/















































































































































































































Text
Р ВУДВОРД, Р ХОФФМАН
СОХРАНЕНИЕ ОРБИТАЛЬНОЙ
СИММЕТРИИ
/
/
-Ч
\
\
/
/
/
\
\
\
ttft
€WWM
ПН
а»
THE CONSERVATION
OF ORBITAL SYMMETRY
R. B. Woodward
Department of Chemistry,
Harvard University
R. Hoffmann
Department of Chemistry,
Cornell University
WEINHEIM
1970
Р. ВУДВОРД, P. ХОФФМАН
СОХРАНЕНИЕ
ОРБИТАЛЬНОЙ
СИММЕТРИИ
Перевод с английского
доктора хим. наук В. А. КРОНГАУЗА
ИЗДАТЕЛЬСТВО «МИР»
Москва 1971
УДК 547.1 : 541
Книга посвящена одной из главнейших проблем
теоретической органической химии — связи между
строением молекулы и направлением ее химических
превращений. Авторы книги внесли значительный
вклад в этот вопрос, обосновав и развив
фундаментальную закономерность, управляющую ходом многих
органических реакций — правило сохранения
орбитальной симметрии. Несмотря на небольшой объем авторам
удалось охватить обширный фактический материал
по реакциям, подчиняющимся этому правилу.
Книга предназначена для научных работников —
химиков-органиков и физико-химиков,
интересующихся вопросами структурной химии.
Редакция литературы по химии
Инд.
2-5-4
66-71
ПРЕДИСЛОВИЕ
В 1965 г. Р. Вудворд и Р. Хоффман
предложили известные правила, основанные на
принципе сохранения в реакции орбитальной
симметрии. Хотя с тех пор прошло лишь немногим
более пяти лет, эти правила стали одним из
важнейших обобщений органической химии. Им
подчиняются все без исключения синхронные
реакции, к которым относятся многие реакции
перегруппировки, изомеризации и циклизации
органических соединений. Применение этих
правил не требует проведения сложных расчетов,
а основано на рассмотрении симметрии
молекул исходных веществ и продуктов реакции.
Правила Вудворда — Хоффмана позволили
объяснить, а в ряде случаев и предсказать стерео-
специфичность различных электроциклических
реакций, реакций циклоприсоединения, сигма-
тропных перегруппировок и т. п. Правила
сохранения орбитальной симметрии позволяют
также предвидеть, в каких случаях реакция идет
термически, а в каких только под действием
света.
Весь этот материал изложен в небольшой
монографии указанных авторов, изданной
одновременно в США и ФРГ. [Русский перевод сде-
лан по идентичной публикации в Angewandte
Chemie, 81, № 21, 797 (1969).]
В первой части книги изложены общие
представления, позволившие сформулировать
правила, и разобраны наиболее простые примеры.
Во второй части рассмотрены отдельные
классы химических реакций, а многочисленные
примеры, часто менее очевидные, обсуждаются на
основе представлений, развиваемых авторами.
В книге собран обширный библиографический
материал.
Книга Вудворда и Хоффмана — первое
издание, посвященное специально правилам
сохранения орбитальной симметрии в
органических реакциях. Материал изложен в форме,
доступной химикам-органикам, снабжен
выразительными схемами и, как нам кажется,
представляет несомненный интерес для широкого
круга читателей.
В. Кронгауз
6
1. ВВЕДЕНИЕ
Метод молекулярных орбиталей справедливо считают
наиболее плодотворным для химика-органика по сравнению
со всеми другими методами теоретического рассмотрения
химической связи. Тем не менее этот метод, если не считать
нескольких известных исключений, использовался главным
образом при исследовании статических свойств молекул
(в основном и возбужденном состояниях). Возможности
метода при рассмотрении реагирующих систем изучались
очень редко.
В 1965 г. в серии предварительных сообщений [1—3]
мы предложили фундаментальную идею для теоретической
трактовки всех синхронных («концертных») реакций.
История происхождения этой идеи была изложена ранее [4].
Основное утверждение состояло в том, что реакция
протекает легко, когда существует соответствие между
характеристиками орбитальной симметрии реагентов и продуктов
реакции. Если такого соответствия нет, реакция идет с
трудом. Кратко этот принцип можно сформулировать так:
в синхронных реакциях сохраняется орбитальная
симметрия. Этот вывод вызвал значительный интерес, выдержал
ряд проверок; в настоящее время уже известны его
применения и основанные на нем предсказания, которые были
подтверждены. В предлагаемой работе мы развиваем наши
взгляды несколько подробнее, рассматриваем ряд
достижений; как наших, так и других авторов, за три года после
публикации первых работ, а также высказываем некоторые
новые предположения.
7
2. ОРБИТАЛИ И ХИМИЧЕСКАЯ СВЯЗЬ
Здесь целесообразно рассмотреть некоторые
элементарные аспекты молекулярно-орбитальной теории химической
связи*. Молекулярные орбитали строятся в виде
комбинаций атомных орбиталей, а затем заселяются электронными
парами. Если комбинируют две эквивалентные атомные
орбитали xi и Х2> т0 они всегда дают связывающую
комбинацию и соответствующую разрыхляющую орбиталь (1).
Связывающая комбинация в этом случае
характеризуем
ется положительным перекрыванием и концентрированием
электронной плотности между ядрами. В
противоположность этому антисвязывающая (разрыхляющая)
комбинация обнаруживает отрицательное перекрывание и узел
между ядрами на поверхности наибольшей электронной
плотности. Если Xi и Х2— s-орбитали, то связывающая
комбинация имеет вид Xi + Ъу а разрыхляющая
комбинация xi— Х2-
О О *i + хг
О • *i - *2
Если xi и Х2—р-орбитали, взаимодействующие с
образованием а-связи и ориентированные так, как показано на
схеме (2)**, то связывающей комбинацией будет Xi+Хг>
а разрыхляющей xi— Х2-
*о о*
(2)
+ В дополнение к классической книге Коулсона [5] мы
рекомендуем работу [5а].
** Здесь и далее незачерненные части электронного облака
означают положительный знак волновой функции, зачерненные—
отрицательный.— Прим. перев.
8
^O 0^*i + *z
фо ■ ^о
(3)
Однако следует подчеркнуть, что если базисные
атомные орбитали произвольно ориентированы первоначально
каким-либо другим способом, например, как показано на
схеме (3), (/2=—х\), то в этом случае связывающая
комбинация xi—Xz и разрыхляющая /i+ Хъ -
Необходимо также иметь в виду, что умножение всей волновой
функции на —1 не влияет на энергию состояния. Например,
перекрывание двух отрицательных лопастей эквивалентно
перекрыванию положительных лопастей, и —xi—Х2
будет точно такой же связывающей орбиталью, как и xi+ Хл-
Описание а-связей углеводородов простое.
Образование каждой химической связи приводит к появлению
а- и а*-орбиталей. В случае связей С—Н и С—С такими
орбиталями являются
w с-с
■G-H ~\^ ^ —О^О*
о<
:-н""^00
с-н ™^AJ бг
-с-*<Х>+
с
Молекулярные орбитали изображаются на схемах как
результат перекрывания двух гибридизованных орбиталей
неспецифической гибридизации. Нужно подчеркнуть, что
это намеренное преувеличение, мнемонический прием;
единственной существенной чертой а-орбитали является ее
приблизительная цилиндрическая симметричность
относительно линии связи. Таким образом, электронная плот-
9
ность сконцентрирована в пространстве между ядрами и
между атомами нет узловой плоскости.
С этой точки зрения простое описание молекулы цикло-
бутана дает четыре а-уровня С—Си восемь а-уровней С—Н,
причем каждому уровню соответствует уровень а* (4).
аС~€
°"ос
(4)
ls-Орбитали атома углерода не рассматриваются. Каждый
из а-уровней заселен двумя электронами.
Спектроскопические исследования показывают, что промежуток между
заселенными и незаселенными уровнями составляет ~10 эв.
Построенные орбитали являются частично
локализованными. Они делокализованы только на двух атомах.
Такие орбитали пригодны для анализа некоторых свойств
молекул, определяющихся полностью заселенными
молекулярными орбиталями (длина связей, энергии, дипольный
момент), но для описания молекулы в целом они не
подходят. В общем случае орбитали делокализованы в
соответствии с полной симметрией молекулы. Для обсуждения
физических свойств, зависящих от двух характерных
молекулярных орбиталей (например, спектры или ионизация),
абсолютно необходимо строить эквивалентные
делокализованные орбитали. Более подробно это будет рассмотрено
в разд. 3.
Кроме а-связей, органические молекулы могут иметь
делокализованные я -орбитали. Например, электронную
структуру этилена можно описывать с помощью четырех
а-связей С—Н и одной а-связи С—С (5).
10
(5)
Каждой из этих связей соответствуют уровни о и а*. На
а-уровнях расположены пять пар электронов. Остаются два
электрона и две атомные р-орбитали, перпендикулярные
плоскости молекулы (6). р-Орбитали комбинируют с обра-
(6)
зованиемя- и я*-орбиталей, отличающихся отсутствием
или наличием узла между атомами (рис. 1).
Существуют две независимые операции симметрии,
которые могут быть использованы для классификации этих
орбиталей: отражение в зеркальной плоскости т,
перпендикулярной плоскости молекулы и рассекающей молекулу
пополам, и поворот вокруг оси вращения второго
порядка С2, проходящей через центр связи углерод—углерод.
Следует подчеркнуть, что свойства симметрии орбиталей
молекулы этилена при каждой из указанных операций
симметрии противоположны. Так, я-орбиталь симметрична (S)
относительно отражения в зеркальной плоскости т и
антисимметрична (А) относительно вращения вокруг оси С2.
Перекрывание двух 2р^-орбиталей значительно меньше,
чем перекрывание при а-взаимодействии, и, таким
образом, я-связь слабее, чем а-связь, а уровни я и я* находятся
выше и ниже уровней с и о* соответственно (рис. 1).
Молекулярные я-орбитали трехорбитальной аллильной
системы показаны на рис. 2. Следует обратить особое
внимание на расположение узлов. Все я-орбитали
антисимметричны по отношению к отражению в плоскости
аллильной системы. Нижняя орбиталь, которая в аллильном
катионе дважды заселена, не имеет узловых точек. Средняя,
несвязывающая орбиталь, которая в аллильном радикале
заселена одним электроном и в анионе — двумя
электронами, имеет одну узловую плоскость, что приводит к исклю-
11
т Со
ы
A S
о
и
Рис. 1. Молекулярные орбитали этилена и свойства симметрии
тс- и т:*-орбиталей.
Горизонтальные линии указывают относительные энергии орбиталей.
12
m Co
S A
A S
S А
с 2. Молекулярные тс-орбиталн аллильноп
m C9
A S
S A
A S
S A
Рис. З. Четыре молекулярные u-орбитали бутадиена в s-
цас-конфигурации.
14
чению участия атомной 2р-орбитали центрального атома
в этой молекулярной орбитали. Орбиталь с еще большей
энергией имеет две узловые плоскости.
Молекулярные орбитали четырехорбитальной
молекулы бутадиена показаны на рис. 3, где изображена s-цис-
структура*'** . Вновь обратим внимание на узловые точки
и альтернирующие свойства симметрии. Корреляция между
высотой энергетического уровня и увеличением числа узлов
не случайна, а является общим следствием классической
и квантовой механики. Огибающие орбиталей полиенов
совпадают с кривыми, описывающими волновую функцию
частицы в одномерном ящике (7). Низшая по энергии ор-
(7)
биталь не имеет узлов, следующая, более высокая, орбиталь
имеет один узел, следующая за ней — два узла и т. д.
Наиболее высокой по энергии орбитали соответствует
наибольшее возможное число узлов. Общее выражение для k-й
молекулярной орбитали полиеновой или полиенильной
* Здесь и в дальнейшем изложении молекулярные орбитали
выражены через атомные орбитали, взаимодействие которых дает
истинные молекулярные орбитали. Ввиду того что, как правило,
нас интересует только направление атомных орбиталей, мы
пренебрегаем тем обстоятельством, что коэффициенты при атомных
орбиталях разные и поэтому различны относительные вклады
атомных орбиталей в молекулярную орбиталь.
** Электронная структура полиенов является, по-видимому,
наиболее разработанным разделом полуэмпирической теории
молекулярных орбиталей. Очень хорошим пособием по этому вопросу
служит книга Салема [7] (см. также [7а]).
15
системы с п атомами углерода можно записать в виде
wk = 2 с
*1ф1>
: = 1
где Фг—
тельно с
нием [7J
атомные орбитали, пронумерованные последова-
одного конца. Коэффициенты задаются выражс-
С
V4
Tiki
sin
+ 1 "'" /г + 1
Орбитали альтернируют по симметрии с увеличением
энергии.
Если п — четное число, то существует я/2 связывающих
и я/2 разрыхляющих я-орбиталей. Нечетному я
соответствуют (я — 1)/2 связывающих, (я — 1)/2 разрыхляющих
орбиталей и одна несвязывающая орбиталь.
Наконец, важен тот факт, что ни одна молекулярная
орбиталь не может быть в одно и то же время симметричной
и антисимметричной по отношению к любому из
имеющихся элементов симметрии. Например, орбиталь (8)
симметрична относительно вращения на 180°, если рассматривать
атомы 2 и 3, но антисимметрична, если рассматривать
атомы 1 и 4. Поэтому такая орбиталь неприемлема в качестве
молекулярной орбитали.
3. КОРРЕЛЯЦИОННЫЕ ДИАГРАММЫ
Впервые построенные в начале 30-х годов Гундом и
Малликеном [8] двухатомные корреляционные диаграммы
разделенные атомы — объединенный атом сыграли важную
роль в теоретической химии. Такие диаграммы
представляют процесс сближения двух атомов из бесконечности.
Справа располагают энергетические уровни разделенных
атомов в порядке увеличения энергии. Затем следует рас-
смотреть процесс сближения атомов, при котором
расстояние между ними уменьшается от физически разумного
молекулярного размера до такого расстояния (реально
невозможного), при котором происходит слияние ядер.
Энергетические уровни образующегося объединенного атома
также известны; их располагают слева на диаграмме. Далее
классифицируют орбитали первоначально разделенных
атомов и объединенного атома по симметрии, сохраняющейся
в ходе гипотетической реакции. Уровни с одинаковой
симметрией соединяют, принимая во внимание квантовомеха-
ническое правило непересечения [пересекаться могут
только уровни неодинаковой симметрии (рис. 4)].
Таким образом, исходя из сравнительно хорошо
известной структуры уровней отдельных атомов и объединенного
атома, можно получить ценную информацию о структуре
уровней промежуточного образования, соответствующего
Is lag _^_-———"""'"
Объединенный Разделенные
атом атомы
Рис. 4 Пример атомной корреляционной диаграммы.
17
молекуле. Именно такого вида диаграмма объясняет
существование молекулы кислорода в основном состоянии
в виде триплета.
Аналогично можно построить корреляционную
диаграмму для синхронной реакции, например для реакции цикло-
присоединения. Слева на диаграмме изображают
приблизительно известные уровни энергии реагентов, справа —
уровни энергии продуктов реакции. Принимая
определенную геометрию сближения, можно классифицировать
уровни на обеих сторонах диаграммы по симметрии,
сохраняющейся во время сближения, а затем соединить уровни с
Рис. 5. Сближение двух параллельно расположенных молекул
этилена.
одинаковой симметрией. Такая молекулярная
корреляционная диаграмма дает ценную информацию о
промежуточной области, которая в данном случае представляет
собой переходное состояние реакции.
Ниже проиллюстрированы некоторые детали построения
молекулярной корреляционной диаграммы. В качестве
первого примера выбрано сближение двух молекул этилена
с сохранением максимальной симметрии, которое приводит
к образованию циклобутана (рис. 5). Обычно при
теоретическом рассмотрении максимальное понимание проблемы
достигается путем наиболее возможного упрощения
картины с одновременным сохранением ее наиболее
существенных физических черт. В данном случае корреляционная
диаграмма строится только из четырех орбиталей —я-ор-
биталей двух молекул этилена. В ходе реакции эти четыре
я-орбитали превращаются в четыре а-орбитали циклобу-
тана. а-Связи С—Н и С—С этиленового скелета можно
исключить из рассмотрения, так как в ходе реакции
меняется лишь гибридизация этих связей, а число,
приблизительное расположение по энергии и, что особенно важно,
симметрия не изменяются.
Первым шагом при построении корреляционной
диаграммы является выделение существенных связей и
расположение их в соответствии с приблизительными величина-
п
с=с __*, с-с
с=с с-с
Рис. 6. Уровни энергии важнейших орбиталей при образовании
циклобутана из двух параллельно расположенных молекул этилена.
ми энергетических уровней реагентов и продуктов.
Результат для рассматриваемого случая показан на рис. 6, где
пунктирная горизонтальная линия означает несвязывающий
уровень — приблизительную величину энергии электрона
на 2р-орбитали свободного атома углерода. Уровни а и а*
разделены большим промежутком, чем уровни я и я*.
Хотя в определении энергии уровней и есть некоторый
смысл, важно иметь в виду, что это не имеет существенного
значения для дальнейшей аргументации. Для того
чтобы установить определенный масштаб вертикальной
энергетической шкалы, можно принять, что промежуток
между уровнями я и я* равен ~5 эв [7].
19
Следующий шаг — построение молекулярных орбиталеи
для реагентов и продуктов реакции. Здесь целесообразно
уделить некоторое внимание построению молекулярных
орбиталей реагирующих систем. Рассмотрим s-mpaнc-бyта-
диен. Один из путей построения четырех молекулярных
орбиталей бутадиена — рассмотрение взаимодействия всех
четырех атомных орбиталей, как показано слева на
схеме (9). Другой путь — рассмотрение бутадиена как обра-
(»)
зования, возникающего в результате взаимодействия двух
частично локализованных двойных связей. Рассмотрим
связывающие я -орбитали двух молекул этилена. В то время
как функции Я1 и я2 удовлетворительно описывают
изолированные двойные связи, они не подходят в качестве
молекулярных орбиталеи бутадиена. Молекулярные орбитали
должны быть либо симметричными, либо
антисимметричными по отношению к любому элементу молекулярной
симметрии. В данном случае главной операцией симметрии
является вращение на 180° вокруг оси второго порядка.
Очевидно, удовлетворять условиям симметрии будут
комбинации Я1± я2 (Ю). Естественно, что они в свою очередь
топологически идентичны двум низшим по энергии орбита-
лям бутадиена, которые можно построить при
рассмотрении прямого взаимодействия всех четырех атомных
орбиталей.
■ ЯТ-Я2
20
Я1
^1 + ^2
Оо)
м
8-я
B-S
^1~^2
Теперь можно обсудить аналогичную проблему четырех
молекулярных орбиталей двух сближающихся молекул
этилена. Диаграмма // (сечение орбиталей в плоскости 3,
ср. рис. 5) представляет локализованные я-связи двух
молекул этилена. Однако эти орбитали не пригодны в качестве
орбиталей комплекса из двух молекул этилена: они не
симметричны и не антисимметричны при отражении в
плоскости 2. И в этом случае подходящими комбинациями будут
Я1~ЬЯ2 И Я1 Я 2 ( 12) •
ь
i
i
i
(//)
21
1
i
ях + яг\ SS
{12)
Я\-Хг\ SA
Комбинация я1+я2 симметрична относительно
отражения в обеих плоскостях 1 и 2 (сокращенно обозначается
SiSg, или SS); комбинация я 1—я2 симметрична при
отражении в плоскости 1 и антисимметрична при отражении в
плоскости 2 (SiAg, или SA)*. Обе орбитали симметричны по
отношению к отражению в плоскости 3. При большом
расстоянии между молекулами этилена уровни Я1+Я2ИЯ1— я 2
вырожденны, однако при малом расстоянии я 1+я2
находится ниже по энергии, чем Я1—я2, так как первая
орбиталь имеет меньше узлов. Подобная комбинация
разрыхляющих молекулярных орбиталей для комплекса
показана на схеме (13).
ь
i
i
t
1
*
а
i
i
л* + л£\ AS
03)
Я*-л£\ A A
Аналогичным образом необходимо проанализировать
молекулу циклобутана. Локализованные а-связи (14) не
* Разумеется, можно ввести обозначения уровней в
соответствии с их симметрией, отвечающей симметрии сближения D2h-
Мы намеренно ввели обозначения S (симметричный) и А
(антисимметричный), так как узловые свойства орбиталей наиболее различны.
22
1
t
(H)
удовлетворяют всем операциям симметрии молекулы цикло-
бутана, поэтому необходимо взять две комбинации ai±a2
(75); так же рассматриваются разрыхляющие а*-орбита-
ли (16).
Gx+ 6Z; SS
(/5)
бх-бг\ AS
G*+G2*;SA
{16)
a*-a*; A A
Учитывая все вышесказанное, можно выяснить
корреляцию орбиталей реагентов с орбиталями продуктов (рис. 7).
Направление изменения различных уровней можно
определить без детального расчета путем выяснения в каждом
случае, является ли данный уровень связывающим или
разрыхляющим при движении вдоль координаты реакции; при
этом нужно иметь в виду определение связывания или
разрыхления. Рассмотрим две молекулярные орбитали
молекулы водорода, образующиеся из двух ls-орбиталей (рис. 8).
о g— связывающая орбиталь, так как заселяющие ее
электроны находятся в пространстве между ядрами; аи—
разрыхляющая орбиталь с более высокой энергией, так как
наличие узла между ядрами изолирует заселяющие
орбиталь электроны (которые находятся в пространстве
отдельных ядер). Существует и другой аспект выявления
связывания или антисвязывания: электроны, находящиеся на
связывающей орбитали, сближают ядра друг с другом
(dEldR> О для og), а электроны, находящиеся на
разрыхляющей орбитали, раздвигают ядра (dE/dR<.0 для au)-
Возвращаясь к корреляционной диаграмме (рис. 7), можно
видеть, что низший уровень SS системы из двух молекул
этилена является связывающим в области сближения двух
молекул этилена, т. е. система стабилизируется при
взаимодействии. Уровень SA имеет узел и, следовательно,
является разрыхляющим в области сближения двух молекул.
При больших расстояниях между реагирующими
молекулами это взаимодействие несущественно, однако при
уменьшении расстояния орбиталь SA дестабилизуется и
сдвигается в область более высоких энёргийТ Аналогично
разрыхляющая я*(А5)-орбиталь становится связывающей в
области, где молекулы сблизились. Эта орбиталь будет
стабилизоваться в ходе реакции, в то время как я*(АА)-ор-
биталь будет дестабилизоваться. Для циклобутана а-уровни
SS и AS являются связывающими в области, где происходит
разрыв циклобутанового кольца. Таким образом, эти связи
препятствуют движению вдоль координаты реакции, так как
они при этом дестабилизуются. Однако а*-уровни SA и АА
являются разрыхляющими вдоль по координате реакции и,
следовательно, сдвигаются в область более низких энергий
при разрыве кольца циклобутана. Полная корреляционная
диаграмма, в которой уровни одинаковой симметрии
соединены между собой (рис. 9), подтверждает эти выводы.
Наиболее отчетливая и удивительная особенность этой диаграм-
24
я* AA«
л* AS"
■АА б*
■SA б4
8-8 8-8
8-8 j-g
ут SA .
.7 SS ■
Л5 б
■SS CJ
» О
Р и с. 7. Связь между орбиталями исходных веществ и продуктов реакции при образовании
циклобутана из двух молекул этилена.
мы — корреляция связывающего уровня реагентов с
разрыхляющим уровнем продукта реакции и наоборот.
Теперь мы приблизились к главному принципу нашей
трактовки синхронных реакций. Если орбитальная
симметрия сохраняется, два основных уровня молекул этилена
не могут комбинировать, давая в синхронной реакции
основное состояние циклобутана. Точно так же циклобутаи
не может распасться на две молекулы этилена в результате
синхронной реакции через переходное состояние, имеющее
Е
*- R
X о о
Р и с. 8. Образование связывающей (а^.) и разрыхляющей (аи)
молекулярных орбиталей молекулы водорода из двух ls-орбиталей.
принятую здесь симметрию. Другими словами, для
рассматриваемой реакции существует в обоих направлениях
большой барьер, обусловленный симметрией. В то же время
такого барьера нет для реакции двух молекул этилена,
в одной из которых электроны переведены на низшую
разрыхляющую орбиталь путем фотохимического возбуждения.
Поэтому реакции первого типа мы назвали запрещенными
по симметрии, а реакции второго типа — разрешенными по
симметрии.
Дальнейшую ясность в этот вопрос можно внести,
рассматривая диаграмму состояния реакции (рис. 10). Основное
состояние электронной конфигурации из двух молекул
этилена коррелирует с дважды возбужденным состоянием
молекулы циклобутана, имеющим очень высокую энергию.
В свою очередь основное состояние молекулы циклобутана
коррелирует с дважды возбужденным состоянием двух мо-
26
ААб*
SAe*
я-*АА
я* AS
Я S-S ...,._
-^ Р и с. 9. Полная корреляционная диаграмма образования циклобутана из двух молекул
этилена.
лекул этилена. Взаимодействие электронов делает
невозможным пересечение линий, соединяющих уровни
одинаковой симметрии, и приводит к тому, что основное
состояние коррелирует с основным состоянием. В физически
реальной ситуации реакция должна «уплатить цену», вы-
Конфигурация Симметрия Симметрия Конфигурация
[положение состояния состояния (положение
уровней) уровней)
Рис. 10. Электронная диаграмма образования циклобутапа из
двух молекул этилена. Симметрия состояния определяется
произведением символов симметрии отдельных электронов в
соответствии со следующими правилами [7]:
SXS->S«-AXA,
S X А -> А <-А X S.
Рассматриваются лишь синглетные возбужденные состояния Как в исходных
молекулах, так и в продуктах эти орбитали можно считать вырожденными.
ражающуюся в энергии активации, за то, чтобы избежать
«предназначенного» ей пересечения. Порядок величины
энергетического барьера, обусловленного симметрией, при
синхронной комбинации двух молекул этилена можно
оценить, учитывая энергию, необходимую для того, чтобы
поднять два связывающих электрона с занятой связываю-
28
щей орбитали на разрыхляющую орбиталь ~ 5 эв
(115 ккал/моль).
Низшее возбужденное состояние двух молекул
этилена — конфигурация (SS)2(SA)1(AS)1 — непосредственно
коррелирует с первым возбужденным состоянием молекулы
циклобутана. Таким образом, для такого превращения не
существует барьера, обусловленного симметрией. Этот факт
определяет соответствующее направление многих
фотохимических превращений. Надо подчеркнуть, что есть некоторая
неопределенность при реакциях тех возбужденных
состояний, которые не имеют соответствия с простейшими
термическими состояниями. Например, реагирующее химически
возбужденное состояние может не быть тем состоянием,
которое возникает при первичном возбуждении. В
частности, синглет-триплетное расщепление для различных
возбужденных состояний так широко изменяется, что
симметрия низшего синглета и низшего триплета может быть
разной. Кроме того, безызлучательная дезактивация может
быть настолько эффективной, что химические изменения,
следующие за облучением, могут происходить в результате
колебательного возбуждения основного состояния.
Наконец, образование переходного состояния в данной
синхронной реакции может конкурировать с релаксацией
компонента, находящегося в возбужденном состоянии, до
равновесной конфигурации. Это делает реакцию невозможной
из геометрических соображений. Следует указать, что ни
одна из вышеприведенных оговорок не делает контроль по
орбитальной симметрии недействительным. Принцип
сохранения орбитальной симметрии остается в силе при условии,
что известно возбужденное состояние, участвующее в
химической реакции. Если состояние продукта реакции,
непосредственно коррелирующее с состоянием реагентов, имеет
более высокую энергию, чем состояние реагентов, то это
изменяет формулировку правила сохранения орбитальной
симметрии, хотя остается еще много неясного в физической
природе процессов, сопровождающих энергетический
каскад от электронно-возбужденного состояния к
основному. _
Многие корреляционные диаграммы резко отличаются
от диаграммы реакции этилен — этилен. Рассмотрим,
например, прототип реакции Дильса — Альдера [4+2]-цик-
лоприсоединение бутадиена к этилену. Наиболее разумный
анализ симметрии системы можно провести с помощью од-
29
ной плоскости симметрии, секущей оба компонента
(рис. 11).
Наиболее важные шесть уровней, участвующие в
данном случае в реакции, изображены на корреляционной
диаграмме (рис. 12). Форма четырех орбиталей бутадиена
и двух орбиталей этилена очевидна. Мы расположили
я-уровни этилена между двумя связывающими орбиталями
диена, однако порядок расположения не очень существен.
Комбинации делокализующихся а-связей продукта
реакции — циклогексена следует строить так, как и для ци-
клобутана.
Рис. 11. Симметричное сближение молекул бутадиена и этилена
при реакции Дильса—Альдера.
Различие между этой корреляционной диаграммой и
диаграммой для комбинации из двух молекул этилена очень
велико. В данном случае, во-первых, каждый связываю;
щий уровень реагентов коррелирует со связывающим
уровнем продукта и, во-вторых, нет корреляций, которые
пересекают большие энергетические разрывы между
связывающими и разрыхляющими уровнями.
Так же, как и в приведенных выше примерах, можно
построить диаграмму состояния (рис. 13). Уровни основного
состояния прямо коррелируют между собой. Диаграмма
показывает, что термический процесс, разрешенный по
симметрии, должен проходить без энергии активации.
Действительно, барьера, обусловленного симметрией, нет, но
экспериментально найдена величина энергии активации
(~20 ккал/моль). Энергия активации не связана прямо с
сохранением орбитальной симметрии. К факторам, обуслов-
30
ХГ А
8-8
тс* А
X* S
*2A —
8-8 —
X S-
■ S л-
-A (j2
' S 6.
и
Рис. 12. Корреляционная диаграмма для реакции Дильса—Альдера между
бутадиеном и этиленом.
Конфигурация Симметрия
[положение состояния
уровней)
(Xi)2(rr)l(X2)2(**)1 A
(Xi)2(jr)I(X2)z(X3*)1 S
<2:
(Xi)2(rr)2(X2)1(X3*)1 A
Симметрия Конфигурация
состояния (положение
уровней)
A (T^fa/OoW)1
A (ox)2la2)2(*)l(of)1
A WHoiftwft**)1
5 (?x)2(o2)2(")l(of)1
(ci)2(72)2(t)1(t*)1
(Xi)V)2(X2)2 S
s (cmWm2
P и с. 13 Электронная диаграмма состояния для реакции Дильса —
Альдера между бутадиеном и этиленом.
ливающим появление энергии активации, относятся
изменение энергии, сопровождающее регибридизацию на тех
уровнях, которые мы не рассматривали, увеличение или
уменьшение длин связей, а также искажение валентных
углов.
Первое возбужденное состояние диен-этиленового
комплекса не коррелирует с первым возбужденным состоянием
(я->я*) циклогексена. Следовательно, в этом случае
возникает обусловленный симметрией барьер для процессов,
протекающих в возбужденном состоянии. Это обстоятельство,
по-видимому, легче понять, если представить, что первое
возбужденное состояние образуется путем перевода
электрона с орбитали, энергия которой уменьшается вдоль
координаты реакции, на орбиталь с увеличивающейся
энергией.
Интуитивно физические корреляции некоторых уровней
могут показаться неправильными, однако они реально
существуют и их можно установить, если тщательно
проанализировать систему. Например, может показаться
странным, что я-уровень этилена переходит в я-уровень
циклогексена. Это соответствие можно объяснить, рассматривая
взаимодействие я-уровня этилена с двумя другими
симметричными уровнями — низшим занятым и низшим
незанятым уровнями диена. В данном случае применимо важное
общее правило квантовой механики: если взаимодействуют
два уровня неодинаковой энергии, то уровень с меньшей
энергией, смешиваясь с волновой функцией более высокой
энергии, становится более «связывающим», а более высокий
уровень, включающий в себя «примесь» более низкого,—
более разрыхляющим. Если взаимодействуют более чем
два уровня, то их смешение можно проанализировать как
суперпозицию таких парных взаимодействий*.
Применение этого правила можно проиллюстрировать на примере
образования а-связи С—Н из гибридной 5/?3-орбитали
углерода и ls-орбитали водорода (17).
В случае бутадиен + этилен я-уровень олефина,
примешивая хг уровень диена, становится более
разрыхляющим, а примешивая уровень ^з,— более связывающим (18).
* Это правило вытекает непосредственно из теории возмущений
и корреляций более высокой энергии с увеличивающимся числом
узлов волновой функции.
2—1556
33
ес-н
о ^у
•о
V
(17)
^-"•ОО
При этом диеновые л-орбитали исчезают у атомов С-1 и С-4
бутадиена и усиливаются у атомов С-2 и С-3. Таким
образом, в переходном состоянии эти орбитали находятся по
существу наполовину на одной и наполовину на другой
реагирующей частице.
и и П
(18)
Если рассмотреть другие случаи реакции циклопрщю-
единения системы из т я-электронов к системе из п
я-электронов с образованием двух новых а-связей (во время
реакции сохраняется плоскость симметрии), то становится
очевидным, что существует только два типа корреляционных
диаграмм:
а. Диаграммы, подобные диаграмме для реакции
Дильса — Альдера, с отсутствием корреляции между
связывающими и разрыхляющими уровнями. Для этого случая
характерно, что превращения разрешены по симметрии
в основном состоянии и запрещены по симметрии в
возбужденном состоянии.
б. Диаграммы, подобные диаграмме для реакции
этилен + этилен, в которых проявляется корреляция связы-
34
вание — разрыхление. Следовательно, превращение
запрещено по симметрии в основном состоянии и разрешено
по симметрии в возбужденном состоянии.
Чтобы сформулировать общее правило, следует
подсчитать, например, связывающие симметричные уровни
реагентов и продуктов реакции. Допустим, реагенты
имеют т я-орбиталей; если т/2 — четное число, то будет т/4
симметричных связывающих я-орбиталей; если т/2—
нечетное число, то таких орбиталей будет (т+2)/4. Продукт
содержит (т—2) я-орбиталей, из которых, если т/2 —
четное число, т/4 — симметричные связывающие; если
т/2 — нечетное число, то связывающих симметричных
орбиталей будет (т—2)/4. Различают три возможных случая
(<7i и q2= 1, 2, 3 ...):
1
2
3
т
4<7i
4<7i + 2
4^i+2
п
4?2
4<72
4?2 + 2
Общее число симметричных
связывающих тс-уровней
до реакции
?1 + ?2
<7l + ?2 + 1
?1 + ?2 + 2
после реакции
Яг + Я2
qi + q?,
Один из новых связывающих а-уровней продукта
реакции всегда симметричный. Таким образом, для термической
реакции, разрешенной по симметрии, общее число занятых
симметричных связывающих я-уровней реагентов на
единицу больше числа этих уровней продукта. Этому
условию удовлетворяет случай 2:
т + п = 4ft + 4</2 + 2 = 4</ + 2 .
Случаи 1 и 3 приводят к корреляции связывание —
антисвязывание, следовательно, термические реакции при этих
условиях запрещены по симметрии
т +п = 4qi + 4g2 или 4qt + 4g2 + 4 = Aq .
Конечно, в каждом случае для реакций в возбужденных
состояниях эти правила обращаются. Кроме того, следует
9 1=
35
подчеркнуть, что эти выводы применимы только, когда
геометрия сближения реагирующих молекул отвечает
указанным выше условиям. В дальнейшем мы покажем, что
при изменении геометрических условий в переходном
состоянии иногда применимы те же правила.
3.1. Общие замечания к построению
корреляционных диаграмм
Решающий этап в построении корреляционной
диаграммы — идентификация включаемых в нее о- и я-уровней и
их делокализация до степени, диктуемой симметрией
переходного состояния. В простейшем случае процедура
построения диаграммы состоит в следующем.
