/


































































































































































































































































Author: Айвазов Б.В.
Tags: другие физико-химические методы анализа (кроме оптических) химия химический анализ хроматография
Year: 1968




Text
il
Больше химической литературы на
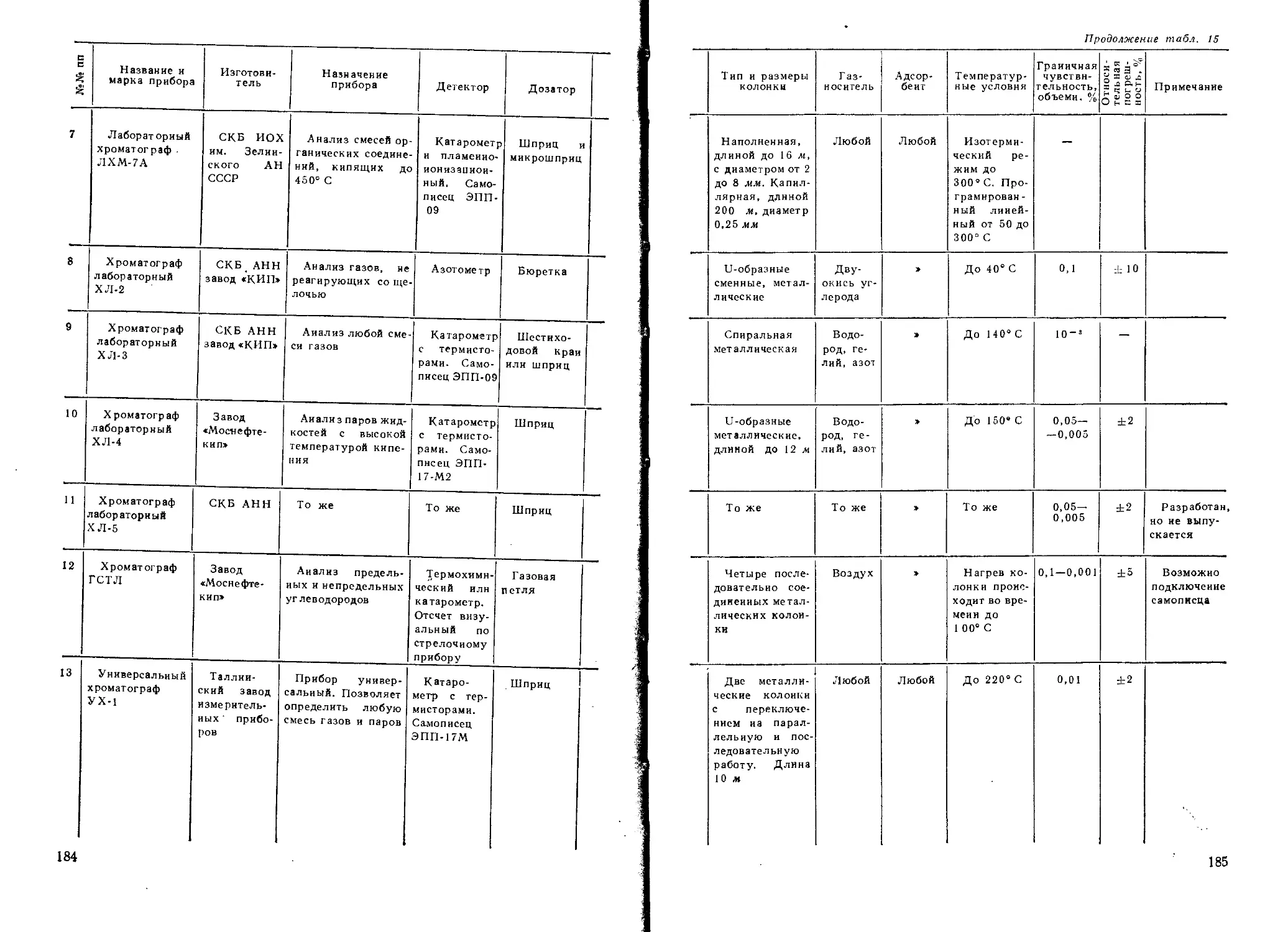
vk.com/chemzone
More chemistry books you can find on
vk.com/chemzone
vk.com/chemzone
Б. В. АЙВАЗОВ
ПРАКТИЧЕСКОЕ РУКОВОДСТВО
ПО ХРОМАТОГРАФИИ
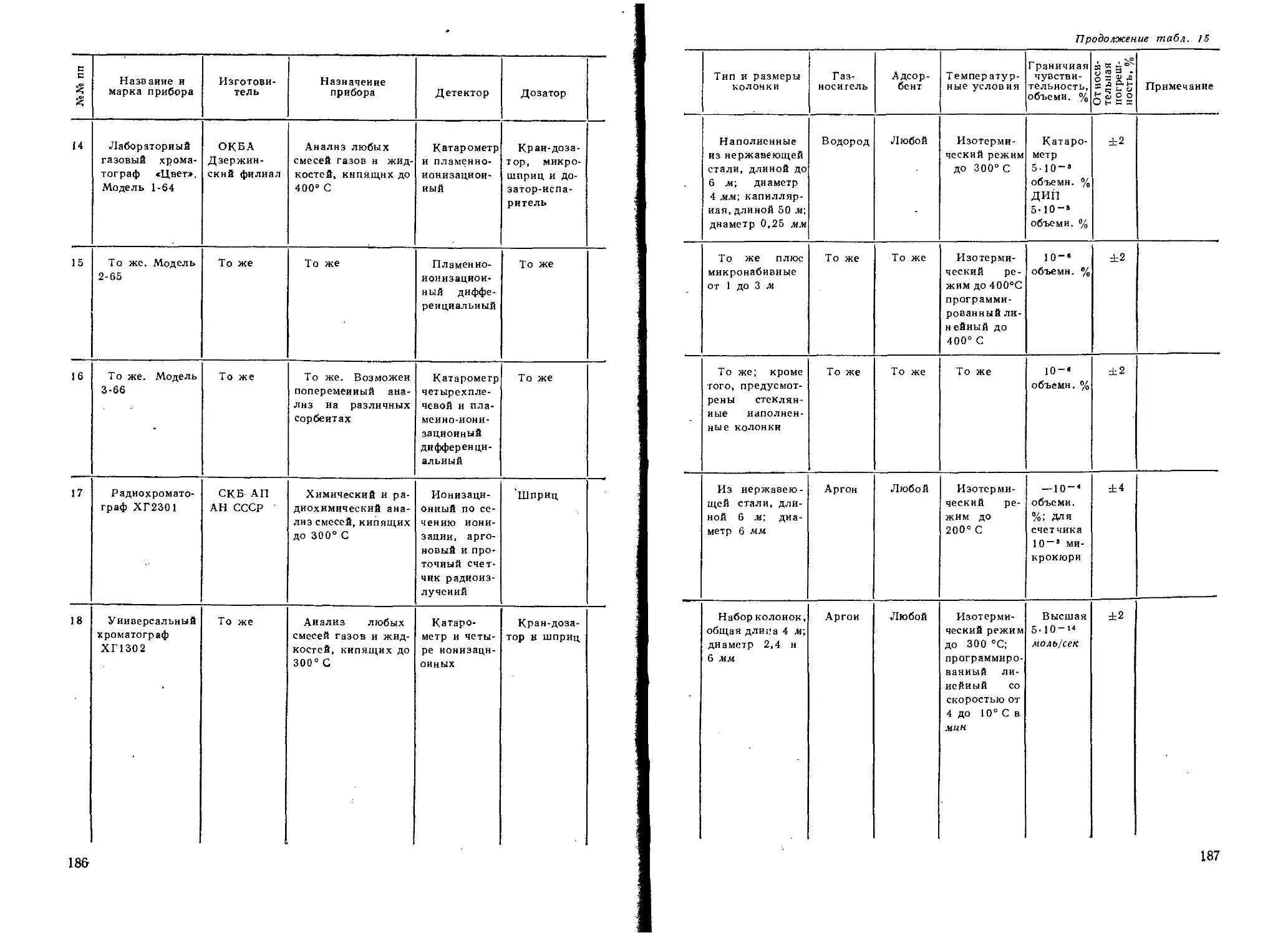
ДОПУЩЕНО МИНИСТЕРСТВОМ ВЫСШЕГО И СРЕДНЕГО СПЕЦИАЛЬНОГО
ОБРАЗОВАНИЯ СССР В КАЧЕСТВЕ УЧЕБНОГО ПОСОБИЯ ДЛЯ СТУДЕНТОВ
ХИМИЧЕСКИХ И ХИМИКО-ТЕХНОЛОГИЧЕСКИХ СПЕЦИАЛЬНОСТЕЙ ВУЗОВ
ИЗДАТЕЛЬСТВО «ВЫСШАЯ ШКОЛА» МОСКВА 196В
УДК 543.544 (022)
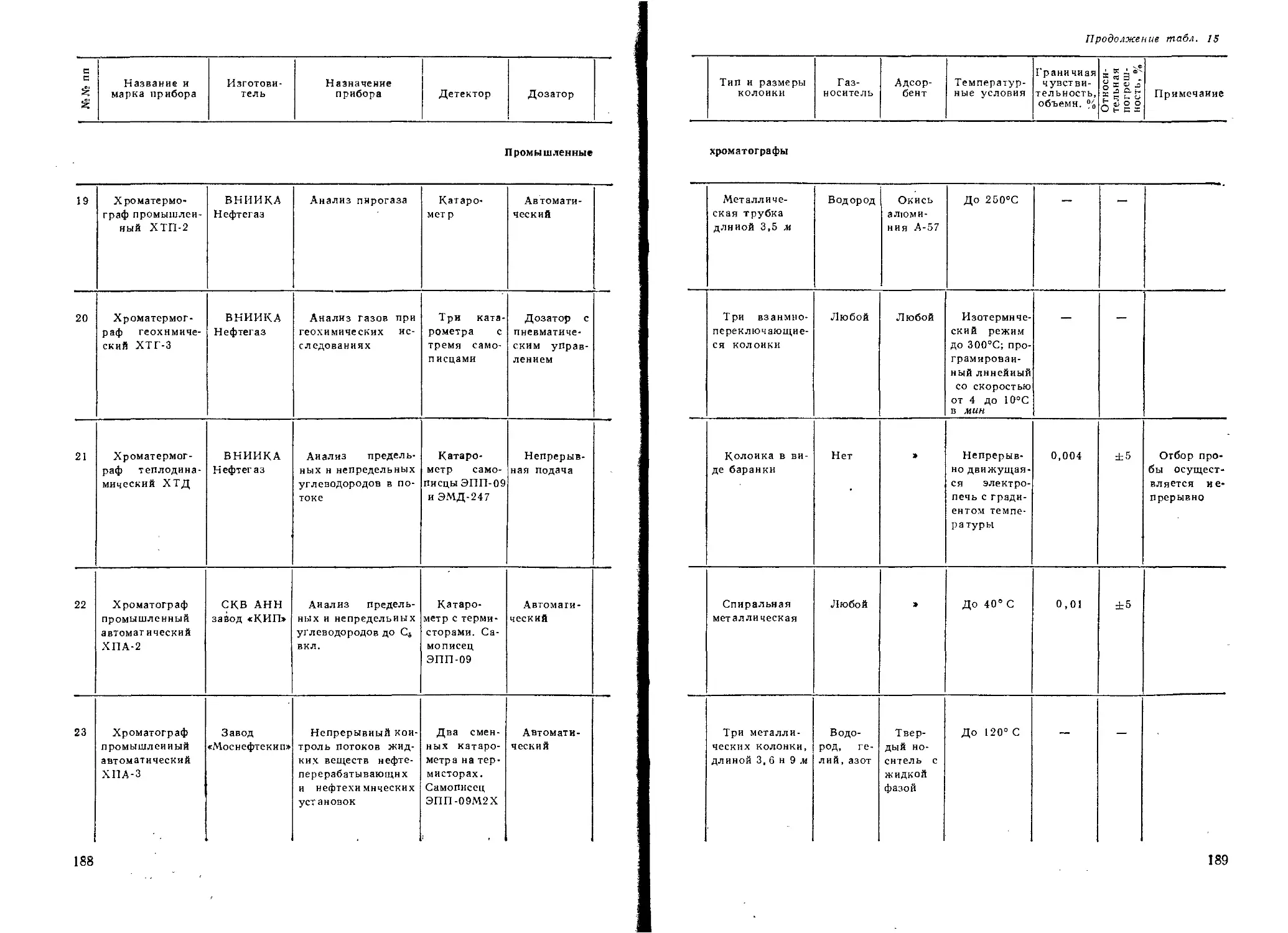
Рецензенты: проф. Жуховицкий А. А. (кафедра
физической химии Московского института стали и
сплавов) и кафедра неорганической и аналитической
химии Московского технологического института мясной
и молочной промышленности (зав. кафедрой проф.
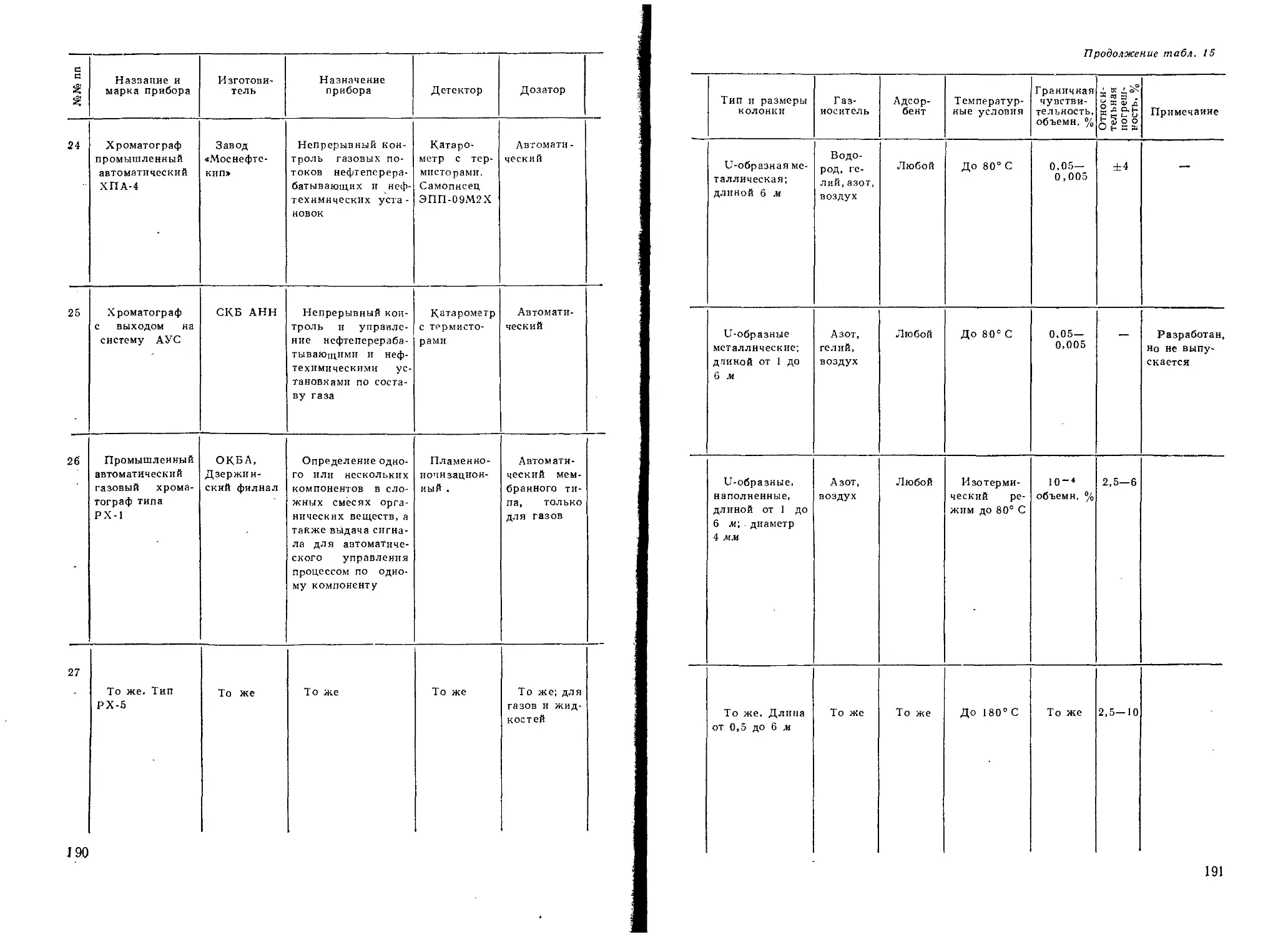
Ольшанова К- М.)
