
ведения этаЛОНН020 раствора титана
(100 ррт Тi) Р 0,1 М раствором кислоты хлори
стоводородной.
Длина волны: длина волны светоиспуска
ния титана 336,12 нм, спектральный фон
336,16 нм.
Проверяют отсутствие титана в использу
емой кислоте хлористоводородной.
Экстраrируемый цинк. Не более 1 ррm.
Атомно абсорбционная спектрометрия
(2.2.23, метод 1).
Испытуемый раствор. Используют paCT
вор S3. <
Раствор сравнения. rотовят путем разведе
ния этаЛОНН020 раствора цинка (10 ррт Zп) Р
0,1 М раствором кислоты хлористоводород
ной.
Источник излучения: лампа с полым цинко
вым катодом.
Длина волны: 213,9 нм.
Атомизатор: воздушно ацетиленовое
пламя.
Проверяют отсутствие цинка в использу
емой кислоте хлористоводородной.
Экстраrируемые тяжелые металлы
(2.4.8, метод А). Не более 2,5 ррm. 50 мл
раствора S3 упаривают до объема около
5 мл на водяной бане и доводят водой Р до
объема 20,0 мл. 12 мл полученною paCT
вора должны выдерживать испытание на тя
желые металлы. Эталон rотовят с использо
ванием 2,5 мл этаЛОНН020 раствора свинца
(10 ррт РЬ) Р.
Сульфатная зола (2.4.14). Не более
1,0 %. Определение проводят из 5,0 r испыту
емоro материала. Эта норма не распространя
ется на материалы, которые содержат титана
диоксид как добавку, придающую непрозрач
ность материалу.
ДОПОЛНИТЕЛЬНЫЕ ИСПЫТАНИЯ
Эти испытания следует проводить пол
ностью или частично только в тех случаях,
К02да эт020 требует заявленный состав или
область применения материала.
Фенольные антиоксиданты. Жидкостная
хроматоrрафия (2.2.29).
Смесь растворителей. Ацетонитрил Р
тетра2идрофуран Р (50:50, об/об).
Испытуемый раствор 821. 50 мл paCT
вора 82 выпаривают досуха в вакууме при TeM
пературе 45 ОС. Остаток растворяют в 5,0 мл
смеси растворителей. Контрольный раствор ro
товят из контрольною раствора, соответствую
щеro раствору S2.
Испытуемый раствор 822. 50 мл paCT
вора S2 выпаривают досуха в вакууме при TeM
пературе 45 ОС. Остаток растворяют в 5,0 мл Me
тиленхлорида Р. Контрольный раствор roтовят
из контрольноro раствора, соответствующеro
раствору 82.
Испытуемый раствор 823. 50 мл раст
вора S2 выпаривают досуха в вакууме при TeM
пературе 45 ОС. Остаток растворяют в 5,0 мл
смеси равных объемов ацетонитрила Р и раст
вора 10 r/л трет бутиЛ2идропероксида Р в тe
тра2идрофуране Р. Закрывают колбу и Bыдep
живают в течение 1 ч. Контрольный раствор
roтовят из контрольноro раствора, COOTBeTCTBY
ющеrо раствору S2.
Из приведенных ниже растворов сравнения
20товят только те, которые необходимы для
анализа фенольных антиоксидантов, входя
щих в состав испытуеМ020 материала.
Раствор сравнения (а). 25,0 Mr ФСО бутил
2идрокситолуола (добавка к пластмассе 07) и
60,0 Mr ФСО добавки к пластмассе 08 paCTBO
ряют в 10,0 мл смеси растворителей. 2,0 мл по
лученноro раствора доводят смесью раствори
телей до объема 50,0 мл.
Раствор сравнения (Ь). 60,0 Mr ФСО дo
бавки к пластмассе 09 и 60,0 Mr ФСО добавки
к пластмассе 10 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro раствора дo
водят смесью растворителей до объема 50,0 мл.
Раствор сравнения (с). 60,0 Mr ФСО дo
бавки к пластмассе 11 и 60,0 Mr ФСО добавки
к пластмассе 12 растворяют в 1 О мл метилен
хлорида Р. 2,0 мл полученноro раствора доводят
метиленхлоридом Р до объема 50,0 мл.
536
rосударственная фармакопея Республики Беларусь
Раствор сравнения (d). 25,0 Mr ФСО добав
ки к пластмассе 07 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro раствора дo
водят смесью растворителей до объема 50,0 мл.
Раствор сравнения (е). 60,0 Mr ФСО добав
ки к пластмассе 08 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro раствора
доводят смесью растворителей Р до объема
50,0 мл.
Раствор сравнения (f). 60,0 Mr ФСО добав
ки к пластмассе 13 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro раствора дo
водят смесью растворителей до объема 50,0 мл.
Раствор сравнения (g). 60,0 Mr ФСО добав
ки клластмассе 09 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro раствора дo
водят смесью растворителей до объема 50,0 мл.
Раствор сравнения (h). 60,0 Mr ФСО добав >
ки к пластмассе 10 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro раствора дo
водят смесью растворителей до объема 50,0 мл.
Раствор сравнения (i). 60,0 Mr ФСО добавки
к пластмассе 11 растворяют в 10,0 мл метилен
хлорида Р. 2,0 мл полученноro раствора доводят
метиленхлоридом Р до объема 50,0 мл.
Раствор сравнения Ш. 60,0 Mr ФСО добав
ки к пластмассе 12 растворяют в 10,0 мл Meти
ленхлорида Р. 2,0 мл полученноro раствора дo
водят метиленхлоридом Р до объема 50,0 мл.
Раствор сравнения (k). 20,0 Mr ФСО добав
ки к пластмассе 18 растворяют в 10,0 мл смеси
равных объемов ацетонитрила Р и раствора
1 О r/л трет бутиЛ2идропероксида Р в тeтpa
2идрофуране Р. Закрывают колбу и выдержива
ют в течение 1 ч. 2,0 мл полученноro раствора дo
водят смесью растворителей до объема 50,0 мл.
Условия хроматоrрафирования:
А. Если испытуемый материал содержит дo
бавку к пластмассе 07 и/или добавку к пластмас
се 08, используют следующие условия:
колонка длиной 0,25 м и внутренним ди
аметром 4,6 мм, заполненная силика2елем OK
тадецилсилильным для хромат02рафии Р с
размером частиц 5 мкм;
подвижная фаза: вода Р aцeтOHи
трил р (30:70, об/об);
скорость подвижной фазы: 2 мл/мин;
детектирование: спектрофотометриче
ский детектор, длина волны 280 нм;
объем вводимой пробы: по 20 мкл испы
туемоro раствора S21, соответствующеro KOH
трольноro раствора, раствора сравнения (а), и
либо растворов сравнения (d) или (е), либо pac
творов сравнения (d) и (е);
время хромат02рафирования: зо мин.
При20дность хромат02рафической системы
разрешение: не менее 8,0 между пиками
добавки к пластмассе 07 и добавки к пластмас
се 08 на xpoMaтorpaMMe раствора сравнения (а);
на xpoMaтorpaMMe испытуемоro раст
вора 821 должны обнаруживаться пики только
тех антиоксидантов, которые входят в состав ис
пытуемоrо материала, кроме этоro MOryT обна
руживаться пики, присутствующие на xpoMaTcr
rpaMMe контрольноro раствора.
Предельное содержание: площади пиков на
xpoMaTorpaMMe испытуемоro раствора S21 не
должны превышать площадей соответствующих
пиков на xpoMaTorpaMMe растворов сравнения
(d) и/или (е).
В. Если испытуемый материал содержит
один или более следующих антиоксидантов:
добавка к пластмассе 09;
добавка к пластмассе 1 о;
добавка к пластмассе 11;
добавка к пластмассе 12;
добавка к пластмассе 13,
проводят испытание, как описано выше, со
следующими изменениями:
подвижная фаза: вода Р тeтpa2идpo
фуран р ацетонитрил Р (10:30:60, об/об/об);
скорость подвижной фазы: 1,5 мл/мин;
объем вводимой пробы: по 20 мкл испы
туемоro раствора 821, соответствующеro KOH
трольноro раствора, раствора сравнения (Ь) и
растворов сравнения антиоксидантов, ВХОДЯЩИХ
в состав испытуемоro материала в соответствии
с перечисленным выше списком.
При20дность хромат02рафической cиcтe
мы:
разрешение: не менее 2,0 между пиками
добавки к пластмассе 09 и добавки к пластмас
се 1 О на xpoMaTorpaMMe раствора сравнения (Ь);
на xpoMaTorpaMMe испытуемоro раст
вора S21 должны обнаруживаться пики только
тех антиоксидантов, которые входят в состав ис
пытуемоro материала, кроме этоro MOryT обна
руживаться пики, присутствующие на xpOMaTO
rpaMMe контрольноro раствора.
Предельное содержание: площади пиков на
xpoMaTorpaMMe испытуемоro раствора 821 не
должны превышать площадей соответствующих
пиков на xpoMaTorpaMMe растворов сравнения
антиоксидантов, входящих в состав испытуемо
ro материала в соответствии с перечисленным
выше списком.
с. Если испытуемый материал содержит
добавку к пластмассе 11 иlили добавку к пласт
массе 12, проводят испытание, как описано для
добавки к пластмассе 07 иlили добавки к пласт
массе 08, со следующими изменениями:
подвижная фаза: вода Р 2 пропа
нол Р метанол Р (5:45:50, об/об/об);
скорость подвижной фазы: 1,5 мл/мин;
объем вводимой пробы: по 20 мкл испы
туемоro раствора 822, соответствующеro KOH
трольноro раствора, раствора сравнения (с), и
либо растворов сравнения (i) или 0), либо pac
творов сравнения (О и 0).
При20дность хромат02рафической cu
стемы:
разрешение: не менее 2,0 между пиками
добавки к пластмассе 11 и добавки к пластмас
се 12 на xpoMaTorpaMMe раствора сравнения (с);
3.1.3. Полиолефины
537
на xpoMaTorpaMMe испытуемоro pacт
вора 522 должны обнаруживаться пики только
тех антиоксидантов, которые входят в состав ис
пытуемоro материала, кроме этоro MOryт обна
руживаться пики, присутствующие на xpOMaTO
rpaMMe контрольноro раствора.
Предельное содержание: площади пиков на
xpoMaTorpaMMe испытуемоro раствора 822 не
должны превышать площадей соответствующих
пиков на xpoMaTorpaMMe растворов сравнения
(i) и/или Ш.
D. Если испытуемый материал содержит дo
бавку к пластмассе 18, проводят испытание, как
описано для добавки к пластмассе 07 и/или дo
бавки к пластмассе 08, со следующими измене
ниями:
подвижная фаза: тетра2идрофуран Р
ацетонитрил Р (20:80, об/об/об);
скорость подвижной фазы: 1 ,5 мл/мин;
детектирование: спектрофотометриче
ский детектор, длина волны 270 нм;
объем вводимой пробы: по 20 мкл испы
TyeMoro раствора 523, соответствующеro KOH
трольноro раствора и раствора сравнения (k).
При20дность хромат02рафической cи
стемы:
разрешение: не менее 6,0 между двумя
основными пиками (приблизительные времена
удерживания 3,5 и 5,8) на xpoMaTorpaMMe paCT
вора сравнения (k);
на xpoMaTorpaMMe испытуемоro раст
вора 823 должны обнаруживаться пики только
тех антиоксидантов, которые входят в состав ис
пытуемоro материала, кроме этоro MOryт обна
руживаться пики, присутствующие на xpOMaTO
rpaMMe контрольноro раствора.
Предельное содержание: площади пиков на
xpoMaтorpaMMe испытуемоro раствора 822 не
должны превышать площадей соответствующих
пиков на xpoMaTorpaMMe раствора сравнения (k).
Нефенольные антиоксиданты. TOHKOC
лойная хроматоrрафия (2.2.27).
Испытуемый раствор 524. 100 мл paCT
вора 82 выпаривают досуха в вакууме при TeM
пературе 45 ос. Остаток растворяют в 2 мл Me
тиленхлорида подкислеНН020 Р.
Раствор сравнения (1). 60 Mr ФСО добав
ки к пластмассе 14 растворяют в 1 О мл Meти
ленхлорида Р. 2 мл полученноro раствора дo
водят метиленхлоридом подкисленным Р до
объема 1 О мл.
Раствор сравнения (т). 60 Mr ФСО добав
ки к пластмассе 15 растворяют в 1 О мл Meти
ленхлорида Р. 2 мл полученноrо раствора дo
водят метиленхлоридом подкисленным Р до
объема 1 О мл.
Раствор сравнения (п). ФСО 60 Mr добав
ки к пластмассе 16 растворяют в 1 О мл Meти
ленхлорида Р. 2 мл полученноro раствора дo
водят метиленхлоридом подкисленным Р до
объема 1 О мл.
Раствор сравнения (о). 60 Mr ФСО добавки
к пластмассе 17 растворяют в 1 О мл метилен
хлорида Р. 2 мл полученноro раствора доводят
метиленхлоридом подкисленным Р до объема
10 мл.
Раствор сравнения (р). 60 Mr ФСО добав
ки к пластмассе 16 и ФСО добавки к пластмас
се 17 растворяют в 1 О мл метиленхлорида Р.
2 мл полученноro раствора доводят метилен
хлоридом подкисленным Р до объема 1 О мл.
Пластинка: ТСХ пластинка со слоем сили
ка2еля GF 254 Р.
Подвижная фаза А: 2ексан Р.
Подвижная фаза В: метиленхлорид Р.
Наносимый объем пробы: по 20 мкл испы
туемоro раствора 824, раствора сравнения (р)
и растворов сравнения, соответствующих всем
фенольным и нефенольным антиоксидантам,
входящим в состав испытуемоro материала.
Фронт подвижной фазы А: 18 см.
Высушивание А: на воздухе.
Фронт подвижной фазы В: 17 см.
Высушивание В: на воздухе.
ПРОЯ8Ление: просматривают в ультрафио
летовом свете при длине волны 254 нм; пластин
ку опрыскивают спиртовым раствором йода Р и
через 1 o 15 мин просматривают в ультрафио
летовом свете при длине волны 254 нм.
При20дность хромат02рафuческой cиcтe
мы: раствор сравнения (р):
на xpoMaTorpaMMe должно обнаруживать
ся два полностью разделенных пятна.
Предельное содержание: на xpoMaтorpaMMe
испытуемоro раствора 524 любое пятно должно
быть не интенсивнее соответствующеro пятна
на xpoMaTorpaMMe раствора сравнения.
Добавка к пластмассе 22. Жидкостная xpo
матоrрафия (2.2.29).
Испытуемый раствор. 25 мл раствора 52
выпаривают досуха в вакууме при температуре
45 ос. Остаток растворяют в 1 О мл толуола Р и
1 О мл раствора 1 О r/л тетрабутиламмония 2и
дроксида Р в смеси из 35 объемов толуола Р и
65 объемов этанола Р. Кипятят с обратным xo
лодильником в течение 3 ч. Охлаждают и, если
необходимо, фильтруют.
Раствор сравнения. 30 Mr ФСО добавки к
пластмассе 22 растворяют в 50 мл толуола Р.
1 мл полученноro раствора прибавляют к 25 мл
контрольноro раствора 82 и выпаривают досуха
в вакууме при температуре 45 ос. Остаток paCT
воряют в 1 О мл толуола Р и 10 мл раствора 1 О r/л
тетрабутиламмония 2идроксида Р в смеси из
35 объемов толуола Р и 65 объемов этанола Р.
Кипятят с обратным холодильником в течение
3 ч. Охлаждают и, если необходимо, фильтруют.
Условия хроматоrрафирования:
колонка длиной 0,25 м и внутренним
диаметром 4,6 мм, заполненная силика2елем
аминопропилсилильным для хромат02рафии Р
с размером частиц 5 мкм;
538
rосударственная фармакопея Республики Беларусь
подвижная фаза: этанол Р 2ексан
(11 :89, об/об);
скорость подвижной фазы: 2 мл/мин;
детектирование: спектрофотометриче
ский детектор, длина волны 227 нм;
объем вводимой пробы: 20 мкл;
время хромат02рафирования: 1 О мин.
При20дность хромат02рафической cиc
темы:
разрешение: не менее 7 между пиком «ди
ольноro» компонента и пиком растворителя на
xpoMaTorpaMMe раствора сравнения.
Предельное содержание: на xpoMaTorpaMMe
испытуемоro раствора площадь пика, COOTBeT
ствующеro «диольному» компоненту добавки к
пластмассе 22, не должна превышать площадь
соответствующеro пика на xpoMaTorpaMMe paCT
вора сравнения.
Амиды и стеараты. Тонкослойная xpOMaTO
rрафия (2.2.27).
Испытуемый раствор. Используют испыту
емый раствор 824, приroтовленный, как указано
в испытании «Нефенольные антиоксиданты».
Раствор сравнения (q). 20 Mr ФСО кислоты
стеариновой (добавки к пластмассе 19) paCTBO
ряют в 1 О мл метиленхлорида Р.
Раствор сравнения ('). 40 Mr ФСО олеамида
(добавки к пластмассе 20) растворяют в 20 мл
метиленхлорида Р.
Раствор сравнения (s). 40 Mr ФСО эрука
мида (добавки к пластмассе 21) растворяют в
20 мл метиленхлорида Р.
Пластинка: ТСХ пластинка со слоем сили
ка2еля GF 254 Р (две пластинки).
А. Подвижная фаза: этанол Р тpиMe
тилпентан Р (25:75, об/об).
Наносимый объем пробы: по 1 О мкл испы
туемоro раствора 824 и раствора сравнения (q).
Фронт подвижной фазы: 10 см.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают pac
твором 2 r/л дихлорфенолиндофенола Haтpи
евой соли Р в этаноле Р и наrревают в cy
шильном шкафу при температуре 120 ос до
появления пятен.
Предельное содержание: на xpoMaTorpaMMe
испытуемоrо раствора 824 пятно, COOTBeTCTBY
ющее добавке к пластмассе 19, должно быть
идентичным по расположению (R, около 0,5) и
быть не интенсивнее соответствующеro пятна
на xpoMaTorpaMMe раствора сравнения (q).
В. Подвижная фаза А: 2ексан Р.
Подвижная фаза В: метанол Р метилен
хлорид Р (5:95, об/об).
Наносимый объем пробы: по 1 О мкл ИСI1Ы
туемоro раствора 824 и растворов сравнения (r)
и (s).
Фронт подвижной фазы А: 13 см.
Высушивание А: на воздухе.
Фронт подвижной фазы В: 10 см.
Высушивание В: на воздухе.
Проявление: пластинку опрыскивают раство-
ром 40 r/л кислоты фосфорномолибденовой Р.
этаноле Р и наrревают в сушильном шкафу при
температуре 120 ос до появления пятен.
Предельное содержание: на xpoMaTorpa
ме испытуемоro раствора 824 пятна, COOTвeт
ствующие добавке к пластмассе 20 или доба
ке к пластмассе 21, должны быть идентичными
по расположению (R, около 0,2) и быть не интен-
сивнее соответствующих пятен на xpoMaTorp
ме раствора сравнения (r) и (s).
01/2013:30104
3.1.4. ПОЛИЭТИЛJ;Н БЕЗ ДОБАВОК
ДЛЯ КОНТЕИНЕРОВ
ДЛЯ ПАРЕНТЕРАЛЬНЫХ
И ОФТАльмолоrИЧЕСКИХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
ОПРЕДЕЛЕНИЕ
Полиэтилен без добавок получают полимери
зацией этилена под высоким давлением в присут
ствии кислорода или инициаторов образования
свободных радикалов в качестве катализаторов.
ОПИСАНИЕ
Шарики, rранулы, порошок или после
трансформации прозрачные пластинки раз
личной толщины или контейнеры.
Практически нерастворим в воде, в этаноле,
в reKcaHe и в метаноле, растворим в roрячих apo
матических уrлеводородах.
Размяrчается при температуре выше 65 ОС.
Относительная плотность: от 0,91 О до 0,937.
ИДЕНТИФИКАЦИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
А. Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
При20товление: к 0,25 r испытуемоro MaTe
риала прибавляют 1 О мл толуола Р и кипятят
с обратным холодильником в течение 15 мин.
Несколько капель раствора помещают на диск
натрия хлорида и выпаривают растворитель в
сушильном шкафу при температуре 80 ОС.
Максимумы П02лощения: при 2920 см."
2850 см." 1465 см.1, 730 см. 1 и 720 см. 1 .
Полученный спектр должен соответствовать
спектру типовоro образца. Если материал имеет
форму пластин, идентификацию можно прове
сти непосредственно на вырезанном фраrменте
пластинки соответствующеro размера.
3.1.4. Полиэтилен без добавок для контейнеров
539
В. Испытуемый материал выдерживает ис
пытание на предельное содержание добавок
(см. раздел «Испытания»).
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
Раствор 51. 25 r испытуемоro материала
помещают в колбу из боросиликатноro стекла
с притертой пробкой. Прибавляют 500 мл воды
для инъекций Р и наrревают с обратным холо
дильником в течение 5 ч. Охлаждают и дeKaH
тируют. Часть раствора оставляют для прове
дения испытаний на прозрачность и цветность.
Оставшуюся часть раствора фильтруют через
стеклянный фильтр (16) (2.1.2). Раствор 81 иc
пользуют в течение 4 ч после при20тоеления.
Раствор 52. 2,0 r испытуемоro материала
помещают в коническую колбу из боросиликат
ноro стекла с притертой пробкой. Прибавляют
80 мл толуола Р и кипятят с обратным холо
дильником при постоянном перемешивании в
течение 1,5 ч. Охлаждают до 60 ос и прибавляют
при продолжительном перемешивании 120 мл
метанола Р. Фильтруют раствор через CTe
клянный фильтр (16) (2.1.2). Промывают колбу
и фильтр 25 мл смеси толуол Р метанол Р
(40:60, об/об), прибавляют промывную жидкость
к фильтрату и доводят до объема 250 мл этой
же смесью растворителей. Параллельно roтовят
контрольный раствор.
Раствор 53.100 r испытуемоro материала
помещают в коническую колбу из боросиликат
ноro стекла с притертой пробкой. Прибавляют
250 мл 0,1 М раствора кислоты хлористово
дородной и кипятят с обратным холодильником
при постоянном помешивании в течение 1 ч. Ox
лаждают и декантируют.
Внешний вид раствора. Раствор S1
должен быть прозрачным (2.2.1) и бесцветным
(2.2.2, метод 11).
Кислотность или щелочность. К 100 мл
раствора 81 прибавляют 0,15 мл раствора иH
дикатора BRP Р. При прибавлении не более
1,5 мл 0,01 М раствора натрия 2идроксида
окраска раствора должна измениться на синюю.
К 100 мл раствора 81 прибавляют 0,2 мл pacт
вора метилов020 оранжев020 Р. При прибавле
нии не более 1,0 мл 0,01 М раствора кислоты
хлористоводородной окраска раствора должна
измениться с желтой на оранжевую.
Оптическая плотность (2.2.25). Оптическая
: плотность раствора S1 в диапазоне длин волн от
: 220 нм до 340 нм не должна превышать 0,2.
Восстанавливающие вещества. К 20 мл
раствора 81 прибавляют 1 мл кислоты серной
разведенной Р и 20 мл 0,002 М раствора калия
пермаН2аната. Кипятят с обратным холодиль
ником в течение 3 мин и немедленно охлаж
дают. Прибавляют 1 r калия йодида Р и He
медленно титруют раствор 0,01 М раствором
натрия тиосульфата, используя в качестве
индикатора 0,25 мл раствора крахмала Р. Па
раллельно проводят контрольный опыт. Раз
ность между объемами титранта не должна
превышать 0,5 мл.
Вещества, растворимые в reKcaHe.
1 О r испытуемоro материала помещают в KO
ническую колбу из боросиликатноro стекла
с притертой пробкой вместимостью 250 мл.
Прибавляют 100 мл 2ексана Р и кипятят с об
ратным холодильником при постоянном пе
ремешивании в течение 4 ч. Охлаждают в
ледяной бане и быстро фильтруют через CTe
клянный фильтр (16) (2.1.2), поддерживая
температуру раствора на уровне О ос (время
фильтрования должно составлять менее
5 мин; при необходимости для ускорения про
цесса допускается фильтрование под давле
нием). 20 мл фильтрата выпаривают в пред
варительно взвешенной стеклянной чашке на
водяной бане. Остаток высушивают в сушиль
ном шкафу при температуре 1 oo 105 ос в Te
чение 1 ч. Масса полученноro остатка должна
находиться в пределах 10 % от массы OCTaT
ка, полученноro для типовоro образца и не
должна превышать 5 %.
Добавки. Тонкослойная хроматоrрафия
(2.2.27).
Испытуемый раствор. 50 мл раствора S2
выпаривают досуха в вакууме при температуре
45 ос. Полученный остаток растворяют в 5 мл
метиленхлорида Р. Контрольный раствор roто
вят из контрольноrо раствора, соответствующе
ro раствору S2.
Раствор сравнения. 20 Mr ФСО добавки к
пластмассе 15 и 20 Mr ФСО добавки к пласт
массе 08 растворяют вметиленхлориде Р и дo
водят до объема 10 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем сили
ка2еля G Р.
Подвижная фаза А: 2ексан Р.
Подвижная фаза В: метанол Р метилен
хлорид Р (5:95, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы А: 13 см.
Высушивание А: на воздухе.
Фронт подвижной фазы В: 1 О см.
Высушивание В: на воздухе.
Проявление: пластинку опрыскивают pac
твором 40 r/л кислоты фосфорномолибдено
вой Р в 96 % спирте Р и наrревают в сушильном
шкафу при температуре 120 ос до появления
пятен на xpoMaTorpaMMe раствора сравнения.
540
rосударственная фармакопея Республики Беларусь
Приzодность хромат02рафической cиcтe
мы: раствор сравнения:
на xpoMaTorpaMMe должно обнаруживать
ся два полностью разделенных пятна.
Предельное содержание: на xpoMaTorpaM
ме испытуемоro раствора не должно обнару
живаться пятен за исключением пятна, обна
руживаемоro на фронте подвижной фазы и
соответствующеro олиrомерам. На xpOMaTO
rpaMMe испытуемоro раствора не учитывают
пятна, обнаруживаемые на xpoMaTorpaMMe
контрольноro раствора.
Экстраrируемые тяжелые металлы (2.4.8,
метод А). Не более 2,5 ррm. 50 мл раствора 83
упаривают до объема около 5 мл на водяной
бане и доводят водой Р до объема 20 мл. 12 мл
полученноro раствора должны выдерживать ис
пытание на тяжелые металлы. Эталон rотовят с
использованием 2,5 мл этаЛОНН020 раствора
свинца (10 ррт РЬ) Р.
Сульфатная зола (2.4. 14). Не более 0,02 %.
Определение проводят из 5,0 r испытуемоro Ma
териала.
01/2013:30105
3.1.5. ПОЛИЭТИЛI;Н С ДОБАВКАМИ
ДЛЯ КОНТЕИНЕРОВ
ДЛЯ ПАРЕНТЕРАЛЬНЫХ
И ОФТАльмолоrИЧЕСКИХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
ОПРЕДЕЛЕНИЕ
Полиэтилен с добавками, полученный поли
меризацией этилена под высоким давлением в
присутствии катализатора или сополимеризаци
ей этилена с не более 25 % rомолоroв высших
алкенов (от С з до С 10 ).
ПРОИЗВОДСТВО
Определенное количество добавок вводит
ся в полимер для улучшения еro химических,
физических и механических свойств, а также
для обеспечения возможности использования
по назначению. Все добавки выбирают из приве
денноro ниже перечня, который определяет для
каждоro вещества максимально допустимое еro
содержание.
Полимер может содержать не более трех
антиоксидантов, один или несколько смазываю
щих или антиадrезивных areHToB, а также титана
диоксид как средство, придающее непрозрач
ность для защиты материала от света:
бутилrидрокситолуол (добавка к пластмас
се 07): не более 0,125 %;
пентаэритритилтетракис[3 (З,5 ди трет
бутил 4 rидроксифенил)пропионат) (добавка к
пластмассе 09): не более 0,3 %;
1 ,3,5 трис(3,5 ди трет бутил 4 rидро
ксибензил ) S триазин 2.4, 6( 1 Н,ЗН,5Н) трион
(добавка к пластмассе 13): не более 0,3 %;
октадецил З (3,5 ди трет бутил--4 rидро
ксифенил )пропионат (добавка к пластмассе 11):
не более 0,3 %;
этиленбис[3.3 бис[3 (1, 1 диметилэтил)--4
rидроксифенил)бутаноат) (добавка к пластмассе
08): не более 0,3 %;
диоктадецилдисульфид (добавка к пласт
массе 15): не более 0,3 %;
4,4' ,4" (2,4,6 триметилбензен 1 ,3,5
триилтрисметилен)трис[2,6 бис(1, 1 диметилэтил)
фенол] (добавка к пластмассе 10): не более 0,3 %;
2,2' бис( октадецилокси ) 5,5' спироби[1.3,2
диоксафосфинан) (добавка к пластмассе 14): не
более 0,3 %;
дидодецил 3,З' тиодипропионат (добавка к
пластмассе 16): не более 0,3 %;
диоктадецил 3,З' тиодипропионат (добав
ка к пластмассе 17): не более 0,3 %;
трис[2,4 бис( 1,1 диметилэтил )фенил ]фос
фит (добавка к пластмассе 12): не более 0,3 %.
Сумма перечисленных выше антиоксидант
ных добавок не должна превышать 0,3 %.
rидротальцит: не более 0,5 %;
алканамиды: не более 0,5 %;
алкенамиды: не более 0,5 %;
натрия алюмосиликат: не более 0,5 %;
кремния диоксид: не более 0,5 %;
натрия бензоат: не более 0,5 %;
эфиры или соли жирных кислот: не
более 0,5 %;
тринатрия фосфат: не более 0,5 %;
вазелиновое масло (парафин жидкий): не
более 0,5 %;
цинка оксид: не более 0,5 %;
тальк: не более 0,5 %;
маrния оксид: не более 0,2 %;
кальция стеарат или цинка стеарат или их
сумма: не более 0,5 %;
титана диоксид только для контейне
ров для офтальмолоrических лекарственных
средств: не более 4 %.
Поставщик материала несет OTBeTCTBeH
ность за то, чтобы качественный и количествен
ный составы типовоro образца COOTBeTCTBOBa
ли качественному и количественному составам
каждой произведенной серии.
ОПИСАНИЕ
Порошок, шарики, rранулы или после
трансформации прозрачные пластинки раз
личной толщины или контейнеры.
Практически нерастворим в воде, в этаноле,
в reKcaHe и в метаноле, растворим в rорячих apo
матических уrлеводородах.
Размяrчается при температуре 70 140 ОС.
Относительная плотность: от 0,890 до 0,965.
3.1.5. Полиэтилен с добавками для контейнеров
541
ИДЕНТИФИКАЦИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
А. Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
При20товление: к 0,25 r испытуемоro MaTe
риала прибавляют 1 О мл толуола Р и кипятят
с обратным холодильником в течение 15 мин.
Несколько капель раствора помещают на диск
натрия хлорида и выпаривают растворитель в
сушильном шкафу при температуре 80 ОС.
Максимумы П02лощения: при 2920 CM 1,
2850 CM 1, 1465 CM \ 1375 CM \ 1170 CM 1, 7ЗО CM 1
И 720 CM 1 .
Полученный спектр должен соответствовать
спектру типовоro образца. Если материал имеет
форму пластин, идентификацию можно прове
сти непосредственно на вырезанном фраrменте
пластинки соответствующеro размера.
В. Испытуемый материал выдерживает ис
пытание на предельное содержание добавок
(см. раздел «Испытания»).
с. 25 Mr испытуемоro материала смешива
ют в платиновом тиrле с 1 r калия 2идросульфа
та Р и наrревают до полною расплавления. Ox
лаждают и прибавляют 20 мл кислоты серной
разведенной Р. Осторожно наrревают и филь
труют полученный раствор. К фильтрату прибав
ляют 1 мл концентрироваНН020 раствора вoдo
рода пероксида Р. Если испытуемый материал
содержит титана диоксид, появляется оранжево
желтое окрашивание.
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
Раствор 51. Раствор 81 используют в тe
чение 4 ч после при20товления. 25 r испытуемо
ro материала помещают в колбу из боросиликат
ноro стекла с притертой пробкой. Прибавляют
500 мл воды для инъекций Р и кипятят с обрат
ным холодильником В течение 5 ч. Охлаждают
и декантирую Часть раствора оставляют для
проведения испытаний на прозрачность и цвет
ность. Оставшуюся часть раствора фильтруют
через стеклянный фильтр (16) (2.1.2).
Раствор 52 2,0 r испытуемоro материала
помещают в коническую КlJлбу из боросиликат
ноro стекла с притертой пробкой. Прибавляют
80 мл толуола Р и кипятят с обратным холо
дильником при постоянном перемешивании в
течение 1,5 ч. Охлаждают до 60 ос и прибавляют
при продолжительном перемешивании 120 мл
метанола Р. Фильтруют раствор через CTe
клянный фильтр (16) (2.1.2). Промывают колбу
и фильтр 25 мл смеси толуол Р метанол Р
(40:60, об/об), прибавляют промывную жидкость
к фильтрату и доводят до объема 250,0 мл этой
же смесью растворителей. Параллельно rотовят
контрольный раствор.
Раствор 53. 100 r испытуемоro материала
помещают в коническую колбу из боросиликат
ноro стекла с притертой пробкой. Прибавляют
250 мл 0,1 М раствора кислоты хлористово
дородной и кипятят с обратным холодильником
при постоянном перемешивании в течение 1 ч.
Охлаждают и декантируют раствор.
Внешний вид раствора. Раствор S1
должен быть прозрачным (2.2.1) и бесцветным
(2.2.2, метод 11).
Кислотность или щелочность. К 100 мл
раствора S1 прибавляют 0,15 мл раствора иH
дика тора BRP Р. При прибавлении не более
1,5 мл 0,01 М раствора натрия 2идроксида
окраска раствора должна измениться на синюю.
К 100 мл раствора S1 прибавляют 0,2 мл pacт
вора метилов020 оранжев020 Р. При прибавле
нии не более 1,0 мл 0,01 М раствора кислоты
хлористоводородной окраска раствора должна
измениться с желтой на оранжевую.
Оптическая плотность (2.2.25). Оптиче
ская плотность раствора S1 в диапазоне длин
волн от 220 нм до 340 нм не должна превы
шать 0,2.
Восстанавливающие вещества. К 20 мл
раствора 81 прибавляют 1 мл кислоты серной
разведенной Р и 20 мл 0,002 М раствора калия
пермаН2аната. Кипятят с обратным холодильни
ком в течение 3 мин и сразу охлаждают. Прибав
ляют 1 r калия йодида Р и немедленно титруют
раствор 0,01 М раствором натрия тиосульфа
та, используя в качестве индикатора 0,25 мл
р.аствора крахмала Р. Параллельно проводят
контрольный опыт. Разность между объемами
титранта не должна превышать 0,5 мл.
Вещества, растворимые в reKcaHe. 10
r испытуемоro материала помещают в KO
ническую колбу из боросиликатноrо стекла
с притертой пробкой вместимостью 250 мл.
Прибавляют 100 мл 2ексана Р и кипятят с
обратным холодильником при постоянном
помешивании в течение 4 ч. Охлаждают в
ледяной бане и быстро фильтруют через
стеклянный фильтр (16) (2.1.2), поддержи
вая температуру раствора на уровне О ос
(время фильтрования должно составлять
менее 5 мин; при необходимости для YCKO
рения процесса допускается фильтрование
под давлением). 20 мл фильтрата выпари
вают в предварительно взвешенной чашке
из боросиликатноrо стекла на водяной бане.
542
rосударственная фармакопея Республики Беларусь
Остаток высушивают в сушильном шкафу
при температуре 100 105 ос в течение 1
ч. Масса полученноro остатка должна Ha
ходиться в пределах 1 О % от массы остат
ка, полученноro для типовоro образца и не
должна превышать 5 %.
Экстраrируемый алюминий. Не более
1 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют paCT
вор 83.
Раствор сравнения. rотовят путем раз
ведения этаЛОНН020 раствора алюминия
(200 ррт А/) Р 0,1 М раствором кислоты хлори
стоводородной.
Длина волны: длина волны светоиспускания
алюминия З96,15 нм, спектральный фон .
396,25 нм.
Проверяют отсутствие алюминия в исполь
зуемой кислоте хлористоводородной.
Экстраrируемый хром. Не более 0,05 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют paCT
вор 83.
Раствор сравнения. r отовят путем разведе
ния этаЛОНН020 раствора хрома (100 ррт с,) Р
смесью из 2 объемов кислоты хлористоводо
родной Р и 8 объемов воды Р.
Длина волны: длина волны светоиспуска
ния хрома 205,55 нм, спектральный фон
205,50 нм.
Проверяют отсутствие хрома в использу
емой кислоте хлористоводородной.
Экстраrируемый титан. Не более 1 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют раст
вор 83.
Раствор сравнения. rотовят путем раз
ведения этаЛОНН020 раствора титана
(100 ррт Ti) Р 0,1 М раствором кислоты хлори
стоводородной.
Длина волны: длина волны светоиспуска
ния титана 336,12 нм, спектральный фон
336,16 нм.
Проверяют отсутствие титана в использу
емой кислоте хлористоводородной.
Экстраrируемый ванадий. Не более
0,1 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют paCT
вор S3.
Раствор сравнения. rотовят путем разве
дения этаЛОНН020 раствора ванадия (1 2/л V) Р
смесью из 2 объемов кислоты хлористоводо
родной Р и 8 объемов воды Р.
Длина волны: длина волны светоиспуска
ния ванадия 292,40 нм, спектральный фон
292,35 нм.
Проверяют отсутствие ванадия в использу
емой кислоте хлористоводородной.
Экстраrируемый цинк. Не более 1 ррт.
Атомно абсорбционная спектрометрия
(2.2.23, метод /).
Испытуемый раствор. Используют paCT
вор 83.
Раствор сравнения. r отовят путем разведе
ния этаЛОНН020 раствора цинка (10 ррт Zn) Р
0,1 М раствором кислоты хлористоводоро
ной.
Источник излучения: лампа с полым цинк<r
вым катодом.
Длина волны: 213,9 нм.
Атомизатор: воздушно ацетиленовое
пламя.
Проверяют отсутствие цинка в использу
емой кислоте хлористоводородной.
Экстраrируемый цирконий. Не более
0,1 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют paCT
вор 83.
Раствор сравнения. rотовят путем разведе
ния этаЛОНН020 раствора циркония (1 2/Л Zr) Р
смесью из 2 объемов кислоты хлористоводо
родной Р и 8 объемов воды Р.
Длина волны: длина волны светоиспускания
циркония 343,82 нм, спектральный фон
343,92 нм.
Проверяют отсутствие циркония в ис
пользуемой кислоте хлористо водородной.
Экстраrируемые тяжелые металлы
(2.4.8, метод А). Не более 2,5 ррm. 50 мл
раствора SЗ упаривают до объема около
5 мл на водяной бане и доводят водой Р до
объема 20,0 мл. 12 мл полученноro paCT
вора должны выдерживать испытание на тя
желые металлы. Эталон rотовят с использо
ванием 2,5 мл этаЛОНН020 раствора свинца
(10 ррт РЬ) Р.
Сульфатная зола (2.4.14). Не более
1,0 %. Определение проводят из 5,0 r испы
туемоro материала. Эта норма не распро
страняется на материалы, которые содержат
титана диоксид как добавку, придающую He
прозрачность материалу.
ДОПОЛНИТЕЛЬНЫЕ ИСПЫТАНИЯ
Эти испытания следует проводить полно
стью или частично только е тех случаях, коrда
этою требует заявленный состав или область
применения материала.
Фенольные антиоксиданты. Жидкостная
хроматоrрафия (2.2.29).
Смесь растворителей. Ацетонитрил Р
тетра2идрофуран Р (50:50, об/об).
3.1.5. Полиэтилен с добавками для контейнеров
543
Испытуемый раствор 821. 50 мл paCT
вора 82 выпаривают досуха в вакууме при TeM
пературе 45 ос. Остаток растворяют в 5,0 мл
смеси растворителей. Контрольный раствор ro
товят из контрольноro раствора, соответствую
щеrо раствору 52.
Испытуемый раствор 822. 50 мл paCT
вора 82 выпаривают досуха в вакууме при TeM
пературе 45 ос. Остаток растворяют в 5,0 мл
метиленхлорида Р. Контрольный раствор roто
вят из контрольноro раствора, соответствующе
ro раствору 82.
Из приведенных ниже растворов cpaвHe
ния 20товят только те, которые необходимы
для анализа фенольных антиоксидантов, вxo
дящих в состав испытуемоао материала.
Раствор сравнения (а). 25,0 Mr ФСО бу
тиЛ2идрокситолуола (добавка к пластмассе
07) и 60,0 Mr ФСО добавки к пластмассе 08
растворяют в 10,0 мл смеси растворителей.
2,0 мл полученноro раствора доводят смесью
растворителей до объема 50,0 мл.
Раствор сравнения (Ь). 60,0 Mr ФСО дo
бавки к пластмассе 09 и 60,0 Mr ФСО добавки
к пластмассе 10 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro раствора
доводят смесью растворителей до объема
50,0 мл.
Раствор сравнения (с). 60,0 Mr ФСО добав
ки к пластмассе 11 и 60,0 Mr ФСО добавки к
пластмассе 12 растворяют в 10,0 мл метилен
хлорида Р. 2,0 мл полученноro раствора ДOBO
дят метиленхлоридом Р до объема 50,0 мл.
Раствор сравнения (d). 25,0 Mr ФСО бу
тиЛ2идрокситолуола (добавка к пластмас
се 07) растворяют в 10,0 мл смеси раствори
телей. 2,0 мл полученноro раствора доводят
смесью растворителей до объема 50,0 мл.
Раствор сравнения (е). 60,0 Mr ФСО добав
ки к пластмассе 08 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro раствора
доводят смесью растворителей Р до объема
50,0 мл.
Раствор сравнения (f). 60,0 Mr ФСО дa
бавки к пластмассе 13 растворяют в 10,0 мл
смеси растворителей. 2,0 мл полученноro paCT
вора доводят смесью растворителей до объема
50,0 мл.
Раствор сравнения (g). 60,0 Mr ФСО дo
бавки к пластмассе 09 растворяют в 10,0 мл
смеси растворителей. 2,0 мл полученноro paCT
вора доводят смесью растворителей до объема
50,0 мл.
Раствор сравнения (h). 60,0 Mr ФСО дo
бавки к пластмассе 10 растворяют в 10,0 мл
смеси растворителей. 2,0 мл полученноro paCT
вора доводят смесью растворителей до объема
50,0 мл.
Раствор сравнения (i). 60,0 Mr ФСО добав
ки к пластмассе 11 растворяют в 10,0 мл Meти
ленхлорида Р. 2,0 мл полученноro раствора дo
водят метиленхлоридом Р до объема 50,0 мл.
Раствор сравнения (j). 60,0 Mr ФСО добав
ки к пластмассе 12 растворяют в 10,0 мл Meти
ленхлорида Р. 2,0 мл полученноro раствора дo
водят метиленхлоридом Р до объема 50,0 мл.
Условия хроматоrрафирования:
А. Если испытуемый материал содержит дo
бавку к пластмассе 07 и/или добавку к пластмас
се 08, используют следующие условия:
колонка длиной 0,25 м и внутренним ди
аметром 4,6 мм, заполненная силика2елем OK
тадецилсилильным для хромат02рафии Р с
размером частиц 5 мкм;
подвижная фаза: вода Р aцeтOHи
трил Р (30:70, об/об);
скорость подвижной фазы: 2 мл/мин;
детектирование: спектрофотометриче--
ский детектор, длина волны 280 нм;
объем вводимой пробы: по 20 мкл испы
туемоro раствора 521, соответствующеro KOH
трольноro раствора, раствора сравнения (а), и
либо растворов сравнения (d) или (е), либо pac
творов сравнения (d) и (е);
время хроматоарафирования: 30 мин.
При2одность хромат02рафической cиcтe
мы:
разрешение: не менее 8,0 между пиками
добавки к пластмассе 07 и добавки к пластмас
се 08 на xpoMaTorpaMMe раствора сравнения (а);
на xpoMaTorpaMMe испытуемоro раст
вора 821 должны обнаруживаться пики только
тех антиоксидантов, которые входят в состав ис
пытуемоro материала, кроме этоro MOryT обна
руживаться пики, присутствующие на xpOMaтo
rpaMMe контрольноro раствора.
Предельное содержание: площади пиков на
xpoMaTorpaMMe испытуемоro раствора 521 не
должны превышать площадей соответствующих
пиков на xpoMaTorpaMMe растворов сравнения
(d) и/или (е).
В. Если испытуемый материал содержит
один или более следующих антиоксидантов:
добавка к пластмассе 09;
добавка к пластмассе 1 о;
добавка к пластмассе 11;
добавка к пластмассе 12;
добавка к пластмассе 13,
проводят испытание, как описано выше, со
следующими изменениями:
подвижная фаза: вода Р тeтpa2идpo
фуран P ацетонитрил Р (10:30:60, об/об/об);
скорость подвижной фазы: 1,5 мл/мин;
объем вводимой пробы: по 20 мкл испы
туемоro раствора 521, соответствующеro KOH
трольноro раствора, раствора сравнения (Ь) и
растворов сравнения антиоксидантов в соответ
ствии с перечисленным выше списком и входя
щих В состав испытуемоro материалэ.
При20дность хромат02рафической cиcтe
мы:
разрешение: не менее 2,0 между пиками
добавки к пластмассе 09 и добавки к пластмас
се 1 О на xpoMaTorpaMMe раствора сравнения (Ь);
544
rосударственная фармакопея Республики БеларуCbj
на xpoMaTorpaMMe испытуемоro раст
вора 821 должны обнаруживаться пики только
тех антиоксидантов, которые входят в состав ис
пытуемоrо материала, кроме этоro MOryт обна
руживаться пики, присутствующие на xpOMaTO
rpaMMe контрольноrо раствора.
Предельное содержание: площади пиков на
xpoMaTorpaMMe испытуемоro раствора 821 не
должны превышать площадей соответствующих
пиков на xpoMaтorpaMMe растворов сравнения
антиоксидантов в соответствии с перечислен
ным выше списком и входящих в состав испыту
емоro материала.
. Если испытуемый материал содержит
добавку к пластмассе 11 и/или добавку к пласт
массе 12, проводят испытание, как описано для
добавки к пластмассе 07 и/или добавки к пласт
массе 08, со следующими изменениями:
подвижная фаза: вода Р 2 пропа
нол Р метанол Р (5:45:50, об/об/об);
скорость подвижной фазы: 1,5 мл/мин;
объем вводимой пробы: по 20 мкл испы
туемоro раствора 822, соответствующеro KOH
трольноro раствора, раствора сравнения (с), и
либо растворов сравнения (i) или (j), либо pac
творов сравнения (i) и (j).
При20дность хромат02рафической cиc
темы:
разрешение: не менее 2,0 между пиками
добавки к пластмассе 11 и добавки к пластмас
се 12 на xpoMaтorpaMMe раствора сравнения (с);
на xpoMaтorpaMMe испытуемою paCT
вора 822 должны обнаруживаться пики только
тех антиоксидантов, которые входят в состав ис
пытуемоro материала, кроме этоro MOryт обна
руживаться пики присутствующие на xpOMaTO
rpaMMe контрольною раствора.
Предельное содержание: площади пиков на
xpoMaTorpaMMe испытуемоro раствора 822 не
должны превышать площадей соответствующих
пиков на xpoMaTorpaMMe растворов сравнения
(i) иlили Ш.
Нефенольные антиоксиданты. TOHKOC
лойная хроматоrрафия (2.2.27).
Испытуемый раствор 823. 100 мл paCT
вора 82 выпаривают досуха в вакууме при TeM
пературе 45 ос. Остаток растворяют в 2 мл Me
тиленхлорида подкислеНН020 Р.
Раствор сравнения (k). 60 Mr ФСО добавки к
пластмассе 14 растворяют в 1 О мл метиленхло
рида Р. 2 мл полученноro раствора доводят Meти
ленхлоридом подкисленным Р до объема 1 О мл.
Раствор сравнения (1). 60 Mr ФСО добавки к
пластмассе 15 растворяют в 1 О мл метиленхло
рида Р. 2 мл полученною раствора доводят Meти
ленхлоридом подкисленным Р до объема 1 О мл.
Раствор сравнения (т). ФСО 60 Mr добавки
к пластмассе 16 растворяют в 1 О мл метилен
хлорида Р. 2 мл полученноro раствора доводят
метuленхлоридом подкuсленным Р до объема
10 мл.
Раствор сравнения (п). 60 Mr ФСО добавки
пластмассе 17 растворяют в 1 О мл метиленхло1
рида Р. 2 мл полученноro раствора доводят Memu-j
ленхлоридом подкисленным Р до объема 1 О мл.
Раствор сравнения (о). 60 Mr ФСО добавi
ки к пластмассе 16 и ФСО добавки к пластмаС1
се 17 растворяют в 1 О мл метиленхлорида Р.I
2 мл полученноrо раствора доводят метилеН-i
хлоридом подкисленным Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем сили
ка2еля GF 254 Р.
Подвижная фаза А: 2ексан Р.
Подвижная фаза В: метиленхлорид Р.
Наносимый объем пробы: по 20 мкл испы
туемоro раствора 823, раствора сравнения (о)
и растворов сравнения, соответствующих всем
фенольным и нефенольным антиоксидантам,
входящим в состав испытуемоro материала.
Фронт подвижной фазы А: 18 см.
Высушивание А: на воздухе.
Фронт подвижной фазы В: 17 см.
Высушивание В: на воздухе.
Проявление: просматривают в ультрафио
летовом свете при длине волны 254 нм; пластин
ку опрыскивают спиртовым раствором йода Р и
через 1 o 15 мин просматривают в ультрафио
летовом свете при длине волны 254 нм.
При20дность хромат02рафической cиcтe
мы раствор сравнения (о):
на xpoMaTorpaMMe должно обнаруживать
ся два полностью разделенных пятна.
Предельное содержание: на xpoMaTorpaMMe
испытуемоro раствора 823 любое пятно должно
быть не интенсивнее соответствующею пятна
на xpoMaTorpaMMe раствора сравнения.
Амиды и стеараты. Тонкослойная xpOMaTO
rрафия (2.2.27).
Испытуемый раствор. Используют испыту
емый раствор 823, приroтовленный, как указано
в испытании «Нефенольные антиоксиданты».
Раствор сравнения (р). 20 Mr ФСО кислоты
стеариновой (добавки к пластмассе 19) paCTBO
ряют в 1 О мл метиленхлорида Р.
Раствор сравнения (q). 40 Mr ФСО олеа
мида (добавки к пластмассе 20) растворяют в
20 мл метиленхлорида Р.
Раствор сравнения ('). 40 Mr ФСО эрука
мида (добавки к пластмассе 21) растворяют в
20 мл метиленхлорида Р.
Пластинка: ТСХ пластинка со слоем сили
ка2еля GF 254 Р (две пластинки).
А. Подвижная фаза: этанол Р тpиMe
тилпентан Р (25:75, об/об).
Наносимый объем пробы: по 1 О мкл испы
туемоro раствора 823 и раствора сравнения (р).
Фронт подвижной фазы: 1 О см.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают pac
твором 2 r/л дихлорфенолиндофенола Haтpи
е80Й соли Р в этаноле Р и наrpевают в сушильном
шкафу при температуре 120 ос до появления пятен.
3.1.6. Полипропилен для контейнеров и укупорочных материалов
545
Предельное содержание: на xpoMaTorpaMMe
испытуемоro раствора S23 пятно, COOTBeTCTBY
ющее добавке к пластмассе 19, должно быть
идентичным по расположению (R, около 0,5) и
быть не интенсивнее соответствующеro пятна
на xpoMaTorpaMMe раствора с авнения (р).
В. Подвижная фаза А: 2ексан Р.
Подвижная фаза В: метанол Р метилен
хлорид Р (5:95, об/об).
Наносимый объем пробы: по 1 О мкл испы
TyeMoro раствора S23 и растворов сравнения
(q) и (r).
Фронт подвижной фазы А: 13 см.
Высушивание А: на воздухе.
Фронт подвижной фазы В: 1 О см.
Высушивание В: на воздухе.
ПРОЯ8Ление: пластинку опрыскивают paCTBO
ром 40 r/л кислоты фосфорномолибденовой Р в
этаноле Р и наrревают в сушильном шкафу при
температуре 120 ос до появления пятен.
Предельное содержание: на xpoMaтorpaM
ме испытуемоro раствора S23 пятна, COOTBeT
ствующие добавке к пластмассе 20 или добав
ке к пластмассе 21, должны быть идентичными
по расположению (R,ОКОЛО 0,2) и быть не интен
сивнее соответствующих пятен на xpoMaTorpaM
ме раствора сравнения (q) и (r).
01/2013:30106
3.1.6. ПОЛИПРОПИЛЕН
ДЛЯ КОНТЕЙНЕРОВ
И УКУПОРОЧНЫХ МАТЕРИАЛОВ
ДЛЯ ПАРЕНТЕРАЛЬНЫХ
И ОФТАльмолоrИЧЕСКИХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
ОПРЕДЕЛЕНИЕ
Полипропилен состоит из roмополиме
ра пропилена или сополимера пропилена с co
держанием не более 25 % этилена или смеси
(сплава) полипропилена с содержанием не
более 25 % полиэтилена. Полимер может coдep
жать добавки.
ПРОИЗВОДСТВО
Определенное количество добавок вводит
ся в полимер для улучшения еro химических,
физических и механических свойств, а также
для обеспечения возможности использования
по назначению. Все добавки выбирают из приве
денноro ниже перечня, который определяет для
каждоro вещества максимально допустимое ero
содержание.
Полимеры MOryт содержать не более трех
антиоксидантов, один или несколько смазыва
lDЩих или антиадrезивных areHToB, а т-акже
. 30. 1712.
титана диоксид как средство, придающее непро
зрачность для защиты материала от света:
бутилrидрокситолуол (добавка к пластмас
се 07): -не более 0,125 %;
пентаэритритилтетракис[3 (3,5 ди трет
бутил-4 rидроксифенил)пропионат) (добавка к
пластмассе 09): не более 0,3 %;
1 ,З,5 трис(3,5 ди трет бутил 4 rидро
ксибензил ) s триазин 2,4,6( 1 H,3H,5H) трион
(добавка к пластмассе 13): не более 0,3 %;
октадецил 3 (3,5 ди трет бутил-4 rидро
ксифенил)пропионат (добавка к пластмассе 11У
не более 0,3 %;
этиленбис[3,3 бис[3 (1, 1 диметилэтил)
4 rидроксифенил)бутаноат) (добавка к пласт
массе 08): не более 0,3 %;
диоктадецилдисульфид (добавка к пласт
массе 15): не более 0,3 %;
,4' ,4" (2,4,6 триметилбензен 1 ,3,5 триил
трисметилен)трис[2,6 бис(1 ,1 диметилэтил)
фенол) (добавка к пластмассе 10): не более
0,3 %;
2,2' бис( октадецилокси) 5,5' спироби[1 ,З,2
ДИОКЕ>афосфинан) (добавка к пластмассе 14): не
более 0,3 %;
дидодецил 3,3' тиодипропионат (добавка к
пластмассе 16): не более 0,3 %;
диоктадецил 3,3' тиодипропионат (добав
ка к пластмассе 17): не более 0,3 %;
трис[2,4 бис(1, 1 диметилэтил)фенил)
фосфит (добавка к пластмассе 12): не более 0,3 %.
Сумма перечисленных выше антиоксидант
ных добавок не должна превышать 0,3 %.
rидротальцит: не более 0,5 %;
алканамиды: не более 0,5 %;
алкенамиды: не более 0 5 %;
натрия алюмосиликат: не более 0,5 %;
кремния диоксид: не более 0,5 %;
натрия бензоат: не более 0,5 %;
эфиры или соли жирных кислот: не
более 0,5 %;
-:-- три натрия фосфат: не более 0,5 %;
вазелиновое масло (парафин жидкий): не
более 0,5 %;
цинка оксид: не более 0,5 %;
тальк: не более 0,5 %;
маrния- оксид: не более 0,2 %;
кальция стеарат или цинка стеарат или их
сумма: не более 0,5 %;
титана ДИОКСИД только для контейне
ров для офтальмолоrических лекарственных
средств: не более 4 %.
Поставщик материала несет OTBeTCTBeH
ность за то, чтобы качественный и количествен
ный составы типовоro образца COOTBeTCTBOBa
ли качественному и КОf1ичественному составам
каждой произведенной серии.
ОПИСАНИЕ
Порошок, шарики, rранулы или после
трансформации полупрозрачные пластинки
. разной толщины или контейнеры.
rосударственная фармакопея Республики Белару1
i
Практически нерастворимы в воде, в этано вании в течение 1,5 ч. Охлаждают до 60 ос
ле, в reKcaHe и в метаноле, растворимы в roря прибавляют при продолжительном перемеши;
чих ароматических уrлеводородах. вании 120 мл метанола Р. Фильтруют paCTBO
Размяrчаютсяпритемпературеокол0120 0 С. через стеклянный фильтр (16) (2.1.2). Пром
вают колбу и фильтр 25 мл смеси толуол Р
метанол Р (40:60, об/об), прибавляют про--
мывную жидкость К фильтрату и доводят до;
объема 250,0 мл этой же смесью растворит 1
лей. Параллельно roтовят контрольный paCT
вор.
546
ИДЕНТИФИКАЦИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
А. Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
При20товление: к 0,25 r испытуемоro MaTe
риала riрибавляют 1 О мл толуола Р и кипятят
с обратным холодильником в течение 15 мин.
Несколько капель раствора помещают на диск
натрия хлорида и выпаривают растворитель в
сушильном шкафу при температуре 80 ОС.
Максимумы П02лощения: при 1З75 CM 1,
1170 CM 1, 995 CM 1 и 970 CM 1.
Полученный спектр должен соответствовать
спектру типовоro образца. Если материал имеет
форму пластин, идентификацию можно прове
сти непосредственно на вырезанном фраrменте
пластинки соответствующеro размера.
В. Испытуемый материал выдерживает ис
пытание на предельное содержание добавок
(см. раздел «Испытания»).
С. 25 Mr испытуемоro материала смешива
ют в платиновом тиrле с 1 r калия 2идросульфа
та Р и наrревают до полноro расплавления. Ox
лаждают и прибавляют 20 мл кислоты серной
разведенной Р. Осторожно наrревают и филь
труют полученный раствор. К фильтрату прибав
ляют 1 мл концентрироваНН020 раствора вoдo
рода пероксида Р. Если испытуемый материал
содержит титана диоксид, появляется оранжево
желтое окрашивание.
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
Раствор 81. Раствор S1 используют в тe
чение 4 ч после при20товления. 25 r испытуемо
ro материала помещают в колбу из боросиликат
ноro стекла с притертой пробкой. Прибавляют
500 мл воды для инъекций Р и кипятят с обрат
ным холодильником В течение 5 ч. Охлаждают
и декантируют. Часть раствора оставляют для
проведения испытаний на прозрачность и ЦBeT
ность. Оставшуюся часть раствора фильтруют
через стеклянный фильтр (16) (2.1.2).
Раствор 82. 2,0 r испытуемоro материала
помещают в коническую колбу из боросили
катноro стекла с притертой пробкой. Прибав
ляют 80 мл толуола Р и кипятят с обратным
холодильником при постоянном перемеши
Раствор 83. 100 r испытуемоro материала
помещают в коническую колбу из боросиликат.
HOro стекла с притертой пробкой. Прибавляют
250 мл 0,1 М раствора кислоты хлористо во.
дородной и кипятят с обратным холодильни.
ком при постоянном перемешивании в течение
1 ч. Охлаждают и декантируют раствор.
Внешний вид раствора. Раствор S1
по степени мутности не должен превышать
эталон 11 (2.2.1) и должен быть бесцветным
(2.2.2, метод 11).
Кислотность или щелочность. К 100 мл
раствора 81 прибавляют 0,15 мл раствора иH
дикатора BRP Р. При прибавлении не более
1,5 мл 0,01 М раствора натрия 2идpOKcи
да окраска раствора должна измениться на '
синюю. К 100 мл раствора S1 прибавляют
0,2 мл раствора метилов020 оранжев020 Р.
При прибавлении не более 1,0 мл 0,01 М pacт
вора кислоты хлористоводородной OKpa
ска раствора должна измениться с желтой на
оранжевую.
Оптическая плотность (2.2.25). Оптиче
ская плотность раствора S1 в диапазоне длин
волн от 220 нм до 340 нм не должна превы
шать 0,2.
Восстанавливающие вещества. К 20 мл
раствора S1 прибавляют 1 мл кислоты серной
разведенной Р и 20 мл 0,002 М раствора калия
пермаН2аната. Кипятят с обратным холодиль
ником в течение 3 мин и сразу охлаждают.
Прибавляют 1 r калия йодида Р и немедлен
но титруют раствор 0,01 М раствором натрия
тиосульфата, используя в качестве индика
тора 0,25 мл раствора крахмала Р. Парал
лельно проводят контрольный опыт. Разность
между объемами титранта не должна превы
шать 0,5 мл.
Вещества, растворимые в reKcaHe. 10 r
испытуемоro материяла помещают в кониче
скую колбу из боросиликатноrо стекла с при
тертой пробкой вместимостью 250 мл. Прибав
ляют 100 мл 2ексана Р и кипятят с обратным
холодильником при постоянном помешивании
в течение 4 ч. Охлаждают в ледяной бане и
быстро фильтруют через стеклянный фильтр
3.1.6. Полипропилен для контейнеров и укупОрОЧНblХ материалов
547
(16) (2.1.2), поддерживая температуру paCT
вора на уровне О ос (время фильтрования
должно составлять менее 5 мин; при необхо
димости для ускорения процесса допускается
фильтрование под давлением). 20 мл филь
трата выпаривают в предварительно взвешен
ной чашке из боросиликатноro стекла на BO
дяной бане. Остаток высушивают в сушильном
шкафу при температуре 1 oo 105 ос в течение
1 ч. Масса полученноro остатка должна Haxo
диться в пределах 1 О % от массы остатка, по
лученноrо для типовою образца и не должна
превышать 5 %.
Экстраrируемый алюминий. Не более
1 ррт.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют paCT
вор S3.
Раствор сравнения. rотовят путем разведе
ния этаЛОНН020 раствора алюминия (200 ррт
А/) р 0,1 М раствором кислоты хлористоводо
родноЙ
Длина волны: длина волны светоиспускания
алюминия 396,15 нм, спектральный фон
396,25 нм.
Проверяют отсутствие алюминия в исполь
зуемой кислоте хлористоводородной.
Экстраrируемый хром. Не более 0,05 ррт.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют раст
вор S3.
Раствор сравнения. rотовят путем разведе
ния этаЛОНН020 раствора хрома (100 ррт С,) Р
смесью из 2 объемов кислоты хлористоводо
родной Р и 8 объемов воды Р.
Длина волны: длина волны светоиспуска
ния хрома 205,55 нм, спектральный фон
205,50 нм.
Проверяют отсутствие хрома в используе
мой кислоте хлористоводородной.
Экстраrируемый титан. Не более 1 ррm. <
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют paCT
вор 83.
Раствор сравнения. rотовят путем раз
ведения этаЛОНН020 раствора титана
(100 ррт Тi) Р 0,1 М раствором кислоты хло
ристоводородной.
Длина волны: длина волны светоиспуска
ния титана 336,12 нм, спектральный фон
336,16 нм.
Проверяют отсутствие титана в используе
мой кислоте хпористоводородной.
Экстраrируемый ванадий. Не более
0,1 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют раст
вор S3.
Раствор сравнения. rотовят путем разве
дения этаЛОНН020 раствора ванадия (1 2/Л V) Р
смесью из 2 объемов кислоты хлористоводо
родной Р и 8 объемов воды Р.
Длина волны: длина волны светоиспуска
ния ванадия 292,40 нм, спектральный фон
292,З5 нм.
Проверяют отсутствие ванадия в использу
емой кислоте хлористоводородной.
Экстраrируемый цинк. Не более 1 ррm.
Атомно абсорбционная спектрометрия
(2.2.23, метод /).
Испытуемый раствор. Используют раст
вор SЗ.
Раствор сравнения. r отовят путем разведе
ния этаЛОНН020 раствора цинка (10 ррт Zn) Р
О, 1 М раствором кислоты хлористоводо
родной.
Источник излучения: лампа с полым цинко
вым катодом.
Длина волны: 213,9 нм.
Атомизатор: воздушно ацетиленовое
пламя.
Проверяют отсутствие цинка в использу
емой кислоте хлористоводородной.
Экстраrируемые тяжелые металлы (2.4.8,
метод А). Не более 2,5 ррm. 50 мл раствора
SЗ упаривают до объема около 5 мл на водя
ной бане и доводят водой Р до объема 20,0 мл.
12 мл полученноro раствора должны выдержи
вать испытание на тяжелые металлы. Эталон
roтовят с использованием 2,5 мл этаЛОНН020
раствора свинца (10 ррт РЬ) Р.
Сульфатная зола (2.4.14). Не более
1,0 %. Определение проводят из 5,0 r испыту
eMoro материала. Эта норма не распространя
ется на материалы, которые содержат титана
диоксид как добавку, придающую непрозрач
ность материалу.
ДОПОЛНИТЕЛЬНЫЕ ИСПЫТАНИЯ
Эти испытания следует проводить пол
ностью или частично только в тех случаях,
К02да эт020 требует заявленный состав или
область применения материала.
Фенольные антиоксиданты. Жидкостная
хроматоrрафия (2.2.29).
Смесь растворителей. Ацетонитрил Р
тетра2идрофуран Р (50:50, об/об).
Испытуемый раствор S21. 50 мл paCT
вора S2 выпарив::!ют досуха в вакууме при TeM
пературе 45 ОС. Остаток растворяют в 5,0 мл
смеси растворителей. Контрольный раствор ro
товят из контрольноro раствора, соответствую
щеro раствору 82. .
Испытуемый раствор S22. 50 мл paCT
вора 82 выпаривают досуха в вакууме при TeM
548
rосударственная фармакопея Республики Беларусь
пературе 45 ос. Остаток растворяют в 5.0 мл Me
тиленхлорида Р. Контрольный раствор roтовят
из контрольноro раствора, соответствующеro
раствору 82. ...
Из приведенных ниже.растворов сравнения
20товят только те, которые необходимы для
анализа фенольных антиоксидантов, входя
щих в состав испытуеМ020 материала.
Раствор сравнения (а). 2.5,0 Mr ФСО бутил
2идрокситолуола (добавка к пластмассе 07) и
60,0 Mr ФСО добавки к пластмассе 08 paCTBO
ряют в 10,0 мл смеси растворителей. 2,0 л по
лученноro раствора доводят смесью растiюри
телей до объ ма 50,0 мл.
Раствор сравнения (Ь). 60,0 Mr ФСО 90
бавки к пластмассе 09 и 60,0 Mr ФСО добавки
к пластмассе 10 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro 'раствора дo
водят смесью растворителей до об -ема 50,0 мл.
Раствор сравнения (с). 60.0 Mr ФСО добав
ки к пластмассе 11 и 60,0 Mr ФСО добавки к
пластмассе 12 растворяют в 10,0 мл метилен
хлорида Р. 2,0 мл полученноro раствора доводят
метиленхлоридом Р до объема 50,0 мл.
Раствор сравнения (d). 25,0 Mr ФСО бу
тиЛ2идрокситолуола (добавка к пластмассе
07) растворяют в 10,0 мл смеси растворителей.
2,0 мл полученноro раствора доводят смесью
растворителей до объема 50,0 мл.
Раствор сравнения (е). 60,0 Mr ФСО добав
ки к пластмассе 08 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro раствора
доводят смесью растворителей Р до объема
50,0 мл.
Раствор сравнения (f). 60,0 Mr ФСО добав
ки к пластмассе 13 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноrо раствора дo
водят смесью растворителей до объема 50,0 мл.
Раствор сравнения (g). 60,0 Mr ФСО добав
ки к пластмассе 09 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученною раствора дo
водят смесью растворителей до объема 50,0 мл.
Раствор сравнения (h). 60,0 Mr ФСО добав
ки к пластмассе 10 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro раствора дo
водят смесью растворителей до объема 50,0 мл.
Раствор сравнения (i). 60,0 Mr ФСО добавки
к пластмассе 11 растворяют в 1.0,0 мл метилен
хлорида Р. 2,0 мл полученноro раствора доводят
метиленхлоридом Р до объема 50,0 мл.
Раствор сравнения (j). 60,0 Mr ФСО добав
ки к пластмассе 12 растворяют в 10,0 мл Meти
ленхлорида Р. 2,0 мл полученноro раствора дo
водят метиленхлоридом Р до объема 50,0 мл.
УСJ10ВИЯ хроматоrрафирования:
А. ЕСЛL1 испытуемый. материал <;одержит дo
бавку к пластмассе 07 иlили добавку к пластмас
се 08, используют следующие условия:
колонка длиной 0,25 м и внутренним ди
аметром 4,6 мм, заполненная силика2елем OK
тадецилсилильным для хромат02рафии Р с
размером частиц 5 мкм;
подвижная- фаза: вода Р ацетонu-'
трил Р (30:70, об/об);
скорость подвижной фазы: 2 мл/мин;
. детектирование: спектрофотометриче--
ский детектор, длина волны 280 нм;
объем вводимой пробы: по 20 мкл испы
туемоro раствора 821, соответствующеro KOH
трольною раствора, раствора сравнения (а), и
либо растворов сравнения (d) или (е), либо pac
творов сравнения (d) и (е);
время хромат02рафирования: 30 мин.
При20дность хромат02рафической cиcтe
мы:
разрешение: не менее 8,0 между пиками
добавки к пластмассе 07 и добавки к пластмас
се 08 на xpoMaтorpaMMe раствора сравнения (а);
на xpoMaTorpaMMe испытуемоro раст
opa 821 должны обнаруживаться пики только
тех антиоксидантов, которые входят в состав ис
пытуемоrо материала, кроме этоro MOryT обна
руживаться пики, присутствующие на xpOMaTO
rpaMMe контрольноro раствора.
Предельное содержание: площади пиков на
xpoMaTorpaMMe испытуемоro раствора S21 не
должны превышать площадей соответствующих
пиков на xpoMaTorpaMMe растворов сравнения
(d) и/или (е).
В. Если испытуемый материал содержит
один или более следующих антиоксидантов:
добавка к пластмассе 09;
добавка к пластмассе 1 о;
добавка к пласtмассе 11;
добавка к пластмассе 12;
добавка к пластмассе 13,
проводят испытание, как описано выше, со
следующими изменениями:
подвижная фаза: вода Р тeтpa2идpo
фуран p ацетонитрил Р (10:30:60, об/об/об);
скорость подвижной фазы: 1,5 мл/мин;
. объем вводимой пробы: по 20 мкл испы
туемоro раствора 821, соответствующеro KOH
трольноro раствора, раствора сравнения (Ь) и
растворов сравнения антиоксидантов в соответ
ствии с перечисленным выше списком и входя
щих В состав испытуемоro материала.
При2одность хромат02рафической системы:
разрешение: не менее 2,0 между пиками
добавки к пластмассе 09 и добавки к пластмас
се 1 О на xpoMaTorpaMMe раствора сравнения (Ь);
на xpOMaTorpaMMe испытуемоro раст
вора S21 должны обнаруживаться пики только
тех антиоксидантов, которые входят в состав ис
пытуемоrо материала, кроме этою Moryт обна
руживаться пики, присутствующие на xpOMaTO
rpaMMe контрольноro раствора.
Предельное содержание: площади пиков на
xpoMaTorpaMMe испытуемоrо раствора S21 не
должны превышать площ"щей соответствующих
пиков на xpoMaTorpaMMe растворов сравнения
антиоксидантов в соответствии с перечислен
ным выше списком и входящих в состав испыту
емоro материала.
3.1.6. Полипропилен для контейнеров и укупорочных материалов
549
с. Если испытуемый материал содержит
добавку к пластмассе 11 и/или добавку к пласт
массе 12, проводят испытание, как описано для
добавки к пластмассе 07 и/или добавки к пласт
массе 08, со следующими изменениями:
подвижная фаза: вода Р 2 пропа
нол Р метанол Р (5:45:50, об/об/об);
скорость подвижной фазы: 1,5 мл/мин;
объем вводимой пробы: по 20 мкл испы
туемоro раствора S22, соответствующеro KOH
трольноro раствора, раствора сравнения (с), и
либо растворов сравнения (i) или 0), либо pac
творов сравнения (i) и 0).
При2одность хромат02рафической системы:
разрешение: не менее 2,0 между пиками
добавки к пластмассе 11 и добавки к пластмас
се 12 на xpoMaTorpaMMe раствора сравнения (с);
на xpoMaTorpaMMe испытуемоro paCT
вора 822 должны обнаруживаться пики только
тех антиоксидантов, которые входят в состав ис
пытуемоro материала, кроме этоro MOryт обна
руживаться пики, присутствующие на xpOMaтo
rpaMMe контрольноro раствора.
Предельное содержание: площади пиков на
xpoMaTorpaMMe испытуемоro раствора S22 не
должны превышать площадей соответствующих
пиков на xpoMaтorpaMMe растворов сравнения
(i) и/или 0).
Нефенольные антиоксиданты. TOHKOC
лойная хроматоrрафия (2.2.27).
Испытуемый раствор S23. 100 мл paCT
вора S2 выпаривают досуха в вакууме при TeM
пературе 45 ос. Остаток растворяют в 2 мл Me
тиленхлорида подкислеНН020 Р.
Раствор сравнения (k). 60 Mr ФСО добавки
к пластмассе 14 растворяют в 1 О мл метилен
хлорида Р. 2 мл полученноro раствора доводят
метиленхлоридом подкисленным Р до объема
10 мл.
Раствор сравнения (1).60 Mr ФСО добавки
к пластмассе 15 растворяют в 1 О мл метилен
хлорида Р. 2 мл полученноro раствора доводят
метиленхлоридом подкисленным Р до объема'
10 мл.
Раствор сравнения (т). ФСО 60 Mr добавки
к пластмассе 16 растворяют в 1 О мл метилен
хлорида Р. 2 мл полученноro раствора доводят
метиленхлоридом подкисленным Р до объема
10 мл.
Раствор сравнения (п). 60 Mr ФСО добавки
к пластмассе 17 растворяют в 1 О мл метилен
хлорида Р. 2 мл полученноro раствора доводят
метиленхлоридом подкисленным Р до объема
1Омл.
РАствор сравнения (о). 60 Mr ФСО добавки
к пластмассе 16 и ФСО добавки к пластмассе
17 растворяют в 1 О мл метиленхлорида Р. 2 мл
полученноrо раствора доводят метиленхлори
дом подкисленным Р до объема 10 мл.
Пластинка: ТСХ пластинка со слоем сили
ка2еля GF Z54 Р.
70 За. 1712
Подвижная фаза А: 2ексан Р.
Подвижная фаза В: метиленхлорид Р.
Наносимый объем пробы: по 20 мкл испы
туемоro раствора 823, раствора сравнения (о)
и растворов сравнения, соответствующих всем
фенольным и нефенольным антиоксидантам,
входящим в состав испытуемоro материала.
Фронт подвижной фазы А: 18 см.
Высушивание А: на воздухе.
Фронт подвижной фазы В: 17 см.
Высушивание В: на воздухе.
ПРОЯ8Ление: просматривают в ультрафио
летовом свете при длине волны 254 нм; пластин
ку опрыскивают спиртовым раствором йода Р и
через 1 (}"",,15 мин просматривают в ультрафио
летовом свете при длине волны 254 нм.
При20дность хромат02рафической cиcтe
мы: раствор сравнения (о):
на xpoMaтorpaMMe должно обнаруживать
ся два полностью разделенных пятна.
Предельное содержание: на xpoMaTorpaMMe
испытуемоro раствора S2З любое пятно должно
быть не интенсивнее соответствующею пятна
на xpoMaтorpaMMe раствора сравнения.
Амиды и стеараты. Тонкослойная xpOMaTO
rрафия (2.2.27).
Испытуемый раствор. Используют испыту
емый раствор 823, приrотовленный, как указано
в испытании «Нефенольные антиоксиданты».
Раствор сравнения (р). 20 Mr ФСО кислоты
стеариновой (добавки к пластмассе 19) paCTBO
ряют в 1 О мл метиленхлорида Р.
Раствор сравнения (q). 40 Mr ФСО олеа
мида (добавки к пластмассе 20) растворяют в
20 мл метиленхлорида Р.
Раствор сравнения (r). 40 Mr ФСО эрука
мида (добавки к пластмассе 21) растворяют в
20 мл метиленхлорида Р.
Пластинка: ТСХ пластинка со слоем сили
ка2еля GF 254 Р (две пластинки).
А. Подвижная фаза: этанол Р тpиMe
тилпентан Р (25:75, об/об).
Наносимый объем пробы: по 10 мкл испы
туемоro раствора S23 и раствора сравнения (р).
Фронт подвижной фазы: 1 О см.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают pac
твором 2 r/л дихлорфенолиндофенола Haтpи
евой соли Р в этаноле Р и наrревают в cy
шильном шкафу при температуре 120 ос до
появления пятен.
Предельное содержание: на xpoMaTorpaM
ме испытуемоro раствора 823 пятно. COOTBeT
ствующее добавке к пластмассе 19, должно
быть идентичным по расположению (R, около
0,5) и быть не интенсивнее соответствующе
ro пятна на xpoMaTorpaMMe раствора cpaBHe
ния (р).
В. Подвижная фаза А: 2ексан Р.
Подвижная фаза В: метанол Р метилен
хлорид Р (5:95, об/об).
550
rосударственная фармакопея Республики Беларусь
Наносимый объем пробы: по 1 О мкл испы
TyeMoro раствора S23 и растворов сравнения
(q) и (r).
Фронт подвижной фазы А: 13 см.
Высушивание А: на воздухе.
Фронт подвижной фазы В: 1 О см.
Высушивание В: на воздухе.
Проявление: пластинку опрыскивают раство--
ром 40 r/л кислоты фосфорномолибденовой Р в
этаноле Р и наrревают в сушильном шкафу при
температуре 120 ос до появления пятен.
Предельное содержание: на xpoMaтorpaM
ме испытуемоro раствора S23 пятна, COOTBeT
ствующие добавке к пластмассе 20 или добав
ке к -пластмассе 21, должны быть идентичными
по расположению (R, около 0,2) и быть не интен
сивнее соответствующих пятен на xpoMaTorpaM
ме раствора сравнения (q) и (r).
01/2013:30107
3.1.7. ПОЛИЭТИЛI;НВИНИЛАЦЕТАТ
ДЛЯ КОНТЕИНЕРОВ
И ТРУБОК ДЛЯ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
ДЛЯ ПАРЕНТЕРАльноrо
ПИТАНИЯ
ОПРЕДЕЛЕНИЕ
Полиэтиленвинилацетат, отвечающий ни
жеперечисленным требованиям, подходит для
изroтовления контейнеров и трубок для лекар
ственных средств для парентеральноro питания.
Полиэтиленвинилацетат получают сополимери
зацией смеси этилена и винилацетата.
Содержание винилацетата:
в материале, предназначенном для пpo
изводства контейнеров: не более 25 %;
в материале, предназначенном для про
изводства трубок: и не более 30 %.
ПРОИ3ВОДСТВО
Определенное количество добавок вводит
ся в полимер для улучшения ero химических,
физических и механических свойств, а также
для обеспечения возможности использования
по назначению. Все добавки выбирают из приве
денноro ниже перечня, который определяет для
каждоro вещества максимально допустимое ero
содержание.
Полиэтиленвинилацетат может содержать
не более трех из следующих антиоксидантов:
бутилrидрокситолуол (добавка к пластмас
се 07): не более 0,125 %;
пентаэритритилтетракис[3 (3,5 ди трет
бутил4 rидроксифенил )пропионат) (добавка к
пластмассе 09): не более 0,2 %;
октадецил 3 (3,5 ди трет бутил4 rидро--
ксифенил)пропионат (добавка к пластмассе 11):
не более 0,2 %;
трис[2,4 ди трет бутилфенил) фосфит
(добавка к пластмассе 12): не более 0,2 %;
2,2',2",6,6',6" rекса трет бутил 4,4',4"
«2,4,6 триметил 1,3,5 бензотриил )трисметилен)
трифенол (добавка к пластмассе 10): не более
0,2%.
Полимер также может содержать:
олеамид (добавка к пластмассе 20): не
более 0,5 %;
эрукамид (добавка к пластмассе 21): не
более 0,5 %;
кальция стеарат или цинка стеарат или
сумму этих компонентов: не более 0,5 %;
кальция карбонат или калия rидроксид: не
более 0,5 % каждоro;
кремния диоксид коллоидный: не более
0,2%.
Поставщик материала несет OTBeTCTBeH
ность за то, чтобы качественный и количествен
ный составы типовоro образца COOTBeTCTBOBa
ли качественному и количественному составам
каждой произведенной серии.
ОПИСАНИЕ
Шарики, rранулы или после трансформа
ции полупрозрачные пластинки разной тол
щины, или трубки разной толщины или образцы
roтовых изделий.
Практически нерастворим в воде, в этаноле,
в метаноле и в reKcaHe, который растворяет низ
комолекулярные полимеры, растворим в rоря
чих ароматических уrлеводородах.
При сжиrании пламя окрашивается в синий
цвет.
Температура размяrчения испытуемоro Ma
териала изменяется в зависимости от coдep
жания винилацетата: она снижается от около
100 ос при содержании нескольких процентов
винилацетата до около 70 ос при содержании
30 % винилацетата.
ИДЕНТИФИКАЦИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
При20товление: к 0,25 r испытуемоro Ma
териала прибавляют 1 О мл толуола Р и ки
пятят С обратным холодильником в течение
15 МИIi. Несколько капель раствора поме
щают на диск натрия хлорида и выпаривают
растворитель в сушильном шкафу при темпе
ратуре 80 ОС.
Максимумы П02лощения винилацетата:
при 1740 см"" 1375 см.,, 1240 см"" 1020 см., и
610 см.'.
3.1.7. Полиэтиленвинилацетат для контейнеров и трубок
551
Максимумы П02лощения этилена: при
292 2850 CM 1, 1470 CM 1, 1460 CM 1, 1375 CM 1,
730 CM 1 и 720 CM 1.
Полученный спектр должен COOTBeTCTBO
вать представленному производителем спектру
типовоro образца. Если материал имеет форму
пластин, идентификацию можно провести He
оосредственно на вырезанном фраrменте пла
стинки соответствующеrо размера.
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
Раствор 51. 2,0 r испытуемоro материала
помещают в коническую колбу из боросиликат
ноro стекла с притертой пробкой. Прибавляют
80 мл толуола Р и кипятят с обратным холо
дильником при постоянном перемешивании в
течение 1,5 ч. Охлаждают до 60 ос и прибавляют
при продолжительном перемешивании 120 мл
метанола Р. Фильтруют раствор через CTe
клянный фильтр (16) (2.1.2). Промывают колбу
и фильтр 25 мл смеси толуол Р метанол Р
(40:60, об/об), прибавляют промывную жидкость
к фильтрату и доводят до объема 250,0 мл этой
же смесью растворителей.
Раствор 52. Раствор 82 используют в тe
чение 4 ч после при20товления. 25 r испытуемо
ro материала помещают в колбу из боросиликат
ноro стекла с притертой пробкой. Прибавляют
500 мл воды для инъекций Р и кипятят с обрат
ным холодильником В течение 5 ч. Охлаждают
и декантирую Часть раствора оставляют для
проведения испытаний на прозрачность и ЦBeT
ность. Оставшуюся часть раствора фильтруют
через стеклянный фильтр (16) (2.1.2).
Внешний вид раствора 52. Раствор 82
должен быть прозрачным (2.2.1) и бесцветным
(2.2.2, метод 11).
Кислотность или щелочность. К 100 мл
раствора S2 прибавляют 0,15 мл раствора иH
дикатора BRP Р. При прибавлении не более
1,0 мл 0,01 М раствора натрия 2идроксида
окраска раствора должна измениться на синюю.
К 100 мл раствора S2 прибавляют 0,2 мл pacт
вора метилов020 оранжев020 Р. При прибавле
нии не более 1,5 мл 0,01 М раствора кислоты
хлористоводородной окраска раствора должна
измениться с желтой на оранжевую.
Оптическая плотность (2.2.25). Оптическая
плотность раствора 82 в диапазоне длин волн от
220 нм до 340 нм не должна превышать 0,2.
Восстанавливающие вещества. К 20 мл
раствора 82 прибавляют 1 мл кислоты серной
разведенной Р и 20 мл 0,002 М раствора калия
пермаН2аната. Кипятят с обратным холодильни
ком в течение 3 мин и сразу охлаждают. Прибав
ляют 1 r калия йодида Р и немедленно титруют
раствор 0,01 М раствором натрия тиосульфа
та, используя в качестве индикатора 0,25 мл
раствора крахмала Р. Параллельно проводят
контрольный опыт. Разность между объемами
титранта не должна превышать 0,5 мл.
Амиды и стеариновая кислота. TOHK(r
слойная хроматоrрафия (2.2.27).
Испытуемый раствор. 100 мл раствора 81
выпаривают досуха в вакууме при температуре
45 ОС. Остаток растворяют в 2 мл метиленхло
рида подкислеНН020 Р.
Раствор сравнения (а). 20 Mr ФСО кислоты
стеариновой (добавки к пластмассе 19) paCTBO
ряют в 1 О мл метиленхлорида Р.
Раствор сравнения (Ь). 40 Mr ФСО олеами
да (добавки к пластмассе 20) растворяют в 10 мл
метиленхлорида Р. 1 мл полученноro раствора
доводят метиленхлоридом Р до объема 5 мл.
Раствор сравнения (с). 40 Mr ФСО эруками
да (добавки к пластмассе 21) растворяют в 1 О мл
метиленхлорида Р. 1 мл полученноro раствора
доводят метиленхлоридом Р до объема 5 мл.
Пластинка: ТСХ пластинка со слоем сили
ка2еля GF 254 Р (две пластинки).
А. Подвижная фаза: этанол Р тpиMe
тилпентан Р (25:75, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: 1 О см.
Высушивание: на воздухе.
Проявление: пластинку опрыскивают pac
твором 2 r/л дихлорфенолиндофенола Haтpи
евой соли Р в этаноле Р и наrревают в сушиль
ном шкафу при температуре 120 ос в течение
нескольких минут до появления пятен.
Предельное содержание: на xpoMaтorpaM
ме испытуемоro раствора пятно, COOTBeTCTBY
ющее добавке к пластмассе 19, должно быть не
интенсивнее соответствующеro пятна на XpoMa
TorpaMMe раствора сравнения (а).
В. Подвижная фаза А: 2ексан Р.
Подвижная фаза В: метанол Р метилен
хлорид Р (5:95, об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы А: 13 см.
Высушивание А: на воздухе.
Фронт подвижной фазы В: 1 О см.
Высушивание В: на воздухе.
Проявление: пластинку опрыскивают paCTBO
ром 40 r/л кислоты фосфорномолибденовой Р в
этаноле Р и наrревают в сушильном шкафу при
температуре 120 ос до появления пятен.
Предельное содержание: на xpoMaTorpaMMe
испытуемоro раствора пятна, соответствующие
добавке к пластмассе 20 или добавке к пласт
массе 21, должны быть не интенсивнее COOT
ветствующих пятен на xpoMaTorpaMMe раствора
сравнения (Ь) и (с).
552
rосударственная фармакопея Республики Беларусь
Фенольные антиоксиданты. Жидкостная
хроматоrрафия (2.2.29).
Смесь растворителей. Ацетонитрил Р
тетра2идрофуран Р (50:50, об/об).
Испытуемый раствор (а). 50 мл paCT
вора S1 выпаривают досуха в вакууме при TeM
пературе 45 ОС. Остаток растворяют в 5,0 мл
смеси растворителей.
Испытуемый раствор (Ь). 50 мл paCT
вора S 1 выпаривают досуха в вакууме при TeM
пературе 45 ОС. Остаток растворяют в 5,0 мл Me
тиленхлорида Р.
Раствор сравнения (а). 25 Mr ФСО бутил
2идрокситолуола (добавка к пластмассе 07),
40 Mr ФСО добавки к пластмассе 10,40 Mr ФСО
добавки к пластмассе 09, 40 Mr ФСО добавки
к пластмассе 11 растворяют в 10,0 мл смеси
растворителей. 2,0 мл полученноro раствора дo
водят смесью растворителей до объема 50,0 мл.
Раствор сравнения (Ь). 40 Mr ФСО добавки
к пластмассе 11 и 40 Mr ФСО добавки к пласт
массе 12 растворяют в 10,0 мл метиленхлори
да Р. 2,0 мл полученноro раствора доводят Me
тиленхлоридом Р до объема 50,0 мл.
Условия хроматоrрафирования:
колонка длиной 0,25 м и внутренним ди
аметром 4,6 мм, заполненная силика2елем OK
тадецилсилильным для хроматО2рафии Р с
размером частиц 5 мкм;
подвижная фаза: вода Р тeтpa2идpo
фуран Р ацетонитрил Р (10:30:60, об/об/об);
скорость подвижной фазы: 1,5 мл/мин;
детектирование: спектрофотометриче
ский детектор, длина волны 280 нм;
объем вводимой пробы: по 20 мкл испы
туемоro раствора (а) и раствора сравнения (а).
При20дность хромат02рафической cиcтe
мы: раствор сравнения (а):
разрешение: не менее 2,0 между пиками
добавки к пластмассе 09 и добавки к пласт
массе 1 о;
число теоретических тарелок: не менее
2500 для пика добавки к пластмассе 07.
Предельное содержание:
на xpoMaTorpaMMe испытуемоro paCT
вора (а) должны обнаруживаться только те oc
новные пики, которые обнаруживаются на
xpoMaTorpaMMe раствора сравнения (а) со Bpe
менем удерживания более 2 мин;
площади пиков на xpoMaTorpaMMe ис
nbITyeMoro раствора (а) не должны превышать
площадей соответствующих пиков на xpOMaTO
rpaMMe раствора сравнения (а), за исключени
ем последнеro элюирующеroся пика на xpOMaTO
rpaMMe раствора сравнения (а).
Если на xpoMaTorpaMMe испытуемоro paCT
вора (а) элюируется пик, соответствующий по
следнему элюирующемуся пику на xpoMaTorpaM
ме раствора сравнения (а), проводят испытание
со следующими изменениями:
подвижная фаза: вода Р 2 пропа
нол Р метанол Р (5:45:50, об/об/об);
объем вводимой пробы: по 20 мкл испы
туемоro раствора (Ь) и раствора сравнения (Ь).
При20дность хромат02рафической систе-
мы: раствор сравнения (Ь):
разрешение: не менее 2,0 между пиками
добавки к пластмассе 11 и добавки к пластмас
се 12.
Предельное содержание:
на xpoMaTorpaMMe испытуемоrо paCT
вора (Ь) должны обнаруживаться только те OC
новные пики, которые обнаруживаются на
xpoMaTorpaMMe раствора сравнения (Ь) со Bpe
менем удерживания более 3 мин;
площади пиков на xpoMaTorpaMMe испыту
eMorO раствора (Ь) не должны превышать пло-
щадей соответствующих пиков на xpoMaTorpaM-
ме раствора сравнения (Ь).
Вещества, растворимые в reKcaHe. 5 r ис
пытуемоrо материала помещают в коническую
колбу из боросиликатноrо стекла с притертой
пробкой. Прибавляют 50 мл 2ексана Р и кипятят
с обратным холодильником при постоянном по
мешивании в течение 4 ч на водяной бане. Ox
лаждают в ледяной бане; при этом может обра
зоваться rель.
К стеклянному фильтру (16}(2. 1. 2), снабжен
ному установкой для фильтрования в вакууме,
присоединяют охлаждающую оболочку, запол
ненную ледяной водой, и охлаждают фильтр в
течение 15 мин. Содержимое конической колбы
фильтруют через охлажденный фильтр под дaB
лением 27 кПа, не промывая осадок; время
фильтрования не должно превышать 5 мин.
20 мл фильтрата выпаривают досуха на водяной
бане и высушивают при температуре 100 ос в Te
чение 1 ч. Масса полученноro остатка не должна
превышать 40 Mr (2 %) для сополимера, исполь
зуемоro для производства контейнеров, и 0,1 r
(5 %) для сополимера, используемоro для про
изводства трубок.
Сульфатная зола (2.4.14). Не более 1,2 %.
Определение проводят из 5,0 r испытуемоro Ma
териала.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
ОТ 0,250 r до 1,000 r испытуемоro матери
ала в зависимости от содержания винил ацетата
в сополимере помещают в коническую колбу с
притертой пробкой вместимостью 300 мл с Mar
нитной мешалкой. Прибавляют 40 мл ксилола Р.
Кипятят с обратным холодильником при переме
шивании в течение 4 ч. Охлаждают при непре
рывном перемешивании до начала образования
осадка и медленно прибавляют 25,0 мл спир
тов020 раствора калия 2идроксида Р1. Снова
кипятят с обратным холодильником при пере
мешивании в течение З ч. Охлаждают при посто
янном перемешивании, промывают холодиль
ник 50 мл воды Р и прибавляют в колбу 30,0 мл
0,05 М раствора кислоты серной. Содержимое
3.1.8. Силиконовое масло
553
колбы переносят в лабораторный стакан вмести
мостью 400 мл. Колбу ополаскивают двумя пор
циями по 50 мл раствора 200 r/л натрия суль
фата безводН020 Р и тремя порциями по 20 мл
воды Р и прибавляют смывы в тот же лабора
торный стакан. Титруют избыток серной кислоты
0,1 М раствором натрия 2идроксида, опреде
ляя точку конца титрования потенциометриче
ским методом (2.2.20). Параллельно проводят
контрольный опыт.
1 мл 0,05 М раствора кислоты серной COOT
ветствует 8,609 Mr винилацетата.
01/2013:30108
3.1.8. СИЛИКОНОВОЕ МАСЛО,
ИСПОЛЬЗУЕМОЕ В КАЧЕСТВЕ
СМАЗЫВАЮЩЕЙ ДОБАВКИ
CH j СН З СНз
Н C i O i O i CH
з I I I з
СН з СН з СН з
п
ОПРЕДЕЛЕНИЕ
Силиконовое масло, используемое в каче
стве смазывающей добавки, представляет собой
полидиметилсилоксан, полученный в результа
те rидролиза и поликонденсации дихлордиме
тилсилана и хлортриметилсилана. Существуют
разные сорта силиконовоro масла, которые xa
рактеризуются 'WIслом, обозначающим величину
еro номинальной вязкости (указывается после
наименования).
Степень полимеризации силиконовых
масел, используемых в качестве смазывающих
добавок (п = 400 1200), должна быть такой,
чтобы их кинематическая вязкость находилась
8 номинальных пределах от 1000 MM2.c 1 ДО
30 000 мм2,с- 1 .
ОПИСАНИЕ
Прозрачная бесцветная жидкость с различ
ной вязкостью.
Практически нерастворимо в воде и в MeTa
ноле, очень мало растворимо 8 этаноле, смеши
вается с этилацетатом, метилэтилкетоном и TO
луолом.
ИДЕНТИФИКАЦИЯ
А. Испытуемый материал выдерживает ис
пытание на кинематическую вязкость при темпе
ратуре 25 ОС, как указано в разделе «Испытания».
В. Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
71 Зак. 1712
Сравнение: ФСО силиконов020 масла.
Область спектра от 850 CM 1 дО 750 CM 1 не
принимают во внимание, поскольку в ней MOryт
выявляться незначительные различия, обуслов
ленные степенью полимеризации.
С. 0,5 r испытуемоro материала помещают в
пробирку и наrревают над небольшим orHeM до
появления белых паров. Пробирку переворачи
вают над друrой пробиркой, которая содержит
1 мл раствора 1 r/л хромотроповой кислоты
натриевой соли Р в кислоте серной Р, таким
образом, чтобы пар достиr раствора. Друryю
пробиpt(у встряхивают 8 течение примерно 1 О с
и наrревают на водяной бане в течение 5 мин.
Раствсч:> становится фиолетовым.
D. В платиновом тиrле получают сульфат
ную золу (2.4.14). 50 Mr полученноro белоro по
рошка дают реакцию на силикаты (2.3.1).
ИСПЫТАНИЯ
Кислотность. К 2,0 r испытуемоro матери
ала прибавляют 25 мл предварительно нейтра
лизованной смеси из равных объемов этано
ла Р и эфира Р, прибавляют 0,2 мл раствора
бромтимолов020 синеео Р1 и взбалтывают. При
прибавлении не более 0,15 мл 0,01 М раствора
натрия еидроксида окраска раствора должна
измениться на синюю.
Вязкость (2.2.10). Динамическую вязкость
определяют при температуре 25 ОС. Кинемати
ческую вязкость рассчитывают, используя OTHO
сительную плотность, равную 0,97. Кинематиче
ская вязкость должна быть не менее 95 % и не
более 105 % от номинальной вязкости, обозна
ченной на этикетке.
Минеральные масла. 2 мл испытуемо
ro материала помещают 8 пробирку и просма
тривают в ультрафиолетовом свете при длине
волны 365 нм. Флуоресценция должна быть не
более интенсивной, чем флуоресценция paCT
вора, содержащеro 0,1 ррm хинина сульфата Р
в 0,005 М растворе кислоты серной, испытыва
eMoro в тех же условиях.
Фенилированные компоненты. Показа
тель преломления (2.2.6) не должен превышать
1,410.
Тяжелые металлы. Не более 5 ррт.
Смесь растворителей. Раствор аммиака
разведенный Р2 раствор 2 r/л 2идроксилами
на 2идрохлорида Р (1 :9, об/об).
1,0 r испытуемоro материала смешивают с
метиленхлоридом Р и доводят до объема 20 мл
этим же растворителем. Прибавляют 1,0 мл
свежеприroтовленноrо раствора 0,02 r/л дити
зона Р вметиленхлориде Р, 0,5 мл воды Р и
0,5 мл смеси растворителей. Одновременно ro
товят раствор сравнения: к 20 мл метиленхло
рида Р прибавляют 1,0 мл свежеприroтовленноro
554
rосударственная фармакопея Республики Беларусь
раствора 0,02 r/л дитизона Р в метиленхло
риде Р, 0,5 мл этаЛОНН020 раствора свинца
(10 ррт РЬ) Р и 0,5 мл смеси растворителей.
Полученные растворы немедленно энерrично
встряхивают в течение 1 мин.
Красное окрашивание испытуемоro paCT
вора не должно быть интенсивнее окрашивания
раствора сравнения.
Летучие вещества. Не более 2,0 %. Опре
деление проводят из 2,00 r испытуемоro матери
ала при наrревании в термостате при темпера
туре 150 ос в течение 24 ч. Испытания проводят,
используя чашку диаметром 60 мм и rлубиной
10мм.
МАРКИРОВКА
На этикетке указывают:
номинальную вязкость в виде числа, pac
положенноro после названия продукта;
информацию о том, что содержимое сле
дует использовать в качестве смазывающей дo
бавки.
01/2013:30109
3.1.9. СИЛИКОНОВЫЙ ЭЛАСТОМЕР
ДЛЯ УКУПОРОЧНЫХ СРЕДСТВ
И ТРУБОК
ОПРЕДЕЛЕНИЕ
Для изroтовления укупорочных средств и
трубок подходит силиконовый эластомер, OTBe
чающий следующим требованиям.
Силиконовый эластомер получают попе
речным сшиванием линейноro полисилокса
на, который состоит, в основном, из диметил
силокси частей с небольшими количествами
метилвинилсилокси rрупп; концы цепей блоки
рованы триметилсилокси или диметилвинилси
локси rруппами.
Общая формула полисилоксана:
{ НЗС\ /СНз
М .........Si
О
n
( СН 2
СН з
/
.........Si
О
М'
о"""'"
п'
МиМ'=
НзС СН З
\/
/Si,
СН з
или
НзС СН з
\/
/Si CH
Поперечное сшивание происходит:
в roрячем состоянии или с использован....
ем: 2,4 дихлорбензоилпероксида для экстру
дированных продуктов; или
2,4 дихлорбензоилпероксида или дику
милпероксида, или OO ( 1 ,1 диметилэтил) О из<r
пропил монопероксикарбоната, или 2,5 бис[(1, 1
диметилэтил )ДИОКСИ] 2,5 диметилrексана для
формованных продуктов;
или
rидросилилированием SiH rруппами по
лисилоксана с использованием платины в каче
стве катализатора.
Во всех случаях используют соответствующие
добавки, такие, как кремния диоксид, и иноrда
небольшое количество орrаносиликоновых дo
бавок (а,w диrидроксиполидиметилсилоксан).
ОПИСАНИЕ
Прозрачный или полупрозрачный материал.
Практически нерастворим в орrанических
растворителях, а некоторые, например, цикло
reKcaH, reKcaH и метиленхлорид, вызывают об
ратимое набухание материала.
ИДЕНТИФИКАЦИЯ
А. Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
Сравнение: ФСО силиконов020 эластомера.
В. 1,0 r испытуемоro материала помещают в
пробирку и наrревают над небольшим orHeM до
появления белых паров. Пробирку переворачи
вают над друrой пробиркой, которая содержит
1 мл раствора 1 r/л хромотроповой кислоты Ha
триевой соли Р в кислоте серной Р, таким об
разом, чтобы пар достиr раствора. Друryю про
бирку встряхивают около 10 с и наrревают на
водяной бане в течение 5 мин. Раствор CTaHO
вится фиолетовым.
D. 50 Mr остатка после сжиrания дают peaK
цию на силикаты (2.3.1).
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части С максимальной длиной стороны не
более 1 СМ.
Раствор 5. 25 r испытуемоro материала по
мещают в коническую колбу из боросиликатноro
стекла с притертой пробкой. Прибавляют 500 мл
воды Р и кипятят с обратным холодильником в
течение 5 ч. Охлаждают и декантируют раствор.
Прозрачность (2.2.1). Раствор S должен
быть прозрачным.
Кислотность или щелочность. К 100 мл
раствора S прибавляют 0,15 мл раствора бром
тимолов020 сине20 Р1. При прибавлении не
более 2,5 мл 0,01 М раствора натрия 2идPOK
сида окраска раствора должна измениться на
3. 1.9. Силиконовый эластомер для укупорочных средств
555
синюю. К 100 мл раствора S прибавляют 0,2 мл
раствора метилов020 оранжев020 Р. При при
бавлении не более 1,0 мл 0,01 М раствора Kиc
лоты хлористоводородной окраска раствора
должна измениться на желтую или оранжевую.
Относительная плотность (2.2.5). От 1,05
до 1,25. Испытание проводят, используя виско
зиметр с этанолом Р в качестве иммерсионной
жидкости.
Восстанавливающие вещества. К 20 мл
раствора S прибавляют 1 мл кислоn7Ы серной
разведенной Р и 20 мл 0,002 М раствора калия
пермаН2аната. Смесь выдерживают в течение
15 мин. Прибавляют 1 r калия йодида Р и He
медленно титруют 0,01 М раствором натрия
тиосульфата, используя в качестве индикато
ра 0,25 мл раствора крахмала Р. Параллель
но проводят контрольный опыт С использовани
ем 20 мл воды Р вместо раствора S. Разность
между объемами титранта не должна превы
шать 1,0 мл.
Вещества, растворимые в reKcaHe. Не
более 3 %. 25 мл раствора, полученноro при ис
пытании «Фенилированные компоненты», BЫ
паривают в стеклянной выпарительной чашке
на водяной бане и сушат в сушильном шкафу
при температуре 100 105 ос в течение 1 ч.
Масса полученноro остатка не должна превы
шать 15 Mr.
Фенилированные компоненты. 2,0 r ис
пытуемоrо материала помещают в колбу из бо
росиликатноro стекла с притертой пробкой и
прибавляют 100 мл 2ексана Р. Кипятят с обрат
ным холодильником В течение 4 ч. Охлаждают,
быстро фильтруют через стеклянный фильтр
(16) (2.1.2) в колбу с притертой пробкой. Полу
ченный фильтрат плотно закрывают для пре
дотвращения испарения. Оптическая плотность
(2.2.25) в области длин волн от 250 нм до 340 нм
не должна превышать 0,4.
Минеральные масла. 2 мл испытуемоro
материала помещают в коническую колбу BMe
стимостью 100 мл, содержащую 30 мл смеси, co
стоящей из 5 объемов раствора аммиака Р и 95
объемов пиридина Р. Смесь выдерживают в Te
чение 2 ч, периодически перемешивая. Получен
ный раствор пиридина декантируют и просма
тривают в ультрафиолетовом свете при длине
волны 365 нм. Флуоресценция должна быть не
более интенсивной, чем флуоресценция paCT
вора, содержащеro 0,1 ррm хинина сульфата Р
в 0,005 М растворе кислоты серной, испытыва
емоro в тех же условиях.
Летучие вещества. Не более 0,5 % для
силиконовоro эластомера, полученноro с ис
пользованием пероксидов; не более 2,0 % для
силиконовою эластомера, полученноro с ис
пользованием платины. 10,0 r испытуемоrо Ma
териала, предварительно выдержанною в экси
каторе в течение 48 ч над кальция хлоридом
безводным Р наrревают при температуре 200 ос
в течение 4 ч, охлаждают в эксикаторе и снова
взвешивают.
Силиконовый эластомер, полученный с иc
пользованием пероксидов, должен выдержи
вать следующее дополнительное испытание.
Остаточные пероксиды. Не более 0,08 %
в пересчете на дихлорбезоила пероксид. 5 r
испытуемоro материала помещают в колбу из
боросиликатноro стекла, прибавляют 150 мл
метиленхлорида Р, закрывают колбу и пере
мешивают при помощи механической мешалки
в течение 16 ч. Затем быстро фильтруют, соби
рая фильтрат в колбу с притертой пробкой. За
мещают воздух в колбе на азот. свободный от
кислорода, Р прибавляют 1 мл раствора 200 r/л
натрия йодида Р в кислоте уксусной безво
дной Р, закрывают колбу, тщательно встряхива
ют и помещают в защищенное от света место на
30 мин. Прибавляют 50 мл воды Р и немедлен
но титруют 0,01 М раствором натрия тиосуль
фата, используя в качестве индикатора 0,25 мл
раствора крахмала Р. Параллельно проводят
контрольный опыт. Разность между объемами
титранта не должна превышать 2,0 мл.
Силиконовый эластомер, иЗ20товленный
с использованием платины, должен выдержи
вать следующее дополнительное испытание.
Платина. Не более 30 ррm. 1,0 r испытуе
моro материала сжиrают в кварцевом тиrле, по
степенно поднимая температуру, до получения
белоro остатка. Остаток пере носят в rрафито
вый тиrель. В кварцевый тиrель прибавляют
1 О мл свежеприroтовленной смеси из 1 объема
кислоты азотной Р и 3 объемов кислоты хло
ристоводородной Р, наrревают на водяной
бане в течение 1 2 мин и переносят в rрафи
товый тиrель. Прибавляют 5 Mr калия хлори
да Р и 5 мл кислоты фтористоводородной Р
и выпаривают досуха на водяной бане. Прибав
ляют 5 мл кислоты фтористоводородной Р и
снова выпаривают досуха; повторяют эту опера
цию дважды. Полученный остаток растворяют в
5 мл 1 М раствора кислоты хлористоводород
ной при наrревании на водяной бане и охлажда
ют. Полученный раствор прибавляют к 1 мл раст
вора 250 r/л олова (/1) хлорида Р в 1 М растворе
кислоты хлористоводородной. промывают rpa
фитовый тиrель несколькими миллилитрами
1 М раствора кислоты хлористоводородной,
присоединяя их к тому же раствору, и доводят
объем раствора этой же кислотой до 10,0 мл.
Параллельно roтовят раствор сравнения: к 1 мл
раствора 250 r/л олова (/1) хлорида Р в 1 М pac
556
rосударственная фармакопея Республики Беларусь
творе кислоты хлористоводородной прибав
ляют 1,0 мл этаЛОНН020 раствора платины
(30 ррт Pt) Р и доводят 1 М раствором кислоты
хлористоводородной до объема 10,0 мл. OKpa
ска испытуемоro раствора должна быть не ин
тенсивнее окраски раствора сравнения.
МАРКИРОВКА
На этикетке указывают, что материал изro
товлен с использованием пероксидов либо с ис
пользованием платины.
01/2013:30110
3.1.10. МАТЕРИАЛЫ НА ОСНОВЕ
НЕПЛАСТИФИЦИРОВАнноrо
ПОЛИВИНИUХЛОРИДА
для КОНТЕИНЕРОВ
ДЛЯ НЕИНЪЕКЦИОННЫХ
ВОДНЫХ РАСТВОРОВ
ОПРЕДЕЛЕНИЕ
Материалы на основе непластифициро
ванноro поливинилхлорида, отвечающие Tpe
бованиям указанных ниже спецификаций, ис
пользуют для изrотовления контейнеров для
неинъекционных водных растворов. Они также
MorYT быть использованы для твердых лекар
ственных форм для пероральноro применения.
В некоторых случаях, Korдa проведены специ
альные исследования на совместимость контей
нера и еro содержимorо, эти материалы MOryт
быть подходящими для изroтовления контейне
ров для суппозиториев. Они состоят из одноro
или более поливинилхлорида/винилацетата или
смеси поливинилхлорида и поливинилацетата
или поливинилхлорида.
Содержание хлора в пересчете на поливи
нилхлорид: не менее 80 %.
Они MOryT содержать не более 15 % сополи
меров на основе акриловой и/или метакриловой
кислот и/или их эфиров, и/или на основе стиро
ла и/или бутадиена.
ПРОИЗВОДСТВО
Материалы на основе непластифициро
ванною поливинилхлорида получают MeToдa
ми полимеризации, которые rарантируют OCTa
точное содержание винилхлорида менее 1 ррm.
Данный метод должен быть валидирован для
получения продукта, выдерживающею испыта
ние на винилхлорид.
Винилхлорид. Не более 1 ррт. Парофаз
ная rазовая хроматоrрафия (2.2.28).
Раствор внутренне20 стандарта. К
20,0 мл диметилацетамида Р с помощью ми
крошприца прибавляют 10 мкл эфира Р, поrpy
жая конец иrлы в растворитель. Непосредствеtr
но перед использованием разводят раствор
диметилацетамидом Р в 1000 раз.
Испытуемый раствор. 1,000 r испытуемоro
материала помещают во флакон вместимостью
50 мл и прибавляют 10,0 мл раствора BHyтpeH
неro стандарта. rерметично закрывают колбу
пробкой. Встряхивают, избеrая контакта жид
кости с пробкой. Помещают флакон в водяную
баню при температуре (60:1:1) ос и выдерживают
в течение 2 ч.
Основной раствор винилхлорида. ,oтo
вят в вытяжном шкафу. 50,0 мл диметилаце
тамида Р помещают во флакон вместимостью
50 мл, rерметично закрывают пробкой и взвеши
вают с точностью до 0,1 Mr. Наполняют полиэти
леновый или полипропиленовый шприц вмести
мостью 50 мл rазообразным еинилхлоридом Р,
оставляют шприц в контакте с rазом в течение
3 мин, затем rаз удаляют и снова наполняют
шприц 50,0 мл rазообразноro винилхлорида Р.
Подбирают для шприца rиподермальную иrлу и
уменьшают объем rаза в шприце до 25 мл. Meд
лен но вводят оставшиеся 25 мл винилхлори
да во флакон, осторожно встряхивая и избеrая
контакта жидкости с иrлой. Опять взвешивают
флакон. Увеличение массы должно составлять
около 60 Mr (1 мкл раствора содержит около
1,2 MKr винилхлорида). Выдерживают в течение
2 ч. Основной раствор хранят в холодильнике.
Стандартный раствор винилхлорида. Oc
нов ной раствор винилхлорида диметилаце
тамид Р (1 :3, об/об).
Растворы сравнения. По 10,0 мл раствора
внутреннеro стандарта помещают в 6 одинако
вых флаконов вместимостью 50 мл. Флаконы
rерметично закрывают пробками. В 5 колб с по
мощью микрошприца прибавляют COOTBeTCTBeH
но 1 мкл, 2 мкл, 3 мкл, 5 мкл И 10 мкл CTaHдapT
ною раствора винилхлорида. Полученные таким
образом 6 растворов содержат соответственно
О мк!; около 0,3 MKr, 0,6 MKr, 0,9 MKr, 1,5 MKr и 3 MKr
винилхлорида. Встряхивают, избеrая контакта
жидкости с пробкой. Помещают колбы в водя
ную баню при температуре (60:t1) ос и выдержи
вают в течение 2 ч.
Условия хроматоrрафирования:
колонка из нержавеющей стали длиной
3 м и внутренним диаметром 3 мм, заполнен
ная диатомитом силанизированным для 2азо
вой хромат02рафии Р, импреrнированным 5 %
(м/м) диметилстеариламида Р и 5 % (м/м) Ma
КР020ла 400 Р;
2аз носитель: азот для хроматоzрафии Р;
скорость 2аза носителя: 30 мл/мин:
детектор: liпаменно ионизационный;
температура: колонка 45 ОС, блок ввода
проб 100 ОС, детектор 150 ОС;
объем вводимой пробы: 1 мл паровой
фазы.
Рассчитывают содержание винилхлорида.
3.1.10. Материалы на основе непластифuцироваННО20 полuвuнилхлорuда
557
Добавки
Для получения необходимых механических
характеристик и характеристик стабильности
материал на основе непластифицированноro
поливинилхлорида может содержать:
эпоксидированное соевое масло с coдep
жанием кислорода в эпоксидной rруппе от 6 % до
8 % и йодным числом не более 6 %: не более 8 %;
кальциевые или цинковые соли алифа
тических жирных кислот, содержащие не более
семи атомов уrлерода: не более 1,5 % или не
более 1,5 % суммы этих веществ;
вазелиновое масло (парафин жидкий): не
более 1,5 %;
воски: не более 1,5 %;
rидроrенизированные масла или эфиры
алифатических жирных кислот: не более 2 %;
эфиры макроroла: не более 1,5 %;
сорбит: не более 1 ,5 %;
2,4 динонилфенилфосфит или ди(4 но
нилфенил)фосфит или трис(нонилфенил)фос
фит: не более 1 %.
Материалы на основе непластифицирован
ноro поливининилхлорида MOryт содержать одну
из rрупп стабилизаторов:
олово в виде ди(изооктил) 2,2' [(диок
тилстаннилен )бис(тио )]диацетата, содержа
щеrо около 27 % три(изооктил)2,2',2" [(моно
октилстаннилидин)трис(тио )]триацетата: не
более 0,25 %;
олово в виде смеси, содержащей не более
76 % ди(изооктил)2,2' [(диметилстаннилен)бис
(тио )]диацетата и не более 85 % три
(изооктил )2,2' ,2" [(монометилстаннилидин )трис
(тио)]триацетата; (изооктил например, 2 этил
rексил): не более 0,25 %;
1-фенилейкозан 1 , дион (бензоилстеароил
метан) или 2 (4 додецилфенил)индол или ди
додецил 1 ,4 диrидропиридин 2,6 диметил 3,5
дикарбоксилат: не более 1 % или не более 1 %
смеси обоих веществ.
Материалы на основе непластифициро
ванноro поливининилхлорида MorYT coдep
жать краситель или пиrмент, а также титана
диоксид для обеспечения непрозрачности Ma
териала.
Поставщик материала несет OTBeTCTBeH
ность за то, чтобы качественный и количествен
ный составы типовоro образца COOTBeTCTBOBa
ли качественному и количественному составам
каждой произведенной серии.
ОПИСАНИЕ
Порошок, шарики, rранулы, пластинки
разной толщины или образцы, отобранные из ro
товых объектов.
Нерастворимы в воде и в этаноле, раствори
мы в тетраrидрофуране, малорастворимы в Me
тиленхлориде.
При сжиrании пламя окрашивается в opaH
жево желтый цвет с зеленой каймой с выделе
нием ryCTOro черноro дыма.
72 За.. 1712
ИДЕНТИФИКАЦИЯ
Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
При20товление: остаток А, полученный при
приrотовлении раствора S2, как указано в раз
деле «Испытания», растворяют в 5 мл тeтpa
2идрофурана Р. Несколько капель полученноro
раствора помещают на диск натрия хлорида и
выпаривают растворитель досуха в сушильном
шкафу при температуре 1 oo 105 ОС.
Максимумы П02лощения: 2975 см. 1 , 291 О см. 1 ,
2865 см.1, 1430 см.1, 13ЗО см.1, 1255 см.1, 69 0 с м. 1
и 615 см. 1 .
Полученный спектр должен соответствовать
спектру типовоro образца.
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
Раствор 51. 25 r испытуемоro материала
помещают в колбу из боросиликатноro стекла.
Прибавляют 500 мл воды Р и закрывают roрло
колбы алюминиевой фольroй или лабораторным
стаканом из боросиликатноro стекла. Наrревают
полученную смесь в автоклаве при температуре
(121:!:2) ос в течение 20 мин, после чеro охлаж
дают и осаждают твердую фазу.
Раствор 52. 5,0 r испытуемоro матери
ала растворяют в 80 мл тетра2идрофурана Р
и доводят до объема 100 мл этим же раствори
телем. Если необходимо, фильтруют (раствор
может иметь опалесценцию). К 20 мл получен
ноro раствора прибавляют по каплям 70 мл 96 %
спирта Р при леrком перемешивании. Охлажда
ют в ледяной бане в течение 1 ч. Потом фильтру
ют или центрифуrируют (остаток А). Остаток А
промывают 96 % спиртом Р, прибавляют еro к
фильтрату или надосадочной жидкости и ДOBO
дят 96 % спиртом Р до объема 100 мл.
Раствор 53. 5 r испытуемоro материала по
мещают в колбу из боросиликатноro стекла с
притертой пробкой. Прибавляют 100 мл О, 1 М
раствора кислоты хлористоводородной и ки
пятят С обратным холодильником в течение 1 ч,
после чеro охлаждают и осаждают твердую фазу.
Внешний вид раствора 51. Раствор S1 по
степени мутности не должен превышать эталон 11
(2.2.1) и быть бесцветным (2.2.2, метод 11).
Оптическая плотность раствора 51
(2.2.25). 100 мл раствора S1 выпаривают досуха.
Остаток растворяют в 5 мл 2ексана Р. Если He
обходимо, фильтруют через фильтр, предвари
тельно промытый 2ексаном Р. Оптическая плот
ность полученноro раствора в области от 250 нм
до 310 нм не должна превышать 0,25.
558
rосударственная фармакопея Республики Беларусь
Оптическая плотность раствора S2
(2.2.25). Оптическая плотность раствора S2 в
области от 250 нм до 330 нм не должна пре
вышать 0,2 для материалов, стабилизиро
ванных оловом, или 0,4 для друrих MaTe
риалов.
Экстраrируемый барий. Не более 2 ррm.
Атомно эмиссионная спектрометрия
(2.2.57).
Испытуемый раствор. Используют paCT
вор S3.
Раствор сравнения. Раствор, содержащий
0,1 ррm бария, rотовят разведением эталонно
20 раствора бария (50 ррт Ва) Р 0,1 М pacтвo
ром кислоты хлористоводородной.
Длина волны: длина волны светоиспуска
ния бария 455,40 нм, спектральный фон
455,30 нм.
Проверяют отсутствие бария в использу
емой кислоте хлористоводородной.
Экстраrируемый кадмий. Не более 0,6 ррm.
Атомно абсорбционная спектрометрия
(2.2.23, метод 1).
Испытуемый раствор. Используют paCT
вор SЗ.
Раствор сравнения. Раствор, содержащий
0,03 ррm кадмия, rотовят разведением эталон
Н020 раствора кадмия (0,1 % Cd) Р 0,1 М pac
твором кислоты хлористоводородной.
Длина волны: 228,8 нм.
Проверяют отсутствие кадмия в использу
емой кислоте ХЛОрИСТ080ДОрОДНОЙ.
Материалы, стабилизированные оловом.
Не более 0,25 % 5п.
Основной раствор олова. 81 Mr ФСО добав
ки к пластмассе 23 растворяют в тeтpa2идpo
фуране Р и доводят до объема 100,0 мл этим же
растворителем.
Стандартный раствор олова. 20,0 мл oc
новноro раствора олова доводят 96 % спир
том Р до объема 100,0 мл.
К 0,10 мл раствора 52 в пробирке прибав
ляют 0,05 мл 1 М раствора кислоты хлористо
водородной, 0,5 мл раствора калия йодида Р и
5 мл 96 % спирта Р, тщательно перемешивают
и оставляют на 5 мин. К полученному раствору
прибавляют 9 мл воды Р и 0,1 мл раствора 5 r/л
натрия сульфита Р и тщательно перемешива
ют. Прибавляют 1,5 мл раствора дитизона Р,
свежеразведенноrо в 100 раз метиленхлори
дом Р, встряхивают в течение 15 с и Bыдep
живают в течение 2 мин. Параллельно rотовят
раствор сравнения, используя 0,1 мл CTaHдapT
ноro раствора олова.
Фиолетовая окраска полученноro нижнеro
слоя раствора 52 должна быть не интенсивнее
окраски раствора сравнения. Зеленовато синее
окрашивание раствора дитизона в присутствии
олова переходит в розовое.
Материалы, не стабилизированные
оловом. Не более 25 ррm 5п. К 5 мл раствора S2
в пробирке прибавляют 0,05 мл 1 М раствора
кислоты хлористоводородной и 0,5 мл pacт
вора калия йодида Р. Тщательно перемешивают
и оставляют на 5 мин. К полученному раствору
прибавляют 9 мл воды Р и 0,1 мл раствора 5 r/л
натрия сульфита Р и тщательно перемешива
ют. Если полученный раствор не окрашен, при
бавляют раствор натрия сульфита порциями по
0,05 мл. Прибавляют 1,5 мл раствора дитизо
на Р, свежеразведенноrо в 100 раз метиленхло
ридом Р, встряхивают в течение 15 с и выдержи
вают в течение на 2 мин. Параллельно roтовят
раствор сравнения, используя 0,05 мл CTaHдapT
ноro раствора олова (приroтовленноrо, как YKa
зано в испытании выше).
> Фиолетовая окраска полученноro нижнеro
слоя раствора 52 должна быть не интенсивнее
окраски раствора сравнения.
Экстраrируемые тяжелые металлы (2.4.8,
метод А). Не более 20 ррm. 12 мл раствора 53
должны выдерживать испытание на тяжелые Me
таллы. Эталон roтовят с использованием 1 О мл
этаЛОНН020 раствора свинца (1 ррт РЬ) Р.
Экстраrируемый цинк. Не более 100 ррт.
Атомно абсорбционная спектрометрия
(2.2.23, метод 1).
Испытуемый раствор. Раствор S3 разво
дят водой Р в 1 О раз.
Раствор сравнения. Раствор, содержащий
0,50 ррm цинка, rотовят разведением эталонно
20 раствора цинка (5 м2Iмл Zп) Р 0,01 М pac
твором кислоты хлористоводородной.
Длина волны: 214,0 нм.
Проверяют отсутствие цинка в использу
емой кислоте хлористоводородной.
Сульфатная зола (2.4.14). Не более 1,0 %.
Определение проводят из 1,0 r испытуемоro Ma
териала. Для материалов, содержащих титана
диоксид как добавку, придающую непрозрач
ность, содержание сульфатной золы не должно
превышать 4,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение проводят методом сжиrания в
колбе с кислородом (2.5.10), используя 50,0 Mr
испытуемоro материала. Продукты сrорания
растворяют в 20 мл 1 М раствора натрия 2и
дроксида. К полученному раствору прибавля
ют 2,5 мл кислоты азотной Р, 10,0 мл 0,1 М
раствора серебра нитрата, 5 мл раствора
железа (111) аммония сульфата Р2. 1 мл дибу
тилфталата Р и титруют 0,05 М раствором
аммония тиоцианата до появления KpaCHOBa
то желтоrо окрашивания. Параллельно прово
дят контрольный опыт.
1 мл 0,1 М раствора серебра нитрата co
ответствует 6,25 Mr поливинилхлорида.
01/2013:30111
3.1.11. Материалы на основе непластифицироваННО20 поливинилхлорида
559
3.1.11. МАТЕРИАЛЫ НА ОСНОВЕ
НЕПЛАСТИФИЦИРОВАнноrо
ПОЛИВИНИUХЛОРИДА
ДЛЯ КОНТЕИНЕРОВ ДЛЯ
ТВЕРДЫХ ЛЕКАРСТВЕННЫХ
ФОРМ дЛЯ ПЕРОРАЛьноrо
ПРИМЕНЕНИЯ
ОПРЕДЕЛЕНИЕ
Материалы на основе непластифицирован
Horo поливинилхлорида для контейнеров для
твердых лекарственных форм для перораль
ноro применения используют для изroтовления
пластинок или контейнеров и состоят из одноro
или более поливинилхлорида/поливинилацета
та или смеси поливинилхлорида и поливинила
цетата или поливинилхлорида.
Содержание хлора в пересчете на поливи
нилхлорид: не менее 80 %.
Они MOryт содержать не более 15 % сополи
меров на основе акриловой иlили метакриловой
кислот и/или их эфиров, и/или на основе стиро
ла и/или бутадиена.
ПРОИЗВОДСТВО
Материалы на основе непластифициро
ванноro поливинилхлорида получают Meтoдa
ми полимеризации, которые rарантируют OCTa
точное содержание винилхлорида менее 1 ррm.
Данный метод должен быть валидирован для
получения продукта выдерживающеro испыта
ние на винилхлорид.
Винилхлорид. Не более 1 ррm. Парофаз
ная rазовая хроматоrрафия (2.2.28).
Раствор внутренне20 стандарта. К
20,0 мл диметилацетамида Р с помощью ми
крошприца прибавляют 1 О мкл эфира Р, norpy
жая конец иrлы в растворитель. Непосредствен
но перед использованием разводят раствор
диметилацетамидом Р в 1000 раз.
Испытуемый раствор. 1,000 r испытуемоrо
материала помещают во флакон вместимостью
50 мл и прибавляют 10,0 мл раствора BHYTpeH
неro стандарта. rерметично закрывают колбу
пробкой. Встряхивают, избеrая контакта жид
кости с пробкой. Помещают флакон в водяную
баню при температуре (60:1:1) ос и выдерживают
в течение 2 ч.
Основной раствор винилхлорида. Toтo
вят в вытяжном шкафу. 50,0 мл диметилаце
тамида Р помещают во флакон вместимостью
50 мл, rерметично закрывают пробкой и взвеши
вают с точностью до 0,1 Mr. Наполняют полиэти
леновый или полипропиленовый шприц вмести
мостью 50 мл rазообразным винилхлоридом Р,
оставляют шприц в контакте с rазом в течение
3 мин, затем rаз удаляют и снова наполняют
шприц 50,0 мл rазообразноro винилхлорида Р.
Подбирают для шприца rиподермальную иrлу и
уменьшают объем rаза в шприце до 25 мл. Meд
ленно вводят оставшиеся 25 мл винилхлори
да во флакон, осторожно встряхивая и избеrая
контакта жидкости с иrлой. Опять взвешивают
флакон. Увеличение массы должно составлять
около 60 Mr (1 мкл раствора содержит около
1,2 MKr винилхлорида). Выдерживают в течение
2 ч. Основной раствор хранят в холодильнике.
Стандартный раствор винилхлорида. Oc
новной раствор винилхлорида диметилаце
тамид Р (1:3, об/об).
Растворы сравнения. По 10,0 мл раствора
внутреннеro стандарта помещают в 6 одинако
вых флаконов вместимостью 50 мл. Флаконы
rерметично закрывают пробками. В 5 колб с по
мощью микрошприца прибавляют COOTBeTCTBeH
но 1 мкл, 2 мкл, 3 мкл, 5 мкл И 10 мкл CTaHдapT
ноro раствора винилхлорида. Полученные таким
образом 6 растворов содержат соответственно
О MKr, около 0,3 MKr, 0,6 MKr, 0,9 мк!; 1,5 MKr и 3 MKr
винилхлорида. Встряхивают, избеrая контакта
жидкости с пробкой. Помещают колбы в водя
ную баню при температуре (60:1:1) ос и выдержи
вают в течение 2 ч.
Условия хроматоrрафирования:
колонка из нержавеющей стали длиной 3 м
и внутренним диаметром 3 мм, заполненная диa
томитом силанизированным для 2азовой xpOMa
т02рафии Р, импреrнированным 5 % (м/м) диMe
тилстеариламида Р и 5 % (м/м) маКР020ла 400 Р;
2аз носитель: азот для хромат02рафии Р;
скорость 2аза носителя: 30 мл/мин;
детектор: пламенно ионизационный;
температура: колонка 45 ОС, блок ввода
проб 100 ос. детектор 150 ОС;
объем вводимой пробы: 1 мл паровой фазы.
Рассчитывают содержание винилхлорида.
Добавки
Для получения необходимых механических
характеристик и характеристик стабильности
материал на основе непластифицированноrо
поливинилхлорида может содержать:
эпоксидированное соевое масло с coдep
жанием кислорода в эпоксидной rруппе от 6 % до
8 % и йодным числом не более 6 % для стаби
лизированноro оловом материала: не более 2 %;
эпоксидированное соевое масло с coдep
жанием кислорода в эпоксидной rруппе от б %
до 8 % и йодным числом не более 6 % для не
стабилизированноro оловом материала: не
более 3 %;
кальциевые или цинковые соли алифа
тических жирных кислот, содержащие не более
семи атомов уrлерода: не более 1,5 % или не
более 1,5 % суммы этих веществ;
вазелиновое масло (парафин жидкий): не
более 1,5 %;
воски: не более 4 %;
rидроrенизированные масла или эфиры
алифатических жирных кислот: не более 2 %;
560
rосударственная фармакопея Республики Беларусь
сумма содержаний трех вышеуказанных
лубрикантов: не более 4 %;
эфиры макроroла: не более 1,5 %;
сорбит: не более 1,5 %;
2,4 динонилфенилфосфит или ди(4 но
нилфенил)фосфит или трис(нонилфенил)фос
фит: не более 1 %;
кальция карбонат: не более 1 %;
кремния диоксид: не более 1 %.
Материалы на основе непластифицирован
ноro поливининилхлорида MOryт содержать одну
из трех rрупп стабилизаторов (rде изооктил, Ha
пример, 2 этилrексил):
олово в виде ди(изооктил) 2,2' [(диок
тилстаннилен )бис(тио )]диацетата, содержаще
ro около 27 % три(изооктил)2,2',2" [(моноок
тилстаннилидин )трис(тио )]триацетата: не более
0,25%;
олово в виде смеси, содержащей не более
76 % ди(изооктил)2,2' [(диметилстаннилен)
бис(тио)]диацетата и не более 85 % три(изо
октил )2,2' ,2" [( монометилстаннилидин )трис
(тио)] триацетата: не более 0,25 %;
1 фенилейкозан 1,З дион (бензоилстеаро
илметан): не более 1 % или не более 1 % смеси
обоих веществ.
Материалы MOryт содержать краситель или
пиrмент, а также титана диоксид для обеспече
ния непрозрачности материала.
Поставщик материала несет OTBeTCTBeH
ность за то, чтобы качественный и количествен
ный составы типовоro образца COOTBeTCTBOBa
ли качественному и количественному составам
каждой произведенной серии.
ОПИСАНИЕ
Порошок, шарики, rранулы, пластинки
разной толщины или образцы, отобранные из ro
товых объектов.
Нерастворимы в воде и в этаноле, раствори
мы в тетраrидрофуране, малорастворимы в Me
тиленхлориде.
При сжиrании пламя окрашивается в opaH
жево желтый цвет с зеленой каймой с выделе
нием ryCTOro черною дыма.
ИДЕНТИФИКАЦИЯ
Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
Пршютовленuе: остаток А, полученный
при приrотовлении раствора S2, как указано в
разделе «Испытания», растворяют в 5 мл тe
тра2идрофурана Р. Несколько капель полу
ченноrо раствора помещают на диск натрия
хлорида и выпаривают растворитель досуха
в сушильном шкафу при температуре 1 oo
105 ОС.
Максимумы П02лощения: 2975 CM 1, 291 О CM 1,
2865 CM \ 1430 CM 1, 1 ззо CM 1, 1255 CM 1, 690 CM 1
и 615 CM 1.
Полученный спектр должен соответствовать
спектру типовоro образца.
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
Раствор 51. 25 r испытуемою материала п<r
мещают в колбу из боросиликатноro стекла. Пр*
бавляют 500 мл воды Р и закрывают roрло колбы
алюминиевой фольюй или лабораторным CTa
каном из боросиликатноro стекла. Наrревают п<r
лученную смесь в автоклаве при температуре
(12Н2) ос в течение 20 мин, после чеro охлаждают
и декантируют раствор.
Раствор 52. 5,0 r испытуемою материала
растворяют в 80 мл тетра2идрофурана Р и д<r
водят до объема 100 мл .этим же. растворителем.
Если необходимо, фильтруют (раствор может
иметь опалесценцию). К 20 мл полученною pacт
вора прибавляют по каплям 70 мл 96 % спирта Р
при леrком перемешивании. Охлаждают в ледяной
бане в течение 1 ч. Потом фильтруют или центри
фуrируют (остаток А). Остаток А промывают 96 %
спиртом Р, прибавляют еro к фильтрату или надо--
садочной жидкости и доводят 96 % спиртом Р до
объема 100 мл.
Раствор 53. 5 r испытуемоro материала поме
щают в колбу из боросиликатноro стекла с притер
той пробкой. Прибавляют 100 мл 0,1 М раствора
кислоты хлористоводородной и кипятят с обрат
ным холодильником В течение 1 ч, после чеro ox
лаждают и осаждают твердую фазу.
Внешний вид раствора 51. Раствор 51 по
степени мутности не должен превышать эталон 11
(2.2.1) и быть бесцветным (2.2.2, метод /1).
Оптическая плотность раствора 51 (2.2.25).
100 мл раствора 51 выпаривают досуха. Остаток
растворяют в 5 мл 2ексана Р. Если необходимо,
фильтруют через фильтр, предварительно промы
тый 2ексаном Р. Оптическая плотность получен
ноro раствора в области от 250 нм до 310 нм не
должна превышать О,з.
Оптическая плотность раствора 52 (2.2.25).
Оптическая плотность раствора 52 в области от
250 нм до 330 нм не должна превышать 1,0 для
материалов, не содержащих 1 фенилейкозан 1 ,3
дион; оптическая плотность раствора 52, разве
денноro в 1 О раз 96 % спиртом Р, в области от
250 нм до ЗЗО нм не должна превышать 0,4 для Ma
териалов, содержащих 1 енилейкозан 1,З дион.
Материалы, стабилизированные оловом.
Не более 0,25 % Sп.
Основной раствор олова. 81 Mr ФСО добав
ки к пластмассе 23 растворяют в тeтpa2идpo
фуране р и доводят до объема 100,0 мл этим же
растворителем.
3.1.13. Добавки к пластмассе
561
Стандартный раствор олова. 20,0 мл oc
новноro раствора олова доводят 96 % спир
том Р до объема 100,0 мл.
К 0,10 мл раствора S2 в пробирке прибав
ляют 0,05 мл 1 М раствора кислоты хлористо
водородной, 0,5 мл раствора калия йодида Р и
5 мл 96 % спирта Р, тщательно перемешивают
и оставляют на 5 мин. К полученному раствору
прибавляют 9 мл воды Р и 0,1 мл раствора 5 r/л
натрия сульфита Р и тщательно перемешива
ют. Прибавляют 1,5 мл раствора дитизона Р,
свежеразведенноro в 100 раз метиленхлори
дам Р, встряхивают в течение 15 с и выдержива
ют в течение 2 мин. Параллельно rотовят paCT
вор сравнения, используя 0,1 мл стандартноro
раствора олова.
Фиолетовая окраска полученноro нижнеro
cnоя раствора S2 должна быть не интенсивнее
окраски раствора сравнения. Зеленовато синее
окрашивание раствора дитизона в присутствии
олова переходит в розовое.
Материалы, не стабилизированные
оловом. Не более 25 ррт Sп. К 5 мл paCT
вора 82 в пробирке прибавляют 0,05 мл 1 М
раствора кислоты хлористоводородной и
0,5 мл раствора калия йодида Р. Тщатель
но перемешивают и оставляют на 5 мин. К по
лученному раствору прибавляют 9 мл воды Р
и 0,1 мл раствора 5 r/л натрия сульфита Р
и тщательно перемешивают. Если получен
ный раствор не окрашен, прибавляют раствор
натрия сульфита порциями по 0,05 мл. При
бавляют 1,5 мл раствора дитuзона Р, свеже
разведенноro в 100 раз метиленхлоридом Р,
встряхивают в течение 15 с и выдерживают в
течение на 2 мин. Параллельно rотовят paCT
вор сравнения, используя 0,05 мл CTaHдapTHO
ro раствора олова (приroтовленноrо, как указа
но в испытании выше).
Фиолетовая окраска полученноro нижнеro
слоя раствора 82 должна быть не интенсивнее
окраски раствора сравнения.
Экстраrируемые тяжелые металлы (2.4.8,
метод А). Не более 20 ррт. 12 мл раствора S3
должны выдерживать испытание на тяжелые Me
таллы. Эталон roтовят с использованием 10 мл
этаЛОНН020 раствора свинца (1 ррт РЬ) Р.
Экстраrируемый цинк. Не более 100 ррm.
Атомно абсорбционная спектрометрия
(2.2.23, метод 1).
Испытуемый раствор. Раствор 83 разво
дят водой Р в 1 О раз.
Раствор сравнения. Раствор, содержащий
0,50 ррт цинка, rотовят разведением эталонно
20 раствора цинка (5 М2/МЛ Zп) Р 0,01 М pac
твором кислоты хлористоводородной.
Длина волны: 214,0 нм.
Проверяют отсутствие цинка в использу
емой кислоте хлористоводородной.
Сульфатная зола (2.4.14). Не более 1,0 %.
Определение проводят из 1,0 r испытуемоro Ma
териала. Для материалов, содержащих титана
диоксид как добавку, придающую непрозрач
ность, содержание сульфатной золы не должно
превышать 4,0 %.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение проводят методом сжиrания в
колбе с кислородом (2.5.10), используя 50,0 Mr
испытуемоro материала. Продукты сrорания
растворяют в 20 мл 1 М раствора натрия 2и
дроксида. К полученному раствору прибавля
ют 2,5 мл кислоты азотной Р, 10,0 мл 0,1 М
раствора серебра нитрата, 5 мл раствора
железа (111) аммония сульфата Р2, 1 мл дибу
тилфталата Р и титруют 0,05 М раствором
аммония тиоцианата до появления KpaCHOBa
то желтоro окрашивания. Параллельно прово
дят контрольный опыт.
1 мл 0,1 М раствора серебра нитрата co
ответствует 6,25 Mr поливинилхлорида.
01/2013:30113
3.1.13. ДОБАВКИ К ПЛАСТМАССЕ
ПРИМЕЧАНИЕ: номенклатура обозначе
на в соответствии справилами 'ИРАС. CиHO
нимы, обозначенные жирным шрифтом, cooт
ветствуют названиям, приведенным в тексте
,лавы З. Даны также синонимы, cooтвeтcтвy
ющие правилам текстов «Chemica/ Abs(,ac(».
Добавка 01. С 24 Н зв 0 4 - [117 81 7]. РМ RN74640.
СНЗ
o СН З
O (СНЗ
О СНз
(2RS) 2 Этилrексилбензол 1 ,2 дикарбо кси
лат.
Синонимы:
ди(2-этилrексил)фталат;
1 ,2 бензолдикарбоновой кислоты бис(2
этилrексил) эфир.
Добавка 02.
РМ RN 54120.
СI6НзоО4ZП'
[1 36 53 8].
о о
H т
СН З СН З
562
rосударственная фармакопея Республики Беларусь
Цинк (2RS) 2 этилrексаноат.
Синонимы:
цинк октаноат;
2 этилrексановой кислоты цинковая соль
(2:1 );
цинк 2 этилкапроат.
Добавка 03. [05518 18 3]/[0011O 30 5].
РМ RN 53440/53520.
1i( д r 1 СН З
НзС N/
л Н т
О
N.N' Этилендиалканамид (п и т = 14 или 16).
Синонимы:
N,N'-диацилэтилендиамины;
N,N' диацилэтилендиамин (в данном
случае «ацил» означает пальмитоил и стеароил).
Добавка 04. [8013 07 8]. РМ RN 88640.
Эпоксидированное соевое масло.
Добавка 05. [8016 11 3]. РМ RN 64240.
Эпоксидированное льняное масло.
Добавка 06. [57455 З7 5](ТSСА)/[101357
30 6](EINECS)/Pigment Ыие 29 (CI 77007).
РМ RN 64240. Пиrмент синий 29.
Ультрамариновый синий.
Д о б а в к а 07. С 15 Н 24 О . [ 1 2 8 3 7 О] .
РМ RN 46640.
Этиленбис[3,3 бис[3 ( 1, 1 диметилэтил '
. 4 rидроксифенил]бутаноат].
Синонимы:
этиленбис[3,3-бис[3-(1, 1-диметилэтил)-
4-rидроксифенил]бутаноат];
3,З бис[3 (1, 1 диметилэтил) 4 rидрокс
фенил]бутановой кислоты 1 ,2 этандииловый'
эфир;
этиленбис[3,3 бис(3 трет бутил-4 rидро-
ксифенил )бутират].
Добавка 09.
РМ RN 71680.
С7ЗН10в012'
[6683 19 8].
. (
ROJ'j .
OR
НзС
R......... =
Мета нтетрилтетра метилтетра кис[3 [3, 5
бис(1, 1 диметилэтил)-4-rидроксифенил]пропаноат].
Синонимы:
пентаэритритилтетракис[3-(3,5-ди-
трет-бутил-4-rидроксифенил)пропионат];
2,2 бис[[[3 [3,5 бис(1, 1 диметилэтил)
4 rидроксифенил ]пропаноил]окси ]метил ]про
пан 1 ,3 диил 3 [3,5 бис( 1, 1 диметилэтил) 4
rидроксифенил]пропаноат;
бензолпропановой кислоты 3,5 бис(1, 1-
ди метилэтил ) 4 rидрокси 2, 2 бис( rидро кси
метил)пропан 1,3 диоловый эфир (4:1);
2,2 бис(rидроксиметил)пропан 1 ,3 диол
тетракис[3 (3,5 ди трет бутил 4 rидрокси фе
нил)пропионат].
НзС СН З
сн з Добавка 10. С 54 Н 7в О з . [1709 70 2]. РМ RN 95200.
2,6 Бис( 1 , 1 диметилэтил )-4 метилфенол.
Синонимы:
бутилrидрокситолуол;
2,6 бис(1, 1 диметилэтил)-4 метилфенол;
2,6 ди трет бутил 4 метилфенол.
Добавка 08. С50Н660в' [32529 66 3].
РМ RN 53670.
0\
о
НО
НзС
НзС
ОН
НзС
R
НЗС * СНЗ
R # R
СН З
НЗ С СНЗ СН
R =НЗС -;::?'I 2
но
СН З
НзС
СН З
4,4',4" [(2,4.6 Триметилбензол 1 ,3,5 триил) ,
трис(метилен)]трис[2,6 бис(1, 1 диметилэтил)
фенол].
Синонимы:
2,2',2",6,6',6" -rекса-трет-бутил-4,4' ,4"-
[(2,4,6-триметил-1,3,5-бензолтриил)трисмети-
лен]трифенол;
1 ,З,5 трис[3,5 ди трет бутил 4 rидрокси
бензил] 2,4,6 триметилбензол;
4,4',4" [(2,4,6 триметил 1 ,3,5 бензолтри
ил )трис( метилен )]трис[2,6 бис( 1 , 1 диметиэтил)
фенол].
3.1.13. Добавки к пластмассе
563
Добавка 11. С з5 Н 6 Рз, [208g..7g..з]. РМ RN 68320.
Добавка 14. С41НВР6Р2' [38Q6...З4-6]. РМ RN50080.
о
Н,С
СН,
Октадецил 3 [3,5 бис(1,1 диметилэтил) 4
rидроксифенил]пропаноа
Синонимы:
октадецил-З-(З,5-ди-трет-бутил-4-rид-
роксифенил)пропионат;
3 [3, бис(1 , 1 диметилэтил )4 rидрокси
фенил] пропановой кислоты октадециловый
эфир.
Добавка 12. С42Н6ЗОзР' [31570 04 4].
РМ RN 74240.
H' ]Cr C СН, Н,С СН,
H' C СН, Н,С 1: СН,
Н,С """ 1 ?
cн /P " СН, СН,
Н,С
СН, I # СН,
СН,
Н,С
СН,
Трис[2,4 бис(1, 1 диметилэтил)фенил]
фосфит.
Синонимы:
трис(2,4-ди-трет-бутилфенил)фосфит;
2,4 бис( 1, 1 диметилэтил )фенола фосфит
(3:1 );
2,4 бис(1, 1 диметилэтил)фенилфосфит.
Добавка 1 З. С 4BH69NP6' [27676 62 61.
РМ RN 95З60.
R
I
0yNyO
/N'l('N,
R 11 R
о
H' C СН,
CH2
R =H,C """1
НО
СН,
Н, СН,
1 ,3,5 Трис[3,5 бис(1.1 диметилэтил)
4 rидроксибензил] 1,3,5 триазин
2,4,6(1 Н,ЗН,5Н) трион.
Синонимы:
1 ,З,5-трис(З,5-ди трет-бутил-4-rидро-
IICИбензил)-s-триазин-2,4,6(1Н,ЗН,5Н)-трион;
1 ,3,5 трис[[3,5--бис(1.1 диметилэтил)4 rидро-
ксифенил]метил] 1 ,З,5 триазин 2,4,6( 1 Н,ЗН,5Н)
трион.
н /..0." =t 0
H'C l J: r
о о
/), CH,
о о
16
З.9 Бис( октадецилокси ) 2,4.8, 1 o TeTpaOKca
3,9 фосфаспиро[5.5]ундекан.
Синонимы:
2,2' -бис( октадецилокси)-5,5' -спироби-
[1,З,2-диоксофосфинан];
3,9 бис( октадецилокси ) 2,4,8, 1 o TeTpaOKca
3,9 дифосфаспиро[5.5]ундекан.
Добавка 15. СЗ6Н74S2' [2500 8 1]. РМ RN 49840.
СН З
СН,
1,1' Дисульфандиилдиоксадекан.
Синонимы:
диоктадецилдисульфид;
1, 1' дитиооктадекан.
Добавка 16. С зо Н 56 О 4 S. [12З 28 ]. РМ RN93120.
H 1.>
Дидодецил 3,З' сульфандиилдипропаноат.
Синонимы:
дидодецил-З,З' -тиодипропионат;
дидодецил 3,3' сульфандиилдипро паноат;
3,3' тиобиспропановой кислоты додецило
вый эфир;
лаурилтиодипропионат.
Д обавка 17 С Н О s.r693 36 7]. РМ RN 93280.
'42B24 L
о о
нзс о s о---i1 нз
Диоктадецил 3,3' сульфандиилдипропаноат.
Синонимы:
ДИОkтадецил-З,З' -тиодипропионат;
диоктадецил З,З' сульфандиилдипропаноат;
3,3' тиобиспропановой кислоты октадеци
ловый диэфир;
стеарилтиодипропионат.
Добавка 18. [119345 01 6]. РМ RN 92560.
564
rосударственная фармакопея Республики Беларусь
Смесь семи продуктов, соответствующих
продукту реакции ди трет бутилфосфонита
с бифосфора трихлоридом, продуктом peaK
ции с бифенилом и 2,4 бис(1, 1 диметилэтил)
фенолом:
.
НзС СН З НзС СН З
компонент /
RO VV OR
\ /
R!\J \J\R
2,4 бис(1, 1 диметилэтил)фенил бифенил
,4' диилдифосфонит;
компонент /1
RO
\
P OR
:хк)
2,4 бис(1, 1 диметилэтил)фенил бифенил
3,4' диилдифосфонит;
компонент 1/1
RO
\
P OR
RO \
OR
2,4 бис(1, 1 диметилэтил)фенил бифенил
3,3' диилдифосфонит;
компонент /V
\r\r <OR
... .OR
2,4 бис(1, 1 диметилэтил)фенил бифенил-4
илфосфонит;
компонент V
OR
/
RO \
OR
2,4 бис(1, 1 диметилэтил)фенил фосфит;
компонент V/
RO VV OR
\ /
/ \,;j \. ;j П'ОR
RO О
2,4 бис( 1 , 1 диметилэтил )фенил 4' [бис[2,4
бис(1, 1 диметилэтил)фенокси]фосфанил]би
фенил-4 илфосфонат;
компонент V/I
R OH: 2,4 бис(1, 1 диметилэтил)фенол.
Добавка 19. С 1в Н З6 О 2 . [57 11-4]. РМ RN 24550.
1с0 2 н
НзС' l "1 в
Октадекановая кислота.
Синонимы:
стеариновая кислота;
октадекановая кислота.
Добавка 20. С 1в Н з5 NО. [301 2 0]. РМ RN 68960.
НЗС О
NH 2
7 7
(Z) Октадец 9 енамид.
Синонимы:
оле амид;
(Z) 9 октадеценамид;
9 цис олеамид.
Добавка21. С 22 Н 4з NО. [112 5]. РМ RN 52720.
НЗС О
NH 2
7 11
(Z) Докоз 9 енамид.
Синонимы:
эрукамид;
(Z) 1 3 докозенамид;
1 3 цис докозенамид.
Добавка 22. [65447 77 0]. РМ RN 60800.
НЗ С 0 1
О НзС Н
НзСО О O C СН з п
Сополимер диметилбутандионата и
1 (2 rидроксиэтил ) 2,2, 6, 6 тетраметил пи пери
дин-4 ола.
Синонимы:
3.1.14. Материалы на основе пластифицироваННО20 полuвинилхлорида
565
cononимер димeтиncyICЦИНё и (4-rмдpo-
к&и-2,2,6,6-тетраметилпиперидин-1-ил)этано-
ва.
Добавка 22.
Смесь компонента I с 27 % компонента 11.
Компонент 1. [26401 97 8].
R S S R
\/
НзСi{SПU СНз
СНЗ
Н,
R = Cyo з СН З
О
Бис[(2R S) 2 этилrексил] 2 ,2' [(диоктил
станнанетрил )бис( сульфанедиил )]диацетат.
Синонимы:
ди(изооктил)-2,2'-[(диоктилстаннилен)
бис(тио)]диацетат;
бис(изооктилоксикарбонилметилтио )ди
октилстаннан.
Компонент 11. [26401 86 5].
R S S R
\/
НзС..,f 1-Sn...... ...--R
IJ7 S
СНЗ
Н,
R = Cyo з СН З
О
Трис[(2RS) 2 этилrексил] 2 ,2' ,2" [( октил
станнанетрил )трис( сульфанедиил )]триацетат.
Синонимы:
ТРИ(ИЗ00ктил)-2,2' ,2" -[( монооктил-
стан нил иди н )трис(тио )]триа цетат;
2,2' ,2" [( октилстаннилидин )трис(тио )]три
уксусной кислоты триизостиловый эфир.
01/2013:30114
3.1.14. МАТЕРИАЛЫ НА ОСНОВЕ
ПЛАСТИФИЦИРОВАнноrо
ПОЛИВИНИЛХЛОРИДА
ДЛЯ КОНТЕЙНЕРОВ ДЛЯ
ВОДНЫХ РАСТВОРОВ
ДЛЯ ВНУТРИВЕнноrо
ПРИМЕНЕНИЯ
ОПРЕДЕЛЕНИЕ
Материалы на основе пластифицирован
Horo поливинилхлорида содержат не менее
55 % высокомолекулярноro полимера по
ливинилхлорида, полученноro полимериза
цией винил хлорида, и разные добавки.
Материалы на основе пластифицированноro
поливинилхлорида для контейнеров для водных
растворов для внутривенноro применения xapaктe
ризуются их природой И пропорцией веществ, KOТO
рые используют при их производстве.
ПРОИЗВОДСТВО
Материалы на основе пластифицированноro
поливинилхлорида получают методами полиме
ризации, которые raрантируют остаточное coдe
жание винилхлорида менее 1 ррm. Данный метод
должен быть валидирован для получения продук
та, выдерживающеro испытание на винилхлорид.
Винилхлорид. Не более 1 ррm. Парофазная
rазовая хроматоrpафия (2.2.28).
Раствор внутренне20 стандарта. К 20,0 мл
диметилацетамида Р с помощью микрошприца
прибавляют 1 О мкл эфира Р, поrpужая конец иrлы
в растворитель. Непосредственно перед использо
ванием разводят раствор диметилацетамидом Р
в 1000 раз.
Испытуемый раствор. 1,000 r испытуемоro
материала помещают во флакон вместимостью
50 мл и прибавляют 10,0 мл раствора внутреннеro
стандарта. rерметично закрывают колбу пробкой.
Встряхивают, избеraя контакта жидкости с пробкой.
Помещают флакон в водяную баню при температу
ре (60:1:1) ос и выдерживают в течение 2 ч.
Основной раствор винилхлорида. tотовят в
вытяжном шкафу. 50,0 мл диметилацетамида Р
помещают во флакон вместимостью 50 мл, rep
метично закрывают пробкой и взвешивают с точ
ностью до 0,1 м!: Наполняют полиэтиленовый или
полипропиленовый шприц вместимостью 50 мл ra
зообразным винилхлоридом Р, оставляют шприц
в контакте с rазом в течение 3 мин, затем rаз yдa
ляют и снова наполняют шприц 50,0 мл rазообраз
ноro винилхлорида Р. Подбирают для шприца rи
подермальную иrлу и уменьшают объем rаза в
шприце до 25 мл. Медленно вводят оставшиеся
25 мл винилхлорида во флакон, осторожно встря
хивая и избеraя контакта жидкости с иrлой. Опять
взвешивают флакон. Увеличение массы должно
составлять около 60 Mr (1 мкл раствора содержит
около 1,2 MKr винилхлорида). Выдерживают в тече
ние 2 ч. Основной раствор хранят в холодильнике.
Стандартный раствор винилхлорида. Oc
новной раствор винилхлорида дuметилацета
мид Р (1 :3, об/об).
Растворы сравнения. По 10,0 мл раствора
внутреннеro стандарта помещают в 6 одинаковых
флаконов вместимостью 50 мл. Флаконы repMe
тично закрывают пробками. В 5 колб с помощью
микрошприца прибавляют соответственно 1 мкл,
2 мкл, 3 мкл, 5 мкл И 10 мкл стандартною paCT
вора винилхлорида. Полученные таким образом
6 растворов содержат соответственно О мк!; около
0,3 мк!; 0,6 мк!; 0,9 мк!; 1,5 MKr и 3 MKr винилхлорида.
Встряхивают, избеraя контакта жидкости с пробкой.
Помещают колбы в водяную баню при температу
ре (60:1:1) ос и выдерживают в течение 2 ч.
566
rосударственная фармакопея Республики Беларусь
Условия хроматоrрафирования:
колонка из нержавеющей стали ДЛИНОЙ 3 м
и внутренним диаметром 3 мм, заполненная ди
атомитом силанизированным для 2азовой XpoMa
т02рафии Р, импреrнированным 5 % (м/м) диMe
тилстеариламида Р и 5 % (м/м) маКРО20ла 400 р;
2аз носитель: азот для хромат02рафии Р;
скорость 2аза носителя: 30 мл/мин;
детектор: пламенно--ионизационный;
температура: колонка 45 ос, блок ввода
проб 100 ос, детектор 150 ос;
объем вводимой пробы: 1 мл паровой фазы.
Рассчитывают содержание винилхлорида.
Добавки
Определенное количество добавок вводится в
полимер для улучшения еro химических, физиче.-
ских и механических свойств, а также для обеспе
чения возможности использования по назначению.
Все добавки выбирают из приведенноro ниже пе
речня. который определяет для каж;дою вещества
максимально допустимое ею содержание:
ди(2 этилrексил)фталат (добавка к пласт
массе 01): не более 40 %;
цинка октаноат (цинка 2 этилrексаноат) (дo
бавка к пластмассе 02): не более 1 %;
кальция стеарат или цинка стеарат: не более
1 % или не более 1 % смеси этих веществ;
N,N' диацилэтилендиамины (добавка к
пластмассе 03): не более 1 %;
не более 1 О % одною из следующих эпокси
дированных масел или не более 1 О % смеси двух
компонентов:
эпоксидированное соевое масло (добав
ка к пластмассе 04) с содержанием кислорода в
эпоксидной rруппе % и кислотным числом не
более 6;
эпоксидированное льняное масло (добавка
к пластмассе 05) с содержанием кислорода в эпок
сидной rруппе не более 1 О % и йодным числом не
более 7.
При добавлении красящих веществ использу
ют ультрамариновый синий. Moryт быть добавле
ны друrие неорrанические пиrменты с условием,
что для компетентною уполномоченною opraHa
будут предоставлены rарантии безопасности Ma
териала при введении таких добавок. В полимере
MOryт быть определены очень малые количества
антиоксидантов, добавленные к используемому
мономеру винилхлорида.
Поставщик материала несет ответственность
за то. чтобы качественный и количественный со--
ставы типовоro образца соответствовали каче.-
ственному и количественному составам каж;дой
произведенной серии.
ОПИСАНИЕ
Бесцветный или бледно желтый материал
в форме порошка, шариков, rранул или после
трансформации полупрозрачные пластинки
разной толщины со слабым запахом.
При сжиrании выделяется ryстой черный дым.
ИДЕНТИФИКАЦИЯ
При необходимости перед использованием
образцы испытуемоао материала разрезают на
части с максимальной длиной стороны не более
1 см.
к 2,0 r испытуемоro материала прибавляют
200 мл эфира, свободН020 от пероксидов Р, и Ha
rpевают с обратным холодильником в течение 8 ч.
Отделяют осадок В от раствора А фильтрованием.
Раствор А выпаривают досуха при понижен
ном давлении на водяной бане при температу
ре 30 ос. Остаток растворяют в 10 мл толуола Р
(раствор А 1). Осадок В растворяют в 60 мл эти
ленхлорида Р, наrревая на водяной бане с обрат
ным холодильником. Фильтруют. Полученный paCT
вор по каплям и при интенсивном встряхивании
прибавляют к 600 мл 2ептана Р, Harpeтoro почти
до кипения. Коаryлят В1 и орraнический раствор
разделяют roрячим фильтрованием. Охлаж;дают
последний, отделяют образующийся осадок В2 и
фильтруют через стеклянный фильтр (40) (2.1.2).
А. Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
При20товпение: коаryлят В1 растворяют в
30 мл тетра2идрофурана Р и прибавляют He
большими порциями при встряхивании 40 мл эта
нола Р. Отделяют осадок ВЗ фильтрованием и
сушат в вакууме над фосфора (V) оксидом Р при
температуре, не превышающей 50 ос. Растворяют
несколько миллиrраммов осадка В3 в 1 мл тe
тра2идрофурана Р, помещают несколько капель
полученноro раствора на диск натрия хлорида и
выпаривают досуха в сушильном шкафу при TeM
пературе 1 0()""",-1 05 ос.
Сравнение: ФСО поливинилхлорида.
В. Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
Получают спектр осадка С, полученною в ис
пытании на добавки к пластмассе 01, 04 и 05.
Сравнение: ФСО добавки к пластмассе 01.
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают на
части с максимальной длиной стороны не более
1 см.
Раствор 51. 5,0 r испытуемоro материала поме
щают в колбу для сжиraния. Прибавляют 30 мл Kиc
лоты серной Р и наrpевают до получения черной,
сиропообразной массы. Охлажд.ают и прибавляют
1 О мл концентрированноао раствора водорода пе
роксида Р. Осторожно наrpeвают. Охлаж;дают paCT
вор и прибавляют 1 мл концентрированноао pacт
вора водорода пероксида Р; повторяют поочередно
выариваниеe и прибавление раствора водорода пе
роксида до получения бесцветной жидкости. YMeHb
шают объем раствора до 1 О мл. Охлажд.ают и дово--
ДЯТ водой Р до объема 50,0 мл.
Раствор 52. 25 r испытуемоro материала по--
мещают в колбу из боросиликатноro стекла. При
бавляют 500 мл воды Р и закрывают roрло колбы
3.1.14. Материалы на основе пластифицироваННО20 поливинилхлорида
567
алюминиевой фольroй или лабораторным CTa
каном из боросиликатноro стекла. Наrpeвают по--
лученную смесь в автоклаве при температуре
(121:1:2) ос в течение 20 мин, после чеro ОХЛa>I\Qают
и декантируют раствор.
Внешний вид раствора 52. Раствор 52
должен быть прозрачным (2.2.1) и бесцветным
(2.2.2, метод 11).
Кислотность или щелочность. К 100 мл
раствора 52 прибавляют 0,15 мл раствора иHди
катора BRP Р. При прибавлении не более 1,5 мл
0,01 М раствора натрия 2идроксида окраска paCT
вора должна измениться на синюю. К 100 мл pacт
вора 52 прибавляют 0,2 мл раствора метилов020
оранжев020 Р. При прибавлении не более 1.0 мл
0,01 М раствора кислоты хлористоводородной
окраска раствора должна измениться с желтой на
оранжевую.
Оптическая плотность (2.2.25). 100,0 мл
раствора 52 выпаривают досуха. Остаток paCTBO
ряют в 5,0 мл 2ексана Р. Оптическая плотность
полученноro раствора в диапазоне длин волн от
250 нм до 310 нм не должна превышать 0,25.
Восстанавливающие вещества. Испыта
ние проводят в течение 4 ч после при20товпе
ния раствора S2. К 20,0 мл раствора 52 прибавля
ют 1 мл кислоты серной разведенной Р и 20,0 мл
0,002 М раствора калия пермаН2аната. Кипя
тят С обратным холодильником в течение З мин и
сразу ОХЛa>I\Qают. Прибавляют 1 r калия йодида Р
и немедленно титруют раствор 0,01 М раствором
натрия тиосульфата, используя в качестве инди
катара 0,25 мл раствора крахмала Р. Параллель
но проводят контрольный опыт С использованием
20 мл воды для инъекций Р. Разность ме>tЩу объе--
мами титранта не должна превышать 0,5 мл.
Первичные ароматические амины. Не
более 20 ррт. К 2,5 мл раствора А 1, полученно
ro при проведении идентификации, прибавля
ют 6 мл воды Р и 4 мл 0,1 М раствора кислоты
хлористоводородной. Интенсивно встряхивают
и удаляют верхний слой. К водному слою при
бавляют 0,4 мл свежеприroтовленноro раствора
1 О r/л натрия нитрита Р. Перемешивают и BЫ
жерживают в течение 1 мин. Прибавляют 0,8 мл
раствора 25 r/л аммония сульфамата Р, Bыдep
живают в течение 1 мин, затем прибавляют 2 мл
раствора 5 r/л нафтилэтилендиамина ди2и
дрохлорида Р. Через 30 мин окраска полученно
ro раствора должна быть не интенсивнее окраски
раствора сравнения, приroтовленноro параллель
но аналоrичным образом, но с использовани
ем вместо водноro слоя смеси из 1 мл раствора
0,01 r/л нафтиламина Р в 0,1 М растворе Kиc
лоты хлористоводородной, 5 мл воды Р и 4 мл
0,1 М раствора кислоты хлористоводородной.
Добавки к пластмассе 01, 04 и 05. TOHKOC
лойная хроматоrpафия (2.2.27).
Растворы сравнения. rотовят растворы
0,1 мr/мл ФСО добавки к пластмассе 01, ФСО дCf-
бавки к пластмассе 04 и ФСО добавки к пласт
массе 05, соответственно, в толуоле Р.
Пластинка: ТСХ пластинка со слоем силика
2еля GF 254 Р.
Подвижная фаза: толуол Р.
Наносимый объем пробы: 0,5 мл раствора А 1,
полученноro при проведении идентификации, в
виде полосы 30 мм х 3 мм и по 5 мкл ка>tЩоro из
растворов сравнения.
Фронт подвижной фазы: 15 см.
Высушивание: на воздухе.
Проявпение А: просматривают в ультрафиоле..-
товом свете при длине волны 254 нм.
Определяют положение зоны, COOTBeTCTBY
ющей добавке к пластмассе 01 (R,ОКОЛО 0,4). Соби
рают силикаrель с этой зоны и встряхивают с 40 мл
эфира Р в течение 1 мин. Фильтруют, промывают
фильтр двумя порциями, ка>tЩая по 1 О мл, эфира Р,
прибавляют их к фильтрату и выпаривают досуха.
Масса остатка С не должна превышать 40 Mr.
Проявпение В: обрабатывают парами йода в
течение 5 мин.
Просматривают xpoMaтorpaMMY, определяя
положение зоны, соответствующей добавкам к
пластмассе 04 и 05 (R, = О). Собирают силикаrель
с данной зоны. Аналоrично собирают с идентич
ной по размеру зоны силикаrель для контрольноro
опыта. Раздельно встряхивают оба образца в тече
ние 15 мин с 40 мл метанола Р. Фильтруют, про
мывают фильтр двумя порциями, каждая по 10 мл,
метанола Р, прибавляя их к фильтрату, и выпари
вают досуха. Разность между массами сухих OCTaT
ков не должна превышать 1 О Mr.
Добавка к пластмассе 03. Осадок В2, полу
ченный при проведении идентификации и находя
щийся на предварительно взвешенном стеклян
ном фильтре (40) (2.1.2), промывают этанолом Р.
Фильтр высушивают ДО постоянной массы над
фосфора (V) оксидам Р и взвешивают. Масса
остатка не должна превышать 20 Mr.
Инфракрасный спектр (2.2.24) остатка должен
соответствовать спектру ФСО добавки к пласт
массе 03.
Барий. Не более 5 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. 1,0 r испытуемоro Ma
териала прокаливают в кварцевом тиrле. Остаток
растворяют в 1 О мл кислоты хлористоводород
ной Р и выпаривают досуха на водяной бане. По
лученный остаток растворяют в 20 мл О. 1 М pacт
вора кислоты хлористоводородной.
Раствор сравнения. Раствор, содержащий
0,25 ррm бария, приroтовленный разведением
эталОНН020 раствора бария (50 ррт Ва) Р 0,1 М
раствором кислоты хлористоводородной.
Длина волны длина волны светоиспускания
бария 455,40 нм, спектральный фон 455,30 нм.
Проверяют отсутствие бария в используемой
кислоте хлористоводородной.
568
rосударственная фармакопея Республики Беларусь
Кадмий. Не более 0,6 ррm.
Атомно абсорбционная спектрометрия (2.2.23,
метод 1).
Испытуемый раствор. 1 О мл раствора S 1 вы...
паривают досуха. Остаток растворяют в 5 мл 1 %
(06/06) раствора кислоты хлористоводородной Р,
фильтруют и доводят объем фильтрата этим же
растворителем до 10,0 мл.
Растворы сравнения. Растворы сравнения ro
товят разведением эталОНН020 раствора кадмия
(0,1 % Cd) Р 1 % (об/о6) раствором кислоты хло
ристоводородной Р.
Источник излучения: лампа с полым кадми--
евым катодом.
ДЛина волны: 228,8 нм.
Атомизатор: воздушно--ацетиленовое пламя.
Проверяют отсутствие кадмия в используемой
кислоте хлористоводородной.
Кальций. Не более 0,07 %.
Атомно--эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют испыту--
емый раствор, приroтовленный для определения
бария.
Раствор сравнения. Раствор, содержащий
50.0 ррm кальция, приroтовленный разбавлени
ем эталОННО20 раствора кальция (400 ррт Са) Р
0,1 М раствором кислоты хлорucтоводородной.
Длина волны: длина волны светоиспуска
ния кальция З15,89 нм, спектральный фон
315,60 нм.
Проверяют отсутствие кальция в использу
емой кислоте хлористоводородной.
Олово. Не более 20 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Непосредственно
перед использованием раствор S1 разводят в 10
раз водой Р.
Раствор сравнения. Непосредственно перед
использованием 2 мл эталОНН020 раствора олова
(5 ррт Sп) Р помещают в мерную колбу вместимо--
стью 50 мл, содержащую 5 мл 20 % (об/о6) pacт
вора кислоты серной р, и доводят водой Р до
объема 50 мл.
Длина волны длина волны светоиспускания
олова 189,99 нм, спектральный фон 190,1 О нм.
Проверяют отсутствие олова в используемой
кислоте хлористоводородной.
Цинк. Не более 0,2 %.
Атомно--абсорбционная спектрометрия (2.2.23,
метод 1).
Испытуемый раствор. Раствор S1 разводят в
100 раз 0,1 М раствором кислоты хлористоводо
родной.
Растворы сравнения. Растворы сравнения ro
товят разведением этаЛОНН020 раствора цинка
(100 ррт Zп) Р 0,1 М раствором кислоты хлори
стоводородной.
Источник излучения: лампа с полым цинко--
вым катодом.
Длина волны 213,9 нм.
Атомизатор: воздушно ацетиленовое пламя.
Проверяют отсутствие цинка в используемой
кислоте хлористоводородной.
Тяжелые металлы (2.4.8, метод А). Не более
50 ррm. К 10.0 мл раствора S1 прибавляют 0,5 мл
раствора фенолфталеина Р и затем KOHцeHтpи
роваНН020 раствора натрия 2идроксида Р до по--
лучения бледно розовоro окрашивания. Доводят
водой Р до объема 25 мл. 12 мл полученноro paCT
вора должны выдерживать испытание на тяжелые
металлы. Эталон roтовят с использованием эта
ЛОНН020 раствора свинца (2 ррт РЬ) Р.
Вещества, экстраrируемые ВОДОЙ. Не более
0,3 %. 50,0 мл раствора S2 выпаривают на BO
дяной бане досуха и высушивают в сушильном
шкафу при температуре 1 O 105 ос до постоян--
ной массы. Параллельно проводят контрольный
опыт, используя 50,0 мл воды для инъекций Р.
Масса сухоro остатка не должна превышать 7,5 Mr
с учетом контрольноro опыта.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение проводят методом сжиraния в
колбе с кислородом (2.5.10), используя 50,0 Mr ис
пытуемоro материала. Продукты сжиraния раство--
ряют в 20 мл 1 М раствора натрия 2идроксида.
К полученному раствору прибавляют 1 мл дибу
тилфталата Р, 2,5 мл кислоты азотной Р, 5 мл
раствора железа (I/Q аммония сульфата Р2 и
10,0 мл 0,1 М раствора серебра нитрата. Титру
ют раствор 0,05 М раствором аммония тиoциa
ната до получения красновато,.желтоro окрашива...
ния. Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора серебра нитрата соот...
ветствует 6,25 Mr поливинилхлорида.
01/2013:30115
3.1.15. ПОЛИЭТИЛЕНТЕРЕФТАЛАТ
ДЛЯ КОНТЕЙНЕРОВ ДЛЯ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
ДЛЯ НЕПАРЕНТЕРАльноrо
ПРИМЕНЕНИЯ
о
0/""'/.-0
о
л
п = 1 00 200
ОПРЕДЕЛЕНИЕ
Полиэтилентерефталат получают поли
меризацией кислоты терефталевой или ди
3. 1. 15. Полиэтилентерефталат для контейнеров для лекарствеННblХ
569
метилтерефталата с этиленrликолем. Для
полимеризации MOryT быть использованы
кислота изофталевая, диметилизофталат,
1 ,4 бис(rидроксиметил)циклоrексан (цикло
reKcaH 1 ,4 диметанол) или диэтиленrликоль.
Он может содержать не более 0,5 % кремния
диоксида или силикатов и краситель, разре
шенный к применению компетентным уполно
моченным opraHoM.
ПРОИЗВОДСТВО
Метод получения должен быть валидирован
для подтверждения, что остаточное содержание
ацетальдеrида в rранулах не превышает 10 ррт.
ОПИСАНИЕ
Прозрачные или опалесцирующие rранулы.
Практически нерастворим в воде Р, в 96 %
спирте Р и в метuленхлориде Р. r идролизуется
сильными основаниями.
ИДЕНТИФИКАЦИЯ
А. Абсорбционная спектрофотометрия в уль--
трафиолетовой и видимой областях (2.2.25).
При20товление: 0,1 О r испытуемоro MaTe
риала помещают в колбу из боросиликатноro
стекла с притертой пробкой. Прибавляют 25 мл
раствора 200 r/л калия 2идроксида Р в paCTBO
ре 50 % (об/об) этанола Р. Кипятят с обратным
холодильником в течение 30 мин, охлаждают и
доводят водой Р до объема 100 мл. Если необ
ходимо, фильтруют. 1,0 мл фильтрата доводят
водой Р до объема 100 мл.
Диапазон длин волн: от 210 нм до ЗЗО нм.
Максимум ПО2Лощения: при 240 нм.
В. Абсорбционная спектрофотометрия в ин
фракрасной области (2.2.24).
При20товление: 0,05 r испытуемоro MaTe
риала растворяют в 2 мл 1, 1, 1,3,3,3 2ексафтор
пропан 2 ола Р. Несколько капель раствора
наносят на стеклянную пластин которая Ha
ходится на водяной бане, в вытяжном шкафу
для получения пластины размером около 15х 15
мм. После выпаривания растворителя пласти <
ну удаляют, используя поток воды и скребок.
Сушат в сушильном шкафу при температуре
100 105 ос в течение 1 2 ч.
Максимумы ПО2Лощения: при 1725 CM 1,
1410 CM 1, 1265 CM 1, 1120 CM 1, 1100 CM 1, 1020 CM 1,
875 см., и 725 CM 1.
Полученный спектр должен соответствовать
спектру типовоro образца.
ИСПЫТАНИЯ
При необходимости перед использованием
образцы испытуеМ020 материала разрезают
на части с максимальной длиной стороны не
более 1 см.
Раствор 51. 10,0 r испытуемоro материала
помещают в колбу из боросиликатноro стекла с
притертой пробкой. Прибавляют 200 мл воды Р
и наrревают при температуре 50 ос в течение
5 ч. Охлаждают и декантируют раствор. Pacт
вор S1 используют в течение 4 ч после при20
товления.
Раствор 52. 10,0 r испытуемоro материала
помещают в колбу из боросиликатноro стекла
с притертой пробкой. Прибавляют 100 мл 96 %
спирта Р и наrревают при температуре 50 ос в
течение 5 ч. Охлаждают и декантируют раствор.
Раствор S2 используют в течение 4 ч после
при20товления.
Раствор 53. 20 r испытуемоro материала по
мещают в колбу из боросиликатноro стекла с при
тертой пробкой. Прибавляют 50 мл 0,1 М раствора
кислоты хлористоводородной Р и наrревают при
температуре 50 ос в течение 5 ч. Охлаждают и дe
кантируют раствор. Раствор S3 используют в тe
чение 4 ч после при20товления.
Раствор 54. 20 r испытуемоro материала по
мещают в колбу из боросиликатноro стекла с при
тертой пробкой. Прибавляют 50 мл 0,1 М раствора
натрия 2идроксида Р и наrpевают при температу
ре 50 ос в течение 5 ч. Охлаждают и декантиру
ют раствор. Раствор S4 используют в течение
4 часов после при20товления.
Внешний вид раствора 51. Раствор S1
должен быть прозрачным (2.2.1).
Внешний вид раствора 52. Раствор S2
должен быть прозрачным (2.2.1) и бесцветным
(2.2.2, метод /1).
Кислотность или щелочность. К 50 мл раст
вора 81 прибавляют 0,15 мл раствора иHдиKaтo
ра BRP Р. Раствор окрашивается в желтый цвет.
При прибавлении не более 0,5 мл 0,01 М раствора
натрия 2идроксида окраска раствора должна из
мениться на синюю. К друrим 50 мл раствора 81
прибавляют 0,2 мл раствора метилов020 opaH
жевО20 Р. Раствор окрашивается в желтый цвет.
При прибавлении не более 0,5 мл 0,01 М раствора
кислоты хлористоводородной окраска раствора
должна измениться с желтой на оранжевую.
Оптическая плотность раствора 51. (2.2.25).
Оптическая плотность раствора S1 в диапазоне
длин волн от 220 нм до 340 нм не должна превы
шать 0,2. Для окрашенноro полиэтилентерефтала
та оптическая плотность раствора 81 в диапазоне
длин волн от 400 нм ДО 800 нм не должна превы
шать 0,05.
Оптическая плотность раствора 52. (2.2.25).
Оптическая плотность раствора S2 в диапазоне
длин волн от 400 нм ДО 800 нм не должна превы
шать 0,05.
Восстанавливающие вещества. К 20,0 мл
раствора S1 прибавляют 2 мл 0,5 М раствора Kиc
лоты серной и 20,0 мл 0,002 М раствора калия
570
rосударственная фармакопея Республики Беларусь
пермаН2аната. Кипятят в течение 3 мин и HeMeд
ленно охлаждают до комнатной температуры. При
бавляют 1 r калия йQдuда Р и немедленно титруют
раствор 0,01 М раствором натрия тиосульфата,
используя в качестве индикатора 0,25 мл раствора
крахмала Р. Параллельно проводят контрольный
опыт, используя 20,0 мл воды Р. Разность между
объемами титранта не должна превышать 0,5 мл.
Вещества, растворимые в диоксане. Не
более 3 %. 2 r испытуемоro материала помеща
ют в колбу из боросиликатноro стекла с притертой
пробкой. Прибавляют 20 мл диоксана Р и кипятят с
обратным холодильником в течение 2 ч. 10 мл по--
лученноro раствора выпаривают на водяной бане
досуха и сушат остаток при температуре 10(}.......
105 ос. Масса полученноro остатка не должна пр&
вышать 30 Mr.
Экстраrируемый алюминий. Не более
1 ррm.
Атомно--эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют pacт
вор S3.
Раствор сравнения. rотовят путем раз
ведения эталонноао раствора алюминия
(200 ррт А/) Р 0,1 М раствором кислоты хлори
стоводородной.
Длина волны: длина волны светоиспуска
ния алюминия 396,15 нм, спектральный фон
396,25 нм.
Проверяют отсутствие алюминия в использу
емой кислоте хлористоводородной.
Экстраrируемая сурьма. Не более 1 ррт.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют pacт
вор S4.
Раствор сравнения. rотовят путем разведе
ния этаЛОНН020 раствора сурьмы (100 ррт Sb) Р
0,01 М раствором натрия 2идроксида.
Длина волны: 231,15 нм или 217,5 нм, спек
тральный фон 231 ,05 нм.
Экстраrируемый барий. Не более 1 ррт.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют раст
вор S3.
Раствор сравнения. rотовят путем разведе
ния эталОНН020 раствора бария (50 ррт Ва) Р
0,1 М раствором кислоты хлористоводородной.
Длина волны длина волны светоиспускания
бария 455,40 нм, спектральный фон 455,30 нм.
Проверяют отсутствие бария в используемой
кислоте хлористоводородной.
Экстраrируемый кобальт. Не более 1 ррт.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют раст
вор S3.
Раствор сравнения. rотовят путем разведения
эталонноао раствора кобальта (100 ррт Со) Р
0,1 М раствором кислоты хлористоводородной.
Длина волны: длина волны светоиспуск
ния кобальта 228,62 нм, спектральный фон
228,50 нм.
Проверяют отсутствие кобальта в использу
мой кислоте хлористоводородной.
Экстраrируемый rерманий. Не более
1 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют раст
вор S4.
Раствор сравнения. rотовят путем раз
ведения эталОННО20 раствора 2ермания
(100 ррт Ge) Р 0,01 М раствором натрия 2и
дроксида.
Длина волны: 206,87 нм или 265,12 нм, спек
тральный фон 206,75 нм.
Экстраrируемый марrанец. Не более
1 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют paCT
вор S3.
Раствор сравнения. rотовят путем раз
ведения эталОНН020 раствора маР2анца
(100 ррт Мп) Р О, 1 М раствором кислоты хлори
стоводородной.
Длина волны: длина волны светоиспуска
ния марrэнца 257,61 нм, спектральный фон
257,50 нм.
Проверяют отсутствие марrанца в использу
емой кислоте хлористоводородной.
Экстраrируемый титан. Не более 1 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют paCT
вор S3.
Раствор сравнения. rотовят путем раз
ведения этаЛОНН020 раствора титана
(100 ррт Тi) Р 0,1 М раствором кислоты хлори
стоводородной.
Длина волны: 323,45 нм или 334,94, спек
тральный фон 323,35 нм.
Проверяют отсутствие титана в использу
емой кислоте хлористоводородной.
Экстраrируемый цинк. Не более 1 ррm.
Атомно эмиссионная спектрометрия (2.2.57).
Испытуемый раствор. Используют paCT
вор S3.
Раствор сравнения. rотовят путем разведе
ния этаЛОНН020 раствора цинка (100 ррт Zп) Р
0,1 М раствором кислоты хлористоводородной.
Длина волны: длина волны светоиспуска
ния цинка 213,86 нм, спектральный фоl-'
21З,75 нм.
Проверяют отсутствие цинка в использу
емой кислоте хлористоводородной.
Сульфатная зола (2.4.14). Не более 0,5 %.
Определение проводят из 1,0 r испытуемоro Ma
териала.
01/2013:30200
3.2.1. Стеклянные контейнеры для фармацевтичеСКО20 использования
01/2013:30201
571
3.2.
КОНТЕЙНЕРЫ
Контейнер для фармацевтическоro ис
пользования представляет собой изделие, KO
торое содержит продукцию или предназначен
ДЛЯ ее хранения и находится или может Ha
ходиться в непосредственном контакте с ней.
Укупорочное средство является частью KOH
тейнера.
Контейнер (см. Общие положения,
статья 1.3.) изrотавливают таким образом,
чтобы содержимое моrло быть извлечено
способом, соответствующим надлежащему
применению лекарственноro средства. Он
обеспечивает разную степень защиты в за
висимости от свойств продукции и действия
факторов окружающей среды и доводит до
минимума потери компонентов. Контейнер
не должен вступать в физическое или хи
мическое взаимодействие с содержимым
таким образом, чтобы изменять показатели
ero качества по отношению к требуемым по
казателям.
Однодозовый контейнер контейнер,
который содержит количество лекарствен
HOro средства, соответствующее полностью
или по частям дозе для одноразовоro приме
нения.
Мультидозовый контейнер контей
нер, который содержит количество лекар
ственноro средства, соответствующее двум
или более дозам.
Плотно укупоренный контейнер KOH
тейнер, который защищает содержимое от
заrрязнения извне твердыми веществами и
жидкостями, а также от потери компонентов в
обычных условиях при применении, хранении
и транспортировке.
Воздухонепроницаемый контейнер
контейнер, который непроницаем для TBep
дых веществ, жидкостей и rазов в оБЫЧIjЫХ
условиях при применении, хранении и TpaHC
портировке. Если контейнер содержит более
одной дозы лекарственноro средства и может
быть открыт неоднократно, то после повтор
ноro закупоривания он должен оставаться
воздухонепроницаемым.
3апаянный контейнер (или 2ерметично
укупоренный контейнер) контейнер, YKY
поренный с помощью расплавления матери
ала контейнера.
Контейнер с контролем первО20 вCKpЫ
тия укупоренный контейнер, обеспечен
ный приспособлением для контроля еro пер
воro вскрытия.
Контейнер. защищенный от вскрытия
детьми контейнер, снабженный системой
укупоривания, исключающей возможность
вскрытия детьми.
3.2.1. СТЕКЛЯННЫЕ КОНТЕЙНЕРЫ
для ФАРМАЦЕВТИЧЕскоrо
ИСПОЛЬЗОВАНИЯ
Стеклянные контейнеры для фармацевти
ческоro использования изделия из стекла, KO
торые непосредственно контактируют с лекар
ственными средствами.
Бесцветное стекло имеет высокую CBeтo
проницаемость в видимой части спектра.
Цветное стекло получают добавлением
небольших количеств оксидов металлов, BЫ
бранных соrласно желаемым спектральным xa
рактеристикам.
Нейтральное стекло представляет собой
боросиликатное стекло, содержащее значитель
ные количества бора оксида, алюминия оксида и
оксидов щелочных и/или щелочноземельных Me
таллов. Блаrодаря своему составу нейтральное
стекло характеризуется высокой термической и
rидролитической устойчивостью.
Натрий кальциевое силикатное стекло
стекло на основе кремния оксида, содержащее
оксиды щелочных металлов, rлавным образом
оксид натрия, и оксиды щелочноземельных Me
таллов, rлавным образом оксид кальция. Бла
rодаря своему составу силикатное стекло xa
рактеризуется только средней rидролитической
устойчивостью.
r идролитическая стабильность стеклянных
контейнеров для фармацевтическоro ИСПОЛIr
зования выражается устойчивостью к выдел
нию растворимых минеральных веществ в воду в
предписанных условиях при контакте внутренней
поверхности контейнера или порошкообразн
ro стекла с водой. rидролитическая устойчивость
оценивается титрованием выделившейся щелочи.
В зависимости от rидролитической устойчи
вости стеклянные контейнеры классифицируют
следующим образом:
контейнеры из стекла класса 1. Изrотовле
ны из нейтральноro стекла и имеют высокую rи
дролитическую устойчивость блаroдаря химиче
скому составу самоro стекла;
контейнеры из стекла класса 11. Изroтов
лены обычно из натрий кальциевоro силикат
ноro стекла и имеют высокую rидролитическую
устойчивость блаrодаря специальной обработке
поверхности;
контейнеры из стекла класса 111. Изroтов
лены обычно из натрий кальциевоro силикат
ноro стекла и имеют среднюю rидролитическую
устойчивость.
Следующие ниже набранные курсивом
общие рекомендации к отдельным классам CTe
клянных контейнеров MOryT быть применены к
разным видам лекарственных средств. Произ
водитель лекарственноro средства несет OTBeT
ственность за обеспечение приroдности выбран
ноro контейнера.
572
rосударственная фармакопея Республики Беларусь
Контейнеры из стекла класса I при20д
ны для хранения большинства лекарственных
средств, предназначенных для парентерально
20 и непарентераЛЬН020 применения.
Контейнеры из стекла класса 11 при20дны
для хранения в основном кислых и нейтраль
ных водных растворов для парентераЛЬН020 и
непарентераЛЬН020 применения.
Контейнеры из стекла класса 111 при20дны
для хранения неводных растворов и порошков
(за исключением лиофилизированных) для па
рентераЛЬН020 применения, а также для ле
карственных средств для непарентераЛЬН020
применения.
Как правило, также MOryт быть использо
ваны стеклянные контейнеры, имеющие rидро
литическую устойчивость выше требуемой для
конкретноro лекарственноro средства.
Материал, из котороro изrотовлен контей
нер для конкретноro лекарственноro средства,
не должен содержать никаких веществ, которые
моrли бы экстраrироваться содержимым контей
нера в количестве, влияющем на стабильность
лекарственноro средства, или быть потенциаль
но опасными в отношении токсичности.
Лекарственные средства для парентераль
ноro применения обычно выпускают в контей
нерах из бесцветноro стекла; для светочув
ствительных веществ MOryт использоваться и
цветные стеклянные контейнеры. Для друrих ле
карственных средств используются как бесцвет
ные, так и цветные стеклянные контейнеры. Pe
комендуется, чтобы все стеклянные контейнеры
для жидких лекарственных средств и порошков
для приroтовления растворов для парентераль
ноro лрименения позволяли проводить визуаль
ный контроль их содержимоrо.
Внутренняя поверхность стеклянных KOH
тейнеров может быть специально обработа
на для увеличения rидролитической устойчи
вости, придания водоотталкивающих свойств и
др. Внешняя поверхность также может быть об
работана для уменьшения трения и увеличения
сопротивления к механическим повреждениям.
Обработка внешней поверхности должна про
водиться так, чтобы исключить заrрязнение BHY
трен ней поверхности контейнера.
Стеклянные контейнеры, за исключением
контейнеров из стекла I класса, не MOryT быть
использованы повторно. Контейнеры для чело
веческой крови и ее компонентов не MOryT быть
использованы повторно.
Стеклянные контейнеры для фармацевти
ческоro использования должны выдерживать ис
пытания на rидролитическую устойчивость. Если
стеклянные контейнеры имеют деТ:::IЛИ, изroтов
ленные из друroro материала, то испытания про
водятся только на стеклянных частях контейнера.
Для определения качества стеклянноro KOH
тейнера в соответствии с еro предполаrаемым
использованием, проводят одно или более из
перечисленных ниже испытаний.
Испытание на rидролитическую устой
чивость проводится для определения класса
стекла (1, 11 или 111) и для контроля еro rидролити
ческой устойчивости.
Кроме этоro, для контейнеров для водных
лекарственных средств для парентеральноro
применения проводят испытания на мышьяк,
для цветных стеклянных контейнеров на спек
тральное пропускание.
rИДРОЛИТИЧЕСКАЯ УСТОЙЧИВОСТЬ
Таблица 3.2.1. 1
Типы стеклянных контейнеров
Тип контейнера Проводимые
испытания
Контейнеры из стекла Испытание А (поверх
КI1acca 1 и класса 11 ностная rидролитиче
(для отличия от KOH ская устойчивость)
тейнеров из стекла
класса 111)
Контейнеры из стекла Испытание В (rидроли
класса 1 (для отли тическая устойчивость
чия от контейнеров измельченноrо в по
из стекла класса 11 и рошок стекла) или ис
класса 111) пытание С (проверка
наличия специальной
обработки поверхно
сти)
Контейнеры из стекла Испытания А и В или
класса 1 и класса 11 в испытания А и С
случае определения,
чем вызвана BЫCO
кая rидролитическая
устойчивость: химиче
ским составом стекла
или специальной об
работкой поверхности
Испытание проводят путем титрования pac
творов, полученных, как указано в испытаниях А,
ВиС.
ОБОРУДОВАНИЕ
Оборудование для испытаний включает:
автоклав, поддерживающий температуру
(121:!::1) ОС, оснащенный термометром или кали
брованным температурным датчиком, клапаном
давления, вентиляционным краном и поддоном,
а также имеющий достаточную емкость для раз
мещения над уровнем воды такоro количества
контейнеров, которое необходимо для проведе
ния испытания; перед испытанием тщательно
промывают сосуд автоклава и вспомоrательное
оборудование водой Р.
бюретка подходящеro объема;
мерные колбы вместимостью 1000 мл;
пипетки и лабораторные стаканы;
конические колбы вместимостью 100 мл И
250 мл;
водяная баня;
металлическая фольrа (например, алюми
ниевая или из нержавеющей стали).
3.2.1. Стеклянные контейнеры для фармацевтическоzо использования
573
Колбы и лабораторные CT KaHЫ должны
ранее быть использованным ' для i1роведения
испытаний либо их предварительно заполняют
водой Р и выдерживают в автоклаве при темпе
ратуре 121 ос в течение не менее 1 ч.
ОПРЕДЕЛЕНИЕ ОБЪЕМА НАПОЛНЕНИЯ
Объем наполнения это объем воды, KO
торой наполняют контейнер для проведения
испытания. Для виал и флаконов объем напол
нения составляет 90 % от объема наполнения
до краев контейнера. Для ампул это объем до
высоты плеча.
Виалы и флаконы. Из серии образцов OT
бирают случайным образом 6 контейнеров (или
3 контейнера, если их вместимость превышает
100 мл) и очищают их от посторонних частиц и
осколков. Взвешивают пустые контейнеры с точ
ностью до 0,1 r. Помещают контейнеры на roри
зонтальную поверхность, заполняют их водой
оостиллированной Р до ободка, избеrая пере
ливан ия и образования пузырьков, и доводят
объем жидкости до краев контейнеров. Взве
wивают заполненные контейнеры, фиксируют
массу воды с точностью ДО сотых (для контей
неров номинальным объемом менее и равном
зо мл) или десятых (для контейнеров номи
нальным объемом более 30 мл) И рассчитыва
IOТ среднее значение. Рассчитывают среднее
sначение объема контейнеров, заполненных до
llpЭев, в миллилитрах, и умножают полученное
значение на 0,9. Полученный объем, указанный
с точностью до десятоro знака, представляет
, собой объем наполнения для данной серии KOH
тейнеров.
Ампулы. Не менее 6 сухих ампул помеща
IOТ на плоскую roризонтальную поверхность и
3аполняют при помощи бюретки водой дистил
лированной Р до точки А (рисунок 3.2.1. 1), в
которой «тело» ампулы переходит в «плечо».
д
;
Рисунок 3.2.1. 1. Объем наполнения ампулы
(до точки А)
Отмечают объем с точностью ДО сотых и pac
считывают среднее значение. Полученный
объем, указанный с точностью до десятоro
знака, представляет собой объем наполнения
для данной серии ампул. Кроме этоro, объем
наполнения может быть определен при помощи
взвешивания.
ИСПblТАНИЕ А. rИДРОЛИТИЧЕСКАЯ
УСТОЙЧИВОСТЬ НАРУЖНОЙ
ПОВЕРХНОСТИ СТЕКЛЯННblХ
КОНТЕЙНЕРОВ (ПОВЕРХНОСТНАЯ
rИДРОЛИТИЧЕСКАЯ УСТОЙЧИВОСТЬ)
Определение проводится на контейнерах,
не используемых ранее. Необходимые объемы
испытуемой жидкости для конечноro испытания
указаны в таблице 3.2.1. 2.
Таблица 3.2.1. 2
Объем испытуемой жидкости и количество
титрований
Объем Объем
испытуемой жид- Количество
наполнения, кости для oднoro титрований
мл
титрования, мл
Д03 25,0 1
Свыше 3 до 50,0 2
30
Свыше 30 до 100,0 2
100
Свыше 100 100,0 3
Очистка. Очищают контейнеры от посторон
них частиц и осколков. Незадолro до проведения
испытания контейнеры ополаскивают не менее
двух раз водой Р и оставляют. Непосредственно
перед проведением испытания контейнеры опу
стошают, ополаскивают один раз водой Р, затем
один раз водой Р1 и оставляют до высыхания.
Полная процедура очистки от первоro опола
скивания не должная длиться меньше 20 мин и
больше 25 мин.
Укупоренные ампулы наrревают на водяной
бане или сушильном шкафу при температуре
около 50 ос в течение 2 мин; дополнительное опо
ласкивание перед испытанием не допускается.
Наполнение и наrревание. Контейнеры
заполняют водой Р1 до достижения объема Ha
полнения. Контейнеры в форме картриджей или
предварительно заполненных шприцев укупори
вают подходящим способом с помощью матери
ала, не оказывающеro влияния на проведение
испытания. Все контейнеры, включая ампулы,
должны быть неплотно прикрыты инертным Ma
териалом, таким как, например, чашка из ней
тральноro стекла или алюминиевая фольrа,
предварительно промытые водой Р. Контейне
ры помещают на подцон автоклава, который
затем располаrают в автоклаве таким образом,
чтобы вода Р не соприкасалась с ним. Автоклав
закрывают и проводят следующие операции:
rосударственная фармакопея Республики Беларусь
Таблица 3.2.1. 3
Допустимые пределы при определении поверхностной 2идролитической устойчивости
574
Максимальный объем 0,01 М раствора кислоты хлористоводо-
Объем наполнения (мл) родной на 100 мл испытуемой жидкости (мл)
Стеклянные контейнеры
Класс стекла 1 и 11 Класс стекла 111
До 1 мл 2,0 20,0
Свыше 1 до 2 1,8 17,6
Свыше 2 до 5 1,3 13,2
Свыше 5 до 10 1,0 10,2
Свыше 1 О до 20 0,80 8,1
Свыше 20. до 50 0,60 6,1
Свыше 50 до 100 0,50 4,8
Свыше 100 до 200 0,40 3,8
Свыше 200 до 500 0,30 > 2,9
Свыше 500 0,20 2,2
подоrревают автоклав до 100 ос в течение
1 О мин, позволяя пару проходить через вентиля
ционный кран;
закрывают вентиляционный кран и повы
шают температуру со 100 ос до 121 ос со CKOpO
стью 1 ос в минуту;
под,церживают температуру 121 ос в тече
ние (60::1:1) мин;
снижают температуру со 121 ос до 100 ос
со скоростью 0,5 ос в минуту с отрытым венти
ляционным краном для предупреждения образо
вания вакуума;
автоклав не открывают до тех пор, пока
температура не снизится до 95 ОС;
вынимают контейнеры из автоклава, co
блюдая обычные меры предосторожности, поме
щают их в водяную баню при температуре 80 ос и
пропускают через нее холодную проточную воду,
обращая внимание на то, чтобы вода не KOHTaK
тировала снеплотно прикрывающим колпачком
(например, из алюминиевой фольrи) и не заrряз
няла экстракционный раствор в контейнерах;
время охлаждения не должно превышать
30 мин.
Экстракционный раствор титруют в COOТBeT
ствии с методикой, описанной ниже.
Методика. Титрование проводят в тече
ние 1 ч после извлечения контейнеров из aBTO
клава. Объединяют жидкости из контейнеров и
перемешивают. В коническую колбу помещают
необходимый объем жидкости (таблица 3.2.1. 2).
В друrую идентичную колбу помещают такой
же объем воды Р1 (контрольный раствор). В
каждую колбу прибавляют по 0,05 мл раствора
метилов020 краСН020 Р на каждые 25 мл жид
кости. Титруют контрольный раствор 0,01 М
раствором кислоты хлористоводородной.
Испытуемую жидкость также титруют 0,01 М
раствором кислоты хлористоводородной до
тех пор, пока цвет раствора не станет аналоrи
чен цвету, полученному в контрольном опыте.
Находят разность объемов титранта, израсхо
дованных на титрование испытуемой жидкости
и контрольною раствора и выражают ее в мил
лилитрах 0,01 М раствора кислоты хлористо
водородной на 100 мл. Значение объема, за
траченною на титрование, в количестве менее
1 мл выражают с точностью до сотых, в количе
стве 1,0 мл И более до десятых.
Пределы. Результаты или средние резуль
таты, если проводилось более одноro титрова
ния, не должны превышать значений, приведен
ных в таблице 3.2.1. 3.
ИСПЫТАНИЕ В. rИДРОЛИТИЧЕСКАЯ
УСТОЙЧИВОСТЬ ИЗМЕЛЬЧЕнноrо
В ПОРОШОК СТЕКЛА
Проверяют, чтобы полученный испытуе
мый материал был подвеР2нут обжи2У до KOM
мерчески приемлеМ020 качества.
Испытание может быть проведено на CTe
клянных стержнях, используемых для получения
трубчатых стеклянных контейнеров, либо на ro
товых контейнерах.
Оборудование:
ступка, пестик (см. рисунок 3.2.1. 2) и MO
лоток из закаленной маrнитной стали;
набор из трех сит из нержавеющей стали с
квадратными отверстиями:
(а) сито NQ 710,
(Ь) сито NQ 425,
(с) сито NQ зоо;
постоянный маrнит;
металлическая фольrа (например, алюми
ниевая или из нержавеющей ст::зли);
сушильный шкаф, поддерживающий TeM
пературу (140::1:5) ОС;
весы для взвешивания до 500 r с точно
стью до 0,005 r;
эксикатор;
ультразвуковая баня.
3.2.1. Стеклянные контейнеры для фармацевтичеСКО20 использования
575
050
о
ф
075
Рисунок З.2.1. 2. Аппарат для измельчения
стекла в порошок
(размеры указаны в миллиметрах)
Методика. Испытуемые контейнеры про
.,ывают водой Р и высушивают в сушильном
wкафу. Не менее трех контейнеров оборачи
вают чистой .бумаroй и разбивают для получе
ния двух образцов массой около 100 r каждый
11 размером кусочков не более 30 мм. 30--------40 r
кусочков первоro образца размером 1 о зо мм
переносят в ступк вводят пестик и сильно
ударяют один раз молотком. Содержимое
ступки переносят на сито (а) с наибольшим ди
аметром пор из комплекта. Повторяют опера
цию необходимое количество раз для полноro
neреноса образца на сито. Быстро просеива
IOT. Остаток, не прошедший через поры сит (а)
11 (Ь), удаляют. Затем фракционируют, повто
ряя операцию до тех пор, пока на сите (а) не
останется около 1 О r стекла. Порцию образца,
оставшуюся на сите (а), и порцию, прошедшую
через поры сита (с), удаляют. Встряхивают KOM
meKT сит в течение 5 мин. Сохраняют порцию
образца, прошедшую чР.рез сито (Ь), но OCTaB
wуюся на сите (с). Повторяют описанную про
цедуру для оставшеrося количества образца
до получения двух порций измельченноro в по
рошок стекла массой более 10 r каждая. PaBHO
мерно распределяют каждую порцию на листе
...стой rлянцевой бумаrи и удаляют какие либо
металлические частички, проводя над порци
ями стекла маrнитом. Каждую порцию перено
сят в лабораторный стакан для промывания. В
каждый стакан прибавляют по 30 мл ацетона Р
и очищают частицы стекла подходящим при
способлением, например, стеклянной палоч
кой с резиновым или пластмассовым покрыти
ем. После очистки дают частицам отстояться и
декантируют максимальное количество aцeTO
на. Прибавляют 30 мл ацетона Р и перемеши
вают вращательным движением, декантируют,
и прибавляют новую порцию ацетона Р.
Наполняют ультразвуковую баню водой при
комнатной температуре, помещают в нее лабо
раторный стакан с образцом на такую rлубину,
при которой уровень ацетона сравняется с YPOB
нем воды в бане. Обрабатывают ультразвуком
в течение 1 мин. Перемешивают вращатель
ным движением, дают частицам отстояться, дe
кантируют максимальное количество ацетона и
повторяют процедуру ультразвуковой очистки.
Повторяют процедуру очистки ацетоном и уль
тразвуковую обработку до получения прозрачно
ro раствора (до исчезновения любой мутности).
Перемешивают вращательным движением, дe
кантируют ацетон и сушат измельченное стекло,
помещая лабораторный стакан сначала на
теплую плитку для удаления остатков ацетона,
а затем в сушильный шкаф при температуре
140 ос в течение 20 мин. Переносят стеклянный
порошок из каждоro лабораторноro стакана в OT
дельную емкость для взвешивания, накрывают
крышками и помещают в эксикатор для OCTЫBa
ния. По 10,00 r высушенных частиц стекла поме
щают в две отдельные конические колбы и при
бавляют по 50 мл воды Р1 при помощи пипетки
(испытуемый раствор). В третью идентичную
колбу пипеткой прибавляют 50 мл воды Р1 (KOH
трольный раствор). При помощи леrкоro встря
хивания равномерно распределяют частички
стекла по дну колбы. Закрывают колбы чашка
ми из нейтральноro стекла или алюминиевой
фольroй, предварительно промытыми водой Р,
либо перевернутыми лабораторными CTaKaHa
ми таким образом, чтобы внутренняя поверх
ность стаканов плотно прилеrала к бокам колб.
Помещают все три колбы в автоклав таким обра
зом, чтобы вода в автоклаве была ниже ПОДQона
с колбами. Проводят процедуру автоклавирова
ния как описано в испытании А на поверхност
ную rидролитическую устойчивость, но ПОДQер
живают температуру (12Н1) сс в течение лишь
(30:t1) мин. Автоклав не открывают до тех пор,
пока температура не снизится до 95 ОС. Насколь
ко возможно быстро и с соблюдением необходи
мых мер предосторожности вынимают roрячие
образцы из автоклава и охлаждают колбы в токе
холодной проточной воды. В каждую колбу при
бавляют 0,05 мл раствора метилов020 KpaCHO
20 Р И титруют контрольный раствор 0,02 М pac
твором кислоты хлористоводородной. Затем
титруют испытуемые растворы 0,02 М pacтвo
.576
rосударственная фармакопея Республики Беларусь
ром кислоты хлористоводородной до появле
ния окрашивания, аналоrичноro полученному
в контрольном опыте. Находят разность между
объемами титра нта , израсходованными на ти
трования испытуемых растворов и контрольно
ro раствора.
ПРИМЕЧАНИЕ: при необходимости для по
tlученuя отчетливой точки конца титрования
прозрачные растворы переносят в отдельные
колбы вместимостью 250 мл. При перемеши
fJaHUU вращательным движением ополаскива
ют частицы секла тремя порциями воды Р1 по
,15 мл каждая, прибавляя промывную жидкость
к основному раствору. Прибавляют 0,05 мл
раствора метилов020 краСН020 р, титруют
и рассчитывают количества титранта, как
описано выше. В этом случае также использу
ют контрольный раствор, к которому прибав
ляют 45 мл воды Р1 и 0,05 мл раствора Meти
лов020 краСН020 Р.
Рассчитывают среднее значение результа
тов титрований в миллилитрах 0,02 М раствора
кислоты хлористоводородной на rpaMM образ
ца и. при необходимости, эквивалент экстраrи
рующейся щелочи в пересчете на количество
натрия оксида в микроrраммах на rpaMM CTe
клянноro порошка.
1 мл 0,02 М раствора кислоты хлористово
дородной соответствует 620 MKr натрия оксида.
Испытание повторяют, если наибольшие и
наименьшие полученные значения отличаются
более чем на 20 %.
Пределы. Для контейнеров из стекла
класса 1 на титрование должно быть израсходо
вано не более 0,1 мл 0,02 М раствора кислоты
хлористоводородной на rpaMM стекла, дЛЯ KOH
тейнеров из стекла класса 11 или класса 111 не
более 0,85 мл 0,02 М раствора кислоты хлори
стоводородной на rpaMM стекла.
ИСПЫТАНИЕ С. ПРОВЕРКА НАЛИЧИЯ
СПЕЦИАЛЬНОЙ ОБРАБОТКИ
ПОВЕРХНОСТИ (ПРОБА ТРАВЛЕНИЕМ)
Если необходимо определить. была ли по
верхность контейнера специально обработана
и/или отличить контейнеры из стекла класса 1 и
класса 11, в дополнение к испытанию А проводят
испытание С. ДЛЯ этих же целей Moryr быть ис
пользованы методы А и В. Испытание С прово
дят на неиспользованнь,х образцах или на об
разцах. использованных для испытания А.
Виалы и флаконы. Необходимые объемы
испытуемой жидкости указаны в таблице 3.2.1. 2.
Контейнеры промывают дважды водой Р,
наполняют до краев смесью из 1 объема Kиc
лоты фтористоводородной Р и 9 объемов
кислоты хлористоводородной Р и оставляют
на 1 О мин. Затем контейнеры освобождают от
содержимоro и тщательно промывают 5 раз
водой Р. Непосредственно перед испытанием
контейнеры еще раз промывают водой Р. Под -
roтовленные таким образом контейнеры BЫ
держивают в автоклаве и титруют аналоrично
тому. как описано для испытания А на поверх
ностную rидролитическую устойчивость.
Если результаты испытания значительно
выше, чем результаты, полученные с необрабо
танной поверхностью (примерно в 5 10 раз).
делают заключение, что поверхность стекла
была подверrнута специальной обработке.
Ампулы
ПРИМЕЧАНИЕ: ампулы, иЗ20товленные
из стеклянных трубок, обычно не подвеР2ают
специальной обработке, так как их высокая xи
мическая устойчивость зависит от химиче
СК020 состава материала контейнера.
Проводят испытание, как описано выше для
виал и флаконов. Если ампулы не подверrнуты
специальной обработке. результаты испытания
будут незначительно ниже, чем результаты, по
лученные в предыдущих испытаниях.
Различие между контейнерами из стекла
класса 1 и класса 11
Результаты, полученные при проведении ис
пытания С, сравнивают с результатами. полу
ченными при испытании А. Интерпретация pe
зультатов приведена в таблице 3.2.1. 4.
Таблица 3.2.1А
Различие между контейнерами из стекла
класса 1 и класса 11
Класс 1 Класс 11
Значения близки к зна Значения значительно
чениям, полученным превышают значения,
при проведении ис полученные при про
пытания на поверх ведении испытания на
ностную rидролити поверхностную rидро
ческую устойчивость литическую устойчи
контейнеров из стекла вость и близки к значе
класса 1 ниям, полученным для
контейнеров из стекла
класса 111, но не превы
шают их
МЫШЬЯК
Испытание проводится для стеклянных KOH
тейнеров для водных лекарственных средств
для парентеральноro применения.
Атомно абсорбционная спектрометрия
(2.2.23, метод 1).
Испытуемый раствор. Используют paCT
вор, полученный для контейнеров из стекла
класса I и класса 11 после автоклавирования
при температуре 121 ос в течение 1 ч, как
описано в испытании А на поверхностную rи
дролитическую устойчивость. 10,0 мл полу
ченноro раствора помещают в мерную колбу
вместимостью 100 мл, прибавляют 10 мл Kиc
лоты хлористоводородной Р, 5 мл раствора
200 r/л калия йодида Р и наrревают на водяной
3.2.1. Стеклянные контейнеры для фармацевтическоао использования
577
бане при температуре 80 ос в течение 20 мин.
Полученный раствор выдерживают до OCTЫBa
ния и доводят водой Р до объема 100,0 мл.
Растворы сравнения. rотовят растворы
сравнения с использованием этаЛОНН020 pacт
вора мышьяка (1 ррт As) Р. Прибавляют 1 О мл
кислоты хлористоводородной Р, 5 мл раствора
200 r/л калия йодида Р и наrревают на водяной
бане при температуре 80 ос в течение 20 мин.
Полученный раствор выдерживают до OCTЫBa
ния и доводят водой Р до объема 100,0 мл. Pac
творы сравнения обычно roтовят с концентраци
ей от 0,005 ррm до 0,015 ррm.
Кислотный реактив. Кислота хлористово
дородная Р.
Восстанавливающий реактив. Натрия тe
тра2идробората восстанавливающий pacт
ворР.
Испытуемый раствор вносят в кювету aTOM
но абсорбционноro спектрометра с использова
нием устройства для получения rидридов. Под
бирают и стандартизируют условия проведения
испытания на используемом оборудовании в
соответствии с ero инструкцией по эксплуата
ЦИИ, настраивают скорость перистальтическоro
насоса, затем присоединяют трубки к резерву
арам с кислотой, восстановителем и испытуе
мым раствором.
Источник излучения: лампа с полым KaTO
дом.
Длина волны: 193,7 нм.
Атомизатор: воздушно ацетиленовое
пламя.
Предельное содержание: 0,1 ррm As.
СПЕКТРАЛЬНОЕ ПРОПУСКАНИЕ ДЛЯ
ЦВЕТНЫХ СТЕКЛЯННЫХ КОНТЕЙНЕРОВ
Оборудование. Спектрофотометр для ви
димой И ультрафиолетовой области, снабжен
ный фотодиодным детектором или оборудован
ный фотоумножительной трубкой, соединенной
с фотометрическим шаром.
Приrотовление образца. Стеклянный KOH
тейнер разбивают или разрезают циркулярной
пилой с диском для влажноro абразивноro шли
фования, например, карборундовым или Me
таллизированным алмазным диском. Выбира
ют осколки с соответствующей толщиной стенок
и обрезают их таким образом, чтобы они моrли
быть вставлены в спектрофотометр. Если об
разец настолько мал, что не может закрыть OT
верстие для образца в спектрофотометре, то
незакрытую часть закрывают непрозрачной бу
маrой или лентой, обеспечивая ширину образца
больше ширины щели. Перед тем, как поместить
образец в спектрофотометр, ero промывают, BЫ
сушивают и протирают тканью для протирания
линз. Закрепляют образец с помощью воска или
друrими подходящими способами, аккуратно,
чтобы не оставить на образце следов пальцев
или друrих следов.
13 За. 1712.
Методика. Образец помещают в спектро
фотометр таким образом, чтобы еro цилиндри
ческая ось была параллельна щели, пучок света
шел перпендикулярно поверхности стекла и
потери в результате отражения были минималь
ны. Измеряют светопропускание образца в cpaB
нении с воздухом в области длин волн от 290 нм
до 450 нм непрерывно или с интервалами 20 нм.
Пределы. Спектральное пропускание для
контейнеров из цветноro стекла для лекарствен
ных средств, не предназначенных для парен
теральноro применения, не должно превышать
1 О % при любой длине волны в интервале от
290 нм до 450 нм независимо от класса и объема
стеклянноro контейнера. Спектральное пропу
скание для контейнеров из цветноro стекла для
лекарственных средств, предназначенных для
парентеральноro применения, не должно пре
вышать пределы, указанные в таблице 3.2.1. 5.
Таблица 3.2.1. 5
Пределы спектраЛЬН020 пропускания для KOH
тейнеров из цветН020 стекла классов 1, 11 и 111
Максимальное спектраль-
Horo пропускание ( %) при
любой длине волны в диа-
Номинальный пазоне 290----450 нм
объем (мл) Контейнеры,
rерметизи- Контейнеры
руемые с пробками
запаиванием
До 1 50 25
Свыше 1 до 2 45 20
Свыше 2 до 5 40 15
Свыше 5 до 10 З5 13
Свыше 1 О до 30 12
20
Свыше 20 15 10
Приложение испытание на поверх
ностную rидролитическую устойчивость
определение методом пламенной
атомно абсорбционной спектрометрии
Поверхностная 2идролитическая устойчu
вость стекла класса 1 и 11 может быть опре
делена методом пламенной атомно абсорб
ционной спектрометрии с использованием
раствора, получеНН020 после выщелачивания
стекла. Некоторые химические элементы,
присутствующие в стекле в виде оксидов и
способствующие выщелачиванию раствора,
М02ут быть определены данным методом и
использованы для выражения щеЛОЧН020 экви
валента. Спектрометрический метод имеет
преимущество, так как позволяет использо
вать 20раздо меньшие объемы образца экс
тракта и может применяться для анализа He
больших индивидуальных контейнеров, что
в свою очередь позволяет оценить oдHopoд
ность контейнеров в серии, 2де такой показа
тель может быть критичным. Результаты,
578
rосударственная фармакопея Республики Беларусь
полученные данным методом, не эквивалент
ны результатам, полученным титpиMeтpи
ческим методом, поэтому они не М02ут быть
взаимозаменяемыми. Корреляция между двумя
методами будет зависеть от класса стекла,
размера и формы контейнера. Титриметриче
ский метод является стандартным фармако
пейным методом; спектрометрический метод
может быть использован в случае, если это
утверждено уполномоченным Ор2аном.
Ниже приведено описание даНН020 метода.
Определение проводится на контейнерах,
не используемых ранее. Количество контейне
ров для испытания подбирают в соответствии с
таблицей 3.2.1. 6.
Таблица 3.2.1. 6
Количество контейнеров, необходимых для
проведения испытания спектрометрическим
методом
Дополни
Количество тельное КО-
Объем контейне личество
наполнения ров для контейнеров
(мл) отдельноrо для предва-
измерения рительных
измерений
Д02 20 2
Свыше 2 до 5 15 2
Свыше 5 до 30 10 2
Свыше 30 до 5 1
100
Свыше 100 3 1
Инструкции по определению объема Ha
полнения, очистке контейнеров, наполнению
и наrреванию даны выше в разделах rидроли
тическая устойчивость и Испытание А. rидроли
тическая устойчивость наружной поверхности
стеклянных контейнеров.
РАСТВОРЬ!
Спектрохимический буферный раствор.
80 r цезия хлорида Р растворяют в около 300 мл
воды Р1, прибавляют 1 О мл 6 М раствора кис
лоты хлористоводородной Р И переносят полу
ченный раствор в мерную колбу вместимостью
1000 мл. Доводят полученный раствор водой Р1
до объема 1000,0 мл И перемешивают.
Основные растворы:
натрия оксид, c(Na 2 0) = 1 мr/мл;
калия оксид, с(К 2 О) = 1 мr/мл;
кальция оксид, с(СаО) = 1 мr/мл.
Допускается использование roтовых, KOM
мерчески доступных, растворов.
Стандартные растворы. rотовят CTaH
дартные растворы путем разведения основных
растворов водой Р1 до получения концентраций,
подходящих для получения растворов cpaBHe
ния по 20 мкr/мл натрия оксида, калия оксида
и кальция оксида. Допускается использование
roтовых, коммерчески доступных, растворов.
Растворы сравнения. rотовят растворы
сравнения для построения rрадуировочноro rpv
фика (набор rрадуировочных растворов) путем
разведения стандартных растворов водой Р1
таким образом, чтобы уложиться в рабочую об.-
ласть определяемых концентраций в том числе
с учетом характеристик используемоro оборудо-
вания. Характерная область концентраций pac
творов сравнения:
для определения натрия оксида и калия
оксида методом атомно эмиссионной спектро-
метрии: до 1 О мкr/мл;
для определения натрия оксида и калия
оксида методом атомно абсорбционной спек
трометрии: до 3 мкr/мл;
для определения кальция оксида MeTO
ДQМ атомно абсорбционной спектрометрии: до
7 мкr/мл.
Растворы сравнения должны содержать 5 %
(об/об) спектрохимическоro буферноro раст
вора.
МЕТОДИКА
Проводят предварительные определения
концентраций калия оксида и кальция оксида на
одном из растворов после экстракции (экстрак
ционный раствор). Если для конт-ейнеров одноro
класса стекла концентрация калия оксида co
ставляет менее 0,2 мкr/мл, а концентрация каль
ция оксида менее 0,1 м кr/мл, то оставшиеся
растворы после экстракции не должны прове
ряться на содержание данных ионов. Экстракци
онные растворы каждоro образца распыляют He
посредственно в пламя атомно абсорбционноrо
или атомно эмиссионноrо спектрометра и опре
деляют приблизительную концентрацию натрия
оксида и. если присутствуют, калия оксида и
кальция оксида путем сравнения с rрадуировоч
ным rрафиком, полученным с растворами cpaB
нения.
ОКОНЧАТЕЛЬНОЕ ОПРЕДЕЛЕНИЕ
При отсутствии необходимости дальнейше
ro разведения к каждому контейнеру прибавля
ют спектрохимический буферный раствор в KO
личестве 5 % от объема наполнения, тщательно
перемешивают и проводят определение натрия
оксида, кальция оксида и калия оксида, исполь
зуя rрадуировочный rрафик. Для определения
концентрации кальции оксида методом пламен
ной атомной спектрометрии необходимо исполь
зовать пламя оксид азота ацетилен.
При необходимости дальнейшеro разведе
ния проводят определение натрия оксида, каль
ция оксида и калия оксида, как указано выше. Из
меряемые растворы должны содержать 5 % (06/06)
спектрохимическоro буферноro раствора. Значе
ния концентраций менее 1,0 мкr/мл выражают с
точностью до сотых, значения, равные или боль
шие 1,0 мкr/мл, до десятых. Корректируют по
лученный результат с учетом прибавленноro бу
ферноrо раствора и проведенных разведений.
3.2.2. Пластмассовые контейнеры и укупорочные средства
РАСЧЕТ
Рассчитывают среднее значение KOHцeH
трации каждоro оксида, полученное для каждо
ro испытуемоro образца, выраженное в микро
rpaMMax оксида в миллилитре экстракционноro
раствора. Затем рассчитывают сумму всех окси
дов, выраженную в микроrраммах натрия оксида
в миллилитре экстракционноro раствора, ис
пользуя следующие коэффициенты пересчета
масс:
1 MKr калия оксида соответствует 0,658 MKr
натрия оксида;
1 MKr кальция оксида соответствует
1,105 MKr натрия оксида.
Пределы. Для каждоro испытанноro контей
нера результат не должен превышать значений,
приведенных в таблице 3.2.1. 7.
Таблица 3.2.1. 7
Предельные значения для испытания на
поверхностную 2идролитическую устойчи
вость, определенную методом пламенной
атомно абсорбционной спектрометрии
Максимальные значения
концентраций оксидов
Объем в пересчете на натрия
наполнения (мл) оксид (мкr/мл)
Стеклянные контейнеры
из стекла класса 1 и 11
До 1 5,00
Свыше 1 до 2 4,50
Свыше 2 до 5 3,20
Свыше 5 до 10 2,50
Свыше 1 О до 20 2,00
Свыше 20 до 50 1,50
Свыше 50 до 100 1,20
Свыше 100 до 200 1,00
Свыше 200 до 500 0,75
Свыше 500 0,50
01/2013:30202
3.2.2.
ПЛАСТМАССОВЫЕ
КОНТЕЙНЕРЫ
И УКУПОРОЧНЫЕ СРЕДСТВА
ДЛЯ ФАРМАЦЕВТИЧЕскоrо
ИСПОЛЬЗОВАНИЯ
Пластмассовый контейнер для фарма
цевтическоro использования изделие из по--
",мерноro материала, которое содержит или
IIQжет содержать фармацевтическую продукцию
11 находится или может находиться в непосред
ственном контакте с ней. Укупорочное средство
-ляется частью контейнера.
579
Пластмассовые контейнеры и укупороч
ные средства для фармацевтическоro ис
пользования изroтавливают из материалов,
которые MorYT содержать определенные дo
бавки. Такие материалы не должны coдep
жать никаких веществ, которые моrли бы
экстраrироваться содержимым контейнера
в количестве, влияющем на эффективность
или стабильность лекарственноro средства,
или быть потенциально опасными в отноше
нии токсичности.
Чаще Bcero используемыми полимерами
являются полиэтилен (содержащий или не co
держащий добавки), полипропилен, поливи
нилхлорид, полиэтилентерефталат и поли
этиленвинилацета
Природа и количество добавок опреде
ляются типом полимера, технолоrией пере
работки полимера в контейнер и предпола
rаемой целью применения. Добавки MorYT
включать антиоксиданты, стабилизаторы,
пластификаторы, смазки, красители и моди
фикаторы ударостойкости. Антистатики и Be
щества, облеrчающие вынимание из формы,
MorYT быть использованы только в составе
полимеров для изrотовления контейнеров,
предназначенных для лекарственных средств
для пероральноro или наружноro примене
ния, для которых разрешено их использова
ние. Допустимые добавки обозначаются в ти
повой спецификации на каждый материал,
описанный в Фармакопее. Друrие добавки
можно использовать с условием, что в каждом
конкретном случае они разрешены к примене
нию компетентным уполномоченным opraHoM.
Чтобы оценить потенциальный риск при
выборе соответствующеro пластмассово
ro контейнера, следует знать полный состав
пластичноro материала при ero производстве,
включая все материалы, используемые в про
цессе формования контейнера. Пластмассо
вый контейнер, выбранный для какоro либо
KOHKpeTHoro лекарственноro средства, должен
соответствовать следующим требованиям:
компоненты лекарственноro средства,
которые находятся в контакте с пластичным
материалом, не должны значительно aдcop
бироваться ero поверхностью и миrрировать
внутрь пластика или через неro;
пластичный материал не должен Bыдe
лять в содержимое контейнера никаких Be
ществ в таком количестве, которое влияет
на эффективность или стабильность лекар
ственноro средства или может быть потенци
алы-ю опасным в отношении токсичности.
ИсrlOЛЬЗУЯ материал или материалы, BЫ
бранные в соответствии с этими критериями,
изroтавливают некоторое количество иден
тичных образцов контейнеров, используя
хорошо отработанную методику, и подверrают
их практическим испытаниям в условиях, co
ответствующих условиям их предполаrаемоrо
580
rосударственная фармакопея Республики Беларусь,
применения, включая, если необходимо, CTe
рилизацию. Для тоro чтобы подтвердить co
вместимость контейнера и ero содержимоro
и убедиться в отсутствии изменений, Hera
тивно влияющих на качество лекарственно
ro средства, проводят различные испытания,
такие как контроль отсутствия изменений фи
зических характеристик, оценка каких либо
потерь или прироста содержимоro контейнера
в связи с проникновением, определение изме
нений рН, оценка изменений, вызванных вли
янием света, химические испытания и, если
необходимо, биолоrические испытания.
Метод производства должен обеспечи
вать возможность ero воспроизводства для
дальнейшеro производства в больших объе
мах, а условия производства подбираются
таким образом, чтобы избежать возможности >
заrрязнения друrими пластичными матери
алами или их инrредиентами. Производитель
продукта обязан rарантировать, что контейне
ры, которые изroтавливаются в условиях про
изводства, по всем показателям идентичны
типовым образцам.
Для TOro, чтобы результаты испытаний
типовых образцов считались достоверными,
важно, чтобы:
не было изменений в составе матери
ала, описанноro для типовых образцов;
не было изменений в производственном
процессе, описанном для типовых образцов,
особенно относительно температуры в про
цессе переработки материала, или в ходе по
следующих процедур, таких как стерилиза
ция;
не использовался материал из отходов
или брака.
Повторная переработка излишка MaTe
риала, природа и состав котороro хорошо из
вестны, может быть разрешена после COOT
ветствующей валидации.
После проведения испытания на COBMe
стимость с положительными результатами
для каждой комбинации контейнера и coдep
жимоrо, материал, описанный в Фармакопее,
признают соответствующим специальным
целям, описанным выше.
01/2013:90003
3.2.2.1. ПЛАСТМАССОВЫЕ
КОНТЕЙНЕРЫ ДЛЯ ВОДНЬJХ
РАСТВОРОВ ДЛЯ ИНФУЗИИ
ОПРЕДЕЛЕНИЕ
Пластмассовые контейнеры для водных
растворов для инфузий изroтавливают из одноro
или нескольких полимеров, которые, если H
ходимо, содержат добавки. Контейнеры, описан-
ные в данном разделе, не обязательно приrодны
для эмульсий. Наиболее часто используемы...
полимерами являются полиэтилен, полипропи-
лен и поливинилхлорид. Спецификации, приве-
денные в данном разделе, следует использовать
вместе с разделом 3.2.2. Пластмассовые K
тейнеры и укупорочные средства для фарм
цевтичеСК020 использования.
Контейнеры MorYT представлять собой
пакеты или флаконы. Они имеют приспос<r
бление для присоединения комплекта для
вливания, сконструированное таким образом,
чтобы обеспечить надежное соединение. Они
MorYT иметь приспособление, позволяющее
проводить инъекцию во время использования. '
Контейнеры обычно снабжены элементом,
обеспечивающим возможность подвешива
ния, стойким к растяrиванию, которое возни
кает во время использования. Контейнеры
должны выдерживать условия стерилизации.
Конструкция контейнера и выбранный метод
стерилизации должны быть такими, чтобы
обеспечить возможность стерилизации всех
элементов контейнера, находящихся в KOH
такте с раствором для вливания. После заку
поривания контейнеры должны быть непро
ницаемыми для микроорrанизмов, а после
наполнения стойкими к повреждениям, об
условленным непредвиденным заморажива
нием, которое может произойти при транспор
тировании roтовоro лекарственноro средства.
Контейнеры должны быть прозрачными, чтобы
обеспечить возможность визуальноro осмотра
содержимоro в любой момент, если нет друrих
указаний.
Пустые контейнеры не должны иметь дe
фектов, которые моrли бы привести к проте
канию, наполненные и закрытые контейнеры
не должны проявлять признаков протечки.
Для соответствующею сохранения He
которых лекарственных средств контейнер
следует упаковывать в защитную упаковку. В
этом случае первичную оценку сохранности
проводят для контейнера, находящеroся в за
щитной упаковке.
ИСПЫТАНИЯ
Раствор S. Раствор S используют в тe
чение 4 ч после при20товления. Контейнер
наполняют водой Р до номинальноro объема
и укупоривают ero, по возможности исполь
зуя обычные укупорочные средства; можно
закрыть контейнер фольroй алюминиевой.
Нэrревают контейнер в автоклаве таким об
разом, чтобы за 20 ЗО мин температура дo
стиrла (12Н2) ОС, и выдерживают при этой
температуре в течение 30 мин. Если HarpeBa
ние при температуре 121 ос ведет к разруше
нию контейнера, наrревание ведут при темпе
ратуре 100 ос в течение 2 ч.
581
3.2.3. Стерильные пластмассовые контейнеры для человеческой крови
Контрольный раствор. Параллельно с
раствором S roтовят контрольный раствор,
используя воду Р и колбу из боросиликатноro
стекла, закрытую фольroй алюминиевой.
Внешний вид раствора S. Раствор S
должен быть прозрачным (2.2.1) и бесцвет
ным (2.2.2, метод 11).
Кислотность или щелочность. К объему
раствора S, соответствующему 4 % номи
нальноro объема контейнера, прибавляют
0,1 мл раствора фенолфталеина Р. Раствор
должен быть бесцветным. При прибавлении
0,4 мл 0,01 М раствора натрия 2идроксида
должно появиться розовое окрашивание. При
прибавлении 0,8 мл 0,01 М раствора кисло
ты хлористоводородной и 0,1 мл раствора
lIетилов020 краСН020 Р должно появиться
оранжево красное или красное окрашивание.
Оптическая плотность (2.2.25). Измеря
IOT оптическую плотность раствора S в диапа
зоне длин волн от 230 нм до 360 нм, исполь
зуя контрольный раствор в качестве раствора
сравнения (см. раствор S). Оптическая плот
ность не должна превышать 0,20.
Восстанавливающие вещества. К 20,0 мл
раствора S прибавляют 1 мл кислоты серной
разведенной Р и 20,0 мл 0,002 М раствора
калия пермаН2аната. Кипятят с обратным холо
дильником в течение З мин и сразу охлаждают.
К полученному раствору прибавляют 1 r калия
iюдида Р и немедленно титруют 0,01 М pacтвo
ром натрия тиосульфата, используя в каче
с:тве индикатора 0,25 мл раствора крахмала Р.
Параллельно проводят контрольный опыт, ис
rюльзуя 20,0 мл контрольноro раствора. Раз
tюсть между объемами титранта не должна пре
8ышать 1,5 мл.
Прозрачность. Контейнер, предвари
тельно использованный для приrотовления
раствора S, наполняют исходной опалесциру
IOщей суспензией (2.2.1), разведенной в COOT
ношении 1 :200 для контейнеров, изrотовлен
ных из полиэтилена или полипропилена, и
1 :400 для друrих контейнеров, в количестве,
равном номинальному объему контейнера.
При просмотре через контейнер и в cpaBHe
нии с контейнером, наполненным водой Р,
должно быть заметно помутнение.
МАРКИРОВКА
Маркировка партии пустых контейнеров
содержит:
наименование и адрес изrотовителя;
номер серии, который позволяет просле
дить историю контейнера и полимерноro MaTe
риала, из котороro он изroтовлен.
74 :за. 1712.
01/2013:30203
3.2.3. СТЕРИЛЬНЫЕ
ПЛАСТМАССОВЫЕ
КОНТЕЙНЕРЫ ДЛЯ
ЧЕЛОВЕЧЕСКОЙ КРОВИ И ЕЕ
КОМПОНЕНТОВ
Пластмассовые контейнеры для сбора,
хранения, обработки и переливания крови и
ее компонентов производят из одноro или He
скольких полимеров, если необходимо, с ис
пользованием добавок. Состав и условия про
изводства контейнеров реrламентируются
соответствующими компетентными орrанами
в соответствии с действующим национальным
законодательством и международными требо
ваниями.
Если состав материалов разных частей
контейнера соответствует конкретным спец
ификациям, их качество контролируется Me
тодами, описанными в данных спецификаци
ях (см. раздел 3.1. Материалы, используемые
для производства контейнеров и ero подраз
делы).
Материалы, отличные от тех, что описаны
в Фармакопее, MOryT быть использованы для
производства контейнеров в том случае, если
их состав утвержден компетентным уполномо
ченным opraHoM, а также, если полученные KOH
тейнеры отвечают требованиям для стерильных
пластмассовых контейнеров для человеческой
крови и ее компонентов.
В нормальных условиях при использовании
материалы не должны выделять мономеры или
друrие вещества в количествах, вредных для че
ловека или вызывающих аномальные измене
ния крови.
Контейнеры MOryT содержать растворы
антикоаryлянтов в зависимости от их предпола
raeMoro использования и должны быть стериль
ными.
Каждый контейнер снабжается приспосо
блениями в зависимости от ero предполаrаемо
ro использования. Контейнер может быть в виде
одноro элемента. Контейнер для отбора крови
может быть присоединен одной или несколь
кими трубками к друrому (одному или несколь
ким контейнерам) для разделения компонентов
крови в закрытой системе.
Форма и размер выходноro отверстия
должны обеспечивать соединение контейнера с
устройством для подачи крови. Защитное покры
тие иrлы для отбора крови и дополнительных
элементов должно обеспечивать сохранение
стерильности. Они должны леrко отсоединяться
и иметь контроль первоro вскрытия.
Емкость контейнеров связана с номиналь
ным объемом, который установлен COOTBeTCTBY
ющим национальным opraHoM в соответствии с
объемом раствора антикоаryлянта. Номиналь
582
rосударственная фармакопея Республики Беларусь
ный объем это объем крови, который следует
собрать в контейнер. Контейнеры должны иметь
такую форму, чтобы после их наполнения можно
было обеспечить возможность центрифуrирова
ния.
Контейнеры должны быть обеспечены co
ответствующим приспособлением для подвеши
вания и фиксации, которое не должно являться
помехой для отбора, хранения, обработки и пе
реливания крови.
Контейнеры должны быть упакованы в rep
метичную (запаянную) защитную упаковку.
ОПИСАНИЕ
Контейнер должен быть достаточно про
зрачным для визуальноro контроля содержимоro
до и после отбора крови и достаточно эластич
ным для обеспечения минимальноro сопротив
ления в процессе наполнения и опустошения от
содержимоro в нормальных условиях. Напол
ненный контейнер может содержать не более
5 мл воздуха.
ИСПЫТАНИЯ
Раствор 51' Наполняют контейнер 100 мл
стерильноro, свободноro от пироrенных веществ
раствора 9 r/л натрия хлорида Р. Закрывают и
наrревают в автоклаве таким образом, чтобы
температура содержимоro поддерживалась на
уровне 110 ос в течение 30 мин.
Если испытуемый контейнер содержит paCT
вор антикоаryлянта, содержимое удаляют, про
мывают контейнер 250 мл воды для инъекций Р
при температуре (20:1:1) ос и сливают промыв
ную жидкость.
Раствор 52' Количество воды для иHЪ
екций Р, соответствующее предполаrаемому
объему раствора антикоаryлянта, помещают
в контейнер. Закрывают контейнер и HarpeBa
ют в автоклаве таким образом, чтобы темпера
тура содержимоro поддерживалась на уровне
110 ос в течение 30 мин. После охлаждения при
бавляют воду для инъекций Р до номинальноro
объема контейнера.
Если испытуемый контейнер содержит paCT
вор антикоаryлянта, содержимое удаляют и об
рабатывают еro, как описано выше.
Устойчивость к центрифуrированию.
Помещают в контейнер количество воды Р,
подкисленной 1 мл кислоты хлористоводо
родной разведенной Р, соответствующее еro
номинальному объему. Заворачивают контей
нер в абсорбирующую бумаrу, пропитанную
разведенным в соотношении 1:5 раствором
бромфенолов020 сине20 Р1 или друrим под
ходящим индикатором и высушенную. ЦeH
трифуrируют при 5000 9 в течение 10 мин. Не
должно наблюдаться протекания раствора на
индикаторную бумаrу, а также каких либо из
менений формы контейнера.
Устойчивость к растяжению. Помещают в
контейнер количество воды Р, подкисленной 1 мл
кислоты хлористоводородной разведенной Р,
соответствующее еro номинальному объему.
Подвешивают контейнер с помощью приспосо-
бления для подвешивания за конец, противопо-
ложный трубке для отбора крови. и вдоль оси
трубки прилаrают усилие в 20 Н (2,05 Krc). Под
держивают усилие растяжения в течение 5 с. По-
вторя ют испытание. прилаrая усилие к каждому
элементу контейнера для наполнения и опусто
шения контейнера. Не должно происходить раз
рывов и друrих повреждений контейнера.
rерметичность. Контейнер, который
прошел испытание на устойчивость к растяже
нию, помещают между двумя пластинами, по
крытыми абсорбирующей бумаrой, пропитан
ной разведенным в соотношении 1:5 раствором
бромфенолов020 сине20 Р1 или друrим под
ходящим индикатором, и высушенной. Посте
пенно прикладывают усилие к пластинам для
сжатия контейнера таким образом, чтобы еro
внутреннее давление (т. е. разность между при
ложенным и атмосферным давлением) достиrло
67 кПа в течение 1 мин. Под,цер ивают давле
ние на этом уровне в течение 1 О мин. На инди
каторной бумаrе, а также в любой из точек при
соединения элементов не должно наблюдаться
следов протекания.
Проницаемость для паров. Если контей
нер содержит раствор антикоаryлянта, напол
няют ero раствором 9 r/л натрия хлорида Р в
объеме, равном объему крови, для котороro он
предназначен.
Если контейнер пустой, то еro наполняют co
ответствующим количеством смеси раствора aH
тикоаryлянта и раствора натрия хлорида. Закры
вают контейнер, взвешивают еro и оставляют
при температуре (5:1:1) ос в атмосфере с относи
тельной влажностью (50:1:5) % в течение 21 дня.
По окончании этоro периода потеря в массе не
должна превышать 1 %.
Удаление содержимоrо под давлением.
Наполняют контейнер водой Р при температуре
(5:1:1) ос в количестве, равном ero номинально
му объему. Присоединяют приспособление для
переливания крови без внутривенной канюли к
одному из элементов. Сжимают контейнер таким
образом, чтобы внутреннее давление (т. е. раз
ность между приложенным и атмосферным дaB
лением) при удалении еro содержимоro достиr
ло 40 кПа. Контейнер должен освобождаться ст
содержимоro в течение не более 2 мин.
Скорость наполнения. С помощью трубки
для отбора крови (снабженной иrлой) присо
единяют контейнер к резервуару, содержащему
раствор, вязкость котороro соответствует вязко
сти крови, например, раствор 335 r/л сахарозы Р
3.2.3. Стерильные пластмассовые контейнеры для человеческой крови
583
с температурой 37 ОС. Поддерживают BHyтpeH
нее давление в резервуаре (т. е. разность между
приложенным и атмосферным давлением) на
уровне 9,З кПа, при этом основа резервуара и
верхняя часть контейнера должны находиться
на одном уровне. Объем жидкости, который Ha
бирается в контейнер в течение 8 мин, не должен
быть меньше номинальноro объема контейнера.
Устойчивость к колебанию температуры.
Контейнер помещают в камеру с температурой
20 2З ОС. Быстро охлаждают с rлубоким замо
раживанием до температуры 80 ос и выдержи
вают при этой температуре в течение 24 ч. Под
нимают температуру до 50 ос и выдерживают в
течение 12 ч. Охлаждают до комнатной темпе
ратуры. Контейнер должен выдерживать испы
тания на устойчивость к центрифуrированию,
устойчивость к растяжению, rерметичность, про
ницаемость для паров, удаление содержимоro
под давлением и скорость наполнения, описан
ные выше.
Прозрачность. Контейнер наполняют до
номинальноro объема исходной опалесциру
ющей суспензией (2.2.1), разведенной таким об
разом, чтобы оптическая плотность (2.2.25) при
длине волны 640 нм составляла от 0,37 до 0,43
(фактор разведения около 1:16). При просмотре
через контейнер и в сравнении с контейнером,
наполненным водой Р, должно быть заметно по
мутнение.
Экстраrируемые вещества. Испытания
проводят такими методами, чтобы контакт между
контейнером и ero содержимым как можно ближе
соответствовал их контакту в реальных услови
ях использования контейнера.
Условия контакта, а также испытания, KOTO
рые следует выполнять для элюатов, описаны, в
зависимости от природы материала, в требова
ниях к конкретному классу контейнера.
rемолитические эффекты в буферных
системах
Основной буферный раствор. 90,0 r натрия
хлорида Р, 34,6 r натрия zидрофосфата Р и
2,43 r натрия ди2идрофосфата Р растворяют
в воде Р и доводят до объема 1000 мл этим же
растворителем.
Буферный раствор Ао, К 30,0 мл основноro
буферноro раствора прибавляют 10,0 мл воды Р.
Буферный раствор ВО' К 30,0 мл основною
буферноro раствора прибавляют 20,0 мл воды Р.
Буферный раствор СО' К 15,0 мл основною
буферноro раствора прибавляют 85,0 мл юды Р.
По 1,4 мл раствора S2 помещают в каждую
из трех пробирок центрифуrи. В пробирку I при
бавляют 0,1 мл буферноro раствора Ао, в пробир
ку 11 0,1 мл буферноro раствора Во' в пробир
ку 111 0,1 мл буферноro раствора СО' В каждую
из пробирок прибавляют по 0,02 мл свежей rепа
ринизированной человеческой крови, тщательно
перемешивают и наrревают на водяной бане при
температуре (30:t1) ос в течение 40 мин. Исполь
зуют кровь, отобранную менее чем за 3 ч, или
кровь, отобранную в раствор антикоаryлянта
раствор цитрат фосфат декстрозы (ЦФД, СРО) ,
менее чем за 24 ч до проведения испытания.
rотовят три раствора, содержащие COOTBeT
ственно:
3,0 мл буферною раствора Ао и 12,0 мл
воды Р (раствор А 1 );
4,0 мл буферноro раствора Во и 11,0 мл
воды Р (раствор В 1 );
4,75 мл буферноro раствора Во и 10,25 мл
воды Р (раствор CJ
В пробирки центрифуrи 1,11, 111 прибавляютсоот
ветственно 1,5 мл раствора А1' 1,5 мл раствора В 1 и
1,5 мл раствора С 1 . Параллельно ютовят три друrие
пробирки центрифуrи, заменяя раствор S2 водой Р.
Центрифуrируют одновременно пробирки с испы
туемым и контрольным растворами в roризонталь--
ной центрифyre при 2500 9 в течение 5 мин. После
центрифуrирования измеряют оптическую плот
ность (2.2.25) растворов при длине волны 540 нм,
используя В качестве раствора сравнения основной
буферный раствор. rемолитическое число (в про-
центах) рассчитывают по формуле:
А
ехр .100,
Аоо
rде:
Аюо оптическая плотность раствора из
пробирки 111;
А оптическая плотность раствора из
ехр
пробирки 1 или 11 или соответствующих контроль
ных растворов.
rемолитическое число раствора из пробир
ки 1 должно быть не более 1 О %, а rемолитиче
ское число раствора из пробирки 11 не должно
отличаться более чем на 1 О % от COOTBeTCTBY
ющеro контрольноro раствора.
Стерильность (2.6.1). Контейнеры должны
выдерживать испытание на стерильность.
В асептических условиях помещают в контейнер
100 мл стерильною раствора 9 r/л натрия хло
рида Р и встряхивают контейнер до тех пор, пока
еro внутренняя стенка не будет полностью CMO
чена. Фильтруют содержимое контейнера через
мембранный фильтр, а затем помещают MeM
брану в соответствующую питательную среду,
как описано в испытании на стерильность.
Пироrенность (2.6.8). Раствор 51 должен
выдерживать испытание на пироrенность. На
1 Kr массы кролика вводят 10 мл раствора.
Аномальная токсичность (2.6.9). PaCT
вор S1 должен выдерживать испытание на aHO
мальную токсичность. Каждой мыши вводят
0,5 мл раствора.
УПАКОВКА
Тосударственная фармакопея Республики Бe/Japyf:.
ИСПЫТАНИЯ
584
Контейнеры упаковывают в защитную упа
ковку.
При удалении контейнера из упаковки не
должно наблюдаться протекания и роста микро
орrанизмов. Защитная оболочка должна быть
достаточно крепкой, чтобы выдерживать xpaHe
ние в обычных условиях.
Защитная оболочка должна быть такой,
чтобы ее невозможно было открыть и повторно
закрыть без видимых следов.
МАРКИРОВКА
Маркировка контейнеров должна соответ
ствовать требованиям национальноro законо
дательства и международным требованиям. На
этикетке указывают:
наименование и адрес производителя;
номер серии, который позволяет просле
дить историю контейнера и полимерноro MaTe
риала, из котороro он был изroтовлен.
Часть этикетки оставляют незаполненной
для:
указания rруппы крови, номера постав
ки и друroй информации в соответствии с наци
ональным законодательством и международны
ми требованиями, а также свободное место для
дополнительной информации.
На этикетке защитной упаковки или этикет
ке контейнера, видимой сквозь оболочку, указы
вают:
дату истечения срока roдности;
«После удаления из защитной оболочки
контейнер необходимо использовать в течение
10 дней».
Чернила или друrие вещества, использу
емые для печати этикеток, не должны проникать
в материал контейнера, надписи должны быть
четкими в течение всеro времени использования
контейнера.
01/2013:30204
3.2.4. ПУСТЫЕ СТЕРИЛЬНЫЕ
КОНТЕЙНЕРЫ
ИЗПЛАСТИФИЦИРОВАнноrо
ПОЛИВИНИЛХЛОРИДА
для ЧЕЛОВЕЧЕСКОИ КРОВИ
И ЕЕ КОМПОНЕНТОВ
Если нет друrих указаний в С09тветствии со
статьей 3.2.3. Стерильные пластмассовые KOH
тейнеры для человеческой крови и ее компонен
тов, при рода и состав материалов, из которых
изroтавливают контейнеры, должны COOTBeT
ствовать требованиям статьи 3.1. 1. Материалы
для контейнеров для человеческой крови и ее
компонентов.
Контейнеры должны выдерживать ИCПbIт.
ния, обозначенные в статье 3.2.3. Стерuльные
пластмассовые контейнеры для человечea«JiJ
крови и ее компонентов, а также следующие ис>-
пытания для определения экстраrируемых ве-
ществ.
Раствор сравнения. Воду для инъекций Р
наrревают в колбе из боросиликатноrо стекла
в автоклаве при температуре 11 О ос в течение
30 мин.
Кислотность или щелочность. К объему
раствора S2 (см. 3.2.3), соответствующему 4 %
номинальноro объема контейнера, прибавля
ют 0,1 мл раствора фенолфталеина Р. Pacт
вор должен быть бесцветным. При прибавл
нии 0,4 мл 0,01 М раствора натрия 2идроксида
должно появиться розовое окрашивание. При
прибавлении 0,8 мл 0,01 М раствора кислоты
хлористоводородной и 0,1 мл раствора Meти
пов020 краСН020 Р должно появиться оранжеВ<r
красное или красное окрашивание.
Оптическая плотность (2.2.25). Оптиче
ская плотность раствора S2 (см. 3.2.3) в области
от 230 нм до 250 нм не должна превышать 0,30;
в области от 251 нм до 360 нм 0,10. В качестве
компенсационноro раствора используют раствор
сравнения.
Восстанавливающие вещества. Свеже
приroтовленный раствор S2 (см. 3.2.3) помещают
в колбу из боросиликатноro стекла в количестве,
соответствующем 8 % от номинальною объема
контейнера. Одновременно с испытуемым pac
твором в друroй колбе из боросиликатноro
стекла rотовят контрольный раствор, используя
такой же объем свежеприroтовленноro раствора
сравнения. К каждому раствору прибавляют по
20,0 мл 0,002 М раствора калия пермаН2аната
и 1 мл кислоты серной разведенной Р. Bыдep
живают в защищенном от света месте в течение
15 мин. К каждому раствору прибавляют 0,1 r
калия йодида Р. Выдерживают в защищенном от
света месте в течение 5 мин и немедленно ти
труют 0,01 М раствором натрия тиосульфата,
используя в качестве индикатора 0,25 мл pacт
вора крахмала Р. Разность между объемами ти
транта не должна превышать 2,0 мл.
Экстраrируемый ди(2 этилrексил)фталат.
В качестве экстраrента используют 96
спирт Р, разведенный водой Р до получения
относительной плотности (2.2.5) от 0,9389 до
0,9395, определенной с помощью пикнометра.
Основной раствор. 0,100 r ди(2 этиЛ2ексил)
фталата Р растворяют в экстраrенте и доводят
до объема 100,0 мл этим же растворителем.
Эталонные растворы. В пять мерных колб
вместимостью 100 мл помещают COOTBeTCTBeH
3.2.5. Стерильные контейнеры из пластифицироваННО20 поливинилхлорида
585
но 1,0 мл, 2,0 мл, 5,0 мл, 10,0 мл И 20,0 мл OCHOB
ноro раствора, доводят экстраrентом до объема
100,0 мл.
Измеряют оптическую плотность (2.2.25)
эталонных растворов в максимуме при длине
волны 272 нм, используя экстраrент в качестве
компенсационноro раствора, и строят кривую за
висимости оптической плотности от KOHцeHTpa
ции ди(2 этилrексил )фталата.
Методика экстракции. Используя ДOHOp
скую трубку и иrлу или адаптер наполняют
контейнер до половины номинальноro объема
экстраrентом, предварительно подоrретым
до температуры 37 ос в плотно укупоренной
колбе. Полностью удаляют воздух из контей
нера и rерметично закрывают (запаивают) дo
норскую трубку. Заполненный контейнер опу
, екают в roризонтальном положении в водяную
баню, в которой поддерживают температуру
(37:t1) ос в течение (60:t1) мин, не встряхи
вая. Вынимают контейнер из водяной бани,
аккуратно переворачивают еro десять раз,
потом пере носят содержимое в стеклянную
колбу. Сразу же измеряют оптическую плот
насть в максимуме при длине волны 272 нм,
мспользуя экстраrент в качестве компенсаци
OHHOro раствора.
Определяют содержание ди(2 этилrексил)
фталата в миллиrраммах на 100 мл экстракта,
мспользуя rрадуировочный rрафик. Содержание
не должно превышать:
1 О Mr на 100 мл для контейнеров с HO
минальным объемом от 300 мл ДО 500 мл;
13 Mr на 100 мл для контейнеров с HO
минальным объемом от 150 мл до 300 мл;
14 Mr на 100 мл для контейнеров с номи
нальным объемом до 150 мл.
Хлориды (2.4.4). Не более 0,4 ррт. 15 мл
раствора 82 должны выдерживать испытание на
хлориды. Эталонный раствор rотовят с исполь
зованием 1,2 мл этаЛОНН020 раствора хлорида
(5 ррт С/) Р и 13,8 мл воды Р
Соли аммония (2.4.1). Не более 2 ррт. 5 мл
раствора S2 доводят водой Р до объема 14 мл.
Сухой остаток. 100 мл раствора 82' пред
варительно подоrретоro до температуры
105 ОС, выпаривают досуха в стакане из бо
росиликатноrо стекла подходящей вместимо
сти. 100 мл раствора сравнения выпаривают
досуха в тех же условиях (контрольный опыт).
Сушат до постоянной массы при температуре
от 100 105 ОС. Масса остатка от раствора 82
не должна превышать 3 Mr с учетом контроль
ноro опыта.
УПАКОВКА
Как обозначено в статье 3.2.3. Стерильные
пластмассовые контейнеры для человеческой
крови и ее компонентов.
75 3.ак 1712.
МАРКИРОВКА
Как обозначено в статье 3.2.3. Стерильные
пластмассовые контейнеры для человеческой
крови и ее компонентов крови.
01/2013:30205
3.2.5. СТЕРИЛЬНЫЕ КОНТЕЙНЕРЫ
ИЗПЛАСТИФИЦИРОВАнноrо
ПОЛИВИНИЛХЛОРИДА ДЛЯ
ЧЕЛОВЕЧЕСКОЙ КРОВИ,
СОДЕРЖАЩИЕ РАСТВОР
АНТИКОАrYЛЯНТА
Стерильные пластмассовые контейне
ры, содержащие раствор антикоаrулянта, KO
торые соответствуют требованиям частной
статьи Растворы антикоа2улянтов и KOH
серваНП10в для человеческой крови, исполь
зуют для сбора, хранения и введения крови.
Перед наполнением они должны COOTBeTCTBO
вать описанию и характеристикам, приве
денным в статье 3.2.4. Пустые стерильные
контейнеры из пластифицироваНН020 поли
винилхлорида для человеческой крови и ее
компонентов.
Если нет друrих указаний в соответствии
со статьей 3.2.3. Стерильные пластмассо
вые контейнеры для человеческой крови и
ее компонентов, при рода и состав матери
алов, из которых изrотавливают контейнеры,
должны соответствовать требованиям статьи
3.1.1. Материалы для контейнеров для че
ловеческой крови и ее компонентов.
ИСПЫТАНИЯ
Контейнеры должны выдерживать ис
пытания, обозначенные в статье 3.2.3. Cтe
рильные пластмассовые контейнеры для че
ловеческой крови и ее компонентов, а также
следующие испытания: определение объема
раствора антикоаrулянта и определение экс
траrируемых веществ.
Объем раствора антикоаrулянта. PaCT
вор антикоаrулянта переносят из контейнера
в rрадуированный цилиндр. Объем не должен
отличаться от обозначенноrо на этикетке
более чем на :t10 %.
Спектрофотометрическое определе-
ние (2.2.25). Измеряют оптическую плотность
раствора антикоаrулянта из контейнера в об
ласти длин волн от 250 нм до 350 нм, исполь
зуя раствор антикоаrулянта тоro же состава,
который не был в контакте с пластическим
материалом, в качестве компенсационноrо
586
rосударственная фармакопея Республики Беларусь
раствора. Оптическая плотность в максиму
ме при длине волны 280 нм не должна пре
вышать 0,5.
Экстраrируемыйди(2 зтилrексил)фталат.
Раствор антикоаryлянта аккуратно удаляют из
контейнера с помощью rибкой соединительной
трубки. Используя воронку, соединенную с труб
кой, наполняют контейнер водой Р, оставляют в
контакте в течение 1 мин, аккуратно сдавливая
контейнер, потом полностью освобождают от co
держимоro. Повторяют промывание.
Контейнер, освобожденный от содержи
моro -и промытый таким образом, должен BЫ
держивать испытания на экстраrируемый ди(2
этилrексил)фталат, как указано в статье 3.2.4.
Пустые стерильные пластмассовые контей
неры из пластифицироваНН020 поливинилхпо
рида для человеческой крови и ее компонентов.
УПАКОВКА И МАРКИРОВКА
Как указано в статье 3.2.3. СтеРlJльные
пластмассовые контейнеры для человеческой
крови и ее компонентов.
01/2013:30206
3.2.6. КОМПЛЕКТЫ ДЛЯ
ПЕРЕЛИВАНИЯ КРОВИ
И КОМПОНЕНТОВ КРОВИ
Комплекты для переливания крови и KOM
понентов крови состоят обычно из пластмассо
вой трубки, к которой присоединены остальные
элементы, необходимые для использования
комплекта для переливания крови надлежа
щим образом. Комплекты включают в себя при
способление для прокалывания пробки, фильтр
для крови, капельницу, реryлятор потока, co
единительный элемент Люэра (Luer) и, как пра
вило, приспособление для инъекций во время
переливания. Если комплекты предназначены
для использования с контейнерами, дЛЯ KOТO
рых необходим воздушный фильтр, еro можно
включить в приспособление для прокалывания
пробки или можно использовать друroе при
способление для введения воздуха. Камера,
включающая фильтр для крови, капельница и
основная трубка должны быть прозрачными.
Используемые материалы и модель комплек
та для переливания выбирают так, чтобы ис
ключить rемолитические эффекты. Комплек
ты должны соответствовать действующим на
данный момент стандартам размеров и эксплу
атационных характеристик.
Все части комплекта, которые MOryт Haxo
диться в контакте с кровью и ее компонентами,
должны быть стерильными и свободными от пи
poreHHbIx веществ. Каждый комплект помещает
ся в индивидуаf1ЬНУЮ упаковку, которая обеспе-
чивает стерильность содержимоrо. Комплекты
нельзя повторно стерилизовать или повторно
использовать.
Комплекты для переливания крови и ее
компонентов должны быть изrотовлены в со--
ответствии с требованиями надлежащей про--
изводственной практики для медицинскоro
оборудования, а также требованиями COOTBeT
ствующих национальных реrламентирующих дo
кументов.
ИСПЫТАНИЯ
Испытания проводятся на стерильных
комплектах.
Раствор S. rотовят замкнутую циркуляци
онную систему из трех комплектов и сосуда из
боросиликатноro стекла вместимостью 300 мл.
Присоединяют к сосуду термостат, который под
держивает температуру жидкости в сосуде на
уровне (37:f:1) ОС. Пропускают через систему в Ha
правлении переливания 250 мл воды для иHъeK
ций Р в течение 2 ч со скоростью 1 л/ч (например,
с помощью перистальтическоrо насоса, присое
диненноro к максимально меньшему участку си
ликоновой трубки). Собирают весь раствор и ox
лаждают.
Внешний вид раствора S. Раствор S
должен быть прозрачным (2.2.1) и бесцветным
(2.2.2, метод 11).
Кислотность или щелочность. К 25 мл
раствора S прибавляют 0,15 мл раствора иH
дикатора BRP Р. При прибавлении не более
0,5 мл 0,01 М раствора натрия 2идроксида
окраска раствора должна измениться на синюю.
К 25 мл раствора S прибавляют 0,2 мл pacт
вора метилов020 оранжев020 Р. При прибавле
нии не более 0,5 мл 0,01 М раствора кислоты
хлористоводородной окраска раствора должна
начать изменяться.
Оптическая плотность (2.2.25). Оптическая
плотность раствора S в диапазоне длин волн от
230 нм до 250 нм не должна превышать 0,30; в
диапазоне длин волн от 251 нм до 360 нм 0,15.
Восстанавливающие вещества. Испыта
ние проводят в течение 4 ч после при20товле
ния раствора S. К 20,0 мл раствора S прибавля
ют 1 мл кислоты серной разведенной Р и 20,0 мл
0,002 М раствора калия пермаН2аната. Кипятят
в течение 3 мин и сразу охла}I(Дают. Прибавляют
1 r калия йодида Р и титруют 0,01 М раствором
натрия тиосульФата, используя в качестве ин
дикатора 0,25 мл раствора крахмала Р. Парал
лельно проводят контрольный опыт, используя
20,0 мл воды для инъекций Р. Разность между
объемами титранта не должна превышать 2,0 мл.
3.2.6. Комплекты для переливания крови и компонентов кровли
587
Этиленоксид. Не более 1 О ррщ если на
этикетке указано, что для стерилизации исполь
зовался этиленоксид.
rазовая хроматоrрафия (2.2.28).
Условия хроматоrрафирования:
колонка из нержавеющей стали длиной
1,5 м и внутренним диаметром 6,4 мм, заполнен
ная диатомитом силанизированным для 2азо
вой хромат02рафии Р, импреrнированным Ma
Кр020ЛОМ 1500 Р (3 r на 1 О r);
zаз носитель: 2елий для хромат02рафии р;
скорость 2аза носителя: 20 мл/мин;
детектор: пламенно ионизационный;
температура: колонка 40 ОС, блок
ввода проб 100 ОС, детектор 150 ОС.
Проверяют отсутствие пиков, выходящих
одновременно спиком этиленоксида, проводя
испытание на нестерилизованных комплектах,
либо выполняют описанное выше определение
со следующими изменениями:
колонка из нержавеющей стали длиной
3 м и внутренним диаметром 3,2 мм, заполнен
ная диатомитом силанизированным для 2a
зовой хромат02рафии Р, импреrнированным
трисцианоэтоксипропаном Р (2 r на 1 О r);
температура: колонка 60 ОС.
Раствор этиленоксидв. rотовят в вы
тяжном шкафу. Во флакон вместимостью
50 мл помещают 50,0 мл диметилацетами
да Р, укупоривают, закрепляют пробку и взве
шивают с точностью до 0,1 Mr. Полиэтиленовый
или полипропиленовый шприц вместимостью
50 мл наполняют rазообразным этиленокси
дом Р, оставляют rаз в контакте со шприцем в
течение 3 мин, освобождают шприц от coдep
жимоrо и снова наполняют ero 50 мл rазообраз
ноro этиленоксида Р. Присоединяют к шприцу
rиподермальную иrлу и уменьшают объем rаза
в шприце от 50 мл до 25 мл. Медленно вводят
эти 25 мл во флакон, аккуратно встряхивая и
предотвращая контакт между иrлой и жидко
стью. Снова взвешивают флакон. Увеличение
массы должно составлять от 45 Mr до 60 Mr;
эта величина используется для расчета точной
концентрации раствора (около 1 r/л).
rрадуировочный 2рафик. В серию из семи
флаконов, аналоrичных флакону для испыта
ния, содержащих по 150 мл диметилацетами
да Р, помещают О мл, 0,05 мл, 0,10 м, 0,20 мл,
0,50 мл, 1,00 мл И 2,00 мл раствора этиленок
сида соответственно. Полученные таким обра
зом растворы содержат О MKr, 50 MKr, 100 MKr,
200 MKr, 500 MKr, 1000 MKr и 2000 MKr этиленок
сида. Флаконы укупоривают, закрепляют пробки
и помещают в сушильный шкаф при температу
ре (701:1) ос на 16 ч. Отбирают из каждоro фла
кона по 1 мл rорячеro rаза, вводят в колонку. По
высоте полученных пиков и содержанию этиле
ноксида в каждом флаконе строят rрадуировоч
ный rрафик.
Испытание. После удаления упаковки взве
шивают комплект. Разрезают ero на части с MaK
симальным размером сторон 1 см и помещают
во флакон вместимостью от 250 мл до 500 мл,
содержащий 150 мл диметилацетамида Р. YKY
поривают флакон и закрепляют пробку. Помеща
ют флакон в сушильный шкаф при температуре
(70:t1) ос на 16 ч. Отбирают из флакона 1 мл ro
рячеro rаза и вводят в колонку.
Содержание этиленоксида рассчитывают
с использованием rрадуировочноro rрафика по
высоте полученных пиков.
Посторонние вещества. Через входное OT
верстие наполняют контейнер раствором 0,1 r/л
натрия лаурилсульфата Р, предварительно
профильтрованным через стеклянный фильтр
(16) (2.1.2) и HarpeTbIM до температуры 37 ОС.
Собирают жидкость, вытекающую из выходноro
отверстия. При просматривании в COOTBeTCTBY
ющих условиях жидкость должна быть прозрач
ной и свободной от каких либо видимых частиц
или волокон (предполаrается, что частицы и BO
локна диаметром 50 мкм И более видны HeBOO
руженным rлазом).
Скорость потока. Через полный комплект с
полностью открытым реryлятором потока пропу
скают 50 мл раствора с вязкостью 3 мПа'с (3 сП)
(например, раствор 33 r/л маКр020па 4000 Р при
температуре 20 ОС) под rидростатическим дaB
лением, соответствующим 1 м. Время, необходи
мое для протекания 50 мл раствора, не должно
превышать 90 с.
Устойчивость к сжатию. rерметизируют
концы комплекта и все входные отверстия для
воздуха. Присоединяют комплект к выходному
отверстию для сжатоro воздуха, снабженноro
реryлятором давления. Помещают контейнер в
резервуар с водой при температуре 20 23 ос.
Постепенно подают избыточное давление вели
чиной 100 кПа и выдерживают в течение 1 мин.
Из комплекта не должны выделяться пузырьки
воздуха.
Прозрачность. В качестве суспензии cpaB
нения используют исходную опалесцирующую
суспензию (2.2.1), разбавленную в соотношении
1:8 для комплектов с трубкой с внешним диа
метром менее 5 мм или в соотношении 1:16
для комплектов с трубкой с внешним диаметром
5 мм и более. Пропускают суспензию сравнения
через комплект и сравнивают с друrим комплек
том из этой серии, заполненным водой Р. Должно
наблюдаться наличие мутности и пузырьков.
Сухой остаток. 50,0 мл раствора S выпа
ривают досуха на водяной бане и высушивают
до постоянной массы в сушильном шкафу при
температуре 1 O 105 ОС. Параллельно прово
дят контрольный опыт, используя 50,0 мл воды
для инъекций Р. Разность между массами сухих
остатков не должна превышать 1 ,5 Mr.
588
rосударственная фармакопея Республики Беларусь
Стерильность (2.6.1). Комплекты должны
выдерживать испытания на стерильность. Если
требуется, чтобы комплект был стерильным
только изнутри, пропускают через нею 50 мл бу
ферноrо раствора с натрия хлоридом и пепто
ном с рН 7,0 (2.6.12), затем анализируют ero Me
тодом мембранной фильтрации.
Если комплект должен быть стерильным
снаружи и изнутри, вскрывают упаковку с соблю
дением требований асептики и:
при использовании метода прямоrо посева
помещают комплект или ero отдельные части в
соответствующую емкость, содержащую ДOCTa
точное количество питательной среды для пол
ноro поrружения комплекта;
при использовании метода мембранной
фильтрации помещают комплект или ею OT
дельные части в соответствующую емкость, co
держащую достаточное количество буферноro
раствора с натрия хлоридом и пептоном с рН 7,0
(2.6.12) для ero полноro промывания в течение
1 О мин.
Пироrенность (2.6.8). Пять комплектов при
соединяют друr к друry и пропускают через по
лученную установку 250 мл стерильноro CBO
бодноro от пироrенных веществ раствора 9 r/л
натрия хлорида Р со скоростью не более
10 мл/мин. В асептических условиях собирают
раствор в контейнер, свободный от пироrенных
веществ. Раствор должен выдерживать испы
тание на пироrенность. На 1 Kr массы кролика
вводят 1 О мл раствора,
МАРКИРОВКА
При необходимости на этикетке указывают,
что стерилизация комплекта проводилась с ис
пользованием этиленоксида.
01/2013:30208
3.2.8. СТЕРИЛЬНЫЕ
ОДНОРАЗОВЫЕ
ПЛАСТМАССОВЫЕ ШПРИЦЫ
Стерильные одноразовые пластмассовые
шприцы это медицинские изделия. использу
емые для непосредственноro введения инъек
ционных лекарственных средств. Они должны
быть стерильными и свободными от пироrен
ных веществ и не должны подверrаться повтор
ной стерилизации и повторному использованию.
Шприцы состоят из цилиндра и поршня, KOTO
рый может быть снабжен эластичным уплотни
тельным кольцом; MOryт комплектоваться иrлой,
которая может быть несъемной. Каждый шприц
должен быть индивидуально упакован для обе
спечен ия стерильности.
Цилиндр шприца должен быть прозрачным
для обеспечения точности дозирования, а также
контроля отсутствия пузырьков и посторонних
частиц.
Пластмассовые и эластичные матери
алы для производства цилиндров и поршней
должны соответствовать определенной специ
фикации или требованиям компетентноro упол
номоченноro opraHa. Наиболее часто использу
емые материалы полипропилен и полиэти
лен. Шприцы должны соответствовать действу
ющим на данный момент стандартам размеров
и эксплуатационных характеристик.
На внутреннюю стенку шприца для обеспе
чения плавной работы поршня может наносить
ся масло силиконовое (3.1.8), но в количестве,
не допускающем заrрязнение содержимоro в
процессе использования.
Чернила, краска и клей, используемые для
маркировки шприцов, нанесения надписи на
упаковку, а также, если необходимо, на комплек
ты шприц/упаковка, не должны проникать через
стенку шприцов.
ИСПЫТАНИЯ
Раствор S. Раствор ютовят таким обра
30М, чтобы предотвратить еro заrрязнение по
сторонними частицами. Используя достаточное
количество шприцов для получения 50 мл paCT
вора, наполняют их до номинальноro объема
водой для инъекций Р и выдерживают при TeM
пературе 37 ос в течение 24 ч. Объединяют co
держи мое шприцов в емкость из боросиликат
ноro стекла.
Внешний вид раствора. Раствор S должен
быть прозрачным (2.2.1), бесцветным (2.2.2,
метод 11) и практически свободным от посторон
них твердых частиц.
Кислотность или щелочность. К 20 мл
раствора S прибавляют 0,1 мл раствора бром
тимолов020 сине20 Р1. При прибавлении не
более 0,3 мл 0,01 М раствора натрия 2идPOKcи
да или 0,01 М раствора кислотbl хлористово
дородной окраска раствора должна измениться.
Оптическая плотность (2.2.25). Опти
ческая плотность раствора S в диапазоне
длин волн от 220 нм до 360 нм не должна пре
вышать 0,40.
Этиленоксид. Не более 10 ррт, если на
этикетке указано, что для стерилизации исполь
зовался этиленоксид.
rазовяя хроматоrрафия (2.2.28).
Условия хроматоrрафирования:
колонка из нержавеющей стали длиной
1,5 м и внутренним диаметром 6,4 мм, заполнен
ная диатомитом силанизироваННblМ для 2азо
вой хромат02рафии Р, импреrнированным Ma
Кр020ЛОМ 1500 Р (3 r на 10 r);
3.2.8. Стерильные одноразовые пластмассовые шприцы
589
2аз носитель: 2елий для хромат02рафии Р;
скорость ааза носителя: 20 мл/мин;
детектор: пламенно ионизационный;
температура: колонка 40 ОС, блок
ввода проб 100 ОС, детектор 150 ОС.
Проверяют отсутствие пиков, выходящих
одновременно спиком этиленоксида, прово
дя испытание на нестерилизованных шприцах,
либо выполняют описанное выше определение
со следующими изменениями:
колонка из нержавеющей стали длиной
3 м и внутренним диаметром 3,2 мм, заполнен
ная диатомитом силанизированным для 2a
зовой хромат02рафии Р, импреrнированным
трисцианоэтоксипропаном Р (2 r на 1 О r);
температура: колонка 60 ОС.
Раствор этиленоксида. Тотовят в вытяж
ном шкафу. Во флакон вместимостью 50 мл по
мещают 50,0 мл диметилацетамида Р, YKY
поривают, закрепляют пробку и взвешивают с
точностью до 0,1 Mr. Полиэтиленовый или поли
пропиленовый шприц вместимостью 50 мл Ha
полняют rазообразным этиленоксидом Р, OCTaB
ляют rаз в контакте со шприцем в течение 3 мин,
освобождают шприц от содержимоro и снова Ha
полняют ero 50 мл rазообразноro этиленокси
да Р. Присоединяют к шприцу rиподермальную
иrлу и уменьшают объем rаза в шприце от 50 мл
до 25 мл. Медленно вводят эти 25 мл во флакон:
аккуратно встряхивая и предотвращая контакт
между иrлой и жидкостью. Снова взвешивают
флакон. Увеличение массы должно составлять
от 45 Mr до 60 Mr; эта величина используется для
расчета точной концентрации раствора (около
1 r/л).
Традуировочный 2рафик. В серию из семи
флаконов, аналоrичных флакону для испыта
ния, содержащих по 150 мл диметилацетами
да Р, помещают О мл, 0,05 мл, 0,10 м, 0,20 мл,
0,50 мл, 1,00 мл И 2,00 мл раствора этиленок
сида соответственно. Полученные таким обра
30М растворы содержат О MKr, 50 MKr, 100 MKr,
200 MKr, 500 MKr, 1000 MKr и 2000 MKr этиленокси
да. Флаконы укупоривают, закрепляют пробки и
помещают в сушильный шкаф при температуре
(70:t1) ос на 16 ч. Отбирают из каждоro флако
на по 1 мл rорячеro rаза, вводят в колонку. По
высоте полученных пиков и содержанию этиле
ноксида в каждом флаконе строят rрадуировоч
ный rрафик.
Испытание. После удаления упаковки взве
шивают шприц. Разрезают еro на части с макси
мальным размером сторон 1 см и помещают во
флакон вместимостью от 250 мл до 500 мл, co
держащий 150 мл диметилацетамида Р. Укупо
ривают флакон и закрепляют пробку. Помеща
ют флакон в сушильный шкаф при температуре
(70:t1) ос на 16 ч. Отбирают из флакона 1 мл ro
рячею rаза и вводят в колонку.
Содержание этиленоксида рассчитывают
с использованием rрадуировочноro rрафика по
высоте полученных пиков.
n; За. 1712.
Масло силиконовое. Рассчитывают пло
щадь внутренней поверхности шприца (см 2 ) по
формуле:
2 .JVnh ,
rде:
V номинальный объем шприца, см З ;
h высота rрадуировки, см.
Берут достаточное количество шприцев для
обеспечения площади внутренней поверхности
от 100 см 2 ДО 200 см 2 . В каждый шприц вводят KO
личество метиленхлорида Р, равное половине
ero номинальноro объема, и воздухом доводят
объем до номинальною. Промывают BHYTpeH
нюю стенку, соответствующую номинальному
объему шприца, растворителем, путем пере
ворачивания еro 1 О раз, закрывая элемент для
присоединения иrлы пальцем, покрыты м пласт
массовой пленкой, инертной к метиленхлори
ду. Собирают экстракты в предварительно BЫ
сушенную до постоянной массы и взвешенную
чашку и повторяют операцию. Выпаривают объе
диненные экстракты досуха на водяной бане.
Высушивают при температуре 1 oo 105 ос в Te
чение 1 ч. Масса полученноro остатка не должна
превышать 0,25 Mr на 1 см 2 площади внутренней
поверхности.
Инфракрасный спектр (2.2.24) полученно
ro остатка должен иметь максимумы при вол
новых числах, соответствующих маслу силико
новому: 805 CM 1, 1020 СМ"1, 1095 CM 1, 1260 CM 1
и 2960 CM 1.
Восстанавливающие вещества. К 20,0 мл
раствора S прибавляют 2 мл кислоты серной Р
и 20,0 мл 0,002 М раствора калия пермаН2ана
та. Кипятят в течение 3 мин и сразу охлаждают.
Прибавляют 1 r калия йодида Р и титруют 0,01 М
раствором натрия тиосульфата, используя в
качестве индикатора 0,25 мл раствора KpaXMa
ла Р. Параллельно проводят контрольный опыт,
используя 20,0 мл воды для инъекций Р Раз
ность между объемами титранта не должна пре
вышать 3,0 мл.
Прозрачность. Наполняют один шприц
водой Р (контрольный опыт), друroй шприц
исходной опалесцирующей суспензией (2.2. 1),
разведенной 1:10. Исходную опалесцирую
щую суспенизию предварительно выдержива
ют при температуре (20:t2) ос в течение 24 ч.
При сравнении с контрольным опытом в pac
сеянном свете на темном фоне невооружен
ным rлазом должна быть заметна мутность cy
спен ии.
Стерильность (2.6.1). Если указано, что
шприцы должны быть стерильными, для них
проводят испытание на стерильность сле
дующим образом. В асептических условиях
вскрывают упаковку, вынимают шприц, разби
590
rосударственная фармакопея Республики Беларусь
рают ero по частям и помещают каждую часть
в емкость, содержащую достаточное количе
ство питательной среды для полноro поrруже
ния шприца. Используют обе peKOMeHДOBaH
ные среды (2.6.1).
Если указано, что шприцы должны быть
стерильными только изнутри, для них прово
дят испытание на стерильность следующим
образом. Для каждоro из испытуемых шприцов
используют 50 мл питательной среды. В асеп
тических условиях снимают защитный колпачок
с иrлы и поrружают иrлу в питательную среду.
Промывают шприц пять раз с помощью поршня,
поло ение котороro должно обеспечивать MaK
симально возможный уровень наполнения.
Пироrенность (2.6.8). Шприцы, имеющие
номинальный объем, равный или более 15 мл,
должны выдерживать испытание на пироrен
ность. Не менее 3 шприцев наполняют дО HO
минальноro объема свободным от пироrенных
веществ раствором 9 r/л натрия хлорида Р и BЫ
держивают при температуре 37 ос в течение 2 ч.
В асептических условиях объединяют растворы
в емкость, свободную от пироrенных веществ, и
выполняют испытание немедленно, используя
10 мл раствора на 1 Kr массы каждоro кролика.
МАРКИРОВКА
На этикетке упаковки указывают:
номер серии;
описание шприца;
«только для одноразовоro использования».
На этикетке вторичной (наружной) упаковки
указывают:
метод стерилизации;
«стерильно» или «стерильно только изнутри»;
информацию о производителе;
«не использовать шприц при нарушении
упаковки и стерильности».
01/2013:30209
3.2.9. РЕЗИНОВЫЕ
УкУпороЧttЫЕ СРЕДСТВА
ДЛЯ КОНТЕИНЕРОВ,
ПРЕДНАЗНАЧЕННЫХ
ДЛЯ ВОДНЫХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
ДЛЯ ПАРЕНТЕРАльноrо
ПРИМЕНЕНИЯ,ПОРОШКОВ
и ЛИОФИЛИЗИРОВАННЫХ
ПОРОШКОВ
Резиновые укупорочные средства дЛЯ KOH
тейнеров, предназначенных для водных лекар
ственных средств для парентеральноro примене
ния, порошков и лиофилизированных порошков,
производят из материалов, полученных методом
вулканизации (поперечным сшиванием) макро--
молекулярных орraнических веществ (эластоме
ров) с соответствующими добавками. Требова
ния данной статьи распространяются также на
укупорочные средства для контейнеров, пред
назначенных для лиофилизированных порош
ков и продуктов, которые растворяют в воде
непосредственно перед использованием. Tpe
бования данной статьи не распространяются на
укупорочные средства, изrотовленные из сили
коновOfО эластомера (который должен отвечать
требованиям статьи 3.1.9. Силиконовый эла
стомер для укупорочных средств и трубок),
ламинированные или лакированные укупороч
ные средства. Эластомеры получают из природ
ных или синтетических веществ, используя по
лимеризацию, аддитивную полимеризацию или
поликонденсацию. Природа основных веществ
и используемых добавок (например, вулканиза
торов, катализаторов, стабилизаторов, красите
лей) зависит от требований, предъявляемых к
конечному продукту.
Резиновые укупорочные средства можно
классифицировать на два типа: тип 1 пробки,
которые соответствуют более строrим требова
ниям и являются предпочтительными для ис
пользования; тип 11 пробки, которые имеют
механические свойства для специальных целей
(например, для мноroразовоro прокалывания) и
не отвечают строrим требованиям, предъявля
емым к пробкам типа 1, из за своеro химическо
ro состава.
К средствам, используемым для укупори
вания конкретноro лекарственноro средства,
предъявляют следующие требования:
компоненты лекарственноro средства, Ha
ходящиеся в контакте с пробкой, не должны aд
сорбироваться на поверхности пробки, а также
проникать в пробку или через ее поверхность в
таком количестве, которое отрицательно влияет
на качество лекарственноro средства;
пробка не должна выделять в лекарствен
ное средство какие либо вещества в количе
ствах, способных повлиять на еro стабильность
или повышающих еro токсичность.
Пробка должна быть совместима с лекар
ственным средством в течение всеro периода
еro хранения и применения.
Производитель лекарственноro средства
должен получить от поставщика укупорочных
средств rарантии тою, что состав укупорочно
ro средства не изменяется и является идентич
ным тому, который подверrался испытаниям на
совместимость. В том случае, если поставщик
litнформирует производителя лекарственноro
средства об изменении в составе, испытания
на совместимость следует проводить повторно
в полном объеме или частично, в зависимости
от характера изменений.
Пробки перед использованием моют и, если
необходимо,стерилизую
3.2.9. Резиновые УКУПОрОЧНblе средства для контейнеров
591
ОПИСАНИЕ
Резиновые укупорочные средства эластич
ны, полупрозрачны или непрозрачны, не имеют
характерноro окрашивания (окрашивание зави
сит от вида используемых добавок). Практиче
ски нерастворимы в тетраrидрофуране, в KOTO
ром, однако, может наблюдаться значительное
обратимое набухание. rOMoreHHbI и практически
не содержат посторонних включений (например,
волокон, механических частиц, отходов резины).
Идентификация типа резины, использу
емой для производства укупорочных средств,
не 02раничиваеrnся rnребованиями данной
статьи. Идентификационные испытания, при
веденные ниже, различают эластомерные и
неэластомерные укупорочные средства, но
не дифференцируют разные виды резины. С
целью выявления различий в сериях по cpaвHe
нию с пробками, используемыми в испытаниях
на совместимость, выполняют дРУ2ие идeH
тификационные испытания. Для эт020 М02ут
использоваться один или несколько аналити
ческих методов: определение относительной
плотности, определение сульфатной золы,
определение содержания серы, тОнкослойная
хромат02рафия экстракта, ультрафиолето
88Я абсорбционная спектрофотометрия экс
тракта, инфракрасная абсорбционная спек
трофотометрия продуктов пиролиза.
ИДЕНТИФИКАЦИЯ
А. Эластичность материала должна быть
taкой, чтобы ленту с поперечной шириной от
1 мм 2 ДО 5 мм 2 можно было растянуть вручную в
2 раза по сравнению с исходной длиной. Будучи
растянутой в 2 раза в течение 1 мин, она должна
сжиматься до не более 1,2 кратноro первона
чальноro размера в течение 30 с.
В. От 1 r до 2 r материала наrревают в Tep
мостойкой пробирке над открытым пламенем
до высушивания образца, продолжают HarpeBa
ние до конденсации паров продуктов пиролиза
у верхнеro края пробирки. Осаждают несколь
ко капель продуктов пиролиза на диске с калия
бромидом. Инфракрасный спектр (2.2.24) полу
ченноrо диска должен соответствовать спектру
типовоro образца.
с. Содержание общей золы (2.4.16) должно
быть в пределах :t10 % от результата, получен
ноro для типовоro образца.
ИСПЫТАНИЯ
Испытуемые образцы М02ут подвер
2аться мойке и стерилизации перед иc
пользованием.
Раствор S. Неразрезанные пробки в количе
стве, соответствующем общей площади поверх
насти около 100 см 2 , помещают в подходящую
стеклянную емкость, заливают водой для иHъeK
ций Р, кипятят в течение 5 мин и промывают пять
раз холодной водой для инъекций Р. Промытые
пробки переносят в широкоrорлую колбу (стекло
класса 1, 3.2.1), прибавляют 200 мл воды для
инъекций Р и взвешивают. Закрывают отверстие
колбы лабораторным стаканом из боросиликат
ноro стекла. Наrревают в автоклаве таким обра
зом, чтобы за 20 30 мин температура достиrла
(12Н2) ОС, и выдерживают при данной темпера
туре в течение 30 мин. Охлаждают до KOMHaT
ной температуры в течение 30 мин и доводят до
первоначальной массы водой для инъекций Р.
Встряхивают и немедленно отделяют раствор от
пробок декантацией. Перед началом каждоro ис
пытания встряхивают раствор S.
Контрольный раствор. rотовят аналоrично
раствору S с использованием 200 мл воды для
инъекций Р.
Внешний вид раствора. Раствор S по CTe
пени мутности не должен превышать эталон
11 для пробок типа 1 и эталон 111 для пробок
типа 11 (2.2.1). Окраска раствора S должна быть
не более интенсивнее эталона GУ(ЗЖ)5 (2.2.1,
метод 11).
Кислотность или щелочность. К 20 мл
раствора S прибавляют 0,1 мл раствора бромти
молов020 сине20 Р1. При прибавлении не более
0,3 мл 0,01 М раствора натрия сидроксида или
не более 0,8 мл 0,01 М раствора кислоты хло
ристоводородной окраска раствора должна из
мениться на синюю или желтую соответственно.
Оптическая плотность (2.2.25). Испытание
выполняют в течение не более 5 ч после при
20товления раствора S. Раствор S фильтруют
через мембранный фильтр с размером пор около
0,45 мкм, отбрасывая первые порции фильтрата.
Измеряют оптическую плотность (2.2.25) филь
трата в области длин волн от 220 нм до 360 нм,
используя в качестве компенсационноro раст
вора контрольный раствор (см. Раствор S). Оп
тическая плотность в данном диапазоне длин
волн не должна превышать 0,2 для пробок типа 1
и О,4 для пробоктипа 11. Если необходимо, раз
водят фильтрат перед измерением и корректиру
ют результат с учетом разведения.
Восстанавливающие вещества. Испыта
ние выполняют в течение не более 5 ч после
при20товления раствора S. К 20,0 мл раствора
S прибавляют 2 мл кислоты серной разведен
ной Р и 20,0 мл 0,002 М раствора калия перман
2аната. Кипятят в течение 3 мин и охлаждают.
Прибавляют 1 r калия йодида Р и немедленно
титруют 0,01 М раствором натрия тиОСУЛЬфf1
та, используя в качестве индикатора 0,25 мл
раствора крахмала Р. Параллельно проводят
контрольный опыт, используя 20,0 мл контроль
ноro раствора. Разность между объемами ти
транта не должна превышать 3,0 мл для пробок
типа I и 7,0 мл для пробок типа 11.
592
rосударственная фармакопея Республики Беларусь
Соли аммония (2.4.1). Не более 2 ррm. 5 мл
раствора S доводят водой Р до объема 14 мл.
Полученный раствор должен выдерживать испы
тание А на аммония соли.
3кстраrируемый цинк. Не более 5 MKr экс
траrируемоro Zп в 1 мл раствора S.
Атомно абсорбционная спектрометрия
(2.2.23, метод /).
Испытуемый раствор. 10,0 мл раствора S
доводят 0,1 М раствором кислоты хлористово
дородной до объема 100,0 мл.
Растворы сравнения. Растворы cpaBHe
ния roтовят разведением этаЛОНН020 раствора
цинка (10 ррт Zп) Р 0,1 М раствором кислоты
хлористоводородной.
Источник излучения: лампа с полым цинко
вым катодом.
Длина волны: 213,9 нм.
Атомизатор: воздушно ацетиленовое
пламя.
3кстраrируемые тяжелые металлы (2.4.8,
метод А). Не более 2 ррm. Раствор S должен
выдерживать испытание на тяжелые металлы.
Эталон roтовят, используя эталонный раствор
свинца (2 ррт РЬ) Р.
Сухой остаток. 50,0 мл раствора S выпари
вают досуха на водяной бане и высушивают до
постоянной массы в сушильном шкафу при TeM
пературе 100 105 ОС. Масса полученноro OCTaT
ка не должна превышать 2,0 Mr для пробок типа I
и 4,0 Mr для пробок типа 11.
Летучие сульфиды. Пробки, если необхо
димо, предварительно разрезанные, с общей
площадью поверхности (20:1:2) см 2 помещают в
коническую колбу вместимостью 100 мл, прибав
ляют 50 мл раствора 20 r/л кислоты лимонной Р.
Над roрлом колбы помещают кусочек cвиHЦO
во ацетатной бума2и Р и удерживают бумаry в
таком положении, помещая сверху перевернутый
флакон для взвешивания. Наrревают в автокла
ве при температуре (121:!:2) ос в течение 30 мин.
Появившееся на бумаrе черное пятно должно
быть окрашено не более интенсивно, чем пятно в
контрольном испытании, выполненном с исполь
зованием 0,154 Mr натрия сульфата Р и 50 мл
раствора 20 r/л кислоты лимонной Р.
Для испытаний на проницаемость, pac
щепление и саМ02ерметизацию используют
пробки, обработанные, как указано при при20
тО8Лении раствора S, и высушенные.
Проницаемость. Для пробок, прокалыва
емых с помощью rиподермальной иrлы, проводят
следующее испытание. 10 флаконов наполняют
до номинальноro объема водой Р, закрывают ис
пытуемыми пробками и укупоривают колпачка
ми. Прокалывают каждую пробку перпендикуляр
но ее поверхности, используя для каждой пробки
новую смазанную rиподермальную иrлу с длин
ным скосом (например, ИСО 7864 «Стериль
ные rиподермальные иrлы для одноразовоro ис
пользования») (уroл скоса (12:1:2n и наружным
диаметром 0,8 мм. Требуемое для прокалыва
ния усилие, измеренное с точностью до :1:0,25 Н
(25 rc), не должно превышать 10 Н (1 Krc) для
каждой пробки.
Фраrментация. Для пробок, прокалыва
емых с помощью rиподермальной иrлы, проводят
следующее испытание. Если пробки предназна
чены для укупоривания водных лекарственных
средств, в 12 чистых флаконов помещают объем
воды Р меньше номинальноro объема на 4 мл,
з?крывают флаконы испытуемыми пробками,
укупоривают колпачками и оставляют на 16 ч.
Если пробки предназначены для укупоривания
сухих лекарственных средств, 12 чистых фла
конов закрывают испытуемыми пробками. С по
мощью чистоro шприца, снабженноro смазанной
rиподермальной иrлой с длинным скосом (напри
мер, ИСО 7864 «Стерильные rиподермальные
иrлы для одноразовоro использования») (уroл
скоса (12:1:2n и наружным диаметром 0,8 мм, ис
пользуя для каждой пробки новую иrлу, вводят во
флакон 1 мл воды Р и удаляют 1 мл воздуха. Опе
рацию повторяют 4 раза для каждоro флакона,
прокалывая еro каждый раз в новом месте. Для
каждой пробки используют новую иrлу и KOHTpO
лируют, чтобы иrла не затупилась в ходе испы
тания. Жидкость во флаконах фильтруют через
фильтр с размером отверстий 0,5 мкм. Считают
количество кусочков резины, видимых HeBOOpy
женным rлазом. Общее их количество не должно
превышать 5. Предполаrается, что частицы ди
аметром 50 мкм И более видны невооруженным
rлазом, в сомнительных случаях частицы про
сматривают под микроскопом для проверки их
природы и размера.
Самоrерметизация. Для пробок, предна
значенных для использования с мультидозо
выми контейнерами, проводят следующее ис
пытание. 1 О флаконов наполняют водой Р до
номинальноro объема, закрывают испытуемыми
пробками и укупоривают колпачками. Прокалы
вают каждую пробку 1 О раз, каждый раз в новом
месте, с помощью rиподермальной иrлы с длин
ным скосом (например, ИСО 7864 «Стерильные
rиподермальные иrлы для одноразовоro исполь
зования») (уroл скоса (12:1:2П и наружным ди
аметром 0,8 мм. Для каждой пробки использу
ют новую иrлу. Помещают флаконы вертикаль
но в раствор 1 r/л метиленов020 сине20 Р и по
нижают внешнее давление до 27 кПа в течение
10 мин. Затем восстанавливают давление до aT
мосферноro и оставляют в течение 30 мин. Про
мывают флаконы снаружи. Ни один из флаконов
не должен иметь каких либо следов окрашенно
ro раствора.
01/2013:40000
4.1. Реактивы, эталонные растворы, буферные растворы
593
4. РЕАКТИВЫ
Дополнительную информацию о peaKти
вах, которые М02ут быть идентифицирова
ны только по тОР20вым наименованиям или дo
ступность которых 02раничена, можно найти
на сайте EDQM в разделе Oatabases: Kпow/
edge Oatabase. Эта информация приводиться
только для обле2чения поиска таких peaKти
вов и не несет никаких особых рекомендаций
или подтверждения их сертификации Европей
ской Фармакопейной Комиссией или Советом
Европы. Допускается использование реактивов
из дРУ2их источников на основании их cooтвeт
ствия требованиям Фармакопеи.
01/2013:40100
4.1.
РЕАКТИВЫ, ЭТАЛОННЫЕ
РАСТВОРЫ, БУФЕРНЫЕ
РАСТВОРЫ
В тех случаях, KorAa название реактива или
раствора реактива сопровождается буквой Р и
выделено курсивом, это указывает на то, что pe
актив включен в нижеприведенный перечень.
В описании каждоro реактива, кроме обо
значенноro значком #, имеется семизначный
код, выделенный курсивом (например, 1002501).
Этот номер остается неизменным для каждо
ro реактива при всех последующих пересмо
трах перечня. Он может быть использован для
идентификации реактива и, например, при учете
и складировании реактивов. Описание может
также включать номер Chemica/ Abstract Service
Registry (CAS), леrко опознаваемый по xapaK
терному обозначению, например, 9002 93 1.
Некоторые из реактивов, включенных в пе
речень, являются токсичными, и при работе с
ними необходимо соблюдать соответствующие
меры предосторожности.
Водные растворы реактивов roтовят с ис
пользованием воды Р. Если раствор реактива
описывают выражением типа «кислота хлористо
водородная (10 r/л HCI)>>, раствор roтовят COOT
ветствующим разведением водой Р более KOHцeH
трированноro раствора реактива, приведенноro в
этом же разделе. Растворы реактивов, использу
емые для испытаний на предельное содержание
бария, кальция и сульфатов, roтовят с использова
нием воды дистиллированной Р. Если раствори
тель не указан, то подразумевают водный раствор.
Реактивы и растворы реактивов должны
храниться в плотно укупоренных контейнерах.
#Максимальный срок roдности реактивов
(при отсутствии информации от производителя
об их сроке roдности) при соблюдении надлежа
щих условий хранения составляет:
сухие (твердые) устойчивые реактивы
(устойчивые к свету, влажности воздуха, не rи
rроскопичные) не более 5 лет;
сухие реактивы (не подходящие к описан
ным выше) не более 3 лет;
жидкие устойчивые реактивы (устойчивые
к свету, влажности воздуха, не rиrроскопичные)
не более 5 лет;
кислоты неорrанические (минеральные)
не более 3 лет;
кислоты орrанические не более 3 лет;
основания (щелочи) не более 3 лет;
азотистые основания не более 1 roда;
орrанические растворители не более 3
лет.
Реактивы MOryт быть использованы после
окончания срока roдности при условии их пере
контроля на предмет соответствия специфика
ции или соответствующей статьи Фармакопеи.
Максимальный срок roдности приroтовлен
ных растворов реактивов (при отсутствии инфор
мации в общих статьях rлавы 4.2, нормативных
документах производителя rOToBoro лекарствен
ноro средства или друrих справочных литератур
ных источниках) при соблюдении надлежащих
условий хранения составляет:
кислоты разведенные не более 1 roда;
основания разведенные не более 1
roда;
водные растворы солей не более 3 лет;
смеси метанол:этанол:вода не более 3
месяцев;
элюенты для жидкостной хроматоrрафии
(вода) не более 1 месяца;
элюенты для жидкостной хроматоrрафии
однокомпонентные не более 6 месяцев;
элюенты для жидкостной хроматоrрафии
мноroкомпонентные не более 1 недели;
элюенты для тонкослойной xpoMaтorpa
фии не более 1 недели;
индикаторы не более 1 юда;
титрованные растворы не более 2 Me
сяцев (после чеrо необходимо повторно YCTaHaB
ливать титр);
приroтовленные титрованные растворы
для титрования по Фишеру не более 2 недель.
Приведенные выше требования к срокам
roдности реактивов носят рекомендательный
характер. В случае обнаружения любых изме
нений внешнеro вида (изменение ЦАета, выпа
дение осадка, образование хлопьев, изменение
физическоro состояния и т. п.) реактивы не под
лежат дальнейшему использованию.#
Маркировка реактивов должна COOTBeTCTBO
вать требованиям национальноro законодатель
ства и международных соrлашений.
594
rосударственная фармакопея Республики Беларусь
#На этикетке реактива ДОЛЖНО быть указано:
наименование предприятия изroтовителя, назва
ние реактива, квалификация реактива (или чист
та реактива), количество реактива (масса, объем),
номер партии (серии), дата изroтовления и срок
rодности (либо продемонстрирована возмож
ность прослеживаемости даты изroтовления или
срока roдности (даты истечения срока rодности) по
номеру серии). При перефасовке реактива друroй
орraнизацией или предприятием на этикетке,
кроме обозначений приведенных выше, указывает
ся намименование орraнизации или предприятия,
перефасовавшеro реактив и дата перефасовки.
На этикетке приroтовленноro раствора YKa
зывают: наименование раствора, идентифика
ционный номер, концентрацию (rде применимо),
дату приroтовления, срок rодности и подпись
лица, приroтовившеro раствор. Требования к
маркировке приroтовленных титрованных pac
творов указаны в статье 4.2.2.#
01/2013:40101
4.1.1. РЕАКТИВЫ
# Arap пищевой.
Пористые пластины толщиной не более
20 мм или пленки толщиной не более 0,5 мм
белоro или светло желтоro цвета; допускается
слеrка сероватый оттенок.
Аrароза ДЛЯ хроматоrрафии. 1001800.
[9012 36 6].
4 % суспензия в воде Р набухших rранул
диаметром от 60 мкм ДО 140 мкм. Используют
в rель хроматоrрафии для разделения белков
с молекулярными массами от 6.104 до 20.106 И
полисахаридов с молекулярными массами от
З.10 З до 5.106.
Аrароза поперечносшитая для xpOMaTO
rрафии. 1001900. [61970 08 9].
Получают из аrарозы реакцией с 2,3 ди
бромпропанолом в сильнощелочной среде.
4 % суспензия в воде Р набухших rранул
диаметром от 60 мкм ДО 140 мкм. Используют
в rель хроматоrрафии для разделения белков с
молекулярными массами от 6.104 до 20.106 И по
лисахаридов с молекулярными массами от З.10 З
до 5.106.
Аrароза поперечносш тая для xpOMaTO
rрафии Р1. 1001901. [65099 79 8].
Получают из аrарозы реакцией с 2,З ди
бромпропанолом в сильнощелочной среде.
4 % суспензия в воде Р набухших rранул
диаметром от 60 мкм ДО 140 мкм. Используют
в rель хроматоrрафии для разделения белков с
молекулярными массами от 7.104 до 40.106 И по
лисахаридов с молекулярными массами от 1 '105
до 2.107.
Аrароза для электрофореза. 1002000.
[9012 36 6].
Нейтральный линейный полисахарид, oc
нов ной компонент котороro получают из arapa.
Порошок белоro или почти белоro цвета.
Практически нерастворима в холодной воде,
очень мало растворима в roрячей воде.
Аrароза ДЭАЭ для ионообменной хрома-
тоrрафии. 1002100. [57407 08 6].
Поперечносшитая аrароза с замещенными
диэтиламиноэтильными rруппами в виде шаро
.образных rранул.
Аrароза/поперечносшитый полиакрила-
мид. 1002200.
Аrароза в поперечносшитой полиакрила
мидной матрице. Используют для разделения
rлобулярных белков с молекулярными массами
от 2.104 до 35.104.
Аденин. 1172800. [73 24 5].
См. статью Аденин.
Аденозин. С10Н1зN504' (М М. 267,2).1001600.
[58 61 7]. 6 Амино 9 f3 D рибофуранозил 9Н пу
рин.
Белый или почти белый кристаллический
порошок. Малорастворим в воде, практически
нерастворим в ацетоне и в 96 % спирте. PaCTBO
ряется в разведенных растворах кислот.
Температура плавления: около 234 ОС.
Адипиновая кислота. С 6 Н 10 О 4 . (М М. 146,1).
1095600. [124 04 9].
Кристаллы в виде призм. Леrкорастворима
в метаноле. растворима в ацетоне, практически
нерастворима в петролейном эфире.
Температура плавления: около 152 ОС.
Азометин Н. C17H12NNaOaS2' (М м. 445,4).
1008700. [5941 07 1]. Натрия rидроА rидро кси
5 (2 rидрокси бензилиденамино ) 2. 7 нафта
линдисульфонат.
Азометина Н раствор. 1008701.
0,45 r азометина Н Р и 1 r кислоты aCKOp
биновой Р растворяют в воде Р при слабом Ha
rревании и доводят до объема 100 мл этим же
растворителем.
Азот. N z . (М м. 28,01). 1059300. [7727 З7 9].
Азот промытый и высушенный.
Азот Р1. 1059400.
Содержит не менее 99,999 % (об/об) N 2 .
Уzлерода монооксид: не более 5 ррm.
Кислород: не более 5 ррm.
4.1.1. Реактивы
595
Азот для хроматоrрафии. 1059500.
Содержит не менее 99,95 % (об/об) N 2 .
Азот, свободный от кислорода. 1059600.
Азот Р очищают от кислорода пропускани
ем через раствор пиР02аллола щелочной Р.
Азота закись. Np. (М. м. 44,01). 1108500.
#Азота (1) оксид.#
Содержит не менее 99,99 % (об/об) Np.
Азота монооксид: не более 1 ррm.
У2лерода монооксид: не более 1 ррm.
Азота диоксид. N0 2 . (М м. 46,01). 1179600.
[10102--44 0]. #Азота (IV) оксид.#
Содержит не менее 98,0 % (об/об) N0 2 .
Азота монооксид. NO. (М м. ЗО,01).
1108300. #Азота (11) оксид.#
Содержит не менее 98,0 % (об/об) NO.
Азотная кислота. НNО з . (М м. 63,0).
1058400. [7697 З7 2J. #Азотная кислота KOHцeH
трированная.#
Содержит не менее 63,0 % (м/м) и не более
70,0 % (м/м) НNО з .
Прозрачная бесцве-тная или почти бесцвет
ная жидкость, смешивается с водой.
d;g: от 1,384 до 1,416.
Раствор 1 О r/л обладает сильнокислотными
свойствами и дает реакцию на нитраты (2.3.1)..
Прозрачность (2.2.1). Кислота азотная
должна быть прозрачной.
Цветность (2.2.2, метод 11). Окраска кисло
ты азотной должна быть не интенсивнее этало
на 'р 6'
Хлориды (2.4.4). Не более 0,00005 % (0,5 ррm).
К 5 r кислоты азотной прибавляют 1 О мл воды Р
и 0,3 мл раствора серебра нитрата Р2 и BЫ
держивают в защищенном от света месте в тече
ние 2 мин. Полученный раствор должен Bыдep
живать испытание на хлориды. Эталон roтовят с
использованием смеси из 13 мл воды Р, 0,5 мл
кислоты азотной Р, 0,5 мл этаЛОНН020 pacт
вора хлорида (5 ррт С/) Р и 0,3 мл раствора ce
ребра нитрата Р2.
Сульфаты (2.4.13). Не более 0,0002 % (2 ррm).
К 1 О r кислоты азотной прибавляют 0,2 r натрия
карбоната Р и выпаривают досуха; остаток paCT
воряют в 15 мл воды дистиллированной Р. Полу
ченный раствор должен выдерживать испытание
на сульфаты. Эталон roтовят с использованием
2 мл этаЛОНН020 раствора сульфата (10 ррт
SO,) Р и 13 мл воды дистиллированной Р.
Мышьяк (2.4.2, метод А). Не более
0,000002 % (0,02 ррm). К 50 r кислоты азотной
прибавляют 0,5 мл кислоты серной Р и осторож
но наrревают до появления белых паров. К OCTaT
ку прибавляют 1 мл раствора 100 r/л 2идpOKcи
ламина 2идрохлорида Р и доводят водой Р до
объема 2 мл. Полученный раствор должен BЫ
держивать испытание на мышьяк. Эталон roто
вят С использованием 1,0 мл эталОНН020 pacт
вора мышьяка (1 ррт As) Р.
Тяжелые металлы (2.4.8, метод А). Не
более 0,0002 % (2 ррт). 10 мл раствора, при
roтовленноrо для испытания на железо, дo
дят водой Р до объема 20 мл. 12 мл полученноro
раствора должны выдерживать испытание на ТЯ
желые металлы. Эталон rотовят с использовани
ем этаЛОНН020 раствора свинца (2 ррт РЬ) Р.
Железо (2.4.9). Не более 0,0001 % (1 ррm).
Осадок, полученный в испытании на сульфат
ную золу, растворяют в 1 мл кислоты хлористо
водородной разведенной Р и доводят водой Р до
объема 50 мл. 5 мл полученноro раствора ДOBO
дят водой Р до объема 10 мл. Полученный paCT
вор должен выдерживать испытание на железо.
Сульфатная зола. Не более 0,001 %. 100 r
кислоты азотной осторожно выпаривают досуха;
остаток смачивают несколькими каплями кислоты
серной Р и наrревают до бледно красноro цвета.
Количественное определение. К 1,50 r кис
лоты азотной прибавляют около 50 мл воды Р и
титруют 1 М раствором натрия 2идроксида, ис
пользуя в качестве индикатора 0,1 мл раствора
метилов020 краСН020 Р.
1 мл 1 М раствора натрия 2идроксида co
ответствует 63,0 Mr НNО з .
Хранят в защищенном от света месте.
Азотная кислота, свободная от свинца.
1058403.
Должна выдерживать испытания для Kиc
лоты азотной Р и следующие дополнительные
испытания.
Свинец. Не более 0,00001 % (0,1 ррm). ATOMHO
абсорбционная спектрометрия (2.2.23, метод /1).
К 100 r кислоты азотной Р прибавляют 0,1 r
натрия карбоната безводН020 Р и выпаривают
досуха; остаток растворяют в воде Р при слабом
наrревании и доводят до объема 50,0 мл paCT
вора этим же растворителем. Интенсивность по
rлощения измеряют при длине волны 283,3 нм
или 217,0 нм, используя в качестве источника
излучения лампу с полым свинцовым катодом и
воздушно ацетиленовое пламя.
Азотная кислота, свободная от свинца, Р1.
1058405.
Кислота азотная Р, содержащая не более
1 MKr/Kr свинца.
Азотная кислота, свободная от свинца,
разведенная. 1058406.
5 r кислоты азотной, свободной от
свинца, Р1 доводят водой дистиллированной
деионизироваНI-IОЙ Р до объема 100 мл.
Азотная кислота, свободная от свинца и
кадмия. 1058401.
Должна выдерживать испытания для Kиc
лоты азотной Р и следующие дополнительные
испытания.
596
rосударственная фармакопея Республики Беларусь
Испытуемый раствор. К 100 r кислоты
азотной Р прибавляют 0,1 r натрия карбона
та безводН020 Р, выпаривают досуха; остаток
растворяют в воде Р при слабом наrревании
и доводят до объема 50,0 мл этим же paCTBO
рителем.
КадмиЙ Не более 0,00001 % (0,1 ррm).
Атомно абсорбционная спектрометрия (2.2.23,
метод 11). Интенсивность поrлощения измеряют
при длине волны 228,8 нм, используя в качестве
источника излучения лампу с полым кадмиевым
катодом и воздушно ацетиленовое или воздуш
но пропановое пламя.
Свинец. Не более 0,00001 % (0,1 ррm).
Атомно абсорбционная спектрометрия (2.2.23,
метод 11). Интенсивность поrлощения измеряют
при длине волны 283,3 нм или 217,0 нм, исполь
зуя лампу с полым свинцовым катодом и воз
душно ацетиленовое пламя.
Азотная кислота, свободная от тяжелых
металлов. 1058404.
Должна выдерживать требования для Kиc
лоты азотной Р и следующие дополнительные
испытания:
As: не более 0,005 ррm;
Cd: не более 0,005 ррт;
Cu: не более 0,001 ррm;
Fe: не более 0,02 ррт;
Hg: не более 0,002 ррт;
Ni: не более 0,005 ррт;
РЬ: не более 0,001 ррт;
Zп: не более 0,01 ррт.
Азотная кислота дымящаяся. 1058500.
[52583A2 3].
Прозрачная жидкость слеrка желтоватоro
цвета, дымящаяся на воздухе.
d;g: около 1,5.
Азотная кислота разведенная. 1058402.
Содержит около 125 r/л НNО з (М м.. 63,0).
20 r кислоты азотной Р доводят водой Р до
объема 100 мл.
# Азотная кислота разведенная Р1.
Смешивают 1 часть кислоты азотной Р и
1 часть воды дистиллированной Р. Плотность
1,18б 1,210 rlcм З . Содержит не менее 31 %
(м/м) и не более 34 % (м/м) НNО з .
# Азотная кислота разведенная Р2.
Смешивают 1 часть кислоты азотной разве
денной Р1 и 1 часть воды дистиллированной Р.
Плотность 1 ,087 1,096 r/CM3. Содержит не
менее 15,5 % (м/м) и не более 17,0 % (м/м) НNО з .
Акриламид. С з Н 5 NО. (М м. 71,1). 1001500.
[79 06 1]. Пропенамид.
Бесцветные или белые хлопья, либо белый
или почти белый кристаллический порошок.
Очень леrко растворим в воде и в метаноле,
леrкорастворим в этаноле.
Температура плавления: около 84 ОС.
Акриламид бисакриламида (29:1) 30 %
раствор. 1001501.
290 r акриламида Р и 1 О r метиленбисакри
ламида Р растворяют в 1 л воды Р и фильтруют.
Акриламид-бисакриламида (36,5:1) 30 %
раствор. 1001502.
292 r акриламида Р и 8 r метиленбисакри
ламида Р растворяют в 1 л воды Р и фильтруют.
Акриловая кислота. С з Н 4 О 2 . (М м. 72,1).
1133700. [79 10 7]. Проп 2 еновая кислота. Ви
нилмуравьиная кислота.
Содержит не менее 99 % С з Н 4 О 2 . Стабили
зирована 0,02 % монометиловоro эфира rидро
хинона.
Едкая жидкость. Смешивается с водой и
96 % спиртом. Леrко полимеризуется в присут
ствии кислорода.
d;g: около 1,05.
20
по: около 1,421.
Температура кипения: около 141 ОС.
Температура плавления: от 12 ос д015 ОС.
Актеозид. С 29 Н З РlS' (М м. 624,6). 1145100.
[61276 17 З]. 2 (З,4 Диrидроксифенил )этил з о
(6 дезокси а l ма н ноп и ранозил ) 4 o [ (2E)
3 (3 ,4 ди rидроксифен ил )проп 2 еноил H3 D
rлюкопиранозид. Вербаскозид.
Светло желтый порошок. Леrкорастворим в
воде и в метаноле.
Температура плавления: около 140 ос с раз
ложением.
Аланин. С з Н 7 N0 2 . (М м. 89,1). 1102900. [5641 7].
(S) 2 Аминопропионовая кислота.
Содержит не менее 98,5 % и не более
101,0 % С з Н 7 N0 2 В пересчете на сухое вещество.
Белый или почти белый кристаллический по
рошок или бесцветные кристаллы. Леrкораство
рим в воде, очень мало растворим в 96 % спирте
и практически нерастворим в эфире.
Хранят в защищенном от света месте.
I3-Аланин. 1004500. [107 95 9]. См. 3 AMи
нопропионовая кислота Р.
Алеуриновая кислота. С 16 Н З Р5' (М м. 304,4).
1095700. [533 87 9]. (9RS, 1 OSR) 9, 1 О, 16 Триrидро
ксиrексадекановая кислота.
Белый I-1ЛИ почти белый порошок, жирный на
ощупь. Растворима в метаноле.
Температура плавления: около 101 ОС.
Ализарин S. C 14 H 7 Na0 7 S'Hp. (М м. 360,3).
1002600. [130 22 3]. Показатель Шульца
NQ 1145. Цветной индекс NQ 58005. Натрия
4.1.1. Реактивы
597
1 ,2 диrидроксиантрахинон 3 сульфонат моноrид
рат. Натрия 3,4 диrидрокси 9, 1 0 диоксcr9.1 О диrид
роантрацен 2 ульфонат моноrидрат. #Ализари
новый красный С.#
Оранжево желтый порошок. Леrкораство
рим в воде и в 96 % спирте.
Ализарина S раствор. 1002601.
Раствор 1 r/л ализарина S Р.
Испытание на чувствительность. Реактив
изменяет окраску от желтой до оранжево крас
ной при установлении титра 0,05 М раствора
бария перхлората (4.2.2).
Изменение окраски. От рН 3,7 (желтое OKpa
шивание) до рН 5,2 (фиолетовое окрашивание).
# Ализариновый желтый Р. См. Протрав
ной желтый 3R Р.
# Ализариновый красный с. См. Ализа
рин S Р.
Альбумин бычий. 1002300. [9048A6 8].
Альбумин бычий сывороточный. Содержит
около 96 % белка.
Порошок от белоro до светлоro желтовато
коричневоro цвета.
Вода (2.5.12). Не более 3,0 %. Определение
проводят из 0,800 r испытуемоro образца.
Альбумин бычий, используемый при коли
чественном определении тетракозактида,
должен быть апиР02енным и не должен про
являть протеолитическую активность при
определении соответствующими методами,
например, при использовании ХРОМ02еНН020
субстрата, а также обладать KopтиKocтepo
идной активностью, определяемой измерени
ем флуоресценции, как указано в статье Te
тракозактид в количественном определении
биОЛ02ической активности.
Альбумин человеческий. 1133800.
Альбумин сыворотки человека, содержащий
не менее 96 % альбумина.
Альбумина человеческоrо раствор.
1002400. [9048 46 8].
Водный раствор белка. полученноro из
плазмы крови человека.
Прозрачная, слеrка вязкая бесцветная,
желтая или зеленая жидкость.
Альбумина человеческоrо раствор Р1.
1002401.
Альбумина человечеСК020 раствор Р разво
дят р8СТВОрОМ 9 r/л натрия хлорида Р до KOH
центрации белка 1 r/л. Доводят кислотой YKCYC
ной ледяной Р до рН 3,5------4,5.
Альдеrиддеrидроrеназа. 1103000.
Фермент, полученный из хлебопекарских
дрожжей, окисляет ацетальдеrид до кислоты YK
сусной В присутствии никотинамид аденина ди
нуклеотида, солей калия и тиолов при рН 8.0.
Альдеrиддеrидроrеназы раствор.
1103001.
Количество альде2идде2идР02еназы Р, co
ответствующее 70 единицам, растворяют в
воде Р и доводят до объема 10 мл этим же paCT
ворителем. Раствор стабилен в течение 8 ч при
температуре 4 ОС.
# Алоrен. См. Барбалоин Р.
Алюминий. AI. (А. м. 26,98). 1118200.
[7429 90 5].
Мяrкий, ковкий металл белоro с roлубова
тым оттенком цвета в виде брусков, листов, по
рошка, ленты или проволоки. На воздухе образу
ется окисная пленка, которая защищает металл
от коррозии.
Аналитической чистоты.
Алюминия калиясульфат.АIК(S04)2 '12Нр.
(м. м. 474,4). 1003000. [7784 24 9]. Квасцы.
rранулированный порошок или бесцветная
прозрачная кристаллическая масса. Леrкорас
творим в воде, очень леrко растворим в кипящей
воде, растворим в rлицерине и практически He
растворим в 96 % спирте.
Содержит 99,0 % 100,5 % AIK(SO 4)2 '12Нр.
Алюминия нитрат. АI(NОз)з '9Нр. (м. м. 375,1).
1002800. [7784 27 2]. Алюминия нитрат нонаrи
драт.
Кристаллы, расплывающиеся на воздухе.
Очень леrко растворим в воде и в 96 % спирте,
очень мало растворим в ацетоне.
Хранят в воздухонепроницаемом контейнере.
Алюминия оксид безводный. 1002900.
[1344 28 1].
Алюминия оксид, состоящий из у АI20з'
обезвоженный и активированный HarpeBoM.
Размер частиц: от 75 мкм до 150 мкм.
Алюминия оксид основный. 1118300.
Алюминия оксид безводный Р основной
формы приroден для хроматоrрафических коло
нок.
рН (2.2.3). От 9 до 10. Измеряют рН суспен
зии, полученной при встряхивании 1 r испыту
емоro образца с 1 О мл воды, свободной от У2ле
рода диоксида, Р в течение 5 мин.
Алюминия оксид нейтральный. 1118400.
См. статью Алюминия оксид 2идpaтиpoвaH
ный.
Алюминия хлорид. АIСl з -6Нр. (м. м. 241.4).
1002700. [7784 13 6]. Алюминия хлорид rексаrи
драт.
598
rосударственная фармакопея Республики Беларусь
Содержит не менее 98,0 % АIСl з '6Н 2 О.
Кристаллический порошок от белоro до
слеrка желтоватоro цвета, rиrроскопичен. Леrко
растворим в воде и в 96 % спирте, растворим в
эфире.
Хранят в воздухонепроницаемом контейнере.
АлЮМИНИЯ хлорида раствор. 1002701.
65,0 r алюминия хлорида Р растворяют в воде Р
и доводят до объема 100 мл этим же растворит
лем. Прибавляют 0,5 2 У2Ля активироваНН020 Р,
перемешивают в течение 1 О мин и фильтруют. К
фильтрату при непрерывном перемешивании при
бавляют достаточное количество раствора 10 r/л
натрия 2идроксида Р (около 60 мл) до рН около 1,5.
Алюминия хлорида реактив. 1002702.
2,0 2 алюминия хлорида Р растворяют в
100 мл раствора 5 % (об/об) кислоты уксусной
ледяной Р в метаноле Р.
Амидcrчeрный 108. C22H14N6NapgS2' (М м.617).
1003100. [1064 48 8]. Показатель Шульца NQ 299.
Цветной индекс NQ 20470. Динатрия 5 амино
4 rидрокси 6 [( 4 нитрофенил )азо] 3 ( фенилазо)
нафталин 2, 7 дисульфонат.
Порошок от темно коричневоro до черноro
цвета. Умеренно растворим в воде, растворим в
96 % спирте.
Амидо-черноrо 10В раствор. 1003101.
Раствор 5 r/л амидо чеРН020 10В Р в смеси
кислота уксусная Р метанол Р (10:90, об/об).
а-Амилаза. 11 00800. 1 ,4 а D rлюкан rлюка
ноrидролаза. (ЕС 3.2.1.1).
Порошок от белою до светло коричневоro
цвета.
а-Амилазы раствор. 1100801.
Раствор а амилазы Р с активностью
800 ФАЕ (франко американских единиц)/r.
# Амилацетат. С 6 Н 14 С0 2 . (м. М. 130,19).
Амиловый эфир уксусной кислоты. Бесцветная
прозрачная жидкость с фруктовым запахом.
# н-Амиловый спирт. См, Пентанол Р.
# трет-Амиловый спирт. См, трет-
Пентиловый спирт Р.
Аминоазобензол. С12Н11Nз. (м. м. 197,2).
1003200. [60 09 3]. Цветной индекс NQ 11000.
4-(Фенилазо )анилин.
Иrол чатые кристаллы коричневато желтоro
с roлубоватым опенком цвета. Малорастворим
в воде, леrкорастворим в 96 % спирте.
Температура плавления: около 128 ОС.
N-(4-Аминобензоил)-L-f'лутаминовая кислота.
C 12 H 14 NP5' (м. М. 266,3). 1141700. [4271 3(} 1]. АБrк.
Белый или почти белый кристалличecкиi
порошок.
Температура"плавления: около 175 ос с p
ложением.
2-Аминобензойная кислота. C 7 H 7 N0 2 .
(м. М. 137,1). 1003400. [118 92 3]. Антраниловая
кислота.
Кристаллический порошок от белоro до
бледно-желтоrо цвета. Умеренно растворима в
холодной воде, леrкорастворима в rорячей воде.
в 96 % спирте и rлицерине. Растворы в 96 %
спирте или эфире и в особенности в rлицерине
обладают фиолетовой флуоресценцией.
Температура плавления: около 145 ОС.
3-Аминобензойная кислота. C 7 H 7 N0 2 .
(м. М. 137,1). 1147400. [99-05 8].
Белые или почти белые кристаллы. Водные
растворы при стоянии на воздухе приобретают
коричневый цвет.
Температура плавления: около 174 ОС.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
4-Аминобензойная кислота. C 7 H 7 N02"
(м. М. 137,1). 1003300. [150 13 O].
Белый или почти белый кристаллический
порошок. Малорастворима в воде, леrкораство-
рима в 96 % спирте, практически нерастворима
в петролейном эфире.
Температура плавления: около 187 ОС.
Хромат02рафия. Тонкослойная xpOMaTO
rрафия (2.2.27), как указано в статье Прокаина
2идрохлорид. На xpoMaTorpaMMe должно обна
руживаться только одно основное пятно.
Хранят в защищенном от света месте.
4-Аминобензойной кислоты раствор.
1003301.
1 r кислоты 4 аминобензойной Р paCTBO
ряют в смеси из 18 мл кислоты уксусной безвод
ной Р, 20 мл воды Р и 1 мл кислоты фосфор
ной Р. Непосредственно перед использованием
смешивают 2 объема полученною раствора с 3
объемами ацетона Р (2:3).
4-Амино6у1анова кисnoта. C/\N0 2 . (М м. 103,1).
1123200. [56-12 2]. у Аминомасляная кислота.
rAMK. #Аминалон#.
При перекристаллизации из метанола и
эфира представляет собой пластинки, из воды и
96 % спирта иroльчатые кристаллы. Леrкорас
творима в воде, практически нерастворима или
малорастворима в друrих растворителях.
ТемпеРепура плавления: около 202 ос (YBe
личивается при высокой скорости HarpeBa).
Аминобутанол. C 4 H 11 NO. (м. м. 89,1).
1003500. [5856 63 3]. 2-Аминобутанол.
Маслянистая жидкость. Смешивается с
водой, растворим в 96 % спирте.
4.1.1. Реактивы
599
d;g: около 0,94.
20
п D : около 1,453
Температура кипения: около 180 ОС.
б-Аминоrексановая кислота. С 6 Н 1з N0 2 .
(м. м. 131,2). 1103100. [60 32 2]. 6 Аминокапро
новая кислота.
Бесцветные кристаллы. Леrкорастворима в
воде, умеренно растворима в метаноле, практи
чески нерастворима в этаноле.
Температура плавления: около 205 ОС.
Аминоrидроксинафталенсульфоновая
кислота.С 1О Н g N0 4 S.(м.м.239,3). 1112400. [116 З--2].
4 амино З rидрокси нафтален 1 сул ьфоно
вая кислота.
Белые или серые иroльчатые кристаллы,
под действием света приобретают розовый цвет,
особенно в присутствии влаrи. Практически He
растворима в воде и в 96 % спирте, растворима
в растворах rидроксидов щелочных металлов и
rорячих растворах натрия метабисульфита.
Хранят в защищенном от света месте.
Аминоrидроксинафталинсульфоновой
кислоты раствор. 1112401.
Смешивают 5,0 r натрия сульфита безводно
20 Р, 94,3 r натрия 2идросульфита Р и 0,7 r кислcr
ты амиН02идроксинафталинсульфоновой Р. 1,5 r
полученной смеси растворяют в ваде Р и доводят
объем раствора тем же растворителем до 10,0 мл.
Срок rодности раствора 1 сут.
Аминоrиппуровая кислота. С g Н,оN 2 О з .
(м. м. 194,2). 1003700. (61 78 9]. (4 Аминобенза
мидо )уксусная кислота.
Белый или почти белый порошок. Умеренно
растворима в воде, растворима в 96 % спирте.
Температура плавления: около 200 ОС.
Аминоrиппуровой кислоты реактив.
1003701.
3 r кислоты фталевой Р и 0,3 r кислоты aMUHcr
2иппуровой Р растворяют в 96 % спирте Р и доводят
до объема раствора 100 мл этим же растворитепем.
# б Аминокапроновая кислота. См. 6 AMи
Н02ексановая кислота Р.
Аминометилализариндиуксусная кислота.
C'9H'5NOa'2Hp. (м. м. 421,4). 1003900. (3952 78 1].
2,2' [(3,4 Диrидроксиантрахинон З ил )метилен
нитрило]диуксусная кислота диrидрат.
Мелкокристаллический порошок от светлою Kcr
ричнеВCIто желтоro до оранжевcrкоричневоro цвета.
Практически нерастворима в воде, растворима в
растворах rидроксидов щелочных металлов.
Температура плавления: около 185 ОС.
Потеря в массе при высушивании (2.2.32).
Не более 10,0 %. Определение проводят из
1,000 r испытуемоro образца.
Аминометилализариндиуксусной кисло-
тыраствор. 1003902
0,192 r кислоты аминометилализарин
диуксусной Р растворяют в 6 мл свежеприro
товленноrо 1 М раствора натрия 2идроксида,
прибавляют 750 мл воды Р, 25 мл CYKциHaтHO
20 буферН020 раствора рН 4,6 Р и по каплям
0,5 М раствор кислоты хлористоводородной
до изменения окраски раствора от фиолетово
красной до желтой (рН от 4,5 до 5). Прибавляют
100 мл ацетона Р и доводят водой Р до объема
1000 мл.
Аминометилализариндиуксусной кисло-
ты реактив. 1003901.
Раствор А. 0,36 r церия (1/1) нитрата Р
растворяют в воде Р и доводят до объема 50 мл
этим же растворителем.
Раствор В. 0,7 r кислоты аминометилали
зариндиуксусной Р суспендируют в 50 мл воды Р,
прибавляют до растворения около 0,25 мл pacт
вора аммиака концентрироваНН020 Р, затем
прибавляют 0,25 мл кислоты уксусной ледя
ной Р и доводят водой Р до объема 100 мл.
Раствор С. 6 r натрия ацетата Р paCTBO
ряют в 50 мл воды Р, прибавляют 11,5 мл Kиc
лоты уксусной ледяной Р и доводят водой Р до
объема 100 мл.
К 33 мл ацетона Р прибавляют 6,8 мл paCT
вора С, 1,0 мл раствора В, 1,0 мл раствора А и
доводят водой Р до объема 50 мл.
Испытание на чувствительность.
К 1,0 мл этаЛОНН020 раствора фторида
(10 ррт F) Р прибавляют 19,0 мл воды Р и
5,0 мл реактива аминометилализариндиуксус
ной кислоты. Через 20 мин должно появиться
roлубое окрашивание.
Срок roдности раствора 5 сут.
Аминонитробензофенон. С 1з Н,оN 2 О з .
(м. м. 242,2). 1 004000. (1775 95 7].2 Амино 5 нитpcr
бензофенон.
Желтый кристаллический порошок. Практи
чески нерастворим в воде, растворим в TeTpa
rидрофуране, малорастворим в метаноле.
Температура плавления: около 160 ОС.
A; : от 690 до 720 при длине волны 233 нм.
Определение проводят с использованием paCT
вора 0,01 r/л аминонитробензофенона в Meтa
ноле Р.
Аминопиразолон. С"Н,зNр. (М м. 203,2).
1004600. [83 07 8]. 4 Амино 2,3 диметил 1--фенил
пиразолин 5 он.
Светло желтые иroльчатые кристаллы или
порошок. Умеренно растворим в воде, леrкорас
творим в 96 % спирте.
Температура плавления: около 108 ОС.
Аминопиразолона раствор. 1004601.
Раствор 1 r/л аминопиразолона Р в буферном
растворе рН 9, О Р.
600
rосударственная фармакопея Республики Беларусь
Аминополиэфир. С 1в Н з6 NР6' (М. м. 376,5).
1112500. [23978 09 8]. 4,7,13, 16,21 ,24 reKcaoKca
1, 1 0 диазабицикло[8,8,8]rексакозан.
Температура плавления: от 70 ос до 73 ОС.
З-Аминопропанол. С з Н 9 NО. (М. м. 75,1).
1004400. [156--87 ].З Аминопропан 1--ол. Пропано
ламин.
Прозрачная бесцветная вязкая жидкость.
d;g: около 0,99
20
п о : около 1,461
Температура плавления: около 11 ос.
З:"Аминопропионовая кислота. С з Н 7 NО 2 .
(М. м. 89,1). 1004500. [107 95 9]. J3 Аланин.
Содержит не менее 99 % С з Н 7 N0 2 .
Белый или почти белый кристаллический
порошок. Леrкорастворима в воде, малораство
рима в 96 % спирте, практически нерастворима
в ацетоне.
Температура плавления: около 200 ос с раз
ложением.
2-Аминоуксусная кислота.
См. статью rлицин.
2-Аминофенол. C 6 H 7 NO. (м. м. 109,1).
1147500. [95 55 6].
Светло желтовато коричневые кристал
лы, которые быстро становятся коричневыми.
Умеренно растворим в воде, растворим в 96 %
спирте.
Температура плавления: около 172 ОС.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
З-Аминофенол. C 6 H 7 NO. (м. м. 109,1).
1147600. [591 27 5].
Светло желтовато коричневые кристаллы.
Умеренно растворим в воде.
Температура плавления: около 122 ОС.
4-Аминофенол. C 6 H 7 NO. (М м. 109,1).
1004300. [123 30 8].
Белый или слеrка окрашенный кристалли
ческий порошок, под действием воздуха и света
приобретает окраску. Умеренно растворим в
воде, растворим в этаноле.
Температура плавления: около 186 ос с раз
ложением.
Хранят в защищенном от света месте.
Аминоxnopбeнэoфeнoн. С 1З Н1QСINO. (М м. 231,7).
1 003600. [719 59 5]. 2 Амино 5 хлор бензофе
нон.
Желтый кристаллический порошок. Прак
тически нерастворим в воде, леrкорастворим в
ацетоне, растворим в 96 % спирте.
Температура плавления: около 97 ОС.
Содержит не менее 95,0 % С 1з Н 1О СINО.
Хранят в защищенном от света месте.
# Аммиака водно-спиртовой раствор.
1 мл раствора аммиака концентрироваННО20 Р
смешивают с 9 мл 96 % спирта Р.
Аммиака раствор. 1004701.
Содержит не менее 170 r/л и не более 180 r/л
NН з (М. м.. 17,03).
67 r раствора аммиака концентрированно--
20 Р доводят водой Р до объема 100 мл.
d;g: от 0,931 до 0,934.
Аммиака раствор Р, используемый в испы
тании на предельное содержание железа, должен
выдерживать следующее дополнительное требо
вание: 5 мл раствора аммиака Р выпаривают на
водяной бане досуха. К остатку прибавляют 1 О мл
воды Р, 2 мл раствора 200 r/л кислоты лимон
ной Р, 0,1 мл кислоты ти02ликолевой Р и раст---
вора аммиака Р до щелочной реакции. Доводят
объем полученноro раствора водой Р до 20 мл.
Раствор не должен окрашиваться в розовый цвет.
Хранят с защитой от уrлерода диоксида при
температуре не выше 20 ОС.
Аммиака раствор концентрированный.
1004700.
См. статью Аммиака раствор KOHцeHтpиpo
ванный.
Аммиака раствор концентрированный Р1.
1004800.
Содержит не менее 32,0 % (м/м) NН з
(М м. 17,03).
Прозрачная бесцветная жидкость.
d;g: от 0,883 до 0,889.
Количественное определение. 50,0 мл 1 М
раствора кислоты хлористоводородной поме
щают в колбу с притертой пробкой, точно взве
шивают, прибавляют 2 мл раствора аммиака
концентрированноro и снова взвешивают. Титру
ют 1 М раствором натрия 2идроксида, исполь
зуя в качестве индикатора 0,5 мл смешаНН020
раствора метилов020 краСН020 Р.
1 мл 1 М раствора кислоты хлористоводо
родной соответствует 17,03 Mr NH3.
Хранят с защитой от уrлерода диоксида при
температуре не выше 20 ос.
Аммиака раствор разведенный Р1.
1004702.
Содержит не менее 100 r/л и не более 104 r/л
NН з (м. м. 17,03).
41 r раствора аммиака KOHцeHтpиpoвaHHO
20 Р доводят водой Р до объема 100 мл.
Аммиака раствор разведенный Р2.
1004703.
Содержит не менее 33 r/л и не более 35 r/л
NН з (м. м. 17,03).
14 r раствора аммиака KOHцeHтpupoвaHHO
20 Р доводят водой Р до объема 100 мл.
4.1.1. Реактивы
601
Аммиака раствор разведенный РЗ. 1004704.
Содержит не менее 1,6 r/л и не более 1,8 r/л
NН з (М м. 17,03).
0,7 r раствора аммиака KOHцeHтpиpoвaHHO
20 Р доводят водой Р до объема 100 мл.
# Аммоний пурпуровокислый. См. MypeK
сидР.
Аммония ацетат. C 2 H 7 N0 2 . (М М. 77,1).
1004900. [6З1 61 8].
Бесцветные кристаллы, очень леrко pac
плывающиеся на воздухе. Очень леrко paCTBO
рим в воде и в 96 % спирте.
Хранят в воздухонепроницаемом контейнере.
Аммония ацетата раствор. 1004901.
150 r аммония ацетата Р растворяют в
воде Р, прибавляют 3 мл кислоты уксусной ле
дяной Р и доводят водой Р до объема 1000 мл.
Срок roдности 7 сут.
Аммония ванадат. NН 4 VО з . (М М. 117,0).
1006800. [7803 55 6]. Аммония триоксо вана
дат (V).
Кристаллический порошок от белоro до
слеrка желтоватоro цвета. Малорастворим в воде,
растворим в растворе аммиака разведенном Р1.
Аммония ванадата раствор. 1006801.
1,2 r аммония ванадата Р растворяют в
95 мл воды Р и доводят кислотой серной Р до
объема 100 мл.
# Аммония ванадата раствор Р1.
0,05 r аммония ванадата Р растворяют в
1 О мл кислоты серной Р.
Аммония rексафторrерманат(IV). (NH 4 )2 GeF б'
(М м. 222,7). 1134000. (16962-47 3].
Белые или почти белые кристаллы. Леrко
растворим в воде.
Аммония rидрокарбонат. NН 4 НСО з .
(М М. 79,1). 1005500. [1066 33 7].
Содержит не менее 99 % NН 4 НСО з .
Аммония диrидрофосфат. (NH 4 )H 2 P0 4 .
(М М. 115,0). 1005400. (7722 76 1]. Аммония
фосфат однозамещенный.
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Леrкорас
творим в воде.
рН (2.2.3). Около 4,2. Измеряют рН раствора
23 r/л испытуемою образца.
(1 RН ) Аммония 1 О камфорсульфонат.
C'OH,gN0 4 S. (М М. 249,3). 1103200.
Содержит не менее 97,0 % (1RН+Аммония
10 камфоросульфоната.
[a] O: 18:!:2. Определение проводят, исполь
зуя раствор 50 r/л испытуемоro образца.
Аммония карбамат. СН Б NР2' (М М. 78,1).
1168400. [1111 78 0]. Аммониевая соль карбами
новой кислоты.
Аммония карбонат. 1005200. [506 87 6].
Смесь аммония rидрокарбоната (NН 4 НСО з ,
М М. 79,1) и аммония карбамата (NH 2 COONH 4 ,
М М. 78,1) в различных количественных COOTHO
шениях.
Белая или почти белая полупрозрачная
масса. Медленно растворим примерно в четы
рех частях воды. Разлаrается в кипящей воде.
Аммония карбонат выделяет не менее 30 % (мlM)
NН з (М м. 17,03).
Количественное определение. 2,00 r испы
туемоro образца растворяют в 25 мл воды Р,
медленно прибавляют 50,0 мл 1 М раствора
кислоты хлористоводородной и титруют 1 М
раствором натрия 2идроксида, используя в Ka
честве индикатора 0,1 мл раствора метилово
20 оранжев020 Р
1 мл 1 М раствора кислоты хлористоводо
родной соответствует 17,03 Mr NН з .
Хранят при температуре не выше 20 ОС.
Аммония карбоната раствор. 1005201.
Раствор 158 r/л аммония карбоната Р.
# Аммония карбонат Р1. Аммоний уrлекис
лый. (NН4)2СОз' (М. М. 96,09).
Бесцветные мелкие кристаллы, в массе
белоro цвета.
# Аммония карбоната раствор Р1.
1 О r аммония карбоната Р1 растворяют в
30 мл воды Р, прибавляют 1 О мл аммиака pacт
вора Р1 и доводят водой Р до объема 100 мл.
Аммония молибдат. (NН4)БМ07024-4Н20,
(М М. 1236). 1005700. [12054 85 2].
Бесцветные кристаллы или кристаллы от
слеrка желтоватоro до зеленоватою цвета. Pac
творим в воде, практически нерастворим в 96 %
спирте.
Аммония молибдата раствор. 1005702.
Раствор 100 r/л аммония молибдата Р.
#Хранят в контейнерах оранжевою стекла.
Срок roдности 6 мес.#
Аммония молибдата раствор Р2. 1005703.
5,0 r аммония молибдата Р растворяют
при наrревании в 30 мл воды Р, охлаждают,
доводят раствором аммиака разведенным Р2
до рН 7,0 и разводят водой Р до объема 50 мл.
Аммония молибдата раствор РЗ. 1005704.
Раствор А. 5 r аммония молибдата Р раст
воряют при наrревании в 20 мл воды Р
Раствор В. Смешивают 150 мл 96 %
спирта Р со 150 мл воды Р При охлаждении
прибавляют 100 мл кислоты серной Р
602
rосударственная фармакопея Республики Беларусь
Непосредственно перед использованием к
20 объемам раствора А прибавляют 80 объемов
раствора В.
Аммония молибдата раствор Р4. 1005705.
1,0 r аммония молибдата Р растворяют в
воде Р, доводят до объема 40 мл этим же раст
ворителем, прибавляют 3 мл кислоты хлори
стоводородной Р, 5 мл кислоты хлорной Р и дo
водят ацетоном Р до объема 100 мл.
Хранят в защищенном от света месте. Срок
roдности 1 мес.
Аммония молибдата раствор Р5. 1005706.
1,0 r аммония молибдата Р растворяют в
40,0 мл 15 % раствора (об/об) кислоты серной Р.
Срок roдности 1 сут.
# Аммония молибдата раствор в серной
кислоте. Реактив Фреде.
0,1 r аммония молибдата Р растворяют в
1 О мл кислоты серной Р.
Хранят в контейнерах оранжевоro стекла.
Срок roдности 6 мес.
# Аммония молибдата раствор в азотной
кислоте.
6,5 r мелко раздробленной кислоты MO
либденовой Р растворяют в смеси из 14 мл
воды Р и 14,5 мл раствора аммиака KOHцeH
трироваНН020 Р. Раствор охлаждают, по
степенно при перемешивании прибавляют к
смеси 32 мл охлажденноro раствора кислоты
азотной Р и 40 мл воды Р и выдерживают в
течение 48 ч. Полученный раствор фильтру
ют через плотный фильтр. Если при хранении
раствора выделяется осадок, ero отделяют
декантацией.
Аммония молибдата реактив. 1005701.
Последовательно смешивают 1 объем paCT
вора 25 r/л аммония молибдата Р, 1 объем paCT
вора 100 r/л кислоты аскорбиновой Р, 1 объем
кислоты серной Р (294,5 r/л H 2 S0 4 ) и прибавля
ют 2 объема воды Р
Срок roдности 1 сут.
Аммония молибдата реактив Р1. 1005706.
Смешивают 10 мл раствора 60 r/л динатрия
арсената Р, 50 мл раствора аммония молибда
та Р, 90 мл кислоты серной разведенной Р и
доводят водой Р до объема 200 мл.
Смесь выдерживают при температуре 37 ос
в течение 24 ч.
Хранят во флаконах оранжевоrо стекла.
Аммония молибдата реактив Р2. 1005708.
50 r аммония молибдата Р растворяют в
600 мл воды Р. К 250 мл холодной воды Р при
бавляют 150 мл кислоты серной Р и охлаждают.
Полученные растворы смешивают.
Срок roдности 1 сут.
Аммония нитрат. NН 4 NО з . (М. м. 80,0).
1005800. [6484 52 2].
Кристаллический порошок белоro цвета или
бесцветные кристаллы. rиrроскопичен, очень
леrкорастворим в воде, леrкорастворим в MeTa
ноле, растворим в 96 % спирте.
Хранят в воздухонепроницаемом контейнере.
Аммония нитрат Р1. 1005801.
Должен выдерживать требования для aMMO
ния нитрата Р и следующие дополнительные
испытания.
Кислотность (2.2.4). Раствор должен иметь
слабо кислую реакцию.
Хлориды (2.4.4). Не более 0,01 % (100 ррm).
0,50 r должны выдерживать испытание на хло
риды.
Сульфаты (2.4.13). Не более 0,015 % (150 ррm).
1,0 r должен выдерживать испытание на сульфа
ты.
Сульфатная зола (2.4.14). Не более 0,05 %.
Определение проводят из 1,0 r.
Аммония оксалат. C2HaNP4'Hp. (М М. 142,1).
1005900. [6009 70 7].
Бесцветные кристаллы. Растворим в воде.
Аммония оксалата раствор. 1005901.
Раствор 40 r/л аммония оксалата Р. #4 r aM
мония оксалата Р растворяют при наrревании в
воде Р, доводят до объема 100 мл этим же раст
ворителем и фильтруют.#
Аммония персульфат. (NH4)2SPB' (М М. 228,2).
1006000. [7727 54 0].
Белый или почти белый кристаллический
порошок или rранулы. Леrкорастворим в воде.
Аммония пирролидиндитиокарбама
C5H12N2S2' (М. М. 164,3). 1006200. [5108 96 3].
Аммония 1 пирролидинилдитиоформиат.
Кристаллический порошок от белоro до свет
ло желтоrо цвета. Умеренно растворим в воде,
очень мало растворим в 96 % спирте.
Хранят в контейнере, содержащем неболь
шое количество аммония карбоната в полотня
ном мешочке.
Аммониярейнекат. NH 4 [cr(NCSMNH)JHp.
(М М. 354.4). 1006300. [13573 16 5]. Аммония
диаминтетракис(изотиоцианато)хромат (111) MOHO
rидрат.
Красный порошок или кристаллы. Умеренно
растворим в холодной воде, растворим в roря
чей воде и в 96 % спирте. #В водном растворе
rазлаrается с выделением свободноro циани
стоro водорода (осторожно!).#
Аммония рейнеката раствор. 1006301.
Раствор 1 О r/л аммония рейнеката Р.
rотовят непосредственно перед использо
ванием.
4.1.1. Реактивы
603
# Аммония роданид. См. Аммония тиoци
анат Р
Аммония сульфамат. NН 2 SО з NН 4 . (м. М. 114,1).
1 006400. [7773 06 0].
Белый или почти белый кристаллический
noрошок или бесцветные кристаллы. rиrроско
nичен. Очень леrко растворим в воде, малораст
варим в 96 % спирте.
Температура плавления: около 130 ОС.
Хранят в воздухонепроницаемом контейнере.
Аммония сульфат. (NH4)2S04" (м. М. 132,1).
1006500. [7783 20 2].
Бесцветные кристаллы или белые или почти
белые rpанулы. Очень леrко растворим в воде,
практически нерастворим в ацетоне и в 96 % спирте.
рН (2.2.3). От 4,5 до 6,0. Измеряют рН pacт
вора 50 r/л испытуемоro образца в воде, свобод
ной от У2лерода диоксида, Р
Сульфатная зола (2.4.14). Не более 0,1 %.
Аммония тиоцианат. NH 4 SCN. (М м. 76,1).
1006700. [1762 95 4]. #Аммония роданид.#
Бесцветные кристаллы, расплывающиеся
на воздухе. Очень леrко растворим в воде, pac
творим в 96 % спирте.
Хранят в воздухонепроницаемом контейнере.
Аммония тиоцианата раствор. 1006701.
Раствор 76 r/л аммония тиоцианата Р.
Аммония формиат. CH 5 N0 2 . (М М. 63,1).
1112600. [540 69 2].
Расплывающиеся кристаллы или rpaHY
лы. Очень леrко растворим в воде, растворим в
96 % спирте.
Температура плавления: от 119 ОС до 121 ОС.
Хранят в воздухонепроницаемом контейнере.
Аммония фосфат. (NH4)2HPO 4' (М м. 132,1).
1006100. [7783 28 0]. Диаммония rидрофосфат.
Белые или почти белые кристаллы или rpa
нулы. rиrроскопичен. Очень леrко растворим в
воде, практически нерастворим в 96 % спирте.
рН (2.2.3). Около 8. Измеряют рН раствора
200 r/л испытуемоro образца. Хранят в воздухо
непроницаемом контейнере.
Аммония хлорид. 1005300. [12125 02 9].
См. статью Аммония хлорид.
Аммония хлорида раствор. 1005301.
Раствор 107 r/л аммония хлорида Р.
Аммония цитрат. C 6 H 14 N 2 0]" (М М. 226,2).
1103300. [З012 65 5]. Диаммония rидроцитрат.
Белый или почти белый кристаллический
noрошок или бесцветные кристаллы. Леrкорас
творим в воде, малорастворим в 96 % спирте.
рН (2.2.3). Около 4,3. Измеряют рН раствора
22.6 r/л испыту моro образца.
Аммония церия (IV) нитрат. (NН4)2Се(NОЗ)6'
(М М. 548,2). 1005000. [16774 21 3].
Оранжево желтый кристаллический поро
шок или прозрачные оранжевые кристаллы. Pac
творим в воде.
Аммония церия (IV) qЛbфar. (NH4)4Ce(S04)4 '2Н,р.
(М. м. 633). 1005100. [10378-47 9].
Оранжево желтый кристаллический по
рошок или кристаллы. Медленно растворим
в воде.
Амоксициллина триrидрат. 1103400.
[6133 70 7]. См. статью Амоксициллина тpи2и
драт.
Анетол. СЮН1Р' (М м. 148,2). 1006900.
[4180 23 8]. 1 Метокси-4 (пропен 1 ил)бензол.
Белая или почти белая кристаллическая
масса при температуре до 20 21 ОС, при TeM
пературе выше 23 ос жидкость. Практически
нерастворим в воде, леrкорастворим в этаноле,
эфире, этилацетате и петролейном эфире.
п 5: около 1,56.
Температура кипения: около 230 ОС.
Анетол, применяемый в 2азовой xpOMaтo
2рафии, должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
анисовое, используя анетол в качестве испыту
емоro раствора.
Содержание анетола (время удерживания
около 41 мин), рассчитанное методом нормали
зации, должно быть не менее 99,0 %.
цис Анетол. С 10 Н 1 р. (М М. 14,2). 1007000.
(Z) 1 Метокси 4 (пропенил 1 )бензол.
Белая или почти белая кристаллическая
масса при температуре до 20 21 ОС, при TeM
пературе выше 23 ос жидкость. Практиче
ски нерастворим в воде, леrкорастворим в
этаноле, эфире, этилацетате и петролейном
эфире.
25
по : около 1,56.
Температура кипения: около 230 ос.
цис Анетол, применяемый в 2азовой X
мат02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rаэовая хро.-
матоrрафия (2.2.28), как указано в статье Масло
анисовое, используя цис анетол в качестве ис
пытуемоro раствора. Содержание цис анетола,
рассчитанное методом нормализации, должно
быть не менее 92,0 %.
п-Анизидин. C 7 H g NO. (м. м. 123,2).1103500.
[1 04 94 9]. 4 Метоксианилин.
Белые или почти белые кристаллы. YMepeH
но растворим воде, растворим в этаноле.
Содержит не менее 97,0 % C 7 H g NO.
604
rосударственная фармакопея Республики Беларусь
Вызывает раздражение кожи; сенсибилиза
тор.
Хранят в защищенном от света месте при
температуре от О ОС до 4 ОС.
При хранении п анизидин темнеет вслед
ствие окисления. Окисленный п анизидин
может быть восстановлен и обесцвечен следу
ющим образом: 20 r п анизидина Р растворяют
в 500 мл воды Р при температуре 75 ОС, при
бавляют 1 r натрия сульфита Р и 1 О r У2ЛЯ
активироваНН020 Р, перемешивают в тече
ние 5 мин и фильтруют. Полученный фильтрат
охлаждают и выдерживают при температуре
около О ос в течение не менее 4 ч и фильтруют.
Полученные кристаллы промывают небольшим
количеством воды Р, охлажденной до темпера
туры О ОС, И сушат в вакууме над фосфора (V)
оксидом Р.
Анилин. C 6 H 7 N. (М м. 93,1). 10071 00. [62 53--3].
Бензоламин.
Маслообразная прозрачная бесцветная или
желтоватоro цвета жидкость. Растворим в воде,
смешивается с 96 % спиртом и эфиром. IIОбра
щаться с осторожностью. 1I
Температура кипения: от 183 ос до 186 ОС.
d;g: около 1,02.
Хранят в защищенном от света месте.
Анилина rидpoхлорид. C 6 H 7 N'HCI. (М м. 129,6).
1147700. [142 04 1]. Бензоламина rидрохлорид.
Кристаллы. Темнеет при воздействии возду
ха и света.
Температура плавления: около 198 ОС.
d 20
20: около 1,02.
Хранят в защищенном от света месте.
Анионообменная смола. 1007200.
Смола в хлоридной форме, содержа
щая четвертичные аммониевые rруппы
[СН 2 N+(СН з )з], присоединенные к полимерной
решетке, состоящей из полистирола попереч
носшитоro 2 % дивинилбензола. Выпускают
в виде rранул, размер которых должен быть
указан в частных статьях.
Смолупромываютнастеклянномфильтре( 40)
1 М раствором натрия 2идроксида до отрица
тельной реакции на хлориды в промывном pac
творе, затем промывают водой Р до получения
нейтральной реакции в промывной воде. Cy
спендируют в свежеприrотовленной воде, cвo
бодной от аммиака, Р и защищают от уrлерода
диоксида.
Анионообменная смола сильнооснов-
ная. 1026600.
rелеобразная смола в ОН форме, co
держащая четвертичные аммониевые rруппы
[СН 2 N+(СН з )з, тип 1], присоединенные к поли
мерной решетке, состоящей из полистирола по
перечносшитоro 8 % дивинилбензола.
Прозрачные rранулы коричневоro цвета.
Размер частиц: от 0,2 мм до 1,0 мм.
Содержание вла2и: около 50 %.
Полная обменная емкость: не менее 1,2 мэкв/мп.
Анионообменная смола сильноосновная
для хроматоrрафии. 1112700.
Смола с четвертичными аммониевыми rруп
пами, присоединенными к решетке латекса по
перечносшитоrо дивинилбензолом.
Анисовый альдеrид. С в Н в 0 2 . (М м. 136,1).
1 007300. [123 11 5]. 4 Метоксибензальдеrид.
Маслянистая жидкость. Очень мало pac
творим в воде, смешивается с 96 % спиртом и
эфиром.
Температура кипения: около 248 ОС.
Анисовый альде2ид, применяемый в 2азо
вой хромат02рафии, должен выдерживать сле
дующее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
анисовое, используя анисовый альдеrид в каче
стве испытуемоro раствора.
Содержание анисовоro альдеrида, рассчи
танное методом нормализации, должно быть не
менее 99,0 %.
Анисовоrо альдеrида раствор. 1007301.
Последовательно смешивают 0,5 мл aHиco
в020 альде2ида Р, 10 мл кислоты уксусной ледя
ной Р, 85 мл метанола Р и 5 мл кислоты серной Р.
Анисовоrо альдеrида раствор Р1.
1007302.
1 О мл анисов020 альде2ида Р смешивают с
90 мл 96 % спирта Р, прибавляют 1 О мл кисло
ты серной Р и перемешивают.
Анолит для изоэлектрофокусировки рН
от 3 до 5. 0,1 М раствор кислоты rлутаминовой
и 0,5 М раствор кислоты фосфорной. 1112800.
14,71 r кислотЫ2лутаминовой Р в paCTBO
ряют в воде Р, прибавляют ЗЗ мл кислоты фос
форной Р И доводят водой Р до объема 1000 мл.
Антимонила калия тартрат. C 4 H 4 K0 7 Sb' %нр.
(М м. 333,9). 1007600. Калия aKBa[TapTpaтo( }-
01,()2,()3] антимониат (111) полуrидрат.
Белый или почти белый rранулированный
порошок или бесцветные прозрачные кристал
лы. Растворим в воде и в rлицерине, леrкорас
творим в кипящей воде, практически HepaCT
ворим в 96 % спирте. Водный раствор имеет
слабокислую реакцию.
Антитромбин 111. 1007800. [90170 80 2].
Антитромбин 111 выделяют из человеческой
плазмы хроматоrрафически с использованием
rепарин аrарозной колонки.
Удельная активность должна быть не менее
6 ME/Mr.
4.1.1. Реактивы
605
# Аравийская камедь. См. rуммиарабик р.
Антитромбина 111 раствор Р1. 1007801.
Антитромбин 111 Р обрабатывают, как YKa
зано производителем, и разводят буферным
раствором триС(2идроксиметил)аминометан
натрия хлорида рН 7,4 Р до активности 1 МЕ/мл.
Антитромбина 111 раствор Р2. 1007802.
Антитромбин 111 Р обрабатывают, как YKa
зано производителем, и разводят буферным
раствором триС(2идроксиметил)аминометан
натрия хлорида рН 7,4 Р до активности 0,5 МЕ}
мл.
# Антраниловая кислота. См. 2 Аминобен
зойная кислота Р.
Антрацен. С 14 Н 10 . (м. м. 178,2). 1007400.
[120 12 7].
Белый или почти белый кристаллический
порошок. Практически нерастворим в воде, Ma
лорастворим в хлороформе.
Температура плавления: около 218 ОС.
Антрон. С 14 Н 10 О. (м. м. 194,2). 1007500.
[90 44 8]. 9 (10H) AHTpaцeHOH.
Светло желтый кристаллический порошок.
Температура плавления: около 155 ОС.
Апиrенин. С15Н1005' (м. м. 270,2). 1095800.
[520 36 5]. 4' ,5, 7 Триrидроксифлавон.
Светло желтоватый порошок. Практически
нерастворим в воде, умеренно растворим в 96 %
спирте.
Температура плавления: около 310 ос с раз
ложением.
Хромат02рафия. Определение проводят,
как указано в статье Цветки римской ромашки,
используя 1 О мкл раствора 0,25 r/л в MeтaHO
ле р. На верхней трети xpoMaтorpaMMbI должно
обнаруживаться основное пятно с желтовато зе
леной флуоресценцией.
Апиrенин 7 rлюкозид. С21Н20010' (м. м.432,6).
1095900.
Светло желтоватый порошок. Практически
нерастворим в воде, умеренно растворим в 96 %
спирте.
Температура плавления: от 198 ос до 201 ОС.
Хромат02рафия. Определение проводят,
как указано в статье Цветки римской ромашки,
используя 10 мкл раствора 0,25 r/л в MeтaHO
ле Р. На средней трети xpoMaTorpaMMbI должно
обнаруживаться основное пятно с желтоватой
флуоресценцией.
Апротинин. 1007900. [9087 70 1].
Полипептид, состоящий из 58 аминокислот.
Инrибирует стехиометрическую активность He
ICOТОРЫХ протеолитических энзимов, таких как
химотрипсин, плазмин, трипсин. Активность не
менее 3,0 ЕФЕ/мr в пересчете на сухое 8еще
crвo.
Арабиноза. С 5 Н 10 О 5 . (м. м. 150,1). 1008000.
[87 72 9]. L (+) Арабиноза.
Белый или почти белый кристаллический
порошок. Леrкорастворима в воде.
[a] O: от +103 до +105. Определение про
водят, используя раствор 50 r/л испытуемоro
образца в воде Р, содержащей около 0,05 %
NН з .
Арбутин. С 12 Н 1 Рт (м. м. 272.3). 1008100.
[497 76 7]. Арбутозид. 4 rидроксифенил J3 D
rлюкопиранозид.
Мелкие белые или почти беЛblе блестя
щие иroльчатые кристаллы. Леrкораство
рим в воде, очень леrко растворим в roрячей
воде, растворим в 96 % спирте.
Хромат02рафия. Тонкослойная xpOMaTO
rрафия (2.2.27), как указано в статье Листья
толокнянки; на xpoMaTorpaMMe должно обна
руживаться только одно основное пятно.
Арrинин. C 6 H I .N.0 2 . (М М. 174,2). 1103600.
[7 4 79 3]. (S) 2 Дмино 5 ryанидинопентановая
кислота.
Арrинин содержит не менее 98,5 % и не
более 101,0 % C e H I .N.0 2 .
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Леrко
растворим в воде, очень мало растворим в
96 % спирте.
[а ] O: от +25,5 до +28.5. Определение про
водят, используя раствор 80 r/л испытуемоro
образца в кислоте хлористоводородной Р1.
Хранят в воздухонепроницаемом контей
нере в защищенном от света месте.
AproH. Ar. (А. м. 39,95). 1008200. [7440 37 1].
Содержит не менее 99,995 % (об/об) Ar.
Оксид У2лерода (2.5.25, метод 1). Не
более 0,6 ррт (об/об). После пропускания
1 О л ар20на Р со скоростью 4 л/ч, на титро
вание должно быть израсходовано не более
0,05 мл 0,002 М раствора натрия тиосуль
фата.
ApOMaAeHApeH.C ls H 24 . (М м. 204,4).1139100.
[489 39A]. (1 R,2S,4R,8R.11R) 3,3, 11 Триметил
7 метилентрицикло[6.3.о. 02 .]ундекан.
Прозрачная, почти бесцветная жидкость.
d 20
4 : около 0,911.
20
по: около 1,497.
[a] O: около +12.
Температура кипения: около 263 ОС.
Аромадендрен, используемый в 2азовой
хромат02рафии, должен выдерживать сле
ду ющее дополнительное испытание.
Количественное определение. rазовая
хроматоrрафия (2.2.28), как указано в статье
Масло чаЙН020 дерева.
606
rосударственная фармакопея Республики Беларусь
Содержание аромадендрена, рассчитан
ное методом нормализации, должно быть не
менее 92 %.
Арсенита раствор. 1008301.
0,50 r мышьяка (111) оксида Р растворяют в
5 мл раствора натрия 2идроксида разведенно
20 Р, прибавляют 2,0 r натрия 2идрокарбона
та Р и доводят водой Р до объема 100,0 мл.
Аскорбиновая кислота. 1008400. [50 81 7].
См. статью Аскорбиновая кислота.
Аскорбиновой кислоты раствор. 1008401.
50 Mr кислоты аскорбиновой Р растворяют
в 0,5 мл воды Р и доводят диметилформами
дом Р до объема 50 мл.
L-Аспартил-L-фенилаланин. С'ЗН'6N205'
(М м. 280,3). 1008500. [13433 09 5]. (S) 3 Амино
N [( S) 1 карбокси 2 фенилэтил]янтарная кислота.
Белый или почти белый порошок.
Температура плавления: около 210 ос с раз
ложением.
Ацеталь. С 6 Н 1 Р2' (М м. 118,2). 1112300.
[1 05 57 7]. Ацетальдеrидадиэтилацеталь. 1, 1 Ди
этоксиэтан.
Прозрачная бесцветная летучая жидкость.
Смешивается с водой и 96 % спиртом.
dig: около 0,824.
20
по: около 1 ,382.
Температура кипения: около 103 ОС.
Ацетальдеrид. С 2 Нр. (М м. 44,1). 1000200.
[75 07 0]. Этаналь.
Прозрачная бесцветная воспламеняющаяся
жидкость. Смешивается с водой и 96 % спиртом.
d 20
20: около 0,788.
20
по : около 1,332.
Температура кипения: около 21 ОС.
Ацетальдеrидаммиака тримера три-
rидрат. С 6 Н ,5 N з '3Нр. (М м. 183,3). 1133500.
[76231 37 3]. 2,4,6 Триметилrексаrидро 1 ,3,5 три
азин триrидрат.
Температура плавления: от 95 ос до 97 ОС.
Ацетилацетамид. C 4 H 7 N0 2 . (М м. 101,1).
1102600. [5977 14 0]. 3 Оксобутанамид.
Температура плавления: от 53 ос до 56 ОС.
Ацетилацетон. С 5 Н в О2' (М м. 100,1).
1000900. [123 54 6]. 2,4 Пентандион.
Бесцветная или слеrка желтоватая леrко
воспламеняющаяся жидкость. Леrкорастворим в
воде, смешивается с ацетоном, 96 % спиртом и
кислотой уксусной ледяной.
П О: от 1,452 до 1,453.
Температура кипения: от 138 ос до 140 ОС.
Ацетилацетона реактив Р1. 1000901.
К 100 мл раствора аммония ацетата Р
прибавляют 0,2 мл ацетилацетона Р.
# Ацетилирующая смесь.
Смешивают 1 объем УКСУСН020 аН2идрида Р и
З объема пиридина Р (точка кипения 115 ОС). Смесь
должна быть бесцветной. Смесь применяют CBEr
жеприroтовленной. Обращаться с осторожностью.
N-АцеТИЛ-Е-капролактам. С в Н 1з N0 2 . (М м.
155,2). 1102700. [1888 91 1]. N Ацетилrексан 6
лактам.
Бесцветная жидкость. Смешивается с эта
нолом.
dig: около 1,100.
20
по: около 1,489.
Температура кипения: около 135 ОС.
N-Ацетилнеураминовая кислота. C'1H,gNOg-
(М м. З09,3). 1001100. [13148 6]. О Сиаловая
кислота.
Белые или почти белые иrольчатые кристал
лы. Растворима в воде и в метаноле, малорас
творима в 96 % спирте, практически нераствори
ма в ацетоне.
[а];О: около 36. Определение проводят, ис
пользуя раствор 1 О r/л испытуемоro образца.
Температура плавления: около 186 ос с раз
ложением.
Ацетилтирозина этиловый эфир.
С ,з Н 17 NО 4 'Нр. (М м. 269,З). 1001200. [36546 50--6].
N Ацетил L тирозина этиловый эфир моноrи
драт. Этил ( S) 2 ацетамидо 3 (4 rидроксифенил)
пропионат моноrидрат_
Белый или почти белый кристаллический
порошок. Приrоден для количественноro опре
деления химотрипсина.
[а];О: от +21 до +25. Определение проводят,
используя раствор 1 О r/л испытуемоro образца в
96 % спирте Р.
A;: : от 60 до 68. Определение проводят при
длине волны 278 нм, в 96 % спирте Р.
Ацетилтирозина этиловоrо эфира 0,2 М
раствор. 1001201.
0,54 r ацетилтирозина этилов020 эфира Р
растворяют в 96 % спирте Р и доводят до
объема 10,0 мл этим же растворителем.
N-Ацетилтриnтофан. С'ЗН'4NРз, (М м. 246,3).
1102800. [1218 344]. 2 Ацетиламино 3 (индол
3 ил)пропионовая кислота.
Белый или почти белый порошок или бес
цветные кристаллы. Малорастворим в воде,
растворим в разведенных растворах rидрокси
дов щелочных металлов.
Температура плавления: около 205 ОС.
Количественное определение. Жидкостная хро-
матоrpафия (2.2.29), как указано в статье Трuптофан.
4.1.1. Реактивы
607
Испытуемый раствор. 10,0 Mr растворяют в
смеси растворителей ацетонитрил Р вода Р
(10:90, об/об) и доводят до объема 100,0 мл этой
же смесью растворителей.
Содержание триптофана, рассчитанное MeTO
дом нормализации, должно быть не менее 99,0 %.
Ацетилхлорид. С 2 Н з СIO. (м. м. 78,5).
1000800. [75 36 5].
Прозрачная бесцветная воспламеняющаяся
жидкость с резким запахом. Разлаrается в воде
и 96 % спирте, смешивается с этиленхлоридом.
Обращаться с осторожностью.
d;g: около 1,10.
Температурные пределы пере20нки
(2.2.11). От 49 ос до 53 ОС; должно переroняться
не менее 95 %.
ДЦетилхапинахлорид. C 7 H 16 C/N0 2 . (М М. 181,7).
1001000. [60 З1 1].
Кристаллический порошок. Очень леrко pac
творим в холодной воде и в 96 % спирте. Разла
rается в rорячей воде и растворах щелочей.
Хранят при температуре 20 ОС.
Ацетилэвrенол. С12Н140з' (м. М. 206,2).
1100700. [93 28 7]. 2 Метокси 4 (2 пропенил)фе
нилацетат.
Маслянистая жидкость желтоro цвета. Леr
корастворим в 96 % спирте, практически HepaCT
ворим в воде.
п O: около 1,521
Температура кипения: от 281 ос до 282 ОС.
Ацетилэв2енол, применяемый в 2азовой
хромат02рафии, должен выдерживать следую
щее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
2воздичное, используя ацетилэвrенол в качестве
испытуемоro раствора.
Содержание ацетилэвrенола, рассчитанное
методом нормализации, должно быть не менее
98,0%.
Ацетоксивалереновая кислота. С 17 Н 24 О 4 .
(м. М. 292,4). 1165800. [81397 67 З]. (2E) 3
[(1 RS,4S, 7R, 7aR) 1 (Ацетилокси) 3, 7 диметил
2,4 ,5 ,6,7, 7 а rесаrидро 1 Н инден 4 ил ] 2 метил
проп 2 еновая кислота.
Бесцветное или бледно желтое вязкое масло.
Спектрофотомерия (2.2.25). Раствор в Me
таноле Р должен иметь максимум поrлощения
при 216 нм.
Ацетон. 1000600. [67 64 1].
См. статью Ацетон.
# Ацетон безводный.
Ацетон Р сушат над натрия сульфатом
безводным Р в течение 12 ч.
Ацетонитрил. С 2 Н з N. (М м. 41,05).1000700.
[75 05 8]. Метилцианид. Этаннитрил.
Прозрачная бесцветная жидкость. CMe
шивается с водой, ацетоном и метанолом.
d 20
20: около 0,78.
п O: около 1,З44.
Раствор 100 r/л ацетонитрила имеет ней
тральную реакцию по лакмусовой бумаrе.
Температурные пределы пере20нки
(2.2.11). От 80 ос до 82 ОС; должно переro
няться не менее 95 %.
Ацетонитрил, используемый для спек
трофотометрии, должен выдерживать сле
дующее дополнительное испытание.
Пропускание (2.2.25). Не менее 98 %.
Определение проводят в области длин волн
от 255 нм до 420 нм, используя в качестве
компенсационной жидкости воду Р.
Ацетонитрил для хроматоrрафии.
1000701. См. Ацетонитрил Р.
Ацетонитрил, используемый в xpOMa
т02рафии, должен выдерживать следующие
дополнительные испытания.
Пропускание (2.2.25). Не менее 98 %.
Определение Пр080ДЯТ при длине волны 240
нм, используя в качестве компенсационной
жидкости воду Р.
Чистота (2.2.28). Не менее 99,8 %.
Ацетонитрил Р1. 1000702.
Должен выдерживать требования для
ацетонитрила Р и следующие дополнитель
ные требования.
Содержит не менее 99,9 % С 2 Н з N.
Оптическая плотность (2.2.25). Не
более 0,1 О при 200 нм. В качестве компенса
ционной жидкости используют воду Р.
БайкаЛИН.С 21 Н 1 Р11' (М м.446,4). 1179200.
[21967 41 9]. 5,6 Диrидрокси 4 оксо 2 фенил
4H 1 бензопиран 7 ил I3 D rлюкопиранозиду
роновая кислота.
Барбалоин. C 21 H2pg'Hp. (М М. 436.4).
1008800. [1415 73 2]. Алоин. 1.8 Диrидрокси
З rидроксиметил 1 О D rл юкопиранозил 1 OH
aHTpaцeH 9 OH.
Кристаллический порошок или иroльча
тые кристаллы от желтоro до теМН<rжелтоro
цвета, под действием воздуха и света TeMHe
ет. Умеренно растворим в воде и 96 % спирте,
растворим в ацетоне, в растворах аммиака и
rидроксидов щелочных металлов.
А 1 1%: около 192 при длине волны 269 нм;
см
около 226 при длине волны 296,5 нм;
около 259 при длине волны 354 нм.
Определение проводят, используя в каче
стве растворителя метанол Р, в пересчете на
безводное вещество.
608
rосударственная фармакопея Республики Беларусь
Хромат02рафия. Тонкослойная xpOMaTO
rрафия (2.2.27), как указано в статье Кора круши
ны; на xpoMaтorpaMMe обнаруживается только
одно основное пятно.
Барбитал. С в Н 12 NРз, (М М. 184,2).1008900.
[57 44 3]. 5,5 Диэтилпиримидин 2,4,6(1 H,3H,5H)
трион.
Содержит не менее 99,0 % и не более 101,0 %
СВН12N20з в пересчете на сухое вещество.
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Мало
растворим в воде, растворим в кипящей воде,
в 96 % спирте. Образует водорастворимые co
единения с едкими щелочами, карбонатами и
аммиаком.
Барбитал натрий. СвН11N2NаОз. (М М. 206,2).
1009000. [144 02 5].
Содержит не менее 98,0 % натриевоro произ
водноro 5,5 диэтил 1 Н,3Н,5Н пиримидин 2,4,6
триона.
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Леrкорас
творим в воде, малорастворим в 96 % спирте.
Барбитуровая кислота. С 4 Н 4 NРз. (М м. 128,1).
1009100. [67 52 7]. 1Н,ЗН,5Н Пиримидин 2,4,6
трион.
Белый или почти белый порошок. Мало
растворима в воде, леrкорастворима в кипящей
воде и разведенных кислотах.
Температура плавления: около 253 ОС.
Бария rидроксид. Ва(ОН)2'8Нр. (М м. 315,5).
1009400. [12230 71 6]. Бария диrидроксид.
Бесцветные или белые кристаллы. PaCTBO
рим в воде.
Бария rидроксида раствор. 1009401.
Раствор 47,3 r/л бария 2идроксида Р.
Бария карбонат. ВаСО з . (М м. 197,3).
1009200. [513 77 9].
Белый или почти белый порошок или pac
сыпчатая масса. Практически нерастворим в
воде.
Бария нитрат. Ва(NОз)т (М м. 261,3).
1163800. [10022 31 8] Барий азотнокислый.
Кристаллы или кристаллический порошок.
Леrкорастворим в воде, очень мало растворим в
96 % спирте и в ацетоне.
Температура плавления: около 590 ос.
# Бария нитрата раствор.
Раствор 50 r/л бария нитрата Р. Ядовит.
Бария сульфат. BaS0 4 . (М м. 233,3).
1009500. [7727 43 7].
Белый или почти белый мелкий 'Тяжелый
порошок, не содержащий крупных частиц.
Практически нерастворим в воде и в орrаниче
ских растворителях. Очень мало растворим в
кислотах и в растворах rидроксидов тяжелых
металлов.
Бария хлорид. BaCI 2 '2Hp. (М. м. 244,3).
1009300. [10326 27 9]. Бария дихлорид.
Бесцветные кристаллы. Леrкорастворим в
воде, малорастворим в 96 % спирте.
Бария хлорида раствор Р1. 1009301.
Раствор 61 r/л бария хлорида Р.
Бария хлорида раствор Р2. 1009302.
Раствор 36,5 r/л бария хлорида Р.
Бензалацетон. С 10 Н 1 р. (М М. 146,2). 1168500.
[122 57 6]. (3Е) 4 Фенилбут 3 ен 2 он.
Белая или бледно желтая масса.
Содержание: не менее 98,0 %.
Температура кипения: около 261 ОС.
Температура плавления: около 39 ОС.
Бензальдеrид. С 7 Н 6 О. (М М. 106,1). 1009600.
[100 52 7].
Бесцветная или слеrка желтоватая жид
кость. Малорастворим в воде, смешивается с
96 % спиртом.
d 20
20: около 1,05.
20
по: около 1 ,545.
Температурные пределы пере20нки
(2.2.11): от 177 ос до 180 ОС; должно переroнять
ся не менее 95 %.
Хранят в защищенном от света месте.
# Бензальдеrида раствор насыщенный.
1 мл бензальде2ида Р встряхивают с 250 мл
воды Р. Смесь выдерживают в течение суток
при периодическом встряхивании. Перед приме
нением сливают прозрачную жидкость. Раствор
применяют свежеприroтовленным.
Бензидин. C6H4NH2'C6H4NH2' (М М. 184,24).
Бифенил 4,4' диамин. #4,4' Диаминодифенил.#
Содержание: не менее95 % C 6 H 4 NH 2 'С6Н4 NH 2 .
Белый или слеrка желтоватый или KpaCHOBa
тый порошок, темнеющий под воздействием воз
духа и света.
Температура плавления: около 120 ОС.
Хранят в защищенном от света месте.
Бензил. С14Н1002' (М М. 210,2). 1117800.
[134 81 6]. Дифенилэтандион. #Дибензоил. Ди
фенилrлиоксаль. #
Желтый кристаллический порошок. Прак
тически нерастворим в воде, растворим в 96 %
спирте, в этилацетате и в толуоле.
Температура плавления: 95 ОС.
Бензилбензоат. 1010800. t120 5141-
См. статью Бензилбензоат.
4.1.1. Реактивы
609
Бензилкоричный эфир. С16Н'402'
(М М. 238,3). 1010900. [103A1 3). Бензил З
фенилпроп 2 еноат. #Бензилциннамат. #
Бесцветные или желтоватые кристаллы.
Практически нерастворим в воде, растворим в
96 % спирте.
Температура плавления: около 39 ОС.
Бензиловый спирт. 1010700. [100 51 6).
См. статью Бензиловый спирт.
Бензиловый эфир. С'4Н'40, (М М. 198,3).
1140900. [103 50 4). Дибензиловый эфир.
Прозрачная бесцветная жидкость. Практи
чески нерастворим в воде, смешивается с aцe
тоном и этанолом.
d 20
20: около 1,043.
20
по: около 1,562.
Температура кипения: около 296 ос с разло
жением.
Бензилпенициллин натрия. 101100Q
[69 57 8).
См. статью Бензилпенициллин натрия.
2-Бензилпиридин. C'2H"N. (М м.. 169,2).
1112900. [101 82 6).
Содержание: не менее 98,0 % C'2H1,N.
Желтая жидкость.
Температура плавления: от 1З ос до 16 ОС.
# Бензин. Бензин авиационный.
Прозрачная бесцветная жидкость.
Температура начала переroнки: не ниже
40 ОС. При температуре 105 ос переrоняется
50 % бензина. При температуре 180 ос переrо
няется 97,5 % бензина. Остаток не более 1,5 %.
Кислотность (количество миллиrраммов
калия rидроксида, идущее на нейтрализацию
100 мл бензина): не более 1,2.
Йодное число (2.5.4): не более 10.
Содержание серы: не более 0,05 %.
Водорастворимые кислоты и щелочи
должны отсутствовать.
Бензоиларrинина этиловоrо эфира rи-
дрохлорид. С'5Н2зС1NРз (М м. 342,8). 1010500.
[2645 08 1). N Бензоил L арrинина этилово
ro эфира rидрохлорид. Этил ( S) 2 бензамидо
5 ryанидиновалерата rидрохлорид.
Белый или почти белый кристаллический
порошок. Очень леrко растворим в воде и в эта
ноле.
[а ] O: от 15 до 18. Определение проводят,
используя раствор 1 О r/л беНЗОИJlарrинина эти
ловоro эфира rидрохлорида.
Температура плавления: около 129 ОС.
А,':;: от 3 1 О до З40. Определение проводят при
длине волны 227 нм, используя раствор 0,01 r/л
бензоиларrинина этиловоro эфира rидрохло
рида.
;7 За. 1712.
N-Бензоил -L-n рол ил -L-фенилалан ил-
L-арrинина 4-нитроанилида ацетат. СЗ5Н42NвОв'
(М М. 703). 1010600.
Бензоилхлорид. С 7 Н 5 СIO. (М. м. 140,6).
1010400. [98 88A).
Бесцветная слезоточивая жидкость. Разла
rается в воде и в 96 % спирте.
d;g: около 1,21.
Температура кипения: около 197 ОС.
Бензоин. С'4Н1Р2' (М. М. 212,3). 1010200.
[579 44 2). 2 rидрокси 1 ,2 дифенилэтанон.
Слеrка желтоватые кристаллы. Очень мало
растворим в воде, леrкорастворим в ацетоне,
растворим в rорячем 96 % спирте.
Температура плавления: около 137 ОС.
Бензойная кислота. 1010100. [65 85 0).
См. статью Бензойная кислота.
Бензол. С6Н6' (М М. 78,1). 1009800. [71A3 2].
Прозрачная бесцветная воспламеняюща
яся жидкость. Практически нерастворим в воде,
смешивается с 96 % спиртом.
Температура кипения: около 80 ОС.
Бензофенон. С,зН,оО. (М м. 182,2).
1010300. [119 61 9). Дифенилметанон.
Призматические кристаллы. Практически
не растворим в воде, леrкорастворим в 96 %
спирте.
Температура плавления: около 48 ОС.
1,4-Бензохинон. С 6 НР2' (М М. 108,1).
1118500. [106 51A). Циклоrекса 2,5 диен 1,4
дион.
Содержание: не менее 98,0 % С 6 Н 4 О 2 .
Бензэтония хлорид. C27H42CIN02'H20.
(М М. 466,1). 1009900. [121 54 0). Бензилдим
тил[2 [2 [4 (1, 1 ,3,3 тетраметилбутил)феноксиJ
этокси)этил)аммония хлорид моноrидрат.
Мелкий белый или почти белый порошок
или бесцветные кристаллы. Растворим в воде и
в 96 % спирте.
Температура плавления: около 163 ОС.
Хранят в защищенном от света месте.
# Берберина бисульфат. C2(jH,aN04'HS04'
(М М. 433,4). [633 66 9]. Берберина сульфат.
Берберина сульфат кислый.
Ярко желтые иrольчатые кристаллы или
темно желтый порошок. Малорастворим в воде
и этаноле.
Берrаптен. С'2НР4' (М М. 216,2). 1103700.
[484 20 8]. 5 Метоксипсорален.
Бесцветные кристаллы. Практически HepaCT
ворим в воде, умеренно растворим в 96 % спирте
и малорастворим в кислоте уксусной ледяной.
Температура плавления: около 188 ОС.
610
rосударственная фармакопея Республики Беларусь
Бисбензимид. С25Н27СlзN60'5НР. (м. м. 624).
11 03800. [2349144 3].4 [5 [5 (4 Метилпиперазин
1 ил )бензимидазол 2 ил ]бензи мидазол 2 ил]
фенола триrидрохлорид пентаrидрат.
Бисбензимида исходный раствор.
1103801.
5 Mr бисбензимида Р растворяют в воде Р и
доводят до объема 100 мл этим же растворите
лем.
Хранят в защищенном от света месте.
Бисбензимида рабочий раствор. 1103802.
Непосредственно перед использованием
100 мкл исходН020 раствора бисбензимида Р
доводят фосфатным забуференным физиоло
2ическим раствором рН 7,4 Р до объема 100 мл.
Бетулин. СЗОН5002' (М. М. 442,7). 1011100.
[473 98 3]. Луп 20(39) ен З ,28 диол.
Белый или почти белый кристаллический
порошок.
Температура плавления: от 248 ос до 251 ОС.
Бибензил. С 14 Н 14 . (м. м. 182,3). 1011200.
[1 03 29 7]. 1 ,2 Дифенилэтан.
Кристаллический порошок белоro цвета.
Практически нерастворим в воде, очень леrко
растворим вметиленхлориде, леrкорастворим в
ацетоне, растворим в 96 % спирте.
Температура плавления: от 50 ос до 53 ОС.
(-)ooQ Бисаболол. С 15 Н 26 О. (м. м. 222,4).
1128800. [23089 26 1]. (2S) 6 Метил 2 [( 1 S) 4
метилциклоrекс 3 енил]rепт 5 ен 2 ол. Лево
менол.
Бесцветная вязкая жидкость со слабым
характерным запахом, практически HepaCTBO
рим в воде, леrкорастворим в 96 % спирте, Me
таноле, толуоле, в жирных маслах и эфирных
маслах.
d 20
20: от 0,925 до 0,935.
20
п о : от 1,492 до 1,500.
[a] O: от 54,5 до 58,0. Определение прово
дят в растворе 50 мr/мл в 96 % спирте Р.
(+а Бисаболол, используемый в 2азовой
хромат02рафии, должен выдерживать следую
щее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
ромашки. Используют раствор 4 r/л в циКЛ02ек
сане Р.
Содержание Н а бисаболола должно быть
не менее 95,0 %, рассчитанное методом HopMa
лизации.
Биурет. C 2 H 5 NP2' (м. м. 103,1). 1011600.
[1 08 19 0].
Белые или почти белые кристаллы. rиrро
скопичен. Растворим в воде, умеренно paCTBO
рим в 96 % спирте.
Температура плавления: от 188 ос до 190 ос
с разложением.
Хранят в воздухонепроницаемом контейнере.
Биуретовый реактив. 1011601.
1,5 r меди (11) сульфата Р и 6,0 r калия на
трия тартрата Р растворяют в 500 мл воды Р,
прибавляют 300 мл раствора 100 r/л натрия 2и
дроксида Р, свободноro от карбонатов, доводят
до объема 1000 мл этим же раствором и пере
мешивают.
Бифенил-4-0Л. С 12 НюО, (м. м. 170,2).
1011300. [9043 7]. 4 Фенилфенол.
Белый или почти белый кристаллический
порошок. Практически нерастворим в воде.
Температура плавления: от 164 ОСдо 167 ОС.
Блокирующий раствор. 1122400.
10 % (об/об) раствор кислоты уксусной Р.
Бора фторид. BF3. (м. м. 67,8). 1012100.
[7637 07 2]. Бора трифторид.
Бесцветный rаз.
Бора фторида раствор в метаноле.
1012101.
Раствор 140 r/л бора фторuда Р в метаноле Р.
Бора хлорид. ВСl з , (м. м. 117,2). 1112000.
[10294 34 5]. Бора трихлорид.
Бесцветный rаз. Бурно реаrирует с водой.
Используют в виде растворов в подходящих
растворителях (2 хлорэтанол, метиленхлорид,
reKcaH, rептан, метанол).
п O: около 1,420.
Температура кипения: около 12,6 ОС.
Токсичен, вызывает коррозию.
Бора хлорида раствор в метаноле.
1112001. Раствор 120 r/л бора хлорида Р в Me
таноле р.
Хранят в защищенном от света месте при
температуре 20 ОС, преимущественно в ампулах.
Борная кислота. 1011800. [10043 35 3].
См. статью Борная кислота.
Борной кислоты раствор, насыщенный,
охлажденный. 1011801.
К 3 r кислоты борной Р прибавляют 50 мл
воды Р и встряхивают в течение 1 О мин. Полу
ченный раствор выдерживают в холодильнике в
течение 2 ч.
Борнеол. С 10 Н 1 р. (м. м.154.3). 1011900.
[507 70 0]. эндо 1,7, 7 Т риметилбицикло[2.2.1 ]rеп
тан 2 ол.
Бесцветные кристаллы. Леrко сублимирует
ся. Практически нерастворим в воде, леrкорас
творим в 96 % спирте и в петролейном эфире.
Температура плавления: около 208 ОС.
4.1.1. Реактивы
611
Хромато2рафuя. Тонкослойная xpoMaтorpa
фия (2.2.27), используя в качестве тонкою слоя cи
лика2ель G Р. На линию старта хроматоrpaфической
пластинки наносят 1 О мкл раствора 1 r/л борнеола
в толуоле Р. Подвижная фаза: хлороформ Р. Korдa
фронт растворителя пройдет 10 см от линии старта,
пластинку вынимают из камеры, сушат на во:щухе,
опрыскивают раствором анисовО20 альдееида Р,
расходуя 1 О мл на пластинку площадью 200 мм 2 ,
И сушат при температуре от 100 ос до 105 ос в T
чение 10 мин. На xpoMaтorpaMMe должно обнаружи
ваться только одно основное пятно.
БорнилацетаТ'С12Н2002.(м.м.196,3).1012000.
(5655 61 8]. эндо 1 ,7,7 Триметилбицикло[2.2.1]
rепт 2 илацетат.
Бесцветные кристаллы или бесцветная жид
кость. Очень мало растворим в воде, растворим
в 96 % спирте.
Температура плавления: около 28 ОС.
Хроматоzрафия. Определение проводят Me
тодом тонкослойной хроматоrрафии (2.2.27), ис
пользуя в качестве тонкоro слоя силика2ель G Р.
На линию старта хроматоrрафической пластинки
наносят 10 мкл раствора 2 r/л борнилацетата в
толуоле Р. Подвижная фаза: хлороформ Р. Korдa
фронт растворителя пройдет 1 О см, пластинку
вынимают из камеры, сушат на воздухе, опры
скивают раствором анисов020 альде2ида Р, pac
ходуя 10 мл на пластинку площадью 200 мм 2 , И
сушат при температуре от 100 ос до 105 ос в Te
чение 10 мин. На xpoMaтorpaMMe должно обна
руживаться только одно основное пятно.
Бриллиантовый синий. 1012200. (6104 59 2].
См. Кислотный синий 83 Р.
Бром. Br 2 . (м. м. 159,8). 1012400. (7726 95 6].
Коричневато красная дымящаяся жидкость.
Малорастворим в воде, растворим в 96 % спирте.
d;g: около 3,1.
Брома раствор. 1012401.
30 r брома Р и 30 r калия бромида Р paCTBO
ряют в воде Р и доводят до объема 100 мл этим
же растворителем.
5 Бром-2'-дезоксиуридин. CgH11BrN205'
(М м. 307,1). 1012500. (59 14 3]. 5 Бром 1 (2
дезокси l3 d эритро пентофуранозил) 1 H,3H
пиримидин 2,4 дион.
Температура плавления: около 194 ОС.
Хромат02рафия. Определениепроводят, как
указано в статье Йодоксиуридин; наносят 5 мкл
раствора 0,25 r/л 5 бром 2' дезоксиуридина; на
полученной xpoMaTorpaMMe должно обнаружи
ваться только одно основное пятно.
Бромелайны. 1012300. [З7189 34 7].
Концентрат протеолитических ферментов,
полученный из Апапаs comosus Мерр.
Тускло желтый порошок.
Активность: 1 r бромелайнов должен BЫ
свобождать около 1,2 r аминноro азота из pacт
вора желатина Р в течение 20 мин при темпера
туре 45 ос и рН 4,5.
Бромелайнов раствор. 1012301.
Раствор 10 r/л 6ромелайнов Р в смеси фос
фатный буферный раствор рН 5,5 Р раствор
9 r/л натрия хлорида Р (1:9, об/об).
Бромистоводородной кислоты 30 % раст-
вор. 1098700. [10035 10 6].
300/0 раствор кислоты бромистоводородной
в кислоте уксусной ледяной Р.
Перед вскрытием содержимое осторожно
деrазируют.
Бромистоводородная кислота разведен-
ная. 1098701.
5,0 мл 30 % раствора кислоты 6pOMиcтo
водородной Р помещают во флаконы из TeMHO
ro стекла, укупоривают в атмосфере ар20на Р
полиэтиленовыми пробками и хранят в защи
щенном отсвета месте. Непосредственно перед
использованием прибавляют 5,0 мл кислоты YK
сусной ледяной Р и перемешивают.
Хранят в защищенном от света месте.
Бромистоводородной кислоты 47 % раст-
вор. 1118900.
Раствор 47 % (м/м) кислоты бромистоводо
родной В воде Р.
Бромистоводородная кислота разведен-
ная Р1. 1118901.
Содержит 7,9 r/л HBr.
16,81 r 47 % раствора кислоты бромисто
водородной Р растворяют в воде Р и доводят до
объема 1000 мл этим же растворителем.
Бромкрезоловый зеленый. C 21 H 1 .Br.OsS.
(м. м. 698). 1012600. [76 60 8]. 3',3",5',5" TeTp&
бром м крезолсульфофталеин. 4,4' (3H 2.1 Бе
зоксатиол 3 илиден )бис(2, 6 дибром 3 метил
фенол ) s, S диоксид.
Коричневато белый порошок. Малорастворим
в воде, растворим в 96 % спирте и в разведенных
растворах rидроксидов щелочных металлов.
Бромкрезоловоrо зеленоro PaCnIOP. 1012601.
50 Mr бромкрезолов020 зеленО2О Р pacтвo
ряют В 0,72 мл 0,1 М раствора натрия 2идpoK
сида и 20 мл 96 % спирта Р и доводят водой Р
до объема 100 мл.
Испытание на чувствитenыюсть. К 100 мл
воды, свободной от У2Лерода дurжcuдa. Р прибав
ляют 0,2 мл раствора бромкрезолoвorо зеленоro.
Появляется синее окрашивание, которое перехо
дит в желтое при прибавлении не более 0,2 мл
0,02 М раствора кислоты хлористоводородной.
Изменение окраски. От рН 3,6 (желтое OKpa
шивание) до рН 5,2 (синее окрашивание).
612
rосударственная фармакопея Республики Беларусь
Бромкрезоловоrо зеленоrо и метиловоrо
KpaCHoro раствор. 1012602.
0,15 r бромкрезолов020 зелеН020 Р и 0,1 r
метилов020 краСН020 Р растворяют в 180 мл
этанола Р и доводят водой Р до объема 200 мл.
Бромкрезоловый пурпуровый. C21H16Brp5S.
(м. м. 540,2). 1012700. [115-40 2]. 3',З" Дибром 0
крезолсульфофталеин. 4,4' (3H 2, 1 Бензоксати
ол 3 илиден )бис(2 бром 6 метилфенол ) s, s
диоксид.
Розоватый порошок. Практически HepaCTBO
рим в воде, растворим в 96 % спирте и в разведен
ных растворах rидроксидов щелочных металлов.
Бромкрезоловоrо nypnypoBoro раствор.
1012701.
50 Mr бромкрезолов020 пурпуров020 Р раст
воряют в 0,92 мл 0,1 М раствора натрия 2идpOK
сида и 20 мл 96 % спирта Р и доводят водой Р
до объема 100 мл.
Испытание на чувствительность. К
100 мл воды, свободной от У2лерода диOKcи
да, Р прибавляют 0,2 мл раствора бромкрезо
ловоro пурпуровоro и 0,05 мл 0,02 М раствора
натрия 2идроксида. Появляется синевато фи
олетовое окрашивание, которое переходит в
желтое при прибавлении не более 0,2 мл 0,02 М
раствора кислоты хлористоводородной.
Изменение окраски. От рН 5,2 (желтое OKpa
шивание) до рН 6,8 (синевато фиолетовое OKpa
шивание).
Бромная вода. 1012402.
3 мл брома Р встряхивают со 100 мл воды Р
до насыщения.
Хранят над избытком брома Р в защищен
ном от света месте.
Бромная вода Р1. 1012403.0,5 мл брома Р
встряхивают со 100 мл воды Р. Хранят в защи
щенном от света месте. Срок roдности 7 сут.
Бромометоксинафтол. C 11 H g BrO. (м. м.237, 1).
1159100. [5111 65 9]. 2 Бром 6--метоксинафтол.
Температура плавления: около 109 ОС.
Бромтимоловый синий. C27H28Br205S,
(М м. 624). 1012900. [76 59 5]. 3',3" Дибромти
молсульфофталеин. 4,4' (3H 2, 1 Бензоксатиол
З илиден )бис(2 бром 6 изоп роп ил 3 метил
фенол ) s, S диоксид.
Порошок от красновато розовоro до корич
неватоro цвета. Практически нерастворим в
воде, растворим в 96 % спирте и в разведенных
растворах rидроксидов щелочных металлов.
Бромтимоловоrо синеro раствор Р1. 1012901.
50 Mr бромтимолов020 сине20 Р paCTBO
ряют в смеси из 4 мл 0,02 М раствора натрия
2идроксида и 20 мл 96 % спирта Р и доводят
водой Р до объема 100 мл.
Испытание на чувствительность. К 100 мл
воды, свободной от уzлерода диоксида, Р при
бавляют 0,3 мл раствора бромтимоловоro синеro.
Появляется желтое окрашивание, которое пере
ходит в синее при прибавлении не более 0,1 мл
0,02 М раствора натрия 2идроксида.
Изменение окраски. От рН 5,8 (желтое OKpa
шивание) до рН 7,4 (синее окрашивание).
Бромтимоловоrо синеrо раствор Р2.
1012902
Раствор 1 О r/л бромтимолов020 сине20 Р в
диметилформамиде Р.
Бромтимоловоrо синеrо раствор РЗ.
1012903.
К 0,1 r бромтимолов020 сине20 Р прибавля
ют 3,2 мл 0,05 М раствора натрия 2идроксида и
5 мл спирта (90 %, об/об) Р и наrревают до раст
ворения. Полученный раствор охлаждают и дo
водят спиртом (90 %, об/об) Р до объема 250 мл.
Бромфеноловый синий. C1gH10Br405S.
(м. м. 670). 1012800. [115 З9 9]. 3',3",5',5" TeTpa
бромфенолсульфофталеин. 4,4' (3H 2,1 Бензо
ксатиол 3 илиден )бис(2,6 дибромфенол ) s, s
диоксид.
Светлый оранжево желтый порошок. Очень
мало растворим в воде, малорастворим в 96 %
спирте, леrкорастворим в растворах rидрокси
дов щелочных металлов.
Бромфеноловоrо синеrо раствор. 1012801.
0,1 r бромфенолов020 сине20 Р paCTBO
ряют в смеси из 1,5 мл 0,1 М раствора натрия
2идроксида и 20 мл 96 % спирта Р и доводят
водой Р до объема 100 мл.
Испытание на чувствительность. К 20 мл
воды, свободной от У2лерода диоксида, Р прибав
ляютО,05 млбромфеноловоroсинеroи 0,05 мл О, 1 М
раствора кислоты хлористоводородной. Появ
ляется желтое окрашивание, которое переходит в
синевато фиолетовое при прибавлении не более
0,1 мл 0,1 М раствора натрия 2идроксида.
Изменение окраски. От рН 2,8 (желтое OKpa
шивание) до рН 4,4 (синевато фиолетовое OKpa
шивание ).
Бромфеноловоrо синеrо раствор Р1.
1012802.
50 Mr бромфенолов020 сине20 Р растворяют
при осторожном наrревании в 3.73 мл 0,02 М
раствора натрия 2идроксида и доводят водой Р
до объема 100 мл.
Бромфеноловоrо синеrо раствор Р2.
1012803.
0,2 r бромфенолов020 сине20 Р растворяют
при наrревании в 3 мл 0,1 М раствора натрия
2идроксида и 1 О мл 96 % спирта Р, полученный
раствор охлаждают и доводят 96 % спиртом Р
до объема 100 мл.
4.1.1. Реактивы
613
Бруцин. С2ЗН26NР4'2Нр. (м. м. 430,5).
1013100. [357 57 3J. 10,11 Диметоксистрихнин.
Бесцветные кристаллы. Малорастворим в
воде, леrкорастворим в 96 % спирте.
Температура плавления: около 178 ос.
# Бура. См. Динатрия тетраборат Р.
Буры раствор. 1033601.
9,55 r динатрия тетрабората Р растворяют
в кислоте серной Р при наrpевании на водяной
бане и доводят до объема 1 л этой же кислотой.
1,4 Бутандиол. С 4 Н 10 О 2 . (м. м.90,12).
Прозрачная бесцветная жидкость. Coдep
жит не менее 99 % С 4 Н 10 О 2 .
d 20
20: около 1,017.
20
по: около 1,52.
Температура кипения: около 121 ос.
Бутанол. С 4 Н 10 О. (м. М. 74,1). 1013200.
[71 36 3J. н Бутанол. 1 Бутанол.
Прозрачная бесцветная жидкость. Смеши
вается с 96 % спиртом.
d;g: около 0,81.
Температура кипения: от 116 ос до 119 ос.
2 Бутанол Р1. С 4 Н I0 О. (М. М. 74,1). 1013301.
[78 92 2J. втор Бутиловый спирт.
Содержание: не менее 99,0 % С 4 Н I0 О.
Прозрачная бесцветная жидкость. PaCTBO
рим в воде, смешивается с 96 % спиртом.
d;g: около 0,81.
Температурные пределы пере20нки
(2.2.11). От 99 ос до 100 ос; должно переroнять
ся не менее 95 %.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Изо
пропиловый спирт.