а) Идентификация всех включаемых орбиталей а, я
или п (несвязывающих пар). Следует иметь в виду, что
каждому о- и я-уровню соответствует а*- и я*-уровень, но
для я-орбиталей это не так. Например, для реакции
выделения окиси углерода из циклопентенона необходимо
учитывать следующие орбитали: в циклопентеноне (19) —
я- и я*-уровни связи С=С, два связывающих а-уровня
связи С—С(О) и соответствующие а*-орбитали и свободная
пара на атоме кислорода; в продуктах (20) и (21) — четыре
диеновых я-уровня, два из которых связывающие, новую
связь СО и соответствующую ей я*-орбиталь, а также
новую орбиталь несвязывающей пары электронов на
углеродном атоме окиси углерода. Другую свободную пару
электронов на кислороде и оставшуюся я-орбиталь С=0
исключают из рассмотрения как несущественные. Правильность
построения корреляционной диаграммы определяется
следующим правилом: число уровней каждого типа симметрии
справа на диаграмме должно быть равно числу уровней
слева.
**"
Ч
(19) (20) (21)
б) Если в реакции участвует пол неновая система, то
следует использовать существующие полиеновые молеку-
36
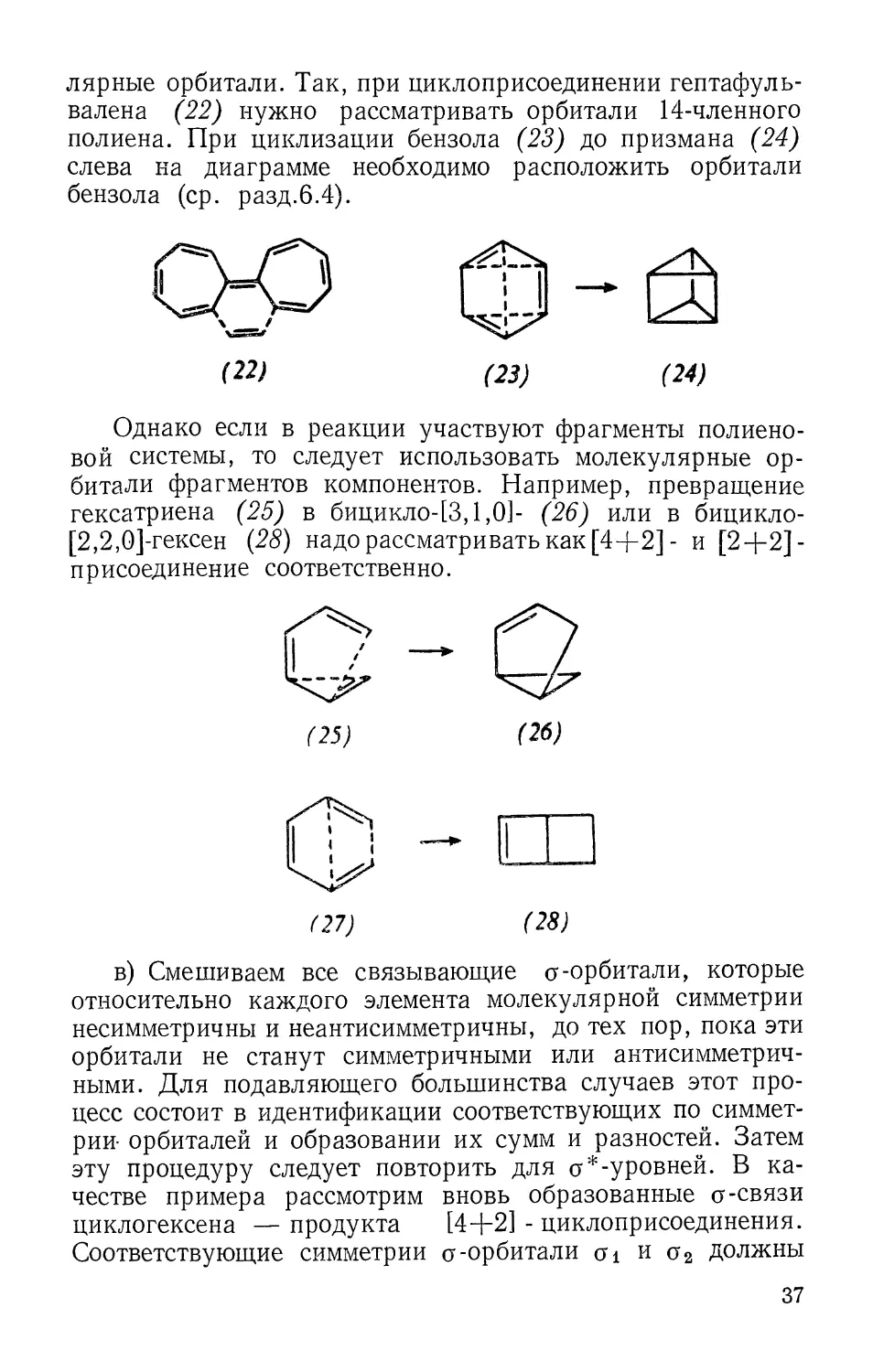
лярные орбитали. Так, при циклоприсоединении гептафуль-
валена (22) нужно рассматривать орбитали 14-членного
полиена. При циклизации бензола (23) до призмана (24)
слева на диаграмме необходимо расположить орбитали
бензола (ср. разд.6.4).
^
ш
(22)
(23)
(24)
Однако если в реакции участвуют фрагменты полиено-
вой системы, то следует использовать молекулярные
орбитали фрагментов компонентов. Например, превращение
гексатриена (25) в бицикло-[3,1,0]- (26) или в бицикло-
[2,2,0]-гексен (28) надо рассматривать как [4+2]- и [2+2]-
присоединение соответственно.
(28)
в) Смешиваем все связывающие а-орбитали, которые
относительно каждого элемента молекулярной симметрии
несимметричны и неантисимметричны, до тех пор, пока эти
орбитали не станут симметричными или
антисимметричными. Для подавляющего большинства случаев этот
процесс состоит в идентификации соответствующих по
симметрии- орбиталей и образовании их сумм и разностей. Затем
эту процедуру следует повторить для а*-уровней. В
качестве примера рассмотрим вновь образованные а-связи
циклогексена —продукта [4+2] - циклоприсоединения.
Соответствующие симметрии а-орбитали о\ и а2 должны
37
быть преобразованы в в[+^2 и <3\—сг2> как показано на
схеме (29).
(29)
(30)
Рассмотрим а*-орбитали циклопентенона (30). Для
несопряженных я-уровней необходимо аналогичное смешение
орбиталей. Так, при рассмотрении реакции [2+2]-цикло-
присоединения 1,5-гексадиена, приводящей к бициклогек-
сану, я-орбитали строятся, как показано на схеме (31).
Л< Л fig Х\-Яъ
г) Следующим удобным, однако не обязательным шагом
является смешение рассматриваемой орбитали со всеми
другими орбиталями данной симметрии. Рассмотрим, на-
38
пример, а-связи при [2+2+2]-циклораспаде циклогексана
до трех молекул этилена. Допустим, молекула
циклогексана имеет конформацию ванны. В этом случае имеем
локализованные а-орбитали а и а2 и аз (32). Единственным
Т) р Y?
<5Х а2 а,
№)
элементом симметрии в переходном состоянии является
плоскость, секущая а4. Орбиталь сч симметрична
относительно отражения в этой плоскости, а а2 и а3
несимметричны. В соответствии с пунктом в) следует образовать делока-
лизованные комбинации а2+аз и а2—аз- Это дает новый
набор орбиталей, связанных с данным элементом
симметрии (33). Теперь орбитали а и а2+аз симметричны
относительно плоскости симметрии и их удобно делокализовать
далее, беря их сумму и разность (34). Дальнейшая дело-
кализация состоит в том, чтобы «примешать» см иа2 —аз
в а2—а3 и о2-\-Сз в симметричные комбинации. Это эффект
второго порядка, однако он может иметь значение для
химии.
(33)
с?! + (>2+<у3) et- (с2+ <y3) <V а3
(34)
39
3.2. Предосторожности, необходимые при построении
корреляционных диаграмм
При построении и применении корреляционных
диаграмм встречаются некоторые трудности, которые могут
привести к ошибкам. Для того чтобы избежать их, следует
соблюдать следующие предосторожности:
1. Каждый базовый процесс необходимо изолировать
и анализировать отдельно. В противном случае наложение
двух запрещенных, но независимых процессов может
привести к неверному выводу: комбинированный процесс
разрешен по симметрии.
2. Выбранные для анализа элементы симметрии
должны пересекать связи, образующиеся или распадающиеся
в рассматриваемом процессе. Отсюда вытекают два
следствия:
а) для анализа реакции бесполезны те элементы
симметрии, по отношению к которым все рассматриваемые
орбитали либо симметричны, либо антисимметричны.
Очевидно, анализ с помощью только таких элементов
(например, плоскость 3 при сближении двух молекул этилена,
ср. рис. 5) должен привести к выводу, что каждая реакция
разрешена по симметрии;
б) если единственный элемент симметрии не пересекает
ни одну образующуюся или распадающуюся связь, то
корреляционная диаграмма, построенная с помощью этого
элемента, может только привести к заключению (часто
неверному), что реакция разрешена по симметрии.
3. В каждом случае должна быть восстановлена
свойственная системе высшая симметрия. Например, если в по-
лиеновых молекулах есть гетероатомы, то их следует
заменить на изоэлектронные им углеродные группировки.
Заместители, обладающие простейшей электронной
структурой, следует заменить атомами водорода. Гетероатомы
создают возможность для новых реакций путем включения
несвязывающих электронных пар или благодаря
доступности низкорасположенных незанятых орбиталей.
Взаимодействия такого типа следует тщательно анализировать.
Значение пункта 1 можно рассмотреть на примере
образования кубана из циклооктатетраена в одну стадию
(рис. 14). Построение корреляционной диаграммы (рис. 15)
без учета пункта 1 приводит к выводу, что суперпозиция
40
Рис. 14. Одностадийное образование кубана из циклооктатетраена.
AXA2
^1-2,5-6 SjAa —
AiA2
^3-4,7-8 AlS2"
AiA2
S iA2 (7з_аг4_7
AiS2 Oi^st 2-6
*l-2, 5-6 AlS2"
SjS2 -
7r3~4,7-8 SjA2 —
S1S2
"""А^г 03-8,4-7
■— S^
S1A2 ^1-6,2-5
— SjS2
Рис. 15. Ложная корреляционная диаграмма образования кубана
из циклооктатетраена.
двух [2+2]-циклоприсоединений должна представлять
собой разрешенный по симметрии термический процесс. На
самом деле диаграмма имеет вид, изображенный на рис. 16.
Орбиталь SjAg, образованная из зтз_4,7-8, первоначально
41
коррелирует с разрыхляющей (S1A2)a3-8, 4-7 -орбиталью.
Небольшое возмущение связывает [3—4, 7—8]-циклопри-
соединение с процессом [1—2, 5—6]. Этот процесс
вследствие симметрии образует Б^-орбиталь кубана. В
результате первоначальная корреляция связывание — разрыхле-
77
1-2,5-6
7^-4, 7-8
""1-2,5-6 A1S2
^3-4,7-8 S jA2
SiS2
— AlA2
■■"■" S iA2 03-8,4-7
AiA2 +
AXS2 01-6,2-5
03-8,4-7
<*l-6, 2-5
Рис 16. Правильная корреляционная диаграмма образования
кубана из циклооктатетраена.
Во всех четырех группах уровни с симметричными символами, так же как и
уровни с антисимметричными символами, расположенные близко друг к другу,
являются вырожденными (индексы здесь не принимаются во внимание). Для
наглядности уровни изображены энергетически разными.
ние не реализуется. Однако реакция не становится менее
запрещенной по симметрии. В действительности разрешен-
ность или запрещенность реакции по симметрии
определяется высотой электронного холма, который должны
«преодолеть» орбитали исходных веществ или продуктов
реакции для достижения переходного состояния. Наличие шги
отсутствие холма зависит от первоначальной корреляции
или от исходного наклона линий, соединяющих
коррелирующие уровни. С этой точки зрения [3—4, 7—8]-комбина-
42
ция никак не облегчается конкурирующим процессом
[1—2, 5—6], и наоборот.
Применение пункта 2 можно проследить, рассматривая
превращение бутадиена (35) в бициклобутан (36). В этом
случае единственный элемент молекулярной симметрии —
ось второго порядка, проходящая через простую связь,
которая не образуется и не рвется в ходе реакции. Подробно
эта реакция обсуждается далее. Другой пример — система
из двух молекул пропилена, обменивающихся атомами
водорода 1(37) -> (38)]\ в этом случае есть только центр
симметрии, расположенный не на связях. Еще одним примером
является разложение пенталена до диацетилена и двух
молекул ацетилена [(39) -> (40)} (образующиеся или
распадающиеся связи не пересекают ни один элемент
симметрии).
\ 2
П.
3 <
(35) (36)
(О
Н. у^Н
н-
(37) (38)
€0
^%
* *
(39) (40)
Целесообразность пункта 3 хорошо иллюстрируется при
сравнении корреляционных диаграмм фронтального
присоединения этилена к этилену и присоединения этилена к
пропилену. В последнем случае, строго говоря, переходное
состояние несимметрично. Таким образом, в тривиальном
смысле все уровни симметричны и можно построить
корреляционную диаграмму так, как показано на рис. 17.
43
н н
><
н н
н2с-сн2
н2с-сн2
с=с
н
н
н н
X
н н
н2с-снсн3
II
н2с-сн2
Рис. 17. Слева: корреляционная диаграмма циклоприсоединения двух молекул этилена. Справа:
ложная корреляционная диаграмма циклоприсоединения этилена к пропилену.
Следовательно, замещение метилом делает термическое
12+2]-циклоприсоединение разрешенным по симметрии.
Этот вывод неправильный. Правильная корреляционная
диаграмма уровней показана на рис. 18.
НзС - С НС Нз
н н н2с-сн2
><
н н
Р и с. 18. Правильная корреляционная диаграмма
циклоприсоединения этилена к пропилену
Вообще говоря, замещение метилом приводит к
одинаковой симметрии всех уровней. Пересечение уровней
становится невозможным. Однако возмущение очень мало и
едва позволяет избежать пересечения. Все же связывающий
уровень исходных реагентов смещается несколько выше,
45
приближаясь к переходному Состоянию. Различие между
соответствующей диаграммой состояния и диаграммой
системы этилен + этилен является тривиальным, и реакция
остается запрещенной по симметрии* .
4. СОХРАНЕНИЕ ОРБИТАЛЬНОЙ СИММЕТРИИ
Ясно, что в общем случае отсутствие или наличие
молекулярной симметрии не может быть окончательной
причиной возможности или невозможности протекания реакции.
Симметрия прерывна. Она может возникать или исчезать,
присутствовать или отсутствовать. Очевидно, что химия
не такова. Слабое возмущение, скажем замещение
метальной группой, может нарушить общую симметрию, однако
нельзя ожидать, что оно «драматически» изменит механизм
реакции. Существенный и решающий фактор,
определяющий запрещенность реакции,— наличие в переходном
состоянии хотя бы одного уровня, который уже не является
связывающим и находится существенно выше остальных.
Мы использовали симметрию для того, чтобы выявить эти
высокие уровни энергии без проведения каких-либо
расчетов. Симметрия может отсутствовать вследствие
обычного замещения или, что более существенно, вследствие
асимметрии компонентов (например, при реакции
непредельных соединений). Однако анализ можно провести, если
изобразить участвующие в реакции орбитали в
соответствии с хорошо известными принципами квантовой механики,
учесть их смешение и проследить за взаимодействующими
в реакции орбиталями. В переходном состоянии уровни
высокой энергии могут возникать в результате
действительного пересечения, или пересечения, которое должно было
произойти. Такие уровни будут отсутствовать, если каждая
связывающая орбиталь продукта (исходных реагентов)
* Аналогичная ситуация возникает и в других областях химии.
Дипольныи п—71*-переход в формальдегиде запрещен по
симметрии. Причина наблюдаемой малой интенсивности остается пока
спорной. В случае ацетальдегида или несимметрично замещенных
кетонов элемент симметрии, который делает дипольный переход
в формальдегиде запрещенным, исчезает. Переход становится
разрешенным. Приводит ли это к скачкообразному повышению
интенсивности поглощения? Нисколько. Интенсивность остается
практически без изменения. Это связано с тем, что истинная симметрия,
т. е. симметрия локального окружения карбонильной группы
остается неизменной.
46
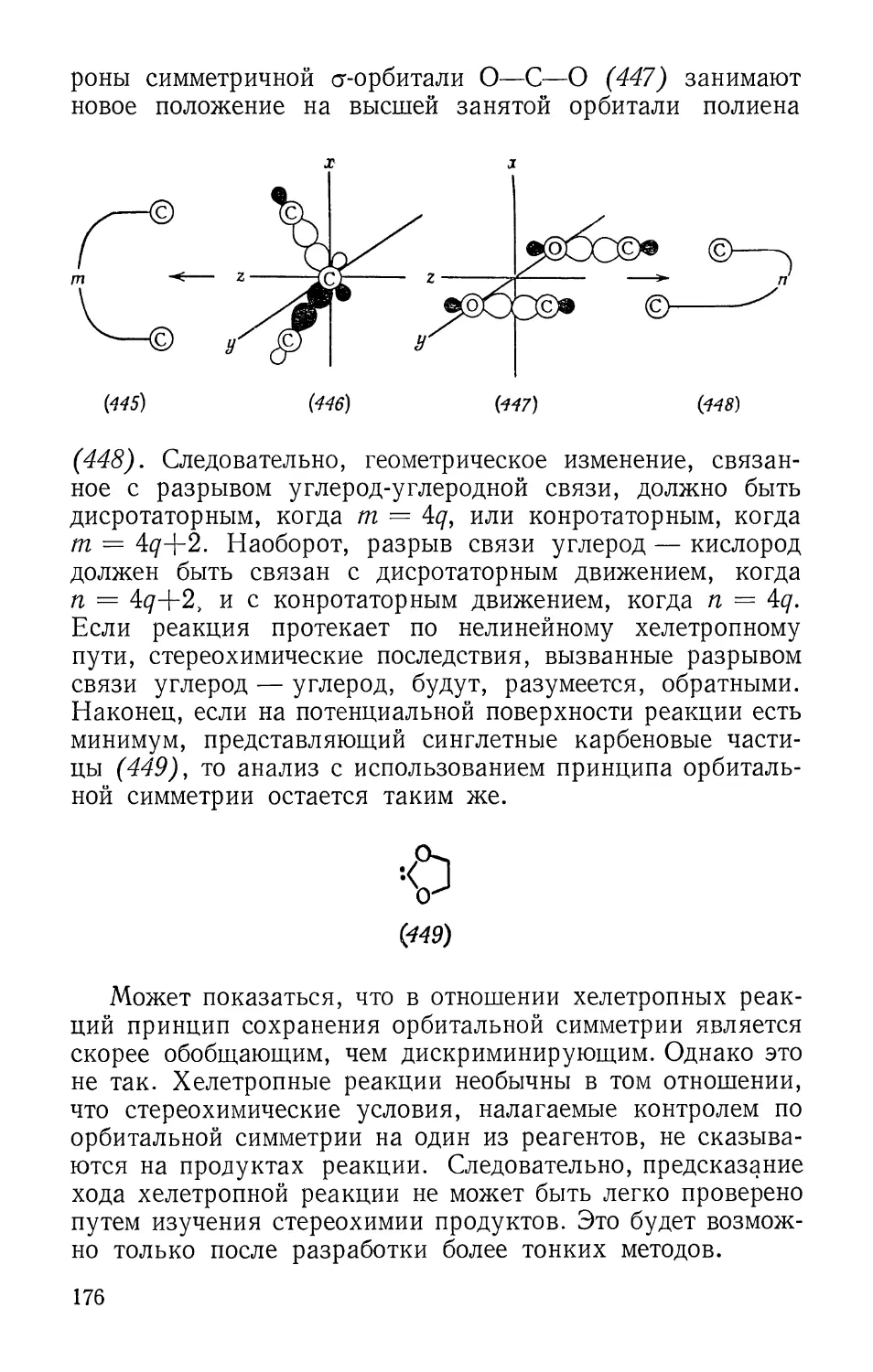
«происходит» из связывающей орбитали исходных
реагентов (продукта). Если какая-нибудь связывающая орбиталь
продукта не «происходит» от некоторой связывающей
орбитали реагентов, то она связана с разрыхляющей орби-
талью реагентов. Корреляция связывающей и
разрыхляющей орбиталей между собой зависит от наличия или
отсутствия общей симметрии. Однако, даже если
первоначальная корреляция нарушается, уровень имеет высокую
энергию и в переходном состоянии.
Таким образом, наиболее общая и в то же время
физически наиболее реальная точка зрения состоит в том, что
контроль химических реакций по орбитальной симметрии
осуществляется путем спецификации молекулярных
орбиталей реагентов и отыскания соответствующих орбиталей
в продуктах реакций, поскольку реакция идет с
сохранением орбитальной симметрии. Для того чтобы показать
наш метод, сначала обсудим некоторые случаи, для
которых можно построить корреляционные диаграммы (один
из них особенно подробно), а затем перейдем к менее
симметричным случаям, при рассмотрении которых анализ
молекулярной симметрии менее полезен.
5. ТЕОРИЯ ЭЛЕКТРОЦИКЛИЧЕСКИХ РЕАКЦИЙ
Реакции внутримолекулярного циклоприсоединения
стимулировали наше исследование симметрии молекулярных
орбиталей и синхронных реакций. Мы называем
электроциклическими реакциями образование простой связи (и
обратный процесс) между концами линейной системы,
содержащей k я-электронов (41). При таких изменениях
определенная геометрическая изомерия, проявляемая системой
с открытой цепью, связана с жесткой тетраэдрической
изомерией циклической структуры. A priori переход из одной
формы в другую может быть дисротаторным или конрота-
торным (42). В первом случае переходное состояние
характеризуется наличием плоскости симметрии, во втором—
сохраняется ось второго порядка.
47
исротаторньил
переход
v—
В >Г^- Конротаторный
переход
(42)
Хг -»
-Н- я
{43)
Рассмотрим основные молекулярные орбитали при
превращении циклобутена в бутадиен: четыре я-орбитали
бутадиена хь Тъ Хз> Х4*, к- и я*-уровни двойной связи
циклобутена и а- и а*-орбитали разрывающейся простой связи (43).
Для наглядности можно представить изменение
а-орбитали в три стадии. Первая стадия — конротаторное
движение, выполняемое а-связью [(44), 1], затем следует
стадия регибридизации [(44), 2]. На этой стадии орбиталь
выглядит как фрагмент орбитали Хг (или Х±) бутадиена.
Последний этап — появление орбиталей на атомах С-2
(44)
и С-3 (стадия 3). Необходимо иметь в виду, что в
действительности стадии 1, 2 и 3 протекают одновременно по мере
смещения системы по координате реакции. Стадия 3 может
показаться загадочной тем, кто не знаком с манипуляциями
с молекулярными орбиталями. В действительности это
универсальное явление, состоящее в том, что по мере
протекания реакции происходит смешение я*- и а-уровней.
Мы проследили за изменением а-орбитали в ходе реакции и
установили корреляцию этой орбитали с другой
связывающей орбиталью Хг- Аналогичным образом можно просле-
49
дить за изменением я-орбитали (45). Следует отметить, что
возникновение стадии 5 — следствие смешения орбиталей я
и а* (ср. стадию 6). Таким образом я-орбиталь
коррелирует с xi-орбиталью.
"f-
f—
(4Ь)
Конечно, корреляцию можно проследить на примере
бутадиена. При конротаторном движении ул превращается
в я (46), а Х2— в о (47).
(41)
50
Этот анализ выявляет процессы исчезновения узловых
плоскостей и орбитальных элементов в результате
смешения с высшими орбиталями соответствующей симметрии,
которые обратны процессам возникновения, рассмотренным
выше. Очень близкие аргументы приводят к корреляции
а* с хз и я* с — /4( = ул\). Таким образом мы получили
корреляцию связывающих уровней исходных реагентов со
связывающими уровнями продукта при условии сохранения
орбитальной симметрии. Следовательно, в этом случае
должна легко протекать термическая реакция.
Для сравнения рассмотрим дисротаторное раскрытие
кольца. Корреляция показана на схеме (48). Как а-, так
-.*._,„—
>- Хъ или Xi
и я -орбитали должны коррелировать с хг или Хз(——Хз-)~°р-
биталями. Однако, так как только одна орбиталь может
коррелировать с Xi, Другая орбиталь должна переходить
в разрыхляющую Хз"°Р^италь. Сохранение орбитальной
симметрии требует в этом случае высоколежащего
переходного состояния, поэтому термическая реакция запрещена
по симметрии. И в этом случае можно получить
корреляцию, исходя из молекулы бутадиена (49). Очевидно, что
в дисротаторном процессе с сохранением орбитальной
симметрии затруднения вызывает Х2"°рбиталь — она не
может переходить ни в одну связывающую орбиталь цикло-
бутена.
На рис. 19 представлены соответствующие описанным
случаям двухуровневые корреляционные диаграммы. Ясно,
51
(49)
что при конротаторном процессе сохраняется ось вращения
второго порядка, в то время как дисротаторное движение
характеризуется неизменной плоскостью симметрии. Эти
диаграммы аналогичны диаграммам для реакций
присоединения я-электронных систем (разд. 3). Диаграмма для кон-
ротаторного процесса характеризует реакцию,
разрешенную по симметрии, и диаграмма для дисротаторного
процесса — реакцию, запрещенную по симметрии.
Следует подчеркнуть, что такой поэтапный анализ был
проведен прежде всего с педагогической целью. Высокая
степень молекулярной симметрии, характерная для
рассмотренных случаев, позволяет получить корреляционные
диаграммы очень простым способом. Однако в дальнейшем
мы встретимся со случаями очень низкой симметрии, когда
поэтапный анализ станет единственно возможным методом
анализа.
Высшие занятые орбитали играют доминирующую роль
в таких корреляциях. Легко убедиться в их важности.
Во-первых, они — носители валентных электронов
молекулы, которые на начальной стадии реакции легче всего
подвержены возмущению [9]. Во-вторых, если молекула
обладает более низкой симметрией и связывающий уровень
в ней, пересекая энергетический промежуток, коррелирует
с разрыхляющим уровнем, то этим связывающим уровнем
обычно бывает высшая занятая орбиталь. Следовательно,
ее изменение определяет ход корреляционной диаграммы
и ее первоначальная форма указывает, является ли про-
б* или УГ*
52
Дйсротаторное Конротаторное
r-R
/
R
R
Плоскость ~л, .
зеркального отражения °сь QrnoP°*° порядка
о* А-
7Г* А
-А Х4 S
•SX3 А —
>А а*
'S **
-AX2S
-—SXXA —
^—— А я-
•S а
Рис. 19. Корреляционные диаграммы дисротаторного и кон-
ротаторного превращений циклобутена в бутадиен.
цесс разрешенным или запрещенным по симметрии.
Рассмотрим дисротаторное и конротаторное превращения /2 в
бутадиене (50).
Дисротаторное
—(---
^
Конро таторное
(50)
При дисротаторном вращении положительная
орбитальная лопасть сближается с отрицательной лопастью. Так как
один конец молекулы «чувствует» фазу волновой функции
на другом конце молекулы, то это разрыхляющее деста-
билизующее отталкивательное взаимодействие. Уровень
изменяется в сторону возрастания энергии при движении
системы по координате реакции. Конротаторное движение
сближает между собой две положительные лопасти (либо
две отрицательные лопасти, что эквивалентно). Такое
связывающее стабилизующее притягивающее взаимодействие
концов молекулы приводит к образованию новой а-связи.
Здесь необходимо сделать оговорку. Смысл контроля
по орбитальной симметрии любой синхронной реакции
можно всегда определить путем рассмотрения поведения
высшей занятой молекулярной орбитали в реагирующей
системе, однако этот анализ часто менее прост, чем в случае
превращения бутадиен ->- циклобутен. При проведении
такого анализа нужно внимательно следить за тем, чтобы не
разместить на одной молекулярной орбитали более чем два
электрона.
Общие правила для электроциклических реакций очень
легко можно получить из узловых свойств полиенов и поли-
енильных ионов. Термические электроциклические
реакции k тг-электронной системы должны быть дисротаторными
при k = 4q + 2 и конротаторными при k = 4q
(q = О, 1,2, ...); в первом возбужденном состоянии имеют
место обратные соотношения. Некоторые следствия из этих.
правил суммированы на рис. 20.
Рассмотрим теперь вторичные факторы, которые могут
влиять на состав продуктов электроциклической реакции,
54
во-первых, стерический фактор. Для каждой реакции могут
быть два конротаторных и два дисротаторных движения,
которые в некоторых случаях можно различить, а в неко-
Реакцыя
U
Основное
состояние
Возбужденное
состояние
Конротаторное Дисротаторное
^*
Дисротаторное Конротаторное
Конротаторное Дисротаторное
3© «— ®\\ Дисротаторное Конротаторное
<<% <«— @<чГ Конротаторное Дисротаторное
(© J Конротаторное Дисротаторное
\|^ "~* \Ы Дисротаторное Конротаторное
Р и с. 20. Конротаторная и дисротаторная электроциклические
реакции.
торых — нельзя. Так, два способа конротаторного
раскрытия ^ас-диметилциклобутена энантиомерны и приводят к
одному и тому же продукту — цис,транс-\,4-Аиметилбута-
диену (51).
55
R.
"Г-
Г-
(51)
Два способа конротаторного движения транс-ддметш-
циклобутена приводят к двум различным изомерам:
цис.цис- и т/?аяс,т/?аяс-1,4-диметилбутадиену (52). В
действительности найден только транс,транс-продукт [10].
По-видимому, это связано с неблагоприятной стерической
ситуацией в переходном состоянии, приводящем к
цис,цис-продукту.
(52)
С электроциклическим раскрытием циклопропильного
катиона в аллильныи катион связан еще более интересный
вопрос, впервые сформулированный Де Пю: если отщепле*
ние группы X циклопропанового кольца и разрыв связи в
электроциклической реакции образования аллильного
катиона являются синхронными, то будет ли какое-либо раз-
56
личие в расположении отщепляющейся группы при двух
a priori возможных дисротаторных вращениях? Расчет с
помощью усовершенствованного метода Хюккеля
показывает, что при встречном вращении заместители и
отщепляющаяся группа X расположены по одну сторону плоскости
трехчленного кольца (53), в то время как при вращении
в разные стороны группа X и заместители располагаются
по разные стороны от плоскости кольца (54).
R Pi
R М
.-Г
X
(54)
Такой же результат можно получить на основании
качественных рассуждений, если представить, что при разрыве
2,3-связи путем вращения в разные стороны (55) электрон-
'---У
X
(55)
ная плотность связи, которая в значительной степени
сосредоточена в плоскости кольца, смещается вверх. Это при-
57
водит к движению отщепляющейся группы в противопо
ложном направлении. Другими словами, это реакция
нормального 5^2-замещения группы X электронами цикло-
пропанового скелета. Отсюда, если R в соединениях (53)
и (54) — объемистые группы, то на основании стерических
соображений следует ожидать более быстрого сольволиза
для соединения (54). С другой стороны, если
^^-положение стянуто короткой метиленовой цепью, то можно
ожидать, что раскрытие цикла (56), где отщепляющаяся
группа находится в ая#ш-положении, будет несколько
затруднено. Это объясняется тем, что в результате вращения
должен образоваться транс,транс-аллильный катион в
малом кольце. Поэтому следует ожидать более легкого
раскрытия кольца только для шя-положения
отщепляющейся группы (57).
X (56) ' (57)
5.1. Примеры электроциклических реакций
Конротаторное электроциклическое превращение цик-
лобутена в бутадиен — хорошо известный процесс.
Стереохимия этого процесса была установлена около десяти лет
назад [11]. Исследованы также энергетические
закономерности и область применимости таких реакций [12]. До тех
пор пока не была установлена решающая роль орбитальной
симметрии в электроциклических реакциях, отсутствовало
и удовлетворительное объяснение удивительной стереоспе-
цифичности реакций. Если предположить, что в
термической реакции конротаторное движение разрешено по
симметрии, то из этого следует, что бициклический циклобута-
диен, содержащий связывающую метиленовую цепочку 'в
Ч^-положении (58), превращается в циклический
цис, транс-диен (59).
58
Т-.
(58) NCH2)n
(?H2)n
(59)
Такой ш/?аяс-олефин становится все более стерически
напряженным по мере уменьшения размера кольца. Эта
тенденция при изменении п прекрасно характеризуется
температурой (7), при которой время полураспада
соединения (58) составляет ~2 час [13]:
7(°С)
;100
2
195
3
>380
4
350
5
335
6
180
Время полураспада соединения (58) уменьшается для
малых п, так как из-за напряжения в кольце реакция сама
по себе становится очень экзотермичной*.
Такие молекулы, как бицикло-[2,2,0]-гексадиен и би-
цикло-[2,1,0]-пентен, стабильны вследствие того, что их
превращение в бензол и циклопентадиен соответственно
запрещено по симметрии.
Криге и Рейнхардт [16] синтезировали производные цик-
лобутена (60) — (63). Эти соединения проявляют
поразительное различие в пиролитической стабильности**. Для
соединений (61) и (63) с аяяш-структурой разрешенное по
симметрии конротаторное движение приводит к
^^-конфигурации двойной связи в шести- или семичленных
кольцах. В этих случаях электроциклическое раскрытие кольца
происходит очень легко. Напротив, разрешенный по
симметрии процесс для шя-изомеров приводит к транс-копфи-
гурации двойных связей в шести- или семичленных
кольцах, и превращения могут происходить только при очень
высоких температурах; естественно, они могут протекать
так же, как несинхронные реакции.
* Значение для /г=1 определено изданных [14]. Более
детальное исследование для случая /г=4 было опубликовано недавно
[15].— Прим. перев.
А-* Аналогичная закономерность отмечалась [17] для
конденсированных девятичленных колец.
59
ш> СЮ
- сю
-сю
Высокозамещенные бутадиены (64) и (66) были
исследованы в изящной работе [18]. Эти соединения могут
превращаться друг в друга через промежуточный циклобутен
(65) и находиться, таким образом, в равновесии. Каждая
молекула циклобутена претерпевает при 124° в течение
51 дня 2,6- 106раз конротаторное раскрытие цикла, при этом
ни одного «ошибочного», дисротаторного раскрытия не
происходит.
Ph Ph Ph
(64) Ph (65) СН3 (66) >снз
Несколько изящных превращений цис- и транс-беизо-
циклобутенов (67) и (68) легко объяснимы, если
трактовать их как электроциклические реакции с последующим
присоединением по Дильсу — Альдеру [19, 20].
н Ph О
(67)
60
Еа, ккал/моль k-№
при °С
42 261
29 87
45 273
ox
(68)
Ph
Ph
Ph
OsAH
Ph
^s.
X
Ph
Ph О
X = О, NR
Можно привести много примеров фотохимического
взаимного превращения циклобутен — бутадиен. В
подавляющем большинстве случаев диены являются той частью
циклической системы, в которой должен протекать
разрешенный по симметрии дисротаторныи процесс. Типичные
примеры таких процессов— реакции (69) и (70) [21, 22].
о°
hv
п/ О "* х^>
(69)
X = Н, С1 (70)
В работе [23] наблюдали превращение, где возможна
большая пространственная свобода [в соответствии с
правилом сохранения орбитальной симметрии реакция идет
по схеме (71)].
оо~сю
(71)
Недавно было подтверждено, что транс,транс-2,4-гек-
садиен фотохимически циклизуется до ^ас-диметилцикло-
бутена [24]. Эта реакция представляет особый интерес,
так как в этом случае отсутствует «геометрическая
необходимость» циклизации.
При фотолизе диазепинона (72) образуется бицикли-
ческий изомер (73), который термически легко переходит
в исходное соединение. Обратимость легко объяснить,
полагая, что цис -> траяс-превращение, которое невозможно
в конденсированных карбоциклических системах, в
соединении (73) представляет собой инверсию у атома азота.
61
СН3
N-N
Н
^етЖ
Н
Г72; (73)
Разрешенная по симметрии фотохимическая конрота-
торная циклизация цис-гексатриенов (и обратная реакция)
была впервые обнаружена при исследовании витамина D.
Превращения (74) — (80), разрешенные по симметрии,
были установлены в большой серии исследований [26].