Практическое руководство по хроматографии.
Айвазов Б. В.
Учебное пособие охватывает все современные раз-
делы хроматографии: адсорбционную, ионообменную,
осадочную, распределительную, бумажную, газо-жид-
костную, капиллярную, в том числе и такие, сравнитель-
но недавно разработанные методы, как вакантная и
ступенчатая хроматография.
Пособие состоит из восьми глав. Каждая глава
содержит краткое изложение теоретических основ дан-
ного раздела хроматографии и подробное описание не-
скольких лабораторных работ. В последней главе опи-
сываются некоторые приемы приготовления адсорбентов,
жидких фаз и других материалов, а также описывается
изготовление простейших узлов хроматографических
колонок.
В конце каждой главы приводится обширная биб-
лиография.
Таблиц 19, иллюстраций 48.
681681^
г'китэськийЧ’.
7и iRLPCMTET;.
2-5 — 5
51—67
П РЕДИСЛОВИЕ
Хроматографический метод анализа находит самое широкое прщ
менение. Он прочно вошел не только в практику научных исследо-
ваний по химии, атомной технике, биологии и медицине, но и в
заводской контроль нефтеперерабатывающей, нефтехимической, хи-
мической и газовой'промышленности. Хроматографический метод
начинают применять для автоматизации технологических процессов,
все шире хроматография становится методом изучения различных
физико-химических констант вещества. Разрабатываются и выпус-
каются промышленностью различные типы хроматографических при-
боров.
Естественно, что в настоящее время потребность в квалифициро-
ванных специалистах, хорошо владеющих хроматографическими
методами, резко возросла. Поэтому в ряде вузов страны ведется
подготовка специалистов в области хроматографии. Хроматография
и хроматографический анализ включены Министерством высшего и
среднего специального образования СССР в список специальных
курсов по физической и аналитической химии химических факуль-
тетов государственных университетов. Тем не менее учебники по
хроматографии в целом или по отдельным разделам курса до сих пор
отсутствуют. Резко также ощущается недостаток в руководствах по
хроматографическим методам анализа. Вышедшие в свет за послед-
ние годы немногочисленные руководства по отдельным разделам
хроматографического метода анализа не восполняют, к сожалению,
создавшийся в этой области пробел. Особенно это касается газовой
хроматографии, по которой, несмотря на обилие различного рода
сборников, отсутствуют руководства.
В связи с изложенным, нам представлялось целесообразным под-
готовить руководство, более или менее полно охватывающее совре-
менные разделы хроматографического метода.
Предлагаемое руководство составлено на основе опыта, накоп-
ленного руководимой автором кафедрой физической и аналитической
химии Башкирского государственного университета, которая в
течение ряда лет готовит специалистов в области хроматографичес-
кого анализа.
Настоящее пособие, предназначенное для студентов старших
курсов химических факультетов университетов и химико-техноло-
1* 3
гических институтов, составлено так, чтобы облегчить усвоение тео-
ретического материала. Поэтому по каждому методу хроматографи-
ческого анализа описанию лабораторных работ предшествует крат-
кое изложение теоретических основ, а список лабораторных работ
составлен таким образом, чтобы наиболее полно охватить рассмат-
риваемые теорией или имеющие практическое значение вопросы.
В конце каждой главы приводится список цитированной и рекомен-
дуемой литературы.
В зависимости от объема преподаваемого курса для выполнения
можно предложить не все приведенные в руководстве работы, а
какую-то часть из них. Выбор должен быть сделан, исходя из воз-
можностей той или иной лаборатории, а также в зависимости от
профиля будущих специалистов.
Автор надеется, что данное руководство принесет также извест-
ную пользу работникам заводских и научно-исследовательских ла-
бораторий, соприкасающихся с хроматографическими методами
анализа.
Автор пользуется случаем, чтобы выразить свою искреннюю при-
знательность профессору, доктору химических наук Александру
Абрамовичу Жуховицкому и профессору, доктору химических наук
Калерии Максимовне Ольшановой, взявшим на себя труд по рецен-
зированию рукописи, за ряд очень ценных советов и указаний,
позволивших значительно улучшить содержание руководства.
Автор также благодарит сотрудников кафедры физической и
аналитической химии БГУ Журенко И. Ф., Кудашеву Ф. X. и
Лапкина Л. М. за сделанные ими ценные замечания по рукописи и
за постановку лабораторных работ.
Все замечания читателей будут приняты с благодарностью.
Автор
ВВЕДЕНИЕ
Хроматографический метод разделения и анализа сложных сме-
сей был открыт русским ботаником М. С. Цветом [1] в 1903 г. Харак-
теризуя принцип своего метода, он писал: «При фильтрации смешан-
ного раствора через столб адсорбента пигменты ... расслаиваются
в виде отдельных, различно окрашенных зон. Подобно световым лу-
чам в спектре, различные компоненты сложного пигмента закономер-
но распределяются друг за другом в столбе адсорбента и становятся
доступными качественному определению. Такой расцвеченный пре-
парат я назвал хроматограммой, а соответствующий метод анали-
за — хроматографическим методом».
В этой формулировке дано четкое определение принципа и наз-
начения хроматографического метода. Однако метод, предложенный
М. С. Цветом, не был по достоинству оценен его современниками.
Лишь в 1931 г., пользуясь методом М. С. Цвета, Р. Куну, А. Вин-
терштейну и Е. Ледереру [2] удалось выделить в кристаллическом
виде а- и 0-каротин из сырого каротина и тем самым продемонстри-
ровать препаративную ценность метода. К этому времени возникла
острая потребность в хорошем методе разделения сложных смесей,
особенно веществ, разлагающихся при нагревании. Хроматографи-
ческий метод был признан и начал развиваться.
Еще большее развитие метод получил после того, как в 1941 г.
в основу разделения смеси веществ А. Дж. П. Мартином и Р. Л. М.
Синджем [3] было положено различие не в адсорбционном сродстве
компонентов разделяемой смеси, а в их коэффициентах распределе-
ния между двумя несмешивающимися жидкостями. Данный метод
был назван распределительной хроматографией, в отличие от ад-
сорбционной, предложенной М. С. Цветом. Наибольших успехов
распределительная хроматография достигла после того, как в ка-
честве носителя неподвижной фазы стали применять полоски бу-
маги — распределительная хроматография на бумаге.
В 1947 г. Т. Б. Гапон, Е. Н. Гапон и Ф. М. Шемякин [4] впервые
осуществили хроматографическое разделение смеси ионов в раст-
воре, причем это разделение было объяснено ими обменом ионов
сорбентов на ионы из раствора. Так возникло одно из новых ответв-
лений метода М. С. Цвета — ионообменная хроматография, полу-
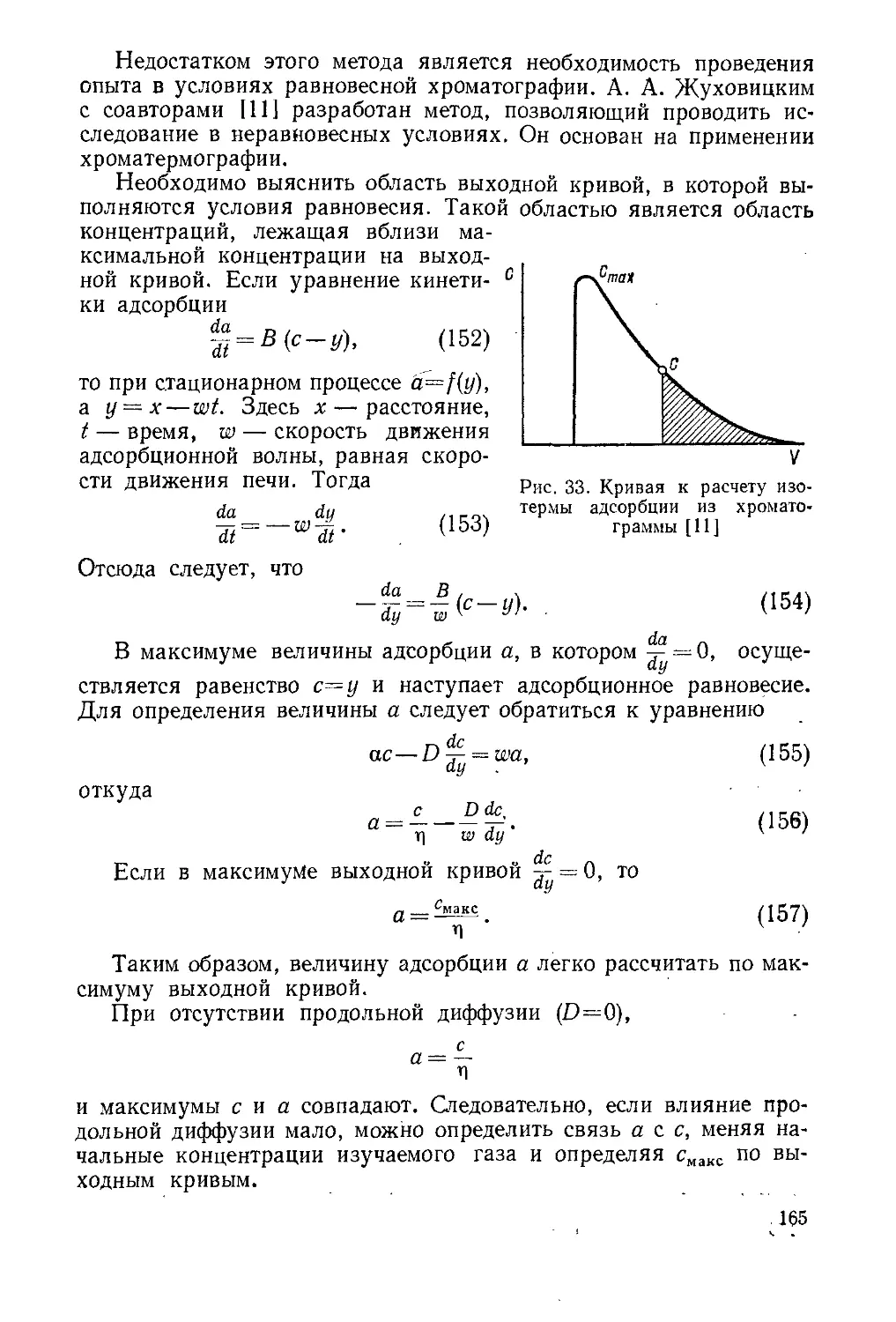
чившая в настоящее время весьма широкое распространение.
5
В 1948 г. Е. Н. Гапон и Т. Б. Гапон [5] предложили осадочную
хроматографию.
Своего расцвета хроматография достигла после того, как А. Дж.
П. Мартин и А. Т. Джеймс [6] в 1952 г. предложили новый метод
хроматографии — газо-жидкостную распределительную хромато-
графию. Метод основан на различии коэффициентов распределения
веществ разделяемой смеси между неподвижной жидкой фазой и
подвижной газообразной или парообразной.
Значительному развитию хроматографии способствовало созда-
ние теории газовой, ионообменной и осадочной хроматографии, а
также разработка в последнее время новых вариантов (хроматермо-
графия, вакантная, ступенчатая, капиллярная, тонкослойная хро-
матография и т. п.).
Разнообразие современных хроматографических методов может
привести на первый взгляд к неправильному представлению о том,
что объединение столь различных методов одним термином «хрома-
тография» является искусственным, неправильным. На самом деле
это различие только кажущееся. Все современные хроматографи-
ческие методы обладают рядом общих, причем весьма существенных
черт. Так, любое хроматографическое разделение включает переме-
щение анализируемой пробы через слой неподвижного вещества
(твердый адсорбент, жидкая неподвижная фаза, нанесенная на твер-
дый порошкообразный носитель или бумагу). Перемещение компо-
нентов смеси осуществляется газом или жидкостью — подвижной
фазой. Вследствие селективного замедления, осуществляемого не-
подвижной фазой, компоненты анализируемой смеси перемещаются
с различными эффективными скоростями. Это обстоятельство при-
водит к образованию отдельных зон или полос, каждая из которых
содержит один компонент разделенной смеси. Задача исследователя
состоит в обнаружении теми или иными способами этих зон и опре-
делении их качественного и количественного состава.
Во всех случаях хроматографирования компоненты анализируе-
мой смеси распределяются между подвижной и неподвижной фаза-
ми. Следовательно, в любом из вариантов хроматографического ме-
тода обязательно наличие двухфазной системы.
Однако последнее требование осуществляется почти во всех
физических методах разделения смесей, например, в экстракции,
ректификации, не относящихся к хроматографическим методам.
Поэтому для определения хроматографического метода необходимо
положить в основу иной принцип. Руководствуясь определением,
данным еще М. С. Цветом, а также изложенными выше особенностя-
ми, характерными для хроматографии, можно дать следующее опре-
деление хроматографического метода:
Хроматографическим методом называется физико-химический
метод разделения смесей, при котором компоненты разделяемой сме-
си распределены между двумя фазами, одной из которых является
неподвижный слой с большой поверхностью контакта, а другая фаза
6
представляет собой поток, фильтрующийся через неподвижный
слой [7].
В процессах дистилляции, экстракции, абсорбции имеет место
противоточное движение обеих фаз, тогда как при фильтрации
движется только одна фаза, а другая остается неподвижной. По-
этому именно слово «фильтрующийся» определяет отличительную
черту хроматографического метода от других физических методов
разделения, основанных на применении двухфазных систем.
Следует отметить, что именно поэтому такие процессы, как рек-
тификация, экстракция и им подобные могут быть непрерывными,
тогда как хроматографический метод является прерывным, перио-
дическим. При этом состав смеси, покидающей хроматографическую
колонку, непрерывно меняется. В процессе ректификации или эк-
стракции можно отбирать в течение всего непрерывного процесса
одну и ту же фракцию или одно и то же вещество, в хроматографи-
ческом же процессе этого делать нельзя. Некоторые варианты хро-
матографического метода, например, теплодинамический метод, ва-
кантохроматография и другие, хотя и требуют непрерывной подачи
анализируемой смеси, однако состав анализируемой смеси и в этом
случае узнается периодически.
Характерной особенностью хроматографического метода явля-
ется многократность повторения процесса сорбции и десорбции. Это
обусловливает высокую эффективность хроматографии как метода
разделения сложных смесей веществ с весьма близкими свойст-
вами.
Многообразие видоизменений и вариантов хроматографического
метода вызывает необходимость их систематизации или классифи-
кации. В настоящее время общепринятыми являются классифика-
ции: по агрегатному состоянию фаз и по методике проведения экспе-
римента.