# 2 Бутанон. См. Метилэтилкетон Р.
Бутиламин. C 4 H I1 N. (м. м. 73,1). 1013600.
[109 73 9J. 1 Бутанамин.
Переroняют и используют в течение 1 мес.
Бесцветная жидкость. Смешивается с водой
и с 96 % спиртом.
п O: около 1,401.
Температура кипения: около 78 ос.
трет Бутиламин. 1100900. [75 64 9J. СМ.
1, 1 Диметилэтиламин Р.
Бутилацетат. С 6 Н I Р2' (м. М. 116,2). 1013400.
[123 86 4J.
Прозрачная бесцветная воспламеняющаяся
жидкость. Малорастворим в воде, смешивается
с 96 % спиртом.
d 20
20: около 0,88.
п O: около 1,395.
Температурные пределы пере20нки (2.2.11).
От 123 ос до 126 ос; должно переroняться не
менее 95 %.
78 Зак.1712
Бутилацетат Р1. 1013401.
Прозрачная бесцветная воспламеняющаяся
жидкость. Малорастворим в воде, смешивается
с 96 % спиртом и эфиром.
d 20
20: около 0,883.
20
по: около 1,395.
Бутанол: не более 0,2 %. Определение nP<r
водят методом rазовой хроматоrрафии.
н Бутилформиат: не более 0,1 %. Onред
ление проводят методом rазовой хроматоrpaфии.
н Бутилпропионат: не более 0,1 %. ОПр&
деление проводят методом rазовой XpoMaтorpa--
фии.
Вода. Не более 0,1 %.
Количественное определение. Не менее
99,5 % С 6 Н 12 О 2 . Определение проводят методом
rазовой хроматоrрафии.
Бутилборная кислота. С 4 Н 11 ВО 2 . (М М. 101.9).
1013700. [442647 5J.
Содержание: не менее 98 % С 4 Н II ВО 2 .
Температура плавления: от 90 ос до 92 ос.
тpem-Бyтилrидpoпepoкc С 4 Н 1О О 2 . (М М oo.1
1118000. [75 91 2J. 1,1 Диметилэтилr
роксид.
Воспламеняющаяся жидкость. Растворим в
орrанических растворителях.
d 20
20: 0,898.
20
по: 1,401.
Температура плавления: 35 ос.
Бутилrидрокситолуол. С 15Н24 О. (М м. 220.4).
1013800. [128 З7 ОJ. 2,6 бис(1,1 ДимеТИЛЭПlЛ
4 метилфенол. Бутилированный rидрокano-
луол.
Белый или желтовато белый кристaJ1I1М-
ческий порошок. Практически нерастворим в
воде, очень мало растворим в ацетоне, леr
корастворим в 96 % спирте и в растительных
маслах.
Температура затвердевания (2.2.18): от
69 ос до 70 ос.
Бутилированный rидpoк 10131ЮО.
[128 37 OJ. СМ. БутиЛ2идрокситoпyon Р.
трет-Бутил метиловый эфмр. 1013900.
[1634 04 4J. СМ. 1, 1 Диметилэтилметиловый
эфир р.
трет Бутиловый спирт. См. 2 Метил
2 пропанол Р.
Бутиловый эфир параrидроксибензой-
ной кислоты. СIIНI40з' (М М. 194,2). 1103900.
[94 26 8J. Бутилпараrидроксибензоат. Бутил
4 rидроксибензоат.
Содержание: не менее 99,0 % и не более
100,5 % СIIНI40з'
614
rосударственная фармакопея Республики Беларусь
Белый или почти белый кристаллический по
рошок или бесцветные кристаллы. Очень леrко
растворим в воде, леrкорастворим в 96 % спирте
и в метаноле.
Бутилпараrидроксибензоат. См. Бутило
вый эфир пара2идроксибензойной кислоты Р.
Бутиролактон. С 4 НР2' (М м. 86,1). 1104000.
[9648 0]. Диrидро 2(3Н) фуранон. у Бутиролак
тон.
Маслянистая жидкость. Смешивается с
водой, растворим в метаноле.
п 5: около 1,435.
Температура кипения: около 204 ОС.
Буферный рабочий раствор для элек-
трофореза в системе натрия додецилсуль-
фат полиакриламидный rель (SDS PAGE).
1114900.
151,4 r триС(2идроксиметил)аминомета
на Р, 721,0 r 2лицина Р и 50,0 r натрия лаурил
сульфата Р растворяют в воде Р и доводят до
объема 5000 мл этим же растворителем. Непо
средственно перед использованием разводят
водой Р в 10 раз и перемешивают. рН (2.2.3) раз
веденноrо раствора должен быть от 8,1 до 8,8.
Буферный образцовый раствор (KOHцeH
трированный) для электрофореза в системе
натрия додецилсульфат полиакриламид
ный rель (SDS PAGE). 1115000.
1,89 r триС(2идроксиметил)аминомета
на Р, 5,0 .r натрия лаурилсульфата Р, 50 Mr
бромфеноловоео сине20 Р растворяют в воде Р.
Прибавляют 25,0 мл 2Лицерина Р и доводят
водой Р до объема 100 мл. Полученный раствор
доводят кислотой хлористоводородной Р до
рН 6,8 и разводят водой Р до объема 125 мл.
Буферный образцовый раствор (концен-
трированный) для электрофореза в системе
натрия додецилсульфат полиакриламид-
ный rель (SDS PAGE) для восстановитель
ных условий. 1122100.
3,78 r триС(2идроксиметил)аминометана Р,
10,0 r натрия додецилсульфата Р, 100 Mr бром
фенолов020 сине20 Р растворяют в воде Р. При
бавляют 50,0 мл 2Лицерина Р и доводят водой Р
до объема 200 мл. К полученному раствору при
бавляют 25,0 мл 2 меркаптоэтанола Р, доводят
кислотой хлористоводородной Р до рН (2.2.3)
6,8 и разводят водой Р до объема 250,0 мл.
Альтернативно в качестве восстанавливающе
ro вещества вместо 2 меркаптоэтанола может быть
использован дити(лреитол. В этом случае образцо
вый буферный раствор roтовят следующим обра
зом: 3,78 r триС(2идроксиметил)аминометана Р,
10,0 r натрия додецилсульфата Р, 100 Mr бром
фенолов020 сине20 Р растворяют в ваде Р. При
бавляют 50,0 мл 2Лицерина Р и доводят водой Р до
объема 200 мл. Полученный раствор доводят Kиc
лотой хлористоводородной Р до рН (2.2.3) 6,8 ..
разводят водой Р до объема 250,0 мл. Неп
ственно перед использованием прибавляют ди
отреитол Р до конечной концентрации 100 мМ.
Быстрый красный В, соль. См. Прочныu
красный В, соль Р.
Быстрый синий В, соль. См. Прочныu
синий В, соль Р.
# Вазелин.
См. статью #Вазелин.
Вазелин белый. См. Парафин МЯ2кий,
белый Р.
Вазелиновое масло. 1062000. [804247 5].
См. статью Парафин жидкий.
Валенцен. С 15 Н 24 . (М. м. 204,4). 1152100.
[4630 07 3]. 4I3Н,5Q Эремофила 1 (10), 11 диен.
(1 R, 7R,8aS) 1 ,8а Диметил 7 (1 метилэтенил)
1,2,3,5,6,7,8,8а--октаrидронафтален.
Маслянистая бесцветная до светло желтоro
цвета жидкость с характерным запахом. Практиче..-
ски нерастворим в воде, растворим в 96 % спирте.
d 20
4 : около 0,918.
п O: около 1,508.
Температура кипения: около 123 ОС.
Валенцен, используемый в 2азовой xpOMa
т02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Апель
синовое масло.
Содержание валенцена, рассчитанное MeTO
дом нормализации, должно быть не менее 90 %.
Валереноваякислота.С 15 Н 2 Р2' (М м. 234,3).
1165700. [3569 10 6]. (2E) 3 [(4S,7R,7aR) 3,7
Диметил 2,4,5,6, 7, 7а rесаrидро 1 Н инден 4 ил]
2 метилпроп 2 еновая кислота.
Температура плавления: от 134 ОС до 138 ОС.
Валериановая кислота. С 5 Н 1О О 2 . (М м. 102,1).
1095200. [109 52 4]. Пентановая кислота.
Бесцветная жидкость. Растворима в воде,
леrкорастворима в 96 % спирте.
d 20
20: около 0,94.
20
п о : около 1,409.
Температура кипения: около 186 ОС.
Ванадиевый анrидрид. См. Ванадия (V)
оксид Р.
Ванадия (V) оксид. VP5' (М. м. 181,9).
1034000. [1314 62 1]. Ванадиевый анrидрид. Ди
ванадия пентоксид.
4.1.1. Реактивы
615
Содержание: не менее 98,5 % V 2 0 5 .
Порошок от желто коричневоro до оранже
вато коричневоro цвета. Малорастворим в воде,
растворим в концентрированных минеральных
кислотах и растворах rидроксидов щелочных Me
таллов с образованием солей.
Прозрачность (2.2.1). 1 r ванадия (V) оксида
наrревают с 1 О мл кислоты серной Р в течение
30 мин, охлаждают и доводят до объема 1 О мл
этой же кислотой. Полученный раствор должен
быть прозрачным.
Чувствительность к водорода пероксиду.
1,0 мл раствора, приroтовленноro для испытания
на прозрачность, осторожно доводят водой Р до
объема50,Омл (испытуемый раствор). КО,5млис
пытуемоrо раствора прибавляют 0,1 мл раствора
водорода пероксида Р (содержит 0,1 r/л НР2)'
Полученный раствор должен иметь четко Bыpa
женную оранжевую окраску в сравнении с OKpa
ской контрольноro раствора, приroтовленноro из
0,5 мл испытуемоro раствора и 0,1 мл воды Р.
Оранжевое окрашивание испытуемоro раствора
должно перейти в оранжево желтое после при
бавления 0,4 мл раствора водорода пероксида
(содержит 0,1 r/л НР2)'
Потеря в массе при прокаливании. Не
более 1,0 %. 1,00 r испытуемою образца прока
ливают при температуре (700ж50) ОС.
Количественное определение. 0,200 r испыту
емоro образца растворяют при наrpeвании в 20 мл
70 % (м/м) раствора кислоты серной Р, прибавля
ют 100 мл воды Р и 0,02 М раствора калия перман
2аната до красноватоro окрашивания. Избыток
калия перманraната обесцвечивают прибавлением
раствора 30 r/л натрия нитрита Р. Прибавляют
5 r мочевины Р и 80 мл 70 % (м/м) раствора Kиc
лоты серной Р, охлаждают и немедленно титруют
0,1 М раствором железа сульфата до появления
зеленовато красною окрашивания, используя в Ka
честве индикатора 0,1 мл ферроина Р.
1 мл 0,1 М раствора железа сульфата co
ответствует 9,095 Mr V 2 0 5 .
Ванадия (V) оксида раствор в кислоте
серной. 1034001.
0,2 r ванадия (V) оксида Р растворяют в 4 мл
кислоты серной Р и доводят водой Р до объема
100 мл.
Ванилин. 1095300. [121 33 5J.
СМ. статью Ванилин.
Ванилина реактив. 1095301.
К 100 мл раствора 10 r/л ванилина Р в 96 %
спирте Р осторожно по каплям прибавляют 2 мл
кислоты серной Р.
Срок roдности 48 ч.
# Ванилина раствор в серной кислоте.
0,1 r ванилина Р растворяют в 10 мл кисло
ты серной Р. Раствор используют свежеприro
товленным.
Ванилина раствор в фосфорной кислоте.
1095302.
1,0 r ванилина Р растворяют в 25 мл 96 %
спирта Р, прибавляют 25 мл воды Р и 35 мл Kиc
лоты фосфорной Р.
# Ванилина раствор в хлористоводород-
ной кислоте.
0,2 r ванилина Р растворяют в 10 мл кисло
ты хлористоводородной Р. Раствор используют
свежеприrотовленным.
# Ван-Урка реактив. К 35 мл воды Р прили
вают при помешивании 65 мл кислоты серной Р
и в еще rорячий раствор вносят 0,03 мл раствора
100 r/л железа (111) хлорида Р. После охлаждения
раствора до 50 ос прибавляют 0,2 r диметила
минобензальде2ида Р. Реактив используют через
24 ч после приroтовления. Срок rодности 7 сут.
Винная кислота. 1087200. [87 694).
См. статью Винная кислота.
# Виннокаменная кислота. См. Винная Kиc
лота р.
Винилацетат. С 4 Н 6 О 2 . (М м. 86,10).1111800.
[108 05 4J. Этенилацетат.
d;g: около 0,930.
Температура кипения: около 72 ОС.
Винилмуравьиновая кислота. См. Акрило
вая кислота Р.
2-Винилпиридин. C 7 H 7 N. (М м. 105,1).
1102200. [100 69 6J.
Жидкость жептою цвета. Смешивается с водой.
d 20 О 9
20: около , 7.
20
П D : около 1,549.
1-8инилпирролидин-2-он.С 6 Н g NО.(Мм.111 ,1).
1111900. [88--12--0). 1 Этенилпирролидин 2--он.
Содержит не менее 99,0 % C 6 H g NO.
Прозрачная бесцветная жидкость.
Вода (2.5.12). Не более 0,1 %. Опредепение
проводят из 2,5 r 1 вилилпирролидин 2--она. ис
пользуя в качестве растворителя смесь 50 мл Me
танола безводН020 Р и 1 О мл бутиролактона Р.
Количественное определение. rазовая xpo
матоrрафия (2.2.28).
Хроматоrрафирование ПрО80ДЯТ на rазовом
хроматоrрафе с пламенно ионизационным дe
тектором в следующих условиях:
колонка кварцевая капиллярная длиной
30 м и внутренниt,,1 диаметром 0,5 мм, покрытая
слоем маКР020ла 20 000 Р толщиной 1,0 мкм;
rаз носитель: 2епий для хромат02рафии Р;
температура блока ввода пробы: 190 ОС;
температуру колонки проrраммируют
следующим образом: выдерживают темпера
туру 80 ос в течение 1 мин, затем повыша
616
rосударственная фармакопея Республики Беларусь
ют до 190 ос со скоростью 1 О ос в минуту и
выдерживают температуру 190 ос в течение
15 мин.
Хроматоrрафируют 0,3 мкл испытуемоro Be
щества, реryлируя скорость потока rаза носи
теля таким образом, чтобы время удерживания
пика, соответствующеro 1 винилпирролидин
2 OHY, составляло около 17 мин. Содержание
C 6 H g NO определяют методом внутренней HOp
мализации.
Винилполимер октадецилсилильный
для хроматоrрафии. 1121600.
Сферические частицы (5 мкм) сополиме
ра виниловоro спирта, связанноro октадецил
силаном.
Содержит 17 % уrлерода.
Винилхлорид. С 2 Н з СI. (М м. 62,5). 1095400.
[75 01A].
Бесцветный rаз. Малорастворим в орrаниче
ских растворителях.
# Висмута нитрат. Вi(NОз)з'5НР. (М м. 485,1).
Висмута (111) нитрат пентаrидрат.
Прозрачные бесцветные кристаллы или
белый или почти белый кристаллический по
рошок.
Висмута нитратосновной.4ВiNОз(ОНh'ВiO(ОН).
(М м. 1462). 1011500. [1304 85 4].
Белый или почти белый порошок. Практиче
ски нерастворим в воде.
Висмута нитрат основной Р1. 1011501.
Содержит не менее 71,5 % и не более
74,0 % висмута (Bi); не менее 14,5 % и не
более 16,5 % нитрата в пересчете на азота (V)
оксид (NP5)'
Висмута нитрата OCHoBHoro раствор.
1011502.
5 r висмута нитрата основН020 Р1 paCTBO
ряют в смеси из 8,4 мл кислоты азотной Р и
50 мл воды Р, доводят водой Р до объема 250 мл
и при необходимости фильтруют.
Кислотность. К 1 О мл прибавляют 0,05 мл
раствора метилов020 оранжев020 Р. При при
бавлении от 5,0 мл до 6,25 мл 1 М раствора
наrприя 2идроксида окраска раствора должна
измениться.
Вода. 1095500. [7732 18 5].
См. статью Вода очищенная.
Вода, свободная от аммиака. 1095501.
[7732 18 5].
К 100 мл воды Р прибавляют 0,1 мл кисло
ты серной Р, переroняют, используя прибор для
определения температурных пределов переrон
ки (2.2.11), отбрасывают первые 10 мл и собира
ют следующие 50 мл.
Вода, свободная от нитратов. 1095506..
[7732 18 5].
К 100 мл воды Р прибавляют несколько миJ1.
лиrраммов калия пермаН2аната Р и бария 2t.J-
дроксида Р. Переroняют, используя прибор для
определения температурных пределов переrо
ки (2.2.11), отбрасывают первые 10 мл и собира
ют следующие 50 мл.
Вода, свободная от уrлерода диоксида.
1 095502. [7732 18 5].
Воду Р кипятят в течение нескольких минут
и охлаждают, защищая от атмосферноro воздей
ствия. Хранят, защищая от атмосферноro воз
действия.
Вода, свободная от частиц. 1095507.
[7732 18 5].
Воду Р фильтруют через мембранный фильтр
с размером пор 0,22 мкм.
Вода дейтерированная. См. Дейтерия
оксид Р.
Вода дистиллированная. 1095504. [7732 18--5].
Вода Р, полученная путем переroнки.
Вода дистиллированная деионизирован
ная. 1095508.
Деионизированную воду Р получают дистил
ляцией. Значение удельноro сопротивления не
менее 18 МОм'м.
Вода для инъекций. 1095505. [7732 18 5].
См. статью Вода для инъекциЙ
Вода ДЛЯ хроматоrpафии. 1095503. [7732 18 5].
Деионизированная вода Р с удельным сопро
тивлением не менее 0,18 МОм'м.
Водород для хроматоrpафии. Н 2 (М м.2,О16).
1043700 [1333 74 0].
Содержит не менее 99,95 % (об/об) Н 2 .
Водорода пероксида раствор концентри
рованный. 1043900. [7722 84 1].
См. статью Водорода пероксида 30 % раствор.
Водорода пероксида раствор разведен
ный. 1043800. [7722 84 1].
См. статью Водорода пероксида 3 % раствор.
Восстанавливающая смесь. 1074700.
Измельчают до получения однородной смеси
вещества, "рибавляемые в следующем порядке:
20 Mr калия бромида Р, 0,5 r 2идразина сулъфа
та Р и 5 r натрия хлорида Р.
rалактоза. С 6 Н 1 Р6' (М М. 180,2). 1039700.
[59 23 4]. D (+) rалактоза.
Белый или почти белый кристаллический
порошок. Леrкорастворима в воде.
4.1.1. Реактивы
617
[a] O: от +79 до +81. Определение проводят,
используя раствор 100 r/л испытуемоro образца
в воде Р, содержащей около 0,05 % NН з .
rалловая кислота. С7Н605'НР' (М м. 188,1).
1039800. [5995 86 8). З,4,5 Триrидроксибензой
ная кислота моноrидрат.
Кристаллический порошок или длинные
иroльчатые кристаллы, бесцветные или слеrка
желтоватоro цвета. Растворима в воде, леrко
растворима в roрячей воде, в 96 % спирте и в
rлицерине.
rалловая кислота теряет кристаллизацион
ную воду при температуре 120 ОС.
Температура плавления: около 260 ос с
разложением.
Хромат02рафия. Тонкослойная xpOMaTO
rрафия (2.2.27), как указано в статье Листья тo
локнянки. На xpoMaTorpaMMe должно обнаружи
ваться только одно основное пятно.
rарпаroзид. С24НЗОО11" (М м. 494,5). 1098600.
Белый или почти белый кристаллический по
рошок. Очень rиrроскопичен. Растворим в воде и
в 96 % спирте.
Температура плавления: от 117 ОС до 121 ОС.
Хранят в во:щухонепроницаемом контейнере.
rвайазулен. С 15 Н 1в . (М м. 198,3). 1041500.
[489 84 9). 1,4 Диметил 7 изопропилазулен.
Кристаллы темно синеro цвета или жид
кость синеro цвета. Очень мало растворим
в воде, смешивается с жирными и эфирны
ми маслами, вазелиновым маслом, умеренно
растворим в 96 % спирте, растворим в paCTBO
ре 500 r/л кислоты серной и 80 % (м/м) paCTBO
ре кислоты фосфорной с образованием бесц
ветноro раствора.
Температура плавления: около 30 ОС.
Хранят в защищенном от света и во:щуха месте.
rваяковая смола. 1041400. Смола, полу
ченная из сердцевины дерева Guaiacum officiпa
'е L. и Guaiacum sапсtит L.
Твердые rладкие частицы KpaCHOBaTO KO
ричневоrо или зеленовато коричневатоro цвета,
блестят на изломе.
rвайякол. С 7 НР2' (М. м. 124,1). 1148300.
[90 05 1]. 2 Метоксифенол. 1 rидрокси 2 мето
ксибензол.
Кристаллическая масса или бесцветная или
желтоватая жидкость. rиrроскопичен. Малораст
ворим в воде, очень леrко растворим в метилен
хлориде, леrкорастворим в 96 % спирте.
Теtv'пература кипения: 205 ОС.
Температура плавления: 28 ОС.
rексадиметрина бромид. (С1зНзоВr2N2)П'
1042300. [28728--55-4]. 1 , Диметил 1, диазаун
декаметилен полиметобромид. Поли(1, 1 ,5, TeTpa
метил 1 ,5--азония ундекаметилен дибромид).
79 За.. 1712.
Белый или почти белый аморфный поро
шок. rиrроскопичен. Растворим в воде.
Хранят в во:щухонепроницаемом контейнере.
rексакозан. С 26 Н 54 . (М м. 366,7). 1042200.
[6ЗО 01 3].
Бесцветные или бепые или почти белые хлопья.
Температура плавления: около 57 ОС.
2,2' ,2" ,6,6' ,6"-rекса(1, 1-диметилэтил)
4,4' ,4" [(2,4,6-триметил-1 ,З,5 бензонитрил)
трисметилен] трифенол. С 54 Н 7 Рз, (М. м. 775).
1042100. 2,2' ,2" ,6,6' ,6" rекса трет бутил-4,4' ,4"
[(2,4.6 триметил 1 ,3,5 бензонитрил)трисмети
лен ]трифенол.
Кристаллический порошок. Практически He
растворим в воде, растворим в ацетоне, мало
растворим в 96 % спирте.
Температура плавления: около 244 ОС.
rексаметилдисилазан. C 6 H 19 NSi 2 . (М м. 161,4).
1042400. [999 97 З].
Прозрачная бесцветная жидкость.
d;g: около 0,78.
20
п о : около 1,408.
Температура кипения: около 125 ОС.
Хранят в во:щухонепроницаемом контейнере.
rексаметилентетрамин. C 6 H 12 N 4 . (М м. 140,2).
1042500. [100 97 O]. rексамин. 1,З.5,7 Тетра аза
трицикло[З.З.1.Р.7]декан. #rексамин.#
Бесцветный кристаллический порошок.
Очень леrко растворим в воде.
# rексамин. См. rексаметилентетрамин р
reKcaH. С 6 Н 14 . (М м. 86,2). 1042600. [110 54 3].
Бесцветная воспламеняющаяся жидкость.
Практически нерастворим в воде, смешивается
с этанолом.
d;g: от 0,659 до 0,663.
20
п о : от 1,375 до 1,З76.
Температурные пределы переzoнки (2.2. 11). от
67 ос до 69 ОС; должно переroняться не менее 95 %.
reKcaH, используемый в спектpoфoirюмfr
трии, должен выдерживать следующее допол
нительное испытание.
Пропускание (2.2.25): Не менее 97 %. Опре.-
деление проводят в области дЛИН волн от 260 нм
до 420 нм, используя в качестве комneнcaцион
ной жидкости воду Р
1,1,1,З,З,з-rексафторпponан-2-ол. С з Н 2 F 60.
(М м. 168,0). 1136000. [92 1].
Содержание: не менее 99.0 %, определен
ное методом rазовой хроматоrpафии.
Прозрачная бесцветная жидкость. Смеши
вается с водой и этанолом.
d;g: около 1,596.
Температура кипения: около 59 ОС.
618
rосударственная фармакопея Республики Беларусь
rексиламин. C6H'5N. (М м. 101,2). 1042700.
[111 26 2]. rексанамин.
Бесцветная жидкость. Малорастворим в
воде, растворим в 96 % спирте.
d 20
20: около 0,766.
20
П D : около 1,418.
Температура кипения: от 127 ос до 131 ОС.
rелий для хроматоrрафии. Не (А. м. 4,003).
1041800. [7440 59 7].
Содержит не менее 99,995 % (об/об) Не.
rемоrлобин. 1041700. [9008 02 0].
Азот: от 15 % до 16 %.
Железо: от 0,2 % до 0,3 %.
Потеря в массе при высушивании (2.2.32):
не более 2 %.
Сульфатная зола (2.4.14): не более 1,5 %.
rемоrлобина раствор. 1041701.
2 r 2еМ02лобина Р помещают в стакан BMe
стимостью 250 мл, прибавляют 75 мл кислоты
хлористоводородной разведенной Р2 и пере
мешивают до полноrо растворения. Доводят
рН (2.2.3) 1 М раствором кислоты хлористо
водородной до 1,6:!:О,1. Полученный раствор
переносят в колбу вместимостью 100 мл С по
мощью раствора кислоты хлористоводород
ной разведенной Р2 и прибавляют 25 Mr тиo
мерсаля Р
Срок roдности 1 сут.
Хранят при температуре (5:!:3) ОС; перед ис
пользованием снова доводят рН до 1,6.
# rенциановый фиолетовый. См. Meти
ловый фиолетовый Р.
rепарин. 1041900. [9041 08 1].
См. статью rепарин натрия.
renTaH. С 7 Н'6' (М м. 100,2). 1042000. [142 2 5].
Бесцветная воспламеняющаяся жидкость.
Практически нерастворим в воде, смешивается
с этанолом и эфиром.
d 20
20: от 0,683 до 0,686.
20
п о : от 1,387 до 1,388.
Температурные пределы пере20нки
(2.2.11). От 97 ос до 98 ОС; должно переroняться
не менее 95 %.
rеранилацетат. С'2Н2002 (М м. 196,3).
1106500. [105 87 3]. (Е) 3,7 Диметилокта 2,6
диен 1 ил ацетат.
Бесцветная или слабо желтая жидкость со
слабым запахом розы и лаванды.
rеранилацетат, используемый в 2азовой
хромат02рафии, должен выдерживать следу
ющее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
цветков померанца, используя rеранилацетат в
качестве испытуемоro раствора.
Содержание rеранилацетата, рассчитанное
методом нормализации, должно быть не менее
99,0%.
rиалуронидазыразбавитель.104330Q
0,140 r желатина 2идролизоваНН020 Р paCT
воряют при температуре 37 ос в 200 мл смеси из
равных объемов фосфатН020 буферН020 pacт
вора рН 6,4 Р и воды Р.
Срок roдности 2 ч.
rидразин. H 4 N 2 . (М м. З2,05). 1136300.
[302 01 2]. Диазан.
Слеrка маслянистая бесцветная жидкость
с сильным запахом аммиака. Смешивается
с водой. Коммерчески доступны водные pac
творы.
Предостережение: токсичный и разъеда
ющий.
п O: около 1,470.
Температура кипения: около 113 ОС.
Температура плавления: около 1,5 ОС.
rидразина сульфат. H6NP4S. (М м. 1ЗО,1).
1043400. [10034 93 2].
Бесцветные кристаллы. Умеренно paCTBO
рим в холодной воде, растворим в rорячей воде
(50 ОС) и леrкорастворим в кипящей воде, прак
тически нерастворим в 96 % спирте.
Мышьяк (2.4.2, методА). Не более 0,0001 %
(1 ррт). 1,0 r должен выдерживать испытание на
мышьяк.
Сульфатная зола (2.4.14). Не более 0,1 %.
# rидразина rидрохлорид. H 4 N 2 '2HCI.
(М м. 104,96). rидразин солянокислый.
Белый или почти белый кристаллический
порошок. Токсичен.
rидрокортизона ацетат. 1098800. [50 03 3].
См. статью rидрокортизона ацетат.
4-rидроксикумарин. С g Н 6 О з . (М м. 162,2).
1169700. [1076 38 6]. 4 rидрокси 2Н 1 бензо
пиран 2 он.
Белый или почти белый порошок. Леrкорас
творим в метаноле.
Содержание: не менее 98,0 %.
rидроксиламина rидрохлорид. NH 4 CIO.
(М м. 69,5). 1044300. [5470 11 1].
Белый или почти белый кристаллический
порошок. Очень леrко растворим в воде, paCTВO
рим в 96 % спирте.
rидроксиламина rидрохлорида раст-
вор Р2. 1044304.
2,5 r 2идроксиламина 2идрохлорида Р paCT
воряют в 4,5 мл rорячей воды Р, прибавляют
40 мл 96 % спирта Р, 0,4 мл раствора бромфе
4.1.1. Реактивы
619
нолов020 сине20 Р2 и достаточное количество
0,5 М раствора калия 2идроксида спиртов020
до появления зеленовато желтоro окрашивания.
Доводят 96 % спиртом Р до объема 50,0 мл.
rидроксиламина раствор спиртовой.
1044301.
З,5 r 2идроксиламина 2идрохлорида Р pacтвo
ряют в 95 мл спирта (60 %, об/об) Р, прибавляют
0,5 мл раствора 2 r/л метилО8ОZO оранжевоzo Р в
спирте (60 %, об/об) Р и достаточное количество
0,5 М раствора калия 2идpoKcUдa в спирте (60 %,
об/об) до получения четкоro жеmоro окрашивания.
Доводят спиртом (60 %, об/об) Р до объема 100 мл.
rидроксиламина раствор щелочной.
1044302.
Непосредственно перед использованием
смешивают равные объемы раствора 139 r/л
2идроксиламина 2идрохлорида Р и раствора
150 r/л натрия 2идроксида Р
rидроксиламина раствор щелочной Р1.
1044303.
Раствор А. 12,5 r 2идроксиламина 2идpo
хлорида Р растворяют в метаноле Р и доводят
до объема 100 мл этим же рt'!створителем.
Раствор В. 12,5 r натрия 2идроксида Р
растворяют в метаноле Р и доводят до объема
100 мл этим же растворителем.
Непосредственно перед использованием
смешивают равные объемы растворов А и В.
# rидроксиламина раствор щелочной РЗ.
Раствор А. 10 r 2идроксиламина 2идрохло
рида Р растворяют в воде Р и доводят до объема
100,0 мл этим же растворителем.
Раствор В. 1 О r натрия 2идроксида Р paCT
воряют в воде Р и доводят до объема 100,0 мл
этим же растворителем.
Непосредственно перед использованием
смешивают растворы А и В (1 :2, об/об).
# rидроксиламина сульфат. N2H602'H2S04'
(м. м. 164,14). rидроксиламин сернокислый.
Бесцветные кристаллы.
4 rидроксибеН30йная кислота. С 7 Н 6 О з .
(М м. 138,1). 1106700. [99 96 7].
Кристаллический порошок. Очень мало pac
творима в воде, очень леrко растворима в 96 %
спирте, растворима в. ацетоне.
Температура плавления: от 214 ос до 215 ОС.
4 rидроксиизофталевая кислота.
С в Н 6 О 5 . (М м. 182,1). 1106900. [636 464].
4 rидроксибензол 1 ,3 дикарбоновая кислота.
Иroльчатые или в виде пластинок кристал
ЛЫ. Очень мало растворима в воде, леrкорас
творима в 96 % спирте и эфире.
Температура плавления: около 314 ос с раз
ложением.
rидроксимет-илфурфурол. С 6 Н 6 О з .
(м. м. 126,1). 1044400. [67 4 7 O]. 5 rидрокси
метилфурфураль.
Иrольчатые кристаллы. Леrкорастворим в
воде, ацетоне и в 96 % спирте.
Температура плавления: около 32 ОС.
rидроксинафтоловоrо синеrо натриевая
соль. С20Н11N2NаР11SЗ' (м. м. 620). 1044500.
[63451 354]. Тринатрия 2,2' диrидрокси 1, 1'
азонафталин 3' ,4,6' трисульфонат.
12 rидроксиолеиновая кислота. См. Pици
нолеиновая кислота Р
12 rидроксистеариновая кислота.
С 1в Н з Рз, (м. м. 300,5). 1099000. [106 14 9].
12 rидроксиоктадекановая кислота.
Белый или почти белый порошок.
Температура плавления: от 71 ос до 74 ОС.
5 rидроксиурацил. С 4 Н 4 NРз, (М м. 128,1).
1044700. [496 764]. Изобарбитуровая кислота.
Пиримидин 2,4,5 триол.
Белый или почти белый кристаллический
порошок.
Температура плавления: около 310 ос с раз
ложением.
Хромат02рафия. Определение проводят,
как указано в статье Фторурацил. На xpo
MaTorpaMMe должно обнаруживаться только
одно основное пятно с R, около 0,3.
Хранят в воздухонепроницаемом контей
нере.
rидроксихинолин. C g H 7 NO. (М м. 145,2).
1 044600. [148 24 3]. 8 rидроксихинолин.
Хинолин 8 ол.
Белый или слеrка желтоватый кристалли
ческий порошок. Малорастворим в воде, леr
корастворим в ацетоне, в 96 % спирте и в раз
веденных минеральных кислотах.
Температура плавления: около 75 ОС.
Сульфатная зола (2.4.14). Не более
0,05 %.
#20 rИДрОКСИЭКДИ30Н. С 27 Н 4 .о 7 . (М М. 480,6).
[5289 74 7]. 213,313, 14а,2013,22,25 rексаrидрокси
7 холестен 6 он. Экдистерон. I3 Экдизон. Поли
подин А.
Содержит не менее 95 % С 27 Н 44 О]"
rидрохинон. С 6 Н 6 О 2 . (М М. 110,1). 1044100.
[123 31 9]. Бензол 1 +диол.
Бесцветные или белоro или почти белоro
цвета иrольчатые мелкие кристаллы, TeMHe
ющие под действием воздуха и света. PaCTBO
рим в воде и в 96 % спирте.
Температура плавления: около 173 ОС.
Хранят в защищенном от света и воздуха
месте.
620
rосударственная фармакопея Республики Беларусь
rидрохинона раствор. 1044101.
0,5 r 2идрохинона Р растворяют в воде Р,
прибавляют 20 мкл кислоты серной Р и доводят
водой Р до объема 50 мл.
rиосциамина сульфат. СЗ4Н4вN201QS'2Н20,
(м. М. 71З). 1044900. [620 61 1]. Бис[(1R,3r,5S)
8 метил 8 азабицикло[3.2.1 ]окт 3 ил (2S)
3 rидрокси 2 фенилпропаноат]сульфат диrидрат.
Содержит не менее 98,0 % и не более 101,0 %
СЗ4Н4вN2010S В пересчете на сухое вещество.
Белый или почти белый кристаллический
порошок или бесцветные иrолки. Очень леrко
растворим в воде, умеренно растворим или pac
творим в 96 % спирте.
Температура плавления: около 203 ос с раз
ложением.
rиосцина rидробромид. C17H22BrN04'3H20,
(м. м.438,3). 1 044800. [6533 68 2]. (1 R,2R,4S,5S, 7 s)-
9 Метил 3 оксо 9 азотрицик ло[3.3.1.02.4]
HOH 7 ил(2S) 3 rидрокси 2 фенил пропаноат
rидробромид триrидрат. #Скополамина rидро
бромид.#
Содержит не менее 99,0 % и не более
101,0 % C 17 H 22 BrN0 4 в пересчете на безводное
вещество.
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Леrкорас
творим в воде, растворим в 96 % спирте.
Температура плавления: около 197 ос с раз
ложением, определяемая после высушивания
под вакуумом в течение 24 ч и затем при темпе
ратуре от 100 ос до 105 ос в течение 2 ч.
rиперозид. C21H2Q012' (м. М. 464,4).1045000.
2 ( 3, 4 Д и rидроксифен ил ) 3 r3 D rала KTO
пиранозилокси 5, 7 диrидроксихромен4 ОН.
Иroльчатые кристаллы светло желтоro
цвета. Растворим в метаноле.
[a] O: 8,3. Определение проводят, исполь
зуя раствор 2 r/л испытуемоro образца в пири
дине Р.
Температура плавления: около 240 ос с раз
ложением.
Раствор в метаноле Р имеет два максиму
ма поrлощения (2.2.25) при длинах волн 259 нм
и 364 нм.
rипоксантин. C 5 H 4 Np. (м. м. 136,1).
1 045300. [68 94 0]. 1 Н Пурин 6 он.
Белый или почти белый кристаллический
порошок. Очень мало растворим в воде, YMe
ренно растворим в кипящей воде, растворим
в разведенных кислотах и разведенных pac
творах rидроксидов щелочных металлов. Pa:.'l
лаrается при температуре около 150 ос (не
плавясь).
Хромат02рафия. Тонкослойная xpOMaTO
rрафия (2.2.27), как указано в статье Меркап
топу рин. На xpoMaTorpaMMe должно обнаружи
ваться только одно основное пятно.
rипофосфита реактив. 1045200.
1 О r натрия 2ипофосфита Р растворяют
при слабом наrревании в 20 мл воды Р, доводят
кислотой хлористоводородной Р до объема
100 мл, отстаивают и декантируют или фильтру
ют через стекловату.
rистамина диrидрохлорид. С 5 Н g N з '2НСI.
(м. м. 184,1). 1 042800. [56 92 8]. 2 (1 Н Имидазол
4 ил )этанамина диrидрохлорид.
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. rиrроско
пичен. Очень леrко растворим в воде, paCTBO
рим в 96 % спирте.
Содержит не менее 98,5 % и не более 101,0%
С5Н11СI2Nз в пересчете на сухое вещество.
rистамина фосфат. С5НgNз'2НзР04'НР'
(М. м. 325,2). 1042900. [2З297 93 0]. 2 (1Н Ими
дазол4 ил)этанамина дифосфат.
Бесцветные длинные призматические кри
сталлы. Леrкорастворим в воде, малорастворим
в 96 % спирте.
Содержит не менее 98,0 % и не более 101,0 %
С5НgNз'2НзР04 в пересчете на безводное вещество.
rистамина раствор. 1042901.
Раствор 9 r/л натрия хлорида Р, содержащий
0,1 мкr/мл rистамина фосфата или rистамина ди
rидрохлорида в пересчете на rистамин снование.
rистидина rидрохлорид моноrидрат.
C6HgNP2'HCI'Hp. (м. м. 209,6). 1043000.
[12ЗЗ33 71 1]. (RS) 2 Амино 3 (имидазол4 ил)
пропионовой кислоты rидрохлорид моноrидрат.
Бесцветные кристаллы или кристалличе
ский порошок. Растворим в воде.
Температура плавления: около 250 ос с раз
ложением.
Хромат02рафия. Тонкослойная xpOMaTO
rрафия (2.2.27), как указано в статье tUCmaMUHa
ди2идрохлорид. На xpoMaTorpaMMe должно об
наруживаться только одно основное пятно.
rитоксин. С 41 Н 6 Р14' (м. М. 781). 1040200.
[4562 36 1]. rликозидDigitаlisриrриrеаL.3r3 ( 0 2,6
Дидезокси r3 D рибо rексопиранозил ( 1 4 ) o
2,6 дидезокси r3 D рибо rексопиранозил ( 1 4)
2, 6 дидезокси r3 D рибо rексопиранозилокси)
14, 16r3 диrидрокси 5r3, 14r3 кард 20(22) енолид.
Белый или почти белый кристаллический
порошок. Практически нерастворим в воде и в
большинстве орrанических растворителей, pac
творим в пиридине.
[a] O: от +20 до +24. Определение проводят,
используя раствор 5 r/л испытуемоro образца
в смеси растворителей хлороформ Р Meтa
нол Р (50:50, об/об).
Хромат02рафия. Тонкослойная xpOMaTO
rрафия (2.2.27), как указано в статье Листья Ha
перстянки. На xpoMaTorpaMMe должно обнару
живаться только одно основное пятно.
4.1.1. Реактивы
621
rликолевая кислота. С 2 Н 4 О з . (М м. 76,0).
1040800. [79 14 1]. 2 rидроксиуксусная кислота.
Кристаллы. Растворима в воде, в ацетоне, в
96 % спирте и в метаноле.
Температура плавления: около 80 ОС.
# rликокол. См. Iлицин Р.
rлиоксальrидроксианил. C14H12N202'
(М м. 240,3). 1041000. [1149 16 2]. rлиоксаль
бис(2 rидроксианил).
Белые или почти белые кристаллы. PaCTBO
рим в rорячем 96 % спирте.
Температура плавления: около 200 ОС.
rлиоксаля раствор. 1098400. [107 22 2].
Содержит около 40 % (м/м) rлиоксаля.
Количественное определение. 1,000 r paCT
вора rлиоксаля помещают в колбу с притертой
стеклянной пробкой, прибавляют 20 мл раст
вора 70 r/л 2идроксиламина 2uдрохлорuда Р и
50 мл воды Р, выдерживают в течение 30 мин
и титруют 1 М раствором натрия 2идроксида
до перехода окраски от красной до зеленой, ис
пользуя в качестве индикатора 1 мл смешаНН020
раствора метилов020 краСН020 Р.
Параллельно проводят контрольный опыт.
1 мл 1 М раствора натрия 2идроксида co
ответствует 29,02 Mr rлиоксаля (С 2 Н 2 О 2 ).
rлицерин. 1040500. [56 81 5].
См. статью Iлицерин.
rлицерин Р1. 1040501.
Должен выдерживать требования статьи
Iлицерин и при использовании для определения
примеси А и сопутствующих примесей, как YKa
зано в статье Iлицерин, должен быть свободным
от диэтиленrликоля.
rлицерин 85 %.1040600.
См. статью Iлицерин 85 %.
rлицерин 85 % Р1. 1040601.
Должен выдерживать требования статьи
Iлицерин 85 % и при использовании для опре
деления примеси А и сопутствующих приме
сей, как указано в статье Iлицерин 85 %, должен
быть свободным от диэтиленrликоля.
# rлицерина раствор.
33 мл 2лицерина Р доводят водой Р до
объема 100 мл И прибавляют крупинку камфо
ры Р или одну каплю фенола жидК020 Р.
rлицин. 1040700. [5640 6].
См. статью Iлицин.
rлицирретиновая кислота. СЗОН4604'
(М м. 470,7). 1040900. [471 53 4]. rлициррети
новая кислота. 12, 1 3 Дидеrидро З/3 rидрокси 11
оксо олеан 30 овая кислота.
80 За"- 1712.
Смесь o.. и /3 rлицирретиновых кислот, в KO
торой преобладает /3 изомер.
Порошок от белою до желтоватcrкоричнево
ro цвета. Практически нерастворима в воде, pac
творима в этаноле и кислоте уксусной ледяной.
[a] O: от +145 до +155. Определение прово-
дят, используя раствор 10,0 r/л испытуемoro
разца в этаноле Р.
Хромат02рафия. Тонкослойная xpoмaтorpcr
фия (2.2.27), используя в качестве ТOHKoro споя
силика2ель GF 254 Р, суспензию котороro roтовят
используя 0,25 % (об/о6) раствор кислоты фос
форной Р. На хроматоrpафическую пластинку Ha
носят 5 мкл раствора 5 r/л кислоты rлициррети
новой в смеси из равных объемов хлороформа Р
и метанола Р. Хроматоrрафируют в смеси раст
ворителей метанол Р хлороформ Р (5:95, об!
06). Korдa фронт растворителей пройдет 10 см,
xpoMaтorpaMMY просматривают в ультрафиоле.-
товом свете при длине волны 254 нм. На xpOMa
тorpaMMe должно обнаруживаться темное пятно
(Rfоколо 0,3), соответствующее J3 rлицирретиновой
кислоте, и меньшее пятно (Rf около 0,5), COOTBeT
ствующее Q rлицирретиновой кислоте. Пластинку
опрыскивают раствором анисов020 альде2ида Р и
наrpевают при температуре от 100 ос до 105 ос в
течение 10 мин. оба пятна должны быть окраше.-
ны в синевато-фиолетовый цвет; ме>IЩУ ними дcr
пускается наличие меньшеro пятна синевато-фиcr
летовоro цвета.
rлутаминовая кислота. 1040400. [56 86 0].
См. статью Iлутаминовая кислота.
rлутаровая кислота. С 5 Н в 0 4 . (М м. 132,1).
1149700. [110 94 1]. Пентандионовая кислота.
Белый или почти белый кристаллический
порошок.
rлутаровыйальдеrид. С 5 Н в 0 2 . (М м. 100,1).
1098300. [111 ЗО 8].
Маслянистая жидкость. Растворим в воде.
п 5: около 1,434.
Температура кипения: около 188 ОС.
rлюкоза. 1025700. [50 99 7].
См. статью Iлюкоза безводная.
rлюкозамина rидрохлорид. C 6 H,.CIN0 5 -
(М м. 215,6). 1040300. (66 84 2].
D rлюкозамина rидрохлорид.
Кристаллы. Растворим в воде, практически
не растворим в эфире.
(a] O: +100, снижающееся до +47,5 через
30 мин. Определение проводят, ИCN>льзуя paCT
вор 100 r/л испытуемоro образца в вoд Р.
D-rлюкуроновая кислота. С 6 Н 10 О 7
(М м. 194,1). 1119700. (6556 12 3].
Содержит не менее 96,0 % С 6 Н 1О О 7 В пере
счете на сухое вещество, высушенное в вaKY
уме (2.2.32).
622
rосударственная фармакопея Республики Беларусь
Растворима в воде и в 96 % спирте.
Обладает мутаротацией: [a] 4: +11,7 ------+ +36,3.
Количественное определение. 0,150 r paCТBO
ряют при перемешивании в метаноле безводном Р
и титруют 0,1 М раствором тетрабутиламмония
2идроксида потенциометрически (2.2.20), защищая
раствор от воздействия уrлерода диоксида воздуха
во время растворения и титрования.
1 мл 0,1 М раствора тетрабутиламмония
zидроксида соответствует 19.41 Mr С 6 Н,о07'
rольмия (111) оксид. Норз. (м. м. 377,9).
1043100. [12055 62 8]. Диroльмия триоксид.
Порошок желтоватоro цвета. Практически
нерастворим в воде.
rольмия перхлората раствор. 1043101.
Раствор 40 r/л 20льмия (111) оксида Р в pac
творе кислоты хлорной Р (141 r/л HCIOJ
rуанидина rидрохлорид. СН 5 N з 'НСI.
(М. м. 95,5). 1098500. [50 01 1].
Кристаллический порошок. Леrкорастворим
в воде и в 96 % спирте.
rуанин. C 5 H 5 Np. (М. м. 151,1). 1041600.
[73-40 5]. 2 Амино 1, 7 диrидро 6Н пурин 6 он.
Аморфный белый или почти белый порошок.
Практически нерастворим в воде, малораство
рим в 96 % спирте, растворим в растворах aM
миака и разведенных растворах rидроксидов
щелочных металлов.
rуммиарабик. 1000100. Аравийская камедь.
Почти полностью, но очень медленно (в Te
чение 2 ч) растворим в двойном от ero массы
объеме воды. На поверхности остаются очень
маленькие кусочки растительных частиц. По
лученная жидкость бесцветная или желтова
тая, плотная, вязкая, клейкая (липкая), просве
чивающаяся и имеющая слабокислую реакцию
по синей лакмусовой бумажке. Практически He
растворим в 96 % спирте.
rуммиарабика раствор. 1000101.
100 r 2уммиарабика Р растворяют в 1000 мл
воды Р при перемешивании механической Me
шалкой в течение 2 ч. Центрифуrируют с YCKope
нием около 2000 9 в течение 30 мин до получе
ния прозрачноrо раствора.
Хранят в полиэтиленовом контейнере BMe
стимостью около 250 мл при температуре от
О ОС до 20 ОС.
Дантрон. С'4НР4' (м. м. 240,2). 1024500.
[117 1 0 2]. 1,8 Диrидроксиантр:зхинон. 1 ,8 Диrид
роксиантрацен 9, 1 О дион.
Оранжевый кристаллический порошок.
Практически нерастворим в воде, малораство
рим в 96 % спирте, растворяется в растворах rи
дроксидов щелочных металлов.
Температура плавления: около 195 ОС.
Дантрон, используемый при количествен
ном определении сесквитерпеновых кислот в
Валерианы корневищах с корнями, должен вы
держивать следующие дополнительные испы
тания.
А;;:: от 355 до 375. Определение проводят
при длине волны 500 нм, используя раствор в
1 М растворе калия 2идроксида.
Количественное определение. Жидкост
ная хроматоrрафия (2.2.29), как указано в статье
Валерианы корневища с корнями в KOHцeHTpa
ции раствора сравнения. Содержание дантрона,
рассчитанное методом нормализации, должно
быть не менее 95 %.
# Деварда сплав. См. Сплав Деварда Р.
2 Дезокси D рибоза. С 5 Н 10 О 4 . (м. м. 134,1).
1163900. [533 67 5]. Тиминоза. 2 Дезокси D
эритро пентоза.
2' Дезоксиуридин. C g H'2NP5' (М. м. 228,2).
1024800. [951 78 ]. Н2 ДезокСи I3 D эритро
пентофуранозил) 1 Н,3Н пиримидин 2,4 дион.
Температура плавления: около 165 ОС.
Хромат02рафия. Определение проводят,
как указано в статье Йодоксиуридин; наносят
5 мкл раствора 0,25 r/л испытуемоro образца. На
xpoMaтorpaMMe должно обнаруживаться только
одно основное пятно.
Дейтерия оксид. 2Нр. (м. м. 20,03).
1025300. [7789 20 0]. Вода дейтерированная.
Степень дейтерирования не менее 99,7 %.
d; : около 1,11.
20
по: около 1,328.
Температура кипения: около 101 ОС.
Дейтерированная уксусная кислота.
С 2 2НР2' (м. м. 64,1). 1101100. [1186 52 3]. Te
традейтероуксусная кислота. Уксусная dз
кислота d.
Степень дейтерирования не менее 99,7 %.
d 20
20: около 1,12.
20
по: около 1,368.
Температура кипения: около 115 ОС.
Температура плавления: около 16 ОС.
Дейтерированный ацетон. С з 2Н 6 О.
(м. м. 64,1). 1024900. [666 52-4]. Ацетон d6' eH6)
Ацетон.
Степень дейтерирования не менее 99,5 %.
Прозрачная бесцветная жидкость. Смеши
вается с водой, с диметилформаl\llИДОМ, с этано
лом и с метанолом.
d 20
20: около 0,87.
20
по: около 1,357.
Температура кипения: около 55 ОС.
Вода и дейтерия оксид: не более 0,1 %.
4.1.1. Реактивы
623
Дейтерированный диметилсульфоксид.
C/H 6 0S. (М. м. 84,2). 1025100. [2206 27 1]. eH6)
Диметилсульфоксид. Диметилсульфоксид d6'
Степень дейтерирования не менее 99,8 %.
Очень rиrроскопичная практически бесцвет
ная вязкая жидкость. Растворим в воде, в aцeTO
не и в этаноле.
d;g: около 1,18.
Температура плавления: около 20 ОС.
Вода и деuтерия оксид: не более 0,1 %.
Хранят в воздухонепроницаемом контейнере.
Дейтерированный метанол. С2Нр. (М М. 36,1).
1025200. [811 98 3). еН) Метанол. Метанол d.
Степень дейтерирования не менее 99,8 %.
Прозрачная бесцветная жидкость. Смешива
ется с водой, с 96 % спиртом и сметиленхлоридом.
d 20
20: около 0,888.
20
по: около 1,326.
Температура кипения: 65,4 ОС.
Дейтерированный хлороформ. С2НСl з ,
(м. м. 120,4). 1025000. [865-49 6]. eH)
Хлороформ. Хлороформ d.
Степень дейтерирования не менее 99,7 %.
Прозрачная бесцветная жидкость. Практи
чески нерастворим в воде, смешивается с aцe
тоном и с 96 % спиртом. Может быть стабилизи
рован серебряной фольroй.
d 20
20: около 1,51.
20
по: около 1 ,445.
Температура кипения: около 60 ОС.
Вода и дейтерия оксид: не более 0,05 %.
Декан. С 10 Н 22 . (м. м. 142,З). 1024600. [12 18 5).
Бесцветная жидкость. Практически HepaCT
ворим в воде.
П О: около 1,411.
Температура кипения: около 174 ОС.
Деканаль. С 10 Н 20 О. (м. м. 156,3). 1149200.
[112 31 2). Дециловый альдеrид.
Маслянистая бесцветная жидкость. Практи
чески нерастворим в воде.
Деканаль, используемый в 2азовой xpOMa
т02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Апель
синовое масло.
Содержание деканаля, рассчитанное Meтo
дом нормализации, должно быть не менее 97 %.
Деканол. С 10 Н 2 р. (М м. 158,3). 1024700.
[112 30 1]. н Дециловый спирт.
Вязкая жидкость, затвердевающая при TeM
пературе 6 ОС. Практически нерастворим в воде,
раство им в 96 % спирте.
п; : около 1,436.
Температура кипения: около 230 ОС.
Декстран 2000 синий. 1011700. [9049 32 5].
rотовят из декстрана, имеющеrо среднюю
молекулярную массу 2'106, введением поли
циклическоro хромофора, окрашивающеro
вещество в синий цвет. Степень замещения
0,017. Лиофилизирован. Быстро и полностью
растворяется в воде Р и водных солевых pac
творах.
Раствор 1 r/л декстрана 2000 синеro в фос
фатном буферном растворе рН 7, О Р имеет
максимум поrлощения (2.2.25) при длине волны
280 нм.
Декстран поперечносшитый для хрома-
тоrрафии Р2. 1025500.
rранулы шарообразной формы. Подхо
дящий для разделения пептидов и белков с
молекулярными массами от 15.102 до 30.103.
Сухие rранулы имеют диаметр от 20 мкм ДО
80 мкм.
Декстран поперечноешитый для хрома-
тоrрафии РЗ. 1025600.
rранулы шарообразной формы. Подхо
дящий для разделения пептидов и белков с
молекулярными массами от 4.103 до 15.104.
Сухие rранулы имеют диаметр от 40 мкм ДО
120 мкм.
Декстроза. 1025700. {50 99 7]. См. rЛЮКQза р.
Демеклоциклина rидрохлорид.
C21H21CINPa'HCI. (М м. 501,3) 1145600. [ 7 3].
(4S,4aS,5aS,6S, 12aS) 7 Хлор 4 (диметил
амино) З,6,1 0,12, 12а пентаrидрокси 1, 11 ди
OKCO 1 ,4,4а,5,5а,6, 11,12а октаrидротетрацен 2
карбоксамида rидрохлорид.
Содержит не менее 89,5 % и не более
102,0 % C21H21CIN20a'HCI В пересчете на безво
дное вещество.
Желтый порошок. Растворим или YMepeH
но растворим в воде, малорастворим в 96 %
спирте, очень мало растворим в ацетоне. Pac
творяется в растворах rидроксидов и карбона
тов щелочных металлов.
Вода (2.5.12): не более 3,0 %.
Хранят в защищенном от света месте.
Диазобензолсульфоновой кислоты раст-
вор Р1. 10265000.
0,9 r кислоты сульфаниловой Р paCTBO
ряют в смеси из 30 мл кислоты хлористо
водородной разведенной Р и 70 мл воды Р.
К 3 мл полученноro раствора прибавляют 3 мл
раствора 50 r/л натрия нитрита Р, охлажда
ют в ледяной бане в течеf-lие 5 мин, прибав
ляют 12 мл раствора натрия нитрита Р,
снова охлаждают и доводят водой Р до объема
100 мл. Полученный раствор помещают в ле
дяную баню. rотовят непосредственно перед
использованием, выдерживая в ледяной бане
в течение 15 мин.
624
rосударственная фармакопея Республики Беларусь
# Диазореактив.
5 мл раствора кислоты сульфаниловой
(4,5 r кислоты сульфаниловой Р и 45 мл Kиc
лоты хлористоводородной Р растворяют
в 500 мл воды Р) помещают в мерную колбу
вместимостью 100 мл, ставят в ледяную баню,
прибавляют 2,5 мл раствора натрия Huтpи
та Р и выдерживают в течение 5 мин. При
бавляют 1 О мл раствора натрия нитрита Р,
встряхивают, выдерживают в ледяной бане в
течение 5 мин и доводят водой Р до объема
100,0 мл.
Используют свежеприroтовленный раствор,
сохраняя в ледяной бане.
# Диазореактив Р1. Диазотированный суль
фацил.
К 7,0 r натрия сульфацетамида Р прибав
ляют 50 мл воды Р, 9 мл кислоты хлорисmoводо
родной Р и доводят водой Р до объема 100,0 мл.
1 мл полученноro раствора помещают в мерную
колбу вместимостью 100 мл, ставят в ледяную
баню, прибавляют 50 мл воды Р, 0,2 мл раствора
100 r/л натрия нитрита Р. Доводят водой Р до
объема 100,0 мл.
Используют свежеприrотовленный раствор.
3,3' Диаминобензидина тетраrидрохло
рид. C'2H'8CI4N4'2Hp. (М м. 396,1). 1098000.
[7411 49 6]. 3,3',4,4' Дифенилтетрамин.
Белый или слеrка розоватый порошок. Pac
творим в воде.
Температура плавления: около 280 ос с раз
ложением.
Диаммония 2,2' азинобис(3 этилбен
зотиазолин-6 сульфонат). C,aH24N606S4'
(М м. 548,7). 1153000. [30931 67 O]. ABTS. Ди
аммония 2,2' (диазандиилиден )бис[3 этил 2,3
диrидробензотиазол 6 сульфонат].
Хромоrенный субстрат, приroдный для им
муноферментноrо анализа (ИФА, ELlSA).
Таблетки зеленою цвета. Леrкорастворим в
воде.
рН (2.2.3). От 4,2 до 5,8 для раствора 0,1 r/л.
Диаммония rидроцитрат. См. Аммония
цитрат Р.
Диатомит. 1025900. [91053 39 3J.
Белый или почти белый мелкий rранулиро
ванный порошок, полученный из кремнистых
оболочек окаменевших диатомовых BOДOpOC
лей или их осколков. Практически нерастворим
в воде и в 96 % спирте. Идентифицируют с помо
щью микроскопа пр'" увеличении х500.
Диатомит дпя rазовой хроматоrрафии.
1026000.
Белый или почти белый мелкий rpaHY
лированный порошок, полученный из KpeM
нистых оболочек окаменевших диатомовых
водорослей или их осколков. Практически He
растворим в воде и в 96 % спирте. Иденти
фицируют с помощью микроскопа при увели
чении х500; очищают посредством обработки
кислотой хлористоводородной Р и промыва
нием водой Р.
Размер частиц. Не более 5 % должно OCTa
ваться на сюе (180) и не более 10 % должно
проходить через сито (125).
Диатомит для rазовой хроматоrрафии Р1.
1026100.
Белый или почти белый мелкий rранулиро--
ванный порошок, полученный из кремнистых
оболочек окаменевших диатомовых BOДOpOC
лей или их осколков. Практически нерастворим
в воде и в 96 % спирте. Идентифицируют с помо
щью микроскопа при увеличении х500; очищают
посредством обработки кислотой хлористово
дородной Р и промыванием водой Р.
Размер частиц. Не более 5 % должно OCTa
ваться на сите (250) и не более 10 % должно
проходить через сито (180).
Диатомит для rазовой хроматоrрафии Р2.
1026200.
Белый или почти белый мелкий rранулиро
ванный порошок с удельной площадью поверх
ности около 0,5 M2fr, полученный из кремнистых
оболочек окаменевших диатомовых BOДOpOC
лей или их осколков. Практически нерастворим
в воде и в 96 % спирте. Идентифицируют с помо
щью микроскопа при увеличении х500; очищают
посредством обработки кислотой хлористово
дородной Р и промыванием водой Р.
Размер частиц. Не более 5 % должно OCTa
ваться на сите (180). Не более 10 % должно про
ходить через сито (125).
Диатомит силанизированный для rазо
вой хроматоrрафии. 1026300.
Диатомит для 28зовой хромат02рафии Р,
силанизированный диметилдихлорсиланом или
друrими подходящими силанизирующими pe
аrентами.
Диатомит силанизированный для rазо-
вой хроматоrрафии Р1. 1026400.
Получают из измельченноro красноro orHe
упорноrо кирпича и силанизируют диметилди
хлорсиланом или друrими подходящими силани
зирующими реаrентами. Очищают посредством
обработки кислотой хлористоводородной Р и
промыванием водой Р.
Дибензил. С '4 Н'4' (М м. 182,3). 1011200.
[103 29 7J. 1,2 Дифенилэтан.
Белый или почти белый кристаллический по
рошок. Практически нерастворим в воде, очень
леrко растворим в метиленхлориде, леrкорас
творим в ацетоне, растворим в 96 % спирте.
Температура плавления: от 50 ос до 53 ос.
4.1.1. Реактивы
625
Дибутиловый эфир. СвН,вО' (М м. 130,2).
1026700. [142 96 1].
Бесцветная воспламеняющаяся жидкость.
Практически нерастворим в воде, смешивается
с этанолом.
d;g: около 0,77.
20
п о : около 1,399.
Не пере20няют, если дибутиловый эфир
не вblдерживает иСПblтания на пероксидbl.
Пероксидbl. 8 мл раствора крахмала с
калия йодидом Р помещают в цилиндр вмести
мостью 12 мл и диаметром около 1,5 см с при
тертой стеклянной пробкой, полностью запол
няют испытуемым эфиром, закрывают пробкой
и перемешивают. Выдерживают в темном месте
в течение 30 мин. Не должно появляться OKpa
шивание.
Название и концентрация любоro добавлен
ноro стабилизатора должны быть указаны на
этикетке.
Дибутилфталат. С'6Н2Р4' (М. м. 278,З).
1026800. [84 74 2]. Дибутилбензол 1 ,2
дикарбоксилат.
Прозрачная бесцветная или слабо окрашен
ная маслянистая жидкость. Очень мало paCTBO
рим в воде, смешивается с ацетоном и с 96 %
спиртом.
d 20
20: от 1,043 до 1,048.
20
п о : от 1,490 до 1,495.
10,11 диrидрокарбамазепин. C'5H'4N20.
(М м. 238). 1028900. [3564 73 6]. 10,11 Диrидро
5Н дибензо[Ь, азепин 5 карбоксамид.
Температура плавления: от 205 ос до 21 О ОС.
5, 7 Диrидрокси-4 метилкумарин. С,оНвО 4'
(М м. 192,2). 1149400. [2107 76 8]. 5,7 Диrи
дроксиА метил 2Н 1 бензопиран 2 он.
Светлый желтоватый порошок, практически
нерастворим в воде, умеренно растворим в 96 %
спирте.
Температура плавления: от 295 ос до З03 ОС.
Диrидроксинафталин. 1029000. [1 З2 86 5].
См. 1,3 Ди2идроксинафталин Р
1 ,З Диrидроксинафталин. С,оН в 0 2 .
(М м. 160,2). 1029000. [1 32 86 5]. Нафталин 1,3
диол.
Кристаллический порошок, обычно коричне
вато фиолетовоro цвета. Леrкорастворим в воде
и в 96 % спирте.
Температура плавления: около 125 ОС.
2,7 Диrидроксинафталин. С,оН в 0 2 . (М м.
160,2). 1029100. [582 17 2]. Нафталин 2,7 Диол.
Иroльчатые кристаллы. Растворим в воде и
в 96 % спирте.
Температура плавления: около 190 ОС.
2, 7 Диrидроксинафталина раствор.
1029101.
1 О Mr 2, 7 ди2идроксинафталина Р paCTBO
ряют в 100 мл кислотbl серной Р и выдержива
ют до обесцвечивания.
Срок юдности 2 сут.
Диrитоксин. С 4 ,Н 6 Р,з, (М м. 765). 1028800.
[71 63 6]. 3IН(0 2,6 Дидезокси J3 D рибо
rексопиранозил (1 4 ) 0 2.6 дидезокси J3 D
риб(нексопиранозил ( 1 4 ) 2.6 дидезокси J3 D
рибснексопиранозил )окси] 14 rидрокси 5J3, 14jЗ
кард 20(22 нолид.
Содержит не менее 95,0 % и не более
103,0 % С.,Н О,з в пересчете на сухое веще
ство.
Белый или почти белый порошок. Практи
чески нерастворим в воде, леrкорастворим в
смеси из равных объемов метанола и метилен
хлорида. малорастворим в 96 % спирте и в Me
таноле.
Хранят в защищенном от света месте.
Диrитонмн. C5I6 029' (м. М. 1229). 1028700.
[11024 24 1]. 3IН лкжonиранозил (1 3)
О J3 D rалактопиранозил ( 1----+2 } О [J3 D кси
лопиранозил ( 1 З) ()..J} D rалактопиранозил
(1 4 ) 0 J3 D rалактonмранозилоксиН25R) 5
спиростан 2а.15 дмon_
Кристаллы. практически нерастворим в
воде, умеренно растворим в этаноле, малораст
ворим в 96 % спирте.
Дидодецил ,3' -Т1IQQМI1POП ионат. С зо Н 58 О 4 S.
(М м. 514,8). 1027700. [123-28-4].
Белый или почти белый кристаллический
порошок. Практически нерастворим в воде, леr
корастворим в ацетоне и в neтponeйном эфире,
малорастворим в 96 % спирте.
Температура плавneния: ОКОЛО 39 ОС.
Диизобутилкетон. CgH"O. (М м. 142,2).
1029200. [1 08 83 8].
Прозрачная бесцветная жидкость. Мало
растворим в воде, смешивается с большинством
орrанических растворителей.
п;О: около 1,414.
Температура кипения: около 168 ОС.
Диизonponиловый эфир. С 6 Н,Р. (М м. 102,2).
1029300. [108 20 3].
Прозрачная бесцветная жидкость. Очень
мало растворим в воде, смешивается с 96 %
спиртом и эфиром.
d;g: от 0,723 до 0,728.
Температура ttипения: от 67 ос до 69 ОС.
Не перееоняюm, если диизопропиловblЙ
эфир не вblдерживает иСПblтания на пероксидbl.
Пероксидbl. 8 мл раствора крахмала с калия
йодидом Р помещают в цилиндр вместимостью
12 мл и диаметром около 1,5 см с притертой CTe
клянной пробкой, полностью заполняют испыту
626
Тосударственная фармакопея Республики Беларусь
емым эфиром, закрывают пробкой и перемеши
вают. Выдерживают в темном месте в течение
30 мин. Не должно появляться окрашивание.
Название и концентрация любоro добавлен
Horo стабилизатора должны быть указаны на
этикетке.
Хранят в защищенном от света месте.
Дикалия rидрофосфат.,\НРО 4 . (м. М. 174,2).
1033000. [7758 11 4].
Белый или почти белый кристаллический
порошок. rиrpоскопичен. Очень леrко растворим
в воде, малорастворим в 96 % спирте.
Хранят в воздухонепроницаемом контейнере.
Дикарбоксидина rидрохлорид.
C20H24NP6'2HCI. (м. м. 461,3). 1026900. [56455-904].
4,4' [(4,4' Диаминодифенил 3,3' диил)диокси]
дибутановойкислотыдиrидрохлорид.
Диметикон. 1105400. [9006 65 9].
Прозрачные бесцветные жидкости различ
ной вязкости. Практически нерастворим в воде,
от очень леrко растворимоro до практически He
растворимоro в эта ноле, смешивается с этила
цетатом, с метилэтилкетоном и с толуолом.
Диметиламинобензальдеrид. CgH"NO.
(Мм.149,2).1029800.[100 10 7].4 Диметиламино
бензальдеrид. -п Диметиламинобензальдеrид.-
Белые или желтовато белые кристаллы. Pac
творим в 96 % спирте и в разведенных кислотах.
Температура плавления: около 74 ос.
Диметиламинобензальдеrидa раствор Р1.
1029801.
0,2 r диметuламинобензальде2ида Р paCT
воряют в 20 мл 96 % спирта Р, прибавляют
0,5 мл кислоты хлористоводородной Р, полу
ченный раствор встряхивают с уzлем aKтиви
рованным Р и фильтруют. Окраска раствора не
должна быть интенсивнее окраски раствора
йода Р3.
rотовят непосредственно перед использо
ванием.
ДиметиламинобензалЬДеrидa раствор Р2.
1029802.
0,2 r диметиламинобензалтде2ида Р paCT
воряют без наrревания в смеси из 4,5 мл воды Р
и 5,5 мл кислоты хлористоводородной Р.
rотовят непосредственно перед использо
ванием.
Диметиламинобензальдеrида раствор Р6.
1029803. -Реактив Оллпорта.-
0,125 r диметиламинобензальде2ида Р
растворяют в охлажденной смеси из 35 мл
воды Р и 65 мл кислоты серной Р, прибавляют
0,1 мл раствора 50 r/л железа (1/1) хлорида Р.
Перед использованием выдерживают 24 ч в
защищенном от света месте.
Хранят при комнатной температуре 7 сут; в
холодильнике в течение нескольких месяцев.
Диметиламинобензальдеrида раствор Р7.
1029804.
1,0 r диметиламинобензальде2ида Р paCT
воряют в 50 мл кислоты хлористоводородной Р
и прибавляют 50 мл 96 % спирта Р.
Хранят в защищенном от света месте. Срок
roдности 1 мес.
Диметиламинобензальдеrида раствор Р8.
1029805.
0,25 r диметиламинобензаптдеzида Р pacт
воряют в смеси из 5 r кислоты фосфорной Р, 45 r
воды Р и 50 r кислоты уксусной безводной Р.
rотовят непосредственно перед использо
ванием.
# п Диметиламинобензальдеrида раст-
вор в кислоте серной.
1 r диметиламинобензальде2ида Р смачи
вают 4 каплями воды Р и прибавляют 3 мл Kиc
лоты серной Р.
4-Диметиламинокоричный альдеrид.
С'1Н,зNО. (М м. 175,2). 1029900. [6203 18 5].
З (4 Диметиламинофенил)проп 2 еналь. 4 Ди
метиламиноциннамальдеrид.
Оранжевые или оранжево коричневые кри
сталлы или порошок. Чувствителен к свету.
Температура плавления: около 138 ос.
4-Диметиламинокоричноrо альдеrида
раствор. 1029901.
2 r 4 диметиламинокориЧН020 альде2ида Р
растворяют в смеси из 100 мл кислоты хлори
стоводородной Р1 и 100 мл этанола Р. Непо
средственно перед использованием раствор
разводят этанолом Р в 4 раза.
Диметиламинонафталинсульфонилхло
рид. C'2H'2CIN02S' (м. М. 269,8). 1 030000. [605 65--2].
5 Диметиламино 1 нафталинсульфонилхлорид.
Желтый кристаллический порошок. Мало
растворим в воде, растворим в метаноле.
Температура плавления: около 70 0 С.
4-Диметиламиноциннамальдеrид. См.
4 Диметиламинокоричный альде2ид Р.
Диметиланилин. CaH"N. (М М. 121,2).
1030100. [121 69 7]. N,N Диметиланилин.
Прозрачная маслянистая жидкость. Свеже
переrнанн!:.IЙ почти бесцветный, при хранении
темнеет до красновато коричневоro цвета. Прак
тически нерастворим в воде, леrкорастворим в
96 % спирте.
п;О: около 1,558.
Температурные пределы пере20нки
(2.2.11). От 192 0 С до 194 0 С; должно переrонять
ся не менее 95 %.
4.1.1. Реактивы
627
N,N Диметиланилин. 1030100. [121 69 7].
См. Диметиланилин Р.
2.З Диметиланилин. CaH'1N. (М. м. 121,2).
1105300. [87 59 2]. 2,3 Ксилидин.
Желтоватая жидкость. Умеренно растворим
в воде, растворим в 96 % спирте.
d 20
20: от 0,99З до 0,995.
20
n D : около 1,569.
Температура кипения: около 224 ос.
2,6 Диметиланилин. C a H 11 N. (М. м. 121,2).
1030200. [87 62 7]. 2, Ксилидин.
Бесцветная жидкость. Умеренно растворим
в воде, растворим в 96 % спирте.
d 20
20: около 0,98.
Диметилацетамид. C 4 H g NO. (М м. 87,1).
1029700. [127 19 5]. N,N Диметилацетамид.
Содержит не менее 99,5 % C 4 H g NO.
Бесцветная жидкость. Смешивается с водой
и со мноrими орrаническими растворителями.
d 20
20: около 0,94.
20
п о : около 1,437.
Температура кипения: около 165 ОС.
Диметилrлиоксим. C 4 H a NP2' (М. м. 116,1).
1030400. [95 45 4]. 2,З Бутандиондиоксим.
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Практиче
ски нерастворим в холодной воде, очень мало
растворим в кипящей воде, растворим в 96 %
спирте.
Температура плавления: около 240 ос с раз
ложением.
Сульфатная зола (2.4.14). Не более 0,05 %.
# Диметилrлиоксима раствор.
1 r диметилелиоксима Р растворяют в 100 мл
раствора 50 r/л натрия zидрокарбоната Р.
Диметилдециламин. C 12 H 27 N. (М. м. 185,4).
1113500. [1120 24 7]. N.N Диметилдециламин.
Содержит не менее 98,0 % (м/м) C'2H27N.
Температура кипения: около 2З4 ОС.
Диметилкарбонат. С з Н 6 О з . (М м. 90,1).
1119300. [616 З8 6]. Диметиловый эфир уrоль
ной кислоты.
Жидкость. Нерастворим в воде, смешивает
ся с 96 % спиртом.
d l1
4 : около 1,065.
20
п D : около 1,368.
Температура кипения: около 90 ОС.
# Диметиловый желтый. С14Н'5Nз.
(М. м. 225,3). 1029600. [60 11 7]. Показатель
Шульца N228. Индекс цветности N2 11020. 4 Ди
метиламиноазобензол. Метиловый желтый.
Мелкие кристаллы желтоro цвета или
хлопья желтоro или оранжевою цвета. Практи
чески нерастворим в воде, очень мало paCTBO
рим в 96 % спирте.
Хромаmo2рафия. Тонкослойная xpoMaтorpa
фия (2.2.27), используя в качестве TOHKorO слоя cи
ликаzeль G Р. На хроматоrpафическую пластинку
наносят 1 О мкл раствора 0,1 r/л диметиловоro жел
Toro в метuлeнхлориде Р и хроматоrpафируют в
этом же растворителе; фронт растворителя должен
пройти не менее 10 см. На xpoMaтorpaMMe должно
обнаруживаться только одно основное пятно.
# Диметиловоrо желтоrо раствор.
0,1 r диметилов020 желтоzо Р растворяют
в 100 мл спирта (95 %, об/об) Р.
Изменение окраски. От рН З,О (красное
окрашивание) до рН 4,0 (желтое окрашивание).
# Диметиловоrо желтоrо раствор Р1.
0,1 r диметилов020 желт020 Р растворяют
в 100 мл бензола Р.
# Диметиловоrо желтоrо и орацетовоrо
синеrо раствор. 1118700.
1 О Mr диметиловоzо желт020 Р и 1 О Mr opa
цетов020 CUHezo В Р растворяют в 300 мл Me
тиленхлорида Р.
N,N-Диметилоктиламин. С,оН 2з N.
(М м. 157,З). 1030500. [7З78 99 6]. Октилдиме
тиламин.
Бесцветная жидкость.
d 20
20: около 0,765.
20
п D : около 1,424.
Температура кипения: около 195 ОС.
1 ,З-Диметил 2 имидазолидинон. C 5 H,oN 2 0.
(М м. 114,2). [80 7З 9]. N,N' Диметилэтиленмоче
вина.
п O: около 1,4720.
Температура кипения: около 224 ОС.
Диметилпиперазин. C 6 H 14 N 2 . (М М. 114.2).
1030700. [1 06 58 1]. 1 ,4 Диметилпиперазин.
Бесцветная жидкость. Смешивается с водой
и с 96 % спиртом.
d 20
20: около 0,85.
20
п D : около 1 ,446.
Температура кипения: около 131 ОС.
Диметилстеарамид. C 2o H 4 ,NO. (М. м. 311,6).
1030800. N,N Диметилстеарамид.
Белая или почти белая твердая масса. Pac
творим во мноrих орrанических растворителях,
включая ацетон.
Температура плавления: около 51 ОС.
Диметилстеариламид. См. Диметилсте
арамид Р.
628
rосударственная фармакопея Республики Беларусь
Диметилсульфоксид. 1029500. [67 68 5J.
СМ. статью Диметилсульфоксид.
Диметилсульфоксид, используемый в спек
трофотометрии должен выдерживать cпeдy
ющее дополнительное испытание.
Пропускание (2.2.25) определяют, используя
в качестве компенсационной жидкости воду Р:
не менее 10 % при 262 нм; не менее 35 % при
270 нм; не менее 70 % при 290 нм; не менее
98 % при З40 нм и более.
Диметилсульфон. C 2 H 6 0 2 S. (М м. 94,1).
1030900. [67 71 OJ.
Белый или почти белый кристаллический
порошок. Леrкорастворим в воде, растворим в
ацетоне и в 96 % спирте.
Температура плавления: от 108 ос до 110 ОС.
# Диметилтетрадециламин. С'6НЗ5N.
(М м. 241,5). 1031000. N,N Диметилтетра
дециламин.
Содержание: не менее 98,0 % (м/м) и не
более 101,0 % (м/м) С'6НЗ5N.
Прозрачная или почти прозрачная бесцвет
ная или желтоватая жидкость. Практически He
растворим в воде, смешивается с ацетоном, с
96 % спиртом и с метанолом.
d;g: около 0.80.
Температура кипения: около 260 ОС.
Вода (2.5.12). Не более 0,3 % (м/м).
Количественное определение. 0,200 r paCT
воряют в 10 мл 96 % спирта Р и титруют 0,1 М
раствором кислоты хлористоводородной до
появления красноro окрашивания, используя в
качестве индикатора 0,1 мл раствора метило
вО20 краСН020 Р
1 мл 0,1 М раствора кислоты хлористово
дородной соответствует 24,15 Mr С'6НЗ5N.
2,6 Диметилфенол. С е Н lО О. (М. м. 122,2).
1030600. [576 26 1].
Бесцветные иroльчатые кристаллы. Мало
растворим в воде, очень леrко растворим в 96 %
спирте.
Температура кипения: около 203 ОС.
Температура плавления: от 46 ос до 48 ОС.
З,4 Диметилфенол. С е Н lО О. (М м. 122,2).
1098100. [95 65 8].
Белые или почти белые кристаллы. Мало
растворим в воде, леrкорастворим в 96 % спирте.
Температура кипения: около 226 ОС.
Температура плавления: от 25 ос до 27 ОС.
Диметилформамид. С з Н 7 NО. (М м. 73,1).
1030300. [68 12 2].
Прозрачная бесцветная жидкость с ней
тральным значением рН. Смешивается с водой
и с 96 % спиртом.
d;g: от 0,949 до 0,952.
Температура кипения: около 153 ОС.
Вода (2.5.12). Не более 0,1 %.
N,N Диметилформамида диметилацеталь.
С5Н,зN02' (М м. 119,2). 1140700. [4637 24 5J. 1, 1 Ди--
метокситриметиламин.
Прозрачная бесцветная жидкость.
d 20
20: около 0,896.
20
ПО : около 1,З96.
Температура кипения: около 103 ОС.
Диметилформамида диэтилацеталь.
C7H'7N02' (М м. 147,2). 1113600. [1188 33 6J. N,N
Диметилформамида диацеталь.
20
по: около 1,40.
Температура кипения: от 128 ОС до 130 ОС.
1,1 Диметилэтиламин. C 4 H"N. (М м. 73,1).
1100900. [75 64 9]. 2 Амино 2 метилпропан. тpeт
Бутиламин.
Жидкость. Смешивается с 96 % спиртом.
d 20
20: около 0,694.
20
по : около 1,378.
Температура кипения: около 46 ОС.
1,1 Диметилэтилметиловый эфир. С 5 Н 12 О.
(М м. 88,1). 1013900. [1634-04-4]. 2 Метокси 2
метилпропан. трет Бутилметиловый эфир.
Бесцветная прозрачная воспламеняющаяся
жидкость.
п O: около 1,376.
Пропускание (2.2.25) определяют, используя
в качестве компенсационной жидкости воду Р: не
менее 50 % при 240 нм; не менее 80 % при 255 нм;
не менее 98 % при 280 нм.
1, 1 Диметилэтилметиловый эфир Р1.
1126400.
Содержит не менее 99,5 % С5Н'20,
d 20 О
20:0КОЛО ,741.
20
по : около 1,369.
Температура кипения: около 55 ОС.
Диметоксипропан. С 5 Н'Р2' (М м. 104,1).
1105200. [77 76 9]. 2,2 Диметоксипропан.
Бесцветная жидкость. Разлаraется под дей
ствием влажноro воздуха или воды.
d 20
20: около 0,847.
п O: около 1,378.
Температура кипения: около 83 ОС.
Димидия бромид. С2ОН1вВrNз. (М м. 380,3).
1031100. [518 67 2]. 3,8 Диамино 5 метил 6
фенилфенантридиния бромид.
TeMHO KpaCHыe кристаллы. Малорастворим в
ооде при температуре 20 ОС, умеренно растворим
в воде при температуре 60 ос и в 96 % спирте.
Димидия бромида и сульфановоrо
синеrо смешанный раствор. 1031101.
0,5 r димидия бромида Р и 0,25 r сульфано
в020 сине20 Р растворяют по отдельности в 30 мл
rорячей смеси этанол Р вода Р (1:9), пере
4.1.1. Реактивы
629
мешивают. Оба раствора смешивают и доводят
до объема 250 мл этой же смесью растворите
лей. 20 мл полученноro раствора смешивают с
20 мл раствора 14,0 % (06/06) кислоты серной Р,
предварительно разведенным примерно 250 мл
вады Р, и доводят вадой Р до объема 500 мл.
Хранят в защищенном от света месте.
Динатрия арсенат. Na 2 HAs0 4 '7H 2 0.
(М м. З12,0). 1102500. [10048 95 0]. Динатрия rи
дроарсенат rептаrидрат.
Кристаллы, выветривающиеся на теплом
воздухе. Леrкорастворим в воде, растворим в rли
церине, малорастворим в 96 % спирте. Водный
раствор имеет щелочную реакцию по лакмусу.
d; : около 1,87.
Температура плавления: около 57 ос (при
быстром наrревании).
Динатрия бицинхонинат. C2oH1oN2Na204'
(М м. 388,3). 1126600. [979 88 4]. Динатрия
2,2' бихинолин44' дикарбоксилат.
Динатрия I"идрофосфат. Na 2 HPO 4 '12Нр.
(М. м. 358,1). 1033300. [10039 324]. Динатрия
фосфат додекаrидрат.
Содержание: не менее 98,0 % и не более
101,0 % Na 2 HP0 4 в пересчете на безводное Be
щество.
Бесцветные прозрачные кристаллы. Леrко
выветриваются. Очень леrко растворим в воде,
практически нерастворим в 96 % спирте.
Хранят в воздухонепроницаемом контейнере.
Динатрия I"идрофосфата раствор.
1033301.
Раствор 90 r/л динатрия 2идрофосфата Р.
Динатрия rидрофосфат безводный.
Na 2 HP0 4 (М м. 142,0). 1033400. [7558 794].
Динатрия rидрофосфат диrидрат.
1033500. [10028 24 7].
См. статью Динатрия фосфат ди2идрат.
Динатрия rидроцитрат. C 6 H 6 Na 2 0 7 '1 ,5Н 2 О.
(М М. 263,1). 1033200. [144 33 2]. Натрия цитрат
кислый. Динатрия rИДРО 2 rидроксипропан 1 ,2,3
трикарбоксилат сесквиrидрат.
Белый или почти белый порошок. PaCTBO
рим менее чем в 2 частях воды, практически He
растворим в 96 % спирте.
Динатрия тетраборат. 1033600. [133043-4].
См. статью Натрия тетраборат.
Динатрия эдетат. 1080600. [6381 92 6].
См. статью Динатрия эдетат.
Динитробензоилхлорид. С 7 Н з СIN 2 О 5 .
(М М. 230,6). 1031400. [99 33 2]. 3,5 Динитро
бензоилхлорид.
Полупрозрачный желтый или зеленовато
желтый порошок или желтоватые кристаллы.
Растворим в ацетоне и толуоле.
Температура плавления: около 68 ос.
Испытание на при20дность. К 1 мл этано
ла Р и 0,1 r динитробензоилхлорида Р прибав
ляют 0,05 мл кислоты серной разведенной Р и
кипятят с обратным холодильником в течение
30 мин. После выпаривания на водяной бане
к остатку прибавляют 5 мл 2ептана Р и Harpe
вают до кипения. rорячий раствор фильтруют.
Кристаллы, образующиеся при охлаждении до
комнатной температуры, промывают неболь
шим количеством 2ептана Р и сушат в эксикато
ре. Температура плавления (2.2.14) полученных
кристаллов: от 94 ос до 95 ос.
Динитробензойная кислота. C 7 H 4 N 2 0 6 .
(М М. 212,1). 1031300. [99 34 3]. 3,5 Динитро
бензойная кислота.
Почти бесцветные кристаллы. Малораство
рима в воде, очень леrко растворима в 96 %
спирте.
Температура плавления: около 206 ос.
Динитробензойной кислоты раствор.
1031301.
Раствор 20 r/л динитробензойной кисло
ты Р в 96 % спирте Р.
Динитробензол. C 6 H 4 NP4' (М. М. 168,1).
1031200. [99 65 0]. 1 ,3 Динитробензол.
Желтоватый кристаллический порошок или
кристаллы. Практически нерастворим в воде,
малорастворим в 96 % спирте.
Температура плавления: около 90 ос.
Динитробензола раствор. 1031201.
Раствор 10 r/л динитр06ензола Р в 96 %
спирте р.
ДинитрофеНИЛl"идразин. C e H E N 4 0..
(М М. 198,1). 1031500. [119 26 6]. 2,4 Динитpcr
фенилrидразин.
Красновато оранжевые кристаллы. Очень
мало растворим в воде, малорастворим в 96 %
спирте.
Температура плавления: около 203 ос
(метод MrHoBeHHoro плавления).
# Динитрофенилrидразина спиртовой
раствор.
К 0,5 r динитрофеНUЛ2идразина Р прибав
ляют 6 мл кислоты хлористоводородной Р и
перемешивают до исчезновения KpaCHo opaH
жевоro окрашивания осадка. Прибавляют
20 мл этанола Р, наrревают смесь на водя
ной бане до получения прозрачноro раствора,
охлаждают и доводят эта нолом Р ДО объема
100,0 мл.
Полученный раствор хранят в холодном
месте. Срок roдности 3 мес.
630
rосударственная фармакопея Республики Беларусь
Динитрофенилrидразина уксусно хлори-
стоводородный раствор. 1031501.
0,2 r динитрофениЛ2идразина Р paCT
воряют в 20 мл метанола Р и прибавляют
80 мл смеси из равных объемов кислоты
уксусной Р и кислоты хлористоводород
ной Р1. rотовят непосредственно перед ис
пользованием.
Динитрофенилrидразина хлористово-
дородный раствор. 1031502.
0,50 r динитрофениЛ2идразина Р paCT
воряют при наrревании в кислоте хлористо
водородной разведенной Р, доводят объем
раствора тем же растворителем до 100 мл,
охлаждают и фильтруют. rотовят непосред
ственно перед использованием.
Динонилфталат. С26Н4204' (М м. 418,6).
1031600. [2855З 12 0].
Бесцветная или бледно желтая вязкая
жидкость.
d 20
20: от 0,97 до 0,98.
20
п о : от 1,482 до 1,489.
Кислотность. 5,0 r динонилфталата
встряхивают с 25 мл воды Р в течение 1 мин.
Выдерживают, водный слой отделяют, филь
труют и прибавляют 0,1 мл раствора фенол
фталеина Р. Окраска раствора должна из
мениться при прибавлении не более 0,3 мл
0,1 М раствора натрия 2идроксида (0,05 % в
пересчете на кислоту фталевую).
Вода (2.5.12). Не более 0,1 %.
Диоксан. С 4 Н в 0 2 . (М м. 88,1). 1032000.
[123 91 1]. 1,4 Диоксан.
Прозрачная бесцветная жидкость. CMe
шивается с водой и большинством орrаниче
ских растворителей.
d;g: около 1,03.
Температура затвердевания (2.2.18). От
9 ос до 11 ОС.
Вода (2.5.12). Не более 0,5 %.
Не пере2оняют, если диоксан не выдep
живает испытания на пероксиды.
Пероксиды. 8 мл раствора крахмала с
калия йодидом Р помещают в цилиндр BMe
стимостью 12 мл и диаметром около 1,5 см
с притертой пробкой, заполняют полностью
диоксаном и перемешивают. Выдерживают в
темном месте в течение 30 мин. Не должно
обнаруживаться окрашивания.
Диоксан, используемый для жидкостной
сцинтилляции, должен быть cooтвeтcтвy
ющей степени чистоты.
Диоксана исходный раствор. 1032001.
1,00 r диоксана Р растворяют в воде Р и
доводят до объема 100,0 мл этим же paCTBO
рителем. 5,0 мл полученноro раствора ДOBO
дят водой Р до объема 50,0 мл (1,0 мr/мл).
Диоксана раствор. 1032002.
50,0 мл исходноао раствора диоксана Р дово-
дят водой Р до объема 100,0 мл (0,5 мr/мл диоксана).
Диоксана раствор Р1. 1032003.
10,0 мл раствора диоксана Р доводят
водой Р до объема 50,0 мл (0,1 мr/мл диоксана).
м Диоксибензол. См. Резорцин Р.
Ди октадецил -3,3' тиоди n роп ионат.
C42HaP4S. (М м. 683). 1031900. [693 36 7].
Белый или почти белый кристаллический
порошок. Практически нерастворим в воде, леr
корастворим в метиленхлориде, умеренно pac
творим в ацетоне, в 96 % спирте и в петролей
ном эфире.
Температура плавления: от 58 ос до 67 ОС.
2,2' -Ди( октадецилокси)-5,5' пироби(1 ,3,2
диоксафосфоринан). С 41 Н в Р6 Р 2' (М м. 733).
1031800.
Белое или почти белое твердое воскообраз
ное вещество. Практически нерастворим в воде,
растворим в растворах rидрокарбонатов.
Температура плавления: от 40 ос до 70 ОС.
Диоктадеци.nдисульфид. С зв Н 74 S 2 . (М М. 571,1).
1031700. [2500 88 1].
Белый или почти белый порошок. Практиче
ски нерастворим в воде.
Температура плавления: от 53 ос до 58 ОС.
2,2'-Дипиридиламин. С 10 Н g N з . (М м. 171,2).
1157700. [1202 34 2]. N (Пиридин 2 ил)пиридин
2 амин.
Температура плавления: около 95 ОС.
Дитизон. С1ЗН12N4S' (М м. 256,3). 1033900.
[60 1 0 6]. 1.5 Дифенилтиокарбазон.
Синевато черный, коричневато черный или
черный порошок. Практически нерастворим в
воде, растворим в 96 % спирте.
Хранят в защищенном от света месте.
Дитизона раствор. 1033901.
Раствор 0,5 r/л дитизона Р в хлороформе Р.
rотовят непосредственно перед использо
ванием.
Дитизона раствор Р2. 1033903.
40,0 Mr дитизона Р растворяют в хлорофор
ме Р и доводят до объема 1000,0 мл этим же
растворителем. 30,0 мл полученноro раствора
доводят хлороформом Р до объема 100,0 мл.
Установка титра. Количество ртути (1/)
хлорида Р, эквивалентное 0,1354 r HgCI 2 , paCTBO
ряют в смеси из равных объемов кислоты серной
разведенной Р и воды Р и доводят до объема
100,0 мл этой же смесью растворителей. 2,0 мл
полученноro раствора доводят смесью из равных
объемов кислоты серной разведенной Р и воды Р
до объема 100,0 мл (раствор содержит 20 ррm Hg).
1,0 мл полученноro раствора помещают в длитель
ную воронку, прибавляют 50 мл кислоты серной
4.1.1. Реактивы
631
разведенной Р, 140 мл воды Р и 10 мл раствора
200 r/л 2идроксиламина 2идрохлорида Р. Титруют
приroтовленным раствором дитизона; после каж
доro прибавления титранта смесь встряхивают 20
раз, к концу титрования смесь оставляют для раз
деления слоев, затем отбрасывают хлороформ
ный слой И продолжают титровать до появления
синевато зеленноro окрашивания.
Количество ртути (Э), в микроrраммах, экви
валентное содержанию дитизона в одном мил
лилитре раствора, рассчитывают по формуле:
э= 20 ,
V
rде V объем раствора дитизона, израсхо
дованный на титрование, мл.
Дитизон Р1. С'ЗН'2N4S' (М М. 256,3).
1105500. [60 10 6]. 1 ,5 Дифенилтиокарбазон.
Содержание: не менее 98,0 % С'ЗН'2N4S'
Синевато черный, коричневато черный или
черный порошок. Практически нерастворим в
воде, растворим в 96 % спирте.
Хранят в защищенном от света месте.
5,5' Дитиобис(2 нитробензойная кислота).
C'4HaNPaS2' (М. М. 396,4). 1097300. [69 78 3].
3 Карбокси--4 нитрофенилдисульфид. Реактив
Эльмана. OTNB.
Желтый порошок. Умеренно растворима в
96 % спирте.
Температура плавления: около 242 ос.
Дитиол. C 7 H a S 2 . (М М. 156,3). 1033800.
[496 74 2]. Толуол 3,4 дитиол. 4 Метилбензол 1 ,2
дитиол.
Белые или почти белые кристаллы. rиrро
скопичен. Растворим в метаноле и в растворах
rидроксидов щелочных металлов.
Температура плавления: около 30 ОС.
Хранят в воздухонепроницаемом контейнере.
Дитиола реактив. 1033801.
К 1 r дитиопа Р прибавляют 2 мл кислоты
ти02ликолевой Р и доводят раствором 20 r/л
натрия 2идроксида Р до объема 250 мл. rотовят
непосредственно перед использованием.
Дитиотреитол. C 4 H 10 0 2 S 2 . (М М. 154.2).
1098200. [27565-41 9]. тpeo 1 ,4--Димеркаптобутан
2,З--диол.
Иroльчатые кристаллы. Слеrка rиrроскопичен.
Леrкорастворим в воде, в ацетоне и в этаноле.
Хранят в воздухонепроницаемом контейнере.
Дифениламин. C'2H"N. (М м. 169,2).
1032100. [122 39 4].
Белые или почти белые кристаллы. Мало
растворим в воде, растворим в 96 % спирте.
Температура плавления: около 55 ОС.
Хранят в защищенном от света месте.
Дифениламина раствор. 1032101.
Раствор 1 r/л дифениламина Р в кислоте
серной Р.
Хранят в защищенном от света месте.
Дифениламина раствор Р1. 1032102.
Раствор 10 r/л дифениламина Р в кислоте
серной Р. Раствор должен быть бесцветным.
Дифениламина раствор Р2. 1032103.
1 r дифениламина Р растворяют в 100 мл
кислоты уксусной ледяной Р и прибавляют
2,75 мл кислоты серной Р.
Раствор используют немедленно.
Дифенилантрацен. С 26 Н,в, (М М. 330,4).
1 032200. [1499 1 o 1]. 9, 1 О Дифенилантрацен.
Кристаллический порошок от желтоватоro
до желтоro цвета. Практически нерастворим в
воде.
Температура плавления: около 248 ОС.
Дифенилбензидин. C24H2oN2' (М М. 336,4).
1032300. [53 1 91 9]. N,N' Дифенилбензидин.
N,N' Дифенилбифенил--4,4' диамин.
Белый или бледно серый кристаллический
порошок. Практически нерастворим в воде, Ma
лорастворим в ацетоне и в 96 % спирте.
Температура плавления: около 248 ОС.
Нитраты. 8 Mr дифенилбензидина Р paCT
воряют в охлажденной смеси из 5 мл воды Р и
45 мл кислоты серной, свободной от азота, Р.
Полученный раствор должен быть бесцветным
или иметь очень бледное roлубое окрашива
ние.
Сульфатная зола (2.4.14). Не более 0,1 %.
Хранят в защищенном от света месте.
Дифенилборной кислоты аминоэтило-
вый эфир. C'4H16BNO. (М м. 225,1). 1032400.
[524 95 8].
Белый или слеrка желтый кристаллический
порошок. Практически нерастворим в воде, pac
творим в 96 % спирте.
Температура плавления: около 193 ОС.
# N,N'-Дифенилrуанидин. С,зН,зNз.
(М м. 211,3).
Белый или светло желтый мелкокристалли
ческий порошок.
Дифенилкарбазид. С.зН, N О. (М м. 242,3).
1032500. [140 22 7]. 1 , Дифенилкарбондиrи
дразид.
Белый или почти белый кристалличе
ский порошок, постепенно розовеет на возду
хе. Очень мало растворим в воде, растворим
в ацетоне, в 96 % спирте и в кислоте уксусной
ледяной.
Температура плавления: около 170 ОС.
Сульфатная зола (2.4.14). Не более 0,1 %.
Хранят в защищенном от света месте.
632
rосударственная фармакопея Республики Беларусь
Дифенилкарбазида раствор. 1032501.
0,2 r дифенилкарбазида Р растворяют в
1 О мл кислоты уксусной ледяной Р и доводят
этанолом Р до объема 100 мл.
rотовят непосредственно перед использо
ванием.
Дифенилкарбазон. С 1з Н 12 Nр. (М м. 240,З).
1032600. [538 62 5]. 1 ,5 Дифенилкарбазон.
Оранжево желтый кристаллический поро
шок. Практически нерастворим в воде, очень
леrко растворим в 96 % спирте.
Температура плавления: около 157 ос с раз
ложением.
Дифенилкарбазон ртутный реактив.
1032601.
Раствор А. 0,1 r дифенилкарбазона Р paCT
воряют в этаноле Р и доводят до объема 50 мл
этим же растворителем.
Раствор В. 1 r ртути (11) хлорида Р раст
воряют в этаноле Р и доводят до объема 50 мл
этим же растворителем.
Смешивают равные объемы растворов А и В.
Дифенилоксазол. C 15 H 11 NO. (М. м. 221,3).
1032700. [92 71 7]. 2,5 Дифенилоксазол.
Белый или почти белый порошок. Практиче
ски нерастворим в воде, растворим в метаноле,
умеренно растворим в диоксане и в кислоте YK
сусной ледяной.
Температура плавления: около 70 ОС.
А;::': около 1260. Определение проводят при
длине волны 305 нм, используя в качестве pac
творителя метанол Р
Дифенилоксазол, используемый для жид
костной сцинтилляции, должен быть cooт
ветствующей степени чистоты.
Дифенилфениленоксида полимер.
1032800. Полимер 2, дифенил--п-фениленоксида.
Белые или почти белые пористые rранулы
шарообразной формы. Размер rранул указыва
ют после названия реактива в испытаниях, в KO
торых он используется.
Дихлорбензол. C 6 H 4 C1 2 . (М М. 147,0).
1027100. [95 50 1]. 1 ,2 Дихлорбензол.
Бесцветная маслянистая жидкость. Практи
чески нерастворим в воде, растворим в этаноле.
d;g: около 1,31.
Температура кипения: около 180 ОС.
Дихлорметан. См. Метиленхлорид Р
Дихлорофос. C 4 H 7 Clpp (М м. 221).
1101200. (62 73 7]. 2,2 Дихлорвинилдиметил
фосфат.
Бесцветная или коричневато желтая жид
кость. Растворим в воде, смешивается с боль
шинством орrанических растворителей.
п 5: около 1,452.
Дихлоруксусная кислота. C 2 H 2 C1 2 0 2 .
(М. М. 128,9). 1027000. (79-43 6].
Бесцветная жидкость. Смешивается с водой
и с 96 % спиртом.
d;g: около 1,566.
20
п о : около 1,466.
Температура кипения: около 19З ОС.
ДИXl'!оруксусной кислоты раствор.
1027001.
67 мл кислоты дихлоруксусной Р ДOBO
дят водой Р до объема 300 мл и нейтрализу
ют раствором аммиака Р по синей лакмусо
вой бума2е Р Охлаждают, прибавляют 33 мл
кислоты дихлоруксусной Р и доводят водой Р
до объема 600 мл.
Дихлорфенолиндофенола натриевая
соль. C'2H6CI2NNa02'2Hp. (М. м. 326,1).
1027300. [620 45 1]. Натриевое произво
дное 2,6 дихлор N (4 rидроксифенил) 1,4
бензохинонмоноимина диrидрат. #2,6 Дихлор
фенолиндофенолят натрия.#
Темно зеленый порошок. Леrкораствори
ма в воде и в этаноле. Водный раствор имеет
темно синюю окраску, которая при подкисле
нии раствора изменяется на розовую.
Дихлорфенолиндофенола титрованный
раствор. 1027301.
50,0 Mr дихлорфенолиндофенола Haтpи
евой соли Р растворяют в 100,0 мл воды Р и
фильтруют.
Установка титра. 20,0 Mr кислоты
аскорбиновой Р растворяют в 1 О мл свеже
приroтовленноro раствора 200 r/л кислоты
метафосфорной Р и доводят водой Р до
объема 250,0 мл. 5,0 мл полученноro paCT
вора быстро титруют приroтовленным paCTBO
ром дихлорфенолиндолфенола из микробю
ретки с ценой деления 0,01 мл до появления
розовоro окрашивания, не исчезающеro в Te
чение 1 О с, время титрования должно быть
не более 2 мин. Раствор дихлорфенолиндол
фенола разводят водой Р до получения paCT
вора, 1 мл KOTOpOro соответствует 0,1 Mr кис
лоты аскорбиновой (С 6 Н в 0 6 ).
Срок rодности 3 сут. Титр устанавливают He
посредственно перед использованием.
Дихлорфлуоресцеин. C2QH 10 CIP5' (м. м. 401,2).
1 027200. [76 54 0]. 2, 7 Дихлорфлуоресцеин.
2 (2, 7 Дихлор 6 rидрокси 3 оксо 3Н ксантен
9 ил)бензойная кислота.
Порошок от желтовато коричневоro до
желто оранжевоro цвета. Малорастворим в
воде, леrкорастворим в 96 % спирте и разве
денных растворах rидроксидов щелочных Me
таллов с образованием раствора с желтова
то зеленой флуоресценцией.
4.1.1. РеактивЬ1
6ЗЗ
Дихлорхинонхлоримид. С 6 Н 2 Сl з NО.
(М м. 210,4). 1027400. [101 38 2]. 2,6 Дихлор N
хлор 1 ,4 бензхинонмоноимин.
Бледно желтый или зеленовато желтый кри
сталлический порошок. Практически HepaCTBO
рим в воде, растворим в 96 % спирте и разведен
ных растворах rидроксидов щелочных металлов.
Температура плавления: около 66 ос.
# 1,2-Дихлорэтан. C 2 H 4 C1 2 . (М м. 98,96).
Этиленхлорид. Этилен хлористый.
Прозрачная бесцветная жидкость. Не CMe
шивается с водой.
Дициклоrексил. С 12 Н 22 . (М. М. 166,3).
1135300. [92 51 3]. Бициклоrексил.
d;g: около 0,864.
Температура кипения: около 227 ос.
Температура плавления: около 4 ос.
Дициклоrексиламин. С 12 Н 2з N. (М м. 181,3).
1027500. [101 83 7]. N.N Дициклоrексиламин.
Бесцветная жидкость. Умеренно растворим
в воде, смешивается с обычными орrаническими
раство ителями.
п : около 1,484.
Температура кипения: около 256 ос.
Температура затвердевания (2.2.18).
От О ОС до 1 ос.
Дициклоrексилмочевина. С1ЗН24N20,
(М м. 224.4). 1027600. [2387 23 7]. 1,3 Дицикло
rексилмочевина.
Белый или почти белый кристаллический
порошок.
Температура плавления: около 232 ос.
Диэтаноламин. C 4 H 11 N0 2 . (М м. 105,1).
1027800. [111 42 2]. 2,2' Иминобисэтанол.
Слеrка желтая прозрачная вязкая жидкость
или кристаллы, расплывающиеся на воздухе и
плавящиеся при температуре около 28 ос. Очень
леrко растворим в воде, в ацетоне и в метаноле.
d;g: около 1,09.
рН (2.2.3). От 10,0 до 11,5. Измеряют рН
раствора 50 r/л диэтаноламина Р.
Диэтаноламин, используемый в испытании
на щелочную фосфатазу, должен выдерживать
следующее дополнительное испытание.
Этаноламин. Не более 1,0 %. Определе
ние проводят методом rазовой хроматоrрафии
(2.2.28), используя в качестве внутреннеro CTaH
дарта 3 аминопро паноламин Р.
Раствор внутренне20 стандарта. 1,00 r
3 аминопропаноламина Р растворяют в aцeтo
не Р и доводят до объема 10,0 мл этим же paCT
ворителем.
Испытуемый раствор (а). 5,00 r диэтилами
на растворяют в ацетоне Р и доводят до объема
10,0 мл этим же растворителем.
Испытуемый раствор (Ь). 5,00 r диэтилами
на растворяют в ацетоне Р, прибавляют 1,0 мл
раствора внутреннеro стандарта и доводят aцe
тоном Р до объема 10,0 мл.
Растворы сравнения. 0,50 r этаноламина Р
растворяют в ацетоне Р и доводят до объема
10,0 мл этим же растворителем. К 0,5 мл, 1,0 мл
и 2,0 мл полученноro раствора прибавляют rю
1,0 мл раствора внутреннеro стандарта и доводят
объем каждоro раствора ацетоном Р до 10,0 МЛ.
Хроматоrрафирование проводят на rаЗ080М
хроматоrрафе с пламенно ионизационным дe
тектором .
Хроматоrрафируют по 1,0 мкл каждоro ис
пытуемоro раствора и по 1,0 мкл каждоro paCT
вора сравнения в следующих условиях:
колонка длиной 1 м и внутренним диаме
тром 4 мм, заполненная полимером дифенил
фениленоксида Р с размером частиц от 180 мкм
до 250 мкм;
rаз носитель азот для хромат02рафии Р;
скорость rаза носителя 40 мл/мин;
температуру колонки поддерживают при
125 ос в течение 3 мин; а затем повышают TeM
пературу до 300 ос со скоростью 12 ОС/мин;
температура блока ввода проб 250 ос;
температура детектора 280 ос.
Хранят в воздухонепроницаемом контейнере.
Диэтиламин. C 4 H 11 N. (М м. 73,1). 1028000.
[109 89 7].
Прозрачная бесцветная воспламеняюща
яся жидкость. Имеет сильнощелочную реакцию.
Смешивается с водой и с 96 % спиртом.
d;g: около 0,71.
Температура кипения: около 55 ос.
Диэтиламиноэтилдекстран. 1028200.
Анионообменная смола в форме rидрохло
рида. Порошок, образующий с водой rель.
N,N Диэтиланилин. C 10 H 15 N. (М м. 149.2).
1028400. [91 66 7].
d;g: около 0,938.
Температура кипения: около 217 ос.
Температура плавления: около 38 ос.
Ди(2 этилrексил)фталат. С24НзеО4'
(М м. 390,5). 1028100. Ди(2 этилrексил)бензол
1 ,2 дикарбоксилат.
Прозрачная маслянистая жидкость. Практи
чески нерастворим в воде, растворим в орrани
ческих растворителях.
d 20 .
20' около 0,98.
20
по: около 1,486.
Вязкость (2.2.9): около 80 мПа'с.
Диэтиленrликоль. С 4 Н 10 О з . (М м. 106,1).
1028300. [111 46 6]. 2,2' Оксидиэтанол.
Содержание: не менее 99,5 % (м/м) С 4 Н 10 О з .
Прозрачная бесцветная rиrроскопичная
жидкость. Смешивается с водой, с ацетоном и с
96 % спиртом.
634
rосударственная фармакопея Республики Беларусь
d;g: около 1,118.
20
п о : около 1,447.
Температура кипения: от 244 ос до 246 ОС.
Хранят в воздухонепроницаемом контей
нере.
Диэтилфенилендиамина сулЬфат. C1OH1BNP4S.
(М. М. 262,3). 1028600. [6283 63 2]. N,N' Диэтил п
фенилендиамина сульфат. N,N Диэтилбензол
1 ,4 диамина сульфат.
Белый или слеrка желтый порошок. PaCTBO
рим в воде.
Температура плавления: около 185 ос с раз
ложением.
Хранят в защищенном от света месте.
Диэтилфенилендиамина сульфата раст-
вор. 1028601.
К 250 мл воды Р прибавляют 2 мл кислоты
серной Р и 25 мл 0,02 М раствора натрия эде
тата. В полученном растворе растворяют 1,1 r
диэтилфенилендиамина сульфата Р и доводят
водой Р до объема 1000 мл.
Раствор не используют, если он окрашен.
Хранят в защищенном от света и тепла
месте.
Срок rодности 1 мес.
N,N-Диэтилэтан-1,2-диамин. 1028500.
[100 З6 7]. См. N,N Диэтилэтилендиамин Р.
N,N-Диэтилэтилендиамин. C 6 H 16 N 2 .
(М. М. 116,2). 1028500. [100 36 7].
Содержание: не менее 98,0 % C 6 H 16 N2"
Бесцветная или слеrка желтая слеrка Mac
лянистая жидкость с сильным запахом амми
ака. Оказывает раздражающее действие на
кожу, rлаза и слизистые оболочки.
d;g: 0,827.
Температура кипения: от 145 ос до 147 ОС.
Вода (2.5.12). Не более 1,0 %. Определение
проводят из 0,500 r испытуемоro образца.
Диэтокситетраrидрофуран. С в Н 16 О з .
(М. М. 160,2). 1027900. [3320 90 9]. 2,5 Диэтокси
тетраrидрофуран. Смесь цис и транс изомеров.
Прозрачная бесцветная или слеrка желтова
тая жидкость. Практически нерастворим в воде,
растворим в 96 % спирте и в большинстве друrих
орrанических растворителей.
d;g: около 0,98.
20
п о : около 1,418.
Докозаrексаеновой кислоты метило
вый эфир. С 2З Н З Р2' (м. м. 342,5). 1142800.
[301 01 9]. Метиловый эфир дrк. (а/l Z) Докоза
4,7,1 О, 13, 16, 19 rексаеновой кислоты метиловый
эфир.
Содержание: не менее 90,0 %; определение
про водят методом rазовой хроматоrрафии.
Докузат натрия. С20НЗ7NаО7S' (М. м. 444,6).
1034100. [577 11 7]. Натрия 1,4 бис[(2 этил
rексил )окси] 1 +диокобутан 2 сульфонат.
Белая или почти белая воскообразная масса
или хлопья. rиrроскопичен. Умеренно paCТBO
рим в воде, леrкорастворим в 96 % спирте и в
метиленхлориде.
Содержит не менее 98,0 % и не более
101,0 % С20НЗ7Nа07S в пересчете на безводное
вещество.
Дотриаконтан. С з2 Н 66 . (м. М. 450,9).
1034200. [544 85 4]. н Дотриаконтан.
Белые или почти белые пластинки. Практи
чески нерастворим в воде, умеренно растворим
в reKcaHe.
Температура плавления: около 69 "С.
Примеси. Не более 0,1 % примесей со
временем удерживания, характерным для
а токоферилацетата. rазовая хроматоrрафия
(2.2.28), как указано в статье a Токофери
лацетат.
# Драrендорфа реактив.
Раствор 1. К 0,85 r висмута нитрата основно--
20 Р прибавляют 40 мл воды Р, 10 мл кислоты YK
сусной ледяной Р и встряхивают в течение 15 мин.
Раствор 11. 8 r калия йодида Р растворяют в
20 мл воды Р.
Смешивают равные объемы растворов 1 и 11.
К 1 О мл полученной смеси прибавляют 100 мл
воды Р и 20 мл кислоты уксусной ледяной Р.
# Драrендорфа реактив модифициро-
ванный.
Раствор 1. К 0,85 r висмута нитрата OCHoв
ною Р прибавляют 40 мл воды Р, 1 О мл кислоты YK
сусной ледяной Р и встряхивают в течение 15 мин.
Раствор 11. 8 r калия йодида Р растворяют в
20 мл воды Р.
Основной раствор. Смешивают растворы I и 11.
Непосредственно перед использованием
5 мл основноro раствора смешивают с 5 мл раст
вора 1 О r/л кислоты аскорбиновой Р и 5 мл 96 %
спирта Р.
Дубильная кислота. См. Таниновая Kиc
лота р
Желатин. 1040000. [9000 70 8].
См. статью Желатин.
Желатин rидролизованный. 1040100.
50 r желатина Р растворяют в 1000 мл
воды Р Обрабатывают насыщенным паром в
автоклаве при температуре 121 ос в течение
90 мин и лиофилизируют.
Железа (11) аммония сульфат.
Fe(NH4MS04)2'6Hp. (м. м. 392,2). 1038200.
[7783 85 9]. Железа диаммония дисульфат reK
саrидрат.
4.1.1. Реактивы
635
Бледные синевато зеленые кристаллы или
rранулы. Леrко растворим в воде, практически
нерастворим в 96 % спирте.
Хранят в защищенном от света месте.
Железа (111) аммония сульфат.
FeNH4(S04)2'12Hp. (м. м. 482,2). 1037700.
[7783 8З 7]. Железа аммония дисульфат дoдe
каrидрат.
Бледно фиолетовые кристаллы, выветри
вающиеся на воздухе. Очень леrко растворим в
воде, практически нерастворим в 96 % спирте.
Железа (111) аммония сульфата раст-
вор Р2. 1037702.
Раствор 100 r/л железа (1/1) аммония сульфата Р.
При необходимости перед использованием
фильтруют.
Железа (111) аммония сульфата раст-
вор Р5. 1037704.
30,0 r железа (111) аммония сульфата Р
встряхивают с 40 мл кислоты азотной Р и дo
водят водой Р до объема 100 мл. Если раствор
мутный, ero центрифуrируют или фильтруют.
Хранят в защищенном от света месте.
Железа (111) аммония сульфата paCT
вор Р6. 1037705.
20 r железа (111) аммония сульфата Р paCT
воряют в 75 мл воды Р, прибавляют 10 мл 2,8 %
(об/об) раствора кислоты серной Р и доводят
водой Р до объема 100 мл.
# Железа (111) аммония сульфата раствор Р7.
0,5 r железа (1/1) аммония сульфата Р помеща
ют в колбу со шлифом, прибавляют 25 мл 2 М pacт
вара хлористоводородной кислоты, встряхивают,
выдерживают в течение 12 16 ч и фильтруют.
Срок roдности 1 мес.
Железа (111) нитрат. Fе(NОз)з'9НР. (м. м. 404).
1106100. [7782 61 8].
Содержит не менее 99,0 % (м/м) Fе(NОз)з '9Н 2 О.
Светло красновато фиолетовые кристаллы
или кристаллическая масса. Очень леrко pac
творим в воде.
Свободная кислота: не более 0,3 % (в виде
HNOJ
Железа салицилата раствор. 1046700.
0,1 r железа (111) аммония сульфата Р paCT
воряют в смеси из 2 мл кислоты серной разве
денной Р и 48 мл воды Р и доводят водой Р до
объема 100 мл. К полученному раствору прибав
ляют 50 мл раствора 11,5 r/л натрия салицила
та Р, 1 О мл кислоты уксусной разведенной Р,
80 мл раствора 136 r/л натрия ацетата Р и дo
водят водой Р до объема 500 мл.
rотовят непосредственно перед использова
нием.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
Железа (11) сульфат. 1038300. [7782 З 0].
См. статью Железа сульфат 2епта2идрат.
Железа (11) сульфата раствор Р2. 1038301.
0,45 r железа (11) сульфата Р растворяют в
50 мл 0,1 М раствора кислоты хлорис
родной и доводят водой, свободной от уzлерода
диоксида, Р до объема 100 мл.
rотовят непосредственно перед испол
ванием.
Железа (111) сульфат. Fe2(S04kxHP.
1037900. [10028 22 5]. Железа (111) трисульфат
водный.
Желтовато белый порошок. Очень rиrроско
пичен. Разлаrается на воздухе. Малорастворим
в воде и в 96 % спирте.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
Железа (111) сульфат пентаrидрат.
Fе2(S04)з'5НР. (м. м.489,9). 1153700. [142906--29-4].
Железа (111) трисульфат водный.
Порошок от белоro до желтоватоro цвета.
Железа (111) хлорид. FеСl з '6Нр.
(м. м. 270,3). 1037800. [10025 77 1]. Железа
трихлорид rексаrидрат.
Желто оранжевая или коричневая кри
сталлическая масса. Расплывается на воздухе.
Очень леrко растворим в воде, растворим в 96 %
спирте. Под действием света железа (111) хлорид
и еro растворы частично восстанавливаются.
Хранят в воздухонепроницаемом контейнере.
Железа (111) хлорида раствор Р1. 1037801.
Раствор 105 r/л железа (111) хлорида Р.
Железа (111) хлорида раствор Р2.1037802.
Раствор 13 r/л железа (111) хлорида Р.
Железа (111) хлорида раствор РЗ. 1037803.
2,0 r железа (111) хлорида Р растворяют в
этаноле Р и доводят до объема 100,0 мл этим
же растворителем.
Железа (111) хлорида, феррицианида м ар-
сенита реактив. 1037805.
Непосредственно перед исполЬЗОВанием сме-
шивают 1 О мл раствора 27 r/л железа xлopuдa Р в
кислоте хористоводородной разведенной Р, 7 мл
раствора калия феррицианuдa Р. 3 мл веды Р и
1 О мл раствора натрия арсениrnа Р.
Железа (111) хлорида и сульфаминовой
кислоты реактив. 1037804.
Раствор содержит 10 r/л железа (111) хлори
да Р и 16 r/л кислоты сульфаминовой Р.
Железо. Fe. (А. м. 55,85). 1046600. [7439 89--6].
Порошок сероro цвета или проволока. Pac
творимо в разведенных минеральных кислотах.
636
rосударственная фармакопея Республики Беларусь
# Желтая кровяная соль. См. Калия фер
роцианид Р.
Желудочный сок, искусственный.
1039900.
2,0 r натрия хлорида Р и 3,2 r порошка пеп
сина Р растворяют в воде Р, прибавляют 80 мл
1 М раствора кислоты хлористоводородной и
доводят водой Р до объема 1000 мл.
Заменитель тромбоцитов. 1066400.
К 0,5 1 r фосфолипидов Р прибавляют
20 мл ацетона Р и встряхивают в течение 2 ч,
затем центрифуrируют в течение 2 мин и сли
вают надосадочную жидкость. Остаток сушат
в вакууме (водяной насос), прибавляют 20 мл
хлороформа Р, встряхивают в течение 2 ч и
фильтруют под вакуумом. Полученный остаток
суспендируют в 5 1 О мл раствора 9 r/л натрия
хлорида Р.
Для использования в количественном опре
делении фактора IX roтовят разведенную cy
спензию в растворе 9 r/л натрия хлорида Р
таким образом, чтобы разность времени Koary
ляции между последовательными разведениями
суспензий БСП составляла около 1 О с.
Хранят разведенные суспензии при темпе
ратуре ЗО ОС. Срок roдности 6 нед.
Изатин. C a H 5 N0 2 . (м. м. 147,1). 1046800.
[91 56 5J. Индолин 2,3 дион.
Мелкие желтовато красные кристаллы. Ma
лорастворим в воде, растворим в rорячей воде,
в 96 % спирте, растворим в растворах rидрокси
дов щелочных металлов с образованием фиоле
товоro окрашивания, переходящеro при стоянии
в желтое.
Температура плавления: около 200 ос с ча
стичной сублимацией.
Сульфатная зола (2.4.14). Не более 0,2 %.
Изатина реактив. 1046801.
6 Mr железа (111) сульфата Р растворяют в
8 мл воды Р, прибавляют при перемешивании
50 мл кислоты серной Р. К полученному paCTBO
ру прибавляют 6 Mr изатина Р и перемешивают
до растворения.
Раствор должен быть светло желтоro цвета,
но не должен иметь оранжевый или красный
цвет.
# Известь жженая. См. Кальция оксид Р.
Изоамиловый спирт. С 5 Н 1 р. (м. м. 88,1).
1046900. [123 51 3J. 3 МеТ!-1лбутан 1 ол.
Бесцветная жидкость. Малорастворим в
воде, смешивается с 96 % спиртом.
Температура кипения: около 130 ОС.
Изоандростерон. С19Нзо02' (м. м. 290,4).
1107100. [481 29 8J. Эпиандростерон. 313 rидро
кси 5а андростан 17 OH.
Белый или почти белый порашок. Практиче
ски нерастворим в воде, растворим в орraнических
растворителях.
Температура плавления: от 172 ОС до 174 ОС.
[a] O: около +88. Определение проводят, ис
пользуя раствор 20 r/л испытуемоro образца в Me
таноле р.
ДА (2.2.41): 14,24-103. Определение право--
дят при длине волны 304 нм, используя раствор
1,25 r/л испытуемоro образца.
# Изобарбитуровая кислота. См. 5--rидрокси
урацил р.
Изокверцитрозид. C21H2Q012' (м. м. 464,4).
1136500. [21637 25--2J. 2 (З,4 Диrидроксифенил}-
3 (J3 D rлюкофуранозилокси ) 5, 7 диrидрокси
4H 1--бензопиран-4--он. 3,3',4' ,5, 7 Пентаrидракси
флавон 3 rлюкозид. Изокверцитрин.
Изомальт. С12Н24011. (М. м. 344,3).1164300.
[64519 82..()J. Смесь из 6--0 а D rлюкопиранозил
D rлюцитола и 1 0 D rлюкопиранозил D манни
тола.
Белый или почти белый порошок или rpанулы.
Леrкорастворим в воде.
Изоментол. С 1О Н 20 О. (м. м. 156,3). 1047000.
[23283 97 8J.
(+ ) Изоментол: (1 S,2R,5R}-2 изопропил 5 ме
тилциклоrексанол.
(:!) Изоментол: смесь из равных частей
(1S,2R,5R)- и (1R,2S,5S) 2 изопропил 5 метил
циклоrексанола.
Бесцветные кристаллы. Практически He
растворим в воде, очень леrко растворим в 96 %
спирте.
[a] O: (+}-Изоментол: около +24. Определение
проводят, используя раствор 100 r/л испытуемоro
образца в 96 % спирте Р.
Температура кипения (+ ) изоментола: около
218 ОС.
Температура кипения (:!) изоментола:
около 218 ОС.
Температура плавления (+) изоментола:
около 80 ОС.
Температура плавления (:!) изоментола:
около 53 ОС.
(+)-Изоментон. С 10 Н 1 р. (м. м. 154,2). 1047100.
(1 R) цис п Ментан 3--он. (1 R}-цис 2 Изопропил 5
метилциклоrексанон.
Содержит различные количества ментона.
Бесцветная жидкость. Очень мало растворим
в воде, растворим в 96 % спирте.
d 20
20: около 0,904.
20
по: около 1,453.
[a] O: около +93,2.
Изоментон, используемый в 2азовой xpOMa
тоарафии, должен выдерживать следующее дo
полнительное испытание.
4.1.1. Реактивы
637
Количественное определение. rазовая
хроматоrрафия (2.2.28), как указано в статье
Масло мяты перечной, с использованием
(+) изоментона В качестве испытуемоro paCT
вора.
Содержание (+ ) изоментона, рассчитанное
методом нормализации, должно быть не менее
80,0%.
# Изопентан. См. 2 Метилбутан Р.
Изопропиламин. С з Н 9 N. (М м. 59,1).
1119800. [75 31 0]. Пропан 2 амин.
Бесцветная сильно летучая воспламеня
ющаяся жидкость.
20
по : около 1,374.
Температура кипения: от 32 ос до 34 ос.
Изопропилйоди . СзН/ (М м. 170,0).
1166600. [75 ЗО 9]. 2 Иодпропан.
Изопропилметансульфонат. С 4 Н 10 О з S.
(М М. 138,2). 1179400. [926 06 7]. 1 Метилэтил
метансульфонат.
Прозрачная, бесцветная жидкость.
Содержит не менее 99,0 % С 4 Н 10 О з S.
Плотность: около 1,129 r/см З (при 20 ОС).
20
по: 1,418 1,421.
Температура кипения: около 82 ос при дaB
лении 6 мм рт. ст.
Изопропилмиристат. 1047200. [110 27 0].
См. статью Изопропилмиристат.
4 Изопропилфенол. С Э Н1Р' (М м. 136,2).
1047300. [99 89 8].
Содержит не менее 98 % С э Н 12 О.
Температура кипения: около 212 ОС.
Температура плавления: от 59 ос до 61 ОС.
Изопулеrол. С 10 Н 1 р. (М М. 154,2).
1139600. [89 79 2]. Н Изопулеroл. (1R,2S,5R) 2
Изопропенил 5 метилциклоrексанол.
d 20
4 : около 0,911.
20
по: около 1,472.
Температура кипения: около 91 ОС.
Изопуле20Л, используемый в 2азовой xpo
мат02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая
хроматоrрафия (2.2.28), как указано в статье
Масло мяты перечной, со следующими изме
нениями:
колонка капиллярная кварцевая длиной
от 30 м (толщина слоя 1 мкм) ДО 60 м (толщи
на слоя 0,2 мкм) и внутренним диаметром 0,25
0.53 мм, покрытая слоем маКР020ла 20 000 Р;
деление потока: 1: 1 00.
Содержание изопулеroла, рассчитанное Me
тодом нормализации, должно быть не менее
99%.
И мидазол. С з Н 4 N 2 . (М М. 68,1). 1045400.
[288 32 4].
Белый или почти белый кристаллический
порошок. Растворим в воде и в 96 % спирте.
Температура плавления: около 90 ОС.
Иминодибензил. С 14 Н 1з N. (М м. 195,3).
1045500. [494 1 9]. 10, 11 диrидродибенз[Ь, aэenин.
Бледно желтый кристаллический порошок.
Практически нерастворим в воде, леrкораство-
рим в ацетоне.
Температура плавления: около 106 ОС.
Индиrокармин. C16HaN2NapaS2' (М М. 466,3).
1045600. [860 22 0]. Показатель Шульца NQ 1309.
Цветной индекс NQ 73015. Динатрия 3,3' диоксо
2,2' бисиндолиден 5,5' дисульфонат. E 1 32.
Обычно содержит натрия хлорид.
Порошок от синеro до фиолетово синеro
цвета или rранулы синеro цвета с медным бле
ском. Умеренно растворим в воде, практически
нерастворим в 96 % спирте. Осаждается из BO
дноro раствора натрия хлоридом.
# Индиrокармина индикаторный раствор.
0,1 r инди20кармина Р растворяют в 100 мл
воды Р.
Изменение окраски. От 11,6 (синее окраши
вание) до 14,0 (желтое окрашивание).
Индиrокармина раствор. 1045601.
0,2 r инди20кармина Р растворяют в смеси
из 1 О мл кислоты хлористоводородной Р и
990 мл раствора 200 r/л кислоты серной, cвo
бодной от азота, Р.
Раствор должен выдерживать следующее
испытание:
1 О мл полученноro раствора прибавляют к
раствору 1,0 Mr калия нитрата Р в 1 О мл воды Р,
быстро прибавляют 20 мл кислоты серной. cвo
бодной от азота, Р и наrревают до кипения
Синее окрашивание раствора должно исчезнуть
в течение 1 мин.
Индиrокармина раствор Р1. 1045602.
4 r инди20кармина Р растворяют в воде Р,
прибавляя воду отдельными порциями до
объема 900 мл, затем прибавляют 2 мл Kиc
лоты серной Р и доводят водой Р до объема
1000 мл.
Установка титра. 10,0 мл зталОНН020
раствора нитрата (100 ррт NO.) Р помеща
ют в коническую колбу с широким roрлом BMe
стимостью 100 мл, прибавляют 10 мл воды Р,
0,05 мл раствора иHди201 apMиHa Р1 и быстро
прибавляют (за один раз. но осторожно) 30 мл
кислоты серной Р Полученный раствор He
медленно титруют раствором иHди20KapMи
на Р1 до появления стабильноro синеro OKpa
шивания.
Количество миллилитров (п), израсходо
ванное на титрование, соответствует 1 Mr NО з .
638
rосударственная фармакопея Республики Беларусь
Индикатора BRP раствор. 1013000.
0,1 r бромтимолов020 сине20 Р, 20 Mr Me
тилов020 краСН020 Р и 0,2 r фенолфталеина Р
растворяют в 96 % спирте Р, доводят до объема
100 мл этим же растворителем и фильтруют.
# Индикаторная бумаrа универсальная.
Используют roтовые тест полоски с соот
ветствующей шкалой значений рН от О до 14.
При определении рН тест полоску поrружают на
1 2 с в испытуемую жидкость так, чтобы она
была равномерно смочена. Быстро вынув TeCT
полоск немедленно сравнивают ее окраску с
цветовой стандартной шкалой.
Индометацин. 1101500. [53 86 1].
См. статью Индометацин.
# Индофеноловый синий. C'BH'6N20.
(М м. 276,3). 1045700. [132 31 0]. Пока
затель Шульца NQ 939. Цветной индекс
NQ 49700. N [4 (Диметиламино)фенил)Н ,4
нафтохинонмоноимин.
Порошок фиолетово черноro цвета. Практи
чески нерастворим в воде.
Хромат02рафия. Тонкослойная xpOMaтo
rрафия (2.2.27), используя в качестве тонкоro
слоя силика2ель G Р. На хроматоrрафическую
пластинку наносят 10 мкл раствора 0,1 r/л ис
пытуемоro образца вметиленхлориде Р и xpo
матоrрафируют в этом же растворителе. Фронт
растворителя должен пройти не менее 1 О см от
линии старта. На xpoMaTorpaMMe должно обна
руживаться только одно основное пятно. Допу
скается пятно на старте.
Инозин. C,oH'2N.D5' (М м. 268,2). 1169900.
[58 63 9]. 9 13 Рибофуранозилrипоксантин. 9 I3 D
Рибофуранозил 1 , диrидро 6Н пурин 6 он.
Температура плавления: от 222 ос до 226 ОС.
Ионообменная смола сильнокислотная.
1085400.
Смола в протонированной форме с rруппа
ми кислоты сульфоновой. присоединенными к
решетке, состоящей из полистирола, поперечно
сшитою 8 % дивинилбензола. Выпускают в виде
rранул шарообразной формы: если нет друrих
указаний, размер частиц составляет от 0,3 мм
до 1,2 мм.
Емкость. От 4,5 ммоль/r до 5 ммоль/r, при
содержании воды от 50 % до 60 %.
При20товление колонки. Если нет друrих
указаний, используют трубку с вплавленным
внутрь диском из пористоro стекла длиной
400 мм, внутренним дияметром 20 мм и BЫCO
той наполнения около 200 мм. Смолу предвари
тельно смешивают с водой Р и вводят получен
ную взвесь в трубк не допуская образования
пузырьков воздуха между частицами. Во время
работы жидкость не должна опускаться ниже по
верхности смолы.
Если смола в протонированной форме, про
мывают водой Р до тех пор, пока для нейтрали
зации 50 мл потребуется не более 0,05 мл 0,1 М
раствора натрия 2идроксида, используя в каче
стве индикатора 0,1 мл раствора метилов020
оранжев020 Р. Если смола в натриевой форме или
нуждается в реrенерации, через колонку медлен
но пропускают около 100 мл смеси из равных обь€--
мов кислоты хлористоводородной Р1 и воды Р,
а затем промывают водой Р, как описано выше.
Йод. 1045800. [7553 56 2]. См. статью Йод.
Йода раствор Р1. 1045801.
К 10,0 мл 0,05 М раствора йода прибавля
ют 0,6 r калия йодида Р и доводят водой Р до
объема 100,0 мл.
rотовят непосредственно перед использо
ванием.
Йода раствор Р2. 1045802.
К 10,0 мл 0,05 М раствора йода прибавля
ют 0,6 r калия йодида Р и доводят водой Р до
объема 1000,0 мл.
rотовят непосредственно перед использо
ванием.
Йода раствор РЗ. 1045803.
2,0 мл раствора йода Р1 доводят водой Р
до объема 100,0 мл.
rотовят непосредственно перед использо
ванием.
Йода раствор Р4. 1045806.
14 r йода Р растворяют в 100 мл раствора
400 r/л калия йодида Р, прибавляют 1 мл кисло
ты хлористоводородной разведенной Р и ДOBO
дят водой Р до объема 1000 мл.
Хранят в защищенном от света месте.
Йода раствор спиртовой. 1045804.
Раствор 1 О r/л йода Р в 96 % спирте Р.
Хранят в защищенном от света месте.
Йода раствор в хлороформе. 1045805.
Раствор 5 r/л йода Р в хлороформе Р.
Хранят в защищенном от света месте.
Йода (V) оксид перекристаллизирован
ный. IP5' (М м. 333,8). 1046000. [12029 98 0].
Ди йода пентоксид. Йодный анrидрид.
Содержит не менее 99,5 % 1205'
Белый или почти белый кристаллический
порошок или rранулы от белоro до серовато бе
лоro цвета. rиrроскопичен. Очень леrко paCTBO
рим в воде с образованием НlO з .
Стабильность при на2ревании. 2 r, предва
рительно выдержанные при температуре 200 ос
в течение 1 ч, растворяют в 50 мл воды Р. PaCT
вор должен быть бесцветным.
Количественное определение. 0,100 r ис
пытуемоro образца растворяют в 50 мл воды Р,
4.1. 1. Реактивы
639
прибавляют 3 r калия йодида Р и 1 О мл кислоты
хлористоводородной разведенной Р Титруют
высвободившийся йод 0,1 М раствором натрия
тиосульфата, используя в качестве индикатора
1 мл раствора крахмала Р
1 мл 0,1 М раствора натрия тиосульфата
соответствует 2,782 Mr 1205'
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
Йода бромид. IBr. (М м. 206,8). 1045900.
[7789 З3 5].
Кристаллы от синевато черноro до корич
невато черноro цвета. Леrкорастворим в воде, в
96 % спирте и кислоте уксусной ледяной.
Температура кипения: около 116 ОС.
Температура плавления: около 40 ОС.
Хранят в защищенном от света месте.
Йода бромида раствор. 1045901.
20 r йода бромида Р растворяют в кисло
те уксусной ледяной Р и доводят до объема
1000 мл раствора этим же растворителем.
Хранят в защищенном от света месте.
Йода хлорид. ICI. (М м. 162,4). 1143000.
[7790 99 9].
Кристаллы черноro цвета. Растворим в воде,
в кислоте уксусной и в 96 % спирте.
Йода хлорида раствор. 1143001.
1,4 r йода хлорида Р растворяют в кислоте
уксусной ледяной Р и доводят до объема 100 мл
этим же растворителем.
Хранят в защищенном от света месте.
# Йода хлорида раствор Р1.
11,06 r калия йодида Р и 7,10 r калия
йодата Р помещают в колбу со шилфом, прибав
ляют 50 мл воды Р и 50 мл кислоты хлористо
водородной Р, закрывают пробкой и встряхивают
до полноro растворения йода, образующеroся в
ходе реакции. Раствор переносят в делительную
воронку и взбалтывают с 10 мл хлороформа Р.
Если хлороформный слой окрасится в фиолето
вый цвет, прибавляют по каплям при энерrичном
встряхивании раствор 1 О r/л калия йодата Р до
исчезновения окраски хлороформноro слоя. Если
хлороформный слой остается бесцветным, при
бавляют по каплям раствор 10 r/л калия йодида Р
до бледно розовоro окрашивания. После отстаи
вания водный слой сливают в мерную колбу BMe
стимостью 1000 мл И доводят водой Р до объема
1000,0 мл. Полученный раствор должен иметь
светло желтое окрашивание.
Йодаткрахмальная бумаrа. 1085101.
Полоски фильтровальной бумаrи поrружают
в 100 мл раствора крахмала, свободН020 от йо
дидов, Р, содержащеrо 0,1 r калия йодата Р Из
быток жидкости удаляют. Сушат в защищенном
от света месте.
Йодкрахмальная бумаra. 1085106.
Полоски фильтровальной бумаrи поrpужа
ют в 100 мл раствора крахмала Р, содержащеrо
0,5 r калия йодида Р Избыток жидкости удаля
ют. Сушат в защищенном от света месте.
Испытание на чувствительность. CM
шивают 0,05 мл 0,1 М раствора натрия Hи
трита и 4 мл кислоты хлористоводо
ной Р и доводят водой Р до объема 100 мл.
1 каплю полученноro раствора наносят на йод
крахмальную бумаrу. Образуется синее пятно.
2 Йодбензойная кислота. С 7 Н 5 1О 2 .
(М м. 248,0). 1046100. [88 67 5].
Кристаллический порошок от белоro до
светло желтоro цвета. Малорастворима в воде,
растворима в 96 % спирте.
Температура плавления: около 160 ОС.
Хромат02рафия. Тонкослойная xpOMaTO
rрафия (2.2.27), используя в качестве тонкоro
слоя целлюлозу для хромат02рафии F 254 Р. На
линию старта хроматоrрафической пластин
ки наносят 20 мкл раствора 2 йодбензойной
кислоты, приroтовленноro растворением 40 Mr
испытуемоro образца в 4 мл 0,1 М раствора
натрия 2идроксида и разведением водой Р
до объема 1 О мл. Хроматоrрафируют, исполь
зуя в качестве подвижной фазы верхний слой,
полученный при встряхивании смеси paCT
ворителей вода Р кислота уксусная ледя
ная Р толуол Р (20:40:40, об/об/об). Korдa
фронт растворителей пройдет 12 см. пластин
ку просматривают в ультрафиолетовом свете
при длине волны 254 нм. На xpoMaTorpaMMe
должно обнаруживаться только одно основное
пятно.
2 Йодrиппуровая кислота. С g Н в INО з '2Н 2 О.
(М м. 341,1). 1046200. [147 58 0]. 2 (2 Йодбенза
мидо )уксусная кислота.
Белый или почти белый кристаллический
порошок. Умеренно растворима в воде.
Температура плавления: около 170 ос.
Вода (2.5.12). От 9 % до 13 %. Определение
проводят из 1,000 r испытуемоro образца.
Хромат02рафия. Тонкослойная xpOMaTO
rрафия (2.2.27), используя в качестве тонкоro
слоя целлюлозу для хромат02рафии F 254 Р. На
линию старта хроматоrрафической пластинки
наносят 20 мкл раствора 2 йодrиппуровой кисло
ты, приrотовленноro растворением 40 Mr испы
туемоro образца в 4 мл О, 1 М раствора натрия
2идроксида и разведением водой Р до объема
1 О мл. Хроматоrрафируют, используя в качестве
подвижной фазы верхний слой, полученный при
встряхивании смеси растворителей вода Р
кислота уксусная ледяная Р толуол Р
(20:40:40, об/об/об). Korдa фронт растворителей
пройдет 12 см, пластинку просматривают в уль
трафиолетовом свете при длине волны 254 нм.
На xpoMaтorpaMMe должно обнаруживаться
только одно основное пятно.
640
rосударственная фармакопея Республики Беларусь
ЙОДИCТQводородная кислота. HI. (М м. 127,9).
1098900. [1 0034 85 2].
Кислоту йодистоводородную переrоняют
над красным фосфором, пропуская во время
переrонки уzлерода диоксид Р или азот Р. Ис
пользуют бесцветную или почти бесцветную, ки
пящую при постоянной температуре смесь (от
55 % до 58 % HI), переюняющуюся при темпера
туре от 126 ос до 127 ОС.
Кислоту помещают в небольшие флаконы
из стекла коричневою цвета, предварительно
продутые У2лерода диоксидом Р или азотом Р,
со стеклянными пробками, rерметизируют пара
фином.
Хранят в защищенном от света месте.
Йодная кислота. Н 5 1О 6 . (м. М. 227,9).
1108900. [10450 60 9].
Кристаллы. Леrкорастворима в воде, pac
творима в 96 % спирте.
Температура плавления: около 122 ОС.
Йодной и уксусной кислоты раствор.
1063000.
0,446 r натрия перйодата Р растворяют в
2,5 мл раствора 25 % (об/об) кислоты серной Р
и доводят кислотой уксусной ледяной Р до
объема 100,0 мл.
Йодплатината реактив. 1046300.
К 3 мл раствора 100 r/л кислоты хлорпла
тиновой Р прибавляют 97 мл воды Р и 100 мл
раствора 60 r/л калия йодида Р.
Хранят в защищенном от света месте.
Йодсернистый реактив. 1046400. #Реактив
Фишера."
Устройство для приютовления реактива, co
стоящее из круrлодонной колбы вместимостью
от 3000 мл ДО 4000 мл С тремя входными OTBep
стиями для мешалки, термометра и трубки, за
полненной осушителем, должно быть закрытым
и сухим в процессе подroтовки. В колбу поме
щают 700 мл пиридина безводН020 Р и 700 мл
монометилов020 эфира этилеН2ликоля Р, при
бавляют при постоянном перемешивании 220 r
мелкоизмельченною йода Р, предварительно
высушенноro над фосфора (V) оксидом Р. Пере
мешивание продолжают до полноrо paCTBope
ния йода (около 30 мин), затем охлаждают колбу
до температуры 10 ос и быстро прибавляют
при постоянном перемешивании 190 r серы ди
оксида Р. Температура реакционной смеси не
должна превышать 30 ОС. Охлаждают.
Установка титра. Около 20 мл MeтaHO
ла безводН020 Р помещают в сосуд для титро
вания и титруют приroтовленным йодсернистым
реактивом (2.5.12). Прибавляют точно взвешен
ное достаточное количество воды Р и повторяют
определение воды. Рассчитывают количество
воды в миллиrраммах соответствующее 1 мл
йодсернистоro реактива.
1 мл йодсернистоro реактива соответствует
как минимум 3,5 Mr воды.
Должны быть приняты меры предосторож
ности для предотвращения воздействия на pac
творы атмосферной влаrи. Титр устанавливают
непосредственно перед использованием.
Хранят в сухом контейнере.
Йодуксусная кислота. С 2 Н з 10 2 " (м. М. 185,9).
1107000. [64 69 7].
Бесцветные или белые кристаллы. PaCTBO
рима в воде и в 96 % спирте.
Температура плавления: от 82 ос до 83 ОС.
S-Йодурацил. С 4 Н з l!'JР2 (м. М. 238,0).
1 046500. [696 7 1]. 5 Иод 1 Н,3Н пиримидин
2,4 дион.
. Температура плавления: около 276 ос с раз
ложением.
Хромат02рафия. Тонкослойная xpOMa
тоrрафия (2.2.27), как указано в статье Йодо
ксиуридин. На хроматоrрафическую пластинку
наносят 5 мкл раствора 0,25 r/л испытуемоro об
разца. На xpoMaTorpaMMe должно быть только
одно основное пятно.
Йодэтан. С 2 Н 5 1. (м. м. 155,9). 1099100.
[75 03 6],
Жидкость от бесцветноro до слеrка желтова
тоro цвета, под действием воздуха и света TeM
неет. Смешивается с 96 % спиртом и большин
ством орrанических растворителей.
d 20
20: около 1,95.
20
по: около 1,513.
Температура кипения: около 72 ОС.
Хранят в воздухонепроницаемом контейнере.
Кадмий. Cd. (А. м. 112,4). 1014100. [10108--64 2].
Серебристо белый блестящий металл.
Практически нерастворим в воде, леrкораство
рим в кислоте азотной и roрячей кислоте хлори
стоводородной.
Казеин. 1016600. [9000 71 9].
Смесь родственных фосфопротеинов, полу
ченных из молока.
Белый или почти белый аморфный порошок
или rранулы. Очень мало растворим в воде и в
неполярных орrанических растворителях, pac
творим в кислоте хлористоводородной KOHцeH
трированной с образованием бледно фиолето
воro окрашивания. Образует соли с кислотами и
основаниями. Изоэлектрическая точка казеина
находится при значении рН около 4,7. Щелочные
растворы являются левовращающими.
# Калий железистосинеродистый. См.
Калия ферроцианид Р.
# Калий железосинеродистый. См. Калия
феррицианид Р.
4.1.1. Реактивы
641
Калия бикарбонат. 1069900. [298 14 6].
См. Калия 2идрокарбонат Р.
Калия бикарбоната раствор насыщенный,
метанольный. 1069901. См. Калия 2идрокарбо
ната раствор насыщенный, метанольный Р.
# Калия бихромат. См. Калия дихромат Р.
Калия бромат. КВrО з , (М м. 167,0). 1068700.
[7758 01 2].
Белый или почти белый rранулированный
порошок или кристаллы. Растворим в воде, Ma
лорастворим в 96 % спирте.
Калияi5ромид. 1068800. [7758 02 3].
См. статью Калия бромид.
Калия бромид, используемый для абсорб
ционной спектрофотометрии в инфракрасной
области (2.2.24), должен выдерживать следу
ющее дополнительное испытание.
ИК спектр диска толщиной 2 мм, полученноro
из калия бромида, предварительно высушенноro
при температуре 250 ос в течение 1 ч, должен
иметь практически ровную базовую линию в ин
тервале от 4000 см., до 620 см.,. Не должен иметь
максимумов с поrлощением более 0,02 над ба
зовой линией, за исключением максимумов для
воды при длинах волн 3440 см., и 1630 см., .
Калия rидрокарбонат. КНСО З ' (М м. 100,1).
1069900. [298 14 6]. Калия бикарбонат.
Прозрачные бесцветные кристаллы. Леrко
растворим в воде, практически нерастворим в
96 % спирте.
Калия rидрокарбоната раствор насыщен-
ный, метанольный. 1069901.
0,1 r калия 2идрокарбоната Р растворяют в
0,4 мл воды Р при наrревании на водяной бане,
прибавляют 25 мл метанола Р и перемешивают
круroвыми движениями, продолжая наrревание
до растворения.
rотовят непосредственно перед использо-
ванием.
Калия rидроксид. КОН. (М м. 56,11).
1070300. [131 0 58 3].
Содержит не менее 85,0 % и не более
100,5 % суммы щелочей в пересчете на КОН.
Белая или почти белая твердая кристалли
ческая масса в виде палочек, пластинок или бес
форменных кусочков. Расплывается на воздухе.
rиrроскопичен. Очень леrко растворим в воде,
леrкорастворим в 96 % спирте.
Хранят 13 воздухонепроницаемом неметал
лическом контейнере.
Калия rидроксида 2 М раствор спирто-
вой. 1070301.
12 r калия 2идроксида Р растворяют в 1 О мл
воды Р и доводят 96 % спиртом Р до объема 100 мл.
81. Зак 1712
Калия rидроксида 0,5 М раствор спирто-
вой (10 %,06/06). 1070302.
28 r калия 2идроксида Р растворяют в 100 мл
96 % спирта Р и доводят водой Р до объема
1000 мл.
Калия rидроксида раствор спиртовой.
1070303.
3 r калия 2идроксида Р растворяют в 5 мл
воды Р и доводят 96 % спиртом, свободным
от альде2идов, Р до объема 100 мл. Деканти
руют прозрачный раствор. Раствор должен быть
почти бесцветным.
Калия rидроксида раствор спиртовой Р1.
1070304.
6,6 r калия 2идроксида Р растворяют в 50 мл
воды Р и доводят этанолом Р до объема 1000 мл.
Калия rидросульфат. KHS0 4 . (М М. 136,2).
1070100. [7646 93 7]. Калия бисульфат.
Прозрачные бесцветные rиrроскопичные
кристаллы. Леrкорастворим в воде с образова
нием сильнокислоro раствора.
Хранят в воздухонепроницаемом контейнере.
Калия rидротартрат. С4Н5КО6' (М М. 188,2).
1070200. [868 14 4]. Калия rидро (2R,3R) 2,3
диrидроксибутан 1 ,4 диоат.
Белый или почти белый кристаллический
порошок или бесцветные, слеrка матовые кри
сталлы. Малорастворим в воде, растворим в ки
пящей воде, практически нерастворим в 96 %
спирте.
Калия rидрофталат. C B H s K0 4 . (М М. 204,2).
1070000. [877 24 7]. Калия rидробензол 1,2
дикарбоксилат.
Белые или почти белые кристаллы. PaCTBO
рим в воде, малорастворим в 96 % спирте.
Калия rидрофталата 0,2 М раствор.
1070001.
Раствор калия 2идрофталата Р, содержа-
щий 40,84 r С в Н 5 К0 4 в 1000,0 мл.
Калия диrидрофосфат. 1 069600. [777 77 -О).
См. статью Калия ди2идрофосфат.
Калия диrидрофосфата 0,2 М раствор.
1069601.
Раствор калия ди2uдрофосфата Р, coдep
жащий 27,22 r КНРО4 в 1000,0 мл.
Калия дихромат. K 2 Crp7' (М М. 294,2).
1069500. [7778 1)Q 9]. Дикалия дихромат. Калия
бихромат.
Калия дихромат, используемый для кали
бровки спектрофотометров (2.2.25), должен co
держать не менее 99,9 % K 2 Cr 2 0 7 , в пересчете
на сухое вещество, высушенное при температу
ре 130 ос.
642
rосударственная фармакопея Республики Беларусь
Оранжево красные кристаллы. Растворим в
воде, практически нерастворим в 96 % спирте.
Количественное определение. 1,000 r
калия дихромата растворяют в воде Р и ДOBO
дят до объема 250,0 мл этим же растворите
лем. 50,0 мл полученною раствора помещают
в колбу вместимостью 500 мл прибавляют CBe
жеприroтовленный раствор, состоящий из 4 r
калия йодида Р, 2 r натрия 2идрокарбоната Р и
6 мл кислоты хлористоводородной Р в 100 мл
воды Р. Колбу закрывают пробкой, выдерживают
в защищенном от света месте в течение 5 мин и
титруют 0,1 М раствором натрия тиосульфа
та, используя в качестве индикатора 1 мл pacт
вора крахмала. свободНО20 от йодидов, Р.
1 мл 0.1 М раствора натрия тиосульфата
соответствует 4,903 Mr K 2 Cr 2 0]'
Калия дихромата раствор. 1069501.
Раствор 106 r/л калия дихромата Р.
Калия дихромата раствор Р1. 1069502.
Раствор 5 r/л калия дихромата Р.
Калия йодат. КIO З ' (м. м. 214,0). 1070400.
[7758 05 6] .
Белый или почти белый кристаллический
порошок. Растворим в воде.
Калия йодид. 1070500. [7681 11 O].
См. статью Калия йодид.
Калия йодида раствор. 1070502.
Раствор 166 r/л калия йодида Р.
# Калия йодида раствор Р1.
Раствор 100 r/л калия йодида Р.
Калия йодида и крахмала раствор.
1070501.
0,75 r калия йодида Р растворяют в 100 мл
воды Р, наrревают до кипения и прибавляют
при перемешивании раствор, содержащий 0,5 r
крахмала раствориМ020 Р в 35 мл воды Р. Кипя
тят В течение 2 мин и охлаждают.
Испытание на чувствительность. Раст
вор из 15 мл раствора калия йодида и крахмала,
0,05 мл кислоты уксусной ледяной Р и 0,3 мл
раствора йода Р2 должен иметь синее окраши
вание.
Калия йодида йодированный раствор.
1070503.
2 r йода Р и 4 r калия йодuда Р растворяют в
1 О мл воды Р. После полноro растворения ДOBO
дят водой р до объема 100 мл.
Калия йодида йодированный раствор Р1.
1070505.
500 Mr йода Р и 1 ,5 r калия йодида Р paCTBO
ряют в воде Р и доводят до объема 25 мл этим
же растворителем.
Калия йодида йодированный раствор Р1.
1070505.
500 Mr йода Р и 1,5 r калия йодида Р paCT
воряют воде Р до объема 25 мл этим же paCTBO
рителем.
Калия йодида насыщенный раствор.
1070504.
Насыщенный раствор калия йодида Р в воде,
свободной от У2лерода диоксида, Р, должен co
держать нерастворенные кристаллы.
Испытание. 0,5 мл насыщенноro раствора
калия йодида смешивают с 30 мл смеси хло
роформ Р кислота уксусная ледяная Р (2:3,
об/об), прибавляют 0,1 мл раствора KpaXMa
ла Р. Появляющееся синее окрашивание должно
быть удалено прибавлением 0,05 мл 0,1 М pacт
вора натрия тиосульфата.
Хранят в защищенном от света месте.
Калия йодовисмутата раствор. 1070600.
К 0,85 r висмута нитрата основН020 Р при
бавляют 40 мл воды Р, 1 О мл кислоты уксусной ле
дяной Р и 20 мл раствора 400 r/л калия йодида Р.
Калия йодовисмутата раствор Р1.
1070601.
100 r кислоты винной Р растворяют в 400 мл
воды Р, прибавляют 8,5 r висмута нитрата oc
новН020 Р. встряхивают в течение 1 ч, прибав
ляют 200 мл раствора 400 r/л калия йодида Р и
интенсивно встряхивают. Выдерживают 24 ч и
фильтруют.
Хранят в защищенном от света месте.
Калия йодовисмутата раствор Р2.
1070602.
Исходный раствор. Суспендируют 1,7 r виc
мута нитрата основН020 Р и 20 r кислоты
винной Р в 40 мл воды Р. К суспензии прибавля
ют 40 мл раствора 400 r/л калия йодида Р, встря
хивают в течение 1 ч и фильтруют.
Срок rодности раствора несколько дней при
хранении во флаконах коричневоro стекла.
Раствор для опрыскивания. Непосред
ственно перед использованием смешивают 5 мл
исходноro раствора с 15 мл воды Р.
Хранят в защищенном от света месте.
Калия йодовисмутата раствор РЗ.
1070604.
0,17 r висмута нитрата основН020 Р paCT
воряют в смеси из 2 мл кислоты уксусной ле
дяной Р и 18 мл воды Р, прибавляют 4 r калия
йодида Р, 1 r йода Р и доводят кислотой серной
разведенной Р до объема 100 мл.
Калия йодовисмутата раствор Р4.
1070605.
1 ,7 r висмута нитрата основН020 Р раст
воряют в 20 мл кислоты уксусной ледяной Р,
прибавляют 80 мл воды дистиллированной Р,
4.1.1. Реактивы
643
100 мл раствора 400 r/л калия йодида Р, 200 мл
кислоты уксусной ледяной Р и доводят водой
дистиллированной Р до объема 1000 мл.
2 объема полученноro раствора смешивают с
1 объемом раствора бария хлорида Р.
Калия йодовисмутата раствор Р5.
1070606.
К 0,85 r висмута нитрата ОСН08Н020 Р при
бавляют 1 О мл кислоты уксусной ледяной Р, слабо
наrревают до полною растворения, прибавля
ют 40 мл воды Р и охлаж.цают. К 5 мл полученноro
раствора прибавляют 5 мл раствора 400 r/л калия
йодида Р, 20 мл кислоты уксусной ледяной Р и
70 мл воды Р.
Калия йодовисмутата раствор разведен-
ный. 1070603.
100 r кислоты винной Р растворяют в 500 мл
воды Р и прибавляют 50 мл раствора калия йо
довисмутата Р1.
Хранят в защищенном от света месте.
Калия карбонат. К 2 СО з . (М М. 138,2).
1068900. [584 08 7]. Дикалия карбонат.
Белый или почти белый rранулированный
порошок. rиrроскопичен. Очень леrко растворим
в воде, практически нерастворим в этаноле.
Хранят в воздухонепроницаемом контейнере.
#Калия метабисульфит. K 2 SP5' (М М. 222,3).
[16731 55 8]. Калия дисульфит.
Белый или почти белый порошок либо бес
цветные кристаллы. Леrкорастворим в воде, Ma
лорастворим в 96 % спирте.
Калия-натрия тартрат. C 4 H 4 KNa0 6 '4H 2 0
(М м. 282,2). 1083500. [6381 59 5]. #Калий на
трий виннокислый. Ce;..JbeToBa соль.#
Бесцветные призматические кристаллы.
Очень леrко растворим в воде.
Калия нитрат. КNО з . (М м. 101,1). 1070700.
[7757 79 1].
Бесцветные кристаллы. Очень леrко paCTBO
рим в воде.
Калия перйодат. КI0 4 . (М М. 230,0).
1070800. [7790 21 8].
Белый или почти белый кристаллический поро
шок или бесцветные кристаллы. Растворим в воде.
Калия nepMaHraHaT. 1070900. [7722 64 7].
См. статью Калия пермаН2анат.
Калия nepMaHraHaTa раствор. 1070902.
Раствор 30 r/л калия пермаН2аната Р.
Калия перманrаната раствор в кислоте
фосфорной. 1070901.
3 r калия пермаН2аната Р растворяют в
смеси из 15 мл кислоты фосфорной Р и 70 мл
воды Р и доводят водой Р до объема 100 мл.
Калия перренат. KRe0 4 . (М м. 289,3).
1071000. 110466 65 6].
Белый или почти белый кристаллический
порошок. Растворим в воде, малорастворим в
96 % спирте, в метаноле и в пропиленrликоле.
Калия персульфат. SPB' (М М. 270,3).
1071100. [7727 21 1]. Дикалия пероксидисульфат.
Бесцветные кристаллы или белый или почти
белый кристаллический порошок. Умеренно pac
творим в воде, практически нерастворим в 96 %
спирте. Водные растворы разлаrаются при KOM
натной температуре, быстрее при наrревании.
Калия пироантимонат. KSb(OH)6' (М М. 262,9).
1071300. [12208 13 8]. Калия rексаrидроксоанти
монат.
Белые или почти белые кристаллы или кри
сталлический порошок. Умеренно растворим в
воде.
Калия пироантимоната раствор. 1071301.
2 r калия пироантимоната Р растворяют в
95 мл roрячей воды Р, быстро охлаждают, при
бавляют раствор, содержащий 2,5 r калия 2и
дроксида Р в 50 мл воды Р. и 1 мл раствора
натрия 2идроксида разведеНН020 Р. Выдержива
ют в течение 24 ч, фильтруют и доводят водой Р
до объема 150 мл.
Калия плюмбита раствор. 1071200.
1,7 r свинца (11) ацетата Р, 3,4 r калия ци
трата Р и 50 r калия 2идроксида Р растворяют
в воде Р и доводят до объема 100 мл раствора
этим же растворителем.
Калия сульфат. K 2 S0 4 . (М М. 174,3).
1033100. [7778 80 5]. Дикалия сульфат.
Бесцветные кристаллы. Растворим в воде.
Калия тартрат. C4H4KP6' Hp. (М м. 235.3).
1071400. [921 53 9]. Дикалия (2R,ЗR) 2,З
диrидроксибутан 1 ,4 дикарбоксилат rемиrидрат.
#Калий виннокислый. Калий щавелевокислый.=
Белый или почти белый rранулированный
порошок или кристаллы. Очень леrко растворим
в воде, очень мало растворим в 96 % спирте.
Калия тетрайодомеркурата раствор.
1071500.
1 ,35 r ртути (11) хлорида Р растворяют в
50 мл воды Р, прибавляют 5 r калия йодида Р и
доводят водой Р до объема 100 мл.
Калия тетрайодомеркурата щелочной
p CTBop.1071600. Реактив Несслера.
11 r калия йодида Р и 15 r ртути (11) йодида Р
растворяют в воде Р и доводят до объема 100 мл
этим же растворителем. Непосредственно перед
использованием полученный раствор смешива
ют с раствором 250 r/л натрия 2идроксида Р
(1:1, об/об).
644
rосударственная фармакопея Республики Беларусь
#К 1 О мл раствора 1000 r/л калия йодида Р пcr
степенно прибавляют при постоянном перемешива
нии насыщенный раствор ртути (11) хлорида Р до
появления неисчезающеro красною осадка. При
бавляют 30 r калия 2идроксида Р и после еro paCTBO
рения еще 1 мл насыщенноro раствора ртути (11)
хлорида Р. Доводят водой Р до объема 200 мл и от
стаивают. Используют прозрачную жидкость.#
Калия тетраоксалат. С 4 Н з КО в '2Н 2 О.
(М М. 254,2). 1071700. [6100 20 5].
Белый или почти белый кристаллический по
рошок. Умеренно растворим в воде, растворим в
кипящей воде, малорастворим в 96 % спирте.
Калия тиоцианат. KSCN. (М м. 97,2).
1071800. [333 20 0].
Бесцветные кристаллы. Расплывается на
воздухе. Очень леrко растворим в воде и в 96 %
спирте.
Хранят в воздухонепроницаемом контейнере.
Калия тиоцианата раствор. 1071801.
Раствор 97 r/л калия тиоцианата Р.
Калия ферриперйодата раствор. 1070801.
1 r калия перйодата Р растворяют в 5 мл
свежеприrотовленноrо раствора 120 r/л калия
2идроксида Р. прибавляют 20 мл воды Р, 1,5 мл
раствора железа (111) хлорида Р1 и доводят CBe
жеприroтовленным раствором 120 r/л калия 2и
дроксида Р до объема 50 мл.
Калия феррицианид. К з [Fе(СN)6]'
(М м. 329.3). 1069700. [13746 66 2]. Калия
rексацианоферрат(III). #Калий железосинероди
стый. Красная кровяная соль.#
Красные кристаллы. Леrкорастворим в воде.
Калия феррицианида раствор. 1069701.
Промывают 5 r калия феррицианида Р He
большим количеством воды Р, растворяют в
воде Р и доводят до объема 100 мл этим же
растворителем.
rотовят непосредственно перед использо
ванием.
Калия ферроцианид. K.[Fe(CN)6].3Hp.
(М м. 422,4). 1069800. [14459 95 1]. Калия reK
сацианоферрат (11). #Калий железистосинероди
стый.Желтая кровяная соль.#
Прозрачные желтые кристаллы. Леrкорас
творим в воде, практически нерастворим в 96 %
спирте.
Калия ферроцианида раствор. 1069801.
Раствор 53 r/л калия ферроцианида Р.
Калия хлорат. КСlO з . (М м. 122,6).1069000.
[3811 04 9].
Белый или почти белый порошок, rранулы
или кристаллы. Растворим в воде.
Калия хлорид. 1069100. [7447 40 7].
См. статью Калия хлорид.
Калия хлорид, используемый для абсорбци
онной спектрофотометрии в инфракрасной
области (2.2.24), должен выдерживать следу
ющее дополнительное требование.
ИК спектр диска толщиной 2 мм, получен
ный из калия хлорида предварительно BЫCY
шенною при температуре 250 ос в течение 1 ч,
должен иметь практически ровную базовую
линию в интервале от 4 000 см., до 620 см.,. Не
должен иметь максимумов с поrлощением более
0,02 над базовой линией, за исключением MaK
симумов для воды при 3 440 см., и 1 630 см.,.
Калия хлорида 0,1 М раствор. 1069101.
Раствор калия хлорида Р содержит 7,46 r
KCI в 1000,0 мл.
Калия хромат. K 2 Cr0 4 . (М М. 194,2).
1069200. [7789 00 6]. Дикалия хромат.
Желтые кристаллы. Леrкорастворим воде.
Калия хромата раствор. 1069201.
Раствор 50 r/л калия хромата Р.
Калия цианид. KCN. (М м. 65,1). 1069400.
[151 50 8].
Белый или почти белый кристаллический
порошок или белая или почти белая масса или
rранулы. Леrкорастворим в воде, малораство
рим в 96 % спирте.
Калия цианида раствор. 1069401.
Раствор 100 r/л калия цианида Р.
Калия цитрат. С6Н5I\зО7'НР' (М м. 324,4).
1069300. [61 00 05 6]. Трикалия2 rидроксипропан
1 ,2,3 трикарбоксилат.
Содержит не менее 99,0 % и не более 101,0 %
C6H5 07 В пересчете на безводное вещество.
Белый или почти белый rранулированный
порошок или прозрачные кристаллы. rиrроско
пичен. Очень леrко растворим в воде, практиче
ски нерастворим в 96 % спирте.
Вода (2.5.12). От 4,0 % до 7,0 %. Определе
ние проводят из 0,250 r испытуемоro образца.
В качестве растворителя используют смесь из
1 объема формамида Р и 2 объемов MeтaHO
па Р. После добавления испытуемою образца
перед титрованием смесь перемешивают в Te
чение 15 мин.
Хранят в воздухонепроницаемом контейнере.
Кальконкарбоновая кислота.С2'Н'4NР7S'3 О,
(М м. 492,5). 1015300. [3737 95 9]. 2 rидрокси 1
(2 rидроксиА сульфо 1 нафтилазо )нафталин
3 карбоновая кислота.
Коричневато черный порошок. Малораство
рима в воде, очень мало растворима в ацетоне
и в 96 % спирте, умеренно растворима в разве
денных растворах натрия rидроксида.
4.1.1. Реактивы
645
Кальконкарбоновой кислоты индикатор
ная смесь. 1015301.
Смешивают 1 часть кальконкарбоновой Kиc
лоты Р с 99 частями натрия хлорида Р.
Испытание на чувствительность. 50 Mr
индикаторной смеси кальконкарбоновой кис
лоты растворяют в смеси из 2 мл раствора
натрия 2идроксида концентрироваНН020 Р
и 100 мл воды Р. Появляется roлубое OKpa
шивание, которое переходит в фиолетовое
при прибавлении 1 мл раствора 1 О r/л ма2ния
сульфата Р и 0,1 мл раствора 1,5 r/л кальция
хлорида Р. При прибавлении 0,15 мл 0,01 М
раствора натрия эдетата вновь появляется
roлубое окрашивание.
Кальция rидроксид. Са(ОН)2' (М м. 74,1).
1015000. [1 305 62 0]. Кальция диrидроксид.
Белый или почти белый порошок. Почти пол
ностью растворим в 600 частях воды.
Кальция rидроксида раствор. 1015001.
Свежеприrотовленный насыщенный раст
вор.
Кальция карбонат. 1014500. [471 34 1].
См. статью Кальция карбонат.
Кальция карбонат Р1. 1014501.
Должен выдерживать требования для каль
ция карбоната Р и следующее дополнительное
испытание.
Хлориды (2.4.4): не более 50 ррm.
Кальция лактат. 1015100. [41372 22 9].
См. статью Кальция лактат пента2идрат.
# Кальция оксид. СаО. (М м. 56,08).
[1305 78 8]. Известь жженая.
Белые или почти белые куски или порошок,
слипающийся в комки. На воздухе поrлощает
воду и уrлерода диоксид.
Кальция сульфат. CaS0 4 '%Hp. (М м. 145,1).
1015200. [10034 76 1]. Кальция сульфат rемиrи
драт.
Белый или почти белый порошок. PaCTBO
рим примерно в 1500 частях воды, практически
нерастворим в 96 % спирте. При смешивании
с водой, масса которой равна половине массы
кальция сульфата, порошок быстро затвердева
ет, превращаясь в твердую пористую массу.
Кальция сульфата раствор. 1015201.
5 r кальция сульqbаrпа Р встряхивают со
100 мл воды Р в течение 1 ч и фильтруют.
Кальция хлорид. 1014600. [10035 04 8].
См. статью Кальция хлорид ди2идрат.
Кальция хлорида раствор. 1014601.
Раствор 73,5 r/л кальция хлорида Р.
82 За.. 1712
Кальция хлорида 0,01 М раствор. 1014602.
0,147 r кальция хлорида Р растворяют в
воде Р и доводят до объема 100,0 мл этим же
растворителем.
Кальция хлорида 0,02 М раствор. 1014603.
2,94 r кальция хлорида Р растворяют в 900 мл
воды Р, устанавливают рН раствора в пределах от
6,0 до 6,2 и доводят водой Р до объема 1000,0 мл.
Хранят при температуре от 2 ос до 8 ОС.
Кальция хлорид безводный. CaCI 2 .
(М м. 111,0). 1014800. [10043 52A].
Содержит не менее 98,0 % CaCI 2 в пересче
те на сухое вещество.
Белые или почти белые rранулы. Расплыва
ется на воздухе. Очень леrко растворим в воде,
леrкорастворим в 96 % спирте и в метаноле.
Потеря в массе при высушивании (2.2.32).
Не более 5,0 %. Испытуемый образец сушат при
температуре 200 ОС.
Хранят в воздухонепроницаемом контейне
ре в защищенном от влаrи месте.
Кальция хлорид Р1. CaCI 2 '4Нр. (М м. 183,1).
1014700. Кальция хлорид тетраrидрат.
Железо: не более 0,05 ррт.
# Кальция хлорид Р6. CaCI 2 '6Нр. (М м. 219,1).
[7774 34 7].
См. статью Кальция хлорид 2екса2идрат.
Камедь бобов рожковоrодерева. 1104500.
Измельченный эндосперм фруктовых косточек
Сеrаtопiа si/iqua L. Taub.
Белый или почти белый порошок, содержа
щий от 70 % до 80 % растворимой в воде смолы,
состоящей в основном из rалактоманноrликона.
Камфен. С 10 Н 16 . (М м. 136,2). 1139200. [7 92 5].
2,2 Диметил 3 метиленбицикло[2.2.1 ]rептан.
Камсрен, используемый в 2азовой xpoMalТКr
2рафии, должен выдерживать следующее до-
полнительное испытание.
Количественное определение. rазовая
матоrрафия (2.2.28), как указано в статье Розм;r
риновое масло.
Содержание камфена, рассчитанное мето-
дом нормализации, должно быть не менее 90 %.
Камфора. 1113000. [76 22 2].
См. статью Камфора рацемuческая.
Камфора, используемая в zaзoвoй xpOMa
т02рафии, должна выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
лавандовое.
Испытуемый раствор. 1 О r/л раствор испы
TyeMoro образца в 2ексане Р.
Содержание камофры, рассчитанное Meтo
дом нормализации, должно быть не менее 95,0 %.
646
rосударственная фармакопея Республики Беларусь
(1 SН+)10 Камфорсульфоновая кислота.
C IO H 16 0 4 S. (М М. 232,3). 1104100. [3144 16 9].
(1 S,4RH + ) 2 OKCO 1 О борненсульфоновая кис
лота. [( 1 S) 7, 7 Диметил 2 оксобицикло[2,2,1]
rептан 1 ил]метансульфоновая кислота. Кисло
та Рейхлера.
Содержит не менее 99,0 % (1SH+)10
камфоросульфоновой кислоты.
Кристаллы в виде призм. rиrроскопична.
Растворима в воде.
Температура плавления: около 194 ос с раз
ложением.
[a] O: +20:1:1. Определение проводят, ис
пользуя раствор 43 r/л испытуемоro образца в
воде Р.
ДА (2.2.41): 10,2'103. Определение прово
дят при длине волны 290,5 нм, используя paCT
вор 1,0 r/л испытуемою образца.
Каолин леrкий. 1047400. [1З32 58 7].
Очищенный природный алюмосиликат rи
дратированный. Содержит подходящий диспер
{"атор.
Леrкий белый или почти белый порошок, не
содержащий твердых спекшихся частиц, масля
нистый на ощупь. Практически нерастворим в
воде и в минеральных кислотах.
Крупные частицы. Не более 0,5 %. 5,0 r Ka
олина помещают в цилиндр со шлифом длиной
около 160 мм и диаметром 35 мм, прибавляют
60 мл раствора 10 r/л натрия пирофосфата Р,
интенсивно встряхивают и отстаивают в течение -
5 мин. С помощью пипетки отбирают 50 мл жид
кости на уровне около 5 см ниже поверхности и
отбрасывают. К оставшейся жидкости прибав
ляют 50 мл воды Р. встряхивают, выдерживают
в течение 5 мин и удаляют 50 мл, как описано
выше. Эту операцию повторяют до тех пор. пока
не будет удалено в общей сложности 400 мл. Пе
реносят оставшуюся суспензию в чашку дЛЯ BЫ
паривания, выпаривают на водяной бане досуха
и сушат до постоянной массы при температуре
от 100 ос до 105 ОС. Масса остатка не должна
превышать 25 Mr.
Мелкие частицы. 5,0 r каолина дисперrиру
ют в 250 мл воды Р при интенсивном встряхи
вании в течение 2 мин и сразу выливают в CTe
клянный цилиндр диаметром 50 мм. С помощью
пипетки отбирают 20 мл, помещают в фарфоро
вую чашку, выпаривают на водяной бане досуха
и сушат до постоянной массы при температуре
от 100 ос до 105 ОС. Остаток суспензии отстаи
вают при температуре 20 ос в течение 4 ч и с по
мощью пипетки удаляют 20 мл на уровне точно
5 см ниже поверхности, не взмучивая осадок.
Остаток помещают в фарфоровую чашку, выпа
ривают досуха и сушат до постоянной массы при
температуре от 100 ос до 105 ОС. Масса BTOpO
ro ОСТalка Аолжна быть не менее 70 % от массы
первоro остатка.
Каприловый спирт. 1024700. См. Деканол Р.
Kap -eH. С 10 Н 1б . (М. М. 1З6,2). 1124000.
[498 15 7]. 3,7,7 Триметилбицикло[4.1.0]rепт 3
ен. 4,7, 7 Триметил 3 норкарен.
Жидкость с характерным запахом, мало
растворим в воде, растворим в орrанических
растворителях.
d 20
20: около 0,864.
20
по: от 1,473 до 1,474.
[a] O: от +15 до +17.
Температура кипения: от 170 ос до 172 ОС.
Kap 3 eH, используемый в 2азовой xpOMaтo
2рафии, должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
мускатН020 ореха.
Содержание Kap 3 eHa, рассчитанное Me
тодом нормализации, должно быть не менее
95,0%.
Карбазол. C 12 H g N. (М. м. 167,2). 1015400.
[86 7 4 8]. Дибензопиррол.
Кристаллы. Практически нерастворим в
воде, леrкорастворим в ацетоне, малораство
рим в этаноле.
Температура плавления: около 245 ОС.
# Карбамид. См. Мочевина Р.
Карбомер. 1015500. [9007 20 9].
Поперечносшитый полимер акриловой
кислоты, после высушивания при темпера
туре 80 ос в течение 1 ч содержит большое
количество карбоксильных rрупп (С0 2 Н, от
56 % до 68 %). Средняя молекулярная масса
около 3.106.
рН (2.2.3). Около 3. Измеряют рН суспензии
1 О r/л испытуемоro образца.
Карбофенотион. С11Н16СЮРSЗ' (М м. 342,9).
1016200. [786 19 6]. 0,0 Диэтил S [[(4 хлорфе
нил)тио]метил] фосфордитиоат.
Жидкость желтоватоro цвета. Практически
нерастворим в воде, смешивается с орrаниче
скими gастворителями.
d 4 5 : около 1,27.
В статье Ланолин, при определении пести
цидов, может быть использован подходящий
сертифицированный раствор сравнения (раст
вор 1 О нr/мкл в изооктане ).
Карвакрол. С 10 Н 1 р. (М М. 150,2). 10164001
[499 75 2]. 5 Изопропил 2 метилфенол.
Коричневатая жидкость. Практически He
растворим в воде, очень леrко растворим в 96 %
спирте.
d 20
20: около 0,975.
20
по: около 1,523.
Температура кипения: около 237 ОС.
4.1.1. Реактивы
647
Карвакрол, используемый в 2азовой xpOMa
т02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
мяты перечной.
Испытуемый раствор. 0,1 r испытуемоro
образца растворяют в 1 О мл ацетона Р.
Содержание карвакрола, рассчитанное Me
тодом нормализации, должно быть не менее
95,0%.
KapBOH.C,oH,p.(MM.150,2).1016500.[2244
16 8). (S) п Мента 6,8 диен 2 он. (+) 2 Метил
5 (1 метилэтенил )циклоrекс 2 енон.
Жидкость. Практически нерастворим в воде,
смешивается с 96 % спиртом.
d;g: около 0,965.
20
по : около 1,500.
[a] O: около +61.
Температура кипения: около 230 ОС.
Карвон, используемый в 2азовой xpOMaтo
2рафии должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано, в статье Масло
мяты перечной, используя испытуемый образец
в качестве испытуемоro раствора.
Содержание карвона, рассчитанное методом
нормализации, должно быть не менее 98,0 %.
I3 Кариофиллен. С 15 Н 24 . (М. м. 204,4).
1101000. [87 44 5). (ЕН1 R,9S) 4,11, 11 триметил
8 метиленбицикло[7 .2.0]YHдeц4 eH.
Маслянистая жидкость, практически HepaCT
ворим в воде, смешивается с 96 % спиртом.
d 17
4 : около 0,905.
20
по : около 1,492.
[a] : около 5,2.
Температура кипеНИЯ'4: от 129 ос до 130 ос.
rз Кариофиллен, используемый в 2азовой
хромат02рафии, должен выдерживать следу
ющее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье lвоз
дичное масло, с использованием испытуемо
ro образца в качестве испытуемоro раствора.
Площадь основноro пика должна составлять не
менее 98,5 % от суммы площадей всех пиков.
# JН{зртин. С40Н56' (М М. 536,9). [72354 7J.
(E) 3, 7, 12, 16 Тетраметил 1, 18 бис(2,6,6 три
метилциклоrекс 1 енил)октадека 1 ,3,5,7,9,11,
13,15, 17 HOHaeH. Провитамин д-
От темно красноro до коричневоro цвета
кристаллический порошок.
Практически нерастворим в воде, paCTBO
рим в хлороформе.
Температура плавления: от 178 ос до 184 ОС.
Катехин. С'5Н'406'ХНР' (М м. 290,3, без
водный). 1119000. [154 234). (+H2R,3S) 2 (3,4
Д и rидроксифен ил ) 3 ,4 ди rидро 2 H хром eH
3,5,7 триол. Катехол. Цианиданол. Цианидол.
Катионит слабоосновный.
См. Катионит слабый Р.
1096000.
Катионит слабый. 1096000.
Смола полиметакриловая, слабокислая, co
держащая карбоксильные rруппы в протониро
ванной форме.
Размер частиц: от 75 мкм до 160 мкм.
Используемые пределы рН: от 5 до 14.
Максимальная температура использова
ния 120 ОС.
Катионообменная смола. 1016700.
Смола в протонированной форме с rруппа
ми сульфоновой кислоты, присоединенными к
решетке полимера, состоящеro из полистирола
поперечносшитоro 8 % дивинилбензола. Выпу
скают в виде rранул, размер которых указывают
после названия реактива в испытаниях, в KOTO
рых он используется.
Катионообменная смола Р1. 1121900.
Смола в протонированной форме с rруппа
ми сульфоновой кислоты, присоединенными к
решетке полимера, состоящеro из полистирола
поперечносшитоro 4 % дивинилбензола. Выпу
скают в виде rранул, размер которых указывают
после названия реактива в испытаниях, в KOTO
рых он используется.
Катионообменная смола сильная.
1156800.
Сильная катионообменная смола в про
тонированной форме с rруппами сульфоновой
кислоты, присоединенными к решетке поли
мера, состоящеro из полистирола поперечнос
шитоrо дивинилбензолом. Выпускают в виде
rранул, размер которых указывают после Ha
звания реактива в испытаниях, в которых он ис
пользуется.
Катионообменная смола сильная (каль
циевая форма). 1104600.
Смола в кальциевой форме с rруппами суль
фоновой кислоты, присоединенными к решетке
полимера, состоящеro из полистирола попереч
но сшитоro 8 % дивинилбензола. Размер частиц
указывают после названия реактива в частных
статьях.
Католит для изоэлектрофокусировки pJ.f
от 3 до 5 (0,1 М раствор l3 ланина). 1113100.
8,9 r fЗ аланина Р растворяют в воде Р и дo
водят до объема 1000 мл этим же растворите
лем.
# Квасцы. См. Алюминия калия сульфат Р.
648
rосударственная фармакопея Республики Беларусь
Кверцетина диrидрат. С15Н1007'2Н20,
(М. м. З38,2). 1138100. 2 (З,4 Диrидроксифенил)
З,5, 7 триrидроксиАН 1 бензопиран 4 он.
Желтые кристаллы или желтоватый поро
шок. Практически нерастворим в воде, paCTBO
рим в ацетоне и метаноле.
Вода (2.5.12): не более 12,0 %. Определе
ние проводят из 0,100 r испытуемою образца.
Количественное определение. Жидкост
ная хроматоrрафия (2.2.29), как указано в статье
Листья 2иНК20.
Содержание безводноro кверцетина, pac
считанное методом нормализации. должно быть
не менее 90 %.
Кизелыур G. 1047600.
Состоит из кизельryра, обработанноro кис
лотой хлористоводородной И кальцинированно
ю прибавлением около 15 % кальция сульфата
rемиrидрата.
Мелкий серовато белый порошок; при pac
тирании с водой серый цвет становится более
выраженным. Средний размер частиц от 1 О мкм
ДО 40 мкм.
Кальция сульфат. Определение проводят
методом, указанным для силика2еля G Р.
рН (2.2.3). От 7 до 8. Измеряют рН суспен
зии, полученной встряхиванием 1 r испытуемою
образца с 1 О мл воды, свободной от У2лерода
диоксида. Р в течение 5 мин.
Хроматоерафическая разделяющая спо
собность. Тонкослойная хроматоrрафия
(2.2.27). Пластинки rотовят, используя взвесь
кизельryра G с раствором 2,7 r/л натрия aцe
тата Р. На линию старта хроматоrрафиче
ской пластинки наносят 5 мкл раствора, coдep
жащею по 0,1 r/л лактозы, сахарозы, rлюкозы
и фруктозы в пиридине Р. Хроматоrрафируют
в системе растворителей вода Р 2 пропа
нол Р этилацетат Р (12:23:65, об/об). Время
прохождения фронта растворителей на paccтo
яние 14 см около 40 мин. Пластинку сушат
на воздухе, опрыскивают раствором анисов020
альде2ида Р, расходуя около 10 мл, И HarpeBa
ют при температуре от 100 ос до 105 ос в тече
ние 5 мин. На xpoMaTorpaMMe должны обнару
живаться четыре четких, хорошо разделенных
без «хвостов» пятна.
Кизелыур для хроматоrрафии. 1047500.
БеI1ЫЙ или желтовато белый леrкий поро
шок. Практически нерастворим в воде, разведен
ных кислотах и в орrанических растворителях.
Скорость фильтрации. Используют xpo
матоrрафическую колонку длиноЙ 0,25 м и
ди аметром 1 О мм с пластинкой из пористоrо
стекла (100) и двумя отметками на высоте 0,1 О м и
0,20 м над пластинкой. Колонку заполняют испы
туемым образцом до первой отметки, а до второй
отметки заполняют водой Р. Коrда первые капли
начнут вытекать из колонки, снова заполняют до
второй отметки водой Р и измеряют время BЫ
текания из колонки первых 5 мл воды. Скорость
потока должна быть не менее 1 мл/мин.
Цветность (2.2.2, метод 1). Элюат, полу
ченный при испытании на скорость фильтрации,
должен быть бесцветным.
Кислотность или щелочность. К 1,00 r ис
пытуемоro образца прибавляют 1 О мл воды Р,
интенсивно встряхивают и выдерживают в тече
ние 5 мин. Суспензию фильтруют через фильтр,
предварительно промытый roрячей водой Р
до нейтральной реакции в промывной воде. К
2,0 мл фильтрата прибавляют 0,05 мл раствора
метилов020 краСН020 Р. Раствор должен иметь
желтое окрашивание. К 2,0 мл фильтрата при
бавляют 0,05 мл раствора фенолфталеина Р1.
Должно появиться слабо розовое окрашивание
раствора.
Водорастворимые вещества. 10,0 r испы
туемоro образца помещают в хроматоrрафиче
скую колонку дллиной 0,25 м и диаметром 1 О мм
и элюируют водой Р. Первые 20 мл элюата выпа
ривают досуха, остаток сушат при температуре
от 100 ос до 105 ос. Масса полученною остатка
не должна превышать 1 О Mr.
Железо (2.4.9). Не более 200 ррт. К 0,50 r
испытуемоro образца прибавляют 1 О мл смеси
из равных объемов кислоты хлористоводород
ной Р1 и воды Р, интенсивно встряхивают, BЫ
держивают в течение 5 мин и фильтруют. 1,0 мл
фильтрата должен выдерживать испытание на
железо.
Потеря в массе при прокаливании. Не
более 0,5 %. Во время прокаливания до темпе
ратуры красною каления «600:1:50) ОС) испыту
емый образец не должен иметь коричневую или
черную окраску.
Кислород. 02' (М м. 32,00). 1108800.
Содержит не менее 99,99 % (об/об) 02'
Азот и ареон: не более 100 ррm.
У2лерода диоксид: не более 10 ррm.
У2лерода монооксид: не более 5 ррm.
# Кислотный синий 1. См. СульфаН06ЫЙ
синий Р.
Кислотный синий 83. С45Н44NзNа07S2'
(М м. 826). 1012200. [6104 59 2]. Цветной индекс
NQ 42660. Бриллиантовый синий Р. Кумасси
бриллиантовый синий R 250.
Коричневый порошок. Нерастворим в xo
лодной воде, малорастворим в кипящей воде и
этаноле, растворим в кислоте серной, кислоте
уксусной ледяной и разведенных растворах rи
дроксидов щелочных металлов.
Кислотный синий 90. С47Н48NзNа07S2'
(М м. 854). 1001300. [6104 58 1]. Цветной индекс
NQ 42655. Натрия [4 [[4 [(4 этоксифенил)амино]
фенил ][[4 ( этил )(3 сульфонатобензил )амино]
фенил )метилен )циклоrекса 2,5 диен 1 илиден]
(этил Н3 сульфонато бензил )аммоний.
4.1.1. Реактивы
649
Темно коричневый порошок с фиолетовым
блеском и вкрапленными частицами, имеющими
металлический блеск. Растворим в воде и эта
ноле.
А;::: более 500 при 577 нм в пересчете на
сухое вещество. Определение проводят, исполь
зуя раствор 0,01 r/л испытуемоro образца в бу
ферном растворе рН 7,0.
Потеря в массе при высушивании (2.2.32).
Не более 5,0 %. 0,500 r испытуемоro образца
сушат при температуре 105 ОС.
Кислотный синий 92. С26Н16NзNаз010SЗ'
(М м. 696). 1001400. [З861 73 2]. Цветной
индекс NQ 13390. Кумасси синий. Аназолен
натрия. Тринатрия 8 rидрокси4' (фениламино)
азонафталин 3,5' ,6 трисульфонат.
Темно синие кристаллы. Растворим в воде,
в ацетоне и в моноэтиловом эфире этиленrлико
ля, малорастворим в 96 % спирте,
Кислотноrо синеrо 92 раствор. 1001401.
0,5 r кислотН020 сине20 92 Р растворяют
в смеси из 1 О мл кислоты уксусной ледяной Р,
45 мл 96 % спирта Р и 45 мл воды Р.
# Кислотный хром черный специальный.
См. Протравной черный 11 Р.
Клобетазола пропионат. С25НЗ2СIF05'
(М м. 467,0). 1097700. [25122 46 7]. 21 Хлор 9
фтор 1113, 17 диrидрокси 1613 метилпреrна 1 ,4
диен 3,20 дион 17 пропионат.
Белый или почти белый кристаллический
порошок. Нерастворим в воде, растворим в 96 %
спирте и в ацетоне.
[a] O: около +104 (в диоксане).
Температура плавления: около 196 ОС.
Кобальта нитрат. Со(NОЗ)2'6Н20, (М м. 291 ,О).
1021700. [10026 22 9].
Мелкие rpaHaToBo KpacHbIe кристаллы.
Очень леrко растворим в воде.
Кобальта хлорид. CoCI 2 '6Hp. (М м. 237,9).
1021600. [7791 13 1).
Красный кристаллический порошок или
кристаллы темно красноro цвета. Очень леrко
растворим в воде, растворим в 96 % спирте.
Кодеин. 1021800. [6059 47 8].
См. статью Кодеин.
Кодеина фосфат. 1021900. [52 28 8).
См. статью Кодеина фосфат 2еми2идрат.
# Конвалатоксин. С 29 Н 4 Рю' (М м. 550,6). [5O 7Ы3).
3IЗ,5а, 14 Триrидрокси 19 оксо 5j3,20[22] кар
денолид 3 [6 дезокси а L маннопиранозид]. CTpO
фантидина a L рамнопиранозид.
8З За.. 1712.
KOHro красный. СЗ2Н22N6NаР6S2' (М м. 697).
1022000. [573 58 0). Показатель Шульца NQ 360.
Цветной индекс NQ 22120. Динатрия (бифенил
4,4' диил бис 2,2' азо )бис( 1 аминонафталин
4 сульфонат).
Коричневато красный порошок. Растворим в
воде.
KOHro KpacHoro бумаrа. 1022002.
Полоски фильтровальной бумаrи поrружают
на несколько минут в раствор КОН20 краСН020 Р.
Высушивают.
KOHro KpacHoro раствор. 1022001.
0,1 r КОН20 краСН020 Р растворяют в смеси
из 20 мл 96 % спирта Р и воды Р и доводят
водой Р до объема 100 мл.
Испытание на чувствительность. К
100 мл воды, свободной от У2лерода диOKcи
да, Р прибавляют 0,2 мл раствора конro красноro
и 0,3 мл 0,1 М раствора натрия 2идроксида. По
является синее окрашивание, которое перехо
дит в розовое при прибавлении не более 0,3 мл
0,1 М раствора натрия 2идроксида.
Изменение окраски. От рН 3,0 (синее OKpa
шивание) до рН 5,0 (розовое окрашивание).
Коричный альдеrид. сэнр. (М м. 1З2,1).
1020700. [104 55 2]. 3 Фенилпропеналь.
Маслянистая жидкость от желтоватоro до
зеленовато желтоro цвета. Малорастворим в
воде, очень леrко растворим в 96 % спирте.
d g: от 1,048 до 1,051.
20
ПО : около 1,620.
Хранят в защищенном от света месте.
Кортизона ацетат. С2зНзо06' (М м. 402,5).
1097800. [50 044]. 17 rидрокси 3, 11 ,20 триоксо
преrн4 ен 21 ил ацетат.
Содержит не менее 97,0 % и не более
103,0 % С2зНзо06 в пересчете на сухое вещество.
Белый или почти белый кристаллический поро-
шок. Практически нерастворим в воде, леrк
рим в метиленхлориде, растворим в диоксане.
ренно растворим в ацетоне, малорастворим в 96 %
спирте и в метаноле. Обладает полиморфизмом.
Хранят в защищенном от света месте.
Кофеин. 1014400. [58 08 2].
См. статью Кофеин.
Кофейная кислота. С;Н е 0 4 - (М. .... 180,2).
1014300. [331 39 5). (Е) 3 (З.4 Диrидpoксифенил)
пропионовая кислота.
Белые или почти белые кристаллы или пла
стинки. Леrкорастворима в roрячей воде и в 96 %
спирте, умеренно растворима в холодной воде.
Температура плавления: около 225 ос с раз
ложением.
Свежеприroтовленный раствор с рН 7,6 имеет
два максимума поrлощения (2.2.25): при 293 нм и
при 329 нм.
650
/осударственная фармакопея Республики Беларусь
# Красная кровяная соль. См. Калия фер
рицианид Р.
Крахмал растворимый. 1085100. [9005 84 9].
Белый или почти белый порошок.
Испытания. rотовят раствор 20 r/л испыту
емоro образца в rорячей воде Р. Раствор должен
иметь слабую опалесценцию и оставаться
жидким при охлаждении.
Крахмала раствор. 1085103.
1,0 r крахмала раствориМ020 Р растирают
с 5 мл воды Р, полученную смесь медленно при
постоянном перемешивании вливают в 100 мл
кипящей воды Р, содержащей 1 О Mr ртути (11)
йодида Р.
При использовании реактива каждый раз
проводят испытание на чувствительность.
Испытание на чувствительность. К смеси
из 1 мл раствора крахмала и 20 мл воды Р при
бавляют около 50 Mr калия йодида Р и 0,05 мл
раствора йода Р1. Раствор должен иметь синее
окрашивание.
Крахмала раствор Р1. 1085105.
1 r крахмала раствориМ020 Р смешивают с
небольшим количеством холодной воды Р. По
лученную смесь прибавляют к 200 мл кипящей
воды Р, прибавляют 250 Mr кислоты салицило
вой Р, кипятят в течение 3 мин и немедленно ox
лаждают.
Срок roдности от 2 до 3 недель при xpaHe
нии раствора при температуре от 4 ос до 1 О ОС.
Свежий раствор крахмала roтовят в том
случае, коrда в точке эквивалентности переход
окраски от синей к бесцветной нерезкий.
Испытание на чувствительность. К
2 мл раствора крахмала Р1 прибавляют 20 мл
воды р, около 50 М2 калия йодида Р и 0,05 мл
раствора йода Р1; полученный раствор должен
иметь синее окрашивание.
Крахмала раствор Р2. 1085107.
1,0 r крахмала раствориМО20 Р растирают
с 5 мл воды Р, полученную смесь медленно при
постоянном леремешивании вливают в 100 мл
кипящей воды Р. Используют свежеприroтовлен
ный раствор.
Испытание на чувствительность. К смеси
из 1 мл раствора крахмала Р2 и 20 мл воды Р
прибавляют около 50 Mr калия йодида Р и 0,05 мл
раствора йода Р1. Раствор должен иметь синее
окрашивание.
Крахмала раствор, свободный от йоди
дов.1085104.
rотовят раствор, как указано для раствора
крахмала Р, но без ртути (11) йодида. rотовят He
посредственно перед использованием.
Крахмала раствор с калия йодидом.
1070501. См. Калия йодида и крахмала раствор Р.
Крезол. С 7 Н в О. (м. м. 108,1). 1022700.
[95-48 7]. о-Крезол. 2 Метилфенол. 4 Окситолуол.
Кристаллы или переохлажденная жид
кость, темнеющая на свету и воздухе. Смеши
вается с этанолом, растворим примерно в 50
частях воды и растворах rидроксидов щелоч
ных металлов.
d 20
20: около 1 ,05.
20
по : от 1,540 до 1,550.
Температура кипения: около 190 ОС.
Температура затвердевания (2.2.18). Не
ниже 30,5 ОС.
Остаток после выпаривания. Не более
0,1 % (м/м). Выпаривают на водяной бане и
сушат при температуре от 100 ос до 105 ОС.
Хранят в защищенном от кислорода, света
и 'влаrи месте.
Перед использованием переrоняют.
п Крезол. С 7 Н в О. (м. м. 108,1). 1153100.
[106-44 5]. 4 Метилфенол.
Бесцветный, или белые кристаллы, или кри
сталлическая масса.
d;g: около 1,02.
Температура кипения: около 202 ОС.
Крезоловый красный. C21H1P5S' (м. м. 382,4).
1 022800. [17ЗЗ 12 6]. Крезолсульфофтале
ин. 4,4' (3H 2, 1 Бензоксатиол 3 илиден)бис (2
метилфенол) S,S диоксид.
Красновато коричневый кристаллический
порошок. Малорастворим в воде, растворим
в 96 % спирте и в разведенных растворах rи
дроксидов щелочных металлов.
Крезоловоrо KpaCHoro раствор. 1022801.
0,1 r крезолов020 краСН020 Р растворяют
в смеси из 2,65 мл 0,1 М раствора натрия
2идроксида и 20 мл 96 % спирта Р, доводят
объем раствора водой Р до 100 мл.
Испытание на чувствительность. К
100 мл воды, свободной от уzлерода диOK
сида, Р прибавляют 0,1 мл раствора крезо
ловоrо красноro и 0,15 мл 0,02 М раствора
натрия 2идроксида; появляется лурпурно
красное окрашивание, которое должно пе
рейти в желтое при прибавлении не более
0,15 мл 0,02 М раствора кислоты хлористо
водородной.
Изменение окраски. От рН 7,0 (желтое
окрашивание) до рН 8,6 (красное окрашива
ние); #от рН 0,2 (красное окрашивание) до рН
1,8 (желтое окрашивание)#.
м-Крезоловый пурпуровый. C21H1B05S'
(м. м. 382,44). 1121700. [2303 01 7].м Крезолсуль
фофталеин.
Оливково зеленый кристаллический поро
шок. Малорастворим в воде, растворим в 96 %
спирте, в кислоте уксусной ледяной и в MeTa
ноле.
4.1.1. Реактивы
651
м Крезоловоrо nypnypoBoro раствор.
1121701.
0,1 r м крезолов020 пурпуров020 Р paCTBO
ряют в 13 мл 0,01 М раствора натрия 2идpOK
сида, доводят водой Р до объема 100 мл и пере
мешивают.
Изменение окраски. От рН 1,2 (красное
окрашиване) до рН 2,8 (желтое окрашивание);
от рН 7,4 (желтое окрашивание) до рН 9,0 (Kpac
но фиолетовое окрашивание).
Кремне вольфрамовая кислота.
H4SiW1P4o'Hp. 1078000. [11130 204].
Белые или желтовато белые кристаллы.
Расплывается на воздухе. Очень леrко paCTBO
рима в воде и в 96 % спирте.
Хранят в воздухонепроницаемом контейнере.
Кристаллический фиолетовый. С25НзоСINз.
(М м. 408,0). 1022900. [548 62 9]. Показатель
Шульца NQ 78. Цветной индекс NQ 42555. reKca
метил парарозанилиния хлорид.
Темно зеленый порошок или кристаллы.
Растворим в воде и в 96 % спирте.
Кристаллическоrо фиолетовоrо раствор.
1022901.
0,5 r кристалличеСК020 фиолетов020 Р
растворяют в кислоте уксусной безводной Р
и доводят до объема 100 мл раствора этим же
растворителем.
Испытание на чувствительность.
К 50 мл кислоты уксусной безводной Р при
бавляют 0,1 мл раствора кристаллическоro
фиолетовоro. Появляется синевао фиолето
вое окрашивание, которое переходит в сине
вато зеленое при прибавлении не более 0,1 мл
0,1 М раствора кислоты хлорной.
Ксантrидрол. С1ЗН1002' (М М. 198,2).
1096100. [90 46 0]. 9 Ксантенол. 9 Оксиксан
тен.
Содержит не менее 90,0 % С1ЗН1002'
Белый или бледно желтый порошок или
мелкие иroльчатые кристаллы. Очень мало pac
творим в воде, растворим в 96 % спирте и кисло
те уксусной ледяной.
Доступен также в виде раствора, содержа
щеro от 90 r/л до 110 r/л ксантrидрола в Meтa
ноле р
Температура плавления: около 123 ОС.
Количественное определение. О,ЗОО r
ксантrидрола помещают в колбу вместимостью
250 мл, растворяют в 3 мл метанола Р (или ис
пользуют 3,0 мл rO"!'OBoro раствора). Прибавляют
50 мл кислоты уксусной ледяной Р и по каплям
при встряхивании 25 мл раствора 20 r/л моче
вины Р Выдерживают в течение 12 ч, фильтру
ют через стеклянный фильтр (ПОР 16). Осадок
на фильтре промывают 20 мл 96 % спирта Р,
сушат при температуре от 100 ос до 105 ос и
взвешивают.
1 r осадка соответствует 0,9429 r ксантrи
дрола.
Хранят в защищенном от света месте.
Если используют метанольный раствор, то
хранят внебольших rерметично закрытых aM
пулах и при необходимости перед использова
нием фильтруют.
Ксантrидрол Р1 1096101.
Должен выдерживать требования для
ксантеидрола Р и следующее дополнитель
ное требование.
Содержание: не менее 98,0 % С1ЗН1002'
Ксантrидрола раствор. 1096102.
К 100 мл кислоты уксусной безводной Р
прибавляют 0,1 мл раствора 100 r/л KcaHт2и
дрола Р в метаноле Р, 1 мл кислоты хлори
стоводородной Р и выдерживают в течение
24 ч.
# Ксантrидроловый реактив. 1 О Mr
ксант2идрола Р растворяют в 100 мл кисло
ты уксусной ледяной Р и прибавляют 1 мл
кислоты хлористоводородной Р. Используют
свежеприroтовленный раствор.
Ксиленоловый оранжеВЫЙ.СЗ1Н2вN2Nа401ЗS'
(М м. 761). 1096300. [З618 43 7]. Тетранатрия
3,3' (3H 2, 1 бензоксати ол 3 илиден )бис[(6
rидрокси 5 метил 3, 1 фенилен)метиленими
нобисацетат] S,S диоксид.
Красновато коричневый кристаллический
порошок. Растворим в воде.
Ксиленоловоrо оранжевоrо индикатор
ная смесь. 1096301.
1 часть ксиленолов020 оранжев020 Р расти
рают с 99 частями калия нитрата Р
Испытание на чувствительность.
К 50 мл воды Р прибавляют 1 мл кислоты YK
сусной разведенной Р, 50 Mr индикаторной
смеси ксиленоловоro оранжевоrо и 0,05 мл
раствора свинца (11) нитрата Р Прибавляют
2ексаметилентетрамин Р до тех пор, пока
окраска раствора не изменится от желтой до
фиолетово красной. При прибавлении 0,1 мл
0,1 М раствора натрия эдетата окраска
раствора должна измениться на желтую.
Ксилоза. С 5 Н'Р5' (М М. 150,1). 1096400.
[58 86 6]. D Ксилоза.
Белый или почти белый кристаллический
порошок или бесцветные иrольчатые кристал
лы. Очень леrко растворима в воде, paCTBO
рима в rорячем 96 % спирте.
[a] O: от +18,5 до +19,5.10,0 r испытуемо
ro образца растворяют в 80 мл воды Р, при
бавляют 1 мл раствора аммиака разведенно
20 Р2, доводят водой Р до объема 100,0 мл и
выдерживают в течение 30 мин.
652
rосударственная фармакопея Республики Беларусь
Ксилол. С в Н 10 , (М. м. 106,2). 1096200. [1330
20 7]. Диметилбензол.
Смесь изомеров. Прозрачная бесцвет
ная воспламеняющаяся жидкость. Практиче
ски нерастворим в воде, смешивается с 96 %
спиртом.
d 20
20: около 0,867.
20
по: около 1,497.
Температура кипения: около 138 ОС.
о Ксилол. СвНю' (М м. 106,2). 1100600.
[95 47 6]. 1 ,2 Диметилбензол.
Прозрачная бесцветная воспламеняющая
ся жидкость. Практически нерастворим в воде,
смешивается с 96 % спиртом.
d 20
20: около 0,881.
20
по: около 1,505.
Температура кипения: около 144 ОС.
Температура плавления: около 25 ОС.
м Ксилол. СвН ю ' (М м. 106.2). 1117700.
[1 08 38 3]. 1 ,3 Диметилбензол.
Прозрачная бесцветная воспламеняющая
ся жидкость. Практически нерастворим в воде,
смешивается с 96 % спиртом.
d 20
20: около 0,884.
20
по: около 1,497.
Температура кипения: около 139 ОС.
Температура плавления: около 47 ОС.
Кукурузное масло. 1050400.
См. статью Кукурузное масло рафиниро
ванное.
Кумарин. СэНе02' (М м. 146,1). 1124900.
[91 64 5]. 2H XpOMeH 2--OH. 2H 1 Бензопиран 2 он.
Бесцветный кристаллический порошок
или призматические либо прямоуrольные кри
сталлы.
Очень леrко растворим в кипящей воде. pac
творим в спирте. Растворяется в растворах ще
лочей.
Температура плавления: от 68 ос до 70 ОС.
Кумарин, используемый в 2азовой xpOMaтo
2рафии, должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
сенны.
Содержание кумарина, рассчитанное Me
тодом нормализации, должно быть не менее
98,0%.
о-Кумаровая кислота. СgНРз, (М м. 164,2).
1157400. [614 60 8]. (Е) 2 rидроксикоричная кис
лота. (2E) 3 (2 r идроксифенил )проп 2 еновая
кислота.
Белый или почти белый порошок.
Температура плавления: около 217 ОС.
п-Кумаровая кислота. СgНРз, (М. м. 164,2).
1157500. [7400 08 0]. 4 rидроксикоричная кис
лота. 3 (4 rидроксифенил)проп 2 еновая кис
лота.
Белые или почти белые иroльчатые кристал
лы, практически нерастворима в воде, раствори
ма в ацетоне и метаноле.
Температура плавления: от 214 ОС до 217 ос.
п Кумаровая кислота, используемая при KO
личественном определении в статье Листья
крапивы, должна выдерживать следующие дo
полнительные испытания.
Потеря в массе при высушивании (2.2.32):
не более 5,0 %. 0,200 r испытуемоro образца
сушат при температуре от 100 ос до 105 ос в Te
чение 2 ч.
Количественное определение. Жидкост
ная хроматоrрафия (2.2.29), как указано в статье
Листья крапивы.
Содержание п кумаровой кислоты, рассчи
танное методом нормализации, должно быть не
менее 95 %.
Кумасси красящий раствор. 1012201.
Раствор 1,25 r/л кислотНО20 сине20 83 Р в
смеси растворителей кислота уксусная ледя
ная Р метанол Р вода Р (1 :4:5, об/об/об).
Фильтруют.
Кумасси красящий раствор Р1. 1173000.
0,275 r бриллиантов020 сине20 Р растворяют
в 200 мл метанола Р и перемешивают до полно
ro растворения (около 2 ч). Прибавляют 750 мл
воды Р, 50 мл кислоты уксусной ледяной Р, пе
ремешивают в течение 16 ч и фильтруют.
Кумасси синий. 1001400. [3861 73 2]. См.
Кислотный синий 92 Р.
Кумасси синеrо раствор. 1001401. См.
КислотН020 сине20 92 раствор Р.
Куркумин. С21Н2006' (М м. 368,4). 1023500.
[458 3 7 7]. 1 ,7 Бис( 4 rидрокси 3 метоксифенил)
rепта 1 ,6 диен 3,5 дион.
Оранжево коричневый кристаллический по
рошок. Практически нерастворим в воде, pac
творим в кислоте уксусной ледяной.
Температура плавления: около 183 ОС.
Лавандулилацетат. С12Н2002' (М м. 196,3).
1114200. [50373 59 6]. 2 Изопропенил 5 метил
reKc 4 eH 1 илацетат.
Бесцветная жидкость с характерным запа
хом.
d 20
20: около 0,911.
20
по: около 1,454.
Температура кипения 1з : от 106 ос до 107 ОС.
Лавандулилацетаm, используемый в 2азо
вой хромат02рафии, должен выдерживать сле
дующее дополнительное испытание.
4.1.1. Реактивы
653
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Лаван
довое масло.
Испытуемый раствор. Испытуемый образец.
Содержание лавандулилацетата, рассчи
танное методом нормализации, должно быть не
менее 93,0 %.
Лавандулол. С 10 Н 1в О. (М м. 154,2). 1114100.
[498 16 8]. (R) 5 Метил 2 ( 1 метилэтенил )A reK
ceH 1 of,J.
Маслянистая жидкость с характерным запа
хом.
Лаванду лол, используемый в zазовой xpo
матоzpафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Лаван
довое масло.
Испытуемый раствор. Испытуемый обра
зец.
Содержание лавандулола, рассчитанное
методом нормализации, должно быть не менее
90,0%.
Лакмус. 1049300. [1393 92 6]. Показатель
Шульца NQ 1386.
Индиro синие фраrменты, полученные из
различных видов Rocella, Lесапоrа или друrих
лишайников. Растворим в воде, практически He
растворим в 96 % спирте.
Изменение окраски. От рН 5 (красное OKpa
шивание) до рН 8 (синее окрашивание).
Лакмусовая бумаrа красная. 1049302.
К синему экстракту лакмуса прибавляют по
каплям кислоту хлористоводородную разве
денную Р до изменения синей окраски на Kpac
ную. Полоски фильтровальной бумаrи пропиты
вают полученным раствором и сушат.
Испытание на чувствительность. Поло
ску фильтровальной бумаrи размером 10х60 мм
поrружают в смесь из 10 мл 0,02 М раствора
натрия 2идроксида и 90 мл воды Р. При встря
хивании бумаrа должна приобрести синее OKpa
шивание в течение 45 с.
Лакмусовая бумаrа синяя. 1049301.
1 О частей rрубо измельченноrо лакмуса Р
кипятят со 100 частями 96 % спирта Р в тече
ние 1 ч. Спирт декантируют, к остатку прибав
ляют смесь из 45 частей 96 % спирта Р и 55
частей воды Р Через 2 дня прозрачную жид
кость декантируют. Полоски фильтровальной
бумаrи пропитывают полученным раствором и
сушат
Испытание на чувствительность. Поло
СКУ фильтровальной бумаrи размером 10х60 мм
поrружают в смесь из 10 мл 0,02 М раствора
кислоты хлористоводородной и 90 мл воды Р
При встряхивании бумаrа должна приобрести
красное окрашивание в течение 45 с.
s.4 З.а" 1712
Лактобионовая киcr.юra. С12Н22012' (м. м.358,3).
1101600. [96 82 2].
Белый или почти белый кристаллический
порошок. Леrкорастворима в воде, практически
нерастворима в 96 % спирте.
Температура плавления: около 115 ОС.
Лактоза. 1047900. [5989 81 1].
См. статью Лактоза МОН02идрат.
I3 Лактоза. С 12 Н 2 Р11' (М м. 342,3). 1150100.
[5965 66 2]. I3 D Лактоза.
Белый или слеrка желтоватый порошок.
Содержит не более 35 % о. D лактозы.
Количественное определение. rазовая xpo
матоrрафия (2.2.28): опредеnение ПрОБОДЮ Me
тодом внутренней нормализации.
Образец вводят в хроматоrраф после полу
чения соответствующеro производноro.
Условия хроматоrрафирования:
колонка капиллярная длиной 30 м и BHY
тренним диаметром 0,25 мм, покрытая слоем
поли{(цианопропил) ( фенил)][диметил]силокса
на Р (толщина слоя 1 мкм);
детектор: 1Jламенно ионизационный;
2аз носитель: 2елий для хромат02рафии Р;
температура:
Время Температура (ОС)
(мин)
Колонка 0 З2,5 20 ------+ 280
Блок ввода проб 250
Детектор 250
Содержание l3 лактозы, рассчитанное
методом нормализации, должно быть не
менее 99 %.
о. Лактоза моноrидрат. С12Н22011'Н20,
(М м. 360,3). 1150000. [5989 81 1]. o. D
Лактоза моноrидрат.
Белый порошок.
Содержит не более 3 % I3 D лактозы.
Количественное определение. rазовая
хроматоrрафия (2.2.28): определение прово
дят методом внутренней нормализации.
Образец вводят в хроматоrраф после по
лучения соответствующеrо производноrо.
Условия хроматоrрафирования:
колонка капиллярная длиной 30 м и BHY
тренним диаметром 0,25 мм, покрытая слоем
поли(диметил)силоксана Р (толщина слоя
1 мкм);
детектор: пламенно ионизационный;
2аз носитель: 2елий для xpOMaт02pa
фииР;
температУР8:
Время Температура (ОС)
(мин)
Колонка 0 12,5 230 ------+ 280
Блок ввода проб 250
Детектор 280
654
rосударственная фармакопея Республики Беларусь
Содержание а лактозы, рассчитанное MeTO
дом нормализации, должно быть не менее 97 %.
Ланатозид с. С4ЭН7602' (м. м. 985). 1163300.
[17575 22 3). 3J3 [I3 D rлюкопиранозил ( 1 4 ) 3
ацетил 2,6 дидезокси I3 D рибо rексопиранозил
(1 4 ) 2,6 дидезокси I3 D рибо rексопиранозил
(1 4 ) 2,6 дидезокси I3 D рибо rексопиранозил)
окси] 12JЗ, 14 диrидрокси 513 кард 20(22) енолид.
Продолroватые плоские призмы, получен
ные после перекристаллизации из 96 % спирта.
Леrкорастворим в пиридине идиоксане.
Лантана (111) нитрат. Lа(NОз)з'6НР.
(м. м. 433,0). 1048000. [10277--43 7]. Лантана
тринитрат rексаrидрат.
Бесцветные расплывающиеся на воздухе
кристаллы. Леrкорастворим в воде.
Хранят в воздухонепроницаемом контейнере.
Лантана (111) нитрата раствор. 1048001.
Раствор 50 r/л лантана (1/1) нитрата Р.
Лантана (111) оксид. Lарз. (м. м. 325,8).
1114000. [1312 81 8). Лантана триоксид.
Почти белый аморфный порошок. Практиче
ски нерастворим в воде, растворяется в разве
денных минеральных кислотах, поrлощает уrле
рода диоксид из воздуха.
Кальций. Не более 5 ррт.
Лантана (111) хлорида раствор. 1114001.
К 58,65 r лантана (1/1) оксида Р медленно
прибавляют 100 мл кислоты хлористоводород
ной Р, наrревают до кипения, охлаждают и ДOBO
дят водой Р до объема 1000,0 мл.
Лауриловый спирт. С'2Н2Р' (м. м. 186,3).
1119900. [112 53 8). 1 Додеканол.
d 20
20: около 0,820.
Температура плавления: от 24 ос до 27 ОС.
Лейцин. 1048500. [61 90 5].
См. статью Лейцин.
Лимонен. С1ОН'6' (м. м. 136,2). 1048600.
[5989 27 5). D Лимонен. (+ ) п Мента 1,8 диен.
(R) 4 Изопропенил 1 метилциклоrекс 1 eH.
Бесцветная жидкость. Практически не pac
творим в воде, растворим в 96 % спирте.
d 20
20: около 0,84.
20
ПО : от 1,471 до 1,474.
[a] O: около +124.
Температура кипения: от 175 се до 177 ОС.
Лимонен, используемый в 2азовой xpOMa
т02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Мяты
перечной масло.
Испытуемый раствор. Испытуемый образец.
Содержание лимонена, рассчитанное Me
тодом нормализации, должно быть не менее
99,0%.
Лимонная кислота. 1021000. [5949 29 1).
См. статью Лимонная кислота МОН02идрат.
При использовании в испытании на железо
кислота лимонная должна выдерживать следу
ющее дополнительное испытание.
0,5 r кислоты лимонной растворяют в 10 мл
воды Р, прибавляют 0,1 мл кислоты ти02лико
левой Р, перемешиваю прибавляют раствор
аммиака Р до щелочной реакции и полученный
раствор доводят водой Р до объема 20 мл. Не
должно появляться розовое окрашивание.
Лимонная кислота безводная. 1021200.
[77 92 9).
См. статью Лимонная кислота безводная.
Лимонное масло. 1101700.
См. статью Лимонное масло.
Линалилацетат. С12Н2002' (м. м. 196,3).
1107200. [115 95 7]. (RS) 1,5 Диметил 1 винил
rекс--4 енилацетат.
Бесцветная или слеrка желтая жидкость с
сильным запахом берrамота и лаванды.
d 25
25: от 0,895 до 0,912.
20
ПО : от 1 ,448 до 1 ,451.
Температура кипения: около 215 ОС.
Линалилацетат, используемый в 2азовой
хромат02рафии, должен выдерживать следую
щее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Поме
ранца цветков масло.
Испытуемый раствор. Испытуемый образец.
Содержание линалилацетата, рассчитанное
методом нормализации, должно быть не менее
95,0%.
Линалол. С 10 Н,Р. (м. м. 154,2). 1048700.
[7 8 70 6]. (R S) 3, 7 Диметилокта 1 , 6 диен 3 ол.
Смесь двух стереоизомеров (ликареола и
кориандрола).
Жидкость. Практически нерастворим в воде.
d 20
20: около 0,860.
20
по: около 1,462.
Температура кипения: около 200 ОС.
Линалол, используемый в 2азовой xpOMaтo
2рафии, должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье AHиco
вое масло.
Испытуемый раствор. Испытуемый образец.
Содержание линалола, рассчитанное Meтo
дом нормализации, должно быть не менее 98,0 %.
4.1.1. Реактивы
655
Литий. Li. (А. м. 6,94). 1048800. [7439 93 2].
Мяrкий металл, на свежем срезе cepe
бристо сероro цвета, при контакте с воздухом
быстро становится тусклым. Бурно реаrирует
с водой с образованием водорода и раствора
лития rидроксида; растворим в метаноле с обра
зованием водорода и раствора лития метилата;
практически нерастворим в петролейном эфире.
Хранят под петролейным эфиром или вазе
лином.
Лития rидроксид. LiOH'Hp. (М м. 41,96).
1049100. [1310 66 3]. Лития rидроксид моноrидрат.
Белый или почти белый rранулированный
порошок. Является сильной щелочью, быстро
поrлощает воду и уrлерода диоксид. Растворим
в воде, умеренно растворим в 96 % спирте.
Хранят в воздухонепроницаемом контейнере.
Лития карбонат. Li 2 СО з . (М. М. 73,9).
1048900. [554 13 2]. Дилития карбонат.
Белый или почти белый леrкий порошок.
Умеренно растворим в воде, очень мало pac
творим в 96 % спирте. Насыщенный раствор при
температуре 20 ос содержит около 13 r/л Li 2 СО з .
Лития метаборат безводный. LiB0 2 .
(М м.49,75). 1120000. [13453 69 5).
Лития сульфат. Li 2 S0 4 'Hp. (М м. 128,0).
1049200. [10102 25 7]. Дилития сульфат MOHO
rидрат.
Бесцветные кристаллы. Леrкорастворим в
воде, практически нерастворим в 96 % спирте.
Лития хлорид. LiCI. (М м. 42,39). 1049000.
[7447 41 8].
Кристаллический порошок, rранулы или
кубические расплывающиеся на воздухе кри
сталлы. Леrкорастворим в воде, растворим в
ацетоне и в 96 % спирте. Водные растворы
имеют нейтральную или слабощелочную pe
акцию.
Хранят в воздухонепроницаемом контейнере.
Лоrанин. С17Н26010' (М М. 390,4). 1136700.
[18524 94 2]. Метил(1 S,4aS,6S,7R,7aS) 1 (13
D rлюкопиранозилокси ) 6 rдрокси 7 метил
1 ,4а,5,6, 7, 7а rексаrидроциклопента[ с]пиран
4 карбоксилат.
Температура плавления: от 220 ос до 221 ОС.
# Люrоля раствор.
0,5 r йода Р и 1 r калия йодида Р растворяют
в небольшом количестве воды Р и доводят до
объема 100 мл этим же растворителем. Перед
употреблением раствор разводят водой Р в 5
раз. Хранят в защищенном от света месте.
Люмифлавин. С 1з Н 12 NР2' (М М. 256,3).
1141000. [1088 56 8]. 7,8,10 Триметилбензо[g]
птеридин 2,4(3Н, 1 ОН) дион.
Желтый порошок или оранжевые кристал
лы. Очень мало растворим в воде, леrко paCTBO
рим вметиленхлориде.
Маrний. Mg. (А. м.24,30). 1049500. [7439 95-4].
Серебристо белая лента, стружка или про
волока или серый порошок.
Маrния ацетат. C 4 H 6 Mg0 4 '4Нр (М м. 214,5).
1049600. [16674 78 5]. Маrния диацетат тетраrи
драт.
Бесцветные кристаллы. Расплывается на
воздухе. Леrкорастворим в воде и в 96 % спирте.
Хранят в воздухонепроницаемом контейнере.
Маrния нитрат. Mg(NOA6Hp. (М м. 256,4).
1049800. [1З446 18 9]. Маrния нитрат rексаrи
драт.
Бесцветные прозрачные кристаллы. Pac
плывается на воздухе. Очень леrко растворим в
воде, леrкорастворим в 96 % спирте.
Хранят в воздухонепроницаемом контейнере.
Маrния нитрата раствор. 1049801.
17,3 r ма2ния нитрата Р растворяют при
осторожном наrревании в 5 мл воды Р, при
бавляют 80 мл 96 % спирта Р, охлаждают и
доводят до объема 100,0 мл этим же paCTBO
рителем.
Маrния оксид. 1049900. [1309 48A].
См. статью Ма2ния оксид, ле2кий.
Маrния оксид Р1. 1049901.
Должен выдерживать требования для
ма2ния оксида Р со следующими изменениями.
Мышьяк (2.4.2, метод А). Не более 0,0002 %
(2 ррm). 0,5 r испытуемоro образца растворяют в
смеси из 5 мл воды Р и 5 мл кислоты хлористо
водродной Р1. Полученный раствор должен BЫ
держивать испытание на мышьяк.
Тяжелые металлы (2.4.8, метод А). Не
более 0,001 % (10 ррm). 0,75 r испытуемоro
разца растворяют в смеси из 3 мл воды Р и 7 мл
кислоты хлористоводородной Р1, прибаВl1ЯlOТ
0,05 мл раствора фенолфталеина Р и pacт
вора аммиака концентрироваНН020 Р дО ПОЯ8
ления розовоro окрашивания. Избыток аммиа
ка нейтрализуют кислотой уксусной ледяной Р,
прибавляют 0,5 мл избытка кислоты. доводят
водой Р до объема 15 мл и при необходимости
фильтруют. 12 мл раствора должны выдержи
вать испытание на тяжелые металлы. Эталон ro
товят с использованием 5 мл эталОНН020 pacт
вора свинца (1 ррт РЬ) Р и 5 мл воды Р
Железо (2.4.9). Не более 0,005 % (50 ррm).
0,2 r испытуемою образца растворяют в 6 мл Kиc
лоты хлористоводродной разведенной Р и ДOBO
дят водой Р до объема 10 мл. Полученный paCT
вор должен выдерживать испытание на железо.
# Маrния оксид леrкий. См. Ма2ния оксид Р
656
rосударственная фармакопея Республики Беларусь
Маrния ОКСИД, тяжелый. 1050000. [1309484].
См. статью Ма2ния оксид, тяжелый.
Маrния сульфат. 1050200. [10034 99 8].
См. статью Ма2ния сульфат 2епта2идрат.
# Маrния сульфата насыщенный раствор.
К 100 r ма2ния сульфата Р прибавляют
100 мл воды Р и оставляют на 24 ч при частом
перемешивании. Раствор фильтруют.
Маrния хлорид. 1049700. [7791 18 6].
См. статью Ма2ния хлорид 2екса2идрат.
# Майера реактив.
1,358 r ртути (11) хлорида Р растворяют в
60 мл воды Р, Пр11бавляют 1 О мл раствора 500' r/л
калия йодида Р и доводят водой Р до объема
100,0 мл.
Макроrол 23 лауриловый эфир. 1129000.
Количество этиленоксидных rрупп, связан
ных с лауриловым спиртом, равно 23 (номиналь
ное значение).
Белая или почти белая жирная масса. Pac
творим или дисперrируется в воде, растворим в
96 % спирте, практически нерастворим в петро
лейном эфире.
Хранят в воздухонепроницаемом контейнере.
Макроrол 200. 1099200. [25322 68 3]. Поли
этиленrликоль 200.
Прозрачная бесцветная или почти бесцвет
ная вязкая жидкость. Очень леrко растворим в
ацетоне и в этаноле, практически нерастворим в
жирных маслах.
d 20
20: около 1.127.
20
по: около 1,450.
Макроrол 200 Р1. 1099201.
500 мл маКр020ла 200 Р помещают в круrло
донную колбу вместимостью 1000 мл, отrоняют
летучие вещества при температуре 60 ос в Te
чение 6 ч, используя ротационный испаритель и
вакуум от 1,5 кПа до 2,5 кПа.
Макроrол 300. 1067100. [25322 68 3]. Поли
этиленrликоль 300.
См. статью МаКР020ЛЫ.
Макроrол 400. 1067200. [25322 68 3]. Поли
этиленrликоль 400.
См. статью МаКр020ЛЫ.
Макроrол 1000. 1067300. [25322 68 3]. По
лиэтиленrликоль 1000.
См. статью МаКр020Лbl.
Макроrол 1500.1067400. [25322 68 3]. По
лиэтиленrликоль 1500.
См. статью МаКр020ЛЫ.
Макроrол 20 000. 1067600. Полиэтиленrли
коль 20 000.
См. статью МаКР020ЛЫ.
Макроrол 20 000 2-нитротерефталат.
1067601. Полиэтиленrликоль 20 000 2 нитрот
рефталат.
МаКр020Л 20 000 Р модифицированный об.-
работкой кислотой 2 нитротерефталевой.
Твердая воскообразная масса белоrо или
почти белоro цвета. Растворим в ацетоне.
Малахитовый зеленый. С2ЗН25СIN2' (м. М. 364,9).
1050500. [12ЗЗ33 61 9]. Показатель Шульца
NQ 754. Цветной индекс NQ 42000. [4 [[4 (Димети
ламино )фенил )фенилметилен ] циклоrекса 2,5
диен 1 илиден ]диметиламмония хлорид.
Зеленые с металлическим блеском кристал
лы. Очень леrко растворим в воде с образова
нием раствора синевато зеленоrо цвета, paCTBO
рим в 96 % спирте и в метаноле.
Раствор 0,01 r/л в 96 % спирте Р имеет макси
мум поrлощения (2.2.25) при длине волны 617 нм.
Малахитовоrо зеленоrо раствор. 1050501.
Раствор 5 r/л в кислоте уксусной безводной Р
#Изменение окраски. От рН 0,1 (желтое
окрашивание) до рН 2,0 (зеленовато rолубое
окрашивание); от рН 11,4 (зеленовато roлубое
окрашивание) до рН 13,0 (бесцветный).
Малеиновая кислота. С 4 Н 4 О 4 . (м. м.116,1).
1050600. [110 16 7]. (Z) Бутендионовая кислота.
Содержит не менее 99,0 % и не более 101,0 %
С 4 Н 4 О 4 В пересчете на безводное вещество.
Белый или почти белый кристаллический по
рошок. Леrкорастворима в воде и в 96 % спирте.
Вода (2.5. 12). Не более 2,0 %. Определение про-
водят из 1,00 r испытуемою образцаикрометодом.
Хранят в стеклянном контейнере в защи
щенном от света месте.
Малеиновый анrидрид. С 4 НРз, (м. м. 98,1).
1050700. [108 31 6]. Бутендионовый анrидрид.
2,5 Фурандион.
Белые или почти белые кристаллы. PaCTBO
рим в воде с образованием кислоты малеино
вой, очень леrко растворим в ацетоне и этила
цетате, леrкорастворим в толуоле, растворим в
96 % спирте с образованием сложноrо эфира,
очень мало растворим в петролейном эфире.
Температура плавления: около 52 ОС.
Любой остаток, нерастворимый в толуоле,
не должен превышать 5 % (кислота малеиновая).
Малеиновоrо анrидрида раствор.
1050701.
5 r малеинов020 аН2идрида Р растворяют в
толуоле Р и доводят до объема 100 мл этим же
растворителем.
Срок roдности 1 мес. Раствор фильтруют в
случае помутнения.
4.1.1. Реактивы
657
Мальтит. С,2Н2Р1,' (М. м. 344,3). 1136800.
(585 88 6]. 4 0 a D r люкопиранозил D rлюцитол
(D мальтитол ).
Белый или почти белый кристаллический
порошок. Очень леrко растворим в воде, практи
чески нерастворим в 96 % спирте.
Маннит. 1051000. (69 65 8].
См. статью Маннит.
# Маннитол. См. Маннит Р.
Манноза. С 6 Н'Р6' (М м. 180,2). 1051100.
(3458 28--4]. D (+ ) Манноза.
Белый или почти белый кристаллический
порошок или мелкие кристаллы. Очень леrко
растворима в воде, малорастворима в этаноле.
[a] O: от +13,7 до +14,7. Определение прово
дят, используя раствор 200 r/л испытуемоro об
разца в воде Р, содержащей около 0,05 % NН з .
Температура плавления: около 132 ос с раз
ложением.
Мар.-анца (11) сульфат. MnSO,,'Hp.
(м. м. 169,0). 1050900. [10034 96 5]. Марrанца
сульфат моноrидрат.
Бледно розовый кристаллический порошок
ми кристаллы. Леrкорастворим в воде, практи
чески нерастворим в 96 % спирте.
Потеря в массе при прокаливании. От
10,0 % до 12,0 %. 1,000 r испытуемою образца
прокаливают при температуре 500 ОС.
Масляная кислота. С 4 Н 8 О 2 . (М м. 88,1).
1014000. [107 92 6]. Бутановая кислота.
Содержит не менее 99,0 % С 4 Н в О 2 .
Маслянистая жидкость. Смешивается с
водой и с 96 % спиртом.
d 20
20: около 0,96.
20
по : около 1,398.
Температура кипения: около 163 ОС.
Меди (11) ацетат. C 4 H 6 Cu0 4 .H 2 0. (М м. 199,7).
1022200. (142 71 2].
Кристаллы или порошок roлубовато зелено
ro цвета. Леrкорастворим в кипящей воде, pac
творим в воде и в 96 % спирте, малорастворим в
rлицерине 85 %.
Меди (11) Hmpaт. сu(NОз12'3НР' (М м. 241,6).
1022400. [10031--43 3]. Меди динитрат триrидрат.
Темно синие кристаллы. rиrроскопичен.
Очень леrко растворим в воде, водный paCT
вор имеет сильнокислую реакцию, леrкорас
творим в 96 % спирте и в кислоте азотной раз
веденной.
Хранят в воздухонепроницаемом контей
нере.
# Меди нитрата раствор. Раствор 50 r/л
меди (11) нитрата Р.
Меди (11) сульфат. CuS0 4 '5Hp. (М м. 249.7).
1022500. [7758 99 8].
Синий порошок или кристаллы. Медленно
выветривается на воздухе. Очень леrко paCTBO
рим в воде, малорастворим в 96 % спирте.
Меди (11) сульфата раствор. 1022501.
Раствор 125 r/л меди (11) сульфата Р.
Меди тетрааммиаката аммиачный paCT
вор. 1022600.
34,5 r меди (11) сульфата Р растворяют в
100 мл воды Р, прибавляют при перемешивании по
каплям раствор аммиака концентрированный Р
до растворения образовавшеroся осадка. Подцер
живая температуру ниже 20 ос, при непрерывном
встряхивании прибавляют по каплям 30 мл pacт
вора натрия 2идроксида концентрироваННО20 Р.
Фильтруют через стеклянный фильтр (ПОР 40)
(2. 1.2), промывают водой Р до получения прозрач
Horo фильтрата. Встряхивают с 200 мл раствора
аммиака концентрироваНН020 Р и фильтруют
через стеклянный фильтр (2.1.2), затем повторно
фильтруют, чтобы уменьшить осадок до минимума.
Меди (11) хлорид. CuCI 2 '2Hp. (М м. 170,5).
1023000. [10125 13 0]. Меди хлорид диrидрат.
Зеленовато синий порошок или кристаллы.
Расплывается на воздухе, выветривается в сухом
воздухе. Леrкорастворим в воде, в 96 % спирте и
в метаноле, умеренно растворим в ацетоне.
Хранят в воздухонепроницаемом контейнере.
Меди эдетата раствор. 1022300.
К 2 мл раствора 20 r/л меди (11) ацетата Р
прибавляют 2 мл 0,1 М раствора натрия эдета
та и доводят водой Р до объема 50 мл.
Меди этилендиамина раствор щелочной.
3008700.
Молярное отношение этилендиамина к
меди составляет 2,OO:tO,04.
Раствор является коммерчески доступным.
Медно тартратный раствор. 1023300.
Раствор А. 34,6 r меди (11) сульфата Р раст
воряют в воде Р и доводят до объема 500 мл
этим же растворителем.
Раствор В. 173 r калия натрия тapтpa
та Р и 50 r натрия 2идроксида Р растворяют в
400 мл воды Р, наrревают до кипения, охлажда
ют и доводят водой, свободной от У2лерода ди
оксида, Р до объема 500 мл.
Непосредственно перед использованием
смешивают равные объемы растворов А и В.
Медно тартратный раствор Р2. 1023302.
1 мл раствора, содержащеro 5 r/л меди (11)
сульфата Р и 1 О r/л калия тартрата Р, смеши
вают с 50 мл раствора натрия карбоната Р1.
rотовят непосредственно перед использо
ванием.
658
rосударственная фармакопея Республики Беларусь
Медно-тартратный раствор РЗ. 1023303.
Смешивают равные объемы раствора 1 О r/л
меди (11) сульфата Р и раствора 20 r/л натрия
тартрата Р. К 1,0 мл полученноro раствора при
бавляют 50 мл раствора натрия карбоната Р1.
rотовят непосредственно перед использо
ванием.
Медно тартратный раствор Р4. 1023304.
Раствор А. Раствор 150 r/л меди (11) суль
фата Р.
Раствор В. 2,5 r натрия карбоната безво
дН020 Р, 2,5 r калия натрия тартрата Р, 2,0 r
натрия 2идрокарбоната Р и 20,0 r натрия суль
фата безводН020 Р растворяют в воде Р и ДOBO
дят до объема 100 мл этим же растворителем.
Непосредственно перед использованием
смешивают 1 часть раствора А и 25 частей paCT
вора В.
Медно-цитратный раствор. 1023100.
25 r меди (11) сульфата Р, 50 r кислоты ли
монной Р и 144 r натрия карбоната безводно
20 Р растворяют в воде Р и доводят до объема
1000 мл этим же растворителем.
Медно цитратный раствор Р1. 1023200.
25 r меди (11) сульфата Р, 50 r кислоты ли
монной Р и 144 r натрия карбоната безводно
20 Р растворяют в воде Р и доводят до объема
1000 мл этим же растворителем (испытуемый
раствор ).
Испытуемый раствор корректируют так,
чтобы он выдерживал следующие требования:
а) К 25,0 мл испытуемоro раствора прибавля
ют 3 r калия йодида Р, затем осторожно неболь
шими порциями прибавляют 25 мл 25 % (м/м)
раствора кислоты серной Р и титруют О, 1 М pac
твором натрия тиосульфата, используя в Ka
честве индикатора 0,5 мл раствора крахмала Р,
который прибавляют в конце титрования.
На титрование должно быть израсходовано
от 24,5 мл до 25,5 мл 0,1 М раствора натрия
тиосульфата.
Ь) 10,0 мл испытуемоro раствора доводят
водой Р до объема 100.0 мл и перемешивают.
К 10,0 мл полученноro раствора прибавляют
25,0 мл 0,1 М раствора кислоты хлористово
дородной, наrревают на водяной бане в течение
1 ч, охлаждают, доводят водой Р до начально
ro объема и титруют 0,1 М раствором натрия
2идроксида, используя в качестве индикатора
0,1 мл раствора фенолфталеина Р1.
На титрование должно быть израсходовано
от 5,7 мл до 6,3 мл 0,1 М раствора натрия 2и
дроксида.
с) 10,0 мл испытуемоro раствора доводят
водой Р до объема 100,0 мл и перемешивают.
10,0 мл полученною раствора титруют 0,1 М
раствором кислоты хлористоводородной, ис
пользуя в качестве индикатора 0,1 мл раствора
фенолфталеина Р1.
На титрование должно быть израсходова
но от 6,0 мл до 7,5 мл 0,1 М раствора кислоты
хлористоводородной.
Медь. Cu. (А. м. 63,55). 1022100. [7440 50 8].
Фольrа очищенная, стружка, проволока или
металлический порошок электролитической
степени чистоты.
Мезитилоксид. С 6 Н 10 О. (М. м. 98,1).
1120100. [141 79 7]. Метилпент З ен 2 он.
Бесцветная маслянистая жидкость. PaCTBO
рим в ЗО частях воды, смешивается с большин
ством орrанических растворителей.
d;g: около 0,858.
Температура кипения: от 129 ос до 130 ОС.
Меклозина rидрохлорид. C25H27CI2N2'2HCI.
(м. м. 463,9). 1051200. [1104 22 9]. H(RS (4
Хлорофенил )фенилметил )] 4 (3 метилбензил)
пиперазина диrидрохлорид.
Содержит не менее 98,0 % и не более
102,0 % C25H27CI2N2'2HCI в пересчете на безво
дное вещество. .
Желтый или желтовато белый кристалличе
ский порошок. Малорастворим в воде, paCTBO
рим в 96 % спирте и вметиленхлориде.
Вода (2.5.12). Не более 5 %. Определение
проводят из 0,200 r испытуемоro образца.
Меламин. С з Н 6 N 6 . (м. м. 126,1). 1051300.
[108 78 1]. 1 ,3,5 Триазин 2,4,6 триамин.
Белый или почти белый аморфный порошок.
Очень мало растворим в воде и в 96 % спирте.
Менадион. С ll НРт (м. м. 172,3). 1051400.
[58 27 5]. 2 Метилнафталин 1 ,4 дион.
Содержит не менее 98,5 % и не более
101,0 % С 11 Н в О 2 в пересчете на сухое вещество.
Бледно желтый кристаллический порошок.
Практически нерастворим в воде, леrкорас
творим в толуоле, умеренно растворим в 96 %
спирте и в метаноле. Светочувствителен.
Температура плавления: от 105 ос до 108 ОС.
Хранят в защищенном от света месте.
Ментилацетат. С 12 Н 2 Р2' (м. м. 198,3).
1051800. [16409 45 3]. 2 Изопропил 5 метил
циклоrексилацетат.
Бесцветная жидкость. Малорастворим в
воде, смешивается с 96 % спиртом.
d 20
20: около 0,92.
20
П D : около 1,447.
Температура кипения: около 228 ОС.
Ментилацетат, используемый в 2азовой
хромат02рафии, должен выдерживать следу
ющее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
мяты перечной, используя ментилацетат в каче
стве испытуемоro раствора.
4. 1. 1. РеактивЬ1
659
Содержание ментилацетата, рассчитанное
методом нормализации, должно быть не менее
97,0%.
Ментол. 1051600. [2216 51 5].
См. статьи Левоментол и Ментол paцeMи
ческий.
Ментол, используемый в 2азовой xpOMaтo
2рафии, должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rазовая
хроматоrрафия (2.2.28), как указано в статье
Ментол рацемический в испытании «Сопутству
ющие примеси» , используя ментол в качестве
испытуемоro раствора.
Содержание ментола, рассчитанное Me
тодом нормализации, должно быть не менее
98,0%.
Ментон. С 10 Н 1в О. (м. М. 154,2). 1051700.
[1407З 97 З]. (2S,5R) 2 Изопропил 5 метилцикло
reKcaHOH. ( ) транс п МентаН 3--0Н.
Содержит различные количества изоментона.
Бесцветная жидкость. Очень мало paCTBO
рим в воде, очень леrко растворим в 96 % спирте.
d 20
20: около 0,897.
20
по: около 1,450.
Ментон, используемый в 2азовой xpOMaтo
2рафии, должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
мяты перечной, используя ментон в качестве
испытуемоro раствора.
Содержание ментона, рассчитанное методом
нормализации, должно быть не менее 90,0 %.
Ментофуран.С 10 Н 1 Р. (м. м. 150,2). 1051500.
[17957 94 7]. 3,9 Эпокси п мента 3,8 диен.
3,6 Диметил 4,5,6, 7 тетраrидробензофуран.
Жидкость слеrка синеватоro цвета. Очень
мало растворим в воде, растворим в 96 % спирте.
d;g: около 0,965
20
по: около 1,480.
[a] O: около +93.
Температура кипения: 196 ОС.
Ментофуран, используемый в 2азовой xpo
мат02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
мяты перечной, ИСПОЛI:::ЗУЯ ментофуран в каче
стве испытуемоro раствора.
Содержание ментофурана, рассчитанное мето--
дом нормализации, должно быть не менее 97,0 %.
2-Меркаптобензимидазол. C 7 H 6 N 2 S. (М М. 150,2).
1170100. [583--39 1]. 1Н Бензимидазол 2 тиол.
Температура плавления: около 302 ОС.
Меркаптопурин. 1051900. [6112 76 1].
См. статью Меркаптопурин.
2-Меркаптоэтанол. C 2 Hps. (м. м. 78,1).
1099300. [60 24 2].
Жидкость. Смешивается с водой.
d;g: около 1,116.
Температура кипения: около 157 ОС.
Метакриловая кислота. С 4 Н 6 О 2 . (м. М. 86,1).
1101800. [7941-4]. 2 Метилпроп 2 еновая кислота.
Бесцветная жидкость.
20
по : около 1,431.
Температура кипения: около 160 ОС.
Температура плавления: около 16 ос.
Метаниловый желтый. С'ВН'4NзNаОзS.
(м. м. 375,4). 1052900. [587 98-4]. Показатель
Шульца NQ 169. Цветной индекс NQ 13065. Натрия
3 [4 ( фенил амино )фенилазо]бензолсульфонат.
Коричневато желтый порошок. Растворим в
воде и в 96 % спирте.
Метаниловоrо желтоrо раствор. 1052901.
Раствор 1 r/л испытуемоro образца в метаноле Р
Испытание на чувствительность. К 50 мл
кислоты уксусной безводной Р прибавляют
0,1 мл раствора метаниловоro желтоrо. Появ
ляется розовато красное окрашивание, которое
должно перейти в фиолетовое при прибавлении
0,05 мл 0,1 М раствора кислоты хлорной.
Изменение окраски. ОТ рН 1,2 (красное
окрашивание) до рН 2,3 (оранжеВ<rжелтое OKpa
шивание).
Метанол. снр. (м. м. 32,04). 105З200. [67 56--1].
Прозрачная бесцветная воспламеняюща
яся жидкость. Смешивается с водой и с 96 %
спиртом.
d 20
20: от 0,791 до 0,793.
Температура кипения: от 64 ос до 65 ОС.
Метанол Р1. 1053201.
Должен выдерживать требования для Me
танола р и следующее дополнительное испы
тание.
Пропускание (2.2.25), определение про
водят используя воду Р в качестве компенса
ционной жидкости: не менее 20 % при 210 нм;
не менее 50 % при 220 нм; не менее 75 % при
230 нм; не менее 95 % при 250 нм; не менее
98 % при 260 нм и более.
Метанол Р2. 1053202.
Метанол Р2, используемый в жидкостной
хромат02рафии. должен выдt;;рживать следу
ющие дополнительные испытания.
Содержит не менее 99,8 % СН 4 О (м. М. 32,04).
Оптическая плотность (2.2.25): не
более 0,17 при 225 нм. Определение прово
дят используя воду Р в качестве компенсаци
ОН ной жидкости.
660
rосударственная фармакопея Республики Беларусь
Метанол подкисленный. 1053203.
1,0 мл кислоты хлористоводородной Р1
доводят метанолом Р до объема 100,0 мл.
Метанол безводный. 1053400. [67 56 1).
1000 мл метанола Р обрабатывают 5 r
ма2ния Р. Если необходимо, инициируют pe
акцию, прибавляя 0,1 мл раствора ртути (1/)
хлорида Р. После прекращения выделения rаза
жидкость переroняют, отroн собирают в сухой
контейнер и защищают от воздействия влаrи.
Вода (2.5.12). Не более 0,3 r/л.
Метанол, свободный от альдеrидов.
1053300.
25 r йода Р растворяют в 1 л метанола Р.
Полученный раствор прибавляют при посто
янном помешивании к 400 мл 1 М раствора
натрия 2идроксида, затем прибавляют 150 мл
воды Р и выдерживают в течение 16 ч. Фильтру
ют И кипятят с обратным холодильником до ис
чезновения запаха йодоформа. Раствор переrо
няют фракционной переroнкой.
Альдеаuды и кетоны: не более 0,001 %.
Метансульфонилхлорид. СН з СI0 2 S.
(М М. 114,6). [124 63 O].
Прозрачная бесцветная или слеrкажелтова
тая жидкость.
Содержание: не менее 99,0 %.
Плотность: 1,48 r/см З .
п : около 1,452.
Температура кипения: около 161 ОС.
Метансульфоновая кислота. СН 4 О з S.
(М М.. 96,1). 1053100. [75 75 2).
Прозрачная бесцветная жидкость, затверде
вающая при температуре около 20 ОС. Смешива
ется с водой, малорастворима в толуоле, прак
тически нерастворима в reKcaHe.
d 20
20: около 1,48.
20
п о : около 1,430.
Метафосфорная кислота. (НРОз)х.
1053000. [37267 86 O].
Стекловидные комочки или палочки; coдep
жащие определенное количество натрия MeTa
фосфата. rиrроскопична. Очень леrко раствори
ма в воде.
Нитраты. 1,0 r испытуемоro образца ки
пятят с 1 О мл воды Р, охлаждают, прибавляют
1 мл раствора инди20кармина Р, 1 О мл кисло
ты серной, свободной от азота, Р и наrревают
до кипения. Синяя окраска не должна полностью
исчезнуть.
Восстанавливающие вещества. Не более
0,01 % в пересчете на НРОз. 35,0 r испытуемо
ro образца растворяют в 50 мл воды Р, прибав
ляют 5 мл раствора 200 r/л кислоты серной Р,
50 Mr калия бромида Р и 5,0 мл 0,02 М pacт
вора калия бромата и наrревают на водяной
бане в течение 30 мин. Охлаждают, прибавля
ют 0,5 r калия йодида Р и титруют выделивший
ся йод 0,1 М раствором натрия тиосульфата,
используя в качестве индикатора 1 мл pacт
вора крахмала Р.
Параллельно проводят контрольный опыт.
1 мл 0,02 М раствора калия бромата COOT
ветствует 4,10 Mr НзРО з .
Хранят в воздухонепроницаемом контейнере.
Метил-4-ацетилбензоат. С 1О Н 1 Рз, (М м. 178,2).
1154100. [3609 5З 8].
Температура плавления: около 94 ОС.
Метил-4 цетилбензоата реактив. 1154101.
0,25 r метил 4 ацетилбензоата Р paCTBO
ряют в смеси из 5 мл кислоты серной Р и 25 мл
охлажденноro метанола Р.
4-Метиламинофенола сульфат. C14H2oNP6S.
(М м. З44,4). 1053800. [55 55 O].
Бесцветные кристаллы. Очень леrко paCTBO
рим в воде, малорастворим в 96 % спирте.
Температура плавления: около 260 ОС.
Метилантранилат. C a H g N0 2 . (М м. 151,2).
1107300. [134 20 3]. Метил 2 аминобензоат.
Бесцветные кристаллы или жидкость от
бесцветноro до желтоватоro цвета. Растворим в
воде, леrкорастворим в 96 % спирте и эфире.
Температура плавления: от 24 ос до 25 ОС.
Температура кипения: от 134 ос до 136 ОС.
Метилантранилат, используемый в 2азо
вой хромат02рафии, должен выдерживать сле
дующее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
цветков померанца, используя метилантрани
лат в качестве испытуемоro раствора.
Содержание метилантранилата, рассчитан
ное методом нормализации, должно быть не
менее 95,0 %.
Метиларахидат. С 21 Н 4 Р2' (М м. 326,6).
1053900. [1120 28 1]. Метилэйкозаноат.
Содержит не менее 98,0 % С 21 Н 4 ;Р2' Опре
деление проводят методом rазовой xpoMaTorpa
фии (2.4.22).
Кристаллическая масса от белоro до желто
ro цвета. Растворим. в 96 % спирте и петролей
ном эфире.
Температура плавления: около 46 ОС.
Метилацетат. С З НР2' (М м. 74,1). 1053700.
[79 20 9].
Прозрачная бесцветная жидкость. PaCTBO
рим в воде, смешивается с 96 % спиртом.
d 20
20: около 0,933.
20
п о : около 1,361.
Температура кипения: от 56 ос до 58 ОС.
4.1.1. Реактивы
661
Метилбеrенат. С2ЗН4602' (м. М. 354,6).
1107500. [929 77 1]. М етилдокозаноат.
Температура плавления: от 54 ос до 55 ОС.
Метилбензол. См. Толуол Р.
Метилбензотиазолонrидразона rидро-
хлорид. С 8 Н g N з S'НСI'НР (м. м. 233,7).1055300.
[38894 11 O]. 3 Метилбензотиазол 2(3Н) он rи
дразона rидрохлорид моноrидрат.
Почти белый или желтоватый кристалличе
ский порошок.
Температура плавления: около 270 ОС.
Испытание на при20дность для определе
ния альде2идов. К 2 мл метанола, свободН020
от альде2идов, Р прибавляют 60 мкл раствора
1 r/л пропионов020 альде2ида Р в метаноле, cвo
бодном от альдееидов, Р и 5 мл раствора 4 r/л
метилбензотиазолонrидразона rидрохлорида,
смешивают и выдерживают в течение 30 мин. ro
товят контрольный раствор, не содержащий про
пионовый альдеrид. К испытуемому и контроль
ному растворам прибавляют по 25,0 мл раствора
2 r/л железа (111) хлорида, доводят ацетоном Р до
объема 100,0 мл И перемешивают. Оптическая
плотность (2.2.25) испытуемоro раствора, изме
ренная при длине волны 660 нм с использова
нием в качестве компенсационной жидкости KOH
трольноro раствора, должна быть не менее 0,62.
2-Метилбутан. С 5 Н 12 . (м. м. 72.2). 1099500.
[78 78A]. Изопентан.
Содержит не менее 99,5 % С 5 Н 12 .
Бесцветная леrковоспламеняющаяся жидкость.
d 20 62
20: около О, 1.
20
по: около 1,354.
Температура кипения: около 29 ОС.
Вода (2.5.12). Не более 0,02 %.
Остаток после выпаривания. Не более
0,0003 %.
Пропускание (2.2.25), определение про
водят используя воду Р в качестве компенса
ционной жидкости: не менее 50 % при 210 нм;
не менее 85 % при 220 нм; не менее 98 % при
240 нм и более.
2-Метилбут-2-ен. С 5 Н ш (м. М. 70.1). 1055400.
[513 35 9].
Очень леrко воспламеняющаяся жидкость.
Практически нерастворим в воде, смешивается
с 96 % спиртом.
Температура кипения: от 37,5 ос до 38,5 ОС.
Метилдеканоат. С 11 Н 2 Р2' (м. М. 186,3).
1054000. [11O 42 9]. Метил н деканоат. Метилка
прат.
Содержит не менее 99,0 % С11Н2202'
Прозрачная бесцветная или желтоватая
жидкость. Растворим в петролейном эфире.
d 20
20: от 0,871 до 0,876.
20
по: от 1,425 до 1,426.
Сопутствующие примеси. rазовая xpo
матоrрафия (2.2.28), хроматоrрафируя равные
объемы каждоro из следующих растворов Be
ществ: раствор 0,02 r/л метилдеканоата в У2ле
рода дисульфиде Р (раствор А), раствор 2 r/л
метилдеканоата в У2лерода дисульфиде Р (раст
вор В), У2лерода дисульфид Р (раствор С). Xpo
матоrрафируют в условиях испытания на бутил
rидрокситолуол, указанных в статье Ланолин.
На xpoMaTorpaMMe раствора В сумма пло
щадей всех пиков, кроме основноro и пика pac
творителя, не должна превышать площадь oc
новноro пика на xpoMaTorpaMMe раствора А.
З-О-Метилдопамина rидрохлорид.
С g Н 1з NО 2 'НСI. (М. м. 203,7). 1055600. [1477 68 5].
4 (2 Амино этил ) 2 метоксифенола rидрохлорид.
Температура плавления: от 21 3 ос до 215 ОС.
4-0-Метилдопамина rидрохлорид.
С g Н 1з N0 2 'НCI. (м. М. 203,7). 1055700. [645 33 0].
5 (2 Амино этил ) 2 метоксифенола rидрохлорид.
Температура плавления: от 207 ос до 208 ОС.
Метилен бис акрилам ид. C7H10N202'
(м. М. 154,2). 1056000. [11O 26 9]. N.N Метилен
биспропенамид.
Белый или почти белый мелкий порошок.
Малорастворим в воде, растворим в 96 % спирте.
Температура плавления: 300 ос с разложе
нием.
Метиленовый синий. С16Н1вСINЗS'ХН20,
(м. М. 319,9, безводный). 1055800. [7220 79 3]. По
казатель Шульца NQ 1038. Цветной индекс NQ 52015.
3, 7 Диметиламинофенотиазина 5 хлорид.
Существует в различных rидратированных
формах и может содержать до 22 % воды.
Темно зеленый или бронзоватый кристалли
ческий порошок. Леrкорастворим в воде, paCTBO
рим в 96 % спирте.
Метиленовоrо синеrо раствор.
0,15 r метиленов020 сине20 Р растворяют
в воде Р и доводят до объема 100 мл этим же
растворителем.
Метиленхлорид. CH 2 CI 2 . (м. М. 84.9).
1055900. [75 09 2]. Дихлорметан.
Бесцветная жидкость. Умеренно растворим
в воде, смешивается с 96 % спиртом.
Температура кипения: от 39 ос до 42 ОС.
Метuленхлорид, используемый в флуори
метрии, должен выдерживать следующее дo
полнительное испытание.
Флуоресценция. ПrИ облучении светом с
длиной волны 365 нм флуоресценция (2.2.21),
измеренная при длине волны 460 нм в кювете
С толщиной слоя 1 см, не должна быть интен
сивнее флуоресценции раствора, содержащеro
0,002 ррm хинина Р в 0,5 М растворе кислоты
серной, измеренной в тех же условиях.
662
rосударственная фармакопея Республики Беларусь
Метиленхлорид подкисленный. 1055901.
К 100 мл метиленхлорида Р прибавляют
1 О мл кислоты хлористоводородной Р и встря
хивают. После разделения слоев используют
нижний слой.
Метилизобутилкетон. С 6 Н 1 р. (М. М. 100,2).
1054300. [108 10 1]. 4 Метил 2 пентанон.
Прозрачная бесцветная жидкость. Мало
растворим в воде, смешивается с большинством
орrанических растворителей.
d; : около 0,80.
Температура кипения: около 115 ОС.
Температурные пределы пере20нки
(2.2.11). Интервал температуры переrонки не
должен превышать 4,0 ОС; должно переroняться
от 1 мл до 95 мл. Переrоняют 100 мл.
Остаток после выпаривания. Не более
0,01 %. Выпаривают на водяной бане, остаток
сушат при температуре от 100 ос до 105 ОС.
Метилизобутилкетон Р1. 1054301.
50 мл свежепереrнанноro метилизобу
тилкетона р встряхивают с 0,5 мл кисло
ты хлористоводородной Р1 в течение 1 мин.
После разделения слоев нижний слой отбра
сывают.
rотовят непосредственно перед использо
ванием.
Метилизобутилкетон РЗ. 1054302.
Должен выдерживать требованиядля метил
изобутилкетона Р и следующие испытания:
Cr: не более 0,02 ррm;
Cu: не более 0,02 ррm;
РЬ: не более 0,1 ррm;
Ni: не более 0,02 ррm;
Sп: не более 0,1 ррm.
1 Метилимидазол. C 4 H 6 N 2 . (М М. 82,1).
1139700. [616--47 7). 1 Метил 1Н имидазол.
Бесцветная или слеrка желтоватая жид
кость.
п O: около 1,495.
Температура капения: от 195 ос до 197 ОС.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
2 Метилимидазол. C 4 H 6 N 2 . (М М. 82,1).
1143400. [693 98 1).
Белый или почти белый кристаллический
порошок.
Температура плавления: около 145 ОС.
Метилкапрат. 1054000. См. Метилдеканоат Р.
Метилкаприлат. С э Н 1в 0 2 . (М М. 158,2).
1120400. [111 11 5). Метилоктаноат.
d 20
20: около 0,876.
20
по: около 1,417.
Температура кипения: от 193 ос до 194 ОС.
Метилкапроат. С 7 Н 14 О 2 . (М М. 130,2).
1120300. [106 70 7]. Метилrексаноат.
d;g: около 0,885.
20
по: около 1,405
Температура кипения: от 150 ос до 151 ОС.
Метиллаурат. С1ЗН2602' (М. М. 214,4).
1054400. [111 82 O]. Метилдодеканоат.
Содержит на менее 98,0 % С1ЗН2602' Опре
деление проводят методом rазовой xpOMaTO
rрафии (2.4.22).
Бесцветная или желтая жидкость. PaCTBO
рим в 96 % спирте и в петролейном эфире.
d;g: около 0,87.
п O: около 1,431.
Температура плавления: около 5 ОС.
Метиллиrноцерат. С25Н5002' (М М. 382,7).
1120600. [557 59 5). Метилтетракозаноат.
Хлопья.
Температура плавления: около 58 ОС.
Метиллинолеат. С 1э Н 34 О 2 . (М М. 294,5).
1120700. [112 63 O]. Метил (9Z,12Z--октадека
9,12 диеноат.
d;g: около 0,888.
п O: около 1,466.
Температура кипения: от 207 ос до 208 ОС.
Метиллиноленат. С19НЗ202' (М. М. 292,5).
1120800. [301 00 8]. Метил (9Z,12Z,15Z)
oктaдeKa 9,12, 15 триеноат.
d;g: около 0,901.
п O: около 1,471.
Температура кипения: около 207 ОС.
Метил-v-линоленат. С19НЗ202' (М. М. 292,5).
1120800. [16326 32 2). Метил (6Z,9Z, 12Z)
OKTaдeKa 6,9, 12 триеноат.
Содержит не менее 99,0 %. Определе
ние проводят методом rазовой хроматоrрафии
(2.2.28).
Метилмарrарат. С 1в Н З Р2' (М М. 284,5).
1120900. [1731 92 6). Метилrептадеканоат.
Белый или почти белый порошок.
Температура плавления: от 32 ос до 34 ОС.
Метилметакрилат. С 5 Н в 0 2 . (М М. 100,1).
1 054500. [80 62 6]. Метил 2 метилпроп 2 еноат.
Бесцветная жидкость.
20
по: около 1,414.
Температура кипения: около 100 ОС.
Температура плавления: около 48 ОС.
Содержит подходящий стабилизирующий
peareHT.
4.1.1. Реактивы
663
МетилметансулЬфонат. С 2 Н 6 О З S' (М м. 110,1).
1179500. [66 27 3].
Прозрачная, бесцветная или желтоватая
жидкость.
Содержит не менее 99,0 % С 2 Н 6 О З S'
Плотность: около 1,3 r/см З (при 25 ОС).
п O: около 1,414.
Температура кипения: около 202 ОС.
Метилмиристат. С15Нзо02' (м. м. 242,4).
1054600. [124 10 7]. Метилтетрадеканоат.
Содержит не менее 98,0 % С15НзоО2' Опре
деление проводят методом rазовой xpoMaTorpa
фии (2.4.22).
Бесцветная или слеrка желтая жидкость.
Растворим в 96 % спирте и в петролейном эфире.
d 20
20: около 0,87.
20
П D : около 1,437.
Температура плавления: около 20 ОС.
# 2 Метил 1 ,4 нафтохинон. С 11 Н в 0 2 .
(М. м. 172,2). Витамин К З ' Метинон.
Желтые иroльчатые кристаллы. Малораст
ворим в воде, растворим в этаноле и в эфире.
Температура плавления: около 106 ОС.
2 Метил 5 нитроимидазол. С 4 Н 5 N з 0 2 ,
(М м. 127,1). 1056100. [88054 22 2].
Порошок от белоro до светло желтоro цвета.
Температура плавления: от 252 ос до 254 ОС.
Содержит не менее 98,0 % С 4 Н 5 N з О 2 ,
Метиловоrо зеленоrо йодомеркуратная
бумаrа.1054201.
Тонкие полоски подходящей фильтроваль
ной бумаrи поrружают в раствор 40 r/л Meти
лов020 зелеН020 Р. сушат на воздухе, затем
поrружают ИХ на 1 ч в раствор, содержащий
140 r/л калия йодида Р и 200 r/л ртути (11)
йодида Р. Полоски промывают водой дистил
лированной Р до тех пор, пока промывные
воды не станут практически бесцветными, и
сушат на воздухе.
Хранят в защищенном от света месте. Срок
roдности 2 сут.
Метиловоrо KpacHoro раствор. 1055102.
50 Mr метuлов020 краСН020 Р растворяют в
смеси из 1,86 мл 0,1 М раствора натрия 2идpOK
сида и 50 мл 96 % спирта Р и доводят водой Р
до объема 100 мл.
Испытание на чувствительность. К 100 мл
воды, свободной от уzлерода диоксида, Р при
бавляют 0,1 мл раствора метиловоro красноro и
0,05 мл 0,02 М раствора кислоты хлористово
дородной. Появляется красное окрашивание, KO
торое должно перейти в желтое при прибавлении
не более 0,1 мл 0,02 М раствора натрия 2идpOK
сида.
Изменение окраски. От рН 4,4 (красное
окрашивание) до рН 6,0 (желтое окрашивание).
Метиловоrо KpacHoro смешанный paCT
вор. 1055101.
0,1 r метилО8ОZO краСН020 Ри 50 Mr метuлен
8020 CUHezo Р растворяют в 100 мл 96 % спирта Р.
Изменение окраски. от рН 5,2 (красно-фиолето-
вое окрашивание) до рН 5,6 (зеленое окрашивание).
Метиловоrо оранжевоrо раствор.
1054802.
0,1 r метилов020 оранжев020 Р растворяют
в 80 мл воды Р и доводят 96 % спиртом Р до
объема 100 мл.
Испытание на чувствительность. К 100 мл
воды, свободной от уzлерода диоксида, Р прибав
ляют 0,1 мл раствора метиловоro оранжевоro. По
является желтое окрашивание, которое должно пе
рейти в красное при прибавлении не более 0,1 мл
0,1 М раствора кислоты хлористоводородной.
Изменение окраски. От рН 3,0 (красное
окрашивание) до рН 4,4 (желтое окрашивание).
Метиловоrо оранжевоrо смешанный
раствор. 1054801.
20 Mr метиловО20 оранжев020 Р и 0,1 r
бромкрезолов020 зелеН020 Р растворяют в 1 мл
0,2 М раствора натрия 2идроксида и доводят
водой Р до объема 100 мл.
Изменение окраски. От рН 3,0 (оранжевое
окрашивание) до рН 4,4 (желтовато зеленое
окрашивание ).
# Метиловоrо фиолетовоrо раствор Р1.
0,1 r метилов020 фиолетов020 Р paCTBO
ряют в 100 мл воды Р.
Изменение окраски. От рН 0,1 (желтое OKpa
шивание) до рН 1,5 (зеленое окрашивание); от
рН 1,5 (зеленое окрашивание) до рН 3,2 (фиоле
товое окрашивание).
# Метиловоrо фиолетовоrо раствор Р2.
0,1 r метилов020 фиолетов020 Р paCTBO
ряют в 100 мл кислоты уксусной ледяной Р.
Раствор используют свежеприrотовленным
Метиловый зеленый. С2БНззС12Nз,
(м. м. 458,5). 1054200. [7114 03 6]. Показа
тель Шульца NQ 788. Цветной индекс NQ 42585.
4 [[4 (Диметиламино ) фен ил ][4 (ди метил им и
нио )циклоrекса 2,5 дие нилиден ) метилфенил)
триметиламмония дихлорид.
Зеленый порошок. Растворим в воде, pac
творим в кислоте серной с образованием жел
тоro окрашивания. переходящеro в зеленое при
разведении водой.
Метиловый красный. С15Н15Nз02'
(М м. 269,3). 1055100. [493 52 7]. Показатель
Шульца NQ 250. Цветной индекс NQ 13020. 2 (4 Ди
метиламинофенилазо )бензойная кислота.
Темно красный порошок или фиолетовые
кристаллы. Практически нерастворим в воде,
растворим в 96 % спирте.
664
rосударственная фармакопея Республики Беларусь
Метиловый оранжевый. С'4Н'4NзNаОзS.
(м. м. 327,3). 1054800. [547 58 0]. Показатель
Шульца NQ 176. Цветной индекс NQ 13025. Натрия
4' (диметиламино )азобензоп-4 сульфонат.
Оранжево желтый кристаллический поро
шок. Малорастворим в воде, практически He
растворим в 96 % спирте.
# Метиловый фиолетовый. С25НзоСINз.
(м. м. 408,0). [548 62 9]. N [4 [Бис[4
(диметиламино )фенил ]метилен ] 2,5 циклоrекса
диен 1 илиден ] N метилметанаминия хлорид.
rексаметил п розанилина хлорид. rенциановый
фиолетовый. Обычно представляет собой смесь
хлоридов rексаметил п розанилина, пентаметил
п розанилина и тетраметил п розанилина, co
держащую не менее 96 % rексаметил п
розанилина хлорида.
Зеленый или темно зеленый кристалличе
ский порошок с металлическим блеском. PaCTBO
рим в воде, в кислотах, в растворах rидроксидов
щелочных металлов.
Метилолеат. С'9НЗ60z' (м. М. 296,4).1054700.
[112 62 9]. Метил (Z) октадек 9 еноат.
Содержит не менее 98,0 % С,gН з6 О z . Опре
деление проводят методом rазовой xpoMaTorpa
фии (2.4.22).
Бесцветная или слабо желтая жидкость.
Растворим в 96 % спирте и в петролейном эфире.
d 20
20: около 0,88.
20
по : около 1,452.
Метилпальмитат. С 17 Н з Рz, (м. м. 270,5).
1 054900. [112 39 0]. Метилrексадеканоат.
Содержит не менее 98,0 % С 17 Н з4 О z . Опре
деление проводят методом rазовой xpoMaTorpa
фии (2.4.22).
Белая или желтая кристаллическая масса.
Растворим в 96 % спирте и в петролейном
эфире.
Температура плавления: около 30 ОС.
Метилпальмитолеат. С 17 Н З Р2' (М М. 268,4).
1121 000. [1120 25 8]. Метил (9Z) rексадек 9
еноат.
dig: около 0,876.
20
по: около 1,451.
Метилпараrидроксибензоат. 1055000.
[99 76 3].
См. статью Метилпара2идроксuбензоат.
4 Метилпентан 2 ол. С6Н'40, (М М. 102.2).
1114300. [108 11 2].
Прозрачная бесцветная летучая жидкость.
d 20
4 : около 0,802.
20
по: около 1,411.
Температура кипения: около 1З2 ОС.
Метилпиперазин. C 5 H 12 N 2 . (м. М. 100.2).
1056300. [74879 18 8]. 1 Метилпиперазин.
Бесцветная жидкость. Смешивается с водой
и с 96 % спиртом.
d 20 : около 0,90.
20
п;О: около 1,466.
Температура кипения: около 138 ОС.
4 (4-Метилпиперидино}пиридин. C1,H'6N2'
(м. М. 176,3). 1114400. [80965 30 6].
Прозрачная жидкость.
п;О: около 1,565.
2-Метилпропанол. С 4 Н 10 О. (м. М. 74,1).
1056400. [78 83 1]. Изобутиловый спирт.
2 Метилпропан 11)Л.
Прозрачная бесцветная жидкость. PaCTBO
рим в воде, смешивается с 96 % спиртом.
d 20 : около 0,80.
20
п 5: от 1,З97 до 1,399.
Температура кипения: около 107 ОС.
Температурные пределы пере20нки
(2.2.11): от 107 ос до 109 ОС; должно переroнять
ся не менее 96 %.
2 Метил-2 пропанол. С 4 Н,оО. (м. М. 74,1).
1056500. [75 65 0]. 1, 1 Диметилэтиловый спирт.
трет Бутиловый спирт.
Прозрачная бесцветная жидкость или кри
сталлическая масса. Растворим в воде, смеши
вается с 96 % спиртом.
Температура затвердевания (2.2.18):
около 25 ОС.
Температурные пределы пере20нки
(2.2.11): от 81 ос и 83 ОС; должно переrоняться
не менее 95 %.
Метилсалицилат. СвНРз, (м. М. 152,1).
1146200. [119 36 8]. Метил 2 rидроксибензоат.
Бесцветная или слеrка желтоватая жид
кость. Очень мало растворим в воде, смешива
ется с 96 % спиртом и с жирными и эфирными
маслами.
20
по : от 1,535 до 1,538.
d 20
4 : от 1,180 до 1,186.
Содержит не менее 99,0 % и не более
100,5 % СвНвО з .
Метилстеарат. С,gНзРz, (м. м. 298,5).
1055200. [112 61 8]. Метилоктадеканоат.
Содержит не менее 98,0 % С'9Нзв02' Опре
деление проводят методом rазовой xpOMaTO
rрафии (2.4.22).
Белая или желтая кристаллическая масса.
Растворим в 96 % спирте и в петролейном
эфире.
Температура плавления: около 38 ОС.
4. 1. 1. Реактивы
665
Метилтридеканоат. С'4Н2802" (М М. 228,4).
1121100. [1731 88 0].
Бесцветная или слабо желтая жидкость.
Растворим в 96 % спирте и в петролейном эфире.
d;g: около 0,86.
п O: около 1,441.
Температура плавления: около 6 ОС.
Метилтрикозаноат. С 24 Н 4 Р2' (М М. 368.6).
1111500. [2433 97 8]. Метиловый эфир трикоза
новой кислоты.
Содержит не менее 99,0 % С24Н4в02'
Белые или почти белые кристаллы. Прак
тически нерастворим в воде, растворим в reK
сане.
Температура плавления: от 55 ос до 56 ОС.
Метилфенилоксазолилбензол. C26H2oN202'
(М м. 392,5). 1056200. [3073 87 8]. 1 +Бис[2 (4
метил 5 фенил )оксазолил ] бензол.
Мелкий зеленовато желтый порошок с
синей флуоресценцией или мелкие кристаллы.
Растворим в 96 % спирте, умеренно растворим
в ксилоле.
Температура плавления: около 233 ОС.
Метилфенилоксазолилбензол, исполь
зуемый для жидкостной сцинтилляции,
должен быть соответствующей степени
чистоты.
# Метилфталеин. См. Фталеиновый пур
пурный Р.
Метилцеллюлоза 450. 1 055500. [9004 67 5].
См. статью Метилцеллюлоза.
Номинальная вязкость: 450 мПа.с.
Метилциннамат. С IО Н,о02' (М М. 162,2).
1099400. [103 26A].
Бесцветные кристаллы. Практически He
растворим в воде, растворим в 96 % спирте.
п O: около 1,56.
Температура кипения: около 260 ос.
Температура плавления: от 34 ос до 36 ОС.
Метилэйкозеноат. С20Нзв02'(М' м. 310,5).
1120500. [2390 09 2]. Метил (11 Z) эйкоз 11
еноат.
Метилэтилкетон. С 4 Н в О. (М М. 72,1).
1054100. [78 93 З]. Этилметилкетон. 2 Бутанон.
Прозрачная бесцветная воспламеняющаяся
жидкость. Очень леrко растворим в воде, CMe
шива'3ТСЯ с 96 % спиртом.
d;g: около 0,81.
Температура кипения: от 79 ос до 80 ос.
Метимазол. См. Тиамазол Р.
DL-Метионин. 1129400. [59 51 8].
См. статью OL MemuoHUH.
L-Метионин. C5H'1N02S. (М М. 149,2).
1053500. [63 68 3]. (S 2 АминоА (метилтио)бу
та новая кислота. Метионин.
Содержит не менее 99,0 % и не более 101,0%
C5H'1N02S в пересчете на сухое вещество.
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Pac
творим в воде, очень мало растворим в 96 %
спирте.
рН (2.2.3): от 5,5 до 6,5. Измеряют рН paCT
вора 25 r/i1 испытуемоro образца в воде, свобод
ной от У2лерода диоксида, Р.
[a] O: от +22,5 до +24,0 в пересчете на сухое
вещество. 1,00 r субстанции растворяют в Kиc
лоте хлористоводородной Р1 и доводят до
объема 50,0 мл этим же растворителем.
Потеря е массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 r испытуемоro образца
сушат при температуре от 100 ос до 105 ОС.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
(1 RS)-(6-Метоксинафтален-2-ил)этанол.
С'ЗН1402' (М М. 202,3). 1159600. [77301 42 9].
6 Метокси а метил 2 нафтолметанол.
Белый или почти белый порошок.
Температура плавления: около 113 ОС.
1-(6-Метоксинафтален-2-ил)этанон.
С'ЗН1Р2' (М. M 200,2). 1159700. [3900А5 б].
6' Метокси 2' ацетонафтон.
Белый или почти белый порошок.
Температура плавления: около 108 ОС.
Метоксифенилуксусная кислота. СgН,оОз.
(М м. 166,2). 1053600. [7021 09 2]. (RS) 2
Метокси 2 фенилуксусная кислота.
Белый кристаллический порошок или белые
или почти белые кристаллы. Умеренно paCTBO
рима в воде, леrкорастворима в 96 % спирте.
Температура плавления: около 70 ОС.
Метоксифенилуксусной кислоты реак-
тив. 1053601.
2,7 r кислоты метоксифенилуксусной Р
растворяют в 6 мл раствора тетраметилам
мония 2идроксида Р и прибавляют 20 мл эта
нола Р.
Хранят в полиэтиленовом контейнере.
(RS)-Метотрексат. C2oH22NaOs' 1120200.
[60388 5З б]. (RS) 2 [4 [[(2 +Даминоптеридин
6 ил )метил ]метиламино ]бензоиламино ]пентан
дионовая кислота.
Содержание: не менее 96,0 %.
Темперспура плавления: около 195 ОС.
# Миллона реактив.
1 О r ртути (1) азотнокислой Р растворяют в
8,5 мл кислоты азотной разведенной Р1 и раз
водят двойным объемом воды Р. Используют
прозрачный раствор.
666
rосударственная фармакопея Республики Беларусь
МИОЗМИН. C g H 10 N 2 . (м. м. 146,2). 1121200.
[532 12 7]. 3 (4,5 Диrидро 3Н пиррол 2 ил )пи
ридин.
Бесцветные кристаллы.
Температура плавления: около 45 ОС.
Миристиловый спирт. С 14 Н зо О. (м. м.214,4).
1121300. 1 Тетрадеканол.
d;g: около 0,823.
Температура плавления: от 38 ос до 40 ОС.
Миристицин. С 11 Н 1 Рз, (м. м. 192,2).
1099600. [607 91 0]. 5 Аплил 1 метокси 2,3
метилендиоксибензол. 4 Метокси 6 (проп 2
енил) 1 ,3 бензодиоксол.
Бесцветная маслянистая жидкость. Практи
чески нерастворим в воде, малорастворим в эта
ноле, смешивается с толуолом и ксилолом.
d;g: около 1,144.
20
по: около 1,540.
Температура кипения: от 276 ос до 277 ОС.
Температура плавления: около 173 ОС.
Хранят в защищенном от света месте.
f3 Мирцен. С 10 Н 16 . (М. м.. 136,2). 1114500.
[123 35 3]. 7 Метил 3 метиленокта 1,6 диен.
Маслянистая жидкость с приятным запахом.
Практически нерастворим в воде, смешивается
с 96 % спиртом, растворим в кислоте уксусной
ледяной, растворяется в растворах rидроксидов
щелочных металлов.
d 20
4 : около 0,794.
20
по : около 1,470.
fЗ Мирцен, используемый в 2азовой xpOMa
т02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
мяты перечной, используя ,В..мирцен в качестве
испытуемою раствора.
Содержание ,В..мирцена, рассчитанное Me
тодом нормализации, должно быть не менее
90,0%.
Молекулярное сито. 1056600.
Молекулярное сито из натрия алюмосилика
та. Имеет вид шариков с размерами пор 0,4 нм И
диаметром 2 мм.
Молекулярное сито для хроматоrрафии.
1129700.
Молекулярное сито из натрия алюмосили
ката. Размер частиц указывают после названия
сорбента в испытаниях, в которых он использу
ется. При необходимости указывают и размер
частиц.
# Молибденовая кислота. Н 2 Мо0 4 .
(м. м. 161,97).
Белый или желтовато белый порошок.
Молибденованадиевый реактив. 1056700.
В лабораторном стакане вместимостью
150 мл смешивают растертые в порошок 4 r aM
мония молибдата Р и 0,1 r аммония вaHaдa
та Р, прибавляют 70 мл воды Р и перемешива
ют стеклянной палочкой до растворения. Через
несколько минут должен получиться прозрачный
раствор, к которому прибавляют 20 мл кислоты
азотной Р и доводят водой Р до объема 100 мл.
Молочная кислота. С з Н 6 О з . (М. М. 90,1).
1 047800. [50 21 5].
Содержит не менее 88,0 % (м/м) и не более
92,0 % (м/м) С з Н 6 О з .
Бесцветная или слеrка желтоватая сиропо
образная жидкость. Смешивается с водой и с
96 % спиртом.
> Молочная кислота смесь 2 rидроксипро
па новой кислоты и продуктов ее конденсации
и воды, соотношение которых зависит от TeM
пературы и концентрации. Имеет энантиомер и
обычно представляет собой рацемат.
Молочной кислоты реактив. 1047801.
Раствор А. К 60 мл кислоты молочной Р
прибавляют 45 мл предварительно профиль
трованной кислоты молочной Р, насыщенной
без наrревания Суданом красным G Р. Кисло
та молочная насыщается медленно без Harpe
вания, поэтому Bcerдa необходим избыток Kpa
сителя.
Раствор В. rотовят 10 мл насыщенноro
раствора анилина Р и фильтруют.
Раствор с. 75 Mr калия йодида Р paCTBO
ряют в воде Р и доводят до объема 70 мл этим
же растворителем. К полученному раствору при
бавляют 1 О мл 96 % спирта Р и 0,1 r йода Р и
встряхивают.
Смешивают растворы А и В, прибавляют
раствор С.
Морфина rидрохлорид. 1056900.
См. статью Морфина 2идрохлорид.
Морфолин. C 4 H g NO. (м. м. 87,1). 1057000.
[11 0 91 8]. Тетраrидро 1 ,4 оксазин.
Бесцветная rиrроскопичная воспламеняю
щаяся жидкость. Растворим в воде и 96 % спирте.
d;g: около 1,01.
Температурные пределы пере20нки
(2.2.11). От 126 ос до 130 ОС; должно переroнять
ся не менее 95 %.
Хранят в воздухонепроницаемом контейнере.
Морфолин для хроматоrрафии. 1057001.
Долwен выдерживать требования для MOp
фолина Р и следующее дополнительное требо
вание.
Содержание: не менее 99,5 %.
Мочевина. 1095000. [57 1 3 6].
См. статью Мочевина.
4.1.1. Реактивы
667
Муравьиная кислота безводная. СН 2 О 2 .
(М м.46,03). 1039300. [64 18 6].
Содержит не менее 98,0 % (м/м) СНР2'
Бесцветная жидкость. Вызывает коррозию,
смешивается с водой и 96 % спиртом.
dig: около 1,22.
Количественное определение. 1 О мл воды Р
помещают в коническую колбу, точно взвешива
ют, быстро прибавляют около 1 мл кислоты MY
равьиной безводной и снова взвешивают. При
бавляют 50 мл воды Р и титруют 1 М раствором
натрия 2идроксида, используя в качестве инди
катора 0,5 мл раствора фенолфталеина Р.
1 мл 1 М раствора натрия 2идроксида co
ответствует 46,03 Mr СНР2'
Мурексид. CaHsNP6'Hp. (М. м. 302,2).
1137200. 5,5' Нитрилобис(пиримидин 2,4,6
(1Н,3Н,5Н) трион) аммония. #Аммоний пурпуро
вокислый.#
Коричневато красный кристаллический по
рошок. Умеренно растворим в холодной воде,
растворим в roрячей воде, практически HepaCT
ворим в 96 % спирте. Растворяется в растворах
калия rидроксида или натрия rидроксида с обра
зованием растворов синеro цвета.
Мышьяка (111) оксид. Аsрз. (М м. 197,8).
1008300. [1327 53 3]. Мышьяковистый анrидрид.
Димышьяка триоксид.
Кристаллический порошок или белая или
почти белая масса. Малорастворим в воде, pac
творим в кипящей воде.
Натрий. Na. (А. м. 22,99). 1078500. [7440 23 5].
Металл, на свежем срезе имеет блестя
щую серебристо серую поверхность. На воздухе
быстро тускнеет и полностью окисляется с обра
зованием натрия rидроксида, который превраща
ется в натрия карбонат. Бурно реаrирует с водой
с образованием водорода и раствора натрия rи
дроксида; растворим в метаноле с образованием
водорода и раствора натрия метилата; практиче
ски нерастворим и петролейном эфире.
Хранят под петролейным эфиром или Ba
зелином.
Натрия азид. NаN з . (М м. 65,0). 1078900.
[26628 22 8].
Белый или почти белый кристаллический
порошок или кристаллы. Леrкорастворим в воде,
малорастворим в 96 % спирте.
Натрия арсенит. NaAs0 2 (М м. 129,9).
1165900. [7784 46 5).
Натрия арсенита раствор. 1165901.
5,0 r натрия арсенита Р растворяют в 30 мл
1 М раствора натрия 2идроксида, охлаждают
до О ОС И прибавляют при постоянном переме
шивании 65 мл кислоты хлористоводородной
разведенной Р.
Натрия аскорбата раствор. 1078800.
[134 03 2].
3,5 r кислоты аскорбиновой Р растворяют в
20 мл 1 М раствора натрия 2идроксида.
rотовят непосредственно перед использо
ванием.
Натрия ацетат. 1078600. [61З1 90-4].
См. статью Натрия ацетат три2идрат.
Натрия ацетат безводный. С 2 Н з Nа0 2 .
(М. м. 82,0). 1078700. [127 09 3].
Бесцветные кристаллы или rранулы. Очень
леrко растворим в воде, умеренно растворим в
96 % спирте.
Потеря в массе при высушивании (2.2.32).
Не более 2,0 %. Определение проводят при TeM
пературе 105 ОС.
# Натрия бензоат.
См. статью Натрия бензоат.
Натрия бикарбонат. См. Натрия 2идpOKap
бонат Р.
# Натрия бисульфит. См. Натрия 2идpo
сульфит р.
Натрия бутансульфонат. С 4 Н 9 NаО з S.
(М м. 160,2). 1115600. [2386 54 1].
Белый или почти белый кристаллический
порошок. Растворим в воде.
Температура плавления: более 300 ОС.
Натрия висмутат. NаВiO з . (М м. 280,0).
1079000. [12232 99 4].
Содержание: не менее 85,0 % NаВiO з .
Желтый или желтовато коричневый по
рошок. Медленно разлаrается под действием
влаrи или высокой температуры, практически
нерастворим в холодной воде.
Количественное определение. 0,200 r суспен
дируют в 10 мл раствора 200 r/л калия йодида Р,
прибавляют 20 мл кислоты серной разведенной Р
и титруют 0,1 М раствором натрия тиосульфата
до появления оранжевоro окрашивания; используя
в качестве индикатора 1 мл раствора крахмала Р.
1 мл 0,1 М раствора натрия тиосульфата
соответствует 14,00 Mr NаВiO з .
Натрия вол ьфра мат. Na 2 W0 4 '2H 2 0.
(М. м. 329,9). 1084700. [10213 10 2]. Динатрия
вольфрамат диrидрат.
Белый или почти белый порошок или бес
цветные кристаллы. Леrкорастворим в воде с об
разованием прозрачноro раствора, практически
нерастворим в 96 % спирте.
Натрия rексансульфонат. С6Н,зNаОзS.
(М м. 188,2). 1081200. [2832-45 3].
Белый или почти белый порошок. Леrкорас
творим в воде.
668
rосударственная фармакопея Республики Беларусь
Натрия rептансульфонат. С 7 Н I5 NаО з S.
(М м. 202,3). 1081000. [22767 50 6].
Белая или почти белая кристаллическая
масса. Леrкорастворим в воде, растворим в Me
таноле.
Натрия rептансульфонат моноrидрат.
С 7 Н I5 NаО з S'НР. (М м. 220,3). 1081100.
Содержание: не менее 96 % С 7 Н 15 NаО з S в
пересчете на безводное вещество.
Белый или почти белый кристаллический
порошок. Растворим в воде, очень мало paCTBO
рим в этаноле.
Вода (2.5.12). Не более 8 %. Определение
проводят из 0,300 r.
Количественное определение. 0,150 r paCT
воряют в 50 мл кислоты уксусной безводной Р и
титруют О, 1 М раствором кислоты хлорной по
тенциометрически (2.2.20).
1 мл О, 1 М раствора кислоты хлорной соот
ветствует 20.22 Mr С 7 Н I5 NаО з S.
Натрия rидрокарбонат. 1081300. [144 55 8].
См. статью Натрия zидрокарбонаm.
Натрия rидрокарбоната раствор. 1081301.
Раствор 42 r/л натрия 2идрокарбонС!.та Р.
Натрия rидроксид. 1081400. [1310 73 2].
См. статью Натрия 2идроксид.
Натрия rидроксида раствор. 1081401.
20,0 r. натрия 2идроксида Р растворяют
в воде Р и доводят до объема 100,0 мл этим
же растворителем. Концентрацию раствора
определяют титрованием 1 М раствором
кислоты хлористоводородной, используя
в качестве индикатора раствор метилово
20 оранжев020. Р; если необходимо, раствор
укрепляют или разбавляют до концентрации
200 r/л.
Натрия rидроксида раствор, свободный
от карбонатов. 1081406.
Навеску натрия 2идроксида Р растворяют
в достаточном количестве воды, свободной от
У2лерода диоксида, Р до получения раствора с
концентрацией 500 r/л и отстаивают. Декантиру
ют прозрачную надосадочную жидкость, продо
храняя ее от попадания уrлерода диоксида.
Натрия rидроксида раствор разведен-
ный. 1081402.
8,5 r натрия 2идроксида Р растворяют в
воде Р и доводят до объема 100 мл этим же
растворителем.
Натрия rидроксида метанольный раст-
вор. 1081403.
40 Mr натрия 2идроксида Р растворяют в
50 мл воды Р. Полученный раствор охлаждают
и прибавляют 50 мл метанола Р.
Натрия rидроксида раствор концентри-
рованный. 1081404.
42 r натрия 2идроксида Р растворяют в
воде Р и доводят до объема 100 мл этим же
растворителем.
Натрия rидросульфит. NаНОзS. (м. м. 104,1).
1115700. [7631 90 5]. #Натрия бисульфит.#
Белый или почти белый кристаллический
порошок. Леrкорастворим в воде, умеренно pac
творим в 96 % спирте.
На воздухе частично теряет серы диоксид и
постепенно окисляется до сульфата.
Натрия rипобромита раствор. 1081500.
20 мл раствора натрия 2идроксида KOHцeH
трированнО2О Р и 500 мл веды Р смешивают на ле.-
дяной бане, прибавпяют 5 мл раствора брома Р и
осторожно перемешивают до образования раствора.
rотовят непосредственно перед использо
ванием.
Натрия rипофосфит. NaH 2 P0 2 'H 2 0.
(м. м. 106,0). 1081700. [10039 56 2]. Натрия фос
финат моноrидрат.
Белый или почти белый порошок или бес
цветные кристаллы. rиrроскопичен. Леrкорас
творим в воде, растворим в 96 % спирте.
Хранят в воздухонепроницаемом контейнере.
# Натрия rипофосфита раствор. Реактив
Тиле. 20 r натрия 2ипофосфита Р растворяют
в 40 мл воды Р. Раствор вливают в 180 мл кисло
ты хлористоводородной Р и выдерживают в Te
чение 24 ч. Сливают прозрачную надосадочную
жидкость. Полученный раствор должен быть
бесцветным. Хранят в стеклянном контейнере с
притертой пробкой.
Натрия rипохлорита раствор концентри-
рованный. 1081600.
Содержание: не менее 25 r/л и не более
30 r/л активноro хлора.
Желтоватая жидкость, имеет щелочную pe
акцию.
Количественное определение. В колбу с
50 мл воды Р последовательно помещают 1 r
калия йодида Р и 12,5 мл кислоты уксусной раз
веденной Р. 10,0 мл концентрированноro paCT
вора натрия rипохлорита доводят водой Р до
объема 100,0 мл. 10,0 мл полученноro раствора
титруют 0,1 М раствором натрия тиосульфа
та, используя в качестве индикатора 1 мл pacт
вора крахмала Р.
1 мл 0,1 М раствора натрия тиосульфата
соответствует 3,546 Mr активноro хлора.
Хранят в защищенном от света месте.
Натрия rлюкуронат. C B H 9 Na0 7 'H 2 0.
(М м. 234,1). 1080900. Натрия D rлюкуронат MO
ноrидрат.
[a] O: около +21,5. Определение проводят,
используя раствор 20 r/л.
4. 1. 1. Реактивы
669
Натрия декансульфонат. С,оН 2 ,NаО з S.
(М м.244,3). 1079800. [13419 61 9].
Белый или почти белый кристаллический
порошок или хлопья. Леrкорастворим в воде,
растворим в метаноле.
Натрия дезоксирибонуклеат. 1079900.
[73049 39 5]. (Около 85 % имеет молекулярную
маесу 2.107 или более).
Белое или почти белое волокнистое веще
ство; получают из тимуса телят.
Испытание на при20дность. 1 О Mr раст
воряют в имидазольном буферном растворе
рН 6,5 Р и доводят до объема 10,0 мл этим же
растворителем (раствор А). 2,0 мл раствора А
доводят имидазольным буферным раствором
рН 6,5 Р до объема 50,0 мл. Оптическая плот
ность (2.2.25) полученноro раствора, измерен
ная при длине волны 260 нм, должна быть от
0,4 до 0,8.
К 0,5 мл раствора А прибавляют 0,5 мл иMи
даЗОЛЬН020 буфеРН020 раствора рН 6,5 Р и 3 мл
раствора кислоты хлорной (25 r/л НСI0 4 ). Об
разуется осадок. Центрифуrируют. Оптическая
плотность надосадочной жидкости при длине
волны 260 нм должна быть не более 0,3. В Ka
честве компенсационной жидкости используют
смесь из 1 мл имидаЗОЛЬН020 буфеРН020 pacт
вора рН 6,5 Р и 3 мл раствора кислоты хлорной
(25 r/л НСI0 4 ).
в каждую из двух пробирок помещают по
0,5 мл раствора А и 0,5 мл раствора сравнения
стрептодорназы, содержащеro 10 МЕ/мл, в иMи
дазольном буферном растворе рН 6,5 Р. В одну
пробирку тотчас прибавляют 3 мл раствора кис
лоты хлорной (25 r/л НСI0 4 ). Образуется осадок.
Центрифуrируют и собирают надосадочную
жидкость (а). Вторую пробирку наrревают при
температуре 37 ос в течение 15 мин, прибав
ляют 3 мл раствора кислоты хлорной (25 r/л
НСI0 4 ), центрифуrируют и собирают надосадоч
ную жидкость (Ь). Оптическая плотность Haдo
садочной жидкости (Ь) при длине волны 260 нм
должна быть не менее 0,15. в качестве компен
сационноro раствора используют надосадочную
жидкость (а).
Натрия дезоксихолат. С24НЗ9Nа04'
(М м. 414,6). 1131800. [302 95A]. Натрия 3a.12a
диrидрокси 5rз холан 2 4 oaT.
Натрия диrидрофосфат. 1080100.
[1 0028 24 7].
См статью Натрия ди2идрофосфат ди2и
драт.
Натрия диrидрофосфат безводный.
NaH P04' (М м. 120,0). 1080200. [7558 80 7].
Белый или почти белый порошок. rиrроско
пичен.
Хранят в воздухонепроницаемом контей
мере.
Натрия диrидрофосфат моноrидрат.
NaH2P04'Hp. (М м. 138,0). 1080300. [10049 21 5].
Белые или почти белые слеrка расплываю
щиеся на воздухе кристаллы или rранулы. Леr
корастворим в воде, практически нерастворим в
96 % спирте.
Хранят в воздухонепроницаемом контейнере.
Натрия дитионит. Na 2 Sp4' (М м. 174,1).
1080400. [7775 14 6].
Белый или серовато белый кристалличе
ский порошок; на воздухе окисляется. Очень
леrко растворим в воде, малорастворим в 96 %
спирте.
Хранят в воздухонепроницаемом контейнере.
Натрия диэтилдитиокарбамат.
C 5 H,oNNaS 2 '3Hp. (М. м. 225,3). 1080000.
[20624 25 3] .
Белые или почти белые или бесцветные
кристаллы. Леrкорастворим в воде, paCTBO
рим в 96 % спирте. Водный раствор бесцвет
ный.
Натрия додецилсульфат. 1080500. [151 21 3].
См. статью Натрия лаурилсульфат за ис
ключением содержания, которое должно быть
не менее 99,0 %.
Натрия йодид. Nal. (М м. 149,9). 1081800.
[7681 82 5].
Кристаллический порошок белоrо цвета или
бесцветные кристаллы. rиrроскопичен. Очень
леrко растворим в воде, леrкорастворим в 96 %
спирте.
Потеря в массе при высушивании (2.2.32).
Не более 3,0 %. 1,00 r вещества сушат при TeM
пературе 105 ос в течение 3 ч.
Хранят в защищенном от света месте.
Натрия карбонат. 1079200. [6132 02 1].
См. статью Натрия карбонат дека2идрат.
Натрия карбонат моноrидрат. Nа 2 СО з 'Н,О.
(М. м. 124,0).
Содержит не менее 83,0 % и не более 87,5 %
Nа 2 СО з .
Белый кристаллический порошок или бес
цветные кристаллы. Леrкорастворим в воде.
практически нерастворим в 96 % спирте.
Хранят в плотно укупоренном контейнере.
Натрия карбонат безводный. Nа 2 СО з .
(М м. 106,0). 1079300. [497 19 ]. Динатрия Kap
бонат.
Белый или почти белый порошок. rиrроско
пичен. Леrкорастворим 8 воде.
После наrревания при температуре около
300 ос потеря в массе не должна превышать 1 %
от массы.
Хранят в воздухонепроницаемом контей
нере.
670
rосударственная фармакопея Республики Беларусь
Натрия карбоната раствор. 1079301.
Раствор 106 r/л натрия карбоната безво
дН020 р.
Натрия карбоната раствор Р1. 1079302.
Раствор 20 r/л натрия карбоната безводно
20 Р В 0,1 М растворе натрия 2идроксида.
Натрия кобал ьти нитрит. Nа з [Со(N0 2 )s].
(М м. 403,9). 1079700. [13600 98 1]. Три натрия
rексанитрокобальтат (111).
Оранжево желтый порошок. Леrкораство
рим в воде, малорастворим в 96 % спирте.
Натрия кобальтинитрита раствор.
1079701.
Раствор 100 r/л натрия кобальтинитри
та Р. rотовят непосредственно перед использо
ванием.
Натриялаурилсульфат. 1081900. [151 21 3].
См. статью Натрия лаурилсульфат.
Натрия метабисульфит. 1082000. [7681 57 -4].
См. статью Натрия метабисульфит.
Натрия метансульфонат. СНзSОзNа.
(М м. 118,1). 1082100. [2386 57A].
Белый или почти белый кристаллический
порошок. rиrроскопичен.
Хранят в воздухонепроницаемом контей
нере.
Натрия молибдат. Na 2 Mo0 4 '2H 2 0.
(М м. 242.0). 1082200. [10102AO 6]. Динатрия
молибдат диrидрат.
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Леrкорас
творим в воде.
Натрия нафтохинонсульфонат. C 10 H 5 Na0 5 S.
(М м. 260,2). 1082300. [521 24A]. Натрия
1 ,2 нафтохинон-4 сульфонат.
Кристаллический порошок от желтоro до
оранжево желтоro цвета. Леrкорастворим в
воде, практически нерастворим в 96 % спирте.
Натрия нитрат. NаNО з . (М м. 85,0). 1082400.
[76З1 99 4].
Белый или почти белый порошок или rpa
нулы или бесцветные прозрачные кристаллы,
расплывающиеся на воздухе. Леrкорастворим в
воде, малорастворим в 96 % спирте.
Хранят в воздухонепроницаемом контей
нере.
Натрия нитрит. NaN0 2 . (М м. 69,0). 1082500.
[7632 00 0].
Содержание: не менее 97,0 % NaN02"
Белый или почти белый rранулированный
порошок или слеrка желтоватый кристалличе
ский порошок. Леrкорастворим в воде.
Количественное определение. 0,100 r Be
щества растворяют в 50 мл воды Р, прибавляют
50,0 мл 0,02 М раствора калия пермаН2аната,
15 мл кислоты серной разведенной Р, 3 r калия
йодида Р и титруют 0,1 М натрия тиосульфата,
используя в качестве индикатора 1 мл раствора
крахмала Р, прибавляя еro в конце титрования.
1 мл 0,02 М раствора калия пермаН2аната
соответствует 3,450 Mr NaN0 2 .
Натрия нитрита раствор. 1082501.
Раствор 100 r/л натрия нитрита Р. rотовят
непосредственно перед использованием.
Натрия нитропруссид. Na 2 [Fe(CNMNO)]-2Нр.
(М м. 298,0). 1082600. [13755 38 9]. Натрия пен
чщианонитрозилферрат (111) диrидрат.
Красновато коричневый порошок или кри
сталлы. Леrкорастворим в воде, малорастворим
в 96 % спирте.
# Натрия нитропруссида окисленноrо
раствор. 1 О r натрия нитропруссида Р раст
воряют в 100 мл воды Р, прибавляют 5 мл paCT
вора 1 :30 калия пермаН2аната Р и 2 мл paCT
вора 100 r/л натрия 2идроксида Р. Полученную
смесь фильтруют и выдерживают в течение
24 ч. Срок roдности раствора 2 мес.
Натрия оксалат. C 2 Nap4' (М м. 134,0).
1082900. [62 76 0].
Белый или почти белый порошок. Растворим
в воде, практически нерастворим в 96 % спирте.
Натрия октансульфонат. С в Н 17 NаО з S.
(М м. 216,3). 1082700. [5З24 84 5].
Содержание: не менее 98,0 % С в Н 17 NаО з S.
Белый или почти белый кристаллический
порошок или чешуйки. Леrкорастворим в воде,
растворим в метаноле.
Оптическая плотность (2.2.25). Оптическая
плотность раствора 54 r/л натрия октансульфона
та при длине волны 200 нм должна быть не более
0,10; при длине волны 250 нм не более 0,01.
Натрия октилсульфат. C a H 17 Na0 4 S.
(М м. 232,3). 1082800. [142 31A].
Белый или почти белый кристаллический
порошок. Леrкорастворим в воде, растворим в
метаноле.
Натрия пентансульфонат. С 5 Н 11 NаО з S.
(М м. 174,2). 1083000. [22767 49 3].
Белое или почти белое твердое кристалли
ческое вещество. Растворим в воде.
Натрия перйодат. Na10 4 . (М м. 213,9).
1083200. [7790 28 5]. Натрия метаперйодат.
Содержание: не менее 99,0 % Na10 4 .
Белый или почти белый кристаллический
порошок или кристаллы. Растворим в воде и ми
неральных кислотах.
4.1. 1. Реактивы
671
Натрия перйодата раствор. 1083201.
1,07 r натрия перйодата Р растворяют в
воде р, прибавляют 5 мл кислоты серной раз
веденной Р и доводят объем раствора водой Р
до 100,0 мл.
rотовят непосредственно перед использо
ванием.
Натрия перхлорат. NaCI0 4 'Hp. (М м. 140,5).
1083100. [7791 07 3].
Содержание: не менее 99,0 % NaCI0 4 'Hp.
Белые или почти белые кристаллы, расплы
вающиеся на воздухе. Очень леrко растворим в
воде.
Хранят в плотно закрытом контейнере.
Натрия пикрата щелочной раствор.
1083300.
Смешивают 20 мл раствора кислоты пи
криновой Р и 1 О мл раствора 50 r/л натрия 2и
дроксида Р, доводят водой Р до объема 100 мл.
Срок roдности 2 сут.
Натрия пирофосфат. NapP7'10Hp.
(М м. 446,1). 1083600. [13472 36 1]. Тетранатрия
дифосфат декаrидрат.
Бесцветные, слеrка выветривающиеся кри
сталлы. Леrкорастворим в воде.
Натрия родизонат. C 6 Nap6' (М м. 214,0).
1122300. [523 21 7]. [(3,4,5,6 Тетраоксоциклоrекс
1 eH 1 ,2 илен )диокси ]динатрий.
Фиолетовые кристаллы. Растворим в воде с
образованием оранжево--желтоro раствора. Раство--
ры нестабильны, их roтовят в день использования.
Натрия салицилат. 1083700. [54 21 7].
См. статью Натрия салицилат.
Натрия сульфат безводный. 1083800.
[7757 82 6].
Прокаливают при температуре от 600 ос до
700 ос натрия сульфат безводный, отвечающий
требованиям статьи Натрия сульфат безво
дный.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. Определение проводят при TeM
пературе 130 ОС.
Натрия сульфат декаrидрат. Na 2 S0 4 '10H 2 0.
(М м. 322,2).
См. статью Натрия сульфат дека2идрат.
# Натрия сульфата насыщенный paCT
вор.
К 60 r натрия сульфата Р Jlрибавляют
100 мл воды Р и выдерживают при перемешива
нии в течение 24 ч.
# Натрия сульфацетамид. Сульфацил на
трий.
См. статью Сульфацетамид натрия.
Натрия сульфид. Na 2 S'9Hp. (М м. 240,2).
1083900. [1313 84 4]. Динатрия сульфид нонаrи
драт.
Бесцветные, быстро желтеющие кристаллы,
расплывающиеся на воздухе. Очень леrко pac
творим в воде.
Хранят в воздухонепроницаемом контейнере.
Натрия сульфида раствор. 1083901.
12 r натрия сульфида Р растворяют при Ha
rревании в 45 мл смеси растворителей вода Р
2лицерuн (85 %) Р (1 0:29, об/об), охлаждают и дo
водят до объема 100 мл этим же растворителем.
Раствор должен быть бесцветным.
Натрия сульфида раствор Р1. 1083902.
rотовят одним из перечисленных ниже Me
тодов.
1) 5 r натрия сульфида Р растворяют в
смеси из 1 О мл воды Р и 30 мл 2лицерина Р.
2) 5 r натрия сульфида Р растворяют в
смеси из 30 мл воды Р и 90 мл 2лицерина Р.
Делят раствор на две равные части. Одну часть
насыщают сероводородом Р с использованием
охлаждения. Смешивают обе части раствора.
Хранение: в заполненном доверху контей
нере в защищенном от света месте. Используют
в течение 3 месяцев.
Натрия сульфит. 1084000. [27610 45 3].
См. статью Натрия сульфит 2епта2идрат.
Натрия сульфит безводный. 1084100.
[7757 83 7].
См. статью Натрия сульфит безводный.
Натрия тартрат. C4H4Nap6'2Hp. (М м. 230,1).
1084200. [6106 24 7]. Динатрия (2R,3R) 2,3
диrидроксибутандиоат диrидрат.
Белые или почти белый кристаллы или rpa
нулы. Очень леrко растворим в воде, практиче
ски нерастворим в 96 % спирте.
Натрия тетраrидроборат. NaBH 4 . (М м. 37,8).
1146900. [16940 66 2]. Натрия борrидрид.
Бесцветные rиrроскопичные кристаллы. Леr
корастворим в воде, растворим в этаноле. Раз
лаrается при высокой температуре или в присут
ствии кислот либо солей некоторых металлов с
образованием буры и водорода.
Хранят в воздухонепроницаемом контейнере.
Натрия тетраrидробората восстанавли-
вающий раствор. 1146901.
100 мл воды Р помещают в мерную колбу
вместимостью 500 мл, содержащую маrнитную
мешалку, добавляют 5,0 r пеллет натрия 2и
дроксида Р и 2,5 r натрия тетра2идробора
та Р. Перемешивают до полоноrо растворения
содержимоrо колбы, доводят водой Р до объема
500,0 мл и снова перемешивают. rотовят непо
средственно перед использованием.
672
rосударственная фармакопея Республики Беларусь
Натрия тетрадейтеродиметилсилапента-
ноат. C 6 H 9 2H 4 Na0 2 Si. (м. м. 172,З). 1084300. TSP.
Натрия (2,2,3,3 тетрадейтеро )--4+диметил4 сила
пентаноат.
Степень дейтерирования не менее 99 %.
Белый или почти белый кристаллический
порошок. Леrкорастворим в воде, в этаноле и в
метаноле.
Температура плавления: около 300 ОС.
Вода и дейтерия оксид: не более 0,5 %.
Натрия тетрафенилборат. NaB(C 6 H 5 )4'
(м. м. 342,2). 1084400. [143 66 8].
Белый или слеrка желтоватый объемный по
рошок. Леrкорастворим в воде и в ацетоне.
Натрия тетрафенилбората раствор.
1084401.
Раствор 1 О r/л натрия тетрафенилбора
та Р. Если необходимо, перед использованием
фильтруют.
Срок roдности 7 сут.
Натрия тиоrликолят. С 2 Н з Nа0 2 S.
(м. м. 114,1). 1084500. [367 51 1]. Натрия Mep
каптоацетат.
Белый или почти белый rранулированный
порошок или кристаллы. rиrроскопичен. Леrко
растворим в воде и в метаноле, малорастворим
в 96 % спирте.
Хранят в воздухонепроницаемом контейнере.
Натрия тиосульфат. 1084600. [10102 17 7].
См. статью Натрия тиосульфат.
Натрия тиосульфат безводный. Nа 2 S 2 О з .
(м. м. 158,1). 1180700. [7772 98 7]. Динатрия ти
осульфат.
Содержание: не менее 98,0 %.
Натрия флуоресцеинат. C2oH1ONa205'
(м. м. 376,3). 1080700. [518 47 8). Показатель
Шульца NQ 880. Цветной индекс NQ 45350. Флу
оресцеин натрия. Динатрия 2 (3 оксо 6 оксидо
3Н ксантен 9 ил)бензоат.
Оранжево красный порошок. Леrкораство
рим в воде. Водные растворы имеют интенсив
ную желтовато зеленую флуоресценцию.
Натрия формиат. CHNa0 2 . (м. м. 68,0).
1122200. [141 53 7).
Белый или почти белый кристаллический
порошок или расплывающиеся rранулы. PaCTBO
рим в воде и в rлицерине, малорастворим в 96 %
спирте.
Температура плавления: окоrю 253 ОС.
Натрия фосфат додекаrидрат. Nap04 '12Н 2 О.
(м. м. 380,1). 1094300. [10101 89 O]. Тринатрия
фосфат додекаrидрат.
Бесцветные или белые или почти белый
кристаллы. Леrкорастворим в воде.
# Натрия фосфорномолибдат.
NaJP(Mop1O)JxHp. (м. м. 1891,2 безводный)).
[131 3 30 0]. Натрий фосфорномолибденовокис-
лый.
Мелкокристаллический порошок.
Натрия фтор ид. NaF. (м. м. 41,99). 1080800.
[7681 49 4].
Содержит не менее 98,5 % и не более
100,5 % NaF в пересчете на сухое вещество.
Порошок белоro цвета или бесцветные кри
сталлы. Растворим в воде, практически HepaCT
ворим в 96 % спирте.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. 1,000 r испытуемоro образца
сушат при температуре 130 ос в течение 3 ч.
Натрия хлорид. 1079500. [7647 14 5).
См. статью Натрия хлорид.
Натрия хлорида раствор. 1079502.
Раствор 20 % (м/м) натрия хлорида Р.
Натрия хлорида насыщенный раствор.
1079503.
1 часть натрия хлорида Р смешивают с 2 ча
стями воды Р, периодически встряхивают и BЫ
держивают до оседания твердых частиц. Перед
использованием раствор декантируют и филь
труют, если необходимо.
Натрия цетостеарилсульфат. 1079400.
Аморфный или кристаллический порошок
белоro или бледно желтоro цвета. Растворим
в rорячей воде с образованием опалесциру
ющеro раствора, практически нерастворим в
холодной воде, умеренно растворим в 96 %
спирте.
Натрия цетостеарилсульфат смесь
натрия цетилсульфата (С'6НззNаО4S' М. м. 344,5)
и натрия стеарилсульфата (С,вНЗ7NаО4S'
М. м. 372,5). Содержит не менее 90,0 % натрия
цетостеарилсульфата и не менее 40,0 % натрия
цетилсульфата оба в пересчете на сухое Be
щество. Может прилаrаться подходящий буфер
ный раствор.
Натрия цитрат. 1079600. [6132 04 3].
См. статью Натрия цитрат.
Натрия эдетат. 1080600. [6381 92 6].
См. статью Динатрия эдетат.
Нафталин. С,ОН в ' (м. м. 128,2). 1057100.
[91 20 3].
Белые или почти белые кристаллы. Прак
тически нерастворим в воде, растворим в 96 %
спирте.
Температура плавления: около 80 ОС.
Нафталин, используемый для жидкостной
сцинтuлляции, должен быть cooтвeтcтвy
ющей степени чистоты.
4.1.1. Реактивы
673
Нафтарзон. C16H11AsN2Nap10S2" (М м. 576,3).
1121400. [3688 924]. Торин.Динатрия4 [(2 арсоно
фенил)азо] 3 rидроксинафталин 2, 7 дисульфонат.
Красный порошок. Растворим в воде.
Нафтарзона раствор. 1121401.
Раствор 0,58 r/л нафтарзона Р.
Испытание на чувствительность. К 50 мл
96 % спирта Р прибавляют 20 мл воды Р, 1 мл
0,05 М раствора кислоты серной, 1 мл раствора
нафтарзона и титруют 0,025 М раствором бария
перхлората. Происходит переход окраски paCT
вора от оранжево желтой к оранжево розовой.
Хранят в защищенном от света месте. Срок
хранения 7 сут.
Нафтиламин. C 10 H g N. (М м. 143,2). 1057700.
[134 32 7]. 1 Нафтиламин.
Белый или почти белый кристаллический поро
шок, под действием света и воздуха розовеет. Ma
лорастворим в воде, леrкорастворим в 96 % спирте.
Температура плавления: около 51 ос.
Хранят в защищенном от света месте.
1 Нафтилуксусная кислота. С12Н1002'
(М М. 186,2). 1148400. [86 87 3]. (Нафтален 1
ил)уксусная кислота.
Кристаллический порошок от белоro до жел
тоro цвета. Очень мало растворим в воде, леrко
растворим в ацетоне.
Температура плавления: около 135 ос.
Нафтилэтилендиамина диrидрохлорид.
C12H16C12N2' (М М. 259,2). 1057800. [1465 254].
N ( 1 Нафтил )этилендиамина диrидрохлорид.
Может содержать кристаллизационный метанол.
Белый или желтовато белый порошок. Pac
творим в воде, малорастворим в 96 % спирте.
Нафтилэтилендиамина диrидрохлорида
раствор. 1057801.
0,1 r нафтилэтилендиамина ди2идрохло
рида Р растворяют в воде Р и доводят до объема
100 мл этим же растворителем.
rотовят непосредственно перед использо
ванием.
а Нафтол. С 10 Нр. (М М. 144,2). 1057300.
[90 15 3]. 1 Нафтол.
Белый или почти белый кристаллический п<r
рошок или бесцветные или белые или почти белые
кристаллы, темнеющие под действием света. Ma
лорастворим в воде, леrкорастворим в 96 % спирте.
Температура плавления: около 95 ос.
Хранят в защищенном от света месте.
а Нафтола раствор. 1057301.
0,10 r а нафтола Р растворяют в 3 мл раст
вора 150 r/л натрия 2идроксида Р и доводят
водой Р до объема 100 мл.
rотовят непосредственно перед использо
ванием.
В5.Зак 1712
# а Нафтола раствор Р1.
2 r а нафтола Р растворяют в 1 О мл 96 %
спирта Р.
Хранят в защищенном от света месте при
температуре от 15 ос до 25 ос не более 7 сут.
J3-Нафтол. СюН в О. (М М.. 144,2). 1057400.
[1 35 19 3]. 2 Нафтол.
Белые или слеrка розовые пластинки или
кристаллы. Очень мало растворим в воде, очень
леrко растворим в 96 % спирте.
Температура плавления: около 122 ос.
Хранят в защищенном от света месте.
J3 Нафтола раствор. 1057401.
5 r свежеперекристаллизованноrо J3 Haqb
тола р растворяют в 40 мл раствора натрия 2и
дроксида разведеНН020 Р и доводят водой Р до
объема 100 мл.
rотовят непосредственно перед использо
ванием.
J3 Нафтола раствор Р1. 1057402.
3,0 Mr нафтола Р растворяют в 50 мл Kиc
лоты серной Р и доводят до объема 100,0 мл
этим же растворителем.
rотовят непосредственно перед использо
ванием.
Нафтолбензеин. С27Н200з' (М М. 392,5).
1057600. [6948 88 5]. а Нафтолбензеин.
Фенилбис( 4 rидроксинафтил )метанол.
Коричневато красный порошок или блестя
щие коричневато черные кристаллы. Практи
чески нерастворим в воде, растворим в 96 %
спирте и кислоте уксусной ледяной.
Нафтолбензеина раствор. 1057601.
Раствор 2 r/л нафтолбензеина Р в кислоте
уксусной безводной Р.
Испытание на чувствительность. К 50 мл
кислоты уксусной ледяной Р прибавляют 0,25 мл
раствора нафтолбензеина. Появляется коричне
вато желтое окрашивание, которое должно перей
ти в зеленое при прибавлении не более 0,05 мл
О, 1 М раствора кислоты хлорноЙ
#а Нафтолфталеин. С 2в Н 1 Р4' (М М. 418.4).
[596 0 1 O]. 3,3 Бис( 4 rидроксинафталенил)
1 (3Н) изобензофуранон.
Мелкокристаллический порошок от зелено
вато сероro до коричневоro или cepoвaTO Kpac
ноro цвета. Практически нерастворим в воде,
леrкорастворим в 96 % спирте и в ледяной YK
сусной кислоте, малорастворим в бензоле.
# а Нафтолфталеина раствор.
0,1 r а нафтолфталеина Р растворяют
в 50 мл 96 % спирта Р и доводят водой Р до
объема 100 мл.
Изменение окраски. От рН 7,4 (желтовато
розовое окрашивание) до рН 8,6 (зеленовато си
нее окрашивание).
674
rосударственная фармакопея Республики Беларусь
# Нейтральный красный. C15H16N4 .НС'
(М м. 288,78). 3 Амино 7 диметиламино 2 ме
тилфеназина rидрохлорид.
Черный или черно зеленый порошок или
кристаллы. Леrкорастворим в воде.
# Нейтральноrо KpacHoro раствор.
0,1 r нейтраЛЬН020 краСН020 Р растворяют в
воде Р и доводят до объема 100 мл этим же раст
ворителем.
Изменение окраски. От рН 6,8 (красное
окрашивание) до рН 8,0 (желтое окрашивание).
Нерилацетат. С12Н2002' (М м. 196,3).
1108000. [141 12 8]. (Z) 3,7 Диметилокта
2,6 диенилацетат.
Бесцветная маслянистая жидкость.
d;g: около 0,907.
20
по: около 1,460.
Температура кипения 25 : 134 ОС.
Нерилацетат. используемый в 2азовой
хромат02рафии, должен выдерживать следу
ющее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Поме
ранца цветков масло.
Испытуемый раствор. Испытуемый образец.
Содержание нерилацетата, рассчитанное
методом нормализации, должно быть не менее
93,0 %.
транс Неролидол. С 15 Н 26 О. (М м. 222,4).
1107900. [40716 66 3]. 3,7,11 Триметилдодека
1 ,6, 10 триен 3 ол.
Слеrка желтая жидкость с леrким запахом
лилии или ландыша. Практически нерастворим в
воде и в rлицерине, смешивается с 96 % спиртом.
d 20
20: около 0,876.
20
по : около 1,479.
Температура кипения 12 : от 145 ос до 146 ОС.
транс Неролидол, используемый в 2азовой
хромат02рафии, должен выдерживать следу
ющее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Поме
ранца цветков масло.
Испытуемый раствор. Испытуемый образец.
Содержание транс неролидола, рассчи
танное методом нормализации, должно быть не
менее 90,0 %.
# Несслера реактив. См. Калия тетрайо
домеркурата щелочной раствор Р
Никель-алюминиевый сплав. 1058100.
Содержит от 48 % до 52 % алюминия
(AI, А. м. 26,98) и от 48 % до 52 % никеля (Ni,
А. м. 58,70).
Перед использованием измельчают до мел
коro порошка (180) (2.9.12).
Практически нерастворим в воде, paCTBO
рим в минеральных кислотах.
Никель алюминиевый сплав, свободный
от rалоrенов. 1118100.
Содержит от 48 % до 52 % алюминия
(AI, А. м. 26,98) и от 48 % до 52 % никеля (Ni,
А. м. 58,70).
Мелкий порошок сероro цвета. Практически
нерастворим в воде, растворим в минеральных
кислотах с образованием солей.
Хлориды. Не более 0,001 % (10 ррm). 0,400 r
испытуемою образца растворяют в смеси из 67
объемов кислоты серной Р и 33 объемов кисло
ты азотной разведенной Р. Полученный paCT
вор упаривают почти досуха, остаток растворяют
в 20,0 мл этой же смеси кислот (раствор А). К по
лови не объема раствора А прибавляют 1,0 мл
0,1 М раствора серебра нитрата. Выдержива
ют в течение 15 мин, фильтруют и прибавляют к
фильтрату 0,2 мл раствора натрия хлорида (co
держащеro 10 MKr ионов хлорида в 1 мл). Через
5 мин полученный раствор по степени MYTHO
сти должен превышать раствор, приroтовлен
ный с использованием второй части раствора А
и 1,0 мл 0,1 М раствора серебра нитрата.
Никеля сульфат. NiS0 4 '7Hp. (М м. 280,9).
1058000. [10101 98 1]. Никеля сульфат rептаrи
драт.
Зеленый кристаллический порошок или кри
сталлы. Леrкорастворим в воде, малорастворим
в 96 % спирте.
Никеля хлорид. NiCI 2 . (М м. 129,6). 1057900.
[7718 54 9]. Никеля хлорид безводный.
Желтый кристаллический порошок. Очень
леrко растворим в воде, растворим в 96 %
спирте. Сублимируется в отсутствие воздуха
и леrко абсорбирует аммиак. Водный раствор
имеет кислую реакцию.
Никотинамид-аденина динуклеотид.
C 21 H 27 NP1PT (М м. 663). 1108100. [53 84 9]. NAD+.
Белый или почти белый порошок. Сильно rи
rроскопичен. Леrкорастворим в воде.
Никотинамид-аденина динуклеотида
раствор. 1108101.
40 Mr HиKoтиHaMид aдeHиHa динуклеоти
да Р растворяют в воде Р и доводят до объема
10 мл этим же растворителем.
rотовят непосредственно перед использо
ванием.
Нильский синийА. С 20 Н 21 NP5 S , (М м. 415,5).
1058200. [3625 57 8]. Показатель Шульца
NQ 1029. Цветной индекс NQ 51180. 5 Амино
9 (диэтила мино )бензо[ а]феноксазинилия rи
дросульфат.
Зеленый кристаллический порошок с бронзо
вым блеском. Умеренно растворим в 96 % спирте,
в кислоте уксусной ледяной и в пиридине.
Раствор 0,005 r/л в спирте (50 %, об/об) Р
имеет максимум поrлощения (2.2.25) при длине
волны 640 нм.
4.1.1. Реактивы
675
Нильскоrо синеrо А раствор. 1058201.
Раствор 10 r/л ниЛЬСК020 сине20 А Р в Kиc
лоте уксусной безводной Р.
Испытание на чувствительность. К 50 мл
кислоты уксусной безводной Р прибавляют
0,25 мл раствора нильскоrо синеro А. Раствор
синий. При прибавлении не более 0,1 мл 0,1 М
раствора кислоты хлорной появляется сине зе
леное окрашивание.
Изменение окраски. От рН 9,0 (синее OKpa
шивание) до рН 13,0 (красное окрашивание).
Нинrидрин. сgнрз,нр. (М м. 178,1).
1058300. [485A7 2]. 1 ,2,3 Индантрион моноrи
драт.
Белый или очень бледный желтый кристал
лический порошок. Растворим в воде и 96 %
спирте. #Ядовит.#
Хранят в защищенном от света месте.
Нинrидрина и олова (11) хлорида реактив.
1058301.
0,2 r ниН2идрина Р растворяют в 4 мл roря
чей воды Р, прибавляют 5 мл раствора 1,6 r/л
олова (11) хлорида Р, выдерживают в течение
30 мин, фильтруют и хранят при температуре от
2 ос до 8 ос.
Непосредственно перед использованием к
2,5 мл полученноro раствора прибавляют 5 мл
воды Р и 45 мл 2 пропанола Р.
Нинrидрина и олова (11) хлорида реак-
тив Р1. 1058302.
4 r ниН2идрина Р растворяют в 100 мл MOHO
этилов020 эфира этилеН2ликоля Р. Осторожно
встряхивают с 1 r смолы катионообменной Р (от
300 мкм ДО 840 мкм) и фильтруют (раствор А).
0,16 r олова (11) хлорида Р растворяют в 100 мл
буферН020 раствора рН 5,5 Р (раствор В).
Непосредственно перед использованием
смешивают равные объемы растворов А и В.
Нинrидрина раствор. 1058303.
Раствор 2 r/л ниН2uдрина Р в смеси кисло
та уксусная разведенная Р бутанол Р (5:95,
об/об).
Нинrидрина раствор Р1. 1058304.
1,0 r ниН2идрина Р растворяют в 50 мл 96 %
спирта Ри прибавляют 10 мл кислоты уксусной
ледяной Р.
Нинrидрина раствор Р2. 1058305.
3 r ниН2идрина Р растворяют в 100 мл paCT
90ра 45,5 r/л натрия метабисульфита Р.
Нинrидрина раствор РЗ. 1058306.
Раствор 4 r/л НИН2идрина Р в смеси кислота
уксусная безводная Р бутанол Р (5:95, об/об).
# Нипаrин. См. Метилпара2идроксибен
зоат р.
# Нипазол. См. Пропилпара2идроксибен
зоат р.
Нитрилотриуксусная кислота. С Б Н g NО 6 .
(М М. 191,1). 1137400. [139 13 9].
Белый или почти белый кристаллический
порошок. Практически нерастворим в воде и в
большинстве орrанических растворителей.
Температура плавления: около 240 ос с раз
ложением.
Нитроанилин. C 6 H 6 NP2' (М М. 138,1).
1058600. [100 01 6]. 4 Нитроанилин. #п Нитро
анилин.#
Ярко желтый кристаллический порошок.
Очень мало растворим в воде, умеренно paCTBO
рим в кипящей воде, растворим в 96 % спирте,
образует водорастворимые соли с сильными ми
неральными кислотами.
Температура плавления: около 147 ос.
Нитробензальдеrид. С]Н 5 NО з . (М м. 151,1).
1058700. [552 89 6]. 2 Нитробензальдеrид.
Желтые иrольчатые кристаллы. Малораст
ворим в воде, леrкорастворим в 96 % спирте,
леrколетучий с паром.
Температура плавления: около 42 ос.
Нитробензальдеrидная бумаrа. 1058701.
0,2 r нитробензальде2ида Р растворяют в
1 О мл раствора 200 r/л натрия 2идроксида Р.
Срок rодности раствора 1 ч. В полученный paCT
вор поrружают нижнюю половину полоски из
медленно фильтрующей бумаrи длиной 1 О см и
шириной 0, 1 см. Избыток реактива удаляют,
промокая полоску между двумя листами филь
тровальной бумаrи.
Используют в течение нескольких минут
после приroтовления.
Нитробензальдеrида раствор. 1058702.
0,12 r порошка нитробензальде2ида Р при
бавляют к 1 О мл раствора натрия 2идроксида
разведеНН020 Р, встряхивают в течение 1 О мин
и фильтруют.
rотовят непосредственно перед использо
ванием.
Нитробензилхлорид. С]НБСINО т (М М. 171,6).
1059000. [100 14 1]. 4 Нитробензилхлорид.
Бледно желтые кристаллы. Вызывает сле
зотечение. Практически нерастворим в воде,
очень леrко растворим в 96 % спирте.
Нитробензоилхлорид. С]Н 4 СINО з . (М м. 185.6).
1058900. [122 04 3]. 4 Нитробензоилхлорид.
Желтые кристаллы или кристаллическая
масса, разлаrающаяся в присутствии влажно
ro воздуха. Полностью растворяется в растворе
натрия rидроксида с образованием желтовато
оранжевоro окрашивания.
Температура плавления: около 72 ос.
676
rосударственная фармакопея Республики Беларусь
Нитробензол. C 6 H 5 N0 2 . (М М. 123,1).
1058800. [98 95 3].
Бесцветная или очень слабо желтая жид
кость. Практически нерастворим в воде, смеши
вается с 96 % спиртом и эфиром.
Температура кипения: около 211 ОС.
Динитробензол. К 0,1 мл нитробензола при
бавляют 5 мл ацетона Р, 5 мл воды Р и 5 мл
раствора натрия 2идроксида KOHцeHтpиpoвaH
Н020 Р и встряхивают. После разделения слоев
верхний слой должен быть почти бесцветным.
4-(4-Нитробензил)пиридин. C12H10N202'
(М М. 214,2). 1101900. [1083A8 3].
Желтый порошок.
Температура плавления: около 70 ос.
# Нитрованадомолибденовый реактив.
См. Нитромолибденованадиевый реактив Р.
Нитрозодипропиламин. C 6 H 14 Np. (М м. 130,2).
1099900. [621 64 7]. Дипропилнитрозамин.
Жидкость. Растворим в этаноле и в KOHцeH
трированных кислотах.
d;g: около 0,915.
Температура кипения: около 78 ОС.
Степень чистоты подходит для определения
хемилюминесценции.
Нитрозодипропиламина раствор. 1099901.
Вводят 78,62 r эта нола Р, прокалывая
инъекционной иrлой пробку сосуда, содержа
щеrо нитрозодипропиламин Р, разбавляют
этанолом Р в соотношении 1: 1 00 и помеща
ют по 0,5 мл в контейнеры с обжатыми крыш
ками.
Хранят в защищенном от света месте при
температуре 5 ОС.
Нитрометан. СН з NО 2 . (М М. 61,0). 1059700.
[75 52 5].
Прозрачная бесцветная маслянистая жид
кость. Малорастворим в воде, смешивается с
96 % спиртом.
d 20
20: 1, 1 32 до 1, 1 34.
20
по: 1,381 до 1,383.
Температурные пределы пере20нки
(2.2.11). От 100 ос до 103 ОС; должно переroнять
ся не менее 95 %.
Нитромолибденованадиевый реактив.
1060100.
Раствор А. 1 О r аммония молuбдата Р раст
воряют в воде Р, прибавляют 1 мл раствора aM
миака Р и доводят водой Р до объема 100 мл.
Раствор В. 2,5 r аммония ванадата Р paCTBO
ряют roрячей ваде Р, прибавляют 14 мл кислоты
азотной Р и доводят водой Р до объема 500 мл.
К 96 мл кислоты азотной Р прибавляют
100 мл раствора А и 100 мл раствора В и ДOBO
дят водой Р до объема 500 мл.
# Нитротерефталевая кислота, модифи-
цированная полиэтиленrликолем. BЫCOKO
молекулярное соединение полиэтиленrликоля
и диэпоксида, этерифицированноrо нитротере
фталевой кислотой. DB FFAP.
Неподвижная фаза для rазовой xpoMaTorpa
фии.
Нитporeтpaзолиевый синий. С4QН зо СI 2 N 1О О 6 .
(М М. 818). 1060000. [298 83 9). 3,3' (3,3' Ди
метокси 4,4' дифенилен )ди[2 ( 4 нитрофенил ) 5
фенил 2Н тетразолия] дихлорид. п Нитротетра
золиевый синий.
Кристаллы. Растворим в метаноле с образо
ванием прозрачноro раствора желтоro цвета.
Температура плавления: около 189 0 С с раз
ложением.
4-Нитрофенол. С 6 Н 5 NО з . (М м. 139,1).
1146400. [100 02 7]. п Нитрофенол.
Содержит не менее 95 % С 6 Н 5 NО з .
Бесцветный или слеrка желтый порошок,
умеренно растворим в воде и в метаноле.
Температура плавления: около 114 ОС.
Нитрофурантоин. 1099700. [67 20 9].
См. статью Нитрофурантоин.
(5-Н итро-2 -фур ил) метиле нди ацетат.
C 9 H 9 N0 7 . (М. М. 243,2). 1099800. [92 55 7]. Ни
трофурфуралдиацетат. 5 Нитрофурфурилиден
диацетат.
Желтые кристаллы.
Температура плавления: около 90 ОС.
Нитрохромовый реактив. 1059100.
0,7 r калия дихромата Р растворяют в Kиc
лоте азотной Р и доводят объем раствора той
же кислотой до 100 мл.
Нитроэтан. C 2 H 5 N0 2 . (М М. 75,1). 1059200.
[79 24 3].
Прозрачная бесцветная маCJiянистая жидкость.
Температура кипения: около 114 ОС.
Нордазепам.С 15 Н 11 СINр. (М м. 270,7).1060200.
[340 57 8] . 7 Хлор 2,3 диrидро 5 фенил 1 H 1 +
бензодиазепин 2 он.
Белый или бледно желтый кристаллический
порошок. Практически нерастворим в воде, Ma
лорастворим в 96 % спирте.
Температура плавления: около 216 ОС.
DL-Норлейцин. С 6 Н 1з N0 2 . (М м. 131,2). 1060300.
[61 6--8]. (RSr2 Аминorексановая кислота.
Блестящие кристаллы. Умеренно растворим
в воде и в 96 % спирте, растворим в кислотах.
Норпсевдоэфедрина rидрохлорид.
С g Н 1з NО'НCI. (М м. 187,7). [53643 20 2]. (1R,2Rr или
(1 S,2Sr2 Амино--1 нилпропанола rидроxnорид.
Кристаллический порошок. Растворим в воде.
Температура плавления: от 180 ОСдо 181 ОС.
4.1.1. Реактивы
677
Носкапина rидрохлорид. С22Н2зN07'НCI'Нр.
(М м. 467,9). 1060500. [912 60 7]. (3S)--6,7 Ди
метокси 3 [ (5R) 4 метоки 6 метил 5, 6,7, 8
тетраrидра 1 ,3 диоксоло[4,5 g]изоквинолин 5 ил]
изобензофуран 1 (3нtOH rидрохлорид.
Содержит не менее 98,5 % и не более 100,5 %
C22H24CIN07 в пересчете на сухое вещество.
Белый кристаллический порошок или бесцвет
ные кристаллы. rиrpoскопичен. Леrкорастворим
в воде и в 96 % спирте. Водные растворы слабо
кислые. При стоянии растворов может выпадать
осадок основания.
Температура плавления: около 200 ос с разло
жением.
Потеря в массе при высушивании (2.2.32). От
2,5 % до 6,5 %. 0,200 r испытуемоro образца сушат
при температуре 105 ОС.
Хранят в во:щухонепроницаемом контейнере в
защищенном от света месте.
Обесцвечивающий раствор. 1012202.
Смесь кислота уксусная ледяная Р Meтa
нол P вoдa Р(1:4:5, 06106/06).
# 9-Оксиксантен. См. Ксант2идрол Р
Окситетрациклина rидрохлорид.
C22H24NP9'HCI. (М м. 496,9). 1146500. [205846-0].
(4S,4аR,5S,5аR,6S,12аS) 4 (Диметиламино )
3,5,6,1 0,12, 12а rексаrидрокси 6 метил 1, 11
диоксо 1 ,4,4а,5,5а,6, 11 , 12а октаrидротетрацен 2
карбоксамид rидрохлорид.
Желтый кристаллический порошок. rиrро
скопичен. Леrкорастворим в воде, умеренно
растворим в 96 % спирте. При стоянии водные
растворы мутнеют из за образования осадка
окитетрациклина.
# 4-Окситолуол. См. Крезол Р
# 8-Оксихинолин. C g H 7 NO. (М м. 145,2).
Q Оксихинолин. Оксин.
Желтоватый мелкокристаллический порошок.
Октадекановая кислота. См. Стеариновая
кислота Р
Октадецил[3 [3,5-бис(1, 1-диметилзтил)-
4-rидроксифенил]пропионат]. СЗ5Н620з'
(М м. 530,9). 1060600. [2082 7 3]. Октадецил 3
(3,5 ди трет-бутил-4 rидроксифенил)пропионат.
Белый или слеrка желтоватый кристалличе
ский порошок. Практически нерастворим в воде,
очень леrко растворим в ацетоне и в reKcaHe, мал<r
растворим в метаноле.
Температура плавления: от 49 ос до 55 ОС.
Октаналь. СвН'60, (М м. 128,2). 1150400.
[124 13 0]. Октиловый альдеrид.
Маслянистая бесцветная жидкость. Практиче
ски нерастворим в воде.
d;O: 0,822.
86. За.. 17/2
п О: 1,419.
Температура кипения: 171 ОС.
Октаналь, используемый в 2азовой xpOMa
т02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Апель
синовое масло.
Содержание октаналя, рассчитанное MeTO
дом нормализации, должно быть не менее 99 %.
Октанол. СвН,Р. (М м. 130,2). 1060700.
[111 87 5]. 1 Октанол. Каприловый спирт.
Бесцветная жидкость. Нерастворим в воде,
смешивается с 96 % спиртом.
d;g: около 0,828.
Температура кипения: около 195 ОС.
3-0ктанон. С в Н 16 О. (М м. 128,2). 1114600.
[106 68 3]. Этилпентилкетон.
Бесцветная жидкость с характерным запахом.
d 20
20: около 0,822.
п;О: около 1,415.
Температура кипения: около 167 ОС.
3 OKтaHOH, используемый в 2азовой xpOMa
т02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Лаван
довое масло.
Испытуемый раствор. Испытуемое вещество.
Содержание 3 OKTaHOHa, рассчитанное
методом нормализации, должно быть не
менее 98,0 %.
Oкroксинол 10. С З4 Н 62 О'1 ( ). (М м. 647).
1 060800. [9002 93 1]. a [4 (1, 1,3,З TeTpaMe
тилбутил )фенил ] ro rидроксиполи ( оксиэтилен).
Прозрачная бледно желтая вязкая жид
кость. Смешивается с водой, с ацетоном и с
96 % спиртом, растворим в толуоле.
Хранят в воздухонепроницаемом контейнере.
Олеамид. С,вНЗ5NО. (М м. 281,5). 1060900.
(Z) Октадек 9 еноамид.
Желтоватый или белый порошок или rpa
нулы. Практически нерастворим в воде, очень
леrко растворим в метиленхлориде, растворим
в этаноле.
Температура плавления: около 80 ОС.
Оливковое масло. 1061000. [8001 25 0].
Оливковое масло масло, полученное Me
тодом холодноro прессования или друrим Me
ханическим методом из зрелых плодов О/еа
еиroраеа L.
Прозрачная желтая или зеленовато жел
тая жидкость с характерным запахом. Практи
чески нерастворимо в 96 % спирте, смешива
ется с петролейным эфиром (50 70 ОС). При
678
rосударственная фармакопея Республики Беларусь
охпаждении до температуры 1 О ос мутнеет, а
при температуре О ос превращается в масло
образную массу.
O
d;o: около 0,913.
Хранят в заполненных доверху контейне
рах, в защищенном от света месте при темпера
туре не выше 2tз "С
Олова (11) хлорид. SnCI 7 '2Hp. (М м. 225,6).
1085000. [1 0025 69 1 J. uлова дихлорид диrидрат.
Содержит не менее 97,0 % sпСI 2 '2Н 2 О.
Бесцветные кристаллы. Очень леrко paCTBO
рим в воде. лerкорастворим в 96 % спирте, в кис
лоте уксусной ледяной. в кислоте хлористоводо
родной разведенной и концентрированной.
Количествеhное определение. 0,500 r
олова (11) хлорида помещают в колбу с притертой
стекляннои пробкой . растворяют в 15 мл кислоты
хлористоводородноtl Р, прибавляют 1 О мл воды Р
и 5 мл хлороформа Р Быстро титруют 0,05 М
раствором калия йодата до обесцвечивания
хлороформноro слоя.
1 мл 0,05 М раствора калия йодата соот
ветствует 22,56 Mr SnCl o '2H 2 0.
Олова (11) хлорида раствор. 1085001.
20 r олова Р наrревают с 85 мл кислоты
хлорuстоводороднои Р до прекращения Bыдe
ления водорода и охлаждают.
Хранят раствор над избытком олова Р с за
щитой от воздуха,
Олова (11) хлорида раствор Р1. 1085002.
Непосредственно перед использованием
раствор олова (/1) хлорида Р разводят кислотой
хлористоводороднои раэведенной Р (1:1 О, об/об).
Олова (11) хлорида раствор Р2. 1085003.
К 8 r олова (т хлорида Р прибавляют 100 мл
раствора 20 % (06/0б I кислоты xпopиcтoвoдopoд
ной Р, встряхивают до растворения, при необходи
мости наrревая на водяной бане при температуре
50 ОС, и пропускают азот Р в течение 15 мин.
rотовят непосредственно перед использо
ванием.
Олово. Sn. (А м. 118,7), 1090800. [7440 31 5],
Серебристо белые rранулы, Растворимо в
кислоте хлористоводородной с выделением BO
дорода '
Мышьяк (2.4.2, метод А). Не более 0,001 %
(10 ррт). 0,1 r должен выдерживать испытание
на мышьяк.
# Оллпорта реактив. См. Диметилаfv1ино
бензальде2ида раствор Р6.
Орацетовый синий 2R. C20H'4Np2' (М М. 314,3),
1061100. [4395 65 7]. Цветной индекс N2 61110.
1 Амино--4 ( фениламино ) aHTpaцeH 9, 1 О дион.
Температура плавления: около 194 ос.
Орацетовый синий В. 1118600. Смесь
1 метиламино 4 анилинантрахинона
(С21Н'БN202: М М. 328,4) и 1 амино--4 анилин
антрахинона (C2oH'4NP2; М М. 314,3).
Сине фиолетовый порошок. Практически
нерастворим в воде, леrкорастворим в ацетоне
и в кислоте уксусной безводной.
Орцин. С7НР2'НР' (М м. 142,2). 1108700.
[6153 39 5]. 5 Метилбензол 1 ,3 диол моноrидрат.
Кристаллический порошок, чувствителен к свету.
Температура кипения: около 290 ОС.
Температура плавления: от 58 ос от 61 <'С.
Осмия (VIII) оксид. 0504' (М М. 254,2).
1061200. [20816 12 0]. Осмия тетраоксид.
Светло желтые иroльчатые кристаллы или
желтая кристаллическая масса. rиrроскопичен.
Чувствителен к свету, Растворим в воде и в 96 %
спирте.
Хранят в воздухонепроницаемом контейнере.
Осмия (VIII) оксида раствор. 1061201.
Раствор 2,5 r/л осмия (VIII) оксида в 0,05 М
растворе кислоты серноЙ
Палладий. Pd. (А м. 106,4). 1114700.
[7 440 05 3].
Металл серовато белоrо цвета. Растворим в
кислоте хлористоводородной .
Палладия хлорид. PdCl c ' (М м. 177 3).
1061500. [7647 10 1].
Красные кристаллы,
Температура плавления: от 678 ос до 680 'С.
Палладия хлорида раствор. 1061501.
1 r палладия хлорида Р растворяют 8 1 О мл
теплой кислоты хлористоводородной Р, полу
ченный раствор доводят смесью из равных объе
мов кислоты хлористоводородной разведен
ной р и воды Р до объема 250 мл.
Непосредственно перед использованием
раствор разводят двумя объемами воды Р.
Пальмитиновая кислота. С'6НЗР2' (М М. 256.4).
1061600. [57 10 3]. rексадекановая кислота.
Белые или почти белые кристаллические
чешуйки. Практически нерастворима в воде, леr
корастворима в roрячем 96 % спирте.
Температура плавления: около 63 ОС.
Хромат02рафuя. Тонкослойная xpOMaтo
rрафия (2.2.27), как указано в статье Хлорам
феникола пальмитат. На полученной xpOMa
TorpaMMe должно обнаружива'!ъся только одно
основное пятно.
ПанаксозидRЫ.С54Н9Р23'знр.(Мм.116З).
1127500. [41753--43 9]. (20S) 3 ди D rлюко
пира нозил 20 ди D rл юкопи ра ноз ил п ротопа
наксадиол. (20S) 3 [(2 0 D rлюко пи
ра н озил О r л юкоп и ра ноз ил)о кси] 2 o
4.1.1. Реактивы
679
[( 6. О f3 D rлюкоп и ранозил р D rлюкопи ранозил )
окси] 5а даммар 24 ен 12р ол. (20S) 3f3 [(2
o p D r л юкоп и ра нозил f3 D rлюкоп и ра ноз ил)
О кси] 20 [( 6 o B о rл ю ко пира ноз ил 13 D
rлюкопиранозил)окси] 4,4,8, 14 тетраметил 18
нор 5а холест 24 eH 12f3 ол. rинзенозид Rbl
Бесцветное твердое вещество. Растворим в
воде, в этаноле и в метаноле.
[а J O: +11,3. Используют раствор 1 О r!л в Me
таноле р.
Температура плавления: около 199 "С
Вода (2.5.12). Не более 6,8 %.
Количественное определение Жидкост
ная хроматоrрафия (2.2.29), как указано в статье
Же.ньшеня корни.
Испытуемый раствор. 3,0 Mr панаксозида
Rbl растворяют в 10.0 мл метанола Р
Содержание панаксозида Rbl рэссчитан
ное методом нормализации, ,СlОЛЖНС бы iЪ не
\i1eHee 95,0 %.
!lанаксозид Re. C,oHEP'e (M М 947.2).
1157800. [52286 !')9 6] (313.6(/, 12r3) 20 H) D
!люкопираНОЗИЛОКС1 ) 3.12 диrидроксидаммар
;),1 ен 6 ил 2 0 (f <;еОКСИ а L маннопиранозил)
Cj rпю!<оr.иранозид rинзенозид Re
5eclIВeTHoe твердое вещество РаСТRОРИМ в
Е'r'де, В 96 % спирте и в метаноле
ПанаксозидRf'С ;Н720'4'2НР (М м-Е(7) 1127700.
[52286 58 5]. (20S) 6 ()..[f3 D r люкопиранозил ( 1 2)
l:\ D rликопиранозид] даммар 24 ен 31}.6и.12r{.20
тетраол. rинзенозид Rf.
Бесцветное твердое вещество. Растворим в
воде, в этаноле и в метаноле.
[a] O: +12,8. Используют раствор 10 r/л Б Me
таноле Р.
Температура плавления: около 198 ос.
Панаксозид Rg1. С42Н72014'2Н20, (М М.
837). 1127600. [22427 39 O]. (20S) 6P D
r л ю ко пира нозил D rл ю ко пира ноз и л п рото п а
наксатриол. (20S) 6а,20 бис(f3 D r люкопира
нозилокси) 5а даммар 24 ен 3р, 1213 диол.
(20 S) 6a ,20 бис( 13 D rл юкоп ира нозилокси)
4.4,8, 14 тетраметил 18 нор 5а холест 24 ен
зр, 12f3 диол. rинзенозид Rg1.
Бесцветное твердое вещество. Растворим в
воде, в этаноле и в метаноле.
[a] O: +31,2. Используют раствор 10 r/л в Me
таноле Р.
Температура плавления: от 188 ОСдо 191 ос.
Вода (2.5.12). Не более 4,8 %.
Количественное определение. Жидкост
ная хроматоrрафия (2.2.29), как указано в статье
Корни женьшеня.
Испытуемый раствор. 3,0 Mr панаксозида
Rg1 растворяют в 10,0 мл метанола Р.
Содержание панаксозида Rg1, рассчитан
ное методом нормализации, должно быть не
менее 95,0 %.
Панкреатина порошок. 1061700.
Панкреатина порошок получают из свежих
или замороженных поджелудочных желез мле
копитающих. Содержит различные ферменты,
обладающие протолитическоi;; пиполитической
и амилолитической активностью. 1 Mr панкре
атина содержит не менее 1,0 ЕФЕ обшей прото
литической активности, 15 ЕФЕ липолитической
активности и 12 ЕФЕ амилолитической активно
сти.
Слеrка коричневый аморфный порошок. Ча
стично t)астворим в воде, практически HepaCTBO
рим в 96 % спирте.
Хранят в воздухонепроницаемом контейне
ре при температуре от 2 ос до 8 ос.
Папаверина rидрохлорид. 1061800.
[61 25 6].
См. статью Папаверина сIJ{ ОО)(ЛОР/Jд.
Парарозанилина rидрохпорид С,эН. ,CIN,.
(М. м. 323,8). 1062200. [ l=;q 61 91 Показа
тель Шульца NQ 779 Цветной индекс NQ 42500.
4 [Бис( 4 ами нофенил )метилен]циклоrексn
2.5 диениминия хлорид.
Синевато красный КРИСТRллический поро
шок. Малорастворим в воде, растворим 8 эта
ноле. Растворы в воде и в эrаноле имеют ИН
тенсивную красную окраску. растворы в кислоте
серной и кислоте хлористоводородной имеют
желтую окраску.
Температура плавления: около 'L70 ос с раз
ложением.
Парарозанилина обесцвечеН:-iЫЙ раст-
вор. 1062201.
0,1 r парарозанилина 2идрохлорида Р поме
щают в колбу с притертой стеклянной пробкой,
прибавляют 60 мл воды Р и раствора 1,0 r натрия
сульфита безводН020 Р, или 2,0 r натрия суль
фита Р, или 0,75 r натрия метабисульфита Р в
1 О мл воды Р, медленно при перемешивании при
бавляют 6 мл кислоты хлористоводородной раз
веденной Р, закрывают колбу пробкой и продолжа
ют перемешивание до растворения. Полученный
раствор доводят водой Р до объема 100 мл.
Раствор используют через 12 ч после при
rотовления.
Хранят в защищенном от света месте.
Парафин жидкий. См. Вазелиновое масло Р.
Парафин мяrкий, белый. 1062100. "Вазе
лин белый.#
Мяrкая мазеобразная масса сосотоящая из
уrлеводородов, полученных при переработке нефти,
отбеленная. Практически нерастворим в воде и в
96 % спирте. растворим в петролейном эфире, при
этом раствор иноrда слеrка опалесцирует.
# Парафин твердый.
См. статью Парафин твердый.
680
rосударственная фармакопея Республики Беларусь
Парацетамол. 1061900. [103 90 2].
См. статью Парацетамол.
Парацетамол, свободный от 4-аминофе-
нола. 1061901.
Парацетамол Р перекристаллизовывают из
воды Р и сушат в вакууме при температуре 70 ОС.
Процедуру повторяют до тех пор, пока парацета
мол не будет выдерживать следующее испыта
ние: 5 r высушенною парацетамола растворяют в
смеси из равных объемов метанола Р и воды Р и
доводят до объема 100 мл этой же смесью paCTBO
рителей. Прибавляют 1 мл свежеприroтовленною
раствора, содержащеro 1 О r/л натрия нитропрус
сида Р и 10 r/л натрия карбоната безводН020 Р,
перемешивают и выдерживают в течение 30 мин
в защищенном от света месте. Не должно появ
ляться синее или зеленое окрашивание.
Пенициллиназы раствор. 1062300.
10 r rидролизата казеина, 2,72 r калия ди
2идрофосфата Р и 5,88 r натрия цитрата Р
растворяют в 200 мл воды Р, доводят раствором
200 r/л натрия 2идроксида Р до рН 7,2 и разво
дят водой Р до объема 1000 мл.
0,41 r ма2ния сульфата Р растворяют в
5 мл воды Р, прибавляют 1 мл раствора 1,6 r/л
железа (11) аммония сульфата Р и доводят водой
р до объема 1 О мл.
Стерилизуют оба раствора наrреванием в
автоклаве, охлаждают, смешивают, распределя
ют тонкими слоями в конических колбах и куль
тивируют С Bacillus ce,eus (NCTC 9946). Bыдep
живают колбы при температуре от 18 ос до 37 ос
до явных признаков роста, а затем выдержива
ют при температуре от 35 ос до 37 ос в тече
ние 16 ч, постоянно встряхивая для обеспечения
максимальной аэрации. Центрифуrируют, Haдo
садочную жидкость стерилизуют методом MeM
бранной фильтрации. 1,0 мл раствора пеницил
линазы содержит не менее 0,4 микрокатал (что
соответствует rидролизу не менее 500 Mr бен
зилпенициллина до бензилпенициллиновой кис
лоты В час) при температуре 30 ос и рН 7, при
условии, что концентрация бензилпенициллина
не опускается ниже уровня, необходимою для
ферментноro насыщения.
Константа Михаэлиса для пенициллиназы
по бензилпенициллину в растворе пеницилли
назы составляет около 12 мкr/мл.
Стерильность (2.6.1). Должен выдержи
вать испытание на стерильность.
Хранят при температуре от О ОС дО 2 ос и ис
пользуют в течение 2 3 сут. Лиофилизирован
ный препарат хранят в запаянных ампулах в Te
чение нескольких месяце13.
Пентан. С 5 Н 1 2' (М М. 72,2). 1062500. [109 66 O].
Прозрачная бесцветная воспламеняющаяся
жидкость. Очень мало растворим в воде, смеши
вается с ацетоном и с этанолом.
d;g: около 0,63.
п;О: около 1,359.
Температура кипения: около З6 ОС.
Пентан, используемый в спектрофотоме
трии, должен выдерживать следующее допол
нuтельное испытание.
Оптическое пропускание (2.2.25) опреде
ляют, используя в качестве компенсационной
жидкости воду Р: не менее 20 % при длине
волны 200 нм; не менее 50 % при длине волны
210 нм; не менее 85 % при длине волны 220 нм;
не менее 93 % при длине волны 230 нм; не
менее 98 % при длине волны 240 нм.
1,2-Пентандиол. С 5 Н 1 Р2' (М м. 104,2).
1155800. [5343 92 O]. (2RS) Пентан 1 ,2 диол.
d;O: около 0,971.
п;О: около 1,439.
Температура кипения: около 201 ОС.
Пентанол. С 5 Н 1 р. (М М. 88). 1062600. [71-41 O].
1 Пентанол. #н Амиловый спирт.#
Бесцветная жидкость. Умеренно растворим
в воде, смешивается с 96 % спиртом.
п;О: около 1,410.
Температура кипения: около 137 ОС.
Пентаэритритилтетракис[З-(З,5-ди(1,1-
ди метил этил) -4-rидроксифе н ил)п ро-
пионат]. С7ЗН10вО12' (М М. 1178). 1062400.
[6683 19 8]. Пентаэритритилтетракис[3 (3,5 ди
трет бутил 4 rидроксифен ил)п роп ионат].
2 ,2' бис(rидроксиметил)пропан 1 ,3 диол
тетра кис[3 [3, 5 ди (1, 1 ди метил этил )
4 rидроксифенил ]]пропионат.
Кристаллический порошок от белою до
слеrка желтоro цвета. Практически нерастворим
в воде, очень леrко растворим 8 ацетоне, pac
творим в метаноле, малорастворим в reKcaHe.
Температура плавления: от 110 ос до 125 ОС.
а форма: от 120 ос до 125 ос.
l3 форма: от 110 ос до 115 ОС.
треm-Пентиловый спирт. С 5 Н 1 р. (М М. 88,1)
1062700. [75 85-4]. трет Амиловый спирт.
2 Метил 2 бутанол.
Летучая воспламеняющаяся жидкость. Леr
корастворим в воде, смешивается с 96 % спир
том и с rлицерином.
d;g: около 0,81.
Температурные пределы пере20нки (2.2. 11).
От 100 ос до 104 ОС; должно переroняться не
менее 95 %.
Хранят в защищенном от света месте.
Пепсина порошок. 1062800. [9001 75 6].
Пепсина порошок получают из слизистой
оболочки желудка свиней, poraтoro скота или
овец. Содержит желудочные протеиназы, aK
тивные в кислой среде (рН 1 5). Имеет акти
ность не менее 0,5 ЕФЕ/мr в пересчете на сухое
вещество.
4.1.1. Реактивы
681
Белый или слеrка желтый кристаллический
или аморфный порошок. rиrроскопичен. Pac
творим в воде, практически нерастворим в 96 %
спирте. Водные растворы имеют слабую опалес
ценцию и слабокислую реакцию.
Потеря в массе при высушивании (2.2.32).
Не более 5,0 %. из навеки 0,500 r испытуемою
образца сушат при температуре 60 ос над фос
фора (V) оксидом Р при давлении не выше 670 Па
в течение 4 ч.
Хранят в воздухонепроницаемом контейне
ре при температуре от 2 ос до 8 ОС.
Пероксидные тест-полоски. 1147800.
Используют roтовые полоски с COOTBeTCTBY
ющей шкалой от О ррm до 25 ррт пероксида.
Песок. 1075800.
Крупинки кремния диоксида белою или
слеrка сероватоro цвета с размером частиц от
150 мкм до 300 мкм.
Петролейный эфир. 1063100. [8032 32--4].
Петролейный эфир (50 70) ос.
Прозрачная бесцветная воспламеняющаяся
жидкость. Не флуоресцирует. Практически He
растворим в воде, смешивается с 96 % спиртом.
d;g: от 0,661 до 0,664.
Температурные пределы пере20нки (2.2.11).
От 50 ос до 70 ОС.
Петролейный эфир Р1. 1063101. Петро
лейный эфир (40 0) ос.
Должен выдерживать требования для пе
тролеЙН020 эфира Р со следующими изменени
ями:
d;g: от 0,630 до 0,656.
Температурные пределы пере20нки (2.2. 11).
От 40 ос до 60 ОС.
Не должен мутнеть при температуре О ОС.
Петролейный эфир Р2. 1063102. Петро
лейный эфир (30-------40) ОС.
Должен выдерживать требования для пе
тролеЙН020 эфира Р со следующими изменени
ями:
d;g: от 0,620 до 0,630.
Температурные пределы пере20нки (2.2. 11).
От 30 ос до 40 ос.
Не должен мутнеть при температуре О ОС.
Петролейный эфир РЗ. 1063103. Петро
лейный эфир (100 120) ОС.
Должен выдерживать требования для пеmро
лейН020 эфира Р со следующими изменениями:
d;g: от 0,659 до 0,671.
Температурные пределы пере20нки (2.2. 11).
От 100 ос до 120 ОС.
Вода (2.5.12). Не более 0,03 %.
Петролейный эфир Р4. 1063104. Петро
лейный эфир 80 100 ОС.
87 За.. 1712
Должен выдерживать требования для пе
тролеЙН020 эфира Р со следующими изменени
ями:
d;g: около 0,70.
Температурные пределы пере20нки (2.2. 11).
От 80 ос до 100 ОС.
Пикриновая кислота. С 6 Н з NР7' (М м. 229,1).
1065800. [88 89 1]. 2,4,6 Тринитрофенол.
Желтые призматические кристаллы или
пластинки. Растворима в воде и в 96 % спирте.
#Ядовита.#
Хранят увлажненной водой Р.
# Пикриновой кислоты насыщенный
раствор в этаноле.
6,25 r пикриновой кислоты Р заливают
100 мл этанола Р и выдерживают при частом
встряхивании в течение 24 ч.
Хранят в стеклянных контейнерах с притер
ты ми пробками в защищенном от света месте
вдали от оrня.
Пикриновой кислоты раствор. 1065801.
Раствор 1 О r/л пикриновой кислоты Р.
Пикриновой кислоты раствор Р1. 1065802.
К 100 мл насыщенноro раствора кислоты
пикриновой Р прибавляют 0,25 мл раствора
натрия 2идроксида концентрироваНН020 Р
а-Пинен. С'ОН'6' (М м. 136,2). 1130800.
[7785 70 8]. (1 R,5R) 2,6,6 Триметилбицикло[3.1.1]
rепт 2 ен.
Не смешивающаяся с водой жидкость.
d;g: около 0,859.
п O: около 1,466.
Температура кипения: от 154 ос до 156 ОС.
а Пинен, используемый в 2азовой xpoMamcr
2рафии, должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rазовая Xp(r
матоrрафия (2.2.28), как указано в статье Масло
цветков померанца, используя а пинен в
стве испытуемоro раствора.
Содержание а пинена, рассчитанное мer
дом нормализации, должно быть не менее 99.0 %.
j3-Пинен. С1ОН'6' (М м. 136.2). 1109000.
[19902 08 0].6,6 Диметил 2 метилeнбмцикno[3.1.1
rептан.
Бесцветная маслянистая 0CТb с запа
ХОМ, напоминающим скипидар. Практически He
растворим в воде, смешивается с 9б % спиртом.
d 20
20: около 0,867.
20
по: около 1,474.
Температура кипения: от 164 ос до 166 ОС.
fЗ Пинен, используемый в 2азовой xpOMaтo
2рафии, должен выдерживать следующее дo
полнительное испытание.
682
rосударственная фармакопея Республики Беларусь
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
цветков померанца, используя l3 пинен в каче
стве испытуемоro раствора.
Содержание l3 пинена, рассчитанное Me
тодом нормализации, должно быть не менее
99,0%.
Пиперазина rидрат. C 4 H 1O N 2 '6H 2 0.
(м. м. 194,2). 1065900. [142 63 2]. Пиперазина
rексаrидрат.
Содержит не менее 98,0 % и не более
101,0 % C4H,r,N2'6H70,
Бесцветные кристаллы. Леrкорастворим в
воде и в 96 % спирте.
Температура плавления: около 43 ОС.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
Пиперидин. CsH"N. (м. м. 85,2). 1066000.
[110 89A]. rексаrидропиридин.
Бесцветная или слеrка желтоватая жид
кость, имеет щелочную реакцию. Смешивается с
водой, с 96 % спиртом и с петролейным эфиром.
Температура кипения: около 106 ОС.
Пиперитон. С 1О Н,Р. (М м. 152,2). 1151200.
[89 81 6]. 6 Изопропил 3 метил циклоrекс 2 ен 1--он
Пирид-2-иламин. C 5 H 6 N 2 . (м. м. 94,1).
1073400. [504 29 0]. 2 Аминопиридин.
Крупные кристаллы. Растворим в воде и в
96 % спирте.
Температура кипения: около 210 ОС.
Температура плавления: около 58 ОС.
Пиридилазонафтол. С 15 Н 11 Np. (м. м.249,3).
1 073500. [85 85 8]. 1 (2 Пиридилазо ) 2 нафтол.
Порошок кирпично красноro цвета. Прак
тически нерастворим в воде, растворим в 96 %
спирте, в метаноле и в roрячих разведенных pac
творах rидроксидов щелочных металлов.
Температура плавления: около 138 ОС.
Пиридилазонафтола раствор. 1073501.
Раствор 1 r/л пиридилазонафтола Р в эта
ноле Р.
Испытание на чувствительность. К 50 мл
воды Р прибавляют 1 О мл ацетатН020 буфер
Н020 раствора рН 4,4 Р, 0,10 мл 0,02 М pacт
вора натрия эдетата и 0,25 мл раствора пи
ридилазонафтола. При прибавлении 0,15 мл
раствора 5 r/л меди (11) сульфата Р окраска
должна измениться от светло желтой до фиоле
товой.
Пиридин. C 5 H 5 N. (М м. 79,1).1073200. [110--86--1].
Прозрачная бесцветная rиrроскопичная
жидкость. Смешивается с водой и с 96 % спир
том. #Ядовит.#
Температура кипения: около 115 ОС.
Хранят в воздухонепроницаемом контейнере.
Пиридин безводный. 1073300. [110 86 1].
Пиридин Р сушат над натрия карбонатом
безводным Р, фильтруют и переroняют.
Вода (2.5.12). Не более 0,01 % (м/м).
Пировиноrpадная кислота. СзНРз, (М м. 88,1).
1109300. [127 17 З]. 2 Оксопропановая кис
лота.
Желтоватая жидкость. Смешивается с водой
и с этанолом.
d 20
20: около 1 ,267.
20
по: около 1,413.
Температура кипения: около 165 ОС.
Пироrаллол. С 6 Н 6 О з . (м. м. 126,1). 1073700.
[87 66 1]. Бензол 1 ,2,З триол.
Белые или почти белые кристаллы, под дей
ствием воздуха и света коричневеют. Очень леrко
растворим в воде и в 96 % спирте, малораство
рим в уrлерода дисульфиде. Под действием воз
духа водные растворы, а еще быстрее щелоч
ные растворы, приобретают коричневую окраску
вследствие абсорбции кислорода.
Температура плавления: около 131 ос.
Хранят в защищенном от света месте.
Пироrаллола щелочной раствор. 1073701.
0,5 r пиР02аллола Р растворяют в 2 мл воды,
свободной от У2лерода диоксида, Р.
12 r калия 2идроксида Р растворяют в 8 мл
воды, свободной от У2лерода дuоксида, Р.
Непосредственно перед использованием
смешивают оба раствора.
Пирокатехин. С 6 Н 6 О 2 . (М м. 110,1).1073600.
[120 80 9]. Бензол 1 ,2 диол.
Бесцветные или слабо желтые кристаллы.
Растворим в воде, в ацетоне и в 96 % спирте.
Температура плавления: около 102 ос.
Хранят в защищенном от света месте.
2-Пирролидон. C 4 H 9 NO. (м. м. 85,1).
1138000. [616A5 5]. Пирролидин 2 он.
При температуре выше 25 ос представляет
собой жидкость.
Смешивается с водой, с эта нолом и с эти
лацетатом.
d 25 16
4 : 1,1 .
Пицеин. С 14 Н 1 Рт (м. м. 298,3). 1130700.
[530 14 3J. Ч4 (J3 D r люкопиранозилокси )фенил J
этанон. п (Ацетилфенил ) I3 D rлюкопиранозид.
Температура плавления: от 194 ос до 195 ОС.
Плазма с пончженным содержанием
тромбоцитов. 106610Q
45 мл человеческой крови отбирают пласт
массовым шприцем вместимостью 50 мл, co
держащим 5 мл стерильноro раствора 38 r/л
натрия цитрата Р, и немедленно центрифу
rируют при 1500 9 при температуре 4 ос в Te
4. 1.1. Реактивы
683
чение 30 мин. Отбирают с помощью пластмас
совоro шприца верхние 2/3 надосадочноro слоя
плазмы и немедленно центрифуrируют с YCKO
рением 3500 9 при температуре 4 ос в течение
30 мин. Отбирают верхние 2/3 слоя жидкости и
быстро замораживают ее в необходимом коли
честве пластмассовых пробирок при температу
ре АО ос или ниже. Используют пластмассовое
оборудование или оборудование. обработанное
силиконом.
Плазмы субстрат. 1066200.
Плазму отделяют из человеческой или
бычьей крови, собирают в раствор 3В r/л натрия
цитрата Р, объем котороro составляет 1/9
объема плазмы или в раствор, содержащий 20 r/л
динатрия 2идроцитрата Р и 25 r/л 2ЛЮКОЗЫ Р,
объем котороro составляет 217 объема плазмы.
В первом случае субстрат rотовят в день сбора
крови; во втором случае субстрат rотовят в тече
ние 2 дней со дня сбора крови.
Хранят при температуре 20 ос.
Плазмы субстрат Р1. 1066201.
Для взятия и обработки крови используют
водоотталкивающее оборудование (изroтовлен
ное из подходящих пластмасс или стекла, обра
ботанноro силиконом).
Необходимый объем крови собирают от
каждой из не менее чем пяти овец. Достаточ
ным объемом является отбор 285 мл крови
в 15 мл раствора антикоаryлянта, но может
быть собран и меньший объем. Кровь берут у
живоro животноro или во время убоя, исполь
зуя иrлу, при соединенную к подходящей канюле
с длиной, достаточной для достижения дна
сосуда для сбора. Отбрасывают первые He
сколько миллилитров и собирают только CBO
бодно текущую кровь. Кровь собирают в ДOCTa
точное количество раствора антикоаrулянта,
содержащеro 8,7 r натрия цитрата Р и 4 Mr
апротинина Р в 100 мл воды Р. Соотношение
крови и раствора антикоаryлянта должно быть
19: 1. Во время сбора и сразу после Hero кровь
слеrка перемешивают, не допуская вспенива
ния. По окончании сбора сосуд закрывают и
охлаждают до температуры от 1 О ос до 15 ОС.
После охлаждения содержимое всех колб объе
диняют, за исключением тех, в которых наблю
дается явный rемолиз или образование CryCT
ков, и хранят собранную кровь при температуре
от 10 ос до 15 ОС.
Как можно скорее, в пределах 4 ч после
сбора, объединенную кровь центрифуrируют с
ускорением от 1000 9 до 2000 9 при температу
ре от 10 ос до 15 ос в течение 30 мин. Haдoca
дочную жидкость отделяют и центрифуrируют с
ускорением 5000 9 в течение ЗО мин (если He
обходимо, для получения прозрачной плазмы
можно центрифуrировать с большим ускорени
ем, например, с ускорением 20 000 9 в течение
30 мин, но фильтрация при этом недопустима).
Отделяют надосадочную жидкость, немедлен
но тщательно перемешивают и помещают суб
страт плазмы в небольшие контейнеры с проб
ками порциями, достаточными для проведения
полноrо количественноro определения rепарина
(например, от 1 О мл ДО 30 мл). Сразу же быстро
охлаждают до температуры ниже 70 ос (Ha
пример, поrружая контейнеры в жидкий азот) и
хранят при температуре не выше зо ОС.
Плазма приroдна в качестве субстрата
плазмы для количественноro определения re
парина, если в условиях количественноro опре
деления она обеспечивает время образования
crYCTKa, соответствующее использованному
методу определения, и обеспечивает получе
ние крутых лоrарифмических кривых доза
отклик.
Перед использованием необходимую
порцию плазмы размораживают на водяной
бане при температуре 37 ОС, осторожно пере
мешивая до полноro размораживания. Размо
роженную плазму содержат при температуре от
1 О ос до 20 ос и немедленно используют. Если
необходимо, размороженный субстрат плазмы
слеrка центрифуrируют, но не фильтруют.
Плазмы субстрат Р2. 1066202.
rотовят из человеческой крови, содержащей
менее 1 % обычноro количества фактора IX. Co
бирают кровь в раствор 38 r/л натрия цитpa
та Р, объем котороro составляет 1/9 объема
плазмы.
Хранят в небольших количествах в пласт
массовых контейнерах при температуре зо ос
или ниже.
Плазмы субстрат Р3. 1066203.
rотовят из человеческой крови, содержащей
менее 1 % обычноro количества фактора XI. Co
бирают кровь в раствор 38 r/л натрия цитpa
та Р, объем котороro составляет 1/9 объема
плазмы.
Хранят в небольших количествах в пласт
массовых контейнерах при температуре зо ос
или ниже.
Плазмы субстрат с недостаточным со-
держанием фактора V. 1066300.
Предпочтительно используют плазму. оолу
ченную от донора с врожденной недостаточно--
стью, или roтовят ее следующим образом: OTдe
ляют плазму от человеческой крови. собранной
в раствор 13,4 r/л натрия оксапата Р, объем
котороro составляет 1/10 объема крови. Культи
вируют при температуре 37 ос от 24 ч до 36 ч.
Время свертыван я, определенное по методу,
описанному для раствора фактора V cвepты
вания крови Р. должно быть от 70 с до 100 с.
Если время свертывания меньше 70 с, то куль
тивируют снова от 12 ч до 24 ч.
Хранят в небольших количествах при темпе
рату ре 20 ос или ниже.
684
rосударственная фармакопея Республики Беларусь
Плазминоrен человеческий. 1109100.
[9001 91 6].
Вещество, присутствующее в крови, которое
может быть активировано до плазмина, фермента,
осуществляющеro лизис фибрина в crycTKax крови.
Повидон. 1068500. [9003 39 8].
См. статью Повuдон.
Подсолнечное масло. 1086900.
См. статью Подсолнечное масло рафиниро
ванное.
# Полиамид. [6342&83--1].
[--СО(СН2)4СОNН(СНZ>6NН ]п. Поли(N,N rексамети
ленадипиндиамид).
# Полиамид для колоночной xpoMaTorpa
фии. Полиамид для колоночной хроматоrрафии, 6.
Насыпная плотность: около 0,25 r/мл.
Размер частиц: от 50 мкм ДО 160 мкм.
# Полиамид для тонкослойной xpOMaTO
rрафии. Полиамид для тонкослойной xpOMaтo
rрафии, 6.
# Поливинилпирролидон. См. Повидон Р.
# Поли(виниловый спирт).
Получают методом полимеризации ви
нилацетата с последующим частичным или
почти полным rидролизом поли(винилацетата)
в присутствии щелочей или минеральных
кислот в качестве катализатора. Средняя OT
носительная молекулярная масса от 20 000 до
150 000; вязкость от 3 до 70 мПа'с; эфирное
число, характеризирующее степень rидроли
за, не более 280.
Желтовато белый порошок или полупрозрач
ные rранулы. Растворим в воде, малорастворим
в этаноле, практически нерастворим в ацетоне.
Потеря в массе при высушивании (2.2.32).
Не более 5,0 %. 1,000 r испытуемоro образца
сушат при температуре 105 ос в течение 3 ч.
Поли(диметил )(85)(дифенил)(15)силок
сан. 11540700.
Содержит 85 % метильных rрупп и 15 % фе
нильных rрупп.
Неподвижная фаза для хроматоrрафии.
Пол и (ди метил )(дифе н ил) сило кса н.
1066900. DB 5, SE52.
Содержит 95 % метильных rрупп и 5 % фе
нильных rрупп.
Неподвижная фаза для raзовой хроматоrpафии.
Пол и(ди метил )(дифен ил )(ди ви Н ил )си
локсан. 1100000. SE54.
Содержит 94 % метильных rрупп, 5 % фе
нильных rрупп и 1 % винильных rрупп.
Неподвижная фаза для raзовой хроматоrpафии.
Поли(диметил)силоксан. 1066800.
Каучук силиконовый (метил). Орrаноси
ликатный полимер, имеющий вид полужидкой
бесцветной смолы.
Характеристическая вязкость, определенная
как указано ниже, должна быть около 115 мл'r 1 .
1,5 r, 1 r и 0,3 r поли(диметил)силоксана взве
шивают с точностью до 0,1 Mr в мерных колбах
вместимостью 100 мл, прибавляют от 40 мл ДО
50 мл толуола Р, встряхивают до растворения
и доводят до объема 100,0 мл этим же paCTBO
рителем. Определяют вязкость (2.2.9) каждою
раствора и вязкость толуола Р в тех же усло
виях. Концентрацию каждоro раствора уменьша
ют вдвое, разбавляя толуолом Р, и определяют
вязкость полученных растворов.
c концентрация, r/100 мл;
t 1 время истечения испытуемоro paCT
вора;
t 2 время истечения толуола;
111 вязкость испытуемоro раствора, в
мПа'с;
112 вязкость толуола, в мПа'с;
d 1 относительная плотность испытуемоro
раствора;
d 2 относительная плотность толуола.
Для получения значений относительной
плотности используют следующие данные:
Концентрация (с), Относительная
r/100 мл плотность (d.)
0 0,5 1,000
0,5 1,25 1,001
1 ,25 2,20 1,002
2,20 2, 75 1,003
2,75 З,20 1,004
3,20 3, 75 1,005
3,75 ,50 1,006
Удельную вязкость (11 уд ) определяют по фор
муле:
11 д = 111 112 == t 1 d 1 1,
У 112 t 2 d 2
приведенную вязкость ( " ) определяют по
"tпpиe
формуле:
YJ уд
YJ прив == .
с
Характеристическую вязкость (11) получают
экстраполяцией предыдущей формулы до с == О
Для этоro строят кривую l1yiJlc или log YJy,,1c как
функцию с. Экстраполяцией до с == О получают 11.
Характеристическую вязкость выражают в мл/r,
поэтому полученное значение должно быть YM
ножено на 100.
Инфракрасный спектр поrлощения (2.2.24),
полученный нанесением вещества. при необхо
димости дисперrированноro в нескольких каплях
4. 1. 1. Реактивы
685
У2лерода тетрахлорида Р, на диск натрия хло
рида, не должен иметь поrлощения при длине
волны 3053 CM 1, соответствующеro винильным
rруппам.
Потеря в массе при высушивании (2.2.32).
Не более 2,0 % при определении из 1,000 r испы
туемоro образца высушиванием в вакууме при
температуре 350 ос в течение 15 мин; не более
0,8 % при определении из 2,000 r испытуемоro
образца высушиванием при температуре 200 ос
в течение 2 ч.
Полимер орrаносиликатный аморфный,
с введенными полярными rруппами, oктaдe
цилсилильный эндкепированный. 1150600.
Синтетические, сферические rибридные
частицы, состоящие из неорrаническоro (си
ликаrель) и орrаническоro (орrаносилоксаны)
компонентов с поверхностью, химически моди
фицированной октадецилсилильными rруппами,
имеющими полярные rруппы в алифатической
цепи. Для дальнейшеro сведения к минимуму Ka
коro либо взаимодействия с основными соедине
ниями тщательно эндкепируют для устранения
большинства оставшихся силанольных rрупп.
Размер частиц указывают после названия cop
бента в испытаниях, в которых он используется.
Полимер орrаносиликатный аморфный
октадецилсилильный. 1144200.
Синтетические, сферические rибридные
частицы, состоящие из неорrаническоro (си
ликаrель) и орrаническоro (орrаносилоксаны)
компонентов с поверхностью, химически моди
фицированной октадецилсилильными rруппами,
имеющими трифункциональные rруппы в али
фатической цепи.
Полимер орrаносиликатный аморфный
октадецилсилильный эндкепированный для
масс-спектрометрии. 1164900.
Синтетические, сферические rибридные ча
стицы, состоящие из неорrаническоro (силика
rель) и орrаническоro (орrаносилоксаны) компо
нентов. Для дальнейшеro сведения к минимуму
какоro либо взаимодействия с основными соеди
нениями тщательно эндкепируют для устранения
большинства оставшихся силанольных rрупп.
Размер частиц указывают после названия cop
бента в испытаниях, в которых он используется.
Полиметилфенилсилоксан. 1067900.
Содержит 50 % метильных rрупп и 50 %
фенильных rрупп. (Средняя молекулярная
масса L1OOO).
Очень вязкая жидкость (вязкость около
1300 мПа'с).
Неподвижная фаза для rазовой xpoMaтorpa
фии.
d;;: около 1,09.
25
по: около 1,540.
ВВ. За. 1712.
Пол и [метил (95 )фен ил (5)]силоксан.
1068000. См. Поли(диметил)(дифенил)силок
сан р.
Поли(метил(94)фенил(5)винил(1 )]силок-
сан. 1068100. См. Поли(диметил)(дифенил)(ди
винил}силоксан р.
Полиоксиэтилированное касторовое
масло. 1068200.
Светло желтая жидкость, становится про
зрачной при температуре около 26 ОС.
Полисорбат 20. 1068300. [9005 64 5].
См. статью Полисорбат 20.
Полисорбат 80. 1068400. [9005 65 6].
См. статью Полисорбат 80.
Полистирол90 1000. 1112200. [9003 53 6].
Орrанический стандарт, используемый для
калибровки в rазовой хроматоrрафии.
М : около 950.
w
MjM n :1,10.
Поли(цианопропил)силоксан. 1066700.
Полисилоксан, замещенный на 100 % циа
нопропильными rруппами.
Поли[( цианоп роп ил)( фенил)][диметил]
силоксан. 1114800.
Содержит 6 % цианопропилфенильных
rрупп и 94 % диметильных rрупп.
Неподвижная фаза для raзовой хроматоrpафии.
Поли( цианопропил )(7)( фенил )(7)( метил)
(86)силоксан. 1109200.
Полисилоксан, замещеный на 7 % циано
пропильными rруппами, на 7 % фенильными
rруппами и на 86 % диметильными rруппами.
Неподвижная фаза для raзовой хроматоrpафии.
Поли(цианопропил)( фенил метил )силок-
сан. 1066600.
Содержит 90 % цианопропильных rрупп и
1 О % фенилметильных rрупп.
Неподвижная фаза для raзовой хроматоrpaфии.
Поли [( цианопропил) метилфенилметил-
силоксан]. 1066500. См. Поли[(цианопропил}
(метил)][(фенил)(метuл)]силоксан Р.
Поли [( цианоп роп ил)( метил)l [( фенил)
(метил)]силоксан. 1066500.
Содержит 25 % цианопропильных rpупп,
25 % фенилы''/ых rрупп и 50 % метильных rрупп.
(Средняя молекулярная масса 8000).
Очень вязкая жидкость (вязкость около
9000 мПа'с).
d 25
25: около 1,10.
25
по: около 1,502.
686
rосударственная фармакопея Республики Беларусь
# Полиэтиленrликоль 200. См. МакfЮ20Л 200 Р.
# Полиэтиленrликоль 300. См. Макро2ОЛ ЗОО Р.
# Полиэтиленrликоль 400. См. МаКро2ОЛ 400 Р.
# Полиэтиленrликоль 1000. См. MaKpo
20Л 1000 Р.
# Полиэтиленrликоль 1500. См. MaKpo
20Л 1500 Р.
# Полиэтиленrликоль 20 000. См. MaKpo
20Л 20 000 Р.
Полиэтиленrликоль, полярно деактиви
рованный.1179000.
Неподвижная фаза для raзовой хроматоrpaфии.
Полиэтиленrликольадипинат. (С в Н 12 О 4)n'
(М М. (172,2». 1067700.
Белая или почти белая воскообразная
масса. Практически нерастворим в воде.
Температура плавления: около 43 ОС.
Полиэтиленrликольсукцинат. (С 6 Н 8 О 4 )n.
(М М. (144,1 Ц. 1067800.
Белый или почти белый кристаллический
порошок. Практически нерастворим в воде.
Температура плавления: около 102 ОС.
Полиэфирный rидроксилированный
rель для хроматоrрафии. 1067000.
rель с небольшим размером частиц, име
ющий rидрофильную поверхность с rидрок
сильными rруппам. Имеет предел эксклюзии по
декстрану с молекулярной массой от 2.105 до
2,5'106.
Прокаина rидрохлорид. 1109400.
См. статью Прокаина 2идрохлорид.
D П ролил -L -фен ил ала н ил -L -а рrин и н-
4-нитроанилидадиrидрохлорид. С26НЗ4Nв05 '2HCI.
(М м. 612). 1072800.
Пропанол. СзНвО, (М М. 60,1). 1072000. [71
23 8]. 1 Пропанол.
Прозрачная бесцветная жидкость. Смеши
вается с водой и с 96 % спиртом.
d;g: около от 0,802 до 0,806.
Температура кипения: около 97,2 ОС.
Температурные пределы пере20нки (2.2.11).
От 96 ос до 99 ОС; должно переroняться не
менее 95 %.
2 Пропанол. СзНвО. (М М. 60,1). 1072100.
[67 63 0]. Изопропиловый спирт.
Прозрачная бесцветная воспламеняющаяся
жидкость. Смешивается с водой и с 96 % спиртом.
d;g: около 0,785.
Температура кипения: от 81 ос до 83 ОС.
2-Пропанол Р1. 1072101.
Должен выдерживать требования для 2 пропа
нола р и следующие дополнительные требования:
20
по: около 1,378.
Вода (2.5.12). Не более 0,05 %. Определе
ние проводят из 1 О r.
Пропускание (2.2.25) определяют, используя
в качестве компенсационной жидкости воду Р: не
менее 25 % при длине волны 21 О нм; не менее 55 %
при длине волны 220 нм; не менее 75 % при длине
волны 230 нм; не менее 95 % при длине волны
250 нм; не менее 98 % при длине волны 260 нм.
# Пропаноламин. С з Н 9 NО. (М м. 75,1).
1072200. [156 87 6]. 3 Амино 1 пропанол.
Прозрачная бесцветная вязкая жидкость.
d 20
20: около 0,99.
п O: около 1,461.
Температура плавления: около 11 ОС.
Пропилацетат. С 5 Н 10 О т (М М. 102,1).
1072600. [109 604].
d;g: около 0,888.
Температура кипения: около 102 ОС.
Температура плавления: около 95 ос.
Пропиленrликоль. 1072900. [57 55 6].
См. статью ПропилеН2ликоль.
Пропиленоксид. С з Н 6 О. (М М. 58,1).
1121800. [75 56 9].
Бесцветная жидкость. Смешивается с 96 %
спиртом.
Пропилпараrидроксибензоат. 1072700.
[94 1 3 3].
См. статью Пропилпара2идроксибензоат.
Пропионовый альдеrид. С з Н 6 О. (М М. 58,1).
1072300. [123 38 6]. Пропаналь.
Жидкость. Леrкорастворим в воде, смеши
вается с 96 % спиртом.
d 20
20: около 0,81.
20
по : около 1,365.
Температура кипения: около 49 ОС.
Температура плавления: около 81 ОС.
Пропионовый анrидрид. С 6 Н 10 О з . (М М. 130,1).
1 072500. [123 62 6].
Прозрачная бесцветная жидкость. PaCTBO
рим в 96 % спирте.
d;g: около 1,01.
Температура кипения: около 167 ОС.
Пропионовоrо анrидрида реактив. 1072501.
1 r кислоты толуолсульфоновой Р paCTBO
ряют в 30 мл кислоты уксусной ледяной Р и при
бавляют 5 мл пропионов020 аН2идрида Р.
Используют через 15 мин после приroтовления.
Срок rодности 1 сут.
4.1.1. Реактивы
687
Протамина сульфат. 1073000. [53597 25A
(сальмин) 9007 31 2 (клупеин)J.
Протамина сульфат продукт белковоro
происхождения, получаемый из спермы разных
видов рыб (обычно Sа/топidае или C/upeidae). 1 Mr
протамина сульфата осаждает не менее 100 МЕ
rепарина в пересчете на сухое вещество.
Белый или почти белый порошок. rиrроско
пичен. Умеренно растворим в воде, практически
нерастворим в 96 % спирте.
Потеря в массе при высушивании (2.2.32).
Максимум 5,0 % из навески 1,000 r при темпера
туре 100 105 ос в течение 3 ч.
Хранят в воздухонепроницаемом контейнере.
Протеаза Staphylococcиs aи,eиs штамм
V8. Тип XVII B. 1115800. [66676--43 5].
Микробиолоrический внеклеточный проте
олитический фермент. Лиофилизированный по
рошок содержит от 500 единиц до 1000 единиц
в 1 Mr раствора.
# Протравной желтый 3R. С,зНвNзNаО5'
(М м. 309,2). Натрия 2 rидрокси 5 [(4 нитрофенил)
азо]бензоат. Ализариновый желтый Р.
Светло коричневый или темно коричневый
или красно коричневый кристаллический поро
шок. Малорастворим в воде и в 96 % спирте, леr
корастворим при наrревании.
# Протравноrо желтоrо 3R раствор.
0,1 r протравН020 желт020 3R Р растворяют
в 100 мл воды Рпри наrревании на водяной бане.
Изменение окраски. От рН 10,2 (желтое
окрашивание) до 12,0 (красное окрашивание).
Протравной черный 11. С20Н'2NзNаО7S'
(М м. 461,4). 1056800. [1787 61 ?]. Показа
тель Шульца NQ 241. Цветной индекс NQ 14645.
Натрия 2 rидрокси Ч(1 rидроксинаФт 2 ил )азо]
6 нитронафталин4 сульфонат. Эриохром черный.
#XpoMoreH черный. Кислотный хром черный специ
альный.#
Коричневато черный порошок. Растворим в
воде и в 96 % спирте.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
Протравноrо черноrо 11 индикаторная
смесь. 1056801.
1 r протравН020 чеРН020 11 Р смешивают с
99 r натрия хлорида Р.
Испытание на чувствительность. 50 Mr
индикаторной смеси растворяют в 100 мл
воды Р. Появляется коричневато фиолетовое
окрашивание, которое должно перейти в синее
при прибавлении 0,3 мл раствора аммиака раз
ведеНН020 Р1. При последующем прибавлении
0,1 мл раствора 1 О r/л ма2ния сульфата Р OKpa
ска должна измениться на фиолетовую.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
Прочный красный В, соль. С17Н,зNЗО9S'"
(М м. 467,4). 1037500. [49735 71 9J. Показа
тель Шульца NQ 155. Цветной индекс NQ 37125.
2 Метокси 4 нитробензол диазония кислый
нафталин 1 ,5 дисульфонат.
Оранжево желтый порошок. Растворим в
воде, малорастворим в 96 % спирте.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте при температу
ре от 2 ос до 8 ОС.
Прочный синий В, соль. C'4H'2C'2N402'
(М. м. 339,2). 1037400. [84633 94 3J. Показа
тель Шульца NQ 490. Цветной индекс NQ 37235,
3,3' Диметокси(бифенил ) 4,4' бисдиазония ди
хлорид.
Темно зеленый порошок. Растворим в воде.
Стабилизирован цинка хлоридом.
Хранят в воздухонепроницаемом контейне
ре при температуре от 2 ос до 8 ОС.
Проявителя раствор. 1122500.
К 2,5 мл раствора 20 r/л кислоты лимон
ной Р прибавляют 0,27 мл формальде2ида Р и
доводят водой Р до объема 500,0 мл.
Птероевая кислота. C14H12N602' (Мм 152,2).
1073100. [119 24 4J. 4 [[(2 Амино--4 оксо 1,4
диrидроптеридин 6 ил )метил ]амино Jбензойная
кислота.
Кристаллы. Леrкорастворима в растворах
щелочей.
Пулеrон. С 10 Н ,6 О. (М. м. 152,2). 1073100.
[89 82 7]. (R)--2 Изопропилиден 5 метилциклоrекса
нон. (+ ) п Мент 4 eH 3 OH.
Бесцветная маслянистая жидкость. Практи
чески нерастворим в воде, смешивается с 96 %
спиртом.
d 20
15: около 0,936.
20
по : от 1,485 до 1,489.
Температура кипения: от 222 ос до 224 ос.
Пуле20Н, используемый в 2азовой xpoMaтcr
2рафии, должен выдержать следущее дonoпHи-
тельное испытание.
Количественное определение.- rаэовая хро--
матоrрафия (2.2.28), как указано в стаТЬе Мяты
перечной масло, используя пулеroн в качестве
испытуемою раствора.
Содержание пулеroна, рассчмтанное M
тодом нормализации, должно бьnъ не менее
98,0%.
Рамноза. C e H,Ps'H 2 0. (М. М. 182,2).
1074900. [615 3 7]. L--(+ мноза. 6--Дезокси
L манноза.
Белый или почти бenый кристаллический
порошок. Леncорастворим в воде.
[a] : от +7.8 l1fJ +8,3. Определение прово
дят. исполЬЗуЯ раствор 50 r/л рамнозы в воде Р,
содержащей oкono 0.05 % NН з .
688
rосударственная фармакопея Республики Беларусь
Рапонтицин. C 21 H 2 Pg' (м. м.420.4). 1075000.
[155 58 8]. з r идрокси 5 [2 (3 rидрокси-4 метокси
фенил)этенил]фенил f3 D rлюкопиранозид.
Желтовато серый кристаллический поро
шок. Растворим в 96 % спирте и в метаноле.
Хромат08рафия. Определение проводят. как
указано в стаТЬе Ревеня корни. На xpoMaтorpaMMe
должно обнаруживаться только одно основное пятно.
Рапсовое масло. 1074600.
Жирное масло, полученное выдавливанием
из семян Brassica париs L. и B,assica campest,is L.
Фракция жирных кислот содержит от 40 %
до 55 % кислоты эруковой.
Прозрачная жидкость от желтоro до TeMHO
желтоro цвета. Практически нерастворимо в 96 %
спирте, смешивается с петролейным эфиром.
Йодное число (2.5.4). От 94 до 120.
Число омыления (2.5.6). От 168 до 181.
Перекисное число (2.5.5). Не более 5.
Кислота эруковая. Тонкослойная xpOMaтo
rрафия (2.4.21), как указано в испытании на по
сторонние жирные кислоты, используя следу
ющие растворы:
Раствор А 20 Mr смеси жирных кислот раст
воряют в 4 мл хлороформа Р.
Раствор В. 2, О мл раствора А доводят хло
роформом Р до объема 50 мл.
На xpoMaTorpaMMe раствора А должно обнару
живаться пять четких пятен. Пятно с самым низким
значением R, около 0,25 должно быть наиболее ин
тенсивным или одним из наиболее интенсивных и
должно соответствовать кислоте эруковой. На хро-
MaтorpaMMe раствора В должно быть четко видно
пятно, соответствующее кислоте эруковой.
# Растворимый красный. См. 1 CyдaH Р.
Красный G Р.
Резорцин. 1074800. [108-46 3].
См. статью Резорцин.
# Резорцина раствор.
2 r резорцина Р растворяют в 1 О мл 96 %
спирта Р.
Хранят в защищенном от света месте при
температуре от 15 ос до 25 ос не более 7 сут.
Резорцина реактив. 1074801.
К 80 мл кислоты хлористоводородной Р
прибавляют 10 мл раствора 20 r/л резорцина Р,
0,25 мл раствора 25 r/л меди (1/) сульфата Р и
доводят водой Р до объема 100,0 мл.
Используют через 4 ч после приroтовления.
Хранят при температуре от 2 ос до 8 ОС. Срок
rодности 7 сут.
Рибоза. С 5 Н IО О 5 . (м. м. 150,1). 1109600.
I50 69 1]. D Рибоза.
Растворима в воде, малорастворима в 96 %
спирте.
Температура плавления: от 88 ос до 92 ОС.
Рицинолеиновая кислота. С lв Н з .О з .
(м. М. 298,5). 1100100. [141 22 0]. 12 rидроксиоле
иновая кислота.
Желтая или желтовато коричневая вязкая
жидкость. Содержит смесь жирных кислот, полу
ченных rидролизом масла касторовоro. Практи
чески нерастворима в воде, очень леrко paCTBO
рима в этаноле.
d;g: около 0,942.
п O: около 1,472.
Температура плавления: около 285 ос с раз
ложением.
Родамин В. С 2в Н з1 СINРз, (м. м. 479,0).
1075100. I81 88 9]. Показатель Шульца NQ 864.
Цветной индекс NQ 45170. [9 (2 Карбоксифенил)
6 (диэтиламино ) 3Н ксантен 3 илиден ]диэти
ламмония хлорид.
Зеленые кристаллы или красновато фиоле
товый порошок. Очень леrко растворим в воде и
в 96 % спирте.
Ртуть. Hg. (А м. 200,6). 1052800. [7439 97 6].
Серебристо белая жидкость, рассыпающа
яся на сферические капли, которые не оставля
ют металлическоro следа при трении о бумаry.
d 20
20: около 13,5.
Температура кипения: около 357 ОС.
Ртути (11) ацетат. C 4 H 6 Hg0 4 . (м. м. 318,7).
1052000. [1600 27 7]. Ртути диацетат.
Белые или почти белые кристаллы. Леrко
растворим в воде, растворим в 96 % спирте.
Ртути (11) ацетата раствор. 1052001.
3,19 r ртути (1/) ацетата Р растворяют
в кислоте уксусной безводной Р и доводят до
объема 100 мл этой же кислотой. При необходи
мости полученный раствор нейтрализуют О, 1 М
раствором кислоты хлорной, используя в каче
стве индикатора 0,05 мл раствора кристалли
чеСК020 фиолетов020 Р.
Ртути (11) бромид. HgBr 2 . (м. м. 360,4).
1052100. [7789 47 1]. Ртути дибромид.
Белые или светло желтые кристаллы или
кристаллический порошок. Малорастворим в
воде, растворим в 96 % спирте.
Ртутно бромидная бумаrа. 1052101.
В прямоуrольную чашку помещают раствор
50 r/л ртути (1/) бромида Р в этаноле Р, поrру
жают в раствор кусочки белой фильтровальной
бумаrи, с плотностью 80 r/M 2 (скорость фильтро
вания, равная времени фильтрования, выражен
ному в секундах, при фильтровании 100 мл воды
при температуре 20 ос через фильтр с поверх
ностью 1 О см 2 И постоянном давлении 6,7 кПа:
от 40 с до 60 с), размером 1,5 х 20 см, сложен
ные вдвое. Бумаry подвешивают на неметалли
ческую нить, позволяя стечь избытку жидкости,
4.1.1. Реактивы
689
сушат в защищенном от света месте. Отрезают
по 1 см с каждоro конца каждой полоски и Hape
зают остальную часть бумаrи на квадратики со
стороной 1,5 см или диски диаметром 1,5 см.
Хранят в контейнере со стеклянной проб
кой, завернутом в черную бумаry.
Ртути (11) йодид. Hg1 2 . (М м. 454,4). 1052300.
[7774 29 0]. Ртути дийодид.
Плотный ярко красный кристаллический по
рошок. Малорастворим в воде, умеренно pac
творим в ацетоне и в 96 % спирте, растворим в
избытке раствора калия йодида Р. #Ядовит. #
Хранят в защищенном от света месте.
# Ртути (1) нитрат. Н92(NОЗ)2'2НР. (М м.561,2).
Бесцветные кристаллы. На воздухе BЫBe
триваются. Ядовит.
Ртути (11) нитрат. Нg(NОЗ)2'НР' (М м. 342,6).
1052400. [7782 86 7]. Ртути динитрат моноrи
драт. #Ртути окисной нитрат моноrидрат.#
Бесцветные или слеrка окрашенные кристал
лы. rиrроскопичен. Растворим в воде в присут
ствии небольшоro количества кислоты азотной.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
Ртути (11) нитрата раствор. 1052801.
3 мл ртути Р осторожно растворяют в
27 мл кислоты азотной дымящейся Р, получен
ный раствор разводят равным объемом воды Р
Хранят в защищенном от света месте. Срок
roдности 2 мес.
Ртути (11) оксид. HgO. (М. м. 216,6). 1052500.
[21908 53 2]. Ртути оксид желтый. Ртути оксид.
Порошок от желтоro до оранжево-желтоro цвета.
Практически нерастворим в воде и в 96 % спирте.
Хранят в защищенном от света месте.
# Ртути роданид. См. Ртути (1/) тиoци
анат Р.
Ртути (11) сульфата раствор. 1052600.
[7783 35 9].
1 r ртути (/1) оксида Р растворяют в смеси
из 20 мл воды Р и 4 мл кислоты серной Р.
Ртути (11) тиоцианат. Hg(SCN)2' (М м. 316,7).
1052700. [592 85 8]. Ртути ди(тиоцианат). #Ртути
роданид.#
Белый или почти белый кристаллический
порошок. Очень мало растворим в воде, мало
растворим в 96 % спирте, растворим в paCTBO
рах натрия хлорида.
Ртути (11) тиоцианата раствор. 1052701.
0,3 r ртути (/1) тиоцианата Р растворяют в
этаноле Р и доводят до объема 100 мл этим же
растворителем.
Срок rодности 7 сут.
Ртути (11) хлорид. HgCI 2 . (М М. 271.5).
1052200. [7487 94 7]. #Сулема.#
Содержит не менее 99,5 % и не более
100,5 % HgCI 2 в пересчете на сухое вещество.
Белый кристаллический порошок или
белые или бесцветные кристаллы или тя
желая кристаллическая масса. Растворим в
воде и в rлицерине, леrкорастворим в 96 %
спирте.