RO
RCT
(79)
RO
RO
(80)
Циклогексадиены (77) и (78) вследствие геометрических
препятствий не могут подвергаться электроциклическому
раскрытию, разрешенному по симметрии. Поэтому
особенно интересно, что фотоизомеризация до циклобутенов (79)
и (80) идет по альтернативному процессу, разрешенному по
симметрии. Недавно [27] были изучены другие примеры
фотохимической реакции циклогексадиен -> гексатриен.
Аналогичная реакция протекает также при
фотоциклизации цис-стилъбепов (81) и родственных соединений [28],
хотя стереохимия продуктов окончательно не установлена.
Более ранние указания на то, что термическая
циклизация триенов является дисротаторной [26], были
подтверждены исследованием простых модельных соединений (82) [29].
С Н3
\гн3
сн
рн3
Ч/ГСНз
н
J2H3 Ц^ЛН
(82)
Сн3
Электроциклическое замыкание (83) цис,цис-октатетра-
ена должно идти термически конротаторным путем, а
фотохимически — дисротаторным путем. Хотя любая конформа-
ция цис,цис-октатетрает далека от планарности,
закономерность расположения узлов в орбиталях сохраняется.
Предсказанная стереохимия термической реакции была
недавно подтверждена [30].
63
Аналогичным образом были интерпретированы
некоторые предварительные эксперименты. Например, для
реакции (84), которая была исследована Майстером [31],
последовательность разрешенных по симметрии изменений
следующая: 8-электронная конротаторная
электроциклическая реакция, 6-электронная дисротаторная реакция,
[4+2]-циклоприсоединение, реверсия его и 4-электронная
конротаторная электроциклическая реакция.
64
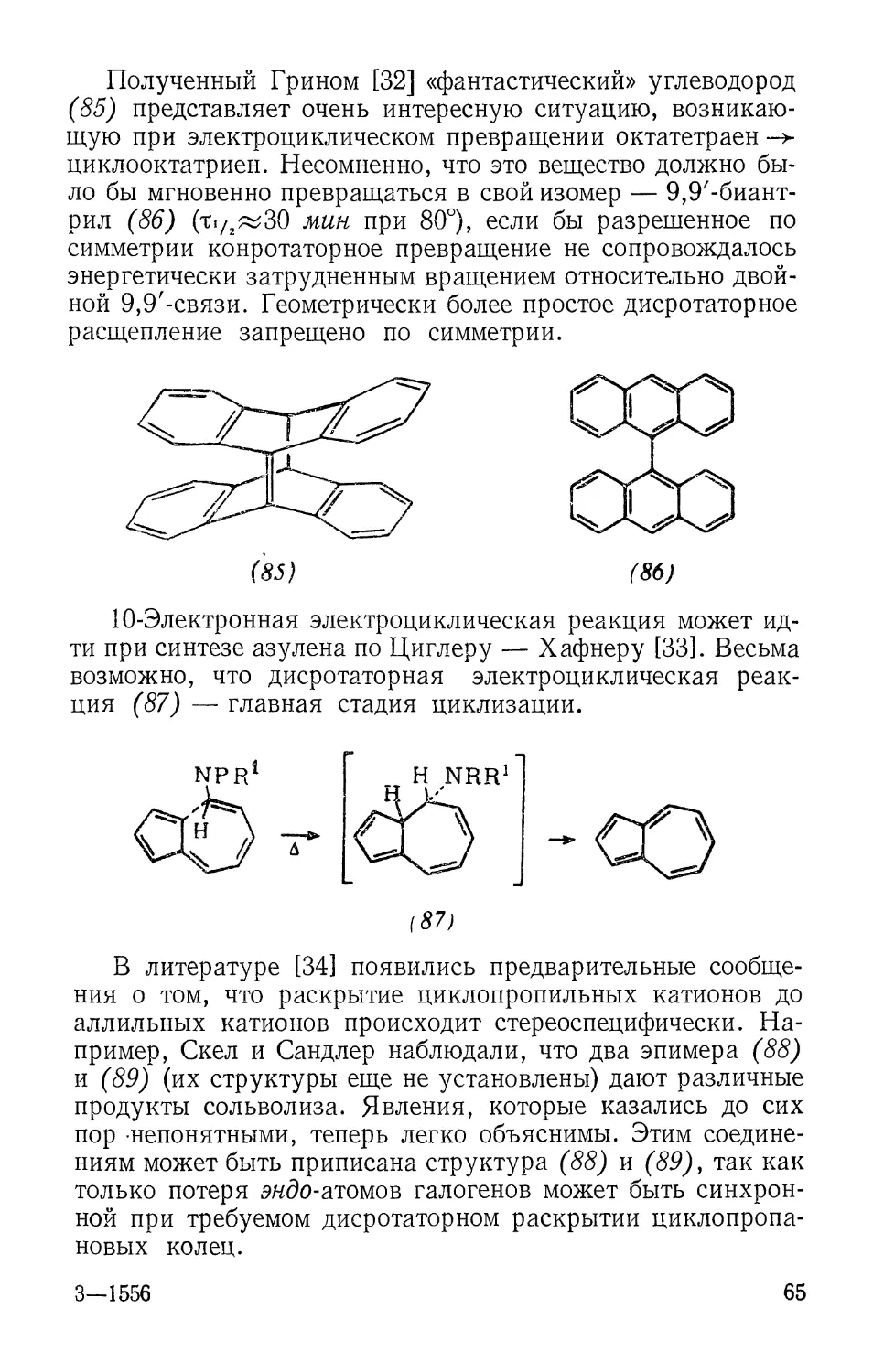
Полученный Грином [32] «фантастический» углеводород
(85) представляет очень интересную ситуацию,
возникающую при электроциклическом превращении октатетраен ->
циклооктатриен. Несомненно, что это вещество должно
было бы мгновенно превращаться в свой изомер — 9,9'-биант-
рил (86) (ti/2^30 мин при 80°), если бы разрешенное по
симметрии конротаторное превращение не сопровождалось
энергетически затрудненным вращением относительно
двойной 9,9'-связи. Геометрически более простое дисротаторное
расщепление запрещено по симметрии.
(85) (86)
10-Электронная электроциклическая реакция может
идти при синтезе азулена по Циглеру — Хафнеру [33]. Весьма
возможно, что дисротаторная электроциклическая
реакция (87) — главная стадия циклизации.
(87)
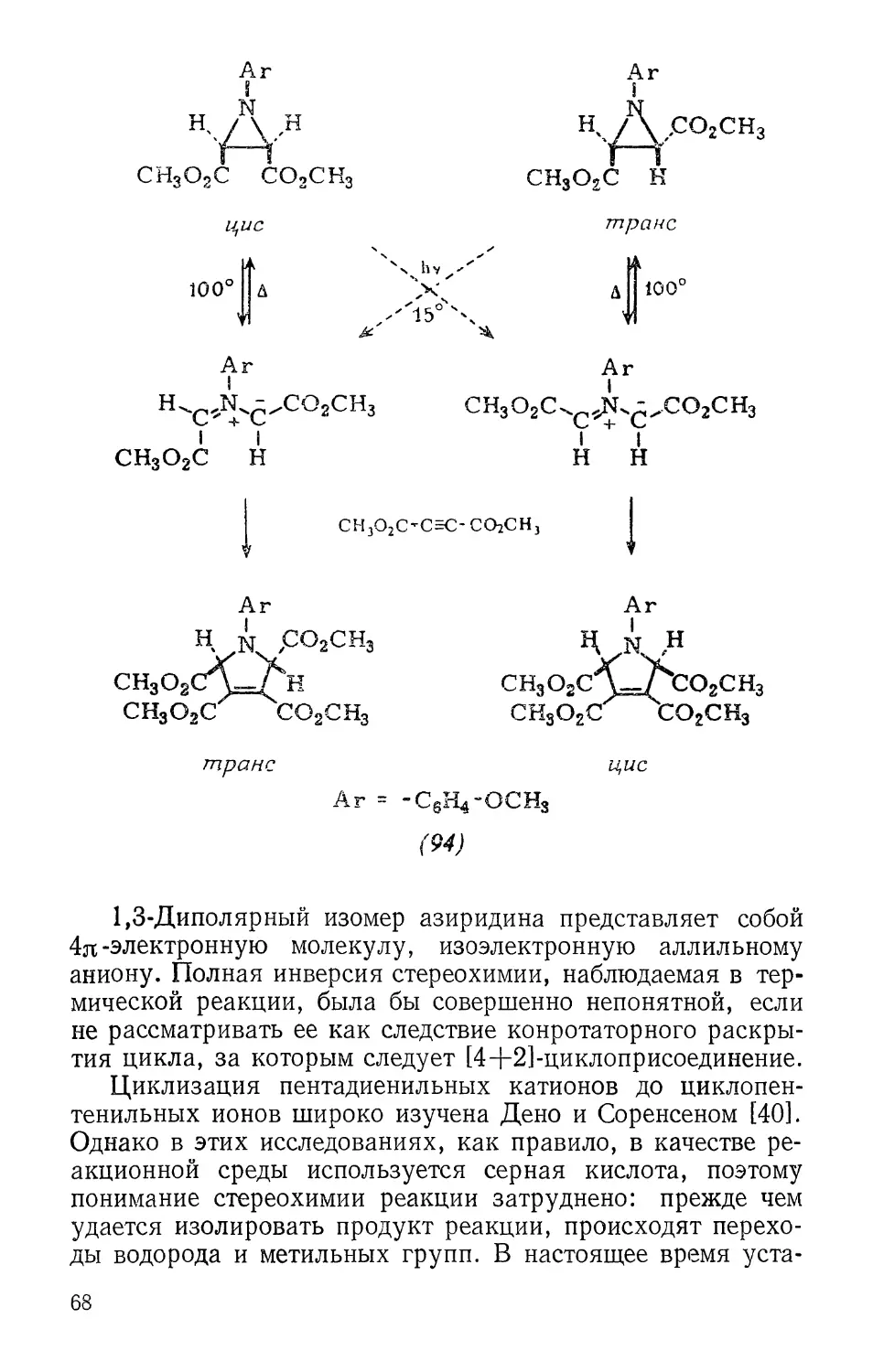
В литературе [34] появились предварительные
сообщения о том, что раскрытие циклопропильных катионов до
аллильных катионов происходит стереоспецифически.
Например, Скел и Сандлер наблюдали, что два эпимера (88)
и (89) (их структуры еще не установлены) дают различные
продукты сольволиза. Явления, которые казались до сих
пор -непонятными, теперь легко объяснимы. Этим
соединениям может быть приписана структура (88) и (89), так как
только потеря зядо-атомов галогенов может быть
синхронной при требуемом дисротаторном раскрытии циклопропа-
новых колец.
3—1556
65
CI Bi
OH
B£ .ci
(89)
Первое достоверное подтверждение наших предсказаний
для электроциклического раскрытия кольца было получено
в 1965 г. В то время как соединение (90) легко подвергается
сольволизу при 125°, его эпимер (91) не изменяется после
продолжительной обработки уксусной кислотой при 210°
[35]*. Детальное исследование метилзамещенных циклопро-
пилтозилатов ясно показывает, что стерическое влияние
метальных групп сводится к их взаимному отталкиванию
из-за стереоэлектронного эффекта [36].
с\ Л
(91)
Позднее появились другие подтверждения наших
выводов. Мы упомянем здесь только два из них. Чозес и
сотр. [37] наблюдали реакции (92)**, аВитхэм [38] показал,
что сольволиз з/сзо-8-бромбицикло-[5,1>0]-октана дает, как
и следует ожидать, траяс-циклооктенол (93) [38].
* Об аналогичных результатах нам сообщил В. Кирмзе (ср..
также поведение при пиролизе очень близких по структуре хлор-
бицикло- [3,1>0]-гексанов [35а]).
** Аналогичные реакции наблюдались также с НСС1 и НСВг
[37а].
66
CI
CI
(92)
,-Br
ОН
(93)
Электроциклическое раскрытие циклопропильного
аниона как такового не изучалось непосредственно, однако
изоэлектронныи пример, взятый из химии азиридина, дает
поразительное подтверждение правила сохранения
орбитальной симметрии. Изящные исследования Хьюсгена, Ши-
ра и Хьюбера [39] суммированы на схеме (94).
3'
67
Аг
I
тт N тт
сн3о2с со2ск3
z^c
100° А
Аг
1
Н"С''^С"С°2СНз
сн3о2с н
\
15° ч*
-
\г
S
НчуД ,С02СН3
сн3о2с к
транс
Д 100°
Аг
сн3о2с^с^^со2сн3
сн3о2ос^о
Аг
н n ро2сн3
СН302сЛ^К
сн3о2с со2сн3
транс
Аг =
-С6Н4
rw;
| i
Н Н
СО2СН3
'
Аг
Д N Н
СНзОгС'Д^СОгСНз
сн3о2с со2сн3
цис
-ОСНз
1,3-Диполярный изомер азиридина представляет собой
4я-электронную молекулу, изоэлектронную аллильному
аниону. Полная инверсия стереохимии, наблюдаемая в
термической реакции, была бы совершенно непонятной, если
не рассматривать ее как следствие конротаторного
раскрытия цикла, за которым следует [4+2]-циклоприсоединение.
Циклизация пентадиенильных катионов до циклопен-
тенильных ионов широко изучена Дено и Соренсеном [40].
Однако в этих исследованиях, как правило, в качестве
реакционной среды используется серная кислота, поэтому
понимание стереохимии реакции затруднено: прежде чем
удается изолировать продукт реакции, происходят
переходы водорода и метильных групп. В настоящее время уста-
68
новлено, что хорошо известная реакция Назарова [41]
включает электроциклическую реакцию пентадиенилкатио-
на, однако и в этом случае обширные исследования,
проведенные ранее, не дают информации о стереохимии
процесса.
Стереохимия реакции установлена недавно в Гарварде
[42]. Обработка дициклогексенильного кетона (95)
фосфорной кислотой дает два кетона (96) и (97) (R = Н).
При R = СН3 образуется соответствующий замещенный
кетон (96)\ в этом случае процесс не осложняется
параллельным образованием стереохимически неинформативного
продукта типа (97). Таким образом, в обоих случаях
предсказанное конротаторное протекание реакции
осуществляется без отклонений. Более того, при облучении кетона (95)
(R = Н) образуется кетон (98) — продукт дисротаторной
циклизации.
ОН
(98)
сДэ
R (97)
R R
Масс-спектрометрические методы позволяют
исследовать реакции катионов в возбужденном состоянии. Джонс-
тоун и Ворд [43] наблюдали, что дифенилметильный
катион (99), образующийся в масс-спектрометре, циклизуется
до частиц гидрофлуорена, которые теряют два атома
водорода в одну стадию. Поэтому весьма вероятно, что цис-иоя
(100) является разрешенным по симметрии продуктом
электроциклизации катиона (99) в первом возбужденном сос-
69
с
I
н
(99)
s?
н н
с
I
н
(100)
4- Н9
тоянии. Напротив, катион-радикал (101) циклизуется до
молекул гидрокарбазола, которые последовательно теряют
атомы водорода. Поэтому возможно, что в этом случае
транс-иоп (102) — результат разрешенного по симметрии
(ХО
н н
I
н
(101)
(102)
процесса в возбужденном состоянии. Чэпмен [44] наблюдал
фотохимическую циклизацию аминов (103). Эти амины
изоэлектронны пентадиениланиону, и их замыкание в
возбужденном состоянии должно идти по конротаторному пути.
Образующийся первоначально промежуточный продукт
(104) возвращается, по-видимому, в основное состояние;
при этом происходит перемещение водорода (разрешенное
по симметрии анионное [1,4]-смещение, см. разд. 7),
приводящее к стабильному конечному продукту (105).
hv
ЧЬч
1
СН3
(103) R = Н, СН3
Н R
(104)
(105)
Совсем недавно обнаружен первый пример
электроциклического замыкания простого циклопентадиенильного
аниона [(105а) -> (1056)] [44а].
70
(105а) (1056)
Винштейн [45] исследовал исключительно интересную
систему, в которой протекают 9- и 10-электронные
электроциклические процессы. Когда к ^ш>бицикло-[6,1,0,]-нона-
[2,4,61-триену (106) добавляют один электрон, образуется
ион-радикал (107). Он образуется путем дисротаторного
(106)
(107)
геометрического перемещения, разрешенного по симметрии.
Более того, при присоединении еще одного — десятого —
электрона образуется 10-электронный двухзарядный анион.
Наоборот, добавление электрона к изомерному транс-
триену (108) не сопровождается делокализацией
электронов циклопропана [46]. В этом случае возникающие
пространственные препятствия не позволяют осуществиться дис-
ротаторному перемещению и образуется 7-электронный
ион-радикал (109).
(108)
(109)
Последовательность электроциклических реакций
(ПО) — (П6) была изучена Фонкеном [47], который
справедливо допустил существование промежуточного
соединения (115) при превращении соединения (114) в (116),
которое он, однако, не наблюдал. Независимым исследова-
71
нием Рэдлик [48] подтвердил приведенный ход
превращений.
Дисрота- I—i /
торное >—-/
Конрота-
торное
Дисрота-
торное
ю
(ПО)
(111)
(112)
(ИЗ)
О
("<;
Конрота-
торное
-С?
Дисрота
торное
^00
гш;
<7ЛУ
Аналогична последовательность электроциклических
реакций при пиролизе бицикло-[6,2,0]-дека-[2,4,6,9]-тетрае-
на в трада-9,10-дигидронафталин (117) [49]. транс-9,10-
&
^
Н
(117)
Дигидронафталин, согласно имеющимся в литературе
данным [50], превращается фотохимически в циклодекапентаен
(при —190°), который термически переходит в ^ш>9,10-ди-
гидронафталин (118). В более поздней работе [50а] была
предложена схема, показанная на диаграмме
am
оо
72
1,5-Алкадиены подвергаются внутримолекулярной
перегруппировке при повышенных температурах, давая диме-
тиленциклобутены (119), (120). Наблюдаемая стереоспеци-
фичность [51] легко объясняется как следствие
разрешенного по симметрии сигматропного [3,3]-сдвига (см. разд. 7)
и 4-электронной конротаторной электроциклической
реакции.
Н3С^
Н3С^
и-"
Н3С.
н3с.
н*
н
Н3С
СН3
^4s
-г
н
(119)
Н3С.
Н'
II.
НзС"
н3с
н
н
Н3С
>=
>^
СН3
СН3
(120)
Недавно синтезированный [16]- аннулен (121)
термически и фотохимически изомеризуется в два различных
изомера [52] — продукты двойного дисротаторного и двойного
конротаторного замыкания цикла.
Особый интерес представляют недавно открытые
электроциклические реакции, катализируемые металлами.
Например, дибензотрициклооктадиен (122) термически изо-
73
меризуется до дибензоциклооктатетраена (124) при
нагревании до 180° в течение 4—5 час [53]. Однако при комнатной
температуре в присутствии ионов серебра изомеризация
заканчивается в течение 10 сек [54]. Запрещенное дисрота-
торное раскрытие цикла (122) ->■ (123), очевидно,
становится здесь разрешенным благодаря добавочным орбиталям
и электронам комплексообразующих металл-ионов.
Аналогичное драматическое обращение правил циклоприсоеди-
нения (и обратных реакций) в рамках сохранения
орбитальной симметрии проанализировано в работе [55].
t^ >^
(122) (123) (124)
Мы закончим перечень примеров электроциклических
реакций описанием двух интересных возможностей,
которые, однако, до сих пор не реализованы. Валентная
таутомерия циклооктатетраена и бицикло-[4,2,0]-октатриена
хорошо известна [56]. Требуемый дисротаторный процесс
можно осуществить термически [раскрытие шестичленного
кольца в (126)] или фотохимически [раскрытие циклобутено-
вого кольца в (126) ]. Электроциклические изменения могут
протекать быстрее, чем осцилляция связей в циклооктатет-
раенах [57], поэтому вполне возможно эти два
альтернативных случая наблюдать экспериментально.
rv*
Дисротатор-
hv
\===/^R ное ^^ R ное
(125) (126) (127)
Не синтезированный до сих пор циклодека-1,2,4,6,7,9-
гексаен (128) может существовать в мезо- и d,
/-модификациях; в обоих изомерах должно отсутствовать напряжение.
Обе молекулы — валентные таутомеры нафталина (129).
(128) (129)
74
Перегруппировка в нафталин формально является
электроциклической конверсией гексатриена в циклогексадиен.
мезо-Форма (130) стерически благоприятней для
превращения в нафталин путем дисротаторного замыкания кольца.
Так как это превращение — электроциклический процесс,
разрешенный по симметрии, молекула (130), по-видимому,
должна иметь мало шансов на существование. Наоборот,
молекула й,/-формы [например, (131)] имеет две интересные
особенности. Во-первых, модель молекулы показывает, что
возможны конформационные переходы без изменения хи-
ральности. Во-вторых, геометрия молекулы такова, что
изомеризация в нафталин может осуществиться только в
результате запрещенного по симметрии конротаторного
замыкания кольца. Следовательно, рацемат должен быть
термически (но не фотохимически) устойчив против
превращения в ароматический изомер. Другими словами, форма
(131) является продуктом конротаторного разрыва 9,10-свя-
зи нафталина и вполне может присутствовать среди
продуктов его фотохимического превращения.
(130) (131)
6. ТЕОРИЯ ЦИКЛОПРИСОЕДИНЕНИЯ И ЦИКЛОРАСПАДА
При обсуждении корреляционных диаграмм мы вывели
правила отбора для одного типа простейшего двухком-
лонентного циклоприсоединения — супраповерхностного*
(suprafacial) присоединения по отношению к каждому
компоненту. Супраповерхностным процессом называют
процесс, при котором образующиеся или разрывающиеся связи
находятся по одну сторону относительно реагирующей
•системы. Например, в этиленовой (132) или цисоидной
диеновой системе (133) образование связей (направление
стрелок указывает место образования связей) соответствует
супраповерхностному процессу.
* Термины супраповерхностный и антараповерхностный были
впервые использованы нами при обсуждении сигматропных
реакций (см. [3]).
75
J L
(132) (133)
A priori можно принять, однако, возможность
альтернативного антараповерхностного* (antarafacial) процесса,
в котором вновь образующиеся или распадающиеся связи
лежат по разные стороны от реагирующей системы [ср. (134)
и (135)1
j
(134) (135)
Мы будем использовать термины супра и антара** для
обозначения этих геометрических альтернатив при
обсуждении типа реакций, а буквы «s» и «а» — как индексы при
обозначении соответствующих реакций. Так, циклоприсое-
динение, упомянутое выше, относят к супра, супра-реакци-
ям, а реакцию Дильса — Альдера — к [4S+ 28]-процессам.
Существуют четыре возможные комбинации соединения
концов двух неодинаковых компонентов в реакции цикло-
присоединения. Каждая комбинация характеризуется
определенными стереохимическими результатами (рис. 21).
* В отечественной литературе встречается другой перевод
этих новых терминов — супраплоскостной и антараплоскостной
соответственно [ С а л е м Л., Усп. хим , т. XXXIX, вып. 8, 1494
(1970)].— Прим. перге.
** Ранее мы и другие авторы [58] использовали обозначения
цис и транс для характеристики геометрических отношений,
обозначаемых здесь супра и антара. Однако использование терминов
цис и транс, которые имеют твердо установленный смысл, для
обозначения разных ситуаций очень неудобно. Это может вызвать
путаницу, особенно в тех случаях, когда эти ситуации
встречаются одновременно, а также при обсуждении многокомпонентных
систем.
76
Ч; X)
"Y Y
Супра
Супра
Г77-2
Y X
m~2
т-г
Рис. 21. Стереохимические результаты циклоприсоединения двух
различных молекул.
Рисунки являются схематичными и ничего не говорят о геометрии переходного
состояния, которая, естественно, различна в разных случаях. Можно заметить,
что для т>2 или п>2 имеется две супра, супра -реакции, которые отличаются
друг от друга способом сближения партнеров (экзо- и эндо-присоединение). То же
самое справедливо и для других случаев. Если в реакции партнеры одинаковы,
то процессы супра, антара и антара, супра не различаются.
77
Для того чтобы определить, разрешено или запрещено
по симметрии циклоприсоединение, необходимо провести
полный анализ. Другими словами, при использовании
принципа сохранения орбитальной симметрии нужно
рассмотреть все орбитали — связывающие и разрыхляющие.
Однако часто может оказаться полезным и упрощенный анализ.
Прежде всего все участвующие в процессе электроны
исходных веществ располагают на полностью делокализованных
связывающих су-орбиталях продукта циклоприсоединения.
Тогда если электроны, занимающие су-орбитали, можно
поместить на связывающих орбиталях продуктов циклораспа-
да, образующихся при разрыве су-связей с сохранением
орбитальной симметрии, то реакция разрешена по симметрии.
Если в системе отсутствует какая-либо молекулярная
симметрия, то знаки орбитальных лопастей определяют путем
сопоставления со всеми точками аналогичной симметричной
системы, которая имеет то же число орбиталей того же типа.
Проиллюстрируем эту процедуру для всех возможных
[2+2]-реакций. В каждом случае четыре рассматриваемых
электрона вначале располагают попарно на двух
обобщенных орбиталях циклобутана (136) и (137).
(136)
(137)
a) [2s-f 28]-Процесс. При разрыве су-связей два
электрона орбитали (136) можно разместить с сохранением
симметрии на связывающих орбиталях (138) молекулы
этилена. Однако электроны орбитали (137) могут попасть
только на разрыхляющую орбиталь (139) другой молекулы
этилена.
8-8
(т)
038)
78
8-8
(137)
039)
Вывод: реакция запрещена по симметрии.
б) [2д+ 2а]-Процесс. При разрыве су-связей два
электрона орбитали (136) (показанной по чисто формальным
соображениям искривленной) могут переходить с
сохранением симметрии на связывающую орбиталь (140) одной из
молекул этилена, а два электрона орбитали (137)
аналогичным образом переходят на связывающую орбиталь (141)
другой молекулы этилена.
{136)
8-8
(140)
&
(137)
Я
(141)
Вывод: реакция разрешена по симметрии.
в) [2а+ 2а]-Процесс. При разрыве су-связей два
электрона орбитали (136) (опять показанной по формальным
причинам искривленной, но несколько другим образом)
могут переходить на разрыхляющую орбиталь (142)
молекулы этилена и два электрона орбитали (137) — на
связывающую орбиталь (143) другой молекулы этилена.
79
(736)
(142)
(137)
О
(ИЗ)
Вывод: реакция запрещена по симметрии.
Следует отметить, что проведенный здесь анализ
говорит о возможности протекания синхронной разрешенной
по симметрии комбинации двух этиленовых молекул в цик-
лобутан. Упрощенный формальный анализ не раскрывает
истинной геометрии сближения при разрешенном [2S+ 2J-
процессе. Очевидно, что она должна отличаться от
геометрии запрещенной по симметрии комбинации. При
подробном рассмотрении максимума перекрывания орбитальных
лопастей в [2S+ 2а]-реакции становится ясно, что
Молекулы этилена должны приближаться одна к другой
ортогонально, как показано на схеме (144).
(М)
Следует также упомянуть, что полную корреляционную
диаграмму для разрешенной реакции [2S+ 2a] можно по-
80
строить, используя оси симметрии второго порядка,
которые проходят через середины этиленовых связей; то же
относится к реакциям типа [2а+ 2а]. Геометрия сближения в
этом случае показана на схеме (145). Реакция запрещена
в основном состоянии, как было показано выше, но
разрешена для возбужденного состояния (ср. рис. 22). Наконец,
важно отметить, что принцип сохранения орбитальной
симметрии действителен независимо от того, можно или нельзя
построить формальную корреляционную диаграмму.
Например, нельзя построить корреляционную диаграмму для
синхронных комбинаций [4S+ 2a] и [6S+ 2a], однако
анализ по орбитальной симметрии показывает, что первая
комбинация запрещена, а вторая разрешена.
/72 + П
4<7
Aq + 2
Разрешено
Запрещено
в основном состоянии,
в возбужденном
состоянии
ms + na
/Иа + ns
ma+ na
Разрешено в возбужденном
состоянии. Запрещено в основном
состоянии
ms + ns
ma + na
ms + na
ma+ns
Рис. 22. Правила отбора для [т+д]-циклоприсоединения.
Теперь легко обобщить правила циклоприсоединения.
При соединении т- и n-электронной систем в синхронном
процессе должны соблюдаться правила, приведенные на
рис. 22 (q = 1,2,3,...). Важной особенностью является то,
что в правилах говорится не об общем числе орбиталей, а о
4—1556
81
числе электронов. Например, [4+2]-присоединение может
идти по каждому из путей, показанных на схемах (146) —
(149).
- О
(146)
- О
(147)
- о-
(.14»)
- О
(Н9)
Следует отметить, что супра, антара- и антара, антара-
циклоприсоединения не так сильно отличаются друг от
друга, как может показаться на основании простых стери-
ческих соображений.
Остается еще указать, что су-связи могут проявлять себя
как компоненты в реакциях циклоприсоединения. В этом
случае следует сделать следующие определения:
а) су-связь участвует в реакции циклоприсоединения
типа супра, если в процессе реакции конфигурация на
обоих концах этой связи либо остается неизменной, либо
инвертирует;
б) cr-связь участвует в реакции циклоприсоединения
типа антара, если в процессе реакции конфигурация
остается постоянной на одном конце связи и инвертирует на
другом.
Рациональная основа этих важных условий сразу
становится очевидной, если рассмотреть присоединение к
молекуле этилена, принимая, что она содержит две су-связи
(150).
4
»->
82
Су приповерхностное присоединение
Сохранение t Сохранение
J I...
1 t
Обращение Обращение
Антараповерхностное присоединение
Сохранение
1
Обращение
Сохранение
. I
(150)
Обращение
Мы предлагаем простой и удобный способ обозначения
типа орбиталей, участвующих в синхронных реакциях,—
предшествующий числу электронов индекс обозначает
соответствующую орбиталь, например а2а, л48 и т. п. Приведем
некоторые поясняющие примеры. Электроциклическое
превращение циклобутенов в бутадиены можно формально
рассматривать как циклоприсоединение а-связи к я-связи
любым из двух эквивалентных способов:
1. Как видно из рис. 23, это превращение является
супраповерхностным по а-связи и антараповерхностным
по я-связи, т. е. [ a2s+ я2а].
4*
83
2. Как показано на рис. 24, превращение является
антараповерхностным по су-связи и супраповерхностным
по я-связи, т. е. [я25 + а2а].
Нетрудно видеть, что, какую бы формальную схему мы
ни приняли, конротаторный процесс разрешен по
симметрии. Дисротаторный процесс, который можно
рассматривать как реакцию [я28 + a2s] или [л2а + а2а], в основном
состоянии запрещен по симметрии.
Рис. 23. Электроциклическое
превращение циклобутена в
бутадиен как [ a2s + те2а]-
циклоприсоединение.
Р и с. 24. Электроциклическое
превращение циклобутена в
бутадиен как [ n2s+ а2а]-цик-
лоприсоединение.
Учитывая все сказанное выше, можно охарактеризовать
синхронный разрыв циклобутана как [a2s-[- а2а]-процесс
(151).
н •.
С \А\
R*lO
R
■■?—к
н-..
R^
н..
R'
.-•R
^Н
.••н
^R
{151)
6.1. Примеры циклоприсоединения и циклораспада
[2+2]-Циклоприсоединение — одна из наиболее
исследованных фотохимических реакций. Более сотни примеров
этой реакции приведено в недавно опубликованном обзо-
84
ре [59] (см. также [60]). Вполне возможно, что многие из
этих реакций не являются синхронными, так как,
по-видимому, с реакцией конкурирует релаксация возбужденного
состояния, сопровождающаяся переходом к равновесной
геометрии, при которой молекулы этилена отклоняются
на 90° от копланарности. Таким образом, для многих цикло-
аддуктов характерно соединение в mpawc-конфигурации
[61]; конечно, продукты траяс-соединения могут
образовываться и при синхронной разрешенной по симметрии
[jt2s+ л2а]-реакции двух колебательно возбужденных
молекул в основном состоянии. Тем не менее недавно
обнаружено два замечательных примера разрешенных по
симметрии фотоиндуцированных [2S+ 28]-реакций (151а) и (1516).
Облучение цис- и транс-2-бутеиов отдельно и в смеси идет
так, как показано на схеме (151а) [61а]. Другой пример
фотохимической [(у28+ ст28]-реакции — недавно изученный
процесс (1516).
Л51а)
&
СЮ
сю
hv (—=—^
■*-»• (СНг)* (СН2)4
-^* (СН2)4 (СН8)«
(1516)
85
Вполне возможно, что пиролиз циклобутана — процесс,
обратный простейшему [2+2]-циклоприсоединению, — не
является синхронной реакцией. Считают [62], что предэкс-
поненциальный множитель в уравнении Аррениуса
указывает на ступенчатое разложение циклобутана, протекающее
через образование тетраметиленового радикала. Кроме того,
тот факт, что пиролиз цис- или траяс-диметилциклобутана
(152) дает среди других продуктов смесь цис- и транс-бу-
тенов [63], подтверждает бирадикальный механизм. Все же,
если циклораспад — синхронная реакция, он должен
относиться к типу [(у2д+ (j2a], и должен наблюдаться тот же
стереохимический результат.
н*.
*н
.'СН3
н1
н.
-н
чн
.н
н3<г
"н
н*с*
{152)
сн3
н3с'
Прекрасный пример разрешенной по симметрии
комбинации [л28+ л2а] — открытая^ Крафтом и Кольтценбургом
[64] реакция спонтанной димеризации олефина (153) до
(154).
(153)
(154)
Из более ранних наблюдений [65] спонтанной
димеризации ^ис,траяс-циклооктадиена-1,5 до циклобутана при
комнатной температуре труднее сделать какие-то выводы,
так как не установлена стереохимия продукта; в настоящее
время для него принята конфигурация (156).
(155)
(156)
Интересно отметить, что, когда двойная связь
закручивается относительно своей оси [по-видимому, это происхо-
86
дит в случае соединений (153) и (155)1, сопровождающее
закручивание орбиталей отчетливо говорит о процессе
[jt2s+ я2а1- При подробном рассмотрении общего случая
(рис. 25) можно обнаружить очень интересное
обстоятельство, которое состоит в том, что закручивающее движение,
облегчающее реакцию, приводит оба этиленовых
компонента к системам с противоположной хиральностью.
Следовательно, можно предсказать, что оптически активные формы
олефинов (153) и (155) будут димеризоваться труднее,
чем рацематы.
*,—ч
то
-+жЬ
-f *-*i*
•*о
Рис. 25. Комбинация систем противоположной хиральности при
[ ir2s+ тг2а ]-циклоприсоединении двух молекул этилена.
Бициклобутаны образуются при облучении некоторых
замещенных бутадиенов [66], а также незамещенного
бутадиена [67]. Можно считать, что бициклобутаны обладают
заметной стабильностью, если принять во внимание
большую энергию напряжения их структур (~69 ккал/моль).
Энергия активации изомеризации в бутадиен составляет
41 ккал/моль [68, 69]. Естественно рассматривать
перегруппировку бициклобутана в бутадиен как цесинхронный
процесс, протекающий через бирадикальную промежуточную
(157)
стадию (157). Однако, если эта реакция является
синхронной, ясно, что она должна быть [a2s+ (у2а]-процессом и
87
R
II
II
(158)
стереохимически идти по схеме (158)*. В настоящее время
получены доказательства того, что реакция является
синхронным [a2s+ а2а]-процессом. Первое, косвенное указание
на это было получено исследованием превращения
соединения (159) в (161) при пиролизе [71]. По-видимому,
(159)
о
(160)
СП
(161)
промежуточный продукт (160) замыкается по конротатор-
ному типу в двойной цикл наблюдаемого продукта (161).
О решающем эксперименте сообщили недавно Клосс и Преф-
фер [72]; их результаты можно суммировать схемой (162).
H.C./W
н
н
Н3С
Н
СНз
Н
Н3С
Н
н
СНз
(162)
Н3С
Н
Н
СН3
Для перехода от бициклобутайа к бутадиену существует
в принципе альтернативный путь с совершенно другой
последовательностью стереохимических превращений: (163)—>
->- (164) -»■ (165). Здесь промежуточным продуктом
является циклобутен, который переходит конротаторным путем
в бутадиен. Термодинамически такая последовательность
вполне возможна [73]. Формально превращение можно пред-
* К аналогичному выводу пришел Виберг [70] на основании
полуэмпирических расчетов МО.
ставить в виде последовательных разрешенных по
симметрии [a2s+ а2а]- и [a2s+ я2а]-реакций. Нельзя исключить
возможность того, что какие-то особые заместители,
которые могли бы вызвать простые стерические эффекты, не
приведут к выявлению этого пути реакции.