Основываясь на первом принципе, методы хроматографии можно
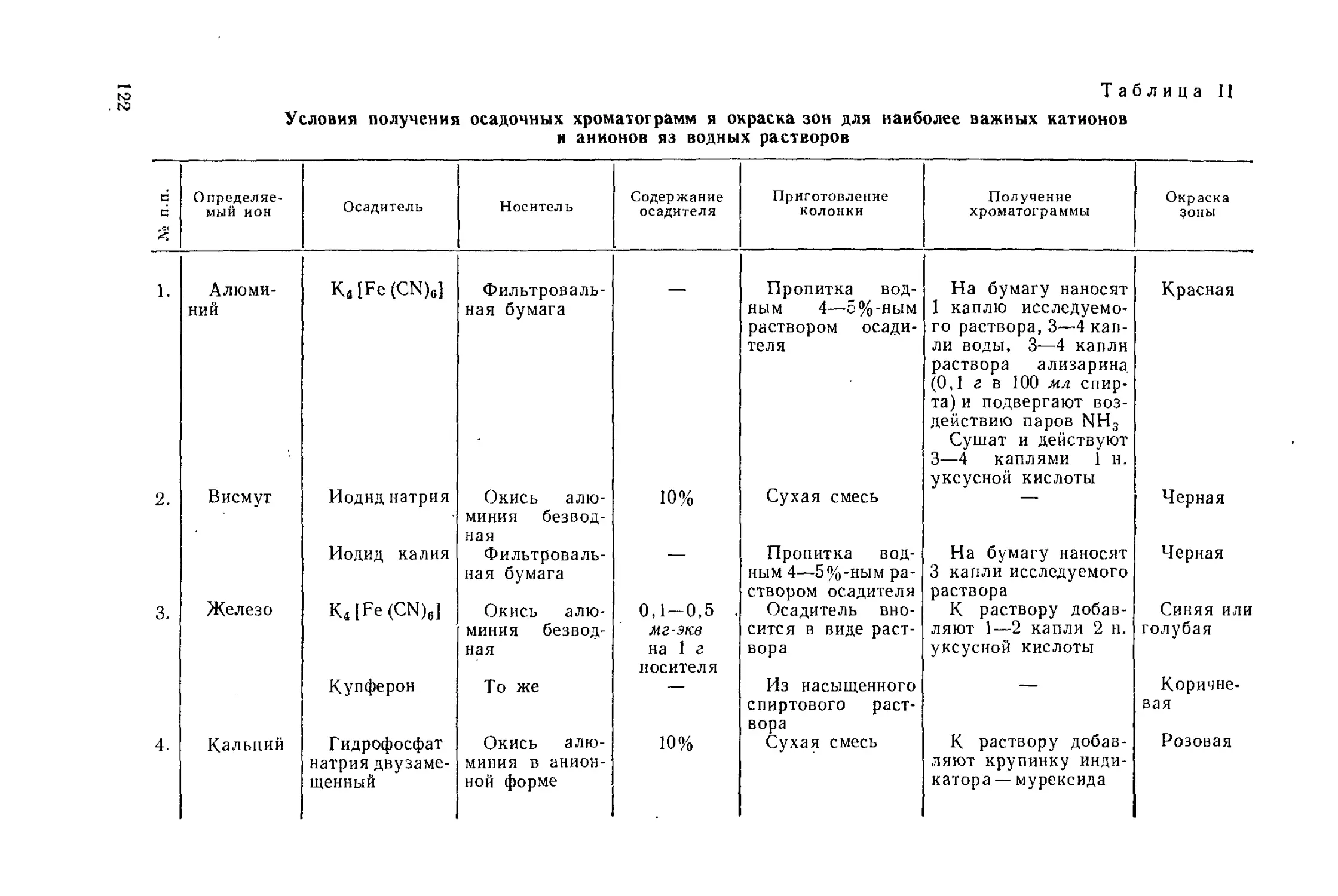
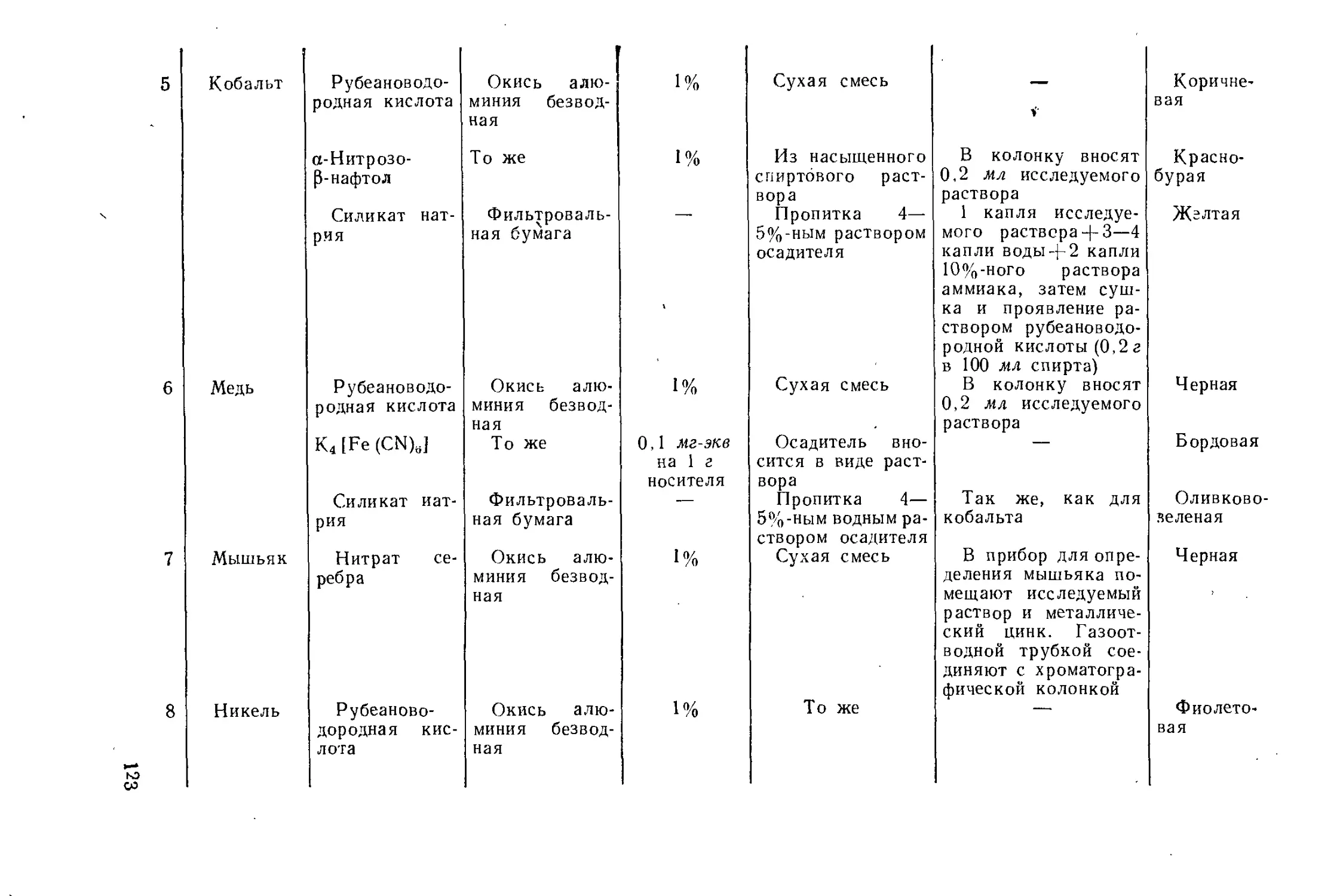
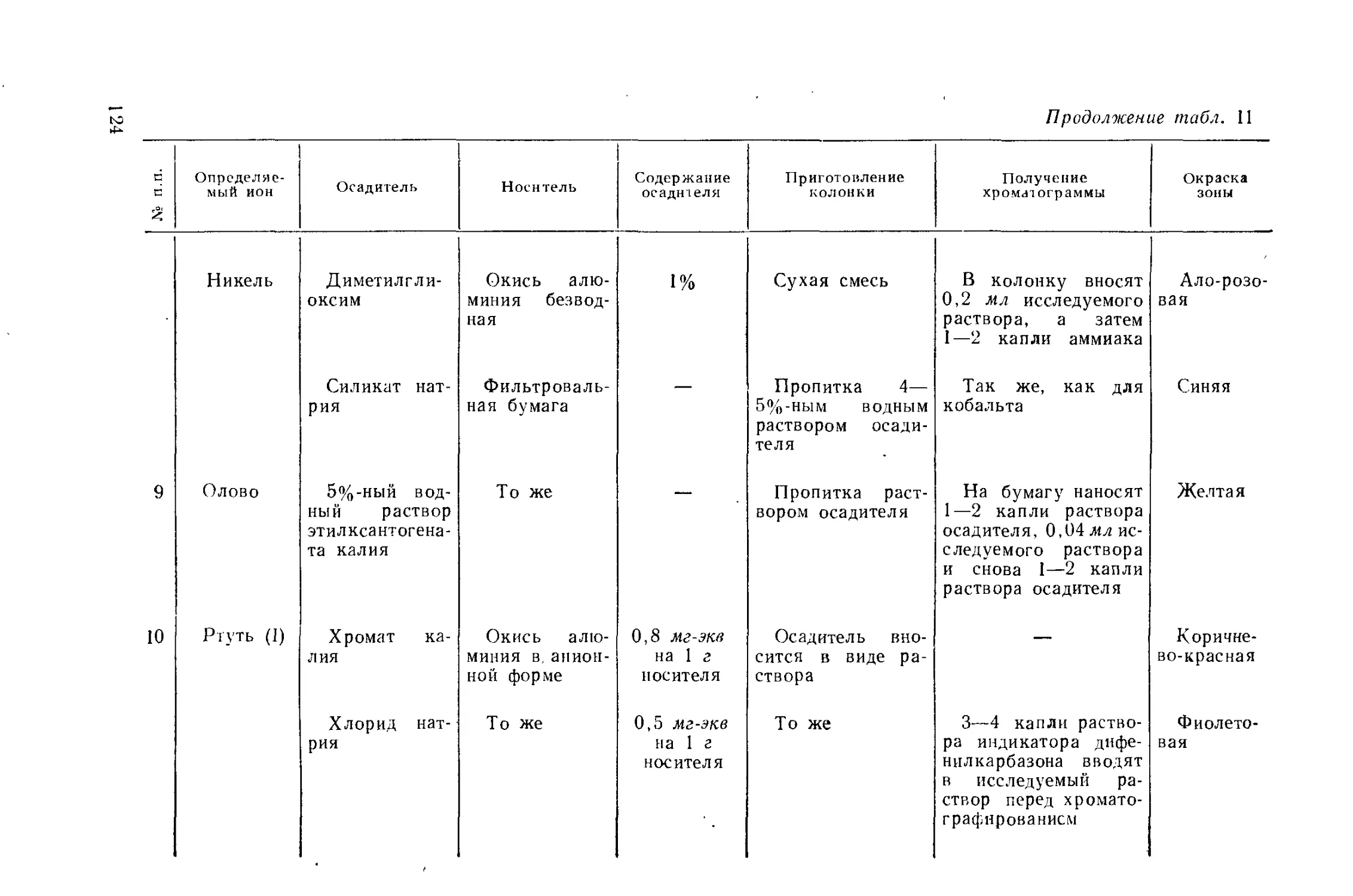
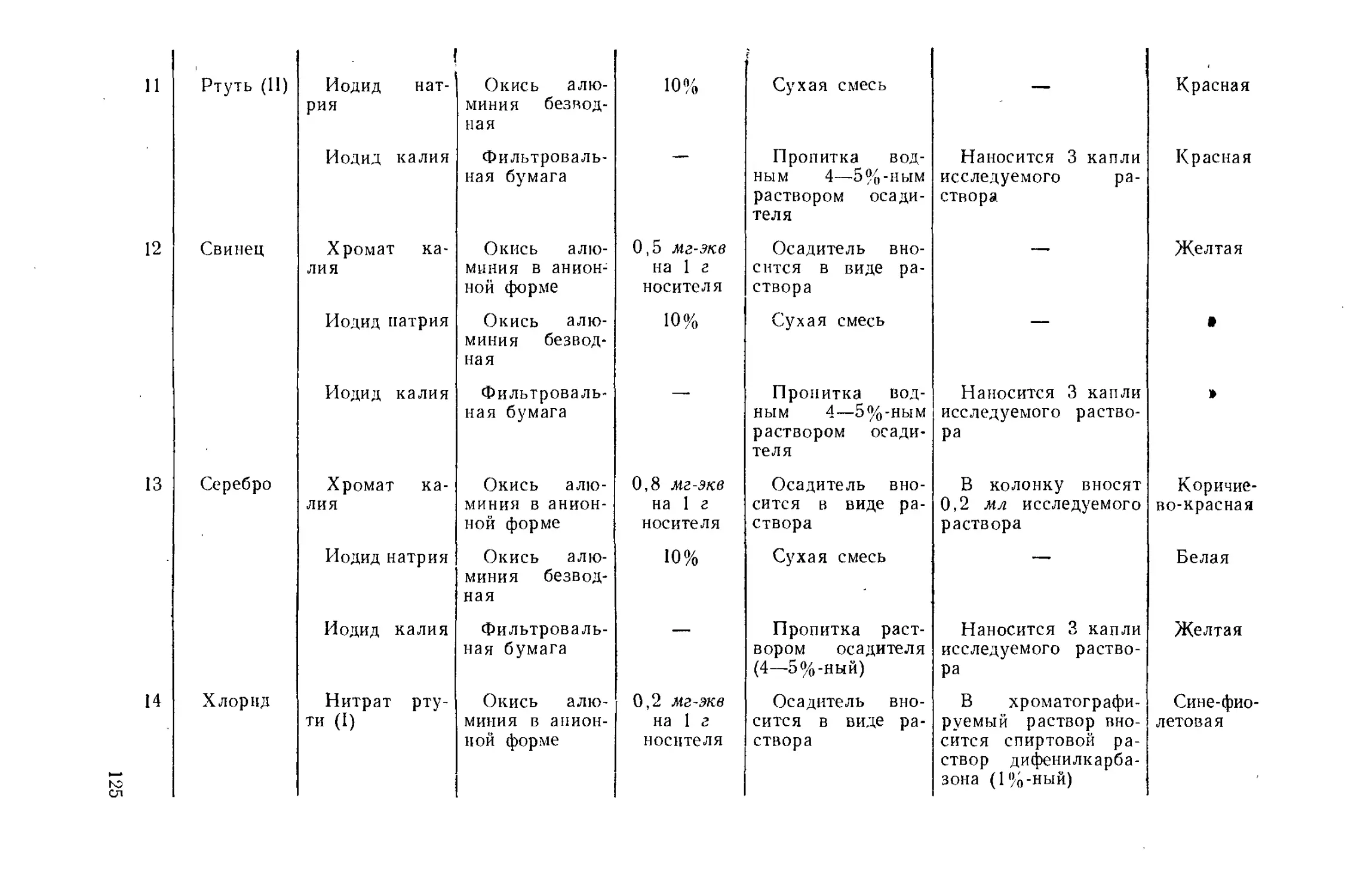
разделить на четыре группы (табл. 1).
Согласно второму принципу мы различаем три вида хромато-
графии: 1) проявительную, или элюентную; 2) фронтальную; 3) вы-
теснительную.
Рассмотрим на простейших примерах применение каждого вида
хроматографии.
Проявительная хроматография. Заполненную сорбентом колон-
ку промывают чистым растворителем Е, жидким или газообразным,
после чего в верхнюю часть колонки вводят порцию анализируемого
раствора веществ А и В в Е. Затем колонку непрерывно промывают
растворителем Е (проявителем). При этом компоненты раствора
А и В перемещаются вдоль слоя сорбента с различными скоростя-
ми, что обусловливает их разделение на зоны. При достаточной
Длине колонки произойдет полное разделение зон, причем менее
сорбирующийся компонент А займет нижнее положение в колонке.
Зона, содержащая более сильно сорбирующийся компонент В, бу-
7
Таблица 1
Классификация хроматографических методов по агрегатному состоянию фаз
Неподвижная фаза Подвижная фаза Наименование метода Возможные варианты
Твердая Жидкая Адсорбционная хро- матография жидко- стей и растворов, ионообменная хрома- тография; осадочная хроматография Окислительно-восстанови- тельная хроматография; ад- сорбционно-комплексообра- зовательная; тонкослойная
Твердая Газооб- разная Газовая адсорбци- онная хроматография Хроматермография, теп- лодинамический метод
Жидкая Жидкая Жидкостная распре- делительная хрома- тография Колоночная; бумажная: одномерная, двумерная, круговая; метод обращен- ных фаз; электрофоретичес- кая; тонкослойная Хроматография газов, жидкостей, вакантная, сту- пенчатая, капиллярная
Жидкая Газооб- разная Газо-жидкостная р асп редел ительна я хроматография
дет расположена в верхней части. Между зонами сорбент будет за-
полнен чистым растворителем (рис. 1,о).