Потеря в массе при высушивании (2.2.32).
Не более 1,0 %. 2,00 r испытуемоro образца
сушат в вакууме в течение 24 ч.
Ртути (11) хлорида раствор. 1052201.
Раствор 54 r/л ртути (/1) хлорида Р.
Рутений красный. [(NНЗ>5RuОRu(NНЗ)40Ru(NНз)J
Сl б '4Нр. (М м. 858). 1075200. [11103 72 3].
Коричневато красный порошок. Растворим в
воде.
Рутения KpacHoro раствор. 1075201.
Раствор 0,8 r/л рутения краСН020 Р в pac
творе свинца (/1) ацетата Р.
Рутин. С27Нзо016'ЗНР' (М м. 665).
1075300. [153 184]. Рутозид. 3 (0 6 Дезок
си и L маннопиранозил ( 1 6Н3 D rлюко
пиранозилокси) 2 (3,4 диrидроксифенил) 5, 7
диrидрокси 4Н хромен 4 он.
Желтый кристаллический порошок, TeM
неет на свету. Очень мало растворим в воде,
растворим примерно в 400 частях кипящей
воды, малорастворим в 96 % спирте, практи
чески нерастворим в эфире, растворим в pac
творах rидроксидов щелочных металлов и aM
миака.
Температура плавления: около 210 ос с раз
ложением.
Раствор в 96 % спирте Римеет два максиму
ма поrлощения (2.2.25) при длинах волн 259 нм
и 362 нм.
Хранят в защищенном от света месте.
Сабинен. С 10 Н 1б . (М м. 136.2). 1109700.
[2009 00 9]. Туй4(1 O) eH. 4 Метилен 1 ИЭOf1)O-f1Мn-
бицикло[3.1.0]rексан.
Бесцветная маслянистая жидкость.
Сабинен, используемый в zaзoвoй JqJOIofaто
2рафии, должен выдерживать cпeдyroщee дcr
полнительное испытание.
Количественное определение. rаэовая x
матоrрафия (2.2.28). как указано в статье пOMe
ранца цветков масло. иaIOЛbЗyЯ сабинен е Ka
честве испытуемоro раствора.
Содержание саб.. leI 18 . рассчитанное Me
тодом нормализации, должно бbrrb не менее
99,0 %.
Салициловая ICМcпoтa. 1075600. [69 72 7].
См. статью Салициловая кислота.
690
rосударственная фармакопея Республики Беларусь
Салициловый альдеrид. С 7 НР2' (М м. 122,1).
1075400. [90 02 8]. 2 rИдроксибензальдеrид.
Прозрачная бесцветная маслянистая жидко," ть.
d;g: около 1,167.
20
по : около 1,574.
Температура кипения: около 196 ОС.
Температура плавления: около 7 ОС.
Салициловоrо альдеrида азин. C14H12N202'
(М м. 240,3). 1075500. [959 36A]. 2,2' Азиноди
метилдифенол.
0,30 r 2идразина сульфата Р растворяют в
5 мл воды Р, прибавляют 1 мл кислоты уксусной
ледяной Р и 2 мл свежеприroтовленноro 20 %
(об/об) раствора салицилов020 альде2ида Р в
2 пропаноле Р. Перемешивают, выдерживают до
образования желтоro осадка, затем встряхива
ют дважды с порциями по 15 мл метиленхлори
да Р. Объединенные орrанические извлечения,
высушенные над натрия сульфатом безво
дным Р, декантируют или фильтруют и выпари
вают досуха. Остаток перекристаллизовывают
при охлаждении из смеси растворителей Me
танол Р толуол Р (40:60, об/об). Кристаллы
сушат в вакууме.
Температура плавления: около 213 ОС.
Хромат02рафия. Определение проводят,
как указано в статье Повидон в испытании на rи
дразин; на полученной xpoMaтorpaMMe должно
обнаруживаться только одно основное пятно.
Салицин. С 1З Н 1 Р7' (М м. 286,3). 1131300.
[1 38 52 3). 2 (rидроксиметил)фенил J3 D rлюко
пиранозид.Саликозид.
[a] O: 62,5:t2.
Температура плавления: от 199 ОС до 201 ОС.
Количественное определение. Жидкост
ная хроматоrрафия (2.2.29), как указано в статье
Кора ивы, с использованием концентрации paCT
вора сравнения.
Содержание салицина, рассчитанное Me
тодом нормализации, должно быть не менее
99,0%.
Сантонин. С 15 Н 1 Рз, (М м. 246,3). 1122000.
[481 06 1]. (+а Сантонин. 3,5а,9 Триметил
За,5,5а, 9b тетраrидро 3Н,4Н нафто[1,2] фуран
2,8 дион.
Бесцветные блестящие кристаллы, желте
ющие под действием света. Очень мало pac
творим в воде, леrкорастворим в roрячем 96 %
спирте, умеренно растворим в этаноле.
[a] O: 173 в этаноле.
Температура плавления: от 174 ОС до 176 ОС.
Хромат02рафия. Определение проводят, как
указано в идентификации С в статье Арники цветки.
На xpoMaтorpaMMe, полученной с использованием
1 О мкл раствора, должна обнаруживаться зона по
rлощения с Rf около 0,5. XpoMaтorpaMMY опрыски
вают раствором анисов020 альде2ида Р, HarpeBa
ют при температуре 105 ос в течение 5--------10 мин.
На xpoMaтorpaMMe при дневном свете наблюда
ется зона с желтым окрашиванием, которое затем
быстро изменяется на фиолетово красное.
# Сапарал. [54692А1 Щ. Сумма аммоний
ных оснований солей тритерпеновых rликозидов
(аралозидов ).
Аморфный порошок белоro цвета с желтова
тым или сероватым оттенком, без запаха. rиrро
скопичен, леrкорастворим в воде.
# Сафранин Т. Смесь 10 фенил 3,6 диами
Ho 2, 7 диметилфеназиния хлорида (C20H19CIN4) и
1 O o толил 3,6 диамино 2, 7 диметилфеназиния
хлорида (C21H21CIN4)'
Красно коричневый порошок. Растворим в
врде и в 96 % спирте. Растворы в 96 % спирте
красноro цвета с желто красной флуоресценци
ей. При прибавлении к водному раствору сафра
нина кислоты хлористоводородной Р появляет
ся сине фиолетовое окрашивание.
# Сафранина раствор.
Приroтовляют в двух отдельных контейнерах
с притертыми пробками. В первом контейнере 1 r
сафранина Р растворяют в спирте (50 % об/об) Р.
Во втором контейнере к 100 мл 96 % спирта Р
прибавляют две капли раствора 10 % (об/об) Kиc
лоты хлористоводородной Р и встряхивают.
# Сахар. См. Сахароза Р.
Сахароза. 1085700. [57 50 1].
См. статью Сахароза.
Свертиамарин. С16Н22010' (М м. 374,3).
1163600. [17388 З9 5]. (4R,5R,6S) 5 Этенил
6 ( rз D rл юкоп иранозил окси ) 4а rидро кси
4,4а ,5 ,6 тетраrидро 1 Н,3Н пирано[3,4 с]пиран
1 OH. Свертиамарозид.
Свинца (11) ацетат. С 4 Н 6 О 4 РЬ-3Нр. (М м. 379,3).
1048100. [6080 56A]. Свинца диацетат.
Бесцветные кристаллы, выветривающиеся
на воздухе. Леrкорастворим в воде, растворим
в 96 % спирте.
Свинца (11) ацетата раствор. 1048103.
Раствор 95 r/л свинца (1/) ацетата Р в воде,
свободной от У2лерода диоксида, Р.
Свинца (11) ацетата OCHoBHoro раствор.
1048400. [1335 32 6). #Свинцовый уксус.#
Содержит не менее 16,7 % (м/м) и не более
17,4 % (м/м) РЬ (А. м. 207,2) в виде соединения
примерноro состава СвН14010РЬЗ'
40,0 r свинца (1/) ацетата Р растворяют
в 90 мл воды, свободной от У2лерода диOKCU
да, Р. Доводят раствором натрия 2идроксида
концентрированным Р рН до 7 ,5, центрифуrиру
ют и используют прозрачный бесцветный Haдo
садочный раствор.
4.1.1. Реактивы
691
При хранении в хорошо закрытом контейне
ре раствор должен быть прозрачным.
Свинца (11) нитрат. РЬ(NО З )2' (М М. 331,2).
1048300. [10099 74 8]. Свинца динитрат.
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Леrкорас
творим в воде.
Свинца (11) нитрата раствор. 1048301.
Раствор 33 r/л свинца (11) нитрата Р.
# Свинца (11) оксид. РЬО. (М м. 223,19).
Мелкокристаллический или аморфный по
рошок от желто зеленоватоro с серебристым OT
тенком до красновато буроro цвета.
Свинца (IV) оксид. РЬО 2 . (М М. 239,2).
1 048200. [1309 60 0]. Свинца диоксид.
Порошок темно коричневоro цвета, Bыдe
ляющий кислород при наrревании. Практически
нерастворим в воде, растворим в кислоте хло
ристоводородной с выделением хлора, paCTBO
рим в кислоте азотной разведенной в присут
ствии пероксида водорода, щавелевой кислоты
или друrих восстанавливающих peareHToB, pac
творим в roрячих концентрированных растворах
rидроксидов щелочных металлов.
Свинцово ацетатная бумаrа. 1048102.
Фильтровальную бумаry, плотность которой
80 r/M 2 , поrружают в смесь кислота уксусная
разведенная Р раствор свинца (11) aцeтa
та Р (1:1 О, об/об), сушат и нарезают на полоски
размером 15 х 40 мм.
Свинцово ацетатная вата. 1048101.
rиrроскопичную вату поrружают в смесь
растворителей кислота уксусная разведен
ная р раствор свинца (11) ацетата Р
(1:10, об/об). Избыток влаrи удаляют, расклады
вая вату, без отжимания, на нескольких слоях
фильтровальной бумаrи. Высушивают на воздухе.
Хранят в воздухонепроницаемом контейнере.
# Свинцовый уксус. См. Свинца (11) aцeтa
та основН020 раствор Р.
Селен. Se. (А. м. 79,0). 1075900. [7782A9 2].
Порошок или rранулы от коричневато красно
ro до черноro цвета. Практически нерастворим в
воде и в 96 % спирте, растворим в кислоте азотной.
Температура плавления: около 220 ОС.
# Селена (IV) оксид. Se02" (М м. 110,96).
Белый кристалличеСI<"ИЙ порошок.
Селенистая кислота. Н 2 SеО з . (М М. 129,0).
1100200. [7783 00 8].
Расплывающиеся на воздухе кристаллы.
Лепюрастворима в воде.
Хранят в воздухонепроницаемом контейнере.
# Сеньетова соль. См. Калия натрия тap
трат Р.
Сера. 1110800. [7704 34 9].
См. статью Сера для наРУЖН020 применения.
Серебрадиэтилдитиокарбамат. C 5 H 10 AgNS 2 .
(М М. 256,1). 1110400. [1470 61 7].
Бледно желтый или серовато желтый поро
шок. Практически нерастворим в воде, paCTBO
рим в пиридине.
Может быть приroтовлен следующим об
разом. 1,7 r серебра нитрата Р растворяют
в 100 мл воды Р. Отдельно растворяют 2,3 r
натрия диэтилдитиокарбамата Р в 100 мл
воды Р. Оба раствора охлаждают до темпера
туры 1 О ОС, затем их смешивают и при переме
шивании собирают осадок желтоro цвета на CTe
клянном фильтре, промывают 200 мл холодной
воды Р и сушат в вакууме в течение 2 3 ч.
Серебра диэтилдитиокарбамат не должен
изменять окраску или иметь сильный запах.
Серебра нитрат. АgNО з . (М м. 169,9).
1078300. [7761 88 8].
Содержит не менее 99,0 % и не более
100,5 % АgNО з .
Белый или почти белый кристаллический поро
шок или прозрачные бесцветные кристаллы. Очень
леrко растворим в воде, растворим в 96 % спирте.
Хранят в неметаллическом контейнере в за
щи щенном от света месте.
Серебра нитрата аммиачный раствор.
1078303.
2,5 r серебра нитрата Р растворяют в 80 мл
воды Р, по каплям прибавляют раствор aMMиa
ка разведенный Р1 до растворения осадка и дo
водят водой Р до объема 100 мл.
rотовят непосредственно перед использо
ванием.
Серебра нитрата раствор Р1. 1078301.
Раствор 42.5 r/л серебра нитрата Р.
Хранят в защищенном от света месте.
Серебра нитрата раствор Р2. 1078302.
Раствор 17 r/л серебра нитрата Р.
Хранят в защищенном от света месте.
Серебра нитрата раствор в пирмдине.
1078304.
Раствор 85 r/л серебра нитрата Р в пири
дине Р.
Хранят в защищенном от света месте.
Серебра нитрата реактив. 1078305.
К смеси из 3 мл раствора аммиака KOHцeH
трироваННО20 Р и 40 мл 1 М раствора натрия
2идроксида прибавляют по каплям при переме
шивании 8 мл раствора 200 r/л серебра Hитpa
та р и доводят водой Р до объема 200 мл.
692
rосударственная фармакопея Республики Беларусь
Серебра оксид. Agp. (М м. 231,7).
1078400. [20667 12 3]. Дисеребра оксид.
Коричневато черный порошок. Практически
нерастворим в воде и в 96 % спирте, леrкорас
творим в кислоте азотной разведенной и в pac
творах аммиака.
Хранят в защищенном от света месте.
Серебряно-марrанцевая бумаrа. 1078200.
Полоски медленно фильтрующей бумаrи
поrружают в раствор, содержащий 8,5 r/л Map
2анца (1/) сульфата Р и 8,5 r/л серебра Hитpa
та р Выдерживают в течение нескольких минут,
сушат над фосфора (V) оксидом Р, защищая от
воздействия паров кислот и щелочей.
Серин. С з Н 7 NО з . (М м. 105,1). 1076000. [5645--1].
(S) 2 Амино 3 rидроксипропановая кислота.
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Леrкорас
творим в воде, практически нерастворим в 96 %
спирте.
Содержит не менее 98,5 % и не более
101,0 % С з Н 7 NО з в пересчете на сухое вещество.
Хранят в защищенном от света месте.
Серная кислота. H 2 S0 4 . (М м. 98.1).
1086800. [7664 93 9].
Содержит не менее 95,0 % (м/м) и не более
97,0 % (м/м) H;SO,.
Бесцветная едкая, маслянистой консистен
ции жидкость. Очень rиrроскопична. Смешива
ется с водой и С 96 % спиртом с интенсивным
выделением тепла.
d;2: от 1,8З4до 1,837.
Раствор 10 r/л кислоты серной является
сильной кислотой и дает реакцию на сульфаты
(2.3.1).
Прозрачность (2.2.1). Кислота серная
должна быть прозрачной.
Цветность (2.2.2. метод 1/). Кислота серная
должна быть бесцветной.
Окисляющиеся вещества. Осторожно при
охлаждении 20 r кислоты серной приливают к
40 мл воды Р и прибавляют 0,5 мл 0,002 М pacт
вора калия пермаН2аната. Фиолетовая окраска
должна сохраняться не менее 5 мин.
Хлориды. Не более 0,00005 % (0,5 ррm).
Осторожно при охлаждении 1 О r кислоты серной
прибавляют к 1 О мл воды Р, охлаждают и ДOBO
дят до объема 20 мл этим же растворителем.
Прибавляют 0,5 мл раствора серебра Hитpa
та Р2 и выдерживают в течение 2 мин в защи
щенном от света месте. Полученный раствор по
степени мутности не должен превышать эталон,
приютовленный в то же самое время с исполь
зованием смеси из 1 мл этаЛОНН020 раствора
хлорида (5 ррт С/) Р, 19 мл воды Р и 0,5 мл
раствора серебра нитрата Р2.
Нитраты. Не более 0,00005 % (0,5 ррm).
Осторожно при охлаждении 50 r или 27,2 мл кис
лоты серной вливают в 15 мл воды Р и прибавля
ют 0,2 мл свежеприroтовленноro раствора 50 r/л
бруцина Р в кислоте уксусной ледяной Р Через
5 мин окраска полученноro раствора должна
быть не интенсивнее эталонною раствора, при
roтовленноro аналоrично испытуемому с исполь
зованием 12,5 мл воды Р, 50 r кислоты серной,
свободной от азота, Р, 2,5 мл этаЛОНН020 pacт
вора нитрата (10 ррт NO) Р и 0,2 мл раствора
50 r/л бруцина Р в кислоте уксусной ледяной Р
Аммоний Не более 0,0002 % (2 ррm). Ocтo
рожно при охлаждении 2,5 r кислоты серной при
бавляют к воде Р, доводят до объема 20 мл этим
же растворителем, охлаждают и по каплям при
бавляют 1 О мл раствора 200 r/л натрия 2uдpOK
сида Р и 1 мл щеЛОЧН020 раствора калия тe
трайодомеркурата Р. Окраска полученноrо
раствора должна быть не интенсивнее эталон
ною раствора, приroтовленноro с использовани
ем 5 мл этаЛОНН020 раствора аммония (1 ррт
NHJ Р, 15 мл воды Р, 10 мл раствора 200 r/л
натрия 2идроксида Р и 1 мл щеЛОЧН020 pacт
вора калия тетрайодомеркурата Р
Мышьяк (2.4.2, метод А). Не более
0,000002 % (0,02 ррm). Осторожно при охлаж
дении к 50 r кислоты серной прибавляют 3 мл
кислоты азотной Р, осторожно упаривают до
объема 10 мл, охлаждают, к полученному OCTaT
ку прибавляют 20 мл воды Р и упаривают до
объема 5 мл. Полученный раствор должен BЫ
держивать испытание на мышьяк. Эталон roто
вят с использованием 1,0 мл этаЛОНН020 pacт
вора мышьяка (1 ррт As) Р
Тяжелые металлы (2.4.8, метод А). Не
более 0,0002 % (2 ррт). 10 мл раствора, при
rотовленноrо для испытания на железо, ДOBO
дят водой Р до объема 20 мл. 12 мл полученноro
раствора должны выдерживать испытание на тя
желые металлы. Эталон roтовят с использовани
ем этаЛОНН020 раствора свинца (2 ррт РЬ) Р
Железо (2.4.9). Не более 0,0001 % (1 ррт).
Зольный остаток, полученный при определении
остатка после прокаливания, растворяют при
слабом наrревании в 1 мл кислоты хлористо
водородной разведенной Р и доводят водой Р
до объема 50,0 мл. 5 мл полученноro раствора
доводят водой Р до объема 10 мл. Получен
ный раствор должен выдерживать испытание на
железо.
Остаток после прокаливания. Не более
0,001 %. Определение проводят из 100 r кисло
ты серной, путем осторожноro выпаривания в
небольшом тиrле над открытым пламенем и Ha
rревания остатка до красноro каления.
Количественное определение. В колбу с
притертой стеклянной пробкой помещают 30 мл
воды Р, точно взвешивают, прибавляют 0,8 мл
кислоты серной, охлаждают и снова точно взве
шивают. Титруют 1 М раствором натрия 2идpOK
сида, используя в качестве индикатора 0,1 мл
раствора метилов020 краСНО20 Р.
1 мл 1 М раствора натрия 2идроксида co
ответствует 49,04 Mr H 2 S0 4 .
4.1.1. Реактивы
693
Хранят в контейнере с притертой стеклян
ной пробкой, изroтовленном из стекла или дpy
roro инертноro материала.
Серная кислота разведенная. 1086804
Содержит 98 r/л Н 2 80 4 .
5,5 мл кислоты серной Р прибавляют к
60 мл воды Р, охлаждают и доводят до объема
100 мл этим же растворителем.
Количественное определение. В колбу
с притертой стеклянной пробкой помещают
30 мл воды Р, прибавляют 10,0 мл кислоты
серной разведенной и титруют 1 М раствором
натрия еидроксида, используя в качестве ин
дикатора 0,1 мл раствора метиловоео Kpac
Н020 р.
1 мл 1 М раствора натрия еидроксида co
ответствует 49,04 Mr Н 2 80 4 .
Серная кислота, свободная от азота.
1086806.
Должна выдерживать требования для Kиc
лоты серной Р и следующее дonолнитenьное
испытание.
Нитраты. К 5 мл воды Р осторожно при
бавляют 45 мл кислоты серной, охлаждают до
температуры 40 ос и прибавляют 8 Mr дифенил
бензидина Р. Полученный раствор должен быть
бледно розовоro или очень бледноro roлубоro
цвета.
Серная кислота, свободная от тяжеЛblХ
металлов. 1086805.
Должна выдерживать требования для Kиc
лоты серной Р и следующие дополнительные
испытания:
As: не более 0,005 ррm;
Cd: не более 0,002 ррm;
Cu: не более 0,001 ррm;
Fe: не более 0,05 ррт;
Hg: не более 0,005 ррт;
Ni: не более 0,002 ррm;
РЬ: не более 0,001 ррт;
Zn: не более 0,005 ррm.
Серной кислоты 2,5 М раствор спирто-
вой. 1086801.
Осторожно при постоянном охлаждении и
перемешивании 14 мл кислоты серной Р при
бавляют к 60 мл этанола Р, охлаждают и ДOBO
дят этанолом Р до объема 100 мл.
rотовят непосредственно перед использо
ванием.
Серной КИСЛОТbl раствор спиртовой.
1086803.
Осторожно при постоянном охлаждении и
перемешивании 20 мл кислоты серной Р при
бавляют к 60 мл 96 % спирта р, охлаждают и дo
водят 96 % спиртом Р до объема 100 мл.
rотовят непосредственно перед использо
ванием.
Сероводород. Н 2 8. (М. м. З4,08) 1044000.
[7783 064].
rаз. Малорастворим в воде.
Сероводород Р1. Н 2 8. (М. м. 34,08) 1106600.
Содержание: не менее 99,7 % (06/06) Н 2 8.
# Сероуrлерод. См. Уелерода дисульфид Р.
Серы диоксид. 802' (М. М. 64,1) 1086700.
[7446 09 5]. Сернистый анrидрид.
Бесцветный raз. При сжатии превращается в
бесцветную жидкость.
СерblдиоксидР1. 802' (М. М. 64,1) 1110900.
Содержание: не менее 99,9 % (об/о6) 802'
# о-Сиаловая кислота. См. N Ацетил
неурамuновая кислота Р.
Силикаrель G. 1076300. [112926 ()o 8].
Содержит около 13 % кальция сульфата re
миrидрата.
Мелкий белый или почти белый roмоreнный
порошок с размером частиц около 15 мкм.
Кальция сульфат. 0,25 r испытуемоro об
разца помещают в колбу с притертой стеклян
ной пробкой, прибавляют 3 мл кислоты хлори
стоводородной разведенной Р и 100 мл воды Р,
энерrично встряхивают в течение 30 мин, филь
труют через стеклянный фильтр (2.1.2) и промы
вают остаток. Фильтрат и промывные воды объе
диняют и ПРОВQДЯт определение содержания
кальция методом комплексометрии (2.5.11).
1 мл 0,1 М раствора натрия эдетата COOT
ветствует 14,51 Mr Са80 4 '%Нр.
рН (2.2.3). Около 7. Измеряют рН суспензии,
полученной взбалтыванием 1 r испытуемоro об
разца с 1 О мл воды, свободной от уелерода ди
оксида, Р в течение 5 мин.
Силикаreль GF 254 . 1076400. [112926 00 8].
Содержит около 1З % кальция сульфата по-
луrидрата и около 1,5 % флуоресцентноro инди
катора, имеющеro оптимальную интенсивность
при длине волны 254 нм.
Мелкий белый или почти белый roмоrенный
порошок с размером частиц около 15 мкм.
Кальция сульфат. Определение проводят
методом, указанным для силuкаееля G Р.
рН (2.2.3). Должен выдерживать требования
для сuликаееля G Р.
Флуоресценция. Тонкослойная xpoMaTorpa
фия (2.2.27) с использованием силикаееля G в
качестве TOHKoro слоя. На хроматоrpафическую
пластинку наносят в десять точек последова
тельно возрастающие объемы от 1 мкл ДО 10 мкл
раствора 1 r/л кислоты бензойной Р в смеси
кислота муравьиная безводная Р 2 пропа
нол Р (10:90, 06/06). Хроматоrpафируют в той
же смеси растворителей. Коrда фронт paCTВO
рителей пройдет около 1 О см, пластинку сушат
694
rосударственная фармакопея Республики Беларусь
и просматривают в ультрафиолетовом свете при
длине волны 254 нм. На верхней трети xpol\.aтo
rpaMMbI на флуоресцирующем фоне должны uб
наруживаться темные пятна кислоты бензойной,
начиная от 2 MKr и более.
Силикаrель Н. 1076500. [112926 00 8]_
Белый или почти белый мелкий roмоrенный
порошок с размером частиц около 15 мкм.
рН (2.2.3): должен выдерживать требования
для силика2еля G Р
Силикаrель Н силанизированный.
1076600.
При20товление тОНК020 слоя. См. силика
2ель HF 254 силанизированный Р
Белый или почти белый мелкий roмоrенный
порошок; после встряхивания с водой всплы
вает на поверхность вследствие rидрофобных
свойств.
Хромат02рафическая разделяющая спо
собность. Должен выдерживать испытание для
силика2еля HF 254 силанизuроваННО20 Р
Силикаrель HF 254 . 1076700.
Содержит около 1,5 % флуоресцентноro ин
дикатора, имеющеro оптимальную интенсив
ность при длине волны 254 нм.
Белый или почти белый мелкий roмоrенный
порошок с размером частиц около 15 мкм.
рН (2.2.3). Должен выдерживать требование
для силuка2еля G Р
Флуоресценция. Должен выдерживать Tpe
бование для силика2еля GF 254 Р
Силикаrель HF 254 силанизированный.
1076800.
Содержит около 1,5 % флуоресцентноro ин
дикатора, имеющеro оптимальную интенсив
ность при длине волны 254 нм.
Белый или почти белый мелкий roмоrенный
порошок; после встряхивания с водой всплы
вает на поверхность вследствие rидрофобных
свойств.
При20товление тОНК020 слоя. 30 r силика
rеля GF 254 силанизированноro энерrично встря
хивают с 60 мл смеси метанол Р вода Р (1 :2,
об/об) в течение 2 мин. Тщательно очищенные
пластинки покрывают слоем толщиной 0,25 мм,
используя устройство для нанесения. Пластинки
с покрытием сушат на воздухе, затем наrревают
при температуре от 100 ос до 105 ос в течение
30 мин.
Хромат02рафическая разделяющая спо
собность. В коническую колбу вместимостью
250 мл помещают по 0,1 r метиллаурата Р,
метилмиристата Р, метилпальмитата Р и
метилстеарата Р, прибавляют 40 мл pacт
вора калия 2идроксида спиртов020 Р и Ha
rревают с обратным холодильником на водя
ной бане в течение 1 ч. Охлаждают, пере носят
раствор в делительную воронку с помощью
100 мл воды Р, подкисляют (рН от 2 до 3) Kиc
лотой хлорuстоводородной разведенной Р и
трижды встряхивают с хлороформом Р пор
циями по 1 О мл каждая. Объединенные хло
роформные извлечения сушат над натрия
сульфатом безводным Р, фильтруют и BЫ
паривают досуха на водяной бане. Остаток
растворяют в 50 мл хлороформа Р Проводят
определение методом тонкослойной xpOMaTO
rрафии (2.2.27), используя в качестве ТOHKO
ro слоя силикаrель HF 254 силанизированный.
На хроматоrрафическую пластинку наносят в
три точки по 1 О мкл хлороформноro раствора
и хроматоrрафируют в системе растворителей
кислота уксусная ледяная Р вода Р ди
оксан Р (10:25:65, об/об/об). Korдa фронт paCT
ворителей пройдет 14 см, пластинку сушат
при температуре 120 ос в течение 30 мин, ox
лаждают, опрыскивают раствором 35 r/л Kиc
лоты фосфорномолибденовой Р в 2 пропа
ноле Р и наrревают при температуре 150 ос
до появления пятен. Пластинку обрабатывай
парами аммиака до получения белоro фона.
На xpoMaTorpaMMax должны обнаруживаться
четыре четко разделенных хорошо выражен
ных пятна.
Силикаrель ОС для хиральных разделе
ний. 1146800.
Силикаrель для хроматоrрафии очень тонко
измельченный с размером частиц 5 мкм, покры
тый следующим производным:
,оь 1
R1 1
(Y y
R/ I
"""- о
Силикаrель OD дЛЯ хиральных разделе
ний. 1110300.
Силикаrель для хроматоrрафии очень тонко
измельченный с размером частиц 5 мкм, покры
тый следующим производным:
j o f: 1oe
R 1
нзе у У
R/ = I
"""- о
ен з
Силикаrель OJ дЛЯ хиральных разделе-
ний. 1179800.
Очень тонко измельченный силикаrель для
хроматоrрафии, состоящий из сферических
4. 1. 1. Реактивы
695
частиц, по крытых трис(4 метилбензоат) целлю
лозой. Размер частиц указывают после назва
ния сорбента в испытаниях, в которых он ис
пользуется.
Силикаrель п акцептор/п донор для хи-
ральных разделений. 1160100.
Очень тонко измельченный сферический
силикаrель для хроматоrрафии ковалент
но связанный с 1 (3,5 динитробензамидо)
1 ,2,3,4 тетраrидрофенантреном, обладаю
щий п электрон акцепторными и п электрон
донорными свойствами. Размер частиц и KOH
фиrурацию указывают после названия cop
бента в испытаниях, в которых он использу
ется.
Силикаrель безводный. 1076100.
[112926 00 8].
Аморфная кремневая кислота, частично
обезвоженная и полимеризованная, поrлоща
ющая при температуре 20 ос около 30 % воды
относительно своей массы. Практически He
растворим в воде, частично растворим в pac
творах натрия rидроксида. Содержит подходя
щий индикатор наличия влаrи, для котороro на
этикетке указано изменение цвета при пере
ходе от rидратированноrо состояния к безво
дному.
Силикаrель для хроматоrрафии. 1076900.
Силикаrель очень тонко измельченный, с
размером частиц от 3 мкм ДО 1 О мкм. Размер
частиц указывают после названия сорбента в
испытаниях, в которых он используется.
Белый или почти белый мелкий rомоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, амино
rексадецилсилильный. 1138400.
Силикаrель очень тонко измельченный, с
размером частиц от 3 мкм ДО 1 О мкм И поверхно
стью, химически модифицированной аминоrек
садецилсилильными rруппами. Размер частиц
указывают после названия сорбента в испыта
ниях, в которых он используется.
Белый или почти белый мелкий rомоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, амино
пропилметилсилильный.1102400.
Силикаrель очень тонко измельченный, с
размером частиц от 3 мкм ДО 1 О мкм И поверхно
стью, химически модифицированной аминопро
пилметилсилильными rруппами. Размер частиц
указывают после названия сорбента в испытани
АХ, в которых он используется.
Белый или почти белый мелкий roмоrенный
nopowок. Практически нерастворим в воде и в
86 % спирте.
Силикаrель для хроматоrрафии, ами
нопропилсилильный. 107700а
Силикаrель очень тонко измельченный,
с размером частиц от 3 мкм ДО 1 О мкм И по
верхностью, химически модифицированной
аминопропилсилильными rруппами. Размер
частиц указывают после названия сорбента в
испытаниях, в которых он используется.
Белый или почти белый мелкий rOMoreH
ный порошок. Практически нерастворим в
воде и в 96 % спирте.
с или каrельдля хроматоrрафии, анионо
обменный сильноосновный. 1077800.
Силикаrель очень тонко измельченный с
размером частиц от 3 мкм ДО 1 О мкм И по-
верхностью, химически модифицирован
ной четвертичноro аммониевыми rруппами.
Размер частиц указывают после названия
сорбента в испытаниях, в которых он ис
пользуется.
Белый или почти белый мелкий rOMoreH
ный порошок. Практически нерастворим в
воде и в 96 % спирте.
рН: используемые пределы от 2 до 8.
Силикаrель для хроматоrрафии, бу-
тилсилильный. 107620а
Силикаrель очень тонко измельченный, с
размером частиц от 3 мкм ДО 1 О мкм И поверх
ностью, химически модифицированной бутил
силильными rруппами. Размер частиц указы-
вают после названия сорбента в испытаниях,
в которых он используется.
Белый или почти белый мелкий roMoreH
ный порошок.
Практически нерастворим в воде и в 96 %
спирте.
Кремния диоксид сфероидальный: 30 нм.
Объем пор: 0,6 смЗ/r.
Удельная площадь поверхности: 80 M2fr.
Силикаrель для хроматоrрафии, reK-
силсилильный. 1077100.
Силикаrель очень тонко измельченный,
с размером частиц от 3 мкм ДО 1 О мкм И по
верхностью, химически модифицированной
rексилсилильными rруппами. Размер частиц
указывают после названия сорбента в испы
таниях, в которых он используется.
Белый или почти белый мелкий roMoreH
ный порошок. Практически нерастворим в
воде и в 96 % спирте.
Силикаrель для хроматоrpафии, rи
дрофильный. 1077200.
Силикаrель очень тонко измельченный, с
размером частиц от 3 мкм ДО 1 О мкм, поверх
ность котороro модифицирована с целью при
дания rидрофильных свойств. Размер частиц
указывают после названия сорбента в испы
таниях, в которых он используется.
696
rосударственная фармакопея Республики Беларусь
Силикаrель для хроматоrрафии, диизо-
пропилцианопропилсилильный. 116Е 100.
Очень тонко измельченный силикаrель, химиче
ски модифицированный диизопропилцианопро
пилсилильными rруппами. Размер частиц указы
вают после названия сорбента в испытаниях, в
которых он используется.
Силикаrель для хроматоrрафии, диме-
тилоктадецилсилильный. 1115100.
Силикаrель очень тонко измельченный, с
размером частиц от 3 мкм ДО 1 О мкм И поверхно
стью, химически модифицированной диметилок
тадецилсилильными rруппами. Размер частиц
указывают после названия сорбента в испыта
ниях, в которых он используется.
Белый или почти белый мелкий roмоrенный
порошок. Практически нерастворим в воде и в
96 % спирте. Размер частиц ирреryлярный.
Удельная площадь поверхности: 300 M2fr.
Силикаrель для хроматоrрафии, диоль-
ный. 1110000.
Сферические частицы кремния диоксида с
привитыми диrидроксипропильными rруппами.
Размер пор 10 нм.
Силикаrель для хроматоrрафии, доде-
цилсилильный, зндкепированный. 1179700.
Силикаrель очень тонко измельченный, с
поверхностью, химически модифицированной
додецилсилильными rруппами. Для сведения
к минимуму какоro либо взаимодействия с oc
новными соединениями тщательно эндкепиру
ют для устранения большинства оставшихся
силанольных rрупп.
Силикаrель для хроматоrрафии, катионо-
обменный сильнокислотный. 1161400.
Силикаrель очень тонко измельченный, с
размером частиц от 5 мкм ДО 1 О мкм И поверхно
стью, химически модифицированной rруппами
сульфоновой кислоты. Размер частиц указыва
ют после названия сорбента в испытаниях, в KO
торых он используется.
Силикаrель для хроматоrрафии, моди-
фицированный амилозой. 1109800.
Силикаrель очень тонко измельченный, с раз
мером частиц 1 О мкм И поверхностью, химиче
ски модифицированной производным амилозы.
Размер частиц указывают после названия cop
бента в испытаниях, в которых он используется.
Белый или почти белый мелкий roмоrенный
порошок.
Практически нерастворим в воде и в 96 %
спирте.
Силикаrель для хроматоrрафии, ни-
трильный.1077300.
Силикаrель очень тонко измельченный, с по
верхностью,химическимодифицированнойциано
пропилсилильными rруппами. Размер частиц
указывают после названия сорбента в испыта
ниях, в которых он используется.
Белый или почти белый мелкий roмоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, ни-
трильный Р1. 1077400.
Силикаrель очень тонко измельченный, co
стоящий из пористых сферических частиц, с хи
мически связанными нитрильными rруппами.
Размер частиц указывают после названия cop
бента в испытаниях, в которых он используется.
Белый или почти белый мелкий rомоrенный
порошок. Практически нерастворим в воде и в
6 % спирте.
Силикаrель для хроматоrрафии, ни-
трильный Р2. 1077400.
Силикаrель сверхчистый с поверхностью, хи
мически модисрицированной цианопропилсилиль
ными rpуппами. Содержит менее 20 ррт метал
лов. Размер частиц указывают после названия
сорбента в испытаниях, в которых он используется.
Белый или почти белый мелкий rомоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, OKTaдe
каноиламинопропилсилильный. 1115200.
Силикаrель тонко измельченный, с размером
частиц от 3 мкм ДО 1 О мкм И поверхностью, хими
чески модифицированной аминопропилсилильны
ми rpуппами, которые ацилированы октадеканоил
rpуппами. Размер частиц указывают после названия
сорбента в испытаниях, в которых он используется.
Белый или почти белый мелкий rомоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, октаде-
цилсилильный.1077500.
Силикаrель очень тонко измельченный, с
размером частиц от 3 мкм ДО 1 О мкм И поверх
ностью, химически модифицированной OKTaдe
цилсилильными rруппами. Размер частиц указы
вают после названия сорбента в испытаниях, в
которых он используется.
Белый или почти белый мелкий rомоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, октаде-
цилсилильный Р1. 1110100.
Силикаrель сверхчистый, очень тонко из
мельченный, с поверхностью, химически моди
фицированной октадецилсилильными rруппами.
Размер частиц, размер пор и содержание уrле
рода указывают после названия сорбента в ис
пытаниях, в которых он используется. Содержа
ние металлов должно быть менее 20 ррm.
4. 1. 1. Реактивы
697
Силикаrель для хроматоrрафии, октаде-
ЦИЛСИЛИЛЬНЫЙ Р2. 1115300.
Силикаrель сверхчистый, очень тонко из
мельченный, с размером пор 15 нм и поверх
ностью, химически модифицированной OK
тадецилсилильными rруппами (содержание
уrлерода 20 %), предназначенный для анализа
полициклических ароматических уrлеводородов.
Размер частиц указывают после названия cop
бента в испытаниях, в которых он используется.
Белый или почти белый мелкий roмоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, октаде-
ЦИЛСИЛИЛЬНЫЙ, деактивированный по отно-
шению к основаниям. 1077600.
Силикаrель очень тонко измельченный, с
размером частиц от 3 мкм ДО 1 О мкм; перед BBe
дением октадецилсилильных rрупп ero предвари
тельно обрабатывают путем тщательноro промы
вания и rидролиза большинства поверхностных
силоксановых мостиков для сведения к миниму
му взаимодействия с основными компонентами.
Размер частиц указывают после названия cop
бента в испытаниях, в которых он используется.
Белый или почти белый мелкий rомоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, октаде-
ЦИЛСИЛИЛЬНЫЙ, деактивированный по отноше-
нию к основаниям, эндкепированный. 1108600.
Силикаrель очень тонко измельченный, с раз
мером частиц от 3 мкм ДО 1 О мкм И размером пор
около 10 нм, содержит около 16 % уrлеродэ. Перед
введением октадецилсилильных rрупп еro предва
рительно обрабатывают путем тщательною промы
вания и rидролиза большинства поверхностных си
локсановых мостиков. Для дальнейшеro сведения
к минимуму какоro либо взаимодействия с OCHOB
ными соединениями тщательно эндкепируют для
устранения большинства оставшихся силанольных
rрупп. Размер частиц указывают после названия
сорбента в испытаниях, в которых он используется.
Белый или почти белый мелкий roмоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, октаде-
цилсилильный, с введенными полярными
rруппами, эндкепированный. 1165100.
Силикаrель очень тонко измельченный, с
размером частиц от 3 мкм ДО 1 О мкм И поверх
ностью, химически модифицированной OKTaдe
цилсилильными rруппами. имеющими полярные
rруппы в алифатической цепи. Поверхность cop
бента дополнительно эндкепирована. Размер
частиц указывают после названия сорбента в ис
пытаниях, в которых он используется.
Мелкий юмоrенный порошок белою или
почти белоrо цвета.
Силикаrель для хроматоrрафии, октаде-
ЦИЛСИЛИЛЬНЫЙ, эндкепированный. 1115400.
Силикаrель очень тонко измельченный, с
размером частиц от 3 мкм ДО 1 О мкм И поверхно
стью, химически модифицированной октадецил
сил ильных rруппами. Для сведения к миниму
му взаимодействия с основными соединениями
тщательно эндкепируют для устранения боль
шинства оставшихся силанольных rрупп. Размер
частиц указывают после названия сорбента в
испытаниях, в которых он используется.
Белый или почти белый мелкий rомоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, октил-
СИЛИЛЬНЫЙ. 1077700.
Силикаrель очень тонко измельченный с
размером частиц от 3 мкм ДО 1 О мкм И поверх
ностью, химически модифицированной октилси
лильными rруппами. Размер частиц указывают
после названия сорбента в испытаниях, в KOTO
рых он используется.
Белый или почти белый мелкий roMoreH
ный порошок. Практически нерастворим в воде
и 96 % спирте.
Силикаrель для хроматоrрафии, октил-
сил ильный Р1. 1077701
Силикаrель очень тонко измельченный, с
размером частиц от 3 мкм ДО 1 О мкм И поверх
ностью, химически модифицированной октил
силильными и метильными rруппами. Размер
частиц указывают после названия сорбента в
испытаниях, в которых он используется.
Белый или почти белый мелкий roмоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, октил-
СИЛИЛЬНЫЙ Р2. 1077702.
Силикаrель сверхчистый, очень тонко измель
ченный, с размером пор 10 нм И поверхностью,
химически модифицированной октилсилильными
rруппами (содержит 19 % уrлерода). Содержание
металлов должно быть менее 20 ррm.
Силикаrель для хроматоrрафии, октил-
СИЛ ильный, деактивированный по отно-
шению к основаниям, эндкепированный.
1148800.
Силикаrель очень тонко измельченный, с раз
мером частиц от 3 мкм ДО 10 мкм. Перед BBeдe
нием октилсилильных rрупп еro предварительно
обрабатывают путем тщательною промывания и
rидролиза большинства поверхностных силокса
новых мостиков. Для дальнейшеro сведения к ми
нимуму какоro либо взаимодействия с основными
соединениями тщательно эндкепируют для ycтpa
нения большинства оставшихся силанольных
rрупп. Размер частиц указывают после названия
сорбента в испытаниях, в которых он используется.
698
rосударственная фармакопея Республики Беларусь
Мелкий roмоrенный порошок белоro или
почти белоro цвета. Практически нераство .им в
воде и 96 % спирте.
Силикаrель для хроматоrрафии, октил-
сил ильный, с введенными полярными rpyn-
пами, эндкепированный. 1152600.
Силикаrель очень тонко измельченный. с
размером частиц от 3 мкм ДО 1 О мкм И поверх
ностью, химически модифицированной октил
силильными rруппами, имеющими полярные
rруппы в алифатической цепи. Поверхность
сорбента дополнительно эндкепирована.
Размер частиц указывают после названия
сорбента в испытаниях, в которых он исполь
зуется.
Мелкий rомоrенный порошок белоro
цвета.
Силикаrель для хроматоrрафии, октил-
силильный, эндкепированный. 1119600.
Силикаrель очень тонко измельченный,
с размером частиц от 3 мкм ДО 1 О мкм И по
верхностью, химически модифицированной
октилсилильными rруппами. Для сведения к
минимуму взаимодействия с основными co
единениями тщательно эндкепируют для
устранения большинства силанольных rрупп.
Размер частиц указывают после названия
сорбента в испытаниях. в которых он исполь
зуется.
Белый или почти белый мелкий roмоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, про-
пилсилильный. 1170700.
Силикаrель очень тонко измельченный, с
размером частиц от З мкм ДО 1 О мкм И поверхно
стью, химически модифицированной пропилси
лильными rруппами. Размер частиц указывают
после названия сорбента в испытаниях, в KOTO
рых он используется.
Силикаrель для хроматоrрафии, три
метилсилильный. 1115500.
Силикаrель очень тонко измелЬЧенный, с
размером частиц от 3 мкм ДО 1 О мкм И noверх
ностью, химически модифицированной трим
тилсилильными rруппами. Размер частиц указы
вают после названия сорбента в испытаниях, в
которых он используется.
Белый или почти белый мелкий roмоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для хроматоrрафии, фе-
нилсилильный. 1110200.
Силикаrель очень тонко измельченный, с
размером частиц от 5 мкм ДО 1 О мкм И поверх
ностью, химически модифицированной фе
нильными rрулпами.
Силикаrель для хроматоrрафии, фенил-
силильный Р1. 1075700.
Силикаrель тонко измельченный, с раз
мером частиц 5 мкм И поверхностью, химиче
ски модифицированной фенильными rруппами.
Размер частиц указывают после названия cop
бента в испытаниях, в которых он используется.
Белый или почти белый мелкий rомоrенный
порошок. Практически нерастворим в воде, 96 %
спирте и вметиленхлориде.
Кремния диоксид сфероидальный: 8 нм.
Удельная площадь поверхности: 180 M 2 /r.
Содержание У2лерода: 5,5 %.
Силикаrель для хроматоrрафии, циано-
силильный. 1109900.
. Силикаrель очень тонко измельченный, с
поверхностью, химически модифицированной
цианосилильными rруппами. Размер частиц YKa
зывают после названия сорбента в испытаниях,
в которых он используется.
Белый или почти белый мелкий rомоrенный
порошок. Практически нерастворим в воде и в
96 % спирте.
Силикаrель для эксклюзионной хромато-
rрафии.1077900.
Силикаrель очень тонко измельченный, с
размером частиц 1 О мкм И очень rидрофильной
поверхностью. Средний диаметр пор около 30 нм.
Он совместим с водными растворами с рН от 2
до 8 и с орrаническими растворителями. Приro
ден для разделения белков с молекулярными
массами от 1.103 до 3.105.
Синенсетин. С20Н2007' (М. м. З72,4). 1110500.
[2З06 27 6]. 3' ,4' ,5,6, 7 Пентаметоксифлавон.
Белый или почти белый кристаллический
порошок. Практически нерастворим в воде, pac
творим в 96 % спирте.
Оптическое 1Ю2Л0щение (2.2.25). Раствор
синенсетина в метаноле Р имеет 3 максимума:
при 243 им, при 268 нм, при 330 нм.
Количественное определение. Жидкостная
хроматоrpафия (2.2.29), как указано в статье Op
mocифона тычиНОЧН020 листья.
Содержание синенсетина, рассчитанное Me
тодом нормализации, должно быть не менее 95 %.
Ситостанол. С 29 Н 5 р. (М. м. 416,7).1140100.
(1946647 8]. ДиrидРо J3 ситостерол. #Ситоста
нил#.
Содержание: не менее 95,0 %.
J3-Ситостерин. C 29 H so O, (М. м. 414,7).
1140200. (8З46 5]. Стиrмаст 5 ен 3J3 ол.
22,23 Диrидростиrмастерол. I3 Ситостерол.
Белый или почти белый порошок, практиче
ски нерастворим в воде, умеренно растворим в
тетраrидрофуране.
Содержание: не менее 75,0 % (м/м) в пере
счете на сухое вещество.
4. 1. 1. Реактивы
699
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Фито
стерин.
Испытуемый раствор. 0,100 r испытуемою 06
разца растворяют в rпеrпра2идроqbуране Р и дo
водят до объема 10,0 мл этим же растворителем.
100 мкл полученноrо раствора помещают в подхо
дящую колбу объемом 3 мл и выпаривают досуха
в потоке азота Р. К остатку прибавляют 100 мкп
свежеприrотовленной смеси из 50 мкл 1 Meти
лимидазола Р и 1.0 мл 2ептафтор N метил N
(триметилсилил)бутанамида Р. Колбv плотно
укупоривают и и наrревают при температуре 100 "С
в течение 15 мин. Выдерживают до охла>IЩения.
Объем вводимой пробы: 1 мкл испы rуемою
раствора.
Сквалан. СЗОН Р ' (М м. 422,8). 1084900.
[111 01 3]. 2,6,1 О, 15.19,2з rексаметилтетракозан.
Бесцветная маслянистая жидкость. Леrко
растворим в жирных маслах, мало растворим в
ацетоне, в 96 % спирте. в кислоте уксусной ледя
ной и в метаноле.
d 20
20: от 0,811 до 0,813.
п O: от 1,451 до 1.453.
Склареол. С 2Q Н З Р2' (М м. 308,5). 1139900.
[515 03 7J. (1 R,2R,4aS,8aS) Н(3R) 3 rидрокси
3 M етил пе HT 4 eH илJ 2 ,5,5. 8a TeTpa м e
тилдекаrидронафтален 2 ол.
Кристаллы без запаха.
] 20
а : 6,7. Раствор в этаноле.
еJпература кипения 19 ....: от 218 ос до 220 ОС.
Температура плавления: от 96 ос до 98 ОС.
Склареол, используемый в 2азовой xpOMa
т02рафии, должен выдерживать следующее
дополниrпельное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28). как указано в статье Масло
шалфея мускатН020.
Содержание склареола, рассчитанное MeTO
дом нормализации, должно быть не менее 97 %.
Скополетин. С 10 Н 8 О 4 . (М м. 192,2).
1158700. [92 61 5]. 7 r идрокси 6 метокси 2Н 1
бензопиран 2 он. 7 r идрокси 6 метоксикумарин.
Бледно серовато желтые мелкие кристаллы.
Температура плавления: от 203 ос до 208 ОС.
# Смесь для спекания. 25 r мелко растертою
натрия карбоната безводН020 Р тщательно CMe
шивают в ступке с 45 r высушенноrо калия карбо
ната Р и с 25 r мелко pacTepтoro калия Hитpa
та Р. Хранят в контейнере с притертой пробкой.
Смола слабокатионитная. 1096000. См.
Анионообменная смола Р
Смола ионообменная сильнокислотная.
1085400. См. Ионообменная смола сильнокис
лотная Р
Сополимер стирол-дивинилбензола.
1085500.
Твердые, пористые rранулы из поперечно
сшитоrо полимера. Существуют различные
марки с разными размерами rранул. Размер
rранул указывают после названия реактива в ис
пытаниях. в которых он используется.
Сополимер этилвинилбензол-дивинил-
бензола. 1036900
Твердые. пористые rранулы шарообразной
формы из поперечносши roю полимера. Суще
ствуют различные марки с разными размерами
rранул. Размер rранул указывают в испытаниях,
е которых он используется.
Сополимер этилвинилбензол-дивинил-
бензола Р1. 1036901.
Твердые, пористь,е rpанvлы шарообразной
формы изпоперечносшитоroполимерасноминаль
ной удельной площадью поверхности от 500 M 2 /r
до 600 M2jr и средним размером пор 7.5 нм.
Существуют различные марки с разными разме
рами rранул. Размер rранул указывают в испы
1аниях, в которых он используется
Сорбитол. См. Сорбит Р
Сорбит. 1084800. [5U 7LJ 4j
См. статью Сорбитол.
96 % спирт. 1002500. [64 17 5].
См. статью Спирт этиловый 96 %.
96 % спирт, свободный от альдеrидов.
1002501.
1200 мл 96 % спирта Р смешивают с 5 мл
раствора 4ОО r/л серебра нитрата Р и 10 мл
охлажденноro раствора 500 r/л калия 2идpoKcи
да Р, встряхивают, выдерживают в течение He
скольких дней и фильтруют.
Фильтрат переroняют непосредственно
перед использованием.
Спирт (Хпроцентов, об/об). 1002502.
Для получения раствора, содержание
спирта в котором соответствует величине Х,
смешивают соответствующие объемы воды Р и
96 % спирта Р, учитывая эффекты наrревания
и уменьшения объема, сопровождающие приro
товление такой смеси.
# Сплав Деварда. Сплав меди Р, алюминия Р
и цинка Р в соотношении 1 :0.9:0.1. Белый хрупкий
металл в виде палочек или сероro порошка.
Стандартный раствор для микроопреде-
ления воды. 1147300.
Коммерчески доступный стандартный
растор для кулонометрическоrо титрования
воды, содержащий сертифицированное количе
ство воды в подходящем растворителе.
700
rосударственная фармакопея Республики Беларусь
Стеариновая кислота. С 1в Н з6 О. (М М. 284,5).
1085200. [57 11-4]. Октадекановая кислота.
Белый или почти белый порошок или че
шуйки. Маслянистая на ощупь, практически He
растворима в воде, растворима в roрячем 96 %
спирте.
Температура плавления: около 70 ОС.
Стеариновая кислота, используемая в KO
личественном определении суммы жирных
кислот в Сабаля мелкопильчат020 плодах,
должна выдерживать следующее дополнитель
ное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Сабаля
мелкопильчат020 плоды.
Содержание стеариновой кислоты, рассчи
танное методом нормализации, должно быть не
менее 98 %.
Стрептомицина сульфат. C42H78N14024 '3H 2 S0 4 .
(М М. 1457). 1085300. [3810 74 O]. Бис[N,N'
бис( аминоиминометил ) 4 0 [5 дезокси 2 0 [2
дезокси 2 ( метиламино ) a L rлюкопиранозил]
з С фо р м ИЛ а L л и ксо фура н оз ил] D
стрептомина] трисульфат.
Стептомицина сульфат антибиотик про
дуцируемый плесневыми штаммами St'eptomy
ces griseus или полученный друrими способами.
Moryт быть добавлены стабилизаторы. Анти
микробная активность должна быть не менее
720 ЕД/мr в пересчете на сухое вещество.
Белый или почти белый порошок. rиrроско
пичен. Очень леrко растворим в воде, практиче
ски нерастворим в этаноле.
Потеря в массе при высушивании (2.2.32). Не
более 7,0 %. 1,000 r испытуемоro образца сушат
над фосфором (V) оксидом Р при температуре
60 ос и давлении не более 0,1 кПа в течение 24 ч.
Хранят в воздухонепроницаемом контейне
ре. Если субстанция стерильна, ее хранят в CTe
рильном воздухонепроницаемом контейнере с
контролем первоro вскрытия.
Стронция карбонат. SrСО з . (М м. 147,6).
1122700. [16ЗЗ 05 2].
Содержит не менее 99,5 % SrСО з .
Белый или почти белый кристаллический
порошок.
Судан красный G. C 17 H 14 NP2' (М М. 278,3).
1085800. Показатель Шульца NQ 149. ЦBeT
ной индекс NQ 12150. Растворимый красный 1.
1 [(2 Метоксифенил )азо]нафталин 2 ол.
Красновато коричневый порошок. Практиче
ски нерастворим в воде.
Хромат02рафия. Тонкослойной xpoMaтorpa
фии -(2.2.27), используя в качестве тонкоro слоя
силикаzель G Р. На хроматоrрафическую пла
стинку наносят 10 мкл раствора 0,1 r/л Судана
красноro G вметиленхлориде Р и xpoMaTorpa
фируют в том же растворителе. Длина пробе
ra фронта растворителя около 1 О см от линии
старта. На полученной xpoMaтorpaMMe должно
обнаруживаться только одно основное пятно.
Судан оранжевый. C 16 H 12 Np. (М м. 248,3).
1110700. [842 07 9]. Цветной индекс NQ 12055.
1 (Фенилазо)нафталин 2 ол. Судан 1.
Оранжево красный порошок. Практически н&
растворим в воде, растворим вметиленхлориде.
Температура плавления: около 131 ОС.
# Судан 1. См. Судан оранжевый Р.
# Судан 111. C 22 H 16 Np. (М м. 352,4).
Коричневый порошок с зеленым металли
чески м блеском. Практически нерастворим в
воде, растворим в хлороформе, в кислоте YKCYC
ной, малорастворим в 96 % спирте, в жирных и в
эфирных маслах.
# Судана 111 раствор. 0,01 r судана 111 Р paCT
воряют в 5 мл 96 % спирта Р и прибавляют 5 мл
2лицерина Р.
# Сулема. См. Ртути (11) хлорид Р.
СулЬфаминовая кислота. НзNОзS. (М м. 97,1).
1085900. [5З29 14 6].
Белый или почти белый кристаллический по
рошок или кристаллы. Леrкорастворима в воде,
умеренно растворима в ацетоне, в 96 % спирте
и в метаноле.
Температура плавления: около 205 ос с раз
ложением.
Сульфаниламид. C6HaNP2S. (М М. 172.2).
10861 00. [63 7 4 1]. 4 Аминобензолсульфонамид.
Белый или почти белый порошок. Малораст
ворим в воде, леrкорастворим в кипящей воде, в
ацетоне, в разведенных кислотах и в растворах
rидроксидов щелочных металлов, умеренно pac
творим в 96 % спирте и в петролейном эфире.
Температура плавления: около 165 ОС.
Сульфаниловая кислота. С 6 Н 7 NО з S.
(М м. 173,2). 1086200. [121 57 3]. 4 Аминобен
золеульфоновая кислота.
Бесцветные кристаллы. Умеренно раствори
ма воде, практически нерастворима в 96 % спирте.
Сульфаниловой кислоты раствор.
1086203.
О,ЗЗ r сульфаниловой кислоты Р, осторож
но наrревая при необходимости, растворяют в
75 мл воды Р и доводят кислотой уксусной ле
дяной Р до объема 100 мл.
Сульфаниловой кислоты раствор диазо-
тированной. 1086202.
0,9 r кислоты сульфаниловой Р растворяют
при наrpевании в 9 мл кислоты хлористоводород
ной Р и доводят водой Р до объема 100 мл. 10 мл
полученноro раствор охлаждают в ледяной бане,
4.1.1. Реактивы
701
прибавляют 1 О мл охлажденноro в ледяной бане
раствора 45 r/л натрия нитрита Р и выдержива
ют при температуре ООС в течение 15 мин (при TeM
пературе О ос раствор устойчив в течение 3 дней).
Непосредственно перед использованием прибав
ляют 20 мл раствора 100 r/л натрия карбоната Р.
Сульфановый синий. С27НЗ1N2NаО6S2'
(М м. 566,6). 1086000. [129 17 9]. Показатель
Шульца NQ 769. Цветной индекс NQ 42045. Ди
сульфин синий. Кислотный синий 1. Синий VS.
Натрия Ш4 (диэтиламино )фенил ](2,4 дисуль
фонатофенил )Me тилен ]циклоrекса 2, 5 диен
1 илиден ]диэтиламмоний.
Фиолетовый порошок. Растворим в воде.
Разведенные растворы имеют синюю окраску, KO
торая переходит в желтую при прибавлении кис
лотыхлористоводороднойконцентрированной.
Сульфатиазол. C9H9NP2S2' (М м. 255,3).
1086300. [72 14 0]. 4 Амино N (тиазол 2 ил)бен
золсульфонамид.
Белый или желтовато белый порошок или
кристаллы. Очень мало растворим в воде, pac
творим в ацетоне, малорастворим в 96 % спирте.
Растворяется в разведенных минеральных кис
лотах, растворах rидроксидов и карбонатов ще
лочных металлов.
Температура плавления: около 200 ОС.
# Сульфацил-натрий. См. # Натрия суль
фацетамид Р.
Сульфомолибденовый реактив Р2.
1086400.
Около 50 Mr аммония молибдата Р paCTBO
ряют в 1 О мл кислоты серной Р.
Сульфомолибденовый реактив РЗ.
1086500.
2,5 r аммония молибдата Р растворяют при Ha
rpевании в 20 мл воды Р. 28 мл кислоты серной Р
разводят в 50 мл воды Р, охлаждают. Оба раствора
смешивают и доводят водой Р до объема 100 мл.
Хранят в полиэтиленовом контейнере.
СулЬфосалициловая кислота. C 7 H 6 0 6 S'2Hp.
(М м. 254,2). 1086600. [5965 83 З]. 2 rидрокси
5 сульфобензойная кислота.
Белый или почти белый кристаллический
порошок или кристаллы. Очень леrко раствори
ма в воде и в 96 % спирте.
Температура плавления: около 109 ОС.
Сурьмы (111) хлорид. SЬСl з . (М м. 228,1).
1007700. [10025 91 9]. Сурьмы трихлорид.
Бесцветные кристаллы или прозрачная кри
сталлическая масса. rиrроскопичен. Леrкорас
творим в этаноле. Сурьмы хлорид rидролизует
ся водой.
Хранят в воздухонепроницаемом контейне
ре, защищают от влаrи.
Сурьмы (111) хлорида раствор. 1007701.
30 r сурьмы (111) хлорида Р быстро промы
вают двумя порциями по 15 мл хлороформа.
свободН020 от этанола, Р, отбрасывают np(r
мывные растворы и тотчас промытые кристал
лы растворяют при слабом наrревании в 100 мл
хлороформа, свободН020 от этанола, Р.
Хранят раствор над несколькими rраммами
натрия сульфата безводН020 Р.
Сурьмы (111) хлорида раствор Р1. 1007702.
Раствор А. 11 О r сурьмы (111) хлорида Р раст
воряют в 400 мл этиленхлорида Р, прибавляют
2 r алюминия оксида безводН020 Р, перемеши
вают и фильтруют через стеклянный фильтр (40)
(2.1.2). Фильтрат доводят этиленхлоридом Р до
объема 500,0 мл и перемешивают. Оптическая
плотность (2.2.25) полученноro раствора, изме
ренная при длине волны 500 нм В кювете с тол
щи ной слоя 2 см, не должна превышать 0,07.
Раствор В. В вытяжном шкафу смешивают
100 мл свежепереrнанноro ацетилхлорида Р и
400 мл этиленхлорида Р.
Смешивают 90 мл раствора А и 1 О мл раст
вора В.
Хранят во флаконах оранжевоro стекла с
притертой пробкой. Срок roдности 7 сут. Реактив
не roден при появлении окрашивания.
Суспензия эритроцитов кролика. 1074500.
1,6 % (обlоб) суспензию эритроцитов кроли
ка roтовят следующим образом: удаляют фибрин
из 15 мл свежеотобранной крови кролика, встря
хивая со стеклянными шариками, затем центри
фуrируют с ускорением 2000 9 в течение 1 О мин
и промывают эритроциты тремя порциями, по
30 мл каждая, раствора 9 r/л натрия хлорида Р
1,6 мл суспензии эритроцитов доводят смесью
растворителей фосфатный буферный pacт
вор рН 7,2 P раствор 9 r/л натрия хлорида Р
(1 :9, об/об) до объема 100 мл.
Таrатоза. С 6 Н 1 Р6' (М м. 180,16). 1111()()().
[87 81 0]. D ликсо rексулоза.
Белый или почти белый порошок.
[a] O: 2,3. Определение проводят. иa1Of1b3yЯ
раствор 21,9 r/л таrатозы в воде Р
Температура плавления: от 1 з4 "С до 135 ос.
Таллия сульфат. Т1 SО.. (М 11. 504.8).
1089100. [7446 18 6]. Диталлия сульфат.
Белые или почти белые ромбс-мдн ые
призмы. Малорастворим в воде. npaктичeacи
растворим в 96 % спирте.
Тальк. 1087000. (14807 96-6).
См. статью Тальк.
Таниновая киcnoтa. 1087100. [1401 55-4].
Дубильная кислота. аниН-.
Блестящие чешуйки или аморфный по
рошок от желтоватоro до светло коричневоro
702
rосударственная фармакопея Республики Беларусь
цвета. Очень леrко растворима в воде, леrкорас
творима в 96 % спирте, растворима в ацетоне.
Хранят в защищенном от света месте.
# Твин 80. См. Полисорбат 80 Р
Теофиллин. C 7 H a NP2' (М м. 180,2).
1 089300. [58 55 9]. 1 ,3 Диметил 3, 7 диrидро
1 Н пурин 2,6 дион.
Содержит не менее 99,0 % и не более
101,0 % C 7 H a N 4 0 2 в пересчете на сухое веще
ство.
Белый или почти белый кристаллический
порошок. Малорастворим в воде, умеренно pac
творим в этаноле, растворяется в растворах rи
дроксидов щелочных металлов, аммиака и ми
неральных кислот.
Температура плавления: от 270 ос до 274 ос
(после высушивания при температуре от 100 ос
до 105 ОС).
а Терпинен. С'СН'б' (М м. 136,2). 1115900.
[99 854]. 1 Изопропил4 метилциклоrекса 1 ,3 диен.
Прозрачная, почти бесцветная жидкость.
d 20
4 : около 0,837.
20
по: около 1,478.
Температура кипения: около 174 ос.
a Терпинен. используемый в 2азовой xpo
мат02рафии. должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Чайно
20 дерева масло. Содержание а терпинена, pac
считанное методом нормализации, должно быть
не менее 90 %.
у Терпинен. С'ОН'б' (М м. 136,2). 1115900. [99
854]. 1 Изопропил4 метилциклоrекса 1 ,4 диен.
Маслянистая жидкость.
у Терпинен. используемый в 2азовой xpOMa
т02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Мяты
перечной масло.
Испытуемый раствор. Испытуемый образец.
Содержание у терпинена, рассчитанное Me
тодом нормализации, должно быть не менее
93,0%.
Терпинен4 ол. с,он,р. (М м. 154,2).
1116000. [562 7 4 3]. 4 Метил 1 (1 метилэтил)
циклоrекс 3 ен 1 ол. п Мент 1 ен 4 ол.
Бесцветная маслянистая жидкость.
Терпинен 4 ол, используемый в еазовой
хромат02рафии, должен выдерживать следу
ющее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Лаван
довое масло.
Испытуемый раствор. Испытуемый образец.
Содержание терпинен4--ола, рассчитанное M
тодом нормализации, должно быть не менее 90,0 %.
а-Терпинеол. с,он,р. (М м. 154,2).
1087300 [98 55 5]. (RS) 2 (4 Метилциклоrекс
3 енил) 2 пропанол.
Бесцветные кристаллы. Практически He
растворим в воде, растворим в 96 % спирте.
d 20
20: около 0,935.
20
П D : около 1,483.
[a] O: около 92,5.
Температура плавления: около 35 ОС.
Может содержать от 1 % до 3 % j) терпинеола.
a Терпинеол, используемый в 2азовой xpo
мат02рафии, должен выдерживать следующее
испытание.
Количественное определение. r азовая xpo
матоrрафия (2.2.28), как указано статье Анисовое
масло.
Испытуемый раствор. 100 r/л а терпинеола в
2ексане Р
Содержание a терпинеола, рассчитанное Meтo
дом нормализации, должно быть не менее 97,0 %.
Не учитывают пик, соответствующий reKcaHY.
Терпинолен. С 10 Н 16 . (М м. 136,2). 1140400.
[586 62 9]. п Мента 1 ,4(8) диен. 4 Изопро
пилиден 1 метилциклоrексен.
Прозрачная, почти бесцветная жидкость.
d 20
4 : около 0,863.
20
ПО : около 1,488.
Температура кипения: около 184 ОС.
Терпинолен, используемый в 2азовой xpo
мат02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Чайно
20 дерева масло.
Содержание терпинолена, рассчитанное
методом нормализации, должно быть не менее
90%.
Тестостерон. С 19 Н 2 Р2' (М м. 288,4).1116100.
[58 22 0]. 17 rидроксиандрост 4 ен 3 он.
Содержит не менее 97,0 % и не более
103,0 % С'9Н2в02 В пересчете на сухое вещество.
Белый или почти белый кристаллический
порошок или бесцветные или желтовато бе
лые кристаллы. Практически нерастворим
в воде, леrкорастворим в спирте, метилен
хлориде, практически нерастворим в жирных
маслах.
Температура плавления: около 150 ос.
Хранят в защищенном от света месте.
ТестостеронапропионаТ'С22НЗ20з.(Мм.344,5).
1087400. [57 85 2]. 3 OKcoaHДpocт 4 eH 17 илпро
паноат.
Содержит не менее 97,0 % и не более
103,0 % С22НЗ20з в пересчете на сухое вещество.
704
rосударственная фармакопея Республики Беларусь
Тетрадекан. С'4НЗО' (М м. 198,4). 1088200.
[629 59-4]. H TeTpaдeKaH.
Содержание: не менее 99,5 % (м/м) С'4НЗО'
Бесцветная жидкость.
d 20
20: около 0,76.
20
по : около 1,429.
Температура кипения: около 252 ОС.
Температура плавления: около 5 ОС.
Тетрадециламмония бромид. C 4o H 64 BrN.
(М м. 659). 1088300. [14937 42 9]. TeTpa
кис(децил)аммония бромид.
Белый или слеrка окрашенный кристалличе
ский порошок или кристаллы.
Температура плавления: от 88 ос до 89 ос.
Тетразолиевый синий. С40НЗ2СI2NвО2'
(М м. 728). 1089000. [1871 22 3]. 3,3' (3,3' Ди
метокси[1 ,1' бифенил] 4,4' диил)бис[2,5
дифенил 2Н тетразолия] дихлорид.
Желтые кристаллы. Малорастворим в воде,
леrкорастворим в 96 % спирте и в метаноле,
практически нерастворим в ацетоне.
Температура плавления: около 245 ос с раз
ложением.
Тетракоз 15 НОВОЙ кислоты метиловый
эфир. С 25 Н 4 Р2' (М м. З80,7). 1144800. [2733
8 2). 15 Тетракозаеновой кислоты метиловый
эфир. Метилтетракоз 15 eHaT.
Содержание: не менее 99,0 %; определение
проводят методом rазовой хроматоrрафии.
Жидкость.
Тетраметиламмонияrидpoксид.С4Н,зNО'5Н20,
(М м. 181,2). 1122800. Тетраметиламмония rи
дроксид пентаrидрат.
Подходящей степени чистоты для В3ЖХ.
Тетраметиламмония rидроксида раствор.
1088600. [75 59 2). Содержит не менее 10,0 %
(м/м) С4Н,зNО'(М м. 91,2).
Прозрачная бесцветная или очень бледная
желтая жидкость. Смешивается с водой и с 96 %
спиртом.
Количественное определение. К 1,000 r
тетраметиламмония rидроксида раствору при
бавляют 50 мл воды Р и титруют 0,05 М pacт
вором кислоты серной, используя в качестве
индикатора 0,1 мл раствора метилов020 Kpac
Н020 р.
1 мл 0,05 М раствора кислоты серной соот
ветствует 9,12 Mr С4Н,зNО.
Тетраметиламмония rидроксида раствор
разведенный. 1088601.
1 О мл раствора тетраметиламмония 2и
дроксида Р доводят 96 % спиртом, свободным
от альде2идов, Р до объема 100 мл.
rотовят непосредственно перед использо
ванием.
ТетраметиламмонияrnдpocулЬфат. С4Н,зNО.S.
(М м. 171,2). 1116400. [80526 82 5].
rиrроскопичный порошок.
Температура плавления: около 295 ОС.
Тетраметиламмония хлорид. C4H'2C1N.
(М м. 109,6). 1100400. [75 57 0].
Бесцветные кристаллы. Растворим в воде и
в 96 % спирте.
Температура плавления: около 300 ос с раз
ложением.
Тетраметилбензидин. C'6H2oN2' (М м. 240,3).
11 32600.[54827 17 7].3,3' ,5,5' Тетраметилбифенил
4,4' диамин.
Порошок. Практически нерастворим в воде,
очень леrко растворим в метаноле.
Температура плавления: около 169 ОС.
Тетра метилдиа м и нодифен ил метан.
C 17 H 22 N 2 . (М м. 254,4). 1088700. [101 61 1].
4,4' Метиленбис (N,N диметиланилин).
Кристаллы или листочки от белою до roлу
бовато белоro цвета. Практически нерастворим
в воде, малорастворим в 96 % спирте, paCTBO
рим в минеральных кислотах.
Температура плавления: около 90 ОС.
Тетраметилдиаминодифенилметана pe
актив. 1088701.
Раствор А. 2,5 r тетраметилдиамино
дифенилметана Р растворяют в 1 О мл кисло
ты уксусной ледяной Р и прибавляют 50 мл
воды Р.
Раствор В. 5 r калия йодида Р растворяют в
100 мл воды Р.
Раствор С. 0,30 r ниН2идрина Р растворяют
в 1 О мл кислоты уксусной ледяной Р и прибав
ляют 90 мл воды Р.
Растворы А и В смешивают, к полученному
раствору прибавляют 1 ,5 мл раствора С.
Тетраметилсилан. C 4 H 12 Si. (М м. 88,2).
1088900. [75 76 3). TMS.
Прозрачная бесцветная жидкость. Очень
мало растворим в воде, растворим в ацетоне и
в 96 % спирте.
d 20
20: около 0,64.
20
ПО : около 1,358.
Температура кипения: около 26 ОС.
Тетраметилсилан, используемый в спек
троскопии ядеРН020 ма2нитН020 резонан
са, должен выдерживать следующее дополни
тельное испытание.
В спектре ЯМР примерно 10 % (об/об) paCT
вора тетраметилсилана в дейтерированном
хлороформе р интенсивность ЛllJбоrо посторон
неro сиrнала, за исключением тех, которые co
ответствуют вращению боковых связей и хло
роформу, не должна превышать интенсивности
боковых линий C 13, расположенных на paCCTO
янии 59,1 rц по обе стороны основноro сиrнала
тетраметилсилана.
4.1.1. Реактивы
705
Тетраметилэтилендиамин. C 6 H 16 N 2 . (Мм 116,2).
1088800. [110 18 9]. N,N,N',N' Тетра метилэти
лендиамин.
Бесцветная жидкость. Смешивается с
водой, 96 % спиртом и эфиром.
d;g: около 0,78.
20 1
п D : около 1,4 8.
Температура кипения: около 121 ОС.
# Тетрахлорметан. См. У2лерода тeтpa
хлорид Р.
Тетрахлорэтан. C 2 H 2 C1 4 . (М м. 167,9).
1088000. [79 З4 5]. 1, 1,2,2 Тетрахлорэтан.
Прозрачная бесцветная жидкость. Мало
растворим в воде, смешивается с 96 % спиртом.
d; : около 1,59.
п O: около 1,495.
Температурные пределы перееонки
(2.2.11). От 145 ос и 147 ос; должно переroнять
ся не менее 95 %.
Тетраэтиламмония rидроксида раствор.
C a H 21 NO. (м. м. 147,3). 1100300. [77 98 5].
Водный раствор 200 r/л тетраэтиламмо
ния rидроксида. Бесцветная жидкость, является
сильной щелочью.
d;g: около 1,01.
20
по: около 1,372.
Подходящей степени чистоты для В3ЖХ.
Тетраэтиламмония rидросульфат.
C e H 21 N0 4 S. (м. м. 227,3). 1116200. [16873 13 5].
rиrроскопичный порошок.
Температура плавления: около 245 ОС.
Тетраэтиленпентамин.С в Н 2з N 5 .(М м. 189,3).
1102000. [112 57 2]. 3,6,9 Триазаундекан 1,11
диамин.
Бесцветная жидкость. Растворим в ацетоне.
п O: около 1,506.
Хранят в защищенном от влаrи и тепла месте.
Тиамазол. C 4 H 6 N 2 S. (М м. 114,2). 1089400.
[60 56 0]. Метимазол. 1 Метил 1 Н имидазол
2 тиол.
Белый или почти белый кристаллический
порошок. Леrкорастворим в воде, растворим в
96 % спирте и вметиленхлориде.
Температура плавления: около 145 ОС.
2-(2-Тиенил)уксусная кислота. C 6 H 6 0 2 S.
(М м. 142,1). 1089500. [1918 77 0].
Коричневый порошок.
Температура плавления: около 65 ОС.
# Тиле реактив. 20 r натрия 2ипофосфита Р
растворяют в 40 мл воды Р. Раствор влива
ют в 180 мл кислоты хлористоводородной Р и
89 За. 1712.
оставляют на 24 ч. По осаждении выделившихся
кристаллов натрия хлорида жидкость сливают с
осадка. Раствор должен быть бесцветным.
Хранят в стеклянном контейнере с притер
той пробкой.
Тимин. C 5 H 6 NP2' (М м. 126,1). 1090400.
[65 714]. 5--Метилпиримидин 2,4(1 Н,3Н) дион.
Короткие иroльчатые кристаллы или пла
стинки. Малорастворим в холодной воде, paCTBO
рим в roрячей воде, растворяется в разведенных
растворах rидроксидов щелочных металлов.
Тимол. с,он,р. (М м. 150,2). 1090500.
[89 83 8]. 5 Метил 2 (метилэтил)фенол
Бесцветные кристаллы. Очень мало pac
творим в воде, очень леrко растворим в 96 %
спирте, леrкорастворим в эфирных и жирных
маслах, умеренно растворим в rлицерине, pac
творяется в разведенных растворах rидрокси
дов щелочных металлов.
Температура плавления: от 48 ос до 52 ос.
Тимол, используемый в 2д3О8Ой XpoMaтo
2рафии, должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rаэовая XP<r
матоrрафия (2.2.28), как указано в стап.е Мяты
перечной масло.
Испытуемый раствор. 0,1 r тимола раство--
ряют в 1 О мл ацетона Р.
Содержание тимола, рассчитанное методом
нормализации, должно быть не менее 95,0 %.
Не учитывают пик, соответствующий ацетону.
Хранят в защищенном от света месте.
# Тимола раствор.
2 r тимола Р растворяют в 1 О мл 96 %
спирта Р.
Хранят в защищенном от света месте при
температуре от 15 ос до 25 ос не более 7 сут.
Тимоловый синий. С27НзоО5S' (М м. 466,6).
1090600. [76 61 9]. Тимолсульфофталеин.
4,4' (3H 2, 1 Бензоксатиол 3 илиден )бис(2 иЗ<r
пропил 5 метилфенол ) s, S диоксид.
Кристаллический порошок от корич
леноro до зеленовато синеro цвета. Малopacтвcr
рим в воде, растворим в 96 % спирте и в разведен
ных растворах rидроксидов щелочных металлов.
Тимоловоrо синеrо раствор. 1090601.
0,1 r тимолов020 сине20 Р растворяют в
смеси из 2,15 мл 0,1 М раствора натрия 2идpOK
сида и 20 мл 96 % спирта Р, доводят водой Р до
объема 100 мл.
Испытание на чувствительность. К 0,1 мл
раствора тимоловоro синеro прибавляют 100 мл
воды, свободной от У2лерода диоксида, Р и
0,2 мл 0,02 М раствора натрия 2идроксида. По
является синее окрашивание, которое перехо
дит в желтое при прибавлении не более 0,1 мл
0,02 М раствора кислоты хлористоводородной.
706
rосударственная фармакопея Республики Беларусь
Изменение окраски. От рН 1,2 (красное
окрашивание) до рН 2,8 (желтое окрашивание);
от рН 8,0 (оливково зеленое окрашивание) до
рН 9,6 (синее окрашивание).
Ти молфталеи н. С2вНзоО4' (М М. 430,5).
1090700. [125 20 2]. 3,3 бис(4 rидрокси 5 изо
пропил 2 метилфенил ) 3Н изобензофуран 1 OH.
Белый или желтовато белый порошок. Прак
тически нерастворим в воде, растворим в 96 %
спирте и в разведенных растворах rидроксидов
щелочных металлов.
Тимолфталеина раствор. 1090701.
Раствор 1 r/л тимолфталеина в 96 %
спирте р
Испытание на чувствительность. К
0,2 мл раствора тимолфталеина прибавляют
100 мл воды. свободной от У2лерода диOKcи
да, Р. Раствор бесцветный. При прибавлении
не более 0,05 мл 0,1 М раствора натрия 2и
дроксида должно появиться синее окрашива
ние раствора.
Изменение окраски. От рН 9,3 (бесцветный)
до рН 10,5 (синее окрашивание).
Тиоацетамид. C 2 H 5 NS. (М м. 75,1). 1089600.
[62 55 5].
Кристаллический порошок или бесцветные
кристаллы. Леrко растворим в воде и в 96 %
спирте.
Температура плавления: около 113 ос.
Тиоацетамида раствор. 1089602.
Раствор 40 r/л тиоацетамида Р.
Тиоацетамида реактив. 1089601.
К 0,2 мл раствора тиоацетамида Р при
бавляют 1 мл смеси из 5 мл воды Р, 15 мл 1 М
раствора натрия 2идроксида и 20 мл 2лицери
на (85 %) Р и наrревают на водяной бане в тече
ние 20 с.
rотовят непосредственно перед использо
ванием.
Тиобарбитуровая кислота. C 4 H 4 N 2 0 2 S.
(М М. 144,2). 1111200. [504 17 6]. 4,6 Диrидрокси
2 сульфанилпиримидин.
Тиоrликолеваякислота.С 2 Н 4 О 2 S.(Мм. 92, 1).
1089700. [68 11 1]. 2 Меркаптоуксусная кислота.
Бесцветная жидкость. Смешивается с
водой, растворима в 96 % спирте.
Тиомерсал. C 9 H g HgNa0 2 S. (М м. 404,8).
1 089800 [54 64 8]. Натрия меркуротиолат.
Натрия 2 [( этилмеркурио )тио ]бензоат.
Леrкий желтовато белый кристаллический
порошок. Очень леrко растворим в воде, леrко
растворим в 96 % спирте.
Тиомочевина. CH 4 N 2 S. (М М. 76,1). 1089900.
[62 56 6].
Белый или почти белый кристаллический
порошок Щ1И кристаллы. Растворима в воде и в
96 % спирте.
Температура плавления: около 178 ос.
Тирамин. C a H 11 NO. (М М. 137.2). 1117600.
[51 67 2]. 4 (2 Аминоэтил)фенол.
Кристаллы. Умеренно растворим в воде.
растворим в кипящем этаноле.
Температура плавления: от 164 ос до 165 ос.
Тирозин. С g Н 11 NО з . (М м. 181,2). 1094800.
[60 18 4]. 2 Амино 3 (4 rидроксифенил)пропи
оновая кислота.
_ Белый или почти белый кристаллический
порошок или бесцветные или белые или почти
белые кристаллы. Малорастворим в воде, практи
чески нерастворим в ацетоне и в этаноле, paCTBO
рим в кислоте хлористоводородной разведенной
и растворах rидроксидов щелочных металлов.
Хромат02рафия. Определение проводят,
как указано в статье Леводопа. На полученной
xpoMaTorpaMMe должно обнаруживаться только
одно основное пятно.
Титан. Ti. (А м. 47,88). 1091000. [7440 32 6].
Содержание: не менее 99 % Ti.
Металлический порошок, или тонкая прово
лока диаметром не более 0,5 мм или ryбка.
Температура плавления: около 1668 ос.
Плотность: около 4,507 r/см З .
Титана диоксид. 1117900. [13463 67 7].
См. статью Титана диоксид.
Титана (111) хлорид. ТiСl з . (М м. 154,3).
1091200. [7705 07 9]. Титана трихлорид.
Красновато фиолетовые кристаллы.
Расплывается на воздухе. Растворим в воде и
96 % спирте.
Температура плавления: около 440 ос.
Хранят в воздухонепроницаемом контейнере.
Титана (111) хлорида раствор. 1091201.
Раствор 150 r/л титана (111) хлорида Р в
растворе кислоты хлористоводородной (100 r/л
HCI).
d 20
20:0КОЛО 1,19.
Титана (111) хлорид и кислоты серной pe
актив. 1091202.
20 мл раствора титана (111) хлорида Р ocтo
рожно смешивают с 13 мл кислоты серной Р,
прибавляют достаточное количество раствора
водорода пероксида концентрироваННО20 Р
до получения желтоro окрашивания, наrревают
до начала выделения белых паров и охлажда
ют. Разводят водой Р, повторяют выпаривание и
прибавление воды Р до получения бесцветноro
раствора. Доводят водой Р до объема 100 мл.
4.1.1. Реактивы
707
Титановый желтый. C2BH'9N5Na206S4'
(м. м. 696). 1090900. [1829 00 1]. Показатель
Шульца NQ280. Цветной индекс NQ 19540. Тиа
золовый желтый. Динатрия 2,2' [(1 триазен 1 ,3
диил )диА, 1 фенилен ]бис (6 метилбензотиазол
7 сульфонат].
Желтовато коричневый порошок. Леп<орас
творим в воде и в 96 % спирте.
Титановоrо желтоrо бумаrа. 1090901.
Полоски фильтровальной бумаrи поrружают
в раствор титанов020 желт020 Р, выдержива
ют несколько минут и сушат при комнатной TeM
пературе.
Титановоrо желтоrо раствор. 1090902.
Раствор 0,5 r/л титанов020 желт020 Р.
Испытание на чувствительность. К
1 О мл воды Р прибавляют 0,1 мл раствора ти
тановоro желтоro, 0,2 мл этаЛОННО20 pacт
вора ма2ния (10 ррт Mg) Р и 1,0 мл 1 М pacт
вора натрия 2идроксида. Полученный раствор
сравнивают с эталонным раствором, приrотов
ленным таким же образом, за исключением
маrния. Должно наблюдаться отчетливое розо
вое окрашивание.
Тозиларrинина метиловоrо эфира rидро-
хлорид. C'4H22NP4S'HCI. (м. м. 378,9). 1092000.
[1784 03 8]. N Тозил L арrинин метилово
ro эфира rидрохлорид. Метил ( S) 5 rуаниди
Ho 2 ( 4 метилбензолсульфонамидо )валерата rи
дрохлорид.
[a] O: от 12 до 16. Определение проводят,
используя раствор 40 r/л тозиларrинина метило
воro эфира rидрохлорида.
Температура плавления: около 145 ОС.
Тозиларrинина метиловоrо эфира rи-
дрохлорида раствор. 1092001.
К 98,5 Mr тозилаР2инина метuлов020
эфира 2идрохлорида Р прибавляют 5 мл бу
феРН020 раствора триС(2идроксиметил)ами
нометана рН 8,1 Р, встряхивают до paCTBope
ния, прибавляют 2,5 мл смешаНН020 раствора
метилов020 краСН020 Р и доводят водой Р до
объема 25,0 мл.
Тозиллизилхлорметана rидрохлорид.
С'4Н22СI2NРзS.(м.м. 369,3).1092100. [4238А 1 9].
N Т озил L лизил хлорметана rидрохлорид. (3S)
7 Амино 1 хлор 3 (4 метилбензол сульФонами
до )rептан 2 он rидрохлорид.
[a] O: от 7 до 9. Определение проводят, ис
пользуя раствор 20 r/л тозил лизил хлорметана
rидрохлорида.
Температура плавления: около 155 ос с раз
ложением.
A ::': от 310 до 340. Определение проводят
при длине волны 230 нм, используя в качестве
компенсационной жидкости воду Р
Тозилфенилаланилхлорметан. С'7Н'8СINОзS.
(м. м. 351,9). 1092200. [402 71 1]. N Тозил
L фенилаланилхлорметан.
[a] O: от 85 до 89. Определение проводят.
используя раствор 10 r/л тозилфенилаланил
хлорметана в 96 % спирте Р
Температура плавления: около 105 ОС.
А 1%: от 290 до 320. Определение проводят
1см
при длине волны 228,5 нм в 96 % спирте Р
а-Токоферилацетат. 1152400. [7695 91 2].
См. статью a Токоферилацетат.
а-Токоферол. 1152300. [10191A1 0]. См.
статью a Токоферол.
о-Толидин. C'4H'6N2' (М М. 212,3). 1123000.
[119 93 7]. 3,3' Диметилбензидин.
Содержание: не менее 97,0 % C14H16N2'
Светлый коричневый кристаллический по--
рошок.
Температура плавления: около 130 ОС.
о-Толидина раствор. 1123001.
0,16 r о толидина Р растворяют в 30,0 мл
кислоты уксусной ледяной Р, прибавляют 1,0 r
калия йодида Р и доводят водой Р до объема
500,0 мл.
# Торин. См. Нафтарзон Р
о-Толуидин. C 7 H g N. (М м. 107,2). 1091700.
[95 53 4]. 2 Метиланилин.
Бледно желтая жидкость, под действием
воздуха и света становится красновато-коричне
вой. Малорастворим в воде, растворим в 96 %
спирте и в разведенных кислотах.
dig: около 1,01.
п O: около 1,569.
Температура кипения: около 200 ОС.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
о-Толуидина rидрохлорид. C H N'HCI.
(М м. 143,6). 1117300. [6З6 21 5J. 2 МеТИJiанилина
rидрохлорид. 2 Метилбензоламина rидрохлорид.
Содержание: не менее 98,0 % C 7 H,oCJN.
Температура плавления: от 215 ос дО 217 ОС.
п-Толуидин. C 7 H 9 N. (М м. 107,2). 1091800.
[10649-0]. 4 Метиланилин.
Блестящие пластинки или хлопья. Мало
растворим в воде, леrкорастворим в ацетоне и
в 96 % спирте.
Температура плавления: около 44 ОС.
Толуидиновый синий. С'5Н'БСINзS.
(М м. 305,8). 1091900. (92 31 9]. Показатель
Шульца N21041. Цветной индекс NQ 52040. Толу
идиновый синий О. 3 Амино 7 диметиламино 2
метилфенотиазина 5 хлорид.
708
rосударственная фармакопея Республики Беларусь
Темно зеленый порошок. Растворим в воде,
малорастворим в 96 % спирте.
Толуол.с 7 н в ,(м. м.92, 1). 1091300. [108 88 3].
Метилбензол.
Прозрачная бесцветная воспламеняющаяся
жидкость. Очень мало растворим в воде, смеши
вается с 96 % спиртом.
d;g: от 0,865 до 0,870.
Температура кипения: около 110 ос.
Толуол, свободный от серы. 1091301.
Должен выдерживать требования для толу
ола Р и следующие дополнительные испытания.
Серосодержащие соединения. К 1 О мл
толуола прибавляют 1 мл эта нола Р, 3 мл
раствора калия плюмбита Р и кипятят с об
ратным холодильником в течение 15 мин и
выдерживают в течение 5 мин. В водном слое
не должно быть потемнения.
Вещества. родственные тиофену. 2 мл
толуола встряхивают с 5 мл реактива изати
на Р в течение 5 мин и выдерживают в тече
ние 15 мин. В нижнем слое не должно наблю
даться синее окрашивание.
о- ТолуoncyлЬфонамид. C 7 H 9 N0 2 S. (М М. 171,2).
1091400. (88 19 7]. 2 Метилбензолсульфонамид.
Белый или почти белый кристаллический
порошок. Малорастворим в воде, растворим в
96 % спирте и в растворах rидроксидов щелоч
ных металлов.
Температура плавления: около 156 ос.
п Толуолсульфонамид. 1091500. См. To
луолсульфонамид Р.
Толуолсульфонамид. C 7 H 9 N0 2 S. (М М. 171,2)
1091500. [70 55 3]. 4 Метилбензолсульфонамид.
п Т олуолсульфонамид.
Белый или почти белый кристаллический
порошок, малорастворим в воде, растворим в
96 % спирте и в растворах rидроксидов щелоч
ных металлов.
Температура плавления: около 136 ос.
Хромат02рафия. Тонкослойная xpOMaTO
rрафия (2.2.27), как указано в статье Толбута
мид. На полученной xpoMaтorpaMMe должно об
наруживаться только одно основное пятно.
Толуолсульqpоновая кислота. С 7 Н в О з S'Н 2 О.
(м. м. 190,2). 1091600. (6192 52 5]. 4 Метилбен
золсульфоновая кислота.
Содержание: не менее 87,0 % С 7 Н в О з S.
Белый или почти белый кристаллический по
рошок или крlt1сталлы. Леrкорастворима в воде,
растворима в 96 % спирте.
TparaKaHT. 1092300. [9000 65 1].
Засохшая на воздухе камедь, вытекающая
из надрезов ствола и ветвей Ast,agalus gummifer
LabilI и друrие виды Ast,aga/us западной Азии.
Состоит из белых или желтовато белых п
чивающихся, часто морщинистых роroвидных кусков
различной формы, ровных и rладких на изломе.
Хранят в защищенном от света месте.
Треонин. 1090000. (72 19 5].
См. статью Треонин.
Триамцинолон. C 21 H 27 F0 6 . (м. М. 394,4).
1111300. (124 94 7]. 9 Фтор 11 13, 16а, 17,21 TeTpa
rидроксипреrна 1 ,4 диен 3,20 дион.
Кристаллический порошок.
Температура плавления: от 262 ос до 263 ос.
Триамцинолона ацетонид. 1133100.
(76 25 5].
См. статью Триамцинолона ацетонид.
Триацетин. С 9 Н 1 Р6' (м. М. 218,2). 1092400
[102 76 1]. Пропан 1 ,2,3 триилтриацетат.
Почти прозрачная жидкость от бесцветной
до желтоватоro цвета. Растворим в воде, смеши
вается с 96 % спиртом.
d 20
20: около 1,16.
п O: около 1,43.
Температура кипения: около 260 ос.
Трибутилфосфат. С12Н2Р4Р' (м. м. 266,3).
1179900. (126 73 8]. Трибутоксифосфина оксид.
Трибутоксифосфана оксид.
Бесцветная жидкость. Малорастворим в
воде, растворим в большинстве орrанических
растворителей.
d;;: около 0.976.
п;5: около 1,422.
Температура кипения: около 289 ос с разло
жением.
Трикозан. С 2з Н 4в . (м. М. 324,6). 1092800.
(638 67 5].
Белые или почти белые кристаллы. Практи
чески нерастворим в воде, растворим в reKcaHe.
Температура плавления: около 48 ос.
# Трилон Б. См. Натрия эдетат Р.
Триметилпентан. С в Н 1в . (М М. 114,2). 1093400.
[540 84-1]. Изооктан. 2,2,4 Т риметилпентан.
Бесцветная воспламеняющаяся жидкость.
Практически нерастворим в воде, растворим в
этаноле.
d 20
20: от 0,691 до 0,696.
20
по : от 1,391 до 1,393.
Температурные пределы пере20нки
(2.2.11). От 98 ос до 100 ос; должно переroнять
ся не менее 95 %
Триметилпентан, используемый в спек
трофотометрии, должен выдерживать следу
ющее дополнительное испытание.
4.1.1. Реактивы
709
Пропускание (2.2.25): Не менее 98 %. Опре
деление проводят в области длин волн от 250 нм
до 420 нм, используя в качестве компенсацион
ной жидкости воду Р.
Триметилпентан Р1. 1093401.
Должен выдерживать требования для
триметилпентана Р со следующим измене
нием.
Оптическая плотность (2.2.25). Не более
0,07. Определение проводят в области длин
волн от 220 нм до 360 нм, используя в качестве
компенсационной жидкости воду Р.
N, О-Бис(триметилсилил )ацетамид.
CaH 21 NOSi 2 . (М. М. 203,4). 1093600. [10416 59 8].
Бесцветная жидкость.
d;g: около 0,83.
Н- Триметисилилимидазол. C 6 H 12 N 2 Si.
(м. м. 140,З). 1100500. [18156 74 6]. 1 Триметил
силилимидазол.
Бесцветная rиrроскопичная жидкость.
d;g: около 0,96.
п : около 1,48.
Хранят в воздухонепроницаемом контейнере.
Трипсин. 1094500. [9002 07 7].
Протеолитический фермент, полученный aK
тивацией трипсиноrена, извлеченноro из подже
лудочной железы быка (Bos taurus L.).
Белый или почти белый кристаллический
или аморфный порошок. Умеренно растворим в
воде.
Трипсин дпя пептидноrо картирования.
1094600. [9002 07 7].
Трипсин высокой чистоты, обработанный с
целью повышения химотрипсиновой активности.
Триптофан. C 11 H 12 NP2' (М. М. 204,2).
1094700. [73 22 3].
Белый или желтовато--белый кристалли е
ский порошок или бесцветные кристаллы. Мало
растворим в воде, очень мало растворим в 96 %
спирте.
[a] O: около зо. Определение проводят, ис
пользуя раствор 1 О r/л триптофана.
1,З,5- Трис[З,5-Ди( 1, 1-диметилэтил)-4-rи-
дрокси-бензил)-1,З,5-триазин-2,4,6(1 Н,ЗН,5Н)-
трион. С4вН6906Nз" (М. М. 784,1). 1094000. [27676
62 6].
Кристаллический порошок белоro цвета.
Температура плавления: от 218 ос до 222 ОС.
Трис[2,4-ди(1,1-диметилэтил)фенил)-
фосфит. С 42 Н 6 Р з Р. (М. м. 647). 1094100.
(31570 044].
Белый или почти белый порошок.
Температура плавления: от 182 ОС до 186 ос.
90. За.. 1712.
Трис(rидрокси метил)а ми номета н.
С 4 Н 11 NО з . (м. м. 121,1). 1094200. [77 86 1].
Аминометилидинетри(метанол). Трометамин.
Содержит не менее 99,0 % и не более
100,5 % С 4 Н 11 NО з в пересчете на сухое вещество.
Белый или почти белый кристалличе
ский порошок или бесцветные кристаллы.
Леrкорастворим в воде, умеренно растворим
в спирте, очень мало растворим в этилаце
тате.
Температура плавления: от 168 ОС до 174 ОС.
Трис(rмдpoксиметил)аминометана раст-
вор. 1094201.
Раствор тРUC(2идроксиметил)аминомета
на Р содержит эквивалент 24,22 r С 4 Н 11 NО з в
1000,0 мл.
Трисцизноэтоксипponан. С12Н7NзОз.
(М.м.251,З). 1093900. 1,2,3 Трис(2 цианоэтокси)
пропан.
Вязкая коричнево желтая жидкость. PaCTBO
рим в метаноле.
Иаюльзуют в качестве неподвижной фазы в
rазовой )(JIOматоrрафии.
d;g: около 1,11.
Вязкость (2.2.9). Около 172 мПа'с.
Трифенилметанол. С 19 Н 16 О. (М. М. 260.З).
1093700. (76 6]. Трифенилкарбинол.
Бесцветные кристаллы. Практически He
растворим в воде, леrкорастворим в 96 % спирте.
Трифенилтетразолия хлорид. C19H15CIN4'
(м. М. 334,8). 1093800. [298 964].2,З,5 Трифенил
2H тетразолия хлорид.
Содержание: не менее 98,0 % C19H15CIN4'
Бледно или тускло желтый порошок. Pac
творим в воде, в ацетоне и в 96 % спирте.
Температура плавления: около 240 ос с раз
ложением.
Количественное определение. 1,000 r
трифенилтетразолия хлорида растворяют в
смеси из 5 мл кислоты азотной разведен
ной Р и 45 мл воды Р, прибавляют 50,0 мл
О, 1 М раствора серебра нитрата и Harpe
вают до кипения. Охлаждаю прибавляют
3 мл дибутилфталата Р, энерrично встря
хивают и титруют 0,1 М раствором аммония
тиоцианата, используя в качестве инди
катора 2 мл раствора железа (111) аммония
сульфата Р2.
1 мл 0,1 М раствора серебра нитрата со--
ответствует 3З,48 Mr C19H15CIN4'
Хранят в защищенном от света месте.
Трифенилтетразолия хлорида раствор.
1093801.
Раствор 5 r/л трифенилтетразолия хло
рида Р в 96 % спирте, свободном от альде
2идов, Р.
Хранят в защищенном от света месте.
710
Jосударственная фармакопея Республики Беларусь
Трифторуксусная кислота. C 2 HF з02'
(М м. 114,0). 1093200. [76 05 1].
Содержание: не менее 99 % C 2 HF з02'
Жидкость, смешивается с ацетоном, 96 %
спиртом и эфиром.
d 20
20: около 1,53.
Температура кипения: около 72 ос.
Используют квалификацию реактива, при
roдную для секвенирования белков.
Хранят в воздухонепроницаемом контейнере.
Трифторуксусный анrидрид. С 4 F Б О з .
(М м. 210,0). 1093300. [407 25 0].
Бесцветная жидкость.
d 20
20: около 1,5.
# Трихлорметан. См. Хлороформ Р
Трихлортрифторэтан. С 2 Сl з F з . (М м. 187,4).
1092700. [76--13--1]. 1,1,2 Трихлор--1,2.2 трифторэтан.
Бесцветная летучая жидкость. Практически
нерастворим в воде, смешивается с ацетоном.
d;g: около 1,58.
Температурные пределы пере20нки
(2.2.11). От 47 =с до 48 ос; должно переroняться
не менее 98 %.
Трихлоруксусная кислота. С 2 НСJ з 0 2 .
(М м. 163.4). 1092500. [76 03 9].
Бесцветные кристаллы или кристалличе
ская масса. Очень леrко расплывается на воз
духе, очень леню растворима в воде и в 96 %
спирте.
Хранят в воздухонепроницаемом контейнере.
Трихлоруксусной кислоты раствор.
1092501.
40.0 r трихлоруксусной кислоты Р paCTBO
ряют в воде Р и доводят до объема 1000,0 мл
этим же раствором. Концентрацию определяют
титрованием О. 1 М раствором натрия 2идpOK
сида и, если необходимо. доводят до KOHцeHTpa
ции (40:t1) r/л.
1,1,1-Трихлорэтан. С 2 Н з Сl з . (М м. 133,4).
1092600. [71 55 6]. Метилхлороформ.
Невоспламеняющаяся жидкость. Практиче
ски нерастворим в воде, растворим в ацетоне и
в метаноле.
d;g: около 1,34.
п O: около 1,438.
Температура кипения: около 74 ос.
Трихлорэтилен. С 2 НСl з . (М м. 131,4).
110210О. [79 01 6].
Бесцветная жидкость. Практически нераст
ворим в воде, смешивается с 96 % спиртом и
эфиром.
d;g: около 1,46.
п O: около 1,477.
Триэтаноламин. 1092900. [102 71 6].
См. статью Троламин.
Триэтиламин. С Б Н 15 N. (М м. 1 01,2).
1093000. [121A4 8]. N,N Диэтилэтанамин.
Бесцветная жидкость. Малорастворим в
воде при температуре ниже 18,7 ОС, смешивает
ся с 96 % спиртом.
d 20
20: около 0,727.
20
по: около 1.401.
Температура кипения: около 90 ос.
Триэтилендиамин. C 6 H 12 N 2 . (М м. 112,2).
1093100. 1 ,4 Диазабицикло[2.2.2]октан.
Кристаллы. Очень rиrроскопичны. Леrко cy
блимируется при комнатной температуре. Леrко
растворимы в воде, в ацетоне и в этаноле.
Температура кипения: около 174 ос.
Температура плавления: около 158 ос.
Хранят в воздухонепроницаемом контейнере.
Тромбин бычий. 1090200. [9002 04A].
Ферментный препарат, полученный из
бычьей плазмы, превращающий фибриноrен в
фибрин.
Желтовато белый порошок.
Хранят при температуре не выше О ос.
Тромбин человеческий. 1090100. [9002 04A].
Сухой тромбин человеческий. Ферментный
препарат, превращающий фибриноrен челове
ческий в фибрин. Получают из жидкой челове
ческой плазмы путем осаждения подходящими
солями и орrаническими растворителями в усло
виях контроля рН, ионной силы и температуры.
Желтовато белый порошок. Леrкорастворим
в растворе 9 r/л натрия хлорида с образованием
мутноro бледно желтоro раствора.
Хранят в стеклянных контейнерах, укупо
ренных в атмосфере азота, при температуре не
выше 25 ос.
Тромбина человеческоrо раствор. 1090101.
Тромбин человеческий Р переводят в paCT
вор, как указано производителем, и разводят
буферным раствором триС(2идроксиметил)
аминометана натрия хлорида рН 7,4 Р дО KOH
центрации 5 МЕ/мл.
Тромбопластина реактив. 1090300.
1,5 r МОЗ2а бычье20, высушеНН020 aцeтo
ном Р, экстраrируют 60 мл воды Р при темпе
ратуре 50 ос в течение 15 мин, центрифуrиру
ют при 1500 об/мин в течение 2 мин и сливают
надосадоччую жидкость. Экстракт сохраняет aK
тивность в течение нескольких суток при xpaHe
нии в холодильнике. Может содержать 3 r/л Kpe
зола р в качестве антимикробноro консерванта.
# Трометамин. См. ТриС(2идроксимеитил)
аминометан Р
4. 1. 1. Реактивы
711
# Тропеолин 00. С1вН14КNРзS, (М м. 391,5).
Калиевая соль 4 [(4 анилинофенил)азо]бензол
сульфокислоты.
Оранжево желтый порошок или золотисто
желтые иroльчатые кристаллы. Растворим в ro
рячей воде и в 96 % спирте.
# Тропеолина 00 раствор.
0,1 r тропеолина 00 Р растворяют в воде Р
при наrревании, охлаждают и доводят водой Р
до объема 100 мл.
Изменение окраски. От рН 1,4 (красное
окрашивание) до рН 3,2 (желтое окрашивание).
Троповая кислота. С g Н 10 О з . (М м. 166,17).
1172000. [529 64 6). (2Rs) з r идрокси 2 фенил
пропановая кислота.
ТСХ пластинка со слоем силикаrеля.
1116700.
Подложка из стекла, металла или пластика.
покрытая слоем силикаrеля с подходящей тол
щиной и размером частиц (обычно от 2 мкм ДО
1 О мкм для пластин с мелким размером частиц
(высокоэффективная тонкослойная xpoMaтorpa
фия, ВЭТСХ) и от 5 мкм ДО 40 мкм для обычных
ТСХ пластин). Если необходимо, размер частиц
указывают после названия сорбента в испытани
ях, rдe он используется.
Сорбент может содержать связующее opra
ническое вещество.
Хромат02рафическая разделяющая спо
собность. На пластинку наносят необходи
мый объем раствора для определения при20д
ности ТСХ пластинок Р (10 мкл для обычной
пластинки и от 1 мкл ДО 2 мкл для пластинки с
мелким размером частиц). Хроматоrрафируют с
использованием подвижной фазы метанол Р
толуол Р (20:80, об/об). Korдa фронт раствори
телей пройдет две трети длины пластинки, ее
вынимают и сушат. Пластинка считается приrод
ной, если на ней видны четыре полностью раз
деленных пятна:
пятно бромкрезоловоro зеленоro с Rf не
более 0,15;
пятно метиловоro оранжевоro с Rf в преде
лах от 0,1 до 0,25;
пятно метиловоro красноro с Rf в пределах
от 0,35 до 0,55;
пятно судана красноro G с Rf в пределах от
0,75 до 0,98.
Раствор для определения приrодности
ТСХ пластинок. 1116600.
Смешивают 1,0 мл раствора 0,5 r/л судана
краСН020 G Р в толуоле Р, 1,0 мл свежеприrо
тоеленноro раствора 0,5 r/л метилов020 opaH
жев020 Р в этаноле Р, 1,0 мл раствора 0,5 r/л
6pJмкрезолов020 зелеН020 в ацетоне Р, 1,0 мл
раствора 0,25 r/л метилов020 краСН020 Р в
ацетоне Р и доводят ацетоном Р до объема
10.0 мл.
ТСХ пластинка со слоем силикаrеля F 254'
1116800.
Должна выдерживать требования для ТСХ
пластинки со слоем силика2еля Р со следующи
ми изменениями.
Содержит флуоресцентный индикатор, име
ющий оптимальную интенсивность при длине
ВОЛНbI 254 нм.
Подавление флуоресценции. На пластинку
наносят в пять точек последовательно возраста
ющие объемы от 1 мкл ДО 1 О мкл для обычной
ТСХ пластинки и от 0,2 мкл до 2 мкл для ВЭТСХ
пластинки раствора 1 r/л кислоты бензойной Р в
смеси этанол Р циКЛ02ексан Р (15:85, об/об).
Хроматоrрафируют в той же смеси растворите
лей. Korдa фронт растворителя пройдет половину
длины пластинки, ее вынимают из камеры и сушат
до испарения растворителей. Пластинку просма
тривают в УФ свете при длине волны 254 нм.
На обычных ТСХ пластинках кислота бензойная
должна обнаруживаться в виде темных пятен
на флуоресцирующем фоне примерно на ce
редине xpoMaTorpaMMbI на нанесенных количе
ствах 2 MKr и более. На ВЭТСХ пластинках кис
лота бензойная должна обнаруживаться в виде
темных пятен на флуоресцирующем фоне при
мерно на середине xpoMaтorpaMMbI для HaHeceH
ных количеств 0,2 MKr и более.
ТСХ пластинка со слоем силикаrеля G.
1116900.
Должна выдерживать требования для ТСХ
пластинки со слоем силика2еля Р со следую
щим изменением.
Содержит кальция сульфат полуrидрат
(rипс) в качестве связующеro вещества.
тех пластинка со слоем силикаrеля GF 2 s.'
1117000.
Должна выдерживать требования для ТСХ
пластинки со слоем силика2еля Р со следующи--
ми изменениями.
Содержит кальция сульфат полуrидpaт
(rипс) в качестве связующеro вещества.
Подавление флуоресценции. Должна вы--
держивать требования для ТСХ пластинки со
слоем силика2еля F 254 Р.
ТСХ пластинка со слоем силикarenя сжта
децилсилильноrо. 1148600.
Подложка из стекла, металла или пластика,
покрытая слоем силикаrеля октадецилсилиль
ноro. Может содержать орraническое связующее
вещество.
тех пластинка со слоем силикаrеля oктa
децилсилильноrо F 254' 1146600.
Подложка из стекла, металла или пласти
ка. покрытая слоем силикаrеля октадецилси
лильноro. Содержит флуоресцентный инди
катор с максимумом поrлощения при длине
волны 254 нм.
712
rосударственная фармакопея Республики Беларусь
ТСХ пластинка со слоем силикаrеля си
ланизированноrо.1117100.
Подложка из стекла, металла или пласти
ка, покрытая слоем силикаrеля силанизиро
ванноro с подходящей толщиной и размером
частиц (обычно от 2 мкм ДО 10 мкм для пластин
с мелким размером частиц (высокоэффектив
ная тонкослойная хроматоrрафия (ВЭТСХ» и
от 5 мкм ДО 40 мкм для обычных ТСХ пластин).
Если необходимо, размер частиц указывают
после названия сорбента в испытаниях, rде он
используется.
Сорбент может содержать связующее op
rаническое вещество.
Хромат02рафическая разделяющая спо
собность. По 0,1 r метиллаурата Р, метил
меристата Р, метилпальмитата Р и метил
стеарата Р помещают в коническую колбу
вместимостью 250 мл, прибавляют 40 мл
раствора калия 2идроксида спиртов020 Р и
наrревают с обратным холодильником на BO
дяной бане в течение 1 ч. Охлаждают, раствор
помещают в делительную воронку с помощью
100 мл воды Р, подкисляют кислотой хлори
стоводородной разведенной Р до рН от 2 до
3 и встряхивают с тремя порциями, по 10 мл
каждая, метиленхлорида Р. Объединенные
метиленхлоридные извлечения сушат над
натрия сульфатом безводным Р, фильтруют
и выпаривают на водяной бане досуха. Сухой
остаток растворяют в 50 мл метиленхлори
да Р (испытуемый раствор). Определение про
водят методом тонкослойной хроматоrрафии
(2.2.27), используя ТСХ пластинку со слоем
силика2еля силанизuроваНН020 Р. На пластин
КУ наносят в три точки необходимый объем ис
пытуемоrо раствора (около 10 мкл для обыч
ной ТСХ пластинки и от 1 мкл ДО 2 мкл для
ВЭТСХ пластинки с мелким размером частиц).
Хроматоrрафируют подвижной фазе кисло
та уксусная ледяная Р вода Р диоксан Р
(10:25:65, об/об/об). Коrда фронт растворите
лей пройдет две трети длины пластинки, ее
вынимают из камеры и сушат при температу
ре 120 ос в течение 30 мин. Пластинку охлаж
дают, опрыскивают раствором 35 r/л кислоты
фосфорномолибденовой Р в 2 пропаноле Р и
наrревают при температуре 150 ос до появ
ления пятен. Затем пластинку обрабатывают
парами аммиака до получения фона белоro
цвета. Пластинка считается приrодной, если
на ней видны четыре полностью разделенных
пятна.
ТСХ пластинка со слоем силикаrеля си
ланизированноrо F 254 . 1117200.
Должна выдерживать требования для ТСХ
пластинки со слоем силика2еля силанизирован
Н020 Р со следующим изменением.
Содержит флуоресцентный индикатор, име
ющий оптимальную интенсивность при длине
волны 254 нм.
Туйон. С 10 Н 16 О. (М м. 152,2). 1116500.
[546 80 5]. 4 Метил 1 (1 метилэтил )бицикло[З.1. O]
reKcaH 3 OH.
Бесцветная или почти бесцветная жидкость.
Практически нерастворим в воде, растворим в
96 % спирте и во мноrих друrих орrанических
растворителях.
# Туши черной раствор. Жидкую черную
тушь разводят водой Р в соотношении 1 :10.
Уrлеводороды с низким давлением паров
(тип L). 1049400.
Маслянистая масса. Растворимы в бензоле
и толуоле.
Уrлерод rрафитированный для xpOMaTO
rрафии. 1015900.
Уrлеродные цепочки с длиной цепи более C g ;
размер частиц от 400 мкм ДО 850 мкм.
Плотность: 0,72.
Удельная площадь поверхности: 1 О M 2 /r.
Не применяют при температуре выше 400 ОС.
Уrлерод rрафитированный для xpOMa
тоrрафии Р1. 1153500. Пористые сфериче
ские частицы уrлерода, содержащие плоские
пластинки rексаroнально выстроенных атомов
уrлерода.
Размер частиц: 5 7 мкм.
Объем пор: 0,7 смЗ/r.
# Уrлерод четыреххлористый. См. У2ле
рода тетрахлорид Р.
Уrлерода диоксид. СО 2 . (М м. 44,01).
1015600. [124 38 9]. #Уrлерода (IV) оксид.#
Содержание: минимум 99,5 % (об/об) С0 2 в
rазообразном состоянии.
Бесцветный rаз. 1 объем rаза растворяет
ся примерно в 1 объеме воды при температуре
20 ос и давлении 101 кПа.
Уrлерода диоксид Р1. СОт (М м. 44.01).
1015700.
Содержание: не менее 99,995 % (об/об) С0 2 .
У2лерода монооксид: менее 5 ррт.
Кислород: менее 25 ррm.
Оксид азота: менее 1 ррт.
Уrлерода дисульфид. CS 2 . (М м. 76,1).
1015800. [75 15 0]. #Сероуrлерод.#
Бесцветная или желтоватая воспламеня
ющаяся жидкость. Практически нерастворим в
воде, смешивается с этанолом.
d 20 : около 1,26.
20
Температура кипения: от 46 ос до 47 ОС.
Уrлерода монооксид. СО. (М м. 28,01).
1016000. [630 08 0]. #Уrлерода (11) оксид.#
Содержит не менее 99,97 % (об/об) СО.
4.1.1. Реактивы
713
Уrлерода тетрахлорид. СС'4' (М м. 153,8).
1016100. [56 23 5]. Тетрахлорметан. #Уrлерод
четыреххлористый.#
Прозрачная бесцветная жидкость. Практи
чески нерастворим в воде, смешивается с 96 %
спиртом.
d;g: от 1,595 до 1,598.
Температура кипения: от 76 ос до 77 ОС.
Уrоль активированный. 1017800. [64365--11 3].
Черный леrкий порошок без твердых частиц.
Практически нерастворим во всех обычных pac
творителях.
Потеря в массе при высушивании (2.2.32).
Не более 15 %. 1,00 r испытуемоro образца
сушат при температуре 120 ос в течение 4 ч.
Адсорбционная способность. В 100 мл кони
ческую колбу помещают 0,300 r активированноro
уrля, прибавляют 25 мл свежеприroтовленноrо
раствора 1 О r/л феназона Р. Тщательно встря
хивают в течение 15 мин. Фильтруют и отбра
сывают первые 5 мл фильтрата. К 10 мл филь
трата прибавляют 1,0 r калия бромида Р, 20 мл
кислоты хлористоводородной разведенной Р и
0,1 мл раствора метилов020 краСН020 Р. Титру
ют 0,0167 М раствором калия бромата до ис
чезновения красноro окрашивания, медленно
прибавляя титрант в конце титрования (1 капля
каждые 15 с). Параллельно проводят контроль
ный опыт, используя 10.0 мл раствора феназо
на. Рассчитывают количество адсорбированною
феназона на 100 r активированноro уrля по фор
муле:
2,35З. (а Ь)
т
rдe:
а объем 0,0167 М раствора калия бро
мата, пошедшеro на титрование контрольноro
опыта, мл;
Ь объем 0,0167 М раствора калия бро
мата, пошедшеro на титрование испытуемоro
раствора, мл;
т масса испытуемой образца, r.
100 r активированноro уrля должны aдcop
бировать не менее 40 r феназона в пересчете на
сухое вещество.
Хранят в воздухонепроницаемом контейнере.
Уксусный анrидрид. С 4 Н 6 О з . (М м. 102,1).
1000500. [108 24 7].
Содержание: не менее 97,0 % (м/м) С 4 Н 6 О з .
Прозрачная бесцветная жидкость. #При pac
творении в воде образуется кислота уксусная,
причем реакция сначала идет медленно, а затем
ускоряется и проходит бурно (возможны выбро
сы). Обращаться с осторожностью.#
Температура кипения: от 136 ос до 142 ОС.
Количественное определение. 2,00 r YKCYC
ноro анrидрида помещают в стеклянную колбу
с притертой пробкой, растворяют в 50,0 мл
1 М раствора натрия 2идроксида, кипятят с
91 3... 1712
обратным холодильником в течение 1 ч и ти
труют 1 М раствором кислоты хлористово
дородной, используя в качестве индикатора
0,5 мл раствора фенолфталеина Р. Вычис
ляют количество миллилитров 1 М раствора
натрия 2идроксида, израсходованное на ти
трование 1 r (п 1 ).
2,00 r помещают в стеклянную колбу с при
тертой пробкой, растворяют в 20 мл циКЛ02ек
сана Р, охлаждают на льду, затем прибавляют
охлажденную смесь 1 О мл анилина Р и 20 мл
циКЛ02ексана Р, кипятят с обратным холодиль
ником в течение 1 ч, прибавляют 50,0 мл 1 М
раствора натрия 2идроксида, перемешивают
и титруют 1 М раствором кислоты хлористо
водородной, используя в качестве индикатора
0,5 мл раствора фенолфталеина Р. Вычис
ляют количество миллилитров 1 М раствора
натрия 2идроксида, израсходованное на титро
вание 1 r (n).
Содержание С 4 Н 6 О з в процентах рассчиты
вают по формуле:
10,2'(n 1 n2)'
YKcycHoro анrидрида раствор Р1. 1000501.
25,0 мл УКСУСН020 аН2идрида Р растворяют
в безводном пиридине Р и доводят до объема
100,0 мл этим же растворителем.
Хранят в защищенном от света и воздей
ствия воздуха месте.
YKcYCHoro анrидрида кислоты серной
раствор. 1000502.
Осторожно смешивают 5 мл УКСУСН020 aH2и
дрида Р и 5 мл кислоты серной Р. Полученную
смесь прибавляют при охлаждении по каплям к
50 мл этанола Р.
rотовят непосредственно перед использо
ванием.
Уксусная кислота безводная. С 2 Н 4 О 2 .
(М м. 60,1). 1000300. [64 19 7].
Содержание: не менее 99,6 % (м/м) С 2 Н 4 О 2 .
Бесцветная жидкость или белые или почти
белые блестящие папоротникообразные кристал
лы. Смешивается с водой или очень леrко pacтВ<r
рима в воде, в 96 % спирте, в rлицерине (85 %) и в
большинстве жирных и эфирных маслах.
d 20 : от 1,052 до 1,053.
20
Температура кипения: от 117 ос до 119 ОС.
Раствор 100 r/л кислоты уксусной безводной
имеет сильно кислую реакцию (2.2.4).
Раствор 5 r/л кислоты уксусной, нейтрали
зованный раствором аммиака разведенным Р2,
дает реакцию (Ь) на ацетаты (2.3.1).
Температура затвердевания (2.2.18). Не
менее 15.8 ОС.
Вода (2.5.12). Не более 0,4 %. Если coдep
жание воды превышает 0,4 %, прибавляют pac
считанное количество УКСУСН020 аН2идрида Р.
Хранят в защищенном от света месте.
714
rосударственная фармакопея Республики Беларусь
Уксусная кислота ледяная. С 2 Н 4 О 2 "
(М м. 60,1). 1000400. [64 19 7].
См. статью Уксусная кислота ледяная.
Уксусная кислота. 1000401.
Содержит не менее 290 r/л и не более 310 r/n
С 2 НР2 (М м. 60,1).
30 r кислоты уксусной ледяной Р доводят
водой Р до объема 100 мл.
Уксусная кислота разведенная. 1000402.
Содержит не менее 115 r/л и не более 125 r/л
С 2 НР2' (М м. 60,1).
12 r кислоты уксусной ледяной Р доводят
водой Р до объема 100 мл.
# Универсальная индикаторная бумаrа.
См. # Индикаторная бума2а универсальная Р
Уридин. C 9 H 12 NP6' (М м. 244,2). 1095100.
[58 96 8]. 1 О Рибофуранозилурацил.
Белый иnи почти белый кристаллический
порошок. Растворим в воде.
Температура плавления: около 165 ОС.
Фактора V свертывания крови раствор.
1021400.
Раствор фактора V свертывания крови
может быть приroтовлен следующим спосо
бом (или любым друrим способом, исключа
ющим фактор VIII).
rотовят реактив фактора V из свежей
оксалатной бычьей плазмы фракционирова
нием при температуре 4 ос с помощью Ha
сыщенноrо раствора аммония сульфата Р,
приrотовленным при температуре 4 ОС. OT
деляют фракцию, которая осаждается в
интервале насыщения от 38 % до 50 % и
содержит фактор V, без существенноro за
rрязнения фактором VIII. Аммония сульфат
удаляют диализом и полученный раствор
разводят раствором 9 r/л натрия хлорида Р
до получения раствора, содержащеro от
1 О % до 20 % количества фактора V, присут
ствующеro в обычной свежей плазме крови
человека.
Определение содержания фактора V.
rотовят два разведения препарата факто
ра V в имидазольном буферном растворе
рН 7,3 Р, содержащих один объем препа
рата в 10 и в 20 объемах буферноro paCT
вора соответственно. Каждый раствор ис
пытывают следующим образом: смешивают
0,1 мл субстрата плазмы без фактора V Р,
0,1 мл испытуемоro раствора, 0,1 мл peaK
тива тромбопластина Р и 0,1 мл раствора
3,5 r/л кальция хлорида Р и измеряют время
свертывания крови, т. е. интервал между MO
ментом прибавления раствора кальция хло
рида и первым признаком образования фи
брина, который можно наблюдать визуально
или при помощи соответствующих приборов.
Таким же образом определяют время CBep
тывания крови (два параллельных определе
ния) четырех растворов обычной плазмы крови
человека в имидазольном буферном pacтвo
ре рН 7,3 Р, содержащих соответственно
1 объем в десяти (эквивалент 100 % фактора V),
1 объем в 50 (20 %), 1 объем в 100 (10 %) и
1 объем в 1 000 (1 %). Используя ДBYCTOPOH
нюю лоrарифмическую бумаrу, наносят cpeд
нее значение времени свертывания крови для
каждоro раствора плазмы человека от эквива
лента процентноro содержания фактора V, в
процентах, и с помощью интерполяции опре
деляют содержание фактора V, в процентах,
для двух разбавленных растворов фактора V.
Среднее значение двух результатов дает co
держание фактора V, в процентах, в испытуе
мом растворе.
Хранят раствор в замороженном COCTO
янии при температуре не выше 20 ОС.
Фактор Ха свертывания крови, бычий.
1037300. [9002 05 5].
Фермент, превращающий протромбин в
тромбин. Полуочищенный препарат получают
из жидкой бычьей плазмы; еro можно получить
также посредством активации проферманта
фактора Х с помощью подходящеro активатора,
такоro как яд rадюки Рассела.
Хранят лиофилизированный препарат при
температуре 20 ос, замороженный раствор
хранят при температуре ниже 20 ОС.
Фактора Ха бычьеrо раствор. 1037301.
Фактор Ха свертывания крови, бычий, Р
переводят в раствор в соответствии с указани
ями производителя и разводят буферным pac
твором триС(2идроксиметил)аминометан на
трия хлорида рН 7,4 Р
Изменение оптической плотности не должно
превышать 0,20 в мин. Измерение проводят при
длине волны 405 нм (2.2.25), используя в Ka
честве компенсационной жидкости буферный
раствор триС(2идроксиметил)аминометан на
трия хлорида рН 7,4 Р
# Фелинrа реактив.
Раствор /. 34,6 r перекристаллизованноro
меди сульфата Р растворяют в воде Р, под
кисленной 2 3 каплями раствора 160 r/л Kиc
лоты серной Р, и доводят водой Р до объема
500 мл.
Раствор 11. 173 r калия натрия тapтpa
та Р и 50 r натрия 2идроксида Р растворяют
в 400 мл воды Р и после охлаждения доводят
водой Р до объема 500 мл.
Непосредственно перед использованием
смешивают равные объемы обоих растворов.
Проверка при20дности реактива. 5 мл pe
актива Фелинrа разводят 5 мл воды Р и HarpeBa
ют до кипения. Раствор должен оставаться про
зрачным и не выделять даже следов осадка.
4.1.1. Реактивы
715
а-Фелландрен. С 10 Н 16 . (М. М. 136,2).
1130400. {4221 98 1]. (R) 5 Изопропил 2 метил
циклоrекса 1 ,3 диен. ( ) п Мента 1,5 диен.
d;g: около 0,839.
20
П D : около 1,471.
[a] O: около 217.
Температура кипения: от 171 ос до 174 ОС.
а Фелландрен, используемый в 2азовой xpo
матоарафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Эвка
липтовое масло, с использованием испытуемо--
ro образца в качестве испытуемоro раствора.
Содержание а-фелландрена, рассчитанное ме.-
тодом нормализации, должно быть не менее 98,0 %.
Феназон. C 11 H 12 Np. (М м. 188,2). 1063400.
[60 80 0]. 1 ,5 Диметил 2 фенил 1 ,2 диrидро 3Н
пиразол З он. #Антипирин.#
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Очень
леrко растворим в воде, в спирте и в метилен
хлориде.
Температура плавления: от 109 ОС до 113 ос.
Содержит не менее 99,0 % и не более 100,5 %
C11H12N20 в пересчете на сухое вещество.
Потеря в массе при высушивании (2.2.32). Не
более 1,0 %. 1,000 r испытуемоro образца сушат в
вакууме при температуре 60 ос в течение 6 ч.
Количественное определение. 0,150 r испы
TyeMoro образца растворяют в 20 мл воды Р. При
бавляют 2 r натрия ацетата Р, 25,0 мл 0,05 М
раствора йода и выдерживают в защищенном
от света месте в течение ЗО мин: Прибавля
ют 25 мл метиленхлорида Р и встряхивают до
растворения осадка. Титруют 0,1 М раствором
натрия тиосульфата, в качестве индикатора
используют раствор крахмала Р, который при
бавляют в конце титрования. Параллельно про
водят контрольный опыт.
1 мл 0,05 М раствора. йода соответствует
9,41 Mr C11H12N20,
Хранят в защищенном от света месте.
Фенантрен. С 14 Н 10 . (М. м. 178,2). 1063200.
[85 01 8].
Белые или почти белые кристаллы. Практи
чески нерастворим в воде, умеренно растворим
в 96 % спирте.
Температура плавления: около 100 ОС.
Фенантролинаrидpoхлорид.С 12 Н в N 2 'НС!'Н 2 О.
(м. м. 234,7). 1 063300. lЗ829 86 5]. 1,1О Фенан
тролина rидрохлорид моноrидрат.
Белый или почти белый кристаллический
порошок. Леrкорастворим в воде, растворим в
96 % спирте.
Температура плавления: около 215 ос с раз
ложением.
Фенилаланин. C g H 11 N0 2 . (м. М. 165,2).
1064100. [63 91 2). (S) 2 Амино 3 фенилпропа
новая кислота.
Белый или почти белый кристалличе
ский порошок или блестящие белые пластин
ки. Умеренно растворим в воде, очень мало
растворим в 96 % спирте. Растворяется в
разведенных минеральных кислотах и раз
веденных растворах rидроксидов щелочных
металлов.
Содержит не менее 98,5 % и не более
101,0 % C g H 11 N0 2 в пересчете на сухое веще
ство.
[a] : от 3З,О до 35,5 в пересчете на сухое
вещество.
Фенилrидразина rидрохлормд. C 6 H g CIN 2 .
(М М. 144,6). 1064500. [59 88--1).
Белый или почти белый кристаллический
noрошок, под действием воздуха приобретает
коричневую окраску. Растворим в воде и в 96 %
спирте.
Температура плавления: около 245 ос с раз
ложением.
Хранят в защищенном от света месте.
Фенилrидразина rидрохлорида раствор.
1064501.
0,9 r фенилаидразина аидрохлорида Р paCT
воряют в 50 мл воды Р, обесцвечивают У2лем aK
тивированным Р и фильтруют. К фильтрату при
бавляют 30 мл кислоты хлорuстоводородной Р
и доводят водой Р до объема 250 мл.
Фенилrидразина раствор в кислоте
серной. 1064502.
65 Mr фенuлаидразина zидрохлорида Р, пред
варительно перекристаллизованноro из спирта
(85 %, об/об) Р, растворяют в смеси вода Р
кислота серная Р (80:170, об/о6) и доводят до
объема 100 мл этим же растворителем. rотовят
непосредственно перед использованием.
а-Фенилrлицин. C a H g N0 2 . (М. м. 151,2).
1 064300. [28З5 06 5]. (RS}-2 Амино 2 фенил
уксусная кислота.
D-Фенилrлицин. C a H g N0 2 . (М м. 151,2).
1144500. [875 7 4 1]. (2R) 2 Амино 2 фенил
уксусная кислота.
Содержит не менее 99 % C a H g N0 2 .
Белый или почти белый кристаллический
порошок.
п-Фенилендиамина диrидрохлорид.
C s H a N 2 '2HCI. (М м. 181,1). 1064200. (615 28 1).
1.4 Диаминобензола диrидрохлорид.
Кристаллический порошок или белые или
слеrка окрашенные кристаллы. На воздухе
краснеет. Леrкорастворим в воде, малораст
ворим в 96 % спирте.
716
rосударственная фармакопея Республики Беларусь
ФеНИЛИ30тиоцианат. C 7 H 5 NS. (М м. 135,2).
1121500. [103 72 0].
Жидкость. Нерастворим в воде, растворим
в 96 % спирте.
d 20
20: около 1,13.
20
п о : около 1,65.
Температура кипения: около 221 ОС.
Температура плавления: около 21 ос.
Используют квалификацию реактива, при
roдную для секвенирования белков.
2 Феноксианилин. C ,2 H"NO. (М м. 185,2).
1165500. [2688 84 8). 2 Феноксибензенамин.
2 Аминофенил фениловый эфир.
Феноксибензамина rидрохлорид.
С,вН2зСI2NО. (М м. 340,3). 1063900. N (2
Хлорэтил ) N ( 1 метил 2 феноксиэтил )бензила
мина rидрохлорид.
Содержание: от 97.0 % до 103,0 % С1вН2зСI2NО
В пересчете на сухое вещество.
Белый или почти белый кристаллический
порошок. Умеренно растворим в воде, леrкорас
творим в 96 % спирте.
Температура плавления: около 138 ОС.
Потеря в массе при высушивании (2.2.32).
Не более 0,5 %. Сушат над фосфора (V) OKcи
дом р при давлении, не превышающем 670 Па,
в течение 24 ч.
Количественное определение. 0,500 r фе
ноксибензамина rидрохлорида растворяют в
50,0 мл хлороформа, свободН020 от этано
ла, Р и экстраrируют тремя порциями, по 20 мл
каждая, 0,01 М раствора кислоты хлористо
водородноЙ Кислотный слой отбрасывают,
хлороформный слой фильтруют через ватный
тампон. 5,0 мл полученноro фильтрата доводят
хлороформом. свободным от этанола, Р до
объема 500,0 мл. Измеряют оптическую плот
ность полученноro раствора в закрытой кювете
в максимуме при 272 нм.
Содержание С'ВН2зСI2NО рассчитывают с
учетом удельноro показателя поrлощения, paB
ноro 56,3.
Хранят в защищенном от света месте.
Феноксиуксусная кислота. СвНРз, (М м. 152,1).
1 063800. [122 59 8). 2 Феноксиэтановая кис
лота.
Почти белые кристаллы. Умеренно paCTBO
рима в воде, леrкорастворима в 96 % спирте и в
кислоте уксусной ледяной.
Температура плавления: около 98 ОС.
ХроматО2рафия. Тонкослойная xpOMa
тоrрафия (2.2.27), как указано в статье Фе
ноксиметилпенициллин. На xpoMaTorpaMMe
должно обнаруживаться только одно OCHOB
ное пятно.
Феноксиэтанол. С в Н ,0 О 2 . (М м. 138,2).
1 064000. [122 99 6]. 2 Феноксиэтанол.
Прозрачная бесцветная маслянистая жид
кость. Малорастворим в воде, леrкорастворим в
96 % спирте.
d 20
20: около 1,11.
20
п о : около 1,537.
Температура затвердевания (2.2.18): не
менее 12 ОС.
Фенол. 1063500. [108 95 2].
См. статью Фенол.
# Фенол жидкий. Кислота карболовая
жидкая. К 100 частей фенола Р, расплавленноro
на водяной бане, прибавляют 1 О частей воды Р
и тщательно перемешивают.
Содержит не менее 89,0 % С Б Н 6 О.
d;g: от 1,058 до 1,065.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте. Обращаются с
осторожностью.
Феноловый красный. 1063600. [143 74 8).
Ярко красный или темно красный кристал
лический порошок. Очень мало растворим в
воде, малорастворим в 96 % спирте.
Феноловоrо KpaCHoro раствор. 1063601.
0,1 r фенолов020 краСН020 Р растворяют в
смеси из 2,82 мл 0,1 М раствора натрия 2идpOK
сида и 20 мл 96 % спирта Р, доводят водой Р до
объема 100 мл.
Испытание на чувствительность. К 0,1 мл
раствора феноловоro KpacHoro прибавляют
100 мл воды, свободной от У2Лерода диOKcи
да, Р. Раствор желтый. При прибавлении не более
0,1 мл 0,02 М раствора натрия 2идроксида OKpa
ска должна измениться на красно фиолетовую.
Изменение окраски. От рН 6,8 (желтое OKpa
шивание) до рН 8,4 (красновато фиолетовое
окрашивание ).
Феноловоrо KpaCHoro раствор Р2. 1063603.
Раствор А. 33 Mr фенолов020 краСН020 Р
растворяют в 1,5 мл раствора натрия 2идpOKcи
да разведеНН020 Р и доводят водой Р до объема
100 мл.
Раствор В. 25 Mr аммония сульфата Р
растворяют в 235 мл воды Р, прибавляют 105 мл
раствора натрия 2идроксида разведеНН020 Р и
135 мл кислоты уксусной разведенной Р.
Раствор В смешивают с 25 мл раствора А.
При необходимости доводят до рН 4,7.
Феноловоrо KpaCHoro раствор РЗ. 1063604.
Раствор А. 33 Mr фенолов020 краСНО20 Р рас т
воряют в 1,5 мл раствора натрия 2идроксида раз
ведеНН020 Р и доводят водой Р до объема 50 мл.
Раствор В. 50 Mr аммония сульфата Р
растворяют в 235 мл воды Р, прибавляют 105 мл
раствора натрия 2идроксида разведеНН020 Р и
135 мл кислоты уксусной разведенной Р.
4.1.1. Реактивы
717
Раствор В смешивают с 25 мл раствора А.
При необходимости доводят до рН 4,7.
Фенолсульфофталеин. См. Феноловый
красный Р.
Фенолфталеин. С 20 Н'Р4' (м. М. 318,3).
1063700. [77 09 8]. 3,3 бис(4 rидроксифенил)
3Н изобензофуран 1 OH.
Порошок от белоro до желтовато белоro
цвета. Практически нерастворим в воде, paCTBO
рим в 96 % спирте.
Фенолфталеина раствор. 1063702.
0,1 r фенолфталеина Р растворяют в 80 мл
96 % спирта Р и доводят водой Р до объема 100 мл.
Испытание на чувствительность. К 0,1 мл
раствора фенолфталеина прибавляют 100 мл
воды, свободной от У2лерода диоксида, Р. При
прибавлении не более 0,2 мл 0,02 М раствора
натрия 2идроксида окраска раствора должна
измениться на розовую.
Изменение окраски. От рН 8,2 (бесцветный)
до рН 10,0 (ярко розовое окрашивание).
Фенолфталеина раствор Р1. 1063703.
Раствор 1 О r/л фенолфталеина Р в 96 %
спирте р.
Фенолфталеиновая бумаrа. 1063702.
Полоски фильтровальной бумаrи поrpужают
в раствор фенолфталеина Р, выдерживают в
течение нескольких минут и сушат.
Фенхон. С'ОН'60, (м. М. 152,2). 1037600.
[7787 20 4]. (1R) 1 ,3,3 Триметилбицикло[2.2.1]
rептан 2 он.
Маслянистая жидкость. Смешивается с
96 % спиртом, практически нерастворим в воде.
п;О: около 1,46.
Температура кипеНИЯ'5ММ: от 192 ОС до 194 ОС.
Фенхон, используемый в 2азовой xpOMaтo
2рафии, должен вblдерживать следующее иСПbl
тание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Фен
хеля 20рЬК020 плоды, используя фенхон в каче
стве испытуемоro раствора.
Содержание фенхона, рассчитанное методом
нормализации, должно быть не менее 98,0 %.
Ферроин. 1038100. [146З4 91 4].
0,7 r железа (11) сульфата Р и 1,76 r фенан
тролина 2идрохлорида Р растворяют в 70 мл
воды Р и доводят до объема 100 мл этим же
растворителем.
Испытание на чувствительносfТIЬ. К 50 мл
кислоты серной разведенной Р прибавляют
0,15 мл раствора осмия (V/II) оксида Р и 0,1 мл
ферроина. После прибавления 0,1 мл 0,1 М
раствора аммония церия нитрата красное
окрашивание раствора должно измениться на
roлубое.
92.Зак. 1712.
Ферроцифен. C26H'6FeN6' (М. м. 468,3).
1038000. [14768 11 7]. Дицианобис(1,10 фенан
тролин )железо(II).
Фиолетово бронзовый кристаллический
порошок. Практически нерастворим в воде и
в 96 % спирте.
Хранят в защищенном от влаrи и света
месте.
Феруловая кислота. С10Ню04' (М. М. 194,2).
1149500. [11 35 24 6]. 4 r идрокси 3 метоксикорич
ная кислота. 3 (4 rидрокси 3 метоксифенил)
пропеновая кислота.
Бледно желтый порошок. Леrкорастворим в
метаноле.
Температура плавления: от 172,9 ос до
173,9 ОС.
Феруловая кислота, используемая при KO
лuчественном определении элеутерозидов в
Корневищах и корнях элеутерококка, должна
выдерживать следующее дополнительное иc
пытание.
Количественное определение. Жидкост
ная хроматоrрафия (2.2.29), как указано в статье
Корневища и корни элеутерококка.
Содержание феруловой кислоты, рассчи
танное методом нормализации, должно быть не
менее 99 %.
Фибрин синий. 1101400.
1,5 r фибрина смешивают с 30 мл paCT
вора 5 r/л индu;юкармина Р в 1 % (об/об) pac
творе кислоты хлористоводородной разведен
ной Р, смесь наrревают до температуры 80 ОС,
выдерживают при этой температуре в течение
30 мин при перемешивании, охлаждают и филь
труют. Осадок тщательно промывают, ресуспен
дируя в 1 % (об/об) растворе кислоты хлорu
стоводородной разведенной Р и перемешивая
в течение 30 мин, и фильтруют. Осадок промы
вают три раза. Сушат при температуре 50 ос и
измельчают.
Фибрин KOHro красный. 1038400.
Промытый фибрин нарезают на маленькие
кусочки и оставляют на ночь в растворе 20 л
конао краСН020 Р в спирте (90 %. об/об) Р и
фильтруют. Фибрин промывают водой Р и хранят
под эфиром Р.
Фибриноrен. 1038500. [9001 32 5].
Фибриноrен получают из плазмы крови
донора. Переход фибриноrена в фибрин, проис
ходящий под влиянием тромбина, обеспечивает
конечную стадию процесса свертывания крови
образование crycTKa.
При растворении в указанном на этикет
ке объеме растворителя раствор содержит не
менее 1 О r/л фибриноrена.
Белый или желтоватый порошок или сухая
пористая масса.
Хранят в защищенном от света месте.
718
rосударственная фармакопея Республики Беларусь
Фиксирующий раствор. 1122600.
К 250 мл метанола Р прибавляют 0,27 мл
формальде2ида Р и доводят водой Р до объема
500,0 мл.
Фиксирующий раствор для изоэлектри-
ческоrо фокусирования в полиакриламид-
ном rеле. 1138700.
Раствор содержит 35 r сульфосалициловой
кислоты Р и 100 r трихлоруксусной кислоты Р
в 1 л воды Р.
# Фитоменадион. С з ,Н 46 О 2 . (м. М. 450,7).
[84 80 0]. 2 Метил 3 фитил 1 +нафтохинон.
3 Фитилменадион. Филлохинон. Витамин К I .
Вязкая жидкость желтоro цвета. Леrкорас
творим в петролейном эфире и в хлороформе,
плохо в этаноле, нерастворим в воде.
20
по: около 1,527.
Температура плавления: около 20 ОС.
# Фишера реактив. См. Йодсернистый
реактив р.
Флороrлюцин. С в Н 6 О з '2Нр. (м. М. 162,1).
1064600. [6099 90 7].
Бензол 1 ,3,5 триол.
Белые или желтоватые кристаллы. Мало
растворим в воде, растворим в 96 % спирте.
Температура плавления: около 223 ос
(метод MrHoBeHHoro плавления).
Флороrлюцина раствор. 1064601.
К 1 мл раствора 100 r/л фЛОР02люцина Р в
96 % спирте Р прибавляют 9 мл кислоты хло
ристоводородной Р.
Хранят в защищенном от света месте.
# Флороrлюцина раствор в эфире.
0,1 r ФЛОР02люцина Р растворяют в
эфире Р и доводят до объема 100 мл этим же
растворителем. Раствор при меняют свежепри
roтовленным.
# Флороrлюцина раствор в кислоте серной.
1,5 r фЛОР02люцина Р растворяют при Ha
rревании на водяной бане в смеси из 75 r воды Р
и 50 r кислоты серной Р.
Флуорантен. С'6Н'О' (м. М. 202,3). 1038600.
[206--44 0]. 1 ,2 (1 ,8 Нафтилен)бензол. 1 ,2 Бенза
ценафтен.
Желтые или желтовато--коричневые кристаллы.
Температура кипения: около 384 ОС.
Температура плавления: от 109 ОС до 110 ос.
ФлуореН'СIЗНIО.(м.м.166,2).112740D.[86 73 7].
Дифениленметан.
Белые или почти белые кристаллы. Леrко
растворим в кислоте уксусной безводной, pac
творим в roрячем 96 % спирте.
Температура плавления: от 113 ос до 115 ОС.
Флуоресцеин. С 20 Н'Р5' (м. М. 332,3).
1106300. [2З21 07 5]. 3' ,6' Диrидроксиспи
ро[ изобензофуран 1 (ЗН), 9' [9Н]KcaHTeH] 3 OH.
Оранжево красный порошок. Практически
нерастворим в воде, растворим в теплом 96 %
спирте, растворим в растворах rидроксидов ще
лочных металлов. В растворе флуоресцеин об
наруживает зеленую флуоресценцию.
Температура плавления: около З15 ОС.
Флуоресцеин сопряженная сыворотка
против бешенства. 1038700.
Иммуноrлобулиновая фракция с высоким
уровнем антител против бешенства, приrотов
ленная из сыворотки подходящих животных, им
мунизированных инактивированным вирусом
бешенства. Иммуноrлобулин сопряжен с флуо
ресцеин изотиоцианатом.
Флуфенаминовая кислота. СI4Н'0FзN02'
(м. М. 281,2). 1106200. [530 78 9]. 2 [[3 (Трифтор
метил )фенил]амино]бензойная кислота.
Бледно желтый кристаллический порошок
или иroльчатые кристаллы. Практически He
растворима в воде, леrкорастворима в 96 %
спирте.
Температура плавления: от 132 ОС до 135 ОС.
Фолиевая кислота. 1039000. [75708 92 8].
См. статью Фолиевая кислота.
# Фолина Дениса реактив. См. # Фосфор
номолибденово вольфрамовый реактив Р1.
# Фол ина реактив.
В круrлодонную колбу помещают 70 мл
воды Р, 1 О r натрия вольфрамата Р, 2,5 r Kиc
лоты фосфорномолибденовой Р, 5 мл раствора
250 r/л кислоты фосфорной Р, кипятят с обрат
ным холодильником в течение 2 ч, охлаждают,
доводят водой Р до объема 100 мл И тщательно
перемешивают.
Хранят в защищенном от света месте в CTe
клянном контейнере с притертой пробкой.
# Фолина-Чокалыеу реактив. См. Фос
форномолибденово вольфрамовый реактив Р.
# Формалин. См. Формальде2ида раствор Р.
Формальдеrид. 1039100. [50 00 0]. См.
Раствор формальде2ида Р.
Формальдеrида раствор. 1039101.
См. статью Формальде2ида 35 % раствор.
Формальдеrида раствор в серной кислоте.
1086805.
2 мл раствора формальде2ида Р смешива
ют с 100 мл кислоты серной Р.
4.1.1. РеактивЬ1
719
Формам ид. СНзNО. (М м. 45,0). 1039200.
[75 12 7].
Прозрачная бесцветная маслянистая жид
кость. rиrроскопичен. Смешивается с водой и с
96 % спиртом. rидролизуется водой.
Температура кипения: около 103 ОС. Опре
деление проводят под давлением 2 кПа.
Хранят в воздухонепроницаемом контейнере.
Формамид Р1. 1039202.
Должен выдерживать требования для фор
мамида Р и следующее дополнительное испы
тание.
Вода (2.5.12). Не более 0,1 %. Определе
ние проводят с равным объемом метанола без
водН020 Р.
Формамид обработанный. 1039201.
1,0 r кислоты сульфаминовой Р дисперrи
руют в 20,0 мл формамида Р, содержащеro 5 %
(об/об) воды Р.
# Фосфолипиды.
Определенное количество мозrа человече
скоro или бычьеro, хорошо отделенноro от KpOBe
носных сосудов, промывают и разжижают в под
ходящем устройстве. Взвешивают от 1000 r до
1300 r разжиженноrо вещества, измеряют объем
(V, мл), затем экстраrируют тремя порциями, по
4 V мл каждая, ацетона Р, и фильтруют при по
ниженном давлении. Полученный остаток сушат
при температуре 37 ос в течение 18 ч. Остаток
экстраrируют смесью растворителей петро
лейный эфир Р2 петролейный эфир Р1 (2:3)
двумя порциями, каждая по 2V мл, фильтруя
каждый экстракт через фильтровальную бумаry,
предварительно увлажненную той же смесью
растворителей. Объединенные извлечения BЫ
паривают досуха при температуре 45 ос под дaB
лением не более 670 Па. Остаток растворяют в
O,2V мл эфира Р и выдерживают при темпера
туре 4 ос до образования осадка. Прозрачную
надосадочную жидкость центрифуrируют, выпа
ривают под пониженным давлением до объема
100 мл на килоrрамм разжиженною вещества
и взвешивают. Выдерживают при температу
ре 4 ос до образования осадка (от 12 ч до 24 ч)
и центрифуrируют. К прозрачной надосадочной
жидкости прибавляют ацетон Р в количестве,
в пять раз превышающем ее объем, центрифу
rируют и отбрасывают надосадочную жидкость.
Осадок сушат.
Хранят в эксикаторе под вакуумом в защи
щенном от света месте.
Фосфора (V) оксид. РР5' (М м. 141,9).
1032900. [1314 56 3]. Дифосфора пентоксид.
Фосфорный анrидрид.
Белый или почти белый аморфный поро
шок, расплывающийся на воздухе. rидратирует
ся водой с выделением тепла.
Хранят в воздухонепроницаемом контейнере.
Фосфорная кислота. 1065100. [7664 38 2].
См. статью Фосфорная кислота KOHцeH
трированная.
Фосфорная кислота разведенная.
1065101.
См. статью Фосфорная кислота разведен
ная.
Фосфорновольфрамовой кислоты раст-
вор. 1065200.
К 1 О r натрия вольфрамата Р прибавляют
8 мл кислоты фосфорной Р и 75 мл воды Р, Ha
rревают с обратным холодильником в течение
3 ч, охлаждают и доводят водой Р до объема
100 мл.
Фосфорномолибденовая кислота.
12МоО з 'Н з Р0 4 'хНР. 1064900. [51429 74A].
ОранжеВ<rжелтые мелкие кристаллы. Леr
корастворима в воде, растворима в 96 % спирте.
Фосфорномоли6деновой кислоты раст-
вор. 1064901.
4 r фосфорномолибденовой кислоты Р
растворяют в воде Р и доводят до объема 40 мл
этим же растворителем. Осторожно при охлаж
дении прибавляют 60 мл кислоты серной Р. ro
товят непосредственно перед использованием.
Фосфорномолибденово-вольфрамовый
реактив. 1065000. #Фолина Чокальтеу реактив.#
100 r натрия вольфрамата Р и 25 r натрия
молибдата Р растворяют в 700 мл воды Р, при
бавляют 100 мл кислоты хлористоводород
ной Р и 50 мл кислоты фосфорной Р. Смесь
наrревают в стеклянной колбе с обратным xo
лодильником в течение 1 О ч, прибавляют 150 r
лития сульфата Р, 50 мл воды Р и несколько
капель брома Р. Кипятят до удаления избытка
брома (15 мин), охлаждают, доводят водой Р до
объема 1000 мл И фильтруют.
Реактив должен иметь желтую окраску. Pe
актив неприroден для использования, если при
обретает зеленый оттенок, но может быть pe
rенерирован путем кипячения с несколькими
каплями брома Р. Избыток брома обязательно
удаляют кипячением.
Хранят при температуре от 2 ос до 8 ОС.
#Фосфорномолибденово-вольфрамо-
вый реактив Р1. Реактив Фолина Дениса.
50 r натрия вольфрамата Р и 1 О r Kиc
лоты фосфорномолибденовой Р растворяют
в 350 мл воды Р и прибавляют и 25 мл кисло
ты фосфорной Р. Смесь кипятят с обратным
холодильником в течение 2 ч. Охлаждают до
комнатной температуры и доводят водой Р до
объема 500,0 мл.
Хранят в защищенном от света месте при
температуре от 2 ос до 8 ОС. Срок rодности 6
мес.
720
rосударственная фармакопея Республики Беларусь
Фосфорномолибденово вольфрамовый
реактив разведенный. 1065001.
Смешивают 1 объем фосфорномолибдено
во вольфрамов020 реактива Р с 2 объемами
воды Р.
Фруктоза. С 6 Н 1 Р6' (М м. 180,2). 1106400.
[57 48 7З. Н D арабино r екс 2 улопираноза.
Белый или почти белый кристаллический
порошок с очень сладким вкусом. Очень леrко
растворима в воде, растворима в 96 % спирте.
Фталазин. C a H 6 N 2 . (М м. 130,1). 1065400.
[253 52 1].
Бледно желтые кристаллы. Леrкорастворим
в воде, растворим в этаноле, в этилацетате и в
метаноле.
Температура плавления: от 89 ос до 92 ос.
Фталевая кислота. С в Н 6 О 4 . (М м. 166,1).
1 065600. [88 99 3З. Бензол 1 ,2 дикарбоновая
кислота.
Белый или почти белый кристаллический
порошок. Растворима в roрячей воде и в 96 %
спирте.
Фталевый альдеrид. С в НР2' (М м. 134,1).
1065300. [643 79 8]. Бензол 1 ,2 дикарбоксаль
деrид.
Желтый кристаллический порошок #или че
шуйки белоro или почти белоro цвета. #
Температура плавления: около 55 ОС.
Хранят в защищенном от света и воздей
ствия воздуха месте.
Фталевоrо альдеrида реактив. 1065301.
2,47 r кислоты борной Ррастворяют в 75 мл
воды Р, доводят раствором 450 r/л калия 2идpOK
сида Р до рН 10,4 и разводят водой Р до объема
100 мл.
1,0 r фталево20 апьде2ида Р растворяют в
5 мл метанола Р, прибавляют 95 мл приroтовлен
ноro раствора кислоты борной и 2 мл кислоты
ти02ликолевой Р и доводят раствором 450 r/л
калия аидроксида Р до рН 10,4.
Хранят в защищенном от света месте. Срок
rодности 3 сут.
Фталевый анrидрид. С 6 НРз, (М м. 148,1).
1065700. [85A4 9]. Изобензофуран 1 ,3 дион.
Содержание: не менее 99,0 % С 8 Н 4 О з .
Белые или почти белые чешуйки.
Температура плавления: от 130 ос до 132 ос.
Количественное определение. 2,000 r
фталевоrо анrидрида растворяют в 100 мл
воды Р, кипятят с обратным холодильником
в течение 30 мин, охлаждают и титруют 1 М
раствором натрия 2идроксида, используя в
качестве индикатора раствор фенолфтале
ина Р.
1 мл 1 М раствора натрия 2идроксида co
ответствует 74,05 Mr С в Н 4 О З '
Фталевоrо анrидрида раствор. 1065701.
42 r фталев020 аН2идрида Р растворяют в
ЗОО мл пиридина безводН020 Р и выдерживают
в течение 16 ч.
Хранят в защищенном от света месте. Срок
roдности 7 сут.
Фталеиновый пурпурный. СЗ2НЗ2N2012 'Н 2 О.
(М м. 637, безводное вещество). 1065500.
[2411 89 4З. Метилфталеин. 2,2',2",2"'
[0 Крезолфталеин 3' ,З" бис( метиленнитрило )]
тетрауксусная кислота. (1 ,З Диrидро З оксо
изобензофуран 1 илиден)бис[(6 rидрокси
5 метил 3.1 фенилен)бис(метиленимино)
диуксусная кислотаJ.
Порошок от желтовато белоro до корич
неватоro цвета. Практически нерастворим в
. воде, растворим в 96 % спирте. Реактив по
ступает в продажу в виде натриевой соли: по
рошок от желтовато белоro до розовоro цвета;
растворим в воде, практически нерастворим в
96 % спирте.
Испытание на чувствительность. 1 О Mr
растворяют в 1 мл раствора аммиака KOHцeH
трироваНН020 Р и доводят водой Р до объема
100 мл. К 5 мл полученною раствора прибав
ляют 95 мл воды Р, 4 мл раствора аммиака
концентрироваНН020 Р, 50 мл 96 % спирта Р
и 0,1 мл 0,1 М раствора бария хлорида. По
является сине фиолетовое окрашивание, KO
торое должно исчезнуть после прибавления
0,15 мл 0,1 М раствора натрия эдетата.
2 Фтор 2 дезокси D rлюкоза. C 6 H 11 F0 5 "
(М м. 182,2). 1113900. [86783 82 6].
Белый или почти белый кристаллический
порошок.
Температура плавления: от 174 ОС до 176 ОС.
Фтординитробензол. С 6 Н з FNР4' (М м. 186,1).
1 038800. [70 34 8]. 1 Фтор 2,4 динитробензол.
Бледно желтые кристаллы. Растворим в
пропиленrликоле.
Температура плавления: около 29 ОС.
1 Фтор 2 нитро-4 (трифторметил)бензол.
С 7 Н з F 4N02' (М м. 209,1). 1038900. [367 86 2З.
Температура плавления: около 197 ОС.
Фтористоводородная кислота. H
(М м. 20,01). 1043600. [7664 39 3].
Содержит не менее 40,0 % (м/м) HF.
Прозрачная бесцветная жидкость.
Остаток после прокаливания. Не более
0,05 % (м/м).
Кислоту фтористоводородную выпаривают
в платиновом тиrле, остаток осторожно прокали
вают до постоянной массы.
Количественное определение. В точно взве
шенную колбу со стеклянной притертой пробкой,
содержащую 50,0 мл 1 М раствора натрия 2и
4.1.1. Реактивы
721
дроксида, помещают 2 r кислоты Фтористоводо
родной И колбу с одержимым взвешивают снова.
Полученный раствор титруют 0,5 М раствором
кислоты серной, используя в качестве индика
тора 0,5 мл раствора фенолфталеина Р.
1 мл 1 М раствора натрия 2идроксида co
ответствует 20,01 Mr НЕ
Хранят в полиэтиленовом контейнере.
Фукоза. С 6 Н 1 Р5' (М. м. 164,2). 1039500.
[669641 9]. 6 Дезокси L rалактоза.
Белый или почти белый порошок. Раствори
ма в воде и в 96 % спирте.
[a] O: около 76. Определение проводят в
растворе 90 r/л фукозы через 24 ч после приro
товления.
Температура плавления: около 140 ОС.
Фуксин основный. 1039400. [632 99 5].
Смесь розанилина rидрохлорида (С20Н20СINз;
М. м. 337,9; цветной индекс NQ 42510; показа
тель Шульца NQ 780) и пара розанилина rидро
хлорида (С19Н1вСINз; М. м. 323,8; цветной индекс
NQ 42500; показатель Шульца NQ 779).
При необходимости очищают следующим об-
разом: 1 r фуксина основноro растворяют в 250 мл
кислоты хлористоводородной разведенной Р, BЫ
держивают в течение 2 ч при комнатной температу
ре, фильтруют, полученный фильтрат нейтрализу
ют раствором натрия 2идроксида разведенным Р
и прибавляют ero избыток от 1 мл ДО 2 мл. Филь
труют через стеклянный фильтр (40) (2.1.2), осадок
промывают водой Р, растворяют в 70 мл MeтaHO
ла Р, предварительно наrpэтоro до кипения, и при
бавляют 300 мл воды Р при температуре 80 ОС. Ox
лаждают и фильтруют; кристаллы сушат в вакууме.
Кристаллы с зеленовато бронзовым бле
ском. Растворим в воде и в 96 % спирте.
Хранят в защищенном от света месте.
Фуксина обесцвеченный раствор.
1039401.
0,1 r фуксина основН020 Р растворяют в
60 мл воды Р, прибавляют раствор, содержа
щий 1 r натрия сульфита безводН020 Рили 2 r
натрия сульфита Р в 1 О мл воды Р. Медлен
но и при постоянном встряхивании прибавляют
2 мл кислоты хлористоводородной Р и доводят
водой Р до объема 100 мл. Выдерживают в за
щищенном от света месте не менее 12 ч, обес
цвечивают У2лем активированным Р и фильтру
ют. Если раствор мутнеет, еro фильтруют перед
использованием. Если при стоянии раствора по
является фиолетовое окрашивание, еro снова
обесцвечивают У2лем активированным Р.
Испытание на чувствительность. К 1,0 мл
прибавляют 1,0 мл воды Р и 0,1 мл 96 % спирта,
свободН020 от альде2идов, Р. Прибавляют 0,2 мл
раствора, содержащеro 0,1 r/л формальдеrида
(СНр, М. м. 30,0). В течение 5 мин должно по
явиться бледно розовое окрашивание.
Хранят в защищенном от света месте.
Фуксина обесцвеченный раствор Р1.
1039402.
К 1 r фуксина основН020 Р прибавляют
100 мл воды Р, наrревают до температуры 50 ос
и охлаждают, периодически перемешивая. Bы
держивают в течение 48 ч, перемешивают и
фильтруют. К 4 мл фильтрата прибавляют 6 мл
кислоты хлористоводородной Р, перемешива
ют и доводят водой Р до объема 100 мл.
Раствор выдерживают в течение не менее 1 ч
перед использованием.
Фурфурол. С 5 НР2' (м. м. 96,1). 1039600.
[98 01 1]. 2 Фуральдеrид. 2 Фуранкарбальде
rид.
Прозрачная маслянистая жидкость от бесц
ветной до коричневато желтоro цвета. Смеши
вается с 11 частями воды, смешивается с 96 %
спиртом.
d : от 1,155до 1,161.
Температурные пределы пере20нки (2.2. 11).
От 159 ос и 163 ОС: должно переrоняться не
менее 95 %.
Хранят в защищенном от света месте.
Хамазулен. С 14 Н 16 . (м. м. 184,3). 1148000.
[529 05 5]. 7 Этил 1 ,4 диметилазулен.
Жидкость синеro цвета, очень мало paCTBO
рим в воде, растворим в 96 % спирте, смешива
ется с жирными маслами, эфирными маслами, с
вазелиновым маслом, растворяется с обесцве
чиванием в фосфорной кислоте (85 %, м/м) и в
серной кислоте (50 %, об/об).
Внешний вид раствора: 50 Mr растворяют
в 2,5 мл 2ексана Р. Полученный раствор синеrо
цвета должен быть прозрачным в тонком слое
наклоненной пробирки.
Хамазулен, используемый в 2азовой XpoMa
т02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
ромашки. Используют раствор 4 r/л хамазулена
в циКЛ02ексане Р.
Содержание хамазулена, рассчитанное Me
ТОДОМ нормализации, должно быть не менее
95,0%.
Хинальдиновый красный. С21Н2з1N2'
(М. м. 430,3). 1073800. [117 92 0]. 2 [2 [4 (Диме
тиламино )фенил ]этенил] 1 этилхинолина йодид.
Темный синевато черный порошок. YMepeH
но растворим в воде, леrкорастворим в 96 %
спирте.
Хинальдиновоrо KpacHoro раствор.
1073801.
0,1 r хинальдинов020 краСН020 Р paCTBO
ряют в метаноле Р и доводят до объема 100 мл
тем же растворителем.
Изменение окраски. От рН 1,4 (бесцветный)
до рН 3,2 (красное окрашивание).
722
rосударственная фармакопея Республики Беларусь
Хинrидрон. С12Н1004' (м. м. 218,2). 1073900.
[106 34 3]. Эквимолекулярное соединение
1 ,4 бензохинона и rидрохинона.
Темно зеленые блестящие кристаллы или
кристаллический порошок. Малорастворим в
воде, умеренно растворим в roрячей воде, pac
творим в 96 % спирте, растворе аммиака KOH
центрированноro.
Температура плавления: около 170 ОС.
ХИНИДИН. C 20 H 24 NP2' (м. м. З24,4). 1074000.
[56 54 2). (SН6 Метоксихинол 4 ил) [(2R,4S,5R)
5 винилхинуклидин 2 ил ]метанол.
Белые или почти белые кристаллы. Очень
мало растворим в воде, умеренно растворим в
96 % спирте, малорастворим в метаноле.
[a] O: около +260. Определение проводят,
используя раствор 1 О r/л хинидина в этаноле Р.
Температура плавления: около 172 ОС.
Хранят в защищенном от света месте.
Хинидина сульфат. C4oH50N40aS'2H20,
(м. м. 783). 1109500. [6591 63 5]. Бис[(S)
[(2R,4S,5R) 5 этинил 1 азабицикло[2.2.2]окт
2 ил ](6 метоксиквинолин4 ил)метанол] сульфат.
Содержит не менее 99,0 % и не более 101,0 %
C4oH50N40aS В пересчете на сухое вещество.
Белый или почти белый кристаллический
порошок или шелковистый бесцветный иroльча
тый порошок. Малорастворим в воде, растворим
в кипящей воде и в 96 % спирте, практически He
растворим в ацетоне.
Потеря в массе при высушивании (2.2.32).
От 3,0 % до 5,0 %. 1,000 r хинидина сульфата
сушат при температуре 130 ОС.
Хранят в защищенном от света месте.
Хинин. C2QH 24 NP2' (М м. 324,4). 1074100.
[1 ЗО 95 0].(RН6 Метоксихинол4 ил )[(2S,45,5R) 5
винил хинуклидин 2 ил]метанол.
Белый или почти белый мелкокристалли
ческий порошок. Очень мало растворим в воде,
малорастворим в кипящей воде, очень леrко
растворим в этаноле.
[a] O: около 167. Определение проводят, ис
пользуя раствор 10 r/л хинина в этаноле Р.
Температура плавления: около 175 ОС.
Хранят в защищенном от света месте.
Хинина rидроxnорид. C2oH25CIN202'2H20,
(м. м. 396,9). 1074200. [611947 7]. (SH(2S,4S,5R)
5 Этенил 1 азабицикло[2.2 .2]окт 2 ил]
(6 метоксиквинолин4 ил )метанола] rидрохлорид.
Содержит не менее 99,0 % и не более
101,0 % C20H25CIN202 в пересчете на сухое Be
щество.
Тонкие шелковистые бесцветные и roл ки ,
часто в пучках. Растворим в воде, леrкораство
рим в 96 % спирте.
Потеря в массе при высушивании (2.2.32).
От 6,0 % до 10,0 %. 1,000 r хинина rидрохлорида
сушат при температуре 105 ос.
Хранят в защищенном от света месте.
Хинина сульфат. C4oH50N40aS'2H20,
(м. м. 783). 1074300. [6119 70 6]. бис[(R)
[(2 S,45, 5R) 5 Этенил 1 азабици кло[2. 2 .2]
окт 2 ил ](6 метоксиквинолин 4 ил )метанола]
сульфат.
Содержит не менее 99,0 % и не более
101,0 % C4oH50N40aS В пересчете на сухое Be
щество.
Белый или почти белый кристаллический
порошок или тонкие бесцветные иroлки. Мало
растворим в воде, умеренно растворим в кипя
щей воде и 96 % спирте.
Потеря в массе при высушивании (2.2.32).
От 3,0 % до 5,0 %. 1,000 r хинина сульфата
сушат при температуре 105 ОС.
Хранят в защищенном от света месте.
2-Хлор-4-нитроанилин.С 6 Н 5 СINР2.(Мм.172,6).
1018800. [121 7 9].
Желтый кристаллический порошок. Леrко
растворим в метаноле.
Температура плавления: около 107 ОС.
Хранят в защищенном от света месте.
Хлоралrидрат. 1017900. [302 17 0].
См. статью ХлораЛ2идрат.
Хлоралrидрата раствор. 1017901.
Раствор 80 r хлораЛ2идрата Р в 20 мл
воды Р.
# Хлоралrидрата раствор Р1.
20 r хлораЛ2идрата Р растворяют при Ha
rревании в 5 мл воды Р и прибавляют 5 мл 2ли
церина Р.
Хлорамин.С 7 Н 7 СINNа0 2 S'3НР.(м. М.281 ,7).
1018000. [127 65 1]. Натрия N хлор4 метил
бензол сульфонимидат триrидрат. Тозилхлора
мид натрия.
Содержит не менее 98,0 % и не более
103,0 % C 7 H 7 CINNa0 2 S'3Hp.
Белый или слеrка желтый кристаллический
порошок. Леrкорастворим в воде, растворим в
96 % спирте.
Хранят в воздухонепроницаемом контейне
ре в защищенном от света месте.
Хлорамина раствор. 1018001.
Раствор 20 r/л хлорамина Р.
rотовят непосредственно перед использо
ванием.
Хлорамина раствор Р1. 1018002.
Раствор 0,1 r/л хлорамина Р.
rотовят непосредственно перед использо
ванием.
Хлорамина раствор Р2. 1018003.
Раствор 0,2 r/л хлорамина Р.
rотовят непосредственно перед использо
ванием.
4.1.1. Реактивы
723
Хлоранилин. C 6 H 6 CIN. (м. м. 127,6).
1018300. [106 47 8]. 4 Хлоранилин.
Кристаллы. Растворим в roрячей воде, леr
корастворим в 96 % спирте.
Температура плавления: около 71 ОС.
Хлорацетанилид. CBHBCINO. (м. м. 169,6).
1018100. [5З9 03 7]. 4' Хлорацетанилид.
Кристаллический порошок. Практиче
ски нерастворим в воде, растворим в 96 %
спирте.
Температура плавления: около 178 ОС.
4-Хлорбензолсульфонамид. C 6 H 6 CIN0 2 S.
(М. м. 191,6). 1097400. [98 64 6].
Белый или почти белый порошок.
Температура плавления: около 145 ОС.
Хлорбутанол. С 4H7C1p. (м. м. 177,5). 1018400.
[57 15 ]. 1,1, 1 Т рихлор 2 метилпропан 2 л.
Содержит не менее 98,0 % и не более
101,0 % С 4 Н 7 Сl з О в пересчете на безводное Be
щество.
Белый или почти белый кристаллический по
рошок или бесцветные кристаллы. Леrко субли
мируется. Малорастворим в воде, очень леrко
растворим в 96 % спирте, растворим в rлицери
не (85 %).
Температура плавления: около 95 ос, опре
деляют без предварительноro высушивания.
Вода (2.5.12). Не более 1,0 %. Определение
проводят из 2,00 r хлорбутанола.
Хлористоводородная кислота. 1043500.
[7647 01 0].
См. статью Хлористоводородная кислота
конценrпрированная.
Хлористо водородная кислота Р1.
1043501.
Содержит 250 r/л HCI.
70 r кислоты хлористоводородной Р ДOBO
дят водой Р до объема 100 мл.
Хлористоводородная кислота бромиро-
ванная. 1043507.
К 100 мл кислоты хлористоводородной Р
прибавляют 1 мл раствора брома Р.
Хлористоводородная кислота разведен-
ная.1043503.
Содержит 73 r/л HCI.
20 r кислоты хлористоводородной Р ДOBO
дят водой Р до объема 100 мл.
Хлористоводородная кислота разведен-
ная Р1. 1043504.
Содержит 0,37 r/л HCI.
1,0 мл кислоты хлористоводородной
разведенной Р доводят водой Р до объема
200,0 мл.
Хлористоводородная кислота разведен-
ная Р2. 1043505.
30 мл 1 М раствора кислоты хлористово
дородной доводят водой Р до объема 1000 мл;
доводят до рН (1,6f:O,1).
Хлористоводородная кислота разве-
денная, свободная от тяжелых металлов.
1043509.
Должна выдерживать требования для Kиc
лоты хлористоводородной разведенной Р и
следующие дополнительные испытания:
As: не более 0,005 ррm;
Cd: не более 0,003 ррт;
Cu: не более 0,003 ррm;
Fe: не более 0,05 ррm;
Hg: не более 0,005 ррт;
Ni: не более 0,004 ррm;
РЬ: не более 0,001 ррm;
Zп: не более 0,005 ррm.
Хлористоводородная кислота, свобод-
ная от свинца. 1043508.
Должна выдерживать требования для Kиc
лоты хлористоводородной Р и следующее дo
полнительное испытание.
Свинец. Не более 0,002 % (20 ррm).
Атомно эмиссионная спектрометрия (2.2.22,
метод 1).
Испытуемый раствор. 200 r кислоты
хлористоводородной помещают в кварцевый
тиrель, испаряют почти досуха, к полученно
му остатку прибавляют 5 мл кислоты азотной,
приroтовленной дистилляцией кислоты азот
ной Р при температуре ниже температуры ки
пения, и выпаривают досуха. К полученному
остатку прибавляют 5 мл кислоты азотной,
приroтовленной дистилляцией кислоты азот
ной Р при температуре ниже температуры ки
пения.
Растворы сравнения. rотовят paCT
ры сравнения, используя эталонный раствор
свинца (0,1 ррт РЬ) Р, разведенный кислотой
азотной, приrотовленной дистилляцией Kи
ты азотной Р при температуре ниже темneра
туры кипения.
Измеряют интенсивность эмиссии при длине
волны 220,35 нм.
Хлористоводородная кислота, свобод-
ная от тяжелых металлов. 1043510.
Должна выдерживать требования для Kиc
лоты хлористоводородной Р и следующие дo
полнительные испытания:
As: не более 0,005 ррт;
Cd: не более 0,003 ppm;
Cu: не более 0,003 ppm;
Fe: не более 0,05 ррт;
Hg: не более 0,005 ррт;
Ni: не более 0,004 ррт;
РЬ: не более 0,001 ррm;
Zn: не более 0,005 ррm.
724
rосударственная фармакопея Республики Беларусь
Хлористоводородной кислоты 2 М paCT
вор. 3001700.
206,0 r кислОП1Ы хлористоводородной Р дo
водят водой Р до объема 1000,0 мл.
Хлористоводородной кислоты 3 м раст-
вор. 3001600.
309,0 r кислоты хлористоводородной Р дo
водят водой Р до объема 1000,0 мл.
Хлористоводородной кислоты 6 М paCT
вор. 3001500.
618,0 r кислоты хлористоводородной Р дo
водят водой Р до объема 1000,0 мл.
Хлористоводородной кислоты раствор в
спирте. 1043506.
5,0 мл 1 М раствора кислоты хлористово
дородной доводят 96 % спиртом Р до объема
500,0 мл.
Хлорная кислота. НСI0 4 . (М м. 100,5).
1062900. [7601 90 3].
Содержит не менее 70,0 % (м/м) и не более
7З,0 % (м/м) НСI0 4 .
Прозрачная бесцветная жидкость. Леrко
смешивается с водой.
d;g: около 1,7.
Количественное определение. 2,50 r кис
лоты хлорной прибавляют к 50 мл воды Р и ти
труют 1 М раствором натрия 2идроксида, ис
пользуя в качестве индикатора 0,1 мл раствора
метилов020 краСН020 Р.
1 мл 1 М раствора натрия 2идроксида co
ответствует 100,5 Mr НСI0 4 .
Хлорной кислоты раствор. 1062901.
8,5 мл кислоты хлорной Р доводят водой Р
до объема 100 мл.
Хлороrеноваякислота'С'6Н,в09' (М.м. 354,3).
1104700. [327 97 9). (1S,3R,4R,5R) З [(3,4 Ди
rидроксициннамоил)окси] 1 ,4,5 триrидрокси
циклоrексанкарбоновая кислота.
Белый или почти белый кристаллический
порошок или иroльчатые кристаллы. Раствори
ма в кипящей воде, в ацетоне и в 96 % спирте.
[a] O: около 35,2.
Температура плавления: около 208 ОС.
Хромат02рафия. Тонкослойная xpOMaтo
rрафия (2.2.27), как указано в статье Беладонны
листьев сухой экстракт стандартизирован
ный в условиях, описанных в идентификации А.
На xpoMaтorpaMMe должно обнаруживаться
только одно основное пятно.
ХЛОР02еновая кислота, используемая в
жидкостной хромат02рафии, должна выдержи
вать следующее дополнительное испытание.
Количественное определение. Жидкостная
хроматоrрафия (2.2.29), как указано в статье Ap
тишока листья.
Содержание: не менее 97,0 %.
Хлороформ. СНСl з . (М м. 119.4). 1018600.
[67 66 3]. Трихлорметан.
Прозрачная бесцветная жидкость. Мало-
раств им в воде, смешивается с 96 % спиртом.
d 2o : от 1,475 до 1,481.
Температура кипения: около 60 ОС.
Хлороформ содержит от 0,4 % (м/м) до
1,0 % (м/м) этанола.
Этанол. 1,00 r (т. r) хлороформа помеща
ют в колбу с притертой стеклянной пробкой, при
бавляют 15,0 мл нитрохромов020 реактива Р,
колбу закрывают, энерrично встряхивают в тече
ние 2 мин и выдерживают в течение 15 мин. При
бавляют 100 мл воды Р и 5 мл раствора 200 r/л
калия йодида Р. Через 2 мин титруют О, 1 М
раствором натрия тиосульфата до появле
ния светло зеленоro окрашивания (п" мл 0,1 М
раствора натрия тиосульфата), используя в
качестве индикатора 1 мл раствора крахмала Р.
Параллельно проводят контрольный опыт
(п 2 , мл 0,1 М раствора натрия тиосульфата).
Содержание этанола, в процентах, рассчи
тывают по формуле:
(п, n2).O,115
т
Хлороформ подкисленный. 1018601.
К 100 мл хлороформа Р прибавляют 1 О мл
кислоты хлористоводородной Р, встряхивают,
отстаивают и разделяют два слоя.
Хлороформ, свободный отзтанола. 1018602.
200 мл хлороформа Р промывают водой Р,
встряхивая с четырьмя порциями, по 100 мл
каждая. Сушат над 20 r натрия сульфата без
водН020 Р в течение 24 ч. Фильтрат переroняют
над 1 О r натрия сульфата безводН020 Р, отбра
СbJвая первые 20 мл дистиллята.
rотовят непосредственно перед использо
ванием.
Хлороформ, стабилизированный амиле-
ном. СНСl з . (М. м. 119,4). 1018700.
Прозрачная бесцветная жидкость. Мало
растворим в воде, смешивается с 96 % спиртом.
Вода. Не более 0,05 %.
Остаток после выпаривания. Не более
0,001 %.
Оптическое пропускание (2.2.25) определя
ют, используя в качестве компенсационной жид
кости воду Р: не менее 50 % при длине волны
255 нм; не менее 80 % при длине волны 260 нм;
не менее 98 % при длине волны 300 нм.
Количественное определение. Не менее
99,8 % СНСl з . Определение проводят методом
rазовой хроматоrрафии.
Хлорплатиновая кислота. H 2 CI 6 Pt'6H 2 0.
(М м. 517,9). 1019000. [18497 13 7]. rексахлор
платиновая (IV) кислота rексаrидрат.
Содержит не менее 37,0 % (м/м) платины
(А м. 195,1).
724
rосударственная фармакопея Республики Беларусь
Хлористоводородной кислоты 2 М paCT
вор. 3001700.
206,0 r кислоты хлористоводородной Р дo
водят водой Р до объема 1000,0 мл.
Хлористоводородной кислоты 3 м раст-
вор. 3001600.
309,0 r кислоты хлористоводородной Р дo
водят водой Р до объема 1000,0 мл.
Хлористоводородной кислоты 6 М раст-
вор. 3001500.
618,0 r кислоты хлористоводородной Р дo
водят водой Р до объема 1000,0 мл.
Хлористоводородной кислоты раствор в
спирте. 1043506.
5,0 мл 1 М раствора кислоты хлористово
дородной доводят 96 % спиртом Р до объема
500,0 мл.
Хлорная кислота. НСI0 4 . (М М. 100,5).
1062900. [7601 90 3].
Содержит не менее 70,0 % (м/м) и не более
7З,0 % (м/м) НСI0 4 .
Прозрачная бесцветная жидкость. Леrко
смешивается с водой.
d;g: около 1,7.
Количественное определение. 2,50 r кис
лоты хлорной прибавляют К 50 мл воды Р и ти
труют 1 М раствором натрия 2идроксида, ис
пользуя в качестве индикатора 0,1 мл раствора
метилов020 краСН020 Р.
1 мл 1 М раствора натрия 2идроксида co
ответствует 100,5 Mr НСI0 4 .
Хлорной кислоты раствор. 1062901.
8,5 мл кислоты хлорной Р доводят водой Р
до объема 100 мл.
Хлороrеноваякислота.С 16Нlв09' (М.м. 354,3).
1104700. [327 97 9J. (1S.3R.4R,5R) З [(3,4 Ди
rидроксициннамоил )оксиJ 1 .4,5 триrидрокси
циклоrексанкарбоновая кислота.
Белый или почти белый кристаллический
порошок или иroльчатые кристаллы. Раствори
ма в кипящей воде, в ацетоне и в 96 % спирте.
[a] O: около 35,2.
Температура плавления: около 208 ос.
Хромат02рафия. Тонкослойная xpOMaтo
rрафия (2.2.27), как указано в статье Беладонны
листьев сухой экстракт стандартизирован
ный в условиях, описанных в идентификации д-
На xpoMaтorpaMMe должно обнаруживаться
только одно основное пятно.
ХЛОР02еновая кислота, lIспользуемая в
жидкостной хромат02рафии, должна выдержи
вать следующее дополнительное испытание.
Количественное определение. Жидкостная
хроматоrрафия (2.2.29), как указано в статье Ap
тишока листья.
Содержание: не менее 97,0 %.
Хлороформ. СНСl з . (М м. 119,4). 1018600.
[67 66 3]. Трихлорметан.
Прозрачная бесцветная жидкость. Мало
раств им в воде, смешивается с 96 % спиртом.
d 2o : от 1,475 до 1,481.
Температура кипения: около 60 ОС.
Хлороформ содержит от 0,4 % (м/м) до
1,0 % (м/м) этанола.
Этанол. 1,00 r (т, r) хлороформа помеща
ют в колбу с притертой стеклянной пробкой, при
бавляют 15,0 мл нитрохромов020 реактива Р,
колбу закрывают, энерrично встряхивают в тече
ние 2 мин и выдерживают в течение 15 мин. При
бавляют 100 мл воды Р и 5 мл раствора 200 r/л
калия йодида Р. Через 2 мин титруют 0,1 М
раствором натрия тиосульфата до появле
ния светло зеленоro окрашивания (п l , мл 0,1 М
раствора натрия тиосульфата), используя в
качестве индикатора 1 мл раствора крахмала Р.
Параллельно проводят контрольный опыт
(П 2 ' мл 0,1 М раствора натрия тиосульфата).
Содержание эта нола , в процентах, рассчи
тывают по формуле:
(П 1 П2).0,115
т
Хлороформ подкисленный. 1018601.
К 100 мл хлороформа Р прибавляют 1 О мл
кислоты хлористоводородной Р, встряхивают,
отстаивают и разделяют два слоя.
Хлороформ, свободный отэтанола. 1018602.
200 мл хлороформа Р промывают водой Р,
встряхивая с четырьмя порциями, по 100 мл
каждая. Сушат над 20 r натрия сульфата без
водН020 Р в течение 24 ч. Фильтрат переroняют
над 1 О r натрия сульфата безводН020 Р, отбра
сывая первые 20 мл дистиллята.
rотовят непосредственно перед использо
ванием.
Хлороформ, стабилизированный амиле-
ном. СНСl з . (М м. 119,4). 1018700.
Прозрачная бесцветная жидкость. Мало
растворим в воде, смешивается с 96 % спиртом.
Вода. Не более 0,05 %.
Остаток после выпаривания. Не более
0,001 %.
Оптическое пропускание (2.2.25) определя
ют, используя в качестве компенсационной жид
кости воду Р: не менее 50 % при длине волны
255 нм; не менее 80 % при длине волны 260 нм;
не менее 98 % при длине волны 300 нм.
Количественное определение. Не менее
99,8 % СНСl з . Определение проводят методом
rазовой хроматоrрафии.
Хлорплатиновая кислота. H 2 C/ 6 Pt'6H 2 0.
(М м. 517,9). 1019000. [18497 13 7]. rексахлор
платиновая (/V) кислота rексаrидрат.
Содержит не менее 37,0 % (м/м) платины
(А. м. 195,1).
4.1.1. Реактивы
725
Коричневато красные кристаллы или кри
сталлическая масса. Очень леrко растворима в
воде, растворима в 96 % спирте.
Количественное определение. 0,200 r кис
лоты хлорплатиновой прокаливают при темпе
ратуре (900I50) ос до постоянной массы и взве
шивают остаток (платина).
Хранят в защищенном от света месте.
З Хлорпропан 1,2 ДИОЛ.СЗН7СI02'(М М.11О,5).
1097600. [96 24 2).
Бесцветная жидкость. Растворим в воде и в
96 % спирте.
d 20
20: около 1,322.
20
по: около 1,480.
Температура кипения: около 213 ОС.
5-Хлорсалициловая кислота. С 7 Н 5 СIO З '
(М м. 172,6). 1019100. [321 14 2].
Белый или почти белый кристаллический
порошок. Растворима в метаноле.
Температура плавления: около 173 ОС.
Хлортиазид. C7H6CINP4S2- (ММ. 295,7).
1112100. [58 94 6]. 6 Хло 2Н 1,2,4 бензотиа
диазин 7 сульфонамид 1,1 диоксид.
Содержит не менее 98,0 % и не более 102,0 %
С7Н6СINз04S2 в пересчете на сухое вещество.
Белый или почти белый кристаллический
порошок. Очень мало растворим в воде, YMepeH
но растворим в ацетоне, малорастворим в 96 %
спирте. Растворяется в разведенных растворах
rидроксидов щелочных металлов.
Потеря в массе при высушивании (2.2.32).
Не более 1,0 %. 1,000 r хлортиазида сушат при
температуре 105 ОС.
Хлортриметилсилан. С з Н 9 СISi. (М м. 108,6).
1019300. [75 77 4].
Прозрачная бесцветная жидкость, дымяща
яся на воздухе.
d 20
20: около 0,86.
20
по: около 1,388.
Температура кипения: около 57 ос.
Хлоруксусная кислота. С 2 Н з СI0 2 . (М м. 94,5).
1018200. [79 11 8].
Бесцветные или белые или почти белые
кристаллы, расплывающиеся на воздухе. Очень
леrко растворима в воде, растворима в 96 %
спирте.
Хранят в воздухонепроницаемом контейнере.
Хлорфенол. С 6 Н 5 СIO. (М м. 128,6). 1018900.
[106A8 9). 4 Хлорфенол.
Бесцветные или почти бесцветные кристал
лы. Малорастворим в воде, очень леrко paCTBO
рим в 96 % спирте и растворах rидроксидов ще
лочных металлов.
Температура плавления: около 42 ос.
2 Хлорэтанол. С 2 Н 5 СIO. (М м. 80,5).
1097500. [107 07 3).
Бесцветная жидкость. Растворим в 96 %
спирте.
d 20 1
20: около 1, 97.
20
по: около 1 ,442.
Температура кипения: около 130 ОС.
Температура плавления: около 89 ОС.
2-Хлорэтанола раствор. 1097501.
125 Mr 2 хлорэтанола Р растворяют в 2 про
паноле р и доводят до объема 50 мл этим же
растворителем. 5 мл полученноro раствора дo
водят 2 пропанолом Р до объема 50 мл.
(2 Хлорэтил)диэтиламина rидрохлорид.
C6H'5CI2N. (М М. 172.1). 1 018500. [869 24 9].
Белый или почти белый кристаллический по
рошок. Очень леrко растворим в воде и в MeTa
ноле, леrкорастворим в метиленхлориде, пра
тически нерастворим в reKcaHe.
Температура плавления: около 211 ос.
(5а) холестан. С 27 Н 4в , (М М. 372,7). 1167900.
[481 21 0).
Малорастворим в безводном этаноле.
Температура плавления: около 81 ОС.
Холестерин. С 27 Н 46 О. (М. м. 386,7).1019400.
[57 88 5].
Содержит не менее 95,0 % холест 5 ен
3 ола и не менее 97,0 % и не более 10З,0 % CTe
ролов в пересчете на сухое вещество.
Белый или почти белый кристаллический
порошок. Практически нерастворим в воде, YMe
ренно растворим в ацетоне и в 96 % спирте. Чув
ствителен к свету.
Хранят в защищенном от света месте.
Холина хлорид. C 5 H 14 CINO. (М м. 139,6).
1019500. [67A8 1]. (2 rидроксиэтил)тримети
ламмония хлорид.
Кристаллы, расплывающиеся на воздухе.
Очень леrко растворим в воде и в 96 % спирте.
Хромат02рафия. Тонкослойная хроматоrpэ
фия (2.2.27), как указано в статье СУКС8метония
хлорид, используя 5 мкл раствора 0,2 r/л холинэ
хлорида в метаноле Р. На xpoMaтorpaMMe должно
обнаруживаться только одно основное пятно.
Хранят в воздухонепроницаемом контейнере.
Хризантемин. С 2 .Н 2 ,СIO". (М. М. 485,5).
1134800. [7084 24 4]. 2 (3.4 Диrдроксифенил)
3 (I3 D rлюкопиранозил )окси 5, 7 диrидрокси
1 бензопирила хлорид. Куроманина хлорид.
Кристаллический порошок KpaCHOBaTO KO
ричневоrо цвета. Растворим в воде и спирте.
Спектрофотомерuя(2. 2. 25). РастворО,01 r/л
в смеси из кислоты хлористоводородной Р и
метанола Р (1:999, об/об) должен иметь макси
мум поrлощения при 528 нм.
726
rосударственная фармакопея Республики Беларусь
Хрома (VI) оксид. СrО з . (М м. 100,0).
1019900. [133З 82 0]. Хрома триоксид.
Темные коричневато красные иroльчатые
кристаллы или rранулы, расплывающиеся на
воздухе. Очень леню растворим в воде.
Хранят в воздухонепроницаемом стеклян
ном контейнере.
Хрома триоксид. См. Хрома (V/) оксид Р.
Хрома (111) трихлорид rексаrидрат.
[Cr(Hp)4CI2]CI'2Hp. (М м. 266,5). 1104800.
[10060 12 5].
Темно зеленый кристаллический порошок.
rиrроскопичен.
Хранят защищая от воздействия влаrи и
окислителей.
Хромазурол s. С2ЗН1ЗСI2NаР9S' (М м. 605).
1019600 [1667 99 8]. Показатель Шульца NQ
841. Цветной индекс NQ 43825. Тринатрия
5 [(3 карбоксилато 5 метил 4 оксоциклоrекса 2,5
диен 1 или ден )(2,6 дихлор 3 сульфонатофенил)
метил] 2 rидрокси З метилбензоат.
Коричневато черный порошок. Растворим в
воде, малорастворим в 96 % спирте.
Хрома-калия сульфат. CrK(SO 4)2 '12Нр.
(М м. 499,4). 1019800. [7788 99 0]. Хромовые
квасцы.
Крупные кристаллы от фиолетово красноro
до черноro цвета. Леrкорастворим в воде, прак
тически нерастворим в 96 % спирте.
Хромовая смесь. 1019700.
Насыщенный раствор хрома (V/) оксида Р в
кислоте серной Р
# XpoMoBoro темно-синеrо индикаторная
смесь.
0,25 r хромов020 тeMHo cиHe20 Р и 25 r
натрия хлорида Р растирают в ступке и пере
мешивают.
# XpoMoBoro темно-синеrо раствор.
0,5 r хромов020 тeMHo иHe20 Р paCTBO
ряют в 1 О мл аммиаЧН020 буферною раствора
рН 10, О Р и доводят спиртом (95 %, об/об) Р до
объема до 100 мл.
Срок roдности 1 мес.
# Хромовые квасцы. См. Хрома калия
сульфат Р
# Хромовый teMHQ-СИНИЙ. C16HgCIN2NapgS2'
(М м. 518,8). [1 058 92 0]. Динатрия 2 [(5 хлор
2 оксифенил)азо] 1 ,8 диоксинафталин 3,6
дисульфонат. Кислотный хромовый темно си
ний. Кислотный синий 13.
Темно коричневый или черный порошок.
Леrкорастворим в воде.
# XpoMoreH черный. См. Протравной
черный 11 Р.
Хромотроп 11 В. С16НgNзNа2010S2'
(М м. 513.4) 1020200. [548 80 1J. Показа
тель Шульца NQ 67. Цветной индекс NQ 16575.
Динатрия 4,5 диrидрокси 3 ( 4 нитрофенилазо)
нафталин 2, 7 дисульфонат.
Красновато коричневый порошок. PaCТBO
рим в воде с образованием желтовато красно
ro раствора, практически нерастворим в 96 %
спирте.
Хромотропа 11 В раствор. 1020201. Раствор
0,05 r/л хромотропа 11 В Р в кислоте серной Р.
_ # Хромотроповая кислота. C10HBOBS2'
(М м. 320,3). 1119100. [148 25 4]. 4,5 Диrидрокси
2, 7 нафтилиндисульфоновая кислота.
Белые или почти белые иroльчатые кристал
лы. Растворима в воде.
Температура плавления: около 300 ОС.
# Хромотроповой кислоты раствор.
0,50 r кислоты хромотроповой Р paCTBO
ряют в 80 мл воды Р и доводят до объема 100 мл
этим же растворителем.
Срок roдности раствора 24 ч.
Хромотроповой кислоты натриевая
соль. C1oH6NaPBS2'2Hp. (М м. 400,3). 1020300.
[5808 22 0]. Показатель Шульца NQ1136. Динат
рия 4,5 диrидроксинафталин 2. 7 дисульфонат
диrидрат. Динатрия 1 ,8 диrидроксинафталин
3,6 дисульфонат диrидрат.
Желтовато белый порошок. Растворима в
воде, практически нерастворима в 96 % спирте.
Хромотроповой кислоты натриевой соли
раствор. 1020301.
0,60 r хромотроповой кислоты натриевой
соли Р растворяют в 80 мл воды Р и доводят до
объема 100 мл этим же растворителем.
Срок roдности раствора 24 ч.
Хромоформный субстрат Р1. 1020000.
N а Бензилоксикарбонил D арrинил L
rлицил L арrинин 4 нитроанилида диrидрохло
рид растворяют в воде Р до получения 0,003 М
раствора. Перед использованием разводят бу
ферным раствором триС(2идроксиметил)
aMиHOMeтaHa EOTA рН 8,4 Р до получения
0,0005 М раствора.
Хромоформный субстрат Р2. 1020100.
D Фен илала н ил L п и пекол ил L a рrи н и H
4 нитроанилида диrидрохлорид растворяют в
воде Р до получения 0,003 М раствора. Перед
использованием разводят буферным pac
твором триС(2идроксиметил)аминометана
ЕDТА рН 8,4 Р до получения 0,0005 М paCT
вора.
4.1.1. Реактивы
727
Цезия хлорид. CsCI. (М м. 168.4). 1014200.
[7647 17 8].
Белый или почти белый порошок. Очень
леrко растворим в воде, леrкорастворим в MeTa
ноле, практически нерастворим в ацетоне.
Целлюлоза для хроматоrрафии. 1016800.
[9004 34 6].
Мелкий белый или почти белый roмоrенный
порошок со средним размером частиц менее
30 мкм.
При20товление тОНК020 слоя. 15 r целлю
лозы для хроматоrрафии суспендируют в 100 мл
воды Р и roмоrенизируют на электромешалке в
течение 60 с. Тщательно очищенные пластины
покрывают слоем толщиной 0,1 мм, используя
прибор для нанесения. Сушат на воздухе.
Целлюлоза для хроматоrрафии Р1.
1016900.
Микрокристаллическая целлюлоза. Мелкий
белый или почти белый roмоrенныи порошок со
средним размером частиц менее 30 мкм.
При20товление тОНК020 слоя. 25 r микро--
кристаллической целлюлозы суспендируют в
90 мл воды Р и rомоrенизируют на электром
шалке в течение 60 с. Тщательно очищенные
пластины покрывают слоем толщиной 0,1 мм,
используя прибор для нанесения. Сушат на воз
духе.
Целлюлоза для хроматоrрафии F 254.
1017000.
Микрокристаллическая целлюлоза F 254'
Мелкий белый или почти белый roмоrенный поро
шок со средним размером частиц менее 30 мкм,
содержащий флуоресцентный индикатор с опти
мальной интенсивностью при длине волны 254 нм.
При20товление тОНК020 слоя. 25 r микро
кристаллической целлюлозы F 254 суспендируют
в 100 мл воды Р и roмоrенизируют на электро
мешалке в течение 60 с. Тщательно очищенные
пластины покрывают слоем толщиной 0,1 мм,
используя прибор для нанесения. Сушат на
воздухе.
Церия (111) нитрат. Се(NОЗ>з'6Нр.
(М м. 434,3). 1017400. [10294 41 4]. Церия три
нитрат rексаrидрат.
Бесцветный или бледно желтый кристалли
ческий порошок. Леrкорастворим в воде и в 96 %
спирте.
Церия (IV) сульфат. Ce(S04)2-4Нр.
(М м.404,3). 1017300. [123333 60 8J. Церий cep
НОКИСJlЫЙ.
Желтый или оранжево желтый кристалли
ческий порошок. Очень мало растворим в воде,
медленно растворим в разведенных кислотах.
Цетилтриметиламмониябромид. С 19H42BrN.
(М м. 364,5). 1017700. [57 09 0]. Цетримония
бромид. N rексадецил N,N,N триметиламмония
бромид.
Белый или почти белый кристаллический
порошок. Растворим в воде, леrкорастворим в
96 % спирте.
Температура плавления: около 240 ОС.
Цетримид. 1017600. [8044 71 1].
Состоит из триметилтетрадециламмония
бромида и может содержать незначительные KO
личества додецил и rексадецилтриметиламмо
ния бромиды.
Содержит не менее 96,0 % и не более
101.0 % алкилтриметиламмония бромидов, pac
считанных как С 17 Н з8 ВrN (М м. 336,4), в пересче
те на сухое вещество.
Белый или почти белый объемный порошок.
Леrкорастворим в воде и в 96 % спирте.
Потеря в массе при высушивании (2.2.32).
Не более 2,0 %. 1,000 r цетримида сушат при
температуре 105 ос в течение 2 ч.
Цефалина реактив. 1017200.
К 0, 1 r мозzа бычье20, 8ысушенноzо в
ацетоне Р, прибавляют 20 мл ацетона Р и BЫ
держивают в течение 2 ч. Центрифуrируют с
ускорением 500 9 в течение 2 мин, жидкость над
осадком сливают. Остаток сушат при понижен
ном давлении, затем прибавляют 20 мл хлоро
форма Р и выдерживают в течение 2 ч, часто
взбалтывая. Фильтруют или центрифуrируют и
выпаривают хлороформ при пониженном дaB
лении. Остаток суспендируют в 5 10 мл paCT
вора 9 r/л натрия хлорида Р.
Растворители, используемые для при20
товления реактива, должны содержать подхо
дящий антиоксидант, например, 0,02 2/Л бути
лироваННО20 2идроксианизола.
Хранят в замороженном или лиофилизиро
ванном виде.
Срок roдности 3 мес.
ЦефалинадиrидРОХЛОРИД.С28Н40СI2N20 ' O.
(М м. 666). 1017100. [5884-43 5]. (R)-1
[(2S,3R, 11ЬS) 3 Этил 1 ,3,4,6,7, 11 b reKca
9, 1 0 диметокси 2Н бензо[а]хинолизин 2--иJW811U1}-
1 ,2,3,4 тетраrидро 7 метоксиизохинолин ла
диrидрохпорид rептаrидрат.
Кристаллический порошок от бenoro до жел
товатоro цвета. Леrкорастворим в ВQQe. paCТBO
рим В ацетоне и в 96 % спирте.
[a] O: около +25. Определение f1)OВQЦЯТ, ис
пользуя раствор 20 r/л цефалинадиrидpoхлорида.
Цианобромида раствор. 1023700.
[506 68 3]
К бромной веде Р прибавляют по каплям
при охлаждении 0.1 М раствор аммония ти
оцианата до исчезновения желтоro окрашива
ния.
rотовят непосредственно перед использd(
ванием.
728
rосударственная фармакопея Республики Беларусь
Цианоryанидин. C 2 H 4 N 4 . (м. М. 84,1).
1023800. [461 58 5]. Дициандиамид. 1 Циа юry
анидин.
Белый или почти белый кристаллический по--
рошок. Умеренно растворим в воде и в 96 % спирте,
практически нерастворим вметиленхлориде.
Температура плавления: около 210 ОС.
Цианокобаламин. 1023600. [68 19 9].
См статью Цианокобаламин.
Цианоуксусная кислота. С з Н з N0 2 . (м. М. 85,1).
1097900. [372 09 8].
rиrроскопичные кристаллы от белоro до
желтовато белоro цвета. Очень леrко раствори
ма в воде.
Хранят в воздухонепроницаемом контей
нере.
Циклоrексан. С 6 Н 12 . (М. М. 84,2). 1023900.
[110 82 7].
Прозрачная бесцветная воспламеняющая
ся жидкость. Практически нерастворим в воде,
смешивается с орrаническими растворителями.
d 20
20: около 0,78.
Температура кипения: около 80,5 ОС.
ЦиКЛ02ексан, используемый в спектрофо
тометрии, должен выдерживать следующее
дополнительное испытание.
Оптическое пропускание (2.2.25) определя
ют, используя в качестве компенсационной жид
кости воду Р: не менее 45 % при длине волны
220 нм; не менее 70 % при длине волны 235 нм;
не менее 90 % при длине волны 240 нм; не менее
98 % при длине волны 250 нм.
Циклоrексан Р1. 1023901.
Должен выдерживать требования для ци
КЛ02ексана Р и следующее дополнительное ис
пытание.
Интенсивность флуоресценции, измеренная
при длине волны 460 нм (при облучении пучком
света с длиной волны 365 НМ), должна быть не
интенсивнее флуоресценции раствора, coдep
жащеro 0,002 ррт хинина Р в 0,05 М растворе
кислоты серной.
Циклоrексиламин. С Б Н 1з N. (М м. 99,2).
1024000. [108 91 8].
Бесцветная жидкость. Растворим в воде,
смешивается с наиболее распространенными
раСТВОfителями.
п : около 1,460.
Температура кипения: от 134 ос до 135 ОС.
Циклоrексилендинитрилтетрауксусная
кислота. C14H22NPB'Hp. (м. м. 364,4). 1024100.
транс Циклоrексилен 1 ,2 динитрило N,N, N', N'
тетрауксусная кислота.
Белый или почти белый кристаллический
порошок.
Температура плавления: около 204 ОС.
Циклоrексилметанол. С 7 Н 14 О. (М М. 114,2).
1135200. [1 0049 2]. Циклоrексилкарбинол.
Жидкость с леrким камфорным запахом,
растворим в спирте.
2 .
по. около 1,464.
Температура кипения: около 185 ОС.
3 Циклоrексилпропионовая кислота.
С 9 Н 1 Р2' (М. М. 156,2). 1119200. [701 97 3].
Бесцветная жидкость.
d 20
20: около 0,998.
20
по : около 1,4648.
Температура кипения: около 130 ОС.
j3-Циклодекстрин модифицированный
ДЛЯ хиральной хроматоrрафии. 1154600.
поли(диметил)(85)(дифенил)(15)силоксан Р,
содержащий 30 % 2,З ди 0 этил 6 0 терт
бутилдиметилсилил l3 циклодекстрина.
п Цимен. С 1О Н 14 . (м. М. 134,2). 1113400. [99
87 6]. 1 Изопропил4 метилбензол.
Бесцветная жидкость, практически нераст
ворим в воде, растворим в спирте.
d;g: около 0,858.
п O: около 1,4895.
Температура кипения: от 175 ос до 178 ОС.
п Цимен, используемый в 2азовой xpOMaтo
2рафии, должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
мяты перечной, с использованием испытуемоro
образца в качестве испытуемоro раствора.
Содержание п цимена, рассчитанное MeTO
дом нормализации, должно быть не менее 96,0 %.
Цинарин. С 25 Н 2 Р12' (м. М. 516,4). 1159300.
[30964 1 3 7]. (1 а,За,4а,5j3) 1 ,З Бис[[3 (3,4 ди
rидроксифенил) 1 оксо 2 пропенил]окси] 4,5
диrидроксициклоrексанкарбоновая кислота.
Белая или почти белая аморфная масса,
без запаха.
Цинеол. С 10 Н 1 р. (м. М. 154,3). 1020600. [470
82 ]. 1 ,8--Цинеол. Эвкалиптол. 1 ,8--Эпокси п ментан.
Бесцветная жидкость. Практически HepaCT
ворим в воде. Смешивается с этанолом.
d 20
20: от 0,922 до 0,927.
20
по: от 1,456 до 1,459.
Температура затвердевания (2.2.18). От
О ОС до 1 ОС.
Температурные пределы пере20нки (2.2.11).
От 174 ос д0177 ОС.
Фенол. 1 r цинеола встряхивают с 20 мл
воды Р. После разделения слоев к 1 О мл BOДHO
ro слоя прибавляют 0,1 М раствора железа (111)
хлорида Р1. Раствор не должен окрашиваться в
фиолетовый цвет.
4.1.1. Реактивы
729
Терпентинное масло. 1 r цинеола paCTBO
ряют в 5 мл спирта (90 %, об/об) Р, по каплям
прибавляют свежеприrотовленную бромную
воду Р Для получения желтоro окрашивания, не
исчезающеrо в течение 30 мин, должно быть из
расходовано не более 0,5 мл.
Остаток после выпаривания. Не более
0,05 %. К 10,0 мл прибавляют 25 мл воды Р, BЫ
паривают на водяной бане, остаток сушат до по
СТОЯН ной массы при температуре от 100 ос до
105 ОС.
Цинеол, применяемый в 2азовой xpOMaтo
2рафии, должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Мяты
перечной масло, используя в качестве испыту
eMoro раствора испытуемое вещество.
Содержание цинеола, рассчитанное Me
тодом нормализации, должно быть не менее
98,0%.
1,4 Цинеол. С 10 Н 1 р. (М м. 154,3). 1142500.
[4 70 67 7]. 1 Метил 4 ( 1 метилэтил 7-,оксаби
цикло[2.2.1]rептан. 1 Изопропил-4 метил 7 l(са
бицикло[2.2.1 ]rептан.
Бесцветная жидкость.
d 20
4 : от 0,922 до 0,927.
20
по : от 1,456 до 1,459.
Температура кипения: около 173 ОС.
Цинк. Zп. (А м. 65,4). 1096500. [7440 66 6].
Содержит не менее 99,5 % Zп.
Серебристо белые цилиндры, rранулы,
шарики или стружка с синим блеском.
Мышьяк (2.4.2, метод А). Не более
0,00002 % (0,2 ррт). 5,0 r цинка растворяют в
смеси 15 мл кислоты хлористоводородной Р и
25 мл воды Р. Полученный раствор должен BЫ
держивать испытание на мышьяк.
Цинк активированный. 1096501.
Цинк в виде цилиндров или шариков поме
щают в коническую колбу, прибавляют ДOCTa
точное количество 50 ррm раствора кислоты
хлорплатиновой Р, чтобы полностью покрыть
металл, через 10 мин металл промывают водой,
удаляют воду и немедленно сушат.
Мышьяк. К 5 r цинка активированноro при
бавляют 15 мл кислоты хлористоводород
ной Р, 25 мл воды Р, 0,1 мл раствора олова
(1/) хлорида Р и 5 мл раствора калия йодида Р
Далее поступают, как указано в испытании на
мышьяк (2.4.2, метод А). На ртутно бромид
ной бума2е Р не должно наблюдаться окраши
вания.
Активность. Повторяют испытание на
мышьяк, используя те же реактивы, прибавляют
раствор, содержащий 1 MKr мышьяка. На pтyт
но бромидной бума2е Р появляется заметное
окрашивание.
Цинка ацетат. (C2Hp)2Zn'2Hp. (М м. 219,5).
1102300. [597045 6]. Цинка ацетат диrидрат.
Ярко белые или почти белые кристаллы,
слеrка выветривающиеся на воздухе. Леrкорас
творим в воде, растворим в 96 % спирте. При TeM
пературе 100 ос теряет кристаллизационную воду.
d;g: около 1,7З5.
Температура плавления: около 237 ОС.
Цинка ацетата раствор. 1102301.
54,9 r цинка ацетата Р растворяют при пе
ремешивании в смеси 600 мл воды Р и 150 мл
кислоты уксусной ледяной Р При перемешива
нии прибавляют 150 мл раствора аммиака KOH
центрироваНН020 Р, охлаждают до комнатной
температуры, доводят раствором аммиака Р до
рН 6.4 и разводят водой Р до объема 1 л.
Цинка оксид. 1096700. [1314 13 2].
См. статью Цинка оксид.
Цинка пopowсж. Zп. (А м. 65,4). 1096800.
[744 ).
Содержит не менее 90.0 % Zп (А. м. 65,4).
Очень мелкий серый порошок. Растворим в
кислоте хлористоводородной разведенной Р
Цинка сульфат. 1097000. [7446 20 0].
См. статью Цинка сульфат 2епта2идрат.
Цинка хлорид. ZnC'2' (М м. 136,3). 1096600.
[7646 85 7].
Содержит не менее 95,0 % и не более
100,5 % ZnCI 2 .
Белый кристаллический порошок или белые
палочки. Расплывается на воздухе.
Очень леrко растворим в воде, леrкораство
рим в 96 % спирте и в rлицерине.
Хранят в неметаллических контейнерах.
Цинка хлорида раствор в кислоте
муравьиной. 1096601.
20 r цинка хлорида Р растворяют в 80 r раст
вора 850 r/л кислоты муравьиной безводной Р.
Цинка хлорида раствор йодированный.
1096602.
20 r цинка хлорида Р и 6,5 r калия йодида Р
растворяют в 10,5 мл воды Р, прибавляют 0,5 r
йода Р и встряхивают в течение 15 мин. При He
обходимости фильтруют.
Хранят в защищенном от света месте.
Цинка йодида и крахмала раствор. 1096502.
К раствору 2 r цинка хлорида Р в 1 О мл
воды Р прибавляют 0,4 r крахмала pacтвopиMO
20 Р И наrревают до растворения крахмала. Ox
лаждают до комнатной температуры, прибавляют
1,0 мл бесцветною раствора, содержащеro 0,10 r
цинка Р в виде опилок и 0.2 r йода Р в воде Р, дo
водят водой Р до объема 100 мл И фильтруют.
Хранят в защищенном от света месте.
730
rосударственная фармакопея Республики Беларусь
Испытание на чувствительность. 0,05 мл
раствора натрия нитрита Р доводят eOC'JiJ Р
до объема 50 мл. К 5 мл полученною раствора
прибавляют 0,1 мл кислоты серной разведен
ной Р и 0,05 мл приroтовленноro раствора цинка
йодида и крахмала и смешивают. Появляется
синее окрашивание.
ЦИНХОНИДИН. C1gH22N20, (м. м. 294,4).
1020400. [485 71 2]. (RНХинолА ил)[(2R,4S,5R)
5 винилхинуклидин 2 ил ]метанол.
Белый или почти белый кристаллический
порошок. Очень мало растворим в воде и в пе
тролейном эфире, растворим в 96 % спирте.
[aJ O: от 105 до 110. Определение прово
дят, используя раствор 50 r/л цинхонидина в
96 % спирте Р
Температура плавления: около 208 ос с раз
ложением.
Хранят в защищенном от света месте.
ЦИНХОНИН. C 1g H 22 Np. (м. м. 294,4).1020500.
[118 1 0 5]. (SНХинол 4 ил)[(2R,4S,5R) 5 винил
хинуклидин 2 ил ]метанол.
Белый или почти белый кристаллический
порошок. Малорастворим в воде, умеренно pac
творим в 96 % спирте и в метаноле.
[a] O: от +225 до +230. Определение прово
дят, используя раствор 50 r/л цинхонина в 96 %
спирте р
Температура плавления: около 263 ОС.
Хранят в защищенном от света месте.
Цирконила нитрат. Основная соль, состав
которой приблизительно соответствует формуле
ZrО(NОЗ)2'2Нр. 1097200. [14985 18 3].
Белый или почти белый порошок или кри
сталлы. rиrроскопичен. Растворим в воде.
Водный раствор прозрачный или слеrка опалес
цирующий.
Хранят в воздухонепроницаемом контейнере.
Цирконила нитрата раствор. 1097201.
Раствор 1 r/л в смеси растворителей вода p
кислота хлористоводородная Р (40:60, об/об).
Цирконила ХЛОРИД. Основная соль, состав
которой приблизительно соответствует формуле
ZrClp'8Hp. 1097100. [15461 27 5].
Содержит не менее 96,0 % ZrClp'8Hp.
Белый или почти белый кристаллический
порошок или кристаллы. Леrкорастворим в воде
и в 96 % спирте.
Количественное определение. 0.600 r цир
конила хлорида растворяют в смеси из 5 мл Kиc
лоты азотной Р и 50 мл воды Р, прибавляют
50,0 мл 0,1 М раствора серебра нитрата, 3 мл
раствора дибутилфталата Р, встряхивают и
титруют 0,1 М раствором аммония тиоцианата
до появления красновато желтоro окрашивания,
используя в качестве индикатора 2 мл раствора
железа (111) аммония сульфата Р2.
1 мл 0,1 М раствора серебра нитрата co
ответствует 16,11 Mr ZrClp'8Hp.
L Цистеин. С з Н 7 NО 2 S. (м. м. 121,1).
1024200. [52 90A].
Порошок. Леrкорастворим в воде, в 96 %
спирте и в кислоте уксусной, практически He
растворим в ацетоне.
Цистеина rидрохлорид. 1024300. [7048 04 6].
См. статью Цистеина 2идрохлорид MOHO
2идрат.
L Цистин. C6H12NP4S2' (м. м. 240,3).
1 024400. [56 89 З].
Белый или почти белый кристаллический
орошок. Практически нерастворим в воде и в
96 % спирте, растворяется в разведенных pac
творах rидроксидов щелочных металлов. Разла
rается при температуре 250 ОС.
[a] O: от 218 до 224. Определение проводят
в 1 М растворе кислоты хлористоводородной.
Цитостеариловый спирт.
См. статью Цитостаериловый спирт
(тип А) эмульсионный.
Цитраль. С 10 Н 1 р. (м. м. 152,2). 1020800.
[5392AO 5]. Смесь (2E) и (2Z) 3,7 диметилокта
2,6 диеналя.
Светло желтая жидкость. Практически He
растворима в воде, смешивается с 96 % спир
том и rлицерином.
Хромат02рафия. Тонкослойная xpOMa
тоrрафия (2.2.27), используя в качестве ТOH
кою слоя силика2ель GF 254 Р На xpoMaTorpa
фическую пластинку наносят 1 О мкл раствора
1 r/л цитраля в толуоле Р Хроматоrрафиру
ют, используя систему растворителей этилаце
тат Р толуол Р (15:85, об/об). Korдa фронт
растворителей пройдет 15 см, пластинку выни
мают из камеры и сушат на воздухе. Просма
тривают в ультрафиолетовом свете при длине
волны 254 нм. На xpoMaтorpaMMe должно обна
руживаться только одно основное пятно.
Цитраль, используемый в 2азовой xpOMa
т02рафии, должен выдерживать следующее
дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Hapдo
вое масло.
Содержание цитраля (нераль+rераниаль),
рассчитанное методом нормализации, должно
быть не менее 95,0 %.
Цитратная плазма кролика. 1020900.
у кролика, не принимавшеro пищу в течение
12 ч, отбирают кровь внутрисердечной пункци
ей, используя пластиковый шприц с иrлой NQ 1,
содержащий соответствующий объем раствора
38 r/л натрия цитрата Р, так, чтобы конечное
соотношение объемов раствора натрия цитрата
4.1.1. РеактивЬ!
731
и крови составляло 1 :9. Отделяют плазму цeH
трифуrированием при ускорении от 1500 9 до
1800 9 и температуре от 15 ос до 20 ос в тече
ние 30 мин.
Хранят при температуре от О ОС до 6 ОС.
Срок roдности 4 ч с момента отбора крови.
Цитронеллаль. с 1О н 1 р. (М м. 154,3).
1113300. [1 06 23 01]. 3, 7 Диметил 6 октеналь.
Очень мало растворим воде, растворяется
в спиртах.
d 20
20: от 0,848 до 0,856.
20
по : около 1,446.
Цитронеллаль, используемый в 28зовой
хромат02рафии, должен выдерживать следу
ющее дополнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье Масло
цитронеллы.
Содержание цитронеллаля, рассчитанное
методом нормализации, должно быть не менее
95,0%.
Цитроптен. С"Н,Р.. (М м. 206,2).1021300.
[487 06 9]. Лиметтин. 5,7 Диметокси 2Н 1
бензопиран 2 он.
Иroльчатые кристаллы. Практически HepaCT
ворим в воде и в петролейном эфире, леrкорас
творим в ацетоне и в 96 % спирте.
Температура плавления: около 145 ос.
Хромат02рафия. Тонкослойная xpOMaтo
rрафия (2.2.27), используя в качестве тонкоro
слоя силика2ель GF 254 Р На хроматоrрафиче
скую пластинку наносят 10 мкл раствора 1 r/л
цитроптена в толуоле Р Хроматоrрафируют,
используя систему растворителей этилаце
тат Р толуол Р (15:85, об/об). Korдa фронт
растворителей пройдет 15 см, пластинку выни
мают из камеры и сушат на воздухе. Просма
тривают в ультрафиолетовом свете при длине
волны 254 нм. На xpoMaтorpaMMe должно обна
руживаться только одно основное пятно.
UЦавелевая кислота. С2НР4'2Нр. (М м. 126.1).
1061400. [6153 56 6]. Этандикарбоновая кисло
та диrидрат.
Белые или почти белые кристаллы. PaCТBO
рима в воде, леrкорастворима в 96 % спирте.
UЦавелевой кислоты и серной кислоты
раствор. 1061401.
Раствор 50 r/л щавелевой кислоты Р в ox
лажденной смеси из равных объемов кислоты
серной Р и воды Р
# Швейцера реактив. 1 О r меди сульфа
та Р растворяют в 100 мл воды Р, прибавляют
раствор 100 r/л натрия 2идроксида Р в ДOCTa
точном для осаждения rидрата окиси меди коли
честве, собирают последний на фильтре и про
мывают водой Р до исчезновения реакции на
сульфаты. Влажный осадок растворяют в мини
мальном количестве раствора аммиака разве
деНН020 Р1, необходимом для полноro paCTBO
рения осадка. Хранят в стеклянном контейнере
с притертой пробкой.
# UЦелочно-аммиачный раствор.
50 r натрия 2идроксида Р растворяют при
перемешивании в 870 мл воды Р. После охлаж
дения раствора прибавляют 80 мл KOHцeHтpи
роваНН020 раствора аммиака Р. Раствор roден
в течение 24 ч.
Эвrенол. С'ОН'Р2' (М м. 164,2). 1037000.
[97 53 O]. 4 Аллил 2 метоксифенол.
Бесцветная или бледно желтая масляни
стая жидкость, под действием воздуха и света
темнеет и становится более вязкой. Практически
нерастворим в воде, смешивается с 96 % спир
том и с жирными и эфирными маслами.
dig: около 1,07.
Температура кипения: около 250 ос.
Эв2енол, используемый в 2азовой xpOMaтo
2рафии, должен выдерживать следующее дo
полнительное испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье rвоз
дичное масло, используя в качестве испытуемо
ro раствора испытуемое вещество.
Содержание эвrенола. рассчитанное Me
тодом нормализации, должно быть не менее
98,0%.
Хранят в защищенном от света месте.
# Эвкалиптол. См. Цинеол Р
Электролитический реактив дnя микро-
определения воды. 1113700.
Коммерчески доступный безводный реактив
или комбинация безводных реактивов для кулоН<r
метрическоro титрования воды, содержащая ПОk
ходящие орrанические основания, диоксид серы и
йод растворенные в подходящем растворителе.
# Эллаrовая кислота. С'4НБОв' (М М. 302,2).
[4 76 664]. 4,4' ,5,5' ,6,6' rексаrидроксидифено
вой кислоты 2,6,2',6' дилактон.
Температура плавления: не менее 350 ОС.
Вода (2.5.12): не более 12 %.
Содержит не менее 96,0 % С'4Н60в' опре
деленной методом высокоэффективноro капил
лярноro электрофореза.
# Эльманта реактив. См. 5,5' Дитиобис
(2 нитробензойная кислота) Р
Эметина диrидрохлорид. C29H42C12N204'
'5Нр. (М м. 643,6). 1034300. [31642 7).
(2S,3R, 11 bS) 2 [[( 1 R) 6, 7 Диметокси 1 ,2,3,4
тетраrидраизоквиналин 1 ил]метил] 3 этил
9, 1 О диметокси 1,3,4,6,7, 11 Ь rексаrидра 2Н
бензо(а]хинализина rидрохлорид. Эметина
rидрохлорид пентаrидрат.
732
rосударственная фармакопея Республики Беларусь
Содержит не менее 98,0 % и не более 102,0 %
C29H42CI2N204 в пересчете на сухое веществе.
Белый или слеrка желтоватый кристалличеСt;ИЙ
порошок. Леrкорастворим в воде и в 96 % спирте.
Потеря в массе при высушивании (2.2.32).
От 11,0 % до 15,0 %.1,00 r эметина диrидрохло
рида сушат при температуре 105 ос в течение 3 ч.
Хранят в защищенном от света месте.
3МОДИН. С'5Н,оО5' (М м. 270,2). 1034400.
[518 82 1]. 1 ,3,8 Триrидрокси 6 метилантрахинон.
Оранжево красные иroльчатые кристаллы.
Практически нерастворим в воде, растворим в
96 % спирте и в растворах rидроксидов щелоч
ных металлов.
Хромат02рафия. Определение проводят,
как указано в статье Ревеня корни. На xpOMaTO
rpaMMe должно обнаруживаться только одно oc
новное пятно.
# ЭОЗИН Н. Смесь динатриевых солей TeTpa
бромфлуоресцеина C 2o H 6 Br 4 Nap5 (М. м. 691,9) и
дибромфлуоресцеина C 2o H B Br 2 Nap5 (М м. 534,1).
Эозин натрий водорастворимый.
Красный или красно коричневый порошок.
Леrкорастворим в воде.
#Эозинатнатрия. C2oH6Br4Na205' (М.м.691 ,9).
[17372 87 1). Динатрия 2',4',5',7' тетрабром
З' ,6' диrидроксиспиро[изобензофуран 1 (3H),9'
[9Н]KcaHTeH] трион. Т етрабромфлуоресцеинат
натрия. Эозин. Эозин у.
Красные кристаллы с синеватым оттенком
или коричневат красный порошок. Леrкорас
творим в воде, малорастворим в 96 % спирте,
практически нерастворим в эфире.
# Эриохром черный. См. Протравной чер
ный 11 Р.
Эритритол. С 4 Н,о04' (М м. 122,1). 1113800.
[149 32 6]. (R",S*) Бутан 1,2,З.4 тетрол. мезо
Эритритол.
Кристаллы в виде тетраroнальных призм.
Очень леrко растворим в воде, растворим в пи
ридине, малорастворим в 96 % спирте.
Температура плавления: около 121,5 ОС.
Эрукамид. С 22 Н 4з NО. (М м. 337,6).1034500.
[112 84 5]. (Z) Докоз 1 З еноамид.
Желтоватый или белый порошок или rранулы.
Практически нерастворим в воде, очень леrко pac
творим в метиленхлориде, растворим в этаноле.
Температура плавления: около 70 ОС.
Эскулин. C'5H'60g'1,5Hp. (М м. 367,3).
1119400. [531 75 9]. 6+D rлюкопиранозилокси)
7 rидрокси 2Н хромен 2 он.
Белый или почти белый порошок или бес
цветные кристаллы. Умеренно растворим в воде
и в 96 % спирте, леrкорастворим в roрячей воде
и в rорячем 96 % спирте.
Хромат02рафия (2.2.27). Определение про
водят, как указано в статье Элеутерококка KOp
невища и корни. На xpoMaTorpaMMe должно об
наруживаться только одно основное пятно.
Эстраrол. С ш Н,Р. (Мм. 148,2). 1034700.
[140 67 O]. 1 МетоксиА проп 2 енилбензол.
Жидкость. Смешивается с 96 % спиртом.
п O: около 1,52.
Температура кипения: около 216 ОС.
Эстра20Л, используемый в 2азовой xpOMa
т02рафии, должен выдерживать следующее
испытание.
Количественное определение. rазовая xpo
матоrрафия (2.2.28), как указано в статье AHи
совое масло, используя в качестве испытуемоro
раствора испытуемое вещество.
Содержание эстраroла, рассчитанное Meтo
дом нормализации, должно быть не менее 98,0 %.
17а-Эстрадиол. С'ВН2Р2' (М м. 272,4).
1034600. [57 91 O].
Белый или почти белый кристаллический
порошок или бесцветные кристаллы.
Температура плавления: от 220 ос до 223 ОС.
Эсцин. 1001700. [11072 93 8].
Смесь родственных сапонинов, полученных
из семян Aescu/us hiрросаstапит L.
Мелкий почти белый или слеrка KpaCHOBa
тый или желтоватый аморфный порошок.
Хромат02рафия. Определение проводят,
как указано в статье Сене2и корни, объем Ha
носимой пробы 20 мкл. После опрыскивания
xpoMaTorpaMMbI раствором анисов020 альде
2ида Р и наrревания на xpoMaTorpaMMe paCT
вора должно обнаруживаться основная зона с R,
около 0,4.
Этанол. 1034800. [64 17 5]. См. Этанол
безводный Р.
Этанол Р1. 1034801.
Должен выдерживать требования для эта
нола безводН020 Р и следующее дополнитель
ное испытание.
Метанол. Не более 0,005 % (об/об). rазовая
хроматоrрафия (2.2.28).
Испытуемый раствор. Испытуемое вещество.
Раствор сравнения. 0,50 мл метанола без
водН020 Р доводят испытуемым этанолом до
объема 100,0 мл. 1,0 мл полученноrо paCTBopaдo
водят испытуемым этанолом до объема 100,0 мл.
Условия хроматоrрафирования:
колонка стеклянная длиной 2 м и BHYTpeH
ним диаметром 2 мм, заполненная сополимером
этилвинилбензол дивинилбензола Р с разме
ром частиц от 75 мкм до 100 мкм;
2аз носитель: азот для хромат02рафии Р;
скорость 2аза носителя: 30 мл/мин;
детектор: пламенно ионизационный;
температура колонки: 130 ОС;
4.1.1. Реактивы
7ЗЗ
температура блока ввода проб: 150 ос;
температура детектора: 200 ос.
Вводят по 1 мкл испытуемоro раствора и
раствора сравнения поочередно три раза. После
каждоro хроматоrрафирования наrревают KO
лонку до температуры 230 ос в течение 8 мин.
Интеrрируют пик метанола.
Содержание метанола в процентах рассчи
тывают по формуле:
а.Ь
,
c b
rдe:
а содержание метанола в растворе cpaB
нения, в процентах (об/об);
Ь площадь пика метанола на xpOMaтo
rpaMMe испытуемоro раствора;
с площадь пика метанола на xpOMaTO
rpaMMe раствора сравнения.
Эта нол безводный. С 2 Н 6 О. (М М. 46,07).
1034800. [64 17 5].
Содержит не менее 99,5 % (об/об) С 2 Н 6 О
(99,2 % М/М), рассчитанных при 20 ос из относи
тельной плотности с использованием алкоroле
метрических таблиц (5.5).
Бесцветная прозрачная летучая воспламе
няющаяся rиrроскопичная жидкость. Смешива
ется с водой и сметиленхлоридом. rорит сине
ватым слабо светящимся бездымным пламенем.
Температура кипения: около 78 ос.
dig: от 0,790 до 0,793.
Хранят в защищенном от света месте.
#Для препаративных целей эта нол безво
дный может быть получен из 96 % спирта Р
одним из перечисленных способов.
1. К 1 л 96 % спирта Р прибавляют 200 r CBe
жепрокаленноrо кальция оксида Р, встряхивают и
кипятят с обратным холодильником в течение 4 ч.
2. 500 r меди (11) сульфата Р, прокаленноro
до изменения цвета на белый с желтоватым oттeH
ком, помещают в колбу со шлифом и прибавляют
1 л 96 % спирта Р и укупоривают. Выдерживают в
течение 3 суток при периодическом встряхивании.
Перед использованием фильтруют.#
Этаноламин. C 2 H 7 NO. (М М. 61,1).1034900.
[14143 5). 2 Аминоэтанол.
Прозрачная бесцветная вязкая rиrроскопичная
жидкость. Смешивается с водой и с метанолом.
d 20
20: около 1,04.
20
по : около 1,454.
Температура плавления: около 11 ос.
Хранят в воздухонепроницаемом контейнере.
Этилакрилат. С 5 НР2' (М М. 100,1). 1035400.
[140 88 5]. Этилпроп 2 еноат.
Бесцветная жидкость.
d 20
20: около 0,924.
20
по: около 1,406.
Температура кипения: около 99 ос.
Температура плавления: около 71 ос.
Этилацетат. С 4 НР2' (М М. 88,1). 1035300.
[141 78 6].
Прозрачная бесцветная жидкость. PaCTBO
рим в воде, смешивается с 96 % спиртом.
d;g: от 0,901 до 0,904.
Температура кипения: от 76 ос до 78 ос.
Этилацетат обработанный. 1035301.
200 r кислоты сульфаминовой Р диспер
rируют в этилацетате Р и доводят до объема
1000 мл этим же растворителем. Полученную cy
спензию перемешивают в течение 3 сут и филь
труют через бумажный фильтр.
Срок roдности 1 мес.
Этилбензол. СвН,о' (М м. 106,2). 1035800.
[10041 4].
Содержит не менее 99,5 % (м/м) СеН,О; опре
деление проводят методом rазовой xpoMaTorpa
фии_
Прозрачная бесцветная жидкость. Практи
чески нерастворим в воде, растворим в ацетоне
и в 96 % спирте.
d 20
20: около 0,87.
20
по: около 1 ,496.
Температура кипения: около 135 .С.
2-Этилrексан-1,З-диол. С В Н'Р2' (1.4.",.146'2).
1105900. [94 96 2).
Слеrка маслянистая жидкость. Растворим в
этаноле, в 2 пропаноле, в пропиленmиколе и в
масле касторовом.
d 20
20: около 0,942.
20
n D : около 1,451.
Температура кипения: около 244 .С.
2-Этилrексановая кислота. С8Н'БО2'
(М М. 144,2). 1036600. [149 57 5]. 2 .Этилкапро
новая кислота.#
Бесцветная жидкость.
d 20
20: около 0,91.
20
n D : около 1,425.
Сопутствующие примеси. rаэовая xpo
матоrрафия (2.2.28). Хроматоrpафируют 1 мкл
раствора, приroтовленноro следующим обра
зом: 0,2 r кислоты 2 этилrексановой суспенди
руют в 5 мл воды Р. прибавляют 3 мл кислоты
хлористоводородной разведенной Р и 5 мл 2eK
сана Р, встряхивают в течение 1 мин, выдержи
вают до разделения слоев и используют верхний
слой. Хроматоrрафируют как указано в испыта
нии «Кислота 2 этилrексановая}) в статье AMOK
сициллин натрия.
Сумма площадей всех пиков, кроме OCHOB
Horo и пика растворителя, не должна превышать
2,5 % площади основноro пика.
734
rосударственная фармакопея Республики Беларусь
Этиленбис[З,З-ди(З-(1,1-диметилэтил)-4-
rидроксифенил)бутират]. С50Н660в' (М м. !95).
1035900. [32509 66 3]. Этиленбис[3,3 ди(3 трет
бутил 4 rидроксифенил )бутират].
Кристаллический порошок. Практически He
растворим в воде и в петролейном эфире, очень
леrкорастворим в ацетоне и в метаноле.
Температура плавления: около 165 ОС.
Этиленбис[З, З-ди(З-трет-бутил -4-rид
роксифенил)бутират]. 1035900. [32509 66 3].
См. Этиленбис[3.3 ди(3 (1.1 диметилэтил) 4
2идроксифенил)бутират] Р.
Этиленrликоль. C 2 H€02' (М м. 62,1).
1036100. [107 21 1]. Этан 1.2 диол.
Содержание: не менее 99,0 %.
Бесцветная, слеrка вязкая rиrроскопич
ная жидкость. Смешивается с водой и с 96 %
спиртом.
d 20
20: от 1,113 до 1,115.
20
п D : около 1 ,432.
Температура плавления: около 12 ОС.
Температура кипения: около 198 ОС.
Кислотность. К 1 О мл прибавляют 20 мл
воды Р и 1 мл раствора фенолфталеина Р; при
прибавлении не более 0,15 мл 0,02 М раствора
наrnрия 2идроксида окраска раствора должна
измениться на розовую.
Вода (2.5.12). Не более 0,2 %.
Этиленrликоля монометиловый эфир.
С З НР2' (М М. 76,1). 1036300. [1 09 86-4]. 2 Me
токсиэтанол.
Содержание: не менее 99,0 %.
Прозрачная бесцветная жидкость. Смеши
вается с водой, с ацетоном и с 96 % спиртом.
d 20
20: около 0,97.
20
п D : около 1 ,403.
Температура кипения: около 125 ОС.
Этиленrликоля моноэтиловый эфир.
С 4 Н 10 О 2 . (Мм. 90,1). 1036200. [110 80 5J. 2 Эток
сиэтанол.
Содержание: не менее 99,0 %.
Прозрачная бесцветная жидкость. Смеши
вается с водой, с ацетоном и с 96 % спиртом.
d;g: около 0,93.
п O: около 1,406.
Температура кипения: около 135 ОС.
Этилендиамин. C 2 H B N 2 . (Мм. 60,1).
1036500. [1 07 15 3]. Этан 1 ,2 диамин.
Прозрачная бесцветная дымящаяся жид
кость; имеет сильнощелочную реакцию. Смеши
вается с водой и с 96 % спиртом.
Температура кипения: около 116 ОС.
(Этилендинитрил)тетрауксусная кислота.
C 10 H 16 NPB' (Мм. 292,2). 1105800. [60 00-4]. N,N'
1 ,2 Этандиилбис[N' (карбоксиметил)rлицин].
Эдетовая кислота.
Белый или почти белый кристаллический
порошок. Очень мало растворима в воде.
Температура плавления: около 250 ос с раз
ложением.
Этиленоксид. С 2 Нр. (М м. 44.05). 1036400.
[75 21 8]. Оксиран.
Бесцветный воспламеняющийся rаз. Очень
леrко растворим в воде и в этаноле.
Температура сжижения: около 12 ОС.
Этиленоксида исходный раствор. 1036401.
Все операции, производимые в ходе при20
товления растворов, выполняют в вытяжном
шкафу. 3ащищают руки и лицо, надевая поли
этиленовые защитные перчатки и подходя
щую маску для лица.
Все растворы хранят в воздухонепроница
емом контейнере в холодильнике при темпера
туре от 4 ос до 8 ос. Все испытания проводят
три раза.
В сухую чистую пробирку, охлажденную в
смеси из 1 части натрия хлорида Р и 3 частей из
мельченною льда, медленно вводят поток rазо
образною этиленоксида Р, позволяя конденсиро
ваться на внутренней стенке пробирки. С помощью
стеклянною шприца, предварительно охлажденно
ю до температуры 1 О ОС, помещают около 300 мкл
(что соответствует приблизительно 0,25 r) жидкоro
этиленоксида Р в 50 мл маКР020ла 200 Р1. Опре
деля ют абсорбированное количество этиленок
сида взвешиванием до и после абсорбции (М ео )'
Доводят маКР020ЛОМ 200 Р1 до объема 100,0 мл.
Перед использованием тщательно перемешивают.
Количественное определение. К 1 О мл cy
спензии 500 r/л ма2ния хлорида Р в этаноле Р
прибавляют 20,0 мл 0,1 М раствора кислоты
хлористоводородной в спирте. Колбу закры
вают пробкой, встряхивают до получения Hacы
щенноro раствора и для достижения равновесия
выдерживают в течение ночи. 5,00 r исходН020
раствора этиленоксида (2,5 2/Л) Р помещают в
колбу, выдерживают в течение 30 мин и титруют
0,1 М раствором калия 2идроксида спиртовым
потенциометрически (2.2.20).
Проводят контрольный опыт, используя
вместо исходною раствора этиленоксида такое
же количество маКР020ла 200 Р1.
Содержание этиленоксида в миллиrраммах
в одном rpaMMe рассчитывают по формуле:
(V o V1).f .4,404
т
rдe:
V o и V 1 объемы 0,1 М раствора калия 2и
дроксида спиртов020, израсходованные на ти
троваl,Jие контрольноro и испытуемоro раствора,
соответственно;
4. 1.1. Реактивы
735
f поправочный коэффициент к О, 1 М pac
твору калия 2идроксида спиртовому;
т масса испытуемою образца, r.
Этиленоксида исходный раствор Р1.
1036406.
Раствор 50 мr/мл этиленоксида Р в Meтa
ноле Р.
Этиленоксида раствор. 1036402.
Взвешивают количество охлажденноro иc
ходН020 раствора этиленоксида Р, соответ
ствующее 2,5 Mr этиленоксида, в охлажденной
колбе, доводят маКр020ЛОМ 200 Р1 до массы
50,0 r и тщательно перемешивают. 2,5 r полу
ченноro раствора доводят маКр020ЛОМ 200 Р1
до объема 25,0 мл (5 MKr этиленоксида в 1 r
раствора).
rотовят непосредственно перед использо
ванием.
Этиленоксида раствор Р1. 1036403.
1,0 мл охлажденною исходН020 раствора
этиленоксида Р (берут точный объем. исполь
зуя взвешивание) доводят макро2ОЛОМ 200 Р1
до объема 50,0 мл и тщательно перемешива
ют. 2,5 r полученною раствора доводят MaKp020
лом 200 Р1 до объема 25,0 мл. Содержание эти
леноксида в ррт вычисляют исходя из объема,
определенноro взвешиванием, принимая плот
ность маКр020ла 200 Р1 равной 1,127.
rотовят непосредственно перед использо
ванием.
Этиленоксида раствор Р2. 1036404.
1,00 r охлажденноro исходН020 раствора
этиленоксида Р (что соответствует 2,5 Mr эти
леноксида) помещают в предварительно взве
шенную колбу, содержащую 40,0 r охлажденноro
маКр020ла 200 Р1. и перемешивают. Определя
ют точную массу и разводят до расчетной массы
таким образом, чтобы получить раствор, coдep
жащий 50 MKr этиленоксида в 1 r раствора. Взве
шивают 10,00 r, помещают в колбу, содержащую
около 30 мл воды Р, перемешивают и доводят
водой Р до объема 50,0 мл (10 мкr/мл этиленок
сида).
rотовят непосредственно перед использо
ванием.
Этиленоксида раствор РЗ. 1036405.
10,0 мл раствора этиленоксида Р2 доводят
водой Р до объема 50,0 мл (2 мкr/мл этиленок
сида ).
rотовят непосредственно перед использо
ванием.
Этиленоксида раствор Р4. 1036407.
1,0 мл исходН020 раствора этиленокси
да Р1 доводят водой Р до объема 100,0 мл.
1,0 мл полученноro раствора доводят водой Р до
объема 25,0 мл.
Этиленхлорид. C 2 H 4 C1 2 . (М М. 99,0).
1036000. [1 07 06 2]. 1 ,2 Дихлорэтан.
Прозрачная бесцветная жидкость. PaCTBO
рим приблизительно в 120 частях воды и в 2
частях 96 % спирта.
d 2G : около 1,25.
тf/мпературные пределы пере20НКU
(2.2.11). От 82 ос до 84 ОС; должно переroняться
не менее 95 %.
N-Этилмалеимид. C 6 H 7 N0 2 . (М М. 125,1).
1036700. [128 53 0]. 1 Этил 1 Н пиррол 2,5 дион.
Бесцветные кристаллы. Умеренно paCTBO
рим в воде. леrкорастворим в 96 % спирте.
Температура плавления: от 41 ос до 45 ОС.
Хранят при температуре от 2 ос до 8 ОС.
Этилметилкетон. 1054100. [78 93 3]. См.
Метилэтuлкетон Р
Этилметансульфонат. С з Нр,S. (М м. 124.2).
1179300. [62-50 ].
Прозрачная, бесцветная жидкость.
Содержит не менее 99,0 % СзНвОзS.
Плотность: около 1,206 r/см З (при 20 ОС).
п O: около 1,418.
Температура кипения: около 213 ОС.
2-Этил-2-метилянтарная кислота. С 7 Н 12 О 4 .
(М М. 160,2). 1036800. [631 31 2]. 2 Этил 2 метил
Iбутандикарбоновая кислота.
Температура плавления: от 104 ОС до 107 ОС.
Этилпараrидроксибензоат. С 9 Н 10 О з .
(М М. 166,2). 1035700. [120A7 8]. ЭтилА rидро
ксибензоат.
Содержит не менее 99,0 % и не более
100,5 % С 9 Н 10 О з .
Белый или почти белый кристаллический по
рошок или бесцветные кристаллы. Очень мало
растворим в воде, леrкорастворим в 96 % спирте
и в метаноле.
Температура плавления (2.2.14): от 115 ос
до 118 ОС.
Этилформиат. С з Н 6 О 2 . (М М. 74,1). 1035600.
[109 94 4]. Этилметаноат.
Прозрачная бесцветная воспламеняющаяся
жидкость. Леrкорастворим в воде, смешивается
с 96 % спиртом.
d 20
20: около 0,919.
п O: около 1,36.
Температура кипения: около 54 ОС.
Этилцианоацетат. C 5 H 7 N0 2 . (М М. 113,1).
1035500. [1 05 56 6J.
Жидкость от бесцветной до светло желто
ю цвета. Малорастворим в воде, смешивается
с 96 % спиртом.
Температура кипения: от 205 ос до 209 ос с
разложением.
736
rосударственная фармакопея Республики Беларусь
Эroксихризоидина rидpoхлорид. С 14 Н 17 CIN 4 О.
(м.м. 292,8). 1035200. [2313 87 3]. 4 [(4 Этокси
фенил)диазенил]фенилен 1 ,3 диамина rидро
хлорид.
Красноватый порошок. Растворим в 96 %
спирте.
Этоксихризоидина раствор. 1035201.
Раствор 1 r/л этоксихризоидина 2идрохло
рида Р в 96 % спирте Р
Испытание на чувствительность. К смеси
из 5 мл кислоты хлористоводородной разве
денной Р и 0,05 мл раствора этоксихризоидина
прибавляют 0,05 мл 0,0167 М раствора бромид
бромата. Окраска раствора должна измениться
от красной до светл желтой в течение 2 мин.
Эуrлобулины бычьи. 1037100.
Используют свежую бычью кровь, собран
ную в раствор антикоаryлянта (например, paCT
вор натрия цитрата). Отбрасывают любую reMo
лизную кровь. Центрифуrируют с ускорением от
1500 9 до 1800 9 при температуре от 15 ос до
20 ос дЛЯ получения супернатанта плазмы с
низким содержанием тромбоцитов.
К 1 л плазмы бычьей прибавляют 75 r бария
сульфата Р, встряхивают в течение 30 мин,
затем центрифуrируют с ускорением от 1500 9 до
1800 9 при температуре от 15 ос до 20 ос и OTдe
ляют прозрачную надосадочную жидкость. При
бавляют 1 О мл раствора 0,2 мr/мл апротинина Р
и встряхивают до смешения. В контейнер с ми
нимальной вместимостью 30 л в камере с TeM
пературой 4 ос помещают 25 л воды дистилли
рованной Р, охлажденной до температуры 4 ОС,
прибавляют около 500 r твердоro уrлерода диок
сида и тотчас при nepeмешивании прибавляют
надосадочную жидкость, полученную из плазмы.
Образуется белый осадок. Для осаждения BЫ
держивают при температуре 4 ос от 10 ч до 15 ч.
Прозрачную надосадочную жидкость отделяют
с помощью сифона. Собирают осадок центри
фуrированием при температуре 4 ОС. Суспенди
руют осадок механическим дисперrированием в
500 мл воды дистиллированной Р при темпера
туре 4 _ОС, встряхивают в течение 5 мин и отделя
ют осадок центрифуrированием при температуре
4 ОС. Осадок механически дисперrируют в 60 мл
раствора, содержащеro 9 r/л натрия хлорида Р
и 0,9 r/л натрия цитрата Р, доводят paCTBO
ром 10 r/л натрия 2идроксида Р до рН 7.2 7,4 и
фильтруют через стеклянный фильтр. Получен
ный осадок измельчают в ступке, фильтр и ступку
промывают 40 мл раствора, содержащею 9 r/л
натрия хлорида Р и 0,9 r/л натрия цитрата Р,
доводят тем же раствором до объема 100 мл И
лиофилизируют. Обычно выход составляет от 6 r
до 8 r эуrлобулинов из 1 л плазмы бычьей.
Испытание на при20дность. rотовят paCT
вор, используя фосфатный буферный pacт
вор рН 7,4 Р, содержащий 30 r/л альбумина бы
чье20 Р
в пробирку диаметром 8 мм, помещенную
в водяную баню при температуре 37 ОС, вносят
0,2 мл раствора сравнения урокиназы, coдep
жащеro 100 МЕ/мл, И 0,1 мл раствора тромби
на человечеСК020 Р, содержащеro 20 МЕ/мл.
Быстро вводят 0,5 мл раствора, содержащею
1 О Mr эуrлобулинов бычьих в миллилитре. За
время менее 1 О с должен образоваться плотный
CryCТOK. Отмечают время, прошедшее между
прибавлением раствора эуrлобулинов бычьих и
разрушением crycTKa. Время лизиса не должно
превышать 15 мин.
Хранят в защищенном от влаrи месте при
температуре 4 ОС.
Срок roдности 1 roд.
Эуrлобулинычеловеческие.103720Q
Для приroтовления используют свежую че
ловеческую кровь, собранную в раствор анти
коаryлянта (например, раствор натрия цитра
та) или человеческую кровь для переливания,
собранную в пластмассовые контейнеры для
крови, с только что истекшим сроком хранения.
Отбрасывают любую rемолизированную кровь.
Центрифуrируют с ускорением от 1500 9 до
1800 9 при температуре 15 ос для получения cy
пернатанта плазмы с низким содержанием TpOM
боцитов.
Можно смешивать плазмы, полученные из
крови одной rруппы.
К 1 л плазмы прибавляют 75 r бария суль
фата Р, встряхивают в течение 30 мин, затем
центрифуrируют при температуре 15 ос с YCKO
рением не менее 1500 9 и отделяют прозрач
ную надосадочную жидкость. Прибавляют 1 О мл
раствора 0,2 мr/мл апротинина Р и встряхивают
до смешения. В контейнер с минимальной BMe
стимостью 30 л в камере с температурой 4 ос
помещают 25 л воды дистиллированной Р, ox
лажденной до температуры 4 ОС, прибавля
ют около 500 r твердоro уrлерода диоксида и
тотчас прибавляют при перемешивании Haдo
садочную жидкость, полученную из плазмы; об
разуется белый осадок. Оставляют для осажде
ния при температуре 4 ос на 1 o 15 ч. Удаляют
прозрачную надосадочную жидкость с помощью
сифонирования. Собирают осадок центрифуrи
рованием при температуре 4 ОС. Суспендиру
ют осадок механическим дисперrированием в
500 мл воды дистиллированной Р при темпера
туре 4 ОС, встряхивают в течение 5 минут и OTдe
ляют осадок центрифуrированием при темпера
туре 4 ОС. Механически дисперrируют осадок в
60 мл раствора, содержащеro 9 r/л натрия хло
рида Р и 0,9 r/л натрия цитрата Р и доводят
раствором натрия 2идроксида Р до рН 7,2 7,4.
Фильтруют через стеклянный фильтр. Получен
ный осадок измельчают с помощью подходяще
ro приспособления. Промывают фильтр и при
способление 40 мл раствора, содержащеro 9 r/л
натрия хлорида Р и 0,9 r/л натрия цитрата Р,
и разводят до объема 100 мл этим же раствором.
4.1.2. Эталонные растворы для испытаний на предельное содержимое примесей 737
Лиофилизируют. Обычно выход составляет от 6 r
до 8 r эуrлобулинов из 1 л плазмы человеческой.
Испытание на при20дность. Для этою ис
пытания rотовят раствор, используя фосфат
ный буферный раствор рН 7,2 Р, содержащий
30 r/л альбумина бычье20 Р.
В пробирку диаметром 8 мм, помещенную в водя
ную баню при температуре 37 ос, вносят 0,1 мл paCT
вора сравнения стрептокиназы, содержащею 10 МEI
мл стрептокиназной активности, и 0,1 мл раствора
тромбина человеческоао Р, содержащеro 20 МElмл.
Быстро прибавляют 1 мл раствора, содержащеro
1 О мr/мл эуrлобулинов человеческих. Должен обра
зоваться плотный CryCТOK за время менее 1 О с. OTMe
чают время, прошедшее между прибавлением pacт
вора эуrлобулинов человеческих и разрушением
crycTKa. Время лизиса не должно превышать 15 мин.
Хранят в воздухонепроницаемом контейне
ре при температуре 4 ос.
Срок roдности 1 roд.
Эфир. С 4 Н 1 р. (М м. 74,1). 1035000. [60 2 7].
Прозрачная бесцветная летучая очень под
вижная, леrковоспламеняющаяся жидкость. rи
rроскопичен. Растворим в воде, смешивается с
96 % спиртом.
d;g: от 0,713 до 0,715.
Температура кипения: от 34 ос до 35 ос.
Не пере20няют, если эфир не выдержива
ет испытания на пероксиды.
Пероксиды. 8 мл раствора крахмала с
калия йодидом Р помещают в цилиндр с притер
той стеклянной пробкой вместимостью 12 мл и
диаметром около 15 мм. Объем цилиндра за
полняют испытуемым эфиром, энерrично встря
хивают и выдерживают в защищенном от света
месте в течение 30 мин. Не должно появляться
окрашивание.
Название и концентрация любоro добав
ленноro стабилизатора должны быть указаны
на этикетке.
Хранят в воздухонепроницаемом контейне
ре, защищенном от света месте при температу
ре не выше 15 ос.
Эфир, свободный от пероксидов.
1035100.
См. статью Эфир анестезирующий.
Эхинакозид. С з5 Н 4 Рш (М м. 786,5).
1159400. (82854 37 3].I3 (З' ,4' Диrидроксифенил)
этил О а L рамнопиранозил( 1 3 ) O I3 D т D
rлюкопиранозил( 1 6 )] ( 4 0 кафеоил)
rлюкопиранозид.
Бледно желтый порошок, без запаха.
Янтарная кислота. С 4 Н 6 О 4 . (М м. 118,1).
1085600. [110 15 6]. Бутандиовая кислота.
Белый или почти белый кристаллический
порошок или бесцветные кристаллы. Раствори
ма в воде и 96 % спирте.
Температура плавления: от 184 ос до 187 ос.
93 з..к 1712
01/2013:40102
4.1.2. ЭТАЛОННЫЕ РАСТВОРЫ
ДЛЯ ИСПЫТАНИЙ
НА ПРЕДЕЛЬНОЕ
СОДЕРЖАНИЕПРИМЕСЕИ
Алюминия эталонный раствор (200 ррm AI).
5000200.
Навеску алюминия калия сульфата Р, co
ответствующую 0,352 r AIK(S04)2'12HP, paCT
воряют в воде Р, прибавляют 1 О мл кислоты
серной разведенной Р и доводят водой Р до
объема 100,0 мл.
Алюминия эталонный раствор (100 ррm AI).
5000203.
8,947 r алюминия хлорида Р растворяют в
воде Р и доводят до объема 1000,0 мл этим же
растворителем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 10 раз.
Алюминия эталонный раствор (10 ррm AI).
5000201.
Навеску алюминия нитрата Р, COOTBeTCТBY
ющую 1,39 r АI(NОз)з'9НР, растворяют в воде Р и
доводят до объема 100,0 мл этим же растворителем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 100 раз.
Алюминия эталонный раствор (2 ррm AI).
5000202.
Навеску алюминия калия сульфата Р, co
ответствующую 0,352 r AIK(S04)2'12HP, paCTBO
ряют в 1 О мл кислоты серной разведенной Р и
доводят водой Р до объема 100,0 мл.
Непосредственно перед использованием
полученный раствор разводят водой Р в 100 раз.
Аммония эталонный раствор (100 ррm NH 4 ).
5000300.
Навеску аммония хлорида Р, COOTBeTCTBY
ющую 0,741 r NH 4 CI, растворяют в воде Р и дo
дят до объема 1000,0 мл этим же растворителем.
Непосредственно перед использованием
1 О мл полученноro раствора доводят водой Р до
объема 25 мл.
Аммония эталонный раствор (2,5 ррт NHJ
5000301.
Навеску аммония хлорида Р, COOTвeтCTBY
ющую 0,741 r NH 4 CI, растворяют в воде Ридово
дят до объема 1000,0 мл этим же растворителем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 100 раз.
Аммония эталонный раствор (1 ррm NH 4 ).
5000302.
Непосредственно перед использованием
эталонный раствор аммония (2,5 ррт NH) Р
разводят водой Р в 2,5 раза.
738
rосударственная фармакопея Республики Беларусь
Ацетальдеrида эталонный раствор
(100 ррm С 2 Н 4 О). 5000100.
1,0 r ацетальде2ида Р растворяют в
2 пропаноле Р и доводят до объема 100,0 мл
этим же растворителем. 5,0 мл полученноro
раствора доводят 2 пропанолом Р до объема
500,0 мл.
rотовят непосредственно перед использо
ванием.
Ацетальдеrида эталонный раствор
(100 ррm с 2 нр) Р1. 5000101.
1,0 r ацетальде2ида Р растворяют в
воде Р и .доводят до объема 100,0 мл этим же
растворителем. 5,0 мл полученноro раствора
доводят водой Р до объема 500,0 мл.
rотовят непосредственно перед использо
ванием
Бария эталонный раствор (50 ррm Ва).
5000600.
Навеску бария хлорида Р, COOTBeTCTBY
ющую 0,178 r ВаС'2'2Н20, растворяют в воде
дистиллированной Р и доводят до объема
100,0 мл этим же растворителем.
Непосредственно перед использованием
полученный раствор разводят водой дистил
лированной Р в 20 раз.
Ванадия эталонный раствор (1 r/л V).
5003300.
Навеску аммония вана дата Р, COOT
ветствующую 0,230 r NН 4 VО з , растворяют в
воде Р и доводят до объема 100,0 мл этим же
растворителем.
Водорода пероксида эталонный раст-
вор (10 ррm НР2)' 5005200.
10,0 мл раствора водорода пероксида
разведеНН020 Р разводят до 300,0 мл водой Р.
10,0 мл полученноrо раствора разводят до
объема 1000, о мл водой Р. Раствор rотовят
непосредственно перед использованием.
rермания эталонный раствор (100 ррm Ge).
5004400.
Навеску аммония 2е/{сафтОр2ермана
ma(/V) Р, соответствующую 0,307 r (NH4)2GeF6'
растворяют в 0,01 % растворе кислоты фто
ристоводородной Р. Прозрачный раствор дo
водят водой Р до объема 1000 мл.
rлиоксаля эталонный раствор (20 ррm
С 2 Н 2 О 2 ). 5003700.
в мерной колбе вместимостью 100 мл
взвешивают количество раствора 2лuокса
ля Р, соответствующее 0,200 r С 2 Н 2 О 2 , и ДOBO
дят эта нолом Р до объема 100,0 мл.
Непосредственно перед использованием
раствор разводят эта нолом Р в 100 раз.
Железа эталонный раствор (0,1 % Fe).
5001605.
0,100 r Fe растворяют в минимально He
обходимом количестве смеси из равных объе
мов кислоты хлористоводородной Р и воды Р
и доводят водой Р до объема 100,0 мл.
Железа эталонный раствор (250 ррm Fe).
5001606.
Навеску железа (/11) хлорида Р, COOTBeT
ствующую 4,840 r FеСl з '6НР растворяют в
растворе 150 r/л кислоты хлористоводород
ной Р и доводят до объема 100,0 мл этой же
кислотой.
Непосредственно перед использованием
полученный раствор разводят водой Р в 40 раз.
Железа эталонный раствор (20 ррm Fe).
5001600.
Навеску железа (/11) аммония сульфата Р,
соответствующую 0,863 r FeNH4(S04)2'12H20,
растворяют в 25 мл кислоты серной разведен
ной Р и доводят водой Р до объема 500,0 мл.
Непосредственно перед использованием
полученный раствор разводят водой Р в 10 раз.
Железа эталонный раствор (10 ррm Fe).
5001601.
Навеску железа (/1) аммония сульфата Р,
соответствующую 7,022 r Fe(NH4MS04)2'6HP,
растворяют в 25 мл кислоты серной разведен
ной Р и доводят водой Р до объема 1000,0 мл.
Непосредственно перед использованием
полученный раствор разводят водой Р в 100 раз.
Железа эталонный раствор (8 ррm Fe).
5001602.
80 Mr железа Р растворяют в 50 мл paCT
вора кислоты хлористоводородной Р (220 r/л
HCI) и доводят водой Р до объема 1000,0 мл.
Непосредственно перед использовани
ем полученный раствор разводят водой Р
в10раз.
Железа эталонный раствор (2 ррm Fe).
5001603.
Непосредственно перед использовани
ем эталонный раствор железа (20 ррт Fe) Р
разводят водой Р в 1 О раз.
Железа эталонный раствор (1 ррm Fe).
5001604.
Непосредственно перед использовани
ем эталонный раствор железа (20 ррт Fe) Р
разводят водой Р в 20 раз.
Исходный эталонный раствор для
атомной спектрометрии (1,000 r/л). 5004000.
Данный раствор roтовят преимуществен
но в кислой среде из элемента или ero соли с
минимальным содержанием не менее 99,0 %.
Концентрация раствора должна составлять
4.1.2. Эталонные растворы для испытаний на предельное содержимое примесей 739
более 0,995 r/л в течение установленноrо
срока rодности в невскрытом контейнере. На
этикетке указывают исходный материал (хи
мический элемент или еro соль) и конечный
растворитель (природа, кислотность и др.).
Йодида эталонный раствор (10 ррm 1).
5003800.
Навеску калия йодида Р, COOTBeTCTBY
ющую 0,131 r КI, растворяют в воде Р и доводят
до объема 100,0 мл этим же растворителем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 100 раз.
Кадмия эталонный раствор (0,1 % Cd).
5000700.
Навеску кадмия Р, соответствующую
0,100 r Cd, растворяют в минимально необхо
димом количестве смеси из 'равных объемов
кислоты хлористоводородной Р и воды Р и
доводят 1 % (об/об) раствором кислоты хло
ристоводородной Р до объема 100, о мл.
Кадмия эталонный раствор (1 О ррm Cd).
5000701.
Непосредственно перед использованием
эталонный раствор кадмия (О, 1 % Cd) Р раз
водят 1 % (об/об) раствором кислоты хлори
стоводородной Р в 100 раз.
Калия эталонный раствор (100 ррm К).
5002400.
Навеску калия сульфата Р, COOTBeTCTBY
ющую 0,446 r K 2 S0 4 , растворяют в воде Р и
доводят до объема 100,0 мл этим же paCTBO
рителем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 20 раз.
Калия эталонный раствор (20 ррm К).
5002401.
Непосредственно перед использованием
эталонный раствор калия (100 ррт К) Р раз
водят водой Р в 5 раз.
Кальция эталонный раствор (400 ррm Са).
5000800.
Навеску кальция карбоната Р, COOTBeT
ствующую 1,000 r СаСО з , растворяют в 23 мл
1 М раствора кислоты хлористоводород
ной Р и доводят водой дистиллированной Р
до объема 100,0 мл.
Непосредственно перед использованием
полученный раствор разводят водой дистил
лированной Р в 1 О раз.
Кальция эталонный раствор (100
ррm Са). 5000801.
Навеску кальция карбоната Р, COOTBeT
ствующую 0,624 r СаСО з , растворяют в 3 мл
кислоты уксусной Р и доводят водой дистил
лированной Р до объема 250,0 мл.
Непосредственно перед использованием
полученный раствор разводят водой дистилли
рованной Р в 1 О раз.
Кальция эталонный раствор (100 ррm Са) Р1.
5000804.
Навеску кальция хлорида безводН020 Р, co
ответствующую 2,769 r CaCI 2 , растворяют в Kиc
лоте хлористоводородной разведенной Р и
доводят до объема 1000,0 мл этим же paCTBO
рителем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 10 раз.
Кальция эталонный раствор спиртовой
(100 ррm Са). 5000802.
Навеску кальция карбоната Р, COOTBeTCTBY
ющую 2,50 r Са СО" растворяют в 12 мл кисло
ты уксусной Р и доводят водой дистиллирован
ной Р до объема 1000,Омл.
Непосредственно перед использованием
полученный раствор разводят 96 % спиртом Р
в 10 раз.
Кальция эталонный раствор (10 ррm Са).
5000803.
Навеску кальция карбоната Р, COOTBeTCTBY
ющую 0,624 r СаСО з , растворяют в 3 мл кисло
ты уксусной Р и доводят водой дистиллирован
ной Р до объема 250,0 мл.
Непосредственно перед использованием
полученный раствор разводят водой дистилли
рованной Р в 100 раз.
Кобальта эталонный раствор (100 ррm Со).
5004300.
Навеску кобальта нитрата Р, COOTBeT
ствующую 0,494 r Со(NОЗ)2'6НР, растворяют в
500 мл 1 М раствора кислоты азотной и ДOBO
дят водой Р до объема 1000 мл.
Маrния эталонный раствор (100 ррm Mg).
5001800.
Навеску ма2ния сульфата Р, COOTBeTcтвy
ющую 1,010 r MgS0 4 '7HP, растворяют в воде Р
и доводят до объема 100,0 мл этим же раство.-
рителем.
Непосредственно перед испольэовзнием
полученный раствор разводят водой Р в 10 раз.
Маrния эталонный раствор (10 ppm Mg).
5001801.
Непосредственно перед использованием
эталонный раствор ма2ния (100 ррт Mg) Р раз
водят водой Р в 1 О раз.
Маrния эталонный раствор (10 ррm Mg) Р1.
5001802.
Навеску 8.365 r ма2ния хлорида Р paCTBO
ряют в кислоте хлористоводородной разведен
ной Р и доводят до объема 1000,0 мл этим же
растворителем.
740
rосударственная фармакопея Республики Беларусь
Непосредственно перед использованием
полученный раствор разводят водой Р в 100 раз.
Марrанца эталонный раствор (1000 ррm Мп).
5005800.
Навеску мар2анца сульфата Р, COOTBeTCTBY
ющую 3,08 r МпS0 4 'нр, растворяют в 500 мл 1 М
раствора кислоты азотной и доводят водой Р до
объема 1000 мл.
Марrанца эталонный раствор (100 ррm Мп).
5005800.
Навеску мар2анца сульфата Р, COOTвeTCTBY
ющую 0,308 r Мпsо 4 -нр, растворяют в 500 мл 1 М
раствора кислоты азотной и доводят водой Р до
объема 1000 мл.
Меди эталонный раствор (0,1 % Cu).
5001100.
Навеску меди (11) сульфата Р, COOTBeTCTBY
ющую 0,393 r CuSO.-5НР, растворяют в воде Р и
доводят до объема 100,0 мл этим же растворителем.
Меди эталонный раствор (10 ррm Cu).
5001101.
Непосредственно перед использованием
эталонный раствор меди (0,1 % Си) Р разводят
водой Р в 100 раз.
Меди эталонный раствор (0,1 ррm Cu).
5001102.
Непосредственно перед использованием эта
лонный раствор меди (10 ррт Си) Р разводят
водой Р в 100 раз_
Мышьяка эталонный раствор (1 О ррm As).
5000500.
Навеску мышьяка (111) оксида Р, COOTBeTCTBY
ющую 0,330 r Аs,Оз, растворяют в 5 мл раствора
натрия 2идроксида разведеНН020 Р и доводят
водой Р до объема 250.0 мл.
Непосредственно перед использованием по
лученный раствор разводят водой Р в 100 раз.
Мышьяка эталонный раствор (1 ррm As).
5000501.
Непосредственно перед использованием эта
лонный раствор мышьяка (10 ррт As) Р разводят
водой Р в 1 О раз.
Мышьяка эталонный раствор (0,1 ррm As).
5000502.
Непосредственно перед использованием эта
лонный раствор мышьяка (1 ррт As) Р разводят
водой Р в 1 О раз.
Натрия эталонный раствор (200 ррm Na).
5002700.
Навеску натрия хлорида Р, соответствующую
0,509 r NaCI, растворяют в воде Р и доводят до
объема 100,0 мл этим же растворителем.
Непосредственно перед использованием по
лученный раствор разводят водой Р в 10 раз.
Натрия эталонный раствор (50 ррm Na).
5002701.
Эталонный раствор натрия (200 ррт Na) Р
разводят водой Р в 4 раза.
Никеля эталонный раствор (10 ррm Ni).
5002000.
Навеску никеля сульфата Р, COOTBeTCTBY
ющую 4,78 r NiSO.-7Нр, растворяют в воде Р и
доводят до объема 1000,0 мл этим же раствори
телем.
Непосредственно перед использованием по
лученный раствор разводят водой Р в 100 раз.
Никеля эталонный раствор (0,2 ррm Ni).
5002002.
Непосредственно перед использованием эта
лонный раствор никеля (10 ррт Ni) Р разводят
водой Р в 50 раз.
Никеля эталонный раствор (0,1 ррm Ni).
5002001.
Непосредственно перед использованием эта
лонный раствор никеля (10 ррт Ni) Р разводят
водой Р в 100 раз.
Нитрата эталонный раствор (100 ррm NО з ).
5002100.
Навеску калия нитрата Р, COOTBeTCTBY
ющую 0,815 r КNО з , растворяют в воде Р и дo
водят до объема 500,0 мл этим же растворите
лем.
Непосредственно перед использованием по
лученный раствор разводят водой Р в 1 О раз.
Нитрата эталонный раствор (10 ррm NО з ).
5002101.
Непосредственно перед использованием эта
лонный раствор нитрата (100 ррт NO) Р разво
дят водой Р в 1 О раз.
Нитрата эталонный раствор (2 ррm NО з ).
5002102.
Непосредственно перед использованием эта
лонный раствор нитрата (10 ррт NO) Р разво
дят водой Р в 5 раз.
Олова эталонный раствор (5 ррm Sn).
5003100.
Навеску олова Р, соответствующую 0,500 r Sn,
растворяют в смеси из 5 мл воды Р и 25 мл кисло
ты хлористоводородной Р и доводят водой Р до
объема 1000,0 мл.
Непосредственно перед использованием по
лученный раствор разводят 2,5 % (об/об) paCTBO
ром кислоты хлористоводородной Р в 100 раз.
Олова эталонный раствор (0,1 ррm Sn).
5003101.
Непосредственно перед использованием
эталонный раствор олова (5 ррт Sп) Р раЗ80
дят водой Р в 50 раз.
4.1.2. Эталонные растворы для испытаний на предельное содержимое примесей 741
Палладия эталонный раствор (500 ррm Pd).
5003600.
50,0 Mr палладия Р растворяют в 9 мл кисло
ты хлористоводородной Р и доводят водой Р до
объема 100,0 мл.
Палладия эталонный раствор (20 ррm Pd).
5003602.
0,333 Mr палладия хлорида Р растворяют в
2 мл теплой кислоты хлористоводородной Р и дo
водят смесью из равных объемов кислоты хлори
стоводородной разведенной Р и воды Р до объема
1000,0 мл.
Непосредственно пер использованием по
лученный раствор разводят водой Р в 1 О раз.
Палладия эталонный раствор (0,5 ррт Pd).
5003601.
1 мл этаЛОНН020 раствора палладия
(500 ррт Pd) Р доводят смесью из 0,3 объема Kиc
лоты азотной Р и 99,7 объема воды Р до объема
1000 мл.
Платины эталонный раствор (ЗО ррm Pt).
5002300.
80 Mr кислоты хлорплатиновой Р растворяют
в 1 М растворе кислоты хлористоводородной и
доводят до объема 100,0 мл этим же растворите
лем.
Непосредственно перед использованием по
лученный раствор разводят 1 М раствором кисло
ты хлористоводородной в 1 О раз.
ртути эталонный раствор (1000 ррm Hg).
5001900.
Навеску ртути (/1) хлорида Р, COOTBeTCTBY
ющую 1,354 r HgCI 2 , растворяют в 50 мл кисло
ты азотной разведенной Р и доводят водой Р до
объема 1000,0 мл.
Свинца эталонный раствор (0,1 % РЬ).
5001700.
Навеску свинца (11) нитрата Р, COOTBeTCTBY
ющую 0,400 r РЬ(NО З )2' растворяют в воде Р и дo
водят до объема 250,0 мл этим же растворителем.
Свинца эталонный раствор (100 ррm РЬ).
5001701.
Непосредственно перед использованием эта
лонный раствор свинца (0,1 % РЬ) Р разводят
водой Р в 10 раз.
Свинца эталонный раствор (10 ррm РЬ).
5001702.
Непосредственно перед использованием эта
лонный раствор свинца (100 ррт РЬ) Р разводят
водой Р в 1 О раз.
Свинца эталонный раствор (10 ррm РЬ) Р1.
5001706.
0,160 r свинца (/1) нитрата Р растворяют в
100 мл воды Р, прибавляют 1 мл кислоты азот
94.3ак 1712.
ной, свободной от свинца, Р и доводят водой Р до
объема 1000,0 мл
Непосредственно перед использованием по
лученный раствор разводят водой Р в 1 О раз.
Свинца эталонный раствор (2 ррm РЬ).
5001703.
Непосредственно перед использованием эта
лонный раствор свинца (10 ррт РЬ) Р разводят
водой Р в 5 раз.
Свинца эталонный раствор (1 ррm РЬ).
5001704.
Непосредственно перед использованием эта
лонный раствор свинца (10 ррт РЬ) Р разводят
водой Р в 10 раз.
Свинца эталонный раствор (0,1 ррm РЬ).
5001705.
Непосредственно перед использованием эта
лонный раствор свинца (1 ррт РЬ) Р разводят
водой Рв 10 раз.
Селена эталонный раствор (100 ррm Se).
5002500.
0,100 r селена Р растворяют в 2 мл кисло
ты азотной Р и полученный раствор выпарива
ют досуха. Полученный остаток растворяют в 2 мл
воды Р и выпаривают досуха. Эту операцию повто
ряют три раза. Остаток растворяют в 50 мл кисло
ты хлористоводородной разведенной Р и доводят
до объема 1000,0 мл этой же кислотой.
Селена эталонный раствор (1 ррm Se).
5002501.
Навеску кислоты селенистой Р, COOTBeTCTBY
ющую 6,54 r Н 2 SеО з , растворяют в воде Р и ДOBO
дят до объема 100,0 мл этим же растворителем.
Непосредственно перед использованием по
лученный раствор разводят водой Р в 40 раз.
Серебра эталонный раствор (5 ррm Ag).
5002600.
Навеску серебра нитрата Р, COOTBeTCTBY
ющую 0,790 r АgNО з , растворяют в воде Р и дoвo
дят до объема 1000,0 мл этим же растворителем.
Непосредственно перед использованием по
лученный раствор разводят водой Р в 100 раз.
Стронция эталонный раствор (1,0 % Sr).
5003900.
Навеску стронция карбоната Р, COOTBeT
ствующую 1,6849 r SrСО з , покрывают водой Р,
прибавляют кислоту хлористоводородную Р до
полноro раСТ90рения и прекращения выделения
пузырьков rаза, фильтруют и доводят водой Р до
объема 100,0 мл.
Сульфата эталонный раствор (1 00 ррm SO 4).
5002802.
Навеску калия сульфата Р, соответствующую
0,181 r S04' растворяют в воде дистиллирован
742
Тосударственная фармакопея Республики Беларусь
ной Р и доводят до объема 100,0 мл этим же paCT
ворителем.
Непосредственно перед использованием
полученный раствор разводят водой дистилли
рованной Р в 1 О раз.
Сульфата эталонный раствор (10 ррm
504).5002800.
Навеску калия сульфата Р, соответствую
щую 0,181 r K 2 S0 4 , растворяют в воде дистилли
рованной Р и доводят до объема 100,0 мл этим
же растворителем.
Непосредственно перед использованием
полученный раствор разводят водой дистилли
рованной Р в 100 раз.
Сульфата эталонный раствор (10 ррm
504) Р1. 5002801.
Навеску калия сульфата Р, соответствую
щую 0,181 r K2S0 . растворяют в спирте (30 %,
об/об) Р и доводят до объема 100,0 мл этим же
растворителем.
Непосредственно перед использованием
полученный раствор разводят спиртом (30 %,
об/об) Р в 100 раз.
Сульфита эталонный раствор (1,5 ррm
502).5002900.
Навеску натрия метабисульфита Р, co
ответствующую 0.152 r Na 2 S 2 0 5 , растворяют в
воде Р и доводят до объема 100,0 мл этим же
растворителем. 5,0 мл полученноro раствора дo
водят водой Р до объема 100,0 мл. К 3 мл по
лученноro раствора прибавляют 4,0 мл 0,1 М
раствора натрия 2идроксида и доводят водой Р
до объема 100,0 мл.
Сурьмы эталонный раствор (100 ррm
5Ь).5000401.
Навеску антимонuла калия тартрата Р,
соответствующую 0,274 r C 4 H 4 K0 7 Sb'Y2HP,
растворяют в 500 мл 1 М раствора кислоты
хлористоводородной и прозрачный раствор дo
водят водой Р до объема 1000 мл.
Сурьмы эталонный раствор (1 ррm 5Ь).
5000400.
Навеску антимонила калия тартрата Р,
соответствующую 0,274 r C 4 H 4 K0 7 Sb'Y2HP,
растворяют в 20 мл кислотbl хлористоводород
ной Р1 и прозрачный раствор доводят водой Р
до объема 100,0 мл. К 10,0 мл полученноro paCT
вора прибавляют 200 мл кислотbl хлористо
водородной Р1 и доводят водой Р до объема
1000,0 мл. К 100,0 мл полученноrо раствора при
бавляют 300 мл кислоты хлористоводород
ной Р1 и доводят водой Р до объема 1000,0 мл.
Разведенные растворы roтовят непосред
ственно перед использованием.
Таллия эталонный раствор (1 О ррm TI).
5003000.
Навеску таллия сульфата Р, COOTBeTCTBY
ющую 0,1235 r TI 2 S0 4 , растворяют в растворе
9 r/л натрия хлорида Р и доводят до объема
1000,0 мл этим же растворителем. 10,0 мл по
лученноro раствора доводят раствором 9 r/л
натрия хлорида Р до объема 100,0 мл.
Титана эталонный раствор (100 ррm Ti).
5003200.
100,0 Mr титана Р растворяют, наrревая
при необходимости, в 100 мл кислоты хло
рисrповодородной Р, разведенной водой Р до
объема 150 мл, охлаждают и доводят водой Р до
объема 1000,0 мл.
Ферроцианида эталонный раствор
(100 ррm Fе(СN)б). 5001200.
Навеску калия ферроцианида Р, соответ
ству ющую 0,20 r K4Fe(CN)6'3Hp, растворяют в
воде Р и доводят до объема 100,0 мл этим же
растворителем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 1 О раз.
Феррицианида эталонный раствор
(50 ррm Fе(СN)б). 5001300.
Навеску калия феррицианида Р, соответ
ству ющую 0,78 r К з Fе(СN)6' растворяют в воде Р
и доводят до объема 100,0 мл этим же раствори
телем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 100 раз.
Формальдеrида эталонный раствор
(5 ррm СНр). 5001500.
Навеску раствора формальде2ида Р, соот
ветствующую 1,0 r СН 2 О, разводят водой Р до
объема 1 л.
Непосредственно перед использованием
полученный раствор разводят водой Р в 200 раз.
Фосфата эталонный раствор (200 ррm
РО 4). 5004200.
Навеску калия ди2идрофосфата Р, соответ
ствующую 0,286 r КН 2 РО 4' растворяют в воде Р
и доводят до объема 1000,0 мл этим же paCTBO
рителем.
Фосфата эталонный раствор (5 ррm Р0 4 ).
5002200.
Навеску калия ди2идрофосфата Р, COOTBeT
ствующую 0,716 r КНРО4' растворяют в воде Р
и доводят до объема 1000.0 мл этим же paCTBO
рителем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 100 раз.
Фтор ида эталонный раствор (10 ррm F).
5001400.
Навеску натрия фторида Р, СООТБетству
ющую 0,442 r NaF, предварительно высушенно
ro при температуре 300 ос в течение 12 ч, paCT
воряют в воде Р и доводят до объема 1000,0 мл
этим же растворителем (1 мл = 0,2 Mr F).
Хранят в полиэтиленовом контейнере.
Непосредственно перед использованием
полученный раствор разводят водой Р в 20 раз.
Фторида эталонный раствор (1 ррm F).
5001401.
4.1.3. Буферные растворы
743
Непосредственно перед использованием
эталонный раствор фторида (10 ррт F) Р раз
водят водой Р в 1 О раз.
Хлорида эталонный раствор (8 ррm CI).
5000900.
Навеску натрия хлорида Р, COOTBeTCTBY
ющую 1,32 r NaCI, растворяют в воде Р и ДOBO
дят до объема 1000,0 мл этим же растворителем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 100 раз.
Хлорида эталонный раствор (5 ррm CI).
5000901.
Навеску натрия хлорида Р, COOTBeTCTBY
ющую 0,824 r NaCl, растворяют в воде Р и ДOBO
дят до объема 1000,0 мл этим же растворителем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 100 раз.
Хрома эталонный раствор (0,1 % Cr).
5001002.
Навеску калия дихромата Р, COOTBeTCTBY
ющую 2,83 r K 2 Crp7' растворяют в воде Р и дo
водят до объема 1000,0 мл этим же растворите
лем.
Хрома эталонный раствор (100 ррm Cr).
5001000.
Навеску калия дихромата Р, COOTBeTCTBY
ющую 0,283 r K 2 Cr 2 0 7 , растворяют в воде Р и дo
водят до объема 1000,0 мл этим же растворите
лем.
Хрома эталонный раствор (0,1 ррт Cr).
5001001.
Непосредственно перед использованием
эталонный раствор хрома (100 ррт С,) Р раз
водят водой Р в 1000 раз.
Цинка эталонный раствор (5 мr/мл Zn).
5003400.
3,15 r цинка оксида Р растворяют в 15 мл
кислоты хлористоводородной Р и доводят
водой Р до объема 500,0 мл.
Цинка эталонный раствор (100 ррm Zn).
5003401.
Навеску цинка сульфата Р, COOTBeTCTBY
ющую 0,440 r ZnS0 4 '7HP, и 1 мл уксусной
кислоты Р растворяют в воде Р и доводят до
объема 100,0 мл этим же растворителем.
Непосредственно перед использованием
полученный раствор разводят водой Р в 1 О раз.
Цинка эталонный раствор (10 ррm Zn).
5003402.
Непосредственно перед использованием
эталонный раствор цинка (100 ррт Zn) Р раз
водят водой Р в 1 О раз.
Цинка эталонный раствор (5 ррm Zn).
5003403.
Непосредственно перед использованием
эталонный раствор цинка (100 ррт Zn) Р раз
водят водой Р в 20 раз.
Циркония эталонный раствор (1 r/л Zr).
5003500.
Навеску цирконила нитрата Р, COOTBeT
ствующую 0,293 r ZrO(NO)2'2HP, растворяют в
смеси из 2 объемов кислоты хлористоводород
ной Р и 8 объемов воды Р и доводят до объема
100,0 мл этой же смесью растворителей.
01/2013:40103
4.1.3. БУФЕРНЫЕ РАСТВОРЫ
Забуференный ацетоновый раствор.
4000100.
8,15 r натрия ацетата Р и 42 r натрия хло
рида Р растворяют в воде Р, прибавляют 68 мл
0,1 М раствора кислоты хлористоводородной,
150 мл ацетона Р и доводят водой Р до объема
500 мл.
Буферный раствор рН 2,0. 4000200.
6,57 r калия хлорида Р растворяют в воде Р,
прибавляют 119,0 мл 0,1 М раствора кисло
ты хлористоводородной и доводят водой Р до
объема 1000,0 мл.
Фосфатный буферный раствор рН 2,0.
4007900.
8,95 r динатрuя 2uдрофосфата Р и 3,40 r
калия ди2идрофосфата Р растворяют в воде Р
и АОВОАЯТ АО объема 1000,0 мл этим же paC'TBO
рителем. При необходимости доводят рН (2.2.3)
кислотой фосфорной Р.
Сульфатный буферный раствор рН 2,0.
4008900.
132,1 r аммония сульфата Р растворяют в
воде Р и доводят до объема 500,0 мл этим же
растворителем (раствор 1).
Осторожно при постоянном охлаждении и
перемешивании 14 мл кислоты серной Р при
бавляют к приблизительно 400 мл воды Р, ox
лаждают и доводят водой Р до объема 500,0 мл
(раствор 11).
Смешивают равные объемы растворов' и 11.
При необходимости доводят рН (2.2.3).
Буферный раствор рН 2,2. 4010500.
Смешивают 6,7 мл кислоты фосфорной Р
с 48,0 мл воды Р и 2,0 мл раствора натрия
2идроксида Р и доводят водой Р до объема
1000,0 мл.
Буферный раствор рН 2,5. 4000300.
100 r калия ди2идрофосфата Р растворяют
в 800 мл воды Р, доводят кислотой хлористово
дородной Р до рН 2,5 (2.2.3) и разводят водой Р
до объема 1000,0 мл.
744
rосударственная фармакопея Республики Беларусь
Буферный раствор рН 2,5 Р1. 4000400.
К 4,9 r кислоты фосфорной разведенной Р
прибавляют 250 мл воды Р, доводят рН (2.2.3)
раствором натрия 2идроксида разведен
ным Р И разводят водой Р до объема 500,0 мл.
Фосфатный буферный раствор рН 2,8.
4010600.
7,8 r натрия ди2идрофосфата Р paCTBO
ряют в 900 мл воды Р, доводят кислотой фос
форной Р ДО рН 2,8 (2.2.3) и разводят водой Р до
объема 1000,0 мл.
Буферный раствор рН 3,0. 4008000.
21,0 r кислоты лимонной Р растворяют в
200 мл 1 М раствора натрия 2идроксида и дов(}-
дят водой Р до объема 1000 мл.
40,3 мл полученноrо раствора доводят 0,1 М
раствором кислоты хлористоводородной до
объема 100,0 мл.
Фосфатный буферный раствор рН 3,0.
4000500.
0,7 мл кислоты фосфорной Р смешивают
со 100 мл воды Р и доводят до объема 900 мл
этим же растворителем. Доводят раствором
натрия 2идроксида концентрированным Р до
рН 3,0 (2.2.3) и разводят водой Р до объема
1000 мл.
Фосфатный буферный раствор рН 3,0 Р1.
4010000.
3,40 r калия ди2идрофосфата Р растворяют
в 900 мл воды Р Доводят кислотой фосфорной Р
до рН 3,0 (2.2.3) и доводят водой Р до объема
1000,0 мл.
Фосфатный буферный раствор рН 3,2.
4008100.
К 900 мл раствора 4 r/л натрия ди2идрофос
фата Р прибавляют 100 мл раствора 2,5 r/л кисло
ты фосфорной Р При необходимости доводят рН
(2.2.3).
Фосфатный буферный раствор рН 3,2 Р1.
4008500.
Раствор 35,8 r/л динатрия 2идрофосфата Р
доводят кислотой фосфорной разведенной Р до
рН 3,2 (2.2.3).
100,0 мл раствора доводят водой Р до объема
2000,0 мл.
Буферный раствор рН 3,5. 4000600.
25,0 r аммония ацетата Р растворяют в 25 мл
воды Р и прибавляют 38,0 мл кислоты хлористо
водородной Р1 и, при необходимости, доводят рН
(2.2.3) кислотой хлористоводородной разведен
ной Р или раствором аммиака разведенным Р.
Разводят водой Р до объема 100,0 мл.
Фосфатный буферный раствор рН 3,5.
4000700.
68,0 r калия ди2идрофосфата Р paCTBO
ряют в воде Р и доводят до объема 1000,0 мл
этим же растворителем. Доводят рН (2.2.3)
кислотой фосфорной Р.
Буферный раствор рН 3,6. 4000800.
К 250,0 мл 0,2 М раствора калия 2идpo
фталата Р прибавляют 11,94 мл 0,2 М pacт
вора кислоты хлористоводородной и доводят
водой Р до объема 1000,0 мл.
Буферный раствор рН 3,7. 4000900.
К 15,0 мл кислоты уксусной Р прибавляют
60 мл 96 % спирта Р и 20 мл воды Р, доводят
раствором аммиака Р до рН 3,7 (2.2.3) и разво
дят водой Р до объема 100,0 мл.
Забуференный раствор меди сульфата
рН 4,0. 4001000.
0,25 r меди (11) сульфата Р и 4,5 r аммония
ацетата Р растворяют в кислоте уксусной раз
веденной и доводят до объема 100,0 мл этим же
растворителем.
Ацетатный буферный раствор рН 4,4.
4001100.
136 r натрия ацетата Р и 77 r аммония
ацетата Р растворяют в воде Р, доводят до
объема 1000,0 мл этим же растворителем, при
бавляют 250,0 мл кислоты уксусной ледяной Р
и перемешивают.
Фталатный буферный раствор рН 4,4.
4001200.
2,042 r калия 2идрофталата Р растворяют
в 50 мл воды Р, прибавляют 7,5 мл 0,2 М pacт
вора натрия 2идроксида и доводят водой Р до
объема 200,0 мл.
0,05 М фосфатный буферный раствор рН
4,5. 4009000.
6,80 r калия ди2идрофосфата Р растворяют
1000,0 мл воды Р.
Значение рН (2.2.3) полученноro раствора
должно быть 4,5.
Ацетатный буферный раствор рН 4,5.
4010100.
63 r натрия ацетата безводН020 Р paCTBO
ряют в воде Р, прибавляют 90 мл кислоты YK
СУСНОЙ Р, доводят до рН 4,5 (2.2.3) и разводят
водой Р до объема 1000 мл.
Ацетатный буферный раствор рН 4,6.
4001400.
5,4 r натрия ацетата Р растворяют в 50 мл
воды Р, прибавляют 2,4 r кислоты уксусной ле
дяной Р И доводят водой Р до объема 100,0 мл.
При необходимости доводят рН (2.2.3).
4.1.3. Буферные растворы
745
Сукцинатный буферный раствор рН 4,6.
400150
11,8 r кислоты янтарной Р растворяют
в смеси из 600 мл воды Р и 82 мл 1 М pacт
вора натрия 2идроксида и доводят водой Р до
объема 1000,0 мл.
Ацетатный буферный раствор рН 4,7.
4001600.
136,1 r натрия ацетата Р растворяют в
500 мл воды Р. 250 мл полученноro раствора
смешивают с 250 мл кислоты уксусной разве
денной Р. Встряхивают дважды со свежепри
roтовленным профильтрованным раствором
0,1 r/л дитизона Р в хлороформе Р. Встряхивают
с уелерода тетрахлоридом Р до обесцвечива
ния экстракта. Водный слой фильтруют для yдa
ления следов уrлерода тетрахлорида.
Ацетатный буферный раствор рН 5,0.
4009100.
К 120 мл раствора 6 r/л кислоты уксусной ле
дяной Р прибавляют 100 мл 0,1 М pacтвop калия
2идроксида и около 250 мл воды Р и перемешива
ют. Доводят до рН 5,0 раствором 6 r/л кислоты YK
сусной Р или 0,1 М раствором калия 2идроксида
и разводят водой Р до объема 1000,0 мл.
Фосфатный буферный раствор рН 5,0.
4011300.
2,72 r калия ди2идрофосфата Р растворяют в
800 мл воды Р, доводят 1 М раствором калия 2и
дроксида и разводят водой Р до объема 1000 мл.
Цитратный буферный раствор рН 5,0.
4010700.
Приroтавливают раствор, содержащий
20,1 r/л кислоты лимонной Р и 8,0 r/л натрия
2идроксида Р. Доводят кислотой хлористово
дородной разведенной Р до рН 5,0.
Буферный раствор рН 5,2. 4001700.
1,02 r калия 2идрофталата Р растворяют
в 30,0 мл 0,1 М раствора натрия 2идроксида и
доводят водой Р до объема 100,0 мл.
Ацетатно-эдетатный буферный раствор
рН 5,5. 4001900.
250 r аммония ацетата Р и 15 r натрия
эдетата Р растворяют в 400 мл воды Р и при
бавляют 125 мл кислоты уксусной ледяной Р.
Буферный раствор рН 5,5. 4001800.
54,4 r натрия ацетата Р растворяют в 50 мл
воды Р, при необходимости наrревая до темпера
туры 35 ОС. После охлаждения медленно прибав
ляют 1 О мл кислоты уксусной безводной Р, пере
мешивают и доводят водой Р до объема 100,0 мл.
Фосфатный буферный раствор рН 5,5.
4002000.
Раствор 1. 13,61 r калия ди2идрофосфа
та Р растворяют в воде Р и доводят до объема
1000,0 мл этим же растворителем.
95. За.. 1712.
Раствор /1.35,81 r динатрия 2идрофосфа
та Р растворяют в воде Р и доводят до объема
1000,0 мл этим же растворителем.
Смешивают 96,4 мл раствора 1 и 3,6 мл
раствора 11.
Фосфатно-цитратный буферный раствор
рН 5,5. 4008700.
Смешивают 56,85 мл раствора 28,4 r/л ди
натрия 2идрофосфата безводН020 Р и 43,15 мл
раствора 21 r/л кислоты лимонной Р.
Фосфатный буферный раствор рН 5,8.
4002100.
1,19 r динатрия 2идрофосфата ди2идpa
та Р и 8,25 r калия ди2идрофосфата Р paCT
воряют в воде Р и доводят до объема 1000,0 мл
этим же растворителем.
Ацетатный буферный раствор рН 6,0.
4002200.
100 r аммония ацетата Р растворяют в
300 мл воды Р, прибавляют 4,1 мл кислоты YK
сусной ледяной Р и при необходимости доводят
рН (2.2.3) раствором аммиака Рили кислотой
уксусной Р. Разводят водой Р до объема 500,0 мл.
Диэтиламмония фосфата буферный
раствор рН 6,0. 4002300.
68 мл кислоты фосфорной Р доводят
водой Р до объема 500 мл. К 25 мл полученно
ro раствора прибавляют 450 мл воды Р и 6 мл
диэтиламина Р и при необходимости доводят
до рН 6:t0,05 (2.2.3) диэтиламином Р или кисло
той фосфорной Р. Разводят водой Р до объема
500,0 МЛ.
Фосфатный буферный раствор рН 6,0.
4002400.
Смешивают 63,2 мл раствора 71,5 r/л диHa
трия 2идрофосфата Ри З6,8 мл раствора 21 r/л
кислоты лимонной Р.
Фосфатный буферный раствор рН 6,0 Р1.
4002500.
6,8 r натрия ди2идрофосфата Ррастворяют
в воде Р и доводят водой Р до объема 1000,0 мл.
Доводят рН (2.2.3) раствором натрия 2идpOKcи
да концентрированным Р.
Фосфатный буферный раствор рН 6,0 Р2.
4002600.
К 250,0 мл 0,2 М раствора калия ди2идpo
фосфата Р прибавляют 28,5 мл 0,2 М pacт
вора натрия аидроксида и доводят водой Р до
объема 1000,0 мл.
Фосфатный буферный раствор рН 6,4.
4002800.
2,5 r динатрия 2идрофосфата Р, 2,5 r
натрия ди2идрофосфата Р, 8,2 r натрия хло
рида Р растворяют в 950 мл воды Р и при He
746
rосударственная фармакопея Республики Беларусь
обходимости доводят 1 М раствором натрия
2идроксида или 1 М раствором кислоты хло
ристоводородной до рН 6,4 (2.2.3). Разводят
водой Р до объема 1000,0 мл.
0,5 М фталатный буферный раствор
рН 6,4. 4009200.
100 r калия 2идрофталата Р растворяют
в воде Р и доводят до объема 1000,0 м:Л этим
же растворителем. При необходимости ДOBO
дят рН (2.2.3) раствором натрия 2идроксида
концентрированным Р.
Буферный раствор рН 6,5. 4002900.
60,5 r динатрия 2идрофосфата Р и 46
r калия ди2идрофосфата Р растворяют в
воде Р, прибавляют 100 мл 0,02 М раствора
натрия эдетата, 20 Mr ртути (1/) хлорида Р
и доводят водой Р до объема 1000,0 мл.
Имидазольный буферный раствор рН
6,5. 4003000.
6,81 r имидазола Р, 1,23 r ма2ния суль
фата Р и 0,73 r кальция сульфата Р paCT
воряют в 752 мл 0,1 М раствора кислоты
хлористоводородной и, при необходимо
сти, доводят рН (2.2.3). Разводят водой Р до
объема 1000,0 мл.
Имидазольный буферный раствор рН
7,3.4004500.
3,4 r имидазола Р и 5,8 r натрия хлори
да Р растворяют в воде Р, прибавляют 18,6 мл
1 М раствора кислоты хлористоводородной
и доводят водой Р до объема 100 мл. При He
обходимости доводят значение рН.
0,1 М фосфатный буферный раствор
рН 6,5. 4010800.
13,80 r натрия ди2идрофосфата MOHO
2идрата Р растворяют в 900 мл дистилли
рованной воды Р. Доводят раствором 400 r/л
натрия 2идроксида Р до рН 6,5 (2.2.3) и раз
водят дистиллированной водой Р до объема
1000,0 мл.
Фосфатный буферный раствор рН 6,6.
4003100.
К 250,0 мл 0,2 М раствора калия ди2идpo
фосфата Р прибавляют 89,0 мл 0,2 М pacт
вора натрия 2идроксида и доводят водой Р
до объема 1000,0 мл.
Фосфатный забуференный физиоло
rический раствор рН 6,8. 4003200.
1,0 r калия дтшдрофосфата Р, 2,0 r ди
калия 2идрофосфата Р и 8,5 r натрия хло
рида Р растворяют в 900 мл воды Р и при He
обходимости доводят рН (2.2.3). Разводят
водой Р до объема 1000,0 мл.
Фосфатный буферный раствор рН 6,8.
4003300.
Смешивают 77,3 мл раствора 71,5 r/л диHa
трия 2идрофосфата Р и 22,7 мл раствора 21 r/л
кислоты лимонной Р.
Фосфатный буферный раствор рН 6,8 Р1.
4003400.
К 51,0 мл раствора 27,2 r/л калия ди2и
дрофосфата Р прибавляют 49,0 мл раствора
71,6 r/л динатрия 2идрофосфата Р. При необхо
димости доводят рН (2.2.3).
Хранят при температуре от 2 ос до 8 ОС.
1 М трис rидрохлорида буферный paCT
вор рН 6,8. 4009300.
60,6 r триС(2идроксиметил)аминомета
на Р растворяют в 400 мл воды Р, доводят рН
(2.2.3) кислотой хлористоводородной Р и раз
водят водой Р до объема 500,0 мл.
Буферный раствор рН 7,0. 4003500.
К 1000 мл раствора, содержащеro 18 r/л
динатрия 2идрофосфата Р и 23 r/л натрия
хлорида Р, прибавляют для установления рН
(2.2.3) достаточное количество (около 280 мл)
раствора, содержащеro 7,8 r/л натрия ди2и
дрофосфата Р и 23 r/л натрия хлорида Р
Растворяют в полученном растворе необходи
мое количество натрия азида Р до получения
раствора 0,2 r/л.
Малеатный буферный раствор рН 7,0.
4003600.
10,0 r натрия хлорида Р, 6,06 r
триС(2идроксиметил)аминометана Р и 4,90 r
малеинов020 аН2идрида Р растворяют в 900 мл
воды Р Доводят раствором 170 r/л натрия 2и
дроксида Р до рН (2.2.3) и разводят водой Р до
объема 1000,Омл.
Хранят при температуре от 2 ос до 8 ОС.
Срок roдности 3 сут.
Фосфатный буферный раствор рН 7,0.
4003700.
Смешивают 82,4 мл раствора 71,5 r/л диHa
трия 2идрофосфата Р и 17,6 мл раствора 21 r/л
кислоты лимонной Р.
0,025 М фосфатный буферный раствор
рН 7,0. 4009400.
Смешивают 1 объем 0,063 М фосфатН020 бу
ферН020 раствора рН 7,0 Р и 1,5 объема воды Р.
0,03 М фосфатный буферный раствор
рН 7,0. 4010300.
5,2 r дикалия 2идрофосфата Р растворяют
в 900 мл воды для хромат02рафии Р Доводят
кислотой фосфорной Р до рН 7,0:tO,1 и раз
водят водой для хроматоарафии Р до объема
1000 мл.
4.1.3. Буферные растворы Эталонные растворы для испытаний на
747
0,063 М фосфатный буферный раствор
рН 7,0. 4009500.
5,18 r динатрия 2идрофосфата безводно
20 Р И 3,65 r натрия ди2идрофосфата MOH02и
драта Р растворяют в 950 мл воды Р, доводят
рН (2.2.3) кислотой фосфорной Р и разводят
водой Р до объема 1000,0 мл.
0,067 М фосфатный буферный раствор
рН 7,0. 4003800.
Раствор 1. 0,908 r калия ди2идрофосфа
та Р растворяют в воде Р и доводят до объема
100,0 мл этим же растворителем.
Раствор 11. 2,38 r динатрия 2идрофосфа
та Р растворяют в воде Р и доводят до объема
100,0 мл этим же растворителем.
Смешивают 38,9 мл раствора I и 61,1 мл
раствора 11. При необходимости доводят рН
(2.2.3).
0,1 М фосфатный буферный раствор
рН 7,0. 4008200.
1,361 r калия ди2идрофосфата Р paCTBO
ряют в воде Р и доводят до объема 100,0 мл
этим же растворителем. Доводят рН (2.2.3) pac
твором 35 r/л динатрия 2идрофосфата Р.
Фосфатный буферный раствор рН 7,0 Р1.
4003900.
Смешивают 250,0 мл 0,2 М раствора калия
диzидрофосфата Р и 148,2 мл раствора 8 r/л
натрия 2идроксида Р и при необходимости дo
водят рН (2.2.3). Разводят водой Р до объема
1000,0 мл.
Фосфатный буферный раствор рН 7,0 Р2.
4004000.
Смешивают 50,0 мл раствора 136 r/л калия
ди2идрофосфата Р и 29,5 мл 1 М раствора
натрия 2идроксида, доводят водой Р до объема
100,0 мл. Доводят до рН 7,0:1:0,1 (2.2.3).
Фосфатный буферный раствор рН 7,0 Р3.
4008600.
5 r калия ди2идрофосфата Р и 11 r дикалия
2идрофосфата Р растворяют в 900 мл воды Р.
Доводят кислотой фосфорной разведенной Р
или раствором натрия 2идроксида разведен
ным Р ДО рН 7,0 (2.2.3), разводят водой Р до
объема 1000 мл И перемешивают.
Фосфатный буферный раствор рН 7,0 Р4.
4010200.
28,4 r динатрия 2идрофосфата безводно
20 Р И 18,2 r калия ди2идрофосфата Р paCTBO
ряют в ваде Р и доводят до объема 500 мл этим
же растворителем.
Фосфатный буферный раствор рН 7,0 Р5.
4011400.
28,4 r динатрия 2идрофосфата безводно
20 Р растворяют в 800 мл воды Р, доводят ЗО %
(М/М) раствором фосфорной кислоты Р и разво
дят водой Р до объема 1000 мл.
Буферный раствор рН 7,2. 4004100.
К 250,0 мл 0,2 М раствора калия ди2идpo
фосфата Р прибавляют 175,0 мл 0,2 М pacт
вора натрия 2идроксида. Доводят водой Р до
объема 1000,0 мл. При необходимости доводят
рН (2.2.3).
Фосфатный буферный раствор рН 7,2.
4004200.
Смешивают 87,0 мл раствора 71,5 r/л диHa
трия 2идрофосфата Ри 1З,0 мл раствора 21 r/л
кислоты лимонной Р.
Забуференный солевой раствор рН 7,2.
4004300.
8,0 r натрия хлорида Р, 0,2 калия хлорида Р,
0,1 r кальция хлорида безводН020 Р, 0,1 r ма2ния
хлорида Р, 3,18 r динатрия 2идрофосфата Р
и 0,2 r калия ди2идрофосфата Р растворяют в
воде Р и доводят водой Р до объема 1000,0 мл.
Фосфатно льбуминовый забуференный
физиолоrический раствор рН 7,2. 4004400.
10,75 r динатрия 2идрофосфата Р, 7,6 r
натрия хлорида Р и 1 О r альбумина бычъе
20 Р растворяют в воде Р и доводят до объема
1000,0 мл этим же растворителем.
Непосредственно перед использованием
устанавливают рН (2.2.3) с помощью раствора
натрия 2идроксида разведеНН020 Р или кисло
ты фосфорной разведенной Р.
Фосфатно льбуминовый забуференный
физиолоrический раствор рН 7,2 Р1. 4009600.
10,75 r динатрия 2идрофосфата р, 7,6 r
натрия хлорида Р и 1 r альбумина бычье20 Р
растворяют в воде Р и доводят тем же раствори
телем до объема 1 000,0 мл.
Непосредственно перед использованием дo
водят рН (2.2.3) раствором натрия 2идроксида
разведенным Р или кислотой фосфорной разве
денной Р.
Имидазольный буферный раствор рН 7,3.
4004500.
3,4 r имидазола Р и 5,8 r натрия хлорида Р
растворяют в воде Р, прибавляют 18,6 мл 1 М
раствора кислоты хлористоводородной и дo
водят водой Р до объема 1000,0 мл. При необхо
димости доводят рН (2.2.3).
Барбитал буферный раствор рН 7,4.
4004700.
Смешивают 50 мл раствора, содержащеro
19,44 r/л натрия ацетата Р и 29,46 r/л барби
тал натрия Р в воде Р, и 50,5 мл 0,1 М pacт
вора кислоты хлорисrnоводородной, прибавлq
ют 20 мл раствора 85 r/л натрия хлорида Р и
доводят водой Р до объема 250 мл.
Буферный раствор рН 7,4. 4004600.
0,6 r калия ди2идрофосфата Р, 6,4 r диHa
rnрия 2идрофосфата Р и 5,85 r натрия хлори
748
rосударственная фармакопея Республики Беларусь
да Р растворяют в воде Р и доводят до объема
1000,0 мл этим же растворителем. При необхо
димости доводят рН (2.2.3).
Фосфатный буферный раствор рН 7,4.
4004800.
К 393,4 мл 0,1 М раствора натрия 2идpOK
сида прибавляют 250,0 мл 0,2 М раствора калия
ди2идрофосфата Р.
Трис(rидроксиметил)аминометан-натрия
хлорида буферный раствор рН 7,4. 4004900.
6,08 r триС(2идроксиметил)аминометана Р
и 8,77 r натрия хлорида Р растворяют в 500 мл
воды дистиллированной Р и прибавляют 10,0 r
альбумина бычье20 Р Доводят рН (2.2.3) кисло
той хлористоводородной Р и разводят водой
дистиллированной Р до объема 1000,0 мл.
Фосфатный забуференный физиолоrи-
ческий раствор рН 7,4. 4005000.
2,38 r динатрия 2идрофосфата Р. 0,19 r
калия ди2идрофосфата Р и 8,0 r натрия хлори
да Р растворяют в воде Р и доводят до объема
1000,0 мл этим же растворителем. При необхо
димости доводят рН (2.2.3).
Боратный буферный раствор рН 7,5.
4005200.
2,5 r натрия хлорида Р, 2,85 r динатрия тeтpa
бората Р и 10.5 r кислоты борной Р растворяют в
воде Р и доводят до объема 1000,0 мл этим же раст
ворителем. При необходимости доводят рН (2.2.3).
Хранят при температуре от 2 ос до 8 ос.
Буферный (HEPES) раствор рН 7,5.
4009700.
2,38 r 2 {4--(2 2идроксиэтил)пиперазин 1 ил]
этансульфоновой кислоты Р растворяют в около
90 мл воды Р. доводят раствором натрия 2идpOKcи
да Р до рН 7,5 и разводят водой Р до объема 100 мл.
Трис( rидроксиметил)аминометан-буфер-
ный раствор рН 7,5. 4005500.
7,27 r триС(2идроксиметил)аминомета
на Р и 5,27 r натрия хлорида Р растворяют в
воде Р и, при необходимости, доводят рН (2.2.3).
Разводят водой Р до объема 1000,0 мл.
0,05 М трис-rидрохлорида буферный
раствор рН 7,5. 4005600.
6,057 r триС(2идроксиметил)аминомета
на Р растворяют в воде Р и, при необходимости,
доводят рН (2.2.3) кислотой хлористоводород
ной Р. Разводят водой Р до объема 1000,0 мл.
0,2 М фосфатный буферный раствор
рН 7,5. 4005400.
27,22 r калия ди2идрофосфата Р paCTBO
ряют в 930 мл воды Р, доводят раствором 300 r/л
калия 2идроксида Р до рН 7,5 (2.2.3) и разводят
водой Р до объема 1000,0 мл.
0,33 М фосфатный буферный раствор
рН 7,5. 4005300.
Раствор /. 119,31 r динатрия 2идрофосфа
та Р растворяют в воде Р и доводят до объема
1000,0 мл этим же растворителем.
Раствор 11. 45,36 r калия ди2идрофосфа
та Р растворяют в воде Р и доводят до объема
1000,0 мл этим же растворителем.
Смешивают 85 мл раствора 1 и 15 мл paCT
вора 11. При необходимости доводят рН (2.2.3).
Натрия цитрата буферный раствор рН 7,8
(0,034 М раствор натрия цитрата и 0,101 М
раствор натрия хлорида). 4009800.
10,0 r натрия цитрата Р и 5,90 r натрия
хлорида Р растворяют в 900 мл воды Р, ДOBO
дят рН (2.2.3) кислотой хлористоводородной Р
и разводят водой Р до объема 1000 мл.
0,0015 М боратный буферный раствор
рН 8,0. 4006000.
0.572 r динатрия тетрабората Р и 2,94 r калЬ
ция хлорида Р растворяют в 800 мл воды Р. Доводят
рН (2.2.3) 1 М раствором кислоты хлористовадо--
родной и разводят вадой Р до объема 1000,0 мл.
Буферный раствор рН 8,0. 4005900.
К 50,0 мл 0,2 М раствора калия ди2идpo
фосфата Р прибавляют 46,8 мл 0,2 М pacт
вора натрия 2идроксида и доводят водой Р до
объема 200,0 мл.
Буферный раствор рН 8,0 Р1. 4010400.
20 r дикалия 2идрофосфата Р растворяют в
900 мл воды Р, доводят рН (2.2.3) кислотой фос
форной Р И разводят водой Р до объема 1000 мл.
0,02 М фосфатный буферный раствор
рН 8,0. 4006100.
К 50,0 мл 0,2 М раствора калия ди2идpo
фосфата Р прибавляют 46,8 мл 0,2 М pacт
вора натрия 2идроксида и доводят водой Р до
объема 500,0 мл.
0,1 М фосфатный буферный раствор
рН 8,0. 4008400.
0,523 r калия ди2идрофосфата Р и 16,73 r
дикалия 2идрофосфата Р растворяют в воде Р
и доводят до объема 1000,0 мл этим же paCTBO
рителем.
1 М фосфатный буферный раствор
рН 8,0. 4007800.
136,1 r калия ди2идрофосфата Р paCTBO
ряют в воде Р и доводят рН (2.2.3) 1 М pacтвo
ром натрия 2идроксида. Разводят водой Р до
объема 1000,0 мл.
Трис(rидроксиметил)аминометан-буфер-
ный раствор рН 8,1. 4006200.
0,294 r кальция хлорида Р растворяют в
40 мл раствора триС(2идроксиметил)аминоме
4.1.3. Буферные растворы
749
тана Р, доводят рН (2.2.3) 1 М раствором Kиc
лоты хлористоводородной и разводят водой Р
до объема 100,0 мл.
# Аминоуксусная буферная смесь с рН 8,3.
8,4 r натрия 2идрокарбоната Р, 1 О r калия
2идрокарбоната Р, 7,7 r 2лицина Р и 4 мл pacт
вора аммиака концентрироваНН020 Р растворяют
в воде Р и доводят водой Р до объема 100,0 мл.
Трис rлицин буферный раствор рН 8,3.
4006300.
6,0 r триС(2идроксиметил)аминометана Р
и 28,8 r 2лицина Р растворяют в воде Р и ДOBO
дят до объема 1000,0 мл этим же растворителем.
Непосредственно перед использованием к
1 объему полученноro раствора прибавляют 1 О
объемов воды Р.
Барбитал-буферный раствор рН 8,4.
4006400.
8,25 r барбитал натрия Р растворяют в
воде Р и доводят до объема 1000,0 мл этим же
растворителем.
Трис-ЕDТА-ВSА буферный раствор
рН 8,4. 4006500.
6,1 r триС(2идроксиметил)аминометана Р,
2,8 r натрия эдетата Р, 10,2 r натрия хлори
да Р и 1 О r альбумина бычье20 Р растворяют в
воде Р, доводят 1 М раствором кислоты хло
ристоводородной до рН 8.4 (2.2.3) и разводят
водой Р до объема 1000,0 мл.
Трис( rидрокси метил )аминометан-ЕDТ А
буферный раствор рН 8,4. 4006600.
5,12 r натрия хлорида Р, 3,03 r
триС(2идроксиметил)аминометана Р и 1,40 r
натрия эдетата Р растворяют в 250 мл воды
дистиллированной Р, доводят кислотой хло
ристоводородной Р до рН 8,4 (2.2.3) и раз
водят водой дистиллированной Р до объема
500,0 мл.
Трис-ацетатный буферный раствор
рН 8,5. 4006700.
0,294 r кальция хлорида Р и 12,11 r
триС(2идроксиметил)аминометана Р paCTBO
ряют в воде Р, доводят рН (2.2.3) кислотой YKCYC
ной Р И разводят водой Р до объема 1000,0 мл.
Барбитал-буферный раствор рН 8,6 Р1.
4006900.
1 ,З8 r барбитала Р, 8,76 r барбитал на
трия Р и 0,38 r кальция лактата Р растворяют
в воде Р и доводят до объема 1000,0 мл этим же
растворителем.
1,5 М трис rидрохлорида буферный paCT
вор рН 8,8. 4009900.
90,8 r триС(2идроксиметил)аминомета
на Р растворяют в 400 мл воды Р, доводят рН
96. Зак. 1712
(2.2.3) кислотой хлористоводородной Р и раз
водят водой Р до объема 500,0 мл.
Буферный раствор рН 9,0. 4007000.
Раствор 1. 6,18 r кислоты борной Р paCT
воряют в 0,1 М растворе калия хлорида Р и дo
водят до объема 1000,0 мл этим же растворите
лем.
Раствор 1/. 0,1 М раствор натрия 2идpOK
сида.
Смешивают 1000,0 мл раствора I и 420,0 мл
раствора 11.
Буферный (фосфатный) раствор рН 9,0.
4008300.
1,74 r калия ди2идрофосфата Р растворяют
в 80 мл воды Р и, при необходимости, доводят
рН (2.2.3) 1 М раствором калия 2идроксида. Раз
водят водой Р до объема 100,Омл.
Буферный раствор рН 9,0 Р1. 4007100.
6,20 r кислоты борной Р растворяют в
500 мл воды Р, доводят рН (2.2.3) 1 М pacтвo
ром натрия аидроксида (около 41,5 мл) и разво
дят водой Р до объема 1000,0 мл.
Аммиачный буферный раствор рН 9,5.
4007200.
З3,5 r аммония хлорида Р растворяют в
150 мл воды Р, прибавляют 42,0 мл раствора
аммиака концентрироваНН020 Р и доводят
водой Р до объема 250,0 мл.
Хранят в полиэтиленовом контейнере.
Аммиачный буферный раствор рН 10,0.
4007300.
5,4 r аммония хлорида Р растворяют в 20 мл
воды Р, прибавляют 35,0 мл раствора аммиака
Р и доводят водой Р до объема 100,0 мл.
Диэтаноламин-буферный раствор
рН 10,0. 4007500.
96,4 r диэтаноламина Р растворяют в
воде Р, доводят до объема 400 мл этим же paCT
ворителем, прибавляют 0,5 мл раствора 186 r/л
ма2ния хлорида Р, доводят рН (2.2.3) 1 М pac
твором кислоты хлористоводородной и разво
дят водой Р до объема 500,0 мл.
Аммония хлорида буферный раствор
рН 10,4. 4011000.
70 r аммония хлорида Р растворяют в 200 мл
воды Р, прибавляют 330 мл раствора аммиака
концентрироваНН020 Р и доводят водой Р до
1000,0 мл. При неоБХОДИМОСТIA доводят pacтвo
ром аммиака Р до рН 10,4.
Боратный буферный раствор рН 10,4.
4011100.
24,64 r кислоты борной Р растворяют в
900 мл дистиллированной воды Р. Доводят pac
твором 400 r/л натрия 2идроксида Р до рН 10,4
750
rосударственная фармакопея Республики Беларусь
(2.2.3). Доводят водой дистиллированной Р до
объема 1000,0 мл.
Буферный раствор рН 10,9.4007600.
6,75 r аммония хлорида Р растворяют в pac
творе аммиака Р и доводят до объема 100,0 мл
этим же растворителем.
Буфер для реryлирования ионной силы.
4007700.
58,5 r натрия хлорида Р, 57,0 мл кислоты
уксусной ледяной Р, 61,5 r натрия ацетата Р
и 5,0 r циКЛ02ексилендинитрилтетрауксусной
кислоты Р растворяют в воде Р и доводят до
объема 500,0 мл этим же растворителем. ДOBO
дЯТ раствором 335 r/л натрия 2идроксида Р до
рН 5,O 5,5 и разводят водой дистиллирован
ной Р до объема 1000,0 мл.
Буфер для реryлирования ионной
силы Р1. 4008800.
Раствор (а). 210 r кислоты лимонной Р
растворяют в 400 мл воды дистиллированной Р,
доводят раствором аммиака KOHцeHтpиpoвaH
ным Р ДО рН 7,0 (2.2.3) и разводят водой диc
тиллированной Р до объема 1000,0 мл.
Раствор (Ь). 132 r аммония фосфата Р
растворяют в воде дистиллированной Р и ДOBO
дят до объема 1000,0 мл этим же растворителем.
Раствор (с). К суспензии 292 r (этилендини
трил)тетрауксусной кислоты Р приблизительно
в 500 мл воды дистиллированной Р прибавляют
около 200 мл раствора аммиака KOHцeHтpиpo
ваНН020 Р. Доводят раствором аммиака KOHцeH
трированным Р до рН 6 7 (2.2.3) и разводят
водой дистиллированной Р до объема 1000,0 мл.
Смешивают равные объемы растворов (а),
(Ь) и (с) и ДОВОДЯТ раствором аммиака KOHцeH
трированным Р до рН 7,5.
4.2. РЕАКТИВЫ,
ТИТРОВАННЫЕ РАСТВОРЫ
ДЛЯ ОБЪЕмноrо
АНАЛИЗА
0112013:40201
4.2.1. ИСХОДНЫЕ
СТАНДАРТНЫЕ ВЕЩЕСТВА
ДЛЯ ТИТРОВдННЫХ РАСТВОРОВ
Исходные стандартные вещества для YCTa
новки титра титрооанных растворов обознача
ют буквами РО и roтовят следующим образом.
Бензойная кислота. С 7 Н 6 О 2 . (М М. 122,1).
2000200. [65 85 O].
Кислоту бензойную Р сублимируют.
Калия бромат. КВrО з . (М м. 167,0).
2000300. [7758 01 2].
Калия бромат Р перекристаллизовывают из
кипящей воды Р. Кристаллы собирают и сушат
до постоянной массы при температуре 180 ОС.
Калия rидрофталат. С в Н 5 К0 4 , (М М. 204,2).
2000400. [877 24 7).
Калия 2идрофталат Р перекристаллизо
вывают из кипящей воды Р Кристаллы соби
рают при температуре выше 35 ос и сушат при
температуре 110 ос до постоянной массы.
#Калия дихромат. K 2 CrPT (М М. 294,2).
1069500. [7778 50 9].
Калия дихромат Р перекристаллизовыва
ют из roрячей воды Р. Кристаллы собирают и
сушат до постоянной массы при температуре
от 130 ОС до 150 ОС.
Мышьяка (111) оксид. Аsрз. (М м. 197,8).
2000100. [1327 5З 3].
Мышьяка (111) оксид Р сублимируют.
Хранят над силика2елем безводным Р.
Натрия карбонат. Nа 2 СО з . (М м. 106,0).
2000500. [497 19 8).
Насыщенный раствор натрия карбоната Р
фильтруют при комнатной температуре. Через
фильтрат медленно пропускают поток уzлерода
диоксида Р при постоянном охлаждении и пере
мешивании. Через 2 ч осадок собирают на CTe
клянном фильтре (2.1.2), промывают фильтр ле
дяной водой Р, насыщенной уrлерода диоксидом.
Сушат при температуре от 100 ос до 105 ос и про
каливают до постоянной массы при температуре
от 270 ос до 300 ос, периодически перемешивая.
Натрия хлорид. NaCI. (М м. 58,44).
2000600. [7647 14 5).
К 1 объему насыщенноro раствора натрия
хлорида Р прибавляют 2 объема кислоты хло
ристоводородной Р. Полученные кристаллы
собирают и промывают кислотой хлористо
водородной Р1, которую удаляют наrреванием
на водяной бане. Прокаливают до постоянной
массы при температуре 300 ОС.
Сульфаниловая кислота. С 6 Н 7 NО з S.
(М м. 173,2). 2000700. [121 57 31).
Кислоту сульфанuловую Р перекристал
лизовывают из кипящей вfJды Р, фильтруют и
сушат при температуре от 100 ос до 105 ос до
постоянной массы.
Цинк. Zп. (А м. 65,4). 2000800. [7440 66 6].
Используют цинк с содержанием не менее
99,9 % Zп.
4.2.2. Титрованные растворы
01/2013:40202
751
4.2.2. ТИТРОВАННЫЕ РАСТВОРЫ
Титрованные растворы должны roто
виться в соответствии с обычными требо
ваниями химическоro анализа: Проверяют
точность используемоrо оборудования для
тоro, чтобы удостовериться в еro приrодно
сти для предполаrаемоrо применения.
Концентрацию титрованных растворов
выражают их молярностью. Молярность BЫ
ражает количество вещества, paCTBopeHHO
ro в 1 л раствора, в виде количества молей.
Раствор, содержащий х молей вещества в
одном литре, обозначают х М раствором.
Концентрация титрованных растворов
не должна отличаться от указанной более
чем на 10 %. Молярность титрованных pac
Т80рОВ определяют с точностью 0,2 %.
Титрованные растворы стандартизуют
описанными ниже методами. Если титро
ванный раствор используют в количествен
ном анализе, в котором конечную точку
определяют электрохимическим методом
(например, амперометрии или потенцио
метрии), раствор стандартизуют тем же Me
юдом. Состав среды, в которой CTaHдap
тизуют титрованный раствор, должен быть
таким же, как и тот, в котором он будет ис
пользован.
Растворы более разведенные, чем опи
санные ниже, получают разведением по
следних водой, свободной от У2лерода ди
оксида, Р Поправочные коэффициенты
полученных растворов такие же, как у ис
ходных растворов.
#Коэффициент поправки рекомендуется
устанавливать при температуре 20 0 С, при
этой же температуре при меняют титрован
ные растворы.
Если устанавливают коэффициент по
правки и применяют раствор при друrих
температурах, то водят температурную по
правку в соответствии с таблицами #4.2.2.
1 и #4.2.2. 2. Коэффициент поправки про
веряют не реже одноrо раза в два месяца,
если раствор устойчив, соблюдены условия
хранения (хранят в помещениях при KOMHaT
ной температуре в местах, защищиенных от
попадания прямых солнечных лучей) и нет
друrих указаний.
Таблица #4.2.2. 1
Приведение объемов растворов при данной температуре к объемам при 20 ос (для 1000 мл)
Температура Вода, растворы Растворы концентрации
(ОС) концентрации 0,1 М 0,5 М HCI 1 М HCI 0,5М 1 М NaOH
0,01 М и раство- (кроме HCI) NaOH
pbIHCI
1 2 3 4 5 6 7
5 +1,5 +1,7 +1,0 +2,3 +2,35 +3,6
6 +1,5 +1,65 +1,85 +2,2 +2,25 +3,4
7 +1,4 +1,6 +1,8 +2,15 +2,2 +3,2
8 +1,4 +1,55 +1,75 +2,1 +2,15 +3,0 I
9 +1,4 +1,5 +1,7 +2,0 +2,05 +2,7
10 +1,3 +1,45 +1,6 +1,9 +1,95 +2.5
11 +1,2 +1,35 +1,5 +1,8 +1,8 +2.3
12 +1,1 +1,3 +1,4 +1,6 +1,7 +2.0
13 +1,0 +1,1 +1,2 +1,4 +1,5 +1.8
14 +0,9 +1,0 +1,1 +1,2 +1,3 +1,6
15 +0,8 +0,9 +0,9 +1,0 +1.1 +1.3
16 +0,6 +0,7 +0,8 +0,8 +0,9 +1,1
17 +0,5 +0,6 +0,6 +0,6 +0.7 +0,8
18 +0,3 +0,4 +0,4 +0,4 +0.5 I +0.6
19 +0,2 +0,2 +0,2 +0,2 I +0.2 I +0,3
20 0,0 0,0 0,0 0,0 I 0.0 I 0,0
21 0,2 0,2 0,2 0,2 ! .2 , ,3
22 0,4 0,4 0,4 0,5 i ,5 ! 0,5
23 0,6 0,6 0,7 0,7 I .8 I 0,9
24 0,8 0,9 0,9 1.0 ! 1,0 i 1 ,2
25 1 ,О 1,1 1,1 I 1.2 1,3 ! 1,5
i
26 1,3 1 ,4 1,4 1.4 1.5 1 ,8
27 1 ,5 1 ,7 1.7 ! 1 ,7 I 1 ,8 2, 1
I 28 1 ,8 2,0 2,0 I 2,0 2, 1 2,4
, 29 2, 1 2,З i 2.3 I 2,3 2,4 2,8
I 30 2,3 2.5 2.5 I 2,6 i 2,8 3,2
752
rосударственная фармакопея Республики Беларусь
ПРИМЕЧАНИЕ:
1. Числа в rрафах 2 7 выражают объемы в
миллилитрах, которые следует прибавить (+) к
1000 мл соответствующей жидкости при tOC или
вычесть Н от 1000 мл, чтобbl получить объем
титрованноro раствора при 20 ОС.
2. Примеры.
2.1. В колбу вместимостью 1000 мл кали
брованную при 20 ОС, необходимо налить при
15 ос 0,1 М раствор серебра нитрата. По
таблице #4.2.2. 1 находят, что при 20 ОС этот
раствор займет объем больший на 0,9 мл.
2.2. Из бюретки, калиброванной при
20 ОС, при 25 ос израсходовано на титрова
ние 34,75 мл 1 М раствора натрия 2идpOKcи
да. При 20 ос расход титрованноro раствора
(в мл) составит:
34 75 34,75 .1,5 34.75 o 05 3470 мл
, 1000 . , ,
ПРИМЕЧАНИЕ:
Таблицей #4.2.2. 2 пользуются при
работе с растворами 0,1 М концентрации
или более разбавленными.
На контейнере с титрованным раствором
указывают: наименование, заданную KOHцeH
трацию, дату приrотовления, коэффициент
поправки. при меняемый индикатор, дату и
температуру установления коэффициента по
правки. Допускается вместо заданной KOHцeH
трации и коэффициента поправки указывать
значение точной концентрации с четырьмя
значащими цифрами после запятой. При элек
трохимическом титровании вместо индикато
ра при водят указание о применении метода
(амперометрическоrо, потенциометрическо
ro). Титрованные растворы, в которых при
хранении появились хлопья или осадок, не
должны применяться.=
1 М раствор азотной кислоты. 3003600.
96,6 r кислоты азотной Р доводят водой Р
до объема 1000.0 мл.
Установка титра. 1,000 r натрия карбо
ната РО растворяют в 50 мл воды Р, прибав
ляют 0,1 мл раствора метилов080 оранж
в080 Р и т труют приrотовленным раствором
кислоты азотной до появления KpaCHOBa
то желтоro окрашивания; кипятят в течение
2 мин. Раствор снова приобретает желтое
окрашивание. Охлаждают и продолжают ти
трование до появления красновато желтоrо
окрашивания.
1 мл 1 М раствора кислоты азотной co
ответствует 53,00 Mr Nа 2 СО з .
0,1 М раствор аммония тиоцианата.
3000500.
7,612 r аммония тиоцианата Р paCTBO
ряют в воде Р и доводят до объема 1000,0 мл
. этим же растворителем.
Установка титра. К 20,0 мл 0,1 М pacт
вора серебра нитрата прибавляют 25 мл
воды Р, 2 мл кислоты азотной разведенной Р,
2 мл раствора железа (/11) аммония сульфата
Р2 и титруют приroтовленным раствором aM
мония тиоцианата до появления KpaCHOBaTO
желтоro окрашивания.
0,1 М раствор аммония церия нитрата.
3000100.
Раствор, содержащий 56 мл кислоты
серной Р и 54,82 r аммония церия (/V) Hи
трата Р, встряхивают в течение 2 мин, при
бавляют последовательно пять порций, по
100 мл каждая, воды Р, перемешивая после
каждоro прибавления. Прозрачный раствор
доводят водой Р до объема 1000,0 мл. Титр
полученноro раствора устанавливают через
1 О сут.
Установка титра. К 25,0 мл приroтов
ленноro раствора аммония церия (IV) нитра
та прибавляют 2,0 r калия йод ида Р и 150 мл
воды Р. Немедленно титруют О, 1 М pacтвo
ром натрия тиосульФата, используя в каче
стве индикатора 1 мл раствора крахмала Р.
Хранят в защищенном от света месте.
Таблица #4.2.2. 2
Поправки объемов разбавленных растворов при разных температурах
Температура Объем,мл
(ОС) 10 20 25 30 40 50
10 +0,01 +0,03 +0,03 +0,04 +0,05 +0,06
12 +0,01 +0,02 +0,03 +0,03 +0,04 +0,06
14 +0,01 +0,02 +0,02 +0,03 +0,04 +0,05
16 +0,01 +0,01 +0,02 +0,02 +0,03 +0,03
18 0,00 +0,01 +0,01 +0,01 +0,01 +0,02
20 0,00 0,00 0,00 0,00 0,00 0,00
22 0,00 0,01 0,01 0,01 0,02 0,02
24 0,01 0,02 0,02 0,02 0,03 0,04
26 0,01 0,03 0,03 0,04 0,05 0,06
28 0,02 0,03 0,04 0,05 0,07 0,09
30 0,02 o,03 0,05 0,07 0,09 0,12
4.2.2. Титрованные растворы
753
0,01 М раствор аммония церия нитрата.
3000200.
К 100,0 мл 0,1 М раствора аммония
церия нитрата прибавляют при охлаждении
30 мл кислоты серной Р и доводят водой Р до
объема 1000,0 мл.
0,1 М раствор аммония церия сульфата.
3000300.
65,0 r аммония церия (/V) сульфата Р
растворяют в смеси из 500 мл воды Р и 30 мл
кислоты серной Р, охлаждают и доводят
водой Р до объема 1000,0 мл.
Установка титра. К 25,0 мл приroтов
ленноro раствора аммония церия (IV) сульфа
та прибавляют 2,0 r калия йодида Р и 150 мл
воды Р. Немедленно титруют О, 1 М pacтвo
ром натрия тиосульфата, используя в каче
стве индикатора 1 мл раствора крахмала Р.
0,01 М раствор аммония церия сульфата.
3000400.
К 100,0 мл 0,1 М раствора аммония
церия сульфата прибавляют при охлаждении
30 мл кислоты серной Р и доводят водой Р до
объема 1000,0 мл.
0,05 М раствор бария перхлората.
3000700.
15,8 r бария 2идроксида Р растворяют в
смеси из 75 мл воды Р и 7,5 мл кислоты хлор
ной Р, доводят кислотой хлорной Р до рН 3
и при необходимости фильтруют. Прибавляют
150 мл 96 % спирта Р, разводят водой Р до
объема 250 мл и доводят буферным pacтвo
ром рН 3,7 Р до объема 1000,0 мл.
Установка титра. К 5,0 мл 0,05 М pacт
вора кислоты серной прибавляют 5 мл
воды Р, 50 мл буферН020 раствора рН 3,7 Р,
0,5 мл раствора ализарина S Р и титруют при
rотовленным раствором бария перхлората до
появления оранжево красноro окрашивания.
Титр устанавливают непосредственно
перед использованием.
0,025 М раствор бария перхлората.
3009600.
500,0 мл 0,05 М раствора бария перхло
рата доводят буферным раствором рН 3,7 Р
до объема 1000,0 мл.
0,1 М раствор бария хлорида. 3000600.
24,4 r бария хлорида Р растворяют в
воде Р и доводят до объема 1000,0 мл этим
же растворителем.
Ycmaf-lОека титра. К 10,0 мл приroтов
ленноro раствора бария хлорида прибавляют
60 мл воды Р, 3 мл раствора аммиака KOH
центрироваНН020 Р, 0,5 1 Mr фталеиново
20 пурпуров020 Р и титруют 0,1 М раствором
натрия эдетата. Коrда окраска раствора
начнет ослабевать, прибавляют 50 мл 96 %
спирта Р и продолжают титрование до исчез
новения сине фиолетовоro окрашивания.
0,004 М раствор бензэтония хлорида.
3000900.
1,792 r бензэтония хлорида Р, предва
рительно высушенноrо до постоянной массы
при температуре от 100 ос до 105 ос, paCTBO
ряют в воде Р и доводят до объема 1000,0 мл
этим же растворителем.
Установка титра. Рассчитывают MO
лярность раствора, исходя из содержания
C 27 H 42 C/N0 2 в высушенном бензэтония хло
риде, определенноro следующим образом.
0,350 r высушенноro вещества растворяют в
30 мл кислоты уксусной безводной Р, прибав
ляют 6 мл раствора ртути (11) ацетата Р
и титруют 0,1 М раствором кислоты хлор
ной, используя в качестве индикатора 0,05 мл
раствора кристалличеСК020 фиолетов020 Р
Параллельно проводят контрольный опыт.
1 мл 0,5 М раствора кислоты хлорной
соответствует 44,81 Mr C27H42CIN02'
0,0167 М раствор бромид-бромата.
3001000.
2,7835 r калия бромата РО и 13 r калия
бромида Р растворяют в воде Р и доводят до
объема 1000,0 мл этим же растворителем.
0,1 М раствор железа аммония сульфата.
3001300.
50,0 r железа (1/1) аммония сульфата Р
растворяют в смеси из 6 мл кислоты серной Р
и 300 мл воды Р и доводят водой Р до объема
1000,0 мл.
Установка титра. К 25,0 мл приroтовлен
Horo раствора железа аммония сульфата при
бавляют кислоты хлористоводородной Р, 2 r
калия йодида Р и через 1 О мин титруют О, 1 М
раствором натрия тиосульфата, используя в
качестве индикатора 1 мл раствора крахмала Р
1 мл 0,1 М раствора натрия тиосульфа
та соответствует 48,22 Mr FeNH 4 (SO 4)2 '12Нр.
0,1 М раствор железа сульфата.
3001400.
27,80 r железа (11) сульфата Р paCTBO
ряют в 500 мл кислоты серной разведенной Р
и доводят водой Р до объема 1000,0 мл.
Установка титра. К 25,0 мл приrотов
ленноro раствора железа сульфата прибавля
ют 3 мл кислоты фосфорной Р и немедленно
титруют 0,02 М раствором калия пермаН2а
ната.
Титр устанавливают непосредственно
перед использованием.
0,5 М раствор йода. 3009400.
127 r йода Р и 200 r калия йод ида Р paCTBO
ряют в воде Р и доводят до объема 1000,0 мл
этим же растворителем.
754
rосударственная фармакопея Республики Беларусь
Установка титра. К 2,0 мл приroтовленно
ro раствора йода прибавляют 1 мл кислоты YK
сусной разведенной Р и 50 мл воды Р и титруют
0,1 М раствором натрия тиосульфата, исполь
зуя в качестве индикатора раствор крахмала Р
Хранят в защищенном от света месте.
0,05 М раствор йода. 3002700.
12,7 r йода Р и 20 r калия йод ида Р paCTBO
ряют в воде Р и доводят до объема 1000,0 мл
этим же растворителем.
Установка титра. К 20,0 мл приroтов
ленноro раствора йода прибавляют 1 мл Kиc
лоты уксусной разведенной Р и ЗО мл воды Р
и титруют О. 1 М раствором натрия тиo
сульфата, используя в качестве индикатора
раствор крахмала Р
Хранят в защищенном от света месте.
0,01 М раствор йода. 3002900.
0,3 r калия йодида Р растворяют в 20,0 мл
0,05 М раствора йода и доводят водой Р до
объема 100,0 мл.
0,033 М раствор калия бромата. 3004200.
5,5670 r калия бромата РО растворяют в
воде Р и доводят до объема 1000,0 мл этим
же растворителем.
0,02 М раствор калия бромата. ЗО04300.
3,340 r калия бромата РО растворяют в
воде Р и доводят до объема 1000,0 мл этим
же растворителем.
0,0167 М раствор калия бромата.
3004400.
rотовят разведением О,ОЗЗ М раствора
калия бромата.
0,083 М раствор калия бромата. 3004500.
rотовят разведением 0,033 М раствора
калия бромата.
1 М раствор калия rидроксида. 3009100.
60 r калия 2идроксида Р растворяют в
воде, свободной от У2лерода диоксида, Р и
доводят до объема 1000,0 мл этим же paCT
ворителем.
Установка титра. 20,0 мл приroтов
ленноro раствора калия rидроксида титруют
1 М раствором кислоты хлористоводород
ной, используя в качестве индикатора 0,5 мл
раствора фенолфталеина Р.
0,1 М раствор калия rидроксида.
3004800.
6 r калия 2идроксида Р растворяют в воде,
свободной от У2лерода диоксида, Р и ДOBO
дят до объема 1000,0 мл этим же раствори
телем.
Установка титра. 20,0 мл приroтов
ленноro раствора калия rидроксида титруют
О, 1 М раствором кислоты хлористоводород
ной, используя в качестве индикатора 0,5 мл
раствора фенолфталеина Р.
0,5 М раствор калия rидроксида в
спирте (60 %, об/об). 3004900.
3 r калия 2идроксида Р растворяют в
спирте (60 %, об/об), свободном от альде2и
до в, Р и доводят до объема 100,0 мл этим же
растворителем.
Установка титра. 20,0 мл приroтовленно
ro раствора калия rидроксида в спирте (60 %,
об/об) титруют 0,5 М раствором кислоты хло
ристоводородной, используя в качестве инди
катора 0,5 мл раствора фенолфталеина Р.
0,5 М раствор калия rидроксида спир
товой. 3005000.
3 r калия 2идроксида Р растворяют в 5 мл
воды Р и доводят 96 % спиртом, свободным
от альде2идов, Р до объема 100,0 мл.
Установка титра. 20,0 мл приroтовлен
Horo раствора калия rидроксида спиртовоrо
титруют 0,5 М раствором кислоты хлористо
водородной, используя в качестве индикатора
0,5 мл раствора фенолфталеина Р.
0,1 М раствор калия rидроксида спир
товой.3005100.
20,0 мл 0,5 М раствора калия 2идроксида
спиртов020 доводят 96 % спиртом, свобод
ным от альде2идов, Р до объема 100,0 мл.
0,01 М раствор калия rидроксида спир
товой. 3009000.
2,0 мл 0,5 М раствора калия 2идроксида
спиртов020 доводят 96 % спиртом, свобод
ным от альде2идов, Р до объема 100,0 мл.
0,1 М раствор калия rидрофталата.
3004700.
Около 800 мл кислоты уксусной безво
дной Р помещают в коническую колбу, при
бавляют 20,42 r калия 2идрофталата РО и
наrревают на водяной бане до растворения,
защищая от действия влаrи. Охлаждают до
температуры 20 ос и доводят кислотой YKCYC
ной безводной Р до объема 1000,0 мл.
0,0167 М раствор калия дихромата.
3004600.
4,90 r калия дихромата Р растворяют в
воде Р и доводят до объема 1000,0 мл этим
же растворителем.
Установка титра. К 20,0 мл приroтов
ленноrо раствора калия дихромата прибавля
ют 1 r калия йодида Р, 7 мл кислоты хлори
стоводородной разведенной Р, 250 мл воды Р
и титруют 0,1 М раствором натрия тиосуль
фата до изменения окраски от синей до CBeT
ло зеленой, используя в качестве индикатора
3 мл раствора крахмала Р.
4.2.2. Титрованные растворы
755
# 0,0167 М раствора калия йодата.
3,567 r калия йодата Р, предварительно
высушенноro при температуре 11 О ос до посто
янной массы и тонко растертоro, растворяют в
воде Р и доводят до объема 1000,0 мл этим же
растворителем.
Установка титра. К 20,0 мл приroтовлен
ноro раствора калия йодата прибавляют 100 мл
воды Р, 25 мл кислоты серной разведенной Р,
2 r калия йодида Р, выдерживают в защищенном
от света месте в течение 1 О мин и титруют О, 1 М
раствором натрия тиосульфата, используя в
качестве индикатора 1 мл раствора крахмала,
свободН020 от йодидов, Р.
0,05 М раствор калия йодата. 3005200.
1 0,70 r калия йодата Р растворяют в воде Р
и доводят до объема 1000,Омл этим же paCTBO
рителем.
Установка титра. 25,0 мл приroтовленно
ro раствора калия йодата доводят водой Р до
объема 100,0 мл. К 20,0 мл полученноro paCT
вора прибавляют 2 r калия йодида Р, 1 О мл Kиc
лоты серной разведенной Р и титруют О, 1 М
раствором натрия тиосульфата, используя в
качестве индикатора 1 мл раствора крахмала Р,
прибавляемый в конце титрования.
0,001 М раствор калия йодида. 3009200.
10,0 мл раствора калия йодида Р (166 r/л)
доводят водой Р до объема 100,0 мл. 5 мл по
лученноro раствора доводят водой Р до объема
500,0 мл.
0,02 М раствор калия nepMaHraHaTa.
3005300.
3,2 r калия пермаН2аната Р растворяют в
воде Р и доводят до объема 1000,0 мл этим же
растворителем. Полученный раствор HarpeBa
ют на водяной бане в течение 1 ч, охлаждают и
фильтруют через стеклянный фильтр (2.1.2).
Установка титра. К 20,0 мл приroтовлен
ноro раствора калия перманrаната прибавляют
2 r калия йодида Р, 1 О мл кислоты серной раз
веденной Р и титруют 0,1 М раствором натрия
тиосульфата, используя в качестве индикато
ра 1 мл раствора крахмала Р, прибавляемый в
конце титрования.
Титр устанавливают непосредственно перед
использованием.
Хранят в защищенном от света месте.
0,1 М раствор лития метилата. 3003300.
0,694 r лития Р растворяют в 150 мл Me
танола безводН020 Р и доводят толуолом Р
оf)ъема до 1000,0 мл.
Установка титра. К 10 мл диметилфор
мамида Р прибавляют 0,05 мл раствора 3 r/л
тимолов020 сине20 Р в метаноле Р и титруют
приroтовленным раствором лития метилата до
появления синеro окрашивания. Немедленно
прибавляют 0,200 r кислоты бензойной РО, пе
ремешивают до растворения и титруют приroтов
ленным раствором лития метилата до повтор
ноro появления синеro окрашивания. Во время
титрования раствор защищают от атмосферноro
уrлерода диоксида. Титр раствора лития мети
лата устанавливают по объему титранта, израс
ходованноrо в повторном титровании.
Титр устанавливают непосредственно перед
использованием.
1 мл 0,1 М раствора лития метилата co
ответствует 12,21 Mr С 7 Н 6 О 2 .
0,1 М раствор маrния хлорида. 3003400.
20,33 r ма2ния хлорида Р растворяют в
воде Р и доводят до объема 1000,0 мл этим же
растворителем.
Установка титра. Проводят комплексоме
трическое определение маrния (2.5.11).
0,02 М раствор меди сульфата. 3001200.
5,0 r меди (1/) сульфата Р растворяют в
воде Р и доводят до объема 1000,0 мл этим же
растворителем.
Установка титра. К 20,0 мл приroтовлен
ноro раствора меди сульфата прибавляют 2 r
натрия ацетата Р, 0,1 мл раствора пириди
лазонафтола Р и титруют 0,02 М раствором
натрия эдетата до перехода окраски от фио
летово синей до ярко зеленой. Вблизи точки эк
вивалентности титруют медленно.
0,1 М раствор натрия арсенита. 3005800.
4,946 r мышьяка (111) оксида РО, в пересчете
на Аs 2 О з , растворяют в смеси из 20 мл раствора
натрия 2идроксида концентрироваНН020 Р и 20
мл воды Р, доводят водой Р до объема 400,0 мл
и нейтрализуют кислотой хлористоводородной
разведенной Р по лакмусовой бума2е Р. PaCTBO
ряют в полученном растворе 2 r натрия 2идpOKap
боната Р и доводят водой Р до объема 500,0 мл.
1 М раствор натрия rидроксида.
3006300.
42 r натрия 2идроксида Р растворяют в воде,
свободной от У2лерода диоксида, Р и доводят до
объема 1000,0 мл этим же растворителем.
Установка титра. 20,0 мл приrотовлен
ноro раствора натрия rидроксида титруют 1 М
раствором кислоты хлористоводородной, ис
пользуя индикатор, указанный в методике KO
личественноrо определения с использованием
1 М раствора натрия 2идроксида.
#Установка титра. 5,000 r калия 2идроф
талата РО растворяют в 100 мл воды Р и титру
ют приroтовленным раствором натрия rидрокси
да (индикатор раствор фенолфталеина Р).
#Титр раствора устанавливают каждый раз
перед применением".
Если необходимо использование paCT
вора натрия rидроксида, свободноro от карбо
натов, ero roтовят следующим образом. PaCTBO
ряют натрия 2идроксид Р в воде Р до получения
раствора от 400 r/л до 600 r/л и отстаивают. Про
756
rосударственная фармакопея Республики Беларусь
зрачную жидкость над осадком сливают, защи
щая от воздействия уrлерода диоксида, разво
дят водой, свободной от У2лерода диоксида, Р
до необходимой молярности. Раствор должен
выдерживать следующее испытание. Титруют
20,0 мл раствора кислоты хлористоводородной
той же молярности, что и приrотовленный paCT
вор натрия rидроксида, используя в качестве ин
дикатора 0,5 мл раствора фенолфталеина Р. В
точке эквивалентности прибавляют небольшое
количество кислоты, необходимое для исчез
новения розовоro окрашивания; концентрируют
раствор до 20 мл кипячением; во время кипяче
ния прибавляют кислоту в количестве, необхо
димом для исчезновения розовоro окрашивания,
которое не должно возобновляться при длитель
ном кипячении. Объем израсходованной кисло
ты не должен превышать 0,1 мл.
0,1 М раствор натрия rидроксида. 3006600.
100.0 мл 1 М раствора натрия 2идроксида
доводят водой, свободной от У2лерода диOKcи
да, Р до объема 1000,0 мл.
Установка титра. Проводят титрование,
как указано для 1 М раствора натрия 2идpOKcи
да, используя 0,1 М раствор кислоты хлористо
водородной.
#Установка титра. 0,500 r калия 2идpo
фталата РО растворяют в 50 мл воды Р и титру
ют приroтовленным раствором натрия rидрокси
да (индикатор раствор фенолфталеина Р).
# 0,1 М раствор натрия rидроксида в
смеси из метанола и бензола.
4,2 r натрия 2идроксида Р растворяют в
100 мл метанола Р и доводят бензолом Р и Me
танолом Р до объема 1000,0 мл, прибавляя их
попеременно при перемешивании. Соотноше
ние (об/об) метанола и бензола при приroтовле
нии раствора должно быть около 1:4. В случае
получения непрозрачноro раствора ero Bыдep
живают в течение 12 ч, после чеro прозрачную
жидкость быстро сливают с осадка.
Установка титра. Установку титра про
водят в тщательно закрытых сосудах. Титро
вание рекомендуется проводить в атмосфе
ре инертноro rаза.
0,100 r кислоты бензойной Р растворяют в 20
мл диметилформамида Р, предварительно ней
трализованноro по тимоловому синему Р в диMe
тилформамиде Р, титруют приrотовленным pac
твором натрия rидроксида в смеси из метанола и
бензола, используя в качестве индикатора раствор
1 r/л тимолов020 сине2О Р в диметилформамиде
Р до перехода окраски из желтой в синюю.
Титр устанавливают непосредственно
перед использованием.
0,1 М раствор натрия rидроксида эта-
нольный. 3007000.
К 250 мл этанола Р прибавляют 3,3 r pacт
вора натрия 2идроксида концентрироваННО20 Р.
Установка титра. 0,200 r кислоты бен
зойной РО растворяют в 2 мл воды Р и 1 О мл
96 % спирта Р и титруют приroтовленным
раствором натрия rидроксида этанольным,
используя в качестве индикатора 0,2 мл pacт
вора тимолфталеина Р.
Титр устанавливают непосредственно
перед использованием.
1 мл 0,1 М раствора натрия 2идроксида
этаНОЛЬН02О соответствует 12,21 Mr С 7 Н 6 О 2 .
0,1 М раствор натрия метилата. 3007100.
175 мл метанола безводноzо Р охлажда
ют в ледяной бане и прибавляют небольши
ми порциями около 2,5 r свеженарезанноro
натрия Р. Korдa металл растворится, разво
дят толуолом Р до объема 1000,0 мл.
Установка титра. К 1 О мл диметилфор
мамида Р прибавляют 0,05 мл раствора 3 r/л
тимолов020 сине2О Р в метаноле Р и титруют
приroтовленным раствором натрия метилата до
появления синеro окрашивания. Тотчас прибав
ляют 0,200 r кислоты бензойной РО, переме
шивают до растворения и титруют приroтовлен
ным раствором натрия метилата до повторноro
получения синеro окрашивания. Во время ти
трования раствор защищают от атмосферноro
уrлерода диоксида. Титр раствора натрия мети
лата устанавливают по объему титранта, израс
ходованноro в повторном титровании.
Титр устанавливают непосредственно
перед использованием.
1 мл 0,1 М раствора натрия метилата
соответствует 12,21 Mr С 7 Н 6 О 2 .
0,1 М раствор натрия нитрита. 3007200.
7,5 r натрия нитрита Р растворяют в
воде Р и доводят до объема 1000,0 мл этим
же растворителем.
Установка титра. 0,300 r кислоты суль
фаниловой РО растворяют в 50 мл кислоты
хлористоводородной разведенной Р и ПРОВОflЯТ
определение первичной ароматической амино
rруппы (2.5.8) электрометрически, используя
приroтовленный раствор натрия нитрита.
Титр устанавливают непосредственно
перед использованием.
1 мл О, 1 М раствора натрия нитрита co
ответствует 17,32 Mr С 6 Н 7 NО з S.
#Установка титра. Около 0,200 r суль
фаниловой кислоты РО помещают в толсто
стенный стакан, прибавляют 60 мл воды Р,
1 О мл кислоты хлористоводородной раз
веденной Р, 1 r калия бромида Р и проводят
определение первичной ароматической ами
ноrруппы (2.5.8).
0,1 М раствор натрия перйодата.
3009500.
21,4 r натрия перйодата Р растворяют в
500 мл воды Р и доводят до объема 1000,0 мл
этим же растворителем.
4.2.2. Титрованные растворы
757
Установка титра. 20,0 мл приroтовлен
HOro раствора натрия перйодата помещают в
колбу с притертой пробкой, прибавляют 5 мл
кислоты хлорной р, колбу закрывают пробкой
и перемешивают. Доводят насыщенным pac
твором натрия 2идрокарбоната Р до рН 6,4
(2.2.3). Прибавляют 1 О мл раствора калия
йодида Р, закрывают пробкой, перемешивают,
выдерживают в течение 2 мин и титруют 0,025
М раствором натрия арсенита до появления
слабо желтоrо окрашивания. затем прибавляют
2 мл раствора крахмала Р и титруют до обесц
вечивания раствора.
0,1 М раствор натрия тиосульфата.
3007300.
25 r натрия тиосульфата Р и 0,2 r натрия
карбоната Р растворяют в воде, свободной
от У2лерода диоксида, Р и доводят до объема
1000,0 мл этим же растворителем.
Установка титра. К 10,0 мл 0,033 М pacт
вора калия бромата прибавляют 40 мл воды р,
1 О мл раствора калия йодида р, 5 мл кислоты
хлористоводородной Р1 и титруют приrотовлен
ным раствором натрия тиосульфата, используя
в качестве индикатора 1 мл раствора KpaXMa
ла Р, прибавляемый в конце титрования.
#Установка титра. 0,150 r калия диxpOMa
та ро растворяют в 50 мл воды Р в колбе с при
тертой пробкой, прибавляют 2 r калия йодида р,
5 мл раствора кислоты хлористоводород
ной Р, закрывают пробкой, смоченной pacтвo
ром калия йодида Р, и выдерживают в защи
щенном от света месте в течение 1 О мин. Затем
титруют приroтовленным раствором натрия тио
сульфата до зеленовато желтоro окрашивания,
прибавляют 2 мл раствора крахмала, свободно
20 от йодидов, Р и продолжают титрование до
перехода синей окраски в светло зеленую.
0,1 М раствор натрия эдетата. 3005900.
37,5 r натрия эдетата Р растворяют в
500 мл воды Р, прибавляют 100 мл 1 М pacт
вора натрия 2идроксида и доводят до объема
1000,0 мл этим же растворителем.
Установка титра. 0,120 r цинка РО paCTBO
ряют в 4 мл кислоты хлористоводородной Р1 и
прибавляют 0,1 мл бромной воды Р. Удаляют из
быток брома кипячением, прибавляют раствор
натрия 2идроксида разведенный Р до слабокис
лой или нейтральной реакции и Пр080ДЯТ KOM
плексометрическое определение цинка (2.5.11).
1 мл 0,1 М раствора натрия эдетата COOT
ветствует 6,54 Mr Zп.
Хранят в полиэтиленовом контейнере.
0,02 М раствор натрия эдетата. 3006000.
7,444 r натрия эдетата Р растворяют в
воде Р и доводят до объема 1000,0 мл этим же
растворителем.
Установка титра. 0,100 r цинка РО раст
воряют в 4 мл кислоты хлористоводород
ной Р1 и прибавляют 0,1 мл бромной воды р.
Удаляют избыток брома кипячением. Перено
сят раствор в мерную колбу и доводят водой Р
до объема 100,0 мл. 25,0 мл полученноro paCT
вора помещают в коническую колбу вмести
мостью 500 мл И доводят водой Р до объема
200 мл. Прибавляют около 50 Mr иHдиKaтop
ной смеси ксиленолов020 оранжев020 Р и 2eK
саметилентетрамина р до появления фио
летово розовоro окрашивания. Прибавляют 2 r
2ексаметилентетрамина Р в избытке. Титру
ют приroтовленным раствором натрия эдетата
до перехода от фиолетово розовоro окрашива
ния до желтоro.
1 мл 0,02 М раствора натрия эдетата co
ответствует 1,З08 Mr Zп.
# 0,02 М раствор ртути нитрата. 3003500.
6,85 r ртути (/1) нитрата Р растворяют в
20 мл 1 М раствора кислоты азотной и ДOBO
дят водой Р до объема 1000,0 мл.
Установка титра. 15,0 Mr натрия хлори
да Р растворяют в 50 мл воды Р и титруют при
rотовленным раствором ртути нитрата потен
циометрически (2.2.20), используя в качестве
индикаторноrо электрода платиновый или PTYT
ный, а в качестве электрода сравнения суль
фатртутный.
1 мл 0,02 М раствора ртути нитрата co
ответствует 2,З38 Mr NaCI.
0,1 М раствор серебра нитрата. 3005600.
17,0 r серебра нитрата Р растворяют в
воде Р и доводят до объема 1000,0 мл этим же
растворителем.
Установка титра. 0,100 r натрия хлорида
РО растворяют в 30 мл воды Р и титруют приro
товленным раствором серебра нитрата потенци
ометрически (2.2.20).
1 мл О, 1 М раствора серебра нитрата co
ответствует 5,844 Mr NaCI.
#Установка титра. Около 0,150 r натрия
хлорида РО растворяют в 50 мл воды Р и титру
ют приrотовленным раствором серебра нитрата
до образования KpacHoBaToro осадка, используя
в качестве индикатора калия хромат Р.
1 мл 0,1 М раствора серебра нитрата co
ответствует 5,844 Mr NaCI.
Хранят в защищенном от света месте.
0,001 М раствор серебра нитрата. 3009300.
5,0 мл 0,1 М раствора серебра нитрата
доводят водой Р до объема 500,0 мл.
0,5 М раствор серной кислоты. 3007800.
28 мл кислоты серной Р смешивают с
водой Р и доводят до объема 1000.0 мл этим же
растворителем.
Установка титра. 1.000 r натрия карбона
та РО растворяют в 50 мл воды р, прибавля
ют 0,1 мл раствора метилов020 оранжев020 Р
и титруют приroтовленным раствором кислоты
серной до появления красновато желтоro OKpa
шивания. Кипятят около 2 мин. Раствор снова
приобретает желтое окрашивание. Охлаждают и
титруют вновь до повторноro появления KpaCHO
вато желтоro окрашивания.
1 мл 0,5 М раствора кислоты серной соот
ветствует 53,00 Mr Nа 2 СО з .
758
rосударственная фармакопея Республики Беларусь
0,05 М раствор серной кислоты.
3008000.
100,0 мл 0,5 М раствора кислоты серной
доводят водой Р до объема 1000.0 мл.
Установка титра. Определение прово
дят, как указано для 0,5 М раствора кисло
ты серной, используя 0,100 r натрия карбо
ната РО, растворенноro в 20 мл воды Р
1 мл 0,05 М раствора кислоты серной
соответствует 5,30 Mr Nа 2 СО з .
0,1 М раствор свинца нитрата. 3003100.
33 r свинца нитрата Р растворяют в
воде Р и доводят до объема 1000,0 мл этим
же растворителем.
Установка титра. К 20,0 мл приroтов
ленноrо раствора свинца нитрата прибавляют
300 мл воды Р и проводят комплексометриче
ское определение свинца (2.5.11).
0,1 М раствор тетрабутиламмония rи
дроксида. 3008300.
40 r тетрабутиламмония йодида Р paCTBO
ряют в 90 мл метанола безводН020 Р, прибав
ляют 20 r тонко измельченноro серебра оксида
Р и энерrично встряхивают в течение 1 ч. ЦeH
трифуrируют несколько миллилитров смеси
и проводят испытание жидкости над осадком
на йодиды. При получении положительной pe
акции дополнительно прибавляют 2 r серебра
оксида Р и встряхивают в течение 30 мин. Эту
процедуру повторяют до тех пор, пока жидкость
не будет свободна от йодидов. Смесь фильтру
ют через стеклянный фильтр (2.1.2) и промыва
ют реакционный сосуд и фильтр тремя порция
ми, по 50 мл каждая, толуола Р. К полученному
фильтрату прибавляют промывной толуол и дo
водят толуолом Р до объема 1000,0 мл. Через
раствор пропускают сухой азот, свободный от
уrлерода диоксида, в течение 5 мин.
Установка титра. К 1 О мл диметилфор
мамида Р прибавляют 0,05 мл раствора 3 r/л
тимолов020 сине20 Р в метаноле Р и титруют
приrотовленным раствором тетрабутиламмо
ния rидроксида до появления чистоro синеro
окрашивания. Тотчас прибавляют 0,200 r Kиc
лоты бензойной РО, перемешивают до paCT
ворения и титруют приroтовленным paCTBO
ром тетрабутиламмония rидроксида снова до
появления синеro окрашивания. Титр paCT
вора тетрабутиламмония rидроксида YCTaHaB
ливают по объему титранта, израсходованно
ro в повторном титровании.
Титр устанавливают непосредственно
перед использованием.
1 мл 0,1 М раствора тетрабутиламмо
ния 2идроксида соответствует 12,21 Mr С 7 Н 6 О 2 .
0,1 М раствор тетрабутиламмония rи
дроксида в 2 пропаноле. 3008400.
rотовят, как указано для 0,1 М раствора
тетрабутиламмония 2идроксида, используя
в качестве растворителя 2 пропанол Р вместо
толуола Р. Титр устанавливают, как указано
для 0,1 М раствора тетрабутиламмония 2и
дроксида.
1 М раствор хлористоводородной кис
лоты. 3001800.
103,0 r кислоты хлористоводородной Р
доводят водой Р до объема 1000,0 мл.
Установка титра. 1,000 r натрия карбо
ната РО растворяют в 50 мл воды Р, прибав
ляют 0,1 мл раствора метилов020 оранже
в020 Р и титруют приroтовленным раствором
кислоты хлористоводородной до появления
красновато желтоro окрашивания. Кипятят в
течение 2 мин. Раствор снова приобретает
желтое окрашивание. Охлаждают и продол
жают титрование до появления KpaCHOBaTO
желтоro окрашивания.
1 мл 1 М раствора кислоты хлористово
дородной соответствует 53,00 Mr Nа 2 СО з .
0,1 М раствор хлористоводородной
кислоты. 3002100.
100,0 мл 1 М раствора кислоты хлори
стоводородной доводят водой Р до объема
1000,0 мл.
Установка титра. Проводят титрование,
как указано для 1 М раствора кислоты хло
ристоводородной, используя 0,100 r натрия
карбоната РО, растворенноro в 20 мл воды Р.
1 мл О, 1 М раствора кислоты хлористо
водородной соответствует 5,30 Mr Nа 2 СО з .
0,1 М раствор хлористо водородной
КИСЛОТbI в спирте. 3008800.
9,0 мл кислоты хлористоводородной Р
доводят 96 % спиртом, свободным от альде
2идов, Р до объема 1000,0 мл.
0,1 М раствор хлорной кислоты.
3003900.
8,5 мл кислоты хлорной Р помещают в
мерную колбу, содержащую около 900 мл Kиc
лоты уксусной ледяной Р, и перемешивают.
Прибавляют 30 мл УКСУСН020 аН2идрида Р,
доводят кислотой уксусной ледяной Р до
объема 1000,0 мл, перемешивают и выдержи
вают в течение 24 ч. Определяют содержание
воды (2.5.12) без прибавления метанола и при
необходимости, доводят содержание воды от
0,1 % до 0,2 % прибавлением УКСУСН020 aH
2идрида Р или воды Р Выдерживают в тече
ние 24 ч.
Установка титра. 0,350 r калия 2идроф
талата РО растворяют в 50 мл кислоты YKCYG
ной безводной Р, при необходимости осторож
но наrревая. Охлаждают, защищая от воздуха,
и титруют приroтовленным раствором кислоты
хлорной, используя в качестве индикатора 0,05
мл раствора кристалличеСК020 фиолетово
20 Р. Измеряют температуру раствора хлорной
4.2.2. Титрованные растворы
759
кислоты во время титрования. Если темпера
тура, при которой проводится количественное
определение, отличается от температуры, при
которой был установлен титр О, 1 М раствора
кислоты хлорной, Torдa объем V c ' необходи
мый для количественноro определения, вычис
ляют по формуле:
V .[1+(t 1 t2).0,0011],
rдe:
t 1 температура, при которой устанавли
вают титр;
t 2 температура, при которой проводят
количественное определение;
V объем, израсходованный на титрова
ние фактически, в миллилитрах.
1 мл О, 1 М раствора кислоты хлорной
соответствует 20,42 Mr С В Н 5 К0 4 .
0,05 М раствор хлорной кислоты.
3004000.
50,0 мл 0,1 М раствора кислоты хлорной
доводят кислотой уксусной безводной Р до
объема 100,0 мл.
0,1 М раствор уксусной кислоты.
3008900.
6,0 r кислоты уксусной ледяной Р ДOBO
дят водой Р до объема 1000,0 мл.
Установка титра. К 25,0 мл приroтовлен
ноro раствора кислоты уксусной прибавляют
0,5 мл раствора фенолфталеина Р и титруют
0,1 М раствором натрия 2идроксида.
0,1 М раствор церия сульфата. 3001100.
40,4 r церия (/V) сульфата Р растворяют в
смеси из 500 мл воды Р и 50 мл кислоты серной
Р. Охлаждают и доводят водой Р до объема
1000,0 мл.
Установка титра. К 25,0 мл приroтовлен
ноro раствора церия сульфата прибавляют 2,0 r
калия йодида Р, 150 мл воды Р и тотчас титру
ют 0,1 М раствором натрия тиосульфата, ис
пользуя в качестве индикатора 1 мл раствора
крахмала Р.
0,05 М раствор цинка хлорида. 3008500.
6,82 r цинка хлорида Р, взвешенноro с co
ответствующими предосторожностями, paCTBO
ряют в воде Р. При необходимости по каплям
прибавляют кислоту хлористоводородную раз
веденную Р до исчезновения опалесценции. Дo
водят водой Р до объема 1000,0 мл.
Установка титра. К 20,0 мл приrотов
ленноrо раствора цинка хлорида прибавляют
5 мл кислоты уксусной разведенной Р и прово
дят комплексометрическое определение цинка
(2.5.11).
0,1 М раствор цинка сульфата. 3008600.
29 r цинка сульфата Р растворяют в воде Р
и доводят до объема 1000,0 мл этим же раство--
рителем.
Установка титра. К 20,0 мл приroтов
ленноro раствора цинка сульфата прибавляют
5 мл кислоты уксусной разведенной Р и прово--
дят комплексометрическое определение цинка
(2.5.11).
5.1.1. Методы ПРU20товленuя стерильных продуктов
761
5. ОБЩИЕ ТЕКСТЫ
5.1. ОБЩИЕ ТЕКСТЫ
по МИКРОБиолоrии
01/2013:50101
5.1.1. МЕТОДЫ приrОТОВЛЕНИЯ
СТЕРИЛЬНЫХ ПРОДУКТОВ
Под стерильностью понимают отсутствие
жизнеспособных микроорrанизмов. Стериль
ность продукции не может rарантироваться ис
пытанием; она должна обеспечиваться про
изводственным процессом, валидированным
надлежащим образом. Важно, чтобы при изуче
нии воздействия выбранной процедуры стери
лизации на продукт (включая контейнер и упа
ковку) была подтверждена ее эффективность,
а также сохранение продукта. Рекомендуется
выбирать такой контейнер, чтобы иметь воз
можность использовать оптимальный режим
стерилизации. Малейшее нарушение валидиро
ванноro процесса стерилизации влечет за собой
риск получения нестерильноro или некачествен
ноro продукта. Повторную валидацию следует
проводить каждый раз при внесении изменений
в процедуру стерилизации, в том числе при из
менении объема продукта, стерилизуемоro за
один раз. При проектировании производствен
Horo процесса следует учитывать принципы Haд
лежащей производственной практики (GMP), в
том числе:
привлечение квалифицированноro персо
нала с соответствующей подrотовкой;
использование соответствующих помеще
ний;
использование соответствующеro произ
водственноro оборудования, сконструированно
ro таким образом, чтобы ero можно было леrко
чистить и стерилизовать;
применение соответствующих мер для
сведения к минимуму биолоrических заrрязне
ний до стерилизации;
использование валидированных процедур
на всех важнейших этапах производства;
мониторинr производспзенной среды и
процесса производства.
Предосторожности, необходимые для сни
жения биолоrической контаминации перед CTe
рилизацией, включают использование KOM
rюнентов с низкой степенью контаминации
IМикроорrанизмами. По отношению к составля
ющим, которые MOryT быть потенциально KOH
таминированными в силу их происхождения,
природы или способа подroтовки, желательно
применение микробиолоrическоro мониторинrа
и установление допустимых пределов микроб
ноro заrрязнения.
Методы, описанные ниже, применимы
2лавным образом к инактивации или удалению
бактерий, дрожжей и плесневых 2рибов. Для
биОЛ02ической продукции животН020 или чело
вечеСК020 происхождения или в случаях, К02да в
производственном процессе участвовал такой
материал, необходимо продемонстрировать
во время валидации, что процесс способен yдa
лить или инактивировать соответствующую
вирусную контаминацию.
По возможности следует выбирать процесс,
при котором продукция стерилизуется в контей
нере (конечная стерилизация). При использо
вании полностью валидированноro метода KO
нечной стерилизации стерилизации паром,
roрячим воздухом или ионизирующим излучени
ем по соrласованию с соответствующим KOM
петентным уполномоченным opraHoM допускает
ся параметрический выпуск серии стерильноro
продукта, при котором заключение о качестве
серии принимается на основании данных о про
цессе стерилизации, а не результатов испыта
ния на стерильность.
В тех случаях, Korдa конечная стерилиза
ция невозможна, используют фильтрацию через
фильтры, способные задержать бактерии, или
производство в асептических условиях. По воз
можности при этом следует проводить дополни
тельную обработку продукта (например, Harpe
вание) в контейнере. Во всех случаях контейнер
и укупорочное средство должны обеспечивать
стерильность продукта на протяжении Bcero
срока roдности.
Уровень rарантии стерильности (SAL
StеrШtу Assuraпce Level)
В тех случаях, коrда это возможно. для Me
тодов, приведенных ниже, указывают «уровень
rарантии стерильности» (SAL). Невозможно дo
казать, что стерильность достиrнута для каждой
из множества единиц, подверrнутых стерилиза
ции. Инактивация микроорrанизмов физически
ми или химическими методами подчиняется экс
поненциальному закону, следовательно, Bcerдa
существует конечная статистическая вероят
ность тоro, что микроорrанизм может выжить в
процессе стерилизации. Для каждоro KOHKpeTHO
ro процесса вероятность выживаl-lИЯ определя
ется количеством, типом и сопротивляемостью
присутствующих микроорrанизмов и средой, в
которой находятся орrанизмы во время обра
ботки. SAL процесса стерилизации представля
ет собой степень rарантии тоro, что процесс, о
котором идет речь, обеспечивает стерильность
762
rосударственная фармакопея Республики Беларусь
rруппы продукции. SAL конкретноro процесса BЫ
ражается как вероятность наличия нестерильно
ro продукта в этой rруппе. Например, SAL=1O 6
означает вероятность наличия не более одноro
жизнеспособноro микроорrанизма в 1.1 06 стери
лизованных единиц конечной продукции. SAL
процесса стерилизации для конкретноro продук
та устанавливается в ходе надлежащеro процес
са валидации.
МЕТОДЫ И УСЛОВИЯ СТЕРИЛИЗАЦИИ
Стерилизация может быть проведена одним
из описанных ниже методов. Возможно использо
вание модификаций или комбинаций этих Meтo
дов при условии проведения валидации выбран
ной процедуры стерилизации как в отношении
эффективности, так и в отношении целостно
сти продукта, в том числе контейнера или упа
ковки. Для всех методов стерилизации ведется
наблюдение за важнейшими стадиями процес
са для Toro, чтобы подтвердить, что установлен
ные ранее необходимые условия стерилизации
выдерживаются для всей серии продукции на
протяжении всеro процесса стерилизации. Это
применимо во всех случаях, включая случаи ис
пользования стандартных условий.
КОНЕЧНАЯ СТЕРИЛИЗАЦИЯ
При конечной стерилизации очень важно
учитывать неоднородность физических и, rдe
это уместно, химических условий внутри стери
лизационной камеры. Для каждоro варианта за
rрузки каждоro типа и размера контейнера или
упаковки следует определить положение в CTe
рилизационной камере, которое наименее дo
ступно для стерилизующеro фактора (напри
мер, самое холодное место в автоклаве). Для
подтверждения тоro, что все заrрузки проходят
предусмотренную обработку равным образом,
необходимо определить минимальную леталь
ность микроорrанизмов в ходе стерилизацион
ноro цикла и подтвердить воспроизводимость
цикла.
После окончательноro выбора режима KO
нечной стерилизации необходимо, rде это воз
можно, собирать сведения о качестве процес
са в повседневном использовании при помощи
мониторинrа и ведения записей о физических и,
если это уместно, химических условиях, полу
ченных в камере при каждом стерилизационном
цикле.
Паровая стерилизация (автоклавирова-
ние). Стерилизация насыщенным паром под
давлением, rде это возможно, в особенности для
rOToBbIx лекарственных средr::тв в виде водных
растворов. Стандартные условия при таком
методе конечной стерилизации rOTOBbIX пекар
ственных средств в виде водных растворов сле
дующие: проrревание при температуре не менее
121 ос в течение 15 мин. Допускается использо
вание друrих сочетаний времени и температуры,
если предварительно доказано, что выбранный
режим обеспечивает необходимый и воспроиз
водимый уровень летальности микроорrаниз
мов в рамках установленных допусков. Исполь
зуемые процедуры и меры предосторожности
должны обеспечивать SAL не более 10 6. PeKO
мендации по проведению валидации с исполь
зованием Fo концепции приведены ниже (5.1.5).
В ходе процесса стерилизации следует pe
rистрировать физические условия (температу
ра и давление) в автоклаве. Температуру, как
правило, измеряют при помощи датчиков TeM
пературы, помещенных в контрольный контей
нер. Дополнительные термоэлементы помеща
ют в области заrруженной камеры с наименьшей
температурой (определенные предварительно).
,Условия в каждом цикле стерилизации должны
соответствующим образом реrистрироваться,
например, в виде температурно временной диа
rpaMMbI или друrим способом.
Если предусмотрена биолоrическая оценка,
ее проводят С использованием подходящих био
лоrических индикаторов (5.1.2).
Сухожаровая стерилизация (стерилиза-
ция rорячим воздухом). Для данноro метода
конечной стерилизации стандартными услови
ями являются: проrревание при температуре не
менее 160 ос в течение не менее 2 часов. Дo
пускается использование друrих сочетаний Bpe
мени и температуры, если предварительно дo
казано, что выбранный режим обеспечивает
необходимый и воспроизводимый уровень ле
тальности микроорrанизмов в рамках YCTaHOB
ленных допусков. Используемые процедуры и
меры предосторожности должны обеспечивать
SAL не более 10 6.
Сухожаровая стерилизация проводится в
сухожаровом шкафу, оснащенном устройством
для принудительной циркуляции Bo yxa, или
при помощи друroro оборудования, специаль
но предназначенноrо для этой цели. Стерилиза
тор заrружают таким образом, чтобы обеспечить
однородность температуры в пределах всей за
rрузки. Температуру, как правило, измеряют при
помощи датчиков температуры, помещенных в
контрольный контейнер. Дополнительные Tep
моэлементы помещают в области заrруженной
камеры с наименьшей температурой (опреде
ленные предварительно). В ходе каждоro цикла
стерилизации реrистрируют температуру подхо
дящим способом.
Если предусмотрена биолоrическая оценка.
ее проводят с использованием подходящих био
лоrических индикаторов (5.1.2).
Сухожаровая стерилизация IlрИ температу
ре более 220 ос часто используются для стери
лизации и депироrенизации изделий из стекла.
В этом случае в качестве замены биолоrиче
ским индикаторам может быть доказано сниже
ние термостойких бактериальных эндотоксинов
на три порядка (5.1.2).
5.1.1. Методы ПРU20товления стериЛЬНblХ продуктов
763
Радиационная стерилизация. Стерилиза
ция этим методом достиrается при помощи воз
действия на продукт ионизирующеro излучения
в форме rамма излучения от соответствующе
ro радиоизотопноro источника (такоro как KO
бальт 60) или пучка электронов, подаваемоro
соответствующим ускорителем электронов.
В некоторых странах существуют распо
ряжения, ре2ламентирующие использование
ионизирующе20 излучения для стерилизации,
например, в соответствующих Руководящих
указаниях ЕвропеЙСК020 Сообщества.
Для этоrо метода конечной стерилиза
ции стандартная поrлощенная доза составляет
25 Krp. Допускается использование друrих доз,
если предварительно доказано, что выбранный
режим обеспечивает необходимый и воспроиз
водимый уровень летальности микроорrаниз
мов в рамках установленных допусков. Исполь
зуемые процедуры и меры предосторожности
должны обеспечивать SAL не более 10 6.
В процессе стерилизации следует постоян
но осуществлять измерения поrлощенноrо про
дуктом излучения при помощи установленных
дозиметрических методов, не зависящих от доз.
Дозиметры должны проходить калибровку по OT
ношению к стандартному источнику на эталон
ной радиационной установке непосредственно
после получения от поставщика, а затем через
определенные промежутки времени с периодич
ностью не реже одноro раза в roд.
Если предусмотрена биолоrическая оценка,
ее проводят с использованием подходящих био
лоrических индикаторов (5.1.2).
rазовая стерилизация. Этот метод CTe
рилизации может использоваться только в
случае, если нет подходящей альтернативы.
При этом должно быть обеспечено проникно
вение rаза и влаrи в стерилизуемый продукт, а
также последующее удаление rаза способом,
позволяющим снизить концентрацию rаза и
продуктов ero превращения в стерилизуемом
продукте до уровня, не вызывающеro токсиче
ских эффектов при использовании продукта.
Соответствующие рекомендации относительно
использования этиленоксида приводятся, Ha
пример, в Руководящих указаниях Европейско
ro Сообщества.
Там, rдe это возможно, необходимо в ходе
стерилизации вести измерение и реrистрацию
концентрации rаза, относительной влажности,
температуры и продолжительности процесса.
Измерения проводятся там, rде наименее Be
роятно достижение условий стерилизации, как
установлено при валидации.
Эффективность процесса для каждой за
rрузки проверяется с помощью использования
подходящеro биолоrическоro индикатора (5.1.2).
Перед выпуском каждой серии необходимо
проводить контроль стерильности на COOTBeT
ствующем количестве образцов (2.6.1).
#Проведение данною вида стерилизации
возможно в следующих условиях:
окись этилена стерилизующая доза
1200 мr/дм З , температура стерилизации не
менее 18 ОС, относительная влажность 80 %,
время стерилизационной выдержки 16 ч (пор
тативный аппарат);
смесь ОБ (смеси окиси этилена и броми
стоro метила в весовом соотношении (1 :2,5):
а) стерилизующая доза 2000 мr/дм З , TeM
пература стерилизации 55 ОС, относительная
влажность 80 %, время стерилизационной BЫ
держки 4 ч;
Ь) стерилизующая доза 2000 мr/дм З , темпе
ратура стерилизации не менее 18 ОС, относи
тельная влажность 80 %, время стерилизацион
ной выдержки 16 ч при той же относительной
влажности (портативный аппарат).
Допускается использование друrих режи
мов rазовой стерилизации, обеспечивающих
стерильность и сохранность объекта. Режим
должен быть обоснован и указан в нормативной
документации.
Стерилизуемые изделия упаковываются в
пакеты из полиэтиленовой пленки толщиной от
0,06 мм до 0,2 мм, перrамента и др. Метод peKO
мендован для изделий из резины, полимерных
материалов, стекла, металла.
В связи с токсичностью окиси этилена и бро
мистоro метила применение стерилизованных
этими rазами изделий допускается только после
их деrазации, т. е. выдержки в вентилируемом
помещении до допустимых остаточных коли
честв, указанных в нормативной документации.
Условия деrазации зависят от назначения,
способа применения, размеров изделий и упа
ковки и указываются в нормативной ДOKYMeHTa
ции на изделие.#
ФИЛЬТРАЦИЯ
Некоторые активные инrредиенты и продук
ты, которые не подлежат конечной стерилиза
ции, MOryт быть подверrнуты процедуре филь
трации через фильтры, приroдность которых
предварительно доказана путем микробиоло
rических испытаний с использованием подхо
дящих тест микроорrанизмов, например, Pseu
dотопаs diтiпиtа (АТСС 19146, NC/MB 11091,
С/Р 103020, #ССМ 4623#). При испытании peKO
мендуется использовать концентрацию микро
орrанизмов не менее 107 КОЭ/см" активной по
верхности фильтра и приrотовление суспензии в
триптон соевом бульоне, который после прохож
дения через срильтр собирается асептическим
способом и инкубируется аэробно при темпера
туре 32 0 С. Такая процедура тrебует соблюдения
мер предосторожности. Орrанизация производ
ственноro процесса и состояние окружающей
среды должны сводить к минимуму риск микроб
ноro заrрязнения, их следует реryлярно подвер
raTb надлежащему мониторинry. Оборудование,
контейнеры, укупорочные средства и, rде это
764
rосударственная фармакопея Республики Беларусь
возможно, инrредиенты должны подверrаться
надлежащей стерилизации. Рекомендуется про
водить процесс фильтрации непосредственно
перед стадией наполнения контейнера. Опера
ции, которые следуют за стадией фильтрации,
необходимо проводить в асептических условиях.
Растворы пропускают через мембранные
фильтры (с номинальным размером пор не
более 0,22 мкм), способные задерживать бакте
рии, или через фильтры друrих типов, не YCTY
пающие им по эффективности. Необходимо
предпринимать надлежащие меры для предот
вращения потерь растворенноro вещества в pe
зультате адсорбции на фильтре, а также BЫCBO
бождения удержанных фильтром заrрязнений.
Следует учитывать уровень биозаrрязнений до
начала фильтрации, пропускную способность
фильтра, объем серии и продолжительность
фильтрации. Фильтр не должен использовать
ся в течение более длительноro времени, чем
утверждено при валидации данноro фильтра в
сочетании с конкретным фильтруемым лекар
ственным средством.
Целостность собранноro стерилизующеrо
фильтра должна проверяться перед использо
ванием и подтверждаться после использования
путем проведения испытаний, соответствующих
типу используемоrо фильтра и этапу испытания,
например, испытания на определение насыще
ния, на удержание давления или скорости диф
фузии.
Так как метод стерилизующей фильтрации
имеет дополнительные потенциальные факто
ры риска по сравнению с друrими процессами
стерилизации, рекомендуется проводить пред
варительную фильтрацию через удерживающий
бактерии фильтр в тех случаях, Korдa низкий
уровень биозаrрязнений не может быть обеспе
чен друrими средствами.
ПРОИЗВОДСТВО В АСЕПТИЧЕСКИХ
УСЛОВИЯХ
Целью производства в асептических усло
виях является сохранение стерильности про
дукта, изroтовленноro из компонентов, каждый
из которых был предварительно стерилизован
одним из методов, описанных выше. Это дости
rается путем использования условий и оборудо
вания, предназначенных для предотвращения
микробноro заrрязнения. В асептических усло
виях MOryT осуществляться такие стадии про
изводственноro процесса, как наполнение KOH
тейнеров и укупорка, смешивание инrредиентов
с последующим асептическим наполнением и
асептической укупоркой.
Для сохранения стерильности компонентов
состава и rOToBoro лекарственноrо средства в
ходе производственноrо процесса особое вни
мание следует уделять:
состоянию производственной среды;
персоналу;
критическим поверхностям;
стерилизации контейнеров/укупорочных
средств и передаточным процедурам;
предельному времени хранения лекар
ственноro средства до наполнения контейнера.
Валидация процесса включает надлежа
щую проверку всеro вышеперечисленноro, а
также систематический контроль при помощи
имитационных испытаний с использованием пи
тательной среды, которую инкубируют и иссле
дуют на наличие микробноro заrрязнения (испы
тания на заполнение питательными средами).
Кроме тоro, перед выпуском серии любоro ле
карственноro средства, стерилизованноro MeTO
дом фильтрации или изroтовленноro в асептиче
ских условиях, следует проводить испытание на
стерильность для соответствующеro количества
образцов (2.6.1).
01/2013:50102
5.1.2. БиолоrИЧЕСКИЕ
ИНДИКАТОРЫ
СТЕРИЛИЗАЦИИ
Биолоrические индикаторы представляют
собой стандартизованные препараты опреде
ленных микроорrанизмов, используемые для
оценки эффективности процедуры стерили
зации. Они обычно представляют собой попу
ляцию спор бактерий, нанесенных на подхо
дящий инертный носитель. Инокулированный
носитель изолируют таким образом, чтобы
предотвратить ero повреждение или заrрязне
ние, но в то же время обеспечить контакт CTe
рилизующеro areHTa с микроорrанизмами. Cy
спензии спор MorYT находиться в rерметично
закрытых ампулах. Биолоrические индикато
ры roтовят таким образом, чтобы существова
ла возможность их хранения при определен
ных условиях, при этом должен быть указан
срок roдности.
Микроорrанизмы тоro же вида, что и бак
терии, используемые в производстве биоло
rических индикаторов, MorYT инокулироваться
непосредственно в жидкий продук предна
значенный для стерилизации, или в жидкий
продукт, аналоrичный тому, который предна
значен для стерилизации. В этом случае He
обходимо доказать, что жидкий продукт не
оказывает инrибирующеrо воздействия на ис
пользуемые споры, особенно на их прораста
ние.
Биолоrический индикатор должен иметь
следующие характеристики: название вида
бактерий, используемых в качестве эталон
ных микроорrанизмов, номер штамма в ис
ходной коллекции, количество жизнеспо
собных спор, приходящееся на носитель, и
5.1.2. БиОЛО2ические индикаторы стерилизации
765
величину О. Величина О параметр стерили
зации (продолжительность или поrлощенная
доза), обеспечивающий снижение числа жиз
неспособных микроорrанизмов до 1 О % от ис
ходноro числа. Данная величина имеет смысл
для точно определенных экспериментальных
условий стерилизации. Биолоrический инди
катор может содержать несколько видов бак
терий на одном носителе. Должна быть указа
на информация о необходимой питательной
среде и условиях инкубации.
Индикаторы рекомендуется размещать
в областях, наименее доступных для стери
лизующеrо areHTa. Эти области определя
ют эмпирически или на основании предва
рительных физических измерений, если это
возможно. Можно использовать биолоrиче
ские индикаторы, которые включают ампулу с
питательной средой, помещенную в упаковку,
защищающую инокулированный носитель.
Выбор тест микроорrанизмов для биоло
rических индикаторов должен проводиться с
учетом следующих требований:
а) сопротивляемость тест штамма должна
быть приroдной для конкретноro метода CTe
рилизации и должна быть более высокой по
сравнению с сопротивляемостью микроорrа
низмов, потенциально контаминирующих про
дукт;
Ь) тест штамм не должен быть патоrен
ным;
с) тест штамм должен леrко культивиро
ваться.
Наличие после инкубации роста эталон
ных микроорrанизмов, подверrшихся проце
дуре стерилизации, указывает на то, что эта
процедура неудовлетворительна.
Паровая стерилизация. Для валида
ции циклов стерилизации рекомендуется ис
пользование биолоrических индикаторов.
предназначенных для стерилизации паром.
Рекомендуются споры Geobacillus stearother
mophilus (например, АТСС 7953, NCTC 10007,
NC/MB 8157 или С/Р 52.81) или иных штам
мов микроорrанизмов, имеющих эквивалент
ные характеристики. Число жизнеспособных
спор должно превышать 5.105 на носитель.
Величина О при температуре 121 ос должна
составлять не менее 1,5 мин. При обработке
биолоrических индикаторов паром при темпе
ратуре (121:t1) ос в течение 6 мин должно Ha
блюдаться сохранение жизнеспособных спор,
а обработка при той же температуре в тече
ние 15 мин должна приводить к полной rибели
эталонных тест микроорrанизмов.
Сухожаровая стерилизация. Для при
rотовления биолоrических индикаторов peKO
мендованы споры Bacillus atrophaeus (напри
мер, АТСС 9372, NC/MB 8058 или С/Р 77.18)
или иных штаммов микроорrанизмов, име
ющих эквивалентные характеристики. Число
жизнеспособных спор должно быть более
1.106 на носитель. Величина О при температу
ре 160 ос должна составлять не менее 2,5 мин.
Для стерилизации и депироrенизации CTe
клянной посуды часто используется rорячий
воздух при температуре более 220 ОС. В этом
случае в качестве замены биолоrическим ин
дикаторам может быть доказано снижение
термостойких бактериальных эндотоксинов
на три порядка.
Радиационная стерилизация. Биоло
rические индикаторы MorYT при меняться для
мониторинrа текущих операций в качестве дo
полнительной возможности оценки эффектив
ности установленной дозы излучения, oco
бенно в случае стерилизации ускоренными
электронами. Рекомендуются споры Bacillus
pumi/us (например, АТСС 27142, NCTC 10327,
NC/MB 10692 или С/Р 77.25) или иных штам
мов микроорrанизмов, имеющих эквивалент
ные характеристики. Число жизнеспособных
спор должно быть более 1 .107 на носитель. Be
личина О должна составлять не менее 1,9 Krp.
Необходимо убедиться, что после облучения
биолоrическоro индикатора дозой 25 Krp (ми
нимальная поrлощенная доза) не наблюдает
ся роста эталонных микроорrанизмов.
rазовая стерилизация. Для всех проце
дур rазовой стерилизации необходимо исполь
зование биолоrических индикаторов как для
валидации циклов, так и для каждодневной
работы. rазовая стерилизация широко приме
няется для стерилизации медицинских приспо
соблений, изоляторов, камер и др., но такое
использование не рассматривается в рамках
Фармакопеи. Для этиленоксида peKOMeHДY
ется использовать споры Bacillus atrophae
us (например, АТСС 9372, NC/MB 8058 или
С/Р 77.18) или иных штаммов микроорrаниз
мов, имеющих эквивалентные характеристики.
#для перекиси водорода и перуксусной кисло
ты споры Geobacillus stearothermophi/us (Ha
пример, АТСС 7953, NCTC 10007, NC/MB 8157
или С/Р 52.81 )#. Число жизнеспособных спор
должно быть более 1.1 Об на носитель. Для
каждой конкретной процедуры должны быть
известны пара метры сопротивляемости TeCT
микроорrанизма, например, для этиленокси
да величина О должна составлять не менее
2,5 мин для цикла стерилизации со следую
щими параметрами: концентрация этиленокси
да 600 мr/л, температура 54 ос и относитель
ная влажность 60 %. Следует убедиться, что
после 25 минутноro цикла стерилизации с YKa
занными параметрами рост эталонных микро
орrанизмов не наблюдается, Torдa как после
50 минутноro цикла при более низкой темпера
туре (600 мr/л, 30 ОС, относительная влажность
60 %) жизнеспособность спор сохраняется.
766
01/2013:50103
rосударственная фармакопея Республики Беларусь
5.1.3. ЭФФЕКТИВНОСТЬ
АНТИМИКРОБНЫХ
КОНСЕРВАНТОВ
Если лекарственное средство само по
себе не обладает антимикробной активностью,
в еro состав MorYT быть введены антимикроб
ные консерванты, в особенности в roтовые ле
карственные средства в виде водных paCTBO
ров для предотвращения размножения или
оrраничения контаминации микроорrанизма
ми, которая может про изойти с продуктами при
нормальных условиях хранения и использова
ния, особенно с контейнерами на большое KO
личество доз, и которая может представлять
опасность для пациента. Антимикробные KOH
серванты не должны при меняться в качестве
альтернативы Надлежащей производственной
практики (GMP).
Эффективность антимикробных KOHcepBaH
тов может усиливаться или ослабляться в pe
зультате взаимодействия с действующим Be
ществом или друrими компонентами roтовою
лекарственноro средства, а также с упаковочны
ми или укупорочными материалами. Поэтому на
протяжении срока roдности следует контролиро
вать антимикробную активность roтовоro лекар
ственною средства, хранящеroся в контейнере,
с целью доказательства тоro, что она не снижа
ется в процессе хранения. Такие исследования
должны проводиться на образцах, извлеченных
из контейнера непосредственно перед испыта
нием.
На стадии разработки лекарственноro cpeд
ства необходимо доказать, что антимикробная
активность самоro лекарственною средства
или, при необходимости, лекарственноro cpeд
ства с консервантом обеспечивает надлежащую
защиту от нежелательных воздействий, которые
MOryт быть результатом микробною заrрязнения
лекарственноro средства или размножения в
нем микроорrанизмов в процессе хранения или
использования.
Эффективность антимикробной активности
может быть продемонстрирована при помощи
приведенноrо ниже испытания. Данное испы
тание не предназначено для использования в
целях повседневноro контроля.
ИСПЫТАНИЕ НА ЭФФЕКТИВНОСТЬ
АНТИМИКРОБНЫХ КОНСЕРВАНТОВ
Испытание состоит во внесении в roтовое
лекарственное средство, по возможности Haxo
дящееся в контейнере, необходимоro количе
ства подходящих микроорrанизмов и хранении
инокулированноro образца при указанной ниже
температуре, извлечении проб из инокулирован
ных образцов через определенный промежуток
времени и определении в нем числа микроорrа
низмов.
Эффективность консерванта в ютовом ле
карственном средстве считают удовлетвори
тельной, если в условиях проведения испыта
ний при хранении инокулированнных образцов
при заданной температуре в течение указан
ных промежутков времени наблюдается значи
тельное уменьшение числа микроорrанизмов.
Критерии оценки, показывающие уменьше
ние числа микроорrанизмов за определенный
период времени, зависят от требуемой степе
ни защиты roтовоro лекарственноro средства
(таблицы 5.1.З. 1 5.1.3. 3).
Тест миКРООР2анизмы
Рsеиdотопаs
ae,ugiпosa
Staphy/ococcus
au,eus
АТСС 9027; NC/MB 8626;
С/Р 82.118; #rиск 453#
АТСС 6538; NCTC 10788;
NC/MB9518; С/Р4.83;
#АТСС 6538 P (FOA 209 P)#
АТСС 10231; NCPF3179;
/Р48.72; #ATCC 885 653#
АТСС 16404; /М/149007;
/Р 143 1.83; #BKMF 1119;
ВКПМ F 824'
Caпdida
а/Ысапs
Aspergillus
brasilieпsis
Необходимо применять один штамм бакте
рий, выбранные микроорrанизмы MorYT допол
няться друrими штаммами или видами, KOTO
рые MorYT представлять вероятное микробное
заrрязнение для лекарственноro средства.
Например, Esche,ichia co/i (АТСС 8739; NC/MB
8545; С/Р 53.126; #АТСС 25922') использует
ся при испытании всех лекарственных средств
для оральноrо применения, а Zygosaccha
roтyces rouxii (NCYC 381; /Р 2021.92)
для лекарственных средств для оральноro
применения с высокой концентрацией сахара.
Подrотовка noceBHoro материала
Предварительно перед испытанием CBe
жевыращенную исходную культуру каж
доrо микроорrанизма из указанных TeCT
микроорrанизмов пересевают на поверхность
аrаризованной среды на основе rидролизата
казеина и соевых бобов (2.6.12) в случае BЫ
ращивания бактерий или декстрозноro arapa
Сабуро без добавления антибиотиков (2.6.12)
в случае выращивания rрибов. Культуры бакте
рий инкубируют при температуре 30 OC 35 ос
в течение 18 24 ч; культуру С. а/Ысапs инку
бируют при температуре 20 OC 25 ос в тече
ние 48 ч и культуру А. ЬrаsШепsis при TeM
пературе 20 OC 25 ос в течение 7 дней или до
получения хорошо развитых спор. Для получе
ния чистой культуры микроорrанизмов MorYT
потребоваться пересевы, однако peKOMeHДY
ется оrраничиться их минимально возможным
числом.
Для приroтовления суспензии бактериаль
ных культур и культуры С. a/bicaпs микробную
массу смывают с питательной среды стериль
5. 1. 3. Эффективность антимикробных консервантов
767
ной суспендирующей жидкостью, содержащей
9 r/л натрия хлорида Р, и переносят в подхо
дящий сосуд. Затем с помощью той же жид
кости доводят содержание микроорrанизмов
дО 10 В в миллилитре. Для приroтовления cy
спензии культуры А. Ьrаsiliепsis используют
стерильную суспендирующую жидкость, co
держащую 0,9 r/л хлорида натрия Р и 0,5 r/л
полисорбата 80 Р, и с ее помощью доводят
содержание спор до приблизительно 10 В в
одном миллилитре.
Из каждой суспензии немедленно отбира
ют подходящий образец и определяют число
колониеобразующих единиц в 1 мл (КОЭ/мл)
каждой суспензии методом прямоrо посева
на чашки или методом мембранной фильтра
ции (2.6.12). Полученное значение служит
для определения числа жизнеспособных ми
кроорrанизмов в инокуляте и сходноrо числа
микроорrанизмов при проведении испытания.
Инокулят следует использовать немедленно
после приroтовления.
МЕТОДИКА
Для подсчета жизнеспособных микроорrа
низмов в инокулированных образцах использу
ют ту же плотную питательную среду, которая
была использована для первоначальноro Bыpa
щивания соответствующих микроорrанизмов.
Необходимо инокулировать каждый из
контейнеров с испытуемым лекарствен
ным средством суспензией одноro из тест
микроорrанизмов до концентрации 1 05 106 КОЭ
в 1 мл или 1 r лекарственноro средства. Объем
суспензии должен составлять не более 1 % от
объема образца. Образец тщательно переме
шивают для обеспечения равномерноro распре
деления микроорrанизмов.
Инокулированные образцы лекарствен
ноro средства выдерживают при температуре
20 ос 25 ос в защищенном от света месте. Из
каждоro испытуемоro образца отбирают пробы
(обычно 1 мл или 1 r) непосредственно после
инокуляции и через подходящие интервалы
времени в соответствии с типом лекарствен
ноro средства и определяют число жизнеспо
собных микроорrанизмов методом посева на
чашки или методом мембранной фильтрации
(2.6.12). Антимикробную активность roтовоro
лекарственноro средства следует устранить
путем разведения, фильтрации или с помощью
подходящеro инактиватора. При использовании
разведения надлежащим образом учитывают
уменьшение чувствительности при обнаруже
нии малых количеств живых микроорrанизмов.
При использовании инактиватора следует под
твердить путем контрольных опытов, что еro
присутствие не влияет на жизнеспособность
микроорrанизмов.
Следует подтвердить приroдность методики
ДЛЯ доказательства требуемоro снижения числа
-изнеспособных микроорrанизмов.
КРИТЕРИИ ПРИЕМЛЕМОСТИ
Критерии оценки антимикробной активности
приведены в таблицах 5.1.3. 1 5.1.3. 3 в виде
лоrарифмическоro уменьшения числа жизне
способных микроорrанизмов относительно зна
чения, полученноro для инокулята.
Таблица 5.1.3. 1
Лекарственные средства для парентералЬН020,
офтаЛЬМОЛ02ичеСК020, внутриматОЧН020
и интрамаммарН020 применения
Ig уменьшение
бч 24ч 7 14 28
СУТ сут сут
Бактерии А 2 3 НВ
В 1 3 НУ
rрибы А 2 НВ
В 1 НУ
НВ нет выявления.
НУ нет увеличения живых микроорrанизмов по сравнению
с предыдущим снятием показаний.
Критерий А соответствует рекомендованной
эффективности. Если обосновано, что критерий А
не может быть достиrнут, например, по причи
не повышенноro риска неблаroприятных воздей
ствий, лекарственное средство должно удовлет
ворять критерию В.
Таблица 5.1.3. 2
Лекарственные средства для УШН020,
назаЛЬН020, местН020 применения, иН2аляци
онные лекарственные средства
19 уменьшение
2сут 7сут 14 28
СУТ сут
А 2 3 НУ
Бактерии В 3 НУ
А 2 НУ
rрибы в 1 НУ
НУ нет увеличения живых микроорrанизмов по
сравнению с предыдущим снятием показаний.