й Н \
(Ш)
(164)
Н
Л1
R
(165)
Соотношения между стереохимией реакций для всех
возможных разрешенных по симметрии реакций бициклобу-
R
«.Ач»
J>v
CR
R
.Av"
Р и с. 26. Разрешенные по симметрии взаимные превращения
бициклобутанов, циклобутенов и бутадиенов и их стереохимия.
реакции, разрешенные в основном состоянии; - реакции,
разрешенные в возбужденном состоянии.
89
танов, циклобутенов и бутадиенов можно установить,
используя рис. 26.
Известно большое число реакций Дильса — Альдера
типа [л4д+ я28]-циклоприсоединения [74, 75]. Несмотря на
то что во многих случаях предложен бирадикальный
механизм (совсем недавно в работе [76]), подавляющее
большинство экспериментальных данных согласуется только с
синхронным механизмом [77].
Растет число фотохимических реакций Дильса —
Альдера (см., например, [78]), однако в большинстве случаев
нет доказательств того, является реакция синхронной или
нет. Вполне возможно, что синхронным разрешенным по
симметрии [я4 + я2]-процессом в возбужденном состоянии
является превращение г^с-гексатриена (166) в бицикло-
в: - О ■ О
(166)
Г3,1,0]-гексен [79]. Если эти реакции синхронные, то они
должны протекать как процессы [я45+ л2а] или [я4а+ я28].
Исключая образование конденсированного /п/шяс-бицикло-
гексена, стереохимическую последовательность для
разрешенных процессов можно представить следующим образом:
(170)
(171)
90
Не было изучено ни одной реакции, включающей
вещество с достаточно хорошими метками, которая могла бы
подтвердить это заключение. В витамине D2 (172) верхняя
и нижняя поверхности триеновой системы не эквивалентны,
и процессы, которые в общем случае приводят к энантиоме-
рам, должны в этом случае приводить к различным
структурам. В действительности облучение витамина D2
приводит к двум бициклогексенам — супрастеролу I (173) [80]
и супрастеролу II (174) [81]. Эти структуры как раз те,
которые должны получаться при синхронных разрешенных
по симметрии [я48+ я2а]-процессах в возбужденном
состоянии. Однако в отсутствие необходимой метки при атоме,
обозначенном звездочкой, невозможно распознать,
участвует ли я4-система в реакции по супраповерхностному
типу.
Н,С R
НО
(172)
НО
(173)
СНо
но.
(174)
Предложенный механизм для наблюдаемого
превращения азепина (175) в фульвен (178) включает разрешенную
по симметрии [я4а+ я2а]-реакцию в основном состоянии
[(175)-+ (176)] [82].
91
НзСООС СНз
ОГСНз
С
Н3СООС
(175)
СНз
Н3СООС СН3
( 3-СООСНз
H-jC^NHCHs
(178)
Н3СООС
СН3
NCH3
Н3СООССН3
НзСООС
СНз
(176)
СООСНз
NCH3
(177)
ЩС СН3
Н3С >НЧ-СНз
При повышенных температурах в присутствии
основания октаметилциклооктатетраен (179) с трудом
превращается в октаметилсемибульвален (180) [83]. По-видимому,
СН3
Н3С СН3
НзС"ЧЛ^/°Нз
СН3| СН3
СН3
(180)
основание служит главным образом для защиты реагентов
от альтернативного кислотно-катализируемого превращения.
Эта реакция простое разрешенное по симметрии
внутримолекулярное циклоприсоединение [я4а+ я2а]. Пет-
тит [84] наблюдал процесс, который лучше всего
интерпретировать, предполагая, что дибензо-[я,с]-циклооктатетра-
ен (181) спонтанно переходит в соединение (182). Особо
следует упомянуть то, что [я4а+ я2а]-реакции
благоприятствует протекающая одновременно потеря хиноидного
характера молекулы (181), а также отклонение геометрии
этой молекулы от нормальной геометрии циклооктатетрае-
на, обусловленное сочлененными шестичленными
кольцами. Трудность протекания процесса —[я4а+ я2а1* в от-
* Здесь и далее знак минус ставится перед квадратными
скобками, если реакция характеризуется по ее продуктам.
92
(181)
(182)
сутствие благоприятствующего стерического напряжения
подтверждается тем фактом, что кетон (183) не вступает в
реакцию циклораспада до бутадиена и соединения (185)
даже при 400°. Соответствующая —[я48+ я28]-реакция
(184) -*- (185) протекает легко [85].
Реакции [4+4]-циклоприсоединения хорошо известны
[86]. В основном это фотохимические реакции; однако
стереохимия таких реакций мало изучена. [8+2]-Циклопри-
соединение встречается сравнительно редко. Гептафуль-
вен (186) легко комбинирует с диметилацетилендикарбок-
аСООСНз /~%-\
il, -* 4JQ-cooCH'
(186) СООСНз (Ж) СООСНз
силатом [87], образуя (187). Фульвен (188) не реагирует
аналогичным образом (отметим, что комбинация [я68+ я28]
93
запрещена по симметрии). Наблюдались аналогичные
реакции для калиценов [88]. Гексафенилпентален (189) также
а
(Ш)
СООСНз
Г
СООСНз
CQ-
СООСНз
СООСНз
комбинирует с диметиловым эфиром ацетилендикарбоновой
кислоты с образованием азулена. При этом промежуточно
образуется аддукт (190) — продукт [8+2]-реакции,
который, однако, не был выделен [89].
Ph-
Ph Ph
Ph Ph
Ph COOCH3
/
Ph Ph /II
(/89) СООСНз
СООСНз
Ph Ph
(190)
СООСНз
Ниже приводятся две необычные реакции [8+2]-цикло-
присоединения, открытые Бёкельхайде [90]:
k^N^J
Н3СООС-СОСООСН3,
N,
и'\ Т"н
Н3СООС СООСН3
(191)
НзСООС^^^СООСНз
PhCO
Ph
Н3СООС - С =С - COOCH3
PhCO
Ph
H
H3COOC
H
COOCH3
PhCO
H3COOC COOCH3
(192)
Реакции [6+4]-циклоприсоединения были не известны"
до тех пор, пока наше открытие принципа сохранения
орбитальной симметрии не стимулировало эти исследования.
Теперь известны некоторые случаи таких реакций:
94
Ph СНз
Ph СНз
Ph
wCH3 s*\
СНз "^
о-О^^Ог
[92]
(195) 5
О+ оО -* 4>~>гэ2'
93]
^°
П«У |
R О
И
0**0-~Л£р
R
(197) О
[94]
+ R-N
w
R = -COOCoH,
(198)
[95]
Известны реакции [6+6]-циклоприсоединения, дающие
я-ксилол в качестве промежуточного продукта [96], однако
нет никакой информации о детальном механизме этих
реакций. Облучение тропона в кислом растворе приводит к
симметричному димеру (199) [97] — продукту синхронной
разрешенной по симметрии комбинации 1я65+ я68] в
возбужденном состоянии. Необычные экспериментальные
условия, а также тот факт, что другие фотоиндуцированные
реакции димеризации тропона могут протекать по
несинхронному механизму [98], указывают на возможность того, что
соединение (199) образуется при синхронном процессе.
О
Гептафульвален (200) комбинирует с тетрацианоэтиле-
ном с образованием аддукта (201), структура которого был.а
надежно установлена кристаллографически с помощью
рентгеновских лучей [99]. Это соединение может быть продуктом
разрешенного по симметрии процесса [я14а+ ^2S],
протекающего в основном состоянии. Закрученная форма
молекулы гептафульвалена служит идеальной основой для
аятара-присоединения к 14-электронной системе.
(200)
96
Циклоприсоединение с участием ионных компонентов
до сих пор несколько необычно. Ионное [4+2]-циклоприсо-
единение, по-видимому, реализуется различными путями:
\? Л — ®Г | Класс А
(202)
(203)
<§ II — o<Q
Класс Б
Класс В
(204)
К циклоприсоединению по классу А можно отнести
реакции циклопропанонов (205), Возможно, циклопропаноны
находятся в равновесии с изомерными диполярными
частицами, которые могут реагировать как 2я-электронные сие-
(205) (206) б
темы, и можно ожидать, что они будут комбинировать с
диенами в процессе [я48+ я28], например реагировать с фу-
ранами, давая аддукты типа (206) [100]. Совсем недавно
была показана [101] прямая комбинация 2-метилаллильного
катиона (207) с циклопентадиеном и циклогексадиеном с
образованием бициклических катионов (208) (Z = —СН2—
или —CHcj—CH3—).
97
(207)
(208)
К реакциям циклоприсоединения класса Б относится
образование пипитзолов (210) из перезона (209) [102].
н--о
НзС
н-о
= н3с,
(209)
(210)
До настоящего времени не обнаружено ни одного
простого случая ионного [4+2]-циклоприсоединения по
классу В. Однако к этому классу может принадлежать очень
важная и многочисленная группа реакций —1,3-диполяр-
ное присоединение [103]. Хотя 1,3-диполярные вещества,
участвующие в реакциях циклоприсоединения, формально
являются нейтральными молекулами, обычно они
функционируют как трехорбитальные 4я-электронные частицы.
Благодаря успешным исследованиям Хьюсгена 1,3-дипо-
лярное присоединение превратилось из непонятного
явления, представленного несколькими случаями, в важный
тип реакций. Этим автором опубликовано несколько
прекрасных обзоров в этой области [103] и проведен анализ
процесса на основании правила сохранения орбитальной
симметрии [104]. Поэтому необходимо только кратко
суммировать основные факторы и осветить некоторые аспекты
вопроса. Подавляющее большинство реагентов в 1,3-дипо-
лярных реакциях изоэлектронно либо озону (211), либо
закиси азота (212) и непременно содержит трехорбиталь-
ную систему, занятую четырьмя я-электронами.
(211)
I/ 1/1/
-N—N—6-
/I ./I S\
(212)
98
Другой класс реагентов представлен лабильными
молекулами, которые могут легко превращаться в частицы,
содержащие необходимые элементы четырехэлектронной
системы. Например, карбонилилиды (214) и азометинилиды
(216) получают электроциклическим превращением окисей
этиленов (213) и этилениминов (215), замещенных таким
образом, чтобы облегчить раскрытие кольца; определяемое
симметрией конротаторное течение этих предшествующих
процессов уже обсуждалось выше (ср. разд. 5).
О
А
+
(213)
I
N
(215)
(214)
(216)
Очень интересную проблему представляет возможность
участия в синхронных реакциях циклоприсоединения
таких неустойчивых соединений, как винилкарбен и его изо-
электронные аналоги. Эти молекулы могут существовать
в трех формах, различающихся числом л-электронов на
аллильной орбитали. Первая форма (217) с двумя
электронами в аллильной системе, как можно ожидать,
комбинирует с диенами в процессе [2+4], однако преобладает
независимая реакция этой молекулы как просто замещенного
карбена. Вторая форма (218) с четырьмя электронами в
аллильной системе может участвовать в нормальной
синхронной комбинации с 2я-электронными молекулами, в то
время как третья форма (219) является аналогом триплет-
ного карбена. Эти системы не были изучены достаточно
широко. Можно ожидать, что их электронные структуры и,
следовательно, ход реакций будут зависеть от природы
атомов скелета и заместителей.
(217)
(218)
(219)
99
Следует упомянуть о возможности аналогий 1,3-дипо-
лярного замещения, когда я-электронная система диполяр-
ного компонента включает большее число электронов. В
качестве мыслимых примеров можно назвать цис-форщ (220)
димерного нитрозосоединения, а-кето- (221) и а-нитрозо-
нитроны (222). Все эти соединения содержат бя-электрон-
ные системы, и синхронная комбинация с диенами
разрешена по симметрии.
N-N +
d- Ъ
(220)
Относительно 1,3-диполярного присоединения следует
сделать дополнительное замечание. Могут наблюдаться
такие реакции, в которых полярный характер компонентов
достаточно сильно выражен для того, чтобы
благоприятствовать двухстадийной комбинации, протекающей через
относительно стабильное диполярное промежуточное
соединение.
6,2. [2-f 2]-Циклоприсоединение в фотохимии
циклогексадиенонов и циклогексенонов
Вслед за пионерской работой Бэртона, Егера и сотр.
очень интересные фотохимические реакции
циклогексадиенонов и циклогексенонов были основательно изучены Чэп-
меном, Шаффнером, Шустером, Циммерманом и сотр.*
Было отмечено три типа первичных процессов
изомеризации: (223), (224), (225).
Вполне возможно, что все эти случаи включают реакции,
протекающие через триплетное п->- я*-состояние, хотя
установлено это было лишь для нескольких реакций.
Квантовые выходы этих процессов изменяются в очень широком
* Прекрасными указателями по структурным работам,
выполненным в этой области, являются два наиболее поздних обзора
[105].
R
R-
N
\
О"
(221)
R +,R
о
о-
(222)
100
о
■о-
о
(223)
О
2 3
~ °-0 ■ °-Q
/Оки* Л [/06]
/Сжкгс £ [707]
5 fioej
6 %U N_/
Г225;
интервале [109]. Циммерман предложил наиболее
подробный и остроумный механизм этих реакций [ПО, 111].
Мы рассмотрим стереохимические напряжения,
вызываемые сохранением орбитальной симметрии при этих
превращениях. Эти напряжения имеют место только при
синхронных реакциях. Известны изящные физические
измерения, которые, как предполагают, подтверждают
несинхронный механизм. Однако мы считаем необходимым указать
на те стереохимические данные, которые согласуются с
синхронным механизмом превращения.
Формально все эти реакции представляют собой
[а2+ я2]-циклоприсоединение. Если это синхронные
превращения возбужденных состояний, то такое циклоприсо-
единение должно протекать по типу [a2s+ n2s] или
1а^а~Г я^а-1-
В классах А и Б супраповерхностное участие в
реакции двойной 2,3-связи стереохимически невозможно, так
как оно приводит к траяс-сочлененным трех- и пяти-
членным кольцам. Следовательно, эти реакции должны идти
по типу [а2а+ я2а], что требует антараповерхностного
присоединения к двойной 2,3-связи и инверсии у атома С-4.
В классе В отсутствуют пространственные напряжения,
возникающие в классах А и Б. Здесь возможны как
[ст2а+ я2а]-, так и [CT2S+ л28]-процессы.
аятара-Присоединение к двойной связи и инверсия при
мигрирующем насыщенном углеродном атоме — это как
раз те явления, которые происходят при классическом пре-
101
вращении сантонина (226) в люмисантонин (227) 1112].
К сожалению, в этом случае протекает альтернативное
запрещенное по симметрии изменение с сохранением
конфигурации при С-4, которое приводит к стерически
маловероятному транс-сочлепеяию кольца. Поэтому едва ли можно
утверждать, что наблюдаемый путь реакции, хотя и
разрешенный по симметрии, во всех случаях обусловлен
геометрией системы.
Простейшая маркировка исходного циклогексадиено-
на (226) центром хиральности могла бы также привести к
решению вопроса. Рассмотрим следствия сохранения
конфигурации или ее инверсии у С-4 в таком хиральном цикло-
гексадиеноне (228)*. Неизменность или инверсия
конфигурации при С-4 возможны в двух случаях в зависимости от
того, происходит ли присоединение к С-3 сверху или снизу.
Если А и В — различные заместители, то эти пути
различаются стерическими факторами. Следует ожидать, что
соответствующие им продукты образуются в разных
соотношениях. Продукты инверсионного процесса диастереомерны
по отношению друг к другу и энантиомерны по
отношению к продуктам сохранения конфигурации.
Действительный ход перегруппировки простейшего хирального цикло-
гексадиенона не установлен, однако он исследуется Шусте-
ром [113], который независимо пришел к выводу, что этот
вопрос очень интересен.
* Мы рассмотрим здесь лишь следствия, касающиеся
присоединения к двойной связи, примыкающей к R. Разумеется, возможно
присоединение к двойной связи, расположенной напротив. Это
приводит к удвоению числа возможных продуктов
102
А В
Сохранение q \i-B А-Ч. О
конфигурации при С-4 %—f^r^R R-^5)—К
А
В
(228)
Обращение
конфигурации при С~4
В
°wfA
/17-R +
СМ
В-4
о
л>
Обширная область химии производных стероидов
позволяет подтвердить наши предположения. Очень
разнообразная фотохимия 1-дегидротестостеронацетата и его
метальных производных [114] начинается с хорошо известного
О
-ОЯ
(229)
(230)
R1
(231)
превращения 2,5-циклогексадиенона (229) в бицикло-
[3,1,01-гексенон (230), а затем в новый циклогексадиенон
(231) с известной стереохимией. При дальнейшем
фотолизе (231) образуются два или три изомера в зависимости
от заместителей. Структуры продуктов (232) — (234)
были установлены в работах [114]. Все эти молекулы —
продукты процессов [я2а+ а2а] в соответствии с
предсказанием, вытекающим из условий о сохранении орбитальной
симметрии в синхронных реакциях. Особо следует отметить
то обстоятельство, что в каждом акте фотоизомеризации
инверсия происходит у С-4. В случае замещенных
молекул (а) и (в) обнаруживают только один продукт (232),
в то время как при изменениях, включающих двойную
103
2,3-связь, по симметрии разрешены два продукта.
Подобным образом при реакциях с участием двойной 5,6-связи (а)
и (в) образуется только (234). В случае реакций (б) были
изолированы лишь три из четырех продуктов, которые
могут образоваться в процессах, разрешенных по симметрии.
Не был обнаружен ни один из четырех продуктов, которые
могли бы образоваться при протекании процессов,
запрещенных по симметрии, или в нестереоспецифических
процессах.
(234)
К сожалению, механизм, предложенный
Циммерманом [111] для реакций класса Л, приводит к тем же
продуктам, что и использование принципов сохранения
орбитальной симметрии в синхронных реакциях. Формулы (235) —
104
кх - $<к ш
(236)
(236а)
Я
(237а)
i
-юс мзх;-°я
(2J5y
(2J7>
R
(238)
(238) показывают сущность механизма, предложенного
Циммерманом [111]. Конечная стадия — перегруппировка
(236) ->• (236а) (разрешенная по симметрии) и обращение
связей (236а) ->- (237) являются формально циклопропил-
(243)
105
карбинильной перегруппировкой. Переход соединения (237)
в исходный циклогексадиенон (236) [ср. стрелки на (237а)]
запрещен по симметрии! Фактически имеются две
возможности для последней стадии всей последовательности
реакций. Их лучше всего представить как сигматропное
смещение [1, 2п] (см. разд. 7). Первая возможность состоит в
двух последовательных [1,2]-смещениях [(239) ->- (240) ->■
->■ (241) = (243)], а вторая — в перегруппировке [1,4]
[(242)-+ (241)].
Сигматропные [1,2]-смещения протекают с сохранением
конфигурации, а вынужденные [1,4]-смещения должны
протекать с инверсией в месте нахождения мигрирующего
углерода, т. е. оба альтернативных пути реакции стереохи-
мически неразличимы. Таким образом, реакции класса Л,
идут ли они по полностью синхронному механизму или по
стадиям, включающим элементы синхронности, приводят
к одним и тем же стереохимическим результатам. Хотя
физические доказательства протекания реакции по механизму
Циммермана очень убедительны, мы не считаем их
окончательными — по нашему мнению возможны оба
механизма.
Фотохимическая перегруппировка циклогексенонов
(реакции класса Б) — в высшей степени стереоспецифический
процесс. Весьма возможно, что он протекает синхронно.
Так, Чэпмен [115] нашел, что в реакциях (244) и (245)
(245)
хиральность сохраняется. Белус и Шаффнер [116]
исследовали реакцию с помощью очень чувствительного теста;
изомеризация (246) протекает с фиксацией при С-1 и
инверсией при С-10. Инверсию при С-10 вызывают стери-
106
ОАс
ОАс
(246)
Me |
D
ческие причины, чего нельзя сказать о фиксации при С-1.
Это противоречит предположению о радикальном разрыве
связи Ci—Ci0.
Обратимся теперь к реакциям класса В. В этом случае
отсутствуют пространственные препятствия для
синхронных процессов [я28+ ст28] или [я2а+ а2а1- Была изучена
[108] реакция (247), в которой образуются два
разрешенных по симметрии продукта циклоприсоединения. Однако
и в этом случае отсутствие траяоконденсированных колец
приводит к тем же стереохимическим последствиям поста-
дийных процессов.
НяС
Н3С
(247
107
Есть еще два фотохимических превращения енонов,
которые можно отнести к категории [2+2]-циклоприсоедине-
ния. Первое превращение — изомеризация несопряженного
циклогексенона (248) [117]:
(248)
О
Ниже показана последовательность стереохимически
разрешенных аятара-реакций двойной связи с фиксацией
или инверсией при С-2:
1 2 ■»- 2 1
Сохранение
конфигурации при С~2
А В
Обращение
конфигурации при С-2
^а
(249)
Был исследован фотолиз производных стероидов этого
класса [117]. Из двух возможных продуктов образуется
только тот, у которого циклопропановое кольцо является
а-ориентированным (250). Так как при С-2 отсутствует
о
О
(250)
метка, то вопрос о сохранении или инверсии конфигурации
остается открытым. Мы полагаем, что впоследствии будет
108
доказана инверсия при С-2. Аналогичная ситуация
существует и во втором случае [118]:
hv
(251)
(252)
В литературе приведен ряд примеров фотохимического
циклоприсоединения [я2а+ 02а1 с участием простых оле-
финов и диенов. Так, Гриффин [119] наблюдал необычную
циклизацию пропилен-> циклопропан [(253), (254)],
Ph
н --ThH
hv
н
Ph
Ph
Н
н
н
(253)
R
Н
R
Ph-CV
П-^- ^>R
R-
I
Н
Н
(254)
сопровождающуюся миграцией групп. Эти реакции вполне
могут быть синхронными, однако пока отсутствует
определенная стереохимическая информация. При фотолизе
4,4-дифенилциклогексенона (255) [120] (реакция такого же
hv
\—' л
Ph
— +
(255)
140
типа) стереохимия реакции была установлена; в этом
случае преобладает продукт антараповерхностного присоеди-
109
нения к двойной связи и инверсии при С-4. Еще один
возможный пример реакции [G2a+ л2а] — недавно изученный
фотолиз 3,19-дикето-4-андростен-17(3-илацетата (256) [121]:
H(D)
hv
(256)
Реакция (257), изученная Сауерсом [122], также проте-
//^
D
D
hv
(257)
D
кает по типу [G2a+ я2а1. Образование соединения (259)
при фотолизе циклооктатриена (258) должно протекать по
hv
(258)
(259)
типу [я2а+ 02а], если реакция синхронная; показано [123],
что этот процесс идет без перемещения водорода.
ПО
S? hv
(260)
Аналогичный случай (260) наблюдал Эдман [124].
Синхронное циклоприсоединение может включать сг-связь
при атомах 3 и 7 (261) или сг-связь при атомах 2 и 3 (262).
Синхронный характер этой реакции можно подтвердить,
используя хиральные исходные соединения.
hv
(261)
(262)
К этому же типу принадлежат превращения дибензоби-
цикло-[2,2,2]-октатриена (263) в дибензосемибульвален
[125] и маркированного бензобицикло-[2,2,2]-октатриена
(264) в бензосемибульвален [126, 127]. Следует
подчеркнуть, что ни одна из этих реакций не должна быть
синхронной. В действительности существует доказательство того,
что в аналогичном случае с барреленом процесс является
стадийным [128].
hv
(263)
111
"^ D
(264)
Теперь мы должны ответить на вопрос: как может идти
по синхронному механизму процесс, протекающий через
триплетное состояние молекулы? По-видимому,
закономерности орбитальной симметрии диктуют молекуле в
возбужденном состоянии определенный характер движения,
которое облегчает одно направление реакции и затрудняет
другое. Система не обязательно должна включать
возбужденное состояние продуктов. В возбужденном состоянии
возникают движения молекул реагирующих веществ,
разрешенные по симметрии. Одновременно протекают безызлу-
чательные переходы продуктов в основное состояние.
Так как рациональная физическая картина таких переходов
отсутствует, то предположение о протекании таких
процессов не должно встретить больше возражений, чем
предположение о любом другом безызлучательном переходе.
6.3. [2+2+2]-Циклоприсоединение
Детальное рассмотрение синхронного [2+2+2]-цикло-
присоединения (или циклораспада) очень полезно для
понимания принципов сохранения орбитальной симметрии.
Подобное рассмотрение выявляет не только некоторые
новые факторы, но позволяет провести некоторые
сопоставления, которые углубляют понимание правил сохранения
орбитальной симметрии как фундаментального дополнения
наших знаний о химической связи.
Рассмотрим распад молекулы циклогексана через
переходное состояние в форме ванны на три молекулы этилена.
Сначала распределим 6 электронов разрывающихся связей
попарно по делокализованным а-орбиталям (265), (266)
и (267) (ср. разд. 3.1). Отметим, что единственный элемент
112
1?
(265)
(266)
симметрии, определяемым геометрией системы,—
зеркальная плоскость т. Показанные орбитали, как и требуется,
либо симметричны, либо антисимметричны относительно
этой плоскости. Особенно важно в этом случае отметить
фундаментальный принцип, состоящий в том, что
несимметричные молекулярные орбитали являются
неприемлемыми. Например, орбиталь (268) нереальна и должна
исчезнуть при добавлении сопряженной с ней орбитали (269).
Кроме того, важно отметить, что орбитали (267) и (265)
можно заменить их суммой (270) и разностью (271). Это неизме-
(270)
(271)
нит результатов, которые можно получить при их
использовании. Однако в общем случае необходимость получить
наибольшую информацию заставляет выбрать те орбитали,
5—1556
113
которые дают максимальную степень делокализации, т. е.
в данном случае (265) и (267), а не (270) и (271). Как бы
то ни было, никакие другие орбитали не используются в
данном случае. Важно подчеркнуть, что полученные
выводы не содержат произвола или неоднозначности.
Шесть электронов я-орбиталей образующегося
продукта — молекул этилена — должны быть распределены по
обобщенным молекулярным орбиталям (272) — (274).
чч чч
(272)
(273)
(274)
Наконец, нужно обратить внимание на один очень
важный момент. Геометрическое соответствие между исходным
соединением и продуктами [см. (275)] требует, чтобы в
(275)
ходе реакции происходило вращение относительно 1,2-
и 3,4-осей. Вращение должно происходить таким образом,
чтобы замещающие группы при С-1 и С-2 с одной стороны
и группы при С-3 и С-4 с другой стороны были расположены
в тех же плоскостях, в которых они находятся в продуктах
реакции — молекулах этилена. A priori, исходя из чисто
геометрических соображений, эти перемещения могут быть
как конротаторными (276), так и дисротаторными (277);
в каждом случае существуют две возможности, которые
показаны стрелками на схемах (276) и (277).
114
{277)
Теперь мы достаточно подготовлены для «разрушения»
нашей молекулы циклогексана. В дальнейшем будет
показано, что при выбранной нами геометрии переходного
состояния имеется два и только два разрешенных по симметрии
пути, по которым может происходить распад.
1. Пусть сначала два электрона переходят с орбитали
(265) на орбиталь (272), что может происходить при
сохранении орбитальной симметрии. Тогда сразу становится
ясным, что два электрона орбитали (267) должны занять
орбиталь продукта (273). Таким образом, из всех
перечисленных выше путей вращения должно происходить
только дисротаторное вращение, направленное внутрь (278).
Остается только разместить на орбитали продукта (274)
оставшуюся пару а-электронов, которая находилась в
начале реакции на орбитали (266). Здесь следует отметить,
что смешение занятой орбитали (266) с незанятой
разрыхляющей сг*-орбиталью скелета (279) также приводит
к направленному внутрь дисротаторному вращению
относительно осей 1,2 и 3,4. Разрешенный по симметрии
процесс, который был сейчас рассмотрен очень детально, можно
назвать реакцией циклораспада —L2S+ ^2S+ jt2sl-
Очевидно, что совершенно аналогичный анализ применим и для
обратного процесса — разрешенной по симметрии реакции
циклоприсоединения [n2s+ я28+ я28].
^2
^
(278)
(279) \/
5*
115
2. Проведем теперь альтернативное рассмотрение. Пусть
по мере протекания циклораспада два электрона с орбита-
ли (267) переходят на орбиталь (272). Тогда у электронов
с орбитали (265) «нет другого выхода», кроме перехода
на орбиталь продукта (273). Следовательно, в отличие от
случая, рассмотренного выше, разрыв связи (2,3)
углеродного скелета должен сопровождаться дисротаторным
смещением, направленным наружу (280). Как и раньше,
(Z80)
оставшаяся пара электронов, занимавшая орбиталь (266),
занимает новое положение — орбиталь (274). Точно так же
смешение орбитали (266) с незанятой разрыхляющей сг*-ор-
биталью скелета (281) согласуется с дисротаторным
движением, направленным наружу, которое обусловлено
соотношениями симметрии между орбиталями (280) и (274).
Рассмотренный процесс можно отнести к разрешенной по
симметрии реакции циклораспада —[n2s+ л2а+ я2а]>
обратной разрешенной по симметрии реакции циклоприсоеди-
нения [я28+ я2а+
^
(281)
Таким образом, мы установили, что существуют два
возможных разрешенных по симметрии циклических
процесса [2+2+2], которые идут через переходное состояние,,
обладающее одной плоскостью симметрии. При различных
конкретных обстоятельствах тому или иному из этих путей
благоприятствуют структурные, стерические или энтропии-
116
ные факторы. Возникает вопрос, существуют ли не
рассмотренные до сих пор стереоэлектронные факторы, которые
всегда благоприятствуют одному из двух путей. Обсудим
более подробно смешение антисимметричной связывающей
орбитали (266) с антисимметричной разрыхляющей орби-
талью (279) [или (281)]. Оказывается, что процесс s, s, s
должен быть предпочтительней процесса s, а, а. Диаграм-
(282) (283)
мы (282) и (283) показывают в плоскости углы между
рассматриваемыми орбиталями в начале реакции. Ясно
видно, что при движении, показанном на схеме (282), легче
происходит более благоприятное перекрывание,
понижающее энергию, и что подъем к переходному состоянию для
процесса s,a,a происходит значительно медленнее*.
Несколько аспектов [2+2+2]-реакций заслуживают
дальнейшего, хотя и краткого рассмотрения. Мы
предоставляем читателю обдумать самостоятельно детали
следующих ниже пунктов и тем самым проверить понимание
применения принципов симметрии:
а) имеется континуум топологически эквивалентных
разрешенных по симметрии термических процессов [n2s+
+ jt2s+ я28]. Их переходные состояния варьируются
от состояния, описанного выше, до состояния, геометрия
которого подобна циклогексановому кольцу в форме кресла;
б) имеются еще два разрешенных по симметрии
термических процесса [я28+ л2а+ я2а], которые отличны по
топологии от процесса, описанного выше. Эти процессы
протекают через энантиомерные переходные состояния,
имеющие ось симметрии второго порядка;
в) имеются неразрешенные по симметрии термические
процессы [я28+ я28+
я^ a J»
* Впоследствии это предположение было экспериментально
подтверждено [128а].
117
г) имеются неразрешенные по симметрии термические
процессы [я2а+ я2а+ я2а1;
д) для реакций с участием занятых разрыхляющих
уровней (например, с участием молекул в электронно-
возбужденных состояниях) отсутствуют разрешенные по
симметрии [2+2+2]-процессы типа s,s,s или s,a,a.
Однако имеются два разрешенных энантиомерных пути s, s, a,
а также два разрешенных и также энантиомерных пути
а,а,а;
е) каждая полностью антара-комбипация [2+2+2+...]
запрещена по симметрии в основном состоянии и разрешена
по симметрии в возбужденном состоянии, в котором
заселена разрыхляющая орбиталь одной из участвующих в
реакциях этиленовых единиц.
Надеясь на то, что не будет возражений против
обсуждения процессов, которые при поверхностном рассмотрении
представляются невозможными, упомянем молекулы (284)
и (286). Эти молекулы в целом являются ненапряженными,
(284)
(286) (287)
однако они до сих пор не известны. Двойные связи этих
молекул расположены в углеродном скелете таким образом,
чтобы благоприятствовать фотохимическому превращению
(285)
118
в (285) и (287) по полностью антара-пролессу (см.
пункт «е»).
Остается только привести несколько известных
примеров, иллюстрирующих перечисленные выше пункты.
Большинство известных реакций принадлежит к разрешенному
по симметрии типу s,s,s. Возможно, что наиболее известно
присоединение олефинов к бициклогептадиену [129]:
(288)
(289)
Другие примеры, относящиеся к той же группе,
приведены ниже:
NCL XCN
(290 )[i30]
NC-^-CN
NC^ZWCN
NC CN
(291)
(293)
119
CN
CN
(294) [132]
(295)
6.4. Призман
Чему обязана эта фантастическая молекула (296) своей
способностью существовать? Ее энергия по крайней мере
на 90 ккал/моль больше, чем энергия ее изомера, бензола
[133]. Для превращения призмана в бензол можно легко
написать процесс атомного перемещения [ср. (297) ->-
-> (298)1
(2%)
(298)
В действительности необходимо еще 33 ккал/моль для
того, чтобы превратить молекулу гексаметилпризмана,
обладающую чрезвычайно напряженной структурой, в
молекулу гексаметилбензола [133]. Без учета ограничений,
которые накладывают соотношения орбитальной симметрии
на образование и разрыв связей, избыток энергии призмана,
образно говоря, напоминает испуганного тигра, который
не может разорвать бумажную клетку.
При подробном рассмотрении с использованием
принципов сохранения орбитальной симметрии выясняется, что
мы имеем дело отнюдь не с бумажной клеткой, а с весьма
надежным барьером. Во-первых, необходимо построить"
обобщенные молекулярные орбитали (299), на которых
надо разместить 6 электронов трех ст-связей, распадаю-
120
Разрыхляюшдя орбиталь
•d*P
•Ofclo
^s
^A
o^>U
^A
•<£>•
Vs
^A
^S
s
to)
s
(°2)
Связывающая орбиталь
(299)
S
<Ръ)
щихся, когда призман превращается в бензол. Разорвем
теперь три ст-связи, сохраняя при этом симметрию орбита-
лей, определяемую геометрией молекулы призмана. Отметим,
что ст2- и ст*2-орбитали одинаковой симметрии при
взаимодействии дают две новые орбитали, которые можно
представить в виде суммы и разности исходных орбиталей
(изменения орбиталей показаны на рис. 27).
Что представляют собой эти новые орбитали? Это как
раз орбитали бензола. В то время как две связывающие
сг-орбитали призмана (ст2 и а3) превращаются в две
связывающие орбитали бензола (я2 и nd), третья орбиталь
превращается в разрыхляющую орбиталь продукта (cti-> я*2).
Физический смысл превращения состоит в том, что эта
корреляция вводит разрыхляющий повышающий энергию
компонент в переходное состояние реакции. Это и есть тот
компонент, который делает превращение призмана (300)
в бензол (301), запрещенным по симметрии, и способствует
существованию призмана.
121
Jo#b
•<$>•
%*
о
*1
Рис. 27. Образование u-орбиталей бензола из а-орбиталей
призмана.
(300)
(301)
Значительно больше сведений о природе обусловленных
симметрией соотношений между связями можно получить
при более подробном обсуждении молекулы призмана.
Прежде всего отметим, что формально превращение
призмана в бензол можно рассматривать как реакцию циклорас-
пада —[K2S+ я2а+ я2а]. Из материала предыдущего
раздела видно, что термические реакции этого класса
разрешены по симметрии. Почему же превращение призман ->-
бензол исключено из класса разрешенных по симметрии
реакций, к которым его можно отнести при поверхностном
рассмотрении? При точном анализе корреляции
связывание — разрыхление орбиталей аг± (302) и я*2 (303) стано-
(302) (303)
вится ясно, что при этом превращении образуются
содержащие узлы разрыхляющие повышающие энергию
зависимости между 1,5-, 5,6-, 2,4- и 3,4-связями, обусловленные
независимостью скелетных ст-связей в молекуле призмана
и отличают превращение призман ->- бензол от других
реакций [2+2+2]-циклораспада. Если бы ст-скелет
отсутствовал, то эти разрыхляющие повышающие энергию
компоненты также отсутствовали бы.
Это утверждение можно проиллюстрировать,
рассматривая некоторые родственные призману молекулы.