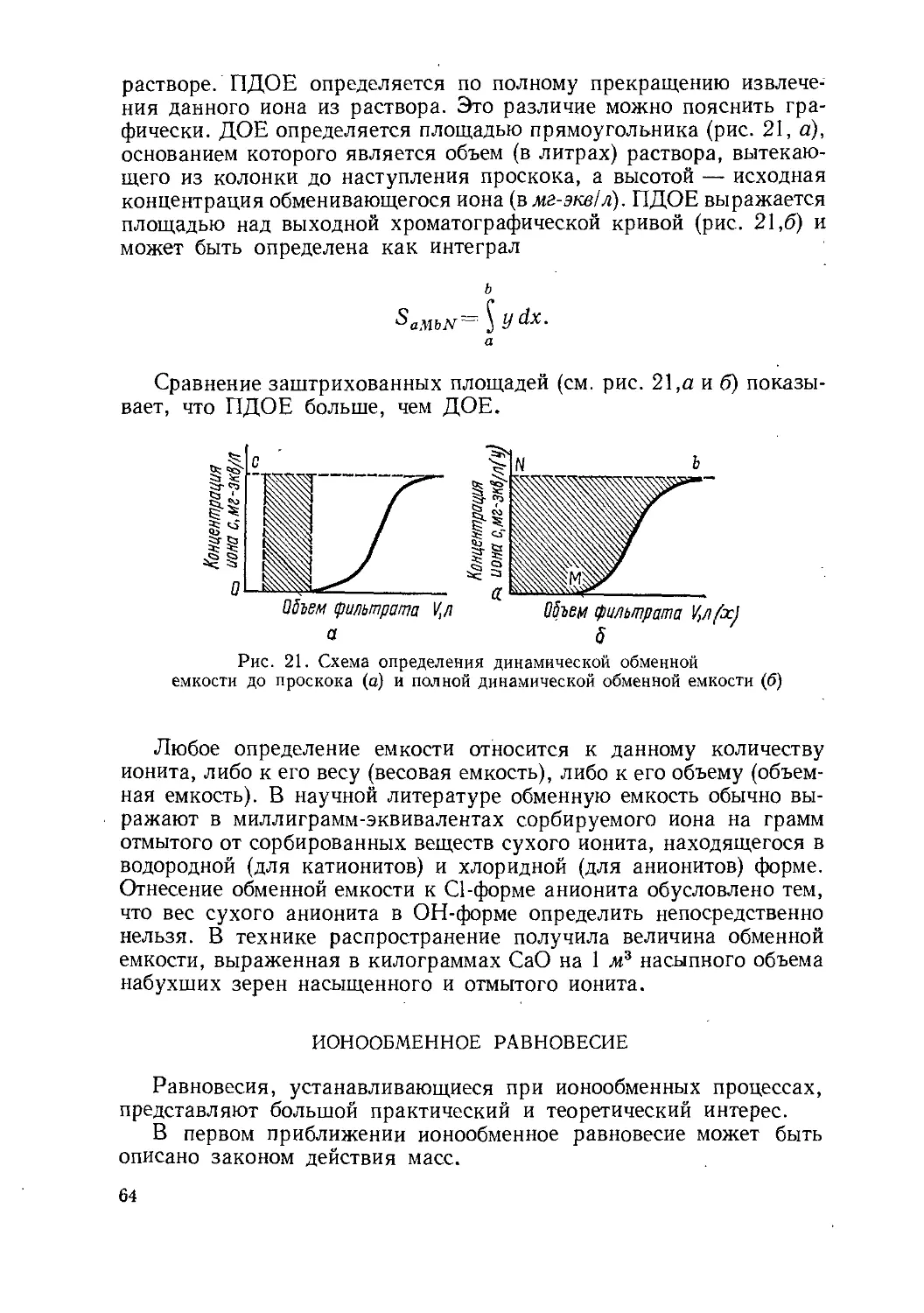
Изменение концентрации вымываемых веществ в вытекающем
растворе изображается кривыми, которые называются хроматограм-
мами, или выходными хроматографическими кривыми (рис. 1,6).
Кажущееся различие порядка выхода компонентов в этих двух ва-
риантах записи зависит только от принятого начала записи и уст-
ройства записывающего приспособления потенциометра.
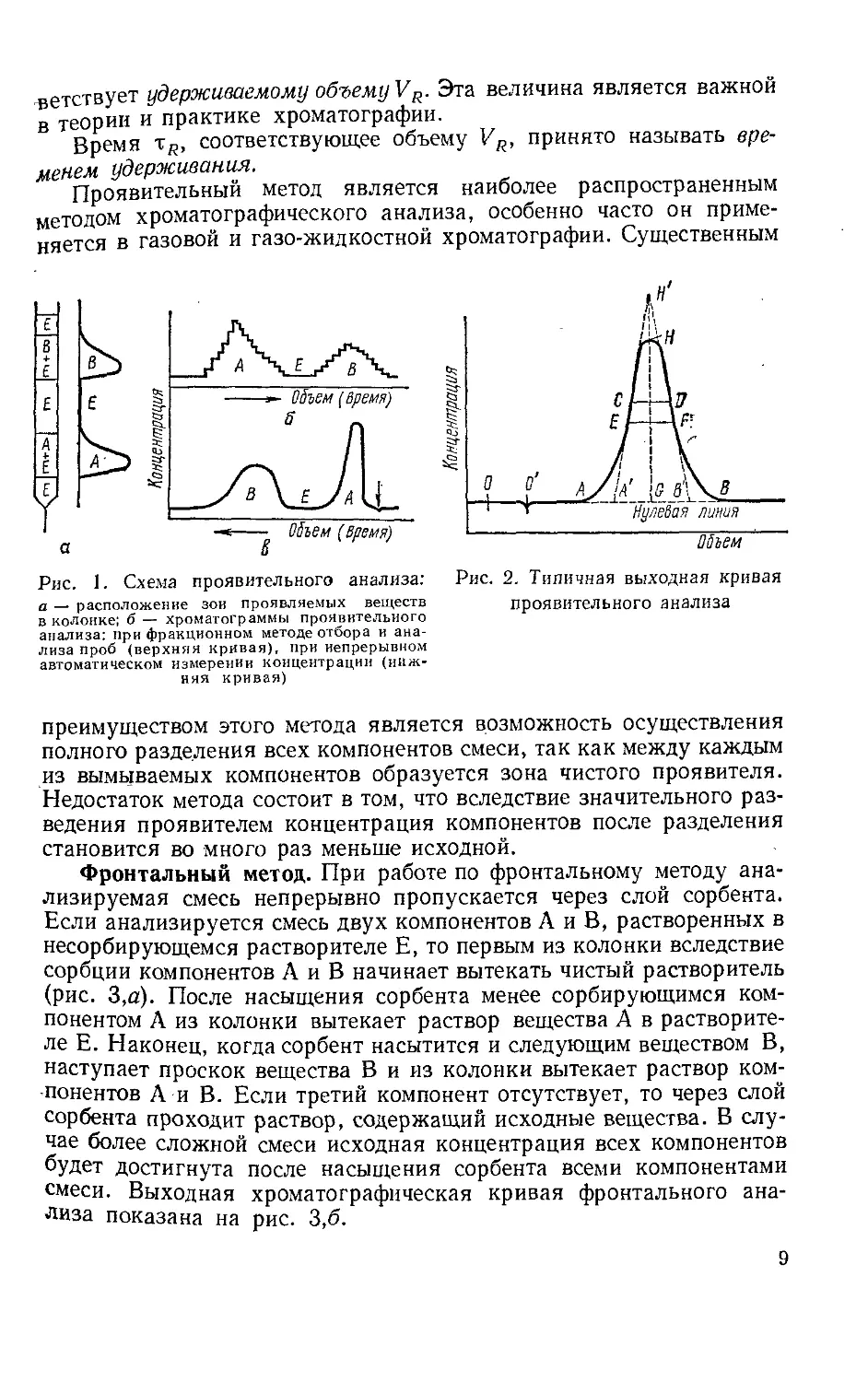
Целесообразно рассмотреть более подробно выходную хромато-
графическую кривую проявительного анализа. На рис. 2 изображе-
на типичная выходная кривая проявительного анализа одного ком-
понента.
По оси абсцисс откладывается значение объема вытекающего
из колонки раствора или выходящего из нее газа, а по оси ор-
динат — концентрация вымываемого вещества. Точка О соответст-
вует вводу пробы анализируемой смеси, а точка О' — появлению
на выходе несорбирующегося в колонке вещества, например, на-
ходившегося в колонке до опыта растворителя или воздуха; таким
образом, отрезок 00'соответствует незаполненному сорбентом объ-
ему колонки. Кривая АНВ носит название хроматографического
пика данного вещества, а расстояние от нулевой линии АВ до
максимума пика Н, т. е. GH — высоты пика. Отрезок АВ называет-
ся шириной пика у основания, CD — шириной в точке перегиба, а
EF — шириной на расстоянии половины высоты. Отрезок O'G соот-
8
веТствует удерживаемому объему VR. Эта величина является важной
в теории и практике хроматографии.
Время TR, соответствующее объему VR, принято называть вре-
менем удерживания.
Проявительный метод является наиболее распространенным
методом хроматографического анализа, особенно часто он приме-
няется в газовой и газо-жидкостной хроматографии. Существенным
Рис. 1. Схема проявительного анализа: Рис. 2. Типичная выходная кривая
а — расположение зои проявляемых веществ
в колонке; б — хроматограммы проявительного
анализа: при фракционном методе отбора и ана-
лиза проб (верхняя кривая), при непрерывном
автоматическом измерении концентрации (ниж-
няя кривая)
проявительного анализа
преимуществом этого метода является возможность осуществления
полного разделения всех компонентов смеси, так как между каждым
из вымываемых компонентов образуется зона чистого проявителя.
Недостаток метода состоит в том, что вследствие значительного раз-
ведения проявителем концентрация компонентов после разделения
становится во много раз меньше исходной.
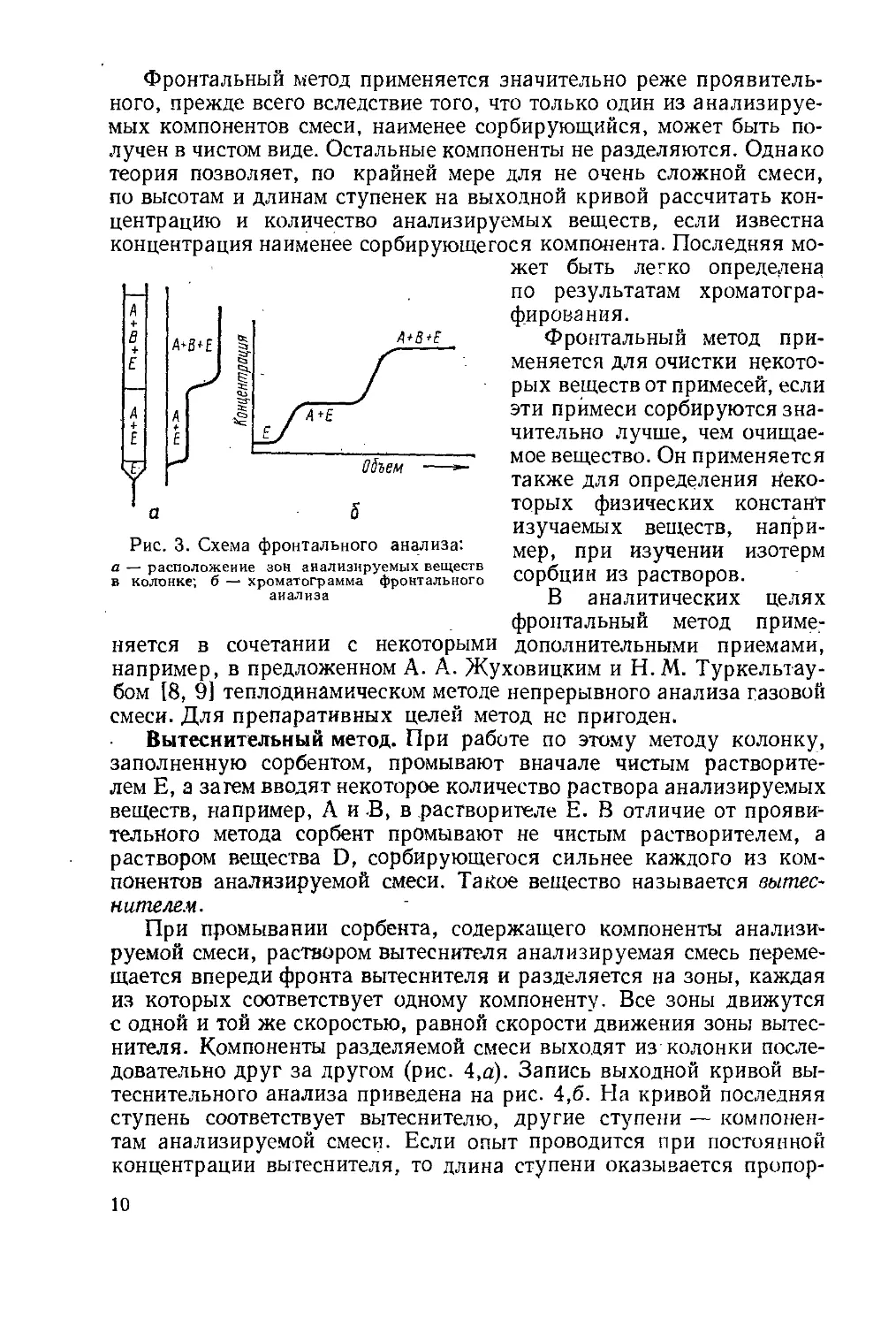
Фронтальный метод. При работе по фронтальному методу ана-
лизируемая смесь непрерывно пропускается через слой сорбента.
Если анализируется смесь двух компонентов А и В, растворенных в
несорбирующемся растворителе Е, то первым из колонки вследствие
сорбции компонентов А и В начинает вытекать чистый растворитель
(рис. 3,а). После насыщения сорбента менее сорбирующимся ком-
понентом А из колонки вытекает раствор вещества А в растворите-
ле Е. Наконец, когда сорбент насытится и следующим веществом В,
наступает проскок вещества В и из колонки вытекает раствор ком-
понентов А и В. Если третий компонент отсутствует, то через слой
сорбента проходит раствор, содержащий исходные вещества. В слу-
чае более сложной смеси исходная концентрация всех компонентов
будет достигнута после насыщения сорбента всеми компонентами
смеси. Выходная хроматографическая кривая фронтального ана-
лиза показана на рис. 3,6.
9
Рис. 3. Схема фронтального анализа:
а — расположение зон анализируемых веществ
в колонке; б — хроматограмма фронтального
анализа
Фронтальный метод применяется значительно реже проявитель-
ного, прежде всего вследствие того, что только один из анализируе-
мых компонентов смеси, наименее сорбирующийся, может быть по-
лучен в чистом виде. Остальные компоненты не разделяются. Однако
теория позволяет, по крайней мере для не очень сложной смеси,
по высотам и длинам ступенек на выходной кривой рассчитать кон-
центрацию и количество анализируемых веществ, если известна
концентрация наименее сорбирующегося компонента. Последняя мо-
жет быть легко определена
по результатам хроматогра-
фирования.
Фронтальный метод при-
меняется для очистки некото-
рых веществ от примесей, если
эти примеси сорбируются зна-
чительно лучше, чем очищае-
мое вещество. Он применяется
также для определения Неко-
торых физических констант
изучаемых веществ, напри-
мер, при изучении изотерм
сорбции из растворов.
В аналитических целях
фронтальный метод приме-
няется в сочетании с некоторыми дополнительными приемами,
например, в предложенном А. А. Жуховицким и Н.М. Туркелыау-
бом [8, 9] теплодинамическом методе непрерывного анализа газовой
смеси. Для препаративных целей метод нс пригоден.
Вытеснительный метод. При работе по этому методу колонку,
заполненную сорбентом, промывают вначале чистым растворите-
лем Е, а затем вводят некоторое количество раствора анализируемых
веществ, например, А и В, в растворителе Е. В отличие от прояви-
тельного метода сорбент промывают не чистым растворителем, а
раствором вещества D, сорбирующегося сильнее каждого из ком-
понентов анализируемой смеси. Такое вещество называется вытес-
нителем.
При промывании сорбента, содержащего компоненты анализи-
руемой смеси, раствором вытеснителя анализируемая смесь переме-
щается впереди фронта вытеснителя и разделяется на зоны, каждая
из которых соответствует одному компоненту. Все зоны движутся
с одной и той же скоростью, равной скорости движения зоны вытес-
нителя. Компоненты разделяемой смеси выходят из колонки после-
довательно друг за другом (рис. 4,а). Запись выходной кривой вы-
теснительного анализа приведена на рис. 4,6. На кривой последняя
ступень соответствует вытеснителю, другие ступени — компонен-
там анализируемой смеси. Если опыт проводится при постоянной
концентрации вытеснителя, то длина ступени оказывается пропор-
10
йиональной количеству данного компонента в смеси, тогда как вы-
соты ступеней могут служить мерой ее качественного состава.
Вытеснительный метод обладает тем преимуществом, что в этом
методе процедура анализа сводится к определению длин и высот
ступенек. Кроме того, в отличие от проявительного метода, компо-
ненты смеси не разбавляются растворителем, вследствие чего их
концентрация не уменьшается при хроматографировании. Вытес-
нительный метод нашел себе широкое применение в жидкостно-ад-
сорбционной и ионообменной
хроматографии.
Недостатком метода яв-
ляется то, что зоны компо-
нентов не разделены зоной
чистого растворителя, по-
этому всегда имеет место
более или менее заметное на-
ложение зоны одного веще-
ства на зону другого. Этот
недостаток особенно резко
проявляется при анализе га-
зов, поэтому вытеснительный
анализ не нашел себе приме-
нения в газовой и газо-жид-
костной хроматографии.