Критерий А соответствует рекомендованной
эффективности. Если обосновано, что критерий
А не может быть достиrнут, например, по причи
не повышенноro риска неблаroприятных воздей
ствий, лекарственное средство должно удовлет
ворять критерию В.
Таблица 5.1.3. 3
Лекарственные средства для слизистой
оболочки полости рта, для ораЛЬН020,
ректаЛЬН020 применения
19 уменьшение
14 СУТ 28 СУТ
Бактерии 3 НУ
rрибы 1 НУ
НУ нет увеличения живых микроорrанизмов по сравнению
с предыдущим снятием показаний.
Приведенные критерии соответствуют pe
комендованной эффективности.
01/2013:50104
rосударственная фармакопея Республики Беларусь
768
5.1.4. МИКРОБиолоrИЧЕСКАЯ
ЧИСТОТА НЕСТЕРИЛЬНЫХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
И ФАРМАЦЕ ТИЧЕСКИХ
СУБСТАНЦИИ
Присутствие некоторых микроорrаниз
мов в нестерильных лекарственных cpeд
ствах потенциально может уменьшить Tepa
певтическую активность продукта или даже
инактивировать ero, кроме этоro существует
возможность неблаroприятноro воздействия
на орrанизм пациента. Поэтому производи
тели должны обеспечить выполнение дей
ствующеrо руководства по Надлежащей про
изводственной практике при производстве,
хранении и распространении лекарственных
средств для rарантирования их низкой биоло
rической заrрязненности.
Микробиолоrическое испытание HeCTe
рильных лекарственных средств проводят в co
ответствии с методами, приведенными в общих
статьях 2.6.12 и 2.6.13. Критерии приемлемости
для нестерильныхлекарственныхсредств, бази
рующиеся на общем количестве аэробов (ОКА)
и на общем количестве rрибов (ОКП,
приведены в таблицах 5.1.4. 1 и 5.1.4. 2.
Критерии приемлемости основываются на OT
дельных результатах или на средних резуль
татах параллельных подсчетов в случае их
проведения (например, в методах чашечноrо
подсчета).
В случае указания критерия приемлемости
для микробиолоrической чистоты, он интерпре
тируется следующим образом:
101 КОЕ: максимально допустимое
число = 20;
102 КОЕ: максимально допустимое
число = 200;
103 КОЕ: максимально допустимое число =
2000 и так далее.
В таблице 5.1.4. 1 присутствует перечень
специфицированных микроорrанизмов, дЛЯ KO
торых установлены критерии приемлемости.
Данный список не является обязательно исчер
пывающим, и для некоторых лекарственных
средств может быть необходимым включение
испытания на друrие микроорrанизмы в зависи
мости от природы исходноro материала и про
цесса производства.
Если показано, что ни одно из описанных ис
пытаний не даст достоверное значение содержа
ния микроорrанизмов на необходимом уровне,
используют валидированный метод с пределом
обнаружения как можно более близким к заяв
ленному критерию приемлемости.
Значимость друrих обнаруженных микро
орrанизмов, кроме перечисленных в таблице
5.1.4. 1, оценивается следующими условиями:
применение продукта: опасность варьиру
ет в зависимости от пути введения (rлаза, нос,
дыхательный тракт);
природа продукта: еro способность под
держивать рост, наличие подходящеro антими
кробноrо консерванта;
способ применения;
Таблица 5.1.4 1
Критерии приемлемости для микробиОЛ02ической чистоты нестерильных лекарственных средств
Общее количе- Общее коли-
Способ применения ство аэробов чество rрибов Специфицированные
(ОКА), KOE/r (ОКП, KOE/r микроорrанизмы
или КОЕ/мл или КОЕ/мл
Неводные лекарственные cpeд 103 102 Отсутствие Esche,ichia co/i
ства для внутреннеro применения (1 r или 1 мл)
Водные лекарственные средства 102 101 Отсутствие Esche,ichia co/i
для внутреннеro применения (1 r или 1 мл)
Ректальный 103 102
Для использования на слизистой
оболочке ротовой полости Отсутствие Staphy/ococcus
Для десенноro использования 102 101 au,eus (1 r или 1 мл)
Для кожноro использования Отсутствие Pseudomoпas
Для назальноro использования ае,иgiпоsа (1 r или 1 мл)
Для ушноro использования
Отсутствие Рsеиdотопаs
ae,ugiпosa (1 r или 1 мл)
Для ваrинальноro применения 102 101 Отсутствие Staphy/ococcus
aureus (1 r или 1 мл)
Отсутствие Сапdidа а/Ысапs
(1 r или 1 мл)
Т рансдермальные пластыри (на Отсутствие Staphy/ococcus
один пластырь, включая клейкую 102 101 au,eus (1 r или 1 мл)
поверхность и подложку) Отсутствие Рsеиdотопаs
ае,иgiпоsа (1 r или 1 мл)
5.1.4. Микробиолоzическая чистота нестерильных лекарственных средств
769
Способ применения
Общее количе
ство аэробов
(ОКА), KOE/r
или КОЕ/мл
Для инrаляционноro применения
(для лекарственных средств для
распыления применяются специ
альные требования)
102
Специальные меры предосторож
ности Фармакопеи для roтовых ле
карственных средств для орально
ro применения, в состав которых
входят фармацевтические суб
станции и вспомоrательные Be
щества природноrо (животноro,
растительноro или минеральноro)
происхождения, для которых пред
варительная антимикробная обра
ботка невозможна и в отношении
которых компетентный уполномо
ченный opraH допускает микроб
ное заrрязнение исходноro MaTe
риала более 103 жизнеспособных
микроорraнизмов в 1 r или в 1 мл
1Q4
предполаrаемый реципиент: риск может
отличаться для новорожденных, детей и ослаб
ленных людей;
использование иммунодепрессантных
средств, кортикостероидов;
наличие заболевания, раны, повреждение
opraHoB.
rде это обосновано, специально подroтов
ленным в области микробиолоrии и интерпре
тации микробиолоrических данных персона
лом проводится оценка рисков релевантных
факторов. В случае сырья, при оценке учи
тывают процессы, которым подверrается про
дукт, действующая технолоrия испытаний и
доступность материалов требуемоro каче
ства.
Таблица 5.1.4. 2
Критерии приемлемости для микробиОЛО2иче
ской чистоты нестерильнЬ1Х субстанций
для фармацевтичеСК020 использования
Общее количе- Общее коли
ство аэробов чество rpибов
(ОКА), KOElr (OKr), KOElr
или КОElмл или КОElмл
Субстанции для
фармацевтиче 103 102
скоro использо
вания
97 З-к 1712.
Общее коли-
чество rpибов
(OKr), KOE/r
или КОЕ/мл
Специфицированные
микроорrанизмы
101
Отсутствие Staphy/ococcus
au,eus (1 r или 1 мл)
Отсутствие Рsеиdотопаs
ae,ugiпosa (1 r или 1 мл)
Отсутствие rрамотрицатель
ных бактерий, толерантных к
желчи (1 r или 1 мл) I/либо OTCYТ
ствие бактерий семейства Eп
terobacteriaceae (1 r или 1 мл)
при испытании в COOTBeT
ствии с альтернативной MeTO
дикой определения#
Не более 102 КОЕ rрамотри
цательных бактерий, толе
рантных к желчи (1 r или 1 мл)
I/либо не более 102 КОЕ бак
терий семейства Eпterobac
teriaceae (1 r или 1 мл) при
испытании в соответствии с
альтернативной методикой
определения#
Отсутствие Sa/moпe//a (1 О r
или 10 мл)
Отсутствие Esche,ichia соП
(1 r или 1 мл)
Отсутствие Staphy/ococcus
au,eus (1 r или 1 мл)
102
#Наличие и количественное содержание
специфицированных микроорrанизмов в суб
станциях для фармацевтическоro использова
ния, если нет друrих указаний в частных статьях,
определяется в следующих случаях.
1. Субстанции для фармацевтичеСК020
использования для производства стериль
НЬ1Х лекарственнЬ1Х средств, а также ле
карственнЬ1Х средств для местН020 и иH2a
ляциОНН020 применения и трансдермальнЬ1Х
пластырей (для указаннЬ1Х субстанций Kpи
терий приемлемости по содержанию обще20
количества жизнеспособнЬ1Х аэробов ОКА
и OKr суммарно не более 102 КОЕ/2 или
КОЕ/мл):
отсутствие rрамотрицательных бактерий,
толерантных к желчи (1 r или 1 мл), либо OTCYT
ствие бактерий семейства Eпterobacteriaceae
(1 r или 1 мл) при испытании в соответствии с
альтернативной методикой определения;
отсутствие Pseudomoпas ае,иgiпоsа (1 r
или 1 мл);
отсутствие Staphy/ococcus au,eus (1 r или
1 мл).
2. Фармацевтические субстанции cиHтe
тичеСК020 происхождения для производства
нестерильнЬ1Х лекарственных средств и вспо
М02атеЛЬНЬ1е вещества:
отсутствие Esche,ichia соП (1 r или 1 мл).
770
rосударственная фармакопея Республики Беларусь
3. Субстанции для фармацевтическо
20 использования для производства лекар
ственных средств, в которых лимитируется
содержание Pseudomoпas ае,иgiпоsа, Staphy
/ococcus au,eus, Sа/топе//а и бактерий семей
ства Eпte,obacte,iaceae:
отсутствие Рsеиdотопаs ае,иgiпоsа (1 r
или 1 мл);
отсутствие Staphy/ococcus aureus (1 r
или 1 мл);
отсутствие Sa/moпe//a (10 r или 10 мл);
не более 102 КОЕ rрамотрицательных
бактерий, толерантных к желчи (1 r или 1 мл),
либо не более 102 КОЕ бактерий семейства
Епtе,оЬасtе,iасеае (1 r или 1 мл) при испыта
нии в соответствии с альтернативной методи
кой определения.
4. Субстанции для фармацевтическо
20 использования природН020 происхождения
(раститеЛЬН020, животН020 или минерально
20), для которых предварительная aHтиMи
кробная обработка невозможна и в отноше
нии которых компетентный уполномоченный
Ор2ан допускает микробное за2рязнение иc
ходН020 материала более 1 03 жизнеспособ
ных миКРООР2анизмов в 1 2 или в 1 мл (не
более 104 КОЕ/2 или КОЕ/мл):
отсутствие Esche,ichia co/i (1 r или 1 мл);
отсутствие Рsеиdотопаs aerugiпosa (1 r
или 1 мл);
отсутствие Staphy/ococcus aureus (1 r
или 1 мл);
отсутствие Sа/топе//а (10 r или 10 мл);
не более 102 КОЕ rрамотрицательных
бактерий, толерантных к желчи (1 r или 1 мл),
либо не более 102 КОЕ бактерий семейства
ЕпtеrоЬасtеriасеае (1 r или 1 мл) при испыта
нии в соответствии с альтернативной методи
кой определения.#
Рекомендуемые критерии приемлемо
сти для микробиОЛ02ической чистоты лекар
ственных средств средств раститеЛЬН020
проиСХОJКдения для внутренне20 применения
приведены в общей статье 5.1.8.
01/2013:50105
5.1.5. ПРИМЕНЕНИЕ Fo КОНЦЕПЦИИ
ПРИ СТЕРИЛИЗАЦИИ ПАРОМ
ВОДНЫХ РАСТВОРОВ
Настоящая стаья носит информацион
ный характер.
Значение Fo процесса стерилизации Hacы
щенным паром это поражающее действие,
выраженное в терминах, эквивалентных Bpe
мени в минутах, при температуре 121 ос, OKa
зываемое процессом на лекарственное cpeд
ство в ero конечном контейнере по отношению
к микроорrанизмам, имеющим теоретическую
величину Z равную 10.
Общее значение Fo процесса, включа
ющеro стадию наrревания и остывания, может
быть рассчитано объединением летальных
норм относительно времени в дискретных
температурных интервалах.
Если процесс паровой стерилизации
выбран на основе Fo концепции, должно быть
уделено большое внимание тому, чтобы по
степенно была достиrнута полная стерилиза
ция. В дополнение к валидации процесса He
обходимо проведение непрерывноro cTpororo
микробиолоrическоrо контроля в ходе Bcero
процесса, чтобы доказать, что микробиоло
rические пара метры находятся в допустимых
пределах не более SAL 10 6.
По отношению к паровой стерилизации
величина Z это величина, связывающая
термостойкость микроорrанизмов и измене
ние температуры. Величина Z это измене
ние температуры, необходимое для измене
ния величины D на 10.
Величина О (или величина десятично
ro уменьшения) параметр стерилизации
(продолжительность или поrлощенная доза),
обеспечивающий снижение числа жизнеспо
собных микроорrанизмов до 1 О % от исход
Horo числа. Это величина, определяемая
только при точных экспериментальных усло
виях.
Для расчетов применяются следующие
математические выражения:
Fo o=0121.(rgN o lgN)o=0121Ig/F,
rдe:
О 121 величина О стандартных спор (5.1.2)
при температуре 121 ОС;
N o начальное число жизнеспособных ми
кроорrанизмов;
N конечное число жизнеспособных ми
кроорrанизмов;
/F коэффициент инактивации.
Z 0= Т 2 Т 1
Ig0 1 lg02
rде:
О 1 величина О микроорrанизмов при TeM
пературе Т I .
02 величина О микроорrанизмов при TeM
пературе Т 2 .
F == N o == 10// О ,
N
rде:
t время экспозиции;
о величина О микроорrанизмов в услови
ях экспозиции.
01/2013:50106
5.1.6. Альтернативные методы контроля микроБИОЛО2ической чистоты
771
5.1.6. АЛЬТЕРНАТИВНЫЕ
МЕТОДЫ КОНТРОЛЯ
МИКРОБиолоrИЧЕСКОЙ
ЧИСТОТЫ
Настоящая статья приводится для иH
формации
1. ОБЩИЕ СВЕДЕНИЯ
Цель данноrо раздела способствовать
внедрению и использованию альтернативных
микробиолоrических методов, Korдa это может
привести к экономически эффективному микро
биолоrическому контролю и повысить обеспече
ние качества лекарственных средств. Данные
альтернативные методы также MOryт применять
ся в мониторинrе окружающей среды.
Микробиолоrические методы, описанные в
Фармакопее, используются на протяжении века
и такие методы, как определение общею коли
чества и идентификация микроорrанизмов, ис
пользуются микробиолоrами до сих пор. На
протяжении десятилетий эти методы являются
незаменимыми при обеспечении контроля и ми
кробиолоrической безопасности производства
лекарственных средств. Однако общепринятые
микробиолоrические методы времязатратны и
результаты доступны только после инкубацион
ноro периода, который обычно составляет до 14
дней. Поэтому результаты стандартных микро
биолоrических методов редко дают возможность
провести профилактические и корректирующие
действия.
Альтернативные методы контроля микро
биолоrической чистоты были введены недавно,
и некоторые из этих методов продемонстриро
вали возможность получения результатов в pe
альном или почти реальном времени с возмож
ностью проведения корректирующеro действия.
Эти новые методы MOryT также привести к значи
тельному улучшению качества испытания.
В данной справочной статье описаны новые
микробиолоrические методы для фармацевтиче
скоro применения. Для каждоro метода сначала
описан основной принцип, и затем обсуждаются
ею преимущества и недостатки. Потенциальное
применение характеризует области применения,
в которых может использоваться описываемый
метод с учетом еro принципов действия: целью
предоставления информации не является пред
ложение использования на практике данноro
метода. Также описаны общие положения о Ba
лидации методов. ПРОИЛЛЮСТРИРlJваны специ
фические примеры для каждоro ТИl1а метода.
Подробный валидационный протокол представ
лен в конце этоro раздела.
Данная статья не предназначена ни для pe
комендации одноro метода вместо друrorо, ни
ДЛЯ представления эксклюзивноro или исчерпы
вающеrо списка альтернативных методов, KOTO
рые MOryT быть использованы для фармацевти
ческоro микробиолоrическоro контроля. Однако
данная справочная статья может быть использо
вана в процессе выбора альтернативноro микро
биолоrическоro метода как дополнения или аль
тернативы общепринятым микробиолоrическим
подходам, а также для предоставления Meтoдo
лоrических принципов процесса валидации BЫ
бранноro метода. Очевидно, что в этой быстро
развивающейся области появятся и новые
методы. Методолоrические принципы, представ
ленные в данной статье, MOryT в равной степени
быть применимы и для них.
Существует 3 основных вида определений,
характерных для микробиолоrических испыта
ний. Они включают:
качественные испытания на присутствие
или отсутствие микроорrанизмов;
количественные испытания по определе
нию общеro числа микроорrанизмов;
испытания на подлинность.
1 1. КАЧЕСТВЕННЫЕ ИСПЫТАНИЯ
НА ПРИСУТСТВИЕ ИЛИ ОТСУТСТВИЕ
микРооРrАНИ3МОВ
В стандартном микробиолоrическом анали
зе такой вид испытания характеризуется исполь
зованием изменения прозрачности или друrих
зависимых от роста изменений в питательной
среде для подтверждения присутствия жизне
способных микроорrанизмов в испытуемом об
разце. Самый распространенный при мер этоro
испытания это испытание на стерильность.
Друrим примером такоro вида определения яв
ляются испытания, предназначенные для оценки
присутствия или отсутствия определенноro вида
жизнеспособных микроорrанизмов в образце.
1 2. КОЛИЧЕСТВЕННЫЕ ИСПЫТАНИЯ
ПО ОПРЕДЕЛЕНИЮ ОБЩЕrо ЧИСЛА
микРооРrАНИ3МОВ
Мембранная фильтрация и чашечный метод
подсчета являются общепринятыми методами
для подсчета числа жизнеспособных микроорrа
низмов, представленных в образце. Метод наи
более вероятноro числа (НВЧ) это еще один
пример этих методов. НВЧ был разработан как
способ подсчета числа жизнеспособных микро
орrанизмов, представленных в образце, не под
лежащих прямому посеву на чашки.
1 3. ИСПЫТАНИЯ НА ПОДЛИННОСТЬ
Биохимическая и морфолоrическая xapaK
теристики неизвестноrо микроорrанизма это
классический метод идентификации, использу
емый в фармакопейных испытаниях. Недавно
разработанные методы ускорили и автоматизи
ровали подходы к идентификации, особенно в
области обработки результатов, анализа и xpa
нения. Некоторые новые подходы, которые были
введены в эти методы, включают биохимические
реакции, поrлощение уrлеродноro субстрата, xa
772
rосударственная фармакопея Республики Беларусь
рактеристику состава жирных кислот, опреде
ление характерной последовательности полос
после рестрикции эндонуклеазой и использова
ние секвенирования 16S рДНк.
2.0БЩИЕ ПРИНЦИПЫ АЛЬТЕРНАТИВНЫХ
МЕТОДОВ
Альтернативн.ые микробиолоrические
методы используют прямые инепрямые
методы детектирования; в некоторых случаях
усиление сиrнала достиrается методами обо
rащения. С учетом этих различий и для удоб
ства их рассмотрения в рамках этоro раздела
альтернативные методы контроля микробио
лоrической чистоты разделены на 3 катеro
рии:
методы, основанные на росте микроорrа
низмов, в которых различимый сиrнал обычно
достиrается по истечении определенноro пери
ода роста;
прямые измерения, в которых индивиду
альные клетки дифференцированы и отчетливо
видны;
анализ клеточных компонентов, при KO
тором экспрессия специфических компонентов
клеток используется для непрямой оценки при
сутствия микроорrанизмов.
В некоторых случаях эти различия искус
ственные, но они дают возможность создать pa
бочую классификацию.
2 1. МЕТОДЫ, ОСНОВАННЫЕ НА РОСТЕ
МИКРООР,АНИ3МОВ
2 1 1. Раннее выявление роста
2 1 1 1. Общие критические аспекты Me
тодов, основанных на раннем выявлении роста
Такие методы в значительной степени
зависят от роста микроорrанизмов для по
лучения обнаруживаемоro количества ми
кроорrанизмов. Для наблюдаемоro в лекар
ственных средствах типично низкоro уровня
микробной контаминации выявление может
занимать 24 ч или более, особенно в случа
ях дрожжевых и плесневых rрибов. Повышен
ная чувствительность может быть достиrну
та с помощью фильтрации образцов. В этом
случае после фильтрации мембрану инкуби
руют в питательной среде, а результат Bыpa
жают как присутствие или отсутствие в коли
честве, соответствующем отфильтрованному
объему. Из за использования стадии инкуби
рования в жидкой среде, данные испытания
не MorYT использоваться для количественноro
определения, а только для оценки отсутствияl
присутствия в анализируемом количестве.
Анализ более одноrо обр зца можно прово
дить полуколичественной оценкой (испытание
на предельное содержание). Основным пре
имуществом таких методов зачастую является
возможность одновременноrо анализа боль
шоrо количества образцов и получение pe
зультатов за минимальные сроки.
2 1 1 2. Электрохимические методы
Принципы измерения. Микроорrанизмы,
которые размножаются и метаболизируют в
подходящей питательной среде, продуциру
ют сильно заряженные ионные метаболиты
из слабо заряженных орrанических питатель
ных веществ, что приводит к изменению элек
трических свойств в этой среде. Эти измене
ния сопротивления (измеряемые с помощью
электропроводности или емкости) измеряют
ся электродами, поrруженными в сосуды с пи
тательной средой и соприкасающимся с ней.
Измеряемая конечная точка это время, за
траченное на определение заранее заданноro
изменения сопротивления; время определения
обратно пропорционально исходному размеру
первичноrо инокулята. Для дрожжевых и плес
невых rрибов, которые вызывают лишь He
значительные изменения электрическоro co
противления, обычно применяют непрямое
измерение электропроводности, используя pe
зервуар с калия rидрохлоридом. Допускается
также и прямое измерение емкости.
Критические аспекты Автоматизирован
ное детектирование с преобразованием в элек
тронные данные, картирование изменения co
противления, соответствующеro кривой роста
микроорrанизмов, и снижение продолжительно
сти времени действия до 48 ч.
Потенциальная область применения. Ми
кробиолоrический анализ антибиотиков, эф
фективность антимикробных консервантов и
определение присутствия/отсутствия микроор
rанизмов в количестве испытуемоro образца при
проведении оценки общеro числа жизнеспособ
ных аэробных микроорrанизмов.
2 1 1 3. Измерение потребляеМ020 или
продуцируеМ020 2аза
Принцип измерения. Активно размножаясь
и метаболизируя, орrанизмы используют COOT
ветствующую питательную среду, что приводит к
образованию метаболитов или удалению специ
фических питательных веществ. В этом случае
MOryT контролироваться изменения в rазовом
составе свободноrо пространства над проду
том в закрытых сосудах с культурой с помощью
датчиков давления, реаrирующих на продуциро
вание rаза (например, С0 2 ) или поrлощение rаза
(например, О). Moryт использоваться и друrие
индикаторы, включая калориметрическое дeTeK
тирование С0 2 .
Критические аспекты. Для медленно pa
стущих микроорraнизмов, таких как микобакте
рии, метод предполаrает более быстрое детекти
рование. Между исходной микробной наrрузкой
и определяемой конечной тоuкой отсутствует
прямая зависимость. Температура инкубации и
алroритм обработки данных значительно влияют
на результаты.
Потенциальная область применения. Про
дукты, в которых MOryт присутствовать медлен
но растущие микроорrанизмы.
5.1.6. Альтернативные методы контроля микробиОЛО2ической чистоты
773
2 1 1 4. Биолюминесценция
Принципы измерения. Аденозинтрифос
фат (АТФ) является хорошо изученным Map
кером жизнеспособности клеток. В данном
методе АТФ должен, прежде всеro, извле
каться из микроорrанизмов с помощью под
ходящеro экстраrента с последующим коли
чественным определением с использованием
люциферин/люциферазной ферментной си
стемы, которая излучает свет пропорциональ
но присутствующему в пробе АТФ. Излуча
емый свет измеряется биолюминометром и
выражается в относительных световых едини
цах (ОСЕ) биолюминисценции в жидкой среде.
Значения ОСЕ, полученные при анализе ис
пытуемоro образца, сравнивают с пороroвой
величиной среды, определенной как 2 или
3 KpaTHoe значение ОСЕ среды для культиви
рования или суспензии образца. Результат ис
следования положительный, если ОСЕ, полу
ченные от испытуемоrо образца, превышают
пороrовую величину. При изменении метода
микроорrанизмы, задержанные на мембране.
инкубируют на аrаровой среде с последую
щим определением микроорrанизмов камерой
прибора с зарядовой связью (ПЗС). Резуль
таты исследования выражают в микроКОЕ.
Данный метод является количественным, но
имеет узкий диапазон линейности.
Критические аспекты. Если испытуемое
средство имеет высокий уровень микробной
контаминации (приблизительно 500 1000 КОЕ
на пробу испытуемоro образца), то определе
ние является быстрым (1 ч). При низком уровне
контаминации (менее 100 КОЕ на пробу испыту
емоro образца) необходимо увеличение числа
микроорrанизмов с помощью стадии инкубации
в питательной среде (жидкий или твердый arap)
в течение 12 8 ч в соответствии с исполь
зуемым методом. После инкубации в жидких
средах одна единичная клетка, способная к
росту, увеличивается от 1 до 1000 и может быть
определена. Выход АТФ варьируется от одноrо
микроорrанизма к друroму, причем бактерии,
содержащие 1 10 фr/клетка, и rрибы, coдep
жащие 100 фr/клетка, а также мноrие друrие
факторы, включая вид, фазу роста клетки, тип
питания, клеточный стресс или возраст клетки,
MorYT повлиять на содержание АТФ в клетке. По
этому определить число микроорrанизмов He
посредственно по значению ОСЕ невозможно.
Кроме тоro, мутность и цвет образца MorYT OKa
зать влияние на реакцию путем или усиления
реакции, или увеличения уровня испускаемоro
света, или привести к понижению уровня испу
скаемоro света. Так как реакция является фер
ментативной, продукты, которые инrибируют
или уменьшают активность ферментов, MorYT
также повлиять на результаты испытания. На
практике такое влияние встречается редко, но
должно быть тщательно изучено при валидации
процесса. Реакция также чувствительна к при
9!; 30. 1712
сутствию нуклеотидфосфатов, таких как АДФ и
ПФ, которые влияют на результаты вследствие
образования АТФ в присутствии аденилаткина
зы. Этот фермент используют для повышения
чувствительности некоторых биолюминисцент
ных методов: добавляют третий реактив, coдep
жащий АДФ, и продуцируется новый АТФ в при
сутствии аденилаткиназы, высвобождаемой из
микроорrанизмов.
Потенциальная область применения.
Определение эффективности антимикроб
ных консервантов, оценка присутствия/отсут
ствия микроорrанизмов в определенном коли
честве испытуемоro образца при определении
общеro числа жизнеспособных аэробных ми
кроорrанизмов (биолюминисценция в пробир
ке или микропланшете), подсчет общеrо числа
жизнеспособных аэробных микроорrанизмов
(биолюминисценция на мембране), мониторинr
окружающей среды и воды. Метод находит при
менение для оценки фильтруемых и нефильтру
емых веществ.
2 1 1 5. Микрокалориметрия
Принципы измерения. Обмен веществ ми
кроорrанизмов происходит с выделением тепла,
которое может быть точно измерено микрока
лори метром. Теплопродукция может быть из
мерена путем помещения контаминированно
ro испытуемоro образца в запаянную ампулу,
содержащую среду для роста, а затем в кало
риметр. При использовании чувствительно
ro оборудования MOryт быть построены кривые
микробноro роста. Большая бионаrрузка может
быть определена проточной калориметрией.
Критические аспекты. Теоретически, для
данноro метода требуется не микробный рост,
а только катаболическая активность. Тем не
менее, необходимо минимальное количество
микроорrанизмов для Toro, чтобы значения Te
плоотдачи были выше базовой линии, и обычно
этоrо можно достиrнуть посредством использо
вания методов обоrащения.
Потенциальная область применения.
Метод для определения эффективности анти
микробных консервантов.
2 1 1 6. Турбидиметрия
Принципы измерения. Рост микроорrаниз
мов вызывает видимое помутнение среды. Это
изменение среды может быть количественно
точно определено путем измерения оптической
плотности при заданной длине волны. В этой
простейшей форме такие измерения проводятся
на стандартном спектрофотометре в диапазоне
длин волн обычно от 420 нм до 615 нм. Исполь
зуются альтернативные автоматизированные
системы с применением ""икропланшетов, по
зволяющие прводить постоянное считывание
данных с ранним детектированием изменения
оптической плотности.
Критические аспекты. Были предприняты
попытки экстраполировать значения исходной
бионаrрузки от длительности периода, необхо
774
lосударственная фармакопея Республики Беларусь
димоro для детектирования, однако данные за
висимости MOryт быть достоверны только для
интактных микроорrанизмов с воспроизводимы
ми характеристиками роста. Эти методы не спо
собны различать жизнеспособные и нежизне
способные микроорrанизмы.
Потенциальная область применения.
Определение размера инокулята в микробной
суспензии с помощью rрадуировочных кривых
для использования в фармакопейном анали
зе. Определение в автоматическом режиме
чувствительности к консервантам испытуемых
микроорrанизмов, извлеченных из roтовых ле
карственных средств.
2 1 1 7. Методы, основанные на бактери
офавах
Принципы измерения. Бактериальные
вирусы (бактериофаr. фаr) MOryT приводить к
инфицированию клеткок хозяев, вызывая либо
лизис, либо встраивание их rенетическоrо Ma
териала и выработку новых белков. Их высокая
специфичность к клетке хозяину может исполь
зоваться в методах детектирования, которые ис
пользуют последствия заражения как конечную
точку. Такие конечные точки включают: форми
рование бляшки на твердом rазоне испытуемых
бактерий; определение внутриклеточноro coдep
жимоrо, высвобожденноro из лизированных бак
терий (возможно колориметрическим методом);
эффекты, вызываемые бактериофаrами, такие
как образование льда или биолюминесценция,
возникающая при заражении rенетически MO
дифицированным бактериофаrом. Колифаrи с
флуоресцентной меткой MOryT быть использо
ваны для селективноro определения жизнеспо
собных Е. coli в комбинации с ПЭФМ (см. раздел
2 2 3).
Критические аспекты. Детектирова
ние, основанное на использовании бактери
офаrов, может при меняться как для одиноч
ных, так и для смешанных культур, в которых
специфичность хозяина допускает и детекти
рование, и идентификацию. Для определения
конечных точек обычно требуется минималь
ное количество клеток мишеней для получе
ния измеряемоrо сиrнала, что приводит К He
обходимости обоrащения в случаях с низкой
бионаrрузкой. Состав испытуемоrо образ
ца может неблаroприятно влиять на процесс
вирусноro заражения. В большинстве случа
ев существует узкий Kpyr клеток хозяев, что
делает затруднительным определение широ
коro спектра микроорrанизмов, которые при
сутствуют в образце.
Потенциальная область применения. В oc
новном эти методы используются в исследова
тельских целях при промышленных разработ
ках, связанных преимущественно с клинической
и пищевой микробиолоrией. Данные методы, Be
роятно, будут использоваться для определения
присутствия/отсутствия указанных микроорrа
низмов.
2 1 2. Разработка питательных сред для
улучшения обнаружения
Принципы измерения. Питательные среды
существуют на протяжении мноrих лет и посто
янно совершенствуются. Современный этап
развития характеризуется появлением xpo
MoreHHbIx субстратов, которые все больше и
больше используются в клинической и пище
вой микробиолоrии. Возможность обнаружи
вать присутствие определенных ферментов,
используя подходящие субстраты, привела
к разработке большоro количества методик
идентификации микроорrанизмов вручную или
с использованием автоматизированных при
боров. Включение таких субстратов в селек
тивную или неселективную первичную среду
для выделения микроорrанизмов исключает
необходимость последующеro пассажа и био
химических тестов для идентификации опре
деленных микроорrанизмов. В результате xpo
MoreHHbIe жидкие или твердые питательные
среды обеспечивают условия для специфиче
ской ферментативной активности, по которой
проводится обнаружение и дифференциация
микроорrанизмов. В состав этих специфиче
ских сред включают определенные субстра
ты, которые в процессе роста rидролизуются
специфическими клеточными ферментами бак
терии или rриба. Выбор субстрата обоснован
искомой диаrностической ферментативной aK
тивностью и окрашенными индикаторами.
Критические аспекты. Использование ин
новационных питательных сред имеет несколько
преимуществ: улучшенное распознавание коло
ний в смешанной культуре, простота использо
вания и леrкость оценки результатов. В дополне
ние, время ответа сокращается, поскольку рост
и идентификация микроорrанизмов осущест
вляются одновременно. Однако должна быть
проведена тщательная валидация среды, под
тверждающая специфичность, селективность
и робастность методики. Качество сиrнала oc
новывается не только на тщательном подборе
ферментов, которые используются в качестве
принципа детектирования, так как эти фермен
ты MorYT присутствовать в различных видах, но
и на физико химических характеристиках среды,
таких как рН.
Потенциальная область применения. Об
наружение определенных микроорrанизмов,
таких как Е. соН. бактерии rруппы кишечной па
лочки, Sa/moпe//a, Staphy/ococcus и Streptococ
cus spp.; особые преимущества MOryT быть по
лучены при использовании для испытания на
присутствие/отсутствие. Дрожжевые rрибы
MoryT быть также обнаружены с использованием
хромоrенной питательной среды.
2 2. ПРЯМОЕ ИЗМЕРЕНИЕ
2 2 1. Твердофазная цитометрия
Принципы измерения. Для выделения ми
кроорrанизмов используют мембранный фильтр.
5.1.6. Альтернативные методы контроля микроБИОЛО2ической чистоты
775
Микроорrанизмы окрашиваются путем маркиро
вания флуорофором в качестве индикатора жиз
неспособности или до, или после фильтрования.
Изначально флуорофор представляет собой He
флуороrенный конъюrированный субстрат, KOTO
рый под влиянием внутриклеточных ферментов
расщепляется с выделением флуоресцентноrо
вещества. Интактная клеточная мембрана He
обходима для удержания флуорофора внутри
цитоплазмы микроорrанизма. Возбуждающее
излучение лазера и автоматизированное CKa
нирование позволяют обнаруживать единичные
жизнеспособные флуоресцентные микроорrа
низмы. Соответствующее проrраммное обеспе
чение позволяет отличать жизнеспособные ми
кроорrанизмы от автофлуоресцентных частиц.
Высокая чувствительность и быстрота детекти
рования делает возможным обнаружение микро
орrанизмов, присутствующих в образце, почти в
реальном времени. Общее число клеток можно
определять с использованием постоянно флуо
ресцирующею красителя.
Критические аспекты. С помощью данноro
метода можно обнаружить метаболически актив
ные, жизнеспособные и сложно культивируемые
или не поддающиеся культивированию микро
орrанизмы. Это может привести к переоценке
микробиолоrических норм, установленных для
испытуемоro образца. Для обнаружения спор
необходима реактивация их роста. Может дo
стиrаться обнаружение одной клетки определен
ною вида, однако, на настоящий момент иден
тификация не входит в стандартный протокол
микробиолоrическоro испытания. Для избира
тельноro обнаружения MOryT быть использованы
флуоресцентные антитела. Автофлуоресцент
ные частицы, которые сложно отличить от ми
кроорrанизмов, MorYT привести к получению
ложно положительных результатов.
Потенциальная область применения. Бы
стрый и чувствительный метод для неспецифи
ческой оценки бионаrрузки. Используется при
контроле воды фармацевтической катеrории.
2 2 2. Проточная цитометрия
Принципы измерения. Микроорrанизмы, по
меченные флуорофором, MOryT обнаруживаться
в суспензии при прохождении через проточную
кювету цитометра. Применение флуорофорноrо
субстрата позволяет дифференцировать жизне
способные микроорrанизмы от нежизнеспособ
ных частиц (смотри 2 2 1).
Критические аспекты. Проточная цитоме
трия может применяться в микробиолоrическом
анализе как фильтруемых, так и нефилыру
емых материалов. Проточный цитометрический
анализ обеспечивает детектирование почти в
реальном масштабе времени, но он не так чув
ствителен, как твердофазная цитометрия. Для
повышения чувствительности при использова
нии в фармацевтических целях, часто бывает
необходимо включить дополнительно стадию
инкубации в питательной среде. В этом случае
метод переходит в катеroрию методов, OCHOBaH
ных на росте. Для оптимизации условий анализа
нефильтруемых образцов может потребоваться
приroтовление серийных разведений образца,
при этом размер механических включений может
значительно повлиять на данный процесс. За ис
ключением возможности использования для об
разца мембранной фильтрации, аналоrичные
проблемы MorYT возникнуть и при твердофазной
цитометрии. Проблемой также может быть ar
rлютинация бактерий (например, S. au,eus).
Потенциальная область применения. В
отличие от твердофазной цитометрии, данный
метод может применяться для определения и
подсчета общеro количества микроорrанизмов в
материалах, содержащих значительное количе
ство механических включений. Если необходи
ма пре инкубационная стадия, метод становится
качественным испытанием.
2 2 3. Метод прямой эпифлуоресцентной
фильтрации (ПЭФМ, DEF1)
Принципы измерения. Данная методи
ка может рассматриваться как предшествен
ник твердофазной цитометрии. Микроорrаниз
мы, сконцентрированные путем фильтрации из
образца, ранее окрашивались флуоресцент
ным красителем акридиновым оранжевым.
В настоящее время более широко используется
4',6 диамидино 2 фенилиндол (ДАФИ, DАР/), KO
торый может определяться путем эпифлуорес
центноro освещения. Методики флуоресцентной
окраски жизнеспособных клеток, при меняемые
в твердофазной цитометрии (смотри 2 2 1),
MorYT дополнять окраску ПЭФМ, а флуоресцент
ные оксислительно восстановительные краси
тел и , такие как 5 циано 2,3 дитолилтетразолия
хлорид (СТС, 5 суапо 2,3 ditоlу/tеtrаzoliит chlo
ride), MOryT использоваться для обнаружения ды
шащих клеток. В сочетании с микроскопией этот
метод позволяет быстро обнаруживать микро
орrанизмы, ею абсолютная чувствительность
зависит от объема фильтруемоro вещества и
числа исследуемых полей зрения. В сочетании С
анализом изображения, полуавтоматические си
стемы автофокусирования способствовали pac
ширению использования данною метода. Суще
ствует модификация метода, при которой отбор
образцов осуществляется с использованием
клейкоro материала, что позволяет собирать
клетки с поверхности с последующим их окраши
ванием и визуальным наблюдением через эпиф
луоресцентный микроскоп.
Критические аспеl(ты. Распределение
микроорrанизмов на мемб[)ане влияет на po
бастность метода На интенсивность флуорес
ценции MorYT повлиять процесс окрашивания
и метаболический статус микроорrанизмов.
Кратковременный период нахождения куль
туры на поверхности фильтра до окрашива
ния способствует формированию микроколо
776
rосударственная фармакопея Республики Беларусь
ний, которые леrко окрашиваются, MorYT быть
леrко подсчитаны и являются доказатель
ством жизнеспособности. Современные раз
работки с использованием флуоресцентной
in situ rибридизации (F/SH, f/иоrеsсепсе iп situ
hybridisation) , являющейся результатом дo
полнительноrо воздействия олиroнуклеотид
ной пробы, маркированной флуоресцентной
меткой, со специфической последовательно
стью рРНК, позволяют проводить селектив
ное детектирование.
Потенциальная область применения. Воз
можности ПЭФМ обычно оrраничены жидкостя
ми с низкой вязкостью, несмотря на то, что пред
варительное разведение или предварительная
фильтрация периодически используются для
вязких или дисперсных веществ. Метод эффек
тивно используется для контроля общеro коли
чества микроорrанизмов в водных лекарствен
ных средствах.
2 3. АНАЛИЗ КЛЕТОЧНblХ КОМПОНЕНТОВ
2 3 1. Фенотипический
2 3 1 1. ИММУНОЛ02ические методы
Принципы измерения. Реакции антитело
антиrен MOryт использоваться для детектирова
ния уникальных клеточных характеристик опре
деленных микроорrанизмов. Эти реакции MOryт
быть связаны с реакцией аrrлютинации, коло
риметрическими или флуориметрическими KO
нечными точками, позволяющими проводить как
качественное, так и количественное определе
ние. Твердофазный иммуноферментный анализ
(ELlSA. епzуте /iпkеd immunosorbent assay) яв
ляется основой для мноrих простых твердофаз
ных методик.
Критические аспекты. Иммунолоrические
методы детектирования зависят от уникальной
экспрессии специфических идентификаторов,
но необязательно подтверждают присутствие
жизнеспособных микроорrанизмов.
Потенциальная область применения. Об
наружение и идентификация определенных ми
кроорrанизмов.
2 3 1 2. Профили жирных кислот
Принципы измерения. Состав жирных
кислот микроорrанизмов стабилен, четко за
фиксирован и характеризуется высокой степе
нью roмоrенности в рамках различных TaKCO
нометрических rрупп. Штамм выращивают на
стандартной питательной среде и собирают.
Жирные кислоты омыляют, метилируют и экс
траrируют. Наличие и количество полученных
метиловых эфиров жирных кислот измеряют
с помощью rазовой хроматоrрафией высокоro
разрешения. Состав }l(ИРНЫХ кислот испытуе
моro штамма сравнивается с базой данных из
вестных штаммов для возможноro совпадения и
идентификации.
Критические аспекты. Использование
профилей жирных кислот для микробиоло
rической идентификации требует высокой
степени стандартизации. Для определения
состава жирных кислот микробных клеток
крайне важно обеспечить культивирование
штаммов с использованием стандартных пи
тательных сред и стандартных условий инку
бации. Для rазовой хроматоrрафии должны
применяться стандартные условия, среди KO
торых большое значение имеют мноroкрат
ные определения калибровочных стандартов
и известных штаммов.
Потенциальная область применения.
Идентификация или характеризирование микро
флоры продукта и окружающей среды дЛЯ KOH
троля контаминации и обнаружения определен
ных микроорrанизмов.
2 3 1 3. Инфракрасная спектроскопия на
С?снове Фурье преобразования (ИК ФП, FТlR,
Fourier transform iпfrаrеd)
Принципы измерения. Фурье преобразо
вание ИК спектра интактных микроорrаниз
мов дает стабильный, опознаваемый образ
(паперн), типичный для таксонометрических
rрупп микроорrанизмов. Анализ ИК ФП спектра
может выполняться на приборах, имеющихся
в продаже. Штамм выращивают на CTaHдapT
ной питательной среде и собирают. Клеточную
массу пере носят на носитель и снимают ИК
спектр. После Фурье преобразования получен
ный спектр сравнивают с базой данных извест
ных штаммов для определения совпадения и
идентификации.
Критические аспекты. Использование об
разцов ИК ФП спектров для микробиолоrиче
ской идентификации требует высокой степени
стандартизации. Для использования ИК ФП
спектров образцов микробиолоrических клеток
крайне важно обеспечить культивирование
штаммов с использованием стандартных пита
тельных сред и стандартных условий инкуба
ции. При анализе клетки должны быть в оди
наковом состоянии ростовоro цикла. Особое
внимание необходимо уделить работам по Ba
лидации.
Потенциальная область применения.
Идентификация или характеризирование микро
флоры продукта и окружающей среды дЛЯ KOH
троля контаминации обнаружения определен
ных микроорrанизмов.
2 3 1 А. Масс спектрометрия
Принципы измерения. rазообразные про
дукты rидролиза, выделяемые при наrревании
выделенных микроорrанизмов под вакуумом,
MorYT быть проанализированы с использова
нием масс спектрометрии с получением xa
рактеристических спектров. Аналоrично ин
тактные микробные клетки, подверrнутые
масс спектрометрическому анализу с иони
зацией лазерной десорбцией при содействии
матрицы и детектированием с использовани
ем времяпролетноro детектора (MALO/ TOF,
matrix assisted /aser desorption iопizаtiоп tiте
of f1ight), дают характерные профили заряжен
5.1.6. Альтернативные методы контроля микробиОЛО2ической чистоты
777
ных частиц. Такие спектры MorYT сравнивать
ся с известными профилями в качестве бы
строro средства идентификации.
Критические аспекты. Перед анализом
требуется культивирование штаммов.
Потенциальная область применения.
Идентификация или характеризирование микро
флоры продукта и окружающей среды дЛЯ KOH
троля контаминации и обнаружения определен
ных микроорrанизмов.
2 3 1 5. Биохимический количественный
анализ, основанный на физиОЛ02ических peaK
циях
Принципы измерения. Биохимическому KO
личественному анализу обычно предшествует
окрашивание по rpaMMY или друroе дифферен
цирующее испытание для определения соответ
ствующеro протокола испытания. Испытуемые
суспензии микробных клеток оценивают с ис
пользованием биохимических наборов peareH
тов. Известно, что микроорrанизмы обладают
специфическими реакциями на эти биохимиче
ские вещества, например, использование специ
фических источников уrлерода. Идентификация
культуры проводится путем сравнения профи
ля биохимической реакции с базой данных. Эти
методы MorYT выполняться вручную или с ис
пользованием автоматических измерительных
приборов.
Критические аспекты. Необходима чистая
колония, возраст которой не должен превышать
3 дней. Выполнение испытания не представля
ет сложности, однако интерпретация результа
тов может оказаться субъективной. В зависи
мости от используемой системы и исследуемых
микроорrанизмов, результаты MOryT быть полу
чены быстро.
Потенциальная область применения.
Идентификация или характеризирование микро
флоры продукта и окружающей среды дЛЯ KOH
троля контаминации и обнаружения определен
ных микроорrанизмов.
2 3 2. rенотипический
2 3 2 1. Методики, основанные на ампли
фикации нуклеиновых кислот (МАНК, NAA Т, пи
c/eic acid атрlifiсаtiоп techпiques)
Общие принципы измерения. МАНК OCHO
вывается на мноroчисленном повторении про
цесса полимеризации ДНК, при водящем к экс
поненциальному увеличению специфическоro
фраrмента нуклеиновой кислоты, т. е. исполь
зованию полимеразной цепной реакции (ПЦР,
PCR, po/ymerase сhаiп reactioп). В этом TepMO
фильном циклическом процессе специфиче
ский фраrмент ДНК реплицируется с исполь
зованием олиroнуклеотидных праймеров (см.
статью 2.6.21). РНК также можно реплициро
вать с помощью ПЦР после транскрибирования
в кДНК с использованием обратной транскрип
тазы. Эта методика известна как ПЦР с обрат
ной транскрипцией (ОТ ПЦР, RT PCR. reverse
99 3.а}.. 1712
trапsс,iрtаsе PCR). Альтернативно, для YBe
личения мноroкратных антисмысловых копий
целевых РНК доступны особые методики, oc
нованные на амплификации РНК, например,
амплификация, основанная на последователь
ности нуклеиновых кислот (NASBA, пuc/eic acid
sequeпce based атрlifiсаtiоп) или транскрип
ционно опосредованная амплификация (ТМА,
trапsсriрtiоп теdiаtеd атрlifiсаtiоп). Фраrмен
ты амплифицированных нуклеиновых кислот
MorYT анализироваться несколькими методами:
анализ размера фраrмента, анализ запроrрам
мированной последовательности, реамплифи
кация со второй парой праймеров или спец
ифическое детектирование rибридизацией с
флуоресцентно меченной пробой. В зависи
мости от выбранноro анализа амплификацион
ная методика может быть качественной, полу
количественной или количественной. С целью
идентификацииtхарактеризации может быть
применен анализ последовательности спец
ифических частей reHoMa (т. е., 16S или 23S
рРНК).
Общие особенности. У МАНК есть мною
преимуществ перед классическими методами
обнаружения микроорrанизмов:
методы являются высокоспецифичными
при обеспечении специфичности выбранных
праймеров для определенных микроорrанизмов
или rрупп микроорrанизмов;
данные методы являются быстрыми, по
зволяют преодолеть проблему длительноro ин
кубационноro периода;
данные методы являются высокочувстви
тельными, позволяют оптимально определить
и амплифицировать один единичный фраrмент
нуклеиновой кислоты в реакционной смеси.
Однако существуют мноroчисленные прак
тические оrраничения по их использованию:
чувствительность методов в значительной
степени зависит от степени концентрации кле
ток мишеней в образце;
присутствие инrибиторов ферментативно
ro процесса при водит к ложноотрицательным pe
зультатам;
начальный объем испытуемою образца
незначительный;
существует высокая вероятность пере
крестной контаминации предыдущими ампли
фицированными фраrментами. что приводит к
получению ложноположительных результатов.
В зависимости от цели исследования,
должен быть сделан выбор РНК или ДНК мишени
для амплификации. Выбор мишени влияет на
корреляцию с )l{изнеспособностью. Использова
ние ДНК в качеСТЕ<е маркера имеет недостаток,
поскольку мертвые микроорrанизмы также co
держат ДНк. в то время как иРНК быстро разру
шается в мертвых бактериях и поэтому она счи
тается лучшим маркером жизнеспособности.
Критические аспекты ОТ ПЦР. ПЦР с об
ратной траскрипцией характеризуется синтезом
778
rосударственная фармакопея Республики Беларусь
кДНК с использованием РНК а качестве Ma
трицы. На этой стадии используется обратная
транскриптаза. Затем специфическая часть
кДНК амплифицируется с помощью ПЦР. В за
висимости от качества выделенной РНК эф
фективность синтеза кДНК может варьировать.
ОТ ПЦР может использоваться специально для
детектирования РНК, если содержание ДНК в
РНК образце минимально.
Критические аспекты методик ампли
фикации рнк. Было установлено, что данные
методы являются высокоэффективными для
специфическою (количественноrо) определения
рнк. Однако их достаточно сложно при менять в
повседневной практике.
Критические аспекты количествеНН020
(полуколuчествеННО20) определения (ПЦР в pe
альном времени). Классические методики ПЦР
основаны на детектировании конечной точки. В
большинстве случаев анализ фраrментов про
водится с использованием аrарозноrо rеля и
особых маркеров размера. Однако отсутствует
корреляция между количеством продукта ПЦР
в конце реакции и исходным количеством моле
кулы мишени. Напротив, количество определя
емою ПЦР продукта в начале экспоненциальной
фазы реакции очень хорошо коррелирует с ис
ходным количеством нуклеиновой кислоты. Co
временные ПЦР методики в реальном времени
разрабатываются для измерения этой экспонен
циальной фазы реакции. В этих методиках полу
чают данные амплификации, по которым можно
установить исходное количество молекул мише
ней. Специфические меченые пробы обеспечи
вают реrистрацию образующеюся продукта ПЦР
в реальном времени, позволяя увидеть наrляд
но экспоненциальную фазу ПЦР реакции. Ko
личественный анализ молекулы мишени может
быть проведен путем сравнения rрафиков aM
плификации серий стандартных разведений. На
рынке доступны автоматические системы ПЦР
в реальном времени. Дополнительным пре
имуществом таких систем является существен
но низкий риск перекрестной контаминации, по
скольку ПЦР продукты сканируются лазером в
закрытых пробирках. Однако существует боль
шая сложность по созданию стандартов.
Критические аспекты амплификации
2енов, кодирующих 165 или 235 рРНК. ПЦР в oc
новном применяется для амплификации и после
дующею анализа последовательности специ
фических частей reHoB, кодирующих 16S или
23S рРНК. Анализ этих специфических ДHK
последовательностей в большинстве случа
ев позволяет идентифицировать микроорrаниз
мы на видовом уровне. Выбор соответствующих
универсальных праймеров или даже видоспеци
фических пар праймеров из международных баз
данных обеспечивает высокую специфичность
амплификации фраrмента. Современная систе
матическая классификация основана на сравни
тельном анализе последовательностей.
Потенциальная область применения.
Вследствие высокой специфичности методик
амплификации, они наиболее приюдны для
целей идентификации. МАНК приroдна для дe
тектирования определенных микроорrанизмов
или конкретных rрупп, таких как микоплазмы.
Для количественноro определения микроорrа
низмов требуется количественная ПЦР в реаль
ном времени.
2 З 2 2. lенетические «отпечатки пальцев»
Принципы измерения. Данный метод опре
деляет характеристики и идентифицирует ми
кроорrанизмы с помощью фраrментов рестрик
ции нуклеиновых кислот reHoMoB бактерий и
rрибов. ДНК экстраrируют из чистоro лизата
микробных клеток и разрезают на фраrменты
с помощью рестриктаз. ДНК фраrменты разде
ляют по размерам с помощью электрофореза,
проявляют и сравнивают результат с друrими
известными профилями изолированных микро
орrанизмов. rенетический «отпечаток» являет
ся стабильным маркером, который делает воз
можным оптимальное распознавание видов
или даже определение характеристик ниже
видовоrо уровня. Типичным примером такоro
метода является риботипирование. Существу
ют также методики rенетической экспертизы.
основанные на ПЦР с использованием прай
меров, которые связываются с несколькими
участками в микробном reHoMe, создавая aM
пликоны с характерным распределением по
размерам.
Критические аспекты. Существует необхо
димость получения чистых колоний, но предва
рительная стадия культивирования не является
необходимой. Условия роста (температура, тип
питательной среды) не влияют на результат aHa
лиза. Для идентификации бактерий в продаже
имеются полуавтоматические системы.
Потенциальная область применения. re
нетическая экспертиза является более инфор
мативным методом для распознавания штам
мов (определения характеристик ниже видовою
уровня), чем для идентификации видов.
3. ОБЩИЕ ТРЕБОВАНИЯ К ВАЛИДАЦИИ
Целью данною раздела является изложе
ние рекомендаций по валидации методик для
альтернативноrо использования Фармакопей
ным микробиолоrическим методикам. ДЛЯ BЫ
деления и идентификации микроорrанизмов
микробиолоrические лаборатории иноrда ис
пользуют альтернативные методики анализа по
ряду причин, таких как экономичность, произво
дительность и удобство. Валидация данных Me
тодик необходима. Некоторые указания по вали
дации представлены в rлаве Общие сведения,
раздел 1.1, использование альтернативных Me
тодик.
При валидации альтернативных микробио
лоrических методик должна приниматься во вни
мание высокая степень вариабельности, KOТO
5.1.6. Альтернативные методы контроля микробиОЛО2ической чистоты
779
рая связана с общепринятыми методами. При
проведении микробиолоrическоro испытания с
помощью традиционноro определения количе
ства микроорrанизмов чашечным методом, Ha
пример, часто получают диапазон результатов
шире, чем в обычно используемых химических
испытаниях.
Korдa специфическое оборудование являет
ся критичным для применения альтернативноro
метода, оборудование, включая компьютерное,
и проrраммное обеспечение, должны быть пол
ностью квалифицированы следующим образом:
квалификация дизайна (ОО) дЛЯ ДOKYMeH
тальноro подтверждения тоro, что конструкция
оборудования является подходящей для KOp
ректноro выполнения метода; должна предо
ставляться поставщиком оборудования:
квалификация монтажа (10) для предо
ставления документальноro подтверждения, что
оборудование было доставлено и установлено в
соответствии с еro спецификацией;
квалификация функционирования (ОО)
дЛЯ документальноrо подтверждения тоro, что
установленное оборудование qpункционирует в
пределах предварительно определенных норм
при использовании в соответствии с инструкци
ями по эксплуатации;
квалификация эксплуатации (ОР) дЛЯ дo
кументальноro подтверждения тоro, что обору
дование, которое было установлено и эксплу
атируется в соответствии с инструкциями по
эксплуатации,постоянноработаетвсоответствии
с предварительно установленными критериями
и поэтому обеспечивает получение корректных
результатов для данной методики. Обычно она
проводится путем использования «модельной»
системы (с тест микроорrанизмами) для под
тверждения тоro, что используемые в лаборато
рии условия удовлетворяют требованиям крите
риев методики, описанным разработчиком.
Некоторые альтернативные методики зави
сят от использования базы данных. При вали
дации должен учитываться объем информации
базы данных в отношении диапазона представ
ляющих интерес микроорrанизмов.
Информативность новой или модифици
рованной методики должна быть доказана в
сравнительном исследовании оqpициальной и
альтернативной методик. Характеристики, опре
деленные в данном разделе, должны использо
ваться для проведения сравнения.
з 1. ТИПЫ МИКРОБиолоrИЧЕСКИХ
ИСПЫТАНИЙ
Для работ по валидации крайне важно
определить '-jасть испытания, проводимоro аль
тернативной методикой. Например, существу
ют разнообразные методы для определения
присутствия жизнеспособных клеток. Данные
методы MOryT при меняться в различных испы
таниях (например. общее число микроорrаниз
мов (бионаrрузка). испытание на стерильность),
однако полностью не MOryT заменить важные
этапы испытания. Например, испытание на CTe
рильность методом мембранной фильтрации
может выполняться в соответствии с фармако
пей ной процедурой до момента объединения
фильтра, подверrшеrося всем указанным мани
пуляциям, с питательной средой, и после этоro
присутствие жизнеспособных клеток может быть
доказано использованием некоторых альтерна
тивных методов. В этом случае при валидации
методики требуется проведение валидации ис
пользуемой системы выделения микроорrаниз
мов, а не испытания в целом.
Общие вопросы. Валидация микробиолоrи
ческой методики это процесс, посредством
котороro экспериментально устанавливают, что
рабочие характеристики методики отвечают Tpe
бованиям предполаrаемой области применения.
Так как микробиолоrические испытания имеют 3
основные области применения, требуются 3 OT
дельных набора валидационных характеристик.
Эти вопросы описаны ниже.
3 2. ВАЛИДАЦИЯ АЛЬТЕРНАТИВНЫХ
КАЧЕСТВЕННЫХ ИСПЫТАНИЙ
ДЛЯ ОПРЕДЕЛЕНИЯ ПРИСУТСТВИЯ
ИЛИ ОТСУТСТВИЯ микРооРrАНИЗМОВ
3 2 1. Правильность и точность
При прямом методе доказательства экви
валентности двух качественных методик они
выполняются одновременно, и определяется
степень их эквивалентности. Примером может
служить сравнение испытаний на стерильность,
при котором проводится сравнение доли поло
жительных и отрицательных результатов, полу
ченных альтернативной методикой относительно
фармакопейной методики для идентичных об
разцов. Однако, в таких случаях, как испытание
на стерильность, незначительное число HeCOB
падающих результатов потребует проведения
тысяч сравнительных испытаний для ycтaHOB
ления эквивалентности методик, что может быть
проблематично.
Более приемлемый способ оценки точно
сти альтернативной качественной методики в
сравнении с фармакопейной методикой заклю
чается в получении степени соrласия результа
тов, полученных при MHoroKpaTHbIx повторах ис
пытаний, выполняемых разными методиками на
разных сериях одноrо и Toro же средства. Пра
вильность и точность альтернативной методи
ки MorYT быть выражены в относительной часто
те ложноположительных и пожноотрицательных
результатов, полученных новой и фармакопей
ной методиками с использованием стандартизи
рованноro обраЗ1..\а. содержащеro небольшое KO
личество микроорrанизмов.
Частота случаев ложноотрицательных pe
зультатов в присутствии образца для двух Me
тодик может оцениваться с использованием
небольших количеств тест микроорrанизмов.
Схема TaKoro испытания похожа на стандартное
780
rосударственная фармакопея Республики Беларусь
бактериостатическое/фунrистатическое испы
тание; однако, количество вносимых в образец
микроорrанизмов должно быть очень малым,
например, около 5 КОЕ на единицу измере
ния. Степень контаминации должна обеспе
чить получение достаточно высокою количества
неудовлетворительных результатов, чтобы
можно было провести статистически значимое
сравнение двух методик. Альтернативная Me
тодика должна обеспечивать получение такоro
же количества положительных результатов или
выше, как и фармакопейная.
3 2 2. Специфичность
Специфичность альтернативной качествен
ной методики заключается в ее способности
обнаружить определенный диапазон микроор
rанизмов, которые MOryT присутствовать в ис
пытуемом образце. Этот параметр адекватно
оценивается путем оценки ростовых свойств пи
тательной среды для качественных методик, KO
торые основаны на наличии роста микроорrаниз
мов, для тою, чтобы подтвердить их присутствие
или отсутствие. Для методик, которые не Tpe
буют роста как индикатора микробноro присут
ствия, оценка специфичности подтверждает. что
постороннее вещество в аналитической системе
не мешает испытанию. В соответствующих слу
чаях для целей испытания в процессе валида
ции используются смеси микроорrанизмов.
3 2 3. Предел обнаружения
Предел обнаружения альтернативной Ka
чественной методики характеризуется мини
мальным числом микроорrанизмов в образце,
которое может быть определено в установлен
ных экспериментальных условиях. Испытание
на предельное содержание микроорrанизмов
определяет присутствие или отсутствие микро
орrанизмов. С учетом природы микробиолоrии
предел обнаружения означает число микроор
rанизмов, присутствующих в исходном образце
до разведения или инкубации, и не относится к
числу микроорrанизмов, присутствующих в об
разце в процессе испытания.
Обе методики (альтернативная и фармако
пейная) должны оцениваться с использованием
инокулята, содержащею незначительное число
тест микроорrанизмов, например, около 5 КОЕ
на единицу измерения, с последующей оценкой
обнаружения микроорrанизмов. Степень KOH
таминации посевноro материала должна быть
доведена до такоro уровня, Korдa рост микро
орrанизмов обнаруживается фармакопейной
методикой не менее чем у 50 % образцов. Необ
ходимо провести такое определение несколько
раз, поскольку предел обнаружения испытания
определяется на соответствующем числе по
второв (для образца не менее 5). Способность
обеих методик обнаруживать присутствие еди
ничных микроорrанизмов может быть оценена с
помощью критерия хи квадрат (',(2).
3 24. Робастность
Робастность альтернативной качествен
ной методики это степень ее способности не
подверrаться влиянию малых задаваемых из
менений в условиях выполнения методики. Po
бастность являться показателем надежности
методики при ее использовании в измененных
стандартных условиях, таких как разные анали
тики, приборы, серии реактивов и лаборатории.
Робастность может быть определена как соб
ственное сопротивление влиянию, оказываемо
му переменными факторами условий испытания
и окружающей среды, на результаты микробио
лоrическоro метода. Робастность это та вали
дационная характеристика, которая определяет
ся разработчиком методики, но если основные
параметры изменены пользователем, их влияние
на робастность должно быть оценено. Робаст
ность качественной методики оценивается по ее
способности определить тест микроорrанизмы
при задаваемых изменениях условий методики.
3 3. ВАЛИДАЦИЯ АЛЬТЕРНАТИВНЫХ
КОЛИЧЕСТВЕННЫХ ИСПЫТАНИЙ
ПО ОПРЕДЕЛЕНИЮ ОБЩЕrо ЧИСЛА
микРооРrАНИЗМОВ
3 3 1. Правильность
Правильность альтернативной количествен
ной методики характеризуется близостью резуль
татов испытания, полученных альтернативной
методикой. к значению, полученному фармако
пей ной методикой. Правильность может быть
доказана рядом практических испытаний. Пра
вильность обычно выражается как процент об
наруженных микроорrанизмов С помощью MeTO
дики.
Правильность может быть оценена путем
приrотовления суспензии микроорrанизмов на
максимальном значении диапазона испытания,
затем серийно разведенной до наименьшеro
значения диапазона испытания. Например, если
считается, что альтернативный метод заменя
ет традиционный чашечный метод подсчета для
определения числа жизнеспособных микроорrа
низмов, Torдa приемлемый диапазон может co
ставлять 1 OO 106 КОЕ/мл. Если альтернатив
ной методикой заменяется НВЧ метод, может
использоваться еще более узкий диапазон. Не
менее 5 суспензий должны быть проанализи
рованы в пределах диапазона испытания для
каждоrо тест микроорrанизма. Если подразуме
вается, что альтернативная методика заменит
общепринятый метод, она должна обеспечивать
проведение оценки жизнеспособных микроорrа
низмов на уровне не менее 70 % результата, по
лучаемоrо фармакопейной методикой.
Методика, используемая для проверки ли
нейности метода (см. раздел 3 3 5), может
также использоваться для проверки правильно
сти: суспензии микроорrанизмов, приroтовлен
ные для оценки альтернативной методики, под
считываются одновременно с использованием
5.1.6. Альтернативные методы контроля микробиоло;шческой чистоты
781
фармакопейной методики. Правильность счи
тается доказанной, если результаты испытания
приюдности показывают, что наклон линии pe
rрессии существенно не отличается от 1 и если
отрезок, отсекаемый на оси У, существенно не
отличается от О.
3 3 2. Точность
Точность альтернативной количественной
методики это степень соrласия результатов
индивидуальных испытаний при выполнении Me
тодики на мноroкратных испытуемых образцах
однородных суспензий микроорrанизмов в YCTa
новленных условиях. Обычно точность выража
ется в виде дисперсии, стандартноro отклонения
или относительноro стандартноro отклонения
(коэффициент вариабельности) серии измере
ний.
Суспензия микроорrанизмов с KOHцeHTpa
цией обычно в середине диапазона измеряет
ся как минимум несколько раз. Число повторов
определяется таким образом, чтобы протокол
оценки точности был выполнен полностью в Te
чение одной рабочей смены, т. е. при одинако
вых рабочих условиях и без изменений в суспен
зии микроорrанизмов. Затем оценка проводится
в друrие рабочие смены при максимально изме
ненных условиях (различные реактивы, испол
нители, дни, и так далее). Рассчитывают диспер
сию результатов, полученных в каждой рабочей
смене (<<rруппе»). Если дисперсия результатов
rруппы незначительна, может быть рассчитана
дисперсия для оценки сходимости результатов.
Рассчитывается межrрупповая дисперсия. Дис
персия для оценки промежуточной точности
это сумма дисперсии для сходимости и меж
rрупповой дисперсии. Затем вычисляются OT
носительные стандартные отклонения (коэф
фициенты вариабельности). Обычно значение
относительноro стандартноro отклонения в диа
пазоне от 10 % до 15 % является приемлемым.
Безотносительно определенных результатов OT
носительное стандартное отклонение, получен
ное для альтернативной методики, не должно
превышать значение, полученное для фармако
пей ной методики.
3 3 3. Специфичность
Специфичность альтернативной количе
ственной методики проверяется при использо
вании ряда подходящих микроорrанизмов. В
определенных случаях для целей испытания в
процессе валидации используют смеси микро
орrанизмов.
3 3 4. Предел количественноrо опреде
ления
Предел количественноro определения аль
тернативной количественной методики это
минимальное число микроорrанизмов, которое
может быть точно подсчитано. Так как невозмож
но получить достоверный образец, содержащий
]ОО.Зак 1712
известное число микроорrанизмов, необходи
мо, чтобы предел количественноro определения
рассчитывался из количества повторов. напри
мер, не менее 5. MOryT быть также использова
ны результаты исследований линейности и пра
вильности. При этом считается, что наименьшая
концентрация в линейном диапазоне является
пределом количественноrо определения MeTO
дики. Предел количественноro определения аль
тернативной методики не должен превышать па
раметры фармакопейной методики.
3 3 5. Линейность
Линейность альтернативной количествен
ной методики это ее способность обеспечи
вать получение результатов, которые пропор
циональны концентрации микроорrанизмов,
присутствующих в образце в пределах опреде
ленноrо диапазона. Линейность должна опре
деляться в пределах, соответствующих назна
чению альтернативной методики. Методика
определения должна включать выбор разных
концентраций каждою тест микроорrанизма и
проведение нескольких повторов для каждой
концентрации. Число повторов выбирается так,
чтобы все испытание моrло выполняться в Te
чение рабочей смены. Затем друrие 2 рабочие
смены выполняются в условиях максимальной
изменчивости (различные реактивы, исполни
тели, дни, и так далее). После оценки OДHOpOД
ности дисперсии результатов, полученных для
каждой концентрации, рассчитывается ypaBHe
ние линейной реrрессии. Линейность считает
ся подтвержденной, если рассчитанный наклон
кривой значителен, и если при проверке откло
нение от линейности несущественно.
3 3 6. Диапазон применения
Диапазоном применения альтернативной
количественной методики является интервал
между минимальным и максимальным количе
ствами микроорrанизмов, которые были опреде
лены с помощью описанной методики с надлежа
щей точностью, правильностью и линейностью.
Диапазон применения устанавливается по pe
зультатам оценки точности, правильности и ли
нейности.
3 3 7. Робастность
Робастность альтернативной количествен
ной методики это степень ее способности не
подверrаться влиянию малых, задаваемых изме
нений в условиях выполнения методики. Робаст
ностьявляться показателем надежности методики
при ее использовании в измененных стандартных
условиях, таких как разные аналитики, приборы,
серии реактивов и лаборатории. Робастность
может быть определена как собственное сопро
тивление влиянию, оказываемому переменны
ми факторами условий испытания и окружаю
щей среды, на результаты микробиолоrическоro
метода. Робастность это та валидационная
782
rосударственная фармакопея Республики Беларусь
характеристика, которая определяется разработ
чиком методики, но если основные параметры
изменены пользователем, их влияние на робаст
ность должно быть оценено. Робастность коли
чественной методики оценивается по ее спо
собности подсчитать тест микроорrанизмы при
задаваемых изменениях условий методики со
статистической значимостью.
3-4. ВАЛИДАЦИЯ АЛЬТЕРНАТИВНЫХ
ИСПЫТАНИЙ НА ПОДЛИННОСТЬ
Существует большое количество доказа
тельств, что разные методики MorYT значительно
различаться по их способности идентифициро
вать микроорrанизмы в лекарственных cpeд
ствах. Необходимо учитывать, что метод клас
сифицирования должен быть совместимым со
своей внутренней системой, но может отличать
ся от друrих способов идентификации микро
орrанизмов. Друrими словами, идентификация
штаммов, основанная на биохимической актив
ности, может привести к одному заключению,
идентификация с помощью анализа жирных
кислот к друroму, идентификация с помощью
ДНК анализа к третьему, и друrие методы
MOryT привести к альтернативным заключениям.
Микробиолоrическая идентификация опреде
ленной системой основана непосредственно на
предыдущем опыте применения данной систе
мы, и, таким образом, может сильно отличаться
от идентификации друroй системой. Крайне He
обходимо, чтобы каждая система обеспечивала
одинаковую идентификацию штаммов из лекар
ственных средств.
3 4 1. Правильность
Правильность альтернативных испытаний
на подлинность это ИХ способность опреде
лить искомый микроорrанизм на требуемом TaK
сономическом уровне и дифференцировать еro
от друrих микроорrанизмов, присутствующих в
образце. Правильность должна быть доказана
на сериях тест микроорrанизмов, полученных из
типичноro образца и предварительно идентифи
цированных друrим методом
З 4 3. Точность
Точность альтернативных испытаний на
подлинность это степень соrласия результа
тов индивидуальных испытаний при выполнении
методики на мноroкратных испытуемых образ
цах суспензий микроорrанизмов на всем диапа
зоне методики.
3 4 4. Робастность
Робастность испытаний на подлинность
это степень их способности не подверrаться вли
янию малых задаваемых изменений в условиях
выполнения методики. Робастность являться по
казателем надежности методики при ее использо
вании в измененных стандартных условиях, таких
как разные аналитики, приборы, серии реактивов
и лаборатории. Робастность может быть опреде
лена как собственное сопротивление влиянию,
оказываемому переменными факторами усло
вий испытания и окружающей среды, на результа
ты микробиолоrическоro метода. Робастность
это та валидационная характеристика, которая
определяется разработчиком методики, но если
основные параметры изменены пользователем,
их влияние на робастность должно быть oцeHe
но. Робастность испытаний на подлинность oцe
нивается по их способности определить тест
микроорrанизмы при задаваемых изменениях
условий методики.
4. ЧАСТНЫЕ ТРЕБОВАНИЯ К ВАЛИДАЦИИ
4 1. ОБЩИЕ СВЕДЕНИЯ
Значение термина «валидация» приводит
СЯ в различных контекстах с некоторыми разли
чиями, но принципиально валидация определя
ется как процесс получения документированных
доказательств тоro, что процесс будет едино
образно удовлетворять критериям своеro пред
назначения. Следовательно, для проведения
корректной валидации новой методики крайне
необходимо понять и определить критерии пред
назначения валидируемой процедуры.
Для общепринятых или альтернативных ми
кробиолоrических методик должны быть предус
мотрены два уровня валидации. Обычно пер
вичная валидация выполняется разработчиком
новой методики, в то время как валидация для
фактическоro использования по предназначе
нию, которая представляет собой подтвержде
ние приroдности или применимости методики в
определенной ситуации, должна рассматривать
ся как ответственность пользователя. Перед Ba
лидацией для фактическоro использования по
предназначению пользователем проводится
квалификация эксплуатации, как описано в раз
деле 3. Общие требования к валидации.
Обычно в качестве индикаторов или основы
для детектирования для более оБLЦИХ задач в
микробиолоrических методиках используются
специфические характеристики микроорrаниз
мов. Получаемая с помощью методики требу
емая информация включает присутствие, коли
чество, жизнеспособность, резистентность или
идентификацию микроорrанизмов в данном ле
карственном средстве или окружаЮLЦей среде.
Определенная методика обычно дает непря
мой или условный ответ на вопросы. Например,
общее число и жизнеспособность микроорrаниз
мов определяются как количество микроорrа
низмов, способных к размножению при опреде
ленном наборе условий приroтовления образца,
культивирования и инкубации. В классической
микробиолоrии размножение, соответственно,
считается общим индикатором жизнеспособ
ности. Однако СУLЦествуют друrие параметры,
которые MorYT быть использованы как индика
торы жизнеспособности. Уровень АТФ, накопле
5.1.6. Альтернативные методы контроля микробиОЛО2ической чистоты
783
ние или метаболизм субстратов в живых клетках
также MOryт служить индикаторами жизнеспо
собности. Результаты различных методов опре
деления жизнеспособности не Bcerдa MOryT быть
одинаковы. Микроорrанизмы не Bcerдa MOryT
размножаться на данной питательной среде, но
MOryT, тем не менее, аккумулировать и метабо
лизировать субстрат. Микроорrанизмы в опре
деленном поврежденном состоянии MOryT быть
неспособны аккумулировать субстрат, но MOryT,
тем не менее, быть способны к восстановлению
и размножению.
Друrим примером являются различные Me
тодики, используемые для идентификации ми
кроорrанизмов. Для видовой идентификации
часто используются характерные особенности
метаболизма микроорrанизмов, а в друroй Meтo
дике проводится сравнение ДНК последователь
ностей. Результаты, полученные с помощью раз
личных методик, MOryT совпадать не полностью,
и один ответ может быть приrодным дЛЯ KOH
струкции корректноro филоrенетическоro KOp
реляционноro древа, друrой ответ может быть
более полезным в контексте патоrенности или
друrих свойств дифференцированных микроор
rанизмов.
4 1 1. Первичная валидация
Принцип детектирования должен быть четко
описан разработчиком, чтобы охарактеризовать
определенную микробиолоrическую методику.
Методика должна содержать подробное опи
сание требуемых условий, необходимых MaTe
риалов и оборудования и ожидаемоro сиrнала.
Принцип применения должен быть описан в pe
цензируемом научном журнале.
Принцип детектирования должен быть oxa
рактеризован на модельной системе и/или с па
нелью тест микроорrанизмов по крайней мере
по следующим параметрам:
предварительная обработка образца или
микроорrанизмов;
тип ответа;
специфичность ответа;
предел определения;
диапазон;
линейность ответа;
правильность и точность ответа;
робастность методики в модельной системе;
пределы приroдности.
Если методика была охарактеризована раз
работчиком по перечисленным параметрам, то
принцип детектирования не подлежит проверке
каждым пользователем.
4 1 2. Валидация альтернативной микро
биолоrической методики
4 1 2 1. Анализ риск/польза
Для валидации специфических алыерна
тивных микробиолоrических методик крайне
важно точно описать назначение испытания. На
основании назначения должны быть определе
ны тип и объем получаемой информации. Об
щепринятая и альтернативная методики должны
быть оценены в отношении получаемой инфор
мации и возможных оrраничений и сопоставле
ны путем анализа отношения риск/польза.
Апьтернативная методика может быть Ha
звана приroдной, если получаемая с ее помо
щью информация дает научно обоснованный
ответ на вопросы, поставленные испытанием, и
если степень оrраничения применения методи
ки не превышает оrраничения стандартной Me
тодики.
4 1 2 2. Валидация фактичеСК020 исполь
зования по назначению
Пользователь должен проверить, что pe
зультаты испытания, полученные с использова
нием альтернативной методики на еro образцах,
сопоставимы с результатами, полученными раз
работчиком в модельной системе.
По возможности должно быть оценено сле
дующее:
сочетаемость ответа с приroтовлением об
разца, необходимым для испытания лекарствен
Horo средства;
предел и диапазон детектирования MeTO
дики с учетом размера имеющейся пробы;
специфичность ответа с учетом меша
ющеro влияния компонентов лекарственноro
средства;
линейность ответа относительно всех
типов испытуемых образцов;
правильность и точность ответа относи
тельно всех типов испытуемых образцов;
робастность методики относительно всех
типов испытуемых образцов.
Необходимо определить критерии приемле
мости методики при повседневном использова
нии в зависимости от цели применения и вали
дационных данных.
4 2. БИОЛЮМИНЕСЦЕНЦИЯ
ДЛЯ ОПРЕДЕЛЕНИЯ КОЛИЧЕСТВА
МИКРООР,АНИ3МОВ
4 2 1. Анализ риск/польза
Обширные научные данные и мноroлет
нее использование подтверждают возможность
использования АТФ как маркера жизнеспо
собности для обнаружения тoro же диапазо
на микроорrанизмов, которые выявляются с ис
пользованием стандартных чашечных методов.
Так как данный метод основан на росте микро
орrанизмов, ero преимуществом по cpaBHe
нию с чашечными методами является быстрое
получение результата (от 5 дней при примене
нии чашечных методов до 24 ч при применен и
биолюминесценции). Существует возможность
идентифицировать микроорrанизмы, обнаружи
ваемые биолюминесценцией, на стадии инкуби
рования в питательной среде, но при этом He
обходимо учитывать, что в смешанной культуре
некоторые микроорrанизмы MOryT yrHeTaTb рост
784
lосударственная фармакопея Республики Беларусь
друrих микроорrанизмов в процессе инкубации.
Этот метод позволяет оценить образцы в тече
ние 24 ч для фильтруемых инефильтруемых
проб (вода, внутрипроизводственный контроль,
образцы окружающей среды, твердые и жидкие
исходные материалы, твердые и жидкие roтовые
лекарственные средства и т. д.) И оценить боль
шое количество образцов, Korдa стадия детекти
рования автоматизирована.
4 2 2. Валидация фактическоrо использо-
вания по назначению
Метод основан на детектировании А ТФ жиз
неспособных микроорrанизмов. Квалификация
эксплуатации проводится с применением TeCT
микроорrанизмов с целью подтверждения тоro,
что в лабораторных условиях пользователя воз
можно соответствие критериям. указанным раз
работчиком, относительно воспроизводимости,
точности и линейности (количественный метод)
или предела обнаружения (качественный и полу
количественный метод) в пределах диапазона,
необходимоrо для использования по назначе
нию. Исходя из этой цели, валидация выполня
ется в три фазы:
фаза 1: оценка ростовых свойств пита
тельной среды в присутствии испытуемоro об
разца (если про водится стадия инкубации);
фаза 2: поиск мешающих воздействий, KO
торые MOryт увеличивать или инrибировать об
разование АТФ (путем добавления стандартноro
раствора АТФ к испытуемому образцу);
фаза 3: сравнительное исследование с
фармакопейной методикой.
Подробный при мер валидации биолюми
несцентноro метода приведен в конце данноro
раздела.
4 3. ЦИТОМЕТРИЯ (ТВЕРДОФАЗНАЯ
И ПРОТОЧНАЯ) ДЛЯ ОПРЕДЕЛЕНИЯ
КОЛИЧЕСТВА микРооРrАНИЗМОВ
4 З 1. Анализ риск/польза
Обширные научные данные подтвержда
ют способность флуоресцентноro маркера жиз
неспособности обнаружить и/или подсчитать
более широкий диапазон микроорrанизмов, чем
при использовании стандартных чашечных Me
тодов. Цитометрия позволяет определить все
жизнеспособные микроорrанизмы. включая те,
что не распознаются методами, основанными на
росте. Несмотря на то, что данный метод обла
дает преимуществом в отношении скорости, еro
возможности по открываемости обнаруженных
микроорrанизмов оrраничены. Таким образом,
для дальнейшей идентификации мо)'{ет потре
боваться обработка альтернативными флуо
ресцентными красителями или использование
альтернативной методики. В настоящее время
невозможно использовать данный метод для
стандартной идентификации микроорrанизмов,
хотя характерные морфолоrические признаки
леrко определяются при твердофазной цитоме
трии С применением флуоресцентных микро
скопов. Данный метод обеспечивает быструю
оценку образцов и, следовательно, позволяет
использование упреждающеro подхода в фар
мацевтическом производстве, способствуя обе
спечению качества производственных операций.
Данный метод не зависит от роста, и, следова
тельно, все метаболически активные микроор
rанизмы будут обнаружены. Однако предел об
наружения методик проточной цитометрии не
позволяет определить общее количество микро
орrанизмов прямым методом для большинства
фармацевтических образцов. Если необходима
предварительная инкубация, оценка становится
полуколичественной (испытание на предельное
испытание ).
4 3 2. Валидация фактическоrо использо-
вания по назначению
Метод основан на детектировании флуорес
центноro сиrнала от маркированных микроорrа
низмов.
Квалификация эксплуатации проводится
для доказательства тоro, что приборы работают
в определенных операционных параметрах. Для
этоro используются флуоресцентные CTaHдap
ты с требуемой интенсивностью и культуры с из
вестным типом и количеством микроорrанизмов.
Этими испытаниями проверяют систему количе
ственноro определения. Для оценки приroдно
сти повседневной методики испытания и степе
ни влияния качества расходных материалов на
получаемые результаты при валидации также
должны быть проведены испытания с реакти
вами и расходными материалами (отрицатель
ные контроли). Испытания на чистой культуре
тест микроорrанизмов проводятся для оценки
системы обнаружения и сравнения результатов
испытания, полученных альтернативной MeTO
дикой, с результатами, полученными с исполь
зованием стандартноro определения количе
ства микроорrанизмов чашечным методом. Для
оценки линейности, правильности, точности, ди
апазона, специфичности, предела количествен
ноro определения (количественный метод) и
предела определения (проточная цитометрия
с предварительной инкубационной стадией)
должны использоваться мноroкратные повторы
(не менее 5) суточных культур, разведенных в
пределах диапазона концентрации (например,
100 %, 75 %, 50 %, 25 % и 10 %). Так как у цито
метрии высокая чувствительность (твердофаз
ная цитометрия может обнаружить единичные
клетки, Torдa как проточная цитометрия чувстви
тельна до уровня 1 0 50 клетокlмл), и обнзруже
ние не основано на росте, линейность приборов
может оцениваться путем сравнения фактиче
ских результатов С ожидаемым значением.
Исходя из задач, валидация выполняется в
две фазы: валидация в отношении испытуемоro
образца и сравнительное испытание. Результаты
каждой фазы должны оцениваться относительно
5.1.6. Альтернативные методы контроля мuкробиолоzической чистоты
785
предварительно установленных критериев при
емлемости с использованием положительных и
отрицательных контролей:
фаза 1: для подтверждения отсутствия
влияния процесса приroтовления образца и
самих образцов на функционирование систе
мы обнаружения в индивидуальные матери
алы, оцениваемые методом цитометрии, должно
быть добавлено определенное количество ми
кроорrанизмов; в частности матрица (среда) об
разца не должна влиять на обнаружение (то есть
не должна содержать эндоrенных хромофоров,
автофлуоресцентных частиц), и в случае про
точной цитометрии для оптимальноrо испыта
ния должны быть определены размер образца/
разведение и скорость потока;
фаза 2: должно быть проведено испыта
ние, в котором сравниваются результаты, по
лученные с помощью цитометрии и фармако
пейноro метода; в протоколе сравнительноrо
испытания должно быть определено число об
разцов и время испытания; число образцов
может быть разным, но должно быть показа
тельным процедуре оценки материала (то есть
время/количество) и должно обеспечивать CTa
тистическую оценку; все образцы должны быть
приroтовлены в соответствии с определенными
процедурами и оценены относительно выбран
ных валидационных характеристик и критериев
приемлемости, подобных тем, что используются
для оценки чистой культуры.
4А. ПРОФИЛИ ЖИРНЫХ КИСЛОТ
ДЛЯ ИДЕНТИФИКАЦИИ
4A 1. Анализ риск/польза
Идентификация с помощью профилей
жирных кислот может быть более точной, чем
идентификационные методы, основанные на Me
таболических профилях в традиционных микро
биолоrических культурных методах. База данных
охватывает больше микроорrанизмов. чем Tpa
диционные культуральные методы. Для дaHHO
ro метода необходима предварительная инкуба
ция, но экстракция и идентификация занимает
меньше времени, чем биохимические методы,
и, следовательно, результаты получаются бы
стрее. Друrие современные методы, такие как
анализ последовательности 16S рРНК или re
нетические «отпечатки пальцев», имеют схожий
широкий диапазон дифференциации и дают pe
зультат так же быстро, как и данный метод.
Разделение близкородственных микроорrа
низмов (например, Е. соН и Salmoпella spp.) с по
мощью профилей жирных кислот может быть за
труднительно. В случае, Korдa идентификация
близкородственных микроорrанизм()в особенно
важна, друrие системы MOryT дать более точные
результаты. Для использования с этой целью
важно определить наиболее критичные типы
микроорrанизмов, подлежащие идентификации.
Если требуется охарактеризовать корректные
филоrенетические виды изолированных микро
орrанизмов, методы идентификации, OCHOBaH
ные на ДНК последовательности, дадут более
точные результаты.
Оrраничения идентификации с помощью
профилей жирных кислот связаны также с необ
ходимостью выращивания микроорrанизмов на
стандартных средах при стандартных темпера
турных условиях и длительности инкубации. Ми
кроорrанизмы, которые невозможно культивиро
вать на таких средах, идентифицировать нельзя.
4 4 2. Валидация фактическоrо использо
вания по назначению
Для подтверждения единообразия получа
емых результатов проводят испытания с исполь
зованием диапазона тест микроорrанизмов и,
по крайней мере, в трех повторах в каждом
случае.
Как минимум должна быть проведена
трехкратная идентификация каждоrо из дo
статочноro количества микроорrанизмов, BЫ
деляемых из типичных анализируемых пользо
вателем образцов. Результаты в каждом случае
должны быть однородны и должны совпадать
с результатами идентификации, полученны
ми с использованием альтернативных методов
идентификации. При получении разных резуль
татов идентификации с помощью друroй систе
мы идентификации причина этоro несходства
должна быть исследована. Различия между си
стемами идентификации MOryT быть прием
лемыми при наличии научно обоснованных
объяснений по распознаванию различных видов.
В таком случае должно быть доказано, что pac
познавание идентифицированных видов являет
ся робастным. Также должно быть доказано, что
система не относит плохо распознанные изоли
рованные микроорrанизмы к одному «виду», си
мулируя таким образом повторяющуюся изоля
цию единичных видов.
4 5. МЕТОДИКИ, ОСНОВАННЫЕ
НА АМПЛИФИКАЦИИ НУКЛЕИНОВЫХ
КИСЛОТ
4 5 1 . Анализ риск/польза
МАНК широко используется в диаrностике.
блаroдаря своей точности и быстроте получения
результатов при относительно низкой стоимо
сти (для анализа, но не для приборов) по cpaB
нению с традиционными методами. При условии
проведения определенных работ по валида
ции и надлежащеro выполнения методик МАНК
MorYT иметь ряд преимушеств в некоторых обла
стях по сравнению с классическими методами,
с друroй стороны. классические методы леrче
стандартизуются. не требуют высококвалифи
цированноrо персонала и MOryт иметь низкую
стоимость Даже коrда выполнение МАНК не яв
ляется более трудным по сравнению с традици
онными методами. интерпретация результатов
обычно требует высокой степени научной KOM
петенции.
786
rосударственная фармакопея Республики Беларусь
При использовании для идентификации Me
тодов, основанных на использовании ДНК, He
обходимо учитывать, что они не позволяют раз
личать мертвые и живые микроорrанизмы. Это
означает, что их невозможно использовать для
прямоro проведения испытания, и требуется
пассаж на традиционной питательной среде,
вследствие чеrо часть преимуществ в быстро
те испытания теряется. Более тoro, при ис
пользовании непосредственно в конце анализа
лекарственноro средства, эти методы не обе
спечивают получение культуры для дальней
шеro исследования и MOryT не иметь ряд пре
имуществ в случае, Korдa обнаруживаемые
микроорrанизмы плохо культивируются или
подверrаются стрессу. Методики амплифика
цИИ РНК (например, полимеразная цепная pe
акция с обратной транскрипцией) MOryT иден
тифицировать живые микроорrанизмы (но не
споры) непосредственно в лекарственных cpeд
ствах, но по сравнению с традиционными MeTO
дами их более сложно использовать реryлярно
(в рабочем порядке). С друrой стороны, при ис
пользовании специфических праймеров иден
тификация (или типирование) с помощью МАНК
является более точной по сравнению с традици
онными методами и в некоторых случаях может
иметь друrие преимущества: например, для
идентификации некоторых вакцин (например,
вакцина для профилактики холеры, вакцина
для профилактики коклюша цельноклеточная)
их использование может заменять специфиче
ские сыворотки и способствовать уменьшению
использования животных или обеспечить BЫCO
коспецифичную идентификацию, rдe она в Ha
стоящее время отсутствует (например, вакцина
БЦЖ).
Как правило, данные методы являются He
количественными (ПЦР) или полуколичествен
ными (ПЦР в реальном времени). Их результа
ты не MOryT быть сопоставлены с результатами
подсчета колоний, rдe требуется точный подсчет
присутствующих микроорrанизмов в образце.
Но даже если подсчет колоний имеет проверен
ную временем достоверность, это положение не
может не подтверждаться для бактерий, KOТO
рые имеют тенденцию к «сливному росту» (ми
кобактерии) или образованию цепочек или CKO
плений (стрептококки, стафилококки), поэтому
точная стандартизация полуколичественных Me
ТОДОВ может давать результаты сравнимой дo
стоверности.
4 5 2. Валидация фактическоrо использо-
вания по назначению
Данный метод валидируетr.я в соответствии
с общей статьей 2.6.21. Сравнение традицион
ных методолоrий и методолоrий, основанных на
ПЦР, которые различаются по чувствительности
и специфичности, является особенно затрудни
тельным и может приводить к противоречивым
заключениям.
Следующий пример приведен для информа
ции и не при20ден для обще20 применения.
При мер валидации альтернативной
методики: подробный протокол, который
выполняется лабораторией для внедрения
биолюминесценции
ОБЩИЕ ПОЛОЖЕНИЯ
Методы, в которых используется стадия пред
варительной инкубации в жидкой среде (биолю
минесценция в пробирке или на микропланшете),
не позволяют получить количественную инфор
мацию и обеспечивают только лишь определе
ние присутствия/отсутствия микроорrанизмов в
анализируемом образце. При использовании He
скольких проб разною объема может быть прове
дено полуколичественное определение (испыта
ние на предельное содержание). Например, для
классическоro испытания на определение общеro
количества жизнеспособных аэробных микроор
rанизмов в нестерильных продуктах, испытуемый
объем образца составляет 0,1 r или 0,1 мл. Это
означает отсутствие микроорrанизмов в 0,1 r или
0,1 мл при получении отрицательноro результата,
то есть менее 1 О микроорrанизмов в 1 r или 1 мл,
И наличие не менее 1 О микроорrанизмов в 1 r или
1 мл при положительном результате. Если ис
пытываются одновременно 0,01 r или 0,01 мл, OT
рицательный результат соответствует количеству
микроорrанизмов менее 100 в 1 r или 1 мл. Комби
нация отрицательною результата для 0,01 r или
0,01 мл и положительною результата для 0,1 r
или 0,1 мл позволяет оценивать уровень контами
нации продукта менее 100, но более или равно 1 О
микроорrанизмам в 1 r или 1 мл.
Как упомянуто в разделе 2, биолюминесцен
ция может использоваться как количественный
метод, если микроорrанизмы выделяют на филь
трационную мембрану и в дальнейшем инкуби
руют в питательной среде (биолюминесценция
на мембране).
Протокол, представленный ниже, описывает
подходыквалидациикачествеННОЙ,полуколиче
ственной и количественной методик.
КВАЛИФИКАЦИЯ ЭКСПЛУАТАЦИИ
АЛЬТЕРНАТИВНОЙ МЕТОДИКИ
Специфичность
Проводят скрининr метода с TeCT
микроорrанизмами, подходящими для данной
методики. Например, для подсчета жизнеспо
собных аэробных микроорrанизмов на HeCTe
рильных лекарственных продуктах, используют
как минимум ряд микроорrанизмор, описанных в
статье 2.6. 12 для оценки ростовых свойств пита
тельных сред в присутствии продукта. Это опре
деление выполняется не менее 3 раз с каждым
микроорrанизмом. Критерий приемлемости: все
тест микроорrанизмы должны быть обнаружены
во всех повторах.
5.1.6. Альтернативные методы контроля микробиолоzической чистоты
787
Предел обнаружения (только для полуко-
личественноrо или качественноrо метода)
rотовят посевной материал с незначитель
ным содержанием (около 5 КОЕ в исходном об
разце) каждоro испытуемоro микроорrанизма.
Выполняют анализ как минимум 5 раз фарма
копейной методикой и соответствующей биолю
минесцентной методикой. Критерий приемле
мости: возможность двух методик определять
присутствие единичноro микроорrанизма может
быть оценена с помощью метода хи квадрат (х 2 ).
Альтернативная процедура: roтовят серию KpaT
ных разведений суспензий микроорrанизмов
(например, 10 КОЕ/инокулят. 2,5 КОЕ/инокулят,
1,25 КОЕ/инокулят, 0.75 КОЕ/инокулят). Выпол
няют испытание на 5 независимых сериях раз
ведений фармакопейной методикой и COOT
ветствующей биолюминесцентной методикой.
Определяют предел обнаружения для каждой
методики. Он соответствует последнему разве
дению, для котороro результаты положитель
ны для всех 5 серий. Критерий приемлемости:
предел обнаружения биолюминесцентной MeTO
дики должен быть равен или меньше предела
обнаружения фармакопейной методики.
Предел количественноrо определения
(количественный метод)
Предел количественноrо определения
может быть оценен одновременно при опре
делении линейности. Предел количественно
ro определения соответствует наименьшей KOH
центрации в выбранном диапазоне, который
отвечает критериям линейности, правильности
и точности. Критерий приемлемости: предел KO
личественноro определения биолюминесцент
ной методики должен быть равен или меньше
предела количественноro определения фарма
копейной методики. .
Точность
Количественная оценка. Для каждоro TeCT
микроорrанизма выполняется как минимум 5
повторных испытаний одной посевной серии
микроорrанизма с концентрацией микроорrаниз
мов, соответствующей, по крайней мере, cepe
дине диапазона. Выполняют три независимых
испытания. Проводят статистический анализ
для сравнения точности 2 методик или вычисля
ют коэффициент вариации (CV). Критерий при
емлемости: CV должен составлять от 15 % до
30 % или точность двух методик должна быть
одинакова при уровне риска ошибки а=5 %. Если
точность различна, то биолюминесцентная Me
тодика превосходит фармакопейную методику.
Качественная или полуколичественная
оценка. Используют альтернативную процеду
ру, описанную для установления предела обна
ружения, и учитывают частоту положительных
результатов альтернативной методики парал
лельно с фармакопейной методикой. Критерий
приемлемости: частота положительных резуль
татов при пределе обнаружения должна COCTaB
лять 100 %, и эта частота выше или эквивалент
на фармакопейной методике.
Линейность
Для каждоro испытуемоro микроорrанизма
rотовят 5 концентраций в диапазоне биолюми
несцентной методики (диапазон обычно опреде
ляется разработчиком). Параллельно образцы
анализируют фармакопейной и валидируемой
методиками. Повторяют это испытание еще 2
раза, чтобы получить результаты 3 независимых
испытаний. Исследуют на линейную реrрессию,
присутствие наклона и на отсутствие соrласия С
критерием Фишера при 0=5 %. Если статистиче
ский анализ невозможен, вычисляют коэффици
ент корреляции (R2) и наклон между 2 методи
ками. Критерий приемлемости: статистический
анализ может подтверждать линейную perpec
сию, присутствие наклона и соrласие данных с
риском ошибки а=5 %. Рассчитывают ypaBHe
ние линейной реrрессии у=а+Ьх, rдe Ь наклон
и а отрезок, отсекаемый на оси. Если стати
стический анализ невозможен, R2 должен быть
не менее 0,9 и наклон не должен отклоняться
более чем на 20 % от 1 (Ь между 0,8 и 1,2). Если
линейность не доказана в таком широком диапа
зоне, диапазон может быть уменьшен, и линей
ность доказывается только на 3 концентрациях
вместо 5.
Правильность
Количественная оценка. Правильность
может быть определена с помощью данных, по
лученных при изучении линейности. Для каждо
ro микроорrанизма используют от 3 до 5 KOHцeH
траций в пределах линейноro диапазона метода.
Выполняют статистический анализ (критерий
Стьюдента с риском 5 %) для исследования по
добия предполаrаемоro наклона (значение=1)
относительно полученноro наклона и для иссле
дования подобия предполаrаемоro отрезка. OT
секаемоro на оси (значение=О), относительно
полученноro отрезка, oTceKaeMoro на оси. Ha
пример, если предполаrаемый наклон это
Ь со стандартным отклонением S t 0,090 с 5 %
концентрациями микроорrанизмов. рассчитыва
ют t=(b 1 )/S(b)' Для отрезка а, oTceKaeMoro на оси.
со стандартным отклонением. эквивалентным
S(a)' t=(a O)/s(a)' Сравнивают эти значения с коэф
фициентом Стьюдента при 5 % для 13 степеней
свободы (3 испытания, 5 концентраций). Крите
рий приемлемости: если полученные значения t
меньше t Стьюдента, метод является точным в
при меняемом диапазоне. В случае, коrда не cy
ществует подобия для наклоча (наклон отлича
ется от 1) или отрезка, отсекаемоro на оси (OTpe
зок, отсекаемый на оси, отличается от О), метод
не точен на всем применяемом диапазоне.
Кжественная и полуколичественная оценка.
Используют альтернативную процедуру, описан
ную для установления предела обнаружения.
788
rосударственная фармакопея Республики Беларусь
Вычисляют частоту ложноотрицательных pe
зультатов для биолюминесцентной и фармако
пей ной методик для всех испытуемых разведе
ний. Сравнивают частоту ложноотрицательных
результатов для 2 или 3 концентраций микроор
rанизмов несколько ниже предела обнаружения
(например, 5 КОЕ/инокулят, 2,5 КОЕ/инокулят
или 1,25 КОЕ/инокулят), которые дают положи
тельный результат. По умолчанию предел об
наружения соответствует О % ложноотрица
тельных результатов. Критерий приемлемости:
процент ложноотрицательных результатов для
биолюминесцентной методики при KOHцeHTpa
циях микроорrанизмов в пробе ниже предела
определения должен быть эквивалентен или
ниже таковоro в фармакопейной методике.
Диапазон
Это интервал между наименьшей и наиболь
шей концентрациями микроорrанизмов при ДOKa
занных линейности, точности и правильности.
Робастность
Информация приводится разработчиком.
ВАЛИДАЦИЯ ДЛЯ ФАКТИЧЕскоrо
ИСПОЛЬЗОВАНИЯ ПО НАЗНАЧЕНИЮ
Для приведенноro примера не было необхо
димости определять правильность и предел об
наружения в присутствии анализируемоrо про
дукта. Валидация состоит из 3 фаз:
фаза 1: ростовые свойства питательной
среды в присутствии продукта;
фаза 2: отсутствие у продукта мешающих
воздействий, которые MOryT увеличивать или ин
rибировать образование АТФ;
фаза 3: сравнительное исследование па
раллельно с фармакопейной методикой.
Эти 3 валидации проводятся в 3 независи
мых испытаниях с использованием, например,
не менее 2 различных серий продукта.
Фаза 1: ростовые свойства питательной
среды в присутствии продукта
Если известно, что у продукта высокий
уровень контаминации (более 500 микроорrа
низмов в 1 rpaMMe или 1 миллилитре), инку
бационная стадия не требуется, допустимо He
посредственное определение микроорrанизмов
в образце. В этом случае испытание ростовых
свойств питательной среды в присутствии ле
карственноro средства не проводится. Однако,
контаминация лекарственных средств обычно
намноro меньше, и культивирование микроор
rанизмов необходимо для их обнаружения с по
мощью биолюминесценции. Поэтому должно
быть подтверждено, что продукт не инrибирует
рост микроорrанизмов в условиях испытания.
Для этоro в порции среды, содержащей про
дукт, раздельно добавляют суспензии каждо
ro тест микроорrанизма в количестве не более
100 КОЕ. ДЛЯ методик с биолюминесценцией в
пробирке или микропланшете выполняют испы
тание биолюминесценции. Для методик с био
люминесценцией на мембране ее инкубиру
ют при температуре от 30 ос до 35 ос или от
20 ос до 25 ос в течение 5 дней и подсчитыва
ют биолюминесцентные колонии на мембране.
Критерий приемлемости: результат испытания
должен быть положительным (биолюминесцен
ция в пробирке или микропланшете); количе
ственное выделение микроорrанизма должно
составлять не менее 70 % (биолюминесценция
на мембране).
Фаза 2: поиск мешающих воздействий
продукта
Цель данной фазы доказать, что лекар
ственное средство не увеличивает рассеянный
свет или содержание немикробной АТФ (не при
водит к ложноположительным результатам: кри
терий А) или не снижает количество обнаружива
ем ой АТФ (не приводит к ложноотрицательным
результатам: критерий В).
Биолюминесценция в пробирке или MиKpO
планшете
А. Выполняют испытание биолюминесцен
ции с питательным бульоном без добавок и с пи
тательным бульоном в присутствии продукта.
Определяют значение RLU (интенсивности сиr
нала люминесценции, относительные световые
единицы) для питательноro бульона и значение
RLU для питательноro бульона в присутствии
продукта.
В. Выполняют определение биолюминес
ценции с питательным бульоном без добавок
и с питательным бульоном в присутствии АТФ.
Определяют коэффициент отклика для KOHцeH
трации АТФ в процентах.
Критерии приемлемости:
критерий А: значение RLU питательноro
бульона в присутствии лекарственноro средства
должно быть меньше удвоенноro значения RLU
питательноro бульона без добавок (если крите
рий А не соответствует требованиям, необходи
мо определить индивидуальную пороroвую Be
личину для этоro лекарственноro средства);
критерий В: значения RLU питательноro
бульона в присутствии лекарственноro средства
и АТФ должно находиться в пределах интервала
от 25 % до 200 % значения RLU питательноro бу
льона С добавлением АТФ.
Биолюминесценция на мембране: выполня
ют полное биолюминесцентное испытание для
поиска мешающеro действия. Критерий при
емлемости: открываемость микроорrанизмов
должна быть выше или равна 70 %, но не более
200%.
Фаза 3: сравнительное исследование
продукта параллельно с фармакопейной ме-
тодикой
Для Toro, чтобы доказать взаимосвязь
между 2 методиками для испытуемоro лекар
5.1.8. Мuкробиолоzuческая чистота лекарственных средств
789
ственноro средства выполняют 3 независимые
испытания не менее двух серий анализируемою
лекарственною средства валидируемой методи
кой параллельно с фармакопейной методикой.
Результат выражают как положительный или OT
рицательный в определенном количестве (био
люминесценция в пробирке или микропланшете)
или как количество на фильтруемый объем (био
люминесценция на мембране). Критерий при
емлемости: результаты валидируемой методики
должны коррелировать с фармакопейной MeTO
дикой.
01/2013:50107
5.1.7. ВИРУСНАЯ БЕЗОПАСНОСТЬ
в данной статье приведены общие требо
вания относительно вирусной безопасности
лекарственных средств, при производстве KO
торых используется материал человеческо
ro или животноro происхождения. Так как ви
русная безопасность это сложный вопрос,
важно, чтобы была проведена оценка рисков.
Требования к специфическим лекарственным
средствам устанавливаются компетентным op
raHOM.
В случае существования риска вирусной
контаминации, при необходимости подтвержде
ния вирусной безопасности используются дo
полнительные подходы, основанные на следу
ющих аспектах:
выбор источника материалов и испытание
на вирусные контаминанты;
исследование способности процесса про
изводства удалять и/или инактивировать вирусы;
исследование вирусной контаминации на
соответствующих стадиях производства.
При необходимости используют одну или
более валидированные процедуры для удале
ния или инактивации вирусов.
Более подробные рекомендации по вирус
ной безопасности, включая изучение валидации,
представлены, в частности, в «Примечаниях К
руководству по валидации исследований виру
сов: разработка, проведение и интерпретация
исследований валидации инактивации и удале
ния вирусов (CPMP/BWP/268195)>> Комитета по
патентованным лекарственным препаратам и в
«Руководстве /СН Q5A: Оценка вирусной безо
пасности биотехнолоrических продуктов, полу
ченных из клеточных линий человеческоro либо
животноro происхождения» (включая любые по
следующие пересмотры этих документов).
Оценка рисков
Оценку рисков, относящихся к вирусной безо
пасности, проводят в случае использования Ma
териалов человеческоro и животною происхож
дения в качестве инrредиентов лекарственных
средств или при производстве фармацевтиче
ских субстанций, вспомоrательных веществ или
лекарственных средств.
Принцип оценки рисков это рассмотрение
различных факторов, способных повлиять на
потенциальный уровень инфицирующих частиц
в лекарственном средстве, и факторов, Kaca
ющихся использования лекарственноro cpeд
ства, с помощью котороro определяют или
влияют на вирусный риск для реципиента.
При оценке рисков принимают к paCCMOTpe
нию релевантные факторы, например:
исходные виды;
исходный opraH, ткань, жидкость;
потенциальные контаминанты с точки
зрения происхождения сырья, истории донора/
доноров. желательно, включая эпидемиолоrиче
ские данные;
потенциальные контаминанты в процессе
производства (например. из материалов rруппы
риска, используемых при производстве);
инвазионная способность и патоrенность
потенциальных контаминантов для предполаrа
емых реципиентов лекарственною средства с
учетом способа введения лекарственноro cpeд
ства;
количество материала, использованно
ro для производства дозы лекарственноro cpeд
ства;
контролирование донора/доноров, сырья
при производстве, а также roтовоro лекарствен
ноro средства;
процесс производства лекарственною
средства и еro способность к удалению и/или
инактивации вирусов.
Если процесс производства включает в себя
стадию инактивации вирусов (например, для же
латина и др., а также для средств, проходящих
конечную стерилизацию паром или сухожаровую
стерилизацию, как описано в общих статьях на
стерильность (5.1», оценка рисков может быть
основана, rлавным образом, на условиях произ
водства.
01/2013:50108
5.1.8. МИКРОБиолоrИЧЕСКАЯ
ЧИСТОТА ЛЕКАРСТВЕННЫХ
СРЕДСТВ РАСТИТЕльноrо
ПРОИСХОЖДЕНИЯ ДЛЯ
BHYTPEHHErO ПРИМЕНЕНИЯ
в статье приводятся рекомендуемые Kpи
терии приемлемости для микробиОЛ02ической
чистоты лекарственных средств раститель
Н020 происхождения.
790
rосударственная фармакопея Республики Беларусь
Присутствие некоторых микроорrанизмов
в нестерильных лекарственных средствах по
тенциально может уменьшить терапевтическую
активность продукта или даже инактивировать
ero, кроме этоro существует возможность He
блаroприятноro воздействия на орrанизм паци
ента. Поэтому производители должны обеспе
чить выполнение действующеro руководства по
Надлежащей производственной практике при
производстве, хранении и распространении ле
карственных средств для rарантирования их
низкой биолоrической заrрязненности.
Микробиолоrическое испытание нестериль
ных лекарственных средств проводят в COOT
ветствии с методами, приведенными в общих
статьях 2.6.12, 2.6.13 и 2.6.31. Критерии при
емлемости для нестерильных лекарственных
средств, основанные на общем количестве
аэробов (ОКА) и на общем количестве rрибов
(OKr), приведены ниже.
Критерии приемлемости основываются на
отдельных результатах или на средних резуль
татах параллельных подсчетов в случае их про
ведения (например, в методах чашечноro под
счета).
Перечень специфицированных микроорrа
низмов, для которых установлены критерии при
емлемости. пред ставлен ниже. Данный список
не является обязательно исчерпывающим, и
для некоторых лекарственных средств может
быть необходимым включение испытания на
друrие микроорrанизмы в зависимости от при
роды исходноro материала и процесса произ
водства.
А. Лекарственные средства раститель-
Horo происхождения, содержащие лекар-
ственное растительное сырье с или без
вспомоrательных веществ, предназначен-
ные для приrотовления настоев и отваров с
использованием кипящей воды (например,
растительные чаи с или без прибавленных
ароматизаторов)
Об I Критерий приемлемости:
щее количество 1 O KOE/r
аэробов (ОКА)
(2612) Максимально допустимое
. . число: 50 000 000 KOE/r
Общее количество Критерий приемлемости:
105 KOE/r
rрибов (ОКП Максимально допустимое
(2.6.12) число: 500 000 KOE/r
Escherichia соН Критерий приемлемости:
(2.6.31) 103 KOE/r
Sа/топе//а (2.6.31) Отсутствие (25 r)
В. Лекарственные средства раститель
Horo происхождения, содержащие, напри
мер, экстракты и/или лекарственное расти-
тельное сырье с или без вспомоrательных
веществ, rAe метод обработки (например,
экстракция) или, rде применимо (в случае
лекарственноrо растительноrо сырья),
метод подrотовки уменьшает уровни coдep
жания микроорrанизмов ниже указанных
для данной катеrории
Общее Критерий приемлемости:
104 KOE/r или КОЕ/мл
количество Максимально допустимое
аэробов (ОКА) число: 50 000 KOE/r или
1(2.6.12) КОЕ/мл
Общее Критерий приемлемости:
количество 102 KOE/r или КОЕ/мл
rрибов (OKr) Максимально допустимое
(2.6.12) число: 500 KOElr или КОЕ/мл
rрамотрица
тельные бакте
рии, толерант
>ные к желчи
(2.6.31) #либо
бактерии семей Критерий приемлемости:
ства Eпterobac
teriaceae (2.6.13, 102 KOE/r или КОЕ/мл
испытание в co
ответствии с
альтернативной
методикой опре
деления)#
Esche,ichia соН Отсутствие (1 r или 1 мл)
(2.6.31)
Sа/топеllа (2.6.31) Отсутствие (25 r или 25 мл)
с. Лекарственные средства раститель-
Horo происхождения, содержащие, напри-
мер, экстракты и/или лекарственное расти-
тельное сырье с или без вспомоrательных
веществ, для которых может быть показа-
но, что метод обработки (например, экс-
Критерий приемлемости:
Общее количество 105 KOE/r или КОЕ/мл
аэробов (ОКА) Максимально допустимое
(2.6.12) число: 500 000 KOE/r или
КОЕ/мл
Критерий приемлемости:
Общее количество 104 KOE/r или КОЕ/мл
rрибов (ОКП Максимально допустимое
(2.6.12) число: 50 000 KOE/r или
КОЕ/мл
rрамотрицатель
ные бактерии, тo
лерантные к желчи
(2.6.31) #либо бак
терии семейства Критерий
Eпterobacte,iaceae приемлемости:
(2.6.13, испытание 104 KOE/r или КОЕ/мл
в соответствии с
альтернативной
методикой опреде
ления)#
Esche,ichia соН Отсутствие (1 r или 1 мл)
(2.6.31)
Sa/moпe//a (2.6.31) Отсутствие (25 r или 25 мл)
5.1.9. Руководство по применению испытания на стерильность
791
тракция этанольным раствором малой KOH
центрации или водой, которая не кипит, или
низкотемпературная концентрация) или,
rAe применимо (в случае лекарственноrо
растительноrо сырья), метод подrотовки не
уменьшает в значительной степени уровни
содержания микроорrанизмов ниже указан
ных критериев для катеrории В
Признано, что для некоторых лекар
ственных средств раститеЛЬН020 проис
хождения критерии кате20рий А, В и С по
общему количеству аэробов и 2рибов, а
также 2рамотрицательных бактерий, тo
лерантных к желчи, не М02ут быть дocти2
нуты из за характеРН020 уровня микробной
зараженности. В таких случаях, на OCHoвa
нии оценки риска, которая учитывает Ka
чественную и количественную xapaKтe
ристики биОЛ02ической за2рязненности и
предпола2аемое использование продукта.
М02ут быть применены более высокие Kpи
терии приемлемости.
Если показано, что ни одно из описанных
испытаний не даст достоверное значение co
держания микроорrанизмов на необходимом
уровне, используют валидированный метод с
пределом обнаружения как можно более близ
ким к заявленному критерию приемлемости.
0112013:50109
5.1.9. РУКОВОДСТВО
ПО ПРИМЕНЕНИЮ
ИСПЫТАНИЯ
НА СТЕРИЛЬНОСТЬ
Цель испытания на стерильность (2.6. п
как и всех фармакопейных испытаний, заклю
чается в предоставлении независимым aHa
литикам средств проверки соответствия дaH
Horo материала требованиям Фармакопеи.
Производителю не вменяется в обязанность
проводить такие испытания и не запрещает
ся использовать модификации установлен
Horo метода или альтернативные методы при
условии, что данная продукция будет COOTBeT
ствовать требованиям Фармакопеи при про
ведении испытания с использованием офици
альноro метода.
УСЛОВИЯ ПРОВЕДЕНИЯ ИСПЫТАНИЙ
Асептические условия проведения испыта
ния MorYT быть достиrнуты, например, использо
ванием камеры с ламинарным потоком воздуха
класса А, расположенной в чистом помещении
класса В, или с помощью изолятора.
РУКОВОДСТВО ДЛЯ ПРОИЗВОДИТЕЛЕЙ
Уровень достоверности, подтверждаемый
удовлетворительным результатом испытания на
стерильность (отсутствие заrрязняющих единиц
в образце), в отношении к качеству партии зави
сит от ее однородности, условий производства
и эффективности принятоro плана отбора проб.
Для целей данноro описания партия определяет
ся как однородный набор запечатанных контей
неров, приrотовленных таким образом, чтобы
риск микробноro заrрязнения был одинаковым
для каждой единицы.
Для продукции, стерилизуемой на послед
ней стадии производства, биолоrически обосно
ванные и автоматически документированные
фактические доказательства правильноro про
текания процесса стерилизации для всей партии
дают большую rарантию по сравнению с испыта
нием на стерильность. Обстоятельства. при KO
торых допустим параметрический выпуск серии,
описаны в разделе 5.1.1. Методы прис.отовле
ния стерильных продуктов. Для подтвержде
ния асептических условий производства может
применяться метод наполнения питательными
средами. За исключением этих ситуаций, испы
тание на стерильность является единственным
аналитическим методом для продукции, произ
водство которой осуществляется в асептических
условиях.
Вероятность определения наличия микро
орrанизмов в ходе испытания на стерильность
возрастает с увеличением их количества в ис
пытуемом образце и варьируется в зависимости
от способности к росту имеющихся микроорrа
низмов. Вероятность обнаружения очень низких
уровней микробноrо заrрязнения, даже если
оно в пределах партии является однородным,
весьма мала. Интерпретация результатов испы
тания на стерильность основывается на пред
положении, что содержимое любоro контейнера
в партии, если он будет подверrнут испытанию.
даст одинаковый результат. Поскольку очевидно.
что каждый контейнер не может быть подверrнут
испытанию, следует использовать COOTBeTCTBY
ющий план отбора проб. В случае асептическоrо
производства рекомендуется отбирать образцы
в начале и в конце выпущенной flартии. а также
после существенноro вмешательства в техноло
rический процесс.
НАБЛЮДЕНИЕ И ИНТЕРПРЕТАЦИЯ
РЕЗУЛЬТАТОВ
Общепринятые микробиолоrические или
биохимические методы в общем случае явля
ются удовлетворительными для идентификации
микроорrанизмоl'J. выделяемых в ходе испыта
ния на стерильность. Однако, если производи
тель желает использовать лишь критерий (d) для
признания результатов испытания на стериль
ность недостоверными, может оказаться необхо
димым использование чувствительных методов
классификации для демонстрации идентично
792
rосударственная фармакопея Республики Беларусь
сти микроорrанизма, выделенноro при испыта
нии продукции, микроорrанизму, выделенному
из материалов и/или окружающей обстановки.
Хотя с помощью рутинных микробиолоrических
или биохимических методов может быть показа
но, что два изолированных штамма не являются
идентичными, эти методы MOryт быть HeДOCTa
точно чувствительными и надежными для полу
чения однозначноro доказательства тоro, что два
штамма имеют один и тот же источник. Для опре
деления тоro, что микроорrанизмы имеют общее
происхождение и клонально связаны, может
оказаться необходимым использование более
чувствительных тестов, например, молекуляр
ной классификации по roмолоrии РНК/ДНК.
01/2013:50110
5.1.10. РУКОВОДСТВО
ПО ПРИМЕНЕНИЮ ИСПЫТАНИЯ
НА БАКТЕРИАЛЬНЫЕ
ЭНДОТОКСИНЫ
1. ВВЕДЕНИЕ
Эндотоксины, источником которых являются
rрамотрицательные микроорrанизмы, являются
наиболее распространенной причиной токсиче
ских реакций. приписываемых наличию в фар
мацевтических продуктах пироrенных примесей;
их пироrенная активность намноro выше, чем у
большинства друrих пироrенных веществ. Эн
дотоксины представляют собой липополисаха
риды. Несмотря на то, что существует незначи
тельное количество пироrенов друroй структуры,
обычно по отсутствию бактериальных эндоток
синов в продукте можно сделать вывод об отсут
ствии пироrенных компонентов при условии, что
наличие пироrенных веществ, не относящихся к
эндотоксинам, можно исключить.
Присутствие эндотоксинов в продукте может
маскироваться факторами, влияющими на про
текание реакции эндотоксинов с лизатом амебо
цитов. Поэтому аналитик, желающий заменить
требуемое в фармакопейной статье испыта
ние на пироrенность С использованием кроли
ков испытанием на бактериальные эндотокси
ны, должен продемонстрировать, что этот тест
может быть выполнен для данноrо продукта; при
этом может оказаться необходимым выполнение
процедуры удаления мешающих факторов.
Как /казано в испытании на бактериаль
ные эндотоксины (2.6.14), результаты испыта
ния образца можно считать достоверными лишь
при наличии информации по следующим двум
аспектам.
Должна быть установлена приroдность
материала, используемоro в испытании. Должно
быть rарантировано отсутствие эндотоксинов в
воде для ИБЭ и в друrих реактивах, а чувстви
тельность лизата амебоцитов должна быть про
верена для подтверждения чувствительности,
заявленной производителем.
В связи с тем, что испытуемый продукт
может влиять на ход испытания, чувствитель
ность лизата амебоцитов должна быть опреде
лена в присутствии и отсутствии испытуемоrо
продукта. Между двумя значениями чувствитель
ности не должно быть существенной разницы.
В испытании на бактериальные эндоток
сины (2.6.14) указаны методы исключения Me
шающих факторов; в случае наличия меша
ющих факторов после применения этоro метода
должно быть проведено дополнительное испы
тание с целью проверки надежности удаления
или нейтрализации мешающих факторов.
В данном разделе поясняются причины Tpe
бований, предъявляемых в испытании на бак
териальные эндотоксины, и даются указания,
касающиеся реrистрации и интерпретации pe
зультатов.
Замена требуемоro в частной статье испыта
ния на пироrенность с использованием кроликов
на испытание с применением лизата амебоци
тов фактически означает использование альтер
нативноro метода анализа и поэтому требует Ba
лидации; в подразделе 11 приведены некоторые
указания, касающиеся практических действий в
данном направлении.
Ссылка на метод, который следует приме
нять для данноro продукта, содержится в COOT
ветствующих частных статьях; если метод не
указан, применяют метод А. Если используется
метод, отличный от предписанноro, должно быть
продемонстрировано, что при меняемый метод
является подходящим для данноro продукта и
дает результат, соrласующийся с результатом,
получаемым предписанным методом (см. также
подраздел 13).
2. МЕТОД
Несмотря на то, что добавление эндоток
си нов к лизату амебоцитов в растворе может
приводить к помутнению, осаждению или rеле
образованию, для определения конечной точки
фармакопейноro испытания первоro типа ис
пользовали лишь образование rелевоro crycTKa.
Преимуществом являлась простота принятия
решения о том, выдерживает ли продукт испы
тание, которое определяется наличием или OT
сутствием rелеобразования, леrко обнаружива
емым невооруженным rлазом. Количественные
методы С, О, Е и F были разработаны позже: они
требуют более сложноrо оборудования, но леrче
поддаются автоматизации для реrулярноro KOH
троля большоro количества образцов одноro и
тоro же продукта.
Эндотоксины MorYT адсорбироваться на по
верхности пробирок или пипеток, выполненных
из некоторых видов пластика и типов стекла.
5.1.10. Руководство по применению иСПblтания на бактериаЛЬНblе эндотоксиНbI 793
MoryT возникать помехи, обусловленные BЫCBO
бождением веществ из пластических матери
алов. Поэтому используемые материалы сле
дует подверrать проверке; последующие партии
пробирок или пипеток MOryт иметь несколько от
личающийся состав, и поэтому рекомендуется
повторять такие испытания при начале работы с
новой партией материала.
Решение об использовании испытания на
бактериальные эндотоксины в качестве теста
на предельное содержание подразумевает, BO
первых, что для испытуемоrо продукта должна
быть определена пороroвая концентрация эн
дотоксина и, BO BТOpЫX, что требуется опреде
лить, выше или ниже этоro пороrа концентрация
эндотоксина в испытуемом продукте. Количе
ственные методы С, О, Е и F дают возможность
определить концентрацию эндотоксина в образ
це, но для определения соответствия Фармако
пее и при рутинном контроле качества основной
вопрос заключается в том, превышает ли эта
концентрация установленный предел.
При установлении пороrовой конuентраuии
эндотоксина в испытуемом продукте следует
уделять должное внимание дозе этоro продук
та: цель должна заключаться в том, чтобы rapaH
тировать, что пока концентрация эндотоксина в
продукте остается ниже TaKoro пороroвоro значе
ния, даже максимальная доза, вводимая указан
ным путем в течение часа, не будет содержать
эндотоксины в количестве, достаточном для воз
никновения токсической реакции.
Как в случае равенства концентрации эндо
токсинов в продукте пороrовому значению, так
и в случае значительно больших концентраций
возникает rелеобразование, и продукт не Bыдep
живает испытание ввиду тоro, что характер ис
пытания «все или ничеro» делает невозможным
различение концентрации. в точности равной
пороroвой, и более высокой. Вывод о том. что
концентрация эндотоксина не превышает поро
rOBoe значение, можно сделать лишь при OTCYT
ствии rелеобразования.
Для продуктов, находящихся в твердом co
стоянии, такая пороroвая концентрация эндо
токсина на единицу массы или Международную
Единицу продукта подлежит переводу в KOHцeH
трацию эндотоксина в миллилитре испытуемоro
раствора, так как испытание может быть выпол
нено только с раствором. Случай, коrда продук
ция уже находится в жидком виде (например, ин
фузионные жидкости), будет оroворен ниже.
Предельное содержание эндотоксинов: для
фармацевтических субстанций, предназначен
ных для парентеральноro введения. определя
ется на основании дозы и равно:
к
,
м
rne:
К допустимая пироrенная доза эндоток
СМНЭ. на килоrрамм массы тела:
м максимальная рекомендованная доза
продукта, на килоrрамм массы тела.
Если продукт вводится через малые проме
жутки времени или если используется в течение
продолжительноro периода времени, в качестве
М используют максимальную общую дозу, BBO
димую В течение одноro часа.
Предельное содержание эндотоксинов за
висит от продукта и пути ero введения и YCTaHaB
ливается в частных статьях. Предлаrаемые зна
чения К приведены в таблице 5.1.10. 1.
В случае друrих путей введения, критерий
приемлемости для содержания эндотоксинов
обычно устанавливается на основании резуль
татов, полученных при разработке roToBoro про
дукта.
Таблица 5.1.10. 1
I
! Способ введения
1
! Внутривенно
/Внутривенно, для
радиофарма
цевтических
препаратов
Интратекально
I К (МЕ ЭНДОТОКСИНОВ
! на килоrрамм массы
; тела)
5,0
2,5
0,2
Какое разведение продукта должно быть ис
пользовано в испытании для получения макси
мальной уверенности в том, что отрицательный
результат свидетельствует о наличии KOHцeHTpa
ции эндотоксина в продукте менее предельной,
а положительный результат означает, что с по
мощью лизата обнаруживается концентрация,
равная или превышающая предельную? Такое
разведение зависит от установленноro предела
и от чувствительности лизата: для Hero применя
ется термин «максимально допустимое разведе
ние» (МДР), и еro значение может быть рассчи
тано по следующей формуле:
!!Еедеf2ьн--- е содержuне эндотоксuн й-,! KOHцeH тpaц ытyeM02C OarmвCDa
Л
Концентрация иСПbfтуеМ020 раствора:
в мr/мл, если предельное содержание эн
дотоксинов выражено в массовых единицах
(ME/Mr);
в Ед/мл, если предельное содержание эн
дотоксинов выражено в пересчете на единицу
биолоrической активности (МЕ/Ед);
в мл/мл, если предельное содержание
эндотоксинов выражено в объемных единицах
(МЕ/м.'l).
А чувствительность лизата (МЕ/мл) в
rель тромб методе. указанная на этикетке, или
низшая концентрация на стандартной кривой в
турбидиметрических или xpOMoreHHbIx методах.
В случаях. Korдa значение максимально дo
пустимоro разведения не является целочислен
ным. для рутинных целей может быть использо
794
rосударственная фармакопея Республики Беларусь
вано подходящее целое число, меньшее МДР
(что означает разведение раствора продукта в
степени, меньшей, чем МДР). В таком случае OT
рицательный результат свидетельствует о том,
что концентрация эндотоксина в продукте ниже
предельноro значения. Однако, Korдa KOHцeH
трация эндотоксина в продукте в таком испыта
нии ниже предельноro значения, но достаточно
высока для тоro, чтобы реакция с лизатом при
вела к образованию crycTKa, испытание в таких
условиях может дать положительный резуль
тат. Поэтому, Korдa испытание с использованием
такой «удобной» степени разведения дает поло
жительный результат, продукт следует развести
дО МДР и повторить испытание. В любых сомни
тельных или спорных случаях следует использо
вать МДР.
ЭТО подчеркивает важность подтверждения
чувствительности лизата.
Пример
Следует провести испытание раствора
50 мr/мл фенитоина натрия, предназначенноro
для внутривенноrо введения. МДР определяют,
используя следующие значения переменных:
М максимальная человеческая доза = 15 Mr
на килоrрамм массы тела;
с 50 мr/мл;
К 5 МЕ эндотоксина на килоrрамм массы
тела;
Л 0.4 МЕ эндотоксина на миллилитр.
MДP 5.50 . 4167.
15 0,4 ,
Для рутинных испытаний этоro продукта
может быть целесообразно 1 мл испытуемоro
раствора разбавить до 20 мл (величина МДР/2,
окруrленная до ближайшеro меньшеrо целоro
числа). Однако. если испытание дает положи
тельный результат следует 1 мл разбавить до
41,67 мл и повторить испытание. Разведение до
41,67 мл необходимо также в тех случаях, Korдa
испытание проводится для разрешения спорной
ситуации.
3. СТАНДАРТНЫЙ МАТЕРИАЛ
БСП стандартНО20 образца эндотокси
на используется в качестве стандартноro образ
ца. Ero количественное определение проводят в
сравнении с Международным стандартным об
разцом эндотоксина ВОЗ, и ero активность BЫ
ражается в МЕ эндотоксина в ампуле. Междуна
родная единица эндотоксина определяется как
специфическая активность определенной массы
Международноrо стандартноrо образца.
Для рутинных целей может быть исполь
зован друrой стандартный образец эндотокси
на при условии проведения еro количественно
ro определения в сравнении с Международным
стандартным образцом эндотоксина или БСП, а
еro активность выражена в МЕ эндотоксина.
Примечание: одна Международная Единица
(МЕ) эндотоксина эквивалентна одной EдиHU
це Эндотоксина (ЕЭ).
4. ВОДА ДЛЯ ИБЭ
Определение отсутствия эндотоксина в про
дукте путем проведения испытания на пироrен
ность на кроликах было исключено по практиче
ским и теоретическим причинам:
испытание на кроликах не обладает ДOCTa
точной чувствительностью для определения эн
дотоксина в воде для ИБЭ, предназначенной для
проведения испытаний продуктов с очень низкой
предельной концентрацией эндотоксинов;
относительно низкая точность темпера
тур ной реакции у кроликов привела бы к необ
ходимости проведения большоro количества по
вторных испытаний;
термины «пироrены» И «эндотоксины» OT
носятся К rруппам веществ, которые не вполне
совпадают друr с друrом.
В описании испытания на бактериальные
эндотоксины (2.6.14) указывается, что для при
rотовления воды для ИБЭ, кроме трехкратной
дистилляции, MorYT быть использованы и друrие
методы. К хорошим результатам приводило ис
пользование метода обратноro осмоса; HeKOTO
рые аналитики MOryT предпочесть переroнять
воду более трех раз. Какой бы метод ни исполь
зовался, полученный продукт не должен coдep
жать поддающихея определению эндотоксинов.
5. ЗНАЧЕНИЕ рН СМЕСИ
Оптимальное rелеобразование смеси при
проведении испытания на бактериальные эн
дотоксины достиrается при значениях рН от 6,0
до 8,0. Однако прибавление лизата к образцу
может привести к понижению рН.
6. ВАЛИДАЦИЯ ЛИЗАТА
При приroтовлении растворов лизата важно
следовать инструкциям производителя.
Факторы положительных конечных разве
дений в методах rелевоro crycTKa А и В перево
дят в лоrарифмы. Причина этоro заключается в
том, что если построить rрафик частотноro pac
пределения этих лоrарифмических значений, то
он обычно намноro ближе к кривой нормально
ro распределения, чем частотное распределе
ние самих факторов разведения; фактически,
они настолько близки, что допускается исполь
зование нормальноrо частотноro распределения
в качестве математической модели и вычисле
ние rраниц доверительноro интервала с помо
щью t критерия Стьюдента.
7. ПРЕДВАРИТЕЛЬНОЕ ИСПЫТАНИЕ
НА МЕШАЮЩИЕ ФАКТОРЫ
Некоторые продукты не MOryT быть подвер
rHYTbI испытанию на наличие эндотоксинов непо
средственно, так как они не смешиваются с peaK
тивами, не MOryT быть доведены до рН 6,0 8,0
5.1.10. Руководство по примененuю испытания на бактериаЛЬНЬ1е эндотоксuнЬ1 795
или являются инrибиторами или активаторами
rелеобразования. Поэтому требуется проведе
ние предварительноro испытания для проверки
наличия мешающих факторов; в случае их обна
ружения должно быть продемонстрировано, что
процедура их удаления была эффективной.
Цель предварительноro испытания cocтo
ит в проверке нулевой rипотезы о том, что чув
ствительность лизата в присутствии продукта
существенно не отличается от чувствительности
лизата в еro отсутствии. В методах А и В исполь
зуется простой критерий: нулевая rипотеза при
нимается, если чувствительность лизата в при
сутствии продукта составляет не менее 50 % и
не более 200 % чувствительности самоro лизата.
В соответствии с классическим подходом
следует вычислить среднее значение лоrариф
ма коэффициента разведения для чувствитель
ности в присутствии И отсутствии продукта и
оценить разницу между двумя средними значе
ниями с помощью t критерия Стьюдента.
Испытание на мешающие факторы в Meтo
дах rелевоro crycTKa А и В требует использова
ния образца продукта, в котором отсутствуют
эндотоксины В поддающейся определению KOH
центрации. Это представляет собой теоретиче
скую проблему при испытании абсолютно новых
продуктов. Поэтому для количественных Meтo
дов С, О, Е и F был разработан друrой подход.
8. ИСКЛЮЧЕНИЕ МЕШАЮЩИХ ФАКТОРОВ
Методика удаления мешающих факторов
не должна приводить к уменьшению или YBe
личению количества эндотоксинов в испыту
емом продукте (например, вследствие адсорб
ции). Корректный способ проверки этоro состоит
в применении методики к обоrащенному образцу
продукта, т. е. к образцу, к которому было добав
лено известное количество эндотоксина, с даль
нейшим измерением ero открываемости.
Методы С и D. Если помехи обусловлены
природой анализируемоrо продукта и не MorYT
быть исключены классическими методами,
может оказаться возможным построение CTaH
дартной кривой с использованием продукта тоro
же типа, разбавленноro или обработанноrо COOT
ветствующим образом для очистки от эндотокси
нов. Испытание на эндотоксины может быть про
ведено с использованием полученной кривой.
Определено, что в большинстве случаев
ультрафильтрация с использованием асимме
трических мембранных фильтров из триацета
та целлюлозы при водит к адекватным результа
там. Фильтры должны пройти соответствующую
валидацию в связи с тем, что производные цел
IlЮЛОЗЫ ( D rлюканы) при некоторых обсто
ятельствах MOryT обусловить получение ложных
положительных результатов.
Установлено, что полисульфоновые филь
тры являются неприroдными, так как при их ис
пользовании были получены ложные положи
тельные результаты.
#В частных статьях должно быть указание о
присутствии мешающих факторов и приведена
методика их устранения.#
9. ЦЕЛЬ ПРОВЕДЕНИЯ КОНТРОЛЬНЫХ
ИСПЫТАНИЙ
Цель контроля, содержащеro воду для ИБЭ
и стандартный образец эндотоксина с KOHцeH
трацией в два раза превышающей чувствитель
ность лизата, указанную на этикетке, состоит в
проверке активности лизата в условиях испы
тания во время ero проведения. Целью отри
цательноrо контроля является подтверждение
отсутствия поддающейся определению KOHцeH
трации эндотоксина в воде для ИБЭ.
Положительный контроль, в котором coдep
жится испытуемый продукт в концентрации, ис
пользуемой при проведении испытания, предна
значен для демонстрации отсутствия мешающих
факторов в условиях испытания во время еro
проведения.
10. РЕrИСТРАЦИЯ И ИНТЕРПРЕТАЦИЯ
РЕЗУЛЬТАТОВ
Незначительные количества эндотоксина в
воде для ИБЭ или в любом друroм peareHTe или
материале, С которым лизат находится в KOHTaK
те в ходе испытания, MorYT не определятся до
тех пор, пока они не достиrнут предела чувстви
тельности лизата. Однако они MOryT увеличить
количество эндотоксина в растворе, содержа
щем испытуемый продукт, до значения, слеrка
превышающеrо предел чувствительности, и BЫ
звать положительную реакцию.
Риск, что это произойдет, может быть YMeHb
шен путем проверки воды для ИБЭ и друrих pe
areHToB и материалов с использованием наи
более чувствительноro лизата из имеющихся в
наличии или, по крайней мере, более чувстви
тельноro, чем лизат, использующийся при прове
дении испытания продукта. Даже в этом случае
риск такоro «ложноro положительноrо результа
та» не может быть полностью исключен Следу
ет, однако, понимать, что в этом отношении MeTcr
дика испытания является безопасной, в отличие
от методики испытания, допускающей ложный
отрицательный результат, который может при
сти к выпуску недоброкачественноro продукта,
опасноro для здоровья пациента
11. ЗАМЕНА ИСПЫТАНИЯ
НА пироrЕННОСТЬ НА КРОЛИКАХ
ИСПЫТАНИЕМ НА БАКТЕРИАЛЬНЫЕ
ЭНДОТОКСИНЫ
В частных статьях. касающихся фармацев
тической продукции. предназначенной для па
рентеральноrо применения, которая может co
держать токсические количества бактериальных
эндотоксинов. содержатся требования о прове
дени и испытания либо на бактериальные эндо
токсины. либо на пироrенность с использовани
ем кроликов. В общем случае:
796
rосударственная фармакопея Республики Беларусь
в любой частной статье при необходимо
сти проведения подобноro испытания содержится
только один тест либо на пироrенность, либо на
бактериальные эндотоксины;
при отсутствии доказательств обратноro,
испытание на бактериальные эндотоксины явля
ется предпочтительным по отношению к испыта
нию на пироrенность, так как обычно оно обеспе
чивает равную или лучшую защиту пациента;
перед включением испытания на бактери
альные эндотоксины в частную статью должны
быть получены доказательства тоro, что один из
методов, описанных в разделе 2.6.14, является
подходящим для испытания данноro продукта;
необходимую информацию предоставляют
производители; приветствуется предоставление
компаниями любых имеющихся подтвержденных
данных, касающихся применимости испытания
на бактериальные эндотоксины к интересующим
субстанциям и roтовым формам; к таким данным
относятся методы подroтовки образцов и проце
дуры, необходимые для исключения мешающих
факторов; кроме тоro. должны быть предоставле
ны любые доступные параллельные данные по
проведению испытания на пироrенность с исполь
зованием кроликов, которые моrли бы подтвер
дить возможность замены испытания на пироrен
ность тестом на бактериальные эндотоксины.
Дополнительные требования приведены в
следующих подразделах.
12. ИСПОЛЬЗОВАНИЕ МЕТОДА
ИСПЫТАНИЯ НА БАКТЕРИАЛЬНЫЕ
ЭНДОТОКСИНЫ, отличноrо
ОТ ПРЕДПИСАнноrо В ЧАСТНОЙ СТАТЬЕ
Если в частной статье предписывается про
ведение испытания на бактериальные эндоток
сины и не указан ни один из методов (A F), опи
санных в разделе 2.6.14, то для данноro продукта
была подтверждена приroдность метода А (пре
дельное испытание методом rелевоro crycTKa).
Если указан один из друrих методов (B F), то
для этоro продукта была подтверждена приroд
ность указанноro метода.
13. ВАЛИДАЦИЯ АЛЬТЕРНАТИВНЫХ
МЕТОДОВ
Замену испытания на пироrенность на KpO
ликах тестом на бактериальные эндотоксины, а
также замену установленноro или подразумева
eMoro метода определения бактериальных эндо
токсинов друrим следует считать использованием
альтернативноro метода взамен фармакопейноro
испытания. Общее положение по использованию
альтернативноro метода взамен фармакопейноro
теста приведено в разделе «Общие положения»:
«Описанные испытания и методики количе
ственноro определения являются официальными
методами, на которых основаны стандарты Фар
макопеи. По соrлашению с компетентными opra
нами в целях контроля MOryт использоваться аль
тернативные методы анализа при условии, что
используемые методы позволяют сделать опре
деленное заключение о том, может ли быть дo
стиrнуто соrласие со стандартами статей, если
бы были использованы официальные методы. В
сомнительных и спорных случаях единственны
ми надежными методами считают методы, приве
денные в Фармакопее».
Для валидации метода испытания на бакте
риальные эндотоксины, отличноro от подразуме
ваемоro или указанноro в частной статье, предла
rаются следующие процедуры.
1 3 1. Процедура, материалы и peareHTbI, ис
пользуемые при проведении испытания с примене
нием лизата амебоцитов, должны пройти валида
цию в соответствии с указаниями, приведенными в
испытании на бактериальные эндотоксины.
13 2. Должно быть проведено испытание на
наличие мешающих факторов (и, при необходи
мости, процедура по их исключению) на образ
цах, отобранных по меньшей мере из трех партий
продукции. Следует иметь в виду, что в методах
О и Е, использующих хромоrенный пептид, требу
ются peareHTbI, не используемые в методах А, В,
С и F, и поэтому соответствие методов А, В, С и F
требованиям, предъявляемым к мешающим фак
торам, не может быть без соответствующеro под
тверждения экстраполировано на методы О и Е.
14. ВАЛИДАЦИЯ ИСПЫТАНИЯ
ДЛЯ НОВЫХ ПРОДУКТОВ
Процедуры, описанные в пунктах 1 3 1 и 1 3 2,
должны быть выполнены для всех новых продук
тов, предназначенных для парентеральноro при
менения, которые, в соответствии с требованиями
Фармакопеи, должны быть подверrнуты испыта
нию на наличие бактериальных эндотоксинов.
5.2. ОБЩИЕ ТЕКСТЫ
ПО БиолоrИЧЕСКИМ
ПРОДУКТАМ
01/2013:50201
5.2.1. ОБЩЕПРИНЯТАЯ
ТЕрминолоrия в СТАТЬЯХ
НА БиолоrИЧЕСКИЕ
ПРОДУКТЫ
В некоторых пунктах настоящей статьи в
скобках приведены альтернативные термины.
Система noceBHoro материала (эталон
ный банк культур микроорrанизмов) [seed
/ot system]. Это система, в соответствии с
5.2.1. Общепринятая теРМUНОЛО2UЯ в статьях на БUОЛО2ические продукты
797
которой последовательные партии (серии) про
дукции производятся из одноro и тоro же rлав
ноro посевноro материала. Для рутинноro про
изводства из rлавноrо посевноro материала
может 'быть приroтовлен рабочий посевной
материал (рабочий банк культур клеток). Про
исхождение и количество пассажей rлавноrо
посевноro материала и рабочеro посевноro Ma
териала реrистрируется.
rлавный посевной материал (контроль.
ный банк культур микроорrанизмов) [master
seed-Iotj. Культура микроорrанизмов, распре
деленная из общей массы по контейнерам, KO
торые обрабатываются вместе в рамках одной
операции таким образом, чтобы обеспечить еди
нообразие, стабильность и предотвратить KOHTa
минацию. rлавный посевной материал в жидком
виде хранится обычно при температуре не выше
70 ОС. Лиофильно высушенный rлавный посев
ной материал хранится при температуре, обе
спечивающей ero стабильность.
Рабочий посевной материал (рабочий
банк культур микроорrанизмов) [working
seed-Iotj. Культура микроорrанизмов, получен
ная из rлавноro посевноro материала и предна
значенная для использования в производстве.
Рабочий посевной материал помещают в KOH
тейнеры и хранят, как описано выше для rлавно
ro посевноro материала.
Система банка клеток (система посев-
ных клеточных культур) [seed-bank system
(cell-seed systeт)]. Это система, по которой
последующие конечные серии (партии) продук
та произведены из культуры клеток из одноrо
и тоro же тавноro банка клеток. Для приroтов
ления рабочеro банка клеток используется He
которое число контейнеров из rлавноro банка
клеток. Систему банка клеток валидируют на
максимально допустимый пассажный уровень,
который может использоваться при текущем
производстве.
rлавный банк клеток (rлавный банк по.
севных клеточных культур) [тaster се" Ьапk
(maste, се" seed)]. Культура клеток, распреде
ленная из общей массы по контейнерам, KOTO
рые обрабатываются вместе в рамках одной
операции таким образом, чтобы обеспечить еди
нообразие, стабильность и предотвратить KOHTa
минацию. rлавный банк клеток хранится обычно
при температуре не выше 70 ОС.
Рабочий банк клеток (рабочий банк по-
севных клеточных культур) [working се" Ьапk
(working се" seed)]. Культура клеток, получен
ная из контрольноro банка и используемая в про
изводстве перевиваемых линий клеток. Рабочий
банк клеток помещают в контейнеры и хранят,
как описано выше для rлавнorо банка клеток
Исходные (первичные) культуры клеток
[p,imary се" cиltи,es]. Культуры клеток, полу
ченные путем трипсинации подходящей ткани
или opraHa. Клетки практически полностью иден
тичны клеткам ткани, из которой они были полу
чены, и подверrаются не более чем 5 пассажам
in vitro из исходноro образца ткани животноro.
Линии клеток (культуры клеток) [се"
lines]. Культуры клеток, обладающие высокой
способностью к размножению in vit,o. В линиях
диплоидных клеток клетки обладают практи
чески теми же характеристиками, что и клетки
ткани, из которой получена линия. В непрерыв
ных линиях клеток клетки способны неоrрани
ченно размножаться в культуре и MOryT быть
получены из здоровой или опухолевой ткани.
Некоторые непрерывные линии клеток при опре
деленных условиях обладают oHKoreHHbIM по
тенциалом.
Производственная культура клеток [рто-
dиction се" cиltиre]. Культура клеток, предна
значенная для использования в производстве;
может быть получена из одноro или нескольких
контейнеров рабочеro банка клеток или может
представлять собой исходную культуру клеток.
Контрольные клетки [сопtrо/ ce"s]. HeKO
торое количество производственных клеток, не
подверrшихся заражению вирусом и использую
щихся В качестве неинфицированной культуры
клеток. Неинфицированные клетки инкубируют
ся в тех же условиях, которые используются для
производственной культуры клеток.
Однократный сбор [single harvestj. Ma
териал, полученный однократно или мноroкрат
но из одной производственной культуры клеток,
зараженной одним рабочим посевным матери
алом, или суспензия, полученная из рабоче
ro посевноro материала, инкубированная и co
бранная за один производственный цикл.
Моновалентный объединенный сбор
[monova/ent poo/ed harvestj. Объединенный
материал, содержащий один штамм, тип микро
орrанизма или антиrен и полученный из HeKOTO
роro количества яиц, контейнеров с клеточными
культурами и т. п., которые обрабатываются oд
новременно.
rотовая нерасфасованная вакцина [final
bиlk vaccine]. Продукт, прошедший все стадии
производства, кроме конечноro заполнения (упа
ковки). Он состоит из одноro или нескольких MO
новалентных объединенных сборов из культур
одноro или нескольких видов микроорrанизма
после очистки, растворения или добавления Ka
коro либо активизирующеro или друrorо вспомо
rательноro вещества. Он обрабатывается для
обеспечения ero rомоrенности и используется
798
rосударственная фармакопея Республики Беларусь
для заполнения контейнеров одной или несколь
ких конечных партий (серий).
Конечная партия (серия) (fiпal lot
(batch)). Совокупность укупоренных конечных
контейнеров или друrих конечных единиц дo
зирования, которые считаются rомоrенными
и равнозначными в отношении риска зараже
ния во время заполнения или приrотовления
конечной продукции. Единицы дозирования
заполняются или ютовятся друrим способом
из одной и той же roтовой нерасфасован
ной вакцины, при необходимости лиофиль
но высушиваются и запечатываются в рамках
одноro непрерывною рабочею процесса. Они
имеют отличительный номер или код, иденти
фицирующий конечную партию (серию). Если
rотовая нерасфасованная вакцина разливает
ся иlили лиофильно высушивается в рамках
нескольких рабочих процессов, это приво
дит К появлению связанных конечных партий
(серий), которые обычно идентифицируются
при помощи использования общей части в OT
личительном номере или коде; эти связанные
конечные партии (серии) иноrда называются
подсериями, подпартиями или фасовочными
партиями.
Комбинированная вакцина (сотЫпеd
vacciпe]. Мноroкомпонентный препарат с рецеп
турой, при которой одновременно вводятся раз
личные антиrены. Различные антиrенные KOM
поненты предназначены для защиты от разных
штаммов или типов одноro и тоro же орrанизма
и/или различных орrанизмов. Комбинированная
вакцина может поставляться производителем
как один жидкий или лиофильно высушенный
препарат или как несколько составляющих с ин
струкцией по смешиванию перед применением.
01/2013:50202
5.2.2. СТАИ КУР, НЕ ИМЕЮЩИХ
КОНКРЕТНЫХ ПАтоrЕНОВ
И ИСПОЛЬЗУЕМЫХ ДЛЯ
ПРОИЗВОДСТВА И КОНТРОЛЯ
КАЧЕСТВА ВАКЦИН
В случае, rде это указано, цыплята, эмбри
оны или культуры клеток, используемые при про
изводстве или контроле качества вакцин, полу
чают из яиц кур, не зараженных конкретными
патоrенами (НЗКП, SPF (,ее (rom speci(ied
раthоgепs). Статус SPF стаи кур обеспечивает
ся посредством описанной ниже системы. При
веденный список микроорrанизмов основан на
имеющихся знаниях и при необходимости будет
обновляться.
Под стаей понимается rруппа птиц, прожива
ющих в одном и том же окружении, за KOТOpЫ
ми ухаживают лица, не имеющие контакта с за
раженными конкретными патоrенами стаями. Как
только стае присваивают катеroрию SPF, к ней
не добавляют птиц, не имеющих катеroрии SPF.
Стаю содержат в таких условиях, чтобы
свести к минимуму вероятность контаминации.
Ее нельзя размещать вблизи стай птиц, не име
ющих катеroрию SPF, за исключением стаи, под
roтавливаемой к процессу присвоения катеroрии
SРF стаи или содержащейся в условиях, COOT
ветствующих условиям содержания SРF стаи.
Стая должна содержаться в изоляторе или в
здании с фильтруемым воздухом, поступающим
под положительным давлением. Следует также
принять соответствующие меры для предотвра
щения доступа rрызунов, диких птиц, насекомых
и лиц, не имеющих соответствующеro разреше
ния.
Персонал, имеющий разрешение на вход в
помещение, не должен иметь контакта с друrи
ми птицами или аrентами, которые MOryт инфи
цировать стаю. Перед входом в контролируемые
помещения персоналу рекомендуется прини
мать душ и переодеваться или надевать защит
ную одежду.
Во всех возможных случаях предметы, KOТO
рые вносят в помещение, стерилизуют. В частно
сти, корм рекомендуется обрабатывать COOTBeT
ствующим образом, чтобы избежать попадания
в неro нежелательных микроорrанизмов, а вода
должна отвечать требованиям питьевой воды,
обрабатываемой, например, хлорированием.
Птицам из SРF стаи не вводят лекарственные
средства, которые моrли бы помешать распоз
наванию болезни в стае.
Данные об общем здоровье стаи постоянно
реrистрирую, все любые отклонения исследуют.
Основные параметры наблюдения включают за
болеваемость, смертность, общее физическое
состояние, потребление корма, ежедневная яй
ценоскость и качество яиц, плодовитость и BЫBO
димость цыплят. Записи хранят не менее 5 лет.
О любых отклонениях от нормы в исследуемых
параметрах или об обнаружении какой либо ин
фекции сообщается потребителям яиц в воз
можно короткие сроки.
Испытания или комбинации тестов, описан
ные ниже, должны обладать соответствующей
специфичностью и чувствительностью к COOT
ветствующим серотипам вирусов. Образцы для
испытаний отбираются случайным образом.
Положительный результат на наличие
вируса анемии цыплят (ВАЦ) не обязательно ис
ключает использование материала, полученно
ro из стаи, но живые вакцины для примене'ния
у птиц в возрасте менее 7 дней должны произ
водиться с использованием материала от птиц
с отрицательной реакцией на ВАЦ. Инактиви
рованные вакцины для применения на птицах в
возрасте менее 7 дней MOryT быть произведены
5.2.2. Стаи кур, не имеющих конкретных патоzенов
799
Таблица 5.2.2. 1
Возбудитель Проводимые Вертикальный Быстрое/медленное
исследования** путь передачи распространение
Аденовирусы птиц, rруппа 1 РДП, ИФА да медленное
Вирус энцефаломиелита птиц РДП, ИФА да быстрое
Вирус инфекционноro PTrA, ИФА нет быстрое
бронхита птиц
Вирус инфекционноro РН, ИФА да медленное
ларинroтрахеита птиц
Вирус лейкоза птиц ИФА для вируса, РН, нет медленное
ИФА для антитела
Вирус нефрита птиц ИО да медленное
Ортореовирусы птиц ИО, ИФА да медленное
Вирус ретикулоэндотелиоза РДП, ИФА да медленное
птиц
Вирус анемии цыплят ИО, ИФА, РН да медленное
Вирус синдрома снижения PTrA, ИФА да медленное
яйценоскости
Серотип 1: РДП, ИФА, I
Вирус инфекционною бурсита РН нет быстрое
птиц Серотип 2: РН
Вирус rриппа А РДП, ИФА, PTrA нет быстрое
Вирус болезни Марека РДП нет быстрое
Вирус болезни Ньюкасла PTrA, ИФА нет быстрое
Вирус ринотрахеита индеек ИФА нет медленное
(птиц)
РА и PTrA для под
Mycop/asma gallisepticum тверждения положи да медленное
тельноro теста, ИФА,
PTrA
РА и PTrA для под
Mycop/asma syпoviae тверждения положи да быстрое
тельноro теста, ИФА,
PTrA
Sа/топеllа pullo,um РА да медленное
РА реакция аrrлютинации;
РДП реакция диффузной преципитации в rеле; данный метод применим для еженедельных исследований;
ИФА иммуноферментный анализ;
PTrA реакция торможения rемаrrлютинации;
ИО иммуноокрашивание;
рн реакция нейтрализации вируса.
.. По соrласованию с компетентным уполномоченным opraHoM возможно использование друrих типов методов при условии. что
они не менее чувствительны и специфичны, чем указанные.
из материала, полученноro из стаи, не давшей
отрицательную реакцию на ВАЦ, при условии
подтверждения возможности инактивации ВАЦ в
процессе инактивации вакцины.
СОЗДАНИЕ СТАИ КАТЕrоРии SPF
Будущую SРF таю создают из птиц, про
верка которых показала отсутствие вертикаль
но передающихся инфекций, возбудители KO
торых приведены в таблице 5.2.2. 1. Проверка
проводится на 2 поколениях птиц, предшеству
ющих формированию SРF стаи. Общая схема
процедуры получения и поддержания SРF стаи
схематически приведена в таблице 5.2.2. 2. Для
формирования новой SРF стаи проводят ряд
испытаний на 3 поколениях птиц. Все птицы в
первом поколении должны быть проверены по
llpaйней мере один раз до достижения возраста
20 недель на наличие rрупповоro антиrена лей
коза птиц и отсутствие антител к вирусу лейкоза
птиц подтипов А, В и J с помощью иммунофер
ментноro анализа (ИФА) или реакции нейтра
лизации вируса (РН). Также все птицы прове
ряются на отсутствие антител к инфекционным
возбудителям, передающимся вертикально, co
rласно таблице 5.2.2. 1. Начиная с возраста 8
недель, создаваемые стаи проверяют на OTCYТ
ствие Sa/moпella. Клинические наблюдения стаи
проводят, начиная с 8 недель; симптомы Иl-!
фекционных заболеваний у птиц должны отсут
ствовать. Методы анализа, используемые для
оценки формируемой стаи, указаны в таблице,
дополнительные рекомендации также приве
дены в разделе по стандартной проверке SPF
стай. Начиная с возраста 20 недель, стаю прове
ряют в соответствии с разделом «Стандартная
800
rосударственная фармакопея Республики Беларусь
проверка SРF стай». Все стадии этоro режима
также применяют к последующим 2 поколениям,
за исключением проверки каждой птицы на Ha
личие возбудителей, передающихся вертикаль
ным путем. Все результаты испытаний должны
показать отсутствие патоrенных микроорrаниз
мов во всех 3 поколениях стаи, из третьеro по
коления которой будет сформирована SРF стая.
Таблица 5.2.2. 2
Схематическое описание процедуры присво
ения и поддерживания у стаи кате20рии SPF
Определяют отсутствие возбу
дителей, передающихея верти
кальным путем
Не позднее 20 недель жизни
проверяют всех птиц на OTCYТ
ствие вируса лейкоза птиц и aH
тител к нему
НОВЫЕ Начиная с 8 недели жизни, про
ПТИЦЫ водят исследование на нали
чие Sa/monella spp. и проводят
общие клинические исследова
ния
Начиная с 20 недельноro воз
раста, проводят стандартные ис
следования на наличие опреде
ленных возбудителей
Не позднее 20 недели жизни
проверяют всех птиц на
отсутствие вируса лейкоза птиц
и антител к нему
Начиная с 8 недели жизни, про
ВТОРОЕ по водят исследование на нали
КОЛЕНИ Е чие Sa/тonella spp. и проводят
общие клинические исследова
ния
Начиная с 20 недельноro воз
раста, проводят стандартные ис
следования на наличие опреде
ленных возбудителей
Не позднее 20 недели жизни
проверяют всех птиц на OTCYТ
ствие вируса лейкоза птиц и aH
ТРЕТЬЕ по тител к нему
КОЛЕНИЕ Начиная с 8 недели жизни, про
водят исследование на нали
чие Sа/топеllа spp. и проводят
общие клинические исследова
ния
ЕСЛИ РЕЗУЛЬТАТЫ ВСЕХ ИССЛЕДОВАНИЙ
ЯВЛЯЮТСЯ УДОВЛЕТВОРИТЕЛЬНЫМИ,
СТАЕ ПРИСВАИВАЮТ КАТЕrоРию SPF
Начиная с 20 недельноro воз
раста, проводят стандартные ис
следования на наличие Оllреде
ленных возбудителей
ТРЕТЬЕ по Проведение исследований
КОЛЕНИЕ после яйцекладки на наличие
возбудителей, передающихся
вертикальным путем
Проверяют две 5 % выборки птиц
в возрасте 12 и 20 недель на OT
сутствие вируса лейкоза птиц и
антител к определенным инфек
ционным возбудителям
Начиная с 8 недели жизни, про
водят исследование на нали
чие Sa/monella spp. и проводят
ПОСЛЕДУ общие клинические исследова
ЮЩИЕ по ния
КОЛЕНИЯ Начиная с 20 недельноro воз
раста, проводят стандартные
исследования на наличие опре
деленных инфекционных возбу
дителей
Проведение исследований
после яйцекладки на наличие
возбудителей, передающихся
вертикальным путем
в стаю можно добавлять эмбрионы SPF KYP,
полученные из друroй SРF стаи, содержавшейся
в отдельных помещениях на той же птицефер
ме. Начиная с возраста 8 недель, эти введенные
на замену птицы считаются стаей и подверrают
ся исследованиям в соответствии с процедура
ми, описанными выше.
ИСХОДНЫЕ ТРЕБОВАНИЯ
ПО ИССЛЕДОВАНИЮ ПОСЛЕДУЮЩИХ
ПОКОЛЕНИЙ, ПОЛУЧЕННЫХ ИЗ СТАЙ
КАТЕrоРии SPF
При условии, что стая получала замены
исключительно из стаи, имеющей катеroрию
SPF, новое поколение предварительно про
веряется для тоro, чтобы получить катеroрию
SPF. В дополнение к исследованиям на нали
чие Sа/топеllа и контролю общеro состояния
здоровья и функционирования стаи, начиная
с возраста 8 недель, необходимо провести дo
полнительные исследования, описанные далее.
Исследования проводятся на двух 5 % выборках
из стаи (не менее 10 и не более 200 птиц); птицы
отбираются в возрасте 12 16 недель и 16 20
недель с интервалом не менее 4 недели.
Все образцы отбирают и проверяют инди
видуально. Отбирают образцы крови для иссле
дований на наличие антител и подходящие об
разцы для проведения теста на наличие вируса
лейкоза. Используемые методики контроля
должны выполняться в соответствии с разделом
«Стандартная проверка SРF стай». При резуль
татах всех исследований, подтверждающих OT
сутствие каких либо инфекций, новому поколе
нию присваивается катеroрия SPF.
СТАНДАРТНАЯ ПРОВЕРКА SРF СТАЙ
Общий осмотр и патОЛ020анатомиче
ское исследование. Клинические исследования
5.2.2. Стаи кур, не имеющих конкретных патО2енов
801
проводят не реже одною раза в неделю в тече
ние жизни стаи для подтверждения отсутствия
вируса сифилиса домашней птицы и признаков
какой либо инфекции. В случае превышения
уровня смертности более чем 0,2 % в неделю про
водят вскрытие по возможности всех поrибших
птиц, чтобы подтвердить отсутствие признаков
какой либо инфекции. При необходимости для
подтверждения диаrноза проводят rистопатоло
rические и/или микробиолоrические/вирусоло
rические исследования. Проводят специальное
исследование на наличие признаков туберкуле
за; rистолоrические образцы из мест поражения
подверrаются специфической окраске для под
тверждения отсутствия Mycobacterium avium.
Содержимое слепой кишки всех поrибших птиц
по возможности подверrают микробиолоrиче
скому исследованию на наличие Sа/топеllа spp.
при помощи методов, описанных ниже. При He
обходимости MOryT быть объединены образцы
содержимоro слепой кишки 5 птиц.
Культуральный метод на наличие MиKpO
ОР2анизмов вида Sа/топеllа spp. Культураль
ный метод на наличие Sa/moпella spp. проводят
на смывах из прямой кишки, или испражнениях
птиц или мазках, полученных с помощью peK
тальноrо тампона. При анализе испражнений
или смывов каждые 4 недели исследуют 60 об
разцов в течение всей жизни стаи. Для проведе
ния исследования можно объединять до 1 О об
разцов. При анализе мазков отбирают не менее
двух образцов, исследования проводят каждые
4 недели в течение всей жизни стаи. Для обна
ружения Sа/топеllа spp. эти образцы предвари
тельно обоrащают, затем культивируют на пита
тельных средах, селективных для Sa/moпella.
Исследование на наличие анти2ена вируса
лейкоза птиц. До начала яйцекладки проводят
исследования ректальных смывов или образ
цов крови (высеивается светлый слой кровяно
ro crycTKa) на наличие специфических rрупповых
антиrенов лейкоза. В общей сложности образ
цы отбирают от 5 % (не менее 10, не более 200
птиц) стаи каждые 4 недели. Во время яйцеклад
ки образцы белка отбирают из 5 % (не менее 10,
не более 200) всех яиц и исследуют каждые 4
недели. Исследования на наличие rрупповых
специсрических антиrенов вируса лейкоза птиц
про водят методом ИФА, используя методики,
обеспечивающие обнаружение антиrена из под
rрупп А, В и J.
Исследование на наличие антител к дРУ2им
инфекционным возбудителям. Исследования
на наличие антител ко всем инфекционным воз
будителям, перечисленным в таблице 5.2.2. 1,
проводят в течение Bcero периода яйцеклад
ки стаи. Каждые 4 недели образцы отбирают у
5 % (не менее 10, не более 200) птиц стаи. Pe
комендуется проведение еженедельной провер
ки 1,25 % стаи, так как некоторые исследования
на отдельные возбудители должны проводиться
каждую неделю. В таблице 5.2.2. 1 приведена
.01 3.1. I I
классификация возбудителей на быстро распро
страняющиеся в стае и медленно распростра
няющиеся или, возможно, не заражающие всю
стаю. На наличие медленно распространя
ющихся возбудителей каждый образец прове
ряют индивидуально. На наличие быстро pac
пространяющихся возбудителей индивидуально
проверяют не менее 20 % образцов, отбирае
мых каждые 4 недели; при использовании peaK
ции нейтрализации сыворотки или твердофаз
ноro иммуноферментноro анализа все образцы
MorYT быть исследованы индивидуально или
объединены по 5 образцов, отобранных OДHO
временно. Методы, приrодные для обнаружения
возбудителей, приведены в таблице 5.2.2. 1. По
соrласованию с компетентным уполномоченным
opraHoM возможно использование друrих типов
тестов при условии, что они не менее чувстви
тельны и специфичны, чем указанные.
ИССЛЕДОВАНИЯ, ПРОВОДИМЫЕ
В КОНЦЕ ПЕРИОДА ЯЙЦЕКЛАДКИ
После последнеro сбора яиц проводят за
ключительные исследования, чтобы подтвер
дить отсутствие возбудителей, передающих
ся вертикальным путем, указанных в таблице
5.2.2. 1. После последнеro сбора яиц не менее
5 % стаи (не менее 10, не более 200 птиц) отби
рают для проведения исследований по крайней
мере в течение 4 недель. Образцы крови отби
рают у каждой птицы в rруппе во время 4 He
дельною периода, при этом по крайней мере у
1,25 % птиц (25 % образцов) образцы отбирают
не ранее чем через 4 недели после заключитель
ноro сбора яиц. Образцы сыворотки проверяют
на наличие возбудителей, передающихся Bep
тикальным путем (таблица 5.2.2. 1), с помощью
указанных методов. Если отбор образцов ocy
ществляют еженедельно, исследованию в этот
период подверrают не менее 1,25 % птиц (25 %
образцов). В качестве альтернативы в течение 4
недель после заключительноro сбора яиц кровь
и/или друrие подходящие для исследований Ma
териалы отбирают по крайней мере у 5 % стаи
и исследуют на наличие возбудителей, переда
ющихся вертикальным путем, с помощью вали
дированных методик на основе на амплифика
ции нуклеиновых кислот (2.6.21).
ДЕЙСТВИЯ, КОТОРЫЕ ДОЛЖНЫ
БЫТЬ ПРЕДПРИНЯТЫ В СЛУЧАЕ
ОБНАРУЖЕНИЯ СПЕЦИФИЧЕскоrо
ВОЗБУДИТЕЛЯ
Если обнаружено заражение стаи возбуди
телем, относящимся соrласно таблице 5.2.2. 1
к медленно распространяющимся, все матери
алы, полученные из стаи в течение 4 недель,
предшествующих дате отбора образца с наличи
ем возбудителя, считаются неудовлетворитель
ными. Аналоrично, если обнаружено заражение
стаи возбудителем, относящимся соrласно Ta
блице 5.2.2. 1 к быстро распространяющимся,
802
rосударственная фармакопея Республики Беларусь
все материалы, полученные из стаи в течение 2
недель, предшествующих дате отбора образца
с наличием возбудителя, считаются неудовлет
ворительными. Любой продукт, про изведенный
с использованием таких материалов, и дЛЯ KO
Toporo обязательно использование материалов
SPF, считают неприroдным и бракуют; все испы
тания по контролю качества, проведенные с ис
пользованием таких материалов, являются He
действительными.
Производители обязаны уведомить потре
бителей всех яиц об обнаружении заражения как
можно скорее после вспышки эпидемии.
Любая стая, для которой подтверждено за
ражение любым определенным возбудителем,
не может повторно получить катеroрию SPF.
Любое потомство стаи, полученное во время или
после 4 недельноro периода до последнеro от
рицательноro образца, не может быть отнесено
к катеroрии SPF.
01/2013:50203
5.2.3. КЛЕТКИ-ПРОДУЦЕНТЫ
ДЛЯ ПРОИЗВОДСТВА ВАКЦИН
ДЛЯ МЕдицинскоrо
ПРИМЕНЕНИЯ
Данный общий раздел распространяется на
линии диплоидных клеток и непрерывные линии
клеток, используемые в качестве клеток проду
центов в производстве вакцин для медицинско
ro применения; специфические вопросы, связан
ные с вакцинами, приroтовленными при помощи
технолоrии рекомбинантной ДНК, описываются
в общей статье Продукты теХНОЛ02ии рекомби
нантной днк. В таблице 5.2.З. 1 указаны испы
тания, которые должны проводиться на различ
ных этапах (посевная культура клеток, rлавный
банк клеток, рабочий банк клеток, клетки на MaK
симальном уровне удвоения популяции или
выше, используемом при производстве). Общие
условия использования культур и методов ис
следования приведены ниже. Требования по ис
пользованию в производстве отдельных вакцин
первичных клеток или клеток, подверrшихся He
скольким пассажам без создания банка культу
ры клеток, при водятся в частных фармакопей
ных статьях.
ЛИНИИ диплоидных клеток (диплоидные
штаммы клеток) [d;plo;d се" liпеs]. Линия ди
плоидных клеток обладает высокой, но оrрани
ченной способностью размножения in vitro.
Непрерывные линии клеток (перевивае-
мые линии клеток) [сопt;пиоиs се" liпеs]. He
прерывные (перевиваемые) линии клеток облада
ют способностью неоrраниченноro размножения
in vit,o; клетки зачастую имеют различия в кари
отипе по сравнению с первичными клетками; их
можно получить из :щоровой ткани или опухоли
либо млекопитающих, либо насекомых.
Процесс очистки вакцин, вводимых инъек
ЦИОННО и производимых С использованием He
прерывных линий клеток, валидируют на спо
собность удаления ДНК клеток субстратов до
уровня, равноro не более 1 О Hr на одну вакцин
ную дозу, если друrое не будет доказано.
Система банка клеток (система банк куль-
тур клеток) [cell-baпk system]. Производство
вакцин с использованием линий диплоидных
клеток и перевиваемых линий клеток основано
на системе банков клеток. Возраст клеток iп vitro
отсчитывают от rлавноro банка клеток. Каждый
рабочий банк клеток rотовят из одноro или He
скольких контейнеров rлавноrо банка клеток. Ис
пользование, происхождение и распределение
контейнеров тщательно документируют.
Среды И вещества животноrо и челове-
ческоrо происхождения. Состав питательных
сред, применяемых для выделения и последу
ющеro культивирования, подробно описывают,
а используемые в них вещества животноro или
человеческоro происхождения не должны co
держать посторонних areHТOB (2.6.16) и должны
соответствовать обuцей qpармакопейной статье
5.1.7. Вирусная безопасность.
При использовании донорскоrо альбумина
необходимо обеспечить еro соответствие част
ной статье Альбумина человека раствор.
Если используется бычья сыворотка, она
должна отвечать требованиям частной статьи
Сыворотка бычья.
Трипсин, используемый для приrотовления
культур клеток, контролируют на стерильность и
отсутствие микоплазм и вирусов, особенно пе
стивирусов, цирковирусов и парвовирусов.
Посевная культура клеток (посев клеток)
[се" seed]. Данные, используемые для оценки
приroдности посевной культуры клеток, включа
ют информацию об источнике, истории и xapaK
теристике, rде это возможно.
Источник посевной культуры клеток Для
линий клеток человека реrистрируют следу
ющие данные о доноре: этническое и rеоrрафи
ческое происхождение, возраст, пол, общее фи
зиолоrическое состояние, использованная ткань
или opraH, результаты всех исследований на Ha
личие патоrенных микроорrанизмов.
Для линий клеток животных реrистрируют
следующие данные об источнике клеток: вид,
линия животных, условия разведения, rеоrрафи
ческое происхождение, возраст, пол, обuцее qpи
зиолоrическое состояние, использованная ткань
или opraH, результаты всех исследований на Ha
личие патоrенных микроорrанизмов.
5.2.3. Клетки продуценты для производства вакцин 803
Таблица 5.2.З. 1
Испытания линий клеток
Клетки на макси-
мальном уровне
Посевная rлавный банк Рабочий банк удвоения по-
Испытание культура пуляции или
клеток клеток
клеток выше, использу-
емые при произ-
водстве
1. ПОДЛИННОСТЬ И ЧИСТОТА
Морфолоrия + + + +
Подлинность: метод «отпе
чатка пальцев» нуклеиновой
кислоты и соответствующий
выбор из следующих MeTO
дов:биохимический(напри + + + +
мер, изоферментный анализ),
иммунолоrический (например,
rистосовместимость), цитоrе
нетические маркеры
Кариотип (диплоидные линии + + +(1) +(1)
клеток)
Продолжительность жизни
(диплоидные линии клеток) + +
2. ПОСТОРОННИЕ ArEHTbI
Заражение бактериями + +
и rрибами
Микоплазмы + +
Спироплазмы (линий клеток, + +
выделенных из насекомых)
Электронная микроскопия
(линий клеток, выделенных +(З) +(З)
из насекомых)
Испытания на внешние +
areHTbI в клеточных культурах
Испытание на клеточных +
культурах
Совместное культивирование +(2) +(2)
Испытания на животных +(2) +(2)
и яйцах
Специфические испытания
на наличие возможных посто
ронних areHTOB в зависимости +(2) +(2)
от происхождения клеток (см.
ниже пункт Посторонние ин
фекционные areHTbI)
Ретровирусы +(З) +(З)
З.КАНЦЕРоrЕННОСТЬ
Канцероrенность +(5) +(4)
(1) Диплоидный характер устанавливают для каждою рабочеrо банка клеток, но с использованием клеток на максимальном
уровне удвоения или свыше ero, используемом для производства.
(2) Испытание проводят для каждоro рабочеrС' банка клеток, но с использованием клеток на максимальном уровне удвоения
популяции, используемом для производства, или свыше нею.
(З) Испытание проводят для каждою rлавною банка клеток, но с использованием клеток на максимальном уровне удвоения
популяции, используемом для производства, или свыше нею.
(4) Линии MRC 5, WI З8 и FRhL 2 признаны неканцероrенными и не требуют исследований Испытания не проводят на колониях
клеток, о которых известно или предполаrается, что они канцероrенны. например. сна и BHK 21_
(5) Испытание проводят на первичных клетках. 'ю с применением клеток на максимальном уровне удвоения популяции,
используемом для производства, или превышающем нею.
804
rосударственная фармакопея Республики Беларусь
Клетки нейрональноro происхождения,
такие нейробластомы и линии клеток Р12, MOryт
содержать вещества, в которых концентриру
ются возбудители ryбчатой энцефалопатии, и
такие клетки не MOryT быть использованы для
производства вакцин.
История посевной культуры клеток Реrи
стрируют следующую информацию: метод, ис
пользованный для выделения первичных клеток,
методы культивирования и любые друrие процеду
ры, использованные для создания rлавноro банка
клеток, в особенности те, при которых в клеточную
культуру MOryт попасть посторонние areHTbI.
Полная информация о компонентах пита
тельных сред, использованных в прошлом для
культивирования клеток, может быть недоступ
на, например, об источнике веществ животною
происхождении; если это обосновано и утверж
дено, уже созданные банки клеток с использова
нием таких культур MOryT применяться в произ
водстве вакцин.
Характеристика посевной культуры клеток
Определяют следующие свойства:
(1) подлинность клеток (например, изофер
менты, серолоrия, последовательность нукле
иновой кислоты);
(2) характеристики роста клеток и их морфо
лоrические свойства (оптическая и электронная
микроскопия );
(3) для линий диплоидных клеток карио
тип;
(4) для линий диплоидных клеток продол
жительность жизни iп vitro, выраженная с учетом
уровней удвоения популяции.
Стабильность клеток продуцентов.
Должна быть подтверждена соответствующая
жизнеспособность линий клеток в предполаrа
емых условиях хранения. Для каждоro препа
рата, производимоro С использованием линий
клеток, следует подтвердить возможность посто
янною производства на клетках различных ypOB
ней пассажа в начале и в конце предполаrаемой
длительности использования.
Посторонние инфекционные areHTbI.
Линии клеток для производства вакцин не
должны содержать посторонних инфекционных
возбудителей. Испытания на выявление посто
ронних areHToB проводятся в соответствии с Ta
блицей 5.2.3. 1, используя методы, описанные
ниже.
Для клеточных линий, выделенных из Hace
комых, применяют испытания на определенные
вирусы, релевантные видам насекомых, из KO
торых были выделены клетки, и Н8 арбовирусы
(артропозные вирусы). Набор испытуемых виру
сов выбирают в соответствии с текущим cocтo
янием научных знаний.
Линии клеток, в которых обнаружены peTpo
вирусы, способные к репликации, не при меняют
для производства вакцин.
Канцероrенность. Культура клеток для про
изводства живых вакцин не должна быть KaH
цероrенной на любом уровне удвоения популя
ции, используемом для производства вакцин.
Если для производства друrих типов вакцин ис
пользуется канцероrенная линия клеток, при Ba
лидации должна быть доказана способность
процесса очистки обеспечивать удаление OCTa
точноro ДНК клеток продуцентов до содержа
ния не более 1 О Hr в одной вакцинной дозе, если
не установлено друroе, и снижение содержания
остаточноro белка клеток продуцентов до при
емлемоro уровня.
Дальнейшие исследования линий клеток не
проводят, если известно об их канцероrенном
действии. Если неизвестно, обладает ли линия
клеток канцероrенным действием, ее либо счита
!ОТ канцероrенной, либо проводят исследования
на наличие канцероrенноro действия с использо
ванием методики iп vivo, описанной ниже, и дo
полнительно испытание in vitro, если необходима
дополнительная информация. Испытания про
водят на клетках на максимальном уровне YДBO
ения популяции или превышающем еro, который
будет использоваться при производстве вакцин.
Линии клеток MRC 5, WI 38 и FRhL 2 при
знаны неканцероrенными, и их дополнительное
исследование не требуется.
Характеристика хромосом. Должно быть
доказано, что линии ДИПЛОИДНЫХ клеток имеют
диплоидный набор хромосом. Если удаление
неразрушенных клеток при обработке биомас
сы после сбора не было валидировано, необхо
димо провести более детальную характеристику
линии диплоидных клеток путем анализа карио
типа. Испытания проводят на образцах четырех
пассажей, полученных через равные промежутки
в течение жизненноro цикла клеток. Для точноro
подсчета хромосом и частоты rиперплоидии, rи
поплоидии, полиплоидии, повреждений и CTPYK
турных аномалий следует исследовать мини
мум 200 клеток в метафазе. Линии клеток MRC 5,
WI 38 и FRhL 2 признаны диплоидными и oxapaK
теризованы в достаточном объеме; если они не
подверrались rенетическим модификациям, их
дополнительная характеристика не требуются.
МЕТОДЫ ИСПЫТАНИЙ КЛЕТОЧНЫХ
КУЛЬТУР
Морфолоrия: морфолоrия клеток должна
быть адекватно описана и задокументирована.
Подлинность. Для идентификации клеток
используют метод «отпечатка пальцев» нукле
иновой кислоты и определяют подходящие из
приведенных ниже параметров:
(1) биохимические характеристики (изофер
ментный анализ);
(2) иммунолоrические характеристики (анти
reHbI rистосовместимости);
(3) цитоrенетические маркеры.
5.2.3. Клетки продуценты для производства вакцин
80Е
Посторонние (контаминирующие) клетки.
Метод «отпечатка пальцев» нуклеиновой кисло
ты используют как для целей идентификации,
так и для доказательства отсутствия посторон
них клеток.
Заrрязнение бактериями и rрибами. rлав
ный банк клеток и каждый рабочий банк клеток
должны быть стерильны (2.6.1), в испытании ис
пользуют 1 О мл супернатанта культур клеток для
каждой питательной среды. Испытанию подвер
rают 1 % контейнеров, но не менее 2 контейне
ров.
Микоплазмы (2.6.7). rлавный банк клеток и
каждый рабочий банк клеток должны выдержи
вать испытание на микоплазмы. Испытание про
водят на одном или более контейнере.
Спироплазмы (линии клеток, выделенные
из насекомых). При проверке валидированным
методом, разрешенным компетентным упол
номоченным opraHoM, rлавный банк клеток и
каждый рабочий банк клеток, выделенных из Ha
секомых, не должны содержать спироплазм. Ис
пытание проводят на одном или более контей
нере.
Электронная микроскопия (линии клеток,
выделенные из насекомых). rлавный банк
клеток исследуют электронной микроскопией на
присутствие посторонних areHToB. Линии клеток
содержат при температуре, обычно используе
мой при производстве, и отбирают их при дости
жении или превышении максимальноro уровня
удвоения популяции. Кроме тоro, линии клеток
содержат при температурах выше или ниже
той, что используется при производстве, и MOryT
также подверrать друrим видам обработки, Ha
при мер, воздействию со стороны химических
стресс факторов. Наряду с температурами xpa
нения и видами обработки компетентный упол
номоченный opraH устанавливает количество ис
следуемых секци онированных клеток.
Испытание на наличие посторонних areH-
тов в культурах клеток. Клетки исследуют на
наличие rемадсорбирующих вирусов и друrих по
сторонних areHТOB в культурах клеток, приведен
ных в статье 2.6.16 в разделе «Производствен
ная клеточная культура: контрольные клетки».
Если клетки взяты от обезьяны, их также вводят
в культуру клеток почки кролика для исследова
ния на наличие вируса rерпеса В (cercopithecid
herpesvirus 1).
Совместное культивирование. Для кле
точных линий, выделенных из млекопитающих
и птиц, отдельно культивируют интактные и/или
разрушенные клетки совместно с друrими кле
точными системами, включая клетки человека
и обезьяны. Для клеточных линий, выделенных
1О2.3"к 1712
из насекомых, инкубируют экстракты разрушен
ных клеток с друrими клеточными системами
включая клетки человека, обезьяны и по край.
ней мере одну клеточную линию, которая отли
чается от той, что используется в производстве
допускаются вирусы насекомых и арбовирусь
человека (например, BHK 21). Проводят иссле
дование для обнаружения морфолоrических из
менений. Для выявления rемаrrлютинирующи>
вирусов проводят испытания на жидкостях кле
точных культур или на клетках для выявле
ния rемадсорбирующих вирусов. Испытание Не
rемаrrлютинирующие вирусы не применяют для
арбовирусов, обнаруженных в клетках HaceKO
мых. Клетки выдерживают испытание, если не
обнаружено доказательства наличия какоro ли-
бо постороннеrо areHTa.
Ретровирусы. Исследования на наЛИЧИf
ретро вирусов проводят с использованием:
(1) количественноro определения образо-
вания продукта под действием обратной транс.
криптазы (РЕRТ) (2.6.21); проводят на супер
натанте из банка клеток, используя клетки не
максимальном уровне удвоения, используемом
для производства. или свыше неro;
(2) трансмиссионной электронной микроско-
пии;
(3) определения инфекционноro титра на
клетках человека с PERT определением (конеч
ной точки) на супернатанте.
Если испытание (1) и/или испытание (2)
дают положительную реакцию, то проводят ис
пытание (3);
Так как чувствительность PERT метода
очень высока, интерпретация положительно-
ro сиrнала может быть ошибочной, и решение
относительно приrодности клето продуцеНТОЕ
принимается с учетом всех имеющихся данных.
Испытания на животных. Вводят внутри-
мышечно (или, для мышей, питающихся моло-
ком матери, подкожно) в каждой из следующи>
rрупп животных 107 жизнеспособных клеток раз-
деленных поровну между животными в каждоv.
rруппе:
(1) два помета мышей в возрасте MeHe€
24 ч, общим числом не менее 1 О животных;
(2) десять взрослых мышей.
Следует ввести интрацеребрально каждой
из десяти взрослых мышей 1 Об жизнеспособных
клеток для обнаружения возможноrо наличия
вируса лимфоцитарноrо хориоменинrита.
За животными наблюдают не менее 4 недель.
Обследуют заболевших живоп"ых или животных,
У которых наблюдаются аномалии, для определе-
ния причины заболевания. Клетки выдерживают
испытание. если не обнаружено доказательств
наличия какоro либо постороннеrо areHTa. Испы-
тание считается недействительным. если менее
80 % животных в каждой rруппе остаются здоро
выми и доживают до конца периода наблюдения.
806
rосударственная фармакопея Республики Беларусь
Испытания на куриных эмбрионах. Жиз
неспособные клетки в количестве 106 на один
эмбрион вводят в аллантоисную полость десяти
куриных эмбрионов катеroрии SPF (5.2.2)
9 11 дневноro возраста и в желточный мешок
десяти куриных эмбрионов катеroрии SPF
6 дневноro возраста. Эмбрионы инкубируют
не менее 5 дней. Аллантоисную жидкость иссле
дуют на наличие rемаrrлютининов, используя
эритроциты млекопитающих и птиц; испыта
ние проводят при температуре (5::t3) ос и (20
25) ОС, результаты считывают спустя 30.. E30
мин. Клетки выдерживают испытание, если не
обнаружено доказательств наличия какоro либо
постороннеro areHTa. Испытание считается He
действительным, если менее 80 % эмбрионов
остаются здоровыми и доживают до конца пери
ода наблюдения.
Специфические испытания на возмож-
ные контаминанты в зависимости от источ-
ника происхождения клеток. Испытания на
специфические патоrены проводят с использо
ванием методов амплификации нуклеиновых
кислот (NAT, пис/еiс acid атрlifiсаtiоп tесhпiqие)
(2.6.21) с проведением или без проведения
предварительной амплификации в клетках.
С друrой стороны, можно использовать COOT
ветствующие серолоrические методы, напри
мер, иммуносорбентный анализ с ферментной
меткой, нейтрализация сыворотки и испытания
по выработке антител на подходящих peKOMeH
дованных животных. Для клеточных линий, BЫ
деленных из rрызунов, используют либо испы
тания по выработке антител на мышах, крысах
или хомяках, либо методы амплификации HY
клеиновых кислот (2.6.21) для обнаружения ви
доспецифичных вирусов. Испытание должно
учитывать происхождение и историю культуры
клеточной линии. Испытания разрабатывают
ся для выявления возможных контаминантов,
особенно тех. для которых известно, что они
инфицируют латентно, например, вакуолизиру
ющий обезьяний вирус (OB 40) у макак резус
или F/ock house вирус в клетках, выделенных
из насекомых.
Испытания на канцероrенность in vivo.
Испытание заключается в сравнительном aHa
лизе исследуемой перевивиной культуры клеток
и соответствующей положительной контрольной
культуры (например, культур клеток HeLa или
Нер2).
Биолоrические модели животных, приroд
ные для этоro теста, включают:
(1) бестимусных мышей (rенотип Nu/Nu);
(2) новорожденных мышей, крыс или хомя
ков, получающих антитимоцитную сыворотку
или иммуноrлобулин;
(3) облученных мышей с удаленной вилоч
ковой железой, получивших костный мозr от здо
ровых мышей (Т ,В').
Вне зависимости от выбранной биолоrич
ской модели испытуемые и контрольные клетки
вводят инъекционно животным, разделенным
на отдельные rруппы по 10 животных в каждой.
В обоих случаях прививочный материал из 107
клеток в виде взвеси объемом 0,2 мл вводят BHY
тримышечно или подкожно. Новорожденным жи
вотным вводят 0,1 мл антитимоцитной сыворотки
или иммуноrлобулина на О, 2,7 и 14 день после
рождения. Сильнодействующая сыворотка или
иммуноrлобулин подавляет иммунные механиз
мы растущих животных до уровня, при котором
последующее введение 107 положительных KOH
трольных клеток реryлярно вызывает опухоли и
метастазы. Животные с тяжелыми поражениями
и симптомами быстро растущих опухолей следу
ет подверrать эвтаназии, не дожидаясь конца ис
пытания, чтобы избежать ненужных страданий.
В конце периода наблюдения всех живот
ных, включая контрольную rруппу (rруппы), под
верrают эвтаназии и исследуют на наличие Ma
кроскопических и микроскопических признаков
пролиферации введенных клеток в месте инъек
ции и в друrих opraHax (например, лимфатиче
ских узлах, леrких, почках и печени).
При использовании любой биолоrической
модели животных наблюдают и пальпируют
через реrулярные промежутки времени для
обнаружения уплотнений в месте инъекции.
Любые сформировавшиеся уплотнения из
меряют в двух перпендикулярных направле
ниях, измеренные размеры реrистрируют для
оценки наличия проrрессивноro увеличения
уплотнений. Животных, у которых в период Ha
блюдения уплотнения начинают уменьшать
ся, подверrают эвтаназии до тоro, как уплот
нения перестанут прощупываться, и проводят
rистолоrическое исследование. За животны
ми с проrрессивно растущими уплотнениями
наблюдают в течение 1 2 недель. За поло
виной животных, у которых уплотнения OTCYT
ствуют, наблюдают в течение 3 недель, а за
второй половиной в течение 12 недель до
тоro, как их подверrают эвтаназии и rистолоrи
ческому исследованию. Каждое животное под
верrают некропсии, которая включает обследо
вание на предмет макроскопических признаков
формирования опухолей в месте инъекции и в
друrих opraHax, таких как лимфатические узлы,
леrкие, мозr, селезенка, почки и печень. Все
опухолевые образования в месте инъекции
подверrают rистолоrическому анализу. Кроме
тоro, так как некоторые линии клеток MorYT BЫ
звать метастазы без признаков местноro роста
опухоли, проводят rистолоrическое исследова
ние всех обнаруживаемых местных лимфати
ческих узлов и леrких животных.
Испытание считается недействительным,
если менее чем у девяти из десяти животных,
которым были введены положительные базовые
клетки, наблюдаются проrрессивно 4 ' растущие
опухоли.
5.2.8. Снижение риска передачи возбудителей еубчатой энцефалопатии
807
Испытания на канцероrенность in vitro.
MorYT быть использованы следующие TeCT
системы:
(1) образование колоний в мяrких arapo
вых rелях;
(2) продуцирование инвазивноro роста
клеток после инокуляции в культуры opraHa;
(3) исследование трансформации актив
ности с использованием, например, системы
3Т3 для количественноro определения актив
ных oHKoreHoB.
01/2013:50208
5.2.8. СНИЖЕНИЕ
РИСКА ПЕРЕДJ\ЧИ
ВОЗБУДИТЕЛЕиrУБЧАТОИ
ЭНЦЕФАЛОПАТИИ
ЖИВОТНЫХ
ПРИ ПРИМЕНЕНИИ
МЕДИЦИНСКИХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
Этот раздел идентичен PeKOMeHдaци
ям по снижению риска передачи возбудите
лей 2убчатой энцефалопатии животных при
применении медицинских и ветеринарных ле
карственных средств, Версия 3, (EMA/410101
версия 3) [Комитет по патентованным ле
карственным средствам (СРМР), Комитет
по ветеринарным лекарственным cpeд
ствам (CVMP), Европейское а2ентство по
оценке лекарственных средств).
Содержание
1. ВВЕДЕНИЕ
1 1. Научная справка
1 2. Соответствие требованиям законода
тельства
2. ОБЛАСТЬ ПРИМЕНЕНИЯ ОБЩЕЙ
СТАТЬИ
3. ОБЩИЕ ПОЛОЖЕНИЯ
3 1. Научные принципы снижения вероятно
ro риска
3 2. Животные источники материала
3 2 1. rеоrрафическое происхождение
3 2 1 1. Материал от крупноro poraToro скота
3 2 1 2. Овцы и козы (мелкие жвачные жи
вотные)
3 2 2. Стада крупноro poraтoro скота
(изолированные) с незначительным риском
rэкРс (BSE)
3 3. Части тела животных, биолоrические
среды и выделения как исходные материалы
3 4. Возраст животных
3 5. Производственный процесс
4. ОЦЕНКА СТЕПЕНИ РИСКА МАТЕРИ
АЛОВ ИЛИ ВЕЩЕСТВ, ИСПОЛЬЗУЕМЫХ В ПРО
ЦЕССЕ ПРОИЗВОДСТВА ЛЕКАРСТВЕнноrо
СРЕДСТВА, ПРОВОДИМАЯ В СООТВЕТСТВИИ
С УСТАНОВЛЕННЫМИ ТРЕБОВАНИЯМИ
5. ОЦЕНКА ПОЛЬЗА/РИСК
6. ПОЛОЖЕНИЯ ПО ОТДЕЛЬНЫМ MATE
РИАЛАМ
6 1. Коллаrен
6 2. Желатин
6 3. Бычья кровь и материалы из бычьей
крови
64. Материалы из твердоro жира
6 5. Животный уrоль
6 6. Молоко и материалы из молока
6 7. Материалы из шерсти
6 8. Аминокислоты
6 9. Пептоны
1. ВВЕДЕНИЕ
1 1. НАУЧНАЯ СПРАВКА
Трансмиссивные ryбчатые энцефалопатии
(тrэ, Т,апsтissiЫеSропgifоrтЕпсерhа/ораthiеs
TSEs) являются хроническими нейродеrе
неративными заболеваниями, характеризу
ющимися накоплением аномальных изоформ
клеточноro rликопротеина, известноrо как PrP
(или белок прион). Аномальная изоформа PrP
(PrPTSE) отличается от нормальной PrP (PrPc)
высокой устойчивостью к протеазе и дeHaTypa
ции при высокой температуре. PrPTSE, как пола
rают, является инфекционным areHToM, вызыва
ющим развитие тrэ.
тrэ болезни животных включают:
ryбчатую энцефалопатию крупноro poraTo
ro скота (rэкРс, BSE),
скрейпи (почесуха) овец и коз,
хроническое истощение у оленей и лосей,
трансмиссивную энцефалопатию норок,
ryбчатую энцефалопатию кошачьих (oco
бенно у домашних кошек и больших животных
семейства кошачьих, содержащихся вневоле),
экзотическую ryбчатую энцефалопатию
копытных, содержащихся в зоопарках.
rубчатая энцефалопатия у людей вклю
чает различные формы болезни Крейцфель
да Якоба (БКЯ), Куру, синдром repcTMaHHa
Штройслерr Шейнкера (rшш), фатальная
(хроническая) семейная бессонница (ФСБ).
Известны случаи развития ятроrенной TpaHC
миссивной rубчатой энцефалопатии. Скрейпи
овец была случайным образом передана при
применении вакцины Lоuрiпg 111, полученной
из объединенной партии обработанноrо фор
мальдеrидом мозrа и селезенки овец, в KO
торую попал материал от овец, страдающих
скрейпи. Также скрейпи была передана овцам
и козам при применении инактивированной
формальдеrидом вакuины против инфекци
онной аrалактии, полученной из roMoreHaToB
мозrа и молочной железы овец, инфициро
ванных Mycop/asma aga/actiae. Сообщалось о
случаях передачи БКЯ у человека вследствие
808
rосударственная фармакопея Республики Беларусь
парентеральною введения соматотропина и ro
надотропина, полученноrо из rипофизов челове
ка. Также случаи возникновения БКЯ связывают
с использованием заrрязненных инструментов
при хирурrических операциях на roловном мозrе
и при трансплантации твердой оболочки мозrа и
роювицы человека.
Межвидовое распространение тrэ оrрани
чивают мноrие естественные барьеры, способ
ность передачи от вида происхождения, штаммы
прионов, дозы, пути воздействия, а для HeKOТO
рых видов, аллели хозяина reHa PRNP. При co
ответствующих обстоятельствах эти барьеры
MorYT быть преодолены.
rубчатая энцефалопатия крупноro poraтoro
скота (rэкРс, BSE) была впервые выявлена в
Великобритании в 1986 r. Заболеванием было
поражено большое количество животных и OT
дельные стада. Известно, что rэкРс (BSE)
заболевание, вызванное приемом пищи, coдep
жащей мясо и костную муку, полученную из жи
вотных, больных тrэ. в друrих странах также
были зареrистрированы случаи rэкРс (BSE)
как у животных, импортированных из Велико
британии, так и у местных животных. Есть убе
дительные доказательства, что вариантная
форма БКЯ вызвана возбудителем, вызываю
..цим rэкРс (BSE) у крупноro poraтoro скота.
Таким образом, необходимо с осторожностью
использовать для производства лекарственных
средств биолоrические материалы от видов, по
;Jажаемых болезнями тrэ, особенно от крупноrо
poraToro скота.
В связи с активными проrраммами Haд
зора две ранее неопознанные формы атипич
чой rэкРс (rЭКРС L, также названная BASE,
rэкРс н) были обнаружены в редких единичных
случаях в Европе, Северной Америке и Японии.
Символы «L» и «Н» указывают на низкое и BЫCO
кое электрофоретическое расположение рези
стентной к протеазе изоформы PrPTSE. Следует
отметить, что атипичные случаи были обнаруже
ны в странах, в которых до сих пор не встреча
лась классическая rэкРс, например, Швеция,
или в которых встречал ось Bcero несколько слу
чаев классической rэкРс. таких как Канада
или СШд. Атипичный areHT rэкРс был экспе
риментально передан TpaHcreHHbIM мышам с
человеческим протеином приона и обезьянам
Cyпomo/gus.
Скрейпи (почесуха) обнаружена во всем
мире и в большинстве европейских стран.
Самый высокий уровень заболеваний отмечен
на Кипре. Хотя люди в течение 250 лет были
подвержены скрейпи, существующей в природе,
нет никаких эпидемиолоrических свидетельств,
непосредственно связывающих скрейпи С ryб
чатой энцефалопатией людей. Однако остается
теоретический и в настоящее время не ПО,DДа
ющийся оценке риск, что некоторые пищевые
белковые добавки, зараженные rэкРс (BSE) ,
возможно, содержатся в корме для овец. Если
такой корм вызывает рекуррентную rэкРс (BSE)
у овец, которая может проявляться в виде скрей
пи, то это может представлять риск передачи
тrэ человеку(1). Далее, необходимо принять во
внимание, что любой возбудитель rэкРс (BSE),
попадающий в маленькую популяцию жвачных
животных через контаминированную пищу, Bepo
ятно, будет переработан и распространен(2).
Существует также интерес в инфицирова
нии клеток возбудителями тrэ для разработки
методов определения и для решения основных
научных вопросов. Было сообщено о HeKOТO
ром успехе при использовании линий как HepB
ных клеток, так и друrих клеток. Не очень понят
ны условия, необходимые для инфицирования
клетки, а также сложен процесс, который требует
комбинации возбудителя и клетки. Должным об
разом не рассматривается внесение специфиче
ских рекомендаций в отношении клеточных суб
стратов, используемых в производстве веществ,
полученных биолоrическим/биотехнолоrическим
путем. Однако при оценке риска должна быть
принята во внимание возможность инфицирова
ния линии клеток возбудителями тrэ.
1 2. СООТВЕТСТВИЕ ТРЕБОВАНИЯМ
3АКОНОДАТЕЛЬСТВА
#Раздел приводится для информации. #
Оценка степени риска. Так как использова
ние материалов, полученных из животных, He
обходимо для производства некоторых лекар
ственных средств, и полное исключение риска
невозможно, меры, принимаемые для управ
ления риском передачи rэкРс (BSE) живот
ных при применении лекарственных средств,
сводятся к минимизации риска, а не еro исклю
чению. Следовательно, выполнение требова
ний законодательства должно быть основано
на оценке степени риска с учетом всех важных
факторов, описанных в этом подразделе (см.
далее).
Правовые аспекты. Настоящие PeKOMeHдa
ции имеют обязательный для исполнения в CTpa
нах Евросоюза юридический статус соrласно:
Приложению 1, часть 1, модуль 3, раздел 3.2.:
Содержание: общие принциПbl и требования,
пункт (9) Директивы 2001/83/ЕС Европейскою
Парламента и Совета от 6 ноября 2001 юда в OT
ношении медицинских лекарственных средств(З';
Приложению 1, Издание 1, раздел С Про
изводство и контроль исходных материалов в
Директиве 2001/82/ЕС Европейскою Парламен
та и Совета от 6 ноября 2001 rода в отношении
ветеринарных лекарственных средств(4).
Соrласно требованиям указанных дирек
тив заявитель при реrистрации ветеринарных
и медицинских лекарственных средств должен
доказать, что лекарственные средства про
изведены в соответствии с последней Bep
5.2.8. Снижение риска передачи возбудителей аубчатой энцефалопатии
809
сией данных Рекомендаций, опубликованной
в Официальном журнале Европейскоro Союза.
Это обязательство продолжается во весь
период действия реrистрации лекарственноrо
средства.
Поопределению, принцип материалов специ
фическою риска, как определено в Постановле
нии ЕС N2 999/2001 Европейскоro парламента и
Совета(5), не применим к лекарственным cpeд
ствам. Однако, Постановление ЕС N2 1774/2002
Европейскоro парламента и Совета(В), которое
вступило в действие 1 марта 2003 юда, YCTaHO
вило правила в отношении побочных продуктов
животною происхождения, не предназначенных
для потребления человеком. Как правило, если
иное не подтверждено, все побочные продук
ты животноro происхождения, используемые в
качестве исходных материалов в производстве
медицинских продуктов, должны относится к Ka
теroрии «Материалы и эквиваленты катеroрии 3
(т. е. безопасные)>>, как указано в Постановле
нии ЕС N2 1774/2002. Использование веществ,
полученных из тканей с высоким риском нали
чия инфекционных areHToB, должно быть BceCTO
ронне обосновано с помощью соответствующей
оценки пользаlриск (см. далее).
Настоящие рекомендации следует при
менять совместно с различными норматив
ными правовыми документами Европейскоro
Союза, включая решения Комиссии, постоян
но вводившиеся в действие с 1991 юда. Если
возможно, ссылки на эти решения при водятся
по тексту. Официальные заявления и поясне
ния, сделанные Комитетом по патентованным
лекарственным средствам (СРМР) и Комите
том по ветеринарным лекарственным cpeд
ствам (CVMP), применимы для выполнения
установленных требований, если COOTBeTC
твующий аспект не рассматривается в PeKO
мендациях.
Общая фармакопейная статья «Продукты,
которые М02ут быть переносчиками 2убчатой
энцефалопатии животН020 происхождения»
Фармакопеи ссылается на этот общий раздел,
который совпадает с Рекомендациями.
Разъяснения по выполнению Рекомендаций.
По мере расширения научных знаний о тrэ, в
частности патоrенеза заболеваний, СНМР и ею
Рабочей rруппе по биолоrическим препаратам в
сотрудничестве с CVMP и ero Рабочей rруппой
по иммунолоrическим препаратам может быть
поручена в будущем разработка дополнитель
ноro руководства в форме официальных заяв
лений и пояснений для разъяснения требований
Рекомендаций. Допnлнительное вспомоrатель
ное руководство будет издано Комиссией, раз
мещено на вебсайте Европейскою Медицин
скоro AreHTcTBa (ЕМА) и будет учитываться при
проведении сертификации веществ Европей
ским Директоратом по качеству лекарственных
средств и здравоохранению (ЕООМ).
103 За. 1712
2. ОБЛАСТЬ ПРИМЕНЕНИЯ ОБЩЕЙ
СТАТЬИ
ВИДbl ЖИВОТНblХ, ПОДВЕРЖЕННblХ тrэ
Крупный роrатый скот, овцы, козы и живот
ные, которые в природе восприимчивы к возбуди
телям трансмиссивной ryбчатой энцефалопатии
или восприимчивы к инфекции, передающей
ся оральным путем, за исключением человека(7)
и приматов, определены как «Виды животных,
подверженных тrэ(в)>>.
МАТЕРИАЛbl
Эта стаья распространяется на материалы,
полученные от «Видов животных, подверженных
тrэ», которые используются для получения:
действующих веществ,
вспомоrательных вещества и адъювантов,
сырья, исходных материалов и реакти
вов, используемых в производстве (например,
бычий сывороточный альбумин; ферменты; пи
тательные среды, включая среды для созда
ния рабочеro банка клеток или новых rлавных
банков клеток для впервые реrистрируемых ле
карственных средств).
Требования этой статьи также распростра
няются на материалы, непосредственно KOH
тактирующие с лекарственным средством или
оборудованием, используемым для производ
ства лекарственноro средства, вследствие
чеrо имеется возможность контаминации про
дукта.
Материалы, используемые при квалифика
ции производственных помещений и оборудо
вания, например, питательная среда, исполь
зуемая при валидации асептических процессов
для имитации процесса розлива, будут COOTBeT
ствовать требованиям данноro раздела, если
компонент или компоненты питательной среды
получены из тканей, инфекционность в которых
не была обнаружена (ткани катеroрии 'С), риск
перекрестной контаминации с потенциаль
но инфекционными тканями (см. раздел З З)
был учтен и материалы были получены из
страны с малым риском возникновения rэкРс
или контролируемым уровнем rэкрс (Катепr
рия А и В соответственно, см. параrраф 3--2).
Такая информация должна быть представле
на в реrистрационном досье и проверена при
стандартной инспекции на соответствие требо
ваний Надлежащей производственной практи
ки (GMP).
Друrие материалы. такие как чистящие
peareHTbI, умяrчители и смазывающие веще
ства, с которыми контактирует лекарственное
средство при стандартнnм технолоrическом про
цессе или на последней стадии, или в первич
ной упаковке, рассматриваются в соответствии с
требованиями данной статьи, если они являют
ся жировыми производными, полученными с ис
пользованием точных физико химических про
цессов, как указано в разделе 6.
810
rосударственная фармакопея Республики Беларусь
ПОСЕВНblЕ МАТЕРИАЛbI, БАНКИ КЛЕТОК
И СТАНДАРТНАЯ ФЕРМЕНТАЦИЯ/
ПРОИЗВОДСТВО(9)
В соответствии с установленными требо
ваниями, rлавные посевные материалы и rлав
ные банки клеток, указанные в реrистрационных
досье, поданных на рассмотрение после 1 июля
2000 r:, должны соответствовать данным PeKO
мендациям. rлавные посевные материалы и
rлавные банки клеток для вакцинных антиrенов,
для биотехнолоrических лекарственных средств
(определенных соrласно Приложению к Поста
новлению Совета (ЕС) NQ 726/2004 Европейскоro
парламента и Совета(10)), а также для друrих ле
карственных средств, в процессе производства
которых используются системы посевных MaTe
риалов и банков клеток, использование которых
для получения компонента зареrистрированноrо
лекарственноro средства уже было разрешено,
будут считаться соответствующими PeKOMeHдa
циям, даже если они включены в реrистрацион
ные досье, поданные к рассмотрению после
1 июля 2000 r.
Для rлавных банков клеток и rлавных посев
ных материалов, созданных до 1 июля 2000 r:,
но еще не разрешенных в качестве компонентов
зареrистрированноrо лекарственноrо средства,
должны быть представлены данные, подтверж
дающие соответствие требованиям PeKOMeH
даций. Если на сырье, исходный материал или
реактив, использованные при получении этих
банков клеток или посевных материалов, нет
полноro документальноro досье, заявитель
должен представить результаты оценки риска
соrласно разделу 4 Рекомендаций.
Созданные рабочие посевные материалы
или банки клеток, используемые в производстве
лекарственных средств, зареrистрированных до
1 июля 2000 r., оценка риска которых была про
ведена компетентным уполномоченным opra
ном rосударства члена Европейскоro Союза или
ЕМА и была сочтена приемлемой, будут также
соответствовать установленным требованиям.
Однако, если в процессах ферментации/произ
водства или при получении рабочих посевных
материалов и рабочих банков клеток использу
ются материалы, полученные из «видов живот
ных, подверженных тrэ», то заявитель должен
представить доказательства их соответствия
требованиям Рекомендаций.
3. ОБЩИЕ ПОЛОЖЕНИЯ
3 1. НАУЧНblЕ ПРИНЦИПbl СНИЖЕНИЯ
ВЕРоятноrо РИСКА
Если у ПРОИЗВО.Qителя есть выбор. то пред
почтительно использование материалов, по
лученных не из «видов животных, подвержен
ных тrэ», или синтетическоrо происхождения.
Должны быть представлены обоснования ис
пользования материалов, полученных из «видов
животных, подверженных тrэ» вместо матери
алов из «видов животных, не подверженных
тrэ» или вместо материалов синтетическоro
происхождения. Если необходимо использова
ние материалов из «видов животных, подвер
женных тrэ», должны быть приняты все необхо
димые меры по снижению риска передачи тrэ.
rOToBbIe диаrностические наборы дЛЯ BЫ
явления инфекционных areHToB тrэ iп vivo пока
еще не доступны. Диаrноз тrэ основывается на
посмертном подтверждении характерных моз
roвых повреждений с помощью rистопатоло
rических данных и/или обнаружением PrPTSE с
помощью Вестерн блоттинrа или иммунолоrи
ческих испытаний. Также для подтверждения
наличия инфекционноrо areHTa ТrЭ использу
ется введение предполаrаемой инфицирован
ной ткани чувствительным к заболеванию лабо
раторным животным. Однако, из за длительных
инкубационных периодов всех areHToB тrэ pe
зультаты испытаний iп vivo получают только по
истечении месяцев или лет.
Для определения PrPTSE в образцах после
забоя было разработано несколько иммунохи
мических испытаний, и некоторые из них сейчас
считаются высокочувствительными. Однако их
способность определять инфицированных жи
вотных зависит от времени отбора образцов
по отношению ко времени экспозиции, типа co
бранных тканей и полученной инфекционной
дозы, наряду со временем начала клиническоrо
заболевания. Сейчас существует недостаточно
информации о влиянии видовой изменчивости
на данное определение.
Несмотря на то, что скрининr животных ис
точников материалов с помощью испытаний in
vit,o может предотвратить использование живот
ных на поздних стадиях инкубационноro пери
ода заболевания и предоставить информацию
об эпидемиолоrическом статусе данной страны
или реrиона, ни один из тестов не является при
roдным для однозначноro подтверждения отри
цательноro статуса животноro.
Снижение риска передачи тrэ основано на
трех взаимодополняющих параметрах:
животные источники материала и их reo
rрафическое происхождение;
при рода материала животноro происхож
дения, используемоro в производстве, и BЫ
полнение процедур по предупреждению пере
крестной контаминации с материалами с более
высоким уровнем риска;
процесс (процессы) производства, вклю
чая наличие системы обеспечения качества,
чтобы rарантировать однородность и прослежи
ваемость продукта.
3 2. ЖИВОТНblЕ ИСТОЧНИКИ
МАТЕРИАЛА
Исходные материалы, используемые для
получения материалов, используемых в произ...
водстве лекарственных средств, должны быть
получены от животных, roдных для использо
5.2.8. Снижение риска передачи возбудителей zубчатой энцефалопатии
811
вания в пищу человеком и выдержавших до
и после забоя проверку в соответствии с Tpe
бованиями Европейскоro Союза или CXOДHЫ
ми (третья страна) условиями, за исключением
материалов, полученных из живых животных, KO
торые должны быть признаны здоровыми по pe
зультатам клиническоro обследования.
З 2 1. rеоrрафическое происхождение
3 2 1 1. Материал от КРУПН020 Р02ат020
скота
Всемирная орrанизация по охране здоро
вья животных (0/Е)(11) устанавливает критерии
по оценке статуса стран в rлаве Международ
ноro Кодекса Здоровья Животных по ryбчатой
энцефалопатии крупноro poraтoro скота. Страны
или реrионы классифицируются следующим об
разом:
А) страны или реrионы снезначительным
риском возникновения rэкРс;
В) страны или реrионы с контролируемым
риском возникновения rэкРс;
С) страны или реrионы снеопределенным
риском возникновения rэкРс.
Как оroворено в Постановлении (ЕС)
NQ 999/2001 Европейскоro Парламента и
Совета(12), классификация стран и реrионов по
риску возникновения rэкРс, основанная на
установленных Всемирной орrанизацией по ox
ране здоровья животных (О/Е) правилах, офици
ально вступила в силу в Европейском Союзе 1
июля 2007r. В Решении комиссии 2007/45З/ЕС(1З)
представлена классификация стран и реrионов
в зависимости от риска возникновения rэкРс.
Ранее Исполнительный Комитет по науке
Европейской Комиссии (Scieпtific Stее,iпg
Committee, SSC)(14) установил временную си
стему классификации стран соrласно их reo
rрафическому риску появления rэкРс (BSE)
(Geog,aphica/ BSE Risk, GBR)<15).
В целях настоящеro раздела должна ис
пользоваться классификация rэкРс, OCHOBaH
ная на правилах Всемирной орrанизации по ox
ране здоровья животных (О/Е). Если страна,
ранее классифицируемая в соответствии с кри
териями SSC GBR, еще не была проклассифи
цирована по правилам О/Е, GBR классификация
может использоваться до классифицирования
Всемирной орrанизацией по охране здоровья
животных при условии отсутствия подтвержде
ния значительных изменений в риске возникно
вения rЭКРС(16).
По возможности животные должны быть из
стран с минимально возможным риском воз
никновения rэкРс (страны снезначительным
риском возникновения rэкрс (Катеroрия А), за
исключением, коrда обосновано использование
материала из стран с высоким риском возникно
вения rэкРс. Некоторые материалы, указанные
в разделе «6. Положения по отдельным матери
алам», MOryT быть из стран с контролируемым
риском возникновения rэкРс (катеroрия В) и в
некоторых случаях из стран снеопределенным
риском возникновения rэкРс (катеroрия С) при
условии, что при меняются указанные в COOT
ветствующих разделах контроль и требования.
Кроме этих исключений животные не должны
быть из стран с неопределенным риском воз
никновения rэкРс (катеroрия С), кроме этоro
должны быть предоставлены обоснования ис
пользования животных из стран с неопределен
ным риском возникновения rэкрс (катеroрия С).
3 2 1 2. Овцы и козы (мелкие жвачные жи
вотные)
О клинических случаях заболевания живот
ных скрейпи в природе сообщалось во мноrих
странах мира. Поскольку rэкРс (BSE) у овец и
коз может быть ошибочно диаrностирована как
скрейпи, в качестве меры предосторожности при
оценке материалов, полученных от мелких жвач
ных животных, учитывается заболеваемость жи
вотных в стране как rэкРс (BSE), так и скрейпи,
а также тип тканей, из которых получены MaTe
риалы.
Для разработки системы по определению
статуса стада мелких жвачных животных в OTHO
шении тrэ MOryт быть использованы принципы,
установленные в разделе «3 2 2. Стада круп
ноro poraToro скота с незначительным риском
rэкРс (BSE)>>. ДЛЯ присвоения стаду овец CTa
туса свободноro от тrэ стада, из за возможно
сти появления rэкРс (BSE) у овец, может быть
использован определенный rенотип(ы), обу
славливающий резистентность к возбудителям
rЭКРС(ВSЕ)/скрейпи(17). Однако, должна быть
принята во внимание возможность, что rеноти
пы, резистентные к скрейпи, MOryт быть воспри
имчивы К rэкРс (экспериментальное воздей
ствие через ротовую полость) или атипичной
скрейпи (случаи в природе). В отношении коз в
настоящее время нет достаточных данных о Ha
личии rенотипной определенной чувствительно
сти к возбудителям rэкРс (BSE).
Материал от мелких жвачных животных, как
правило, должен быть получен из стран с мноro
летним статусом отсутствия скрейпи. При проис
хождении материала из друrих источников необ
ходимо соответствующее обоснование.
З 2 2. Стада KpYnHoro poraToro скота
(изолированные) с незначительным риском
rэкРс (В5Е)
Самый безопасный источник материала
страны или реrионы снезначительной уrрозой
возникновения rэкРс (страны катеroрии А). В
друrих странах MOryT быть выявлены или уже
выявлены случаи заболевания rэкРс (BSE)
в какой то период времени, в связи с чем SSC
была разработана практическая концепция
«Стада крупноro poraToro скота (изолированно
ro) с незначительным риском», одобренная
СНМР и CVMP. Критерии по созданию и под
держанию «стада крупноro poraтoro скота (изо
812
rосударственная фармакопея Республики Беларусь
лированноro) с незначительным риском rэкРс
(BSE)>> изложены в заключении SSC от 22 23
июля 1999 roда(16).
В настоящее время невозможно изме
рить степень снижения rеоrрафическоro риска
rэкРс (BSE) для животных из стад крупною po
raToro скота (изолированных) снезначительным
риском rэкРс (BSE). Однако, предполаrается
что это снижение достаточно существенное. Сле
довательно, получение исходных материалов
от таких изолированных стад крупноro poraToro
скота при оценке риска будет рассматриваться с
учетом катеrории страны по классификации О/Е.
3 3. ЧАСТИ ТЕЛА ЖИВОТНЫХ,
БиолоrИЧЕСКИЕ СРЕДЫ и ВЫДЕЛЕНИЯ
КАК ИСХОДНЫЕ МАТЕРИАЛЫ
Различные opraHbI и биолоrические среды
животноro, страдающеrо rэкРс (BSE), имеют
различную степень инфекционной активности.
Если используются материалы от видов живот
ных, для которых характерно тrэ, приоритет
должен быть отдан материалам, относящим
ся к катеroрии с наименьшим риском. Табли
цы, представленные в Приложении к данному
разделу(19), резюмируют современные данные
о распределении инфекционности и PrPTSE у
крупною роrатою скота с rэкРс и у овец и коз
со скрейпи t2О ).
Информация, приведенная в таблице, по
лучена исключительно при изучении случаев
заболевания в природе или эксперименталь
ных моделях при заражении оральным путем
первичным возбудителем (у крупноro poraToro
скота) и не содержит данные от моделей забо
левания, в которых используются штаммы тrэ,
адаптированные к лабораторным животным,
так как фенотипы штаммов, подверrнутых пе
ресевам, MOryT значительно отличаться от име
ющихся в природе. Поскольку было доказано,
Ткани с высокой инфекционной
активностью
ткани центральной нервной си
Катеrория стемы (ЦНС), в которых обнару
IA живаются высокие инфекционные
титры на более поздних стадиях
тrэ, и некоторые ткани, которые
анатомически связаны с ЦНС
Ткани с более низкой инфекцион
ной активностью
Катеrория периферические ткани, результаты
IB исследования которых подтверди
ли наличие инфекционной актив
ности и/или PrPTSE по крайней мере
одной из форм тrэ
Ткани, в которых не обнаружена
инфекционная активность
Катеrория ткани, результаты исследования
IC которых на наличие инфекционной
активности и/или PrPTSE были отри
цательными
что иммуноrистохимическое исследование и/
или Вестерн блопинr неправильно упакован
ноro белка хозяина (PrPTSE) является условным
маркером инфекционной активности, резуль
таты исследования PrPTSE были представлены
вместе с данными биолоrических исследований.
Ткани сrруппированы в три основные катеroрии
в отношении инфекционной активности незави
симо от стадии болезни:
Ткани катеroрии 'А и материалы, получен
ные из них, не должны использоваться в произ
водстве лекарственных средств, за исключени
ем случаев, Korдa возможность их использования
обоснована (см. Раздел 5).
Несмотря на то, что в rруппу тканей с более
низким уровнем риска (ткани катеroрии 18) почти
Bcerдa относятся некоторые ткани с более низ
ким уровнем риска (например, кровь), чем
друrие (например, лимфоретикулярные ткани),
данные об уровнях инфекционной активности в
этих тканях слишком оrраничены, чтобы Bыдe
лить в этой катеroрии подкатеroрии с различным
уровнем риска. Также очевидно, что отнесение
определенной ткани к той или иной катеrории
может зависеть от заболевания или вида живот
ных И подлежит пересмотру по мере появления
новых данных.
При оценке риска (см. раздел 4) произво
дители и/или заявители/держатели реrистраци
онных удостоверений должны учитывать клас
сификацию тканей, приведенную в таблицах
Приложения к этому общему разделу(12 J .
Катеrории в таблице являются приблизи
тельными, поэтому важно учитывать следующие
аспекты.
в определенных ситуациях может прои
зойти перекрестная контаминация тканей с раз
личной катеroрией инфекционной активности.
На вероятность риска будут влиять условия, при
которых ткани были получены, в частности Ha
личие контакта тканей с более низкой инфек
ционной активностью или тканей, в которых не
было обнаружено инфекционной активности
(ткани катеroрии IB и IC) с тканями с высокой ин
фекционной активностью (ткани катеroрии IA).
Степень пере крестной контаминации HeKOTO
рых тканей может быть увеличена, если в про
цессе убоя зараженных животных используется
оrлушение мозrа (проникающее или непроника
ющее) или перерезание спинноro мозrа и/или
roловноro мозrа. Риск пере крестной контами
нации будет меньше при сборе биолоrических
жидкостей с минимальным повреждением ткани
и удалении клеточных компонентов, при сборе
эмбрионалы-юй крови без контаминации друrи
ми материнскими или эмбриональными тканя
ми, включая плаценту, амниотическую и аллан
тоисную жидкости. Перекрестную контаминацию
определенных тканей с тканью катеroрии IA
очень трудно или невозможно предотвратить
(например, череп). Это должно быть учтено при
оценке риска.
5.2.8. Снижение риска передачи возбудителей аубчатой энцефалопатuu
813
Для определенных классов веществ ис
пользуемая техника оrлушения/убоя может
быть критична для определения потенциально
ro риска(21) из за вероятности попадания частиц
мозrа в периферические opraHbI, в частно
сти, в леrкие. Техника оrлушения/убоя должна
быть описана, так же как и процедуры удале
ния тканей с высокой инфекционной активно
стью. Должны быть подробно описаны проце
дуры сбора тканей/орrанов животных, которые
будут использоваться, и при ни маемые меры по
предупреждению перекрестной контаминации с
материалом более высокоro риска.
На вероятность контаминации тканей и op
raHoB возбудителями rэкРс (BSE), потенциаль
но присутствующими в материале из ЦНС, при
использовании техники оrлушения, использу
емоro для убоя крупноro poraToro скота, влияют
следующие факторы:
степень инфекционной активности ТЭКРС
(BSE) в мозrе умерщвленноro животноrо,
степень повреждения roловноro мозrа,
распространение мозroвых частиц по телу
животноro.
Эти факторы следует оценивать вместе с
катеroрией места происхождения животных по
классификации O/E/GBR, возрастом животных в
случае использования крупноro poraToro скота и
посмертными исследованиями крупноro poraTo
ro скота валидированными методиками. OCHOB
ные принципы, описанные выше, также приме
ни мы к овцам и козам.
Риск возникновения перекрестной KOHTa
минации будет зависеть от некоторых дополни
тельных факторов, включая:
меры, предпринятые для предупрежде
ния контаминации во время сбора тканей (см.
выше ),
уровень контаминации (количество KOHTa
минированных тканей),
количество и тип одновременно получа
емых материалов.
Производители и заявители/держатели
реrистрационноro удостоверения должны oцe
нивать риск с учетом вероятности пере крестной
контаминации.
3 4. ВОЗРАСТ ЖИВОТНblХ
Поскольку инфекционная активность при
тrэ у крупноro poraтoro скота нарастает в ин
кубационный период, длящийся нескольких лет,
рекомендуется использовать в качестве источ
ника материалов молодых животных.
Подтверждено присутствие инфекционноro
материала больше всеro в центральной HepB
ной системе и прилеrающих тканях, а также в
лимфоретикулярной системе, в зависимости от
тrэ areHTa (rэкРс у крупноro poraтoro скота или
скрейпи у овец и коз). Точный период действия
инфекционности, дата начала инфекции в COOT
ветствующих частях тела и тканях не известны в
обоих случаях и по сути сложно четко дать опре
104. За.. 1712
деление возрасту, выше котороro различные
ткани MOryT быть инфицированы и не должны
быть собраны. До сих пор действует rлавная pe
комендация по сбору тканей в самом молодом
возрасте. Также очевидно, что возрастной кри
терий зависит также от rеоrрафическоro проис
хождения. Возраст является более важным кри
терием для материалов из стран, rдe риск выше
(страны Катеrории В и С), чем для стран с незна
чительным риском rэкРс (страны Катеroрии А).
3 5. ПРОИЗВОДСТВЕННblЙ ПРОЦЕСС
Оценка общеro снижения потенциальноro
риска лекарственных средствах в отношении тrэ
должна проводится С учетом мер по контролю:
источника сырья/исходных материалов,
производственноro процесса.
Контролируемые источники материалов яв
ляются очень важным критерием по обеспе
чению приемлемой безопасности продукта
вследствие имеющихся данных о резистентно
сти возбудителей тrэ к большинству процедур
инактивации.
Контроль производственноro процесса и еro
серийности (то есть формирование серии, разде
ление партий, процедуры очистки между произ
водством разных серий) должен осуществляться
в рамках соответствующей системы обеспече
ния качества, например, /SO 9000, НАССр22) или
GMP. Должны выполняться процедуры, обеспе
чивающие прослеживаемость материалов, про
ведение внутренних аудитов и аудитов постав
щиков сырья/исходных материалов.
Определенные технолоrические процеду
ры MorYT значительно снизить риск контами
нации тrэ, например, процедуры, использу
емые при производстве производных жиров
(см. раздел 6). Так как такие жесткие про
цессы химической обработки не MorYT быть
применены ко мноrим продуктам, более при
емлемыми для них MorYT быть процессы фи
зическоro удаления материала, боrатоrо при
онами, такие как осаждение и фильтрация.
Должно быть представлено подробное опи
сание производственноrо процесса, включая
внутрипроизводственный контроль, а также
обсуждены технолоrические стадии. которые
MorYT снизить или исключить риск контаминации
тrэ. в случае участия в производственном про
цессе нескольких производственных площадок.
должны быть четко указаны стадии, выполня
емые на каждой площадке. Должны быть опи
саны все принимаемые меры по обеспечению
прослеживаемости каждой производственной
серии до исходноrо материала.
Процесс очистки. Процедуру очистки
технолоrическоro оборудования может быть
трудно валидировать на способность удаления
возбудителей тrэ. Имеются сообщения о том, что
после работы с препаратами с высоким титром
814
rосударственная фармакопея Республики Беларусь
возбудителей тrэ поддающееся обнаружению
количество инфекционных areHToB может быть
адсорбировано на поверхности нержавеющей
стали. Удаление всех адсорбирующих белков
при помощи 1 М раствора натрия rидрокси
да или хлорсодержащими дезинфицирующими
средствами (например, 20 000 ррт хлора в Te
чение 1 ч) считается приемлемым методом в
случаях, Korдa оборудование (которое не может
быть заменено) подверrалось воздействию по
тенциально контаминированноrо материала.
Более мяrкая обработка с оrраниченными KOH
центрациями щелочи или стабилизированной
хлорной извести при соответствующем смеши
вании с очищающими средствами и при исполь
зовании указанной температуры была проведе
на для демонстрации подобной эффективности
для удаления прионов, как и при использовании
классическоro NaOH или хлорирования. Систе
ма, основанная на превращенном в пар перокси
де водорода, также является эффективной для
инактивации возбудителей тrэ. Эти новые спо
собы обработки более совместимы с чувстви
тельными материалами и MorYT быть приюдны
для практическоro применения(2З).
При использовании в производстве про
дукта материалов с риском тrэ, для снижения
риска перекрестной контаминации между про
изводственными сериями должны выполняться
процедуры очистки, включая внутрипроизвод
ственный контроль. Это особенно важно, если
обработка материалов различных катеroрий
риска проводится на одной производственной
площадке с использованием одноro и тоro же
оборудования. Если при производстве продукта
используются материалы катеroрии 'А, то произ
водство должно осуществляться на оборудова
нии специальноro назначения, если иное не под
тверждено.
Дальнейшие исследования необходимы для
развития и валидации новых методов обеззара
живания для уменьшения риска перекрестной
контаминации для материалов и устройств, KO
торые не совместимы с рекомендованными ВОЗ
методами.
Валидация процедур удаления/инак
тивации. Валидационное изучение процедур
удаления/инактивации тrэ достаточно трудно
интерпретированть. Необходимо учитывать при
роду добавляемоro к образцам материала и еro
применимость к обычной ситуации, дизайн ис
следования (включая уменьшение масштаба
технолоrических процессов) и методики обна
ружения возбудителя (количественное опреде
лени in vitro или in vivo). Для выработки peKO
мендаций по наиболее приемлемому для работ
по валидации способу подroтовки образцов Meтo
дом добавок необходимы дальнейшие исследо
вания. Поэтому в настоящее время проведение
работ по валидации не требуется. Однако, если
в реrистрационном досье имеются утверждения
о безопасности продукта в отношении тrэ, oc
нованные на способности производственных
процессов удалять или инактивировать возбу
дителей тrэ, они должны быть подтверждены
соответствующими испытаниями(24).
В дополнение к контролю за происхождени
ем материалов производителям рекомендуется
продолжать изучение методов удаления и инак
тивации инфекционных areHToB, используемых
для идентификации операций/процессов, KOTO
рые моrли бы более эффективно обеспечить
удаление или инактивацию возбудителей тrэ. В
любом случае производственный процесс про
изводства всеrда, Korдa это возможно, должен
быть разработан с учетом имеющейся инфор
мации о методах, считающихся приroдными для
инактивации или удаления возбудителей тrэ.
Для определенных типов продуктов (см.
раздел «6 3. Бычья кровь и материалы из бычьей
крови»), rдe валидированные методы удаления/
инактивации трудно применимы, может быть He
обходима оценка процесса. Она должна быть
основана на исходном материале и любых опу
бликованных данных о риске тrэ.
4. ОЦЕНКА СТЕПЕНИ РИСКА
МАТЕРИАЛОВ ИЛИ ВЕЩЕСТВ,
ИСПОЛЬЗУЕМЫХ В ПРОЦЕССЕ
ПРОИЗВОДСТВАЛЕКАРСТВЕнноrо
СРЕДСТВА,ПРОВОДИМАЯ
В СООТВЕТСТВИИ С УСТАНОВЛЕННЫМИ
ТРЕБОВАНИЯМИ
При оценке риска, связанноro с тrэ, необ
ходимо тщательное рассмотрение всех параме
тров, описанных в общих чертах в разделе «3 1.
Научные принципы снижения вероятноro риска».
Как было указано во введении данной
общей статьи, соответствие установленным
требованиям основывается на получении бла
roприятноro результата при оценке риска. Про
водимые производителями и/или заявителями
или держателями реrистрационных YДOCTOBe
рений оценки риска различных материалов или
веществ, полученных от «видов животных, под
верженных тrэ» и используемых при произ
водстве лекарственных средств, должны пока
зать, что все факторы риска тrэ были учтены и,
при возможности, что риск был минимизирован
путем выполнения принципов, описанных в этой
общей фармакопейной статье. Сертификаты
приroдности по тrэ, выданные ЕDОМ, MOryт
использоваться заявителями или держателя
ми реrистрационных удостоверений в качестве
основ оценки риска.
Всесторонняя оценка риска для лекарствен
ною срР.дства, проведенная заявителем или
держателем реrистрационноro удостоверения,
должна содержать оценку риска для всех раз
личных материалов, полученных от «видов жи
вотных, подверженных тrэ», и, rдe возможно,
оценку снижения содержания areHToB тrэ или
их инактивации на технолоrических стадиях про
5.2.8. Снижение риска передачи возбудителей zубчатой энцефалопатии
815
изводства действующеro вещества и/или лекар
ственноro средства.
Окончательное решение о соответствии
установленным требованиям принимает компе
тентный уполномоченный opraH.
Выбор и обоснование порядка KOHTpO
ля определенною материала, полученною из
«видов животных, подверженных тrэ», ocy
ществляется производителями и/или заявителя
ми или держателями реrистрационных YДOCTOBe
рений на медицинские лекарственные средства
с учетом последних данных о научном и TeXHO
лоrическом проrрессах.
5. ОЦЕНКА СООТНОШЕНИЯ
ПОЛЬЗNРИСК
В дополнение к параметрам, указанным в
разделе 3 (которые MorYT быть описаны в Cep
тификате приroдности тrэ, изданном EOQM)
и 4, при оценке приемлемости определенноrо
лекарственноro средства, содержащеrо матери
алы, полученные от «видов животных, подвер
женных тrэ», или на финальных стадиях произ
водства котороro используются эти материалы,
учитывают следующие факторы:
путь введения лекарственноro средства,
количество материала животноrо проис
хождения, включенноro в состав лекарственно
ro средства,
максимальная терапевтическая доза (cy
точная доза и продолжительность лечения),
показания к применению лекарственною
средства и еro клинический эффект,
присутствие видовоro барьера.
Ткани с высокой инфекционной активно
стью (ткани катеroрии 'А) и вещества, получен
ные из них, не должны использоваться в про
изводстве лекарственных средств, исходных
материалов для них и полупродуктов (включая
действующие и вспомоrательные вещества и
реактивы), если иное не разрешено. Должно
быть представлено обоснование необходимо
сти использования исключительно данных Ma
териалов. В этих исключительных и оправдан
ных обстоятельствах использование тканей
с высокой инфекционной активностью в про
изводстве действующих веществ может быть
разрешено при предоставлении заявителем
положительной оценки соотношения польза/
риск, основанной на результате оценки риска в
соответствии с разделом 4 и предполаrаемых
показаниях к применению. Вещества, произво
димые из материалов катеroрии 'А, если их ис
пользование обосновано, должны быть полу
чены от животных из стран снезначительным
риском rэкРс (Катеroрия А).
6. ПОЛОЖЕНИЯ ПО ОТДЕЛЬНЫМ
МАТЕРИАЛАМ
Описанные ниже материалы, полученные
от «видов животных, подверженных тrэ», счи
таются соответствующими требованиям Ha
стоящей общей статьи Фармакопеи в случае
их соответствия, по крайней мере, условиям,
определенным ниже. Соответствующая инфор
мация или сертификат приюдности, выданный
EDQM, должны быть представлены держателя
ми реrистрационных удостоверений или заяви
телями.
6 1. КОЛЛАrЕН
Коллаrен это фибриллярный белковый
компонент соединительной ткани млекопита
ющих.
С целью подтверждения соответствия Be
щества требованиям данной общей статьи дo
кументация на коллаrен должна быть представ
лена с учетом положений разделов 3 5. Кроме
тоro, должно быть рассмотрено следующее.
Для коллаrена. полученноrо из костей, при
меняют требования, установленные для желати
на (см. ниже). В процессе производства колла
reHa предполаrается более низкая способность
к инактивации, чем в процессе производства
желатина. Поэтому источник становится более
важным критерием, принимаемым во внимание.
Коллаrен, произведенный из таких тканей,
как шкура, кожа, сухожилие и спилок шкур,
обычно не обладает существенным риском в от
ношении тrэ, если возможность контаминации
потенциально зараженными материалами, Ha
пример, попадание крови и/или тканей ЦНС, при
их заroтовке исключена. Таким образом, шкуры
являются наиболее безопасным источником
материала среди производных коллаrена для
вживления человеку. Однако, перекрестная KOH
таминация с полученными во время убоя MaTe
риалами мозrа, которые MOryT быть высушены
на поверхности шкур, может быть трудно пре
одолимой. Это друroй критерий, который учиты
вается в процессе оценки безопасности источни
ка материала.
Процесс производства коллаrена может
иметь некоторые общие стадии с процессом про
изводства желатина, такие как обработка щело
чью и натрия сульфатом, кальция rидроксидом
и натрия rидроксидом или ферментами. Однако,
даже эти общие стадии MOryT различаться в дли
тельности и значениях рН, что может привести
к существенным отличиям в их способности к
инактивации. Производители должны по крайней
мере провести оценку, основанную на сходствах
стадий производства коллаrена по сравнению
с известными стадиями инактивации в процес
се производства желатина. для доказательства
безопасности продукта. Кроме производства
отличия также присутствуют при последнем ис
ПОЛЫlовании материала и, следовательно, в их
оценке степени риска, в то время как желатин
широко используется для пероральноro приема,
мноrие коллаrеновые компрессы представлены
в виде хирурrических имплантов. Данный аспект
также должен быть учтен при конечной оценке
степени риска.
816
rосударственная фармакопея Республики Беларусь
6 2. ЖЕЛАТИН
Желатин это природный растворимый
белок, способный или не способный к rелеобра
зованию, образующийся при частичном rидро
лизе коллаrена, полученноro из костей, шкуры и
кожи животных.
С целью подтверждения соответствия Be
щества требованиям данной общей статьи ДOKY
ментация на желатин должна быть представле
на с учетом положений разделов 3 5. Кроме
тоro, должно быть рассмотрено следующее(25).
Используемый исходный материал
Желатин, входящий в состав лекарственных
средств, может быть произведен из костей или
шкур.
Шкуры как исходный материал. Соrлас
но имеющимся в настоящее время данным
шкуры, используемые для производства же
латина, представляют собой более безопас
ный исходный материал, чем кости. При этом
должны быть обеспечены условия, преду
преждающие во время заrотовки перекрест
ную контаминацию потенциально зараженны
ми материалами.
Кости как исходный материал. При полу
чении желатина из костей качество исходноrо
материала должно быть проверено на допол
нительный параметр для подтверждения без
опасности конечноro продукта. Таким образом,
должны быть учтены следующие ниже критерии.
1. Череп и спинной мозr должны быть yдa
лены из собранных костей (сырой/исходный Ma
териал) в независимости от возраста или страны
происхождения крупноrо poraToro скота.
2. Позвоночник должен быть удален из
сыроrо/исходноro материала у крупноro poraTo
ro скота старше 30 месяцев из стран с KOHTpO
лируемым или неопределенным риском rэкРс
(Катеroрии В и С).
3. Желатин для парентеральноro примене
ния должен быть произведен только из костей
из стран с незначительным или контролиру
емым риском rэкРс (Катеroрия А и В COOTBeT
ственно). Желатин для пероральноro примене
ния должен быть произведен из костей из стран
с незначительным, контролируемым или He
определенным риском rэкРс (Катеrории А, В и
С соответственно).
4. Желатин должен быть произведен с ис
пользованием одноro из методов производства,
описанных ниже.
Методыпроизводства
Шкуры. При производстве желатина из шкур
не требуется особых способов и условий пере
работки, если выполняются все контрольные
операции по предупреждению перекрестной
контаминации при заroтовке шкуры и в ходе про
изводственноro процесса.
Кости. При использовании костей в каче
стве исходноro материала, процесс производ
ства будет вторым параметром для подтвержд
ния безопасности желатина.
Кости для производства желатина должны
быть получены из стран с незначительным, KOH
тролируемым или неопределенным риском
rэкРс (Катеroрии А, В и С), в соответствии с yc
ловиями, описанными в разделе 6 2 (подраздел
«Используемый исходный материал»), исполь
зуя В процессе производства кислоту, щелочь
или наrревание/давление.
Процесс производства должен учитывать
ся при проведении оценки степени риска, как
указано в разделе 4 данной статьи. Как кислот
ный, так и щелочной методы производства про
демонстрировали схожую общую инактивацию/
удаление тrэ инфекционности в желатине при
проведении валидации. Исследования пока
. зали, что дополнительная обработка щелочью
(рН 13, в течение 2 ч) костей/оссеина повыша
ет способность процесса производства к инакти
вации/удалению тrэ. Друrие стадии процесса,
такие как фильтрация, ионообменная xpoMaтorpa
фия и высокотемпературная стерилизация, также
MOryт способствовать безопасности желатина.
При стандартном щелочном способе про
изводства кости тщательно измельчают, обе
зжиривают rорячей водой и деминерализуют
кислотой хлористоводородной разведенной (не
менее 4 % и рН < 1,5) в течение по крайней мере
2 дней для получения оссеина. Потом проводит
ся еro щелочная обработка насыщенным pac
твором кальция rидроксида (рН не менее 12,5)
в течение по крайней мере 20 дней. Желатин
экстраrируют, промывают, фильтруют и KOHцeH
трируют.
Для бычьих костей может применять
ся кислотный способ. При этом обработка Ha
сыщенным раствором кальция rидроксида заме
няется на предварительную обработку кислотой,
при которой оссеин вымачивается при рН < 3,5
как минимум 1 О часов.
«Краткая» термообработка (стерилиза
ция) при 138 ос как минимум 4 с применяется
как при кислотном, так и при щелочном способе
производства.
в процессе наrревания/давления, BЫCY
шенные обезжиренные сломанные кости aBTO
клавируются с насыщенным паром при давле
нии более 3 бар и минимальной температуре
133 ос в течение не менее 20 мин с последу
ющей экстракцией протеина roрячей водой.
Конечные стадии одинаковые для щелоч
ноro, кислотноro способов и наrревания/давле
ния и включают экстракцию желатина, очистку,
фильтрацию и концентрацию.
6 3. БblЧЬЯ КРОВЬ И МАТЕРИАЛbI
ИЗ БblЧЬЕЙ КРОВИ
Эмбриональная (фетальная) бычья cы
воротка широко используется при работе
с клеточными культурами. Эмбриональная
бычья сыворотка должна быть получена из
5.2.8.
Снижение риска передачи возбудителей 2убчатой энцефалопатии
817
плодов, извлеченных на скотобойнях у здо
ровых коров, приrодных для потребления в
пищу человеком; матка должна быть полно
стью удалена; эмбриональная кровь соби
рается в закрытую систему для сбора крови
в специально предназначенном для этоrо
месте или зоне в асептических условиях
путем сердечной пункции.
Сыворотку новорожденноro теленка полу
чают у телят в возрасте менее 20 дней; CЫBO
ротку телят у животных в возрасте менее 12
месяцев. Донорскую бычью сыворотку полу
чают у животных в возрасте не более 36 Me
сяцев, при этом отрицательный статус стада
в отношении тrэ должен быть четко опреде
лен и зареrистрирован. Во всех случаях для
предупреждения перекрестной контаминации
тканями с более высоким риском сыворотка
должна быть собрана соrласно определенным
процедурам обученным этим процедурам пер
соналом .
С целью подтверждения соответствия
бычь€й крови и производных бычьей крови
требованиям данной общей статьи ДOKYMeHTa
ция на них должна быть представлена с уче
том положений разделов 3 5. Кроме тоro,
должно быть рассмотрено следующее.
Прослеживаемость
Должна быть обеспечена прослеживаемость
в каждой серии сыворотки или плазмы дО CKO
тобой ни. У скотобоен должны храниться списки
ферм. откуда поступили животные. Если CЫBO
ротка получена у живых животных, для каждой
серии сыворотки должны вестись записи, обе
спечивающие ее прослеживаемость до COOTBeT
ствующей фермы.
rеоrрафическое происхождение
Так как уровень инфекционной активности
rэкРс (BSE) тканей крупноro poraToro скота
более высокий. чем в отношении скрейпи, в Ka
честве меры предосторожности бычья кровь
должна быть получена из стран Катеroрии А.
Бычья кровь из стран Катеroрии В также при
меняется, если обеспечивает отсутствие риска
перекрестной контаминации крови материалом
мозrа. полученным при убое животных в возрас
те старше 21 месяца(2б).
Методы оrлушения
Если образцы получают от убитых живот
ных, метод оrлушения иrрает важную роль в
обеспечении безопасности материала. Было
установлено, что оrлушение пистолетом
Принцип принятия бычьей крови/сыворотки и их производных
Таблица 5.2.8. 1
Феталь Бычья Бычья Матери- Матери- Матери
Донор- Сыво- алы из алы из алы из
ная сыворот- CЫBOpOT бычьей бычьей бычьей
Продукт бычья ская сы- ротка ка/плазма
ка взрос-
CЫBOpOT воротка лоrо телен- взросло- сыворот- сыворот- сыворот-
теленка ка ки взрос- ки взрос- ки взрос
ка донора ro
лоrо лоrо лоrо
reorpa
фическое
происхож Кат А и В Кат. А и В Кат. А и В1 Кат. А Кат. А Кат. В Кат. А Кат. В
дение круп иВ
ноro pora
Toro скота
Возраст новорож < 1 юда < 36 меся < 1 roдa Нет преде < 21 Нет пре < 30 Me
денный цев ла месяца 2 дела сяцев I
Убой/ !
перекрест
ная KOHTa Нет риска
минация Риск перекрестной контаминации
крови Ma перекрестной контаминации
териалом
ЦНС
ДeMOHCTpa I
ция YMeHb
шения KO I
личества Нет Нет Да З
прионов во
время про
изводства
1. Если крупный роrатый скот источник из стран Катеrории В. то он дол;-кен быть вз>,т из опредепенноrо. подтвержденноro ДOKY
ментально. стада.
2. Если перекрестная контаминация крови материалами ЦНС может быть rолностью исключена (например. специальный убой).
то возраст может быть увеличен.
З. Доказательства уменьшения количества пр.юнов может быть необs;зате"ьным. если перекрестная контаминация крови MaTe
риалами ЦНС может быть полностью исключена (наприr:.ер. спеllиаЛсl--ЫЙ убой).
rосударственная фармакопея Республики Беларусь
6 4. МАТЕРИАЛЫ ИЗ ТВЕРДО/О ЖИРА
Твердый жир получают из тканей подкож
ных, брюшных и межмышечных областей и
костей. Твердый жир, используемый в качестве
исходноro материала для изroтовления произ
водных жира, должен быть материалом катеro
рии 3 или аналоrичным материалом соrласно
Постановлению (ЕС) NQ 1774/2002 Европейско
ro парламента и Совета от 3 октября 2002 r.,
устанавливающему правила, касающиеся суб
продуктов животноro происхождения, не пред
назначенных для потребления в пищу.
Считается, что материалы из твердоro жира,
такие как rлицерин и жирные кислоты, произво
димые с использованием жестких процессов,
не MOryT стать источником инфекции; данная
тема была предметом специальноro анализа
со стороны СНМР и CVMP. Поэтому такие Ma
териалы, произведенные в условиях по крайней
мере столь же жестких, как описано ниже, будут
соответствовать требованиям данной общей
статьи, независимо от rеоrрафическоro проис
хождения и природы тканей, из которых они по
лучены. При меры жестких технолоrических про
цессов:
переэтерификация или rидролиз при TeM
пературе не ниже 200 ос в течение не менее 20
минут под давлением (производство rлицерина,
жирных кислот и сложных эфиров жирных кислот);
омыление с помощью 12 М раствора NaOH
(производство rлицерина и мыла):
серийный процесс: при температуре не
ниже 95 ос в течение не менее 3 ч;
непрерывный процесс: при температу
ре не ниже 140 ОС, под давлением в течение не
менее 8 мин или эквивалентная обработка.
переroнка при температуре 200 ОС.
Материалы из твердоro жира, произведен
ные в соответствии с этими условиями, не будут
представлять риск в отношении передачи тrэ
и, следовательно, будут соответствовать Tpe
бованиям данной общей статьи. Соответствие
материалов из твердоro жира, произведенных
в друrих условиях, требованиям данной статьи
должно быть доказано.
818
с выдвиrающимся ударным стержнем с или
без прокалывания спинноro мозrа, так же как
пневматическим ударным аппаратом, особен
но с введением воздуха, может разрушить мозr
и привести к распространению тканей мозrа
через кровоток. Непроникающее оrлушение
более не рассматривается как альтернатива
проникающему оrлушению. так как происходит
контаминация крови материалами мозrа(27).
Незначительный риск может ожидаться при
электронаркозе(2S), однако даже это не обеспе
чивает полную безопасность, потому что при
неудаче животных необходимо оrлушить дo
полнительно. Методы оrлушения должны быть
обязательно описаны для процесса сбора
бычьей крови.
Там, rдe нельзя избежать риска перекрест
ной контаминации крови мозroм при рутинном
убое скота в странах с контролируемым риском
rэкРс (Катеroрия В). должны быть приняты
меры безопасности, такие как оrраничение воз
раста крупноro poraToro скота и/или сокращение
возбудителей инфекции во время производства.
Возраст
Для стран с контролируемым риском
rэкРс (Катеrория В) для бычьей крови или
материалов из бычьей крови должен быть
установлен предупредительный возрастной
предел равный 21 месяцу, если при производ
стве существенно не уменьшается количество
возбудителей тrэ. Возрастной предел в 30 Me
сяцев рассматривается как обязательный для
материалов из крови при значительном YMeHb
шении количества возбудителей тrэ, как опи
сано выше.
Уменьшение количества возбудителей
тrэ при производстве
Для производных крови способность про
цесса производства уменьшать/удалять воз
будителей тrэ должна быть оценена посред
ством специальных исследований. Оценка
может быть основана на опубликованных
или внутрипроизводственных (собственных)
данных в том случае, если такие данные
важны для специфичноro процесса производ
ства. Если не может быть сделано заключе
ние, что способность уменьшения соизмери
ма, производителям рекомендуется провести
продукт специфичные специальные исследо
вания. Исследований с использованием био
химическоro анализа может быть достаточно,
если существует научное подтверждение KOp
реляции данноro анализа с данными по инфек
ционности. Существует общее руководство по
специальным исследованиям по уменьшению
количества возбудителей тrЭ( 29 i. Препарат,
обоrащенный компонентами мозrа, является
подходящим для специальных исследований
риска, обусловленноro кровью, контаминиро
ванной мозroм.
6 5. ЖИВОТНЫЙ У/ОЛЬ
Животный уroль получают путем обуrлива
ния тканей животных, таких как кости, при BЫ
сокой температуре (> 800 ОС). Если нет друrих
указаний, исходный материал для производства
животноro уrля должен быть катеroрии 3 или эк
вивалентным, соrласно Постановлению (ЕС)
NQ 1774/2002 Европейскоro парламента и COBe
та от 3 октября 2002 r., устанавливающему пра
вила, касающиеся субпродуктов животноro
происхождения, не предназначенных для по
требления в пищу. Независимо от rеоrрафиче
скоro происхождения и природы ткани, с целью
выполнения требований законодательства, жи
вотный уroль необходимо оценивать в соответ
ствии с данной общей статьей.
5.2.8. Снижение риска передачи возбудителей zубчатой энцефалопатии
819
Животный уroль, произведенный соrлас
но этим условиям, маловероятно представля
ет риск передачи тrэ, и, соответственно, будет
отвечать требованиям данной статьи. COOTBeT
ствие животноro уrля, произведенноro в друrих
условиях, требованиям данной статьи должно
быть подтверждено.
6 6. МОЛОКО И МАТЕРИАЛЫ ИЗ МОЛОКА
Соrласно имеющимся научным данным вне
зависимости от rеоrрафическоro происхождения
молоко крупноro poraToro скота не представляет
риск контаминации тrэ(зО).
Некоторые материалы, включая лактозу,
извлекают из молочной сыворотки или отрабо
танной жидкости после коаryляции в процессе
производства сыра. При коаryляции может ис
пользоваться ренин теленка, экстракт желудка
(сычуr) или ренин, полученный у друrих жвач
ных животных. CHMP/CVMP провели оценку
риска лактозы и друrих материалов из молоч
ной сыворотки, полученной с использованием
ренина теленка, и сделали вывод, что риск тrэ
незначителен, если ренин теленка произведен в
соответствии с процессом, описанным в отчете
оценки риска(З1). Данное заключение было под
тверждено 55032), который также провел общую
оценку риска рен ина в отношении тrэ зз ).
Материалы из молока крупноro poraToro
скота, произведенные с выполнением описан
ных ниже условий, не представляют риска пере
дачи тrэ и, следовательно, будут COOTBeTCTBO
вать требованиям данной общей статьи:
молоко, получают от здоровых животных
в тех же самых условиях, что и молоко, получа
емое для потребления в пищу человеком,
никакие материалы от друrих жвачных
животных, за исключением ренина теленка, не
используются при получении таких материа
лов (например, панкреатический rидролизат
казеина).
Соответствие производных молока, по
лученных при помощи друrих процессов, или
ренина, полученноro от друrих видов жвачных
животных, требованиям данной статьи должно
быть подтверждено.
6 7. МАТЕРИАЛЫ ИЗ ШЕРСТИ
Материалы из шерсти и волос жвачных жи
вотных, такие как ланолин и спирты шерстяноro
воска, полученные из волос, будут COOTBeTCTBO
вать требованиям данной статьи, если шерсть и
волосы получают от живых животных.
Материалы из шерсти, полученные от
убитых животных, признанных «предназначен
НЫМИ дЛЯ употребления в пищу» и С использо
ванием технолоrическоro процесса С рН, TeM
пературой и продолжительностью обработки,
соответствующими как минимум одному из YKa
занных ниже условий, не будут представлять
риск передачи тrэ и, следовательно, COOTBeT
ствуют требованиям данной статьи.
Обработка при рН > 13 (начальная; KOH
центрации NaOH соответствует по крайней мере
0,1 М концентрации NaOH) при температуре
60 ос в течение не менее 1 ч. Это стандартные
условия на стадии промывки шерсти при орrани
ческо щелочной обработке.
Молекулярная дистилляция при темпера
туре> 220 ос при пониженном давлении.
Соответствие материалов из шерсти, полу
ченных в друrих условиях, требованиям Hacтo
ящей общей статьи должно быть подтверждено.
6 8. АМИНОКИСЛОТЫ
Аминокислоты MOryT быть получены путем
rидролиза материалов из различных источников.
Если не указано иное. исходный матери
ал для получения аминокислот должен быть
катеroрии 3 или эквивалентным. соrласно По
становлениию (ЕС) NQ 1774/2002 Европейскоro
парламента и Совета от 3 октября 2002 r., YCTa
навливающему правила, касающиеся субпро
дуктов животноro происхождения. не предназна
ченных для потребления в пищу.
Аминокислоты маловероятно представляют
риск передачи тrэ и будут соответствовать Tpe
бованиям общей статьи, если полученные при
выполнении следующих условий:
аминокислоты, полученные из шкур и кожи
с помощью технолоrическоro процесса, включа
ющеro обработку исходных материалов paCTBO
ром при рН 1 2, затем при рН > 11, с последу
ющей термообработкой при температуре 140 ос
в течение 30 мин при давлении 3 бар,
полученные аминокислоты или пепти
ды должны быть отфильтрованы после обра
ботки,
анализ чистоты проводится с помощью
валидированной и чувствительной методики,
позволяющей оценить содержание всех OCTa
точных неrидролизованных макромолекул в
установленных допустимых пределах.
Соответствие аминокислот, полученных в
друrих условиях, требованиям настоящей общей
статьи должно быть подтверждено.
6 9. ПЕПТОНЫ
Пептоны это частично rидролизованные
протеины, полученные ферментативным или
кислотным расщеплением. Они используются в
микробиолоrической культуральной среде для
поддержания питательных нужд микроорrаниз
мов, которые MorYT быть использованы как ce
менной фонд, или в промышленной системе
ферментации для производства медицинских
и ветеринарных лекарственных средств, вклю
чая вакцины. Существует значительный интерес
в использовании растительноrо белка в каче
стве альтернативы животному источнику белка.
Однако:
если в качестве материала источника про
теина используется желатин, ссылка делается
на раздел «6 2. Желатин» данной статьи;
820
rосударственная фармакопея Республики Беларусь
если в качестве материала источника про
теина используется казеин, ссылка делается на
раздел «6 6. Молоко и материалы из молока»
данной статьи;
если в качестве материала источника про
теина используются ткани животных, подвер
женных тrэ, ткани должны быть получены от
животных, приroдных для употребления в пищу
(смотри раздел «3 2. Животные источники Ma
териала» данной статьи) с максимальным воз
растом ЗО месяцев для крупнороrатоro скота из
стран с контролируемым риском rэкРс (KaTe
roрия В); возраст животных не иrрает роли для
животных. из стран с незначительным риском
rэкРс (Катеrория А).
ПРИМЕЧАНИЕ
(1) В настоящее время это оценивается
EFSA и ЕСОС. Обновленные данные можно
найти по адресу httр:llrеgistеrofquеstiопs.еfsа.
eu roра. eu/roq F rontend/ q uestionsListLoader? m
andate=M 2009 0221.
(2) В январе 2005 roда после подтверж
дения rэкРс у коз во Франции были приня
ты дополнительные законодательные меры,
касающиеся мониторинrа и еще большеro
изучения мелких жвачных животных. Повы
шенное наблюдение не идентифицировало
друrие случаи rэкРс у овец и коз вЕС.
(3) OJ L 311,28.11.2001, р.67.
(4) OJ L 311,28.11.2001, р.1.
(5) OJ L 147, 31.05.2001, р.1.
(6) OJ L 273, 10.10.2002, р.1. Постановле
ние (ЕС) 1774/2002 было отменено Постанов
лением (ЕС) 1069/2009, которое вступило в
силу 4 Марта 2011 (OJL300, 14. 11.2009,р.1).
(7) Руководства и информационные
письма были выпущены Комитетом по лекар
ственным средствам для использования че
ловеком (СНМР) и ero Рабочей rруппой по
биолоrическим средствам в отношении лекар
ственных средств, получаемых с использова
нием тканей человека, в БКЯ и вариантной
БКЯ. Такое руководство может быть найдено
на http://www.emea.europa.eu.
(8) Свиньи и птицы, представляющие
собой наиболее часто используемые виды
животных при производстве лекарственных
средств, обладают естественной невосприим
чивостью к инфекциям, передающихся перо
ральным путем. Соответственно, положения
данноro раздела к ним не относится. Собаки,
кролики и рыба не являются видами живот
ных, подверженных тrэ.
(9) См. также: Информационное письмо
об оценке риска передачи возбудителей rуб
чатой энцефалопатии животных через rлав
ные посевные материалы, используемые в
производстве вакцин для ветеринарноro при
менения (EMEA/CVMPI019101 февраль
2001 r., принятое Комитетом по лекарствен
ным средствам для ветеринарноro при
менения (CVMP) в июле 2001, (OJ С 286,
12.10.2001. р. 12».
(1 О) OJ L 136, 30.4.2004, р.1.
(11) httр://оiе.iпt/епg/Stаtus/ВSЕ/еп ВSЕ
free.html.
(12) Постановление (ЕС) NQ722/2007 (OJ
L.164, 26.6.2007, р.7).
(13) OJ L 172,30.6.2007, р.84.
(14) Исполнительный Комитет по науке,
созданный Решением Комиссии 97/404/ЕС
(OJ L 169, 27.6.1997, р. 85), должен помоrать
Комиссии в получении наилучших научных
рекомендаций, касающихся здоровья потре
бителей. С мая 2003 эти функции были пере
даны Европейскому AreHTcTBY по безопасно
сти пищи (EFSA): http://www.efsa.eu.int.
(15) Классификация rеоrрафическоro
риска rэкРс (BSE) (GBR) Исполнительно
ro Комитета по науке Европейской комиссии
предназначена для оценки уровня вероятно
сти присутствия одноro или более животных
с клиническими проявлениями или латентным
течением rэкРс (BSE) в данной стране или
реrионе. Определение этих четырех катеroрий
представлено в приведенной ниже таблице.
Присутствие одноrо или более
Уровень животных с клиническими про-
GBR явлениями или латентным те-
чением rэкРс (BSE) в данной
стране или реrионе
I Очень маловероятно
11 Маловероятно, но не исключено
111 Вероятно, но не подтверждено или
подтверждено на низком уровне
Подтверждено на более высоком
IV уровне ( 1 00 случаев/1 миллион
взрослоro крупноro poraToro скота
в roд)
Отчеты об оценке стран по классификации
GBR доступны на вебсайте SSC (http://ec.europa.
eu/food/fs/sc/ssc/outcome еп.html).
(16) Эксперты рассматривают систему клас
сификации GBR как стабильную, поэтому она
может использоваться далее в течение проме
жутка времени, пока данная статья не проде
монстрирует приroдность к использованию.
(17) Взrляд на Научный Список био
лоrических уrроз о «проrрамме выращи
вания тrэ резистентных овец» (http://
WWW.efsa. е u ropa.e u/E F SA/efsa loca I e
1178620753812 1178620775678.htm).
(18) SSC научное заключение о «Бычьих
стадах с незначительным риском rэкРс»,
принятое на совещании 22 23 Июня 1999
roда (http://ec.europa.eu/food/fs/sc/ssc/out56
еп.htтl).
5.2.8. Снижение риска передачи возбудителей 2убчатой энцефалопатии
821
(19) Таблицы классификации тканей oc
нованы на самых последних указаниях ВОЗ о
распространении инфекционности в тканях
при трансмиссивной ryбчатой энцефало
патии (201 О) (httр:l/whо.iпtlЫооdрroduсts.
tаЫеstissuеiпfесtivitу. pdf).
(20) Сейчас EFSA пересматривается Ha
учное заключение о rЭКРСlТrэ инфекци
онности в тканях мелких жвачных животных
(Вопрос NQ EFSA Q 2012 052). С последними
данными можно ознакомиться на сайте http://
reg isterofq uеstiопs .efsa. europa. eu/roq F rопtеd/
quеstiопsListLоаdеr?тапdаtе=М 201 0 0041.
(21) SSC заключение о методах оrлу
шения и риске rэкРс (BSE) (риск попада
ния частиц мозrа в кровь и тушу животных
при использовании определенных методов
оrлушения), принятое на встрече от 10 11
января 2002 rода (httр://еurора.еu.iпtlсоmm/
fооd/fs/sс/ssс/оut245 еп.рdf). Отчет рабочей
rруппы EFSA о риске rэкРс от распростра
нения частей мозrа в крови и туше. Вопрос
NQ EFSA Q 2003 112, принятый 21 октября
2004 roда (http://efsa.europa.eu/EFSA/efsa
locale 1178620753812 1178620777397.htm).
(22) Hazard Aпa/ysis Апd Critica/ Сопtrо/
Points (Анализ рисков и критические KOH
трольные точки).
(23) Таблицы классификации тканей oc
нованы на самых последних указаниях ВОЗ
о распространении инфекционности в тканях
при трансмиссивной rубчатой энцефалопа
тии (2006) (http://who.int/bIoodproducts/tse/
WH O%20TS Е %20Gu id еl iпеs % F I NAL 22%20
juпеuрdаtеdNL.рdf).
(24) Указания об изучении производствен
ноro процесса для медицинских лекарствен
ных средств из материалов плазмы с учетом
риска вариантной БКЯ CPMP/BWP/5136/03.
(25) На основании взrляда на Научный
список биолоrических уrроз Европейско
ro управления по безопасности продуктов
(EFSA) «Количественная оценка риска чело
веческоrо rэкРс, полученноro от желатина,
с учетом остаточноro риска rэкрс». Журнал
EFSA, 312, (1 28) (http://efsa.europa.eu/EFSAI
efsa locale 1178620753812 11786207761 07.htт).
Требования для источника выбранноrо
материала и метода производства подходят
для пероральноrо и парентеральноrо желати
на для использования в медицинских лекар
ственных средствах.
(26) Взrляд на Научный список биоло
rических уrроз при оценке возрастноrо пре
дела у крупно poraToro скота для удале
ния определенных материалов рИСКА (ОМР,
SRM). Вопрос NQ EFSA Q 2004 146, принятый
28 апреля 2005 roда.
(27) Таблицы классификации тканей oc
нованы на самых последних указаниях ВОЗ
о распространении инфекционности в тканях
при трансмиссивной rубчатой энцефало
патии (201 О) (http://who.int/bIoodproducts.
ta Ыеstissuеiпfесtivitу. pdf).
(28) Отчет рабочей rруппы EFSA о риске
rэкРс от распространения частей мозrа в
крови и туше. Вопрос NQ EFSA Q 2003 112,
принятый 21 октября 2004 roда (http://efsa.
eu ropa. е u/e п/sсiепсеЫоhаz/Ыоhаz opi п ion/
орiпiоп а ппехеsП33. html).
(29) Указания об изучении производствен
HOro процесса для медицинских продуктов из
материалов плазмы с учетом риска вариант
ной БКЯ CPMP/BWP/5136103.
(30) Ознакомиться с мнением EFSA о
молоке и материалах из молока мелких жвач
ных животных можно В Вопросе NQ EFSA
Q 2008 310, принятом 22 октября 2008 roда
(http://efsa.europa.eu/en/scdocs/scdoc849.htm ).
(31) Комитет по лекарственным средствам
для использования человеком и ero Рабочая
rруппа по биолоrическим препаратам выпол
нили оценку риска лактозы, произведенной С
использованием ренина телят. Оценка риска
включала оценку источника животных и нали
чие надлежащих процедур обеспечения каче
ства. Качество любых заменителей молока,
используемых для кормления животных, из
которых получают сычуr, особенно важно.
Текст доступен по адресу: http://ema.europa.
eu/pdfs/h u тап/рrеss/рusI05 7102. pdf.
(32) Предварительное заключение о безо
пасности ренина, полученноro от телят, для
производства лактозы, принятое на собрании
SSC 4 5 Апреля 2002 roда (http://ec.europa.
eu/food/fs/sc/ssc/out255 еп. pdf).
(З3) SSC высказал мнение о безопасно
сти животноro рен ина в отношении риска от
животных с тrэ и rэкРс в частности, при
нятое на собрании 15 мая 2002 roда (http://
ес. eu ropa .eu/food/fs/sc/ssc/out265 еп. pdf).
Приложение: основные катеroрии
инфекционности тканей
Приведенные ниже таблицы взяты из Py
ководства ВОЗ по распределению инфек
ционности по тканям при трансмuссивных
2убчатых энцефалопатиях (201 О).
Используемые в Таблицах обозначения:
«+» наличие инфекционности или
PrpTSE;
« » отсутствие признаков инqpекцион
ности или PrpTSE;
Н П исследования не проводились;
НИ исследование не используется;
? неопределенная интерпретация;
О оrраниченные или предварительные
данные;
[] инфекционность или данные PrPTSE,
основанные исключительно на испытани
ях на транпенных (Тg) мышах, экспрессиру
ющих РrР кодирующий reH или PrPTSE методы
амплификации.
822
rосударственная фармакопея Республики Беларусь
Кате20рия IA: ткани с высокой инфекционностью
Крупный роrатый скот Овцы и козы Лоси и олени
Ткани rэкРс (BSE) скрейпи CWD
Инфекци PrPTSE Инфекци- PrPTSE Инфекци- PrPTSE
онность 1 онность' онность 1
Мозr + + + + + +
Спинной мозr + + + + НП +
Сетчатка, + НП НП + НП +
зрительный нерв
Зрительный нерв 2 + НП НП + НП +
Спинномозroвой + + + + НП +
rанrлий
rанrлий тройничноro + + НП + НП
нерва
rипофиз з НП + + НП +
Твердая мозroвая НП НП НП НП НП НП
оболочка
Кате20рия IB: ткани более низкой инфекционности
Крупный роrатый скот Овцы и козы Лоси и олени
Ткани rэкРс (BSE) скрейпи CWD
Инфекци- PrPTSE Инфекци- PrPTSE Инфекци- PrPTSE
онность 1 онность' онность 1
Периферическая нервная система
Периферийные [+] + + + НП +
нервы
Веrетативный НП + НП + НП +
rанrлий 4 Лимфоретикулярные ткани
Селезенка + + НП +
Лимфатические узлы + + НП +
Миндалевидная + + + НП +
железа
Миrательная + [+] + НП +
перепонка
Тимус НП + + НП
Пищеварительный тракт
Пищевод НП [+] + НП +
Преджелудок6
(только у жвачных НП [+] + НП +
животных
Желудокlсычуr НП [+] + НП +
Двенадцатиперстная [+] + НП +
кишка
Тощая кишка 7 + [+] + НП НП
Подвздошная кишка 7 + + + + НП +
Аппендикс НИ НИ НИ НИ НИ НИ
Толстая кишкаl + + НП +
слепая кишка 7
Прямя кишка НП НП НП + НП +
Репродуктивные ткани
Плацента В НП + + НП
Яичник З НП НП
Матка З НП НП
Друrие ткани
Молочная железа/ НП + НП НП
вымя 9
5.2.8. Снижение риска передачи возбудителей еубчатой энцефалопатuu
823
Крупный роrатый скот Овцы и козы Лоси и олени
Ткани rэкРс (BSE) скрейпи CWD
Инфекци- PrPTSE Инфекци- PrPTSE Инфекци- PrPTSE
онность 1 онность 1 онность 1
Кожа З ,10 НП + [+] [+]
Жировые ткани НП НП НП [+] НП
Сердцеlперикардий НП НП НП +
Леrкие НП НП +
Печень З НП + НП
Почки З . 11 [+] + НП +
Надпочечник [+] + + НП +
Поджелудочная НП + НП НП +
железаЗ
Костный мозr 12 [+] НП + НП НП
Скелетная мышца 1З [+] НП [+] + [+]
Язык 14 НП [+] + НП
Кровеносные сосуды НП НП + НП
Слизистая НП + + НП +
носоrлотки 15
Слюнная железа НП + НП
Роroвица 16 НП НП НП НП НП НП
Жидкости тела, секреция и экскреция
Цереброспинальная НП + НП НП
жидкость
Кровь 17 ? + ? + ?
Слюна НП НП НП + Н
Молоко lВ + [+] НП НП
Моча 19 НП [+] [+]
Фекалии 19 НП НП [+] НП
Кате20рия /с: Ткани без детектируемой инфекционности
Крупный роrатый скот Овцы и козы Лоси и олени
Ткани rэкРс (BSE) скрейпи CWD
Инфекци- PrPTSE Инфекци- PrPTSE Инфекци- PrPTSE
онность 1 онность 1 онность 1
Репродуктивные ткани
Яички НП НП
Предстательная
железа/3пидидимиd НП НП
Семенные пузырьки
Сперма НП НП НП
Плацента и
амниотическая НП НП НП НП НП
жидкость
Плод (зародыш)20 НП НП Н
3мбрион 2О НП ? НП НП НП
Скелетно-мышечные ткани -
-
Кость НП НП НП НП НП
Сухожилие НП НП НП НП НП
Друrие Ткани
Десенные ткани НП НП НП НП НП НП
Пульпа зуба НП НП НП НП НП НП
Трахея НП НП НП НП
824
rосударственная фармакопея Республики Беларусь
Крупный роrатый скот Овцы и козы Лоси и олени
Ткани rэкРс (BSE) скрейпи CWD
Инфекци PrPTSE Инфекци- PrPTSE И нфекци- PrP TSE
онность 1 онность 1 онность 1
Щитовидная железа НП НП НП НП
Биолоrические жидкости, секреты и выделения
Молозив0 21 Н (?) НП НП НП
Пуповинная кровь 21 НП НП НП НП НП
Пот НП НП НП НП НП НП
Слезы НП НП НП НП НП НП
Носовая слизь НП НП НП НП НП НП
Желчь НП НП НП НП НП НП
1. Исследования инфекционности тканей человека проводилось или на приматах, или на мышах (или на тех и друrих), исследования ин
фекционности тканей крупною роraтою скота проводились или на крупном poraТOM скоте, или на мышах (или на тех и друrих); большин
ство количественных исследований тканей овец и/или коз проводилось только на мышах. В отношении овец и коз не все результаты со-
поставимы для обоих видов, например, две козы (но не овцы) заразились rэкРс естественным путем [Еиrosuтуеiflапсе, 2005, Jeffreyet
а/., 2006]. Подобным образом большинство результатов, описанных для CWO, были получены при исследованиях на оленях, и данные
MOryт отличаться от таковых для лосей и друrих плотнороrих.
2. На экспериментальных образцах тrэ было продемонстрировано, что зрительный нерв является путем к нейровторжению и содержит
высокие титры инфекционности.
3. Публикации о каких либо экспериментальных данных о наличии инфекционности в rипофизе или твердой мозювой оболочке челове
ка со всеми формами человеческою тrэ отсутствуют, но отмечены мноroчисленные случаи передачи трансмиссивной инфекции сотням
людей при использовании участков твердой трупной мозroвой оболочки и соматотропина. полученноro из трупноro материала rипофиза
человека, поэтому данный материал должен быть включен в катеroрию тканей с высоким риском заражения. PrP TSE было выявлено им
муноблоттинroм в твем<>й оболочке мозra пациента с вариантной БКЯ, который умер в США после необычно длинноro инкубационноro
периода (см. так же та&1ицу 'В для друrих положительных тканей: кожа, почки. печень. поджелудочная железа, яичники и матка) [Notari
et а/., 2010]. Так же должны быть отмечены ранее проведенные в Великобритании исследования мнoroчисленных случаев, которые BЫ
явили отрицательные ткани [/roпside et а/. 2002, Head et а/. 2004].
4. У крупноro poraтoro скота PrP TSE оrpаничено дистальным отделом подвздошной кишки, но иммунноrистохимический анализ тканей из
одиночною случая с rэКРс в Японии показал (хотя и сомнительно) вовлечение межмышечноro сплетения через маленькую и большую
кишки [Кimura аnd Нaпtaт. 2008].
5. У вариантной БКЯ PrPTSE orpаничен связанной с кишечником лимфоидной и нервной тканями (слизистая оболочка, мышцы и серозная
оболочка отрицатeJ1bНЫ).
6. Преджелудсж жвачных животных (ретикулум, рубец и книжка) широко используется, как и сам желудок (сычуr). Сычуr крупноro poraтoro
скота (и инorдa овцы) является таюке источником реннина.
7. Korдa болЬШаЯ nepopaльная доза rэкРс используется для экспериментальноro инфицирования крупноro poraToro скота, инфекцион
ность была выявлена в тонкой кишке и в месте соединения подвздошной и слепой кишок TpaHcreHHbIx мышей, экспрессирующих PrP
[правило Dr М Groschup]. PrP TSE было выявлено в низких концентрациях в лимфоидной ткани подвздошной кишки [Тепу et а/., 2003] и
было выявлено даже в более низких количествах в лимфоидной ткани тонкой кишки крупноro poraToro скота, таюке инфицированноro
пероральным путем [EFSA. 2009]
8. Не было подтверждено ни oднoro случая передачи спорадическоro инфекционноro БКЯ через плаценту человека, поэтому рассматри
вается как невозможное.
9. PrP TSE было выявлено у инфицированных скрейпи овец с хроническим маститом, но не у инфицированных овец без мастита [Ligios et
а/., 2005].
10. В ходе исследования у перорально инфицированных скрейпи хомяков было выявлено, что скопление PrP TSE в коже находилось
внутри маленьких нервных волокон Таюке сообщается, что поверхностная кожа «бархат» оленьеro pora СWD--инфицированных оленей
содержит PrpTSE и инфекционность [Aпgers et а/., 2009].
11. Выявленное иммуноцитохимически PrP TSE в почечной лоханке инфицированных скрейпи овец [Siso et а/., 2006) и в лимфоидных фол
ликулах внутри связывающей ткани соседствующей с почечной лоханкой у СWD--инфицированных олень мулов [Fox et а/., 2006).
12. Единичный положительный костный мозr в мноroчисленных попытках передачи от крупноro poraтoro скота, перорально зараженноro
rэкрс инфицированным мозrом [Wells et а/., 1999,2005, Sohп et а/.. 2009).
13. Мышечные roMoreHHaTbI не передают болезнь от приматов человеку с помощью спорадическоro БКЯ или от крупнороrатоro скота
крупнороraтому скоту с rэкРс. Однако. внутрицеребральное введение roMoreHaтa semiteпdiпosus мышцы (включая нервные и лимфо--
тические компоненты) от единичной коровы с клиническим rэкРс передало заболевание трансrенетическим мышам, которые экспрес
сируют PrP с частотой. показывающей степень присутствия инфекционности [Buschmaпп aпd Groschup. 2005]. Таюке недавно опублико-
ванные и неопубликованные исследования продемонстрировали присутствие PrP TSE в скелетной мускулатуре подопытных rpызунов со
скреипи и вариантной БКЯ [Beekes et а/.. 2005). в экспериментальных и природных инфекциях срейпи у овец и коз (Aпdreo/etti et а/., 2004),
у овец, перорально зараженных rэv.рс [Aпdreo/etti. неопубликованные данные). и у людей со спорадической. ятроreнной и вариантной
формами БКЯ [G/atze/ et а/., 2003, Kovacs et а/., 2004, Pede/ et а/., 2006). Исследования мышц тpaHcreHHbIx мышей, экспрессирующих PrP
оленеобразных, документально подтвердили инфекционность у СWО;1НфИЦИРОванных олень мулов [Aпgers et а/., 2006), и в скором Bpe
мени исследования позволят определить, связаны ли с инфекционностю выявленные PrPTSE в друrих формах тrэ.
14. у крупноro poraтoro скота результат определения инфекционности в языке был отрицательным, но присутствие инфекционности в
небной миндалине подняло вопрос о возможной инфекционности В ткани язычной миндалины в основании языка, которая моrла быть не
удалена при убое [Wells et а/., 2005, EFSA, 2008). у 7 из 10 естественно инфицированных скрейпи овец животных было выявлено PrP TSE
в языке [Casa/oпe et а/., 2005, Соroпа et а/., 2006).
15. Оrраниченная rлавным образом областями, вовлеченными в рецепторы обоняния.
5.3.
Статистический анализ результатов биолосюческих испытаний
825
16. Поскольку только один случай ятроrенной БКЯ был обоснованно приписан трансплантации роroвицы среди сотен тысяч про
оперированных (еще один случай рассматривается как вероятный, и еще один только возможен), роroвица отнесена к катеroрии
тканей более низкоro риска; исследование друrих тканей передней камеры rлаза (хрусталик, внутриrлазная жидкость, радужная
оболочка, конъюнктива) дали отрицательный результат в обоих случаях передачи вариантноro БКЯ и друrих заболеваний тrэ
человека. и эпидемиолоrические данные об их связи с ятроrенным путем передачи отсутствуют.
17. OrpoMHoe количество данных об инфекционности крови у подопытных rрызунов с тrэ было увеличено с помощью недавних
исследований, подтвердивших документально инфекционность в крови естественно инфицированных скрейпи овец и овец, KO
торым перелили кровь от rЭКРС инфицированноro KpYnHoro poraToro скота [Houstoп et а/., 2008]. естественно инфицированных
CWD оленей [Mathiasoп et а/., 2006] и (при эпидемиолоrических исследованиях) фракции красных кровяных клеток (которая вклю
чает orpoMHoe количество плазмы и лейкоцитов) от четырех доноров в пре клинической фазе вариантной БКЯ [пересмотрено в
Browп, 2006, Hewitt et а/., 2006]. Введение плазменноro фактора VIII таюке влечет за собой субклинический случай вариантной
БКЯ у пациента с rемофилией [Pedeп et а/., 2010]. Было продемонстрировано, что заболевание не передается с помощью крови
человека с любой формой «классической» тrэ [Dorsey et а/.. 2009] или крови крупноro poraтoro скота с rэкРс (включая кровь
эмбрионов телят). Некоторые лаборатории, которые используют новые высокочувствительные методы для определения PrpTSE,
предоставили отчет об успешных результатах в различных человеческих и животных ТrЭ. Однако у некоторых возникли TPYДHO
сти В получении воспроизводимых результатов в плазме. и до сих пор неизвестно предоставляют ли положительные результаты
возможность передачи заболевания как из за наличия ложно положительных. так и из за «правдивых» положительных резуль
татов, которые возникают из за слабо передающихся концентраций PrpTse. Вследствие данных положений (и фактически до сих
пор недоступны данные о слепых тестах образцов от естественно инфицированных людей или животных) экспертная rpynna счи
тает, что еще очень рано делать вывод о достоверности данных исследований. чтобы с достаточной уверенностью сказать о по
ложительном или отрицательном заключении.
18. Данные об отсутствии инфекционности в молоке от rЭКРС инфицированных коров включают длюельные эпидемиолоrиче
ские наблюдения, не выявившие передачу заболевания телятам через молоко' клинические наблюдения за более чем 100 теля
тами, вскармливавшимися инфицированными коровами без симптомов rэкрс (BSE): данные экспериментальных исследований
об отсутствии передачи заболевания мышам через молоко инфицированных коров. выращенных до возраста. равною мини
мальному инкубационному периоду. при интрацеребральном или пероральном введении [M/dd/etoп aпd Bar/ow. 1993. Tay/or et
а/., 1995]. Таюке PrP TSE не было выявлено в молоке крупноro poraToro скота. вынашивающеro rэкрс после экспериментальноrо
пероральноro введения [SEAC 2005]. Однако низкий уровень (от мкr/л до нr/л) нормальноro PrP был определен в молоке как у
животных, так и у человека [Fraпsch/f1/ et а/" 2006). PrP se был выявлен в молочных железах инфицированных скрейпи овец с xpo
ническим маститом [Ltgios et а/" 2005J. и совсем недавно было сообщено, что молоко (в некоторых случаях содержит молозиво)
от инфицированных скрейпи овец передает заболевание здоровым животным [Koпo/d et а/., 2008, Lacroux et а/., 2008].
19. Смешанный инокулят мочи и фекалий 01 естественно инфицированных CWD оленей не передает заболевание в течение
18 месячноrо наблюдения после введения ero здоровым оленям с rетерозиroтным (96G/S) PRNP rенотипом [Mathiasoп et а/.,
2006]. Однако последние биопробы транпенных мышей передали болезнь как при помощи мочи [На/еу et а/., 2009]. так и при
помощи фекалий [Taтguпey et а/.. 2009J. Кроме тою. мыши с лимфотическим нефритом, которые были экспериментально ин
фицированы скрейпи. избавились и от PrPTSE. и от инфекционности в моче при биоанализе в TpaHcreHHbIx мышах [Seegeret et а/.,
2005]. Очень низкие уровни инфекционности были выявлены в моче (и rистолоrически нормальных почках) хомяков, эксперимен
тально инфицированных скрейпи [Gregori aпd Rohwer. 2007, Goпza/ez Roтero et а/., 2008]. В заключение, в экспериментальной
модели с зараженными скрейпи хомяками пероральное введение вылилось в инфекционные фекалии при биоанализе в TpaHC
reHHbIx мышах, экспрессирующих PrP [Safar et а/.. 2008].
20. Эмбрионы крупноro poraToro скота с rэкРс (BSE) не вызвали заболевание у мышей, однако исследования инфекционности
тканей эмбрионов телят rэкРс сделаны не были, кроме крови (отрицательная биопроба мыши) [Fraser aпd Foster 1994]. Телята,
родившиеся от коров, которым вводили эмбрионы с rэкРс (BSE). выжили в течение периода наблюдений до семи лет. и иссле
дование roловною мозrа здоровых коров и их телят не выявило наличие ryбчатой энцефалопатии или PrPTSE [Wratha/l et а/.. 2002].
21. Единичные сообщения о передаче спорадической инфекционности БКЯ через человеческую пуповинную кровь и молозиво
так и не были подтверждены и считаются недостоверными Биопроба от коров с rэкрс у TpaHcreHHbIx мышей, экспрессирующих
бычиЙ PrP, дает отрицательный результат [Buschтaпп aпd Groschup. 2005]. и PrPTSE не было выявлено в молозиве от KpYnHoro
poraToro скота, вынашивающеro rэкРс после экспериментальноro пероральноro введения [SEAC. 2005].
01/2013:50300
5.3.
СТАТИСТИЧЕСКИЙ
АНАЛИЗ РЕЗУЛЬТАТОВ
БиолоrИЧЕСКИХ
ИСПЫТАНИЙ И ТЕСТОВ
1. ВВЕДЕНИЕ
Настоящая часть является руководством
для планирования испытзний и тестов биоло
rическими методами. описанными в настоящей
Фармакопее, и последующей обработки их pe
зулыатов. Она предназначен для специали
стов, не обладающих специальным образова
нием и не работающих в области статистики. в
том случае, если они несут ответственность за
проведение количественноrо анализа или ин
терпретацию результатов этих анализов. как
правило, без помощи и советов специалистов
в области статистики. Приведенные в данной
rлаве методы расчета не являются обязательны
ми при выполнении количественных определе
ний биолоrическими методами. предусмотрен
ными Фармакопеей. Допускается использование
альтернативных методов при условии, что они
MorYT быть соrласованы компетентным opraHoM
и имеются соответствующие данные, подтверж
дающие приюдность меТОДИI{И при ее валида
ции. Существует широкий спектр проrраммноro
обеспечения. которое может быть полезно в за
8ИСИМОСТИ от средств, доступных аналитику, от
ею знаний и опыта.
Следует обращаться за помощью к профес
сионалам в ситуациях, Korдa:
826
rосударственная фармакопея Республики Беларусь
требуется детальная проработка плана и
методов анализа, подходящих для научноro ис
следования или разработки новоro лекарствен
ноro средства;
нельзя придерживаться оrраничений, Ha
кладываемых данной rлавой на план количе
ственноro определения, например, необходи
мость корректировки планов количественноrо
определения может быть вызвана специфиче
скими лабораторными условиями или невозмож
ностью применения равных количеств paBHO
мерно распределенных доз;
требуется анализ для расширенных нели
нейных rрафиков зависимости «доза эффект»,
например, при про ведении количественных им
мунолоrических определений. Основные по
ложения анализа расширенных кривых зави
симости «доза эффект» с помощью одной из
распространенных методик, тем не менее, вклю
чены в раздел 3.4, а простейший пример пред
ставлен в разделе 5.4.
1.1. ОБЩИЕ ПОЛОЖЕНИЯ И ТОЧНОСТЬ
Биолоrические методы анализа применя
ют для количественноro определения таких
субстанций и препаратов, биолоrическая aK
тивность которых не может быть корректно oцe
нена при помощи химических или физических
методов анализа. Везде, rдe это возможно, в
основу количественных определений положен
принцип сравнения со стандартным препара
том для тоro, чтобы определить, какое количе
ство анализируемой субстанции вызовет такой
же биолоrический эффект, как определенное
количество (Единица действия) стандартноro
препарата. Существенным условием проведе
ния таких биолоrических количественных опре
делений является то, что анализ стандартноro
препарата и исследуемой субстанции должен
быть выполнен в одно и тоже время и при иден
тичных условиях.
Для некоторых количественных определе
ний (например, определения титра вируса) aK
тивность испытуемоro образца не выражается
относительно стандарта. Такой тип количествен
ноro определения рассмотрен в разделе 4.5.
Любая оценка активности, полученная в
ходе биолоrическоro количественноro определе
ния, включает случайную поrрешность, обуслов
ленную свойственной биолоrическим реакциям
изменчивостью, и, если это возможно, следует
провести вычисление поrрешности результатов
каждоro количественноro определения даже в
случае использования официальной методи
ки определения. В этой связи ниже приведены
методы планирования кпличественных опреде
лений и вычисления их поrрешностей. Каждый
раз перед выбором Toro или иноro статистиче
скоro метода, следует провести предваритель
ное исследование с определенным числом по
вторений, чтобы убедиться в применимости
этоro метода.
Доверительный интервал активности при
водят для указания точности, с которой была
установлена эта активность при количествен
ном определении. Он рассчитывается исходя
из плана эксперимента и объема выборки. При
количественных определениях биолоrическими
методами обычно выбирается 95 % доверитель
ный интервал. Для обеспечения справедливости
утверждения о том, что действительное значе
ние активности находится в данном интервале с
вероятностью 95 %, расчет rраниц доверитель
ноro интервала проводится методами MaTeMa
тической статистики. Является ли эта точность
приемлемой для Фармакопеи, зависит от требо
ваний, установленных в частной статье на KOH
кретный препарат.
Термины «среднее значение» и «CTaHдapT
ное отклонение» используются в данном раз
деле в соответствии с их определением в боль
шинстве руководств по биометрии.
Термины «заявленная активность» или «aK
тивность, указанная в маркировке», «YCTaHOB
ленная(аттестованная)активность»,«предпола
rаемая активность», «отношение активностей»
и «расчитанная активность» используются в
данном разделе для обозначения следующих
понятий:
«заявленная активность» или «активность,
указанная в маркировке»: для roтовоro препара
та номинальное значение, установленное на
основании известной активности исходноro (He
расфасованною) материала; для исходноro (He
расфасованною) материала активность, YCTa
новленная производителем;
«установленная (аттестованная) актив
ность» активность стандартноro препарата;
«предполаrаемая активность» BpeMeH
но установленная активность испытуемоrо пре
парата, на основании которой рассчитываются
дозы, которые были бы эквивалентны дозам ис
пользуемоro стандартноro препарата;
«отношение активностей» для испытуемо
ro препарата отношение равноактивных доз
стандартноro препарата и испытуемоro препара
та в условиях количественноro определения;
«рассчитанная активность» активность,
вычисленная на основании данных количествен
ноro анализа.
В разделе 9 (словарь символов) приведены
наиболее важные используемые в данной rлаве
символы. Если в тексте используется символ,
не приведенный в этом разделе, или символ ис
пользуется в друroм значении, то еro определе
ние приводится в соответствующей части текста.
2. РАНДОМИЗАЦИЯ И НЕЗАВИСИМОСТЬ
ОТДЕЛЬНЫХ ИССЛЕДОВАНИЙ
Порядок введения препаратов, различных
воздействий на экспериментальные объекты (жи
вотные, пробирки и т. д.) должен быть осущест
влен определенным cTporo случайным образом.
Любой друroй выбор экспериментальных условий,
5.3. Статистический анализ результатов биОЛО2ических испытаний
827
который преднамеренно не учитывается в плане
эксперимента, также должен быть выполнен слу
чайным образом. Например, выбор размещения
боксов в лаборатории и порядок введения пре
па ратов. В частности, rруппа животных, получа
ющих одинаковую дозу какоro нибудь препарата,
не должна одновременно подверraться исследо
ванию (в то же самое время или в том же самом
месте) до тех пор, пока не будет весомых доказа
тельств тоro, что источник вариации (например,
между временем или между положениями) явля
ется незначительным. Рандомизация может быть
осуществлена с использованием компьютера при
помощи встроенноro reHepaтopa случайных чисел.
Каждый раз после запуска проrраммы аналитик
должен убедиться, что rенерируется новая после
довательность случайных чисел.
Препараты, назначаемые каждому экспери
ментальному объекту, должны быть настолько
независимы, насколько это возможно. В преде
лах каждой экспериментальной rруппы назначен
ные разведения должны быть не только разведе
ниями одной и той же дозы, но и должны быть
приroтовлены индивидуально. Без выполнения
данноrо условия вариабельность, свойственная
препарат не будет полностью представлена в
дисперсии ошибки эксперимента. В результате
будет недооценена остаточная дисперсия, что
может привести к:
1) необоснованному ужесточению критерия
при дисперсионном анализе (см. разделы 3.2.3 и
3.2.4 );
2) недооценке истинных доверительных ин
тервалов при испытании, которые, как показано
в разделе 3.2.5, рассчитаны исходя из оценки S2
(ошибка среднеквадратической разности или дис
персия остаточной поrрешности).
з. ИСПЫТАНИЯ, ОСНОВАННЫЕ
НА КОЛИЧЕСТВЕННЫХ ЭФФЕКТАХ
3.1. СТАТИСТИЧЕСКИЕ МОДЕЛИ
3.1.1. ОБЩИЕ ПРИНЦИПЫ
Количественные определения биолоrической
активности, включенные в Фармакопею, основаны
на «принципе разведения». Это означает, что aHa
лизируемый испытуемый препарат предположи
тельно содержит то же самое активное вещество,
что и стандартный препарат, но отличается от по
следнеro соотношением активноro и неактивноro
компонентов. В этом случае испытуемый препа
рат можно теоретически получить из стандартноro
путем ero разведения неактивными компонентами.
Для тoro, чтобы провеrить, подчиняется ли какой
либо конкретный тест количественноro определе
ния «принципу разведения», необходимо сравнить
зависимость «доза эффект» стандартноrо и испы
туемоro препаратов. Если эти зависимости раз
личаются статистически значимо, то теоретиче
ская модель «принципа разведения» не является
достоверной. Статистически значимые различия
в зависимостях «доза эффект» для cтaндapTHO
ro и испытуемоro препаратов позволяют предпо--
ложить, что один из препаратов, кроме активною
компонента, содержит друrие компоненты, кото--
рые обладают активностью или влияют на измеря
емые результаты.
Для тоro, чтобы в теоретической модели до--
стиrнуть хорошо выраженноro эффекта раЗ8€--
дения, следует преобразовать зависимость «до--
за эффект» в линейную функцию в наибольшем
возможном диапазоне доз. В качестве модели
для рассматриваемых количественных определе
ний биолоrической активности интерес представ
ляют две модели: модель параллельных линий и
модель уrловых коэффициентов.
Применение той или иной модели зависит от
выполнения следующих условий:
1) различные воздействия на эксперимен
тальные объекты были выполнены случайным об
разом;
2) результаты (эффект) каждоro воздействия
подчиняются закону нормальноro распределения;
3) стандартные отклонения эффектов в
каждой исследуемой rруппе как для стандартноro,
так и для испытуемоro препаратов статистически
достоверно не отличаются друr от друrа.
При разработке методики количественноro
определения аналитик должен убедиться, что по
лученные данные от различных количественных
определений удовлетворяют этим теоретическим
условиям.
Условие 1 может быть выполнено при
помощи использования раздела 2.
Условие 2 является предположением, KOTO
рое на практике почти Bcerдa выполняется. Незна
чительные отклонения от этоro предположения в
основном не вносят серьезных ошибок в анализ,
поскольку исследование включает несколько по
вторений. В случае сомнения может быть выпол
нена проверка на наличие отклонений от HO
мальноro распределения (например, при помощи
критерия Шапиро Уилка (Shapiro Wilk)1.
Для проверки выполнения Условия 3 MOryт
быть использованы тесты проверки OДHO
сти дисперсий (например, критерий БартлеттaL
(Bart/ett), критерий Кохрена З (Cochraп». для этих
целей также может быть использовано rpaфич
ское представление результатов анализа (см. при
меры в разделе 5).
Если условия 2 и/или 3 не выnaпняются. то
преобразование результатов может привести к
улучшению выполнения этих условий. Примерами
таких преобразований 1!ВЛЯЮТСЯ лoraрифмическое
(/п у), квадратичное ( "У , у).
, Wi/k. МВ aпd 5hap,ro S S (1968) The joiпt assesтeпt о(
пorтa/ity of severa/ тdependeпt saтp/es. Techпoтetrics 10.
825-839
2 Bar1/ett. М 5. (1937). Proper1les о( sufficieпcy aпd statistica/
tests. Proc. Roy Soc. Loпdoп. 5eries А 160. 280.282.
, Cochraп. WG (1951). Testiпg а /теа, re/atioп aтoпg
vапапсеs. Bioтetrics 7. 17.32.
828
rосударственная фармакопея Республики Беларусь
Лоrарифмическое преобразование зна
чений у в /п у, может выполняться, если OДHO
родность дисперсий неудовлетворительна. Оно
также может улучшить нормальность распреде
ления, если оно смещено вправо.
Преобразование у в .JY можно исполь
зовать в случае, если результаты подчиняются
распределению Пуассона, то есть если они по
лучены путем подсчета числа событий.
Преобразование у в у2 может быть исполь
зовано, если, например, доза в большей мере
пропорциональна площади инrибирования зоны
роста микроорrанизмов, чем измеренному ди
аметру этой зоны роста.
Для некоторых тестов количественноro опре
деления, которые зависят от количественных
реакций. например. иммунолоrический анализ
или iп vitro методы количественноro определе
ния с использованием клеточных банков, ис
пользуется большое число доз. Эти дозы вызы
вают эффекты. которые полностью охватывают
возможный диапазон ответов/реакций и образу
ют расширенную нелинейную кривую зависимо
сти «доза эффект». Такие кривые типичны для
всех количественных определений биолоrиче
скими методами. но для мноrих испытаний ис
пользование большою числа доз не отвечает
критериям биоэтики (например, определения
iп vivo) или непрактично, и цели такоro количе
ственноro определения MorYT быть достиrнуты
на оrраниченном числе доз. Поэтому допускает
ся оrраничение количества тестируемых доз до
такой части диапазона кривой «доза эффект»,
которая была бы линейна после соответствую
щеro преобразования, чтобы можно было при
менять методы, описанные в разделах 3.2 и 3.3.
Однако в некоторых случаях может быть необ
ходим анализ всей расширенной кривой «доза
эффект». Краткое изложение одной из моделей,
которая моrла бы быть применена для TaKoro
анализа, представлено в разделе 3.4, а простой
пример описан в разделе 5.4.
Существует друrая катеroрия тестов, Korдa
при анализе результат не может быть количе
ственно измерен для каждою эксперименталь
ноro объекта, а определяется как часть выборки,
у которой получен ответ на воздействие. Эта Ka
теroрия рассмотрена в разделе 4.
3.1.2. ПОВСЕДНЕВНblЕ (РУТИННblЕ)
КОЛИЧЕСТВЕННblЕ ОПРЕДЕЛЕНИЯ
При рутинном проведении количественноro
определения практически отсутствует возмож
ность систематически контролировать выполне
ние условий 1 3, так как оrраниченное число
измерений в каждом анализе может отрицатель
но влиять на чувствительность статистических
тестов. Однако специалистами по статистиче
скому анализу было доказано, что при симме
тричных сбалансированных испытаниях неболь
шие отклонения от однородности дисперсий
и нормальности распределения не оказывают
существенноro влияния на результаты количе
ственноro определения. Вопрос оприменимости
статистической модели следует ставить под co
мнение только в том случае, если ряд получен
ных результатов испытаний имеет сомнитель
ную достоверность. При этом может возникнуть
необходимость выполнить новую серию пред
варительных испытаний, как описано в разделе
3.1.1.
В зависимости от применяемой статистиче
ской модели следует контролировать выполне
ние двух дополнительных условий.
Для модели параллельных линий:
4А) отношение между лоrарифмом дозы и
реrистрируемым результатом может быть пред
ставлено в виде прямой линии во всем диапазо
не исследованных доз;
. 5А) при анализе прямая линия для любоro
испытуемоro препарата параллельна соответ
ствующей прямой линии для стандартноro пре
парата.
Для модели уrловых коэффициентов:
4В) во всем диапазоне исследованных доз
отношение между дозой и полученным эффек
том для каждоro препарата может быть пред
ставлено в виде прямой линии;
5В) при анализе для любою испытуемою
препарата прямая линия пересекает ось у (при
дозе, равной нулю) в той же точке, что и прямая
линия для стандартноro препарата (друrими
словами, при анализе rрафики результирующей
функции эффектов всех испытуемых препара
тов должны пересекаться с rрафиками результи
рующей функции эффектов стандартноro препа
рата в одной и той же точке).
Условия 4А и 4В MOryT быть подтверждены
при количественных определениях, в которых ис
пользовано как минимум по три разведения для
каждою протестированноro препарата. Исполь
зование результатов количественноro определе
ния, полученных при одном или двух разведени
ях, может быть обосновано лишь в том случае,
если накопленный опыт свидетельствует, что yc
ловия линейности, параллельности или совпаде
ние точек пересечения Bcerдa выполняются.
После получения результатов количествен
Horo определения и перед вычислением OTHO
сительной активности каждоro препарата BЫ
полняется дисперсионный анализ с целью
про верки выполнения условий 4А и 5А (или 4В
и 5В). ДЛЯ этоro общую дисперсию представля
ют в виде определенноro числа сумм диспер
сий, соответствующих каждому выполняемому
условию. Оставшаяся дисперсия представля
ет собой неисключенную (остаточную) поrреш
ность определения. ДJlЯ нее при помощи расче
та F отношений можно оценить отсутствие или
наличие значимых источников вариации.
После валидации методики анализа актив
ность каждоrо испытуемоro препарата по OTHO
шению к стандартному может быть рассчита
на и выражена как отношение активностей или
5.3.
Статистический анализ результатов биОЛО2ических испытаний
829
преобразована в определенные единицы актив
ности, например, в Международные Единицы.
Также для каждоro ряда данных анализа MOryт
быть установлены доверительные интервалы.
В разделе 3.2 рассматриваются методики
количественных определений, основанные на
использовании модели параллельных линий, а
в разделе 3.3 основанные на использовании
модели уrловых коэффициентов.
Если хотя бы одно из перечисленных пяти
условий (1,2,3, 4А, 5А или 1,2,3, 4В, 5В) не BЫ
полняется, применение приведенных в данной
статье методов вычисления не может быть обо
сновано и следует провести специальный анализ
результатов количественноro определения.
Аналитик не должен применять друюй
метод преобразования до тех пор, пока не убе
дится, что невыполнение условий является не
случайным, а обусловлено систематическим из
менением условий испытания. В этом случае,
прежде чем принять новое преобразование для
постоянных (рутинных) количественных опреде
лений, следует повторить исследования, опи
санные в разделе 3.1.1.
Высокое число недостоверных результа
тов анализа, возникающих вследствие непарал
лельности или нелинейности, при выполнении
постоянных (рутинных) количественных опреде
лений в результате сравнения со стандартным
материалом чаще всею свидетельствует о He
правильном планировании эксперимента и He
достаточном числе повторений. Обычно это обу
словлено недостаточно полной идентификацией
всех источников вариации, воздействующих на
количественное определение, что в результате
может привести к заниженной оценке остаточ
ной поrрешности и, соответственно, к увеличе
нию значения F отношений.
В рамках однократноro количественноro
определения не Bcerдa можно принять во вни
мание все возможные источники вариации (Ha
пример, внутрилабораторная вариация в серии
«изо дня в день»). В этом случае доверитель
ные интервалы повторных результатов количе
ственных определений одноro и тоro же препа
рата MorYT не совпадать, и следует соблюдать
осторожность при оценке отдельных довери
тельных интервалов. Для получения более Ha
дежной оценки доверительноrо интервала He
обходимо выполнить несколько независимых
количественных определений, объединить по
лученные результаты и получить одну оценку
активности и доверительный интервал (см.
раздел 6).
Для контроля качества постоянных (рутин
ных) количественных определений peKOMeHДY
ется заносить в контрольные статистические
карты результаты оценки уrловых коэффициен
тов и оценки остаточной поrрешности.
Довольно высокая неисключенная (OCTa
точная) поrрешность может свидетельствовать
об определенной технической проблеме. Эти
случаи должны расследоваться и, при выявле
нии ошибок в ходе выполнения количественноro
определения, испытание следует повторить. He
обычно высокая остаточная поrрешность также
может указывать на наличие случайною выбро
са или неправильноro учета результата. Эффект,
достоверность котороro ставится под сомнение
из за неправильноro выполнения количествен
ноro определения, следует исключить. Обосно
ванным также можно считать исключение из
общей оценки аномальноro эффекта после за
вершения количественноro определения, если
удается проследить причину отклонения и убе
диться, что она обусловлена нарушениями в
ходе количественноro определения. Произволь
ное отбрасывание или сохранение очевидных He
достоверных результатов может оказаться cepb
езным источником поrрешности измерения. В
целом не рекомендуется отбрасывать результа
ты лишь на основании статистической значимо
сти исследования на наличие выбросов.
Время от времени может возникать доволь
но низкая остаточная поrрешность, что приводит
к превышению F тношениями критических зна
чений. В этом случае может быть оправданной
замена остаточной поrрешности отдельною KO
личественноro определения на среднюю OCTa
точную поrрешность, полученную на основании
архивных данных, зафиксированных в контроль
ных статистических картах.
3.1.3. ВЫЧИСЛЕНИЯ И оrРАНИЧЕНИЯ
Соrласно общим принципам надлежащеro
планирования эксперимента на план анализа
обычно накладываются следующие три оrрани
чения (они обладают преимуществами как в про
стоте вычислений, так и в обеспечении их точ
ности):
а) число разведений должно быть одинако
вым для каждоro из испытуемых препаратов;
Ь) при использовании модели параллель
ных линий отношение двух последователь
ных доз должно быть Bcerдa постоянным для
всех введений, проводимых в рамках испыта
ния; при использовании модели уrловых J(ОЭф
фициентов должна быть постоянной разница
(интервал) между двумя последовательными
дозами;
с) число испытуемых объектов должно быть
одинаковым во всех исследованиях..
Если используемый план удовлетворя
ет этим условиям, то вычисления упрощаются.
Формулы вычислений приведены в разделах 3.2
и 3.3. Рекомендуется использовать проrраммное
обеспечение, РClЗработанное специально для
этих целей. СущеСТАует ряд статистических про
rpaMM, при помощи которых можно леrко обра
батывать планы количественных определений,
приведенные в частных статьях. Не все про
rpaMMbI MOryт использовать одни и те же форму
лы и алroритмы, но они все должны приводить к
одинаковым результатам.
830
rосударственная фармакопея Республики Беларусь
Планы количественных определений, не OT
вечающие вышеуказанным требованиям, MOryт
быть также допустимы и корректны. Однако He
обходимые для таких вычислений формулы
слишком сложны и поэтому в данной части не
приводятся. Краткое описание методов расче
та приводится в Разделе 7.1. Эти методы MOryт
также использоваться для оrраниченных планов,
в этом случае они эквивалентны упрощенным
формулам.
Формулы для оrраниченных планов, приве
денные в данной статье, MoryT быть использова
ны, например, для создания специальных про
rpaMM с использованием электронных таблиц.
Для лучшеro понимания статистики и проверки
правильности результатов таких nporpaMM MOryт
быть использованы примеры, приведенные в
Разделе 5.
3.2. МОДЕЛЬ ПАРАЛЛЕЛЬНЫХ ЛИНИЙ
3.2.1. ВВЕДЕНИЕ
На рисунке 3.2.1. I показана модель парал
лельных линий. Лоrарифмы доз откладывают по
оси абсцисс (х) таким образом, чтобы их значе
ния возрастали слева направо. Наблюдаемые
эффекты откладывают по оси ординат (у). OT
дельные значения эффектов для каждоro из ис
следований показаны в виде черных точек. Две
линии представляют собой рассчитанную зави
симость «/п(доза}-эффект» для стандартноro и
испытуемоro препаратов.
Примечание: в данной часть во всех случаях
используется натуральный лоraрифм (/п или /oge>.
Везде, rдe ИCfЮЛЬЭуется термин «антилоraрифм»,
подразумевается значение ЭХ. Тем не менее, также
можно полЬЗОваТЬСЯ лоrарифмом Бриrrca или дe
сятичным лоraрифмом (/g или /og10)' В этом случае
соответствующий «антилоrарифм» равен 1 ОХ.
ДЛЯ удовлетворительноrо количественноrо
определения предполаrаемая активность испы
туемоro препарата должна быть близка к дей
ствительной активности. Исходя из предпола
rаемой активности испытуемоro препарата и
аттестованной активности стандартноro пре
парата, roтовятся разведения с одинаковой (по
возможности) активностью. То есть COOTBeTCTBY
ющие дозы стандартноro и испытуемоro препа
ратов должны вызывать одинаковый эффект.
Если нет информации о предполаrаемой актив
ности, выполняют серию предварительных опре
делений в широком диапазоне доз для YCTa
новления области, в которой эта зависимость
является линейной.
Чем точнее определена принятая предпола
rаемая актиl3НОСТЬ испытуемоro препарата к aT
тестованной активности стандартноro препарата,
тем ближе одна к одной будут эти линии, посколь
ку они должны давать одинаковые эффекты при
одинаковых дозах. rоризонтальное расстояние
между линиями представят собой «истинное»
значение активности испытуемоro препарата по
отношению к еro предполаrаемой активности.
Чем больше расстояние между линиями, тем
менее точен проrноз в отношении предполаrа
емой активности испытуемоro препарата. Если
линия, соответствующая испытуемому препара
ту, смещена вправо от линии, соответствующей
стандартному препарату, то оценка предполаrа
емой активности была завышена и при расчетах
рассчитанная активность будет ниже предполаrа
емой. Аналоrично, если линия, соответствующая
испытуемому препарату, расположена слева от
линии, соответствующей стандартному препара
ту, то предполаrаемая активность была заниже
на и при определении рассчитанная активность
будет выше предполаrаемой.
.
(1)
.
In Дозы (х)
Рисунок 3.2.1. 1. Модель параллельных линий
в случае анализа (3+3)
3.2.2 ПЛАН МЕТОДА КОЛИЧЕСТВЕННО/О
ОПРЕДЕЛЕНИЯ
Для оптимизации точности плана анализа
будет полезным соблюдение следующих требо
ваний:
1) отношение между уrловым коэффици
ентом и остаточной поrрешностью должно быть
как можно большим;
2) диапазон доз должен быть максимально
широким;
3) линии должны располаrаться как можно
ближе друr к друry, т. е. предполаrаемая aK
тивность должна с хорошей точностью давать
оценку «истинной» активности.
Распределение экспериментальных объек
тов (животные, пробирки и т. д.) для различных
исследований t./Iожет быть осуществлено разны
ми способами.
3.2.2.1. Дизайн (схема) полной рандомизации
Если вся совокупность экспериментальных
объектов считается достаточно однородной и
нет никаких оснований полаrать, что разброс pe
5.3. Статистический анализ результатов биОЛО2ических испытаний
831
зультатов будет меньшим внутри определенным
образом сформированных подrрупп, распреде
ление объектов в разные rруппы выполняется
случайным образом.
Если объекты, распределенные в таких под
rруппах, как положение в пространстве или дата
исследования, распределены более однородно,
чем вся совокупность в целом, точность коли
чественноro определения может быть повыше
на путем введения в план исследований одноro
или нескольких оrраничений. Тщательно проду
манное распределение объектов с использова
нием этих оrраничений позволяет исключить He
значимые источники вариации.
3.2.2.2. Дизайн (схема) рандомизирован-
ных блоков
В основе этой схемы лежит возможность BЫ
делить идентифицируемый источник вариации
(например, различная чувствительность у He
скольких приплодов экспериментальных живот
ных или различие между чашками Петри в случае
микробиолоrическоro определения активности Me
тодом диффузии в arape). В соответствии с данной
схемой каждое исследование должно быть повто
рено одинаковое число раз в каждом из блоков
(приплод или чашка Петри). Данная схема при
менима только Torдa, Korдa блок достаточно боль
шой для осуществления всех видов воздействий.
При мер использования такой схемы приведен в
Разделе 5.1.3. Также допускается использовать
схемы рандомизированных блоков с повторно
стями. В пределах каждоro блока воздействия
должны распределяться случайным образом.
В Разделе 8.5 приведен алrоритм, при
помощи котороro можно получить случайные пе
рестановки.
3.2.2.3. Дизайн (схема) латинскоrо квадрата
Данная схема применяется, Korдa на наблю
даемый результат воздействует два различных
источника вариации, каждый из которых может
характеризоваться k различными уровнями или
позициями. Например, в случае количественно
ro определения антибиотиков с использовани
ем планшетов исследования MOryт быть орrани
зованы на большом планшете в виде матрицы
kxk, причем каждое воздействие встречается по
одному разу в каждом столбце и в каждой строке.
Схема применяется в случае, Korдa число строк,
столбцов и число исследований одинаково. Pe
зультаты записываются в виде квадрата, назы
ваемою латинским. Вариации, обусловленные
различием в результатах между k строками и k
столБL:ами, MOryт быть сrруппированы, что по
зволит уменьшить поrрешность. Пример исполь
зования схемы латинскоro квадрата приведен в
Разделе 5.1.2. В Разделе 8.6 дан алroритм для по
строения латинских квадратов. В некоторых слу
чаях MOryT быть полезны более сложные построе
ния, rдe в рамках латинскою квадрата повторяется
одно или больше воздействий (введений препара
та). Упрощенные формулы, приведенные в Hacтo
ящем разделе, не подходят для таких планов, по
этому требуется совет специалиста.
3.2.2.4. Перекрестный дизайн (схема)
Данную схему полезно применять, Korдa
эксперимент может быть разделен на блоки, но
к каждому блоку возможно применить только
два вида воздействия. Например, таким блоком
может быть один экспериментальный объект,
который может быть протестирован дважды.
Данная схема предназначена для повышения
точности путем исключения различий резуль
татов между объектами за счет их взаимной
компенсации при двух экспериментах с общим
уровнем эффекта. Если исследуются две дозы
испытуемою и стандартноro препаратов, такую
схему называют двойной пере крестной схемой.
Эксперимент разбивают на две стадии, раз
деленные достаточным промежутком времени.
Объекты подразделяют на четыре rруппы, и на
первой стадии эксперимента в каждой rруппе BЫ
полняется одно из четырех воздействий. Объек
ты, которые на первой стадии получали один
препара на второй стадии получают второй
препарат. Объекты, которые на первой стадии
получали меньшие дозы, на второй стадии по
лучают большие. Распределение доз приведено
в Таблице 3.2.2. 1 Пример использования схемы
показан в Разделе 5.1.5.
Таблица З.2.2. 1
Распределение доз при использовании
перекрестной схемы
rруппа единиц Период I
1 81
2 82
3 Т 1
4 Т 2
Период 11
Т 2
Т,
82
81
3.2.3. ДИСПЕРСИОННЫЙ АНАЛИЗ
(ANOVA)
В данном разделе приведены формулы,
которые необходимы при выполнении диспер
сионноro анализа. В Разделе 5.1 рассмотрены
конкретные примеры, позволяющие понять Ha
значение этих формул. Если необходимо, можно
также обратиться к списку терминов и условных
обозначений (Раздел 9).
Приведенные формулы приrодны для сим
метричных количественных определений, в KO
торых один или более испытуемых препара
тов (7; И И др.) сравниваются со стандартным
препаратом (8). Следует подчеркнуть, что фор
мулы MorYT быть использованы только в том
случае, если дозы отличаются одна от друroй в
одинаковое число раз, если каждый из препара
тов исследуется в одинаковом числе доз и если
каждая доза применяется одинаковое число
раз. Если эти условия не выполняются, данные
формулы не должны применяться для анализа.
832
rосударственная фармакопея Республики Беларусь
За исключением некоторых уточнений OCTa
точной поrрешности, основной статистический
анализ данных количественноro определения
одинаков для схем полной рандомизации, paH
домизированных блоков и латинских квадратов.
Формулы для перекрестной схемы исследова
ния значительно отличаются и приведены в при
мере 5.1.5.
Исходя из рассмотренных в Разделе 3.1 пун
ктов и преобразовав, если это необходимо, эф
фекты, для каждой rруппы и для каждой дозы
препарата следует вычислить среднее значе
ние, как описано в таблице 3.2.3. 1. Также сле
дует вычислить линейные контрасты, связанные
с наклоном прямых «Iп(доза) эффект». В табли
це 3.2.3. 11 приведены три дополнительные фор
мулы, которые необходимы для проведения дис
персионноro анализа.
Общая вариация результатов, вызванная
различными последовательностями введения
препаратов, подразделяется далее, как опи
сано в таблице 3.2.3. III; сумму квадратов при
этом рассчитывают с использованием данных из
таблиц 3.2.3. 1 и 3.2.3. 1I. Сумма квадратов, обу
словленная нелинейностью, может быть рассчи
тана только в том случае, если при количествен
ном определении использовались по крайней
мере по три дозы каждоro из препаратов.
Остаточная поrрешность (вариация) коли
чественноro определения рассчитывается путем
вычитания вариаций, обусловленных схемой
эксперимента, из общей вариации для эффекта
(таблица 3.2_3. IV). В этой таблице j7 среднее
значение всех результатов, полученных при KO
личественном определении. Следует отметить,
что для латинскоro квадрата число результатов
в повторностях (п) равно числу строк, столбцов
и видов воздействий (последовательности доз)
(dh).
Дисперсионный анализ завершают следу
ющим образом. Находят дисперсию (средний
квадрат отклонения) путем деления каждой
суммы квадратов на соответствующее число
степеней свободы. Далее оценивают статисти
чес кую значимость отношения дисперсии для
каждой переменной к остаточной поrрешности
(82) (так называемое F отношение), для этоro
можно использовать таблицу 8.1 или COOTBeT
ствующую функцию компьютерноro проrрамм
ною обеспечения.
3.2.4. ПРОВЕРКА ДОСТОВЕРНОСТИ
Результаты количественноro определения
считаются «статистически обоснованными»,
если при дисперсионном анализе выполняются
следующие условия:
1 ) проведенные вычисления свидетель
ствуют о значимости линейной реrрессии, т. е.
. рассчитанная вероятность меньше 0,05; если
это условие не выполняется, вычислить довери
тельный интервал с вероятностью 95 % не пред
ставляется возможным;
2) проведенные вычисления свидетель
ствуют, что непараллельность незначима, т. е.
рассчитанная вероятность не меньше 0,05;
это означает, что выполняется условие 5А
(Раздел 3.1 );
3) проведенные вычисления свидетель
ствуют, что нелинейность незначима, т. е. рассчи
танная вероятность не меньше 0,05; это означа
ет, что выполняется условие 4А (Раздел 3.1 ).
Значимое отклонение от параллельности
при мноroкратных количественных определе
ниях может быть обусловлено тем, что уrловой
коэффициент кривой зависимости «Iп(доза)
эффект» для одноrо из препаратов, включенных
в исследование, отличается от аналоrичноrо
значения для друrих препаратов. В этом случае
допускается не признавать несостоятельность
всеro анализа, а исключить результаты, име
ющие отношение к этому препарату, и повторить
статистический анализ заново.
Таблица 3.2.3. I
Формулы для количественных определений с d дозами кажд020 из препаратов, при использовании
модели параллельных линий
Стандартный 1 й испытуемый 2-й испытуемый
препарат (5) препарат (Т) препарат
(U и т. д.)
Средний эффект от минималь SI Т I U 1
ной дозы
Средний эффект от второй I S2 I Т 2 I U 2
дозы
.. . I .. . I ... I ...
Средний эффект от макси I Sd I Td I U d
мальной дозы
Суммарный эффект препарата I Ps ==S1+ S 2+",+ S d I Р т == Т 1 + Т 2 + ... + Td I РИ == ...и т. д.
Линейный контраст Ls 15, + 25, + ... + d8, 2(d + 1)Р, LT 1T1 + 2Т 2 + .. dTd \d+ 1JPr Lu == ...и т. д.
2 2
8ЗЗ
5.3. Статистический анализ результатов биолоаических испытаний
Таблица 3.2.3. 11
п
Нр ==
d
Доп олнительные формулы для дисперс иОНН02О анализа
1 . н 1 .
. L d З d .
к == п(рs + Р7 + .../
hd
Таблица 3.2.ЗАII
Формулы для расчета суммы квадратов и числа степеней свободы
ИСТОЧНИК вариации (размаха) Степени свобо Сумма квадратов
AbI(f)
Препараты h 1 2 2
SSprep == Hp(P s + Р Т + ...) K
1 2 2 2
Линейная реrрессия 1 SSreg == HL (Ls + L T + ...)
h
Непараллельность h 1 SSpar == HL(L; + L + ...) SSreg
Нелинейность(*) h(d 2) SS'т == SStmat SSprep SSreg SSpar
rруппы(последовательность hd 1 SStrear == п(S + ... т S: т Т,2 ... + Т; + ...) к
доз)
П Не рассчитывается для двухдозовых количественных определений.
Оценка остаточной П02решности (вариации)
Таблица З.2.З. IV
Источник вариаций Степени Сумма квадратов
свободы
2 2
Блоки (строки)(') п 1 SSbIock == hd(R f + ... + Rn) К
2 2
Столбцы(") п 1 SSco! == hd(C f + ... + Cп) К
{ Схема полной рандоми hd(п 1 ) SSres == SStot SStreat
зации
Остаточная вари Рандомизированный блок (hd 1)(п 1) SSres == SStot SStreat SSbIock
ация("')
Латинский квадрат (hd 2)(п 1) SSres == SStot SStreat SSbIock SSco/
Общая вариация пhd 1 SStot == L(Y у I
Для схемы латинскоro квадрата эти формулы применимы только в случае, если п==hd.
(') Не рассчитывается для схемы полной рандомизации
(") Рассчитывается только для схемы латинских квадратов.
("') Зависит от схемы рандомизации.
После тоro, как установлена статистическая
достоверность, MOryT быть рассчитаны оценки
активности препаратов и rраницы доверитель
ных интервалов при помощи приведенных в сле
дующем разделе методов.
3.2.5. ОЦЕНКА АКТИВНОСТИ И ,РАНИЦ
ДОВЕРИТЕЛЬНbfХ ИНТЕРВАЛОВ
Если обозначить через I натуральный лоrа
рифм отношения между ближайшими значения
ми доз любоro из препаратов, то общий для всех
прямых уrловой коэффициент (Ь) в случае коли
чественноro определения, которое включает d
доз для каждоro препарата, вычисляется по фор
муле:
Ь == HL(L s + L T + ...) . (3.2.5. 1)
/пh
105.3_к 1712
Лоrарифм отношения активности испыту
емоro препарата, например, Т, вычисляют по
формуле:
М Т Р7 p s . ( 3.2.5. 2 )
db
Рассчитанная активность представляет
собой оценку «истинной активности» каждоro из
испытуемых препаратов. rраницы доверитель
ноro интервала MOryT быть рассчитаны как анти
лоrар&.-1фМ выражения:
CM i: (C 1)(CM/ +2V), (3.2.5. 3)
. SS SSreg
rдe. с == 2 2' а V == .
SSreg s t Ь dn
834
rосударственная фармакопея Республики Беларусь
Значение t может быть найдено из табли
цы 8.2 при р=О,05 и числе степеней свободы,
равном числу степеней свободы остаточной по
rрешности. Оценка активности (Rr) и COOТBeT
ствующий доверительный интервал вычисляют
путем умножения результатов на величину А7
после антилоrарифмирования. Если выяснится,
что активности исходных растворов, приroтов
ленных исходя из ожидаемой предполаrаемой и
установленной (аттестованной) активностей, не
равны между собой, следует ввести поправоч
ный коэффициент (см. примеры 5.1.2 и 5.1.3).
3.2.6. ПРОПУЩЕННЫЕ РЕЗУЛЬТАТЫ
(ЗНАЧЕНИЯ)
В случае сбалансированноrо количествен
ноro определения существует вероятность слу
чайной потери одноrо или нескольких резуль
татов, абсолютно не связанной с процедурой
количественноro определения, например, вслед
ствие смерти животноrо. Если будет доказано,
что смерть животноro никоим образом не свя
зана с составом введенноro препарата, возмож
ность выполнения точных расчетов остается, но
формулы значительно усложняются и MOryт быть
представлены только в рамках общих линейных
моделей (см. Раздел 7.1). Тем не менее, суще
ствует приблизительный метод, в котором про
стота сбалансированноro плана сохраняется
путем замены потерянноro результата рассчитан
ным значением. Потерю информации учитывают
следующим образом: число степеней свободы
для общей суммы квадратов и для остаточной
поrрешности уменьшают на число потерянных
результатов, а для вычисления отсутствующих
значений используют одну из приведенных ниже
формул. Следует иметь в виду, что этот метод яв
ляется приблизительным, и предпочтение следу
ет отдавать точным расчетам.
Если потеряно более одноro результа
та, MOryT быть использованы эти же формулы.
В этом случае для всех потерянных значений,
кроме одноro, проводят rрубые приближен
ные оценки. Для этоrо значения вычисляют при
помощи соответствующей формулы с учетом
всех имеющихся данных, включая сделанные
первичные предположения. Это рассчитанное
значение включают в общий массив данных и
аналоrичным образом проводят вычисление
значений для первой из rрубых оценок. После
вычисления всех пропущенных значений по
вторяют весь цикл сначала, используя более
точные приближения или рассчитанные значе
ния для каждоro результата, для котороro при
менялась формула. Такой цикл вычислений по
вторяют до тех пор, пока два последовательных
цикла не дадут те же самые значения. Обычно
сходимость достиrается очень быстро.
При условии, что число замененных резуль
татов невепико по отношению к общему числу
данных в эксперименте (например, меньше 5 %),
приближения, используемые при этих заменах,
и уменьшение степеней свободы на число по
терянных данных, замененных таким образом,
обычно являются удовлетворительными. Полу
ченные результаты следует интерпретировать
с большой осторожностью, особенно если име
ются пропущенные результаты в одном виде доз
или блоке. В случае неясностей либо в непред
виденных ситуациях следует проконсультиро
ваться со специалистом по биостатистике. Ka
теroрически недопустима замена пропущенных
результатов в случае испытаний без повторений.
Схема полной рандомизации
В данном случае пропущенное значение
может быть заменено средним арифметическим
всех друrих результатов в пределах одной дозы
(вида воздействия).
Схема рандомизированных блоков
Пропущенное значение заменяют величи
ной у', рассчитанной по формуле (3.2.6. 1):
пB'+kT' G'
y' (п 1)(k 1)' (3.2.6. 1)
rдe:
B' сумма результатов для блока, содержа
щеrо пропущенное значение;
Т' соответствующее общее количество
видов воздействий (доз препаратов);
G' сумма всех результатов (эффектов), по
лученных при количественном определении.
Схема латиНСКО20 квадрата
Пропущенное значение у' рассчитывают по
формуле (З.2.6. 2):
, k(B'+C'+T ) 2G' , (З.2.6. 2)
у (k 1)(k 2)
rдe:
В' и с' суммы результатов, COOTвeTCTBeH
но, в строках и столбцах, содержащих потерян
ные значения. В данном случае k=п.
Перекрестная схема
Если при использовании перекрестной
схемы результат был случайно потерян, следу
ет обратиться к пособию по статистике (напри
мер, к руководству D. J. Fiппеу, см. Раздел 10),
поскольку применение той или иной формулы
зависит от конкретной комбинации видов воз
действий (доз).
3.3. МОДЕЛЬ Уrловых КОЭФФИЦИЕНТОВ
3.3.1. ВВЕДЕНИЕ
Данная модель может при меняться, напри
мер, для некоторых микробиолоrических количе
ственных определений, в которых независимая
переменная представляет собой концентрацию
эссеl-lциальноrо фактора роста для микроорrа
низмов в питательной среде ниже оптимальной
концентрации для роста. На рисунке 3.3.1 А по
казана модель уrловых коэффициентов.
По rоризонтальной оси абсцисс откладыва
ются значения доз, начало оси координат соот
ветствует нулевой дозе; значения доз возрастают
5.3. Статистический анализ результатов биолоzических испытаний
835
слева направо. По вертикальной оси ординат OT
кладываются полученные эффекты. Индивидуаль
ные результаты каждоro исследования показаны в
виде черных точек. Две линии представляют собой
зависимости «доза эффект», рассчитанные для
стандартноro и испытуемоrо препаратов исходя
из предположения, что они пересекаются друr с
друrом в точке, соответствующей нулевой дозе. В
отличие от модели параллельных линий, значения
доз не представляются в виде лоraрифмов.
.
1--
'"
ф
-8
с')
Испытуемый
препарат
Доза (х)
Рисунок З.3.1. I. Модель У2ловых коэффициен
тов для количествеНН020 определения 2х3+ 1
Так же, как и в случае количественных опре
делений, основанных на модели параллельных
линий. важно, чтобы предполаrаемая активность
была как можно ближе к истинной и, если возмож
но, желательно приroтовить разведения испыту
емоro и стандартноro препаратов с одинаковыми
активностями. Чем точнее будет предполаrаемая
активность, тем ближе линии будут располаrать
ся друr к друry. Отношение уrловых коэффици
ентов представляет собой отношение «истинно
ro» значения активности испытуемоro препарата
к еro предполаrаемой активности. Если уroл Ha
клона линии испытуемоro препарата больше уrла
наклона линии стандартноro препарата, это оз
начает, что оценка активности была занижена и
полученная в ходе испытания рассчитанная aK
тивность будет выше предполаrаемой активно
сти. Аналоrично если уroл наклона линии испы
TyeMoro препарата меньше уrла наклона линии
стандартноrо препарата, то активность была за
вышена, и расчеты покажут, что рассчитанная aK
тивность будет ниже предполаrаемой.
При планировании эксперимента все резуль
таты следует проверить на выполнение условий
1. 2 и З, приведенных в Разделе 3.1. Процедура
проведения дисперсионноrо анализа описана в
Разделе 3.3.3, таким образом, он предполаrает
возможность про верки выполнения условий 4В
и 5В, приведенных в Разделе 3.1.
3.3.2. ПЛАН КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ
Использование методов статистическоrо
анализа, приведенных в данном разделе, накла
дывает на методику количественноro определ
ния следующие оrраничения:
а) как стандартный, так и испытуемый пре
параты должны анализироваться при одинако
вом числе равномерно распределенных разве
дений;
Ь) возможно наличие дополнительной
rруппы экспериментальных объектов, которым
не вводят препарат (плацебо rруппа или KOH
трольная rруппа);
с) во всех rруппах должно быть одинаковое
число экспериментальных объектов.
Как уже было отмечено в Разделе 3.1.3,
планы количественных определений, которые
не отвечают требованиям этих оrраничений,
MOryT быть также корректными и обоснованны
ми. Однако описанные здесь простые методы
статистическоro анализа будут в данном случае
неприменимы, и следует либо обратиться за по
мощью к специалисту либо использовать COOT
ветствующее проrраммное обеспечение.
Обычно предпочтительным является ис
пользование плана исследования с двумя
дозами каждоro препарата и одной плацебо
rруппой. Так называемая «(2h+1) точечная схема
с общей нулевой точкой», которая обеспечивает
наибольшую точность наряду с возможностью
проверки достоверности с учетом оrраничений,
упомянутых выше. Однако рассчитанная линей
ность зависимостей не Bcerдa может наблюдать
ся вплоть до нулевой дозы. Если допускается He
большая потеря точности, можно использовать
план, который не предполаrает проведение пла
цебо исследования. В этом случае предпочти
тельно использовать по три дозы каждоro пре
парата, так называемая «(Зh) точечная схема с
общей нулевой точкой». Эти дозы вводятся сле
дующим образом:
1) стандартный препарат вводится в MaK
симальной дозе, которая должна быть близка
к наивысшей дозе, воспроизводящей средний
эффект на линейном отрезке зависимости «дo
за эффект», но не превышать ее:
2) две друrие дозы равномерно распреде
ляются в промежутке между максимальной и HY
левой дозами;
3) испытуемый препарат вводится в COOT
ветствующих дозах, определенных исходя из
предполаrаемой активности вещества.
Возможно использование схем полной paH
домизации, рандомизированных блоков или ла
ТI-1НСКИХ квадратов. описанных в Разделе 3.2.2.
Так же, как и для модели параллельных линий,
использование любой из этих схем требует KOp
ректировки на ошибку суммы квадратов. Ниже
описана схема статистическоrо анализа в случае,
если со стандартным препаратом сравнивается
один или несколько испытуемых препаратов.
836
rосударственная фармакопея Республики Беларусь
3.3.3. ДИСПЕРСИОННЫЙ АНАЛИЗ
3.3.3.1. (hd+1} cxeMa
Результаты проверяют соrласно требова
ниям, описанным в Разделе 3.1 и, если необхо
димо, проводят их преобразование. Затем для
каждоro эксперимента и каждоro препарата BЫ
числяют среднее значение, как описано в табли
це 3.3.3.1 I. Дополнительно рассчитывают cpeд
нее значение для плацебо исследований (В).
В таблицах 3.3.3.1. 1 3.3.3.1. 1II приведен
расчет суммы квадратов для дисперсионноro aHa
лиза. Сумма квадратов, обусловленная нелинейно-
стью, может быть рассчитана только в том случае,
если в план количественною определения было
включено по крайней мере по три дозы каждо
ю из препаратов. Остаточную поrpeшность нахо-
дят путем вычитания вариаций, предусмотренных
планом эксперимента, из общей вариации для pe
зультата исследования (таблица 3.3.3.1. IV).
Далее дисперсионный анализ завершают
следующим образом: каждую сумму квадратов
отклонений делят на соответствующее число
степеней свободы для вычисления дисперсии
(среднею квадрата отклонений). Затем оцени
вают статистическую значимость отношения
дисперсии (среднеro квадрата отклонений) для
каждой переменной к остаточной поrрешности
(S2) (F отношение). Для этою можно воспользо
ваться таблицей 8.1 или соответствующей функ
цией компьютерною проrраммноro обеспечения.
3.3.3.2. (hd) cxeMa
Для этой схемы используются в OCHOB
ном те же самые формулы, что и для (hd+1)
схемы, за исключением некоторых небольших
различий:
переменная В исключается из всех
формул;
K == n(P s +Р Т +..У ;
hd
55 bIaпk исключается из дисперсионноro
анализа;
число степеней свободы для вариаций,
обусловленных rруппами, равно hd 1;
число степеней свободы для остаточной
поrрешности и общей вариации рассчитывают,
как описано для модели параллельных линий
(см. таблицу 3.2.3AV).
Обоснованность (достоверность) количе
ственноro определения, активность и rраницы
доверительноro интервала определяют, как опи
сано в Разделах 3.3.4 и 3.3.5.
3.3.4. ПРОВЕРКА ДОСТОВЕРНОСТИ
Результаты количественноro определения
считаются «статистически обоснованными», если
результаты, полученные при дисперсионном aHa
лизе, удовлетворяют следующим условиям:
1) вариация, обусловленная плацебо в (hd+1)
схеме, не является статистически значимой, т. е.
рассчитанная вероятность не меньше 0,05; это
означает, что результаты, полученные при плаце
бо исследовании, несущественно отличаются от
общей точки пересечения и линейная зависимость
сохраняется вплоть до нулевой дозы;
2) вариация, связанная с точкой пересече
ния, не является значимой, т. е. рассчитанная
вероятность не меньше 0,05; это означает, что
выполняется условие 5В, Раздел З.1;
Таблица 3.3.3.1. '
ФОРМУЛbl для d дозовblХ количествеННblХ определений с использованием модели У2ловblХ
коэффициентов для кажд020 препарата и плацеБО UСПblтания
Стандартный 1-й испытуемый 2 й испытуемый
препарат (S) препарат (7) препарат (и и т. д.)
Среднее значение для 51 Т I U 1
минимальной дозы
Среднее значение для 52 Т 2 U 2
второй дозы
... ... .. . ...
Среднее значение для 5d Td U d
максимальной дозы
Суммарный результат Ps == S,+ S 2+"'+ S d Р Т == + Т 2 +...+ Td
для препарата Ри=='"
Линейное произведение Ls == 1S , + 2S 2 + ... + dSd L T == 1Т, +2Т 2 + ... +dT d Lu == ...
Точка пересечения a s == (4d +2}P s 6Ls а 7 == (4d + 2}Р т 6L7 а и = '"
Уrловой коэффициент b s == 2Ls (d +1}P s Ь Т == 2L7 ( d + 1 }Р 7 Ь и = ...
Значения доз для rрупп G s == S; +... + S G T == Т/ +... + Т; G u = ...
Нелинейность(О) J == G Р; 3b J == G р 7 2 3Ь; J u = '"
S s d d З d 7 7 d d З d
(') Не рассчитывается для двухдозовых количественных определений.
5.3. Статистический анализ результатов биОЛО2ических испытаний
837
Таблица З.3.3.1. 11
Дополнительные формулы для проведения дисперсиОННО20 анализа
nhd 2 nhd Н п a s +а т +... n(B+P s +Р Т +..,/
Н а== К==
в hd 2 hd +4d +2 I 4d З 2d2 2d h(d2 d) hd+1
Формулы для расчета сумм квадратов и степеней свободы
Таблица 3.З.3.1. 111
Источник вариаций Степени свободы (" Сумма квадратов
Реrрессия h SSreg == SStreat SSbIaпk S.s;nt SStiп
Плацебо исследования 1 SSbIaпl< == HB(B a?
Точка пересечения h 1 ssтt ==H,(a +а; +...) h(d2 dYa2)
Нелинейность(*) h(d 2) SS/,п == п( J s + J T + ...)
rруппы hd SSrrear ==п(в 2 +G s +G T +...) K
(') Не рассчитывается для двухдозовых количественных определений
Оценка остаточной П02решности (вариации)
Таблица 3.3.3.1. IV
Источник вариаций Степени Сумма квадратов
свободы
Блоки (строкиi.) п 1 SSbIOCI< ==hd(R +...+R ) K
Столбцы(**) п 1 SSCO/ == hd(C 1 2 + --. + C ) К
I Полная рандомизация (hd+1 )(п 1) SSres == SStot SStreat
Остаточная вариа Рандомизированный блок hd(п 1) SSres == SStot SS/rea/ SSbIoek
ция{*'*)
Латинский квадрат (hd 1 )(п 1) SSres == SS/ot SS/rea/ SSbIOCk SSeo!
Общая вариация nhd+n 1 SStot = L(Y у/
Для схемы латинскою квадрата эти формулы применимы только в случае, если n=hd.
(') Не рассчитывается для схемы ПОлной рандомизации.
П Рассчитывается только для схемы латинских квадратов.
('.') Зависит от схемы рандомизации.
3) в количественных определениях, KOTO
рые включают по крайней мере по три дозы
для каждоro из препаратов, вариация, об
условленная нелинейностью, не является
значимой, т. е. рассчитанная вероятность не
меньше 0,05; это означает, что выполняется
условие 4В, раздел 3.1.
Статистически значимая вариация, обу
словленная проведением плацебо исследо
вания, свидетельствует о том, что предполо
жение о линейности не подтверждается для
диапазона доз возле нулевой точки. Если это
отклонение носит скорее систематический,
чем случайный характер, наиболее подходя
щей является (hd) cxeMa. При этом любые pe
зулыаты, связанные с плацебо исследовани
ями, должны быть исключены.
Если все выполненные статистические ис
следования свидетельствуют о достоверности
106 Зак. 1712
(обоснованности) количественноro определения,
активность и rраницы доверительноro интервала
рассчитываются, как описано в Разделе 3.3.5.
3.3.5. ОЦЕНКА АКТИВНОСТИ И rРАНИЦ
ДОВЕРИТЕльноrо ИНТЕРВАЛА
3.3.5.1. (hd+1)-схеМа
Общая точка пересечения а' для препаратов
может быть вычислена по формуле:
а'= (2d 1)В (2d З)f1а ,(3.3.5.1. 1)
h(2d З) 2d 1
уrловой коэффициент для стандартноro
препарата и, аналоrично, для каждоro из друrих
препаратов, рассчитывают по формуле:
b = 6Ls Зd( d + 1 )а' . (3.3.5.1. 2)
2с1' + Зd 2 + d
838
rосударственная фармакопея Республики Беларусь
Отношение активностей для каждоro из ис
пытуемых препаратов теперь может быть pac
считано следующим образом:
. ЬТ
R T = . (3.3.5.1. 3)
b s
Для определения рассчитанной активности R T
полученное по формуле 3.3.5.1. 3 значение нужно
умножить на предполаrаемую активность А т
испытуемоro препарата. Если интервал между по
следовательными дозами стандартноro и испы
тyeMoro препаратов не был одинаковым, актив
ность следует умножить на величину 'I/r Следует
помнить, что, в отличие от модели параллельных
линий, антилоrарифмы при этом не вычисляются.
Доверительный интервал для переменной R T '
вычисляется по формуле:
. . ! '2 .
CRT K :tV(C 1)(CRT 1)+K'(K 2CRT)' (3.3.5.1 А)
'2
b s '
rдe: С= и К (C 1)V2.
bJ s2t21;.J
V I И V 2 связаны с дисперсией и ковариацией
числителя и знаменателя R T . Их рассчитывают
по формулам:
6 1 3 )
111 = п(2d 1) d (d 1) + 2(2d+1)+hd(d 1) ,(3.З.5.1. 5)
v Зd(d+1)
2 (Зd 1)(d+2)+hd(d 1)' (3.3.5.1. 6)
Доверительный интервал умножается на Be
личину А т и. если необходимо, на величину /I/T'
3.3.5.2. (hd)-схеМа
Для этой схемы используются те же форму
лы, что И для (hd+1 ) cxeMЫ, за исключением сле
дующих изменений:
а' = а. (3.З.5.2. 1 )
v.; 6 ( 1 3 )
1 п(2 d+1) d(d 1) h(d 1) ,(3.3.5.2. 2)
L'2 3d(d + 1) . (3.3.5.2. 3)
(3d + 1 )+ h(d 1 )
3.4. РАСШИРЕННЫЕ НЕЛИНЕЙНЫЕ
(сиrМОИДНЫЕ) КРИВЫЕ ЗАВИСИМОСТИ
«ДОЗА ЭФФЕКТ»
Эта модель подходит, например, дЛЯ KO
личественных определений, выполненных им
мунохимическими методами, коrда требует
ся анализ вытянутых кривых «доза эффект».
Данная модель показана на Рисунке 3.4. 1.
Лоrарифмы доз откладывают на rоризон
тальной оси, начиная слева направо с мини
/у
//8 /"'.
I / /
Стандартный Образе . J./ I
! ,I I
/ / ;естируемый образец
./ .
I' /-:
i . // . //
/ /
L .
Ln дозы (х)
Рисунок З.4. I. Четbfрехпараметрическая
модель Л02истической кривой
мальной концентрации и заканчивая макси
мальной. Величины эффектов откладывают на
вертикальной оси. Индивидуальные эффекты
на каждую дозу указывают в виде черных точек.
Для стандартною и испытуемоro препарата
строят 2 кривые зависимости эффектов от Ha
туральноro лоrарифма дозы «/п(доза) эффект».
Общую форму кривых можно леrко описать
лоrистической функцией, но возможны и друrие
формы кривых. Каждую кривую можно oxapaктe
ризовать 4 параметрами: верхняя асимптота (а),
нижняя асимптота (8), уrловой коэффициент или
коэффициент наклона (13), расстояние между
кривыми (у). Поэтому данная модель часто назы
вается четырехпараметрической. Математиче
ское представление кривой зависимости эффек
тов от натуральноro лоrарифма дозы выrлядит
следующим образом:
a 8
и 8+
1+e P(X Y)
Для достоверноro испытания необходимо,
чтобы кривые стандартноro и испытуемоro пре
паратов имели одинаковый уrловой коэффициент
и максимальный и минимальный уровни эффек
та в крайних частях кривых. У кривых может раз
личэться только положение в пространстве (расстоя
ние между ними) у. rоризонтальное расстоя
ние между кривыми связано с «истинной актив
ностью» испытуемоro препарата. Если испытание
проводится реryлярно, то на стадии разработки
методики достаточно будет проверить условие pa
венства верхнеro и нижнеro уровней эффекта и
затем реryлярно проверять выполнение данною
условия через установленные промежутки BpeMe
ни, а также при изменении используемых матери
алов или условий проведения испытаний.
Оценки максимальною правдоподобия па
раметров и их доверительных интервалов можно
получить, используя соответствующие компью
5. 3. Статистический анализ результатов биолоzических испытаний
839
терные проrраммы. Эти компьютерные проrрам
мы MOryT включать некоторые статистические
функции, обеспечивающие проверку ДOCТOBep
ности полученных результатов. Например, если
оценка максимальноrо правдоподобия показы
вает достоверные отклонения от аппроксими
рованной модели при допустимых условиях pa
венства верхней и нижней асимптот и уrловых
коэффициентов, Torдa одно или все эти условия
возможно не удовлетворяются.
При использовании лоrистической модели
MOryт возникнуть различные статистические пробле
мы, которые возможно потребуют различных реше
ний для различных типов количественных опреде
лений, и простое суммирование результатов будет
неприемлемо. В литературе, посвященной данной
тематике, описано большое разнообразие возмож
ных подходов. Поэтому для данноro типа анализа
рекомендуется воспользоваться профессиональ
ным советом. Тем не менее, в Раздел 5.4 включен
простой пример, чтобы показать «возможный» путь
анализа представленных данных. Краткое 06сужд,е
ние альтернативных подходов и друrих стаТИСТИЧЕ7
ских соображений представлено в Разделе 7.5.
Если нет возможности получить професси
ональный совет или воспользоваться COOTBeT
ствующим проrраммным обеспечением, то име
ются следующие альтернативные подходы:
1) если имеется «приемлемая» оценка
BepxHero (а) и нижнеro пределов (8), то выбира
ют для всех препаратов дозы со средним значе
нием эффекта (и), приблизительно попадающим
в диапазон от 20 % до 80 % максимальной вели
чины пределов, преобразуя эффекты, возникаю
щие при введении выбранных доз, по формуле
у /п ( и д ) , и для анализа используют модель
a и
параллельных линий (см. Раздел 3.2);
2) отбирают диапазон доз, для которых эф
фекты (и) или соответствующим образом пре
образованные эффекты, например. /п(и), явля
ются приближенно линейными при построении
rрафика зависимости эффектов от натураль
Horo лоrарифма дозы; затем для анализа ВОЗ
можно применить модель параллельных линий
(см. Раздел 3.2).
4. КОЛИЧЕСТВЕННЫЕ ОПРЕДЕЛЕНИЯ
С АЛЬТЕРНАТИВНЫМ ТИПОМ ЭФФЕКТА
4.1. ВВЕДЕНИЕ
В ряде случаев невозможно или слишком
трудоемко количественно оценить эффект для
каждоro экспериментальноro объекта. В то же
время, такие результаты, как смерть И!lИ сим
птомы rипоrликемии, MorYT реrистрироваться
только по тому, наступили они или нет у каждоro
из экспериментальных объектов, и результат ис
пытания будет зависеть от количества объектов,
для которых такой эффект наступил. Такой тип
испытания называется количественным опреде
лением с альтернативными (дискретными) pe
зультатами или количественным определением
типа «все или ничеro».
Ситуация при этом в целом очень похожа
на описанную в разделе 3.1, но вместо п инди
видуальных результатов для каждой дозы запи
сывают одно значение, т. е. процент объектов в
каждой испытуемой rруппе, у которых проявил
ся эффект. Если эти результаты представить в
виде rрафика зависимости доли прореаrиро
вавших объектов от лоrарифма дозы, получим
не линейную, а сиrмоидную кривую (s образной
формы). Для анализа сиrмоидной кривой «доза
эффект» применяют математическую функцию,
описывающую данную кривую. Обычно исполь
зуют кумулятивную функцию нормальноro pac
пределения (характеристическую кривую). Эта
функция имеет преимущества с теоретической
точки зрения и. очевидно. является предпочти
тельной, если результат отражает толерант
ность экспериментальных объектов к оказыва
емому воздействию. Если результаты связаны
с процессами роста, предпочтительной являет
ся лоrистическая функция распределения, хотя
различие в результатах, полученных при исполь
зовании этих двух функций, как правило, незна
чительно.
Максимально правдоподобное оценивание
уrловых коэффициентов и расположения кривых
может быть получено только при использовании
итерационных процедур. Существует множество
таких статистических процедур, которые при
водят к одинаковым результатам, но отличают
ся по эффективности вследствие разной CKOpO
сти конверrенции. Одним из наиболее быстрых
методов является метод прямой оптимизации
функции максимальной правдоподобности (см.
Раздел 7.1). Этот метод можно леrко выполнить
при помощи компьютерноro проrраммноro обе
спечения, имеющеrо соответствующую BCTpoeH
ную функцию. К сожалению, большинство этих
процедур не дает оценки rраниц доверительно
ro интервала, а методики их вычисления слиш
ком сложны для рассмотрения в данном разде
ле. Методика, приведенная ниже, не является
самой быстрой, но достаточно проста по cpaBHe
нию с друrими методиками. Эта методика может
при меняться для количественных определений,
в которых один или более испытуемых препара
тов сравниваются со стандартным препаратом.
Кроме этою, должны выполняться следующие
условия:
1) зависимость между лоrарифмом дозы
и полученным эффектом должна быть пред
ставлена в виде кумулятивной кривой HOp
мальноro распределения (характеристическая
кривая);
2) кривые для стандартноro и испытуемо
ro препаратов должны быть параллельны, Т. е.
иметь одинаковую форму, и MOryT различаться
только расположением по оси абсцисс;
3) теоретически не должно наблюдаться
эффекта от введения чрезвычайно малой дозы,
840
rосударственная фармакопея Республики Беларусь
Таблица 4.2.1. 1
Первая рабочая таблица
(1) (2) (3) (4) (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15)
доза п , х р У Ф Z у w шх шу wr шу шху
S
L L L L L L
Т
L L L L L L
ит.д.
и невозможно отсутствие эффекта от введения
чрезвычайно большой дозы.
4.2. МЕТОД ПРОБИТ АНАЛИ3А
Сиrмоидную кривую можно преобразовать в
прямую линию путем замены каждоro эффекта
(т. е. доли объектов, у которых появился эффект)
в rруппе С соответствующими значениями KYMY
лятивноro нормальноro эквивалентноro отклоне
ния от среднеro эффекта. Эти значения, часто
называемые «нормиты»4, теоретически находят
ся в интервале от OO дО +00. Ранее было принято
к каждому «нормиту» прибавлять число 5, таким
образом, получали так называемые «пробиты»5.
Это облеrчало проведение расчетов вручную,
потому что в этом случае исключались отри
цательные результаты. С появлением компью
теров необходимость в прибавлении 5 отпала.
Поэтому метод, описываемый ниже, правиль
нее было бы назвать «метод нормит анализа».
Однако, поскольку термин «метод пробит ана
лиза» широко распространен и используется
в литературе, он сохраняется в данной статье
исходя из исторических соображений.
На первый взrляд кажется, что после лине
аризации результатов следует применять метод
параллельных линий, описанный в Разделе 3.2.
Однако это не так, поскольку для каждой дозы не
выполняется условие однородности дисперсий.
Дисперсия минимальна, если нормит равен нулю
и возрастает как для позитивных, так и для Hera
тивных значений нормита. Следовательно, необ
ходимо придать больший вес значениям в cpeд
ней части кривой и меньший ее крайним частям.
Ниже приведено описание TaKoro метода, а также
процедура дисперсионноrо анализа, оценка aK
тивности и rраниц доверительноro интервала.
4.2.1. подrОТОВКА РАБОЧИХ ТАБЛИЦ
РЕ3УЛЬТАТОВ
В Таблицу 4.2.1. I заносятся данные по столб
цам, номера которых обозначают следующее:
(1) доза стандартноro или испытуемоro пре
парата;
4 от 8нrл. «norтaJity иnit»
5 0т анrл. «probability иnit»
(2) число п объектов, подверrшихся иссле
дованию;
(3) число, объектов, у которых получен по
зитивный эффект в ответ на введение данной
дозы;
(4) лоrарифм Х дозы;
(5) доля позитивных эффектов р=r/п в
rруппе.
Далее начинается первый цикл:
(6) столбец У при первой итерации заполня
ют нулями;
(7) соответствующие значения Ф=Ф( у)
функции кумулятивноro стандартноro (xapaKTe
ристической кривой) распределения (см. также
Таблицу 8.4).
Значения, заносимые в столбцы (8) (10),
вычисляют по формулам:
y212
(8) Z е ,(4.2.1. 1)
v27[
( р ф
9) у у + , (4.2.1. 2)
Z
пz 2
(1 О) {J} ==- 2 . (4.2.1. 3)
ф ф
Значения ШХ, шу, шх 2 , шу2 И шху, заносимые
в столбцы с (11) по (15), можно леrко вычислить
при помощи данных, находящихся в столбцах
(4), (9) и (10), затем вычисляют сумму ( ] этих
значений для каждоro из препаратов.
Полученные результаты суммирования пе
реносят из таблицы 4.2.1. I в столбцы (1 Н6) Ta
блицы 4.2.1. 1I, а значения, заносимые в 6 дo
полнительных столбцов таблицы 4.2.1. 1I с (7) по
(12), вычисляют следующим образом:
2 (I (Ох)2
(7) Sxx I(йX I ,(4.2.1.--4)
(о
(L шх}I соу)
(8) Sxy LШХУ ", -, (4.2.1. 5)
/ ш
2 (Lcoy?
(9) Sxy Lcoy L , (4.2.1. 6)
со
5.3. Статистический анализ результатов биолоzических испытаний
841
Таблица 4.2.1. 1I
Вторая рабочая таблица
(1) (2) (З) (4) (5) (6) (7) (8) (9) (10) (11) (12)
5<0 5roх 5(оу 5roх2 5<оу 2 5roху 5 5ху 5 х у а
хх уу
S
Т
и т. д.
I= I=
I OJХ
(10) Х == IOJ ' (4.2.1. 7)
L OJУ
(11) у== LOJ . (4.2.1. 8)
Теперь можно вычислить общий уrловой KO
эффициент Ь спрямленной характеристической
кривой:
Ь= LSxy . (4.2.1. 9)
LSxx
Точку пересечения а с осью ординат опреде
ляют одинаково как для стандартноro, так и для
испытуемоro препаратов:
(12) 8==y bx. (4.2.1. 1O)
Значения, приведенные в столбце (6) первой
рабочей таблицы, теперь заменяют на значения
У=а+Ьх и повторяют цикл вычислений до тех
пор, пока разница между двумя последователь
ными циклами не станет достаточно малой (Ha
пример, максимальная разница У между двумя
соседними циклами будет меньше 10 B).
4.2.2. ПРОВЕРКА ДОСТОВЕРНОСТИ
Перед тем, как начать вычисление активно
стей и rраниц доверительных интервалов, сле
дует оценить достоверность (обоснованность)
результатов количественноrо определения. Если
для каждоro из препаратов было проведено ис
следование не менее чем 3 доз, то отклонение
от линейности может быть рассчитано следу
ющим образом: к таблице 4.2.1. 1I добавляют
тринадцатый столбец и заполняют значениями:
2
Syy = Sxy . (4.2.2. 1)
Sxx
Суммарное значение данных этоro столбца
является мерой отклонения от линейности и при
близительно подчиняется х2 распределению со
степенями свободы, равными N 2h. Значимость
этой величины можно оценить при помощи Ta
блицы 8.3 или соответствующей функции KOM
пьютерноrо проrраммноro обеспечения. Если
при уровне вероятности 0,05 рассчитанная Be
личина является значимой, результаты количе
107. Зак. 1712
ственноro определения следует отклонить (см.
Раздел 4.2.4).
Если не выявлено значимоro отклонения от
линейной реrрессии, проверяют отклонение от
параллельности при уровне значимости 0,05,
для чеrо рассчитывают значение при числе CTe
пеней свободы. равном h 1:
2 S (ISxy)2
Z L
s 'S .(4.2.2. 2)
хх k.. ХХ
4.2.3. ОЦЕНКА АКТИВНОСТИ И ,РАНИЦ
ДОВЕРИТЕЛЬНО,О ИНТЕРВАЛА
Если нет никаких признаков значительноrо
отклонения от линейности и параллельности, BЫ
числяют натуральный лоraрифм отношения aK
тивностей M по формуле:
. 8т 8S
МТ == . (4.2.3. 1)
ь
Вычисляют антилоrарифм этоro значения.
Далее, приняв значения 1=1,96 и s=1, рассчиты
вают rраницы доверительноro интервала как aH
тилоrарифм значения:
CM (C 1)(>:5 >:т )=\(C 1)(VLSxx+C(M x5 + XTI) (4.2.3. 2)
b2LSxx 1 1
rде: с== ,а v== +
b2LSxx s2t2 LOJ Lcv
s т
4.2.4. НЕДОСТОВЕРНЫЕ
КОЛИЧЕСТВЕННblЕ ОПРЕДЕЛЕНИЯ
Если отклонения от линейности, описанные
в Разделе 4.2.2, являются статистически значи
мыми, результаты количественноro определе
ния обычно признают недостоверными. Если
существуют основания для сохранения резуль
татов количественноro определения, формулы
несколько изменяются. Величину t берут равной
t значению (р=0,05) с тем же числом степеней
свободы, которое использовал ось при провер
ке линейности. Значение s2 берут равным значе
нию Z2, деленному на то же число степеней CBO
боды (обычно это значение больше 1).
Испытание на параллельность также He
сколько видоизменяется. Значение х 2 для непа
раллельности делят на соответствующее число
степеней свободы. Полученное значение делит
ся на рассчитанное выше значение s2, для опре
842
rосударственная фармакопея Республики Беларусь
деления F отношения с h 1 и N 2h степенями
свободы, которое оценивают обычным способом
при уровне значимости 0,05.
4.3. МЕТОД лоrИТ АНАЛИЗА
Как отмечалось в Разделе 4.1, в HeKOTO
рых случаях метод лоrит анализа является
наиболее приемлемым. Название метода про
исходит от названия функции лоrит преобра
зования, которая является обратной функции
лоrистическоrо распределения. Процедура в
этом случае аналоrична той, которая описана
для метода пробит анализа, за исключением
тоro, что видоизменяются две формулы для
расчета значений Ф и Z.
1
Ф == ,(4.3. 1)
1 y
e
e Y
z= у 2. (4.З. 2)
( 1. e )
4.4. друrИЕ ФОРМЫ КРИВОЙ
Для анализа количественных результатов
тестов, предусмотренных Фармакопеей, почти
Bcerдa приемлемыми являются методы пробит
и лоrит анализа. Однако, если форма кривой
зависимости «Iп(доза) эффект» отличается от
форм этих двух кривых, следует взять друrую
зависимость Ф. Z в этом случае берется как
первая производная от Ф.
Например, если выявляется, что кривая
несимметрична, приемлемым может быть pac
пределение rомпертца (Gompertz) (метод rOM
пит анализа). В этом случае Ф == 1 e eY, а
Z == е у е У
4.5. МЕДИАННАЯ ЭФФЕКТИВНАЯ ДОЗА
Для некоторых типов количественных
определений необходимо определить медиан
ную эффективную дозу, т. е. дозу, позитивный
эффект на которую дают 50 % эксперимен
тальных объектов (ЕД50)' Для определения
этой дозы можно применить метод пробит
анализа, но поскольку при этом нет необхо
димости сравнивать эту дозу со стандартным
препаратом, формулы несколько отличаются.
Примечание: стандартный препарат
может эпизодически при меняться при исполь
зовании данноro метода С целью проверки дo
стоверности количественноrо определения.
Обычно результаты количественноro опреде
ления считаются достоверными, если рассчи
танное значение ЕД50 дЛЯ стандартноro пре
парата достаточно близко к аттестованному
значению ЕД50' Значение термина «достаточ
но близко» в данном контексте зависит от Tpe
бований, предъявляемых в конкретной MOHO
rрафии на лекарственный препарат.
Подroтовка рабочих таблиц результатов,
полученных для испытуемых препаратов. и,
при необходимости, для стандартноro, описана
в Разделе 4.2.1. Проверка линейности описана
в Разделе 4.2.2. Проверка параллельности для
данноro типа количественноro определения
не требуется. Значение ЕД50 дЛЯ испытуемоro
препарата Т (и аналоrично для друrих тестиру
емых образцов) рассчитывается, как описано в
Разделе 4.2.3, при этом изменяются формулы
4.2.3. 1 и 4.2.3. 2.
. aT
М Т == , (4.5. 1)
Ь
CM (C 1)xT:t ( C 1)( vl:s xx + C(M + хт)2). (4.5. 2)
rде V= , а значение С остается без изме
неНИЙа (О
5. ПРИМЕРЫ
В данном разделе приведены при меры, ил
люстрирующие применение вышеописанных
формул. Примеры подбирались rлавным обра
зом с целью проиллюстрировать статистиче
ские методы обработки результатов. Их выбор
в каждом случае не означает преимущество
тоro или иноro метода количественноro опреде
ления перед альтернативными методами, KOTO
рые допускаются конкретной частной статьей.
Для тоro, чтобы сделать примеры подходящими
для проверки достоверности работы компьютер
ных проrрамм, в них приводится большее коли
чество десятичных знаков, чем это обычно He
обходимо на практике. Следует также отметить,
что существуют и альтернативные эквивалент
ные методики расчетов. Результаты, полученные
при использовании этих методик, должны быть
точно такими же, как и те, которые приводятся в
данных примерах.
5.1. МОДЕЛЬ ПАРАЛЛЕЛЬНЫХ ЛИНИЙ.
5.1.1. ДВУХДОЗОВОЕ MHOrOKPATHOE
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ С
ИСПОЛЬ30ВАНИЕМ СХЕМЫ ПОЛНОЙ
РА НДОМИ3А ции.
Количественное определение активности
кортикотропина путем подкожной инъекции у
крыс.
Стандартный препарат вводили в дозах от
0,25 до 1,0 ЕД на 100 r массы тела. Предполаrа
лось, что оба испытуемых препарата имели aK
тивность 1 ЕД/мr и вводились в тех же количе
ствах, что и стандартный. Значения эффектов и
средние значения приведены в таблице 5.1.1. 1.
rрафическое представление результатов
(рисунок 5.5.1. 1) не дает поводов сомневаться
в однородности дисперсий и нормальности pac
пределения данных, но возникают некоторые co
мнения относительно параллельности результа
тов для препарата И.
- """'""'Т""" - . .
400
5.3. Статистический анализ результатов биолоzических испытаний
843
.
380
.
.
360 . .
Ьз40 . .
.... .
'"
Q) .
-е-320 .
-е- .
'" .
,,.300 .
1i
280
'"
CI.
260
м
:s:
240 . .
. .
I .
220 . . .
. .
. .
200
81 82 Т1 Т2 U1 U2
180 .
Рисунок 5.1.1. 1
Расчет по формулам, приведенным в табли
цах 3.2.3. ' и 3.2.3. 11, дал следующие результаты:
Ps = 580,4, Ls = 41,8,
Р т = 567,9, = 39,95,
Pu=532,2, Lu= 16,1,
Н = 1 О =5 Н = 120 =20
р 2' L 6 .
Далее про водят дисперсионный анализ по
формулам, приведенным в таблицах 3.2.3. III и
3.2.3. IV. Результаты приведены в таблице 5.1.1. 11.
Анализ подтвердил высокую значимость ли
нейной реrрессии. Отклонение от параллель
ности для препарата и по сравнению со CTaH
дартным препаратом, которое ожидалось на
основании анализа rрафическоro представления
результатов, тем не менее также оказалось зна
чимым (р=0,0075). По этой причине препарат и
исключают и анализ повторяют только для препа
рата Т и стандартноro препарата.
Анализ, проведенный без препарата и, сви
детельствует о соответствии условиям как ли
нейности реrрессии, так и параллельности, что
позволяет перейти к расчету активности. Ис
пользуя формулы, приведенные в Разделе 3.2.5,
получаем следующие результаты:
Таблица 5.1.1. I
Измеряемый эффект у масса аскорбиновой кислоты (М2) на 1002 надпочечника
Стандартный препарат 5 Испытуемый препарат Т Испытуемый препарат U
5, 50 Т Т, и, и о
300 289 310 230 250 236
310 221 290 210 268 213
330 267 360 280 273 283
290 236 341 261 240 269
364 250 321 241 307 251
328 231 370 290 270 294
390 229 303 223 317 223
360 269 334 254 312 250
З42 233 295 216 320 216
306 259 315 235 265 265
Среднее 332,0 248,4 323,9 244,0 282,2 250,0
Дисперсионный анализ
Таблица 5.1.1. "
Источник вариации Степени Сумма квадратов Дисперсия (средний F-отношение Вероят i
свободы отклонений квадрат отклонения) ность
Препараты 2 6256,6 3128,3
Линейная реrрессия 1 63830,8 63 830,8 83,38 I 0,000
Непараллельность 2 8218,2 4109,1 5.37 i 0,007
rруппы (дозы) 5 78 305,7 i
Остаточная вариация 54 41 340,9 765,57 i
Общая вариация 59 119646,6
Дисперсионный анализ без препарата и
Таблица 5.1.1. 111
Источник вариации Степени Сумма квадратов Дисперсия (средний I f:.aтнowe Вероят-
свободы отклонений ) ние
квцдратотклонении . ность
Препараты 1 390,6 390.6 I
Линейная Реrрессия 1 66 830,6 66 830.6 90,5 0,000
Непараллельность 1 34,2 34.2 I 0,05 0,831
rруппы (дозы) 3 67 255,5
Остаточная вариация 36 26587,3 738,54
Общая вариация 39 93 842,8 i
844
rосударственная фармакопея Республики Беларусь
общий уrловой коэффициент:
b 20( 41,8 39,95) 58,970:
/п4.10.2 .
натуральный лоrарифм отношения актив
ностей:
м' 567 ,9 580,4 o 1060
т 2. ( 58,970) , ,
c 66830.6 1.o476,
66830,6 738,54 . 2,0282
v = 668 o 9609'
( 58.970)2'2'10 ' ,
натуральный лоrарифм доверительных
интервалов:
1,0476' 0,1060= \ 0.0476' ('.0476' 0.10602 т 2. 0,9609)= О.1110:!: 0.3034.
Вычислив антилоrарифм, мы найдем OTHO
шение активностей. равное 1.11 при 95 % ДOBe
рительном интервале от 0,82 до 1,51.
Умножение на предполаrаемую активность
препарата Тдаетистиннуюактивность 1,11 EДlMr
при 95 % доверительном интервале от 0,82 до
1,51 EДlMr.
5.1.2. ТРЕХДОЗОВОЕ КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ С ИСПОЛЬ30ВАНИЕМ
СХЕМЫ ЛАТИНСКИХ КВАДРАТОВ
Количественное определение aKтивHO
сти антибиотика методом диффузии в а2ар
с использованием ПРЯМОУ20ЛЬН020 планшета.
Стандартный препарат имел YCTaHOB
ленную (аттестованную) активность, равную
4855 ME/Mr. Предполаrаемая активность ис
пытуемоro препарата принималась равной
5600 ME/Mr. Исходные растворы roтовились
следующим образом: 25,2 Mr стандартноro
препарата растворяли в 24,5 мл растворите
ля и 21,4 Mr испытуемоro препарата paCTBO
ряли в 23,95 мл растворителя. Для анализа
исходные растворы сначала разводили в co
отношении 1/20, а затем, используя степень
разведения 1,5.
Латинские квадраты строились при
помощи метода, описанноrо в Разделе 8.6
(см. таблицу 5.1.2. 1). Результаты, полученные
в ходе данноro количественноro определе
ния, приведены в таблице 5.1.2. 11 (зоны инrи
бирования роста в мм х1 О). Средние значе
ния для rрупп приведеl-!Ы в таблице 5.1.2. 111.
rрафическое представление данных (см. Ри
сунок 5.1.2. 1) не дает поводов сомневаться в
нормальности распределения и OДHOpOДHO
сти дисперсий.
Используя формулы, приведенные в табли
цах 3.2.3. 1 и 3.2.3. 11, получаем следующие pe
зультаты:
Ps = 529,667,
Р т = 526,333,
6
Нр = 3 = 2,
Ls = 35,833,
L T = 39,З33,
72
H L = 24 = 3.
Далее проводят дисперсионный анализ по
формулам, приведенным в таблицах 3.2.3. III и
3.2.3. IV. Результаты приведены в таблице 5.1.2. IV.
210
0200
::<
90
'"
s:
I
!\J
2518о.....
cl
s:
ID
s:
170
:;;
I
О
C')160
15о..... .
.
81
140
.
.
.
.
.
.
83
82
U1
U2
UЗ
Рисунок 5.1.2. 1.
Анализ показывает значительные различия
между строками. Это означает, что использова
ние схемы латинских квадратов обеспечивает в
данном случае более высокую точность по cpaB
нению со схемой полной рандомизации. Высокая
значимость реrрессии, а также отсутствие значи
Moro отклонения индивидуальных линий perpec
сии от параллельности и линейности подтвержда
ют, что результаты количественною определения
приюдны для расчета активности.
Используя формулы, приведенные в Разде
ле 3.2.5, получаем следующие результаты.
общий уrловой коэффициент:
ь з . (З 5,ВЗ З +З9,ЗЗЗ) 46,З46,
/п(1,5) . 6 . 2
натуральный лоrарифм отношения актив
ностей:
м' 526 ,ззз 5 29 ,667 o 02З974 ,
Тз. 46,346 '
c В475 ,0417 1,010B ,
В475,0417 20,7667 . 2,ОВ6 2
v 475,0417 02192;
46,3462 . 3 . 6 '
натуральный лоrарифм доверительных
интервалов:
1.0108 х ( 0.0240)" \ 0.0108 х ( ОЩ40)< + 2. 0.2192 = 0,02423 с 0,06878 .
5.3. Статистический анализ результатов биолоzических испытаний
845
Таблица 5.1.2. I
1
2
3 Т 1
4 Т
5 $1
6 $, Т 1 $2
Таблица 5.1.2А/
ИзмереННblе ЗОНbI иН2ибирования роста в ммх 1 О
1
2
3
4
5
6
Среднее по
строкам
175,2 = Rl
171,2 = Rz
172,7 = R з
178,5 = R.
1П,5=R s
181,0= R б
1
2
3
4
5
6
Среднее
столбцов
161 160 178 187 171 194
151 192 150 172 170 192
162 195 174 161 193 151
194 184 199 160 163 171
176 181 201 202 154 151
193 166 161 186 198 182
172,8
= С 1
179,7
= С 2
177,2
= С З
178,0
= С 4
174,8
= С 5
173,5
=С 6
Таблица 5.1.2. 1I1
Средние значения
Стандартный препарат 5 Испытуемый препарат Т
51 52 5з Т , Т 2 Т3
Среднее 158,67 176,50 194,50 156,17 174,67 195,50
Таблица 5.1.2. IV
Дисперсионный анализ
Степе- Сумма квадра- Дисперсия (сред- Вероят-
Источник вариации ни сво- ний квадрат от- F-отношение
боды тов отклонений клонения) ность
Препараты 1 11,1111 11,1111
Линейная реrрессия 1 8475,0417 8475,0417 408,1 0,000
Непараллельность 1 18,3750 18,3750 0,885 0,358
Нелинейность 2 5,4 722 2,7361 0,132 0,877
rруппы (дозы) 5 8510
Строки 5 412 82,40 3,968 0,012
Столбцы 5 218,6667 43,73 2,106 0,107
Остаточная вариация 20 415,3333 20,7667
Общая вариация 35 9556
Таблица 5.1.3. I
Стандартный препарат 5 Испытуемый препарат Т Среднее
Блок 5. 5, 5, 5d Т. Т, Т, Td
1 252 207 168 113 242 206 146 115 181,1
2 249 201 187 107 236 197 153 102 179,0
3 247 193 162 111 246 197 148 104 176,0
4 250 207 155 108 231 191 159 106 175,9
5 235 207 140 98 232 186 146 95 167,4
Среднее 246,6 203,0 162,4 107,4 237,4 195,4 150,4 104,4
Показатель оптической плотности суспензий (х1000)
108. За.. 1712.
846
rосударственная фармакопея Республики Беларусь
Вычислив антилоraрифм, мы найдем отноше
ние активностей, равное 0,9763 при 95 % довери
тельном интервале от 0,9112 до 1,0456.
Поскольку разведения, приroтовленные
исходя из предполаrаемой активности, не были
точно эквивалентными, следует ввести попра
4855 . 25,2 /245
вочныи коэффициент ' O,99799.
5600.21,4/23,95
Перемножая данный коэффициент и предпо
лаrаемую активность препарата 5600 ME/Mr,
получаем рассчитанную активность 5456 МЕ/
Mr при 95 % доверительном интервале от 5092
до 5843 ME/Mr.
5.1.3. ЧЕТЫРЕХДОЗОВАЯ СХЕМА
РАНДОМИЗИРОВАННЫХ БЛОКОВ
Количественное определение aKтивHO
сти антибиотика турбидиметрическим Me
тодом
Данное количественное определение
проводят с целью установления активности
антибиотика в международных единицах на
флакон. Стандартный препарат имеет YCTa
новленную активность 670 ME/Mr. Предпо
лаrаемая активность испытуемоrо препара
та принимается равной 20 000 ЕД/флакон.
Исходя из этих данных, исходные растворы
roтовились следующим образом: 16,7 Mr CTaH
дартноrо препарата растворяли в 25 мл pac
творителя, а содержимое одноro флакона ис
пытуемоrо препарата растворяли в 40 мл
растворителя. Для анализа исходные paCTBO
ры сначала разводили в соотношении 1/40,
а затем, используя степень разведения 1,5.
Пробирки размещали в водяной бане в co
ответствии со схемой рандомизированноrо
блока (см. Раздел 8.5). Полученные результа
ты приведены в таблице 5.1.3. 1.
Анализ рисунка 5.1.3. 1 не дает оснований
сомневаться всправедливостипредположения
онормальности распределенияданныхиоднород
ности дисперсий. Стандартное отклонение S3
несколько велико, но не является причиной
для беспокойства.
Используя формулы, приведенные в Ta
блицах 3.2.3. I и 3.2.3. 11, получаем следую
щие результаты:
275
225
ф
:s;
I
20
о
t;
L..
175
ф
о
15
Q)
:F
:s;
1: 125
о
100
75
S1
50
S2
Рисунок 5.1.3. 1.
Ps = 719,4,
Р 7 = 687,6,
5
Н р ="4 = 1,25,
.
.
.
.
.
83 ; U1
U2
UЗ
Ls = 229,1,
L T = 222,
60
H L = 60 = 1.
Далее проводим дисперсионный анализ по
формулам, приведенным в таблицах 3.2.3. "1 и
3.2.3. IV. Полученные результаты приведены в
таблице 5.1.3. 11.
Следует отметить значительное различие
между блоками. Это означает, что применение
схемы рандомизированных блоков позволяет
добиться более высокой точности оценки. Bы
сокая значимость реrрессии, а также отсутствие
значимоro отклонения от параллельности и ли
нейности подтверждают, что результаты количе
ственноro определения приroдны для расчета
активности.
Используя формулы, приведенные в Разде
ле 3.2.5, получаем следующие результаты:
общий уrловой коэффициент:
Ь= 1. ( 229,1 222) = 111255.
'п (1,5) . 5 . 2 "
Дисперсионный анализ
Таблица 5.1.3. "
Источник вариации Степе Сумма квадра- Дисперсия F-отношение Вероят-
ни CBO тов отклонений (средний квадрат ность
боды отклонения)
Препараты 1 632,025 632,025
Линейная реrрессия 1 101 745,6 101 745,6 1887,1 0,000
Непараллельность 1 25,205 25,205 0,467 0,500
Нелинейность 4 259,14 64,785 1,202 О,3З2
rруппы 7 102662
Блоки 4 876,75 219,188 4,065 0,010
Остаточная вариация 28 1509,65 53,916
Общая вариация 39 105048,4
5.3. Статистический анализ результатов бuолоzuческих испытаний
847
натуральный лоrарифм отношения актив
настей:
м = 687.6 719,4 0071457
т 4.( 111,255)' ,
с= 101745,6 =100223
101745.6 5З,916' 2,0482' ,
v = 101745,6 О 4110'
( 111.255)2 .4.5 . ,
натуральный лоrарифм доверительных
интервалов:
1,00223' 0,0715:t .J 0,00223 .0,07152 + 2' 0,4110 =
= 0,07162 :t 0,04293.
Вычислив антилоraрифм, мы найдем отноше
ние активностей, равное 1,0741 при 95 % довери
тельном интервале от 1,0291 до 1,1214. Поскольку
разведения, приroтовленные исходя из предпола
raемой активности, не были точно эквиактивны
ми, следует ввести поправочный коэффициент
670 . 16 7/ 25
, = 0,89512. Перемножив данныи коэф
20000 . 1/40
фициент и предполаrаемую активность препарата
20 000 МElфлакон, получаем рассчитанную актив
ность 19 228 МЕ/флакон при 95 % доверительном
интервале от 18423 до 20 075 МЕ/флакон.
5.1.4. ПЯТИДОЗОВОЕ MHOrOKPATHOE
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
С ИСПОЛЬ30ВАНИЕМ СХЕМЫ ПОЛНОЙ
РАНДОМИ3АЦИИ
Количественное определение iп vitro aK
тивности трех вакцин против rепатита В
по сравнению со стандартным препаратом.
Для каждой из вакцин подroтавливали по
три независимые серии разведен ий, каждая из
которых состоит из пяти двукратных разведе
ний. После определенных подrотовительных
операций, предусмотренных методикой коли
051 I I
1. i
001 . 1.
i
i I
0.5-r i
:I ; I
ф I
:r i
i
1.o-: I
L. !
О :
1: I
151
u
ф
,.
2.O-
1:
О
2.
зо-
S т u
З5
v
Рисунок 5.1.4. 1.
чественноrо определения, измеряли оптиче
ские плотности растворов. Результаты приве
дены в таблице 5.1.4. 1.
Как известно, натуральные лоrарифмы
оптических плотностей раствора имеют ли
нейную зависимость от лоrарифмов доз pac
TBopeHHoro вещества. Средние результаты ло
rарифмическоrо преобразования оптических
плотностей приведены в таблице 5.1.4. 1I. rpa
фическое представление результатов не выя
вило каких либо необычных закономерностей
расположения данных (см. рисунок 5.1.4. 1).
Используя формулы, приведенные в табли
цах З.2.3. 1 и 3.2.3. 1I, получаем следующие pe
зультаты:
Ps = 9,108,
Р т = 5,586,
Р u = 6,544,
Pv = 6,027,
з
Нр = '5= 0,6,
Ls = 6,109,
L T = 6,264,
Lu = 6,431,
Lv = 6,384,
36
H L = 120 = 0,3.
Оптические плотности растворов вакцин
Таблица 5.1.4. I
Разведение Стандартный препарат S Испытуемый препарат Т
1:16000 0,043 0,045 0,051 0,097 0,097 0,094
1:8 000 0,093 0,099 0,082 0,167 0,157 0,178
1:4 000 0,159 0,154 0,166 0,327 0,355 0,345
1:2 000 0,283 0,295 0,362 0,501 0,665 0,576
1 : 1 000 0,514 0,531 0,545 1,140 1,386 1,051
Разведение Испытуемый препарат и Испытуемый препарат V
1 : 16 000 0,086 0,071 0,073 0,082 0,082 0,086
1 :8 000 0,127 0,146 0,133 0,145 0,144 0,173
1 :4 000 0,277 0,268 0,269 0,318 0,306 0,316
1 :2 000 0,586 0,489 0,546 0,552 0,551 0,624
1:1 000 0,957 0,866 1,045 1,037 1,039 1,068
848
rосударственная фармакопея Республики Беларусь
Далее проводим дисперсионный анализ по
формулам, приведенным в таблицах 3.2.З. 1I1
и 3.2.3. IV. Полученные результаты приведены
в таблице 5.1.4. I/I.
Высокая значимость линейной реrрессии,
а также отсутствие значимоrо отклонения от
параллельности и линейности подтверждают,
что результаты данноro количественноro опре
деления приrодны для 'расчета активности.
Используя формулы, приведенные в Разделе
3.2.5, получаем следующие результаты:
общий уrловой коэффициент:
b 0,3. (6,109+ 6264 6,431+ 6,384) 0,90848;
/п 2 . 3 . 4
натуральный лоrарифм отношения актив
ностей:
м' 5,586 ( 9,108) 07752
т 5' 0,90848 '
c 47,58 1,00057'
47 ,58 0,0067 . 2,0212 '
V = 47 ,58 3 8436'
О 90852 . 5 . 3 ' ,
натуральный лоrарифм доверительных
интервалов для испытуемой вакцины Т:
1,00057 х 0,7752 :": 'Ii 0,00057 х (1,00057 х 0,77522 + 2 х 3,8436) =
= О,7756:!: 0,0689.
Вычислив антилоrарифм, мы найдем OT
ношение активностей вакцин, равное 2,171
при 95 % доверительном интервале от 2,027
до 2,327. Все образцы имеют установленную
(аттестованную) активность 20 MKr протеина/
мл и, следовательно, найденная активность
испытуемоro препарата Т равна 43,4 MKr про
теина/мл при 95 % доверительном интервале
от 40,5 до 46,5 MKr протеина/мл.
Аналоrичным образом вычисляют актив
ность и доверительные интервалы для друrих
испытуемых препаратов. Полученные резуль
таты приведены в таблице 5.1.4. IV.
Таблица 5.1.4. IV
Окончательные результаты оценки aKтивHO
стей испытуемых вакцин (МК2 протеина/мл)
и 95 % доверительные интервалы
Нижняя Оценка Верхняя
rраница rраница
Вакцина Т 40,5 43,4 46,5
Вакцина и 32,9 35,2 37,6
Вакцина V 36,8 39,4 42,2
5.1.5. ДВОЙНАЯ ПЕРЕКРЕСТНАЯ СХЕМА
Количественное определение aKтивHO
сти инсулина путем подкожной инъекции у
кроликов
Стандартный препарат вводился в дозах
1 и 2 ЕД/мл. Эквивалентные дозы испытуе
Moro препарата неизвестной активности Ha
значались исходя из предполаrаемой актив
ности 40 ЕДlмл. Кроликам подкожно вводили
0,5 мл соответствующих растворов в COOT
ветствии со схемой, приведенной в табли
це 5.1.5. 1. Полученные результаты приве
дены в таблице 5.1.5. 11 и на рисунке 5.1.5 1.
Высокое значение дисперсии свидетельствует
о наличии статистически значимых различий
между кроликами и необходимости использо
вать перекрестную схему исследования.
Таблица 5.1.4. 11
Средние значения натуральных П02арифмов оптических плотностей растворов
SI 3,075 Т I 2,344 И I 2,572 V I 2,485
S2 2,396 Т 2 1,789 И 2 2,002 V 2 1,874
SЗ 1,835 Тз 1,073 ИЗ 1,305 V З 1,161
S4 1,166 Т4 0,550 И 4 0,618 V 4 0,554
S5 0,635 Т. 0,169 И 5 0,048 V 5 0,047
Дисперсионный анализ
Таблица 5.1.4. 111
Степени Сумма квадра- Дисперсия (cpeд Вероят
Источник вариации ний квадрат OT F-отношение
свободы тов отклонений клонений) ность
Препараты 3 4,475 1,492
Линейная реrрессия 1 47,58 47,58 7126 0,000
Непараллельность 3 0,0187 0,006 0,933 0,434
Нелинейность 12 0,0742 0,006 0,926 0,531
rруппы 19 52,152
Остаточная вариация 40 0,267 0,0067
Общая вариация 59 52,42
5.3. Статистический анализ результатов биОЛО2ических испытаний
849
Таблица 5.1.5. 1
Планирование исследования
rруппа кроликов
1 2 3 4
День 1 8, 80 Т, Т?
День 2 Т? Т, 80 8,
1БО .
.
.
140
I .
120 . . .
I .
. . .
100 .
::!'
о . I
о
80
. . . .
'" I .
,. . .
60 . . .
.... . .
о .
I
40 .
.
.
20
S1 S2 Т1 Т2
о
Рисунок 5.1.5. '
Для рассматриваемой схемы количествен
Horo определения применение дисперсионно
ro анализа является более сложным, чем для
рассмотренных ранее схем, потому что KOM
понент вариации, обусловленный параллель
ностью, не является независимым от KOM
понента, обусловленноro различием между
кроликами. Поэтому проверка параллельно
сти линий реrрессий требует дополнительноro
введения BTOpOro поправочноro коэффициен
та для ошибки среднеro квадрата отклонений
(дисперсии), который вычисляется путем вычи
тания компонента параллельности и пар «вза
имодействующих» компонентов из компонента,
связанноrо с различием между кроликами.
Засчетповторенийвкаждойизrруппвдиспер
сионном анализе присутствуют три пары «взаи
модействующих» компонентов: днихпрепараты;
дни хреrрессия; дни хпараллельность.
Эти коэффициенты характеризуют TeH
денцию компонентов (препараты, реrрессия
и параллельность) изменяться в разные дни
испытаний (в серии «изо дня в день»). Таким
образом, соответствующие F отношения обе
спечивают проверку этих компонентов оценки
достоверности количественноro определения.
Если статистическая значимость полученных
оценок F отношений является высокой, сле
дует с большой осторожностью интерпретиро
вать результаты количественноro определения
и, если это возможно, необходимо повторить
количественное определение активности инсу
лина.
Дисперсионный анализ проводит
ся с использованием формул, приведенных в
Таблица 5.1.5. 1I
Эффект у: сумма результатов содержания 2ЛЮКОЗЫ в крови (М2I1О0 мл) через 1 час и 2,5 часа
rpynna 1 rpynna 2 rpynna 3 rpynna 4
51 Т 2 52 Т 1 Т 1 52 Т 2 51
112 104 65 72 105 91 118 144
126 112 116 160 83 67 119 149
62 58 73 72 125 67 42 51
86 63 47 93 56 45 64 107
52 53 88 113 92 84 93 117
110 113 63 71 101 56 7З 128
116 91 50 65 66 55 39 87
101 68 55 100 91 68 31 71
Среднее 95,6 82,8 69,6 93,3 89,9 66,6 72,4 106,8
Дисперсионный анализ
Таблица 5.1.5. 111
Источник вариации Степени Сумма Дисперсия (средний F OTHO- Вероят
свободы квадратов квадрат отклонений) шение ность
Непараллельность 1 1453,5 1453,5 1.064 0,311
Днихпрепарат 1 31,6 31,6 I 0.023 0,880
Днихреrрессия 1 50,8 50,8 0.037 0,849
Остаточная вариация между 28 38 258,8 1366,4 I
rPvппами кроликов
Кролики 31 39794,7 1283,7
Препараты 1 0,14 0,14 0,001 0,975
Реrрессия 1 8859,5 8859.5 64,532 0,000
Дни 1 478,5 478.5 3,485 0,072
Днихнепараллельность 1 446,3 446,3 3,251 0,082
Остаточная вариация внутри 28 3844,1 I 137.3
rPVПП кроликов I
Общая вариация 63 53423.2 i
850
rосударственная фармакопея Республики Беларусь
таблицах 3.2.3А 3.2.3. III, отдельно как для ка>IЩО-
ro дня, так и для объединенноro набора данных. Ис
пользуя формулы, приведенные в таблицах З.2.З. 1
и 3.2.3AI, получаем следующие результаты:
День 1:
Ps = 165,25, Ls = 13,
Р Т = 162,25, L7 = 8,75,
8 96
Нр = =4 H L = =16.
2 6
Ps = 173,38, Ls = 20,06,
Р Т = 176,00, L T = 5,25,
8 96
Н р = =4, H L = =16.
2 6
Ps = 196,31. Ls = 16,53,
Р7 = 169,13, L T = 7,00,
16 192
Н Р =2=8' H L =6=32.
День 2:
Объединенные
данные:
Используя формулы, приведенные в табли
це 3.2.3. 1II, получаем следующие результаты:
День 1 День 2 Объединенные
данные
SS = 18,000 SS = 13,781 SS = 0,141
prep ртер ртер
SS = 3784,5 SS = 5125,8 SS = 8859,5
reg reg reg
SSPд' = 144,5 SSpar = 1755,3 SSPд' = 1453,5
Коэффициенты взаимодействия рассчи
тывают как: «День 1 +День2 Объединенные
данные» .
SS = 31 64
dауsч)( р t 1
SS = 50 77
aays-reg "
SS d = 446,27_
ays"pa r
Дополнительно, рассчитывают сумму KBa
дратов отклонений вследствие вариации, об
условленной разными днями испытаний (в
серии «изо дня в день»):
1 2 2
SSdays ='jN(D 1 02) K=478,52
и сумму квадратов отклонений вследствие
вариаций в блоках (различие между кроликами):
SSbIock = 2IBJ K= 39794,7,
rдe В; среднее значение наблюдаемоro
эффекта в пересчете на одноro кролика.
Далее про водят дисперсионный анализ, pe
зультаты котороro приведены в таблице 5.1.5. 1II.
ДисперсионныЙ анализ подтверждает, что по
лученные данные удовлетворяют необходимым
условиям достоверности оценки активности инсу
лина: высокая статистическая значимость линей
ной реrрессии, отсутствие статистически значимых
отклонений от параллельности инезначимость
всех трех коэффициентов взаимодействия.
Используя формулы, приведенные в Разде
ле 3.2.5, получаем следующие результаты:
общий уrловой коэффициент:
ь = 32' ( 16,5З 7) = 33.95;
'п2 . 16 . 2
натуральный лоrарифм отношения актив
ностей:
м' = 169,13 169,З1 =000276
т 2. ( З3,95) . ,
с= 8859.5 2 =1,0695,
8859,5 1З7,З' 2,048
v = 8859,5 0,2402'
( зз,95)2. 2 '16 '
натуральный лоrарифм доверительноro
интервала:
1,0695 . О,ОО276:!: 0.0695 . (1,0695 . 0,002762 + 2 . 0,2402) =
= О,ОО295:!: 0,18279.
Вычислив антилоrарифм, мы найдем OTHO
шение активностей, равное 1,003 при 95 % ДOBe
рительном интервале от 0,835 до 1,204.
Умножив на А т =40, получим активность
40,1 единиц на миллилитр при 95 % довери
тельном интервале от 33,4 до 48,2 единиц на
миллилитр.
5.2. МОДЕЛЬ уrловых КОЭФФИЦИЕНТОВ
5.2.1. СХЕМА ПОЛНОЙ РАНДОМИЗАЦИИ
(0,3,3)
Количественное определение активности
фактора VIII.
В лаборатории выполняют количественное
определение активности фактора VIII в KOHцeH
тратах по образованию окрашенноro продукта.
Предположим, что лаборатория не имеет опыта
в проведении количественноro определения
подобноro рода, но, тем не менее, попыталась
выполнить эту методику. rотовят по три экви
валентных разведения для стандартноro и ис
пытуемоro препаратов. Дополнительно roтовят
препарат «плацебо», хотя и не ожидается нали
чия линейной зависимости эффекта от дозы в
области малых доз. Число повторений для каж
доro из разведений равно восьми, что несколь
ко больше, чем требуется при выполнении по
вседневных количественных определений.
rрафическое представление данных (рис.
5.2.1. 1) показывает, что зависимость результа
та от дозы действительно нелинейна в области
малых доз. На этом основании результаты, по
лученные при анализе препарата «плацебо», не
будут использоваться при расчетах (в дальней
шем для обоснования данноro решения, безус
ловно, нужно будет провести повторные опре
5.3. Статистический анализ результатов биолоzических испытаний
851
0.З50
sз
О.зоо
J) 0.250
<.J
о
:r
Ь
с; 0.200
<::
'"
IC
0.150
:т
:s:
....
<::
00.100
0.050
в
0.000
Рисунок 5.2.1. 1.
деления с препаратом «плацебо»). Используя
формулы, приведенные в таблицах 3.3.3.1 1 и
3.3.3.1. \I, получаем следующие результаты:
Ps = 0,6524
Ls = 1,4693
a s = 0,318
b s = 0,329
G s = 0,1554
J s = 4, 17x10 B
И
Н, = 0,09524
Рт= 0,5651
L T = 1 ,2656
а/ = 0,318
Ь 7 = 0,271
G r = 0,1156
J r = 2,84 х 10- 6
а' = 0,05298
к = 1,9764.
Далее выполняют дисперсионный анализ по
формулам, приведенным в таблицах 3.3.3.1 AII и
3.3.3.1. IV.
Высокая статистическая значимость perpec
сии и отсутствие значимых отклонений от линей
ности и точки пересечения с осью ординат пока
зывают достоверность данных и дают основание
для расчета активности.
Уrловой коэффициент стандартноro
препарата:
b = 6 '1,469 36' 0,0530 =0.0822.
84
Уrловой коэффициент испытуемоro препа
рата:
ь' = 6 . 1.266 36. 0,0530 = 00677.
т 84 '
По формуле 3.3.5.1. 3 получаем:
R = о ,0677 ==о 823
0,0822 ' ,
2
с= 0,0822 =1,000083,
0,08222 З,86.10 6 - 2,0182 . 0,0357
к' = 0,000083 . 0,75 = 0,000062,
Показатели оптической плотности
Таблица 5.2.1. 1
Плацебо Стандартный препарат 5 Испытуемый препарат Т
(в МЕ/мл) (в МЕ/мл)
KOHцeHTpa В 51 52 5 з Т 1 Т 2 Тз
ция 0,01 0,02 0,03 0,01 0,02 0,03
0,022 0,133 0,215 0,299 0,120 0,188 0,254
0,024 0,133 0,215 0,299 0.119 0,188 0,253
0,024 0,131 0,216 0,299 0,118 0,190 0,255
0,026 0,136 0,218 0,297 0,120 0,190 0,258
0,023 0,137 0,220 0,297 0,120 0,190 0,257
0,022 0,136 0,220 0,305 0,121 0,191 0,257
0,022 0,138 0,219 0,299 0,121 0,191 0,255
0,023 0,137 0,218 0,302 0,121 0,190 0.254
Среднее 0,0235 0,1351 0,2176 0,2996 0,1200 0,1898 0,2554
Дисперсионный анализ
Таблица 5.2.1. 11
Степени Сумма KBaдpa Дисперсия(сред Вероят
Источник вариации свобо тов отклонений ний квадрат OT F отноwение ность
ды клонения)
Линейная реrреССИ!=l 2 0,1917 0,0958 I 24 850 0,000
Т очки пересечения 1 3.10 9 3'1 0 9 7-1 0 4 0,978
Нелинейность 2 2-10 5 1-1 0 5 2,984 0,061
rруппы 5 0,1917
Остаточная вариация 42 1 ,62.10 4 3,86-10 6
Общая вариация 47 0,1919
852
rосударственная фармакопея Республики Беларусь
а 95 % доверительный интервал равен
0823:!: J O.000083 х 1,678 + 0,000062 х ( 1,646) = О,823:!: 0,006.
Таким образом, полученная активность
равна 0,823 с 95 % доверительным интервалом
от 0,817 до 0,829.
5.2.2. СХЕМА ПОЛНОЙ РАНДОМИ3АЦИИ
(0,4,4,4)
Количественное определение активности
вакцины против 2риппа iп vitro.
Содержание антиrена reмаrлютинина (НА) в
двух вакцинах против rpилпа определяли методом
радиальной иммунодиффузии. На этикетках обеих
вакцин была указана активность 15 Mкr НА в одной
дозе, что эквивалентно содержанию 30 MKr НAlмл.
Стандартный препарат имел установленную (aттe
стованную) активность 39 MKr НAlмл.
Исследовали четыре концентрации стандарт
ной и испытуемой вакцин, рассчитанных исходя из
предполаraемых и обозначенных на этикетке актив
ностей; в Ka>t<дOM случае число повторений равно
двум. После установления равновесия М€>fЩУ BHY
тренним и внешним реаrентами измерялась пло
щадь кольцевых зон преципитации. Результаты
приведены в таблице 5.2.2. 1.
Таблица 5.2.2. '
Площадь кольцевых зон преципитации (мм 2 )
KOH CTaндapT Испыту Испыту
цен- ный препа емый пре емый "ре-
тра- рат 5 парат Т парат U
ция
(мкrfмn) I 11 I 11 1 11
7,5 18,0 18,0 15,1 16,8 15,4 15,7
15,0 22,8 24,5 23,1 24,2 20,2 18,6
22,5 30,4 30,4 28,9 27,4 24,2 23,1
30,0 35,7 36,6 34,4 37,8 27,4 27,0
r рафическое представление данных не BЫ
явило никаких необычных особенностей распо
ложения данных (см. рисунок 5.2.2. 1). Исполь
зуя формулы, приведенные в таблицах 3.3.3.1 I
и З.3.3.1. II, получаем следующие результаты:
40 84
.
35
3
m
]25
r:;
а
"
20
ro
з-
а Т1
е 15
U1
10
5
О
Ps = 108,2
Ls = 301,1
а 5 =141,0
b s = 61,2
G s = 3114,3
J s = 0,223
и
Н, = 0,0093
Рисунок 5.2.2А
Р т = 103,85
L T = 292,1
а т = 116,7
Ь т = 64,95
G T = 2909,4
J 7 = 2,227
Ри = 85,8
Lu = 234,1
а и = 1З9,8
Ь и = 39,2
G u = 1917,3
J u = 0,083
а'=11,04
к = 14785.8.
Далее выполняют дисперсионный анализ по
формулам, приведенным в таблицах 3.3.3.1. 1II
и 3.3.3.1. IV. Результаты представлены в табли
це 5.2.2. 1I.
Высокая значимость реrрессии и отсутствие
значимых отклонений от линейности и точки пе
ресечения свидетельствуют о достоверности по
лученных результатов и позволяют рассчитать
активность.
Дисперсионный анализ
Таблица 5.2.2. 1I
Степени Сумма квадра- Дисперсия Вероят-
Источник вариации (средний квадрат F-oтноwение
свободы тов отклонений отклонения) насть
Линейная реrрессия 3 1087,7 362,6 339,5 0,000
Точки пересечения 2 3,474 1,737 1,626 0,237
Нелинейность 6 5,066 0,844 0,791 0,594
rруппы 11 1096,2
Остаточная вариация 12 12,815 1,068
Общая вариация 23 1109,0
Оценка содержания НА (МК2/доза)
Таблица 5.2.2. JII
Нижняя rраница Оценка Верхняя rраница
Вакцина Т 13,4 14,3 15,3
Вакцина и 8,9 9,7 10,6
5. 3. Статистический анализ результатов биолоzических испытаний
853
Уrловой
препарата:
стандартноro
коэффициент
ь' бх 301,1 60х 11 ,04 6356.
s 180 '
Уrловой коэффициент испытуемой вакци
ны Т;
ь' =' 6 х292,1 60 х11 ,04 =- 6056.
т 180 '
Уrловой коэффициент испытуемой вакци
ны И;
Ь ' 6х 234,1 60х 11,04 4123
и 180 ,.
В результате получаем следующие от
ношения активностей 6,056/6,356=0,953 для
вакцины Ти 4,123/6,356=0,649 для вакцины И.
2
С 6,356 =-10056,
6,3562 1,068 х 2,17g2 х 0,0444 '
к' =- 0,0056 хО,625 0,0035.