Рассмотрим сначала квадрициклен (304) и трициклогексан
5
2Lz^ ^^з
(304) (305)
123
(305). На первый взгляд можно ожидать, что симметрия
разрешает этим молекулам переход в циклогептатриен (306)
;0 С:
(306) (307)
и гексатриен (307) соответственно. Однако анализ в
соответствии с указанными выше принципами показывает, что
эти реакции из-за ст-скелета принадлежат к группе
запрещенных по симметрии процессов, так же как в случае приз-
мана. Фактически квадрициклен не превращается в
циклогептатриен даже при очень высоких температурах [134].
В связи с этим представляет особый интерес
кислородный аналог этого соединения (308). Продукт его
(308) ^ (309)
—[jt2s+ я2а+ я2аЬциклораспада — оксепин (309).
Орбитальная корреляция, аналогичная той, которая приводит
в других случаях к разрыхляющему уровню, дает теперь
высшую занятую орбиталь основного состояния, которая,
по-видимому, является слабо связывающей. В соответствии
с этим производные окса-аналогов гладко переходят в ок-
сепияы при нагревании [135, 136].
Другое родственное призману соединение — бицикло-
[2,2,0]-гексан (310). В этом соединении отсутствуют ст-связи
1,5 и 2,4; поэтому при приближении молекулы к
переходному состоянию в реакции циклораспада —[л28+я2а+я2а1
не возникает узловой плоскости симметрии, что в свою
очередь делает термическое превращение соединения (310)
в этилен и бутадиен (311) разрешенным по симметрии
124
(310) (311)
В действительности этого до сих пор не наблюдали. При
нагревании незамещенный бициклогексан превращается в
1,5-гексадиен (312) [137] либо по двухстадийному
механизму [с промежуточной частицей (313)], либо по механизму
[a 2S + a 2а], разрешенному по симметрии.
С: О
з
(312) (313)
6.5. [2+ 2+2+2]-Циклоприсоединение
Реакция циклоприсоединения [тс 2 + тс 2 + т. 2 + г2]
разрешена по симметрии:
а) для реагентов в основном состоянии, когда нечетное
число компонентов реагирует супраповерхностно;
б) когда один компонент находится в возбужденном
состоянии, а четное число компонентов реагирует
супраповерхностно.
Очевидно, что энтропийные факторы приводят к тому,
что полимолекулярные реакции этого типа очень
маловероятны. Так же очевидно, что этих трудностей нет, если
все реагирующие компоненты объединены в одной
молекуле в геометрически благоприятную форму. Мы уже
приводили соответствующие примеры [(284) и (286)].
Ряд факторов препятствует также многокомпонентному
распаду цикла на фрагменты, разрешенному по симметрии.
Каждое превращение а-связи в тг-связь является
эндотермическим и требует ~ 20 ккал.
Некоторые особенности циклоприсоединения к 2 + к 2 +
+к 2 + тг 2] маскируются другими процессами. Рассмотрим,
например, двойное циклоприсоединение ацетилена к двум
молекулам этилена (314). Анализ орбиталей показывает,
125
{314)
что две я-связи ацетилена взаимодействуют независимо.
Наблюдали по крайней мере две такие реакции — (315)-+
-+ (316) и (317) -+ (318) [138, 139].
Р
О
hv
О
н3с— ^=—сн3
(315)
hv
ноос—==-соон ноо£ соон
(317) (318)
Очень интересен распад на фрагменты (319).
Аналогичное разложение пенталена до диацетилена и двух молекул
ацетилена рассматривалось в литературе как возможное
[140]. В настоящее время можно сказать, что это
запрещенный по симметрии термический процесс.
со
(319)
се
7. ТЕОРИЯ СИГМАТРОПНЫХ РЕАКЦИЙ
Сигматропным изменением порядка [i,j] называют не-
катализируемую внутримолекулярную миграцию ст-связи,
к которой присоединены одна или несколько я-электронных
систем, в новое положение, отделенное от исходного поло-
126
жения i — 1 и / — 1 атомами. Так, хорошо известные
перегруппировки Клайзена и Коупа являются сигматропными
изменениями порядка [3,3].
A priori существует две топологически разные
возможности сигматропной миграции. Они проиллюстрированы на
рис. 28 для [1,5]-миграции атома водорода. В первом, супра-
поверхностном, процессе мигрирующий атом водорода
находится все время на одной и той же стороне я-системы.
Во-втором, антараповерхностном, процессе мигрирующий
атом переходит с верхней стороны углеродного конца
молекулы на нижнюю сторону другого конца молекулы. Нет
необходимости использовать корреляционную диаграмму
С у пра поверхностное %//
смещение C^^D
А
Ля тара поверхностное // ! >ь
смещение ОуЧ}/
/А' в
Р и с. 28. Супраповерхностные и антараповерхностные
сдвиги атома водорода.
[1,5]-
лс=с
%^ %^%ц
Изолированные
орбитами
а-Ь
^ %ц
а + Ь
Взаимодействующие
ордитсили до перехода
водорода
Вза имодействующие
орбитами после
перехода водорода
Рис. 29. Сохранение орбитальной симметрии при супраповерх-
ностном [1,3]-сдвиге атома водорода.
127
для анализа этих реакций, так как элементы молекулярной
симметрии есть только у переходного состояния и их нет у
молекул исходных соединений и продуктов. Мы опишем
несколько эквивалентных методов для анализа этих
реакций.
1. Использование принципа сохранения орбитальной
симметрии иллюстрируется здесь супраповерхностным
[1,3]-перемещением водорода. Соответствующие корреляции
показаны на рис. 29. Ясно, что если симметрия
сохраняется, то два электрона могут занять связывающую орби-
таль (ст или я) продукта, однако два других электрона
должны занять орбиталь ст* или я*. В этом случае реакция
запрещена по симметрии.
2. При сигматропной [1,/]-миграции водорода в
пределах полностью ^ис-полиенового скелета 1(320) -> (321)]
R2C = СН — (СН = СН)* — CHR2 —
{320)
-+ R2CH — (СН = СН)* — СН = CR2
(321)
переходное состояние можно представить состоящим из
комбинации орбитали атома водорода с той орбиталью
радикала, которая содержит (2&+3) я-электронов. Высшая
занятая орбиталь углеродного скелета системы является несвя-
зывающей аллильной орбиталью, обладающей симметрией
(322). Пусть атом водорода присоединен к концу такой
системы. Для того чтобы в переходном состоянии не было
орбиталей высокой энергии, необходимо сохранение
положительного перекрывания между орбиталью углеродного
скелета и орбиталью мигрирующего атома. В результате
возникают разрешенные по симметрии супраповерхностная
миграция (для нечетных k) и антараповерхностная миграция
(для четных k).
1 2 3 Л 5 6 7 • j
(322)
128
3. Теперь рассмотрим переходное состояние с другой,
но эквивалентной точки зрения. Сначала изобразим трех-
центровую связь, которая включает концы углеродной цепи
и мигрирующий атом водорода. Трехцентровая связь имеет
набор уровней, типичная схема которого изображена на
•о
о*
(>•
•о о о*
Рис. 30. Взаимосвязь между орбиталями в трехцентровой связи.
Знаки симметрии не зависят от того, используется ли для классификации
зеркальная плоскость или ось вращения второго порядка.
S-
А-
-А
- S
-А
А
S
А
S
S-H-
S
-А
s-«-
Р и с . 31.
Взаимодействие несвязывающих
антисимметричных
уровней трехцентровой
связи и остатка молекулы
при антараповерхност-
ном [1,7]-сдвиге атома
водорода (k=2)
Рис. 32. Отсутствие
взаимодействия между нес-
вязывающими уровнями
трехцентровой связи и
остатка молекулы при суп-
раповерхностном [1,7]-
сдвиге атома водорода
(*=2).
рис. 30. Остается еще система из 2&+1 тс-орбиталей
углеродной цепи. Проведем классификацию молекулярных орби-
талей остатка цепи относительно элементов симметрии
переходного состояния — оси второго порядка и
зеркальной плоскости. Здесь возможны два случая, различающих-
129
ся по симметрии несвязывающей орбитали остатка цепи.
В первом случае (рис. 31) эта орбиталь антисимметрична
и сильно взаимодействует с центральной орбиталью
трехцентровой связи. Во втором случае (рис. 32) орбиталь
остатка симметрична и не может взаимодействовать
аналогичным образом. Обычно при взаимодействии один
уровень стабилизуется, а другой дестабилизуется. Всего мы
рассматриваем 2£+4 электронов, из которых 2k размещены
на связывающих уровнях полиенила и 2 — на низшей
орбитали трехцентровой связи. Таким образом, остается еще
2 электрона для размещения на низшей из двух орбиталей,
возникающих при взаимодействии. Отсюда следует, что
при реакции в основном состоянии первый случай
разрешен по симметрии. В общем, для того чтобы несвязывающая
орбиталь (2&+1) орбитального остатка л-системы была бы
симметрична при соответствующей операции симметрии,
реакции должны протекать через переходное состояние,
имеющее зеркальную плоскость (при нечетном k) или ось
второго порядка (при четном k).
Для реакций в возбужденном состоянии, как в случае
электроциклических реакций и циклоприсоединения,
правила отбора обратны тем, которые применимы для реакций
в основном состоянии.
i+i
Aq
4?+ 2
Основное состояние
антара, супра
супра, антара
супра, супра
антара, антара
Возбужденное состояние
супра, супра
антара, антара
антара, супра
супра, антара
Р и с. 33. Правила отбора для сигматропных реакций порядка
[/, /] при i>\ и /> 1.
Для сигматропных изменений порядка U',/], в которых
i и / больше 1, мигрирующая группа состоит более чем из
одного атома. Поэтому необходимо установить
топологическое различие между двумя л-системами, по которым
мигрирует сг-связь. Правила отбора суммированы на рис. 33,
J 30
При обсуждении [1,/]-сигматропных изменений были
рассмотрены только те случаи, в которых or-орбиталь
мигрирующей группы взаимодействует с л;-системой в
переходном состоянии, а миграция происходит с сохранением
конфигурации при мигрирующем центре. Если мигрирующая
группа имеет доступную я-орбиталь и не имеет таких
заместителей, которые создали бы в переходном состоянии сте-
рически невозможную ситуацию, необходимо
рассматривать также возможность превращения с участием этой
л-орбитали. Ясно, что такое превращение должно
протекать с инверсией при мигрирующем центре [ср. (323)].
В этом случае правила отбора должны быть обратны тем,
которые приведены на рис. 33.
(323)
Следует отметить ряд особенностей сигматропных
изменений:
а) антараповерхностные процессы не возможны для
малых или средних циклов;
б) сопротивление углеродного скелета замыканию
зт-электронных систем может привести к тому, что
превращение станет затрудненным или невозможным. Это
справедливо, например, для антараповерхностного
[1,Замещения и не влияет на [1,7]-миграцию;
в) циклопропановое кольцо может заменить л-связь в
углеродном скелете при сигматропном изменении;
г) для сигматропных изменений в ионных частицах
также применимы аргументы, основанные на орбитальной
симметрии. Так, супраповерхностный [1,2]-сдвиг в кар-
бониевом ионе разрешен по симметрии и хорошо известен.
Предсказанный [1,4]-сдвиг с инверсией мигрирующей
группы в бутен-2-1-ильном катионе недавно удалось обнаружить.
Можно ожидать, что [1,6]-сдвиг в
гексадиен-2,4-1-ильном катионе идет через легко достижимое супраповерх-
ностное переходное состояние.
Остается добавить, что результаты обсуждения
материала предыдущих разделов позволяют рассматривать сигмат-
131
ропные изменения как реакции циклоприсоединения.
Например, [1,3]-сигматропный процесс (324) -> (325)
является реакцией [а2+я2], а [3,3]-сигматропная
перегруппировка (326) -> (327) — синхронным циклоприсоединением
[я2+02+я2].
-*- с о - о
(324) (325) (326) (327)
Исходя из этой точки зрения, рассмотрим подробно сте-
реохимические аспекты [1,3]-сигматропного изменения.
Известно, что процессы, разрешенные по симметрии,
принадлежат либо к типу [а28+л2а]} либо к типу [а2а+я28]. На
рис. 34 показаны все возможные варианты. Сразу ясно, что
Супраповерхностный
[1,3]-сдвиг с
инверсией при R
Супраповерхностный
[1,?>]-сдвиг с
инверсией при R
Антарапоеерхнсстный
/
/
1
1
Jtssss
/
I
1
1
ь
Y^"*
N
\
R
f
\
К
*\
[1,3]- сдвиг с сохране- \
нием конфигурации при R '
Рис. 34. Стереохимические возможности при сигматропных
[1,3]-сдвигах,
две первые альтернативы представляют собой просто
разные способы рассмотрения одного и того же физического
процесса. Другими словами, как и следовало ожидать^
такой анализ дает те же самые выводы, что и предыдущее
рассмотрение сигматропных реакций.
132
7.1. Примеры сигматропных реакций
Известно немного термических некатализируемых
[1,3]-смещений*. Те, которые можно формально отнести
к этой категории, например перегруппировка винилцикло-
пропан -> циклопентен, протекают с такой высокой
энергией активации (~50 ккал/моль), что энергетическая
поверхность для синхронной реакции не далеко отстоит от
энергетической поверхности ступенчатого протекания процесса.
Тем не менее Берсон и Нельсон [142] недавно сделали
сенсационное наблюдение того, что (328) в результате
синхронного разрешенного по симметрии супраповерхностно-
го [1,3]-смещения переходит в (329) при 307° с инверсией
при мигрирующем центре, обозначенном звездочкой.
(328) (329)
Относительная стабильность триена (330) обусловлена
[143] тем, что для некатализируемой перегруппировки его
в толуол необходимо запрещенное по симметрии
[1,Замещение.
О - Осн'
(330)
Перегруппировка эфира (331) особенно интересна как
пример [1,3]-сигматропного превращения. Исследования
оптически активного вещества показали, что реакция очень
стереоспецифична [144]. Из двух a priori возможных
разрешенных по симметрии процессов в углеродном скелете —
супраповерхностной [1,3]-миграции с инверсией при миг-
* Например, [1Л4С]-пропилен не перегруппировывается в
[3-14С]-пропилен [141].
133
рирующем атоме и антараповерхностном сдвиге с
сохранением конфигурации — последний представляется
невозможным из-за напряжения в сг-скелете. Так как в кольцо
н3соос
н
н
н
н
н н
н
СООСНз
Н3СООС
СООСНо
(331)
н
(332)
н
н
соосн.
н
Н3СООС
(333)
введены заместители, разрешенный процесс может быть
реализован двумя электронно идентичными, однако сте-
рически разными путями. Следовательно, можем
предсказать, что эфир (331) должен переходить в (332) и (или)
(333). В настоящее время получено доказательство [144],
что два образующихся в действительности изомера
являются как раз предсказанными веществами.
Выше упоминалось, что энергия активации
конверсии винилциклопропан->-циклопентен [145, 146] равна
~50 ккал/моль [146]. Сам процесс разрыва связи в
циклопропане требует энергии активации ~63 ккал/моль [147],
аллильная стабилизация дает ~13 ккал/моль [148].
Следовательно, если полностью используется энергия аллильнои
стабилизации бирадикальной промежуточной частицы, то
термодинамически возможен двухстадииныи несинхронный
путь превращения винилциклопропана в циклопентен.
Некоторые предварительные исследования подтвердили [149]
такой механизм. Тем не менее недавно было установлено,
что разрешенный по симметрии синхронный процесс
происходит, несмотря на геометрические искажения,
препятствующие процессу. Целесообразно рассмотреть стереохи-
134
мические последствия возможных синхронных процессов
для превращения винилциклопропан ->- циклопентен.
Рассмотрим максимально помеченное производное (334).
Разрешены по симметрии следующие реакции:
а) антараповерхностное [1,3]-смещение 1,2-связи к С-5
с сохранением конфигурации при С-2, которое приводит
к изомеру (335);
б) супраповерхностное [1,3]-смещение 1,2-связи к С-5
с инверсией при С-2, которое приводит к изомеру (336).
Изомер (337) нельзя получить в разрешенном по
симметрии процессе.
R /A. R R /==\ . R /==\ R
^: .<р^ К^>.
■ i
R
(334) (335) (336)
(337)
Фотохимические [1,3]-перегруппировки, разрешенные по
симметрии как супраповерхностные смещения,
наблюдались во многих случаях [150]. Характерные термические
[1,5]-сдвиги наблюдали во многих случаях*. Простейший
случай [1,5]-сдвига в 1,3-пентадиене был изучен Ротом и
Кёнигом [152]. Эти авторы сравнили перегруппировки
соединений (338) и (339), при этом они наблюдали большой
D2C CH3 H2C CD3
(338) (339)
* Ссылки на литературу можно найти в [151]. Недавно был
открыт интересный [1,5]-сдвиг метильной группы [151а].
135
кинематический изотопный эффект (1,22 при 25°), что
согласуется с высокой степенью симметрии переходного
состояния в синхронном процессе. Энергия активации
перегруппировки составляет 35 ккал/моль. Прямое подтверждение
синхронного супраповерхностного характера сигматроп-
ного [1,5]-сдвига атома водорода очень изящно было
получено теми же исследователями [153], которые показали
полную стереоспецифичность превращения соединения (340)
в (341) и (342). Многочисленные стереоспецифические
н5с2
СН,
Н5С2
СН3
Н,С
(342)
сигматропные [1,5]-смещения наблюдали [154—156] в ци-
клопентадиенах и циклогептатриенах.
Абсолютную предпочтительность [1,5]-сдвигов по
сравнению с [1,3]-сдвигами в циклических системах очень
изящно показал Рот. Так, дейтерий в соединении (343) при
повышенной температуре распределяется равномерно по всем
неароматическим положениям вследствие [1,51-сдвигов, хотя
это должно происходить через стадию образования
нестабильного изоиндена (344). Однако под действием основных
катализаторов происходит [1,3]-смещение [157].
<f
Н D
г
н
(343)
ОУ1
(344)
136
Еще один тест на возможность протекания термического
[1,3]-с^ещения оказался отрицательным [158]. В 7,8-дидей-
тероциклооктатриене-1,3,5 (345) дейтерий должен
распределяться статистически по всем положениям вследствие
серии обратимых сигматропных [1,3]-сдвигов с участием
изомерного циклооктатриена-1,3,6. С другой стороны,
последовательные [1,5]-смещения должны перемещать метки
только в положения 3, 4, 7 и 8; последний результат
наблюдали экспериментально.
(D) (D)
(D)^==V(D) 1,,ь| (/=>уа>) [1,з1^ (Р)^/==>у(р>
(D) (D)
(345)
Фотохимические [1,51-сдвиги неизвестной пока
стереохимии наблюдали для соединений с открытой цепью, но
не наблюдали для циклических углеводородов [159] и гете-
роциклов [160].
9
Я
I
U.SL ^ NJ
D
Рис. 35. Реакции изомеризации метилзамещенных циклогептат-
риенов, проходящие через серию сигматропных [1
^-перегруппировок и электроциклических реакций.
Берсон и Виллкот [161] наблюдали заслуживающие
внимания серии изомеризации метилзамещенных циклогептат-
риенов. Их результаты, суммированные на рис. 35,
представляют собой серию сигматропных [1,5]-перегруппиро-
вок и электроциклических реакций. Супраповерхностные
[1,51-сдвиги, по-видимому, должны протекать с сохранени-
137
ем конфигурации при смещающемся центре. Это условие
означает, что заместитель циклопропанового кольца
поворачивается при каждом сдвиге на 180°. Берсон и Виллкот
[161] предложили прекрасную экспериментальную проверку
этого предположения, основанную на пиролизе оптически
активного соединения (346). Если реакцию контролируют
А В
(346)
принципы минимально возможного движения, то должны
происходить запрещенные по симметрии [1,5]-сдвиги с
инверсией, которые приводят к рацемизации; в то же время
предсказанные сдвиги с сохранением конфигурации не
должны приводить к потере оптической активности (рис. 36).
х^
W
Рис. 36. Возможные положения заместителей в кольце
циклопропана, которое превращается в циклогексадиеновое кольцо
через серию сигматропных [1,5]-смещений.
Слева: с сохранением конфигурации; справа: с инверсией. Циклопропановое
кольцо расположено перпендикулярно плоскости чертежа и поэтому невидимо.
Перегруппировки Коупа и Клайзена являются сигмат-
ропными [3,3]-смещениями; они изучены на столь многих
примерах [162], что здесь нет возможности их перечислить.
Обе перегруппировки разрешены как супра,супра- и ан-
тара,антара-реак1хии. С первого взгляда кажется
очевидным, что переходное состояние для антара,антара-иро-
цесса недостижимо по стерическим причинам. Тем не менее
Мукаи [163] сообщил о стереоспецифической
перегруппировке соединения (347) в (348). Это удивительный пример
того, как напряжение в скелете делает возможной
разрешенную по симметрии реакцию, которая в отсутствие этого
напряжения не могла бы произойти [ср. переходное
состояние (349)].
138
H3co4jJ
D
p сн(сн3)2
!d
(348)
(349)
Недавно стало известно [164] антараповерхностное сиг-
матропное превращение порядка [1,7], известное для
реакции прекальциферол ->- кальциферол.
В циклогептатриене антараповерхностный [ 1,7]-сдвиг
невозможен. Следовательно, [1,7]-сдвиги в этой системе
должны индуцироваться фотохимически или происходить
термически с инверсией при смещающемся центре.
Известно [165] несколько последовательностей таких [1,7]-пере-
группировок, индуцируемых светом.
Мурэй и Каплан [166] наблюдали последовательность
превращений (350) — (353), представляющих собой сиг-
матропные сдвиги водорода в изомерах 1,4-ди(циклогеп-
татриенил)бензола, которые термически происходят как
[1,5]-сдвиги, а фотохимически — как [1,7]-сдвиги. Шмид
[166а] описал другие интересные примеры [1,7]-сдвигов.
(350)
(351)
(352)
им
Н н (353)
Н Н
139
Разрешенная по симметрии сигматропная [3,5]-пере-
группировка должна идти в возбужденном состоянии, если
она супраповерхностна относительно обоих компонентов.
До сих пор ее удалось наблюдать, по-видимому, только в
одном случае [167].
Первый сигматропный [5,5]-сдвиг был осуществлен Фра-
тером и Шмидом [168] при проведении легко протекающей
стереоспецифической перегруппировки:
(354)
(355)
Часто [1,2]-сдвиги происходят в карбониевом ионе*.
Стереоспецифические сдвиги водорода или метильной
группы в катионах бензенониума [170] можно рассматривать
как [1,2]- или [1,6]-сдвиги.
Сигматропные реакции порядка [1,4], например
миграция в бутен-2-1 -ильном катионе, не наблюдались до
недавнего времени, когда Светтон и Харт [171] опубликовали
НзС^^сНз Н3С^^СН3
НО
'■|-СН3 >__^.|-ОН
СНз
Н3С
СН3
НзС^^СНз
Н3С
Н3С
СИ,
(356)
Смотри обзоры Покера и Берсона [169].
140
сообщение о быстро протекающей дегенерирующей
перегруппировке (356). При супраповерхностном сигматропном
[1,4]-смещении должна происходить инверсия при
мигрирующей группе. Совершенно другая ситуация возникает
при [1,5]-смещении в подобном соединении (357).
з
(357)
Предсказанная инверсия при сигматропном [1,4]-сдвиге
была недавно установлена независимо несколькими
авторами. Хилл [172] и Циммерман [173] показали, что этот
процесс происходит при реакции Фаворского в бицикло-
[3,1,0]-гексаноне:
(358) (359) (360)
Харт [174] установил, что открытая им ранее
перегруппировка (356) [171] в действительности протекает с
инверсией. Наиболее ясно природу этой перегруппировки
показали Винштейн и Чилдз [175]. Эти авторы получали в
очень кислой среде различные метилированные бицикло-
[3,1,0]-гексенилкатионы (362), так что можно было
наблюдать непосредственно при разных температурах ЯМР-спектр
катиона. Катионы образовывались в разрешенной по
симметрии фотохимической дисротаторной циклизации
соответствующих циклогексадиенильных катионов (361).
Полученные таким образом катионы сравнительно устойчивы
вследствие обусловленного симметрией барьера на пути
обратной термической реакции. При повышении
температуры, при которой измеряют сигналы ЯМР, пять
помеченных звездочками метальных групп в гептаметильном
катионе становятся эквивалентными, а две оставшиеся метиль-
141
ные группы дают отчетливые резонансные сигналы как при
низкой, так и при высокой температурах.
Н3С СН3
HaCv^X. ^CH3
Н3С
СН3
hv
Н3С
В опубликованных недавно элегантных исследованиях
Шмида [176] приведено несколько примеров катионных сиг-
матропных [3,41-реакций. В этих диенолбензольных (363)
и аналогичных диенонфенольных перегруппировках сигмат-
ропные [3,4]-реакции конкурируют с разрешенными по
симметрии сдвигами [1,2] и [3,3].
НО н
Н3С
Н3С
(363)
Примером анионных сигматропных [2,3]-реакций
является, по-видимому, перегруппировка Виттига
(катализируемое щелочью превращение бензиловых эфиров в
спирты). Так, Макисуми и Натцумото [177] наблюдали
катализируемое щелочью следующее превращение:
R
N
СН
l
ОН
R1
(365)
Аналогичные результаты на меченых 9-фторенилаллильных
эфирах получил Шёлькопф [178]. Было найдено [179], что
142
аналогичная изоэлектронная реакция 1(366) -> (367)]
формально нейтральных частиц происходит очень легко и
является весьма распространенным типом реакций.
(366) (367)
Анионные [1,2]-сдвиги должны протекать, по-видимому,
с инверсией при мигрирующем атоме. Такие сдвиги
наблюдали [180] в системах, полностью состоящих из углеродных
атомов, однако стереохимия этих реакций не известна. В
перегруппировке Стивенса алкильная группа мигрирует от
четвертичного аммониева атома азота к соседнему карба-
нионному центру. Таким образом, эта реакция изоэлект-
ронна карбанионной [1,2]-перегруппировке и является
внутримолекулярной [181], причем миграция происходит
с сохранением конфигурации при смещающемся углеродном
атоме [182]. Есть различные предположения относительно
механизма этой реакции [180—183]; предложены
механизмы, рассматривающие ионные пары и ион-радикалы [182,
183], которые снимают возражения против предположения
о синхронном процессе. Возможная альтернатива,
кажущаяся несколько необычной, состоит в том, что решающую
роль играют ионы металла, которые всегда присутствуют
в таких системах. Если ионы металла сильно ассоциируют
с карбанионоидным центром и связываются атомом азота
при окончании миграции (368), то возможен разрешенный
Ph
РЬ^Й^ /СН = СН2
(368)
по симметрии процесс. В этом случае происходит инверсия
как при атоме азота, так и при карбанионоидном атоме
углерода; в реакции участвует /?-орбиталь металла.
143
Хорошо известен [184] сигматропный [1,5]-гомосдвиг:
(369)
7.2. Последовательные сигматропные смещения
Толчком для общего рассмотрения последовательных
сигматропных смещений послужили наблюдения Берсона
и Виллкота [161] последовательности сигматропных [1,5]-
смещений в норкарадиенах (370). Такие процессы должны
(370)
протекать с сохранением конфигурации при мигрирующем
атоме углерода; последовательность перемещений с
сохранением конфигурации показана на схеме (371).
Иллюстрация, по существу, представляет суперпозицию
«моментальных фотографий» всех возможных положений меток,
присоединенных к метиленовому атому углерода, когда он
мигрирует по шестичленному кольцу (ср. рис. 36). '
(VI)
Допустим, хотя это энергетически маловероятно, что
метиленовая группа мигрирует (372) при
последовательных сигматропных [1,3]-сдвигах по промежуточным
144
ф
/ 3 \
[1,3] [1,3]
частицам норборнадиена. Супраповерхностное сигматроп-
ное [1,5]-смещение протекает с сохранением конфигурации;
[1,31-смещение под влиянием напряжения в углеродном
скелете является супраповерхностным и происходит с
инверсией. Возникает вопрос: приведет ли случайная
последовательность [1,3]- и [1,5]-сдвигов к такому же
расположению меток, как и ранее проанализированная
последовательность [1,5]-сдвигов (371)? В данном случае следует
ответить положительно. Это видно из диаграммы рис. 37,
в которой все возможные частицы норкарадиена и норбор-
Рис. 37. Сигматропные [1,3]- ( ) и [1,5]-смещения (----) в
замещенных норкарадиенах и норборнадиенах.
6—1556
145
надиена связаны между собой всеми возможными
процессами. Информация, содержащаяся на рис. 37, суммирована
на схемах (373) и (374). Аналогичные вопросы можно сфор-
(373)
(374)
мулировать и в случае других [2/г,1,0]-бициклов. Для
[2,1,01-систем возможны только [1,3]-смещения;
расположение меток суммировано на схеме (375).
(375)
Для систем [6,1,0] возможны сдвиги [1,3], [1,5] и [1,7].
Диаграмма, которая связывает все изомеры, очень сложна,
однако ее смысл передан на схемах (376) и (377). В этом
случае любая последовательность [1,3]-, [1,5]- и [1,7]-сдви-
гов, которая связывает два изомера, также дает одну и ту
же конфигурацию.
(376)
(377)
Можно показать, что полученные выше результаты
представляют общие топологические свойства бициклических
[2я,1,0]-систем. Доказательство состоит из двух частей.
146
Во-первых, ясно, что [1,5]-сдвиг с сохранением
конфигурации при мигрирующем центре неотличим от
последовательности из двух [1,3]-сдвигов, каждый из которых
протекает с инверсией при мигрирующем центре. Аналогичным
образом [1,7]-сдвиг с инверсией стереохимически
неотличим от серии из трех [1,3]-сдвигов, каждый из которых
протекает с инверсией. В общем случае
[1,(2? + l)] = [l,3p.
Следовательно, достаточно рассмотреть статистическую
последовательность [1,3]-смещений.
Мы покажем, что в бицикло-[(2п—2),1,0]-системе каждая
возможная последовательность [1,3]-смещений, которая
превращает (378) в (379), состоит из нечетного числа
смещений (п — четное число) и из четного числа смещений (п —
нечетное число). Это означает, что для четного п происхо-
2/7-3
(38/)
- Q
(383)
дит нечетное число инверсий, т. е. в итоге имеем инверсию
[(380) ->• (381)]; при нечетном п конфигурация сохраняется
2ц-з
(378)
(380)
(382)
6*
147
1(382) ->- (383)]. Циклическая симметрия
перегруппировки приводит к соотношению между изомерами,
показанному на схеме:
Четное и
Нечетное п
1
(384)
Сформулируем теперь математическую задачу,
отражающую последовательность перегруппировок. Рассмотрим
многоугольник (385) с 2/г углами, которые мы пронумеру-
148
ем, начиная с некоторой произвольной точки. Затем
разместим в углах 1 и 2 метки А и В и сыграем в игру со
следующими правилами:
а) путем произвольного числа последовательных
перемещений поместим метки в новые углы;
б) при каждом перемещении либо А, либо В должны
перемещаться на два положения по часовой стрелке или
против нее. Например, если А находится в положении k, то
при одном смещении оно может оказаться либо в
положении k — 2, либо в положении k-\-2\
в) не разрешены перескоки через метки. Это означает,
что если метка А находится в положении k, а метка В —
в положении k—1, то А не может сразу переместиться в
положение k—2, а должна попасть в положение k-\-2.
Теорема. При этих правилах игры каждая произвольная
последовательность перемещений,, которая начинается с А
в положении 1 и с В в положении 2 и оканчивается на А
в положении 3 и на В в положении 2, состоит из четного
числа перемещений, если п — нечетное и нечетного числа
перемещений при четном п.
Прежде чем доказывать теорему, убедимся, что игра и
теорема эквивалентны проблеме последовательных
перемещений. Кольцо, состоящее из 2/г атомов, вокруг которого
мигрирует атом углерода, соответствует многоугольнику.
Метки А и В представляют связи, идущие от вершин
многоугольника к мигрирующему атому.
Доказательство. Измерим расстояние между А и В по
периметру многоугольника, начиная с В, по часовой
стрелке. Тогда если В находится в положении 2, а А — в
положении 1, то расстояние между ними максимально и равно
149
2п—1. Определим теперь число перемещений, которое
необходимо произвести, чтобы это расстояние сократить до 1,
так, чтобы В находилось в положении 2, а А — в
положении 3. Для этого сначала установим, сколько необходимо
перемещений, чтобы сократить расстояние до 1, если В
находится в любом месте. Первое перемещение уменьшает
расстояние между А и В с 2п—1 до 2п—3. Каждое
следующее перемещение можно классифицировать либо как
эффективное, либо как неэффективное. Эффективное
перемещение уменьшает расстояние между А и В по сравнению с
предыдущим минимальным расстоянием, неэффективное
перемещение не уменьшает. Все неэффективные перемещения
происходят попарно: если в какой-то момент расстояние
было равно / и перемещение увеличило его до /+2, тогда
при другом перемещении расстояние вновь станет равным /.
Только эффективное перемещение уменьшает расстояние
между А и В. Так как каждое эффективное перемещение
уменьшает расстояние на 2, то совокупность перемещений
произвольной последовательности, необходимой для
уменьшения расстояния с 2п—1 до 1, следующая:
(2п — 2)/2 = (п — 1) + Некоторое четное число.
Эффективное Неэффективное
Таким образом, можно вычислить число перемещений,
необходимых для уменьшения расстояния до 1.
Однако В может находиться и в вершине некоторого
угла, имеющего четный номер. Теперь А и В могут быть
расположены на любой соседней вершине, и достаточно
четного числа неэффективных перемещений для того,
чтобы В переместить в положение 2, а А — в положение 3.
Итак, мы показали, что общее число необходимых
перемещений составляет
(п — 1) + Некоторое четное число.
Следовательно, число перемещений нечетно при
четном п и четно при нечетном п.
Для катионных сигматропных перегруппировок
топологические соотношения могут существенно отличаться от
только что выведенных соотношений для нейтральных
частиц. Рассмотрим сначала сигматропные перегруппировки
бицикло-[3,1,0]-гексенильного катиона:
150
(386)
В этой системе могут происходить сдвиги [1,2], [1,4] и [1,3].
Экспериментально показано, что ионный [1,2]-сдвиг должен
происходить гораздо быстрее, чем нейтральные [1,(2^+1)1-
перегруппировки. Используем предыдущие условия и
примем во внимание условия, определяемые симметрией,
которые требуют, чтобы [1,2]-сдвиги происходили с сохра-
Р и с. 38. Сигматропные [1,4]- ( ), [1,2]- (....) и [1,3]-смещения
(----) в катионе (386)
нением конфигурации, а сдвиги [1,3] и [1,4] — с инверсией.
Тогда мы получим для превращения катиона (386)
зависимости, которые показаны на рис. 38 и суммированы на
схемах (387) и (355).
(387) (388)
Может показаться, что ряд сигматропных реакций в
циклических полиенил-ионах всегда имеет ту же
последовательность, что и в циклических полиенах. На самом деле
это не так. Рассмотрим, например, простейшую систему,
в которой возможны [1,2]-смещения — бицикло-[1,1,0]-бу-
тилкатион (389). В противоположность всем рассмотрен-
4
А
(389)
ным выше случаям последовательность превращений
приводит к эпимеризации, т. е. серия перегруппировок
превращает (390) в (391). Нетрудно заметить, что
аналогичная ситуация возникает для (7,11...)-членных колец, так же
как и для трехчленного кольца (ср. рис. 39).
•&
ф.
ж
152
(390) (392)
Превращениябицикло-[(2/г+1),1,0]-катионов можно
разделить на две группы. В первой группе сигматропные
[1,(2/г+2)]-сдвиги происходят с сохранением конфигурации,
и эпимеризация должна протекать как серия из 2/г+З
таких сдвигов, например в случае [1,2]-, [1,6]- и [1,10]-сдви-
0--Q
Рис. 39. Сигматропные [1,б]-смещения в бицикло-[5,1,0]
октадиенилкатионе.
гов, т. е. всегда, когда п — четное число. Вторая
категория объединяет те случаи, когда п — нечетное число;
сигматропные сдвиги происходят с инверсией, и эпимериза-
цию осуществить не удается, каково бы ни было число
разрешенных по симметрии инверсий, например в случае
циклических полиенов.
8. ПЕРЕДАЧА И ЭЛИМИНИРОВАНИЕ ГРУПП
Рассмотрим синхронную реакцию, в которой два атома
водорода переходят от этана к этилену (392). Этот процесс
является супраповерхностным для обоих реагирующих ве-
153
°с-н
JC-H
тт*
А тг*
S ir
On.