Области применения
Рис. 4. Схема вытеснительного анализа:
а — расположение зон вытесняемых веществ н
вытеснителя в колонке; б — хроматограмма
вытеснительного анализа
Кроме главного своего
хроматографии.
применения — качественного и количественного анализа сложных
смесей — хроматографические методы позволяют решать ряд дру-
гих не менее важных задач. К ним относятся следующие:
- 1) идентификация веществ и установление различия между
ними;
. 2) разделение сложной смеси на отдельные компоненты с пре-
паративными целями;
3) испытание вещества на однородность, на чистоту;
4) очистка веществ от примесей;
5) концентрирование вещества и его выделение из разбавленных
растворов или смесей;
6) контроль и автоматизация производственных процессов.
В последнее время хроматографический метод начали применять
Для определения ряда физических и физико-химических свойств
Индивидуальных веществ. К числу таких свойств относятся, напри-
мер, относительная скорость движения хроматографических полос,
положение вещества в сорбционном ряду, теплоты сорбции, изотер-
мы сорбции, удерживаемые объемы и др. Многие из этих свойств
связаны с другими важными физическими характеристиками ве-
щества и структурой молекул и поэтому могут быть использованы
Для определения этих характеристик.
11
ГЛАВА I
МОЛЕКУЛЯРНАЯ АДСОРБЦИОННАЯ
ХРОМАТОГРАФИЯ ЖИДКИХ ВЕЩЕСТВ
В основе молекулярной адсорбционной хроматографии лежит
различие в адсорбционных свойствах компонентов разделяемой
смеси.
В состоянии равновесия каждой концентрации адсорбируемого
вещества отвечает определенное количество его на адсорбенте.
В связи с тем, что такое равновесие зависит от температуры, его изу-
чение должно проводиться при постоянной температуре. Зависи-
мость количества адсорбированного вещества от его концентрации в
растворе при состоянии равновесия и при постоянной температуре
может быть выражена изотермой адсорбции.
Изотерма адсорбции является важной характеристикой системы
адсорбент — адсорбат, так как знание формы изотермы адсорбции,
а также взаимного расположения изотерм адсорбции различных ве-
ществ может помочь правильному выбору условий хроматографи-
ческого разделения сложных смесей.
Наиболее общей теорией адсорбции является теория Лэнгмюра.
В основе ее лежит предположение, что на поверхности твердого
тела — адсорбента — находятся активные участки, свободное си-
ловое поле которых способно так или иначе фиксировать молекулы
посторонних веществ. Второе предположение сводится к тому, что
каждый элементарный участок поверхности адсорбента способен
фиксировать только одну молекулу. Поэтому при адсорбции на
твердых поверхностях образуется мономолекулярный слой, кото-
рый экранирует силовое поле адсорбента.
Однако на поверхности адсорбента происходит не только адсорб-
ция молекул из окружающей среды, но и их возврат в окружающую
среду — испарение, или десорбция. В результате между поверх-
ностью адсорбента и средой устанавливается подвижное равнове-
сие, определяемое равенством скоростей прилипания (адсорбции)
и испарения (десорбции) молекул.
Для вывода уравнения изотермы адсорбции предположим, что
для того чтобы молекула вещества адсорбировалась, она должна
12
„париться о поверхность адсорбента и попасть на незанятое место.
Так как число ударов о поверхность адсорбента пропорционально
концентрации вещества в растворе с, а вероятность попасть на неза-
нятое место равна числу этих мест, то для скорости процесса ад-
сорбции ыадс можно написать уравнение
«аде = ^(1-0)» (1)
где _ постоянная; 0 — доля занятых мест на адсорбенте.
С другой стороны, какая-то часть адсорбированных молекул
покидает-поверхность адсорбента и уходит в раствор. Число таких
молекул пропорционально общему числу адсорбированных молекул.
Поэтому скорость десорбции равна
«дес = &20> (2)
где k2 — постоянная.
В состоянии равновесия скорости ыадс и цдес равны между со-
бой, поэтому
fcxc(l — 0) = Л20. (3)
Решая уравнение (3) относительно 0 и полагая k-Jk2~b, получим
0 = г^-. (4)
1 + be v ’
Если максимальное число мест на адсорбенте, которое может
быть занято молекулами адсорбата, обозначить через а*, а коли-
чество адсорбированного вещества, соответствующее равновесному
состоянию при заданной концентрации, обозначить через а, то а=
== а„0. Подставляя эти значения в уравнение (4), получим уравне-
ние изотермы адсорбции Лэнгмюра для одного компонента
Ьс ....
Из уравнения (5) следует, что существует предел адсорбции, т. е.
увеличение концентрации раствора выше определенного значения
не приводит к дальнейшему увеличению количества адсорбирован-
ного вещества. Теоретически этот предел наступает при ассимптоти-
ческом приближении кривой изотермы адсорбции к линии насыще-
ния, так как изотерма адсорбции представляет собой гиперболу
(рис. 5).
Уравнение Лэнгмюра вполне удовлетворительно описывает за-
висимость величины адсорбции от концентрации для очень большого
Писла экспериментальных данных.
Изотермы адсорбции Лэнгмюра в общем по своему виду анало-
гичны как изотермам адсорбции газов и жидкостей, так и изотермам
вДсорбции из растворов. Однако адсорбция из растворов по сравне-
нию с газовой адсорбцией представляет собой значительно более
^ложное явление. Это объясняется тем, что на адсорбцию твердым
ом веществ, находящихся в растворе, оказывает влияние не
13
Только природа адсорбента и растворенного вещестьа, но также
и свойства растворителя, которого, как правило, всегда значи-
тельно больше, чем растворенного вещества. Поэтому простейший
случай адсорбции из раствора должен рассматриваться как случай
адсорбции двух видов молекул.
Молекулы растворителя, адсорбируясь на поверхности адсор-
бента, уменьшают адсорбируемость растворенного вещества. Поэ-
тому при выборе растворителя в хроматографическом анализе
необходимо отдавать преимущество тому из них, который обла-
дает наименьшей адсорбцией на данном адсорбенте. Кроме того,
при сравнительном изучении изо-
терм адсорбции различных веществ,
необходимо иметь в виду, что все
они должны быть получены с одним
и тем же растворителем.
Сложность процесса адсорбции из
растворов часто приводит к разного
рода искажениям обычного типа изо-
терм адсорбции. Так, например, если
поверхность адсорбента покрыта про-
чно удерживаемым слоем адсорбиро-
ванного вещества, молекулы которого
Рис. 5. Изотерма адсорбции
Лэнгмюра:
а — количество адсорбированного
вещества, мМ./г\ — величина пре-
дельной адсррбции; с — концентра-
ция раствора в равновесии, мМ/л
поляризованы, то такая поверхность
в свою очередь может притягивать
свободные молекулы из раствора, по-
ляризуя их. Так может возникнуть
второй и последующие адсорбционные слои. В этом случае изо-
термы адсорбции принимают S-образную форму, причем величина
адсорбции при возрастании концентрации бесконечно возрастает.
Из сказанного следует, что для правильного выбора условий
хроматографического разделения и анализа 'смеси веществ большое
значение имеет знание изотерм адсорбции каждого из компонентов
разделяемой смеси. Получение изотерм адсорбции можно осущест-
вить с помощью фронтального хроматографического метода.
1. ФРОНТАЛЬНЫЙ МЕТОД
Как уже указывалось во введении, фронтальный метод состоит
в непрерывном пропускании смеси анализируемых веществ через
слой адсорбента до тех пор, пока адсорбент не насытится всеми ком-
понентами изучаемой смеси веществ. При этом насыщение наступает
последовательно для каждого компонента в порядке возрастания
адсорбционного сродства компонентов. При достижении насыщения
на выходе из колонки наступает проскок данного вещества.
Очевидно, что для случая раствора одного вещества количество
адсорбированного компонента может быть получено из величины
удерживаемого объема VR, так как при объеме, большем, чем удер-
живаемый объем, раствор будет проходить через адсорбент без
14
изменения концентрации. Если а — количество вещества, адсорби-
рованного в данных условиях, ас — концентрация вещества в раст-
воре при равновесии, то
а = ЕдС,
или, в расчете на 1 г адсорбента
а° = V°Rc.
(6)
Так как а=[(с), то уравнение (6) может быть представлено в виде
(7)
Следовательно, измеряя величины удерживаемых объемов, по-
лучаемых при фронтальном хроматографировании растворов одного
и того же вещества, взятых в различных концентрациях, можно
получить данные для построения
изотермы адсорбции.
Как следует из . сказанного,
фронтальный анализ очень удобен
для экспериментального определе-
ния изотерм адсорбции, т. е. для
вычисления функции /(с) по урав-
нению (6). Задаваясь различными
концентрациями растворов, можно
заранее определить концентрации
с
Рис. 6. Диаграмма фронтального
анализа двух веществ
растворенного вещества, находящегося в состоянии равновесия
с адсорбированным веществом, чего нельзя сделать при встря-
хивании раствора с адсорбентом при статическом способе определе-
ния изотерм адсорбции.
При проведении фронтального анализа раствора, содержащего
два растворенных вещества, на выходной хроматографической кри-
вой возникает две ступени, соответственно проскоку каждого из
компонентов раствора.
Первая ступень соответствует проскоку раствора одного наиме-
нее адсорбирующегося вещества, вторая — проскоку раствора обоих
веществ исходной концентрации (рис. 6).
При наличии в растворе двух и более веществ имеет место адсорб-
ционное вытеснение, вследствие чего высоты ступенек на хромато-
графических кривых фронтального анализа не соответствуют кон-
центрации первоначального раствора.
Адсорбционным вытеснением называется явление, возникающее
при одновременной адсорбции нескольких веществ на одном адсор-
бенте. В стремлении занять активные центры на поверхности ад-
сорбента адсорбирующиеся вещества взаимно уменьшают величину
адсорбции. Однако имеют место случаи, когда адсорбционное вы-
теснение не происходит и различные вещества из смеси адсорби-
15
руются в той же степени, как и из растворов индивидуальных ве-
ществ.
Рассмотрим фронтальный анализ раствора, содержащего два
компонента. Обозначим через а? количество первого вещества, ад-
сорбированное 1 г адсорбента из раствора с концентрацией первого
вещества с1 и второго с2- Для первого вещества получим
ai=;/i(ci> сг)> (8)
и, аналогично, для второго
a°s=f2(cv q). (9)
В случае адсорбционного равновесия величина fx(q) будет боль-
ше, чем //q, с2). Следовательно, при фронтальном анализе двухком-
понентных систем концентрация первого, менее адсорбирующегося
вещества, в растворе, соответствующем первой ступеньке хроматог-
рафической кривой, будет больше, чем в исходном растворе. Это
объясняется тем, что движущееся за первым веществом второе час-
тично вытесняет с адсорбента первое.
Величины а[ и а° легко вычислить. Так как после достижения
насыщения адсорбента как первым, так и вторым компонентами
(вторая ступенька) раствор проходит через колонку без изменения,
то мы имеем
aS = V^,2q. (10)
После прохождения через адсорбент миллилитров раствора,
содержащего вначале Vr,2 q граммов первого вещества, этот раствор
будет содержать только (Vr,2—Vr,i)ci,i граммов первого вещества
(если концентрацию первого вещества в растворе, соответствующем
первой ступеньке, обозначить qa). Отсюда получим выражение для
а?
(Н)
Таким образом нетрудно определить изотермы адсорбции каж-
дого компонента двухкомпонентной системы. Для этого необходимо
составить ряд растворов с различными, но известными концентра-
циями определяемых компонентов и провести с этими растворами
фронтальный анализ на избранном адсорбенте. При этом измеряе-
мыми величинами будут удерживаемые объемы и 2, а также
концентрация первого вещества в первой ступеньке q г
Определение изотерм адсорбции для двухкомпонентной системы
статическим методом, без применения фронтального анализа, пред-
ставляет значительные трудности.
В случае трех и более растворенных веществ вычисление адсор-
бированного количества по данным хроматографической кривой
становится невозможным, так как при этом число неизвестных ока-
зывается больше числа уравнений.
16
2. ВЫТЕСНИТЕЛЬНЫЙ МЕТОД
Рассмотрим простейший случай вытеснительного анализа одно-
компонентного раствора [10]. Фронт анализируемого вещества и
фронт вытеснителя, раствор которого введен в колонку с адсорби-
рованным веществом, перемещаются вдоль слоя адсорбента с одной
и той же скоростью. Предположим, что через слой адсорбента про-
шел объем раствора ДК При этом оба фронта переместятся на одно
и то же расстояние \xD=\xr, где Дхо —расстояние, на которое пе-
реместится фронт вытеснителя, а \хг — фронт анализируемого ве-
щества. Тогда мы получим
ДУ ДУ
Дхх Дхд ’
Г (с)
Так как V--—- > гДе /(с) есть уравнение изотермы адсорбции, то
на основании уравнения (12) мы можем написать, что
(12)
fo(CD) fl (с1) 17 /1 п\
~D (13)
Это уравнение показывает, что для успешного вытеснения необ-
ходимо выбрать подходящую концентрацию вытеснителя cD в соот-
ветствии с уравнением (13). Если в течение всего опыта концентра-
ция вытеснителя будет оставаться постоянной, то будет постоянна и
концентрация анализируемого вещества сх, независимо от количест-
ва этого вещества, прошедшего через слой адсорбента.
Если неизвестная смесь содержит несколько компонентов, обла-
дающих различными изотермами адсорбции /г^г); /3(с3), •••
и, соответственно, разными равновесными концентрациями q; с2;
с3....то, основываясь на вышеприведенных рассуждениях, можно
написать для сложной системы
fD (ср) _ f 1 (с1) _ f2 (сг) _ fз (сз) . _ у о /14)
сг с2 с3 - R.D- V >
Такое уравнение легко решить графическим путем, если известны
изотермы адсорбции для каждого компонента смеси и вытеснителя.
Как показывает рис. 7, прямая линия проводится от начала коор-
динат до выбранной точки на изотерме адсорбции вытеснителя,
соответствующей концентрации cD. Уравнение этой прямой имеет вид
a0 = /(c) = Vbc, (15)
где V^,d — удельный удерживаемый объем вытеснителя. Так как
все члены уравнения (14) равны V# D, то искомую равновесную
концентрацию любого компонента находят в точке пересечения
прямой с соответствующей изотермой адсорбции.
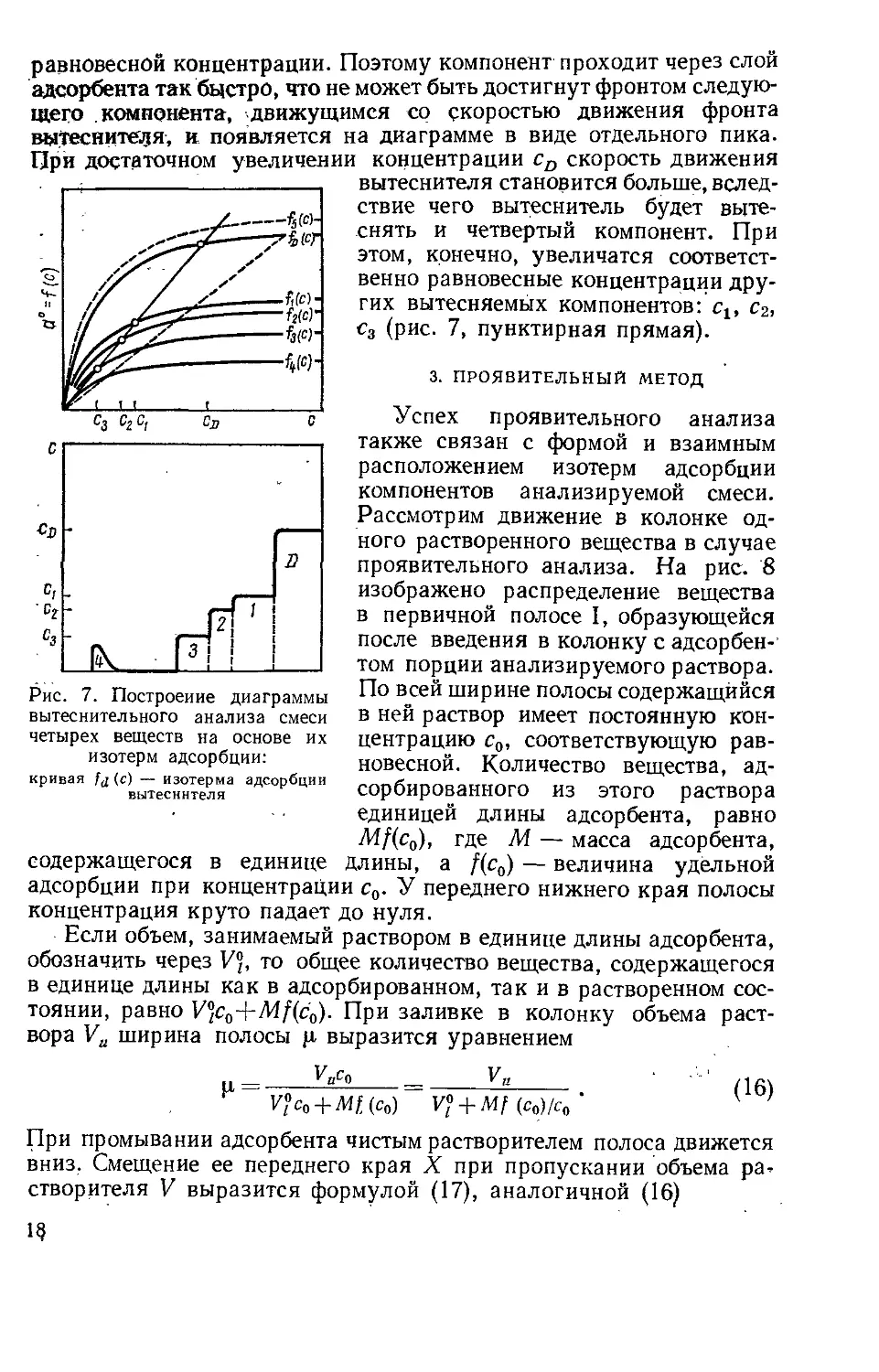
Из рис. 7 следует, что если один из компонентов, например,
Четвертый, адсорбируется настолько слабо, что его изотерма ад-
сорбции не пересекается прямой, то для него не может получиться
17
равновесной концентрации. Поэтому компонент проходит через слои
адсорбента так быстро, что не может быть достигнут фронтом следую-
щего компонента, движущимся со скоростью движения фронта
вытеснителя, и появляется на диаграмме в виде отдельного пика.