Доверительные интервалы вычисляют по
формуле 3.3.5.1.4.
Для вакцины Т:
0,955 :t ) 0.0056'1 ,913 + 0,0035 . ( 1,913) =O,955:t 0,063.
Для вакцины И:
0,649 :t .J o ,0056 . 1,423 + 0.0035 . ( 1,30 1 ) = 0,649 :t 0,058.
Содержание НА в MKr в 1 мл вакцины Haxo
дят путем умножения отношения активностей
и доверительных интервалов на предполаrа
емое содержание НА 15 мкr/доза. Результаты
приведены в таблице 5.2.2. 1I1.
5.3. АЛЬТЕРНАТИВНЫЕ ЭФФЕКТЫ
5.3.1. ИСПОЛЬ30ВАНИЕ МЕТОДА
ПРОБИТ АНАЛИ3А ПРИ СРАВНЕНИИ
ИСПЫТУЕМО/О ПРЕПАРАТА
СО СТАНДАРТНЫМ
Количественное определение aKтивHO
сти вакцины против дифтерии in vivo.
Выполняли количественное определение
вакцины против дифтерии (преД!lолаrаемая
активность 140 МЕ/флакон) по ОТНО'1Jению к
стандартному препарату (установленная aK
тивность 132 МЕ/флакон). На основании этих
данных rотовили эквивалентные дозы и BBO
дили случайным образом rруппам морских
свинок. Через определенный период времени
после введения препарата животным вводи
ли дифтерийный токсин, что вызывало rибель
животных. Подсчитывали число выживших
свинок. Полученные результаты приведены в
таблице 5.3.1. 1.
Затем эти результаты переносили в
первую рабочую таблицу, остальные ее столб
цы заполняли в соответствии с peKOMeHдa
циями, приведенными в Разделе 4.2.1. В Ta
блице 5.З.1. 1I приведены результаты первоro
цикла процедуры итерационноro анализа.
Далее суммируют данные последних
шести столбцов для каждоro из препаратов и
результаты заносят во вторую рабочую табли
цу (см. таблицу 5.3.1. 1I1). Остальные столб
цы заполняют данными, полученными при
использовании формул 4.2.1. 4. 4.2.1. 10.
В результате получают значение общеro уrло
BOro коэффициента Ь, равное 1,655.
Далее значения У первой рабочей табли
цы заменяют значениями а+Ьх и выполняют
второй цикл (см. таблицу 5.3.1. IV).
Циклы повторяют до тех пор, пока разни
ца между результатами двух последователь
ных циклов не станет достаточно маленькой.
В результате получают вторую рабочую Ta
блицу 5.З.1. V.
Проверку линейности проводят, как опи
сано в Разделе 4.2.2. Значение х 2 для 4 CTe
пеней свободы равно 0,851+1,070=1,921,
чему соответствует значение р=о, 750, KOTO
рое не является статистически значимым.
Поскольку отклонение от линейности не
является статистически значимым, проверку
отклонения от параллельности можно выпол
нить, как описано в том же разделе. Значение
х 2 для 1 степени свободы равно
14,152
(16,71 + 17,27) 0,001, чему соответствует
5,89
значение р=0,974, которое также не является
статистически значимым.
Теперь натуральный лоrарифм отношения
активностей может быть рассчитан, как описа
но в Разделе 4.2.3.
(16,71+17,27) 4,152 =0.001
5,89
Далее:
c 2,4012 , З =1127,
2,401 2 х 5,893 Р.1,960 2 '
1 1
V = . .. - =- 0.110.
18.37 17.96
Натуральный лоrарифм доверительноro ин
тервала:
2
0,155 0.013 :: \ 0.127(0,649 +1,127 . 0,036 )
0,142::0,288.
854
rосударственная фармакопея Республики Беларусь
Таблица 5.3.1. I
Исходные необработанные данные, полученные при количественном определении активности
вакцины для профилактики дифтерии методом летальной пробы у морских свинок
Стандартный препарат (5) Испытуемый препарат (Т)
Установленная активность 132 МЕ/флакон Предполаrаемая активность 140 МЕ/флакон
Доза Число зара- Число выживших Доза Число зара- Число выживших
(МЕ/мл) женных (МЕ/мл) женных
1,0 12 О 1,0 11 О
1,6 12 3 1,6 12 4
2,5 12 6 2,5 11 8
4,0 11 10 4,0 11 10
Таблица 5.3.1. 11
Первая рабочая таблица для перв020 цикла итерации
Вакци Доза п r х р У Ф Z У UJ (йХ (йУ (йх 2 (йу 2 (йху
на
1,0 12 i О 0,000 0,000 о 0,5 0,399 1 ,253 7,64 0,00 9,57 0,00 12,00 0,00
1,6 I 12 ! 3 0,470 0,250 О 0,5 0,399 0,627 7,64 3,59 4,79 1,69 3,00 2,25
S I
2,5 I 12 I 6 0,916 0,500 О 0,5 0,399 0,000 7,64 7,00 0,00 6,41 0,00 0,00
I !1О
4,0 I 11 1,386 0,909 О 0,5 0,399 1,025 7,00 9,71 7,18 13,46 7,36 9,95
1,0 I 11 О 0,000 0,000 О 0,5 0,399 1 ,253 7,00 0,00 8,78 0,00 11,00 0,00
1,6 I 12 4 0,470 0,333 О 0,5 0,399 0,418 7,64 3,59 3,19 1,69 1,3З 1,50
Т I 0,3991 0,570
2.5 I 11 8 0,916 0,727 О 0,5 7,00 6,42 3,99 5,88 2,27 3,66
4,0 I 11 10 1,386 0,909 О 0,5 0,399 i 1,025 7,00 9,71 7,18 13,46 7,36 9,95
Таблица 5.3.1. 111
Вторая рабочая таблица для перв020 цикла итерации
Вакцина I (,) LШХ LUJY LUJX 2 LUJy2 LUJXY 5 5 5 У
х а
JO( ку уу
S 29,92 20,30 7,18 21,56 22,36 7,70 7,79 12,58 20,64 0,68 0,24 1,36
Т 28,65 19,72 0,80 21,03 21,97 12,11 7,46 12,66 21,95 0,69 0,03 1,17
Таблица 5.3.1. IV
Первая рабочая таблица для втОр020 цикла итерации
Вакци Доза п r Х р У Ф Z У UJ (йХ (йУ (йх 2 (йу 2 (йху
на
1,0 12 О '0,000 0,000 1,36 0,086 0,158 1 ,911 3,77 0,00 7,21 0,00 13,79 0,00
1,6 12 3 0.470 0,250 0,58 0,279 0,336 0,672 6,74 3,17 4,53 1,49 3,04 2,13
S
2,5 12 6 0,916 0,500 0,15 0,561 0,394 0,001 7,57 6,94 0,01 6,36 0,00 0,01
4,0 11 10 1,386 0,909 0,93 0,824 0,258 1,260 5,07 7,03 6,39 9,75 8,05 8,86
1,0 11 О 0,000 0.000 1,17 0,122 0,202 1,769 4,20 0,00 7,43 0,00 13,14 0,00
1,6 12 4 0,470 0,333 O,39 0,349 0,370 0,430 7,23 3.40 3,11 1,60 1,34 1,46
Т
2,5 11 8 0,916 0,727 0,35 0,637 0,375 0,591 6,70 6,14 3,96 5,62 2,34 3,63
4,0 11 10 1,386 0,909 1,13 0,870 0,211 1,311 4.35 6,03 5,70 8,36 7,48 7,90
Таблица 5.3.1. V
Вторая рабочая таблица после достатОЧН020 количества циклов итерации
Вакцина LШ LШХ LUJY Lшх 2 Lшу 2 L(UXY 5 5 S х У а
кк ку уу
s 18,37 14,80 2,14 14,85 17,81 5,28 2,93 7,00 17,56 0,81 0,12 2,05
Т 17,96 12,64 0,55 11,86 18,35 6,76 2,96 7,15 18,34 0,70 0,03 1,72
5.3. Статистический анализ результатов биолоzических испытаний
855
Вычислив антилоrарифм и умножив полу
ченное значение на предполаrаемую актив
ность 40 МЕlфлакон, получим оценку актив
ности 160,6 МЕlфлакон и 95 % доверительный
интервал от 121,0 до 215,2 МЕ/флакон.
0.6
u
О
r
1;:05
о
Q.
0.4
1.0
0.9
0.0
Рисунок 5.3.1. 1.
5.3.2. ИСПОЛЬЗОВАНИЕ МЕТОДА лоrит
АНАЛИ3А И друrих АНАлоrИЧНblХ
МЕТОДОВ КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ ПРИ СРАВНЕНИИ
ИСПblТУЕмоrо ПРЕПАРАТА
СО СТАНДАРТНЫМ ПРЕПАРАТОМ
В данном разделе описаны результаты об
работки данных, представленных в разделе
5.3.1 методом лоrит анализа и друrими «клас
сическими» методами данноrо типа. В данной
конкретной ситуации приведенный матери
ал следует рассматривать как пример, а не в
качестве альтернативы методу пробит анали
за. Друrая форма кривой может быть приня
та только в том случае, если необходимость
замены подтверждается экспериментальными
данными или теоретическими расчетами (см.
таблицу 5.3.2А).
5.3.3. ОПРЕДЕЛЕНИЕ ЕД50 ВЕЩЕСТВА.
С ИСПОЛЬЗОВАНИЕМ МЕТОДА
ПРОБИТ АНАЛИ3А
Количественное определение iп vitro aK
тивности вакцины против полиомиелита, при
меняемой орально.
Для количественноro определения ЕД5Q BaK
цины против полиомиелита было исследова
но 1 О различных разведений 50 мкл исходной
вакцины на ИФА планшетах по 8 повторений в
каждом случае. Полученные результаты приве
дены в таблице 5.3.3. 1.
Полученные результаты заносят в первую pa
бочую таблицу, а остальные ее столбцы заполня
ют, как описано в Разделе 4.2.1. В таблице 5.3.3. II
приведены результаты первоro цикла процедуры
итерации, описанной в данном разделе.
Затем данные последних 6 столбцов CYM
мируют для каждоro из препаратов и результаты
заносят во вторую рабочую таблицу (см. табли
цу 5.3.3. 111). Остальные столбцы заполняют дaH
ными, рассчитанными по формулам 4.2.1.A
4.2.1. 10. В результате получают общий уrловой
коэффициент Ь, равный 0,295.
'.0
0.8
0.7
оз
02
о.,
o.g
08
07
Об
о
I
1;:05
о
Q.
0.4
оз
0.2
01
00
Рисунок 5.3.3А.
Таблица 5.3.2. I
Результаты, полученные при использовании альтернативных методов анализа
Лоrит rомпит Уrловой l .'
Ф 1 I e e' 1. 1
1 + e Y SlпY
2 2
Z е-У eY eY I
cos У
(1 + е У )2 .,
Уrловой 4,101 2,590 1,717
коэффициент Ь
х 2 (линейность) 2,15 3,56 1,50
х 2 (параллельность) 0,0066 0,168 0,0010
Активность 162,9 158,3 155,8
Нижняя rраница 121,1 118,7 i 122,6
Верхняя rраница 221,1 213,3 200,7
(') 1
ф=о и Z=O, приУ< п
2
Ф=1 Z=O, 1
и при У> п
2
856
rосударственная фармакопея Республики Беларусь
Таблица 5.3.3. 1
3,5 4,0 4,5
+ + +
+ + +
+ +
+ + +
+ + +
+ + +
+ + +
+ + +
Разведения (10" мкл исходной вакцины)
5,0 5,5 6,0 6,5 7,0
7,5
8,0
+
+
+
+ +
+ + +
+ +
Таблица 5.3.3. "
Первая рабочая таблица для перв020 цикла
Вакци I I
Доза П I r Х р У Ф Z У w ыХ соу юх 2 щ,2 соху
на
т 1О з,5 8 :0 8,06 0,000 0,00 0,5 0,399 1,253 5,09 41 ,04 6,38 330,8 8,00 51,4
I
1O O 18 'о 9,21 0,000 0,00 0,5 0,399 1,253 5,09 46,91 6,38 432,0 8,00 58,8
I
10 5 8 11 10,36 0,125 0,00 0,5 0,399 0,940 5,09 52,П 4,79 546,8 4,50 49,6
10---50 8 12 11,51 0,250 0,00 0,5 0,399 0,627 5,09 58,63 3,19 675,1 2,00 36,7
,
10---55 8 16 12,66 0,750 0,00 0,5 0,399 0,627 5,09 64,50 3,19 816,8 2,00 40,4
1 0 8 17 13,82 0,875 0,00 0,5 0,399 0,940 5,09 70,36 4,79 972, 1 4,50 66,1
I
1 5 8 7 14,97 0,875 0,00 0,5 0,399 0,940 5,09 76,23 4,79 1140,8 4,50 71,7
1O 7C 18 8 16,12 1,000 0,00 0.5 0,399 1,253 5,09 82,09 6,38 1323,1 8,00 102,9
1O 75 !8 8 17,27 1,000 0,00 0,5 0,399 1,253 5,09 87,95 6,38 1518,9 8,00 110,2
1O % 18 8 18,42 1,000 0,00 0,5 0,399 1,253 5,09 93,82 6,38 1728,2 8,00 117,6
I
Таблица 5.3.3. 1II
Вторая рабочая таблица для перв020 цикла
Вакцина I'w I'wx I'wy I'шх 2 I'wy2 I'wxy S S S х у а
хх ху уу
т 50,93 674,3 11,17 9484,6 57,50 312,32 556,92 164,43 55,05 13,24 0,219 3,690
Таблица 5.3.3. IV
Вторая рабочая Таблица после проведения достатОЧНО20 числа циклов
Вакцина S(» I S(!)X Swy Scox 2 S wy 2 Scoxy S хх S S х У а
ху уу
т 19,39 I 238,2 0,11 2981 ,1 26,05 37,45 55,88 36,11 26,05 12,28 0,006 7,931
Таблица 5.4.1. 1
Наблюдаемые эффекты
Стандартный препарат (5) Испытуемый препарат (1)
Разведение 1 2 Разведение 1 2
1/10 2,912 2,917 1/10 3,017 2,987
1/20 2,579 2,654 1/20 2,801 2,808
1/40 2,130 2,212 1/40 2,401 2,450
1/80 1,651 1,638 1/80 1,918 1,963
1/160 1,073 0,973 1/160 1,364 1,299
1/320 0,585 0,666 1/320 0,861 0,854
1/640 0,463 0.356 1/640 0,497 0,496
1/1280 0,266 0,234 1/1280 0,340 0,344
1/2560 0,228 0,197 1/2560 0,242 0,217
1/5120 0,176 0,215 1/5120 0,178 0,125
5.3. Статистический анализ результатов биОЛО2ических испытаний
857
Далее значения У первой рабочей таблицы
заменяют на значения а+Ьх и выполняют второй
цикл итерации. Циклы повторяют до тех пор,
пока разница между результатами двух последо
вательных циклов не станет достаточно малой.
В результате получают вторую рабочую таблицу
5.3.3. IV.
Проверку линейности проводят. как описано в
разделе 4.2.2. Значение х 2 для 8 степеней свободы
равно 2,711, чему соответствует значение р=0,951.
которое не является статистически значимым.
Теперь оценка отношения активностей
может быть получена, как описано в разделе 4.5.
м' ( 7 ,931) 12,273.
т 0,646
Далее:
С = ( 0,646 t х55,883 = 1197
( 0,646tx 55,883 12x 1,9602 ' ,
v 0052.
19.39 '
Натуральный лоrарифм доверительноro ин
тервала:
14,692 ( 2,420 ):t 0,197 х (2,882 + 1,197 xo,oog2 =
= 12,272 :tO,754.
Эта оценка все еще выражена в виде HaTY
ральноro лоrарифма разведений. Для тоro, чтобы
выразить аКТИВНОСТr в /п(ЕД50)/МЛ' результаты пре
образуют в M Tlпl 1 gO ). Поскольку активность
такою типа вакцин обычно выражают с исполь
зованием десятичною лоrарифма 109,о(ЕД50)/МЛ'
полученный результат следует разделить на
Iп(10). В результате получим оценку активности
6,36 109,о(ЕД50)/мл при 95 % доверительном ин
тервале 6,30 6,96 109,о(ЕД50)/МЛ'
5.4. РАСШИРЕННЫЕ КРИВЫЕ
сиrмоидной ЗАВИСИМОСТИ «ДОЗА
ЭФФЕКТ»
5.4.1. АНАЛИЗ ЧЕТЫРЕХ
ПАРАМЕТРИЧЕСКОЙ ЛО/ИСТИЧЕСКОЙ
КРИВОЙ
СерОЛ02ическое количественное определе
нив противостолбнячной сыворотки.
Как уже было указано в Разделе 3.4, этот
пример представлен только в качестве упраж
нения для «возможноro» способа анализа полу
ченных данных, но не как «единственный» или
«наиболее приемлемый» способ. Литературные
источники предлаrают мною иных подходов, и
они, в большинстве случаев, не приводят к He
приемлемой разнице в конечных результатах.
Короткое обсуждение альтернативных способов
анализа данных и друrих статистических реше
ний приведены в Разделе 7.5.
С помощью твердофазноro иммунофермент
ноro анализа (ELlSA) проведено количествен
ное определение антисыворотки, полученной от
морских свинок, в сравнении со стандартной cы
вороткой (0,4 МЕ/мл). Для каждой сыворотки ro
товили десять двукратных разведений и вноси
ли В лунки 96 Луночноro ИФА планшета. Число
повторностей для каждою разведения равно 2.
Наблюдаемый эффект представлен в таблице
5.4.1. 1.
Данный пример приводится с таким дo
пущением, что лаборатория, в которой прово
дится исследование, имеет документальное
подтверждение выполнения условий 1 3 (см.
Раздел 3.1.1), полученное в ходе разработки
методики для проведения повседневноro (py
тинною) выполнения. Кроме тою, лаборатория
также должна иметь документальное подтверж
дение тоro, что верхние и нижние пределы при
оценке активности испытуемоro препарата co
впадают с пределами стандартною препарата.
rрафическое представление результатов не
выявило каких либо необычных закономерно
стей расположения данных. Для приведения по
лученных данных в соответствие лоrистической
функции применяли подходящую компьютерную
проrрамму, используя rипотезу о том, что OCTa
точная поrрешность является независимой и ее
распределение является нормальным случай
ным распределением. В таком случае три пара
метра (а, J3 и Б) должны быть описаны с приме
нением общею уrловою коэффициента и общих
верхней и нижней асимптот. Для описания roри
зонтальноro расположения двух кривых требует
ся два дополнительных пара метра (Ys И у т ).
Далее представлены параметры, получен
ные с помощью упомянутой компьютерной про
rpaMMbI
а=3,196,
13= 1,125,
8= 0,145.
Is= А,307,
У Т = А,684,
Кроме тою, остаточная вариация (s ) равна
0,001429 при 20 степенях свободы (внутриrруп
повая вариация). Для расчета доверительноro
интервала, а также для проверки на параллель
ность и линейность наблюдаемые эффекты (и)
были подверrнуты линейному преобразованию
и затем введены в компьютер для анализа MeTO
дом взвешенных параллельных линий. Эта MeTO
дика очень похожа на методику пробит анализа,
которая приведена в разделе 4.2, со следующи
ми изменениями:
Y=/ p )')
( и /i )
ф
\' = 1" + ,{( (
z
1
ф=
1 ..с. C '
t.. l
Z = ,.. о
\ 1 + t' . )"
Z2\{( l '):
Н' =
о
s.....
858
rосударственная фармакопея Республики Беларусь
Результаты взвешенноro дисперсионно
ro анализа эффектов (у), преобразованных с
учетом их веса (w), приведены в таблице 5.4.1. 11.
Таблица 5.4.1. 11
Взвешенный дисперсионный анализ
ИСТОЧНИК Степени х 2 Вероят-
вариации свободы НОСТЬ
Препараты 1 ! 0,529653 0,467
Линейная 1 6599,51 0,000
реrрессия
Непараллель 1 0,0458738 0,830
ность
Нелинейность 16 8,89337 0,918
Уровни I 19 6608,98 0,000
воздействия !
(rруппы)
i
Остаточная I 20 20,0000
вариация i
Общая 39 6628,98
вариация
Отсутствие значимых статистических OT
клонений от параллельности и линейности под
тверждает, что результаты количественноro
определения приroдны для расчета активности.
Если условие соответствия верхней и нижней
асимптот не было выявлено, не следует ожидать
статистически значимых отклонений от линейно
сти и/или параллельности ввиду тоro, что про
верка линейности и параллельности подтверж
дает приrодность экспериментальных данных
предлаrаемой модели четырех параметриче
ской лоrистической кривой. Остаточная поrреш
ность в данном дисперсионном анализе Bcerдa
будет равна 1. как результат произведенных пре
образований. Однако. можно рассчитать фактор
неоднородности (по аналоrии с пробит моде
лью). Относительная активность испытуемоro
препарата представляет собой антилоrарифм
YS Y7' Умножив рассчитанную активность испы
TyeMoro препарата на активность стандартноrо
препарата, получают оценку активности, равную
1,459.0,4=0.584 МЕ/мл. Применив формулу
4.2.3. 2, вычисляют 95 % доверительный интер
вал от 0,557 до 0,612 МЕ/мл.
6. ОБЪЕДИНЕНИЕ РЕЗУЛЬТАТОВ
КОЛИЧЕСТВЕнноrо ОПРЕДЕЛЕНИЯ
6.1. ВВЕДЕНИЕ
С целью удовлетворения требованиям Фар
макопеи часто необходимо повторение нескольких
независимых количественных определений и объ
единение полученных результатов. В связи с этим
возникает вопрос: в каких случаях возможно 06ъe
динение результатов и каким образом это осущест
вляется.
Два количественных определения MOryт быть
рассмотрены как действительно независимые,
если выполнение любоrо из них не влияет на Be
роятности появления возможных результатов дpy
roю. Это означает, что случайные ошибки всех cy
щественных факторов, которые MOryт повлиять на
результат в ходе выполнения одноro количествен
ною определения (например, разведения cтaндapT
Horo и испытуемою препаратов, чувствительность
биолоrических индикаторов) не зависят от соответ
ствующих случайных ошибок этих факторов в ходе
выполнения друroro количественною определения.
Таким образом, количественные определения, KO
торые проводятся в последующие дни с использо--
ванием одинаковых сохраненных разведений CTaH
дартною препарата, не являются независимыми.
Существует несколько методов объединения
результатов независимых количественных опреде
лений и наиболее приемлемый из них с теоретиче
ской точки зрения является довольно сложным при
практическом использовании. Ниже описаны три
простых метода аппроксимации, однако MOryт ис
пользоваться и друrие методы, если выполнены He
обходимые условия объединения результатов.
Если данные количественною определе
ния основываются на использовании модели па
раллельных линий или модели пробит анализа,
полученные значения активностей перед объе
динением необходимо представить в лоrарифми
ческом виде; значения активностей, полученные
при использовании модели уrловых коэффициен
тов, объединяют без преобразований. Посколь
ку модель параллельных линий используют чаще,
чем модель уrловых коэффициентов, в данном
разделе в формулах используется символ М, обо
значающий натуральный лоrарифм отношения aK
тивностей. Подставляя вместо М отношения уrло
вых коэффициентов R, можно применять те же
формулы для определения активностей, найден
ных при использовании модели уrловых коэффи
циентов. Перед объединением значения активно
стей всех исследованных препаратов должны быть
скорректированы по отношению к установленной
активности.
6.2. ВЗВЕШЕННОЕ ОБЪЕДИНЕНИЕ
РЕЗУЛЬТАТОВ КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ
Данный метод может быть использован
только в том случае, если выполняются следу
ющие условия:
1) оценки активностей получены в ходе He
зависимых количественных определений;
2) для каждою из количественных опреде
лений значение С близко к 1 (друrими словами,
меньше чем 1,1);
3) число степеней свободы конкретных OCTa
точных поrрешностей (вариаций) не меньше 6,
но желательно, чтобы оно было больше 15;
4) значения индивидуальных активностей об
разуют однородное множество (см. Раздел 6.2.2).
Если эти условия не выполняются, метод
взвешенноro объединения результатов нельзя
использовать. В этом случае для нахождения
наилучшей оценки средней активности, KOТO
5.3. Статистический анализ результатов биОЛО2ических испытаний
859
рая затем будет применяться в последующих
количественных определениях в качестве пред
полаrаемой активности, может использоваться
метод, описанный в Разделе 6.3.
6.2.1. РАСЧЕТ ВЕСОВЫХ
КОЭФФИЦИЕНТОВ
Предположим, что проведено п' количе
ственных определений, в ходе которых получено
п' значений М с соответствующими доверитель
ными интервалами. Для каждоro количественно
ro определения рассчитывается лоrарифмиче
ский доверительный интервал L путем вычитания
нижнеro значения из верхнеro. Вес W для каждо
ro значения М вычисляется по формуле 6.2.1. 1,
rдe t имеет то же значение, которое используется
при расчете доверительных интервалов.
4е
w == . (6.2.1. 1)
е
6.2.2. ОДНОРОДНОСТЬ ОЦЕНОК
АКТИВНОСТИ
Однородность оценок активностей опре
деляют следующим образом. Отклонение каж
доro из значений М от взвешенноro среднеro
возводят в квадрат, умножают на COOTBeTCTBY
ющий вес и суммируют по всем количествен
ным определениям. В результате получают
статистический ряд, приблизительно распре
деленный как х 2 (см. таблицу 8.3), который
может использоваться для оценки OДHOpOД
ности множества натуральных лоrаРИфМ08
оценок активностей:
х 2 ,., L:W(M M )2, rдe: М == WM . (6.2.2.1)
п' W
Если рассчитанное значение х 2 меньше Ta
бличноro значения, соответствующеrо (п' 1) степе
ней свободы, то активности однородны, и среднее
значение активности, а также доверительные ин
тервалы, рассчитанные при помощи метода, опи
санною в Разделе 6.2.3, будут обоснованными.
Если рассчитанное значение статистическо
ro ряда больше табличноro, активности HeOДHO
родны. Это означает, что отклонение между ин
дивидуальными оценками М больше, чем можно
было бы ожидать исходя из оценок rраниц дo
верительных интервалов, то есть между коли
чественными определениями существует значи
тельная вариабельность. В этом случае условие 4
не выполняется и формулы, приведенные в
Разделе 6.2.3, неприменимы. Вместо них MOryт
быть использованы формулы, приведенные в
Разделе 6.2.4.
6.2.З. РАСЧЕТ ВЗВЕШЕнноrо
СРЕДНЕrо и rРАНИЦ ДОВЕРИТЕЛЬНЫХ
ИНТЕРВАЛОВ
ДЛЯ каждоro количественноro определе
ния рассчитывают значения WM, а их сумму
делят на суммарный вес всех количествен
ных определений. В результате получают зна
чение лоrарифма взвешенной средней актив
ности:
М == L WM , (6.2.3. 1)
IW
Стандартная ошибка натуральноro лоrа
рифма средней активности вычисляется как KBa
дратный корень из величины, обратной CYMMap
ному весу:
! 1
sM == \1 L W ' (6.2.3. 2)
а приблизительные rраницы доверительно
ro интервала равны антилоrарифмам значений,
полученным по формуле
м t-t.s м , (6.2.3. 3)
rAe число степеней свободы t равно
сумме чисел степеней свободы средних KBa
дратов ошибок отдельных количественных
определений.
6.2.4. ВЗВЕШЕННОЕ СРЕДНЕЕ
И rРАНИЦЫ ДОВЕРИТЕльноrо
ИНТЕРВАЛА,ОСНОВАННЬ
НА ДИСПЕРСИИ МЕЖДУ ИСПЫТАНИЯМИ
И ВНУТРИ НИХ
При объединении результатов нескольких
повторных количественных определений вели
чина х 2 может быть статистически значимой. В
этом случае полученная вариация имеет две
компоненты:
вариация в пределах одной серии количе
ственных определений S == 1/ W;
вариация между различными сериями KO
личественных определений
2 l)M M /
S == ,
м п'( п' 1)
rдe М невзвешенное среднее значение.
Первый компонент вариации изменяется от
одноro количественноro определения до друro
ro, Torдa как второй компонент является общим
для всех М.
Torдa для каждоro значения М рассчитывают
весовой коэффициент w' = 2 5 ' который заме
sM SЛ:,
няет значение W в Разделе 6.2.3; при этом t при
близительно равно 2.
6.3. НЕВЗВЕШЕННОЕ ОБЪЕДИНЕНИЕ
РЕЗУЛЬТАТОВ КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ
Наиболее простым способом объединения
п' оценок для значений М при п' количествен
860
rосударственная фармакопея Республики Беларусь
ных определениях является вычисление cpeДHe
ro значения и оценка стандартноro отклонения по
формуле:
52 L(M M/ ,(6.3. 1)
м п'(п' 1)
а rраницы доверительною интервала:
М If,sM ' (6.3. 2)
rде t имеет (п' 1) степеней свободы. Число
п' оценок значений М обычно мало, а значение t,
соответственно, довольно велико.
6.4. ПРИМЕР ОПРЕДЕЛЕНИЯ
ВЗЕШЕННОЙ СРЕДНЕЙ АКТИВНОСТИ
С ДОВЕРИТЕЛЬНЫМ ИНТЕРВАЛОМ
В таблице 6.4. 1 приведено шесть независи
мых оценок активности одною и тоro же препара
та, а также их 95 % доверительные интервалы и
число степеней свободы для их дисперсий ошибки.
Условия 1, 2 и 3. приведенные в Разделе 6.2,
выполнены. Натуральный лоraрифм активностей
и веса рассчитаны. как описано в Разделе 6.2.
Однородность оценок активностей рассчи
тывают по формуле 6.2.2. 1, в результате полу
чают значение "1..2. равное 4,42 при 5 степенях
свободы. Этот результат не является статисти
чески значимым (р=0.49) и, следовательно, все
условия для применения оценки взвешенной
средней активности выполняются.
Взвешенную среднюю активность вычисля
ют по формуле 6.2_3_ 2, в результате получают
значение 9,8085.
По формуле 6.2.3. 2 рассчитывают cтaHдapT
ное отклонение. равное 0,00673, а по формуле
6.2.3. 3 вычисляют 95 % доверительный интер
вал 9,7951 9.8218, rдe tимеет 120 степеней CBO
боды.
Взяв антилоrарифм, получают значение aK
тивности, равное 18 187 МЕ/флакон при 95 %
доверительном интервале от 17 946 до 18431
МЕ/флакон.
макопейных целей. В данном разделе сделана
попытка представить в большей степени краткий
обзор альтернативных или наиболее общих Me
тодов статистическоro анализа. 3аинтересован
ные лица MOryT также обратиться к специальной
литературе по этой теме. В случае -использова
ния более специализированных методов стати
стическоro анализа следует обратиться за помо
щью к квалифицированным специалистам.
7.1. ОБЩИЕ ЛИНЕЙНЫЕ МОДЕЛИ
Методы, изложенные в данном разделе,
Moryт быть описаны в рамках общих линей
ных моделей (или обобщенных линейных MO
делей для тоro, чтобы включить методы пробит
и лоrит анализа). Принцип всех методов OCHO
ван на построении линейной матрицы CTPYКТY
ры Х (или матрицы плана), в которой каждая
строка представляет результаты наблюде
ния, а каждый столбец один из линейных
факторов (препарат, блок, столбец, дозу). Ha
при мер, в случае схемы латинскоro квадрата,
рассмотренной в Разделе 5.1.2, такая матри
ца состояла бы из 36 строк и 13 столбцов
по одному столбцу на каждый из препаратов,
один столбец для доз, пять столбцов на каждый
из блоков, за исключением первоrо, и пять столб
цов для каждой строки, за исключением первой.
Все столбцы, за исключением одноro для доз,
заполняют О или 1 в зависимости от TOro, свя
за но данное наблюдение с данным фактором
или нет. Вектор У заполняют результатами Ha
блюдений (преобразованными). Искомые пара
метры вычисляют по формуле (XtXY)(IY, после
чеro оценка активности т может быть леrко по
лучена как отношение соответствующих пара
метров. Доверительные интервалы рассчитыва
ются на основе теоремы Филлера (Fielle'):
[ gV12 ts I 1
т 1: 'v 2тV 12 т2v v
v 22 Ь V 11 22
тL,тU
(1 g)
Vf2 )]
v22
,
t 2 s 2 V
r: д еg 22
Ь 2
а V 11 , V 22 множители дисперсии знамена
теля и числителя, соответственно; У 12 множи
тель ковариации. Эти множители можно непо
средственно вычислить из матрицы ()(IХ)-1 или
косвенным методом, приняв во внимание, что
Var(a 1 a)=Var( а 1 )+Var(a2) 2Cov( а 1 ,а 2 ),
а Cov(a1 a2,b)=Cov(a1,b) Cov(a2,b).
Таблица 6.4. I
Оценки активностей и доверительные интервалы 6 независимых количественных определений
7. ДОПОЛНЕНИЕ
Невозможно дать исчерпывающий обзор
статистических методов, используемых при про
ведении фармакопейных исследований. Тем не
менее, методы, изложенные в данном разделе,
удовлетворяют требованиям большинства фар
Оценка активности Нижняя rраница Верхняя rраница Степени Lп активности М Вес
(МЕ/флакон) (МЕ/флакон) (МЕ/флакон) свободы W
18 367 17 755 19 002 20 9,8183 3777,7
18 003 17 415 18610 20 9,7983 3951,5
18064 17 319 18 838 20 9,8017 2462,5
17 832 17 253 18 429 20 9,7887 4003,0
18 635 17 959 19 339 20 9,8328 3175,6
18 269 17 722 18 834 20 9,8130 4699,5
5.3. Статистический анализ результатов биОЛО2ических испытаний
861
Полный дисперсионный анализ с полным
разделением компонентов несколько более
сложен, поскольку он предполаrает пере
смотр матрицы Х, которая дополняется столб
цами для ослабления предположений о па
раллепьности и линейности, после чеro может
быть проверена rипотеза о линейности. В
случае количественных определений, OCHO
ванных на альтернативных эффектах, фак
торы линейности (точки пересечения кривых
с осью ординат a s ' а т и т. д. общий УfЛовой
коэффициент Ь) находят путем максимиза
ции суммы по rруппам воздействий препа
ратов (дозы) пlпФ(а,+Ьх)+(п r)lп(1 Ф(а/Ьх»,
rде х натуральный лоrарифм дозы
(/п(доза», Ф определяет форму распреде
ления, i e:{S, Т,...}.
7.2. НЕОДНОРОДНОСТЬ ДИСПЕРСИИ
Проблема неоднородности дисперсии
не Bcerдa может быть решена путем просто
ro преобразования эффектов. В этом случае
один из возможных способов решения данной
проблемы состоит в применении метода взве
шенной линейной реrрессии. Чтобы получить
объективную оценку, веса результатов наблю
дений берутся как величины, обратно пропор
циональные дисперсии ошибок. Так как ис
тинное значение дисперсии ошибок не всеrда
известно, веса MorYT подбираться с исполь
зованием линейной итерактивной процедуры.
Однако при расчете доверительны)( интерва
лов при этом возникают дополнительные про
блемы.
7.3. РЕЗКО ВЫДЕЛЯЮЩИЕСЯ ЗНАЧЕНИЯ
(ВЫБРОСЫ) И УСТОЙЧИВОСТЬ МЕТОДОВ
В РАБОТЕ (РОБАСТНОСТЬ)
Недостатком метода наименьших KBaдpa
тов, описанноro в данном разделе, являет
ся ero довольно высокая чувствительность к
резко отклоняющимся от среднеrо значения
данным. Очевидный выброс может целиком
исказить результаты вычислений. Эту про
блему часто решают путем исключения таких
выбросов из набора данных. Такой подход
может привести к субъективному исключению
данных не всеrда корректному и безопасно
му. Довольно сложно дать общие peKOMeHдa
ции, касающиеся решения относительно TOro.
является ли конкретный результат наблюде
ния резко выделяющимся выбросом или нет.
и с этим связано появление и развитие ряда
робастных (устойчивых в работе) методов
анализа. Эти методы менее чувствительны к
выбросам за счет Toro, что результатам, KOTO
рые в большей мере отличаются от проrнози
pyeMoro значения, придается меньший вес. В
данном случае возникает ряд новых проблем,
связанных с расчетом доверительных интер
валов, а также с определением подходящей
функции для минимизации ошибок.
7.4. ОШИБКИ КОРРЕЛЯЦИИ
С практической точки зрения полная paH
домизация не Bcerдa бывает осуществима или
крайне нежелательна. Поэтому для последова
тельных доз в пределах серии разведен ий часто
характерны ошибки корреляции, которые при
водят к чрезмерному сужению доверительных
интервалов. Сущес.вует несколько методов,
которые позволяют оценить такой эффект «aB
токорреляции» .
7.5. РАСШИРЕННЫЕ НЕЛИНЕЙНЫЕ
КРИВЫЕ ЗАВИСИМОСТИ
«ДОЗА ЭФФЕКТ»
Статистический анализ расширенных He
линейных кривых зависимости «доза эффект»
приводит к возникновению специфических TPYД
ностей, для решения которых необходим совет
специалиста по биостатистике. Некоторые из
таких трудностей перечислены ниже.
1) Ранее приведен при мер анализа с при
менением четырехпараметрической лоrисти
ческой функции. Однако для этоro случая
применимы и модели, которые подчиняются
друrим функциям, имеющим rрафик сиrмоид
ной формы. Может быть рекомендована, Ha
пример, модель, включающая дополнитель
ные параметры асимметрии.
2) Неоднородность дисперсии характерна
для случая, Korдa эффект распределяется в ши
роком диапазоне значений. В том случае, если
при статистическом анализе неоднородность
будет проиrнорирована, полученная оценка ин
терпретированных результатов может быть He
правильной и отяющенной предвзятостью. Ис
пользование взвешенных обратных величин
ошибки дисперсии не может быть применено
для случая оrраниченноro числа повторностей в
испытании. Для такоro случая лучше будет при
менение функции, описывающеЙ зависимость
дисперсии от среднеro значения эффекта.
3) Процедура приведения rрафика зависи
мости к соответствию статистической модели
дает различные оценки, в зависимости от при
меняемых предположений относительно дис
персии и диапазона значений эффекта.
4) В принципе, эквивалентность нижнею
и верхнеro пределов эффекта для различных
препаратов при количественном определении
может быть проверена непосредственно в ходе
количественноro определения каждой серии.
Однако интерпретация результатов этих опре
делений может быть непрямой. Так, например,
проверка линейности и параллельности, пред
принятая по упрощенной методике анализа (см.
пример 5.4.1) включает косвенную оценку экви
валентности и правильности нижнеro и верхнеro
пределов эффекта.
5) Мноrие методы количественноro опре
деления предполаrают применение «KOH
трольноrо опыта», результаты которою пред
назначаются для определения верхнею и/или
862
rосударственная фармакопея Республики Беларусь
нижнеro пределов эффекта. Однако, значе
ния этих rраниц MorYT быть несопоставимы со
значениями пределов, определенных в ходе
применения статистических моделей, OCHO
ванных на расширенных нелинейных rрафи
ках зависимости «доза эффект».
6) Упрощенный методстатистическоroана
лиза, приведенный, как при мер 5.4.1, предус
матривает приблизительную оценку ДOBe
рительноro интервала результата. В этом
случае MorYT применяться друrие методы, Ha
пример, метод интервалов с несоrласованны
ми значениями для полностью определенной
модели. Так для типичных результатов коли
чественноro определения с эффектами, pac
пространяющимися на весь диапазон значе
ний для каждоro испытуемоro препарата, все
методы анализа дают сходные результаты.
7.6. НЕПАРАЛЛЕЛЬНОСТЬ КРИВЫХ
ЗАВИСИМОСТИ «ДОЗА ЭФФЕКТ»
Сходство зависимостей «доза эффект»
является фундаментальным требованием при
оценке результатов количественноro опре
деления, которые MorYT рассматриваться как
основные на «принципе разведения) и, COOT
ветственно. оценка результатов относитель
ной активности которых является достоверной
(см. Раздел 3.1.1). Соответствие этому требо
ванию часто определяют по отсутствию CTa
тистически значимоro различия rрафически
представленных кривых зависимости «доза
эффект» для стандартноro и испытуемоro пре
паратов. Недооценка остаточной поrрешности
(вариации) может привести к избыточному ис
ключению результатов количественноro опре
деления из за статистически значимых откло
нений от параллельности и/или линейности.
Такие случаи часто представляют собой apTe
факт при выборе несоответствующеro плана
исследований или несоответствующеro aHa
лиза результатов. Небольшие изменения, BHO
симые в план исследования, в большинстве
случаев MorYT существенно исправить оценку
остаточной поrрешности. Также в этой ситу
ации может помочь проведение достаточноro
числа повторных количественных определе
ний, если это возможно. Если оценка величи
ны соответствующей остаточной поrрешности
(вариации) не может быть проведена для каж
доro конкретноro количественноrо определе
ния, например, в том случае, коrда это Heocy
ществимо или практически нецелесообразно,
может быть проведена более точная оценка
остаточной поrрешности в ходе валидации Me
тодики количественноrо определения. Также
MorYT встречаться случаи, коrда аналитиче
екая система количественноro определения
обладает необходимой точностью для обнару
жения небольших отклонений от непараллель
ности, обусловленных природой эффекта.
Если непараллельность является истинной,
тоща следует разработать и применить подхо
дящее решение проблемы. Для решения этой
проблемы, например, может потребоваться
использование стандартноro препарата, по co
ставу соответствующеro (соответственно «па
раллельноro») испытуемому препарату. Если
же аналитическая система количественноro
определения дает неспецифический ответ на
неопределяемые компоненты стандартноro
или испытуемоro препаратов, тоща решени
ем проблемы может стать солее специфиче
ская аналитическая система, которая не будет
реаrировать на несущественные компоненты
препаратов. Для решения этих фундаменталь
ных проблем не существует простых общепри
менимых статистических методов. Подходя
щие методы MorYT быть выбраны в каждом
конкретном случае, опираясь на помощь спе
циалистов статисти ков.
8. ТАБЛИЦЫ И ПРОЦЕДУРЫ
rЕНЕРИРОВАНИЯ
В данном разделе приведены таблицы. в
которых указаны критические значения для
наиболее часто встречающихся чисел CTe
пеней свободы. Если необходимое значе
ние отсутствует в таблице, следует обратить
ся к более полным таблицам. Статистические
функции включены во мноrие компьютерные
проrраммы, которые MorYT быть использованы
вместо таблиц. В качестве альтернативы MorYT
использоваться приведенные после каждой Ta
блицы процедуры rенерирования б , которые по
зволяют рассчитать вероятность, COOTBeTCTBY
ющую заданному статистическому массиву и
числу степеней свободы.
8.1. F РАСПРЕДЕЛЕНИЕ
Если полученное значение превышает при
веденное в таблице 8.1. 1, оно рассматривается
как значимое (верхние строки, р=0,05) либо как
высокозначимое (нижние строки, р=0,01). df1
число степеней свободы числителя, df2 число
степеней свободы знаменателя.
Процедура 2енерирования. Пусть F
есть F отношение для dfl и df2 степе
ней свободы, как описано выше. Положим
рi=л=3,14159265358979... Тоща процедура,
описанная в Таблице 8.1. 11 будет rенерировать
р значение статистики.
8.2. t РАСПРЕДЕЛЕНИЕ
Если полученное значение превышает зна
чение в таблице 8.2. 1, оно рассматривается как
б Процедуры rенерирования представляют собой про
rpaMMHbIe коды, которые написаны для редактора VBA
(Visua/ Basic for App/icatioп) и позволяют осуществлять pac
четы указанных статистических параметров в прикладных
проrраммах типа Ехсе/ (Мiсrоsоft Offjce). Ряд дополнитель
ных процедур rенерирования и управления данными для
биолоrических испытаний и тестов можно найти в PYKOBOД
стве Лапача с. Н. и соавт. (2001).
5.3. Статистический анализ результатов БИОЛО2ических испытаний 863
Таблица 8.1. I
Критические значения F распределения
':IZ 1 2 3 4 5 6 8 10 12 15 20 Q()
df2
10 4,965 4,103 3,708 3.4 78 3,326 3,217 3,072 2,978 2,913 2,845 2,774 2,538
10,044 7,559 6,552 5,994 5,636 5,З86 5.057 4,849 4,706 4,558 4.405 3,909
12 4,747 3,885 3,490 3,259 3,106 2,996 2,849 2,753 2,687 2,617 2,544 2,296
9,330 6,927 5,953 5,412 5,064 4,821 4,499 4,296 4,155 4,010 3,858 3,361
15 4,543 3,682 3,287 3,056 2,901 2,790 2,641 2,544 2.475 2,403 2,328 2,066
8,683 6,359 5,417 4,893 4,556 4,318 4,004 3,805 3,666 3,522 3,372 2,868
20 4,351 3,493 З,098 2,866 2,711 2,599 2.447 2,348 2,278 2,203 2,124 1,843
8,096 5,849 4,938 4,431 4,103 3.871 3,564 3,368 3.231 3,088 2,938 2.421
25 4,242 3,385 2,991 2,759 2,603 2.490 2,337 2,236 2,165 2,089 2.007 1,711
7,770 5,568 4,675 4,177 3,855 3.627 3,324 3,129 2,993 2,850 2,699 2,169
30 4,171 3,316 2,922 2,690 2,534 2.421 2,266 2,165 2,092 2,015 1,932 1,622
7,562 5,390 4,510 4,018 3,699 3,473 3,173 2,979 2,843 2,700 2,549 2,006
50 4,034 3,183 2,790 2,557 2,400 2,286 2,130 2,026 1,952 1,871 1,784 1,438
7,171 5,057 4,199 3,720 3.408 3,186 2,890 2,698 2,563 2,419 2,265 1,683
Q() 3,841 2,996 2,605 2,372 2,214 2,099 1,938 1,831 1,752 1,666 1,571 1,000
6,635 4,605 3,782 3,319 3,017 2,802 2,511 2,321 2,185 2,039 1,878 1,000
Таблица 8.1. 11
Процедура 2енерирования для F распределения
Если df1 четное Если df1 нечетное, а df2 четное Если df1 и df2 нечетные
x dfl/(dfl+df2/F) x df2/(df2+dfl*F) x atn(sqr(dfl*F/df2)}
з 1 S l cs cas(x}
t l t l sn sin(x)
far i 2 ta (df1 2) step 2 far i;;;::2 tc (ci::2 2) step 2 x x/2
t t*x*(df2+i 2)/i t t*x*(dflTi 2i/i s o
s s+t s s+t t sn*cs/2
next i next i v o
р s*(1 х}Л(df2/2) р 1 s*(1 х)Л(dfl/2} w l
far i 2 to (d:2 1) step 2
s s+t
t t*i/(iTl)*cs*cs
next i
for i l to (df1 2) step 2
V==\T+\\I
w w*(df2+i)/(i+2)*sn*sn
next i
p 1+(t*df2*v x s)/pi*4
864
rосударственная фармакопея Республики Беларусь
значимое (р=0,05), либо как высокозначимое
(р=0,01 ).
Процедуры 2енерирования. Для заданно
ro значения t при числе степеней свободы df
значение р может быть найдено при помощи
процедур, описанных в Разделе 8.1, rде F=t:;,
df1=1, а df2=df.
Для заданноrо числа степеней свободы df
значение t (при р=0.05) может быть найдено
с помощью следуюшей процедуры, приведен
ной в таблице 8.2. 11. (В этом случае следует
обеспечить точность до 6 десятичноro знака):
8.З. РАСПРЕДЕЛЕНИЕ у}
Если полученное значение превышает
значение в таблице 8.3. 1, оно рассматрива
ется как значимое (р=О.05), либо как BЫCOKO
значимое (р=0.01)
Процедура 2енерuрования. Обозначим
значение z:; через :': ., а df присвоим значе
ни€, как это указанно выше. Выполнение сле
дующей процедуры. представленной в табли
це 8.3. 11. rенерирует значение р статистики.
В этой процедуре phi является функци
ей кумулятивноro стандартноro нормальноrо
распределения Ф (см. Раздел 8.4).
8.4 Ф РАСПРЕДЕЛЕНИЕ
(КУМУЛЯТИВНАЯ СТАНДАРТНАЯ
ФУНКЦИЯ НОРМАльноrо
РАСПРЕДЕЛЕНИЯ)
Для отрицательных х Ф значение находят
из таблиuы 8.4. I как 1 Ф( х).
Процедура 2енерироеания. Примем зна
чение х равным . Следующая процедура
(см. таблицу 8.4 II) позволяет получить зна
чения Ф при 0""х....В.15. Если х больше 8,15,
ф значение можно принять равным 1. Для
отрицательных х может быть использована
формула. привепенная выше. Данная про
цедура предполаrает. что компьютер может
представлять около 15 десятичных знаков.
Если число дес5'Н ЧНЫХ знаков меньше или
больше, в процедуру необходимо внести
определенные простые преобразования.
8.5. СЛУЧАЙНЫЕ ПЕРЕСТАНОВКИ
Необходимость В случайных перестанов
ках возникает в случае использования схемы
рандомизированных блт:ов. Приведенный
ниже алrоритм позволяет получить случай
ные перестановки N воздеЙствиЙ (доз), ис
пользуя встроенный в компьютерное 06еспе
чение reHepaTop псевдослучайных чисел:
1) записывают N возможных ВИДОВ воз
действия (доз) в ряд;
2) rенерируют случаЙное целое число (,
чтобы 1 r N;
3) меНЯЮl местами ( Toe и N Toe исследо
вания В ряду;
4) уменьшают N на единицу (N=N 1) и повторя
ют шаrи 2 до тех пор. пока N не станет равно 1.
Например, случай из 6 видов воздействий
(доз) иллюстрирует этот алrоритм.
1. N=6 5, 52 5, Т, Т, Тз
о <
2. [= 2
3. 5, Тз 5 з Т, Т 2 52
4. N=5
2. (= 4
3. 5, Тз 5 з Т 2 Т, $2
4. N=4
2. (= 4 1
3. 5, Т, 5 з Т 2 Т, $2
о
4. N=3
2. (= 1
3. 5 з Т" 5, Т 2 Т, 52
о
4. N=2
2. (= 1
3. ТЗ 5з 5, Т 2 Т, 52
4. N=1
Таблица 8.2. 1
Критические значения t распределенuя
df р = 0,05 Р = 0,01 df Р = 0,05 Р = 0,01
1 12,706 63,656 22 2,074 2,819
2 4,303 9,925 24 2,064 2,797
3 3,182 5,841 26 2,056 2,779
4 2,776 4,604 28 2,048 2,763
5 2,571 4,032 30 2,042 2.750
6 2,447 3,707 I 35 2,030 2,724
7 2,365 3,499 I 40 2,021 2,704
I
I
8 2,306 3,355 I 45 2,014 2,690
9 2,262 3,250 50 2,009 2,678
10 2.228 3.169 I 60 2.000 2.660
12 2,179 3,055 I 70 1,994 2,648
14 2,145 2,977 I 80 1,990 2,639
16 2,120 2,921 I 90 1,987 2,632
I
,
I
18 2.101 2,878 1100 1.984 2,626
i
20 2,086 2,845 <$о 1,960 2,576
. .
Таблица 8.2. 11
Процедура 2енерирования для t распределения
---
:, '59ПЕ .
., ..-' ;2:28/ jf...
L ';: : :-';;;: f(i "
-: '= jL 4 ! '.... ,
; .i --
f :: l S Е .: : 1 ":, ,:. -..
} I C,2 '7 ; ,:;т/>:,
, 4 ? 2 С / с; f' "- ;
. - - - ._ - ... - -
5. З. Статистический анализ результатов биолоеических испытаний
865
Таблица 8.3. I
Критические значения распределения х 2
df Р = 0,05 Р = 0,01 df Р = 0,05 Р = 0,01
1 3,841 6,635 11 19,675 24,725
2 5,991 9,210 12 21,026 26,217
3 7,815 11,345 13 22,362 27,688
4 9,488 13,277 14 23,685 29,141
5 11,070 15,086 15 24,996 30,578
6 12,592 16,812 16 26,296 32,000
7 14,067 18,475 20 31,410 37,566
8 15,507 20,090 25 37,652 44,314
9 16,919 21,666 30 43,773 50,892
10 18,307 23,209 40 55,758 63,691
Таблица 8.3. 11
Процедура 2енерации для распределения х 2
Если df четное
s O:
t exp ( x2/2)
for i 2 to df step 2
s s+t
t t*x2/i
next i
p l s
Если df нечетное
x sqr(x2)
s O
t==x*exp( x2, 7\ ;:;:-==- !;:.
for i 3 tc a S 2
S==S---t-t
?,..
..
!:: X .:..
P< S 2'p .l (х)
Таблица 8.4. 1
3начения Ф распределения
х I ф х I ф х I ф
0,00 0,500 1,00 0,841 2,00 0,977
0,05 0,520 1,05 0,853 2,05 0,980
0,10 0,540 1,10 0,864 2,10 0,982
0,15 0,560 1,15 0,875 2,15 0,984
0,20 0,579 1,20 0,885 2,20 0,986
0,25 0,599 1,25 0,894 2,25 0,988
0,30 0,618 1,30 0,903 2,30 0,989
0,35 0,637 1,35 0,911 2,35 0,991
0,40 0,655 1,40 0,919 2,40 0,992
0,45 0,674 1,45 0,926 2,45 0,993
0,50 0,691 1,50 0,933 2,50 0,994
0,55 0,709 1,55 0,939 2,55 0,995
0,60 0,726 1,60 0,945 2,60 0,995
0,65 0,742 1,65 0,951 2,65 0,996
0,70 0,758 1,70 0,955 2,70 0,997
0,75 0,773 1,75 0,960 2,75 0,997
0,80 0,788 1,80 0,964 2,80 0,997
0,85 0,802 1,85 0,968 2,85 0,998
0,90 0,816 1,90 0,971 2,90 0,998
0,95 0,829 1,95 0,974 2,95 0,998
Таблица 8.4. 11
Процедура 2енерирования для Ф распределен ия
s o
t x
i l
repeat
s s+t
i i+2
t t.*x*x/i
until t<lE 16
phi O.5+s*exp( x*x/2)/sqr(2*pi)
109 Зак. 1712.
8.6. ЛАТИНСКИЕ КВАДРАТЫ
Приведенный ниже пример показывает,
как можно построить латинский квадрат, ис
пользуя 3 независимых перестановки.
1. rенерируют случайную перестановку
N возможных видов воздействия (доз) (см.
раздел 8.5):
Тз
5 з
51
Т 2
Т I
52
2. Используя эту перестановку, можно по
строить простой латинский квадрат. Для этоro
перестановку «поворачивают» по часовой
стрелке следующим образом. Полученную в
ходе выполнения шаrа 1 перестановку записы
вают в первой строке. Во вторую строку запи
сывают те же значения. но смещенные на один
столбец вправо. При этом крайнее правое зна
чение записывают в левую пустую ячейку. Про
цедуру повторяют до тех пор, пока каждое ис
следование не встретится по одному разу в
каждом столбце:
ТЗ 5 з 51 Т 2 Т I 52
52 Тз 5 з 51 Т 2 Т I
Т I 52 Тз 5 з 51 Т 2
Т 2 Т l 52 Тз 5 з 51
51 Т 2 Т7 52 ТЗ 5 з
5 з 51 Т 2 Т I 52 Тз
3. rенерируют две независимых случайных
перестановки натуральных чисел от 1 до N:
одну для строк:
236 1 4 5
и одну для столбцов:
3 4 6 2
5
1
4. Теперь можно построить латинский KBa
драт, сортируя строки и столбцы простоro латин
скоro квадрата в порядке возрастания для этих
двух перестановок для строк и столбцов:
2
3
Тз
1
52
6
51
4
5 з
2
Т 2
5
Т l
3
52
Т l
Тз
5 з
51
Т 2
6
52
Т 2
Т I
Тз
5 з
51
1
Т 2
51
Т l
52
Тз
5 з
4
51
5 з
52
Т 2
Т l
Тз
5
5 з
51
ТЗ
Т 2
!
Т I
52
866 rосударственная фармакопея Республики Беларусь
! СИМВОЛ Определение
1 2 3 4 5 6 S2 Оценка дисперсии остаточной по
1 SI Тз Т 2 Т I SЗ S2 rрешности (вариации). При OДHO
факторном дисперсионном анализе
2 S2 Т 2 ТЗ SЗ Т I SI рассчитывается как ошибка cpeд
неквадратической разности
3 Т I SI S2 ТЗ Т 2 SЗ t Критерий Стьюдента (таблица 8.2)
V11,V12,V22 (Ко)вариационные множители для
4 SЗ S2 SI Т 2 ТЗ Т I числителя и знаменателя отноше
ния т в теореме Филлера
5 ТЗ Т, SЗ SI S2 Т 2 W Весовой коэффициент (вес)
х Натуральный лоrарифм дозы
6 Т 2 Sз Т I S2 SI ТЗ Индивидуальный полученный pe
у
зультат или преобразованный pe
зультат
9. СЛОВАРЬ СИМВОЛОВ А Предполаrаемые активности испы
СИМВОЛ Определение туемых препаратов на стадии под
Точка пересечения с осью ординат бора доз
а
кривой линейной реrрессии зависи В Средний результат в плацебо ис
мости полученных результатов (эф пытании (контрольном испытании)
фектов) от дозы или натуральноro для модели уrловых коэффициен
лоrарифма дозы тов
Ь Уrловой коэффициент линейной С Статистика, используемая при pac
реrрессии зависимости полученных чете доверительных интервалов:
c=
результатов (эффектов) от дозы 1 9
или натуральною лоrарифма дозы С I "'" Сп Среднее значение для каждоro
столбца латинскоro квадрата
d Число уровней дозы для каждоro из Среднее значение для периода 1 и
препаратов (кроме плацебо препа 01,02
рата для модели уrловых коэффи периода 2 для двойной перекрест
циентов) ной схемы
F Отношение двух независи
е Основание натуральною лоrариф мых значений дисперсии при
ма (=2,71828182845905...) F распределении (таблица 8.1)
9 Статистика, при меняемая в Teope G s ' G]'... Усредненные значения доз, исполь
ме Филлера (Fieller): 9 с 1 зуемые при дисперсионном анали
С зе для модели уrловых коэффици
h Число препаратов, используемых ентов
при количественном определении, Множители, используемые при дис
включая стандартный препарат Нр H L
персионном анализе для модели
т Оценка активности, рассчитанная параллельных линий
как отношение эффектов в общей НВ' Н, Множители, используемые при дис
линейной модели персионном анализе для модели
п Число повторений для каждоrо уrловых коэффициентов
вида воздействия (дозы) Натуральный лоrарифм отноше
Вероятность тоro, что данная CTa ния между соседними дозами для
р модели параллельных линий или
тистика будет больше полученноro интервал между соседними дозами
значения. Используется также для для модели уrловых коэффициен
обозначения отношения r/п в про тов
бит MeToдe J s ' J]'...
Характеристики линейности, ис
r Число экспериментальных объек пользуемые при дисперсионном
тов в rруппе, у которых наблюдает анализе для модели уrловых коэф
ся эффект в ходе количественноro фициентов
определения в модели альтерна
тивных эффектов К Поправочный коэффициент, ис
пользуемый для расчета сумм KBa
s Оценка стандартноro отклонения дратов отклонений при дисперси
(== р) онном анализе
5.3. Статистический анализ результатов биолоаических испытаний
867
СИМВОЛ
L
Ls' Lp'..
м'
N
Ps,Pp...
R
R'
R1'"'' R n
S
SI..... Sd
SS
т, и, \1, ...
Т1 ,..., Td
V
w
х
У
z
а
f3
Определение
Ширина доверительноrо интервала,
выраженная в лоrарифмах
Линейные контрасты стандартноro
И испытуемоro препаратов
Натуральный лоrарифм отношения
активностей для данноro испыту
емоro препарата
Суммарное число испытаний
(видов воздействий или доз) в ходе
количественноro определения (=dh)
Сумма стандартноro (S) и испыту
емоro (т) препаратов
Рассчитанная(оцененная)актив
ность данноro испытуемоrо препа
рата
Отношение активностей данноrо
испытуемою препарата
Среднее значение данных в каждой
строке от 1 до п для схемы латин
скоro квадрата или в каждом блоке,
для схемы рандомизированных
блоков
Стандартный препарат
Среднее значение дозы (от мини
мальной дозы 1 до максимальной
дозы d) стандартноro препарата S
Сумма квадратов, обусловленная
данным ИСТОЧНИКОМ вариации
Испытуемые препараты
Среднее значение дозы (от мини
мальной дозы 1 до максимальной
дозы d) испытуемоro препарата Т
Коэффициент вариации, использу
емый при расчете rраниц довери
тельноrо интервала
Весовой коэффициент, применя
емый при объединении результатов
количественноro определения
Линейная структура или матри
ца планирования, используемая в
общих линейных моделях
Вектор, представляющий данные
(преобразованные) в общих линей
ных моделях
Первая производная от Ф
Верхняя асимптота кривой
«/п(доза) эффект» в четырех пара
метрическом анализе
Коэффициент наклона кривой
«/п(доза) эффект» в четырех пара
метрическом анализе
Натуральный лоrарифм дозы, обе
спечивающий 50 % эффекта в че
тырех параметрическом анализе
СИМВОЛ Определение
{) Нижняя асимптота кривой
«/п(доза) эффект» в четырех пара
метрическом анализе
тr 3,141592653589793238...
Ф Функция кумулятивноro CTaHдapT
ноro нормальноro распределения
характеристической кривой (табли
ца 8.4)
J! Критерий хи квадрат (таблица 8.3.)
10. ЛИТЕРАТУРА
В данном разделе приведен список peKOMeH
дуемой литературы для дополнительноro изучения
Finney, O.J. Probit Апа'уsis / O.J. Finney, з rd
Ed. Cambridge: Cambridge University Press, 1971
Ne'der. J.A. Genera'ized 'iпеаr mode's / J.A.
Ne'der, Wedderbuт. R. WM /1 Jouтa' о' the Roya'
Statistica' Society, Series А. 1972. Vo/. 135,
NQ3. Р 370 384.
ОеLеап, А. Siти'tапеоиs апа'уsis о' fami/ies
о' sigmoida' curves: App'ication to bioassay,
rаdiоligапd assay, апd physio'ogica/ dоsе rеsропsе
curves / А. OeLean, pJ. Мипsоп, О. Rodbard 1/ Ат.
J. Physio'. 1978. Vo/. 235, NQ2. PE97 E102.
Fiппеу, O.J. Statistica' Method iп Bi%gica/ Assay
/O.J. Fiппеу. :rd Ed. London: Griffiп, 1978.
Soka/, R.R. Biometry: Рriпсiр/еs and Practice о'
Statistics in Bio'ogica/ Research / R.R. Soka/, F.R. Roh/f.
L'd Ed. New York: WH Frеетапп & СО, 1981.
Реасе, КЕ. Biopharmaceutica/ Statistics for
Orug Оеvе/ортепtю New York/Base': Marce/
Oekker /пс., 1988.
Воwеrтап, B.L. Linear Statistica/ Mode/s ап
App'ied Approach / B.L. Воwеrтап, R. Т. О'Соппе/l
PubIishing Сотрапу 1990. 2nd Ed. Boston:
PWS KENT
Govindaraju'u, Z. Statistica' Тесhпiqиеs iп Bio
assay / Z. Gоviпdа,аjи/и. 2nd ,evised апd eп
'arged еditiоп. New York: Karger, 2001.
#Беленький, м.л. Элементы количественной
оценки фармаколоrическоro эффекта 1 МЛ. Бе
ленький. Л.: Медrиз, 1963. 152 с.
rланц, С. Медико биолоrическая статисти
ка / С. rланц. М.: Практика, 1998. 459 с.
Серrиенко, В.И. Математическая статисти
ка в клинических исследованиях / В.И. Серrи
енко, И.Б. Бондарева. М.: rэотар Медицина,
2000. 256 с.
Лапач, С.Н. Статистические методы в Me
дико биолоrических исследованиях с исполь
зованием Ехсе' / С.Н. Лапач, А.В. Чубенко,
П.Н. Бабич. К: МОРИОН, 2001. 408 с.
Юнкеров, В.И. Математико статистическая
оGработка данных медицинских исследований /
В.И. Юнкеров, C.r. rриrорьев. С. Пб.: ВМА,
2002. 267 с.
Реброва О.Ю. Статистический анализ меди
цинских данных. Применение пакета приклад
ных проrрамм STAТlSТlCA. / О.Ю. Реброва.
М.: Медиа Сфера, 2002. 305 с.#
868
rосударственная фармакопея Республики Беларусь
01/201З:РБ50З01
#5.3.1. СТАТИСТИЧЕСКИЙ АНАЛИЗ
РЕЗУЛЬТАТОВ ХИМИЧЕскоrо
ЭКСПЕРИМЕНТА
ПРИНЯТЫЕ ОБОЗНАЧЕНИЯ
В настоящей статье приняты следующие
обозначения:
А
Измеряемая величина
Свободный член линейной
зависимости
а
ь
Уrловой коэффициент
линейной зависимости
F
f(x,j.1,a)
Критерий Фишера
Функция плотности вероятно
сти нормальноro распределе
ния
Но
Н 1
Нулевая rипотеза
Альтернативная rипотеза
Порядковый номер варианты
Фактор, используемый при
оценке сходимости результатов
параллельных определений
Объемы выборки
Доверительная вероятность
без конкретизации постановки
задачи
Доверительная вероятность co
ответственно при ДBYX и OДHO
сторонней постановке задачи
Контрольные критерии для
идентификации rрубых поrреш
ностей
L
т,п
Р
Р 2 , Р,
Q1' Qn
Размах варьирования
Общий индекс корреляции
(линейный) коэффициент
корреляции
RSD S, .100% Относительное стандартное
отклонение, %
R
R
с
,
RSD x Sii., .100% Относительное стандартное OT
клонение среднеrо значения, %
8
Стандартное отклонение
Относительное (по отношению
к среднему результату) CTaH
дартное отклонение
Дисперсия
Относительная дисперсия
Стандартное отклонение
среднеro результата
8
,
82
82
,
8
х
8
Х,'
8'9
2
819
819)(
222
SO,8 b ,Sa
и
Х,у
Х;, v;
Х,у
Х" у,
х :tДХ
Х; :tдх
Д
а
13
у
Дх
Дх
!1 Х,'
Относительное (по отношению
к среднему результату) CTaH
дартное отклонение среднеro
результата
Лоrарифмическое стандартное
отклонение
Лоrарифмическая дисперсия
Лоrарифмическое стандартное
отклонение среднеro результата
Общая дисперсия и дисперсия
коэффициентов линейной зави
симости
Критерий Стьюдента
Коэффициент для расчета
rраниц среднеro результата ra
рантии качества анализируемо
ro продукта
Текущие координаты в ypaBHe
нии линейной зависимости
Вычисленные исходя из ypaB
нения линейной зависимости
значения переменных х и у
Средние значения выборки (KO
ординаты центра линейной за
висимости)
i--ая варианта (i ая пара экспери
ментальных значений х и у)
rраНИ4ные значения довери
тельноro интервала среднеro
результата
rраничные значения довери
тельноro интервала результата
единичноro определения
Разность некоторых величин
Уровень значимости, степень Ha
дежности; поrрешность перво
ro рода (вероятность принятия
rипотезы Н 1 в то время, как на
самом деле верна rипотеза Но)
Поrрешность BToporo рода (Be
роятность принятия rипотезы Но
в то время, как на самом деле
верна rипотеза Н 1 )
Критическая статистика
Полуширина доверительноro
интервала неопределенности
единичноro определения
Полуширина доверительноro
интервала неопределенности
среднеro результата
Полуширина относительноro
доверительноro интервала He
определенности единичноrо
определения
#5. 3. 1. Статистический анализ результатов химичеСКО20 эксперимента
869
11 ".r
Полуширина относительноro
доверительноro интервала He
определенности среднеro pe
зультата
Суммарная неопределенность
анализа
11 As.r
11 FAO r
Неопределенностьконечной
аналитической операции
Неопределенность аттестации
стандартною образца
Неопределенность
пробоподroтовки
Относительная величина
систематической поrрешности
Относительные неопределен
ности, соответственно, резуль
тата отдельноro определения и
среднеro результата
Истинное значение измеряемой
величины
!1 RS,r
[t"SP,r
д
&,&
11
/
Число степеней свободы; пере
менный объем выборки при по
следовательном анализе
«Эффективное» число степе
ней свободы в подходе Уэлча
Сатертуэйта
Знак суммирования (сумма)
Дисперсия rенеральной
совокупности
Критерий хи квадрат
У е "
I
(J"2
;(2
Метролоrические характеристики методик
и результатов, получаемых при статистической
обработке данных эксперимента, позволяют
про водить оценку и сравнение как эксперимен
тальных методик, так и изучаемых объектов, и
на этой основе решать ряд прикладных задач,
связанных с определением статистической дo
стоверности результатов исследования. В част
ности, описанные ниже статистические подходы
и метролоrические характеристики используют
ся при валидации разрабатываемых методик и
оценке корректности полученных результатов
анализа.
В rлавах 1 9 описаны подходы, применя
емые при статистическом анализе результатов,
являющихся функцией одной случайной пере
менной. Применение этих подходов для функ
ции нескольких случайных переменных описано
в rлаве 10. В rлаве 11 приведены необходимые
статистические таблицы.
При изложении материала используются
термины, принятые в статье #5.3.2. Валидация
аналитических методик и испытаний.
110З.,1712
1.ВЫБОРКА
Термином «выборка» обозначают совокуп
ность статистически эквивалентных результатов
(вариант). В качестве такой совокупности можно,
например, рассматривать ряд результатов, по
лученных при параллельных определениях co
держания какою либо вещества в однородной
по составу пробе. Отдельные значения вариант
выборки объема п принято обозначать через Х"
(1 :s; i:S; п). Упорядоченная в порядке возрастания
выборка может быть представлена в виде:
Х 1 ; Х 2 ; ...Х,; "'Xп 1; Х п . (1. 1)
Результаты. полученные при статистической
обработке выборки. будут достоверны лишь в
том случае, если эта выборка однородна. Про
верка однородности выборки обсуждается в
rлаве 1.2. Однако если целью испытаний явля
ется проверка однородности серии препарата
(например, при про ведении испытаний по опре
делению однородности содержания действу
ющеro вещества в единице дозированноro ле
карственноro средства (2.9.6) и однородности
дозированных единиц (2.9.40», то оцениваются
все полученные результаты (значения вариант)
без предварительной проверки однородности
выборки.
1.1. СРЕДНЕЕ ЗНАЧЕНИЕ И ДИСПЕРСИЯ
В большинстве случаев среднее выборки
является наилучшей оценкой истинною значе
ния измеряемой величины р, если ero вычисля
ют как среднее арифметическое всех вариант:
п
LX,
x .(1.1. 1)
п
При этом разброс вариант Х, BOKpyr cpeДHe
ro Х характеризуется величиной стандартною OT
клонения S. В количественном химическом aHa
лизе величина s часто рассматривается как мера
случайной поrрешности, свойственной данной
методике анализа. Квадрат этой величины S<
зывают дисперсией. Величина дисперсии может
рассматриваться как мера воспроизводимости
(сходимости) результатов, представленных в
данной выборке. Вычисление величин sz и s п
водят по уравнениям 1.1 А и 1.1. S Иноrдa для
этою предварительно определяют значения от
клонений d; и число степеней свободы (число He
зависимых вариант) ':
d, Х x ,(1.1. 2)
l' = П 1 . (1.1. З)
Ld,2 LX пx2
S2 ,(1.1А)
\' \'
s ,, . (1.1. 5)
870
rосударственная фармакопея Республики Беларусь
Стандартное отклонение среднеro резулыа
та Sx рассчитывают по уравнению:
S
Sx == .Jп . (1.1. 6)
Во мноrих случаях при контроле качества
лекарственных средств целесообразно исполь
зовать относительные (по отношению к х) вели
чины относительное стандартное отклонение
8,. относительную дисперсию s; и относитель
ное стандартное отклонение среднею резулыа
та Sx,,' Их рассчитывают по формулам:
2 82
S, == ,(1.1Аа)
х'
8
8, == -= ,(1.1. 5a)
Х
S,
S.. =
"п
. (1.1. 6a)
Эти относительные величины в зависи
мости от решаемой задачи MorYT выражать
ся также и в ilроцентах к Х. В этом случае они
часто обозначаются, соответственно, как RSO
и RSD" :
RSD == 8, .100% ,(1.1. 5b)
RSD, ==8х., .100% . (1.1. 6b)
В фармакопейном анализе абсолютные Be
личины обычно используют для прямых, а OTHO
сительные для косвенных методов анализа.
При мер расчетов приведен в rлаве 9.1.
Если при измерениях получают лоrариф
мы искомых вариант, среднее выборки вычисля
ют как среднее rеометрическое, используя лоrа
рифм вариант:
L1gx,
Igx = ,(1.1. 7)
9 п
откуда
х = X"X2 ""'Х п =aпtilg(lgxJ. (1.1. 8)
Значения 82, 8 И S" В этом случае также pac
считывают исходя из лоrарифмов вариант и обо
значают соответственно через SI ' 5 1g и 5,gx'
1.2. ПРОВЕРКА ОДНОРОДНОСТИ
ВЫБОРКи. ИСКЛЮЧЕНИЕ ВЫПАДАЮЩИХ
ЗНАЧЕНИЙ ВАРИАНТ
Как было указано выше, значения Х, S2, S И
81< MorYT быть признаны достоверными, если ни
одна из вариант выборки не отяrощена rрубой
поrрешностью, т. е. если выборка однородна.
Выявление rрубых поrрешностей (выбросов)
это весьма деликатная задача, относительно
которой в литературе нет единоro устоявше
roся мнения (смотрите, например, Раздел 7.3
статьи 5.3. Статистический анализ результа
тов биОЛ02ических испытаний и тестов). Oco
бенно это относится к выборкам совсем малоro
объема (3 5 измерений). Проверку таких выбо
рок на однородность целесообразно проводить
только в том случае. если методика метроло
rически аттестована (см. rлаву 6.1). Ниже при
водятся наиболее часто используемые подхо
ды для проверки однородности выборок малоro
(п::>10) и большоro (п>10) объемов.
Про верка однородности выборок малоro
объема (п::>1 О) осуществляется без предвари
тельною вычисления статистических xapaK
теристик. С этой целью после представления
выборки в виде (1. 1) для крайних вариант Х,
и Х п (которые предполаrаются выпадающими)
рассчитывают значения контрольною крите
рия Q исходя из величины размаха варьиро
вания R:
{ IX, Х п ! при п = 3... 7 (1.2. 1)
R = 'х, Xп ,1 при п == 8...10
'х, X21
о, == R ' (1.2. 2a)
' Х x I
Оп == п п 1 . (1.2. 2b)
R
Выборка признается неоднородной, если
хотя бы одно из вычисленных значений О, или
Оп превышает табличное значение Q(Р"п), най
денное для доверительной вероятности Р, (Ta
блица 11. 1). Варианты Х, или Х п , которым COOT
ветствует значение Q>Q(Р"п), отбрасываются, и
для полученной выборки уменьшенноro объема
выполняют новый цикл вычислений по ypaBHe
ниям 1.2. 1 и 1.2. 2 с целью проверки ее OДHO
родности.
При :х, х 2 1 < /Х 2 хзl и 'Х п Xп ,1 < 'Xп , х п2 1
уравнения 1.2. 2a и 1.2. 2b принимают вид:
Q == !Х 2 хзl ; Q == Xп ' Xп.2 . (1.2. 3)
, R п R
Полученная в конечном счете однородная
выборка используется для вычисления Х, S2, S
И Sx'
При мер расчетов приведен в rлаве 9.2.
Для выборок большоro объема (п>1 О) про
верку однородности проводят после предвари
тельноro вычисления статистических характери
стик Х, 82, S И Sii' При этом выборка признается
однородной, если для всех вариант (1.1. 2) BЫ
полняется условие:
i d ;l::>зs . (1.2А)
Если выборка признана неоднородной, то
варианты, для которых Id,1 > 3S, отбрасывают
#5. З. 1. Статистический анализ результатов химическоzо эксперимента
871
ся как отяrощенные rрубыми поrрешностями с
доверительной вероятностью Р 2 >99,0 %. В этом
случае для полученной выборки сокращенноro
объема повторяют цикл вычислений статисти
ческих характеристик по уравнениям (1 .1. 1
1.1. 6) и снова проводят проверку однородности.
Вычисление статистических характеристик счи
тают законченным, Korдa выборка сокращенноro
объема оказывается однородной.
1.3. ДОВЕРИТЕЛЬНЫЕ ИНТЕРВАЛЫ
И ОЦЕНКА ИХ ВЕЛИЧИНЫ
Если случайная однородная выборка конеч
ноro объема п получена в результате последо
вательных измерений некоторой величины А,
имеющей истинное значение /1, то среднее этой
выборки х следует рассматривать лишь как
приближенную оценку А. Достоверность этой
оценки характеризуется величиной доверитель
Horo интервала х 1: Дх' для которой с заданной
доверительной вероятностью Р выполняется
условие:
(x д, )<::р<::(х+д,). (1.3. 1)
Следует отметить. что данный доверитель
ный интервал не характеризует (как это Hepeд
ко считается) поrрешность определения величи
ны t, поскольку найденная величина Х может
быть в действительности очень близка к истин
ному значению /1. Но мы этоrо истинноro зна
чения не знаем. Полученный доверительный
интервал характеризует степень неопределен
ности наших знаний об истинном значении /l Be
личины А по результатам последовательных из
мерений выборки конечноro объема п. Поэтому
правильно roворить (и далее это будет исполь
зоваться) о «неопределенности результатов
анализа» (которая характеризуется доверитель
ным интервалом) вместо выражения «П02реш
ность результатов анализа», которое нередко
используется не совсем корректно.
Расчет rраничных значений доверительно
ro интервала при известном значении CTaHдapT
Horo отклонения S или для выборок большою
объема проводят по уравнению
( ) U(Р)'5 (1 3 2)
Х1:Дх =х:!: Jп ' ..
предполаrая, что варианты, входящие в выбор
ку, распределены нормально. Здесь U(Р) Ta
бличное значение функции нормальноro распре
деления.
Для выборок небольшоro объема rраничные
значения доверительноro интервала рассчиты
вают с использованием критерия Стьюдента:
((Р 1')'5
(X:tt'1,)=X1: " (1.3. 3)
'-Jn
или с использованием относительных вели
чин:
( 1+ д х ) = ( 1+ д ) = 1+ t(P,I')'S( , ( 1.3. 3a )
Х xr Jп
rдe:
(Р, \/) табличное значение критерия
Стьюдента (таблица 11. 2). Распределение
Стьюдента (Р, v) является обобщением HOp
мальноro распределения U(Р) и переходит в неro
при достаточно большом числе степеней CBO
боды V, т. е. (Р. \/ ) U(Р). С учетом этоro для
единообразия далее везде будут использовать
ся более часто употребляемые соотношения
(1.3. 3) и (1.3. 3a). даже если речь идет об об
работке выборок достаточно большою объема.
Полуширины относительных доверитель
ных интервалов единичною (/'..,,) и среднеro
(Д, () результатов часто выражают в процентах
к Х. В этом случае в выражении (1.3. 3a) вместо
величины S. используют RSO. а вместо 1 берут
100 %. т- е.:
(100 z: дж' %)= 100 z: t(P.I') RSO (1.3. 3b)
"п
Если при измерении одной и той же методи
кой двух близких значений А были получены две
случайные однородные выборки с объемами п и
т, то при т<п для выборки объема т справед
ливо выражение:
+ t(p, V(nJ s(n) ( 1 3 4 )
Хlт):tll.жiтl Xlтl Jт . .
(индекс указывает принадлежность величин к
выборке объема т или п).
Выражение (1.3.4) позволяет оценить Be
личину доверительноrо интервала среднеrо
X 1тl , найденноro исходя из выборки объема т.
Иными словами, доверительный интервал cpeд
Hero выборки относительно малою объема т
может быть сужен блаroдаря использованию из
вестных величин S\n) И (Р, V(п)' найденных ранее
для выборки большеro объема п. Более общим
подходом является объединение выборок с pac
четом объединенноrо стандартною отклонения
и степеней свободы по уравнениям (2.1.1. 1
2.1.1. 2). Это стандартное отклонение и COOTBeT
ствующий объединенному числу степеней CBO
боды критерий Стьюдента подставляются затем
в выражение (1.3А).
Пример расчетов в процентах приведен в
rлаве 9.3.
Аналоrично (1.3. 1 1.3. 3) определяется
доверительный интереал результата отдельно
ro определения. Подставляя п=1 в выражение
1.3. 3, получаем:
Х, 1:Д, = Х. :tt(P,I')'S (1.3. 5)
или, с использованием относительных вели
чин:
872
rосударственная фармакопея Республики Беларусь
Х, Х; ((Р )
х :!:Дх., = х :!: ,V 'S, . (1.3. 5a)
Этот интервал является доверительным ин
тервалом результата отдельноrо определения.
Для неro с доверительной вероятностью Р BЫ
полняются взаимосвязанные условия:
Х, Дx J1 х, + 1'>.. , (1.3. 6)
J1 Llx X, J1+.')... (1.3. 7)
Значения 1'>., и 1'>.. из выражений (1.3. 3) и
(1.З. 5) используют при вычислении относитель
ных неопределенностей отдельной варианты
(Б-) И среднеro результата (с), выражая эти Be
личины в процентах:
&=I'>.xr .100 % .100 %, (1.3. 8)
х
z = I'>.xr .100 % = .100 %. (1.3. 8a)
х
Если при измерениях получают лоrарифмы
исходных вариант. выражения (1.3. 3) и (1.3. 5)
принимают вид:
. t(P.I')'SI
Igx:!:.').,,> =Igx:: 9 , (1.3. 9)
"';п
Igx :: .').'9' Igx :: t(P,I')'SI9' (1.3. 10)
Потенцирование выражений (1.3. 9) и
(1.3. 10) приводит к несимметричн м довери
тельным интервалам для значений Х ИХ,:
aпti IgIIg х . -;. ' х aпti Ig(lg х + 1'>.19.) , (1.3. 11)
aпti Ig(lg х. \<;., х aпti Ig(lg Х, + 1'>.'9.) , (1.З. 12)
rдe:
л . = tIP.I.j.s.; , ( 1.3. 13 )
L.1 lg >:
,п
1'>.19 X =t(P.I')'So _ (1.3.14)
При этом для нижних и верхних rраниц ДOBe
рительных интервалов Х и химеем:
[ lantilg(19X:!: 1'>.19X) x i ]
if = .100 %, (1.3. 15a)
х
[ iaпtiI9(IYX, :!:1'>.19J X'! j
& = I .100 %. (1.3. 15b)
х,
1.4. ОДНОСТОРОННИЕ И ДВУСТОРОННИЕ
ДОВЕРИТЕЛЬНЫЕ ИНТЕРВАЛЫ
Соотношения (1.3. 1 1.3. 15) характеризу
ют так называемые «двусторонние» доверитель
ные интервалы. Они основаны на двустороннем
t распределении и широко при меняются при
оценке точности методик и представлении pe
зультатов. Однако при решении вопросов rapaH
тии качества продукции (rлава 6), а также при
контроле серийной продукции, в частности, при
контроле качества лекарственных средств, He
редко возникает необходимость использования
так называемых «односторонних» доверитель
ных интервалов. Например, для какоro нибудь
roтовоro лекарственноro средства допуски co
держания активноro компонента установлены от
90 % до 11 О % от номинальноro. В процессе aHa
лиза получено среднее значение содержания
Х =94 % от номинальноro значения. Нас инте
ре сует, не выходит ли доверительный интервал
за допуски содержания (90 11 О %). Очевидно,
что в данном случае этот доверительный интер
вал может выйти за пределы только нижнеro дo
пуска (90 %), но не нижнеro и верхнеro (110 %)
одновременно. Вопрос о возможности выхода
истинной величины J1 за пределы верхнеro допу
ска нас в данном случае не интересует (в связи
с еro крайне низкой вероятностью). Таким обра
зом, истинное значение J1 находится в интервале
х Дх J1 S ос! . (1.4. 1a)
Аналоrичное выражение можно записать
для случая, Korдa Х превышает 100 % (напри
мер, Х =105 %):
OC J1 x+Llx .(1.4. 1b)
Соотношения (1.4 1 a 1.4. 1 Ь) характери
зуют односторонние доверительные интерва
лы, поскольку величина J1 ими оrраничивается
только с одной стороны. Это отличает их от COOT
ношения (1.3. 1), rдe величина J1 оrраничивается
с обеих сторон. Табличные значения критерия
Стьюдента для одностороннеro и двустороннеro
распределения приведены в таблице 11. 2. Cy
ществует следующее соотношение между ДBY
сторонним (Р 2 ) И односторонним (Р 1 ) критериями
Стьюдента:
t[P2.1'] = t[(2P1 1).1']. (1.4. 2)
в частности, односторонний критерий Стью
дента для вероятности 0,95 (т. е. 95 %) совпада
ет с двусторонним критерием Стьюдента для Be
роятности 0,90 (т. е. 90 %).
Таким образом, Р 2 это вероятность тоro,
что математическое ожидание (или истинное
значение) оцениваемой величины находится в
двусторонне оrраниuенных пределах (1.3. 1
1.3. 15), а Р 1 это вероятность тоro, что оно Ha
ходится в односторонне оrраниченных пределах
(1.4. 1 1.4. 2). В литературе (в частности, в Ta
блицах) нередко используются обозначаемые
по разному величины (1 P) и (1 P1)' которые xa
рактеризуют вероятность тоro, что математиче
#5. 3.1. Статистический анализ результатов химичеСКО20 эксперимента
873
ское ожидание (или истинное значение) оцени
ваемой величины выходит за вышеуказанные
пределы. Во мноrих случаях такие величины яв
ляются более удобными.
2.МЕтРолоrИЧЕСКИЕ
ХАРАКТЕРИСТИКИ МЕТОДИКИ АНАЛИЗА
Метролоrические характеристики методи
ки устанавливают путем статистической обра
ботки одной выборки или совместной стати
стической обработки нескольких выборок из
одной и той же rенеральной совокупности. В
качестве таких выборок MorYT использоваться
как данные аналитическоro архива лаборато
рии, так и результаты, полученные специально
при анализе образцов с известным содержа
нием определяемоro компонента /1. Результа
ты статистической обработки MorYT быть пред
ставлены в виде таблицы 2. 1.
Таблица 2. 1
МетРОЛ02ические характеристики методики
анализа
Jl V Х S Р t(P,,'1 .... Е:
1 2 3 4 5 6 7 8
Во мноrих случаях проще использовать OTHO
сительные (по отношению к р) величины. Резуль
таты статистической обработки MOryт быть пред
ставлены в этом случае в виде таблицы 2. 1a.
Таблица 2. 1a
МетРОЛ02ические характеристики методики
анализа
Jl V x/f.1 S S r Р t(p, \1 ) l1н [;
1 2 3 4 5 6 7 8 9
2.1. ОБЪЕДИНЕНИЕ ВЫБОРОК
2.1.1. Объединенная дисперсия и объе-
диненное среднее
Если имеется 9 выборок из одной rенераль
ной совокупности с порядковыми номерами k
(1 k g), расчет дисперсии S2 целесообразно
проводить по формуле:
rId: r[(п; 1)s:] I ( 'fx: п;x;.1
52= ; 1,=1 = ;=1 = ;=, ,=1 (2.1.1. 1)
У, О', ",
или для относительных величин, принимая
во внимание, что п k 1 = Il k
k=g
L>k . S:.r
S2 = k=, ,(2.1.1. 1a)
r
\'
,
k g
L>k . RSO:
RSOt , = ы . (2.1.1. 1b)
У,
111 Зак 1712
При этом объединенное число степеней
свободы V/ равно
9
V t = L>k' (2.1.1. 2)
k='
X k среднее k ой выборки;
П k число вариант в k ой выборке;
V/ число степеней свободы в k ой BЫ
борке;
X'k i ая варианта k ой выборки;
8 дисперсия k ой выборки;
8t r относительная дисперсия k ой BЫ
борки;
d ik отклонение i ой варианты k ой BЫ
борки.
Если 9 выборок из одной rенеральной co
вокупности С порядковыми номерами k (1 k g)
характеризуются выборочными средними значе
ниями Х". rюлученными из п. риант, то объе
диненное среднее значение х по всем выбор
кам рассчитывают по формуле:
"=9
L П k . X k
Х = k=:=9 . (2.1_1. 3)
LП k
k=1
Необходимым условием совместной CTa
тистической обработки нескольких выборок яв
ляется отсутствие статистически значимой раз
ницы между отдельными значениями S: (т. е.
справедливость rипотезы равенства дисперсий).
В простейшем случае можно оrраничиться cpaB
нением крайних значений S: с использовани
ем критерия Фишера F, как указано в rлаве 3. В
более общем случае используют критерии Барт
лена и Кохрена.
2.1.2. Критерий Бартлетта
Для проверки rипотезы, что все S; принад
лежат одной rенеральной совокупности, исполь
зуют выражение, приближенно распределенное
как ;(2:
2 ( 2 -f 2\
х =2,303. v,.lgs t{k.19Sk J(2.1.2. 1)
При этом величины S2 и 1', рассчитывают
ся по уравнениям (2.1.1. 1) и (2.1.1. 2). Найден
ная таким образом величина ;(2 сравнивается с
процентной точкой хи квадрат распределения
;( 2 (Р" v )') (таблица 11. 3). Если имеется 9 выбо
рок, то число степеней свободы для ;(2 (р" V х )
берется равным \. = 9 1. Проверяемая rипо
теза принимается / при условии ;(2 < ;(2 (Р" v х ).
в противном случае вычисленное значение ;(2
корректируют по формуле
. 2
1"2 = L , (2.1.2. 2)
1'. С
874
rосударственная фармакопея Республики Беларусь
rдe
9
L (1/ V k ) 1 f У,
С = k 1 + 1
3(g 1) ,
и снова сравнивают с процентной точкой хи квадрат
распределения X 2 (P1,VJ. Если x"Z > ./(Pl'l'.i)' то
ме>IЩУ некоторыми стандартными отклонениями
имеются значимые различия. В этом случае необ
ходимо провести анализ имеющихся данных, OT
бросить одно или несколько значений дисперсии,
наиболее сильно отличающиеся от остальных, и
снова провести тест Бартлетта. Нужно иметь в виду,
что критерий Бартлетта (так же, как и критерий Kox
рена) очень 4увствителен к нарушению требования
нормальности. Но именно по--этому он может быть
весьма полезен при формировании надежных aHa
литических архивов.
Описанный критерий Бартлетта применим
только при условии, что число степеней свободы у
всех объединяемых ДИOlерсий больше 3 (т- е. все
v k >3). Однако именно этот случай, Korдa v k s3, He
редко и представляет наибольший интерес. Поэто
му Бартлеттом была предложена более сложная
модификация данною критерия, применимая при
любых степенях свободы. Однако использование
ее на практике достаточно затруднительно без при
менения ЭВМ
2.1.3. Критерий Кохрена
В том случае. KorAa все объединяемые дис
персии имеют одинаковое число степеней CBO
боды (т. е. .! = "2 = ... = V 9 = V), дЛЯ про
верки rипотезы равенства дисперсий можно
при менять значительно более простой критерий
Кохрена со статистикой:
G = :", ,(2_1.2. З)
t s:
S ". = maxls I
Критические точки критерия Кохрена приведе
ны в таблице 11.--4. Рассчитанное значение G на
выбранном уровне значимости (95 % или 99 %) не
должно превосходить табличное значение. В про
тивном случае rипотеза равенства дисперсий не
может быть принята и формулы (2.1.1. 1 2.1.1. 2)
объединения выборок не являются корректными.
В формулах (2.1.1. 1) и (2.1.2. 3) вместо аб
солютных величин s; MorYT использоваться OT
носительные величины S;k и RSO k .
При меры использования критериев Барт
лена и Кохрена приведены в rлаве 9.4.
2.2. ПРОВЕРКА НАЛИЧИЯ ЗНАЧИМОЙ
СИСТЕМАТИЧЕСКОЙ поrРЕШНОСТИ
При известном содержании определяемоrо
компонента J1 в образце следует решить вопрос
о наличии статистически значимой систематиче
ской поrрешности. Для этоro вычисляют крите
рий Стьюдента t:
'р :x1..Jт
t= (2.2. 1)
s
или, в относительных величинах:
! :x1
11 I.Jт
t = 1 . (2.2. 1a)
Sr
Если, например, при Р=95 % и v = т 1, pe
ализуется неравенство
t > ((Р, у), (2.2. 2)
то полученные данной методикой результаты
отяroщены систематической поrрешностью, OT
носительная величина которой д может быть
оценена по формуле:
I I
, Х,
д =:1 : .100 % . (2.2. 3)
PI
Значимые систематические поrрешности
(т. е. поrрешности, для которых реализуется He
равенство (2.2. 2) должны быть обязательно ис
ключены из результатов анализа.
При мер расчета приведен в rлаве 9.4.
При проведении статистической обработ
ки нескольких выборок, полученных при анали
зе образцов с разным содержанием определя
емоro компонента р, данные в rрафах 1, 2, 3,
4, 7 и 8 таблицы 2. 1 при водят непосредствен
но для каждой выборки. При этом в rрафах 2,
4, 6, 7 в последней строке под чертой приводят
средние значения V, s, t, Д х .
Также для расчета метролоrических xapaK
теристик методики используются данные из aHa
литическоro архива, значение /-1 неизвестно и,
соответственно, заполняются не все rрафы Ta
блицы 2. 1.
3. СРАВНЕНИЕ ДВУХ МЕТОДИК АНАЛИЗА
ПО ВОСПРОИЗВОДИМОСТИ
Данное сравнение про водят путем выяс
нения значимости различия выборочных дис
персий анализа этих двух методик. В более
общем случае данный подход применяется
для оценки значимости различия двух выбо
рочных дисперсий, например, с целью BЫ
яснения, можно ли их считать выборочными
оценками одной и той же дисперсии reHe
ральной совокупности.
При сравнении воспроизводимости (сходи
мости) двух методик анализа с оценками дис
персий s; и s (s; > S ) вычисляют критерий
Фишера F:
2
F= . (3. 1)
S2
2
Критерий F характеризует при s; > S дocтo
верность различия между s; и S .
#5.3.1. Статистический анализ результатов химическосо эксперимента
875
Вычисленное значение F сравнивают с Ta
бличным значением F(Ppl',,v2)' найденным при
Р,=99 % (таблица 11. 5).
Если
F> F(P"I/,,1'2)' (3. 2)
то различие дисперсий и признается статистиче
ски значимым с вероятностью Р" что позволяет
сделать заключение о более высокой воспроиз
водимости второй методики. При
F F(p" ',,lf2) (З. 3)
различие дисперсий S; и S; не может быть при
знано значимым, и заключение о различии BOC
производимости (сходимости) методик сделать
нельзя ввиду недостаточною объема информа
ции.
Если
F(P, O,95'\'1, f7)< F < F(P. O.99y..\' ', (3.4)
целесообразно провести дальнейшие
экспериментальные исследования для MeTO
дики С лучшей воспроизводимостью.
При сравнении двух методик анали
за результаты статистической обработки
MorYT быть представлены в виде таблицы
(3. 1). Сравнение желательно проводить при
11, 112, 1/, > 10 и 1'2 > 10. Если точные значе
ния J.1, и J.1 2 неизвестны, величины б и t выч не
определяют.
Если при измерениях получают лоrариф
мы исходных вариант, вместо величин 11, х и
S в таблице З. 1 приводят величины Ig,u, Igx g и
Sg. При этом в rрафу 8 вносят величину Дlgх, а
в rрафу 9 максимальное по абсолютной Be
личине значение Е . Аналоrичные замены про
водят при вычислении t по уравнению (2.2. 1)
и F по уравнению (3. 1).
Пример расчетов приведен в rлаве 9.5.
4. МЕтРолоrИЧЕСКАЯ ХАРАКТЕРИСТИКА
СРЕДНЕrо РЕЗУЛЬТАТА
Если с помощью данной методики анализа
(измерения) следует определить значение He
которой величины А, то для эксперименталь
но полученной однородной выборки объема т
рассчитывают величины, необходимые для за
полнения таблицы 4. 1. Если методика имеет
метролоrическую аттестацию, rрафы 2, 4,5,7,
8 и 9 таблицы 4. 1 заполняются на основании
данных таблицы 2. 1. Это позволяет значи
тельно сузить rраницы доверительноro интер
вала за счет большеro числа степеней свобо
т+п
ды (уравнение (1.3-А). Если п:515, а > 1,5,
n
величины S и \/ целесообразно вычислять по
формулам (2. 1) и (2. 2).
Таблица 4. 1
МетРОЛ02ические характеристики средне20
результата
т l' ! Х S Sx Р ((Р, 11 ) fl.x fl.x или с
X::t6,
,
1 21з ,4 5 6 7 8 9 10
I
Во мноrих случаях проще использовать OT
носительные (по отношению к х) величины. В
этом случае целесообразно проводить расчеты
по таблице 4. 1a.
Таблица 4. 1a
МетРОЛ02ические характеристики средне20
результата
I т! \. I Р 1 t(P,I') 6.. "i
I Х S S s :
! х.!
1 , 2 3 4 5 6 7 8 9 10
,
i i I , I
I i
Таким образом. на основании выражения
(1.3. 1) для измеряемой величины А при незна
чительности систематической поrрешности с Be
роятностью Р выполняется условие:
X [-..x ="А="Х+6 х , (4. 1)
т. е.
А == х::!: 6" (4. 2)
или с использованием относительных вели
чин:
А
-= == 1::!: [-.." r . (4. 2a)
х .
Если при измерениях получают лоrарифмы
исходных вариант, в rрафе 9 таблицы 4. 1 приво
дят величину [-..g х ' а каждую из rраф 3, 9 и 1 О раз
бивают на две (а, б). В rрафе За приводят значе
ние Х 9 , в rрафе 3б значение Ig х g' в rрафах 9а
и 9б соответственно значения нижней и Bepx
ней rраниц доверительною интервала для Х 9
(уравнения (1.3. 11) и (1.3. 12)). Наконец. в rрафе
1 О при водят максимальное по абсолютной вели
чине значение Е (уравнение (1.3. 15a)).
5. СРАВНЕНИЕ СРЕДНИХ РЕЗУЛЬТАТОВ
ДВУХ ВЫБОРОК
Если в результате измерений одной и той же
величины А получены две выборки объема n. и
п , причем х, * х? может возникнуть необходи
мость проверк статистической достоверности
rипотезы:
Х. .. Х. . (5. 1)
т. е. значимости разности ,х, х 2 1.
Такая проверка необходима, если величи
на А определялась двумя разными методиками
876
rосударственная фармакопея Республики Беларусь
Таблица 3. 1
Данные для сравнительной метРОЛ02ической оценки двух методик анализа
Методика Jl v х s Р t(P.I' ) "". t F(P, , 1',,1'2) F Приме
NQ п/п [; еыч (табл.) Р=99 % еыч i5 чания
1 2 3 4 5 6 7 8 9 10 11 12 13 . 14
1
2
с целью их сравнения или если величина А опре
делялась одной и той же методикой для двух
разных объектов, идентичность которых требу
ется доказать. Для проверки rипотезы (5. 1) сле
дует установить, существует ли статистически
значимое различие между дисперсиями и s; и
s; . Эта проверка проводится так, как указано в
rлаве 3.
Рассмотрим три случая.
5.1. РА3ЛИЧИЕ ДИСПЕРСИЙ s; И s;
СТАТИСТИЧЕСКИ НЕЗНАЧИМО
Различие дисперсий S; и S; статистически
незначимо, Korдa справедливо неравенство (3. З).
В этом случае средневзвешенное значение 52
вычисляют по уравнению (2.1.1. 1), а дисперсию
S разности .х, Х 2 по уравнению (5.1. 1):
s;= J!7 пJ ,(5.1. 1)
П, 'П;
Sp=\.S (5.1. 2)
Далее вычисляют критерий Стьюдента:
Х. Х; Х. Х;: . : П, 'П 2 (5 1 3)
t . ,
Sc S \' П, + п 2 ' ..
1'=П' П2 2 . (5.1,А)
Если при выбранном значении Р 2 (напри
мер, при Р 2 =95 %)
t > ((Р 2 .1") . (5.1. 5)
то результат про верки положителен: разность
(х, х 2 ) является значимой, и rипотезу х, = )(2
отбрасывают. В противном случае надо при
знать, что эта rипотеза не противоречит экспе
риментальным данным.
5.2. РАЗЛИЧИЕ ДИСПЕРСИЙ s; И s;
СТАТИСТИЧЕСКИ ЗНАЧИМО
Различие дисперсий S; и S; статистически
значимо, Korдa справедливо неравенство (3. 2).
Если s; > S; , дисперсию s разности !х, )(21 Ha
ходят по уравнению (5.2. 1), а число степеней
свободы по уравнению (5.2. 2):
2 2
8 = + ,(5.2. 1)
П, П;
1" = (п, + П 2 2)J 0,5 + S;2 . 8;. . ] . (5.2. 2)
l S, + 82
Следовательно, в данном случае
t JX1 )(;1 = 'Х )( 1. П , 'П 2 . (5.2. 3)
Sp 1 2 П 2 . S; + п, . S;
Вычисленное по уравнению (5.2. 3) значение
t сравнивают с табличным значением t(P2'V'), как
это описано выше для случая 1.
Рассмотрение проблемы упрощается, Korдa
П'==П 2 и 8;»8;. Torдa в отсутствие систематиче
ской поrрешности среднее )( выборки объема
П 2 принимают за достаточно точную оценку Be
личины А, т. е. принимают )(2 = fJ. Справедли
вость rипотезы )(, = fJ эквивалентной rипотезе
(5. 1) проверяют с помощью выражений (2.2. 1) и
(2.2. 2), принимая У, = П, 1. rипотеза (5. 1) OT
клоняется как статистически недостоверная,
если выполняется неравенство (2.2. 2).
5.3. И3ВЕСТНО ТОЧНОЕ 3НАЧЕНИЕ
ВЕЛИЧИНbI А
Если А=т, проверяют две rипотезы: )(, = fJ
и )(2 = fJ. Проверку выполняют так, как описа
но в rлаве 2 с помощью выражений (2.2. 1) и
(2.2. 2), отдельно для каждой из rипотез. Если
обе проверяемые rипотезы статистически дo
стоверны, то следует признать достоверной и
rипотезу (5. 1). В противном случае rипотеза
(5. 1) должна быть отброшена.
Если при измерениях получают лоrарифмы
исходных вариант, при сравнении средних исполь
I 2 В
зуют величины 9 Х g' SI9 И S19' тех случаях, Korдa
разность 1)(1 )(21 оказывается значимой, опреде
ляют доверительный интервал для разнос:rи соот
ветствующих rенеральных средних/х, х 2 1:
Ix, х) t(P;, 1')' Sp $IX, х 2 1 $IX, х 2 1 + t(P 2 ,1')' Sp. (5.3. 1)
Пример расчетов приведен в rлаве 9.6.
6. ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ
АНАЛИЗА, ПОЛУЧЕННЫХ С ПОМОЩЬЮ
МЕтРолоrИЧЕСКИАТТЕСТОВАННОЙ
МЕТОДИКИ
Данная интерпретация основывается на
том, что для метролоrически аттестованной Me
тодики известна принятая оценка стандартноrо
отклонения.
6.1. ОЦЕНКА СХОДИМОСТИ РЕЗУЛЬТАТОВ
ПАРАЛЛЕЛЬНblХ ОПРЕДЕЛЕНИЙ
При рутинных анализах аналитик обычно
проводит 2 3, реже 4 параллельных опреде
#5.3.1. Статистический анализ результатов химичеСКО20 эксперимента
877
ления. Варианты полученной при этом упорядо
ченной выборки объема т, как правило, доволь
но значительно отличаются друr от друrа. Если
методика анализа метролоrически аттестована,
то максимальная разность результатов двух па
раллельных определений должна удовлетво
рять неравенству:
IX, xпl < L(P,m).s. (6.1. 1)
rдe:
s п-ринятая оценка стандартноro отклоне
ния;
ЦР,т) фактор. вычисленный по Пирсону
при Р=95 %.
Если неравенство (6.1. 1) не выполняется,
необходимо провести дополнительное опреде
ление и снова проверить, удовлетворяет ли Be
личина IX 1 xпl неравенству (6.1. 1).
т
L
2
2,77
4
3,65
3
3,31
Если для результатов четырех naраллелtr
ных определений неравенство (6.1. 1) не выпол
няется, следует считать, что конкретные условия
анализа привели к снижению воспроизводимости
методики и принятая оценка величины s примени
тельно к данному случаю является заниженной. В
этом случае поступают, как указано в rлаве 1.2.
6.2. ОПРЕДЕЛЕНИЕ НЕОБХодимоrо
ЧИСЛА ПАРАЛЛЕЛЬНЫХ ОПРЕДЕЛЕНИЙ
Если необходимо получить средний резуль
тат с относительной неопределенностью & rp
(rдe ер некоторое число, например, 2 %),
причем методика анализа метролоrически ат
тестована, необходимое число параллельных
определений т находят из уравнения:
т? ( Дх - o ) 2. (6.2. 1)
(j)-X
6.3. rАРАНТИЯ КАЧЕСТВА ПРОДУКЦИИ
Описанный ниже подход применим к Me
тролоrически аттестованной методике. В друrих
случаях MOryт применяться иные подходы (см.
общую статью #5.3.2. Валидация аналитиче
ских методик и испытаний).
Предположим, что качество продукции pe
rламентируется предельными значениями a m1r и
а тах величины А, которую определяют на OCHO
вании результатов анализа. Примем, что вероят
ность соответствия качества продукта условию
B min < А < Вт.. (6.3. 1)
должна составлять Р,.
Пусть величину А находят эксперименталь
но как среднее выборки объема т, а методика ее
определения метролоrически аттестована. Torдa
112 За. 1712
условие (6.3. 1) будет выполняться с вероятностью
Р" если значение х 0= А будет лежать в пределах
B min + ДА < А < В та . ДА ' (6.3. 2)
rдe:
u( ).s (6 3 3)
ДА .Jт'"
Значения коэффициента U для вероятности
Р,=95 % и Р,=99 % соответственно равны 1,65
и 2,33.
Иными словами, для rарантии качества Ha
блюдаемые пределы изменения величины А на
практике следует оrраничить значениями:
U(P,)-s
А."," В...., + A =8"",. . (6.3. 4)
"т
ИIР. ) s
А.... в-- A 8.... . (6.3. 5)
"т
Наоборот. если заданы значения Ат," и
Аmзх' значения a min и а т . х , входящие в HepaBeH
ство (6.3. 1). MOryт быть найдены путем реше
ния уравнений (6.3.4) и (6.3. 5). Наконец, если
заданы пары значений A min , a min и Атах' а тах , то
уравнения (6.3.4) и (6.З. 5) MOryT быть решены
относительно т. Это может быть использовано
для оценки необходимоro числа параллельных
определений величины А.
Если при измерениях получают лоrариф
мы исходных вариант, описанные в rлаве 6, BЫ
числения проводят с использованием величин
Igx g , Igx i , 519 И Т. п. Примеры расчетов приведе
ны в rлаве 9.7.
7. РАСЧЕТ И СТАТИСТИЧЕСКАЯ
ОЦЕНКА ПАРАМЕТРОВ ЛИНЕЙНОЙ
ЗАВИСИМОСТИ
При использовании ряда химических и физи
ко химических методов количественноro анализа
непосредственному измерению подверrается He
которая величина у, которая является линейной
функцией искомой концентрации (количества) х
определяемоro вещества или элемента. Иными
словами, в основе таких методов анализа лежит
существование линейной зависимости:
у 0= Ьх + а , (7. 1)
rде:
у измеряемая величина;
Х концентрация (количество) определя
eMoro вещеСТ13а или элемента;
Ь уrловой коэффициент линейной зави
симости;
а свободный член линейной зависимости.
Для использования зависимости (7. 1) в
аналитических целях, т. е. для определения
878
rосударственная фармакопея Республики Беларусь
конкретной величины х по измеренному значе
нию У. необходимо заранее найти числовые зна
чения констант Ь и а, т. е. провести калибровку.
Иноrда константы функции (7. 1) имеют тот или
иной физический смысл, и их значения должны
оцениваться с учетом соответствующеro ДOBe
рительноro интервала. Если калибровка прове
дена и значения констант а и Ь определены, Be
личину х находят по измеренному значению У,:
1 а
Х; bY' b' (7. 2)
При калибровке величину х рассматривают
как apryMeHT, а величину y как функцию. Нали
чие линейной зависимости между х и У не Bcerдa
является очевидным. По этой причине экспери
ментальные данные, полученные при калибров
ке, в первую очередь используют для оценки
жесткости, т. е. степени неслучайности линей
ной связи между х и у. и лишь затем определяют
значения констант а и Ь и их доверительные ин
тервалы. В первом приближении судить о жест
кости линейной связи между переменными х и
У можно по величине линейноro коэффициента
корреляции (или просто коэффициента корреля
ции)" который вычисляют по формуле:
fТ'
т-Ixy LX,'LY,
r I [ m '", 2 ' m 1 ( т ) 2 ]
, / т-Ix,; ; Ix, .: т-Ly,2 LY,
. .. '1 1
(7. 3)
исходя из экспериментальных данных.
Линейный коэффициент корреляции r изме
няется в пределах от 1 до +1. Положительные
значения r указывают на рост, а неrативные
на уменьшение у с ростом Х.
Линейный коэффициент корреляции r явля
ется частным случаем общеro индекса корреля
ции Rc' который применим также и для нелиней
ных зависимостей между величинами У и х:
53
R 1 ......:... ,(7 . 3a)
с \ 5
rдe:
So остаточное стандартное отклонение
(уравнение (7. 7»;
Sy стандартное отклонение величин У; OT
носительно среднеro значения У (уравнение
(7. 15»); рассчитывают с использованием ypaB
нения (1.1 А).
Уравнение (7. 3a) в силу своей простоты и
наrлядности нередко используется вместо COOT
ношения (7. 3) в том случае, Korдa знак коэффи
циента корреляции не имеет значения.
Чем ближе абсолютная величина Irl к едини
це, тем менее случайна наблюдаемая линейная
зависимость между переменными х и у.
Коэффициент корреляции r используется
обычно для выявления стахостической взаимо
связи между величинами, функциональная зави
симость между которыми может и отсутствовать.
Коэффициент корреляции является значимым,
если еro величина для данной вероятности Р и
числа степеней свободы v превышает значе
ния, приведенные в таблице 11. 6. В противном
случае нельзя rоворить о существовании значи
мых зависимостей (7. 1 7. 2).
Значимость коэффициента корреляции яв
ляется обязательным, но не достаточным усло
вием использования уравнений (7. 1 7. 2) для
аналитических целей (см. ниже). В аналитиче
ской химии в большинстве случаев используют
линейные зависимости с коэффициентом Koppe
ляции Irl 0,98 (при соответствии требованиям
таблицы 11. 6), и только при анализе следовых
количеств рассматривают линейные зависимо
сти с коэффициентом корреляции Irl 0,9.
Коэффициенты а и Ь и друrие метролоrиче
ские характеристики зависимости (7. 1) рассчи
тывают с использованием метода наименьших
квадратов по экспериментально измеренным
значениям переменной У для заданных значе
ний apryMeHTa Х. Пусть в результате эксперимен
та найдены представленные в таблице 7. 1 пары
значений apryMeHTa х и функции у.
Таблица 7. 1
i х У,
,
1 Х 1 У1
2 Х 2 У2
. .. .. . . ..
т х ут
т
Torдa, если величины У; имеют одинаковую
неопределенность (а такое допущение обычно
выполняется для достаточно узкоro диапазона
варьирования величин У). то:
т. fx;y, fx;. fy;
Ь 1 1 1 , (7 А)
т ( т ) 2
т. x; X;
fy; b. fx,
a ' 1
т
, (7. 5)
v т 2 . (7. 6)
Если полученные значения коэффициентов
а и Ь использовать для вычисления значений У
по заданным в таблице 7. 1 значениям apryMeHTa
х соrласно зависимости (7. 1), то вычисленные
значения У обозначают через У " У 2 ' ... у;, ... У n '
Разброс значений относительно значений у;
характеризует величина остаточной дисперсии
s; , которую вычисляют по формуле:
f{y, У;)
5 2 1
О
fy: a. fy, b. fx,y,
1 1 1
l'
. (7. 7)
l'
#5.3.1. Статистический анализ результатов химичеСКО20 эксперимента
879
Таблица 7. 2
Результаты статистической обработки экспериментальных данных, полученных при изучении
линейной зависимости вида у == Ьх + а
' х У Ь а ((Р 2 , у) t:,. f1b S2 R Sx при t:,. /'1.", .100
а о ,
при n j == 1 х
Р 2 =95 % Y j == У
1 2 3 4 5 6 7 8 9 10 11 12 13
Примечание 1: если целью экспериментальной работы являлось определение констант Ь и а, rрафы
11, 12 и 13 таблицы 7. 2 не заполняются.
Примечание 2: если у == bIgx + а, вычисления, описанные в rлаве 7, выполняют с использованием
уравнений (1.1. 7), (1.1. 6), (1.3. 9 1.3. 12).
Примечание 3: сравнение дисперсий S , полученных в разных условиях для двух линейных зави
симостей, может быть проведено, как указано в rлаве 3.
Для тоro чтобы уравнения (7. 1 7. 2) aдeKBaT
но описывали экспериментальные данные, необхо
2
димо, чтобы остаточная дисперсия 80 не отлича
лась значимо по критерию Фишера (соотношения
3. 1 3.-4) от дисперсии воспроизводимости (cxo
димости) величин У,. Последняя может быть най
дена экспериментально или спроrнозирована (см.
rлаву 10) из паспортных данных оборудования.
В свою очередь, дисперсии констант Ь и а
находят по уравнениям:
2
2 тs o (7 8)
Sb m ( m ) 2' .
т. x; X,
s2 m
S2 == . " х2 . (7. 9)
а т '
Стандартные отклонения Sb и Sa И величины ДЬ
и Да' необходимые для оценки доверительных ин
тервалов констант, рассчитывают по уравнениям:
So == R . (7. 10)
Sa == R , (7. 11)
/'I. b == t(P2Y)'Sb , (7. 12)
/'"а == t(P2,I-)-Sа. (7. 13)
Коэффициенты а и Ь должны значимо отли
чаться от нуля, т. е. превышать, соответственно,
величины Да И Дь,
Уравнению (7. 1) с константами а и Ь обяза
тельно удовлетворяет точка с координатами Х и
у, называемая центром калибровочноro rрафика:
m
Ix,
х == , (7. 14)
т
m
LY,
Y== .(7. 15)
т
Наименьшие отклонения значений У; от зна
чений V; наблюдаются в окрестностях центра
rрафика. Стандартные отклонения Sy и S, вели
чин У и х, рассчитанных соответственно по ypaB
нениям (7. 1) и (7. 2) исходя из известных зна
чений х и у, определяются с учетом удаления
последних от координат центра rрафика:
i 1
. I
/21 1 т(x x)2
Sy == ;sO I -+- 2 ' ( 7 16 )
\ т m 'т \ .
. т. >.2 ; LX, I
L 1 ", J
i s; [ '; 1 т . (у J У f I
". о 11 + т + ь+ х; [ хлJ (7. 17)
rдe:
У J среднее значение;
П j чис о вариант, использованных при
определении У J .
При Х == Х и Yj == У
Sy == , (7. 16a)
S, == S [ + ]
Ь 2 п. т'
}
С учетом значений Sy и S, MOryT быть найде
ны значения величин Д и Д :
у х
/'" у == S у . ((Р2 ' l' ), (7. 18)
/'1., ==S, .t(P..v), (7. 19)
Значения S, и Д" найденные при П} == 1, яв
ляются характеристиками воспроизводимости
(сходимости) аналитической методики, если х
концентрация, а У функция х.
Обычно результаты статистической обра
ботки по методу наименьших квадратов сводят
в таблицу 7. 2.
8. ПОСЛЕДОВАТЕЛЬНАЯ СХЕМА
СТАТИСТИЧЕскоrо АНАЛИЗА
РЕЗУЛЬТАТОВ ХИМИЧЕСКИХ ИЗМЕРЕНИЙ
Традиционно в фармакопейном анали
зе преобладают методы статистическоro
880
rосударственная фармакопея Республики Беларусь
анализа с фиксированным объемом выбор
ки. На(?яду с этим в последние roAbI все шире
при меняются методы так называемоro после
довательноrо (секвенциональноro) анализа 7 .
Использование этих методов имеет смысл в
тех случаях, Korдa выполнение каждоro aHa
лиза дороro, трудоемко или отнимает мноro
времени и при этом имеется возможность
анализировать результаты последовательно,
по мере их поступления.
Частным случаем последовательной
схемы является метод проверки с ДBYKpaT
ной выборкой. Такой метод применяется в
фармакопейном анализе, например, при KOH
троле однородности дозирования: берется
первая выборка, и по полученным резулыа
там партия либо проходит, либо бракуется,
либо принимается решение взять вторую BЫ
борку. Такая схема позволяет сэкономить (в
среднем) число наблюдений, необходимое
для принятия решения. Еще более экономич
на последовательная схема в общем виде.
По данным В коэффициент выrоды при сопо
ставлении с традиционными схемами (фикси
рованный объем выборки) колеблется между
двумя и тремя.
Последовательный критерий для разли
чения двух простых rипотез:
Но: выборка извлечена из rенеральной
совокупности ((Х,Ро,О');
Н,: выборка извлечена из rенеральной
совокупности ((х, р" а);
предложен Вальдом 9 (здесь f(x,jJ,O')
функция плотности вероятности нормальноrо
распределения). Критическая статистика у(п)
(п число наблюдений) задается в виде:
у,п! = I f(x"jJ"O') = 1 2 (8 1)
, L n ( ) ' п , ... .
, 1 f x"jJo,O'
Область возможных значений критиче
ской статистики разбивается на три (а не на
две, как в случае выборок фиксированноrо
объема) части:
1) область принятия rипотезы НО
ylп) 51nL; (8. 2)
1 a
2) область принятия rипотезы Н,
ylп) In 1 fЗ ; (8. 3)
а
3) область продолжения наблюдений
InL < r 1пl < In 1 fЗ . (8А)
1 a а
7 См., например, D. Siegтuпd. Sequeпtia/ Aпa/ysis. Spriпger
Verlag. 1985.
8 С. А. Айвазян. Теория вероятностей и ее применения.
1959. Т.4. NQ 1. с. 87 9З.
9 А. Вальд. Последовательный анализ. М.: Физматrиз. 1960.
Здесь:
а поrрешность первоro рода (вероят
ность принятия rипотезы Н 1 в то время, как на
самом деле верна rипотеза Но);
fJ поrрешность второro рода (вероят
ность принятия rипотезы Но в то время, как на
самом деле верна rипотеза Н,).
Если результаты п испытаний рассматри
вать как случайную выборку из rенеральной
совокупности, поДчиняющейся нормальному
распределению с дисперсией 0'2 (которая пред
полаrается известной из предыдущих экспери
ментов), то:
у(п) = jJ, 2jJO . LX, + (.u jJ ). (8. 5)
а 20'
Если значение критической статистики, BЫ
численное на шаrе п, попадает в область 1, то
принимается rипотеза Но; если оно попадает в
область 2, то принимается rипотеза Н,; если зна
чение критической статистики попадает в об
ласть 3, то производится еще одно измерение.
Доказано, что с вероятностью 1 этот процесс за
канчивается принятием одной из двух альтерна
тивных rипотез.
Критерий Вальда является оптималь
ным в том смысле, что среди всех последова
тельных критериев он требует минимально
ro среднеro числа наблюдений при заданных
значениях поrрешности первоro и второro
рода.
На практике вычисления MOryт быть орrани
зованы следующим образом. На rрафик наносят
четыре прямые, задаваемые уравнениями, в KO
торых П номер испытания.
то = 80 + Ьп, (8. 6a)
т, = а, + Ьп, (8. 6b)
т; = a -t Ь'п, (8. 6c)
т,' = а; + Ь'п. (8. 6d)
В этих уравнениях
а о =a = .lnL, (8. 7a)
8" 1 а
2
а 1 fЗ
а =а' = .In ( 8 7b )
1 1 8 а"
jJ
2
rдe:
а стандартное отклонение метода, KOТO
рое предполаrается известным;
Ь и Ь' верхний и нижний пределы coдep
жания анализируемоro вещества в образце;
д'" = i,uo ,u,1 разность rенеральных cpeд
них rенеральных совокупностей ((Х,ро,а) и
f(x,,u,,a); б" задается экспериментатором и xa
рактеризует способность метода различать эти
rенеральные совокупности.
#5. 3.1. Статистический анализ результатов химичеСКО20 эксперимента
881
Прямые (8. 6a 8. 6d) разбивают пло
скость на 5 областей (рисунок 8. 1). Область 3
это область принятия rипотезы Но; области
1 и 5 области принятия rипотезы Н" обла
сти 2 и 4 области продолжения наблюдений.
Чем меньше СУ и больше б l' ' тем более узкими
являются области 2' и 4 и тем быстрее сходит
ся метод.
Испытания про водятся последовательно.
После каждоro испытания по оси ординат откла
дывается накопительная сумма получеНF-IЫХ pe
зультатов. В зависимости от тоro, куда попадает
очередная точка, принимается одно из трех воз
можных решений: попадание точки в область 3
означает, что образец выдерживает испытание;
попадание в область 1 или 5 означает, что обра
зец не выдерживает испытание; если точка по
падает в область 2 или 4, то испытания должны
быть продолжены.
о 1 2 3 4
Рисунок 8. 1. Практическая Ор2анизация схемы
последовательных испытаний
Более конкретно применение секвенци
ональноro анализа описано в rлаве 9.8.
9. ПРИ МЕРЫ
9.1. ВblЧИСЛЕНИЕ CPEДHErO ЗНАЧЕНИЯ
И ДИСПЕРСИИ
При определении содержания стрептоци
да в образце линимента были получены следу
ющие данные:
п=5; ,'=п 1=5 1=4,
п
L X '
5( = = 9,52 + 9,55 + 9,83 + 10,12 + 10,33 = 9.87
п 5
d, = Ix, 5(/ = /Х ; 9,87/,
т. е.
d, = 19,52 9,871 = 0,35
и т. д.
fd,2 fx; пx2
S2 = = 1
V V
= (9,522 + 9.552 + 9,8з2 + 10,122 + 1 0,332 ) 5. 9,872 01252
4 ' ,
5 ="\ 52 = .JO,1252 = 0,3538,
5 = = 0,3538 = 003585,
, 5( 9,87 '
RSD = 5. 100 = 3,59 %,
s = = 0.3 = 0,1582
, ,п ,5
s = S. = 0,1582 = 0.01603
,. х 9.87 '
RSD. = s" .100 = 1.60 %.
9.2. ПРОВЕРКА ОДНОРОДНОСТИ
ВblБОРКИ МАлоrо ОБЪЕМА
При проведении девяти (п=9) определений
содержания общеro азота в плазме крови крыс
были получены следующие данные (в порядке
возрастания ):
По уравнениям 1.2. 1 и 1.2. 2a находим:
R = 'х, Xп 11 = 10,62 0,981 = 0,36,
/XI X21 10,62 0,811
о, = = = 053.
R 0,36 '
По таблице 11. 1 находим:
0(95 %,9) = 0.46 < О, = 0,53,
0(99 %,9) = 0,55 > О, = 0,53.
Следовательно, rипотеза о том, что значение
х,=О,62 должно быть исключено из рассматрива
емой совокупности результатов измерений как
отяroщенное rрубой поrрешностью, может быть
принята с доверительной вероятностью 95 %, но
должна быть oT8eprHYTa, если выбранное значе
ние доверительной вероятности равно 99 %.
9.3. ВblЧИСЛЕНИЕ ДОВЕРИТЕЛЬНblХ
ИНТЕРВАЛОВ И НЕОПРЕДЕЛЕННОСТЕЙ
ИЗМЕРЕНИЙ
В результате определения содержания
хинона в стандартном образце хинrидрона были
получены следующие данные (п=10).
882
rосударственная фармакопея Республики Беларусь
Расчеты по формулам (1.1. 1, 1.1. 3 1.1. 6)
дали следующие результаты:
)(==49,96; 1' 9; 52 0,O1366; 5 0,1169;
5, 0,03696.
Доверительные интервалы результата OT
дельноro определения и среднеro результа
та при Р 2 =90 % получаем соrласно (1.3. 5) и
(1.3. За):
Х ; :и,х Х, = t(P"I')'5 Х, ::tt(90 %, 9)'5
Х ; :t 1,83. 0,1169 Х, :t 0,21,
x:t t- ji х :t ap,, 1.5 = 49.96:t 1,83 169 49,96 :t 0,07.
п 10
Torдa относительные неопределенности Е: и
"& , соrласно (1.З. 8) и (1.3. 8a), равны:
[; "" 100% 0' .100% 042%
х 49,96 "
z "', -100 % 0?--.100 % 014 %
х 49,96 ,.
Обозначая истинное содержание хинона в
хинrидроне через J.1, можно считать, что с 90 %
доверительной вероятностью справедливы He
равенства:
р 021 :s: Х, :s: J1 + 0,21,
Х, 0,21 :s: l' :s: Х 0,21 (при любом i);
Ji 0,07 $ х $ Ji -+ 0,07; х 0,07 $ J1 $ х -+ 0,07 (при п=10)
9.4. ПРОВЕРКА rИПОТЕ3Ы РАВЕНСТВА
ДИСПЕРСИЙ
9.4.1. Объединение результатов выборок
разноrо объема
В процессе проведения внутрилабора
торных исследований неопределенности Me
тодики титрования субстанции ацетилсали
циловой кислоты четырьмя (Т. е. g=4, v х =3)
разным!,! аналитиками получены средние зна
чения X k и относительные стандартные OT
клонения (RSO k %) для указанноro числа
опытов (п k ), представленные в таблице
9.4.1. 1.
Можно ли считать данные RSO k выборка
ми из одной rенеральной совокупности и каковы
объединенное х и RSO,o?
Вначале проверим rипотезу равенства дис
персий, т. е. что все RSO k являются выборками
из одной rенеральной совокупности.
Рассчитаем величины v t по формуле
(2.1.1. 2):
9
V, LVk 4+6+8+7 25.
k 1
По формуле (2.1.1. 1) рассчитываем RSO ,:
'-о
L\', .RSO: 2 2 2 2
RS02 = ,., 4 - 0,3 + 6 - 0,8 -+ 8.0,7 -+ 7.0,9 0552.
'01 \', 25 '
Теперь по формуле (2.1.2. 1) рассчитаем х 2 :
х 2 = 2,303 - (\-" .19 RSO , l' k .19 RSO; J =
= 2,303 . (25 .19 0,552 (4 .190,09 + 6 .190,64 +
+ 8 .190,49 + 7 .190,81)) = 4,62.
Табличное значение (таблица 11. 4)
х 2 (Р1 0,95%, V = 3)= 7,815> 4,62.
у 2
Таким образом. значение х меньше крити
ческоro, поэтому можно принять rипотезу о pa
венстве дисперсий.
2
Значение Х меньше критическоro, и по
этому нет необходимости в расчете KoppeK
тирующеro фактора С и величины х'2. Для
иллюстрации рассчитаем и эти величины по
формуле (2.1.2. 2):
С = [(1-0- 4)+ (1-0- 6)+ (1-0- 8)+ (1-0- 7)] (1-0- 25) + 1 1072
3. (4 1) , ,
'2 L 4,62 =431
Х С 1,072 ,.
'2
Как видно, значение Х еще меньше крити
ческоro значения (7,815), что подтверждает rи
потезу о равенстве дисперсий.
Таблица 9.4.1. 1
RSD k RSD, , RSD,O' х 2 '2 Табличное
NQ x k П k l' l' С Х х 2 (р, 95, % l' j 3)
k ,
% %
1 99,9 0,3 5 4
2 99,4 0,8 7 6 25 0,552 0,74 4,62 1,072 4,31 7,815
3 99,2 0,7 9 8
4 99,3 0,9 8 7
#5.3.1. Статистический анализ результатов химичеСКО20 эксперимента
883
Рассчитаем объединенное RSD rot :
RSD,o' RSD t .JO,552 0,74 %.
Рассчитаем объединенное среднее х по
всем четырем выборкам по формуле (21.1. 3):
)( 99,9.5 + 99,4.7 +99,2 .9+99,3.8 99,4 %.
5+7+9+8
Таким образом, по данным внутрилабора
торных исследований относительное CTaHдapT
ное отклонение титрования субстанции aцe
тилсалициловой кислоты равно 0,74 %, а ее
содержание 99,4 %.
9.4.2. Объединение результатов выборок
одинаковоrо объема
При анализе методом ВЭЖХ пяти различ
ных серий (т. е. g=5) лекарственноro средства
получены следующие значения относительных
стандартных отклонений (RSO i %) площадей
пиков при трехкратном (т. е. п=3) хроматоrрафи
ровании раствора каждой серии:
1,08 %; 0,60 %; 0,43 %,1,59 % и 0,71 %.
Можно ли объединять данные выборки (pe
зультаты анализа пяти серий) и каково объеди
ненное относительное стандартное отклонение?
Поскольку в нашем случае число степеней
свободы для всех пяти выборок (серий) одинако
во и равно v '" П 1 '" 3 1 '" 2 , то для проверки rи
потезы равенства дисперсий при мени м критерий
Кохрена (см. rлаву 2.1.3 и таблицу 11.-4). В нашем
случае smax 1,59 %, ' 2, 9 '" 5, и соотношение
(2.1.2. 3) дает:
G = S ax = 1,592
9 1 , 082 0 , 602 + 0 , 4з2 т 1.592 0,712
I:S:
k==1
0,533 < 0,684 = G(P 0,95;2;5).
Как видно, рассчитанное значение G меньше
табличноro на 95 % уровне значимости. Следо
вательно, данные выборки можно объединить.
Рассчитаем по уравнению (2.1.1. 2) объеди
ненное число степеней свободы:
9
1/, L>k 5.2 10.
k=,
Рассчитаем по формуле (2. 1.1. 1 Ь) объеди
ненное относительное стандартное отклонение
(RSO ro ,):
k=g
'1' .RSD 2
L... k k
RSDr t '" k=,
У,
'" 2. (1,082 + 0,602 + 0,4з2 + 1.592 + 0,712) '" 0,9510.
10
Отсюда
RSD,O' ...10,951 О 0,98.
9.5. СРАВНЕНИЕ ДВУХ МЕТОДИК
АНАЛИЗА ПО ВОСПРОИЗВОДИМОСТИ
Пусть для двух выборок аналитических
данных (1 и 2), характеризующих, например, раз
личные методики анализа, получены метролоrи
чески е характеристики, приведенные в rрафах
1 10 таблицы 9.5. 1.
Для заполнения rрафы 1 О вычислим значе
ния t, и t 2 :
iJ1 x,I' 1100 100,131. 20 +1
t '" 128
, s, 0,464 ' ,
1J1 xJ 1100 98,011. 15+1
t 2 72,36.
82 0,110
Поскольку t,=1 ,28 < t,(95 %,20)=2,09, rипоте
за 11', )(,17' О может быть oTBeprHYTa, что позво
ляет считать результаты выборки 1 свободными
от систематической поrрешности.
Напротив, поскольку t 2 =72,36» t 2 (95 %, 15)=2,13,
rипотезу 11'2 )(217' О приходится признать стати
стически достоверной. что свидетельствует о Ha
личии систематической поrрешности в резулыа
тах выборки 2. В rрафу 13 вносим:
: x! 1100 9801 1
Ii ;J1, 21 .100% I ' .100% 199%.
2 J1, 100 '
Заполним rрафы 11 и 12:
F(99 %;20;15) 3,36,
F '" s; 0,215 1792
S 0,012 .,
F 17,92 > F(99 %:20:15) 3,36.
Таблица 9.5. 1
Р2' t(P2 ' l' ) F(P,,1',,1' 2 )
N!! J.1 t-' Х ,% s /:о"х [; t (табл.) F <5
% (табл.) выч 8ЫЧ
Р=99 %
1 2 3 4 5 6 7 8 9 10 11 12 13
1 100 20 100,13 0,464 95 2,09 0,97 0,97 1,28 3,36 17,92
2 100 15 98,01 0,110 95 2,13 0,23 0,23 72,36 3,36 17,92 1,99
884
rосударственная фармакопея Республики Беларусь
Следовательно, при Р, =99 % rипотезу о раз
личии дисперсий s: и s; следует признать CTa
тистически достоверной.
Выводы:
а) результаты, полученные при использова
нии первой методики, являются правильными,
т. е. они не отяroщены систематической поrреш
ностью;
б) результаты, полученные при использова
нии второй методики, отяroщены систематиче
ской поrрешностью;
в) по воспроизводимости вторая методика
существенно лучше первой методики.
9.6. СРАВНЕНИЕ СРЕДНИХ РЕЗУЛЬТАТОВ
ДВУХ ВblБОРОК
При определении содержания основноro
вещества в двух образцах препарата, изrотов
ленных по разной технолоrии, получены MeTpo
лоrические характеристики средних результа
тов, приведенные 8 таблице 9.6. 1. Требуется
решить, является ли первый образец по дaHHO
му показателю лучшим по сравнению со вторым
образцом.
Поскольку
F 0.31 t24 < F(99 %;5;7) 7,46,
5; 025
соrласно неравенству 3. 3 статистически дo
стоверное различие величин s: и s; отсутствует.
Следовательно. rипотеза )(, == )(2 (5. 1) прове
ряется с помощыо уравнений (2.1.1. 1), (2.1.1. 2),
(5. 2) и (5. 3).
I[(п, 1)-5 ] _ 2 _ 2
.=1 \.5. \2S2 7.0,25T5-0,31 0275
s 1 ,
I(п. 1) \-' '-2 7+5
к=1
5 '52 ,0.275 0,524 ,
5 "" 52 . (п 1 + п 2 ) 0.275 - (8 + 6) == 0,0802,
П 1 'П 2 8.6
с2
5 р == ...;5 р ,-0,0802 0,283 ,
v П 1 + П 2 2 == 8 6 2 12,
1)(1 )(21 199,1 о 98,331
t== == ==272
sp 0,283 "
t 2,72 > t(95 %;12) 2,18,
t == 2,72 < t(99 %;12) == 3,08.
Следовательно, с доверительной вероятно
стью Р 2 =95 % rипотеза х, Х 2 может быть при
нята (т. е. первый образец лучше второro по
содержанию основноro вещества). Однако с дo
верительной вероятностью Р 2 =99 % принять эту
rипотезу нельзя из за недостатка информации.
Если rипотеза )(1 *-)(2 принята, то определя
ют доверительный интервал разности rенераль
ных средних Х 1 X2 (уравнение 5.3. 1):
1)(, )(21 t(P2"+ sp $IX 1 х 2 1 $
$1)(1 )(21 + t(p 2 , v). sp,
(Р 2 =95 %; v =12);
199,10 98,33: 2,18 - 0,283 $1)(1 )(21 $
$199,1 О 98,33i + 2,18 . 0,283,
0,15 $IX, х 2 1 $1,39.
9.7. ОЦЕНКА КАЧЕСТВА ПРОДУКЦИИ
Рассмотрим данны-е таблицы 9.6. 1, OT
носящиеся к выборке 1, как метролоrическую
характеристику используемоrо меТРОЛQrиче
ски аттестованной методики анализа.
а) Пусть a min =98 %, а mах =100,50 %. Torдa
для испытуемоro образца продукта средний
результат анализа А при проведении трех па
раллельных определений (т=З) должен Haxo
диться в пределах:
И(Р 1 )'5
В m1Л + .Jт < А < Втах
И(Р 1 ).5
.Jт
При Р 1 =99 %:
98 + 2,33 . 0,464 < А < 1 00,5 2'331464 ,
3 3
98,62 < А < 99,88.
При Р,=95 %:
98 + 1,65. 464 < А < 1 00,5 1,65;464 ,
3 3
98,44 < А < 100,06.
Таблица 9.6. 1
N2 образца v s Р 2 , % t(P2 ' v) /'t,,)( /'t,,
п Х,% s )( )( Е ,%
1 2 3 4 5 6 7 8 9 10 11
1 8 7 99,10 0,50 0,18 95 2,36 1,18 0,42 0,42
2 6 5 98,33 0,56 0,23 95 2,57 1,44 0,59 0,60
#5. 3.1. СтатистичесК'ий анализ результатов химическоzо эксперимента
885
б) Реальный средний результат анали
за образца испытуемоro продукта А=99 %
(при т=3). Torдa определение пределов а m1п
и а тах , rарантированно характеризующих Ka
чество данноro образца с заданной довери
тельной вероятностью Р1' проводим исходя
из уравнения -(6.З. 4) или (6.3. 5), полаrая
А =А =А.
mlП тах
8 m1п
А U( ),s ,
-Jт
8тах
А + U(pJ. S .
-Jт
При Р 1 =99 %:
a min
99 2,33 . 0,464 983 8 %,
.J3 '
99 2,33 . 0,464 99 62 0/ .
а тах +.J3 ,/0
При Р1=95 %:
а тll1
99 1,65. 464 98,56 %,
,:з
8тах
99 + 1,65. 0,464 9964 %.
.J3 '
Полученные оценки а т1п и а тах близки к rpa
ницам доверительноro интервала
A:tL'.x A:t L'.x 99:t О,;; 99:tO,56.
т ,,3
9.8. КОНТРОЛЬ СОДЕРЖАНИЯ
САЛИЦИЛОВОЙ КИСЛОТЫ
В САЛИЦИЛОВОМ СПИРТЕ
ПОСРЕДСТВОМ
СЕКВЕНЦИОНАльноrо АНАЛИЗА
Определение содержания кислоты салицило
вой (С К, М. м.=138,12) в спирте салициловом 2 %
проводили путем титрования спиртовым раствором
щелочи с молярной концентрацией 0,1000 моль/л.
Для титрования берут пробы по 5,00 МЛ- На титро
вание идет V мл щелочи. Результаты титрования
(х найденное содержание СК в процентах к HO
минальному содержанию) двух различных образ
цов приведены ниже в таблице 9.8. 1.
Предварительные исследования показали,
что а = 0,5 %. Допуски содержания салициловой
кислоты Ь 90 % и Ь' 110 %.
120
'"
::Е
::Е
>-
u 100
о:
'"
:1:
...
<:;
Q) 80
...
:s:
J::
е
'" 60
I
40
20
О
2
з "
20
Номер испытания
Рисунок 9.8. 1. Результаты титрования сали
цилов020 спирта (2 %) спиртовым раствором
щелочи: крестиками показаны результаты
для образца 1. кружками для образца 2
Обычно принимают а = fJ = 0,05. Зададим
ся также различием rенеральных средних 8. 2.
Уравнения (8. 7a) и (8. 7b) дают:
а a' .lnL= 0,5 .In 0,05
о о др 1 а 2 1 0,05
2 2
= 0,25 .lпО,0513 = 0,25.2,97 = O,743,
а = а' .Iп 1 /3 = 0,5 .Iп 1 0,05 =
1 1 д а 2 0,05
JI -
2 2
= 0,25 .In 38 = 0,25.3,64 = 0,909.
Для удобства представления на rрафике
(чтобы накопительные суммы изменялись в неши
роком диапазоне) вычтем из величин Ь и Ь' HeKO
торую величину, например, 85%. Эту же величину
вычтем и из каждоro результата х (таблица 9.8. 1).
Тоща уравнения (8. 6a 8. 6d) примут вид:
ТО 0,743+5.п,
т., =0,909+5.п,
T 0,743 + 25. п.
т; 0,909 + 25 . п.
Данные уравнения образуют коридоры, по
казанные на рисунке 9.8. 1. Накопительные
суммы результатов титрований (см. таблицу
9.8 1) показаны крестиками.
Таблица 9.8. 1
Образец 1 Образец 2
N2 V,мл х,% x 85 IJx 85) V, мл х, О/О x 85 l)x 85)
1 6,71 92,68 7,68 7,68 6,40 88,26 3,26 3,26
2 7,20 99,45 14,45 22,12 6,40 88,40 3,40 6,66
3 7,08 97,79 12,79 34,91 6,20 85,77 0,77 7,43
886
rосударственная фармакопея Республики Беларусь
Для образца 1 первый результат попада
ет в нижний коридор, и испытания необходи
мо продолжить. Но уже второй результат по
падает в область положительных решений,
что дает основания заключить, что образец
выдерживает испытание. Третье испытание в
этом случае не требуется: соответствующий
результат показан для большей наrлядно
сти рисунка. Первый результат для образца
2 (кружки) указывает на необходимость про
должить испытания; второй снова оказыва
ется в той же области у нижней rраницы.
Третий результат попадает в область отрица
тельных решений образец испытания не
выдерживает.
10. РАСЧЕТ НЕОПРЕДЕЛЕННОСТИ
ФУНКЦИИ НЕСКОЛЬКИХ СЛУЧАЙНЫХ
ПЕРЕМЕННЫХ
Описанные в предыдущих rлавах расчеты
доверительных интервалов результатов MeTO
диканализа применимы лишь в том случае, если
измеряемая величина (концентрация, co
держание и т. Д.) является функцией только
одной случайной переменной. Такая си
туация обычно возникает при использова
нии прямых методов анализа (титрование,
определение сульфатной золы, тяжелых Me
таллов и т. Д.). Однако большинство MeTO
дик количественноro определения в фарма
копейном анализе являются косвенными.
т. е. используют стандартные образцы. Сле
довательно. измеряемая величина является
функцией как минимум двух случайных пере
менных аналитических сиrналов (оптическая
плотность. высота или площадь пика и т. д.) ис
пытуемоrо и стандартноro образцов. Кроме
Toro, нередко возникает проблема проrнозиро
вания неопределенности аналитической MeTO
дики, состоящей из нескольких стадий (взвеши
вание, разбавление. конечная аналитическая
операция), каждая из которых является по OT
ношению к друrой случайной величиной.
Таким образом, возникает общая пробле
ма оценки неопределенности косвенно измеря
емой величины, зависящей от нескольких изме
ряемых величин. В частности, как рассчитывать
неопределенность всей аналитической методи
ки, если известны неопределенности отдельных
ее составляющих (стадий)?
Если измеряемая на опыте величина у яв
ляется функцией п независимых случайных Be
личин Х" т. е.
у = ((х 1 ,х 2 ,...Х п ), (10. 1)
и число степеней свободы величин оди
наково или достаточно велико (более 30, чтобы
можно было при менять статистику raycca, а не
Стьюдента), то дисперсия величины у связана с
дисперсиями величин Х, соотношением (правило
распространения неопределенностей):
( ) 2
2 п Ы 2
Sy = L . Sx;. (10. 2)
; 1 ОХ;
Однако на практике степени свободы величин
Х, обычно невелики и не равны друr друry. Кроме
тоro, обычно интерес представляют не сами дис
персии (стандартные отклонения), а доверитель
ные интервалы, рассчитать которые, используя
уравнение (10. 2), при небольших инеодинаковых
степенях свободы невозможно. Поэтому для pac
чета неопределенности величины у (д ) предложе
у
ны различные подходы, среди которых можно BЫ
делить два основных: линейную модель и подход
Уэлча Сатертуэйта (Welch Satterthwaite).
10.1. ЛИНЕЙНАЯ МОДЕЛЬ
Если случайные переменные Х ; статистически
независимы, то доверительный интервал функции
ду связан с доверительными интервалами пере
менных д., соотношением (доверительные интер
валы берутся для одной и той же вероятности):
п r '" J 2
/'12 = . д2
у L Х'. (10.1. 1)
'==1,СХ;
Данное соотношение является обобщением
соотношения (10. 2).
В фармакопейном анализе измеряемая
величина у представляет собой обычно про
изведение или частное случайных и постоян
ных величин (масс навесок, разбавлений, оп
тических плотностей или площадей пиков и
т. д.), т. е. (К некая константа):
y К'Х 1 'Х 2 '''..Х т .(10.1. 2)
Xт 1 . Xт 2 ..... Х п
в этом случае соотношение (10. 2) прини
мает вид:
п
2 "2
"'р = L.. '" xi.p (1 O.1. 3)
; 1
rде использованы относительные довери
тельные интервалы.
Соотношение (10.1. 3) применимо при
любых (разных) степенях свободы (в том числе
и бесконечных) для величин Х,- Еro преимуще
ством является простота и наrлядность. Ис
пользование абсолютных доверительных ин
тервалов приводит к rораздо более rромоздким
выражениям, поэтому рекомендуется использо
вать относительные величины.
При проведении фармакопейноro анализа
в суммарной неопределенности (/'1 As ) анализа
обычно Bcerдa можно выделить такие типы He
определенностей: неопределенность пробопод
roтовки (Qsp,)' неопределенность конечной aHa
литической операции (/'1 FAo ,) инеопределенность
аттестации стандартноrо образца (ДRS.). Величи
на ДRS.r обычно столь мала, что ею можно пре
#5.3.1. Статистический анализ результатов химическоао эксперимента
887
небречь. Учитывая это, а также то, что анализ
проводится и для испытуемоrо раствора (индекс
«smp»), И для раствора сравнения (индекс «st»),
выраж ение (10.1. 3) можно представит ь в виде:
1 , 2 2 ] r 2 21
llAH= llll ;p') +(ll p.r) +Y\ ;6r) +(ll Aor) J' (10.1.А)
При этом каждое из слаrаемых рассчитыва
ется из входящих в нею компонентов по форму
ле (10.1. 3).
В том случае, Korдa числа степеней свобо
ды величин х, одинаковы или достаточно велики
(более 30), выражение (10.1. 3) дает:
п
S:,r == IS;j,r' (10.1,А)
j 1
Это же соотношение получается при тех же
условиях и из выражения (10. 2).
10.1.1. Взвешенное среднее
Следует отметить, что в рамках линейной
модели (1 0.1. 1) можно получить взвешенное
среднее нескольких неравноточных выборок
разных rенеральных совокупностей, используя
в качестве весов квадраты соответствующих дo
верительных интеевалов. Если имеются 9 выбо
рочных средних Х k разных rенеральных COBO
купностей, полученных с неопределенностями
!1 x,k ' то среднее этих выборок х определяется
из соотношения:
9
I(1: ll .k)' Х .
х == k==1 (1 О 1 1 1 )
9 .. . .
2::(1: ll .k)
" 1
Абсолютный доверительный интервал этой
взвешенной средней определяется из COOTHO
шения:
2 1
1'1., = 9 . (10.1.1. 1a)
I(1: ll2 x . k )
k==1
В том случае, Korдa числа степеней CBO
боды выборочных средних х k одинаковы или
достаточно велики (более 30), выражение
(10.1.1. 1) дает:
9
I (1: 8;.k)' Х .
х= k 1g ,(10.1.1. 2)
2:: (1 : 8; .)
"о1
82 =
х
1
9 . (10.1.1. 2a)
I(1: 8;.k)
"-1
Здесь S;k дисперсия единичною резуль
тата k ой выборки. Отметим, что частный случай
(1 0.1.1. 2) roраздо менее применим, чем общее
соотношение (1 0.1.1. 1).
Выборочные средние X k обычно изки
между собой и к взвешенному среднему Х, по
этому в соотношениях (10.1.1. 1) и (10.1.1. 1a)
вместо абсолютных доверительных интерва
лов MorYT быть использованы относительные
доверительные интервалы, а в соотношениях
(10.1.1. 2) и (1 0.1.1. 2a) вместо абсолютных дис
персий относительные дисперсии.
В том случае, Korдa выборки представляют
собой, например, результаты анализа одноrо и
тoro же вещества в разных лабораториях, иноrда
возникает необходимость оценить среднюю He
определенность анализа этою вещества по всем
лабораториям. В этом случае не совсем KoppeK
тно каким либо образом усреднять доверитель
ные интервалы Д х . поскольку они зависят от
числа анализов и теоретически MOryт быть cдe
ланы как уroдно малыми посредством увеличения
числа опытов. Более корректно оценивать меж
выборочное относительное стандартное откло
нение. Для этoro мoryт быть использованы соот
ношения (2.1.1. 1b) и (2.1.1. 2). Следует отметить,
что в этом случае полученное межвыборочное от
носительное стандартное отклонение не является
оценкой некоею «rенеральноro стандартноro OT
клонения», а представляет собой просто HeKOTO
рую среднюю величину.
10.2. ПОДХОД УЭЛЧА САТЕРТУЭЙТА
В этом подходе дисперсию величины у (s2)
у
рассчитывают по соотношению (10. 2), не об
ращая внимания на различие в степенях свобо
ды (V j ) величин Х,. ДЛЯ полученной дисперсии
рассчитывают некое «эффективное» число CTe
пеней свободы V eff (которое обычно является
дробным), на основе которою затем по табли
цам для заданной вероятности находят интерпо
ляцией коэффициент Стьюдента. На еro основе
далее рассчитывают обычным путем довери
тельный интервал величины у (д ):
у
l.1 eff ==
84
У
'(10.2. 1)
( N ) 4
f X;
1==1 \/,
'8:
В фармакопейном анализе для определя
емой величины у обычно выполняется ypaBHe
ние (10.1. 2). В этом случае в подходе Уэлча
Сатертуэйта соотношение (10. 2) переходит в
выражение (10.1.А), и соотношение (10.2. 1)
принимает более простой вид:
84
l' ff = . .(10.2. 2)
е "8"
I"
\'
1-=1 I
Здесь величина 8;., рассчитывается из co
отношения (10.1,А).
Подход Уэлча Сатертуэйта обычно дает
более узкие доверительные интервалы, чем ли
888
rосударственная фармакопея Республики Беларусь
нейная модель. Однако он roраздо сложнее в при
менении и не позволяет выделить так просто He
определенности разных этапов (с последующими
рекомендациями по их минимизации), как линей
ная модель в форме выражения (10.1..-4).
При проrнозе неопределенности анализа ис
пользуются rенеральные величины (с бесконечным
числом степеней свободы). В этом случае подход
Уэлча Сатертуэйта совпадает с линейной моделью.
10.3. ПРИМЕРЫ РАСЧЕТОВ
НЕОПРЕДЕЛЕННОСТИ ФУНКЦИИ
НЕСКОЛЬКИХ ПЕРЕМЕННЫХ
10.3.1. Расчет неопределенности ВЭЖХ
анализа rOToBoro лекарственноrо средства
В таблетке со средней массой 0,50 r coдep
жится 0,050 r вещества А. В процессе проведения
количественноro определения методом ВЭЖХ
брали навеску т=0,5052 r порошка растертых Ta
блеток в мерную колбу вместимостью 50 мл и дo
водили растворителем до метки. Параллельно ro
товили раствор сравнения: 0,0508 r стандартноro
образца помещали в мерную колбу вместимо
стью 50 мл И доводили объем раствора раствори
телем до метки. Попеременно хроматоrрафиро
вали испытуемый раствор и раствор сравнения,
получая по 5 xpoMaтorpaMM. Ниже представлены
площади полученных пиков (табл. 10.З.1. 1).
10.3.1.1. Конечная аналитическая операция
Рассчитаем средние значения, содержание
вещества А в одной таблетке и относительные
стандартные отклонения площадей пиков для
испытуемоro раствора и раствора сравнения в
конечной аналитической операции.
а) Средние значения площадей пиков:
Испытуемый раствор:
(13 957 605 13806 804+ 13 924 245+
+ 13 715195 14059478)/5 =о 13892665.
Раствор сравнения:
(14240777 + 14 102 192.14316388+
+ 14 205 217 + 144095851/5 =о 14254 832.
Ь) Содержание анализируемоrо вещества А,
в rpaMMax, в пересчете на среднюю массу
таблетки:
А =о 5.т .50.0,5 1 3892665.0,0508. 5 . 05 00490.
55' . Щ/ .50 14254 832.0,5052.50' ,
с) Относительные стандартные отклонения
площадей пиков:
Испытуемый раствор:
(13957 605 13 892 665)2+
+ (13 806 804 13 892 665)2+
+ (13 924 245 1З 892 665)2+
1 + (13 715 195 13 892 665)2+
RSD = 100 . + (14 059 478 13 892 665У = 097 %.
13892665 4 .
Аналоrично для раствора' сравнения:
R5D 5 ' 0,81 %.
10. З. 1.2. Суммарная неопределенность
пробопод20товки t.Sp.r
Неопределенности:
неопределенность для мерной колбы BMe
стимостью 50 мл не более 0,17 %;
неопределенность взвешивания на анали
тических весах не более 0,2 Mr (0,0002 r), что
составляет:
100 . 0,0002
0,04 % для испытуемоro образца
0,5052 '
100 . 0,0002
0,0508 0,39 % для стандартноro образца.
Данные неопределенности можно считать дo
верительными интервалами для вероятности 95 %.
Суммарная неопределенность пробоподro
товки рассчитывается по формуле (1 0.1. 3):
ДSР,r = ( o,042 + 0,392 ) + ( 0.172 + 0,172 ) 0,46 %,
Отметим, что такой расчет является Koppeкт
ным для обоих подходов линейной модели и
подхода Уэлча Сатертуэйта, поскольку число
степеней свободы для каждоro члена здесь бес
конечно, то используется статистика raycca.
10.3. 1.3. Расчет суммарной неопределен
ности анализа {l"AS.r
Данный расчет различается для линейной
модели и подхода Уэлча Сатертуэйта.
а) Линейная модель
Общий случай. Рассчитаем неопределенно
сти конечной аналитической операции t.FAO.r дЛЯ
испытуемоro раствора и раствора сравнения.
При расчете доверительных интервалов исполь
Таблица 10.3.1 1
Площади пиков (S и Sst) для xpoMaTorpaMMbI N2
1 2 3 4 5
Испытуемый 13957605 13806804 13924245 13715195 14059478
раствор
Раствор 14240 777 14102192 14316388 14205217 14409585
сравнения
#5.3.1. Статистический анализ результатов химическоzо эксперимента
889
зуем односторонний коэффициент Стьюдента
для вероятности 95 % (равен 90 % для ДBYCТO
роннеro распределения), который для числа CTe
пеней свободы 5 1=4 равен 2, 13. Доверитель
ные интервалы рассчитываются для среднеro из
5 результатов, поэтому в знаменателе стоит .J5 :
ДS;::O r . 1(90 %,4). RSD . 2,13. 0,97 0,92 %,
. ,,5 ,,5
Д AOc ,1(90%,4).RSDst .2,1З,О,81 0,77%.
. ,,5 ,,5
Суммарная неопределенность конечной
аналитической операции:
ДFАО r (дS;::O У + (Д AO У (0,92)2 + (0,77)2 1,20 %.
Используя уравнение (10.1,А), рассчитаем
суммарную неопределенность анализа ДА' ,:
ДАs.r = .1 0,462 + 1,202 = 1,29 %.
Использование объединеНН020 cтaHдapт
Н020 отклонения
Суммарную неопределенность анализа
можно уменьшить за счет использования объе
диненноrо стандартноrо отклонения дЛЯ KO
нечной аналитической операции. Для этоrо
надо учесть, что RSO и RSO S ' являются выбо
рочными величинами одной и той же rенераль
ной совокупности.
Проверим вначале по Фишеру (rлава 2.1 ) rи
потезу о равенстве дисперсий:
RSD: 0,97: 1,434 < 6,388 F(PI 95 %;4;4).
RSD st 0,81
Как видно, расчетное значение отношения
дисперсий rораздо ниже табличноrо значения
F критерия на 95 % уровне значимости. Поэтому
можно принять rипотезу о равенстве дисперсий
и использовать формулы rлавы 2 для объедине
ния выборок.
Рассчитаем объединенное стандартное OT
клонение по формуле (2.1.1. 1 Ь):
RSD ,O' ( 0,972 + 0,812 ) /2 0,89 %.
Соrласно (2.1.1. 2), RSD tot имеет число CTe
пеней свободы 2. (5 1) = 8 . Коэффициент Стью
дента для данноro числа степеней свободы и oд
носторонней вероятности 0,95 равен 1,86.
Torдa доверительные интервалы резулыа
тов конечной аналитической операции для испы
туемоro и стандартноrо растворов будут равны:
,"},";';,'Q, O=L'I AOr .t(90%:8).RSO '1,86.0,89 0,74%_
",5 ",5
Суммарная неопределенность конечной
аналитической операции равна:
ДFАО., = (д % У + (д AO у = (0,74 У + (0,74 у 1,05 %.
Используя уравнение (1 0.1,А), рассчитаем
суммарную неопределенность анализа
/),Asr 0,462 + 1,052 0= 1,15 %.
Как можно увидеть, данная величина меньше
полученной для обычноro случая (1,29 %).
Ь) Подход Уэлча Сатертуэйта
Найдем стандартное отклонение про
боподrотовки из доверительноrо интервала
L'.SP.r=0,83 %, используя коэффициент raycca 1,65
для односторонней вероятности 0,95 (поскольку
число степеней свободы бесконечно, как для re
неральной совокупности):
s SP. 0= 0.39/1.65 = 0.24 %.
Из соотношения (10.1. З) найдем CTaHдapT
ное отклонение всей аналитической методики
(RSfY и (RSOS')2 делим на 5, как для дисперсий
среднеro результата):
S.... = S p, "'i-' SD2 + (RSD st r] 0,242 '" i-. [0.972 +0,812] 0.61
Найдем эффективное число степеней CBO
боды V eff . При этом для RSO и RSOS' число
степеней свободы равно 5 1 = 4, а для SSP.r
бесконечность.
S s.r
V eff S;p, RS0 4 (Rso,,)4
+ - +
х 524 524
0,614 10,5.
0+ _1 _ (0,974 + 0,814)
100
По таблице 11.2 находим коэффици
ент Стьюдента для числа степеней свободы
10,5 и односторонней вероятности 0,95. Это
1.81 (интерполяция). Torдa доверительный
интервал всей аналитической методики будет:
ДАн 1,77. 0,61 1,10 %.
Как можно увидеть, доверительный ин
тервал получается меньше, чем для линейной
модели (1,29 % или 1,15 %).
10.3.2. Проrноз неопределенности спек
трофотометрическоrо анализа rOToBoro ле
KapcTBeHHoro средства
При таких проrнозах Bcerдa используются
rенеральные величины. поэтому применяется
статистика raycca. Обычно используется коэф
фициент raycca 1.65 для односторонней вероят
ности 0,95.
При проведении спектрофотометриче
cKoro количественноrо определения roтовоro
лекарственноro средства берутся номиналь
ные навески около т=0,50 r (rлс) и m S ,=0,050
890
rосударственная фармакопея Республики Беларусь
r (стандартный образец). Используются оди
наковые разбавления для испытуемоro pacт
вора и раствора сравнения: навеска 50,0
мл (мерная колба); 1,0 мл (пипетка) получен
ноro раствора 100,0 мл (мерная колба).
Спектрофотометрическая неопределенность
оптической плотности (по паспорту прибора)
SA,,=0,2 %, кюветная неопределенность (экс
периментально найдена) SC€' .=0,1 %. Предпо
лаrается, что будет проводиться трехкратное
измерение оптической плотности испытуемо
ro раствора и раствора сравнения с вынима
нием кюветы. Необходимо провести проrноз
неопределенности анализа.
1) Вначале найдем неопределенность про
боподroтовки:
неопределенность взвешивания на анали
тических весах не более 0,2 Mr (0,0002 r), что
составляет 100.0.0002/ 0.5 0,04 % для испыту
емоro образца и 100 .0.0002/ 0,05 0,40 % для
стандартноro образца:
мерная колба вместимостью 50 мл 0,17 %;
мерная колба вместимостью 100 мл о, 12 %;
пипетка вместимостью 1 мл 0,6 %.
Полная неопределенность пробоподrотов
ки составляет (учитывая испытуемый раствор и
раствор сравнения):
'\5".,1004' c: '= = < : . =, '01 7') :Tri 2' , 0,12' )+lo,6' o.6'lol,06%
Отсюда ЕЩДНО. что основной вклад в неопреде
ленность пpoбonoдroтовки вносит пипетка малоro
объема (0.6 %) и маленькая навеска 0,05 r (0,4 %).
2) Найдем неопределенность конечной aHa
литической операции (спектрофотометрии).
Коэффициент 2 учитывает наличие испытуемоro
раствора и раствора сравнения:
, .: З ' : J 1 .65 , 12' (0,22 з + 0,12) 0, 30 0' 0
.'\FAO, 1.65 /(
3) Найдем теперь по формуле (10.1.-4) полную
проrнозируемую неопределенность анализа:
c. AS ' \ 1.06' 0.30' '= 1.10 %.
Как видно, основной вклад в полную He
определенность анализа вносит пробоподrо
товка (1,06 %).
10.3.3. Расчет cpeAHero значения несколь-
ких неравноточных выборок
В результате межлабораторноro экспери
мента получены следующие результаты коли
чественноro анализа некотороro roтовоro лекар
ственноro средства:
N 1 2 3 4 5 6 7 8 9 10
x k
% 10,8 10,6 11,2 11,1 10,9 11,1 10,5 10,8 11,0 11,2
абс.
Дх,k
% 0,32 0,21 0,65 0,45 0,25 0,32 0,19 0,34 0,42 0,58
абс.
Какое среднее значение содержания можно
приписать лекарственному средству по данным
межлабораторноro эксперимента?
Результаты анализа в разных лаборато
риях нельзя считать выборками из одной reHe
ральной совокупности, даже если они получе
ны с использованием одноro метода, например,
ВЭЖХ. Это связано с тем, что точностные ха,
рактеристики при боров в разных лабораториях
разные. В частности, хроматоrрафы MOryT обла
дать существенно разными rенеральными дис
персиями сходимости хроматоrрафическоrо сиr
нала, связанными как с приборными факторами.
так и с различием колонок и условий анализа.
Поэтому уравнение (2,1.1. 3) здесь непримени
мо. Неприменимо и уравнение (10.1.1. 2): число
опытов, как правило, невелико инеодинаково.
Поэтому применим соотношение (10.1.1. 1):
10.8 + 10,6 , 11.2 , 11.1 , 10.9 11,1 , 10,5 , 10,8 + 11,0 + 11,2
0.32' 0,21' 0,65' 0.45' 0.25" 0,32' 0.19' 0,34' 0.42' 0.58'
х == - - . - -
1 . 1 1 1 1 1 1 1. 1 1
0.32' '0.21' 0.65' 0,45" 025' . 032' + 0.19' 0,34" 0,42' ' 0,58'
.о21 .8 10П %
110.51
Для сравнения: обычное (невзвешенное) cpeд
нее значение по формуле (1.1. 1) будет 11,92 %.
т. е. на 100. /11,92 10,77)/10,77 1,4 % выше.
В соответствии с соотношением (10.1.1. 1a)
абсолютный доверительный интервал этоrо
взвешенноrо среднеro равен:
LL 0095 О/
с.;; " , /0
1110,51
Как видно, эта величина существенно меньше
любою частною доверительноro интервала Д x,k .
11. ПРИЛОЖЕНИЯ IО
Таблица 11. 1
Числовые значения контРОЛЬН020 критерия О/Р"п)
п Q
Р=90 % Р=95 % Р=99 %
3 0.89 0,94 0,99
4 0,68 0,77 0,89
5 0,56 0.64 0,76
6 0,48 0,56 0,70
7 0,43 0,51 0,64
8 0,40 0,48 0,58
9 0,38 0,46 0,55
15 Л. Н. Большев, Н. В. Смирнов. Таблицы математической статистики. М.: Наука. 1983. 415 с.
#5. 3.1. Статистический анализ результатов химическоео эксперимента
891
Таблица 11. 2
Числовые значения коэффициента Стьюдента t(p, 1/)
Вероятность Р 1 или Р 2
P 95% 97,5% 99% 99,5% 99,9% 99,95 %
1
P2 90% 95% 98% 99% 99,8% 99,90 %
Число
степеней Значения t(p, ,)
свободы V J
1 6,3138 12,7062 31,8205 63,6567 318,31 636,619
2 2,9200 4,3027 6,9646 9,9248 22,3271 31,5991
3 2,3534 3,1824 4,5407 5,8409 10,2145 12,9240
4 2,1318 2,7764 3,7469 4,6041 7,1732 8,6103
5 2,0150 2,5706 3,3649 4,0321 , 5,8934 6,8688
6 1,9432 2,4469 3,1427 3,7074 i 5.2076 5,9588
7 1,8946 2,3646 2,9980 3,4995 , 4,7853 5.4079
,
I
8 1 ,8595 2,3060 2,8965 3,3554 4,5008 5,0413
9 1,8331 2,2622 2,8214 3,2498 4,2968 4,7809
10 1 ,8125 2.2281 2,7638 3,1693 4,1437 5,5869
11 1 ,7956 2,2010 2,7181 3,1058 4,0247 4,4370
12 1 ,7823 2,1788 2,6810 3,0545 3,9296 4,3178
13 1 ,7709 2,1604 2,6503 3,0123 З,8520 4,2208
14 1,7613 2,1448 2,6245 2,9768 3,7874 4,1405
15 1,7530 2,1314 2,6025 2,9467 3,7328 4,0728
16 1,7459 2,1199 2,5835 2,9208 3,6862 4,0150
17 1,7396 2,1098 2,5669 2,8982 3,6458 3,9651
18 1 ,7341 2,1009 2,5524 2,8784 3,6105 3,9216
19 1,7291 2,0930 2,5395 2,8609 3,5794 3,8834
20 1,7247 2,0860 2,5280 2,8453 3,5518 3,8495
21 1,7207 2,0796 2,5176 2,8314 3,5272 3,8193
22 1,7171 2,0739 2,5083 2,8188 3,5050 3,7921
23 1,7139 2,0687 , 2,4999 I 2,8073 3,4850 3,7676
24 1,7109 2,0639 2,4922 2,7969 3,4668 3,7454
25 1,7081 2,0595 2,4851 2,7874 3,4502 3,7251
26 1,7056 2,0555 2,4786 2,7787 3,4350 3,7066
27 1 ,7033 2,0518 2,4727 2,7707 3,4210 3.6896
28 1,7011 2,0484 2,4671 2,7633 3,4082 3,6739
1,6991 , 3,3962 I 3,6594
29 2,0452 2,4620 2,7564 I
30 1,6973 2,0423 2.4573 2,7500 3,3852 I 3,6460
40 1,6839 2,0211 2,4233 2,7045 3,3069 3,5510
50 1,6759 2,0086 2,4033 2,6778 3,2614 3,4960
100 1,6602 1,9840 2,3642 2,6259 3,1737 3,3905
00 1,6479 1,9647 2,3338 2,5857 3,1066 3,3101
Р, вероятность нахождения истинноro значения величины (11) в интервале х ""-, :5 Р :5 ос или
Х :<; JJ :<; Х + ""-, (одностороннее распределение);
Р2 вероятность нахождения истинноro значения величины (/1) в интервале х ""-, :<; 1':5 Х + ""-,
(двустороннее распределение)_
892
rосударственная фармакопея Республики Беларусь
Таблица 11. 3
Процентные точки распределения x 2 (p"v)
v Р1=95 % Р1=99 %
1 3,841 6,635
2 5,991 9,210
3 7,815 11,345
4 9,488 13,277
5 11,070 15,086
6 12,592 16,812
7 14,067 18,475
8 15,507 20,090
9 16,919 21,666
10 18,307 23,209
11 19,675 24,725
12 21,026 26,217
13 22,362 27,688
14 23,685 29,141
15 24,996 30,578
16 26,296 32,000
17 27,587 33,409
18 28,869 34,805
19 30,144 36,191
20 31,410 37,566
21 32,671 38,932
22 33,924 40,289
23 35,172 41,638
24 36,415 42,980
25 37,652 44,314
26 38,885 45,642
27 40,113 46,963
28 41,337 48,278
29 42,557 49,588
за 43,773 50,892
40 55,758 63,691
50 67,505 76,154
" 2
Р, вероятность тоro. что оцениваемое значение х не превышает табличное. Это оцениваемое
значение рассматривается как значимое (Р,=95 %) или высоко значимое (Р,=99 %).
2 Таблица 11.А
Критерий Кохрена. Критические точки статистики G = Smax ,построенной по 9 независимым
9
'2Js; )
. 1
оценкам дисперсии (S:), каждая из которых обладает ' степенями свободы
v 1 2 3 4 5 6 7 8 9 10 16 36 00
g!
G(P=95 %)
2 0,9985 0,9750 0,9392 0,9057 0,8772 0,8534 0,8332 0,8159 0,8010 0,7880 0,7341 0,6602 0,5000
3 0,9669 0,8709 0,7977 0,7457 0,7071 0,6771 0,6530 0,6333 0,6167 0,6025 0,5466 0,4748 0,3333
4 0,9065 0,7679 0,6841 0,6287 0,5895 0,5598 0,5365 0,5175 0,5017 0,4884 0,4366 0,3720 0,2500
5 0,8412 0,6838 0,5981 0,5440 0,5063 0,4783 0,4564 0,4387 0,4241 0,4 18 0,3645 0,3066 0,2000
6 0,7808 0,6161 0,5321 0,4803 0,444 7 0,4184 0,3980 0,3817 0,3682 0,3568 0,3135 0,2612 0,1667
7 0,7271 0,5612 0,4800 0,4307 0,3974 0,3726 0,3535 0,3384 0,3259 0,3154 0,2756 0,2278 0,1429
8 0,6798 0,5157 0,4377 0,3910 0,3595 0,3362 0,3185 0,3043 0,2926 0,2829 0,2462 0,2022 0,1250
9 0,6385 0,4775 0,4027 0,3584 0,3286 0,3067 0,2901 0,2768 0,2659 0,2568 0,2226 0,1820 0,1111
10 0,6020 0,4450 0,3733 0,3311 0,3029 0,2823 0,2666 0,2541 0,2439 0,2353 0,2032 0,1655 0,1000
#5.3.1. Статистический анализ результатов химичеСКО20 эксперимента
893
v........ 1 2 3 4 5 6 7 8 9 10 16 36 00
g!
G(P=95 %)
12 0,5410 0,3924 0,3264 0,2880 0,2624 0,2439 0,2299 0,2187 0,2098 0,2020 0,1737 0,1403 0,0833
15 0,4709 0,3346 0,2758 0,2419 0,2195 0,2034 0,1911 0,1815 0,1736 0,1671 0,1429 0,1144 0,0667
20 0,3894 0,2705 0,2205 0,1921 0,1735 0,1602 0,1501 0,1422 0,1357 0,1303 0,1108 0,0879 0,0500
24 0,3434 0,2354 0,1907 0,1656 0,1493 0,1374 0,1286 0,1216 0,1160 0,1113 0,0912 0,0743 0,0417
30 0,2929 0,1980 0,1593 0,1377 0,1237 0,1137 0,1061 0,1002 0,0958 0,0921 0,0771 0,0604 0,0333
40 0,2370 0,1576 0,1259 0,1082 0,0968 0,0887 0,0827 0,0780 0,0745 0,0713 0,0595 0,0462 0,0250
60 0,1737 0,1131 0,0895 0,0765 0,0682 0,0623 0,0583 0,0552 0,0520 0,0497 0,0411 0,0316 0,0167
00 О О О О О О О О О О О О О
v........ 1 2 3 4 5 6 7 8 9 10 16 36 00
g!
G(P=99 %)
2 0,9999 0,9950 0,9794 0,9586 0,9373 0,9172 0,8988 0,8823 0,8674 0,8539 I 0.7949 0,7067 0,5000
3 0,9933 0;9423 0,8831 0,8335 0,7933 0,7606 0,7335 0,7107 0,6912 0.6743 0,6059 0.5153 0,3333
4 0,9676 0,8643 0,7814 0,7212 0,6761 0,6410 0.6129 0,5897 0,5702 0,5536 0,4884 0,4057 0,2500
5 0,9279 0,7885 0,6957 0,6329 0,5875 0,5531 0.5259 0,5037 0,4854 0.4697 0,4094 0,3351 0,2000
6 0,8828 0,7218 0,6258 0,5635 0,5195 0,4866 0,4608 0,4401 0,4229 0,4081 0,3529 0,2858 0,1667
7 0,8376 0,6644 0,5685 0,5080 0,4659 0,4347 0,4105 0,3911 0,3751 0,3616 0,3105 0,2494 0,1429
8 0,7945 0,6152 0,5209 0,4627 0,4226 0,3932 0,3704 0,3522 0,3373 0,3248 0,2779 0,2214 0,1250
9 0,7544 0,5727 0,4810 0,4251 0,3870 0,3592 0,3378 0,3207 0,3067 0,2950 0,2514 0,1992 0,1111
10 0,7175 0,5358 0,4469 0,3934 0,3572 0,3308 0,3106 0,2945 0,2813 0,2704 0,2297 0,1811 0,1000
12 0,6528 0,4751 0,3919 0,3428 0,3099 0,2861 0,2680 0,2535 0,2419 0,2320 0,1961 0,1535 0,0833
15 0,5747 0,4069 0,3317 0,2882 0,2593 0,2386 0,2228 0,2104 0,2002 0,1918 0,1612 0,1251 0,0667
20 0,4799 0,3297 0,2654 0,2288 0,2048 0,1877 0,1748 0,1646 0,1567 0,1501 0,1248 0,0960 0,0500
24 0,4247 0,2871 0,2295 0,1970 0,1759 0,1608 0,1495 0,1406 0,1338 0,1283 0,1060 0,0810 0,0417
30 0,3632 0,2412 0,1913 0,1635 0,1454 0,1327 0,1232 0,1157 0,1100 0,1054 0,0867 0,0658 0,0333
40 0,2940 0,1915 0,1508 0,1281 0,1135 0,1033 0,0957 0,0898 0,0853 0,0816 0,0668 0,0503 0,0250
60 0,2151 0,1371 0,1069 0,0902 0,0796 0,0721 0,0668 0,0625 0,0594 0,0567 0,0461 0,0344 0,0167
о() О О О О О О О О О О О О О
2 ( 2 ) "
Smax тах Sk '
Р вероятность тоro, что все 9 оценок дисперсии являются выборочными значениями одной и
той же rенеральной совокупности. rипотеза равенства дисперсий может быть значимой (Р=95 %) или
высоко значимой (Р=99 %).
Таблица 11. 5
Процентные точки распределения Фишера (F(P',I',.1'2 ) распределения)
v 1 ........ 1 2 3 4 5 6 7 8 9 10 12 15 20 24 I зо I о()
V 2 !
Р1 =95 %
1 161,5 199,5 215,7 224,6 230,2 234,0 236,8 238,9 240,5 241,9 243,9 246,0 248,0 i 249,1 250,1 254,3
2 18,51 19,00 19,16 19,25 19,30 19,33 19,35 19,37 19,39 19,4 19,41 19,43 19,45! 19,45 19,46 19,50
3 10,13 9,552 9,277 9,117 9,014 8,941 8,887 8,845 8,812 8,786 8,745 8,703 8,660 8,639 8,617 8,527
4 7,709 6,944 6,591 6,388 6,256 6,163 6,094 6,041 5,999 5,964 5,912 5,858 5,803 5,774 5,746 5,628
5 6,608 5,786 5,410 5,192 5,050 4,950 4,876 4,818 4,773 4,735 4,678 4,619 4.558 4,527 4,496 4,365
6 5,987 5,143 4,757 4,534 4,387 4,284 4,207 4,147 4,099 4,060 4,000 3,938 3,874 3,842 3,808 3,669
7 5,591 4,737 4,347 4,120 3,972 3,866 3,787 3,726 3,677 3,637 3,575 3,511 3,445 3,411 3,376 3,230
8 5,318 4,459 4,066 3,838 3,688 3,581 3,501 3,438 3,388 3,347 3.284 3,218 3,150 3,115 3,079 2,928
9 5,117 4,257 3,827 3,633 3,482 3,374 3,293 3,230 3,179 3,137 3,073 3,006 2,937 2,901 2,864 2,707
10 4,965 4,103 3,708 3,478 3,326 3,217 3,136 3,072 3,020 2,978 2,913 2,845 2,774 2,737 2,700 2,538
12 4,747 3,885 3,490 3,259 3,106 2,996 2,913 2,849 2,796 2,753 2,687 2,617 2,544 2,506 2,466 2,296
15 4,543 3,682 3,287 3,056 2,901 2,790 2,707 2,641 2,588 2,544 2,475 2,403 2,328 2,288 2,247 2,066
894
rосударственная фармакопея Республики Беларусь
V1 1 2 3 4 5 6 7 8 9 10 12 15 20 24 30 00
v 2 J
Р, =95 %
20 4,351 3,493 3,098 2,866 2,711 2,599 2,514 2,447 2,393 2,348 2,278 2,203 2,124 2,083 2,039 1,843
25 4,242 3,385 2,991 2,759 2,603 2,490 2,405 2,337 2,282 2,236 2,165 2,089 2,007 1,964 1,919 1,711
30 4,171 3,316 2,922 2,690 2,534 2,421 2,334 2,266 2,211 2,165 2,092 2,015 1,932 1,887 1,841 1,622
50 4,034 3,183 2,790 2,557 2,400 2,286 2,208 2,130 2,082 2,026 1,952 1,871 1,784 1,747 1,697 1,438
00 3,841 2,996 2,605 2,372 2,214 2,099 2,010 1,938 1,880 1,831 1,752 1,666 1,571 1,517 1,459 1,000
V1 1 2 I 3 4 5 6 7 8 9 10 12 15 20 24 30 00
v 2 J i
Р. =99 %
1 4052 5000 1 5403 I 5625 5764 5859 5928 5981 6023 6056 6106 6157 6209 6235 6261 6366
2 98.50 99.00 i 99.17 i 99.25 99,30 99,33 99,36 99,37 99,39 99,40 99.42 99,43 99,45 99,46 99,47 99,50
3 34,12 30.82 I 29.46 28.71 28,24 27,91 27,67 27,49 27,35 27.23 27,05 26,87 26,69 26,60 26,51 26,13
4 21,20 18.00 116_69 15,98 15,52 15,21 14,98 14,80 14,66 14,55 14,37 14,20 14,02 13,93 13,84 13,46
5 16,26 13.27! 12.06111,39 10,97 10,67 10,46 10,29 10,16 10,05 9,888 9,722 9,553 9,467 9,379 9,020
6 13,75 10.93: 9.780 9.148 8,746 8,466 8,260 8,102 7,976 7,874 7,718 7,559 7.396 7,313 7,229 6,880
7 12.25 9.547 ; 8.451 7,847 7,460 7,191 6,993 6,840 6,719 6,620 6,469 6,314 6,155 6,074 5,992 5,650
8 11,26 8.649 : 7,591 7,006 6,632 6,371 6,178 6,029 5,911 5,814 5,667 5,515 5,359 5,279 5,198 4,859
9 10.56 8.022 : 6.992 6,422 6,057 5,802 5,613 5,467 5,351 5,257 5,111 4,962 4,808 4,729 4,649 4,311
10 10.04 7.559! 6.552 5,994 5,636 5,386 5,200 5,057 4,942 4,849 4,706 4,558 4,405 4,327 4,247 3.909
12 9.330 6.927 : 5.953 5,412 5,064 4,821 4,640 4,499 4,388 4,296 4,155 4,010 3,858 3,781 3,701 3,361
15 8.63816.359 i 5.417 4,893 4,556 4,318 4,142 4,004 3,895 3,805 3,666 3,522 3,372 3,294 3,214 2,868
20 I 8_0961 5.849 ! 4.938 4,431 4,103 3,871 3,699 3,564 3,457 3,368 3,231 3,088 2,938 2,859 2,779 2,421
25 I 7.770: 5.568 i 4.675 4,177 3,855 3,627 3,457 3,324 3,217 3,129 2,993 2,850 2,699 2,620 2,538 2,169
30 : 7.562: 5.390 I 4.510 4,018 3,699 3,473 3,305 3,173 3,067 2,979 2,843 2,700 2,549 2,469 2,386 2,006
50 i 7.171 ; 5.05714.199 3,720 3,408 3,186 3,038 2,890 2,803 2,698 2,536 2,419 2,265 2,202 2,116 1,683
о:; : 6.63514.6051 3.782 3,319 3,017 2,802 2,639 2,511 2,407 2,321 2,185 2,039 1,878 1,791 1,696 1,000
Р1 вероятность тоro, что оцениваемое значение F не превышает табличное. Это оцениваемое
значение рассматривается как значимое (Р1=95 %) или высоко значимое (Р1=99 %).
Таблица 11. 6
Процентные точки выБОрОЧН020 коэффициента корреляции r
I Вероятность Р1 или Р2
P1 95 % 97,5% 99% 99,5% 99,75 % 99,95 %
P2 90 % ! 95% 98% 99% 99,5% 99,9%
I
Число
степеней
свободы v Значения R(P,I')
1
1 0,9877 0,99692 0,999507 0,999877 0,999969 0,999999
2 0,900 0,9500 0,9800 0,99000 0,99500 0,999000
3 0,805 0,878 0,9343 0,9587 0,9740 0,99114
4 0,729 0,811 0,882 0,9172 0,9417 0,9741
5 0,669 0,754 0,833 0,875 0,9056 0,9509
6 0,621 0,707 0,789 0,834 0,870 0,9249
7 0,582 0,666 0,750 0,798 0,836 0,898
8 0,549 0,632 0,715 0,765 0,805 0,872
9 0,521 0,602 0,685 0,735 0,776 0,847
10 0,497 0,576 0,658 0,708 0,750 0,823
#5. 3. 2. Валидация аналитических методик и испытаний
895
Вероятность Р 1 или Р 2
Р 1 ---4 95% 97,5% 99% 99,5% 99,75 % 99,95 %
Р 2 ---4 90% 95% 98% 99% 99,5% 99,9%
Число
степеней
свободы v Значения R(Py)
!
11 0,476 0,553 0,634 0,684 0,726 0,801
12 0,457 0,532 0,612 0,661 0,703 0,780
13 0,441 0,514 0,592 0,641 0,683 0,760
14 0,426 0,497 0,574 0,623 , 0,664 0,742
15 0,412 0,482 0,558 0,606 0,647 0,725
16 0,400 0,468 0,543 0,590 0,631 0,708
17 0,389 0,456 0,529 0,575 i 0,616 0,693
18 0,378 0,444 0,516 0,561 I 0,602 0,679
19 0,369 0,433 0.503 0,549 ! 0,589 0,665
20 0,360 0,423 0,492 0,537 I 0.576 I 0,652
25 0,323 0,381 0,445 0,487 0,524 I 0,597
30 0,296 0,349 0,409 0,449 0,484 0,554
35 0,275 0,325 0,381 0,418 0,452 0,519
40 0,257 0,304 0,358 0,393 0,425 0,490
45 0,243 0,288 0,338 0,372 0,403 0,465
50 0,231 0,273 0,322 0,354 0,384 0,443
60 0,211 0,250 0,295 0,325 0,352 0,408
70 0,195 0,232 0,274 0,302 0,327 0,380
80 0,183 0,217 0,257 0,283 0,307 0,357
90 0,173 0,205 0,242 0,267 0,290 0,338
100 0,164 0,195 0,230 0,254 0,276 0,321
Р1 вероятность тоro, что r < R или R < r (односторонний критерий);
Р2 вероятность тоro, что коэффициент корреляции, находится в интервале R < r < R (ДBYCTO
ронний критерий);
Выборочный коэффициент корреляции r значимо (с надежностью Р 1 или Р 2 ) отличается от нуля,
если еro абсолютная величина Irl превышает табличное значение R(P,I/).
01/2013: РБ50302
#5.3.2. ВАЛИДАЦИЯ
АНАЛИТИЧЕС!<ИХ МЕТОДИК
И ИСПЫТАНИИ
В данной статье описываются процедуры,
при меняемые для валидации методик и испы
таний (испытание это аналитическая методи
ка, описанная в частной статье, в совокупности
с требованиями к получаемым по ней результа
там; результатом проведения испытания являет
ся ответ на вопрос, соответствует или нет данное
лекарственное средство требованиям частной
статьи), включаемых в общие статьи Фармако
пеи и аналитическую нормативную ДOKYMeH
тацию на лекарственные средства и вспомоrа
тельные вещества (частные статьи). Поскольку
в частные статьи включаются различные инстру
ментальные и не инструментальные испытания
(определение подлинности, контроль примесей,
количественное определение и др.), требования
к валидации испытания зависят от ero типа и
применяемоro аналитическоrо метода.
А. ТЕРМИНЫ И ОПРЕДЕЛЕНИЯ.
ИСПОЛЬЗУЕМЫЕ ПРИ ВАЛИДАЦИИ
АНАЛИТИЧЕСКИХ МЕТОДИК
1. ВВЕДЕНИЕ
Валидация аналитической методики это
а) экспериментальное доказательство тоro, что
методика приroдна для решения предполаrа
емых задач; Ь) документированное подтвержде
ние обоснованности (правильности) выбора Me
тодики испытаний, rарантирующее получение
ожидаемых и воспроизводимых результатов, co
ответствующих поставленной цели.
896
rосударственная фармакопея Республики Беларусь
В данном разделе рассматриваются xapaK
теристики аналитических методик (испытаний),
подлежащие валидации (далее «валидацион
ные характеристики»).
Валидационные характеристики методик,
применяемых в целях идентификации, контроля
примесей и количественноro определения, при
ведены в таблице 5.3.3 1.
2. АНАЛИТИЧЕСКИЕ ИСПЫТАНИЯ
И МЕТОДИКИ, ПОДЛЕЖАЩИЕ ВАЛИДАЦИИ
В статье рассматривается проведение вали
дации для таких испытаний, как:
испытания для идентификации;
количественные испытания для определе
ния примесей;
испытания для определения предельноro
содержания примесей;
количественные испытания для определе
ния действующеro вещества и друrих компонен
тов (например, консервантов) в фармацевтиче
ских субстанциях и лекарственных средствах.
Все аналитические методики и испытания,
входящие в общие статьи Фармакопеи и в анали
тическую нормативную документацию, должны
быть валидированы. Для валидации некоторых
испытаний, например. таких как «Растворение»
или «Определение размера частиц», MOryT по
требоваться друrие валидационные процедуры,
не описанные в общей статье.
Краткая характеристика испытаний
Испытания для идентификации предна
значены для ПQДтверждения наличия анализи
руемоro вещества в образце. Обычно это дo
стиrается путем сравнения каких либо свойств
(например, спектральных характеристик, xpOMa
тоrрафическоro поведения, химической реакци
онной способности и т. д.) испытуемоro и CTaH
дартноro образцов.
Испытания, предназначенные для KOHтpo
ля примесей, MOryT быть как количественными,
так и предельными. Назначение обоих испыта
ний характеризовать чистоту образца. Для
валидации .количественных и предельных испы
таний необходимы различные валидационные
характеристики.
Количественное определение предназначе
но для определения анализируемоro вещества
в образце. Такая же валидационная процедура
может быть применена к методике количествен
Horo определения, связанной с друrим испыта
нием (например, в испытании «Растворение»).
3. ВАЛИДАЦИОННЫЕ ХАРАКТЕРИСТИКИ
И ТРЕБОВАНИЯ
Набор исследуемых валидационных xapaK
теристик зависит от назначения аналитической
методики. Типичные валидационные характери
стики:
правильность;
точность (прецизионность);
сходимость;
внутрилабораторная точность;
специфичность;
предел обнаружения;
предел количественноro определения;
линейность;
Таблица 5.3.3. 1
Валидационные характеристики, рассматриваемые для различных испытаний и методик
Типы аналитических методик
Испытания на при меси Количественное
определение
Характеристики Идентификация Растворение,
Количественные Предельные определение
только содержа-
ния, активности
Правильность + +
Точность:
Сходимость + +
Внутрилабораторная +* +*
точность
Специфичность ** + + + +
Предел обнаружения *** +
Предел количествен +
ноro определения
Линейность + +
Диапазон применения + +
<Н> характеристика обычно не исследуется;
«+» характеристика обычно исследуется;
«*» В тех случаях, Korдa проводится исследование воспроизводимости, исследование внутрилабораторной точности не Tpe
буется;
«**» недостаток специфичности испытания можно компенсировать друrим (друrими) дополнительным испытанием (см. п. 4.2);
«***» может потребоваться в некоторых случаях (например, Korдa предел определения и нормируемый предел содержания
определяемой примеси близки).
#5.3.2. Валидация аналитических методик и испытаний
897
диапазон применения.
Этот список следует рассматривать как ти
повой для указанных испытаний (аналитических
методик). Как правило, на стадии разработки Me
тодики изучается также валидационная xapaKTe
ристика «робастность».
Повторное проведение валидации может
потребоваться в следующих случаях:
внесение изменений в синтез фармацев
тической субстанции;
внесение изменений в состав лекарствен
ноro средства;
изменение аналитической методики.
Объем проведения повторной валидации
определяется спецификой изменений. Повторная
валидация может требоваться и в иных случаях.
4. СЛОВАРЬ
Аналитическая методика (апа/ytiса/ proce
du,e) это способ проведения анализа, т. е. дe
тальное изложение всех операций, необходи
мых для выполнения испытания. Она включает
в себя описание подroтовки испытуемых образ
цов, стандартов и реактивов, описание исполь
зуемоro оборудования с указанием параметров,
условия получения калибровочных кривых, ис
пользуемые расчетные формулы и т. д.
Специфичность (specificity) способность
однозначно оценивать анализируемое веще
ство в присутствии друrих компонентов, которые
MOryT присутствовать в образце (примесей, про
дуктов разложения, вспомоrательных веществ
и т. д.). Недостаток специфичности испытания
может быть компенсирован друrим (друrими) дo
полнительным испытанием.
Специфичность для различных типов испы
таний означает следующее:
испытания для идентификации ДOKa
зательство тоro, что идентифицировано именно
анализируемое вещество;
испытания для определения содержания
примесей доказательство тоro, что каждое
испытание для определения содержания при
месей позволяет однозначно характеризовать
содержание примесей в образце (например, ис
пытания «Сопутствующие примеси», «Тяжелые
металлы», «Содержание остаточных количеств
орrанических растворителей» и др.);
количественное определение (содержа
ния или активности) доказательство тою, что
методика позволяет точно и правильно YCTaHO
вить содержание или активность именно анали
зируемоro вещества в образце.
Правильность (accuracy, t,ueness) xapaKTe
ризует степень соответствия между известным
истинным значением или справочной величиной
и значением, полученным по данной методике.
Точность (прецизионность) (р,есisiоп) aHa
литической методики выражает степень бли
зости (или степень разброса) результатов для
серии измерений, выполненных по данной Me
тодике на различных пробах одноro и тоro же
113. За.. 1712
однородноro образца. Точность может paCCMa
триваться на трех уровнях: сходимость (повто
ряемость), внутрилабораторная точность и BOC
производимость.
Точность необходимо изучать на ДOCТOBep
но однородных образцах. Однако если OДHOpOД
ный образец получить невозможно, можно ис
пользовать еro раствор или модельные смеси.
Точность аналитической методики обычно
характеризуют отклонением, стандартным OT
клонением или относительным стандартным OT
клонением для серии измерений.
Сходимость (повоторяемость) (,epeata
bility) характеризует точность методики при ее
выполнении в одних и тех же условиях (в част
ности, одним и тем же аналитиком или rруппой
аналитиков) в течение небольшоro промежутка
времени.
Внутрилабораторная точность (intermedi
ate рrесisiоп) характеризует влияние внутрила
бораторных вариаций: различные дни, различ
ные аналитики, различное оборудование и т. п.
изменения.
Воспроизводимость (,eprodиcibility) xapaK
теризует точность в межлабораторном экспери
менте.
Предел обнаружения для конкретной aHa
литической методики представляет собой мини
мальное количество анализируемоrо вещества
в образце, которое может быть обнаружено (при
этом не обязательно должно быть определено
точное значение).
Предел количествеНН020 определения для
аналитической методики представляет собой
минимальное количество анализируемоro Be
щества в образце, которое может быть коли
чественно определено с требуемой правиль
ностью и точностью. Предел количественноro
определения является валидационной xapaKTe
ристикой методик количественноro определения
малых концентраций веществ в образце и pac
сматривается в основном при определении при
месей и/или продуктов разложения.
Линейность (/inea,ity) это способность
методики (в пределах диапазона применения)
давать величины, прямо пропорциональные KOH
центрации (количеству) анализируемоro веще
ства в образце.
Диапазоном применения (rапgе) аналитиче
ской методики является интервал между мини
мальной и максимальной концентрациями (коли
чествами) анализируемоro вещества в образце
(включая эти концентрации), для котороro пока
зано, что аналитическая методика имеет требу
емую точность, правильность и линейность.
Робастность (robustness) это способ
ность аналитической методики быть устойчивой
к влиянию малых задаваемых (контролируемых)
аналитиком изменений в условиях выполнения
методики. Робастность является показателем
надежности методики при ее использовании в
указанных условиях.
898
rосударственная фармакопея Республики Беларусь
В. ПРОВЕДЕНИЕ ВАЛИДАЦИИ
АНАЛИТИЧЕСКИХ МЕТОДИК
1. ВВЕДЕНИЕ
rлавной задачей валидации аналитической
методики является экспериментальное доказа
тельство тоro, что данная методика приroдна для
достижения тех целей, для которых она пред
назначена. В отчет по валидации должны быть
включены все данные, полученные в процессе
валидации, и использованные для расчетов фор
мулы с соответствующим их обсуждением.
Подходы к проведению валидации методик
анализа биолоrических и биотехнолоrических
препаратов MOryт быть иными, чем указано в
данной статье.
При проведении валидации необходимо ис
пользовать только стандартные образцы с из
вестными характеристиками, подтвержденными
документально. Необходимая степень их чисто
ты зависит от задач, которые решаются при их
использовании.
Последовательность рассмотрения вали
дационных характеристик отражает процесс, по
которому может разрабатываться и валидиро--
ваться аналитическая методика. Однако целе
сообразно планировать эксперимент так, чтобы
соответствующие валидационные характеристи
ки изучались одновременно, например: специ
фичность, линейность, диапазон применения,
правильность и точность.
2. СПЕЦИФИЧНОСТЬ
Исследование специфичности проводится
при валидации испытаний на идентификацию,
контроль примесей и количественное определе
ние. Способ подтверждения специфичности за
висит от задач. для решения которых предназна
чена аналитическая методика.
В тех случаях. коrда методика недостаточ
но специфична. применяют сочетание двух или
более аналитических методик для достижения
необходимоrо уровня избирательности.
2.1. Идентификация
Испытания на идентификацию должны обе
спечивать возможность различать соединения
близкоro строения, которые MorYT присутство
вать в образце совместно с определяемым KOM
понентом. Избирательность методики может
быть подтверждена получением положитель
ных результатов (возможно, путем сравнения с
известным стандартным образцом) для образ
цов, содержащих определяемый компонент, и
отрицательных результатов, полученных для об
разцов, не содержащих еro. Для подтвержде
ния отсутствия ложноположительных резулыа
тов испытание на идентификацию может быть
проверено для веществ с близким строением
или сопутствующих анализируемому веществу.
Выбор потенциально мешающих проведению
испытания веществ должен быть обоснован.
2.2. Количественное определение и испы
тания для определения содержания примеси
При валидации хроматоrрафических Meтo
дик для подтверждения специфичности должны
использоваться характерные xpoMaTorpaMMbI с
указанием индивидуальных веществ. Аналоrич
ный подход используют И для друrих методов
разделения.
Для хроматоrрафических методик степень
разделения должна быть исследована для co
ответствующих концентраций веществ. Для
подтверждения специфичности может быть ис
пользована степень разделения двух наиболее
близко элюирующихся веществ.
В случае использования неспецифичноro
метода количественноro определения необходи
мо при менять дополнительные аналитические
методики и подтверждать специфичность всеro
комплекса методик. Например, если количе
ственное определение проводится титриметри
ческим методом, то ero можно дополнить COOT
ветствующим испытанием на примеси.
Для количественноro определения и для
испытаний на при меси применяют одинаковые
подходы, описанные ниже.
2.2.1. Образцы примесей имеются
Для метода количественноro определения
необходимо подтвердить избирательность опре
деления анализируемоro вещества в присут
ствии примесей иlили друrих компонентов об
разца. Это можно сделать внесением в образец
(субстанцию или лекарственное средство) при
месей и/или друrих компонентов образца в co
ответствующей концентрации и последующим
доказательством тоro, что это не отразил ось на
получаемом результате (путем сравнения pe
зул, ьтатов полученных на исходном и заrрязнен
ном образцах).
Для испытаний на чистоту подтверждение из
бирательности проводят путем заrрязнения суб
станции или roтовоro лекарственноro средства
соответствующими количествами примесей и дo
казательства разделения этих примесей как друr
от друrа, так и от друrих компонентов образца.
2.2.2. Образцы примесей отсутствуют
В случае если образцы примесей или про
дуктов разложения отсутствуют, подтверждение
специфичности проводят путем сравнения pe
зультатов анализа образцов, содержащих при
меси или продукты разложения, предлаrаемой
методикой и друroй арбитражной методикой.
В качестве последней может быть использова
на фармакопейная или друrая валидированная
методика. Этот подход предполаrает предвари
тельное заrрязнение образца продуктами раз
пожения путем выдерживания еro в стрессовых
условиях: воздействие света, тепла, влажности,
rидролиза, окисления и т. п.
При валидации количественноro определе
ния следует сравнить результаты анализов, по
лученных с использованием валидируемой и ap
битражной методик.
#5.3.2. Валидация аналитических методик и испытаний
899
При валидации испытания на чистоту следу
ет сравнить результаты определения примесей,
полученные с использованием валидируемой и
арбитражной методик
Для доказательства тоro, что пик анализи
руемоro вещества соответствует только одному
компоненту, используют тесты на чистоту пиков,
например, с использованием диодно матрично
ro детектирования, масс спектрометрии и др.
3. ЛИНЕЙНОСТЬ
Линейная зависимость должна быть ис
следована в пределах диапазона примене
ния аналитической методики. Она может быть
подтверждена непосредственно на субстан
ции (путем разбавления исходноro раствора)
или, для лекарственных средств, на модель
ных смесях с использованием COOTBeTCTBY
ющей процедуры.
По полученным данным строят rрафик зави
симости сиrнала как функции концентрации или
количества определяемою компонента и i3изу
ально оценивают еro линейность. Если линейная
зависимость наблюдается, то результаты обра
батывают подходящим статистическим методом,
например, методом наименьших квадратов. В
некоторых случаях для получения линейности
данные следует подверrнуть предварительно
му математическому преобразованию. Должны
быть определены и представлены: коэффициент
корреляции, точка пересечения с осью ординат,
TaHreHc уrла наклона прямой и остаточная сумма
квадратов отклонений, а также rрафик со всеми
экспериментальными данными. Для оценки ли
нейности MOryт понадобиться отклонения экспе
риментальных данных от прямой.
Некоторые аналитические методики, напри
мер, иммуноаналитические, не показывают ли
нейности ни при каких математических преобра
зованиях. В таких случаях аналитический отклик
должен быть описан подходящей функцией KOH
центрации анализируемоro вещества в образце.
Для подтверждения линейности использу
ют не менее пяти концентраций. Друrие подходы
должны быть обоснованы.
4. ДИАПАЗОН ПРИМЕНЕНИЯ
Диапазон применения методики зависит от
ее предназначения и определяется при изуче
нии линейности. В пределах диапазона приме
нения методика должна обеспечивать требу
емую линейность, правильность и точность.
Минимальные допустимые диапазоны при
менения методик:
для количественноro определения лекар
ственных субстанций или лекарственных форм
от 80 % до 120 % от номинальноro содержания;
для однородности дозирования от 70 %
до 130 % от номинальноro содержания, если для
испытания не требуется более широкий интер
вал (например, для дозированных аэрозолей);
для испытаний по тесту «Растворение»
::1:20 % (абсолютных) от нормируемой величины
высвобождения; например, если при контроле
высвобождения пролонrированных лекарствен
ных средств нормируемая величина высвобож
дения составляет от 20 % за первый час и до
90 % за 24 ч, то диапазон применения должен
быть от О % до 11 О % от номинальноro coдep
жания;
для определения примесей от KOHцeH
трации, в которой примесь обычно обнаружива
ется, д0120 % от нормируемоro содержания.
Для примесей, обладающих чрезвычай
но сильным действием или имеющих токсиче
ский или непредсказуемый фармаколоrический
эффект, предел детектирования/количествен
ноro определения должен соответствовать тому
уровню концентрации. на котором эти при меси
должны контролироваться.
Примечание: при проведении валидации иc
Пblтания для определения содержания приме
сей непocpeдcmвенно в процессе разработки
методики нео6ходимо определить диапазон
применения, внутри копюро20 находится пред
полаzаемый предел нормирования примесей.
В случае если количественное определение
и испытание по определению содержания при
месей выполняются совместно, как одно испыта
ние, и используется только стандарт основноro
вещества, соответствующий ero номинальному
содержанию, диапазон применения должен ox
ватывать диапазон концентрации от нормиру
емоro содержания примеси до 120 % от номи
нальноro содержания основноro вешества.
5. ПРАВИЛЬНОСТЬ
Правильность изучают в пределах диапазо
на применения аналитической методики.
5.1. Количественное определение
5. 1. 1. Субстанции
MOryT использоваться следующие способы
определения правильности:
применение аналитической методики к об
разцу с известной степенью чистоты, например,
к стандартному образцу;
сравнение результатов анализа, получен
ных с использованием валидируемой методики и
арбитражноro метода, правильность и точность
котороro известны (использование независимо
ro метода, см. п. 2.2);
заключение о правильности можно cдe
лать после тою, как установлены точность, ли
нейность и специфичность.
5.1.2. /отовые лекарствеННblе средства
MoryT использоваться следующие способы
определения правильности:
применение методики к искусственным
смесям, к которым были добавлены известные
количества анализируемоro вещества;
в случае, Korдa невозможно получить об
разцы всех компонентов лекарственноro cpeд
ства, возможно применение метода добавок или
900
rосударственная фармакопея Республики Беларусь
арбитражной методики, правильность которой
доказана (см. п. 2.2);
заключение о r ,j)авильности можно cдe
лать после тоro, как установлены точность, ли
нейность и специфичность.
5.2. Примеси (количественное содержа-
ние)
Правильность изучают на образцах (суб
станции или roтовоro лекарственноro средства) с
добавленным известным количеством примесей.
Если примеси или продукты разложения He
доступны, применяют арбитражный метод (см.
п. 2.2). Если примеси неизвестны, то чувстви
тельность их определения может быть принята
равной чувствительности определения субстан
ции. В случае если чувствительность определе
ния субстанции и примеси существенно отлича
ется, вводят коэффициент пересчета.
Должен быть указан конкретный способ HOp
мирования содержания примесей, например, в
массовых процентах, в процентах по отношению
к площади пика rлавноro компонента или др.
5.3. Представление данных
Правильность оценивают не менее чем для
девяти определений для трех различных KOH
центраций. охватывающих весь диапазон при
менения, т. е. три концентрации и три опреде
ления для каждой концентрации. Определения
должны включать все стадии методики.
Правильность выражают в процентах най
денноro значения от введенноro или как разность
между средним и истинным значениями с учетом
соответствующих доверительных интервалов.
6. ТОЧНОСТЬ
Валидационная характеристика «точность»
изучается для методик количественноro опреде
ления.
6.1. Сходимость
Сходимость изучают. выполняя:
не менее девяти определений, OXBaTЫBa
ющих диапазон применения методики (три KOH
центрации/три повтора), или
не менее шести определений для образ
цов с содержанием анализируемоro вещества,
близким к номинальному.
6.2. Внутрилабораторная точность
Устанавливают влияние случайных факто
ров на точность валидируемой аналитической
методики. Типичными исследуемыми факторами
являются различные дни, различные аналитики,
различное оборудование и п. При изучении
влияния различных факторов предпочтительно
использовать планирование эксперимента.
6.3. Воспроизводимость
Воспроизводимость оценивают путем про
ведения межлабораторных исследований. Boc
производимость должна быть изучена при вклю
чении методики в Фармакопею.
6.4. Представление данных
При изучении точности должны представ
ляться: стандартное отклонение, относительное
стандартное отклонение и доверительный ин
тервал.
7. ПРЕДЕЛ ОБНАРУЖЕНИЯ
В зависимости от тоro, является методи
ка инструментальной или неинструментальной,
возможны различные подходы для определения
предела обнаружения (ПО, detectioп Iimit (OL)
или Iim;t о( dеtесtiоп (LOO)). Используются сле
дующие подходы.
7.1. Визуальная оценка
Визуальную оценку используют как для He
инструментальных, так и для инструментальных
методов.
Предел обнаружения устанавливают путем
анализа образцов с известными концентраци
ями анализируемоro вещества и оценкой мини
мальноro содержания, при котором анализиру
емое вещество надежно определяется.
7.2. Соотношение сиrнал/шум
Этот подход применим только к тем Me
тодам, для которых наблюдается шум базо
вой линии. Для определения соотношения
сиrнал/шум сравнивают величины сиrналов,
полученные для контрольноro опыта и для
образцов с низкими концентрациями анали
зируемоrо вещества. На основании получен
ных данных устанавливают минимальную
концентрацию, для которой величина OTHO
шения сиrнал/шум составляет обычно от трех
до двух.
7.3. Использование калибровочной
прямой и cTaHAapTHoro отклонения аналити-
ческоrо сиrнала
Предел обнаружения (ПО) может быть BЫ
раже н как:
по зз. б ,(1)
, S
rдe:
о стандартное отклонение сиrнала;
S TaHreHC уrла наклона калибровочной
прямой.
Значение TaHreHca уrла наклона калибро
вочной прямой вычисляют из калибровочной
прямой для анализируемоro вещества. Оценка
стандартноro отклонения сиrнала может быть
проведена мноrими способами, например, сле
дующими.
Использование стандартН020 отклонения
си2нала для контРОЛЬН020 Dпыrnа. Измеряют
величину аналитическоro сиrнала для необходи
Moro числа образцов, не содержащих анализи
#5.3.2. Валидация аналитических методик и испытаний
901
руемое вещество, и вычисляют стандартное от
клонение.
Использование калибровочной прямоЙ По
лучают калибровочную прямую для образцов с
содержанием анализируемоrо вещества, близ
ким к пределу обнаружения, и вычисляют ее па
раметры. В качестве стандартноro отклонения б
в формуле (1) может быть использовано CTaH
дартное отклонение свободноro члена линейной
зависимости.
7.4. Представление данных
Представляют значение предела обнаруже
ния с указанием способа, использованноro для
еro определения. Если определение предела об
наружения основывается на отношении сиrналl
шум, представляют соответствующие xpOMaTO
rpaMMbI.
Если значение предела обнаружения полу
чено путем вычислений или экстраполяции. еro
оценка должна быть подтверждена анализом
необходимоrо числа образцов с содержанием
анализируемоro вещества, близким к пределу
обнаружения.
8. ПРЕДЕЛ КОЛИЧЕСТВЕННО/О
ОПРЕДЕЛЕНИЯ
Возможно несколько подходов для опреде
ления предела количественноro определения
(ПКО, quaпtitatioп Iimit (QL) или Iimit о' qиапtitа
tioп (LOQ)), зависящих от тоro, является методи
ка инструментальной или неинструментальной.
Moryт быть использованы следующие подходы.
8.1. Визуальная оценка
Визуальную оценку используют как для He
инструментальных, так и для инструментальных
методов.
Предел количественноro определения YCTa
навливают путем анализа образцов с известны
ми концентрациями анализируемоro вещества и
оценкой минимальноro содержания, при котором
анализируемое вещество определяется количе
ственно с требуемой правильностью и точностью.
8.2. Соотношение сиrнал/шум
Этот подход применим только к тем MeTO
дам, для которых наблюдается шум базовой
линии (см. п. 7.2.). Для определения соотноше
ния сиrнал/шум сравнивают величины сиrналов,
полученные для холостоro опыта и для образцов
с низкими концентрациями анализируемоro Be
щества. На основании полученных данных YCTa
навливают минимальную концентрацию, для
которой величина отношения сиrнал/шум co
ставляет около 1 о: 1.
8.3. Использование калибровочной
прямой и cTaHAapTHoro отклонения сиrнала
ПКО ==10. t5 (2)
S
114 З.. 1712
rдe:
б стандартное отклонение сиrнала;
S TaHreHc уrла наклона калибровочной
прямой.
Значение TaHreHca уrла наклона калибро
вочной прямой может быть определено из Ka
либровочной прямой для анализируемоrо веще
ства. Оценка стандартноro отклонения сиrнала
может быть проведена мноrими способами, Ha
пример, следующими.
Использование стандартН020 отклонения
си2нала для контРОЛЬН020 опыта. Измеряют
величину аналитическоro сиrнала для необходи
моro числа образцов, не содержащих анализи
руемое вещество, и вычисляют стандартное OT
клонение.
Использование калибровочной прямой.
Строят калибровочную прямую для образцов с
содержанием анализируемоro вещества, близ
ким к пределу количественноro определения,
и вычисляют ее параметры. В качестве CTaH
дартноrо отклонения в формуле (2) может
быть использовано стандартное отклонение
свободноro члена линейной зависимости.
8.4. Представление данных
Представляют значение предела количе
CТBeHHoro определения с указанием способа, ис
пользованноro для ero определения. Значение
предела количественноro определения должно
быть подтверждено анализом необходимоrо
числа образцов с содержанием анализируемо
ro вещества, близким к пределу количественно
ro определения.
9. РОБАСТНОСТЬ
Оценку робастности проводят на стадии
разработки методики с учетом типа изучаемой
методики. Эта оценка должна доказать надеж
ность результатов анализа при небольших изме
нениях пара метров методики.
Если на результаты анализа влияют усло
вия ero проведения, то эти условия должны быть
стандартизированы и в текст методики вносят
соответствующие предостережения.
Типичные примеры изучаемых параметров:
устойчивость во времени аналитических
растворов;
время экстракции.
В случае жидкостной хроматоrpафии:
рН подвижной фазы;
состав подвижной фазы;
колонки (различные серии и/или постав
щики);
температура;
скорость подвижной фазы.
В случае raзо8ОЙ хроматоrpафии:
колонки (различные серии и/или постав
щики);
температура;
скорость raза носителя.
902
rосударственная фармакопея Республики Беларусь
10. ПРОВЕРКА ПРИiОДНОСТИ
ХРОМАТОiРАФИЧЕСКОЙ СИСТЕМЫ
Проверка приrодности системы является co
ставной частью мноrих аналитических методик.
Этот тест основан на представлении о том, что
оборудование, электроника, аналитические опе
рации и анализируемые образцы составляют
единую систему, которую можно исследовать как
целое. Параметры, вводимые в тест «Проверка
приюдности хроматоrрафической системы», за
висят от используемоro метода анализа и обо
сновываются исследованиями по робастности.
С. ВАЛИДАЦИЯ АНАЛИТИЧЕСКИХ
МЕТОДИК. ОСОБЕННОСТИ
ЕЕ ПРИМЕНЕНИЯ К МЕТОДАМ,
ИСПОЛЬЗУЕМЫМ В ФАРМАКОПЕЕ
1. ОПТИЧЕСКОЕ ВРАЩЕНИЕ (2.2.7)
1.1. Введение
Выбирают растворитель, позволяющий по
лучать максимально возможный уroл вращения.
Исследуют стабильность уrла вращения испы
туемоro раствора в течение не менее 2 ч. При
необходимости указывают, что раствор исполь
зуют свежеприroтовленным. В необходимых слу
чаях указывают время достижения стабильною
значения уrла вращения. Там, rдe возможно, ис
пользуют D линию натрия.
1.2. Идентификация
Если испытуемое вещество представляет
собой энантиомер, то для идентификации ис
пользуют удельный показатель оптическоro Bpa
щения.
Если удельный показатель оптическоro Bpa
щения используют только для целей идентифи
кации, то еro значение можно не пересчитывать
на сухое вещество. Реrламентируемые пределы
величины удельноro показателя должны учиты
вать допустимые пределы количественноro co
держания анализируемоrо вещества и чистоту
образцов различноrо происхождения, которые
выдерживают требования соответствующей MO
ноrрафии.
Если удельный показатель оптическо
ro вращения используется также и для KOHTpO
ля чистоты энантиомеров, то испытание разде
ла «Идентификация» может содержать ссылку:
выдерживает требования испытания «Удельное
оптическое вращение».
1.3. Испытания
Удельный показатель оптическоrо вращения
(в отдельных случаях уюл вращения) может
быть использован для подтверждения оптиче
ской чистоты энантиомера. Этот метод менее
чувствителен, чем метод жидкостной xpOMaTO
rрафии (ЖХ). В случае, Korдa измерение удель
ноro оптическою вращения предназначено для
тoro, чтобы нормировать содержание одноro из
энантиомеров, необходимо показать, что в yc
лов иях методики анализируемый энантиомер
имеет достаточную величину оптическоro Bpa
щения, чтобы быть обнаруженным. Результат
определения представляют в пересчете на сухое
вещество. Там, rдe это возможно, представля
ют данные о влиянии потенциальных примесей.
Пределы удельноro показателя оптическоro Bpa
щения устанавливают с учетом возможноro co
держания примесей. При отсутствии информа
ции об оптическом вращении примесей обычно
устанавливают пределы отклонения :1:5 % от
среднею значения, полученноro на образцах,
отвечающих всем остальным требованиям част
ной статьи. Там, rдe это возможно, исследуют
образцы различноrо происхождения. Полезно
также исследовать образцы со сроками roдно
сти, близкими к предельному.
Измерение оптическоrо вращения может
использоваться для подтверждения тоro, что
субстанция является рацематом. В таких слу
чаях обычно устанавливают пределы от 0,100
до +0,100.
1.4. Количественное определение
Оптическое вращение может использо
ваться для количественноrо определения суб
станции. При этом необходимо использовать
стандартный образец с известной оптической
чистотой.
2. АБСОРБЦИОННАЯ
СПЕКТРОФОТОМЕТРИЯ
В УЛЬТРАФИОЛЕТОВОЙ И ВИДИМОЙ
ОБЛАСТЯХ СПЕКТРА (2.2.25)
Должна быть доказана приroдность выбран
ных условий определения, таких как использу
емые растворители и их качество, значение рН
раствора и т. д.
Обычно ультрафиолетовая спектрофотоме
трия имеет оrраниченную специфичность, KOTO
рую можно повысить использованием первой и
второй производных спектра.
2.1. Идентификация
Ультрафиолетовая спектрофотометрия
сама по себе редко используется для идентифи
кации. Korдa этот метод включают в набор испы
таний для идентификации, необходимо изучить
еro специфичность путем сравнения спектров
анализируемою вещества со спектрами подоб
ных соединений. Специфичность метода можно
повысить, если использовать не абсолютные
значения оптических плотностей, а спектраль
ные отношения.
2.2. Испытания на предельное содержа-
ние примесей
Если ультрафиолетовая спектрофотоме
трия используется в испытаниях на допустимые
пределы содержания примесей, следует пока
зать, что анализируемые примеси дают ДOCTa
#5.3.2. Валидация аналитических методик и испытаний
903
точный вклад в измеряемую оптическую плот
ность. При выбранной длине волны должна быть
установлена оптическая плотность, COOTBeTCTBY
ющая нормируемой концентрации анализиру
емой примеси.
2.3. Количественное определение
Если ультрафиолетовая спектрофотоме
трия используется для количественноro опреде
ления, то следует оценить влияние примесей на
светопоrлощение. При количественном опреде
лени и не рекомендуется использовать удельный
показатель поrлощения. Если удельный показа
тель поrлощения все же при меняется, то еro зна
чение следует устанавливать на основании меж
лабораторноrо исследования, используя серии
с известной чистотой. Чистота этих образцов
должна оцениваться с использованием различ
ных методов, включающих как методы разделе
ния, так и абсолютные методы (не требующие
использования стандартноro образца).
3. НЕИНСТРУМЕНТАЛЬНblЕ ИСПblТАНИЯ
НА ЧИСТОТУ И ПРЕДЕЛЬНОЕ
СОДЕРЖАНИЕ ПРИМЕСЕЙ
3.1. Внешний вид раствора (прозрач-
ность (2.2.1) И цветность (2.2.2))
Это визуальные испытания, предназначен
ные для оценки общей чистоты субстанции и
основанные на сравнении окраски (или опалес
ценции) испытуемоro раствора и серии этало
нов. Часто неизвестно, какие примеси и в какой
концентрации обуславливают окраску или опа
лесценцию. В этом случае валидация OCHOBЫBa
ется на сопоставлении данных, полученных для
разных серий, представленных производителем
(или производителями). Если примеси извест
ны и доступны, валидацию этоro метода прово
дят путем сравнения с более совершенным Me
тодом.
3.2. Кислотность или щелочность
Это неспецифичное испытание является
одной из характеристик чистоты образца и ис
пользуется для контроля протеолитических при
месей.
Для контроля протеолитических примесей в
субстанциях используют два испытания:
полуколичественное титрование с исполь
зованием индикатора или потенциометрическо
ro определения точки титрования испытание
«Кислотность или щелочность»;
измерение значения рН.
Если вещество имеет буферную емкость,
преимущественным является измерение значе
ния рН. В обратном случае рекомендуется ти
три метрическая методика. Испытание «Кислот
ность или щелочность» применяют в случае,
если испытуемая субстанция не rидролизуется
или нерастворима в воде.
Вопрос выбора испытания «Кислотность
или щелочность» или «рН» при разработке HOp
мативной документации или частной статьи на
субстанцию может быть определен на подступе
оценки буферных емкостей самой субстанции.
Для оценки буферных емкостей субстанции
строят кривую потенциометрическоro титрова
ния водноro раствора (или, в случае нераствори
мых в воде веществ, экстракта (водной вытяжки))
необходимой концентрации (от 10 r/л до 50 r/л),
используя в качестве титранта 0,01 М раствор
кислоты хлористоводородной или 0,01 М pacт
вор натрия 2идро/(сида соответственно. Точка
изrиба на кривой титрования является HaCTO
ящим рН раствора и для чистой субстанции Ha
ХОДИТСЯ на изrибе с пределом рН. Ступенью бу
ферной емкости испытуемоro раствора является
величина суммарноro сдвиrа рН (8рН), рассчи
танная исходя из кривой титрования в результа
те прибавления к 1 О мл испытуемоro раствора
0,25 мл 0,01 М раствора натрия 2идроксида, а
затем к друrим 1 О мл этоro же раствора 0,25 мл
0,01 М раствора кислоты хлористоводород
ной. Чем больше величина 8рН, тем меньше бу
ферная емкость раствора.
Величина 8рН испытуемоro раствора опре
деляет выбор метода для реrламентации прото
литических примесей в соответствии с приведен
ной схемой (таблица 5.5.3. 2). Классификация
Классификация субстанций в соответствии с величиной f1pH
Таблица 5.3.З. 2
Класс pH Название предполаrаемоrо испытания
буферности
Класс А 8рН>4 Испытание «Кислотность или щелочность» с использованием двух COOT
ветствующих индикаторов
Класс В 2<8рН<4 Испытание «Кислотность или щелочность» с использованием одноro co
ответствующеro индикатора
Класс С 0,2<8рН<2 Прямое измерение рН
Класс D 8рН<0,2 Протолитические при меси невозможно удовлетворительно контролиро
вать. К таким субстанциям относят вещества, которые являются солями
и состоят из ионов с более чем одной кислотной и/или основной функци
ональными rруппами. Для них измерение рН может способствовать обе
спечению обозначенноro хранения субстанции, если пределы рН являют
ся достаточно узкими
904
rосударственная фармакопея Республики Беларусь
субстанций базируется на том, что для большин
ства индикаторов переход окраски происходит в
пределах 2 единиц рН.
Путем изменения концентрации испытуемо
ro раствора можно изменить класс буферности,
к которому относится испытуемая субстанция, в
соответствии со схемой, приведенной в табли
це 5.5.3. 2. При этом будет изменяться и форма
кривой титрования. Если возможно, не следует
выходить за пределы обозначенных выше KOH
центраций. Однако, если вещество очень мало
растворимо в воде, можно использовать раствор
с концентрацией меньше 1 0 50 r/л.
В некоторых случаях испытание «Кислот
ность или щелочность» нельзя провести при
помощи индикатора или из за окрашивания
самоro индикатора. или из за разложения самоro
вещества. В таком случае испытание проводят
при помощи потенциометрическоrо титрования.
Если прибавление кислоты или щелочи вызы
вает равновесие молекулы субстанции или BЫ
падения осадка, следует, не обращая внимания
на буферную емкость, отказаться от проведения
испытания «Кислотность/щелочность» на пред
мет измерения рН.
Растворы roтовят с использованием воды,
свободной от У2лерода диоксида, Р.
3.3. Испытания на предельное содержа
ние анионов и катионов (2.4)
Приroдность этих испытаний следует ДOKa
зать путем использования метода добавок иlили
сравнения с друrими, более совершенными Me
тодами.
Сульфатная зола (2.4.14). Это испытание
предназначено для определения суммы кати
онов металлов, присутствующих в орrанических
субстанциях. но не подходит для неорrаниче
ских солей орrанических соединений. Обычно
предел не должен превышать 0,1 %. Этот метод
не требует валидации.
Тяжелые металлы (2.4.8). Применяют раз
личные способы проведения этою испытания.
Обычно предел содержания тяжелых метал
лов составляет 0,001 % (10 ррт) или 0,002 %
(20 ррт), но иноrда и 0,0005 % (5 ррm), что Haxo
дится вблизи предела обнаружения.
Для валидации испытания на тяжелые Me
таллы анализируют испытуемый образец и об
разец, специально заrрязненный свинцом в
соответствующей концентрации. Окраска испы
туемоro образца должна быть меньше, а заrряз
ненноro образца равной или большей, чем
окраска эталона.
Для ряда методик, требующих сжиrания об
разца, существует опасность потерь некоторых
тяжелых металлов (таких как, например, ртуть
и свинец в присутствии хлоридов). В таких слу
чаях, Korдa это возможно, контролируют co
держание тяжелых металлов методом aTOM
но абсорбционной спектрометрии или друrим
инструментальным методом.
Если известно, что при синтезе субстанции
используется катализатор, например, палла
ДИЙ, никель или родий, то их содержание целе
сообразно контролировать колориметрическими
или инструментальными методами (например,
атомно абсорбционной спектрометрии и др.).
Цветные реакции или реакции осаждения.
Для отдельных катионов и анионов описаны пре
дельные испытания, основанные на визуальном
сравнении окраски или опалесценции. При этом
необходимо доказать, что:
окраска или опалесценция для нормиру
емых концентраций отчетливо видна;
найденная концентрация добавленноro
иона одинакова как для испытуемоro раствора,
так и для раствора сравнения (как визуально,
так и с помощью методов, основанных на изме
рении поrлощения);
значения оптической плотности paCTBO
ров, содержащих 50 %, 100 % и 150 % анализи
руемой примеси от нормируемой концентрации,
должны значимо различаться;
определение примеси на уровне норми
руемой концентрации проводят не менее шести
раз и вычисляют стандартное отклонение. Най
денная концентрация должна составлять не
менее 80 % от введенной, а относительное CTaH
дартное отклонение не более 20 %.
Целесообразно провести сравнение резуль
татов предельноrо испытания с результатами
количественноrо определения с использовани
ем независимоro метода, например, атомно аб
сорбционной спектрометрии для катионов или
ионной хроматоrрафии для анионов. Результа
ты, полученные двумя методами, должны быть
близки.
4. АТОМНО АБСОРБЦИОННАЯ
СПЕКТРОМЕТРИЯ (2.2.23)
Метод атомно абсорбционной спектроме
трии (МС) применяют для испытаний по опре
делению содержания отдельных элементов,
присутствующих в образце.
4.1. Специфичность
Специфичность данноro метода определяет
ся тем, что атомы анализируемоro элемента по
rлощают характеристическое излучение от ис
точника со строro дискретными длинами волн,
соответствующими данному элементу. Однако
возможны помехи вследствие как оптических, так
и химических эффектов. Перед началом валида
ции необходимо выявить помехи такоro рода и,
если возможно, снизить их влияние путем ис
пользования соответствующих приемов.
Эти помехи MOryT привести к систематиче
ской поrрешности при использовании метода
прямой калибровки или к изменению чувстви
тельности метода. Основным источником по
rрешности в методе МС являются поrрешности,
связанные с процессом калибровки и меша
ющим влиянием матрицы.
#5.3.2. Валидация аналитических методик и испытаний
905
4.2. rрадуировка
Использование линейной модели rрадуи
ровки описано в общей статье 2.2.23. AтOM
но абсорбционная спектрометрия. Для
доказательства применимости линейной perpec
сионной модели рекомендуется использовать не
менее пяти rрадуировочных растворов. В HeKO
торых случаях возможно использование парабо
лической модели rрадуировки. При этом также
используют не менее пяти rрадуировочных pac
творов. Рекомендуется использовать KOHцeHTpa
ции rрадуировочных растворов с равномерным
распределением внутри диапазона применения.
Для каждой концентрации рекомендуется
выполнять не менее пяти измерений.
Проблемы с калибровкой часто MOryT быть
выявлены визуально. Однако rрадуировочные
rрафики сами по себе нельзя использовать как
доказательство приroдности метода калибровки.
Калибровочный rрафик представляют в следу
ющем виде:
а) на rрафике откладывают измеренные оп
тические плотности как функции концентраций и
строят кривую, описывающую эту rрадуировоч
ную функцию вместе с ее доверительными ин
тервалами; экспериментальные точки должны
находиться в пределах доверительноro интерва
ла построенноro rрафика;
Ь) на rрафике откладывают остаточные OT
клонения (разности между измеренными и BЫ
численными по rрадуировочному rрафику
оптическими плотностями) как функции KOHцeH
трации; эти разности должны распределяться
BOKpyr оси абсцисс случайным образом.
В некоторых случаях разброс значений сиr
нала возрастает с ростом концентрации, что
может быть выявлено из rрафика остаточных OT
клонений или статистическими методами. При
этом наибольшая точность может быть достиr
нута при использовании rрадуировки с BeCOBЫ
ми множителями. Может быть применена как ли
нейная, так и квадратичная весовая функция.
В случае использования весовой модели
строится rрафик взвешенных разностей (т. е.
разностей, умноженных на веса) как функции
концентрации следующим образом:
а) на rрафике откладывают измеренные оп
тические плотности как взвешенные функции
концентраций и строят кривую, описывающую
эту rрадуировочную функцию вместе с ее ДOBe
рительными интервалами;
Ь) на rрафике откладывают взвешенные
остаточные отклонения (т. е. взвешенные раз
ности между измеренными и вычисленными по
rрадуировочному rрафику оптическими плотно
стями) как функции концентрацl.1И.
Необходимо показать, что модель достаточ
но точно описывает экспериментальные данные.
4.3. Эффекты матрицы
Если для получения rрадуировочной функ
ции используют метод rрадуировочноro rрафи
1153_"1712
ка, то необходимо по казать , что чувствитель
ность для раствора анализируемоro образца и
rрадуировочных растворов одинакова.
Если при меняется rрадуировка в виде
прямой линии, различия в чувствительности
MOryT быть обнаружены путем сравнения накло
нов rрадуировочных rрафиков, полученных с ис
пользованием эталонных растворов и растворов,
полученных внесением стандартной добавки к
испытуемому раствору. Точность оценки накло
нов обеих прямых зависит от числа и распреде
ления точек измерения. Поэтому для построения
обеих реrрессионных линий рекомендуется ис
пользовать достаточное число точек (не менее
пяти) и выбирать концентрации преимуществен
но на rраницах диапазона rрадуировки.
Обоснованием для возможности использо
вания метода rрадуировочноro rрафика являет
ся незначимость различий наклонов полученных
прямых по критерию Стьюдента. Если различия
значимы, то используют метод стандартных дo
бавок.
4.4. Предел обнаружения и предел коли
чественноrо определения (метод, основан-
ный на стандартном отклонении контрольно
ro опыта см. раздел В, пп. 7.3.1 и 8.3.1)
Выполняют контрольные опыты, дЛЯ KOТO
рых предпочтительно использовать растворы
«плацебо», содержащие все компоненты образ
ца, за исключением определяемоro. Если такие
контрольные опыты выполнить невозможно, то
допустимо использовать холостые растворы, co
держащие все peareHTbI и приroтовленные так
же, как и испытуемый раствор.
5. ХРОМАТО,РАФИЧЕСКИЕ МЕТОДЫ
Различные хроматоrрафические методы
MOryT использоваться для идентификации, KOH
троля примесей и количественноrо определе
ния. Ниже описаны особенности валидации
данных методов, при этом необходимо учиты
вать требования статьи 2.2.46. Хромат02рафи
ческие методы разделения.
5.1. Тонкослойная хроматоrрафия (2.2.27)
Специфичность. Для тестов идентифика
ции обычно нельзя добиться специфичности,
используя только тонкослойную хроматоrpaфию
(ТСХ) саму по себе, однако достаточная избира
тельность может быть достиrнута при сочетании
ТСХ с друrими методами. Если для испытания
на допустимые пределы примесей избиратель
ность недостаточна, то используют дополнитель
ное испытание (испытания) для контроля приме
си (примесей), зона которой не была отделена
от друrих зон. Необходимо доказать избиратель
ность совокупности используемых методик. Для
испытаний идентификации улучшение избира
тельности может быть достиrнуто при использо
вании опрыскивания реактивом, который позво
ляет различать близкие соединения по цвету.
906
rосударственная фармакопея Республики Беларусь
Неподвижная фаза. Необходимо показать,
что данное испытание приroдно для проведения
анализа на пластинках одноro типа, но различ
ноro происхождения.
Тест «Проверка при20дности xpOMaт02pa
фической системы». Такой тест обычно про
водится для подтверждения разделения двух
близко элюирующихся соединений, одним из KO
торых является анализируемое вещество (см.
статью 2.2.46. Хромат02рафические методы
разделения). Необходимо доказать, что разде
ление выбранных соединений rарантирует при
roдность системы для достижения поставлен
ных целей.
Для испытаний на предельное содержание
примесей необходимо учитывать следующее.
Обнаружение. Необходимо избеrать ис
пользования особых опрыскивающих peareHToB,
если в методике не используется стандарт HOp
мируемой примеси.
Предел обнаружения. Если используют KO
личественную инструментальную методик для
определения предела обнаружения используют
подходы, описанные в пункте 7 раздела В. Если
используют визуальную методику, то необходи
мо показать, что обнаруживается количество,
соответствующее указываемому пределу обна
ружения.
Коэффициент пересчета. Если приме
си определяемы, то необходимо по казать, что
чувствительности определения примеси и oc
новноro вещества близки. Для испытаний на
допустимые пределы примесей различия в чув
ствительностях MOryT быть показаны путем
сравнения пределов обнаружения.
Предел количествеНН020 определения, ли
нейность, диапазон и сходимость. Эти данные
необходимо представлять при использовании
инструментальной количественной ТСХ.
5.2. Жидкостная хроматоrрафия (2.2.29)
5.2.1. Идентификация
Специфичность. Для тестов идентифика
ции обычно нельзя добиться специфичности,
используя только жидкостную хроматоrрафию
саму по себе, однако достаточная избиратель
ность может быть достиrнута при сочетании жид
костной хроматоrрафии с друrими методами.
Необходимо предоставлять данные о времени
удерживания, относительном времени удержи
вания или коэффициенте емкости дЛЯ OCHOBHO
ю вещества и близких по строению веществ для
нескольких стационарных фаз одною типа.
5.2.2. Испытания на предельное содержа
ние примесей
Специфичность. Избирательность разде
ления. Должно быть показано разделение из
вестных и возможных примесей с основным
веществом и, если возможно, между собой.
Специфичность может быть подтверждена
при использовании для детектирования Macc
спектрометра. Примеси, которые не отделяются
от основноro вещества, должны контролировать
ся друrим методом. Необходимо предоставлять
данные о времени удерживания, относительном
времени удерживания или коэффициенте eM
кости для основноro вещества и примесей. Эти
данные должны предоставляться для несколь
ких неподвижных фаз одноro типа.
Избирательностьдетектирующейсистемы.
Выбор детектора и условий детектирования
должен быть обоснован. Специфичность может
быть подтверждена, например, при использова
нии для детектирования масс спектрометра.
Коэффициент пересчета. Если примеси
определяемы, то необходимо по казать, что чув
ствительности определения примеси и OCHOB
ноro вещества близки (при использовании Уф
детектирования при выбранной длине волны
детектирования; это необходимо показать и для
друrих типов детекторов, например, рефракто
метрическою или кондуктометрическоro). Если
отношение чувствительностей известной при
меси и основною вещества выходит за пределы
0,8 1,2 и если допустимый предел содержания
этой примеси больше 0,1 %, то необходимо ис
пользовать коэффициент пересчета либо CTaH
дарт нормируемой примеси в варианте внешне
ro стандарта.
Предел обнаружения или количественно
20 определения. Эти пределы должны опре
деляться для метода внешнеro стандарта при
использовании разведен ий испытуемой субстан
ции или при использовании стандарта при меси.
Если пик при меси выходит внепосредственной
близости от пика субстанции (особенно если He
посредственно за ним), то предел обнаружения
или количественноro определения должен YCTa
навливаться по этой примеси. Метод, описанный
в пункте 7 раздела В, приroден для вычисления
обоих указанных пределов.
Стабильность. Необходимо представлять
данные, подтверждающие срок roдности испы
туемоro раствора и раствора сравнения. Также
должны представляться данные о стабильности
подвижной фазы.
Степень извлечения. При использовании
экстракции необходимо изучить степень извле
чения известных и доступных примесей при оп
тимальных условиях. Необходимо представить
данные, подтверждающие, что экстракция обе
спечивает достаточную точность.
Получение производных. Если использу
ют пред или постколоночное получение произ
водных, необходимо установить оптимальные
условия реакции (время, температура и др.) и
исследовать стабильность полученных произво
дных.
Тест «Проверка при20дности xpOMaтo
2рафической системы». Как описано для ТСХ.
Использование соотношения сиrнал/шум Tpe
буется только Torдa, Korдa предел определения
и нормируемый предел содержания примеси
близки.
#5.3.2. Валидация аналитических методик и испытаний
907
5.2.3. Количественное определение
Специфичность. Это желательное, но не
основное требование. Если метод не специфи
чен, то возможность еro использования обеспе
чивается низким уровнем содержания примесей,
которые контролируются друrим испытанием.
Тест «Проверка при20дности xpOMaт02pa
фической системы». Как описано для тех.
5.3. rазовая хроматоrрафия (2.2.28)
5.3.1. Идентификация
Специфичность. Как описано для жидкост
ной хроматоrрафии.
5.3.2. Испытания на предельное содержа
ние примесей
Специфичность. Как описано для жидкост
ной хроматоrрафии.
Коэффициент пересчета. Как описано для
жидкостной хроматоrpафии. Должны представлять
ся коэффициенты пересчета нормируемой прим
си относительно основноro вещества. Это особенно
важно при использовании селективных детекторов.
таких как детектор электронноro захвата и т. п.
Пределы обнаружения и количествеНН020
определения. Как описано для жидкостной xpo
матоrрафии.
Стабильность. Как описано для жидкост
ной хроматоrрафии.
Получение производных. Как описано для
жидкостной хроматоrрафии.
Внутренний стандарт. Необходимо пока
зать, что при выбранных условиях пик BHYTpeH
неro стандарта не перекрывается с пиками воз
можных примесей или основноro вещества.
Степень извлечения. Как описано для жид
костной хроматоrрафии.
Тест «Проверка при20дности xpOMaт02pa
фической системы». Ниже приводятся HeKOTO
рые особенности, которые необходимо учиты
вать для данноro теста (см. также статью 2.2.46.
Хромат02рафические методы разделения):
а) отношение си2нал/шум обычно опреде
ляют для сиrналов, равных или несколько боль
ших, чем предел обнаружения и предел количе
ственноro определения;
Ь) разрешение определяют для пика aHa
лизируемоro вещества и ближайшеro пика при
меси или для пика анализируемоro вещества и
пика внутреннеro стандарта.
с) если фактор асимметрии отличается от
принятоro диапазона (0,8 1,2), целесообразно
нормировать еro пределы (2.2.28); это особенно
важно для тех случаев, Korдa используются Ha
бивные колонки или пик нормируемой примеси
элюируется непосредственно за пиком основною
вещества; там, rдe ВОЗМОЖНО, подтверждают BЫ
полнение испытания с использованием колонок
подобноro типа.
Метод анализа равновесной паровой фазы.
Этот метод применяют для анализа леrколетучих
веществ. Необходимо показать, что выбранные
температура и время предварительноrо HarpeBa
сосудов с анализируемым образцом обеспечива
ют установление равновесия. Валидация метода
заключается в проведении повторных анализов об
разца. При этом после каждоro введения пробы ra
зовая фаза в сосуде замещается и сосуды снова
уравновешиваются перед новым вводом raзовой
фазы. По полученным данным строят rрафик зави
симости лоrарифмов площадей пиков анализиру
eMoro вещества от числа экстракций и рассчиты
вают ero характеристики. Близость коэффициента
корреляции полученной зависимости к 1,0 свиде
тельствует о правильности выбора условий испыта
ний. Эффекты влияния матрицы можно устранить
при использовании метода стандартных добавок.
5.3.3. Количественное определение
Специфичность. Как описано для жидкост
ной хроматоrрафии.
Тест «Проверка при20дности системы».
Как описано для жидкостной хроматоrрафии.
6. ОПРЕДЕЛЕНИЕ ВОДЬ!
ПОЛУМИКРОМЕТОДОМ (2.5.12)
Для подтверждения корректности использо
вания йодсернистых реактивов, которые отлича
ются по составу от йодсернист020 реактива Р
(например, реактив Карла Фишера), следу
ет провести валидацию одним из приведенных
ниже методов.
6.1. Метод добавок
Определяют содержание воды (т н о' Mr) в
2
субстанции в соответствии с методикой, указан
ной в частной статье. Определение проводят не
менее пяти раз. Затем к испытуемому образцу
прибавляют, избеrая попадания атмосферной
влаrи, подходящий объем стандартизированно
ro раствора воды в метаноле Р и определяют
содержание воды (m i , Mr). Определение прово
дят не менее пяти раз для разных объемов CTaH
дартизированноrо раствора в принятом диапа
зоне употребления методики.
Методом наименьших квадратов рассчи
тывают параметры линейной зависимости най
денноro содержания воды от количества при
бавленной воды: TaHreHc уrла наклона (Ь), точку
пересечения с осью ординат (а) и точку пере
сечения экстраполированной калибровочной
прямой с осью абсцисс (d).
Значение TaHreHca уrла наклона Ь должно
находиться в пределах от 0,975 до 1,025 (:t2,5 %).
Относительные отклонения е 1 и е 2 , в процентах,
рассчитывают по формулам:
a т
е 1 н,о .100 %,
тн,о
d т
e1 o .1000/0,
тн,о
е 1 и е 2 не MOryT превышать :t2,5 %.
Для каждоro из определений рассчиты
вают найденное количество в процентах от
908
lосударственная фармакопея Республики Беларусь
прибавленноrо количества. Среднее значе
ние пяти определений должно быть от 97,5 %
до 102,5 %.
6.2. Сравнение с арбитражным методом
Определяют содержание воды полумикро
методом и друrим подходящим валидированным
методом, например, методом rазовой xpOMaTO
rрафии (2.2.28), методом термоrравиметриче
скоro анализа (2.2.34) и др.
Средние значения. полученные полумикро
методом и арбитражным методом, не должны
отличаться статистически значимо.
01/2013:50400
5.4. ОСТАТОЧНЫЕ
КОЛИЧЕСТВА
оРrАНИЧЕСКИХ
РАСТВОРИТЕЛЕЙ
РЕrЛАМЕНТАЦИЯ СОДЕРЖАНИЯ
ОСТАТОЧНЫХ РАСТВОРИТЕЛЕЙ
В СУБСТАНЦИЯХ, вспомоrАТЕЛЬНЫХ
ВЕЩЕСТВАХ И rOTOBbIX
ЛЕКАРСТВЕННЫХ СРЕДСТВАХ
На Международной конференции по rapMo
низации технических требований для реrистра
ции лекарственных средств для использования
человеком (/СН) было принято Руководство по
примесям остаточных растворителей, в KOTO
ром указаны конкретные предельные содержа
ния растворителей. которые MorYT оставаться
в фармацевтических субстанциях, вспомоrа
тельных веществах и roтовых лекарственных
средствах после обработки. Данное PYKOBOД
ство, текст KOToporo приведен ниже, не распро
страняется на продукты, уже присутствующие
на рынке. Фармакопея использует те же самые
принципы к существующим фармацевтическим
субстанциям, вспомоrательным веществам и
roтовым лекарственным средствам вне зави
симости от тоro, являются они объектом Фар
макопеи или нет. Все субстанции или roтовые
лекарственные средства должны быть прове
рены на содержание возможных остаточных
растворителей в них.
Если используемые пределы совпадают
с приведенными ниже, испытание на coдep
жание конкретных остаточных растворителей
обычно не указывается в частной статье, так
как используемые растворители MorYT отли
чаться в зависимости от производителя, а Tpe
бования данной общей статьи применяются
посредством общей статьи «Субстанции для
фармацевтичеСК020 использования». Уполно
моченный opraH должен быть информирован
об используемых в процессе производства pac
творителях. Эта информация также предостав
ляется в реrистрационном досье и указывается
в сертификате.
Если используются растворители только
класса 3, то для контроля их содержания может
быть использовано либо испытание «Потеря в
массе при высушивании», либо специфичное
испытание на содержание конкретноro paCTBO
рителя. Если для растворителя класса 3 обо
сновано и разрешено предельное содержание
более 0,5 %, то специфичное испытание на co
держание растворителя является обязатель
ным.
Для контроля остаточных растворителей
класса 1 или 2 (или класса 3 с содержанием
более 0,5 %) следует использовать, по возмож
ности, методолоrию, описанную в общей статье
2.4.24. В противном случае следует применять
подходящую валидированную методику.
Если проводят количественное определе
ние содержания остаточных растворителей,
полученный результат учитывают при расчете
количественноro содержания субстанции, за
исключением случаев, Korдa проводят опреде
ление потери в массе при высушивании.
1. ВВЕДЕНИЕ
Целью данноrо руководства является peKO
мендация приемлемых для безопасности боль
ноro количеств остаточных растворителей в
лекарственных средствах. В руководстве pe
комендуется использование менее токсичных
растворителей и описываются токсиколоrически
обоснованные предельные содержания HeKOТO
рых из них.
Остаточные растворители в лекарствен
ных средствах определяются соrласно дaHHO
му руководству как летучие орrанические Be
щества, используемые или образующиеся при
производстве субстанций, вспомоrательных
веществ или rOToBbIx лекарственных средств.
Эти растворители полностью не удаляются
в при меняемом технолоrическом процессе.
Выбор подходящеro растворителя для синте
за субстанции может увеличить ее выход или
обусловить такие характеристики, как кристал
лическая форма, чистота и растворимость.
Таким образом, растворитель иноrда может
быть критическим параметром в процессе син
теза. Данное руководство не распространяет
ся на растворители, используемые в качестве
вспомоrательных веществ или относящиеся по
составу к сольватам. Однако содержание paCT
ворителей в таких препаратах должно обосно
вываться и определяться.
Поскольку остаточные растворители не
имеют терапевтическоro действия, aдeKBaT
ноro действию лекарственноro вещества, они
должны удаляться до такой степени, чтобы
удовлетворять требованиям спецификации,
5.4. Остаточные количества ореанических растворителей
909
надлежащей производственной практики (GMP)
или друrим требованиям к качеству. Лекарствен
ные средства не должны содержать остаточные
растворители выше нормы, установленной дaH
ными по безопасности. При производстве суб
станций, вспомоrательных веществ и roтовых
лекарственных средств следует избеrать ис
пользования растворителей, имеющих высокую
токсичность (класс 1, таблица 1), если их приме
нение не может быть достаточно обоснованным
с точки зрения оuенки «риск польза». Содержа
ние некоторых растворителей, имеющих MeHb
шую токсичность (класс 2, таблица 2), должно
быть оrраничено, чтобы исключить потенциаль
ный побочный эффект. В идеале на практике
должны использоваться менее токсичные pac
творители (класс 3, таблица 3). Полный список
растворителей, включенных в данное PYKOBOД
ство. представлен в Приложении 1.
Данный перечень не является исчерпыва
ющим, он может дополняться друrими исполь
зуемыми растворителями. Рекомендуемые пре
дельные содержания остаточных растворителей
классов 1 и 2, а также классификация paCT
ворителей MOryT изменяться по мере появле
ния новых данных относительно их безопасно
сти. Обоснование безопасности применения на
рынке новоro лекарственноro средства, coдep
жащеro новый растворитель, отсутствующий в
перечне растворителей (Приложение 1), может
строиться на концепциях данноro руководства.
2. ОБЛАСТЬ ПРИМЕНЕНИЯ
Область применения руководства OCTa
точные растворители в фармацевтических
субстанциях, вспомоrательных веществах и
лекарственных средствах. Определение OCTa
точных растворителей необходимо проводить,
если известно, что процесс производства или
очистки при водит К их появлению. Следу
ет определять те растворители, которые ис
пользуются или производятся В процессе из
rотовления или очистки фармацевтических
субстанций, вспомоrательных веществ или ле
карственных средств. Для определения coдep
жания остаточных растворителей возможно как
проведение испытания лекарственноro cpeд
ства, так и использование совокупноro метода
расчета, исходя из информации об их coдep
жании во входящих инrредиентах, использо
ванных в процессе производства лекарствен
ноro средства. Если на основании результатов
вычисления концентрация остаточных paCTBO
рителей не превышает предела, peKOMeHДY
емоro в настоящей статье, нет необходимости
проводить испытаl-lИЯ roTOBOro лекарственноro
средства на содержание остаточных раствори
телей. Однако, если расчетная концентрация
выше рекомендуемоro предела, лекарствен
ное средство должно быть проверено, чтобы
установить, способствует ли процесс изroтов
ления уменьшению количества данноro pac
116.3а.1712.
творителя до приемлемоro уровня. Испытание
roтовоro лекарственноro средства также необ
ходимо проводить, если растворитель исполь
зуется в процессе ero производства.
Данная статья не относится ни к потенци
ально новым фармацевтическим субстанциям,
вспомоrательным веществам и roтовым лекар
ственным средствам, находящимся на стадии
клинических испытаний, ни к зареrистрирован
ным лекарственным средствам.
Статья применима ко всем дозированным
формам и способам применения. В некоторых
случаях, таких как краткосрочное (30 дней или
меньше) или местное применение, MOryT быть
приемлемы более высокие уровни остаточных
растворителей. Оценка таких уровней должна
проводиться в каждом конкретном случае.
Дополнительную информацию по остаточ
ным растворителям см. в Приложении 2.
3. ОБЩИЕ ПОЛОЖЕНИЯ
3.1. КЛАССИФИКАЦИЯ ОСТАТОЧНЫХ
РАСТВОРИТЕЛЕЙ ПО СТЕПЕНИ РИСКА
Международная проrрамма по химической
безопасности (IPCS, lпtematioпal Program оп
Chemical Safety) для описания пределов воздей
ствия токсических химических peareHТOB использу
ет термин «максимально допустимое ежедневное
потребление» (ТО/, to/erabIe daily iпtаkе, или ДЕП),
а Всемирная орrанизация здравоохранения (ВОЗ)
и друrие национальные и международные opraни
зации здравоохранения и институты используют
термин «приемлемое ежедневное потребление»
(ADI, acceptabIe dai/y iпtаkе, или ПЕП). Во из
жание путаницы с ПЕП некоторых субстанций,
в данном руководстве определен новый термин
«допустимая (разрешенная) суточная доза» (РОЕ
perтitted dai/y exposure, или ДСД) это макси
мально приемлемое суточное потребление OCTa
точноro растворителя в лекарственном средстве.
Остаточные растворители, которым дает
оценку данное руководство, перечислены в При
ложении 1 под общими названиями и CTPYКТYP
ными формулами. Они оценивались по степени
возможноro риска для здоровья человека и раз
делены на 3 класса:
Класс 1: Растворители, использования KO
торых нужно избе2ать
Известные канцероrены для человека; пред
полаrаемые канцероrены для человека; paCTBO
рители. опасные для окружающей среды.
Класс 2: Растворители, использование KO
торых нужно 02раничивать
Неrенотоксичные канцероrены для живот
ных или растворители, которые MOryт вызвать
друrие необратимые эффекты, такие как нейро
токсичность или TepaToreHHocTb.
Растворители с предполаrаемой друroй cy
щественной, но обратимой токсичностью.
910
rосударственная фармакопея Республики Беларусь
Класс 3: Малотоксичные растворители
Растворители с низким потенциалом токсич
ности для человека; для них не требуется YCTaHaB
ливать предельное содержание, обусловленное
информацией о риске для здоровья человека. Pac
творители класса 3 имеют ДСД от 50 Mr/cyт и выше.
3.2. МЕТОДЫ УСТАНОВЛЕНИЯ
ПРЕДЕЛЬНО,О СОДЕРЖАНИЯ
В Приложении 3 представлен метод для
установления предельной суточной дозы OCTa
точных растворителей.
3.3. ПОДХОДЫ УСТАНОВЛЕНИЯ
ПРЕДЕЛЬНО,О СОДЕРЖАНИЯ
РАСТВОРИТЕЛЕЙ КЛАССА 2
Для устанuвления преДБJ1ЬНОro содержания
растворителей класса 2 MOryт использоваться два
подхода.
Подход 1: Иt;ПользУюТ Iтредельные KOHцeH
трации в ррт из таблицы 2, которые были pac
считаны с помощью уравнения (1) исходя из
предположения, что суточное потребление ле
карственноro средства составляет 1 О r.
1000.ДСД
Концентрация (ррт)=- , (1)
доза
rде дед выражается в Mr/cyт, а доза в r/cyт.
Эти предельные содержания остаточных
растворителей рассматриваются как приемле
мые для всех субстанций, вспомоrательных Be
ществ и roтовых лекарственных средств. Поэ
тому этот метод может быть использован, если
суточная доза неизвестна или не установлена.
Если содержание остаточных растворителей во
всех вспомоrательных веществах и фармацев
тических субстанциях, которые входят в состав
roтовоro лекарственною средства, удовлетворя
ет пределам, приведенным в таблице 2, то все
эти компоненты можно использовать в любой
пропорции. Если суточная доза лекарственноro
средства не превышает 1 О r, никакие дальней
шие вычисления не требуются. Определение
предельноro содержания остаточных раствори
телей в лекарственных средствах, которые при
нимаются в дозах, превышающих 1 О r, должно
проводиться с использованием Подхода 2.
ПОДХОД 2. Нет необходимости, чтобы
содержание остаточных растворителей в
каждом компоненте лекарственноrо средства
соответствовало пределам, реrламентиро
ванным с использованием Подхода 1. Опре
делить предеЛhное содержание остаточно
ro растворителя в лекарственном средстве
можно по формуле (1), используя ДСД (Mr/cYT),
приведенное в таблице 2, и известное зна
чение максимальной суточной дозы лекар
ственноro средства. Такие пределы являются
приемлемыми при условии, что показано сни
жение содержания остаточною растворителя
до минимума, который достиrается практиче
ски. Пределы должны быть реалистичными в
отношении аналитической точности, произ
водственной возможности, разумною изме..
нения производственноrо процесса и COOT
ветствовать современным производственным
стандартам.
Подход 2 предусматривает суммирование
количеств остаточноro растворителя, присут
ствующеro в каждом из компонентов roтовоro
лекарственноro средства. Суммарное содержа
ние растворителя в сутки должно быть меньше,
чем ДСД.
Рассмотрим использование Подхода 1 и
Подхода 2 для расчета предельноro содержа
ния ацетонитрила в лекарственном средстве.
ДСД дЛЯ ацетонитрила 4,1 мr/сутки. Таким
образом, еro предельное содержание с ис
пользованием Подхода 1 41 О ррm. Макси
мально потребляемая масса лекарственноro
средства в сутки 5,0 r. Лекарственное cpeд
ство содержит два вспомоrательных вещества.
Состав лекарственноro средства и расчетное
предельное содержание ацетонитрила приве
дены в таблице:
Коли Содержание Суточное
Компонент чество в ацетони воздейст
составе, трила, ррm вие,мr
... I
Фармацев
тическая 0,3 800 0,24
субстанция
Вспомоrа
тельное 0,9 400 0,36
вещество 1
Вспомоrа
тельное 3,8 800 3,04
вещество 2
r отовое
лекарст 5,0 728 3,64
венное
средство
Концентрация ацетонитрила во всп.омоrа
тельном веществе 1 удовлетворяет предель
ному значению, установленному с исполь
зованием Подхода 1, но еro концентрация в
фармацевтической субстанции, вспомоrатель
ном веществе 2 и roтовом лекарственном cpeд
стве не удовлетворяе Тем не менее, coдep
жание ацетонитрила в roтовом лекарственном
средстве, установленное с использованием
Подхода 2, не превышает предела 4,1 мr/сутки
и, следовательно, соответствует рекомендаци
ям данноro руководства.
Рассмотрим друroй пример для ацетонитри
ла в качестве остаточноro растворителя. Макси
мальная потребляемая масса лекарственноro
средства в сутки 5,0 r, и лекарственное cpeд
ство содержит два вспомоrательных вещества.
5.4. Остаточные количества ораанических растворителей
911
Состав лекарственноro средства и расчетное
максимальное содержание ацетонитрила приве
дены в следующей таблице:
I Коли- Содержание Суточное
чество в
! Компонент составе, ацетони в ейст-
i r трила, ррт вие,мr
I Фармацев I
!тическая 0,3 800 0,24
I
! субстанция
I Вспомоrа 0,9 2000 1,80
I тельное
вещество 1
I Вспомоrа
!тельное 3,8 800 3,04
I вещество 2
r отовое
лекарст 5,0 1016 5,08
венное
. средство .. . ..
В данном случае концентрация ацетонитри
ла в лекарственном средстве не УДОВIlетворя
ет предельному значению ни с использовани
ем Подхода 1, ни с использованием Подхода 2.
Производитель может провести испытания ле
карственноro средства, чтобы определить, сни
жает ли процесс производства содержание aцe
тонитрила. Если же содержание ацетонитрила
не уменьшается в процессе производства до
допустимоro предела, производитель должен
предпринять друrие шаrи для уменьшения KOH
центрации ацетонитрила в лекарственном cpeд
стве. Если все предпринятые меры не приведут
к уменьшению содержания остаточною paCTBO
рителя, в исключительных случаях производи
тель может подroтовить резюме о предпринятых
усилиях, направленных на уменьшение coдep
жания растворителя до норм данноro PYKOBOД
ства, и провести анализ «риск польза», чтобы
получить разрешение на использование лекар
cTBeHHoro средства, содержащею более BЫCO
кий уровень остаточноro растворителя.
3.4. АНАЛИТИЧЕСКИЕ МЕТОДИКИ
Остаточные растворители, как правило,
определяются с использованием xpoMaTorpa
фических методов, в частности rазовой xpOMa
тоrрафии. Для определения содержания OCTa
точных растворителей MOryT использоваться
любые подходящие методики, описанные в
фармакопеях. Иначе roворя, производители
должны быть свободны в выборе наиболее под
ходящей валидированной аналитической Me
тодики для конкретноro применения. Если при
сутствуют ТOJ1bKO растворители класса 3, MorYT
быть использованы неспецифические методы
контроля, такие как, например, потеря в массе
при высушивании.
Валидация методов контроля остаточных
растворителей должна соответствовать PYKO
водству /СН «Валидация аналитических MeTO
дик» #(см. статью #5.3.2. Валидация аналити
ческих методик и испытаний (разделы А, В»,
а также требованиям соответствующих действу
ющих ТНПN.
3.5. ИНФОРМАЦИЯ О СОДЕРЖАНИИ
ОСТАТОЧНЫХ РАСТВОРИТЕЛЕЙ
Для тоro, чтобы соответствовать требовани
ям данною руководства, производителям юто
вых лекарственных средств необходима точная
информация о содержании остаточных paCTBO
рителей во вспомоraТАЛЬНЫХ веществах или фар
мацевтических субстанциях. Информация о co
держании остаточных растворителей, которая
может передаваться производителям лекарствен
ных средств поставщиками фармацевтических
субстанций или вспомоrательных веществ, может
быть представлена в следующих вариантах:
вероятно присутствуют остаточные pac
творители только класса 3; потеря в массе при
высушивании менее 0,5 %:
вероятно присутствуют остаточные pac
творители только класса 2; содержание каждою
из них не превышает пределов в соответствии с
Подходом 1; указывают наименование каждоrо
из остаточных растворителей;
вероятно присутствуют остаточные pac
творители классов 2 и 3; содержание каждою
из растворителей класса 2 не превышает преде
лов в соответствии с Подходом 1, а содержание
растворителей класса 3 менее 0,5 %.
Если существует вероятность присутствия
растворителей класса 1, то каждый из них
должен быть идентифицирован и определен KO
личественно.
«Вероятно присутствуют» относится К pac
творителям, используемым на заключительном
этапе производства, и тем, которые используют
ся на ранних стадиях производства и полностью
не удаляются утвержденным (валидированным)
процессом.
Если присутствуют остаточные раствори
тели класса 2 выше предельной концентрации
в соответствии с Подходом 1, а содержание
остаточных растворителей класса 3 превышает
0,5 %, то каждый из них должен быть идентифи
цирован и определен количественно.
4. ПРЕДЕЛЬНЫЕ СОДЕРЖАНИЯ
ОСТАТОЧНЫХ РАСТВОРИТЕЛЕЙ
4.1. РАСТВОРИТЕЛИ, ИСПОЛЬ30ВАНИЯ
КОТОРЫХ НУЖНО И3БЕrАТЬ
Растворители класса 1 не должны исполь
зоваться в производстве фармацевтических суб
станций, вспомоrательных веществ и roтовых
лекарственных средств из за их высокой токсич
ности И вредноro воздействия на окружающую
среду. Однако, если их использование неизбеж
но для производства лекарственноro средства,
которое имеет сильно выраженный терапев
тический эффект, их количества должны быть
912
rосударственная фармакопея Республики Беларусь
оrраничены в соответствии с таблицей 1, если
иное не обосновано. 1,1.1 Трихлорэтан включен
в таблицу 1, потому что он опасен для окружа
ющей среды. Установленный предел в 1500 ррт
основан на обзоре данных о безопасности.
Таблица 1
Растворители класса 1 в лекарственных
средствах и субстанциях для фармацевтиче
СК020 использования (растворители, примене
ния которых нужно избе2ать)
Растворитель Концентрацион Влияние
ный предел, ррт
Бензол 2 канцероrен
Четырех 4 токсичен и
хлористый опасен для
уrлерод окружа
ющей среды
1,2 Дихлорэтан 5 токсичен
1 , 1 Дихлорэтен 8 токсичен
1, 1,1 Трихлор 1500 опасен для
этан окружающей
среды
4.2. РАСТВОРИТЕЛИ, ИСПОЛЬ30ВАНИЕ
КОТОРЫХ НУЖНО оrРАНИЧИВАТЬ
Содержание растворителей, приведенных
в таблице 2. должно быть оrраничено в лекар
ственных средствах в связи с их токсичностью.
Данные ДСД даны с точностью до 0,1 Mr/cyт, а
концентрации с точностью до 10 ррт. YCTa
новленные значения не отражают необходимую
аналитическую точность определения. Точность
должна быть определена как часть валидации
(проверки правильности) методики.
Таблица 2
Растворители класса 2 в лекарственных
средствах и субстанциях для фармацевтиче
СК020 использования
ДСД, KOHцeHTpa
Растворитель Mr/ ционный
сут предел, ррт
Ацетонитрил 4,1 410
reKcaH 2,9 290
N,N Диметилацетамид 10,9 1090
N,N Диметилформамид 8,8 880
1 ,2 Диметоксиэтан 1,0 100
1 ,4 Диоксан 3,8 380
1,2 Дихлорэтен 18,7 1870
Ксилол* 21,7 2170
Метанол 30,0 3000
Метилбутилкетон 0,5 50
Метиленхлорид 6,0 600
N Метилпирролидон 5,3 530
Метилциклоrексан 11,8 1180
2 Метоксиэтанол 0,5 50
Нитрометан 0,5 50
Пиридин 2,0 200
ДСД, KOHцeHTpa
Растворитель Mr/ ционный
сут предел, ррт
Сульфолан 1,6 160
Тетраrидросруран 7,2 720
Тетралин 1,0 100
Толуол 8,9 890
1,1 ,2 Трихлорэтен 0,8 80
Формамид 2,2 220
Хлорбензол 3,6 360
Хлороформ 0,6 60
Циклоrексан 38,8 3880
Этиленrликоль 6,2 620
2 Этоксиэтанол 1,6 160
. Обычно 60 % м ксилола. 14 % п ксилола. 9 % о ксилола
и 17 % этилбензола.
4.3. МАЛОТОКСИЧНblЕ РАСТВОРИТЕЛИ
Растворители класса 3 (представлены в Ta
блице 3) MOryт быть отнесены к менее токсичным
и обладающим меньшим риском для здоровья че
ловека растворителям. Класс 3 не включает pac
творители, известные как опасные для здоровья
человека в концентрациях, которые обычно допу
скаются в лекарственных средствах. Однако для
мноrих растворителей класса 3 не проводилось
долroсрочное изучение токсичности или KaHцepo
rенности. Доступные данные указывают на то, что
они менее токсичны в острых или краткосрочных
испытаниях и дают отрицательный результат в
испытаниях на rенотоксичность (не проявляют re
нотоксичность). Считается, что содержание этих
остаточных растворителей, равное 50 Mr/cyт или
меньше (соответствует 5000 ррт или 0,5 % по
Подходу 1), приемлемо без обоснования. Более
высокие значения также MOryт быть приемлемы
при условии, что они определяются возможностя
ми производства, которое отвечает требованиям
надлежащей производственной практики (GMP).
Таблица 3
Растворители класса 3, которые должны
быть 02раничены требованиями GMP
или дРУ2ими требованиями к качеству
Анизол 2 Метил 1 пропанол
Ацетон Метилэтилкетон
1 Бутанол Муравьиная кислота
2 Бутанол Пентан
Бутилацетат 1 Пентанол
трет Бутилметиловый 1 Пропанол
эфир
rептан 2 Пропанол
Диметилсульсроксид Пропилацетат
Изобутилацетат Т етраrидрофуран
Изопропилацетат Уксусная кислота
Кумол Эта нол
Метилацетат Этилацетат
3 Метил 1 бутанол Этиловый эфир
Метилизобутилкетон Этилформиат
5.4. ОстатОЧНbJе количества орzаническuх растворителей
913
4.4. РАСТВОРИТЕЛИ, ДЛЯ КОТОРЫХ
ОТСУТСТВУЮТ НЕОБХОДИМЫЕ ДАННЫЕ
О ТОКСИЧНОСТИ
Растворители, представленные в таблице 4,
MOryт также представлять интерес для произво
дителей вспомоrательных веществ, фармацевти
ческих субстанций или лекарственных средств.
Однако для них отсутствуют обоснованные
данные о токсичности. Производители должны
сами обосновывать остаточные содержания этих
растворителей в лекарственных средствах.
Таблица 4
Растворители, для которых отсутствуют
обоснованные данные о токсичности
1, 1 Диметоксиметан Метилизопропилкетон
2,2 Диметоксипропан Метилтетраrидрофуран
1, 1 Диэтоксипропан Петролейный эфир
Изооктан Триерторуксусная
кислота
Изопропиловый спирт Трихлоруксусная
кислота
ТЕРМИНЫ
rенотоксuчные канцеР02ены канцероrены.
которые вызывают появление злокачественных
опухолей. воздействуя на reHbI или хромосомы.
Коэффициент корреляции (поправочный
коэффициент) (тоdifуiпg facto,) коэффици
ент, определенный в соответствии с обоснован
ным заключением токсиколоrа и основанный на
экстраполяции на человека данных по безопас
ности, полученных при испытаниях на животных.
Минимальный уровень, при котором Ha
блюдается эффект (LOEL, /owest observed
effect /eve/, МУНЭ) минимальная доза (веще
ства) в испытании или серии испытаний, которая
вызывает биолоrически существенное увеличе
ние в частоте или серьезности любых эсрфектов
у людей или животных.
Нейротоксuчность способность веще
ства оказывать неблаroприятное воздействие на
нервную систему.
Обратимая токсичность возникновение
вредных эффектов, которые вызваны данным
веществом и исчезают после прекращения дей
ствия вещества.
Предпола2аемый канцер02ен человека
вещество, для KOTOpOro нет эпидемиолоrической
очевидности канцероrенеза, но есть положи
тельные данные по rенотоксичности и очевид
ность канцероrенеза у rрызунов.
Разрешенная (допустимая) суточная доза
(РОЕ, permitted daily exposure, ДСД) макси
мально приемлемое суточное потребление OCTa
точноro растворителя в лекарственном cpeд
стве.
Терат02енность возникновение CTPYK
турных уродств В развивающемся зародыше,
Korдa вещество принимается в период беремен
ности.
Уровень, при котором не наблюдается
эффект (NOEL, по оЬsеrvеd еffесt exposure,
УНН3) максимальная доза вещества, при KO
торой нет биолоrически существенных увеличе
ний в частоте или серьезности любых эффектов
у людей или животных.
ПРИЛОЖЕНИЕ 1. СПИСОК РАСТВОРИТЕЛЕЙ, ВКЛЮЧЕННЫХ В РУКОВОДСТВО
Растворитель Второе название Структура Класс
UO CH '
Анизол Метоксибензол '# Класс 3
I
Ацетон 2 Пропанон, пропан 2 он СНзСОСН з Класс 3
Ацетонитрил СНзСN Класс 2
Бензол О Класс 1
1 Бутанол н Бутиловый спирт, бутан 1 ол СН з [СН 2 JРН Класс 3
2 Бутанол втор Бутиловый спирт, бутан 2 СН з СН 2 СН(ОН )СН:; Класс 3
ол
Бутилацетат Бутиловый эфир уксусной СН з СОО[СН 2 J з СН з Класс 3
кислоты
трет Бутилметиловый 2 Метокси 2 метилпропан (СН)з СОСН :; Класс З
эфир
reKcaH H reKcaH СН З [СН 2 ]4 СН З Класс 2
rептан н rептан СН з [СН 2 ]sСН з Класс 3
N,N Диметилацетамид ДМА СН з СОN(СН З )2 Класс 2
Диметилсульфоксид Метилсульфинилметан, (СН З )2 S0 КлассЗ
метилсульфоксид, ДМСО
N,N Диметилформамид ДМФА НСОN(СН З )2 Класс 2
914
rосударственная фармакопея Республики Беларусь
- I
Растворитель BTOPOt' 4эза. "t!ме Структура Класс
Диметиловый эФ'IIР ;' , енrли I
1 ,2 Диметоксиэтан fСЛЯ' моноrлим, ДИМС".',,,,еллtr Н з СОСН 2 СНРСН з Класс 2 i
зольв !
I (О, I
I
I
11 ,4 Диоксан п Диоксан, [1.4]диоксзН Класс 2 I
"'-''0"'/ I
i
--
Дихлорметан Метиленхлорид CH 2 CI 2 Класс 2 I
I'-------- - -
1 ,2 Дихлорэтан I сим Дихлорэтан, CH 2 CICH 2 C1 I Класс 1 !
I этилендихлорид, этv.':5" н хлорид
-- r --- -- ---- -- ------ I
1 , 1 Дихлорэтен 1 .1 Дихлорэтилен, H 2 C=CCI 2 Класс 1 I
l/3инилиденхлорид I
11 2 Дихлорэтилен, !
1 ,2 Дихлорэтен ,ацетилендихлорид CIHC=CHCI Класс 2 I
Изобутилацетат I Изобутиловый эфир - усной СН з СООСН 2 СН(СН З )2 Класс 3 j
I кислоты I
-- l
Изопропилацетат I Изопропиловый эфир '-, ксусной СН з СООСН(СН З )2 Класс 3 i
; кислоты I
I Дим"",,лбензол ((С Н,
Ксилол* НзС I Класс 2
СН З
Кумол Изопропилбензол, I CH' Класс 3
(1 метилэтил )бензол
- -
I Метанол l!v1етиловый спирт снрн Класс 2 1
-- . I
I Метилацетат Метиловый эфир уксус ,ой СНзСООСН з Класс 3
кислоты
3 Метил 1 бутанол Изоаtv l ИЛОВЫЙ спирт, изопропило (СНЗ)2СНСН2СНРН Класс 3 i
вый спирт, 3 метилбу!ati 1 ол i
, Метилбутилкетон 2 reKcaHoH, reKcaH 2 OH СНJСН2]ЗСОСНз Класс 2
Метилизобутилкетон 4 Метилпентан 2 он, СН з СОСН 2 СН(СН З )2 Класс 3
4 метил 2 пентанон, МИ6К
СН з
1 Метилпирролидин 2 он, I
N Метилпирролидон LJ Класс 2
1 метил 2 пирролидинон
2 Метил 1 пропанол Изобутиловый спирт, (СНЗ)2СНСНРН Класс 3
2 метилпропан 1 ол
Метилциклоrексан Циклоrексилметан асн' Класс 2
Метилэтилкетон 2 Бутанон, МЭК, бутан 2 он СН з СН 2 СОСН з Класс 3
2 Метоксиэтанол Метилцеллозольв СНрСН 2 СНрН Класс 2
Муравьиная кислота НСООН Класс 3
Нитрометан СН з NО 2 Класс 2
Пэнтан н Пентан СН з [СН 2 ]з СН з Класс 3
1 Пентанол Амиловый спирт, пентан 1 ол СН з [СН 2 ]з СН РН Класс 3
Пиридин О Класс 2
5.4. ОстатОЧНblе количества орzанических растворителей
915
...
Растворитель Второе название Структура Класс
1 Пропанол Пропан 1 ол, пропиловый спирт СН з СН 2 СНрН Класс 3
2 Пропанол Пропан 2 ол, изопропиловый (СНз)СНОН Класс 3
спирт
Пропилацетат Пропиловый эфир уксусной ! СН з СООСН 2 СН 2 СН з Класс 3
кислоты
, O /?
\
Сульфолан Тетраrидротиофен 1,1 диоксид slj Класс 2
О
Тетраметиленоксид, О
Т етраrидрофуран Класс 2
оксациклопентан
Тетралин 1 ,2,3,4 Тетраrидронафтш ,ИН I I Класс 2
I
I
асн, I
Толуол Метилбензол Класс 2 I
1#
1,1, 1 Трихлорэтан Метилхлороформ CH,CCI, Класс 1
1,1.2 Т рихлорэтен Трихлорэ т зн HCIC=CCI? Класс 2
. "
Уrлерод т етрахлорметан CCl 4 Класс 1
четыреххлористый
Уксусная кислота Этановая кислота СН СООН Класс 3
Формамид Метанамид HCONH? Класс 2
ас'
Хлоробензол '# Класс 2
Хлороформ Трихлорметан CHCI, Класс 2
Циклоrексан rексаметилен О Класс 2 I
I
I
Эта нол Этиловый спирт CH,CH OH Класс 3
. _.
Этилацетат Этиловый эфир уксусной СН з СООСН 2 СН з Класс 3 I
кислоты
Этиленrликоль 1 ,2 Диrидроксиэтан, НОСН 2 СНрН Класс 2
1 ,2 этандиол
Этиловый эфир Диэтиловый эфир, этоксиэтан, СН з СНРСН 2 СН з Класс 3
1, 1 ' оксибисэтан
Этилформиат Этиловый эфир муравьиной НСООСН 2 СН з Класс 3
кислоты
2 Этоксиэтанол Целлозольв СН з СНрСН 2 СНрН Класс 2
* Обычно 60 % м ксилола, 14 % п ксилола, 9 % о ксилола и 17 % этилбензола
ПРИЛОЖЕНИЕ 2. ДОПОЛНИТЕЛЬНЫЕ
ДАННЫЕ
мических соединений в статьях «Критерии
здоровья окружающей среды» (ЕНС) и «Объ
единенная информационная система риска»
(/R/S). Цели таких орrанизаций, как Между
народная проrрамма по химической безопас
ности (/PSC) , Управление по охране OKPy
жающей среды США (USEPA), Управление
лекарственных средств и пищевых продуктов
(USFDA), включают определение предельно
А2.1. ВЛИЯНИЕ оРrАНИЧЕСКИХ ЛЕТУЧИХ
РАСТВОРИТЕЛЕЙ НА ОКРУЖАЮЩУЮ
СРЕДУ
Некоторые из остаточных растворителей,
часто используемых в фармацевтическом про
изводстве, внесены в перечень токсичных хи
916
rосударственная фармакопея Республики Беларусь
ro содержания химических веществ. OCHOB
ная их цель защита человеческоro зцо
ровья и окружающей среды от возможноrС?
неrативноrо влияния химических соединений
в резулыате длительноro воздействия.
Методы, используемые для оценки макси
мальных, безопасных пределов воздействия,
обычно основываются на долroсрочных ис
следованиях. Коrда данные долroсрочных
испытаний недоступны, MorYT быть исполь
зованы данные краткосрочных испытаний с
модификацией подхода, например, использо
вание более высоких коэффициентов Koppe
ляции. Подход, описанный здесь, относится,
прежде всеro, к долroсрочным или пожизнен
ным воздействиям окружающей среды на Ha
селение, т. е. к воздействиям воздуха, пищи,
питьевой воды и друrих сред.
А2.2. ОСТАТОЧНЫЕ РАСТВОРИТЕЛИ
В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ
Пределы воздействия в этом руководстве
установлены в соответствии с методолоrией и
данными токсичности, приведенными в MOHO
rрафиях ЕСН и /R/S. Однако при установлении
пределов воздействия должны быть приняты
некоторые допущения относительно остаточ
ных растворителей, которые используются в
процессе синтеза и изrотовления лекарствен
ных средств, а именно:
1) пациенты (не все население) использу
ют лекарственные средства для лечения бо
лезней или для профилактики с целью избе
жать инфекции или болезни;
2) предположение о воздействии на про
должительность жизни пациента не обязатель
но для большинства лекарственных средств,
но может рассматриваться как рабочая rипо
теза, чтобы уменьшить риск для здоровья че
ловека;
3) остаточные растворители неизбеж
ные компоненты фармацевтическоro произ
водства, которые зачастую являются COCTaB
ной частью лекарственных средств;
4) остаточные растворители не должны
превышать рекомендуемые концентрации,
кроме исключительных обстоятельств;
5) данные о токсиколоrических испытани
ях, которые используются для определения
приемлемых концентраций остаточных раст
ворителей, должны быть зафиксированы с ис
пользованием соответствующих протоколов,
описанных, например, в документах ОЕСD и
FDA Red Book.
ПРИЛОЖЕНИЕ 3. МЕТОДЫ
УСТАНОВЛЕНИЯ ДОПУСТИМОЙ
СУТОЧНОЙ ДОЗЫ
Для оценки риска канцероrенных paCTBO
рителей Класса 1 используют метод rейлора
Коделя (Gay/or, D. W апd Kode/l, R./.: Liпеа, /п
tеrро/аtiоп a/gorithm (or /ow dose аssеssтепt о(
toxic sиЬstапсе. J. Eпviroп. Path%gy, 4, 305,
1980). Для установления ДСД экстраполяцию
с использованием математических моделей
следует при менять только 8 тех случаях, Korдa
есть достоверные данные о канцероrенности.
ДСД дЛЯ растворителей класса 1 также может
определяться с использованием высокоro зна
чения коэффициента корреляции (например,
от 1 О 000 до 100 000) для уровня, при котором
эффект не наблюдается. Обнаружение и коли
чественное определение этих растворителей
следует проводить валидированными анали
тическими методиками.
Предельные содержания для растворите
лей класса 2 в этом руководстве были YCTa
новлены путем вычисления значений ДСД co
rласно методикам определения предельноrо
содержания растворителей в лекарственных
средствах (Pharmacopeia/ Fo,um, ноябрь де
кабрь 1989 r.) и методам, принятым /PCS для
оценки риска химических веществ в отноше
нии здоровья человека (Eпvi,oпmeпta/ Health
C,iteria 170, ВОЗ, 1994). Эти методы подоб
ны тем, которые используют USEPA (/R/S) ,
USFDA (Red Book) и др. Метод описан ниже,
чтобы пояснить происхождение значений
ДСД. Чтобы использовать значения ДСД, при
веденные в таблице раздела 4 этоro ДOKYMeH
та, нет необходимости производить эти BЫ
числения.
При экспериментах на животных значе
ния ДСД рассчитывают, исходя из уровня,
при котором эффект не наблюдается (УННЭ),
или уровня, при котором наблюдается самый
низкий эффект (МУНЭ), по формуле:
ДСД = УННЭ. масса тела .
F1. F2. FЗ. F4. F5
Значение ДСД преимущественно получа
ют на основании УННЭ. Если значения УННЭ
неизвестны, MorYT быть использованы значе
ния МУНЭ. Коэффициенты корреляции, пред
ложенные здесь, для экстраполяции на чело
века данных, полученных на животных, это
те же «коэффициенты неопределенности», KO
торые использовались в моноrрафии «Крите
рии здоровья окружающей среды» (Епviroп
meпta/ Hea/th C,iteria 170, ВОЗ, Женева, 1994),
и «коэффициенты корреляции» или «коэф
фициенты безопасности» в «Pha,macopeia/
Fo,um». Во всех расчетах принимается пред
положение о 100 % системном воздействии
независимо от способа применения лекарств.
Коэффициенты корреляции:
F1 коэффициент корреляции для расче
та экстраполяции между видами;
F1 =5 при экстраполяции на человека
данных, полученных при исследованиях на
крысах;
F1 =12 при экстраполяции на человека данных,
полученных при исследованиях на мышах;
5.4. Остаточные количества ораанических растворителей
917
F1 =2 при экстраполяции на человека данных,
полученных при исследованиях на собаках;
F1 =2,5 при экстраполяции на человека
данных, полученных при исследованиях на KpO
ликах;
F1 =3 при экстраполяции на человека данных,
полученных при исследованиях на обезьянах;
F1=10 при экстраполяции на челове
ка данных, полученных при исследованиях на
друrих животных.
F1 принимает во внимание отношение пло
щади поверхности тела к весу тела COOTBeTCTBY
ющих видов животных и человека. Площадь
поверхности рассчитывается следующим
образом:
5 == kmO,67,
rдe:
т масса тела;
k константа, принята равной 10.
Массы тела, используемые в уравнении,
представлены в таблице A3. 1.
Таблица A3. 1
Значения, использованные при расчетах
в данном документе
Масса крысы 425 r
Масса беременной крысы 330 r
Масса мыши 28 r
Масса беременной мыши 30 r
Масса морской свинки 500 r
Масса макаки резус 2,5 Kr
Масса кролика 4 Kr
(беременноrо или нет)
Масса roнчей собаки (биrль) 11,5 Kr
Дыхательный объем крысы 290 л/сут
Дыхательный объем мыши 43 л/сут i
Дыхательный объем кролика 1440 л/сут i
Дыхательный объем морской свинки 430 л/сут
Дыхательный объем человека 28 800 л/сут i
Дыхательный объем собаки 9000 л/сут
Дыхательный объем обезьяны 1150 л/сут
Потребление воды мышью 5 мл/сут
Потребление воды крысой 30 мл/сут
Потребление пищи крысой 30 r/cYT
F2 коэффициент, учитывающий индивиду
альную изменчивость. Для всех орrанических paCT
ворителей, приведенных в данном руководстве, KO
эффициент обычно принимают равным 10.
FЗ переменный коэффициент для расчета ис
пытаний токсичности кратковременных воздействий.
F3=1 для испытаний, которые длятся по
меньшей мере в течение периода, равноro по
ловине продолжительности жизни животных
(1 rод для rрызунов и кроликов; 7 лет для собак,
котов и обезьян).
F3=1 для репродуктивных (воспроизводи
тельных) испытаний, которые охватывают весь
период орrаноrенеза.
F3=2 для испытаний в течение 6 месяцев на
rрызунах, или 3,5 лет не на rрызунах.
F3=5 для 3 месячных испытаний на rрызу
нах, или 2 летних не на rрызунах.
F3=10 для испытаний более короткой про
должительности.
Для всех промежуточных испытаний необхо
димо использовать более высокий коэффициент
(например, для 9 месячных испытаний на rрызу
нах используется коэффициент=2).
F4 коэффициент, который может приме
няться при высокой токсичности растворителя,
например, неrенотоксичной канцероrенности,
нейротоксичности или тератоrенности. В испы
таниях репродуктивной токсичности использу
ются следующие коэффициенты:
F4=1 для эмбриональной токсичности, связан
ной С материнской токсичностью (интоксикацией);
F4=5 для эмбриональной токсичности (ин
токсикацией), не связанной с материнской;
F4=5 для TepaToreHHoro эффекта, связанно
ro с материнской интоксикацией;
F4=1 О для TepaтoreHHoro эффекта, не свя
занноro с материнской интоксикацией.
F5 переменный коэффициент, который
может применяться, если УННЭ (уровень, не
вызывающий эффекта) не был установлен.
Коrда доступны только данные уровня МУНЭ
(уровень, вызывающий минимальный эффект),
то, в зависимости от уровня токсичности, может
использоваться коэффициент вплоть до 10.
Допускается, что масса тела взрослоro чело
века любоro пола равна 50 Kr: Этот относитель
но низкий вес обеспечивает дополнительный
коэффициент безопасности стандартному весу
человека 60 или 70 Kr, который часто использу
ется в таких вычислениях. Известно, что мноrие
взрослые пациенты весят менее 50 Kr, поэтому в
этом случае при определении ДСД используют
ся друrие коэффициенты. Если лекарственное
средство, содержащее растворитель, предна
значено для педиатрии, то необходимо сделать
корректировку на более низкую массу тела.
Как пример применения этоrо уравнения
рассмотрим испытание токсичности ацетонитри
ла на мышах, которое было описано в Pharmeu
юра, т. 9, NQ 1. Дополнение. Апрель 1997, с. 524.
Установлено, что значение УННЭ 50, 7 Mr/Kr'cYT.
ДСД дЛЯ ацетонитрила при этом рассчитывали
следующим образом:
ДСД 50, 7 Mr'Kr 1 сут . . 50 Kr 4 22 .1
== ==, Mr'CYT.
12.10.5.1.1
в этом при мере:
F1=12, учитывает экстраполяцию на человека
данных, полученных при исследованиях на мышах;
F2=10, учитывает индивидуальную измен
чивость;
918
rосударственная фармакопея Республики Беларусь
F3=5, так как продолжительность испыанийй
составила только 13 недель;
F4=1, так как с серьезной токсичностью не
сталкивались;
F5=1, так как был определен уровень, не BЫ
зывающий эффекта.
Для перерасчета концентраций rазов, ис
пользуемых вдыхательных (инrаляторных) ис
пытаниях из ррт в мr/л или мr/м З , использова
ли уравнение для идеальноro rаза: PV = пRТ
Рассмотрим в качестве при мера испытание
репродуктивной токсичности крысы в резуль
тате вдыхания четыреххлористоrо уrлерода
(М м. 153.,84), описанное в Pharmeurope, т. 9,
NQ 1, Дополнение, апрель 1997, стр. S9.
!2", 300.10<;атм.153 840 Mr моль' '" 46,15 Mr '" 1.89 мr/л.
V RT 0,082 л атм К ' моль' 298 К 24,45 л
Для перевода в мr/м З используют отношение
1000 л = 1 м З .
01/2013:50500
5.5. АЛкоrОЛЕМЕТРИЧЕСКИЕ
ТАБЛИЦЫ
Таблица #5.5. 1
Соотношение между плотностью водно спир
тов020 раствора и содержанием безводН020
спирта в растворе
Содержание этанола
в водно пиртовом растворе
Плот- в процентах миллили
ность rpaMMOB тpo8B100r
Р20 по по В 100 мл при взве
массе объему при 20 ос шивании
В воздухе
0,99823 0,00 0,00 0,00 0,00
80 12 16 13 16
0,9978 23 29 23 29
6 34 43 34 43
4 44 56 44 56
2 55 70 55 70
О 66 83 66 83
0,9968 77 97 77 97
6 87 1,10 87 1,10
4 98 24 98 24
2 1,09 38 1,09 38
О 20 51 19 51
0,9958 31 65 32 66
6 42 79 41 80
4 52 92 52 93
2 63 2,06 63 2,07
О 74 20 74 21
Содержаниеэтанола
в водно-спиртовом растворе
Плот- в процентах миллили-
ность rpaMMo8 тpoBB100r
Р20 по по В 100 мл при 8зве-
массе объему при 20 ос шивании
в воздухе '
0,9948 85 34 85 35 I
6 96 48 96 50
4 2,07 62 2,07 64
2 19 76 18 78
О 29 90 29 92
0,9938 41 3,04 40 3,06
6 52 18 51 20
4 63 32 62 34
2 75 46 73 48
О 86 60 84 63
0,9928 97 74 95 77
6 3,09 89 3,07 92
4 20 4,03 18 4,06
2 32 17 29 20
О 44 32 41 36
0,9918 55 46 52 50
6 67 61 64 65
4 78 75 75 80
2 90 90 87 95
О 4,02 5,05 99 5,10
0,9908 14 20 4,10 25
6 26 35 22 41
4 38 50 34 56
2 50 65 46 71
О 62 80 58 87
0,9898 75 95 70 6,02
6 87 6,10 81 17
4 99 26 94 34
2 5,11 41 5,06 49
О 24 57 19 65
0,9888 37 73 31 81
6 49 88 43 97
4 62 7,04 56 7,13
2 75 20 68 29
О 87 36 81 46
0,9878 6,00 52 94 62
6 13 67 6,05 77
4 26 83 18 94
2 39 99 31 8,10
О 52 8,15 43 27
0,9868 65 32 57 44
6 78 48 69 61
4 92 64 82 77
2 7,05 80 95 93
О 18 97 7,08 9,11
0,9858 32 9,13 21 27
6 45 30 34 45
4 58 47 47 62
2 72 63 60 78
5.5. АЛКО20леметрические таблицы
919
Содержаниеэтанола
в водно-спиртовом растворе
Плот- в процентах миллили-
ность rpaMMoB тров в 100 r
Р20 по по В 100 мл при взве-
массе объему при 20 ос wивании
В воздухе
О 85 80 73 96
0,9848 99 97 87 10,13
6 8,12 10,13 8,00 30
4 26 30 13 47
2 39 47 26 65
О 53 63 39 82
0,9838 67 80 52 99
6 80 97 66 11,17
4 94 11,14 79 34
2 9,08 31 93 52
О 22 48 9,06 70
0,9828 35 65 19 87
6 49 82 33 12,04 I
4 63 99 46 22
2 77 12,16 60 40 !
О 91 34 74 I 58
0,9818 10,05 51 87 75
6 19 68 10,01 93
4 34 85 14 13,11
2 48 13,03 28 29
О 62 20 42 47
0,9808 76 38 56 66
6 91 55 69 83
4 11,05 73 84 14,02
2 20 90 97 20
О 34 14,08 11,11 38
0,9798 49 26 25 57
6 64 44 40 76
4 78 62 54 94
2 93 79 67 15,12
О 12,07 97 82 31
0,9788 22 15,15 96 50
6 37 34 12,11 69
4 52 52 25 88
2 67 70 39 16,07
О 81 88 53 26
0,9778 96 16,06 68 44
6 13,11 25 83 66
4 27 43 97 83
2 42 61 13,11 17,01
О 57 80 26 21
0,9768 72 98 40 40
6 87 17,17 55 ео
4 14,02 35 69 79
2 18 54 84 99
О 33 73 99 18,19
0,9758 49 91 14,14 38
6 64 18,10 29 58
4 80 29 44 78
! Содержаниеэтанола
в водно-спиртовом растворе
Плот- в процентах миллили-
ность rpaMMoB тров в 100 r
Р20 по по В 100 мл при взве-
массе объему при 20 ос wивании
В воздухе
2 I 96 48 59 97
О 15,11 67 74 19,17
0,9748 27 86 89 37
6 43 19,05 15,04 57
4 58 24 19 77
2 74 43 34 97
О 90 62 49 20,16
0,9738 ' 16.05 81 64 36
6 21 20,00 79 56
4 37 19 94 76
2 52 I 37 16,08 95
,
I О 68 56 23 21,15
0.9728 84 75 38 35
I 6 : 99 93 52 54
!
4 17,15 21,12 67 74
2 30 31 82 94
О 45 49 96 22,13
0,9718 61 68 17,11 33
6 76 86 25 52
4 92 22,05 40 72
2 18,07 23 55 91
О 22 41 69 23,10
0,9708 37 60 84 31
6 52 78 98 50
4 67 96 18,12 69
2 83 23,14 26 88
О 98 32 41 24,07
0,9698 19,13 50 55 26
6 28 68 69 45
4 43 86 83 64
2 58 24,04 97 83 I
О 73 22 19,12 25,02 :
0,9688 88 40 26 21 !
6 20,03 57 39 40
4 18 75 53 59
2 33 93 68 77
О 47 25,11 82 96
0,9678 62 28 95 26,15
6 77 46 20,09 I з4
4 92 64 24 53
2 21,07 81 I 37 72
О 21 99 I 51 91
0,9668 36 26.16 ' 65 27,09
6 50 34 79 28
4 65 51 92 47
2 80 68 21,06 65
О 94 85 19 83
0,9658 22,09 27,03 33 28,02
6 23 20 47 20
920
rосударственная фармакопея Республики Беларусь
Содержание эта нола I
в водно-спиртовом раСТВОрЕ:
Плот- в процентах миллили- I
ность rpaMMOB тров в 100 r
Р20 по по В 100 мл при взве- I
массе объему при 20 ос' шивании
в воздухе
4 37 37 60 38
2 52 54 74 I 56
О 66 71 87 75
0,9648 81 88 22.00 93
6 95 28,05 14 29,12
4 23,09 22 i 27 29
!
2 23 38 I 40 47
О 38 55 i 53 65
0,9638 52 72 i 67 83
6 66 I 88 79 30,00
4 80 29,05 I 93 18
2 94 21 23,05 36
О 24,08 38 19 54
0,9628 22 54 32 71
6 36 71 45 90
4 50 87 58 31,07
2 64 30,03 70 24
О 78 19 83 42
0,9618 92 35 95 60
6 25,05 52 24,09 78
4 19 68 21 95
2 32 84 з4 32,12
О 46 31,00 47 30
0,9608 59 16 59 48
6 73 31 71 63
4 86 47 84 81
2 26,00 63 96 98
О 13 78 25,08 33,14
0,9598 26 94 21 31
6 39 32,09 33 48
4 52 ! 24 45 64
2 65 39 56 81
О 78 54 68 96
0,9588 92 69 80 34,13
6 27,04 84 92 29
4 17 99 26,04 46
2 30 33,14 16 62
О 43 29 27 79
0,9578 55 44 39 95
6 68 59 51 35,11
4 81 73 62 26
2 94 88 74 43
О 28,06 34,03 86 59
0,9568 19 17 97 75
6 31 31 27,08 90
4 43 45 19 36,06
2 56 60 31 22
О 68 74 42 37
0,9558 80 88 53 53
Содержаниеэтанола
в водно спиртовом растворе
ПЛОТ- в процентах миллили-
ность rpaMMoB тровв 100r
Р20 по ПО В 100 мл при взве
массе объему при 20 ос шивании
в воздухе
6 93 35,02 64 68
4 29,05 16 75 84
2 17 30 86 99
О 29 44 97 37,15
0,9548 41 58 28,07 30
6 53 72 19 46
4 65 85 30 51
2 77 99 41 76
О 89 36,13 52 92
0,9538 30,01 26 62 38,06
6 13 40 73 21
4 25 53 83 36
2 36 67 94 51
О 48 80 29,05 66
0,9528 60 94 16 81
6 72 37,07 26 96
4 84 20 36 39,10
2 95 з4 47 25
О 31,07 47 57 40
0,9518 18 60 68 55
6 30 73 78 69
4 41 86 88 84
2 53 99 98 98
О 64 38,12 30,09 40,12
0,9508 76 25 19 27
6 87 38 29 42
4 99 51 39 56
2 32,10 64 50 70
О 21 77 60 85
0,9498 33 90 70 41,00
6 44 39,03 81 14
4 55 15 90 28
2 66 28 31,00 42
О 78 40 10 56
0,9488 89 53 20 71
6 33,00 66 30 86
4 11 78 40 99
2 22 91 50 42,14
О 33 40,04 60 28
0,9478 44 46 70 42
6 55 28 79 56
4 66 41 89 70
2 77 53 99 84
О 88 65 32,08 98
0,9468 99 78 18 43,12
6 34,10 90 28 26
4 21 41,02 38 39
2 32 15 48 54
О 43 27 57 68
5.5. АЛКО20леметрические таблицы
921
Содержание этанола
в водно пиртовом растворе
Плот- в процентах миллили-
ность rpaMMOB тpoBB100r
Р20 по по В 100 мл при взве
массе объему при 20 ос шивании
В воздухе
0,9458 54 39 67 81
6 65 51 76 95
4 76 63 86 44,08
2 86 75 95 22
О 97 87 33,05 35
0,9448 35,08 99 14 49
6 19 42,11 24 63
4 29 23 33 76
2 40 35 43 90
О 50 46 51 45,03
0,9438 61 58 61 17
6 71 70 70 30
4 82 82 80 44
2 93 94 89 58
О 36,03 43,05 98 71
0,9428 13 17 34,07 84
6 24 28 16 97
4 34 39 25 46,10
2 45 51 34 23
О 55 62 43 36
0,9418 65 74 52 49
6 76 85 61 62
4 86 97 70 75
2 96 44,08 79 88
О 37,07 19 88 47,01
0,9408 17 30 96 14
6 27 42 35,06 27
4 37 53 15 41 ,
2 47 64 23 53
О 58 75 32 66
0,9398 68 86 41 79
6 78 98 50 93
4 88 45,09 59 48,06
2 98 20 68 18
О 38,09 31 76 31
0,9388 19 42 85 43
6 29 53 94 56
4 39 64 36,02 69
2 49 75 11 82
О 59 86 20 95
0,9378 69 97 28 49,07
6 79 46,08 37 20
4 89 19 46 33
2 99 30 54 46
О 39,09 41 63 58
0,9368 19 52 72 71
6 29 63 80 84
4 39 73 88 96
2 49 84 97 50,08
Содержание этанола
в водно-спиртовом растворе
Плот- в процентах миллили-
ность rpaMMOB тров в 100 r
Р20 по по В 100 мл при взве
массе объему при 20 ос шивании
в воздухе
О 59 95 37,06 21
0,9358 69 47,06 14 34
6 79 17 23 47
4 89 27 31 59
2 99 38 40 72
О 40,09 49 48 85
0,9348 19 59 56 97
6 29 70 65 51,10
4 38 81 73 22
2 48 92 82 35
О 58 48,02 90 47
0,9338 68 13 99 60
6 78 23 38,07 72
4 88 33 15 84
2 98 44 23 97
О 41,07 54 31 52,09
0,9328 17 65 40 22
6 27 75 48 34
4 36 86 56 46
2 46 96 64 58
О 56 49,07 73 71
0,9318 65 17 81 83
6 75 27 89 95
4 85 38 97 53,08
2 94 48 39,05 20
О 42,04 58 13 32
0,9308 13 69 22 45
6 23 79 30 56
4 33 89 38 68
2 42 99 46 80
О 52 50,10 54 93
0,9298 61 20 62 54,05
6 71 30 70 17
4 80 40 78 29
2 90 50 86 41
О 43,00 60 94 53
0,9288 09 71 40,02 i 66
;
6 18 81 10 78
4 28 91 18 90
2 37 51,01 26 55,02
О 47 11 I 34 14
0,9278 56 21 I 42 26
i
6 66 31 I 50 38
I
4 75 41 58 50
2 85 51 66 92
О 94 61 73 74
0,9268 44,04 71 81 86
6 13 81 89 98
4 23 91 97 56,10
922
rосударственная фармакопея Республики Беларусь
Плот-
ность
Р20
i
I
I 2
, О
0,9258
6
4
2
О
0,9248
6
4
2
О
0,9238
6
4
2
О
0,9228
6
4
2
О
0,9218
6
4
2
О
0,9208
6
4
2
О
0,9198
6
4
2
О
0,9188
6
4
2
О
0,9178
6
4
2
О
0,9168
6
Содержание этанола J
в водно-спиртовом растворе .
в процентах миллили- I
rpaMMoB трав в 100 r
по по в 100 мл при взве- I
массе объему при 20 ос шивании I
в воздухе i
32
41
51
60
70
79
88
98
45,07
16
26
35
44
53
63
72
81
91
46,00
09
18
28
37
46
55
65
74
83
92
47,01
10
20
29
38
47
56
65
74
83
93
48,02
11
20
29
38
47
56
65
75
52,00
10
20
30
40
50
60
69
79
89
99
53,09
18
28
38
48
57
67
77
86
96
54,06
15
25
34
44
54
63
73
82
92
55,01
11
20
30
39
48
58
67
77
86
95
56,05
14
23
33
42
51
61
41,04
12
20
28
36
44
52
59
67
74
82
90
97
42.05
13
21
28
36
44
21
59
67
74
82
89
97
43,05
12
20
27
35
42
50
57
65
72
79
87
94
44,02
09
16
24
31
38
46
53
60
68
21
33
45
57
69
81
93 !
57,04 i
16
28
40
52
63
75
88
58,00
11
23
35
46
58
70
81
93
59,05
17
29
40
52
63
75
86
98
60,10
22
33
44
56
67
79
91
61,02
14
25
37
49
60
71
83
I
Содержаниеэтанола
в водно-спиртовом растворе
Плот- в процентах миллили-
ность rpaMMoB тpoBB100r
Р20 по ПО в 100 мл при взве-
массе объему при 20 ос шивании
в воздухе
4 84 70 75 95
2 93 79 82 62,06
О 49,02 89 90 18
0,9158 11 98 97 29
6 20 57,07 45,04 40
4 29 17 12 53
2 38 26 19 64
О 47 35 26 76
0,9148 56 44 34 87
6 65 53 41 98
4 74 62 48 63,09
2 83 72 56 21
О I 92 81 63 32
I
0.9138 ; 50.01 90 70 44
6 10 99 77 55
4 19 58,08 84 66
2 28 17 91 77
О 37 26 98 89
0,9128 46 35 46,05 64,00
6 55 44 12 11
4 64 54 20 23
2 73 63 27 35
О 82 72 35 46
0,9118 91 81 42 57
6 51,00 90 49 68
4 09 99 56 80
2 18 59,08 63 91
О 27 17 70 65,02
0,9108 36 26 77 14
6 45 35 84 25
4 54 44 91 36
2 63 53 99 48
О 71 62 47,06 59
0,9098 80 71 13 70
6 89 80 20 82
4 98 89 27 93
2 52,07 98 34 66,05
О 16 60,07 41 16
0,9088 25 16 48 27
6 34 25 55 39
4 43 34 62 50
2 52 43 70 61
О 60 52 77 72
0,9078 69 60 83 83
6 78 69 90 95
4 87 78 97 67,06
2 96 87 48,04 17
О 53,05 96 11 29
0,9068 14 61,05 18 41
i
..
!
----,
i
5.5. АЛКО20леметрические таблицы
923
Содержание зтанола
в водно-спиртовом растворе
Плот- в процентах миллили-
ность rpaMMoB трав в 100 r
Р20 по по В 100 мл при взве-
массе объему при 20 ос шивании
В воздухе
6 22 14 26 52
4 31 22 32 62
2 40 31 39 73
О 49 40 46 85
0,9058 58 49 53 97
6 67 57 60 68,07
4 75 66 67 19
2 84 75 74 30
О 93 84 81 41
0,9048 24,02 92 87 52
6 11 62,01 94 63
4 19 10 49,01 75
2 28 19 08 87
О 37 27 15 96
0,9038 46 36 22 69,08
6 54 45 29 19
4 63 53 35 30
2 72 62 42 42
О 81 71 50 53
0,9028 89 79 56 63
6 98 88 63 74
4 55,07 97 70 86
2 16 63,05 76 97
О 25 14 83 70,08
0,9018 33 22 90 19
6 42 31 97 30
4 51 40 50,04 42
2 60 48 10 52
О 68 57 17 64
0,9008 77 65 24 75
6 86 74 31 86
4 95 82 37 97
2 56,03 91 44 71,08
О 12 64,00 51 20
0,8998 21 08 58 30
6 30 17 65 42
4 38 25 71 53
2 47 34 78 64
О 56 42 84 75
0,8988 65 51 92 86
6 73 59 99 97
4 82 68 51,05 72,08
2 91 76 11 19
О 57,00 85 18 30
0,8978 08 93 25 41
6 17 65,02 32 53
4 26 10 38 63
2 34 18 44 73
О 43 27 52 85
Содержание этанола
в водно-спиртовом растворе
Плот- в процентах миллили- I
ность rpaMMoB трав в 100 r I
Р20 по ПО В 100 мл при взве- i
массе объему при 20 ос шивании '
В воздухе i
0,8968 52 35 58 96 i
6 60 43 64 73,06
4 69 52 71 18
2 78 61 78 30 i
О 87 69 85 41
0,8958 95 77 91 51
6 58,04 86 99 63
4 13 94 52,05 73
2 21 66,02 11 84
О 30 11 18 95
0,8948 39 19 24 74,06
6 47 27 30 17
4 56 36 38 29
2 65 44 44 39
I О 74 53 51 51
0,8938 82 61 57 61
6 91 69 64 72
4 59,00 77 70 83
2 08 86 77 95
О 17 94 83 75,05
0,8928 26 67,02 90 16
6 34 11 97 27
4 43 19 53,03 39
2 52 27 09 49
О 60 36 17 61
0,8918 69 44 23 72
6 77 52 29 83 I
4 86 61 36 94
2 95 69 43 76,05 I
О 60,03 77 49 15 i
0,8908 12 85 55 26 i
,
6 21 94 62 38
4 29 68,02 69 49
2 38 10 75 59 I
О 47 18 81 70 !
0,8898 55 26 88 ! 81 !
6 64 35 95 I 93
I
4 72 43 54.01 , 77,04 i
2 81 51 07 14 !
О 90 59 14 : 25
0,8888 98 67 I 20 36
6 61,07 75 , 26 ! 47
4 15 83 ! 33 57
2 24 91 i 39 68
О 33 69,00 46 80
0,8878 41 08 52 91
6 50 16 59 78,02
4 58 24 65 12
2 67 32 71 23
924
rосударственная фармакопея Республики Беларусь
Содержаниеэтанола I
в водно-спиртовом раСТВоре
Плот- В про центах миллили-
ность rpaMMoB трав в 100 r
Р20 по по В 100 мл при взве-
массе объему при 20 ос шивании
в воздухе
О 76 40 78 34
0,8868 84 48 84 45
6 93 56 i 90 56
4 62,01 64 96 66
2 10 72 55,03 77
О 18 80 09 88 I
0,8858 27 88 15 99
6 36 96 22 79,10
4 44 70,05 29 21
2 53 12 34 31
О 61 20 41 42
0,8848 70 28 47 53
6 79 36 53 64
4 87 45 60 75
2 96 53 67 86
О 63,04 61 73 97
0,8838 13 69 79 80,08
6 21 77 86 19
4 30 85 92 30
2 39 93 98 40
О 47 71,01 56,05 51
0,8828 56 09 11 62
6 64 17 17 73
4 73 25 24 84
2 82 35 30 95
О 90 41 36 81,06
0,8818 99 49 42 17
6 64,07 57 49 28
4 16 65 55 39
2 24 72 61 49
О 33 80 67 60
0,8808 41 88 73 70
6 50 96 80 81
4 59 72,04 86 93
2 67 12 92 82,04
О 76 20 99 15
0,8798 84 28 57,05 25
6 93 36 11 36
4 65,01 44 17 47
2 10 51 23 57
О 18 59 29 68
0,8788 27 67 36 79
6 35 75 42 90
4 44 83 48 83,01
2 52 91 55 12
О 61 98 60 22
0,8778 69 73,06 66 33
6 78 14 73 45
4 86 22 79 56
Содержание этанола
в водно-спиртовом растворе
Плот- В процентах миллили-
ность rpaMMOB трав В 100 r
Р20 по ПО В 100 мл при взве-
массе объему при 20 ос шивании
В воздухе
2 95 29 85 66
О 66,03 37 91 77
0,8768 12 45 97 87
6 20 53 58,03 98
4 29 60 09 84,08
2 37 68 15 19
О 46 76 22 30
0,8758 54 84 28 42
6 63 91 33 51
4 71 99 40 63
2 80 74,07 46 74
О 88 15 52 85
0,8748 97 22 58 95
6 67,05 30 64 85,06
4 14 37 70 16
2 22 45 76 27
О 31 53 82 38
0,8738 39 61 89 49
6 47 68 94 59
4 56 76 59,01 70
2 64 84 07 81
О 73 91 12 91
0,8728 81 99 19 86,03
6 90 75,06 24 12
4 98 14 31 24
2 68,07 22 37 35
О 15 29 42 45
0,8718 24 37 49 56
6 32 45 55 67
4 41 52 61 77
2 49 60 67 89
О 58 68 73 87,00
0,8708 66 75 79 10
6 75 83 85 21
4 83 90 91 31
2 92 98 97 42
О 69,00 76,06 60,03 53
0,8698 08 13 09 63
6 17 21 15 74
4 25 28 21 85
2 34 36 27 96
О 42 43 32 88,06
0,8688 51 51 39 17
6 59 58 44 27
4 68 66 51 38
2 76 74 57 50
О 84 81 62 60
0,8678 93 89 69 71
6 70,01 96 74 81
5.5. АЛКО20леметрические таблиЦbl
925
Содержаниеэтанола
в водно-спиртовом растворе
Плот- в процентах миллили-
ность rpaMMoB трав в 100 r
Р20 по по В 100 мл при взве- I
массе объему при 20 ос шивании
в воздухе
4 10 77,04 81 93
2 18 11 86 89,02
О 26 19 92 14
0,8668 35 26 998 24
6 43 33 61,03 34
4 52 41 10 46
2 60 48 15 56
О 68 56 22 67
0,8658 77 63 27 77
6 85 70 33 88
4 94 78 39 99
2 71,02 85 44 90,09
О 10 93 51 21
0,8648 19 78,00 56 31 I
6 27 07 62 41
4 36 15 68 53
2 44 22 74 63
О 52 29 79 73
0,8638 61 37 86 84
6 69 44 91 95
4 77 51 97 91,05
2 86 59 62,03 16
О 94 66 08 26
0,8628 72,03 73 14 36
6 11 81 20 47
4 19 88 26 57
2 28 95 31 68
О 37 79,03 38 79
0,8618 44 10 43 90
6 53 17 49 92,00
4 61 24 54 10
2 69 32 60 22
О 78 39 66 32
0,8608 86 46 72 . 42
6 95 53 77 52
4 73,03 61 83 64
2 11 68 89 74
О 20 75 94 84
0,8598 28 83 63,01 96
6 36 90 06 93,06
4 45 97 12 16
2 53 80,04 17 27
О 61 1 23 37
0,8588 70 19 29 49
6 78 26 35 59
4 86 33 40 70
2 95 40 46 80
О 74,03 47 51 90
0,8578 11 54 57 94,01
.
Содержаниеэтанола
в водно-спиртовом растворе
Плот- в процентах миллили-
ность rpaMMoB трав в 100 r
Р20 по по В 100 мл при взве-
массе объему при 20 ос шивании
В воздухе
6 20 62 63 13
4 28 69 69 23
2 36 76 74 33
О 44 83 80 43
0,8568 53 90 85 54
6 61 97 91 64
4 69 81,05 97 76
2 78 12 64,03 87
О 86 19 08 97
0,8558 94 26 14 95,07
6 75,02 33 ; 19 17
I 4 11 i 40 25 28
;
, 2 19 47 30 38
: О 27 54 36 49
0,8548 35 61 41 59
6 44 68 47 70
4 52 75 52 80
2 60 82 58 90
О 69 89 63 96,01
0,8538 77 96 68 11
6 85 82,03 74 21
4 93 10 80 32
2 76,01 17 85 43
О 10 24 91 53
0,8528 18 31 96 63
6 26 38 65,02 74
4 35 45 08 85
2 43 52 13 95
О 51 59 19 97,06
0,8518 59 66 24 16
6 67 73 30 27
4 76 80 35 38
2 84 87 41 48 1
О 92 94 46 59
0,8508 77,00 83,01 52 69
6 09 08 57 80
4 17 14 62 89
2 25 21 68 I 99
О 33 28 73 i 98,10
0,8498 42 35 I 79 20
6 50 42 ! 84 31
4 58 49 ; 90 42
2 66 56 95 53
О 74 63 66,01 63
0,8488 83 69 05 73
6 91 76 11 83
4 99 I 83 16 93
2 78,07 90 22 99,04
О 16 97 28 15
926
rосударственная фармакопея Республики Беларусь
Содержаниеэтанола
в водно-спиртовом paCTBopt:
Плот- в процентах миллили-
ность rpaMMoB трав в 100 r
Р20 по ПО В 100 МЛ при взве
массе объему при 20 ос шивании
в воздухе
0,8478 24 84,04 33 25
6 32 10 38 34
4 40 17 43 45
2 48 24 49 56
О 56 31 54 67
0,8468 64 38 ! 60 78
6 73 44 65 87
4 81 , 51 70 98
2 89 I 58 76 100,08
О 97 65 81 19 I
0,8458 79,05 71 I 86 28 I
6 13 ! 78 91 39
4 22 i 85 97 50
2 30 i 91 I 67,02 60
О 38 I 98 07 70
0,8448 46 85,05 13 80 ,
6 54 ! 12 18 91 ;
4 62 18 23 101,01
2 i 70 ! 25 29 12
О 78 ! 32 34 23
0,8438 ; 87 i 38 39 32
6 i 95 45 44 42
4 80.0З 51 49 52
2 ! 11 58 55 63
О 19 65 60 74
0,8428 i 27 71 65 83
6 35 I 78 70 94
4 I 43 I 85 76 102,04
2 51 91 81 14
О 60 98 86 25
0,8418 68 86,05 92 36
6 76 11 96 45
4 84 18 68,02 56
2 92 24 07 65
О 81,00 31 12 76
0,8408 08 37 17 85
6 16 44 22 96
4 24 50 27 103,06
2 32 57 33 17
О 40 63 37 26
0,8398 48 70 43 37
6 56 76 48 47
4 64 83 53 58
2 72 89 58 67
О 80 96 63 78
0,8388 88 87,02 68 87
6 96 09 74 98
4 82,04 15 78 104,08
2 12 21 83 18
Содержание эта нола
в водно-спиртовом растворе
ПЛОТ- в процентах миллили-
ность rpaMMoB тров в 100 r
Р20 по по В 100 МЛ при взве-
массе объему при 20 ос шивании
в воздухе
О 20 28 89 29
0,8378 28 34 93 38
6 36 41 99 49
4 44 47 69,04 59
2 52 53 08 68
О 60 60 14 79
0,8368 68 66 19 89
6 76 72 23 98
4 84 79 29 105,09
2 92 85 34 19
О 83,00 92 39 30
0,8358 08 98 44 40
6 16 88,04 49 49
4 24 11 54 61
2 32 17 59 70
О 40 23 64 80
0,8348 48 29 68 89
6 56 36 74 106,01
4 64 42 79 10
2 72 48 83 20
О 80 54 88 30
0,8338 88 61 94 42
6 96 67 98 51
4 84,04 73 70,03 61
2 11 79 08 70
О 19 86 1З 82
0,8328 27 92 18 91
6 35 98 23 107,01
4 43 89,04 28 10
2 51 10 32 20
О 59 16 37 30
0,8318 67 23 43 42
6 74 29 47 52
4 82 35 52 61
2 90 41 57 71
О 98 47 62 80
0,8308 85,06 53 66 91
6 14 59 71 108,00
4 21 65 76 10
2 29 71 81 20
О 37 77 85 29
0,8298 45 83 90 39
6 53 90 96 51
4 61 96 71,00 61
2 68 90,02 05 70
О 76 08 10 81
0,8288 84 14 14 90
6 92 20 19 109,00
4 86,00 26 24 10
5.5. АЛКО20леметрическuе таблиЦbl
927
Содержание этанола
в водно-спиртовом растворе
Плот в процентах миллили-
ность rpaMMoB тpoBB100r
Р20 по ПО В 100 мл при взве
массе объему при 20 ос шивании
в воздухе
2 07 32 29 20
О 15 38 33 30
0,8278 23 43 37 38
6 31 49 42 48
4 38 55 47 58
2 46 61 52 68
О 54 67 56 78
0,8268 62 73 61 88
6 69 79 66 98
4 77 85 71 110,08
2 85 91 75 18
О 93 97 80 28
0,8258 87,00 91,03 85 38
6 08 09 89 48
4 16 15 94 58
2 24 20 98 66
О 31 26 72,03 76
0,8248 39 32 08 86
6 47 38 12 96
4 54 44 18 111,06
2 62 50 23 16
О 70 55 27 25
0,8238 78 61 32 35
6 85 67 36 45
4 93 73 41 55
2 88,01 79 45 65
О 08 85 50 75
0,8228 16 90 53 83
6 24 96 58 93
4 31 92,02 63 112,03
2 39 08 68 14
О 47 13 72 22
0,8218 54 19 76 32
6 62 25 81 42
4 69 30 85 51
2 77 36 90 61
О 85 42 94 71
0,8208 92 47 98 81
6 89,00 53 73,03 91
4 08 58 07 113,00
2 15 64 12 10
О 23 70 17 20
0,8198 30 75 20 29
6 38 81 25 39
4 45 87 30 49
2 53 92 34 58
О 60 98 39 68
0,8188 68 93,04 43 78
6 75 09 47 86
Содержаниеэтанола
в водно спиртовом растворе
Плот в процентах миллили
ность rpaMMoB тров в 100 r
Р20 по ПО В 100 мл при взве-
массе объему при 20 ос шивании
В воздухе
4 83 14 51 95
2 91 20 56 114,06
О 98 25 60 15
0,8178 90,06 31 65 25
6 13 36 69 33
4 21 42 73 44
2 28 47 77 53
О 35 53 82 63
0,8168 43 58 86 72
6 50 63 90 80
4 58 69 95 91
2 65 74 99 115,00
О 73 80 ! 74,03 10
0,8158 I 80 85 07 19
I I
6 i 88 91 12 30
4 95 96 16 38
, 2 91,03 94,02 21 49
О 10 07 25 58
0,8148 17 12 29 67
6 25 17 33 76
4 32 23 37 86
2 39 28 41 95
О 47 33 45 116,03
0,8138 54 38 49 12
6 61 43 53 21
4 69 49 58 32
2 76 54 62 41
О 83 59 66 50
0,8128 91 64 70 59
6 98 70 74 70
4 92,05 75 78 78 I
2 13 80 82 87
О 20 85 86 96
0,8118 27 91 91 117,07
6 35 96 95 I 16
1
4 42 95,01 99 25
2 49 06 75,03 ! 34
О 56 11 07 I 43
0,8108 64 16 11 52
6 71 21 15 61
4 78 26 19 70
2 85 31 ! 22 80
О 93 36 26 89
0,8098 93,00 I 41 I 30 98
6 07 46 I 34 118,07
4 14 52 39 18
2 22 57 43 27
О 29 62 47 36
0,8088 36 67 51 45
928
rосударственная фармакопея Республики Беларусь
Содержание этанола
в водно-спиртовом растворе
Плот- в процентах миллили-
ность rpaMMoB трав в 100 r
PZO по по в 100 мл при взве-
массе объему при 20 ос шивании
в воздухе
6 43 72 55 54
4 50 77 59 63
2 58 82 63 73
О 65 87 67 82
0,8078 72 92 71 91
6 79 97 75 119,00
4 86 ; 96,02 79 09
2 I 94 07 83 18
О 194,01 12 86 27
0,8068 08 16 90 35
6 15 21 94 44
4 22 26 98 53
2 29 31 76,02 63
О 36 36 05 72
0,8058 43 41 09 81
6 50 45 13 89
4 57 50 16 98
2 65 55 20 120,08
О i 72 , 60 24 17
i
0,8048 79 I 65 28 26
6 86 70 32 35
4 93 74 35 43
2 95,00 79 39 52
О 07 84 43 61
0,8038 14 89 47 70
6 21 94 51 80
4 28 99 55 89
2 35 97,03 58 97
О 42 08 62 121,06
0,8028 49 12 65 14
6 56 17 69 23
4 63 22 74 33
2 70 26 77 40
О 77 31 81 50
0,8018 84 35 85 58
6 91 40 88 67
4 98 44 92 75
2 96,04 49 96 84
О 11 54 77,00 94
0,8008 18 58 03 122,01
6 25 63 07 11
4 32 67 10 19
2 39 72 14 29
О 46 76 17 36
0,7998 52 81 21 46
6 59 86 25 55
4 66 90 28 63
2 73 95 32 72
О 80 99 35 80
Содержаниеэтанола
в водно--спиртовом растворе
Плот- в про центах миллили-
ность rpaMMoB трав в 100 r
PZO по ПО в 100 мл при взве-
массе объему при 20 ос шивании
в воздухе
0,7988 87 98,04 38 90
6 93 08 41 98
4 97,00 12 44 123,06
2 07 17 48 16
О 14 21 53 24
0,7978 20 25 56 332
6 27 29 59 40
4 34 34 63 50
2 41 38 66 58
О 47 42 69 66
0,7968 54 47 73 76
6 61 51 76 84
4 67 55 79 92
2 74 59 83 99
О 81 64 86 124,09
0,7958 88 68 90 17
6 94 72 93 25
4 98,01 77 97 35
2 08 81 78,00 43
О 14 85 03 51
0,7948 21 89 06 59
6 27 94 09 69
4 34 98 12 77
2 41 99,02 15 85
О 47 06 19 93
0,7938 54 10 22 125,02
6 60 14 25 10
4 67 18 28 18
2 74 22 31 26
О 80 26 34 34
0,7928 87 30 37 43
6 93 34 41 51
4 99,00 38 44 59
2 06 42 47 67
О 13 46 50 75
0,7918 19 50 53 84
6 26 54 56 92
4 32 58 60 126,01
2 38 62 63 09
О 45 66 66 17
0,7908 51 70 69 25
6 58 74 72 33
4 64 78 75 42
2 70 82 78 50
О 77 86 82 58
0,7898 83 89 84 64
6 89 93 87 72
4 96 97 90 81
0,78927 100,00 100,00 78,93 87
Таблица #5.5. 2
Количество (в 2раммах при температуре 20 ОС) воды и спирта разной концентрации, которые необходимо смешать, чтобы получить 1 К2 спирта
концентрации 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 % и 92 %
'"
Концентрация 30% 40% 50% 60% 70% 80% 90% 92%
взятоrо
спирта, % спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода
96 \ 262 738 355 645 452 548 555 445 665 335 783 217 913 87 941 59
95 266 734 360 640 459 541 564 436 675 325 795 205 927 73 955 45
94 270 730 366 634 466 534 572 428 686 314 807 193 941 59 970 ЗО
93 275 725 371 629 473 527 581 419 696 304 820 180 956 44 985 15
92 279 721 377 623 481 519 590 410 707 293 832 168 970 30
91 283 717 383 617 488 512 599 401 717 283 845 155 985 15
90 287 713 389 611 495 505 608 392 728 272 858 142
89 292 708 395 605 503 497 617 383 739 261 871 129
88 296 704 401 599 511 489 627 373 751 249 884 116
87 301 699 407 593 518 482 636 364 762 238 898 102
86 305 695 413 587 526 474 646 354 774 226 911 89
.
85 310 690 419 581 534 466 656 344 786 214 925 75
84 315 685 426 574 543 457 666 334 798 202 f . . 940 60
83 320 680 432 568 551 449 676 324 810 190 954 46
.
82 325 675 439 561 560 440 687 313 823 177 969 31
81 330 670 446 554 568 432 698 302 836 164 984 16
80 335 665 453 547 577 423 709 291 849 151
79 340 660 460 540 587 413 720 280 863 137
78 346 654 468 532 596 404 732 268 876 124
77 351 649 475 525 605 395 743 257 890 110
76 357 643 483 517 615 385 755 245 905 95
75 363 637 491 509 625 375 768 232 920 80
74 369 631 499 501 636 364 781 219 935 65
73 375 625 507 493 646 354 794 206 951 49
72 381 619 516 484 657 343 807 193 967 33
71 388 612 525 475 669 3З1 821 179 983 17
70 394 606 534 466 680 320 835 165
69 401 599 543 457 692 308 849 151
68 408 592 553 447 704 296 864 136
67 416 584 562 438 716 284 879 121
66 423 577 572 428 729 271 895 105
65 431 569 583 417 742 258 911 89
):,.
8
с\)
g
ф
:s::
ф
.g
с:
ос
с!>
с')
е
с!>
:3
Q)
gt
с:
J::
!!:
<D
N
<D
Концентрация 30% 40% 50% 60% 70% 80% 90% 92%
взятоrо
спирта, % спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода
64 438 562 593 407 756 244 928 72
. , _. -- - - . . .-
63 447 553 604 396 770 230 945 55 - -
I---- - - .- - - - --- - -- _._. ._ -- --
62 455 545 616 384 784 215 963 37 __ш_ - - --
_. - - --- - --- - - .. . f-----------
61 464 536 627 З73 799 201 981 19 ,
1 1f -- 1 - 1-85 - - _. .. -_. - -- - - -- - _. --- ---- - ---- -- -
60 472 528 815
- -
59 482 518 830 170
. - -
58 491 509 847 153
1---- - 1-------- - --- ------- . .
57 501 499 678 I 322 864 136 i
- - -
56 511 489 692 308 881 119
55 522 478 706 294 899 101
54 532 468 720 280 918 82 !
-- I
53 544 456 736 264 937 63
_ш
52 555 445 751 249 958 42
51 567 433 768 232 978 22
50 580 420 785 215 I --
-
49 593 407 803 197
48 607 393 821 179
47 621 379 840 160
46 636 364 860 140
45 651 349 881 119
44 667 333 902 98
43 684 316 925 75
42 701 299 949 51
41 720 280 974 26
40 739 261
39 759 241
. --
38 781 219
37 803 197
36 826 174
35 851 149
34 878 122 i
33 905 95
32 935 65
с.о
(.v
о
Q1
Q,
Q)
"ь
(")
:3
ф
CD
::t
::t
Q)
::а
-е-
Q)
"ь
s:
Q)
8
:::J
CD
::а
"1J
CD
(")
t::
"
t::
01
Q)
(")
t1"
Таблица #5.5. 3
Количество (в миллилитрах при температуре 20 ОС) воды и спирта разной концентрации, которые необходимо смешать, чтобы получить
1 л спирта концентрации 30 %. 35 %, 40 %, 45 %, 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %
Концен- 30% 35% 40% 45% 50% 55% 60% 65% 70% 75% 80% 85% 90%
трация
взятоro
спирта, спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода
% I
. , . .. .
i
95 316 707 З68 658 421 607 474 556 526 504 579 451 632 397 684 343 737 288 789 233 842 176 I 895 119 947 61
90 333 687 389 634 444 581 500 526 556 470 611 414 667 357 722 299 778 240 833 182 89 122 944 62
85 353 665 412 609 471 551 529 493 588 434 647 374 706 313 765 252 824 190 882 127 941 64
:: J::: 1:::f :J f . .
80 375 641 438 581 500 519 262 457 625 394 688 ЗЗО 67
417 rб67 . .
75 400 614 367 549 533 483 600 349 733 280
+ 1 ! . ..
70 429 584 500 514 571 443 643 371 714 298 786 225 857 150 929 J 76 i
. Ш
65 462 549 538 473 615 396 692 319 769 240 846 161 923 81
... ..
60 500 509 583 426 667 343 750 258 833 173 916 87
i
55 545 462 536 371 727 279 818 187 909 94 I
i !
I . .
I
50 600 405 700 305 800 204 900 103 I I
I
- ... . I
I , ,
I I
45 667 336 778 225 889 113 I I
+ I
. . ...
I I
40 750 252 875 126 I ....
,
35 857 143 I J
I
. -- _. .
:ь.
::з
а
(\)
g
ф
ф
t::
..с
ф
(')
е
ф
:3
Q)
t::
J::
!:?::
<о
w
Таблица #5.5,А
Количество (в миллилитрах при температуре 20 ОС) воды и спирта разной концентрации, которые необходимо смешать, чтобы получить 1 л
спирта концентрации 30 %, 35 %, 40 %, 45 %, 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 95 %
Концен- 30 % 35 % 40% 45% 50% 55% 60% 65% 70 % 75 % 80 % 85 % 90% 95 %
трация
взятоro - - . - - - - + -
спирта, % спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода спирт вода
- - I------- - , - - - >--------- - -
96,5 310,9 713,1 362,7 664,7 414,5 615,3 466.3 565,0 518,1 513,8 569,9 461,8 621.8 409,1 673,6 355,8 725.4 301.8 777,2 247,2 829,0 192,0 880,8 135,8 932,6 78,2 984,5 18,6
-
96,4 311,2 712,7 363,1 664,2 414.9 614,8 466,8 564,4 518,7 513,1 570,5 461,1 622,4 408,3 674,3 354.9 726.1 300.9 778.0 246.3 829.9 190.9 881.7 134,7 933,6 77,1 985,5 17,3
с------
96,3 311,5 712,3 363,4 663,8 415,4 614,3 467,3 563,8 519,2 512,5 571,1 460,4 623,1 407,6 675,0 354,1 726,9 300,0 778,8 245,3 830.7 189,9 882.7 133,6 934,6 75,9 986,5 16,1
96,2 311,9 712,0 363,8 663,3 415,8 613,7 467,8 563,2 519,8 511,8 571,7 459,7 623,7 406,8 675,7 353,2 727,7 299,1 779,6 244,3 831,6 188,8 883,6 132,4 935,6 74,7 987,5 14,9
"
96,-1- 312,2 711,6 364.2 662,9 416,2 613.2 468,2 562,6 520,3 511,2 572,3 458,9 624,3 406,0 676,4 352,4 728,4 298,2 780,4 243,3 832,5 187,8 884,5 131,3 936,5 73,6 988,6 13,6
96,0 312,5 711,2 364,6 662,4 416,7 612,7 468,8 562,0 520,8 510,5 572,9 458,2 625,0 405,2 677,1 351,5 729,2 297,2 781,3 242,4 833,3 186,8 885,4 130,2 937,5 72,4 989,6 12,4
--
95,9 312,8 710,8 365,0 662,0 417,1 612,2 469,2 561,5 521,4 509,9 573,5 457,5 625,7 404,4 677,8 350,7 729,9 296,3 782,1 241.4 834,2 185,7 886,3 129,1 938,5 71,2 990,6 11,2
95,8 313,2 710,4 365.3 661,5 417,5 611,7 469,7 560,9 521,9 509,2 574,1 456,8 626,3 403,7 678,5 349,8 730,7 295,4 782,9 240,4 835,1 184,7 887,3 128,0 939,5 70.0 991,6 9,9
95,7 313,5 710,0 365,7 661,1 418,0 611,1 470,2 560,3 522,5 508,6 574,7 456,1 627,0 402,9 679,2 349,0 731,5 294,5 783,7 239,4 835,9 183,6 888,2 126,9 940,4 68,9 992,7 8,7
95,6 313,8 709,6 366,1 660,6 418,4 610,6 470,7 559,7 523,0 507,9 575,3 455,4 267,6 402,1 679,9 348,2 732,2 293,6 784,5 238,5 836,8 182,6 889,1 125,8 941,4 67,7 993,7 7,5
95,5 314,1 709,2 366,5 660,1 418,8 610,1 471,2 559,1 523,6 507,3 575,9 454,7 628,3 401,3 680,6 347,3 733,0 292.7 785,3 237,5 837,7 181,6 890,1 124,7 942,4 66,5 994,8 6,2
95,4 314,5 708,8 366,9 659,7 419,3 609,6 471,7 558,5 524,1 506,6 576,5 453,9 628,9 400,5 681,3 346,5 733,7 291,8 786,2 236,5 838,6 180,5 891,0 123,6 943,4 65,4 995,8 5,0
95,3 314,8 708,4 367,3 659,2 419,7 609,1 472,2 558,0 524,7 506,0 577,1 453,2 629,6 399,7 682,1 345,6 734,5 290,9 787,0 235,5 839,5 179,5 891,9 122,5 944,4 64,2 996,8 3,7
95,2 315,1 708,0 367,6 658,8 420,2 608,5 472,7 557,4 525,2 505,3 577,7 452,5 630,3 399,0 682,8 344,8 735,3 290,0 787,8 234,5 840,3 178,4 892,9 121,4 945,4 63,0 997,9 2,5
95,1 315,5 707,6 368,0 658,3 420,6 608,0 473,2 556,8 525,8 504,7 578,3 451,8 630,9 398,2 683.5 343,9 736,1 289,0 788,6 233.6 841.2 177,4 893,8 120,3 946,4 61.8 998,9 1,3
CD
W
N
Q1
!l)
(")
:3
а>
(1)
::х:
::х:
!l)
::а
-е-
!l)
!l)
8
:::J
(1)
::а
(")
:::J
""
с:
е
01
!l)
'"
Таблица #5.5. 5
Таблица для получения спирта различной концентрации при 20 ос
-->
""
Крепость Желаемая концентрация разведенноrо спирта
разводимоrо
спирта 30% 35% 40% 45% 50% 55% 60% 65% 70% 75% 80% 85% 90%
(1000 объе-
мов), %
---- -
35 167
- --
40 335 144
45 505 290 127
50 674 436 255 114
55 845 583 384 229 103
60 1017 730 514 344 207 95
65 1189 878 644 460 311 190 88
- f-------- - -
70 1360 1027 774 577 417 285 175 81
75 1535 1177 906 694 523 382 264 16з_ -t_ 76
- ._- - _.
80 1709 1327 1039 812 630 480 353 246 , 153 72
-- - . - ." t- -
85 1884 1478 1172 932 738 578 443 329 231 144 68
90 2061 1630 1306 1052 847 677 535 414 310 218 138 65
95 2239 1785 1443 1174 957 779 629 501 391 295 209 133 64
Примечание: цифра в месте пересечения roризонтальной и вертикальной строк указывает объем воды при 20 ос. который спедует припить к 1000 объемам спирта имеющейся концентрации при
20 ос дЛЯ попучения разведения.
Таблица #5.5. 6
Количество (в миллилитрах при температуре 20 ОС) воды и спирта концентрации 96,6 % 97,O %. которые необходимо смешать, чтобы получить
1 л (при температуре 20 ОС) спирта концентрации 40 %, 70 %. 80 %. 90 %, 95 %
Концентрация 40% 70% 80% 90% 95%
взятоrо
спирта, % спирт вода спирт вода спирт вода спирт вода спирт вода
96,6 414,1 615.8 724,6 З02,7 828,2 193,0 931,7 79,4 983,4 19,8
96,7 413,7 616,3 723,9 303,6 827,3 194,0 930,7 80,6 982,4 21,0
96,8 413,2 616,8 723,1 304,5 826,5 195,7 929,7 81,7 981 ,4 22,3
96,9 412,8 617,4 722.4 305,4 825,6 196,1 928,8 82,9 980,4 23,5
97,0 412,4 617,9 721,7 306,3 824,7 197,1 927,8 84,0 979,4 24,7
}::.
::э
8
(\)
g
(!)
(!)
.g
с:
.с:
(!)
(')
:>;
с:
(!)
:3
Q)
с:
J:::
!l:
с.о
(J.)
(J.)
01/2013:50600
rосударственная фармакопея Республики Беларусь
934
5.6. КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
ИНТЕРФЕРОНОВ
Данная статья публикуется для информациu.
1. ВВЕДЕНИЕ
Частные статьи на интерфероны человека
описывают rлавным образом биоанализ, OCHOBaH
ный на инrибиторной активности интерферона в OT
ношении цитопатическоro действия вируса в куль
туре клеток Однако если статья описывает более
одноro подкласса интерферона, то в большинстве
случаев сам вирус, культура клеток и детали иссле
дования не указаны точно для обеспечения ДOCTa
точной rибкости.
Данный текст представляет собой OCHOB
ную информацию для исследователя относитель
но тоro, как планировать. оптимизировать и вали
дировать подобные количественные поределения
после Toro, как будет определена комбинация куль
туры клеток и цитопатическоro вируса. Подробная
процедура для цитопатическоro антивирусноro aHa
лиза приводится в качестве примера подходящеro
метода наряду с информацией по друrим комбина
циям вирус еточная линия и руководством по
адаnтированию и валидированию данной процеду
ры для аналоrичных комбинаций.
2. АНТИВИРУСНОЕ (СНИЖАЮЩЕЕ
ЦИТОПАТИЧЕСКИЙ ЭФФЕКТ)
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Антивирусное количественное определе
ние человеческих интерферонов основывается
на индукции t\Jlеrочной реакции в клетках чело
века. которая предотвращает или снижает цито
патический эффек-т инфекционною вируса. Сила
действия интерферона оценивается путем cpaB
нения ею защитною эффекта против вирусноro
цитопатическоrо эффекта с подобным эффек
том соответствующеrо препарата сравнения, Ka
либрованноro в Международных единицах.
3. ИССЛЕДОВАНИЕ ИНТЕРФЕРОНА
С ИСПОЛЬЗОВАНИЕМ КЛЕТОК
НЕР2С И ВИРУСА ИНФЕкционноrо
ЭНЦЕФАЛОМИОКАРДИТА
Данное антивирусное количественное опре
деление интерферонов человека описывает ци
топатический эффект редукционноrо типа. В нем
используются клетки человека Нер2с, инфици
рованные вирусом энцефаломиокардита (ЭМКВ,
EMCV), для определения эффективности разлlt1ч
ных экспериментальных препаратов человече
скою интерферона. Этот анализ использовался
в трех международных исследованиях под эrидой
Всемирной орrанизации здравоохранения (ВОЗ)
как вариант международных стандартов для че
ловеческоro интерферона альфа, человеческо
ro интерферона бета и человеческоro интерфе
рона raMMa и проявил себя как чувствительный,
надежный и воспроизводимый метод для оценки
эффективности различных типов человеческоro
интерферона.
Для культур клеток млекопитающих все про
цедуры выполняются с использованием стандарт
ных операционных процедур по поддержанию
клеточных линий в культуре. Объемы peareHТOB
обозначены для клеточных культур, выполня
емых в колбах вместимостью 75 см 2 . MOryT ис
пользоваться друrие типы контейнеров (колбы
или планшеты), но при этом необходимо COOTBeT
ствующим образом откорректировать объем.
3.1. ХРАНЕНИЕ И подrОТОВКА КЛЕТОК
НЕР2С.
Клетки Нер2с содержатся и пересеваются в
культуральной среде А.
Клетки хранятся в замороженном виде co
rласно стандартным операционным процеду
рам. Растущие клетки MOryT пересеваться в
культуре дО ЗО пассажей, после чеro новую куль
туру получают из замороженноro материала.
В начале количественноro определения из
колб берут клетки, образующие 90 % сплошной
монослой, используя трипсиновую обработку,
описанную ниже.
Из колб удаляют культуральную среду.
В каждую колбу прибавляют по 5 мл раст
вора трипсина, HarpeToro до 37 ос (исходный
раствор трипсина содержит 4 мr/мл трипсина Р
и 4 мr/мл натрия эдетата Р; непосредствен
но перед использованием раствор разводят в
50 раз фосфатным забуференным физиолоrи
чески м раствором). Закрытую пробкой колбу
взбалтывают для промывки клеточноro MOHO
слоя. Излишки трипсиновоrо раствора убирают.
Колбы выдерживают в течение 1 О мин при
температуре 37 ОС. Визуально или через микроскоп
наблюдают за клетками на предмет появления при
знаков разделения. При наблюдении в микроскоп
видно, как клетки окруrляются или разделяются и
свободно плавают. Энерrично встряхивают колбы
для разделения всех клеток, добавляют приблизи
тельно 5 мл культуральной среды А, еще раз встря
хивают для получения суспензии отдельных клеток
Для приrотовления суспензии для процеду
ры количественноro определения необходимо тща
тельно разделить клетки с помощью пипетки, чтобы
разрушить клеточные композиты. Подсчитывают
количество клеток и ресуспендируют их в культу
ральной среде до концентрации 6.105 клетокlмл.
3.2. ВОСПРОИЗВЕДЕНИЕ ВИРУСА
ЭНЦЕФАЛОМИОКАРДИТА
Вирус энцефаломиокардита воспроизводит
ся в культуре клеток мышей L 929. Клетки L 929
получают с использованием трипсинизации, как
это описывалось для клеток Нер2с (примечание:
при слабом росте клеток может возникнуть
необходимость замены неонатальной теля
5.6. Количественное определение интерферонов
935
чьей сыворотки на эмбриональную коровью cы
воротку).
Берут несколько колб со сплошным моносло
ем культуры клеток L 929. Удаляют среду из колб.
Добавляют 2 мл суспензии ЭМКВ, разбавленной в
кулыуральной среде В таким образом, чтобы она
содержала приблизительно 2,5'108 бляшкообразу
ющих единиц (БОЕ) на миллилитр. Каждая колба
должна содержать 4 '107 L 929 клеток и, таким
образом, инфекционность будет составлять около
10 БОЕ/клетка. Осторожно взбалтывают вирус
ную суспензию, распределяя по клеточному MOHO
слою, и ставят колбы в термостат приблизительно
на один час. Поддерживают рН среды от 7,4 до 7,8.
После адсорбции ЭМКВ прибавляют прибли
зительно 40 мл культуральной среды в каждую
колбу и возвращают колбы в инкубатор с темпе
оатурой 37 ос приблизительно на 30 ч. Поддержи
вают рН среды от 7,4 до 7,8 с целью получения
максимальною количества вируса. По окончании
инкубации удаляют культуральную Жl-'1дкость и
хранят при температуре приблизительно 4 .с
Колбы охлаждают до температуры 20 ОС.
чтобы заморозить клеточный монослой. Затем Ha
rревают до комнатной температуры. Добавляют
приблизительно 5 мл кулыуральной среды и встря
хивают колбу, чтобы разрушить стенки клеток. Co
держимое каждой колбы переносят в контейнер с
культуральной жидкостью. Затем кулыуральную
жидкость, содержащую ЭМКВ, переносят в 50 мл
пластиковые центрифужные пробирки и цeH
трифуrируют при ускорении 500 9 около 1 О мин.
Очищенную кулыуральную жидкость распреде
ляют по стеклянным контейнерам с закручива
ющейся пробкой в количествах 20 мл, 1 О мл, 5 мл,
1 мл, 0.5 мл или 0,2 мл, в соответствии с необхо
димостью. Хранят при 70 ОС. При необходимости
большие объемы можно размораживать и распре
делять по меньшим объемам и повторно замора
живать. При постоянном хранении приблизитель
но при температуре 70 ос ЭМКВ сохраняет свой
первоначальный титр, но повторяющиеся циклы
замораживания размораживания или хранение
при более высоких температурах, например, при
20 ОС, при водят к проrрессирующей потери титра.
3.3. МЕТОДИКА КОЛИЧЕСТВЕнноrо
ОПРЕДЕЛЕНИЯ
3.3.1. Определение уровня доза-ответ
При20товление растворов
Стандарт интерферона (например, специфиче.-
ский подтип стандарта ВОЗ интерферона) разводят
в 10 ти кратных разведениях кулыуральной среды
А, таким образом, чтобы доза была в диапазоне
100 ,001 МElмл. Процедура исследования BЫ
полняется в 9 ячеечном микротитровом планшете.
В Кa>lЩую ячейку добавляют 100 мкл кулыуральной
среды А Затем в КЭ>fЩYЮ ячейку, за исключением тех,
которые предназначены для вирусных контролей, до--
бавляют около 100 мкл Кa>lЩоro раствора препарата
сравнения, используя мноroканальные пипетки на
100 мкл. Содержимое ячеек перемешивают.
Распределение клеточной суспензии
Суспензию клеток Нер2с, содержащую при
близительно 6.105 клетокlмл клеточной культу
ры А, разливают в пластиковые чашки Петри.
Используя мноroканальную пипетку на 100 мкл,
переносят клеточную суспензию из чашек Петри
в каждую ячейку мноrотитровых планшетов. По
мещают планшеты приблизительно на 24 ч в ин
кубатор, установленный на 37 сс и содержащий
5 % уrлерода диоксида.
Вирусная инфекция
На этом этапе, используя микроскоп, про
веряют, чтобы монослои клеток Нер2с были
сплошными, имели правильную морфолоrию и
были живыми.
Удаляют большую часть культуральной
среды, перевернув планшет, встряхнув еro и
промокнув бумажным попотенцем (процедура
идентична той. что описывается ниже по yдa
пению жидкости из микротитровых планшетов).
Разводят ЭМКВ свежей культуральной средой А
до титра приблизительно 3.1n" БОЕ/мл (прuме
uaHue: в каждом планшете должно быть окопа
20 мл вирусной суспензии плюс от 5 % до 1 О %
сверх объема). Разведенную суспензию, получен
ную из 9 см стерильных чашек Петри, распреде
ля ют, используя мноroканальные пипетки, YCTa
новленные на 200 мкл, во все s:lчейки, включая
вирусные контроли, за исключением клеточных
контролей. Добавляют приблизительно 200 мкл
культуральной среды А без виоуса в каждую
ячейку с клеточным контролем.
Планшет помещают в термостат, YCTaHOB
ленный на 37 ос и содержащий 5 % уrлерода ди
оксида на 24 ч.
Окрашивание
Планшеты проверяют через микроскоп на
предмет тоro, что ЭМКВ вызвал цитопатический
эффект (ЦПЭ) в вирусных контролях. Временной
интервал для максимальноro ЦПЭ может варьи
ровать от одноro исследования к друюму вслед
ствие характерных изменений клеток Нер2с, BЫ
зываемых введением вируса в течение данноro
периода непрерывноro культивирования.
Большую часть культуральной среды yдa
ляют из ячеек в соответствующий дезинфициру
ющий раствор (например, натрия rипохлорит).
В каждую ячейку прибавляют фосфатный забу
ференный физиОЛ02ический раствор рН 7,4 Р.
Сливают фосфатный забуференный физиоло
2ический раствор рН 7.4 Р в дезинфицирующий
раствор. В каждую ячейку прибавляют 150 мкл
окрашивающею раствора. Клетки окрашивают в
течение 30 мин при комнатной температуре. Сли
вают окрашивающий раствор в дезинфицирую
щий раствор. Распределяют приБЛИ1ительно 150
мкл фиксирующею раствора. Фиксируют в тече
ние 1 О мин при комнатной температуре. Сливают
фиксирующий раствор в дезинфицирующий paCT
вор и промывают клеточный монослой, поrрузив
планшеты в пластиковые контейнеры с проточ
ной водой. Удаляют воду и промокают планшеты
936
rосударственная фармакопея Республики Беларусь
бумажными полотенцами. Высушивают планше
ты при температуре от 20 ос до 37 ос до тех пор.
пока не испарится вся влаrа.
В каждую ячейку добавляют 150 мкл 0,1 М
раствора натрия 2идроксида. Элюируют краску
аккуратным встряхиванием планшетов или по
стукиванием ладонью руки. Перед тем, как
снимать спектрометрические показания, He
обходимо убедиться, что краска равномерно
распределилась по всем ячейкам.
Снимают показания при длине волны от
61 О нм ДО 620 нм, используя считыватель ми
кротитровоrо планшета. а в качестве холостой
пробы ячейку или ряд ячеек, содержащих
около 150 мкл О, 1 М натрия 2идроксида и не co
держащих клеток.
Определяют концентрации интерферон
стандарта, которые вызывают максимальное
и минимальное снижение цитопатическоro эф
фекта. Это и есть дозовая зависимость, COOTBeT
ствующая рабочему интервалу количественноro
определения.
3.3.2. Методика количественноrо опреде
ления
Исследование проводят, как описано выше,
используя в качестве растворов:
испытуемый образец. разведенный двумя
объемами культуральной среды А для достиже
ния номинальных концентраций, охватывающих
рабочий диапазон количественноro определения;
соответствующий стандарт интерферона
(например, специфический подтип интерферо
на ВОЗ) в качестве растворов сравнения, разве
денный в двух объемах культуральной среды А
для достижения номинальных концентраций, ox
ватывающих рабочий диапазон количественноro
определения.
3.3.3. Анализ данных
Если функция представлена rрафически за
висимостью концентрации интерферона (ло
rарифм обратной величины разведения интер
ферона) от поrлощения красителя, результаты
определения снижения цитопатическоro эффек
та обычно соответствуют сиrмоидальному rpa
фику доза ответ.
Строят rраqpик зависимости концентрации
интерферона (лоrарифм обратной величины раз
ведения) от поrлощения красителя для CTaHдapT
ноro препарата интерферона и для испытуемых
растворов интерферона. Используя линейный
участок rрафика, рассчитывают концентрацию
интерферона в испытуемом образце путем cpaB
нения реакций для испытуемоro раствора и paCT
вора сравнения, используя обычные статистиче
ские методы для параЛJ1ельноro анализа.
4. ВАЛИДАЦИЯ друrих ПРОЦЕДУР
4.1. ВblБОР КЛЕТОЧНblХ культур
И ВИРУСА
В антивирусных исследованиях интерфе
рона использовался целый ряд друrих KOM
бинаций клеточных культур и вирусов. Ha
при мер, ЭМКВ использовался в комбинации
с эпителиальной клеточной культурой А549
карциномы леrких, вирус Semliki Forest или
вирус Sindbis использовался с фибробласта
ми человека, вирус клеточной культурой амни
она человека с клеточной линией W/SH или
почечной клеточной линией Маdiп Dа,Ьу круп
ноro poraToro скота. В каждом случае выбор
комбинации клеточная культура/вирус OCHO
вывается на том, какая из них дает наиболее
чувствительный ответ на препарат интерфе
рона, предназначенный для исследования, и
дает параллельные реакции при сравнении
экспериментальноro препарата и интерферо
HOBoro стандарта.
4.2. ВblБОР РЕАКЦИИ
Процедура окрашивания, описанная выше,
оценивает оставшиеся жизнеспособные клетки.
Кроме тоro, применялся целый ряд друrих peaK
ций, включая окрашивание метиленовым синим
или кристаллическим фиолетовым или преобра
зование тиазолиновоro синеrо (МТТ). В каждом
случае выбор метода основывался на получе
нии подходящих линейной и чувствительной
зависимостей между реакцией цвета и количе
ством жизнеспособных клеток.
4.3. СТАТИСТИЧЕСКОЕ ПОДТВЕРЖДЕНИЕ
ДОСТОВЕРНОСТИ
Как и в случае с параллельными биоанали
зами, данное исследование должно COOTBeTCTBO
вать обычным статистическим критериям линей
ности реакции, параллелизма и дисперсии.
4.4. ВАЛИДАЦИЯ ПЛАНИРОВАНИЯ
КОЛИЧЕСТВЕнноrо ОПРЕДЕЛЕНИЯ
Как и в случае с процедурой микротитровоro
планшета, необходимо уделять внимание пла
нированию исследования. В частности, необ
ходимо предусмотреть и исключить системные
ошибки в результате неслучайноro порядка пи
петирования и друrих факторов.
PEArEHTbI И КУЛЬТУРАЛЬНАЯ СРЕДА
Культуральная среда А (10 % неонаталь
ной телячьей сыворотки)
RPMI1640 культуральная среда, 450 мл
дополненная при необходимости
антибиотиками (пенициллин
10000 МЕ/мл; стрептомицин 10 нr/мл)
L rлутамин, 200 мМ, стерильный 5 мл
Неонатальная телячья сыворотка 50 мл
Культуральная среда В (2 % эмбриональ
ной коровьей сыворотки)
RPMI1640 культуральная среда, 490 мл
дополненная при необходимо
сти антибиотиками (пеницил
лин 1 О 000 МЕ/мл; стрептомицин
10 нr/мл)
5.7. Таблица физических характеристик радионуклидов
937
L rлутамин, 200 мМ, стерильный
Эмбриональная коровья
сыворотка
Окрашивающий раствор
Нафталин черный
Кислота уксусная ледяная
Натрия ацетат безводный
Вода
Фиксирующий раствор
Формальдеrид 40 %
Кислота уксусная ледяная
Натрия ацетат безводный
Вода
5 мл
10 мл
0,5 r
90 мл
8.2 r
до 1000 мл
100 мл
90 мл
8,2мл
ДО 1000 мл
01/2013:50700
5.7. ТАБЛИЦА ФИЗИЧЕСКИХ
ХАРАКТЕРИСТИК
РАДИОНУКЛИДОВ,
УПОМИНАЕМЫХ
В ФАРМАКОПЕЕ
Следующая таблица приведена в дополне
ние к общей статье Радиофармацевтические
препараты.
Данные взяты из базы данных Nаtiопа/ Nu
c/ear Data Cente, (NNDC) Brookhaven Nаtiопа/
Laboratory, Uрtоп. N. У., USA, а также разме
щены в Интернете по адресу: httр://www.ппdс.
Ьnl.gоv/ппdс/nиdаtlrаdfоrт. html.
В том случае, коrда был использован
друrой источник информации (более COBpe
менные значения), это указывается отдельно.
Друrие источники информации:
* DAMRI (Dераrtетепt des App/ications et
de la Metr%gie des Rауоnпетеnts /onisants,
СЕА Gif sur Yvette, France);
** РТВ (Physika/isch Тесhпisсhе Випdеs
ansta/t, Вrаипsсhwеig, Gеrтапу);
*** NPL (Nаtiопа/ Physica/ Laborato,y, Ted
dington, Midd/esex, ИК).
Отклонение в периоде полураспада
дается в круrлых скобках. Цифра в круrлых
скобках является стандартным отклонением
соответствующей последней цифры указан
HOro числовоro значения (<<Руководство по
выражению отклонения в измерениях», Меж
дународная орrанизация по стандартизации
(/SO), 1993, ISBN 92 67 10188 9).
В разделе используются следующие co
кращения:
е д возбужденные электроны;
се конверсионные электроны;
13" электроны;
13+ позитроны;
у rамма излучение;
Х peHTreHoBcKoe излучение.
Эмиссия электронов Эмиссия фотонов
Вероят- Вероят-
Период ность ность
Радио нуклид Энерrия, эмиссии, Энерrия, эмиссии,
полураспада Вид МэВ на 100 Вид МэВ на 100
распа- распа-
дов дов
Тритий (ЗН) *12,33 (6) roда *13" *0,006(1) *100
(макс: 0,019)
Уrлерод 11 (11С) 20,385 (20) мин 13+ 0,386(\) 99,8 у 0,511 199.5111)
(макс: 0,960)
Азот 13 (1ЗN) 9,965 (4) мин 13+ 0,492(1) 99,8 У 0,511 I 199,6(11)
(макс: 1,198)
Кислород 15 (150) 122,24 (16) с 13+ 0,735(1) 99,9 У 0,511 199,8(11)
(макс: 1,732)
Фтор 18 CBF) 109,77 (5) мин 13+ 0,250(1) 96,7 У 0,511 193,5(11)
(макс: 0,663)
Фосфор 32 (32Р) 14,26 (4) сут 13" 0,695(\) 100
(макс: 1,71)
Фосфор 33 (3ЗР) 25,34 (12) сут 13" 0,076(1) 100
(макс: 0,249)
Cepa 35 (35S) 87,51 (12)сут 0,049(1) 100
(макс: 0,167)
XpOM 51 (51Cr) 27,7025 (24) сут е д 0,004 67 Х 0,005 22,3
у 0,320 9,9
119.3,к 1712.
938
rосударственная фармакопея Республики Беларусь
Эмиссия электронов Эмиссия фотонов
Вероят- Вероят-
Период ность ность
Радионуклид Энерrия, эмиссии, Энерrия, эмиссии,
полураспада Вид МэВ на 100 Вид МэВ на 100
распа- распа-
дов ДО В
Кобальт 56 (56Со) 77,27 (3) сут е д 0,006 47 Х 0,00 ,007 25
I I I I I
р+ I 0,179(1) I 0,9 I у I 0,511 I 38,0(11)
0,631(1) 18,1 0,847 I 100
I 1,038 I 14,1
1,175 I 2,2
i 1,238 I 66,1
I I 1,360 I 4,3
I I
I I 1,771 I 15,5
I 2,015 I 3
I 2,035 I 7,8
I 2,598 I 17
I 3,202 I 3,1
3,253 7,6
Кобалы 57 (57Со) I 271,79 (9) сут е д +се 0,00 ,007 177,4 Х 0,006 0,007 j 57
I I I I I I
I
I се I 0,014 I 7,4 I у I 0,014 I 9,2 ,
I I 0,115 I 1,8 0,122 I 85,6
I
I I 0,129 I 1,3 0,136 I 10,7
I
0,692 0,15
Кобальт 58 (58Со) 70,89 (7) сут е д 0,006 I 49,4 Х 0,006........0,007 26,3
I I I I I
р+ I 0,201(1) I 14,9 I у I 0,511 I 29,9(11)
0,811 I 99,4
I
I 0,864 I 0,7
i
1,675 0,5
Кобальт 60 ( 5С Со) I 5,2714 (5) r p 0,096(1) 99,9 У 1,173 100
I (макс: 0,318) 1,333 100
I
rаллий 66 (6БGа) , 9,49 (7) ч е д 0,008 21 Х 0,009 0,010 19,1
I
I I I I I I
i
р+ I 0,157(1) I 1 I у I 0,511 I 112(11)
I 0,331(1) I 0,7 0,834 I 5,9
I 0,397(1) I 3,8 1,039 I 37
I 0,782(1) I 0,3 1,333 I 1,2
I 1,90(1) I 50 1,919 I 2,1
2,190 I 5,6
2,423 I 1,9
2,752 I 23,4
3,229 I 1,5
3,381 I 1,5
3,792 I 1,1
4,086 I 1,3
4,295 I 4,1
4,807 1,8
5.7. Таблица физических характеристик радионуклидов
939
Эмиссия электронов Эмиссия фотонов
Вероят Вероят
Период ность насть
Радионуклид полураспада Вид I Энерrия, эмиссии, Вид Энерrия, эмиссии,
I I МэВ на 100 МэВ на 100
i распа распа
алЛИЙ 7 ("Ga) I дов дов
! 3.2612 (6) сут е д I 0,008 62 Х o.o08 O,01O r 57
I I
rерманий 68
(6BGe) в paBHOBe
сии с rаллием 68
(68Ga)
r аллий 68 (БВGа)
i
I
I Крип тон 81 m
(81 "'Kr)
I
i I
се J 0,082 0,084 I 30,4 у
! 0,o90 O,092 I 3.6
I 0,175 I 0.3
I
I
270,82 (27) СУТ е д I 0,008 42,4 Х
I I i I
I 1 3 '! 0,353[1) I 1,2 ! у
(68Ga: 67,629 L ' i . 0,836';' I 88,0 I
(24) мин). I + ' I
67,629 (24) мин I e: n ,008 I 11 х
I I I I
I [3'! 0,353(1) I 1,2 I у
: 0.836(1) J 88 J
13,10 (3) с се' 0,17 X
: 0,189 I 4,6 I
i
I 1
Х
Рубидий 81 (81Ru) 4,576 (5) ч
в равновесии с
криптоном 81 m
(81mKr)
(81mKr: 13,10 (3) с)
Стронций 89 ( B9 Sr) 50,53 (7) СУТ
в равновесии В9mу: 16,06 (4) с
с иттрием 89m 1\
( В9 m У)
Стронций 90 ( 9 0S r ) 28,74 (4) r
в равновесии (90У: 64,10 (8) ч)
с иттрием 90m
(90mу)
Иттрий 90 (90У) 64,10 (8) ч
Молибден 99 65,94 (1) ч
(99Мо) в paBHOBe
сии с технецием
99т (99mТ с)
i (99mтс: 6,01 (1) ч)
е д I 0,011 31,3
I I
се I 0,176 I 25
I 0,188 I 4,3
[3+ I I
I 0,253(1) I 1,8
I 0,447(1) I 25,0
[3. 0,583(1) 99,99
(макс: 1,492)
[3. 0,196(1) 100
(макс: 0,546)
I
I у
I
I
I
I
[3. 0,934(1) 100
(макс: 2,280)
[3. 0,133(1) 16,4 Х
0,290(1) I 1,1 I
I 0,443(1) I 82,4 I
I
I
I
i
I
у
I I
I 0,091 .O,093 I 42,4
I 0,185 l ' 21,2
0,209 2,4 I
0,300 I 16,8
0,394 I 4,7
0,888 .
O.009 0.010 I 44,1 I
! I
I I .
, I
I 0,511 i 178,3
I I
I 1,077 I 3
I O,009 ,010 I 4,7
I I
I 0,511 I 178,3
i 1,077 l.
I 0,012 0,014 I 17 i
I I I
0,190 67,6 I
0,01 3 O,014 57,2
I I
I 0,190 I 64
I 0,446 I 23,2
I 0,457 I 3
I 0,510 I 5,3
I 0,511 l ' 54,2
0,538 2.2
0,909 I 0.01
I 0,018 .021 3,6
I I
I 0,041 I 1,1
;
I 0,141 I 4,5
I 0,181 I 6
I 0,366 I 1,2
I
0,740 I 12,1
0,778 4,3
940 rосударственная фармакопея Республики Беларусь
Эмиссия электронов Эмиссия фотонов
Вероят- Вероят-
Период ность ность
Радионуклид Энерrия, эмиссии, Энерrия, эмиссии,
полураспада Вид МэВ на 100 Вид МэВ на 100
распа- распа-
дов дов
Технеций 99m 6,01 (1) ч се 0,002 74 Х 0,018 ,021 7.3
(99mТ с) I I I I
е д I 0,015 2,1 I у I 0,141 I 89,1
I I I I
се I 0,12 I 9,4 I I I
0,137 0,140 1,3
Технеций 99 (99Тс) 2. 11 .1 05 r 13 0,085(1) 100
(макс: 0,294)
Рутений 103 39,26 (2) сут ед+се 0,017 12 Х 0,020 0,023 9
C0 3 Ru) в paBHO I I I I I
весии с родием
103т C0 3m Rh) се I 0,030 ,039 I 88,3 I у I 0,497 I 91
I I I I 0,610 I 5.8
13 I 0,031(1) I 6,6 I I I
с озm Rh: 56,114 0,064(1) 92,2
(20) мин)
Индий 110 C 1 Oln) 4,9 (1) ч е д 0,019 13,4 Х 0,o23 ,026 70,5
I I
у I 0,642 I 25,9
I 0,658 I 98,3
I 0,885 I 92,9
I 0,938 I 68,4
0,997 10,5
Индий 11 От 69,1 (5) мин е д 0,019 5,3 Х 0,023 ,026 27,8
(' 1Gm lп) I I
13+ 1,015(1) 61 у I 0,511 I 123,4(11)
I 0,658 I 97,8
2,129 2,1
Индий 111 ('11Iп) : 2,8047 (5) сут е д 0.019 0,003 6,9
I I I O,023 ,026 I 82,3
I се I 0,145 I у I I
I I О, 167 0, 171 I I 0,171 I 90,2
I
I 0,219 I I 0,245 I 94
0,241 245
Индий 114т 49,51 (1) сут се 0,162 0,023 0,027 36,3
С 14m lп) В paBHOBe I 0,186 0,190 I I I
сии с индием 114 I I "( I 0,190 I 15,6
С 14 1п) *13 I 0777(1) I 95 I 0,558 I 3,2
(MaK : 1,985)
С 14 1п: 71,9 (1) с) 0,725 3,2
Теллур 121 т 154,0 (7) СУТ е д 0,003 0,026 ,031 50,5
С 21m Те) В paBHO I I 0,022 0,023 I I I
весии с теллу I I I I 0,212 I 81,4
pOM 121 С 21 Те) у
I се I 0,050 I I 1,102 I 2,5
I I 0,077 I I I
('21Те: 19,16 (5) 0,180
сут)
5.7. Таблица физических характеристик радионуклидов 941
Эмиссия электронов Эмиссия фотонов
Вероят- Вероят-
Период ность ность
Радионуклид Энерrия, эмиссии, Энерrия, эмиссии,
полураспада Вид МэВ на 100 Вид МэВ на 100
распа- распа-
дов дов
Теллур 121 С 21 Те) **19,16 (5) сут е А 0,022 11,6 Х 0,026 0,030 75,6
I I I I I
i I i у I 0,470 I 1,4
1
I I I 0,508 I 17,7
0,573 80,3
Йод 123 С 23 1) 13,27 (8) ч е А 0,023 0,004 9,3
I I 0,027 0,031 I 86,6
се 0,127 13,6 I у I I
0,154 1,8 I I 0,159 I 83,3
0,158 0,4 I I 0,346 I 0,1
I I 0,440 I 0,4
I I 0.505 I 0,3
I I I 0,529 I 1,4
I
i 0,538 0,4
Йод 125 (1251) 59,402 (14) СУТ е +00 I о 004 0,004 15,5
А '
I I O,023 ,035 I 0,027 114
I I I 0,ОЗ1 26
I I I
0,035 6,7
Йод 126 (1261) 13,11 (5)сут е А 0,023 0,027 0,031 I 42,2
I I I
се 0,354 0,5 I у I 0,388 I 34
0,634 0,1 I I 0,491 I 2,9
I
I I 0,511 I 2,3(11)
p 0,109(1) 3,6 I I 0,666 I зз
0,290(1) 32,1 I I 0,754 I 4,2
0,459(1) 8,0 I I 0,880 0,8
I I 1,420 I 0,3
0,530(1)
Йод 131 С 3 Ч) 8,02070 (11) сут 0,46 0,029 0,030 3,9
I 0,330 I I
I у I 0,080 I 2,6
I p 0,069(1) 2,1 I 0,284 I 6,'1
I 0,097(1) 7,3 I 0,365 I 81,7
I 0,192(1) 89,9 I 0,637 I 7,2
0,723 1,8
KceHOH 131т 11,84 (7) сут 0,025 0,004 8,3
C 31m Xe) 0,030 44
се 0,129 0,034 10,2
0,159
0,163 0,164 2
Йод 133 С 33 1) (до 20,8 (1) ч 0,140(1) 0,530 87
радиоактивноro 0,162(1) 0,875 4,5
KceHOHa 1 33) 0,299(1) 1,298 2,4
0,441(1)
120 За. 1712
942
rосударственная фармакопея Республики Беларусь
Эмиссия электронов Эмиссия фотонов
I I Вероят- Вероят-
i
Период 1 ность ность
, Радионуклид I Энерrия, I эмиссии, Энерrия, эмиссии,
полураспада Вид I МэВ 1 на 100 Вид МэВ на 100
i распа распа-
дов дов
! 0,026 5,8 Х 0,004 6,3
KceHOH 133 С 3З Хе) I 5,243 (1) сут е д
1 I I I I
I I 0,031 40,3
I се I 0,045 I 55,1 I 0,035 I 9,4
I
I I
I KceHOH 133т
с ззт Хе) (до радио
активною KceHO
Ha 133)
I 0,075 0,080 I
I I
13 ' 0,101(1) I
е д 1. 0,025 I
I I
се I 0,199 I
I 0,228 I
0,232
13 0,140(1)
0,237(1)
0,307(1)
0,352(1)
0,399(1)
0,444(1)
0,529(1)
2.19(1)сут
Йод 135 (1351) (до
радиоактивноro
KceHOHa 135)
6,57 (2) ч
99,0
7
64,0
20,7
4,6
7,4
8
8,8
21,9
8
7,5
23,8
9,9
I
I у
I х
I
I
I
у
0,080 38,3
0,004 7,8
0,030 45,9
0,034 10,6
0,233 10
*0,527 13,8
0,547 7,2
0,837 6,7
1,039 8
1,132 22,7
1,260 28,9
1 ,458 8,7
1,678 9,6
1,791 7,8
KceHOH 135 СЗ 5 Хе) 9,14 (2) ч се 0,214 5,5 Х 0,031 0,035 5
I I I I I I
I 13 I 0,171 I 3,1 I у I 0,250 I 90,2
1
0,308 96,0 0,608 2,9
Цезий 1З7 C Ts) I 30,04 (3) r е д 0,026 0,8 Х 0,005 1
в равновесии с 1 I I I I I 0,032 0,036 I 7
барием 1 37т
С 37т Ва) I се I 0,624 I 8,0 I I I
I I 0,656 I 1,4 I у I 0,662 I 85,1
I I 13 I 0,174(1) I 94,4 I I I
I С'? Ва.2.552(1)мин) 0,416(1) 5,6
Таллий 200 (200ТI) 26.1 (1) ч се 0,285 3,4 Х 0,010 32
I 0,353 I 1,4 I I 0,069 0,071 I 63,3
I I I I 0,08 I 17,5
I 13+ 0,495(1) I 0,3 I I I
I I I у I 0,368 I 87,2
I I I I 0,579 I 13,8
I I I I 0,828 I 10.8
I I I I 1,206 I 29,9
I I I I 1,226 I 3,4
I I I I 1,274 I 3,3
I I I I 1,363 I 3,4
I I 1,515 4
5.9. Полиморфизм
943
I I
! Радионуклид I Период
I I полураспада
! J . .
'Свинец 201 ! 9,33 (3) ч
i (201рЬ) (до радио i
I активноro таллия 1
;201) i
I
I
i
I
I
I
I
,
I
!
.
!Таллий 201 (2О1Т1) 72,912 (17) ч
l
I Таллий 202 (2О2Т1) 12,23 (2) сут
I
i
Свинец 203 51,873 (9) ч
(2ОЗРЬ)
F Эмиссия электронов Эмиссия фотонов
I '1 Вероят . .. I Вероят-
ность I I ность
Энерrия, эмиссии, I Энерrия. i эмиссии,
I Вид I МэВ на 100 Вид I МэВ I на 100
I I распа I I распа-
h + 055 д; Х I 0.070=О .iJ7З + .Д6 В .
I t I I i 0,083 I 19
I се I 0,246 8,5 I i
I 0,276 2 1" 0,331 i 79
I 0,316 2,3 I 0,361 I 9,9
I 0,406 I 2
I 0,585 I 3,6
I i
0,69:' i 4,3
I 0.767 i 3,2
I 0.82 ! 2.4
i i !
I i 0,90& i 5,7
I I 0,946 7,9
: I 1,099 I 1,8 I
i 1,277 ' 1,6 I
cei 0.016=. ('! .:О17 j 17,7 х I 0,010 i 46.
: O,027.. {J,0291 4,1 I 0,069 O,071 I 73,7
i 0,052 I 7,2 I 0,080 I 20,4
i 0.084 I 15,4 I I
i i I I
I 0,153 I 2,6 ., i 0.135
i L . I .. i '
е А I 0,054 0,010
I ,
I I O,069 OД71
I I 0,080
0,440
0,010
I 0,071 0,073 I
I 0,083 I
у I 0,279 I
0,401
О:.бi
2,6
10
31
61,6
17,1
91,4
37
69,6
19,4
80,8
3,4
се
0.357
2,4
у
е А
0,055
3,0
се
0.194
13,3
111 Имеется в виду 13.спекр.
(11) Максимальная вероятность эмиссии, относящейся к суммарной анниrиляции в источнике на 100 распадов.
5.9. ПОЛИМОРФИЗМ
0112013:50900
в случае если это явление наблюдается у
химическоro элемента (например, серы), вместо
термина «полиморфизм» используется термин
«аллотропия» .
При описании сольватов (включая rидраты),
в которых растворитель присутствует в кристал
лической решетке в стехиометрической пропор
ЦИИ, используют термин «псевдополиморфизм»;
этот термин также может быть применен к Be
ществам, в которых растворитель присутству
ет внепостоянных пропорциях. Так как термин
Полиморфизм (или кристаллический поли
морфизм) это явление, характерное для TBep
дых веществ; это способность вещества в твердом
состоянии существовать в различных кристалли
ческих формах при одном и том же химическом co
ставе. Твердые вещества, находящиеся в некри
сталлической форме, называются аморфными.
944
rосударственная фармакопея Республики Беларусь
«псевдополиморфизм» допускает двоякое тол
кование в зависимости от обстоятельств, при
описании таких веществ предпочтительнее ис
пользовать только термины «сольваты» И «rи
драты».
В тех случаях, Korдa в частной статье YKa
зано, что субстанция обладает полиморфиз
мом, это означает, что наблюдается истинный
кристаллический полиморфизм, либо нали
чие сольватов, либо аллотропия либо наличие
аморфной формы.
Идентичность химическоro состава пред
полаrает, что все кристаллические и аморфные
формы данноro вещества проявляют одинако
вые химические свойства в растворах и распла
вах, однако в твердом состоянии их физико хими
ческие и физические свойства (растворимость,
твердость, сжимаемость, плотность, температу
ра плавления и др.). а соответственно и их pe
акционная способность и биодоступность, MOryT
быть различными.
Если соединение обладает полиморфиз
мом, форма. для которой значение свободной
энтальпии является самым низким при данных
температуре и давлении. будет наиболее TepMO
динамически стабильной, а остальные формы
будут находиться в так называемом MeTacтa
бильном состоянии. При нормальных темпера
туре и давлении метастабильная форма способ
на как оставаться в неизменном состоянии, так и
переходить в термодинамически более стабиль
ную форму.
Если имеются несколько кристаллических
форм, то одна из них, при заданных темпера
туре и давлении. термодинамически будет яв
ляться более стабильной. Такая кристалличе
ская форма способна составлять фазу, которая
может достиrать равновесия с друrими TBepды
ми, а также с жидкими и rазообразными фазами.
Если каждая из кристаллических форм яв
ляется наиболее стабильной в определен
ном интервале температур, переход из одной
формы в друrую является обратимым и называ
ется энантиотропным. Переход из одной фазы
в друrую является фазовым переходом первоro
рода, поэтому при заданном давлении это COCTO
яние будет определяться изменением темпера
туры. Если только одна из форм стабильна во
всем интервале температур, переход является
необратимым или монотропным.
Путем варьирования условий кристаллиза
ции(температура,давление, растворитель, KOH
центрация, скорость кристаллизации, внесение
затравок кристаллизации, присутствие и KOHцeH
трация примесей и др.) можно получить различ
ные кристаллические формы или сольваты.
Для изучения полиморфизма MorYT быть ис
пользованы следующие методы:
дифракция peHTreHoBcKoro излучения на
порошке (2.9.33);
дифракция peHTreHoBcKoro излучения на
отдельных кристаллах;
термический анализ (2.2.34) (дифферен
циальная сканирующая калориметрия, TepMO
rравиметрия, термомикроскопия);
микрокалориметрия;
анализ поrлощения влаrи;
оптическая и электронная микроскопия;
ядерный маrнитный резонанс в твердом
теле;
инфракрасная спектрофотометрия (2.2.24);
рамановская спектрометрия (2.2.48);
измерение растворимости и характеристи
ческой скорости растворения;
измерение плотности.
Эти методы часто дополняют друr друrа, по
этому при исследовании важно использовать He
сколько из них.
Диаrраммы давлениеlтемпература и
знерrия/температура, основанные на анали
тических данных, являются ценными инстру
ментами для более полноro понимания энер
rетических взаимодействий (энантиотропия,
монотропия) и термодинамической стабиль
ности отдельных модификаций полиморфноro
вещества.
Для исследований сольватов предпочти
тельны дифференциальная сканирующая кало
риметрия и термоrравиметрия, используемые
вместе с определением растворимости, xapaK
теристической скорости растворения и peHTre
ноrрафией.
При исследовании rидратов для обнаруже
ния зон относительной стабильности определя
ют изотермы сорбции/десорбции воды.
В общем случае rидраты меньше раствори
мы, чем безводные формы, подобно как и соль
ваты меньше растворимы в их растворителе,
чем несольватированные формы.
01/2013:51000
5.10. КОНТРОЛЬ ПРИМЕСЕЙ
В СУБСТАНЦИЯХ ДЛЯ
ФАРМАЦЕВТИЧЕскоrо
ИСПОЛЬЗОВАНИЯ
Введение
Частные статьи Фармакопеи на субстан
ции для фармацевтическоro использования
разработаны с целью обеспечения их при
емлемоro качества для потребителей. Роль
Фармакопеи в защите общественноro здоро
вья заключается в адекватном контроле при
месей, реrламентируемом частными фарма
копейными статьями. Требования к качеству
определяют с учетом научноro проrресса,
технической обеспеченности и реrуляторных
требований.
5.10. Контроль примесей в субстанциях
945
Требования, касающиеся примесей, даются
в частных статьях и в общей статье Субстанции
для фармацевтичеСК020 использования, KOTO
рые дополняют друr друrа: частные статьи опре
деля ют критерии приемпемости для примесей, а
общая статья указывает на необходимость KBa
лификации, идентификации и описания любых
орrанических примесей, которые присутствуют в
активных субстанциях (фармацевтических суб
станциях).
Пределы для учитывания, идентификации
и квалификации, содержащиеся в общей статье
Субстанции для фармацевтичеСК020 исполь
зования, применяются ко всем сопутствующим
примесям. Однако если частная статья не co
держит испытания по количественному опреде
лению сопутствующих примесей, любые новые
примеси, присутствующие выше установленных
пределов, Moryт быть не выявлены, так как тест
не позволяет их определять.
Требования раздела «Сопутствующие при
меси» общей статьи Субстанции для фарма
цевтичеСК020 использования, особенно Kaca
ющиеся предельноrо содержания примесей,
не применяются к вспомоrательным веще
ствам; также эти требования не распростра
няются на: биолоrические и биотехнолоrиче
ские продукты; пептиды; олиrонуклеотиды;
радиофармацевтические препараты; продукты
ферментации и полученные из них полусинте
тические продукты; лекарственные средства
растительноro происхождения инеочищенные
продукты животноro и растительноro проис
хождения. Несмотря на неприменимость норм
предельноro содержания примесей, YCTaHOB
ленных в общей статье, общая концепция опи
сания, идентификации (rдe возможно) и квали
фикации примесей для этих продуктов также
приемлема.
Основания ДЛЯ внесения уточнений
в частные статьи Фармакопеи
Частные статьи Фармакопеи разработаны
для субстанций, которые входят в состав лекар
ственных средств, зареrистрированных компе
тентным уполномоченным opraHoM Республи
ки Беларусь, поэтому эти статьи не обязательно
охватывают все субстанции для фармацевтиче
CKOro использования, представленные на миро
вом рынке.
Присутствие орrанических и неорrанических
примесей в субстанциях, качество которых KOH
тролируется соответствующим компетентным
уполномоченным opraHoM, оценивается на oc
новании данных о безопасности максимальноro
допустимоro содержания (максимальная суточ
ная доза), за исключением новых данных о безо
пасности, которые становятся действительными
при последующем доказательстве более низких
норм.
Основная часть частных статей Фармако
пеи на субстанции для фармацевтическоro ис
пользования rармонизирована с требованиями
частных статей Европейской Фармакопеи, KO
торые, в свою очередь, разрабатываются rруп
пами экспертов и рабочими rруппами, COTPYД
ничающими с европейскими национальными
фармакопейными орrанами, компетентными
орrанами по маркетинry, национальными KOH
трольными лабораториями и лабораторией EB
ропейской Фармакопеи при содействии исполь
зующих эти субстанции производителей и/или
поставщиков.
Контроль примесей в субстанциях для
фармацевтическоrо использования
Качество субстанции, связанное с coдep
жанием примесей, контролируется определен
ным числом испытаний, приведенных в част
ной статье. Эти испытания предназначены для
оценки как орrанических, так и неорrанических
примесей, которые важны с точки зрения спосо
бов получения активных субстанций, для лекар
ственных средств.
Контроль остаточных растворителей
предусматривается в общей статье Субстан
ции для фармацевтичеСК020 использования
и статье 5.4. Остаточные количества op2a
нических растворителей. Сертификат при
rодности частной статьи Фармакопеи для
субстанции, полученной определенным спо
собом, указывает контролируемые остаточ
ные растворители, установленные критерии
приемлемости и валидированый метод KOH
троля в случае, если он отличен от описанно
ro в общей статье 2.4.24. Идентификация и
контроль содержания остаточных pacтвo
рителей.
Испытание «Сопутствующие примеси»,
описанное в частных статьях для орrанических
веществ, проводится в отношении родственных
орrанических примесей. Если испытание «Co
путствующие примеси» не позволяет KOHTpO
лировать конкретную примесь или есть особые
причины (например, безопасность), требующие
специальноro контроля, то оно может допол
няться специфическими испытаниями.
Если в частной статье отсутствует испы
тание «Сопутствующие примеси» (или экви
валентное испытание), то использующая суб
станцию орrанизация должна. тем не менее,
rарантировать, что обеспечивается COOTBeT
ствующий контроль орrанических примесей.
При обнаружении орrанических примесей
выше предела идентификации они должны
быть идентифицированы (если это возмож
но), и, при их обнаружении выше предела
квалификации, квалифицированны (если это
не установлено ранее) (см. также «PeKOMeH
дации по использованию частных статей на
активные субстанции»).
Если частная статья применима для суб
станции с различными профилями примесей, то
она может иметь либо единственное испытание
946
rосуiJарственная фармакопея Республики Беларусь
для контроля всех сопутствующих примесеи.
упоминающихся в разделе «Примеси», ЛИL j H(;
сколько испытаний для контроля всех извесп ых
профилей примесей. Ссответствие может быть
установлено с ИСПОЛЬЗОt:3анием только Vlспыта
ний, относящихся к известным профилям при
месей, в зависимости от спос06а получения суб
станции.
Инструкции по контролю ПРviмесей MorYT
быть включены в раздел частной статьи <,Про
изводство», например, Korдa единственный aHa
литический метод. подхо.цящий для контроля
данной примеси, должен осуществляться Пр(J
изводителем, так как является технически СЛОЖ
ным для общеro использования, или Korдa метод
не может быть применен к субстанции в rOTOBOM
виде, и/или Korдa валидаL\ИЯ ПРОVlзводственноrо
процесса (включая этап очистки) будет обеспе
чивать достаточный контроль.
Раздел «Примесю> В частных статьях на
активные субстанции
Раздел частной статьи «Примеси» включа
ет примеси (с указанием структурной форму
лы И химическою назваН 1Я, если это возможно)
обычно орrаническоro происхождения, обнару
живаемые в испытании, описанном в частной
статье. Раздел основан на информации, доступ
ной на время разработки или пересмотра част
ной статьи, и не обязательно является исчерпы
вающим. Раздел включает специфицироваhные
и, rдe это указано, друrие обнаруживаемыe прVl
меси.
Специфицированные ПрLJмеси имеют крите"
рии приемлемости, не превышающие таковых,
утвержденных компетентным уполномоченным
opraHoM.
ДРУ2ие обнаруживаемые примеси по
тенциальные примеси с определенной CTPYКТY
рой, но С недоказанным обычным содержанием
выше предела идентификации в субстанциях,
используемых при изroтовлении лекарственных
средств. В разделе «Примеси» они приводятся
для информации.
Если пюбые друrие при меси, отличные от
специфицированных, обнаруживаются в актив
ной субстанции, ответственность за проверку
предположения о необходимости идентифика
ции/квалификации примесей в зависимости от
их содержания, природы, максимально допусти
мой суточной дозы и соответствующих предель
ных значений идентификации/квалификации в
соответствии с разделом «Сопутствующие при
меси» общей статьи Субстанции для фарма
цевтичеСК020 использования несет ИСПОJ1ЬЗУ
ющая эту субстанцию орrанизация.
Трактовка испытания по определению
сопутствующих примесей в частных статьях
на активные субстанции
Частные статьи на субстанции для фар
мацевтическоro использования следует читать
и трактовать, ориентируясь на общую статью
Субстанции для фармацевтичеСК020 исполь
зования.
В случае если указывается общий крите
рий приемлемости для примесей (<<любая друrая
примесь», «друrие примеси», «любая при
месь» ), эквивалентный номинальному coдep
жанию, больший чем соответствующий предел
идентификации (см. общую статью Субстанции
для фармацевтическосю использования), то он
применим только для специфицированных при
месей. указанных в разделе «При меси». Необ
ходимость идентификации (KOrдa это представ
ляется возможным), описания, спецификации и
квалификации друrих примесей, которые присут
с!вуют в субстанции, должна рассматриваться в
соответствии с требованиями общей статьи. OT
ветственность за обоснованность критериев при
емлемости для примесей, не указанных в разде
ле «Примеси», и для примесей, обозначенных
как «друrие обнаруживаемые примеси», несет
IIIспользующая эту субстанцию орrанизация.
Критерии приемлемости для испытания на
сопутствующие при меси представлены в раз
личных вариантах в существующих част ных CTa
тьях; схема ПРИНЯП-1Я решений (рисунок 5.1 O. 1)
может использоваться Kar< вспомоrательнu
средство в трактовке общих критериев при
емлемости и их отношения к разделу частной
статьи «Примеси».
Общие критерии приемлемости для
{{друrих» примесей в '-IаСТНbIХ статьях выража
k.JТСЯ различными способами: «любая друrая
примесь», «друrие примесю>, «любая примесь»,
«любое пятно», и т. д. Общий критерий приемле
мости может применяться либо только к опреде
ленным специфицированным примесям, либо к
неспецифицированным и определенным специ
фицированным примесям в зависимости от при
роды активной субстанции и приемлемоrо пре
дела идентификации. В случае обнаружения
двусмысленных формулировок в частных CTa
тьях до их переиздания можно использовать
схему принятия решений (рисунок 5.10. 1) для
определения критериев приемлемости.
Рекомендации по использованию част-
ных статей на активные субстанции
Частные статьи реrламентируют подхо
дящее качество субстанций с теми профиля
ми примесей, которые учитывались в процессе
разработки и/или пересмотра этих статей. OT
ветственность за проверку обеспечения част
ной статьей соответствующеro контроля при
месей в субстанции для фармацевтическою
использования с определенным способом по
лучения rлавным образом с помощью про
цедуры утверждения частной статьи Фарма
копеи несет использующая эту субстанцию
орrанизация.
Частная статья с испытанием «Сопутству
ющие при меси», основанным на количествен
5.10.
Контроль примесей в субстанциях
947
ном методе определения (таком как жидкостная
хроматоrрафия, rазовая хроматоrрафия и Ka
пиллярный электрофорез), обеспечивает COOT
ветствующий контроль примесей в субстанции
для фармацевтическоro использования с опре
деленным способом получения, если примеси,
присутствующие в количествах, превышающих
соответствующий предел идентификации, яв
ляются специфицированными примесями, YKa
занными в разделе «Примеси».
Если субстанция содержит примеси, отлич
ные от указанных в разделе «Примеси», необ
ходимо проверить возможность обнаружения
этих примесей при помощи методики, описан
ной в частной статье; в противном случае He
Применим ли раздел
«Сопутствующие примеси» статьи
Субстанции для фармацевтическоzо
использования*?
Имеется ли в частной статье
раздел «Примеси>>?
Нет
Применяется раздел «Сопутствующие примеси»
статьи Субстанции для фармацевтическоzо использования
обходимо разработать новую методику опре
деления и внести предложение опересмотре
частной статьи. В зависимости от найденноro
содержания и предложенных предельных зна
чений необходимо рассмотреть вопрос об иден
тификации и/или квалификации этих примесей,
Если испытание «Сопутствующие приме
си» применимо для различных профилей при
месей. то в сертификате анализа необходимо
указывать только при меси с известным про
филем для данноro способа получения суб
станции, за исключением случаев, Korдa про
изводитель лекарственных средств использует
активные субстанции с различными профилями
примесей.
r' '< '
I Общий критерий приемлемости применяется:
I < O всем неспецифицированным примесям;
..; . {'.;1ецифицированны'' примеся '. pOMe тех,
,'то имеют собственные специфические.
.Jдобренные в частной статье критерии
Да
()бщий критерий приемлемости при меняется:
ко всем неспецифицированным примесям;
... . к специфицированным примесям, кроме тех,
i что имеют собственные специфические,
I одобренные в частной статье критерии
l .
Да
I
.,сщий критерий приемлемости применяется:
I . К СПЕщифицированным примесям, кроме тех,
I ', -:J имеют собственные специфические,
! одобренные в частной статье критерии
I Для неспецифицированных примесей применяется
I
i раздел «Сопутствующие примеси» статьи Субстанции
; для фармацевтическоzо использования..
* Требования ЭТОЮ раздела применяются к активным субстанциям. за исключением биолоrических и биотехнолоrических продук,
тов, пептидов; олиrонуклеотидов; радиофармацевтических препаратов; продуктов ферментации и полученных из них полусинте.
тических продуктов; неочищенных продуктов животноro и растительною происхождения; лекарственных средств растительною
происхождения
*. К разделу «Сопутствующие примеси» общей статьи Субстанции для фармацевтичеСКО20 использования:
индивидуальный критерий приемлемости должен быть определен для любой примеси. которая может присутствовать в коли.
чествах выше предела идентификации;
любая примесь с критерием приемлемости выше предела идентификации должна быть идентифицирована, если ЭТО возможно;
любая примесь с критерием приемлемости выше предела квалификации должна быть квалифицирована.
Рисунок 5.1 O. 1. Схема принятия решений для трактовки общих критериев приемлемости
для «друrих» примесей в частных статьях
948
rосударственная фармакопея Республики Беларусь
Идентификация примесей (соотнесение
пиков)
Если для при меси в частной статье YKa
зано индивидуальное предельное значение,
то обычно описывается способ ее идентифи
кации, например, с использованием CTaHдapT
ноro образца, образца xpoMaTorpaMMbI или
относительноrо удерживания. Орrанизации,
использующие субстанции для производства
лекарственных средств, MorYT дополнитель
но идентифицировать примеси, отличные от
идентифицируемых в частной статье. Так как
Фармакопея не предусматривает использова
ние для этих целей стандартных образцов, об
разцов xpoMaTorpaMM или информации об OT
носительном удерживании, кроме указанных в
частной статье, такие орrанизации должны ис
пользовать доступные научные подходы для
идентификации примесей, отличных от иден
тифицируемых в частной статье.
Новые примеси/специфицированные
при меси с содержанием выше специфициро
BaHHoro предела
В случае если новый производственный
процесс или изменение в утвержденном про
цессе ведет к возникновению новой примеси,
необходимо выполнить условия общей статьи
Субстанции для фармацевтичеСК020 исполь
зованuя относительно идентификации и квали
фикации, а также проверить приrодность част
ной статьи для контроля такой примеси. Если
методика определения примесей, описанная в
частной статье, не подходит для COOTBeTCTBY
ющеrо контроля такой примеси, необходимо
разработать новую методику, указать критерий
приемлемости и подать запрос на пересмотр
частной статьи.
В случае если новый производственный
процесс или изменение в утвержденном про
цессе ведет к увеличению содержания специ
фицированной примеси выше заданноro пре
дела, необходимо выполнить условия общей
статьи Субстанции для фармацевтuчеСК020
использования относительно квалификации.
Выражение критериев приемлемости
Критерии приемлемости по содержанию co
путствующих примесей в частных статьях Bыpa
жаются либо в виде сравнения площадей пиков
(сравнительные испытания), либо в виде число
вых значений.
Хроматоrрафические методы
Общая статья 2.2.46. Хромат02рафические
методы разделения касается различных J3спек
тов контроля примесей.
На сайте ЕDОМ (www.pheu,.org) доступна
информация о торroвых названиях колонок, pe
активов и оборудования, которые рекомендуют
ся для использования при усовершенствовании
частных статей.
rЛОССАРИЙ
Друrие обнаруживаемые при меси по
тенциальные примеси с определенной CTPYКТY
рой, обнаруживаемые испытанием, описанным
в частной статье, но с недоказанным обычным
содержанием выше предела идентификации в
субстанциях, используемых при изroтовлении
лекарственных средств. Такие примеси явля
ются неспецифицированными и оrраничивают
ся общим критерием приемлемости.
Идентифицируемая примесь любая
примесь с установленной химической CTPYK
турой.
Квалификация процесс получения
и оценки данных, на которых основывается
биолоrическая безопасность индивидуаль
ной примеси или данноro профиля примесей
при заданных (специфицированных) преде
лах.
Неидентифицируемая примесь любая
примесь, для которой структурная формула не
установлена и которая определяется исключи
тельно по качественным аналитическим xapaK
теристикам (например, относительное удержи
вание).
Неспецифицированная примесь
любая примесь, содержание которой оrраниче
но общим критерием приемлемости и не имеет
собственноro специальноro критерия приемле
мости.
Неучитываемый предел в xpoMaTorpa
фических тестах незначительное содержание,
при котором или ниже котороro пики/сиrналы
не учитываются при расчете суммы примесей.
Численное значение неучитываемоrо предела
обычно совпадает со значением учитываемоro
предела.
Номинальная концентрация KOHцeH
трация, которая рассчитана на основании KOH
центрации предписанноro вещества сравнения
с учетом предписанноro поправочноro коэффи
циента.
Потенциальная примесь любая при
месь, которая теоретически может возник
нуть в ходе производственноrо процесса или
при хранении. Она может присутствовать
либо отсутствовать в субстанции. Если из
вестно, что потенциальная примесь опреде
ляется испытаниями, описанными в частной
статье, но ее постоянное присутствие в суб
станциях, используемых при изroтовлении
лекарственных средств, не доказано, то она
включается в раздел «Примеси» В подраздел
«Друrие обнаруживаемые при меси» для ин
формации.
5.11. Раздел «Описание (Свойства)>> в частНblХ статьях
949
Предел идентификации предел.
выше KOTOpOro примесь должна быть иденти
фицирована.
Предел квалификации предел, выше
котороro примеси должны быть квалифициро
ваны.
Примесь любой компонент субстан
ции для фармацевтическоro использования,
химическая структура KOTOpOro отличается от
химической структуры субстанции.
Сопутствующие при меси заrоловок,
используемый в частных статьях для общих
испытаний по контролю орrанических при
месей.
Специфицированная примесь
любая примесь, содержание которой в част
ных статьях индивидуально оrраничено
специальными критериями приемлемости.
Специфицированная примесь может быть
как идентифицируемой, так и неидентифи
цируемой.
Учитываемый предел предел, выше
котороro примеси должны быть отражены в
отчете об испытании.
01/2013:51100
5.11. РАЗДЕЛ «ОПИСАНИЕ
(СВОЙСТВА)>> В ЧАСТНЫХ
СТАТЬЯХ
В разделе «Общие сведения» указано, что
информация раздела «Описание (Свойства)>>
носит рекомендательный характер. В качестве
информации ниже приведены методики опреде
ления rиrроскопичности, кристалличности и pac
творимости.
rиrроскопичность
Методика используется для субстанций,
которые выдерживают испытание «Потеря В
массе при высушивании» или «Вода», указан
ные в частной статье. Она позволяет опреде
лить степень rиrроскопичности.
В предварительно взвешенный (т 1 ) CTe
клянный сосуд (внешний диаметр 50 мм,
высота 15 мм) с подходящей проб кой поме
щают испытуемый образец в количестве, YKa
занном в испытании «Потеря В массе при BЫ
сушивании» или «Вода», и взвешивают (т 2 ).
Открытый сосуд помещают в эксикатор при
температуре 25 "С, содержащий насыщен
ный раствор аммония хлорида или аммония
сульфата, либо в климатическую камеру при
температуре (25:1:1) "С и относительной влаж
ности (80:1:2) %. Выдерживают в течение 24
ч. Сосуд укупоривают пробкой и взвешивают
(тз).
Увеличение в массе в процентах рассчи
тывают-по формуле:
тз т2 .100.
т2 т1
Полученные результаты трактуют следу
ющим образом:
расплывается на воздухе: абсорбирует
достаточное количество воды для образования
жидкости;
очень 2иzроскопичен: увеличение в массе
равно или более 15 %;
2U2роскопичен: увеличение в массе менее
15 % и равно или более 2 %;
сле2ка 2иzроскопичен: увеличение в массе
менее 2 % и равно или более 0,2 %.
КРИСТАЛЛИЧЕСКИЕ СВОЙСТВА
Методика используется для выявления
кристаллической природы испытуемоro об
разца.
Несколько кристаллов испытуемоro образ
ца в минеральном масле помещают на чистое
предметное стекло и просматривают с исполь
зованием поляризационноrо микроскопа. Кри
сталлические частицы проявляют двойное
лучепреломление и зоны поrлощения при Bpa
щении предметноro столика микроскопа.
РАСТВОРИМОСТЬ
Для проведения испытания используют не
более 111 Mr испытуемоro образца (для каждо
ro растворителя) и не более 30 мл каждоro pac
творителя.
Процесс растворения
Энерrично встряхивают в течение 1 мин и
выдерживают при температуре (25,0:1:0,5) "С
в течение 15 мин. Если испытуемый обра
зец не полностью растворился, повторяют
встряхивание в течение 1 мин и выдержива
ют при температуре (25,0:1:0,5) ос в течение
15 мин.
Методика
100 Mr тонко измельченноro (90) испы
TyeMoro образца помещают в пробирку (BHY
тренний диаметр 16 мм, длина 160 мм)
с пробкой, прибавляют 0,1 мл раство!)ителя
и проводят процесс растворения, как описано
выше. Если испытуемый образец полностью
растворился, он считается очень ле2КО pac
творимым.
Если испытуемый образец растворил
ся не полностью, прибавляют 0,9 мл paCTBO
950
rосударственная фармакопея Республики Беларусь
рителя и проводят процесс растворения, как
описано выше. Если испытуемый об dзец
полностью растворился, он считается леGКО
растворимым.
Если испытуемый образец растворился
не полностью, прибавляют 2,0 мл раствори
теля и проводят процесс растворения. Если
испытуемый образец ПОЛt-ЮСТЬЮ растворился,
он считается растворимым
Если испытуемый образец растворил.