OH
(?C-H
Рис. 40. Корреляционная диаграмма для синхронного перехода
двух атомов водорода от этана к этилену.
он s
S °с-н
JC-H S
А
--S ис-н
Рис. 41. Корреляционная диаграмма для синхронного перехода
двух атомов водорода от молекулы этана к концам молекулы
бутадиена.
а + н -* J ♦ V
Ai а А нх<
ществ. Легко построить корреляционную диаграмму
(рис. 40) для этой реакции, используя плоскость симметрии,
секущую обе молекулы. Процесс разрешен по симметрии.
Совершенно иной вид (рис. 41) имеет диаграмма для
синхронного перехода двух атомов водорода от молекулы
этана к концам молекулы бутадиена (393); эта реакция
запрещена по симметрии.
х-» A- v ">v
Л. Vе "* A xJ
(393)
Для общего случая (394) легко вывести следующее
правило отбора: переход двойной группы разрешен по
симметрии для основного состояния, еслит+я = 4^+2, и для
возбужденного состояния, если т-\-п = 4q (т и п — числа
jx-электронов, a q = 0, 1, 2 ...). Правило применимо к анта-
раповерхностному по отношению к обоим компонентам
процессу; оно обращается, если процесс антараповерх-
ностен только для одного компонента. Если возникает
возможность инверсии при R, то правило должно быть
модифицировано (ср. разд. 11).
^—<R >—^ /—( R>—^
/77 п —* т + 2 п - 2
^ <R > ^ ^ < R> '
(394)
Наиболее известный пример реакции этого типа (т = 0,
п = 2) — переход водорода от диамида к олефинам [185].
Существуют также углеводородные аналогии, например,
реакция (395), которую наблюдали Дюринг и Розенталь
[186]. Наблюдали [187] также процесс ст = 2,я = 4 (396).
155
Ф * ::о - оо * ::b
н н
(395)
000*0-000*0
(396)
Выведенное выше правило применимо к предельному
случаю, когда п = 0, т. е. происходит синхронное
элиминирование групп (397). Мы можем предсказать, таким об-
S ^R ^ < R
т —* /п+2 +|
4 f» V ( R
(397)
разом, что синхронное некаталитическое гидрирование
диена или обратный процесс элиминирования водорода
должны быть скорее процессами [1,4], чем [1,2]. В то время
когда было сформулировано это предсказание, в
литературе имелось лишь два отрывочных сообщения по этому
вопросу:
а) 2,5-дигидрофуран теряет водород в
мономолекулярном процессе с ER= 48,5 ккал/моль и отрицательной
энтропией активации, в то время как 2,3-дигидрафуран не
реагирует таким образом [188];
б) 3,3,6,6-тетраметил-1,4-циклогексадиен при
пиролизе в пентан дает этан (и n-ксилол) с выходом значительно
большим, чем при радикальной цепной реакции [189].
Впоследствии наши предсказания были подтверждены
в ряде исследований. Белдвин [190], исследуя отщепление
водорода от меченого циклопентена, нашел, что
отщепление 1,4 имеет преимущество (10 : 1) по сравнению с отщеп-
156
лением 1,2. Эллис и Фрей [191], Бенсон и Шоу [192]
исследовали пиролиз 1,3- и 1,4-циклогексадиена. Последний
разлагается мономолекулярно на бензол и водород с Еа=
= 42,7 ккал/моль; при этих же температурах 1,3-циклогек-
садиен термически устойчив, а при более высоких
температурах разлагается главным образом по цепному
радикальному механизму. Фрей сообщил о других интересных
примерах синхронного элиминирования водорода; с другой
стороны, его работа показала, что процессы образования
метана или этана идут по цепному свободнорадикальному
механизму [194].
Синхронная потеря двух гем-групи (398) в
возбужденном состоянии должна идти при условии, что она протекает
через переходное состояние, имеющее симметрию C2v (ср.
разд. 10.1). Таким образом, эта реакция конкурирует с
,R v R
*\ * R
(398)
вицинальным отщеплением [(397), т = 0], которое тоже
разрешено по симметрии в возбужденном состоянии. Оба
случая известны*.
9. ВТОРИЧНЫЕ ЭФФЕКТЫ
Соображения, основанные на орбитальной симметрии,
были использованы также для того, чтобы понять
происхождение вторичных конформационных эффектов в
реакциях синхронного циклоприсоединения.
Рассмотрим реакцию Дильса — Альдера между
молекулами бутадиена как синхронную реакцию [n4s+n2s]-
циклоприсоединения. A priori эта реакция может идти
через одно из двух переходных состояний (399) или (400).
зядо-Сближение (399) отличается от з/сзо-сближения (400)
* Фотолиз циклогексадиена [195], фотолиз этана и этилена
[195а], циклогексана [1956]; в газовой фазе фотолиз азометана.
Установлено, что преобладающим первичным процессом является
разложение до азота и двух метильных радикалов. Однако были
получены некоторые указания на то, что в небольшой степени
происходит отщепление этана [195в].
157
(399) (400)
главным образом близостью орбиталей (3 и (3'. Все
вторичные обменные взаимодействия между занятыми
молекулярными орбиталями диена и диенофила дают лишь
небольшой вклад в общую энергию переходного состояния. Это
связано с тем, что такие взаимодействия будут увеличивать
энергию одних уровней и понижать энергию других.
Существенные взаимодействия обусловлены разрешенным по
симметрии смешением занятых и незанятых уровней (это
подчеркивает также Фукуи [196]). В рассматриваемом
случае из диаграмм (401) и (402) следует, что любая возмож-
(402)
Смешение низшей незанятой
„диеновой" орбитали (верх)
С высшей занятой „олефи-
новой" орбиталью {низ)
(401)
Смешение высшей занятой
„ диеновой " орбитали {верх)
с низшей незаполненной ,,оле-
финовой"орбиталью {низ)
158
ность такого смешения приводит к связыванию, т. е.
понижающему энергию взаимодействию сблизившихся орби-
талей (3 и |3' (роль других вторичных взаимодействий была
выявлена Салемом [197]). Таким образом, переходное эндо-
состояние (399) для синхронного [л45+ л25]-циклоприсо-
единения стабилизовано по сравнению с з/сзо-формой (400)
благодаря контролируемому симметрией вторичному
орбитальному обменному взаимодействию.
Показанные здесь соотношения орбитальной симметрии
дают простую квантовохимическую основу для понимания
многих экспериментальных наблюдений, которые были
обобщены в правиле для зядо-присоединения,
сформулированным Альдером [198]. Наша точка зрения отличается от
взглядов других авторов, которые подчеркивали роль
индуктивных сил [199], электростатических сил, возникающих
вследствие переноса заряда между диеном и диенофилом
[200], а также максимального накопления двойных связей
[198]. В настоящее время установлено, что орбитальное
обменное взаимодействие между ненасыщенными центрами,
участвующими в синхронном циклоприсоединении, может
повысить энергию переходного зядо-состояния вместо того,
чтобы понизить его. Таким образом, з/сзо-присоединение
может оказаться более предпочтительным, если
доминируют факторы симметрии. Рассмотрение соответствующих
орбитальных диаграмм (403) и (404) для разрешенных по
симметрии [л63+ л48]-комбинаций показывает, что в
данном случае эта ситуация реализуется.
(403) (404)
159
Это предсказание было сделано до открытия [6+4]-цик-
лоприсоединения и впоследствии было подтверждено [201].
В противоположность этому разрешенные по симметрии
реакции циклоприсоединения [я88+ я28] и [я28+я28+я28]
должны быть подобны процессу [4+2] и протекают
преимущественно через переходные зядо-состояния. Уже
появились доказательства преимущественно такого протекания
для процесса [я28+ я28+ я28] [202].
Димеризация циклобутадиена является особым случаем,
представляющим большой интерес в связи с правилами
сохранения орбитальной симметрии. Если двойные связи в
этой исключительно реакционноспособной молекуле
действительно локализованы в значительной степени
(по-видимому, это так и есть), то следует a priori учитывать
возможность реакций циклоприсоединения [я28+ я28], [я28+я48]
и [jt4s+ д48]. Наши правила отбора требуют преимущества
для синхронного [я28+ я48]-процесса. В настоящее время
получены данные, указывающие на правильность этого
вывода [203]. Изучение вторичного орбитального
взаимодействия в рамках развитых здесь представлений
показывает, что зядо-присоединение, ведущее к шя-димеру (405),
предпочтительней зкзо-присоединения, при котором
образуется анти-лииер (406). Опубликованы сведения об
образовании как син-, так и аяяш-димеров в реакциях,
инициируемых различными возможными промежуточными
продуктами образования циклобутадиена. Однако только там,
где наиболее вероятным промежуточным продуктом
является свободный циклобутадиен, наблюдают предсказанный
зядопроцесс [204].
(405j (406)
Сигматропный [3,3]-сдвиг в 1,5-гексадиенах, как было
показано, с наибольшей легкостью идет через четырех-
центровое креслоподобное переходное состояние (407),
160
а не через ванноподобную конфигурацию (408) [205]*.
/ /
^—/ (407)
I Г
i i
i i
*~р? (408)
Мы предполагаем, что соотношения орбитальной симметрии
могут играть определяющую роль в таком
преимущественном протекании реакции. Корреляционная диаграмма для
G* А'
А 0*
;А ,.
Рис. 42. Корреляционная диаграмма для сигматропного [3,3]-
превращения 1,5-гексадиена.
молекулярных орбиталей, участвующих в
перегруппировке, показана на рис. 42. Для ванноподобного переходного
состояния уровни классифицированы как симметричные
или антисимметричные относительно зеркальной
плоскости т, а для переходного состояния типа кресла —
относительно оси вращения второго порядка. Рис. 42
соответствует форме ванны. Диаграмма для формы кресла
качественно та же. Корреляция связывающих уровней реагента
* Недавно опубликована теоретическая работа на эту тему
[205а]. Аналогичные исследования по перегруппировке Клайзена
ароматических веществ были проведены Фратером и др. [2056].
161
со связывающими уровнями продукта характерна для
диаграммы разрешенной по симметрии термической
реакции. В точке, отвечающей половине пути реакции, серия
уровней соответствует уровням двух сильно
взаимодействующих аллильных радикалов. Истинное поведение
уровней вдоль координаты реакции получено из расчетов по
расширенному методу Хюккеля.
Ф-т
Рис. 43. Корреляционная диаграмма сближения двух
аллильных радикалов из бесконечности в состояние квазиванны
и квазикресла.
(1) и (2) в верхней части центрального кольца обозначают элементы симметрии
(см. текст).
Если корреляционные диаграммы качественно подобны
для реакций с переходным состоянием как формы ванны,
так и формы кресла, то почему наблюдается
преимущественное протекание реакции через одну из этих форм? Ответ
на этот вопрос требует построения двух других
корреляционных диаграмм (рис. 43) для альтернативных гипотети-'
ческих процессов, в которых два аллильных радикала
приближаются друг к другу из бесконечности. Эти радикалы
сближаются в параллельных плоскостях и ориентированы
162
относительно друг друга таким образом, что образуют в
одном случае форму квазиванны, а в другом случае форму
квазикресла. В каждом из этих процессов присутствуют два
элемента симметрии:
а) для обеих форм зеркальная плоскость ть которая
проходит через С-2 и С-5 и сечет углы С^Сз и С6С5С4;
б) при сближении радикалов по типу ванны вторая
зеркальная плоскость т2, параллельная плоскостям, в
которых находятся сближающиеся радикалы, и расположенная
посередине между ними; при сближении по типу кресла
ось симметрии второго порядка, находящаяся на
пересечении т2 и плоскости, проходящей через C^QCe.
Для полной корреляции надо дать спецификацию
конечных продуктов такого типа гипотетического движения: при
сближении по типу ванны — бициклогексан; при
сближении по типу кресла — 1,4-циклогексилиденовый бирадикал.
При сравнении поведения заселенных уровней SiA2
проявляется решающее различие между обоими путями. В случае
ванноподобного сближения этот уровень коррелирует с
разрыхляющим а-уровнем, а в процессе по типу кресла он
переходит в несвязывающий бирадикальный уровень.
Основной пункт этих рассуждений состоит в том, что реакция,
протекающая в соответствии с диаграммой рис. 42, должна
проходить в середине пути реакции через точку на
корреляционной диаграмме, которая аналогична той или иной
точке на диаграмме рис. 43. Таким образом, эта точка должна
иметь приблизительно ту же горизонтальную координату,
что и точка, лежащая между вертикальными
штрихованными линиями на альтернативных диаграммах рис. 43.
Любая такая точка переходного состояния типа кресла
соответствует меньшей энергии, чем точка для переходного
состояния типа ванны вследствие разницы корреляционных
свойств занятой орбитали S^. Основной вывод состоит
в том, что орбитальные взаимодействия, включающие С-2
и С-5, вызывают лишь антисвязывание и повышение
энергии. В этом смысле приведенные здесь соображения
развивают предложенную Дюрингом [205] идею о простом
эффекте орбитального отталкивания.
Следует подчеркнуть, что рассмотренные эффекты, как
и ожидалось, являются очень малыми, так что сигматропные
[3,3]-смещения легко происходят и в системах, геометрия
которых допускает образование ванноподобного
переходного состояния [206].
163
В заключение проиллюстрируем ход реакции [4+2]-цик-
лоприсоединения молекул бутадиена к аллильному
катиону, для которой еще нет экспериментальных данных.
Схемы (409) и (410) показывают важнейшие взаимодействия.
Вторичными взаимодействиями в (409) можно пренебречь,
а взаимодействия в (410) являются разрыхляющими.
Таким образом, можно ожидать преобладания переходного
жзо-состояния (впоследствии это предположение было
подтверждено [206а]).
(4Ю)
Смешение низшей незанятой
„диеновой" орбитали (верх)
с высшей занятой
„аллильной" орбиталью (низ)
10. ДИВЕРТИСМЕНТ
В предыдущих разделах обсуждалось, каким образом
орбитальная симметрия контролирует большинство
важнейших типов синхронных органических реакций, и
приводились соответствующие примеры. Теперь мы вновь вер?
немея к обсуждению двух особых типов реакций, каждый
из которых обладает очень интересными особенностями.
Эти реакции дополнительно иллюстрируют важность
принципов сохранения орбитальной симметрии для понимания
и предсказания деталей протекания химических
превращений.
10.1. Хелетропные реакции
Мы называем хелетропными реакциями такие процессы, .
в которых синхронно образуются или разрываются две
cr-связи, оканчивающиеся на одном атоме. Рассмотрим
хелетропную реакцию (411), в которой образуется малая
Смешение^ высшей занятой
„диеновой" орбитали (верх)
с низшей незанятой„аллиль-
ной"орбиталью (низ)
164
молекула X(YZ...) и полиен, содержащий т я-электронов.
/^ СЛ f
т-2 X(YZ...) —* т
-С-1
V
/
-С-т
V.
+ X (YZ...)
-С-т
(411)
Обсудим геометрические аспекты реакции относительно
инвариантной системы координат (412), начало которой
(412)
находится в точке X, а ось z пересекает линию между С-1
и С-т. Тогда разрывающиеся а-связи лежат в плоскости xz,
а их четыре электрона распределены, как обычно, попарно
на двух обобщенных орбиталях (413) и (414). В общем
W3)
(414)
165
случае по два электрона должны перейти с этих орбиталей
на каждый из образующихся продуктов с сохранением
орбитальной симметрии. Здесь как раз наступает решающий
момент в наших рассуждениях: для перехода двух
электронов с разрывающихся а-связей на новые орбитали молекулы
продукта X(YZ...) возможны два совершенно разных пути:
1. Два электрона с симметричной орбитали (413)
[=(415)] занимают в виде свободной пары новое положение
в X(YZ...) на орбитале, симметричной относительно z,
или становятся частью я-системы, антисимметричной
относительно плоскости ху.
2е
2е
2. Два электрона с антисимметричной орбитали (414)
[=(416)] могут занять новое положение на орбитали
свободной пары, симметричной относительно х, в молекуле
X(YZ...) или становятся частью я-системы, симметричной
относительно плоскости yz.
В свете этого анализа становится ясно, что
геометрические изменения вблизи С-1 и С-т зависят от деталей
геометрии отрыва X(YZ...) и особенно от движения
атомов YZ..., связанных с X. Для того чтобы пояснить этот
важнейший момент, рассмотрим отделение расположенной
углом молекулы X—Y—Z, которая в момент своего
образования приобретает симметричную свободную пару элект-
166
ронов на центральном атоме. Реакция может протекать по
одному из двух путей, изображенных на схемах:
а) рассмотрим схему (417). X получает необходимую
свободную пару с симметричной а-орбитали (415), в то
время как X, Y и Z остаются колинеарными в плоскости уг.
Мы будем называть такие процессы линейными хелетроп-
ными реакциями. Следует отметить, что формально
электронная пара продукта ведет себя по супраповерхностному
типу, так как две связи, которые дают два электрона,
находятся на одной стороне узловой плоскости (совпадающей
приблизительно с плоскостью ху) орбитали продукта,
которую занимает электронная пара.
б) рассмотрим схему (418). X получает необходимую
электронную пару с антисимметричной а-орбитали (416).
В то время как X отделяется колинеарно, оставаясь в
плоскости yz, Y я Z перемещаются как в направлении х, так
и в направлении г, что приводит к тому, что молекула
продукта находится в плоскости ху. Мы назовем такие
процессы нелинейными хелетропными реакциями. В этом случае
электронная пара продукта X ведет себя по антараповерх-
ностному типу, так как обе связи, которые дают два
электрона пары, находятся по разные стороны узловой плоскости
167
(совпадающей приблизительно с плоскостью yz) орбитали
продукта, которую занимают электроны.
Теперь ясно, что при линейных хелетропных реакциях
типа (411) два электрона должны переходить от
антисимметричной а-орбитали [(416)=(420)=(423)] на высшую
занятую я-орбиталь полнена (419). Следовательно,
смещения при атомах С-1 и С-т должны быть дисротаторными,
когда т = 4q [(420) -> (421)] и конротаторными, когда
т = 4?+2 [(423) -> (424)].
В противоположность этому если отщепление (411)
протекает как нелинейная хелетропная реакция, то два
электрона должны переходить с симметричной а-орбитали
[(415)=(422)=(425)] на высшую занятую я-орбиталь
полнена (419). В этом случае смещения при С-1 и С-т
должны быть конротаторными, если т = 4q [(422) -> (421)],
и дисротаторными, если т = 4д+2 [(425) -> (424)].
т
Aq
4<7 + 2
Реакции, разрешенные в основном состоянии
линейная
дисротаторная
конротаторная
нелинейная
конротаторная
дисротаторная
Рис. 44. Правила отбора для хелетропных реакций.
Правила отбора суммированы на рис. 44. В этих
хелетропных превращениях каждой реакции в основном
состоянии соответствует процесс в возбужденном состоянии,
характеризующийся противоположными стереохимическими
свойствами. Выделяемая малая молекула X(YZ...) часто
имеет много заселенных орбиталей свободных пар. Из этого
вытекает возможность многих п -> я*-состояний, что
обусловливает в свою очередь возможность протекания в
возбужденном состоянии процесса с такими же
стереохимическими характеристиками, как и для соответствующего
процесса в основном состоянии.
Реакции линейного хелетропного элиминирования
фрагментов, приводящие к образованию азота или окиси
углерода, очень хорошо изучены; это позволяет построить
формальные корреляционные диаграммы, что и было сделано
168
о
J
ЧЮ
* %
q?
£>
&?
Л
I
с?»
4- ^
С>«
Лемалом [207], Болдвиным [208] и авторами этой книги.
Стереохимия выделения окиси углерода не известна.
Однако известно, что циклопентен-З-оны-1 (т = 4) легко
декарбонилируются, по-видимому, в результате
синхронного процесса [208]. В связи с этим примечательна неудача
попыток получить норборнадиенон-7 (426), что
контрастирует со стабильностьюнорборнанонов (см., например, [209]).
О
(426)
3,5-Циклогептадиенон представляет случай, когда т = 6
(427). Это один из немногих известных примеров декарбо-
нилирования в конденсированной фазе, которое протекает
при фотолизе с образованием гексатриена и окиси
углерода [210].
4/° (y=N
(427) (428)
Выделение азота из диазенов (428) было тщательно
изучено Лемалом [207]. Это, как и следовало ожидать, стерео-
специфический дисротаторный процесс. Ясно, что в этом
случае линейное конротаторное превращение происходит
с меньшим напряжением структуры, чем альтернативный
нелинейный конротаторный процесс.
Карпино [211] описал интересные последовательности
реакций, в которых дисротаторное выделение азота
завершается конротаторным электроциклическим процессом
1(429) и (430)1
Ph Ph H
Ph Ph Ph H
(429)
170
?hH Ph Ph H
Ph jbh jih H
(430)
Хорошо известно обратимое, гладко проходящее
присоединение двуокиси серы (характеристику МО двуокиси
серы и сульфинов можно найти в [212]) к диену [213];
интересные примеры фотохимического выделения двуокиси
серы были описаны в [213а]. Было установлено [214] дисро-
таторное выделение двуокиси серы из бутадиенсульфона,
что соответствует правилам симметрии. Соответствующий
фотохимический процесс не полностью стереоспецифичен,
однако было показано [215] явное преобладание конрота-
торного пути. Двуокись серы также легко вступает в
реакцию [1,6]-присоединения к 3-^с-гексатриену; обратный
процесс, как было недавно показано, протекает по конрота-
торному типу, поэтому его можно считать линейной хелет-
ропной реакцией [216].
Хелетропные реакции, в которых образуются или
распадаются трехчленные циклы, т. е. идет распад на
фрагменты типа (411) (т = 2), представляют особый интерес.
В этом случае геометрические факторы обусловливают
дисротаторный или супраповерхностный тип реакции,
следовательно, превращения должны проходить по
нелинейному хелетропному пути.
Хорошо известная [217] стереоспецифическая
комбинация синглетных карбенов с олефинами, дающая
циклопропаны, безусловно, принадлежит к нелинейному
хелетропному классу [218] (полуэмпирические расчеты, выполненные
для этой реакции, дают поразительное согласие с
приведенным анализом нелинейных хелетропных процессов).
Циклопропеноны и циклопропаноны теряют окись
углерода термически в мягких условиях [219, 220] и еще легче
при'облучении [219, 221]. Стереохимическая информация
о декарбонилировании отсутствует. Однако было показано
[222], что атом азота N-2 стереоспецифически выделяется
по дисротаторному механизму из алкилзамещенного
трехчленного кольца диазена (431).
171
X
l\+ t
l/N = N"
1 2
(431)
Соответствующие арилзамещенные соединения реагируют,
однако, нестереоспецифически (согласно частному
сообщению Карпино). Это изменение должно идти по нелинейному
пути, причем N-2 должен смещаться в направлении х в
плоскости азиридинового кольца. Эписульфоны выделяют
двуокись серы по стереоспецифическому дисротаторному
механизму [223, 224]. По-видимому, наибольшая трудность
при объяснении состоит в том, что линейный хелетропный
распад на фрагменты в этом случае запрещен по симметрии.
Для того чтобы преодолеть эту трудность, было высказано
предположение [224], что реакция несинхронна и может
протекать через определенные промежуточные стадии.
Материал этого раздела показывает, однако, что выделение
двуокиси серы можно объяснить также протеканием
разрешенной по симметрии нелинейной хелетропной
реакции 1(418)1
Легкое выделение окиси азота (433) из N-нитрозоази-
ридинов [225] происходит, вероятно, также по нелинейной
хелетропной реакции:
(432) &33)
4луСистема остается неизменной в ходе реакции, а из
связывающей антисимметричной ст-орбитали азиридинового
кольца образуется симметричная относительно оси х ор-
биталь свободной электронной пары азота (433). Здесь,
однако, следует рассмотреть одну очень интересную
альтернативу. Если нитрозосоединение принимает конформа-
цию (434), то несвязывающая электронная пара на
пирамидальном атоме азота азиридина становится одной из
J 72
z-симметричных свободных пар окиси азота (435). В то же
время электронная пара связывающей антисимметричной
or-орбитали азиридинового кольца становится частью
4л^-системы. Учитывая то, что распаривание двух
электронов 4я3,-системы (432) требует ~23 ккал/моль (эта величина
(434) (435)
оценена по вращательному барьеру в N-нитрозоаминах
[226]), кажется невероятным, что при распаде молекулы
реализуется альтернативный запрещенный по симметрии
процесс.
Гипотетическая реакция отрыва кислорода (436) — еще
один пример хелетропного изменения. Если отрыв
происходит по линейному типу, то неподелеиная пара электронов
амина и две неподеленные пары электронов атома кислорода
образуют связь N—О и неподеленные пары N-окиси,
направленные по осям z и у (437). Остающаяся неподелеиная
{437)
173
пара N-окиси, направленная по оси х, образуется из
антисимметричной or-орбитали распадающихся связей углерод —
кислород. Два электрона соответствующей симметричной
or-орбитали должны переходить к полиеновому продукту.
Таким образом, реакция должна проходить по дисрота-
торному пути, когда т = 4^+2, и по конротаторному
пути, когда т = 4q.
Если амин реагирует нелинейно, т. е. в плоскости xz,
но в направлении х, то стереохимические последствия
реакции противоположны. Возможно, линейной реакции
аналогичен стереоспецифический отрыв серы от цис- и транс-
бутенэписульфидов фосфинами [227].
Интересную комбинацию хелетропного распада на
фрагменты и вицинального элиминирования представляет
разложение спирокеталя до двуокиси углерода и двух молекул
олефина. До сих пор эту реакцию (438) наблюдали [228]
только на этиленкеталях, образующихся из замещенных
/-"V
0+со'+1
(438)
норборнадиенонов. Между тем стереохимические аспекты
этой реакции чрезвычайно интересны. Диаграмма (439)
показывает общий случай; тип соответствуют числу
электронов в я-системах образующихся полиенов. В дополнение
к 4 электронам, которые скелетные ст-связи расходуют на
174
двуокись углерода (440), требуется еще 12 электронов:
8 заселяют две перпендикулярные 4я-системы,
расположенные в плоскостях ху и yz соответственно, и 4 — две
орбитали свободных электронных пар, симметричные
относительно оси у. Если обсуждаемая реакция линейна и хе-
летропна, то 8 электронов свободных пар на атомах
кислорода (439) легко переходят на симметричную относительно
оси у орбиталь свободной пары и 4л; .^-систему двуокиси
углерода (440). Кроме того, орбитали ортогональной
4я -системы могут быть заполнены путем перехода 2
электронов с симметричной ст-орбитали С—С—С (441) на
нижнюю занятую симметричную я^-орбиталь (442) и 2
электронов с антисимметричной ст-орбитали О—С—О (444) на
высшую занятую яУ/г-орбиталь (443). Поэтому электроны
(442)
{443)
^МШР
(444)
-z
антисимметричной ст-орбитали С—С—С (446) должны
перейти на высшую занятую орбиталь полнена (445), а элект-
175
роны симметричной ст-орбитали О—С—О (447) занимают
новое положение на высшей занятой орбитали полиена
<ё>-
^©бЬэФ &
(445)
(446)
(447)
(448)
(448). Следовательно, геометрическое изменение,
связанное с разрывом углерод-углеродной связи, должно быть
дисротаторным, когда т = Aq9 или конротаторным, когда
т = iq+2. Наоборот, разрыв связи углерод — кислород
должен быть связан с дисротаторным движением, когда
п = 4^+2, и с конротаторным движением, когда п = 4q.
Если реакция протекает по нелинейному хелетропному
пути, стереохимические последствия, вызванные разрывом
связи углерод — углерод, будут, разумеется, обратными.
Наконец, если на потенциальной поверхности реакции есть
минимум, представляющий синглетные карбеновые
частицы (449), то анализ с использованием принципа
орбитальной симметрии остается таким же.
•о
Может показаться, что в отношении хелетропных
реакций принцип сохранения орбитальной симметрии является
скорее обобщающим, чем дискриминирующим. Однако это
не так. Хелетропные реакции необычны в том отношении,
что стереохимические условия, налагаемые контролем по
орбитальной симметрии на один из реагентов, не
сказываются на продуктах реакции. Следовательно, предсказание
хода хелетропной реакции не может быть легко проверено
путем изучения стереохимии продуктов. Это будет
возможно только после разработки более тонких методов.
176
10.2. Реакции циклоприсоединеяия кетенов
Комбинация кетенов с олефинами с образованием цикло-
бутанов известна уже давно [229]. В последнее время эта
реакция тщательно изучалась. Установлено, что это
синхронная реакция; она характеризуется большой
отрицательной энтропией активации [230]; положение равновесия
очень слабо зависит от природы растворителя [230, 231],
а стереохимические условия для реагентов сохраняются и
для продуктов реакции [232]. Наиболее яркий пример —
образование различных аддуктов при реакции дихлорке-
тена с цис- и /праяс-циклооктенами [233]. Самым
замечательным является наблюдение, что реакция кетенов с
диенами дает только циклобутаны, но не циклогексены даже
тогда, когда строение диена благоприятствует
присоединению к концам цепи [234]. Например, циклопентадиен и ди-
фенилкетен реагируют, образуя соединение (450)] (451) не
образуется.
Ph Ph
{450) (451)
Синхронная комбинация этиленов с образованием цик-
лобутанов должна идти по типу [я28+ я2а]; этот случай
показывает, что олефины реагируют супраповерхностно.
Есть ли в структуре кетенов какая-то особенность, которая
приводит к тому, что они играют роль антараповерхност-
ных партнеров в реакции циклоприсоединения?
Процесс [я28+я2а] включает ортогональное сближение
реагирующих частиц (ср. разд. 6), т. е. я-системы
ориентируются друг относительно друга, как показано на схеме
(452). При отсутствии специальных факторов (ср. рис. 25)
этот путь нельзя считать вполне доступным. Для
нормальных олефинов непреодолимые трудности возникают из-за
пространственных препятствий и напряжения в
переходном состоянии, связанного с нарушением скелета, которое
необходимо для сохранения эффективного перекрывания
орбиталей.
7—1556
177
# т
(452)
Кетены — сильные электрофилы. Химически
реакционным центром служит атом углерода их очень реакционно-
способной карбонильной группы. Основные аспекты
структуры кетенов кратко характеризуются валентной схемой
(453), которая показывает, что мы анализируем
комбинацию винильного катиона (454). Другими словами, мы
рассматриваем реакцию винилиум-иона с олефином, исходя из
принципа сохранения орбитальной симметрии.
(453) (454)
Соответствующие орбитальные диаграммы показаны на
схемах (455) и (456). Основное отличие от простой
комбинации олефин — олефин (452) состоит в существенно более
благоприятной стерической ситуации, и прежде всего в
наличии ортогональной вакантной р-орбитали в винилиум-
ионе [зачерненные области в (455) и заштрихованная
область в (456)]. Сразу же можно заметить, что эта орбиталь
ш
(455) (456)
участвует в двух сильных связывающих взаимодействиях
[пунктирные линии на схеме (455)], которые отсутствуют
в [п2Б-\-п2д]-реакции двух молекул простых олефинов.
Отметим, что на схеме (455) вакантная р-орбиталь показана
сверху; нижняя часть орбитали взаимодействует с
заселенной я-системой простого олефинового реагента. Другими
словами, нормальная разрешенная по симметрии комбина-
178
ция катиона с этиленом в этой исключительной ситуации
«прокладывает дорогу» циклоприсоединению типа [я2д+я2а]
и совпадает с ним.
В свете проведенного здесь анализа становится ясно,
что винилиум-ион следует рассматривать прежде всего как
я2а-компонент в синхронном [я28+ я2а]-циклоприсоеди-
нении. Предсказанная реакция наблюдалась в
действительности, однако она не рассматривалась с интересующей нас
точки зрения. Грисбаум [235] нашел, что аллен реагирует
с хлористым водородом при —70°, давая смесь стереоизо-
мерных дихлордиметилциклобутанов (457).
Первоначально образующийся катион (458) комбинирует с молекулой
аллена по типу [я28+ я2а] с образованием катиона (459),
С1
СН3
С1
(457)
/>С-СН3
Н
0
н2с
(458) (459)
который в свою очередь реагирует с хлорид-ионом и
хлористым водородом, образуя наблюдаемые продукты.
Теперь легко понять ход не менее удивительного синтеза
1,2-дихлор-1,2,3,4-тетраметилциклобутена (460). Этот
синтез проводят, обрабатывая хлором 2-бутин в присутствии
трехфтористого бора [236, 237]. Комбинация 2-бутина с
(3-хлорвинилиум-ионом типа [я28+ я2а] (461) прямо дает
катион (462), который нейтрализуется ионом хлора в
соединение (460). Эти примеры не оставляют сомнения в том,
что винилиум-ионы в большой степени обладают
способностью вступать в синхронные комбинации с я23-системами.
С1С1
сн3
Нзс~й~
Н3С СН3
(460)
7*
179
C1- ■ н3с#4-сн3
/с=с-сн3
н3с н3С сн:
(461) (462)
Учитывая все вышесказанное, можно ответить на
вопрос, поставленный в начале обсуждения. Кетены, которые
могут реагировать как винилиумилиды (453),— идеальные
партнеры в антараповерхностных реакциях с
^-системами, и они играют эту разрешенную симметрией роль во
многих наблюдаемых синхронных комбинациях с олефино-
выми системами. Конечно, кетены и винилиум-ионы не
идентичны простым винилиум-ионам, однако детальный
анализ показывает, что обстоятельства меняются не очень
существенно и в более сложных случаях. Диаграмма
орбиталей кетена изображена на схемах (463) и (464). Место
(463) {464)
вакантных р-орбиталей простого винилиум-иона здесь
занимает незанятая зх*с=о-орбиталь молекулы кетена [ср.
(463')]; чрезвычайно низкое положение этой орбитали
объясняет в рамках теории МО большую электрофильную
реакционноспособность кетенов. Как и в случае
простейших винилиум-ионов, в этом случае нет изменения в
связывании из-за взаимодействия внешней я-системы с я*-ор-
биталью я28-компонента [ср. (456) и (464)]. Наконец,
отметим, что взаимодействие зтс=с-системы молекулы
кетена с антисимметричной орбиталью неподеленной пары на
атоме кислорода не имеет отношения к рассматриваемому
вопросу. Энергия этого взаимодействия не велика; в
результате взаимодействия образуются три новые аллильные
орбитали (рис. 45), из которых две заселены. Низшая из
этих орбиталей занимает место, которое обычно занимает
двухорбитальная зх-система [ср. (464) и (456)]. Электроны
180
средней орбитали по мере протекания реакций принимают
свой первоначальный статус свободной пары.
При димеризации кетенов, которую также относят к
синхронным реакциям [238], одна из молекул кетена ведет
себя как л2а-компонент, а другая проявляет себя обычным
образом по типу л28. Следует отметить, что участие в
реакции кетена в качестве ^-компонента не облегчается
наличием ортогональной яс=о-системы и что взаимодействие
#с=с Орбиталь
Орбит аль
не поделенной
электронной
пары на кис
лороде
Аллилъная орбиталь
Рис. 45. Аллильные орбитали молекулы кетена.
Вследствие относительной стабильности занятой атомной орбитали атома
кислорода узел средней аллильной орбитали находится не на центральном атоме
углерода, а между углеродным и кислородным атомами. На центральном атоме
углерода узел должен находиться в случае системы, состоящей из трех атомов
углерода.
со свободной парой на атоме кислорода не оказывает
значительного влияния в этом случае. Таким образом, при
участии в реакциях циклоприсоединения в качестве л28-
компонента кетены не должны заметно отличаться от
нормальных алкилэтиленов. Следовательно, нельзя ожидать,
что в реакциях кетенов с диенами нормальная разрешенная
по симметрии [л48+ я28]-реакция будет протекать легко.
Кроме того, [я48+ л2а]-реакция запрещена по симметрии,
и наличие ортогональной вакантной р-орбитали или
низко расположенной я*с=о-системы не может облегчить
[я4а+ л2а]-процесс. Взятые вместе эти обстоятельства
объясняют образование циклобутанонов и отсутствие цикло-
гексенонов при реакциях кетенов с простейшими диенами.