При достаточном увеличении концентрации cD скорость движения
вытеснителя становится больше, вслед-
ствие чего вытеснитель будет выте-
снять и четвертый компонент. При
этом, конечно, увеличатся соответст-
венно равновесные концентрации дру-
гих вытесняемых компонентов: с1( Сг,
с3 (рис. 7, пунктирная прямая).
Рис. 7. Построение диаграммы
вытеснительного анализа смеси
четырех веществ на основе их
изотерм адсорбции:
кривая (с) — изотерма адсорбции
вытеснителя
3. ПРОЯВИТЕЛЬНЫЙ МЕТОД
Успех проявительного анализа
также связан с формой и взаимным
расположением изотерм адсорбции
компонентов анализируемой смеси.
Рассмотрим движение в колонке од-
ного растворенного вещества в случае
проявительного анализа. На рис. 8
изображено распределение вещества
в первичной полосе I, образующейся
после введения в колонку с адсорбен-
том порции анализируемого раствора.
По всей ширине полосы содержащийся
в ней раствор имеет постоянную кон-
центрацию с0, соответствующую рав-
новесной. Количество вещества, ад-
сорбированного из этого раствора
единицей длины адсорбента, равно
где М — масса адсорбента,
длины, а /(с0) — величина удельной
содержащегося в единице
адсорбции при концентрации с0. У переднего нижнего края полосы
концентрация круто падает до нуля.
Если объем, занимаемый раствором в единице длины адсорбента,
обозначить через V”, то общее количество вещества, содержащегося
в единице длины как в адсорбированном, так и в растворенном сос-
тоянии, равно Е?с0+Л1/(с0). При заливке в колонку объема раст-
вора Vu ширина полосы р выразится уравнением
„ _ Уис0 _ уа
У'сь + МЦсь) У« + М/(со)/Со •
(16)
При промывании адсорбента чистым растворителем полоса движется
вниз. Смещение ее переднего края X при пропускании объема ра-
створителя V выразится формулой (17), аналогичной (16)
1$
У1 + М1(с0)1са '
(17)
При. этом передний край полосы остается довольно резким, задний
же постепенно размывается. Смещение зоны полосы (У), находящей-
ся в «хвосте», выразится формулой
Y =---------------------------,
+ (с)
где f(c) является первой производной функ-
ции /(с).
Отдельные зоны полосы, соответствующие
различной концентрации, будут двигаться с раз-
ной . скоростью, зависящей от функции /(с).
Для зон с концентрацией меньшей, чем с1(
f’(c)>f(c0)/c0 и, следовательно, Y<X, т. е. зад-
ние зоны будут двигаться медленнее передних.
Зоны, для которых с>сх, нагонят передние зоны.
В результате этого полоса по мере ее промывания
и продвижения вдоль слоя адсорбента растяги-
вается, появляется «хвост» (зона III на рис. 8).
Такой тип полос наблюдается в случае вы-
пуклой, Лэнгмюровской изотермы адсорбции.
При наличии вогнутой изотермы картина будет
обратной. В случае прямолинейной изотермы
Рис. 8. Схема рас-
пределения веще-
ства вдоль слоя ад-
сорбента по мере
вымывания его про-
явителем
(18)
адсорбции распределение концентрации в зоне
будет близким к симметричному. Однако случаи прямолинейных
изотерм адсорбции весьма редки.
В соответствии с приведенными выше формулами, чем больше
адсорбируемость вещества, т. е. чем круче его изотерма адсорбции,
тем медленнее оно движется по слою адсорбента. Поэтому при вы-
мывании нескольких адсорбированных веществ, обладающих раз-
личными изотермами адсорбции, зоны этих веществ будут двига-
ться с различными скоростями, что и обусловливает их полное
разделение при достаточно длинном слое адсорбента.
Мы не рассматриваем здесь вопроса о скорости адсорбции. Бе-
зусловно, однако, что все адсорбционные процессы хроматографиро-
вания должны проводиться в условиях, близких к равновесным.
Равновесие же может быть достигнуто лишь при условии соблюдения
требований кинетики адсорбции, т. е. при подборе таких ско-
ростей потока, которые обеспечивают время контакта раствора с ад-
сорбентом, необходимое как на перенос вещества из объема раствора
к поверхности адсорбента, так и на собственно процесс адсорбции,
а также на диффузию или миграцию молекул адсорбирующегося
вещества по поверхности адсорбента. Все эти вопросы требуют
специального рассмотрения [11] и выходят за рамки настоящего
руководства.
19
АДСОРБЕНТЫ И РАСТВОРИТЕЛИ
1. АДСОРБЕНТЫ
Правильный выбор адсорбента и растворителя имеет очень боль-
шое значение при решении задач хроматографического разделения
сложных смесей. Поэтому экспериментатору необходимо знать ос-
новные принципы такого подбора, а также наиболее важные свойст-
ва адсорбентов и предъявляемые к ним требования.
Главным требованием, предъявляемым к любому адсорбенту,
применяемому в хроматографии, должно быть отсутствие
химического взаимодействия между адсорбентом
и анализируемыми веществами. Он не должен также оказывать
каталитического воздействия как на раствори-
тель, так и на вещества разделяемой смеси. Последнее требование
часто бывает трудно выполнимым вследствие того, что важнейшие
адсорбенты, как правило, являются активными катализаторами.
Поэтому нередки случаи, когда при хроматографировании имеют
место процессы изомеризации, полимеризации, окисления и других
химических превращений, что приводит к образованию новых ве-
ществ, не присутствовавших в исследуемой смеси, и может навести
исследователя на ложный вывод.
Для устранения каталитического эффекта существует несколько
способов. В связи с тем, что каталитическое действие адсорбентов
обусловливается наличием посторонних примесей кислот, щелочей,
солей, окислов и т. п., одним из средств уменьшения или полного
устранения каталитической активности может служить тщательная
очистка адсорбента от примесей или нейтрализации кислых или ос-
новных его свойств. Каталитическое окисление можно устранить,
проводя процесс в атмосфере инертного газа. В некоторых случаях
приходится прибегать к искусственному отравлению адсорбентов,
например, полярные адсорбенты снижают свою каталитическую ак-
тивность в присутствии влаги. Возможна также температурная об-
работка адсорбента.
Вторым важнейшим требованием к адсорбенту является его и з-
бирательность, т. е. возможно большее различие в адсор-
бируемости веществ разделяемой смеси. Одним из возможных кри-
териев такого различия может служить величина теплоты адсорб-
ции или смачивания. Поэтому при выборе адсорбента бывает по-
лезно предварительное определение теплот адсорбции веществ
разделяемой смеси из данного растворителя. Различие в величинах
теплот адсорбции изучаемых веществ связано с различием их ад-
сорбируемости и может поэтому дать ценные указания при выборе
адсорбента.
К сожалению, в настоящее время еще довольно трудно устано-
вить прямую связь между адсорбируемостью того или иного веще-
ства и его химическим строением, а также между химическим строе-
нием адсорбента и его адсорбционной емкостью. Полезным следует
20
признать деление адсорбентов на две основные группы: полярные
и аполярные. Адсорбционное сродство полярных веществ к поляр-
ным адсорбентам значительно выше, чем аполярных к полярным.
Этим различием следует пользоваться при выборе адсорбентов.
Выбор определенной степени дисперсности адсор-
бента также имеет немаловажное значение. Чем меньше частицы ад-
сорбента, тем быстрее устанавливается адсорбционное равновесие
и тем лучше работает колонка. С другой стороны, увеличение сте-
пени дисперсности влечет за собой возрастание сопротивления
колонки течению жидкости. Одним из средств борьбы с этим нежела-
тельным явлением может служить применение адсорбента с одина-
ковыми по размерам частицами, т. е. изодисперсных адсорбентов.
Изодисперсные порошки обладают значительно меньшим сопротив-
лением течению, чем полидисперсные. Поэтому изодисперсность
адсорбента следует признать одним из его существенных достоинств.
Наконец, чрезвычайно важным требованием к адсорбенту долж-
но быть постоянство, стандартность свойств.
Если это требование не будет соблюдено, то при воспроизводстве
или сопоставлении разных опытов могут возникнуть существенные
затруднения вследствие неодинаковости условий эксперимента.
К сожалению, установление спецификации на адсорбенты, подобно
тому, как это делается для химических реактивов (ч., ч. д. а., х. 4J,
до сих пор оказывается невозможным вследствие зависимости ад-
сорбционных свойств адсорбента от способа его приготовления, а
также последующей его обработки и влагосодержания. В связи
с этим возникает необходимость не только приготовления адсорбен-
та по строго соблюдаемой единой методике и из одинакового сырья,
но и установления единых методов характеристики и сравнения ад-
сорбционной емкости адсорбентов.
Для проверки адсорбционной активности адсорбентов предло-
жено немало различных методик, которые могут применяться в
зависимости от дальнейшего назначения адсорбента, т. е. от того,
для решения какой задачи используется тот или иной адсорбент.
Экспериментальное выполнение некоторых методик описано ниже.
Переходя к рассмотрению важнейших адсорбентов, следует
указать, что в настоящее время в хроматографических работах приме-
няются почти исключительно адсорбенты заводского производства,
вследствие того, что получение их в лабораторных условиях свя-
зано со значительными трудностями, затратой большого количества
времени, а также с невозможностью соблюдения требований стан-
дартизации.
Окись алюминия. Одним из наиболее часто прйменяемых адсор-
бентов является окись алюминия, на которой удается хроматографи-
чески разделить весьма широкий круг смесей веществ как из поляр-
ных, так и аполярных растворителей. Это свойство окиси алюминия
как адсорбента определяется тем, что она обладает амфотерным
характером.
21
Активность окиси алюминии зависит от ее влагосодержания,
что имеет практическое значение для хроматографии, так как поз-
воляет заменить набор адсорбентов различной адсорбционной ем-
кости одним адсорбентом. Увлажняя наиболее активную форму окиси
алюминия различным количеством воды, можно получить набор ад-
сорбентов с различной емкостью.
Отечественная промышленность выпускает в настоящее время
активированную окись алюминия двух марок (ГОСТ 8136—56).
Она представляет собой у-модификацию окиси алюминия, содер-
жащую кристаллизационную воду и имеющую высокоразвитую по-
верхность. Марка А-1 отличается от марки А-2 меньшим насыпным
весом и несколько меньшей механической прочностью [12]. Выпус-
кается также окись алюминия для хроматографии по ТУ 2962—52.
Основные данные об окиси алюминия по ГОСТ 8136—56 приведены
в табл. 2.
Таблица 2
Характеристика активированной окиси алюминия
для хроматографии (у-модификация) (ГОСТ 8136—56)
Показатели
Внешний вид ............................
Марка
A-I | А-2
Цвет......................................
Каталитическая активность (константа скорости
дегидратации этилового спирта до этилена):
при 360° С, не менее....................
при 420° С, не менее....................
Насыпной вес, г/л.........................
Механическая прочность, %, не менее . . . .
Потери при прокаливании при 800° С, %, не
более.......................................
Содержание железа, %, не более............
Диаметр пор, А............................
Объем пор, мл[ г. ........................
Адсорбционная способность по бензолу, л.г/100г
Цилиндрики длиной
4—25 мм и диаметром
4—6 мм или шарики
того же диаметра
Белый. Допускается
кремовый оттенок
6,2
37
400—550
95
6
0,1
6,2
37
550—750
98
6
0,1
60—98
0,17—0,45
1,9—7,1
Силикагели. Очень широко применяются в хроматографии сили-
кагели различных марок. Силикагель оказался лучшим адсорбентом
для хроматографического разделения смесей нефтяных углево-
дородов, смолистых веществ, содержащихся в нефтепродуктах, выс-
ших жирных кислот и их сложных эфиров, нитро- и нитрозопроиз-
водных, ароматических аминов и других органических соединений.
Получается силикагель действием минеральных кислот на кон-
центрированные растворы силиката натрия. Продуктом реакции
при этом является золь гидратированной кремниевой кислоты, дис-
22
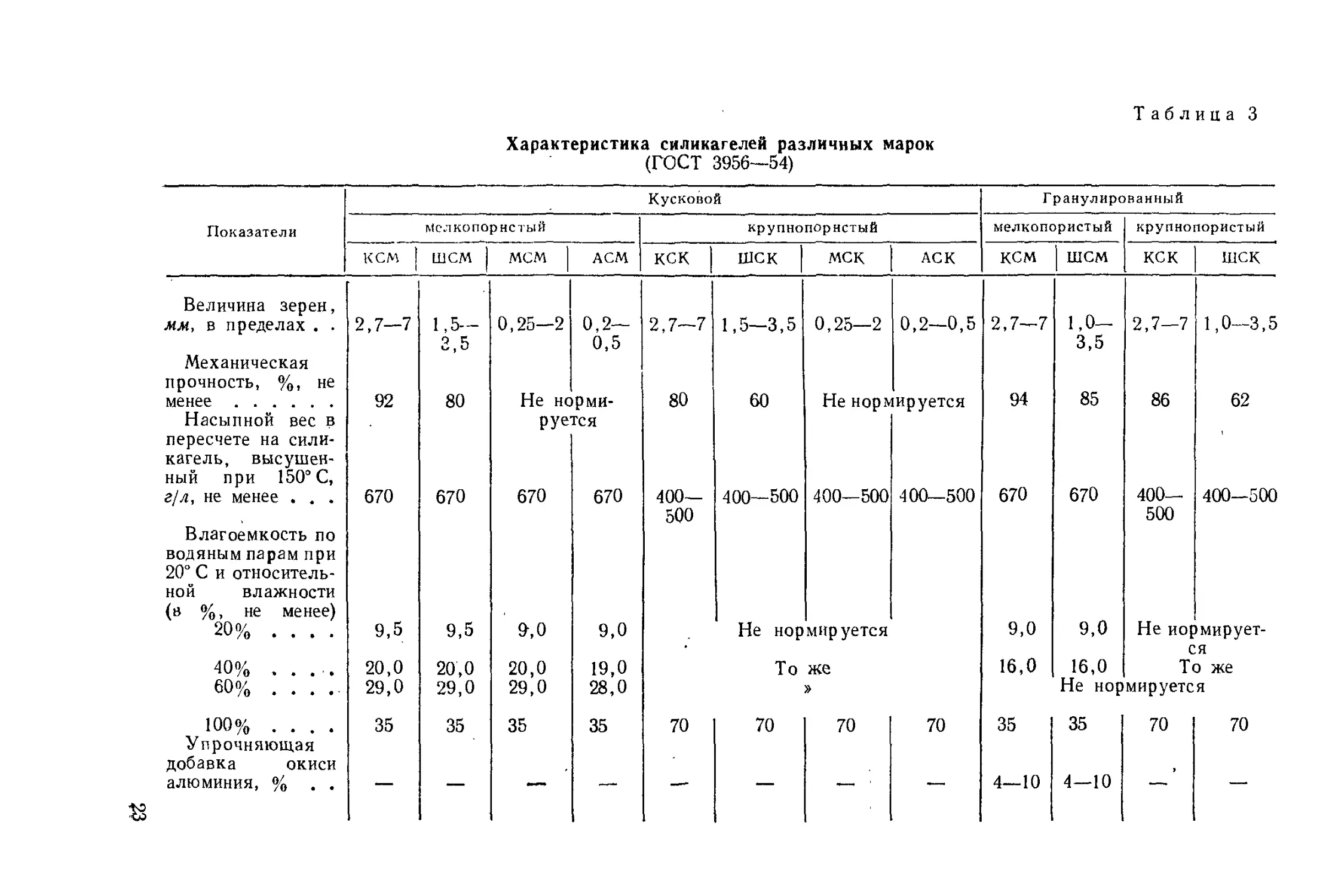
Характеристика силикагелей различных марок
(ГОСТ 3956—54)
Таблица 3
Показатели Кусковой Гранулированный
Мслкопорнстый крупнопористый мелкопористый крупнопористый
кем | шем | мем | АСМ кек | шск мск АСК кем | шем кек шск
Величина зерен, 1,0- 3,5 1,0—3,5
мм, в пределах . . Механическая прочность, %, не 2,7—7 1,5— 2,5 0,25—2 0,2— 0,5 2,7—7 1,5—3,5 0,25—2 0,2—0,5 2,7—7 2,7—7
менее Насыпной вес в пересчете на сили- кагель, высушен- ный при 150° С, 92 80 Не нс руе рми- гся 80 60 Не нор^ ируется 94 85 86 62
г/л, не менее . . . Влагоемкость по водяным парам при 20° С и относитель- ной влажности (в %, не менее) 670 670 670 670 400— 500 400—500 400—500 400—500 670 670 400— 500 400-500
20% .... 40% ..... 60% 9,5 20,0 29,0 9,5 20,0 29,0 9,0 20,0 29,0 9,0 19,0 28,0 Не нормируется То же » 9,0 16,0 9,0 16,0 Не нор Не нормирует- ся То же мируется
100% .... Упрочняющая добавка окиси 35 35 35 35 70 70 70 70 35 35 70 70
алюминия, % . . & — — —— — — — 4—10 4—10 —- —‘
пергированный в растворе нейтральной соли. После созревания гель
освобождают от примесей минеральных солей и сушат при 115—
130° С до остаточной влажности 5—7%. В этом случае оставшаяся
влага представляет собой структурную воду поверхностного слоя,
что, -по-видимому, обусловливает наибольшую адсорбционную ак-
тивность силикагеля при данном содержании влаги.