ся не полностью, прибавляют 7,0 мл paC'TBO
рителя и проводят процесс растворения, как
описано выше. Если испытуемый образец
полностью растворился, он считается YMe
peHHo pacтвopиMЫM .
Если испытуемый образец растворился
не полностью, 1 О Mr тонко измельченноrо (90)
испытуемоro образца помещают в пробирку
с пробкой, прибавляют 10.0 мл растворителя
и проводят процесс растворения, как описа
но выше. Если испытуемый образец полно
стью растворился. он считается малораство
римым.
Если испытуемый образец растворился
не полностью, 1 Mr тонко измельченноrо (90)
испытуемоro образца помещают в пробирку с
пробкой, прибавляют 10,0 мл растворителя и
проводят процесс растворения, как описано
выше. Если испытуемый образец полностью
растворился, он считается очень мало рас-
творимым.
01/2013:51200
5.12. СТАНДАРТНЫЕ ОБРАЗЦЫ
Статья приводится для информации.
1. ВВЕДЕНИЕ
Термин «стандартный образец» приме
няется в данной статье как обобщенное по
нятие, включающее стандартные вещества,
стандартные препараты и эталонные спек
тры.
Стандартные образцы необходимы для
проведения адекватноrо контроля качества
субстанций для фармацевтическоro исполь
зования и лекарственных средств.
Стандартные образцы утверждаются в
установленном порядке, а их приroдность для
последующеro применения проверяется co
rласно предписанной проrрамме. Необходи
мость использования стандартноrо образца
предусматривается в фармакопейной статье
или нормативном документе по контролю Ka
чества. Если в частной или общей статье
указан стандартный образец Европейской
Фармакопеи #(или друrой подходящий CTaH
дартный образец)#, то только он принимается
за официальный стандарт, т. е. единственно
надежный в спорных случаях эталон.
Ниже даны определения стандартным Ma
териалам и аттестованным стандартным Ma
териалам.
В нескольких частях данной статьи при
водится подробная информация о химиче
ских стандартных образцах. Общие принципы
для биолоrических стандартных препаратов
приводятся ниже, однако подробная инфор
мация по применению, нормированию и про
rpaMMaM пере контроля не включается в связи
с различной природой и зачастую сложностью
сравнения с химическими стандартными об
разцами. Для стандартных образцов пепти
дов и белков в некоторых случаях требуется
оtобый подход, особенно для количествен
Horo определения; в данной статье такая ин
формация не приводится.
2. ТЕрминолоrия
Первичный стандартный образец. CTaH
дартный образец, обладающий подходящими
свойствами для целенаправленноro использо
вания, проверка приroдности котороro осущест
вляется без сравнения с существующим CTaH
дартным образцом.
Вторичный стандартный образец. CTaH
дартный образец, утвержденный путем cpaBHe
ния с первичным стандартным образцом.
Международный стандартный образец.
Международный стандартный образец это
первичный стандартный образец, который опре
деляет Международную Единицу. Эквивалент
ность международноro стандартноro образца
в Международных Единицах устанавливается
Всемирной орrанизацией здравоохранения.
Стандартный образец Европейской Фар
макопеи (Еиroреап Pharтacopoeia ,еfе,епсе
staпda,d). Стандартный образец, утвержденный
и одобренный Европейской Фармакопейной Ko
миссией.
Фармакопейный стандартный образец
(ФСО, chemica/ ,еfе,епсе sиЬstапсе CRS).
Вещество или смесь веществ, предназначен
ные для использования, как указано в частной
или общей статье Фармакопеи. Фармакопейные
стандартные образцы это первичные CTaH
дартные образцы. Исключение составляют фар
макопейные стандартные образцы (особенно
антибиотики), активность которых выражается в
Международных Единицах. Такие образцы явля
ются вторичными стандартными образцами, co
измеримыми с международными стандартными
образцами.
БиОЛ02ический стандартный препарат
(БСП, bi%gica/ rеfеrепсе рrера,аtiоп BRP).
Вещество или смесь веществ, предназначен
ные для использования, как указано в частной
или общей статье Фармакопеи. Биолоrические
стандартные препараты это либо вторич
5.12. Стандартные образцы
951
ные стандартные образцы, активность KOTO
рых выражается в Международных Единицах,
либо первичные стандартные образцы, KOTO
рые MorYT использоваться для определения EB
ропейской Фармакопейной Единицы. MoryT ис
пользоваться и друrие подходящие единицы
измерения, например, вирусный титр или число
бактерий.
Стандартный материал (СМ, rеfеrепсе
materia/ RM). Материал или вещество, одно
или более свойств котороro в достаточной мере
однородны и установлены, чтобы использовать
ся для оценки прибора, методики измерения или
материалов.
Аттестованный стандартный материал
(АСМ, certified ,efe,eпce mate'ia/). Стандартный
материал, сопровождаемый сертификатом каче
ства, одно или более свойств котороro aттeCTY
ются процедурой, устанавливающей их просле
живаемость по отношению к ТОЧНОМУ значению
единицы, в которой это свойство выражается, и
для котороro каждое значение, указанное в cep
тификате, сопровождается неопределенностью
с установленным уровнем достоверности.
Примечание: фармакопейные cтaHдapт
ные образцы необходимо отличать от cтaH
дартных материалов и аттестованных
стандартных материалов, которые М02ут иc
пользоваться в некоторых случаях в различных
аналитических методиках для оценки количе
ственных результатов. Применение cтaHдapт
ных материалов необходимо или рекомендовано
в частных или общих статьях Фармакопеи в oc
новном для калибровки и контроля удовлетво
рительной эффективности приборов.
Специфичность фармакопейных стандарт
ных образцов была официально признана во
введении Руководства ИСО 34 Общие тpe
бования к компетентности производителей
стандартных материалов (Второй выпуск,
2000): «Фармакопейные стандартные образ
цы и вещества создаются и распространяются
уполномоченными орrанами фармакопеи. co
блюдающими основные принципы данноro PYKcr
водства. Однако следует отметить, что для сооб
щения пользователю информации сертификата
анализа образца и сроки rодности уполномочен
ными орrанами фармакопеи используются раз
личные подходы. Неопределенность указанных
значений не приводится, так как она является
незначительной в сравнении с установленными
пределами метод специфических количествен
ных определений, предписанных фармакопеей,
для которых они предназначены».
3. ИСПОЛЬЗОВАНИЕ СТАНДАРТНЫХ
ОБРАЗЦОВ
Стандартные образцы используются для
идентификации, в испытаниях на чистоту и
при количественном определении субстанций
для фармацевтическоro использования и ле
карственных средств. Стандартные образцы
должны соответствовать непосредственному
предназначению; они не обязательно должны
быть приrодны для друrих целей. Если CTaHдapT
ные образцы используются для целей, отличных
от тех, для которых они были созданы, их при
roдность должна быть установлена. Установлен
ное значение любоro показателя CTaHдapTHO
ro образца приroдно только для предписанною
использования и не является обязательным для
друrих целей.
Стандартные образцы с установленным co
держанием/активностью для количественно
ro определения фармацевтической субстанции
MOryT быть приrодны для количественноro опре
деления содержания данной субстанции в ле.-
карственном средстве при соблюдении следу
ющих условий:
применим хроматоrрафический метод KO
личественноro определения фармацевтической
субстанции, описанный в частной статье;
подтверждена применимость данноro
метода для конкретноro лекарственноro cpeд
ства (отсутствие взаимодействий и друrих Me
шающих влияний);
любая пробоподroтовка образца (напри
мер, экстракция) валидирована для конкретноro
лекарственноro средства;
использование стандартных образцов ут
верждено компетентным уполномоченным opra
ном.
Стандартные образцы используются для
определения содержания компонентов в лекар
ственном растительном сырье и лекарствен
ных средствах растительноro происхождения.
Это MOryT быть: непосредственно активные Be
щества; маркеры, используемые для количе
ственноro определения; экстракты. Создание
стандартных образцов, представляющих собой
экстракты, осуществляется с использованием
образцов активных веществ или маркеров с из
вестными характеристиками.
Как правило, стандартные образцы по
ставляются в соответствующих количествах
для немедленноrо использования после
вскрытия контейнера. За использование CTaH
дартных образцов в друrих условиях ответ
ственность несет аналитик. Если закрытый
контейнер хранится в рекомендуемых услови
ях, он остается приroдным в течение срока rод
ности данной серии. Например, информация о
номерах серий стандартных образцов EBpO
пейской Фармакопеи имеется в каталоrе EBpo
пейскоro управления по качеству лекарствен
ных средств и здравоохранению при Совете
Европы (Europeaп Di,ecto,ate for the Quality о'
Меdiсiпеs (Couпcil о' Europe). ХРАнение pac
творов стандартных образцов не peKOMeHДY
ется за исключением случаев, коrда предва
рительно подтверждена их приrодность для
использования.
Вторичные стандартные образцы. BTO
ричные стандартные образцы MoryT быть ис
952
rосударственная фармакопея Республики Беларусь
пользованы для рутинноro контроля качества во
всех случаях, описанных выше для перви .-Iныx
стандартных образцов, при условии их утверж
дения по первичному стандартному образц
Вторичные стандартные образцы предназначе
ны для сокращения использования первичных
стандартных образцов, требующих более тща
тельноro описания и оценки и доступных в orpa
ниченном количестве. Вторичные стандартные
образцы используются в тех же целях, что и пер
вичные стандартные образцы, по которым они
утверждены.
4. СОЗДАНИЕ СТАНДАРТНЫХ ОБРАЗЦОВ
4 1. ПЕРВИЧНЫЙ СТАНДАРТНЫЙ
ОБРА3ЕЦ
Для подтверждения приroдности использо
вания веществ или препаратов в качестве пер
вичных стандартных образцов используются
различные аналитические методики.
Для фармацевтических субстанций и их
примесей обычно при меняются COOTBeTCTBY
ющие части следующей проrраммы испытаний.
Описание вещества (структурная xapaK
теристика) с помощью подходящих химических
атрибутов, таких как структурная формула, эм
пирическая формула и молекулярная масса.
При этом MOryт быть использованы различные
методы, включая:
спектрометрию ядерноro маrнитноro
резонанса;
масс спектрометрию;
инфракрасную спектрометрию;
элементный анализ.
Определение чистоты:
определение содержания орrаниче
ских примесей подходящим методом разделе
ния или спектрометрическим методом;
определение содержания воды;
определение содержания остаточных
растворителей;
определение потери в массе при BЫ
сушивании, которое может в некоторых случаях
заменить определение воды и остаточных paCT
ворителей;
определение неорrанических при
месей (тяжелые металлы, сульфатная зола,
атомная спектрометрия, спектрометрия индук
тивно связанной плазмы, рентrеновская флу
оресценция); результаты не используются при
количественном определении устанавлива
емоro содержания основноro компонента, за
исключением значительноro влияния на это
содержание;
определение чистоты абсолютным
методом (например, дифференциальная CKa
нирующая калориметрия или фазовая pac
творимость; результаты этих определений ис
пользуются для подтверждения результатов,
полученных С использованием методов разде
ления; они не используются при количественном
определении устанавливаемоro содержания oc
новноro компонента).
Для использования первичных химических
стандартных образцов в количественном анали
зе устанавливаемое содержание основноro KOM
понента обычно рассчитывают исходя из зна
чений, полученных при определении примесей
(орrанические при меси, неорrанические приме
си, вода и остаточные растворители), используя
принцип материальноro баланса; MOryT исполь
зоваться и друrие приroдные методы.
Отчет о создании стандартноro образца ro
товится и утверждается уполномоченным лицом.
4 2. СТАНДАРТНЫЕ ОБРА3ЦЫ
ЕВРОПЕЙСКОЙ ФАРМАКОПЕИ
Испытуемые стандартные образцы про
веряются с использованием большоro количе
ства аналитических методов. Рамки испытаний
и число привлеченных лабораторий зависит от
предназначения стандартноro образца. Если не
установлено иноro, обычно необходимо соответ
ствие частной статье.
При проведении совместноro лабораторно
ro испытания в ходе установления физико хи
мических характеристик стандартноro образца,
каждым участником должен быть представлен
протокол испытания. Только обоснованные pe
зультаты, указанные в протоколах, используют
ся для установления количественноro содержа
ния основноro компонента или для какоro либо
друrоro подтверждения приroдности.
Для химических стандартных образцов
обычно используются соответствующие части
приведенной ниже проrраммы.
4 2 1. ПОДЛИННОСТЬ (идентификация).
Серия, отобранная при обычном производстве
субстанции, является удовлетворительной при
использовании в качестве стандартноro образ
ца. Она должна выдерживать требования част
ной статьи. Для первой серии про водится полное
структурное описание.
4 2 2. Испытание на сопутствующие при
меси. Стандартный образец, соответствующий
примеси, характеризуется показателями «под
линность» И «чистота». В случае использова
ния стандартноro образца для определения co
держания заданной при меси предпочтительно,
чтобы содержание этой примеси в стандартном
образце составляло не менее 95,0 %; если пре
делы не указываются, содержание принимается
за 100,0 %; такое приближенное значение допу
стимо, если оно не будет оказывать существен
ноro влияния на определение примесей. Если
такое минимальное содержание не может быть
получено, используют заявленное процентное
содержание примеси в стандартном образце.
Если примесь недоступна в достаточном
для получения стандартноro образца количе
стве, используются следующие подходы:
5.12. Стандартные образцы
953
приroтовление стандартноro образца, KO
торый содержит смесь основноro компонента
(компонентов) и примеси или примесей;
приroтовление стандартноro образца, co
держащеro смесь специфических примесей.
В случае если такая смесь используется и
для определения содержания данной при меси,
содержание при меси в стандартном образце
устанавливается при помощи стандартных Meтo
дов разделения, а полученное значение припи
сывается стандартному образцу.
4 2 3. Количественное определение
4 2 3 1. Химический метод количествен
НО20 определения. Если стандартный образец
используют для количественноro определения
фармацевтической субстанции или вспомоrа
тельноro вещества, рамки испытания TaKoro
стандартноro образца расширяются. В общем
случае несколько при влеченных и совместно
работающих лабораторий испытывают пред
ложенную субстанцию и выдают детализи
рованный протокол испытаний. Полученные
результаты используют для расчета YCTaHaB
ливаемоrо содержания. В случае использо
вания селективноro метода количественно
ro определения особенно важно определить
содержание примесей в стандартном образ
це; при этом лучше всеro использовать дo
полнительные аналитические научно обосно
ванные методы, включая, rдe это возможно,
абсолютные методы определения #(абсолют
ный метод основан на использовании только
фундаментальных физических постоянных и/
или универсальных величин (молярная масса,
атомное число, стехиометрические коэффи
циенты»#.
Если стандартные образцы требуются для
методов количественною определения, не
включающих хроматоrрафию (например, коло
риметрии или спектрофотометрии в ультрафи
олетовой области), то должна быть проверена
относительная реакционная способность или OT
носительное поrлощение примесей, присутству
ющих в стандартизируемой субстанции, чтобы
удостовериться, что эти характеристики значи
тельно не отличаются от таковых для OCHoBHoro
компонента. Протокол испытаний обычно coдep
жит следующие показатели:
определение содержания воды (или
потеря в массе при высушивании);
определение содержания орrанических
примесей (включая остаточные растворители) с
использованием предписанных методов разде
ления;
по возможности, определение процентно
ro содержания основною компонента с исполь
зованием абсолютноro метода; это подтвержда
ющее определение не обязательно выполняется
всеми участниками, и полученные результаты не
будут использоваться для расчета устанавлива
eMoro содержания.
Протокол также содержит испытания при
roдности системы и критерии приемлемости для
каждою из выполненных испытаний.
Если не указано иное, установленное coдep
жание для субстанции или препарата принима
ется таким, как указано на контейнере (<<as is»
«как есты», и стандартный образец не требует
высушивания перед использованием. Для CTaH
дартных образцов, использующихся при количе
ственном определении и приютовленных путем
лиофилизации, содержание чистой субстанции
указывается в миллиrраммах или в Междуна
родных Единицах на контейнер.
4 2 3 2. МикробиОЛ02ический метод KO
личествеНН020 определения. Прежде всеro,
стандартный образец для микробиолоrическо
ro метода количественноro определения про
веряют на соответствие требованиям частной
статьи. В случае получения удовлетворитель
ных результатов Пр080ДИТСЯ совместное (с
при влечением нескольких участников) коли
чественное определение микробиолоrическим
методом с использованием международноrо
стандартноrо образца. Результат выражается
в Международных Единицах. Если отсутствует
международный стандартный образец, исполь
зуются Европейские Фармакопейные Единицы.
Установленная активность рассчитывается,
исходя из результатов объединенноro испыта
ния. Применяются различные валидационные
критерии соrласно обычным статистическим
методикам (5.3). Установленная активность с
доверительным интервалом рассчитывается
на основании статистически достоверных pe
зультатов.
4 2 З З. Количественное определение
компонентов в лекарственном раститель
ном сырье и в лекарственных средствах pac
титеЛЬН020 происхождения. Рамки испыта
ний стандартных образцов, используемых в
частных статьях на лекарственные средства
растительноrо происхождения, варьируют
в зависимости от типа этих стандартных об
разцов.
Активные вещества или маркеры oцe
ниваются по показателям подлинности и чи
стоты; значение количественноrо содержания
устанавливается независимо от чистоты.
Экстракт используется в качестве CTaH
дартноro образца, если отсутствует ДOCTa
точное количество соответствующеro актив
HOro вещества или маркера. Установленное
содержание для экстракта рассчитывают по
результатам количественноrо определения,
полученным при совместном определении
разными лабораториями, с использованием
образца активноro вещества или маркера с
известными характеристиками.
4 2A. Отчет о создании cTaHAapTHoro
образца. Подrотовленный отчет о создании
стандартноro образца содержит результаты
954
rосударственная фармакопея Республики Беларусь
исследований, полученные при еro раз:,абот
ке, а также информацию об ИСПОЛЬЗОВ-JНИИ
стандартноro образца. В отчете по химичес.<о
му методу количественноro определения CTaH
дартноro образца указывают установленное
содержание с ero лоrическим обоснованием.
Рассчитывают неопределенность установлен
ноro содержания и. если эта неопределен
ность меньше предписаннои величины, KOTO
рая обоснованно является незначительной по
отношению к критериям приемлемости для
количественноrо определения. то результаты
испытаний принимают. В противном случае
MorYT быть проведены повторные испытания
(целиком или частично) либо расширены пре
делы количественноro содержания фарма
цевтической субстанции. Неопределенность
полученноro установленноrо содержания не
приводится как часть информации, предо-'
ставляемой со стандартным образцом, так как
точность метода и неопределенность YCTaHOB
ленноro содержания для стандартноro образ
ца учитываются при установлении предела
(пределов) в частной статье.
4 3. ВТОРИЧНЫЙ СТАНДАРТНЫЙ
ОБРА3ЕЦ
Вторичный стандартный образец должен
проявлять те же значимые для испытания (ис
пытаний) свойства что и первичный CTaHдapT
ный образец. Объем испытаний вторичною
стандартноro образца не такой большой, как
требуется при создании первичноrо CTaHдapTHO
ro образца. Вторичный стандартный образец YT
верждается путем сравнения с первичным CTaH
дартным образцом.
Идентификация
Для использования в абсорбционной
спектрофотометрии в инфракрасной области:
полосы поrлощения соответствуют по располо
жению и относительной величине полосам по
rлощения первичноrо стандартноro образца.
Для использования в методах разделения:
расстояние, пройденное веществом (тонкослой
ная хроматоrрафия, электрофорез), время ми
rрации вещества (капиллярный электрофорез)
и время удерживания (rазовая или жидкостная
хроматоrрафия) для вторичноro стандартною
образца соответствуют таковым для первичноrо
стандартноro вещества.
Испытание на чистоту. Для использования
в методах разделения: требования такие же, как
для идентификации, но при использовании для
количественных расчетов должно быть YCTaHOB
лено содержание, соответствующее аналитиче
скому сиrналу, полученному при использовании
первичноro стандарта.
Количественное определение. ДЛЯ BTO
ричных стандартных образцов количественное
определение проводят относительно первичных
стандартных образцов с установленным coдep
жанием или активностью. Свойство, величину
KOToporo необходимо установить для вторично
ro стандартноro образца, должно быть близким
по значению этому же свойству первичноro CTaH
дартноro образца, с которым сравнивается BTO
ричный стандартный образец. Количество неза
висимых повторных определений, так же как и
применяемые критерии приемлемости, заранее
определены.
5. ПРОИЗВОДСТВО, МАРКИРОВКА,
ХРАНЕНИЕ И РАСПРОСТРАНЕНИЕ
5 1. ПРОИЗВОДСТВО
Все производственные операции осущест
вляются в соответствии с существующими HOp
мами надлежащей практики для тоro, чтобы ra
рантировать прослеживаемость и сохранность
стандартноro образца. Производственная дo
кументация включает информацию относитель
но упаковки, маркировки и хранения. Для обе
спечения сохранности стандартных образцов их
помещают в контейнеры при соблюдении COOT
ветствующих условий наполнения и укупорки.
Применяемые для этих целей контейнеры MOryT
быть одноразовоro или мноroразовоro ИСПОЛЬЗО
вания; при этом для уменьшения риска разложе
ния. контаминации или попадания воды более
предпочтительны одноразовые контейнеры.
5 2. МАРКИРОВКА
Маркировка включает название CTaHдapTHO
ro образца, название поставщика, номер серии,
а также любую друrую информацию, необходи
мую для правильноro использования стандарт
ноro образца. Если стандартный образец ис
пользуется для количественноro определения,
дополнительно указывают:
установленное процентное содержание;
либо концентрацию химическоro вещества
в миллиrраммах или миллилитрах на контейнер;
либо установленную активность (для био
лоrическоrо или микробиолоrическоrо метода
количественноro определения) в единицах aK
тивности на миллиrрамм или на контейнер.
Маркировка стандартных образцов coдep
жит дату пере контроля или дату истечения
срока roдности. Для стандартных образцов
Европейской Фармакопеи дату переконтроля
или дату истечения срока rодности не приво
дят, так как в плане переконтроля (см. ниже)
предусмотрен контроль непрерывной приrод
ности стандартных образцов для использова
ния.
Пояснительный листок (вкладыш). Дo
полнительно может предоставляться сопрово
дительный пояснительный листок (вкладыш),
содержащий информацию, необходимую
для правильноrо использования CTaHдapTHO
5.14. Лекарственные средства для zенной терапии
955
ro образца. Пояснительный листок считается
частью маркировки. Если это указано в част
ной статье, пояснительный листок содержит
xpoMaтorpaMMY.
5 3. ХРАНЕНИЕ И РАСПРОСТРАНЕНИЕ
Стандартные образцы должны храниться и
распространяться в условиях, rарантирующих
наилучшую стабильность.
Стандартные образцы Европейской
Фармакопеи. Стандартные образцы Европей
ской Фармакопеи rлавным образом хранятся
в помещениях с контролируемым темпера
турным режимом при температуре (5:t3) ОС.
Однако ряд стандартных образцов, которые
являются относительно нестабильными, xpa
нятся при температуре ( 20:t5) ОС; или, в He
которых случаях (например, препараты живых
вирусов), при температуре ( 80:t10) ОС; кле
точные культуры хранятся в жидком азоте (при
температуре 180 ОС).
ДЛЯ минимизации риска повреждения при
транспортировке используют специальную упа
ковку.
Стандартные образцы, которые обычно
хранятся при температуре (5:t3) ОС, отправля
ются обычной почтой, так как KpaTKOBpeMeH
ное отклонение от температурных условий дол
roвременноro хранения для таких образцов не
опасно. Стандартные образцы, хранящиеся при
температуре 20 ОС, упаковываются в контейне
ры со льдом и отправляются экспресс-почтой.
Стандартные образцы, хранящиеся при темпе
ратуре 80 ос или в жидком азоте, упаковывают
ся в контейнеры с «сухим льдом» и отправляют
ся экспресс почтой.
6. ПЛАН ПЕРЕКОНТРОЛЯ
Система переконтроля разработана и BЫ
полняется для rарантирования непрерывной
приroдности стандартных образцов для исполь
зования. Обычно в плане переконтроля учиты
вают известные физико химические свойства и
данные по стабильности стандартною образца.
Стандартные образцы периодически проверяют
на стабильность при хранении. Проrрамма мони
торинrа призвана обнаруживать любые призна
ки разложения на ранней стадии с использова
нием соответствующих аналитических методик.
Используемые методы должны быть выбраны из
уже использованных в процессе создания стан-
дартных образцов.
Периодичность и полнота пере контроля
стандартных образцов зависят от ряда следу
ющих факторов:
стабильность;
контейнер и система укупоривания;
условия хранения;
rиrроскопичность;
физическое состояние;
предполаrаемое использование;
однократное или мноroкратное исполь
зование.
Стандартные образцы обычно пред
ставляют собой порошки, в некоторых слу
чаях растворы. Предпочтительно, чтобы
стандартные образцы были представлены в
контейнере для однократноro использования.
Если стандартные образцы представлены в
контейнерах для мноroкратноro использова-
ния, переконтроль rиrроскопичных или чув
ствительных к кислороду веществ может про
водиться чаще. Методы испытаний включают
определение воды и продуктов распада (если
они известны). Период переконтроля может
быть увеличен при наличии обоснованных
данных. Максимально допустимое отклоне
ние от установленноro содержания должно
быть заранее определено: в случае ero пре
вышения серию необходимо перепроверить
или заменить.
Стандартные образцы Европейской
Фармакопеи. Проrрамма мониторинrа EBpO
пейскоrо управления по качеству лекарствен
ных средств и здравоохранению при Совете
Европы (ЕООМ) включает набор следующих
испытаний, отличающихся быстротой, чув
ствительностью и применимостью для малых
количеств:
определение воды, потеря в массе при
высушивании и/или термоrравиметрический
анализ;
оценка содержания примесей с исполь
зованием методов разделения;
в некоторых случаях определение
молярной чистоты с помощью дифференци
альной сканирующей калориметрии;
применение друrих специфических ис
пытаний для определения примесей.
Любые обнаруженные значимые разли
чия, выявленные последним испытанием,
ведут к более тщательному изучению серии
и, в случае необходимости, к созданию новой
серии.
01/2013:51400
5.14. ЛЕКАРСТВЕННЫЕ
СРЕДСТВД ДЛЯ
rЕННОЙ ТЕРДПИИ
ДЛЯ МЕдицинскоrо
ПРИМЕНЕНИЯ
Данная общая статья приводится для иH
формации.
Настоящая общая статья содержит ряд
текстов по лекарственным средствам для
956
rосударственная фармакопея Республики Беларусь
2енной терапии для медициНСК020 применения,
которые формируют набор требований, ПI:;tJ.\1е
няемых для производства и контроля качества
таких продуктов. Применение этих требований
и необходимость использования любых дРУ2их
текстов для конкретН020 лекарствеНН020 cpeд
ства определяются компетентным уполномо
ченным Ор2аном. Положения настоящей статьи
не исключают возможность использования аль
тернативных способов производства и Meтo
дов контроля при условии С02ласования их KOM
петентным уполномоченным Ор2аном.
Более подробные рекомендации по ле
карственным средствам для 2енной тepa
пии для медициНСК020 применения приведены
в Рекомендациях руководства по качеству,
доклинических и клинических аспектах ле
карственных средств для 2енной терапии
(Note for Gиidапсе оп the Оиаlitу, Р,есliпiса/
апd Clinica/ Aspects о' Gепе Тrапsfе, Меdiсiпа/
Products (CPMP/BWP/3088/99)) и Руководстве
по разработке и производству лентиви
ральных векторов (Guideliпe оп Dеvе/ортепt
апd Мапиfасturе о' Leпtivira/ Vectors (СНМР/
BWP/2458/03)) Комитета по лекарственным
средствам для медициНСК020 применения
(включая все последующие пересмотры этих
документов).
ОПРЕДЕЛЕНИЕ
В рамках настоящей общей статьи лекар
ственное средство для rенной терапии означа
ет продукт, полученный с помощью ряда про
изводственных процессов и предназначенный
для доставки производимоro in vivo или ех vivo
профилактическоro, диаrностическоrо или Te
рапевтическоro reHa (например, набора HY
клеиновых кислот) в клетки человека и ero
последующей экспрессии iп vivo. Перенос reHa
включает систему экпрессии, называемую
вектором, который имеет вирусное или He
вирусное происхождение. Вектор также может
быть включен в клетку человека или животных.
Рекомбинантые векторы, в частности
вирусные векторы и плазм иды. Рекомбинант
ные векторы вводятся или непосредственно в
орrанизм больноro (пере нос reHa iп vivo), или
в клетки хозяина с последующим введением
этих rенетически модифицированных клеток
больному (перенос reHa ех vivo). Вирусные
векторы получают из различных вирусов (Ha
при мер, аденовирусов, поксвирусов, ретрови
русов, аденоассоциированных вирусов, лен
тивирусов, rерпесвирусов). Эти векторы MorYT
существовать в репликативной, нерепликатив
ной или условно репликативной формах. Плаз
мидные векторы содержат нуклеиновые кис
лоты в чистом виде (например, «roлой» ДНК)
или в виде комплекса с друrими молекулами
(синтетические векторы, например, лип ИДЫ
или полимеры). rенетический материал, пе
реносимый лекарственными средствами для
rенной терапии, состоит из последовательно
сти нуклеотидов, которая может представлять
продукт с кодирующим reHoM, антисмысловые
транскрипты или рибозимы. Олиroнуклеотиды,
полученные методом химическоro синтеза, не
входят в область применения данноro общеro
раздела. После переноса rенетический MaTe
риал может находиться или в цитоплазме или
эписоме, или встраиваться в reHoM клетки ре
ципиента. в зависимости от способности BeK
тора интеrрироваться в reHoM.
lенетически модифицированные клетки.
rенетически модифицированные эукаритиче
ские или бактериальные клетки модифициру
ются с помощью векторов для экспрессии Tpe
буемоrо продукта.
ПРОИЗВОДСТВО
Вещества, используемые в производ-
стве. Исходные материалы, используемые в
ходе производственноro процесса, включая по
лучение посевноro вирусноro материала и соз
дание банка клеток, если применимо, подле
жат контролю. Если не установлено иное, все
используемые вещества должны производить
ся В условиях признанной системы обеспече
ния качества с использованием COOTBeTCTBY
ющих промышленных мощностей. Имеются
спецификации, содержащие как минимум ис
пытание на подлинность, определение биоло
rической активности (если применимо), испыта
ния на чистоту и безопасность в отношении
микробиолоrической чистоты и контаминации
бактериальными эндотоксинами. В производ
стве должна использоваться вода COOTBeTCTBY
ющеrо качества, отвечающая требованиям co
ответствующих частных фармакопейных статей
Вода очищенная, Вода высокоочищенная, Вода
для инъекций. Использующаяся в производ
стве сыворотка крупноro poraToro скота должна
удовлетворять требованиям частной фармако
пей ной статьи Сыворотка КРУПН020 p02aтo
20 скота. По возможности использование aH
тибиотиков в процессе производства должно
быть исключено.
Вирусная безопасность. Применяются
требования статьи 5.1.7.
Трансмиссивные ryбчатые энцефалопа-
тии (5.2.8). Проводится оценка риска продукта
в отношении трансмиссивных ryбчатых энцефа
лопатий, и предпринимаются соответствующие
меры для минимизации такоro риска.
Рекомбинантные векторы
ПРОИЗВОДСТВО
ОБЩИЕ ПОЛОЖЕНИЯ
Производство вирусных векторов основано
на системах банка клеток и вирусноro посевно
5.14. Лекарственные средства для 2енной терапии
957
ro материала. В производстве плазмидных BeK
торов используется система банка бактериаль
ных клеток.
Должно быть продемонстрировано, что BЫ
бранный метод производства обеспечивает по
лучение вектора требуемою качества. Если
иное не доказано и не утверждено, вектор в ro
товом продукте подверrается такому количеству
пассажей или пересевам из rлавноro посевноro
материала, как и вектор, чья эффективность и
безопасность была установлена в клинических
исследованиях.
КЛЕТКИ ПРОДУЦЕНТbI
ДЛЯ КУЛЬТИВИРОВАНИЯ ВЕКТОРА
Используемые в производстве клетки про
дуценты должны УДО6Злетворять COOTBeTCTBY
ющим требованиям Фармакопеи (5.2.2, 5.2.3, и
подразделу «Бактериальные клетки, использу
емые в производстве плазмидных векторов для
медицинскоro применения» данной статьи).
ХАРАКТЕРИСТИКА ВЕКТОРА
Все данные по конструированию вектора
документируются, включая происхождение BeK
тора и последующие манипуляции с ним, в част
ности, удаление или модификация участка BeK
тора.
Вектор характеризуется с помощью приroд
ных для этою валидированных методик. reHe
тическая стабильность вектора оценивается co
ответствующими методиками при максимально
допустимом или меньшем количестве пассажей
или на максимальном количестве удвоенных
клеток клеточной линии, используемой при про
изводстве вектора.
КУЛЬТИВИРОВАНИЕ И СБОР БИОМАССbI
Все манипуляции с банками клеток и
полученными культурами клеток проводятся
в зоне, свободной от одновременной работы с
друrими клетками или векторами. Все матери
алы человека или животною происхождения,
используемые при подroтовке клеточных куль
тур, и питательные среды подлежат KOHTpO
лю. Чистота биомассы оценивается с помощью
предназначенных для этоro испытаний, указан
ных в соответствующих специальных разделах.
ОЧИЩЕННАЯ БИОМАССА
Серия очищенноro рекомбинантноrо BeK
тора (вирусною вектора, «юлой» ДНК или KOM
плексной плазм иды) является нефасованной
(<<bu/k») активной субстанцией.
rОТОВblЙ ПРОДУКТ
Если иное не обосновано и не утвержде
но, розлив и распределение активной суб
станции проводятся в асептических условиях
в стерильные контейнеры (3.2). Стабильность
rOToBoro продукта оценивается по протоколам
оценки стабильности, включающим длитель
ность И условия хранения, количество серий,
подлежащих оценке, rрафик испытаний и ис
пользуемые методики количественноro опре
деления.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
И дРУrИЕ ИСПЫТАНИЯ
Лекарственные средства для rенной Tepa
пии должны соответствовать требованиям, YKa
занным для количественноro определения и для
друrих испытаний, описанных в COOTBeTCTBY
ющих частных статьях.
rенетически модифицированные клетки
Данные по рекомбинантным векторам, с по
мощью которых модифицируются клетки, реrи
стрируются в порядке, описанном выше в под
разделе «Рекомбинантные векторы».
ПРОИЗВОДСТВО
КЛЕТКИ ПРОДУЦЕНТbI
При использовании линий KceHoreHHbIx
клеток, включая бактериальные клетки, создает
ся система банка клеток, состоящая из rлавноrо
банка клеток и рабочеro банка клеток.
При использовании аутолоrических и алло
reHHbIx клеток создается система банка клеток,
состоящая из rлавноro банка клеток и рабочею
банка клеток, если это возможно.
ТРАНСФЕКЦИЯПРАНСДУКЦИЯ
Процесс трансфекцииlтрансдукции pe
комбинантноrо вектора (плазмидноro или ви
pycHoro), контролируемоro в соответствии с
разделом «Рекомбинантные векторы», вали
дируется. Операции с клетками проводятся в
асептических условиях в зоне, свободной от oд
новременной работы с друrими клетками или
векторами. Все реактивы, используемые при
работе с клетками, полностью проконтролиро
ваны. Использование антибиотиков не допуска
ется, если иное не обосновано и разрешено.
Трансфекцияlтрансдукция про водятся в асеп
тических условиях.
rОТОВblЙ ПРОДУКТ
При хранении клеток в замороженном COCTO
янии жизнеспособность rенетически модифици
рованных клеток оценивается до замораживания
и после опаивания. Если клетки не исполь
зуются в течение KOpOTKOro периода времени,
стабильность оценивается по подтверждению
жизнеспособности клеток и экспрессии rенети
ческоro материала.
Если больным вводятся инкапсулированные
rенетически модифицированные клетки, все ин
капсулированные компоненты считаются частью
roтовою продукта, и, соответственно, их качество
контролируется и должно быть полностью oxapaK
теризовано (например, физическая целостность,
селективная проницаемость, стерильность).
958
rосударственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
И друrИЕ ИСПЫТАНИЯ
Контроль KceHoreHHbIx, аллоrенных jли
аутолоrичных клеток включает следующие испы
тания:
определение подлинности. подсчет и
оценка жизнеспособности клеток;
общая целостность. функuиональность,
количество копий на клетку, перенос и эффек-
тивность экспрессии rенетическоro материала;
микробиолоrическая оценка (2.6.1 или
2.6.27), содержание эндотоксинов, контамина
ция микоплазмами (2.6.9), контаминация посто-
ронними вирусами и, если применимо, reHepa
ция репликативноro вируса.
Компетентный уполномоченный opraH
может соrласовать сокращенную проrрамму ис-
пытаний, в связи с оrраниченным количеством
клеток. В связи с оrраничениями по времени,
продукт может быть разрешен к использованию
до завершения определенных испытаний.
Плазмидные векторы
для медицинскоro применения
ОПРЕДЕЛЕНИЕ
Плазмидные вепоры для медицинскоro при-
менения представляют собой двойную кольце
видную бактериальную ДНК, которая содержит
требуемый reH или последовательность нуклео
тидов, антисмысловую последовательность или
рибозимы и их экспресионную оболочку; они
амплифицируются в бактерии экстрасомаль
но. Плазмиды используются для переноса reHe
тическоro материала в соматические клетки
человека in vivo или в rенетически модифициро-
ванные аутолоrичные, аллоrенные, KceHoreHHbIe
или бактериальные клетки до введения боль
ному. Плазмидные векторы MOryт представлять
собой чистую (<<rолую») ДНК, или комплекс син
тетической системы доставки, например, липиды
(липоплексы), полимеры (полиплексы) и/или
пептидные лиrанды, которые облеrчают перенос
rенетическоro материала через клеточную обо-
лочку и ero доставку в клетку, или направленную
доставку через специфические рецепторы.
Плазм иды , представляющие синтетиче-
ские системы доставки, не рассматриваются в
данном подразделе.
ПРОИЗВОДСТВО
КОНСТРУКЦИЯ ПЛАЗМИДbI
Типичный плазмидный вектор состоит из:
скелета плазмидноro вектора, который
содержит мноroчисленные сайты распознава
ния эндонуклеазной рестриктазы для включе-
ния rенетическоrо материала и элементы reHo
типа бактерий, необходимые для производства
плазм ид, например, определенные rенетиче-
ские маркеры для идентификации клеток, KOTO
рые содержат рекомбинантный вектор;
требуемых реryлирующих rенетических
элементов, необходимых для обеспечения экс
прессии rенетическоro материала;
rенетическоro материала;
сиrнала полиаденилирования.
Полное описание плазмидной ДНК, включая
ее последовательности нуклеотидов, проводится
путем идентификации источников, способов вы-
деления и последовательности нуклеотидов re-
нетическоro материала. Описываются источник и
функции компонентов плазмиды, такие как источ-
ник репликации, вирусные и эукаРИОТlАческие про
моутеры, селективные rен-кодирующие маркеры.
ОБЩИЕ ПОЛОЖЕНИЯ
Банки клеток. Производство плазмидных
векторов основано на системе банка бактери
альных клеток с получением rлавноro банка
клеток, рабочеro банка клеток и посевноro кле
точноro материала, которые соответствуют по
ложениям подраздела «Бактериальные клетки,
используемые для производства плазмидных
векторов для медицинскоro применения». Ис-
ходные материалы, используемые в производ-
ственном процессе, включая создание банка
клеток, подлежат контролю.
Методы селекции. Если иное не обоснова
но и не утверждено, reHbI антибиотикорезистент
ности, используемые в качестве селективных
reHHbIx маркеров, в частности для терапевтиче-
ских антибиотиков, не включаются в KOHCTPYK
цию вектора. Предпочтительно использование
друrих методов селекции для рекомбинантных
плазмид.
Стандартные образцы. Приемлемая серия
roтовой плазмиды, преимущественно из исполь
зованных в клинических исследованиях, полно-
стью характеризуется и используется в качестве
стандартноro образца в повседневном контроле
качества, если необходимо.
КУЛЬТИВИРОВАНИЕ И СБОР БИОМАССbI
Плазмидная ДНК переносится в культу-
ру бактериальных клеток-реципиентов; единый
клон трансформированной бактерии выращи-
вается для создания rлавноro банка клеток.
Затем из rлавноrо банка клеток получают рабо
чий банк клеток. Посевной клеточный материал
получают из рабочеro банка клеток с помощью
ферментации в производственных условиях.
Плазмидная ДНК выделяется из биомассы с по-
мощью экстракции и очищается до получения ro
товоro ...ерасфасованноro продукта.
Если ИI-Jое не обосновано и разрешено, для
производства не используются rрадиенты плот-
HocTи по цезия хлорид итидия бромиду.
ОЧИЩЕННАЯ ПЛАЗМИДА
Производственный процесс оптимизирует
ся с целью адекватноro удаления примесей при
5.14. Лекарственные средства для zенной терапии
959
сохранении биолоrической активности. Требова
ния по проведению оценки содержания опреде
ленной примеси зависят от следующеro:
доказанная в процессе валидации с при
менением определенных количественных MeTO
дов способность процессов культивирования и
очищения удалять или инактивировать примеси;
возможная токсичность примеси:
возможное снижение эффективности BBO
димоro rенетическоro материала, связанное с
примесью.
Если для селекции была использована из
бирательная резистентность к определенным
антибиотикам, данные из работ по валидации
процессов очистки необходимы для подтверж
дения очистки продукта от остаточных антибио
тиков.
Для обеспечения постоянноro контроля про
цесса при внутрипроизводственном контроле
проводятся соответствующие испытания, напри
мер, определение количества и формы плаз
миды, содержание бактериальных эндотокси
нов после стадий экстракции. К дальнейшему
использованию допускаются серии очищен
ной плазм иды , удовлетворяющие нижеперечис
ленным требованиям.
Подлинность и целостность очищенной
плазмиды. Подлинность и целостность очищен
ной плазмиды определяется соответствующими
методами, например, секвенирования или aM
плификации нуклеиновых кислот (2.6.2); может
быть использован рестрикционный фермента
тивный анализ, если еro возможности позволя
ют выявить возможные критические модифика
ции в плазмиде и подтвердить подлинность.
Плазмидная днк. Нижеприведенные Tpe
бования даны в качестве при мера.
Концентрация ДНК выше 500 нr/мл может
быть определена путем измерения величины
поrлощения при 260 нм. Величина поrлощения
раствора двойной кольцевой ДНК в KOHцeHTpa
ции 50 мкr/мл составляет 1 (удельное поrлоще
ние 200).
Содержание ДНК в концентрации менее
500 нr/мл определяется после инкубирования с
флуоресцентным красителем, который избира
тельно связывается с двойной кольцевой ДНк.
и использованием стандартноro образца ДНК
дЛЯ построения калибровочной кривой.
Для определения плазмидной ДНК также
может быть использована жидкостная xpOMaTO
rрафия с применением стандартноro образца. В
некоторых случаях используется капиллярный
элепрофорез.
Формы днк. Плазмидная ДНК характери
зуется в терминах соотношения сверхскручен
ных, мноroмерных, растянутых мономеров и
линейных форм с использованием приroдных
для этоro аналитических методов, некоторые
ИЗ которых указаны далее. Для количествен
ноro определения сверхскрученных форм может
быть использована высокоэффективная ани
онообменная жидкостная хроматоrрафия или
капиллярный электрофорез. Капиллярный элек
трофорез также приroден для количественноro
определения друrих форм.
Остаточная ДНК клетки хозяина. Coдep
жание остаточной ДНК клетки хозяина опреде
ляется с использованием приroдноro для этоro
метода, если валидацией процесса не было по
казано достижение требуемой степени очист
ки продукта. Учитывая чувствительность и
специфичность, для этоro рекомендуется
количественный метод полимеразной цепной
реакции (ПЦР), друrие методы также MOryт быть
использованы.
Остаточная РНК. Содержание остаточной
РНК контролируется, если валидацией процес
са не было показано достижение требуемой
степени очистки продукта. Для этоro может ис
пользоваться обращенно фазовая ВЭЖХ или
количественный метод обратно транскриптаз
ной полимеразной цепной реакции (2.6.21), если
требуется определить нижний предел количе
ственноro определения.
Остаточные белки клетки-хозяина. KOH
центрация остаточных белков клетки хозя
ина определяется с использованием CTaHдapT
ных методов количественноro определения
белков (2.5.33), электрофореза (SDS PAGE)
после окрашивания серебром или специфиче
ских иммунобиолоrических методов, например.
Вестерн блот или тИФА (EUSA), если валидаци
ей процесса не было показано достижение Tpe
буемой степени очистки продукта.
Микробиолоrический контроль. В зависи
мости от контролируемоro продукта должны BЫ
держиваться испытания на стерильность (2.6.1)
или на микробиолоrическую чистоту (2.6.12).
Бактериальные эндотоксины (2.6.14).
Менее установленноro значения для KOHKpeTHO
ro продукта.
rотовый НЕФАСОВАННЫЙ ПРОДУКТ
Несколько очищенных биомасс может быть
объединено при получении rOТOBOro нерасфасо
BaHHoro продукта. На этой стадии допускается дo
бавлять стабилизатор и друrие вспомоrательные
вещества. После завершения смешивания про
дукт подверrается стерилизующей фильтрации.
Для получения roтовоro лекарственно
ro средства используется продукт, COOTBeTCTBY
ющий нижеуказанному требованию.
Стерильность (2.6.1). Продукт должен быть
стерильным.
960
rосударственная фармакопея Республики Беларусь
rотовый ПРОДУКТ
К выпуску разрешается roтовый прс,д'/кт,
удовлетворяющий требованиям разделов «Под
линность», «Количественное определение»,
«Испытания», описанных далее.
ПОДЛИННОСТЬ
Плазмидный вектор идентифицируется с
помощью рестрикционноrо ферментативноro
анализа или секвенирования. Также для испыта
ния на подлинность может использоваться испы
тание на биолоrическую активность.
ИСПЫТАНИЯ
Описание.
рН (2.2.3). Значение рН должно находить
ся в установленных для конкретноro продукта
пределах.
Извлекаемый объем (2.9.17). Испытуемый
образец должен выдерживать испытание на из
влекаемый объем.
Вода (2.5.12). Содержание влаrи должно Ha
ходиться в установленных для конкретною лио
филизированноrо продукта пределах.
Формы ДНК. Процент специфических MO
номерных сверхскрученных форм определяется
в соответствии с рекомендациями по очищенной
плазм иде.
Стерильность (2.6.1). Испытуемый образец
должен выдерживать испытание на стерильность.
Бактериальные эндотоксины (2.6.14).
Менее установленною предельноro значения
для конкретноro продукта.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Плазмидная ДНК: не меньше количества,
указанноro на этикетке, определенноro, напри
мер, с помощью одной из нижеприведенных Me
тодик.
Концентрации ДНК более 500 нr/мл MOryт
быть измерены путем определения величины
поrлощения при 260 нм. Величина поrлощения
раствора двойной кольцевой ДНК концентрации
50 мкr/мл равна 1 (удельный показатель поrло
щения 200).
Содержание ДНК в концентрации менее
500 нr/мл определяется после инкубирования с
флуоресцентным красителем, который избира
тельно связывается с двойной кольцевой ДНК, и
использованием стандартноro образца ДНК дЛЯ
построения калибровочной кривой.
Для определения концентрации плазмид
ной ДНК также может использоваться жидкост
ная хроматоrрафия с использованием стандарт
ноro образца. В некоторых случаях применяется
капиллярный электрофорез.
Биолоrическая активность. Если воз
можно, биолоrическая активность оценива
ется с помощью методик определения биоло
rической активности iп vitro или iп vivo. Для
этих определений требуется хорошо oxapaK
теризованный репрезентативный стандарт
ный образец. В методах оценки биолоrической
активности, применяющихся для определения
содержания плазмидных векторов, обычно
используют трансфекцию соответствующей
линии клеток iп vitro с последующим изме
рением экспрессированноrо rенетическо
ro материала по какой либо функции. Такие
функциональные методы количественною
определения обеспечивают информацией об
активности продукта, кодированноro rенети
чески м материалом, вместо уровня экспрес
сии caMoro rенетическоrо материала. Может
потребоваться проведение дополнительных
определений биолоrической активности с по
мощью Вестерн блота или твердофазноro им
муноферментноrо анализа для оценки цe
лостности и количества экспрессированноrо
продукта.
МАРКИРОВКА
На этикетке указывается:
концентрация плазмидной ДНК;
рекомендуемая для медицинскоro приме
нения доза;
для лиофилизированных продуктов:
название и объем добавляемоro раствори
теля;
время, в течение котороro продукт должен
быть использован после растворения.
Бактериальные клетки, используемые
в производстве плазмидных векторов
для медицинскоro применения
Получение плазмидных векторов для Me
дицинскоrо применения основано на исполь
зовании системы банка бактериальных клеток
с получением и характеристикой rлавноro
банка клеток, рабочею банка клеток и посев
ноro материала. Банк бактериальных клеток,
используемый для производства плазмидных
векторов, представляет собой набор ампул,
содержащих бактериальные клетки, хранящих
ся в определенных условиях, с одинаковым co
ставом и полученных из пула клеток, произо
шедших от одноro клона трансформированной
клетки хозяина. У rлавноrо банка клеток име
ется четкая документированная история ero
получения; rлавный банк клеток, как прави
ло, получают из проконтролированноro источ
НИl(а. Рабочий банк клеток получают путем
культивирования клеток из одной или более
ампул rлавною банка клеток. Реrистрируются
методики и реактивы, использованные для по
лучения банка, и условия хранения. rлавные
и рабочие банки клеток контролируются путем
проведения испытаний на аликвоте храняще
5.14. Лекарственные средства для zенной терапии
961
roся в банке материала или полученноro путем
субкулыивирования банка клеток.
В данной таблице приведены испытания,
проводящиеся на каждой производственной
стадии.
I Количе I Рабо- j ПОС В-
I rлав-
Клетка- ный нои
I ственное чии
хозяин банк б I клеточ-
i опреде- анк 'ныи ма-
ление клеток клеток *
териал
Подлин
I ность И
I чистота
Жизнеспо
собные + + + +
клетки !
I I
I XapaK I
теристи i
ки линии + + ,
бактери i +
j
i !
альных I I I
;
клеток I !
I
rенотипи ! i
рование/ I I
фенотипи + + + I
рование I
Наличие
I
плазмиды I
после
дователь + +
ность ДНК
плазмиды
номер + + +
копии
! рестрик I
ционная I + + +
iKapTa I
процент I
ное co
держание + + +
клеток, co
держащих
плазм иды
Посто-
ронние
areHTbI
Посторон
няя ми + + + +
крофлора
Присут
ствие бак + + +
териофа
roв
. Посевной материал это клетки, прошедшие по крайне
мере такое же или меньшее количество пассажей, что и
клетки. использованные для производства. Анализ прово
дится один раз для про верки каждою новою рабочеrо банка
клеток. за исключением чистоты, которая проверяется при
каждой ферментации.
).!. _ .;}\ 1712.
ИСПЫТАНИЯ НА ПОДЛИННОСТЬ
И ЧИСТОТУ
Жизнеспособность клеток. Количество
жизнеспособных клеток определяется путем
посева разведения аликвоты бактериальных
клеток на соответствующую питательную среду
и подсчета отдельных колоний.
Биохимическая и физиолоrическая ха-
рактеристики бактериальной линии. В зави
симости от используемой в производстве линии
бактерий, проводятся соответствующие опре
деления биохимических и физиолоrических
свойств клеток для подтверждения принадлеж
ности клеток к определенному виду.
rенотипирование/фенотипирование. re
нотип бактериальных клеток верифицируется
путем определения признанных фенотипиче
ских маркеров или соответствующим rенетиче
ским анализом.
Наличие плазмиды.
Секвенирование. Подтверждается полная
нуклеотидная последовательность плазм иды.
Номер копии. Плазмидная ДНК выделяется
и очищается из определенноrо количества бак-
терий, номер копии определяется с помощью
приroдноrо для этоro метода количественноro
ПЦР (2.6.21).
Рестрикционная карта. rидролиз рестрик
ционными эндонуклеазами проводится с ДOCTa
точным разрешением, чтобы подтвердить, что
структура плазмиды, находящаяся в бактери
альной клетке, не повреждена.
Процент клеток. содержащих плазмиду.
Бактериальные элементы, присутствующие в
плазмиде. такие как селективные rенетические
маркеры. используются для определения
процентноro количества бактерий, содержащих
плазм иду.
ПОСТОРОННИЕ ArEHTbI И эндоrЕННЫЕ
ВИРУСЫ
Посторонняя микрофлора. Бактериаль
ные клетки высевают на соответствующую пи-
тательную среду методом штриховоro посева
и термостатируют в требуемых условиях, необ
ходимых для обнаружения возможных бактери-
альных посторонних areHТOB. Для определения
инrибирования роста посторонних микроорrа
низмов проводят дополнительные испытания
в присутствии определенноrо количества кон-
трольных бактерий в качестве положительноro
контроля. Оценивается требуемое количество
колоний; посторонней микрофлоры не должно
обнаруживаться.
Наличие бактериофаrов. Для выявления
наличия бактериофаrов бактериальные клетки
высевают и термостатируют в питательной
среде, обеспечивающей рост и размножение
962
rосударственная фармакопея Республики Беларусь
бактериофаrов. Приroдность испытания под
тверждается путем использования референт
ной линии бактериофаroв и факультативных
клеток в качестве положительных контролей.
Оценивается требуемое количество колоний;
посторонней микрофлоры не должно обнару
живаться.
Аденовирусные векторы
для медицинскою применения
ОПРЕДЕЛЕНИЕ
Аденовирусные векторы для медицинско
ro применения производятся в лиофилизиро
ванном виде или в виде жидких препаратов
рекомбинантных аденовирусов, rенетически MO
дифицированных для обеспечения переноса re
нетическоrо материала в соматические клетки
человека iп vivo или ех vivo.
ПРОИЗВОДСТВО
КОНСТРУКЦИЯ ВЕКТОРА
Существуют различные подходы для раз
работки и конструирования аденовирусноro BeK
тора. Выбор подхода определяется целью кли
ническоro использования. Выбирается метод,
который минимизирует риск rенерирования pe
пликационно компетентных аденовирусных BeK
торов или при котором эффективно удаляются
вирусы помощники. которые моrли бы использо
ваться при производстве.
ПРОИЗВОДСТВО ВЕКТОРА
Должно быть показано, что методы про
изводства векторов обеспечивают постоянное
получение вектора требуемоro качества. Если
иное не обосновано и разрешено, вектор в ro
товом продукте не должен подверrается боль
шему количеству пассажей из rлавноro банка
клеток, чем вектор, чья эффективность и безо
пасность была доказана в клинических иссле
дованиях. rенетическая и фенотипическая
стабильность вектора. использованноro для
производства на максимальном количестве
пассажей или менее, оценивается приroдными
для этоro методами.
КЛЕТКИ ПРОДУЦЕНТЫ
ДЛЯ КУЛЬТИВИРОВАНИЯ ВЕКТОРА
Вектор культивируется в постоянно культи
вируемой линии клеток (5.2.3) с помощью си
стемы банка клеток. Наличие репликацион
но компетентных аденовирусов может быть
значительным, Korдa существуют большие об
пасти, roмолоrичные между вирусным reHoM и
reHoMoM клетки реципиента. Эта вероятность
может быть уменьшена путем уменьшения roмо
лоrизации двух reHoMoB. Для производства pe
комендуется использовать клетки, не имеющие
нуклеотидной последовательности, rомолоrич
ной вектору.
ПОСЕВНОЙ МАТЕРИАЛ
Производство вектора проводится с ис
пользованием системы посевноro материала.
Линия аденовируса идентифицируется с помо
щью данных получения вектора, включая ин
формацию о ero происхождении и последующих
манипуляциях, в частности, об исключенных
или модифицированных областях. Разраба
тывается подробное описание rенетическоro
материала и использующихся для контроля
участков конструкции, включая нуклеотидную
последовательность. Описывается метод вклю
чения rенетическоro материала в вектор.
Для производства вектора используется по
севной материал, отвечающий приведенным
далее требованиям.
Подлинность. Вектор идентифицирует
ся в образцах rлавноrо посевноro материала и
каждой серии рабочеro посевноro материала с
помощью иммунохимических методов (2.7.1),
методами амплификации нуклеиновых кислот
(2.6.21) или ферментативным рестрикционным
анализом.
rенетические и фенотипические характе-
ристики. Выполняются следующие испытания:
полный reHoM вектора секвенируется на
пассаже. соответствующем производственной
серии; инструментально подтвержденная после
довательность сравнивается с теоретической
последовательностью, определенной, исходя из
конструкции вектора и имеющихся баз данных;
рестрикционный ферментативный анализ
выполняется на ДНК вектора rлавноro посевно
ro материала, каждоro рабочеro посевноro MaTe
риала и производственной серии; вирусная ДНК
выделяется, очищается и rидролизуется с дo
статочным разрешением; rидролизованные cer
менты разделяются с помощью электрофореза
в rеле или капиллярноro электрофореза, полу
ченные рестрикционные картины сравниваются
с теоретическими рестрикционными картинами,
рассчитанными по конструкции вектора;
приемлемое количество изолированных
суб клонов исследуется на наличие признаков
экспрессии rенетическоrо материала и биоло
rической активности на уровне пассажа, COOT
ветствующеro производственной серии; при BЫ
явлении суб клонов с более низким уровнем
экспрессии или биолоrической активности Tpe
буются дальнейшие испытания.
Концентрация вектора. Титр инфекцион
Horo вируса или концентрация векторных частиц
определяется в rлавном посевном материале и
каждом рабочем посевном материале.
Посторонние areHTbI (2.6.16). В rлавном
посевном материале и каждом рабочем посев
ном материале должны отсутствовать посторон
ние areHTbI.
5.14. Лекарственные средства для 2енной терапии
963
Репликационно-компетентные аденови-
русы. Репликационно компетентные вирусы
образуются вследствие rомолоrичной рекомби
нации между рекомбинантной вирусной ДНК и
аденовирусной последовательностью, интеrри
рованной в reHoM вспомоrательных клеток реци
пиентов.
Обнаружение репликационно компетентных
аденовирусов проводится с помощью подходя
щеro для этоro метода, соrласованноro уполно
моченным компетентным opraHoM. Как правило,
для этоrо используется определение инсрекци
онной активности с использованием чувстви
тельной линии клеток детекторов. которые
не способны комплементироваться с rенами,
удаленными из вектора. MOryT быть использо
ваны друrие приroдные индикаторы вирусной
репликации. Если в испытуемом образце не
предполаrается наличие репликационно компе
тентных вирусов, учитывая конструкцию вектора
и использованные линии клеток, на линии I\ле
TOK дeTeKTopOB про водится по крайней мере 2,
но предпочтительнее 3 или 4 последовательных
пассажа, если применимо. Наблюдаемый цито
патический эффект в конце пассажей означа
ет наличие репликационно компетентных виру
сов в продукте. В каждое определение включают
положительные контроли для мониторинrа чув
ствительности методики.
Если предполаrается, что в продукте MorYT
присутствовать репликационно компетентные
вирусы, на линии клеток детекторов может быть
проведен анализ реакции бляшкообразования
или определение пределов разведения.
КУЛЬТИВИРОВАНИЕ И СБОР БИОМАССbI
Все операции с банком клеток и последу
ющими культурами клеток проводятся в зоне с
приемлемым уровнем контаминации при отсут
ствии одновременной работы с друrими клетками
или векторами. Контролируется любой матери
ал, полученный от человека, или животноro про
исхождения, используемый в приrотовлении кле
точных суспензий и питательных сред. Среда для
культивирования клеточных КУЛЬТУР может coдep
жать рН индикатор, например, феноловый Kpac
ный, и подходящие антибиотики в наименьшей
эффективной концентрации, однако более пред
почтительно использовать в процессе производ
ства клеточную культуру, не содержащую антиби
отики. Если иное не обосновано и разрешено, ни
на одной из технолоrических стадий не исполь
зуется пенициллин или стрептомицин. Часть
производственной культуры клеток (неинфици
рованная клеточная культура) отбирается для ис
пользования в качестве контрольных клеток.
Последующая очистка партии биомассы
проводится, если она соответствует указанным
далее требованиям.
Подлинность. Вектор идентифицируется
с помощью иммунохимических методов (2.7.1),
методов амплификации нуклеиновых кислот
(2.6.21) или рестрикционным ферментативным
анализом.
Концентрация вектора. В каждой партии
биомассы определяется титр инфекционноro
вектора и концентрация векторных частиц.
Посторонние areHTbI (2.6.16). Каждая
партия биомассы не должна содержать посто
ронних areHToB.
Контрольные клетки. Контрольные клетки
должны выдерживать испытание на подлин
ность (5.2.3) и на отсутствие посторонних areH
тов (2.6.16).
ОЧИЩЕННАЯ БИОМАССА
Несколько партий биомассы Moryт быТ!-.
объединены перед процессом очистки. Процесс
очистки валидируется с целью демонстрации co
ответствующеro удаления примесей.
Для производства roтовоro нефасованноrо
продукта используется очищенная биомасса, co
ответствующая указанным далее требованиям.
Подлинность. Вектор идентифицируется
с помощью иммунохимических методов (2.7.1),
методов амплификации нуклеиновых кислот
(2.6.21) или рестрикционным ферментативным
анализом.
Целостность rеномз. Целостность reHoM3
вектора подтверждается соответствующими Me
тодами, например. рестрикционным фермента
тивным анализом.
Концентрация вектора. В каждой партии
биомассы определяется титр инфекционною
вектора и концентрация векторных частиц.
Остаточные белки клетки-хозяина. KOH
центрация остаточноrо белка клетки хозяина
определяется приrодным для этоrо иммуно
химическим методом (2.7.1), если при валида
ции процесса не было показано достижение
требуемой степени очистки продукта.
Остаточная ДНК клетки-хозяина. Coдep
жание остаточной ДНК клетки хозяинз опреде
ляется с использованием приrодноrо для этоrо
метода, если валидацией процесса не было пока
зано достижение требуемой степени очистки про
дукта. Учитывая чувствительность и специфич
ность, для этою рекомендуется количественный
метод полимеразной цепной реакции (ПЦР),
друrие методы также MOryт быть использованы.
Остаточные реактивы. Если в производ
ственном процессе используются реактивы. ис
пытания на наличие этих веществ проводятся на
очищенной биомассе, если валидациеЙ процес
964
rосударственная фармакопея Республики Беларусь
са не было показано достижение требуемой CTe
пени очистки продукта.
Остаточные антибиотики. Если в произ
водственном процессе используются антиби
отики, их остаточная концентрация определя
ется микробиолоrическим методом (адапти
рованным общим методом 2.7.2) или друrими
приrодными методами (например, жидкостной
хроматоrрафией), если валидацией процесса не
было показано достижение требуемой степени
очистки продукта
rОТОВbfЙ НЕФАСОВАННbfЙ ПРОДУКТ
Несколько партий очищенной биомассы
MOryT быть объединены перед приroтовлением
roТOBoro продукта. MoryT быть добавлены CTa
билизатор и друrие вспомоrательные вещества.
rотовый продукт подверrается стерилизующей
фильтрации.
Для получения roтовоro лекарственно
ro средства используется продук COOTBeTCTBY
ющий нижеуказанному требованию.
Стерильность (2.6.1). Продукт должен быть
стерилен.
rОТОВbfЙ ПРОДУКТ
К выпуску разрешается roтовый продукт,
удовлетворяющий требованиям в разделах
«Подлинность» «Количественное определе
ние», «Испытания», описанным далее. Если
испытания на наличие бычьеro сывороточноrо
альбумина (если он используется в производ
стве вектора) и репликационно компетентных
аденовирусов были проведены на roтовом He
расфасованном ПРО.1укте с положительными pe
ЗУJlьтатами, они MOryт не проводиться при KOH
троле rOТOBoro продукта.
ПОДЛИННОСТЬ
Подлинность вектора подтверждается с
помощью иммунохимических методов (2.7.1),
методов амплификации нуклеиновых кислот
(2.6.21) или рестрикционным ферментатив
ным анализом.
ИСПЫТАНИЯ
Осмоляльность (2.2.35). Значение OCMO
ляльности должно находиться в установленных
для конкретноro продукта пределах.
рН (2.2.3). Значение рН должно находиться в
установленных для конкретноro продукта пределах.
Извлекаемый объем (2.9.17). Испытуемый
образец должен выдерживать испытание на из
влекаемый объем.
Вода (2.5.12). Содержание влаrи должно Ha
ходиться в установленных для конкретноro лио
филизированноro продукта пределах.
Бычий сывороточный альбумин. Если в
производстве использовалась сыворотка крупноro
poraToro скота, содержание бычьеro сывороточно
ro альбумина не должно превышать установленно
ro для KOHKpeTHoro продукта предельноrо значения
при определении подходящим иммунохимическим
методом (2.7.1).
Концентрация репликационно компе
тентных аденовирусов. Концентрация должна
находиться в установленных для конкретноro
продукта пределах.
ArperaTbI вектора. ArperaTbI вектора опре
деляются подходящим методом (например, CBe
торассеянием ).
Стерильность (2.6.1). Испытуемый обра
зец должен быть стерильным.
Бактериальные эндотоксины (2.6.14).
Менее установленноro предельноro значения
для конкретноro продукта.
Термическая стабильность. Образцы ro
товоro препарата вектора выдерживают при TeM
пературе и в течение времени, установленных и
одобренных для конкретноro продукта. Определя
ется общая концентрация инфекционною Beктo
ра после наrревания в соответствии с методикой,
приведенной в ра еле «Количественное опре
деление». В параллельных испытаниях опреде
ляется концентрация вектора в образце, не под
верrавшемся наrреванию. Выявленная разница
концентраций вектора в образце, не подверrшем
ся наrреванию, и образце, подверrшемся HarpeBa
нию, должна находиться в установленных дЛЯ KOH
кретноro продукта пределах.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Концентрация векторных частиц. Физи
ческое титрование проводится приroдным для
этоro методом (например, жидкостной xpOMa
тоrрафией, измерением величины поrлоще
ния или амплификацией нуклеиновых кислот
(2.6.21)). Для валидации каждоro испытания
при меняется соответствующий стандартный
образец вектора.
Концентрация векторных частиц в испыту
емом образце должна быть не менее количе
ства, указанноro на этикетке.
Титр инфекционноrо вектора. Титр испы
туемоro образца определяется по еro влиянию
при внесении в культуру клеток. Для валидации
каждоro испытания применяется COOTBeTCTBY
ющий стандартный образец вектора.
Результаты испытания являются Heдocтo
верными, если:
доверительный интервал (Р = 0,95) лоrариф
ма концентрации вектора больше нормы, YCTaHOB
ленной компетентным уполномоченном opraHoM;
5.14. Лекарственные средства для 2енной терапии
965
титр инфекционноro вектора в стандарт
ном образце выходит за рамки значений, YCTa
новленных в контрольном rрафике.
Соотношение концентрации векторных
частиц с титром инфекционноrо вектора.
Должно соответствовать норме, установленной
для конкретноro продукта.
Экспрессия rенетическоrо материала
в продукте. Экспрессия rенетическоro MaTe
риала (материалов) в продукте определяет
ся Bcerдa, если это возможно, после введения
испытуемоrо образца в культуры клеток в за
ранее определенной инфицирующей дозе с
помощью приroдных для этоro иммунохимиче
ских (2.7.1) или биохимических методов коли
чественноro определения или проточной цито
\i1етрией (2.7.24).
Биолоrическая активность. Если иное не
обосновано и не утверждено, биолоrИl'еская ак'
тивность определяется с помощью COOTBeTCT8Y
ющеro испытания in vit,o или in vivo.
МАРКИРОВКА
На этикетке указывается:
содержание действующеro вещества;
рекомендуемая терапевтическая доза, BЫ
раженная в единицах концентрации векторных
частиц;
для лиофилизированных продуктов:
название и объем растворителя, с помо
щью котороro проводят растворение продукта;
время, в течение котороro продукт должен
бы',ъ использован после растворения.
Поксвирусные векторы
для медицинскою применения
ОПРЕДЕЛЕНИЕ
Поксвирусные векторы для медицинско
ro применения производятся в лиофилизиро
ванном виде или в виде жидких препаратов
рекомбинантных поксвирусов, rенетически MO
дифицированных для обеспечения переноса re
нетическоrо материала в соматические клетки
человека in v;vo или ех vivo.
ПРОИЗВОДСТВО
КОНСТРУКЦИЯ ВЕКТОРА
Общий дизайн поксвирусноro вектора в Ha
стоящее время заключается во введении rенети
ческоro материала по ходу транскрипции промо
утера поксвируса. Эта экспрессионн-зя кассета
вводится в reHoM поксвируса таким образом, что
она блокирует вирусный reH, не участвующий в
репликации, или вставляется между двумя OT
крытыми для считывания рамками вируса. В
большинстве используемых таким образом
стратеrий конструирования вектора экспресси
.2:<. 3а.. 1712.
онная кассета вначале вставляется между цe
левым сайтом фраrмента вирусной ДНК, кло
нированной в бактериальной плазмиде. Затем
плазмида вводится в клетку реципиент, культи
вируемую in vit,o, которая одновременно инфи
цируется исходным поксвирусом. Рекомбина
цИЯ ДНК происходит в инфицированных клетках,
между roмолоrичными последовательностями
в вирусном reHoMe и вирусной последователь
ностью в плазмиде, что обеспечивает включе
ние rенетическоro материала в целевой сайт
вирусноro reHoMa. Правильность включения
ДНК в reHoM проверятся методом рестрикцион
ной карты, амплификацией нуклеиновых кислот
(2.6.21) и секвенированием. Для выделения pe
комбинантных вирусов из смеси исходных и pe
комбинантных вирусов выполняются последо
вательные операции по бляшкообразующему
клонированию. Для облеrчения распознавания
и/или селекции рекомбинанп-юro поксвируса из
':::меси с исходным вирусом используется целый
,)яд методов (например, чужеродные reHbI Map
r;epbI, rибридизация ДНК, ИММУНОJlоrическая
детекция, фенотипические изменения вируса).
Если для этоro используются временные чуже
родные reHbI MapKepbI, они ВllOследствии удаля
tOтся из окончательноro reHoMa рекомбинантно
ro вируса.
Альтернативная стратеrия получения пок
свирусноro вектора заключается в предвари
тельном конструировании полноrо вирусноrо
reHOMa in vitro с помещением экспрессионной
кассеты в выбранный целевоЙ сайт. Затем этот
рекомбинантный reHOM ВВСJ4ИТСЯ в клетку хо
зяина с одновременным инфицированием пок
свирусом помощником, который не способен
размножаться. Вирус помощник может быть
поксвирусом TOro же вида, чья способность
размножаться была инактивирована, или пок
свирусом друrоro вида, который не может
размножаться в клетке хозяине. KOHCTPYK
цИЯ неспособных к репликации поксвирус
ных векторов зависит от свойств специфи
ческой линии клетки хозяина или первичных
клеток, которые по своей природе толерантны
к вирусу, или линии клетки хозяина, которая
была модифицирована для обеспечения экс
прессии требуемоro reHa поксвируса. Эти клет
ки обеспечивают выполнение основных требо
ваний к производству лекарственных средств
(5.2.3) и препятствуют размножению реплика
ционных векторов.
ПРОИ3ВОДСТВО ВЕКТОРА
Должно быть показано, что методы про
изводства векторов обеспечивают п()стоян
ное получение вектора требуемоrо качества.
Если иное не обосновано и разрешено, вектор
в roтовом продукте не должен подверrаеться
большему количеству пассажей из rлавноro
банка клеток, чем вектор, чья эффективность
и безопасность была доказана в клинических
966
rосударственная фармакопея Республики Беларусь
исследованиях. rенетическая и фенотипиче
ская стабильность вектора, использованноrо
для производства на максимальном количе
стве пассажей или менее, оценивается приrод
ными для этоro методами.
КЛЕТКИ ПРОДУЦЕНТbI
ДЛЯ КУЛЬТИВИРОВАНИЯ ВЕКТОРА
Вектор культивируется в асептических yc
ловиях в диплоидных клетках человека (5.2.3),
постоянно культивируемой линии клеток (5.2.3)
или в культуре клеток куриноro эмбриона, полу
ченных из стада птиц, свободных от специфиче
ской патоrенной микрофлоры (5.2.2). Если вирус
культивируется в постоянно культивируемой
культуре клеток или диплоидных клетках чело
века, создается система банка клеток.
ПОСЕВНОЙ МАТЕРИАЛ
Производство вектора проводится с ис
пользованием системы посевноro материала.
Линия поксвируса идентифицируется с помо
щью данных получения вектора, включая ин
формацию о еro происхождении и последу
ющих манипуляциях. в частности об исключен
ных или модифицированных областях. Разра
батывается подробное описание rенетическоro
материала и использующихся для контроля
участков конструкции. включая нуклеотидную
последовательность. Описывается метод вклю
чения rенетическоrо материала в вектор.
Для производства вектора используется по
севной материал. отвечающий приведенным
далее требованиям.
Подлинность. Вектор идентифициру
ют в образцах rлавноrо посевноro материала и
каждой серии рабочеro посевноro материала с
помощью иммунохимических методов (2.7.1),
методами амплификации нуклеиновых кислот
(2.6.21).
rенетические и фенотипические характе-
ристики. Выполняются следующие испытания:
полный reHOM вектора секвенируется на
пассаже, соответствующем производственной
серии, инструментально подтвержденная после
довательность сравнивается с теоретической
последовательностью. определенной по KOH
струкции вектора и имеющимся базам данных;
рестрикционный ферментативный
анализ выполняется на ДНК вектора rлавно
ro посевноro материала, каждоrо рабочеro по
севноro материала и производственной серии;
вирусная ДНК выделяется, очищается и rидро
лизуется с достаточным разрешением; rидро
лизованные cerMeHTbI разделяются с помо
щью электрофореза в rеле или капиллярноrо
электрофореза, полученные рестрикционные
картины сравниваются с теоретическими pe
стрикционными картинами, рассчитанными по
конструкции вектора;
приемлемое количество изолированных
суб клонов исследуется на наличие признаков
экспрессии rенетическоro материала (матери
алов) и биолоrической активности на уровне
пассажа, соответствующеrо производствен
ной серии; при выявлении суб клонов с более
низким уровнем экспрессии или биолоrичес
кой активности требуются дальнейшие испы
тания;
допустимая вариабельность клеток хо
зяина оценивается путем подтверждения pe
пликационных свойств вектора и их сравнения
с исходным вирусом на уровне пассажа, COOT
ветствующеro производственной серии.
Титр инфекционноrо вируса. Титр инфек
ционноro вируса определяется в rлавном посев
ном материале и каждом рабочем посевном Ma
териале.
Посторонние areHTbI (2.6.16). В rлавном
посевном материале и каждом рабочем по
севном материале должны отсутствовать по
сторонние areHTbI, за исключением случаев,
Korдa цитопатические линии не MorYT быть
нейтрализованы и вектор вызывает интерфе
ренцию. Если испытание не может быть про
ведено, допускается использование альтер
нативной валидированной методики.
КУЛЬТИВИРОВАНИЕ И СБОР
БИОМАССbI
Все операции с банком клеток и последу
ющими культурами клеток проводятся в зоне
с приемлемым уровнем контаминации при
отсутствии одновременной работы с друrи
ми клетками или векторами. Любой матери
ал, полученный от человека, или животноro
происхождения, используемый в приroтовле
нии клеточных суспензий и питательных сред,
контролируется. Среда для культивирования
клеточных культур может содержать pH
индикатор, например, феноловый красный,
и подходящие антибиотики в наименьшей
эффективной концентрации, однако более
предпочтительно использовать в процессе
производства клеточную культур не coдep
жащую антибиотики. Если иное не обоснова
но и разрешено, ни на одной из технолоrиче
ских стадий не используется пенициллин или
стрептомицин. Часть производственной куль
туры клеток (неинфицированной клеточной
культуры) отбирается для использования в
качестве контрольных клеток.
Последующая очистка партии биомассы
проводится, если она соответствует указанным
далее требованиям.
Подлинность. Вектор идентифицируют с
помощью иммунохимических методов (2.7.1),
методов амплификации нуклеиновых кислот
(2.6.21).
5.14. Лекарственные средства для zенной терапии
967
Титр инфекционноrо вектора. В каждой
партии биомассы определяется титр инфекци
онноro вектора.
Посторонние areHTbI (2.6.16). В каждой
партии биомассы должны отсутствовать посто
ронние areHTbI, за исключением случаев, Korдa
цитопатические линии не MOryт быть нейтрали
зованы и вектор вызывает интерференцию. Если
испытание не может быть проведено, допуска
ется использование альтернативной валидиро
ванной методики.
Контрольные клетки. Если для производ
ства используются диплоидные клетки человека
или перевиваемая клеточная линия, контроль
ные клетки должны удовлетворять требовани
ям испытания на подлинность (5.2.З). Клетки не
должны содержать посторонних areHTOB (2.6.16).
ОЧИЩЕННАЯ БИОМАССА
Операции выполняются в асептических yc
ловиях. Несколько партий биомассы MOryT быть
объединены перед процессом очистки. Биомас
са сначала очищается путем удаления клеток, а
затем, если применимо, очищается валидиро
ванными методами.
Для производства roтовоro нефасованноrо
продукта используется очищенная биомасса, co
ответствующая указанным далее требованиям.
Подлинность. Вектор идентифицируют с
помощью иммунохимических методов (2.7.1),
методов амплификации нуклеиновых кислот
(2.6.21).
Целостность reHoMa. Целостность reHoMa
вектора подтверждается соответствующими Me
тодами, например, рестрикционным фермента
тивным анализом.
Титр инфекционноrо вектора. В каждой
партии биомассы определяется титр инфекци
онноro вектора.
Отношение титра инфекционноrо вектора
к концентрации общеrо белка. Концентрация
общеro белка определяется соответствующим
методом (2.5.3З). Рассчитывается соотношение
титра инсрекционноrо вектора к концентрации
общеro белка.
Остаточные белки клетки хозяина. KOH
центрация остаточноro белка клетки хозяина
определяется приroдным для этоro иммунохими
ческим методом (2.7.1), если I1рИ валидации про
цесса не было показано достижение требуемой
степени очистки продукта.
Остаточная ДНК клетки-хозяина. Coдep
жание остаточной ДНК клетки хозяина опреде
ляется с использованием приroдноrо для этоro
метода, если валидацией процесса не было пока
зано достижение требуемой степени очистки про
дукта. Учитывая чувствительность и специфич
ность, для этоro рекомендуется количественный
метод полимеразной цепной реакции (ПЦР),
друrие методы также MOryт быть использованы.
Остаточные реактивы. Если в производ
ственном процессе используются реактивы, ис
пытания на наличие этих веществ проводятся на
очищенной биомассе, если валидацией процес
са не было показано достижение требуемой CTe
пени очистки продукта.
Остаточные антибиотики. Если в произ
водственном процессе используются антибио
тики, их остаточная концентрация определяется
микробиолоrическим методом (адаптированным
общим методом 2.7.2) или друrими приrодными
методами (например, жидкостной хроматоrрафи
ей), если валидацией процесса не было показано
достижение требуемой степени очистки продукта.
rотовый НЕФАСОВАННЫЙ ПРОДУКТ
Несколько партий очищенной биомассы
MOryT быть объединены перед приroтовлени
ем rOToBoro продукта. MorYT быть добавлены
стабилизатор и друrие вспомоrательные Be
щества.
Для получения roтовоro лекарственно
ro средства используется продукт, COOTBeTCTBY
ющий нижеуказанному требованию.
Стерильность (2.6.1). Продукт должен быть
стерильным.
rотовый ПРОДУКТ
К выпуску разрешается roтовый продукт,
удовлетворяющий требованиям разделов «Под
линность», «Количественное определение»,
«Испытания», описанным далее. Если испы
тание на наличие бычьеro сывороточноro аль
бумина (если он используется в производстве
вектора) было проведено на roтовом нефасо
ванном продукте с положительными результата
ми, оно может не про водиться при контроле ro
TOBoro продукта.
ПОДЛИННОСТЬ
Подлинность вектора подтверждают с по
мощью иммунохимических методов (2.7.1),
методов амплификации нуклеиновых кислот
(2.6.21).
ИСПЫТАНИЯ
Осмоляльность (2.2.35). Зl1ачение OCMO
ляльности должно находиться в установленных
для конкретноro продукта пределах.
рН (2.2.3). Значение рН должно находиться
в установленных для конкретноro продукта пре
делах.
968
rосударственная фармакопея Республики Беларусь
Извлекаемый объем (2.9.17). Испытуемый
образец должен выдерживать испытание на из
влекаемый объем.
Вода (2.5.12). Содержание влаrи должно Ha
ходиться в установленных для конкретноro лио
филизированною продукта пределах.
Бычий сывороточный альбумин. Если в
производстве использовалась сыворотка круп
ноro poraToro скота. содержание бычьеrо cы
вороточною апьбумина не должно превышать
установленноro для конкретноro продукта пре
дельноro значения при определении подходя
щим иммунохимическим методом (2.7.1).
Стерильность (2.6.1). Испытуемый обра
зец должен быть стерильным.
Бактериальные эндотоксины (2.6.14).
Менее установленною предельноro значения
для конкретноro продукта.
Термическая стабильность. Образцы ro
товоro препарата вектора выдерживают при
температуре и в течение времени, установлен
ных и одобренных для конкретноro продукта.
Определяется общая концентрация инфекци
oHHoro вектора после наrревания, в COOTBeT
ствии с методикой, приведенной в разделе
«Количественное определение». В парал
лельных испытаниях определяется KOHцeH
трсщия вектора в образце, не подверrавшемся
наrреванию. Выявленная разница KOHцeHTpa
ций вектора в образце, не подверrшемся Ha
rреванию, и образце, подверrшемся HarpeBa
нию, должна находиться в установленных для
конкретноro продукта пределах.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Титр инфекционноrо вектора. Не менее
3 флаконов испытуемоro образца титруют и
вносят в клеточную культуру. Для валидации
каждоro испытания используется титрование
флакона соответствующеro стандартною образ
ца вектора.
Титр вектора в испытуемом образце должен
быть не менее титра, указанноro на этикетке.
Результаты испытания являются HeДOCTO
верными, если:
доверительный интервал (Р = 0,95) лоrа
рифма концентрации вектора больше нормы,
установленной уполномоченном компетентным
opraHoM;
титр инфекционноro вектора в стандарт
ном образце выходит за рамки значений, YCTa
новленных в контрольном rрафике.
Экспрессия rенетическоrо материала
в продукте. Экспрессия rенетическою MaTe
риала (материалов) в продукте определяет
ся Bcerдa, если это возможно, после введения
испытуемою образца в культуры клеток в за
ранее определенной инфицирующей дозе с
помощью приroдных для этоro иммунохимиче
ских (2.7.1) или биохимических методов коли
чественноrо определения или проточной цито
метрией (2.7.24).
Биолоrическая активность. Если иное не
обосновано и не утверждено, биолоrическая aK
тивность определяется с помощью COOTBeTCTBY
ющеrо испытания in vitro или in vivo.
МАРКИРОВКА
На этикетке указывается:
минимальный титр вектора в одной дозе;
рекомендуемая терапевтическая доза;
для лиофилизированных продуктов:
название и объем растворителя, с помо
щью KOToporo проводят растворение продукта;
время, в течение котороro продукт должен
быть использован после растворения.
Векторы на основе
семейства ретровирусов
для медицинскою применения
ОПРЕДЕЛЕНИЕ
Векторы на основе семейства ретровирусов,
предназначенные для медицинскоro примене
ния, производятся в лиофилизированном виде
или в виде жидких препаратов рекомбинантных
ретровирусов, лентивирусов или спумавирусов,
rенетически модифицированных с целью лиwе
ния их способности к репликации, которые ис
пользуются для переноса rенетическоrо матери
ала в соматические клетки человека iп vivo или
ех vivo. Этот подраздел относится к нереплика
ционным векторам.
ПРОИЗВОДСТВО
КОНСТРУКЦИЯ ВЕКТОРА
Типичный вектор содержит:
минимальный reHoM исходных вирусов,
содержащий структурные rенетические элемен
ты, необходимые для производства вектора;
требуемые реryляторные rенетические
элементы для экспрессии целевою rенетическо
ro материала (например, длинные концевые по
вторы);
введенный целевой rенетический матери
ал. Конструкция вектора должна предотвращать
появление репликационно компетентных виру
сов.
ПРОИЗВОДСТВО ВЕКТОРА
Должно быть показано, что методы про
изводства векторов обеспечивают постоянное
получение вектора требуемоro качества. Если
иное не обосновано и разрешено, клетки упа
ковщики или клетки продуценты не должны
подверrаеться большему удвоению клеток из
5.14. ЛекарствеННblе средства для zенной терапии
969
rлавноro банка клеток, чем клетки, использо
ванные для получения вектора, чья эффек
тивность и безопасность была доказана в
клинических исследованиях. rенетическая и
фенотипическая стабильность клеток упаков-
щиков или клеток продуцентов на уровне или
ниже максимальноrо количества удвоения
клеток, использованных для производства BeK
тора, оценивается приroдными для этоro MeTO
дами. Вектор получают на постоянно культиви
руемой линии клеток (5.2.3) с использованием
системы банка клеток. В производстве MorYT
использоваться или постоянно, или временно
трансфицированные клетки.
ОПРЕДЕЛЕНИЯ
Клетки упаковщики: исходная клеточная
линия, постоянно трансфицированная плазми
дами, содержащими вирусные reHbI, необходи
мые для производства пустых векторных частиц:
gag, ро/, епv.
Клетки продуценты: содержат вирусные
reHbI и экспрессионную кассету, необходимые
для производства вектора.
В постоянной производственной системе,
клетки продуценты получают путем устойчивой
трансфекции линии клеток упаковщиков с помо
щью трансферной плазмиды, содержащей Tepa
певтические последовательности нуклеотидов.
Во временной производственной системе
клетки продуценты получают при производстве
однократно путем одновременной трансфекции
исходной клеточной линии вирусными rенами
и трансrенной экспрессионной плазм идой или
путем временной трансфекциии линии клеток
упаковщиков трансферной плазмидой, содержа
щей терапевтические последовательности HY
клеотидов.
ПРОИЗВОДСТВО ПОЛУПРОДУКТОВ
Клетки упаковщики
Номер копии. Из определенноro количе-
ства клеток выделяется и очищается rеномная
ДНК и reHbI gag, ро/ и епу. Номер копии опреде-
ляется приroдным для этоro методом, например,
количественной ПЦР (2.6.21).
Целостность последовательности вирус-
ных reHoB. Проводится полное секвенирование
введенных вирусных reHoB и их реryляторных
элементов.
rенетическая стабильность. rенетическая
стабильность клеток упаковщиков подтвержда
ется на уровне или ниже максимальноro количе
ства удвоения клеток, использованных для про
изводства вектора.
Плазмиды
При производстве вектора требуется ис
пользование промежуточных плазмид. Для
123. За. 1712.
каждой ДНК плазмиды, используемой в про-
изводстве, приводится полное описание,
включая идентификацию, источник получе
ния, способы выделения и нуклеотидную
последовательность. Описываются источник и
функции компонентов этих плазм ид, такие как
происхождение репликации, вирусные и эука-
риотические промоутеры, reHbI, кодирующие
селекционных маркеров. Производство плаз
мид полупродуктов основано на системе
банка бактериальных клеток. rлавный банк
клеток должен соответствовать требованиям
подраздела «Бактериальные клетки, исполь-
зуемые для производства плазмидных BeKTO
ров для медицинскоro применения». Плазми
ды очищаются с помощью соответствующих
методов.
Для производства вектора используют
ся серии плазмид. отвечающие приведенным
далее требованиям.
Подлинность. Плазмиды идентифицируют
с помощью рестрикционноro ферментативноro
анализа, секвенирования или методами ампли
фикации нуклеиновых кислот (2.6.21).
Целостность reHoMa. Целостность плаз
мидноrо reHoMa подтверждается COOTBeTCTBY
ющими методами, например, рестриционным
ферментным анализом вирусных reHoB, введен-
ноro rенетическоro материала и их реryляторных
элементов соответственно.
Плазмидная ДНк. Нижеприведенные YKa
зания даны в качестве примера.
Концентрация ДНК выше 500 нr/мл может
быть определена путем измерения величины
поrлощения при 260 нм. Величина поrлощения
раствора двойной кольцевой ДНК в KOHцeHTpa
ции 50 мкr/мл составляет 1 (удельное поrлоще-
ние 200).
Содержание ДНК в концентрации менее
500 нпмл определяется после инкубирования
с флуоресцентным красителем, который из
бирательно связывается с двойной кольцевой
ДНК, и использованием стандартноrо образ-
ца ДНК дЛЯ построения калибровочной кривой.
Для определения плазмидной ДНК также может
быть использована жидкостная хроматоrрафия
с применением стандартноro образца. В HeKO
торых случаях используется капиллярный элек
трофорез.
Остаточная ДНК клетки-хозяина. Coдep
жание остаточной ДНК клетки хозяина опреде
ляется с использованием приrодноrо для этоro
метода, если валидацией процесса не было по
казано достижение требуемой степени очистки
продукта. Учитывая чувствительность и специ-
фичность, для этоro рекомендуется количествен-
ный метод полимеразной цепной реакции (ПЦР),
друrие методы также MOryт быть использованы.
970
rосударственная фармакопея Республики Беларусь
Бактериальные эндотоксины (2.6.14).
Менее установленноrо предельноro значения
для конкретноro продукта.
Стерильность (2.6.1). Плазмида должна
быть стерильной.
Клетки продуцентbl. используемые
в постОЯННblХ производствеННblХ
системах
Номер копии. Номера копии интеrрирован
ных вирусных reHoB и экспрессионной кассеты
определются приrо,оным для этоro методом.
rенетическая стабильность. rенетическая
стабильность клеток упаковщиков подтвержда
ется на уровне или ниже максимальноro количе
ства удвоения клеток, использованных для про
изводства вектора.
Целостность последовательности вирус
ных reHoB и экспрессионной кассеты. Про
водится полное секвенирование введенных ви
русных reHoB. экспрессионной кассеты и их
реryляторных элементов соответственно (Ha
пример, длинный концевой повтор, промоутеры,
psi последовательность, сиrнал полиаденилиро
вания).
Репликационно компетентные вирусы.
Обнаружение репликационно компетентных ви
русов проводится С помощью подходящеrо для
этою метода. Метод детектирования может
быть основан на совместном культивировании
нескольких поспедовательно удвоенных куль
тур клеток продуuентов с чувствительной кле
точной линией с последующим обнаружением
репликации (или по наличию цитопатическо
ro или rемоадсорбционною эффекта вируса
на клетки индикаторы типа PG4 S+L , или с ис
пользованием линий клеток индикаторов и амп
лификацией нуклеиновых кислот (2.6.21) или
путем количественною испытания на спасение
reHa MapKepa). В каждое определение включают
положительные контроли для мониторинrа чув
ствительности методики. Репликационно компе
тентные вирусы должны отсутствовать.
КУЛЬТИВИРОВАНИЕ И СБОР БИОМАССbI
Все операции с банком клеток и последу
ющими культурами клеток проводятся в зоне с
приемлемым уровнем контаминации при OTCYT
ствии одновременной работы с друrими клетка
ми или векторами. Любой материал, получен
ный от человека, или животною происхождения,
используемый в приroтовлении клеточных cy
спензий и питательных сред, контролируется.
Предпочтительно использовать в процессе про
изводства субстрат, не содержащий антибиоти
ки. Если иное не обосновано и разрешено, ни на
одной из технолоrических стадий не использует
ся пенициллин или стрептомицин.
Последующая очистка партии биомассы
проводится, если она соответствует указанным
далее требованиям.
Подлинность. Вектор идентифицируют с
помощью иммунохимических методов (2.7.1),
методов амплификации нуклеиновых кислот
(2.6.21) или рестрикционным ферментативным
анализом.
Концентрация вектора. В каждой партии
биомассы определяется титр инфекционноro
вектора и/или концентрация векторных частиц.
Посторонние areHTbI. Каждая партия био
массы не должна содержать посторонних areH
тов (2.6.16).
Контрольные клетки. Контрольные клетки
должны выдерживать испытание на подлин
ность (5.2.3) и на отсутствие посторонних areH
тов (2.6.16).
ОЧИЩЕННАЯ БИОМАССА
Несколько партий биомассы MOryT быть
объединены перед процессом очистки. Для про
изводства roтовоro нефасованноro продукта ис
пользуется очищенная биомасса, COOTBeTCTBY
ющая указанным далее требованиям.
Подлинность. Вектор идентифицируют с
помощью иммунохимических методов (2.7.1),
методов амплификации нуклеиновых кислот
(2.6.21) или рестрикционным ферментативным
анализом.
Целостность reHoMa. Целостность reHoMa
вектора подтверждается соответствующими Me
тодами.
Концентрация вектора. В каждой партии
биомассы определяется титр инфекционных
частиц приroдным для этоro метода, напри
мер, инфицированием чувствительных клеток
с последующим количественным методом aM
плификации нуклеиновых кислот (например,
количественной ПЦР), Саузерн блотом или
определением экспрессированноro белка. Для
лентивирусных векторов определяется физиче
ский титр, например, с помощью твердофазноrо
иммуноферментноrо анализа (р24).
Репликационно компетентные вирусы.
Обнаружение реппикационно компетентных ви
русов про водится С помощью подходящеro для
этою метода. Как правило, для этоro использу
ется амплификация н::з чувствительных клетках
с последующей амплификацией нуклеиновых
кислот (2.6.21), с помощью обнаружения вирус
ноro антиrена (например, р24 методом TBepдo
фазною иммуноферментноro анализа (тИФА,
ELlSA)) или количественным испытанием на
спасение reHa MapKepa. В каждое определение
5.14. ЛекарствеННblе средства для 2енной терапии
971
включают положительные контроли для мони
торинrа чувствительности методики. Испытание
проводится на очищенной биомассе или на ro
товом продукте. Репликационно компетентные
вирусы должны отсутствовать.
Остаточные белки клетки-хозяина. KOH
центрация остаточных белков клетки хозяина
определяется с использованием приrодных им
мунохимических методов (2.7.1), если валидаци
ей процесса не было показано достижение Tpe
буемой степени очистки продукта.
Остаточная ДНК клетки-хозяина. Coдep
жание остаточной ДНК клетки хозяина опреде
ляется с использованием приrодноrо для этоro
метода, если валидацией процесса не было пока
зано достижение требуемой степени очистки про
дукта. Учитывая чувствительность и специфич
ность, для этоro рекомендуется количественный
метод полимеразной цепной реакцир (ПЦР),
друrие методы также MOryт быть использvваl-iЫ.
Остаточные реактивы. Если в производ
ственном процессе используются реактивы, ис
пытания на наличие этих веществ проводятся на
очищенной биомассе, если валидацией процес
са не было показано достижение требуемой CTe
пени очистки продукта.
Остаточные антибиотики. Если в производ
ственном процессе используются антибиотики. их
остаточная концентрация определяется микро
биолоrическим методом (адаптированным общим
методом 2.7.2) или друrими приroдными Meтo
дами (например, жидкостной хроматоrрафией),
если валидацией процесса не было показано дo
стижение требуемой степени очистки продукта.
Остаточные плазмиды. При использова
нии временноro производственноro процесса,
должна быть определена допустимая KOHцeH
трация остаточных плазм ид, представляющих
собой посторонние примеси.
,ОТОВЫЙ НЕФАСОВАННЫЙ ПРОДУКТ
Несколько партий очищенной биомассы
MOryT быть объединены перед приrотовлением
roТOBOro продукта. MoryT быть добавлены CTa
билизатор и друrие вспомоrательные вещества.
rотовый продукт подверrается стерилизующей
фильтрации.
Для получения roTOBOro лекарственно
ro средства используется продукт, COOTBeTCTBY
ющий нижеуказанным требованиям.
Стерильность (2.6.1). Продукт должен быть
стерильным.
,ОТОВЫЙ ПРОДУКТ
К выпуску разрешается roтовый
удовлетворяющий требованиям
продукт,
разделов
«Подлинность», «Количественное определе
ние», «Испытания», описанным далее. Если
испытания на наличие бычьеro сывороточно
ro альбумина (если он используется в про
изводстве вектора) и репликационно компе
тентных вирусов были проведены на roTOBOM
нефасованном продукте с положительными
результатами, они MorYT не проводиться при
контроле roTOBOro продукта.
ПОДЛИННОСТЬ
Подлинность вектора, полученноro на
основе семейства ретровирусов, подтверж
дают с помощью иммунохимических методов
(2.7.1), методов амплификации нуклеиновых
кислот (2.6.21) или рестрикционным фермента
тивным анализом.
ИСПЫТАНИЯ
Осмоляльность (2.2.35). Значение OCMO
ляльности должно находиться в установленных
для KOhKpeTHoro продукта пределах.
рН (2.2.3). Значение рН должно находиться в
установленных для KOHKpeTHoro продукта пределах.
Извлекаемый объем (2.9.17). Испытуемый
образец должен выдерживать испытание на из
влекаемый объем.
Вода (2.5.12). Содержание влаrи должно Ha
ходиться в установленных для кон ;ретноro лио
филизированноro прuдукта пределах.
Бычий сывороточный альбумин. Если
в производстве использоваЛ8СЬ сыворотка
крупноro poraToro скота, содержание бычье
ro сывороточноrо альбумина не должно пре
вышать установленноro для конкретноro про
дукта предельноro значения при определении
подходящим иммунохимическим методом
(2.7.1).
Репликационно-компетентные вирусы.
Обнаружение репликационно компетентных
вирусов проводится с помощью подходящеro
для этоro метода. Как правило, для этоro ис
пользуется амплификация на чувствительных
клетках с последующей амплификацией HY
клеиновых кислот (2.6.21), с помощью обна
ружения вирусноro антиrена (например, р24
методом твердофазноro иммуноферментно
ro анализа) или количественным испытанием
на спасение reHa MapKepa. В каждое опреде
ление включают положительные контроли для
мониторинrа чувствительности методики. Ис
пытание проводится на очищенной биомассе
или на roтовом продукте. Репликационно ком
петентные вирусы должны отсутствовать.
Стерильность (2.6.1). Испытуемый обра
зец должен быть стерильным.
972
rосударственная фармакопея Республики Беларусь
Бактериальные эндотоксины (2.6.14).
Менее установленноro предельноrо значения
для конкретноro продукта.
Концентрация векторных частиц. Фи
зическое титрование про водится приrодным
для этоro методом (например, иммунохими
чески ми методами (2.7.1) или амплификацией
нуклеиновых кислот (2.6.21». Для валидации
каждоro испытания применяется COOTBeTC
твующий стандартный образец вектора.
Титр инфекционноrо вектора. Титр испы
TyeMoro образца определяется по еro влиянию
при внесении в культуру клеток. Для валида
ции каждоro испытания применяется COOTBeT
ствующий стандартный образец вектора.
Титр инфекционноro вектора испыту
емоro образца должен быть не меньше мини
мальноro титра, указанноro на этикетке.
Результаты испытания являются HeДOCTO
верными, если:
доверительный интервал (Р = 0,95)
лоrарифма концентрации вектора больше
нормы, установленной компетентным уполно
моченном opraHoM;
титр инфекционноro вектора в CTaH
дартном образце выходит за рамки значений,
установленных контрольной кривой.
Соотношение концентрации векторных
частиц с титром инфекционноrо вектора.
Должно соответствовать норме, установлен
ной для KOHKpeTHoro продукта.
Экспрессия rенетическоrо материала
в продукте. Экспрессия rенетическоrо MaTe
риала (материалов) в продукте определяет
ся всеrда, если это возможно, после BBeдe
ния испытуемоro образца в культуры клеток
в заранее определенной инфицирующей дозе
с помощью приrодных для этоro иммунохими
ческих (2.7.1) или биохимических методов KO
личественноro определения или проточной
цитометрией (2.7.24).
Биолоrическая активность. Если иное
не обосновано и не утверждено, биолоrиче
ская активность определяется с помощью co
ответствующеrо испытания in vitro или in vivo.
МАРКИРОВКА
На этикетке указывается:
минимальный титр вектора в терапев
тической дозе;
реКОМl3ндуемая терапевтическая доза;
для лиофилизированных препаратов:
название и объем растворителя, с по
мощью KOToporo проводят растворение про
дукта;
время, в течение котороro продукт
должен быть использован после растворения.
Аденоассоциированные
вирусные векторы
для медицинскоro применения
ОПРЕДЕЛЕНИЕ
Аденоассоциированные вирусные (AAV)
векторы для медицинскою применения произ
водятся в лиофилизированном виде или в виде
жидких препаратов рекомбинантных aдeHoacco
циированных вирусов (rAAV), rенетически моди
фицированных для обеспечения переноса re
нетическоrо материала в соматические клетки
человека in vivo или ех vivo.
ПРОИЗВОДСТВО
КОНСТРУКЦИЯ ВЕКТОРА
(АА V векторы получают путем замещения
(ер и сар reHoB терапевтическим rенетическим
материалом. Инвертированные терминальные
повторы (/TR) последовательности сохраняют
ся в rAAV векторе, так как они являются един
ственными АА V последовательностями, которые
необходимы для осуществления cis функции как
основы репликации. reHbI 'ер и сар требуются
для t,ans и функций репликации и упаковки COOT
ветственно. Таким образом, ,AAV вектор coдep
жит /TR последовательности и встроенный reHe
тический материал.
Дикие типы АА V векторов, как правило,
реплицируются только в присутствии вирусов
хелперов, в частности сопутствующим aдeHO
вирусом или вирусом rерпеса. В связи с этим,
существуют различные подходы к производ
ству AAV векторов. Производственная CTpaTe
rия выбирается с учетом минимизации риска
возникновения репликационно компетентных
AAV векторов и эффективноro удаления виру
сов хелперов, которые MorYT быть использо
ваны при производстве.
ПРОИЗВОДСТВОВЕКТОРА
Должно быть продемонстрировано, что
методы производства векторов обеспечивают
постоянное получение вектора требуемоro каче
ства.
Для получения AAV векторов в настоящее
время существует несколько стратеrий, напри
мер:
временная совместная трансфекция кле
точной линии плазмидами, содержащими /TRs и
rенетический материал, (ер и сар reHbI и компо
ненты, выполняющие функции хелперов;
инфицирование линии клеток продуцен
тов, содержащих 'ер и сар reHbI, /TRs и терапев
тический rенетический материал, репликацион
но дефицитными вирусами хелперами;
инфицирование линии чувствительных
клеток одним или несколькими производствен
ными вирусами с кодами 'ер и/или сар и/или
терапевтическим rенетическим материалом и
/TRs, что может обеспечивать или не обеспе
5.14. Лекарственные средства для zенной терапии
973
чивать выполнение функций хелперов (вирусы
хелперы и бакуловирусы соответственно).
В зависимости от выбранной стратеrии
производства АА V векторов требуются раз
личные полупродукты (плазмиды, вирусы, ис
пользуемые для производства, клетки упаков
щики).
Риск появления репликационно компе
тентных АА V векторов может быть значитель
ным, KorAa существуют roмолоrичные области
между reHoMoM производственных полупродук
тов и ,AAV вектором. Эта вероятность может
быть уменьшена путем снижения roмолоrично
сти этих reHoMoB до минимума. Рекомендуется
использовать в производстве полупродукты без
roмолоrичных последовательностей.
rенетическая и фенотипическая стабильность
вектора, использованноro для производства на
максимальном количестве пассажей или менее,
оценивается приroдными для этоro методами.
ПРОИЗВОДСТВО ПОЛУПРОДУКТОВ
Вирусы, используемые в производстве, и
,АА V вектор производятся в постоянно культи
вируемой линии клеток (5.2.3) с использованием
системы посевноro материала и системы банка
клеток.
Клетки упаковщики и клетки продуценты
Номер копии. Из определенноro количе
ства клеток выделяется и очищается rеномная
ДНК, номера копии введенных вирусных reHoB и
экспрессионной кассеты определяются приroд
ным для этоro методом, например, количествен
ной ПЦР (2.6.21).
Целостность последовательности вирус-
ных reHoB и экспрессионной кассеты. Прово
дится полное секвенирование введенных вирус
ных reHoB, их реryляторных элементов и, если
применимо, экспрессионной кассеты.
rенетическая стабильность. rенетическая
стабильность клеток подтверждается на уровне
или ниже максимальноro количества удвоения
клеток, использованных для производства BeK
тора.
Дикие типы ААУ векторов. Отсутствие
диких типов АА V векторов подтверждается MeTO
дом амплификации нуклеиновых кислот (2.6.21).
Плазмиды
При производстве АА V вектора методом co
bmeCT!-IОЙ трансфекции требуется использова
ние ПРОr./lежуточных плазмид. Для каждой ДНК
плазмиды, используемой в производстве, при
водится полное описание, включая идентифи
кацию,источникполучения,способывыделения
и нуклеотидную последовательность. Описыва
ются источник и функции компонентов этих плаз
мид, такие как происхождение репликации. ви
:24.За 17J2
русные и эукариотические промоутеры, reHbI,
кодирующие селекционных маркеров.
Производство плазмид полупродуктов oc
новано на системе банка бактериальных клеток.
rлавный банк клеток должен. соответствовать
требованиям раздела «Бактериальные клетки,
используемые для производства плазмидных
векторов для медицинскоro применения». Плаз
миды очищаются с помощью соответствующих
методов.
Для производства вектора используют
ся серии плазм ид, отвечающие приведенным
далее требованиям.
Подлинность. Плазм иды идентифицируют
с помощью рестрикционноro ферментативноro
анализа, секвенирования или методами ампли
фикации нуклеиновых кислот (2.6.21).
Целостность reHOMa. Целостность плаз
мидноro reHoMa подтверждается COOTBeTCTBY
ющими методами, например, рестрикционным
ферментативным анализом области, COOTBeT
ствующей ,ер, сар и экспрессионной кассете.
Плазмидная ДНк. Нижеприведенные YKa
зания даны в качестве примера.
Концентрация ДНК выше 500 нr/мл может
быть определена путем измерения величины
поrлощения при 260 нм. Величина поrлощения
раствора двойной кольцевой ДНК 8 KOHцeHTpa
ции 50 мкr/мл составляет 1 (удельное поrлоще
ние 200).
Содержание ДНК в концентрации менее
500 нпмл определяется после инкубирования
с флуоресцентным красителем, который из
бирательно связывается с двойной кольцевой
ДНк. и использованием стандартноro образ
ца ДНК дЛЯ построения калибровочной кривой.
Для определения плазмидной ДНК также может
быть использована жидкостная хроматоrрафия
с применением стандартноro образца. В HeKO
торых случаях используется капиллярный элек
трофорез.
Остаточная ДНК клетки-хозяина. Coдep
жание остаточной ДНК клетки хозяина опреде
ляется с использованием приrодноrо для этою
метода, если валидацией процесса не было по
казано достижение требуемой степени очистки
продукта. Учитывая чувствительность и специ
фичность, для этоro рекомендуется количест
венный метод полимеразной цепной реакции
(ПЦР), друrие методы также MOryт быть исполь
зованы.
Бактериальные эндотоксины (2.6.14).
Менее установленноro предельноro значения
для конкретною продукта.
Стерильность (2.6.1). Плазм ида должна
быть стерильной.
974
rосударственная фармакопея Республики Беларусь
Вирусы, используемые для производства
Производство вирусов основано на систе
ме посевноro материала и банке клеток или,
если применимо (например, для бакуловиру
сов), на временной системе. Используемая
линия вирусов идентифицируется по данным
разработки, включая информацию о ее проис
хождении и последующих операциях, в част
ности об удалении или модификации областей
reHoMa. Нуклеотидная последовательность ви
русов описывается.
Для производства используется вирус,
соответствующий приводимым далее требова
ниям.
Подлинность. Вирусы, используемые для
производства, идентифицируют с помощью
иммунохимических методов (2.7.1), методами
амплификации нуклеиновых кислот (2.6.21) или
ферментативным рестрикционным анализом.
Целостность reHoMa. Целостность reHoMa
вируса, использующеroся для производства,
подтверждается приroдным для этоro методом,
например, рестрикционным ферментативным
анализом. Если вирусы модифицированы для
экспрессии reHoB rep или сар или экспрессион
ной кассеты, целостность rHHoMa оценивается
секвенированием или количественной ПЦР этих
областей.
rенетическая стабильность. При исполь
зовании стабильной производственной систе
мы rенетическая стабильность подтверждается
на уровне или ниже максимальноro количества
удвоения клеток, использованных для производ
ства вируса.
Титр вируса. Инфекционный титр определя
ется соответствующим количественным методом.
Дикие типы AAV векторов. Если примени
мо, отсутствие диких типов АА V векторов в по
севном материале вирусов хелперов подтверж
дается методом амплификации нуклеиновых
кислот (2.6.21).
Репликационно-компетентные вирусы.
Обнаружение репликационно компетентных ви
русов проводится соответствующими методами.
Репликационно компетентные вирусы должны
отсутствовать.
Посторонние areHTbI (2.6.16). Посторонние
areHTbI должны отсутствовать. В дополнение
должно проводиться определение возможной
контаминации специфическими вирусами Hace
комых, если применимо.
ПРОИ3ВОДСТВО И СБОР БИОМАССЬ1
Все операции с банком клеток и после
дующими культурами клеток про водятся
в зоне с приемлемым уровнем контамина
ции при отсутствии одновременной работы с
друrими клетками или векторами. Любой Ma
териал, полученный от человека, или живот
ноro происхождения, используемый в приro
товлении клеточных суспензий и питательных
сред, контролируется. Среда для кулыивиро
вания клеточных культур может содержать pH
индикатор, например, феноловый красный,
и подходящие антибиотики в наименьшей
эффективной концентрации, однако более
предпочтительно использовать в процессе
производства субстрат, не содержащий анти
биотики, ни на одной из технолоrических CTa
дий не используется пенициллин или стреп
томицин. Часть производственной культуры
клеток (неинфицированная клеточная кулыу
. ра) отбирается для использования в качестве
контрольных клеток.
Последующая очистка партии биомассы
проводится, если она соответствует указанным
далее требованиям.
Подлинность. Вектор идентифицируют с
помощью иммунохимических методов (2.7.1),
методов амплификации нуклеиновых КИСЛОТ
(2.6.21) или рестрикционным ферментативным
анализом.
Концентрация вектора. В каждой партии
биомассы определяется титр инфекционноro
вектора и концентрация векторных частиц.
Посторонние areHTbI (2.6.16). Посторонние
areHTbI в партии биомассы должны OTCYTCTBO
вать.
Контрольные клетки. Контрольные клетки
должны выдерживать испытания на подлин
ность (5.2.3), на наличие посторонних areHТOB
(2.6.16) и специфических вирусов насекомых,
если в производстве используются линии клеток
насекомых.
ОЧИЩЕННАЯ БИОМАССА
Несколько партий биомассы MorYT быть
объединены перед процессом очистки. Процесс
очистки валидируется с целью подтверждения
эффективноro удаления примесей.
Для производства roтовоro нефасованно
ro продукта используется очищенная биомас
са, соответствующая указанным далее требо
ваниям.
Подлинность. Вектор идентифицируют с
помощью иммунохимических методов (2.7.1),
методов амплификации нуклеиновых кислот
(2.6.21) или рестрикционным ферментативным
анализом.
rенетическая характеристика. ПРОВОДЯТСЯ
следующие испытания.
5.14. ЛекарствеННblе средства для zенной терапии
975
Полный reHoM вектора, полученноro из
приемлемоro количества производственных
циклов, подверrается секвенированию. Испы
тание проводится или на очищенной биомас
се, или на roтовом нерасфасованном продукте.
Определенная при анализе последовательность
сравнивается с теоретической последователь
ностью, рассчитанной по данным о конструкции
вектора и имеюшимся данным.
Целостность reHoMa проверяется на BeK
торе ДНК. Для этоro может использоваться ПЦР.
Концентрация вектора. Определяется титр
инфекционноro вектора и концентрация Beктop
ных частиц.
Остаточные вирусы, использующиеся в
производстве. Наличие остаточных вирусов,
использующихся в производстве, оценивается
количественным методом бляшкообразовачия
или по дозе заражения 50 % КУЛЬТУР I л!ани
(ТСID50) на чувствительных к вирусам клеточ
ных линиях или количественным ПЦР в зави
симости от используемой производственной
системы.
Остаточные белки. Концентрация OCTa
точноrо белка клетки хозяина и/или вирусных
белков определяется приroдным для этоro им
мунохимическим методом (2.7.1), если при в ли
дации процесса не было показано достижение
требуемой степени очистки продукта.
Остаточная ДНК клетки хозяина. Coдep
жания остаточной ДНК клетки хозяина и OCTa
точной ДНК полупродуктов, таких как плазм иды
и производственные вирусы, определяются с
использованием приrодноrо для этоro метода,
если валидацией процесса не было показано
достижение требуемой степени очистки поо
дукта. Учитывая чувствительность и специфич
ность, для этоro рекомендуется количествен
ный метод полимеразной цепной реакции
(ПЦР), друrие методы также MorYT быть ис
пользованы.
Остаточные реактивы. Если в производ
ственном процессе используются реактивы, ис
пытания на наличие этих веществ проводятся на
очищенной биомассе, если валидацией процес
са не было показано достижение требуемой CTe
пени очистки продукта.
Остаточные антибиотики. Если в произ
водственном процессе используются антибио
тики, их остаточная концентрация определяется
микробиолоrическим методом (адаптированным
общим методом 2.7.2) или друrими приrодными
методами (например, жидкостной xpoMaтorpa
фией), если валидацией процесса не было по
казано достижение требуемой степени очистки
продукта.
,ОТОВЫЙ НЕФАСОВАННЫЙ ПРОДУКТ
Несколько партий очищенной биомассы
MOryт быть объединены перед приroтовлением
roтовоro продукта. Moryт быть добавлены CTa
билизатор и друrие вспомоrательные вещества.
rотовый продукт стерилизуется с помощью MeM
бранной фильтрации.
Для получения roтовоro лекарственно
ro средства используется продукт, COOTBeTCTBY
юший нижеуказанному требованию.
Стерильность (2.6.1). Продукт должен быть
стерильным.
,ОТОВЫЙ ПРОДУКТ
К выпуску разрешается roтовый продукт,
удовлетворяющий требованиям разделов «Под
линность», «Количественное определение»,
«Испытания», описанным даЛ61О>.
Еспи испытание на наличие бычьеro CЫBO
роточноro альбумина (если он используется в
'lроизводстве вектора). репликационно компе
,ентных AAV и остаточных вирусов, использу
ющихся В производстве, было проведено на roто
вом нефасованном продукте с положительными
результатами, оно может не про водиться при
кочтроле roтовоro продукта.
ПОДЛИННОСТЬ
Подлинность вектора подтвержд.ают с помо
щью иммунохимичеСI(ИХ методов (2. 7.1), MeTO
ДОВ амплификации нуклеиновых кислот (2.6.21)
или рестриктивноro ферментативноro анализа.
ИСПЫТАНИЯ
Осмоляльность (2.2.35). Значение OCMO
ляльности должно находиться в установленных
для конкретноro продукта пределах.
рН (2.2.3). Значение рН должно находиться в
установленных для конкретноro продукта пределах.
Извлекаемый объем (2.9.17). Испытуемый
образец должен выдерживать испытание на из
влекаемый объем.
Вода (2.5.12). Содержание влаrи должно Ha
ходиться в установленных для /(oHKpeTHoro лио
филизированноro продукта пределах.
Бычий сывороточный альбумин. Если в
производстве использовалась сыворотка КРУП
ноro poraToro скота. содержание бычьеro cы
вороточноro альбумина не должно превышать
установленноro для конкретноro продукта пре
дельноro значения при определении подходя
щим иммунохимическим методом (2.7.1).
Концентрация репликационно компетент
ных AAV векторов. Концентрация должна Haxo
диться в установленных для конкретноro продукта
пределах.
976
rосударственная фармакопея Республики Беларусь
Обнаружение репликационно компетентных
АА V векторов про водится с помощью реплика
ционноrо количественноrо анализа на чувстви
тельной линии клеток, предварительно инсрици
рованных вирусом хелпером. с последующим
анализом репликационных форм с помощью Cay
зерн блота на низкомолекулярной ДНК или путем
обнаружения rep reHa количественной ПЦР.
ArperaTbI вектора. ArperaTbI вектора
определяются приroдным для этоrо методом
(например, по светорассеянию).
Стерильность (2.6.1). Испытуемый обра
зец должен быть стерильным.
Бактериальные ЭНДОТОКСИНЫ (2.6.14).
Менее установленноro предельноro значения
для конкретноro продукта.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Концентрация векторных частиц. Опре
деление концентрации векторных частиц прово
дится приroдным для этоro методом, например,
количественной ПЦР по сравнению с калибро
вочным rрафиком, полученным С использова
нием рекомбинантной АА V плазмиды или CTaH
дартноro образца АА V вектора. Концентрация
векторных частиц в испытуемом образце должна
находиться в установленных для конкретноro
продукта пределах.
Титр инфекционноrо вектора. Титр испы
туемоro образца определяется по ero влиянию
при внесении в культуру клеток. Для валидации
каЖдОro испытания применяется COOTBeTCTBY
ющий стандартный образец вектора.
Тиrр инфекционноro вектора в испытуемом
образце должен быть не меньше минимальноro
количества, указанноro на этикетке.
Результаты испытания являются HeДOCTO
верными, если:
доверительный интервал (Р = 0,95) лоrа
рифма концентрации вектора больше нормы,
установленной компетентным уполномоченном
opraHoM;
титр инфекционноro вектора в стандарт
ном образце выходит за рамки значений, YCTa
новленных в контрольном rрафике.
Соотношение концентрации векторных
частиц с титром инфекционноrо вектора.
Должно соответствовать норме, установленной
для определенноro продукта.
Экспрессия rенетическоrо материала в
продукте. Экспрессия rенетическоro матери
ала в продукте определяется Bcerдa, если это
возможно, после введения испытуемоro образ
ца в культуры клеток в заранее определенной
инфицирующей дозе с помощью приroдных
для этоro иммунохимических (2.7.1) или био
химических методов количественноro опреде
ления или проточной цитометрии (2.7.24).
Биолоrическая активность. Если иное не
обосновано и не утверждено, биолоrическая aK
тивность определяется с помощью COOTBeTCTBY
ющеro испытания iп vitro или in vivo.
МАРКИРОВКА
На этикетке указывается:
содержание действующеro вещества;
рекомендуемая терапевтическая доза, BЫ
раженная в единицах концентрации векторных
частиц;
для лиофилизированных препаратов:
название и объем растворителя, с помо
щью KOTOpOro проводят растворение продукта;
время, в течение котороro продукт должен
быть использован после растворения.
01/2013:51500
5.15. ФУНКЦИОНАЛьно.
ОБУСЛОВЛЕННЫЕ
ХАРАКТЕРИСТИКИ
вспомоrАТЕЛЬНЫХ
ВЕЩЕСТВ
Данная общая статья и разделы частных
статей Функционально обусловленные xapaK
теристики не являются обязательными и пу
бликуются для информации и руководства.
ВВЕДЕНИЕ
Вспомоrательные вещества, предва
рительно исследованные на предмет безо
пасности, используются в рецептуре roто
вых лекарственных средств для придания
им определенных функций. Целью исполь
зования вспомоrательноrо вещества являет
ся rарантирование заданных физических и
биофармацевтических свойств roтовоro ле
KapcTBeHHoro средства.
Функциональные характеристики вспо
моrательных веществ определяются их фи
зическими и химическими свойствами и, в
некоторых случаях, содержанием побочных
продуктов и добавок, использованных для
улучшения желаемых функциональных xa
рактеристик. Кроме тоro, эти характеристи
ки MorYT зависеть от сложных взаимодей
ствий между составляющими лекарственноro
средства и от внешних воздействий во время
производственноrо процесса. Поэтому функ
циональные характеристики вспомоrатель
ных веществ MorYT быть установлены только
5. 15. Функцuонально обусловленные характеристики ВСПОМО2атеЛЬНbfХ веществ 977
в контексте конкретной рецептуры и процес
са производства, зачастую с использованием
нескольких аналитических методик. Знание
этих характеристик может облеrчить приме
нение процессно аналитической технолоrии
(Process Aпa/ytica/ Тесhпо/оgу РАТ).
Однако некоторые свойства BcnoMora
тельных веществ (такие как размер частиц,
требующийся для приroтовления твердых ле
карственных форм, или молекулярная масса
полимерноro материала, используемоro в Ka
честве добавки, увеличивающей вязкость)
MorYT относиться к функциональным xapaKTe
ристикам в более широком понимании. Если
на этапе разработки была продемонстриро
вана критическая роль таких функциональ
но обусловленных характеристик (ФОХ, fипс
tioпa/ity reJated characte,istics FRCs) для
процесса производства и пара метров каче
ства roTOBOro лекарственноro средства, эти
характеристики MorYT быть отслежены, и для
них MorYT быть установлены продукт специ
фические требования к качеству.
Частные статьи Фармакопеи на вспомо
rательные вещества предназначены для обе
спечения их приемлемоrо качества для по
требителей. Информация по внешнему виду
и описанию вспомоrательных веществ, Tpe
бования к идентификации, химической и ми
кробиолоrической чистоте и сризическим xa
рактеристикам, связанным с химической
структурой, таким как оптическое враще
ние, приведены в частных статьях и в общей
статье Субстанции для фармацевтичеСК020
использования.
ФОХ включены в статьи на вспомоrатель
ные вещества с целью помочь производите
лям лекарственных средств установить требо
вания к качеству, основанные на стандартных
аналитических методиках. Эти характеристи
ки позволяют найти взаимопонимание произ
водителям и потребителям вспомоrательных
веществ с целью осуществления поставок
вспомоrательных веществ со специфициро
ванными свойствами. ФОХ MorYT быть YKa
заны (например, в сертисрикате) производи
телем вспомоrательных веществ со ссылкой
на фармакопейную статью, указывая, таким
образом, метод, использованный для YCTa
новления данноro свойства. Раздел «Функ
ционально обусловленные характеристики»
частных статей описывает ФОХ, влияющие на
функциональность вспомоrательных веществ
для предполаrаемоrо использования. Из за
широкоro применения мноrих BcnoMoraT"тb
ных веществ и разработки новых способов их
использования, перечисленные ФОХ не явля
ются исчерпывающими.
РЕrYЛЯТОРНЫЕ РУКОВОДСТВА
В соответствии с действующими PYKOBOД
ствами, например, /СН О8 «Фармацевтиче
екая разработка», в заявлении на реrистрацию
должны быть рассмотрены выбранные BcnoMcr
rательные вещества и их концентрация, а также
должны быть указаны свойства, которые MOryт
влиять на качество продукта и на процесс про
изводства, в зависимости от функций каждоro
вспомоrательноro вещества. Также должна быть
продемонстрирована способность вспомоrа
тельных веществ выполнять свои функции в Te
чение необходимоro срока roдности лекарствен
ноro средства. Информация о характеристиках
вспомоrательных веществ при необходимо
сти может быть использована для обоснования
выбора таких веществ, а также их показателей
качества.
Вспомоrательные вещества производят
ся сериями, поэтому у одноro и тоro же произ
водителя существует возможность варьирова
ния характеристик вспомоrательных веществ
от серии к серии. Вспомоrательные вещества
от различных производителей MorYT не иметь
идентичных свойств, иrрающих важную роль
при использовании этих вспомоrательных Be
ществ в конкретной рецептуре. Неизбежное Ba
рьирование химических и физических свойств
является одной из наиболее важных перемен
ных, влияющих на фармацевтический произ
водственный процесс, так как вспомоrательные
вещества обычно составляют основную часть
roтовоro лекарственноro средства. Мноrие
вспомоrательные вещества являются веще
ствами природноro происхождения и представ
ляют собой сложную смесь химически близ
ких соединений. Некоторые вспомоrательные
вещества про изводятся на химических заво
дах, из начально нацеленных на производство
химических веществ не для фармацевтической
промышленности. Таким образом, процесс про
изводства может быть сфокусирован на полу
чении химических характеристик и некоторых
физических свойств, необходимых для первич
ноro рынка сбыта производителя. Очень часто
производители вспомоrательных веществ не
имеют достаточных знаний о фармацевтиче
ском использовании их продукта.
Понимание химической и физической
при роды фармацевтической субстанции и
вспомоrательных веществ по отдельности,
а также тоro, каким образом их свойства
влияют на друrие инrредиенты и на процесс
производства, является ключевым аспектом
создания успешной и надежной рецептуры.
При фармацевтической разработке иденти
фицируют свойства инrредиентов, критиче
ские для процесса производства и для xa
рактеристик лекарственноro средства. После
идентификации критических свойств вспо
моrательных веществ, основываясь на под
ходе оценки рисков, в процессе фармацев
тической разработки может быть установлен
диапазон приемлемых значений критических
пара метров, включая варьирование как фи
978
rосударственная фармакопея Республики Беларусь
зических, так и химических свойств. И' тере
сующие ФОХ MorYT не контролироваться rl':Ю
изводителем вспомоrательноro вещества и,
таким образом. быть непостоянными. Поэто
му предпочтительным является планИ[юва
ние устойчивоrо процесса производства roто
воro лекарственноro средства. чтобы эффект
воздействия на неro нормальной изменчиво
сти вспомоrательных веществ был оrраничен.
СОРТА С РАЗЛИЧНЫМИ ФИЗИЧЕСКИМИ
ХАРАКТЕРИСТИКАМИ
Сыпучие твердые вспомоrательные Be
щества MOryт быть доступны в виде большо
ro количества различных сортов с различны
ми физическими характеристиками (например,
распределением частиц по размерам). которые
обычно контролируются поставщиком. Однако
ФОХ этих веществ MOryT затраrивать друrие
свойства, обусловленные твердым состоянием
вещества, а также обусловленные сыпучим co
стоянием твердоro вещества, которые MOryT не
контролироваться поставщиком вспомоrатель
ноro вещества.
Примерами свойств, учитываемых при раз
работке твердой лекарственной формы и об
условленных твердым состоянием вещества,
MOryT служить полиморфизм, кристалличность
или плотность. Дополнительные методики изу
чения кристаллических форм и сольватов при
ведены в общих статьях:
5.9. Полиморфизм:
2.2.34. Термический анализ;
2.9.ЗЗ. Определение параметров Kpи
сталлических и частично кристаллических
твердых веществ при помощи дифракции
рент2еновСК020 излучения на порошке;
2.2.42. Плотность твердых тел;
2.9.23. Определение плотности твердых
частиц при помощи 2азов020 пuкнометра.
Свойства сыпучих твердых веществ вклю
чают в себя, например, распределение частиц
по размерам, удельную площадь поверхно
сти, насыпную плотность, текучесть, смачи
ваемость и сорбцию воды. В зависимости
от диапазона распределение частиц по раз
мерам может быть установлено с помощью
аналитическоro просеивания (2.9.38. Опре
деление размера частиц методом аналити
чеСК020 просеивания) либо с использованием
инструментальноro метода, например, 2.9.31.
Определение размера частиц методом диф
ракции лазеРН020 излучения. Метод, приве
денный в общей статье 2.9.26. Определение
удельной площади поверхности методом 2a
зовой адсорбции, основан на теории Брунау
эра Эмметта Телера (БЭТ). Методы, исполь
зуемые для описания текучести и насыпной
плотности, описаны в статьях 2.9.36. TeKY
честь порошков и 2.9.34. Насыпная плот
ность и плотность после усадки. Свойства,
обусловленные твердым состоянием сыпучих
веществ, MorYT влиять на смачиваемость и на
взаимодействие «твердое тело вода». Для
определения этих характеристик доступен
ряд инструментальных методов, например,
методы определения статическоro и динами
ческоro краевых уrлов и rравиметрическая
сорбция пара и/или методы rравиметрическо
ro анализа.
СОРТА С РАЗЛИЧНЫМИ ХИМИЧЕСКИМИ
ХАРАКТЕРИСТИКАМИ
Доступные вспомоrательные вещества
представлены в виде сортов с различными хи
мическими характеристиками и имеют при
родное, полусинтетическое или синтетическое
происхождение. В частных статьях обычно KOH
тролируется химический состав вспомоrатель
HblX веществ, которые представляют собой
смесь близких соединений, например, состав
жирных кислот растительных масел или поверх
ностно активных веществ. Однако в Фармако
пее присутствуют статьи, описывающие класс
полимерных материалов, которые MOryт варьи
ровать по составу в зависимости от структуры
roмополимеров, блоков полимеров и сополиме
ров. степени полимеризации, а, следовательно,
по массе и массовому распределению, степени
замещения и, в некоторых случаях, даже по за
местителям на основной цепи полимера. Эта Ba
риабельность может иметь сильное влияние на
Функциональные характеристики вспомоrатель
ных веществ и должна учитываться при фарма
uевтической разработке; желательно установить
диапазон приемлемых значений для каждоro па
раметра, критическоro как для процесса произ
водства, так и для характеристик roтовоro лекар
cTBeHHoro средства.
Несмотря на то, что в прошлом обязатель
ная часть статей на полимерные вспомоrатель
ные вещества мота содержать некоторые ис
пытания физических или химических свойств,
например, испытание «Вязкость» С критери
ями приемлемости, такие испытания будут
постепенно переводиться внеобязательный
раздел «Функционально обусловленные xa
рактеристики», за исключением случаев, коrда
рассматриваемые свойства являются HeOTъeM
лемой частью испытаний на подлинность.
Данный процесс должен вестись в рамках py
ководящих документов по фармацевтической
разработке с заданной реrуляторной rибко
стью, основанной на установлении диапазона
приемлемых значений данных параметров ис
пытуемоro образца для желаемоro использо
вания. Таким образом, разработка химических
характеристик и, rдe это необходимо, устано13
ление специальных требований для критиче
ских характеристик, являются частями фарма
цевтической разработки, вне зависимости от
тоro, является ли раздел «Функционально об
условленные характеристики» обязательным
или нет.
5. 15. Функционально обусловленные характеристики вСПОМО2ательных веществ 979
РАЗДЕЛ«ФУНКЦИОНАЛЬНО
ОБУСЛОВЛЕННЫЕ ХАРАКТЕРИСТИКИ»
В ЧАСТНЫХ СТАТЬЯХ
Частные статьи на вспомоrательные веще
ства MoryT содержать раздел «Функциональ
но обусловленные характеристики», который
при водится для информации и не является
обязательным. В этом разделе перечисляют
ся характеристики, которые существенны для
конкретноro использования вспомоrательно
ю вещества. При этом указывают вид исполь
зования, для которою данная характеристика
важна, на применение в друrих целях данные xa
рактеристики MoryT не влиять. По этой причине
данный раздел не должен рассматриваться как
просто дополнение к частной статье. Решение о
том, как приведенная в разделе «Функциональ
но обусловленные характеристики» информа-
ция будет использована в процессе производ-
ства и применения вспомоrательноro вещества с
учетом данных, полученных при фармацевтиче-
ской разработке, принимает производитель ле-
карственноro средства.
Информация о функционально-обусловлен-
ных характеристиках может быть приведена раз
личными способами:
название функционально обусловленной
характеристики;
название функционально обусловленной
характеристики и рекомендованный метод для
ее определения со ссылкой, rде это возможно,
на статьи Фармакопеи;
название функционально обусловленной
характеристики с рекомендованным методом
для ее определения и типичным критерием при
емлемости, который может быть представлен в
виде отклонений от номинальноro значения.
Одна и та же характеристика может присут
ствовать как в обязательном разделе частной
статьи, так и в разделе «Функционально обу-
словленные характеристики». Степень полиме
ризации встречается в обязательном разделе
«Идентификация» частных статей на микро
кристаллическую целлюлозу и на целлюлозу в
виде порошка для тою, чтобы можно было OT
личить эти два типа. Степень полимеризации
микрокристаллической целлюлозы не должна
превышать 350, в то время как для целлюлозы
в виде порошка она составляет от 440 до 2250.
Знание действительной степени полимеризации
важно для некоторых видов использования, и
RO этой причине она указывается как ФОХ, KO
торую производитель лекарственноro средства
имеет право конкретизировать для точноro YKa
зания качества, требуемоro для конкретноro ле
карственноro средства.
Раздел, включающий ФОХ, предназна
чен для отражения текущих знаний о наиболее
важных использованиях вспомоrательноro Be
щества. В виду широкой области применения
некоторых вспомоrательных веществ и продол-
жающейся разработки новых типов их исполь
зования, данный раздел может быть неполным.
Кроме этоro, методы, указанные для определе-
ния конкретных характеристик, приведены в Ka
честве рекомендации и не исключают возможно
сти использования друrих методов.
МЕЖДУНАРОДНАЯ rАРМОНИЗАЦИЯ
Количество частных фармакопейных
статей на вспомоrательные вещества являет-
ся предметом rармонизации между Фармакопе-
ями Европы, Японии и США. Введение разде-
ла «Функционально-обусловленные характери-
стики» в Европейскую Фармакопею означает,
что вид rармонизированных статей отличается.
Испытания физических и химических xapaктe
ристик, относящихея к ФОХ В Европейкой Фар
макопее, в остальных двух фармакопеях вклю
чены в текст частной статьи. Такой различный
формат не влияет на спецификацию вспомоrа-
тельноro вещества для производителя лекар-
cTBeHHoro средства. Руководящие документы
в настоящее время рекомендуют идентифици-
ровать и специфицировать только такие кри-
тические свойства, которые влияют на произ-
водственный процесс и на качество roтовоro
лекарственною средства. Различная законо-
дательная база в случае трех фармакопей до-
пускает существование различных форматов
частных фармакопейных статей без вреда для
статуса международной rармонизации.
rЛОССАРИЙ
Критическая характеристика (c,itica/ cha-
racteristic): любая физическая или химическая ха-
рактеристика вещества, которая в значительной
степени влияет на технолоrичность и/или качество
лекарственною средства.
Пространство проектных пара метров
(desigп space): мноroмерная комбинация и вза
имодействие вводимых переменных (например,
характеристики вещества) и параметров процес-
са, которые обеспечивают надлежащее каче
ство.
Функционально обусловленная xapaK
теристика (fипсtiопа/itу rеlаtеd characte'istic):
контролируемая физическая или химическая
характеристика вспомоrательною вещества,
влияющая на еro функциональность.
Испытание функциональности (fипсtiопа-
Iitу tеstiпg): непосредственное испытание инте-
ресующих функций вспомоrательноro вещества
в конкретном лекарственном средстве и про-
изводственном процессе для подтверждения
тою, что вспомоrательное вещество обеспечи-
вает необходимую функциональность.
Испытания приrодности (реrfо,тапсе
tests): аналитическое испытание критических
свойств лекарственноro средства.
980
rосударственная фармакопея Республики Беларусь
Робастность процесса (process robust
пеss): способность процесса допускать . ари
абельность веществ и изменений процеСС<l и
оборудования без отрицательноrо влияния на
качество.
01/2013:51600
5.16. КРИСТАЛЛИЧНОСТЬ
в данной 2лаве приведена общая инфор
мация о кристалличности и указаны ссылии на
различные фармакопейные методы, использу
емые для ее определения.
ВВЕДЕНИЕ КОНЦЕПЦИЯ
КРИСТАЛЛИЧНОСТИ
Большинство орrанических и неорrани
ческих веществ, относящихся к субстанциям
для фармацевтическоro использования, яв
ляются твердыми веществами, которые MorYT
быть описаны структурами, находящими
ся в диапазоне от идеальноrо кристалла до
аморфноro вещества.
Реальные кристаллы имеют структуры, за
ни мающие положения между идеальным кри
сталлом и аморфным состоянием. Место кри
сталла на шкале, расположенной между этими
двумя противоположными состояниями, назы
вается кристалличностью.
Идеальный кристалл это идеальное
состояние вещества, которое достиrается
редко, если вообще достиrается. CTPYKTYP
ные единицы кристалла, называемые эле
ментарными ячейками, равномерно и He
оrраниченно повторяются в трех измерениях
пространства. Элементарная ячейка имеет
определенную ориентацию и форму, которые
определяются векторами переноса а, Ь и с и
уrлами а, и у, и, таким образом, она имеет
определенный объем V, в котором находятся
атомы и молекулы, необходимые для форми
рования кристалла. Кристаллическая систе
ма определятся тремя операторами дальней
симметрии (трансляционным, ориентацион
ным и конформационным); различные мезо
фазы (жидкие кристаллы, кристаллы и пла
стичные кристаллы) имеют один или два
оператора дальней симметрии; в идеальном
аморфном состоянии отсутствуют все три
оператора.
Каждый кристалл может быть классифи
цирован как представитель одной из семи
возможных кристаллических систем, которые
определяются отношением между индивиду
альными размерами а, Ь и с, а также между
индивидуальными уrлами а, и у элементар
ной ячейки. Структура KOHKpeTHoro кристал
ла может быть классифицирована в COOTBeT
ствии с одной из семи систем, в соответствии
с одной из четырнадцати пространственных
решеток Браве и в соответствии с одной из
230 пространственных rрупп. Все 230 воз
можных пространственных rрупп, их симме
трии и симметрии их дифрактоrрамм собраны
в Международных таблицах кристаллоrрафии
(/пtеrпаtiопа/ Tables (о, C,ysta//ography).
При кристаллизации мноrие вещества
MorYT иметь более чем один тип кристалли
ческой решетки; это явление называется по
лиморфизмом. Среди орrанических молекул
полиморфизм это распространенное явле
ние, что при водит к различиям в физико хи
мических свойствах, так как кристаллические
полиморфы имеют одинаковый химический
состав, но различную кристаллическую CTPYK
туру и, В результате, обладают различными
физико химическими свойствами. Существо
вание различных кристаллических структур
возможно блаrодаря различным способам
атомарной упаковки иlили различным KOH
формациям молекул (см. rлаву 5.9 Полимор
физм).
Еще одно крайнее кристаллическое COCTO
яние это идеальное или истинное аморф
ное состояние, при котором отсутствует сим
метрия дальнеrо порядка. Для большинства
орrанических систем сохраняется симметрия
близкоro порядка, но она распространяет
ся не далее, чем на ближайшее окружение
либо на расположенные рядом с ближайшим
окружением структуры, что составляет менее
2 2,5 нм для малых орrанических молекул.
Аморфный материал характеризуется OT
сутствием четких пятен на дифрактоrрамме
peHтreHoBcKoro излучения, полученной с ис
пользованием порошка (2.9.33).
Кристалличность реальных порошков
может быть рассмотрена с использовани
ем двух моделей кристалличности. В OДHO
фазной модели все частицы имеют одинако
вую кристалличность, а в двухфазной модели
каждая частица может быть кристаллической
либо аморфной, и, таким образом, реаль
ная кристалличность порошка является взве
шенным средним этих двух крайних кристал
личностей. Такой порошок образуется при
физическом перемешивании чистых кристал
лической и аморфной фаз. В действительно
сти порошок может содержать частицы с раз
личной степенью кристалличности, также как
он может содержать частицы различных раз
меров и форм.
Протяженность неупорядоченности в кри
сталлическом твердом теле может влиять
на мноrие физико химические свойства суб
станций для фармацевтическоro использова
ния. Из за высокой значимости этих свойств,
5.16. Кристалличность
981
важно иметь возможность определения He
упорядоченности или кристалличности TBep
дых веществ подходящим количественным
методом.
МЕТОДЫ монитоРинrА
И ОПРЕДЕЛЕНИЯ КРИСТАЛЛИЧНОСТИ
ДЛЯ определения кристалличности TBep
дых веществ используют различные методы.
Мноrие подходы по отдельности не способны
обнаруживать или измерять эти свойства; по
этой причине полезно объединять несколь
ко методов, описанных ниже. Эти методы
часто не дают точных результатов и имеют
пределы количественноro определения, KO
торые обычно значительно превышают TaKO
вые для определения химических примесей.
Кроме тоro, должны быть сделаны опреде
ленные допущения о связи между CTaHдapT
ными образцами, использованными для Ka
либровки и которые обычно представляют
собой смеси кристаллических и аморфных
частиц (двухфазная модель), и испытуемы
ми образцами, которые MorYT содержать He
большую часть вещества, ведущую себя в
соответствии однофазной моделью. В итоrе,
недостаток хорошо изученных стандартных
образцов для полностью кристаллическоro
или для полностью аморфноro вещества yc
ложняет валидацию таких методов. Как сле
дует из объяснений, приведенных выше, оче
видно, что в твердом порошке существуют и
даже сосуществуют различные аморфные и
некристаллические фазы. Эти различные He
кристаллические формы твердоro вещества
MorYT приводить к получению различных от
кликов, в зависимости от методики, исполь
зуемой для определения степени кристаллич
ности.
Дифракция peHTreHoBcKoro излучения
на порошке (2.9.33). Данный метод все еще
является наиболее часто используемым Me
тодом для определения степени кристаллич
ности, хотя он имеет некоторые оrраничения,
связанные суширением пиков, аморфным
rало и предпочтительной ориентацией, что
делает сложным интерпретацию и количе
ственное определение.
Использование только дифракции peHT
reHoBcKoro излучения на порошках часто яв
ляется недостаточным для тоro, чтобы раз
личить отличающиеся некристаллические
фазы. Дифрактоrрамма peHTreHoBcKoro из
лучения чисто аморфной и нанокристалли
ческой фазы представл ет собой широкое
диффузное rало. Тщательный анализ диф
paKTorpaMMbI peHTreHoBcKoro излучения по
казывает, что диффузное rало на дифрак
TorpaMMe нанокристаллическоrо материала
проявляет некоторую корреляцию с дифрак
тоrраммой родительской кристаллической
фазы, Torдa как в случае чистой аморфной
фазы такой корреляции не существует. Для
установления истинной природы материала,
определенной с помощью peHTreHoBcKoro из
лучения как аморфная, может потребоваться
дополнительная процедура.
Термический анализ. Термический
анализ (2.2.34) кристаллических материалов
показывает переходы при плавлении, сопро
вождающиеся разложением или испарением
растворителей. В случае истинных аморфных
веществ термический анализ обнаруживает
переход в стеклянное состояние, в то время
как для нанокристаллических веществ будет
наблюдаться только плавление.
Микрокалориметрия. Это очень чувстви
тельный подход, который позволяет опреде
лять скорость и rлубину химических реакций,
фазовых изменений и изменений структуры.
Аморфная часть вещества может быть pe
кристаллизована путем воздействия на об
разец высокой относительной влажности или
атмосферы, содержащей пары орrанических
веществ. Измерение тепла рекристаллизации
позволяет определить содержание аморф
ной части исходя из энтальпии рекристалли
зации. С помощью соотнесения данных ми
крокалориметра для испытуемоro образца с
данными, полученными для аморфноro CTaH
дартноrо образца, становится возможным KO
личественное определение аморфной части
в испытуемом образце. Диапазон содержания
аморфной части, который можно определить
с помощью этоrо подхода, зависит от KOHKpeT
ноro испытуемоro вещества; в блаrоприятных
условиях может быть достиrнут предел обна
ружения ниже 1 %.
Калориметрия растворов. Калориме
трия растворов является средством опреде
ления энтальпии раствора для твердоrо веще
ства. Кристалличность твердоro испытуемоro
образца устанавливается с помощью энталь
пии раствора твердоro образца (f>,H:) минус
энтальпия раствора выбранноro CTaHдapTHO
ro образца этоro же вещества (j,H:) при про
ведении испытания в одних и тех же услови
ях. По причине тоro, что стандартный образец
обычно выбирают исходя из ero воспринима
емой высокой степени кристалличности, эн
тальпия еro раствора обычно алrебраически
выше (более эндотермический или менее эк
зотермический), чем энтальпия твердоro ис
пытуемоrо образца в том )l(е растворителе.
Следовательно, определенная степень кри
сталличности представляет собой отрицатель
ную величину в единицах СИ кДж/моль или
Дж/r (Дж/кr избеrают из за еro rромоздкости
и возможности совершения ошибок). Отрица
тельное значение степени кристалличности
982
rосударственная фармакопея Республики Беларусь
большинства образцов rоворит о том, что они
имеют более низкую степень кристаЛЛИЧI:'JСТИ,
чем стандартный образец.
Спектрофотометрия ближнеrо ИК-
диапазона. Еще одним методом, использу
емым для определения степени кристаллич
ности, является спектрофотометрия ближнеro
ИК диапазона (БИК) (2.2.40); он также полезен
при исследовании полиморфизма. БИК спектр
содержит информацию как о химическом COCTa
ве, так и о физическом состоянии. Будучи неин
вазивным, неразрушающим методом, примени
мым при комнатной температуре, БИК является
ценным инструментом для оценки изменений в
аморфном и кристаллическом состояниях.
Абсорбционная спектрофотометрия в
ИК-диапазоне и рамановская спектроме-
трия. Методами, используемыми для опре
деления степени кристалличности. являют-
ся также абсорбционная спектрофотометрия в
ИК диапазоне (2.2.24) и рамановс/(ая спектро
метрия (спектрометрия комбинационноro рас-
сеяния) (2.2.48); они также полезны при иссле
довании полиморфизма. ИК спектр и спектр
комбинационноrо рассеяния содержат инфср
мацию как о химическом составе, так и о физи
ческом состоянии.
ЯМР твердых тел. Ядерный маrнитный
резонанс (ЯМР) (2.2.33) твердых тел может ис
пользоваться для получения информации о по
ли морфизме и связанных родственных молеку
лярных конформациях. Однако интерпретация
результатов должна проводиться с осмотритель
ностью, так как не Bcerдa с леrкостью можно раз
личить образцы, которые представляют собой
смесь различных физических форм (двухфаз
ная модель), и образцы, включающие в себя
кристаллы, имеющие дефекты замещения. По
добно этому, образцы, содержащие дефекты,
возникающие из за различных молекулярных
конформаций или немною отличающихся моле
кулярных упаковок (однофазная модель), MOryT
давать дополнительные сиrналы. ЯМР твердых
тел может быть достаточно чувствительным к
этим случаям, даже если параметры кристалли
ческой решетки едва подверrлись воздействию
и, следовательно, с помощью дифракции peHT
reHoBcKoro излучения на порошке изменения не
наблюдаются или наблюдаются очень слабо.
Очевидно, что кристалличность субстанций для
фармацевтическоro использования может быть
комплексной, при этом кристаллические дефек
ты и аморфное вещество Moryт присутствовать
одновременно.
Оптическая микроскопия. Кристаллич
ность или некристалличность веществ опре
деляют с использованием поляризационноro
микроскопа (2.9.37) по появлению двойноro лу
чепреломления и зон поrлощения при повороте
предметноro столика микроскопа.
5.17. РЕКОМЕНДАЦИИ
ПО ПРОВЕДЕНИЮ
ИСПЫТАНИЙ
ДОЗИРОВАННЫХ
ЛЕКАРСТВЕННЫХ
СРЕДСТВ
01/2013:51701
5.17.1. РЕКОМЕНДАЦИИ
ПО ПРОВЕДЕНИЮ ТЕСТА
«РАСТВОРЕНИЕ»
Данная статья не является обязатель-
ной: в данной статье прuводuтся информа
ция по проведению испытания «Растворение».
по рекомендованным средам растворения и по
выражению требований в спецификациях для
оральных дозированных форм по тесту «Pac
творение» (см. статью 2.9.3. Тест «Pacтвope
ние) для твердых дозированных форм). Данная
информация представляет собой общие при
емлемые параметры касательно испытания
«Растворение».
При описании метода определения скорости
растворения действующеro вещества/веществ в
твердой дозированной форме должны быть YKa
заны следующие параметры:
используемый прибор и, в случае прибора
с проточной кюветой, используемая кювета;
состав, объем и температура среды paCT
ворения;
скорость вращения или скорость потока
среды растворения;
время, способ и количество отбираемоro
испытуемоro раствора или условия непрерывно
ro наблюдения;
метод анализа;
критерии приемлемости.
Выбор используемоrо прибора зависит
от физико химических свойств дозированной
формы. При необходимости использования
большою объема среды растворения для co
блюдения условий поrружения или при необхо
димости смены рН предпочтительнее использо-
вать прибор с проточной кюветой.
ЭКСПЕРИМЕНТАЛЬНЫЕ УСЛОВИЯ
ПРОВЕДЕНИЯ ИСПЫТАНИЙ
Использование прибора с корзинкой или с
лопастью-мешалкой, а также прибора с поршне-
5.17.1. Рекомендации по проведению теста «Растворение»
983
выми цилиндрами обычно основано на принци
пе проведения испытания в условиях поrруже
ния, то есть в таких условиях, Korдa вещество,
уже находящееся в растворе, не влияет в зна
чительной степени на скорость растворения
остатка. Эти условия обычно подразумевают ис
пользование количества среды растворения как
минимум в 3 1 О раз большеrо, чем необходимо
для получения насыщенноrо раствора.
Обычно используют водную среду. Состав
среды выбирают на основании физико химиче
ских свойств действующеro вещества/веществ и
вспомоrательноro вещества/веществ в услови
ях, В которых лекарственное средство предполо
жительно будет находиться после еro приема. В
частности это касается рН и ионной силы среды
растворения.
Обычно используют среды растворения со
значениями рН от 1 до 8. При соответствующем
обосновании может использоваться среда paCTBO
рения с более высоким значением рН. Для полу
чения низких значений рН в кислой среде обычно
используют О, 1 М раствор кислоты хлористово
дородной. Рекомендованные среды растворения
приведены далее в настоящей статье.
Использование воды в качестве среды раст
80рения может быть рекомендовано, только
если доказано, что изменение рН не влияет на
характеристики растворения.
В отдельных случаях при соrласовании с
уполномоченным opraHoM среда растворения
может содержать ферменты, поверхностно ак
тивные вещества, друrие неорrанические и opra
нические вещества. При испытании лекарствен
ных средств, содержащих плохо растворимые
в воде вещества, модификация среды paCTBO
рения может быть необходима. В таких обсто
ятельствах рекомендуется использовать низкую
концентрацию поверхностно активноro веще
ства; рекомендуется избеrать использования op
rанических растворителей.
Растворенные в среде растворения rазы
MorYT влиять на результаты испытания. В част
ности это справедливо для прибора с проточ
ной кюветой, rдe деаэрация среды необхо
дима для избежания образования пузырьков
в кювете. Может быть использован следу
ющий способ деаэрации: среду наrревают при
аккуратном перемешивании до температуры
около 41 ОС, немедленно фильтруют под BaKY
умом через фильтр с размером пор 0,45 мкм
или менее при интенсивном перемешивании и
продолжают перемешивать под вакуумом в Te
чение около 5 мин. Также может быть исполь
зован иной валидированный метод удаления
растворенных rазов.
При использовании прибора с лопастью ме
шалкой или с корзинкой объем среды paCTBope
ния обычно составляет 500 1000 мл. Скорость
вращения обычно выбирают в диапазоне от
50 об/мин до 100 об/мин; она не должна превы
шать 150 об/мин.
При использовании прибора с проточной
кюветой скорость потока среды растворения
обычно находится в диапазоне от 4 мл/мин ДО
50 мл/мин.
РЕКОМЕНДУЕМЫЕ СРЕДЫ
РАСТВОРЕНИЯ
MOryT быть использованы следующие среды
растворения.
Таблица 5.17.1. 1
Примеры сред растворения
рН Среда растворения
рН 1,0 HCI
рН 1,2 NaCI, НС'
IpH 1,5 NaCI, НСI
\рН4,5 Фосфатный или ацетатный
буферный раствор
рН 5,5 и рН 5,8 Фосфатный или ацетатный
буферный раствор
,рН6,8 Фосфатный буферный
раствор
i рН 7,2 и рН 7.5 I Фосфатный буферный
раствор
Состав и приroтовление сред указаны ниже.
Среды схлористоводородной кислотой
0,2 М раствор кислоты хлористоводо
родной.
0,2 М раствор натрия хлорида. 11,69 r
натрия хлорида Р растворяют в воде Р и ДOBO
дят до объема 1000,0 мл этим же растворителем.
Для приrотовления сред со значениями рН,
указанными в таблице 5.17 .1. 2, 250,0 мл 0,2 М
раствора натрия хлорида смешивают с указан
ным объемом 0,2 М раствора кислоты хлори
стоводородной и доводят водой Р до объема
1000,0 мл.
Таблица 5.17.1. 2
Среды с хлористоводородной кислотой
i рН НСI (мл)
!
I 1,2 425,0
I 1,3 336.0
! 1,4 266,0
1,5 207,0
1,6 162,0
1,7 130,0
1,8 102,0
1,9 ! 81,0
2,0 65,0
2,1 51,0
2,2 39,0
Среды с хлористоводородной кислотой
также MorYT быть приroтовлены с использовани
ем калия хлорида вместо натрия хлорида.
984
rосударственная фармакопея Республики Беларусь
Ацетатные буферные растворы
2 М раствор кислоты уксусной. 1200 r
кислоты уксусной ледяной Р доводят водой Р
до объема 1000,0 мл.
Ацетатный буферный раствор
рН 4,5. 2,99 r натрия ацетата Р paCTBO
ряют в воде Р, прибавляют 14,0 мл 2 М paCT
вора кислоты уксусной и доводят водой Р до
объема 1000,0 мл.
Ацетатный буферный раствор рН 5,5.
5,98 r натрия ацетата Р растворяют в
воде Р, прибавляют 3,0 мл 2 М раствора кис
лоты уксусной И доводят водой Р до объема
1000,0 мл.
Ацетатный буферный раствор рН 5,8.
6,23 r натрия ацетата Р растворяют в воде Р,
прибавляют 2,1 мл 2 М раствора кислоты YKCYC
ной И доводят водой Р до объема 1000,0 мл.
Фосфатные буферные растворы
Для приroтовления буферов со значениями
рН, указанными в таблице 5.17.1. 3, 250.0 мл
0,2 М раствора натрия диеидрофосфата Р
смешивают с указанным объемом 0,2 М pacт
вора натрия 2идроксида и доводят водой Р до
объема 1000.0 мл.
Таблица 5.17.1. 3
Фосфатные буферные растворы
рН I 5,8 6,0 6,2 6,4 6,6 6,8 I
NaOH (мл) 18.0 28.0 40,5 58,0 82.0 112.0
рН 7,0 7;2. 7,4 7,6 7,8 8,0
. NaOH (мл) 145,5 172,5 195,5 212,0 222,5 230,5
Иные фосфатные буферные растворы
Фосфатный буферный раствор рН 4,5.
13,61 r натрия диzидрофосфата Р paCTBO
ряют в 750 мл воды Р. При необходимости KOp
ректируют значение рН (2.2.З) 0,1 М раствором
натрия 2идроксида или 0,1 М раствором кисло
ты хлористоводородной и доводят водой Р до
объема 1000,0 мл.
Фосфатный буферный раствор рН 5,5 Р.
Фосфатный буферный раствор рН 6,8 Р1.
Буферный раствор рН 7.2 Р.
О,ЗЗ м фосфатный буферный раствор
рН 7,5 Р.
Искусственный кишечный сок рН 6,8
Смешивают 77,0 мл 0,2 М раствора натрия
2идроксида, 250,0 мл раствора, содержаще
ro 6,8 r калия ди2идрофосфата Р, и 500 мл
воды Р, прибавляют 10,0 r порошка панкреати
на Р, перемешивают, при необходимости KoppeK
тируют значение рН (2.2.З) и доводят водой Р до
объема 1000,0 мл.
Искусственный желудочный сок
2,0 r натрия хлорида Р и 3,2 r порошка пепси
на Р растворяют в воде Р, прибавляют 80 мл 1 М
раствора кислоты хлористоводородной и дово--
дят водой Р до объема 1000,0 мл. При необходи
мости порошок пепсина может быть исключен.
Повышение рН
В случае проведения испытания с повыше
нием рН может быть использована одна из сле
дующих последовательностей:
Время (}""",1 1 2
(ч)
рН 1,0
рН 1,2
рН 1,2 2,5 7,5
рН 1,5 7,2
Для достижения указанных изменений рН
необходимо:
или заменять один буферный раствор
друrим (полная замена);
или каждый раз убирать только половину
буфернoro раствора (способ неполной замены)
и заменять ero буферным раствором с более BЫ
соки м значением рН: рН начальноrо буферноrc
раствора 1.2. а второro 7.5:
или в начальный буферный раствор с
рН 1 ,5 прибавлять сухую смесь, состоящую из
триС(2идроксиметил)аминометана Р и натрия
ацетата безводН020 Р, сначала до получения
значения рН 4,5, а затем до получения значе
ния рН 7.2, как указано ниже:
раствор кислоты хлористоводород
ной рН 1,5: 2 r натрия хлорида Р растворяют в
воде Р, прибавляют 31,6 мл 1 М раствора кисло
ты хлористоводородной и доводят водой Р до
объема 1000,0 мл;
буферный раствор рН 4,5: смешивают
2,28 r триС(2идроксиметил)аминометана Р с
1.77 r натрия ацетата безводН020 Р; получен
ную смесь растворяют в описанном выше pac
творе кислоты хлористоводородной рН 1,5;
буферный раствор рН 7,2: смешивают
2,28 r трuс(аидроксиметил)аминометана Р с
1 ,77 r натрия ацетата безводН020 Р; получен
ную смесь растворяют в описанном выше бу
ферном растворе рН 4,5.
Проточная кювета может быть использована
для непрерывной смены рН.
КВАЛИФИКАЦИЯ И ВАЛИДАЦИЯ
ПО причине природы метода испытания, Ka
чество конструктивноro исполнения является
важным аспектом при квалификации оборудо
вания для испытания растворения iп vit,o. Необ
ходимо избеrать любых нарушений, например,
вибрации или нежелательноrо перемешивания
из за механических дефектов.
Квалификация оборудования для испытания
растворения должна учитывать размеры и допу
ски используемоro оборудования. Критические
параметры испытания, такие как температура и
объем среды растворения, скорость вращения
5.17.1. Рекомендации по проведению теста «Растворение»
985
или скорость потока жидкости, отбор проб, MeTO
дики, должны периодически контролироваться.
Функционирование оборудования может
быть проверено путем испытания CTaHдapTHO
ro образца, чувствительноro к rидродинамиче
ским условиям. Такие испытания MOryT прово
диться периодически или постоянно в целях
сравнения получаемых результатов различных
лабораторий.
Во время испытания требуется выполне
ния критическою осмотра и наблюдения. Этот
подход особенно важен при объяснении резуль
татов, выходящих за рамки спецификации.
При валидации автоматизированных
систем, включающих отбор проб, анализ или
приroтовление среды растворения и проведение
испытания, необходимо принимать во внимание
точность, правильность, а также предотвраще
ние заrрязнений при различных разведениях;
перемещениях, очистке и отборе проб или при
rотовлении растворителя.
СПЕЦИФИКАЦИИ ТЕСТА «РАСТВОРЕНИЕ»
ДЛЯ ДОЗИРОВАННЫХ ФОРМ
ДЛЯ BHYTPEHHErO ПРИМЕНЕНИЯ
в спецификации на растворение указывают
количество (О) действующеro вещества, paCTBO
рившеrося за определенный промежуток BpeMe
ни, выраженное в процентах от номинальноro
содержания.
Твердые дозированные формы с обыч-
ным высвобождением
В большинстве случаев при испытании в
обоснованных и доказанных условиях критерий
приемлемости на уровне S, таков, что не менее
80 % действующеrо вещества должно высво-
бодиться в течение указанною периода BpeMe
ни, составляющеro обычно 45 мин или менее
Это соответствует значению О, равному 75 %,
таким образом, как указано в таблице 2.9.3. 1,
для уровня S, индивидуальные значения каждоCi
из 6 испытуемых единиц должны быть не менее
О+5 %, т. е. не менее 80 %.
Обычно для демонстрации полноты BЫ
свобождения действующеro вещества ДOCTa
точным является установка критерия в одной
временной точке, хотя в некоторых случаях
для доказательства соответствующеro раст
ворения может быть необходимым про водить
анализ в дополнительный момент (моменты)
времени.
Твердые дозированные формы с про-
лонrированным высвобождением
Обычно в спецификации на дозированные
формы с пролонrированным высвобождени
ем указывают 3 или более контрольных точек.
Первая контрольная точка предназначена
для непреднамеренно быстроro высвобожде
ния действующеro вещества (<<сбрасывание
дозы»). Поэтому она устанавливается через
период времени, соответствующий обычно
высвобождению от 20 % до 30 % действую
щеrо вещества. Вторая контрольная точка
характеризует картину растворения и COOT
ветствует высвобождению примерно 50 %
действующеro вещества. Конечная контроль
ная точка предназначена для подтверждения
почти полноro высвобождения, под которым
обычно понимают высвобождение более 80 %
действу ющеrо вещества.
Твердые дозированные формы с замед-
ленным высвобождением
Дозированные формы с замедленным BЫ
свобождением способны высвобождать дей
ствующее вещество (вещества) порционно
или полностью в соответствии с разработан
ны"!. составом при растворении 8 различных
средах растворения, например, в условиях
увеличения значения рН. Поэтому специфи
кации должны быть обозначены для каждоro
случая.
Для дозированных форм с оболочкой.
устойчивой к действию желудочноrо сока, He
обходимы как минимум 2 контрольных точки
при последовательном испытании и 2 pa
личных спецификации при параллельном
исr.ытаI--fИИ. При последовательном испыта
нии первая контрольная точка представляет
собой верхний предел, и ее устанавливают
через 1 чили 2 ч испытания в кислой среде;
а вторую контрольную точку через YCTaHOB
ленное время испытания в подходящем бу
ферном растворе (предпочтительно рН 6,8).
В большинстве случаев критерий прием
лемости на уровне В, соответствует тому, что
высвобождается не менее 80 % действующе
ro вещества. Это соответствует значению О,
равному 75 %, так как в соответствии с Ta
блицей 2.9.3. 4 на уровне В, индивидуаль
ные значения для каждой из 6 испытуемых
дозированных единиц должны быть не менее
О+5 %, т. е. не менее 80 %.
#6. 1. 1. Жидкие лекарственные средства
01/201З:РБ60000
987
#6. ЭКСТЕМПОРАЛЬНЫЕ
ЛЕКАРСТВЕННЫЕ
СРЕДСТВА
Нормы и требования rлавы #6. Экстемпо
ральные лекарственные средства применимы
к экстемпоральным лекарственным средствам,
т. е. приroтовленным в условиях аптек по рецеп
там врачей либо требованиям учреждений здра
воохранения, а также в порядке внутриаптечной
заroтовки.
#6.1. приrОТОВЛЕНИЕ
ЭКСТЕМПОРДЛЬНЫХ
ЛЕКАРСТВЕННЫХ
СРЕДСТВ
01/201З:РБ60101
#6.1.1. ЖИДКИЕ ЛЕКАРСТВЕННЫЕ
СРЕДСТВА
Жидкие лекарственные средства в услови
ях аптеки изrотавливают следующими MeToдa
ми: массо объемным, 'по массе и по объему с
соответствующим выражением концентраций
(м/об, м/м, об/об). В таблице #6.1.1. 1 приведе
ны примеры обозначения концентраций фар
мацевтических субстанций и вспомоrательных
веществ (далее веществ) в жидких лекар
ственных средствах в прописях рецептов (Tpe
бований) и методы их изroтовления.
Массо объемным методом изroтавливают:
водные и водно спиртовые растворы TBep
дых веществ;
водные и водно спиртовые суспензии с
содержанием твердых веществ до 3 %;
растворы водорода пероксида, изroтов
ленные из 30 % раствора водорода пероксида.
При изroтовлении жидких лекарственных
средств массо объемным методом использу
ют мерную посуд rрадуированную на «налив»
(мерные колбы, цилиндры, стаканы, мензурки,
rрадуированные пробирки) и на «вылив» (аптеч
ные бюретки, каплемеры и пипетки) и откалибро
ванную в соответствии с нормативными ДOKYMeH
тами.
Методом по массе изroтавливают:
растворы твердых и жидких веществ в
вязких и летучих растворителях, дозируемых по
массе;
суспензии с содержанием твердых Be
ществ 3 % и более и эмульсии;
roмеопатические жидкие лекарственные
средства.
Кроме тоro, по массе дозируют:
жидкости вязкие (жирные и минеральные
масла, rлицерин, полиэтиленrликоли, полиэти
леноксиды, силиконовые жидкости);
жидкости летучие (диметилсульфоксид,
масло терпентинное очищенное, метилсалици
лат, эфир, хлороформ, масла эфирные и друrие);
жидкости с плотностью, значительно отли
чающейся от плотности воды очищенной (бен
зилбензоат, валидол, винилин, деrоть березо
вый, ихтиол, кислота молочная, эфирные масла,
масло терпентинное очищенное, метилсалици
лат, нитроrлицерин, 30 % раствор водорода пе
роксида и друrие).
Методом по объему изroтавливают:
растворы спирта этиловоrо различной KOH
центрации;
растворы кислоты хлористоводородной,
хлорrексидина биrлюконата и стандартные фар
макопейные растворы (кроме растворов перrи
дроля) (см. таблицу #6.1.1. 8).
Кроме тоro, по объему дозируют:
воду очищенную и воду для инъекций;
водные растворы веществ (в том числе ca
харный сироп, сироп алтея и друrие);
продукты из лекарственноro растительно
ro сырья (настойки, жидкие экстракты, адонизид
и друrие).
В случае если требуется установить объем
жидкости, прописанной в рецепте (требова
нии) и дозируемой по массе, или массу жидко
СТИ, прописанной в рецепте (требовании) и дo
зируемой по объему, используются значения их
плотности, указанные в частных фармакопей
ных статьях или документах, подтверждающих
качество лекарственноro средства (сертификат
качества предприятия производителя или про
токол испытаний испытательной лаборатории).
При изroтовлении водных растворов для
инъекций, rлазных жидких лекарственных
средств и концентрированных растворов, coдep
жащих в составе rлюкозу моноrидрат, пересчет
ее количества проводят с учетом содержания
кристаллизационной воды. Для изroтовления
жидких лекарственных средств rиrроскопичные
вещества MOryT использоваться в виде KOHцeH
трированных растворов (например, 50 % paCT
вор кальция хлорида rексаrидрата).
Спиртовые растворы веществ, при OTCYT
ствии указания в рецепте (требовании) KOHцeH
трации этиловоro спирта, изroтавливают в COOT
ветствии с таблицей #6.1.1. 2.
Если в рецепте (требовании) не указан paCT
воритель, изroтавливают водный раствор. Под
названием «вода» при отсутствии особых указа
ний понимают воду очищенную. Под названием
«спирт» понимают этиловый спирт. При OTCYT
ствии указаний о концентрации спирта в рецепте
iосударственная фармакопея Республики Беларусь
ПОСЛЕДОВАТЕЛЬНОСТЬ РАСТВОРЕНИЯ
И СМЕШИВАНИЯ ВЕЩЕСТВ
При изrотовлении жидких лекарственных
средств с водной дисперсионной средой в первую
очередь отмеривают рассчитанный объем воды
(воды очищенной, воды для инъекций или воды
ароматной), в котором последовательно paCTBO
ряют твердые вещества с учетом растворимости
и возможноro их взаимuдействия.
Первыми в отмеренном объеме воды раст
воряют вещества списка А, затем списка Б и
далее общеro списка с учетом их растворимости.
Для повышения растворимости веществ их
предварительно измельчают, в процессе изro
товления наrревают с учетом их физико химиче
ских свойств и перемешивают.
При использовании малорастворимых или
практически нерастворимых веществ может ис
пользоваться получение растворимых произво
дных (комплекс006разование, образование pac
творимых солей) и солюбилизация.
988
(требовании) используют этиловый спирт 90 0/0,
за исключением стандартных спиртовых pa;-TBO
ров, указанных в таблице #6.1.1. 2. Под наЗLjа
нием «эфир» понимают эфир диэтиловый. Под
названием «rлицерин» понимают rлицерин 85 %
(плотность около 1 ,223 1,233 r/см З ).
изrОТОВЛЕНИЕ ЛЕКАРСТВЕННЫХ
СРЕДСТВ В АСЕПТИЧЕСКИХ УСЛОВИЯХ
В асептических условиях изroтавливают.
растворы для инъекций и инфузий; растворы
для орошениv.; жидкие пекарственные средства
для новорожденных детей и детей до OAHorc
roда; жидкие лекарственные средства, coдep
жащие антибиотики и друrие антимикробные
вещества, предназначенные для нанесеНiIIЯ
на раневые и ожоroвые поверхности; rлазные
капли, rлазные примочки; концентрированные
растворы, в том числе roмеопатические разве
дения; ароматные воды; внутриаптечную заro-
товку (а также осуществляют ее фасовку).
Обозначение кон-
центрации веществ
Пример
Таблица #6.1.1. 1
l
Метод изrотовления I
массо-объемный
по массе I
!
I
по объему I
Sо/иtiопis Natrii Ьru?Й..:fj 2 % 200 т/
-
So/utioпis Camphorae oieosae 2 %
В процентах (%)
Rp.:
I Rp.:
,50,0
i Rp.:
Sо/иtiопis Acidi hyc!roch/orici 2 %
200 т/
- - . I массо объемный l
I Rp.: Natrii bromidi 4,0 i
I Aquae purificatae 200 т/ i
I Раздельное пере Rp.: Camphorae 1,0 по массе J
,числение веществ и О/е; Неliапthi 49, О
растворителя
Rp.: Acidi hydroch/o,ici 4 т/ по объему i
Aquae purificatae 196 т/ I
Rp.: Nat,ii bromidi 4,0 массо объемный I
Aquae pu,ificatae ad 200 т/
С указанием paCTBO Rp.: Caтpho,ae 1,0 по массе
рителя до заданноro О/е; Неliапthi ad 50, (]
объема или массы
Rp.: Acidi hydroch/orici 4 mi по объему
Aquae pu,ificatae ad 200 т/
С Rp.: Sо/иtiопis Nat,ii broт/di ех 4, О 200 т/ массо объемный
указанием соот (seu 1 :50 200 т/)
ношения массы или
объема растворя Rp.: Sо/иtiопis Camphorae o/eosae ех 1, О по массе
eMoro вещества и 50,0 (seu 1:50)
объема или массы Rp.: Sо/иtiопis Acidi hydroch/orici ех 4 т/ по объему
раствора 200 т/ (seu 1:50 200 т/)
Прuмечанuя:
1. При массо-объемном методе изютовления обозначение концентрации, например. 1:10 или 1:20. означает содержание веще-
ства по массе (r) в указанном объеме изютавливаемою жидкою лекарственною средства (мл). то есть для получения жидкою
лекарственною средства следует взять 1 r вещества и растворителя до получения 10 мл или 20 мл раствора.
2. При изютовлении методом по массе обозначение концентрации 1:10 или 1:20 означает содержание вещества по массе (r) в
указанной массе жидкою лекарственною средства (r), то есть для получения жидкою лекарственною средства следует взять 1 r
вещества и 9 r или 19 r растворителя.
3. При изrотовлении методом по объему обозначение концентрации 1 :10 или 1 :20 означает содержание вещества по объему (мл)
в указанном объеме лекарственною средства (мл), то есть для получения жидкою лекарственною средства следует взять 1 мл
вещества и растворителя до получения 10 мл или 20 мл раствора.
4. Отклонение общею объема или массы жидкою лекарственною средства от указанною в прописи рецепта (требовании) не
должно превышать норму допустимою отклонения, указанною в статье #6.3.2.
#6.1.1. Жидкие лекарственные средства
989
Таблица #6.1.1. 2
Перечень стандартных спиртовых растворов
I N!! Наименование Состав спиртовоrо раствора
1. Бриллиантовоro зеленоro 1 % и 2 % Бриллиантовый зеленый 1 r или 2 r
растворы Этиловый спирт 60 % * до 100 мл
2. Йода 1 % и 2 % растворы Йод 1 О r или 20 r
Этиловый спирт 96 % до 1000 мл
.
3. Йода 5 % раствор Йод 50 r
Калия йодид 20 r
Вода очищенная и этиловый спирт 95 % (1:1, об/об)
до 1000 мл
4. Йода 1 О % раствор Йод 100 r
Этиловый спирт 95 % до 1000 мл
5. Кислоты борной 0,5 %, 1 %, 2 % и Кислота борная 5 r, 1 О r, 20 r и 30 r
3 % растворы Этиловый спирт 70 % до 1000 мл
6. Кислоты салициловой 1 % и 2 % Кислота салициловая 1 О r и 20 r
растворы ЭтиловыJ спирт 70 % до 1000 мл
7. Кислоты салициловой и левомицети Кислота салициловая 2 r
на поровну по 2 % раствор Хлорамфеникол 2 r
Эиловый спирт 95 % до 100 мл
8. Левомицетина 0,25 %, 1 %, 3 % и 5 % Хлорамфеникол 0,25 r, 1 r, 3 r и 5 r
растворы Этиловый спирт 70 % до 100 мл
9. Левомицетина 2 % и новокаина 1 % Хлорамфеникол 2 r
раствор Прокэuна 2идрохлорид 1 r
Этиловый спирт 70 % до 100 мл
10. Меновазин I Ментол рацемuческий 2,5 r
I ПрОК8Ut J З аидрохлорид 1 r
БеНЗОJ<А:n.'; 1 r
! Этилов!:::.1 спирт 70 % до 100 мл
11. Ментола 1 % и 2 % растворы I МенПJ<; :.. цемический 10 r и 20 r
Этuловыu спирт 90 % до 1000 мл
12. Метиленовоrо синеro 1 % раствор Метилтuониния хлорид 1 О r
Этиловый спирт 95 % 600 мл
Вода очutЦенная 400 мл
13. Новокаина 2 % и кислоты борной 3 % Прокаuна 2идрохлорид 2 r
раствор КиСЛОff!д борная 3 r
Этиловый спирт 70 % до 100 мл I
- -
14. Водорода пероксида 1 ,5 % раствор Boдopo , пероксuда 3 % раствор 50 мл
Этиловь/U спирт 95 % 50 мл
15. Резорцина 1 % и 2 % растворы Резорцu.'i 1 О r и 20 r
Натрия метабисульфит 1 r
Этиловый спирт 70 % до 1000 мл
16. Танина 4 % раствор Танин 40 r
Этиловый спирт 70 % до 1000 мл
17. Фурацилина раствор 1: 1500 Нитрофурал 1 r
Этиловый спирт 70 % до 1500 мл
18. Цитраля 1 % раствор Цитраль 1 r
Этиловый спирт 96 % до 100 мл
. Здесь и далее курсивом отмечены субстанции. описанные в частных статьях Фармакопеи
Изroтовленный раствор освобождают от Me
ханических включений (процеживание или филь
трование). Для (\чистки изroтовленноro раствора
от различных нерастворимых механических
включений ero процеживают с помощью CTe
клянной воронки через небольшой тампон ваты
или несколько слоев марли либо фильтруют
через бумажный фильтр с подложенным комоч
ком ваты или через стеклянный фильтр. Проце
живание (фильтрование) производится в зара
нее подroтовленчый контейнер.
Твердые вещества MoryT быть BBeдe
ны в состав лекарственной формы в виде за
ранее приroтовленных жидких лекарственных
средств и концентрированных растворов (табли
цЫ #6.1.1. З, #6.1.1,А, #6.1.1. 5), прибавляемых
после растворения твердых веществ и процежи
вания (фильтрования) раствора.
990
rосударственная фармакопея Республики Беларусь
Таблица #6.1.1. 3
Перечень рекомендованных для иСПОЛЬЗС:3dНИЯ в Аптеке концентрированных растворов и жидких
лекарств< '"ных средств
. - - .--
i Концентрация, % Срок rодности (сутки) при тем-
N!! Наименование пературе хранения
(разведение) не выше 25 ос I от 2 ос дО 8 ос
._ - --
1. Аммония хлорид 20 15
12. /ексаметилентетрамин' 10.20.40 20
,
3. /люкоза безводная 5 2
' 1
4. /люкоза безводная iC :.30 40 : 0 шt 4
. -
5. Димексид *
- .
6. Калия бромид' 20 20 i
7. Калия йодид' 20 15 I
- -
8. Кальция хлорид , 5.10,20 10
I
_._- r ._
9. Кальция хлорид 50 30
--
10. Кислота аскорбиновая' 5 5
.
11. Кислота хлористоводородная 2 10 (1:10) 30
12. Кофеина натрия 6ензоат 5 i 7 15
- -
13. Кофеина натрия бензоат 20 I 20 I r
- + 1
14. Ма2ния сульфат 10. 25. 50 I 15
- - -
15. Натрия бензоат 10 20 I
-
16. Натрия бромид' 20 20 .
. - - ....-- -
17. Натрия zидрокарбонаm 5 4 10
.
18. Натрия салицuлат I 40 20
19. Настойка валерианы' ' ' 1 . *
i
.
20. . Настойка календулы' * *
,
, * *
21. Настойка красавки' ,
h 22 . Настойка ландыша' - * *
,
Настойка ландыша и валерианы поровну'l -=--
23. * *
. .-
24. ХлораЛ2идрат' 10 5
25. ХлораЛ2идрат' 20 15
26. Экстракт (концентрат) валерианы' (1 :2) * *
27. Экстракт (концентрат) rорицвета 1 (1 :2) * .
28. Экстракт (концентрат) пустырника 1 (1 :2) . *
29. Вода очищенная 3
30. Вода мятная 15 30
31. Вода укропная 30
1 Хранят в защищенном от света месте;
2 rотовят из 8,3 % раствора КИСЛОТЫ хлористоводородной, принимая ее за 100 %;
* Используют roToBbIe лекарственные средства промышленноro производства.
Таблица #6.1.1 А
Перечень растворов u жидких лекарственных средств, рекомендуемых для отмеривания
из пипетки, откалиброванной по каплемеру
N!! Наименование растворов и лекарственных средств Концентрация
1. Раствор адреналина rидрохлорида 1 :1000
2. Раствор фурацилина 1 :5000
3. Раствор этакридина лактата 1:500,1:1000
4. Раствор цитраля спиртовой 1:100
5. Настойка мяты перечной
6. Настойка полыни
7. Настойка пустырника
8. Нашатырно анисовые капли I
#6. 1. 1. Жидкие лекарственные средства
991
Таблица #6.1.1. 5
Данные для uзсотовления 1 л концентрироваНН020 раствора
Наименование концентрирован К ТПлотность l Ко ичество:
N2 OHцeH pa I раствора, I веществ а ВОДЫ
Horo раствора ция, о , r/мл или r/cM 3 r очищенной, мл
1. Раствор аммония хлорида 20 1.055 200,0 855
2. Раствор rексаметилентетрамина 10 1,021 100,0 921
3. Раствор rексаметилентетрамина 20 1,042 200,0 842
4. Раствор rексаметилентетрамина 40 1,088 400,0 688
5. Раствор rлюкозы 5 1,018 50,0* 968
6. Раствор rлюкозы 10 1,034 100,0* 934
7. Раствор rлюкозы 20 I 1,068 200,0* 868
8. Раствор rлюкозы 40 1,150 400,0* 749
9. Раствор rлюкозы . . f 1,186 . 500,0* 685
1 O Раствор калия бромида ,20 1,144 200,0 944
11. Раствор калия йодида . .. 200,0 848
2Q. 1,148
12. Раствор кальция rлюконата , 10 i 1,044 100,0 944
13. Раствор кальция хлорида 5 1,020 50,0 970
I
14. Раствор кальция хлорида 10 : 1,041 100,0 941
15. Раствор кальция хлорида 20 i 1,078 200,0 878
16. Раствор кальция хлорида 50 1,207 500,0 707
17. Раствор кислоты аскорбиновой 5 i 1,018 50,0 968
18. Раствор кислоты борной 3 1,008 30,0 978
19. Раствор кислота борной I 4 1,010 40,0 970
20. Раствор кофеин натрия бензоата 10 I 1,034 100,0 934
121. Раствор кофеин натрия бензоата! 20 I 1,073 200,0 I 873
22. .. . 1,048 100,0 948
Раствор маrния сульфата i 1 О i
23. . .. . 1,093 200,0 893
Раствор маrния сульфата i 20.. . I
24. Раствор маrния сульфата ! 25 1,116 250,0 866
,
25. Раствор маrния сульфата 1,221 500,0 , 721
, 50
26. Раствор натрия бензоата 10 I 1,038 I 100,0 938
27. Раствор натрия бромида 20.J. 1,149 200.0 949
28. Раствор натрия rидрокарбоната . . J 1,033 50,0 988
29. Раствор натрия салицилата . .+ 1,030 100,0 940
30. Раствор натрия салицилата 1,083 I 200,0 883
I
31. Раствор натрия салицилата 40 I 1,160 400,0 760
32. Раствор сульфацила натрия 20 1,072 200,0 872 I
33. Раствор сульфацила натрия 30 , 1,108 300,0 808 ,
I
* в пересчете на rлюкозу безводную.
в случае если в состав жидкоrо лекар
cTBeHHoro средства входят друrие жидкие ле
карственные средства, их прибавляют к BOДHO
му раствору в следующей последовательности:
водные нелетучие и непахучие жидко
сти;
иные нелетучие жидкости, смешива
ющиеся с водой;
водные летучие жидкости;
жидкие лекарственные средства, coдep
жащие этанол, соrласно таблице #6.1.1. 6, в
порядке возрастания ero концентрации:
летучие и пахучие жидкости.
При изrотовлении растворов с использо.
ванием вязких и летучих растворителей не.
посредственно в сухой контейнер для отпуска
отвешивают фармацевтические субстанции,
вспомоrательные вещества, затем отвешива
ют растворитель (спирт отмеривают).
При использовании вязких растворите
лей (rлицерин, масла) при меняют наrревание с
учетом физико химических свойств веществ.
При растворении веществ в спирте с KOH
центрацией до 70 % (об/об) раствор наrревают
только в случае необходимости и с соблюдени
ем мер предосторожности. При использовании
спирта с концентраuией выше 70 % (об/об) Ha
rревание растворов не допускается.
Растворы, содержащие летучие вещества,
наrревают при температуре не более 40 ос
45 ос, но не наrревают жидкости, содержащие
эфир и ею смеси со спиртом.
Растворы. изroтовленные на основе вязких
и летучих растворителей. процеживают по мере
необходимости через сухой фильтрующий MaTe
риал (вата, марля и друrие), который подбирают
с учетом вязкости и летучести растворителя, co
блюдая меры предосторожности для снижения
потерь. связанных с испарением.
992
rосударственная фармакопея Республики Беларусь
ОПРЕДЕЛЕНИЕ ОБЩЕrо ОБЪЕМА
жидкоrо ЛЕКАРСТВЕнноrо СРЕДС7fЗ.А.
ПРИ изrОТОВЛЕНИИ MACCO
ОБЪЕМНЫМ МЕТОДОМ ИЛИ МЕТОДОМ
ПО ОБЪЕМУ
При раздельном выписывании фармацевти
ческих субстанций и вспомоrательных веществ
в прописи рецепта (требовании) общий объем
жидкоrо лекарственноrо средства определяют
суммированием объемов всех компонентов, BXO
дящих в пропись, например:
Rp.: So/utionis G'ucosi 10 % 200 т'
Sо/иtiопis Citra/i spirituosae 1 % 2 т'
Маgпеsii sulfatis 4,0
Nat,ii bromidi 2, О
Sirupi simp/icis
Тincturae Va/eriaпae апа 1 О т/
Общий объем жидкоro лекарственноroсред
ства равен 222 мл (200+2+10+10).
Если в состав лекарственноro средства
входит жидкость, выписанная по массе (т). ее
объем (\1) определяют с учетом плотности (р) по
формуле:
v т .
р
например:
Rp.: So/utioпis Kalii acetatis 10 % 100 т'
G'ycero/i 10,0
В прописи присутствует жидкость, выпи
санная по массе rлицерин. Для определения
объема rлицерина используют среднее значе
ние ero плотности. Объем 10 r rлицерина равен
8 мл. Общий объем жидкоro лекарственноro
средства 108 мл (100+8).
Общий объем жидкоro лекарственноro cpeд
ства указан в прописи рецепта (требовании), Ha
при мер:
Rp.: Тinctu'ae Vа'еriапае 5 т'
Sо/иtiопis Natrii bromidi 3 % ad 100 т'
Общий объем жидкоro лекарственноro cpeд
ства указан в прописи и равен 100 мл.
ОПРЕДЕЛЕНИЕ ОБЩЕЙ МАССЫ
жидкоrо ЛЕКАРСТВЕнноrо СРЕДСТВА
ПРИ изrОТОВЛЕНИИ МЕТОДОМ
ПО МАССЕ
При раздельном выписывании фармацевти
ческих субстанций и вспомоrательных веществ
в прописи рецепта (требовании) общую массу
жидкоro лекарственноro средства определяют
суммированием масс всех компонентов, входя
щих в пропись, например:
Rp.: Pheno/i 0,1
О'е; Не/iапthi 10,0
Общая масса жидкоro лекарственноro cpeд
ства равна сумме массы фенола и массЬ! масла
подсолнечноro и составляет 10,1 r.
Общая масса жидкоro лекарственноro cpeд
ства может быть указана в прописи рецепта (Tpe
бовании), например, «ad 200,0», «5 % 200,0»,
«1 :20 200,0»:
Rp.: Natrii tetraboratis 20,0
G/ycero/i ad 80, О
Общая масса жидкоro лекарственноro cpeд
ства обозначена и составляет 80,0 r.
Если в прописи рецепта (требовании) при
сутствует жидкость, выписанная по объему, ее
массу определяют с учетом плотности. т = V' р,
например:
Rp.: Р/итЫ acetates
Ammonii ch/oridi апа 3,0
G'ycero'i 25,0
Spi,itus aethy/ici 95 % 25 т'
Su/furis p,aecipitati 4, О
Aquae pu,ificatae 180 т/
Общая масса жидкоro лекарственно
ro средства равна сумме масс всех Be
ществ и массы 25 мл этиловоro спирта 95 %
(т = 25 . 0.8114 = 20,29) и составляет 235,29 r
(3+3+25+20,29+4+180).
ОПРЕДЕЛЕНИЕ ИЗМЕНЕНИЯ ОБЩЕrо
ОБЪЕМА ЖИДКИХ ЛЕКАРСТВЕННЫХ
СРЕДСТВ И КОНЦЕНТРИРОВАННЫХ
РАСТВОРОВ ПРИ МАССО ОБЪЕМНОМ
МЕТОДЕизrОТОВЛЕНИЯ
Изменение общеrо объема жидкоro лекар
ственноro средства при растворении твердых
веществ с получением концентрации до 3 %
можно не учитывать, так как оно укладывается 8
норму допустимоro отклонения.
Для каждоro лекарственноro вещества MaK
симальная концентрация (Ста,' %), при KOTO
рой изменение общеro объема укладывается в
норму допустимоro отклонения, рассчитывается
по формуле:
N
С таХ = I
КУО
rдe:
N норма допустимоro отклонения для
данноro общеro объема, %;
КУО коэффициент увеличения объема
(таблица #6.1.1. 7), мл/r.
Коэффициент увеличения объема (КУО) по
казывает увеличение объема раствора в милли
литрах при растворении 1 rpaMMa вещества при
температуре 20 ОС.
При изroтовлении жидкоro лекарственноro
средства путем растворения нескольких TBep
дых веществ изменение общеro объема учиты
вают, если их суммарное содержание составля
ет 3 % и более.
РАЗВЕДЕНИЕ СТАНДАРТНЫХ
ФАРМАКОПЕЙНЫХ РАСТВОРОВ
Перечень стандартных фармакопейных pac
Т80рОВ и их концентрации указаны в таблице
#6.1.1. 8.
#6.1.1. Жидкие лекарственные средства
993
Таблица #6.1.1. 6
Содержание этанола в некоторых жидких лекарственных средствах
N!! Наименование жидкоrо Содержание этанола, % (06/06)
лекарственноrо средства
1. rрудной эликсир Не менее 14
2. Йода 5 % раствор не менее 46
3. Настойка аралии 70
4. Настойка боярышника 70
5. Настойка валерианы 70
6. Настойка женьшеня 70
7. Настойка заманихи 70
8. Настойка зверобоя 40
9. Настойка календулы 70
10. Настойка красавки 40
11. Настойка ландыша 70
12. Настойка лимонника 95
13. Настойка мяты 90
14. Настойка полыни 70
15. Настойка пустырника 70
16. Настойка стручковоro перца 90
17. Настойка эвкалипта 70
18. Нашатырно анисовые капли 7 80
19. Цитраля 1 % раствор 96
20. Экстракт жидкий боярышника 70
21. Экстракт roрца перечноrо (водяноrо перца) 70
22. Экстракт калины 50
23. Экстракт крапивы 50
24. Экстракт тимьяна 20
25. Экстракт тысячелистника 40
26. Экстракт элеутерококка 40
27. Экстракты жидкие стандартизованные (концентраты) 2 ЗО
Таблица #6.1.1. 7
Водные Спиртовые растворы Водные
растворы суспензии
N!! Вещество KOHцeHTpa
КУО, млlr КУО, мл/r Iция этанола, КУО, мл/r
% (об/о6)
1. Аминокапроновая кислота 0,79
2. Аммония хлорид 0,72
3. Аскорбиновая кислота 0,61
4. Ацетилсалициловая кислота 0,72 90
5. Бензойная кислота 0,87 70,90,96
6. Бензокаин(анестезин) 0,85 70,90,96
7. Бензилпенициллин натрия 0,68
8. Борная кислота 0,68 0,65 70,90,96
9. Бромкамфора 0,80 70
10. Висмута нитрат основной 0,19
11. tексаметuлентетрамин 0,78 0,79 70,90
12. tлюкоза безводная 0,64
13. tлюкоза МОН02идрат 0,69
14. tлютаминовая кислота 0,62
15. Ди6азол 0,82 0,86 30
16. ДифеН2идрамина 2идрохлорид 0,86 0,87 70,90,96
(димедрол)
17. Желатин 0,75
125 Зак. 1712
994
rосударственная фармакопея Республики Беларусь
Водные Спиртовые растворы Водные
растворы суспензии
N2 Вещество KOHцeHTpa
КУО, мл/r КУО, мл/r ция этанола, КУО, мл/r
% (об/о6)
18. Изониазид 0,72
19. Йод 0,23' 0,22 70,90,96
20. Калия бромид 0,27 0,36 70
21. Калия йодид 0,25
22. Калия перманванат 0,36
23. Калия хлорид 0,37
24. Кальция 2лицерофосфат 0,46
25. Кальция 2люконат 0,50
26. Кальция карбонат 0,38
27. Кальция лактат 0,67
28. Кальция хлорид 2екса2идрат 0,58
29. Камфора 1,ОЗ 70,90,96
30. Каолин тяжелый (rлина белая) 0,39
31. Кофеин натрия бензоат 0,65
32. Крахмал картофельный 0,68 0,67
33. Лимонная кислота МОН02идрат I 0.62
34. Ма2ния оксид, ле2кий I 0,34
35. Ма2ния сульфат 2епта2идрат 0,50
36. Ментол 1,10 70,90,96
37. Метамизол натрия (анальrин) 0,68 0,67 30
38. Метилурацил 0,692
39. Метилцеллюлоза 0,61
40. Натрия аминосалицилат ди2ират 0,64
41. Натрия ацетат три2идрат 0,71
42. Натрия ацетат (безводный) 0,52
43. Натрия бензоат 0,60
44. Натрия бромид 0,26 0,30 70
45. Натрия 2идрокарбонат 0,30
46. Натрия rидроцитрат 0,46
47. Натрия йодид 0,38
48. Натрия нитрат 0,38
49. Натрия нитрит 0,З7
50. Натрия нуклеинат 0,55
51. Натрия салицилат 0,59
52. Натрия сульфат дека2идрат 0,53
53. Натрия тетраборат 0,47
54. Натрия тиосульфат 0,51
55. Натрия хлорид 0,33
56. Натрия цитрат 0,48
57. Папаверина2идрохлорид 0,77 0,81 30
58. Пепсин 0,61
59. Пилокарпинаrидрохлорид 0,77
60. Пиридоксина 2идрохлорид 0,71
61. Повuдон(поливинилпирролидон) 0,81
62. Поливиниловый спирт 0,77
63. Прокаина 2идрохлорид (новокаин) 0,81 0,81 70,90
#6.1.1. Жидкие лекарствеННblе средства
995
Водные Спиртовые растворы Водные
растворы суспензии
N2 Вещество KOHцeHTpa
КУО, мл/r КУО, мл/r ция этанола, КУО, мл/r
% (об/об)
64. Прокаинамида2идрохлорид 0,83
( новокаинамид)
65. Резорцин 0,79 0,77 70,90,96
66. Салициловая кислота 0,77 70,90,96
67. Свинца ацетат 0,30
68. Сера для наРУЖН020 применения 0,483
69. Серебра нитрат 0,18
70. Серебра протеинат (протарroл) 0,64
71. Серебро коллоидное для наРУЖН020 при 0,61
менения(колларrол)
72. Стрептомицина сульфат 0,58
73. Сульфаниламид (стрептоцид) 0,69
74. Сульфацетамид натрия (сульфацил 0,62 0,65 70
натрия)
75. Тальк 0,34
76. Танин 0,65 0,60 70,90,96
77. Т еофиллин этилендиамин (эуфиллин) 0,70 0,71 12
78. Терпинrидрат 0,77 96
79. Тетракаина 2идрохлорид (дикаин) 0,86
80. Тиамина бромид 0,61
81. Тимол 1,01 70,90,96
82. Тримекаин 0,89
83. Фенилэфрина 2идрохлорид (мезатон) 0,77
84. Фенол 0,90
85. Хинина rидрохлорид 0,81
86. ХлораЛ2идрат 0,76 0,59 70,90,96
87. Хлорамфеникол (левомицетин) 0,66 70,90,96
88. Цинка оксид 0,21
89. Цинка сульфат 2епта2идрат 0,41
90. Экстракт (концентрат) roрицвета сухой 0,60 I
стандартизованный 1: 1
91. Экстракт (концентрат) алтея сухой CTaH 0,61 0,61 12
дартизованный 1: 1
92. Эфедрина 2идрохлорид 0,84
1 В растворе калия йодида;
2 Суспензия в этиловом спирте (ЗО %, об/об);
3 Суспензия в 70 %, 90 %, 96 % этиловом спирте.
Таблица #6.1.1. 8
Перечень стандартных фармакопейных растворов и их концентрации
Химическое название раствора Концентрация 1 , % Условное название раствора
Хлористоводородной кислоты 25 % раствор 24,8 25,2
Хлористоводородной кислоты 8,3 % раствор 8,2 8,4
Аммиака 1 О % раствор 9,5 10,5
Уксусной кислоты 30 % раствор 29,5 30,5
Водорода пероксида 30 % раствор 29,0 31,0 Перrидроль
Формальде2ида 35 % раствор 34,5 38,0 Формалин 2
1 Указана концентрация химическою вещества (М/М):
2 Номинальная концентрация формальдеrида для расчетов принимается равной З5 %.
996
rосударственная фармакопея Республики Беларусь
Растворыхлористоводороднойкислоты
Растворы кислоты хлористоводородной
любой концентрации изroтавливают в аптеках
из 8,3 % раствора кислоты хлористоводород
ной, принимая ее за единицу (100 %); 8,3 %
раствор кислоты хлористоводородной исполь
зуют также для получения 10 % (1 :10) paCT
вора (таблица #6.1.1. 3) в качестве внутриап
течной заrотовки (концентрация HCI при этом
будет от 0,82 % до 0,84 %).
Если в прописи рецепта (требовании) KOH
центрация раствора не указана, то отпускают
8,3 % раствор кислоты хлористо водородной.
25 % раствор кислоты хлористоводородной
отпускается только в тех случаях, коrда в пропи
си рецепта имеется соответствующее указание.
Без дополнительноro указания 25 % paCT
вор кислоты хлористоводородной использует
ся при изroтовлении раствора 2 по прописи
Демьяновича.
Растворы аммиака и уксусной кислоты
Растворы аммиака и уксусной кислоты
изrотавливают, исходя из фактическоrо co
держания вещества в стандартном фармако
пейном растворе. При расчетах используют
формулу разведения:
v = v. . С , ,
С
rде:
V объем стандартною фармакопейно
ro раствора, мл;
V, требуемый объем изrотавливаемоrо
раствора, мл;
С, требуемая концентрация раствора, %;
С концентрация стандартною фарма
копейноrо раствора, %.
Если в прописи рецепта (требовании)
концентрация раствора не указана, то отпу
скают 30 % раствор кислоты уксусной и 1 О %
раствор аммиака.
Растворы формальдеrида и водорода
пероксида
При выполнении расчетов для разведе
ния стандартных фармакопейных растворов
водорода пероксида и формальдеrида до
требуемой концентрации учитывают назва
ние (химическое или условное) выписанно
ro раствора в прописи рецепта (требовании),
например:
1) Rp.: Sо/иtiопis Forma/dehydi 5 % 200 т/
В прописи рецепта раствор выпи
сан под химическим названием, но в Ha
личии имеется раствор формальдеrида с
концентрацией 34 %. Количество милли
литров раствора формальдеrида, требу
емое для разведения, рассчитывают с учетом
ero фактическою содержания в растворе:
200 мл . 5 % : 34 % = 29,4 мл, воды очищен
ной 170,6 мл (200 29,4).
2) Rp.: Sо/иtiопis FоrтаJiпi 5 % 200 т/
В прописи рецепта (требовании) раствор
выписан под условным названием. При pac
четах стандартный фармакопейный раствор
принимают за единицу (100 %), то есть берут
1 О мл формалина и 190 мл воды очищенной.
В случае использования раствора формаль
деrида с концентрацией 34 % ero объем pac
считывают с учетом номинальноro содержа
ния (35 %) формальдеrида в «формалине»:
10. 35: 34 = 10,3.10,З мл 34 % раствора фор
мальдеrида и 189,7 мл воды очищенной.
Для изrотовления разведенных растворов
формальдеrида и водорода пероксида разре
шается использовать растворы с содержани
ем формальдеrида менее 34,5 % и содержа
нием водорода пероксида более 31,0 %.
Если в прописи рецепта (требовании) KOH
центрация раствора не указана, то отпускают
3 % раствор водорода пероксида и 35 % pacт
вор формальдеrида (формалин).
Соотношения плотности и концентрации
водорода пероксида в растворе следующие:
Плотность
водорода пе
роксида, r/см З
Концентрация водорода
пероксида, %
по массе Macco
объемная
29,18
30,72
31,94
33,15
34,36
35,59
36,82
38,05
39,29
40,55
41,81
43,07
44,34
45,62
1,096
1,098
1,101
1,105
1,109
1,112
1,116
1,119
1,123
1,126
1,130
1,134
1,137
1,141
27,5
28
29
30
31
32
33
34
35
36
37
38
39
40
изrОТОВЛЕНИЕАРОМАТНЫХВОД
Изrотовление ароматных вод проводит
ся В асептических условиях. Для приrотовле
ния мятной воды берут 0,05 r масла мятноrо,
а для укропной 0,44 r масла фенхелевоro и
энерrично смешивают с 1 л воды очищенной
стерильной до растворения в течение 1 мин.
Сроки хранения:
вода укропная 30 суток;
вода мятная:
в виде фасовки (200 мл) 30 суток;
#6.1.1. Жидкие лекарственные средства
997
в виде полуфабриката по 500 мл И
1000 мл 15 суток.
изrОТОВЛЕНИЕ ЖИДКИХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ,
СОДЕРЖАЩИХ АРОМАТНЫЕ ВОДЫ
Ароматные воды дозируют по объему. При
растворении твердых веществ объем воды
ароматной, указанный в прописи рецепта, не
уменьшают на величину изменения объема,
так как изменяется концентрация ароматной
воды. Изroтовление растворов проводят в
мерной посуде.
В случае точноro указания объема воды
ароматной в прописи рецепта изменение
объема при растворении твердых веществ
учитывают при контроле качества изroтов
ленноro лекарственноro средства. При расче
те общеro объема используют значения КУО
веществ.
При изroтовлении жидких лекарственных
средств, в которых основной дисперсионной
средой является ароматная вода, концентри
рованные растворы веществ не используют.
ОСОБЕННОСТИ изrОТОВЛЕНИЯ
НЕКОТОРЫХ ЖИДКИХ ЛЕКАРСТВЕННЫХ
СРЕДСТВ
Раствор по Демьяновичу NQ 1
Раствор по Демьяновичу NQ 1 имеет сле
дующий авторский состав при ero изrотовле
нии по массе:
Натрия тиосульфата 60 r
Воды очищенной 40 r
Изrотовление раствора массо объемным
методом путем растворения 60 r натрия ти
осульфата и доведения водой очищенной
объема до 100 мл запрещается, так как в
этом случае масса раствора увеличится на
29,4 r (масса раствора 100 r; объем paCT
вора 70,6 мл (40 + (60 . 0,51 куо ))); KOHцeH
трация по массе вместо 60 % будет 46,37 %
(100 . 60 : 129,4).
При изrотовлении раствора массо объем
ным методом необходимо учитывать коэффи
циент увеличения объема для сохранения co
ответствия авторской прописи, например:
Rp.: So/utioпis Nat,ii thiosulfati 60 %
100 т/ (раствор по Демьяновичу NQ 1)
Для изrотовления 100 мл 60 % раствора Mac
со обьемным методом следует взять 85 r натрия
тиосульфата и воды очищенной до 100 мл.
Водные растворы Люrоля
Состав:
Йода 1,0 r (5,0 r)
Калия йод ида 2,0 r (10,0 r)
Воды очищенной до 100 мл
Водные растворы (1 % и 5 %) Люrоля из
roтавливают массо объемным методом, Ha
пример:
Rp.: Sо/иtiопis /odi 1 % (5 %) 100 т/
126 Зак. 1712.
В мерной посуде растворяют калия йодид
в приблизительно равном количестве воды
очищенной. В насыщенном растворе калия
йодида растворяют йод. Объем раствора дo
водят водой очищенной до требуемоro. Если
приrотовление ведется не в мерной посуде,
то объем воды очищенной рассчитывают с
использованием КУО.
Применение:
1 % и 5 % раствор внутрь в виде
капель;
1 % раствор наружно.
rлицериновые растворы Люrоля
rлицериновые растворы (0,25 % и 1 %)
Люrоля изrотавливают методом по массе, Ha
пример:
Rp.: /odi 1,0 (0,25)
Ka/ii iodidi 2,0 (0,5)
Aquae purificatae 3 т/ (0,75 т/)
G/ycero/i 94.0 (98,5)
При изroтовлении rлицериновых paCTBO
ров в предварительно взвешенном контей
нере растворяют калия йодид в указанном в
прописи рецепта количестве воды очищен
ной. В насыщенном растворе калия йод ида
растворяют йод. сюда же отвешивают rлице
рин и смешивают.
Применение: наружно.
РАСЧЕТЫ И ПРАВИЛА ДОЗИРОВАНИЯ
этиловоrо СПИРТА
Выписанное в рецепте количество этилово
ro спирта соответствует объемным единицам из
мерения.
При разведении спирта этиловоro использу
ют таблицы #5.5. 1 5.5. 6, #6.1.1. 9 .1.1. 20.
Норму отпуска этиловоro спирта учетной
концентрации в пересчете на массу учитыва
ют в соответствии с действующими норматив
ными документами.
При изrотовлении лекарственных средств
этиловый спирт дозируют по объему. не
уменьшая объем. указанный в рецепте (Tpe
бовании), на величину ero увеличения при
растворении веществ. Исключение COCTaB
ляют спиртовые растворы, приведенные в
таблице #6.1.1. 2. Общий объем учитывают
при контроле качества лекарственноrо cpeд
ства. Изменение объема при растворении Be
ществ, учитываемое при контроле, рассчиты
вают, используя значения КУО веществ.
Если в прописи рецепта (требовании)
без указания концентрации выписан раствор,
представленный несколькими концентраци
ями вещества, отпускают раствор с меньшей
концентрацией, то есть: бриллиантовоro зе
леноro 1 % раствор: йода 1 % раствор; кисло
ты борной 1 % раствор; кислоты салициловой
1 % раствор; левомицетина 0,25 % раствор;
ментола 1 % раствор; резорцина 1 % раствор;
камфоры 2 % раствор.
998
rосударственная фармакопея Республики Беларусь
Таблица #6.1.1. 9
Разведение спирта этиловоzо по массе
. 30% 40% 50% 60% 70% 80% 90% 95% 96%
ж
ф .... .... .... ....
::1'0:: IU .... IU IU .... IU IU IU IU .... IU .... IU ....
Ж S Q. q Q. q Q. q Q. q Q. Q. q Q. q Q. q Q.
0::1' S S S S S S S S S
::.::[ 1: о 1: О 1: О 1: О 1: 1: О 1: О 1: О 1:
IXI U IXI U IXI U IXI U IXI U IXI U IXI U IXI U IXI U
....
96,1 738 262 646 354 548 452 446 554 336 664 218 782 88 912 17 983 2 998
96,2 739 261 646 354 549 451 447 553 337 663 219 781 90 910 18 982 3 997
96,3 739 261 647 353 550 450 447 553 338 662 221 779 91 909 20 980 5 995
96,4 739 261 647 353 551 449 448 552 339 661 222 778 93 907 21 979 7 994
96,5 740 260 648 352 551 449 449 551 340 660 222 777 94 906 23 977 8 992
96,6 740 260 648 352 552 448 450 550 341 659 224 776 96 904 24 976 9 991
96,7 741 259 649 351 553 447 451 549 342 658 225 775 97 903 26 974 11 989
96,8 741 259 650 350 553 447 452 548 343 657 226 773 98 902 27 973 12 988
96,9 741 259 650 359 554 446 453 547 344 656 228 772 100 900 29 971 14 986
Примечание: количества воды очищенной и этиловоro спирта концентрации от 96,1 % до 96,9 % в rpaM
мах, которые необходимо смешать при 20 ос, чтобы получить 1000 r этиловоro спирта концентрации:
30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 %, 95 %, 96 %.
Таблица #6.1.1. 1 О
Соответствие объемов (мл) этилово,ю спирта различной концентрации
массе (сl этиловоzо спирта 95 % при температуре 20 ос
Концентрация ! Объем (мл) спирто-водноrо раствора
i и соответствие ero массе (r) этиловоrо спирта
этиловоrо
спирта, % 5 10 15 20 25 30 40 50 100
95 4,06 8,11 12,17 16,23 20,29 24,34 32,46 40,57 81,14
90 3,84 7,69 11,53 15,37 19,22 23,06 30,75 38,44 76,87
80 3,42 6,83 10,25 13,66 17,08 20,50 27,33 34,16 68,32
70 2,99 5,98 8,97 11,95 14,94 17,93 23,91 29,89 59,77
60 2,56 5,13 7,69 10,26 12,82 15,38 20,51 25,64 51,28
50 2,14 4,27 6,41 8,54 10,68 12,81 17,08 21,35 42,70
40 1,71 3,41 5,12 6,83 8,53 10,24 13,65 17,07 34.13
30 1,28 2,56 3,84 5,12 6,40 7,68 10,24 12,80 25,60
20 0,85 1,70 2,56 3,41 4,26 5,11 6,82 8,52 17,04
Концентрация Объем (мл) спирто-водноrо раствора
этиловоrо и соответствие ero массе (r) этиловоrо спирта
спирта, % 5 10 15 20 25 30 40 50 100
96 4,04 8,08 12,11 16,15 20,19 24,23 32,30 40,38 80,75
90 3,79 7,57 11,36 15,14 18,93 22,71 30,28 37,86 75,71
80 3,37 6,73 10,09 13,46 16,82 20,19 26,92 33,65 67,29
70 2,95 5,89 8,83 11,78 14,72 17,67 23,56 29,45 58,89
60 2,52 5,05 7,57 10,09 12,62 15,14 20,18 25,23 50,46
50 2,10 4,20 6,31 8,41 10,51 12,61 16,82 21,02 42,04
40 1,68 3,37 5,05 6,73 8,42 10,10 13,46 16,83 33,66
30 1,26 2,52 3,78 5,04 6,30 7,56 10,08 12,61 25,21
20 0,84 1,68 2,53 3.37 4,21 5,03 6,74 8,42 16,84
Таблица #6.1.1. 11
Соответствие объемов (мл) этиловоzо спирта различной концентрации
массе (с) 96 % этиловоzо спирта при температуре 20 ос
#6.1.1. Жидкие лекарственные средства
999
Таблица #6.1.1. 12
Соответствие объемов (мл) этилов020 спирта различной концентрации
массе (с) 96,1 % этиловоzо спирта при температуре 20 ос
Концентрация Объем (мл) спирто-водноrо раствора
этиловоrо и соответствие ero массе (r) этиловоrо спирта
спирта, % 5 10 15 20 25 ЗО 40 50 100
96,1 4,04 8,07 12,11 16,14 20,18 24,12 32,28 40,35 80,71
96 4,03 8,06 12,09 16,12 20,16 24,19 32,25 40,31 80,62
95 3,99 7,98 11,97 15,96 19,95 23,94 31,92 39,90 79,79
90 3,78 7,56 11,34 15,12 18,90 22,68 30,24 37,80 75,59
80 3,36 6;72 10,08 13,44 16,80 20.16 26,88 33,60 67,19
70 2,94 5,88 8,82 11,76 14,70 17.64 23,52 29,40 58,80
60 2,52 5,04 7,56 10,08 12,60 15.12 20,16 25,20 50,40
50 2,10 4,20 6,30 8,40 10,50 12,60 16,80 21,00 42,00
40 1,68 3,36 5,04 6,72 8,40 10,08 13,44 16,80 33,59
30 1,26 2,52 3,78 5,04 6,30 I 7.56 10,08 12,60 25,20
20 0,84 1,68 2,52 3,36 4,20 5.04 6,72 8,40 16,79
Таблица #6.1.1. 13
Соответствие объемов (мл) этиловоzо спирта различной концентрации
массе (с) 96,2 % этиловоzо спирта при 20 ос
Концентрация Объем (мл) спирто-водноrо раствора
этиловоrо и соответствие ero массе (r) этиловоrо спирта
спирта, % 5 10 15 20 25 ЗО 40 50 100
96,2 4,03 8,07 12,10 16,13 20,17 24,20 32,27 40,33 80,67
96 4,02 8,05 12,07 16,10 20,12 24,14 32,19 40,24 80,48
95 З,98 7,97 11,95 15,93 19,92 23,90 31,86 39,83 79,65
90 3,77 7,55 11,32 15,09 18,87 22,64 30,18 37,73 75,45
80 3,35 6,71 10,06 13,41 16,77 20,12 26,83 33,54 67,07
70 2,94 5,87 8,81 11,74 14,68 17,61 23,48 29,35 58,69
60 2,52 5,03 7,55 10,06 12,58 15,09 20,12 25,15 50,30
50 2,10 4,19 6,29 8,38 10,48 12,58 16,77 20,96 41,92
40 1,68 3,35 5,03 6,71 8,39 10,06 13,41 16,77 33,53
30 1,26 2,52 3,77 I 5,03 6,29 7,55 10,06 12,58 25,15
20 0,84 1,68 2,52 3,35 4,20 5,03 6,71 8,39 16,77
Таблица #6.1.1. 14
Соответствие объемов (мл) этиловоzо спирта различной концентрации
массе (с) 96,3 % этиловоzо спирта при температуре 20 ос
Концентрация Объем (мл) спирто водноrо раствора I
этиловоrо и соответствие ero массе (r) этиловоrо спирта
спирта, % 5 10 15 20 25 ЗО I 40 50 100
96,3 4,03 8,06 12,09 16,12 20,16 24,19 I 32.25 40,31 80,62
96 4,02 8,04 12,05 16,07 20,09 24,11 32.14 40,18 80,36
95 3,98 7,95 11,93 15,91 19,89 23.86 31.82 : 39,77 79,54
90 3,7r 7,54 11,30 15,07 18,84 22.61 30.14 37,68 75,35
80 3,35 6,70 10,05 13,40 16,75 20.09 26,79 i 33,49 66,98
.
70 2,93 5,86 8,79 11,72 14,65 I 17.58 23.44 ! 29,31 58,61
60 2,51 5,02 7,54 10,05 12.56 15.07 , 20.09 i 25,12 50,23
.. I , , i
50 2,09 4,19 6,28 , 8.37 10.47 12.56 16.74 20,93 41,86
40 1,68 3,35 t 8.37 i 10.05 13.40 16,75 33,49
r 5,03...........t... 6.70
30 1,26 2,51 I 3.77 5.02 6.28 7.54 10.05 12,56 25,12
! 20 ! О 84 1.67 I 2.51 3.35 4,19 I 5,02 6,70 I 8,37 16,74
I .. . . ....1 ' I I
,
1000
rосударственная фармакопея Республики Беларусь
Таблица #6.1.1. 15
Соответствие объемов (мл) этиловоzо спирта различной концентрации
массе (с) 96,4 % этиловоzо спирта при температуре 20 ос
Концентрация Объем (мл) спирто водноrо раствора
этиловоrо и соответствие ero массе (r) этиловоrо спирта
спирта, % 5 10 15 20 25 30 40 50 100
96,4 4,03 8,06 12,09 16,12 20,15 24,17 32,23 40,29 80,58
96 4,01 8,03 12,04 16,05 20,06 24,08 32,10 40,1З 80,25
95 3,97 7,94 11,91 15,88 19,85 23,82 31,76 39,71 79,41
90 3,76 7,53 11,29 15,05 18,81 22,58 30,10 37,63 75,25
80 3,34 6,69 10,03 13,37 16,72 20,06 26,75 33,44 66,87
70 2,93 5,85 8,78 11,70 14,63 17,56 23,30 29,26 58,52
60 2,51 5,02 7,52 10,03 12,54 15,05 20,06 25,08 50,16
50 2,09 4,18 6,27 8,36 10,45 12,54 16,72 20,90 41,80
40 1,67 3,34 5,02 6,69 8,36 10,03 13,38 16,72 33,44
30 1,25 2,51 3,76 5,02 6,27 7,52 10,03 12,54 25,08
20 0,84 1,67 2,51 3,34 4,18 5,02 6,69 8,36 16,72
Таблица #6.1.1. 16
Соответствие объемов (мл) этиЛО80ZO спирта различной концентрации
массе (с) 96,5 % этилов020 спирта при температуре 20 ос
Концентрация Объем (мл) спирто водноrо раствора
этиловоrо и соответствие ero массе (r) этиловоrо спирта
спирта, % I 5 10 15 20 25 30 40 50 100
96,5 i 4,03 8,05 12,08 16,11 20,14 24,16 32,22 40,27 80,54
96 4,01 8,01 12,02 16,02 20,03 24,04 32,05 40,06 80,12
95 3,97 7,93 11,90 15,86 19,82 23,79 31,72 39,65 79,29
90 3,76 7,51 11,27 15,02 18,78 22,53 30,04 З7,56 75,11
80 3,34 6,68 10,02 13,35 16,69 20,03 25,71 33,39 66,77
70 2,92 5,84 8,77 11,69 14,61 17,53 23,37 29,22 58,43
60 2,50 5,01 7,51 10,02 12,52 15,02 20,03 25,04 50,08
50 2,09 4,17 6,26 8,35 10,44 12,52 16,70 20,87 41,74
40 1,67 3,34 5,01 6,86 8,35 10,01 13,35 16,69 33,38
30 1,25 2,50 3,76 5,01 6,26 7,51 10,02 12,52 25,04
20 0,84 1,67 2,51 3,34 4,17 5,01 6,68 8,.35 16,69
Таблица #6.1.1. 17
Соответствие объемов (мл) этиловоzо спирта различной концентрации
массе (с) 96,6 % этиловоzо спирта при температуре 20 ос
Концентрация Объем (мл) спирто водноrо раствора
этиловоrо и соответствие ero массе (r) этиловоrо спирта
спирта, % 5 10 15 20 25 30 40 50 100
96,6 4,03 8,05 12,07 16,10 20,12 24,15 32,20 40,25 80,50
96 4,00 8,00 12,00 16,00 20,00 24,00 32,00 40,00 79,99
95 3,96 7,92 11,87 15,83 19,79 23,75 31,66 39,58 79,16
90 3,75 7,50 11,25 15,00 18,75 22,50 30,00 37,50 75,00
80 3,33 6,67 10,00 13,33 16,67 20,00 25,67 33,34 66,67
70 2,92 5,83 8,75 11,67 14,59 17,50 23,34 29,17 58,34
60 2,50 5,00 7,50 10,00 12,50 15,00 20,00 25,00 50,00
50 2,08 4,17 6,25 8,33 10,42 12,50 16,67 20,84 41,67
40 1,67 3,33 5,00 6,67 8,33 10,00 13,33 16,67 33,33
30 1,25 2,50 3,75 5,00 6,25 7,50 10,00 12,50 25,00
20 0,83 1,67 2,50 3,33 4,17 5,00 6,66 8,33 16,66
#6.1.1. Жидкие лекарственные средства
1001
Таблица #6.1.1. 18
Соответствие объемов (мл) этиловоzо спирта различной концентрации
массе (с) 96,7 % этиловоzо спирта при температуре 20 ос
Концентрация Объем (мл) спирто водноrо раствора
этиловоrо и соответствие ero массе (r) этиловоrо спирта
спирта, % 5 10 15 20 25 30 40 50 100
96,7 4,02 8,05 12,07 16,09 20,11 24,14 32,18 40,23 80,46
96 3,99 7,99 12,11 15,97 19,97 23,96 31,95 39,94 79,87
95 3,95 7,91 11,86 15,81 19,76 23,72 31,62 39,53 79,05
90 3,74 7,49 11,23 14,98 18,72 22,46 29,95 37,44 74,88
80 3,33 6,66 9,98 13,31 16,64 19,97 26,62 33,28 66,56
70 2,91 5,83 8,74 11,65 14,56 17 ,48 23,30 29,13 58,25
60 2,50 4,99 7,49 9,98 12,48 14,98 19,97 24,96 49,92
50 2,08 4,16 6,24 8,32 10,40 12,48 16,64 20,81 41,61
40 1,66 3,33 4,99 6,66 8,32 9,98 13,31 16,64 33,28
30 1,25 2,50 3,74 4,99 6,24 7,49 9,98 12,48 24,96
20 0,83 1,66 2,50 3,33 4,16 4,99 6,66 8,32 16,64
Таблица #6.1.1. 19
Соответствие объемов (мл) этиловоzо спирта различной концентрации
массе (с) 96,8 % этиловоzо спирта при температуре 20 ос
Концентрация Объем (мл) спирто водноrо раствора
этиловоrо и соответствие ero массе (r) этиловоrо спирта
спирта, % 5 10 15 20 25 30 40 50 100
96,8 4,02 8,04 12,06 16,08 20,11 24,13 32,17 40,21 80,42
96 3,99 7,98 11,96 15,95 19,94 23,93 31,90 39,88 79,75
95 3,95 7,89 11,84 15,78 19,73 23,68 31,57 39,46 78,92
90 3,74 7,48 11,22 14,95 18,69 22,43 29,91 37,39 74,77
80 3,32 6,65 9,97 13,29 16,62 19,94 26,58 33,23 66,46
70 2,91 5,82 8,72 11,63 14,54 17,45 23,26 29,08 58,16
60 2,49 4,99 7,48 9,97 12,46 14,96 19,94 24,93 49,85
50 2,08 4,15 6,23 8,31 10,39 12,46 16,62 20,77 41,54
40 1,66 3,32 4,99 6,65 8,31 9,97 13,29 16,62 33,23
30 1,25 2,49 3,74 4,98 6,23 7,48 9,97 12,46 24,92
20 0,83 1,66 2,49 3,32 4,15 4,98 6,64 8,31 16,61
Таблица #6_1_1. 20
Соответствие объемов (мл) этl)ловоzо спирта различной концентрации
массе (с) 96,9 % этиловоzо спирта при температуре 20 ос
Концентрация Объем (мл) спирто-водноrо раствора
этиловоrо и соответствие ero массе (r) этиловоrо спирта
спирта, % 5 10 15 20 25 30 40 i 50 100
,
96,9 4,02 8,04 12,06 16,08 20,10 24,11 32,15 i 40,19 80,38
96 3,98 7,96 11,95 15,93 19,91 23,89 31.85 39,82 79,63
95 3,94 7,88 11,82 15,76 19,70 23,64 31.52 39,41 78,81
90 3,73 7,47 11,20 14,93 18,67 22.40 29,86 37,33 74,66
80 3,32 6,64 9,94 13,27 16,59 19,91 26,55 33,19 66,37
70 2,90 5,81 8,71 11,61 14,52 17,42 23,22 29,04 58,07
60 2,49 4,98 7,48 9,96 12,45 14.93 19,91 24,94 49,78
50 2,07 4,15 6,22 8,30 10,37 12,44 16,59 20,74 41,48
40 1,66 3,32 4,98 6,64 8.30 9,95 13,27 16,59 З3,18
30 1,24 2,49 3,73 4,98 6,22 7,46 9,95 12,44 24,88
20 0,83 1,66 2,49 3,32 4,15 4,98 6,64 8,30 16,59
127. За.. 1712.
1002
rосударственная фармакопея Республики Беларусь
изrОТОВЛЕНИЕ ЖИДКИХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ,
СОДЕРЖАЩИХ ВОДНЫЕ ИЗВЛЕЧЕНИЯ
Водные извлечения (настои и отвары) из
roтавливают в соответствии с требованиями
Фармакопеи экстракцией лекарственноro pac
тительноro сырья водой очищенной, а также
растворением сухих или жидких экстрактов (KOH
центратов) в рассчитанном объеме воды очи
щенной.
Запрещается изrотовление в аптеках и ис
пользование водных извлечений заведомо
более высокой концентрации с целью после
дующеro разведения, так как при изroтовлении
концентрированных извлечений из сырья не дo
стиrается полнота экстракции биолоrически aK
тивных веществ.
При изroтовлении настоев и отваров запре
щается заменять лекарственное растительное
сырье настойками, эфирными маслами и экс
трактами, не предназначенными для изroтовле
ния водных извлечений.
При расчете требуемоro для экстракции
объема воды очищенной и количества сырья ис
пользуют значения коэффициентов водопоrло
щения или расходных коэффициентов в соот
ветствии с Фармакопеей.
При изroтовлении водных извлечений обе
спечивают оптимальные условия экстракции,
учитывая стандартность лекарственною расти
тельною сырья; ero измельченность и rистоло
rическую структуру; соотношение массы сырья
и объема экстраrента; физико химические свой
ства действующих и сопутствующих веществ;
материал аппаратуры и друrие факторы, вли
я ющие на качество водноro извлечения.
Изroтовленные водные извлечения после
отжатия сырья и процеживания доводят водой
очищенной до объема, указанноro в прописи pe
цепта.
Продукты из лекарственноro растительно
ro сырья (настойки, жидкие экстракты, адонизид
и друrие) следует прибавлять к изrотовленно
му водному извлечению в последовательности,
установленной в разделе «Последовательность
растворения и смешивания лекарственных Be
ществ».
Мноroкомпонентные водные извлечения
из лекарственноro растительноro сырья, Tpe
бующеro одинаковоro режима экстракции, обу
словленноro физико химическими свойствами
действующих и сопутствующих веществ, изro
тавливают в одном инфундирном стакане без
учета rистолоrической структуры сырья.
изrОТОВЛЕНИЕ ВОДНЫХ ИЗВЛЕЧЕНИЙ
ИЗ ЭКСТРАКТОВ (КОНЦЕНТРАТОВ)
Стандартизованные сухие (1:1) и жидкие
(1 :2) экстракты (концентраты) вводят в состав
жидких лекарственных средств по правилам
растворения твердых веществ и введения
продуктов из лекарственноro растительноro
сырья (настойки, жидкие экстракты, адонизид и
друrие).
При изroтовлении водных извлечений из
экстрактов (концентратов) Moryт быть использо
ваны концентрированные растворы лекарствен
ных веществ.
изrОТОВЛЕНИЕ СУСПЕНЗИЙ
И ЭМУЛЬСИЙ
Суспензии и эмульсии для внутреннеro, Ha
ружноro и парентеральноro применения изroтав
ливают в соответствии с требованиями Фарма
копеи.
Суспензии с содержанием нерастворимых
твердых веществ 3 % и более, а также эмульсии.
независимо от концентрации веществ изroтав
ливают по массе, концентрированные растворы
водорастворимых веществ, при изroтовлении cy
спензий не используют.
изrОТОВЛЕНИЕРДСТВОРОВ
для ПДРЕНТЕРдЛьноrо ПРИМЕНЕНИЯ
При изrотовлении растворов для паренте
ральноro применения (инъекции, инфузии) сле
дует соблюдать особые требования к их изroтов
лению и контролю качества.
Контроль качества растворов для паренте
ральноro применения должен охватывать все
стадии их изroтовления. Результаты постадий
ноro контроля изroтовления парентеральных
растворов реrистрируют в Журнале реrистра
ции отдельных стадий изroтовления стерильных
растворов в соответствии с требованиями HOp
мативных документов.
Бутылки и флаконы со стерильными лекар
ственными средствами после укупорки маркируют
путем надписи или штамповки на крышке с исполь
зованием металлических жетонов с указанием
наименования, концентрации и номера серии.
Стерилизация лекарственных средств
должна проводиться в соответствии с требова
ниями статей 5. 1. 1 и #6. 1.4 и не позднее 3 ч с
момента изroтовления под контролем провизора
или фармацевта. Реrистрацию пара метров CTe
рилизации производят в журнале реrистрации
отдельных стадий изrотовления стерильных pac
творов. Повторная стерилизация растворов для
парентеральноro применения не допускается.
Разrрузка стерилизатора должна про водиться
при снижении температуры раствора от 120 ос
до 70 ос и выравнивания давлений (время ox
лаждения более 60 мин).
Контроль растворов для парентеральноrо
применения на отсутствие механических вклю
чений до и после стерилизации проводят в COOT
ветствии с требованиями статьи 2.9.20.
Одновременно до стерилизации проводят
проверку объема наполнения флаконов, каче
ства укупорки флаконов (металлический кол
пачок «под обкатку» не должен прокручиваться
при проверке вручную и раствор не должен BЫ
ливаться при опрокидывании флакона).
#6.1.1. Жидкие лекарствеННblе средства
1003
Микробиолоrический контроль стериль
ных лекарственных средств на стерильность
(2.6.1) и испытания на пироrенность (2.6.8)
или бактериальные эндотоксины (2.6. 14) про
водят в соответствии с требованиями Фарма
копеи.
Стерильные растворы считаются забра
кованными при несоответствии по внешне
му виду; величине рН; подлинности и коли
чественному содержанию входящих веществ;
наличию видимых механических включений;
недопустимым отклонениям от номинальноro
объема раствора; нарушению фиксированно
сти укупорки; по оформлению лекарственных
средств, предназначенных к отпуску.
Изrотовление стерильных лекарственных
средств запрещается при отсутствии данных
о химической совместимости входящих в них
веществ; о технолоrии изrотовления и режиме
стерилизации; о проведенном анализе входя
щих инrредиентов; при отсутствии методик их
полноro химическоro контроля.
Запрещается одновременное изютовление
на одном рабочем месте нескольких растворов
для парентеральноro применения, содержащих
вещества с различными наименованиями или
одноrо наименования, но в различных KOHцeH
трациях.
Стерильные лекарственные средства
должны храниться в соответствии с физико хи
мическими свойствами входящих в них веществ
и использоваться в течение установленною
срока юдности. По истечении сроков roдности
растворы для парентеральною применения под
лежат изъятию.
изrОТОВЛЕНИЕ ЛЕКАРСТВЕННЫХ
СРЕДСТВ ДЛЯ НОВОРОЖДЕННЫХ
И ДЕТЕЙ ДО 1 rОДА
Лекарственные средства для новорожден
ных и детей до 1 roда для наружноrо и BHY
треннеro применения и друrие, не подлежащие
стерилизации, изютавливают в аптеках в асеп
тических условиях без прибавления стабилиза
торов и консервантов.
изrОТОВЛЕНИЕ rЛАЗНЫХ КАПЕЛЬ
И ПРИМОЧЕК
Изroтовление rлазных капель и примочек
проводят массо объемным методом в соответ
ствии с правилами, изложенными при изrотовле
нии растворов для парентеральноro применения.
Растворы термолабильных веществ roто
вят в асептических условиях с использованием
воды очищенной стерильной. Для сохранения
стерильности rлазных капель и при мочек в их
состав по назначению врача MOryT быть введены
консерванты (метилпараrидроксибензоат, про
пилпараrидроксибензоат, бензалкония хлорид и
друrие ).
При изrотовлении rлазных капель в He
больших количествах (10 15 мл) растворитель
делят на две части, одну из которых используют
для растворения веществ, друryю для смыва
адсорбированноro на фильтре вещества (Be
ществ).
Обеспечение изотоничности rипотоничных
растворов осуществляется путем добавления в
состав натрия хлорида, натрия нитрата, натрия
сульфата и друrих веществ с учетом их COBMe
стимости с остальными компонентами paCT
вора.
Расчет изотонических концентраций произ
водят с помощью изотонических эквивалентов
веществ по натрия хлориду в соответствии с Ta
блицей #6.1.1. 21. Изотоничной считается KOH
центрация веществ в растворе, равноценная
концентрации 0,7 1,1 % натрия хлорида.
При изroтовлении rлазных капель, офталь
молоrических растворов и примочек, в состав KO
торых входят антибиотики, прописанные в еди
ницах действия, определение массы навески
порошка антибиотика производят с учетом зави
симости между массой и активностью антибио
тика в соответствии с таблицей #6.1.1. 22.
изrОТОВЛЕНИЕ КОНЦЕНТРИРОВАННЫХ
РАСТВОРОВ
Концентрированные растворы (KOHцeHTpa
ты) заранее изroтовленные растворы Be
ществ более высокой концентрации, чем та,
в которой эти вещества выписываются в pe
цептах. К концентратам относят также KOH
центрированные экстракты из некоторых ле
карственных растений, изroтовленные на
фармацевтических предприятиях, например,
экстракты (концентраты) валерианы, roрицве
та, пустырника и друrие. Концентраты предна
значены для быстрою и качественноro изrотов
ления жидких лекарственных средств.
Рекомендуется изroтавливать концентраты
из веществ rиrроскопичных, выветривающихся
на воздухе, содержащих значительное количе
ство кристаллизационной воды. Номенклату
ра концентрированных растворов определяет
ся спецификой рецептуры и объемом работы
аптеки и утверждается в соответствии с Tpe
бованиями нормативных документов. KOHцeH
траты изroтавливают по мере необходимости с
учетом срока их roдности.
Концентраты изroтавливают массо объем
ным методом в мерной посуде в асептических
условиях, используя свежеприrотовленную
воду очищенную. Если приroтовление BeдeT
ся не в мерной посуде, то объем воды очищен
ной рассчитывают с использованием значения
плотности концентрата или КУО веществ.
ИзroТОl3ленные растворы подверrают пол
ному химическому контролю, фильтруют и
проверяют на отсутствие механических вклю
чений.
При изroтовлении концентрированных pac
творов следует избеrать концентраций, близких
к насыщенным, так как при понижении темпера
rосударственная фармакопея Республики Беларусь
Таблица #6.1.1. 21
Соответствие изотонических эквивалентов веществ по натрия хлориду
1004
N!! Наименование вещества (1 r) 3квивалентноеколичество
натрия хлорида (r)
1. Аминокапроновая кислота 0,27
2. Аскорбиновая кислота 0,18
3. Атропина сульфат 0,10
4. Борная кислота 0,53
5. rлюкоза безводная 0,18
6. Динатрия фосфат ди2идрат (натрия rидрофосфат диrидрат) 1,0
7. ДифеН2ирамина 2идрохлорид (димедрол) 0,20
8. Калия йодид 0,З5
9. Калия хлорид 0,76
10. Кальция 2люконат 0,16
11. Кальция хлорид 2екса2идрат 0,36
12. Кодеина фосфат 0,12
13. Кофеин натрия бензоат 0,23
14. Ма2ния сульфат 2епта2идрат 0,14
15. Меди сульфат пента2идрат 0,13
16. Натрия аминосалицилат ди2идрат 0,27
17. Натрия ацетат три2идрат 0,46
18. Натрия бензоат 0,40
19. Натрия бромид 0,62
20. Натрия 2идрокар60нат 0,65
21. Натрия йодид 0,38
22. Натрия метабисульфит 0,65
23. Натрия салицилат 0,35
24. Натрия сульфат дека2идрат 0,23
25. Натрия тетраборат 0,34
26. Натрия тиосульфат 0,30
27. Натрия хлорид 1,0
28. Натрия цитрат 0,30
29. Никотинамид 0,20
30. Никотиновая кислота 0,25
31. Папаверина 2идрохлорид 0,10
32. Пилокарпинаrидрохлорид 0,22
33. Платифиллина rидротартрат 0,13
34. Прозерин 0,19
35. Прокаина 2идрохлорид (новокаин) 0,18
36. Прокаинамида2идрохлорид(новокаинамид) 0,22
37. Теофиллин этилендиамин (эуфиллин) 0,17
38. Тетракаина 2идрохлорид (дикаин) 0,18
З9. Тиамина 2идрохлорид 0,21
40. Фенилэфрина 2идрохлорид (мезатон) 0,28
41. Цинка сульфат 2епта2идрат 0,12
42. Эфедрина rидрохлорид 0,28
туры возможна кристаллизация растворенноro
вещества. Отклонение в концентрации paCTBO
ров допускается в пределах, установленных в
статье #6.3.1. В случае превышения нормы дo
пустимоro отклонения производят исправление
концентрации раствора.
Емкости с концентратами оформляют эти
кетками с указанием наименования и KOHцeH
трации раствора, номера серии и анализа,
даты изroтовления, срока rодности.
Концентраты хранят в зависимости от фи
зико химических свойств веществ, входящих в
их состав, в стерилизованных плотно укупорен
ных I{онтейнерах (баллонах, штанrласах) в за
щищенном от света месте, при температуре от
2 ос до 8 ос или не выше 25 ос.
Изменение цвета, помутнение, появление
хлопьев, налетов раньше истечения YCTaHOB
ленноro срока roдности являются признаками
неприroдности растворов.
#6.1.1. Жидкие лекарственные средства
1005
Таблица #6.1.1. 22
Соответствие массы и 1 миллиона единиц для некоторых антибиотиков
N2 Наименование аН1"ибиотика rpaMMbI
1. Амоксициллин 1,0
2. Ампициллин 1,0
3. Ампициллин натрия 1,0
4. Ампициллина триrидрат 1,0
5. Амфотерицин В 1,38
6. Бензилпекициллин калия 0,625
7. Бензилпенициллин натрия 0,65
8. Бензилпенициллина новокаиновая соль 1,0
9. rентамицина сульфат 1,7
10. rрамицидин 1,1
11. Доксициклина rиклат 1,15
12. Канамицина сульфат 1,0
13. Карбенициллин динатрия 1,3
14. Клиндамицинаrидрохлорид 1,1
15. Клиндамицина фумарат 1,1
16. Леворин 0,02
17. Леворин натрия 0,02
18. Линкомицинаrидрохлорид 1,1
19. Мономицин 1,0
20. Неомицина сульфат 1,56
21. Нистатин 0,25
22. Оксациллин натрия 1,1
23. Окситетрациклина диrидрат 1,0
24. Олеандомицина фосфат 1,3
25. Олететрин 1,0
26. Полимиксина В сульфат 0,1
27. Полимиксина М сульфат 0,125
28. Ристомицина сульфат 1,25
29. Стрептомицина сульфат 1,0
30. Тетрациклин 1,0
31. Тетрациклинаrидрохлорид 1,0
32. Феноксиметилпенициллин 0,6
33. Цефазолин 1,0
34. Цефазолин натрия 1,0
35. Цефалексин 1,0
36. Цефокситин 1,0
37. Цефотаксим 1,0
3В. Цефоперазон 1,0
39. Цефтазидим 1,0
40. Цефтриаксон 1,2
41. Цефуроксим 1,0
42. Эритромицин 1,11
43. Эритромицина фосфат 1,0
128. Зак. 1712.
1006
rосударственная фармакопея Республики Беларусь
ПРИМЕРыизrОТОВЛЕНИЯ
КОНЦЕНТРИРОВАННЫХ РАСТВОРОВ
При изroтовлении 50 % (м/об) раствора
rлюкозы необходимо учитывать содержа
ние влаrи в веществе. Количество rлюкозы,
рассчитанное с учетом влаrи, помещают в
мерную колбу и растворяют в части rорячей
воды при перемешивании. После охлаждения
доводят водой очищенной до метки и филь
труют.
При изroтовлении 50 % (м/об) раствора
кальция хлорида не в мерной посуде для pac
чета количества прибавляемой воды очищен
ной необходимо учитывать пибо плотность
получаемоrо 50 % раствора (плотность
1,207 r/мл), либо КУО кальция хлорида
(0,58 мл/r).
ИСПРАВЛЕНИЕ КОНЦЕНТРАЦИИ
РАСТВОРОВ
Если концентрация раствора выше Tpe
буемой, то объем воды очищенной (мл), необ
ходимый для разведения полученноro paCT
вора, рассчитывают по формуле:
A-(C B)
В
rде:
А объем изrОТ08ленноrо раствора, мл;
С фактическая концентрация paCT
вора, %;
В требуемая концентрация раствора, %.
Общий объем полученноro раствора YBe
личится на прибавленный объем воды очи
щенной.
Если концентрация раствора ниже требу
емой, то массу вещества (r) для укрепления
полученноrо раствора рассчитывают по фор
муле:
А-(С B)
100p B
rдe:
А объем изroтовленноrо раствора, мл;
в требуемая концентрация раствора, %;
С фактическая концентрация paCT
вора, %;
р плотность раствора при 20 ОС, r/мл.
Общий объем полученноro раствора увели
чится с учетом КУО вещества, взятоro для YKpe
пления.
В случае укрепления растворов rлюкозы
расчеты про водят с учетом про цента влажно
сти.
Концентрированные растворы после их раз
ведения или укрепления следует анализировать
повторно.
01/2013:РБ60102
#6.1.2. ТВЕРДЫЕ ЛЕКАРСТВЕННЫЕ
СРЕДСТВА
ПОРОШКИ
Приroтовление простых и сложных порош
ков из веществ, прописанных в равных и резко
отличающихся количествах, с трудноизмельча
емыми и леrковесными веществами.
Для расчета количества инrредиентов при
распределительном способе прописывания по
рошков необходимо однократные дозы, указанные
в рецепте (требовании), умножить на число доз.
При разделительном способе прописывания
порошков следует взять количества инrредиен
тов, указанные в рецепте.
Приroтовление порошков начинают с выбора
ступки. Общая масса должна быть близка к оп
тимальной заrрузке и не превышать максималь
ную заrрузку. Номер ступки находят по диаметру
ступки. Характеристики ступок приведены в Ta
блице #6.1.2. 1.
Для порошков, в состав которых входят леr
ковесные вещества, при выборе номера ступки
масса леrковесноro компонента теоретически
удваивается. Леrковесными веществами яв
Таблица #6.1.2. 1
Рабочая по-
N!! Диаметр, верхность Рабочий Время Максимальная Оптимальная
мм объем, см 3 измельчения, с заrрузка, r заrрузка, r
см 2 К
1. 50 45 1 20 60 1,0 0,5
2. 75 90 2 80 90 4,0 1,5
3. 86 90 2 80 90 4,0 1,5
4. 110 135 3 160 120 8,0 3,0
5. 140 225 5 320 150 16,0 6,0
6. 184 450 10 960 210 48,0 18,0
7. 243 765 17 2240 300 112,0 42,0
1006
rосударственная фармакопея Республики Беларусь
ПРИМЕРыизrОТОВЛЕНИЯ
КОНЦЕНТРИРОВАННЫХ РАСТВОРОВ
При изroтовлении 50 % (м/об) раствора
rлюкозы необходимо учитывать содержа
ние влаrи в веществе. Количество rлюкозы,
рассчитанное с учетом влаrи, помещают в
мерную колбу и растворяют в части rорячей
воды при перемешивании. После охлаждения
доводят водой очищенной до метки и филь
труют.
При изrотовлении 50 % (м/об) раствора
кальция хлорида не в мерной посуде для pac
чета количества прибавляемой воды очищен
ной необходимо учитывать либо плотность
получаемоro 50 % раствора (плотность
1,207 r/мл), либо КУО кальция хлорида
(0,58 мл/r).
ИСПРАВЛЕНИЕ КОНЦЕНТРАЦИИ
РАСТВОРОВ
Если концентрация раствора выше Tpe
буемой, то объем воды очищенной (мл), необ
ходимый для разведения полученноro paCT
вора, рассчитывают по формуле:
A.(C B)
В
rдe:
А объем изrотовленноrо раствора, мл;
С фактическая концентрация paCT
вора, %;
В требуемая концентрация раствора, %.
Общий объем полученноrо раствора YBe
личится на прибавленный объем воды очи
щенной.
Если концентрация раствора ниже требу
емой, то массу вещества (r) для укрепления
полученноro раствора рассчитывают по фор
муле:
A-(C B) ,
100p B
rдe:
А объем изrотовленноrо раствора, мл;
в требуемая концентрация раствора, %;
С фактическая концентрация paCT
вора, %;
р плотность раствора при 20 ОС, r/мл.
Общий объем полученноro раствора увели
чится с учетом КУО вещества, взятоro для YKpe
пления.
В случае укрепления растворов rлюкозы
расчеты про водят с учетом про цента влажно
сти.
Концентрированные растворы после их раз
ведения или укрепления следует анализировать
повторно.
01/201З:РБ60102
#6.1.2. ТВЕРДЫЕ ЛЕКАРСТВЕННЫЕ
СРЕДСТВА
ПОРОШКИ
Приroтовление простых и сложных порош
ков из веществ, прописанных в равных и резко
отличающихся количествах, с трудноизмельча
емыми и леrковесными веществами.
Для расчета количества инrредиентов при
распределительном способе прописывания по
рошков необходимо однократные дозы, указанные
в рецепте (требовании), умножить на число доз.
При разделительном способе прописывания
порошков следует взять количества инrредиен
тов, указанные в рецепте.
Приroтовление порошков начинают с выбора
ступки. Общая масса должна быть близка к оп
тимальной заrрузке и не превышать максималь
ную заrрузку. Номер ступки находят по диаметру
ступки. Характеристики ступок приведены в Ta
блице #6.1.2. 1.
Для порошков, В состав которых входят леr
ковесные вещества, при выборе номера ступки
масса леrковесноro компонента теоретически
удваивается. Леrковесными веществами яв
Таблица #6.1.2. 1
Рабочая по-
N2 Диаметр, верхность Рабочий Время Максимальная Оптимальная
мм объем, см з измельчения, с заrрузка, r заrрузка, r
см 2 К
1. 50 45 1 20 60 1,0 0,5
2. 75 90 2 80 90 4,0 1,5
3. 86 90 2 80 90 4,0 1,5
4. 110 135 3 I 160 120 8,0 3,0
5. 140 225 5 320 150 16,0 6,0
6. 184 450 10 960 210 48,0 18,0
7. 243 765 17 2240 300 112,0 42,0
#6.1.2. Твердые лекарственные средства
1007
ляются ликоподий, маrния карбонат основной
леrкий, кремния диоксид коллоидный, тальк и
др. В зависимости от размера ступки подбира
ют пестик.
Взвешивание рассчитанноro количества ин
rредиентов производят в зависимости от массы.
В соответствии с массой инrредиента следует
выбирать весы, у которых минимальная и MaK
симальная наrрузка соответственно не больше
и не меньше массы взвешиваемоro компонента
порошка.
Приroтовление однокомпонентных порош
ков сводится к измельчению вещества, еro дози
рованию и упаковке.
Вещество, измельчаемое в ступке первым,
теряется в ее порах и порах пестика. Количе
ство теряемоro вещества зависит от структуры
самоro вещества.
Учитывая потери при затирании пор, в
ступку первыми вносят:
вспомоrательное вещество или вещество
общеro списка;
при отсутствии вспомоrательноro веще
ства и вещества общеro списка то веще
ство, котороro прописано HaMHoro больше по
сравнению с друrими инrредиентами;
если в рецепте прописаны два и более
веществ в равных количествах, поры ступки
затирают веществом, имеющим наименьшие
абсолютные потери в ступке NQ 1 (таблица
#6.1.2. 2);
если вещества в данном варианте от
личаются по размеру кристаллов, измельче
ние начинают с крупнокристаллическоrо Be
щества.
В остальных случаях учитывают относи
тельную потерю вещества.
Относительная потеря вещества это
абсолютная потеря вещества в рабочей
ступке, выраженная в про центах. Рассчиты
вают по формуле:
а . к .100
т
Потери веществ при растирании в ступке NQ 1
Таблица #6.1.2. 2
Вещество Потери, Mr Вещество Потери, Mr
Аскорбиновая кислота 12 Метилурацил 10
Ацетилсалициловая кислота 33 Натрия бензоат 20
Бензойная кислота 34 Натрия 2идрокарбонат 11
Бензокаин(анестезин) 24 Натрия салицилат 23
Бромкамфора 15 Никотиновая кислота 15
Висмута нитрат основной, тяже 42 Папаверина 2идрохлорид 10
лый
/ексаметилентетрамин 26 Резорцин 27
/люкоза моноеидрат 7 Салициловая кислота 55 i
!
Дибазол 18 Сахароза 21 I
Калия бромид 15 Сера для наРУЖН020 применения 24 !
Калия йодид 21 Синтомицин 30
Кальция 2лицерофосфат 25 Сульфадимидин (сульфадимезин) 18
Кальция карбонат 14 Сульфаниламид (стрептоцид) 23
Кальция лактат пента2идрат 12 Сульфатиазол (норсульфазол) i 22
Камфора 24 Танин 11
Каолин тяжелый (rлина белая) 14 Теобромин 18
Кодеин, кодеина фосфат ceCKви2и 7 Т еофиллин 16
драт
Кофеин 15 Терпинrидрат 15
Кофеин натрия бензоат 16 Фенилсалицилат 24
Ксероформ 57 Фенобарбитал 18
Ма2ния оксид, леzкий 16 Фталилсульфатиазол (фталазол) 19
Ма2ния сульфат zепта2идрат 17 ! Хинидина сульфат 21
I
Ментол 17 1 Хлорамфеникол (левомицетин) 29
I
Метамизол натрия (анальrин) I 22 I Цинка оксид 36
1008
rосударственная фармакопея Республики Беларусь
rдe:
а абсолютная потеря вещества в ступке
NQ 1 (таблица #6.1.2. 2), r;
К коэффициент рабочей поверхности
ступки (показывает, во сколько раз рабочая поверх
ность данной ступки больше, чем рабочая поверх
ность ступки NQ 1). Находят по таблице #6.1.2. 1 ;
т масса вещества по прописи, r.
Порядок смешивания веществ зависит не
только от их свойств, но и от количества. Пер
выми после затирания пор в ступку помещают
труднопорошкуемые вещества: ментол, камфо
ра, тимол и др. Их измельчают отдельно со вспо
моrательной жидкостью (этиловый спирт, эфир),
количество которой зависит от формы кристал
лов инrpедиентов. Обычно на 1,0 r камфоры,
ментола, тимола прибавляют 10 капель этило
воro спирта 9б % или 15 капель эфира. Для из
мельчения 1,0 r борной кислоты, натрия TeTpa
бората прибавляют 5 капель этиловоro спирта
96 % или 8 капель эфира. С помощью этих жид
костей измельчают также йод, салициловую ки{;
лоту (ее частицы раздражают слизистую обо
лочку носоrлотки).
Леrковесные вещества имеют высокую CTe
пень дисперсности. и поэтому их можно прибав
лять к измельченной смеси кристаллических Be
ществ без дополнительноrо измельчения.
Остальные порошки в этом случае вводят
ся до испарения спирта этиловоro или эфира,
чтобы избежать укрупнения частиц. Инrреди
енты полностью помещают в ступку и смеши
вают друr с друюм, если их соотношение не
превышает 1 :5. Если же соотношение больше,
то из ступки необходимо отсыпать часть по
рошка, внести входящие инrредиенты, соблю
дая правило «от меньшею количества к боль
шему» .
Порошки должны быть измельчены до мел
коro порошка (2.9.12) и быть однородными при
рассмотрении невооруженным rлазом, если нет
друrих указаний в частных статьях.
Вещества для присыпок растирают до очень
мелкою порошка (2.9.12).
Разделение на дозы однокомпонентных и
мноroкомпонентных порошков производится по
массе с помощью весов и по объему с помощью
дозаторов.
Для упаковки отдельных доз порошков ис
пользуют бумажные капсулы. В зависимости
от свойств входящих инrредиентов пользуются
простыми, парафинированными, перrаментны
ми капсулами.
Изrотовление порошков с Rеществами
списка А и списка Б
При изrотовлении порошков С вещества
ми списка А или списка Б в количестве меньше
0,05 r на всю массу порошка используют триту
рации. Тритурация смесь вещества списка А
или списка Б со вспомоrательным наполнителем
(как правl.1ЛО с лактозы моноrидратом (молоч
ный сахар» в соотношении 1:10 или 1:100. Три
турация 1:10 содержит 1 часть вещества списка
А или списка Б и 9 частей лактозы моноrидрата.
Она используется, как правило, коrда в рецепте
(требовании) общее количество вещества списка
А илl.1 списка Б достиrает десятых долей rpaMMa.
Тритурация 1 :1'00 содержит 1 часть вещества
списка А или списка Б и 99 частей лактозы и ис
пользуется, как правило, тorдa, Korдa общее KO
личество вещества списка А или списка Б в pe
цепте не превышает тысячных долей rpaMMa.
Тритурации roтовят в аптеке в количестве, дo
статочном для обеспечения примерно месячной
потребности. Каждые 15 дней тритурации вновь
перемешивают в отдельной ступке для YMeHb
шения расслаивания.
Пример приroтовления тритурации атропи
на сульфата 1 :100. 4,95 r лактозы моноrидрата
помещают в отдельную ступку, тщательно из
мельчают, отсыпают часть еro на капсулу, OCTa
вив в ступке около 0,05 r. На специальных весах
из шкафа «А» отвешивают 0,05 r атропина суль
фата, помещают в ступку, тщательно растирают
до получения однородной смеси, затем в 7 9
приемов при тщательном перемешивании при
бавляют остальное количество лактозы MOHO
rидрата, тритурацию помещают в небольшой
штанrлас с этикеткой:
« Trituratio Atropihi sulfatis 1: 1 00 сит Saccharo
lactis (0,001 Аtroрiпi sulfatis = 0,1 tritиrаtiопis)
Дата. Подпись лица, изroтовившеrо тритура
цию».
Ценность лактозы как разбавителя заклю
чается в том, что она не rиrроскопична и имеет
плотность (1,52 r/CM 3 ), близкую к плотности
солей алкалоидов и друrих веществ списка А
или списка Б.
Если в состав рецепта кроме вещества
списка А или списка Б, выписанных в дозе
меньше 0,05 r (т. е. в случае использования
тритурации), входит сахароза, то, чтобы не
увеличивать массу одноro порошка, peKOMeH
дуется уменьшить количество сахара на массу
тритурации. Если в рецепте отсутствует caxa
роза, то развеска порошка увеличится за счет
тритурации.
Приrотовление порошков с пахучими,
красящими веществами и экстрактами
В технолоrии порошков в основном исполь
зуют экстракт белладонны (красавки). Исполь
зуют два экстракта белладонны: rустой, coдep
жащий 1,4--------1,6 % алкалоидов в пересчете на
rиосциамин, и сухой, содержащий 0,? 0,8 % ал
калоидов в пересчете на rиосциамин.
Для удобства работы в аптеках из экс
тракта rустою roтовят еro раствор 1:2 по сле
дующей прописи: 100 частей экстракта ry
стоro растворяют в смеси из 60 частей воды,
1 О частей этиловоro спирта 90 % и 30 частей
rлицерина 85 %. Такие растворы rycTbIx экс
#6.1.2. Твердые лекарственные средства
1009
трактов хранят не более 16 дней. На этикетках
штанrласов обозначают название раствора и
число капель, которое соответствует 0,1 r ис
ходноro rycToro экстракта.
Если в рецепте нет точноro указания о
форме экстракта, следует использовать ryстой.
Сухой экстракт белладонны и раствор ryCTOro
экстракта белладонны применяются в двойном
количестве. Способ приroтовления лекарствен
ных средств с экстрактами зависи.т от конси
стентных свойств экстракта, входящеro в их
состав. Если в состав сложных порошков входит
сухой экстракт 1 :2, то roТ08ЯТ по общим прави
лам приrотовления сложных порошков. Перед
началом работы следует проверить разовую и
суточную дозы экстракта белладонны как лекар
ственноro средства списка Б.
rycTbIe экстракты, обладая вязкой конси
стенцией, плохо распределяются в общей массе
порошка и требуют специальных приемов при
взвешивании: экстракт взвешивают на филь
тровальной бумаrе. Для последующеro отделе
ния бумаrи наружную поверхность ее смачивают
растворителем, который применяется для экс
тракции, несколько капель 20 % (06/06) эти
ловоro спирта.
rустой экстракт вносят в порошковую массу
путем растирания. rотовят порошковую смесь
по правилам сложных порошков, в ступке OCTaB
ляют небольшое количество смеси (двойное по
отношению к навеске rycToro экстракта), ryстой
экстракт с roловки пестика переносят на часть
порошка, находящеrося в ступке, путем OCTO
рожноro, без сильноro надавливания на пестик,
вращения и растирания. Порошковую массу
растирают до равномерноro окрашивания, а
затем смешивают с остальной массой порошка.
Раствор rycтoro экстракта (1 :2) отмеривают
каплями равномерно в порошковую смесь в co
ответствии с указаниями на этикетке флакона
капельницы.
При использовании сухоro или раствора ry
стоro экстракта белладонны масса одноro по
pOUJKa Bcerдa будет больше, чем при использо
вании rycToro экстракта. Порошки с экстрактами
отпускают в парафинированной или вощеной
бумаrе.
Сложные порошки, в состав которых входят
красящие вещества (бриллиантовый зеленый,
метиленовый синий, калия перманrанат, фура
ципин, этакридина лактат, рибофлавин, акрихин
и др.) или вещества с резким стойким запахом
(тимол. камфора. ментол, ксероформ и др.), ro
товят на отдельном рабочем месте, при меняя
весы и ступку.
Красящие вещества леrко втираются в поры
ступки, и поэтому при приrотовлении порошков
соблюдается особый прием: красящее вещество
вносят в лунку roтовой порошковой смеси и тща
тельно с ней смешивают
Аналоrично roтовят порошки с пахучими Be
ществами. Некоторые пахучие и летучие лекар
ственные средства одновременно представляют
собой трудноизмельчаемые инrредиенты; их цe
лесообразно измельчать в присутствии вспомо
rательных жидкостей.
СУППОЗИТОРИИ
Различают ректальные (Suppositoria recta
/ia), ваrинальные (Supposilo,ia vagiпa/ia) суппо
зитории и палочки (Bacifll).
Ректальные суппозитории MOryT иметь
форму конуса, цилиндра с заостренным концом
или иную форму с максимальным диаметром
1,5 см. Масса одноro ректальноro суппозито
рия должна находиться в пределах от 1 r до
4 r. Масса одноro ректальноro суппозитория для
детей должна быть от 0,5 r до 1,5 r.
8аrинальные суппозитории MorYT быть сфе
рическими. яйцевидными, в виде плоскоro тела
с закруrленным концом. Масса их должна Haxo
диться в пределах от 1,5 r до 6 r.
Палочки имеют форму цилиндра с заострен
ным концом и диаметром не более 1 см. Масса
палочки должна быть от 0,5 r до 1 r.
Однородность суппозиториев определяют
визуально на продольном срезе по отсутствию
вкраплений, на срезе допускается наличие воз
душноro стержня или воронкообразноro уrлу
бления.
Суппозитории MOryT быть рассчитаны на
местное и резорбтивное действие.
Скорость всасывания при ректальном BBe
ден-ии сравнима с подкожным и внутримышеч
ным введением. поэтому необходима проверка
доз фармацевтических субстанций списка А и
списка Б. Дозы в этом случае сравнивают с BЫC
шими разовыми и суточными дозами для BHY
TpeHHero применения.
Для приroтовления суппозиториев использу
ют основы липофильные масло какао, сплавы
масла какао с парафином твердым и rидроrени
зированными жирами, сплавы rидроrенизиро
ванных жиров с воском, парафином твердым;
rидрофильные желатиноrлицериновый rель,
сплавы полиэтиленоксидов разной молекуляр
ной массы и друrие.
Суппозитории roтовят по массе.
Если в рецепте не указано иное. то ректаль
ные суппозитории roтовят массой 3 r, ваrиналь
ные суппозитории не менее 4 r.
Если в состав суппозиториев входят
фармацевтические субстанции списка А и
списка Б, то необходимо проверять дозы.
Дозы в этом случае сравнивают с высшими
разовыми и суточными дозами для BHYTpeH
Hero применения.
Суппозитории изrотавливают выливани
ем расплавленной массы в формы и BЫKaTЫ
ванием.
При использовании метода выливания
проводят расчет количества основы, учитывая
прямой и обратный заместительные коэффи
циенты.
1010
rосударственная фармакопея Республики Беларусь
Введение действующих веществ в суппо
зитории
Вещества вводят в суппозитории, учитывая
их растворимость в основе и воде, а также выпи
санные количества.
1. Вещества, растворимые в жирах (фенол,
хлоралrидрат, камфора, тимол и др.), вводят в
суппозиторные основы, растворяя в расплав
ленной основе. При необходимости для повы
шения температуры плавления массы вводят
парафин твердый, воск в количестве до 5 % от
массы основы.
2. Вещества, растворимые в воде (соли ал
калоидов, этакридина лактат, колларroл, про
тарroл, новокаин, rексаметилентетрамин, калия
йодид и др.), предварительно растворяют в ми
нимальном количестве воды. Если BoдopaCTBO
римоro вещества выписано мною и требуется
большое количество воды, вещество вводят cy
спензионно.
Колларroл, протарroл и танин вводят в суп
позиторную основу только В виде ВОДНЫХ или во--
дно rлицериновых растворов.
3. Вещества, нерастворимые ни в воде. ни
в основе (дерматол, ксероформ, висмута нитрат
основной, стрептоцид и дp ), вводят в основы cy
спензионно.
Если указанные вещества прописаны в He
больших количествах (до 5 %), их растирают с
несколькими каплями жирноro масла, а затем
смешивают с измельченной основой.
Если вещества прописаны в значительных
количествах, их измельчают в ступке, а затем
смешивают с измельченной основой.
01/2013: РБ601 03
#6.1.3. мяrКИЕ ЛЕКАРСТВЕННЫЕ
СРЕДСТВА
ЛИНИМЕНТЫ
Различают roMoreHHbIe, суспензионные,
эмульсионные и комбинированные линименты.
rOMoreHHbIe линименты, или линименты
растворы, представляют собой жидкие про
зрачные смеси жирных масел с эфирными Mac
лами, хлороформом, скипидаром, метилсали
цилатом. rOMoreHHbIe линименты rотовят по
общим правилам растворения и смешивания
жидкостей: твердые растворимые вещества
предварительно растворяют в той жидкости, в
которой они лучше растворимы, приroтовление
ведут в контейнере для отпуска, пахучие и ле
тучие вещества вводят в линименты в послед
нюю очередь.
Суспензионные линименты представляют
собой взвеси нерастворимых в воде, rлицери
не, маслах и друrих жидкостях веществ (суль
фаниламидные производные, ксероформ, цинка
оксид, крахмал, rлина белая). При изroтовлении
суспензионных линиментов вещества растира
ются в мельчайший порошок, а затем смешива
ются с одним из имеющихся в прописи наименее
вязким и нелетучим растворителем. Суспензи
онные линименты характеризуются невысокой
седиментационной устойчивостью, для повы
шения которой можно использовать заryстители
(аэросил в количестве З 5 % от общей массы).
Эмульсионные линименты представля
ют собой эмульсии первоro или второю рода.
Эмульсионные линименты нуждаются в исполь
зовании эмульrаторов, которые или вводятся дo
полнительно к двум несмешивающимся фазам,
или образуются в результате взаимодействия
инrредиентов. Например, линимент бензилбен
зоата и линимент полисульфидный.
Rp.: Linimenti Вепzуlii Ьепzоаtis 100,0
Состав:
для взрослых:
Бензилбензоат 20,0 r
Мыло зеленое 2,0 r
Вода очищенная 78,0 r
ДЛЯ детей до 3 лет:
Бензилбензоат 10,0 r
Мыло зеленое 2,0 r
Вода очищенная 88,0 r
При изroтовлении линимента бензилбен
зоата в качестве эмульrатора целесообразно
использовать мыло зеленое, так как оно раз
рыхляет кожу и усиливает фармаколоrический
эффект бензилбензоата. В контейнер дЛЯ OT
пуска помещают 78 мл (88 мл) воды очищен
ной, растворяют 2,0 r мыла зеленоro, прибавля
ют 20,0 r (10,0 r) бензилбензоата и тщательно
встряхивают.
Для повышения стабильности допуска
ется заменять 1,0 r мыла зеленоro на 1,0 r
эмульrатора T 2. В контейнер для приroтов
ления помещают 76 мл воды очищенной и
растворяют 1,0 r мыла зеленою. В подоrре
той ступке расплавляют 1,0 r эмульrатора
T 2, прибавляют 2 мл rорячей воды очищен
ной, эмульrируют до потрескивания и разбав
ляют при энерrичном перемешивании водным
раствором мыла зеленою, прибавляя ero по
частям. К полученной смеси эмульrаторов по
частям прибавляют 20,0 r бензилбензоата и
перемешивают.
Комбинированные линименты представля
ют собой сочетание двух и более систем раст
вор, суспензия, эмульсия.
При оценке качества приroтовленно
ro линимента проверяют соответствие цвета
и запаха инrредиентам. Отклонения общей
массы и массы отдельных инrредиентов
должны укладываться в нормы допустимых OT
клонений, как указано в статье #6.3.1. Нормы
отклонений, допустимые при иЗ20товлении
лекарственных средств (в том числе 20Meo
патических) в аптеках.
#6.1. З. МЯ2кие лекарственные средства
МАЗИ
По типу дисперсных систем различают ro
MoreHHbIe (сплавы, растворы, экстракционные
мази) и reTeporeHHbIe (суспензионные, эмульси
онные и комбинированные) мази.
rOMoreHHbIe мази мази, в которых дей
ствующие вещества распределены в основе по
типу раствора, т. е. доведены до молекулярной
дисперсности.
reTeporeHHbIe мази характеризуются нали
чием межфазной поверхности между действу
ющими веществами и основой.
Для приrотовления мазей иcnольэуют
основы rидрофильные rели метилцеллюлозы
и натрия карбоксиметилцеллюлозы, комбинации
полиэтиленоксидов и друrие полимеры; rидро
фобные природные жиры (свиной, roвяжий),
rидроrенизаты растительных масел (соевоro,
подсолнечноro, касторовоro и друrих), сплавы
rидроrенизированных жиров с растительными
маслами и жироподобными веществами; yre
ловодородные вазелин, силиконовые (эси
лон аэросильная) основы; абсорбционные
сплавы вазелина с ланолином безводным в раз
личных соотношениях, ланолин безводный и
друrие; водосмывные эмульсионные основы
типа «масло вода», приroтовленные С использо
ванием поверхностно активных веществ, BЫCO
коrидрофильных неорrанических (бентониты),
орrанических (водорастворимые эфиры целлю
лозы) веществ и их смесей.
Мази roтовят по массе. Масса мази опреде
ляется как сумма количеств инrредиентов, BXO
дящих в пропись.
Если нет друrих указаний, то в качестве
основы используют:
для rлазных мазей стерильный сплав вa
зелина, не содержащею восстанавливающих вe
ществ, с ланолином безводным в соотношении 9:1;
для мазей с антибиотиками стерильный
сплав вазелина с ланолином безводным 6:4;
для друrих мазей основу подбирают с
учетом физико химической совместимости KOM
понентов мази.
Если масса мази меньше либо равна 30,0 r.
компоненты основы можно расплавлять в подо
rретой ступке. Если масса мази более 30,0 r, то
компоненты основы расплавляют в выпаритель
ной чашке на водяной бане. Сплавление начина
ют с наиболее туroплавких компонентов.
При отсутствии указаний концентрации
действующеro вещества следует roтовить мазь
1 О %, кроме мазей, содержащих фармацевтиче
ские субстанции списка А и списка Б.
Введение действующих веществ в мази
Вещества вводят в мази, учитывая их paCTBO
римость в основе и воде, а также выписанные KO
личества. Различают следующие rруппы веществ.
1. Вещества, растворимые в основе.
2. Вещества, нерастворимые в основе, но
растворимые в воде:
1011
протарroл, колларrол, танин растворяют в
равном количестве воды (к протарroлу для облеr
чения растворения можно прибавить 1 2 капли
rлицерина 85 % и затем растереть с водой);
резорцин, цинка сульфат вводят в мази
(кроме rлазных) по типу суспензий;
антибиотики вводят в мази по типу суспен
зий;
rycTbIe и сухие экстракты растворяют в
равном количестве либо смеси этиловый спирт
90 % вода очищенная rлицерин 85 %
(1:6:3, об/об/об).
При изroтовлении эмульсионных мазей ис
пользуют воду. Вода может быть:
в прописи рецепта, либо прописан водный
раствор друrих веществ; в этом случае действу
ющие вещества растворяют в прописанном pac
творе;
в составе ланолина водноro (если в pe
цепте не указан тип ланолина, то используют
водный); в таком случае водный ланолин заме
няют на безводный, а в воде растворяют веще
ства;
если в прописи рецепта не содержится
вода, ее берут минимальное количество, требу
емое для растворения действующеro вещества.
При приroтовлении эмульсионных мазей
на вазелине, не содержащих в своем составе
эмульrатора, используют до 5 % воды (вазелин
инкорпорирует до 5 % воды).
3. Вещества, нерастворимые ни в основе, ни
в воде.
Способ приrотовления суспензионных
мазей зависит от процентноro содержания дей
ствующих веществ:
до 5 % вещества растирают с половин
ным количеством жидкости, родственной основе
(для уrлеводородных основ это вазелиновое
масло, для жировых жирные масла, для rи
дрофильных вода, этиловый спирт 90 %, rли
церин 85 %);
от 5 % до 25 % вещества растирают с
половинным количеством (от массы веществ)
расплавленной основы;
25 % и более (пасты) вещества растира
ют вначале с половинным количеством расплав
ленной основы, а затем по частям вводят OCTaB
шуюся расплавленную основу.
Летучие вещества вводят в состав мазей в
последнюю очередь при температуре не выше
40 ос.
Комбинированные мази это мноroфазные
системы, представляющие собой сочетание раз
личных типов дисперсных систем (растворов,
эмульсий, суспензий). В такие мази OДHOBpeMeH
но прописываются вещества с различными фи
зико химическими свойствами.
Для приroтовления комбинированных
мазей MOryT использоваться мазевые основы,
относящиеся к различным rруппам (rидрофоб
ные, водорастворимые. абсорбционные, BOДO
смывные ).
1012
rосударственная фармакопея Республики Беларусь
При приroтовлении комбинированных мазей
придерживаются следующеro порядка: внача
ле сплавляют компоненты основы (мазь сплав),
затем в основе растворяют вещества (мазь
раствор), следующей roтовят мазь суспензию и
в последнюю очередь мазь эмульсию. Если
вещество, образующее мазь раствор, является
пахучим и летучим, ею вводят в последнюю оче
редь.
rлазные мази
rлазные мази предназначены для HaHece
ния на конъюнктиву rлаза путем закладывания
за веко при помощи специальных шпателей. Они
выделяются в отдельную rруппу и к ним предъяв
ляются следующие требования:
не должны содержать твердых частиц с
острыми rранями, способными травмировать
конъюнктиву;
должны леrко, а лучше самопроизволь
но распределяться по влажной слизистой обо
лочке;
rлазные мази roтовят в асептических yc
ловиях.
rлазные мази должны изroтавливаться на
основах высокоro качества и содержать TBejr
дую фазу в состоянии тончайшей дисперсности.
Основа должна быть нейтральной, стерильной,
равномерно распределяющейся по слизистой
оболочке rлаза и не содержащей посторонних
примесей.
В качестве компонента основы для rлаз
ных мазей применяют вазелин, не содержащий
восстанавливающих веществ. Он может быть
получен в аптечных условиях путем обработ
ки вазелина по следующей методике. Вазелин
расплавляют, прибавляют 2 % активированно
ro уrля, смесь наrревают до 150 ос. Наrревание
при этой температуре продолжают при переме
шивании в течение 1 ч. При этом происходит
удаление летучих примесей и адсорбция крася
щих и посторонних орrанических веществ. Затем
вазелин фильтруют через складчатый фильтр,
используя воронки для rорячею фильтрования,
или в сушильном шкафу при температуре от
90 ОС до 100 ОС в стерильные воздухонепрони
цаемые контейнерах. Определение BOCCTaHaB
ливающих веществ проводят по методике, опи
санной в статье Вазелин.
Основу для rлазных мазей получают путем
сплавления ланолина безводноro и вазелина,
не содержащеro восстанавливающих веществ,
в фарфоровой чашке при наrреван.ии на водя
ной бане. Расплавленную основу процеживают
через несколько слоев марли, фасуют в сухие
стерильные стеклянные контеЙl-fеры, обвязыва
ют перrаментной бумаroй и стерилизуют в воз
душном стерилизаторе при температуре 180 ос в
течение 30-------40 мин или при температуре 200 ос
в течение 15 20 мин в зависимости от объема.
В ряде случаев для приroтовления rлазных
мазей используют rидрофильные основы.
Все водорастворимые вещества (соли алка
лоидов, азотистых оснований, протарroл, цинка
сульфат, резорцин и др.) предварительно paCT
воряют в стерильной воде очищенной.
При изroтовлении rлазных суспензион
ных мазей особое внимание обращают на CTe
пень дисперсности веществ. Нерастворимые
или труднорастворимые вещества (ртути оксид
желтый, ртути амидохлорид, ксероформ и др.)
вводят в виде мельчайших порошков после тща
тельною растирания их со вспомоrательной
жидкостью (родственной основе и веществу),
взятой в половинном количестве от массы TBep
доro вещества.
Мази с антибиотиками
Мази с антибиотиками ютовят в аселтиче
ских условиях по общим правилам изroтовления
мазей. rлазные мази с антибиотиками roтовят на
основе для rлазных мазей.
Мази с бензилпенициллином применяют в
rлазной практике и дерматолоrии. Мази roтовят
из расчета 1 О 000 ЕД в 1 r. В аптечных УCilOвиях
следует roтовить мази с солями бензилпеницил
лина только тритурационноro типа, т. е. вводить
бензилпенициллин в основу внерастворенном
виде, так как в водном растворе пенициллин
быстро инактивируется.
Отпускают rлазные мази и мази с антибио
тиками в стерильных воздухонепроницаемых
контейнерах.
01/2013:РБ60104
#6.1.4. РЕЖИМЫ СТЕРИЛИЗАЦИИ
ЭКСТЕМПОРАЛЬНЫХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
Общие требования к стерилизации указа
ны в статье 5. 1. 1. Методы при20товления cтe
рильных продуктов.
В условиях аптеки используются паровая
стерилизация (автоклавирование) и сухожаровая
стерилизация (стерилизация юрячим воздухом).
Паровая стерилизация проводится при
температуре (121:t2) ос (давление 0,11 МПа
(1,1 кrс/см 2 ),если нет друrих указаний, позво
ляющих использовать друrие температуры CTe
рилизации, например, (132:t2) ос при давлении
0,20 МПа (2 Krc/cM 2 ), а также стерилизацию «Te
кучим паром» (при температуре 100 ОС).
Паровая стерилизация при температуре
(121:t2) ос рекомендуется для жидких лекар
ственных средств (таблица #6.1.4. 1). Время
стерилизационной выдержки зависит от физико
химических свойств лекарственноro средства,
объема раствора и используемоrо оборудова
ния.
Таблица #6.1.4. 1
#6.2. Экспресс анализ экстемпоральных лекарственных средств
1013
Минимальное время
Объем образца, мл стерилизационной
выдержки, мин
До 100 8
От 100 до 500 12
От 500 до 1000 15
Стерилизацию лекарственных средств в
виде водных растворов проводят в rерметично
укупореНttых, предварительно стерилизованных
флаконах или ампулах.
Жиры и масла в rерметично укупорен
ных сосудах рекомендуется стерилизовать при
(121:t2) ос в течение 2 ч.
Этим методом стерилизуются также изде
лия из стекла, фарфора, металла, перевязоч
ные и вспомоrательные материалы (вата, марля,
бинты, фильтровальная бумаrа, перrамент,
спецодежда и др.). Время стерилизационной BЫ
держки при температуре (121:t2) ос 45 мин,
при температуре (132:t2) ос 20 мин. Для CTe
рилизации изделий из резины следует использо
вать первый из указанных режимов.
Стерилизацию перечисленных объектов
проводят в стерильных коробах (биксах) или в
двухслойной упаковке из бязи, перrамента и др.
В исключительных случаях допускается стери
лизация при температуре ниже 121 ОС. KOHKpe
тизация отдельных режимов применительно к
определенному объекту стерилизации должна
быть обоснована и указана в нормативной дo
кументации.
Сухожаровая стерилизация проводится при
температуре tte менее 160 ОС. Эффективность
сухожаровой стерилизации зависит от темпера
туры, времени, степени теплопроводности CTe
рилизуемых объектов и правильности располо
жения их внутри стерилизационной камеры для
обеспечения свободной циркуляции roрячеro
воздуха.
Данный метод используют для стерилиза
ции термостойких порошкообразных веществ
(натрия хлорида, цинка оксида, талька, rлины
белой и друrих (таблица #6.1.4. 2.)) или мине
ральных и растительных масел, жиров, ланоли
на, вазелина, воска и друrих (таблица #6.1.4. 3.).
Таблица #6.1.4. 2
РежиМbI стерилизации термостойких
порошкообраЗНblХ веществ
Минимальное
Масса Температура, время стерили-
образца,r ос зационной вы-
держки,МИН
До 25 180:t2 30
200:t2 10
От 25 до 100 180:t2 40
200:t2 20
От 100 до 180:t2 60
200 200:t2 30
Таблица #6.1.4. 3
РежиМbI стерилизации минералЬНblХ
и растительных масел, жиров, ланолина,
вазелина, воска и дРУ2их
Минимальное
Масса Температура, время стерили-
образца,r ос зационной вы-
держки, МИН
До 100 180:t2 30
200:t2 15
От 100 до 180:t2 40
500 200:t2 20
Изделия из стекла, металла, силиконовой
резины, фарфора, установки для стерилизу
ющеro фильтрования с фильтрами и приемни
I<И фильтрата стерилизуются при температуре
(180:t2) ос в течение 60 мин или при температу
ре 160 ос в течение 2,5 ч. Допускается исполь
зование более высоких температур HarpeBa при
соответствующем уменьшении времени CTe
рилизационной выдержки, обеспечивающих
стерильность и сохранность объекта. Режим
должен быть обоснован и указан в нормативной
документации.
01/201З:РБ60200
#6.2. ЭКСПРЕСС..ДНДЛИЗ
ЭКСТЕМПОРДЛЬНЫХ
ЛЕКАРСТВЕННЫХ
СРЕДСТВ
в оценке качества лекарственных средств
аптечноrо изroтовления основную роль иrрает
химический контроль. В условиях аптек прово
дят экспресс анализ лекарственных средств,
который отличается быстротой проведения,
простотой используемых методик. требует мини
мальноro количества испытуемоrо образца и pe
активов.
Данный раздел содержит методики экс
пресс анализа часто встречающихся прописей
экстемпоральных лекарственных средств. Экс
темпоральные лекарственные средства должны
выдерживать требования статьи #6.3.1. Нормы
отклонений, допустимые при иЗ20товлении
лекарственных средств (в том числе 20меопа
тических) в аптеках.
Аминокапроновой кислоты 0,5 %, 1 %, 2 %,
3 %. 5 % растворы (001)
Аминокапроновой кислоты 5 % раствор для
инъекций (002)
Аскорбиновой кислоты 2 % раствор (003)
Аскорбиновой кислоты порошок (004)
1014
rосударственная фармакопея Республики Беларусь
Ацетилсалициловой кислоты порошок (005)
Борной кислоты 2 %, 3 %, 4 % растворы (006)
rлутаминовой кислоты 1 % раствор
для инъекций (007)
rлюкозы 5 % раствор (008)
rлюкозы 5 %, 1 О %, 20 %, 30 % растворы без
стабилизатора (009)
rлюкозы 5 %, 1 О %, 20 %, 25 %, 40 % paCTBO
ры со стабилизатором (01 О)
rлюкозы 1 О % раствор с калия хлоридом
(011 )
Дибазола 0,5 %, 1 %, 2 % растворы (012)
Дибазола порошок (013)
Дикаина 0,5 %, 1 % растворы (014)
Димедрола 0,1 %, 1 % растворы (015)
Димедрола 0,25 %, 0,5 % растворы (016)
Димедрола 1 %,2 % растворы (017)
Димедрола 1 %,2% растворы для инъекций
(018)
Димедрола и кальция rлюконата порошок
(019)
Димедрола и папаверина rидрохлорида по
рошок (020)
Димедрола порошок (021)
Жидкость Петрова кровезамещающая (022)
Йода 1 %, 2 % раствор (023)
Йода 5 % раствор (024)
Йода 10 % раствор (025)
Калия бромида и маrния сульфата раствор
с rлюкозой (026)
Калия йодида 0,1 %, 0,25 %, 0,5 %, 1 %, 2 %,
3 %, 5 % растворы (027)
Калия йодида 0,25 %,1 %,3 % раствор (028)
Калия йодида 20 % раствор (029)
Калия йодида и новокаина раствор (030)
Калия йодида и эуфиллина раствор (031)
Калия перманrаната 0,0125 %,1 %,5 % pac
творы (ОЗ2)
. Калия хлорида 4 %, 5 %, 7,5 % растворы (033)
Кальция хлорида 1 %, 2 %, 2,5 %, 3 %, 5 %,
10 % растворы (034)
Кальция хлорида 1 %, 1 О % растворы для
инъекций (035)
Кальция хлорида 50 % раствор (ОЗ6)
Кардиоплеrические растворы NQ 1, NQ 2, NQ 3
(037)
Колларroл с rлицерином (038)
Колларroла 1 %,2 %, 3 % раствор (039)
Колларroла 1 %,2 %, 3 %,5 % раствор (040)
Маrния сульфата 1 %, 5 %, 14 %, 25 %, 33 %
растворы (041)
Маrния сульфата 25 %, 33 % раствор для
инъекций (042)
Натрия бромида 1 %,2 %, 3 % раствор (043)
Натрия бромида 3 % раствор с настойкой
валерианы (044)
Натрия бромида 20 % раствор (045)
Натрия бромида и аскорбиновой кислоты
раствор с rлюкозой (046)
Натрия бромида и маrния сульфата раствор
с rлюкозой (047)
Натрия rидрокарбоната 4 % раствор для
инъекций (048)
Натрия rидрокарбоната 4 % раствор для
инъекций стабилизированный (049)
Натрия тетраборат с rлицерином (050)
Натрия тетрабората и натрия rидрокарбона
та раствор (051)
Натрия тиосульфата 0,25 %, 2 %, 10 %,
15 %, 20 %, 30 % раствор (052)
Натрия хлорида 0,9 %, 2 % раствор (05З)
Натрия цитрата двузамещенноrо 3,2 % paCT
вор (054)
Натрия цитрата двузамещенноrо З,8 %, 5 %
растворы (055)
Натрия цитрата для инъекций 5 % раствор
(056)
Никотинамида 1 % раствор для инъекций
(057)
Никотиновой кислоты 0,1 %, 0,5 %, 1 % pac
творы (058)
Никотиновой кислоты 1 % раствор для инъек
ций (059)
Новокаина 2 % раствор (060)
Пероксида водорода 3 % раствор с натрия
6ензоатом (061)
Пиридоксина rидрохлорида порошок (062)
Протарrола 1 %,2 %, 3 %, 5 % растворы (063)
Раствор по Демьяновичу NQ 1 (064)
Раствор Ринrера (065)
Рибофлавина 0,02 % раствор (066)
Рибофлавина, аскорбиновой кислоты и ни
котиновой кислоты раствор (067)
Рибофлавина и аскорбиновой кислоты раст
вор (068)
Сульфацила натрия 15 %, 20 %,30 % paCT
вор (069)
Фурацилина 0,02 % раствор (070)
Фурацилина 0,02 % раствор изотонический (071)
Хлорrексидина биrлюконата 0,02 %, 0,05 %
растворы (072)
Хлористоводородной кислоты 1 %, 2 % pac
творы (073)
Хлористоводородной кислоты 1 О % раствор
(074)
Цинка сульфата 0,25 %, 1 %, 2 % раствор с
борной кислотой (075)
Цинка сульфата 2 % раствор (076)
Эуфиллина 1 % раствор (077)
Эуфиллина 1 %, 2 %, 2,4 % раствор изото
нические (078)
Эуфиллина порошок с сахарозой (079)
АМИНОКАПРОНОВОЙ КИСЛОТЫ
0,5 %, 1 %, 2 %, 3 %, 5 %
РАСТВОРЫ (001)
СОСТАВ
Аминокапроновая кислота (С 6 Н 1з N0 2 ; М М.
131 ,2) 5,0 r, 10,0 r, 20,0 r, 30,0 r, 50,0 r;
Вода очищенная до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
Аминокапроновой кислоты 5 % раствор для инъекций (002)
1015
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 3 5 каплям испытуемоro образца при
бавляют 2 3 капли раствора ниН2идрина Р и Ha
rревают до кипения. Появляется сине фиолето
вое окрашивание.
В. К 2 каплям испытуемоro образца прибав
ляют 1 мл раствора хлорамина Р, 1 мл раст
вора 1 О r/л фенола Р и наrревают на водяной
бане в течение 2 мин. Появляется синее OKpa
шивание.
С. 2 мл испытуемоrо образца нейтрализу
ют 0,1 М раствором натрия 2идроксида до по
явления отчетливой красной окраски, используя
в качестве индикатора 0,1 мл раствора фенол
фталеина Р, прибавляют 2 капли раствора
формальде2ида Р, предварительно нейтрали
зованноro О, 1 М раствором натрия 2идроксида
до появления слаборозовой окраски, используя
в качестве индикатора 0,1 мл раствора фенол
фталеина Р, и встряхивают. Раствор обесцвечи
вается.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 2,0 мл 0,5 % и 1 % испытуемых pac
творов или к 0,5 мл 2 %, З % и 5 % испы
туемых растворов прибавляют 5 мл воды Р,
5 мл раствора формальде2ида Р, предвари
тельно нейтрализованноro по раствору фе
нолфталеина Р, встряхивают и через 2 мин
титруют О, 1 М раствором натрия 2идpOKcи
да до появления розовоro окрашивания, ис
пользуя в качестве индикатора раствор фе
нолфталеина Р1.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия 2идроксида co
ответствует 13,12 Mr С 6 Н 1з N0 2 .
АМИНОКАПРОНОВОЙ КИСЛОТЫ 5 %
РАСТВОР ДЛЯ ИНЪЕКЦИЙ (002)
СОСТАВ
Аминокапроновая кислота (С Б Н 1з NО 2 ; м. м.
131 ,2) 50,0 r;
Натрия хлорид (NaCI; М. м. 58,44) 9,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная прозрачная жидкость со значе
нием рН от 7,0 до 8,0.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 3 5 каплям испытуемоro образца при
бавляют 2 3 капли раствора ниН2идрина Р и
наrревают до кипения. Появляется сине фиоле
товое окрашивание.
В. К 2 каплям испытуемоro образца при
бавляют 1 мл раствора хлорамuна Р, 1 мл
раствора 1 О r/л фенола Р и наrревают на
водяной бане в течение 2 мин. Появляется
синее окрашивание.
С. 2 мл испытуемоro образца нейтрализуют
0,1 М раствором натрия 2идроксида до появле
ния отчетливой красной окраски, используя в каче
стве индикатора 0,1 мл раствора фенолфталеина
Р, прибавляют 2 капли раствора формальде2ида
Р, предварительно нейтрализованноro О, 1 М pac
твором натрия 2идроксида до появления слабо
розовой окраски, используя в качестве индикатора
0,1 мл раствора фенолфталеина Р, и встряхива
ют. Раствор обесцвечивается.
D. К 2 мл испытуемоro образца дают peaK
цию (а) и (с) на натрий (2.3.1).
Е. К 3 каплям испытуемоro образца прибав
ляют 3 капли кислоты азотной разведенной Р,
3 капли раствора серебра нитрата Р1, переме
шивают и отстаивают. Образуется белый творожи
стый осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Натрия хлорид
МЕТОД 1
К 1,0 мл испытуемоro образца прибавляют
1 2 капли раствора бромфенолов020 сине20 Р,
по каплям кислоту уксусную разведенную Р до
зелено желтоro окрашивания и титруют О, 1 М
раствором серебра нитрата до появления фи
олетовоro окрашивания.
1 мл 0,1 М раствора серебра нитрата co
ответствует 5,844 Mr NaCI.
МЕТОД 2
1,0 мл испытуемоro образца титруют О, 1 М
раствором серебра нитрата до появления opaH
жево желтоrо окрашивания, используя в качестве
индикатора 0.2 мл раствора калия хромата Р.
1 мл 0,1 М раствора серебра нитрата co
ответствует 5,844 Mr NaCI.
Аминокапроновая кислота
МЕТОД 1
К 0,5 мл испытуемоro образца прибавляют
5 мл воды Р, 5 мл раствора формальдеzида Р,
предварительно нейтрализованноrо по pacrnвo
ру фенолфталеина Р, встряхивают и через 2 мин
титруют 0,1 М раствором натрия 2идроксида до
появления розовоro окрашивания, используя в Ka
честве индикатора раствор фенолфталеина Р1.
Параллельно проводят контрольный опыт.
1 мл О, 1 М раствора натрия 2идроксuда co
ответствует 13,12 Mf СБН,зNО2'
МЕТОД 2
Определяют показатель преломления (2.2.6).
Содержание кислоты аминокапроновой
(мr/мл) рассчитывают по формуле:
п (пc +0,00170.с).10
0,00178
rдe:
п показатель преломления испытуемоro
образца;
rосударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый однородный поро
шок кисло сладlюrо вкуса.
1016
ПО показатель преломления воды;
0,00170 величина прироста показате
ля преломления при увеличении концентрации
натрия хлорида на 1 %;
0,00178 величина прироста показателя
преломления при увеличении концентрации кис
лоты аминокапроновой на 1 %;
с концентрация натрия хлорида в испыту
емом образце, %.
АСКОРБИНОВОЙ КИСЛОТЫ 2 %
РАСТВОР (003)
СОСТАВ
Аскорбиновая кислота (С 6 НР6; м. м. 176,1)
2,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или со слабым жел
товатым опенком жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоro образца прибавляют
0,5 мл раствора серебра нитрата Р2. Образу
ется темно серый осадок.
В. К 0,2 мл испытуемоro образца прибав
ляют по каплям раствор 1 r/л дихлорфенолин
дофенола натриевой соли Р в 96 % спирте Р.
Синяя окраска последнеro исчезает.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
К 2,0 мл испытуемоro образца прибавляют
0,5 мл раствора калия йодида Р1, 2 мл раствора
крахмала, свободН020 от йодидов, Р, 1 мл paCT
вора 274 r/л кислоты хлористоводородной раз
веденной Р и титруют 0,0167 М раствором калия
йодата до появления слабо синеro окрашивания.
1 мл 0,0167 М раствора калия йодата COOT
ветствует 8,806 Mr С 6 Н в О 6 .
МЕТОД 2
К 2,0 мл испытуемоro образца прибавляют
2 капли раствора фенолфталеина Р1 и титру
ют 0,1 М раствором натрия 2идроксида до по
явления розовоro окрашивания.
1 мл 0,1 М раствора натрия 2идроксида co
ответствует 17,61 Mr С 6 Н в 0 6 .
АСКОРБИНОВОЙ КИСЛОТЫ
ПОРОШОК (О04)
СОСТАВ
Аскорбиновая кислота (С 6 НР6; м. м. 176,1)
0,05r;
rлюкоза моноrидрат (С6Н1206'Н20; м. м.
198,2) 0,1 r:
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 0,15 rиспытуемоroобразца растворяют в 9 мл
воды Р (раствор А). К 2 мл раствора А прибавпяют 3
капли раствора серебра нитрата Р2. Образуется
осадок сероro цвета.
В. К 2 мл раствора А прибавпяют по каплям
раствор 1 r/л дихлорфенолиндофенола натриевой
соли Р в 96 % спирте Р. Синее окрашивание по--
следнеro исчезает.
с. К 5 мл раствора А прибавляют 1 каплю
раствора водорода пероксuда концентрированно--
20 Р, 2 капли раствара аммиака разведеННО20 Р2,
кипятят в течение 1 мин и прибавляют 0,5 мл pacт
вара медно--тартратнО2О Р. Образуется коричне
вато--красный осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Аскорбиновая кислота. 0,05 r испытуемоro
образца растворяют в 5 мл вады Р, прибавляют
2 капли раствора фенолфталеина Р1 и титруют
О, 1 М раствором натрия 2идроксида до появле
ния розовоro окрашивания.
1 мл 0,1 М раствора натрия 2идроксида co
ответствует 17,61 Mr С 6 Н в 0 6 .
rлюкоза
МЕТОД 1
0,05rиспытуемоroобразцарастворяютв 7 ,5мл
воды Р. 1,0 мл полученноro раствора титруют
0,05 М раствором йода до появления желтоro
окрашивания, прибавляют 4,0 мл 0,05 М pacт
вора йода, 0,3 мл раствора натрия 2идpOKcи
да разведеНН020 Р и выдерживают в течение
5 мин в защищенном от света месте. Прибавля
ют 0,5 мл кислоты серной разведенной Р и из
быток йода титруют 0,1 М раствором натрия
тиосульфата до обесцвечивания раствора.
1 мл 0,1 М раствора натрия тиосульфата
соответствует 9,91 Mr С 6 Н 12 О 6 'Н 2 О.
МЕТОД 2
0,15 r испытуемоro образца растворяют в
2 мл воды Р и определяют показатель прелом
ления (2.2.6).
Содержание rлюкозы в rpaMMax рассчиты
вают по формуле:
п (пo +0,00160,с).2.Ь
0,00129. т .100
rде:
п показатель преломления испытуемоro
образца;
по показатель преломления воды;
0,00160 величина прироста показателя
преломления при увеличении концентрации кис
лоты аскорбиновой на 1 %;
rлутамuновой кислоты 1 % раствор для инъекций (007)
1017
0,00129 величина прироста показате
ля преломления при увеличении концентрации
rлюкозы на 1 %;
с концентрация кислоты аскорбиновой в
растворе, %;
т масса навески испытуемоrо образца, r;
Ь средний вес порошка, r.
АЦЕТИЛСАЛИЦИЛОВОЙ КИСЛОТЫ
ПОРОШОК (005)
СОСТАВ
Ацетилсалициловая кислота (СэНвО 4;
м. м. 180,2) 0,01 r, 0,02 r, 0,03 r;
rлюкоза моноrидрат (С 6 Н 12 О 6 ' Н 2 О;
М. М. 198,2) 0,2 r.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический
порошок.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 0,5 r испытуемоro образца кипятят в
течение 3 мин с 5 мл раствора 2идроксида
натрия разведеНН020 Р, охлаждают и подкис
ляют кислотой серной разведенной Р. Выпа
дает белый кристаллический осадок. Haдoca
дочную жидкость сливают в друrую пробирку
и прибавляют к нему 2 мл 96 % спирта Р и
2 мл кислоты серной Р. Появляется запах
этилацетата. К осадку nрибавляют 1 2 капли
раствора железа (1/1) хлорида Р1. Появляется
фиолетовое окрашивание.
В. 0,2 r испытуемоro образца помещают в
фарфоровую чашку, прибавляют 0,5 мл кисло
ты серной Р, перемешивают и прибавляют 1 2
капли воды Р. Появляется запах уксусной кисло
ты. Прибавляют 1 2 капли раствора формаль
де2ида Р. Появляется розовое окрашивание.
С. 0,1 r испытуемоro образца растворяют в
1 О мл воды Р, прибавляют 3 мл раствора Meд
Ho тapтpaтHoeo Р и наrревают. Образуется KO
ричневато красный осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 r испытуемоroобразца растворяют в 1 О мл
96 % спирта Р, нейтрализованноro по раствору
фенолфталеина Р (5--------6 капель) и охлажденноrо
до температуры о. 8 ос до 10 ОС. Раствор титруют
с тем же индикатором 0,1 М раствором натрия 2и
дрок сида до розовоrо окрашивания.
1 мл 0,1 М раствора натрия 2идроксида COOT
ветствуе. 18,02 Mr С э Н в 0 4
БОРНОЙ КИСЛОТЫ 2 %, 3 %, 4 %
РАСТВОРЫ (006)
СОСТАВ
Борная кислота (НзВОз; М. м. 61 ,8) 2,0 r,
3.0 r. 4,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
0,5 мл испытуемоro образца выпаривают на
водяной бане досуха. Oc:raToK растворяют в 2 мл
96 % спирта Р. Раствор roрит пламенем, окайм
ленным зеленым цветом.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,5 мл испытуемоro образца прибавля
ют 5 мл 2лицерина (85 %) Р, предварительно
нейтрализованноro 0,1 М раствором натрия
2идроксида по фенолфталеину до устойчиво
ro розовоro окрашивания, перемешивают и ти
труют О, 1 М раствором натрия 2идроксида
до появления розовоro окрашивания, исполь
зуя в качестве индикатора 2 капли раствора
фенолфталеина Р1. Затем к раствору при
бавляют еще 5 мл 2лицерина (85 %) Р, пред
варительно нейтрализованноro О, 1 М pacтвo
ром натрия 2идроксида по фенолфталеину
до устойчивоro розовоro окрашивания. Если
окраска при этом исчезает, снова титруют
0,1 М раствором натрия 2идроксида до появ
ления розовой окраски. Прибавление нейтра
лизованноro 2лицерина (85 %) Р и титрование
0,1 М раствором натрия 2идроксида продол
жают до появления розовой окраски, не исчеза
ющей в течение 30 с.
1 мл 0,1 М раствора натрия 2идроксида co
ответствует 6,183 Mr НзВО з .
rЛУТАМИНОВОЙ КИСЛОТЫ 1 %
РАСТВОР ДЛЯ ИНЪЕКЦИЙ (007)
СОСТАВ
rлутаминовая кислота (C 5 H s N0 4 ; Мм. 147,1)
10,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость со значе
нием рН от 3,4 до 3,6.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 0,5 мл испытуемоro образца прибавля
ют 2 3 капли раствора ниН2идрина Р1 и Harpe
вают в водяной бане в течение 1 мин. Появляет
ся сине фиолетовое окрашивание.
В. 2 капл испытуемоro образца выпа
ривают досуха. прибавляют несколько крупи
нок резорцина Р, 5 капель кислоты серной Р
и наrревают до появления зелено коричнево
ro окрашивания. Охлаждают, прибавляют 5 мл
воды Р и 5 мл раствора аммиака разведенно
20 Р1. Появляется красно фиолетовое окраши
вание с зеленой флуоресценцией.
1018
rосударственная фармакопея Республики Беларусь
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
2,0 мл испытуемоro образца титруют 0,1 М
раствором натрия 2идроксида до изменения
окраски раствора от желтой до roлубовато зеле
ной, используя в качестве индикатора раствор
бромтимолов020 сине20 Р1.
1 мл 0,1 М раствора натрия 2идроксида co
ответствует 14,71 Mr C 5 H g N0 4 .
rлюкозы 5 % РАСТВОР (008)
СОСТАВ
rлюкоза моноrидрат (С6Н1206'Н20;
М. М. 198,2) 5,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
К 1 мл испытуемоrо образца прибавляют
1 2 мл реактива ФелиН2а Р и наrpевают до ки
пения. Образуется оранжев красный осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
1,0 мл испытуемоrо образца помеща
ют в колбу с притертой пробкой, прибавляют
10,0 мл 0,05 М раствора йода, 0,5 мл pacт
вора натрия 2идроксида Р. Колбу закрыва
ют пробкой и выдерживают в защищенном
от света месте в течение 5 мин. Прибавля
ют 3 5 мл кислоты серной разведенной Р
и выделившийся йод титруют 0,1 М pacтвo
ром натрия тиосульфата до обесцвечива
ния раствора.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
9,009 Mr С 6 Н 1 Р6'
МЕТОД 2
Определяют показатель преломления
(2.2.6); FСБН120Б HP = 0,00129.
rлюкозы 5 %, 1 О %, 20 %, 30 %
РАСТВОРЫ
БЕЗ СТАБИЛИЗАТОРА (009)
СОСТАВ
rлюкоза безводная (С 6 Н 1 Р6; м. М. 180,2)
50,0 r, 100,0 r, 200,0 r, 300,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или слеrка желтова
тая жидкость со значением рН от 3,8 до 6,5.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
К 1 мл испытуемоro образца прибавляют
1 2 мл реактива ФелиН2а Р и наrревают до ки
пения. Образуется оранжево красный осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
Объем испытуемоro образца, COOTвeTCTBY
ющий 0,05 r rлюкозы, помещают в колбу с притер
той пробкой, прибавляют 10,0 мл 0,05 М раствора
йода, 0,5 мл раствора натрия 2идроксида Р. Колбу
закрывают пробкой и выдерживают в защищенном
от света месте в течение 5 мин. Прибавляют 3--------5 мл
кислоты серной разведенной Р и титруют Bыдe
лившийся ЙОД 0,1 М раствором натрия тиосуль
фата до обесцвечивания раствора.
Параллельно проводят контрольный опыт.
1 мл 0.05 М раствора йода соответствует
9.009 Mr С 6 Н 12 О 6 .
МЕТОД 2
Определяют показатель преломления
(2.2.6); F сбн . д = 0,00142.
rлюкозы 5 %, 10 %, 20 %, 25 %, 40 %
РАСТВОРЫ
СО СТАБИЛИЗАТОРОМ (010)
СОСТАВ
rлюкоза безводная (С 6 Н 1 Р6; м. М. 180,2)
50,0 r, 100,0 r, 200,0 r, 250,0 r, 400,0 r;
Натрия хлорид (NaCI; М. м. 58,44) 0,26 r;
Хлористоводородной кислоты 0,1 М
раствор 5 мл;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или слеrка желтова
тая жидкость со значением рН от 3,0 до 4,1.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоro образца прибавляют
1 2 мл реактива ФелиН2а Р и наrревают до ки
пения. Образуется оранжево красный осадок.
В. 2 мл испытуемоro образца подкисляют
кислотой азотной разведенной Р, прибавляют
0.4 мл раствора серебра нитрата Р1. Образу
ется белый творожистый осадок, растворимый в
растворе аммиака Р.
С. 20 мл испытуемоro образца упаривают до
объема 2 мл. Полученный раствор дает реакцию
(а) на натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
rлюкоза
МЕТОД 1
Объем испытуемоro образца, COOTBeTCTBY
ющий 0,05 r rлюкозы, помещают в колбу с при
Дибазола 0,5 %, 1 %, 2 % растворы (012)
1019
тертой пробкой, прибавляют 10,0 мл 0,05 М
раствора йода, 0,5 мл раствора натрия 2и
дроксида Р. Колбу закрывают пробкой и Bыдep
живают в защищенном от света месте в течение
5 мин. Прибавляют З 5 мл кислоты серной раз
веденной Р и выделившийся йод титруют 0,1 М
раствором натрия тиосульфата до обесцве
чивания раствора.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
9,009 Mr С 6 Н 12 О 6 .
МЕТОД 2
Определяют показатель преломления
(2.2.6); F C Н 0 =0,00142.
6 12 б
Хлористоводородная кислота. 5,0 мл ис
пытуемоro образца титруют 0,01 М раствором
натрия 2идроксида до появления желтоrо OKpa
шивания (V , ' мл), используя В качестве индика
тора раствор метиленов020 краСН020 Р.
1 мл 0,01 М раствора натрия 2идроксuда
соответствует 0,3646 Mr HCI.
Натрия хлорид. К 5,0 мл испытуемоro об
разца прибавляют 3 капли раствора бромфено
лов020 сине20 Р, 2 капли кислоты уксусной раз
веденной Р и титруют 0,01 М раствором серебра
нитрата до появления фиолетовоro окрашива
ния (V 2 ' мл).
Количество 0,01 М раствора серебра Hи
трата, израсходованное на титрование натрия
хлорида, рассчитывают по формуле V 2 V , .
1 мл 0,01 М раствора серебра нитрата co
ответствует 0,5844 Mr NaCI.
rлюкозы 1 О % РАСТВОР С КАЛИЯ
ХЛОРИДОМ (011)
СОСТАВ
rлюкоза безводная (С 6 Н ' Р6; м. м. 180,2)
100,0 r;
Калия хлорид (KCI; М. м. 74,6) 12,0 r, 18,0 r,
24,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или слеrка желтова
тая жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоro образца прибавляют
1 2 мл реактива ФелиН2а Р и наrревают до ки
пения. Образуется оранжево красный осадок.
В. 2 мл испытуемоro образца подкисляют
кислотой азотной разведенной Р, прибавляют
0,4 мл раствора серебра нитрата Р1. Образу
ется белый творожистый осадок, растворимый в
растворе аммиака Р.
с. Испытуемый образец дает реакцию (Ь) на
калий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Калия хлорид. 1,0 мл раствора титруют 0,1 М
раствором серебра нитрата до появления
желтовато коричневоro окрашивания, исполь
зуя в качестве индикатора раствор калия xpo
мата Р.
1 мл 0,1 М раствора серебра нитрата co
ответствует 7,456 Mr KCI.
rлюкоза
МЕТОД 1
5,0 мл испытуемоro образца доводят
водой Р до объема 50,0 мл. 5,0 мл получен
ноro раствора помещают в колбу с притертой
пробкой, прибавляют 10,0 мл 0,05 М раствора
йода, 0,5 мл раствора натрия 2идроксида Р.
Колбу закрывают пробкой и выдерживают в
защищенном от света месте в течение 5 мин.
Прибавляют 3 5 мл кислоты серной разве
денной Р и выделившийся йод титруют 0,1 М
раствором натрия тиосульфата до обесцве
чивания раствора.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
9,009 Mr С 6 Н ,2 О 6 .
МЕТОД 2
Определяют показатель преломления
(2.2.6).
Содержание rлюкозы в rpaMMax рассчиты
вают по формуле:
п (пo +0,00127 .с)
0,00142.100
rдe:
п показатель преломления испытуемоrо
образца;
по показатель преломления воды;
0,00127 величина прироста показате
ля преломления при увеличении концентрации
калия хлорида на 1 %;
0,00142 величина прироста показате
ля преломления при увеличении концентрации
rлюкозы безводной на 1 %;
с концентрация калия хлорида. опреде
ленная, как указано в разделе «Количественное
определение. Калия хлорид», %.
ДИБАЗОЛА 0,5 %, 1 %. 2 %
РАСТВОРЫ (012)
СОСТАВ
Дибазол (C'4H'2N2'HCI; М. м. 244,7) 5,0 r,
10,0 r, 20,0 r;
ХлористоводороднойкислотыО, 1 MpaCTBOp
10 мл;
Вода для инъекций до 1000 мл.
1020
rосударственная фармаК0f7ея Республики Беларусь
ОПИСАНИЕ
Прозрачная бесцветная ЖИДКОСТЬ со значе
нием рН от 2,8 до 3,5.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоro образца прибавляют
3 капли кислоты хлористоводородной разведен
ной Р. 2 3 капли 0,05 М раствора йода и встряхи
вают. Образуется I<расновато серебристый осадок
В. 2 мл испытуемоro образца подкисляют
кислотой азотной разведенной Р, прибавляют
0,4 мл раствора серебра нитрата Р1. Образу
ется белый творожистый осадок, растворимый в
растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Дибазол. К 10,0 мл 0,5 % испытуемоro paCT
вора или 5,0 мл 1 % и 2 % испытуемоrо раствора
прибавляют 3 капли раствора бромфеноло
в020 сине20 Р, по каплям кислоту уксусную раз
веденную Р до появления зеленовато желтоro
окрашивания и титруют 0,1 М раствором cepe
бра нитрата до появления фиолетовоro oкpa
шивания.
1 мл 0,1 М раствора серебра нитрата co
ответствует 24,47 Mr C'4H'2N2'HCI.
Хлористоводородная кислота. 5,0 мл ис
пытуемоro образца титруют 0,01 М pacтвo
ром натрия 2идроксида до появления жеmоro
окрашивания, используя в качестве индикатора
раствор метиленов020 краСН020 Р.
1 мл 0,01 М раствора натрия 2идроксида
соответствует 0.3646 Mr HCI.
ДИБАЗОЛА ПОРОШОК (013)
СОСТАВ
Дибазол (CI4H'2N2'HCI; М. м. 244,7} 0,001 r,
0,003 r, 0,005 r, 0,008 r;
Сахароза (С ,2 Н 22 О,,; М. м. 342,3) [rлюкоза
моноrидрат (С 6 Н ' Р6"НР; М. м. 198,2)] 0,2 r.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический
порошок
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
При использовании сахарозы в качестве Ha
полните ля: А, В.
При использовании 2ЛЮКОЗЫ в качестве Ha
полнителя: А, С.
А. 0,2 rиспытуемоroобразца растворяютв5мл
воды Р, прибавляют 3 капли кислоты хлористо
водородной разведенной Р, 2 3 капли 0.05 М
раствора йода и встряхивают. Образуется Kpac
новато серебристый осадок.
В. К 0,01 r испытуемоro образца прибавля
ют 1 2 мл кислоты хлористоводородной раз
веденной Р, несколько кристаллов резорцина Р
и кипятят в течение 1 мин. Появляется красное
окрашивание.
с. К 0,02 r испытуемоro образца прибавляют
1 мл воды Р, 1 мл реактива ФелиН2д Р и Harpe
вают до кипения. Образуется коричневато крас
ный осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,2 r испытуемоro образца растворяют в
2 мл воды Р, прибавляют 3 капли раствора
бромфенолов020 сине20 Р, по каплям кислоту
уксусную разведенную Р до появления зелено
вато желтоro окрашивания и титруют 0,02 М pac
твором серебра нитрата до появления фиоле
товоro окрашивания.
1 мл 0,02 М раствора серебра нитрата co
ответствует 4,89 Mr CI4H'2N2'HCI.
ДИКдИНА 0,5 %, 1 % РАСТВОРЫ (014)
СОСТАВ
Тетракаина rидрохлорид (C'5H24N202; М. м.
300,8) 0,05 r, 0,1 r;
Натрия хлорид (NaCI; М. м. 58,44) 0,081r,
O,072r;
Вода очищенная 10 мл.
ОПИСАНИЕ
Бесцветная прозрачная жидкость
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 0,5 мл испытуемою образца выпарива
ют на водяной бане досуха. После охлаждения
к сухому остатку прибавляют 3 5 капель Kиc
лоты азотной дымящейся Р и выпаривают на
водяной бане досуха. Охлаждают, растворяют
полученный остаток в 5 мл ацетона Р и при
бавляют 1 мл раствора 0,1 % (м/об) калия 2и
дроксида в 96 % спирте Р. Появляется фиоле
товое окрашивание.
В. К 0,5 мл испытуемою образца прибав
ляют 0,5 мл кислоты азотной разведенной Р2
и 0,5 мл раствора серебра нитрата Р1. Об
разуется белый творожистый осадок, Hepac
творимый в кислоте азотной разве.денной Р2
и растворимый в растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Тетракаина rидрохлорид. К 0,5 мл испыту
емоrообразца прибавляют2 3 мл хлороформа Р
и титруют при встряхивании 0,01 М раствором
натрия 2идроксида до появления розовоro Ol<pa
шивания водноro слоя, используя в качестве ин
дикатора раствор фенолфталеина Р.
1 мл 0,01 М раствора натрия 2идpOKcи
да соответствует 3,008 мr тетракаина rидро
хлорида.
Димедрола 0,25 %,0,5 % растворы (016)
1021
Натрия хлорид. К 0,5 мл испытуемоro об
разца прибавляю-т 1 2 капли раствора бром
фенолов020 сине20 Р и по каплям кислоту YK
сусную разведенную Р до зелен()вато желтоro
окрашивания и. титруют О, 1 М раствором cepe
бра нитрата до сине фиолетовоro окрашива
ния осадка.
1 мл 0,1 М раствора серебра нитрата co
о-тветствует 5,844 Mr натрия хлорида.
Количество 0,1 М раствора серебра Hитpa
та, израсходованное на титрование натрия хло
рида, рассчитывают по формуле:
х V V NBOH .
АgNОЗ 10
ДИМЕДРОЛА 0,1 %, 1 %
РАСТВОРЫ (015)
СОСТАВ
Дифенrидрамина rидрохлорид
(C 17 H 21 NO'HCI; м. м. 291 ,8) 0,1 r, 1,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл 1 % испытуемоro образца прибав
ляют 3 5 капель 0,1 М раствора кислоты хло
ристоводородной, 3 5 капель раствора 10 r/л
меди (/1) сульфата Р, 8 1 О капель раствора
1 О r/л аммония тиоцианата Р. Появляется KO
ричневое окрашивание.
В. 1 мл 0,1 % испытуемоro раствора и 0,2 мл
1 % испытуемоro раствора выпаривают на
водяной бане досуха. После охлаждения к
сухому остатку прибавляют 4 5 капель Kиc
лоты серной Р. Появляется желтое окрашива
ние, исчезающее при прибавлении 2 3 капель
воды Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
К 5,0 мл 0,1 % испытуемоro раствора или
1,0 мл 1 % испытуемоrо раствора прибавляют
1 2 капли раствора бромфенолов020 сине20
Р, по каплям кислоту уксусную Р до зеленова
то желтоro окрашивания и титруют 0,02 М pac
твором серебра нитрата до появления фиоле
товоro окрашивания.
1 мл 0,02 М раствора серебра нитрата co
ответствует 5,836 Mr C 17 H 21 NO'HCI.
МЕТОД 2
К 5,0 мл 0,1 % испытуемоrо раствора или
1.0 мл 1 % испытуемоro раствора прибавляют
3 4 мл хлороформа Р и титруют 0,02 М pac
твором натрия 2идроксида при встряхивании
до появления розовоro окрашивания водноro
слоя, используя в качестве индикатора pacт
вор фенолфталеина Р.
1 мл 0,02 М раствора натрия 2идроксида
соответствует 5,836 Mr C 17 H 21 NO'HCI.
ДИМЕДРОЛА 0,25 %, 0,5 %
РАСТВОРЫ (016)
СОСТАВ
Дифенrидрамина rидрохлорид
(C 17 H 21 NO'HCI; м. м. 291 ,8) 0,025 r; 0,050 r;
Натрия хлорид (NaCl; М. м. 58,44) 0,085 r;
0,080 r;
Вода для инъекций до 1 О мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 0,5 мл испытуемоro раствора выпарива
ют на водяной бане досуха. После охлаждения к
сухому остатку прибавляют 5 капель кислоты
серной Р. Появляется желтое окрашивание, исче
зающее при прибавлении 2 3 капель воды Р.
В. 0,5 мл испытуемоro раствора дают peaK
цИИ (Ь) и (с) на натрий (2.3.1).
с. К 2 мл испытуемоro образца прибавляют
0,5 мл кислоты азотной разведенной Р2, 0,5 мл
раствора серебра нитрата Р1. Образуется
белый творожистый осадок, нерастворимый в
кислоте азотной разведенной Р2 и раствори
мый в растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Дифенrидрамина rидрохлорид. К 2,0 мл
0,25 % испытуемоro раствора или 1,0 мл 0,5 %
испытуемоro раствора прибавляют 3 мл хло
роформа Р и титруют при встряхивании 0,02 М
раствором натрия 2идроксида до появления
розовоro окрашивания водноro слоя (Ао 25 "< или
А о . 5 % соответственно, мл), используя в качестве
индикатора раствор фенолфталеина Р.
1 мл 0,02 М раствора натрия 2идроксида
соответствует 5,836 Mr C 17 H 21 NO'HCI.
Натрия хлорид. К 1 ,О мл испытуемоro раст
вора прибавляют 1 2 капли раствора бромфе
нолов020 сине20 Р, по каплям уксусную кислоту Р
до зеленовато жел-тоro окрашивания и титруют
0,1 М раствором серебра нитрата до появле
ния фиолетовоro окрашивания (В, мл).
Количество 0.1 М раствора серебра Hитpa
та (Х 025 о, или Х С5 <.' мл), израсходованное на
титрование натрия хлорида в 0,25 % или 0,5 %
испытуемом растворе соотве-тс-твенно, рассчи
-тываю-т по формулам:
А
Х = B 0,25'% ,
0.25%
10
1022
rосударственная фармакопея Республики Беларусь
х в А о ,5% .
0,5% 5
1 мл 0,1 М раствора серебра нитрата co
ответствует 5,844 Mr NaCI.
ДИМЕДРОЛА 1 %, 2 % РАСТВОРЫ (017)
СОСТАВ
Дифенrидрамина rидрохлорид (C 17 H 21 NO'HCI;
м. м. 291 ,8) 0,1 r; 0,2 r;
Вода очищенная до 1 О мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоro образца прибавляют
5 капель 0,1 М раствора кислоты хлорuсто
водородной, 3 5 капель раствора 10 r/л меди
(11) сульфата Р, 8 1 О капель раствора 1 О r/л
аммония тиоцианата Р. Появляется коричне.-
вое окрашивание.
В. 0,2 мл испытуемоro раствора выпарива
ют на водяной бане досуха. После охлаждения
к сухому остатку прибавляют 4 5 капель Kиc
лоты серной Р. Появляется желтое окрашива
ние, исчезающее при прибавлении 2 3 капель
воды Р.
с. К 2 мл испытуемоrо образца прибавляют
0,5 мл кислоты азотной разведенной Р2, 0,5 мл
раствора серебра нитрата Р1. Образуется
белый творожистый осадок, нерастворимый в
кислоте азотной разведенной Р2 и раствори
мый в растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
К 1 ,О мл 1 % испытуемоro раствора или 0,5 мл
2 % испытуемоro раствора прибавляют 1 2
капли раствора бромфенолов020 сине20 Р, по
каплям кислоту уксусную Р до зеленовато жел
тoro окрашивания и титруют 0,02 М раствором
серебра нитрата до появления фиолетовою
окрашивания.
1 мл 0,02 М раствора серебра нитрата co
ответствует 5,836 Mr C 17 H 21 NO'HCI.
МЕТОД 2
К 1 ,О мл 1 % испытуемоro раствора или 0,5 мл
2 % испытуемою раствора прибавляют 3 мл
хлороформа Р и титруют 0,02 М раствором
натрия 2идроксида при встряхивании до появ
ления розовоro окрашивания водною слоя, ис
пользуя в качестве индикатора раствор фенол
фталеина Р.
1 мл 0,02 М раствора натрия еидроксида
соответствует 5,836 Mr C 17 H 21 NO'HCI.
ДИМЕДРОЛА 1 %, 2 % РАСТВОРЫ
ДЛЯ ИНЪЕКЦИЙ (018)
СОСТАВ
Дифенrидрамина rидрохлорид (C 17 H 21 NO'HCI;
м. м. 291 ,8) 10,0 r; 20,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость со значе
нием рН от 5,0 до 6,5.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоro образца прибавляют
3 5 капель 0,1 М раствора кислоты хлористо
водородной, 5 капель раствора 10 r/л меди (11)
сульфата Р, &-------1 О капель раствора 1 О r/л
аммония тиоцианата Р. Появляется коричне
вое окрашивание.
В. 0,2 мл испытуемоro раствора выпари
вают на водяной бане досуха. После охлажде
ния к сухому остатку прибавляют 4 5 капель
кислоты серной Р. Появляется желтое OKpa
шивание, исчезающее при прибавлении 2 3
капель воды Р.
с. К 2 мл испытуемоro образца прибавляют
0,5 мл кислоты азотной разведенной Р2, 0,5 мл
раствора серебра нитрата Р1. Образуется
белый творожистый осадок, нерастворимый в
кислоте азотной разведенной Р2 и раствори
мый в растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
К 1,0 мл 1 % испытуемоro раствора или
0,5 мл 2 % испытуемою раствора прибавляют
1 2 капли раствора бромфенолов020 сине20
Р, по каплям кислоту уксусную Р до зеленовато
желтою окрашивания и титруют 0,02 М pacтвo
ром серебра нитрата до появления фиолетово
ro окрашивания.
1 мл 0,02 М раствора серебра нитрата co
ответствует 5,836 Mr C 17 H 21 NO'HCI.
МЕТОД 2
К 1,0 мл 1 % испытуемою раствора или 0,5 мл
2 % испытуемою раствора прибавляют з.......-4 мл
хлороформа Р и титруют 0,02 М раствором натрия
2идроксида при встряхивании до появления розо
вою окрашивания водною слоя, используя в каче
стве индикатора раствор фенолфталеина Р.
1 мл 0,02 М раствора натрия 2идроксида
соответствует 5,836 Mr C 17 H 21 NO'HCI.
ДИМЕДРОЛА И КАЛЬЦИЯ
rЛЮКОНАТА ПОРОШОК (019)
СОСТАВ
Дифенrидрамина rидрохлорид (C 17 H 21 NO'HCI;
м. м. 291 ,8) 0,005 r;
Димедрола порошок (021)
1023
Кальция rлюконат (С12Н22СаО14'Н20;
М. М. 448,4) 0,25 r;
Сахароза (С12Н22011; м. М. 342,3) [rлюкоза
моноrидрат (С6Н1Р6'НР; м. м. 198,2)] 0,1 r.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический
порошок.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
При использовании сахарозы в качестве Ha
полните ля: А, В, С, D.
При использовании 2ЛЮКОЗЫ в качестве Ha
полните ля: А, В, С, Е.
А. К 0,02 r испытуемоro образца прибавля
ют 2 3 капли кислоты серной Р. Появляется
желтое окрашивание, исчезающее при прибав
лени и нескольких капель воды Р.
В. К 0,02 r испытуемоro образца прибавля
ют 1 мл кислоты уксусной Р и наrревают до ки
пения. Полученный раствор дает реакцию (с) на
кальций (2.3.1)
С. К 0,02 r испытуемоro образца прибавляют
1 мл воды Р и 1 каплю раствора железа (11/) хпори
да Р1. Появляется светло зеленое окрашивание.
О. К 0,01 r испытуемою образца прибавляют
1 2 мл кислоты хпорисmoвoдoродной разведен
ной Р, несколько кристаллов резорцина Р и кипятят
в течение 1 мин. Появляется красное окрашивание.
Е. К 0,02 r испытуемоro образца прибавляют
1 мл воды Р, 1 мл реактива ФелиН2а Р и Harpe
вают до кипения. Образуется коричневато крас
ный осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Дифенrидрамина rиДрОХЛОрИД. К 0,5 r ис
пытуемоro образца прибавляют 5 мл воды Р, 2 мл
кислоты азотной разведенной Р, 3,0 мл 0,02 М
раствора серебра нитрата, 1 мл раствора
железа (11/) аммония сульфата Р5. Избыток 0,02 М
раствора серебра нитрата титруют 0,02 М pac
твором аммония тиоцианата до появления жел
товато розовоro окрашивания.
1 мл 0,02 М раствора серебра нитрата co
ответствует 5,836 Mr C 17 H 21 NO.HCI.
Кальция rлюконат. 0,05 r испытуемо
ro образца растворяют при наrревании в 5 мл
воды Р. Охлаждают, прибавляют 5 мл aMMи
аЧН020 буфеРН020 раствора рН 1 О, О Р, 3 5
капель раствора хромов020 тeMHo cиHe20 Р
и титруют 0,05 М раствором натрия эдетата
до появления сине фиолетовоro окрашивания.
1 мл 0,05 М раствора натрия эдетата co
ответствует 22,42 Mr С12Н22СаО14'Н20,
ДИМЕДРОЛА И ПАПАВЕРИНА
rИДРОХЛОРИДА ПОРОШОК (020)
СОСТАВ
Дифенrидрамина rидрохлорид (С 17 Н 21 NO' HCI;
М. м. 291,8) 0,005 r, 0,01 r, 0,015 r;
Папаверина rидрохлорид (C2oH21N04'HCI;
м. м. 375,9) 0,005 r, 0,01 r, 0,015 r;
rлюкоза моноrидpaт (С6Н1Р6'НР; м м 100,2)
0,2 r.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический
порошок.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 0,02 r испытуемоro образца прибавля
ют 2 3 капли кислоты серной Р. Появляется
желтое окрашивание, исчезающее при прибав
лении нескольких капель воды Р.
В. 0,05 r испытуемоro образца помещают
в фарфоровую чашку и смачивают 2 каплями
азотной кислоты Р. Появляется желтое OKpa
шивание, которое при наrревании на водяной
бане переходит в оранжевое.
С. К 0,05 r испытуемоro образца прибавляют
1 мл кислоты серной Р и наrревают. Появляется
фиолетовое окрашивание.
О. 0,02 r испытуемоro образца растворяют
в 0,5 мл воды Р, подкисляют кислотой азотной
разведенной Р и прибавляют 0,4 мл раствора
серебра нитрата Р1. Образуется белый TBO
рожистый осадок, растворяющийся в растворе
аммиака Р.
Е. К 0,02 r испытуемою образца прибавляют
1 мл воды Р, 1 мл реактива ФелиН2а Р и Harpe
вают до кипения. Образуется коричневато крас
ный осадок.
КОЛИЧЕСТВЕНОЕ ОПРЕДЕЛЕНИЕ
Сумма дифенrидрамина rидрохлорида
и папаверина rидрохлорида. 0,200 r испы
туемоro образца растворяют при наrревании
в воде Р. После охлаждения прибавляют 3
капли раствора бромфенолов020 сине20 Р, по
каплям кислоту уксусную разведенную Р до
появления зеленовато желтоro окрашивания и
титруют 0,02 М раствором серебра нитрата
до окрашивания осадка в сиреневато фиоле
товый цвет.
1 мл 0,02 М раствора серебра нитрата co
ответствует 6,677 Mr суммы дифенrидрамина rи
дрохлорида и папаверина rидрохлорида.
ДИМЕДРОЛА ПОРОШОК (021)
СОСТАВ
Дифенrидрамина rидpoхлорид (C 17 H 21 NO'HCI;
м. м. 291,8) 0,001 r, 0,002 r. 0,005 r;
Сахароза (С 12 Н 22 О" ; М. м. 342,3) [rлюкоза MO
ноrидрат (С 6 Н,Ре 'НР: М. м. 198.2)] 0,1 r; 0,2 r.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический
порошок.
1024
rосударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
При использовании сахарозы в качестве Ha
полните ля: А, В, С.
При использовании 2ЛЮКОЗЫ в качестве Ha
полнителя: А, D.
А. К 0,02 r испытуемоro образца прибавля
ют 2 3 капли кислоты серной Р. Появляется
желтое окрашивание, исчезающее при прибав
лени и нескольких капель воды Р.
В. К 0,02 r испытуемою образца прибавляют
3 5 капель воды Р, 2 капли раствора натрия
2идроксида разведеНН020 Р и 1 каплю раствора
50 r/л кобальта нитрата Р. Появляется сине
фиолетовое окрашивание.
С. К 0,01 r испытуемоro образца прибавля
ют 1 2 мл кислоты хлористоводородной раз
веденной Р, несколько кристаллов резорцина Р
и кипятят в течение 1 мин. Появляется красное
окрашивание.
о. К 0,02 r испытуемоro образца прибавля
ют 1 мл воды Р, 1 мл реактива Фелинеа Р ина
rревают до кипения. Образуется коричневато
красный осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
Навеску испытуемоro образца, COOTBeT
ствующую 5 Mr дифенrидрамина rидрохлорида,
растворяют в 2 3 мл воды Р, прибавляют 1 2
капли раствора бромфенолов020 сине20 Р, по
каплям кислоту уксусную Р до зеленовато жел
тоro окрашивания и титруют 0,02 М раствором
серебра нитрата до появления фиолетовоro
окрашивания.
1 мл 0,02 М раствора серебра нитрата co
ответствует 5,836 Mr C 17 H 21 NO'HCI.
МЕТОД 2
Навеску испытуемоro образца, COOTBeT
ствующую 5 Mr дифенrидрамина rидрохлори
да, растворяют в 2 3 мл воды Р, прибавляют
2 3 мл хлороформа Р и титруют 0,02 М pacтвo
ром натрия 2идроксида при встряхивании до по
явления розовою окрашивания водноro слоя,
используя в качестве индикатора раствор фе
нолфталеина Р.
1 мл 0,02 М раствора натрия 2идроксида
соответствует 5,836 Mr C 17 H 21 NO'HCI.
ЖИДКОСТЬ ПЕТРОВА
'КРОВЕЗАМЕЩАЮЩАЯ (022)
СОСТАВ
Натрия хлорид (NaCI; М. м. 58,44) 15,0 r;
Калия хлорид (KCI; М. м. 74,6) 0,2 r;
Кальция хлорид rексаrидрат (CaCI 2 '6HP;
м. м. 219,1) 1,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная прозрачная жидкость
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 2 3 мл испытуемоro образца упарива
ют до объема около 0,2 мл. Полученный paCT
вор дает реакцию (Ь) на калий (2.3.1).
В. 0,5 мл испытуемоro образца дают peaK
цИИ (Ь) и (с) на натрий (2.3.1).
С. 1 мл испытуемою образца дает peaK
цию (с) на кальций (2.3.1).
о. К 2 мл испытуемою образца прибавnя
ют 0,5 мл кислоты азотной разведенной Р2,
0,5 мл раствора серебра нитрата Р1. Обра
зуется белый творожистый осадок, HepaCTBO
римый в кислоте азотной разведенной Р2 и
растворимый в растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Кальция хлорид. К 2,0 мл испытуемоro
образца прибавляют 5 мл аммиаЧН020 буфер
. Н020 раствора рН 10,0 Р, 0,05 r индикаторной
смеси хромов020 тeMHo cиHeeo Р и титруют
0,05 М раствором натрия эдетата до появ
ления сине фиолетовоro окрашивания.
1 мл 0,05 М раствора натрия эдетата
соответствует 10,95 Mr CaCI 2 '6Hp.
Сумма хлоридов. 1,0 мл испытуемоro об
разца титруют О, 1 М раствором серебра пи
трата до оранжевоro окрашивания, исполь
зуя в качестве индикатора 0,5 мл раствора
калия хромата Р.
1 мл 0,1 М раствора серебра нитрата co
ответствует 6,034 Mr суммы хлоридов.
ЙОДА 1 %, 2 % РАСТВОРЫ (023)
СОСТАВ
Йод (12; М. м. 253,8) 10,0 r, 20,0 r;
Этиловый спирт 96 % до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная жидкость красно коричневоro
цвета с характерным запахом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
К 1 2 каплям испытуемоro образца при
бавляют 2 мл воды Р и 2 3 капли раствора
крахмала, свободН020 от йодидов, Р. Появля
ется синее окрашивание.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 2,0 мл 1 % или 1,0 мл 2 % испытуемоro
раствора прибавляют 0,8 мл раствора калия
йод ида Р и титруют О, 1 М раствором натрия
тиосульфата до обесцвечивания.
1 мл 0,1 М раствора натрия тиосульфата
соответствует 12,69 Mr 1.
Калия бромида и ма2ния сульфата раствор с 2ЛЮКОЗОЙ (026)
1025
ЙОДА 5 % РАСТВОР (024)
СОСТАВ
Йод (12; м. м. 253,8) 50.0 r;
Калия йодид (КI; м. м. 166,0) 20,0 r;
Вода очищенная и этиловый спирт 95 % по
ровну ДО 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная жидкость красно коричневоro
цвета с характерным запахом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 2 каплям испытуемоro образца при
бавляют 2 мл воды Р и 2 З капли раствора
крахмала, свободН020 от йодидов, Р. Появляет
ся синее окрашивание.
В. К 2 мл испытуемоro образца прибавляют
2 3 капли кислоты уксусной разведенной Р, 1 2
капли раствора натрия кобальтинитрита Р и
2 мл хлороформа Р. Хлороформный слой OKpa
шивается в фиолетовый цвет; в водном слое об
разуется желтый кристаллический осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Йод. 0,5 мл испытуемоro образца титру
ют 0,1 М раствором натрия тиосульфата до
обесцвечивания (раствор А) (V 1 , мл).
1 мл 0,1 М раствора натрия тиосульфата
соответствует 12,69 Mr 1.
Калия йодид. К раствору А, полученно
му при количественном определении йода,
прибавляют 2 мл воды Р, 0,5 мл кислоты YK
сусной разведенной Р, 2 капли раствора 1 r/л
эозина Н Р и титруют 0,1 М раствором cepe
бра нитрата до ярко розовоro окрашивания
осадка (V 2 , мл).
Количество 0,1 М раствора серебра Hи
трата, израсходованное на титрование калия
йод ида , рассчитывают по формуле V 2 V 1 .
1 мл 0,1 М раствора серебра нитрата co
ответствует 16,6 Mr КI.
ЙОДА 1 О % РАСТВОР (025)
СОСТАВ
Йод (12; М. м. 253,8) 100,0 r;
Этиловый спирт 95 % до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная жидкость красно коричневоro
цвета с характерным запахом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
К 1 2 каплям испытуемоro образца при
бавляют 2 мл воды Р и 2 3 капли раствора
крахмала, свободН020 от йодидов, Р. Появля
ется синее окрашивание.
129 30. 1712.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 2,0 мл испытуемоro образца прибавляют
8.0 мл раствора калия йодида Р и титруют О, 1 М
раствором натрия тиосульфата до обесцвечи
вания.
1 мл 0,1 М раствора натрия тиосульфата
соответствует 12,69 Mr 1.
КАЛИЯ БРОМИДА И МАrния
СУЛЬФАТА РАСТВОР С rлюкозой
(026)
СОСТАВ
Калия бромид (KBr; М. м. 119,0) 0,5 r;
Маrния сульфат rептаrидрат (MgS0 4 '7H 2 0;
м. м. 246,5) 0,5 r;
rлюкозы 10 % раствор 200 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоro образца прибавляют 2 мл
реактива ФелиН2а Р и наrpевают до кипения. Об--
разуется оранжево красный осадок.
В. 2 мл испытуемоro образца дают реакцию на
маrний (2.3.1).
с. 1 мл испытуемоro образца дает реакцию (а)
на сульфаты (2.3.1).
D. 1 мл испытуемоro образца подкисляют
кислотой азотной разведенной Р, прибавляют
0,4 мл раствора серебра нитрата Р1. Образу
ется светло желтый творожистый осадок.
Е. 5 мл испытуемоro образца дают реакцию (Ь)
на калий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Калия бромид. К 10,0 мл испытуемоro об
разца прибавляют 2 3 капли раствора бромфе
нолов020 сине20 Р, по каплям кислоту уксусную
разведенную Р до появления зеленовато желто
ro окрашивания и титруют О, 1 М раствором ce
ребра нитрата до появления фиолетовоro oкpa
шивания.
1 мл 0,1 М раствора серебра нитрата COOT
ветствует 11,90 Mr KBr.
Маrния сульфат. К 10,0 мл Иalытуемorо об
разца прибавляют 10 мл воды Р, 10 мл аммиачно--
20 буферн020 раствора рН 10,0 Р, 50 Mr иHдиKa
торной смеси протравнО2О ЧеРНО2О 11 Р и титруют
0,05 М раствором натрия здетата до перехода
окраски от фиолетовой до синей.
1 мл 0,05 М раствора натрия эдетата COOT
ветствует 12,32 Mr MgS04-7Нр.
rлюкоза
МЕТОД 1
1,0 мл испытуемоro образца помещают в
колбу со шлифом, прибавляют 10,0 мл 0,05 М
1026
rосударственная фармакопея Республики Беларусь
раствора йода и 0,5 мл раствора натрия 2и
дроксида Р. Колбу закрывают пробкой и Bыдep
живают в защищенном от света месте в течение
5 мин. Прибавляют 5 мл кислоты серной раз
веденной Р и титруют выделившийся ЙОД О, 1 М
раствором натрия тиосульфата до обесцве
чивания.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
9,01 Mr С 6 Н 1 Р6'
МЕТОД 2
Определяют показатель преломления
(2.2.6).
Содержание rлюкозы в про центах рассчиты
вают по формуле:
п (по + 0,00119. с, + 0,00130. c z )
0,00142
rдe:
п показатель преломления испытуемоro
образца;
по показатель преломления воды;
0,00130, 0,00119 и 0,00142 величины
прироста показателя преломления при увели
чении концентрации растворов на 1 °k калия
бромида, маrния сульфата и rлюкозы COOTBeT
ственно;
с, и c z концентрации калия бромида и
маrния сульфата соответственно, определен
ные в разделе «Количественное определе
ние», %.
КАЛИЯ ЙОДИДА 0,1 %, 0,25 %, 0,5 %,
1 %, 2 %, 3 %, 5 % РАСТВОРЫ (027)
СОСТАВ
Калия йодид (КI; м м. 166,0) 0,1 r, 0,25 r,
0,5 r, 1,0 r, 2,0 r, 3,0 r, 5,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец дает реакцию (а) на
калий (2.3.1)
В. Испытуемый образец дает реакцию (Ь) на
йодиды (2.3.1)
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
К 10,0 мл 0,1 %,5,0 мл 0,25 %, 2,0 мл 0,5 %,
1,0 мл 1 %, 2 %, 3 %, 5 % испытуемых растворов
прибавляют 5 капель кислоты уксусной раз
веденной Р и титруют О, 1 М раствором серебра
нитрата до окрашивания осадка в розовый цвет,
используя в качестве индикатора раствор 1 r/л
эозина Н Р.
1 мл 0,1 М раствора серебра нитрата co
ответствует 16,6 Mr КI.
МЕТОД 2
Используют только для 5 % раствора калия
йодида.
Определяют показатель преломления
(2.2.6); F кt . 5 %= 0,00130.
КАЛИЯ ЙОДИДА 0,25 %, 1 %, 3 %
РАСТВОРЫ (028)
СОСТАВ
КалИЯЙQQИД(КI; М. M.166,0) O,025r, 0,1 r. 0,3r;
Вода для инъеКЦИЙ до 10 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец дает реакции на
калий (2.3.1).
В. Испытуемый образец дает реакции (а) и
(Ь) на йодиды (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 5,0 мл 0,25 % испытуемоro раствора
либо к 1,0 мл 1 % или 3 % испытуемоro paCT
вора прибавляют 3 5 капель кислоты YK
сусной разведенной Р, 2 капли раствора 1 r/л
эозина Н Р и титруют 0,1 М раствором cepe
бра нитрата до окрашивания осадка в розо
ВЫЙ цвет.
1 мл 0,1 М раствора серебра нитрата co
ответствует 16,60 Mr КI.
КАЛИЯ ЙОДИДА 20 % РАСТВОР (029)
СОСТАВ
Калия йодид (КI; м м. 166,0) 20,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или слеrка желтова
тая жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец дает реакции на
калий (2.3.1).
В. Испытуемый образец дает реакции (а) и
(Ь) на йодиды (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определяют показатель преломления
(2.2.6); F кt . 20 %= 0,00130.
Калия йодида u эуфиллина раствор (031)
КАЛИЯ ЙОДИДА И НОВОКАИНА
РАСТВОР (030)
СОСТАВ
Калия йодид (КI; м. м. 166,0) 0,75 r;
Прокаина rидрохлорид (С'ЗН20N202'НСI;
М. м. 272,8) 4,0 r;
Вода очищенная 300 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 5 капель испытуемоro образца помеща
ют в пробирку, прибавляют 2 З капли pacт
вора кислоты хлористоводородной разве
денной Р, 3 капли раствора 100 r/л натрия
нитрита Р, 0,5 1 мл хлороформа Р и встря
хивают. Хлороформный слой окрашивается
в фиолетовый цвет. Водный слой отделяют и
используют для идентификации В.
В. Водный слой, полученный в идентифи
кации А, помещают в пробирку и прибавляют
1 2 мл раствора fЗ нафтола Р. Образуется
оранжево красный осадок.
С. 2 мл испытуемоro образца дают peaK
ции (а) и (Ь) на калий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Прокаина rидрохлорид
МЕТОД 1
К 2,0 мл испытуемоro образца прибавля
ют 1 мл кислоты хлористоводородной раз
веденной Р, 5 6 мл воды Р, 0,1 r калия бро
мида Р, 2 капли раствора тропеолина 00 Р,
1 каплю раствора метиленов020 сине20 Р и
про водят определение аминноro азота (2.5.8).
Параллельно про водят контрольный
опыт.
1 мл О, 1 М раствора натрия нитрита
соответствует 27,28 Mr С'ЗН2оN202'НСI.
МЕТОД 2
К 2,0 мл испытуемою образца прибав
ляют 5 7 мл смеси 96 % спирт Р хло
роформ Р (1 :2, об/об), нейтрализованной по
раствору фенолфталеина Р, 3 5 капель
раствора фенолфталеина Р и титруют О, 1 М
раствором натрия 2идроксида при встря
хивании до слабо розовоrо окрашивания BO
дноrо слоя (V,).
1 мл О, 1 М раствора натрия 2идроксида
соответствует 27,28 Mr С'ЗН2оN202.НСI.
Калия йодид. К 2,0 мл испытуемоro об
разца прибавляют 2 3 капли раствора бром
фенолов020 сине20 Р, по каплям кислоту YK
сусную разведенную Р до зеленовато желтоro
окрашивания и титруют О, 1 М раствором cepe
бра нитрата до появления сине фиолетовоrо
окрашивания осадка и раствора (V).
1027
Объем 0,1 М раствора серебра нитрата,
пошедшеro на титрование калия йодида, pac
считывают по формуле V 2 V,.
1 мл 0,1 М раствора серебра нитрата co
ответствует 16,6 Mr КI.
КАЛИЯ ЙОДИДА И ЭУФИЛЛИНА
РАСТВОР (031)
СОСТАВ
Калия йодид (КI; м. м. 166,0) 0,25 r, 0,5 r,
1,0 r, 3,0 r, 5,0 r;
Теофиллин этилендиамин (C2HaN2'(C7Ha02N4)2;
М. М. 420,4) от 0,04 r до 1,5 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или слеrка желтова
тая жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 З мл испытуемою образца при
бавляют 2 мл кислоты хлорuстоводород
ной разведенной Р и 2 мл раствора вoдo
рода пероксида концентрироваНН020 Р и
выпаривают на водяной бане. Выделяются
пары йода.
В. К охлажденному OCTaTK полученному
в идентификации А, прибавляют 2 5 капель
раствора аммиака разведеНН020 Р1. Появляет
ся фиолетово красное окрашивание.
С. 2 5 мл испытуемоro образца дают peaK
ции (а), (Ь) на калий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Теофиллин этилендиамин. Для лекар
ственной формы, содержащей от 0,04 r до 0,2 r
теофиллина этилендиамина, используют от
25,0 мл до 5,0 мл испытуемоro образца (или раз
веденные титрованные растворы); для лекар
ственной формы, содержащей от 0,2 r до 1,5 r Te
офиллина этилендиамина, используют от 5,0 мл
ДО 1,0 мл испытуемоro образца. Титруют 0,1 М
раствором кислоты хлористоводородной до
изменения окраски раствора от желтой до Kpac
ной, используя в качестве индикатора 1 2
капли раствора метиловО20 оранжев020 Р.
1 мл О, 1 М раствора кислоты хлористо
водородной соответствует 3,005 Mr C2HaN2' при
пересчете на теофиллин этилендиамин резуль
тат умножают на коэфициент 7.02 (COOTBeTCTBY
ет 14,25 % этилендиамина в субстанции).
Калия йодид. к 5.0 мл, 2,0 мл, 1,0 M/l или
0,5 мл испытуемоro раствора, содержащею
0.25 %. 0.5 %. 1 % или 3 % и 5 % калия йодида
соответственно. прибавляют 2 5 мл воды Р,
0.1.........Q.5 мл 0.1 М раствора аммония тиoциa
ната. 0. 2 мл раствора 100 r/л железа (111) aM
мония сульфата Р2 и титруют 0,1 М раствором
1028
rосударственная фармакопея Республики Беларусь
ОПИСАНИЕ {СВОЙСТВА)
Прозрачная бесцветная жидкость со значе
нием рН от 5,0 до 6,5.
серебра нитрата до исчезновения красноro
окрашивания.
1 мл 0,1 М раствора серебра нитрата co
ответствует 16,6 Mr КI.
КАЛИЯ ПЕРМАнrАНАТА 0,0125 %,
1 %, 5 % РАСТВОРЫ (032)
СОСТАВ
Калия перманrанат (КМп0 4 ; М. м. 158,0)
0,0125 r, 1,0 r, 5,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная фиолетовая (0,0125 % paCT
вор) или темно фиолетовая (1 % и 5 % раство--
ры) жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
К 0,5 мл 0,0125 % испытуемоro раствора
или к 2 каплям 1 % и 5 % испытуемых paCT
вора прибавляют 0,5 мл кислоты серной
разведенной Р и 0,5 мл раствора водорода
пероксида разведеНН020 Р. Раствор обесцве
чивается.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытуемый раствор. В зависимости от исход
ной концентрации roтовят следующие разведения.
0,0125 % раствор. Используют 20,0 мл
0,0125 % раствора.
1 % раствор. 10,0 мл испытуемоro образца
доводят водой Р до объема 100,0 мл. Использу
ют 5,0 мл полученноro раствора.
5 % раствор. 5,0 мл испытуемоro образца
доводят водой Р до объема 100,0 мл. Использу
ют 5,0 мл полученноro раствора.
Испытуемый раствор помещают в колбу
с притертой пробкой, прибавляют 2 мл paCT
вора 166 r/л калия йод ида Р и 1 мл кислоты
серной разведенной Р, закрывают пробкой,
смоченной раствором 166 r/л калия йодида Р.
Выдерживают в течение 1 О мин в защищенном
от света месте и титруют 0,1 М раствором
натрия тиосульФата до обесцвечивания, ис
пользуя в качестве индикатора раствор Kpax
мала, свободный от йодидов, Р.
1 мл 0,1 М раствора натрия тиосульФата
соответствует 3,161 Mr КМп0 4 .
КАЛИЯ ХЛОРИДА 4 %, 5 %, 7,5 %
РАСТВОРЫ (033)
СОСТАВ
Калия хлорид (KCI; М. м. 74,6) 40,0 r; 50,0 r;
75,0 r;
Вода для инъекций до 1000 мл.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 2 мл испытуемоro образца дают реакции
(а) и (Ь) на калий (2.3.1).
В. К 2 мл испытуемоro образца прибавляют
0,5 мл кислоты азотной разведенной Р2, 0,5 мл
раствора серебра нитрата Р1. Образуется
белый творожистый осадок, нерастворимый в
кислоте азотной разведенной Р2 и раствори
мый в растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
5,0 мл испытуемоro образца доводят
водой Р до объема 25,0 мл. 1,0 мл получен
ноro раствора титруют О, 1 М раствором cepe
бра нитрата до появления оранжево желтоro
окрашивания, используя в качестве индикато
ра 0,5 мл раствора калия хромата Р.
1 мл 0,1 М раствора серебра нитрата co
ответствует 7,46 Mr KCI.
КАЛЬЦИЯ ХЛОРИДА 1 %, 2 %, 2,5 %,
3 %, 5 %, 1 О % РАСТВОРЫ (034)
СОСТАВ
Кальция хлорид rексаrидрат (СаС'2'6Н20;
м. м. 219,1) 1,0 r, 2,0 r, 2,5 r. 3,0 r. 5.0 r. 10,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 1 мл испытуемоro образца дает реакцию
(с) на кальций (2.3.1).
В. К 2 мл испытуемоro образца прибавляют
0,5 мл кислоты азотной разведенной Р2, 0,5 мл
раствора серебра нитрата Р1. Образуется
белый творожистый осадок, нерастворимый в Kиc
лоте азотной разведенной Р2 и растворимый в
растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,5 мл 1 %, 2 %, 2,5 % или 3 % испы
туемоro раствора прибавляют 5 мл аммиачно
20 буфеРН020 раствора рН 10,0 Р, 2 3 капли
раствора хромов020 тeMHo cиHe20 Р и титру
ют 0,05 М раствором натрия эдетата до по
явления сине фиолетовоro окрашивания.
1 мл 0,05 М раствора натрия эдеmaта co
ответствует 10,95 Mr CaCI 2 '6Hp.
Определяют показатель преломления
5 % и 10 % растворов (2.2.6); FcacI2.5% = 0,00117;
F caCl ,10% = 0,00116.
Кардuоплеzический раствор NQ 1, NQ 2, Ng 3 (037)
1029
КАЛЬЦИЯ ХЛОРИДА 1 %, 10 %
РАСТВОРЫ ДЛЯ ИНЪЕКЦИЙ (035)
СОСТАВ
Кальция ХЛОРИД rексаrидрат (CaCI 2 '6HP;
м. м. 219,1) 10,0 r, 100,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость со значе
нием рН от 5,5 до 7,0.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 1 мл испытуемоro образца дает реакцию
(с) на кальций (2.3. 1).
В. К 2 мл испытуемоro образца прибавляют
0,5 мл кислоты азотной разведенной Р2, 0,5 мл
раствора серебра нитрата Р1. Образуется
белый творожистый осадок, нерастворимый в
кислоте азотной разведенной Р2 и раствори
мый в растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
КО,5мл 1 %испытуемоroраствора прибавляют
5 мл аммиаЧН020 буферН020 раствора рН 10, О Р,
2 З капли раствора хромов020 тeMHo cиHe
20 Р И титруют 0,05 М раствором натрия эде
тата до появления сине фиолетовоro окраши
вания.
1 мл 0,05 М раствора натрия эдетата co
ответствует 10,95 Mr CaCI 2 '6Hp.
МЕТОД 2
Используют только для 1 О % раствора каль
ция хлорида.
Определяют показатель преломления
(2.2.6); F CaCl2 .1O"/o= 0,00116.
КАЛЬЦИЯ ХЛОРИДА 50 % РАСТВОР
(036)
СОСТАВ
Кальция хлорид rексаrидрат (CaCI 2 '6Нр;
М. м. 219,1) 50,O r;
Вода очищенная до 1-00 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 1 мл испытуемоro образца дает реакцию
(с) на кальций (2.3.1).
В. К 1 мл испытуемоro образца прибавляют
0,5 мл кислоты азотной разведенной Р2, 0,5 мл
раствора серебра нитрата Р1. Образуется
белый творожистый осадок, нерастворимый в
lЗО За. 1712
кислоте азотной разведенной Р2 и раствори
мый в растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определяют показатель преломления
(2.2.6); Fcac/2.50%= 0,00108.
КАРДИОПЛЕrИЧЕСКИЙ РАСТВОР
N!! 1, N!! 2, N!! 3 (037)
СОСТАВ
Наименование Количество (r)
вещества Раствор Раствор Раствор
N21 N22 N!3
Маrния суль 1,92 1,92 1,92
фат rептаrидрат
(MgSO 4' 7НР;
М. м. 246,5)
Натрия хлорид 7,БJ3 7,66 7,66
(NaCI; М. м.
58,44 )
Калия хлорид 8.0 4,5 8,0
(KCI; М. м.
74,56)
rлюкоза безвод 14,5 8,0 8,0
ная (С 6 Н 12 О 6 ;
М. м. 180.2)
Кальция хлори 0,44 мл 0,44 мл 0,44 мл
да 50 % раствор
Вода для инъек до 1000 до 1000 до 1000
ций мл мл мл
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость со значе
нием рН от З,8 до 6,5.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоro образца прибавляют
2 мл реактива ФелиН2а Р и наrревают до кипе
ния. Образуется оранжево красный осадок.
В. 0,5 мл испытуемоro образца дают peaK
цию на маrний (2.3.1).
С. 0,5 мл испытуемоro образца даЮ1 peaK
цию (а) на сульфаты (2.3.1).
О. 5 мл испытуемоrо образца дают реакцию
(а) на натрий (2.3.1).
Е. 1 мл испытуемоro образца дает реакцию
(Ь) на калий (2.3.1).
F. 2 мл испытуемоro образца дают реакцию
(с) на кальций (2.3.1).
G. 1 мл испытуемоro образца подкисляют
кислотой азотной разведенной Р, прибавляют
0,4 мл раствора серебра нитрата Р1. Образу
ется белый творожистый осадок, растворимый в
растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Кальция хлорид. К 10,0 мл испытуемоro об
разца прибавляют 4 мл раствора натрия 2идpOK
сида Р, 20 Mr индикаторной смеси мурексида Р
1030
rосударственная фармакопея Республики Беларусь
и титруют 0,05 М раствором натрия эдетата
(V 1 ) до появления фиолетовоro окрашивания.
1 мл 0,05 М раствора натрия эдетата co
ответствует 10,95 Mr CaCI 2 '6Hp.
Маrния сульфат. К 5,0 мл испытуемоro об
разца прибавляют 5 мл аммиачН020 буфеРН020
раствора рН 10,0 Р, 50 Mr индикаторной смеси
протравН020 чеРН020 11 Р и титруют 0,05 М pac
твором натрия эдетата (V) до перехода OKpa
ски от фиолетовой до синей.
1 мл 0,05 М раствора натрия эдетата co
ответствует 12,32 Mr MgS0 4 '7Hp.
Содержание маrния сульфата в rpaMMax
рассчитывают по формуле:
( V 1 V ) .1232
2 2 1 '
5,0
Сумма хлоридов калия и натрия. 1,0 мл
испытуемоro образца титруют О, 1 М раствором
серебра нитрата до появления желтовато--ко--
ричневоro окрашивания, используя в качестве ин
дикатора раствор калия хромата Р (V).
1 мл 0,1 М раствора серебра нитрата co
ответствует 6,67 Mr суммы хлоридов калия и
натрия для испытуемых растворов NQ 1 и NQ 3
или 6,44 Mr суммы хлоридов калия и натрия для
испытуемоrо раствора NQ 2.
Содержание суммы хлоридов калия и
натрия в испытуемых растворах NQ 1 и NQ 3 pac
считывают по формуле:
( V 1 V ) . 6 67 .1000
з 10 1 '
1,0
Содержание суммы хлоридов калия и
натрия в испытуемом растворе NQ 2 рассчитыва
ют по формуле:
( V 1 V j '- 6 44 .1000
з 10 1 '
1,0
rлюкоза. 1,0 мл испытуемоro образца поме
щаютв колбу со шлифом, прибавляют3,0 мл 0,05М
раствора йода и 0,3 мл раствора натрия 2идpOK
сида Р. Колбу закрывают пробкой и выдерживают
в защищенном от света месте в течение 5 мин.
Прибавляют 5 мл кислоты серной разведенной
Р и титруют выделившийся йод О, 1 М раствором
натрия тиосульфата до обесцвечивания.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
9,01 Mr С 6 Н 12 О 6 .
КОЛЛАрrол с rЛИЦЕРИНОМ (038)
СОСТАВ
Серебро коллоидное для наружноro приме
нения (колларroл) 0,3 r;
rлицерин 85 % 39,0 r.
ОПИСАНИЕ (СВОЙСТВА)
rустая жидкость темно коричневоro цвета.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 2 3 каплям испытуемоro образца при
бавляют 1 2 капли раствора водорода перок
сида концентрироваНН020 Р. Выделяются пу
зырьки rаза и образуется обильная пена.
В. Несколько капель испытуемоro образца
выпаривают и наrревают до обуrливания. Появ
ляется запах >юкенноro pora.
С. К 5------6 каплям испытуемоro образца при
бавляют 1 мл воды Р и 2 3 капли кислоты хло
ристоводородной разведенной Р. Образуется
темно коричневый осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
К 2,00 r испытуемоro образца прибавля
ют 1 мл воды Р, 0,5 мл кислоты азотной раз
веденной Р, 0,5 мл раствора железа (111) aM
мония сульфата Р2 и наrревают на водяной
бане до обесцвечивания. Охлаждают и титру
ют О, 1 М раствором аммония тиоцианата до
появления желтовато розовоro окрашивания.
1 мл 0,1 М раствора аммония тиоцианата
соответствует 15,41 Mr колларrола.
МЕТОД 2
К 2,00 r испытуемоro образца прибавляют
1 2 капли кислоты уксусной разведенной Р,
5,0 мл 0,05 М раствора йода и встряхивают в
течение 2 3 мин. Избыток йода титруют 0,1 М
раствором натрия тиосульфата, используя
в качестве индикатора раствор крахмала, cвo
бодный от йодидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
15,41 Mr колларroла.
КОЛЛАРrОЛА 1 %, 2 %, 3 %
РАСТВОРЫ (039)
СОСТАВ
Серебро коллоидное для наружноro приме
нения (колларroл) 0,1 r, 0,2 r, 0,3 r;
Вода очищенная до 1 О мл.
ОПИСАНИЕ (СВОЙСТВА)
Жидкость темно коричневоro цвета.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 2 3 каплям испытуемоro образца при
бавляют 1 2 капли раствора водорода перок
сида концентрироваНН020 Р. Выделяются пу
зырьки rаза и образуется обильная пена.
Маzния сульфата 1 %, 5 %, 14 %, 25 %, ЗЗ % растворы (041)
1031
В. К 2 3 каплям испытуемоro образца при
бавляют 1 О капель кислоты азотной Р, Ha
rревают до обесцвечивания, прибавляют 1 2
капли кислоты хлористоводородной разведен
ной Р. Появляется белый осадок.
С. Несколько капель испытуемоro образца
выпаривают и наrревают до обуrливания. Появ
ляется запах жженноro pora.
О. К 5 6 каплям испытуемоro образца при
бавляют 1 мл воды Р и 2 3 капли кислоты хло
ристоводородной разведенной Р. Образуется
темно коричневый осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
К 2,0 мл 1 %,1,0 мл 2 % или 0,5 мл 3 % испы
туемоro раствора прибавляют 1 мл воды Р, 0,5 мл
кислоты азотной разведенной Р, 0,5 мл раствора
железа (111) аммония сульфата Р2 и наrревают на
водяной бане до обесцвечивания. Охлаждают и ти
труют 0,1 М раствором аммония тuоцианата до
появления желтовато розовоro окрашивания.
1 мл 0,1 М раствора аммония тиоцианата
соответствует 15,41 Mr колларroла.
МЕТОД 2
К 1,0 мл испытуемоro образца прибавляют
1 2 капли кислоты уксусной разведенной Р,
5,0 мл 0,05 М раствора йода и встряхивают в
течение 2 3 мин. Избыток йода титруют 0,1 М
раствором натрия тиосульфата, используя
в качестве индикатора раствор крахмала, cвo
бодный от йодидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
15,41 Mr колларrола.
КОЛЛАРrОЛА 1 %, 2 %, 3 %, 5 %
РАСТВОРЫ (040)
СОСТАВ
Серебро коллоидное для наружноro приме
нения (колларroл) 1,0 r, 2,0 r, 3,0 r, 5,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Жидкость темно коричневоro цвета.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 2 3 каплям испытуемоrо образца
прибавляют 1 2 капли раствора вoдopo
да пероксида концентрироваНН020 Р. Bыдe
ляются пузырьки rаза и образуется обильная
пена.
В. К 2 3 каплям испытуемоro образца при
бавляют 1 О капель кислоты азотной Р, Ha
rревают до обесцвечивания, прибавляют 1 2
капли кислоты хлористоводородной разведен
ной Р. Появляется белый осадок.
С. Несколько капель испытуемоro образца
выпаривают и наrревают до обуrливания. Появ
ляется запах жженноro pora.
О. К 5 каплям испытуемоro образца при
бавляют 1 мл воды Р и 2 3 капли кислоты хло
ристоводородной разведенной Р. Образуется
темно коричневый осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
К 2,0 мл 1 %, 1,0 мл 2 % или 0,5 мл 3 % и 5 %
испытуемых растворов прибавляют 1 мл воды Р,
0,5 мл кислоты азотной разведенной Р, 0,5 мл
раствора железа (111) аммония сульфата Р2 и
наrpeвают на водяной бане до обесцвечивания.
Охлаждают и титруют 0,1 М раствором аммония
тиоцианата до появления желтовато розовоro
окрашивания.
1 мл 0,1 М раствора аммония тиоцианата
соответствует 15,41 Mr колларroла.
МЕТОД 2
К 1,0 мл испытуемоro образца прибавляют
1 2 капли кислоты уксусной разведенной Р,
5,0 мл 0,05 М раствора йода и встряхивают в
течение 2 3 мин. Избыток йода титруют 0,1 М
раствором натрия тиосульфата, используя в
качестве индикатора раствор крахмала, cвo
бодный от йодидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
15,41 Mr колларroла.
МАrния СУЛЬФАТА 1 %, 5 %, 14 %,
25 %, ЗЗ % РАСТВОРЫ (041)
СОСТАВ
Маrния сульфат rептаrидрат (MgSO . 7Н,0:
М. м. 246,5) 1,0 r, 5,0 r, 14,0 r, 25,0 r. 33,0 r:
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 5 мл испытуемоrо образца дают реакции
на сульфаты (2.3.1).
В. 2 мл испытуемоro образца дают реакцию
на маrний (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
Испытуемый раствор. В зависимости от
исходной концентрации roтовят следующие раз
ведения.
1 % и 5 % растворы. К 1,0 мл испытуемоro
образца прибавляют 3 мл аммиаЧН020 буферно
20 раствора рН 10.0 Р.
1032
rосударственная фармакопея Республики Беларусь
14 %, 25 % и зз % растворы. 1,0 мл ис
пытуемоro образца доводят водой Р до объема
50,0 мл. К 5,0 мл полученноro раствора прибав
ляют 5 мл аммиаЧН020 буфеРН020 раствора рН
10,ОР.
Испытуемый раствор титруют при энерrич
ном перемешивании 0,05 М раствором натрия
эдетата до появления синеro окрашивания,
используя в качестве индикатора проrправной
черный 11 Р.
Параллельно про водят контрольный опыт.
1 мл 0,05 М раствора натрия эдетата co
ответствует 12,32 Mr MgS0 4 '7Hp.
МЕТОД 2
Используют только для растворов маrния
сульфата 5 %, 14 %, зз %.
Определяют показатель преломления
(2.2.6).
F M9 so.. 5 % = 0,00095;
FМgSO..14% = 0,00093;
FM9SO..25% = 0,00089;
FМ9SО..зз% = 0,00086.
МАrния СУЛЬФАТА 25 %, ЗЗ %
РАСТВОРЫ ДЛЯ ИНЪЕКЦИЙ (042)
СОСТАВ
Маrния сульфат rептаrидрат (MgSO 4' 7Нр;
М. м. 246,5) 250,0 r, 330,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость со значе
нием рН от 6,2 до 8,0.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 5 мл испытуемоro образца дают реакции
на сульфаты (2.3.1).
В. 2 мл испытуемоro образца дают реакцию
на маrний (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
1,0 мл испытуемоro образца доводят
водой Р до объема 50,0 мл. К 5,0 мл получен
ноro раствора прибавляют 5 мл аммиаЧН020
буферН020 раствора рН 10,0 Р и титруют при
энерrичном перемешивании 0,05 М p8cтвo
ром натрия эдетата до появления синеro
окрашивания, используя в качестве индикато
ра протравной черный 11 Р.
Параллельно проводят контрольный
опыт.
1 мл 0,05 М раствора натрия эдетата
соответствует 12,32 Mr MgS0 4 '7Hp.
МЕТОД 2
Определяют показатель преломления
(2.2.6).
FMgSO..250/,= 0,00089;
FМ9SО..зз%= 0,00086.
НАТРИЯ БРОМИДА 1 %,2 %, 3 %
РАСТВОРЫ (043)
СОСТАВ
Натрия бромид (NaBr; М. м. 102,9) 1,0 r;
2,0 r; 3,0 r;
Вода очищенная дo 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец дает реакцию (с) на
бромиды (2.3.1).
В. Испытуемый образец дает реакции (а) и
(с) на натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,5 мл испытуемоro образца при
бавляют 2 мл воды Р, 2 3 капли pacт
вора бромфенолов020 сине20 Р и по каплям
кислоту уксусную разведенную Р до по
явления зеленовато желтоro окрашива
ния. Полученный раствор титруют 0,1 М
раствором серебра нитрата до окрашива
ния осадка в фиолетовый цвет.
1 мл 0,1 М раствора серебра нитрата co
ответствует 10,29 Mr NaBr.
НАТРИЯ БРОМИДА 3 % РАСТВОР С
НАСТОЙКОЙ ВАЛЕРИАНЫ (044)
СОСТАВ
Натрия бромид (NaBr; М. м. 1 02,9) 3,0 r;
Вода очищенная до 100 мл;
Настойка валерианы 8 мл или 1 О мл.
ОПИСАНИЕ (СВОЙСТВА)
Коричневатая жидкость со специсрическим
запахом.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец дает реакцию (с) на
бромиды (2.3.1).
В. Испытуемый образец дает реакции (а) и
(с) на натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,5 мл испытуемоro образца прибавля
ют 2 мл воды Р, 2 3 капли раствора бром
Натрия бромида и аскорбиновой кислоты раствор с zлюкозой (046)
1033
феноловоzо сине20 Р и по каплям кислоту
уксусную разведенную Р до появления зеле
новато желтоrо окрашивания. Полученный
раствор титруют О, 1 М раствором серебра
нитрата до окрашивания осадка в фиолето
вый цвет.
1 мл 0,1 М раствора серебра нитрата co
ответствует 10,29 Mr NaBr.
НАТРИЯ БРОМИДА 20 % РАСТВОР
(045)
СОСТАВ
Натрия бромид (NaBr; М. м. 102,9) 20,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец дает реакцию (с) на
бромиды (2.3.1).
В. Испытуемый образец дает реакции (а) и
(с) на натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
5,0 мл испытуемоro образца доводят водой Р
до объема 100,0 мл. 5,0 мл полученноrо paCT
вора титруют 0,1 М раствором серебра Hитpa
та до оранжево желтоro окрашивания, исполь
зуя в качестве индикатора калия хромат Р.
1 мл 0,1 М раствора серебра нитрата co
ответствует 10,29 Mr NaBr.
МЕТОД 2
Определяют показатель преломления
(2.2.6); F NaBr .20%= 0,00130.
НАТРИЯ БРОМИДА
И АСКОРБИНОВОЙ КИСЛОТЫ
РАСТВОР С rлюкозой (046)
СОСТАВ
Натрия бромид (NaBr; М. м. 102,9) 6,0 r;
Аскорбиновая кислота (С 6 Н в О б ;
м. М. 176,1) 8,0 r;
rлюкоза моноrидрат (С6Н1206'Н20; М. М.
198,2) 80,0 r;
Вода очищенная 420 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемою образца прибавляют
2 мл воды Р, 2 3 капли раствора водорода пе
131 За". 1712.
роксида концентрироваНН020 Р, кипятят в тече
ние 2 мин, прибавляют 1 мл реактива ФелиН2а Р
и наrревают до кипения. Образуется оранжево
красный осадок.
В. 4 5 капель испытуемоro образца выпа
ривают на водяной бане, охлаждают, прибав
ляют 0,02 r тимола Р, 5---------6 капель кислоты
серной Р и 1 2 капли воды Р. Появляется Kpac
но фиолетовое окрашивание.
с. К 1 2 каплям испытуемоro образца при
бавляют 1 2 капли раствора серебра Hитpa
та Р1. Образуется светло желтый осадок, по
степенно приобретающий серый оттенок.
D. К 1 мл испытуемоro образца прибавляют
1 мл хлороформа Р, 1 О капель раствора хло
рамина Р и встряхивают. Хлороформный слой
окрашивается в желто коричневый цвет.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Натрия бромид. К 1,0 мл испытуемою paCT
вора прибавляют 0,5 мл кислоты азотной разве
деннойР, 1 млрастворажелеза(III)аммониясуль
фата Р5 и перемешивают. Прибавляют О, 1 мл (V o )
0,1 М раствора аммония роданида и титруют
0,1 М раствором серебра нитрата (у) до ис
чезновения красноro окрашивания.
1 мл 0,1 М раствора серебра нитрата co
ответствует 10,29 Mr NaBr.
Содержание натрия бромида в rpэммах pac
считывают по формуле:
(V V o ).10,29 . k . 478 ,
1000
rдe:
k поправочный коэффициент к МОЛЯРН(r
сти титрованноro раствора;
4 78 объем roтовой лекарственной формы,
рассчитанный с учетом коэффициентов увели
чения объема (#6.1.1).
Аскорбиновая кислота. 1,0 мл испытуемоro
образца помещают в колбу со шлифом и титруют
0,05 М раствором йода (V 1 ) до появления слабо
желтоro окрашивания (раствор А. сохраняют для
количественноro определения rлюкозы).
1 мл 0,05 М раствора йода соответствует
8,81 Mr С 6 НР6 .
rлюкоза
МЕТОД 1
К раствору А, полученному при количе
ственном определении аскорбиновой кисло
ты, прибавляют 25,0 мл 0,05 М раствора йода
и 1 мл раствора натрия 2идроксида Р. Колбу
закрывают пробкой и выдерживают в защи
щенном от света месте в течение 5 мин. При
бавляют 5 мл кислоты серной разведенной Р
и титруют выделившийся йод О, 1 М pacтвo
ром натрия тиосульфата (V 2 ) до обесцвечи
вания.
1034
rосударственная фармакопея Республики Беларусь
Параллельно проводят контрольный опыт (V K ).
1 мл 0,05 М раствора йода соответствует
9,91 Mr С6Н1206'Н20,
Содержание rлюкозы моноrидрата в rpaM
мах рассчитывают по формуле:
(V K Vl V2).9,01.478
1000
МЕТОД 2
Определяют показатель преломления (2.2.6).
Содержание rлюкозы моноrидрата в rpaM
мах рассчитывают по формуле:
[п (по + 0,00130. С 1 +0,00160. с 2 )]. 478 ,
0,00129.100
rдe:
п показатель преломления испытуемоro
образца;
по показатель преломления воды:
0,00130, 0,00160 и 0,00129 величины
прироста показателя преломления при увеличе
нии концентрации растворов на 1 % натрия бро
мида, аскорбиновой кислоты и rлюкозы моноrи
драта соответственно;
С 1 и С 2 концентрации натрия бромида и
аскорбиновой кислоты соответственно, опреде
ленные в разделе «Количественное определе
ние», %.
НАТРИЯ БРОМИДА И МАrния
СУЛЬФАТА РАСТВОР С rлюкозой
(047)
СОСТАВ
Наимено- Количество
вание N!!1 N!!2 N!!З N!!4 N!!5 N!!6 N!!7
Натрия
бромид 0,5r 1,5 r 1,0 r 0,6r O,7r 4,Or 2,Or
(NaBr; М. м.
102,9)
Маrния
сульфат
rептаrидрат 0,25 0,5r 2,Or 1,5 r 1.7 r 2,Or 5,Or
(MgS0 4 ' r
7НР; М. м.
246,5)
rлюкозы
раствор 5% 5% 10% 10% 10% 10% 10%
200 мл
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоro образца прибавляют
2 мл реактива ФелиН2а Р и наrревают до кипе
ния. Образуется оранжево красный осадок.
В. 1 мл испытуемых растворов NQ 1 и NQ 2
или 0,5 мл испытуемых растворов NQNQ 3 7
дают реакцию на маrний (2.3.1).
С. 0,5 мл испытуемоro образца дают peaK
цию (а) на сульфаты (2.3.1).
О. 5 мл испытуемоro образца дают реакцию
(а) на натрий (2.3.1).
Е. 1 мл испытуемоro образца подкисляют
кислотой азотной разведенной Р, прибавляют
0,4 мл раствора серебра нитрата Р1. Образу
ется светло желтый творожистый осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Натрия бромид. К 5,0 мл испытуемоro paCT
вора NQN2 1 5 или 1,0 мл испытуемых paCTBO
ров NQ 6 и NQ 7 прибавляют 2 3 капли раствора
бромфенолoвozo сине20 Р, по каплям кислоту
уксусную разведенную Р до появления зелено
вато-желтоro окрашивания и титруют 0,1 М pac
твором серебра нитрата до появления фиоле
TOВOro окрашивания.
1 мл 0.1 М раствора серебра нитрата co
ответствует 10,29 Mr NaBr.
Маrния сульфат. К 10,0 мл испытуемых pac
творов NQ 1 и NQ 2 или 2,0 мл испытуемых paCTBO
ров NQNQ 3 7 прибавляют 10 мл воды Р, 10 мл
аммиаЧН020 буфеРН020 раствора рН 10,0 Р,
50 Mr индикаторной смеси протравН020 чер
Н020 11 Р и титруют 0,05 М раствором натрия
эдетата до перехода окраски от фиолетовой до
синей.
1 мл 0,05 М раствора натрия эдетата co
ответствует 12,32 Mr MgS0 4 '7Hp.
rлюкоза
МЕТОД 1
1,0 мл испытуемоro раствора NQ 1 и NQ 2 или
0,5 мл испытуемых растворов NQNQ 3 7 помеща
ют в колбу со шлифом, прибавляют 10,0 мл 0,05 М
раствора йода и 0,5 мл раствора натрия 2идpOK
сида Р. Колбу закрывают пробкой и выдерживают
в защищенном от света месте в течение 5 мин.
Прибавляют 5 мл кислоты серной разведенной Р
и титруют выделившийся йод О, 1 М раствором
натрия тиосульфата до обесцвечивания.
Параллельно про водят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
9,01 Mr С Б Н 12 О Б .
МЕТОД 2
Определяют показатель преломления (2.2.6).
Содержание rлюкозы в процентах рассчиты
вают по формуле:
п (по + 0,00090. С 1 т 0,00130. С 2 )
0,00142
rдe:
П показатель преломления испытуемоro
образца;
по показатель преломления воды;
Натрия тетраборат с 2лицерином (050)
1035
0,00130,0,00090 и 0,00142 величины при
роста показателя преломления при увеличении
концентрации растворов на 1 % натрия броми
да, маrния сульфата и rлюкозы соответственно;
С 1 и С 2 концентрации натрия бромида и
маrния сульфата соответственно, определенные
в разделе «Количественное определение», %.
НАТРИЯ rИДРОКАРБОНАТА 4 %
РАСТВОР для ИНЪЕКЦИЙ (048)
СОСТАВ
Натрия rидрокарбонат (NаНСО з ; М. м.
84,0) 40,0 r;
Вода для инъекций дo 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость со значе
нием рН от 8,1 до 8,9.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 2 мл испытуемою образца дают реакции
(а) и (с) на натрий (2.3.1).
В. К 3 5 каплям испытуемоrо образца при
бавляют 2 3 капли кислоты уксусной разведен
ной Р. Наблюдается бурное выделение пузырьков
rаза.
с. К 4 5 каплям испытуемоro образца при
бавляют 5 капель насыщенноro раствора ма2ния
сульфата Р (к 100 r ма2ния сульфата Р прибав
ляют 100 мл воды Р, встряхивают в течение 24 ч и
фильтруют) и кипятят. Образуется белый осадок.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
1,0 мл испытуемоro образца титруют 0,1 М
раствором кислоты хлористоводородной до по
явления розовоro окрашивания, используя в Ka
честве индикатора раствор метилов020 оранже
в020 Р.
1 мл О, 1 М раствора кислоты хлористово
дородной соответствует 8,40 Mr NаНСО з .
МЕТОД 2
Определяют по казатель
(2.2.6); F Na CD,.4% = 0,00125.
преломления
НАтрияrИДРОКАРБОНАТА
4 % РАСТВОР ДЛЯ ИНЪЕКЦИЙ
СТАБИЛИЗИРОВАННЫЙ (049)
СОСТАВ
Натрия rидрокарбонат(NаНСО з ; М. м. 84,O)
40,0 r;
Динатрия эдетат (C1CH14N Na208'2H O: М. м.
372,2) 0,2 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость со значе
нием рН от 8,1 до 8,9.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 2 мл испытуемоro образца дают реакции
(а) и (с) на натрий (2.3.1).
В. К 3 5 каплям исnытуемоro образца при
бавляют 2 3 капли кислоты уксусной разве
денной Р. Наблюдается бурное выделение пу
зырьков rаза.
с. К 4 5 каплям испытуемоro образца при
бавляют 5 капель насыщенноro раствора ма2ния
сульфата Р (к 100 r ма2ния сульфата Р при
бавляют 100 мл воды Р, встряхивают в течение
24 ч и фильтруют) и кипятят. Образуется белый
осадок.
О. Испытуемый образец выдерживает Tpe
бование раздела «Количественное определе
ние. Динатрия эдетат».
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Натрия rидрокарбонат
МЕТОД 1
1,0 мл испытуемоro образца титруют 0,1 М
раствором кислоты хлористоводородной до
появления розовою окрашивания, используя
в качестве индикатора раствор метилов020
оранжев020 Р.
1 мл О, 1 М раствора кислоты хлористово
дородной соответствует 8.40 Mr NаНСО з .
МЕТОД 2
Определяют показатель
(2.2.6); F Nащ 4', = 0,00125.
преломления
Динатрия эдетат. К 5,0 мл испытуемоro
образца прибавляют 1 каплю раствора Meти
ловО20 оранжев020 Р и нейтрализуют кисло
той хлористоводородной разведенной Р, при
бавляя ее постепенно, небольшими порциями
до появления розовоro окрашивания. Раствор
перемешивают до прекращения выделения пу
зырьков rаза, прибавляют 1 мл аммuаЧН020 бу
феРН020 раствора рН 10,0 Р, 0,01 r иHдиKaтop
ной смеси протравН020 чеРН020 11 Р и титруют
0,1 М раствором сульфата цинка до измене
ния окраски раствора от сине зеленой до фио
летовой.
1 мл О, 1 М раствора цинка сульфата соот
ветствует 3,722 Mr C. H. N:Na208 '2Н 2 О.
НАТРИЯ ТЕТРАБОРАТ
С rЛИЦЕРИНОМ (050)
СОСТАВ
Натрия тетраборат декаrидрат
(Na,Bp7'10Hp: М. м. 381,4) 10,0 r;
rлицерин 85 % 90 r.
1036
rосударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная почти бесцветная сиропоо
бразная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоrо образца прибав
ляют 5 6 капель кислоты серной Р, 1 2 мл
96 % спирта Р и поджиrают. Пламя окаймле
но зеленым цветом.
В. К 2 3 каплям испытуемоro образца
прибавляют 4 5 капель раствора натрия
2идроксида разведеНН020 Р и 4 5 капель
раствора меди (11) сульфата Р. Появляется
интенсивное синее окрашивание.
С. 3 мл испытуемоro образца дают peaK
цию (с) на натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,5 r испытуемоro образца прибавля
ют 5 мл воды Р и титруют О, 1 М раствором
кислоты хлористоводородной до появления
розовоro окрашивания, используя в качестве
индикатора раствор метилов020 оранжево
20 Р.
1 мл 0,1 М раствора кислоты хло
ристоводородной соответствует 19.07 Mr
Na 2 B 4 0 7 '10H 2 0.
НАТРИЯ ТЕТРАБОРАТА И НАТРИЯ
rИДРОКАРБОНАТА РАСТВОР (051)
СОСТАВ
Натрия тетраборат декаrидрат
(Na2Bp7'lOHp; М. м. 381,4) O,1 r;
Натрия rидрокарбонат (NаНСО з ; М. м.
84,0) 0,1 r;
Вода очищенная до 1 О мл.
ОПИСАНИЕ (СВОЙСТВА)
Бесцветная прозрачная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 3 5 каплям испытуемоro образца при
бавляют 2 3 капли кислоты хлорuстоводород
ной разведенной Р. Выделяются пузырьки rаза.
В. 1 мл испытуемоrо образца помещают в
фарфоровую чашку и выпаривают на водяной
бане. К сухому остатку прибавляют 5 6 капель
кислоты серной разведенной Р, 1 2 мл 96 %
спирта Р и поджиrают. Пламя окаймлено зеле
ным цветом.
С. 3 мл испытуемоro образца дают реакцию
(Ь) на натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 1,0 мл испытуемоrо образца прибавляют
2 3 мл воды Р и титруют О, 1 М раствором KUC
лоты хлористоводородной до появления розо
воro окрашивания (А, мл), используя В качестве
индикатора 0,05 мл раствора метилов020 opaH
жев020 Р. Опитрованный раствор наrревают на
водяной бане в течение 10 мин. После охлаж
дения прибавляют 5--------6 мл 2лицерина (85 %) Р,
предварительно нейтрализованноro по pacтвo
ру фенолфталеина Р, и титруют 0,1 М pacтвo
ром натрия 2UдpoKcидa до появления розовоro
окрашивания (В, мл).
1 мл 0,1 М раствора натрия 2идроксида co
ответствует 9,54 Mr Na 2 B 4 0 7 '1 ОН 2 О.
Количество О, 1 М раствора кислоты хло
ристоводородной, израсходованноro на титро
вание натрия rидрокарбоната, раССЧИ'fывают по
формуле:
в
A '
2
1 мл 0,1 М раствора кислоты хлористово
дородной соответствует 8,4 Mr NаНСО з .
НАТРИЯ ТИОСУЛЬФАТА 0,25 %, 2 %,
10 %,15 %,20 %, 30 % РАСТВОРЫ (052)
СОСТАВ
Натрия тиосульфат пентаrидрат
(Nа2Sрз'5Нр; М. м. 248,2) 0,25 r, 2,0 r, 10,0 r,
15,0 r, 20,0 r. 30,0 r;
Вода очищенная дo 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 1 мл испытуемоro образца дает реакции
(Ь) и (с) на натрий (2.3.1).
В. К 0,5 2 мл испытуемоro образца прибав
ляют 1 мл кислоты хлористоводородной Р. По
является осадок серы и выделяется rаз, окраши
вающий в синий цвет йодкрахмальную бума2У Р.
С. К 0,5 1 мл испытуемоro образца прибав
ляют 2 мл раствора серебра нитрата Р2. Об
разуется белый осадок, окраска котороro быстро
переходит в желтоватую, а затем в черную.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Испытуемый раствор. В зависимости от ис
ходной концентрации roтовят следующие разве
дения:
0,25 % раствор: используют 5,0 мл испыту
емоro образца;
2 % раствор: используют 1,0 мл испыту
емоro образца;
1 О % и 15 % растворы: 2,0 мл испытуемоro
образца доводят водой Р до оБЪf ма 25,0 мл; ис
пользуют 2,0 мл полученноrо раствора;
20 % и 30 % растворы: 1,0 мл испытуемоrо
образца доводят водой Р до объема 25,0 мл; ис
пользуют 2,0 мл полученноrо раствора.
Испытуемый раствор титруют 0,05 М pac
твором йода до появления синеrо окрашива
Натрия цитрата для инъекций 5 % раствор (056)
1037
ния, используя В качестве индикатора 1 мл
раствора крахмала, свободН020 от йоди
дов, Р.
1 мл 0,05 М раствора йода соответствует
24,82 Mr Nа2Sрз'5Нр.
НАТРИЯ ХЛОРИДА 0,9 %, 2 %
РАСТВОРЫ (053)
СОСТАВ
Натрия хлорид (NaCI; М. м. 58,44) 0,9 r; 2,0 r;
Вода очищенная до 100 МЛ.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 5 мл испытуемоro образца упаривают
до объема 1 мл. Полученный раствор дает
реакцию (с) на натрий (2.3.1).
В. К2 мл испытуемоroобразца прибавляют
0,5 мл кислоты азотной разведенной Р2, 0,5 мл
раствора серебра нитрата Р1. Образуется
белый творожистый осадок, нерастворимый в
кислоте азотной разведенной Р2 и paCTBO
римый в растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 1,0 мл испытуемоrо образца прибавля
ют 5 мл воды Р, 2 капли раствора калия xpo
мата Р и титруют О, 1 М раствором серебра
нитрата до оранжево желтоro окрашивания
осадка.
1 мл 0,1 М раствора серебра нитрата co
ответствует 5,844 Mr NaCI.
НАТРИЯ ЦИТРАТА ДВУЗАМЕЩЕнноrо
3,2 % РАСТВОР (054)
СОСТАВ
Натрия цитрат двузамещенный
(C6H6Nap7'1 %НР; М. м. 263,1) 0,32 r;
Вода очищенная до 10 мл.
ОПИСАНИЕ
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый раствор дает реакцию (с) на
натрий (2.3.1).
В. Испытуемый раствор дает реакции (Ь) и
(с) на цитраты (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 0,5 мл испытуемоro образца прибавля
ют 2 капли раствора фенолфталеина Р и ти
труют 0,1 М раствором натрия 2идроксида
до появления слабо роэовоro окрашивания.
132 30. 1712
1 мл 0,1 М раствора натрия 2идроксида co
ответствует 26,31 Mr натрия цитрата двузаме--
щенноro.
НАТРИЯ ЦИТРАТА ДВУЗАМЕЩЕнноrо
3,8 %, 5 % РАСТВОРЫ (055)
СОСТАВ
Натрия цитрат двузамещенный
(C6H6Nap7'1%HP; М. м. 263,1) 38,0 r, 50,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ
Прозрачная бесцветная жидкость со значе
нием рН от 4,5 до 5,0.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 1 мл испытуемоro образца дает реакции
(Ь) и (с) на натрий (2.3.1).
В. 0,5 мл испытуемоro образца дает peaK
цию (Ь) на цитраты (2.3.1).
С. 0,5 мл испытуемоro образца выпарива
ют на водяной бане. После охлаждения к сухому
остатку прибавляют 1 2 капли раствора вa
нилина в серной кислоте Р и наrревают на BO
дяной бане в течение 5 мин. Затем добавляют
1 2 мл воды Р. Появляется зеленое окрашива
ние.
О. К 1 мл испытуемоro образца прибавляют
0,02 r натрия zидрокарбоната Р выделяются
пузырьки rаза без цвета и запаха.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
К 1 мл испытуемоro образца прибавляют
20 мл воды, свободной от У2лерода диоксида,
Р и титруют 0,1 М раствором натрия 2идpOK
сида до появления розовоro окрашивания, ис
пользуя в качестве индикатора раствор фе
нолфталеина Р1.
1 мл 0,1 М раствора натрия 2идроксида СООТ
ветствует 26,31 Mr натрия цитрата двузамещенноro.
МЕТОД 2
Используют только для 5 % раствора натрия
цитрата двузамещенноrо.
Определяют показатель преломления
(2.2.6); F 1 = 0.00140.
C.H.Na20,1 2Н ,О
НАТРИЯ ЦИТРАТА ДЛЯ ИНЪЕКЦИЙ
5 % РАСТВОР (056)
СОСТАВ
Натрия цитрат для инъекций
(CfH5Nap7'5 20; М. м. 357,2) (в пересчете на
сухое вещество) 50 r;
вода для инъекций до 1000 мл.
1038
rосударственная фармакопея Республики Беларусь
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость со значе
нием рН от 7,8 до 8,3.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 1 мл испытуемоro образца дает реакцию
(а) на натрий (2.3.1).
В. 0,5 мл испытуемоro образца дает peaK
цию (а) на цитраты (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определяют показатель преломления
(2.2.6); Fc н Na 0 =0,0016.
6 5 3 7
НИКОТИНАМИДА 1 % РАСТВОР
ДЛЯ ИНЪЕКЦИЙ (057)
СОСТАВ
Никотинамид (C 6 H 6 NP; М. м. 122,1) 10,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или слеrка желтова
тая жидкость со значением рН от 5,0 до 7,0.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 2 мл испытуемоrо образца выпаривают
на водяной бане досуха, прибавляют 2 мл pacт
вора натрия 2идроксида Р и наrревают. Bыдe
ляется аммиак, обнаруживаемый по запаху и
по посинению красной лакмусовой бума2и Р.
В. 2 мл испытуемоro образца выпарива
ют на водяной бане досуха, прибавляют 0,1 r
натрия карбоната Р и наrревают. Выделяет
ся пиридин, обнаруживаемый по xapaKTepHO
му запаху.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 6 мл испытуемоro образца прибавляют
20 мл УКСУСН020 аН2идрида Р, наrревают на
водяной бане 8 течение 30 мин и охлаждают.
Капли влаrи с колбы удаляют фильтровальной
бумаroй, к раствору прибавляют 1 О мл кислоты
уксусной безводной Р, 3 капли раствора Kpи
сталличеСК020 фиолетов020 Р и титруют 0,1 М
раствором кислоты хлорной до появления зе
леноro окрашивания.
Параллельно проводят контрольный опыт.
1 мл О, 1 М раствора кислоты хлорной соот
ветствует 12,21 Mr C 6 H 6 N 2 0.
НИКОТИНОВОЙ КИСЛОТЫ 0,1 %,
0,5 %, 1 % РАСТВОРЫ (058)
СОСТАВ
Кислота никотиновая (C 6 H 5 N0 2 ; М. м. 123, 1)
1,0 r; 5,0 r; 10,0 r;
Вода очищенная до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
К 1 мл испытуемоro образца прибавляют
5 капель раствора меди (11) сульфата Р. Обра
зуется осадок синеro цвета. При прибавлении
1 мл раствора аммония тиоцианата Росадок
окрашивается в зеленый цвет.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
5,0 мл испытуемоro раствора помещают
в мерную колбу вместимостью 50 мл, прибав
ляют 2 капли раствора фенолфталеина Р,
затем по каплям 0,1 М раствора натрия 2и
дроксида до появления розовоro окрашива
ния. К полученному раствору прибавляют 5,0
мл 5 % раствора меди (1/) сульфата Р, Bыдep
живают в течение 1 О мин, доводят водой Р до
объема 50,0 мл, перемешивают и фильтруют,
отбрасывая первые 15 мл фильтрата. 25,0 мл
фильтрата помещают в колбу со шлифом, при
бавляют 5,0 мл кислоты хлористоводородной
разведенной Р и 1,0 r калия йодида Р. Колбу
закрывают пробкой и выдерживают в течение
1 О мин в защищенном от света месте. Bыдe
лившийся йод титруют:
для 0,1 % и 0,5 % растворов кислоты нико
тиновой 0,01 М раствором натрия тиосуль
фата;
для 1 % раствора кислоты никотиновой
0,1 М раствором натрия тиосульфата.
В качестве индикатора используют раствор
крахмала, свободный от йодидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,01 М раствора натрия тиосульфа
та соответствует 2,462 Mr C 6 H 5 N0 2 ; 1 мл 0,1 М
раствора натрия тиосульфата соответствует
24,62 Mr C 6 H 5 N0 2 .
НИКОТИНОВОЙ КИСЛОТЫ 1 %
РАСТВОР ДЛЯ ИНЪЕКЦИЙ (059)
СОСТАВ
Кислота никотиновая (C 6 H 5 N0 2 ; М. м. 123, 1)
10,0 r;
Натрия rидрокарбонат (NаНСО з ; М. м. 84,0)
7,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость со значе
нием рН от 5,0 до 7,0.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоro образца прибавля
ют 5 капель раствора меди (11) сульфата Р. Об
разуется осадок синеro цвета. При прибавлении
1 мл раствора аммония тиоцианата Росадок
окрашивается в зеленый цвет.
Пероксида водорода 3 % раствор с натрия бензоатом (061)
1039
В. К 0,5 мл испытуемоro образца прибавля
ют 5 7 капель кислоты хлористоводородной
разведенной Р. Выделяются пузырьки rаза.
с. Испытуемый образец дает реакции (а) и
(с) на натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
ДО СТЕРИЛИЗАЦИИ
Натрия rидрокарбонат. К 2,0 мл испыту
емоro образца прибавляют 1 каплю раствора
метилов020 оранжев020 Р и титруют 0,1 М pac
твором кислоты хлористоводородной до розо
воro окрашивания (раствор А).
1 мл 0,1 М раствора кислоты хлористово
дородной соответствует 8,40 Mr NаНСО з .
Кислота никотиновая. К раствору А, по
лученному при количественном определении
натрия rидрокарбоната, прибавляют 5 капель
раствора фенолфталеина Р и титруют 0,1 М
раствором натрия 2идроксида до перехода
окраски от зеленовато желтоrо до розовато
красноro цвета.
1 мл О, 1 М раствора натрия 2идроксида co
ответствует 12,31 Mr C 6 H 5 N0 2 .
ПОСЛЕ СТЕРИЛИ3АЦИИ
Кислота никотиновая. 5,0 мл испытуемо
ro раствора помещают в мерную колбу вмести
мостью 50 мл, прибавляют 2 капли раствора
фенолфталеина Р, затем по каплям 0,1 М
раствора натрия 2идроксида до появления
розовоro окрашивания. К полученному paCTBO
ру прибавляют 5,0 мл 5 % раствора меди (11)
сульфата Р, выдерживают в течение 10 мин,
доводят водой Р до объема 50,0 мл, перемеши
вают и фильтруют, отбрасывая первые 15 мл
фильтрата. 25,0 мл фильтрата помещают в
колбу со шлифом, прибавляют 5,0 мл кисло
ты хлористоводородной разведенной Р и 1,0 r
калия йодида Р. Колбу закрывают пробкой и BЫ
держивают в течение 1 О мин в защищенном от
света месте. Выделившийся йод титруют 0,1 М
раствором натрия тиосульфата, используя
в качестве индикатора раствор крахмала. cвo
бодный от йодидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия тиосульфата
соответствует 24,62 Mr C 6 H 5 N0 2 .
НОВОКАИНА 2 % РАСТВОР (060)
СОСТАВ
Прокаина rидрохлорид (C13H2oN202 'HCI;
М. м. 272,8) 2,0 r;
Вода очищенная 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 1,0 мл испытуемою образца дает peaK
цию на первичные ароматические амины (2.3.1).
В. К 1,0 мл испытуемоro образца прибавляют
0,5 мл кислоты серной разведенной Р и 1,0 мл
0,02 М раствора калия пермаН2аната. Фиоле
товая окраска исчезает.
С. 1,0 мл испытуемою образца подкисляют
кислотой азотной разведенной Р и прибавляют
0,4 мл раствора серебра нитрата Р1. Образу
ется белый творожистый осадок, растворяющий
ся в растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
К 1,0 мл испытуемою образца прибавля
ют 1 мл раствора кислоты хлористоводород
ной разведенной Р, !)..........6 мл воды Р, 0,1 r калия
бромида Р, 2 капли раствора тропеолина 00 Р,
1 каплю раствора метиленов020 сине20 Р и
проводят определение аминноro азота (2.5.8).
1 мл 0,1 М раствора натрия нитрита соот
ветствует 27,28 Mr прокаина rидрохлорида.
МЕТОД 2
К 1 ,О мл испытуемою образца прибавляют
2 капли раствора бромфенолов020 сине20 Р, по
каплям кислоту уксусную разведенную Р до зе
леновато желтоro окрашивания и титруют О, 1 М
раствором серебра нитрата до фиолетовоro
окрашивания.
1 мл 0,1 М раствора серебра нитрата co
ответствует 27,28 Mr прокаина rидрохлорида.
ПЕРОКСИДА ВОДОРОДА 3 %
РАСТВОР С НАТРИЯ БЕНЗОАТОМ
(061 )
СОСТАВ
Водорода пероксида 30 % раствор 10,0 r
(в зависимости от фактическою содержания BO
дорода пероксида в растворе);
Натрия бензоат (C 7 H 5 Na0 2 ; М. м. 144,1)
0,05 r;
Вода очищенная дo 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 2 мл испытуемоro образца прибавля
ют 0,2 мл кислоты серной разведенной Р и
1 мл 0,02 М раствора калия пермаН2аната.
Раствор обесцвечивается с выделением пу
зырьков rаза.
В. К 1 мл испытуемоrо образца прибавляют 0.2
мл кислоты серной разведенной Р, 2 мл эфира Р,
0,2 мл раствора калия дихромата Р и встряхива
ют. Эфирный слой окрашивается в синий цвет.
1040
rосударственная фармакопея Республики Беларусь
С. 20 мл испытуемоro образца упаривают на
водяной бане до 2 мл. К 1 мл полученноro paCT
вора прибавляютО,5мл раствора 30 r/лжелеза (111)
хлорида Р. Образуется розовато желтый осадок,
растворимый в эфире Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Водорода пероксид. 5,0 мл испытуемоro
образца доводят водой Р до объема 50,0 мл. К
1,0 мл полученноro раствора прибавляют 0,5 мл
кислоты серной разведенной Р и титруют 0,02 М
раствором калия пермаН2аната до появления
слабо розовоro окрашивания.
1 мл 0,02 М раствора калия пермаН2аната
соответствует 1,701 Mr Н 2 О 2 .
Натрия бензоат. К 10,0 мл испытуемоro об
разца прибавляют 1 o 15 мл эфира Р, 2 капли
раствора метилов020 оранжев020 Р, 1 каплю
раствора метиленов020 сине20 Р и титруют
при перемешивании 0,1 М раствором кислоты
хлористоводородной до появления фиолетово
ro окрашивания водноro слоя.
1 мл 0,1 М раствора кислоты хлористово
дородной соответствует 14,41 Mr C 7 H 5 Na0 2 .
ПИРИДОКСИНАrИДРОХЛОРИДА
ПОРОШОК (062)
СОСТАВ
Пиридоксина rидрохлорид (С в Н 11 NО з 'НСI;
м. м. 205,6) 0,0002 r, 0,01 r;
rлюкоза моноrидрат (С6Н1206'Н20;
м. м. 198,2) 0,1 r, 0,2 r, 0,3 r.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 0,02 r испытуемоrо образца прибавля
ют 1 мл воды Р, 2 капли раствора 30 r/л железа
(111) хлорида Р. Появляется красно коричневое
окрашивание, исчезающее при прибавлении
нескольких капель кислоты серной разведен
нойР.