181
И. ОБОБЩЕННЫЕ ПРАВИЛА ОТБОРА
ДЛЯ ПЕРИЦИКЛИЧЕСКИХ РЕАКЦИЙ
Рассматривая контроль по орбитальной симметрии
синхронных химических изменений, мы создали основу для
общего рассмотрения всех перициклических реакций. Пери-
циклическими мы называем такие реакции, в которых все
изменения первого порядка соотношений между связями
происходят синхронно по замкнутым кривым. Выше
подробно обсуждались особенности отдельных четко
разграниченных типов реакций, однако было показано, что
различные классы имеют много общего. Особенно отмечено, что
электроциклические реакции и сигматропные изменения
также можно трактовать как синхронные
внутримолекулярные процессы циклоприсоедииения. Теперь мы
предложим дальнейшее важное обобщение, состоящее в том, что
все перициклические реакции можно рассматривать как
синхронные процессы циклоприсоедииения, которые
должны подчиняться правилам отбора для таких изменений.
Правила отбора для двухкомпонентных реакций
циклоприсоедииения (ср. рис. 22) можно легко обобщить методом
индукции, вводя любое число компонентов. Таким образом,
мы вывели следующее общее правило для всех
перициклических изменений: перициклическое изменение в основном
состоянии разрешено по симметрии, если общее число
(4<7+2) s- и (4г) ^-компонентов нечетное.
Очевидно, если общее число компонентов четное, то
реакция запрещена по симметрии. Системы с нечетным
числом электронов по своему поведению соответствуют
системам с четным числом, содержащим на один электрон
больше. Каждой реакции в основном состоянии соответствует,
вообще говоря, процесс в возбужденном состоянии, для
которого правило отбора обращается.
Для того чтобы завершить обобщенное описание
перициклических реакций, необходимо ввести еще одно,
последнее условие. Ясно, что если q или г равны 0, то 4г
-компонент соответствует одной атомной орбитали [также для
(4<7+2)-компонента]. Это означает, что такой одноорбиталь-
ный компонент может быть незаполненным или занятым.
Подобно любым другим компонентам, одноорбитальныё
компоненты могут участвовать либо в супраповерхностных,
либо в антараповерхностных процессах. Для их
обозначения необходим специальный символ со, который сочетается
182
с символами а- и я-компонентов, например a0s, ^0а, ^й89
Используя эти обозначения, легко классифицировать
любую перициклическую реакцию и определить, разрешена
ли она по симметрии. Теперь мы вновь возвратимся к
примерам, рассмотренным ранее, а также поясним процедуру
на нескольких новых примерах. Отметим, что пунктирные
линии на всех схемах представляют связывающее
взаимодействие в переходном состоянии.
а) Конротаторное электроциклическое раскрытие цикло-
бутенов (ср. рис. 23 и 24) можно рассматривать либо как
[jt2s+ a2a]-, либо как [л2а+а28]-процесс. Общее правило
сразу показывает, что процесс разрешен по симметрии.
б) Супраповерхностное сигматропное [1,3]-смещение с
инверсией при мигрирующем центре (ср. рис. 34) можно
также рассматривать как разрешенную по симметрии
реакцию типа [л28+ а2а] или [я2а+а28].
в) Дисротаторное электроциклическое превращение цик-
лопропилкатиона в аллильный катион можно
рассматривать либо как [coOs+(j2s]- (465), либо как [ш0а+а2аЬпро-
цесс (466). Оба процесса разрешены по симметрии. В
формальном отношении когда один со-компонент вступает в
перициклическую реакцию, то со-орбиталь перемещается
в новое положение и одна из пунктирных линий,
используемых для анализа, исчезает в изображении продукта.
{465) (166)
г) Сигматропный [1,2]-сдвиг в катионе не может
происходить с инверсией мигрирующей группы, так как
процессы L0a+(j2a] (467) и [coOs+(j2a] (468) запрещены по
симметрии. Отметим, что антараповерхностное связывающее
взаимодействие между двумя атомами, которые связаны
183
друг с другом не участвующей в химическом
взаимодействии связью и одним атомом, образующим мостик, физи-
Ufla + rfs]
(467)
(468)
чески невозможно. Таким образом, процессы [coOs+ CT2S],
изображенные на схемах (469) и (470), представляют собой
разрешенные по симметрии, однако физически
нереализуемые антараповерхностные сигматропные [1,3]-сдвиги с
инверсией при мигрирующем центре.
(469)
(470)
д) Супраповерхностный сигматропный [1,4]-сдвиг с
инверсией при мигрирующем центре можно рассматривать
как разрешенный по симметрии [л2а+ соОа+ а2а]-процесс
(471)
(472)
184
(471). Любая альтернативная формальная классификация
из тех, которые изображены ниже (473), представляет собой
разрешенную по симметрии реакцию, требующую инверсии
при R. Нет ни одного разрешенного супраповерхностного
процесса, в котором R мигрирует с сохранением
конфигурации. Например, перициклическая [я28+ o)0s+ а28]-ре-
акция (472), очевидно, запрещена по симметрии
L k^s г ш^а \ cr^s J
[ „2, + ш08 f a2a ] (473)
L u^a ~T~ cu^s ~T a^s J
e) Нелинейное хелетропное выделение двуокиси серы
из эписульфона 1(418), разд. 10.1] — разрешенная по
симметрии [а28+а2а]-реакция (474), а обратный процесс
должен быть изменением типа [n2s+(02a] (475).
(474) (47S)
ж) Реакция Дильса — Альдера является, очевидно,
разрешенным по симметрии [я48+л28]-превращением, но
она может быть проанализирована и как разрешенный
[ш08+ш28+ш0а+ш2а+ш08+ш28]-процесс (476). Этот при-
(476)
185
;so2
У
мер включен сюда частично из спортивного интереса,
однако мы преследуем и серьезные цели — показать,
что: 1) участие в перициклической реакции любого числа
и типа со-компонентов не создает трудностей в анализе
процесса с помощью общего правила и 2) поляризация связей,
участвующих в перициклической реакции, не влияет на
контроль этой реакции принципом сохранения
орбитальной симметрии.
Следует подчеркнуть, что использование общего
правила отбора для перициклических реакций требует
соблюдения двух предосторожностей.
1. В каждом индивидуальном случае должна быть
тщательно исследована геометрия системы для того, чтобы
заключить, насколько физически реализуем
рассматриваемый процесс. Пример, приведенный в пункте г), и
обсуждение превращения эфиров в разд. 7 (331) показывают,
что процесс, который формально разрешен по симметрии,
геометрически невозможен.
2. В тех случаях, в которых реагирующие компоненты
непосредственно соединены не участвующими в химическом
превращении связями, разрешенные реакции могут стать
запрещенными по симметрии из-за создающих напряжения
разрыхляющих взаимодействий, возникающих вне системы
связей, непосредственно участвующих в реакции. Так, при-
зман не может быть превращен в результате процесса
t(j2s+(j2a+(j2a] в три изолированные невзаимодействующие
частицы этилена, находящиеся в основном состоянии.
Геометрические напряжения, вызываемые неучаствующим в
реакции ст-скелетом, требуют, чтобы частицы этилена,
образующиеся в такой реакции, сильно взаимодействовали
по разрыхляющему типу. Электронная конфигурация
возникающего образования, обусловленная орбитальной
симметрией, является дважды возбужденным состоянием
образующегося бензола (разд. 6.4). Когда возникают подобные
обстоятельства, необходимо проводить полный анализ,
используя принцип сохранения орбитальной симметрии.
12. ИСКЛЮЧЕНИЯ
Исключений нет!
Нельзя также ожидать исключений из такого
фундаментального принципа, как принцип максимального
связывания. Тем более важно рассмотреть те реакции, которые
186
йа первый взгляд противоречат принципу сохранения
орбитальной симметрии.
Прежде всего нужно особенно подчеркнуть, что
разрешенная по симметрии синхронная реакция не обязательно
должна соответствовать истинному поведению реагирующих
молекул. Вполне возможен другой путь, при котором
процесс идет через образование реакционноспособных
промежуточных продуктов сравнительно низкой энергии. При
этом в некоторых случаях могут образовываться продукты,
которые соответствуют запрещенному по симметрии
синхронному процессу. В этих случаях действительный путь
реакции является несинхронным, включающим дискретные
промежуточные частицы. Принцип сохранения
орбитальной симметрии очень полезен для определения условий,
при которых следует ожидать протекания таких
ступенчатых процессов. Это справедливо, например, для
комбинации двух молекул этилена с образованием циклобутана.
Прямая синхронная комбинация [^2S+^2S1 запрещена по
симметрии, а разрешенная [я28+я2а]-реакция может быть
реализована только в особых условиях. Тем не менее
существует много случаев, когда отсутствуют специальные
предварительные требования для протекания реакции
[л23+л2а], и комбинация двух молекул этилена приводит
к образованию циклобутана (прекрасный обзор по этому
вопросу см. в [239]). В таких случаях реакции протекают
через бирадикальные или ионные промежуточные
частицы [240] и только тогда, когда реагирующие олефины
содержат заместители, которые стабилизуют необходимые
промежуточные образования.
Остается сделать только несколько замечаний о
запрещенных по симметрии реакциях, которые не могут
протекать постадийно. В принципе, если имеется достаточно
энергии и способ перевода молекулы в энергетически
богатую конфигурацию, можно вызвать реакцию,
запрещенную по симметрии. Очень сильный демон Максвелла может
получить молекулу циклобутана, если он возьмет в
каждую руку по молекуле этилена и, не без того, чтобы
проявить свое могущество, заставит их сблизиться «лицом к
лицу». Он мог бы также схватить молекулу циклобутена
за метиленовые группы, разорвать кольцо дисротаторным
путем и получить таким образом бутадиен. Однако без такого
демонического вмешательства молекулы очень редко ведут
себя подобным образом, так как почти всегда доступен тот
187
путь реакции, который соответствует более низкой
энергетической поверхности. Поэтому запрещенная по
симметрии реакция не происходит. Тем не менее всегда
остается возможность такого строения молекулы, что для нее
не существует другой альтернативы. Тогда, сообщая
молекуле достаточную энергию, можно провести реакцию
запрещенным по симметрии путем. Производные цис-бицикло-
[2,2,01-гексадиена (477) представляют, вероятно, пример
такого процесса. Эти соединения действительно
превращаются в соответствующие бензоидные изомеры с энергией
Н
ф
н
(477)
активации~37 ккал/моль [241]. Простое дисротаторное
раскрытие центральной связи запрещено по симметрии,
однако (477) уже находится по сравнению с бензолом в
напряженном состоянии. Это напряжение выражается величиной
~60 ккал [242]; добавление еще 37 ккал может оказаться
достаточным для того, чтобы обойти запрет на
дисротаторное раскрытие цикла. Однако даже в этом случае нельзя
полностью исключить возможность протекания реакции
через дискретные промежуточные состояния. Например,
если распад центральной связи в соединении (477)
сопровождается квазиконротаторным скошенным разложением
[(478); превращение ванна -»- закрученная ванна], то воз:
da «&<$>•
(478) (379)
никающее образование (479) содержит две скошенные
аллильнорадикальные системы [С5—С6—Q и С2—С3—C4],t
которые соединены ортогонально своими концами. Если
все связывание между С-1 и С-4 утрачивается одновременно
с этим движением, то обусловленные симметрией
разрыхляющие компоненты в переходном к бензолу состоянии
188
должны исчезнуть. Тогда реакция сможет закончиться бла^
годаря разрешенной по симметрии комбинации
изолированных аллильных радикалов. Однако даже если 1,4-связывание
сохраняется в какой-то степени (это приводит, конечно,
к возникновению связывания между С-2 и С-5), то
соответствующие разрыхляющие компоненты в переходном
состоянии (479), обусловленные симметрией, должны быть
меньше, чем в переходном состоянии простого,
запрещенного по симметрии дисротаторного раскрытия цикла.
Особенно интересно то, что если (479) представляет собой
переходное состояние превращения бициклогексадиенов в
ароматические вещества, то бициклогексадиен должен изоме-
ризироваться по схеме:
£jjj Ш Щ
5 4 3 6 3 4 16 3
(480) (481) (482)
Наконец, мы должны еще раз отметить, что заметная
стабильность ^нс-бицикло-[2,2,0]-гексадиена обусловлена
барьерами, связанными с симметрией. Наши аспиранты
в шутку предложили оценить время жизни изомера (483),
который синтезирован, вероятно, все тем же
разносторонним демоном.
|1 J Ц
Н
(483)
13. ДРУГИЕ ТЕОРЕТИЧЕСКИЕ РАБОТЫ
Если учесть, что принцип сохранения орбитальной
симметрии, как оказалось, является исключительно мощным
орудием предсказания и интерпретации химических
реакций, нет ничего удивительного в том, что были предложены
некоторые другие альтернативные теоретические
концепции. Во всех случаях основные выводы по необходимости
совпадают во всех отношениях с нашими. Здесь необходимо
189
сослаться на интересный вклад в теорию таких авторов,
как Фукуи [243], Салем [244], Циммерман [245] и Дьюр
[246], Остерхов и Ван-дер-Люгт [247] и, естественно, Лон-
ге-Хиггинс и Абрахамсон [248]. Уместно также
упомянуть некоторые ранние исследования, связанные с нашими
работами, и предшествующие им. Корреляционные
диаграммы для простейших химических реакций используются не
впервые. Лайдлер [249] описал некоторые интересные
применения. Гриффинг [250] широко использовал такие
диаграммы, однако он неправильно их истолковал. В важной
работе Герцберг [251] показал, что для группы 2/?-орбита-
лей число узлов в молекулярной системе сохраняется при
сближении атомов друг с другом. Этот автор показал
также, что два энергетически различных состояния не могут
иметь одинаковое число узлов. Бейдер [252] по-новому
рассмотрел и интересно обсудил некоторые элементарные
реакции.
Мы уже упоминали предположение [253] о том, что
орбитальная симметрия может играть определенную роль
в электроциклических реакциях. Сыркин [254] отметил
преимущество шестиэлектронного переходного состояния.
Его работы и работы Балабана [255] и Матью и Валлса
[256] служат богатым источником ссылок на литературу
по синхронным реакциям.
Появились многочисленные обзоры, которые суммируют
различные аспекты применения принципа сохранения
орбитальной симметрии [257].
Фукуи [258] развил теорию граничных электронов,
разработал и интенсивно изучал различные методы расчета,
связанные с проблемой химической реакционноспособностй.
В некоторых работах мы применяли расширенные расчеты
по Хюккелю [259] для того, чтобы подтвердить
качественные выводы. Преимущество этих приближенных расчетов
состоит в том, что их можно применять как для а-, так и
для я-систем. Метод был первоначально развит в
совместной работе с Липскомбом, также были использованы
предшествующие исследования Лонге-Хиггинса по боргидридам.
Специальный интерес представляют некоторые недавние
применения принципа сохранения симметрии в новых
направлениях. Особого внимания заслуживает распростра-'
нение правил отбора на реакции изомеризации и
замещения комплексов металлов переменной валентности [260].
Высказано предположение, что орбитальная симметрия
190
определяет хемилюминесцентные реакции [261]. Проведен
даже анализ роли орбитальной симметрии при переносе
энергии от возбужденных состояний [262].
14. ЗАКЛЮЧЕНИЕ
Теперь мы разъяснили принцип сохранения
орбитальной симметрии и проиллюстрировали примерами его
применение. По-видимому, следует подчеркнуть, что главное
содержание принципа основано на безусловном
утверждении, что химическая реакция происходит тем легче, чем
в большей степени связывание сохраняется в ходе
превращения. Следовательно, можно не сомневаться, что
принцип сохранится независимо от того, каким образом он будет
формулироваться и какова будет точность его применения
и формулировок.
Несомненно, будет происходить развитие принципа как
в фундаментальном направлении, так и в деталях. Так как
каждая элементарная стадия в любой химической реакции
представляет собой синхронный процесс, то
корреляционные идеи можно применить ко всем реакциям. Их
применимость в неорганической химии едва только начали
исследовать. Только недостаток воображения создает пределы
числу и разнообразию новых и замечательных молекул,
которые можно изобразить. Все это должно способствовать
поискам реакций, которые еще не открыты, но о которых
известно, что они возможны.
191
ЛИТЕРАТУРА
1. W о о d w a r d R. В., Н о f f m a n n R., J. Amer. chem. Soc.
87, 395 (1965).
2. Hoffmann R., Woodward R., J. Amer. chem. Soc,
87, 2046 (1965).
3. Woodward R. В., Н о f f m a n n R., J. Amer. chem. Soc,
87, 2511 (1965).
4. Woodward R. В., Aromaticity, Special Publication № 21.
The Chemical Society, London, 1967, p. 217.
5. Коулсон Ч., Валентность, изд-во «Мир», М., 1965.
5а. С о и 1 s о п С. A., Stewart E. Т. in Patai S., The
Chemistry of Alkenes, Wiley-Interscience, New York, 1964.
6. Heilbronner E., Bock H., Das HMO-Modell und
seine Anwendung, Verlag Chemie, Weinheim, 1968; M u 1 1 i-
ken R. S., Science, 157, 13 (1967); Angew. Chem., 79, 541
(1967).
7. Salem L., The Molecular Orbital Theory of Conjugated
Systems, Benjamin, New York, 1966.
7a. Стрейтвизер Э., Теория молекулярных орбит, изд-во
«Мир», М., 1965.
8. Н и п d F., Z. Phys., 40, 742 (1927); 42, 93 (1927); 51, 759 (1928);
Mu 1 1 i k ё n R. S., Phys. Rev., 32, 186 (1928); Rev. Mod-.
Phys., 4, 1 (1932).
8a. Herzberg G., The Electronic Structure of Diatonic Mole-
kules, 2 end. Van Nostrand, Princeton, 1950.
9. F u k u i K-, Y о n e z a w а Т., S h i n g u H., J. Chem. Phys.,
20, 722 (1952); F u k u i K-, Y о n e z a w а Т., N a g a t a C,
S h i n g u H., ibid., 22, 1433 (1954); Фукуи К.,
Современная квантовая химия, изд-во «Мир», М., 1968, т. 1, стр. 59.
10. W i п t e r R. Е. К., Tetrahedron Letters, 1965, 1207.
11. Vo gel E., Liebigs Ann., 615, 14 (1958); CriegeeR.,
Noll K., ibid., 627, 1 (1959).
12. Criegee R.,SeebachD., Winter R. E., В о г г е t-
z e n В., В r u n e H.-A., Chem. Ber., 98, 2339 (1965); В г a n-
tonG. R., FreyH.M., Skinner R.F., Trans.
Faraday Soc, 62, 1546 (1966); F г е у H. M., W a 1 s h R.f Chem.
Rev., 69, 103 (1969).
13. CriegeeR., BolzG., неопубликованные данные; S e e-
b а с h D., частное сообщение.
192
14. BraumanJ. I., Ellis L. E., van Tamelen E. E.,
J. Amer. chem. Soc, 88, 846 (1966).
15. В 1 о о m f i e 1 d J. J., M с С о n a g h у J. S., Jr., Hort-
m a n n A. G., Tetrahedron Letters, 1969, 3723.
16. С r i e g e e R., R e i n h a r d t H.G., Chem. Ber.,101,102(1968).
17. Untch K. G., Martin D. J., J. Amer. chem. Soc, 87,
4501 (1965).
18. D о о г а к i a n G. A., F r e e d m a n H. H., J. Amer. chem.
Soc, 90, 5310, 6896 (1968).
19. HuisgenR., Seidel H., Tetrahedron Letters, 1964,
3381.
20. Quinkert G., Opitz K., Wiersdorff W. W.,
F i n к е М., ibid., 1965, 3009.
21. С о г е у E. J., S t r e i t h J., J. Amer. chem. Soc, 86, 950
(1964).
22. P a q u e t t e L. А., В a r r e t t J. H., S p i t z R. P.,
Pitcher R., J. Amer. chem. Soc, 87, 3417 (1965).
23. Daub en W. G., Car gill R. G., Coates R. M.,
S a 1 t i e 1 J., J. Amer. chem. Soc, 88, 2742 (1966);
Crowley K. J-, Tetrahedron, 21, 1001 (1965).
24. S r i n i v a s a n R., J. Amer. chem. Soc, 90, 4498 (1968).
25. T h eu e r W. J.., Moo r e J. A., Chem. Commun., 1965, 468.
26. HavingaE., de Ко с к R. J., Rappold M. P.,
Tetrahedron, 11, 278 (1960); HavingaE., Schlatt-
m a n n J. L. M. A., ibid., 16, 146 (1961); SandersG.M,
H a v i n g a E., Rec trav. chim., 83, 665 (1964); I n h о f-
fenH. H., Irmscher K., Fortschr. Chem. org.
Naturstoffe, 17, 70 (1959); Inhof f en H. H., Angew.
Chem., 72, 875 (I960); L у t h g о е В., Proc Chem. Soc.
(London), 1959, 141; Daub en W. G., Fonken G. J.,
J. Amer. chem. Soc, 81, 4060 (1959).
27. CourtotP., RuminR., Tetrahedron Letters, 1968,
1091.
28. Muszkat K. A., Fisher E., J. Chem. Soc. (London),
B, 1967, 662; M a 1 1 о г у F. В., WoodC. S., Gordon
J. Т., J. Amer. chem. Soc, 86, 3094 (1964); ScholzM.,
DietzF., M о h 1 s t a d t M., Z. Chem., 7, 329 (1967).
29. V о g e 1 E., G r i m m e W., Dinne E., Tetrahedron
Letters, 1965, 391; M a r v e 1 1 E. N., С a p 1 e G., S с h a t z В.,
ibid., 1965, 385; G 1 a s s D. S., W a t t h e у J. W. H.,
WinsteinS., ibid., 1965, 377.
30. H u i s g e n R., D a h m e n A., H u b e r H., J. Amer. chem.
Soc, 89, 7130(1967); Marvel 1 E. N., SeubertJ.,
ibid., 89, 3377 (1967).
31. Meister H., Chem. Ber., 1963, 1688.
32. WeinshenkerN. M., Greene F. D., J. Amer. chem.
Soc, 90, 506 (1968).
33. .Ziegler K-, H a f n e г К-, Angew. Chem., 67, 301 (1955);
H a f ne r K., ibid., 67, 301 (1955).
34a) S к e 1 1 P. S., S a n d 1 e r S. R., J. Amer. chem. Soc, 80,
2024 (1958); 6) Schweizer E. E., P a r h a m W. E.,
ibid., 82, 4085 (1960); в) Р e t t i t R., ibid., 82, 1972 (1960).
35. CristolS. F., S e q u e i r a R. M., De Puy С. Н.,
J. Amer. chem. Soc, 87, 4007 (1965).
193
35а. В a i r d M. S., ReeseC. В., Tetrahedron Letters, 1967,
1379.
36. S с h 1 е у е г P. v. R., Van Dine G. WM S с h 6 1 1-
k о p f U., P a u s t J., J. Amer. chem. Soc, 88, 2868 (1966);
S с h 6 1 1 к о р f U., Angew. Chem., 80, 603 (1968); Angew.
Chem. internat. Edit., 7, 588 (1968).
37. ChosezL, Slinckx G., GlineurM., H о е t P.,
L a r о с h e P., Tetrahedron Letters, 1967, 2773.
37a. J e f f о r d С W., H u a n g Y e n E., M e d a r у R., ibid.,
1966, 6317.
38. WhithamG. H., WrightM., Chem. Commun., 1967,
294.
39. Huisgen R., Scheer W., H u b e r H., J. Amer. chem.
Soc, 89, 1753 (1967).
40. DenoN.C, P i t t m a n С V., Jr., T u r n e r J. O.,
J. Amer. chem. Soc, 87, 2153 (1965); Sorensen T. S„
Canad. J. Chem., 42, 2768 (1964); 43, 2744 (1965); J. Amer.
chem. Soc, 89, 3782, 3794 (1967).
41. N a z a r о v I. N., Z a r e t s к а у a I. I., Z. obsc. Chim.,
27, 693 (1957).
42. L e h r R., К u r 1 a n d D., Wood.ward R. В.,
неопубликованные данные; К u г 1 a n d D., Dissertation, Harvard
University, 1967; Lehr R., Dissertation, Harvard
University, 1968.
43. J о h n s t о n e R. A. W., W a r d S. D., J. Chem. Soc
(London), C, 1968, 1805; В i s h о p M. J., F 1 e m i n g I.,
ibid., 1969, 1712.
44. С h a p m a n O. L., E i a n G. L., J. Amer. chem. Soc, 90,
5329 (1968).
44a. В a t e s R. В., М с С о m b s D. A., Tetrahedron Letters,
1969, 977.
45. Rieke R., Ogliaruso M., McClung R., Win-
s t e i n S., J. Amer. chem. Soc, 84, 4729 (1966); К a t z T. J.,
TalcottC, ibid., 88, 4732 (1966).
46. Moshuk G., Petrowski G., Winstein S., J.
Amer. chem. Soc, 90, 2179 (1968).
47. F о n k e n G. J., частное сообщение; Schumate К. М.<,
Fonken G. J., J. Amer. chem. Soc, 87, 3996 (1965); 88,
1073 (1966).
48. R a d 1 i с k P., F e n i с a 1 W.,Tetrahedron Letters, 1967,4901.
49. M a s a m u n e S., С h i n С G., H о j о К., Sei dner
R. Т., J. Amer. chem. Soc, 89, 4804 (1967).
50. van T a m e 1 e n E. E., В u r k о t h T. L., J. Amer. chem.
Soc, 89, 151 (1967); С о о k s о n R. C, HudecJ., Mar-
s d e n J., Chem. Ind., 1961, 21; Vogel E., Meckel W.,
G r i m m e W., Angew. Chem., 76, 786 (1964); Angew. Chem.
internat. Edit., 3, 643 (1964).
50a. M a s a m u n e S., S e i d n e r R. Т., Chem. Commun., 1969,
542.
51. Huntsman W. D., Wr isters H. J., J. Amer. chem.
Soc, 89, 342 (1967).
52. SchroderG., Martin W., Oth J. F. M., Angew.
Chem., 79, 861 (1967); Angew. Chem. internat. Edit., 6, 870
(1967).
194
53. A v г a rn M., Dinu D., MateescU G., N е n i t z e s-
cu С. D., Chem. Ber., 93, 1789 (I960).
54. Merk W.,Pettit R.,J. Amer. chem. Soc, 89, 4788 (1967).
55. M a n g о F. D., S с h а с h t s с h n e i d e r J. H., J. Amer.
chem. Soc, 89, 2484 (1967); HogeveenH., Volger
H. C, ibid., 89, 2486 (1967).
56. VogelE., KieferH., R о t h W. R., Angew. Chem.,
76, 432 (1964); Angew. Chem. internat. Edit., 3, 442 (1964);
HuisgenR. MietzschF., Ange,w. Chem., 76, 36
(1964); Angew. Chem. internat. Edit., 3, 83 (1964).
57. AnetF. A.L, BournA. J. R., Lin Y. S., J. Amer.
chem. Soc, 86, 3576 (1964).
58. Chem. Res., 1, 17 (1968).
59. Warrener R. NM В r e m n e r J. В., Rev. Pure Appl.
Chem., 16, 117 (1966).
60. D i 1 1 i n g W. L., Chem. Rev., 66, 373 (1966).
61. Corey E. J., BassJ. D., LeMahieuR., Mitra
R. В., J. Amer. chem. Soc, 86, 5570 (1964); E a t о n P. E.,
Lin K-, ibid., 86, 2087 (1964); С о x A., d e M а у о P.,
Y i p R. W., ibid., 88, 1043 (1966); Robson R., Grubb
P. W., В a r 1 t г о p J. A., J. Chem. Soc. (London), 1964,
2153.
61a. Y a m a z а к i H., С v e t a n о v i с R. J., J. Amer. chem.
Soc, 91, 520 (1969).
616. S a 1 t i e 1 J., N g L i m L.-S., J. Amer. chem. Soc, 91, 5404
(1969).
62. BensonS. W, NangiaP.S., J. Chem. Phys., 38, 18
(1963); F г е у H. M. in Gold V., Advances in Physical Organic
Chemistry, Academic Press, New York, 1966, Vol. 4, p.
170.
63. G e r b e r i с h H. R., Walters W. D., J. Amer. chem.
Soc, 83, 3935, 4884 (1961).
64. Kraft K-, К о 1 t z e n b u r g G., Tetrahedron Letters,
1967, 4357, 4723.
65. Ziegler K-, S a u e r H., BrunsL., Froitzheim-
KuhlhornH., Schneider J., Liebigs Ann., 589,
122 (1954); Ziegler K-WilmsH., ibid., 567, 1 (1950);
С о p e A. C, HowellC. F., К n о w 1 e s A., J. Amer.
chem. Soc, 83, 3190 (1962).
66. DaubenW.G., Wipke W.T., Rev. Pure Appl. Chem.,
9, 539 (1964).
67. S r i n i v a s a n R., J. Amer. chem. Soc, 85, 4045 (1963).
68. FreyH. M., Stevens I. D. R., Trans. Faraday Soc,
61, 90 (1965).
69. S r i n i v a s a n R., L e v i A. A., H a 1 1 e r I., J. Phys.
Chem., 69, 1775 (1965).
70. W i b e r g К. В., Tetrahedron, 24, 1083 (1968).
71. Wiberg К. В,, Szeimies G,, Tetrahedron Letters,
1968, 1235.
72. Closs G. L., Pfeffer P. E., J. Amer. chem. Soc, 90,
2452 (1968).
73. T u r n e r R. В., G 0 e b e 1 P., v 0 n D 0 e r i n g E. W.,
Cob urn J. F., Jr., Tetrahedron Letters, 1965, 997.
74. HuisgenR., GrasheyR., SauerJ. in Patai S.,
195
The Chemistry of Alkenes, Interscience, New York, 1964,
p. 739.
75. В а с с е р м а н А., Реакция Дильса—Альдера, изд-во «Мир»,
М., 1968.
76. В е n s о n S. W., J. Chem. Phys., 46, 4920 (1967).
77. Woodward R. В., К a t z Т. J., Tetrahedron, 5, 70
(1959).
78. H a m m о n d G. S., T u г г о N. J., L i u R. S. H., J. Org.
Chem., 28, 3297 (1963); Hammond G. S., Liu R. S. H.,
J. Amer. chem. Soc, 85, 477 (1963); Valentine D., T u r-
roN. J., Hammond G. S., ibid., 85, 5202 (1964);
L i u R. S. H., T u г г о N. J., H a m m о n d G. S., ibid.,
87, 3406 (1965); SchenckG. O., Mannsfeld S.-P.,
S с h о m b u r g G., К r a u с h С. Н., Z. Naturforsch.,.
19b, 18 (1964).
79. M e i n w a 1 d J., M a z z о с с h i P. H., J. Amer. chem. Soc,
88, 2850 (1966); M e i n w a 1 d J., E с k e 1 1 A., Erick-
s о n K. L., ibid., 87, 3532 (1965); P r i n z b а с h H., Ha-
g e m a n n H., Angew. Chem., 76, 600 (1964); Angew. Chem.
internat. Edit., 3, 653 (1964); Prinzbach H., Druck-
r e у E., Tetrahedron Letters, 1965, 2959; Crowley K. J.,
ibid., 1965, 2863.
80. D a u b e n W. G., частное сообщение; D a u b e n W. G.,
Bell I., H u t t о n T. W., L a ws G. F., R h e i n e r A.,
Urscheler H., J. Amer. chem. Soc, 80, 4116 (1958).
81. D a u b e n W. G., В a u m a n n P., Tetrahedron Letters,
1961, 565; S a u n d e r s о п С. Р., Н о d g k i n D. C,
ibid., 1961, 573.
82. Childs R. F., Grigg R., Johnson A. W., J. Chem.
Soc. (London), C, 1967, 201.
83. С r i e g e e R., A s k a n i R., Angew. Chem., 80, 531 (1968);
Angew. Chem. internat. Edit., 5, 537 (1968).
84. E m e r s о n G. F., Watts L., P e t t i t R., J. Amer. chem.
Soc, 87, 131 (1965); MerkW., P e t t i t R., ibid., 89,
4787 (1967).
85. Nace H. R., частное сообщение.
86. P a q u e t t e L. A., S 1 о m p G., J. Amer. chem. Soc, 85,
765 (1963); d e M а у о P., Y i p R. W., Proc Chem. Soc
(London), 1964, 84; A p p 1 e q u i s t D. E., SearleR.,
J. Amer. chem. Soc, 86, 1389 (1964); В r a d s h a w J. S.,
Hammond G. S., ibid., 85, 3953 (1963); Kraft K.,
KoltzenburgG., Tetrahedron Letters, 1967, 4357.
87. D о e r u n g W. v. E., W i 1 e у D. W., Tetrahedron, 11, 183
(1960).
88. Prinzbach H., S e i p D., Englert G., Liebigs Ann.,
698, 57 (1966); Prinzbach H., S e i p D., К n о t h e L.,
FaisstW., ibid., 698, 34 (1966).
89. Le Goff E., J. Amer. chem. Soc, 84, 3975 (1962).
90. G a 1 b r a i t h A., S m a 1 1 Т., В a r n e s R. A., Boe-
kelheidue V., J. Amer. chem. Soc, 84, 453 (1961); Boe-
k e 1 h e i d e V., F e d о r u k N. A., Proc. Nat. Acad. ScL,
55, 1385 (1966).
91. Houk K-, Dissertation, Harvard University, 1968.
92. С о о k s о n R. C, D r a k e B. V., HudecJ., Morri-
196
s о n A., Chem. Commun., 1966, 15; I t 6 S., F u j i s e Y.
Oku da Т., Inoue Y., Bull. Chem. Soc. Japan, 39, 135
(1966).
93. NozoeT., M u к a i Т., Takase K-, Takase Т.,
Proc. Japan. Acad., 28, 477 (1952).
94. I t 6 S., Fujise Y., W о о d s M. C, Tetrahedron Letters,
1967, 1059.
95. P a q u e t t e L. А., В a r r e t t J. H., J. Amer. chem. Soc,
88, 2590 (1966).
96. WinbergH. E., FawcettF. S., Mo с he 1 W. E.,
T h e о b a 1 d С W., J. Amer. chem. Soc, 82, 1428 (1960);
Cram D. J., Montgomery С S., Knox G. R., ibid.,
88, 515 (1966); С r am D. J., Da Hon С. К.Дпох G. R.,
ibid., 85, 1088 (1963); LongoneD. Т., Boettcher
F.-P., ibid., 85, 3436 (1963).
97. M u k a i Т., T e z u k а Т., A k a s a k i Y., J. Amer. chem.
Soc, 88, 5025 (1966).
98. К e n d e A. S., Lancaster J. E., J. Amer. chem. Soc,
89, 5283, (1967).
99. D о e r i n g W. v. E., частное сообщение.
100. CooksonR. С, Nye M. J., SubrahmanyamG.,
J. Chem. Soc. (London), C, 1967, 473; 1965; 2009; Fort A. W.,
J. Amer. chem. Soc, 84, 2620, 2625, 4979 (1964); T u г г о N. J.,
H a m m о n d W. В., ibid., 87, 3258 (1965); 88, 3672 (1966);
H a m m о n d W. В., T u г г о N. J., ibid., 88, 7880
(1966).
101. H о f f m a n n H. M. R., J о у D. R., Suter A. K. J.
Chem. Soc (London), B, 1968, 57.
102. W a 1 1 s F., Pa dill a J., Joseph-Nathan P., Gi-
r a 1 F., R о m о J., Tetrahedron Letters, 1965, 1577.
103. H u i s g e n R., Angew. Chem., 75, 604 (1963); Angew. Chem.
internat. Edit., 2, 565 (1963); H u i s g e n R., G r a s h e у R.,
S a u e r J. in Patai S., The Chemistry of Alkenes, Interscience,
New York, 1964, p. 739; H u i s g e n R., Helv. Chim. Acta,
50, 2421 (1967).
104. E с k e 1 1 A., H u i s g e n R., S u s t m a n n R., W a 1 1-
b i 1 1 i с h G., G r a s h e у D., Spindler E., Chem.
Ber., 100, 2192 (1967).
105. a) Schaffner K-, Advances in Photochemistry, 4, 81
(1966); б) К г о p p P. J., Organic Photochemistry, 1, 1 (1967).
106. Barton D. H. R., McGhie J., Rosenberger R., J.
Chem. Soc. (London), 1961, 1215; В a r t о n D. H. R., d e
M а у о P., S h a f i q M., ibid., 1958, 140, 3314; A r i g o-
niD., Bosshard H., Bruderer H., Biichi G.,
J e g e г О., К r e b a u m L. J., Helv. Chim. Acta, 40,
1732 (1957).