Отечественная промышленность выпускает крупнопористые и
мелкопористые силикагели различных марок. Эти марки отлича-
ются друг от друга как химическим составом, пористостью, так и
внешней формой [12]. Характеристика силикагелей, выпускаемых
согласно ГОСТ 3956—54, приведена в табл. 3. Набор силикагелей
специально для хроматографии выпускается «Союзреактивом» по
ТУ 382р—61 (табл. 4).
Таблица 4
Характеристика силикагелей для хроматографии
(ТУ 382р—61)
Марка । Насыпной вес с утряской,г/см* Поверхность, м2/г гл , Истинная плот- 1 НОСТЬ, c’/CJW3 Кажущаяся плотность, г/см* Объем пор, смя/г Средний радиус пор, А Пористость, % Влагоемкость (вес. %) при относительной влажности воздуха:
20% 40% 60% 100%
КСК № 2 0,39 338 2,240 0,611 1,19 70 72,7 2,5 4,6 7,8 119
КСК № 2,5 0,46 376 2,244 0,706 0,971 51,6 67,4 2,2 4,6 8,7 97,9
КСК № 3 0,50 522 2,236 0,729 0,925 35,4 67,4 2,9 5,7 13,5 87,1
КСК № 4 0,58 650 2,235 0,831 0,760 23,4 62,8 2,4 7,4 20,1 70,4
КСМ № 5 0,66 715 2,250 0,980 0,575 16,1 56,4 4,4 15,5 34,9 56,8
КСМ № 6п 0,87 527 2,255 1,353 0,296 11,2 40,0 5,7 15,2 24,7 26,9
КСМ № 6с 0,87 624 2,179 1,218 0,362 11,6 44,1 н,з 20,5 33,1 34,8
।
Активированные угли. Для хроматографического анализа сме-
сей веществ, принадлежащих к одному гомологическому ряду, наи-
более подходящим адсорбентом являются активированные угли, вы-
пускаемые под различными марками. Для повышения дисперснос-
ти адсорбирующей поверхности и освобождения пор адсорбента от
смолистых веществ угли подвергаются специальной обработке, ко-
торая и называется активированием. Уголь прокаливается при тем-
пературе около 900° С, затем экстрагируются смолы органическими
растворителями с] последующим удалением растворителей прокали-
ванием и окислением поверхности угля и органических веществ в
его порах газообразными окислителями.
Адсорбционная способность активированных углей в 50—60 раз
выше обычных. Их удельная поверхность достигает 1300—1700 м2/г.
В хроматографии применяют различные марки активированных
углей, приготовленных из древесных, костных, лигнинных, камен-
24
ных и других углей. «Союзреактив» выпускает набор активирован-
ных углей для хроматографии согласно ТУ 74В—60 (табл. 5) [121.
Таблица 5
Характеристика активированных углей для хроматографии
(ТУ 74В—60)
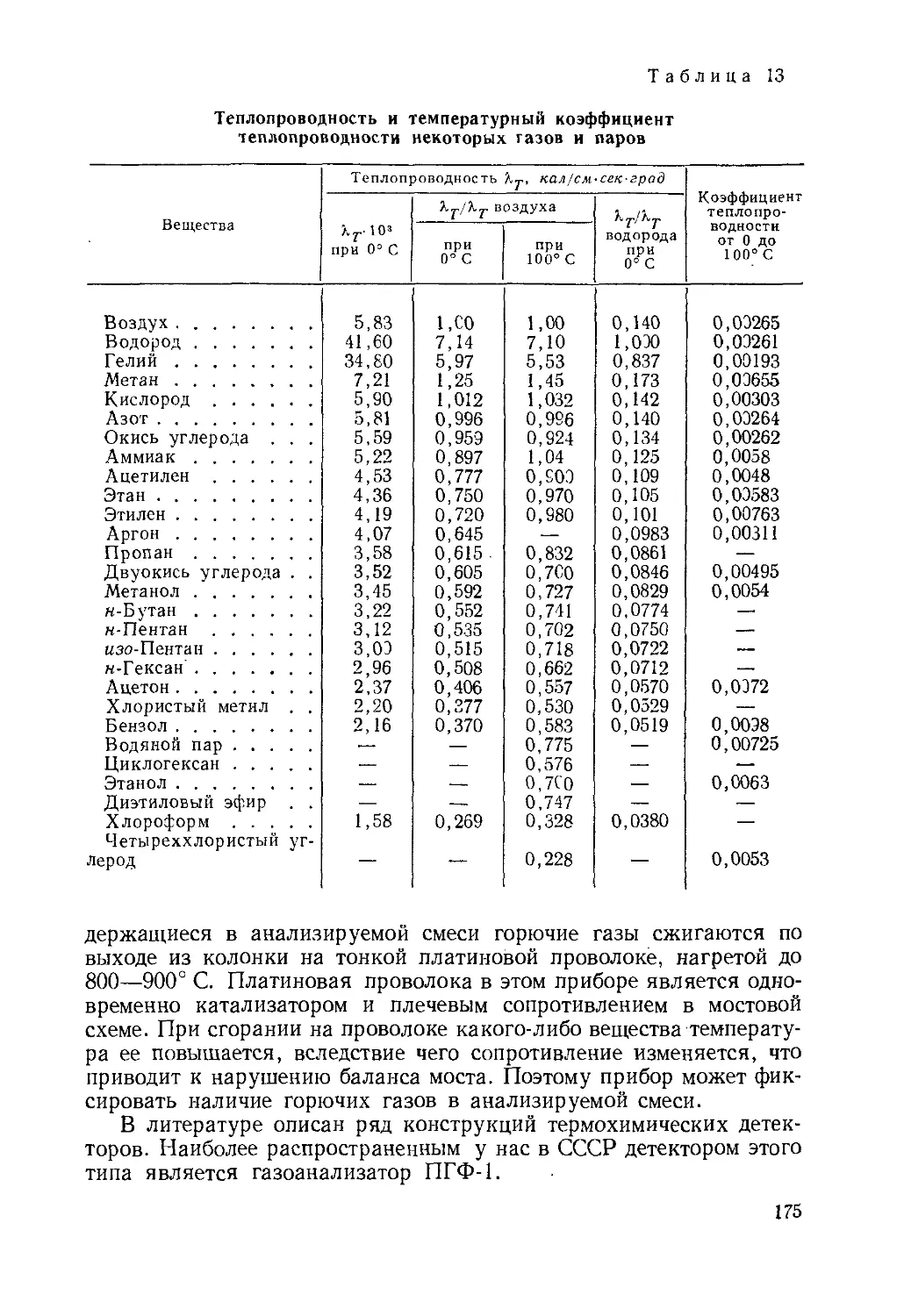
Марка углей Динамическая активность по бензолу, не менее еская ость по з раство- >ез 90 мин, ее Размер зерен, мм 1 Масса 1 л угля, 1г Влажность, %, не более Прочность, %, не менее X о.
Статич активн ноду и ров чер X X
БАУ, газовый, дроб-
леный орешек — 35* 1—5 350 10 70 7-8
СКТ гранулированный торфяной, газовый . . . 71 мм ,— 1,5—2,7 380—450 8 70 6
АР-3 гранулирован- ный, рекуперационный 115 г 1л — 2—5 не более 6С0 400 15 90 7—8
КАД иодный — 55 1 — 1,5 10 90 7-8
КАД молотый .... АТ-3 гранулирован- — — 1,5—2,7 — 10 — 7—8
иый, газовый ОУ осветляющий: 38 мин — 440—460 5 75 7-8
А—сухой щелочной — — 1,5—2,7 .— 10 — 8
Б—влажный кислый АГН дробленый оре- — — 1,5—2,7 0,5—10 •— 58 — 4—6
шек — 75 .— 10 65 7-8
АГ-5 гранулированный, газовый мелкий .... 48 мин — 1—1,5 440—460 5 75 7—8
* Статическая активность по хлору.
Молекулярные ситаПЗ]. К числу особенно интересных сорбен-
тов относятся природные и синтетические вещества, получившие
название молекулярных сит. Это мелкие пористые кристаллы при-
родных или синтетических минералов — цеолитов. Поры таких
кристаллов имеют размеры, близкие к размерам молекул жидких
или газообразных веществ. Поэтому те вещества, молекулы которых
по своим размерам могут проникнуть в эти поры, сорбируются в
кристаллах цеолитов; более же крупные молекулы остаются несор-
бированными. Вследствие того, что существующие в природе и син-
тезируемые цеолиты могут обладать порами разных размеров, воз-
никает возможность очень четкого разделения на цеолитах смесей
различных веществ.
К природным цеолитам относится довольно боль-
шая группа минералов, являющихся водными алюмосиликатами
кальция, натрия и некоторых других металлов.
25
Среди природных цеолитов обычно различают три группы:
группу шабазита, группу натролита и группу гейландита. Типичным
представителем первой группы является минерал шабазит
(Ca,Na2) [AlSi2Oe]-6H2O
Ко второй группе относится минерал натролит
Na2[Al2SisO10]-2H2O
В этом минерале в качестве катиона имеется только натрий.
Наиболее характерными для третьей группы являются минералы
гейландит
(Ca,Na2)[AlSi3O8]2.5H2O
и морденит
(Са,Na2,K2)[AlSi4O12] -6Н2О
Каждый тип цеолита имеет свои особенности, обусловленные
его составом и строением, и может поглощать только те вещества,
размеры молекул которых не превышают размеров пор данного цео-
лита. Однако природные цеолиты встречаются сравнительно редко.
Кроме того, чаще всего они бывают загрязнены примесью других
минералов. Все это затрудняет применение природных цеолитов
для получения однородных по свойствам молекулярных сит. В свя-
зи с этим возник вопрос о получении цеолитов с определенными
свойствами и размерами пор синтетическим путем. В настоящее
время такой синтез осуществляется в промышленных масштабах,
причем синтезируются не только аналоги природных цеолитов, но
и совершенно новые их типы.
Наибольшее практическое применение получили синтети-
ческие молекулярные сита под марками NaA, СаА,
NaX, СаХ. Первая буква обозначает преобладающий в цеолите ка-
тион (Na, Са и т. д.), вторая — тип решетки цеолита А или X.
Синтетические цеолиты имеют следующие размеры пор:
Марка цеолита По классифика- ции США Размеры пор, X
NaA 4А около 4
СаА •; 5А » 5
СаХ 10Х » 8
NaX 13Х » 9—10
Природные и синтетические цеолиты превосходят по величине
внутренней удельной поверхности некоторые широко применяемые
адсорбенты. Так, внутренняя удельная поверхность (в м2/г) равна:
шабазита 750, молекулярных сит типа NaA и СаА—750—800, типа
СаХ—1030, тогда как активированная окись алюминия обладает
поверхностью всего лишь равной 230—380, а силикагели 500—600.
Особенностью молекулярных сит, существенно отличающей их
от других типов адсорбентов, является строгая однородность струк-
26
туры и размеров их пор. Механизм прохождения молекул сорбирую-
щегося вещества через «окна», соединяющие полости цеолитов, яв-
ляется сложным, поскольку здесь одновременно проявляются силы
притяжения и отталкивания между отдельными молекулами, а
также оказывают существенное влияние особенности строения мо-
лекул и 'структуры цеолита. Многочисленными исследованиями,
например,о установлено, что молекулярные сита с размером «окон»
около 5 А хорошо сорбируют парафиновые углеводороды нормаль-
ного строения, тогда как изомеры этих же углеводородов, имеющие
разветвленное строение, не сорбируются. Особенностью молекуляр-
ных сит, имеющих малые размеры «окон», является также и тот
факт, когда-то казавшийся необъяснимым, что легкие вещества,
обладающие небольшими размерами молекул, сорбируются, а тя-
желые остаются не сорбированными, тогда как на обычных адсор-
бентах имеет место обратное явление.
Таким образом, структура цеолитов и размеры тех отверстий,
«окон», через которые молекулы сорбирующихся веществ могут про-
никнуть к .развитой поверхности и сорбироваться, определяют осо-
бые свойства цеолитов. Эти свойства позволили применить цеолиты
для «просеивания» веществ сложных смесей и добиться четкого раз-
деления их на индивидуальные компоненты;
Наряду с рассмотренными выше важнейшими адсорбентами, в
хроматографии также применяются и другие, такие как окись каль-
ция, окись магния, углекислый кальций, тальк, крахмал, а также
природные адсорбенты: глины, диатомит, фуллерова земля, кизель-
гур, отбеливающие земли и др. Однако значение этих адсорбентов
значительно меньше, чем значение окиси алюминия, силикагелей,
синтетических молекулярных сит и активированных углей.
2. РАСТВОРИТЕЛИ
В молекулярной адсорбционной хроматографии из растворов
существенное значение имеет правильный выбор растворителя, осо-
бенно в проявительном анализе, в котором растворитель является
проявляющим веществом. Выбор растворителя тесно связан как с
природой выбранного адсорбента, так и со свойствами компонен-
тов анализируемой смесцПРастворители должны прежде всего удов'-
летворять следующим основным требованиям: они должны хорошо
растворять все компоненты анализируемой смеси, минимально
адсорбироваться на выбранном адсорбенте, не реагировать хими-
чески ни с анализируемыми веществами, ни с адсорбентом.
Часто практикуется последовательное вымывание веществ ря-
дом растворителей с постепенно увеличивающейся десорбционной
способностью. При этом отдельные компоненты смеси десорбиру-
ются и вымываются из колонки последовательно. В связи с этим
представляет интерес элюотропный ряд Траппе [14], в котором наи-
более часто применяемые в хроматографии растворители располо-
жены в порядке убывания их десорбирующей способности с поляр-
27
ных адсорбентов (табл. 6). Следует заметить, что десорбирующая
способность приведенных в табл. 6 растворителей, за немногими
исключениями, находится в зависимости от величины их диэлект-
рической постоянной. Очевидно, что для аполярных адсорбентов
десорбирующая способность приведенных в табл. 6 растворителей
будет изменяться в обратном порядке.