В. К 0,01 r испытуемоro образца прибавляют
1 мл воды Р, 1 мл реактива ФелиН2а Р и Harpe
вают до кипения. Образуется коричневато крас
ный осадок
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,5 r испытуемою образца, содержащею
0,0002 r пиридокеина rидрохлорида, или 0,2 r испы
туемоro образца, содержащеro 0,01 r пиридоксина
rидрохлорида, растворяют в 2 мл воды Р, прибавля
ют 4 капли раствора бромтимолов020 сине20 Р1
и титруют до появления roлубоro окрашивания:
для порошков, содержащих 0,0002 r пи
ридоксина rидрохлорида, 0,01 М раствором
натрия еидроксида;
для порошков, содержащих 0,01 r пиридок
сина rидрохлорида, 0,1 М раствором натрия
еидроксида.
1 мл 0,01 М раствора натрия 2идроксида
соответствует 2,056 Mr С в Н 11 NО з 'НСI; 1 мл 0,1 М
раствора натрия 2идроксида соответствует
20,56 Mr С в Н 11 NО з 'НСI.
ПРОТАРrОЛА 1 %, 2 %, 3 %, 5 %
РАСТВОР (063)
СОСТАВ
Серебра протеинат 1.0 r, 2,0 r, З,О r, 5,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Жидкость темно коричневоro цвета.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
К O, 1 мл испытуемоro образца прибавля
ют З 5 капель кислоты хлористоводородной
разведенной Р, наrревают до кипения и филь
труют. К фильтрату прибавляют капель
раствора натрия 2идроксида Р и 1 каплю pacт
вора меди (11) сульфата Р. Появляется фиолето
вое окрашивание.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К 2,0 мл 1 % или 1,0 мл 2 %, 3 % и 5 % ис
пытуемых растворов прибавляют 0,5 мл Kиc
лоты азотной разведенной Р, 0,5 мл pacт
вора железа (111) аммония сульфата Р2 и
титруют 0,02 М раствором аммония тиoциa
ната до появления желтовато розовоrо OKpa
шивания.
1 мл 0,02 М раствора аммония тиoциaHa
та соответствует 27,00 Mr серебра протеината.
РАСТВОР ПО ДЕМЬЯНОВИЧУ N 1
(064)
СОСТАВ
Натрия тиосульфат пентаrидрат
(Nа2Sрз'5Нр; М. м. 248,2) 60,0 r;
Вода очищенная 40,0 r.
Авторская пропись предусматривает изrо
товление 60 % (м/м) раствора. При изroтовлении
массо объемным методом состав следующий:
Натрия тиосульфат пентаrидрат
(Nа2Sрз'5Нр; М. м. 248,2) 85,0 r;
Вода очищеl-'ная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 1 мл испытуемоro образца дает реакции
(Ь) и (с) на натрий (2.3.1).
Рибофлавина 0,02 % раствор (066)
1041
В. К 0,5 мл испытуемоro образца прибав
ляют 1 мл кислоты хлористоводородной Р.
Появляется осадок серы и выделяется rаз,
окрашивающий в синий цвет йодкрахмальную
бума2У Р.
С. К 0,5 мл испытуемоro образца прибав
ляют 2 мл раствора серебра нитрата Р2.
Образуется белый осадок, окраска KOTOpO
ro быстро переходит в желтоватую, а затем в
черную.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,5 мл испытуемоro образца доводят
водой Р до объема 25,0 мл. 1,0 мл полученноrо
раствора титруют 0,05 М раствором йода до по
явления синеro окрашивания, используя в каче
стве индикатора 1 мл раствора крахмала, cвo
бодН020 от йодидов, Р.
1 мл 0,05 М раствора йода соответствует
24,82 Mr Nа2Sрз'5Нр.
РАСТВОР РинrЕРА (065)
СОСТАВ
Натрия хлорид (NaCI; М. м. 58,44) 9,0 r;
Калия хлорид (KCI; М. м. 74,6) 0,2 r;
Кальция хлорид rексаrидрат (CaCI 2 '6HP;
м. м. 219,1) 0,2 r;
Натрияrидрокарбонат(NаНСО з ; М. м. 84,0)
0,2 r;
Вода для инъекций д01000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость со значе
нием рН от 7,5 до 8,2.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 2 мл испытуемоro образца дают реакции
(Ь) и (с) на натрий (2.3.1).
В. 1 О мл испытуемоro образца упаривают до
объема 1 мл. Полученный раствор дает реакцию
(Ь) на калий (2.3.1).
С. 1 О мл испытуемоro образца упаривают до
объема 1 мл. Полученный раствор дает реакцию
(с) на кальций (2.3.1).
О. К 2 мл испытуемоro образца прибавляют
0,5 мл кислоты азотной разведенной Р2, 0,5 мл
раствора серебра нитрата Р1. Образуется
белый творожистый осадок, нерастворимый в
кислоте азотной разведенной Р2 и раствори
мый в растворе аммиака Р.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Сумма хлоридов. 1,0 мл испытуемоro об
разца титруют О, 1 М раствором серебра Hитpa
та до появления оранжевоro окрашивания. ис
пользуя в качестве индикатора раствор калия
хромата Р
1 мл О. 1 М раствора серебра нитрата co
ответствует 5.96 Mr суммы хлоридов.
Кальция хлорид. К 10,0 мл испытуемоrо
образца прибавляют 5 мл аммиаЧН020 буфер
Н020 раствора рН 10,0 Р, 0,05 r индикаторной
смеси кислотНО20 хромов020 тeMHo cиHe20 Р и
титруют 0,05 М раствором натрия эдетата до
появления сине фиолетовоro окрашивания.
1 мл 0,05 М раствора натрия эдетата co
ответствует 10,95 Mr CaCI 2 '6Hp.
Натрия rидрокарбонат. 10,0 мл испыту
eMoro образца титруют О, 1 М раствором кисло
ты хлористоводородной до появления красноro
окрашивания, используя в качестве индикатора
раствор метилов020 краСН020 Р.
1 мл 0,1 М раствора кислоты хлористово
дородной соответствует 8,40 Mr NаНСО з .
РИБОФЛАВИНА 0,02 % РАСТВОР (066)
СОСТАВ
Рибофлавин (C 17 H 20 NP6; М. м. 376,4)
0,002 r;
Натрия хлорид (NaCI; М. м. 58,44) 0,09 r;
Вода для инъекций до 1 О мл.
ОПИСАНИЕ (СВОЙСТВА)
Зеленовато желтая прозрачная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец имеет зеленую
флуоресценцию в ультрафиолетовом свете
(при отсутствии источника ультрафиолетовоrо
света наблюдают флуоресценцию в растворе в
пробирке при боковом освещении настольной
лампы). При прибзвлении кислоты хлористо
водородной Р1 или раствора натрия 2идpOK
сида концентрироваНН020 Р флуоресценция
исчезает.
В. К 2 каплям испытуемоro образца прибав
ляют 3 капли кислоты азотной разведенной Р2
и 3 капли раствора серебра нитрата Р2. Обра
зуется белый творожистый осадок, растворимый
в растворе аммиака разведеНН020 Р1.
С. Испытуемый образец дает реакцию (с) на
натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Рибофлавин. 10,0 мл испытуемоro образ
ца помещают в колбу со шлифом, прибавляют
25 мл свежеприroтовленноro 0.00167 М pacт
вора калия йода та. встряхивают и выдерживают
в течение 25 ЗО мин. Прибзвляют 5 мл кисло
ты хлориСfYIоводородной разведенной Р и 5 мл
раствора 100 r/л калия йодида Р и титруют BЫ
делившийся йод 0.01 М раствором натрия ти
осульфата. используя в качестве индикатора
раствор крахмала. свободный от йодидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,00167 М раствора калия йодата co
ответствует 0,6273 Mr CI;,H2oN406'
1042
rосударственная фармакопея Республики Беларусь
Натрия хлорид. К 1 ,О мл испытуемоro образ
ца прибавляют 2 3 капли раствора бромфено
лов020 сине20 Р, по каплям кислоту уксусную Р
до появления желтовато зеленоro окрашивания
и титруют 0,1 М раствором серебра нитрата
до окрашивания осадка в фиолетовый цвет.
1 мл 0,1 М раствора серебра нитрата co
ответствует 5,844 Mr NaCI.
РИБОФЛАВИНА,АСКОРБИНОВОЙ
КИСЛОТЫ И НИКОТИНОВОЙ
КИСЛОТЫ РАСТВОР (067)
СОСТАВ
Рибофлавин (C 17 HzoNP6; М. м. 376,4)
0,002 r;
Аскорбиновая кислота (С 6 Н в 0 6 ;
М. м. 176,1) 0,02 r:
Никотиновая кислота (C 6 H 5 N0 2 ;
М. м. 123,1) 0,01 r;
rлюкоза бeзвQцнaя (С 6 Н 1 Р6; м. м. 180,2) 0,2 r;
Натрия хлорид (NaCI; М. м. 58,44) 0,02 r;
Вода для инъекций до 1 О мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная зеленовато желтая жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец имеет зеленую
флуоресценцию в ультрафиолетовом свете
(при отсутствии источника ультрафиолетовоro
света наблюдают флуоресценцию в растворе
в пробирке при боковом освещении настоль
ной лампы).
В. К 2 каплям испытуемоro образца прибав
ляют 3 капли кислоты азотной разведенной Р2
и 3 капли раствора серебра нитрата Р2. Об
разуется белый осадок, постепенно приобрета
ющий серое окрашивание.
С. К 2 3 каплям испытуемоro образца при
бавляют 5---------6 капель раствора водорода перок
сида разведеНН020 Р и наrревают. Прибавляют
4 капли реактива ФелиН2а Р и наrревают до ки
пения. Образуется коричневато красный осадок.
О. 1 2 капли испытуемою образца выпари
вают, прибавляют 1 каплю раствора 1 О r/л Kиc
лоты фосфорномолибденовой Р в кислоте
серной Р. Появляется серо зеленое окрашивание.
Е. Испытуемый образец дает реакцию (с) на
натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Рибофлавин. 10,0 мл испытуемоro образца
помещают в колбу со шлифом, прибавляют 25 мл
свежеприroтовленноro 0,00167 М раствора
калия йодата, встряхивают и выдерживают в
течение 25 30 мин. Прибавляют 5 мл кисло
ты хлористоводородной разведенной Р и 5 мл
раствора 100 r/л калия йодида Р и титруют BЫ
делившийся йод 0,01 М раствором натрия ти
осульфата, используя в качестве индикатора
раствор крахмала, свободный от йодидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,00167 М раствора калия йодата co
ответствует 0,6273 Mr C17H20N406'
Аскорбиновая кислота. 2,0 мл испытуемо
ro образца титруют 0,05 М раствором йода до
появления синеro окрашивания (А, мл), исполь
зуя В качестве индикатора раствор крахмала,
свободный от йодидов, Р.
1 мл 0,05 М раствора йода соответствует
8,81 Mr С 6 НР6'
Никотиновая кислота. К 2,0 мл испыту
eMoro образца прибавляют 2 капли раствора
фенолфталеина Р и титруют 0,1 М раствором
натрия 2идроксида до появления розовою OKpa
шивания (В, мл).
Количество О, 1 М раствора натрия 2идpOK
сида, израсходованноro на титрование никоти
новой кислоты, рассчитывают по формуле:
А
B '
2
1 мл О, 1 М раствора натрия 2идроксида co
ответствует 12,31 Mr C 6 H 5 N0 2 .
rлюкоза. к 0,5 мл испытуемоro образ
ца прибавляют 2,0 мл 0,05 М раствора йода,
6 капель раствора 100 r/л натрия 2идpOKcи
да Р и выдерживают в защищенном от света
месте в течение 5 10 мин. Прибавляют 7 10
капель раствора 160 r/л кислоты серной Р и
титруют выделившийся йод 0,1 М раствором
натрия тиосульфата до перехода окраски от
синей до желтой, используя в качестве инди
катора раствор крахмала, свободный от йо
дидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
9,0 Mr С 6 Н 12 О 6 .
Натрия хлорид. К 1,0 мл испытуемо
ro образца прибавляют по каплям 1 мл кисло
ты азотной разведенной Р2 и 1 мл раствора
железа (11/) аммония сульфата Р5, 0,1 мл 0,1 М
раствора аммония тиоцианата и титруют О, 1 М
раствором серебра нитрата до исчезновения
красной окраски.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора серебра нитрата co
ответствует 5,844 Mr NaCI.
РИБОФЛАВИНА И АСКОРБИНОВОЙ
КИСЛОТЫ РАСТВОР (068)
СОСТАВ
Рибофлавин (C 17 H 20 NP6; м. м. 376,4) 0.002 r;
Сульфацила натрия 15 %,20 %,30 % растворы (069)
1043
Аскорбиновая кислота (С 6 Н в 0 6 ;
м. м. 176,1) 0,02 r;
rлюкоза безводная (С 6 Н 1 Р6; М. м. 180,2) O,2 r;
Натрия хлорид (NaCI; М. м. 58,44) 0,05 r;
Вода для инъекций до 1 О мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная зеленовато желтая жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец имеет зеленую флу
оресценцию в ультрафиолетовом свете (при OT
сутствии источника ультрафиолетовоro света Ha
блюдают флуоресценцию в растворе в пробирке
при боковом освещении настольной лампы).
В. К 2 каплям испытуемоro образца прибав
ляют 3 капли кислоты азотной разведенной Р2
и 3 капли раствора серебра нитрата Р2. Об
разуется белый осадок, постепенно приобрета
ющий серое окрашивание.
с. К 2 3 каплям испытуемоro образца при
бавляют 5..........б капель раствора водорода перок
сида разведеНН020 Р и наrревают. Прибавляют
4 капли реактива Фелuнzа Р и наrревают до ки
пения. Образуется коричневато красный осадок.
D. Испытуемый образец дает реакцию (с) на
натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Рибофлавин. Проводят количественное
определение рибофлавина в растворе рибофла
вина до прибавления остальных инrредиентов.
10,0 мл испытуемоro образца помещают
в колбу со шлифом, прибавляют 25 мл свеже
приroтовленноro 0,00167 М раствора калия
йодата, встряхивают и выдерживают в течение
25 30 мин. Прибавляют 5 мл кислоты хлори
стоводородной разведенной Р и 5 мл раствора
100 r/л калия йодида Р и титруют выделившийся
йод 0,01 М раствором натрия тиосульфата,
используя в качестве индикатора раствор Kpax
мала, свободный от йодидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,00167 М раствора калия йодата co
ответствует 0,6273 Mr C17H2oN406'
Аскорбиновая кислота. 2,0 мл испытуемо
ro образца титруют 0,05 М раствором йода до
появления синеrо окрашивания, используя в Ka
честве индикатора раствор крахмала, свобод
ный от йодидов, Р.
1 мл 0,05 М раствора йода соответствует
8,81 Mr С 6 НР6'
rлюкоза. К 0,5 мл испытуемоro образца при
бявляют 2,0 мл 0,05 М раствора йода, 6 капель
раствора 100 r/л натрия 2идроксида Р и Bыдep
живают в защищенном от света месте в течение
5 10 мин. Прибавляют 7 10 капель раствора
160 r/л кислоты серной Р и титруют выделив
шийся йод 0,1 М раствором натрия тuосуль
фата до перехода окраски от синей до желтой,
используя в качестве индикатора раствор Kpax
мала, свободный от йодидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
9,0 Mr С 6 Н 12 О 6 .
Натрия хлорид. К 0,5 мл испытуемоro образ
ца прибавляют 2 З капли раствора бромфено
лов020 сине20 Р, 5 капель кислоты уксусной Р
и титруют 0,1 М раствором серебра нитрата
до окрашивания осадка в фиолетовый цвет.
1 мл 0,1 М раствора серебра нитрата co
ответствует 5,844 Mr NaCI.
СУЛЬФАЦИЛА НАТРИЯ 15 %, 20 %,
ЗО % РАСТВОРЫ (069)
СОСТАВ
Сульфацетамид натрия (СвНgN2NаОЗS'Н20;
м. м. 254,2) 1,5 r, 2,0 r, 3,0 r;
Вода для инъекций до 1 О мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная или слеrка желтова
тая жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 0,2 мл испытуемоro образца прибавля
ют 0,2 мл раствора 100 r/л меди (11) сульфата Р.
Образуется осадок roлубовато зеленоro цвета,
который не изменяется при стоянии.
В. К 0,5 мл испытуемоro образца прибавля
ют 2 3 капли кислоты хлористоводородной
разведенной Р, 2 3 капли раствора натрия Hи
трита Р и 3 капли раствора {3 нафтола Р.
Появляется красное окрашивание.
с. Испытуемый образец дает реакцию (с) на
натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
1,0 мл испытуемоro образца доводят водой Р
до объема 10,0 мл. К 2,0 мл полученноro paCT
вора прибавляют 2 капли раствора метилов020
оранжев020 Р и 1 каплю раствора метиленовО20
сине20 Р и титруют 0,1 М раствором кислоты хло
ристоводородной до появления фиолетовоro oкpa
шивания.
1 мл О, 1 М раствора кислоты хло
ристоводородной соответствует 25,42 Mr
С в Н g N 2 NаО з S'НР.
МЕТОД 2
Определяют показатель преломления (2.2.6).
F'5% = 0,00197;
F 20 % = 0,00199;
F ЗО % = 0,00200.
1044
rосударственная фармакопея Республики Беларусь
ФУРАЦИЛИНА 0,02 % РАСТВОР (070)
СОСТАВ
Нитрофурал (C 6 H 6 NP 4; М. м. 198,1) 0,2 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВд)
Прозрачная желтая жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
К 2 мл испытуемоro образца прибавляют
0,1 мл раствора натрия 2идроксида разведен
Н020 Р. Появляется оранжево красное окраши
вание.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
5,0 мл испытуемою образца помещают в
колбу со шлифом, прибавляют 2,0 мл 0,05 М
раствора йода. 0,4 мл натрия 2идроксида
раствора разведеНН020 Р, перемешивают,
выдерживают в течение 2 мин и прибавляют
2 мл кислоты серной разведенной Р. Выделив
шийся йод титруют 0,1 М раствором натрия
тиосульфата до исчезновения синею OKpa
шивания, используя в качестве индикатора
1 мл раствора крахмала, свободН020 от йо
дидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
4,954 Mr C 6 H 6 N.O..
ФУРАЦИЛИНА 0,02 % РАСТВОР
ИЗОТОНИЧЕСКИЙ (071)
СОСТАВ
Нитрофурал (С Б Н 6 N 4 О 4 ; М. м. 198,1) 0,2 r;
Натрия хлорид (NaCI; М. м. 54,88) 9,0 r;
Вода для инъекций до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная желтая жидкость со значением
рН от 5,2 до 6,8.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 2 мл испытуемоro образца прибавляют
0,1 мл раствора натрия 2идроксида разведен
Н020 Р. Появляется оранжево красное окраши
вание.
В. 2 мл испытуемоro образца подкисляют
кислотой азотной разведенной Р и прибавляют
0,4 мл раствора серебра нитрата Р1. Образу
ется белый творожистый осадок, растворимый в
растворе аммиака Р.
с. 1 мл испытуемою образца дает реакции
(Ь) и (с) на натрий (2.3.1).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Нитрофурал. 5,0 мл испытуемоro образ
ца помещают в колбу со шлифом, прибавляют
2,0 мл 0,05 М раствора йода, 0,4 мл натрия
2идроксида раствора разведеНН020 Р, пере
мешивают, выдерживают в течение 2 мин и
прибавляют 2 мл кислоты серной разведен
ной Р. Выделившийся йод титруют 0,1 М pac
твором натрия тиосульфата до исчезнове
ния синеro окрашивания, используя в качестве
индикатора 1 мл раствора крахмала, свобод
Н020 от йодидов, Р.
Параллельно проводят контрольный опыт.
1 мл 0,05 М раствора йода соответствует
0,4954 Mr C 6 H 6 NP4'
Натрия хлорид. 0,5 мл испытуемоro об
разца титруют 0,1 М раствором серебра Hи
трата до появления красноватоro осадка, ис
пользуя в качестве индикатора раствор калия
хромата Р.
1 мл 0,1 М раствора серебра нитрата co
ответствует 5,844 Mr NaCI.
хлорrЕКСИДИНА БиrЛЮКОНАТА
0,02 %, 0,05 % РАСТВОРЫ (072)
СОСТАВ
Хлорrексидина диrлюконата раствор
(C 34 H 54 CI 2 N 1 P14; м. м. 898) 1,0 мл, 2,5 мл;
Вода очищенная до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость без
запаха.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 50 мл 0,02 % испытуемоrо раствора упа
ривают на водяной бане до объема 20 мл. К 1 О мл
полученноro раствора или 1 О мл 0,05 % испы
туемоro раствора прибавляют 0,5 мл раствора
меди (/1) сульфата Р. Появляется светло толу
бое окрашивание. Наrревают на водяной бане ,8
течение 1 О мин. В верхней части пробирки об
разуется светлый розовато фиолетовый хлопье
видный осадок.
В. 50 мл 0,02 % испытуемоro раствора упа
ривают на водяной бане до объема 20 мл. К 1 О мл
полученноro раствора или 10 мл 0,05 % испы
туемоro раствора прибавляют 1 мл раствора
железа (111) хлорида Р1 и наrревают до кипения.
Окраска раствора должна измениться от CBeT
ло желтой до темно оранжевой. Прибавляют
1 мл кислоты хлористоводородной Р. Окраска
раствора изменяется на желтую.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
5,0 мл 0,02 % испытуемоro раствора или
2,0 мл 0,05 % испытуемоro раствора доводят
водой Р до объема 100,0 мл (раствор А). Изме
ряют оптическую плотность (2.2.25) раствора А
при 253 нм.
Цинка сульфата 0,25 %, 1 %, 2 % растворы с борной кислотой (075)
1045
Содержание хлорrексидина диrлюконата в
процентах рассчитывают по формуле:
А-100
,
ЗЗО.V
rдe:
ЗЗО удельный показатель поrлощения
хлорrексидина диrлюконата;
А оптическая плотность раствора А;
V объем испытуемоro образца, взятый
для приroтовления раствора А.
ХЛОРИСТО ВОДОРОДНОЙ КИСЛОТЫ
1 %, 2 % РАСТВОР (073)
СОСТАВ
Хлористо водородной кислоты 8,3 % pa{;Т
вор 10,0 мл, 20,0 мл;
Вода очищенная дo 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоro образца прибавляют
0,5 мл раствора серебра нитрата Р1. Образу
ется белый творожистый осадок, растворимый в
растворе аммиака Р.
В. Испытуемый образец имеет сильнокис
лую реакцию раствора.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
5,0 мл испытуемоrо образца титруют
О, 1 М раствором натрия 2идроксида до по
явления желтоro окрашивания, используя в
качестве индикатора раствор метиленов020
краСН020 Р.
1 мл О, 1 М расспвора натрия 2идроксида co
ответствует 43,82 Mr раствора 8,3 % хлористово
дородной кислоты.
ХЛОРИСТО ВОДОРОДНОЙ КИСЛОТЫ
10 % РАСТВОР (074)
СОСТАВ
Хлористо водородной кислоты 8,3 % paCT
вор 100,0 мл;
Вода очищенная до 1000 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоrо образца прибавляют
0,5 мл раствора серебра нитрата Р1. Образу
ется белый творожистый осадок, растворимый в
растворе аммиака Р.
В. Испытуемый образец имеет сильнокис
лую реакцию раствора.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,0 мл испытуемоro образца титруют 0,1 М
раствором натрия 2идроксида до появления
желтоro окрашивания, используя в качестве ин
дикатора раствор метиленов020 краСН020 Р.
1 мл 0,1 М раствора натрия 2идроксида co
ответствует 43,82 Mr раствора 8,3 % хлористово
дородной кислоты.
ЦИНКА СУЛЬФАТА 0,25 %, 1 %,2%
РАСТВОРЫ С БОРНОЙ КИСЛОТОЙ (075)
СОСТАВ
Цинка сульфат rеmаrидрат (ZnSO 4' 7НР;
М. м. 287 ,5) 0,025 r, 0,1 r, 0,2 r;
Борная кислота (НзВОз; М. м. 61 ,8) 0,2 r;
Вода для инъекций до 1 О мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 0,5 мл испытуемоrо образца прибав
ляют 1 мл 96 % спирта Р и поджиrают. Пламя
окаймлено зеленым цветом.
В. К 2 каплям испытуемоro образца прибав
ляют 0,5 мл воды Р и 2 капли раствора калия
ферроцианида Р. Образуется белый студени
стый осадок, нерастворимый в кислоте хлори
стоводородной разведенной Р.
с. К 2 3 каплям испытуемоro образца при
бавляют 1 мл воды Р, 2 капли кислоты хлори
стоводородной разведенной Р и 2 капли paCT
вора 50 r/л бария хлорида Р. Образуется белый
осадок, нерастворимый в разведенных кислотах.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Цинка сульфат. 2,0 мл испытуемою образца
помещают в коническую колбу, прибавляют 5 мл
воды Р, 2 мл аммиачН020 буфеРН020 раствора
рН 10, О Р и титруют 0,05 М раствором натрия
эдетата до перехода окраски от фиолетовой до
синей, используя индикаторную смесь протрав
леНН020 черН020 11 Р в качестве индикатора.
1 мл 0,05 М раствора натрия эдетата COOT
ветствует 14,38 Mr ZпSО '7Н;;0.
Кислота борная. 0.5 мл испытуемоro образ
ца помещают в колбу. прибавляют 4 мл 2лицери
на (85 %) Р, предварительно нейтрализованною
по раствору фенолфталеина Р, и 4 мл раствора
калия ферроцианида Р, перемешивают и титруют
О. 1 М раствором натрия 2идроксида до появле
ния розовоro окрашивания, используя в качестве
индикатора 1 мл раствора фенолфталеина Р1.
1 мл О. 1 М раствора натрия 2идроксида co
ответствует 6,183 Mr НзВО з .
1046
rосударственная фармакопея Республики Беларусь
ЦИНКА СУЛЬФАТА 2 % РАСТВОР (076)
СОСТАВ
Цинка сульфат rептаrидрат (ZnS0 4 '7HP;
м. м. 287,5) 2,0 r;
Вода очищенная до 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 2 каплям испытуемоro образца прибав
ляют 0,5 мл воды Р и 2 капли раствора калия
ферроцианида Р. Образуется белый студени
стый осадок, нерастворимый в кислоте хлори
стоводородной разведенной Р.
В. К 2 3 каплям испытуемоro образца
прибавляют 1 мл воды Р, 2 капли кислоты
хлористоводородной разведенной Р и 2 капли
раствора 50 r/л бария хлорида Р. Образуется
белый осадок, нерастворимый в разведенных
кислотах.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1,0 мл испытуемоrо образца помещают
в коническую колбу. прибавляют 5 мл воды
Р, 2 мл аммидЧН020 буферН020 раствора
рН 10,0 Р и титруют 0.05 М раствором натрия
эдетата до перехода окраски от фиолето
вой до синей. используя индикаторную смесь
протравленнО2О черН020 11 Р в качестве ин
дикатора.
1 мл 0.05 М раствора натрия эдетата co
ответствует 14.38 Mr ZnS0 4 '7H 2 0.
ЭУФИЛЛИНА 1 % РАСТВОР (077)
СОСТАВ
Т еофиллин этилендиамин
(C2HaN2'(C7HP2NJ2: М. М. 420,4) 1,0 r;
Вода очищенная дo 100 мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1 мл испытуемоrо образца прибавляют
1 О капель кислоты хлористоводородной раз
веденной Р и 1 О капель раствора водорода пе
роксида концентрироваНН020 Р и выпаривают
на водяной бане. После охлаждения к сухому
остатку прибавляют 3 5 капель раствора aM
миака разведеНН020 Р1. Появляется фиопетово
красное окрашивание.
В. К 2 мл испытуемоro образца прибавляют
1 каплю раствора 50 r/л меди (11) сульфата Р.
Появляется фиолетовое окрашивание.
С. 1 мл испытуемою образца выпаривают
на водяной бане досуха. После охлаждения к
сухому остатку прибавляют 3 капли ацетона Р
и 2 капли раствора 50 r/л натрия нитропрусси
да Р. Постепенно появляется фиолетовое OKpa
шивание.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
МЕТОД 1
2,0 мл испытуемоrо образца титруют 0,1 М
раствором кислоты хлористоводородной до
появления розовоro окрашивания, используя
в качестве индикатора раствор метилов020
оранжев020 Р.
1 мл О, 1 М раствора кислоты хлористо
водородной соответствует 3,005 Mr C 2 H a N 2 ; при
пересчете на теофиллин этилендиамин pe
зулыат умножают на коэффициент 7,02 (COOT
ветствует 14,25 % этилендиамина в субстан
ции).
МЕТОД 2
5,0 мл испытуемоrо образца титруют 0,02 М
раствором серебра нитрата при встряхивании
до появления оранжево желтоro окрашивания,
используя в качестве индикатора раствор калия
хромата Р.
1 мл 0,02 М раствора серебра нитрата co
ответствует 3,604 Mr C 7 H a 0 2 N 4 ; при пересчете на
теофиллин этилендиамин результат умножают
на коэффициент 1,166 (соответствует 85,75 %
теофиллина в субстанции).
ЭУФИЛЛИНА 1 %, 2 %, 2,4 % РАСТВОР
ИЗОТОНИЧЕСКИЙ (078)
СОСТАВ
Т еофиллин этилендиамин
(C2HaN2'(C7Ha02N4)2; М. м. 420,4) 0,1 r, 0,2 r,
0,24 r;
Натрия хлорид (NaCI; М. м. 58,44) 0,073 r,
0,056 r, 0,049 r;
Вода очищенная до 1 О мл.
ОПИСАНИЕ (СВОЙСТВА)
Прозрачная бесцветная жидкость.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 1,0 мл испытуемоro образца прибав
ляют 10 капель кислоты хлористоводородной
разведенной Р и 1 О капель раствора водорода
пероксида концентрироваНН020 Р и выпарива
ют на водяной бане. После охлаждения к сухому
остатку прибавляют 3 5 капель раствора aM
миака разведеНН020 Р1. Появляется фиолетово
красное окрашивание.
В. К 2 мл испытуемоro образца прибавляют
1 каплю раствора 50 r/л меди (11) сульфата Р.
Появляется фиолетовое окрашивание.
С. 1 мл испытуемоro образца выпаривают
на водяной бане досуха. После охлаждения к
сухому остатку прибавляют 2 3 капли aцeтo
#6.3. Оценка качества экстемпоральных лекарственных средств
1047
на Р и 1 2 капли раствора 50 r/л натрия Hи
тропруссида Р. Постепенно появляется фиоле
товое окрашивание.
D. К 1 2 каплям испытуемоro образца при
бавляют 3 капли кислоты азотной разведен
ной Р2 и 3 капли раствора серебра нитрата
Р2. Образуется белый творожистый осадок, pac
творимый в растворе аммиака разведеНН020 Р1.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Теофиллин этилендиамин. 2,0 мл испыту
емоro образца титруют О, 1 М раствором кисло
ты хлористоводородной до появления розовоro
окрашивания, используя в качестве индикатора
раствор метилов020 оранжевоео Р.
1 мл 0,1 М раствора кислоты хлористово
дородной соответствует 3,005 Mr C 2 H 6 N 2 ; при пе
ресчете на теофиллин этилендиамин результат
умножают на коэффициент 7,02 (соответствует
14,25 % этилендиамина в субстанции).
Натрия хлорид. К 0,5 мл испытуемоrо об
разца последовательно прибавляют 4 мл воды
Р, 0,1 мл 0,1 М раствора аммония тиоцианата,
0,5 млрастворажелеза (///) аммония сульфата Р5
и титруют 0,1 М раствором серебра нитрата
до исчезновения красноrоокрашивания.
1 мл 0,1 М раствора серебра нитрата co
ответствует 5,844 Mr NaCI.
ЭУФИЛЛИНА ПОРОШОК
С САХАРОЗОЙ (079)
СОСТАВ
Т еофиллин этилендиамин
(C2HsN2'(C7H602N4)2; м м. 420,4) 0,003 r;
Сахароза (С'2Н2Р"; М м. 342,3) 0,2 r.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. К 0,1 r испытуемою образца прибавляют
10 капель кислоты хлористоводородной
разведенной Р и 1 О капель раствора вo
дорода пероксида концентрироваНН020 Р
и выпаривают на водяной бане. После ox
лаждения к сухому остатку прибавляют 3 5
капель раствора аммиака разведеНН020 Р1.
Появляется фиолетово красное окрашивание.
В. К 0,01 r испытуемоro образца прибавляют
2 мл кислоты хлорuстоводородной разведен
ной Р, несколько кристаллов резорцина Р и кипятят
в течение 1 мин. Появляется красное окраШl1вание.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,400 r испытуемоro образца растворяют в
5 мл воды, свободной от У2лерода диоксида. Р и
титруют 0,02 М раствором кислоты хлористо
водородной до появления розовоro окрашива
ния, используя В качестве индикатора раствор
метилов020 оранжев020 Р
1 мл 0,02 М раствора кислоты хлористо
водородной соответствует 0,601 Mr C 2 H s N 2 ; при
пересчете на теофиллин этилендиамин pe
зулыат умножают на коэффициент 7,02 (cooт
ветствует 14,25 % этилендиамина в субстан
ции).
01/201З:РБ60ЗОО
#6.3. ОЦЕНКА КАЧЕСТВА
ЭКСТЕМПОРАЛЬНЫХ
ЛЕКАРСТВЕННЫХ
СРЕДСТВ
Качество экстемпоральных лекарственных
средств (в том числе roмеопатических) устанавли
вается по характеристическим показателям. Ypo
вень качества лекарственных средств оценивается
в соответствии с требованиями, реrламентирован
ными Фармакопеей, приказами и друrими HopMa
тивными документами Министерства здравоохра
нения Республики Беларусь.
Для оценки качества экстемпоральных лекар
ственных средств применяются два термина «YДOB
летворяет» (<<rодная продукция») или «Не удовлет
воряет» (<<Забракованная продукция» ). Качество
изroтовленных лекарственных средств определя
ется орrанолептическим и измерительными Meтo
дами.
Неудовлетворительность изroтовленных ле
карственных средств устанавливается по следу
ющим показателям их качества:
несоответствие по описанию (внешний вид.
цвет, запах);
несоответствие растворов по прозрачности
или цветности;
неоднородность смешения порошков, мазей,
суппозиториев, roмеопатических тритураций:
несоответствие степени измельченности по--
рошков;
несоответствие размера частиц в тритураци
онных мазях;
наличие видимых механических включений;
несоответствие пponиси по rюдлинности:
замена одною вещества друrим, OTCYТ
ствие прописанноrо или нanжие непрописанноro
вещества;
замена веществ на аналоrичные по фар
маколоrическому действию без обозначения той
замены на требовании, рецепте (копии рецепта,
этикетке ):
отклонения от пponиси по массе или объему:
O'П<I1OНeНия по общей массе (объему);
O'П<I1OНeНия по массе отдельных доз и их
количеству;
1048
rосударственная фармакопея Республики Беларусь
отклонения по массе (или по KOHцeH
трации) входящих в пропись рецепта (требова
ния) веществ;
несоответствие по значению рН (кислотно
сти или щелочности);
несоответствие по стерильности;
несоответствие по микр06иолоrической
чистоте;
нарушение rерметичности укупорки (для
стерильных лекарственных форм);
нарушение действующих правил оформ
ления лекарственных средств, предназначен
ных к отпуску.
Изменения в составе лекарственных форм
(если необходимо) должны производиться
только С соrласия врача, за исключением слу
чаев, установленных Фармакопеей, приказами и
друrими нормативными документами Министер
ства здравоохранения Республики Беларусь,
и должны отмечаться на требовании, рецепте
(копии рецепта. этикетке). При отсутствии YKa
занной отметки на требовании, рецепте (копии
рецепта, этикетке) качество изroтовленноro ле
карственноro средства оценивается как HeYДOB
летворителыIOe.
Изменения в количестве oTnyuцeHHoro ле
KapcTBeHHoro средства или отпуск таблеток
вместо порошков должны также отмечаться на
требовании. рецепте (копии рецепта, этикетке).
Определение отклонений в лекарственных
средствах следует проводить с использованием
измерительных средств TOro же типа (с одинако
выми метpoлorическими характеристиками), что
и при их иэroтовлении.
Нормы отклонений, допустимые при из
rотовлении экстемпоральных лекарственных
средств (в том числе roмеопатических), и нормы
отклонений, допустимые при фасовке в аптеках
лекарственных средств промышnеннorо произ
водства, описаны в статьях #6.3.1 и #6.3.2.
01/201 З:РБб0301
#6.3.1. НОРМЫ ОТКЛОНЕНИЙ,
ДОПУСТИМЫЕ
ПРИ изrОТОВЛЕНИИ
ЛЕКАРСТВЕННЫХ
СРЕДСТВ (В ТОМ ЧИСЛЕ
rОМЕОПА ТИЧЕСКИХ)
В АПТЕКАХ
1. Отклонения, допустимые в массе OT
дельных доз (8 том числе при фасовке) порош
ков (в том числе порошковыми дозаторами) и
в общей массе rомеопатических тритураций,
указаны в таблице #6.3.1. 1. Отклонения. дo
пустимые в массе отдельных доз порошков (в
том числе при фасовке), определяются на про
писанную дозу одноro порошка. Отклонения,
допустимые в общей массе roмеопатических
тритураций, определяются на ПрОП14санную
массу тритураций.
Таблица #6.3.1. 1
Прописанная Отклонения, %
Macca,r
до 0,1 :t15
свыше 0,1 до 0,3 :t10
свыше 0,3 до 1 :t5
свыше 1 до 10 :t3
свыше 10 до 100 :tЗ
свыше 100 до 250 :t2
свыше 250 :tO,3
2. Отклонения, допустимые в общей массе
rранул roмеопатических (в том числе при фа
совке) для одной упаковки, указаны в таблице
#6.3.1. 2.
Таблица #6.3.1. 2
Прописанная Отклонения, %
Macca,r
до 1 :t5
свыше 1 до 100 :t3
3. Отклонения, допустимые в массе отдель
ных доз суппозиториев и пилюль:
для суппозиториев :t5 %;
для пилюль массой до 0,3 r :t1 О %;
для пилюль массой свыше 0,3 r :t5 %.
Среднюю массу определяют взвешиванием
с точностью до 0,01 r не менее 10 сynпозитори
ев или пилюль. При изroтовлении суппозитори
ев менее 1 О штук взвешивают все суппозитории.
Отклонения в массе суппозиториев и пилюль от
средней массы определяют взвешиванием каж
доro суппозитория или пилюли В количестве не
менее 5 штук.
4. Отклонения, допустимые в массе HaBe
ски отдельных фармацевтических субстанций
в порошках, пилюлях и суппозиториях (при из
roтовлении методом выкатывания или выли
вания), определяются на дозу каждой субстан
ции, входящей в эти лекарственные средства.
Отклонения указаны в таблице #6.3.1. 3.
Таблица #6.3.1_ 3
Прописанная Отклонения, %
Macca,r
до 0,02 :t20
свыше 0,02 до 0,05 :t15
свыше 0,05 до 0,2 :t10
свыше 0,2 до 0,3 :t8
свыше 0,3 до 0,5 :t6
свыше 0,5 до 1 :t5
свыше 1 до 2 :t4
свыше 2 до 5 :t3
#6.3.1. Нормы отклонений, допустимые при иЗ20товлении
1049
Прописанная Отклонения, %
Macca,r
свыше 5 до 10 :t2
свыше 1 О :t1
5. Отклонения, допустимые в общем объеме
жидких лекарственных средств при изrотовле
нии массо объемным методом с использовани
ем как концентрированных растворов, так и фар
мацевтических субстанций, указаны в таблице
#6.3.1 А.
Таблица #6.3.1 А
Прописанный Отклонения, %
объем,мл
до 10 :t10
свыше 1 О до 20 :t8
. свыше 20 до 50 :t4
свыше 50 до 150 :t3
свыше 150 до 200 :t2
свыше 200 :t1
6. Отклонения, допустимые в общем объеме
растворов для инъекций, изrотовляемых в виде
серийной внутриаптечной заrотовки при фа
совке (розливе) в rрадуированные бутылки для
крови, указаны в таблице #6.3.1. 5.
При отмеривании (и фасовке) жидкостей
после слива струей дается выдержка на слив
капель:
для невязких жидкостей в течение 1 мин;
для вязких жидкостей в течение 3 мин.
Таблица #6.3.1. 5
Прописанный Отклонения, %
объем,мл
до 50 :t10
свыше 50 :t5
7. Отклонения, допустимые в массе HaBe
ски отдельных фармацевтических субстанций в
жидких лекарственных средствах при изroтов
лении массо объемным методом с использова
нием как концентрированных растворов, так и
фармацевтических субстанций, указаны в Ta
блице #6.З.1. 6. Отклонения определяются не на
концентрацию в процентах, а на массу навески
каждой субстанции, входящей в эти лекарствен
ные средства.
Таблица #6.3.1. 6
Прописанная Отклонения, %
масса, r
до 0,02 :1:20
свыше 0,02 до 0,1 :1:15
свыше 0,1 до 0,2 :1:10
свыше 0,2 до 0,5 :1:8
Прописанная Отклонения, %
Macca,r
свыше 0,5 до 0,8 :t7
свыше 0,8 до 1 :t6
свыше 1 до 2 :t5
свыше 2 до 5 :1:4
свыше 5 :1:3
8. Отклонения, допустимые в массе жидких
лекарственных средств при изrотовлении MeTO
дом по массе с использованием как концентри
рованных растворов, так и фармацевтических
субстанций, указаны в таблице #6.З.1. 7. OT
клонения определяются не на концентрацию в
про центах, а на массу навески каждой субстан
ции, входящей в эти лекарственные средства.
Таблица #6.3.1. 7
Прописанная Отклонения, %
Macca,r
до 10 :t10
свыше 1 О до 20 :t8
свыше 20 до 50 :1:5
свыше 50 до 150 :tЗ
свыше 150 до 200 :t2
свыше 200 :t1
9. Отклонения, допустимые в массе навески
отдельных фармацевтических субстанций в мазях
и в жидких лекарственных средствах при изroтов
лении методом по массе с использованием как
концентрированных растворов, так и фармацев
тических субстанций, указаны в таблице #6.3.1. 8.
Отклонения определяются не на концентрацию
в процентах, а на массу навески каждой субстан
ции, входящей в эти лекарственные средства.
Таблица #6.3.1. 8
Прописанная Отклонения, %
масса, r
до 0,1 :1:20
свыше 0,1 до 0,2 :t15
свыше 0,2 до 0,3 :1:12
свыше 0,3 до 0,5 :1:10
свыше 0,5 до 0,8 :1:8
свыше 0,8 до 1 :1:7
свыше 1 до 2 :1:6
свыше 2 до 10 :t5
свыше 1 О ! :1:3
;
10. Отклонения. допустимые в общей массе
мазей, указаны в таблице #6.3.1. 9.
Таблица #6.3.1. 9
Прописанная
масса, r
Отклонения, %
!до 5
I свыше 5 до 10
:1:15
:1:10
1050
rосударственная фармакопея Республики Беларусь
Прописанная Отклонения, %
Macca,r
свыше 1 О до 20 :t8
свыше 20 до 30 :t7
свыше 30 до 50 :t5
свыше 50 до 100 :t3
свыше 100 :t2
11. Отклонения, допустимые нормативной
документацией при изrотовлении фармакопей
ных лекарственных средств в аптеках, указаны
в таблице #6.3.1. 10.
Примечания:
При определении допустимых отклонений
в проверяемых лекарственных средствах, изro
товленных в виде серий внутриаптечной заroтов
ки, следует пользоваться нормами отклонений,
приведенными в пунктах 1 10, а также в дей
ствующей нормативной документации, реrла
ментирующей изroтовление и контроль качества
различных лекарственных средств в аптеках.
При изrотовлении лекарственных средств в
виде серий внутриаптечной заroтовки отклоне
ния, допустимые в массе отдельных субстанций,
определяются на массу отдельных субстанций,
взятых для изroтовления требуемоro объема
(или массы) данной серии (один контейнер или
одна заrрузка). Например, при изroтовлении 2 л
0,9 % раствора натрия хлорида отвешивают Ha
веску 18 r, для которой допускается отклоне
ние:t3 %. При химическом контроле достаточно
установить, что было взято не менее 17,46 r и не
более 18,54 r натрия хлорида.
Отклонения. допустимые в массе отдельных
субстанций в лекарственных средствах, изroтов
ленных в виде серий внутриаптечной заroтовки
и изъятых из аптеки для проверки, определяют
ся, как указано в пункте 4 и пункте 7. Например,
если для контроля изымают 200 мл 0,9 % paCT
вора натрия хлорида. то при химическом KOHTpO
ле достаточно установить, что в растворе coдep
жится не менее 1,71 r и не более 1,89 r натрия
хлорида (отклонение:t5 %, см. пункт 7).
12. Отклонения, допустимые в концентриро
ванных растворах (в процентах), изroтовленных
массо объемным методом:
при концентрации до 20 % не более
:t2 % от нормируемоro процента;
при концентрации свыше 20 % не более
:t1 % от нормируемоro процента.
13. При проверке лекарственных средств,
изroтовляемых в roмеопатических аптеках по ин
дивидуальным прописям, используют нормы OT
клонений, приведенные в данной статье.
14. Отклонения, допустимые в rомеопати
ческих тритурациях, растворах и разведениях
жидких лекарственных средств при изroтовле
нии их в виде концентрированных растворов и
flолуфабрикатов:
при содержании субстанции 10 % (первое
десятичное разведение 01) не более:t5 %
от нормируемоro процента;
при содержании субстанции 1 % (второе
десятичное разведение 02) не более :t5 %
от нормируемоro процента;
при содержании фармацевтической суб
станции 0,1 % (третье десятичное разведение
03) не более:t1 0% от нормируемоro процента.
01/201З:РБ60ЗО2
#6.3.2. НОРМЫ ОТКЛОНЕНИЙ,
ДОПУСТИМЫЕ ПРИ ФАСОВКЕ
В АПТЕКАХ ЛЕКАРСТВЕННЫХ
СРЕДСТВ ПРОМЫШЛЕнноrо
ПРОИЗВОДСТВА
1. Отклонения, допустимые при фасовке
по массе таблеток, драже, капсул (aHrpo), для
одной упаковки указаны в таблице #6.3.2. 1. При
Таблица #6.3.1. 10
Наименование Содержание Нормативная
Плотность действующеrо
лекарственноrо средства вещества, % документация
Этиловый спирт 95 % 0,8114 0,8075 95 96
Этиловый свыше 90 % 0,8292 0,8259 90 91 # Этиловый спирт 95 %,
Этиловый свыше 70 % 0,8855 0,88ЗО 70 71 90 %, 80 %, 70 %, 60 % и
40%
Этиловый свыше 40 % 0,9487 0,9473 39,5 0,5
Водорода пероксида 3 % 2,5 3,5 Водорода пероксида
раствор 3 % раствор
Водорода пероксида 30 % 29,0 31,0 Водорода пероксида
раствор (перrидроль) 30 % раствор
Водорода пероксида 27,5 % ,
27,225 27,775 I
раствор* I
,
Аммиака 1 О % раствор 9,5 10,5 ,
I
I
.Отклонение, допустимое в концентрации раствора, составляет :1:1 %. что соответствует отклонениям, допустимым при изroтое
лении концентрированных растворов с концентрацией свыше 20 % (см. пункт 12).
#6.3.2. Нормы отклонений, допустимые при фасовке в аптеках
1051
фасовке поштучно таблеток, драже, капсул в ин
дивидуальную упаковку допустимые отклонения
не устанавливаются. Недовложение единиц ле
карственноro средства не допускается.
Таблица #6.3.2. 1
Измеряемая масса, r Отклонения, %
свыше 10 до 100 :t3
свыше 100 до 250 :t2
свыше 250 :t0,3
2. Отклонения, допустимые при фасовке
жидких лекарственных средств по объему (для
одной упаковки), указаны в таблице #6.3.2. 2.
Таблица #6.3.2. 2
Измеряемый Отклонения, %
объем,мл
д05 :t8
свыше 5 до 25 :t5
свыше 25 до 100 :t3
свыше 100 до 300 :t1,5
свыше 300 до 1000 :t1,O
свыше 1000 :t0,5
3. Отклонения, допустимые при фасовке
жидких лекарственных средств по массе (для
одной упаковки), указаны в таблице #6.3.2. 3.
Таблица #6.3.2. 3
Измеряемая масса, r Отклонения, %
до 5 :t4
свыше 5 до 100 :t2
свыше 100 до 5000 :t0,6
4. Отклонения, допустимые при фасовке
мазей, линиментов и порошков по массе (для
одной упаковки), указаны в таблице #6.3.2.А.
Таблица #6.3.2А
Измеряемая масса, r Отклонения, %
до 5 :t5
свыше 5 до 50 :t4
свыше 50 до 100 :t2,5
свыше 100 до 5000 :t1
5. Отклонения, допустимые при фасовке ле
карственноro растительноrо сырья (для одной
упаковки), указаны в таблице #6.3.2. 5.
Таблица #6.3.2. 5
Измеряемая масса, r Отклонения, %
до 100 :t5
свыше 100 до 200 :t3
свыше 200 до 1000 :t2
свыше 1000 :t1
Вакцины для медUЦUНСКО20 прuмененuя
1053
ОБЩИЕ СТАТЬИ
01/2013:0153
ВАКЦИНЫ для МЕдицинскоrо
ПРИМЕНЕНИЯ
Vacciпa ad usum hитапит
ОПРЕДЕЛЕНИЕ
Вакцины для медицинскоro применения
это лекарственные средства, содержащие aH
тиrенные вещества, способные индуцировать
специфический и активный иммунитет у челове
ка против возбудителя инфекции, или токсина,
или антиrена, вырабатываемоro возбудителем.
Иммунный отклик включает в себя индукцию
врожденной и адаптивной (клеточной, ryMO
ральной) частей иммунной системы. Вакцины
для медицинскоro применения должны обла
дать приемлемой иммуноrенной активностью
и безо пасностью при предполаrаемом плане
вакцинации.
Вакцины для медицинскоro применения
MOryT содержать: целые микроорrанизмы (бак
терии, вирусы или паразиты), инаf{тивирован
ные химическим или физическим способом
таким образом, что они сохраняют достаточные
иммуноrенные свойства; целые живые микро
орrанизмы, которые по своей природе невиру
лентны или которые были обработаны для тоro,
чтобы ослабить вирулентность, в то время как
у них сохраняются достаточные иммуноrенные
свойства; антиrены, экстраrированные из ми
кроорrанизмов, или секретированные микроор
rанизмами, или полученные с помощью rенной
инженерии или химическоro синтеза. Эти анти
reHbI MorYT использоваться в обычном состоянии
или MorYT быть детоксифицированы различны
ми химическими или физическими способами,
MOryT быть аrреrированы, полимеризованы или
конъюrированы с носителем для усиления их
иммуноrенности. Вакцины MorYT иметь адъю
вант. Если антиrен адсорбирован на минераль
ном адъюванте, вакцина называется «адсорби
рованной».
Терминолоrия. используемая в частных CTa
тьях для вакцин для медицинскоro применения,
определяется в CT;;JTbe 5.2. 1.
Бактериальные вакцины, содержащие
целые клетки, представляют собой или лиофи
лизат, или различной степени мутные суспензии
в бесцветных или почти бесцветных жидкостях.
Они MorYT быть адсорбированными. KOHцeH
трация живых или инактивированных бактерий
выражается в Международных Единицах мут
ности или, rAe это приемлемо, определяется по
средством прямоro подсчета клеток или же, для
живых бактерий, подсчетом количества жизне
способных микроорrанизмов.
Бактериальные вакцины, содержащие бак
териальные компоненты, представляют собой
суспензии или лиофилизат. Они MOryT быть aд
сорбированными. Содержание антиrена опреде
ляют с использованием подходящей валидиро
ванной методики количественноro определения.
Бактериальные анатоксины rотовятся из
токсинов посредством уменьшения их токсично
сти ниже приемлемоro уровня или посредством
полноr устранения ,оксичности физическими
или химическими методами с сохранением им
муноrенных свойств. Токсины получают из COOT
ветствующих штаммов микроорrанизмов. Метод
производства должен быть таким, чтобы aHa
токсин не превращался в токсин. Анатоксины
должны быть очищенными. Очистка должна про
водиться до иlили после детоксификации. AHa
токсины MOryт быть адсорбированными.
Вирусные вакцины roтовятся из вирусов,
выращенных в орrанизме животных, на куриных
эмбрионах, в подходящих тканях, в COOTBeTCTBY
ющих клеточных культурах или с помощью куль
туры rенно модифицированных клеток. Они яв
ляются жидкостями, имеющими разную степень
мутности в зависимости от производственно
ro метода, или лиофилизатами. Они MorYT быть
адсорбированными. Если в питательной среде
использовался кислотно основный индикатор,
типа феноловоro KpacHoro, жидкие средства и
лиофилизаты после восстановления MOryT быть
окрашены.
ВакцинbI. содержащие синтетические aH
ти2ены, как правило, прозрачные или бес
цветные жидкости. Концентрацию компонентов
обычно выражают в единицах содержания специ
фическоro антиrена.
Комбинированные вакцины представля
ют собой мультикомпонентные лекарственные
средства, изroтовленные таким образом. что
различные антиrены вводятся одновременно.
Различные антиrенные компоненты предназна
чены для защиты от различных штаммов или
типов одних и тех же орrанизмов и/или против
различных орrанизмов. Комбинированные BaK
цины MOryT выпускаться производителем либо
в виде oAHoro раствора или лиофилизата, либо
в виде нескольких составляющих с инструкци
ей по их смешиванию перед использованием.
При отсутствии частной статьи на комбиниро
ванную вакuин она должна соответствовать
требованиям частных статей для каждоrо KOH
KpeTHoro компонента С соответствующими MO
дификациями. утвержденными компетентными
орrанами.
Адсорбированные вакцины представляют
собой суспензии и MOryт образовывать осадок
на дне контейнера.
1054
rосударственная фармакопея Республики Беларусь
ПРОИЗВОДСТВО
Общие положения. Необходимо проде
монстрировать, что метод производства дaHHO
ro продукта постоянно приводит к получению
серий, сравнимых по эффективности, иммуно
rенности и безопасности для людей, что и серия,
прошедшая клинические испытания. Необходи
мо, чтобы были созданы спецификации на про
дукт, включая испытания на этапе производства.
Специфические требования для продукции,
включая испытания на этапе производства,
включаются в частные статьи. При COOTBeTCTBY
ющем обосновании и утверждении некоторые
испытания MOryт быть опущены, если проде
монстрировано, например, при валидационных
исследованиях, что процесс производства обе
спечивает постоянное соответствие данноro ис
пытания требованиям.
Как правило, вакцины производятся с ис
пользованием эталонноro банка культур микро
орrанизмов (see(Mot systeт). Методы приroтов
ления должны обеспечивать соответствующие
иммуноrенные свойства, обезвреживание и пре
дотвращение заrpязнения лекарственноro cpeд
ства посторонними аrентами.
Если вакцины производятся С использова
нием материалов человеческоro или животно
ro происхождения, должны выполняться Tpe
бования статьи 5.1.7. Вирусная безопасность,
а также требования более специфических
статей, касающихся вирусной безопасности в
данной общей статье, 5.2.2. Стаи кур, не иMe
ющих конкретных пат02енов и используемых
для производства вакцин и контроля их каче
ства, 5.2.3. Субстраты клеток для производ
ства вакцин для медициНСК020 применения и
2.6.16. Испытания на посторонние а2енты в
вирусных вакцинах для медициНСК020 приме
нения.
Как правило, при производстве конечной
партии вакцины число пассажей вируса или
бактерий из контрольноro банка культур микро
орrанизмов не должно превышать то, которое
использовалось для производства вакцины,
представленной для клинических испытаний.
Вакцины, по возможности, не должны co
держать токсичные, аллерrенные или друrие He
желательные реакции в орrанизме человека. В
состав вакцин MorYT входить необходимые дo
бавки, включая стабилизаторы и адъюванты.
Пенициллин и стрептомицин не должны исполь
зоваться при производстве и не должны при
бавляться в конечный продукт; однако, может
использоваться контрольный банк микроорrа
низмов, приroтовленный на среде, содержащей
пенициллин или стрептомицин, если это обосно
вано и разрешено.
Важной деталью производства вакцин яв
ляется постоянство качества получаемоro про
дукта. В частных статьях на вакцины для Me
дицинскоro применения указываются пределы
для различных испытаний, которые проводят
на этапе производства и на конечной партии
вакцины. Эти пределы должны быть в форме
максимально допустимых значений, мини
мально допустимых значений или максималь
ных и минимальных допусков для данноro
значения. Соответствие данным пределам яв
ляется необходимым, но не достаточным yc
ловием для подтверждения постоянства про
изводства данной вакцины. Таким образом,
производитель должен определить для каждо
ro продукта подходящее действие или предел
или пределы для выпуска (с учетом резулыа
тов, полученных на партии, прошедшей кли
нические испытания), которые будут исполь
зоваться для подтверждения постоянства
качества продукта. Впоследствии эти преде
лы должны быть уточнены с использованием
статистических данных, полученных при про
изводстве.
Субстраты для размножения. Субстра
ты для размножения должны выдерживать co
ответствующие требования Фармакопеи (5.2.2,
5.2.3) или, в отсутствие таких требований, пред
писания уполномоченных opraHoB. Подroтовка
банков клеток и последующих клеточных куль
тур осуществляется в асептических условиях,
в помещении, rдe не ведутся работы с друrи
ми клеточными культурами. Сыворотка и трип
син, используемые в приroтовлении клеточных
суспензий, не должны содержать посторонних
areHToB.
Банки культур микроорrанизмов. Штаммы
бактерий или вирусов, используемые в контроль
ном банке микроорrанизмов, идентифицируются
посредством архивных записей, которые coдep
жат информацию о происхождении штамма и о
последующих манипуляциях с ним. Необходи
мо предпринять меры, чтобы rарантировать, что
никакой друroй микроорrанизм, кроме исходноro
стандартноrо штамма, и никакое нежелательное
вещество не присутствуют в банке культур ми
кроорrанизмов.
Питательная среда. Питательная среда,
по возможности, не должна содержать токсич
ные и аллерrенные инrредиенты. Если включе
ние таких компонентов необходимо, то нужно
убедиться, что их количество в конечном про
дукте на выходе безопасно. Сыворотка животно
ro (но не человеческоro) происхождения может
использоваться в питательной среде для под
держания роста клеточных культур. Однако во
время накоплеl-lИЯ вирусов среда не должна co
держать такую сыворотку, если не утверждены
друrие требования. Питательные среды MorYT
содержать индикатор рН, например, феноловый
красный, и разрешенные антибиотики в самой
низкой эффективной концентрации, хотя пред
почтительно иметь среду, не содержащую анти
биотиков.
Вакцины для медициНСКО20 прuменения
1055
Размножение и сбор. Исходные стандарт
ные культуры размножаются и собираются при
определенных условиях. Чистота сбора прове
ряется посредством проведения испытаний, YKa
занных в частной статье.
Контрольные культуры клеток. Контроль
ные клетки для вакцин, производимых на кле
точной культуре, должны содержаться и ис
пытываться, как предписано. Для тоro, чтобы
обеспечить эффективный контроль, эти клетки
должны содержаться в таких же условиях, в KO
торых осуществляется производство клеточной
культуры, включая использование той же самой
партии питательной среды и смены питательной
среды.
Контрольные куриные эмбрионы. KOH
трольные куриные эмбрионы для живых вакцин,
производимых в куриных эмбрионах, инкуби
руются и проверяются, как указано в частной
статье.
Очистка. При необходимости MOryт приме
няться утвержденные процедуры очистки.
Инактивация. Инактивированные вакци
ны производятся в ходе эффективноro валиди
рованноro процесса инактивации. Если очевид
но присутствие посторонних areHToB в продукте,
например, в вакцинах, произведенных с исполь
зованием куриных эмбрионов от здоровых, за
раженных конкретными патоrенами (поп SРF)
стай, процесс инактивации валидируется также
с учетом их присутствия. Испытание на эффек
тивность инактивации выполняется сразу же
после процесса инактивации.
Конечный нефасованный продукт (final
bu/k). Конечный нефасованный продукт roто
вится путем смешивания компонентов вакцины
в асептических условиях. Для вакцин, не пред
ставляющих собой жидкость и не предназначен
ных для парентеральноro введения, конечный
нефасованный продукт roтовят смешиванием
компонентов в подходящих условиях.
Адъюванты. С целью потенцирования или
корректировки иммунноro ответа на антиrен
(антиrены), в состав вакцины MorYT быть вклю
чены один или более адъювантов. Адъюван
ты MorYT быть включены в состав вакцины или
присутствовать отдельно. Для производства
продукции постоянноro качества важно прово
дить надлежащее описание и контроль каче
ства адъювантов как отдельно, так и в комби
нации с антиrенами. Создают спецификации
качества на адъюванты отдельно и в комбина
ции с антиrенами.
Адсорбенты и адъюванты. Вакцины MorYT
быть адсорбированными на rидроксиде алюми
ния, на фосфате алюминия, фосфате кальuия
или друrом подходящем адсорбенте. Для при
дания определенной формы и адсорбционных
свойств, адсорбенты подroтавливают в специ
альных условиях.
Если адсорбент используется в качестве
адъюванта и образуется iп situ при производстве
вакцины, должны быть созданы спецификации
качества на каждый инrредиент и на образу
ющийся в вакцине адъювант. Спецификации Ka
чества предназначены, в частности, для KOHTpO
лирования:
качественноro и количественноro химиче
скоro состава;
физической формы и связанной с ней aд
сорбирующей способности, rде это важно, и oco
бенно, если адъювант будет выступать в каче
стве адсорбента;
взаимодействия между адъювантом и aH
тиrеном;
чистоты, в том числе содержания бактери
альных ЭНДОТОКСИНОВ и микробиолоrической чи
стоты;
любых друrих критических для функциони
рования пара метров.
При разработке устанавливают стабиль
ность каждоro адъюванта, отдельноro или в
смеси с антиrенами, особенно для критических
параметров.
Антимикробные консерванты. Антими
кробные консерванты используются для пре
дотвращения порчи или для предотвраще
ния побочных эффектов при использовании
вакцины, вызываемых микробным заrрязне
нием. Антимикробные консерванты не дo
бавляют в лиофилизированные продукты. В
однодозовые лекарственные средства анти
микробные консерванты обычно не вносят.
Необходимость использования эффектив
Horo антимикробноro консерванта дЛЯ MHO
rОДОЗ08ЫХ лекарственных средств YCTaHaB
ливают, принимая в расчет подверженность
вакцины контаминации во время использо
вания и максимальный рекомендованный
период использования после вскрытия KOH
тейнера. Если используется антимикробный
консервант, он не должен ухудшать безопас
ность и эффективность вакцины. Использо
вание для этих целей антибиотиков не жела
тельно.
На стадии разработки должна быть доказа
на эффективность антимикробноro консерванта
в течение срока roдности вакцины.
Эффективность антимикробноro KOHcep
ванта оценивается соrласно статье 5.1.3. Если
не MorYT быть выполнены ни критерий А, ни
критерий В то для оценки эффективности aH
тимикробноro консерванта используются сле
дующие критерии:
бактерии: 24 часа и 7 дней нет увеличе
ния, 14 дней Ig уменьшения 3, 28 дней нет
увеличения;
rpибы: 14 дней и 28 дней нет увеличе
ния.
1056
rосударственная фармакопея Республики Беларусь
Стабильность промежуточных продук-
тов. При производстве вакцин на разных CTa
диях образуются и хранятся, иноrда в течение
долrоrо периода времени, промежуточные про
дукты. Эти промежуточные продукты включают
в себя:
банки культур микрооpraнизмов;
живые или инактивированные сборы;
очищенные сборы, которые MOryT coдep
жать токсины или анатоксины, полисахариды,
бактериальные или вирусные суспензии;
очищенные антиreны;
адсорбированные антиrены;
конъюrированные полисахариды;
вакцины в виде нефасованноro конечноro
продукта;
вакцины в конечных закрытых контейне
рах, хранящиеся при температуре ниже, чем
температура, ИСПОЛЬЗОВанная при изучении CTa
бильности и предназначенная для выпуска без
повторноrо проведения количественноrо опре
деления.
За исключением случаев, коrда промежу
точные продукты хранятся в течение KOpOTKO
ro периода времени, должно быть проведено
исследование стабильности в предполаrа
емых условиях хранения для установления
ожидаемой степени деструкции. Для вакцин
в виде конечноro нефасованноro продукта,
изучение стабильности может быть проведе
но на репрезентативных образцах при услови
ях, эквивалентных предполаrаемым условиям
хранения. При необходимости на основании
данных изучения стабильности для всех про
межуточных продуктов (кроме банков куль
тур микроорrанизмов) устанавливают период
приroдности, применимый к данным условиям
хранения.
Конечная партия. Конечную партию
асептически расфасовывают по стерильным,
устойчивым к внешним воздействиям контей
нерам, которые после лиофилизации, если
необходимо, закрывают таким образом, чтобы
предотвратить контаминацию. Для вакцин, не
представляющих собой жидкость и не пред
назначенных для парентеральноro введения,
конечную партию расфасовывают при подхо
дящих условиях в стерильные, устойчивые
к внешним воздействиям контейнеры. Если
обосновано и утверждено, некоторые испыта
ния, предписанные для проведения на конеч
ной партии, MorYT быть проведены на конеч
ном нефасованном продукте, если доказано,
что последующий производственный процесс
не влияет на полученный результат.
Описание. Если иное не обосновано и не
утверждено, каждый контейнер (флакон, шприц
или ампула) в каждой конечной партии должен
быть проверен визуально или механически на
соответствие описанию.
Степень адсорбции. Для адсорбированных
вакцин, если иное не обосновано и не утверж
дено, должна быть создана спецификация на
выпуск, содержащая требования к степени aд
сорбц&#!, основанные на результатах, получен
ных для партии, прошедшей клинические ис
пытания. Из данных по изучению стабипьности
вакцины должно прослеживаться, что в конце
срока rодности степень адсорбции не ниже, чем
степень адсорбции серии, использованной в
клинических испытаниях.
Термическая устойчивость. Если в статье
на живую ослабленную вакцину присутствует
указание на проведение испытания термической
устойчивости, данное испытание проводят на
конечной партии с целью прослеживания посто
янства содержания от партии к партии чувстви
. тельных к температуре вирусных/бактериальных
частиц в лекарственном средстве. Подходящие
условия указывают в частной статье. По соrла
сованию с уполномоченным opraHoM данное ис
пытание может не проводиться рутинно при yc
ловии, что стабильность производственноrо
процесса была подтверждена по таким параме
трам, как постоянство выхода, отношение coдep
жания инфекционных вирусов (живых бактерий)
перед и после лиофильной сушки, стабильность
активности на выпуске и в реальном времени в
предписанных условиях, а также термическая
устойчивость. При внесении существенных из
менений в производственный процесс антиrена
(антиrенов) или в рецептуру должна быть pac
смотрена необходимость повторноrо введения
испытания.
Стабильность. На этапе разработки
должна быть продемонстрирована сохранность
активности конечноro продукта в течение срока
roдности; при этом оценивают потерю актив
ности в рекомендованных условиях хранения.
Чрезмерное снижение активности, даже в дo
пустимых пределах, может указывать на то, что
вакцина неприrодна.
Срок 20дности. Если иноro не указано, срок
rодности исчисляют от начала проведения KO
личественноro определения или от начала про
ведения первоro количественноro определения
для комбинированной вакцины. Для вакцин, xpa
нящихся при температуре ниже, чем температу
ра во время изучения стабильности, и предназна
ченных для выпуска без повторноro проведения
количественноro определения, срок roдности ис
числяют от даты окончания хранения при пони
женной температуре. Если для данной вакцины
количественное определение не проводят, срок
rодности конечной партии отсчитывают с MOMeH
та проведения испытания, характеризующеro
стабильность или, за неимением такоro, от даты
лиофилизации или заполнения в конечные KOH
тейнеры. Для комбинированных вакцин, у KOTO
рых компоненты находятся в разных контейне
рах, сроком roдности является дата компонента,
у котороro срок rодности истекает первым.
Вакцины для медицинскосо применения
1057
Во время срока rодности вакцины должны
храниться в указанных условиях.
Испытания на животных. В COOTBeT
ствии с положениями Европейской Конвенции
о защите позвоночных животных, использу
емых для экспериментальных и друrих Ha
учных целей, испытания должны быть про
ведены таким образом, чтобы задействовать
минимальное количество животных и чтобы
причинять им минимальную боль, страдания,
стресс и длительный урон. Оценка испытаний
частной c-rаТbIlI должна проводиться с учетом
этих положений. Например, если животное
проявляет положительную реакцию, инфи
цировано и так далее, коrда проявляется ти
пичная клиническая картина или наступает
смерть, после тоro, как получено достаточное
количество положительных результатов, за
раженные животные должны быть как можно
скорее умерщвлены или должны получить ле
чение для предотвращения необязательных
страданий. В соответствии с Общими указани
ями, MorYT быть использованы альтернатив
ные методы для демонстрации соответствия
требованиям частной статьи. Использование
таких испытаний поощряется, если это ведет
к прекращению или уменьшению использова
ния животных или уменьшению их страданий.
ИСПЫТАНИЯ
Вакцины должны отвечать требованиям,
описанным в частных статьях, включающим, rдe
это применимо, следующие:
рН (2.2.3). Жидкие вакцины, при необходи
мости после восстановления, должны иметь зна
чения рН в установленных в каждом конкретном
случае пределах.
Адъювант. Если вакцина содержит адъю
вант, еro количество определяют с использова
нием указанной методики; содержание должно
быть в допустимых пределах относительно заяв
ленноro значения (см. также испытания на алю
миний и кальций).
Алюминий (2.5. 13). Если использовался aд
сорбент, содержащий алюминий, то ДQпускается
не более 1,25 Mr алюминия (AI) в одной дозе для
человека, если иное не обосновано и не утверж
дено.
Кальций (2.5.14). Если использовался aд
сорбент, содержащий кальций, то допускается не
более 1,25 Mr кальция (Са) в одной дозе для чело
века, если иное не обосновано и не утверждено.
Свободный формальдеrид (2.4.18).
Если при приroтовлении вакцины использо
вался формальдerид, то допускается не более
0.2 r/л свободноro формальдеrида в конечном
133 з.. 1 12
продукте, если иное не обосновано и не YT
верждено.
Фенол (2.5.15). Если при приroтовлении
вакцины использовался фенол, то допускается
не более 2,5 r/л фенола в конечном продукте,
если иное не обосновано и не утверждено.
Вода (2.5.12). Для лиофилизированных
вакцин допускается не более 3,0 % (М/М), если
иное не обосновано и не утверждено.
Извлекаемый объем (2.9.17). Лекарствен
ное средство должно выдерживать испытание
на извлекаемый объем, если иное не обоснова
но и не утверждено.
Бактериальные эндотоксины. Испыта
ние на бактериальные эндотоксины проводят
на конечной партии, если иное не обосновано и
не утверждено. Если в частной статье не указа
Нbl специфицированные пределы, содержание
бактериальных эндотоксинов, определенное с
использованием подходящеro метода (2.6.14),
должно быть менее предела, установленноro
для отдельноro продукта.
ХРАНЕНИЕ
Хранят в защищенном от света месте. Если
нет друrих указаний, температура хранения
должна быть (5:1:3) ОС; адсорбированные жидкие
вакцины нельзя подверrать замораживанию.
Срок 20дности. Если нет друrих указаний,
срок roдности рассчитывается с момента начала
испытаний.
МАРКИРОВКА
На этикетке указывают:
наименование лекарственноro средства;
контрольный номер, идентифицирующий
конечный продукт;
рекомендуемую дозировку для человека и
способ применения:
условия хранения;
срок roдности;
наименование и количество антимикроб
ноro консерванта;
наименование антибиотика. адъюванта,
ароматизатора или стабилизатора. содержа
щихся в вакцине;
при необходимости. что вакцина адсорби
рована;
наименование любоro элемента, KOTO
рый может вызвать неблаronриятные реакции и
любые противопоказания к использованию BaK
цины;
для лиофилизированных вакцин:
название или состав и объем жидкости
для приroтовления (восстановления);
время, в пределах котороro вакцина
должна использоваться после приrотовления
( восстановления).
1058
rосударственная фармакопея Республики Беларусь
01/2013:0084
ИММУННЫЕ СЫВОРОТКИ
животноrОПРОИСХОЖДЕНИЯ
для МЕдицинскоrо ПРИМЕНЕНИЯ
/ттипоsеrа ех aпima/e ad usum huтапит
ОПРЕДЕЛЕНИЕ
Иммунные сыворотки животноro проис
хождения для медицинскоro применения
это жидкие или лиофилизированные лекар
ственные средства, содержащие очищенные
иммунопiобулины или фраrменты иммуно
rлобулинов, полученные из сыворотки или
плазмы иммунизированных животных различ
ных видов.
Иммуноrлобулины или фраrменты имму
ноrлобулинов обладают способностью специ
фической нейтрализации или связывания aH
тиrенов, использованных для иммунизации.
Антиrены включают в себя микробные или
иные токсины. человеческие антиrены, суспен
зии бактериальных или друrих антиrенов. яды
змей, скорпионов и пауков. Лекарственные
средства предназначены для внутривенноro
или внутримышечноrо введения, будучи при
необходимости разведенными.
ПРОИЗВОДСТВО
ОБЩИЕ ПОЛОЖЕНИЯ
Производственный метод должен приво
дить к получению иммунных сывороток с при
емлемыми безопасностью, активностью и CTa
бильностью.
Любой реактив биолоrическоro происхож
дения, используемый в производстве иммунной
сыворотки, должен быть свободным от бакте
риальных, вирусных заrрязнений и заrрязне
ний rрибами. К процессу производства иммун
ных сывороток для медицинскоro применения
вместе с более специфичными требованиями
по вирусной безопасности, указанными в данной
общей статье, должны применяться общие Tpe
бования статьи 5.1.7. Вирусная безопасность.
Производственный метод должен включать
стадию или стадии, предназначенные для yдa
ления или инактивации известных инфициру
ющих факторов.
Методы, используемые для производства,
должны быть валидированными, эффект в
ными, воспроизводимыми и не должны пони
жать биолоrическую активность продукта.
Метод производства должен быть вали
дирован для подтверждения тоro, что продукт
будет выдерживать испытание, при необходи
мости ero проведения. на аномальную токсич
ность иммунных сывороток И вакцин для Me
дицинскоro применения (2.6.9).
Стандартный образец (препарат cpaв
нения). Серию, приroдность которой была под
тверждена клиническими исследованиями, или
образец такой серии используют в качестве пре
парата сравнения для испытания на содержание
белков с высокой молекулярной массой и для
испытания на чистоту.
ЖИВОТНЫЕ
Используемые животные должны принад
лежать утвержденному компетентным opraHoM
виду; они должны быть здоровыми и быть пред
назначенными исключительно для целей произ
водства иммунной сыворотки. Они не должны
содержать инфицирующих факторов из YCTa
новленноro списка. Ввод животных в закрытое
стадо должен осуществляться в соответствии с
утвержденными процедурами, включая опреде
ление карантинных мер. При необходимости, в
зависимости от rеоrрафическоrо расположения
орrанизации, занимающейся разведением и BЫ
ращиванием животных, принимают во внимание
дополнительные специфические areHTbI. Источ
ник корма для животных должен контролиро
ваться и в неro не должны добавляться белки жи
BOTHOro происхождения. Поставщики животных
должны быть сертифицированы компетентным
opraHoM. Если животное лечили с применением
антибиотиков, перед отбором плазмы должен
выдерживаться период времени COOTBeTCTBY
ющей продолжительности. Лечение животных
не должно осуществляться с использованием
пенициллиновых антибиотиков. Если вводят
живую вакцину, между вакцинацией и отбо
ром сыворотки или плазмы для производства
иммунной сыворотки должен выдерживаться
период времени соответствующей продолжи
тельности.
ИММУНИ3АЦИЯ
При необходимости используемые антиrе
ны идентифицируют и характеризуют; rдe это
важно, удостоверяются, что они не содержат по
сторонних инфекционных факторов. Их иденти
фицируют по их наименованиям и номеру серии;
отмечают информацию об источнике и о приro
товлении.
Выбранных животных изолируют как ми
нимум за 1 неделю перед иммунизацией в co
ответствии с установленным расписанием с бу
стер инъекциями через подходящие интервалы
времени. Может быть использован адъювант.
Животных содержат под общим наблюдени
ем и при контролировании продуцирован ия специ
фических антител в каждый цикл иммунизации.
Перед отбором крови или плазмы животных
тщательно проверяют. Если у животноro прояв
ляются патолоrические изменения, не связан
ные с процессом иммунизации, данное живот
ное не используют, а также не используют все
остальные животные данной rруппы до тех пор,
пока не будет ясно, что они не повлияют на безо
пасность продукта.
Иммунные сыворотки животНО20 происхождения для медициНСКО20 применения
1059
ОТБОР КРОВИ ИЛИ ПЛАЗМЫ
Отбор крови проводят с помощью венопунк
ции или плазмофереза. Место прокола выбрива
ют, очищают и дезинфицируют. Животные MorYT
быть анестезированы при условии, что это не
повлияет на качество продукта. Если нет друrих
указаний, может быть добавлен антимикробный
консервант. Кровь или плазма должны отбирать
ся таким образом, чтобы поддерживалась CTe
рильность продукта. Отбор крови или плазмы
должен проводиться отдельно от мест, rдe жи
вотные содержатся или выращиваются, и OT
дельно от мест, rдe происходит очистка иммун
ных сывороток Если кровь или плазма должна
храниться перед дальнейшим производствен
ным процессом, должны быть приняты меры
по предотвращению микробной контаминации.
Перед очисткой несколько отдельных образцов
плазмы или сыворотки MOryT быть объедине
ны. Перед очисткой с образцами сыворотки или
плазмы должны быть проведены следующие ис
пытания.
Испытания на вирусное заrрязнение.
Если был добавлен антимикробный KOHcep
вант, то перед испытаниями он должен быть
удален, либо испытания должны быть прове
дены на образцах, взятых перед добавлением
антимикробноro консерванта. Каждый пул про
веряют на вирусное заrрязнение с помощью
подходящих методов in vitro.
Каждый пул проверяют на содержание ви
русов путем инокуляции в клеточные культуры,
способные детектировать широкий ряд виру
сов, релевантных для конкретноro продукта.
Активность. При необходимости про водят
биолоrическое определение активности, как
указано в частной статье, и выражают резуль
тат в Международных Единицах на миллилитр.
Кроме этоro, может быть использован валиди
рованный метод iп vitro.
Содержание белка. Испытуемый образец
разводят раствором 9 r'л натрия хлорида Р
таким образом, чтобы получить раствор с co
держанием белка около 15 Mr в 2 мл. В Kpyr
лодонную центрифужную пробирку помещают
2 мл полученноro раствора, прибавляют 2 мл
раствора 75 r'л натрия молибдата Р и 2 мл
смеси из 1 объема кислоты серной. свобод
ной от азота, Р и 30 объемов воды Р, встряхи
вают, центрифуrируют в течение 5 мин, дeKaH
тируют надосадочную жидкость и оставляют
перевернутую на фильтровальной бумаrе про
бирку подсохнуть. В полученном остатке опре
деляют содержание азота после минерализа
ции серной кислотой (2.5.9) и рассчитывают
содержание белка путем умножения получен
ною результата на коэффициент 6,25. Coдep
жание белка должно быть в рамках утвержден
ных пределов.
ОЧИСТКА И ИНАКТИВАЦИЯ ВИРУСОВ
Иммуноrлобулины концентрируют и очища
ют с помощью фракционноro осаждения, xpo
матоrрафии, иммуноадсорбции или с помощью
иных химических или физических методов. В
дальнейшем они MOryT быть подверrнуты фер
ментативной обработке. Методы выбирают и Ba
лидируют таким образом, чтобы на всех стади
ях производства предотвратить заrрязнение и
чтобы избежать аrреrации белков, что способно
влиять на иммунобиолоrические свойства про
дукта. Для продуктов, состоящих из фраrментов
иммуноrлобулинов, метод должен быть валиди
рован для rарантирования полной фраrмента
ции. Используемые методы очистки не должны
при водить к появлению дополнительных компо
нентов и не должны подверrать риску качество и
безопасность продукта.
Если иное не обосновано и не утверждено,
вирусы должны быть удалены и'или инактивиро
ваны с применением валидированных процедур.
Процедуры выбирают таким образом, чтобы из
бежать образования полимеров или arperaToB и,
если продукт не должен состоять из фраrментов
Fab', минимизировать расщепление фраrментов
F(ab')2 на фраrменты Fab'.
После очистки и обработки по удалению и,
или инактивации вирусов к промежуточному про
дукту может быть добавлен стабилизатор, и про
межуточный продукт может храниться в течение
времени, установленноro на основании данных
по изучению стабильности.
Для изroтовления конечноro нефасованноrо
продукта может использоваться только тот про
межуточный продукт, который выдерживает сле
дующее испытание.
Чистота. Испытание проводят с помощью
электрофореза в полиакриламидном rеле в He
восстанавливающих условиях (2.2.31), сравни
вая с результатами, полученными для препарата
сравнения. Полосы должны быть сопоставимы
по интенсивности и не должны наблюдаться дo
полнительные полосы.
КОНЕЧНЫЙ НЕФАСОВАННЫЙ ПРОДУКТ
Конечный нефасованный продукт (fiпа/ bu/k)
изroтавливают из отдельною промежуточноro
продукта или из объединенных промежуточных
продуктов, полученных от животных одною и тoro
же вида. Допускается объединять в пул промежу
точные продукты с различной специфичностью.
MorYT быть добавлены антимикробные KOH
серванты и стабилизаторы. Если в кровь или
плазму добавлялся антимикробный консервант,
то же самое вещество используют в качестве aH
тимикробноro консерванта и в конечном нефа
сованном продукте.
Для изютовления конечною продукта может
использоваться только тот конечный нефасо
ванный продукт. который выдерживает следу
ющие испытания.
1060
rосударственная фармакопея Республики Беларусь
Антимикробные консерванты. При He
обходимости определяют количество антими
кробноro консерванта с использованием под
ходящеrо физико химическоrо метода. Должно
содержаться не менее 85 % и не более 115 % от
количества, указанноro на этикетке.
Стерильность (2.6.1). Должен выдержи
вать испытание на стерильность.
КОНЕЧНАЯ ПАРТИЯ
Конечную партию иммунной сыворотки
асептически расфасовывают по стерильным,
устойчивым к внешним воздействиям контей
нерам. Контейнеры закрывают таким образом,
чтобы предотвратить контаминацию.
К применению допускается только та конеч
ная партия, которая выдерживает требования,
приведенные в разделах ниже «Идентифика
ция», «Испытания» и «Количественное опреде
ление». При условии, что конечный нефасован
ный продукт был проверен на осмоляльность,
содержание белка, молекулярно массовое pac
пределение, антимикробный консервант, стаби
лизатор, чистоту, посторонние белки, альбумин
и количественное определение, эти показатели
MoryT не проверяться в конечной партии.
Лекарственное средство восстанавлuва
ют, как указано на этикетке, непосредствен
но перед проведением идентификации, испы
таний (кроме растворимости и определения
содержания воды) и количествеНН020 опреде
ления.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Подлинность устанавливают с помощью им
мунолоrических испытаний и, при необходимо--
сти, путем установления биолоrической активно
сти. Количественное определение также может
служить для подтверждения подлинности.
ОПИСАНИЕ (СВОЙСТВА)
Иммунные сыворотки это прозрачные
или опалесцирующие жидкости, бесцветные
или имеющие очень бледный желтый цвет. Они
не должны быть мутными. Лиофилизирова1-iные
продукты представляют собой белые или слеrка
желтоватые порошки или твердую рыхлую массу.
После восстановления они должны COOTBeTCTBO
вать описанию жидких лекарственных средств.
ИСПЫТАНИЯ
Растворимость. В контейнер с испыту
емым лекарственным средством прибавляют
объем жидкости, необходимый для BOCCTaHOB
ления, как указано на этикетке. Лекарственное
средство должно полностью растворяться за
время, указанное на этикетке.
Извлекаемый объем (2.9. 17). Лекарствен
ное средство должно выдерживать испытание
на извлекаемый объем.
рН (2.2.3). Значение рН должно находиться
в рамках пределов, обоснованных для каждоro
конкретною продукта.
Осмоляльностъ (2.2.35). Не менее 240 MOC
молыкrпосле разведения, rдe это необходимо.
Содержание белка. От 90 % до 11 О % от
количества, указанноro на этикетке, и, если обо
сновано иное, не более 100 r'л.
Испытуемый образец разводят раствором
9 r'л натрия хлорида Ртаким образом, чтобы по
лучить раствор с содержанием белка около 15 Mr
в 2 мл. В круrлодонную центрифужную лробирку
помещают 2 мл полученноro раствора, прибав
ляют 2 мл раствора 75 r'л натрия молuбдата Р
и 2 мл смеси из 1 объема кислоты серной, cвo
. бодной от азота, Р и 30 объемов воды Р, встря
хивают, центрифуrируют в течеl-Ме 5 мин, дe
кантируют надосадочную жидкость и оставляют
перевернутую на фильтровальной бумаrе npo
бирку подсохнуть. В полученном остатке опре
деляют содержание азота после минерализации
серной кислотой (2.5.9) и рассчитывают coдep
жание белка путем умножения полученноro pe
зулыата на коэффициент 6,25.
Молекулярно-массовое распределение.
Жидкостная хроматоrрафия (2.2.29 или 2.2.30).
Должны выдерживаться требования специфика
ции, обоснованной для каждоro конкретноro про
дукта.
Антимикробный консервант. При необхо
димости определяют содержание антимикроб
ною консерванта с использованием подходя
щеro физико химическою метода. Количество
должно быть не ниже минимальною количества,
которое является эффективным, и не более
115 % от количества, указанноro на этикетке.
Фенол (2.5.15). Не более 2,5 r'л для лекар
ственноro средства, содержащеro фенол.
Стабилизатор. Определяют содержание
стабилизатора с использованием подходящею
физико химическоro метода. Количество должно
быть не ниже 80 % и не более 120 % от количе
ства, указанною на этикетке.
Чистота. Испытание проводят с помощью
электрофореза в полиакриламидном rеле в He
восстанавливающих условиях (2.2.31), сравни
вая с результатами, полученными для препарата
сравнения. Полосы должны быть сопоставимы
по интенсивности и не должны наБJlюдаться дo
полнительные полосы.
Посторонние белки. Если нет друrих YKa
заний, наприМ€р, Korдa в процессе производ
ства используются вещества человеческо
ro происхождения, при реакции осаждения со
Лекарственное раститеЛЬНQе сырье
1061
специфическими антисыворотками, должны об
наруживаться белки только заявленных видов
ЖIпВОТНIbIХ.
Альбумин. Если нет друrих указаний в
частной статье, при электрофоретическом ис
пытании содержание альбумина не должно пре
вышать значение, установленное для каждо
ro конкретноro продукта, и в любом случае не
должно превышать З %.
Вода (2 5. 12). Не более 3 %.
Стерильность (2.6.1). Лекарственное cpeд
СТВО должно выдерживать испытание на CTe
рильность.
Пироrенность (2.6.8). Если иное не обосно
вано и не утверждено, лекарственное средство
должно выдерживать испытание на пироrен
ность. Тест доза 1 мл испытуемоrо образца
на 1 Kr массы тела кролика.
КОЛИЧЕСТВЕННОЕ ОПРЕдЕЛЕНИЕ
При необходимости проводят биолоrиче
ское определение активности, как указано в
частной статье, и выражают результат в Между
народных Единицах на миллилитр. Кроме этоrо
может быть использован валидированный метод
in vitro.
ХРАНЕНИЕ
В защищенном от света месте при темпе
ратуре, указанной на этикетке. Жидкие лекар
ственные средства не замораживают.
Срок 2одности. Срок roдности отсчитывают от
начала определения количественноro содержания.
МАРКИРОВКА
На этикетке указывают:
при необходимости, количество Междуна
родных Единиц на миллилитр;
содержание белка в контейнере;
для лиофилизированных лекарственных
средств:
наименование и объем жидкости, ис
пользующейся для приroтовления (восстановле
ния);
что иммунная сыворотка должна быть
использована немедленно после приroтовления
(восстановления );
время, необходимое для полноro paCTBO
рения;
способ введения;
условия хранения;
срок rодности, за исключением контейне
ров объемом менее 1 мл, которые упаковывают
ся индивидуально; срок rодности можно не YKa
зывать на этикетке контейнера при условии, что
он указан на упаковке и что этикетка на упаковке
указывает на то, что контейнер должен хранить
ся в упаковке до самоro использования;
134 Зак 1712
вид животноro источника сыворотки;
наименования и количества всех антими
кробных консервантов, стабилизаторов и друrих
вспомоrательных веществ.
01/2013:1433
ЛЕКАРСТВЕННОЕ РАСТИТЕЛЬНОЕ
СЫРЬЕ
P'antae medicina/es
"ОПРЕДЕЛЕНИЕ
Лекарственное растительное сырье ис
пользуемые для промышленноro производ
ства, аптечноro изroтовления лекарственных
средств цельные' лекарственные растения
или части лекарственных растений, на KOTO
рые имеются соответствующие фармакопей
ные статьи.
Лекарственное растительное сырье вклю
чает в себя цельные растения, фраrменты pac
тений или измельченные растения, части pac
тений, морские водоросли, rрибы, лишайники
в непереработанной форме, обычно высушен
ные, а иноrда и в свежем виде. Некоторые эксу
даты (например: rуммиарабик, камеди), которые
не были подверrнуты специальной обработке,
также MoryT относиться к лекарственному расти
тельному сырью. Лекарственное растительное
сырье точно определяется ero ботаническим Ha
учным названием в соответствии с имеющейся
биноминальной системой (род, вид, разновид
ность и автор).
Цельное лекарственное растительное
сырье лекарственное растительное сырье,
которое не проходило стадию измельчения и
представлено, в сухом или свежем виде, таким,
каким оно было собрано (например: шиповни
ка плоды, фенхеля ropbKoro плоды и фенхеля
сладкоro плоды); цельное лекарственное расти
тельное сырье включает в себя и обмолоченное
сырье.
Измельченное лекарственное раститель
ное сырье включает в себя резаное, дробленое
сырье и сырье в виде порошка.
Измельченное лекарственное растительное
сырье должно отвечать требованиям частной
статьи на это же цельное лекарственное расти
тельное сырье, за ИCКJ1ючением макроскопиче
ских признаков. если не установлены иные Tpe
бования.
ПРОИЗВОДСТВО
Лекарственное растительное сырье получа
ют из специально выращиваемых или дикора
стущих растений. Чтобы rарантировать качество
1062
rосударственная фармакопея Республики Беларусь
лекарственноro растительноro сырья, важно co
блюдать соответствующие правила выращива
ния, сбора урожая, сушки, измельчения и усло
вий хранения.
Лекарственное растительное сырье не
должно иметь признаков rниения. Оно должно
быть по возможности тщательно очищено от
примесей, таких как остатки почвы. пыли, rрязи,
заrрязнений наподобие rрибов. амбарных Bpe
дителей и друrих примесей животною происхож
дения. Проверку на наличие живых и мертвых
вредителей осуществляют при приемке и пе
риодически при хранении путем осмотра HeBO
оруженным rлазом и с помощью лупы (5 10 х)
при внешнем осмотре, а также при определе
нии степени измельчения и содержания приме
сей. При этом обращают внимание на наличие
частей сырья, поврежденных амбарными вреди
телями. Кроме сырья, тщательно просматрива
ют швы, складки упаковочноro материала, щели
в ящиках.
При обнаружении в сырье амбарных Bpe
дителей определяют степень еro зараженно
сти. Для этоro соответствующую аналитиче
скую пробу сырья (2.8.20) просеивают сквозь
сито (500) (2.9.12). В сырье, прошедшем сквозь
сито, проверяют С использованием лупы нали
чие клещей; в СЫрье, оставшемся на сите, про
веряют невооруженным rлазом и с использо
ванием лупы наличие моли, точильщика и их
личинок и дрyrиx живых и мертвых вредителей.
Количество найденных вредителей и их личинок
пересчитывают на 1000 r растительноro сырья и
устанавливают степень ею зараженности.
При наличии в 1 Kr сырья не более 20
клещей (клещ мучной (Tyrog'yphus fariпae L.),
клещ волосатый (G/yciphagus destructor
Sсhrапk.), клещ хищный (Chey/etus 'actis L.),
сухофруктовый клещ (Carpog/yphus 'actis L.) и
др.) зараженность сырья относят к I степени;
при наличии более 20 клещей, свободно пере
двиrающихся по поверхности сырья и не обра
зующих сплошных масс, ко 11 степени; если
клещей мноro, они образуют сплошные войлоч
ные массы, движение их затруднено к 111 CTe
пени. При наличии в 1 Kr сырья амбарной моли
(Тiпеа g,aпella L.) и ее личинок, а также хлеб
ноro точильщика (Sidotrepa paпicea L.) и друrих
вредителей в количестве не более 5 заражен
ность относят к I степени, при наличии 6 10
вредителей ко 11 степени, более 1 О вредите
лей к 111 степени.
При I степени зараженности сырье после
надлежащей деконтаминации может быть дo
пущено к производству лекарственных средств.
При 11 степени и в исключительных СЛУ'iаях при
111 степени зараженности сырье после надлежа
щей деконтами нации может быть использовано
для переработки с целью получения индивиду
альных веществ.
После про ведения деконтаминации необ
ходимо убедиться, что части растения не по
страдали и что после обработки в сырье не
осталось вредных примесей. Для деконтами
нации лекарственноro растительноro сырья
запрещается использование этиленоксида.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Лекарственное растительное сырье иденти
фицируют с использованием еro макроскопиче
ских и микроскопических признаков; кроме Toro,
при идентификации применяют и друrие методы
испытаний (например, тонкослойную xpOMaTO
rрафию ).
ИСПЫТАНИЯ
Примеси (2.8.2). Если иноro не указано
в частной статье, про водят испытание на дo
пустимые при меси. Количество всех допусти
мых примесей должно составлять не более
2 % (М/М), если иноro не указано в частной
статье.
Потеря в массе при высушивании (2.2.32).
Если иноro не указано в частной статье, прово
дят испытание на потерю в массе при высуши
вании.
Вода (2.2.13). Определение воды может
проводиться вместо испытания «Потеря В массе
при высушивании» для лекарственною расти
тельноro сырья с высоким содержанием эфир
ных масел.
Общая зола (2.4.16).
Пестициды (2.8.13). Лекарственное pac
тительное сырье должно соответствовать Tpe
бованиям в отношении остаточных количеств
пестицидов. Эти требования при меняются с
учетом происхождения растения, ero дальней
шеrо использования с учетом протокола полных
данных по обработке определенной партии pac
тения.
Микробиолоrическая чистота. PeKOMeHдa
ции по исследованию микробиолоrической чи
стоты продуктов, состоящих из одноro или He
скольких видов лекарственноro растительноro
сырья, приводятся в статьях 5.1.8. Микробио
Л02ическая чистота лекарствеННblХ средств
раститеЛЬН020 происхождения для ораЛЬН020
применения или 5. 1.4. МикробиОЛ02ическая чи
стота нестериЛЬНblХ лекарственных средств
и фармацевтических субстанций (например,
для наружною применения).
Тяжелые металлы (2.4.27). Необходимо
учитывать также риск заrрязнения лекарствен
ною растительною сырья тяжелыми металла
ми. Если в частной статье не указывается пре
дельное содержание тяжелых металлов или
друrих токсичных элементов, такие предельные
нормы при необходимости MOryT быть YCTaHOB
#Лекарственное растительное сырье цельное или измельченное фасованное
1063
лены. Если иноro не указано в частной статье,
лекарственное растительное сырье должно co
ответствовать нормам и требованиям действу
ющеro законодательства по содержанию тяже
лых металлов и друrих токсичных элементов.
Радиоактивное заrрязнение. Если иноro
не указано в частной статье, лекарственное
растительное сырье должно соответствовать
нормам и требованиям действующеro законода
тельства по содержанию радионуклидов.
При необходимости лекарственное расти
тельное сырье подверrают, например, нижесле
дующим испытаниям.
Зола, нерастворимая в хлористоводо-
родной кислоте (2.8.1).
Экстрактивные вещества.
Коэффициент набухания (2.8.4).
Показатель rоречи (2.8.15).
Афлатоксин 81 (2.8.18). При необходимо
сти MOryT быть установлены нормы по содержа
нию афлатоксина.
Охратоксин А (2.8.22). При необходимости
MOryт быть установлены нормы по содержанию
охратоксина.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Если нет специальных требований, коли
чественное содержание веществ, обуславли
вающих фармаколоrическую активность лекар
cTBeHHoro растительноro сырья или их маркеров,
устанавливают с использованием подходящеrо
метода.
ХРАНЕНИЕ
В защищенном от влаrи и света месте при
температуре от 15 ос до 25 ОС, если иноro не
указано в частной статье.#
01/201З:РБОО04
#ЛЕКАРСТВЕННОЕ РАСТИТЕЛЬНОЕ
СЫРЬЕ ЦЕЛЬНОЕ
ИЛИ ИЗМЕЛЬЧЕННОЕ ФАСОВАННОЕ
ОПРЕДЕЛЕНИЕ
Лекарственное растительное сырье цель
ное или измельченное фасованное представ
ляет собой измельченное, реже цельное, ле
карственное растительное сырье, иноrда с
добавлением солей, эфирных масел, с опреде
ленным действием, предназначенное для при
менения в лечебных целях после изroтовле
ния водных извлечений, как указано в статье
#Настои, отвары и чаи, расфасованное в COOT
ветствующую товарную упаковку.
Лекарственное растительное сырье цель
ное или измельченное фасованное может быть
дозированным инедозированным.
ПРОИЗВОДСТВО
Используемое сырье должно COOTBeTCTBO
вать требованиям частной фармакопейной
статьи.
При необходимости сырье измельчают и
просеивают. Если иноrо не указано в частной
статье, то сырье, используемое для приroтов
ления настоев и отваров, должно иметь степень
измельчения (5600) (2.9.12); сырье, использу
емое для приroтовления чаев, должно иметь
степень измельчения (2000) (2.9.12).
Листья толокнянки и брусники, использу
емые для приroтовления настоев и отваров,
измельчают до степени измельчения (2800)
(2.9.12).
Побеrи баryльника болотноro, используемые
для приroтовления настоев и отваров, измельча
ют до степени измельчения (4000) (2.9.12).
В тех случаях, Korдa добавляют соль, из нее
roтовят насыщенный раствор и опрыскивают им
лекарственное растительное сырье при переме
шивании, после чеro высушивают при темпера
туре не выше 60 ОС. Эфирное масло вносят в
лекарственное растительное сырье в виде спир
товоro раствора (1 :10) опрыскиванием при пере
мешивании.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Необходимо подтвердить соответствие MOp
фолоrических и анатомических признаков ле
карственных растений требованиям, указанным
в частной статье.
Подлинность определяют подходящим Me
тодом (качественные реакции, ТСХ. жх. rX).
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Степень измельчения (2.9.12). примеси
(2.8.2), потерю в массе при высушивании (2.2.32),
общую золу (2.4.16). золу. нерастворимую в хло
ристоводородной кислоте (2.8.1) и друrие число
вые показатели определяют. как указано в част
ной фармакопейной стап.е.
Если иноrо не указано в частной фар
макопейной статье. при определении степе
ни измельчения лекарственное растительное
сырье должно выдерживать следующие Tpe
бования:
сырье. используемое для приroтовле
ния настоев и отваров: частиц, проходящих
сквозь сито (5600), должно быть не менее 90 %,
1064
rосударственная фармакопея Республики Беларусь
Таблица РБОО04. 2
а частиц, проходящих сквозь сито (355), не
более 5 %;
сырье, используемое для приrотовления
чаев: частиц, проходящих сквозь сито (2000),
должно быть не менее 90 %, а частиц, проходя
щих сквозь сито (250), не более 10 %.
Листья толокнянки и брусники. использу
емые для приrотовления отваров, должны BЫ
держивать следующее требование: частиц, про
ходящих сквозь сито (2800), должно быть не
менее 95 %.
Побеrи баryльника болотною, использу
емые для приroтовления настоев, должны BЫ
держивать следующие требования: частиц, про
ходящих' сквозь сито (4000), должно быть не
менее 95 %, а частиц. проходящих сквозь сито
(З55), не более 5 %.
Однородность массы для дозированно
ro сырья. Определяют среднюю массу 20 слу
чайно выбранных дозированных единиц упа
ковки (фильтр пакет. пакетик, брикет и друrие)
следующим образом: взвешивают одну дози
рованную единицу упаковки, затем открывают
ее таким образом. чтобы не были утеряны Ka
кие либо фраrменты. Упаковку полностью очи
щают от содержимоro при помощи щеточки.
Взвешивают пустую упаковку и рассчитывают
массу содержимоrо упаковки путем вычита
ния. Повторяют данные действия -на девятнад
цати оставшихся единицах упаковки. Если нет
друrих указаний. то не более чем две из ДBaд
цати индивидуальных масс содержимоro OT
клоняются от средней массы содержимоro
больше чем на процентное отклонение, приве
денное в таблице РБОО04. 1, но не более чем
вдвое.
Таблица РБООО4. 1
Средняя масса Допустимое отклонение
с учетом влаrи
до 1 ,5 r 15%
от 1,5 r до 2,0 r 10%
включительно
более 2,0 r 7,5%
Однородность массы для недозиро
BaHHoro сырья. Определяют среднюю массу
1 О выбранных случайно недозированных
единиц упаковки (пачка, пакет и друrие) сле
дующим образом: взвешивают одну упаков
ку с содержимым, затем открывают ее таким
образом, чтобы не были утеряны какие либо
фраrменты. Упаковку полностью очищают от
содержимоro при помощи щеточки. Взвешива
ют пустую упаковку и рассчитывают массу co
держимоro упаковки путем вычитания. Повто
ряют данные действия на девяти оставшихся
упаковках. Допустимые отклонения массы co
держимоro упаковки приведены в таблице
РБОО04. 2.
Допустимое отклонение
Средняя масса с учетом влаrи
для одной для десяти
упаковки 'упаковок
до 100 r 5% 1,6%
от 101 r до 200 r 3% 0,9%
от 201 r до 1000 r 2% 0,6%
При определении микробиолоrической чи
стоты (5. 1.8) необходимо учитывать nрименя
емый метод приroтовления.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Содержание фармаколоrически активных
веществ, обусловливающих фармаколоrическое
действие лекарственноro растительноro сырья,
определяют методом, указанным в частной фар
макопейной статье.
ХРАНЕНИЕ
В контейнере, предохраняющем от воз
действия влаrи и света, при температу
ре от 15 ос до 25 ОС, если в частной статье
нет особых указаний. Не допускается xpaHe
ние лекарственною растительноro сырья в
местах, не защищенных от воздействия по
сторонних запахов.
МАРКИРОВКА
Указывают:
массу содержимою контейнера с учетом
влажности (потеря в массе при высушивании
либо содержание воды) лекарственноro расти
тельноro сырья;
информацию о проведении радиационно
ro контроля.
01/2013:1063
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
НА ОСНОВЕ АЛЛЕРrЕНОВ
Producta а//еrgепiса
Данная общая статья не распространя
ется: на химические вещества, используемые
только для диа2ностики контактных дepMa
титов; на химически синтезированные про
дукты; на аллеР2ены, полученные с ПОМОЩЬЮ
теХНОЛ02ии рекомбинантной ДНк. Лекар
ственные средства на основе аллеР2енов для
ветеринаРНО20 использования не обязатель
но должны отвечать требованиям данной
общей статьи.
Лекарственные средства на основе аллерzенов
1065
ОПРЕДЕЛЕНИЕ
Лекарственные средства на основе аллер
reHoB это лекарственные средства, получен
ные из экстрактов материалов природноro про
исхождения, содержащих аллерrены, то есть
вещества, приводящие к аллерrическим pe
акциям и/или провоцирующие аллерrические
реакции. В большинстве случаев аллерrиче
ские комлоненты имеют белковую природу. Ле
карственные средства на основе аллерrенов
предназначены для диаrностики в условиях iп
WVO или лечения аллерrических заболеваний
(реакции rиперчувствительности), вызванных
данным аллерrеном.
Лекарственные средства на основе аллер
reHoB MorYT выпускаться как в форме roтовых
лекарственных средств, так и в форме roтовых
лекарственных средств, применяемых у KOH
кретных пациентов. Лекарственные средства
на основе аллерrенов обычно применяются для
парентеральноrо введения, rлазноro использо
вания, инrаляций, оральноro и сублинrвальноro
применения и для проведения кожных скарифи
кационных проб.
Лекарственные средства на основе аллер
2енов для диа2ностики в условиях iп vivo обычно
roтовятся как немодифицированные экстракты
в 50 % (об/об) растворе rлицерина для прове
дения кожных скарификационных проб. Для ди
аrностики путем подкожноro введения или для
диаrностики с выполнением эндоназальных,
конъюнктивальных или эндобронхиальных про
вокационных тестов соответствующие растворы
аллерrенов MOryT быть изroтовлены путем раз
ведения водноro или rлицериновоrо экстрактов
или же путем растворения немодифицирован
ных экстрактов, высушенных сублимацией, He
посредственно перед введением.
Лекарственные средства на основе ал
леР2енов, используемые для специфической
иммунотерапии, MorYT быть в виде либо He
модифицированных экстрактов, либо экс
трактов, модифицированных химически иlили
путем адсорбции на различные носители (Ha
пример, rидроксид алюминия, фосфат каль
ция или тирозин).
ПРОИЗВОДСТВО
ИСХОДНblЕ МАТЕРИАЛbl
Источником сырья для приroтовления ле
карственных средств на основе аллерrенов яв
ляются продукты животноro или растительноro
происхождения, rлавным образом пыльца, плес
невые rрибы, клещи, эпителий и покровы живот
ных (такие как волосы и перья) и/или перхоть,
яды перепончатокрылых, насекомые и HeKOTO
рые пищевые продукты.
При использовании в качестве исходноro Ma
териала продуктов животноro или человеческоrо
происхождения, Д(>лжны выдерживаться требо
вания статьи 5. 1. 7. Вирусная безопасность.
135. Зак. 1712
Исходные материалы характеризуются про
исхождением, природой, методом получения
или производства и предварительной обработ
кой. Для проверки их подлинности и чистоты
должны быть разработаны методы контроля Ka
чества и критерии приемлемости. Критерии при
емлемости должны обеспечивать постоянство
качества исходных материалов с точки зрения
качества и количественноro содержания. Исход
ные материалы должны храниться в контроли
руемых условиях, подтвержденных данными по
стабильности.
Пыльца. Содержание потенциально опас
ных химических примесей, таких как пестици
ды, тяжелые металлы и растворители, должно
быть минимизировано. В пыльце допускается
содержание не более 1 % суммарной чужерод
ной пыльцы, и не более 0,5 % индивидуальной
чужеродной пыльцы, что определяется микро
скопическим исследованием. Количество обна
руживаемых спор плесневых rрибов не должно
превышать 1 %. Заrрязнения частицами pac
тительноro происхождения, иными, чем споры,
должно быть сведено к минимуму. Максимальное
содержание примесей должно быть обосновано.
Плесневые rрибы. Биолоrически активные
примеси, такие как микотоксины, в плесневых
rрибах должны быть минимизированы, и любое
их наличие должно нормироваться. Должны при
ниматься необходимые меры для предотвра
щения заrрязнения чужеродными штаммами
плесневых rрибов. Следует принимать все необ
ходимые меры предосторожности, чтобы свести
к минимуму присутствие любых аллерrенных
компонентов в средах, используемых для куль
тивирования плесневых rрибов как источников
исходных материалов. Применение питательных
сред, которые содержат вещества человеческо
ro или животноro происхождения, должно быть
обосновано, и такие среды, при необходимости
их использования, должны быть COOTBeTCTBY
ющим образом обработаны, чтобы rарантиро
вать инактивацию или удаление потенциально
патоrенных areHToB.
Производство должно быть валидирова
но для подтверждения Toro, что лекарственные
средства на основе аллерrенов. получаемые с
использованием плесневых rрибов и предназна
ченные для парентеральноro введения, будут
выдерживать испытание на аномальную токсич
ность для иммунной сыворотки и вакцин для Me
дицинскоro применения при проведении данноro
испытания.
Клещи. Должны приниматься необходи
мые меры для предотвращения заrрязнения
чужеродными видами клещей. Следует прини
мать все необходимые меры предосторожности,
чтобы свести к минимуму присутствие любых ал
лерrенных компонентов в средах, используемых
1066
rосударственная фармакопея Республики Беларусь
для культивирования клещей как источников ис
ходных материалов. Применение питательных
сред, которые содержат вещества человеческо
ro или животноro происхождения, должно быть
обосновано, и такие среды, при необходимости
их использования, должны быть COOTBeTCTBY
ющим образом обработаны, чтобы rарантиро
вать инактивацию или удаление потенциально
патоrенных areHToB.
Эпителий и покровы животных и/или
перхоть. Эпителий животных должен быть по
лучен от здоровых специально отобранных жи
вотных, чтобы исключить попадание возможных
возбудителей антропозоонозных заболеваний.
Яды перепончатокрылых. Виды перепон
чатокрылых, от которых получают яды, должны
быть идентифицированы и специфицирова
ны. Способы сбора насекомых и получения
яда должны быть описаны, и должно быть под
тверждено, что получаемый источник материала
имеет надлежащее качество.
Пищевые продукты. Если возможно,
должно быть указано научное название (вид,
сорт, штамм и т. д.) животноro или растения, а
так же должна быть обозначена используемая
часть. Пищевые продукты должны быть подхо
дящеro для использования человеком качества.
Отмечают источник пищевых продуктов и еro
производственную стадию.
ПРОИ3ВОДСТВЕННЫЙ ПРОЦЕСС
Лекарственные средства на основе аллер
reHoB получают в основном путем экстракции,
возможно с последующей очисткой, из исходных
материалов с использованием подходящих MeTO
дов, сохраняющих аллерrенные свойства компо
нентов. Для аллерrенов, для которых отсутствует
достаточное количество пациентов для опреде
ления их общей аллерrенной активности in vivo
или in vitro, как минимум необходимо указывать
экстракционное соотношение, отображающее
относительные количества (м/об) исходных Ma
териалов аллерrенов и растворителей. Лекар
ственные средства на основе аллерrенов, пред
назначенные для парентеральноro введения,
rлазноro использования, инrаляций и для прове
дения кожных скарификационных проб, должны
производиться в асептических условиях.
При производстве, упаковывании, хранении
и распространении лекарственных средств на
основе аллерrенов, предназначенных для BBe
дения иными путями, проводят необходимые
измерения для подтверждения их микробиоло
rическоro качества; рекомендации приведены в
статье 5. 1.4 МикробиОЛ02ическая чистота He
стериЛЬНblХ лекарственных средств и фарма
цевтических субстанций.
Лекарственные средства на основе аллер
reHoB производятся в условиях, позволяющих
минимизировать экзоrенную и эндоrенную фер
ментные деrрадации.
Процедуры очистки предназначены для
тоro, чтобы свести к минимуму наличие любых
потенциальных раздражающих компонентов с
низкой молекулярной массой или друrих неал
лерrенных компонентов.
Лекарственные средства на основе аллер
reHoB MOryт содержать соответствующий анти
бактериальный консервант, вид и концентрация
котороro должны быть обоснованы.
Производственный процесс включает раз
личные стадии:
исходный материал;
активная субстанция: это обычно модифи
цированный или немодифицированный экстракт
аллерrена; при необходимости она хранится в
условиях, обеспечивающих ее стабильность, Ha
пример, в лиофилизированном виде;
roтовое лекарственное средство.
Все остальные стадии производственноro
процесса рассматриваются как промежуточные.
ВНУТРЕННИЙ ПРЕПАРАТ СРАВНЕНИЯ
В качестве внутреннеro препарата cpaBHe
ния (ВПС, /HRP iп hоиsе ,efe,ence рrераrаtiоп)
выбирают соответствующий репрезентативный
образец, который обладает определенным Ha
бором характеристик и используется дЛЯ KOH
троля постоянства состава лекарственных
средств на основе аллерrенов от серии к серии.
Хранение ВПС осуществляется в виде подходя
щих по размеру аликвот в условиях, rарантиру
ющих еro стабильность, например, в лиофили
зированном виде.
Характеристика BHYTpeHHero препарата
сравнения
Диапазон характеристик ВПС зависит от
природbl исходН020 материала, сведений о е20
аллеР2еННblХ компонентах и наличия подходя
щих реактивов, а также от предназначения.
КвалифицироваННblЙ по отличитеЛЬНblМ xapaK
теристикам ВПС используется как cтaHдapт
НblЙ образец при проверке партии нативных
природНblХ аллеР2еННblХ компонентов или про
межуточных продуктов на основе аллеР2енов,
и, если возможно, при проверке партии 20тo
вых лекарствеННblХ средств на основе аллер
2енов.
Внутренний препарат сравнения xapaKTe
ризуется посредством определения содержа
ния белка и белковоro профиля, для чеro ис
пользуются подходящие методы (например,
изоэлектрическая фокусировка, ДСН ПАr элек
трофорез, иммуноэлектрофорез, капиллярный
электрофорез, хроматоrрафические методы и
масс спектрометрия ).
Аллерrенные компоненты MOryт быть обна
ружены посредством применения COOTBeTCTBY
ющих методов (например, иммуноблоттинrа или
перекрестноro радио иммуноэлектрофореза).
Лекарственные средства на основе аллеР2енов
1067
Характеристика аллерrенных компонентов может
включать идентификацию отдельных аллерrе
нов, основанную на серолоrической или друroй
методике, с использованием общих или индиви
дуальных аллерroсывороток, а также аллерrен
спеuифичных поликлональных или моноклональ
ных антител.
При возможности проводят определение
содержания релевантных аллерrенов. Выбор
релевантных аллерrенных компонентов, co
держание которых проверяют, должен быть
обоснован. При возможности индивидуальные
аллерrены идентифицируют и называют в co
ответствии с принятой международной HOMeH
клатурой.
Если имеются аллерrенные контрольные
вещества, то может быть определено содержа
ние конкретных отдельных аллерrенов. Korдa
это возможно, конкретные отдельные аллерrе
ны идентифицируются в соответствии с меж
дуна родной установленной номенклатурой.
Биолоrическая сила ВПС определяется у
пациентов iп vivo с использованием таких Me
тодик, как кожные скарификационные тесты,
при этом результаты выражаются в единицах
биолоrической активности, кроме тех случаев,
коrда для таких испытаний отсутствует ДOCTa
точное количество пациентов. В этом случае
силу первоro ВПС определяют iп vitro. Далее
биолоrическую активность последующих ВПС
проверяют iп vit,o путем сравнения результатов,
полученных при исследовании первоro ССП.
Исследования iп vitro MOryT быть проведены с
использованием иммунолоrическоro анализа
(например, основанноro на определении инrи
бирования связывающей способности опреде
ленных типов антител иммуноrлобулина Е).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Испытание на подлинность проводят на как
можно более поздней стадии производственно
ro процесса. В случае лекарственных средств,
применяемых у конкретных пациентов, испыта
ние проводят на активной субстанции и/или на
промежуточной стадии при изroтовлении roтово
ro лекарственноro средства.
Идентичность лекарственноrо средства на
основе аллерrенов подтверждается с использо
ванием соответствующих методов идентифика
ции белковоro профиля (например, изоэлектри
ческа я фокусировка, ДСН ПАr электрофорез,
иммуноэлектрофорез, иммуноблоттинr, жидкост
ная хроматоrрафия или масс спектрометрия).
В отдельных случаях, Korдa ВПС недоступ
ны, для подтверждения подлинности может быть
использована репрезентативная серия.
ИСПЫТАНИЯ
Испытание проводят на как можно более
поздней стадии производственноrо процесса. В
случае лекарственных средств, при меняемых у
конкретных пациентов. испытание проводят на
активной субстанции и/или на промежуточной
стадии при изroтовлении roтовоro лекарственно
ro средства.
Для качественноro и количественноro опи
сания аллерrенов разработаны различные био
химические и иммунолоrические испытания. В
случае если такие методы не MOryт быть исполь
зованы, в частности для определения аллерrен
ной активности и аллерrенноro и/или белковоro
профиля, должно быть проведено обоснование
этоro.
Вода (2.5.12 или 2.5.32). Не более 5 % для
лиофилизированных продуктов.
В случае лиофилизированных таблеток,
при соответствующем обосновании содержание
воды может превышать 5 %.
Стерильность (2.6.1). Лекарственные cpeд
ства на основе аллерrенов, предназначенные
для парентеральноro, инrаляционноro, конъюн
ктивальноro введения, и лекарственные cpeд
ства для проведения кожных скарификационных
проб должны выдерживать испытание на CTe
рильность.
Микробиолоrическая чистота. PeKOMeHдa
ции для нестерильных лекарственных средств
приведены в разделе 5.1.4. МикробиОЛ02иче
ская чистота нестериЛЬНblХ лекарствеННblХ
средств и фармацевтических субстанций.
Содержание белка (2.5.33). От 80 % до
120 % от указанноro количества, если не обо
снованы иные нормы. Если может быть опреде
лена биолоrическая активность аллерrена, то
испытание на содержание белка проводят для
подтверждения однородности продукта от серии
к серии, при этом содержание белка должно co
ставлять от 50 % до 150 % от установленноro
значения. Если roтовый продукт содержит бел
ковоподобные вспомоrательные вещества. ис
пытание на содержание белка проводится на
наиболее поздней стадии приrотовления про
дукта до прибавления в неro таких белковопо
добных веществ.
Белковый профиль. Белковый профиль,
полученный с использованием подходящих Me
тодов, должен соответствовать белковому про
филю ВПС. По возможности контролируют
присутствие релевантных аллерrенных компо
нентов. В большинстве случаев выбор контроли
руемых аллерrенных компонентов должен быть
обоснован.
в зависимости от представляюще20 иH
терес аллерzеНН020 продукта моеут исполь
зоваться разлиЧНblе дополнитеЛЬНblе иСПbl
тания. например. с большей селективностью,
но. в любом случае, для аллеР2еННblХ продук
тов. предназначеННblХ для терапевтических
1068
rосударственная фармакопея Республики Беларусь
01/2013:2031
целей, должно применяться валидирован
ное испытание по определению содержания
действующих веществ (общая аллеР2ен
ная активность, определение отдельных ал
лерzенов или любое дРУ20е обоснованное иc
пытание).
АлЮМИНИЙ (2.5.13). Не менее 80 % и не
более 120 % от указанноro количества (в случае
если в качестве адсорбента используется rи
дроксид алюминия или фосфат алюминия), но в
любом случае не более 1.25 Mr в одной челове
ческой дозе, если не предписано иначе.
Кальций (2.5.14). Не менее 80 % и не более
120 % от указанноro количества, в случае если
в качестве адсорбента используется фосфат
,
кальция.
Аллерrенный профиль. Соответствующие
аллерrенные компоненты идентифицируются
посредством надлежащих методов с использо
ванием соответствующих антиrен специфичных
антител человека.
Общая аллерrенная активность. От 50 %
до 200 % от указанноro количества при опре
делении методом инrибирования связывающей
способности определенных типов антител им
муноrлобулина Е или иным подходящим эквива
лентным методом in vitro.
Отдельные аллерrены. От 50 % до 200 %
от указанноro количества каждоro релевантноro
аллерrеннoro компонента, определенноro Haд
лежащим методом.
ХРАНЕНИЕ
Адсорбированное лекарственное средство
на основе аллерrенов не должно подверrаться
замораживанию, если иное не обосновано.
МАРКИРОВКА
На этикетке указывают:
наименование аллерrенноro продукта;
биолоrическую активность (силу), и/илисо
держание белка и/или концентрацию экстракта;
способ применения и назначение;
условия хранения;
наименование и количество добавленноro
антимикробноro консерванта, если необходимо;
для лиофилизированных средств, если
необходимо:
наименование, состав и объем жидко
сти для приroтовления (восстановления), KOTO
рая будет добавлена:
период времени, в течение котороro
должен использоваться приroтовленный (BOC
становленный) раствор;
«стерильно» , если необходимо;
наименование и количество адсорбента,
если необходимо.
МОНОКЛОНАЛЬНЫЕ АНТИТЕЛА
ДЛЯ МЕдицинскоrо ПРИМЕНЕНИЯ
Anticorpo,a monoc/onalia ad usum humanиm
ОПРЕДЕЛЕНИЕ
Моноклональные антитела для медицин
CKOro применения это препараты иммуно
rлобулина или фраrмента иммуноrлобулина,
например, F(ab')2, с определенной специ
фичностью, выработанные одним клоном
клеток. Они MorYT быть конъюrированы с
друrими веществами, включая радиоактив
ные метки.
Они MorYT быть получены из линии «бес
смертных» В лимфоцитов, кО'Юрые клони
рованы и воспроизведены как непрерывные
линии клеток. Также моноклональные антите
ла получают из линии клеток с рекомбинант
ной ДНК.
При подходящих условиях визуальноro
осмотра должны быть практически свободны
ми от частиц.
В настоящее время выпускаются следу
ющие rенно инжинерные рекомбинантные aH
титела.
Химерные моноклональные aHтитe
ла: вариабельные домены тяжелых и леrких
цепей антител человека заменяют на домены
антител животных, которые обладают необхо
димоi1 антиrенной специфичностью.
Iуманизированные моноклональные aH
титела: 3 короткие rипервариабельные по
следовательности (комплементарные дeTep
минантные области) вариабельных доменов
каждой цепи антитела друrих видов животных
встраивают в структуру вариабельных ДOMe
нов антител человека; друrие изменения по
следовательности MorYT быть сделаны для
улучшения связывания антиrена.
Рекомбинантные человеческие монокло
нальные антитела: вариабельные домены
тяжелых и леrких цепей человеческих антител
комбинируют с константной областью антите
ла человека.
Моноклональные антитела, получаемые с
использованием линий клеток, модифициро
ванных с помощью rенной инженерии, также
должны соответствовать требованиям общей
статьи Продукты теХНОЛ02ии рекомбинант
ной ДНК.
Эта общая статья распространяется на
моноклональные антитела, включая конъюrа
ты, для лечебноrо и профилактическоrо при
менения и для использования в диаrностике in
vivo. Она не применяется к моноклональным
антителам, используемым в качестве peareH
тов при производстве лекарственных средств.
Также ее не применяют к моноклональным aH
Моноклональные антитела для медицинскоzо применения
1069
тителам, производящимся с накоплением в
аСЦИ1"ической жидкости, требования к которым
устанавливаются компетентным уполномочен
ным opraHoM.
ПРОИЗВОДСТВО
ОБЩИЕ ПОЛОЖЕНИЯ
Производство моноклональных антител oc
новано на системе серий посевноro материала
с использованием rлавноro банка клеток и, если
применимо, рабочеro банка клеток, получен
ных из клонированных клеток. Метод производ
ства валидируется в ходе разработки продукта
с целью предупреждения передачи инфекцион
ных возбудителей roтовым продуктом. Все био
лоrические материалы и клетки, используемые
в производстве, характеризуются и должны co
ответствовать требованиям статьи 5.2.8. CHи
жение риска передачи возбудителей 2убчатой
энцефалопатии животных при применении
медицинских лекарственных средств. При ис
пользовании в производстве моноклональных
антител для медицинскоro применения матери
алов животноro происхождения или от человека
также при меняются требования статьи 5. 1. 7. Bи
русная безопасность. Если исrюльзуют иммуни
зирующий антиrен, то еro характеризуют и опи
сывают метод иммунизации.
Валидация процесса
В ходе процесса разработки
средства метод производства
следующим критериям:
воспроизводимость процесса производ
ства, включая культуру клеток/ферментацию,
очистку и, rде применимо, метод фраrментации;
удаление или инактивация инфекционных
возбудителей;
адекватное удаление примесей, связан
ных с исходными материалами и образующих
ся в процессе производства (например. белки и
ДНК клетки хозяина, белок А, антибиотики, KOM
поненты культур клеток);
специфичность и биолоrическая актив
ность моноклональных антител;
отсутствие пироrенов, отличных от эндо
токсинов, rдe применимо;
мноroкратность использования компонен
тов системы очистки (например, материал KO
лонки), предельное содержание или критерии
приемлемости устанавливают как валидацион
ный критерий;
используемые методы конъюrирования,
rдe применимо.
производства,
лекарственноrо
валидируют по
Характеристика продукта. Продукт xapaK
теризуют для получения адекватной информа
ции, включая: структурную целостность, изотип,
аминокислотную последовательность, вторич
ную структуру, уrлеводородную часть молекулы.
дисульфидные связи, конформацию. специфич
136 З.. 1712
ность, сродство, биолоrическую активность и re
TeporeHHocTb (характеристика изоформ).
Используют целый ряд соответствующих
аналитических методов, включая химические,
физические, иммунохимические и биолоrиче
ские испытания (например, пептидное карти
рование, последовательность N и C KOHцeBЫX
аминокислот, масс спектрометрия, xpoMaтorpa
фические, электрофоретические и спектроско
пические методы). Дополнительные испытания
выполняют для получения информации о пере
крестной чувствительности с тканями человека.
Для тех продуктов, которые модифицирова
ли путем фраrментации или конъюrации, xapaK
теризуют влияние используемых методов на aH
титела.
Промежуточные продукты. В случае xpa
нения промежуточных продуктов для каждоro
устанавливают срок roдности или период xpa
нения, подтвержденный данными по стабиль
ности.
Количественное определение биолоrи-
ческим методом. Для количественною опре
деления выбирают биолоrический метод, OTpa
жающий предполаrаемый механизм действия
моноклональноrо антитела.
Стандартный образец. В качестве CTaH
дартноro образца для целей идентификации,
количественноro определения и друrих испыта
ний применяют использованную в клиническ х
исследованиях серию лекарственноro средства
с доказанной стабильностью или реперезента
тивную ей. Стандартный образец характери
зуют -соответствующим образом, как указано в
параrрафе «Характеристика продукта», за ис
ключением, если нет необходимости, определе
ния перекрестной чувствительности для каждой
партии стандартноro образца.
Определение партии. Требуется полное
определение партии в течение Bcero процесса
производства.
ИСТОЧНИК КЛЕТОК
К источникам клеток относятся партнеры
слияния, лимфоциты, миеломные клетки, фи
дерные клетки и клетки хозяина для экспрессии
рекомбинантных моноклональных антител.
Документируют происхождение и xapaKTe
ристики родительских клеток, включая инфор
мацию о состоянии здоровья доноров и исполь
зуемых партнеров слияния (наПРlAмер, линия
миеломных клетOI<. линия лифобластоидных
В клеток человека).
По возможности. источники клеток подвер
rаются соответствующему скрининry на наличие
посторонних экэoreнных и эндоrенных areHTOB.
Выбор определяемых вирусов зависит от видов
и происхождения тканей.
1070
rосударственная фармакопея Республики Беларусь
ЛИНИЯ КЛЕТОК, ПРОДУЦИРУЮЩИХ
МОНОКЛОНАЛЬНblЕ АНТИТЕЛА
Приroдность линии клеток, продуцирующих
моноклональные антитела, подтверждают сле
дующим:
документацией об истории линии клеток,
включая описание слияния клеток, иммортали
зацию или трансфекцию и процедуры клониро
вания;
характеристикой линии клеток (например,
фенотип, анализ изоферментов. иммунохимиче
ские маркеры и цитоrенетические маркеры);
характеристикой соответствующих oco
бенностей антител;
стабильностью критических качественных
свойств антитела до или после удвоения попу
ляции или количества rенераций, используемых
при повседневном (рутинном) производстве;
для rенно инженерных рекомбинантных
продуктов стабильностыо кодирующей после
довательности конструкции в клетках, культиви
рованных до предельноro in vitro клеточноro воз
раста, для использования в производстве или
запредельноro возраста путем либо контроля
нуклеиновой кислоты, либо анализа продукта.
БАНКИ КЛЕТОК
rлавный банк клеток представляет собой ro
моrенную суспензию линии клеток, продуциру
ющих моноклональные антитела, расфасован
ных в равных объемах в рамках одной операции
по индивидуальным контейнерам для хранения.
Рабочий банк клеток представляет собой ro
моrенную суспензию линии клеток, полученных
из rлавноro банка клеток при установленном
уровне пассажа и расфасованных в равных объе
мах в рамках одной операции по индивидуаль
ным контейнерам для хранения.
Послепроизводственные клетки это
клетки, культивированные до или выше YДBO
ения популяции клеток или числа rенераций, ис
пользуемых в повседневном производстве.
rлавный банк клеток контролируют по следу
ющим параметрам: жизнеспособность, идентич
ность, отсутствие бактерий. rрибов и микоплазм,
характеристика продуцируемых моноклональ
ных антител. Дополнительно определяют KOHTa
минацию неэндоrенными вирусами с помощью
подходящеro ряда испытаний iп vivo и in vitro.
Определяют контаминацию ретровирусами и
друrими эндоrенными вирусами с использовани
ем ряда подходящих испытаний in V/tro.
Рабочий банк клеток проверяют по следу
ющим параметрам: жизнеспособность, идентич
ность, отсутствие бактерий, rрибов, микоплазм.
Дополнительно оценивают наЛИЧ"1е посторонних
вирусных areHТOB с помощью ряда подходящих
испытаний in vivo и iп vitro. Для первичноro рабо
чеro банка клеток эти исследования выполняют
на послепроизводственных клетках, полученных
из рабочеro банка клеток. Для последующеrо pa
бочеro банка клеток MorYT быть проведены еди
ничные испытания iп vitro и in vivo либо непо
средственно на клетках рабочеro банка, либо на
послепроизводственных клетках.
Испытания на наличие определенных ви
русо в проводятся на rлавном и рабочем банках
клеток, Korдa при приroтовлении банков клеток
используется потенциально контаминирован
ный биолоrический материал, учитывая проис
хождение этоro материала. Испытание можно
не проводить, если этот материал подверrает
ся валидированной процедуре инактивации ви
русов.
Послепроизводственные клетки контроли
руются по следующим параметрам: отсутствие
бактерий, rрибов, микоплазм. Дополнительно
определяют наличие вирусов в клетках или куль
туре клеток супернатантов. Контаминация по
сторонними вирусами определяется с помощью
ряда подходящих испытаний iп vivo и in vitro. Ha
личие ретровирусов и друrих эндоrенных виру
сов оценивают с помощью ряда COOTBeTCTBY
ющих испытаний in vitro.
КУЛЬТИВИРОВАНИЕ И СБОР БИОМАССbI
Производство с использованием orpa-
ниченноrо количества пассажей (однократ-
ный сбор). Клетки культивируют до YCTaHOB
ленноro максимальноro количества пассажей,
или удвоения популяций или до установленно
ro времени сбора (в соответствии со стабиль
ностью линии клеток). Собирают биомассу в
один производственный цикл.
Производство С непрерывным культи-
вированием (мноrократный сбор). Клетки
непрерывно культивируют в течение YCTaHOB
ленноro периода времени (в соответствии со
стабильностью системы и стабильностью про
изводства). Необходимо проводить монито
ринr на протяжении всеro периода культивиро
вания. Требуемая частота и тип мониторинrа
зависят от при роды продуцирующей системы.
Каждый сбор испытывают на содержание
антител, бионаrрузку, содержание эндотокси
нов и наличие микоплазм. Общие или специ
фические испытания на посторонние вирусы
проводятся на соответствующей технолоrиче
ской стадии в зависимости от при роды про
изводственноro процесса и используемых
материалов. При производственном процес
се с использованием оrраниченноro количе
ства пассажей (однократный сбор) посторон
ние вирусы определяют по крайней мере в 3
сборах с помощью ряда подходящих испыта
ний in vitro.
Критерии приемлемости для сборов био
массы в отношении последующей обработки
четко определены и включены в установлен
ный rрафик мониторинrа. Если обнаружены
любые посторонние вирусы, то процесс тща
тельно изучают для определения причины KOH
таминации, и дальнейшую обработку собран
Моноклональные антитела для медuцинско;ю прuменения
1071
ной биомассы не проводят. Сборы биомассы,
в которых обнаружены эндоrенные вирусы,
не используют для очистки, если не разрабо
тан план соответствующих действий по преду
преждению пере носа инфекционных areHToB.
ОЧИСТКА
Сборы или промежуточные пулы биомас
сы MorYT быть объединены перед дальнейшей
обработкой. Процесс очистки включает этапы
по удалению и/или инактивированию безобо
лочечных и оболочечных вирусов. Используют
валидированный процесс очистки, эффектив
ность KOTOpOro по удалению и/или инактива
ции инфекционных areHToB и удалению приме
сей (связанных с продуктом и технолоrическим
процессом) была подтверждена. Четко разра
ботанные этапы процесса обеспечивают по
лучение очищенных моноклональных антител
(активных субстанций) с постоянным каче
ством и биолоrической активностью.
АКТИВНАЯ СУБСТАНЦИЯ
Объем испытаний активной субстанции
зависит от валидации технолоrическоrо про
цесса, подтверждения стабильности процесса
и ожидаемоro уровня примесей, обусловлен
ных продуктом и технолоrическим процес
сом. Очищенные активные субстанции испы
тывают соответствующими аналитическими
методиками на внешний вид (прозрачность,
цветность), подлинность, степень микробио
лоrической контаминации (бионаrрузка) и co
держание бактериальных эндотоксинов, Ha
личие веществ, обусловленных продуктом,
наличие примесей, обусловленных продуктом
и технолоrическим процессом (включая ис
пытания на белки из клетки хозяина и ДНК из
клетки хозяина и вектора), а также на CTPYK
турную целостность, содержание белка и био
лоrическую активность, при необходимости,
сравнивая со стандартным образцом. Если
активная субстанция представляет собой
конъюrированное или модифицированное aH
титело, соответствующие испытания должны
быть проведены до и после конъюrации/моди
фикации.
Если предусмотрено хранение полупродук
тов, то проводится оценка надлежащей стабиль
ности этих препаратов и влияние этоro этапа на
качество или срок roдности roToBoro продукта.
rотовый НЕФАСОВАННЫЙ ПРОДУКТ
rотовый нефасованный продукт получают
из одной или нескольких партий очищенной aK
тивной субстанции. Соответствующие стаби
лизаторы и друrие вспомоrательные вещества
MOryT быть добавлены в процессе приrотовле
ния roтовоro нефасованноro продукта.
rотовый нефасованный продукт должен
храниться при валидированных условиях xpaHe
ния с учетом бионаrрузки и стабильности.
СЕРИЯ rOTOBOrO ЛЕКАРСТВЕнноrо
СРЕДСТВА
rотовый нефасованный продукт подверrа
ют стерильной фильтрации и расфасовывают в
асептических условиях в стерильные контейне
ры, в которых может проводиться дальнейшая
лиофилизация.
Как часть внутрипроизводственноro KOH
троля, каждый контейнер (флакон, шприц или
ампула) после наполнения должен проверять
ся для исключения контейнеров, содержащих
видимые частицы. При разработке продукта
должно быть продемонстрировано, что процесс
производства не приводит к появлению видимых
белковоподобных частиц либо такие частицы
должны измельчаться до более низкоro уровня,
который обоснован и утвержден.
ОПИСАНИЕ (СВОЙСТВА)
Жидкие лекарственные средства представ
ляют собой прозрачные или слеrка опалесци
рующие, бесцветные или слеrка окрашенные
жидкости. Лиофилизированные продукты
это белые или слеrка окрашенные порошки или
твердая рыхлая масса. После восстановления
(получения раствора) имеют те же характери
стики, что и жидкие лекарственные средства.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Подлинность устанавливают COOTBeTCTBY
ющими валидированными методиками, при He
обходимости сравнивая продукт со стандартным
образцом. Количественное определение также
учитывают при подтверждении подлинности.
ИСПЫТАНИЯ
Внешний вид #(прозрачность, цвет-
ность)#. Жидкие или восстановленные лиофи
лизированные лекарственные средства должны
выдерживать соответствующие требования для
конкретноro продукта по прозрачности (2.2.1) и
цветности (2.2.2). Они не должны содержать ви
димых частиц, если иное не обосновано и не YT
верждено.
Растворимость. Лиофилизированные ле
карственные средства должны полностью pac
творяться в указанном объеме растворителя в
течение определенноrо времени, как утвержде
но для KOHKpeTHoro продукта.
рН (2.2.3). Должен соответствовать Tpe
бованиям, установленным для конкретноro
продукта.
Осмоляльность (2.2.35). Не менее
240 мОсмол/кr. если иное не доказано и не YT
верждено.
Извлекаемый объем (2.9.17). Должен BЫ
держивать испытание на определение извлека
емоro объема.
1072
rосударственная фармакопея Республики Беларусь
Общий белок (2.5.33). Должен COOTBeTCTBO
вать требованиям, установленным для KOHKpeT
Horo продукта.
Определение размера молекул. Опреде
ление размера молекул проводят COOTBeTCTBY
ющим методом, например, эксклюзионной xpo
матоrрафией (2.2.30). Должен соответствовать
требованиям, установленным для конкретноro
продукта.
Молекулярная идентичность и CTPYKTYP
ная целостность. В зависимости от при роды
моноклональных антител, их микроrетероrенно
сти и изоформ может быть использован целы.й
ряд различных испытаний для подтверждения
молекулярной идентичности и структурной цe
лостности. Эти испытания MOryT включать пеп
тидное картирование, изоэлектрическое <POKY
сирование, ионнСК>бменную хроматоrрафию,
rидрофобную интерактивную хроматоrрафию,
олиrосахаридное картирование, содержание MO
носахаридов и масс<пектрометрию.
Чистота. Подходящим методом проводят
испытание по определению примесей, обуслов
ленных процессом производства и самим про
дуктом. При представлении данных, доказы
вающих проведение испытаний для активной
субстанции или roтовоro нефасованноro продук
та по определению примесей, обусловленных
процессом производства, с получением YДOB
летворительных результатов, такие испытания
MOryT не проводиться для roтовоro продукта.
Стабилизатор. rде применимо, еro coдep
жание должно соответствовать требованиям,
установленным для определенноro продукта.
Вода (2.5.12). Лиофилизированные лекар
ственные средства должны соответствовать Tpe
бованиям, установленным для конкретноro про
дукта.
Стерильность (2.6.1). Должен быть CTe
рильным.
Бактериальные эндотоксины (2.6.14).
Должны соответствовать требованиям, YCTaHOB
ленным для конкретноro продукта.
Испытания, при меняемые для модифи-
цированных антител. Выполняют COOTBeT
ствующие испытания, зависящие от типа моди
фикации.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Выполняют соответствующее биолоrиче
ское количественное определение с использова
нием стандартноro образца. План испытания и
расчет результатов осуществляют с использова
нием общепринятых принципов (например, 5.3).
ХРАНЕНИЕ
В соответствии с указаниями на этикетке.
Срок 20дности. Срок roдности. YCTaHaB
ливают от даты стерилизующей фильтрации,
даты розлива (для жидких лекарственных
средств) или даты лиофильной сушки (rдe при
менимо ).
МАРКИРОВКА
На этикетке указывают:
количество единиц действия на милли
литр, rде применимо;
содержание белка в контейнере;
содержание моноклональных антител на
контейнер;
для жидких лекарственных средств
объем лекарственноro средства в контейнере;
для лиофилизированных лекарственных
средств:
название и объем растворителя для
приroтовления восстановленноro раствора;
период времени, в течение котороro
моноклональные антитела должны быть исполь
зованы после восстановления;
разведение, которое необходимо сделать
перед использованием лекарственною cpeд
ства, rде применимо.
01/2013: РБОО02
#НАСТОИ, ОТВАРЫ И ЧАИ
/пfusa, decocta et theae
ОПРЕДЕЛЕНИЕ
Настои, отвары и чаи свежеприroтовлен
ные водные извлечения из лекарственноro pac
тительноro сырья, сборов, растительных чаев,
а также водные растворы сухих или жидких экс
трактов (концентратов).
изrОТОВЛЕНИЕ
При изroтовлении настоев, отваров и
чаев используют измельченное лекарствен
ное растительное сырье, сборы и раститель
ные чаи, отвечающие требованиям статей
Лекарственное растительное сырье, #Ле
карственное растительное сырье цельное
или измельченное фасованное, #СБОрbl, Pac
титеЛЬНblе чаи.
Для приroтовления настоев и отваров
измельченное лекарственное растительное
сырье заливают водой (водой Р при изrотов
лении в аптеках) комнатной температуры, Ha
стаивают в инфундирном аппарате или в co
ответствующей емкости в водяной бане при
частом помешивании: настои в течение
#Настои, отвары и чаu
1073
15 мин, отвары в течение 30 мин, затем ox
лаждают при комнатной температуре: настои
не менее 45 мин, отвары 1 О мин, проце
живают (отжимая растительное сырье) и при
бавляют воду до требуемоrо объема извлече
ния.
Для приrотовления чаев указанное количе
ство измельченноro лекарственноro раститель
ноro сырья заливают указанным количеством
кипящей воды и выдерживают в течение указан
ноro промежутка времени.
При отсутствии указаний о количестве ле
карственноro растительноro сырья настои и
отвары roтовят в соотношении 1: 1 о; из травы
rорицвета, корневищ валерианы с корнями
1 :зо; из лекарственноro растительноro сырья,
содержащеroсильнодействующие вещества,
1 :400. Для приroтовленИ'я настоев и отваров
измельченное лекарственное растительное
сырье заливают водой Р комнатной темпера
туры, взятой с учетом коэффициента водопо
rлощения (таблица РБОО02. 1).
Примечания:
1. Коэффициент водопоrлощения соответ
ствует количеству жидкости (мл), удержива
емому 1,0 r лекарственноro растительною сырья
после еro отжатия в перфорированном стакане
инфундирки.
2. Если коэффициент водопоrлощения для
сырья отсутствует, используют следующие еro
значения:
для корней и корневищ 1,5 млlr;
для коры, травы и цветков 2,0 мл/r;
для семян 3,0 мл/r;
для брикетов 2,3 мл/r.
3. Из корня алтея roтовят водное извлече
ние методом холодноro настаивания, даже если
прописан настой или отвар. Расходные коэффи
циенты для изroтовления водноro извлечения
корней алтея различной концентрации:
1 % 1,05;
2 % 1,1 о;
3 % 1,15;
4% 1,20;
5 % 1,30.
Расходный коэффициент, используемый при
изroтовлении водноro извлечения корней алтея, по
казывает, во сколько раз следует увеличить массу
сырья и объем экстраrента, чтобы получить задан
ный объем извлечения необходимой концентрации.
4. Для водноro извлечения корней алтея с
концентрацией более 5 % расходный коэффици
ент (К ) рассчитывают по формуле:
р
100
100 (C.V)
rдe:
С выписанная в рецепте концентрация
водноro извлечения, %;
V объем водноro извлечения, удержива
емый 1 r сырья (4,6 мл).
При изrотовлении настоев, содержащих
сердечные rликозиды или алкалоиды, приме
няют лекарственное растительное сырье в co
ответствии с биолоrической активностью или
с определенным содержанием алкалоидов.
Если используют сырье с большей биолоrиче
ской активностью или большим содержанием
алкалоидов, то вместо прописанноro берут KO
личество сырья, рассчитанное по формуле:
А-С
,
в
rдe:
А прописанное количество лекарственно
ro растительноro сырья;
Таблица РБОО02. 1
Коэффициенты водОП02лощения лекарствеННО20 раститеЛЬН020 сырья
Наименование лекарствен- Коэффициент Наименование лекарстве н- Коэффициент
Horo растительноrо сырья водопоrлощения Horo растительноrо сырья водопоrлощения
Кора дуба 2,0 Листья шалфея 3,3
Кора калины 2,0 Плоды рябины 1,5
Кора крушины 1,6 Плоды шиповника 1,1
Корни аира 2,4 Трава roрицвета ! 2,8
Корни солодки 1,7 Трава зверобоя 1,6
Корневища змеевика 2,0 Трава ландыша 2,5
Корневища с корнями 2,9 Трава полыни 2,1
валерианы
Корневища лапчатки 1,4 Трава пустырника 2,0
Листья брусники 1,5 Трава сушеницы 2,2
Листья крапивы 1,8 Трава хвоща полевоro 3,0
Листья мать и мачехи 3,0 Трава череды 2,0
Листья мяты 2,4 Цветки липы 3,4
Листья подорожника 2,8 ; Цветки ромашки 3,4
Листья сены 1,8 Шишки хмеля 3,2
Листья толокнянки 1,4
1074
rосударственная фармакопея Республики Беларусь
в фактическое количество единиц дей
ствия или алкалоидов в 1 r сырья;
С стандартное содержание rликозидов
или алкалоидов в 1 r сырья.
Сырье с меньшей биолоrической активно
стью или с меньшим содержанием алкалоидов
для изroтовления настоев не применяют.
При изroтовлении настоев и отваров из
лекарственноrо растительноrо сырья, co
держащеrо алкалоиды. прибавляют кисло
ту хлористоводородную Р (в пересчете на
HCI), причем кислоты берут по массе столь
ко, сколько содержится алкалоидов во взятом
количестве лекарственноrо растительноro
сырья.
Отвары из листьев толокнянки, брусники и
сырья, содержащеro дубильные вещества (кора
дуба, корневище змеевика и др.), процежива
ют без охлаждения, отвары из листьев сенны
после полноro охлаждения.
Лекарственные вещества растворяют в
процеженном извлечении. Сиропы, настойки и
жидкие экстракты прибавляют к полученному
настою, отвару или водному извлечению.
При изroтовлении водных растворов экс
трактов (концентратов) последние берут в KO
личестве, соответствующем количеству лекар
ственноro растительноro сырья.
При необходимости к водным извлечениям
прибавляют консерванты (метилпараrидрокси
бензоат. пропилпараrидроксибензоат, кислоту
сорбиновую и друrие. разрешенные компетент
ным уполномоченным opraHoM).
ХРАНЕНИЕ
Настои и отвары хранят в течение указан
ноro срока roдности при температуре от 8 ос до
15 ОС. Чаи используют свежеприroтовленными.
МАРКИРОВКА
Перед употреблением взбалтывать.
01/2013:0784
ПРОДУКТЫТЕхнолоrии
РЕКОМБИНАНТНОЙ ДНК
Producta аЬ arte ADN ,есотЫпапdоrит
Данная общая статья содержит общие Tpe
бования к разработке и производству проду
тов технолоrии рекомбинантной ДНК (reHHo
инженерных рекомбинантных лекарственных
средств). В определенных случаях эти требо
вания MorYT не быть исчерпывающими, допол
нительные и расширенные требования, помимо
приведенных в настоящей статье, приводятся в
частной статье и устанавливаются компетент
ным уполномоченным opraHoM.
Данная статья не применяется к модифи
цированным живым орrанизмам, которые пред
назначены для непосредственноrо примене
ния у человека, например, в качестве живых
вакцин.
ОПРЕДЕЛЕНИЕ
Продукты технолоrии рекомбинантной ДНК
получают путем rенной модификации, при KO
торой В подходящий микроорrанизм или клетку
посредством плазм иды или вирусноro Beктo
ра вводят ДНК, кодирующую требуемый про
дукт. В этих клетках хозяина ДНК экспрессиру
ется и транслируется в белок. Затем требуемый
продукт получают путем выделения и очистки.
Клетки или микроорrанизмы, в которые вводит
ся вектор, называются клетками хозяина, а их
стабильная ассоциация, используемая в про
цессе производства, называется системой xo
зяин вектор.
ПРОИЗВОДСТВО
Производство основано на валидированной
системе серий посевноro материала, в которой
используется комбинация хозяин вектор, COOT
ветствие требованиям компетентноro уполномо
ченноro opraHa которой было доказано. В систе
ме посевноro материала используется rлавный
банк клеток и рабочий банк клеток, полученный
из исходной rлавной серии комбинации хозяин
вектор. Должно иметься подробное описание
стадий культивирования, выделения и порядок
установления серии продукта.
Если продукты технолоrии рекомбинант
ной ДНК производят с использованием матери
алов животноro происхождения и от человека,
то также применяют требования статьи 5. 1. 7. Bи
русная безопасность.
Определение приемлемости комбинации
хозяин вектор и валидация системы серий по
севной культуры включают описанные далее
элементы.
КЛОНИРОВАНИЕ И ЭКСПРЕССИЯ
Приемлемость системы реципиент вектор,
в частности микробиолоrической чистоты, ДOKa
зывают путем:
характеристики клеток хозяина, вклю
чая происхождение, фенотип и 2енотип, и xa
рактеристики питательной среды для клеток;
документированной страте2ии клониро
вания 2ена и характеристики рекомбинантно
20 вектора, включая:
происхождение и характеристику reHa;
анализ нуклеотидной последователь
ности клонированноro reHa и промежуточные
реrуляторные участки вектора для экспрессии;
количество клонированных последователь
ностей сводят к минимуму и все COOTBeTCTBY
ющие экспрессивные последовательности
Продукты технолоаии рекомбuнантной днк
1075
четко идентифицируют и подтверждают на
уровне РНК; последовательность ДНК кло
нированных reHoB обычно подтверждают на
стадии серии посевной культуры, до или выше
нормальноrо уровня удвоения популяции для
полномасштабной ферментации; в HeKOТO
рых системах, например, Korдa мноroчислен
ные копии reHa вставляют в reHoM непрерыв
ной линии клеток, определение нуклеотидной
последовательности клонированноrо reHa на
промышленном уровне может быть неприем
лемым; при данных обстоятельствах, может
быть проведен анализ общей клеточной ДНК
методом Саузерн блота или анализ последова
тельности матричной (информационной) РНК
(мРНК), особое внимание уделяют характери
стике экспрессивноro белка;
конструкцию, rенетические особенно
сти и структуру полноro вектора экспрессии;
характеристики системы хозяин век
тор, включая:
механизм введения вектора в клетки
хозяина;
номер копии, физическое состояние и
стабильность вектора внутри клетки хозяина
механизмы, используемые для обеспе
чения и контроля экспрессии.
СИСТЕМА БАНКА КЛЕТОК
/лавный банк клеток это однородная cy
спензия исходных клеток, уже с введенным BeK
тором для экспрессии, содержащим требуемый
reH, расфасованная в равных объемах в индиви
дуальные контейнеры для хранения (например,
в жидком азоте). В некоторых случаях может
потребоваться создание отдельных rлавных
банков клеток для вектора экспрессии и клеток
хозяина.
Рабочий банк клеток это однородная
суспензия клеточноrо материала, полученная
из rлавноro (rлавных) банка (банков) клеток при
установленном уровне пассажа, расфасован
ная в индивидуальные контейнеры в равных
объемах для хранения (например, в жидком
азоте ).
В обоих банках клеток все контейнеры
хранят в одинаковых условиях и после извлече
ния контейнеры не возвращают в место xpaHe
ния.
Банк клеток может быть использован для
производства с установленным уровнем пасса
жа или с непрерывным культивированием.
Производство с установленным уровнем
пассажа
При этом методе культивирования YCTaHaB
ливается опредепенное количество пассажей
или удвоений популяции, которое нельзя пре
вышать в ходе производства. Должно быть YCTa
новлено максимальное количество удвоения
клеток или пассажей, при которых повседневный
производственный процесс соответствует крите
риям, указанным ниже.
Производство с непрерывным культивиро
ванием
При этом методе культивирования не orpa
ничивают количество пассажей или удвоений по
пуляции от начала производства. Критерии для
сбора полученноro продукта, а также для пре
кращения культивирования определяются про
изводителем. Необходимо проводить монито
ринr в течение всеro процесса культивирования;
требуемая частота и тип мониторинrа зависят от
природы системы производства и продукта.
После длительною культивирования требу
ется собрать данные о молекулярной целостно
сти reHa, который был экспрессирован, и о фе
нотипических и rенотипических характеристиках
клеток хозяина. Требования к собираемой био
массе для последующей обработки должны
четко коррелировать с rрафиком внутрипроиз
водственноro контроля. Также требуется опреде
лить порядок формирования «серии)} продукта,
подлежащеro последующей обработке.
ВАЛИДАЦИЯ БАНКОВ КЛЕТОК
Валидация банков клеток включает:
оценку стабильности, устанавливаемую по
жизнеспособности и удержанию вектора;
подтверждение подлинности клеток по фе
нотипическим особенностям;
подтверждение, rде применимо, OTCYT
ствия в банках клеток потенциально oHKoreH
ных или посторонних инфекционных areHToB
(вирусов, бактерий, rрибов или микоплазм);
особое внимание должно быть уделено виру
сам, которые обычно присутствуют в видах жи
вотных, от которых была получена линия клеток;
определенные линии клеток содержат эндоrен
ные вирусы, например, ретровирусы, которые
сложно удалить; экспрессия этих орrанизмов
должна быть изучена при различных условиях.
в которых она может происходить;
получение дпя клеток млекопитающих данных
о потенциальной канцероrенности банка клеток
КОНТРОЛЬ КЛЕТОК
Должны быть полностью документированы
данные о происхождении, форме. хранении,
рядке использования для всех банков клеток в yc
ловиях хранения и восстановления. Новые банки
клеток должны быть полностью валидированы.
ВАЛИДАЦИЯ ПРОЦЕССА ПРОИЗВОДСТВА
Выделение и очистка
Должна быть валидирована эффективность
каждоro этапа выделения и очистки в отношении
удаления и/или инактивации контаминирующих
веществ. которые пpt>ИCходят от клеток хозяина
или из питательной среды, включая, в частно
сти. вирусные частицы, белки, нуклеиновые кис
лоты и добавленные вещества.
Работы по валмдации выполняют с целью
подтвер>IЩения соответствия стандартною про
цесса производства следующим критериям:
1076
rосударственная фармакопея Республики Беларусь
удаление посторонних areHToB из про
дукта; в изучение включают, например, опре
деление вирусов с соответствующими физико
химическими параметрами и оценку снижения
содержания таких контаминантов на каждой
стадии очистки;
адекватное удаление из продукта вектора,
клеток хозяина, питательной среды и примесей,
образующихся из реактивов; уменьшение co
держания ДНК устанавливают методом добавок;
уменьшение содержания белков животноro про
исхождения может быть определено иммунохи
мическими методами;
поддержание в установленных пределах
выхода продукта из культуры;
адекватная стабильность всех полупро
дуктов иlили полуфабрикатов, если технолоrией
предусмотрено их промежуточное хранение.
Характеристика вещества
В начале производства устанавливают под
линность, чистоту. активность и стабильность
roтовоro нерасфасованноro продукта с помо
щью широкоrо ряда химических, физических,
иммунохимических и биолоrических испытаний.
Перед выпуском в обращение каждая серия про
дукта испытывается производителем на подлин
ность и чистоту. а также соответствующее коли
чественное определение.
Стабильность производства
Проводят соответствующие исследования
по подтверждению стабильности производства
и очистки. В частности, исследования включают
испытания по характеристике продукта, внутри
производственный контроль и испытания roтово
ro продукта, как показано далее.
АМИНОКИСЛОТНЫЙ СОСТАВ
Частичный анализ аминокислотной после
довательности. Данные анализа последова
тельности позволяют подтвердить правильность
сборки N конца и обнаружить потерю аминокис
лот C KOHцa.
Пептидное картирование. Результаты пеп
тидноro картирования с использованием хи
мическоrо и/или ферментативноro rидролиза
белковоro продукта и с применением приroдно
ro метода оценки, например, двумерноro элек
трофореза или жидкостной хроматоrрафии, не
должны выявить значительной разницы между
испытуемым белком и стандартным образцом.
Пептидное картирование также может быть ис
пользовано для подтверждения правильных
дисульфидных связей.
ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОЙ СВЯЗИ
Удержание клонироваНН020 2ена. Мини
мальный процент клеток, содержащих вектор
или клонированный reH после культивирования,
соrласовывается компетентным уполномочен
ным opraHoM.
Общий белок. Определяют выход получа
емоro белка.
Химическая чистота. Чистоту белковоro
продукта оценивают в сравнении со CTaHдapT
ным образцом при помощи соответствующеro
метода, например, жидкостной хроматоrрафи
ей, капиллярным электрофорезом с натрия дo
децилсульфатом в полиакриламидном rеле.
Белки клеток хозяина. Белки клеток хозяина
обнаруживают иммунохимическими методами, ис
пользуя, например, поликлональную антисыво
ротку к компонентам системы хозяин вектор, ис
пользуемой для производства продукта, если не
указано иначе в частной статье. Следующие типы
методик MOryт быть использованы: количествен
ное определение с замещением в жидкой фазе
(например, радио---иммунолоrическое количе
ственное определение, количественное определе
ние с прямым связыванием в жидкой фазе и коли
чественное определение с прямым связыванием
с использованием антиrенов, иммобилизованных
на нитроцеллюлозных (или подобных) мембранах
(например, дот анализ, Вестерн блот). Общие Tpe
бования для валидации методик иммунолоrиче
скоro количественноro определения даны в статье
2.7.1. Иммунохимические методы. Дополнитель
но методики иммунолоrическоro количественноro
определения контаминантов, связанных с клетка
ми хозяина, соответствуют следующим критериям.
Препараты анти2енов. Получают анти
сыворотки к препарату антиrенов, полученных
от орrанизма хозяина, в который был введен ис
пользуемый в процессе производства вектор, в
котором отсутствует специфический reH, кодиру
ющий продукт. Эти клетки хозяина культивиру
ют и выделяют белки в условиях стандартноro
производственноro процесса. При приroтовле
нии антисыворотки можно также использовать
частично очищенные препараты антиrенов, по
лученные при использовании некоторых стадий
очистки производственноro процесса.
Калибровка и стандартизация. Количе
ственные результаты получают при сравнении с Ka
либровочной кривой зависимости ответа от дозы,
полученной для стандартных образцов, белков
антиrенов клетки хозяина. Поскольку эти препара
ты представляют собой смесь плохо очищенных
белков, стандартный образец roтовят и калибру
ют с помощью подходящеro метода определения
белка. Стандартный образец хранят в стабильном
состоянии, обеспечивающем еro использование в
течение продолжительноro времени.
Антисыворотки. Антисыворотки coдep
жат высокоактивные антитела со сродством, по
возможности, 1{ большому ряду белков антиrен
ной смеси и не св зываются с продуктом.
ДНК клеток хозяина и вектора. Остаточную
ДНК обнаруживают с помощью rибридизационно
ro анализа, используя соответствующую чувстви
тельную не зависящую от последовательности
аналитическую методику или друrие подходящие
чувствительные аналитические методы.
Продукты ферментации
1077
01/2013:1468
rибридизационный анализ
ДНК в испытуемом образце денатуриру
ют для получения одноцепочечной ДНК, KOTO
рую наносят на нитроцеллюлозный или друroй
подходящий фильтр и rибридизируют с меченой
ДНК, полученной из производственной системы
хозяин вектор (ДНК зонды). Несмотря на нали
чие разнообразных экспериментальных Meтo
дов, все rибридизационные методики для изме
рения ДНК клеток хозяина отвечают следующим
требованиям.
ДНК зондbl. Получают очищенную ДНК из
системы хозяин вектор в условиях CTaHдapTHO
ro производственноro процесса. Хромосомные
ДНК хозяина и вектора MOryт быть приroтовле
ны отдельно и использованы в качестве зондов.
Калибровка и стандартизация. Коли
чественные данные получают при сравнении с
ответом, полученным для стандартных образ
цов. Используются ДНК зонды хромосом и ДНК
зонды вектора и стандарты ДНК хромосом и ДНК
вектора соответственно. Стандартные образцы
калибруют с помощью спектроскопических изме
рений и хранят в состоянии, позволяющем ис
пользовать их продолжительное время.
Условия 2ибрuдизации. Жесткие условия
rибридизации обеспечивают специфическую rи
бридизацию между зондами и стандартными
образцами ДНк. Используемые концентрации
лекарственных веществ не должны мешать rи
бридизации.
Методы, не зависящие от последова
тельности
К приrодным методам относятся: обнару
жение сульфонированных остатков цитозина
в одноцепочечной ДНК (ДНК иммобилизуют на
фильтр и цитозины расщепляют in situ перед
обнаружением и количественным определени
ем с использованием антител к сульфониро
ванным rруппам); обнаружение одноцепочечной
ДНК с использованием фраrмента одноцепо
чечной ДНК, связанной с белком, и антите
ла к этому белку. Для испытания не требуется
специфическая ДНК клетки хозяина или BeKТO
ра в качестве стандартноro образца. Однако ис
пользуемые методы должны быть валидированы
для обеспечения параллельности с использу
емым ДНК стандартом, линейности ответа и OT
сутствия мешающеro влияния или лекарствен
Horo вещества, или вспомоrательных веществ в
разведениях, используемых в количественном
определении.
ПОДЛИННОСТЬ, ИСПЫТАНИЯ
И КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Требования, которым должен COOTBeTCTBO
вать rотовый продукт (нерасфасованный MaTe
риал или лекарственная форма) на протяжении
CBoero срока roдности, а также специфиче
ские методики испытаний приведены в частной
статье.
ПРОДУКТЫ ФЕРМЕНТАЦИИ
Producta аЬ fеrтепtаtiопе
Данная общая статья касается побоч
ных (неПРЯМblХ) 2енетических продуктов, по
лучаеМblХ посредством ферментации. Она
не имеет отношения к:
статьям Фармакопеи, касающимся
вакцин для медициНСКО20 применения;
продуктам, производным от перевива
емых клеточных линий животН020 или чело
вечеСК020 происхождения;
непосредствеННblМ 2енетическим про
дуктам, которые являются результатом
транскрипции 2ена и преобразования инфор
мации нуклеиновой кислоты в лротеин, вне
зависимости от mozo, подлежат эти про
теины модификации после преобразования
или нет;
полусинтетuческим продуктам, полу
чаеМblМ из продуктов ферментации и про
дуктов, получаемых посредством биоката
литичеСК020 преобразования;
цельным концентратам или сырьевым
ферментативным продуктам.
Эта статья содержит общие требова
ния к разработке и производству продуктов
ферментации. Эти требования не являются
всеобъемлющими для кажд020 конкретН020
случая. Дополнительные требования к пред
писаниям данной статьи М02ут бblть изло
жены в частной статье или компетентНbI
ми Ор2анами.
ОПРЕДЕЛЕНИЕ
Исходя из задач данной статьи, продук
ты ферментации определяются как активные
или неактивные фармацевтические веще
ства, получаемые в процессе контролиру
емой ферментации как непрямые продук
ты reHa. Они представлены первичными или
вторичными метаболитами микроорrанизмов
типа бактерий, дрожжей, rрибов и микроско
пических морских водорослей. модифици
рованными либо немодифицированными в
соответствии с традиционными технолоrи
ческими процессами или в соответствии с
технолоrиями рекомбинантной ДНК (рДНК).
Такие метаболиты включают витамины, ами
нокислоты, антибиотики, алкалоиды и поли
сахариды.
Они MorYT быть получены в ходе фермен
тативноrо процесса по производству одной
партии (серии) либо в ходе непрерывноro фер
ментативноro процесса, который сопровожда
ется такими технолоrическими процедурами,
как экстракция, концентрирование, очистка и
изолирование.
1078
rосударственная фармакопея Республики Беларусь
ПРОИ3ВОДСТВО
Производство должно быть основано на
воспроизводимом процессе, который прошел
процедуру подтверждения. Степень воспроиз
водимости определяется по критическому (ли
митирующему) этапу соответствующеro TeXHO
лоrическоro процесса.
ХАРАКТЕРИСТИКА микроорrАНИ3МА
ПРОИЗВОДИТЕЛЯ
Должна документироваться история раз
вития микроорrанизма, используемоro в про
изводственном процессе. Характеристика
микроорrанизма может включать в себя опре
деление ero фенотипа, макроскопических и
микроскопических признаков, биохимических
свойств. Если необходимо. проводится опре
деление rенотипа микроорrанизма и ero MO
лекулярных rенетических свойств.
ПРОЦЕССЫ С ИСПОЛЬЗОВАНИЕМ
СИСТЕМЫ ПОСЕвноrо МАТЕРИАЛА
/лавНblй банк клетОЧНblХ культур MиKpO
ОР2анизмов это rомоrенная суспензия или
лиофилизат ориrинальных микроорrанизмов,
распределенных по индивидуальным KOH
тейнерам для хранения. Жизнеспособность
и производительность микроорrанизмов при
выбранных условиях хранения и их приroд
ность для ввода в производственный процесс
после хранения должны быть очевидными и
надлежащим образом подтвержденными.
Промежуточный rлавный банк клеточных
культур микроорrанизмов, который может
быть получен с использованием системы по
севноro материала. используется для созда
ния рабочеro банка клеточных культур микро
орrанизмов
Рабочий банк клетОЧНblХ культур MиKpO
ОР2анизмов это rомоrенная суспензия или
лиофилизат микроорrанизмов, полученных
из rлавноrо банка культур микроорrанизмов,
распределенный в равных объемах по инди
видуальным контейнерам для хранения (Ha
пример, в жидком азоте).
Производство может происходить в виде
одноro законченноro цикла или быть непре
рывным производственным процессом, KOTO
рый может заканчиваться при определенных
условиях.
Все контейнеры банка клеточных культур
микроорrанизмов хранятся в одинаковых yc
ловиях. После изъятия ампул или флаконов
из мест хранения они не возвращаются об
ратно в банк клеток.
ПРОЦЕССЫ С ИСПОЛЬЗОВАНИЕМ
СТУПЕНЬЧАтоrо РОСТА КУЛЬТУР
микроорrАНИ3МОВ
Содержимое емкости рабочеro банка кле
точных культур микроорrанизмов (при He
обходимости после ресуспендирования) ис
пользуется для приrотовления прививочноrо
материала в подходящую питательную среду.
После соответствующеro периода роста куль
туры используются для инициации фермен
тативноro процесса (если это необходимо,
после этапа прекультивирования в префер
ментаторе). Условия. которые необходимы на
каждой стадии процесса, строro определяют
ся и ДОЛЖНЫ выполняться при каждом произ
водственном цикле.
КОНТРОЛЬ ИЗМЕНЕНИЙ
Если в ходе производственноrо процесса
происходят какие либо изменения, которые cy
щественным образом MorYT изменить профи
ли примесей продукта, критические этапы про
изводства, связанные с этими изменениями,
ДОЛЖНЫ утверждаться повторно.
Если существенное изменение затраrи
вает сам микроорrанизм, используемый для
производства, и влечет за собой существен
ное изменение в профиле примесей продук
та, все критические этапы производственно
ro процесса, связанные с этим изменением,
в частности, процедура очистки и изолирова
ния, также утверждаются повторно.
Соrласование произошедших измене
ний включает в себя подтверждение Toro.
что новые примеси в продукте, появивши
еся вследствие изменения, MorYT COOTBeT
ствующим образом контролироваться в ходе
производственноrо процесса. Если возни
кает необходимость в дополнительных или
альтернативных испытаниях, то они должны
вводится с определенными оrраничениями.
Если изменение в процессе или в микроор
rанизмах приводит к увеличению уровня уже
присутствующих примесей, то приемлемость
такоro увеличения должна быть четко YCTa
новлена.
При замене контрольноro банка культур
микроорrанизмов критические этапы произ
водственноrо процесса должны повторно co
rласовываться таким образом, чтобы было
очевидно, что никаких отрицательных изме
нений в качестве продукта и ero соответствии
стандарту не произошло. Если в процесс BBO
дится видоизмененный или новый микроор
rанизм, то особое внимание необходимо об
ратить на изменения в профиле примеси
продукта.
СЫРЬЕ
Сырье, используемое на всех этапах
процесса ферментации и/или последующих
этапах обработки, должно иметь COOTBeTCTBY
ющее качество. Оно проверяется на COOTBeT
ствие письменным спецификациям.
Уровни биолоrической наrрузки в среде
или входящем для аэрации воздухе YMeHb
шаются до уровня, rарантирующеrо, что в
случае возникновения микробной контамина
Радиоактивные фармацевтические препараты
1079
01/2013:1483
ции не возникнет неrативноrо воздействия на
качество, чистоту и безопасность продукта.
Добавка таких компонентов, как питательные
вещества, прекурсоры и субстраты, во время
ферментации проводится в асептических yc
ловиях.
КОНТРОЛЬ В ПРОЦЕСС Е ФЕРМЕНТАЦИИ
Контроль в ходе процесса ферментации
осуществляется для тоro, чтобы rарантиро
вать постоянство условий во время caMoro
процесса и последующих этапов обработки, а
также для обеспечения качества изолирован
ноro продукта. Особое внимание должно быть
уделено способности используемых методов
контроля обнаружить любое микробное за
rрязнение, которое может отрицательно CKa
заться на качестве, чистоте и безопасности
продукта.
Производственные условия MorYT KOHTpO
лироваться путем выполнения COOTBeTCTBY
ющих процедур, например, посредством pery
лирования и проверки:
температуры;
значения рН;
нормы аэрации;
скорости перемешивания;
давления;
а также посредством контроля KOHцeHTpa
ции требуемоro продукта.
ОБРАБОТКА НА ВЫХОДЕ
В конце ферментации микроорrанизм
продуцент инактивируется или удаляется.
Дальнейшая обработка проводится с целью
удаления остатков питательной среды до при
емлемоro уровня, чтобы rарантировать посто
янное качество требуемоrо продукта.
MorYT использоваться различные процес
сы очистки, например, обработка активиро
ванным уrлем, улырафилырация и экстрак
ция растворителем. Необходимо убедиться.
что выбранный процесс или процедура YMeHb
шает до минимума или удаляет:
остатки микроорrанизма продуцента,
питательной среды, субстратов и прекурсо
ров;
нежелательные продукты трансформа
ции субстратов и прекурсоров.
Если необходимо, соответствующие ис
пытания выполняются либо в ходе процесса
ферментации, либо на изолированном про
дукте ферментации.
ИДЕНТИФИКАЦИЯ, ИСПЫТАНИЯ
И ПРОБЫ
Требования и специальные методы испы
таний. которым продукт должен COOTBeTCTBO
вать в течение Bcero периода проверки Ka
чества. указываются в конкретных частных
статьях.
ПРОДУКТЫ, КОТОРЫЕ MOrYТ
БЫТЬ ПЕРЕНОСЧИКАМИ rYБЧАТОЙ
ЭНЦЕФАЛОПАТИИ животноrо
ПРОИСХОЖДЕНИЯ
P,oducta сит possibifi trапsтissiопе vectorium
enkepha/opatiarum spoпgifo,mium апiтаfiит
ОПРЕДЕЛЕНИЕ
К продуктам, которые MOryT быть перенос
чиками ryбчатой энцефалопатии, относятся про
дукты, полученные из тканей или секретов жи
вотных, которые MOryT быть переносчиками
ryбчатой энцефалопатии, кроме случаев науч
ных исследований. Эта статья относится ко всем
субстанциям или лекарственным средствам:
полученным от таких животных;
в состав которых входят продукты, полу
ченные от таких животных;
в производстве которых использовались
такие продукты, например, в качестве сырья, ис
ходных материалов или реактивов.
ПРОИЗВОДСТВО
Производство должно осуществляться в co
ответствии со статьей 5.2.8. Снижение риска пе
редачи возбудителей 2убчатой энцефалопа
тии животных при прuменении медицинских
лекарственных средств.
01/2013:0125
РАДИОАКТИВНЫЕ
ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ
Radiopha,maceuиca
ОПРЕДЕЛЕНИЕ
В данной статье под радиоактивными фар
мацевтическими препаратами понимают:
радиоактивное лекарственное средство
любое лекарственное средство, roToBoe к меди
цинекому применению, которое содержит один
или более радионуклидов (радиоактивные изо
топы);
изотопный rенерэтор любая система,
включающая установленный материнский pa
дионуклид, от котороro производится дочерний
радионуклИД, удаляемый элюированием или
любым друrим методом и используемый для
приroтовлении радиоактивноro лекарственноro
средства;
1080
rосударственная фармакопея Республики Беларусь
комплект для приroтовления радиоак
тивноro лекарственноro средства любой
препарат, который восстанавливают и/или
объединяют с радионуклидом при заключи
тельном приroтовлении радиоактивноro ле
карственноro средства, обычно перед ero при
менением;
радиоактивный фармацевтический пре
курсор радионуклид, используемый для
радиоактивной метки друrorо вещества, име
ющеrо первичное назначение.
Нуклид это разновидность атома, xa
рактеризующаяся числом протонов и нейтро
нов в ero ядре (т. е. ero атомным номером Z
и массовым числом А), а также ero потенци
алом ядерной энерrии. Изотопы элемента
это нуклиды с одинаковым атомным номером,
но с различным массовым числом. Нуклиды,
содержащие нестабильные комбинации про
тонов и нейтронов. MorYT преобразовывать
ся спонтанно в устойчивые или друrие HeCTa
бильные комбинации протонов и нейтронов
с постоянной статистической вероятностью.
Такие нуклиды являются радиоактивными и
называются радионуклидами. Начальный He
стабильный нуклид называется материнским
радионуклидом, а конечный нуклид дочер
ним нуклидом.
Радиоактивный распад или преобразо
вание может повлечь излучение заряженных
частиц, электронный захват (ЭЗ) или изомер
ный переход (ИП). Заряженные частицы, ис
ходящие от ядра, MorYT быть альфа частица
ми (ядрами rелия с массовым числом 4) или
бета частицами (отрицательно заряженные
электроны или положительно заряженные
позитроны). Эмиссия заряженных частиц от
ядра может сопровождаться rамма излуче
нием. rамма излучение также происходит в
процессе изомерноro перехода. Эта эмис
сия может быть частично заменена выбро
сом электронов (конверсионные электроны).
Данное явление, подобно процессу электрон
ноro захвата, влечет вторичное peHTreHoBcKoe
излучение из за реорrанизации электронов в
атоме. Вторичная эмиссия может быть частич
но заменена выбросом электронов (оже элек
тронов). Радионуклиды с дефицитом нейтро
нов MorYT распадаться, излучая позитроны.
Эти радионуклиды называют эмиттерами по
зитрона. Позитроны уничтожаются при KOHTaK
те с электронами. Этот процесс, обычно co
провождаемый эмиссией двух rамма фотонов,
каждый с энерrией 511 кэВ, обычно с излуче
нием под уrлом 1800 друr к друrу, называется
анниrиляционным излучением.
Распад радионуклида происходит соrлас
но законам вероятности с характерным посто
янством и находится в экспоненциальной за
висимости. Время, в течение котороro число
радиоактивных ядер уменьшается вдвое, Ha
зывают периодом полураспада (Т 1I2 ),
Проникающая способность каждоro излу
чения существенно изменяется в зависимости
от ero природы и энерrии. Альфа частицы пол
ностью поrлощаются объектом толщиной от
нескольких микрометров до нескольких десят
ков микрометров. Бета частицы полностью по
rлощаются объектом толщиной от нескольких
миллиметров до нескольких сантиметров. raM
ма излучение полностью не поrлощается, а
только ослабляется, и, например, для десяти
кратноro уменьшения может потребоваться пла
стина свинца толщиной несколько сантиметров.
В большинстве случаев с увеличением плот
ности объекта уменьшается проникающая
способность альфа и бета частиц и rамма из
лучения.
Каждый радионуклид характеризуется по
стоянным периодом полураспада, выражен
ным в единицах времени, а также природой и
энерrией еro излучения. Энерrия выражается
в электронвольтах (эВ), килоэлектронвольтах
(кэВ) или меrаэлектронволыах (МэВ).
Термин «радиоактивность» использует
ся для описания феномена радиоактивноro
распада и для количественноro физическо
ro выражения (активности) этоro феномена.
Радиоактивность вещества это количество
ядерных распадов или преобразований в еди
ницу времени.
В Международной Системе единиц (СИ)
радиоактивность выражается в беккерелях
(Бк); один беккерель равен одному ядерному
превращению в секунду. Точные (абсолютные)
измерения радиоактивности MorYT быть cдe
ланы только в специализированной лаборато
рии. Идентификация и относительное измере
ние уровня радиации MorYT быть выполнены
в обычной лаборатории путем сравнения со
стандартизированными веществами. прове
ренными компетентными орrанами.
Радионуклидная чистота отношение
радиоактивности определенноrо радионукли
да к общей радиоактивности радиоактивноro
лекарственноrо средства, выраженное в про
центах. Радионуклидные примеси, которые
MorYT встречаться в лекарственном средстве,
с их предельными значениями должны быть
указаны в частной статье.
Радиохимическая чистота отношение
радиоактивности определенноrо радионукли
да, который присутствует в радиоактивном
лекарственном средстве в заявленной хими
ческой форме, к общей радиоактивности pa
дионуклида, присутствующеrо в радиоактив
ном лекарственном средстве, выраженное в
про центах. Радиохимические при меси, KOTO
рые MorYT встречаться в лекарственном cpeд
стве, с их предельными значениями должны
быть указаны в частной статье.
Радиоактивные фармацевтические препараты
1081
Химическая чистота химические при
меси, которые MOryT встречаться в радиоактив
ном лекарственном средстве, с их предельны
ми значениями должны быть указаны в частной
статье.
Переносчик (носитель) изотопа CTa
бильный изотоп соответствующеro элемента,
присутствующий или добавленный в радиоак
тивное лекарственное средство в той же самой
химической форме, что и сам радионуклид.
Удельная радиоактивность радиоак
тивность радио нуклида на единицу массы эле
мента или определенной химической формы.
Радиоактивная концентрация радио
активность радионуклида на единицу объема.
Общая радиоактивность радиоактив
ность радионуклида на единицу измерения
(флакон, капсула, ампула, reHepaTop, и т. д.).
Стартовые (исходные) материалы все
составляющие, которые используются при при
rотовлении радиоактивноrо лекарственноrо
средства.
Срок rодности время, в течение KOTOpO
ro должны выдерживаться все показатели, YKa
занные в частной статье. Срок roдности и, при
необходимости, время должны указываться в
частной статье.
ПРОИ3ВОДСТВО
Частная статья должна описывать метод
производства радионуклида настолько точно,
насколько это возможно. Радиоактивное ле
карственное средство содержит радиону
клид:
как элемент в атомной или молекулярной
форме, например, [133XeJ, [150]02;
как ион, например, [1 31 1]йодид, [99mТс]пер
технетат;
включенный в орrанические молекулы с
образованием координационною комплекса. Ha
пример, [111IпJоксин, или ковалентноro соедине
ния, например, 2 [16F]фтор 2 дезокси D rлюкоза.
Практические способы производства pa
дионуклидов для использования в радиоак
тивных лекарственных средствах или как He
посредственно радиоактивное лекарственное
средство:
бомбардировка материала мишени ней
тронами (обычно в ядерных реакторах);
бомбардировка материала мишени заря
женными частицами (в акселераторах типа ци
клотронов);
ядерное деление тяжелых нуклидов Ma
териала мишени (обычно после бомбардировки
нейтронами или частицами);
от изотопноro reHepaTopa.
БОМБАРДИРОВКА НЕЙТРОНАМИ
ИЛИ ЗАРЯЖЕННbJМИ ЧАСТИЦАМИ
Ядерная реакция и вероятность ее возник
новения в единицу времени зависят от природы
и физических свойств материала мишени и при
роды, энерrии и количества бомбардирующих
частиц.
Ядерное преобразование, появляющееся
в результате бомбардировки частицами, может
быть записано в следующей форме:
ядра мишени (бомбардирующая частица,
излученная частица или излучение) произведен
ные ядра.
Примеры: 58Fе(п,у)59Fе,
16О(р,п)16Е
в дополнение к желаемой ядерной реакции
MOryт произойти случайные преобразования. На
них будет оказывать влияние энерrия бомбар
дирующих частиц и чистота материала мишени.
Такие побочные преобразования MOryт приво
дить К возрастанию уровня примесей в лекар
ственном средстве.
ДЕЛЕНИЕ ЯДРА
Небольшое количество нуклидов с высоким
атомным номером являются делимыми, и наи
более часто используемой реакцией является
реакция расщепления ypaHa 235 нейтронами в
ядерном реакторе. При ядерном расщеплении
ypaHa 235 MOryт быть получены йод 131, молиб
дeH 99 и KceHOH 133. Их выделение из смеси,
содержащей более 200 друrих радионуклидов,
должно тщательно контролироваться, чтобы ми
нимизировать количество радиоактивных при
месей.
ИЗОТОПНЬ1Е rEHEPATOPb1
Система изотопных reHepaTopoB использует
относительно долroживущий материнский радио
нуклид, который распадается до дочернеrо pa
дионуклида, обычно обладающеro более KOpOT
ким периодом полураспада.
Используя химический или физический про
цесс, отделяют дочерний радионуклид от MaTe
ринекою, что позволяет использовать дочерний
радионуклид, несмотря на еro малы период
полураспада, на значительном расстоянии от
места ero получения.
ВЕЩЕСТВА МИШЕНИ
Процентное соотношение основноro радио
нуклида и радиоактивных примесей зависит от
изотопноro состава и чистоты бомбардируемою
вещества мишени. Использование искусствен
но обоrащенноrо изотопами вещества мишени
может помочь увеличить выход и повысить чи
стоту продукта на выходе.
Химическая форма, чистота, физическое
состояние. химические добавки, условия бом
бардировки, непосредственная физическая и
1082
rосударственная фармакопея Республики Беларусь
химическая среда определяют химическое co
стояние и химическую чистоту произведенных
радионуклидов.
При производстве радионуклидов и, в част
ности, короткоживущих радионуклидов, перед
обработкой и производством радиоактивно
ro лекарственноro средства может OTCYTCTBO
вать возможность определить какой либо из Ka
чественных критериев. Поэтому каждая серия
бомбардируемоro вещества мишени должна
проверяться в опытных производственных за
пусках перед ero использованием в основном
производственном процессе, чтобы обеспечить
при указанных условиях выход радионуклидов
в требуемом количестве с соответствующим Ka
чеством.
Бомбардируемый материал в rазообразном,
жидком или твердом виде должен находиться в
емкости, обеспечивающей возможность облуче
ния. Для бомбардировки нейтронами бомбарди
руемый материал обычно помещают в KBapцe
вые ампулы или в алюминиевые или титановые
контейнеры высокой степени очистки. Необхо
димо убедиться. что никакоro взаимодействия
между контейнером и еro содержимым в усло
виях облучения (температура, давление, время)
произойти не может
Для бомбардировки заряженными частица
ми емкость для бомбардируемых материалов
обычно делают из алюминия или друroro COOT
ветствующеro металла, с входным и выходным
отверстиями, системой охлаждения и окном из
тонкой металлической фольrи. При рода и тол
щина окна оказывают влияние на продукт ядер
ной реакции и MOryт также повлиять на радиоак
тивную чистоту.
Описание производственноro процесса
включает:
бомбардируемое вещество мишень;
тип емкости для вещества мишени;
способ заrрузки вещества мишени в си
стему облучения;
метод облучения (бомбардировки);
выделение требуемоro радионуклида;
влияние всех условий производства на Ka
чественный и количественный выход roтовоro
радионуклида.
Химическое состояние изолированноro pa
дионуклида может иrрать основную роль в даль
нейшей обработке.
ПРЕКУРСОРЫ ДЛЯ СИНТЕЗА
Обычно прекурсоры не производятся в
больших масштабах. Некоторые прекурсоры
синтезируются в лаборатории для производства
радиоактивных лекарственных средств, друrие
поставляются специализированными произво
дителями или лабораториями.
Испытания на подлинность, химическую чи
стоту и количественное определение должны
проводиться в соответствии с валидированными
процедурами.
Korдa партии прекурсоров выбираются по
данным сертификатов качества производи
теля, то должна быть установлена процеду
ра подтверждения стабильной надежности pe
зультатов анализов производителя и должно
про водиться по крайней мере одно испытание
на подлинность. Перед использованием в oc
новном производственном процессе по изroтов
лению радиоактивных лекарственных средств
рекомендуется проверять вещества прекурсоры
в опытных производственных запусках, чтобы
убедиться, что при указанных условиях произ
водства прекурсор дает на выходе радиоактив
ное лекарственное средство в требуемом коли
честве и соответствующеro качества.
ПРОИ3ВОДСТВЕННАЯ СИСТЕМА
Все операции, от подroтовки бомбардиров
ки до распределения rOToBoro радиоактивноrо
лекарственноro средства, включая их влияние
на чистоту roтовоro продукта и эффективность
процесса, должны быть документированы. По
возможности необходимо выполнять контроль
в ходе производственноrо процесса с реrистра
цией результатов на каждом этапе производства
для тоro, чтобы иметь возможность идентифи
цировать, на какой стадии может наблюдаться
отличие от нормальноro процесса производства.
а) При производстве радиоактивноro лекар
cTBeHHoro средства MOryT использоваться Me
ханические и автоматизированные процессы,
которые применимы в фармацевтической про
мышленности и адаптированы к специфике pa
диоактивноro производства.
Ь) ДЛЯ изrотовления радиоактивных лекар
ственных средств, содержащих короткоживу
щие радионуклиды, типа некоторых эмиттеров
позитрона, обычно используются дистанцион
ное управление производством и автоматизи
рованный радиоактивный синтез. Для радиону
клидов с очень коротким периодом полураспада
(меньше 20 мин) контроль работы производ
ственной системы является очень важной про
цедурой, так как это позволяет обеспечить каче
ство радиоактивноro лекарственноro средства
перед еro выходом.
с) Любая производственная процедура
должна быть валидирована в опытных произ
водственных запусках перед ее использованием
в основном производственном процессе, чтобы
убедиться, что при указанных условиях про
изводства производственная система дает на
выходе радиоактивное лекарственное средство
в требуемом количестве с соответствующим Ka
чеством.
d) В практике медицинской радиолоrии при
roтовление дозированных форм roтовоro ради
оактивноro лекарственноro средства обычно
включает оrраничение радиоактивности начи
ная с roтовых к использованию радиоактивных
лекарственных средств, reHepaTopoB, комплек
тов и радиоактивных прекурсоров. Все условия,
Радиоактивные фармацевтические препараты
1083
которые MOryT отразиться на качестве продукта
(например, радиохимическая чистота и стериль
ность) должны быть четко установлены, а также
должны быть приняты соответствующие меры
по защите от облучения.
ИДЕНТИФИКАЦИЯ
Радиоактивный распад. Радиоактивный
распад находится в экспоненциальной зависи
мости от констант распада каждоro отдельною
радионуклида.
Кривая экспоненциальною распада (кривая
распада) выражается уравнением:
А, Aoe A' ,
rдe:
А, радиоактивность во время t;
Ао радиоактивность во время t = о;
А. константа распада, характерная для
каждоro радионуклида;
е основание натуральноro лоrарифма.
Период полураспада (Т 1I2 ) связан с KOHCTaH
той распада (л.) уравнением:
т In2 (lп2::::: 0,693).
112 ...
А
Радионуклид обычно идентифицируют по
ero периоду полураспада, природе и энерrии
ero излучения или радиации, как указано в
частной статье.
Измерение периода полураспада.
Период полураспада измеряется. с помо
щью подходящей аппаратуры обнаружения,
такой как ионизационная камера, счетчик
rейrера Мюллера, сцинтилляционный счет
чик (твердый кристалл, жидкость) или полу
проводниковый детектор. Исследуемый об
разец используется в том виде, в каком есть.
или растворяется либо высушивается в кап
суле после соответствующеro растворения.
Выбранная радиоактивность, используемая
в экспериментальных условиях, должна быть
достаточноro уровня для обнаружения в тече
ние нескольких периодов полураспада, но не
должна быть слишком высока, чтобы мини
мизировать потери при подсчете, например,
из за мертвоro времени.
Исследуемое радиоактивное вещество
подrотавливается таким образом, чтобы избе
жать потери материала в период обработки.
Жидкости (растворы) помещают во флаконы
или запечатанные тубы, твердые вещества
(остаток при высушивании в капсуле) поме
щают в упаковку из клейкой ацетилцеллюло
зы или какоrо либо друrоrо материала.
Период полураспада исследуемоrо радио
активноro вещества измеряется при посто
янных rеометрических условиях с промежут
ками, обычно соответствующими половине
установленноro периода полураспада, в Te
чение времени, приблизительно равноro трем
периодам полураспада. Правильное функци
онирование аппарата проверяется путем ис
пользования вещества с длинным периодом
полураспада и при необходимости корректи
руется (см. Измерение радиоактивности).
Строится rрафик зависимости времени
(ось абсцисс) от лоrарифма относительноro
показания прибора (ось ординат). Если нет
иных указаний в частной статье, рассчитан
ный период полураспада может отличаться
не более чем на 5 % от периода полураспада,
указанноro в Фармакопее.
Определение природы и энерrии излу-
чаемой радиации. Природа и энерrия излу
чаемой радиации MorYT определяться путем
построения кривой ослабления и использова
ния метода спектрометрии. Кривая ослабле
ния используется для анализа электронноrо
излучения; спектрометрия используется для
идентификации rамма лучей и детектируемо
ro ренпеновскоrо излучения.
Кривая ослабления строится:
для чистых электронных эмиттеров,
Korдa нет в распоряжении спектрометра для
бета лучей;
для бета/rамма эмиттеров, Korдa нет в
распоряжении спектрометра для rамма лучей.
Этот метод оценки максимальной энерrии
бета радиации дает только приблизительное
значение. Исследуемое радиоактивное Be
щество помещается в постоянные rеометри
чески е условия впереди тонкоro окна счетчи
ка rейrера Мюллера или пропорциональноrо
счетчика. Исследуемое радиоактивное веще
ство подrотавливается, как описано выше. Из
меряют число разрядов исследуемоrо радио
активноro вещества. Между исследуемым
веществом и счетчиком, сохраняя постоян
ные rеометрические условия, последователь
но помещают не менее шести алюминиевых
экранов с увеличением массы каждоro после
дующеro на единицу площади таким образом,
чтобы добавление каждоro последующеro
экрана не сказывал ось на числе разрядов чи
стоro бета излучения. Строят rрафик зависи
мости массы экрана на единицу еro площади,
выраженной в миллиrраммах на квадратный
сантиметр (ось абсцисс), от лоrарифма числа
разрядов, полученных для этоro экрана (ось
ординат). Таким же образом строят rрафик за
висимости для стандартизированноro веще
ства. Массовые коэффициенты ослабления
рассчитываются из срединных (прямолиней
ных) частей кривых.
Массовый коэффициент ослабления 11т'
выраженный в квадратных сантиметрах на мил
лиrрамм, зависит от спектра энерrии бета из
лучения и от природы и физических свойств
1084
rосударственная фармакопея Республики Беларусь
экрана. Это позвопяет идентифицировать бета
эмиттеры. Массовый коэффициент ослабления
рассчитывают по формуле:
InA 1 lnA2
J.1 т ==
т 2 т 1
rдe:
т 1 масса на единицу площади самоro
леrкоro экрана;
т 2 масса на единицу площади самоro
тяжелоro экрана, т 1 и т 2 должны находиться в
пределах прямолинейной части кривой;
А 1 число разрядов для т 1 ;
А 2 число разрядов для т т
Рассчитанный таким образом массовый KO
эффициент ослабления J1 m может отличаться
не более чем на 10 % от коэффициента, полу
ченноro при идентичных условиях с использова
нием стандартизированной подrотовки тоro же
самоro радионуклида.
Амплитуда бета частиц это дополни
тельный параметр, который может исполь
зоваться для определения бета энерrии. Ее
определяют как массу на единицу площади, co
ответству ющую пересечению экстраполяций
спускающейся прямолинейной части кривой
и roризонтальной линии фоновой радио
активности из кривой ослабления, описанной
выше.
Жидкостный сцинтилляционный счетчик
может использоваться для получения спектров
a и J3 эмиттеров (см. Измерение радиоактивно
сти).
tамма--спектрометрия используется для
идентификации радионуклидов по энерrии и ин
тенсивности peHTreHoBcKoro и raMMa излучений.
Наиболее подходящим детектором для
рентrеновской и raмма спектрометрии являет
ся rерманиевый полупроводниковый детектор.
Также используется активизированный таллием
йодисто натриевый сцинтилляционный дeTeK
тор, но он имеет более низкую энерrетическую
разрешающую способность.
rамма детектор должен быть откалиброван
посредством использования стандартных радио
активных веществ, потому что эффективность
детектирования является функцией как энерrии
raMMa и peHTreHoBcKoro излучения, так и формы
источника и расстояния от источника до дeTeK
тора. Эффективность обнаружения может быть
измерена путем использования калиброван
ноro радиоактивноro вещества или, для общих
целей, по rрафику зависимости эффективности
от энерrии peHTreHoBcKoro и raMMa излучения,
который строится с помощью калиброванных pa
диоактивных веществ источников различных
радионуклидов.
raMMa и рентrеновские спектры радиону
клидов являются специфическими для данноro
нуклида и характеризуются энерrией и количе
ством фотонов конкретной энерrии, излучаемых
при переходе из одноro энерrетическоro уровня
на друroй. Это свойство используется для иден
тификации присутствующих радио нуклидов в
радиоактивном веществе и определения их KO
личества, а также позволяет оценить количество
друrих радионуклидных примесей.
Так как пики уменьшаются в зависимости
от периода полураспада, становится возмож
ным определить скорость падения радиоактив
ности с использованием спектрометрии raMMa
излучения. Если в таком источнике присутствует
радиоактивная примесь с друrим периодом по
лураспада, ее можно обнаружить путем иденти
фикации характеристическоro пика или пиков,
чья амплитуда затухает со скоростью, отлича
ющейся от скорости затухания для конкретноro
радионуклида. Определение периода полурас
пада дополнительных пиков путем нескольких
повторных измерений образца поможет иденти
фицировать примесь.
Таблица физических характеристик paди
онуклидов, упоминаемых в Фармакопее (5. 7),
содержит наиболее общие физические xapaK
теристики радионуклидов, применяемых в Me
дицинской практике и описанных в частных
статьях. Кроме тоro, таблица содержит физиче
ские характеристики основных потенциальных
примесей радионуклидов, описанных в частных
статьях.
Под термином «вероятность перехода» под
разумевается вероятность преобразования ядра
в данном состоянии энерrии через определен
ный переход. Вместо термина «вероятность»
MOryT использоваться термины «интенсивность»
И «распространенность».
Под термином «вероятность эмиссии» под
разумевают вероятность атома радионуклида
вызывать эмиссию частиц или определенноro
излучения.
Вне зависимости от тоro, в котором из двух
случаев употребляется термин «вероятность»,
она обычно измеряется за период 100 распадов.
ИЗМЕРЕНИЕ РАДИОАКТИВНОСТИ
Радиоактивность препарата устанавливает
ся на определенный день и, если необходимо,
на определенное время.
Абсолютное измерение радиоактивности
конкретноro образца может быть выполнено,
если известна схема распада радионуклида,
но на практике приходится учитывать мноro по
правок, чтобы получить точные результаты. По
этой причине обычно выполняют измерение при
помощи первичноrо, cTaHAapTHoro, радиоактив
ноro вещества. Короткоживущие радионуклиды,
например, эмиттеры позитрона, MoryT не иметь
первичных стандартов. Измерительные при
боры калибруются с использованием CTaHдap
тов определенных радионуклидов. Стандарты
получают из аттестованных для этих целей ла
бораторий. Ионизационные камеры и счетчики
rейrера Мюллера MOryт использоваться для из
мерения бета и бета/rамма эмиттеров; сцинтил
Радиоактивные фармацевтические препараты
1085
ляционые или полупроводниковые счетчики или
ионизационные камеры MOryT использоваться
для измерения rамма эмиттеров; для измерения
низкоэнерrетических бета эмиттеров использу
ется жидкостный сцинтилляционный счетчик.
Для обнаружения и измерения альфа эмитте
ров необходимы специальные оборудование и
методы. Для получения точных результатов He
обходимо выполнять испытания исследуемых
радиоактивных веществ и стандартов в одина
ковых условиях.
Низкоэнерrетические бета эмиттеры MOryт
измеряться с помощью жидкостноro сцинтилля
ционноro счетчика. Для этоro образец paCTBO
ряют в растворе, содержащем один или более
часто два орrанических флуоресцентных веще
ства (первичные и вторичные сцинтилляторы),
которые преобразовывают часть энерrии pac
пада в фотоны света, которые обнаруживаются
фотоумножителем и преобразовываются в элек
трические импульсы. При использовании жид
костноro сцинтилляционноrо счетчика сравни
тельные измерения корректируются с поправкой
на подавляющие свет эффекты. Прямые (непо
средственные) измерения проводятся BcerAa в
одинаковых условиях для исследуемою и CTaH
дартноrо радиоактивных веществ (например,
объемы и тип растворов).
Все измерения радиоактивности должны
корректироваться вычитанием фона, возникше
ro из за радиоактивности окружающей среды и
из за побочных сиrналов, rенерируемых самим
оборудованием.
При работе с оборудованием, коrда измере
ния производятся при высоких уровнях радиоак
тивности, иноrда необходимо сделать поправку
на конечное время отклика (<<мертвое» время)
детектора и связанноro с ним друroro электрон
ною оборудования. Для системы подсчета с
фиксированным «мертвым» временем t расчет
производят по формуле:
N = N Ндб
1 NНдБТ
rде:
N истинное число распадов в секунду;
N Ндб наблюдаемое число распадов в ce
кунду;
T «мертвое» время, в секундах.
Эта корректировка должна быть сделана
перед корректировкой фоновоro излучения, для
некоторою оборудования это корректировка BЫ
полняется автоматически.
Если время индивидуальноro измерения t m
незначительно короче по сравнению с периодом
полураспада T'12' должен учитываться распад,
происходящий во время измерения. После учета
фона и электронных эффектов прибора (число
распадов, поток ионизации, и т. д.) корректиров
ка распада в течение времени измерения опре
деляется по формуле:
R t", In2
R""" ('1' I 2]'
1 exp f1J n
Т" 2
rдe:
R KOPP показания прибора, откорректиро
ванные на начало отдельною измерения;
R показания прибора до корректировки
распада, но уже откорректированные с учетом
фона, и т.Д.
Результаты определений радиоактивности
представляют собой колебания, которые явля
ются результатом случайности процессов ядер
Horo преобразования. Поэтому должно быть
зареrистрировано достаточное количество pac
падов за единицу времени. Стандартное OT
клонение это квадратный корень количества
распадов, поэтому при минимуме в 1 О 000 pac
падов относительное стандартное отклонение
составит не более 1 % (доверительный интер
вал: 1 сиrма).
Результаты измерения радиоактивности co
провождаются указанием даты и, если необхо
димо, времени измерения. Эти данные должны
быть сделаны со ссылкой на часовой пояс. Pa
диоактивность в друroе время может быть pac
считана из экспоненциальноro уравнения или из
таблиц.
Радиоактивность раствора выражается pa
диоактивной концентрацией. Радиоактивная
концентрация это радиоактивность единицы
объема раствора.
РАДИОНУКЛИДНАЯ ЧИСТОТА
В большинстве случаев для определе
ния радионуклидной чистоты радиоактив
ною фармацевтическоro препарата должен
быть идентифицирован каждый присутству
ющий радио нуклид и должна быть известна
еro радиоактивности. Наиболее подходящим
методом для проверки радионуклидной чи
стоты является rамма спектрометрия. Однако
этим методом не Bcerдa леrко обнаруживают
ся альфа и бета излучения, и, если использу
ется йодисто натриевый детектор, пики при
месей с rамма излучением часто затеняются
спектром основноro радионуклида.
Требования по радионуклидной чистоте
(например, спектр rамма излучения не должен
значительно отличаться от спектра CTaHдap
тизированноrо радиоактивною вещества) и
нормы содержания определенных радиону
клидных примесей (например, кобалы 60 '3 KO
балые 57) указывают в частных статьях. Хотя
эти требования необходимы, они не являются
достаточными, чтобы определять радиоактив
ное лекарственное средство rодным к меди
цинекому применению. Изrотовитель должен
проверить радионуклидные препараты
1086
rосударственная фармакопея Республики Беларусь
с коротким периодом полураспада на coдep
жание примесей с долrим периодом полурас
пада после соответствующеro периода pac
пада. Таким образом получают информацию
относительно приrодности производствен
ных процессов и адекватности процедур ис
пытания. В случаях, коrда два или более
радионуклидов, излучающие позитроны, He
обходимо идентифицировать и/или диффе
ренцировать как, например. 18F примеси в
1 3N препаратах, определение периода полу
распада должно быть выполнено дополни
тельно к методу rамма спектрометрии.
Из за различий в периодах полураспада
различных радионуклидов. присутствующих в
радиоактивном лекарственном средстве, pa
дионуклидная чистота может изменяться со
временем, но требования к ней должны BЫ
полняться В течение Bcero срока rодности. В
случае, коrда период полураспада радиону
клида в радиоактивном лекарственном cpeд
стве очень мал, такие испытания выполнить
трудно. Поэтому перед разрешением выпуска
серии радиоактивноro лекарственноro cpeд
ства проводят контроль производственноro
процесса.
РАДИОХИМИЧЕСКАЯ ЧИСТОТА
Определение радиохимической чистоты pa
диоактивноrо лекарственноrо средства требует
разделения входящих в еro состав химических
веществ, содержащих радионуклид, и оценки
их радиоактивности. Радиохимические примеси
MOryт возникать при:
производстве радионуклида;
последующих химических процессах;
неполном подrотовительном разделении;
химических изменениях в период xpaHe
ния.
Требование радиохимической чистоты
должны выполняться в течение всеro срока roд
ности.
Для определения радиохимической чисто
ты можно использовать любой метод анали
тическоrо разделения. например, бумажную
хроматоrрафию (2.2.26), тонкослойную xpOMa
тоrрафию (2.2.27), электрофорез (2.2.31), экс
клюзионную хроматоrрафию (2.2.30), rазовую
хроматоrрафию (2.2.28) и жидкостную xpOMa
тоrрафию (2.2.29). Конкретное описание MeTO
дики определения описано в частной статье.
При этом необходимо принять COOTBeTCTBY
ющие меры предосторожности для защиты от
облучения.
В условиях клиник наиболее часто исполь
зуемыми являются методы бумажной и TOH
кослойной хроматоrрафии. При использова
нии этих методов объем, равный указанному
объему в частной статье, наносят на линию Ha
несения, как указано в описании общих MeTO
дов хроматоrрафии. Испытуемый образец же
лательно не разводить, однако нужно избеrать
нанесения таких количеств радиоактивности,
которые бы приводили к невозможности изме
рений из за совпадений импульсов при изме
рении радиоактивности. По причине использо
вания очень малых количеств радиоактивноrо
вещества, в частной статье может содержать
ся указание на прибавление переносчика (HO
сителя). После проведения хроматоrрафии
подложку сушат, расположение радиоактив
ных пятен детектируют с помощью авторадио
rрафии или с помощью измерения радиоак
тивности по длине xpoMaTorpaMMbI, используя
подходящие коллимированные счетчики или
разрезая xpoMaTorpaMMY на полоски и изме
ряя радиоактивность каждой. Обнаруженные
пятна идентифицируют путем сравнения с pac
творами тех же соединений (не радиоактив
'ных) с использованием подходящеro способа
проявления.
Радиоактивность может быть измерена ин
теrрально с использованием автоматическоro
самописца или цифровою счетчика. Отноше
ния площадей пиков дают отношения радио
активной концентрации химических веществ. В
случае если полоски разрезаны на порции, OTHO
шения количеств измеренной радиации COOTBeT
ствуют отношению концентраций радиоактив
ных химических соединений.
УДЕЛЬНАЯ РАДИОАКТИВНОСТЬ
Удельная радиоактивность обычно рассчи
тывается из радиоактивной концентрации (pa
диоактивность на единицу объема) и KOHцeHTpa
ции химическоro вещества после определения
радионуклидной и радиохимической чистоты
радиоактивноro лекарственною средства.
Удельная радиоактивность изменяется со
временем, поэтому необходимо указывать дату
(и время) определения. Требуемый уровень
удельной радиоактивности должен выдержи
ваться в течение срока roдности радиоактивноro
лекарственноro средства.
ХИМИЧЕСКАЯ ЧИСТОТА
ДЛЯ определения химической чистоты He
обходимо определить количество индивидуаль
ных химических примесей, указанных в частной
статье.
ЭНАНТИОМЕРНАЯ ЧИСТОТА
При необходимости должна быть проверена
стереоизомерная чистота.
ФизиолоrИЧЕСКОЕРАСПРЕДЕЛЕНИЕ
Если необходимо, для некоторых радио
активных лекарственных средств проводят ис
пытание физиолоrическоro распределения.
Картина распределения радиоактивности, Ha
блюдаемая в отдельных opraHax, тканях или
друrих отделах тела животною подходящею
вида (обычно крысы или мыши), может служить
надежным индикатором ожидаемою распреде
Радиоактивные фармацевтические препараты
1087
ления в орrанизме человека и, таким образом,
приroдности для предполаrаемоro назначения.
В частных статьях описываются детали, Ka
сающиеся проведения испытания и требований
к физиолоrическому распределению, которые
должны выполняться для радиоактивноro ле
карственноro средства. Физиолоrическое pac
пределение, соответствующее требованиям,
будет обеспечивать подходящее распределение
радиоактивных соединений в предназначенных
биолоrических мишенях в человеческом теле и
оrраниченное распределение в областях, KOТO
рые не являются целевыми.
В общем случае испытание выполняется
следующим образом.
Исследуемое лекарственное средство BBO
дится внутривенно каждому из трех животных.
Вид, пол, породу, вес и/или возраст животных
при необходимости указывают в частной статье.
Испытуемой инъекцией является радиоактивное
лекарственное средство, приютовленное анало
rично, как для медицинскоro использования. При
необходимости лекарственное средство BOCCTa
навливают соrласно рекомендациям производи
теля. В некоторых случаях может требоваться
разведение непосредственно перед введением.
Обычно лекарственное средство вводится в
хвостовую вену животною. В отдельных случаях
используют введение в подкожные вены НО!; бе
дренные, яремные или относящиеся к мужскому
половому члену вены. Животные с признаками
внесосудистой инъекции (обнаруживаемой при
введении или по последующему COOTBeTCTBY
ющему обнаружению радиоактивности) исклю
чаются из испытания.
После инъекции каждое животное HeMeд
лен но помещается в отдельную клетку, условия
содержания в которой позволяют собирать BЫ
деления и предотвращать заrрязнение поверх
ности тела животноro.
По истечении определенною времени после
инъекции животные умерщвляются и расчле
няются соответствующим способом. ИЗ OTO
бранных opraHoB и тканей берутся пробы для
определения их радиоактивности с применени
ем подходящеro метода, описанною в данной
статье. Физиолоrическое распределение pac
считывают и выражают в процентном значении
радиоактивности, обнаруженной в каждом из
отобранных opraHoB или тканей. Для этой цели
радиоактивность в opraHe соотносится с BBeдeH
ной радиоактивностью, рассчитанной из радио
активною содержания шприца, измеренною до
и после инъекции. Для некоторых радиоактив
ных лекарственных средств требуется опреде
ление соотношения радиоаКТИ8НОСТИ к массе
образцов отобранных тканей (радиоактивность/
масса).
Радиоактивное лекарственное средство
должно выдерживать требования физиолоrиче
cKoro распределения по крайней мере у двух из
трех животных.
СТЕРИЛЬНОСТЬ
Радиоактивное лекарственное средство
для парентеральноro применения должно
производиться в асептических условиях. Ис
пытание на стерильность выполняется, как
описано в соответствующей статье (2.6.1).
Трудности возникают с радиоактивными ле
карственными средствами с коротким пери
одом полураспада, малым размером серии
и из за радиационной опасности. При невоз
можности проведения испытания стерильно
сти roтовоro радиоактивною лекарственноro
средства перед разрешением выпуска серии,
пара метрический выпуск (5.1.1) является Me
тодом выбора. Контроль стерильности должен
проводиться при контроле качества лекар
ственною средства в случае асептическоro
производства.
Если размер выпускаемой серии радио
активноrо лекарственноrо средства оrраничен
одним или несколькими образцами (например,
терапевтическое или очень недолrовечное pa
диоактивное лекарственное средство), исследо
вание на стерильность может не проводиться.
Если радиоактивное лекарственное средство
стерилизуют с помощью фильтрации и/или про
изводят в асептических условиях (5.1.1), про
цесс валидации является критическим.
Если период полураспада радионуклида
очень короткий (например, менее 20 мин), прием
радиоактивноro лекарственноro средства паци
ентом обычно происходит в месте производства
сразу после еro изrотовления в соответствии с
валидированной системой.
Из соображений безопасности (высокий
уровень радиоактивности) исследование необ
ходимоro для стерильности (2.6.1) количества
образца невозможно. Для тоro, чтобы оrрани
чить облучение персонала, необходимо исполь
зовать метод мембранной фильтрации.
Метод мембранной фильтрации являет
ся предпочтительным для уменьшения воздей
ствия излучения на персонал.
Несмотря на требования по использова
нию антимикробных консервантов. указанные
в общей статье Лекарственные средства для
парентераЛЬН020 применения. их прибавление
в радиоактивные лекарственные средства, фа
сованные в мноroдозовые контейнеры, не явля
ется обязательным, если нет друrих указаний в
частной статье.
БАКТЕРИАЛЬНЫЕ ЭНДОТОКСИНЫ
пироrЕНЫ
Для некоторых радиоактивных лекарствен
ных средств проводится испытание на бактери
альные эндотоксины. Испытание выполняется,
как указано в общем методе (2.6.14), прини
мая необходимые меры предосторожности по
защите персонала от облучения.
Предельные показатели для бактериальных
эндотоксинов указывают в частной статье.
1088
rосударственная фармакопея Республики Беларусь
01/2013:1579
Если природа радиоактивноro лекарствен
ноro средства при водит к интерференции путем
инrибирования или активации и удаление ин
терферирующих факторов не представляется
возможным, испытание на пироrенность (2.6.8)
может быть описано отдельно.
Из за невозможности проведения испыта
ний радиоактивных лекарственных средств с
коротким периодом полураспада перед разре
шением выпуска серии, проводится контроль
производственноrо процесса.
ХРАНЕНИЕ
Хранят в воздухонепроницаемых контейне
рах в местах. обеспечивающих защиту персонала
от облучения первичным или вторичным излуче
нием, в соответствии с национальными и меж
дуна родными правилами по хранению радио
активных веществ. Во время хранения контейне
ры из за облучения MOryT потемнеть, что не обя
зательно означает ухудшение качества лекар
cTBeHHoro средства.
Радиоактивные лекарственные средства
должны быть использованы в течение KOpOTKO
ro периода времени, конец срока их roдности
должен указываться четко.
МАРКИРОВКА
#Маркировка радиоактивных лекарственных
средств должна соответствовать требованиям,
установленным уполномоченным opraHoM.#
На этикетке контейнера указывают:
наименование лекарственноro средства и,
или ссылку на Hero;
наименование предприятия изroтовителя;
идентификационный номер;
для жидких и raзообразных лекарственных
средств: общую радиоактивность в контейнере
или радиоактивную концентрацию на миллилитр
с указанием даты и, если необходимо, времени;
объем жидкости в контейнере;
для твердых лекарственных средств
(таких, как лиофилизаты): общую радиоактив
ность с указанием даты и, если необходимо,
времени; после прибавления соответствующеro
растворителя твердое лекарственное средство
считают жидким;
для капсул: радиоактивность в капсуле с
указанием даты и, если необходимо, времени;
число капсул в контейнере.
В некоторых случаях на этикетки помещают
дополнительную информацию (например, коrда
радиоактивные лекарственные средства coдep
жат короткоживущие радионуклиды).
Кроме вышеперечисленноro на вторичной
упаковке указывают:
способ применения;
срок roдности;
наименование и концентрацию всех дo
бавленных антимикробных консервантов;
rдe применимо, любые специальные усло
вия хранения.
РАСТИТЕЛЬНЫЕ ЖИРНЫЕ МАСЛА
О/еа Herba,ia
ОПРЕДЕЛЕНИЕ
Растительные жирные масла представля
ют собой в основном твердые или жидкие три
rлицериды жирных кислот. Они MorYT coдep
жать небольшие количества друrих липидов,
таких как воски, свободные жирные кислоты,
частичные rлицериды или неомыляемые Be
щества. Растительные жирные масла получа
ют из семян, плодов или косточек'зерен'ядер
различных растений посредством отжима и,
или экстракции растворителями, затем, по
возможности, подверrают рафинированию и
rидрированию. При необходимости добавля
ют антиоксидант.
Неочищенное масло масло, получаемое
из сырья определенноro качества посредством
механической обработки (например, холодноro
отжима или центрифуrирования).
Очищенное (рафинированное) масло
масло, получаемое посредством отжима и'или
экстракции растворителями и последующим
щелочным (сопровождается обесцвечиванием
и дезодорацией) или физическим рафинирова
нием.
,идР02енизированное масло масло, по
лучаемое посредством отжима и'или экстракции
растворителями и последующим щелочным или
физическим рафинированием, обесцвечивани
ем (по возможности), сопровождаемым высуши
ванием, rидрированием и последующим обес
цвечиванием и дезодорацией.
Для приrотовления парентеральных дозиро
ванных форм используются только масла, рафи
нированные с использованием щелочи.
ПРОИЗВОДСТВО
Проводят измерения для подтверждения
тоro, что масло не содержит бенз[а]пирен выше
пределов, установленных компетентным opra
ном.
ПОЛУЧЕНИЕ СЫРО,О МАСЛА
Из растений с высоким содержанием масла
в большинстве случаев масло получают посред
ством roрячеro отжима с последующей экстрак
цией; из растений с низким содержанием масла
путем прямой экстракции.
Механические способы
А.Отжим
Отжим при высоком давлении. Состоит из
всех или некоторых нижеследующих стадий:
очистка, сушка, лущение или вышелушивание,
размол, тепловая обработка и отделение от
чешуи.
РаститеЛЬНblе жирНblе масла
1089
На стадии очистки удаляют инородные при
меси. Сушка может быть необходима, если co
держание влаrи в зернах превышает допусти
мое содержание влаrи для процесса отжима под
давлением. Лущение используют для получения
продукта с большим содержанием белков путем
уменьшения волокон и для уменьшения приме
сей в масле. Тепловая обработка применяет
ся для различных целей, таких как разрушение
клеток, содержащих масло, снижение вязкости
масла, коаryляция белков в продукте, реryлиро
вание влажности, стерилизация семян, YCTpa
нение лишних компонентов семян (rоссипол из
семян хлопчатника) и фиксирование опреде
леННblХ фосфатидов в использованном сырье.
Таким образом уменьшаются потери при очист
ке. Эффективность процесса выделения такова,
что в использованном сырье остается только от
3 %до 6 % масла.
Влажный отжим. Плоды заrружаются в ceT
чатые барабаны (например, для плодов пальмы)
и помещаются в roризонтальный стерилизатор
для обработки паром и теплом с целью инакти
вации ферментов, разрыхления плодов, Koary
ляции белков и т. д. После проrревания в aBТO
клаве массу заrружают в пресс. Масло очищают
в центрифуrе и сушат в вакуумной сушилке.
Прессование с последующей экстракци
ей растворителем. Состоит из вышеописан
ных стадий. Основное назначение процесса
предварительноrо прессования это получе
ние массы с высокой проницаемостью для по
следующей стадии экстракции растворителями.
Экстракция выполняется в перколяционных или
иммерсионных аппаратах. Эффективность экс
тракции растворителями такова, что остаточный
уровень масла в использованном сырье обычно
менее 1 %.
В. Центрифуrирование
С помощью центрифуrирования отделяют
маслянистую фракцию от воды и растворимых в
воде компонентов и остаточных твердых частиц.
Этот способ производства про водят с использо
ванием:
самоочистных роликовых или дисковых
центрифуr;
супер декантаторов, которые являются ro--
ризонтальными турбинами с цилиндрической
чашей, постепенно сужающейся к одному концу
и содержащей постоянно вращающуюся резьбу,
которая очищает стороны чаши; резьба и чаша
вращаются с различной скоростью; твердые ча
стицы выходят с заостренноro конца чаши, а
масло вытекает с противоположноro.
Экстракция растворителями. Перед экс
тракцией производятся следующие действия:
семена выдерживаются около недели при TeM
пературе ниже 24 ОС, чтобы отделить шелуху
от семени и позволить семенной влаrе достичь
балланса. Затем семена очищаются, шлифу
! 17 З ..: 1712
ются, лущатся и отделяются от чешуи. Наи
более часто используемый растворитель
смесь H reKcaHa и метилпентанов (точка
кипения: (65 70) ОС), как правило, относя
щихся к «reKcaHaM». Из за высокой опасности
воспламенения или взрыва этой смеси MOryT
использоваться сжиженные rазы или rазы в
сверхкритическом состоянии.
РАФИНИРОВАНИЕ
Очистку (рафинирование) осуществляют с
целью удаления из масла примесей и проводят
таким образом, чтобы как можно меньше разру
шить триrлицериды и обойтись минимальными
потерями масла. В результате очистки удаляют
ся следующие примеси:
свободные жирные кислоты, которые MOryт
вызвать ухудшение качества масла, проявля
ющееся в окислении, появлении вкуса дыма при
наrревании и остром запахе (щелочное рафини
рование);
вода, способствующая ферментативному
rидролизу (щелочное рафинирование, сушка);
частичные rлицериды, которые MOryт BЫ
звать вспенивание и обусловливать roрький вкус
(нейтрализация, промывка);
фосфатиды и соединения фосфора, обла
дающие эмульrирующими свойствами, которые
мoryт вызвать выпадение осадка, потемнение
масла при наrревании, помутнение и ухудшение
орrанолептических свойств (щелочное рафини
рование );
красящие вещества, такие как хлоро
филл (щелочное рафинирование), каротиноиды
(обесцвечивание );
rликолипиды, которые MOryт образовывать
коллоодные растворы с водой;
свободные уrлеводороды, парафины,
воски и смолистые вещества;
металлы (Fe, Cu, РЬ, Sп, Pt, Pd и т. д.), яв
ляющиеся сильными катализаторами окисл
ния;
пиrменты, такие как roссипол (в хлопковом
масле), или микотоксины, такие как афлатоксин
(8 основном, в арахисовом масле);
пестициды;
продукты окисления (альдеrиды, пероксиды);
белки, которые MOryт вызвать аллерrиче
ские реакции;
неомыляемые вещества (например, CTe
ролы, токоферолы и друrиe витамины);
полициклические аро ические уrлево
дороды.
UЦелочное рафинирование. Включает в
себя следующие стадии: обессмоливание (при
необходимости), щелочная нейтрализация, про
мывка и сушка.
ОбесCI.ЮЛиванue. На этой стадии проводит
ся обработка водой, и'или фосфорной кислотой
и'или натрия хлоридом, в результате чеrо yдa
ляются фосфатиды, фосфорные соединения и
1090
rосударственная фармакопея Республики Беларусь
металлы. Использование данноro процесса за
висит от природы масла.
Щелочная нейтрализация. На этой стадии
уменьшается содержание жирных кислот ДО
уровня менее 0,1 %; жирные кислоты преобра
зуются в нерастворимые в масле мыла. Друrие
вещества MOryT быть удалены посредством aд
сорбции на этих мылах: слизистые субстанции,
фосфатиды, продукты окисления, красящие Be
щества, и т. д. Все вещества, нерастворимые в
масле, при rидратации удаляются. Недостатком
щелочной нейтрализации является возможность
омыления части нейтральноro масла.
Промывка. Эта стадия заключается в yдa
лени и избытков мыл и щелочей, а также следов
металлов, фосфатидов и иных примесей при
помощи roрячей воды.
Сушка. Остатки воды устраняются под Ba
куумом перед последующими процедурами,
такими как обесцвечивание.
Физическое рафинирование. Оно вклю
чает паровую обработку масла при низком дaB
лении и температуре выше 235 ОС. Этот способ
подходит только для масел с низким содержани
ем фосфатидов и металлов (пальмовое, KOKOCO
вое, оливковое масла) или для масел, из которых
фосфатиды и металлы удалены посредством
кислотной обработки концентрированной фос
форной кислотой И последующей адсорбционной
отбеливающей обработкой (подсолнечное, рап
совое и соевое масла). Этот способ не применя
ется для чувствительных к HarpeBY масел (хлоп
ковое масло), которые темнеют при обработке.
Обесцвечивание. Общим методом отбе
ливания является адсорбционная обработка
масла, выдержанноro при температуре 90 ос
в течение 30 мин под вакуумом в присутствии
обесцвечивающеro сорбента (природноrо или
активированноro) или уrля (активированноro
или нет); также MOryт быть добавлены синтети
ческие силикаrели. Вещества, такие как хлоро
филл и каротиноиды, оставшиеся после очист
ки, на данной стадии удаляются полностью.
Дезодорирование. Дезодорация устраняет
запахи, летучие вещества и остатки от раствори
телей после экстракции; она заключается во BBe
дении сухоro пара в масло, которое находится в
вакууме при высокой температуре. В зависимо
сти от вида масла при меняются различные TeM
пературные режимы: от 200 ос до 235 ос в тече
ние 1,5 3 ч или выше 240 ос в течение 0,5 ч.
Одна из основных побочных реакции это
термическое обесцвечивание путем разрушения
каротиноидов при температуре выше 150 ос. Этот
метод приводит к потере веществ, которые MOryт
быть oтorHaHbI (свободные жирные кислоты, CTe
ролы, токоферолы и часть очищенноro масла),
и может привести к цис транс изомеризации He
насыщенных двойных связей жирных кислот.
ФРАКЦИОНИРОВАНИЕ
РАФИНИРОВАнноrо МАСЛА ПУТЕМ
ОХЛАЖДЕНИЯ
ЭТО устранение твердых частиц и восков по
средством фильтрации при низких температу
рах (также называется депарафинизация). Эти
частицы и воски MOryT повлиять на внешний вид
масла и выпадать в осадок.
rИДРИРОВАНИЕ
rидрирование высушенных и'или обесцве
ченных масел проводиться с помощью катали
заторов (например, Ni, Pt, Pd) при температурах
от 100 ос до 200 ос под давлением водорода. Ka
тализаторы впоследствии удаляются фильтра
цией при температуре 90 ос. Водород должен
быть чистым, т. е. свободным от ядов для KaTa
лизатора, воды с низким содержанием двуокиси
уrлерода, метана и азота. При этом может полу
чаться небольшое количество полимеров. При
частичном rидрировании образуются тpaHC
жирные кислоты.
ХРОМАтоrРАФИЧЕСКАЯ ОЧИСТКА
При необходимости получения особо чи
стоro масла, rлавным образом для паренте
ральноrо применения, ero очищают посред
ством пропускания через колонку, содержащую
активированный сорбент. Для повышения эф
фективности иноrда используют растворители.
При этом удаляются преимущественно сильно
полярные молекулы, такие как оксиды, кисло
ты, спирты, частичные rлицериды и свободные
стеролы.
Если масло используется в приroтовлении
парентеральных дозированных форм, требова
ния по предельным значениям кислотноro и пе
рекисноro чисел и содержанию воды в частных
статьях MOryT быть различными.
МАРКИРОВКА
На этикетке указывают:
rде применимо, способ получения (отжим
или экстракция);
rдe применимо, что масло подходит для про
изводства парентеральных дозированных форм.
01/2013:1435
РАСТИТЕЛЬНЫЕ ЧАИ
Р/апtае ad рtisапат
ОПРЕДЕЛЕНИЕ
Растительные чаи состоят из одноro или
более измельченноro, реже цельноrо, лекар
ственноro растительноro сырья и предназначе
#Сборы
1091
ны для приrотовления жидких лекарственных
средств для оральноro применения после из
roтовления водных извлечений, #как указано в
статье #Настои, отвары и чаи.
Лекарственное растительное сырье, BXO
дящее в состав растительноro чая, должно co
ответствовать требованиям общей и частной
статей.
Растительные чаи должны выдерживать
требования статей #Лекарственное pacти
тельное сырье цельное или измельченное
фасованное (если в состав входит одно лекар
ственное растение) и #Сборы (если в состав
входит смесь лекарственных растений).#
01/201 З:РБОООЗ
#СБОРЫ
Species
ОПРЕДЕЛЕНИЕ
Сборы представляют собой смеси He
скольких видов измельченноrо, реже цельно
ro, фасованноrо лекарственноrо раститель
ноro сырья, иноrда с добавлением солей,
эфирных масел, с определенным действием,
предназначенные для применения в лечеб
ных целях после изrотовления водных извле
чений, как указано в статье #Настои, отвары
и чаи.
Сборы MOryT быть дозированными и Heдo
зированными.
ПРОИЗВОДСТВО
Сырье, используемое для приroтовления
сборов, должно соответствовать требованиям
частной фармакопейной статьи.
Внешний вид, цвет, запах сборов должны
быть характерными для сырья, входящеro в
их состав. Сборы не должны издавать посто
роннеro запаха.
Сырье, входящее в состав сборов, из
мельчают и просеивают по отдельности. Если
иноro не указано в частной статье, то сырье,
используемое для приrотовления настоев и
отваров, должно иметь степень измельчения
(5600) (2.9.12); сырье, используемое для при
roтовления чаев, должно иметь степень из
мельчения (2000) (2.9.12).
Листья толокнянки и брусники, исполь
зуемые для приrотовления отваров, должны
иметь степень измельчения (2800) (2.9.12).
Побеrи баrульника болотноro, исполь
зуемые для приrотовления настоев. должны
иметь степень измельчения (4000) (2.9.12).
Цветы, семена и плоды не измельчают,
если нет друrих указаний в частной фармако
пейной статье.
Компоненты, входящие в состав сбора,
перемешивают до получения равномерной
смеси. Смешивание сырья следует начинать с
основноro составляющеro, добавляя осталь
ные в порядке очередности соrласно пропи
си. В тех случаях, KorAa в состав сбора входит
соль, из нее rотовят насыщенный раствор и
опрыскивают им сбор при перемешивании,
после чеrо высушивают при температуре не
выше 60 ОС. Эфирное масло вносят в сбор в
виде спиртовоro раствора (1: 1 О) опрыскива
нием при перемешивании.
rиrроскопичное и леrко портящееся от YB
лажнения сырье следует прибавлять в сбор
после опрыскивания друrих компонентов pac
твором соли и высушивания с последующим
перемешиванием.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Необходимо подтвердить соответствие MOp
фолоrических и анатомических признаков OT
дельных лекарственных растений (составля
ющих сбора) требованиям, указанным в частной
статье. Испытание проводят из аналитической
пробы массой 1 О r.
Подлинность сбора и еro составляющих, пе
речисленных в разделе «Состав», определяют
подходящим методом (качественные реакции,
ТСХ, ЖХ, rX).
ИСПЫТАНИЯ (ЧИСЛОВЫЕ ПОКАЗАТЕЛИ)
Степень измельчения (2.9.12), приме
си (2.8.2), потерю в массе при высушива
нии (2.2.32), общую золу (2.4.16), золу, He
растворимую в хлористоводородной кислоте
(2.8.1) и друrие числовые показатели опре
деляют, как указано в частной фармакопей
ной статье.
Однородность массы для дозирован
Horo сырья. Определяют среднюю массу 20
случайно выбранных дозированных единиц
упаковки (фильтр пакет, пакетик, брикет и
друrие) следующим образом: взвешивают
одну дозированную единицу упаковки, затем
открывают ее таким образом, чтобы не были
утеряны какие либо фраrменты. Упаковку пол
ностью очищают от содержимоro при помощи
щеточки. Взвешивают пустую упаковку и pac
считывают массу содержимоrо упаковки путем
вычитания. Повторяют данные действия на
девятнадцати оставшv.хся единицах упаков
ки. Если нет друrих указаний, то не более чем
две из двадцати индивидуальных масс coдep
жимоrо отклоняются от средней массы coдep
жимоro больше чем на процентное отклоне
ние. приведенное в таблице РБОО03. 1, но не
более чем вдвое.
Таблица РБООО3. 1
rосударст6енная фармакопея Республики Беларусь
01/201З:РБООО5
1092
Средняя масса Допустимое отклонение
с учетом влаrи
до 1,5 r 15%
от 1,5 r до 2,0 r 10%
включительно
более 2,0 r 7,5%
Однородность массы для недозирован
ното сырья. Определяют среднюю массу 1 О
случайно выбранных недозированных единиц
упаковки (пачка, пакет и друrие) следующим об
разом: взвешивают одну упаковку с содержи
мым, затем открывают ее таким образом, чтобы
не были утеряны каки либо фраrменты. Упа
ковку полностью очищают от содержимоro при
помощи щеточки. Взвешивают пустую упаков
ку и рассчитывают массу содержимоro упаковки
путем вычитания. Повторяют данные действия
на девяти оставшихся упаковках. Допустимые
отклонения массы содержимоro упаковки приве
дены в таблице РБОО03. 2.
Таблица РБОО03. 2
Допустимое отклонение
с учетом влаrи
Средняя масса
для одной для десяти
упаковки упаковок
до 100 r 5% 1,6%
от101rд0200r 3% 0,9%
от 201 r до 1000 r 2% 0,6%
При определении микробиолоrической чи
стоты (5.1.8) необходимо учитывать применя
емый метод приrотоеления.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определяют содержание фармаколоrиче
ски активных веществ, обусловливающих фар
маколоrическое действие сбора, методом, YKa
занным в частной фармакопейной статье.
ХРАНЕНИЕ
В контейнере, предохраняющем от воздей
ствия влаrи и света, при температуре от 15 ос до
25 ОС, если в частной статье нет особых указа
ний. Не допускается хранение сборов в местах,
не защищенных от воздействия посторонних за
пахов.
МАРКИРОВКА
Указывают:
массу содержимоro контейнера с учетом
влажности (потеря в массе при высушивании
либо содержание воды) лекарственноro расти
тельноro сырья.
информацию о проведении радиационно
ro контроля.
#РЕrИСТРАЦИОННЫЕ ТРЕБОВАНИЯ
И ПРАВИЛА ПРО ВЕДЕНИЯ
ИССЛЕДОВАНИЙ БИОДОСТУПНОСТИ
И БИОЭКВИВАЛЕНТНОСТИ
rЕНЕРИЧЕСКИХ ЛЕКАРСТВЕННЫХ
СРЕДСТВ
1. ВВЕДЕНИЕ
rенерические лекарственные средства
должны удовлетворять тем же стандартам Ka
чества, эффективности и безопасности, что и
ориrинальныe лекарственные средства, но
при этом дополнительно должно быть предо
ставлено убедительное подтверждение тою,
что они эквивалентны ранее эареrистриро
ванным аналоrичным лекарственным cpeд
ствам и клинически взаимозаменяемы с ними.
Оценка биоэквивалентности rенериче
ских лекарственных средств (средств, coдep
жащих одно и то же лекарственное вещество
в одинаковой дозе и в той же лекарственной
форме, что и ориrинальное лекарственное
средство) считается основным видом меди
ко биолоrическоrо контроля их эффективно
сти. Оценка биодоступности rенерических ле
карственных средств выполняется в случаях,
если средство содержит одно и то же лекар
ственное вещество в одинаковой дозе, но при
этом имеет отличную лекарственную форму
от ориrинальноrо лекарственноro средства.
Исследования биоэквивалентности и био
доступности позволяют сделать обоснован
ный вывод об их эффективности по меньше
му объему первичной информации и в более
сжатые сроки, чем проведение клинических
испытаний.
Несмотря на то, что для некоторых rрупп
лекарственных средств, в первую очередь
парентеральны,, содержащих компонен
ты, хорошо растворимые в воде, не требу
ется проведение биоэквивалентных иссле
дований, тем не менее, для большинства
номинально эквивалентных лекарствен
ных средств (включая большинство твердых
оральных дозированных форм) должна быть
проведена демонстрация терапевтической
эквивалентности.
Таким образом, положения правил pac
пространяются на rенерические лекар
ственные средства, произведенные разны
ми предприятиями, но которые должны быть
биоэквивалентны и клинически взаимоза
меняемы, и не распространяются на лекар
ственные средства биолоrическоrо проис
хождения, такие как вакцины, сыворотки
животных, препараты человеческой крови
и плазмы и препараты, произведенные при
помощи биотехнолоrии.
#Реаистрационные требования и правила проведения исследований биодоступности 1093
Хотя понятие биоэквивалентности может
рассматриваться в отношении растительных ле
карственных средств, основные принципы, BЫ
деленные в данных указаниях, не применимы к
растительным лекарственным средствам, для
которых активные составляющие определены
недостаточно по сравнению с химическими co
единениями.
Рекомендации по моделированию и про
ведению исследований для изучения биоэкви
валентности, приведенные в данном разделе,
MOryT также при меняться для сравнительноro
исследования биодоступности с целью оценки
различных рецептур, используемых в процессе
разработки новоro лекарственноro средства, co
держащеro новое химическое соединение, и для
сравнительноro изучения биоэквивалентности
лекарственных средств при изменении их лекар
ственной формы на форму модифицированноro
высвобождения или разработке комбинирован
ных средств.
Для rарантии взаимозаменяемости reHe
рическое лекарственное средство должно яв
ляться терапевтически эквивалентным лекар
ственному средству, которое использовано для
сравнения. Прямое доказательство на практике
терапевтической взаимозаменяемости при кли
нических исследованиях требует участия боль
шоro количества пациентов. Соrласно основным
положениям клинической фармаколоrии Tepa
певтическую равнозначность можно rарантиро
вать, если rенерическое лекарственное cpeд
ство является одновременно фармацевтически
эквивалентным и биоэквивалентным. Допуская,
что у одноro и тоro же пациента эквивалентные
показатели биодоступности и концентрации ле
карственных средств в плазме крови создают эк
вивалентные их концентрации в MeCTe( ax) дей
ствия, а следовательно вызывают по существу
подобные терапевтические эффекты, можно ис
пользовать данные фармакокинетики вместо Te
рапевтических результатов для доказательства
эквивалентности rенерических лекарственных
средств. В отдельных случаях для установле
ния признаков равнозначности может считаться
достаточным сравнение iп vitro профиля paCT
ворения rенерическоro лекарства с таковым у
компаратора или «тест изучения сравнительной
кинетики растворения».
Следует отметить, что концепция взаимоза
меняемости предлолаrает эквивалентность дo
зировок, а также показаний и инструкций по при
менению. Допускаются альтернативные подходы
к принципам и методам, описанные в данном
разделе, при условии их соответствующеro науч
ноro обоснов<'IНИЯ. Данные рекомендации следу
ет толковать и при менять, не оrраничивая смысл
обязательств, возникающих в связи с действу
ющими соrлашениями по торroвым аспектам
прав на интеллектуальную собственность.
Настоящие правила подroтовлены на основе
следующих нормативных документов:
138 ЗаК.1712.
Mu'tisource (gепеriс) pharmaceutica'
products: guideliпes оп registratioп rеquirетепts
to estabIish iпtе,сhапgеаЫ'itу / WHO Techпica'
Report Series, NQ.937, 2006. P347 390;
Проведение качественных исследований
биоэквивалентности лекарственных средств ,
И.Б. Бондарева, В.Б. rерасимов, АЛ. Дрожжин
и др. 11 Методические указания, утвержденные
Минздрасоцразвития Российской Федерации
10.08.2004. Москва, 2004. 43с.;
Руководство по клиническим исследо
ваниям. Лекарственные средства. Исследова
ние биодоступности и биоэквивалентности, 42
7.1 :2005 Киев, 2005. 22с.;
CPMP/EWP/QWP/1401/98 Rev.1/Corr
Guidaпce оп the /пvestigatioп о' Bioequiva/eпce.
Lопdоп, 2010. 27р.;
<1090> 'П vivo Вiоеqиivа/епсе Guidaпces
U.S. Pharmacopia 24th ed, М2 F 2, Supp'. 2.
2000. Р 2056 2098,
с учетом современных требований и особен
ностей законодательства Республики Беларусь.
Основной целью правил является предо
ставление техническоrо руководства специали
стам при про ведении экспертизы документации
на предмет возможности реrистрации и прове
дения испытаний rенерических лекарственных
средств и обеспечения rарантий пациентам по
защите их здоровья, безопасности и прав.
2. ОСНОВНЫЕ ТЕРМИНЫ
И ОПРЕДЕЛЕНИЯ
Приведенные ниже определения касают
ся терминов, использованных в настоящем раз
деле, так как они MOryT иметь иные значения в
друrих документах и нормативных актах.
Биолоrическая доступность (биодоступ-
ность, Ыоауаilаыlitу)
Скорость и степень, с которыми действу
ющее вещество или еro активные компоненты
из дозированной лекарственной формы вcaCЫ
ваются и становятся доступными в месте дей
ствия. Эти пара метры определяют с помощью
кривой зависимости «концентрация время» в
системном кровообращении или по выделению
активноro компонента в мочу. слюну и друrие
биожидкости. При этом считают. что (1) веще
ство а prio,i в системном кровотоке находится в
динамическом равновесии с веществом в месте
действия; (2) если два лекарственных средства
у одноro и тоro же человека имеют одинаковые
скорость всасывания и концентрации в плазме
(сыворотке) крови. то их концентрации в месте
действия также одинаковы.
Абсолютную биодоступность данной лекар
ственной формы определяют путем сравнения
с биодоступностью этоro лекарственноro cpeд
ства при условии еro внутрисосудистоro BBeдe
ния (100 %) (например, раствор для оральноro
применения в сравнении с раствором для BHY
тривенноro введения).
1094
rосударственная фармакопея Республики Беларусь
Относительную биодоступность данной ле
карственной формы определяют путем cpaB
нения с биодоступностью друrой лекарствен
ной формы, введенной тем же или друrим (но
не внутривенным) путем (например, таблетки в
сравнении с раствором для оральноro приме
нения).
Основой проведения исследований биоэ
вивалентности и биодоступности rенерических
лекарственных средств является определение
относительной биодоступности.
Биолоrическая эквивалентность (биоэк
вивалентность, bioeqиivaleпce)
Два лекарственных средства биолоrиче
ски эквивалентны, если они фармацевтически
эквивалентны или они фармацевтически вза
имозаменяемы и их биолоrические доступ
ности (выраженные как максимальная KOH
центрация С та. И степень доступности в виде
площади под фармакокинетической кривой
АUС) после приема в одной и той же моляр
ной дозе похожи до такой степени, что можно
предполаrать, что их терапевтические эффек
ты и показатели безопасности будут по суще
ству одинаковыми.
Биофармацевтическая система класси
фикации (БСК. Biopharmaceutics Classifica
tiоп System, BCS)
Научный подход, позволяющий разделить
активные компоненты лекарственных средств
на основании степени их растворимости в воде
и кишечной проницаемости. Вместе с тестом
«Растворение» для лекарственноrо средства
БСК учитывает три основных фактора, вли
яющих на скорость и степень абсорбции ле
карственных форм для пероральноro приема:
растворение, растворимость и кишечную про
ницаемость.
«Биовейвер» (Biowaive,)
Обозначение процедуры оценки биолоrиче
ской эквивалентности лекарственноro средства
без проведения исследования еro эквивалентно
сти in vivo.
Препарат сравнения (эталонное лекар-
ственное средство, компаратор, comparator
product)
Лекарственное средство, с которым rенери
ческое лекарственное средство предполаrает
ся взаимозаменяемым в клинической практике.
Препаратом сравнения (эталонным лекарствен
ным средством) обычно является ориrинальное
лекаРСТВl3нное средство, для котороro уже YCTa
новлены эффективность, безопасность и каче
ство. В тех случаях, Korдa ориrинальное лекар
ственное средство неизвестно, в еro роли может
выступать препарат, который выбран на OCHO
вании процедуры, предусмотренной разделом
6.5.2 данной статьи.
Лекарственная форма (dosage form)
Форма, которую придают лекарственному
средству для удобства еro практическоro при
менения и оказания оптимальноro терапевти
ческоro действия, например, таблетки, капсулы,
свечи, растворы и т. п.
Нормативы эквивалентности (eqиiva
'епсе rеqиirетепts)
Допустимые rраницы колебаний резуль
татов iп vivo и'или iп vitro тестирования для ro
сударственной реrистрации rенерическоro
лекарственноro средства.
Исследование биоэквивалентности
(eqиivaleпce test)
Испытание, которое определяет эквива
лентность между rенерическими лекарственны
ми средствами и продуктами сравнения, исполь
зуя подход тестирования iп vivo иlили in vit,o.
Фиксированная комбинация дОЗ (ФКД,
fixed dose сотЫпаtiоп, FDC)
Комбинация двух и более активных KOM
понентов с установленным соотношением доз.
Используется для обозначения конкретной
комбинации активных фармацевтических инrре
диентов в независимости от состава или TOBap
ноro знака roтовоro лекарственноro средства.
В рамках испытаний биоэквивалентности в каче
стве препарата сравнения данная комбинация
может применяться как в виде совокупности MO
нокомпонентных лекарственных средств, вводи
мых параллельно, так и в виде roтовоro мноro
компонентноro лекарственноro средства.
Фиксированная комбинация доз rOToBo
ro лекарственноrо средства (Фкд rлс, fixed
dose сотЫпаtiоп fiпished pharmaceиtica/
prodиct, FDC FPP)
rOToBoe лекарственное средство, содержа
щее два и более активных фармацевтических
инrредиентов.
rенерическое лекарственное средство
(geпe,ic prodиct)
Фармацевтически эквивалентные или фар
мацевтически взаимозаменяемые лекарственные
средства, которые MOryт являться терапевтиче
ски эквивалентными либо не являются таковыми
с ориrинальным лекарственным средством. Tepa
певтически эквивалентные лекарственные cpeд
ства являются взаимозаменяемыми.
Ориrинальное лекарственное средство
(iппovatorpharl11aceи6calprodиc
Лекарственное средство, которое было
первым зареrистрировано и размещено на ми
ровом фармацевтическом рынке на основании
представления полноro досье, то есть досье, co
держащеro документальное подтверждение еro
качества, эффективности и безопасности.
#Ре2истрационные требования и правила проведения исследований биодоступностu 1095
Взаимозаменяемое лекарственное cpeд
ство (iпtеrсhапgеаЫе pharmaceutical product)
Лекарственное средство, которое Tepa
певтически эквивалентно препарату cpaBHe
ния (эталонному лекарственному средству) и
может взаимно заменяться с ним в клиниче
ской практике.
Исследование фармацевтической экви
валентности iп vitro (in vitro equivalence test)
Лабораторное исследование эквивалентно
сти в тесте кинетики растворения, включающем
сравнение профилей растворения rенерическо
ro лекарственною средства и препарата cpaBHe
ния в трех средах со значениями рН 1,2, рН 4,5
и рН 6,8.
Тест «Растворение» для контроля
качества iп vitro (in vitro quality contro/ disso
lution test)
Определенная Фармакопеей процедура
теста «Растворение» в виде испытания на pac
творение с одним контрольным моментом Bpe
мени для лекарств с немедленным высвобож
дением и испытание на растворение с тремя и
более контрольными моментами времени для
препаратов с модифицированным высвобожде
нием.
Фармацевтически взаимозаменяемые
лекарственные средства (pha,maceutica/ a/
terпatives)
Лекарственные средства являются фар
мацевтически взаимозаменяемыми, если они
содержат один и тот же активный компонент
(или компоненты), но различаются еro химиче
ской формой (соль, эфир и др.), лекарствен
ной формой (таблетки, капсулы, суспензии) или
силой действия (активностью). Фармацевтиче
ски взаимозаменяемые лекарственные средства
доставляют одинаковый активный компонент с
помощью аналоrичноro способа введения, но в
остальном фармацевтически равнозначными не
считаются. По отношению к препарату cpaBHe
ния они MorYT являться биоэквивалентными или
нет, либо быть терапевтически эквивалентными.
Фармацевтическая эквивалентность
(pha,maceutical equivalence)
Лекарственные средства являются фар
мацевтически эквивалентными, если они co
держат одинаковое количество одноro и тоro
же активноro вещества (или веществ) в одной
и той же лекарственной форме, COOTBeTCTBY
ют одним и тем же сопоставимым стандартам
и применяются одинаковым способом. Однако,
фармацевтическая эквивалентность не обяза
тельно предполаrает биолоrическую или Tepa
певтическую эквивалентность, так как различия
в наполнителях и'или в процессе производства
MOryT при водить К различиям в эффективности
препарата.
Терапевтическая эквивалентность (thera
peutic equivalence)
Два лекарственных средства являются Te
рапевтически эквивалентными, если они фар
мацевтически эквивалентны или фармацевтиче
ски взаимозаменяемы, при этом после приема
в одинаковой молярной дозе одним и тем же
способом (соrласно инструкции по медицинско
му применению) их клиническое воздействие с
точки зрения эффективности и безопасности,
будет по существу одинаковым, о чем свиде
тельствуют данные соответствующих исследо
ваний (биоэквивалентности, фармакодинамиче
ские, клинические или иследования iп vitro).
С практической точки зрения доказательство
биоэквивалентности, как правило, является наи
более подходящим способом доказательства Te
рапевтической эквивалентности лекарственных
средств при условии, что эти средства содержат
вспомоrательные вещества. которые известны
как компоненты, не влияющие на безопасность
и эффективность, а также при соблюдении Tpe
бований к маркировке относительно вспомоrа
тельных веществ.
В некоторых случаях, коrда при подобной
степени абсорбции наблюдается различная
скорость всасывания, лекарственные средства
MOryT быть признаны терапевтически эквива
лентными, если такие различия не являются
важными в терапевтическом отношении (для
подтверждения этоro MOryT потребоваться дo
полнительные клинические исследования).
Дозировка лекарственноrо средства
(dose)
Это количество (r, М!; MKr) лекарственною
средства в единице лекарственной формы (таб
летка, капсула, драже и т. д.).
Доза лекарственноrо средства (posology)
Это количество (!; М!; MKr) лекарственною
средства на один прием (однократное или MHO
roкратное применение).
3. РЕrИСТРАЦИОННАЯ ОЦЕНКА
ЭКВИВАЛЕНТНОСТИ ВЗАИМО
ЗАМЕНЯЕМЫХ ЛЕКАРСТВЕННЫХ
СРЕДСТВ
Процедура реrистрации должна rаранти
ровать, что все лекарственные средства, в том
числе и rенерические. прошедшие реrистрацию,
соответствуют установленным стандартам каче
ства, безопасности и эффективности и что все
условия при производстве, хранении и реали
зации соответствуют стандартам Надлежащей
производственной практики (GMP).
Номинально эквивалентные взаимозаменя
емые (rенерические) лекарственные средства
должны содержать одинаковые количества оди
наковых терапевтически активных инrредиен
тов и должны соответствовать фармакопейным
стандартам. Однако они обычно не являются
1096
rосударственная фармакопея Республики Беларусь
идентичными, и при некоторых обстоятельствах
их клиническая взаимозаменяемость может BЫ
зывать сомнения. Несмотря на то, что различия
в цвете, форме и вкусе очевидны и это вызыва
ет сомнения у пациентов, они, как правило, не
оказывают никакоro существенноro ВЛI11ЯНИЯ на
действие лекарственноro средства. Однако раз
личия в потенциальной чувствительности из за
использования различных наполнителей и раз
личия в стабильности и биолоrической доступ
ности MOryT иметь очевидные клинические по
следствия. Поэтому необходимо следить не
только за качеством, эффективностью и безо
пасностью таких лекарственных средств, но и за
их взаимозаменяемостью. Концепция взаимоза
меняемости применима не только к лекарствен
ным формам, но и к инструкциям по их примене
нию и даже к техническим пара метрам упаковок,
если они имеют решающее значение для CTa
бильности и срока roдности.
Для тоro чтобы считать ориrинальное и re
нерическое лекарственные средства взаимо
заменяемыми, следует прямо или косвенно
продемонстрировать терапевтическую эквива
лентность rенерическоro лекарственноrо cpeд
ства по отношению к препарату сравнения. Под
ходящими исследовательскими методами для
оценки такой равнозначности являются:
сравнительные фармакокинетические ис
следования на людях, при которых активный
фармацевтический инrpeдиент (АФИ) и'или еro
метаболит( ы) представляются в виде функции
зависимости от времени фармакокинетических
показателей, таких как Аис и С тах ' которые OT
ражают системное воздействие, в доступной
биолоrической ЖИДКОСТИ, такой как, например,
кровь, плазма, cывopo1l<a или моча;
сравнительные фармакодинамические ис
следования на людях;
сравнительные клинические испытания;
сравнительные испытания in vitro.
Ниже обсуждается применимость каждой из
этих четырех методик. Представлена подробная
информация по проведению оценки испытаний
на эквивалентность с ИСПОЛЬЗО8анием фармако
кинетических показателей и in vit,o методов, KO
торые в настоящее время являются наиболее
адекватными способами документальноro под
тверждения равнозначности для большинства
вводимых перорально лекарственных средств
системноro воздействия.
Приемлемость любой из вышеописанных
исследовательских процедур при официальном
подтверждении эквивалентности двух лекар
ственных средств зависит от мноrих факторов,
включая характеристики АФИ и лекарственноro
средства. В случае если АФИ создает измери
мые концентрации в доступных биолоrических
жидкостях (например, плазме), MoryT проводить
ся сравнительные фармакокинетические иссле
дования. В некоторых случаях по отношению к
лекарственным средствам с немедленным BЫ
свобождением тестирование iп vit,o и базиру
ющиеся на БСК процедуры «биовейвер.а» MOryT
rарантировать равнозначность между rенериче
ским лекарственным средством и препаратом
сравнения (разделы 5 и 9). Если АФИ не обра
зует поддающихся измерениям концентраций в
доступных биолоrических жидкостях, то сравни
тельные фармакодинамические испытания яв
ляются альтернативным способом документаль
ноro подтверждения эквивалентности. Иноrда
фармакокинетический профиль не поддается
определению или невозможно найти подходя
щие фармакодинамические конечные точки, в
таких случаях можно считать оправданным про
ведение сравнительных клинических I11спытаний.
В следующих двух разделах требований об
суждаются условия, которые указывают, коrда
необходимо выполнять исследования эквива
лентности.
4. ОТСУТСТВИЕ НЕОБХОДИМОСТИ
В ПРОВЕДЕНИИ ИССЛЕДОВАНИЯ
ЭКВИВАЛЕНТНОСТИ ДЛЯ РЕrИСТРАЦИИ
ЛЕКАРСТВЕнноrо СРЕДСТВА
Следующие rруппы лекарственных средств
считаются эквивалентными и для них не Tpe
буется предоставление специальноrо ДOKYMeH
тальноro подтверждения эквивалентности у че
ловека.
1) Лекарственные средства вводимые па
рентерально (например, внутри вен но, внутримы
шечно, подкожно или внутритрахеально) в виде
водных растворов, которые содержат то же aK
тивное вещество (вещества), что и инновацион
ные эталонные средства, в одинаковой с ними
молярной концентрации (концентрациях) и те
же самые вспомоrательные вещества в сопоста
вимых концентрациях. Определенные вспомо
rательные вещества (а именно, буфер, KOHcep
вант и антиокислитель) MOryT различаться при
условии, что можно доказать отсутствие вли
яния изменения( ий) данных субстанций на безо
пасность и'или эффективность лекарственноro
средства.
2) Растворы для пероральноro примене
ния (т. е. сиропы, эликсиры и настойки), KOTO
рые содержат активное вещество в той же MO
лярной концентрации, что и эталонное средство,
а также по существу одинаковые вспомоrатель
ные вещества в сопоставимых концентрациях.
При этом лекарственные средства не содержат
наполнителей, которые заведомо или предполо
жительно воздействуют на желудочно кишечный
тракт или абсорбцию активноro вещества. Поло
жечия указанноro пункта не касаются суспензий,
эмульсий и прочих жидких лекарственных форм.
3) Порошки для приroтовления растворов,
если раствор соответствует вышеприведенным
критериям 1) или 2).
4) rазы.
5) Ушные или rлазные лекарственные
средства, приroтовленные в виде водных pac
#Реаистрационные требования и правила проведения исследований биодоступности 1097
творов, которые содержат одно и то же активное
вещество (вещества) в одинаковой молярной
концентрации (концентрациях) и по существу те
же самые вспомоrательные вещества в сопоста
вимых концентрациях. Определенные вспомо
rательные вещества (как то: консервант, буфер,
субстанция для реryлирования изотоничности
или заryститель) MOryT различаться при усло
вии, если их применение предположительно не
влияет на безопасность и'или эффективность
лекарственноro средства.
6) Лекарственные средства для местноro
применения, приroтовленные в виде водных pac
творов, которые содержат одно и то же активное
вещество (вещества) в одинаковой молярной
концентрации (концентрациях) и по существу те
же наполнители в сопоставимых концентрациях.
7) Инrаляционные лекарственные cpeд
ства или назальные спреи, которые применя
ются с помощью практически одинаковых при
способлений (или без них), приютовлены в виде
водноro раствора, содержат одно и то же актив
ное вещество (вещества) в одной и той же KOH
центрации (концентрациях) и содержат в OCHOB
ном одни И те же вспомоrательные вещества в
сопоставимых концентрациях. По решению Ми
нистерства здравоохранения может быть назна
чено специальное лабораторное тестирование в
отношении устройства для применения инrаля
ционных лекарственных средств.
В отношении требований пунктов 2), 3), 5),
6) и 7) на заявителя возлаrается обязанность дo
кументально доказать, что состав наполнителей
в лекарственных средствах качественно и коли
чественно идентичен эталонным лекарствен
ным средствам, либо, в отдельных ситуациях
(например, 5) и 7) позиции), по казать, что их при
менение не должно повлиять на безопасность и,
или эффективность препарата. При отсутствии
возможности у заявителя предоставить такую
информацию, а также при отсутствии доступа
к достоверным данным у Министерства здра
воохранения, в обязанность заявителя входит
проведение соответствующих испытаний для
подтверждения тою, что различия во вспомоrа
тельных веществах или устройствах доставки не
изменяют действия лекарственноro средства.
5. НЕОБХОДИМОСТЬ ПРОВЕДЕНИЯ
ИССЛЕДОВАНИЯ ЭКВИВАЛЕНТНОСТИ
НА ЧЕЛОВЕКЕ
За исключением случаев, указанных в раз
деле 4, сравнительные исследования по оценке
биоэквивалентности'биодоступности, осущест
вляемые на людях, проводятся для следующих
rрупп лекарственных средств и форм 1 .
5.1. ИССЛЕДОВАНИЯ /N V/VO
ДЛЯ определенных лекарственных средств
и лекарственных форм документальное под
тверждение эквивалентности in vivo путем aHa
лиза фармакокинетической биоэквивалентно
139 Зак. 1712.
сти, сравнительноro фармакодинамическоro
изучения или сравнительноro клиническоro ис
пытания считается особо важным. Документаль
ное подтверждение эквивалентности in vivo Tpe
буется в случаях, коrда имеется риск тоro, что
возможные различия 8 биодоступности MOryT
привести к терапевтической неравнозначности 2 .
Исследования in vivo Пр080ДЯТСЯ для следующих
rрупп лекарственных средств и форм:
1) любые лекарственные формы, пред
назначенные для пероральноro приема, с He
медленным высвобождением, содержащие ле
карственные средства с системным действием
(таблетки, капсулы, драже, суспензии, эмульсии
и проч.), если выполняется одно или более из
указанных ниже условий:
а) предназначены для интенсивной терапии
и лечения тяжелых состояний, требующих
rарантированной терапевтической эффек
тивности;
Ь) имеют узкий терапевтический диапа
зон (соотношение эффективность'безопас
ность) действия и узкие rраницы безопасности
(крутая кривая зависимости «доза реакция»);
с) имеются документальные доказательства
изменчивости биолоrической доступности
или биоэквивалентности, связанные как с
самим АФИ, так и с лекарственной формой
(вне зависимости от растворимости лекар
ственноro средства), а также средствами
сходной химической структуры и состава;
d) имеются документальные научные ДOKa
зательства, позволяющие предположить, что
полиморфные модификации АФИ, вспомоrа
тельные вещества и'или технолоrические про
цессы, используемые в производстве, MOryт
оказать воздействие на биоэквивалентность;
2) непероральные и непарентеральные ле
карственные средства, предназначенные для воз
действия посредством системной абсорбции (Ha
пример, трансдермальные пластыри, мази, rели,
биодеrрадирующие таблетки, суппозитории);
3) лекарственные формы с модифици
рованным высвобождением активноro веще
ства, которые предназначены для воздействия
посредством системной абсорбции (включая
трансдермальные терапевтические системы )3;
4) средства с фиксированной комбинаци
ей системноro действия, в которых как минимум
один из ДФИ требует исследования iп vivo.
1 Следует проводить исследования тoro препарата, который
предназначается для продажи (см. также подраздел 6.5).
L HHS/FDA Guidance for ;ndustry: bioavailability and
bioequiva/eпce studies for ora//y adт;n;stered drug products
geпera/ coпs/deratlOns. Departтent о' Hea/th and Нитап
Services US Food and Drug Adтinistratioп. Rockvil/e, 2003.
23p.
3 В некоторых случаях для лекарственных средств модифи
цированноrо высвобождения заключение об эквивалентно
сти может основываться на сведениях об ;п vitro ;п v;vo KOp
реляции (/V//VC) и на данных опытов ;п vitro при условии, что
эта лекарственная форма с модифицированным высвобож
дением проходит не первую (ориrинальную) процедуру rocy
дарственной реrистрации.
1098
rосударственная фармакопея Республики Беларусь
Для лекарственных форм, предназначенных
для местноro применения (оральноro, назально
ro, rлазноro, кожноro, ректальноro, ваrинально
ro и др. пути назначения), которые не являются
растворами и действие которых должно осущест
вляться без системной абсорбции, доказатель
ство эквивалентности осуuцествляется путем
проведения сравнительных клинических или
фармакодинамических, фармакокинетических
исследований и'или iп vitro исследований. В опре
деленных случаях может требоваться определе
ние концентрации АФИ с целью оценки нежела
тельной системной абсорбции.
5.2. ИССЛЕДОВАНИЯ 'N V'TRO
В отношении определенных лекарственных
средств и лекарственных форм может подой
ти документальное подтверждение эквивалент
ности iп vitro. Этим исследованиям посвящен
раздел 9 требований.
6. РЕrЛАМЕНТ И ПРОВЕДЕНИЕ
ИССЛЕДОВАНИЙ БиолоrИЧЕСКОЙ
ЭКВИВАЛЕНТНОСТИ
И БИОДОСТУПНОСТИ НА ЛЮДЯХ
6.1. ОБЩИЕ ТРЕБОВАНИЯ
6.1.1. Условия проведения исследования
на людях
В последующих разделах сформулированы
требования к реrламенту и проведению иссле
дований биодоступности и биоэквивалентности.
Реrламент исследования должен быть основан
на достаточном знании фармакодинамики и'или
фармакокинетики действующеro вещества.
Исследование биоэквивалентности по cy
uцecTBY представляет собой сравнительное
изучение биодоступности, предназначенное для
установления эквивалентности между испыту
емым препаратом и референтным препаратом.
Последующие разделы посвящены в основном
исследованиям биоэквивалентности. Посколь
ку исследования биодоступности являются по
характеру сравнительными. содержание после
дующих разделов также применимо к этим ис
следованиям с необходимыми поправками в
соответствии с задачей каждоro конкретноro ис
следования.
Методолоrия исследований биоэквивалент
ности может быть использована для оценки раз
личий фармакокинетических параметров при
фармакокинетических исследованиях, таких как
изучение взаимодействий «лекарство лекар
ство» или «лекарство пища», или для оценки
различий в подrруппах популяции. В этом случае
необходимо следовать соответствующим PYKO
водствам и надлежащим образом реryлировать
выбор субъектов исследования, дизайн иссле
дования и статистический анализ.
Исследования биолоrической эквивалент
ности должны быть проведены в строroм соот
ветствии с положениями Техническоro кодекса
установившейся практики «Надлежащая клини
ческая практика» (ТКП 184 2009 (02040)).
Участие здоровых испытуемых и больных в
исследованиях биоэквивалентности лекарствен
ных препаратов является добровольным. Добро
волец (волонтер) имеет право отказаться от уча
стия в Пр080ДИМЫХ исследованиях на любой
стадии. Этические нормы проведения иссле
дований, биоэквивалентности реrламентирова
ны соответствующими национальными ДOKYMeH
тами, а также положениями, содержащимися в
действующей редакции Хельсинской деклара
ции, включая принцип уважения свободы и He
прикосновенности личности, принцип пользы
(<<оказать наибольшую пользу при наименьшем
вреде и ошибках») и принцип непричинения зла
(<<не навреди»), и действующей редакции Меж
дународных этических принципов для биомеди
цинских исследований С вовлечением человека,
изданных Советом международных орrаниза
ций по медицинской науке (CIOMS). При пла
нировании исследования законодательное обе
спечение соблюдения этических норм должно
быть выбрано исходя из тоro, какие из перечис
ленных выше документов предоставляют наи
высший уровень защиты добровольца. Эти
ческую экспертизу клинических исследований
биоэквивалентности лекарственных препаратов
про водит Комитет по этике клинической базы
исследований. Добровольцы, включенные в
исследование биоэквивалентности, подписы
вают письменное информированное соrласие,
один экземпляр котороro выдается добровольцу
наряду с «Информацией для добровольцев, уча
ствующих в исследовании биоэквивалентности
лекарственноro средства» (Приложение 3).
6.1.2. Обоснование исследований биоэк-
вивалентности на человеке
Большинство исследований фармакокине
тической и фармакодинамической эквивалент
ности являются нетерапевтическими, при KO
торых пациенты не извлекают никакой прямой
клинической пользы, участвуя в них.
При планировании исследования лекар
ственных средств на человеке важно, чтобы
тщательно рассматривались специфические
цели, проблемы и риски или выroды предложен
ноro исследования на людях и чтобы выбран
ный дизайн был этически обоснованным и тща
тельно продуманным с научной точки зрения.
Предполаrается, что люди, при влеченные к пла
нированию исследования, знакомы с фармако
кинетическими теориями, особенно с исследо
ваниями биодоступности и биоэквивалентности.
Полный дизайн изучения биоэквивалентности
должен основываться на знании фармакокине
тики, фармакодинамики и терапевтическоrо при
менения действующеro вещества лекарственно
ro средства. Сведения о способах производства
и данные, полученные при анализе серии ле
#Ре2истрационные требования и правила проведения исследований биодоступности 1099
карств, которая будет изучаться, должны под
тверждать соответствующее качество испыту
емоro препарата.
Оценка эквивалентности может основываться
как на данных, полученных при однократном BBe
дении препаратов, так и при их мноroкратном (кyp
совом) применении. В последнем случае необхо
димо, чтобы испытуемые получали препараты в
одинаковой разовой дозе с одинаковым интерва
лом дозирования (в соответствии с инструкцией
по медицинскому применению данноro ориrиналь
ноro лекарственноro средства) вплоть до достиже
ния стационарноro состояния их концентраций в
исследуемых биолоrических жидкостях.
6.1.3. Выбор врачей исследователей
Каждый исследователь должен обладать co
ответствующим опытом, квалификацией и KOM
петенцией для проведения предлаrаемоro ис
следования. Прежде чем начать исследование,
заявитель должен достиrнуть с клиническим цeH
тром письменной доroворенности (заключить дo
roвор) по протоколу исследования, процедурам
и частоте мониторинrа, аудита, применяемым
стандартным операционным процедурам и pac
пределению различных видов ответственности у
врачей исследователей.
Для проведения исследований биоэквива
лентности клиническим центром должны быть
выделены сотрудники:
контролирующие состояние здоровья
добровольцев и оказывающие при необходи
мости экстренную медицинскую помощь;
контролирующие соблюдение режима и
орrанизацию питания;
контролирующие установку катетеров и
отбор образцов крови, а также их обработку.
В состав rруппы обязательно должны BXO
дить 1 2 врача исследователя и 1 2 медицин
ские сестры.
Клинический блок, rде будут находиться дo
бровольцы, должен включать помещения для
пребывания добровольцев, процедурную, место
приема пищи и отдыха, туалетную комнату. Перед
началом исследования в указанных помещениях
должна быть проведена санитарная обработка.
6.1.4. Протокол проведения исследова-
ний
Проrрамма (протокол) исследования COCTaB
ляется в строroм соответствии с Техническим KO
дексом установившейся практики «Надлежащая
клиническая практика» (ТКП 184 2009 (02040») и
должна содержать:
сведения об исследуемых лекарственных
средствах (лекарственная форма, дозировка,
фирма производитель);
сведения об испытуемых и их числе;
план рандомизации;
дозу и режим дозирования;
интервал времени между приемом исследу
емоro препарата и препарата сравнения;
биоматериал, в котором предполаrается
определять концентрацию лекарственноro Be
щества;
схему отбора проб и условия их хранения;
сведения об аналитическом методе;
сведения о методах фармакокинетическо
ro анализа;
сведения о допустимых rраницах критери
ев биоэквивалентности.
Исследование биоэквивалентности следу
ет проводить в соответствии с соrласованным
и подписанным исследователем и заявителем
протоколом. В протоколе и приложениях и'или
дополнениях к нему должна указываться цель
испытания и методика, которая будет использо
вана, обоснования для проведения эксперимен
та на людях, сущность и степень любых извест
ных рисков, а также средства rарантии тоro, что
перед принятием решения добровольцы будут
достаточно информированы. Исследователь
несет ответственность за обеспечение выпол
нения протокола. Любое( ые) необходимое( ые)
изменение( ия) обязательно соrласовывается и
подписывается исследователем и заявителем,
а затем прилаrается в виде поправок, за исклю
чением случаев необходимости предотвратить
явный непосредственный риск или опасность
для испытуемоro субъекта.
Протокол и приложения/дополнения к нему
должны оцениваться с научной и этической
точек зрения комитетом по этике, УП «Центр
экспертиз и испытаний в здравоохранении» и YT
верждаться Министерством здравоохранения.
Подписанный и датированный протокол ис
следования вместе с отчетом об исследовании
должен представляться заявителем при roсу
дарственной реrистрации rенерическоro лекар
cTBeHHoro средства.
6.2. ОБЩАЯ СХЕМА ИССЛЕДОВАНИЙ
Исследования биоэквивалентности разра
батываются для сравнения in vivo действия reHe
рическоro лекарственноrо средства и препарата
сравнения.Исследованияqpармакокинетической
биоэквивалентности лекарственных средств,
предназначенных для реализации системноro
воздействия АФИ, служат двум задачам:
выступают в качестве заменителя клини
ческоro доказательства эквивалентности;
дают оценку фармацевтическоro качества
iп vivo.
Схема исследования должна свести к ми
нимуму вариабельность, не связанную с Tex
нолоrическими особенностями состава лекар
CТBeHHoro средства, и исключить, насколько это
возможно, систематические ошибки, связанные
с ходом эксперимента. Вариабельность peaK
ций у различных субъектов и в популяции сле
дует уменьшить с помощью соответственно по
добранноro дизайна исследования. В целом,
для исследования фармакокинетической био
эквивалентности с использованием одноro
1100
rосударственная фармакопея Республики Беларусь
rенерическоro лекарственною средства и одноro
препарата сравнения достаточным будет пр
вести на здоровых добровольцах пере крестное
однодозовое исследование с двумя периодами
применения лекарственных средств. Однако, в
определенных условиях можно принять альтер
нативную, хорошо статистически обоснованную
адекватную модель эксперимента. Такое изме
нение определяется исследователем по соrла
сованию с заявителем и вносится в проrрамму
(протокол) исследования, после чеro утвержда
ется Министерством здравоохранения.
Наиболее предпочтительным вариантом
для испытаний на фармакокинетическую био
эквивалентность считается рандомизирован
ное перекрестное однодозовое исследование
с двумя периодами и двумя последовательно
стями лечения. Каждый испытуемый получает в
случайном порядке rенерическое средство (т) и
препарат сравнения (R) схема TR'RT. После
приема каждоro лекарства должен пройти co
ответствующий период еro вымывания. Между
дозами каждою лекарственною средства сле
дует соблюдать достаточно длительный интер
вал (период вымывания), чтобы дать возмож
ность вывести из орrанизма практически всю
предыдущую принятую дозу. Период вымывания
должен быть одинаков для всех добровольцев и
в нормальных условиях превышать пятикратный
конечный период полуэлиминации АФИ (5tJ.J
В случае образования активных метаболитов с
более продолжительными периодами полувыве
дения и при друrих обстоятельствах необходи
мо рассмотреть увеличение данноro периода.
Например. если скорость выведения препа
рата сильно варьируется в популяции испыту
емых, период вымывания может увеличивать
ся с тем, чтобы raрантировать более медленное
выведение у пациентов с меньшими скоростя
ми элиминации. Непосредственно перед при
емом лекарства во втором периоде исследова
ния у добровольцев отбираются образцы крови
и определяется концентрация в них АФИ и'или
метаболитов. Минимальный период BЫMЫBa
ния должен составлять не менее 7 дней. AдeK
ватность периода вымывания можно оценить,
исходя из концентрации АФИ в крови перед BBe
дением второй дозы. Ее значение не должно
превышать 5 % С тах '
Период отбора проб крови для построения
фармакокинетической кривой должен COCTaB
лять не менее 4ty,' Однако для лекарственных
средств с периодом полуэлиминации более 24 ч
нет необходимости в сборе проб крови на протя
жении более 72 часов.
6.2.1. Альтернативные дизайны исследо
ваний
В отношении АФИ, которые высокоактивны
или слишком токсичны для их введения здоро
вым добровольцам в обычной дозировке (т. е.
из за вероятности серьезных нежелательных яв
лен ий, если испытание проводится с приемом
большой дозы), рекомендуется проводить ис
следования с меньшими дозами. В то же время,
если фармакокинетика лекарственноro cpeд
ства не имеет пропорциональной зависимости
(нелинейна) либо растворимость АФИ являет
ся проблематичной, экстраполяция результатов
испытаний биоэквивалентности при меньшем
содержании на их значения при более BЫCO
ких концентрациях неприменима. В отношении
АФИ. оказывающих нежелаемое фармаколо
rическое действие на здоровых добровольцев,
может потребоваться пере крестное испытание
на пациентах, которые проходят курс лечения
данным лекарственным средством в условиях
стационарноro состояния концентрации лекар
ственноro средства с приемом множественных
доз, или может быть использована модель ис
следования в параллельных rрупnaх пациентов.
Апьтернативный дизайн исследования должен
быть обоснован заявителем, которому необхо
димо предусмотреть набор пациентов со CTa
бильным течением заболевания во время про
ведения испытания на фармакокинетическую
биоэквивалентность.
6.2.2. Планирование испытаний лекар
ственных средств с длительным периодом
полуэлиминации
Перекрестное однодозовое испытание на
фармакокинетическую биоэквивалентность для
пероральноrо лекарственноrо средства с дли
тельным периодом полуэлиминации (24 ч и
более) можно проводить при условии соблюде
ния между приема ми лекарств адекватноro пе
риода вымывания. Промежуток между днями
проведения исследования должен быть ДOCTa
точным, чтобы позволить вывести из орrаниз
ма практически всю предыдущую дозу. В идеале
следует установить интервал не менее 5 конеч
ных периодов полуэлиминации АФИ или еro aK
тивноro метаболита, если измеряется послед
ний. В нормальных условиях промежуток между
периодами исследования не должен превышать
3 недели. В случае не возможности орrани
зовать перекрестное испытание более подходя
щим может оказаться исследование фармакоки
нетической биоэквивалентности в параллельных
rруппах.
Для перекрестноro эксперимента, как и для
испытания с параллельной моделью, следует
подобрать подходящий момент отбора пробы,
rарантирующий завершение транзита лекар
ственноro препарата по ЖКТ (приблизительно
2 3 дня) и всасывание АФИ. Если в ПРОТОКI)
ле не будет обоснован более короткий период,
достаточным будет провести последний отбор
крови не позднее 72 часов после приема лекар
ственноro средства.
Количество испытуемых, участвующих в ис
следовании, определяется на основании CTa
тистических расчетов, но, как правило, для па
#Ре2истрационные требования и правила проведения исследований биодоступности 1101
раллельной модели исследования требуется
большее число субъектов, чем для перекрест
ной схемы испытания 4 .
6.2.3. Исследования с множественными
(повторными) приема ми доз лекарственноrо
средства
В определенных случаях приемлемыми
MOryT считаться исследования с введением MHO
жественных (повторных) доз. Наиболее обосно
ванными испытания с множественными дозами
являются в ситуациях, коrда испытуемое ле
карственное средство оценивается как BЫCO
коактивное иlили высокотоксичное для приема
здоровыми добровольцами даже в однократ
ной дозировке (см. раздел 6.2.1). В таких случа
ях может проводиться пере крестное исследова
ние с множественными дозами на пациентах без
прекращения основной и сопутствующей Tepa
пии. Оценка таких испытаний может OCHOBЫBaTb
ся в равной мере на фармакокинетических или
фармакодинамических конечных точках, однако
использование фармакодинамических конеч
ных точек, вероятнее всеro, потребует большеro
числа пациентQi3, чем использование фармако
кинетических конечных точек.
Схема приема лекарства при исследовани
ях С множественными дозами должна COOTBeT
ствовать обычным указаниям по дозированию в
инструкции по медицинскому применению пре
парата сравнения.
Испытания с множественными дозами MOryT
оказаться приroдными в иных ситуациях, в част
ности:
для лекарственных средств, демонстриру
ющих нелинейную кинетику в стационарном co
стоянии (например, метаболизм с насыщением,
активная секреция);
для случаев, Korдa чувствительность aHa
лиза слишком низка, чтобы после однократной
дозы должным образом описать фармакокине
тический профиль;
для лекарственных форм продленноro
действия со склонностью к кумуляции (как дo
полнение к однодозовому исследованию )5;
для трансдермальных лекарственных
форм (при этом про водят испытания с формой,
имеющей максимальную дозировку в услови
ях однократной .и мноrократной аппликации на
идентичные области тела).
В испытаниях в стационарном состоянии
элиминация последней дозы предыдущеro ле
карственноro средства может перекрываться с
наступлением стационарноrо состояния paBHO
4 При использовании параллельноrо дизайна peKOMeHдyeT
ся, чтобы число включаемых в исследование добровольцев
было не менее чем в 2 раза большим, чем для дизайна с пе
рекрестными rруппами.
5 В этом случае биоэквивалентность следует проверять в yc
ловиях OAHOKpaTHOro приема всех дозировок. а с наиболь
шей дозировкой дополнительно выполняют исследования в
условиях MHoroKpaTHoro приема (достижения стационарноro
состояния).
140 За. 1712
весия второro лекарственноro средства, что Tpe
бует достаточно длительноro периода насыще
ния (не менее трехкратноro конечноro периода
полуэлиминации). Чтобы документально зафик
сировать достижение стационарноro состояния,
следует должным образом выполнять введение
лекарственноro средства и отбор проб.
6.2.4. Планирование исследований для
лекарственных средств с модифицирован
ным высвобождением
Препараты с модифицированным BЫCBO
бождением включают в себя лекарственные
средства продленноro (пролонrированноrо) дей
ствия и лекарственные средства с высвобожде
нием действующеro вещества в определенных
отделах ЖКТ (например, формы с кишечнора
створимым покрытием).
Для установления биоэквивалентности ле
карственных средств с модифицированным
высвобождением рекомендуется выполнять
нерепликативное (невоспроизводимое) пере
крестное исследование с приемом одной дозы
натощак, при котором сравниваются наивыс
шие концентрации rенерическоrо лекарственно
ro средства и компаратора. Предпочтительнее
про водить однодозовые испытания, чем иссле
дования с множественными дозами, поскольку
первые обеспечивают более чувствительные KO
личественные показатели оценки высвобожде
ния АФИ из лекарственной формы в системный
кровоток. Однако может понадобиться проведе
ние испытаний с множественными дозами (как
дополнение к однодозовым испытаниям) приме
нительно к лекарственным формам продленноro
действия со склонностью к кумуляции.
В качестве препарата сравнения в таком
эксперименте следует выбирать фармацевти
чески эквивалентный препарат с модифициро
ванным высвобождением. Условия фармакоки
нетической биоэквивалентности для лекарств с
модифицированным высвобождением являются
в основном теми же, что и для лекарственных
форм с обычным высвобождением.
Одновременный прием пищи вместе с
пероральными лекарственными cpeДCTBa
ми может повлиять на их биодоступность, а
также в некоторых случаях на фармакокине
тическую биоэквивалентность. Помимо фи
зиолоrических изменений в ЖКТ, еда может
воздействовать на высвобождение АФИ из ле
карственноro средства. Проблемы для лекар
ственных средств с модифицированным BЫ
свобождением связаны с вероятностью тоro,
что пища может спровоцировать неожидан
ное и резкое высвобождение АФИ, ведущее к
«демпинrу доз». Это можно установить в виде
преждевременноro и MrHoBeHHoro скачка в
профиле «концентрация В плазме время».
Потому, как правило, требуется проводить ис
спедование фармакокинетической биоэкви
валентности на пациентах в прандиальном
1102
rосударственная фармакопея Республики Беларусь
состоянии (после еды) по отношению к BBO
димым перорально лекарственным cpeд
ствам с модифицированным высвобожде
нием. Заявитель должен документально
обосновать отказ от исследований в пран
диальном состоянии или натощак. Испыта
ние на фармакокинетическую биоэквивалент
ность в прандиальном состоянии следует
про водить после приема соответствующей
стандартной порции пищи за оrоворенный
промежуток времени (обычно не более
30 минут) перед приемом лекарственноro
средства (см. раздел 6.4). Пища с высоким
содержанием жира часто создает наиболь
шие трудности для обеспечения устойчивоro
процесса высвобождения АФИ из лекарствен
ноro средства. В протоколе и отчете исследо
вания необходимо указать состав и распре
деление калорий в порции для испытуемых.
6.3. ИСПblТУЕМblЕ
Оценка биоэквивалентности всех ле
карственных средств, за исключением пси
хотропных, онкохимиотерапевтических и
средств, применяемых при ВИЧ инфекции,
как правило, проводится на здоровых добро
вольцах. Соответствующие исследования
психотропных средств. а также средств, при
меняемых при онколоrических заболеваниях
и ВИЧ инфекции, MorYT выполняться на боль
ных соответствующеro профиля в период pe
миссии заболевания и ВИЧ инфицированных
соответственно. В этом случае следует обе
спечить выполнение 2 следующих условий:
1) поддержание постоянства курса OCHOB
ной терапии в rруппе добровольцев;
2) отсутствие взаимодействия лекар
ственных средств, входящих в основной курс,
с испытуемыми средствами.
Если известно, что активное вещество
имеет неблаrоприятные побочные реакции,
а фармаколоrические эффекты или риск яв
ляются неприемлемыми для здоровых добро
вольцев, то допустимо использовать крупных
лабораторных животных (соrласно приложе
нию 2). Эта альтернатива должна быть обо
снована заявителем, внесена в проrрамму
(протокол) исследования и утверждена Мини
стерством здравоохранения.
Популяция субъектов для исследова
ния биолоrической эквивалентности должна
быть как можно более однородна, чтобы
снизить вариабельность, не обусловленную
лекарственным средством. Должны быть
установлены ясные критерии ВКЛl'.)чения и ис
ключения испытуемых. ПО ВОЗМОЖI-IQСТИ ис
пытуемые должны быть разнополые, однако
риск для женщин следует рассматривать на
индивидуальной основе и, если это необхо
димо, они должны быть предупреждены о
любой опасности для плода, в случае если
они беременны.
Если в исследование включены умеренно
курящие субъекты (менее 10 сиrарет в день), их
следует соответствующим образом идентифи
цировать как таковых; в отчете по результатам
исследования необходимо обсудить возможные
последствия включения данных добровольцев в
отношении результатов исследования.
6.3.1. Число испытуемых
В исследование должны быть включены ис
пытуемые в количестве, достаточном для обе
спечения статистической значимости исследо
вания. Необходимое количество испытуемых
для надежноro исследования фармакокинетиче
ской биоэквивалентности определяется:
априорной дисперсией ошибок (коэффи
циентом вариации) основных показателей cpaB
нения (АUС и Cma)' связанной с анализируемыми
первичными параметрами, которая рассчитыва
ется на основании пилотною эксперимента, на
основании предыдущих исследований или опу
бпикованных данных;
желаемым уровнем значимости (5 %);
желаемой статистической мощностью (не
менее 80 %. если не обосновано иное);
средним отклонением rенерическоro
средства, сходноro по биоэквивалентности,
безопасности и эффективности, от эталонно
ro препарата;
потребностью расчета 90 % доверитель
ноro интервала для среднею rеометрическо
ro значения соотношения параметров биоэк
вивалентности, который должен находиться
в rраницах 80 125 % биоэквивалентности
для лоrарифмически трансформированных
данных.
Расчет числа субъектов, которых потребует
ся рекрутировать для испытания, следует прово
дить с учетом вышеуказанных нормативов. При
этом необходимо пользоваться COOTBeTCTBY
ющими статистическими методами расчета (см.
раздел 6.8). Количество набранных испытуемых
должно Bcerдa обосновываться вычислением
объема выборки, которое включается в протокол
и отчет по исследованию.
Минимальное рекомендуемое число вклю
ченных испытуемых не менее 18 человек.
Большее число испытуемых может потребовать
ся для сравнения препаратов, обладающих зна
чительной вариабельностью фармакокинетиче
ских параметров.
Предварительная оценка необходимоro
числа испытуемых может быть получена с помо
щью таблицы 6.3.1. 1 или HOMorpaMMbI приложе
ния 4.
Если при проведении статистическоro cpaB
нения уровень мощности теста оказался ниже
80 %, в тех случаях, Korдa сравниваемые пре
параты оказались небиоэквивалентными, для
принятия обоснованноro заключения о небиоэк
вивалентности необходимо включить в исследо
вание большее число испытуемых.
#Ре2истрационные требования и правила проведения исследований биодоступности 1103
Таблица 6.3.1 А
Число испытуемых, необходимое для обеспечения 80 % мощности
статистичеСК020 критерия в случае Л02 нормаЛЬН020 распределения
(2раницы доверитеЛЬН020 интервала 0,8 1,25; уровень значимости 5 %)
C\I" (%) J.!lJ.!R
0,85 0,90 0,95 1,00 1,05 1,10 1,15 1,20
5,0 12 6 4 4 4 6 8 22
7,5 22 8 6 6 6 8 12 44
10,0 36 12 8 6 8 10 20 >48
12,5 >48 16 10 8 10 14 30 >48
15,0 >48 22 12 10 12 20 42 >48
17,5 >48 30 16 14 16 26 >48 >48
20,0 >48 38 20 16 18 32 >48 >48
22,5 >48 46 24 20 24 40 >48 >48
25,0 >48 >48 28 24 28 48 >48 >48
27,5 >48 >48 34 28 34 >48 >48 >48
30,0 >48 >48 40 32 38 >48 >48 >48
'Примечание:
Cv коэффициент вариации, CV e(,,2) 1 , rAe
а 2 средний квадрат «ошибки» или остаточная внутри индивидуальная вариация, определяемая при дисперсионном анализе
после лоrарифмической трансформации значений показателя;
J.!T rенеральное среднее показателя сравнения для исследуемоro средства;
J.!R rенеральное среднее показателя сравнения для референтноro средства
6.3.2. Выбывание добровольцев из ис-
следования и их дублеры
Заявитель должен обеспечить отбор дo
статочноrо количества испытуемых субъектов с
учетом возможных их выбытий или исключений.
Поскольку замена испытуемых во время прове
дения исследования может осложнить статисти
ческую модель и анализ, то, как правило, не сле
дует заменять выбывших субъектов. Опричинах
исключения участника (как то: нежелательная
лекарственная реакция или личные мотивы)
следует сообщить. В этой связи целесообразно
в ходе подroтовки к исследованию осуществить
также подбор дублеров. Дублеры до начала ис
следования должны подписать информирован
ное соrласие и пройти обследование в том же
объеме, что и добровольцы. Число дублеров co
ставляет 4 6 человек. При проведении каждо
ro из периодов приема лекарственных средств
дублеры, наряду с добровольцами основноro
состава, осуществляют прием лекарственноro
средства и сдачу образцов биолоrической жид
кости, которые консервируются и включаются в
анализ только в случае преждевременноro BЫ
бытия из испытания добровольцев основноro
состава. В протоколе обязательно указывается,
анализируются ли образцы, взятые у дополни
тельных субъектов, если тоro не требуют усло
вия статистическоro анализа.
Заявителям, желающим заменять выбыв
ших во время проведения исслеДОВ;jНИЯ лиц или
рассматривающим данный дизайн, следует оro
ворить свое намерение в протоколе.
Если исследование биоэквивалентно
сти проводил ось на соответствующем коли
честве испытуемых, но биоэквивалентность
невозможно установить из за больших, чем
ожидал ось, случайных колебаний или OT
носительной разности, разрешается осуще
ствить исследование на дополнительных дo
бровольцах, используя в качестве BToporo
состава добровольцев не менее половины
от числа участников исходноro исследования
при условии, что эта ситуация проrнозирова
лась и предусматривалась в протоколе ис
следования. Суммирование данных допуска
ется лишь в случае применения одинаковоrо
протокола и лекарственных средств из той же
самой серии. Дизайны дополнительных испы
таний должны осуществляться строro в co
ответствии с планом исследования и с сQп.
При этом следует провести полаrающуюся
статистическую обработку результатов.
6.3.3. Отбор испытуемых (добровольцев)
Исследования фармакокинетической биоэк
вивалентности в основном следует проводить с
привлечением здоровых добровольцев. В прото
коле исследования необходимо устанавливать
четкие критерии включения иневключения.
Критерии включения добровольцев в иc
следование. В качестве здоровых добровольцев
MOryT привлекаться лица обоеro пола в возрасте
от 18 до 55 лет, если не обосновано иное, OTBe
чающие следующим критериям:
верифицированный диаrноз: «здоров» по
данным клинических, лабораторных и инстру
ментальных методов обследования;
масса тела не выходит за пределы 1:15 %
по Beco pOCТOBOMY индексу Кетле;
для женщин отрицательный тест на бе
ременность и соrласие придерживаться aдeK
ватных методов контрацепции; в случае ис
пользования roрмональных контрацептивов,
1104
rосударственная фармакопея Республики Беларусь
они ДОЛЖНЫ быть отменены не менее чем за 2
месяца до начала исследования.
Если лекарственное средство предназнача
ется для приема представителями двух полов,
заявитель может пожелать включить в выборку и
мужчин, и женщин. Опасность исследований для
женщин необходимо рассматривать в индивиду
альном порядке, и при необходимости их пред
упреждают о потенциальной уrрозе для разви
тия плода в случае наступления беременности.
Исследователи обязаны удостовериться, что
добровольцы женскоro пола не находятся в co
стоянии беременности или беременность не Ha
ступила во время проведения испытаний. Под
тверждение получается с помощью анализов
мочи, взятых непосредственно перед приемом
первой и последней доз испытуемоro препарата.
Критерии невключения добровольцев в иc
следование:
отяroщенный аллерroлоrический анамнез;
лекарственная непереносимость;
хронические заболевания сердечно сосу
дистой, бронхолеrочной, нейроэндокринной си
стемы а также заболевания желудочно кишечно
ro тракта, печени, почек, системы крови;
хирурrические вмешательства на желудоч
но кишечном тракте (за исключением аппендэк
томии);
острые инфекционные заболевания менее
чем за 4 недели до начала исследования;
реryлярный прием лекарственных препа
ратов менее чем за 2 недели до начала иссле
дования;
прием лекарственных препаратов, оказы
вающих выраженное влияние на rемодинамику,
функцию печени и др. (барбитураты, омепразол,
циметидин, эритромицин, рифампицин и т. д.),
менее чем за 30 дней до начала исследования;
донорство (450 мл крови или плазмы и
более) менее чем за 2 месяца до начала иссле
дования;
прием более чем 10 ед. алкоroля в неделю
(1 ед. алкоroля эквивалентна 0,5 л пива, 200 мл
сухоro вина или 50 мл спирта), а также aHaMHe
стические сведения об алкоrолизме, HapKOMa
нии, злоупотреблении лекарственными cpeд
ствами;
выкуривание более 1 О сиrарет в день.
Доброволец не может одновременно быть
участником двух исследований биоэквивалент
ности. После завершения исследования вклю
чение добровольца в следующее исследование
возможно через 3 недели, но не менее чем через
6t% препарата, в исследованиях котороro уча
ствовал доброволец.
Банк добровольцев фОРfv1ируется в соот
ветствии с критериями включения в исследова
ния и невключения в исследование, указанными
выше.
В беседе с врачом исследователем добро
волец должен получить следующую информа
цию:
цель исследования;
наличие разрешения на проведение ис
следования;
сведения о лекарственном средстве (фар
маколоrическая rруппа, механизм действия, по
казания к применению, возможные нежелатель
ные эффекты);
путь введения и доза лекарственноro
средства;
длительность исследования и условия
отбора проб крови;
время прибытия в исследовательский
центр;
условия, в которых будет находиться дo
броволец во время исследования, требования к
соблюдению пищевоro и водноro режима;
оrраничения в приеме лекарственных
средств во время исследования;
возможность оказания медицинской
помощи во время и после исследования;
условия страхования и вознаrраждения.
Если доброволец включается а банк данных,
на неro заводится индивидуальная карта, rдe
указываются:
ФИО, возраст, адрес, телефон, паспорт
ные данные;
медицинский анамнез (с указанием xpo
нических заболеваний, а также состояний, по
поводу которых доброволец находился на CTa
ционарном лечении) и аллерroлоrический aHaM
нез.
В индивидуальной карте реrистрируется
участие добровольца во всех клинических ис
следованиях лекарственных средств.
Не более чем за 1 неделю до начала испы
таний добровольцы, привлекаемые к исследо
ваниям конкретноro лекарственноro препара
та, приrлашаются в исследовательский центр.
Врач исследователь проводит с ними беседу, в
ходе которой повторно собирается медицинский
анамнез и проводится оценка соответствия дo
бровольцев критериям включения в исследова
ние.
Добровольцу rарантируют, что при необхо
димости ему будет оказана квалифицированная
медицинская помощь как во время, так и после
проведения исследования биоэквивалентности,
что информация о нем, полученная в ходе ис
следований, будет иметь конфиденциальный xa
рактер. После этоro доброволец для участия в
исследовании должен подписать «Информиро
ванное соrласие доброВоЛЬЦа», копия котороro
выдается добровольцу наряду с «Информацией
для добровольцев, участвующих в исследовани
ях биоэквивалентности лекарственноrо препа
рата».
После подписания информированноro co
rласия перед исследованием проводятся спе
циальные медицинские обследования добро
вольцев. Объем обследований определяется в
зависимости от фармаколоrических особенно
стей исследуемоro лекарственноro средства.
#Реzистрационные требования и правила проведения исследований биодоступности 1105
Рекомендуется проведение следующих лабора
торных и инструментальных тестов:
клинический анализ крови (эритроциты,
rемоrлобин, лейкоциты, лейкоцитарная форму
ла, тромбоциты);
биохимический анализ крови (общий
белок, креатинин, мочевина, билирубин, АЛТ,
АСТ, y rrт, щелочная фосфатаза, rлюкоза,
калий, натрий, хлориды);
клинический анализ мочи (белок, rлюкоза,
лейкоциты, эритроциты);
анализ крови на ВИЧ, вирусные rепатиты
В,С;
электрокардиоrрафическое исследова
ние.
Если результаты всех исследований в пре
делах нормы, доброволец включается в иссле
дование. Если результаты анализов выходят
за пределы нормы, исследования повторяют,
при этом возможны следующие варианты pe
шений:
если результаты повторноro анализа поло
жительные, доброволец включается в исследо
вание;
если результаты повторноro исследова
ния выходят за пределы нормы, но отклонение
от нормы не имеет клиническою значения, дo
броволец может быть включен в исследование;
если результаты повторною анализа BЫXO
дЯТ за пределы нормы и отклонение имеет кли
ническое значение, доброволец исключается из
исследования.
Не более чем за 7 дней до начала исследо
вания доброволец осматривается терапевтом,
который проводит:
оценку лабораторных данных и данных
электрокардиоrрафическоrо обследования;
сбор анамнеза (медицинскоrо, аллерrоло
rическою );
измерение артериальноro давления, ча
стоты пульса;
физикальный осмотр по системам.
Результаты осмотра вносятся в индивидуаль
ные реrистрационные карты добровольцев. По
результатам клиническоro осмотра и лаборатор
ноro тестирования врач исследователь делает
заключение, на основании котороro добровольцы
либо допускаются либо не допускаются к иссле
дованию. Врач исследователь составляет список
добровольцев и передает еro лицу, OTBeTCTBeHHO
му за проведение исследований. После включе
ния добровольцев в исследование производится
их рандомизация с использованием методики oд
номоментной простой рандомизации, результаты
которой реrистрируются в еro карте.
6.3.4. Динамическое наблюдение за до-
бровольцами во время исследования
Динамическое наблюдение за добровольца
ми в период отбора образцов крови осуществля
ется врачом исследователем и включает как ми
нимум:
клинический осмотр каждые з.........а ч (в за
висимости от фармаколоrических особенностей
препарата);
измерение уровня артериальноrо давле
ния и частоты сердечных сокращений.
Результаты обследования заносятся в инди
видуальные карты добровольцев.
По окончании первою периода исследо
вания после удаления катетера проводится за
ключительный врачебный осмотр доброволь
цев. При отсутствии отклонений в состоянии
здоровья добровольцев их отпускают домой до
начала второro периода исследования.
Перед вторым периодом исследования про
водится повторное обследование доброволь
цев, включающее врачебный осмотр, и, при
необходимости, клинико инструментальные ис
следования (экr и друrие), а также лаборатор
ные тесты (клинический анализ крови, клиниче
ский анализ мочи, биохимический анализ крови,
тест на беременность для женщин).
На основании результатов повторноro об
следования врач исследователь допускает или
не допускает добровольцев ко второму периоду
исследования.
После завершения второro периода иссле
дования биоэквивалентности проводится заклю
чительный врачебный осмотр. При отсутствии
отклонений в состоянии здоровья добровольцев
их отпускают домой.
Для обеспечения безопасности исследо
вания биоэквивалентности врачом исследова
телем про водится мониторинr нежелательных
явлений. Случаи возникновения нежелатель
ных явлений реrистрируются в индивидуальной
карте добровольца и соответствующей форме.
Необходимо сообщать о частоте возникнове
ния, тяжести и продолжительности любой неже
лательной реакции и побочных эффектов, обна
руженных во время испытания. Исследователем
оценивается вероятность тою, что нежелатель
ная реакция вызвана приемом лекарства.
6.3.5. Проведение rенотипирования или
фенотипирования добровольцев
Определение активности метаболизиру
ющих ферментов методом фенотипирования
или rенотипирования может иметь существен
ное значение для испытаний с лекарственными
средствами, обладающими высоким клиренеом,
которые метаболизируются энзимами, подвер
женными rенетическому полиморфизму, таких
как пропанолол. В таких случаях медленные Me
таболизаторы будут показывать большую биодо
ступность, чем истинная для данною АФИ, при
этом биодоступность возможных активных Me
таболитов будет ниже. Фенотипирование или re
нотипирование субъектов можно рассматривать
как необходимую меру в отношении испытаний
лекарственных средств, которые проявляют свя
занный с фенотипом метаболизм и для которых
будет при меняться параллельная модель испы
1106
rосударственная фармакопея Республики Беларусь
тания, так как это позволяет быстрым и медлен
ным метаболизаторам распределяться paBHO
мерно в пределах двух rрупп испытуемых.
Фенотипирование может оказаться также
полезным по соображениям безопасности, для
определения моментов отбора проб и периодов
вымывания при исследованиях с перекрестной
моделью.
6.3.7. Причины исключения доброволь-
цев
Для объективной оценки результатов paHДO
мизированноro исследования требуется, чтобы
все субъекты наблюдались и принимали иссле
дуемые средства в соответствии с одинаковы
ми правилами. Данные правила должны быть
независимыми от принимаемоro лекарственно
ro средства или исхода этоrо приема. Впослед
ствии перед биоанализом должно быть принято
решение об исключении субъектов из статисти
ческоro анализа. если данное требование было
нарушено.
Для исключения подходит основание при
условии, что оно предварительно указано в про
токоле и что решение об исключении принято
перед биоанализом. Тем не менее, необходи
мо избеrать исключения данных, так как эффект
исследования будет снижен, а также потому что
требуется минимум 18 субъектов для проведе
ния полноценноrо статистическоro анализа.
При меры оснований исключения результа
тов, полученных от субъекта в определенный
период: рвота и диарея, которые MOryT приве
сти к недостоверности временной кривой KOH
центрации плазмы. В исключительных случаях
причиной для исключения субъекта может стать
прием недопустимых сопутствующих лекар
ственных средств.
Разрешенные основания для исключения
должны быть oroBopeHbl в протоколе. Если слу
чается одно из этих событий, это должно быть
зафиксировано в индивидуальной реrистраци
онной карте добровольца в процессе проведе
ния исследования. Исключение субъектов на
основании этих предопределенных критериев
должно быть ясно описано и внесено в отчет об
исследовании.
Не допускается исключение данных только
на основании статистическоrо анализа или по
фармакокинетическим причинам. так как невоз
можно отличить эффект состава лекарственно
ro средства от остальных эффектов, воздейству
ющих на ero фармакокинетику. Исключениями из
этоro являются:
1) субъект с недостаточным уровнем
любой измеряемой концентрации по всем
временным точкам или очень низкой KOHцeH
трацией в плазме для ориrинальноrо лекар
ственноro средства; считается, что у субъекта
низкая концентрация плазмы, если еro АUС
составляет менее 5 % от средней rеометри
ческой АUС ориrинальноrо лекарственноrо
средства (которое должно рассчитываться
без включения данных, полученных от этоro
удаляемоro субъекта); исключение данных
по этим причинам допустимо только в исклю
чительных случаях и может поставить под
вопрос достоверность данноro испытания;
2) субъекты с ненулевой базовой KOHцeH
трацией >5 % С тах ' Такие данные должны ис
ключаться из расчета биоэквивалентности
(<<эффект пере носа» ).
Для лекарственных средств с HeMeд
ленным высвобождением вышеперечислен
ное может быть результатом невыполнения
субъектом условий приема и недостаточно
ro периода отмены препарата COOTBeTCTBeH
но. По мере возможности этоro нужно избе
raTb, проверяя полость рта субъектов после
приема исследуемоro лекарственноro cpeд
ства, чтобы убедиться, что субъекты проrло
тили исследуемое лекарственное средство, и
моделируя исследования с достаточным пе
риодом отмены препарата. Образцы, полу
ченные от субъектов, исключенных из стати
стическоro анализа, тем не менее, должны
быть количественно оценены, а результаты
внесены в таблицу концентраций.
Как указано в разделе 6.6.2, AUC O _ t
должна составлять не менее 80 % АUСо_ .
Субъекты не должны исключаться из стати
стическоro анализа, если AUC O _ t составляет
менее 80 % АUСо_ , но если процентное co
отношение таких профилей составляет более
чем 20 % наблюдений, необходимо оценить
(проверить) достоверность исследования.
Это не касается тех случаев, коrда период
отбора образцов составляет 72 часа и более
и вместо АUС о _ ! используется АUС о _ 72 .
6.4. СТАНДАРТИЗАЦИЯ ИССЛЕДОВАНИЯ
Исследование должно быть спланировано
так, чтобы условия испытаний способствова
ли снижению внутри и межсубъектной вари
абильности и позволили избежать искажения
результатов. Стандартизация физических Ha
rрузок, питания, потребления жидкостей и по
ложения тел, оrраничение в употреблении ал
коrоля, кофеина, некоторых фруктовых соков
и лекарственных средств, ненужных для ис
следования, в период до исследования и в
еro процессе является важным для сведения
к минимуму вариабильности всех факторов,
исключая факторы, связанные с тестируемым
лекарственным средством.
Добровольцам следует воздерживаться на
протяжении определенноrо протоколом и кри
териями включенияiнееключения периода до
начала испытания или в еro ходе от приема Ka
ких либо лекарственных средств, алкоroльных
напитков или отпускаемых без рецепта лекар
ственных средств (ОТС препаратов). В случае
чрезвычайных обстоятельств необходимо сооб
щить врачу исследователю об имевшем место
#Ре2истрационные требования и правила проведения исследований биодоступности 1107
приеме какоro либо постороннеro лекарствен
ною средства (дозировка и время введения).
Если цель исследования биолоrической эк
вивалентности ставит особые вопросы (напри
мер, биолоrическая эквивалентность у специ
фических rрупп населения), то критерии отбора
должны быть соответствующим образом CKOp
ректированы.
Требуется, насколько это возможно, при
вести к стандартной форме физическую актив
ность и положение тела, чтобы оrраничить их
влияние на кровообращение и моторику >ККТ.
Каждый день в ходе испытания следует при
держиваться примерно одинаковоro положения
тела в период отдыха и характера двиrательной
и физической активности. В протоколе необхо
димо оroворить время суток для приема испыту
емоro препарата.
Как правило, лекарственные средства BЫ
даются и применяются после ночноrо воздер
жания от пищи, длившеюся не менее 1 О часов,
при этом участники получают свободный доступ
к воде в неоrраниченных количествах. Утром
накануне испытания в течение часа до начала
приема препарата запрещается потреблять
воду. Порцию лекарства следует запивать CTaH
дартным объемом воды (обычно 150 250 мл).
Через два часа после приема лекарственно
ro средства снова разрешается пить воду в He
оrраниченных количествах. Нормальная порция
пищи (первый завтрак), как правило, предостав
ляется добровольцу через четыре часа после
введения препарата.
Все порции пищи следует нормировать, а в
протоколе и отчете испытания указывается их
состав.
Определенные лекарственные средства
принимаются вместе с пищей для снижения по
бочноro действия на >ККТ. В некоторых случа
ях совмещение с приемом пищи увеличива
ет биоэквивалентность вводимых перорально
препаратов. Если в инструкции по медицинско
му применению ориrинальною лекарственною
средства, которое используется в виде компара
тора, указано, что еro необходимо принимать с
пищей, для оценки биоэквивалентности исполь
зуется исследование в прандиальном состоя
нии (после стандартноro завтрака). Данный тип
испытаний также требуется для определения
биоэквивалентности лекарственных средств с
модифицированным высвобождением. В этих
случаях стоит задача выбрать питание, KOТO
рое проверит устойчивость rенерическоro ле
карственноro средства к влиянию пищи на по
казатели биоэквивалентности (см. раздел 6.2.4).
Питание в периnд исследования выбирается в
пределаходноrоиздиетическихстоловиупотреб
ляется не позднее чем через 20 минут после
приroтовления. При выполнении исследований
в прандиальном состоянии лекарственное cpeд
ство следует принимать в соответствии с прото
колом и не позднее 30 минут после еды.
6.5. ИССЛЕДУЕМblЕ ЛЕКАРСТВЕННblЕ
СРЕДСТВА
6.5.1. Испытуемое rенерическое лекар
ственное средство
Образцы rенерическоro лекарственноro
средства, используемые в исследовании био
лоrической эквивалентности для реrистраци
онных целей, должны быть идентичны разра
батываемому коммерческому лекарственному
средству. По этой причине не только состав и Ka
чественные характеристики (включая стабиль
ность), а также и методы производства должны
быть идентичны тем, которые будут применять
ся при их дальнейшем серийном производстве.
Испытуемые препараты изroтавливаются С при
менением стандартов GMP. Должны быть указа
ны результаты контроля серии rенерическоro ле
KapcTBeHHoro средства, а также номер партии и
срок хранения и для rенерическоro средства, и
для компаратора.
Образцы должны быть взяты из промыш
ленных партий. Korдa это невозможно, допуска
ется использовать партии из опытноro или опыт
но промышленноro производства при условии,
что они будут составлять не менее 1/10 (10 %)
объема максимальной ожидаемой партии ce
рийноro производства или не менее 100 тысяч
единиц (в зависимости от тою, какой из этих двух
показателей выше), если не обосновано ДOKY
ментально иное. Данные серии изroтавливают
ся с помощью одинаковоro оснащения, оборудо
вания и процессов, запланированных для серий,
произведенных в коммерческих целях. Если про
дукция будет в дальнейшем масштабироваться,
то это следует соrласовать должным образом в
проrрамме испытания и в разделе «Обоснова
ние фармацевтической разработки препарата»
реrистрационноrо досье.
Рекомендуется до проведения испытания
на биоэквивалентность установить количествен
ное содержание и характеристики растворения
in vitro для rенерическоrо лекарственноro cpeд
ства и компаратора. Содержание АФИ в двух ис
следуемых препаратах не должно отличаться
более чем на 5 %. Серия, в которой содержание
АФИ отличается более чем на 5 % от эталонно
ro средства с заявленным содержанием 100 %,
должна быть исключена из испытания.
В случае, если про водится оценка биоэк
вивалентности/биодоступности для средств
с фиксированной комбинацией компонен
тов, rенерическое комбинированное средство
должно сравниваться с фармацевтически экви
валентным инновационным комбинированным
средством. Только в случае еro отсутствия на
фармацевтическом рынке MorYT быть исполь
зованы в качестве средств сравнения MOHO
препараты, назначаемые одновременно в KOM
бинации. Биоаналитические методы в этом
случае должны быть подверrнуты валидации в
отношении определения всех измеряемых co
единений.
1108
rосударственная фармакопея Республики Беларусь
6.5.2. Выбор эталонноrо лекарственноrо
средства
Ориrинальное лекарственное средство
обычно является наиболее приемлемым эта
лонным препаратом для rенерических препа
ратов, поскольку в целом ero качество хорошо
проверено, а еro эффективность и безопас
ность надежно подтверждены в клинических
испытаниях и в проrраммах дo и послереrи
страционноro надзора. rенерическое лекар
ственное средство нельзя использовать в Ka
честве компаратора до тех пор, пока можно
приобрести ориrинальный препара так как
это может привести к непрерывно уменьша
ющейся достоверности подобия будущеro re
нерическоrо лекарственноrо средства и воз
можно К отсутствию взаимозаменяемости с
ориrиналом.
Для некоторых лекарственных средств
невозможно определить ориrинальный пре
парат, а в отдельных случаях на рынке нет в
наличии TaKoro препарата. Заявитель должен
обосновать в данном случае выбор компара
тора. Следует указать страну происхождения
препарата сравнения, а также номер партии
и срок хранения. Выбором препарата cpaBHe
ния проверяется yn _Центр экспертиз и испы
таний в здравоохранении» и одобряется Ми
нистерством воохранения Республики
Беларусь.
Принцип выбора обусловлен следующи
ми критериями (neречисленными в порядке их
предпочтительности):
(i) выбор в качестве эталонноro иннова
ционноro лекарственноro средства, качество,
безопасность и эффективность котороro были
установлены. если оно прошло roсударствен
ную реrистрацию в Республике Беларусь (<<YT
вержденный на национальном уровне ориrи
нал» );
(ii) выбор в качестве эталонноro препа
рата сравнения из перечня ВОЗ (который
получил реrистрационное удостоверение
блаrодаря своему качеству, безопасности
и эффективности «препарат сравнения
ВОЗ»); .основной производственный участок
указывается в списке компараторов ВОЗб,
при этом компаратор необходимо закупать в
данной стране;
(iii) выбор в качестве эталонноro лекар
cTBeHHoro средства, получившеro блаroда
ря своему качеству, безопасности и эффек
тивности реrистрационное свидетельство в
roсударстве с хорошо реrулируемым фар
мацевтическим рынком (roсударство член
Международной конференции по rармониза
цИИ (/СН) или ассоциированные roсударства)
(<<ориrинал /СН»), и ero следует закупать на
данном рынке;
(iv) в случае невозможности установить
ориrинальный препарат с учетом вышеука
занных пунктов (i) (iii) заявитель обязан тща
тельно выбрать компаратор и документально
обосновать свой выбор. Наиболее важными
критериями TaKoro отбора в порядке предпо
чтения являются:
наличие реrистрации компаратора в ro
сударствах /СН и ассоциированных roсудар
ствах;
прохождение лекарственным средством
«преквалификации» ВОЗ;
наличие сообщений в рецензируемых Ha
учных журналах о документально подтверж
денном широком применении данноrо лекар
cTBeHHoro средства в клинических испытаниях;
длительный период контроля результа
тов клиническоrо использования без серьез
ных нежелательных реакций и сообщений о Te
рапевтической неэффективности (<<правильно
выбранный компаратор»7).
Дополнительно «правильно выбранный
компаратор» должен соответствовать CTaH
дартам качества rосударственной Фармако
пеи.
Для выбора референтноro средства можно
также руководствоваться рекомендуемым ВОЗ
алroритмом подбора компаратора 6 .
Если невозможно выбрать референтное
средство аналоrичной лекарственной формы
и дозировки из тех, что имеются на рынке, то
в качестве средства сравнения допустимо ис
пользовать фармацевтически взаимозаменяе
мое средство (иной лекарственной формы и
дозировки). в этом случае выполняют иссле
дование сравнительной биодоступности, в
рамках котороro рассчитывают, по возможно
сти, параметры ожидаемой максимальной и
минимальной стационарной концентрации и
режимов равноэквивалентноro дозирования.
6.5.3. Запасные образцы
Достаточное количество (необходимое для
проведения полноro анализа) образцов каждой
серии лекарственных средств, используемых
для исследования, вместе с результатами их
анализов и характеристик должны храниться у
спонсора для справочных и арбитражных целей
с соблюдением соответствующих условий xpa
нения не менее одноrо rода после окончания
испытаний. По специальному требованию Ми
б Guidaпce оп the se/ectioп о( coтparator pharтaceutica/
products (от equiva/eпce assessтeпt о( iпterchaпgeabIe
тu/tisource (geпeric) products: WHO Expert Coттittee оп
Speci{icatioпs {о, Pharтaceutica/ Preparatioпs. Thirty sixth
report // Wor/d Hea/th Orgaпizatioп. Geпeva, 2002.
7 rенерическое лекарственное средство, прошедшее после
испытаний процедуру roсударственной реrистрации исходя
из сопоставления с зарубежным препаратом сравнения,
может быть или может не быть взаимозаменяемым с отече
ственными лекарственными средствами, маркетируемыми
в настоящее время. С целью расширения доступа к лекар
ственным средствам эффективным может оказаться опреде
ление локальноro препарата сравнения, качество, безопас
ность и эффективность KOТOpOro были установлены.
в /bid.
#Реаистрационные требования и правила проведения исследований биодоступности 1109
нистерства здравоохранения эти запасные об
разцы MOryT выдаваться им для повторной про
верки этих лекарственных средств.
6.6. ПРОВЕДЕНИЕ ИССЛЕДОВАНИЯ
6.6.1. Подбор ДОЗЫ
В исследованиях на биоэквивалентность
следует использовать эквивалентные молярные
дозы rенерическоro лекарственноro средства и
компаратора.
Должна применяться дозировка лекар
ственноro средства, которая обеспечивает наи
большую чувствительность оценки биоэкви
валентности. Как правило, это наибольшая из
зареrистрированных дозировок компаратора
в дозе, равной единице лекарственной формы
(одна капсула, таблетка, драже, порция). Более
высокая доза (т. е. больше одной единицы ле
карственной формы) может применяться, если
существуют проблемы с аналитической точки
зрения. В данном случае совокупная OДHOKpaT
ная доза не должна превышать максимальной cy
точной дозы для терапевтической схемы приема
лекарственноro средства. В определенных слу
чаях проблему отсутствия чувствительности
анализа можно исключить, если использовать
усеченную площадь под фармакокинетической
кривой (АUС) препарата сравнения, оrраничен
ной промежутком до 3 x средних t max '
В некоторых ситуациях исследование с
более низкой дозировкой может считаться при
емлемым, если такая дозировка выбирается по
соображениям безопасности.
6.6.2. Время отбора проб биоматериала
Схема отбора проб определяется формой
кривой «концентрация действующеro вещества
время». Образцы крови необходимо брать с ча
стотой, достаточной для корректной оценки С ,
АUС и друrих параметров. Временные тo.:;'
отбора проб должны включать 1 пробу до приме
нения лекарственноro средства, не менее 1 2
образцов до достижения С тах ' 2 образца в обла
сти С таХ И 3 в фазе выведения. СоответствеН7
но для оценки обязательных фармакокинетиче
ских показателей понадобится не менее 7 точек
отбора проб. Для большинства лекарственных
средств необходимое количество образцов будет
выше, чтобы компенсировать различия в CKOpO
сти всасывания и выведения в популяции субъек
тов и чтобы дать возможность точно определить
максимальный уровень концентрации в крови
АФИ (Ста.> и коэффициент скорости элиминации
(k e \) у всей популяции испытуемых.
Общая продолжительность наблюдения
за ко центрацией действующеro вещества при
однократном приеме лекарственноrо средства
должна быть не менее чем в 4 раза больше пери
ода полувыведения, а при мноroкратном приеме
препарата равной интервалу дозирования.
Однако при однократном приеме длительность
наблюдения считается удовлетворительной,
если для усредненноro фармакокинетическоrо
профиля величина площади ПОД кривой «KOH
центрация время» в пределах от нуля дО MO
мента отбора последней пробы составляет не
менее 80 % от площади под кривой, экстраполи
рованной в бесконечность (AUCo J. Для лекар
ственных средств с периодом полуэлиминации
более 24 ч нет необходимости в отборе образ
цов более чем в течение 72 ч. Точная продолжи
тельность отбора проб зависит от природы АФИ
и цели применения лекарственной формы (см.
раздел 6.11.4).
Интервал времени между приема ми препа
ратов зависит от длительности циркуляции ле
карственноro средства в орrанизме, определя
емой периодом полуэлиминации (ty), и должен
составлять не менее 5t. .
6.6.3. Анализируемые биоматериалы и
процедуры их отбора
При обычных обстоятельствах биолоrиче
ской жидкостью, отбираемой для измерения KOH
центрации АФИ, должна быть кровь. В большин
стве случаев измеряется содержание АФИ или
ero метаболитов в сыворотке или плазме. Можно
делать отбор мочи, если АФИ выделяется из op
rанизма преимущественно в неизменном COCTO
янии с мочой. Объем каждой пробы следует из
учать по возможности незамедлительно после
сбора и вносить результаты в отчет. Должно
быть достаточное количество образцов, чтобы
провести расчет фармакокинетических параме
тров. Тем не менее. в большинстве случаев сле
дует избеrать использования только данных о
выделении АФИ с мочой, так как это не позво
ляет рассчитать t max и максимальную KOHцeHTpa
цию вещества в системной циркуляции.
Образцы крови необходимо обрабатывать
и хранить в условиях, при которых ранее не об
наруживалось разложение определяемых KOM
понентов (в большинстве случаев, приемлемым
является хранение при температуре не выше
20 ОС). Это можно подтвердить, контролируя в
течение анализируемоro периода сдвоенные об
разцы контроля качества. Образцы контроля Ka
чества следует приroтовить из представляющей
аналитический интерес биожидкости (например,
плазмы), включая концентрации как минимум в
минимальной, средней и максимальной частях
диапазона значений. используемых при вали
дации аналитической методики. Их необходимо
хранить вместе с испытуемыми пробами и aHa
лизировать с каждым комплектом испытуемых
проб для каждоro аналитическоro этапа.
Методолоrия сбора образцов оroваривает
ся в ПРОТQколе исследования. При отборе проб
крови должны соблюдаться следующие условия:
кровь, как правило, отбирается из локте
вой вены через кубитальный катетер;
первая порция крови (исходная, т. е. до
приема препарата) отбирается утром, натощак,
через 5 1 О минут после установки катетера;
1110
rосударственная фармакопея Республики Беларусь
время отбора последующих проб соответ
ствует проrрамме исследования;
пробирки для отбора проб должны иметь
маркировку с указанием шифра испытуемоro,
номера пробы и названия препарата;
при возникновении экстремальной ситу
ации (ухудшение самочувствия. психические Ha
рушения, желание испытуемоro выйти из иссле
дования) отбор проб прекращается;
при возникновении непредвиденных ситу
аций, исключающих возможность отбора крови в
установленном временном интервале, работа с
данным испытуемым продолжается, но шифро
ванная пробирка остается пустой.
Пробы крови с сопроводительным направ
лением, в котором указываются ФИО испыту
емоro, пол, возраст. масса тела, рост, COOTBeT
ствующие шифру на пробирке, предоставляются
в фармакокинетическую лабораторию.
6.6.4. Анализируемые параметры
На основании первичных результатов pac
считывают необходимые параметры биодо
ступности, а именно: АИС" АИС ., С тах , (тах' Ае"
Ае, (в зависимости от ситуации) или любые
друrие подтверждаемые параметры (см. при
ложение 1).
В исследованиях биоэквивалентности
форма и площадь под кривыми «KOHцeHTpa
ция время,. чаще всеro используются для
оценки скорости (C a.' t ma .> и степени (АИС)
всасывания. Моменты или периоды отбора
проб должны выбираться так, чтобы ДOCTOBep
но устанавливался профиль «концентрация
время», позволяя рассчитать релевантные по
казатели.
Для ОДНОДОЭО8ых испытаний требуется из
мерить или рассчитать как минимум следующие
параметры.
Площадь под кривой «концентрация В
плазме/сыворотке/крови время» с нуле
вой точки до времени t (AUC t ), rдe ( является
моментом последнеro отбора проб с измери
мым содержанием АФИ конкретноro анали
зируемоro лекарственноro средства. Следу
ет оroворить метод расчета значений АИС,.
Индивидуальные значения площади под кри
выми «концентрация время» АИС (как
в пределах длительности наблюдения за
концентрацией лекарственноrо средства
АИС" так в пределах от О до 00 AUCJ сле
дует оценить внемодельными методами по
данным «концентрация время». YCTaHOB
ленным у каждоro испытуемоro для каждоro
из изучаемых препаратов. Как правило. АИС
рассчитывается с помощью интеrрирования
линейно лоrарифмическим методом трапе
ций. Не рекомендуется использовать только
показатели, основанные на компартментном
подходе.
С тах означает максимальную или наблю
даемую пиковую концентрацию в плазме, CЫBO
ротке или цельной крови в стационарном COCTO
янии, отражая воздействие АФИ (или метаболи
та) в стационарном состоянии. Значения пара
метра С тах оценивают как наибольшее из изме
ренных значений концентрации.
Наиболее релевантными показателями для
оценки биоэквивалентности считаются АИС, и
С тах . В качестве дополнения рекомендуется oцe
нить следующие параметры.
Площадь под кривой «концентрация В
плазме'сыворотке'крови время» с нулево
ro времени до бесконечности (AUCJ, отража
ющую общее действие. Значения АИС опре
деля ют по формуле: AUC =AUC,+C/ket' rдe С,
и k eJ расчетные значения концентрации ле
KapcTBeHHoro средства в последней пробе и
константы элиминации соответственно. Для
> вычисления С, и k el конечный (моноэкспоненци
альный) участок фармакокинетической кривой,
как правило, описывают с помощью нелиней
ноro реrрессионноro анализа;
(тах время после приема лекарства.
при котором наблюдается С тах .
В качестве дополнительной информации
можно определить параметры выведения веще
ства:
(у, время полуэлиминации из плазмы
(сыворотки, цельной крови);
MRT среднее время удержания ле
карственноro средства в центральном компар
тменте.
Последующий анализ фармакокинетиче
ских данных предусматривает вычисление ин
дивидуальных отношений АИС, или АИС", (co
ответственно " и f оценки относительной
степени всасывания) и С тех (f'j для любых ле
карственных форм. а для форм пролонrирован
ноro действия продолжительность периода
времени, в течение котороro концентрация ле
карственноro средства превышает 75 % от С тах
(Т>75 % Ста.).
В стационарных условиях (ss), реализу
ющихся при повторяющемся введении ле
карственных препаратов в одинаковой дозе с
одним и тем же интервалом дозирования (т),
индивидуальные фармакокинетические профи
ли следует охарактеризовать такими показате
лями, как:
AUC t . ss значения площади под кривой
«концентрация время» в пределах интервала
дозирования после установления стационарно
ro распределения препарата;
С тах '
С mlп значения минимальной KOHцeHTpa
Цl--'и (концентрация в конце интервала дозирова
ния);
значения флуктуации (разности между зна
чениями С тах И Стоп)' выраженные в процентах от
Ста>' а также флуктуации, отнесенной к средней
стационарной концентрации (C ss = АИС.)т).
Также вычисляют индивидуальные значе
ния отношений АИС и С для исследуемо
.. .S тах
#Реzистрационные требования и правила проведения исследований биодоступности 1111
ro препарата и препарата сравнения (COOTBeT
ственно (' и Р).
ДЛЯ форм пролонrированною действия
рассчитываются продолжительность пери
ода времени, в течение которою KOHцeHTpa
ция лекарственноro средства превышает cpeд
нее стационарное значение C ss (T>C ss )' а также
Т>75 % С
тах
При использовании образцов мочи вместо
АUС и С таХ берут показатели суммарною BЫBe
дения с мочой (Ае) и максимальной скорости BЫ
ведения с мочой.
6.6.5. Исследование метаболитов
Использование данных о продуктах обмена
в исследовании биолоrической эквивалентно
сти требует осторожноro подхода. Оценка био
лоrической эквивалентности основывается в
основном на измерении концентраций фарма
колоrически активною вещества и еro активных
метаболитов, если таковые имеются. Как пра
вило, оценка фармакокинетической биоэквива
лентности должна основываться на измеренных
концентрациях исходноro АФИ, выделившеroся
из лекарственной формы, а не еro метаболита.
Профиль «концентрация АФИ время» являет
ся более чувствительным к изменениям в дей
ствии препарата, чем метаболит, который лучше
отражает процесс образования, распределения
и выведения метаболита. Важно оroворить зара
нее в протоколе исследования, какие молекулы
будут количественно определяться в образцах
(пролекарство, лекарство АФИ или метаболит).
Измерение концентрации метаболита ис
ходноro АФИ может потребоваться в следующих
случаях:
если исследуемое лекарственное cpeд
ство содержит исходный фармацевтический ин
rредиент в виде пролекарства;
если метаболит вносит весомый вклад (не
менее 50 %) в профиль безопасности или клини
ческой эффективности данноro лекарственною
средства;
если невозможно измерить концентрацию
активною лекарственноro вещества ввиду слиш
ком низких еro концентраций в крови, плазме
или сыворотке в течение требуемоro времени с
точки зрения возможности достоверною анали
тическоro определения или если исходный фар
мацевтический инrредиент является нестабиль
ным в биолоrических жидкостях.
Если производится измерение в моче, то
определяемый метаболит должен предостав
лять собой основную фракцию дозы. Если из
мерения концентрации активных метаболитов
обычно считается допустимым, то определение
концентрации неактивноro метаболита лишь в
редких случаях можно считать обоснованным.
Следует заметить, что измерение един
ственноro анализируемоro вещества АФИ
или метаболита с вероятностью 5 % несет
риск совершения ошибки первоro рода (риск
потребителя). Однако выбор двух и более раз
пичных анализируемых субстанций в качестве
детерминанты биоэквивалентности ретроспек
тивно изменет риски и потребителя, и произ
водителя 9 .
При оценке активных метаболитов для aдeK
ватной характеристики фармакокинетическоrо
профиля может понадобиться корректировка пе
риода вымывания и моментов отбора проб.
6.6.6. Исследование индивидуальных
энантиомеров
В настоящее время для большинства ис
следований фармакокинетической биоэквива
лентности допускается осуществление HeCTe
реоселективною анализа. Если энантиомеры
обладают весьма отличными фармаколоrи
ческими или метаболическими профилями,
может быть обосновано испытание с опреде
лением различных энантиомеров хиральноrо
АФИ. Стереоселективное определение счита
ется также более предпочтительным в случае,
Korдa системная доступность разных энанти
омеров характеризуется нелинейными свой
ствами.
6.6.7. Исследование в прандиальном со-
стоянии при изучении биоэквивалентности
6.6.7. 1. Лекарственные средства с HeMeд
ленным высвобождением.
Более предпочтительно в целом исследо
вание натощак. Если известно, что препарат
вызывает у субъектов желудочно кишечные
расстройства или нежелательные явления при
приеме на пустой желудок, либо в инструкции
по применению ориrинальноro лекарственноro
средства накладывается оrраничение на необ
ходимость ero использования после еды, пред
почтительным подходом становится испытание
на фармакокинетическую биоэквивалентность
у добровольцев в прандиальном состоянии
(после еды). Состав принимаемой пищи должен
быть традиционным для восточноевропейскою
реrиона (см. раздел 6.4).
6.6.7.2. Лекарственные средства с Moди
фицироваННblМ вblсвобождением
Исследования воздействия пищи необходи
мо проводить в отношении всех rенерических
лекарственных средств с модифицированным.
высвобождением, чтобы rарантировать OTCYT
ствие «демпинrа доз». Последнее свидетель
ствует о неудачной разработке лекарственноrо
средства, поскольку вся доза высвобождается
сразу, а не через длительный промежуток BpeMe
ни. Это приводит К преждевременному и резко
му скачку в профиле «концентрация В плазме
время». Как правило, пища с высоким coдep
жанием жира наиболее критична для обеспече
, Midha. К-К- Coттentary: The Ro/e о{ Metabo/ites in
Bioequivalence / К-К- Midha, MJ. Rawson. J. W Hubbard //
Pharтaceutica/ Research. , 2004. Vo/. 21. р. 1ЗЗ1 1З44.
1112
rосударственная фармакопея Республики Беларусь
ния устойчивости процесса высвобождения АФИ
из лекарственноro средства. При составлении
меню следует также учитывать традиционные
требования для восточноевропейскоrо реrиона
(см раздел 6.2.4).
6.6.8. 3HAoreHHbIe вещества
Если вещество исследуется как эндоrен
ное, расчёт фармакокинетических параметров
должен осуществляться с использованием ба
зовых коррекций так, чтобы рассчитанные
фармакокинетические параметры относились
к дополнительным концентрациям, предпола
raeMbIM вследствие приема лекарственноro
средства. Можно рассматривать назначение
сверхтерапевтических доз при исследовании
биоэквивалентности эндоrенных лекарствен
ных средств при условии, что доза хорошо
переносится, чтобы дополнительная KOHцeH
трация, выше основной, предполаrаемой Te
рапевтическим приемом лекарственною cpeд
ства, моrла быть надёжно определена. Если
ранее не была установлена продолжитель
ность периода «отмывки» после назначения
различных доз определенноro эндоrенноro Be
щества, это необходимо доказать либо в пред
варительном исследовании, либо в течение oc
HOBHoro исследования биоэквивалентности с
использованием различных доз исходных ле
карственных средств с целью убедиться, что
доза, используемая для сравнения биоэквива
лентности. чувствительна к определению по
тенциальных различий между составами ле
карственных средств.
Точный метод для коррекции фоновоro
уровня эндоrенной субстанции должен быть
определен и обоснован в протоколе исследова
ния. В общем случае, предпочтителен CTaHдapT
ный субтрактивный метод коррекции фоновоro
уровня, означающий либо вычитание из каждой
временной точки значения индивидуальных эн
доrенных концентраций. которые наблюдались
перед приемом очередной дозы, либо вычи
тание индивидуальной эндоrенной АUС, KOТO
рая была построена накануне приема дозы. В
редких случаях, Korдa наблюдается существен
ное превышение концентрации эндоrенных фо
новых уровней, коррекция фона может не пона
добиться.
При исследовании биоэквивалентности эн
доrенных веществ ее невозможно оценить Ha
прямую, если произошел перенос, поэтому He
обходимо уделять особое внимание тому, чтобы
период отмены препарата длился необходимое
количество времени.
6.7. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
АКТИВНО/О ФАРМАЦЕВТИЧЕСКО/О
ИН/РЕДИЕНТА (ТРЕБОВАНИЯ
К АНАЛИТИЧЕСКОМУ МЕТОДУ)
Все аналитические контрольно испыта
тельные методы, применяемые для количе
cTBeHHoro определения активноrо компонента
и'или продукта еro биотрансформации в био
лоrической жидкости, должны быть хорошо
изученными, полностью валидированными и
документированными. К задачам валидации OT
носится предоставление доказательств тою,
что конкретный метод и аналитическая методи
ка, применяемые для количественноro измере
ния анализируемых веществ в данной биоло
rической среде (кровь, плазма, сыворотка или
моча), являются надежными и воспроизводи
мыми при целевом использовании. Биоанали
тическую часть испытаний биоэквивалентности
следует проводить в соответствии с принципа
ми надлежащей лабораторной практики (GLP) и
требованиями к испытательным лабораториям
(СТБ ИСО 17025).
Для определения концентрации действу
ющих веществ в плазме, сыворотке или цель
ной крови, а также моче MOryT быть использо
ваны различные методы (физико химические,
иммунолоrические, микробиолоrические и др.),
обеспечивающие возможность получения дo
стоверных лабораторных данных о KOHцeHTpa
ции действующеro вещества при выбранных yc
ловиях фармакокинетическоrо исследования, в
частности,еrодлительности.Биоаналитические
методы должны соответствовать установлен
ным критериям приемлемости по специфично
сти, чувствительности, правильности, точности
и воспроизводимости. Условием получения дo
стоверных результатов является изученность
стабильности АФИ и'или продуктов еro био
трансформации.
С учетом требований, сформулированных
на Международной конференции по параме
трам валидации биоаналитических методов 1О ,
валидация аналитической методики должна
удовлетворять следующим минимальным кри
териям.
Валидация должна включать в себя Ha
чальную фазу и фазу проведения исследова
ния. Во время начальной фазы исследования
следует подтвердить стабильность в биолоrи
ческой среде рабочеrо раствора и образцов с
добавлением вещества, специфичность, чув
ствительность, правильность, точность и BOC
производимость. Валидация в ходе исследо
вания доказывает стабильность собранных на
протяжении клиническоro испытания образцов в
условиях их хранения и подтверждает правиль
ность и точность опытов.
Валидация должна распространяться на
целевое применение анализа.
Диапазон значений, используемых при
проверке, должен соответствовать диапазо
ну концентраций исследуемых образцов. rpa
дуировочный rрафик следует составлять в той
же самой биолоrической среде, которая будет
10 Bi08п8/ytic8/ тethod v8lid8tioп 8 revisit with 8 deC8de о(
prog ess / VP Sh8h [et 8/] // Ph8rт8ceutic8/ Rese8rch. 2000
vo/. 17. P.1551 1557.
#РеzистраЦUОННbtе требования и правила проведения исследований биодоступности 111 3
использоваться в качестве образцов в заплани
рованном исследовании, посредством добавле
ния в среду заданной концентрации анализиру
емоro вещества. Он должен состоять из холо
стой пробы, нулевой пробы и 6 ненулевых
проб, охватывающих предполаrаемый диапазон.
Концентрацию стандартов следует выбирать на
основании интервала концентрации, ожидаемо
ro в конкретном испытании.
Если анализ должен использоваться при
менительно к различным лабораториям, еro сле
дует проверить в каждой из них и определить
перекрестную сопоставимость результатов на
различных базах.
Методика, не применяемая реryлярно (py
тинно), требует соответствующей ревалидации
для доказательства тоro, что она все еще рабо
тает соrласно исходным проверенным анали
тическим процедурам. Следует документально
подтвердить проведение ревалидационноro ис
следования, как правило, в форме приложения к
отчету об испытании.
Настоятельно не рекомендуется в рамках
исследования при менять две и более методики
для количественноrо определения образцов в
одинаковой среде в пределах одноro и тоro же
rрадуировочноro rрафика.
Если будут сравниваться различные ис
следования, в которых пробы анализировались
разными методами (методиками), затраrива
ющими подобный интервал концентрации и по
добные аналитические среды, следует провести
перекрестную валидацию данных методов.
Для принятия или отклонения серии aHa
литических исследований требуется исполь
зовать сдвоенные пробы для контроля каче
ства с добавлением вещества не менее чем в
трех различных концентрациях (<<контроли Ka
чества» ).
По возможности все образцы, полученные
от одноro субъекта (за все периоды), следует
изу чать в одной серии аналитических опытов.
В аналитическом протоколе валидации
методики и'или в СОП лаборатории необходи
мо указать способы валидации, методолоrию
и критерии приемлемости. В отчете (отчет о
валидации методики) следует описать все ис
следования, использованные для подтвержде
ния доводов или представления заключений
о валидности этоro метода. Любая модифи
кация методики в ходе анализа исследуемых
проб требует соответствующей ревалидации.
В аналитическом отчете представляются pe
зультаты количественноro определения ис
пытуемых образцов, а также резулыаты для
образцов rрадуировки и контроля качества,
повторные анализы (если были), сведения
по аналитическим проrонам и расположению
в них контролей качества, а также репрезен
тативное количество xpoMaTorpaMM образцов
(как правило, не менее 5 % от общеro числа
анализов).
6.8. СТАТИСТИЧЕСКИЙ АНАЛИЗ
РЕЗУЛЬТАТОВ
Первостепенное значение для оценки био
эквивалентности имеет минимизация риска
ложноположительноro результата биоэквива
лентности. Статистический анализ испытания
биоэквивалентности должен подтвердить Ma
ловероятность клинически значимоrо различия
между биодоступностью rенерическоro лекар
ственноro средства и препарата сравнения. CTa
тистические процедуры следует оrоворить в про
токоле перед началом сбора данных.
Статистический метод для анализа фармако
кинетической биоэквивалентности основывается
на определении 90 % доверительноro интерва
ла для отношений лоrарифмически преобразо
ванных средних арифметических (rенерический'
эталонный) рассматриваемых фармакокинетиче
ских параметров, а таюке на выполнении двух oд
носторонних тестов при 5 % уровне значимости.
Для установления фармакокинетической биоэк
вивалентности рассчитанный доверительный ин
тервал должен находиться в rраницах заранее
установленноrо интервала биоэквивалентности.
Эти статистические процедуры должны приво
дить К симметричному заключению относительно
двух изучаемых лекарственных средств (напри
мер, позволяя получить одинаковый вывод о том,
является ли эквивалентным rенерическое лекар
ственное средство по отношению к эталонному
или эталонное лекарственное средства по OTHO
шению к rенерическому).
Все фармакокинетические параметры, KOTO
рые непосредственно зависят от концентрации
(АUС и Ста)' следует преобразовать лоrариф
мированием, используя десятичные или HaTY
ральные лоrарифмы. Выбор вида лоrарифмов
(десятичные или натуральные) должен OCTa
ваться неизменным и указываться в отчете ис
следования.
Преобразованные лоrарифмированием
фармакокинетические параметры, зависимые
от концентрации, необходимо оценивать с помо
щью дисперсионноro анализа (ANOVA). Обычно
модель дисперсионноro анализа включает в Ka
честве независимых вариант лекарство, период
исследования, остаточную концентрацию или
«эффект пере носа» и субъективные факторы.
Параметрические методы, т. е. основанные
на законе нормальноro распределения, peKO
мендуются для анализа показателей биоэквива
лентности, преобразованных лоrарифмировани
ем. Общий принцип заключается в построении
90 % доверительноro интервала для величины
J.1T J.1R' который позволяет сделать вывод о фар
макокинетической биоэквивалентности, если
данный доверительный интервал находится в
rраницах принятых предельных значений. Прин
цип параметрических доверительных интерва
лов означае что их определение равнозначно
проведению двух односторонних тестов для rи
потезы при 5 % уровне статистической значимо
1114
rосударственная фармакопея Республики Беларусь
сти. Полученные антилоrарифмы доверитель
ных интервалов составляют 90 % доверительный
интервал для соотношения среднеrеометриче
ских значений rенерическоro и эталонноro ле
карственных средств.
Такая же процедура должна использоваться
для изучения параметров, полученных в резуль
тате испытаний в стационарном состоянии или
суммарноro выведения с мочой, если это требу
ется.
Следует представить также данные по опи
сательной статистике для показателя 'тах' Если
параметр 'тах будет подверrаться статистиче
скому анализу, то изучение должно OCH08ЫBaTb
ся на непараметрических методах и проводить
ся С использованием нетранеформированных
данных. Для повышения точности оценки 'тах He
обходимо взять достаточное количество образ
цов, близких к ожидаемым максимальным KOH
центрациям. Для показателей, описывающих
фазу выведения (t.). требуется только описа
тельная статистика.
В протоколе следует оroворить методы
идентификации и обработки возможных «резко
выделяющихся» (<<outliers») значений. Следу
ет попытаться обнаружить и представить кли
ническое или фармакокинетическое объясне
ние данных наблюдений. Резко выделяющиеся
наблюдения Moryт не приниматься в расчет при
оценке биоэквивалентности при условии, что
справедливость исключения этих данных дo
казана. Однако поскольку выбросы MOryт YKa
зывать на недо6рокачественность изучаемоro
лекарственноro средства. то, как правило. не pe
комендуется апостериорное удаление выделя
ющихся значений. Для обработки данных, coдep
жащих выбросы. применяются независимые от
распределения (неnaраметрические) статисти
ческие методы. Доnyacается для демонстрации
наличия таких наблюдений при водить rрафики
индивидуальных стандартизованных различий
(центрированных по среднему значению и HOp
мированных по стандартному отклонению).
Непараметрическую статистическую MeTO
дику обработки результатов можно выбрать в
случае, если распределение преобразованных
лоrарифмированием результатов не подчиняет
ся нормальному закону. Следует в протоколе за
ранее обосновать свои намерения использовать
непараметрические статистические методы.
6.9. ДОПУСТИМЫЕ ,РАНИЦЫ
БИОЭКВИВАЛЕНТНОСТИ
6.9.1. Отношение площадей ПОД фармако-
кинетическими кривыми
Для данной оценки относитеЛЬНQЙ биодо
ступности 90 % доверительный интервал должен
находиться в rраницах диапазона биоэквива
лентности, равною 0,80 1,25 (или 80 125 %,
что отвечает симметричному лоrарифмически
преобразованному интервалу :1::0,223). Если Te
рапевтический диапазон особенно узок, может
потребоваться уменьшение диапазона допу
стимости, исходя из клинически обоснованных
причин. Более широкий диапазон допустимости
может быть приемлемым в исключительных слу
чаях при документальном подтверждении кли
нической обоснованности.
6.9.2. Отношение показателей С
тах
К отношению С таХ следует при менять rpa
ницы приемлемости, равные 0,80 1,25 (или
80 125 %, что отвечает симметричному ло
rарифмически преобразованному интервалу
:1::0,223). Однако данный показатель относитель
ной биодоступности является более вариабель
ным, чем отношение Аис, и в некоторых случа
ях может оказаться приемлемым более широкий
диапазон допустимости. Используемый диапа
. зон должен определяться при планировании
исследования, до начала статистической обра
ботки данных и обосновываться с учетом сооб
ражений безопасности и эффективности. В ис
ключительных ситуациях при соответствующем
документальном обосновании в отношении безо
пасности и эффективности можно допустить
простое условие о нахождении точечной оценки
(среднеro rеометрическоro показателя отноше
ний Ста) В rраницах предельных значений био
эквивалентности 0,80 1,25.
6.9.3. Отклонение t
тах
Статистический анализ параметра 'тах целе
сообразен только при наличии документальноro
доказательства клинически значимою быстро
ro начала действия вещества или опасений по
поводу ero нежелательноrо действия. Непара
метрический 90 % доверительный интервал для
данной оценки относительной биодоступности
должен находиться в клинически обоснованном
диапазоне приемлемости.
Описанные выше положения при меняются и
в отношении друrих фармакокинетических пока
зателей.
6.10. ПРЕДСТАВЛЕНИЕ ОТЧЕТНОСТИ
ПО РЕ3УЛЬТАТАМ ИССЛЕДОВАНИЯ
Отчет об исследовании биоэквивалентно
сти должен содержать полную документацию по
протоколу ею проведения, ходу и анализу в co
ответствии справилами Техническоrо кодекса
установившейся практики «Надлежащая клини
ческа я практика» (ТКП 184 2009 (02040»). При
составлении акта можно использовать соответ
ствующее приложение ТКП 184 2009 (02040).
ОТ8етственный( ые) исследователь( и) должен
подписать разделы документа, соответствующие
еro компетенции. Следует указать имена jI1 долж
ности ответственноro( ых) исследователя( ей).
клиническую базу исследования и период ero
осуществления.
Заявителю необходимо включить в отчет
ную документацию по испытанию документ с
наименованиями и номерами серий лекарствен
#Ре,шстрационные требования и правила проведения исследований биодоступностu 1115
ных средств, использованных в исследовании,
а также COCTaB( Ы) исследованных лекарствен
ных средств. Следует предоставить результаты
тестов сравнительной кинетики растворения in
vitro. В дополнение заявитель должен предста
вить подписанное заявление, подтверждающее,
что испытуемый препарат является идентичным
с лекарственным средством, реrистрация KOТO
роro про водится.
К отчету необходимо приложить биоанали
тический отчет о валидации методики (см раздел
6.7). Данный документ должен содержать CBeдe
ния о rрадуировке и образцах для контроля Ka
чества. Следует включить репрезентативное KO
личество (как правило, не менее 20 % от общеro
числа анализов) xpoMaTorpaMM или друrие пер
вичные данные аналитическоro оборудования,
охватывающие весь калибровочный диапазон, а
также анализ проб контроля качества и образ
цов из клинических испытаний.
Все результаты излаrаются четко. Для каж
дою лекарства в виде таблицы следует пред
ставить все измеренные у каждою субъекта KOH
центрации и время отбора проб. В сведенные в
таблице данные следует включить дату проведе
ния серии анализов, индикацию субъекта, период
исследования, вводимый препарат (rенериче
ский или эталонный) и прошедшее время между
приемом лекарства и забором крови в открытой
форме. Также должны присутствовать система
тизированные результаты, которые показывают
оценки содержания АФИ соrласно конкретною
аналитическоrо проюна (в том числе серий, ис
ключенных из дальнейших расчетов, включая
все калибровочные стандарты и образцы для
контроля качества из соответствующеro проro
на). Обязательно указывается процедура расчета
по исходным данным производных пара метров
(АUС, t). Любое исключение данных следует
объяснить. Также обосновываются использован
ные теория и метод расчета, если результаты
вычислялись не путем модельно независимых
расчетов, а при помощи фармакокинетических
модельных теорий. Индивидуальные кривые
«концентрация В крови'сыворотке'плазме
время» должны строиться в линейно линейной и
линейно лоrарифмической системах координат.
Следует указывать все индивидуальные данные
и результаты, включая сведения о субъек
тах, выбывших из исследования. Необходимо
сообщать и отчитываться за выбывших и исклю
ченных из исследования испытуемых.
Результаты измерений и рассчитанные фар
макокинетические параметры сводятся в табли
цы по каждой комбинации «субъект лекарство»
вместе с описательной статистикой. Статистиче
ский отчет должен быть достаточно детальным,
чтобы при необходимости имелась возможность
повторить статистические анализы. В случае
если примененные статистические методы отли
чаются от оroворенных в протоколе испытания,
необходимо указать причины отклонений.
Таким образом, отчет должен содержать
следующую документацию:
утвержденную проrрамму (протокол) ис
следований;
одобрение комитета по этике на проведе
ние исследований;
клинический отчет по всем периодам ис
пытания с анализом соблюдения стандартиза
ции испытания (по питанию, режиму пребывания
добровольцев, приему лекарственных средств и
отбору проб крови) и безопасности лекарствен
ноro средства;
оформленные и подписанные информиро
ванные соrласия добровольцев на участие в ис
следовании;
индивидуальные карты испытаний, coдep
жащие демоrрафические данные добровольцев
(при проведении исследований на больных
также диаrноз и номер истории болезни);
образец памятки добровольца:
документальное описание аналитическою
метода, включающеro метролоrические xapaK
теристики и первичные аналитические данные
(например, демонстрационные xpoMaтorpaM
мы, если использованы хроматоrрафические
методы);
отчет по валидации аналитической MeTO
дики;
индивидуальные фармакокинетические
профили в обычных и полулоrарифмических KO
ординатах;
усредненные фармакокинетические про
фили в обычных и полулоrарифмических KOOp
динатах;
отчет по статистическому анализу и pac
четам индивидуальных значений параметров
фармакокинетики, содержащий COOTBeTCTBY
ющие средние значения и величины стандарт
ных отклонений, средние rеометрические значе
ния лоr нормально распределенных параметров
фармакокинетики, соответствующие интерваль
ные оценки.
Дополнительно отдельные части отчета
MOryT представляться на электронном носителе
в форматах *.pdf, *.doc, *.docx, *.rtf. .xJs.
6.11. СПЕЦИАЛЬНblЕ ПОЛОЖЕНИЯ
6.11.1. Лекарственные средства с фикси
рованной комбинацией доз
Если фармакокинетическая биоэквива
лентность лекарственных средств с фиксиро
ванной комбинацией дОЗ (ФКД) оценивается с
помощью исследований iп vivo, то дизайн экс
пери мента должен подчиняться тем же общим
принципам. которые описаны в Пр13дыдущих раз
делах. rенерическое лекарственное средство с
ФКД следует сравнивать с фармацевтически
эквивалентным компараторным лекарствен
ным средством с ФКД. В некоторых случаях
(например, если на рынке отсутствует компара
торный препарат с ФКД) в качестве компарато
ра может при меняться свободная комбинация
1116
rосударственная фармакопея Республики Беларусь
нескольких монокомпонентных лекарственных
средств. Моменты отбора проб следует выби
рать так, чтобы можно было оценить должным
образом фармакокинетические параметры всех
АФИ. Необходимо проводить валидацию био
аналитическоro метода в отношении всех изме
ряемых компонентов. Статистический анализ
следует выполнять на основании фармакоки
нетических данных, собранных по всем АФИ.
90 % доверительные интервалы соотношения
всех активных компонентов испытуемоro'ком
параторноro лекарственных средств должны
находиться в пределах допустимости.
6.11.2. Клинически значимые вариации
биодоступности
Разработчики rенерическоrо лекарствен
ноro средства должны приложить максимум
усилий для фармацевтическоro обеспечения ле
карственному средству заданных качественных
характеристик биодоступности. Если со BpeMe
нем производителем ориrинальноro лекарствен
ноro средства разрабатывается еro улучшенный
состав или технолоrия, Torдa в качестве компа
ратора должно Вblступать данное лекарственное
средство. Новое лекарственное средство с био
доступностью, находящейся вне диапазона при
емлемости, по определению не является взаи
мозаменяемы" относительно существующеro
эталонноro лекарственною средства. При этом
решение данной проблемы путем изменения
концентрации или дозировки в единице лекар
ственной формы с целью компенсации HeДOCTa
точной или чрезмерной биодоступностью новою
лекарственнoro средства по сравнению с компа
раторным препаратом не может рассматривать
ся в рамках Иalытаний биэквивалентности, по
скольку в этом случае не выполняется условие
фармацевтической э«вивалентности.
6.11.3. Лекарственные средства с высо-
кой вариабельностью
«Высоко вариабельными лекарственны
ми средствами» признаются АФИ с внутриин
дивидуальной вариабельностью 2:30 % У одноro
субъекта при расчете коэффициента вариации
в рамках дисперсионною анализа. Кроме тоro,
«высоко вариабельные лекарственные cpeд
ства» в целом должны являться доказано безо
пасными лекарственными средствами с полоrи
ми кривыми «доза эффект». Доказательство
биоэквивалентности лекарственных средств, co
держащих «высоко вариабельные АФИ», счи
тается затруднительным, так как чем выше KO
эффициент вариации, тем шире должен быть
90 % доверительный интервал. Следовательно,
чтобы получить соответствующую статистиче
скую мощность, требуется набрать большое KO
личество субъектов для исследования с высоко
вариабельными препаратами. В настоящее
время допустимым считается применение сле
дующих подходов.
Использование масштабирования для
расширения пределов биоэквивалентности.
При двухэтапной модели предельные значения
rраниц 90 % доверительноro интервала MorYT
приводиться в соответствие с остаточным CTaH
дартным отклонением или в репликативном ди
зайне в соответствие со стандартным ОТКЛО
нением компаратора у одной из rрупп субъектов.
Степень расширения определяется на oc
новании остаточной внутрииндивиуальной Ba
риации, установленной при исследовании
биоэквивалентности, с использованием Mac
штабируемоrо среднеrо биоэквивалентности
исходя из соотношения [и, [] == e=kX->"'R, rдe
и верхний предел интервала эквивалентно
сти, L нижний предел интервала эквивалент
ности, k реryлирующая константа для уровня
0,760 и CV остаточная внутрииндивидуаль
ная вариация от лоrарифмически трансфор
мируеМbJХ значений С таХ ориrинальноro лекар
ственноro средства. Ниже приведены примеры
различных уровней вариабельности и соответ
ствующее им различное расширение пределов
биоэквивалентности с использованием данной
методолоrии:
Внутренний
CV (0/0)*
30
35
40
45
2:50
Нижний
предел
80,00
77,23
74,62
72,15
69,84
Верхний
предел
125,00
129,48
134,02
138,59
143,19
. Для расчета CV (0/0) СМ. раздел 6.3.1.
в исключительных, документально обо
снованных случаях, если доверительный интер
вал выходит за указанные rраницы, испытуемые
препараты признаются биоэквивалентными,
если 90 % доверительный интервал для cpeд
них значений отношений АUС и С таХ испытуемо
ro препарата к АUС и С таХ препарата сравнения
находится в допустимом диапазоне 0,8 1,25,
при этом неукоснительно исполняются следу
ющие три условия:
1) размер общей выборки исходноro ис
следования биоэквивалентности составляет не
менее 20 (п2:10 человек в rруппе) или размер
объединенной выборки исходноro исследования
и испытаний дополнительных субъектов равня
ется не менее 30 добровольцев;
2) соотношение пределов rеометриче
ских средних значений АUС и С таХ испытуемо
ro препарата и препарата сравнения составляет
0.90 1.11 ;
3) скорости растворения испытуемоrо пре
парата и препарата сравнения оцениваются как
равнозначные при всех режимах тестирования
на растворение.
Данное правило нельзя применять к Meд
ленно растворяющимся лекарственным cpeд
#Реzистрацuонные требования и правила проведения исследований биодоступности 1117
ствам, т. е. к тем, у которых в течение 2 ч BЫCBO
бождается менее 80 % лекарственною средства
в среде с рН 1,2 и в течение 6 часов в Аруrих
средах при любых режимах, описанных в тесте
сравнительной кинетики растворения.
6.11.4. Использование для установления
биоэквивалентности усеченной площади под
фармакокинетической кривой
В исследованиях биодоступности в целом
рекомендуется отслеживать концентрацию Be
щества в плазме на протяжении не менее 4
периодов полувыведения после приема ле
карства. Анализ в плазме или сыворотке силь
нодействующих лекарственных средств в низких
концентрациях обычно требует сложноro и дopo
roстоящеrо оборудования, позволяющеro зафик
сировать АФИ в терминальных отделах кривой
«концентрация в плазме время». При pac
смотрении биоэквивалентности лекарств с He
медленным высвобождением для системно
ro действия самым значимым отделом кривой
«концентрация в плазме время» является
отрезок до завершения фазы всасывания. С
друroй стороны, в процессе принятия решения о
биоэквивалентности фаза распределения не от
ражает различий между составом rенерическо
ro лекарственноro средства и препарата cpaBHe
ния. При использовании для анализа частичной
(усеченной) АUС с помощью моделирования
методом Монте Карло была показана BЫCO
кая степень корреляции между выводом о био
эквивалентности, основанном на такой непол
ной АUС, усеченной до четырех периодов tтax'
и площадью, экстраполированной до бесконеч
ности. Экспериментальные данные 11 позволяют
предположить, что для лекарств снемедленным
высвобождением нет необходимости отбирать
образцы крови более четырех периодов t max '
Существует два преимущества в использовании
усеченных площадей:
можно сrруппировать в области t max боль
шее количество проб крови, чтобы обеспечить
большую точность расчета и t max , ИСтах;
для определения фазы распределения не
требуется высокой чувствительности анализа.
Применимость метода с усеченной АUС
может быть обоснована в следующих случаях:
если в терминальных отделах кривой «KOH
центрация время» появляются низкие KOH
центрации АФИ, которые MOryт не поддаваться
количественному определению средствами про
BepeHHoro должным образом, чувствительноro
аналитическоro метода;
в отношении АФИ для лекарственных
средств с длительными периодами полуэлими
нации.
11 Truпc8ted 8re8 uпder the ситуе 8S 8 тe8sure о( re/8tive
exteпt о( bi08V8i/8bi/ity: EV8/U8tioп usiпg experiтeпt8/ d8t8 8пd
Moпte C8r/o Siтu/atioпs / Gaudre8u/t J, (et а/.] Pharтaceutic8/
Rese8rch. 1998. Vo{. 15. Р.1б21 1б29.
7.ФАРМАКОДИНАМИЧЕСКИЕ
ИССЛЕДОВАНИЯ
Фармакодинамические исследования на
здоровых добровольцах или пациентах MorYT
быть использованы для установления экви
валентности между двумя лекарственными
средствами. Исследования на здоровых дo
бровольцах и пациентах с помощью фармако
динамических измерений MOryT использовать
ся для установления эквивалентности между
двумя лекарственными средствами. Фармако
динамические испытания не рекомендованы
для принимаемых перорально лекарственных
средств с системным действием, если АФИ
абсорбируются в систему кровообращения, и
при этом для оценки системноro воздействия и
установления биоэквивалентности можно BЫ
брать фармакокинетический подход. Это свя
зано с тем, что фармакодинамические показа
тели имеют Bcerдa большую вариабельность,
чем фармакокинетические данные. Кроме тою,
фармакодинамические пара метры часто под
вержены значительным плацебо эффектам,
которые добавляют к вариабельности сложный
дизайн испытания. В результате, для дости
жения соответствующей статистической мощ
ности зачастую может потребоваться набор
большоro числа пациентов для фармакоди
намических исследований. Испытания фар
макодинамической биоэквивалентности MorYT
оказаться необходимыми, если невозможно с
достаточными достоверностью и чувствитель
ностью выполнить количественный анализ co
держания АФИ и'или еro метаболита( ов) в
плазме или моче (см. раздел 6.11.4). Иссле
дования фармакодинамической биоэквива
лентности на людях требуются в случае, есЛill
для доказательства эффективности и безо
пасности конкретноro лекарственноro cpeд
ства нельзя при менять в роли замещающих
конечных точек количественные показатели
концентраций АФИ. В некоторых rруппах ле
карственных форм, например, лекарствен
ные средства, разработанные для местноro
действия, отсутствует реальная альтернатива
проведению испытания на фармакодинамиче
скую биоэквивалентность. Анализ фармакоди
намической биоэквивалентности. таким обра
зом, может подойти для лекарственных форм,
применяемых местно, и для инrаляционных
лекарственных форм.
При фармакодинамических исследовани
ях условия, в которых они проводятся, должны
строю контролироваться, так же как и при иссле
довании биолоrической эквивалентности. Эти
условия должны отвечать требованиям Техни
ческоro кодекса установившейся практики «Haд
лежащая клиническая практика» (ТКП 184 2009
(02040»).
При планировании, про ведении и оценке pe
зультатов исследования должны соблюдаться
следующие требования:
1118
rосударственная фармакопея Республики Беларусь
измеряемая реакция должна представ
лять собой фармаколоrический или терапев
тический эффект, подтверждающий эффек
тивность и'или безопасность лекарственноrо
средства;
методика должна быть валидирована с
точки зрения точности, воспроизводимости,
специфичности и достоверности;
ни испытуемое, ни эталонное лекар
ственные средства не должны вызывать MaK
симальную реакцию в ходе исследования,
поскольку может оказаться невозможным BЫ
явить различия между действующими Be
ществами, применяемыми в дозах, которые
вызывают максимальные или близкие к макси
мальным эффекты; изучение взаимодействия
«доза реакция» может стать необходимой
частью исследования;
реакция должна измеряться количе
ственно двойным слепым методом и резуль
таты должны записываться с помощью соот
ветствующеrо прибора с воспроизведением,
чтобы обеспечить реrистрацию фармакоди
намических явлений, которые заменяют KOH
центрацию в плазме. Если такие измерения
невозможны, можно провести реrистрацию
по визуально аналоroвым шкалам, а если
данные оrраничены качественными показате
лями (катеrоризированными данными), требу
ется выполнение специальноro статистическо
ro анализа;
испытуемые, не реаrирующие на ле
карственное средство, должны быть исключе
ны из исследования после предварительноro
скрининrа, и в проrрамме (протоколе) должны
быть указаны критерии, по которым идентифи
цируются реаrирующие и не реаrирующие ис
пытуемые;
если возможен существенный плацебо
эффект, то сравнение между лекарственными
средствами можно проводить только на основе
априорноro предположения в дизайне иссле
дования потенциальноrо плацебо эффекта,
при этом в схему испытания можно внести по
правку на данный эффект посредством вклю
чения в лечение плацебо (в качестве третьей
фазы в этой схеме);
в схеме исследования должна быть
предусмотрена лежащая в основе патолоrия и
анамнез болезни и приведена информация о
воспроизводимости исходных условий;
следует использовать перекрестную
модель, однако, если не приrоден перекрест
ный метод исследования, необходимо исполь
зовать метод параллельных rрупп.
Для rенерическоro и ориrинальноro лекар
ственных 'средств принципы формирования
выборки должны оставаться такими же, как
описано в разделе 6.5.
В исследованиях, в которых реrистрируют
непрерывные переменные, изменение интен
сивности действия лекарственноrо средства,
наблюдаемое в течение некотороro промежут
ка времени, может быть описано таким же об
разом, что и в исследовании для измерения
концентрации препарата в плазме. Можно BЫ
вести параметры, описывающие площадь под
кривой «эффект время», максимальную pe
акцию и время, Korдa происходит эта реакция.
Статистические методы, применяемые для
оценки результатов исследования, в основном
те же, что и при исследованиях биолоrиче
ской эквивалентности. Однако рекомендуется
сделать поправку на потенциальную нелиней
ность взаимосвязи между дозой и площадью
под кривой «эффект время», построенной по
результатам исследования «доза реакция».
Следует отметить, что обычный допустимый
диапазон для оценки биолоrической эквива
лентности, который применяется в фармако
кинетических исследованиях, в данном случае
неприменим, поскольку, как правило, слишком
широк; в связи с этим еro необходимо опреде
лять на основании анализа конкретной ситуа
ции и описывать в проrрамме (протоколе).
8. КЛИНИЧЕСКИЕ ИСПЫТАНИЯ
ДЛЯ некоторых лекарственных средств
и форм (например, случай д) в разделе 5.1)
кривые зависимости «концентрация В плазме
время» не приroдны для оценки эквивалентно
сти двух действующих веществ. Также не MorYT
быть проведены и фармакодинамические ис
следования в случае отсутствия значимых и
количественно измеримых фармакодинамиче
ских параметров. В этих обсто ятельствах для
демонстрации эквивалентности двух препара
тов необходимо проведение клинических ис
пытаний. При таком клиническом испытании
применяются те же статистические принципы,
что и при исследовании биолоrической экви
валентности. Число пациентов, включенных в
исследование, будет зависеть от вариабель
ности планируемых пара метров и допустимо
ro диапазона и обычно намноro больше, чем
это требуется при исследовании биолоrиче
ской эквивалентности. В частности, требуется
8600 пациентов для достижения адекватной
статистической мощности, позволяющей об
наружить 20 % повышение реакции на испы
туемое лекарственное средство по сравнению
с плацебо. Точно так же необходимо привлечь
2600 пациентов с инфарктом миокарда, чтобы
продемонстрировать 16 % снижение риска за
болевания. Еще большее число испытуемых
требуется для сравнения двух составов с оди
наковым АФИ на основании подобных конеч
ных точек 12 .
12 Yusif The Studies о' Left Ventricu/ar Dysfunctioп (SOL VD)
/nvestigators. Yusif [et а/.] // Jourпa/ о' the Aтerican Medica/
Associatioп. 1988. Vo/. 260. р. 2259 22б3.
Effect о' ena/april оп surviva/ in patients with reduced /eft
ventricu/ar ejection fractions and congestive heart fai/ure. New
Eng/and Jourпa/ о' Medicine. 1991. Vo/. 325 Р 293 ЗО2
#Реаистрационные требования и правила проведения исследований биодоступности 1119
В проrрамме (протоколе) проведения таких
испытаний должны быть четко определены сле
дующие положения:
в качестве контрольных параметров выби
рают значимые конечные клинические точки, на
основании которых MOryт быть рассчитаны ин
тенсивность и начало проявления реакции opra
низма, если это необходимо;
rраницы диапазона эквивалентности сле
дует определять на основе анализа ситуации,
принимая во внимание специфические клиниче
ские условия, например, естественное течение
заболевания, эффективность доступных Meтo
дов лечения и выбранные искомые параметры.
В отличие от фармакокинетическоrо исследо
вания биолоrической эквивалентности (rдe ис
пользуется стандартный допустимый диапазон).
масштаб допустимоro диапазона в клинических
испытаниях не может быть стандартным для
всех rрупп лекарственных средств и определя
ется для каждоro терапевтическоro класса и по
казания;
в настоящее время общепринятым для
данноro типа испытаний является использова
ние статистических методов, основанных на
определении доверительною интервала. OCHOB
ная задача заключается в том, чтобы исследу
емый препарат не уступал эталонному более
чем на строю заданную величину. Поэтому цe
лесообразен расчет одностороннею довери
тельноro интервала (для эффективности и'или
безопасности). Доверительный интервал может
быть рассчитан параметрическим или непара
метрическим методом;
в дизайн исследования при возможности
следует включать этап плацебо;
целесообразно включить в сравнитель
ные испытания анализ конечных результатов по
оценке безопасности;
правила формирования выборки для reHe
рическоrо и компараторноro препаратов должны
быть такими же, как описанные в разделе 6.5.
9. ТЕСТ СРАВНИТЕЛЬНОЙ КИНЕТИКИ
РАСТВОРЕНИЯ /N V'TRO (ИСПЫТАНИЕ
НА РАСТВОРЕНИЕ)
Сравнительные исследования по оценке
растворимости in vit,o (<<вне живоro орrанизма»)
должны выполняться как дополнение к иссле
дованию iп vivo в качестве предварительноro
этапа при разработке новоro rенерическоro ле
карственноro средства и в качестве доказатель
ства возможности распространения результатов
испытаний биоэквивалентности для одной дози
ровки на I3сю линейку дозировок.
Тест сравнительной кинетики растворения
(ТСКР) развивается сейчас как замещающий
анализ биоэквивалентности для определенных
катеroрий принимаемых внутрь лекарственных
средств. В отношении данных лекарственных
средств (в основном твердые пероральные ле
карственные формы, содержащие АФИ со спе
циальными свойствами) для документальноro
подтверждения равнозначности rенерическоro
и компараторноro препаратов (см. раздел 6.5)
может использоваться сравнительное подобие
профиля растворимости iп vit,o.
Необходимо отметить, что хотя тесты «Pac
творение», рекомендованные Международной
и Национальными фармакопеями для прове
дения контроля качества, разрабатывались как
подобные тесту сравнительной кинетики paCT
ворения, они не соответствуют всем требо
ваниям по оценке эквивалентности rенериче
cKoro лекарственноrо средства в сравнении с
компараторами и их не следует использовать в
данных целях.
В ходе разработки лекарственноro средства
испытание на растворение используют в каче
стве средства идентификации фактороs COCTa
ва, которые влияют и MOryT оказывать реша
ющее влияние на биодоступность лекарствен
ноro средства. Как только определены состав
и процесс производства, испытание на paCTBO
рение используют при контроле качества серий,
произведенных при масштабировании процесса,
и промышленных серий, чтобы обеспечить YBe
ренность как в постоянстве от серии к серии, так
и в том, что профили растворения остаются по
добными профилям растворения серий, исполь
зуемых для проведения клинических испытаний.
Кроме тоro, испытание на растворение может
быть использовано для подтверждения биодо
ступности новоro лекарственною средства, био
эквивалентности rенерическоro средства или
при внесении изменений в состав и технолоrию
производства.
Таким образом, исследования сравнитель
ной кинетики растворения iп vit,o MOryT служить
для нескольких целей.
1. Обеспечение качества:
для получения информации об испыту
емых сериях, используемых в исследованиях
биодоступности'биоэквивалентности и OCHOB
ных клинических исследованиях, чтобы подтвер
дить показатели с целью контроля качества;
для использования при контроле качества
как способ доказательства постоянства при про
изводстве;
для получения информации о препарате
сравнения, используемом в ходе исследований
биодоступности'биоэквивалентности и OCHOB
ных клинических исследований.
2. В качестве дополнительной информации
к заключению о биоэквивалентности
для доказательства подобия референтных
препаратов;
для дока тельства подобия между раз
личными составами препарата (с отдельными
изменениями и новым качественным составом)
и референтным лекарственным средством;
для сбора информации о постоянстве от
серии к серии препаратов (испытуемоro и pe
ферентноro), которые будут использованы при
1120
rосударственная фармакопея Республики Беларусь
выборе приемлемых серий для исследования
in vivo.
9.1. ИСПЫТАНИЯ /N V/TRO
И БИОФАРМАЦЕВТИЧЕСКАЯ СИСТЕМА
КЛАССИФИКАЦИИ
9.1.1. Биофармацевтическая система
классификации
Биофармацевтическая система классифи
кации (БСК) основывается на водорастворимо
сти и проницаемости лекарственноro вещества
через слизистые оболочки желудочно кишечно
ro тракта. Сотасно этой классификации, АФИ
делятся на четыре класса:
растворимость проницаемость
Класс 1: хорошая хорошая
Класс 11: плохая хорошая
Класс 111: хорошая плохая
Класс IV: плохая плохая
Сочетая степень растворения лекарственно
ro средства с двумя данными свойствами АФИ,
принимают 80 внимание три основных фактора,
реryлирующие скорость и степень абсорбции ле
карственнoro вещества из ТВеРДЫХ дозирован
ных пекарственных форм с немедленным BЫ
свобожденмем. Исходя из свойств растворения
лекарственные формы с немедленным BЫCBO
бождением можно классифицировать как являю
щиеся «очень быстрорастворимыми», «быстро
растворимыми- или «не быстрорастворимыми»
с позиции характеристики растворения.
На основании растворимости и проница
емости АФИ. а также свойств растворения ле
карственной формы. принцип БСК позволяет
отказаться от испытаний in vivo на фармакоки
нетическую биоэквивалентность в отношении
некоторых катеroрий лекарственных средств с
немедленным высвобождением. Пероральные
лекарственные средства, не подходящие для
так называемой процедуры «биовейвера», KO
торая основывается на правилах БСк. описаны
в разделе 5.1. (а).
9.1.1.1. Хорошая растворимость
АФИ считается хорошо растворимым, Korдa
максимальная разовая терапевтическая доза,
рекомендованная ВОЗ (если АФИ присутствует
в «Типовом перечне ВОЗ жизненно важных ле
карственных средств»). или максимальная Te
рапевтическая доза лекарственноrо средства,
соrласно инструкции по медицинскому приме
нению ориrинальноro лекарственноro cpeд
ства в наибольшей из дозировок (если АФИ OT
сутствует в «Типовом перечне ВОЗ жизненно
важных лекарственных средств» ), растворя
ется в объеме водной среды 250 мл и меньше
с рН в диапазоне 1,2 6,81З. Профиль АФИ
«рН растворимость» определяется при TeM
пературе водной среды (37:1:1) ОС. Необходи
мо выполнить не менее трех количественных
определений растворимости с повторами при
каждом изменении рН. В исходных peKOMeHдa
циях Руководства БСК14 предлаrается измерять
растворимость на протяжении диапазона рН
1 ,2 7,5, однако диапазон рН 1.2-------6'8 также яв
ляется удовлетворительным.
9.1.1.2. Хорошая проницаемость
АФИ считается хорошо проникающим,
коrда степень всасывания у людей составля
ет 85 % и более, исходя из количественноro
определения баланса веществ или в cpaBHe
нии с внутривенной компараторной дозой. Дo
пустимым альтернативным методом анализа
для определения проницаемости АЛК может
'служить:
(i) кишечная перфузия in vivo на людях,
Если данный метод используется для иссле
дований проникновения вещества, необходи
мо по казать приroдность методолоrии, вклю
чая определение проницаемости субстанции
по сравнению с ее значением у референтно
ro компонента, у котороro документально под
тверждено, что абсорбированная фракция
дозы составляет не менее 85 %, а также вклю
чая применение неrативноro контроля.
Вспомоrательные сведения можно предо
ставить с помощью следующих дополнитель
ных методов испытания:
(ii) кишечная перфузия iп vivo или iп situ с
использованием животных моделей;
(iii) проницаемость iп vit,o сквозь монослой
культивированных эпителиальных клеток (Ha
пример. Caco 2) с помощью метода, проверен
ноro на АФИ с известными значениями прони
цаемости.
Однако, вне связи с данными (i) результа
ты, полученные способом (ii) или (iii), не будут
считаться приемлемыми. В данных исследо
ваниях хорошая проницаемость оценивается
в отношении хорошей проницаемости серии
референтных компонентов с официально под
твержденными значениями проницаемости
и абсорбированной доли вещества, включая
такие, у которых коэффициент абсорбции co
ставляет не менее 85 %.
Рекомендации по приroтовлению сред для
растворения и проведению испытания приве
дены в статье 5. 17. 1. Рекомендации по прове
дению теста «Растворение».
13 Иными r.ловами, если отношение наибольшей вводимой перо--
рально дозы лекарственноro средства (в Mr) к ero растворимости
(в мr/мл) равно 250 и менее.
14 HHS/FDA Gu;dance for /ndustry. Wa;ver о' ;п v;vo bioavailabi/ity and
bioequ;va/ence stud;es for immediate--re/ease solid от/ dosage forтs
based оп а biopharтaceut;cs c/assificatioп system: Dераптепl о'
Health and Нитап SelVices, US Food and Drug Administration.
Rockville, МD, 2000. (htlp:l/www.fda.gov/cder/guidance/;ndex.htm).
Уи. LX. Вiopharтaceut/cs C/assif;catioп Systeт: The scieпtific basis for
biowaiver extens;ons. / Уи, LX [et а/.] // Pharтaceut;ca/ Research.
2002. Vo/. 19. Р 921 925.
#Реzистрационные требования и правила проведения исследований биодоступности 1121
9.1.2. Определение свойств растворе-
ния rенерических лекарственных средств с
учетом процедуры «биовейвера», основан-
ной на БСК
ДЛЯ отказа от исследования iп vivo фарма
кокинетической биоэквивалентности rенериче
ское лекарственное средство снемедленным
высвобождением должно отвечать критери
ям очень быстрорастворимоro лекарственноro
средва или быстрорастворимоro лекарствен
ноro средва iп vit,o (смотрите далее), незави
симо от свойств класса, к которому относится
входящий в неro АФИ по БСК. Данные анализа
iп vit,o должны доказывать подобие профилей
растворения испытуемоro и компараторноro
препаратов.
9.1.2.1. Очень быстрорастворимое ле
карственное средство
rенерическое лекарственное средство
считается очень быстрорастворимым, если
не менее 85 % заявленноro количества лекар
ственной субстанции растворяется через 15
минут в приборе для растворения с лопастной
мешалкой при частоте вращения 75 об'мин или
в приборе для растворения с вращающейся
корзинкой при частоте вращения 100 об'мин,
В объеме 900 мл и менее каждой из следу
ющих сред (см также раздел 9.2):
раствор кислоты хлористоводородной с
рН 1,2;
ацетатный буферный раствор с рН 4,5;
фосфатный буферный раствор с рН 6,8.
9.1.2.2. Быстрорастворимоелекарствен
ное средство
rенерическое лекарственное средство при
знается быстрорастворимым, если не менее
85 % заявленноro количества лекарственной
субстанции растворяется через 30 минут в при
боре для растворения с лопастной мешалкой
при частоте вращения 75 об'мин или в приборе
для растворения с вращающейся корзинкой при
частоте вращения 100 обlмин, в объеме 900 мл
и менее каждой из следующих сред:
раствор кислоты хлористоводородной С
рН 1,2;
ацетатный буферный раствор с рН 4,5;
фосфатный буферный раствор с рН 6,8.
9.2. ПОДТВЕРЖДЕНИЕ ПРАВОМЕРНОСТИ
(АТТЕСТАЦИЯ) ПРИМЕНЕНИЯ
ПРОЦЕДУРЫ «БИОВЕЙВЕРА»
НА ОСНОВАНИИ БСК
ДЛЯ вынесения решения о возможности
применения процедуры «биовейвера», OCHO
ванной на БСК, оцениваются:
(а) документальные подтверждения
растворимости и проницаемости АФИ
(см. раздел 9.1);
(б) документальное подтверждение подобия
профилей растворения rенерическоro и компа
раторноro лекарственных средств в средах с рН
1,2,4,5 и 6,8 (см. далее);
141.32K.1712
(в) документальное подтверждение COOTBeT
ствия качественноrо состава и сохранения про
порциональности вспомоrательных веществ, ис
пользованных в составе лекарственной формы
(см. далее);
(r) документальная оценка риска, связанно
ro с вероятностью принять ошибочное заключе
ние о возможности процедуры «биовейвера»,
с учетом терапевтическоro индекса и клиниче
ских показаний к применению АФИ (см. раздел
5.1 для случаев, Korдa для доказательства био
эквивалентности может потребоваться исследо
вание iп vivo).
Разрешение о проведении испытания на
биоэквивалентность соrласно представленным
в этом пункте правилам допускается исключи
тельно в том случае, коrда установлено прием
лемое соотношение профиля «польза риск»
для обеспечения охраны здоровья населения в
целом и опасности для конкретноro пациента.
Снuженuе риска U оценка вСПОМ02атель
ных веществ в лекарственной форме
Опасность принятия несостоятельноrо
вывода о том, что rенерическое лекарственное
средство является равнозначным препарату
сравнения, можно уменьшить с помощью пра
вильной классификации АФИ и следующих pe
комендаций для испытания на растворение и
сравнения профилей растворения. Необходи
мо доказать, что вспомоrательные вещества,
входящие в состав rенерическоrо лекарствен
ноro средства, допускаются для изroтовления
лекарственных средств, содержащих данный
АФИ. Также документально подтверждается,
что использованные вспомоrательные веще
ства (i) не приведут к различиям между reHe
рическим и эталонным препаратами в том, что
касается процессов, которые влияют на Bcacы
вание (например, посредством воздействия на
перисталыику ЖКТ или путем взаимодействия
с процессами транспорта) или (ii) не приводят к
взаимному влиянию, нарушающему фармакоки
нетику АФИ.
Доказательством тоro, что каждое вспомо
rательное вещество, присутствующее в rенери
ческом лекарственном средстве, является дo
пустимым и не оказывает влияния на моторику
ЖКТ или иные процессы, воздействующие на
всасывание, являются следующие документаль
но установленные факты:
(i) вспомоrательное вещество присутству
ет в препарате сравнения, или оно присутству
ет в целом ряде друrих продуктов, содержащих
тот же самый АФИ, как и в rенерическом ле
карственном средстве и лекарственных cpeд
вах, зареrистрированных в roсударствах членах
Международной конференции по rармонизации
(MKr, /СН) или ассоциированных roсударствах;
(ii) вспомоrательное вещество имеется в re
нерическом лекарственном средстве в том же
количестве, как и в компараторе, либо оно при
сутствует в rенерическом лекарственном cpeд
1122
rосударственная фармакопея Республики Беларусь
стве в количестве, стандартно применяемом для
данноro вида лекарственных форм 15 .
Как правило, чем ближе состав rенерическо
ro лекарственноro средства к составу препара
та сравнения в части вспомоrательных веществ,
тем ниже риск ложноro заключения о равнознач
ности при использовании процедуры «биовейве
ра», основанной на БСк.
Препараты с недостаточной u uзбыточ
ной бuодоступностью
В отношении оценки потенциальных рисков
для здоровья населения и конкретноro пациента
при вынесении ошибочноro заключения относи
тельно биоэквивалентности существует два воз
можных отрицательных следствия.
Первое возникает, если rенерическое лекар
ственное средство является недостаточно био
доступным. В таком случае замещение компа
ратора rенерическим лекарственным средством
может привести к сниженной терапевтической
эффективности. АФИ. которым необходимо дo
стиrнуть определенной концентрации для эф
фективноro воздействия (например, антибио
тики), проявляют наибольшую уязвимость в
отношении недостаточной биодоступности.
Второе следствие возникает, если reHe
рическое лекарственное средство является
сверхбиодоступным. В данном случае заме
щение компаратора rенерическим лекарствен
ным средством может привести к токсичности.
АФИ, обладающие токсическим действием при
концент.рациях, близких к терапевтическому ди
апазону, чаще всеro подвержены риску чрез
мерной биодоступности. По этим причинам важ
ными факторами, которые следует учитывать,
определяя возможность применения процеду
ры «биовейвера», основанной на БСК, являют
ся показания к применению препарата и Tepa
певтический индекс.
Сравнение профuлей растворенuя
Оценка возможности roсударственной pe
rистрации rенерических лекарственных средств
с помощью исследований растворения ;п vitro
должна основываться на составлении сравни
тельных профилей растворения, а не на OДHOТO
чечном тесте «Растворение». При сопоставлении
rенерическоrо лекарственноrо средства и KOM
параторноro препарата профили растворения
можно сравнивать посредством коэффициента
подобия ('). В данной ситуации для соотнесения
профилей растворения двух продуктов приме
няется модельно независимый математический
метод. Профили растворения обоих средств
rенерическоro (испытуемоro) и компараторно
ro (референтноro) или профили растворения
15 Сведения о составе зареrистрированных лекарствен
ных средств находятся на Интернет сайтах некоторых наци
ональных opraHoB по реryлированию лекарственных средств
Примеры вспомоrательных веществ, достоверно вызыва
ющих бионеравнозначность, которую невозможно предска
зать на основании испытаний на растворение, включают: все
поверхностно активные вещества, маннитол и сорбитол.
лекарственных средств двух дозировок данноrо
производителя следует сопоставлять при одина
ковом режиме тестирования. Профили paCTBope
ния rенерическоro и компараторноro лекарствен
ных средств количественно определяются при
аналоrичном режиме тестирования при помощи
прибора, который соответствует техническим
условиям rосударственной фармакопеи Pe
спублики Беларусь, либо, используя прибор с
лопастной мешалкой при частоте вращения
75 об'мин или при бор с вращающейся корзин
кой при частоте вращения 100 обlмин при TeM
пературе (37:!:0,5) ос при значении рН 1,2, 4,5 и
6,8 (рекомендуется использовать буферные pac
творы, описанные в rосударственной фармако
пее Республики Беларусь; однако допускается
выбор альтернативных фармакопейных буфе
ров с тем же уровнем рН и буферной емкостью).
Следует отбирать пробы с достаточным
числом интервалов, чтобы в полной мере опи
сать профиль растворения лекарственноro
средства, например, через 1 О, 15, 20, 30, 45 и 60
минут. Для каждоro лекарства (rенерическоro и
компараторноro) необходимо изучить как мини
мум 12 единиц лекарственной формы.
Профили растворения rенерическоro и KOM
параторноro препаратов можно сравнивать с по
мощью коэффициента подобия ('). Для расчета
'2 можно использовать только данные, удовлет
воряющие следующим условиям:
дисперсия не более 20 % в первой Bpe
менной точке и дисперсия не более 1 О % в по
следующих моментах времени;
максимальное значение высвобождения
АФИ более 85 % достиrается только в 1 из Bpe
менных точек;
для вычисления '2 требуется не менее трех
временных точек (исключая нулевой момент),
для препаратов с контролируемым высвобожде
нием действующеro вещества не менее 5.
Величина '2 от 50 до 100 свидетельствует
о тождественности или эквивалентности двух
кривых, а следовательно о равнозначности xa
рактеристик iп vitro обоих препаратов. Коэффи
циент подобия ('2) должен рассчитываться с по
мощью формулы:
{[ 1 n ] O.5 )
'2 50.lg 1+п' ( Rt =т,у .100'
rдe Rt и составляют общий процент АФИ,
перешедшеro в раствор в каждый выбранный
момент времени п для компараторноro (рефе
рентноro) и rенерическоro (испытуемоro) препа
ратов соответственно.
Если компаратор и rенерическое лекар
ственное средство являются очень быстрора
створимыми, а именно не менее 85 % АФИ пере
ходит в раствор через 15 минут и раньше во всех
трех средах при использовании peKOMeHдyeMO
ro метода тестирования, то нет необходимости в
сравнении профилей.
#Ре2истрационные требования и правила проведения исследований биодоступности 1123
Можно использовать иные подходящие CTa
тистические методы сравнения профилей раст
ворения при условии, что используются одина
ковые rраницы эквивалентности (максимальное
различие между профилями в 1 О %), например,
с помощью анализа линейной реrрессии коли
чества вещества (в процентах), растворенноro
к определенным моментам времени, с помощью
статистическоro сравнения пара метров функции
Вейбулла (Weibu/l 'ипсtiоп).
9.2.1. Критерии растворения для приме-
нения процедуры «биовейвера», основанной
на БСК, в соответствии со свойствами АФИ,
для полной замены испытаний фармакоки-
нетической биоэквивалентности
Процедуру «биовейвера» в отношении TBep
дых пероральных лекарственных форм с HeMeд
ленным высвобождением можно допустить при
следующих условиях.
1. Лекарственные формы с АФИ, являющи
мися хорошорастворимыми, хорошо проника
ющими (Класс I БСК) и быстрорастворимыми,
MOryT проходить процедуру «биовейвера», OCHO
ванную на БСК, при условии, что:
(i) лекарственное средство является дo
казано быстрорастворимым (как определено
в 9.1.2.2), а профиль растворения rенериче
скоro средства является подобным с профи
лем компараторноro препарата при значениях
рН 1,2, 4,5 и 6,8 при использовании прибора
с лопастной мешалкой при частоте вращения
75 об'мин или прибора с вращающейся KOp
зинкой при частоте вращения 100 об'мин (как
определено в разделе 9.2), а также COOTBeT
ствует критерию подобия профиля paCTBope
ния 50 (или равнозначной статистической
оценке );
(ii) если и компараторные, и rенерические
лекарственные средства признаются очень бы
строрастворимыми (как определено в 9.1.2.1),
то они рассматриваются как равнозначные без
необходимости в сравнении профилей.
2. Лекарственные средства, содержаЩl1е
АФИ, относящиеся к Классу 111 БСК, если reHe
рическое и компараторное лекарственные cpeд
ства являются очень быстрорастворимыми (не
менее 85 % растворения через 15 минут при зна
чении рН 1,2, 4,5 и 6,8) при использовании при
бора с лопастной мешалкой при частоте Bpa
щения 75 об'мин или прибора с вращающейся
корзинкой при частоте вращения 100 об'мин (как
определено в разделе 9.2).
З. Лекарственные формы с АФИ, являю
щимися хорошорастворимыми и плохопроника
ющими (Класс 111 БСК), MOryT проходить процР.
дуру «биовейвера», если выполняются все кри
терии (a r), перечисленные в разделе 9.2, и дo
полнительно документально подтвержден факт
изучения корреляции «риск/польза» относитель
но степени, места и механизма всасывания для
данноrо состава лекарственноro средства.
Риски получения ошибочноro вывода о
процедуре «биовейвера» требуют критическо
ro изучения, если степень абсорбции низкая
(особенно при значениях '2<50 %), если места
всасывания оrраничены верхним отделом ЖКТ
и/или если механизм абсорбции связан с уча
стием переносчиков или является конкурентным
процессом. Korдa применимы эти варианты, по
требуется также тщательно проверить исполь
зованные вспомоrательные вещества в части их
качественноro и количественноro состава: чем
больше отклонение от состава компаратора,
тем выше риск неправомерноro применения за
ключения о процедуре «биовейвера». .
Если предполаrается, что риск принятия
ошибочноro решения о процедуре «биовейве
ра» и ассоциированные с ним риски для здо
ровья населения и для конкретных пациентов
являются приемлемыми, то rенерическое лекар
ственное средство проходит процедуру «био
вей вера», основанную на БСК, при условии, что
компаратор и rенерическое лекарственное cpeд
ство являются очень быстрорастворимыми (не
менее 85 % растворения через 15 мину как
определено в 9.1.2.1).
4. Лекарственные формы с АФИ с хорошей
растворимостью при значении рН 6,8, но не при
рН 1,2 или 4,5, и с хорошей проницаемостью
(данному условию удовлетворяют некоторые co
единения из класса слабых орrанических кислот,
относящиеся к Классу 11 БСК) считаются приroд
ными для процедуры «биовейвера», основанной
на БСК, при условии, что:
(i) выполняются описанные в разделе 9.2
критерии (б r);
(ii) АФИ обладает хорошей проницаемостью
(т. е. абсорбированная доля составляет 85 % и
больше) и соотношением «доза'растворимость»
250 мл и менее при значении рН 6,8;
(iii) rенерическое лекарственное cpeд
ство является быстрорастворимым (85 % через
30 минут и меньше) в буфере с рН 6,8 при ис
пользовании процедуры тестирования, соrласу
ющейся с разделом 9.2;
(iv) демонстрирует при трех значениях рН
(1,2, 4,5 и 6,8) подобие профилей растворения,
исходя из количественноro определения '2 или
равнозначной статистической оценки, профилям
препарата сравнения.
В отношении rенерических лекарственных
средств, содержащих АФИ Класса 11 с COOTHO
шением «доза'растворимость>} 250 мл и меньше
при рН 6,8, следует дополнительно критически
изучить вспомоrательные вещества в части их
качественноrо и количественноro состава, Ha
пример, поверхностно активных веществ, в pe
цептуре. Если Ста> имеет критическое для Te
рапевтической эффективности АФИ значение,
может признаваться недопустимым риск полу
чить ошибочный вывод о применении процедуры
«биовейвера» и ассоциированные с ним уrрозы
здоровью населения и KOHKpeTHoro пациента.
rосударственная фармакопея Республики Беларусь
1124
9.3 ПРОЦЕДУРЫ «БИОВЕЙВЕРА»,
ОСНОВАННЫЕ НА ПРОПОРЦИ
ОНАЛЬНОСТИ ДОЗИРОВКИ
ЛЕКАРСТВЕННО,О СРЕДСТВА
При проведении испытаний фармакокине
тической биоэквивалентности на человеке для
одной дозировки лекарственноro средства, pe
rистрация друrих дозировок rенерическоrо ле
карственноro средства может рассматривать
ся на основании оценки профилей кинетики
растворения, если лекарственные средства
имеют пропорционально близкие составы.
9.3.1. Пропорционально близкие лекар-
ственные средства
Пропорционально близким лекарствен
ным средствам можно дать два определения,
исходя из величины дозировки.
Все активные и неактивные компонен
ты присутствуют в точно таких же пропорци
ях в различных дозировках (например, Ta
блетка с дозировкой 50 Mr содержит ровно
половину от всех активных инеактивных
компонентов таблетки с дозировкой 100 Mr,
и их в два раза больше, чем в таблетке с дo
зировкой 25 Mr).
В случае высокоактивных АФИ, если
количество АФИ в единице лекарственной
формы относительно мало (до 1 О Mr на едини
цу дозирования) и общая масса единицы ле
карственной формы для всех дозировок OCTa
ется практически одинаковой (отклонение в
пределах :t10 % от всей массы), те же самые
неактивные вспомоrательные вещества ис
пользуются для всех дозировок в одинаковых
количествах, а изменение в активности лекар
ственноro средства достиrается по существу
путем изменения только объема АФИ.
9.3.2. Процесс отбора для процедуры
«биовейвера», основанной на пропорци
ональности дозировки препарата
Можно применить процедуру реrистрации
«биовейвер» для линейки дозировок лекар
ственной формы данноro rенерическоrо ле
карственноro средства при выполнении всех
следующих условий:
в исследованиях iп vivo на человеке
подтверждено, что данное rенерическое ле
карственное средство с данной одной из дози
ровок биоэквивалентно препарату сравнения
с соответствующей дозировкой;
прочие последующие дозировки reHe
рическоrо лекарственноro средства являются
пропорционально подобными по составу изу
ченной дозировке;
профили растворения прочих последу
ющих дозировок проявляют, исходя из взаи
мосвязи «процент высвобождения время»,
сходство с профилями растворения дозиров
ки, изученной в испытаниях биоэквивалентно
сти на человеке.
Как и в случае процедуры «биовейвера»,
основанной на БСК, процедура, основанная
на пропорциональности дозировки препара
та, должна применяться только Torдa, Korдa
есть допустимое равновесие соотношения
«польза'риск» В том, что касается здоровья
населения и опасности для конкретноro паци
ента, как указано в разделе 9.2.
9.3.3. Сравнение профилей paCTBope
ния для процедур «биовейвера», OCHOBaH
ных на пропорциональности дозировки
препарата
Так же, как и для процедуры «биовейве
ра», основанной на БСК, для сравнения про
филей растворения двух лекарственных
средств можно использовать модельно неза
висимый математический подход (например,
определение (2)' Профиль растворения двух
лекарственных средств, rенерическоro (испы
туемоro) и компараторноrо (референтноro),
необходимо измерять в одинаковом режиме
тестирования.
Моменты отбора образцов растворения
для профилей rенерическоro и компараторно
ro лекарственных средств должны быть теми
же самыми:
для лекарственных средств с немедлен
ным высвобождением 1 О, 15, 20, 30, 45 и 60
минут;
для лекарственных средств с продлен
ным на 12 часов высвобождением 1, 2, 4, 6
и 8 часов;
для лекарственных средств с продлен
ным на 24 часа высвобождением 1, 2, 4, 6,
8 и 16 часов.
После растворения 85 % компараторно
ro препарата следует учитывать только один
момент отбора проб. Величина '2' равная от
50 до 100, свидетельствует об эквивалентно
сти двух кривых (отклонение не более 10 %) и,
следовательно, о равнозначности характери
стик обоих препаратов iп vitro. При этом можно
использовать средние показатели, если коэф
фициент вариации не превышает 20 % в самой
ранней временной точке и 1 О % в остальных
временных точках.
9.3.3.1. Таблетки с немедленным выcвo
божденuем
Процедура применима для различных дo
зировок rенерическоrо лекарственноrо cpeд
ства, если они изroтавливаются одним произ
водителем на одних и тех же лроизводственных
площадях, rдe:
(i) все дозировки являются пропорци
онально одинаковыми по составу;
(ii) с хотя бы одной концентрацией лекар
ственноro средства было проведено COOTBeT
ствующее исследование биоэквивалентности
(обычно с самой высокой дозировкой, если
только из соображений безопасности не выби
рается более низкая концентрация);
#Реаистрационные требования и правила проведения исследований биодоступности 1125
(iii) профили растворения для различных
концентраций признаны подобными.
Так же, как и в случае процедуры «биовей
вера», основанной на БСК, если во всех дози
ровках при использовании всех сред paCTBO
рения через 15 минут высвобождается 85 % и
более заявленноrо содержания АФИ, как peKO
мендуется в разделе 9.2, сравнение профилей
посредством определения величины '2 не яв
ляется необходимым.
9.3.3.2. Таблеткu u капсулы с модuфuцu
рованным высвобожденuем
Для таблеток с модифицированным BЫ
свобождением при условии, что rенерическое
лекарственное средство представлено в той
же самой лекарственной форме, но в отличной
дозировке, а также является пропорциональ
но подобным по еro активным инеактивным
инrредиентам и обладает тем же самым Mexa
низмом модифицированноro высвобождения,
к процедуре «биовейвера» может допускаться
более низкая дозировка при условии, что она
демонстрирует подобный профиль paCTBO
рения, значение fi .50, в рекомендуемом для
препарата с модифицированным высвобожде
нием режиме тестирования: тест изучения ки
нетики растворения в кислотной среде (рН 1,2)
в течение 2 часов со сменой среды при рН 6,8.
В отношении капсул с модифицированным
высвобождением, rде различные дозировки
получаются исключительно путем реrулирова
ния количества пеллет (rранул), содержащих
АФИ, достаточным условием для процеду
ры «биовейвера» является подобие профиля
растворения новой (более низкой) дозировки
профилю зареrистрированной дозировки (Be
личина fi :50) в рекомендуемом для препа
рата с модифицированным высвобождением
режиме тестирования (см. выше).
9.3.3.3. Капсулы с пеллетамu (2рануламu)
с продленным высвобожденuем
Для капсул с пеллетами с продленным
высвобождением, если различные KOHцeH
трации получаются исключительно путем pe .
rулирования количества пеллет, содержащих
АФИ, достаточным для применения процеду
ры «биовейвера», основанной на пропорци
ональности дозировки препарата, является
сравнение профиля растворения (величина
fi 50) при одном из рекомендуемых условий
тестирования.
9.3.3.4. Таблеткu с продленным выcвo
божденuем
Для таблеток с продленным высвобожде
нием, если rенерическое лекарственное cpeд
ство представлено в той же caMo.;j лекарствен
ной форме, но в отличной дозировке, а также
является пропорционально подобным по ero
активным инеактивным инrредиентам и обла
дает тем же самым механизмом высвобожде
ния лекарственноro вещества, более низкая
дозировка может допускаться к процедуре
142.3_.1712.
«биовейвера» при условии, что в peKOMeHДO
ванном способе тестирования она демонстри
рует в буферах с тремя уровнями рН (значение
рН от 1,2 до 7,5) подобные профили paCTBope
ния с '2 50.
9.4. ПРОЦЕДУРbI «БИОВЕЙВЕРА»
В ОТНОШЕНИИ МАСШТАБИРОВАНИЯ
ПРОИЗВОДСТВА
ИПОСТРЕrИСТРАЦИОННЬ
ИЗМЕНЕНИЙ В изrОТОВЛЕНИИ
ЛЕКАРСТВЕнноrо СРЕДСТВА
В настоящих правилах в первую очередь
сформулированы требования для реrистрации
rенерических лекарственных средств. Следует
отметить, что для подтверждения подобия каче
ства препарата и характеристик эффективности
в определенных условиях, исходя из незначи
тельных изменений в технолоrии изroтовления
или производственных процессах, произошед
ших после roсударственной реrистрации лекар
cTBeHHoro средства, может использоваться Te
стирование на растворение in vitro.