107. К w i e W. W., S h о u 1 d e r s B. A., G a r d n e r P. D.,
• J. Amer. chem. Soc, 84, 2268 (1962); С h a p m a n O. L.,
R e t t i g T. A., G r i s w о 1 d A. A., D u t t о n A. I.,
F i t t о n P., Tetrahedron Letters, 1963, 2049; N a n n В.,
CravelD., S с h о r t a R., Wehrli H., S с h a f f.
n e r K., J e g e r O., Helv. Chim. Acta, 46, 2473 (1963).
108. NannB., Grave ID., SchortaR., Wehrli H,
Schaffner K-, J e g e r O., Helv. Chim. Acta, 46, 2473
197
(1963); N a n n В., W e h r 1 i H., S с h a f f n е г К., J е-
ger О., ibid., 48, 1680 (1965).
109. Zimmerman H. E., Lewis R. G., McCullough
J. J., P a d w a A., St a ley S., S e m m e 1 h а с к M.,
J. Amer. chem. Soc, 88, 159 (1966); ChapmanO.L,
S i e j a J. В., W e 1 s t e a d W. J., Jr., ibid., 88, 161 (1966)
110. Zimmerman H. E., Advances in Photochemistry, 1, 183
(1963); ChapmanO.L, ibid., 1, 323 (1963).
111. Zimmerman H. E., 17th National Organic Symposium
of the American Chemical Society, Bloomington, Indiana,
1960, Abstracts, p. 31; Zimmerman H. E.,
Schuster D. I., J. Amer. chem. Soc, 83, 4486 (1961); Zimmer-
m a n H. E., Tetrahedron, 19, Suppl. 2, 393 (1963).
112. Barton D. H. R., de M а у о P., ShafiqM., J.
Chem. Soc.(London), 1958,140; Ar i gon i D., Bos shardM.,
Bruderer H., В ii с h i G., Jeger O., Krebaum
L. J., Helv. Chim. Acta, 40,1732 (1957); С о с к e r W.,
Crowley К-, Edward J. Т., McMurry Т. В. Н.,
Stuart E. R., J. Chem. Soc. (London), 1957, 3416;
Barton D. H. R., Gilham P. Т., ibid., 1960, 4596.
113. Schuster D. I., частное сообщение.
114. D u t 1 e г Н., В о s s h a r d M., J e g e г О., Helv. Chim.
Acta, 40, 494 (1957); Weinberg K-, Utzinger E. C,
A r i g о n i D., Jeger O., ibid., 43, 236 (1960); Dut-
1 e r H., G a n t e r C, R у f H., U t z i n ger E. C.,We i n-
b e r g K-, Schaffner K., Arigoni D., Jeger O.,
ibid., 45, 2346 (1962); GanterC, GreuterF., К а -
g i D., S с h a f f n e r K., J e g e r O., ibid., 47, 627 (1964);
FreiF., GanterC, К a g i D., К о с s i s K., M i 1 j-
kovicM., SiewinskiA., WengerR.*,
Schaffner K-, Jeger O., ibid., 49, 1049 (1966).
115. ChapmanO.L, S i e j a J. В.. Welstead W. J.,
Jr., J. Amer. chem. Soc, 88, 161 (1966).
116. BellusD., KearnsD.R., Schaffner K. Helv.
Chim. Acta, 52, 971 (1969).
117. Williams J. R., Ziffer H., Chem. Commun., 1967,
194, 469; Tetrahedron, 24, 6725 (1968).
118. P a q u e t t e L. A., E i z e m b e r R. F., Cox O., J. Amer.
chem. Soc, 90, 5153 (1968).
119. Griffin G. W., С о v e 1 1 J., P e t t e r s о n R. C, D о d-
s о n R. M., К 1 о s e G., J. Amer. chem. Soc, 87, 1410 (1965);
К r i s t i n s s о n H., Griffin G. W., ibid., 88, 378
(1966).
120. Zimmerman H. E., Hancock K. G., J. Amer. chem.
Soc, 90, 3749 (1968); Z i m m e r m a n H. E., M о r s e R. L.,
ibid., 90, 954 (1968).
121. Pfenninger E., Poel D. E., Berse C, Wehr-
1 i H., Schaffner K-, Jeger O., Helv. Chim. Acta,
51, 772 (1968).
122. SauersR. R., Shur p ik A..J. Org. Chem., 33, 799 (1968).
123. R о t h W. R., P e 1 t z e г В., L i e b i g s Ann., 685, 56
(1965).
124. E d m a n J. R., J. Amer. chem. Soc, 88, 3454 (1966).
125. С i g a n e к E., J. Amer. chem. Soc, 88, 2882 (1966).
126. Zimmerman H. E., G i v e n s R. S., P a g n i R. M.,
J. Amer. chem. Soc, 90, 4192 (1968).
127. Brewer J. P. N., H e a n e у Н., Chem. Commun., 1967,
811; R a b i d e a u P. W., H a m i 1 t о n J. В., F r i e d-
m a n L., J. Amer. chem. Soc, 90, 4465 (1968).
128. Zimmerman H. E., BinkleyR. W., Givens
R. S., S h e r w i n M. A., J. Amer. chem. Soc, 89, 3932
(1967).
128a. В e r s о n J. А., О 1 i n S. S., J. Amer. chem. Soc, 91, 777
(1969).
129. В 1 о m q u i s t А. Т., M e i n w a 1 d Y. C, J. Amer. chem.
Soc, 81, 667 (1959); С о о к s о n R. С, G i 1 a n i S. S. H.,
Stevens I. D. R., Tetrahedron Letters, 1962, 615;
Hall H. K., J. Org. Chem., 25, 42 (1960).
130. Will i a ms J. K-, Benson R. E., J. Amer. chem. Soc,
84, 1257 (1962).
131. S t a a b H. A., G r a f F., J u n g e В., Tetrahedron Letters,
1966, 743.
132. S m i t h C. D., J. Amer. chem. Soc, 88, 4273 (1966); P r i n z-
b а с h H., Rev. Pure Appl. Chem., 16, 24 (1968); P r i n z-
bachH., RivierJ., Angew. Chem., 79, 1102 (1967);
Angew. Chem. internat. Edit., 6, 1069 (1967).
133. О t h J. F. M., Angew. Chem., 80, 633 (1968); Angew. Chem.
internat. Edit., 7, 646 (1968); Rec trav. chim., 87, 1185 (1968).
134. P r i n z b а с h H., RivierJ., Tetrahedron Letters, 1967,
3713.
135. PrinzbachE., A r g u ё 1 1 e s M., Druckrey E.,
Angew. Chem., 78, 1057 (1966); Angew. Chem. internat. Edit.,
5, 1039 (1966).
136. P r i n z b а с h H., V о g e 1 P., A u g e W., С h i m i a, 21,
469 (1967).
137. S t e e 1 C, Z a n d R., H u r w i t z P., С о h e n S. G., J.
Amer. chem. Soc, 86, 679 (1964).
138. A s к a n i R., Chem. Ber., 98, 3618 (1965).
139. T а к a n a s h i M., Kitahara Y., M u r a t a L, N i t-
t а Т., W о о d s M. C, Tetrahedron Letters, 1968, 3387.
140. Лонге-Хиггинс Г. К., в сб. Теоретическая
органическая химия. Доклады, представленные на симпозиуме,
посвященном памяти Кекуле, организованном Химическим
Обществом, Лондон, сентябрь, 1958 г., изд-во «Мир», М., 1963,
стр. 17.
141. Sublett В., В о w m a n N. S., J. Org. Chem., 26, 2594
(1961).
142. В е г s о n J. A., Nelson G. L., J. Amer. chem. Soc, 89,
5303 (1967); В e r s о n J. A., Accounts chem. Res., 1, 152
(1968).
143. Bailey W. J., В а у 1 о u n у R. A., J. Org. Chem., 27,
" 3476 (1962).
144. D о e r u n g W. v. E., частное сообщение; U 1 1 m a n E. F.,
J. Amer. chem. Soc, 82, 505 (1960).
145. О v e r b e r g e г С G., В о г с h e r t A. E., J. Amer. chem.
Soc, 82, 1007 (1960).
146. Flowers M. C, F г е у H. M., J. Chem. Soc, 1961, 3547;
E 1 1 i s R. J., F г е у H. M., ibid., 1964, 959, 4188; E 1-
199
1 i о t С. J., F г е у H. M., ibid., 1965, 345; А, 1966, 553;
W e 1 1 i n g t о п С. A., J. Phys. Chem., 66, 1671 (1962);
F г е у H. M., Marshall D. C, J. Chem. Soc, 1962,
3981; В r a n t о n G. R., F г е у Н. M., ibid., 1966, 1342.
147. R a b i n о w it с h B. S., S с hi a g E. W., W i b er g К. В.,
J. Chem. Phys., 28, 504 (1958); Rabinowitch B. S.,
Schlag E. W., J. Amer. chem. Soc, 86, 5996 (1960).
148. EggerK. W., Golden D. M., BensonS.W., J.
Amer. chem. Soc, 86, 5420 (1964).
149. W i 1 1 с о t t M. R., С a r g 1 e V. M., J. Amer. chem. Soc,
89, 723 (1967); Roth W. R., частное сообщение.
150. DaubenW. G., W i p к e W. Т., Rev. Pure Appl. Chem.,
9, 539 (1964); H u r s t J. J., W h i t h a m G. M., J. Chem.
Soc, 1960, 2864; С о о к s о n R. С, G о g t e V. N.. H u-
d e с J., M i r z a N. A., Tetrahedron Letters, 1965, 3955;
Erman W. F., KretschmarH. C.,J. Amer. chem.
Soc, 89, 3842 (1967); BrownR. F.C., С о о к s о n
R. С, H u d e с J., Tetrahedron, 24, 3955 (1968); Cook-
son R. C, Chem. in Britain, 5, 6 (1969); В a g g 1 i o-
1 i n i E., HemlowH. P., S с h a f f n e r K., J e g e r
O., Chimia, 23, 181 (1969).
151. G 1 a s s D. S., Boikess R. SM Winstei n S.,
Tetrahedron Letters, 1966, 999.
151a. Grigg R., Johnson A. W., Richardson K-,
S h e 1 t о n K. W., Chem. Commun., 1967, 1192; Boekel-
h e i d e V, SturmE., J. Amer. chem. Soc, 91, 902
(1969).
152. RothW. R., К б n i g J., Liebigs Ann., 699, 24 (1966);
kloosterziel H., ter Borg A. P., Rec, trav.
chim., 84, 1305 (1965).
153. Roth W. R., KonigJ., частное сообщение.
154. M i г о n о v V. A., S о b о 1 е v Е. V., Е 1 i z a r о v a A. N.,
Tetrahedron, 19, 1939 (1963); McLean S.,Haynes R.,
Tetrahedron Letters, 1964, 2385.
155. RothW. R., Tetrahedron Letters, 1964, 1009.
156. ter Borg A. P., Kloosterziel H., vanMeurs N.,
Rec trav. chim., 82, 717, 741, 1189 (1963); Weth E,,
Dreiding A. S., Proc Chem. Soc. (London), 1964,
59; E gger K. W., J. Amer. chem. Soc, 89, 3688 (1967).
157. Bergsson G., Weidler A., Acta Chem. Scand., 17,
1798 (1963); W e i d 1 e r A., ibid., 17, 2724 (1963);
Weidler A., Bergsson G., ibid., 18, 1484 (1964); A 1 m у J.,
U у e d a R. Т., С r a m D. J., J. Amer. chem. Soc, 89,
6768 (1967).
158. RothW. R., Liebigs Ann., 671, 25 (1964).
159. Srinivasan R., J. Amer. chem. Soc, 84, 3982 (1962);
Crowley K. J., Proc Chem. Soc. (London), 1964, 17;
Prinzbach H., Druckrey E., Tetrahedron Letters,
1964 2959.
160. Zwicker E.VF., Grossweiner L. I., Yang N. C.,'
J. Amer. chem. Soc, 85, 2671 (1963); W e t t e r m a r к G.,
Photochem. and Photobiol., 4, 621(1965); Huffman K- R.,
L о у M., U 1 1 m a n E. F., J. Amer. chem. Soc, 87, 5417
(1965); В u с h i G., Y a n g N. C, ibid., 79, 2318 (1957).
200
161. В e r s о n J. A., W i 1 1 с о t t M. R., Ill, J. Amer. chem
Soc, 87, 2751, 2752 (1965); 88, 2494 (1966).
162. D о e r i n g W. v. E., R о t h W. R., Tetrahedron, 18, 67
(1962); Angew. Chem., 75, 27 (1963); Angew. Chem. internat.
Edit., 2, 115 (1963); R h о a d s S. J., in de Mayo P.,
Molecular Rearrangements, Interscience, New York, 1963, Vol. 1,
p. 655; Jefferson A., ScheinmannF., Quart.
Rev. (Chem. Soc, London), 22, 391 (1968).
163. M i у a s h i Т., N i t t a M., M u к a i Т., Tetrahedron
Letters 1967 3433
164. S с h'l a t m a n n J. L. M. A., P о t J., H a v i n g a E.,
Rec, trav. chim., 83, 1173(1964); AkhtarM., Gibbons
С J., Tetrahedron Letters, 1965, 509.
165. ter Borg A. P., Kloosterziel H., Rec. trav. chim.,
84, 241 (1965); D oer i ng W. v. E., G a s p a r P. P.,
J. Amer. chem. Soc, 85, 3043 (1963); Roth W. R., Angew.
Chem., 75, 921 (1963); Angew. Chem. internat. Edit., 2, 688
(1963); Jones L. В., J о n e s V. K-, J. Amer. chem. Soc,
90, 1540 (1968).
166. Murray R. W., К a p 1 a n M. L., J. Amer. chem. Soc,
88, 3527 (1966).
166a. Hug R., Hansen H.-J., S с h m i d H., Chimia, 23, 108
(1969).
167. S с h m i d K., S с h m i d H., Helv.Chim.Acta, 36, 687 (1953).
168. Frater Gy., S с h m i d H., Helv.Chim.Acta, 51, 190 (1968).
169. PockerY., BersonJ. A. in de Mayo P., Molecular
Rearrangements, Vol. 1. Interscience, New York, 1963.
170. M с С a u 1 а у D. A., L i e n A. P., J. Amer. chem. Soc, 79,
5953 (1957); Steinberg H., S i x m a F. L. J., Rec.
trav. chim., 81, 185 (1962); MacLeanC, Mackor
E. L., J. Chem. Phys., 34, 2208 (1961); Коптун В. А.,
Шубин В. Г., Р е з в а к и н Э. И. Изв. АН СССР, 1965,
201.
171. SwattonD. W., H a r t H., J. Amer. chem. Soc, 89,
5075 (1967).
172. BrennanT. M., HillR. К., J. Amer. chem. Soc, 90,
5614 (1968).
173. Z i m m e r m a n H. E., С r u m r i n e D. S., J. Amer. chem.
Soc, 90, 5612 (1968).
174. HartH, R о d g e r s T. R., G r i f f i t h s J., J. Amer.
chem. Soc, 91, 754 (1969).
175. С h i 1 d s R. F., W i n s t e i n S., J. Amer. chem. Soc, 90,
7146 (1968).
176. Hans en H.-J., S u t t e г В., Schmid H., Helv.
Chim. Acta, 51, 828 (1968).
177. MakisumiY., NotzumotoS., Tetrahedron Letters,
1967, 6393.
178. Schol lkopf U.,Fellenberger K., Liebigs Ann,,
698, 80 (1966); Zimmerman H. E. in de Mayo P.,
Molecular Rearrangements, Interscience, New York, 1963, Vol. 1,
p. 372.
179. В a 1 d w i n J. E., H а с k 1 e r R. E., К e 1 1 у D. P., Chem.
Commun., 1968, 537, 538; В 1 а с k b u r n G. M., О 1 1 i s
W. D., P 1 а с k e t t J. D., S m i t h C, Sutherland
201
I. О., ibid., 1968, 186; В a t e s R. В., F e 1 d D.,
Tetrahedron Letters, 1968, 417; T г о s t В. М., LaRochelle R.,
ibid., 1968, 3327; В a 1 d w i n J. E., P e a v у R. E., ibid.,
1968, 5029; KirmseW, К а р р s M., Chem. Ber., 101,
994, 1004 (1968).
180. Grovenstein E., Jr., Wentworth G., J. Amer.
chem. Soc, 89, 1852, 2348 (1967).
181. StevensT. S..J. Chem. Soc. (London), 1930, 2107; John-
stoneR. A. W., StevensT. S., ibid., 1955, 4487;
Hill R. К., Chan T.-H., J. Amer. chem. Soc, 88, 866
(1966).
182. Brewster J. H., Kline M. W., J. Amer. chem. Soc,
74, 5179, (1952); Mi 1 1 ar d B. J., StevensT. S., J.
Chem. Soc. (London), 1963, 3397; J e n n у E. F., D r u e у J.,
Angeiw. Chem., 74, 152 (1962); Angew. Chem. internat. Edit.,
1, 155 (1962).
183. Крам Д., Основы химии карбанионов, изд-во «Мир», М.,
1967; LansburyP. Т., Р a t t i s о п V. A., Sidler
J. D., В i e b e r J. В., J. Amer. chem. Soc, 88, 78 (1966);
L e p 1 e у A. R., ibid., 91, 1237 (1969); В a 1 d w i n J. E.,
частное сообщение.
184. Glass D. S., Z i r n e r J., Winstein S., Proc Chem.
Soc, 1963, 276; Ellis R. J., Frey H. M., ibid., 1964,
221; J. Chem. Soc, 1964, Suppl. 1, 5578; R о t h W. R., Lie-
bigs Ann., 671, 10 (1964); R о t h W. R., К 6 n i g J., ibid.,
688, 28 (1965); Grim me W., Chem. Ber., 98, 758 (1965);
Crandall J. K-, Watkins R. J., Tetrahedron
Letters, 1967, 1717; RobertsR.M., L a n d о 1 t R. G.,
Greene R. N., H e у e r E. W., J. Amer. chem. Soc,
89, 1404 (1967); О h 1 о f f G., Tetrahedron Letters, 1965,
3795; R о t h W. R., С h i m i a, 20, 229 (1966); McGre-
e r D. E., С h i u N. W. К., М с D a n i e 1 R. S., Proc.
Chem. Soc, 1964, 415; Marvell E. N., Anderson
D. R., О n g J., J. Org. Chem., 27,1109 (1962); Lauer W. M,
J о h n s о n T. A., ibid., 28, 2913 (1963); H a b i с h A.,
Barner R.,von Philipsborn W., SchmidH.,
Helv. Chim. Acta, 48, 1297 (1964); Roberts R. M., L a n-
d о 1 t R. G., J. Amer. chem. Soc, 87, 2281 (1964);
RobertsR.M, Greene R. N., Landol t R. G., He-
у e r E. W., ibid., 87, 2282 (1965); Roberts R. M., L a n-
d о 1 t R. G., Greene N., HeyerE. W., ibid., 89,
1404 (1967); С о n i a J. M., Leyendecker R, Du-
bois-FagetC, Tetrahedron Letters, 1966, 129, Roues-
sacF., ConiaJ. M., ibid., 1965, 3313.
185. HiinigS., M u 1 1 e r H. R., Thier W., Angew. Chem.,
77, 368 (1965); Angew. Chem. internat. Edit., 4, 271 (1965);
С о г е у E. J., P a s t о D. J., Mock W. L, J. Amer.
chem. Soc, 83, 2957 (1961).
186. Doering W. v. E., Rosenthal J. W., J. Amer. chem.
Soc, 89, 4535 (1967).
187. D о w d P'., частное сообщение; Fleming I., частное
сообщение.
188. WellingtonC. A., Walters W. D., J. Amer. chem.
Soc, 83, 4888 (1961).
202
189. Reusch W., R u s s e 1 1 M., D z u г е 1 1 а С, J. Org. Chem.,
29, 2446 (1964).
190. BaldwinJ. E., Tetrahedron Letters, 1966, 2953.
191. EllisR. J., FreyH. M., J. Chem. Soc. (London), A,
1966, 553.
192. В e n s о n S. W., ShawR., Trans. Faraday Soc, 63, 985
(1967); J. Amer. chem. Soc, 89, 5351 (1967).
193. Walters W. D., частное сообщение.
194. FreyH. M., Lister D. H., J. Chem. Soc (London), A,
1967, 609, 1800; FreyH. M., WalshR, Chem. Rev.,
69, 103 (1969).
195. S r i n i v a s a n R., J. Amer. chem. Soc, 83, 2806 (1961).
195a. О к a b e M., M с N e s b у J. R., J. Chem. Phys., 34, 668
(1962).
1956. D о е р к e r R. P., A u s 1 о о s P., ibid., 42, 3746 (1965).
195b. R e b b e r t R. E., A u s 1 о о s P., J. Phys. Chem., 66, 2253
(1962).
196. Fukui K. in Lowdin P.-O., Pullman В., Molecular
Orbitals in Chemistry, Physics and Biology. Academic Press,
New York, 1964, p. 525.
197. Salem L., J. Amer. chem. Soc, 90, 543, 553 (1968).
198. Alder K., SteinG., Angew. Chem., 50, 514 (1937); A 1-
d e r K., Liebigs Ann., 571, 157 (1951); Al der K.,
Schumacher M., Fortschr. Chem. Org. Naturstoffe, 10, 1 (1953);
В e r s о n J. A., HamletZ., Mueller W. A., J.
Amer. chem. Soc, 84, 297 (1962).
199. Wassermann A., J. Chem. Soc. (London), 1935, 825, 1511;
1936, 432; Trans. Faraday Soc, 35, 841 (1939).
200. W о о d w a r d R. В., В a e r H., J. Amer. chem. Soc, 66,
645 (1944).
201. CooksonR.C, D г а к e B. V., H u d e с J.,
Morrison A., Chem. Commun., 1966, 15; H о u к К.,
Dissertation, Harvard University, 1968.
202. CooksonR.C, D a n с e J., H u d e с J., J. Chem. Soc,
1964, 5416.
203. W i t t i g G., W e i n 1 i с h J., Chem. Ber., 98, 471 (1965).
204. С r i e g e e R., Angew. Chem., 74, 703 (1962); Ange/w. Chem.
internet. Edit., 1, 519 (1962); S к e 1 1 P. S., Peterson
R. J., J. Amer. chem. Soc, 86, 2530 (1964); Watts L.,
F i t z p a t r i с к J. D., P e t t i t R., ibid., 88, 623 (1966);
H e d а у a E., Miller R. D., McNeil D. W., D'A n-
g e 1 о P. F., S с h i s s e 1 P., J. Amer. chem. Soc, 91, 1875
(1969).
205. DoeringW. v. E., R о t h W. R., Tetrahedron, 18, 67
(1962); Angew. Chem., 75,27 (1963); Angew. Chem. internet.
Edit., 2, 115 (1963); HiilR. R., Gilman N. W., Chem.
Commun., 1967, 619.
205a.. Simonetta M., F a v i n i G., Mariani C, G r a -
maccioni P., J. Amer. chem. Soc, 90, 1280 (1968).
2056. Frater G., H a b i с h A., H an sen H.-J., S chmi d H.,
Helv. Chim. Acta, 52, 335 (1969).
206. BrownJ.M, Proc Chem. Soc, 1965, 226; D о e r i n g W.
v. E., Roth W. R., Tetrahedron, 19,715 (1963); Mere-
n у i R., Oth J. F. M„ Schroder G., Chem. Ber., 97,
203
3150(1964); S t a a b H. A., V б g t 1 е F., Tetrahedron
Letters 1965 54
206a. H о f f m a n n H.' M. R., J о у D. R., J. Chem. Soc.
(London), B, 1968, 1182.
207. L e m a 1 D. M., M с G r e g о r S. D., J. Amer. chem. Soc,
88, 1335 (1966).
208. BaldwinJ. E., Canad. J. Chem., 44, 2051 (1966).
209. YankelevichS., FuchsB., Tetrahedron Letters,
1967, 4945.
210. ChapmanO.L, BordenG. W..J. Org. Chem., 26,
4185 (1961); ChapmanO.L, P a s t о D. J., Borden
G. W., G r i s w о 1 d A. A., J. Amer. chem. Soc, 84, 1220
(1962); Schus ter D. I., S с к о 1 n i с к В. R., Lee
F.-T. H., ibid., 90, 1300 (1968).
211. С a r p i n о L. A., Chem. Commun., 1966, 494.
212. Moff itt W., Proc Roy. Soc, A200, 409 (1950).
213. Stau dinger H., RitzenhalerB., Ber. dtsch. chem.
Ges., 68, 455 (1935); Hesse G., Reicho 1 d E., Maj-
mudarS., Chem. Ber., 90, 2106 (1957); Cava M. P.,
D e a n a A. A., J. Amer. chem. Soc, 81, 4266 (1959).
213a. С a v a M. P., S с h 1 e s s i n g e r R. H., Van Meter
J. P., J. Amer. chem. Soc, 86, 3173 (1964).
214. Mock W. L., J. Amer. chem. Soc, 88, 2857(1966);
McGregor S. D., Lemal D. M., ibid., 88, 2858 (1966).
215. S a 1 t i e 1 J., M e t t s L., J. Amer.chem.Soc, 89, 2232 (1967).
216. Mock W. L., J. Amer. chem. Soc, 89, 1281 (1967); 91, 5682
(1969).
217. S к e 1 1 P. S., W о о d w о r t h R. C, J. Amer. chem. Soc,
78, 4496 (1956); 81, 3383 (1959); S к e 1 1 P. S., Garner
A. Y., ibid., 78, 5430 (1956); D о e r i n g W. v. E., La
F 1 a m m e P., ibid., 78, 5447 (1956); F г е у Н. М., ibid.,
80, 5005 (1958); Moore W. R., M о s e r W. R., LaPra-
d e J. E., J. Org. Chem., 28, 2200 (1963).
218. Hoffmann R., J. Amer. chem. Soc, 90, 1475 (1968).
219. В r e s 1 о w R., Eicher Т., KrebsA., Peterson
R. A., P о s n e r J., J. Amer. chem. Soc, 87, 1320 (1965);
Breslow R., Altman L. J., Krebs A., Mohac-
si E., MurataL, Peterson R. A., PosnerJ.,
ibid., 87, 1326 (1965); Breslow RM Ryan G, ibid., 89,
3073 (1967).
220. T u г г о N. J., L e e r m а к e r s P. A., Wilson H. R.,
NeckersD. C, ByersG. W., Vesley G.F., J.
Amer. chem. Soc, 87, 2613 (1965).
221. ZecherD. C, WestR., J. Amer. chem. Soc, 89, 153
(1967).
222. Freeman J. P., Graham W. H., J. Amer. chem. Soc,
89, 1761 (1967); С a r p i n о L. А., частное сообщение.
223. N e u r e i t e г N. P., Bordwell F. G.,J. Amer. chem.
Soc, 85, 1210 (1963); T о к u r a N.. N a g a i Т., Matsu-
тага S., J. Org. Chem., 31, 439 (1966); С a r p i n о L. A.-,
Mc Adams L. V., Ill, J. Amer. chem. Soc, 87, 5804
(1965); Neureiter N. P., ibid., 88, 558 (1966); P a -
quette L. A., Accounts chem. Res., 1, 209 (1968).
224. В о r d w e 1 1 F. G.f Williams J. M., Jr., H о у t E. В.,
204
Jr., J a r v i s В. В., J. Amer. chem. Soc, 90, 429 (1968).
225. С 1 a r к R. D., H e 1 m к a m p G. K-, J. Org. Chem., 29,
1316 (1964).
226. L о о n e у С. E.„ P h i 1 1 i p s W. D., R e i 1 1 у E. L., J.
Amer. chem. Soc, 79, 6136 (1957).
227. D e n n e у D. В., В о s к i n M. J., J. Amer. chem. Soc, 82,
4736 (1960).
228. L e m a 1 D. M., G о s s e 1 i n к E. P., M с G r e g о r S. D.,
J. Amer. chem. Soc, 88, 528 (1966).
229. S t a u d i n g e r H., Die Ketene, Enke, Stuttgart, 1912.
230. HuisgenR., F e i 1 e r L. А., О t t о P., Tetrahedron
Letters, 1968, 4485.
231. Brady W. T.,0'N e a 1 H. R., J. Org. Chem., 32, 612(1967).
232. BinschG, Feiler L. A.,Huisgen R., Tetrahedron
Letters, 1968, 4497; MartinJ.C, Goodlett V. W.,
BurpittR. D., J. Org. Chem., 30, 4309 (1965);
HuisgenR., F e i 1 e r L., BinschG., Angew. Chem., 76,
892 (1964); Angew. Chem. internat. Edit., 3, 753 (1964).
233. Montaigne R.( GhosezL, Angew. Chem., 80, 194
(1968); Angew. Chem. internat. Edit., 7, 221 (1968).
234. HuisgenR., О t t о P., Tetrahedron Letters, 1968, 4491.
235. G r i e s b a u m K-, N a e g e 1 e W., W a n 1 e s s G. G., J.
Amer. chem Soc, 87, 3151 (1965); Griesbaum K., Angew.
Chem., 78, 953 (1966); Angew. Chem. internat. Edit., 5, 933
(1966).
236. С r i e g e e R., Moschel A., Chem. Ber., 92, 2181 (1959).
237. Смирнов-Замков И. В., ДАН СССР, 83, 869 (1952);
Смирнов-Замков И. В., Кострамина Н. А.,
Укр. хим. ж., 21, 233 (1953).
238. Huisgen R., О t t о P., J. Amer. chem. Soc, 90, 5342 (1968)
239. Roberts J. D., S h a r t s С. М., Organic Reactions, 12,
1 (1962).
240. В a r t 1 e t t P. D., Montgomery L.K., S e i d e 1 В.,
J. Amer. chem. Soc, 86, 616 (1964); M о n t g о m e г у L. K.,
S с h u e 1 1 e г К., В a r t 1 e t t P. D., ibid., 86, 622 (1964);
Bartlett P. D., M о n t g о m e г у L. K-, ibid., 86,
628 (1964); Bartlett P. D., Science, 159, 833 (1968).
241. Oth J. F. M.f Angew. Chem., 80, 633(1968); Angew. Chem.
internat. Edit., 7, 646 (1968); Rec trav. chim., 87, 1185 (1968);
VolgerH.C, H о g e v e e n H., ibid., 86, 830 (1967).
242. Schafer W., Angew. Chem., 78, 716 (1966); Angew. Chem.
internat. Edit., 5, 669 (1966).
243. F u k u i K., Tetrahedron Letters, 1965, 2009, 2427; Bull. Chem.
Soc Japan, 39, 498 (1966); F u k u i K., Fujimoto H.,
Tetrahedron Letters, 1966, 251; Bull. Chem. Soc. Japan, 39,
2116 (1966); 40, 2018 (1967).
244. SalemL, J. Amer. chem. Soc, 90, 543, 553 (1968).
245. .Z i m m e r m a n n H. E., J. Amer. chem. Soc, 88, 1563, 1566
(1966).
246. D e w а г M. J. S., Tetrahedron, Suppl. 8, p. 75 (1966); Aroma-
ticity, Special Publication of The Chemical Society № 21,
London, 1967, p. 177.
247. van d e r Lugt W. Th. A. M., О о s t e r h о f f L. J.,
Chem. Commun., 1968, 1235.
205
248. Longuet-Higgins H. С, Abrahamson E. W.,
J. Amer. chem. Soc, 87, 2045 (1965).
249. L a i d 1 er K- J-, The Chemical Kinetics of Excited States,
Oxford University Press, Oxford, 1955.
250. Gr i f f i ng V., J. Chem. Phys., 25, 1015 (1955); J. Phys. Chem.,
61, 11 (1957).
251. H e r z f e 1 d K. F., Z. Naturforsch., 3a, 457 (1948); Rev. mod.
Physics, 41, 527 (1949).
252. В a d e r R. F. W., Canad. J. Chem., 40, 1164 (1962); Pear-
s о n R. G., J. Amer. chem. Soc, 91, 1252 (1969); Salem L.,
Chem. Physics Letters, 3, 99 (1969).
253. H a v i n g a E., Schlatmann J. L. M. A., Tetrahedron,
16, 151 (1961).
254. СыркинЯ. К, Изв. АН СССР, 1959, 238, 389, 401, 600.
255. В a 1 a b a n А. Т., Rev. Roum. Chim., 11, 1097 (1966); 12,
875 (1967).
256. Ma t h i e u J., Vails J., Bull. Soc. Chim. France, 1957,
1509.
257. V о 1 1 m e r J. J., S e r v i s K. L., J. Chem. Educat., 45,
214 (1968); Miller S. I., Advances in Physical Organic
Chemistry, 6, 185 (1968); G i 1 1 G. В., Quart. Rev. (Chem.
Soc, London), 22, 338 (1968); S e e b а с h D., Fortschr.
chem. Forsch., 11, 177 (1969); M i 1 1 i e P., Bull. Soc. Chim.
France, 1966, 4031; О р ч и н М., Д ж а ф ф е Г.,
Разрыхляющие орбитали, изд-во «Мир». М., 1958; Kosower E.M.,
An Introduction to Physical Organic Chemistry. Wiley, New
York, 1968; CervinkaO., Kfiz O., Chem. Listy, 61,
1036 (1967); 62, 321 (1968); H о u b i e r s J. P. M., Chem.
Weekbl., 62, 61 (1966); HorigC, Z. Chem., 7, 298 (1967);
D e w a r M. J. S., The Molecular Orbital Theory of Organic
Chemistry, McGraw-Hill, New York, 1969.
258. Fukui K. in Lowdin P.-O., Pullman В., Molecular
Orbitals in Chemistry, Physics and Biology. Academic Press,
New York, 1964, p. 513; Фукуи К., Современная
квантовая химия, изд-во «Мир», М., т. 1, стр. 59.
259. Н о f f m a n n R., Lipscomb W. N., J. Chem. Phys.,
36, 2179 (1962); 37, 2872 (1962); Hoffmann R., ibid., 39,
1397 (1963); 40, 2474, 2480, 2745 (1964); Tetrahedron,
22, 521, 539 (1966).
260. E a t о n D. R., J. Amer. chem. Soc, 90, 4272 (1968)
261. M с С a p r a F., Chem. Commun., 1968, 155.
262. UllmanE. F..J. Amer. chem. Soc, 90, 4158 (1968).
СОДЕРЖАНИЕ
Предисловие 5
1. Введение 7
2. Орбитали и химическая связь 8
3. Корреляционные диаграммы 16
3.1. Общие замечания к построению корреляционных
диаграмм 36
3.2. Предосторожности, необходимые при построении
корреляционных диаграмм 40
4. Сохранение орбитальной симметрии 46
5. Теория электроциклических реакций 47
5.1. Примеры электроциклических реакций .... 58
6. Теория циклоприсоединения и циклораспада .... 75
6.1. Примеры циклоприсоединения и циклораспада . . 84
6.2. [2+2] - Циклоприсоединение в фотохимии цикло-
гексадиенонов и циклогексенонов 100
6.3. [2+2+2] - Циклоприсоединение 112
6.4. Призман 120
6.5. [2+2+2+2] - Циклоприсоединение 125
7. Теория сигматропных реакций 126
7.1. Примеры сигматропных реакций 133
7.2. Последовательные сигматропные смещения . . . 144
8. Передача и элиминирование групп 153
9. Вторичные эффекты 157
10. Дивертисмент 164
10.1. Хелетропные реакции 164
10.2. Реакции циклоприсоединения кетенов .... 177
11. Обобщенные правила отбора для перициклических реакций182
12. Исключения 186
13. Другие теоретические работы 189
14. Заключение 191
Литература 192
Р. Вудворд, Р. Хоффман
СОХРАНЕНИЕ ОРБИТАЛЬНОЙ СИММЕТРИИ
Редактор Т. Румянцева. Художник В. А. Медников. Художественный редактор
Н. Блинов. Технический редактор Г. Алюлина. Корректор В. Постнова
Сдано в набор 21/XII 1970 г. Подписано к печати 30/IV 1971 г. Бумага № 1
84x108732=3,25 бум. л. 10,92 усл. печ. л. Уч.-изд. л. 9,64. Изд. №3/5877.
Цена 97 коп. Зак. 1556
ИЗДАТЕЛЬСТВО «МИР»
Москва, 1-й Рижский пер., 2
Ярославский полиграфкомбинат Главполиграфпрома Комитета по печати при
Совете Министров СССР. Ярославль, ул. Свободы, 97.
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании книги, ее
оформлении, качестве перевода просим
присылать по адресу: Москва, И-278,
1-й Рижский пер., д. 2, Издательство «Мир».
97 коп.
чЯШ