10. КЛИНИЧЕСКИ ЗНАЧИМЫЕ
КОЛЕБАНИЯ БИОДОСТУПНОСТИ,
ОБУСЛАВЛИВАЮЩИЕ ОТКАЗ
В РЕrИСТРАЦИИ ЛЕКАРСТВЕнноrо
СРЕДСТВА
Новое лекарственное средство, биолоrи
ческая доступность KOTOpOro находится за пре
делами допустимых rраниц колебаний при co
поставлении с существующим лекарственным
средством, не является взаимозаменяемым. Pe
rистрация TaKoro средства с более низкой био
доступностью не может быть произведена по
соображениям эффективности. Напротив, реrи
страция лекарственноro средства с более BЫCO
кой биодоступностью (сверхбиодоступность) не
может быть разрешена из соображений безопас
ности. В этом случае возможны два варианта pe
шения проблемы:
1. Если состав лекарственноro средства со
сверхвысокой доступностью изменен таким об
разом, что он стал биоэквивалентным существу
ющему лекарственному средству, он может счи
таться взаимозаменяемым препаратом. Однако
это решение может оказаться неидеальным, по
скольку действенность лекарственных форм с
более низкой биодоступностью имеет склон
ность к изменениям.
2. Лекарственная форма с повышенной био
доступностью, в которой содержание действу
ющеro вещества снижено, может рассматривать
ся как новая (усовершенствованная) лекарствен
ная форма, но это решение обычно требует под
тверждения данными клинических испытаний.
Такое лекарственное средство не должно счи
таться взаимозаменяемым с существующим пре
паратом, и обычно оно становится эталонным
продуктом для будущих взаимозаменяемых ле
карственных средств.
1126
rосударственная фармакопея Республики Беларусь
11. ПРИЛОЖЕНИЯ
С
тах
С.
топ
С
55
АИС
АИС t
АИС,
АИС т ,55
Ае
Ае !
Ае о
Ае т ,55
t
t
тах
t,/,
MRT
f
('
("
р
т
т
Приложение 1
Обозначения u сокращения
наблюдаемый максимум или пик концентрации лекарственноro средства (или метаболи
та) в плазме, сыворотке или крови;
минимальная концентрация в плазме;
концентрация лекарственноrо вещества при стационарных (равновесных) условиях;
площадь под кривой зависимости концентрации лекарственноro средства (или метаболи
та) в плазме (или сыворотке) или в цельной крови от времени. Величина АИС может быть
отнесена к конкретному промежутку времени, например, АИС для периода от О до 12 ч изо
бражается как АИС 12 ;
АИС от нуля до последней количественно оцениваемой концентрации;
АИС от нуля до бесконечности, определяется экстраполяцией;
АИС дЛЯ интервала между приемами лекарственноro средства (т) В стационарном cocтo
янии;
степень кумулятивноro извлечения исходноro лекарственноro средства (или метаболита)
из мочи. Величина Ае может быть определена для конкретноro промежутка времени, напри
мер, Ае от О до 12 ч может быть выражена как Ае 12 ;
Ае от нуля до последней количественно оцениваемой концентрации;
Ае от нуля до бесконечности, определяется экстраполяцией;
Ае для интервала между приемами лекарственноro средства в стационарном состоянии;
время от момента приема (введения) лекарственноro средства;
время после введения лекарственноro средства, в которое отмечается С тах ;
nepиQЦ полувыведения лекарственноrо вещества из плазмы (сыворотки, цельной крови);
среднее время удержания;
относительная степень всасывания (относительная биодоступность) лекарственноro Be
щества. определяемая отношением АИСt/АИС.'R (в процентах), для испытуемоro препарата
(7) и пpenaрата сравнения (R); .
относительная степень всасывания лекарственноro вещества, определяемая отношени
ем АИС :lАИС .R (в процентах). для испытуемоro препарата (7) и препарата сравнения (R);
относительная степень всасывания лекарственноro вещества, определяемая отношением
Сma.jСт-иR (в процентах), для испытуемоro препарата (7) и препарата сравнения (R);
уровень вероятности, при котором статистическая rипотеза верна;
длительность наблюдения за концентрацией лекарственноro вещества;
интервал дозирования при мноroкратном приеме (введении) лекарственноro средства.
Приложение 2
Перечень лекарственных средств, для которых допускается проведенuе исследованuй
бuоэквuва.лентностu на крупных лабораторных жuвотных
N!! Фармакотерапев- МНН Доза тin , Mr Доза maх , Mr Т тах' Ч ТII,Ч
тическая rpynna
1. Нейролептики хлорпромазин* 25 75 2 61 сут
левомепромазин 25 50 1 3 1 78
rалоперидол 1,5 4,5 2 13 0
сульпирид 200 600 4,5 7
2. Антиконвульсанты этосуксимид 250 1 60
карбамазепин* 100 200 4 15
вальпроеваякислота 150 2 3 8 20
З. Снотворные фенобарбитал 100 10 1 2 2 10
нитразепам* 5 15 1 26
зопиклон 7,5 15 1 3 3,5 6
мидазолам 7,5 100 0,5 1, 2,5
#Ре2истрационные требования и правила проведения исследований биодоступности 1127
N!! Фармакотерапев- МНН Доза miп , Mr Доза mах , Mr Т тах' Ч Ту,'Ч
тическая rpynna
4. Миорелаксанты толперизон 50 25 0.5 1,0 1,5
баклофен 10 25 2 3 4
5. Антидепрессанты амитриптилин* I 10 25 2 7,7 9 25
!
мапротилин* i 10 20 8 43 5
6. Транквилизаторы феназепам 10 10 18
Примечание: средства, помеченные знаком ("), исследуются также по активному метаболиту.
Приложение 3
Образец формы uнформироваНН020 С02ласuя добровольца на участие в uспытанuях
бuоэквuвалентностu/бuодоступностu
Я,
паспорт: серия
NQ
выдан «
»
r.
проживающий по адресу
внимательно ознакомился с информацией о целях и правилах проведения исследования и доброволь
но соrлашаюсь участвовать в исследовании биоэквивалентности лекарственноro средства
, производства , в сравнении с лекарственным
средством
, производства
я получил полное объяснение врача исследователя относительно целей и
продолжительности исследования, а также Toro, что от меня требуется.
Я информирован о том, что на момент проведения исследования лекарственный препарат не
имеет номера roсударственной реrистрации.
Я предупрежден о том, что данное исследование может сопровождаться некоторым дискомфор
том, и В еro процессе не исключено какое либо вредное воздействие на мое здоровье или самочув
ствие.
Я получил возможность задать любые интересующие меня вопросы врачу исследователю по всем
аспектам исследования. Я понял все данные мне рекомендации и в меру моих знаний осознал полу
ченную информацию.
Я соrласен, чтобы врач исследователь обратился к моему лечащему врачу и известил последнеrо
о моем участии в исследовании. Я разрешаю моему лечащему врачу сообщить в конфиденциальном
порядке о состоянии моеro здоровья, вредных привычках и перенесенных заболеваниях врачу иссле
дователю.
Я соrласен подчиняться инструкциям, получаемым в течение исследования, добросовестно co
трудничать с врачом исследователем и немедленно сообщать ему о любоro рода нарушениях со CTO
роны моею здоровья, изменениях моеro самочувствия и информировать еro обо всех неожиданных или
необычных симптомах, коrда бы они не возникли.
Я соrласен, чтобы информация, полученная в ходе исследования, использовалась в полной мере и
передавалась в Комиссию по лекарственным средствам и Министерство здравоохранения Республики
Беларусь, а также в орrанизацию, по заказу которой проводится исследование.
Я извещен, что имею полное право в любой момент прекратить свое участие в исследовании без
необходимости обосновывать свое решение. Мой отказ от участия в исследовании не повлечет за
собой изменения отношения ко мне медицинскоro персонала.
В случае моеro решения о прекращении участия в исследовании биоэквивалентности, я обязуюсь
информировать об этом врача исследователя для тоro, чтобы предоставить ему возможность оценить
мое состояние и дать необходимые рекомендации.
Если моему здоровью или блаюполучию будет причинен ущерб, непосредС!венно связанный с моим
участием в исследовании, спонсор исследования выплатит мне компенсацию. Если виновность заказчи
ка доказана, денежное выражение этой компенсации должно устанавливаться белорусскими судебными
орrанами в соответствии с существующими нормами. Сумма компенсации может быть пересмотрена в
случае моей частичной вины в возникновении данною ущерба.
Мне была предоставлена копия данноrо информированноro соrласия. Я соrлаСЕ:Н принять участие в
исследовании.
Ф. И. О. добровольца:
Подпись
Дата
Подтверждаю, что подробно объяснил сущность. цель и возможный риск данноro исследования добровольцу.
Ф. И. О. врача исследователя: Подпись Дата
1128
rосударственная фармакопея Республики Беларусь
Приложение 4
НОМ02рамма для опредеJ енuя (JостатОЧН020 чuсла добровольцев
по результатам прс",, деНН020 uсследованuя*
!:)
,, \ъ
b CS !:)
D-CS !:)
"CS !:)
1- CS :P
,,[>; !:)
"CS !:)
€fp !:)
b
') s::p
[>; !:)
"CS
D-
'}:
!:)
,, R>
"tx
,, н
,,
ro\J
"I
'b
<-
,
D-
,,
1-
1-
"Ь
"D-
,,1-
,,
ro
0.05
0.0
0.1
со 0.2
::r
s
:I:
,." 0.3
со
Q.
0.4
:r
:I:
со 0.5
QJ
О
,."
0.6
Q.
со
r:r 0.7
:r
со
UO.8
0.9
1.0
1.1
1.2
0.995
0.97
0.95
0.9
0.85
n
s
::J
D.J
0.05
Уровень значимости
.по. Gore. Мтап Statistics in Practice. Brit. Med. Assoc.London, 1982. Р. 7
Пример: При проведении исследования
биоэквивалентности на 18 добровольцах по
казатели АUС ! для испытуемоro препарата co
ставили (288,53::t55,86) нr'ч'мл, а для препара
та сравнения (259,93::t40,78) нr,ч'мл.
Таким образом, ожидаемое выявляемое
отличие составляет:
IAUC',R Аис"т I = 1259.93 288,5з1 = 28,6,
стандартное отклонение для этоro отличия:
CYR + СУТ 55,86 + 40,78 = 4832
2 2 ' ,
стандартизированная разница, таким обра
зом, составит:
С д выявл. отличие 28,6 О 59
тан . разница= = , .
ст. отклонение 48,32
Выбирая 0,8 за значение мощности (силы),
следует соединить значения 0,59 слева и 0,8
справа. Линия пересечет диаrональ для а=0,05
на числе 90. Таким образом, 90 минимальное
число пациентов, которое должно быть включено
в исследование для выявления данноro различия.
Для расчета мощности настоящеro исследо
вания необходимо соединить линией CTaHдap
тизированную разницу 0,59 слева и реальное
число участников 18 человек для настояще
ro исследования на диаroнали. Продолжение
линии пересечет шкалу для мощности (силы) в
точке 0,22. Таким образом, мощность настояще
ro исследования 22 %.
#Ре2истрационные требования и правила проведения исследований бuодоступности 1129
Приложение 5
Дополнительная литература
1} Соловьев, в.н. Фармакокuнетuка. / В.Н Соловьев, А.А. Фuрсов, В.А. Фuлов М: Медицина,
1980. 423 с.
2} Атidоп. G.L. А theoretica' basis (or а biopha,maceutic drug с'аssifiсаtiоп: The соrrе'аtiоп о( in vitro
drug product disso'ution апd in vivo bioavailability / Н Lеппетаs, v.P. Shah, J.R. C,ison // Pharm. Res.
1995. Vo'. 12. Р. 413 420.
3) Stаgпi, G. Bioavailiability assessment (rom pha,maco'ogic data: Method апd сliпiса' еvа'uаtiоп / G.
Stagni, А.ММ Shepherd, Уапjиап ии. WR. Gillespie // J. Рhаrтасоkiпеt. Biopha,m. 1997. Vo'. 25,
NQ3. Р. 349 362.
4) Stеiпijапs, V. W Bioequiva'ence Assesment. Methods апd Applications / V. W Stеiпijапs // 'пt. J. Сliп.
Pharmaco'. Ther. & Toxico'. Vo'. 30 (Supp'. 1). 1992. Р. 1 66.
5} Steinijans, V. W Bioequiva'ence Studies: Sing'e vs Mu1tip'e Dose / V. W. Steinijans, R. Sauter,
HG. Jonkman [et а'.] // 'nt. J. Сliп. Pharmaco'. Ther. & Toxico'. 1989. Vo'. 27. Р. 261 266.
6} Уи, L.X. Biopha,maceutics С'аssifiсаtiоп System: The sсiепti(iс basis (о, biowaive, ехtепsiопs /
LX Уи, G.L. Amidon, J.E. РоЩ Eet а'.] // Pharт. Res. 2002. Vo'. 19. Р. 921 925.
7) Biowaiver monographs re(erences: examp'es // J. Pha,m. Sci. 2004. Vo'. 93. NQ 8. Р. 1 94
1956; J. Pha,m. Sci. 2005. Vo'. 94, NQ 7. Р. 1389 1395; J. Pharm. Sci. 2005. Vo'. 94, NQ 8.
Р. 1611 1617.
8} CPMP/EWP/QWP/1401/98 Rev.1/Corr Guidапсе оп the 'пvеstigаtiоп о( Вiоеquivа'епсе. 2010.
9} Diletti, Е. Samp'e size dеtеrтiпаtiоп (or Ыоеquivа'епсе аssеssтепt Ьу теапs о( confidence interva's /
Е. Di'etti, D. Hauschke, V. W Stеiпijапs // /пt. J. С'iп. Pharmaco'. Ther. & Toxico/. 1 991. Vo'. 29. Р. 1 8.
10) Guideline (or bioequiva'ence studies о( generic products (Jарап). httр:l/www.пihs.gо.jр/drug/Ье
guide( е }/Gene,ic/be97E.htm'
11) Hauschke, D. Distribution (,ee Procedure (orthe Statistica' Ana/ysis о( Вiоеquivа'епсе Studies/ D. Hauschke,
V. W Stеiпijапs, Е.А. Di'etti // 'пt. J. С/iп. Pharmaco'. Ther. & Toxico'. 1 990. Vo'. 28. Р 72 78.
12) HHS/FDA Gиidапсе (or 'ndustry: Waiver о( iп vivo bioavailability and Ыоеquivа'епсе studies (or
immediate release so/id ora' dosage (orms based оп а biopharmaceutics c'assification system. Aug. 2000.
http://www.(da.gov/cder/guidance/index.htm
13) Shah, v.P. 'П vitro dissо'utiоп profile сотра,isоп. Statistics and Ana'ysis о( the Simila,ity (actor, (2/
v.P. Shah, У. Tsong, Р. Sathe, J.P. ии // Pharm. Res. 1998. Vo'. 15. Р. 889 896.
14}Jаwiеп, W Оп сопtiпиitу о( iпtеgrаtiоп methods (or АUС: А note / W Jawien // J. Рhаrтасоkiпеt.
Biopha,т. 1998. Vo'. 26, NQ 1. Р. 12 130.
15) LiпdепЬеrg, М С'аssifiсаtiоп о( o,ally administe,ed d,ug оп the Wo,'d Hea'th О'gапizаtiОП mode' /ist
о( essentia' medicines ассоrdiпg to the biopharтaceutics c'assi(ication systeт / М LiпdепЬеrg, К S. орр,
D,essman J.B. // Eur. J. Pha,maceutics and Biopharm. 2004. Vol. 58. Р. 26 278.
16) Lund, R.E. TabIes (or ап App,oximate Test (or Outlie,s iп Linear Mode's / R.E. Luпd // Тесhпотеtпсs.
1975. Vo'. 17. Р. 473 476.
17) Midha, к-к. Соттепtаry: The Ro'e о( Metabolites in Вiоеquivа'епсе / К-К- Midha, MJ. Rаwsоп,
J. W Hubba,d // Pharmaceutica' Resea,ch. 2004. Vo'. 21. Р. 1 331 1 344.
18} Kasim, N.A. Molecular Properties о( WHO Essentiaf Drugs апd Рrovisiопаf Biopharmaceutica'
С'аssifiсаtiоп / N.A. Kasim, М Whitehouse, С. Ramachandran [et а']. // Мо'еси'а, Pharmaceutics. 2004.
Vo'. 1. Р. 8 96.
19} Moore, J.W Mathematica' comparison о( curves with ап emphasis оп in vitro dissofution profiles /
J. W Moore, НН F'аппеr // Pharm. Tech. 1996. Vo'. 20, Мl 6. Р. 64--------74.
20} Sсhui,тапп, D.J. А comparison о( the two one side tests procedure and the power approach (о,
аssеssiпg the еquivа'епсе о( average bioavaifabifity / D.J. Schuirmaпп // J. Рhаrтасоkiпеt. Biopha,m.
1987. Vo'. 15. Р. 657 680.
21) Stеiпijапs, V. W Statistica' Апа'уsis о( Bioavaifability Studies: Paramet,ic aпd Nonpa,amet,ic
Сопfidепсе 'ntervafs / V. W Stеiпijапs, Е. Difetti // Eur. J. Cfin Pharmacof. 1983. Vo'. 24. Р. 127 136.
22} Tothfa'usi, L. Limits for sca'ed average Ыоеquivаlепсе of high'y va,iabIe drugs апd d,ug p,oducts /
L. Tothfa'usi, L. Епdrепуi// Pharmaceutica' Research. 2003. Vo'. 20. Р. 382 389.
23} Tothfafusi, L. Evafuation of bioequivafence of high/y variabIe drugs / L. Tothfalusi, L. Епd,епуi,
К-К- Midha, MJ. Rawson, J. W Hubbard // Pharmaceutica' Research. 2001. Vo'. 18. Р. 728------733.
24} Waive, о( iп vivo Bioavai'ability апd Bioequiva'ence Studies (о, 'mmediate Re'ease Solid О,а/ Dosage
Forms Based оп а Biopharтaceutics C'assification System. и S. Department of Hea,th and Нuтап Services
Food апd D,ug Administartion, Сепtе, (or D,ug Eva'uation and Resea,ch. Aug. 2000.
143 За" 1712
1130
rосударственная фармакопея Республики Беларусь
01/2013:2034
СУБСТАНЦИИ
ДЛЯ ФАРМАЦЕВТИЧЕскоrо
ИСПОЛЬЗОВАНИЯ
Co,pora ad usum pharmaceuticum
ОПРЕДЕЛЕНИЕ
Субстанции для фармацевтическоrо ис
пользования (субстанции) это орrанические
или неорrанические вещества, которые исполь
зуются как активные субстанции (действующие
вещества. фармацевтические субстанции) или
вспомоrательные вещества при производстве
лекарственных средств, предназначенных для
медицинскоro применения. Они MOryт быть при
родноro происхождения или полученны путем
экстракции, ферментации или синтеза.
Эта общая статья не распространяется
на лекарственное растительное сырье, лекар
ственное растительное сырье для roмеопа
тических лекарственных средств, экстракты,
матричные настойки для roмеопатических ле
карственных средств, на которые распростра
няются отдельные общие статьи (Лекарствен
ное растuтельное сырье, Лекарственное
растuтельное сырье для 20меопатuческuх
лекарственных средств, Экстракты, Ma
трuчные настойкu для 20меопатuческuх ле
карственных средств). Общая статья не pac
пространяется на сырье для roмеопатических
лекарственных средств, за исключением тех
субстанций, которые описаны в неroмеопати
ческой части Фармакопеи.
Если субстанция, на которую в Фармако
пее отсутствует частная статья, используется в
составе лекарственноrо средства, изroтавлива
емоro для специальных нужд отдельных паци
ентов, необходимость ее соответствия данной
общей статье принимается на основании оценки
рисков, при которой учитывают качество доступ
ной субстанции и ее предполаrаемое примене
ние.
Если лекарственные средства производят с
использованием субстанции человеческоrо или
животноro происхождения, должны выдержи
ваться требования статьи 5. 1. 7. Вирусная безо
пасность.
Субстанции MOryT применяться в том виде,
в котором они находятся, или как исходные Ma
териалы для производства лекарственных
средств. В зависимости от состава лекарствен
ных средств, некоторые субстанции MorYT ис
пользоваться как действующее или вспомоrа
тельное вещество. Твердые субстанции MorYT
быть уплотненные, покрытые оболочкой, в виде
rранул, измельченные до необходимоro разме
ра или обработанные друrим способом. Част
ная статья применима для субстанции, обра
ботанной с использованием вспомоrательных
веществ, только в том случае, Korдa такая обра
ботка указана в разделе «Определение» част
ной статьи.
Субстанцuu специальной квалuфикацuu.
Если нет друrих указаний в частной статье, суб
станции для фармацевтическоro использования
предназначены для использования людьми и
имеют соответствующее качество для производ
ства всех лекарственных форм, для которых они
MOryT быть использованы.
Полuморфuзм. В частных статьях обычно
не указывают кристаллическую или аморфную
форму, если только это не влияет на биодо
ступность. Если нет друrих указаний в частной
статье, все формы субстанций для фармацев
тическоro использования должны выдерживать
требования частных статей.
ПРОИЗВОДСТВО
Субстанции производят в условиях, которые
обеспечивают качество и соответствие требова
ниям частной статьи или утвержденной HopMa
тивной документации.
При контроле примесей в субстанциях для
фармацевтическоro использования применяют
положения общей статьи 5. 10.
Независимо от тоro, указано или не указано
в частной статье, в приведенных ниже случаях
субстанции для фармацевтическоro использо
вания должны отвечать соответствующим Tpe
бованиям:
рекомбинантные белки или друrие суб
станции, полученные при помощи rенной моди
фикации, должны соответствовать требованиям
общей статьи Продукты теХНОЛ02UU рекомбu
нантной ДНК;
субстанции, полученные из животных, BOC
приимчивых К инфекционному ryбчатому эн
цефалиту, за исключением экспериментально
вызванных случаев, должны соответствовать
требованиям общей статьи Продукты, которые
М02ут быть переносчикамu 2убчатой энцефа
лопатuu жuвотН020 проuсхожденuя;
субстанции, полученные в результате про
цесса ферментации, независимо от тоro, моди
фицируются или не модифицируются исполь
зованные микроорrанизмы обычным способом
или технолоrией рекомбинантной ДНК (p ДHK),
должны соответствовать требованиям общей
статьи Продукты ферментацuu.
Растворители, используемые в производ
стве субстанций, должны быть соответствующе
ro качества. Также следует принять во внимание
токсичность растворителей и их остаточные KO
личества (5.4). Вода, используемая в производ
стве субстанций, должна быть соответствующе
ro качества.
Субстанции, изroтовленные или обработан
ные с целью получения необходимой формы
или качества, должны выдерживать требования
частных статей. Для контроля показателей, KO
торые MorYT влиять на приroдность субстанции
Субстанции для фармацевтическоао использования
1131
Таблица 2034. 1
Учuтыванuе, uдентuфuкация и квалификацuя ОР2анuческuх прuмесей в активных субстанциях.
Применение Максимальная Учитываемый Предел для иден I Предел для
дневная доза предел тификации квалификации
> 0,10 % или сута+ > 0,15 % или cyт
Для медицинскоro 2 r/cYT > 0,05 % ная доза > 1,0 Mr ная доза > 1,0 Mr
применения (в зависимости (в зависимости
от тоro, что ниже) от тоro, что ниже)
Для медицинскоro > 2 r/cYT > 0,03 % > 0,05 % > 0,05 % I
применения
Таблица 2034. 2
Учuтыванuе, uдентuфuкацuя u квалuфuкацuя ОР2анuческuх прuмесей в пептидах,
полученных с помощью хuмичеСК020 сuнтеза
Учитываемый предел Предел для идентификации Предел для квалификации
> 0,1% > 0,5% > 1,0%
и таким образом на показатели приroтовленных
из них лекарственных форм, должны быть опи
саны необходимые функционально обусловлен
ные характеристики.
Измельченные субстанциu MOryT обрабаты
ваться для получения необходимой степени из
мельчения (2.9.35).
Уплотненные субстанцuu получают для YBe
личения размера частиц или для получения частиц
специфической формы иlили получения субстан
ций с более высокой насыпной плотностью.
Покрытые оболочкой активные субстан
ции состоят из частиц действующею вещества,
по крытых одним или несколькими вспомоrатель
ными веществами.
Транулuрованные активные субстанциu
представляют собой частицы специфическоro
размера и/или формы, изrотовленные из дей
ствующеro вещества путем прямой rрануляции
или rрануляцией с использованием одноrо или
нескольких вспомоrательных веществ.
Если субстанции обрабатывают вспомоrа
тельными веществами, последние должны BЫ
держивать требования соответствующей част
ной статьи или, если частная статья OTCYТCTBY T,
соответствующей утвержденной нормативной
документации (спецификации).
Если субстанции обрабатывают вспомоrа
тельными веществами для получения, например,
покрытых оболочкой или rранулированных суб
станций, процесс должен выполняться в COOТBeT
ствии с требованиями Надлежащей производ
ственной практики и обработанные субстанции
рассматриваются как промежуточные продукты
производства лекарственною средства.
ОПИСАНИЕ (СВОЙСТВА)
Положения, приведенные в данном разделе
(например, растворимость или температура раз
ложения), не являются обязательными требова
ниями. Они носят информационный характер.
Если субстанция обладает полиморфиз
мом, это должно быть указано в разделе «Опи
сание (свойства)>> с целью учета обозначенной
характеристики при производстве лекарствен
ных средств.
ИДЕНТИФИКАЦИЯ
В некоторых частных статьях есть подразде
лы «Первая идентификация» и «Вторая иденти
фикация». Испытания, описанные в подразделе
«Первая идентификация», MOryT быть исполь
зованы во всех случаях. Испытания, описанные
в подразделе «Вторая идентификация», MOryT
быть использованы в аптеках, если есть ДOKa
зательства Toro, что данная серия субстанции
была сертифицирована на соответствие всем
друrим требованиям частной фармакопейной
статьи.
В некоторых частных статьях для первой
идентификации при водятся ДВа или более
вида испытаний, которые являются эквива
лентными и MorYT использоваться независимо
друr от друrа. Один или более из этих рядов
обычно содержат перекрестные ссылки на ис
пытания, приведенные в разделе «Испытания»
частной статьи. Это может быть использовано
для облеrчения работы аналитика, проводяще
ro идентификацию и предписанные испытания.
Например, один ряд испытаний для идентифи
кации имеет ссылку на определение знантио
мерной чистоты, в то время как в друroм ряду
проводится определение удельноro вращения:
оба ряда преследуют одну и ту же цель под
твердить присутствие необходимоrо энантио
мера.
ИСПЫТАНИЯ
Полиморфизм (5.9). Если кристалличе
ская или аморфн::!я форма субстанции накла
дывает оrраничения на ее использование в
лекарственных средствах, то при роду специ
фической кристаллической или аморфной
формы идентифицируют, ее морфолоrию тща
тельно контролируют и обозначение указыва
ют на этикетке.
1132
rосударственная фармакопея Республики Беларусь
Сопутствующие примеси. Если нет друrих
указаний в частной статье или не обосновс'но и
не утверждено иное, орrанические примеси в 3K
тивных субстанциях должны учитываться, rдe
это возможно, идентифицироваться и квалифи
цироваться, как указано в таблице 20З4. 1 и Ta
блице 20З4. 2 (для пептидов, полученных с по
мощью химическоro синтеза).
Для примесей, которые характеризуют
ся сильной токсичностью или непредвиденным
фармаколоrическим действием, устанавливают
индивидуальные предельные значения.
Если в частной статье отсутствует методика
определения новой примеси, необходимое ис
пытание должно быть разработано и включено в
нормативную документацию.
Эти требования не распространяются на
биолоrические и биотехнолоrические продук
ты, олиroнуклеотиды, радиофармацевтические
препараты, продукты ферментации и получен
ные из них полусинтетические продукты или на
сырье животноro или растительноro происхож
дения.
Остаточные количества орrанических
растворителей. Содержание реrламентируют
в соответствии с требованиями статьи 5.4, ис
пользуя общий метод статьи 2.4.24 или друrие
подходящие методы. Если проводят количе
ственное определение остаточных количеств
орrанических растворителей и не проводят ис
пытание «Потеря В массе при высушивании»,
содержание остаточных количеств орrанических
растворителей принимают во внимание при pac
чете количественноro содержания субстанции,
удельноro вращения и удельноro лоrлощения.
Микробиолоrическая чистота. При необ
ходимости контроля микробиолоrической чи
стоты, критерии приемлемости при водятся в
частной статье. В таблице 5.1.4. 2. Критерии
приемлемости для микробиолоrической чистоты
нестерильных субстанций для фармацевтиче
скоro использования статьи 5. 1.4. Мuкробuоло
2ическая чuстота нестерuльных лекарствен
ных средств и фармацевтuческuх субстанцuй
приведены рекомендации по микробиолоrиче
ской чистоте субстанций, подверженных микро
биолоrическому заражению. В зависимости от
природы субстанции и предполаrаемоro исполь
зования MOryт быть установлены друrие нормы.
Стерильность (2.6.1). Если субстанция
предназначена для производства стерильных
лекарственных средств, которые не подверrают
ся дальнейшей стерилизации, или субстанция
обозначена «стерильно», она должна выдержи
вать испытание на стерильность.
Бактериальные эндотоксины (2.6.14). Если
субстанция обозначена как свободная от бактери
альных эндотоксинов, она должна выдерживать
требования на бактериальные эндотоксины. Пре
дельная концентрация эндотоксинов и метод ис
пытания (если это не метод rелеобразования А)
указывают в частной статье. Предельную KOHцeH
трацию рассчитывают в соответствии с разделом
«Испытание на бактериальные эндотоксины. Pe
комендации» общей статьи 2.6.14. Бактерuаль
ные эндотоксuны, если меньшая концентрация
не установлена на основании результатов анали
за промышленных серий или требований уполно
моченноrо opraHa. Если проводят испытание на
бактериальные эндотоксины, испытание на пиро
reHHoCТb не требуется.
Пироrенность (2.6.8). Если испытание на
пироrенность более необходимо, чем испыта
ние на бактериальные эндотоксины, или если
субстанция обозначена как «апироrенная», она
должна выдерживать испытание на пироrен
ность. Критерии определения и метод испыта
ния указывают в частной статье. На основании
соответствующей валидации испытаний на бак
териальные эндотоксины и на пироrенность ис
пытание на бактериальные эндотоксины может
заменить испытание на пироrенность.
Дополнительные определения. Контроль
дополнительных определений (например, фи
зические характеристики, функционально обус
ловленные характеристики) может быть необ
ходим для KOHKpeTHoro производственноro про
цесса или конкретноro состава лекарственно
ro средства. При производстве лекарственных
средств для парентеральноro применения или
друrих лекарственных форм MOryт использо
вать субстанции разных квалификаций (таких
как «стерильно» , «не содержит бактериальных
эндотоксинов», «апироrенно»), И COOTBeTCTBY
ющие требования должны быть конкретизирова
ны в частных статьях.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
В субстанциях проводят количественное
определение подходящим аналитическим MeTO
дом, если нет друrих указаний в частной статье.
МАРКИРОВКА
#Как правило, требования к маркировке YCTa
навливаются уполномоченным opraHoM.# Таким
образом, требования, приведенные в разделе
«Маркировка», не являются полными. Они ориен
тированы в первую очередь на фармакопейные
цели, и обязательными являются только те по
ложения, которые необходимы для подтвержде
ния соответствия продукта требованиям статьи.
Вся друrая Иl-!формация носит рекомендатель
ный характер. В случае, Korдa в Фармакопее ис
пользуется термин «этикетка», соответствующая
информация может быть обозначена на контей
нере, на упаковке, в листке вкладыше, утверж
денном уполномоченным opraHoM, или сертифи
кате анализа, сопровождающем продукт.
Экстракты
1133
rде это необходимо, на этикетке указывают,
что субстанция:
предназначена для специфическоrо ис
пользования;
имеет определенную кристаллическую
форму;
имеет специальную степень измельчения;
уплотненная;
покрыта оболочкой;
rранулированная;
стерильная;
не содержит бактериальных эндотоксинов;
апироrенна;
содержит скользящие вещества.
rде применимо, на этикетке дополнительно
указывают:
степень rидратации;
наименование и концентрацию всех вспо
моrательных веществ.
01/2013:0765
ЭКСТРАКТЫ
Extracta
ОПРЕДЕЛЕНИЕ
Экстракты продукты жидкой (жидкие
экстракты и настойки), мяrкой (rycTbIe экстрак
ты) или твердой (сухие экстракты) консистен
ции, получаемые из лекарственноro раститель
ноro сырья или животноro материала, которые
обычно используются в высушенном виде.
Лекарственные средства, в производстве
которых использовались экстракты животноro
происхождения, должны выдерживать требова
ния статьи 5.1.7. Вuрусная безопасность.
Известно несколько разных типов экстрак
тов. Стандартизованные экстракты это экс
тракты, в которых содержание компонентов с
известной терапевтической активностью pery
лируется в определенных пределах. CTaHдap
тизация достиrается смешиванием экстракта
с инертным материалом или друrими сериями
экстракта. Количественные экстракты это
экстракты, имеющие определенный состав. Их
стандартизацию проводят смешением разных
серий экстракта. Друrие экстракты характери
зуются процессом их производства (в зависи
мости от вида используемоro лекарственноro
растительноro сырья или животноro материала,
растворителя, условий экстракции) и их специ
фикациями.
ПРОИЗВОДСТВО
Экстракты изroтавливают соответствующи
ми методами, используя спирт или друroй под
144 Зак 1712
ходящий растворитель. Разные серии лекар
cTBeHHoro растительноro сырья или животноro
материала перед экстракцией MOryT быть CMe
шаны. Материал, который подверrается экстра
rированию, может быть предварительно обрабо
тан путем, например, инактивации ферментов,
измельчения или обезжиривания. После экс
тракции ненужные вещества, если необходимо,
MOryT быть удалены.
Лекарственное растительное сырье, живот
ные материалы и орrанические растворители.
использующиеся при изrотовлении экстрактов,
должны выдерживать требования COOTBeTCTBY
ющих статей Фармакопеи. Для ryCTbIX и сухих экс
трактов, в которых растворители удаляют выпа
риванием, можно использовать переrнанные или
рецеркулированные растворители при условии,
что процесс переroнки контролируется и раство--
ритель проверяют на соответствие стандартам
перед повторным использованием или смешива
нием с друrими соrласованными материалами.
Вода, которую используют при экстраrировании,
должна быть соответствующеro качества. Под
ходящей считается вода, которая выдерживает
требования частной статьи Вода очuщенная, за
исключением испытания на содержание бакте
риальных эндотоксинов. Питьевую воду можно
использовать при условии, что она выдержива
ет требования соответствующей спецификации,
обеспечивающие надлежащее качество воды
для производства соответствующеro экстракта.
При необходимости ПрО80ДЯТ концентри
рование до желаемой консистенции, используя
подходящие методы, обычно под пониженным
давлением и с использованием температуры.
при которой разрушение компонентов сводит
ся к минимуму. Эфирные масла, выделенные
в процессе обработки, MOryт быть добавлены
в экстракт на определенной стадии производ
ственноro процесса. На разных стадиях произ
водственноro процесса MOryт быть добавлены
вспомоrательные вещества, например, для обе
спечения roмоrенности или консистенции. Также
MorYT быть добавлены стабилизаторы и антими
кробные консерванты.
Экстраrирование определенным раствори
телем проводят до стандартных соотношений
характерных компонентов в материале, который
экстраrируется; однако в процессе производ
ства СТ,андартизованных или количественных
экстрактов MOryT при меняться процедуры очист
ки, что приводит К увеличению этих пропорций
по сравнению с предполаrаемыми значениями.
Такие экстракты называют «очищенными».
ИДЕНТИФИКАЦИЯ
Идентификацию экстрактов проводят, ис
пользуя подходящие методы.
ИСПЫТАНИЯ
С учетом анализа лекарственноro расти
тельноro сырья или животноro материала, KOTO
1134
rосударственная фармакопея Республики Беларусь
рый используют в производстве, и oцeHK ' про
цесса производства для экстрактов MOryT 61,ITb
проведены испытания на микробиолоrическую
чистоту (5.1.4), тяжелые металлы, афлатоксины,
содержание пестицидов (2.8.13).
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
rде возможно, определяют количественное
содержание компонентов экстрактов COOTBeT
ствующими методами.
МАРКИРОВКА
На этикетке указывают:
использованное растительное сырье или
животный материал;
является ли данный экстракт жидким,
ryCTbIM или сухим, либо это настойка;
для стандартизованных экстрактов co
держание компонентов с известной терапевти
ческой активностью;
для количественных экстрактов coдep
жание компонентов (маркеров), которые опреде
ляют количественно;
соотношение исходноro материала и полу
ченноrо экстракта (экстракта без вспомоrатель
ных веществ) (DER);
использованный при экстракции раствори
тель или растворители;
если необходимо, указывают, что исполь
зовалось свежее растительные сырье или жи
вотный материал;
если необходимо, указывают, что экстракт
«очищенный» ;
наименование и количество всех вспомо
rательных веществ, в том числе стабилизаторов
и антимикробных консервантов;
если необходимо, содержание сухою
остатка в процентах.
Жидкие экстракты extracta f/иida
ОПРЕДЕЛЕНИЕ
Жидкие экстракты это жидкие продукты, в
которых обычно одна часть по массе или объему
эквивалентна одной части по массе исходноro BЫ
сушенноro лекарственноro растительноro сырья
или животноro материала. При необходимости их
стандартизируют таким образом, чтобы они COOT
ветствовали требованиям по содержанию paCTBO
рителя и, rдe возможно, действующих веществ.
ПРОИЗВОДСТВО
Жидкие экстракты MOryT быть изroтовле
ны экстракцией лекарственноro растительною
сырья или животноro материала спиртом эти
ловым определенной концентрации, или водой,
или растворением rycтoro или сухоro экстракта,
полученноro путем использования тех же раст
ворителей в тех же концентрациях, что и при из
rотовлении жидкою экстракта, изroтовленноrо
путем прямой экстракции. При необходимости
жидкие экстракты фильтруют.
При хранении может образовываться He
большой осадок при условии отсутствия суще
ственноro изменения состава.
ИСПЫТАНИЯ
Относительная плотность (2.2.5). При He
обходимости, экстракт должен выдерживать
требования, указанные в частной статье.
Содержание этанола (2.9.10). Для спирто
содержащих жидких экстрактов проводят опре
деление содержания этанола. Содержание
этанола должно соответствовать пределам, YKa
занным в частной статье.
Метанол и 2-пропанол (2.9.11). В спирто
содержащих жидких экстрактах допускается co
держание не более 0,05 % (об/об) метанола и
не более 0,05 % (об/об) 2 пропанола, если нет
друrих указаний в частной статье.
Сухой остаток (2.8.16). При необходимости,
экстракт должен выдерживать требования, YKa
занные в частной статье, учитывая, если необ
ходимо, содержание вспомоrательных веществ.
# Тяжелые металлы (2.4.8, метод А). Не
более 0,01 % (100 ррт).
К 1,0 мл жидкоro экстракта прибавляют 1 мл
кuслоты серной Р, осторожно сжиrают и прока
ливают. К полученному остатку прибавляют при
наrревании 5 мл раствора 615 r/л аммонuя aцe
тата Р, фильтруют через обеззоленный фильтр,
промывают 5 мл воды Р и доводят объем филь
трата водой Р до 100 мл.
12 мл полученноro раствора должны Bыдep
живать испытание на тяжелые металлы. Эталон
roтовят с использованием этаЛОНН020 pacт
вора свинца (1 ррт РЬ) Р.
В лекарственных средствах, содержащих
железо в количестве 0,05 % и более, определе
ние тяжелых металлов проводят после отделе
ния железа соrласно указаниям в частной статье.
# Количественное определение. Содержа
ние определяемых веществ для жидких экстрак
тов выражают в процентах (м/об).
ХРАНЕНИЕ
В защищенном от света месте.
МАРКИРОВКА
На этикетке дополнительно к вышеперечис
ленным требованиям указывают:
rAe применимо, содержание этанола в
процентах (об/об) в roтовом экстракте.
Настойки tiпctиrae
ОПРЕДЕЛЕНИЕ
Настойки это жидкие продукты, которые
обычно изroтавливают, используя одну часть
Экстракты
1135
лекарственноro растительноro сырья или жи
вотноro материала и десять частей экстраrента
либо одну часть лекарственноro растительноro
сырья или животноro материала и пять частей
экстраrента.
ПРОИЗВОДСТВО
Настойки изrотавливают мацерацией или
перколяцией, #или друrими валидированны
ми методами (например, реперколяцией, про
тивоточной экстракцией)#, используя только
спирт этиловый соответствующей KOHцeHTpa
ции для экстракции лекарственноro расти
тельноro сырья или животноro материала, или
разведением в спирте этиловом COOTBeTCTBY
ющей концентрации rycTbIx или сухих экстрак
тов, при изroтовлении которых использовали
те же растворители и в тех же концентраци
ях, что и при изroтовлении жидкоro экстракта,
изroтовленноro путем прямой экстракции. При
необходимости настойки фильтруют.
#Полученные извлечения обычно отстаива
ют не менее 2 суток при температуре не выше
1 О ос до получения прозрачной жидкости и
фильтруют.#
Настойки обычно прозрачные. При xpaHe
нии может образовываться небольшой осадок
при условии отсутствия существенноro измене
ния состава.
Метод мацерации. Если нет друrих YKa
заний, экстраrируемое лекарственное pac
тительное сырье или животный материал из
мельчают до частиц определенноro размера,
тщательно смешивают с указанным экстра
reHToM и выдерживают в закрытом контейне
ре необходимое время. Остаток отделяют от
экстраrента и, если необходимо, отжимают.
В последнем случае обе жидкости объеди
няют.
Метод перколяции. Если необходимо,
экстраrируемое лекарственное растительное
сырье или животный материал измельчают
до частиц определенноro размера, тщательно
смешивают с порцией указанноro экстраrента
и оставляют на необходимое время. Затем пе
реносят смесь в перколятор и медленно перко
лируют при комнатной температуре, следя за
тем, чтобы сырье все время было полностью
покрыто слоем экстраrента. Остаток может
быть отжат, а полученную жидкость объединя
ют сперколятом.
ИСПЫТАНИЯ
Относительная плотность (2.2.5). При He
обходимости, настойка должна выдерживать
требования, указанные в частной статье.
Содержание эта нола (2.9.10). Содержание
эта нола должно соответствовать пределам, YKa
занным в частной статье.
Метанол и 2-пропанол (2.9.11). В настойках
допускается содержание не более 0,05 % (об/об)
метанола и не более 0,05 % (об/об) 2 пропанола,
если нет друrих указаний в частной статье.
Сухой остаток (2.8.16). При необходимости
настойка должна выдерживать требования, YKa
занные в частной статье, учитывая, если необ
ходимо, содержание вспомоrательных веществ.
# Тяжелые металлы (2.4.8, метод А). Не
более 0,001 % (10 ррm), если нет друrих указа
ний в частной статье.
5,0 мл настойки выпаривают досуха, прибав
ляют 1 мл кuслоты серной Р, осторожно сжиrа
ют и прокаливают. К полученному остатку при
бавляют при наrревании 5 мл раствора 615 пл
аммония ацетата Р, фильтруют через обеззо
ленный фильтр, промывают 5 мл воды Р и ДOBO
дят объем фильтрата водой Р до 50 мл.
12 мл полученноro раствора должны Bыдep
живать испытание на тяжелые металлы. Эталон
rотовят с использованием этаЛОНН020 pacт
вора свинца (1 ррт РЬ) Р.
# Количественное определение. Coдep
жание определяемых веществ в настойках BЫ
ражают в процентах (м/об).
ХРАНЕНИЕ
В защищенном от света месте.
МАРКИРОВКА
На этикетке дополнительно к вышеперечис
ленным требованиям указывают:
для настоек, за исключением стандарти
зованных и количественных, соотношение ис
ходноro сырья и экстраrента или исходноro Ma
териала и roтовой настойки;
концентрацию спирта этиловоro в процен
тах (об/об) в roтовой настойке.
rYCTbIe экстракты extracta spissa
ОПРЕДЕЛЕНИЕ
rycTbIe экстракты это мяrкие продукты,
изroтовленные путем упаривания или частично
ro упаривания растворителя, использованноro
для экстракции.
ИСПЫТАНИЯ
Сухой остаток (2.8.16). rустой экстракт
должен выдерживать требования, указанные в
частной статье.
Растворители. Остаточные количества op
rанических растворителей контролируют в co
ответствии с требованиями статьи 5.4, если не
обоснованы и не утверждены иные требования.
# Тяжелые металлы (2.4.8, метод А). Не
более 0,01 % (100 ррm).
1136
rосударственная фармакопея Республики Беларусь
к 1,00 r rycToro экстракта прибавляют 1 мл
кислоты серной Р, осторожно сжиrают и про
каливают. К полученному остатку прибавляют
при наrревании 5 мл раствора 615 пл aMMO
нuя ацетата Р, фильтруют через обеззолен
)..fbIЙ фильтр. промывают 5 мл воды Р и доводят
объем фильтрата водой Р до 100 мл.
12 мл полученноro раствора должны BЫ
держивать испытание на тяжелые металлы.
Эталон roтовят с использованием этаЛОНН020
раствора свинца (1 ррт РЬ) Р.
В лекарственных средствах, содержащих
железо в количестве 0,05 % и более, определе
ние тяжелых металлов проводят после отделе
ния железа соrласно указаниям в частной статье.
# Количественное определение. Содержа
ние определяемых веществ для ryCTbIX экстрак
тов выражают в процентах (м/м).
ХРАНЕНИЕ
В защищенном от света месте.
Смолы o/eoresiпa
ОПРЕДЕЛЕНИЕ
Смолы это rycTbIe экстракты, состоящие
из камеди, находящейся в растворе в эфирном
масле и/или в жирном масле, получаемые путем
испарения растворителя (растворителей), ис
пользуемоrо в процессе производства.
Данная СТ8ТЬЯ распространяется на смолы,
получаемые путем экстракции, и не распростра
няется на природные смолы.
ИСПЫТАНИЯ
Вода (2.2.13). Бальзам должен выдержи
вать требования, указанные в частной статье.
Растворители. Остаточные количества op
rанических растворителей контролируют в co
ответствии с требованиями статьи 5.4, если не
обоснованы и не утверждены иные требования.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в за
щищенном от света месте.
Сухие экстракты extracta sicca
ОПРЕДЕЛЕНИЕ
Сухие экстракты это твердые продук
ты, получаемые удалением растворителя, ис
пользованноrо для их приrотовления. Потеря
в массе при высушивании сухоro экстракта не
должна превышать 5 % (м/м), если в частной
статье не указаны друrие требования или если
не указано определение содержания воды.
ИСПЫТАНИЯ
Вода (2.2.13). Если необходимо, содержа
ние воды должно соответствовать пределам,
указанным в частной статье.
Потеря в массе при высушивании (2.8.17).
Если необходимо, потеря в массе при высуши
вании должна соответствовать пределам, YKa
занным в частной статье.
Растворители. Остаточные количества op
rанических растворителей контролируют в соот
ветствии с требованиями rлавы 5.4, если не обо
снованы и не утверждены иные требования.
# Тяжелые металлы (2.4.8, метод А). Не
более 0,01 % (100 ррm).
К 1,00r сухоro экстракта прибавляют 1 мл
кuслоты серной Р, осторожно сжиrают и прока
ливают. К полученному остатку прибавляют при
наrревании 5 мл раствора 615 r/л аммонuя aцe
тата Р, фильтруют через обеззоленный фильтр,
промывают 5 мл воды Р и доводят объем филь
трата водой Р до 100 мл.
12 мл полученноro раствора должно Bыдep
живать испытание на тяжелые металлы. Эталон
rотовят с использованием этаЛОНН020 pacт
вора свинца (1 ррт РЬ) Р.
В лекарственных средствах, содержащих
железо в количестве 0,05 % и более, определе
ние тяжелых металлов проводят после отделе
ния железа соrласно указаниям в частной статье.
# Количественное определение. Содержа
ние определяемых веществ для сухих экстрак
тов выражают в процентах (м/м).
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в за
щищенном от света месте.
01/2013:2098
ЭФИРНЫЕ МАСЛА
Aethe,o/ea
Данную статью необходuмо чuтать вместе
с частнымu статьями на эфирные масла. Реше
ние о прuмененuu данной статьи к неопuсанным
в Фармакопее эфuрным маслам может быть вы
несено уполномоченным Ор2аном.
ОПРЕДЕЛЕНИЕ
Пахучий продукт, обычно сложноro состава,
получаемый из определенною растительноro
сырья путем переroнки с водяным паром, сухой
переrонки или подходящею механическоro про
цесса без наrревания. Эфирные масла обычно
отделяются от водной фазы с помощью физиче
скоro процесса, который не влияет в значитель
ной степени на их состав.
Эфирные масла MOryT впоследствии быть
подверrнуты дальнейшей обработке. KOMMep
Эфирные масла
1137
ИСПЫТАНИЯ
чески доступные эфирные масла MOryт быть дe
терпенированными, десесквитерпенированны
ми, ректифицированными или «х» свободными.
Детерпенuрованное эфuрное масло
это эфирное масло, из котороro частично или
полностью были удалены монотерпеновые уrле
водороды.
Детерпенированное u десесквuтерпенu
рованное эфирное масло это эфирное масло,
из котороro частично или полностью были yдa
лены монотерпеновые и сесквитерпеновые
уrлеводороды.
Ректuфuцuрованное эфuрное масло
это эфирное масло, подверrшееся фракцион
ной переrонке с целью удалить определенные
составляющие или с целью модифицировать
состав.
«х» свободное эфирное масло это
эфирное масло, из котороro частично или пол
ностью были удалены одна или несколько co
ставляющих.
ПРОИЗВОДСТВО
В зависимости от фармакопейной статьи,
растительное сырье может быть свежим, под
вявшим, сухим, цельным, ломаным или моло
тым.
Пере20нка с водяным паром. Эфирное
масло получают путем пропускания пара через
растительное сырье в подходящем приборе.
Пар может быть подведен от внешнеro источ
ника или может быть rенерирован кипячени
ем воды, расположенной ниже растительноro
сырья, или кипячением воды с поrруженным
в нее растительным сырьем. Пар и эфирное
масло конденсируют. Воду и эфирное масло от
деляют с помощью декантации.
Сухая пере20нка. Эфирное масло получа
ют путем наrревания до высокой температуры
стеблей или коры в подходящем приборе без ис
пользования воды или пара.
Механuческuй процесс. Эфирное масло,
обычно называемое холоднопрессованным, по
лучают с помощью механическоro процесса
без использования подоrревания. Этот метод
обычно используют при получении эфирноrо
масла из фруктов Cit,us. Он включает в себя BЫ
деление масла из перикарпия и частичное раз
деление физическим способом.
В некоторых случаях к эфирному маслу
может быть добавлен антиоксидант.
ОПИСАНИЕ
Определяют внешний вид и запах эфирно
ro масла.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Эфирные масла идентифицируют с исполь
зованием их rазохроматоrрафическоrо профиля
или, за неимением этоro, с применением друrих
методов, которые MOryт потребоваться (напри
мер, тонкослойная хроматоrрафия).
ОБЩИЕ ИСПblТАНИЯ
Эфирные масла должны выдерживать Tpe
бования следующих испытаний.
Относительная плотность (2.2.5).
Коэффициент преломления (2.2.6).
Оптическое вращение (2.2.7).
Жирные масла и минеральные масла в
эфирных маслах (2.8.7).
ДОПОЛНИТЕЛЬНblЕ ИСПblТАНИЯ
При необходимости эфирные масла должны
выдерживать требования следующих испытаний.
Температура затвердевания (2.2.18).
Кислотное число (2.5.1).
Перекисное число (2.5.5).
Посторонние эфиры (2.8.6).
Остаток после выпаривания (2.8.9).
Вода (2.8.5).
Растворимость в спирте (2.8.10).
Фальсификация. При необходимости
может быть проведено испытание на один или
более фальсификатов с помощью тонкослой
ной хроматоrрафии (2.2.27), rазовой xpOMaTO
rрафии (2.2.28) с использованием хиральных
колонок, при необходимости, или с помощью
любоrо Apyroro подходящеro метода.
Хроматоrрафический профиль. rазовая
хроматоrрафия (2.2.28): определение прово
дят методом внутренней нормализации.
Кроме условий приroдности хроматоrрафи
ческой системы, указанных в частной фарма
копейной статье, приrодность системы необхо
димо проверять с использованием следующеro
испытания, которое периодически проводят по
схеме квалификации приroдности метода.
XpoMaTorpaMMa на рисунке 2098 1 приве
дена в качестве при мера.
Раствор сравнения: ФСО эфUРН020
масла. При необходимости раствор сравнения
может быть разведен 2ептаном Р.
Условия хроматоrрафирования:
колонка: кварцевая капиллярная длиной
60 м и диаметром 0,25 мм, покрытая слоем Ma
Кр020ла 20 000 Р (толщина слоя 0,25 мкм);
283 носитель: 2елuй для хроматоерафuи Р;
скорость потока: 1.5 мл/мин;
детектор: пламенно ионизационный;
объем вводuмой пробы: 1 мкл;
1138
rосударственная фармакопея Республики Беларусь
деление потока: 1 :500; деление потока
может быть отреryлировано в зависимости от
исnoльзуемоrо прибора при условии, что Ha
rрузка на колонку останется без изменений;
температура:
Время (мин) Температура (ОС)
Колонха ()"""",15 70
1 100 70------+240
1 0()"""",1 05 240
Блок ввода 250
проб
Детектор 270
Идентификация компонентов: компонен
ты идентифицируют, используя xpoMaTorpaM
му, прилаraемую к ФСО эфирН020 масла.
Приеодность хроматоерафuческой cи
стемы: раствор сравнения:
разрешение: не менее 1,5 между пиками
линалола илиналилацетата;
12
I
,
1
с
516
7
;
I1
11 3
; 11 I
.....I ....... ....ii... _. . . 1...
отношение си2Нал/шум: не менее 100
для пика деканаля;
предельное содержание: процентtЮе со--
держание каждоro из 9 компонентов должно
находиться в пределах, указанных в сопрово
дительных документах ФСО эфиРН020 масла.
ХРАНЕНИЕ
В полностью наполненных плотноукупорен
ных контейнерах в защищенном от света месте.
МАРКИРОВКА
На этикетке указывают:
научное наименование использованною
растительноro сырья;
при необходимости, тип и/или хемотип
эфирноro масла;
. при необходимоcrи, способ получения;
при необходимocrи, наименование и кон-
центрацию всех добавпенных антиоксидантов;
при необходимости, дополнительные
стадии процесса, не указанные в разделе «Опре
деление» .
6
9
,
4 I t I
.LJt...
30 ... ..40... . 50' ,. 7'. 60...... .
i
t
. ш. . .......
70 .. 80
I
-.. ... -- - .
о
10 20
1 а пинен; 3,",,",,: reкС8НОЛ; 5 линалол;
2 циеол; 4 деканаль; 6 линалилцетат;
90
7 l3 кариофиллен;
8 эвreнол;
9 бензилсалицилат.
Рисунок 2098 1. Хромат02рафический профиль эфирН020 масла
Основные термины и определения
1139
ДОЗИРОВАННЫЕ
ЛЕКАРСТВЕННЫЕ ФОРМЫ
01/2013:1502
ОСНОВНЫЕ ТЕРМИНЫ
И ОПРЕДЕЛЕНИЯ
G/ossa
Данная статья содержuт определенuя
u/uлu объясненuя терминов, которые вcтpe
чаются в тексте uлu uспользуются в общuх
статьях на дозированные формы U cooт
ветствующuх статьях с фармацевтuко
теХНОЛ02uческuмu uспытанuямu (2.9). Прu
необходuмостu дается ссылка на друеие
равнозначные термины, которые uспользу
ются в дРУ2их публuкацuях uлu контекстах.
Данная статья предназначена для
справкu.
Действующее вещество
Синонимы: активный инrредиент, актив
ное вещество, активная субстанция, фар
мацевтическая субстанция, лекарственное
вещество, активный фармацевтический ин
rредиент.
Дозированные формы с обычным
высвобождением
Дозированные формы с обычным BЫCBO
бождением лекарственные средства с BЫ
свобождением действующеro вещества/ве
ществ, которое не меняется преднамеренно
с помощью создания специальноro COCTa
ва и/или способа производства лекарствен
HOro средства. Для твердоro лекарственноro
средства характеристики растворения дей
ствующеrо вещества зависят, rлавным об
разом, от ero внутренних свойств. Синоним:
дозированная форма с моментальным BЫ
свобождением.
Дозированные формы с модифици-
рованным высвобождением
Дозированные формы с модифициро
ванным высвобождением лекарственные
средства, у которых скорость иlили место
высвобождения действующеro веществаl
веществ отличаются от скорости BЫCBO
бождения в дозированных формах с обыч
ным высвобождением, назначаемых тем
же способом. Такое преднамеренное изме
нение достиrается при помощи специаль
но разработанноrо состава и/или способа
производства лекарственноro средства. Дo
зированные формы с модифицированным
высвобождением включают дозированные
формы с пролонrированным высвобождени
ем, замедленным высвобождением и преры
вистым высвобождением.
Дозированные формы с ПРОЛОНl'"иро-
ванным высвобождением
Дозированные формы с пролонrиро
ванным высвобождением лекарственные
средства с модифицированным высвобожде
нием с более медленным высвобождением
действующеro вещества/веществ по cpaBHe
нию с дозированными формами с обычным
высвобождением, назначаемыми тем же спо
собом. Пролонrированное высвобождение
достиrается при помощи специально разра
ботанноrо состава и/или способа производ
ства лекарственноro средства. Синонимы:
дозированные формы с длительным BЫCBO
бождением.
Дозированные формы с замедлен-
ным высвобождением
Дозированные формы с замедленным
высвобождением лекарственные cpeд
ства с модифицированным высвобождением,
в которых высвобождение действующеro Be
щества/веществ происходит с задержкой. За
медленное высвобождение достиrается при
помощи специально разработанноro состава
и/или способа производства лекарственно
ro средства. К дозированным формам с за
медленным высвобождением относятся ки
шечнорастворимые лекарственные средства,
описанные в общих статьях на твердые дози
рованные лекарственные формы.
Дозированные формы с прерыви-
стым высвобождением
Дозированные формы с прерывистым
высвобождением лекарственные средства
с модифицированным высвобождением с по
следовательным высвобождением действую
щеro вещества/веществ. Последовательное
высвобождение достиrается при помощи спе
циально разработанноro состава иlили спо
со ба производства лекарственноro средства.
Коллоидная дисперсия
Коллоидная дисперсия это система, в
которой частицы коллоидноro размера (при
мерно от 1 нм ДО 500 нм) любой природы
(твердые, жидкие или rазообразные) диспер
rированы в непрерывной фазе различноro co
става и/или состояния.
1140
rосударственная фармакопея Республики Беларусь
Лекарственные средства для паренте-
ральноrо применения большоrо объема
Растворы для инфузий и инъекций, BЫ
пускаемые в контейнерах с номинальным co
держанием более 100 мл.
Лекарственные средства для паренте-
ральноrо применения малоrо объема
Растворы для инфузий и инъекций, BЫ
пускаемые в контейнерах с номинальным co
держанием 100 мл или менее.
Носитель
Носитель одно или более вспомоrатель
ных веществ, которые доставляют действу
ющее вещество/вещества в жидком лекар
ственном средстве.
Основа
Основа одно или более вспомоrатель
ных веществ, которые доставляют действу
ющее вещество/вещества в мяrком и твердом
лекарственных средствах.
Раствор
Раствор это смесь, образующая единую
фазу, содержащую одно или более paCTBopeH
ных веществ, то есть веществ, дисперrирован
ных в молекулярном состоянии в растворителе
или в смешивающихся растворителях.
Стандартный термин
Стандартный термин, который использу
ется для описания лекарственной формы ле
карственноro средства. способов приема и
контейнеров. устанавливается Фармакопеей и
при водится в отдельной публикации по CTaH
дартным терминам.
Суспензия
Суспензия это дисперсная система, co
держащая твердые частицы, дисперrированные в
жидкой или мяrкой непрерывной фазе, в которой
твердые частицы практически нерастворимы.
Сфероиды
Сфероиды это сферические или прак
тически сферические rранулы, обычно с более
высокой механической прочностью по cpaBHe
нию с традиционными rранулами (как описано
в соответствующей общей статье на дозиро
ванную форму). Они имеют rладкую ровную по
верхность и размер, лежащий обычно в диапа
зоне от 200 мкм до 2,8 мм. Сфероиды получают
любым подходящим методом.
Эмульсия
Эмульсия это дисперсная система, пред
ставляющая собой смесь по крайней мере двух
несмешивающихся друr с друroм жидкостей.
Одна из жидкостей дисперrирована во второй
в виде капелек.
01/2013:1163
rЛАЗНЫЕ ЛЕКАРСТВЕННЫЕ
СРЕДСТВА
Ophtha/mica
ОПРЕДЕЛЕНИЕ
rлазные лекарственные средства CTe
рильные жидкие, мяrкие или твердые лекар
ственные средства, предназначенные для
нанесения на rлазное яблоко и/или конъюн
ктиву или для введения в коньюнктивальный
мешок.
Если это применимо, контейнеры для rлаз
ных лекарственных средств должны соответ
ствовать требованиям статей разделов 3.1. Ma
териалы, используемые для проuзводства
контейнеров и 3.2. Контейнеры.
rлазные лекарственные средства можно
классифицировать как:
rлазные капли;
rлазные примочки;
порошки для приrотовления rлазных
капель и при мочек;
rлазные мяrкие лекарственные средства;
ПРОИЗВОДСТВО
При разработке rлазных лекарственных
средств, в состав которых входят антимикроб
ные консерванты, уполномоченному opraHY
представляют данные, подтверждающие He
обходимость использования и эффективность
выбранных консервантов. Подходящий метод
определения и критерии оценки эффективности
консервантов представлены в статье 5. 1.3. Эф
фектuвность антuмuкробных консервантов.
rлазные лекарственные средства произво
дят с использованием материалов и методов,
обеспечивающих стерильность и предотвраща
ющих заrрязнение лекарственных средств и раз
витие микроорrанизмов в соответствии с требо
ваниями статьи 5. 1. 1. Методы ПРU20товленuя
стерильных продуктов.
При производстве rлазных лекарственных
средств, содержащих дисперrированные части
цы, принимают меры, обеспечивающие необхо
димый размер частиц и их контроль.
При разработке rлазных лекарственных
средств должно быть доказано, что из OДHOДO
зовоro контейнера, содержащеro жидкое или
мяrкое лекарственное средство, может быть
извлечено номинальное ero содержимое.
#В случае отсутствия у производителя ле
карственноro средства сертификата C()OT
ветствия GMP, а также данных о валидации
процесса производства KOHKpeTHoro лекар
ственноro средства испытание по определе
нию извлекаемоro объема должно быть вклю
чено в спецификацию ютовоro продукта.#
rлазные лекарственные средства
1141
ИСПЫТАНИЯ
Стерильность (2.6.1). rлазные лекарствен
ные средства должны выдерживать испытание
на стерильность. Аппликаторы, прилаrаемые OT
дельно, также должны выдерживать испытание
на стерильность. Их извлекают из контейнера в
асептических условиях и помещают в емкость с
питательной средой до полноro поrружения. Ин
кубацию посевов и оценку результатов про водят
в соответствии с требованиями испытания на
стерильность.
ХРАНЕНИЕ
Если иное не обосновано и не утвержде
но, хранят в стерильных воздухонепроницаемых
контейнерах с контролем первоro вскрытия.
МАРКИРОВКА
На этикетке указывают названия всех дo
бавленных антимикробных консервантов.
rлазные капли
ОПРЕДЕЛЕНИЕ
rлазные капли стерильные водные или
масляные растворы или суспензии, содержащие
одно или более действующих веществ и предна
значенные для инстилляции в rлаз.
rлазные капли MOryT содержать вспомоrа
тельные вещества, например, для обеспечения
необходимоro осмотическоro давления, вязко
сти, создания или стабилизации необходимоro
значения рН, увеличения растворимости дей
ствующих веществ, обеспечения стабильности
лекарственноro средства. Эти вещества в ис
пользуемых концентрациях не должны отри
цательно влиять на основное терапевтическое
действие лекарственноro средства и оказывать
нежелательноro местноro раздражения.
Водные лекарственные средства, выпуска
емые в мноroдозовых контейнерах, должны co
держать подходящие антимикробные KOHcep
ванты в необходимых концентрациях, за исклю
чением тех случаев, Korдa само лекарственное
средство обладает достаточным антимикроб
ным действием. Выбранные антимикробные
консерванты должны быть совместимы с дpy
rими инrредиентами лекарственноro средства и
сохранять эффективность в течение всеro пери
ода использования rлазных капель.
Если rлазные капли не содержат антими
кробных консервантов, они должны быть упа
кованы в однодозовые контейнеры либо в MHO
rодозовые контейнеры, предотвращающие
микробное заrрязнение после их в,:;крытия.
rлазные капли, предназначенные для ис
пользования при хирурrических процедурах, не
должны содержать антимикробных KOHcepBaH
тов и должны выпускаться в однодозовых KOH
тейнерах.
rлазные капли, представляющие собой pac
творы, в соответствующих условиях наблюдения
должны быть практически прозрачными и прак
тически свободными от частиц.
rлазные капли в виде суспензий MOryT об
разовывать осадок, который должен быстро pe
суспендироваться при взбалтывании, образуя
суспензию, которая должна быть достаточно
стабильной и обеспечивать необходимую дозу
при введении.
Мноroдозовые лекарственные средства BЫ
пускают в таких контейнерах, которые позволяют
дозировать по каплям. Если иное не обосновано
и не утверждено, контейнер должен содержать
не более 10 мл лекарственноro средства.
ИСПЫТАНИЯ
Размер частиц. Если иное не обосновано
и не утверждено, rлазные капли в виде суспен
зий должны выдерживать следующее испыта
ние: определенное количество суспензии вносят
в счетную камеру или с помощью микропипет
i{и наносят на предметное стекло и просматри
вают под микроскопом площадь, COOTBeTCTBY
ющую 1 О MKr твердою действующеro вещества.
Исходя из практических соображений, сначала
образец просматривают при малом увеличении
(например, х50), отмечая частицы с максималь
ным размером более 25 мкм. Затем производят
измерение этих частиц при большем увеличении
(например, от х200 до х500). Для каждоro образ
ца, содержащеro 1 О MKr твердоro действующе
ro вещества, должно быть не более 20 частиц с
максимальным размером более 25 мкм, и из них
не более двух частиц с максимальным размером
более 50 мкм. Не допускается наличие частиц с
максимальным размером более 90 мкм.
МАРКИРОВКА
На этикетке указывают:
для лекарственных средств в мноroдозо
вых контейнерах: срок хранения после BCKpЫ
тия. Если иное не обосновано и не утверждено,
этот срок не должен превышать 4 недели.
rлазные при мочки
ОПРЕДЕЛЕНИЕ
rлазные примочки стерильные водные
растворы, предназначенные для смачивания и
промывания rлаз, а также для пропитывания Ma
териалов, накладываемых на rлаза.
rлазные примочки MOryт содержать вспомо
rательные вещества, например, для обеспече
ния необходимоro осмотическоro давления, вяз
кости, создания или стабилизации необходимою
значения рН. Эти вещества в используемых KOH
центрациях не должны отрицательно влиять на
действие лекарственною средства и не должны
оказывать нежелательное местное раздражение.
Водные растворы rлазных лекарственных
средств, выпускаемые в мноroдозовых контейне
рах, должны содержать подходящие антимикроб
ные консерванты в необходимых концентрациях,
1142
rосударственная фармакопея Республики Беларусь
за исключением тех случаев, Korдa само лекар
ственное средство обладает достаточным анти
микробным действием. Выбранные антимикроб
ные консерванты должны быть совместимы с
друrими инrредиентами лекарственноro cpeд
ства и сохранять эффективность в течение всеro
периода использования rлазных примочек.
Если rлазные примочки не содержат анти
микробных консервантов, они должны быть упа
кованы в однодозовые контейнеры. rлазные
примочки, предназначенные для использования
при хирурrических процедурах и для оказания
первой медицинской помощи, не должны содер.-
жать антимикробных консервантов и должны BЫ
пускаться только в однодозовых контейнерах.
rлазные примочки в соответствующих усло
виях наблюдения должны быть практически про
зрачными и практически свободными от частиц.
Если иное не обосновано и не утверЖдено.
мноroдозовый контейнер должен содержать не
более 200 мл rлазной при мочки.
МАРКИРОВКА
На этикетке указывают:
для однодозовых контейнеров указывают,
что содержимое должно использоваться только
один раз;
для лекарственных средств в мноroдозо
вых контейнерах: срок хранения после BCKpЫ
тия. Если иное не обосновано и не утвеРЖдено,
этот срок не должен превышать 4 недели.
Порошки для приroтовления rлазных
капель и примочек
ОПРЕДЕЛЕНИЕ
Порошки для приroтовления rлазных капель
и примочек это сухие стерильные лекар
ственные средства, которые перед применени
ем растворяют или суспендируют в предписан
ной стерильной жидкости. Они MOryT содержать
вспомоrательные вещества, которые облеrча
ют растворение или дисперrирование и предот
вращают аrреrацию частиц, обеспечивают He
обходи мое осмотическое давление, создают
или стабилизируют необходимое значение рН
или обеспечивают стабильность лекарственно
ro средства.
После растворения или дисперrирования
они должны соответствовать требованиям для
rлазных капель или примочек соответственно.
ИСПЫТАНИЯ
Однородность дозированных единиц
(2.9.40). Однодозовые порошки для приroтов
ления rлазных капель и примочР.к должны BЫ
держивать испытание на однородность дози
рованных единиц или, если это обосновано и
утверждено, испытания на однородность coдep
жания и/или однородность массы, как указано
ниже. На лекарственное растительное сырье и
лекарственные средства из неro, представлен
ные в дозированной форме, требования данною
раздела не распространяются.
Однородность содержания (2.9.6). Порош
ки для приютовления rлазных капель и примо
чек в однодозовых контейнерах с содержанием
действующеro вещества менее 2 Mr или менее
2 % от общей массы содержимою должны BЫ
держивать испытание на однородность coдep
жания действующеro вещества в единице дo
зированноrо лекарственноro средства (тест В).
если нет друrих указаний в частной статье. Если
лекарственное средство содержит более одноro
действующеro вещества, это требование pac
пространяется только на те вещества, содержа
ние которых соответствует вышеуказанным yc
ловиям.
Однородность массы (2.9.5). Порошки
для приrотовления rлазных капель и примочек
в однодозовых контейнерах должны выдержи
вать испытание на однородность массы для
единицы дозированноro лекарственноro cpeд
ства. Испытание на однородность массы не
требуется, если испытание на однородность
содержания предусмотрено для всех действу
ющих веществ.
rлазные мяrкие лекарственные средства
ОПРЕДЕЛЕНИЕ
rлазные мяrкие лекарственные средства
это однородные стерильные мази, кремы или
rели, предназначенные для нанесения на
конъюнктиву или веки. Они содержат одно или
более действующих веществ, растворенных или
дисперrированных в подходящей основе.
rлазные мяrкие лекарственные средства
должны соответствовать требованиям статьи
МЯ2кие лекарственные средства для наружно
20 прuмененuя. Основа не должна раздражать
конъюнктиву.
rлазные мяrкие лекарственные средства
упаковывают в стерильные необратимо сжи
маемые мелкоемкие тубы со встроенным или
приложенным стерильным наконечником. Если
иное не обосновано и не утверждено, coдep
жимое тубы должно быть не более 10 r. Тубы
должны быть плотно укупорены, чтобы пре
дотвращать микробное заrрязнение. rлазные
мяrкие лекарственные средства также можно
выпускать в специально предназначенных OДHO
дозовых контейнерах. Контейнеры или наконеч
ники туб должны быть такой формы, чтобы обе
спечить введение без заrрязнения.
ИСПЫТАНИЯ
Размер частиц. rлазные мяrкие лекар
ственные средства, содержащие дисперrиро
ванные твердые частицы, должны выдерживать
следующее испытание: пробу, содержащую не
менее 10 MKr твердоro действующеro вещества,
rранулы
1143
осторожно наносят тонким слоем и просматри
вают под микроскопом всю площадь образца.
Исходя из практических соображений, сначала
образец просматривают при малом увеличении
(например, >(50), отмечая частицы с максималь
ным размером более 25 мкм. Затем производят
измерение этих частиц при большем увеличе
нии (например, от х200 до х500). Для каждо
ro образца, содержащеro 1 О MKr твердоro дей
ствующеro вещества, должно быть не более
20 частиц с максимальным размером более
25 мкм, и из них не более двух частиц с макси
мальным размером более 50 мкм. Не допуска
ется наличие частиц с максимальным размером
более 90 мкм.
МАРКИРОВКА
На этикетке указывают:
для лекарственных средств в мноrодозо
вых контейнерах: срок хранения после BCKpЫ
тия. Если иное не обосновано и не утверждено,
этот срок не должен превышать 4 недели.
rлазные вставки
ОПРЕДЕЛЕНИЕ
rлазные вставки стерильные твердые или
мяrкие лекарственные средства COOTBeTCTBY
ющеro размера и формы, предназначенные для
введения в конъюнктивальный мешок для соз
дания окулярноro эффекта. Они обычно состоят
из матрицы, в которую включено действующее
вещество, или действующее вещество окруже
но мембраной, контролирующей скорость BЫ
свобождения. Действующее вещество должно
иметь достаточную растворимость в физиоло
rической жидкости и высвобождаться за опреде
ленный период времени.
Каждая rлазная вставка выпускается в инди
видуальном стерильном контейнере.
ПРОИЗВОДСТВО
Производство rлазных вставок должно обе
спечивать необходимое высвобождение деl1
ствующеro вещества.
ИСПЫТАНИЯ
Однородность дозированных единиц
(2.9.40). rлазные вставки должны выдержи
вать испытание на однородность дозированных
единиц или, если это обосновано и утвержде
но, испытание на однородность содержания, как
указано ниже. На лекарственное растительное
сырье и лекарственные средства из неro, пред
ставленные в дозированной форме, требования
данноro раздела не распространяются.
Однородность содержания (2.9.6). rлаз
ные вставки должны выдерживать испытание на
однородность содержания действующеro веще
ства в единице дозированноrо лекарственноrо
средства (тест А).
МАРКИРОВКА
На этикетке указывают:
если это необходимо, общее количество
действующеro вещества в одной вставке;
при необходимости, дозу, высвобождаемую
за единицу времени.
01/2013:0499
rрднулы
G,aпu/ata
Требования к 2ранулам, uспользуемым для
ПРU20товленuя растворов uлu суспензuй для
внутренне20 (оралЬН020) прuменения, прuведе
ны в статье Жuдкuе лекарственные средства
для внутренне20 прuмененuя.
ОПРЕДЕЛЕНИЕ
rранулы лекарственные средства, co
стоящие из твердых сухих, достаточно прочных
arperaToB частиц порошка. rранулы предна
значены для приема внутрь. Некоторые из них
предназначаются для rлотания в целом виде,
некоторые подлежат разжевыванию, HeKOTO
рые растворяют или дисперrируют перед при
менением в воде или друroй подходящей жид
кости.
rранулы содержат одно или более действу
ющих веществ с или без наполнителей и, при
необходимости, MOryт содержать красители и
ароматизаторы, разрешенные к применению
уполномоченным opraHoM.
rранулы выпускают в однодозовых или MHO
roдозовых контейнерах. Каждую дозу rранул
из мноroдозовоrо контейнера извлекают при
помощи соответствующеro приспособления для
отмеривания предписанноro количества. Каждая
доза однодозовоro лекарственноro средства
должна быть упакована в отдельный контейнер,
например, пакетик или флакон.
Если это применимо, контейнеры для rранул
должны соответствовать требованиям статей
разделов 3. 1. Матерuалы, uспользуемые для
проuзводства контейнеров и 3.2. Контейнеры.
rранулы можно классифицировать как:
rранулы «шипучие»;
rранулы, покрытые оболочкой;
rранулы кишечнорастворимые;
rранулы с модифицированным высвобож
дением.
ПРОИЗВОДСТВО
При производстве, упаковке, хранении и
реализации rранул предпринимают меры, обе
спечивающие микробиолоrическую чистоту
в соответствии с требованиями статьи 5.1.4.
1144
rосударственная фармакопея Республики Беларусь
МuкроБUОЛ02uческая чuстота нестерuльнЬ1Х
лекарственнЬ1Х средств u фармацевтuческuх
субстанцuЙ
ИСПЫТАНИЯ
Однородность дозированных единиц.
Однодозовые rранулы должны выдерживать ис
пытание на однородность дозированных единиц
(2.9.40) или, если это обосновано и утверждено,
испытания на однородность содержания и/или
однородность массы, как указано ниже. На ле
карственное растительное сырье и лекарствен
ные средства из Hero, представленные в дозиро
ванной форме, требования данноro раздела не
распространяются
Однородность содержания (2.9.6). rpaHY
лы в однодозовых контейнерах с содержанием
действующеro вещества менее 2 Mr или менее
2 % от общей массы содержимоro должны BЫ
держивать испытание на однородность coдep
жания действующеro вещества в единице дo
зированноro лекарственноro средства (тест В),
если нет друrих указаний в частной статье. Если
лекарственное средство содержит более одноro
действующеro вещества, это требование pac
пространяется только на те вещества, содержа
ние которых соответствует вышеуказанным yc
ловиям.
Однородность массы (2.9.5). rранулы в oд
нодозовых контейнерах, за исключением rранул,
покрытых оболочкой, должны выдерживать ис
пытание на однородность массы для единицы
дозированноro лекарственноro средства. Ис
пытание на однородность массы не требуется,
если испытание на однородность содержания
предусмотрено для всех действующих веществ.
Однородность массы одной дозы, BЫ
свобожденной из мноrодозовоrо контейнера
(2.9.27). rранулы в мноroдозовых контейнерах
должны выдерживать испытание на OДHOpOД
ность массы одной дозы, высвобожденной из
мноroдозовоro контейнера.
ХРАНЕНИЕ
Если лекарственное средство содержит ле
тучие вещества или содержимое должно быть
защищено, хранят в воздухонепроницаемом
контейнере.
rранулы «шипучие»
ОПРЕДЕЛЕНИЕ
rранулы «шипучие» rранулы, не покры
тые оболочкой, rлавным образом содержащие
кислоты и карбонаты или rидрокарбонаты, KOТO
рые при наличии воды быстро вступают в peaK
цию с выделением уrлекислоro rаза. Они предна
значены для растворения или дисперrирования
в воде перед применением.
ИСПЫТАНИЯ
Распадаемость. Одну дозу rранул «шипучих»
помещают в лабораторный стакан, содержащий
200 мл водЬ1 Р при температуре от 15 ос до 25 ОС;
выделяются мноroчисленные пузырьки rаза. Korдa
выделение rаза BOKpyr отдельных частиц прекра
щается, rранула считается распавшейся в резуль
тате растворения или дисперrирования в воде. По
вторяют процедуру на пяти друrих дозах. r ранулы
выдерживают испытание, если каждая из шести
доз распадается в течение не более 5 мин.
ХРАНЕНИЕ
Хранят в воздухонепроницаемых контейне
рах.
rранулы, покрытые оболочкой
ОПРЕДЕЛЕНИЕ
rранулы, покрытые оболочкой лекар
ственные средства в мноroдозовых контейне
рах, обычно состоящие из rранул, по крытых
одним или несколькими слоями смеси из раз
личных вспомоrательных веществ.
ПРОИЗВОДСТВО
Вещества, используемые для получения
оболочки, обычно наносят в виде раствора или
суспензии в условиях, в которых происходит ис
парение растворителя.
ИСПЫТАНИЯ
Растворение. Испытание проводят для
подтверждения соответствующеro высвобожде
ния действующеro вещества или веществ, Ha
пример, одним из способов, описанных в статье
2.9.3. Тест ((Растворение» для твердЬ1Х дози
рованнЬ1Х форм.
rранулы с модифицированным BЫCBO
бождением
ОПРЕДЕЛЕНИЕ
r ранулы с модифицированным высвобожде
нием rранулы, покрытые оболочкой или без обо
лочки, полученные с использованием специаль
ных вспомоraтельных веществ или специальных
способов, которые, отдельно или вместе, предна
значены для изменения скорости, места или Bpe
мени высвобождения действующеro вещества.
К rранулам с модифицированным BЫCBO
бождением относятся rранулы с пролонrирован
ным и замедленным высвобождением.
ПРОИЗВОДСТВО
Проводят испытание, подтверждающее co
ответствующее высвобождение действующеro
вещества или веществ.
ИСПЫТАНИЯ
Растворение. Проводят испытание для под
тверждения соответствующеro высвобождения
ЖеватеЛЬНblе резинки лекарственные
1145
действующеro вещества или веществ, например,
одним из способов, описанных в статье 2.9.3. Тест
«Растворение» для твердых дозированных форм.
r ранулы кишечнораСТ80римые
ОПРЕДЕЛЕНИЕ
rранулы кишечнорастворимые rpанулы с
модифицированным высво60>tЩением, которые
устойчивы к действию желудочноro сока и спосо6--
ны высвобождать действующее вещество или Be
щества в кишечном соке. Это достиraется покрыти
ем rранул материалом, устойчивым к желудочному
соку, или друrими подходящими способами.
ПРОИЗВОДСТВО
Проводят испытание, которое подтверждает,
что технолоrия обеспечивает необходимое BЫCBO
бождение действующеro вещества или веществ.
ИСПЫТАНИЯ
Растворение. Проводят испытание для
подтверждения соответствующеro высвобожде
ния действующеro вещества или веществ, Ha
при мер, одним из способов, описанных в статье
2.9.3. Тест «Растворение» для твердых дози
рованных форм.
01/2013:РБОО06
#ДРАЖЕ
Dragee
ОПРЕДЕЛЕНИЕ
Драже твердые дозированные лекар
ственные средства, содержащие одно или более
действующих веществ, полученные путем MHO
rOKpaTHoro наслаивания действующих и вспо
моraтельных веществ и предназначенные для
оральноrо применения.
ПРОИЗВОДСТВО
Драже обычно получают путем мноroкрат
ноro наслаивания (дражирования) действующих
и вспомоrательных веществ на сахарную крупку
или друroй подходящий носитель.
ИСПЫТАНИЯ
Однородность содержания (2.9.6). Драже
должны выдерживать испытание на OДHOpOД
ность содержания действующеro вещества в
единице дозированноro лекарственноrо cpeд
ства (тест А) независимо от содержания в них
действующеro вещества или веществ, если нет
друrих указаний в частной статье. Данное ис
пытание не проводится для поливитаминных
лекарственных средств и для лекарственных
средств, содержащих микроэлементы, если нет
друrих указаний в частной статье.
Однородность массы. Испытание прово
дят по методике, указанной в статье 2.9.5. Oд
нородность массы для единицы дозuрованно
20 лекарствеНН020 средства. Лекарственное
средство считают выдержавшим испытание,
если не более двух индивидуальных масс откло
няются от средней массы более чем на 1:15 %,
если нет друrих указаний в частной статье. При
этом ни одна индивидуальная масса не должна
отклоняться от средней массы более чем на
1:30 % или на величину, в два раза превыша
ющую установленную в частной статье норму
отклонения.
Испытание на однородность массы не Tp
буется, если испытание на однородность coдep
жания предусмотрено для всех действующих Be
ществ или если нет д:-,vrих указаний в частной
статье.
Распадаемость. Драже должны выдержи
вать испытание, описанное в общей статье 2.9.1.
Распадаемость таблеток и капсул. В качестве
жидкой среды используют воду Р. В каждую CTe
клянную трубку помещают диск. Прибор включа
ют на 60 мин, если нет друrих указаний в част
ной статье, и исследуют состояние драже. Если
драже не выдержали испытание вследствие при
липания к дискам, испытание повторяют на сле
дующих шести драже без дисков. Лекарствен
ное средство выдерживает испытание, если все
шесть драже распались.
Драже для разжевывания испытанию на
распадаемость не подлежат.
Растворение (2.9.3). Драже должны Bыдep
живать испытание, если нет друrих указаний в
частной статье. Испытание проводят для под
тверждения высвобождения действующеro Be
щества или веществ.
Если проводят испытание по показателю
«Растворение», испытание по показателю «Pac
падаемость» не требуется.
01/2013:1239
ЖЕВАТЕЛЬНЫЕ РЕЗИНКИ
ЛЕКАРСТВЕННЫЕ
Masticabilia gummis medicata
ОПРЕДЕЛЕНИЕ
Лекарственные жевательные резинки
твердые дозированные лекарственные средства
1146
rосударственная фармакопея Республики Беларусь
с основой, состоящей rлавным образом из
резины, предназначенные для жевания, но не
rлотания.
Лекарственные жевательные резинки co
держат одно или несколько действующих Be
ществ, которые высвобождаются при жевании.
В результате растворения или дисперrирования
действующих веществ в слюне, жевательные
резинки используют:
для местною лечения заболеваний поло
сти рта;
для системноro лечения после BcaCЫBa
ния через слизистую щек или желудочно кишеч
ноro тракта.
ПРОИЗВОДСТВО
Лекарственные жевательные резинки изю...
тавливают из основы безвкусной жеватель---
ной резины, которая состоит из натуральных
или синтетических эластомеров. Они MOryт co
держать вспомоrательные вещества, такие как
наполнитеЛИ,смяrчитеЛИ,подсластители,ВКУСО
вые добавки, стабилизаторы, пластификаторы и
красители, разрешенные к применению уполно
моченным opraHoM.
Лекарственные жевательные резинки из
roтавливают путем или прессования, или раз
мяrчения, или сплавления основы резины с
друrими веществами, которые добавляют по
следовательно. В последнем случае жеватель
ные резинки обрабатывают для придания резин
ке товарноro вида. Жевательные лекарственные
резинки MOryT быть покрыты, например, если He
обходима защита от влаrи и света.
Если иное не обосновано и не утверждено,
проводят испытание, подтверждающее BЫCBO
бождение действующею вещества или веществ.
Для этоro может быть использован метод 2.9.25.
Высвобождение действующе20 вещества uз
лекарственных жевательных резuнок.
В процессе производства, упаковки, xpaHe
ния и реализации лекарственных жевательных
резинок предпринимают меры, обеспечивающие
микробиолоrическую чистоту в соответствии с
требованиями раздела 5.1.4. МuкроБUОЛ02uче
ская чuстота нестерuльных лекарственных
средств u фармацевтических субстанцuй.
ИСПЫТАНИЯ
Однородность дозированных единиц.
Лекарственные жевательные резинки должны
выдерживать испытание на однородность дози
рованных единиц (2.9.40) или, если это обосно
вано и утверждено, испытания на однородность
содержания и/или однородность массы, как YKa
зано ниже. На лекарственное растительное
сырье и лекарственные средства из неro, пред
ставленные в дозированной форме, требования
данною раздела не распространяются.
Однородность содержания (2.9.6). Если
иное не обосновано и не утверждено, лекарствен
ные жевательные резинки с содержанием дей
ствующеro вещества менее 2 Mr или менее 2 % от
общей массы содержимоro должны выдерживать
испытание на однородность содержания действу
ющеro вещества в единице дозированною лекар
ственноro средства (тест А). Если лекарственное
средство содержит более одноro действующе
ro вещества, это требование распространяется
только на те вещества, содержание которых co
ответствует вышеуказанным условиям.
Однородность массы (2.9.5). Непокры
тые оболочкой жевательные резинки и, если нет
друrих указаний в частной статье, покрытые обо
лочкой жевательные резинки должны выдержи
вать испытание на однородность массы для еди
ницы дозированноro лекарственноro средства.
Испытание на однородность массы не требует
ся, если испытание на однородность содержа
ния предусмотрено для всех действующих Be
ществ.
ХРАНЕНИЕ
Непокрытые оболочкой. лекарственные же
вательные резинки хранят в защищенном от воз
действия влаrи и света месте, если нет друrих
указаний в частной статье.
01/2013:0672
ЖИДКИЕ ЛЕКАРСТВЕННЫЕ
СРЕДСТВА ДЛЯ BHYTPEHHErO
ПРИМЕНЕНИЯ
Рrаераrаtiопеs Iiquidae ad usum peroraliae
ОПРЕДЕЛЕНИЕ
Жидкие лекарственные средства для BHY
треннеro (оральноro) применения представляют
собой растворы, эмульсии или суспензии, coдep
жащие одно или более действующих веществ в
соответствующем носителе; они MorYT также co
стоять только из жидких действующих веществ
(жидкости для внутреннеro применения).
Некоторые лекарственные средства для
внутреннеro применения изroтавливают раз
ведением жидких концентратов, порошков или
rранул для приютовления растворов или cy
спензий для внутреннею применения, капель
для внутреннеro применения или сиропов, ис
пользуя соответствующий растворитель.
Растворитель для всех лекарственных
средств для внутреннеro применения выбирают
исходя из при роды действующеro вещества или
веществ: он должен обеспечивать орrанолепти
ческие свойства лекарственному средству в за
висимости от еro предназначения.
Жидкие лекарственные средства для внутреннеао применения
1147
Жидкие лекарственные средства для BHY
TpeHHero применения MorYT содержать подхо
дящие антимикробные консерванты, антиок
сиданты и друrие вспомоrательные вещества,
которые обеспечивают дисперrирование, cy
спендирование, а также заrустители. эмуль
raTopbI, вещества, предназначенные для соз
дания или стабилизации рН, для обеспечения
смачивания и растворимости, стабилизато
ры, ароматизаторы, вкусовые добавки и Kpa
сители, разрешенные к применению уполно
моченным opraHoM.
Эмульсии MorYT расслаиваться, однако
быстро восстанавливаются при взбалтыва
нии. Суспензии MorYT образовывать осадок,
который быстро ресуспендируется при взбал
тывании, образуя суспензию достаточно CTa
бильную, чтобы обеспечить однородность ле
KapcTBeHHoro средства при дозировании.
Если это применимо, контейнеры для
жидких лекарственных средств для BHYTpeH
Hero применения должны соответствовать Tpe
бованиям статей разделов 3.1. Матерuалы,
uспользуемые для проuзводства контейне
ров и 3.2. Контейнеры.
Жидкие лекарственные средства для BHY
треннеro применения можно классифициро
вать как:
растворы, эмульсии и суспензии для
внутреннеro применения;
порошки и rранулы для приrотовления
растворов и суспензий для внутреннеro при
менения;
капли для внутреннеro применения;
порошки для приroтовления капель для
BHYTpeHHero применения;
сиропы;
порошки И rранулы для приrотовления
сиропов.
ПРОИЗВОДСТВО
При разработке жидких лекарствен
ных средств для внутреннеro применения, в
состав которых входят антимикробные KOH
серванты, уполномоченному opraHY пред
ставляют данные, подтверждающие необ
ходимость использования и эффективность
выбранных консервантов. Подходящий метод
определения и критерии оценки эффективно
сти консервантов описаны в статье 5.1.3. Эф
фективность антuмuкробных KOHcepвaH
тов.
При разработке жидких лекарственных
средств для внутреннеro применения должно
быть доказано, что из однодозовоro контей
нера, содержащеro лекарственное средство,
может быть извлечено номинальное еro co
держимое. #В случае отсутствия у производи
теля лекарственноro средства сертификата
соответствия GMP, а также данных о валида
ции процесса производства KOHKpeTHoro жид
Koro лекарственноrо средства для BHYTpeH
неro применения испытание по определению
извлекаемоro объема должно быть включено
в спецификацию roTOBOro продукта.#
При производстве, упаковке, xpaHe
нии и реализации жидких лекарственных
средств для внутреннеro применения прини
мают меры, обеспечивающие микробиолоrи
ческую чистоту в соответствии с требовани
ями статьи 5.1.4. МuкроБUОЛ02uческая чuсто
та нестерuльных лекарственных средств u
фармацевтuческuх субстанций.
При производстве лекарственных средств
для BHYTpeHHero применения, содержа
щих дисперrированные частицы, принимают
меры, обеспечивающие необходимый размер
частиц и еro контроль.
ИСПЫТАНИЯ
Однородность дозированных единиц.
Растворы, суспензии и эмульсии в однодозо
вых контейнерах должны выдерживать испы
тание на однородность дозированных единиц
(2.9.40) или, если это обосновано и утвержде
но, испытания на однородность содержания
и/или однородность массы, как указано ниже.
На лекарственное растительное сырье и ле
карственные средства из Hero, представлен
ные в дозированной форме, требования дaH
ноro раздела не распространяются.
Однородность содержания (2.9.6). Если
нет друrих указаний в частной статье, жидкие
лекарственные средства в виде суспензий в
однодозовых контейнерах должны выдержи
вать следующее испытание: после взбалты
вания освобождают каждый контейнер как
можно полнее и проводят испытание на опре
деление содержания действующеro веще
ства в каждом контейнере. Лекарственное
средство должно выдерживать испытание
на однородность содержания действующе
ro вещества в единице дозированноro лекар
cTBeHHoro средства (тест В).
Однородность массы. Жидкие лекар
ственные средства в виде растворов или
эмульсий в однодозовых контейнерах должны
выдерживать следующее испытание: OCBO
бождают каждый из 20 контейнеров как можно
полнее, взвешивают содержимое каждоro
контейнера и определяют среднюю массу co
держимоrо. Масса содержимоro не более чем
двух контейнеров может отклоняться более
чем на 10 % от средней массы, масса coдep
жимоrо ни одноro контейнера не должна OT
клоняться более чем на 20 %.
Доза и однородность дозирования
капель для BHYTpeHHero применения. Ko
личество капель, соответствующее одной
дозе, с помощью капающеro или дозирующе
ro устройства помещают в мерный цилиндр.
1148
rосударственная фармакопея Республики Беларусь
ИСПЫТАНИЯ
Скорость капания не должна превышать 2
капли в секунду. Жидкость взвешивают, при
бавляют еще одну дозу и вновь взвешива
ют; повторное прибавление с последующим
взвешиванием проводят до тех пор, пока не
будет взвешено 1 О доз. Определяют среднюю
массу дозы. Масса ни однои дозы не должна
отклоняться более чем на 1 О % от средней
массы. Суммарная масса 1 О доз не должна
отличаться более чем на 15 % от номиналь
ной массы 1 О доз. Если необходимо, измеря
ют общий объем 1 О доз. Объем не должен OT
личаться более чем на 15 % от номинальноro
объема 10 доз.
Однородность массы доз в MHoroAo
зовых контейнерах (2.9.27). Жидкие лекар
ственные средства для внутреннеro приме
нения в мноrодозовых контейнерах должны
выдерживать испытание на однородность
массы одной дозы, высвобожденной из MHO
roдозовоro контейнера. Данный раздел не
распространяется на капли для BHYTpeHHero
применения.
МАРКИРОВКА
На этикетке указывают названия всех дo
бавленных антимикробных консервантов.
Растворы, эмульсии и суспензии
для BHYTpeHHero применения
ОПРЕДЕЛЕНИЕ
Растворы, эмульсии и суспензии для BHY
треннеro применения выпускают в однодозовых
или мноroдозовых контейнерах. Каждая доза из
мноroдозовоro контейнера применяется с помо
щью подходящеro дозирующеro приспособле
ния, предназначенноro для измерения пред
писанноro объема. Приспособление обычно
представляет собой мерную ложку или CTaKaH
чик вместимостью 5 мл или кратной обозначен
ному объему, либо оральный шприц для друroro
объема.
Порошки И rранулы для приютовления
растворов и суспензий для внутреннею
применения
ОПРЕДЕЛЕНИЕ
Порошки и rранулы для приroтовления pac
творов и суспензий для внутреннеro применения
в основном соответствуют определениям статьи
Порошкu для внутренне20 прuмененuя или
статьи rранулы соответственно. Они MOryT co
держать также вспомоrательные вещества, KOТO
рые способствуют дисперrированию или paCTBO
рению или предотвращают аrреrацию частиц.
После растворения или суспендирования
они должны соответствовать требованиям, KOТO
рые предъявляют к растворам или суспензиям
для внутреннеro применения соответственно.
Однородность дозированных единиц.
Однодозовые порошки и однодозовые rранулы
должны выдерживать испытание на OДHOpOД
ность дозированных единиц (2.9.40) или, если
это обосновано и утверждено, испытания на
однородность содержания и/или однородность
массы, как указано ниже. На лекарственное pac
тительное сырье и лекарственные средства из
неro, представленные в дозированной форме,
требования данноrо раздела не распространя
ются.
Однородность содержания (2.9.6). По
рошки или rранулы в однодозовых контейнерах
С содержанием действующеro вещества менее
2 Mr или менее 2 % от общей массы содержи
моro должны выдерживать испытание на OДHO
родность содержания действующеro вещества
в единице дозированноro лекарственноro cpeд
ства (тест В), если нет друrих указаний в част
ной статье. Если лекарственное средство coдep
жит более одноro действующеro вещества, это
требование распространяется только на те Be
щества, содержание которых соответствует BЫ
шеуказанным условиям.
Однородность массы (2.9.5). Порошки и
rранулы в однодозовых контейнерах должны BЫ
держивать испытание на однородность массы
для единицы дозированноro лекарственноrо
средства. Испытание на однородность массы не
требуется, если испытание на однородность co
держания предусмотрено для всех действующих
веществ.
МАРКИРОВКА
На этикетке указывают:
способ приrотовления раствора или cy
спензии;
условия и срок хранения после приroтов
ления.
Капли для внутреннею применения
ОПРЕДЕЛЕНИЕ
Капли для внутреннеro применения это
растворы, эмульсии или суспензии, которые
принимают малыми объемами (каплями) с по
мощью подходящеro дозирующеrо устройства.
МАРКИРОВКА
На этикетке указывают количество капель в
одном миллилитре или rpaMMe лекарственноro
средства, если доза измеряется в каплях.
Порошки для приютовления капель
для внутреннею применения
ОПРЕДЕЛЕНИЕ
Порошки для приrотовления капель для
внутреннеro применения в основном COOT
Жидкие лекарствеННblе средства для внутреннесо примененuя
1149
ветствуют определениям статьи Порошки для
внутренне20 применения. Они MorYT coдep
жать вспомоrательные вещества, облеrча
ющие растворение или дисперrирование в
соответствующем растворителе или предот
вращающие аrреrацию частиц.
После растворения или суспендирова
ния они должны соответствовать требовани
ям, предъявляемым к каплям для BHYTpeHHe
ro применения.
ИСПЫТАНИЯ
Однородность дозированных единиц.
Однодозовые порошки для приroтовления
капель для внутреннеro пименения должны
выдерживать испытание на однородность
дозированных единиц (2.9.40) или, если это
обосновано и утверждено, испытания на oд
нородность содержания иlили однородность
массы, как указано ниже. На лекарственное
растительное сырье и лекарственные cpeд
ства из Hero, представленные в дозирован
ной форме, требования данноro раздела не
распространяются.
Однородность содержания (2.9.6). По
рошки для приrотовления капель для BHY
TpeHHero применения в однодозовых KOH
тейнерах с содержанием действующеro
вещества менее 2 Mr или менее 2 % от общей
массы содержимоro должны выдерживать ис
пытание на однородность содержания дей
ствующеrо вещества в единице дозированно
ro лекарственноro средства (тест В), если нет
друrих указаний в частной статье. Если ле
карственное средство содержит более одноro
действующеro вещества, это требование pac
пространяется только на те вещества, coдep
жание которых соответствует вышеуказан
ным условиям.
Однородность массы (2.9.5). Порош
ки для приrотовления капель для BHYTpeHHe
ro применения в однодозовых контейнерах
должны выдерживать испытание на OДHO
родность массы для единицы дозированноrо
лекарственноro средства. Испытание на oд
нородность массы не требуется, если испы
тание на однородность содержания предус
мотрено для всех действующих веществ.
Сиропы
ОПРЕДЕЛЕНИЕ
Сиропы это жидкие rycToBaTbIe ле
карственные средства, характеризующиеся
сладким вкусом. Они MorYT содержать caxa
розу в концентрации не менее 45 % (м/м).
Сладкий вкус может быть достиrнут исполь
зованием друrих полиолов или подсласти
телей. Сиропы обычно содержат ароматиза
торы или друrие вкусовые добавки. Каждая
доза из мноroдозовоro контейнера применя
ется при помощи подходящеro дозирующеrо
приспособления, предназначенноro для из
мерения предписанноrо объема. Приспосо
бление обычно представляет собой мерную
ложку или стаканчик вместимостью 5 мл или
кратной обозначенному объему.
МАРКИРОВКА
На этикетке указывают наименование и
концентрацию полиола или подсластителя.
Порошки И rранулы для приrотов
ления сиропов
ОПРЕДЕЛЕНИЕ
Порошки и rранулы для приrотовления
сиропов в основном соответствуют определе
ниям статьи Порошкu для внутренне20 прu
мененuя или статьи Транулы соответственно.
Они MorYT содержать вспомоrательные веще
ства, облеrчающие растворение.
После растворения они должны COOTBeT
ствовать требованиям, предъявляемым к си
ропам.
ИСПЫТАНИЯ
Однородность дозированных единиц.
Однодозовые порошки и rранулы для приrо
товления сиропа должны выдерживать испы
тание на однородность дозированных единиц
(2.9.40) или, если это обосновано и утвержде
но, испытания на однородность содержания
и/или однородность массы, как указано ниже.
На лекарственное растительное сырье и ле
карственные средства из Hero, представлен
ные в дозированной форме, требования дaH
ноro раздела не распространяются.
Однородность содержания (2.9.6). Oд
нодозовые порошки и rранулы для приroтов
ления сиропа с содержанием действующеro
вещества менее 2 Mr или менее 2 % от общей
массы содержимоro должны выдерживать ис
пытание на однородность содержания дей
ствующеrо вещества в единице дозированно
ro лекарственноro средства (тест В), если нет
друrих указаний в частной статье. Если ле
карственное средство содержит более одноro
действующеro вещества, это требование pac
пространяется только на те вещества, coдep
жание которых соответствует вышеуказан
ным условиям.
Однородность массы (2.9.5). OДHOДO
зовые порошки и rранулы для приroтовления
сиропа должны выдерживать испытание на
однородность массы для единицы дозирован
Horo лекарственноro средства. Испытание на
однородность массы не требуется, если ис
пытание на однородность содержания предус
мотрено для всех действующих веществ.
01/2013:0927
rосударственная фармакопея Республики Беларусь
1150
ЖИДКИЕ ЛЕКАРСТВЕННЫЕ
СРЕДСТВА ДЛЯ НАРУЖноrо
ПРИМЕНЕНИЯ
Р,аераrаtiопеs Iiquidae ad usum dermicum
Требованuя данной статьи не распростра
няются на лекарственные средства, предна
значенные для систеМН020 действuя, еслu не
обосновано u не утверждено uначе.
ОПРЕДЕЛЕНИЕ
Жидкие лекарственные средства для Ha
ружноro применения это различные по вязко
сти лекарственные средства, предназначенные
для местноro или трансдермальноro высвобож
дения действующих веществ. Это растворы,
эмульсии или суспензии, которые содержат одно
или более действующих веществ в COOTBeTCTBY
ющем носителе. Они MOryт содержать COOTBeT
ствующие антимикробные консерванты, антиок
сиданты и друrие вспомоrательные вещества,
такие как стабилизаторы, ЭМУЛbrаторы и заry
стители.
Эмульсии MOryT расслаиваться, но леrко
восстанавливаются при взбалтывании. Суспен
зии MOryT образовывать осадок, который быстро
ресуспендируется при взбалтывании, образуя
суспензию достаточно стабильную, чтобы обе
спечить однородность лекарственноro средства
при дозировании.
Если это применимо, контейнеры для
жидких лекарственных средств должны COOTBeT
ствовать требованиям статей разделов 3. 1. Ma
терuалы, uспользуемые для проuзводства KOH
тейнеров и 3.2. Контейнеры.
Жидкие лекарственные средства, выпуска
емые в контейнерах под давлением, должны co
ответствовать требованиям статьи Лекарствен
ные средства, находi1щuеся под давленuем.
Лекарственные средства, предназначен
ные для применения на сильно поврежденных
кожных покровах, должны быть стерильными.
Лекарственные средства для наружноro
применения можно классифицировать как:
шампуни;
пены для кожи.
ПРОИЗВОДСТВО
При разработке жидких лекарственных
средств для наружноrо применения. в состав
которых входят антимикробные KOHcepBaH
ты, уполномоченному opraHY представляют
данные, подтверждающие необходимость
использования и эффективность выбран
ных консервантов. Подходящий метод опре
деления и критерии оценки эффективности
консервантов представлены в статье 5.1.3.
Эффектuвность антuмuкробных KOHcep
вантов.
При разработке жидких лекарственных
средств для наружноro применения должно
быть доказано, что из однодозовоro контейнера,
содержащеro лекарственное средство, может
быть извлечено номинальное ero содержимое.
#В случае отсутствия у производителя лекар
cTBeHHoro средства сертиqpиката соответствия
GMP, а также данных о валидации процесса
производства конкретноro жидкоro лекарствен
Horo средства для наружноro применения ис
пытание по определению извлекаемоro объема
должно быть включено в спецификацию roтово
ro продукта.#
При производстве, упаковке, хранении и pe
ализации жидких лекарственных средств для
наружноro применения принимают меры, обе
спечивающие микробиолоrическую чистоту в
'"Соответствии с требованиями статьи 5. 1.4. Mи
кроБUОЛ02uческая чuстота нестерuльных ле
карственных средств u фармацевтическuх
субстанций.
Стерильные жидкие лекарственные cpek
ства для наружноro применения производят С
использованием материалов и методов, обеспе
чивающих стерильность и предотвращающих
заrрязнение лекарственных средств и развитие
микроорrанизмов в соответствии с требовани
ями статьи 5.1.1. Методы ПРU20товленuя cтe
рuльных продуктов.
При производстве жидких лекарственных
средств, содержащих дисперrированные части
цы, принимают меры, обеспечивающие необхо
димый размер частиц и еro контроль.
ИСПЫТАНИЯ
Стерильность (2.6.1). Если на этикетке YKa
зано, что лекарственное средство стерильно,
оно должно выдерживать испытание на стериль
ность.
ХРАНЕНИЕ
Если лекарственное средство стерильно,
еro хранят в стерильных воздухонепроницаемых
контейнерах с контролем первоro вскрытия.
МАРКИРОВКА
На этикетке указывают:
названия всех добавленных антимикроб
ных консервантов;
«стерильно», если необходимо.
Шампуни
ОПРЕДЕЛЕНИЕ
Шампуни это жидкие или иноrда мяrкие
лекарственные средства, предназначенные для
применения на коже roловы и последующеro
смывания их водой. При растирании с водой они
обычно образуют пену.
Шампуни являются эмульсиями, суспензи
ями или растворами. Они обычно содержат по
верхностно активные вещества.
Капсулы
Пены для кожи
1151
ОПРЕДЕЛЕНИЕ
Пены для кожи должны соответствовать
требованиям статьи Пены медицинские.
01/2013:0016
КАПСУЛЫ
Capsu/ae
Требованuя данной статьи необязательны
для лекарствеННbfХ средств, иЗ20товленных в
виде капсул, предназначенных к прuмененuю
не оральным, а дРУ2ИМ способом. Требованuя к
таким лекарствеННbfМ средствам прuведены в
дРУ2их общuх статьях, напрuмер Лекарствен
ные средства для ректаЛЬН020 прuменения
uлu Лекарственные средства для ва2uнально
20 прuмененuя.
ОПРЕДЕЛЕНИЕ
Капсулы твердые лекарственные cpeд
ства с твердой или мяrкой оболочкой разной
формы и вместимости. Капсула содержит
одну дозу действующею вещества или Be
ществ и предназначена для оральною приме
нения.
Оболочки капсул изroтавливают из желати
на или друrих веществ, консистенция оболочки
может быть обеспечена путем добавления таких
веществ, как rлицерин или сорбит. В состав обо
лочки MOryт входить такие вспомоrательные Be
щества, как поверхностно активные вещества,
непрозрачные наполнители, антимикробные
консерванты, подсластители, красители, разре
шенные к применению уполномоченным opra
ном, ароматизаторы и др. Поверхность капсул
может быть маркирована.
Содержимое капсул может быть твердым,
жидким или пастообразным. Оно состоит из
одноro или более действующих веществ и вспо
моrательных веществ, таких как растворители,
разбавители, смазывающие, разрыхляющие Be
щества и др., или без вспомоrательных веществ.
Содержимое капсул не должно разрушать обо
лочку. Однако под воздействием пищеваритель
ных соков оболочка, напротив, должна BЫCBO
бождать содержимое капсул.
Контейнеры для капсул должны соответ
ствовать требованиям статей разделов 3. 1. Ma
терuалы, uспользуемые для проuзводства KOH
тейнеров и 3.2. Контейнеры, если нет друrих
указаний в частной статье.
Капсулы MoryT быть классифицированы как:
капсулы твердые;
капсулы мяrкие;
капсулы кишечнорастворимые;
капсулы с модифицированным высвобож
дением;
облатки.
ПРОИЗВОДСТВО
В процессе производства, упаковки, xpaHe
ния и реализации капсул принимают меры, 06e
спечивающие микробиолоrическую чистоту в
соответствии с требованиями статьи 5. 1.4. Mи
кроБUОЛ02uческая чистота нестеРUЛЬНbfХ ле
карственных средств u фармацевтuческuх
субстанцuЙ
ИСПЫТАНИЯ
Однородность дозированных единиц
(2.9.40). Капсулы должны выдерживать испыта
ние на однородность дозированных единиц или,
если это обосновано и утверждено, испытания
на однородность содержания и/или OДHOpOД
ность массы, как указано ниже. На лекарствен
ное растительное сырье и лекарственные cpeд
ства из неro, представленные в дозированной
форме, требования данною раздела не распро
страняются.
Однородность содержания (2.9.6). Кап
супы с содержанием действующеro вещества
менее 2 Mr или менее 2 % от общей массы co
держимоro должны выдерживать испытание на
однородность содержания действующею веще
ства в единице дозированноro лекарственно
ro средства (тест В), если нет друrих указаНИI1
в частной статье. Если лекарственное средство
содержит более одноro действующеro веще
ства, это требование распространяется только
на те вещества, содержание которых COOTBeT
ствует вышеуказанным условиям.
Однородность массы (2.9.5). Капсулы
должны выдерживать испытание на OДHOpOД
ность массы для единицы дозированноro ле
карственною средства. Испытание на OДHOpOД
ность массы не требуется, если испытание на
однородность содержания предусмотрено для
всех действующих веществ.
Растворение. Испытание проводят для
подтверждения высвобождения действующеro
вещества или веществ, например, одним из спо
собов, описанных в статье 2.9.3. Тест «Pacтвo
рение» для твердых дозuрованных форм.
Если проводится испытание по показателю
«Растворение», испытание по показателю «Pac
падаемость» не требуется.
ХРАНЕНИЕ
Хранят при температуре не выше 30 ОС.
МАРКИРОВКА
На этикетке указывают названия всех дo
бавленных антимикробных консервантов.
1152
rосударственная фармакопея Республики Беларусь
Капсулы твердые
ОПРЕДЕЛЕНИЕ
Капсулы твердые имеют оболочку. cocтo
ящую из двух предварительно изroтовленных
частей цилиндрической формы, один конец KO
торых закруrлен и закрыт, а друroй конец открыт.
ПРОИЗВОДСТВО
Действующее вещество или вещества,
обычно в твердом состоянии (в виде порошка
или rранул), засыпают в одну из частей оболочки
и закрывают второй частью. Надежность закры
тия капсулы может быть усилена COOTBeTCTBY
ющими способами.
ИСПЫТАНИЯ
Распадаемость. Капсулы твердые должны
выдерживать испытание на распадаемость Ta
блеток и капсул (2.9.1). В качестве жидкой среды
используют воду Р. Если указано в частной
статье, в качестве жидкой среды может быть ис
пользована 0.1 М кuслота хлорuстоводородная
или uскусственный желудочный сок Р. Если кап
сула всплывает на поверхность воды, следует
использовать диск. Прибор включают на 30 мин,
если нет друrих указаний в частной статье.
Капсулы мяrкие
ОПРЕДЕЛЕНИЕ
Капсулы мяrкие обычно имеют более тол
стую оболочку, чем капсулы твердые. Оболочка
цельная и имеет различные формы.
ПРОИЗВОДСТВО
Капсулы мяrкие обычно формируют, запол
няют и запечатывают в одной операции, но для
экстемпоральноro изroтовления MOryT быть ис
пользованы предварительно изroтовленные
оболочки. Оболочка может содержать действу
ющее вещество.
Жидкости MOryT быть заключены в капсу
лу непосредственно; твердые вещества обычно
растворяют или дисперrируют в подходящем
растворителе для получения раствора или cy
спензии пастообразной консистенции.
Возможна частичная миrрация компонентов
содержимоro капсулы в оболочку и наоборот.
обусловленная природой контактирующих MaTe
риалов и поверхностей.
ИСПЫТАНИЯ
Распадаемость. Капсулы мяrкие должны
выдерживать испытание на распадаемость Ta
блеток и капсул (2.9.1). В качестве жидкой среды
используют воду Р. Если указано в частной
статье, в качестве жидкой среды может быть ис
пользована О, 1 М кuслота хлорuстоводородная
или uскусственный желудочный сок Р. В каждую
стеклянную трубку помещают диск. Жидкие дей
ствующие вещества, распределенные в мяrкой
капсуле, MOryT обволакивать диск; в таких случа
ях или если указано в частной статье, диск не ис
пользуют. Прибор включают на 30 мин, если нет
друrих указаний в частной статье, и исследуют
состояние капсул. Если капсулы не полностью
распались из за прилипания к дискам, испыта
ние повторяют на следующих шести капсулах
без дисков.
Капсулы с модифицированным
высвобождением
ОПРЕДЕЛЕНИЕ
Капсулы с модифицированным высвобож
дением твердые или мяrкие капсулы, которые
имеют в составе содержимоrо или оболочки,
или в том и друrом одновременно специальные
вспомоrательные вещества или изroтовлены
специальным методом, предназначенные для
изменения скорости, места или времени BЫCBO
бождения действующеro вещества или веществ.
К капсулам с модифицированным BЫCBO
бождением относятся капсулы с пролонrирован
ным и замедленным высвобождением.
ПРОИЗВОДСТВО
Проводят испытание, подтверждающее co
ответствующее высвобождение действующеro
вещества или веществ.
Капсулы кишечнорастворимые
ОПРЕДЕЛЕНИЕ
Капсулы кишечнорастворимые капсу
лы с замедленным высвобождением, которые
должны быть устойчивыми к действию желудоч
ноro сока и высвобождать действующее веще
ство или вещества в кишечном соке. Как прави
ло, они изroтовлены путем заполнения капсул
rранулами или частицами, покрытыми кислото
устойчивой оболочкой или, в некоторых случаях,
путем покрытия твердых или мяrких капсул кис
лотоустойчивой оболочкой (кишечные капсулы).
ПРОИЗВОДСТВО
Для капсул, заполненных rранулами или ча
стицами, покрытыми кислотоустойчивой обо
лочкой, проводят испытание, подтверждающее
высвобождение действующеro вещества или Be
ществ.
ИСПЫТАНИЯ
Распадаемость. Для капсул с кислото
устойчивой оболочкой проводят испытание на
распадаемость (2.9.1) со следующими изме
нениями: в качестве жидкой среды используют
0,1 М кuслоту хлорuстоводородную и при бор
включают на 2 ч, если нет друrих указаний в
частной статье, без дисков. Исследуют состоя
ние капсул. Время устойчивости в кислой среде
колеблется в зависимости от состава. Обычно
оно составляет от 2 ч до 3 ч, но в любом случае
ЛекарствеННbtе средства для ваzинальноzо применения
1153
оно ДОЛЖНО быть не менее 1 ч. Ни одна из
капсул не должна обнаруживать признаков pac
пада или разрывов, через которые возможен
выход содержимоro. Кислоту заменяют фос
фатным буферным раствором рН 6,8 Р. Если
указано в частной статье, может быть исполь
зован буферный раствор рН 6,8 с добавлени
ем порошка панкреатuна Р (например, 0,35 r
порошка панкреатuна Р на 100 мл буферноro
раствора). В каждую стеклянную трубку поме
щают диск. Прибор включают на 60 мин и ис
следуют капсулы. Если капсулы не выдержали
испытание из за прилипания к дискам, испыта
ние повторяют на следующих шести капсулах
без дисков.
Растворение. Для капсул, заполненных
rранулами или частицами, покрыты м и кислото
устойчивой оболочкой, проводят испытание,
подтверждающее высвобождение действующе
ro вещества или веществ, например, одним из
способов, описанным в статье 2.9.3. Тест «Pac
творение» для твердых дозuрованных форм.
Облатки
ОПРЕДЕЛЕНИЕ
Облатки твердые лекарственные cpeд
ства с твердой оболочкой, содержащие одну
дозу действующеro вещества или веществ.
Оболочка облатки изrотавливается из пресноro
хлеба, изroтовленноro обычно из рисовой муки,
и состоит из двух предварительно изrотовлен
ных плоских цилиндрических частей. Перед
применением облатку поrружают в воду на He
сколько секунд, затем проrлатывают, запивая
водой.
МАРКИРОВКА
На этикетке указывают способ применения
облатки.
01/2013:1164
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
для ВАrИНАльноrо ПРИМЕНЕНИЯ
Vаgiпаliа
ОПРЕДЕЛЕНИЕ
Лекарственные средства для ваrинаllЬНО
ro применения это жидкие, мяrкие или TBep
дые лекарственные средства, предназначенные
для применения во влаrалище обычно с целью
обеспечения местноro действия. Они содержат
одно или более действующих веществ в подхо
дящей основе.
145. За.. 1712.
rде это применимо, контейнеры для ле
карственных средств для ваrинальноro при
менения должны соответствовать требо
ваниям статей разделов 3.1. Матерuалы,
uспользуемые для производства контейне
ров и 3.2. Контейнеры.
Лекарственные средства для ваrинальноro
применения можно классифицировать как:
пессарии;
ваrинальные таблетки;
ваrинальные капсулы;
ваrинальные растворы, эмульсии и cy
спензии;
таблетки для приroтовления ваrинальных
растворов и суспензий;
мяrкие лекарственные средства для ваrи
нальноro применения;
ваrинальные пены;
ваrинальные тампоны.
ПРОИЗВОДСТВО
При разработке должно быть доказано,
что из однодозовоro контейнера, содержаще
ro жидкое или мяrкое лекарственное средство,
может быть извлечено номинальное ero coдep
жимое. #В случае отсутствия у производителя
лекарственноro средства сертификата соответ
ствия GMP, а также данных о валидации про
цесса производства конкретноro лекарственноro
средства для ваrинальноrо применения испы
тание по определению извлекаемоrо объема
должно быть включено в спецификацию rOToBo
ro продукта.#
При производстве, упаковке, хранении и
реализации лекарственных средств для ваrи
нальноro применения принимают меры, обе
спечивающие микробиолоrическую чистоту в
соответствии с требованиями статьи 5. 1.4. Mи
кроБUОЛ02uческая чuстота нестерuльных ле
карственных средств u фармацевтuческuх
субстанцuй.
ИСПЫТАНИЯ
Однородность дозированных единиц
(2.9.40). Жидкие и мяrкие лекарственные cpeд
ства в однодозовых контейнерах должны BЫ
держивать испытание на однородность дo
зированных единиц. Твердые лекарственные
средства в однодозовых контейнерах должны
выдерживать испытание на однородность дози
рованных единиц или, если это обосновано и
утверждено, испытание на однородность массы
или на однородность содержания, как указано
ниже. На лекарственное растительное сырье и
лекарственные средства из неro, представлен
ные в дозированной форме, требования дaHHO
ro раздела не распространяются.
Однородность содержания (2.9.6).
Твердые лекарственные средства в OДHOДO
зовых контейнерах с содержанием действу
ющеro вещества менее 2 Mr или менее 2 %
1154
rосударственная фармакопея Республики Беларусь
от общей массы содержимоro должны ВL,lдер
живать испытание на однородность coдep)'{a
ния действующеro вещества в единице дo
зированноrо лекарственноrо средства (тест
А для ваrинальных таблеток или тест В для
пессариев и ваrинальных капсул), если нет
друrих указаний в частной статье. Если ле
карственное средство содержит более одноro
действующеro вещества, это требование pac
пространяется только на те вещества, co
держания которых соответствуют указанным
выше условиям.
Однородность массы (2.9.5). Твердые
лекарственные средства в однодозовых KOH
тейнерах должны выдерживать испытание
на однородность массы для единицы дози
рованноro лекарственноro средства. Испы
тание на однородность массы не требуется,
если испытание на однородность содержания
предусмотрено для всех действующих веществ.
Растворение. Для подтверждения co
ответствующеro высвобождения действу
ющеrо вещества (веществ) ,из однодозовых
твердых лекарственных средств проводят ис
пытание, например, одним из способов, опи
санных в статьях 2.9.3. Тест' «Pacтвope
ние» для твердых дозированных форм или
2.9.42. Тест «Растворение» для лuпофuль
ных твердых дозuрованных форм.
Если проводят испытание по показате
лю «Растворение», испытание по показателю
«Распадаемосты) не требуется.
Пессарии
ОПРЕДЕЛЕНИЕ
Пессарии это твердые однодозовые
лекарственные средства. Пессарии MorYT
быть различной формы, обычно яйцевидной;
по объему и консистенции должны COOTBeT
ствовать ваrинальному применению.
Пессарии содержат одно или более дей
ствующих веществ, дисперrированных или
растворенных в подходящей основе, KOTO
рая растворяется или дисперrируется в воде
или расплавляется при температуре тела. В
состав пессариев, при необходимости, MorYT
входить вспомоrательные вещества, такие
как разбавители, адсорбенты, поверхност
но активные и смачивающие вещества, анти
микробные консерванты, а также красители,
разрешенные к применению уполномочен
ным opraHoM.
ПРОИЗВОДСТВО
Пессарии обычно изrотавливают методом
выливания. При производстве пессариев при
нимают меры, обеспечивающие необходимый
и контролируемый размер частиц. При необ
ходимости действующее вещество (веще
ства) предварительно измельчают и просеи
вают через подходящее сито.
Если пессарии изroтавливают методом
выливания, приroтовленную массу предва
рительно расплавляют при наrревании и раз
ливают в соответствующие формы. Пессарии
затвердевают при охлаждении. Чтобы обе
спечить процесс затвердевания, вводят такие
вспомоrательные вещества, как твердый жир,
макроroлы, масло какао, различные rеле
образующие смеси, например, состоящие из
желатина, воды и rлицерина.
Для подтверждения соответствующеro
высвобождения действующеro вещества или
веществ из пессариев, предназначенных для
пролонrированноro MecTHoro действия, про
водят подходящее испытание.
ИСПЫТАНИЯ
Распадаемость (2.9.2). Если пессарии не
предназначены для MecTHoro пролонrирован
ноro действия, они должны выдерживать ис
пытание. Если иное не соrласовано и не YT
верждено, состояние пессариев исследуют
через 60 мин.
8аrинальные таблетки
ОПРЕДЕЛЕНИЕ
Ваrинальные таблетки это твердые oд
нодозовые лекарственные средства, в общем
сходные определениям таблеток без оболоч
ки или таблеток, покрытых оболочкой, приве
денным в статье Таблеткu.
ПРОИЗВОДСТВО
Для подтверждения соответствующеro
высвобождения действующеro вещества или
веществ из ваrинальных таблеток, предна
значенных для пролонrированноro местноro
действия, проводят подходящее испытание.
ИСПЫТАНИЯ
Распадаемость (2.9.2). Если ваrиналь
ные таблетки не предназначены для MeCTHO
ro пролонrированноrо действия, они должны
выдерживать испытание (специальный метод
для ваrинальных таблеток). Если иное не
обосновано и не утверждено, состояние Ba
rинальных таблеток исследуют через 30 мин.
8аrинальные капсулы
ОПРЕДЕЛЕНИЕ
Ваrинальные капсулы (пессарии с оболоч
кой) это твердые однодозовые лекарствен
ные средства, в общем сходные с мяrкими
капсулами, описанными в статье Капсулы, OT
личающиеся только формой и размером. Ba
rинальные капсулы MorYT иметь различную
форму, обычно яйцевидную. Они должны быть
rладкими и иметь однородный внешний вид.
Лекарственные средства для ва2uнаЛЬНО20 прuменения
1155
ПРОИЗВОДСТВО
Для подтверждения соответствующеro
высвобождения действующеro вещества или
веществ из ваrинальных капсул, предназна
ченных для пролонrированноro MecTHoro дей
ствия, проводят подходящее испытание.
ИСПЫТАНИЯ
Распадаемость (2.9.2). Если ваrиналь
ные капсулы не предназначены для MecTHoro
пролонrированноrо действия, они должны BЫ
держивать испытание. Если иное не соrласо
вано и не утверждено, состояние ваrинальных
капсул исследуют через 30 мин.
8аrинальные растворы, эмульсии
и суспензии
ОПРЕДЕЛЕНИЕ
Ваrинальные растворы, эмульсии и cy
спензии это жидкие лекарственные cpeд
ства, предназначенные для местноro дей
ствия, орошения или для применения с
диаrностической целью. Они MOryT содержать
вспомоrательные вещества, например, для
обеспечения необходимой вязкости, создания
или стабилизации необходимоro значения рН,
повышения растворимости действующеrо Be
щества или веществ или обеспечения CTa
бильности лекарственноro средства. Вспомо
rательные вещества не должны отрицательно
влиять на основное действие лекарственноrо
средства или, в используемых концентрациях,
оказывать нежелательноro MecTHoro раздра
жающеrо действия.
Ваrинальные эмульсии MorYT рассла
иваться, однако должны быстро BOCCTaHaB
ливаться при взбалтывании. Ваrинальные cy
спензии MorYT образовывать осадок, который
быстро ресуспендируется при взбалтывании,
образуя суспензию достаточно стабильную,
чтобы обеспечить однородность лекарствен
ноro средства при введении.
Ваrинальные растворы, эмульсии и cy
спензии обычно выпускают в однодозовых
контейнерах, которые приспособлены для BBe
дения лекарственноro средства во влаrалище
или снабжены подходящей насадкой.
ПРОИЗВОДСТВО
При производстве ваrинальных суспен
зий принимают меры, обеспечивающие необ
ходимый и контролируемый размер частиц, с
учетом способа применения лекарственноro
средства.
Таблетки для приrотовления
ваrинальных растворов или суспензий
ОПРЕДЕЛЕНИЕ
Таблетки для приroтовления ваrинальных
растворов и суспензий это однодозовые ле
карственные средства, которые растворяют
или дисперrируют в воде непосредственно
перед применением. Они MorYT содержать
вспомоrательные вещества, которые способ
ствуют растворению или дисперrированию
или предотвращают аrреrацию частиц.
За исключением испытания на распада
емость, таблетки для приrотовления paCTBO
ров или суспензий для влаrалищноrо приме
нения должны соответствовать определению,
приведенному в статье Таблеткu.
После растворения или дисперrирования
они должны выдерживать требования, KOТO
рые предъявляют к ваrинальным растворам
или суспензиям соответственно.
ИСПЫТАНИЯ
Распадаемость (2.9.1). Таблетки для при
roтовления ваrинальных растворов или cy
спензий должны выдерживать испытание, при
этом используют воду Р при температуре от
15 ос до 25 ос и 6 таблеток. Состояние табле
ток исследуют через 3 мин. Должны распасть
ся все 6 таблеток.
МАРКИРОВКА
На этикетке указывают:
способ приroтовления ваrинальных pac
творов или суспензий;
условия и срок хранения раствора или
суспензии после приrотовления.
Мяrкие лекарственные средства
для ваrинальноro применения
ОПРЕДЕЛЕНИЕ
К мяrким лекарственным средствам для
ваrинальноro применения относятся мази,
крема или rели.
Они обычно представляют собой OДHOДO
зовые лекарственные средства в контейнерах,
снабженных подходящей насадкой.
Мяrкие лекарственные средства для Ba
rинальноrо применения должны выдерживать
требования статьи МЯ2кuе лекарственные
средства для наРУЖН020 прuмененuя.
8аrинальные пены
ОПРЕДЕЛЕНИЕ
Ваrинальные пены должны соответствовать
требованиям статьи Пены медицинские.
8аrинальные тампоны
ОПРЕДЕЛЕНИЕ
Ваrинальные тампоны это твердые oд
нодозовые лекарственные средства, пред
назначенные для введения во влаrалище на
определенное время.
Они должны соответствовать требовани
ям статьи Тампоны медицинские.
1156
01/2013:0671
rосударственная фармакопея Республики Беларусь
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
для инrАЛЯЦИЙ
/пhа/апdа
ОПРЕДЕЛЕНИЕ
Лекарственные средства для инrаляций
это жидкие или твердые лекарственные средства
в виде паров или аэрозолей, предназначенные
для получения местноro или системноro эффек
та при поступлении в дыхательные пути. Ле
карственные средства для инrаляции содержат
одно или более активных веществ, которые paCT
воряют или дисперrируют в соответствующем
носителе.
Лекарственные средства для инrаляций, в
зависимости от типа лекарственноro средства,
содержат пропелленты, вспомоrательные pac
творители, разбавители, антимикробные KOH
серванты, солюбилизирующие и стабилизиру
ющие areHTbI и др. Эти наполнители не должны
оказывать неблаroприятное воздействие на
функции слизистой дыхательных путей и ее
ресничек.
Суспензии и эмульсии должны быстро BOC
станавливаться при встряхивании и оставаться
достаточно стабильными, чтобы обеспечить oд
нородность дозирования.
Лекарственные средства для инrаля
ций выпускают в мноroдозовых или однодозо
вых контейнерах. Лекарственные средства для
инrаляций, выпускаемые в контейнерах под дaB
лением, должны соответствовать требованиям
статьи ЛекарствеННblе средства, находящuеся
под давлением.
Лекарственные средства для инrаляций в
виде аэрозолей (дисперсии твердых или жидких
частиц в rазовой среде) применяют при помощи
одноro из специальных приспособлений:
распылителя (небулайзера);
инrалятора (дозированноro и нrалятора ,
находящеroся под давлением, дозированноro
инrалятора, находящеroся без давления, cyxo
порошковоro инrалятора).
Лекарственные средства для инrаляций
MOryт быть классифицированы как:
лекарственные средства, которые перево
дятся в парообразное состояние;
жидкие лекарственные средства для pac
пыления;
дозированные лекарственные средства
для инrаляций, находящиеся под давлением;
дозированные лекарствеl-lные средства
для инrаляций, находящиеся без давления;
порошковые инrаляции.
ПРОИЗВОДСТВО
При разработке лекарственных средств для
инrаляций, в состав которых входят антими
кробные консерванты, уполномоченному opraHY
представляют данные, подтверждающие эф
фективность выбранных консервантов. Метод
определения и критерии оценки эффективно
сти консервантов должны соответствовать Tpe
бованиям статьи 5.1.3. Эффектuвность aHти
микроБНblХ консервантов.
При производстве, упаковке, хранении и pe
ализации лекарственных средств для инrаляций
принимают меры, обеспечивающие микробио
лоrическую чистоту в соответствии с требовани
ями статьи 5. 1.4. МикроБUОЛ02uческая чuстота
нестериЛЬНblХ лекарствеННblХ средств u фар
мацевтuческuх субстанциЙ
При оценке однородности высвобождаемой
дозы из мноroдозовоro инrалятора недостаточно
оценить только один инrалятор. Производители
должны предоставить методику оценки OДHOpOД
ности высвобождаемых доз из разных инrалято
ров (межинrаляторная однородность доз) и Me
тодику оценки однородности высвобождаемых
доз из одноro инrалятора (внутриинrалятор
ная однородность доз). Процедура для внутри
инrаляторноrо испытания должна оценивать 1 О
специфицированных доз, собранных в начале,
середине и конце количества доз, указанных на
маркировке инrалятора.
МАРКИРОВКА
На этикетке дозированных инrаляторов
должно быть указано:
высвобождаемая доза; если доза являет
ся отмеренной или предварительно выделен
ной, на этикетке указывают отмеренную или
предварительно выделенную дозу;
при необходимости, количество нажатий
на инrалятор для получения минимальной peKO
мендованной дозы;
число доз в инrаляторе.
Если это применимо, на этикетке также YKa
зывают названия всех входящих в состав лекар
ственноro средства антимикробных KOHcepBaH
тов.
Лекарственные средства, которые
переводятся в парообразное состояние
ОПРЕДЕЛЕНИЕ
Лекарственные средства, которые перево
дятся в парообразное состояние, представляют
собой растворы, дисперсии или твердые лекар
ственные средства. Обычно в них добавляют ro
рячую воду и образующийся пар вдыхают.
Жидкие лекарственные средства
для распыления
ОПРЕДЕЛЕНИЕ
Жидкие лекарственные средства для pac
пыления преобразуются в аэрозоли при помощи
распылителей или дозированных распылителей
для растворов, суспензий и эмульсий.
Лекарственные средства для иН2аляцuй
1157
Жидкие лекарственные средства для pac
пыления, которые представляют собой KOHцeH
Tpa перед применением разводят в предпи
санном объеме указанной жидкости. Жидкие
лекарственные средства для распыления
MorYT также быть приroтовлены из порошков.
Значение рН жидких лекарственных
средств в распылителях непрерывноro дей
ствия должно быть не менее 3 и не более 10.
Жидкие лекарственные средства для pac
пыления, которые выпускают в мноroдозовых
контейнерах, Moryт содержать антимикробные
консерванты, за исключением лекарственных
средств, которые сами обладают антимикроб
ными свойствами.
Выпускаемые в мноrодозовых контейнерах
жидкие лекарственные средства для распыле
ния, которые не содержат антимикробные KOH
серванты и которые не обладают антимикроб
ными свойствами, должны быть стерильными и
выпускаться в контейнерах, предотвращающих
микробное заrрязнение содержимоro при xpa
нении и использовании.
Если иное не обосновано и не утверждено,
жидкие лекарственные средства для распыле
ния, выпускаемые в однодозовых контейнерах,
должны быть стерильными и не должны coдep
жать антимикробных консервантов.
Распылители непрерывноro действия
представляют собой приспособления, преоб
разующие жидкости в аэрозоли при помощи
высокоro давления rаза, ультразвука или под
друrим воздействием. Они позволяют вдыхать
необходимую дозу лекарственноro средства с
определенной скоростью. При этом образуют
ся частицы определенноro размера, что позво
ляет распределяться лекарственному средству
в леrких.
Распылители MorYT быть инициализиро
ваться вдохом или MOryT включать в состав
приспособления для синхронизации или MO
дификации действий распылителя с дыханием
пациента.
ПРОИЗВОДСТВО
Скорость высвобождения действующеro
вещества и общее количество высвобожден
ноro вещества определяют с использованием
методов, описанных в общей статье 2.9.44. Ле
карственные средства для распыления: опре
деление параметров. Если обосновано и YT
верждено, MOryт использоваться иные прибор и
методика.
Для жидких лекарственных средств для pac
пыления, которые представляют собой paCTBO
ры или суспензии, устанавливают распреде
ление частиц по размер используя прибор и
методику, описанные в общем разделе 2.9.44.
Лекарственные средства для распыления:
определенuе параметров. Если обосновано и
утверждено, Moryт использоваться иные прибор
и методика.
146. Зак. 1712.
ИСПЫТАНИЯ
Жидкuе лекарственные средства для pac
пыления под20тавлuвают, как указано в ин
струкции по прuмененuю.
Аэродинамическая оценка распылен-
ных аэрозолей. Для жидких лекарственных
средств для распыления, которые представляют
собой суспензии, устанавливают массу мелких
частиц, используя прибор и методику, описан
ные в общем разделе 2.9.44. Лекарственные
средства для распыления: определенuе пара
метров. Если обосновано и утверждено, MOryT
использоваться иные прибор и методика.
Дозированные лекарственные средства
для инrаляций, находящиеся
под давлением
ОПРЕДЕЛЕНИЕ
Дозированные лекарственные средства для
инrаляций, находящиеся под давлением, пред
ставляют собой растворы, суспензии или эмуль
сии, которые выпускают в специальных контей
нерах, снабженных дозирующим клапаном, и
которые находятся под давлением с подходящи
ми пропеллентами или подходящими смесями
растворенных пропеллентов, которые MOryт BЫ
ступать в роли растворителей.
Высвобождаемая доза это доза, высвобож
денная из инrалятора. Для некоторых лекарствен
ных средств устанавливается доза, отмеренная
дозатором. Доза, отмеренная дозатором, опреде
ляется путем прибавления количества, отложив
шеroся в инrаляторе, к высвобожденной дозе. Она
также может быть установлена непосредственно.
ПРОИЗВОДСТВО
Размер частиц вдыхаемоro аэрозоля KOH
тролируют таким образом, чтобы постоянная их
часть откладывалась в леrких. Характеристики
мелких частиц дозированноro лекарственноro
средства для инrаляций, находящеrося под дaB
лением, устанавливают, используя метод, опи
санный в общем разделе 2.9.18. Лекарственные
средства для иН2аляцuй: аэродuнамuческое иc
пытание мелкuх частuц.
Дозированные инrаляторы, находящиеся
под давлением, испытывают на rерметичность.
ИСПЫТАНИЯ
Для дозuрованных лекарственных средств
для UН2аляцuй, находящuхся под давленuем,
uнициалuзuруемых вдохом, опuсанные нuже yc
ловuя М02ут потребовать модuфuкацuu.
ИН2алятор под20тавлuвают, как указано в
инструкции по прuмененuю.
Однородность высвобождаемой дозы.
Контейнеры, как правило, используют в пе
ревернутом виде (клапаном вниз). Для KOH
1158
rосударственная фармакопея Республики Беларусь
тейнеров, которые применяются веРТИК<1ЛЬНО
(клапаном вверх), используют методы Иt-ПЫ
тания, позволяющие определять высвобож
денную дозу.
Прибор для сбора дозы должен обеспечи
вать ее количественный сбор.
С этой целью может быть использован сле
дующий прибор (рисунок 0671 1) и методика.
в..УТ JI p€.Ibfu
.;38.1
.:З55
:;::' @ ' o ' ,
.;272
_ 267 ."
,'251
.'71
....
I IT
r\
1
Vр.- ш а
I-!dру>I<Н [)(!.JbOO
Е..уIDe'М"R!O pe.J<>oo
i=:" , 2C'H .
'.'.....]
o ,,
Сna.з J.1TDO' a К! pe
ПР "",,*,IoIeВfl"УУм;'j
lМPЖВТtlno,фоо"".р"
"""".,
\...f\C......,.е о::олыю
....
, (J.
J
, ,. qj
f@ i.
Рисунок 0671 1. Прuбор для сбора дозы
в дозированных лекарственных средствах,
находящuхся под давлением
(размеры указаны в мuллuметрах)
Прибор состоит из держателя фильтра в
виде стальноro экрана, трубки для сбора об
разца. которая прижата или прикручена к дep
жателю фильтра, и адаптера мундштука, KOTO
рый rерметично соединяет трубку и мундштук
Используют адаптер мундштука, позволяющий
лицевой стороне мундштука инrалятора Haxo
диться на одном уровне с лицевой стороной
трубки для сбора образцов или на 2,5 мм ее
нарезной части, в зависимости от инrалятора.
Вакуумный ниппель соединяет систему, вклю
чающую в себя источник вакуума и реryлятор
потока. Источник должен быть отреrулирован
таким образом, чтобы воздух проходил через
всю конструкцию, включа фильтр и инrалятор,
со скоростью 28,3 л/мин (:t5 %). Во избежание
потерь действующеro вещества в атмосферу
воздух должен проходить через прибор посто
янно. Основание держателя фильтра пред
назначено для фильтров диаметром 25 мм.
Фильтр и друrие составляющие части KOHCTPYK
ции должны быть совместимы с действующим
веществом и растворителями, которые исполь
зуются для извлечения действующеro вещества
из фильтра. Один конец соединительной трубки
сделан таким образом, чтобы прочно прижимать
фильтр к держателю фильтра. В собранном
виде соединения компонентов прибора должны
быть rерметичны, чтобы при воздействии BaKY
ума на держатель фильтра весь воздух прохо
дил через собирательную трубку инrалятора.
Если нет особых указаний в инструкции
по применению, перед использованием инrа
лятор встряхивают в течение 5 с и выпускают
и отбрасывают одну дозу. Перевернутый инrа
лятор разряжают в прибор, нажимая клапан на
некоторое время, чтобы полностью выпустить
порцию лекарственноro средства. Повторяют
процедуру необходимое количество раз, KOTO
рое бы обеспечило отбор минимальной peKO
мендованной дозы. Количественно собирают
содержимое прибора и определяют содержа
ние действующею вещества.
Процедуру повторяют для двух друrих доз.
Сбрасывают несколько доз с интервалом
не менее 5 с до тех пор, пока не останется
(пI2)+1 доз, rдe п количество доз, указан
ное на этикетке, и собирают 4 дозы по описан
ной выше методике.
Опять сбрасывают несколько доз с ин
тервалами не менее 5 с, пока не останется З
дозы. Собирают три последние дозы по опи
санной выше методике.
Для лекарственных средств, содержащих
более одноro действующеro вещества, испы
тание на однородность высвобождаемой дозы
выполняют для каждоro.
Если иное не обосновано и не утвержде
но, лекарственное средство выдерживает ис
пытание, если 9 из 10 результатов находятся
в пределах от 75 % до 125 % среднею значе
ния, а все результаты находятся в пределах от
65 % до 135 %. Если 2 или 3 результата BЫ
падают из пределов 75 125 %, испытание
повторяют с использованием двух друrих ин
rаляторов. Не более 3 из 30 значений MorYT
выходить за пределы 75 125 % и все значе
ния должны быть в пределах 65 135 %.
Размер частиц. Используют прибор и
методику, описанные в общей статье 2.9.18.
Лекарственные средства для UН2аляцuй:
аэродuнамuческое uспытанuе мелкuх частuц
(прибор С, О или Е), рассчитывают дозу мелких
частиц.
Число ДОЗ В инrаляторе. Берут один ин
rалятор и выпускают еro содержимое. Клапан
нажимают с интервалом не менее 5 с. Полу
ченное количество доз должно быть не меньше
значения, указанноro на этикетке (это испыта
ние можно выполнять параллельно с испыта
нием на однородность высвобожденной дозы).
Лекарственные средства для иН2аляций
Дозированные лекарственные средства
для инrаляций, находящиеся без давления
Дозированные лекарственные средства для
инrаляций, находящиеся без давления пред
ставляют собой растворы, суспензии или эмуль
сии для использования в инrаляторах, которые
превращают жидкости в аэрозоли с использова
нием одной или нескольких форсунок, ультра
звуковых колебаний или иных методов. Пред
варительно отмеренный или отмериваемый
инrалятором объем жидкости, превращаемой в
аэрозоль, имеет такой размер, что доза, BЫCBO
бождаемая из инrалятора, может быть поrлоще
на за один или более вдох.
Дозированные лекарственные средства для
инrаляций, находящиеся без давления, которые
выпускают в мноroдозовых контейнерах, MOryT
содержать антимикробные консерванты, за ис
ключением лекарственных средств, которые
сами обладают антимикробными свойствами.
Выпускаемые в мноroдозовых контейнерах
дозированные лекарственные средства для инrа
ляций, находящиеся без давления, которые не co
держат антимикробные консерванты и которые не
обладают антимикробными свойствами, должны
быть стерильными и выпускаться в контейнерах,
предотвращающих микробное заrрязнение co
держимоro при хранении и использовании.
Если иное не обосновано и не утверждено,
дозированные лекарственные средства для инrа
ляций, находящиеся без давления, выпускаемые
в однодозовых контейнерах, должны быть CTe
рильными и не должны содержать антимикроб
ных консервантов.
ПРОИЗВОДСТВО
Размер частиц вдыхаемоro аэрозоля KOH
тролируют таким образом, чтобы значительная
их часть откладывалась в леrких. Характеристи
ки мелких частиц дозированноro лекарственно
ro средства для инrаляций, находящеroся без
давления, устанавливают, используя метод, опи
санный в общей статье 2.9.18. Лекарственные
средства для UН2аляций: аэродинамuческое иc
пытанuе мелкuх частиц. В качестве альтерна
тивы может быть использован метод лазерной
дифракции при условии, что он надлежащим
образом валидирован по сравнению с методом
2.9.18 (прибор С, О или Е).
ИСПЫТАНИЯ
ДЛЯ дозuрованных лекарственных средств
для UН2аляцuй, находящuхся без давленuя, ини
цuалuзuруемых вдохом, опuсанные нuже усло
вuя М02ут потребовать модuфикацuu.
ИН2алятор под20тавлuвают, как указано в
инструкции по прuмененuю.
Однородность высвобождаемой дозы.
Прибор для сбора дозы должен обеспечивать
ее количественный сбор. Может использовать
147 За.. 1712.
1159
ся прибор, описанный в испытании на OДHOpOД
ность высвобождаемой дозы дозированных ин
rаляторов, находящихся под давлением.
Инrалятор разряжают в прибор. Повторяют
процедуру необходимое количество раз, которое
бы обеспечило отбор минимальной peKOMeHДO
ванной дозы. Количественно собирают содержи
мое прибора и определяют содержание действу
ющеro вещества.
Процедуру повторяют для двух друrих доз.
Сбрасывают несколько доз до тех пор, пока
не останется (п/2)+1 доз, rде п количество
доз, указанное на этикетке, и собирают 4 дозы
по описанной выше методике.
Опять сбрасывают несколько доз, пока не
останется 3 дозы. Собирают три последние дозы
по описанной выше методике.
Для лекарственных средств, содержащих
более одноro действующеro вещества, испыта
ние на однородность высвобождаемой дозы BЫ
полняют для каждою.
Если иное не обосновано и не утверждено,
лекарственное средство выдерживает испыта
ние, если 9 из 1 О результатов находятся в пре
делах от 75 % до 125 % среднеro значения, а
все результаты находятся в пределах от 65 %
до 1З5 %. Если 2 или 3 результата выпадают
из пределов 75-------125 %, испытание повторяют
с использованием двух друrих инrаляторов. Не
более 3 из 30 значений MOryт выходить за пре
делы 75 125 % и все значения должны быть в
пределах 65 135 %.
Если обосновано и утверждено, MOryT быть
использованы иные прибор и методика.
Размер частиц. Используют прибор и MeTO
дику, описанные в общей статье 2.9.18. Лекар
ственные средства для UН2аляцuй: аэродина
мuческое uспытанuе мелкuх частuц (прибор
С, О или Е), рассчитывают дозу мелких частиц.
Испытание ПРОВОДЯТ по методике, описанной
для дозированных лекарственных средств для
инrаляций, находящихся под давлением, с co
ответствующей адаптацией для инrаляторов,
находящихся без давления. В зависимости от
характеристик дозированных лекарственных
средств для инrаляций, находящихся без давле
ния, может понадобиться контроль относитель
ной влажности и/или температуры.
Число доз в инrаляторе. Выпускают co
держи мое одною инrалятора. Полученное KO
личество доз должно быть не меньше значения,
указанноro на этикетке (это испытание можно
выполнять параллельно с испытанием на OДHO
родность высвобожденной дозы).
Порошки для инrаляций
ОПРЕДЕЛЕНИЕ
Порошки для инrаляций выпускают в oд
нодозовых или мноrодозовых контейнерах.
1160
rосударственная фармакопея Республики Беларусь
Для более эффективноro их ИСПОЛЬЗОL1ания
действующие вещества MorYT быть в сме.::: '1 с
носителями. Порошки вводят с помощью по
рошковых инrаляторов. В системах, имеющих
дозаторы, порошок используется в капсулах
или в друrих удобных дозированных формах.
В приспособлениях, содержащих резервуар
для порошка, доза лекарственноro средства
отмеряется автоматически.
Высвобождаемая доза это доза, BЫ
свобожденная из инrалятора. Для некоторых
лекарственных средств указанная на этикет
ке доза представляет собой дозу, отмеренную
дозатором, или предварительно дисперrиро
ванную дозу. Доза, отмеренная дозатором,
определяется путем прибавления части дозы,
отложившейся на инrаляторе. к высвобож
денной дозе. Она также может быть YCTaHOB
лена непосредственно.
ПРОИЗВОДСТВО
Размер частиц вдыхаемоro аэрозоля KOH
тролируют таким образом, чтобы постоянная
их часть откладывалась в леrких. Характери
стики мелких частиц дозированноro лекар
cTBeHHoro средства для инrаляций, находяще
roся без давления, устанавливают, используя
метод, описанный в общей статье 2.9.18. Ле
карственные средства для UН2аляцuй: аэро
дuнамuческое uспытанuе мелкuх частuц.
ИСПЫТАНИЯ
ИН2алятор под20тавлuвают, как указа
НО в инструкции по прuмененuю.
Однородность высвобождаемой дозы.
Прибор для сбора дозы должен обеспечи
вать ее количественный сбор. Может исполь
зоваться прибор, описанный в испытании на
однородность высвобождаемой дозы дозиро
ванных инrаляторов, находящихся под дaB
лением, при условии, что размеры трубки и
фильтра позволят обеспечить измеренную
скорость потока. Подходящая трубка описа
на в таблице 0671 1. Трубку присоединяют к
системе в соответствии со схемой, указанной
на рисунке 0671 2 и в таблице 0671 1.
А
Т;эуб\ol,а ДЛS:; сбора образца
............... ВХОД
ДвухсторонниЙ r Клапан 1
электромаrНИТНЫЙ I реrулироваl1И
клаП(iН рPiсхода
Е Н
Соединитель ВЭКУУМI-<ВЯ
трубка
D
Фильтр
В
Адаптер
мундштука
с
Рисунок 0671 2. Прuбор для определенuя oдHO
родности дозы в порошковых UН2аляторах
Если нет друrих указаний в частной
статье, следует определить скорость потока
и продолжительность с помощью собиратель
ной трубки, присоединенной к системе, под
ходящеro дифференциальноro манометра и
подходящеro волюметрическоro измерителя
потока, калиброванноro для измерения BЫ
ходноro потока, в соответствии со следующей
методикой.
Инrалятор rотовят, как указано в инструк
ции по применению, и соединяют ero с прибо
ром при помощи адаптера, убедившись в rep
метичности соединения. Используют адаптер
мундштука, который позволяет лицевой CTO
роне мундштука инrалятора находиться на
одном уровне с лицевой стороной собирающей
трубки. Соединяют одно отверстие MaHOMe
тра со шкалой давления Р1 (рисунок 0671 2),
а второе оставляют открытым. Включают
насос, открывают двухсторонний электромаr
нитный клапан. С помощью контрольноro кла
па на устанавливается давление инrалятора на
уровне 4,0 кПа (40,8 см Н 2 О). Отсоединяют ин
rалятор от адаптера мундштука и, не нажимая
контрольный клапан, присоединяют измери
тель расхода к выходному отверстию прибора.
Используют объемный счетчик, откалиброван
ный для измерения выходноro потока, либо
рассчитывают выходной поток (Qou/) , исполь
зуя закон для идеальноro rаза. Для измери
тельных приборов, калиброванных на входной
поток (От)' используют следующее выражение:
Q = От . РО
ои/ Ро дР'
rде:
Ро атмосферное давление;
др падение давления на измеритель
ном приборе.
Если скорость потока составляет более
100 л/мин, с помощью контрольноro клапана
реryлируют этот показатель в пределах 100 л/мин
(:t5 %). Замечают объемную скорость потока
воздуха (тестируемая скорость потока, Оои/'
л/мин). Замеряют продолжительность потока
т в секундах время, в течение котороro 4 л
воздуха проходят через инrалятор.
Следует проверить критическую скорость
потока. При скорости потока Оои/ измеряют аб
солютное давление с обеих сторон контроль
ноro клапана (точки замера давления Р2 и Р3
представлены на рисунке 0671 2). Если OT
ношение РЗ/Р2 s 0,5 это критическая CKO
рость потока. Если критическая скорость не
достиrнута, следует переключиться на более
мощный насос и повторить измерение CKOpO
сти потока.
Системы предварuтеЛЬН020 дuспеР2U
рования. Инrалятор присоединяют к прибо
ЛекарствеННblе средства для иН2аляций
1161
Таблица 0671 1
Специфuкацuu прuбора, uспользуеМО20 для порошковых иН2аляторов u опuсаНН020
на рисунке 0672 2
Код Приспо- Описание
собление
А Трубка Приспособлена для количественноrо захвата высвобождаемой дозы, например, подобная
для сбора трубке, описанной на рисунке 0671 1 с размерами 34,85 мм (внутренний диаметр) х 12 см
образца (длина) (например, продукт номер ХХ40 047 00, Millipore Соrpоrаtiоп, Bedford, МА 01732,
США, с модифицированной отводящей трубкой с внутренним диаметром не менее 8 мм,
снабженной продуктом Ge/maп, номер 61631), или аналоrичная
В Фильтр 47 мм фильтр, например, А/Е фильтр из стеЮlOволокна (Ge/тaп Scieпces, Апп Arbor, М/
48106, США, или аналоrичный)
С Соедини Внутренний диаметр не менее 8 мм, например, короткая металлическая муфта с короткой
тель отводной трубкой к Р3
О BaKY Трубка подходящей длины с внутренним диаметром не менее 8 мм и внутренним объемом
умная (25:1:5) мл
трубка
Е ДBYXCTO Двухсторонний электромаrнитный клапан с двумя выходами, с отверстием с минималь
ронний ной сопротивляемостью воздуху, внутренний диаметр не менее 8 мм, время открытия не
электро более 100 мс (например, тип 256 A08, BOrkert GmbH, 74653 /пgеffiпgеп или аналоrичный)
Mar
нитный
клапан
F BaKYYM Насос должен быть способным поддерживать необходимый поток воздуха через
ный насос собранный прибор с порошковым инraлятором (например, тип 1023, 1423 или 2565, Gast
Мапufасturiпg fпе., Вепtоп Harbor, М/ 49022, США или аналоrичный). Соединение насоса
и электромаrнитноro клапана осуществляется с помощью короткоro иlили широкою (BHY
тренний диаметр не менее 1 О мм) вакуумноro шланrа и соединителей для уменьшения I
требований к емкости насоса
G Таймер Таймер, способный управлять электромаrнитным клапаном в течение необходимою Bpe
мени (например, тип G814, RS Сотропепts /пtетаtiопа/, Соту, NN17 9RS, Великобрита
ния, или аналоrичный)
Р1 Реryлятор Внутренний диаметр 2,2 мм, внешний диаметр З,1 мм, находящийся на одном уровне с
давления внутренней стороной трубки для сбора образца, отцентрированный, без неровностей, Ha
ходящийся на расстоянии 59 мм от входноro отверстия. Реryлятор давления Р1 никоrда не I
должен открываться в атмосферу. Давление относительно атмосФеРною измеряется Р1 I
Р2 MaHOMe Абсолютное давление
Р3 тры
Н KOH Настраиваемый реryлирующий клапан с максимальным Cv 1 (например, тип 8FV12LNSS,
трольный Pa,ke, Наппifiп р/с., ЕХЗ1 1 NP, Великобритания, или аналоrичный)
клапан
ру, используя адаптер для rерметичноrо co
единения. Используя ранее установленные
параметры, пропускают воздух через инrаля
тор. Повторяют процедуру необходимое KO
личество раз, которое бы обеспечило отбор
минимальной рекомендованной дозы. Коли
чественно собирают содержимое прибора и
определяют содержание действующеro Be
щества.
Процедуру повторяют для следующих
девяти доз.
Системы с резервуаром. Инrалятор присо
единяют к прибору, используя адаптер для rep
метичноro соединения. Используя ранее YCTa
новленные параметры, пропускают воздух через
инrалятор. Повторяют процедуру необходимое
количество раз, которое бы обеспечило отбор
минимальной рекомендованной дозы. Количе
ственно собирают содержимое прибора и опре
деляют содержание действующеro вещества.
Процедуру повторяют для следующих двух
доз.
Сбрасывают несколько доз до тех пор, пока
не останется (п /2) 1 доз, rдe п количество
доз, указанное на этикетке и, при необходимо
сти, оставляют контейнер для снятия электро
статическоro заряда. Собирают 4 дозы по опи
санной выше методике.
Опять сбрасывают несколько доз, пока не
останется 3 дозы. При необходимости, OCTaB
ляют контейнер для снятия электростатиче
CKoro заряда. Собирают три последние дозы
по описанной выше методике.
1162
rосударственная фармакопея Республики Беларусь
Для лекарственных средств, содерхащих
более одноro действующеro вещества, ИСП':>lта
ние на однородность высвобождаемой дозы BЫ
полняют для каждоro.
Если иное не обосновано и не утвержде
но, лекарственное средство выдерживает ис
пытание, если 9 из 1 О результатов находятся в
пределах от 75 % до 125 % среднеro значения,
а все результаты находятся в пределах от 65 %
до 135 %. Если 2 или 3 результата выпадают
из пределов 75 125 %, испытание повторяют
с использованием двух друrих инrаляторов. Не
более 3 их 30 значений MorYT выходить за пре
делы 75 125 % и все значения должны быть в
пределах 65 135 %.
При соответствующем обосновании и YT
верждении rраницы MOryт быть расширены, но
не должны превышать 150 % и быть менее 50 %
среднеro значения.
Размер частиц. Используют прибор и MeTO
дику, описанные в общей статье 2.9.18. Лекар
ственные средства для UН2аляций: аэродuна
мическое uспытанuе мелкuх частuц (прибор С,
О или Е), рассчитывают дозу мелких частиц.
Число доз в мноrодозовом инrаляторе.
Высвобождают дозы в предварительно YCTa
новленных условиях до полноro опустошения
инrалятора. Полученное количество высвобож
денных доз должно быть не менее значения,
указанною на этикетке (это испытание можно
выполнять параллельно с испытанием на OДHO
родность высвобожденной дозы).
01/2013:1116
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
ДЛЯ ОРОШЕНИЯ
Р,аераrаtiопеs ad ir,igаtiопет
ОПРЕДЕЛЕНИЕ
Лекарственные средства для орошения
стерильные водные лекарственные средства
большоro объема, предназначенные для ороше
ния пораженных участков тела, ран и поверхно
стей, например, во время хирурrическоro BMe
шательства.
Лекарственными средствами для ороше
ния являются или растворы, приroтовленные
растворением одною или более действующих
веществ, электролитов или осмотически aK
тивных веществ в воде, которая COOTBeTCTBY
ет требованиям статьи «Вода для uнъекций»,
или лекарственные средства, состоящие
только из такой воды. В последнем случае ле
карственные средства должны быть промар
кированы «вода для орошений». Обычно ле
карственные средства для орошения должны
быть изотоничными.
Лекарственные средства для орошения, ис
пытываемые в подходящих условиях, должны
быть прозрачными и практически свободными
от частиц.
Лекарственные средства для орошения
выпускают в однодозовых контейнерах. KOH
тейнеры и укупорочные средства должны co
ответствовать требованиям, предъявляемым к
контейнерам для лекарственных средств для
парентеральноro применения (З.2.1 и З.2.2).
Однако приспособление к контейнеру для BBe
дения лекарственноro средства не может быть
совместимо с приспособлением для внутривен
ных инъекций и не может позволять использо
вание лекарственных средств для орошения
для внутривенных инъекций.
ПРОИ3ВОДСТВО
Лекарственные средства для орошения
производят с использованием материалов и
методов, обеспечивающих их стерильность и
исключающих возможность их последующе
ro заrрязнения и развития микроорrанизмов
в соответствии с требованиями статьи 5.1.1.
Методы при20товленuя стерuльных продук
тов.
При разработке лекарственноro cpeд
ства необходимо доказать, что из контейнера
может быть извлечен ero номинальный объем.
#В случае отсутствия у производителя лекар
cTBeHHoro средства сертисриката соответствия
GMP, а также данных о валидации процесса
производства конкретною лекарственноro cpek
ства для орошения испытание по определению
извлекаемоro объема должно быть включено в
спецификацию ютовоro продукта.#
ИСПЫТАНИЯ
Стерильность (2.6.1). Лекарственные cpeд
ства для орошен ий должны выдерживать испы
тание на стерильность.
Бактериальные эндотоксины (2.6.14).
Менее 0,5 МЕ/мл.
Пироrенность (2.6.8). Лекарственные
средства для орошения, для которых невоз
можно провести валидированное испытание
на бактериальные эндотоксины, должны BЫ
держивать испытание на пироrенность. Если
иное не доказано и не утверждено, вводят
1 О мл лекаРСТАенноro средства на 1 Kr массы
кролика.
МАРКИРОВКА
На этикетке указывают:
лекарственное средство не может быть
использовано для инъекций;
Лекарственные средства для парентераЛЬНО20 применения
1163
лекарственное средство должно быть ис
пользовано за один раз, а любая неиспользован
ная часть уничтожена.
01/2013:0520
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
ДЛЯ ПАРЕНТЕРАЛьноrо
ПРИМЕНЕНИЯ
Pa,eпfe,alia
Требованuя данной статьи не обязатель
но должны распространяться на лекарствен
ные средства, UЗ20товленные из человече
ской крови, UММУНОЛ02uческuе лекарственные
средства UЛU радиофармацевтuческuе лекар
ственные средства.
ОПРЕДЕЛЕНИЕ
Лекарственные средства для парентераль
ноro применения это стерильные лекарствен
ные средства, предназначенные для введения
путем инъекций, инфузий или имплантаций в op
rанизм человека.
Для приroтовления лекарственных средств
для парентеральноrо применения MorYT исполь
зоваться вспомоrательные вещества, например,
обеспечивающие изотоничность лекарственных
средств относительно крови, реryлирующие рН,
улучшающие растворимость действующих Be
ществ, предотвращающие их разложение, обе
спечивающие соответствующие антимикробные
свойства. Эти вещества не должны отрицатель
но влиять на основное действие лекарственно
ro средства или, в используемых концентраци
ях, вызывать токсичность или нежелательное
местное раздражающее действие.
Контейнеры для лекарственных средств для
парентеральноro применения должны быть из
roтовлены из материалов, которые как можно
более прозрачны и позволяют визуально про
смотреть содержимое контейнера, за исключе
нием имплантатов и друrих обоснованных и ут
вержденных случаев.
Если это применимо, контейнеры для ле
карственных средств для парентеральноro при
менения должны соответствовать требованиям
статей разделов 3. 1. Матерuалы, uспользуемые
для проuзводства контейнеров и 3.2. Контей
неры.
Лекарственные средства для парентераль
ною применения выпускают в стеклянных KOH
тейнерах (3.2.1) или в друrих, таких как пласт
массовые контейнеры (3.2.2, 3.2.2.1 и 3.2.9) и
предварительно наполненные шприцы. repMe
тичность контейнера обеспечивают COOTBeT
ствующими способами. Укупорочные средства
148 За. 1712.
должны обеспечивать надежную изоляцию, пре
дотвращать доступ микроорrанизмов и друrих
заrрязнений и обычно позволяют извлекать
часть или все содержимое контейнера без yдa
ления укупорочноro средства. Пластиковые Ma
териалы или эластомеры (3.2.9), из которых из
roтавливают укупорочные средства, должны
быть достаточно прочными и эластичными,
чтобы при прохождении иrлы выделялось наи
меньшее количество частиц. Укупорочные cpeд
ства контейнеров, содержащих несколько доз
лекарственною средства, должны быть ДOCTa
точно эластичными, чтобы обеспечивать repMe
тизацию контейнера при извлечении иrлы.
Лекарственные средства для парентераль
ноro применения можно классифицировать как:
инъекции;
инФузии;
концентраты для приroтовления инъекций
и инфузий;
порошки для приrотовления инъекций и
инфузий;
rели для инъекций
импланты;
# салфетки#.
ПРОИЗВОДСТВО
При разработке лекарственных средств для
парентеральноro применения, в состав которых
входят антимикробные консерванты, уполно
моченному opraHY представляют данные, под
тверждающие необходимость использования и
эффективность выбранных консервантов. Под
ходящий метод определения и критерии оценки
эффективности консервантов представлены в
статье 5.1.3. Эффективность антимикробных
консервантов.
Лекарственные средства для парентераль
Horo применения изrотавливают с использова
нием материалов и методов, обеспечивающих
стерильность и предотвращающих заrрязнение
лекарственных средств и развитие микроорrа
низмов, в соответствии с требованиями статьи
5.1.1. Методы при20товления стерuльных про
дуктов.
Вода, используемая при производстве ле
карственных средств для парентеральноrо
применения, должна соответствовать требо
ваниям, указанным в частной статье Вода для
uнъекцuй.
ИСПЫТАНИЯ
'Заrрязнение механическими включени-
ями: невидимые частицы (2.9. 19). Лекарствен
ные средства, предназначенные для введения
в орrанизм человека и представляющие собой
растворы для инфузий и инъекций, должны BЫ
держивать данное испытание.
В случае лекарственных средств, пред
назначенных для подкожноro или внутримы
шечноro введения, MOryт быть применены
более строrие пределы. Данное испытание
1164
rосударственная фармакопея Республики Беларусь
не распространяется на радиоактивные q,'jpMa
цевтические лекарственные средства. Да,iное
испытание не распространяется на лекарствен
ное средство, на маркировке котороro указано,
что оно используется с фильтром, если показа
но, что фильтр обеспечивает получение pacт
вора, выдерживающеro данное испытание.
Стерильность (2.6.1). Лекарственные
средства для парентеральноrо применения
должны выдерживать испытание на стериль
ность.
ХРАНЕНИЕ
Хранить в стерильном воздухонепроница
емом контейнере с контролем первоro BCKpЫ
тия.
МАРКИРОВКА
На этикетке указывают:
наименование и концентрацию всех дo
бавленных антимикробных консервантов;
если необходимо, использование филь
тра для раствора;
если необходимо, «не содержит бактери
альных эндотоксинов» или «апироrенно».
Инъекционные лекарственные
средства
ОПРЕДЕЛЕНИЕ
Инъекционные лекарственные cpeд
ства (инъекции) это стерильные paCTBO
ры, эмульсии или суспензии. Их rотовят путем
растворения, эмульrирования или суспенди
рования действующеro вещества или веществ
и вспомоrательных веществ в воде, в подхо
дящей неводной жидкости, которая, если обо
сновано, может быть нестерильна, или в смеси
этих растворителей.
Растворы для инъекций в соответствующих
условиях наблюдения должны быть прозрач
ными и практически свободными от частиц.
Эмульсии для инъекций не должны обна
руживать признаков расслоения. В суспензи
ях для инъекций может наблюдаться осадок,
который должен быстро дисперrироваться при
взбалтывании, образуя суспензию, достаточно
стабильную, чтобы обеспечить необходимую
дозу при введении.
МН020дозовые лекарственные средства.
Мноroдозовые водные инъекционные лекар
ственные средства содержат соответствующий
антимикробный консервант в необходимой
концентрации, за исключением лекарственных
средств, обладающих соответствующими про
тивомикробными свойствами. При выпуске ле
карственноro средства для парентеральноrо
применения в мноroдозовом контейнере необ
ходимо указывать меры предосторожности по
ero введению и особенно по хранению между
отборами доз.
Антuмuкробные консерванты. Водные ле
карственные средства, которые roтовят в асеп
тических условиях и которые не MOryT быть под
BeprHYTbI термической стерилизации, должны
содержать подходящий антимикробный KOHcep
вант в соответствующей концентрации.
Антимикробные консерванты не применяют,
если:
объем, вводимый в одноразовой дозе, пре
вышает 15 мл, если иное не обосновано;
лекарственное средство предназначено
для введения такими способами, при которых
по медицинским показателям не допускается
присутствие антимикробных консервантов, Ha
пример, для внутриполостноro, эпидуральноro,
интратекальноro введения или друrоro спосо
ба введения, дающеro доступ к спинномозroвой
жидкости, или интра или ретробульбарноro BBe
дения.
Такие лекарственные средства выпускают в
однодозовых контейнерах.
ПРОИЗВОДСТВО
При производстве инъекционных лекар
ственных средств, содержащих дисперсные ча
стицы, принимают меры, обеспечивающие необ
ходимый размер частиц и еro контроль.
Однодозовые лекарственные средства.
Объем инъекционноro лекарственноro средства
в однодозовом контейнере должен быть ДOCTa
точным для отбора и введения номинальной
дозы при использовании обычноro способа BBe
дения (2.9.17).
ИСПЫТАНИЯ
Однородность дозированных единиц.
Однодозовые суспензии для инъекций должны
выдерживать испытание на однородность дози
рованных единиц (2.9.40) или, если это обосно
вано и утверждено, испытание на однородность
содержания, как указано ниже. На лекарствен
ное растительное сырье и лекарственное
сырье из неro, представленные в дозированной
форме, требования данноro раздела не распро
страняются.
Однородность содержания (2.9.6). Oд
нодозовые суспензии для инъекций с coдep
жанием действующеro вещества менее 2 Mr
или менее 2 % от общей массы содержимоro
должны выдерживать испытание на OДHOpOД
ность содержания действующеro вещества в
единице дозированноrо лекарственноrо cpeд
ства (тест А), если в частной статье нет друrих
указаний. Если лекарственное средство coдep
жит более одноro действующеro вещества, это
требование распространяется только на те Be
щества, содержание которых соответствует BЫ
шеуказанным условиям.
Бактериальные эндотоксины пи-
poreHHocTb. Инъекционные лекарствен
Лекарственные средства для парентераЛЬНО20 примененuя
1165
ные средства должны выдерживать испыта
ние на бактериальные эндотоксины (2.6.14)
или, если обосновано и утверждено, испы
тание на пироrенность (2.6.8). Рекомендации
по предельно допустимым нормам содержа
ния бактериальных эндотоксинов приведены
в статье 2.6.14.
Инфузионные лекарственные
средства
ОПРЕДЕЛЕНИЕ
ИнФузионные лекарственные средства
(инфузии) это стерильные водные растворы
или эмульсии с водой в качестве дисперсион
ной среды. Они обычно изотоничны с кровью.
Инфузии преимущественно предназначаются
для применения в больших объемах. Инфузии
не содержат антимикробных консервантов.
Растворы для инфузий оценивают в COOT
ветствующих условиях наблюдения, при этом
они должны быть прозрачными и практически
свободными от частиц.
Эмульсии для инфузий не должны обна
руживать признаков расслоения.
ПРОИЗВОДСТВО
При производстве инфузий, содержа
щих дисперсные частицы, предпринимают
меры, обеспечивающие необходимый размер
частиц и ero контроль.
Объем инфузий в однодозовом контейне
ре должен быть достаточен для отбора и BBe
дения номинальной дозы при использовании
обычноro способа введения (2.9.17).
ИСПЫТАНИЯ
Бактериальные эндотоксины пи
poreHHoCTb. Инфузионные лекарственные
средства должны выдерживать испытание
на бактериальные эндотоксины (2.6.14) или,
если обосновано и утверждено, испытание на
пироrенность (2.6.8). При проведении испы
тания на пироrенность вводят 1 О мл лекар
ственноro средства на килоrрамм веса кроли
ка, если в частной статье нет друrих указаний.
Концентраты для приrотовления
инъекционных или инфузионных
лекарственных средств
ОПРЕДЕЛЕНИЕ
Концентраты для приroтовления инъек
ционных лекарственных средств или инфузий
представляют собой стерильные растворы,
предназначенные для инъекций или инфузий
после разведения. Концентраты перед приме
нением разводят до указанноro объема COOT
ветствующей жидкостью. После разведения
полученный раствор должен соответствовать
требованиям, предъявляемым к инъекционным
или инфузионным лекарственным средствам.
ИСПЫТАНИЯ
Бактериальные эндотоксины пироrен-
ность. Лекарственное средство должно Bыдep
живать требования. предъявляемые к инъек
ционным или инфузионным лекарственным
средствам соответственно, после разведения до
определенноro объема.
Порошки для приroтовления инъекци
онных или инфузионных лекарственных
средств
ОПРЕДЕЛЕНИЕ
Порошкидля приrотовления инъекционных
или инфузионных лекарственных средств
это твердые стерильные вещества, поме
щенные в контейнеры, которые при встряхи
вании с указанным объемом соответствующей
стерильной жидкости быстро образуют или
прозрачный, практически свободный от
частиц раствор, или однородную суспензию.
После растворения или суспендирования
они должны соответствовать требованиям,
предъявляемым к лекарственным средствам
для инъекций или инфузий соответственно.
Лиофилизированные лекарственные
средства для парентеральноrо применения
рассматривают как порошки для приroтовле
ния инъекционных или инфузионных лекар
ственных средств.
ПРОИЗВОДСТВО
Однородность содержания и OДHOpOД
ность массы лиофилизированных лекар
ственных средств для парентеральноrо
применения обеспечивают в ходе внутри
производственноrо контроля за количеством
раствора перед лиофилизацией.
ИСПЫТАНИЯ
Однородность дозированных единиц.
Порошки для приrотовления инъекционных или
инфузионных растворов должны выдерживать
испытание на однородность дозированных
единиц (2.9.40) или, если это обосновано и YT
верждено, испытания на однородность coдep
жания и/или однородность массы, как указано
ниже. На лекарственное растительное сырье и
лекарственные средства из неro, представлен
ные в дозированной форме, требования дaH
ноro раздела не распространяются.
Однородность содержания (2.9.6). Порош
ки для приrотовления инъекционных или инфу
зионных растворов с содержанием действующе.-
ro Beu \ества менее 2 Mr или менее 2 % от общей
массы содержимоro или с массой дозированной
единицы, равной 40 Mr либо менее, должны BЫ
держивать испытание на однородность coдep
жания действующеro вещества в единице дo
зированноro лекарственноro средства (тест А),
если нет друrих указаний в частной статье. Если
1166
rосударственная фармакопея Республики Беларусь
лекарственное средство содержит более nдноrо
действующеro вещества, это требование раt.поо
страняется только на те вещества, содержание ,{o
торых соответствует вышеуказанным условиям.
Однородность массы (2.9.5). Порошки для
приrотовления инъекционных или инфузионных
растворов должны выдерживать испытание на
однородность массы для единицы дозированно
ro лекарственноro средства. Испытание на OДHO
родность массы не требуется, если испытание
на однородность содержания предусмотрено
для всех действующих веществ.
Бактериальные эндотоксины пироrен
ность. Лекарственное средство должно Bыдep
живать требования, предъявляемые к инъекцион
ным или инфузионным лекарственным средствам
соответственно, после разведения или суспенди
рования до определенноro объема жидкости.
МАРКИРОВКА
На этикетке указывают способ приroтовления
лекарственною средства для инъекций и инфузий.
rели для инъекций
ОПРЕДЕЛЕНИЕ
rели для инъекций это стерильные rели
с вязкостью, обеспечивающей модифицирован
ное высвобождение действующеro вещества
или веществ в месте инъекции.
Импланты
ОПРЕДЕЛЕНИЕ
Импланты (имплантаты) представляют собой
стерильные твердые лекарственные средства,
имеющие подходящие для парентеральной им
плантации размеры, форму и высвобождение
действующеro вещества или веществ в течение
длительноro периода времени. Они упакованы в
индивидуальные стерильные контейнеры.
#Салфетки
ОПРЕДЕЛЕНИЕ
Салфетки это стерильная лекарственная
форма, имеющая фиксированные rеометриче
ские размеры, предназначенная для наружноro
и имплантационноrо применения.
ПРОИЗВОДСТВО
Салфетки получают путем сорбции лекар
ственных веществ на полотне. Стерилизация
салфеток осуществляется у лучами, что должно
быть указано в частной статье.
ХРАНЕНИЕ
Хранят в стерильном воздухонепроница
емом, неповрежденном контейнере, если нет
друrих указаний в частной статье.
01/2013:1145
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
ДЛЯ РЕКТАльноrо ПРИМЕНЕНИЯ
Rectalia
ОПРЕДЕЛЕНИЕ
Лекарственные средства для ректаль
ноro применения предназначены для BBeдe
ния в прямую кишку с целью получения си
cTeMHoro или местноro эффекта. Они MorYT
быть также использованы для диаrностиче
ских целей.
rде это применимо, контейнеры для peK
тальных лекарственных средств должны co
. ответствовать требованиям статей разделов
3.1. Матерuалы, uспользуемые для проuз
водства контейнеров и 3.2. Контейнеры.
Ректальные лекарственные средства можно
классифицировать как:
суппозитории;
ректальные капсулы;
ректальные растворы, суспензии и эмуль
сии;
порошки И таблетки для приrотовления
ректальных растворов или суспензий;
мяrкие лекарственные средства для peK
тальноro применения;
ректальные пены;
ректальные тампоны.
ПРОИЗВОДСТВО
При разработке лекарственных средств
для ректальноro применения, в состав KO
торых входят антимикробные KOHcepBaH
ты, уполномоченному opraHY представляют
данные, подтверждающие необходимость ис
пользования и эффективность выбранных
консервантов. Метод определения и критерии
оценки эффективности консервантов должны
соответствовать требованиям статьи 5.1.3.
Эффективность антuмuкробных KOHcep
вантов.
При производстве, упаковке, хранении и
реализации лекарственных средств для peK
тальноro применения принимают меры, обе
спечивающие микробиолоrическую чистоту
в соответствии с требованиями статьи 5.1.4.
МuкробиОЛ02uческая чuстота нестерuль
ных лекарственных средств u фармацевти
ческuх субстанцuй.
При производстве мяrких и жидких лекар
ственных средств для ректальноro примене
ния, содержащих дисперrированные частицы,
принимают меры, обеспечивающие необхо
димый и контролируемый размер частиц.
ИСПЫТАНИЯ
Однородность дозированных единиц
(2.9.40). Жидкие и мяrкие лекарственные
ЛекарствеННblе средства для ректаЛЬНО20 прuмененuя
1167
средства в однодозовых контейнерах должны
выдерживать испытание на однородность дo
зированных единиц. Твердые лекарственные
средства в однодозовых контейнерах должны
выдерживать испытание на однородность дo
зированных единиц или, если это утверждено
уполномоченным opraHoM, испытания на oд
нородность содержания и/или однородность
массы, как указано ниже. На лекарственное
растительное сырье и лекарственные cpeд
ства из Hero, представленные в дозирован
ной форме, требования данноro раздела не
распространя ются.
Однородность содержания (2.9.6). Если
нет друrих указаний в частной статье или если
не доказано и не утверждено иное, твердые
лекарственные средства в однодозовых KOH
тейнерах с содержанием действующеro Be
щества менее 2 Mr или менее 2 % от общей
массы содержимоro должны выдерживать ис
пытание на однородность содержания дей
ствующеrо вещества в единице дозированноro
лекарственноro средства (тест А ректаль
ные таблетки или тест В суппозитории, peK
тальные капсулы). Если лекарственное cpeд
ство содержит более одноro действующеrо
вещества, это требование распространяется
только на те вещества, содержание которых
соответствует вышеуказанным условиям.
Однородность массы (2.9.5). Твердые
лекарственные средства в однодозовых KOH
тейнерах должны выдерживать испытание на
однородность массы для единицы дозирован
Horo лекарственноrо средства. Испытание на
однородность массы не требуется, если испы
тание на однородность содержания предус
мотрено для всех действующих веществ.
Растворение. Для подтверждения COOT
ветствующеrо высвобождения действующеro
вещества или веществ из твердых однодозо
вых лекарственных средств проводят COOT
ветствующее испытание, например, 2.9.42.
Тест «Растворение» для лuпофиЛЬНblХ
твердых дозuроваННblХ форм.
Если проводят испытание по показателю
«Растворение», проведение испытания по по
казателю «Распадаемость» не требуется
МАРКИРОВКА
На этикетке указывают названия всех дo
бавленных антимикробных консервантов.
Суппозитории
ОПРЕДЕЛЕНИЕ
Суппозитории твердые однодозовые ле
карственные средства. Форма. объем и конси
стенция должны соответствовать ректальному
применению.
149 Зак 1712
Они содержат одно или более действу
ющих веществ, дисперrированных или pac
творенных в подходящей основе, которая
растворяется или дисперrируется в воде или
плавится при температуре тела. При соrла
совании с уполномоченным opraHoM в состав
суппозиториев, если необходимо. MorYT BXO
дить вспомоrательные вещества, такие как
разбавители, адсорбенты, поверхностно ак
тивные и смазывающие вещества, антими
кробные консерванты, а также красители.
ПРОИЗВОДСТВО
Суппозитории изroтавливают прессова
нием или методом выливания.
#8 условиях аптеки наряду с вышеуказан
ными методами может использоваться метод
ручноrо формования.#
Если необходимо, действующее веще
ство или вещества предварительно измель
чают и просеивают через соответствующие
сита. Если суппозитории изrотавливают MeTO
дом выливания, приroтовленную массу пред
варительно расплавляют при наrревании и
разливают в соответствующие формы. Суп
позитории затвердевают при охлаждении.
Чтобы обеспечить процесс затвердевания,
вводят такие вспомоrательные вещества, как
твердый жир, макроrолы, масло какао, раз-
личные rелеобразующие смеси, которые co
держат, например, желатин, воду и rлицерин.
Проводят определение времени деформации
липофильных суппозиториев (2.9.22).
Проводят испытание для подтверждения
соответствующеro высвобождения действу
ющеrо вещества или веществ из суппозито
риев, предназначенных для модифицирован-
HOro высвобождения или пролонrированноro
местноro действия.
При производстве суппозиториев, coдep
жащих дисперrированные частицы действу
ющих веществ, принимают меры, обеспе
чивающие необходимый и контролируемый
размер частиц.
ИСПЫТАНИЯ
Распадаемость (2.9.2). Если суппозито
рии не предназначены для модифицирован
HOro высвобождения или пролонrированноrо
MecTHoro действия, они должны выдерживать
испытание. Если иное не обосновано и не YT
верждено, состояние суппозиториев на rи
дрофобной основе исследуют через 30 мин,
а суппозиториев на rидрофильной основе
через 60 мин.
Ректальные капсулы
ОПРЕДЕЛЕНИЕ
Ректальные капсулы (суппозитории с
оболочкой) это твердые однодозовые ле
карственные средства, в основном сходные
1168
rосударственная фармакопея Республики Беларусь
с мяrкими капсулами, описанными в (.. татье
Капсулы, за исключением Toro, что они MO YT
иметь скользящую оболочку. Они должны
быть rладкими, однородными по внешнему
виду и иметь удлиненную форму.
ПРОИЗВОДСТВО
Проводят испытание для подтверждения
соответствующеro высвобождения действу
ющеrо вещества или веществ из ректальных
капсул, предназначенных для модифициро
BaHHoro высвобождения или пролонrирован
HOro MecTHoro действия.
ИСПЫТАНИЯ
Распадаемость (2.9.2). Если ректальные
капсулы не предназначены для модифициро
ванноro высвобождения или пролонrирован
ноro местноro действия, они должны выдержи
вать испытание. Если иное не обосновано и не
утверждено, состояние капсул исследуют через
30 мин.
Ректальные растворы, эмульсии
и суспензии
Ректальные растворы, эмульсии и суспен
зии это жидкие лекарственные средства,
предназначенные для введения в прямую
кишку с целью оказания системноro или MeCT
ноro действия. Они MorYT быть также исполь
зованы для диаrностических целей.
Ректальные растворы, эмульсии и суспен
зии выпускаются в однодозовых контейнерах
и содержат одно или более действующих Be
ществ, растворенных или дисперrированных в
воде, rлицерине, макроroлах или в друrих под
ходящих растворителях. Ректальные эмульсии
MorYT расслаиваться, однако быстро BOCCTa
навливаются при взбалтывании. Ректальные
суспензии MorYT образовывать осадок, KOTO
рый быстро ресуспендируется при взбалты
вании, образуя суспензию достаточно CTa
бильную, чтобы обеспечить однородность
лекарственноro средства при дозировании.
Ректальные растворы, эмульсии и суспен
зии MorYT содержать вещества, предназначен
ные для обеспечения необходимой вязкости,
создания или стабилизации необходимоro зна
чения рН, для улучшения растворимости дей
ствующеro вещества или веществ либо для
стабилизации лекарственноrо средства. Они
не должны отрицательно влиять на основное
действие лекарственноro средства или, в ис
пользуемых концентрациях, не должны оказы
вать нежелательное местное раздражающее
действие.
Ректальные растворы, эмульсии и суспен
зии выпускают в контейнерах объемом от 2,5 мл
до 2000 мл. Контейнер должен быть приспосо
блен для введения лекарственноrо средства
в прямую кишку или снабжен подходящей Ha
садкой.
Порошки И таблетки для приroтовления
ректальных растворов или суспензий
ОПРЕДЕЛЕНИЕ
Порошки и таблетки для приroтовления peK
тальных растворов и суспензий это OДHOДO
зовые лекарственные средства, которые paCTBO
ряют или дисперrируют в воде непосредственно
перед применением. Они MOryT содержать вспо
моrательные вещества, способствующие pac
творению или дисперrированию либо предот
вращающие аrреrацию частиц.
После растворения или дисперrирования
они должны выдерживать требования, которые
предъявляют к ректальным растворам или cy
спензиям соответственно.
ИСПЫТАНИЯ
Распадаемость (2.9.1). Таблетки для приro
товления ректальных растворов или суспензий
должны выдерживать испытание с использова
нием воды Р при температуре от 15 ос до 25 ос в
качестве жидкой среды и 6 таблеток. Состояние
таблеток проверяют через 3 мин. Все 6 таблеток
должны распасться.
МАРКИРОВКА
На этикетке указывают:
способ приroтовления ректальных paCTBO
ров или суспензий;
условия и срок хранения раствора или cy
спензии после приroтовления.
Мяrкие лекарственные средства
для ректальноro применения
ОПРЕДЕЛЕНИЕ
К мяrким лекарственным средствам для
ректальноro применения относятся линименты,
мази, кремы и rели.
Они обычно представляют собой OДHOДO
зовые лекарственные средства в контейнерах,
снабженных подходящей насадкой.
Мяrкие лекарственные средства для peK
тальноro применения должны выдерживать Tpe
бования статьи МЯ2кuе лекарственные cpeд
ства для местН020 прuмененuя.
Ректальные пены
ОПРЕДЕЛЕНИЕ
Ректальные пены должны соответствовать
требованиям статьи Пены медицинские.
Ректальные тампоны
ОПРЕДЕЛЕНИЕ
Ректальные тампоны это твердые OДHO
дозовые лекарственные средства, предназна
ченные для введения в нижний отдел прямой
кишки на определенное время.
Они должны соответствовать требованиям
статьи Тампоны медицинские.
ЛекарствеННblе средства для слизистой оболочки полости рта
1169
01/2013:1807
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
ДЛЯ СЛИЗИСТОЙ ОБОЛОЧКИ
ПОЛОСТИ РТА
P,aeparatioпes bucca/es
Требованuя данной статьи не распро
страняются на лекарственные средства для
стоматОЛ02UU uлu на жевательные таблет
ки, лекарственные жевательные резинки, тa
блеткu лuофилuзаты u дРУ2ие твердые uлu
МЯ2кuе лекарственные средства, предназна
ченные для разжевыванuя илu дuспеР2ированuя
в слюне перед 2лотанuем.
ОПРЕДЕЛЕНИЕ
Лекарственные средства для слизистой
оболочки полости рта это твердые, мяrкие
или жидкие лекарственные средства, содержа
щие одно или более действующих веществ и
предназначенные для введения в полость рта
и/или roрло для получения местноro или си
стемноro эффекта. Лекарственные средства,
предназначенные для получения местноro эф
фекта, MorYT применяться путем нанесения на
определенную область внутри полости рта, Ha
пример, на десну (лекарственные средства для
десен) или roрла (орофаринrиальные лекар
ственные средства). Лекарственные средства,
предназначенные для получения системноro
эффекта, применяют для всасывания в одной
или нескольких областях слизистой оболоч
ки полости рта (например, сублинrвальные ле
карственные средства). Слизистые адrезивные
лекарственные средства предназначены для
удержания в полости рта путем адrезии к эпи
телию слизистой оболочки и MorYT изменять си
стемную абсорбцию лекарственноro средства в
месте применения. Для мноrих лекарственных
средств для слизистой оболочки полости рта
существует вероятность тоro, что некоторое KO
личество действующеro вещества или веществ
будет проrлатываться и абсорбироваться желу
дочно кишечным трактом.
Лекарственные средства для слизистой
оболочки полости рта MorYT содержать COOT
ветствующие противомикробные консерванты
и друrие вспомоrательные вещества, такие как
дисперrирующие, суспендирующие вещества,
заrустители, эмульrаторы, буферы. смачива
ющие areHTbI, солюбилизаторы, стабилизато
ры, ароматизаторы и подсластители. Твердые
лекарственные средства дополнительно MorYT
содержать вещества для скольжения и смазки,
а также наполнители, способные влиять на BЫ
свобождение действующеro вещества или Be
ществ.
rде это применимо, контейнеры для лекар
ственных средств для слизистой оболочки поло
сти рта должны соответствовать требованиям
статей разделов 3. 1. Матерuалы, uспользуемые
для проuзводства контейнеров и 3.2. Контей
неры.
Лекарственные средства для слизистой обо
лочки полости рта можно классифицировать как:
растворы для полоскания roрла;
растворы для полоскания полости рта;
растворы для десен;
растворы и суспензии для слизистой обо
лочки полости рта;
мяrкие лекарственные средства для сли
зистой оболочки полости рта (включая, напри
мер, rель для десен, пасту для десен, rель и
пасту для слизистой оболочки полости рта);
капли для слизистой оболочки полости
рта, спреи для слизистой оболочки полости рта
и подъязычные спреи (включая орофаринrиаль
ные спреи);
леденцы и пастилки;
прессованные леденцы;
таблетки подъязычные и таблетки защеч
ные;
капсулы для слизистой оболочки полости рта;
адrезивные лекарственные средства для
слизистой оболочки полости рта;
пленки, дисперrируемые в полости рта.
ПРОИЗВОДСТВО
При разработке лекарственных средств для
слизистой оболочки полости рта, в состав KOTO
рых входят антимикробные консерванты, упол
номоченному opraHY представляют данные, под
тверждающие необходимость использования и
эффективность выбранных консервантов. Под
ходящий метод определения и критерии оценки
эффективности консервантов представлены в
статье 5.1.3. Эффектuвность антuмuкроБНblХ
консервантов.
При производстве, упаковке, хранении и
реализации лекарственных средств для сли
зистой оболочки полости рта принимают меры,
обеспечивающие микробиолоrическую чисто
ту в соответствии с требованиями статьи 5.1.4.
МuкроБUОЛ02ическая чuстота нестеРUЛЬНblХ
лекарственных средств u фармацевтuческих
субстанцuй.
При производстве твердых и мяrких ле
карственных средств для слизистой оболоч
ки полости рта, содержащих дисперrированные
частицы, принимают меры, обеспечивающие He
обходимый размер частиц и ero контроль.
ИСПЫТАНИЯ
Однородность дозированных единиц.
Однодозовые лекарственные средства для сли
зистой оболочки полости рта должны выдержи
вать испытание на однородность дозированных
единиц (2.9.40) или, если это обосновано и YT
верждено, испытания на однородность содержа
ния и/или однородность массы, как указано ниже.
На лекарственное растительное сырье и лекар
ственные средства из неro, представленные в дo
1170
rосударственная фармакопея Республики Беларусь
зированной форме, требования данноro рС\Здела
не распространяются.
Однородность содержания (2.9.6).
Однодозовые лекарственные средства
с содержанием действующеrо вещества
менее 2 Mr или менее 2 % от общей массы
должны выдерживать испытание на OДHO
родность содержания действующеrо веще
ства в единице дозированноrо лекарствен
ноro средства (тест А прессованные и
отлитые лекарственные средства; тест В
капсулы), если нет друrих указаний в частной
статье. Если лекарственное средство coдep
жит более одноro действующеrо вещества,
это требование распространяется только на
те вещества, содержание которых COOTBeT
ствует вышеуказанным условиям.
Однородность массы (2.9.5). Твердые oд
нодозовые лекарственные средства должны BЫ
держивать испытание на однородноC'tb массы
для единицы дозированноro лекарственноro
средства. Испытание на однородность массы не
требуется, если испытание на однородность co
держания предусмотрено для всех действующих
веществ.
МАРКИРОВКА
На этикетке указывают названия всех дo
бавленных антимикробных консервантов.
Растворы для полоскания roрла
ОПРЕДЕЛЕНИЕ
Растворы для полоскания rорла это
водные растворы, предназначенные для по
лоскания rорла с целью достижения MeCTHO
ro эффекта. Эти растворы не предназначены
для rлотания. Они выпускаются в виде rOToBbIx
к применению растворов или концентрирован
ных растворов для последующеro разбавле
ния; MorYT изroтавливаться из порошков или
таблеток, которые перед применением paCTBO
ряют в воде.
Растворы для полоскания rорла MorYT co
держать вспомоrательные вещества для обе
спечен ия уровня рН, который в большинстве
случаев является нейтральным.
Растворы для полоскания рта
ОПРЕДЕЛЕНИЕ
Растворы для полоскания рта это водные
растворы, предназначенные для обработки сли
зистой оболочки полости рта. Эти растворы не
предназначены для rлотания. Они выпускаются
в виде roтовых к применению растворов или KOH
центрированных растворов для последующеro
разбавления; MOryT изroтавливаться из порош
ков или таблеток, которые перед применением
растворяют в воде.
Растворы для полоскания рта MOryT coдep
жать вспомоrательные вещества для обеспече
ния уровня рН, который в большинстве случаев
является нейтральным.
Растворы для десен
ОПРЕДЕЛЕНИЕ
Растворы для десен предназначаются для
нанесения на десну посредством COOTBeTCTBY
ющеro аппликатора.
Растворы и суспензии для слизистой
оболочки полости рта
ОПРЕДЕЛЕНИЕ
Растворы и суспензии для слизистой обо
почки полости рта это жидкие лекарственные
средства, предназначенные для введения в по
лость рта посредством соответствующеro ап
пликатора.
Суспензии MOryT образовывать осадок, KO
торый быстро ресуспендируется при взбалтыва
нии, образуя суспензию достаточно стабильную,
чтобы обеспечить однородность лекарственноro
средства при дозировании.
Мяrкие лекарственные средства
для слизистой оболочки полости рта
ОПРЕДЕЛЕНИЕ
Мяrкие лекарственные средства для слизи
стой оболочки полости рта это rидрофильные
мази, rели или пасты, предназначенные для BBe
дения в полость рта или нанесения на ее отдель
ную область, такую как десна (rель для десен,
паста для десен). Они MOryT выпускаться в OДHO
дозовых контейнерах.
Мяrкие лекарственные средства для слизи
стой оболочки полости рта должны COOTBeTCTBO
вать требованиям, изложенным в статье МЯ2кие
лекарственные средства для наРУЖН020 при
мененuя.
Капли и спреи для слизистой оболочки
полости рта и подъязычные спреи
ОПРЕДЕЛЕНИЕ
Капли и спреи для слизистой оболочки по
лости рта и подъязычные (сублинrвальные)
спреи это растворы, эмульсии или суспен
зии, предназначенные для достижения MeCT
ноro или системноrо эффекта. Они применя
ются путем инстилляции или распыления в
полость рта или на определенную еro область,
например, распыления под язык (подъязыч
ный (сублинrвальный) спрей) или в roрло (opo
фаринrиальный спрей).
Эмульсии MOryT расслаиваться, но быстро
восстанавливаются путем взбалтывания. Cy
спензии MOryT образовывать осадок, который
быстро ресуспендируется при взбалтывании, об
ЛекарствеННblе средства для слизистой оболочки полости рта
1171
разуя суспензию достаточно стабильную, чтобы
обеспечить однородность лекарственноro cpeд
ства при дозировании.
}Кидкие спреи для слизистой оболочки по
лости рта выпускаются в контейнерах с распы
лителем или контейнерах, находящихся под
давлением и снабженных соответствующим
адаптером с или без дозатора, которые должны
соответствовать требованиям, изложенным в
статье Лекарственные средства, находящuеся
под давленuем.
Размер капелек спрея должен быть таким,
чтобы оrраничить депонирование их в полости
рта или roрла в соответствии с предназначением
лекарственноro средства.
ИСПЫТАНИЯ
Если нет друrих указаний в частных
статьях, капли для слизистой оболочки полости
рта, спреи для слизистой оболочки полости рта
и сублинrвальные спреи, предназначенные для
получения системноro эффекта, выпускаемые
в однодозовых контейнерах, должны выдержи
вать следующие испытания.
КАПЛИ ДЛЯ СЛИЗИСТОЙ ОБОЛОЧКИ
ПОЛОСТИ РТА В ОДНОДОЗОВЫХ
КОНТЕЙНЕРАХ
Однородность дозированных единиц.
Капли для слизистой оболочки полости рта в
однодозовых контейнерах должны выдержи
вать испытание на однородность дозированных
единиц (2.9.40) или, если это обосновано и YT
верждено, испытание на однородность массы
или на однородность содержания, как указано
ниже. На лекарственное растительное сырье и
лекарственные средства из неro, представлен
ные в дозированной форме, требования дaHHO
ro раздела не распространяются.
Однородность массы. Каплu для СЛU
зистой оболочкu полостu рта, представля
ющuе собой растворы, должны выдержuвать
следующее испытание. Освобождают 1 О KOH
тейнеров как можно полнее и взвешивают co
держи мое каждоro из них, определяют cpeд
нюю массу содержимоrо. Масса содержимоro
не более чем двух контейнеров может откло
няться более чем на 10 % от средней массы,
и масса содержимоro ни одноro контейнера не
должна отклоняться более чем на 20 % от cpeд
ней массы.
Однородность содержания (2.9.6). Каплu
для слuзuстой оболочкu полостu рта, пред
ставляющuе собой суспензuu uлu эмульсuu,
должны выдержuвать следующее uспытанuе.
Освобождают каждый контейнер как можно
полнее и определяют содержание действу
ющеrо вещества для каждоro контейнера. Ле
карственное средство должно выдерживать ис
пытание на однородность содержания (тест В).
ДОЗИРОВАННЫЕ СПРЕИ
ДЛЯ СЛИЗИСТОЙ ОБОЛОЧКИ ПОЛОСТИ
РТА И СУБлинrВАЛЬНЫЕ СПРЕИ
Однородность дозированных единиц. Дo
зированные спреи для слизистои оболочки поло
сти рта и сублинrвальные спреи должны Bыдep
живать испытание на однородность дозированных
единиц (2.9.40) или, если это обосновано и yт
верждено, испытание на однородность массы или
на однородность содержания, как указано ниже.
На лекарственное растительное сырье и лекар
ственные средства из Hero, представленные в дo
зированной форме, требования данноro раздела
не распространяются.
В случае дозuрованных спреев для слuзu
стой оболочкu полостu рта u суБЛUН2вальных
спреев, которые являются растворами, посту
пают следующим образом. Выпускают и отбрасы
вают одну дозу. Ожидают не менее 5 с, встряхи
вают В течение 5 с выпускают и отбрасывают еще
одну дозу. Повторяют эту операцию еще 3 разэ.
Взвешивают контейнер, выпускают и отбрасывают
одну дозу и снова взвешивают контейнер. Находят
разницу между двумя массами. Повторяют про
цедуру для следующих 9 контейнеров. Проводят
определение расчетно весовым методом (2.9.40).
В случае дозированных спреев для слuзu
стой оболочки полостu рта u суБЛUН2вальных
спреев, которые являются суспензuямu uлu
эмульсuями, поступают следующuм образом.
Используют прибор, позволяющий количественно
удерживать дозу после нажатия распылительно
ro устройства. Встряхивают контейнер в течение
5 с, выпускают и отбрасывают одну дозу. Ожида
ют не менее 5 с, встряхивают в течение 5 с и BЫ
пускают и отбрасывают еще одну дозу. Повторя
ют эту операцию еще 3 раза. Через две секунды
выпускают одну дозу дозированноro сп рея в co
бирающий сосуд путем активации распылитель
ноro устройства. Собирают содержимое сосуда
достаточным количеством смывов. Определяют
содержание действующеro вещества в объеди
ненных смывах. Процедуру повторяют для следу
ющих 9 контейнеров. Испытание проводят MeTO
дом прямоro определения (2.9.40).
Однородность массы. Дозuрованные
спреu для слuзuстой оболочкu полостu рта u
суБЛUН2вальные спреu, которые являются pac
творами, должны выдержuвать следующее
uспытание. Выпускают и отбрасывают одну
дозу. Ожидают не менее 5 с, встряхивают в Te
чение 5 с и выпускают и отбрасывают еще одну
дозу. Повторяют эту операцию еще 3 раза. Взве
шивают контейнер, выпускают и отбрасывают
одну дозу и снова взвешивают контейнер. Haxo
дят разницу между двух масс. Повторяют проце
дуру для следующих 9 контейнеров.
Индивидуальные значения не более чем
двух контейнеров Moryт отклоняться более чем
на 25 % от среднеro значения, и ни одно значе
ние не должно отклоняться более чем на 35 %.
1172
rосударственная фармакопея Республики Беларусь
Однородность высвобождаемой дозы.
Дозuрованные спреu для слuзuстой оболочкu
полостu рта u суБЛUН2вальные спреu, которые
являются суспензuямu uли эмульсuямu, должны
выдерживать следующее uспытанuе. Исполь
зуют прибор, позволяющий количественно yдep
живать дозу после нажатия распылительноro
устройства. Встряхивают контейнер в течение
5 с, выпускают и отбрасывают одну дозу. Ожи
дают не менее 5 с, встряхивают в течение 5 с,
выпускают и отбрасывают еще одну дозу. Повто
ряют эту операцию еще 3 раза. Через две ceKYH
ды выпускают одну дозу дозированноro сп рея в
сосуд для сбора путем активации распылитель
ноro устройства. Собирают содержимое сосуда
достаточным количеством смывов. Определяют
содержание активноro вещества в объединен
ных смывах. Процедуру повторяют для следу
ющих 9 контейнеров.
Если иное не обосновано и не утверждено,
лекарственное средство удовлетворяет требо
ваниям, если содержание действующеro веще
ства в дозе не более чем одноro контейнера не
укладывается в пределы от 75 % до 125 %, но не
выходит за пределы от 65 % до 135 % от cpeДHe
ro значения.
Если содержание действующеro вещества
в дозе двух или трех отдельных контейнеров
не укладывается в пределы от 75 % до 125 %,
но укладывается в пределы от 65 % до 135 %,
повторяют испытание еще для 20 контейнеров.
Лекарственное средство выдерживает испыта
ние, если содержание действующеro вещества
в дозе не более чем трех контейнеров не укла
дывается в пределы от 75 % до 125 %, но не BЫ
ходит за пределы от 65 % до 1 З5 % от среднеro
значения.
Леденцы и пастилки
ОПРЕДЕЛЕНИЕ
Леденцы и пастилки это твердые OДHO
дозовые лекарственные средства, предназна
ченные для рассасывания с целью достижения
обычно местноro эффекта в полости рта и rорле.
Содержат одно или более действующих Be
ществ, обычно на ароматной и сладкой основе,
и предназначаются для медленноrо paCTBope
ния или разрушения во рту в результате pacca
сывания.
Леденцы это твердые лекарственные
средства, изroтавливаемые путем формовки.
Пастилки это мяrкие эластичные лекарствен
ные средства, изroтавливаемые путем формовки
из смесей, содержащих природные или синтети
ческие полимеры или камеди и подсластители.
Прессованные леденцы
ОПРЕДЕЛЕНИЕ
Прессованные леденцы это твердые oд
нодозовые лекарственные средства, предназна
ченные для рассасывания с целью получения
местноro или системноro эффекта. Они изro
тавливаются путем сжатия, часто имеют форму
ромба.
Прессованные леденцы соответствуют
общему определению таблеток.
ПРОИЗВОДСТВО
При производстве прессованных леденцов
следует принять меры, rарантирующие, что ле
денцы обладают соответствующей механиче
ской прочностью, не крошатся при разламыва
нии рукой. Такие характеристики определяют,
как указано в статье 2.9.7. Прочность таблеток
без оболочкu на истирание и статье 2.9.8. Проч
ность таблеток на сжатuе.
ИСПЫТАНИЯ
Растворение. Для прессованных леден
цов, предназначенных для получения систем
ноro эффекта, проводят испытание для под
тверждения высвобождения действующеro
вещества или веществ.
Таблетки подъязычные и таблетки
защечные
ОПРЕДЕЛЕНИЕ
Таблетки подъязычные и таблетки защеч
ные это твердые однодозовые лекарственные
средства для введения под язык или в полость
рта соответственно с целью получения систем
ноro эффекта. Они изrотавливаются путем прес
сования из смеси порошков или rранул в таблет
ки подходящей для их использования формы.
Таблетки подъязычные и таблетки защечные
соответствуют общему определению таблеток.
ПРОИЗВОДСТВО
При производстве таблеток подъязычных и
таблеток защечных принимают меры, rарантиру
ющие, что таблетки обладают соответствующей
механической прочностью, чтобы не крошиться
или не разламываться при транспортировке. Это
может быть подтверждено путем проведения ис
пытаний, как указано в статье 2.9.7. Прочность
таблеток без оболочкu на истирание и статье
2.9.8. Прочность таблеток на сжатuе.
ИСПЫТАНИЯ
Растворение. Если иное не обосновано и
не утверждено, проводят испытание для под
тверждения высвобождения действующеro Be
щества или веществ.
Капсулы для слизистой оболочки
полости рта
ОПРЕДЕЛЕНИЕ
Капсулы для слизистой оболочки полости
рта это мяrкие капсулы, предназначенные для
жевания или рассасывания.
Лекарственные средства, находящuеся под давлением
Адrезивные лекарственные средства
ОПРЕДЕЛЕНИЕ
Адrезивные лекарственные средства
это лекарственные средства. содержащие
одно или более действующих веществ и пред
назначенные для системной абсорбции через
слизистую оболочку щеки в течение длитель
ноro периода времени. Они MorYT выпускаться
в виде адrезивных защечных таблеток, защеч
ных пленок или друrих адrезивных твердых
или мяrких лекарственных средств. Обычно
такие лекарственные средства содержат rи
дрофильные полимеры, которые при увлаж
нении слюной образуют rель, прилипающий к
слизистой оболочке щечноro кармана; кроме
этоro, защечные пленки MorYT растворяться.
Адrезивные защечные таблетки изroтав
ливают путем прессования MOHO или муль
тислойных таблеток.
Защечные пленки представляют собой
OДHO и мноroслойные пластинки из подходя
щеrо материала.
ПРОИЗВОДСТВО
При производстве адrезивных защечных
таблеток и пленок принимают меры, rаранти
рующие их соответствующую механическую
прочность, чтобы не крошиться и не ломать
ся при транспортировке. Для адrезивных за
щечных таблеток это может быть подтвержде
но путем проведения испытаний, как указано в
статье 2.9.7. Прочность таблеток без оболоч
ки на истирание и статье 2.9.8. Прочность тa
блеток на сжатuе.
ИСПЫТАНИЯ
Растворение. Если иное не обосновано и
не утверждено, проводят испытание для под
тверждения высвобождения действующеro Be
щества или веществ.
Пленки, дисперrируемые в полости рта
ОПРЕДЕЛЕНИЕ
Пленки, дисперrируемые в полости рта
представляют собой OДHO и мноrослойные пла
стинки из подходящеro материала, которые при
помещении в ротовую полость быстро диспер
rируются.
ПРОИЗВОДСТВО
При производстве пленок, дисперrируемых
в полости рта, принимают меры, rарантирующие
соответствующую механическую прочность для
возможности транспортировки без повреждений.
ИСПЫТАНИЯ
Растворение. Если иное не обосновано и
не утверждено, проводят испытание для под
тверждения высвобождения действующеro Be
щества или веществ.
1173
01/2013:0523
ЛЕКАРСТВЕННЫЕ СРЕДСТВА,
НАХОДЯЩИЕСЯ ПОД ДАВЛЕНИЕМ
Praeparat;ones pharтaceut;cae ;n vas;s сит
pressu
к nекарственным средствам. находя
щuмся под давленuем, предъявляют допол
нuтельные требованuя, указанные в дРУ2их
статьях, например, Лекарственные cpeд
ства для UН2аляцuй, JКuдкuе лекарственные
средства для наРУЖН020 примененuя, Порош
ки для наРУЖН020 прuмененuя, Назальные ле
карственные средства u Ушные лекарствен
ныв средства, еслu нет дРУ2их указанuй в
частных статьях.
ОПРЕДЕЛЕНИЕ
Лекарственные средства, находящиеся
под давлением, это лекарственные CDeд
ства, находящиеся в специальных контейне
рах под давлением rаза и содержащие одно
или более действующих веществ. Данные
лекарственные средства при выходе из KOH
тейнера при нажатии на клапан представля
ют собой аэрозоль (дисперсию твердых или
жидких частиц в rазе, размер которых зависит
от назначения лекарственноro средства), жид
кость или мяrкую пену. Давление, необходи
мое для выхода лекарственноrо средства из
контейнера, обеспечивают соответствующие
пропелленты.
Лекарственные средства, находящиеся
под давлением, представляют собой раствор,
эмульсию или суспензию. Они предназначены
для местноro нанесения на кожу, на слизистые
оболочки или для инrаляции. В состав лекар
ственных средств MOryT входить такие вспомо
rательные вещества, как эмульrаторы, суспен
дирующие вещества, растворители, а также
скользящие вещества, предотвращающие засо
рение клапана.
Пропелленты. Пропелленты представляют
собой сжиженные под давлением rазы, сжатые
rазы или низкокипящие жидкости. Сжиженны
ми rазами являются, например, фторирован
ные уrлеводороды и уrлеводороды с низкой MO
лекулярной массой, такие как пропан или бутан.
Сжатые rазы это уrлерода диоксид, азот или
азота (1) оксид.
Для получения лекарственноro средства
с оптимальными свойствами и заданными xa
рактеристиками давления, дозы и распыления
MoryT быть ИСflользованы смеси этих пропел
лентов.
Контейнеры. Контейнеры должны быть
прочными И устойчивыми по отношению к BHY
трен нему давлению. Они MOryт быть изroтовле
ны из металла, стекла, пластика или комбинации
этих материалов и не должны взаимодействовать
1174
rосударственная фармакопея Республики Беларусь
с содержимым. Стеклянные контейнеры должны
иметь защитное пластиковое покрытие.
Распыляющее устройство. Клапан должен
rерметично закрывать контейнер внерабочем
положении и обеспечивать необходимую дозу
лекарственноro средства в процессе использо
вания. На характеристику распыления влияют
размеры, количество и расположение OTBep
стий, а также тип распыляющеro устройства.
Клапаны обеспечивают непрерывный выход
(клапан непрерывною действия) или выдают OT
меренное количество лекарственноrо средства
при каждом нажатии (клапан дозирующею дей
ствия).
Материалы, используемые для производ
ства клапанов, не должны взаимодействовать с
содержимым контейнера.
Требования, предъявляемые к лекар
ственным средствам, находящимся под дав-
лением. Лекарственные средства, находящи
еся под давлением, должны быть оснащены
соответствующим распыляющим устройством
соrласно еro назначению.
Специальные требования MOryT предьяв
ляться к выбору пропеллентов, размеру частиц,
дозе, получаемой при одном нажатии на дозиру
ющий клапан.
МАРКИРОВКА
На этикетке указывают:
способ применения;
меры предосторожности;
для лекарственных средств, находящих
ся в контейнере с клапаном дозирующею дей
ствия, указывают количество действующеro Be
щества в одной дозе.
01/2013:0132
мяrКИЕ ЛЕКАРСТВЕННЫЕ
СРЕДСТВА ДЛЯ НАРУжноrо
ПРИМЕНЕНИЯ
Рrаераrаtiопеs mo//es ad usum dermicum
Требованuя данной статьи распростра
няются на все МЯ2кuе лекарственные cpeд
ства для наРУЖН020 прuмененuя. К МЯ2КUМ
лекарственным средствам, предназначеl-'
ным для прuмененuя на определенных по
верхностях тела uлu слuзuстых оболочках,
должны быть предъявлены дополнuтельные
требованuя в соответствующuх статьях
(Ушные лекарственные средства, Назаль
ные лекарственные средства, Лекарствен
ные средства для ректаЛЬН020 прuмененuя,
/лазные лекарственные средства u Лекар
ственные средства для ва2инаЛЬН020 прu
мененuя).
ОПРЕДЕЛЕНИЕ
Мяrкие лекарственные средства для Ha
ружноrо применения предназначены для MeCT
ноro или трансдермальноrо высвобождения
действующих веществ или для смяrчающеro
или защитноro действия. По внешнему виду
они однородны.
Мяrкие лекарственные средства для наруж
Horo применения состоят из простой или слож
ной основы, в которой растворены или дисперrи
рованы действующие вещества. В зависимости
от состава основа может влиять на активность
лекарственноrо средства.
. Основа может состоять из природных или
синтетических веществ и представлять собой
однофазную или мноroфазную систему. В за
висимости от природы основы лекарственное
средство может обладать как rидрофильными,
так и rидрофобными свойствами. В их состав
MOryт входить подходящие вспомоrательные Be
щества, такие как антимикробные консерванты,
антиоксиданты, стабилизаторы, эмульrаторы,
заryстители и areHTbI, усиливающие проникнове
ние действующею вещества.
Мяrкие лекарственные средства для наруж
ноro применения, предназначенные для HaHece
ния на раневую поверхность, должны быть CTe
рильными.
rде это применимо, контейнеры для мяrких
лекарственных средств для наружноro приме
нения должны соответствовать требованиям
статей разделов 3. 1. Материалы, uспользуемые
для проuзводства контейнеров и 3. 2. Контей
неры.
Мяrкие лекарственные средства для наруж
ноro применения можно классифицировать как:
мази;
кремы;
rели;
пасты;
припарки;
пластыри медицинские;
пластыри кожные;
# линименты#;
# rидроrелевые пластины#.
В зависимости от структуры, мази, кремы и
rели обычно обладают вязкоупруrими свойства
ми и являются неньютоновскими по своему xa
рактеру, например, с пластическим, псевдопла
стическим или тиксотропным типом потока при
высокой скорости сдвиrа. Пасты обладают спо
собностью к расширению.
ПРОИЗВОДСТВО
При разработке мяrких лекарственных
средств для наружноro применения, в состав
которых входят антимикробные консерванты,
уполномоченному opraHY представляют данные,
МЯ2кuе лекарственные средства для наРУЖНО20 применения
1175
подтверждающие необходимость использова
ния и эффективность выбранных KOHcepBaH
тов. Подходящий метод определения и критерии
оценки эффективности консервантов представ
лены в статье 5.1.3. Эффектuвность aHти
мuкробных консервантов. При производстве,
упаковке, хранении и реализации мяrких лекар
ственных средств для наружноro применения
принимают меры, обеспечивающие микробиоло
rическую чистоту в соответствии с требованиями
статьи 5. 1.4. МuкроБUОЛ02uческая чuстота He
стерuльных лекарственных средств u фарма
цевтuческuх субстанцuй. Стерильные мяrкие
лекарственные средства для наружноro при
менения производят с использованием матери
алов и методов, обеспечивающих стерильность,
предотвращающих заrрязнение лекарственных
средств и развитие микроорrанизмов в COOTBeT
ствии с требованиями статьи 5. 1. 1. Методы при
20товленuя стерuльных продуктов.
При разработке лекарственноrо средства
должно быть доказано, что из контейнера может
быть извлечено номинальное количество OДHO
дозовоro мяrкоro лекарственноro средства для
наружноro применения. #В случае отсутствия у
производителя лекарственноro средства cep
тификата соответствия GMP, а также данных о
валидации процесса производства KOHKpeTHoro
мяrкоro лекарственноro средства для наружноro
применения испытание по определению извле
KaeMoro объема должно быть включено в специ
фикацию roтовоro продукта.#
В процессе производства мяrких лекар
ственных средств для наружноro применения
принимают меры, обеспечивающие определен
ные реолоrические свойства. При необходимо
сти выполняют следующие испытания: изме
рение консистенции методом пенетрометрии
(2.9.9), определение вязкости (кажущейся вяз
кости) (2.2.10) и подходящее испытание, позво
ляюшее оценить высвобождение действующеro
вещества или веществ.
В процессе производства мяrких лекар
ственных средств для наружноrо применен\.1Я,
содержащих действующее вещество или веще
ства, которые не растворяются в основе (напри
мер, эмульсии или суспензии), принимают меры,
обеспечивающие однородность лекарственноro
средства.
При производстве мяrких лекарственных
средств для наружноro применения, содержа
щих дисперrированные частицы, принимают
меры по обеспечению и контролю необходимо
ro размера частиц в соответствии с назначением
данноrо лекарственноro средства.
ИСПЫТАНИЯ
Однородность дозированных единиц.
Мяrкие лекарственные средства, выпускаемые
либо в однодозовых контейнерах, либо в дози
рующих контейнерах, и которые предназначе
ны для трансдермальной доставки действующеro
вещества или веществ с целью получения си
стемноro эффекта, должны выдерживать испы
тание на однородность дозированных единиц
(2.9.40). Мяrкие лекарственные средства, coдep
жащие действующее вещество или вещества в
растворенном виде, должны выдерживать испы
тание расчетно--весовым методом; мяrкие лекар
ственные средства, содержащие действующее
вещество или вещества в суспензии, должны
выдерживать испытание методом прямоro опре
деления. Испытание про водят соrласно про
цедуре, указанной для жидких дозированных
форм. На лекарственное растительное сырье и
лекарственные средства из неro, представлен
ные в дозированной форме, требования данноro
раздела не распространяются.
Мяrкие лекарственные средства, выпуска
емые в дозирующих контейнерах, содержащие
действующее вещество или вещества в paCTBO
ренном виде, испытывают следующим образом.
Выпускают и отбрасывают одну дозу. Не менее
чем через 5 с при необходимости встряхивают
в течение 5 с, снова выпускают и отбрасывают
одну дозу. Эти действия повторяют еще 3 раза.
Взвешивают контейнер, выпускают и отбрасыва
ют одну дозу и снова взвешивают контейнер. Ha
ходят разницу между полученными значениями
масс. Повторяют описанную процедуру для сле
дующих 9 контейнеров. Определяют OДHOpOД
ность дозированных единиц с помощью расчет
но весовоro метода.
Мяrкие лекарственные средства, выпуска
емые в дозирующих контейнерах, содержа
щие действующее вещество или вещества в cy
спендированном виде, испытывают следующим
образом. Используют прибор, позволяющий
количественно удерживать отмерянную дозу, BЫ
пускаемую из дозирующеro контейнера. Встря
хивают один контейнер в течение 5 с, выпускают
и отбрасывают одну дозу. Не менее чем через 5
с встряхивают в течение 5 с, снова выпускают и
отбрасывают одну дозу. Эти действия повторя
ют еще 3 раза. Через 2 с выпускают из дозирую
щеro контейнера одну дозу в собирающий сосуд.
Собирают содержимое собирающеro сосуда с
помощью ополаскиваний. В полученном смыве
определяют содержание действующеro веще
ства. Повторяют описанную процедуру для сле
дующих 9 контейнеров. Определяют OДHOpOД
ность дозированных единиц с помощью метода
прямоro определения.
Стерильность (2.6.1). Если на этикетке YKa
зано, что лекарственное средство стерильно,
оно должно выдерживать испытание на стеРИJlb
ность.
ХРАНЕНИЕ
Если в состав лекарственноrо средства
входит вода или друrие испаряющиеся Be
щества, ero хранят в воздухонепроницаемых
контейнерах. Если лекарственное средство
1176
rосударственная фармакопея Республики Беларусь
стерильно, ero хранят в стерильных возду
хонепроницаемых контейнерах с контролем
первоro вскрытия.
МАРКИРОВКА
На этикетке должно быть:
названия всех добавленных вспомоrа
тельных веществ;
для стерильныхлекарствен ных cpeДCTB
указание о стерильности.
Мази
ОПРЕДЕЛЕНИЕ
Мазь состоит из однофазной основы, в
которой дисперrированы твердые вещества
или жидкости.
/uдрофобные мазu
rидрофобные мази MorYT абсорбировать
лишь небольшое количество воды. Основы,
которые используют для приroтовления таких
мазей, представляют собой твердые, жидкие
и леrкие жидкие парафины, растительные
масла, животные жиры, синтетические rлице
риды, воски и жидкие полиалкилсилоксаны.
Водоэмульсионные мазu
Водоэмульсионные мази MorYT абсорби
ровать большее количество воды и при ro
моrенизации образуют эмульсии типа вода/
масло или масло/вода в зависимости от при
роды эмульrатора. Эмульсии вода/масло об
разуются при использовании таких эмульrа
торов, как спирты шерстноro воска, эфиры
сорбита на, моноrлицериды и жирные спирты.
Эмульсии масло/вода образуются при ис
пользовании таких эмульrаторов, как суль
фатные жирные спирты, полисорбаты, цeTO
стеариловый эфир макроrола или сложные
эфиры жирных кислот с макроrолами. Основа
этих мазей такая же, как при приrотовлении
rидрофобных мазей.
/uдрофuльные мазu
rидрофильные мази представляют собой
лекарственные средства. имеющие основу, KO
торая смешивается с водой. В качестве основы
чаще всеro используют смеси жидких и TBep
дых макроroлов (полиэтиленrликолей). Эти
мази MorYT содержать некоторое количество
воды.
Кремы
ОПРЕДЕЛЕНИЕ
Кремы представляют собой мноrофазные
лекарственные средства, состоящие из липо
филь ной фазы и водной фазы.
Лuпофuльные кремы
В липофильных кремах в качестве посто
янной фазы используют липофильную фазу.
Они содержат эмульrаторы типа вода/масло,
такие как спирты шерстноrо воска, эфиры
сорбитана и моноrлицериды.
/uдрофuльные кремы
В rидрофильных кремах в качестве посто
янной фазы используют водную фазу. Они co
держат эмульrаторы типа маслоlвода, такие
как натриевые или троламиновые мыла,
сульфатные жирные спирты, полисорбаты и
полиоксильные жирные кислоты или эфиры
высших жирных спиртов, смешанные при He
обходимости с водно жировыми эмульrато
рами.
rели
ОПРЕДЕЛЕНИЕ
rели состоят из жидкостей, превращен
ных в rели с помощью rелеобразователей.
Липофuльные 2елu
Липофильные rели (олеоrели) представ
ляют собой лекарственные средства, COCTO
ящие из основы, содержащей вазелиновое
масло с полиэтиленом или жирными масла
ми, и rелеобразователей, таких как кремния
диоксид коллоидный, алюминиевое или цин
ковое мыло.
/идрофuльные 2елu
rидрофильные rели (rидроrели) представ
ляют собой лекарственные средства, приrо
товленные на основах, состоящих из воды,
rлицерина или пропиленrликоля, и rелеобразо
вателей, таких как крахмал, производные цел
люлозы, карбомеры и маrний алюминиевые
силикаты.
Пасты
ОПРЕДЕЛЕНИЕ
Пасты представляют собой мяrкие лекар
ственные средства для наружноrо примене
ния, содержащие значительное количество
твердых веществ #(обычно более 25 % м/м)#,
дисперrированных в основе.
Припарки
ОПРЕДЕЛЕНИЕ
Припарки состоят из rидрофильной, co
храняющей высокую температуру основы, в
которой дисперrированы твердые или жидкие
действующие вещества. Как правило, при
парки распределяют на подходящем перевя
зочном материале и наrревают перед нало
жением на кожу.
Пластыри медицинские
ОПРЕДЕЛЕНИЕ
Пластыри медицинские это rибкая
лекарственная форма, содержащая одно
или более действующих веществ и предна
значенная для нанесения на кожу. Пласты
ри медицинские предназначены для тоro,
чтобы удерживать действующее вещество
МЯ2кuе лекарственные средства для наРУЖНО20 прuмененuя
1177
или вещества в непосредственном контакте
с кожей, в результате чеrо они моrли бы Meд
ленно абсорбироваться или действовать как
защитные или кератолитические вещества.
Пластыри медицинские состоят из кле
евой основы, которая может быть окраше
на, и содержат одно или более действующих
веществ, нанесенных однородным слоем
на подложку, изrотовленную из природноro
или синтетическоro материала. Материал не
должен раздражать кожу или вызывать ал
лерrическую реакцию. Клеевой слой должен
быть защищен лайнером, который удаляется
перед нанесением пластыря на кожу. Защит
ный лайнер при удалении не должен OTдe
лять лекарственное средство от поддержи
вающеrо слоя.
Пластыри медицинские выпускают раз
личных размеров, в зависимости от пред
назначения, или в виде больших листов,
которые перед применением разрезают
ся. Пластыри медицинские при нанесении
на кожу должны плотно прилеrать к ней при
леrком надавливании, а при удалении не
должны повреждать кожу и не должны OTдe
лять лекарственное средство от поддержи
вающеro слоя.
ИСПЫТАНИЯ
Растворение. Проводят испытание, под
тверждающее соответствующее высвобож
дение действующеro вещества или веществ,
например, одним из испытаний, описанным
в статье 2.9.4. Тест «Растворение» для
трансдермальных пластырей.
Пластыри кожные
ОПРЕДЕЛЕНИЕ
Кожные пластыри это rибкая лекар
ственная форма, содержащая одно или более
действующих веществ и предназначенная для
нанесения на кожу. Пластыри предназначе
ны для тоro, чтобы удерживать действующ е
вещество или вещества внепосредственном
контакте с кожей, в результате чеrо они моrли
бы оказывать местное действие.
Кожные пластыри состоят из клеевой
основы, которая может быть окрашена, и co
держат одно или более действующих Be
ществ, нанесенных однородным слоем на
подложку, изroтовленную из природноro или
синтетическоro материала. Материал не
должен раздражать кожу или вызывать ал
лерrическую реакцию. Клеевой слой должен
быть защищен лайнером, КОТОI1ЫЙ удаляется
перед нанесением пластыря на кожу. 3ащит
ный лайнер при удалении не должен отделять
лекарственное средство от поддерживающе
ro слоя.
Кожные пластыри выпускают различных
размеров, в зависимости от предназначения,
или в виде больших листов, которые перед
применением разрезаются. Кожные пластыри
при нанесении на кожу должны плотно при
леrать к ней при леrком надавливании, а при
удалении не должны повреждать кожу и не
должны отделять лекарственное средство от
поддерживающеrо слоя.
ИСПЫТАНИЯ
Растворение. Проводят испытание, под
тверждающее соответствующее высвобож
дение действующеro вещества или веществ,
например, одним из испытаний, описанным
в статье 2.9.4. Тест «Растворение» для
трансдермальных пластырей.
#Линименты
ОПРЕДЕЛЕНИЕ
Линименты мяrкие лекарственные
средства для наружноro применения, плавя
щиеся при температуре тела. К линиментам
MorYT быть отнесены мази, кремы, rели и
пасты, характеризующиеся этим признаком.
#rидроrелевые пластины
ОПРЕДЕЛЕНИЕ
rидроrелевые пластины стерильная
эластичная лекарственная форма для наруж
HOro применения, имеющая фиксированные
rеометрические размеры. rидроrелевые пла
стины содержат одно или более действующих
веществ и предназначаются для аппликации
на пораженные кожные покровы с целью соз
дания защитноrо барьера и высвобождения
действующеrо вещества для наружноro при
менения.
ПРОИЗВОДСТВО
rидроrелевые пластины получают путем
специальной обработки водных растворов
полимеров, содержащих одно или более дей
ствующих веществ. Стерилизация rидроrеле
вых пластин осуществляется rамма лучами.
ИСПЫТАНИЯ
Стерильность (2.6.1). rидроrелевые
пластины должны выдерживать испытание на
стерильность.
МАРКИРОВКА
На этикетке указывают:
количественное содержание действу
ющеrо вещества;
«стерильно».
ХРАНЕНИЕ
Хранят в стерильном воздухонепроница
емом неповрежденном контейнере при TeM
пературе от 15 ос до 25 ОС, если нет друrих
указаний в частной статье.
01/2013:0676
rосударственная фармакопея Республики Беларусь
1178
НАЗАЛЬНЫЕ ЛЕКАРСТВЕННЫЕ
СРЕДСТВА
Nasalia
ОПРЕДЕЛЕНИЕ
Назальные лекарственные средства
жидкие, мяrкие или твердые лекарственные
средства, предназначенные для введения в HO
совую полость С целью получения системно
ro или местноro действия. Они содержат одно
или более действующих веществ. Назальные
лекарственные средства не должны оказывать
раздражающеro и друrоrо неблаroприятноro
воздействия на слизистую носа и ее волоски.
Водные назальные лекарственные средства
обычно изотоничны и MorYT содержать вспомо
rательные вещества, например, для обеспече
ния вязкости, создания или стабилизации рН,
повышения растворимости действующеrо Be
щества или обеспечения стабильности лекар
cTBeHHoro средства.
Назальные лекарственные средства выпу
скают в контейнерах. содержащих одну или He
сколько доз лекарственноrо средства и снаб
женных, если необходимо, приспособлением,
которое обеспечивает удобство применения и
предотвращает заrрязнение.
Если иное не обосновано и не утверждено,
водные назальные лекарственные средства в
мноroдозовых контейнерах содержат подходя
щий антимикробный консервант в необходимой
концентрации, за исключением лекарственнь
средств, обладающих достаточным антими
кробным действием.
Если это применимо, контейнеры для Ha
зальных лекарственных средств должны COOT
ветствовать требованиям статей разделов 3.1.
Матерuалы, используемые для проuзводства
контейнеров и 3.2. Контейнеры.
Назальные лекарственные средства можно
классифицировать как:
назальные капли и жидкие назальные
спреи;
назальные порошки;
назальные мяrкие лекарственные cpeд
ства;
назальные промывки;
назальные палочки.
ПРОИЗВОДСТВО
При раЗр'3ботке назальных лекарствен
ных средств, в состав которых входят антими
кробные консерванты, уполномоченному opraHY
представляют данные, подтверждающие He
обходимость использования и эффективность
выбранных консервантов. Подходящий метод
определения и критерии оценки эффективности
консервантов представлены в статье 5. 1.3. Эф
фективность антuмuкробных консервантов.
При производстве, упаковке, хранении и pe
ализации назальных лекарственных средств
принимают меры, обеспечивающие микробио
лоrическую чистоту в соответствии с требовани
ями статьи 5.1.4. МикроБUОЛ02uческая чuстота
нестерuльных лекарственных средств u фар
мацевтuческuх субстанцuЙ
Стерильные назальныелекарственныесред
ства производят с использованием материалов
и методов. обеспечивающих стерильность и
предотвращающих заrрязнение лекарственных
средств и развитие микроорrанизмов, в COOT
ветствии с требованиями статьи 5. 1. 1. Методы
ПРU20товленuя стерuльных продуктов.
При производстве назальных лекарствен
ных средств, содержащих дисперrированные ча
стицы, принимают меры, обеспечивающие необ
ходимый размер частиц и ero контроль.
ИСПЫТАНИЯ
Стерильность (2.6.1). Если на этикетке YKa
зано, что лекарственное средство стерильно, оно
должно выдерживать испытание на стерильность.
ХРАНЕНИЕ
Если лекарственное средство стерильно,
ero хранят в стерильных воздухонепроницаемых
контейнерах с контролем первоro вскрытия.
МАРКИРОВКА
На этикетке указывают:
названия всех добавленных антимикроб
ных консервантов;
«стерильно», если необходимо.
Назальные капли и жидкие назальные
спреи
ОПРЕДЕЛЕНИЕ
Назальные капли и жидкие назальные
спреи это растворы, эмульсии или суспензии,
предназначенные для закапывания или распы
ления в носовую полость.
Эмульсии MOryт расслаиваться, однако
быстро восстанавливаются при взбалтывании.
Суспензии MOryт образовывать осадок, который
быстро ресуспендируется при взбалтывании, об
разуя суспензию достаточно стабильную, чтобы
обеспечить однородность лекарственноro cpeд
ства при дозировании.
Назальные капли обычно выпускают в MHO
roдозовых контейнерах, снабженных подходя
щей насадкой.
Жидкие назальные спреи выпускают в контей
нерах с дозирующим устройством или в контейнерах
под давлением, снабженных подходящей насадкой,
а TalOКe с дозирующим клапаном или без нею. Если
сп реи выпускают в контейнерах под давлением, они
должны выдерживать требования статьи Лекар
ственные средства, нахадящuеся пад давпенuем.
Назальные лекарственные средства
1179
Размер распыленных капелек должен быть
таким, чтобы обеспечивать их осаждение в по
лости носа.
ИСПЫТАНИЯ
Если нет друrих указаний в частных статьях
или если иное не обосновано и не утвержде
но, назальные капли в однодозовых контейне
рах и отдельные дозы дозированных назальных
спреев, предназначенные для системноro дей
ствия, должны выдерживать следующие испы
rания.
НА3АЛЬНЫЕ КАПЛИ В ОДНОДОЗОВЫХ
КОНТЕЙНЕРАХ
Однородность дозированных единиц.
Назальные капли в однодозовых контейнерах
должны выдерживать испытание на OДHOpOД
ность дозированных единиц (2.9.40) или, если это
обосновано и утверждено, испытание на OДHO
родность массы или на однородность содержа
ния, как указано ниже. На лекарственное расти
тельное сырье и лекарственные средства из неro,
представленные в дозированной форме, требо
вания данноro раздела не распространяются.
Однородность массы. Назальные капли,
представляющuе собой растворы, должны вы
держuвать следующее uспытание. Освобожда
ют 1 О контейнеров как можно полнее и взвеши
вают содержимое каждоro из них, определяют
среднюю массу содержимоrо. Масса содержимоrо
не более чем двух контейнеров может отклонять
ся более чем на 10 % от средней массы, и масса
содержимоro ни одноro контейнера не должна от
клоняться более чем на 20 % от средней массы.
Однородность содержания (2.9.6). Ha
зальные каплu, представляющuе собой cy
спензиu uлu эмульсиu, должны выдержuвать
следующее испытанuе. Освобождают каждый
контейнер как можно полнее и определяют co
держание действующеro вещества для каждо
ro контейнера. Лекарственное средство ДОЛЖIiО
выдерживать испытание на однородность co
держания (тест В).
ДОЗИРОВАННЫЕ НА3АЛЬНЫЕ СПРЕИ
Однородность дозированных единиц.
Дозированные назальные спреи должны Bыдep
живать испытание на однородность дозирован
ных единиц (2.9.40) или, если это обосновано и
утверждено, испытание на однородность массы
или на однородность содержания, как указано
ниже. На лекарственное растительное сырье и
лекарствечные средства из неro, представлен
ные в дозированной форме, требования данноro
раздела не распространяются.
В случае дозuрованных назальных спреев,
которые являются растворами, поступают
следующuм образом. Выпускают и отбрасыва
ют одну дозу. Ожидают не менее 5 с, встряхива
ют в течение 5 с, выпускают и отбрасывают еще
одну дозу. Повторяют эту операцию еще 3 раза.
Взвешивают контейнер, выпускают и отбрасыва
ют одну дозу, снова взвешивают контейнер. Ha
ходят разницу между двумя массами. Повторяют
процедуру для следующих 9 контейнеров. Про
водят определение расчетно весовым методом
(2.9.40).
В случае дозuрованных назальных спреев,
которые являются суспензuямu uлu эмульсu
ямu, поступают следующuм образом. Исполь
зуют прибор, позволяющий количественно yдep
живать дозу после нажатия распылительноro
устройства. Встряхивают контейнер в течение
5 с, выпускают и отбрасывают одну дозу. Ожида
ют не менее 5 с, встряхивают в течение 5 с, BЫ
пускают и отбрасывают еще одну дозу. Повторя
ют эту операцию еще 3 раза. Через две секунды
выпускают одну дозу дозированноro назальноro
сп рея в собирающий сосуд путем активации pac
пылительноro устройства. Собирают содержи
мое сосуда достаточным количеством смывов.
Определяют содержание действующеro веще
ства в объединенных смывах. Процедуру по
вторя ют для следующих 9 контейнеров. Испы
тание проводят методом прямоro определения
(2.9.40).
Однородность массы. Дозuрованные Ha
зальные спреu, которые являются pacтвo
рами, должны выдержuвать следующее испы
тание. Выпускают и отбрасывают одну дозу.
Ожидают не менее 5 с, встряхивают в течение
5 с выпускают и отбрасывают еще одну дозу. По
вторя ют эту операцию еще 3 раза. Взвешивают
контейнер, выпускают и отбрасывают одну дозу
снова взвешивают контейнер. Находят разни
цу между двумя массами. Повторяют процедуру
для следующих 9 контейнеров.
Индивидуальные значения не более чем
двух контейнеров MOryт отклоняться более чем
на 25 % от среднеro значения, и ни одно значе
ние не должно отклоняться более чем на З5 %.
Однородность высвобождаемой дозы.
Дозuрованные назальные спреu, которые яв
ляются суспензиямu uлu эмульсuямu, должны
выдерживать следующее uспытанuе. Исполь
зуют прибор, позволяющий количественно yдep
живать дозу после нажатия распылительноro
устройства. Встряхивают контейнер в течение 5 с,
выпускают и отбрасывают одну дозу. Ожидают
не менее 5 с, встряхивают в течение 5 с, выпу
скают и отбрасывают еще одну дозу. Повторя
ют эту операцию еще 3 раза. Через две секунды
выпускают одчу дозу дозированноro назальноro
спрея в сосуд для сбора путем активации pac
пылительноro устройства. Собирают содержи
мое сосуда достаточным количеством смывов.
Определяют содержание действующеrо веще
ства в объединенных смывах. Процедуру повто
ряют для следующих 9 контейнеров.
1180
rосударственная фармакопея Республики Беларусь
Если иное не обосновано и не утвержде
но, лекарственное средство удовлетворяет
требованиям, если содержание действующе
ro вещества в дозе не более чем одноro KOH
тейнера не укладывается в пределы от 75 %
до 125 %, но не выходит за пределы от 65 %
до 135 % от среднеrо значения.
Если содержание действующеro веще
ства в дозе двух или трех отдельных контей
неров не укладывается в пределы от 75 % до
125 %, но укладывается в пределы от 65 %
до 135 %, повторяют испытание еще для 20
контейнеров. Лекарственное средство Bыдep
живает испытание. если содержание действу
ющеrо вещества в дозе не более чем трех
контейнеров не укладывается в пределы от
75 % до 125 %, но не выходит за пределы от
65 % до 135 % от среднеro значения.
Назальные порошки
ОПРЕДЕЛЕНИЕ
Назальные порошки это порошки, пред
назначенные для введения в носовую полость
посредством подходящеro приспособления.
Они должны выдерживать требования
статьи Порошкu для наРУЖН020 примененuя.
Размер частиц, которые осаждаются в
носовых полостях, подтверждают COOTBeT
ствующими методами определения размера
частиц.
Назальные мяrкие лекарственные
средства
ОПРЕДЕЛЕНИЕ
Мяrкие назальные лекарственные cpeд
ства должны выдерживать требования статьи
МЯ2кuе лекарственные средства для наруж
Н020 прuмененuя.
Контейнер должен иметь подходящее
приспособление для нанесения лекарствен
ноro средства.
Назальные промывки
ОПРЕДЕЛЕНИЕ
Назальные промывки обычно представляют
собой водные изотонические растворы, предна
значенные для очистки носовых полостей.
Назальные промывки, предназначенные
для применения на поврежденных участках носа
или используемые перед проведением хирурrи
ческих операций, должны быть стерильными.
ПРОИЗВОДСТВО
При разработке назальных промывок
должно быть доказано, что может быть из
влечено номинальное содержимое OДHO
дозовоro контейнера, содержащеro лекар
ственное средство. #В случае отсутствия у
производителя лекарственноro средства cep
тификата соответствия GMP, а также данных
о валидации процесса производства KOHKpeT
Horo лекарственноro средства испытание по
определению извлекаемоro объема должно
быть включено в спецификацию roTOBOro
продукта.#
Назальные палочки
ОПРЕДЕЛЕНИЕ
Назальные палочки должны выдерживать
требования статьи Палочкu.
01/2013:0671
ПАЛОЧКИ
Sty/i
к палочкам М02ут быть предъявлены дo
полнuтельные требованuя, указанные в дРУ2их
статьях, напрuмер Назальные лекарственные
средства.
ОПРЕДЕЛЕНИЕ
Палочки твердые лекарственные cpeд
ства, предназначенные для наружноro приме
нения. Они имеют форму коническоro стерж
ня и состоят из одноro или более действующих
веществ, распределенных в соответствующей
основе, которая распадается или расплав
ляется при температуре тела. Палочки, KOTO
рые вставляют в уретру или помещают в раны,
должны быть стерильными.
При производстве, упаковке, хранении и
реализации палочек принимают меры, обеспе
чивающие микробиолоrическую чистоту в co
ответствии с требованиями статьи 5. 1.4. Mи
кроБUОЛ02uческая чuстота нестерuльных
лекарственных средств и фармацевтuческuх
субстанцuй.
Стерильные палочки производят С исполь
зованием материалов и методов, обеспечива
ющих стерильность и предотвращающих за
rрязнение лекарственных средств и развитие
микроорrанизмов, в соответствии с требовани
ями статьи 5.1.1. Методы ПРU20товления cтe
рuльных продуктов.
Производство палочек обеспечивает OДHO
родность массы и/или однородность содержа
ния действующеro вещества.
ИСПЫТАНИЯ
Стерильность (2.6.1). Палочки, KOTO
рые вставляют в уретру или помещают в раны,
должны выдерживать испытание на стериль
ность.
Пенымедицинскuе
1181
МАРКИРОВКА
На этикетке указывают:
количество действующеro вещества,
содержащеrося в палочке;
«стерильно», если необходимо.
01/2013:1105
ПЕНЫ МЕДИЦИНСКИЕ
Musci madicati
К пенам медицинским М02ут быть
предъявлены дополнuтельные требованuя,
указанные в дРУ2их статьях, например, Ле
карственные средства для ректаЛЬН020
прuмененuя, Лекарственные средства для
ва2uнаЛЬН020 прuмененuя u >.кидкие лекар
ственные средства для наРУЖН020 прuмене
нuя.
ОПРЕДЕЛЕНИЕ
Пена медицинская это лекарственная
форма, состоящая из большоrо объема дис
перrированноrо в жидкости rаза и содержа
щая обычно одно или более действующих
веществ, поверхностно активные вещества,
обеспечивающие образование пены, а также
друrие вспомоrательные вещества. Пены Me
дицинские обычно предназначаются для Ha
несения на кожные покровы или слизистые
оболочки.
Пены медицинские обычно образуются
непосредственно во время применения из
жидкоro лекарственноro средства, находяще
rося в контейнере под давлением. Контейнер
должен быть снабжен устройством, состоящим
из клапана и насадки нажимноro типа, обе
спечивающим высвобождение пены.
Пены медицинские, предназначенные для
использования на сильно пораженных участ
ках кожных покровов или на больших OTKpЫ
тых ранах, должны быть стерильными.
Пены медицинские, выпускаемые в
контейнере под давлением, должны COOT
ветствовать требованиям статьи Лекар
ственные средства, находящuеся под дaв
ленuем.
ПРОИЗВОДСТВО
Стерильные пены медицинские произво
дят с использованием материалов и методов,
обеспечивающих их стерильность и исключа
ющих возможность их последующеro заrряз
нения и развитие микроорrанизмов в COOTBeT
ствии с требованиями статьи 5.1.1. Методы
ПРU20товленuя стерuльных продуктов.
ИСПЫТАНИЯ
Относительная плотность пены меди
цинекой. Контейнер выдерживают при TeM
пературе около 25 ос в течение как минимум
24 ч. Следя за тем, чтобы не HarpeTb KOH
тейнер, устанавливают на насадку нажимно
ro типа жесткую трубку длиной 70 100 мм с
внутренним диаметром отверстия около 1 мм.
Встряхивают контейнер для получения oд
нородноro содержимоrо, выпускают и отбра
сывают 5 10 мл пены. Взвешивают плоско
донную чашу вместимостью около 60 мл И
высотой около 35 мм. Помещают конец жест
кой трубки, установленной на насадке нажим
ноro типа, в уrол емкости, нажимают насадку
и равномерно, круrовыми движениями, запол
няют емкость. После тоro, как пена расширит
ся, сrлаживают ее уровень, удалив излишки
пены подходящим плоским предметом, Ha
пример. предметным стеклом. Взвешивают.
Определяют массу такоro же объема воды Р,
наполнив ту же чашу водой Р.
Относительную плотность пены медицин
ской рассчитывают по формуле:
т
е
rдe:
т масса испытуемоro образца пены, r;
е масса такоro же объема воды Р, r.
Проводят три определения. Ни одно из ин
дивидуальных значений не должно отклоняться
от среднеro значения более чем на 20 %.
Время расширения. Прибор (рисунок 1105.
1) состоит из бюретки вместимостью 50 мл,
внутренним диаметром 15 мм и ценой деления
0,1 мл, снабженной запорным краном с одним
отверстием диаметром 4 мм. Отметка, COOTBeT
ствующая 30 мл, должна находиться на paCCTO
янии не менее 210 мм от оси запорноro крана.
Нижняя часть бюретки соединена при помощи
пластмассовой трубки длиной не более 50 мм и
внутренним диаметром 4 мм с пенообразующим
контейнером, оборудованным подходящей для
данноro соединения насадкой нажимноro типа.
Контейнер выдерживают при температуре около
25 ос в течение как минимум 24 ч. Следя за тем,
чтобы не HarpeTb, встряхивают контейнер для
получения однородноrо содержимоro, выпуска
ют и отбрасывают 5 1 О мл пены. Соединяют
насадку и носик бюретки. Открывают запорный
кран, нажимают на насадку и выпускают одну
дозу пены объемом около 30 мл. Закрывают за
поrный кран и одновременно включают XpOHO
метр. Наблюдают за объемом пены в бюретке,
каждые 1 О с отмечают увеличение объема пены
до тех пор, пока не будет достиrнут максималь
ный объем. Проводят три определения. Время
достижения максимальноro объема в каждом
определении не должно превышать 5 мин.
1182
rосударственная фармакопея Республики Беларусь
50 Внутренний диаметр 15 мм
20
10
о
Насадка нажимноrо типа
Пластиковая трубка
внутренний диаметр 4 .....
маУ,сималы.,ая длиt-iа 50 мм
Контейнер.
находящийся
ПОД давлением
Рисунок 1105. 1 Прuбор для определенuя
времени расширения
Стерильность (2.6.1). Если на этикетке YKa
зано, что лекарственное средство стерильно,
оно должно выдерживать испытание на стериль
ность.
МАРКИРОВКА
При необходимости на этикетке указывают
«стерильно» .
01/2013:1165
ПОРОШКИ для BHYТPEHHEro
ПРИМЕНЕНИЯ
Pulve,es perorales
Требованuя к порошкам, uспользуемым для
при20товленuя растворов uлu суспензuй для
ораЛЬН020 прuмененuя, прuведены в статье
Жuдкuе лекарственные средства для вHyтpeH
не20 прuмененuя.
ОПРЕДЕЛЕНИЕ
Порошки для внутреннеro применения
это лекарственные средства, состоящие из TBep
дых отдельных сухих частиц различной степе
ни измельчения. Они содержат одно или более
действующих веществ со вспомоrательными Be
ществами или без них и, если необходимо, Kpa
сители, разрешенные к применению, и apOMa
тизаторы. Порошки обычно принимают с водой
или друroй подходящей жидкостью. Их можно
также rлотать непосредственно. Порошки MOryт
содержать одну или несколько доз лекарствен
ноro средства.
Если это применимо, контейнеры для по
рошков для внутреннею применения должны
соответствовать требованиям общих статей раз
делов 3. 1. Матерuалы, используемые для про
uзводств8 контейнеров и 3.2. Контейнеры.
Каждую дозу порошка из мноroдозовоrо KOH
тейнера отбирают с помощью соответствующеro
приспособления для отмеривания предписанно
ro количества. Каждая доза однодозовоro по
рошка должна быть упакована в отдельный KOH
тейнер, например, в пакетик или флакон.
ПРОИЗВОДСТВО
При производстве порошков для внутреннеro
применения принимают меры, обеспечивающие
получение необходимоro размера частиц.
При производстве, упаковке, хранении и
реализации порошков для внутреннеro приме
нения принимают меры, обеспечивающие ми
кробиолоrическую чистоту в соответствии с
требованиями статьи 5. 1.4. МuкробиОЛ02uче
ская чuстота нестерuльных лекарственных
средств u фармацевтuческих субстанцuй.
ИСПЫТАНИЯ
Однородность дозированных единиц. Oд
нодозовые порошки для внутреннеro примене
ния должны выдерживать испытание на OДHOpOД
ность дозированных единиц (2.9.40) или, если это
обосновано и утверждено, испытания на OДHO
родность содержания иlили однородность массы,
как указано ниже. На лекарственное раститель
ное сырье и лекарственные средства из неro.
представленные в дозированной форме, требо
вания данноro раздела не распространяются.
Однородность содержания (2.9.6). По
рошки для внутреннеro применения в однодозо
вых контейнерах с содержанием действующеro
вещества менее 2 Mr или менее 2 % от общей
массы содержимоro должны выдерживать ис
пытание на однородность содержания действу
ющеro вещества в единице дозированноro ле
карственноro средства (тест В), если нет друrих
указаний в частной статье. Если лекарственное
средство содержит более одною действующе
ro вещества. это требование распространяется
только на те вещества, содержание кnторых co
ответствует вышеуказанным условиям.
Однородность массы (2.9.5). Порошки для
BHYTpeHHero применения в однодозовых контей
нерах должны выдерживать испытание на oд
нородность массы для единицы дозированноrо
Порошкu для наружноео прuменения
1183
лекарственноro средства. Испытание на OДHO
родность массы не требуется, если испытание
на однородность содержания предусмотрено
для всех действующих веществ.
Однородность массы одной дозы, BЫ
свобожденной из мноrодозовоrо контейнера
(2.9.27). Порошки для внутреннеro применения
в мноroдозовых контейнерах должны выдержи
вать испытание на однородность массы дозы.
высвобожденной из мноrодозовоro контейнера.
ХРАНЕНИЕ
Если лекарственное средство содержит ле
тучие вещества или содержимое должно быть
защищено. хранят в воздухонепроницаемом
контейнере.
Порошки «шипучие»
Порошки «шипучие» это однодозовые
или мноroдозовые порошки, содержащие rлав
ным образом кислоты и карбонаты или rидро
карбонаты, которые при наличии воды быстро
вступают в реакцию с выделением уrлекислоro
rаза. Они предназначены для растворения или
дисперrирования в воде перед применением.
ХРАНЕНИЕ
Хранят в воздухонепроницаемом контейнере.
01/2013:1166
ПОРОШКИ ДЛЯ НАРУжноrо
ПРИМЕНЕНИЯ
Pu/veres ad usum dermicum
Еслu иное не обосновано и не утверждено,
требованuя данной статьи не распространя
ются на порошкu для наРУЖН020 прuмененuя,
uспользуемые в ветеринарии.
ОПРЕДЕЛЕНИЕ
Порошки для наружноro применения это
лекарственные средства, состоящие из TBep
дых отдельных сухих частиц различной степе
ни измельчения. Они содержат одно или более
действующих веществ со вспомоrательными
веществами или без них и, если необходимо,
красители, разрешенные к применению уполно
моченным opraHoM.
Порошки для наружноro применения MOryT
содержать одну или несколько доз лекарствен
Horo средства. Они не должны проявлять зерни
стость. Порошки, предназначенные для наложе
ния на большие открытые раны или на сильно
поврежденные участки кожи, должны быть CTe
рильными.
Мноroдозовые порошки упаковывают в KOH
тейнеры, снабженные фильтром, контейнеры,
снабженные механическим приспособлением
для распыления, или в контейнеры под давле
нием.
Порошки, выпускаемые в контейнерах под
давлением, должны соответствовать требовани
ям статьи Лекарственные средства, находящu
еся под давлением.
Если это применимо, контейнеры для по
рошков для наружноro применения должны co
ответствовать требованиям общих статей раз
делов 3. 1. Матерuалы, uспользуемые для
производства контейнеров и 3.2. Контейнеры.
ПРОИЗВОДСТВО
При производстве порошков для наружноro
применения принимают меры, обеспечивающие
получение необходимоrо размера частиц.
При производстве, упаковке, хранении
и реализации порошков для наружноro при
менения принимают меры, обеспечивающие
микробиолоrическую чистоту в соответствии
с требованиями статьи 5. 1.4. МикроБUОЛ02U
ческая чuстота нестерuльных лекарствен
ных средств u фармацевтuческих субстан
ций.
Стерильные порошки для наружноro приме
нения производят с использованием материалов
и методов, обеспечивающих стерильность и
предотвращающих заrрязнение лекарственных
средств и развитие микроорrанизмов, в COOTBeT
ствии с требованиями статьи 5. 1. 1. Методы прu
20товленuя стерuльных продуктов.
ИСПЫТАНИЯ
Степень измельчения. Степень измельче
ния порошка определяют аналитическим просе
иванием (2.9.35) или друrим подходящим MeTO
дом, указанным в частной статье.
Однородность дозированных единиц.
Однодозовые порошки для наружноro приме
нения должны выдерживать испытание на oд
нородность дозированных единиц (2.9.40) или,
если это обосновано и утверждено, испытания
на однородность содержания и/или OДHOpoД
ность массы, как указано ниже. На лекарствен
ное растительное сырье и лекарственные cpeд
ства из неro, представленные в дозированной
форме, требования данною раздела не распро
страняются.
Однородность содержания (2.9.6). По
рошки для наружноro примеl--!ения в однодозо
вых контейнерах с содержанием действующеro
вещества менее 2 Mr или менее 2 % от общей
массы содержимоro должны выдерживать ис
пытание на однородность содержания действу
ющеrо вещества в единице дозированноro ле
карственноro средства (тест В), если нет друrих
указаний в частной статье. Если лекарственное
1184
rосударственная фармакопея Республики Беларусь
средство содержит более одноrо действующе
ro вещества, это требование распространяется
только на те вещества, содержание которых co
ответствует вышеуказанным условиям.
Однородность массы (2.9.5). Порошки для
наружною применения в однодозовых контей
нерах должны выдерживать испытание на oд
нородность массы для единицы дозированноrо
лекарственноro средства. Испытание OДHOpOД
ности массы не требуется, если испытание на
однородность содержания предусмотрено для
всех действующих веществ.
Стерильность (2.6.1). Если на этикетке YKa
зано, что лекарственное средство стерильно,
оно должно выдерживать испытание на стериль
ность.
МАРКИРОВКА
На этикетке указывают:
лекарственное средство предназначено
для наружноro применения;
«стерильно» , если необходимо.
01/2013:0478
ТАБЛЕТКИ
Compress/
Требованuя данной статьи не обяза
теЛЬНbI для таблетuроваННblХ лекарствен
НЫХ средств. предназначеННblХ к примененuю
не ораЛЬНblМ, а дРУ2ИМ способом. Требованuя
к таким лекарственным средствам М02ут
быть прuведены в дРУ2их статьях, напрu
мер, Лекарственные средства для ректаль
Н020 применения, Лекарственные средства
для ва2uнаЛЬН020 прuмененuя U Лекарствен
ные средства для слuзuстой оболочкu поло
сти рта. Требованuя данной статьи не pac
пространяются на леденцы, оральные пасты
U жевательные резuнкu. Требования данной
статьи не распространяются на таблет
ки для прuмененuя в ветеринарии, если нет
дРУ2их указанuй в частных статьях.
ОПРЕДЕЛЕНИЕ
Таблетки твердые лекарственные cpeд
ства, содержащие одну дозу одною или более
действующих веществ, полученные обычно
прессованием определенноro объема частиц
или arperaToB частиц или иным подходящим
методом производства, например, экструзией,
формованием или лиофилизацией, и предна
значенные для оральною применения. HeKOTO
рые таблетки проrлатывают целыми, некоторые
предварительно разжевывают, друrие же pacт
воряют или дисперrируют в воде перед приме
нением, оставляют во рту, rдe действующее Be
щество высвобождается.
Частицы состоят из одноro или более дей
ствующих веществ и вспомоrательных Be
ществ, таких как разбавляющие, связыва
ющие, разрыхляющие, скользящие, способные
изменить поведение лекарственной формы в
пищеварительном тракте, красители, разре
шенные к применению уполномоченным opra
ном, и ароматизаторы, или без вспомоrатель
ных веществ.
Таблетки обычно представляют собой цель
ные правильные круrлые твердые цилиндры,
верхняя и нижняя поверхности которых плоская
или выпуклая, края поверхностей MOryT быть
кошены. На поверхности таблеток MOryT быть
нанесены риски для деления, надписи и друrие
обозначения. Таблетки MOryT быть покрыты обо
лочкой.
Контейнеры для таблеток должны COOTBeT
ствовать требованиям общих статей разделов
3. 1. Матерuалы, uспользуемые для производ
ства контейнеров и 3.2. Контейнеры, если нет
друrих указаний в частной статье.
Таблетки для приема внутрь можно класси
фицировать как:
таблетки без оболочки;
таблетки, покрытые оболочкой;
таблетки «шипучие»;
таблетки растворимые;
таблетки дисперrируемые;
таблетки, дисперrируемые в полости рта;
таблетки с модифицированным высвобож
дением;
таблетки кишечнорастворимые;
таблетки для применения в полости рта;
таблетки лиофилизаты.
ПРОИЗВОДСТВО
Таблетки обычно получают прессовани
ем определенноro объема частиц или arpera
тов частиц, полученных методом rрануляции.
При производстве таблеток предпринимают co
ответствующие меры, обеспечивающие необхо
димую механическую прочность и устойчивость
таблеток к раздавливанию и истиранию. Это
подтверждается испытаниями 2.9.7. Прочность
таблеток без оболочкu на истирание и 2.9.8.
Прочность таблеток на сжатuе. Таблетки же
вательные изrотавливают, обеспечивая леrкое
разрушение при жевании.
#Если не обосновано и не утверждено иначе,
количество твина 80, кислоты стеариновой, каль
ция или маrния стеарата в таблетках не должно
превышать 1 %, талька 3 %, аэросила
10 % от массы таблетки.#
Деленuе таблеток. Таблетки MorYT иметь
одну или несколько рисок и MOryT быть разде
лены на части в целях более удобною приема
лекарственноro средства, либо для обеспече
Таблетки
1185
ния принятия определенной дозы. В последнем
случае деление таблеток должно быть oцeHe
но и утверждено уполномоченным opraHoM. Для
тоro чтобы убедиться в том, что пациент будет
получать необходимую ДОЗУ, на этапе разработ
ки лекарственноro средства должна быть oцe
нена эффективность риски/рисок в отношении
однородности массы частей таблетки. Каждая
заявленная доза должна быть проверена в соот
ветствии со следующим испытанием.
30 таблеток, выбранных случайным обра
зом, делят вручную, из полученных частей Ta
блетки берут одну для проведения испытания,
отбрасывая оставшуюся часть/части. Взвешива
ют каждую из 30 частей по отдельности и рассчи
тывают среднюю массу. Таблетки выдерживают
испытание, если не более чем одна индивиду
альная масса выходит за пределы от 85 % до
115 % от полученной средней массы. Таблет
ки не выдерживают испытание, если более чем
одна индивидуальная масса выходит за YCTaHOB
ленные пределы либо если хотя бы одна инди
видуальная масса выходит за пределы от 75 %
до 125 % от полученной средней массы.
В процессе производства, упаковки, xpaHe
ния и реализации таблеток принимают меры,
обеспечивающие микробиолоrическую чисто
ту в соответствии с требованиями статьи 5.1.4.
МuкроБUОЛ02uческая чuстота нестерuльных
лекарственных средств u фармацевтическuх
субстанцuй.
ИСПЫТАНИЯ
Однородность дозированных единиц
(2.9.40). Таблетки должны выдерживать испыта
ние на однородность дозированных единиц или,
если это обосновано и утверждено, испытания
на однородность содержания и/или OДHOpOД
ность массы, как указано ниже. На лекарствен
ное растительное сырье и лекарственные cpeд
ства из неro, представленные в дозированной
форме, требования данноro раздела не распро
страняются.
Однородность содержания (2.9.6). Ta
блетки с содержанием действующеro вещества
менее 2 Mr или менее 2 % от общей массы co
держимоrо должны выдерживать испытание на
однородность содержания действующеrо веще
ства в единице дозированноro лекарственно
ro средства (тест А), если нет друrих указаний
в частной статье. Если лекарственное средство
содержит более одноro действующеro веще
ства, это требование распространяется только
на те вещества, содержание которых COOTBeT
ствует вышеуказ:знным условиям.
Таблетки, покрытые оболочкой, за исключе
нием пленочноro покрытия, должны выдержи
вать испытание на однородность содержания
действующеro вещества в единице дозирован
ноro лекарственноro средства (тест А) независи
мо от содержания в них действующеro вещества
или веществ, если нет друrих указаний в част
ной статье.
Однородность массы (2.9.5). Таблетки без
оболочки и, если нет друrих указаний в част
ной статье, таблетки, покрытые пленочной обо
лочкой, должны выдерживать испытание на oд
нородность массы для единицы дозированноrо
лекарственноro средства. Испытание на OДHO
родность массы не требуется, если испытание
на однородность содержания предусмотрено
для всех действующих веществ или если нет
друrих указаний в частной статье.
Растворение. Испытание про водят для
подтверждения высвобождения действующеro
вещества или веществ, например, одним из спо
собов, описанных в статье 2.9.3. Тест «Pacтвo
рение» для твердых дозuрованных форм.
Если проводят испытание по показателю
«Растворение», испытание по показателю «Pac
падаемость» не требуется.
# Тальк и аэросил. Если не обосновано и
не утверждено иначе, про водят определение co
держания в таблетках талька и аэросипа. 1,000 r
порошка растертых таблеток обрабатывают в
сосуде 200 мл теплой воды Р. Полученную жид
кость фильтруют через беззольный фильтр и
сосуд тщательно ополаскивают водой Р. Остаток
на фильтре несколько раз промывают теплой
водой Р порциями по 1 О мл до отсутствия види
моro остатка после выпаривания капли промыв
ной воды на часовом стекле. Фильтр с остатком
высушивают, сжиrают, прокаливают и взвешива
ют с точностью до 0,0001 r.
Если таблетка содержит HecropaeMbIe или
нерастворимые в теплой воде Р вещества, Ha
веску таблеток после сжиrания и прокаливания
обрабатывают при наrревании 30 мл кuслоты
хлорuстоводородной разведенной Р Получен
ный раствор фильтруют и остаток на фильтре
промывают rорячей водой Р до отсутствия в
промывной воде реакции на хлориды. Фильтр с
остатком высушивают, сжиrают. прокаливают и
взвешивают с точностью до 0,0001 r::
Содержание талька и аэросила не должно
превышать пределы, указанные в частной
статье.
Таблетки без оболочки
ОПРЕДЕЛЕНИЕ
Таблетки без оболочки однослойные Ta
блетки, полученные однократным прессованием
частиц, или мноroСJlойные таблетки, состоящие
из концентрических или параллельных слоев,
полученные последовательным прессованием
частиц различноro состава. Используемые вспо
моrательные вещества специально не предна
значены для высвобождения действующеro Be
щества в желудочно кишечном тракте.
1186
rосударственная фармакопея Республики Беларусь
Таблетки без оболочки соответствуют
общему определению таблеток. На разломе при
рассмотрении под лупой видна та или иная OTHO
сительно однородная структура (однослойные
таблетки) или послойная структура (мноrослой
ные таблетки), но не признаки оболочки.
ИСПЫТАНИЯ
Распадаемость (2.9.1). Таблетки без
оболочки должны выдерживать испытание на
распадаемость таблеток и капсул. В качестве
жидкой среды используют воду Р. В каждую
стеклянную трубку помещают диск. Прибор
включают на 15 мин, если нет друrих указа
ний в частной статье, и исследуют состояние
таблеток. Если таблетки не выдержали испы
тание вследствие прилипания к дискам, ис
пытание повторяют на следующих шести Ta
блетках без дисков.
Таблетки жеватетельные uспытанuю
на распадаемость не подлежат.
Таблетки, покрытые оболочкой
ОПРЕДЕЛЕНИЕ
Таблетки, покрытые оболочкой, Ta
блетки, покрытые одним или нескольки
ми слоями смеси различных веществ, таких
как натуральные или синтетические смолы,
камеди, желатин, неактивные и нераствори
мые наполнители, сахара, пластификаторы,
полиспирты, воски, красители, разрешенные
к применению уполномоченным opraHoM, и
иноrда ароматизаторы и действующие веще
ства. Вещества, используемые для покрытия
таблеток, обычно наносят в виде растворов
или суспензий в условиях, позволяющих pac
творителю испариться. Korдa оболочка пред
ставляет собой очень тонкое полимерное по
крытие, таблетки определяют как таблетки,
покрытые пленочной оболочкой.
Таблетки, покрытые оболочкой, имеют
rладкую поверхность, которая часто окраше
на и может быть отполирована; на разломе
при рассматривании под лупой видно ядро,
окруженное одним или несколькими сплош
ными слоями различной структуры.
ПРОИЗВОДСТВО
Однородность содержания или OДHOpOД
ность массы таблеток, покрытых оболочкой,
за исключением таблеток, покрытых пленоч
ной оболочкой, достиrается путем контроля
ядер таблеток.
ИСПЫТАНИЯ
Распадаемость (2.9.1). Таблетки, покры
тые оболочкой, за исключением пленочной,
должны выдерживать испытание на распада
емость таблеток и капсул. В качестве жидкой
среды используют воду Р. В каждую стеклян
ную трубку помещают диск. При бор включают
на 60 мин, если нет друrих указаний в част
ной статье, и исследуют состояние таблеток.
Если не распалась хотя бы одна из шести Ta
блеток, испытание повторяют на следующих
таблетках, заменив воду Р в сосуде на О, 1 М
кuслоту хлористоводородную.
Таблетки, покрытые пленочной оболоч
кой, должны выдерживать испытание, как
указано выше, при этом прибор включают на
30 мин, если нет друrих указаний в частной
статье.
Если таблетки, покрытые оболочкой, или
таблетки, покрытые пленочной оболочкой, не
выдержали испытание вследствие прилипа
ния к дискам, испытание повторяют на следу
ющих шести таблетках без дисков.
Таблеткu жевательные, покрытые обо
почкой, испытанuю на распадаемость не
подлежат.
Таблетки «шипучие»
ОПРЕДЕЛЕНИЕ
Таблетки «шипучие» таблетки без обо
лочки, основную массу которых составляют кис
лоты и карбонаты или rидрокарбонаты, быстро
реаrирующие в присутствии воды с выделени
ем уrлерода диоксида. Эти таблетки предназна
чены для растворения или дисперrирования в
воде перед применением.
ИСПЫТАНИЯ
Распадаемость. Одну таблетку помещают в
лабораторный стакан, содержащий 200 мл воды Р
при температуре от 15 ос до 25 ОС; выделяются
мноroчисленные пузырьки rаза. Таблетка счита
ется распавшейся, если после прекращения BЫ
деления rаза BOKpyr таблетки или ее фраrментов
она растворилась или дисперrировалась в воде
без аrломератов частиц. Повторяют процедуру
на пяти друrих таблетках.
Лекарственное средство выдерживает ис
пытание, если каждая из шести таблеток распа
лась в течение 5 мин, если нет друrих указаний
в частной статье.
Таблетки растворимые
ОПРЕДЕЛЕНИЕ
Таблетки растворимые таблетки без
оболочки или таблетки, покрытые пленочной
оболочкой. Эти таблетки перед применением
растворяют в воде. В полученном растворе дo
пускается леrкая опалесценция из за вспомоrа
теЛЫ.IЫХ веществ, использованных при изroтов
лении тяблеток.
ИСПЫТАНИЯ
Распадаемость (2.9.1). Таблетки раствори
мые должны распадаться в течение 3 мин. В Ka
честве жидкой среды используют воду Р с TeM
пературой от 15 ос до 25 ОС.
Таблетки
1187
Таблетки дисперrируемые
ОПРЕДЕЛЕНИЕ
Таблетки дисперrируемые таблетки без
оболочки или таблетки, покрытые пленочной
оболочкой. Перед применением их дисперrиру
ют в воде до образования roмоrенной суспеl-lЗИИ.
ИСПЫТАНИЯ
Распадаемость (2.9.1). Таблетки дисперrи
руемые должны распадаться в течение 3 мин. В
качестве жидкой среды используют воду Р с TeM
пературой от 15 ос до 25 ОС.
Степень дисперrирования. Две таблетки
помещают в колбу, содержащую 100 мл воды р,
и перемешивают до полноro дисперrирования.
Должна образовываться однородная суспензия,
проходящая через сито с номинальным разме
ром отверстий 710 мкм.
Таблетки, дисперrируемые в полости рта
ОПРЕДЕЛЕНИЕ
Таблетки, дисперrируемые в полости рта,
таблетки без оболочки, которые помещают в po
товую полость, rдe они быстро дисперrируются
прежде, чем будут проrлочены.
ИСПЫТАНИЯ
Распадаемость (2.9.1). Таблетки, дисперrи
руемые в полости рта, должны распадаться в Te
чение 3 мин.
Таблетки с модифицированным BЫCBO
бождением
ОПРЕДЕЛЕНИЕ
Таблетки с модифицированным высвобож
дением таблетки, покрытые оболочкой или
без оболочки, содержащие специальные вспо
моrательные вещества или изютовленные спе
циальными способами, которые отдельно или
вместе предназначены для изменения скорости
или места высвобождения действующею веще
ства или веществ.
К таблеткам с модифицированным BЫCBO
бождением относятся таблетки с пролонrиро
ванным, замедленным и прерывистым BЫCBO
бождением.
ПРОИЗВОДСТВО
Проводят испытание, подтверждающее co
ответствующее высвобождение действующеro
вещества или веществ.
Таблетки кишечнорастворимые
ОПРЕДЕЛЕНИЕ
Таблетки кишечнорастворимые таблет
ки с замедленным высвобождением, которые
должны быть устойчивыми в желудочном соке
и высвобождать действующее вещество или Be
щества в кишечном соке. Такие таблетки изro
тавливают, покрывая ядра таблеток оболочкой,
устойчивой к желудочному соку (таблетки, по
крытые кишечнорастворимой оболочкой), или
изroтавливают из rранул или частиц с HaHeceH
ной на них ранее оболочкой, устойчивой к желу
дочному соку.
Таблетки, покрытые кишечнорастворимой
оболочкой. относят к rруппе таблеток, по крытых
оболочкой.
ПРОИЗВОДСТВО
Для таблеток, приrотовленных из rранул или
частиц, предварительно покрытых кишечнора
створимой оболочкой. проводят необходимое
испытание, подтверждающее соответствующее
высвобождение действующеro вещества или Be
ществ.
ИСПЫТАНИЯ
Распадаемость (2.9.1). Для таблеток, по
KpblTbIX кишечнорастворимой оболочкой, про
водят испытание на распадаемость со следу
ющими изменениями: в качестве жидкой среды
используют 0,1 М кuслоту хлорuстоводород
ную. Прибор включают на 2 ч, если нет друrих
указаний в частной статье, без дисков и иссле
дуют состояние таблеток. Время устойчивости
таблеток в кислой среде может быть различ
ным и зависит от состава испытуемых табле
ток. Обычно оно составляет от 2 ч до 3 ч, но,
даже если приведены друrие указания в част
ной статье, время устойчивости в кислой среде
должно быть не менее 1 ч. Ни одна из таблеток
не должна обнаруживать признаков распада (не
считая фраrментов покрытия) и иметь трещины,
через которые возможен выход содержимоro.
Кислоту заменяют фосфатным буферным pac
теором рН 6,8 Р и в каждую стеклянную трубку
вносят диск. Прибор включают на 60 мин и ис
следуют состояние таблеток Если таблетки не
выдержали испытания вследствие прилипания к
дискам, испытание повторяют на шести следу
ющих таблетках без дисков.
Растворение (2.9.3). Для таблеток, изro
товленных из rранул или частиц, уже покрытых
кишечнорастворимой оболочкой, проводят He
обходимое испытание, подтверждающее co
ответствующее высвобождение действующеro
вещества или веществ, например, одним из спо
собов, описанных в статье 2.9.3. Тест «Pacтвo
рение» для твердых дозuрованных форм.
Таблетки для применения в полости рта
ОПРЕДЕЛЕНИЕ
Таблетки для применения в полости рта
обычно таблетки без оболочки. Состав обеспе
чивает медленное высвобождение и местное
действие действующеro вещества или веществ
1188
rосударственная фармакопея Республики Беларусь
или высвобождение и всасывание действующе
ro вещества или веществ в определенных обла
стях рта. Таблетки для применения в полости
рта должны выдерживать требования статьи Ле
карственные средства для слuзuстой оболоч
ки полостu рта.
т аблетки лиофилизаты
ОПРЕДЕЛЕНИЕ
Таблетки лиофилизаты твердые лекар
ственные средства, предназначенные для поме
щения в рот или для дисперrирования (или paCT
ворения) в воде перед применением.
ПРОИЗВОДСТВО
Таблетки лиофилизаты получают с помо
щью лиофилизации, включающей в себя разде
ление на дозы, замораживание, сублимацию и
высушивание обычно водных жидких или полу
жидких лекарственных средств.
ИСПЫТАНИЯ
Распадаемость. Одну таблетку лиофили
зат помещают в лабораторный стакан, coдep
жащий 200 мл воды Р при температуре от 15 ос
до 25 ОС. Таблетка должна распасться в тече
ние 3 мин. Повторяют процедуру на пяти друrих
таблетках лиофилизатах. Лекарственное cpeд
ство выдерживает испытание, если распалась
каждая из шести таблеток.
Вода (2.5. 12). Таблетка лиофилизат должна
выдерживать требование по содержанию воды,
указанное в частной статье.
01/2013:1155
ТАМПОНЫ МЕДИЦИНСКИЕ
Татропае medicate
К тампонам медицинским М02ут быть
предъявлены дополнuтельные требованuя,
указанные в дРУ2их статьях, напрuмер, Ле
карственные средства для ректаЛЬНО20 при
мененuя, Лекарственные средства для вa2и
наЛЬН020 прuмененuя и Ушные лекарственные
средства.
ОПРЕДЕЛЕНИЕ
Тампоны медицинские это твердые ле
карственные средства, содержащие одну дозу
лекарственноro вещества и предназначен
ные для введения в полости на оrраниченный
период времени. Тампоны медицинские состоят
из подходящеro материала, TaKoro как целлюло
за, коллаrен или силикон, пропитанноro одним
или более действующим веществом.
ПРОИЗВОДСТВО
При производстве, упаковке, хранении и
реализации тампонов медицинских принимают
меры, обеспечивающие микробиолоrическую
чистоту в соответствии с требованиями статьи
5.1.4. МикроБUОЛ02uческая чuстота Hecтe
рuльных лекарственных средств U фармацев
тuческuх субстанцuЙ
МАРКИРОВКА
На этикетке указывают количество действу
ющеro вещества (веществ), содержащеroся в
тампоне.
01/2013:1011
ТРАНСДЕРМАЛЬНЫЕ ПЛАСТЫРИ
Emp/astra trапsсutапеа
ОПРЕДЕЛЕНИЕ
Трансдермальные пластыри это эластич
ные лекарственные средства различных разме
ров, содержащие одно или более действующих
веществ. Они предназначаются для апплика
ции на непораженные кожные покровы с целью
высвобождения действующеro вещества или
веществ в системное кровообращение через
кожный барьер.
Трансдермальные пластыри обычно co
стоят из внешней оболочки, которая удержива
ет лекарственное средство с действующим Be
ществом или веществами. Трансдермальные
пластыри покрыты на участке высвобождения
действующеro вещества защитным слоем, KO
торый удаляют перед аппликацией пластыря
на кожу.
Внешней оболочкой является непроница
емая для действующеro вещества или веществ
и обычно для воды пластинка, предназначен
ная для удержания и защиты лекарственно
ro средства. Внешняя оболочка может иметь
те же размеры, что и само лекарственное
средство, или по размерам она может быть
больше. В последнем случае выступающий
край внешней оболочки покрыт прикрепля
ющимися путем надавливания клейкими Be
ществами, которые обеспечивают аппликацию
пластыря на кожу.
Лекарственное средство содержит действу
ющее вещество или вещества и вспомоrатель
ные вещества, такие как стабилизаторы, солюби
лизаторы или вещества, изменяющие скорость
высвобождения действующеro вещества или
Ушные лекарствеННblе средства
1189
веществ или увеличивающие трансдермаль
ную абсорбцию. Это может быть однослойная
или мноrослойная твердая или пастообразная
матрица, и в этом случае ее состав и CTPYКТY
ра определяют диффузию действующеro веще
ства или веществ на кожных покровах. Матрица
может содержать прикрепляющиеся путем Haдa
вливания клейкие вещества, которые обеспечи
вают аппликацию лекарственною средства на
коже. Лекарственное средство может существо
вать в виде полутвердой пластинки, одна CTO
рона которой является оболочкой, контролиру
ющей высвобождение и диффузию действую
щеro вещества или веществ из лекарственно
ro средства. В этом случае прикрепляющиеся
путем надавливания клейкие вещества MOryT
располаrаться на отдельных участках оболочки,
или на всей оболочке или только по краю внеш
ней оболочки.
При аппликации на сухую, чистую и непо
врежденную кожу пластырь прочно прилипает
к коже путем леrкоro надавливания рукой или
пальцами и удаляется, не вызывая ощутимоro
повреждения кожи или вытеснения препарата из
внешней оболочки. Пластырь не должен вызы
вать раздражения или повышенной чувствитель
ности кожи даже после повторных аппликаций.
Защитный слой обычно изroтавливают из
пластика или фольrи. При еro удалении лекар
ственное средство (матрица или резервуар) или
клейкое вещество не должны отделяться от пла
стыря.
Трансдермальные пластыри обычно выпу
скают в отдельных запечатанных пакетиках.
ПРОИ3ВОДСТВО
При производстве, упаковке, хранении и pe
ализации трансдермальных пластырей предпри
нимают меры, обеспечивающие микробиолоrи
ческую чистоту в соответствии с требованиями
статьи 5.1.4. МикроБUОЛ02uческая чuстота He
стерuльных лекарственных средств u фарма
цевтических субстанцuй.
ИСПЫТАНИЯ
Однородность дозированных единиц.
Трансдермальные пластыри должны выдержи
вать испытание на однородность дозированных
единиц (2.9.40) или, если это обосновано и YT
верждено, испытание на однородность coдep
жания, как указано ниже. На лекарственное pac
тительное сырье и лекарственные средства из
неro, представленные в дозированной форме,
требования данноrо раздела не распространя
ются.
Однородность содержания (2.9.6). TpaHC
дермальные пластыри должны выдерживать
испытание на однородность содержания дей
ствующеro вещества в единице дозированно
ro лекарственноro средства (тест С), если нет
друrих указаний в частной статье.
J50.Зак 1712.
Растворение. ПРОВQДЯт испытание для
подтверждения соответствующеrо высвобо,!<
дения действующею вещества или веществ,
например, одним из способов, описанных в
статье 2.9.4. Тест «Растворение» для тpaHC
дермальных пластырей. В зависимости от
состава, размеров и формы пластыря при
меняют или метод сборною диска, или метод
ячеек, или метод вращающеюся цилиндра.
Может использоваться мембрана, изro
тавливаемая из таких материалов, как инерт
ная пористая целлюлоза или силиконы, она
не должна оказывать влияние на кинетику BЫ
свобождения действующеro вещества или Be
ществ из пластыря. Кроме тою, мембрана не
должна содержать веществ, которые MOryT
оказывать влияние на ее действие (например,
жир). Мембрана может соответствующим об
разом обрабатываться перед проведением ис
пытания, например, путем выдерживания ее
в среде для проведения испытания в течение
24 ч. Мембрану накладывают на выступающий
край пластыря, избеrая образования пузырь
ков воздуха.
Условия проведения испытания и требо
вания к нему должны быть указаны в частной
статье.
ХРАНЕНИЕ
Хранят при комнатной температуре, если
нет друrих указаний в частной статье.
МАРКИРОВКА
На этикетке указывают:
общее количество действующеro веще
ства или веществ в одном пластыре;
дозу, высвобождающуюся за единицу Bpe
мени;
площадь высвобождения действующеro
вещества или веществ.
01/2013:0652
УШНЫЕ ЛЕКАРСТВЕННЫЕ
СРЕДСТВА
Au,icula,ia
ОПРЕДЕЛЕНИЕ
Ушные лекарственные средства это
жидкие, мяrкие или твердые лекарственные
средства, предназначенные для закапывания,
распыления, вдувания или аппликации в слухо-
вое отверстие или для промывания уха.
Ушные лекарственные средства обычно co
держат одно или более действующих веществ
в подходящем носителе. Они MOryт содержать
1190
rосударственная фармакопея Республики Беларусь
вспомоrательные вещества, например, для обе
спечения необходимоrо осмотическоro давления
или вязкости, создания или стабилизации необ
ходимоro значения рН, увеличения раствори
мости действующих веществ, обеспечения CTa
бильности или соответствующих антимикробных
свойств. Вспомоrательные вещества в использу
емых концентрациях не должны отрицательно
влиять на действие лекарственноro средства, не
должны оказывать токсичноro или нежелатель
ноro местноro раздражающеro действия.
Лекарственные средства, применяемые
при повреждениях уха, особенно при перфора
ции барабанной перепонки, или перед хирурrи
ческой операцией, должны быть стерильными,
и, если иное не обосновано и не утверждено, не
должны содержать антимикробных KOHcepBaH
тов и должны выпускаться в однодозовых KOH
тейнерах.
Ушные лекарственные средства выпускают
как в мноroдозовых, так и в однодозовых контей
нерах, которые снабжены, при необходимости,
приспособлением, которое обеспечивает удоб
ство применения и предотвращает заrрязнение.
Если иное не обосновано и не утверждено,
водные ушные лекарственные средства выпу
скают в мноroдозовых контейнерах, которые co
держат подходящий антимикробный консервант
в необходимой концентрации, за исключением
лекарственных средств, обладающих достаточ
ным антимикробным действием.
rде это применимо, контейнеры для ушных
лекарственных средств должны соответствовать
требованиям статей разделов 3. 1. Матерuалы,
uспользуемые для производства контейнеров
и 3.2. Контейнеры.
Ушные лекарственные средства можно
классифицировать как:
ушные капли, спреи;
ушные мяrкие лекарственные средства;
ушные порошки;
ушные промывки;
ушные тампоны.
ПРОИЗВОДСТВО
При разработке ушных лекарственных
средств, в состав которых входят антимикроб
ные консерванты, уполномоченному opraHY
представляют данные, подтверждающие He
обходимость использования и эффективность
выбранных консервантов. Подходящий метод
определения и критерии оценки эффективности
консервантов представлены в разделе 5.1.3. Эф
фективность антимuкробных консервантов.
При разработке ушных промывок должно
быть доказано, что из однодозовоro контей
нера, содержащеro лекарственное средство,
может быть извлечено номинальное еro coдep
жимое. #В случае отсутствия у производителя
лекарственноro средства сертификата COOТBeT
ствия GMP, а также данных о валидации про
цесса производства конкретноro ушноro лекар
ственноro средства испытание по определению
извлекаемоro объема должно быть включено в
спецификацию roтовоro продукта.#
При производстве, упаковке, хранении и pe
ализации ушных лекарственных средств при
нимают меры, обеспечивающие микробиолоrи
ческую чистоту в соответствии с требованиями
статьи 5.1.4. МuкроБUОЛ02uческая чuстота He
стерильных лекарственных средств u фарма
цевтических субстанций.
Стерильные ушные лекарственные cpeд
ства производят с использованием матери
алов и методов, обеспечивающих стерильность,
предотвращающих заrрязнение лекарственных
средств и развитие микроорrанизмов в соответ
ствии с требованиями статьи 5.1. 1. Методы при
20товленuя стерильных продуктов.
При производстве ушных лекарственных
средств, содержащих дисперrированные части
цы, принимают меры, обеспечивающие необхо
димый размер частиц и их контроль.
ИСПЫТАНИЯ
Однородность дозированных единиц.
Однодозовые ушные лекарственные средства
должны выдерживать испытание на OДHOpOД
ность дозированных единиц (2.9.40) или, если это
обосновано и утверждено, испытания на OДHO
родность содержания иlили однородность массы,
как указано ниже. На лекарственное раститель
ное сырье и лекарственные средства из неro,
представленные в дозированной форме, требо
вания данноro раздела не распространяются.
Однородность содержания (2.9.6). Ушные
лекарственные средства в однодозовых контей
нерах с содержанием действующеro вещества
менее 2 Mr или менее 2 % от общей массы co
держимоro должны выдерживать испытание на
однородность содержания действующеro веще
ства в единице дозированноro лекарственно
ro средства (тест В), если нет друrих указаний
в частной статье. Если лекарственное средство
содержит более одноro действующеro веще
ства, это требование распространяется только
на те вещества, содержание которых COOTBeT
ствует вышеуказанным условиям.
Однородность массы (2.9.5). Ушные ле
карственные средства в однодозовых контей
нерах должны выдерживать испытание на oд
нородность массы для единицы дозированноro
лекарственноro средства. Испытание на OДHO
родность массы не требуется, если испытание
на однородность содержания предусмотрено
для всех действующих веществ.
Стерильность (2.6.1). Если на этикетке YKa
зано, что лекарственное средство стерильно,
оно должно выдерживать испытание на стериль
ность.
Ушные лекарственные средства
1191
ХРАНЕНИЕ
Если лекарственное средство стериль
но, хранят в стерильных воздухонепроница
емых контейнерах с контролем первоro BCKpЫ
тия, если нет друrих указаний в частной статье.
МАРКИРОВКА
На этикетке указывают:
названия всех добавленных антимикроб
ных консервантов;
«стерильно», если необходимо;
для лекарственных средств в MHOro
дозовых контейнерах: срок хранения после
вскрытия. Если иное не обосновано и не YT
верждено, этот срок не должен превышать
4 недели.
Ушные капли и спреи
ОПРЕДЕЛЕНИЕ
Ушные капли и сп реи растворы, эмуль
сии или суспензии, содержащие одно или
более действующих веществ в подходя
щих жидкостях (например, вода, rликоли или
жирные масла) и предназначенные для BBeдe
ния в слуховое отверстие без оказания опас
ноro давления на барабанную перепонку. Они
также MOryт быть введены в слуховое OTBep
стие посредством тампона, пропитанноro ле
карственным средством.
Ушные эмульсии MOryT расслаиваться, но
при взбалтывании леrко восстанавливаются.
Ушные суспензии Moryт образовывать осадок,
который должен быстро ресуспендироваться
при взбалтывании, образуя суспензию, ДOCTa
точно стабильную, чтобы обеспечить необхо
димую дозу введения.
Ушные капли обычно выпускают в MHoro
дозовых контейнерах из стекла или подходя
щеrо пластиковоro материала, снабженных
встроенной капельницей или закручивающей
ся крышкой из подходящеro материала с Ka
пельницей и резиновым или пластмассовым
соском. Комплект такой крышки может при
лаrаться отдельно. Спреи обычно выпускают
ся в мноrодозовых контейнерах, снабженных
подходящим аппликатором. Если спреи BЫ
пускаются в контейнерах под давлением, то
они должны выдерживать требования статьи
Лекарственные средства, находящuеся под
давлением.
Ушные мяrкие лекарственные средства
ОПРЕДЕЛЕНИЕ
Ушные мяrкие пекарственные средства
предназначены для введения в наружное слу
ховое отверстие. Если необходимо, закладыва
ют или вводят тампон, пропитанный лекарствен
ным средством.
Ушные мяrкие лекарственные средства
должны соответствовать требованиям статьи
МЯ2кuе лекарственные средства для наружно
20 прuмененuя.
Их выпускают в контейнерах, снабженных
подходящим аппликатором.
Ушные порошки
ОПРЕДЕЛЕНИЕ
Ушные порошки предназначены для ап
пликации или инсуффляции (вдувания) во BHY
тренний слуховой проход. Они должны COOT
ветствовать требованиям статьи ПороU1КU для
наРУЖН020 применения.
Их выпускают в контейнерах, снабженных
подходящим устройством для аппликации или
вдувания.
Ушные промывки
ОПРЕДЕЛЕНИЕ
Ушные промывки лекарственные cpeд
ства, предназначенные для очистки слухово
ro отверстия. Они обычно представляют собой
водные растворы со значением рН, COOTBeTCTBY
ющим физиолоrическим пределам.
Ушные промывки, при меняемые при по
вреждениях уха или перед хирурrическими опе
рациями, должны быть стерильными.
Ушные тампоны
ОПРЕДЕЛЕНИЕ
Ушные тампоны предназначены для BBeдe
ния в наружное слуховое отверстие. Они должны
соответствовать требованиям статьи Тампоны
медицинские.
rомеопатические лекарственные средства
1193
rОМЕОПАТИЧЕСКИЕ
ЛЕКАРСТВЕННЫЕ
СРЕДСТВА
ВВЕДЕНИЕ
Для характеристики 20меопатuческuх
лекарственных средств М02ут uспользо
ваться все общие статьи rосударственной
фармакопеu Республuкu Беларусь, примени
мые к 20меопатиu.
tлава «tомеопатическuе лекарственные
средства» содержuт общие статьи, опuсы
вающuе исходные вещества U лекарственные
средства, uспользуемые uсключuтельно в 20
меопатuческой медицине. Ссылкu на част
ные фармакопейные статьи для дРУ2их целей
должны быть подтверждены компетентным
уполномоченным Ор2аном.
01/2013:1038
rОМЕОПАТИЧЕСКИЕ
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
P,aepa,atioпes homoeopathicae
ОПРЕДЕЛЕНИЕ
rомеопатические лекарственные средства
roтовят из фармацевтических субстанций, про
дуктов или лекарственных средств, называ
емых базисными препаратами, в соответствии
с обычной производственной rомеопатической
практикой. rомеопатические лекарственные
средства обычно обозначаются латинским Ha
званием базисноro препарата с дальнейшим
указанием степени разведения.
Сырье
Сырье для производства roмеопатических
лекарственных средств может быть природноrо
#(минеральноro, растительноro, животноro, че
ловеческоro)# или синтетическоro происхожде
ния #(MOryт использоваться и аллопатические
лекарственные средства)#.
К сырью животноro или человеческоro
происхождения должны предъявляться COOT
ветствующие требования, сводящие к мини
муму риск инфицирования, включая вирусно
ro (5.1.7), roмеопатических лекарственных
средств. Для этоro следует продемостриро
вать, что:
методы производства включают стадию
или стадии, которые удаляют либо инактивиру
ют факторы инфицирования;
\5\ Зак 1712
в некоторых случаях сырье животноro про
исхождения должно соответствовать требова
ниям статьи Продукты, которые М02ут быть
переносчuками 2убчатой энцефалопатuu ЖU
вотН020 проuсхожденuя;
в некоторых случаях животные или их
ткани, используемые для получения сырья,
должны выдерживать требования компетентно
ю уполномоченною opraHa к здоровью живот
ных, используемых для употребления челове
ком;
в ряде материалов человеческоro проис
хождения донор должен соответствовать peKO
мендациям, предъявляемым к донорам крови
человека и донорской крови (см. частную статью
Плазма человеческая для фракцuонuрования),
если иное не обосновано и не утверждено.
Сырье растительноro, животною и челове
ческоrо происхождения может использоваться
в свежем или высушенном виде. В некоторых
случаях допускается хранить свежий матери
ал в замороженном виде. Сырье растительноro
происхождения должно соответствовать требо
ваниям статьи Лекарственное растuтельное
сырье для 20меопатuческих лекарственных
средств.
Если обосновано и утверждено, в целях
транспортирования или хранения свежее расти
тельное сырье может храниться в 96 % спирте
или этаноле подходящей концентрации при yc
ловии, что для дальнейшей переработки будет
использован весь материал, включая среду для
хранения.
Сырье должно отвечать требованиям соот
ветствующих частных фармакопейных статей.
Разбавители
Разбавители это вспомоraтельные веще
ства, используемые для приroтовления первичных
базисных препаратов или потенций. Разбавителя
ми MOryт быть: вода очищенная, спирт необходи
мой концентрации, rлицерин или лактоза.
Разбавители должны отвечать требовани
ям соответствующих частных фармакопейных
статей.
#Вспомоrательные вещества
В качестве вспомоrательных веществ ис
пользуют разбавители, указанные в разде
ле «Разбавители», растительные масла, Ba
зелин, ланолин, масло какао, кальциевый
бентонит, твердый жир, мед, маrния стеарат,
натрия хлорид, крахмал и друrие.
Вспомоrательные вещества должны OTBe
чать требованиям соотвеТСТВУЮЩIIIХ частных
фармакопейных статей.
Базисные препараты
Базисные препараты (stocks) это фар
мацевтические субстанции, продукты или ле
карственные средства, используемые в каче
стве исходною материала для производства
1194
rосударственная фармакопея Республики Беларусь
roмеопатических лекарственных средств. Ба
зисные препараты обычно представляют собой:
для сырья растительноro, животноro'и человече
скоro происхождения матричную настойку или
rлицериновый мацерат; для сырья химическоro
или минеральноro происхождения непосред
ственно само вещество.
Матричные настойки должны выдерживать
требования статьи Матричные настойки для
20меопатических лекарственных средств.
rлицериновые мацераты #(вымачивания)#
это жидкие лекарственные средства, получен
ные из сырья растительною, животноro и че
ловеческоrо происхождения с использованием
rлицеРИl;lа или смеси rлицерина со. спиртом He
обходимой концентрации или раствором натрия
хлорида необходимой концентрации.
Потенцирование
Разведения и тритурации получают из ба
зисноro препарата при помощи процесса потен
цирования в соответствии с roмеопатической
производственной практикой: это означает по
следовательное разведение и встряхивание,
или последующие соответствующие тритурации,
или объединение этих двух процессов.
Обычно используют следующие степени по
тенцирования:
1 часть базисноro препарата плюс 9 частей
растворителя; обозначают «ак «О», «ОН» или
«Х» (десятичное разведение);
1 часть базисноro препарата плюс 99
частей растворителя; обозначают как «С» или
«СН» (сотенное разведение).
Число степеней потенций определяет меру
разведения, например, «DЗ», «ЗDН» или «ЗХ»
обозначает третью десятичную потенцию, а
«СЗ», «ЗСН» или «ЗС» третью сотенную по
тенцию "(таблица #10З8. 0)".
«LM » (или «a ))) потенцию rотовят в COOT
ветствии со специфическими процедурами "(пя
тидесятитысячное разведение '1/50 000 с самой
высокой степенью потенцирования LМЗО)".
"Низкими разведениями 8 roмеonатической
практике считаются разведения от исходных
субстанций или тинктур до С6 разведения, cpeд
ними от С6 до СЗО, высокими более СЗО
разведения."
ЛекарствеНН I форМ,ы
Лекарственная форма roмеопатическоrо
лекарственноro средства должна выдерживать
требования статьи Фармакопеи на COOTBeTCTBY
ющую лекарственную форму со следующими
особенностями:
«активными субстанциями» лекарствен
ных форм для roмеопатическоro применения
являются разведения или тритурации roмеопа
тических базисных препаратов;
эти лекарственные формы rотовят с ис
пользованием соответствующих вспомоrатель
ных веществ;
испытание на однородность содержа
ния (2.9.6) или на однородность дозированных
форм (2.9.40) обычно не проводят; однако в
некоторых случаях эти показатели MOryT быть
введены в специфи«ацию по требованию KOM
петентноro уполномоченною opraHa.
rомеопатuческая лекарственная форма
«Пилюли»
Пилюли для roмеопатическоro примене
ния это твердые лекарственные формы, по
лученные с использованием сахарозы, лактозы
или друrих необходимых вспомоrательных Be
ществ. Они MOryт быть получены смачиванием
предварительно сформированных пилюль, раз
ведением/разведениями roмеопатических ба
зисных препаратов или путем постепенноro co
единения указанных вспомоrательных веществ
и разведения/разведений rомеопатических ба
зисных препаратов. Они пРедназначены для
оральною или сублинrвальноrо применения.
rомеопатическая лекарственная форма
« Таблетки»
Таблетки для юмеопатическоro примене
ния это твердые лекарственные формы, по
лученные с использованием сахарозы, ЩIКТО
зы или друrих неоБХОДIiIМЫХ вспомоraтельных
веществ в соотвеТСТВIiIИ с общей статьей Ta
блетки. Они MOryт быть получены прессова
нием одноro или нескольких твердых активных
Обозначения еомеопатических разведении
Таблица #10З8. 0
Буквенно-цифровые Математические (1110) Математические(1 о"')
десятичные сотенные десятичные сотенные десятичные сотенные
1Х, О1 ,1ОН 1/10 , 10-1
2Х, О2, ЮН С1, 1СН, 1С 1/100 1/100 10-2 10-1
ЗХ, DЗ, ЗDН 1/1000 1(t3
4Х, О4, 4ОН С2, 2СН, 2С 1/10000 1/10000 1(Т4 10-2
5Х, О5, 5ОН 1/100 000 10.5
6Х, Dб, 6ОН 'СЗ, зсн, ЗС 1/1000000 1/1 000000 10-6 10-3
7Х, Р7, 7оН 1/10000000 1(t7
8Х, О8, 8ОН C4.4GH, 4С 1/10ОООО ООО 1/100000000 1О-В 10'"
rомеопатическuе лекарственные средства
1195
субстанций со вспомоrательными веществами
или смачиванием предварительно сформиро
ванных таблеток разведениемlразведениями
rомеопатических базисных препаратов. Пред
варительно сформированные для смачивания
таблетки получают из сахарозы, лактозы или
друrих необходимых вспомоrательных веществ
в соответствии с общей статьей Таблеткu. Они
предназначены для оральноro или сублинr
вальноro применения.
#/омеопатuческая лекарственная
форма «Настои U отвары»
Настои и отвары rомеопатические это
водные извлечения из лекарственноrо расти
тельноro сырья, полученные в соответствии
со статьей «#Настои, отвары u чаu». Их при
меняют для изrотовления roмеопатических
лекарственных средств в различных лекар
ственных формах. Для изrотовления настоев
и отваров при меняют свежее или высушенное
лекарственное растительное сырье, разре
шенное в roмеопатии и отвечающее требова
ниям фармакопейной статьи. В roмеопати
ческие водные извлечения и их разведения
консерванты, как правило, не добавляют.
#/омеопатuческая лекарственная
форма «Растворы u разведенuя (потенцuu)>>
Растворы и разведения (потенции) roMe
опатические это жидкие лекарственные
формы для изrотовления roмеопатических
тритураций, rранул и друrих лекарственных
форм. MorYT использоваться и как лекар
ственные средства для внутреннеro и наруж
ноro применения.
Разведения получают:
разбавлением и потенцированием roMe
опатических растворов;
из тритураций, потенцируя их в COOTBeT
ствующем разбавителе;
разбавлением и потенцированием Ma
тричных настоек.
Растворы и разведения (потенции) rOMeo
патические представляют собой жидкую OДHO
родную систему в соответствующем paCTBO
рителе (вода очищенная, вода для инъекций,
спирт этиловый различной концентрации,
rлицерин).
Если нет указаний в соответствующей
частной фармакопейной статье, растворы не
следует HarpeBaTb.
Процесс изroтовления разведений за
ключается в последовательном разведе
нии в десятичной или сотенной пропорции в
водно спиртовом растворе исходной субстан
ции (тинктуры, раствора) со встряхиванием
(динаминизацией, потенцированием) после
каждоrо разведения. Разведение может про
должаться до уровня, Korдa все исходное Be
щество устраняется из среды, но при этом ле
карственная активность отчетливо выражена.
#/омеопатuческuе лекарственные формы
«Растворы для uнъекций. Каплu 2лазные. Каплu
для носа»
Растворы для инъекций, капли rлазные и
капли для носа roмеопатические изroтавливают
из свежеприroтовленных разведений с использо
ванием воды для инъекций (растворы для инъ
екций), воды очищенной стерильной (капли rлаз
ные) и воды очищенной (капли для носа).
В качестве разбавителей применяют также
0,9 % раствор натрuя хлорuда Р или буферные
растворы, указанные в частных фармакопейных
статьях.
При изroтовлении разведен ий в малых коли
чества (менее 3 r) исходные растворы и препа
раты дозируют каплями, используя стандартные
каплемеры или эмпирический каплемер пипет
ку, откалиброванную по массе для данноro пре
парата.
Растворы для инъекций и rлазные капли
изroтавливают в асептических условиях, и они
должны быть стерильными, а капли назальные
должны выдерживать требования статьи 5.1.4.
МuкробиОЛ02ическая чuстота нестерuльных
лекарственных средств u фармацевтических
субстанциЙ
#/омеопатuческая лекарственная форма
«Тритурации 20меопатuческие»
Тритурации roмеопатические это TBep
дая дозированная лекарственная форма в виде
порошка, состоящая из смеси порошков лекар
ственных веществ, жидких препаратов (настоек,
эссенций, растворов) или их разведен ий со вспо
моrательным веществом (лактозой (молочным
сахаром) или друrим индифферентным веще
ством, разрешенным уполномоченным opraHoM).
Тритурации MOryт быть использованы как
roтовая лекарственная форма для внутреннеro
применения, а также для изroтовления друrих ro
меопатических лекарственных форм (обычно из
rотовление тритураций заканчивают С3, последу
ющие разведения roтовят как жидкие).
Концентрацию лекарственноro вещества в
тритурации выражают соотношением 1: 1 О или
1:100.
Порошкообразные субстанции для фарма
цевтическоro использования (в том числе Me
таллы) и лактоза должны быть предварительно
измельчены до порошка с размером частиц не
более 63 мкм, если нет друrих указаний в част
ных фармакопейных статьях. Степень измельче
ния (дисперсность) исходных веществ оценива
ют ситовым анализом (2.9.12) или определяют
размер частиц методом МИI{РОСКОПИИ (2.9.37) или
по величине их удельной площади поверхности
(2.9.14).
При изroтовлении тритураций вручную ис
пользуют фарфоровые ступки (применение Me
таллических ступок не допускается).
rиrроскопичные лекарственные вещества
следует растирать только в подоrретых ступках.
1196
rосударственная фармакопея Республики Беларусь
#rомеопатическая лекарственная
форма «tранулы»
rранулы roмеопатические это твеРАая
дозированная лекарственная форма для BHY
треннеro применения (пилюли, крупинки, rло
були). rомеопатические тритурации и rpa
нулы обычно применяют сублинrвально для
обеспечения быстроro всасывания через сли
зистую оболочку полости рта.
Изroтавливают их путем нанесения
жидких rомеопатических разведений лекар
ственных веществ или их смесей на исходные
rранулы вспомоrательных веществ (caxapo
зы, лактозы и др.), разрешенных к медицин
скому применению.
Получают юмеопатические rранулы,
нанося на rранулы сахара жидкие препараты,
растворы, лекарственные вещества или их
смеси. Количество лекарственноro вещества,
нанесенною на исходные rранулы сахара, cy
щественно не влияет на их размер и друrие
физико химические показатели, поэтому ro
меопатические rранулы по размеру, среднему
диаметру оценивают по исходным rранулам.
#tомеопатическая лекарственная
форма «Мазш>
Мази это мяrкая лекарственная форма
для наружною применения, которая COCTO
ит из основы С равномерно распределенны
ми юмеопатическими лекарственными cpeд
ствами.
В качестве основ для мазей используют
rлавным образом вазелин и ланолин. Для из
rотовления мазей, содержащих более 1 О %
настоек, применяют основу: 85 % вазелина и
5 % ланолина. В rлазных мазях абсорбцион
ная основа вазелин и ланолин безводный в
соотношении 9:1. На мазь определенноro co
става основа должна быть указана в частной
фармакопейной статье.
Из несильнодействующих веществ и тин
ктур изrотавливают 1 О % мази. Концентрация
ядовитых и сильнодействующих веществ в
составах юмеопатических мазей обязатель
но должна быть указана. Если концентрация
сильнодействующих веществ не указана, то
изютавливают мази 5 % концентрации.
При изroтовлении мазей, содержащих
настойки на вазелиновой основе в KOHцeH
трации более 5 % от массы мази, их предва
рительно концентрируют, выпаривая до по
ловины взятой массы, или добавляют от 5 %
д01 О % безводною ланолина.
В юмеопатические мази обычно не вводят
стабилизаторы, антипксиданты и KOHcepBaH
ты. Последние добавляют только Korдa в Ka
честве основы используют rели, содержащие
воду или эмульсионные основы типа «масло/
вода» .
Мази юмеопатические rлазные должны
быть стерильными.
#tомеопатическая лекарственная форма
на основе масла
rомеопатическая лекарственная форма
на основе масла это жидкая лекарственная
форма для наружноro применения, состоящая
из roмеопатическоro средства, растительных
масел (персиковоro, абрикосовоro, миндально
ro, оливковою, подсолнечноro, разрешенных к
медицинскому применению уполномоченным
opraHoM и указанных в частных фармакопей
ных статьях) или минеральноro (вазелиновоro)
масла.
Получают масла или экстракцией (Maцe
рацией), или растворением лекарственных
средств в соответствующих маслах в соотноше
ниях 1:10 или 1:20.
#tомеопатuческая лекарственная форма
«Жuдкий оподельдок»
Жидкий оподельдок это мяrкая лекар
ственная форма для наружноro применения
roмоrенная дисперсная система смесь roмео
патических средств с основой в соотношении по
массе, как правило, 1: 10. Оподельдок представ
ляет собой этанольный линимент, основа KOTO
роro состоит из 2 x массовых частей мыльно
ro спирта (раствора калийноro (зеленоro) мыла
в спирте этиловом: мыла калийноro 63,953 r,
спирта этuловоzо Р (86 %, об/об) 27,836 r,
воды Р 8,115 r, масла эфирноro лавандово
ro 0,096 r), 1 массовой части воды Р и 1 части
спирта этиловоro (93,9 %, об/об).
#tомеопатuческая лекарственная форма
«Суппозиториu»
Суппозитории roмеопатические это TBep
дые при комнатной температуре и расплавля
ющиеся при температуре тела дозированные
лекарственные формы, применяемые для BBe
дения в полости тела. Они состоят из roмеопа
тических лекарственных средств, равномерно
распределенных в основе (масло какао, TBep
дый жир типов «А» и «В» и друrие), разрешен
ной уполномоченным opraHoM.
Суппозитории изroтавливают в соответствии
с общими правилам и соrласно статье Суппозu
тории.
Как правило, в суппозитории не вводят CTa
билизаторы. Тинктуры (настойки матричные),
растворы и тритурации в соответствующих раз
ведениях (потенциях) вводят в суппозиторные
массы в соотношении 1:10. Жидкие экстракты
и тинктуры предварительно концентрируют BЫ
париванием. Термолабильные лекарственные
вещества в случае изroтовления способом BЫ
ливания в формы добавляют к основе непосред
ственно перед формованием суппозиториев.
Методы проuзводства
rомеопатические лекарственные средства
изютавливают с применением ряда методов
приroтовления и в различных лекарственных
Методы при20товления 20меопатических базиСНblХ препаратов
1197
формах (как описано в общих статьях на лекар
ственные формы). Методы изroтовления опи
саны в общей статье Методы прuzотовления
zомеопатическuх базuсных препаратов u по
тенцирование. Использование определенных
лекарственных средств, полученных с использо
ванием методов, перечисленных ниже, оrрани
чено определенными лекарственными форма
ми, приведенными в таблице 1038. 1.
Компетентный уполномоченный .opraH имеет
право принять либо отклонить конкретные KOM
бинации метода производства и субстанции.
#Контроль качества
rомеопатические лекарственные средства
отличаются от соответствующих лекарственных
форм, описанных в Фармакопее, условиями,
предъявляемыми к контролю качества лекар
ственных средств в зависимости от содержания
в них активных веществ. Рекомендуется:
к лекарственным средствам, содержащим
активные вещества нижеразведенийС2, предъяв
лять те же требования, что и к лекарственным
средствам, описанным в Фармакопее;
лекарственные средства, содержащие aK
тивные вещества в разведен иях от С2 до С3,
контролировать после проведения специальных
приемов концентрирования (выпаривание, сжи
rание, сплавление веществ, переведение их в
нелетучий В'-ид) одним из подходящих методов,
исходя из ero приroдности;
лекарственные средства, содержащие aK
тивные вещества в разведениях от С4 до С6,
контролировать в пробе, соответствующей про
писанной дозе, в частном случае в пробе, co
ответствующей дозе, прописанной на курс лече
ния;
для лекарственных веществ, содержащих
активные вещества в разведениях выше С6, Ka
чество обеспечивать соблюдением технолоrиче
скоro процесса.
01/2013:2371
МЕтодыприrОТОВЛЕНИЯ
rОМЕОПАТИЧЕСКИХ БАЗИСНЫХ
ПРЕПАРАТОВ И ПОТЕНЦИРОВАНИЕ
Via рrаера,апdi sti,pes homoeopathicas et
роtепtifiсапdi
rомеопатические исходные компоненты
roтовят подходящими методами из исходноro
сырья, которое должно соответствовать требо
ваниям общей статьи tомеопатическuе лекар
ственные средства. Методы, описанные ниже
и совмещенные с широко применяющимися Me
тодами для потенцирования, приведены в Ka
честве при мера; друrие методы, описанные
в официальных национальных фармакопеях
друrих стран, MOryT быть использованы как paB
нозначные.
При использовании сырья животноro про
исхождения обязательно учитывают требо
вания, установленные в общей статье 'OMe
опатuческuе лекарственные средства на
использование исходных материалов живот
Horo происхождения или от человека. При
приrотовлении жидких разведений использу
ют эта нол в концентрации, указанной в MeTO
дике, при необходимости, еro можно заменить
на эта нол (36 %, об/об) [эта нол (30 %. м/м)]
или этанол (18 %, об/об) [эта нол (15 %, м/м)].
Korдa в частной статье указано, что матричная
настойка приютовлена из более чем из одною pac
тения, ее rотовят из указанных частей и COOTBeT
ствующих видов растений или из любой их смеси.
Если иное не указано, матричную настойку
roтовят путем М8церации. Продолжительность
мацерации должна составлять не менее 1 О дней
и не более 30 дней.
Можно проводить более длительную Ma
церацию (не .более 60 дней) или очень дли
Таблица 1ОЗ8. 1
Методыпроизводства Лекарственная форма
2.1.2 rлазные капли
Растворы для инъекций
Назальные лекарственные средства
2.2.1, 2.2.2, 2.2.3 rлазные капли
Пилюли (globuli ve/at/)
Растворы для инъекций
Назальные лекарственные средства
Мази, крема, rели
Порошки для приема внутрь (тритурации)
Суппозитории
2.2.4 Растворы для инъекций
3.1.2, 3.2.2 rлазные капли
Пилюли (g/obuli velati)
Растворы для инъекций
Назальные лекарственные средства
Мази, крема, rели
Суппозитории
152 За" 1712
1198
rосударственная фармакопея Республики Беларусь
тельную мацерацию (не более 180 дней) при
условии подтверждения, что качество образу
ющейся матричной настойки сопоставимо с Ka
чеством продукта, получаемом при CTaHдapT
ной мацерации.
Если иное не указано, термином «часть
(части)>> обозначают «массовую часть (Macco
вые части)>>. Если в методике не указано иначе,
то при приroтовлении максимальная температу
ра должна составлять 25 ОС.
1. МАТРИЧНЫЕ НАСТОЙКИ
МЕТОД 1.1.
МЕТОД 1.1.1 (СООТВЕТСТВУЕТ номБОРАТНI
SCHES ARZNEIBUCH (НАВ) 1а: МАТРИЧНЫЕ HA
СТОЙКИ И ЖИДКИЕ РАЗВЕДЕНИЯ)
Метод 1.1.1 используют для свежеro лекар
ственноro растительноro сырья, содержащеro
обычно более 70 % выжатоro сока, в котором OT
сутствует эфирное масло или смола, или слизь.
Матричные настойки, приrотовленные в COOT
ветствии с методом 1.1.1, представляют собой
смеси равных частей выжатоro сока и этанола
(90 %, об/об) [этанола (86 %, м/м)].
Отжимают измельченное лекарственное
растительное сырье. Немедленно смешивают
выжатый сок с равной массой этанола (90 %,
об/об) [этанола (86 %. м/м)]. Выдерживают в за
крытой емкости при температуре, не превыша
ющей 20 ос, в течение не менее 5 дней, затем
фильтруют.
Приведение в соответствие со значени-
ем, указанным в частной статье
Определяют процентное содержание сухоro
остатка (2.8.16) или. если указано, процентное
количественное содержание описанноro выше
фильтрата. Рассчитывают количество (А 1 ), в ки
лоrраммах, требуемоro этанола (50 %, об/об)
[этанола (43 %, м/м)] по следующей формуле:
m (N No) ,
N o
rдe:
т масса фильтрата, Kr;
N o содержание cyxoro остатка или коли
чественное содержание, указанное в частной
статье, %;
N содержание сухоro остатка или количе
CTBeH oe содержание фильтрата, %.
Смешивают фильтрат с рассчитанным коли
чеСТВОМЭТ8нола (50%, об/об)[этанола (43 %, м/м)].
Выдерживают при температуре не выше 20 ос в
течение не менее 5 дней, затем при необходимо
сти фильтруют.
Потенцирование
Первое десятичное разведение (01) roтовят
следующим образом:
2 части матричной настойки;
8 частей этанола (50 %, об/об) [этанола
(43 %, м/м)].
Второе десятичное разведение (02) roтовят
следующим образом:
1 часть первоrо десятичноrо разведе
ния;
9 частей этанола (50 %, об/об) [эта нола
(43 %, м/м)].
Последующие десятичные разведения roто
вят, как указано для 02.
Первое сотенное разведение (С1) roтовят
следующим образом:
2 части матричной настойки;
98 частей этанола (50 %, об/об) [этано
ла (43 %, м/м)].
Второе сотенное разведение (С2) roтовят
следующим образом:
1 часть первоro сотенноro разведения;
99 частей этанола (50 %, об/об) [этанол
(43 %, м/м)].
Последующие сотенные разведения roто
вят, как указано для С2.
МЕТОД 1.1.2 (СООТВЕТСТВУЕТ НАВ 1 Ь: MA
ТРИЧНЫЕ НАСТОЙКИ И ЖИДКИЕ РАЗВЕДЕНИЯ)
Метод 1.1.2 используют, Korдa в процессе
производства используется лекарственное pac
тительное сырье, содержащее латекс.
Матричные настойки, приroтовленные в co
ответствии с методом 1.1.2, представляют собой
смесь свежеro растительноro латекса с этано
лом (36 %, об/об) [этанолом (30 %, м/м)]. Свежий
латекс смешивают с 2 массовыми частями эта
нола (36 %, об/об) [этанола (ЗО %, м/м)] и филь
труют.
Приведение в соответствие со значени-
ем, указанным в частной статье
Определяют процентное содержание сухоro
остатка (2.8.16) или, если указано, процентное
количественное содержание описанноro выше
фильтрата. Рассчитывают количество (А,), в ки
лоrраммах, требуемоro этанола (36 %, об/об)
[этанола (30 %, м/м)] по следующей формуле:
m. ( N N )
х о ,
N o
rдe:
т масса фильтрата, Kr;
N o содержание сухоro остатка или коли
чественное содержание, указанное в частной
статье, %;
N x содержание сухоro остатка или количе
ственное содержание фильтрата, %.
Смешивают фильтрат с рассчитанным коли
чеством эта нола (36 %, об/об) [эта нола (30 %, м/м)].
Выдерживают при температуре не выше 20 ос в
течение не менее 5 дней, затем при необходимо
сти фильтруют.
Методы приzотовления zомеопатических базисных препаратов
1199
Потенцирование
Первое десятичное разведение (01) roтовят
следующим образом:
3 части матричной настойки;
7 частей этанола (36 %, об/об) [эта нола
(30 %, м/м)].
Второе десятичное разведение (02) roтовят
следующим образом:
1 часть первоrо десятичноrо разведе
ния;
9 частей этанола (18 %, об/об) [эта нола
(15 %, м/м)].
Последующие десятичные разведения roто
вят, как указано для 02.
МЕТОД 1.1.3 (СООТВЕТСТВУЕТ НАВ 2а: MA
ТРИЧНЫЕ НАСТОЙКИ И ЖИДКИЕ РАЗВЕДЕНИЯ)
Метод 1.1.3 используют для свежеro лекар
ственноro растительноro сырья, содержащеro
обычно менее 70 % выжатоro сока и имеюще
ro влажность (потерю в массе при высушива
нии) более 60 %, в котором отсутствует эфирное
масло или смола.
Матричные настойки, приroтовленные в co
ответствии с методом 1.1.3 (содержание эта нола
приблизительно 50 % об/об или 43 % м/м), полу
чают мацерацией, как указано ниже.
Лекарственное растительное сырье измель
чают. Отбирают пробу и определяют потерю
в массе при высушивании (2.2.32). Если иное
не указано, потерю в массе при высушивании
определяют с использованием 2,00 5,00 r из
мельченноrо исходноro сырья, которое поме
щают в плоскодонный предварительно BЫCY
шенный (как указано для сырья) и взвешенный
сосуд диаметром 45 55 мм. Сушат при темпе
ратуре 105 ос в течение 2 ч и охлаждают в экс
икаторе.
К измельченному лекарственному расти
тельному сырью немедленно прибавляют не
менее половины массы этанола (90 %, об/об)
[этанола (86 %, м/м)] и хранят в плотно укупо
ренных контейнерах при температуре не выше
20 ОС.
По следующей формуле рассчитывают KO
личество (А 2 ), в килоrраммах, этанола (90 %, об/
об) [этанола (86 %, м/м)], требуемоro для массы
(т) исходноrо сырья; затем вычитают количество
уже добавленноro эта нола (90 %, об/об) [этанола
(86 %, м/м)] и прибавляют разницу к смеси.
т.Т
,
100
rдe:
т масса исходноrо сырья, Kr;
т потеря в массе при высушивании, %.
Выдерживают при температуре не выше
20 ос в течение не менее 1 О дней при периоди
ческом взбалтывании, отжимают смесь и полу
ченную жидкость фильтруют.
Приведение в соответствие со значе-
нием, указанным в частной статье
Определяют процентное содержание сухоro
остатка (2.8.16) или, если указано, процентное
количественное содержание описанноro выше
фильтрата. Рассчитывают количество (А,), в ки
лоrраммах, требуемоro этанола (50 %, об/об)
[этанола (43 %, м/м)] по следующей формуле:
т. ( N N ) .
х о ,
N o
rдe:
т масса фильтрата, Kr;
N o содержание сухоro остатка или коли
чественное содержание, указанное в частной
статье, %;
N содержание сухоro остатка или количе
CTBeH oe содержание фильтрата, %.
Смешивают фильтрат с рассчитанным KO
личеством этанола (50 %. об/об) [этанола (43 %,
м/м)]. Выдерживают при температуре не выше
20 ос в течение не менее 5 дней, затем при He
обходимости фильтруют.
Потенцирование
Первое десятичное разведение (01) rотовят
следующим образом:
2 части матричной настойки;
8 частей этанола (50 %, об/об) [эта нола
(43 %, м/м»).
Второе десятичное разведение (02) roтовят
следующим образом:
1 часть первоrо десятичноrо разведе
ния;
9 частей этанола (50 %, об/об) [эта нола
(43 %, м/м)].
Последующие десятичные разведения roто
вят, как указано для 02.
Первое сотенное разведение (С1) roтовят
следующим образом:
2 части матричной настойки;
98 частей эта нола (50 %, об/об) [этано
ла (43 %, м/м)].
Второе сотенное разведение (С2) rотовят
следующим образом:
1 часть первоrо сотенноro разведения;
99 частей эта нола (50 %, об/об) [этано
ла (43 %, м/м)].
Последующие сотенные разведения roто
вят. как указано для С2.
МЕТОД 1.1.4 (СООТВЕТСТВУЕТ НАВ 2Ь: MA
ТРИЧНЫЕ НАСТОЙКИ И ЖИДКИЕ РАЗВЕДЕНИЯ)
Метод 1.1.4 используют для свежеro лекар
cTBeHHoro растительноro сырья, содержащеro
обычно менее 70 % выжатоro сока и имеюще
ro влажность (потерю в массе при высушива
нии) более 60 %, в котором отсутствует эфир
ное масло или смола.
1200
rосударственная фармакопея Республики Беларусь
Матричные настойки, приroтовленные в co
ответствии с методом 1.1.4 (СQAержание этано
ла приблизительно 36 % об/об или 30 % м/м), по
лучают мацерацией, как указано ниже.
Лекарственное растительное сырье измель
чают. Отбирают пробу и определяют потерю в
массе при высушивании (2.2.32). Если иное не
указано, потерю в массе при высушивании опре
деляют с использованием 2.00 5,OO r измель
ченноro исходноro сырья, которое помещают
в плоскодонный предварительно высушенный
(как указано для сырья) и взвешенный сосуд
диаметром 45 55 мм. Сушат при температуре
105 ос в течение 2 ч и охлаждают в эксикаторе.
К измельченному лекарственному расти
тельному сырью немедленно прибавляют не
менее половины массы этанола (70 %, об/об)
[эта нола (62 %, м/м)] и хранят в плотно YKY
поренных контейнерах при температуре не
выше 20 ОС.
По следующей формуле рассчитыва
ют количество (А 2 ), в килоrраммах, эта нола
(70 %, об/об) [эта нола (62 %, м/м)], требуемо
ro для массы (т) исходноro сырья; затем BЫ
читают количество уже добавленноro эта нола
(70 %, об/об) [этанола (62 %, м/м)] и прибав
ляют разницу к смеси.
т.Т
,
100
rдe:
т масса исходноro сырья, Kr;
т потеря в массе при высушивании, %.
Выдерживают при температуре не выше
20 0 С в течение не менее 1 О дней при периоди
ческом взбалтывании, отжимают смесь и полу
ченную жидкость фильтруют.
Приведение в соответствие со значени
ем, указанным в частной статье
Определяют процентное содержание сухоro
остатка (2.8.16) или, если указано, процентное
количественное содержание описанноro выше
фильтрата. Рассчитывают количество (A j ), в ки
лоrраммах, требуемоro этанола (36 %, об/об)
[этанола (30 %, м/м)] по следующей формуле:
m. ( N N )
х о ,
N o
rде:
т масса фильтрата, Kr;
N o содержание cyxoro остатка или KO
личР.ственное содержание, указанное в част
ной статье, %;
N содержание cyxoro остатка или KO
личеdтвенное содержание фильтрата, %.
Смешивают фильтрат с рассчитанным
количеством этанола (36 %, об/об) [этанола
(30 %, м/м)]. Выдерживают при температуре не
выше 20 0 С в течение не менее 5 дней, затем
при необходимости фильтруют.
Потенцирование
Первое десятичное разведение (01) roTO
БЯТ следующим образом:
2 части матричной настойки;
8 частей этанола (36 %, об/об) [этано
ла (30 %, м/м)].
Второе десятичное разведение (02) roто
вят следующим образом:
1 часть первоro десятичноro разведе
ния;
9 частей эта нола (36 %, об/об) [этано
ла (ЗО %, м/м)].
Последующие десятичные разведения ro
т6вят, как указано для 02.
МЕТОД 1.1.5 (СООТВЕТСТВУЕТ НАВ За: MA
ТРИЧНЫЕ НАСТОЙКИ И ЖИДКИЕ РАЗВЕДЕНИЯ)
Метод 1.1.5 используют для свежеro ле
карственноro растительноro сырья, содержа
щеro эфирное масло или смолу или обычно
имеющеro влажность (потерю в массе при BЫ
сушивании) менее 60 %.
Матричные настойки, приrотовленные в
соответствии с методом 1.1.5 (содержание
этанола приблизительно 68 % об/06 или 60 %
м/м), получают мацерацией, как указано ниже.
Лекарственное растительное сырье из
мельчают. Отбирают пробу и определяют
потерю в массе при высушивании (2.2.32).
Если иное не указано, потерю в массе при
высушивании определяют с использованием
2,00 5,00 r измельченноro исходноro сырья,
которое помещают в плоскодонный предвари
тельно высушенный (как указано для сырья)
и взвешенный сосуд диаметром 45 55 мм.
Сушат при температуре 105 ос Б течение 2 ч
и охлаждают в эксикаторе.
К измельченному лекарственному расти
тельному сырью немедленно прибавляют не
менее половины массы этанола (90 %, об/об)
[этанола (86 %, м/м)] и хранят в плотно YKY
opeHHЫX контейнерах при температуре не
выше 20 ос.
По следующей формуле рассчитыва
ют количество (Аз), в килоrраммах, этанола
(90 %, об/об) [этанола (86 %, м/м)], требуемо
ro для массы (т) исходноro сырья; затем BЫ
читают количество уже добавленноro этанола
(90 %, об/об) [этанола (86 %, м/м)] и прибав
ляют разницу к смеси.
2.т.Т
100
rдe:
т масса исходноro сырья, Kr;
т потеря в массе при высушивании, %.
Методы ПРU20товленuя 20меопатическuх базисных препаратов
1201
Выдерживают при температуре не выше
20 ос в течение не менее 1 О дней при пери
одическом взбалтывании, отжимают смесь и
полученную жидкость фильтруют.
Приведение в соответствие со значе-
нием, указанным в частной статье
Определяют процентное содержание
cyxoro остатка (2.8.16) или, если указано, про
центное количественное содержание описан
ноro выше фильтрата. Рассчитывают количе
ство (А 1 ), в килоrраммах, требуемоro эта нола
(70 %, об/об) (эта нола (62 %, м/м)] по следу
ющей формуле:
m. ( N N )
х о ,
N o
rдe:
т масса фильтрата, Kr;
N o содержание сухоro остатка или KO
личественное содержание, указанное в част
ной статье, %;
N x содержаНV1е сухоro остатка V1ли KO
личественное содержание фильтрата, %.
Смешивают фильтрат с рассчитанным
количеством этанола (70 %, об/об) (этанола
(62 %, м/м)]. Выдерживают при температуре
не выше 20 ос в течение не менее 5 дней,
затем при необходимости фильтруют.
Потенцирование
Первое десятичное разведение (01) roто
вят следующим образом:
3 части матричной настойки;
7 частей этанола (70 %, об/об) (этано
ла (62 %, м/м)].
Второе десятичное разведение (02) roто
вят следующим образом:
1 часть первоro десятичноro разведе
ния;
9 частей эта нола (70 %, об/об) (эта
нола (62 %, м/м)].
Последующие десятичные разведения ro
товят, как указано для 02. Для приroтовления
дальнейших разведений из 04 используют
этанол (50 % об/об) (этанол (43 % м/м)].
Первое сотенное разведение (С1 ) rотовят
следующим образом:
3 части матричной настойки;
97 частей этанола (70 %, об/об) (эта
нола (62 %, м/м)].
Второе сотенное разведение (С2) roтовят
следующим образом:
1 часть первоrо cOTeHHoro разведе
ния;
99 частей эта нола (50 %, об/об) (эта
нола (43 %, м/м)].
Последующие сотенные разведения roто
вят, как указано для С2.
15., Зак 1712
МЕТОД 1.1.6 (СООТВЕТСТВУЕТ НАВ ЗЬ: МА-
ТРИЧНЫЕ НАСТОЙКИ И ЖИДКИЕ РАЗВЕДЕНИЯ)
Метод 1.1.6 используют для свежеro ле
карственноro растительноro сырья, содержа
щеrо эфирное масло или смолу или обычно
имеющеro влажность (потерю в массе при BЫ
сушивании) менее 60 %.
Матричные настойки, приrотовленные
в соответствии с методом 1.1.6 (содержа
ние эта нола приблизительно 50 % об/об или
43 % м/м), получают мацерацией, как указа
но ниже.
Лекарственное растительное сырье из
мельчают. Отбирают пробу и определяют
потерю в массе при высушивании (2.2.32).
Если иное не указано, потерю в массе при
высушивании определяют с использованием
2,00 5,00 r измельченноrо исходноrо сырья,
которое помещают в плоскодонный предвари
тельно высушенный (как указано для сырья)
и взвешенный сосуд диаметром 45 55 мм.
Сушат при температуре 105 ос в течение 2 ч
и охлаждают в эксикаторе.
К измельченному лекарственному раститель
ному сырью немедленно прибавляют не менее
половины массы этанола (80 %, об/об) (эта нола
(73 %, м/м)] и хранят в плотно укупоренных KOH
тейнерах при температуре не выше 20 ОС.
По следующей формуле рассчитыва
ют количество (Аз), в килоrраммах, эта нола
(80 %, об/об) (эта нола (73 %, м/м)], требуемо
ro для массы (т) исходноro сырья; затем BЫ
читают количество уже добавленноro этанола
(80 %, об/об) (этанола (73 %, м/м)] и прибав
ляют разницу к смеси.
2.т.Т
100
rдe:
т масса исходноro сырья. Kr:
т потеря в массе при высушивании, %.
Выдерживают при температуре не выше
20 0 С в течение не менее 1 О дней при периоди
ческом взбалтывании, отжимают смесь и полу
ченную жидкость фильтруют.
Приведение в соответствие со значени-
ем, указанным в частной статье
Определяют процентное содержание сухоro
остатка (2.8.16) или, если указано, процентное
количественное содержание описанноro выше
фильтрата. Рассчитывают количество (А 1 ), в ки
лоrраммах, требуемоro этанола (50 %, об/об)
(эынола (43 %. м/м)] по следующей формуле:
m. ( N N )
х о ,
N o
rдe:
т масса фильтрата, Kr;
1202
rосударственная фармакопея Республики Беларусь
N o содержание сухою остатка или коли
чественное содержание, указанное в частной
статье, %;
N x содержание сухоro остатка или количе
ственное содержание фильтрата, %.
Смешивают фильтрат с рассчитанным KO
личеством эта нола (50 %, об/об) [этанола (43 %,
м/м)]. Выдерживают при температуре, не выше
20 ос в течение не менее 5 дней, затем при He
обходимости фильтруют.
Потенцирование
Первое десятичное разведение (01) roтовят
следующим образом:
3 части матричной настойки;
7 частей эта нола (50 %, об/об) [этано
ла (43 %, м/м)].
Второе десятичное разведение (02) rотовят
следующим образом:
1 часть первоrо десятичноrо разведе
ния;
9 частей этанола (36 %, об/об) [этанола
(30 %, м/м)].
Третье десятичное разведение (03) rотовят
следующим образом:
1 часть второю десятичноro разведе
ния;
9 частей эта нола (18 %, об/об) [этанола
(15 %. м/м)].
Последующие десятичные разведения roто
вят, как указано для 03.
МЕТОД 1.1.7 (СООТВЕТСТВУЕТ НАВ Зс: MA
ТРИЧНЫЕ НАСТОЙКИ И ЖИДКИЕ РАЗВЕДЕНИЯ)
Метод 1.1.7 используют для свежеro лекар
ственноro растительною сырья, обычно име
ющеro влажность (потерю в массе при высуши
вании) менее 60 %.
Матричные настойки, приrотовленные в co
ответствии с методом 1.1.7 (содержание этанола
приблизительно 36 % об/об или 30 % м/м), полу
чают мацерацией, как указано ниже.
Лекарственное растительное сырье из
мельчают. Отбирают пробу и определяют
потерю в массе при высушивании (2.2.32). Если
иное не указано, потерю в массе при высушива
нии определяют с использованием 2,00 5,00
r измельченноro исходноro сырья, которое по
мещают в плоскодонный предварительно BЫCY
шенный (как указано для сырья) и взвешенный
сосуд диаметром 45 55 мм. Сушат при темпе
ратуре 105 ос в течение 2 ч и охлаждают в экс
икаторе.
К измельченному лекарственному расти
тельному сырью немедленно прибавляют не
менее половины массы этанола (50 %, об/об)
[этанола (43 %, м/м)] и хранят в плотно укупо
ренных контейнерах при температуре не выше
20 ОС.
По следующей формуле рассчитывают коли
чество (А), в килоrраммах, этанола (50 %, об/об)
[этанола (43 %, м/м)], требуемоro для массы (т)
исходноro сырья; затем вычитают количество
уже добавленноro этанола (50 %, об/об) [этано
ла (43 %, м/м)] и прибавляют разницу к смеси.
2.т.Т
100
rдe:
т масса исходноro сырья, Kr;
т потеря в массе при высушивании, %.
Выдерживают при температуре не выше
20 0 С в течение не менее 1 О дней при периоди
ческом взбалтывании, отжимают смесь и полу
ченную жидкость фильтруют.
Приведение в соответствие со значени-
ем, указанным в частной статье
Определяют процентное содержание сухою
остатка (2.8.16) или, если указано, процентное
количественное содержание описанноro выше
фильтрата. Рассчитывают количество (А 1 ), в ки
лоrраммах, требуемоro этанола (36%, об/об)
[эта нола (30%, м/м)] по следующей формуле:
m./N N )
\ х о ,
N o
rдe:
т масса фильтрата, Kr;
N o содержание сухоro остатка или коли
чественное содержание, указанное в частной
статье, %;
N, содержание сухоro остатка или количе--
ственное содержание фильтрата, %.
Смешивают фильтрат с рассчитанным коли-
чеством этанола (36 %, об/об) [эта нола (30 %, м/м)]
Выдерживают при температуре не выше 20 0 С н
течение не менее 5 дней, затем при необходимо-
сти фильтруют.
Потенцирование
Первое десятичное разведение (01) roТОВЯ'!
следующим образом:
3 части матричной настойки;
7 частей эта нола (36 %, об/об) [этаНОЛё!
(30 %, м/м)].
Второе десятичное разведение (02) roтовя r
следующим образом:
1 часть первоrо десятичноro разведе-
ния;
9 частей этанола (18 %, об/об) [этанола
(15 %, м/м»).
Последующие десятичные разведения юто-
вят, как указано для 02.
МЕТОД 1.1.8 (СООТВЕТСТВУЕТ НАВ 4а: МА-
ТРИЧНЫЕ НАСТОЙКИ И ЖИДКИЕ РАЗВЕДЕНИЯ)
Метод 1.1.8 обычно используют для высу-
шенноrо лекарственноro растительноrо сырья.
Матричные настойки, приroтовленные в СОО1
ветствии с методом 1.1.8, получают путем Maцepa
Методы при;ютовления zомеопатических базисных препаратов
1203
ции или перколяции, как указано ниже, используя
1 часть высушенноro лекарственною раститель
ноro сырья и 1 О частей этанола COOTBeTCTBY
ющей концентрации (безводный, 96 % об/об
94 % м/м, 90 % об/об 86 % м/м, 80 % об/об
73 % м/м, 70 % об/об 62 % м/м, 50 % об/об
43 % м/м, 36 % об/об 30 % м/м, 18 % об/об
15 % м/м), если иное не указано в частной статье.
Получение путем мацерации. Если иное
не указано, измельчают лекарственное расти
тельное сырье, тщательно смешивают с эта
нолом соответствующей концентрации и BЫ
держивают в закрытом контейнере в течение
подходящеro времени. Остаток отделяют от
эта нола и при необходимости отжимают. В по
следнем случае объединяют две полученные
жидкости.
Получение путем перколяции. При необ
ходимости измельчают лекарственное расти
тельное сырье. Тщательно перемешивают с
частью этанола соответствующей KOHцeHTpa
ции и выдерживают в течение подходящеro
времени. Пере носят в перколятор и позволяют
перколяту свободно вытекать при комнатной
температуре, при этом необходимо следить
за тем, чтобы экстраrируемое лекарственное
растительное сырье было полностью покры
то остающимся этанолом. Остаток может быть
отжат и полученную жидкость объединяют с
перколятом .
При необходимости доведения до опреде
ленной концентрации, рассчитывают количе
ство (А , ), в килоrраммах, эта нола подходящей
концентрации, необходимоro для достижения
концентрации, которая задана или исполь
зовалась при приrотовлении, по следующей
формуле:
m. ( N N )
х о ,
N o
rдe:
т масса фильтрата, Kr;
N o содержание cyxoro остатка или коли
чественное содержание, указанное в частной
статье, %;
N x содержание cyxoro остатка или количе
С1венное содержание фильтрата, %.
Смешивают мацерат или перколят с рассчи
танным количеством эта нола подходящей KOH
центрации. Выдерживают при температуре, не
выше 20 0 С в течение не менее 5 дней, затем при
необходимости фильтруют.
Ilотенцирование
Матричная настойка соответствует первому
,:\есятичному раствору (0 = 01).
Второе десятичное разведение (UL) ютовят
.:педvющим образом:
1 часть матричной настойки (1);
9 частеи этанола той же концеНТРВ11ИИ.
Третье десятичное разведение (03) ютовят
следующим образом:
1 часть Bтoporo десятичноro разведе
ния;
9 частей эта нола той же концентрации.
Если в спецификации не приведена
друrая концентрация этанола, то используют
этанол (50 %, об/об) [эта нол (43 %, м/м)] для
последовательных десятичных разведений
из 04 и далее поступают, как указано для 03.
Первое сотенное разведение (С1) roтовят
следующим образом:
10 частей матричной настойки (01);
90 частей эта нола той же концентрации.
Второе сотенное разведение (С2) rотовят
следующим образом:
1 часть первorо сотенноro разведения;
99 частей этанола (50 %, об/об) [этано
ла (43 %, М/М)], если в спецификации не
указана друrая концентрация эта нола.
Последующие сотенные разведения юто
вят, как указано для С2.
МЕТОД 1.1.9 (СООТВЕТСТВУЕТ НАВ 4Ь: MA
ТРИЧНЫЕ НАСТОЙКИ И ЖИДКИЕ РАЗВЕДЕНИЯ)
Метод 1.1.9 обычно используют для сырья
животноro происхождения.
Матричные настойки, приrотовленные в co
ответствии с методом 1.1.9, получают путем
мацерации или перколяции, как указано ниже,
используя 1 часть материала животною проис
хождения и 1 О частей этанола соответствующей
концентрации (безводный, 96 % об/об 94 % М/М,
90 % об/об 86 % м/м, 80 % об/об 73 % М/М,
70 % об/об 62 % м/м, 50 % об/об 43 % м/м,
36 % об/об 30 % м/м, 18 % об/об 15 % М/М),
если иное не указано в частной статье.
Получение путем мацерации. Если иное
не указано, измельчают материал животноrо
происхождения, тщательно смешивают с эта
нолом соответствующей концентрации и BЫ
держивают в закрытом контейнере в течение
подходящею времени. Отделяют остаток от
этанола и при необходимости отжимают. В по
следнем случае объединяют две полученные
жидкости.
Получение путем перколяцuи. При необ
ходимости измельчают материал животноrо
происхождения. Тщательно перемешивают с
частью этанола соответствующей KOHцeHTpa
ции и выдерживают в течение подходящею
времени. Переносят в перколятор и позволяют
перколяту свободно вытекать при комнатной
температуре, при этом экстраrируемый MaTe
риал животною происхождения должен быть
Bcerдa полностью покрыт остающимся этано
лом. Остаток может быть отжат, и полученную
жидкость объединяют сперколятом.
rlри необходимости ДОВtщения до опреде
ленной концентрации, раL:СЧИI ывают количество
A,), в килоrраммах, этаНОJlа пvдходящеи KOH
1204
rосударственная фармакопея Республики Беларусь
центрации, необходимоro для достижения KOH
центрации, которая задана или использовалась
при приroтовлении, по следующей формуле:
m. ( N N )
х о ,
N o
rдe:
т масса фильтрата, Kr;
N o содержание сухоro остатка или коли
чественное содержание, указанное в частной
статье, %;
N x содержание сухоro остатка или количе
ственное содержание фильтрата. %.
Смешивают мацерат или перколят с рассчи
танным количеством этанола подходящей KOH
центрации. Выдерживают при температуре не
выше 20 ос в течение не менее 5 дней, затем
при необходимости фильтруют.
Потенцирование
Матричная настойка соответствует первому
десятичному раствору (0 = 01).
Второе десятичное разведение (02) roтовят
следующим образом:
1 часть матричной настойки (01);
9 частей этанола той же концентрации.
Третье десятичное разведение (03) roтовят
следующим образом:
1 часть BToporo десятичноrо разведе
ния;
9 частей этанола той же концентрации.
Если в спецификации не приведена друrая
концентрация эта нола, то используют эта нол
(50 %, об/об) [эта нол (43 %, м/м)] для последова
тельных десятичных разведений из 04 и далее
поступают, как указано для 03.
Первое сотенное разведение (С1) rотовят
следующим образом:
1 О частей матричной настойки (01);
90 частей этанола той же концентрации.
Второе сотенное разведение (С2) rотовят
следующим образом:
1 часть первоro cOTeHHoro разведения;
99 частей этанола (50 %. об/об) [этано
ла (43 %, м/м)], если в спецификации не
указана друrая концентрация этанола.
Последующие сотенные разведения roто
вят, как указано для С2.
МЕТОД 1.1.10 (ФРАНЦУЗСКАЯ ФАРМАКОПЕЯ)
Метод 1.1.1 О обычно используют для ле
KapcTBeHHoro растительноrо сырья. COCTO
яние лекарственноro растительноrо сырья
(свежее или высушенное) указывают в част
ных статьях.
Матричные настойки, приroтовленные в co
ответствии с методом 1.1.10, получают путем
мацерации.
Подходящим образом измельчают лекар
ственное растительное сырье. Отбирают об
разец и определяют потерю в массе при BЫCY
шивании при температуре 1 GS ос в течение 2 ч
(2.2.32) или содержание воды (2.2.13). Принимая
во внимание полученное значение, рассчитыва
ют и прибавляют к лекарственному растительно
му сырью количество этанола соответствующей
концентрации, необходимое для I1риroтовле
ния, если иное не указано, матричной настойки
1 часть в 10 частях (матричная настойка 1:10)
с подходящим содержанием этанола. Проводят
мацерацию в течение 1 О дней при достаточном
встряхивании.
Остаток отделяют от этанола и при необ
ходимости процеживают под давлением. Объ
единенные жидкости выдерживают в течение
48 ч и фильтруют. Матричные настойки с реrла
ментированным количественным содержани
ем при необходимости MorYT быть приведены в
соответствие с требованиями путем прибавле
ния этанола той же концентрации, что исполь
зовалась при приrотовлении настойки.
Потенцирование
Первое десятичное разведение (01) roтовят
следующим образом:
1 часть матричной настойки;
9 частей этанола соответствующей KOH
центрации.
Второе десятичное разведение (02) roтовят
следующим образом:
1 часть первоrо десятичноrо разведе
ния;
9 частей этанола соответствующей KOH
центрации.
Последующие десятичные разведение по
лучают, как указано для 02, используя этанол
соответствующей концентрации.
Первое сотенное разведение (С1) roтовят
следующим образом:
1 часть матричной настойки;
99 частей этанола соответствующей
концентрации.
Второе сотенное разведение (С2) rотовят
следующим образом:
1 часть первоro сотенноro разведения;
99 частей этанола соответствующей
концентрации.
Последующие сотенные разведения roто
вят, как указано для С2, используя эта нол COOT
ветствующей концентрации.
МЕТОД 1.1.11 (ФРАНЦУЗСКАЯ ФАРМАКОПЕЯ)
Метод 1.1.11 обычно используют для сырья
животноro происхождения.
Матричные настойки, приroтовленные в co
ответствии с методом 1.1.11, получают путем Ma
церации.
Соотношение массы исходноro сырья к
матричной настойке обычно составляет 1 к 20.
К исходному материалу, соответствующим
образом измельченному, прибавляют эта нол
необходимой концентрации в количестве,
Методы при20товления 20меопатических базисных препаратов
1205
требующемся для получения матричной Ha
стойки 1 :20. Проводят мацерацию в течение
не менее 1 О дней при достаточном встряхива
нии. Декантируют и фильтруют. Выдерживают
в течение 48 ч и снова фильтруют. Матричные
настойки с реrламентированным количествен
ным содержанием при необходимости MorYT
быть приведены в соответствие с требовани
ями путем прибавления этанола той же KOH
центрации, что использовалась при приrотов
лении настойки.
Потенцирование
Первое десятичное разведение (01) roтовят
следующим образом:
1 часть матричной настойки;
9 частей этанола соответствующей KOH
центрации.
Второе десятичное разведение (02) roтовят
следующим образом:
1 часть первоrо десятичноrо разведе
ния;
9 частей этанола соответствующей KOH
центрации.
Последующие десятичные разведения по
луча ют, как указано для 02, используя эта нол
соответствующей концентрации.
Первое сотенное разведение (С1) rотовят
следующим образом:
1 часть матричной настойки;
99 частей этанола соответствующей
концентрации.
Второе сотенное разведение (С2) roтовят
следующим образом:
1 часть первоro сотенноro разведения;
99 частей эта нола соответствующей
концентрации.
Последующие сотенные разведения roто
вят, как указано для С2, используя этанол COOT
ветствующей концентрации.
2. rЛИЦЕРИНОВЫЕ МАЦЕРАТЫ
МЕТОД 2.1
Метод 2.1 используют для мацерации сырья
животноro или растительноrо происхождения в
rлицерине 85 % или смеси rлицерина/этанола
соответствующей концентрации. Патолоrиче
ский материал исключается.
При необходимости перед использованием
сырье тонко измельчают.
МЕТОДЫ 2.1.1, 2.1.2 (СООТВЕТСТВУЮТ НАВ
42а И 42Ь: МАТРИЧНЫЕ НАСТОЙКИ И ЖИДКИЕ
РАЗВЕДЕНИЯ ИЗ НИХ)
ИспользуютсырьеживотноroпроисхождР.ния
только что забитые животные или их части. Живот
ных перерабатывают немедленно после забоя.
Мацерация
Дисперrируют 1 часть тонко измельченноrо
животноrо материала в:
9 частях (десятичные разведения) или
99 частях (сотенные разведения) rлицерина
85 % для метода 2.1.1.
или 2,1 части rлицерина 85 % для
метода 2.1.2.
Проводят мацерацию не менее 2 часов,
затем интенсивно встряхивают. При необходи
мости фильтруют.
Если доказано, перед измельчением к 1
части животноrо материала может быть прибав
лена 1 часть rлицерина 85 %. В случаях исполь
зования очень малых количеств животноro Ma
териала разведение может быть произведено
дисперrированием 1 части тонко измельченно
ro животноro материала в 99 частях rлицерина
85 % (С1 или 02 если используется для даль
нейшеrо десятичноro разведения).
Потенцирование
МЕТОД 2.1.1
Второе десятичное разведение (02) roтовят
следующим образом:
1 часть rлицериновоro мацерата 01;
9 частей rлицерина 85 % или этанола
(18 %, об/об) [этанола (15% м/м)].
Последовательные десятичные разведе
ния выполняют, как указано для 02. но в каче
стве разбавителя используют этанол (18 %, об/
об) [этанол (15 %, м/м)].
Второе сотенное разведение (С2) rотовят
следующим образом:
1 часть rлицериновоro мацерата С 1 ;
99 частей этанола (18 %, об/об) [этанола
(15 %, м/м)].
Последовательные сотенные разведения
выполняют, как указано для С2.
МЕТОД 2.1.2
Первое «десятичное» разведение (01) roто
вят следующим образом:
3 части rлицериновоro мацерата:
7 частей воды для инъекций.
Второе десятичное разведение (02) roтовят
следующим образом:
1 часть 01;
9 частей воды для инъекций.
Последовательные десятичные разведения
выполняют, как указано для 02.
МЕТОД 2.1.3 (ФРАНЦУЗСКАЯ ФАРМАКОПЕЯ)
Используют сырье растительноro или жи
вотноro происхождения.
Мацерация
Подходящим образом измельчают исходное
сырье. Отбирают образец и определяют потерю в
массе при высушивании при температуре 105 ос
в течение 2 ч (2.2.32) или содержание воды
(2.2.13). Принимая во внимание полученное зна
чение, рассчитывают и прибавляют к лекарствен
ному растительному сырью количество смеси
этанол/rлицерин соответствующей KOHцeHTpa
ции, необходимое для приrотовления, если иное
1206
rосударственная фармакопея Республики Беларусь
не указано, мацерата 1 часть в 20 частях. Прово
дят мацерацию в течение не менее 3 недель при
достаточном встряхивании. Сливают надосадоч
ную жидкость и при необходимости процеживают
под давлением. Объединенные жидкости Bыдep
живают в течение 48 ч и фильтруют.
Потенцирование
Первое десятичное разведение (01) rотовят
следующим образом:
1 часть rлицериновоro мацерата;
9 частей смеси вода эта нол rлице
рин соответствующей концентрации.
Второе десятичное разведение (02) rотовят
следующим образом:
1 часть первоrо десятичноrо разведе
ния;
9 частей смеси вода этанол rлице
рин соответствующей концентрации.
Последующие десятичные разведения по
лучают, как указано для 02, используя иной под
ходящий разбавитель.
Первое сотенное разведение (С1) roтовят
следующим образом:
1 часть rлицериновоrо мацерата;
99 частей смеси вода эта нол rли
церин соответствующей концентрации.
Второе сотенное разведение (С2) ютовят
следующим образом:
1 часть первоro сотенною разведения:
99 частей смеси вода этанол rли
церин соответствующей концентрации.
Последующие сотенные разведения roто
вят, как указано для С2, используя иной подхо
дящий разбавитель.
МЕТОД 2.2
МЕТОДЫ 2.2.1, 2.2.2, 2.2.3,
ВЕТСТВУЮТ НАВ 41а, 41Ь, 41с
ЦЕРИНОВЫЕ МАТРИЧНЫЕ
И ЖИДКИЕ РАЗВЕДЕНИЯ ИЗ НИХ)
Метод 2.2 используют для мацерации сырья
животноro происхождения в rлицериновом pac
творе, содержащем натрия хлорид. Патолоrиче
ский материал исключается.
В методах 2.2.1, 2.2.2 и 2.2.3 используют
сырье из только что забитых животных или их
частей или секретов. Низших животных умерщ
вляют уrлерода диоксидом в закрытом сосуде.
Животных перерабатывают немедленно после
забоя.
В методе 2.2.4 используются компоненты
крови живых лошадей.
2.2.4 (СООТ
И 41d: rЛИ
НАСТОЙКИ
Сбор образцов и/или предварительная
обработка.
При необходимости, сырье для методов
2.2.1, 2.2.2 и 2.2.3 перед использованием тонко
измельчают.
Кровь для метода 2.2.4, должен отбирать
ветеринар. Кровь, полученную от животных, за
битых кровопусканием, использовать нельзя.
Берут 200 мл крови И на 1 мл ее прибавляют
15 МЕ rепарина натрия и 0,625 мл раствора 9 r/Kr
натрия хлорида. Компоненты крови отделя
ют фракционным центрифуrированием и pecy
спендируют каждый отдельный осадок клеток в
1,1 мл раствора 9 r/Kr натрия хлорида. Эти кле
точные суспензии обрабатывают для получения
rлицериновоro мацерата.
Мацерация
1 часть тонко измельченноro животноrо Ma
териала, секретов или суспензий клеток крови
смешивают в соответствии с применяемым Me
тодом с 5 частями раствора натрия хлорида co
ответствующей концентрации (таблица 2371. 1)
и 95 частями rлицерина. Выдерживают в за
щищенном от света месте не менее 7 суток,
затем сливают. При необходимости, для MeTO
дов 2.2.1, 2.2.2 и 2.2.3 перед декантированием
используют центрифуrирование, затем Haдoca
дочную жидкость, при необходимости, фильтру
ют. Декантированная жидкость или фильтрат,
соответственно, представляют собой rлицери
новый мацерат.
Перед работой с rлицериновым мацератом
весь имеющийся осадок должен быть ресуспен
дирован.
I Методы 2.2.1 I М 2 2 2
I и 2.2.4 i етод..
раствор 15 r/Kr i . 1 раствор 40 r/Kr
натрия хлори натрия хлори
да в во!\е очи I да в во!\е очи
щеннои I щеннои
Таблица 2371. 1
I Метод 2.2.3
раствор 80 r/Kr
натрия хлори
да в воде очи
щенной
Разбавитель
0,2 части натрия rидрокарбоната и
8,8 частей натрия хлорида в 991 частях воды для
инъекций или воды очищенной соответственно.
Потенцирование
rлицериновый мацерат соответствует BTO
рому десятичному разведению (02) или пер
вому сотенному разведению (С1). Третье дe
сятичное разведение (03) rотовят следующим
образом:
1 часть BToporo десятичноrо разведе
ния;
9 частей соответствующеrо разбави
теля.
Последовательные десятичные разведения
выполняют, как указано для 03.
При необходимости четвертое десятичное
Р<Jзведение (04) rотовят из 1 части TpeTber..) де
сятичноro разведения, 5,6 частей разбавителя VI
3,4 частей воды для инъекц; й.
Второе сотенное разведение (С2) roТОВЮ
следующим образом:
1 часть первоro сс.тенноro раЗБt.дения,
99 частей соотвеТСТВУЮll,еrо раЗС5с:v"
теля.
Методы при20товленuя 20меопатuческих базисных препаратов
1207
Последовательные сотенные разведения
выполняют, как указано для С2.
3. ЖИДКИЕ РАЗВЕДЕНИЯ
МЕТОД 3.1
Методы 3.1.1, 3.1.2 и 3.1.3 используют для
растворения любоro подходящеro неорrаниче
скоro или орrаническоro исходноro материала,
например, минералов или ядов.
Если не установлено иное, 1 часть исходно
ro материала растворяют в 9 частях (01) или 99
частях (С1) жидкоro разбавителя и интенсивно
встряхивают.
Если обосновано и утверждено, в случае
недостаточной растворимости исходноrо Ma
териала в указанном разбавителе, сразу по
лучают первое возможное разведение. Напри
мер, если исходный материал мало растворим.
1 часть исходноro материала растворяют в 99
частях разбавителя (С1 или 02, если будет ис
пользоваться для дальнейших десятичных раз
ведений).
МЕТОДЫ 3.1.1, 3.1.2 (СООТВЕТСТВУЮТ НАВ
5а, 5Ь: РАСТВОРЫ, ВОДНЫЕ РАСТВОРЫ)
Разбавители
MorYT быть использованы разбавители, YKa
занные в таблице 2371. 2.
При использовании этанола (18 %, об/об)
[эта нола (15 %, м/м)] для метода 3.1.1 для дe
сятичных разведений исходный материал может
быть растворен в 7,58 частях воды очищенной
и концентрация этанола может быть ДOBeдe
на прибавлением к раствору 1,42 частей этано
ла (96 %, об/об) [эта нол (94 %, м/м)]. Для COTeH
ных разведений используют 83,4 частей воды
очищенной и 15,6 частей эта нола (96 %. об/об)
[этанол (94 %, м/м)].
Для метода 3.1.2, если исходный материал
нестабилен и/или растворим в воде, может быть
прибавлен rлиuерин 85 % в качестве разбавите
ля в концентрации не более 35 % для ПОТei-ЩИ
рования вплоть до 04.
Потенцирование
Если не указано иное, второе десятичное
разведение (02) rотовят следующим образом:
1 часть первоrо десятичною разведе
ния (01);
9 частей эта нола (50 %, об/об) [этано
ла (43 %, м/м)] для метода 3.1.1 или
9 частей воды для инъекций (или воды
очищенной, как необходимо) для
метода 3.1.2.
Последовательные десятичные разведения
выполняют, как указано для 02.
Если не указано иное, второе сотенное раз
ведение (С2) rотовят следующим образом:
1 часть первоrо сотенноro разведения (С1);
99 частей этанола (50 %, об/об) [этано
ла (43 %, м/м)] для метода 3.1.1 или
99 частей воды для инъекций (или воды
очищенной, как необходимо) для
метода 3.1.2.
Последовательные сотенные разведения
выполняют, как указано для С2.
Добавки
Если при использовании метода 3.1.1 в KO
нечном разведении наблюдается преципитация,
для улучшения стабильности и/или раствори
мости, если иное не указано, можно при менять
следующие добавки:
уксусную кислоту ледяную;
хлороводородную кислоту концентриро
ванную;
молочную кислоту;
натрия rидроксид.
В тех случаях, коrда рН растворов и разве
дений был скорректирован, дальнейшему потен
цированию их не подверrают.
МЕТОД 3.1.3
Разбавители
Подходящие разбавители, например, этанол
соответствующей концентрации, rлицерин или
вода очищенная, MOryT быть использованы сами
по себе или в комбинации.
Потенцирование
Если иное не указано, второе десятич
ное разведение (02) roтовят следующим об
разом:
1 часть первоrо десятичноrо разведе
ния (01);
9 частей соответствующеrо разбавителя.
Последовательные десятичные разведения
выполняют, как указано для 02.
Если не указано иное, второе сотенное раз
ведение (С2) rотовят следующим образом:
1 часть первоro сотенною разведения (С1);
99 частей соответствующеro разбавителя.
Последовательные сотенные разведения
выполняют, как указано для С2.
МЕТОД 3.2
Метод 3.2 обычно используют для получе
ния жидких разведений тритураций веществ,
растворимость которых по большей части коле
блется от умеренно растворимых до практиче
ски нерастворимых.
МЕТОДЫ 3.2.1, 3.2.2 (СООТВЕТСТВУЮТ НАВ
8а, 8Ь: ЖИДКИЕ ЛЕКАРСТВЕННЫЕ СРЕДСТВА,
приrОТОВЛЕННЫЕ ИЗ ТРИТУРАЦИЙ, ВОДНЫЕ
ЛЕКАРСТВЕННЫЕ СРЕДСТВА, пРиrОТОВЛЕН
НЫЕ ИЗ ТРИТУРАЦИЙ)
В соответствии с методом 3.2.1 и MeTO
дом 3.2.2 лекарственные средства получают
1208
rосударственная фармакопея Республики Беларусь
из тритураций 04, 05 и 06 или тритураций С4,
С5 и С6, приroтовленных в соответствии с Me
тодом 4.1.1 путем не менее 2 стадий потенци
рования.
Разбавители
Можно использовать разбавители, приве
денные в таблице 2371. 3.
Потенцирование
Для первоrо жидкоro потенцирования
1 часть тритурации растворяют в 9 частях (дe
сятичные разведения) или 99 частях (COTeH
ные разведения) указанноro разбавителя (Ta
блица 2371. 3) и потенцируют. Для дальнейших
потенцирований поступают точно так же, как с
1 частью предыдущеro разведения.
Разведения 06, 07, С6 и С7, получаемые
вышеуказанным методом, для приrотовления
дальнейших разведений не применяются.
МЕТОД 3.2.3
В соответствии с методом 3.2.3 лекарствен
ные средства получают из тритураций, начиная
с 02, и С1, С2, С3 и С4, приroтовленных в COOT
ветствии с методом 4.1.2.
Разбавители
MoryT быть использованы подходящие раз
бавители, например, этанол соответствующей
концентрации или вода очищенная.
Потенцирование
Если иное не указано, первое жидкостное
десятичное разведение (Оп 1) roтовят следу
ющим образом:
1 часть десятичной тритурации Оп 2;
9 частей воды очищенной или иноro
подходящеro разбавителя в соответ
ствующей пропорции.
Следующее десятичное разведение (Оп) ro
товят следующим образом:
1 часть первоro жидкостноro десятично
ro разведения Оп 1 ;
9 частей подходящеro разбавителя.
Последовательные десятичные разведения
выполняют, как указано для Оп.
Если иное не указано, первое жидкостное
сотенное разведение (Сп 1) roтовят следующим
образом:
1 часть сотенной тритураций Сп 2;
99 частей воды очищенной или иноro
подходящеro разбавителя в COOTBeT
ствующей пропорции.
Следующее сотенное разведение (Сп) roто
вят следующим образом:
1 часть первоro жидкостноro сотенноro
разведения Cn 1 ;
99 частей подходящеro разбавителя.
Последовательные сотенные разведения
выполняют, как указано для Сп.
4. ТРИТУРАЦИИ
МЕТОД 4.1
Метод 4.1 применяют для тритураций,
то есть твердых разведений, сырья или три
тураций, приroтовленных в соответствии с
методами 4.2.1 или 4.2.2. Длительность и
интенсивность тритураций должны быть дo
статочны для достижения однородности и по
тенцирования.
Таблица 2371. 2
Метод 3.1.1 Метод 3.1.2
Безводный этанол Вода для инъекций
Этанол (96 %, об/об) [этанол (94 %, м/м)] Вода очищенная
Этанол (90 %, об/об) [эта нол (86 %, м/м)]
Этанол (80 %, об/об) [эта нол (73 %, м/м)]
Этанол (70 %, об/об) [этанол (62 %, м/м)]
Этанол (50 %, об/об) [этанол (43 %, м/м)]
Этанол (36 %, об/об) [этанол (30 %, м/м)]
Этанол (18 %, об/об) [этанол (15 %, м/м)]
Вода очищенная
rлицерин 85 %
Таблица 2371. 3
Метод 3.2.1 Метод 3.2.2
1 e потенцирование: Все потенцирования:
вода очищенная вода для инъекций
2 e потенцирование: вода очищенная
этанол (36 %, об/об) [этанол (30 %, м/м)]
Дальнейшие потенцирования:
эта нол (50 %, об/об) [этанол (43 %, м/м)]
Методы ПРU20товленuя 20меопатических базисных препаратов
1209
Разбавитель
Если иное не указано, используют лактозу
моноrидрат.
МЕТОД 4.1.1 (СООТВЕТСТВУЕТ НАВ б: ТРИТУ
РАЦИИ)
Тритурации rотовят вручную или механи
чески. Механическую тритурацию необходимо
использовать для количеств, превышающих
1 Kr. Окончательный размер частиц сырья в
первом десятичном или сотенном разведении
не должен превышать 100 мкм, если иное не
указано в частной статье.
Соотношения сырья и разбавителя
Десятичные Сотенные
разведения разведения
Первое десятичное Первое сотенное раз
разведение (01) roто ведение (С1) roтовят
вят следующим обра следующим образом:
зом: 1 часть сырья;
1 часть сырья; 99 частей разбавите
9 частей разбавителя. ля.
Последовательные дe Последовательные co
сятичные тритурации тенные тритурации
(Оп) выполняют, как (Сп) выполняют, как
указано для 01, ис указано для С1, ис
пользуя 1 часть пре пользуя 1 часть пре
дыдущей тритурации дыдущей тритурации
(On 1). (Сп 1).
в тех случаях, коrда используется свежее
растительное сырье, количество прибавленноro
разбавителя должно быть таким, чтобы получи
лось 1 О частей тритурации (десятичной) или 100
частей тритурации (сотенной) из 1 части сырья
(заменить массу потери воды из свежеro pac
тительноro сырья на эквивалентное количество
разбавителя). Для твердоro разведения может
понадобиться соответствующий щадящий про
цесс сушки.
Если обосновано и утверждено, может БЬ1ТЬ
необходимо непосредственное приroтовление
С1 или 02, если они используются для дальней
ших десятичных тритураций в качестве первой
твердой тритурации, из 1 части сырья и 99
частей разбавителя.
Тритурация
Если не обосновано и утверждено иное,
метод включает в себя разделение разбавителя
на 3 равных части, прибавление сырья к первой
части и последующиf' прибавления второй и
третьей частей разбавителя, выполняя тща
тельную тритурацию после каждоro прибавле
ния.
Для механической тритурации используют
установку, обеспечивающую соответствие Tpe
бованиям относительно размера частиц первой
десятичной или сотенной твердой тритура
ции. Для обеспечения равномерной тритура
ций установка может быть снабжена скребком.
Если не обосновано и утверждено иное, время,
необходимое для приroтовления одной триту
рации, должно быть не менее 1 ч.
Для тритурации, полученной вручную, раз
бавитель делят на 3 равные части и быстро три
турируют первую часть в фарфоровой ступке.
Прибавляют сырье, тритурируют смесь в тече
ние 6 мин, соскабливают в течение 4 мин co
ответствующим неметаллическим приспосо
блением (например, фарфоровым шпателем).
Продолжают тритурацию еще 6 мин, снова co
скабливают в течение 4 мин, затем прибавля
ют вторую часть разбавителя и продолжают,
как описано выше. Таким же образом поступа
ют с оставшейся частью разбавителя. Таким об
разом, минимальное время, затрачиваемое на
весь процесс, должно составлять 1 час. Для каж
доro последующею твердоro разведения весь
процесс выполняют снова.
Тритурации, начиная с 05 или С5, можно
так же приютовить путем интенсивной механи
ческой обработки на подходящей смешивающей
установке следующим образом: твердую триту
рацию прибавляют к одной трети разбавителя
и смешивают. При6авляют вторую треть разба
вителя, смешивают и таким же образом обраба
тывают последнюю треть разбавителя. Если не
обосновано и утверждено иное, весь процесс
должен длиться не менее 1 ч.
В любом случае на жидкую среду возможно
перейти с четвертой, пятой и шестой десятичной
или сотенной тритураций, как описано в методах
3.2.1 и 3.2.2.
МЕТОД 4.1.2 (ФРАНЦУЗСКАЯ ФАРМАКОпея)
Тритурация
Тритурации roтовят следующим образом:
Десятичные тритурации
1 часть roмеопатическоrо базисноro пре
парата измельчают в порошок. Тщательно три
турируют с небольшим количеством разбави
теля. Небольшими количествами прибавляют
разбавитель до тех пор. пока не израсходует
ся 9 ею частей. Результирующая тритурация
представляет собой первую десятичную триту
рацию (01).
Тритурируют 1 часть полученной тритурации
с 9 частями разбавителя, как описано выше. Pe
зультирующая тритурация представляет собой
вторую десятичную тритурацию (02).
В любом случае на жидкую среду возможно
перейти после седьмой десятичной тритурации
(07), как описано в методе 3.2.3.
Сотенные тритурации
Получают таким же образом, но следуя co
тенным разведен иям.
В любом случае на жидкую среду возможно
перейти после третей сотенной тритурации (С3),
как описано в методе 3.2.3.
1210
rосударственная фармакопея Республики Беларусь
МЕТОД 4.2
Метод 4.2 применяют для тритураций, то
есть твердых разведений, жидких препара
тов, например, матричных настоек и paCTBO
ров, их разведений, смесей и потенцирован
ных смесей.
ПостеlJенно пропитывают все количество
разбавителя, осторожно сушат увлажненную
смесь, растирают и при необходимости просе
и вают, затем смешивают и тритурируют до дo
стижения однородности и потенцирования.
Дальнейшие тритурации выполняют, как описа
но для методов 4.1.1 или 4.1.2.
Разбавитель
Если иное не указано, используют лакто
зу моноrидрат.
МЕТОД 4.2.1 (СООТВЕТСТВУЕТ НАВ 7: ТРИТУ
РАЦИИ)
Соотношения сырья и разбавителя
Количество прибавляемоrо разбавителя
Bcerдa должно быть таким, чтобы получить 1 О
частей тритурации (десятичная тритурация)
или 100 частей тритурации (сотенная триту
рация) из требуемоrо числа частей жидкоro
препарата (таблица 2371. 4), учитывая массу
сухоro остатка. В тех случаях, Korдa сухим
остатком пренебреrают, количество прибав
ленноro разбавителя равняется 1 О частям
тритурации (десятичная тритурация) или 100
частям тритурации (сотенная тритурация) на
1 часть жидкоro лекарственноro средства.
МЕТОД 4.2.2
Соотношения сырья и разбавителя
Матричные настойки, приrотовленные
в соответствии с методами 1.4.3 и 1.4.4
Первую десятичную Первую сотенную три
тритурацию (01) roто турацию (С1) roтовят
вят следующим обра следующим образом:
зом: 1 часть матричной Ha
1 часть матричной Ha стойки
стойки; 100 частей разбавите
1 О частей разбавителя ля
5. ИНЫЕ ЛЕКАРСТВЕННЫЕ СРЕДСТВА
МЕТОД 5.1
Метод 5.1 используют для приrотовления
roмеопатических лекарственных средств копо
тенцированием 2 или более базисных препара
тов и/или разведений из них, rдe копотенциро
вание включает в себя смешивание нескольких
Таблица 2371. 4
Десятичные тритурации Сотенные тритурации
Матричные настойки, приrотовленные в соответствии с методами 1.1.1, 1.1.3 и 1.1.4
Первую десятичную тритурацию (01) roтовят сле Первую сотенную тритурацию (С1) roтовят следу
дующим образом: ющим образом:
2 части матричной настойки; 2 части матричной настойки;
максимум 1 О частей разбавителя, учитывая массу максимум 100 частей разбавителя, учитывая
сухоro остатка массу сухоro остатка
Матричные настойки, приrотовленные в соответствии с методами 1.1.2, 1.1.5, 1.1.6 и 1.1.7
Первую десятичную тритурацию (01) roтовят сле Первую сотенную тритурацию (С1) roтовят следу
дующим образом: ющим образом:
3 части матричной настойки; 3 части матричной настойки;
максимум 10 частей разбавителя, учитывая массу максимум 100 частей разбавителя, учитывая
сухоro остатка массу cyxoro остатка
Матричные настойки, приrотовленные в соответствии с методами 1.1.8 и 1.1.9
Матричная настойка соответствует первому десятичному разведению (01)
Вторую десятичную тритурацию (02) roтовят сле Первую сотенную тритурацию (С1) rотовят следу
дующим образом: ющим образом:
1 часть матричной настойки; 10 частей матричной настойки;
максимум 10 частей разбавителя, учитывая массу максимум 100 частей разбавителя, учитывая
сухоro остатка массу сухоro остатка
Растворы, приrотовленные в соответствии с методом 3.1.1 или жидкие разведения, смеси и
потенцированные смеси
Десятичную тритурацию п+1 (Оп+1) roтовят сле Сотенную тритурацию п+1 (Сп+1) roтовят следу
дующим образом: ющим образом: I
1 часть разведения (Оп); 1 часть разведения (Сп); I
максимум 1 О частей разбавителя, учитывая массу максимум 100 частей разбавителя, учитывая I
сухоro остатка массу сухоro остатка
Матричные настойки для 20меопатических лекарствеННblХ средств
1211
базисных препаратов или разведений базис
ных препаратов, последующее потенцирова
ние их вместе в одну или более стадий.
МЕТОДЫ 5.1.1, 5.1.2, 5.1.3 (COOTBEТCTBY
ЮТ НАВ 40а, 40Ь, 40с: КОПОТЕНЦИРОВАННЫЕ
СМЕСИ)
MOryT быть использованы следующие ба
зисные препараты и/или разведения (таблица
2371. 5).
Разбавители
Выбор разбавителя определяется и
должен соответствовать любому специально
му требованию для конкретною базисноrо пре
парата, а также лекарственной формы (табли
ца 2371. 6).
Для метода 5.1.1, если начинают с тритура
ции и если обосновано, для первой стадии по
тенцирования используют воду очищенную.
Для метода 5.1.2, если начинают с rлицери
новоro мацерата, содержащею натрия хлорид и
если иное не обосновано и не утверждено, ис
пользуют следующий разбавитель: 0,2 части
натрия rидрокарбоната и 8,8 частей натрия хло
рида в 991 части воды для инъекций.
Потенцирование
На каждой стадии потенцирования объеди
няют и потенцируют или тритурируют 1 часть
данной смеси с 9 частями (десятичные разведе
ния) или 99 частями (сотенные разведения) под
ходящеro разбавителя.
МЕТОД 5.1.4
Разбавители
Может быть использован, например, этанол
подходящей концентрации, вода очищенная или
лактоза моноrидрат.
Потенцирование
Потенцирование можно проводить, как опи
сано для методов 5.1.1, 5.1.2 и 5.1.3, либо на по
следней стадии, либо на нескольких последова
тельных стадиях.
МЕТОД 5.1.5
Разбавители
Может быть использован, например, эта нол
подходящей концентрации, вода очищенная или
лактоза моноrидрат.
Потенцирование
Для копотенцирования сотенных разведен ий
каждое разведение (Сп 1) представляет 1 % roто
вою препарата, и доля прибавляемоro разбавите
ля уменьшается за счет доли активных веществ [т. е.
100 % (1 % х число активных веществ)]. Такую же
процедуру применяют в соответствующих пропорци
ях при копотенцировании десятичных разведений.
01/2013:2029
МАТРИЧНЫЕ НАСТОЙКИ
ДЛЯ rОМЕОПАТИЧЕСКИХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
Тiпсtи,ае mateтae ad рrаера,аtiопеs homoeo
pathicas
ОПРЕДЕЛЕНИЕ
Матричные настойки для юмеопатических
лекарственных средств (матричные настойки)
Таблица 2371. 5
Метод 5.1.1 Метод 5.1.2 Метод 5.1.3
Базисные препараты I Водные лекарственные средства ! Тритурации
Растворы I rлицериновые мацераты и водные!
разведения из них !
Тритурации I Тритурации I
Жидкие разведения I I
Матричные настойки, чьи методы при
roтовления дают разведения 1/10 (или
1/100)
Метод 5.1.1 Метод 5.1.2
Этанол (96 %, об/об) [этанол (94 %, м/м)] Вода для инъекций
Эта нол (90 %, об/об) [этанол (86 %, м/м)] I Вода очищенная
Эта нол (80 %, об/об) [этанол (73 %, м/м)] I Сахарный сироп (сахароза, вода
Этанол (70 %, об/об) [этанол (62 %, м/м)] очищенная (64:36)
I
Этанол (50 %, об/об) [этанол (43 %, м/м)] I
Этанол (36 %, об/об) [этанол (30 %, м/м)] I
Этанол (18 %, об/об) [этанол (15 %, м/м)]
Таблица 2371. 6
Метод 5.1.3
Лактоза моноrидрат
I
I
I
I
rосударственная фармакопея Республики Беларусь
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
При возможности следует провести хотя бы
одно испытание идентичности хроматоrрафиче
ским методом.
1212
жидкие лекарственные средства, полученные
экстракцией сырья соответствующим paCTBO
рителем. Обычно используют свежее сырье,
но можно использовать и высушенное сырье.
Матричные настойки MorYT быть также получе
ны из растительных соков с добавлением или
без добавления растворителя #(эссенции)#. В
некоторых случаях экстраrируемый материал
может подверrаться предварительной обра
ботке.
ПРОИЗВОДСТВО
Матричные настойки получают мацераци
ей, настаиванием, перколяцией, ферметацией
или друrими способами, описанными в частных
статьях, обычно с использованием спирта необ
ходимой концентрации.
Матричные настойки roтовят, используя YKa
занные соотношения сырья и растворителя, с
учетом влажности сырья, если нет друrих указа
ний.
При использовании свежих растений при
меняют соответствующие методы, обеспечи
вающие их свежесть. Компетентный уполномо
ченный opraH может потребовать проведения
необходимых испытаний. подтверждающих CBe
жесть сырья.
#Эссенции изroтавливаются путем CMe
шивания сока растения со спиртом этиловым
86 % (м/м) или 90 % (м/об). В некоторых слу
чаях может быть использован спирт этиловый
друrой концентрации, что указывается в част
ной статье. Эссенции не используют для изro
товления разведений и лекарственных форм.
Из них roтовят только настойки матричные ro
меопатические. "
Матричные настойки обычно прозрачные.
При хранении может образовываться неболь
шой осадок, допускаемый при условии ОТСУТ
ствия существенноro изменения состава.
Производственный процесс должен бblТЬ
воспроизводимым.
Метод мацерации . Если нет друrих указа
ний, лекарственное растительное сырье, под
верrающееся экстракции, измельчают до частиц
необходимоro размера, тщательно перемеши
вают, экстраrируют указанным способом указан
ным растворителем и выдерживают в закрытом
контейнере указанное время. Осадок отделяют
от экстраrента и, при необходимости, отжимают.
В последнем случае обе полученные жидкости
объединяют.
Корректировка количественноrо coдep
жан ия. При необходимости может быть прове
дена корректировка содержания компонентов
либо путем прибавления экстраrента подходя
щей концентрации, либо путем прибавления
друroй матричной настойки для roмеопатиче
ских лекарственных средств растительноro или
животноro происхождения.
ИСПЫТАНИЯ
В частных статьях пределы содержания YCTa
новлены для официальных методов производ---
ства. Для каждоro определенноro метода произ
водства MOryT быть установлены свои пределы.
При проведении испытания относитель
ной плотности определение содержания эта
нола не проводят, и наоборот.
Относительная плотность (2.2.5). 3наче
ние относительной плотности матричной Ha
стойки должно соответствовать значению, YKa
занному в частной статье.
Содержание этанола (2.9.10). Содержание
этанола должно соответствовать пределам, YKa
занным в частной статье.
Метанол и 2 пропанол (2.9.11). Не более
0,05 % (об/об) метанола и не более 0,05 % (об/об)
2 пропанола, если нет друrих указаний в част
ной статье.
Сухой остаток (2.8.16). При необходимости
содержание сухоro остатка матричной настойки
должно соответствовать значениям, указанным
в частной статье.
Остаточные количества пестицидов
(2.8.13). При необходимости матричные настой
ки должны выдерживать испытание на пестици
ды. Условия считаются выполненными, если ле
карственное растительное сырье выдерживает
испытание.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
При возможности проводят количественное
определение с установлением пределов coдep
жания.
ХРАНЕНИЕ
В защищенном от света месте. Может быть
указана максимальная температура хранения.
МАРКИРОВКА
На этикетке указывают:
что продукт представляет собой матрич
ную настойку для rомеопатических лекарствен
ных средств (обозначают «ТМ» или «0»);
название сырья на латинском языке в co
ответствии с частной статьей Фармакопеи, если
таковая имеется;
метод приroтовления;
содержание в матричной настойке этанола
или друroro растворителя, в процентах (об/об);
'соотношение сырья к матричной настойке;
при необходимости условия хранения.
01/2013:2045
Лекарственное растительное сырье для 20меопатических лекарственных
1213
ЛЕКАРСТВЕННОЕ РАСТИТЕЛЬНОЕ
СЫРЬЕ дЛЯ rОМЕОПАТИЧЕСКИХ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
Р/апtае теdiсiпа/еs
homoeopathicas
ad
p,aepa,atioпes
ОПРЕДЕЛЕНИЕ
Лекарственное растительное сырье для
rомеопатических лекарственных средств
в основном цельные, измельченные или дpo
бленые растения либо части растений, в том
числе водоросли, rрибы или лишайники в
необработанном виде обычно в свежем,
иноrда в высушенном. Растительным
сырьем для rомеопатических лекарственных
средств являются также соки растений, не
подверrшиеся специальной обработке. Ha
звание растительноro сырья roмеопатических
лекарственных средств обозначается ботани
ческим научным названием исходноro расти
тельноro сырья в соответствии с биноминаль
ной системой (род, вид, тип и автор).
ПРОИ3ВОДСТВО
Растительное сырье для roмеопатических
лекарственных средств получают кулыивиро
ванием или сбором дикорастущих растений.
Для rарантии качества растительноro сырья
для roмеопатических лекарственных средств
существенными являются условия кулыиви
рования, сбора, сортировки, сушки, измель
чениf1 и хранения.
Растительное сырье для roмеопатических
лекарственных средств должно быть по воз
можности свободным от заrрязнений почвой,
пылью, мусором, а также rрибами, HaceKOMЫ
ми и друrими заrрязнениями животноro про
исхождения. В сырье должны отсутствовать
признаки rниения.
Если проводилась деконтаминация, Heoo
ходимо продемонстрировать, что компоненты
растительноro сырья не повреждены и что в
сырье не содержится вредных примесей. При
проведении деконтаминации лекарственно
ro растительноro сырья для roмеопатических
лекарственных средств запрещается исполь
зование этиленоксида.
Свежее лекарственное растительное
сырье должно быть переработано как можно
скорее после ero сбора. Если обосновано и
утверждено, в целях транспортировки или
хранения свежее растительное сырье может
быть подверrнуто заморозке; так же ero можно
хранить в 96 % этаноле или этаноле подходя
щей концентрации при условии, что для даль
нейшей переработки будет использован весь
материал, включая среду для хранения.
Должны быть приняты соответствующие
меры, обеспечивающие необходимую микро
биолоrическую чистоту roмеопатических ле
карственных средств, содержащих один или
более видов лекарственноro растительноro
сырья, соrласно рекомендациям статьи 5.1.4.
Микробиолоаическая чистота нестериЛЬНblХ
лекарствеННblХ средств и фармацевтиче
ских субстанций.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Подлинность растительноro сырья для ro
меопатических лекарственных средств опре
деляют, используя макроскопический и, при He
обходимости, микроскопический метод, а также
друrие необходимые испытания (например, TOH
кослойную хроматоrpафию).
ИСПЫТАНИЯ
При меси (2.8.2). Если для производства
roмеопатических лекарственных средств в
качестве исходноro материала используют
свежие растения, содержание посторонних
примесей в них должно быть минимальным.
При необходимости предельное содержание
этих примесей указывают в частной статье.
При использовании высушенных растений в
качестве исходноro материала для производ
ства roмеопатических лекарственных средств
необходимо проведение испытания на co
держание примесей, если нет друrих указа
ний в частной статье. Содержание допусти
мых примесей должно быть не более 2 % м/м,
если нет друrих указаний в частной статье.
Фальсификация. Растительное сырье для
roмеопатическоro применения, которое может
быть фальсифицировано, должно подверrаться
соответствующим специфическим испытаниям.
Потеря в массе при высушивании (2.2.32).
Проводят испытание по определению потери
в массе при высушивании высушенноro лекар
cTBeHHoro растительноrо сырья для rомеопати
ческих лекарственных средств.
Вода (2.2.13). Определение воды проводят
для растительноro сырья с высоким содержани
ем эфирноro масла. Определение содержания
воды в свежем растительном сырье для roме
опатических лекарственных средств проводят
подходящим методом.
Пестициды (2.8.13). Растительное сырье
для roмеопатических лекарственных средств
должно соответствовать требованиям на co
держание остаточных количеств пестицидов.
При этом учитывают индивидуальные особен
ности растения, в каком виде оно будет ис
пользоваться и, при наличии, исчерпывающие
данные по обработке данной серии расти
тельноro сырья.
1214
rосударственная фармакопея Республики Беларусь
Афлатоксины (2.8.18).
При необходимости к лекарственному pac
тительному сырью для roмеопатических лекар
ственных средств MOryT быть применены следу
ющие требования, например:
Общая зола (2.4.16).
Показатель rоречи (2.8.15).
Тяжелые металлы. Следует учитывать
риск заrрязнения растительноrо сырья для
roмеопатических лекарственных средств тя
желыми металлами. Если в частной статье
не указаны пределы содержания тяжелых Me
таллов или отдельных элементов, указание
таких пределов может иметь место в обосно
ванных случаях.
Радиоактивное заrрязнение. В HeKO
торых специфических случаях должен быть
принят во внимание риск радиоактивноro за
rрязнения.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
При необходимости про водят количе
ственное определение растительноro сырья
подходящим методом.
ХРАНЕНИЕ
Сухое растительное сырье хранят в за
щищенном от света месте.
Опечатки, допущенные в З М томе
1215
ОПЕЧАТКИ, ДОПУЩЕННЫЕ В 3..М ТОМЕ ПЕРвоrо ИЗДАНИЯ
rОСУДАРСТВЕННОЙ ФАРМАКОПЕИ РЕСПУБЛИКИ БЕЛАРУСЬ
Номер страницы, Старая редакция Новая редакция
абзаца,пункта,ра ела
стр. 146 #Микробиолоrическая #Микробиолоrическая
чистота (2.6.12,2.6.13, 5.1.4). чистота (2.6.12, 2.6.13, 5.1.4).
Аминотриптилинаrидрохлорид Аминотриптилинаrидрохлорид
в условиях испытания не в условиях испытания обладает
обладает антимикробным антимикробным действием. Для
действием. устранения антимикробноro действия
посев на питательные среды проводят
методом мембранной фильтрации.
..
стр. 166, Раствор сравнения (Ь). 10,0 Mr Раствор сравнения (Ь). 10,0 Mr ФСО
Сопутствующие ФСО артикаина примеси А и артикаина примеси А и 5.0 Mr ФСО
примеси 5,0 Mr ФСО артикаина примеси артикаина примесu Е растворяют в
Е растворяют в подвижной фазе подвижной фазе и доводят до объема
и доводят до объема 50,0 мл 100,0 мл этим же растворителем.
этим же растворителем.
стр. 187, ОПИСАНИЕ Очень леrко растворим в воде, Очень мало растворим в воде,
(СВОЙСТВА) леrкорастворим в 96 % спирте леrкорастворим в 96 % спирте
стр. 255, 257, 259, .., На титрование должно пойти ... На титрование должно пойти не
Азотсодержащие не более 0,15 мл 0,1 М раствора более 0,15 мл 0,01 М раствора
вещества кислоты хлористоводородной. кислоты хлористоводородной.
стр.269 Оптическая плотность (2.2.25). Оптическая плотность (2.2.25). Не
Не более 0,05 при 450 нм... более 0,05 при 440 нм...
стр. 363 КСИЛОМЕТАЗОЛИНА КСИЛОМЕТАЗОЛИНАrидрохлорид
rидрохлорид (# НАФТИЗИНА
rидрохлорид)
стр. 397, C5HcNcS2-Н20 CsHcNcS-Н20
МЕРКАПТОПУРИН
стр. 398, 1 мл О,1 М раствора 1 мл 0,1 М раствора
КОЛИЧЕСТВЕННОЕ твтрабутиламмония твтрабутиламмония 2идроксида
ОПРЕДЕЛЕНИЕ аидрокеида соответствует 15,22 соответствует 15,22 Mr C s H 4 N 4 S.
Mr C s H 4 N 4 S 2 .
с
стр. 403 РИСУНОК1.Ин4>ракраеный Рисунок 1. Инфракрасный спектр
спектр ПО2Лощения ФСО пропускания ФСО метилурацила в
метилурацuла в дисках с калия дисках с калия бромидам Р.
бромидом Р.
етр. 426, НАТРИЯ « «
АМИНОСАЛИЦИЛАТ I . 2Н 2 О I . 2H
диrИДРАТ N он H 2 N ОН
стр. 436, абзац 1 Нитрать.. К 10 мл... Нитриты. К 10 мл...
стр.447, ...Отношение оптической ... Отношение оптической плотности
ПОДЛИННОСТЬ плотности в максимуме при 219 в максимуме при 291 нм к оптической
(ИДЕНТИФИКАЦИЯ) нм к оптической плотности в плотности в максимуме при 305 нм: от
максимуме при 305 нм: от 0,61 0,61 до 0,73.
до 0 73.
стр. 447, ИСПЫТАНИЯ Оптическая плотность (2.2.25). Оптическая плотность (2.2.25). Не
Не более 0,60 в при 305 нм. менее 0,60 в при 305 нм.
=.....
=сп
=сп
iz
:0
=V1
=0
--...1
::.
-i:"-
o
::00
!!!ОО
1216
rосударственная фармакопея Республики Беларусь
Номер страницы,
абзаца,пункта,раздела
стр. 472, ПАРАФИН
мяrкий, ЖЕЛТЫЙ
стр. 509, ОПРЕДЕЛЕНИЕ
стр. 529
стр.530,UЦелочность
стр. 564,
Сопутствующие
при меси
стр. 572,
Сопутствующие
при меси
стр. 589,
Примесь А и друrие
сопутствующие
примеси
стр. 591, ИСПЫТАНИЯ
стр.615,
Сопутствующие
примеси
Старая редакция
#Микробиолоrическая чистота
(2.6.12, 2.6.13, 5.1.4). Парафин
мяrкий, белый в условиях
испытания не обладает
антимикробным действием.
Рибофлавина натрия фосфат
представляет собой смесь,
содержащую в качестве
основноro компонента 5' (натрия
rидрофосфат), а также друrие...
Продукты разложения белка.
К 4 мл раствора S прибавляют
10 мл воды Р И 0,5 мл раствора
100 r/л натрия сидроксида Р
и наrревают до кипения. Не
должен выделяться аммиак,
обнаруживаемый по запаху или
посинению красной лакмусовой
бумаzи Р.
. .. При прибавлении не более
1,5 мл 0,1 М раствора натрия
сидроксида окраска раствора
должна измениться на розовую.
Испытуемый раствор. 0,35 Mr
испытуемоro образца...
Предельное содержание
примесей:
примесь А: не более 0,5 %;
примесь В: не более 1,5 %;
примесь с: не более 0,5 %;
сумма примесей О и Е: не
более 0,25 %;
сумма примесей: не более
2,5 %.
Приzодность
хроматоzрафической системы:
разрешение: не менее 0,8
между пиками. ..
Тяжелые металлы (2.4.8,
метод F). Не более 0,001% (10
ррm).
Предельное содержание
примесей:
примесь А (не более 0.4%):
на xpoMaTorpaMMe испытуемоro
раствора площадь пика,
соответствующеrо при меси
А, не должна превышать 0,8
площади OCHOBHoro пика на
xpoMaTorpaMMe раствора
сравнения (Ь);
Новая редакция
#Микробиолоrическая чистота
(2.6.12, 2.6.13, 5.1.4). Парафин мяrкий,
желтый в условиях испытания не
обладает антимикробным действием.
Рибофлавина натрия фосфат
представляет собой смесь,
содержащую в качестве основноro
компонента рибофлавин 5' (натрия
rидрофосфат), а также друrие...
Продукты разложения белка. К 4 мл
раствора S прибавляют 1 О мл воды
Р и 0,5 мл раствора 100 r/л натрия
сидроксида Р и наrревают до кипения.
Не должно обнаруживаться продуктов
разложения белка ни по запаху
аммиака, ни по посинению влажной
красной лакмусовой бумаzи Р.
.. .при прибавлении не менее 1,5 мл
0,1 М раствора натрия сидроксида
окраска раствора должна измениться
на розовую.
Испытуемый раствор. 0,35 r
испытуемоro образца. ..
Предельное содержание примесей:
примесь А и с: не более 0,5 %
каждой;
примесь В: не более 1,5 %;
сумма примесей О и Е: не более
1,0 %;
любая друzая примесь: не более
0,25 % каждая;
сумма примесей: не более 2,5 %.
Приzодность хроматоzрафической
системы:
разрешение: не менее 8,0 между
пиками.. .
Тяжелые металлы (2.4.8, метод F).
Не более 0,002% (20 ррт).
Предельное содержание примесей:
примесь А (не более 0,4%): на
xpoMaTorpaMMe испытуемоro раствора
площадь пика, соответствующеro
при меси А, не должна превышать
0,8 площади основноro пика на
xpoMaTorpaMMe раствора сравнения
(а);
Опечатки, допущенные в З М томе
1217
Номер страницы, Старая редакция Новая ,,-.д... ...
абзаца,пункта,ра ела
стр. 625, ИСПЫТАНИЯ Нитрофурфураля диацетат. Нитрофурфуроладиацenп.Не
Не более 1,0 %... более 1,0 %...
... ФСО нитрофурфураля ... ФСО нитрофурфурола
диацетата.. . диацетата.. .
стр. 626, ИСПЫТАНИЯ Результаты: на xpoMaTorpaMMe Результаты: на xpoMaTorpaMMe
испытуемоro раствора пятно испытуемоro раствора пятно
соответствующее нитрофураля соответствующее нитрофурола
диацетату.. . диацетату.. .
стр. 632, Хлориды (2.4.4). Не более 0,01 % (100
ХЛОРАМФЕНИКОЛ, ррт). К 1,00 r испытуемоro образца
ИСПЫТАНИЯ прибавляют 20 мл воды Р, 10 мл
кислоты азотной Р и встряхивают
в течение 5 мин. Фильтруют
через фильтровальную бумаry
предварительно промытую путем
пропускания порций по 5 мл воды
Р до тех пор, пока 5 мл фильтрата
перестанут давать опалесценцию при
прибавлении 0,1 мл кислоты азотной
Р и 0,1 мл раствора серебра нитрата
Р1. 15 мл полученноro фильтрата
должны выдерживать испытание на
хлориды.
стр. 660, Отношение оптических Отношение оптических плотностей:
ПОДЛИННОСТЬ плотностей: Ay,/ 7 559 от 3,15 до 3,45;
(ИДЕНТИФИКАЦИЯ) Ay,/A547 559 от 3,15 до 3,45; А З6 /А П6 от 1,70 до 1,90.
6/ 76 от 5,4 до 5,7.
стр. 701 WATER PIPER HERBA* WATER PIPER HER8*
стр. 706, ЗЕМЛЯНИКИ Собранные в фазу цветения Собранные в фазу цветения и
ЛЕСНОЙ ЛИСТЬЯ и высушенные листья высушенные листья дикорастущеrо
дикорастущеro мноrолетнеrо мноroлетнеro травянистоro растения
травянистоro растения F,aga,iae F,aga,ia vesca L.
vescae L.
стр. 709 Потеря в массе при Потеря в массе при высушивании
высушивании (2.2.32). Не (2.2.32). Не более 13,0%...
более 14,0%...
стр.713 LANCELEAF THERMOPSIS LANCELEAF THERMOPSIS HERB*
HERBA*
Справочное издание
rОСУДАРСТВЕННАЯ ФАРМАКОПЕЯ
РЕСПУБЛИКИ БЕЛАРУСЬ
(rФ. РБ 11)
Общие методы контроля качества
лекарственных средств
в двух томах
Том 1
Редактор А. А. Шеряков
Компьютерный дизайн и верстка К. А. Журавский
Корректор Е. А. Кулешова
1
Подписано в печать 28.05.2012. Формат 60х84 18.
Бумаrа офсетная. rарнитура Arial.
Печать офсетная. Уел. печ. л. 163,175. Уч. изд. л. 93,7.
Тираж 1200. 3аказ 1712.
Выпущено по заказу Министерства здравоохранения Республики Беларусь
Издатель и полиrрафическое оформление:
Республиканское унитарное предприятие «Типоrрафия «Победа,,_
ЛИ 02330/0494282 от 08.04.2009. ЛП 02330/0494182 от 03.04.2009.
Ул. Тавлая, 11, 22231 О, r. Молодечно.