Таблица 6
Элюотропный ряд растворителей
Растворитель Диэлект- рическая постоян- ная Растворитель Диэлект- рическая постоян- (ая
Вода....................
Метиловый спирт.........
Этиловый спирт..........
я-Пропиловый спирт . . .
Ацетон..................
Дихлорэтан..............
Этилацетат..............
Амилацетат..............
Этиловый эфир...........
81,0
31,2
25,8
22,8
21,5
10,4
6,1
4,4
Диоксаи.................
Хлороформ ..............
Хлористый метилен . . . .
Бензол..................
Толуол .................
Трихлорэтилен...........
Четыреххлористый углерод
Циклогексан.............
Петролейиый эфир (фрак-
ция 35—50’ С).........
5,2
2,3
2,3
3,4
2,2
2,0
1,9
При выборе растворителей следует также обращать серьезное
внимание на их чистоту. Особенно важно, чтобы аполярные раство-
рители не содержали бы примесей полярных веществ (воды, спирта
и т. п.), резко снижающих адсорбцию из растворителей. Применение
в качестве вымывающего растворителя чистого индивидуального
вещества желательно также в том случае, когда наблюдение за хо-
дом разделения компонентов смеси производится путем измерения
какого-либо физического свойства растворителя, изменяющегося в
ходе процесса (показатель преломления, плотность и др.).
В табл. 7 приведен список растворителей и адсорбентов, наибо-
лее часто применяемых при разделении жидких смесей веществ ме-
тодом молекулярной адсорбционной хроматографии.
АППАРАТУРА
1. ХРОМАТОГРАФИЧЕСКИЕ КОЛОНКИ
Правильный выбор размеров колонок для хроматографического
разделения анализируемых смесей имеет большое значение для обес-
печения четкости разделения, а также влияет на скорость анализа.
Излишне длинные колонки могут чрезмерно затянуть время опыта,
наоборот, слишком короткие не смогут обеспечить достаточного
разделения смеси на составляющие компоненты. Немаловажное
значение имеет также соотношение между длиной и диаметром ко-
лонки.
28
Таблица 7
Растворители и адсорбенты, наиболее часто применяемые
в жидкостной адсорбционной хроматографии
Разделяемые смеси веществ Растворители Адсорбенты
Углеводороды Пентан, петролейный эфир, бензин, изооктан, хлороформ, четыреххлористый углерод, хлорбензол, бензол, этиловый спирт, ацетон ' Активированная окись алюминия, силикагели различных марок, алю- мосиликатный катализа- тор
Галоидопроиз- Пентан, изооктан, петролей- ный эфир, четыреххлористый Силикагели различных
водные углеводо- марок, активированная
родов углерод окись алюминия
Спирты Изопропиловый спирт, бути- ловый спирт, этиловый эфир, хлороформ, диоксан, бензол, петролейный эфир Активированный уголь, окись алюминия, силика- гели
Фенолы Петролейный эфир, бензол, этиловый эфир, этиловый спирт Окись алюминия, окись кальция
Альдегиды и ке- Петролейный эфир, бензол, Окись алюминия, окись
ТОНЫ этиловый эфир, четыреххлори- стый углерод, сероуглерод магния, тальк, силика- гели
Карбоновые кис- Бензол, петролейный эфир, Тальк, активированный
ЛОТЫ этиловый спирт, гептан, иитро- пропан, вода (для низших кис- лот) уголь, окись алюминия, силикагели
Сложные эфиры Петролрйиый эфир, бензол, этиловый эфир, четыреххлори- стый углерод, хлороформ, гек- сан Окись алюминия, си- ликагели, активирован- ный уголь
Хиноны Бензол, гексан, этиловый спирт, ацетои, метиловый спирт Окись алюминия, ки- зельгур
Амины, амиды Петролейный эфир, бензол, четыреххлористый углерод, этиловый эфир Силикагели, окись алюминия
Нитро- и нитро- Бензол, петролейный эфир, Тальк, гидроокись
зосоединения хлористый метилен кальция, карбонат каль- ция, силикагели, окись алюминия
Сульфокислоты Вода Окись алюминия
Углеводы Вода, изопропиловый спирт, этиловый спирт, бутиловый спирт, диоксан, петролейный эфир, бензол, хлороформ Боксит, активирован- ный уголь, силикагели, окись алюминия
Аминокислоты Вода, метиловый спирт, вод- ный раствор формальдегида, этиловый эфир, раствор фено- ла, л-крезол, хлороформ Активированный уголь, силикагели, окись алю- миния, двуокись титана, крахмал
29
Продолжение
Разделяемые смеси веществ Растворители Адсорбенты
Г етероцикли чес- кие соединения Бутиловый спирт, этиловый эфир, хлороформ, петролейный эфир, бензол, этиловый спирт, ацетон, 0,004 н. раствор соля- ной кислоты, вода, уксусная кислота Окись алюминия, гид- роокись кальция, крах- мал, силикагели, кизель- гур, карбонат кальция, тальк, сахар
Алкалоиды Вода, бензол, хлороформ, этиловый эфир, этиловый спирт, ацетон, раствор фенола Окись алюминия, си- ликагели, фуллерова земля
Витамины Петролейный эфир, бензол, вода, эгилацетат, этиловый спирт Окись алюминия, гид- роокись кальция, окись магния
Терпены Четыреххлористый углерод, петролейный эфир, гексан, бен- зол, этиловый эфир, ацетон, метиловый спирт, этилацетат, хлороформ Окись алюминия
Сераорганиче- ские соединения ПетроЛейный эфир, изоок- тан, бензол, спиртобензольная смесь, этиловый спирт, ацетон Силикагели, окись алюминия
К сожалению, в жидкостной адсорбционной хроматографии все
еще отсутствуют какие-либо определенные и теоретически обосно-
ванные критерии для выбора оптимальных размеров и форм хро-
матографических колонок. Поэтому такой выбор обычно произво-
дится опытным путем.
Применяют колонки цилиндрической, конической и телескопичес-
кой формы. Высота их обычно, в зависимости от поставленных це-
лей, колеблется от нескольких сантиметров до 10—20 м, диаметр —
от нескольких миллиметров до 8—20 см. В качестве материала в
лабораторных работах чаще всего применяется стекло, реже сталь,
алюминий, латунь. Использование металла позволяет изготовлять
колонки достаточно больших размеров.
Существенное влияние на качество разделения оказывает состоя-
ние внутренней поверхности материала колонки. Шероховатая по-
верхность уменьшает так называемый стеночный эффект, т. е. про-
никновение анализируемой смеси вдоль стенки колонки без разде-
ления. В этом отношении металлические колонки с необработанной
поверхностью стенок оказываются более выгодными, чем стеклянные.
Выбор материала колонок связан также со свойствами анализи-
руемой смеси и применяемых растворителей. Ни те, ни другие не
должны химически реагировать со стенками колонки.
Решающее значение для четкости разделения компонентов смеси
имеет правильный выбор отношения длины колонки I к ее диаметру d.
30
Многочисленные опытные данные показывают, что наилучшие резуль-
таты обычно достигаются при отношении Hd в пределах 40—100.
Если для разделения смеси необходимо иметь достаточно боль-
шой слой адсорбента, то целесооб-
разно сооружать колонки телеско-
пического типа со ступенчатым
уменьшением диаметра в направ-
лении потока разделяемой смеси
(рис. 9). Число ступеней может
быть различным. Некоторые иссле-
дователи рекомендуют не ступен-
чатое, а плавное уменьшение диа-
метра, т. е. применение конусооб-
разных колонок (рис. 10). Счи-
тается, что лучшие результаты раз-
деления достигаются на колонках
с углом сужения в пределах
15—18°.
Проведение хроматографиче-
ского разделения возможно на ко-
лонках с нисходящим потоком раз-
деляемой жидкой смеси (рис. 11,а),
а также с восходящим потоком
(рис. 11,6). Более четкое разделе-
ние вследствие меньшего влияния
стеночного эффекта достигается
Рис. 10. Хро-
матографиче-
ская колонка
с конусооб-
разным су-
жением:
1, 3 — стек-
лянная вата;
2 — адсорбент
на колонках последнего типа.
Однако более простая конструкция
колонок с нисходящим потоком
обусловила большую распростра-
ненность колонок первого типа.
Работа хроматографических ко-
лонок может осуществляться либо
под давлением, либо в вакууме.
В первом случае подаваемая в ка-
честве промывающего вещества
жидкость находится под давлением
выше атмосферного, тогда как на
выходе из колонки она находит-
ся при атмосферном давлении
(рис. 12, а). Во втором случае ва-
куумный насос подключается к
Рис. 9. Хромато-
графическая колон-
ка телескопическо-
го вида (размеры
в мм):
1—патрубок для при-
соединения к баллону
с газом; 2 — крепле-
ние; 3 — резервуар
для проявителя; 4,
10 — спаи; 5 — ши-
рокая часть колонки;
6 — рубашка для во-
ды; 7 — крепление;
8 — узкая часть ко-
лонки; 9 — пористая
стеклянная перего-
родка; 11, 15 — кра-
ны; 12— шлиф; 13-
копчик. колонки; 14—
прнемннк вытекающе-
го раствора
нижней части колонки, а подаваемая сверху жидкость находится
под атмосферным давлением (рис. 12, б). Опытные данные свиде-
тельствуют о том, что четкость разделения в колонках, работаю-
щих под вакуумом, вследствие более полного удаления воздуха из
пор адсорбента, оказывается выше.
31
При всем многообразии форм применяемых в хроматографии
колонок следует отметить, что во многих случаях нет необходимости
в особом усложнении их конструкции. Очень часто вполне удовлет-
ворительный эффект разделения может быть достигнут при исполь-
Рис. 11. Цилиндрические
хроматографические ко-
лонки:
а — с нисходящим потоком
жидкости; б — с восходящим
потоком жидкости. / — на-
порный. сосуд; 2 — стеклян-
ная вата; 3 — сорбент; 4 —
патрубок для выхода жид-
кости
а 5
Рис. 12. Хроматографические
колонки, работающие при по-
вышенном (а) и пониженном (б)
давлении:
J — от баллона с сжатым газом;
2 — к вакуумному насосу
зовании самых простейших колонок, состоящих из стеклянной
трубки с краном на ее конце, типа бюретки для титрования, укреп-
ленной на обычном штативе. В сложных случаях применение коло-
нок специальной конструкции безусловно необходимо.
При хроматографировании жидких веществ колонки необходимо
устанавливать строго вертикально, так как наличие даже неболь-
шого наклона существенно увеличивает стеночный эффект.
2. УСТРОЙСТВА ДЛЯ РАВНОМЕРНОЙ ПОДАЧИ ЖИДКОСТИ В КОЛОНКУ
Во всех случаях хроматографирования жидкостей необходимым
условием является равномерная подача в хроматографическую ко-
лонку как анализируемой смеси, так и жидкости, применяемой в
32
качестве проявителя или вытеснителя. Случайные изменения ско-
рости течения жидкости вдоль слоя адсорбента, возникающие либо
вследствие потери напора жидкости в питающей емкости, либо по
причине набухания зерен адсорбента или попадания пузырька воз-
духа между зернами адсор-
бента, могут привести к зна-
чительному ухудшению раз-
деления анализируемой сме-
си, размыванию зон отдель-
ных компонентов, а также
затрудняют сравнение резуль-
татов опыта с данными пред-
варительной калибровки. Все
это требует не только осуще-
ствления на протяжении всего
опыта постоянства скорости
потока жидкости, но и непре-
рывного контроля за величи-
ной скорости потока.
В большинстве аналитиче-
ских исследований при хро-
матографировании жидкостей
скорость потока измеряется
несколькими миллилитрами
в минуту. Поэтому постоян-
ство скорости потока жидко-
сти в колонке можно легко
обеспечить поддержанием
столба жидкости над адсор-
бентом на постоянном уров-
не. Это условие может быть
достигнуто несколькими спо-
собами: 1) периодическим под-
ливанием жидкости в верх-
нюю часть колонки, имею-
щую достаточное расширение
(см. рис. 11); 2) при помощи сосуда Мариотта (рис. 13); 3) шприцем
с непрерывным и равномерным движением поршня (рис. 14); 4)
специальными микронасосами.
Периодическое подливание жидкости требует постоянного наб-
людения за ее уровнем в напорном сосуде, не обеспечивает должно-
го постоянства скорости потока и может поэтому применяться толь-
ко в опытах, не требующих высокой степени разделения и тщатель-
ности.
Применение сосудов Мариотта обеспечивает должное постоян-
ство скорости потока и делает систему достаточно «жесткой», так
как создает неразрывный столб жидкости и исключает образование
Рис. 13. Хромато-
графическая колон-
ка со склянкой
Мариотта для не-
прерывной подачи
жидкости под по-
стоянным давле-
нием
Рис. 14. Приспособ-
ление для подачи
жидкости при по-
мощи шприца:
1 — электромотор с
редуктором; 2 — чер-
вячная передача; 3 —
поршень шприца; 4—
шпрнц; 5 — трехходо-
вой кран; 6 — хрома-
тографическая колон-
ка
2 Б. В. Айвазов
33
воздушных буферов в системе. Такое устройство создает, кроме
того, удобство при регулировании скорости потока. Последнее
может быть осуществлено при помощи крана, находящегося на вы-
ходе жидкости из колонки. Однако применение сосуда Мариотта
целесообразно только в тех случаях, когда используются достаточно
большие колонки, требующие для проведения опыта значительных
объемов жидкости (не менее 1 л). Кроме того, при работе с сосудом
Мариотта нельзя применять легко окисляющиеся жидкости. В этом
случае приходится работать в атмосфере инертного газа, что созда-
ет дополнительные трудности при проведении эксперимента.
Для опытов, требующих небольших количеств жидкости и малых
размеров Колонки, целесообразно применение медицинских шпри-
цев, лучше со стеклянным поршнем (см. рис. 14). В этом случае
щприц 4, заполненный промывающей жидкостью, устанавливается
в верхней части колонки 6, причем наличия крана в нижней части
колонки в этом случае не требуется. Равномерное движение поршня
3 шприца осуществляется при помощи синхронного электромотора
1 с червячной передачей 2. Скорость потока регулируется измене-
нием скорости вращения электромотора или передаточного числа
Червячной передачи.
В простейших случаях, а также при незначительных скоростях
потока контроль за величиной скорости может осуществляться либо
путем измерения времени, в течение которого вытекающая из колон-
ки жидкость заполняет сосуд определенного объема, либо путем
подсчета на выходе из колонки числа капель жидкости в единицу
времени. Такой прием, однако, дает возможность узнавать о вели-
чине скорости потока только после того, как пройдет определенное
время опыта, необходимое для заполнения измерительного сосуда
или для подсчета числа капель.
Значительно более удобными являются индикаторы скорости
потока: реометры и ротаметры. Простейшим прибором для изме-
рения скорости потока жидкости может служить обращенный рео-
метр (рис. 15). Такой прибор может быть легко изготовлен из стекла
в любой лаборатории. Применение сменных капилляров позволяет
пользоваться прибором в широком . диапазоне скоростей, кроме
того, он не требует заполнения его специальной измерительной жид-
костью, так как последней является сама жидкость потока. Разность
уровней жидкости в коленах реометра зависит от скорости ее потока,
а также от величины сопротивления капилляра (его диаметра и
длины) и плотности жидкости. Поэтому для каждой жидкости, при-
меняемой при хроматографировании, и для каждого капилляра рео-
метр должен быть предварительно откалиброван. Чтобы устранить
влияние температуры на плотность жидкости, реометр требуется
термостатировать. Устанавливается реометр на пути жидкости
•между напорным сосудом и колонкой.
Более удобным является обращенный ротаметр (рис. 16). При-
менение этого прибора в хроматографии существенно упрощает уст-
34 ’
ройство установки и позволяет измерять скорость потока жидкости'
с достаточной степенью точности. Обращенный ротаметр состоит из
вертикальной трубки с коническим, расширяющимся книзу каналом
2, в котором находится поплавок 3 с диаметром, равным диаметру
верхней узкой части канала. Поплавок обладает меньшей плотно-
стью, чем жидкость, скорость потока которой измеряется. Поэтому
в отсутствие потока поплавок находится в верхней части ротаметра.
Рис. 16. Обращенный
ротаметр (разрез):
/ — вход жидкости; 2 —ко-
нический канал; 3 — по-
плавок; 4 — выход жид-
кости
При наличии тока жидкости поплавок опускается вниз и останавли-
вается на некотором расстоянии от своего первоначального положе-
ния. Расстояние, на которое опустится поплавок, зависит от ско-
рости потока жидкости, угла конуса канала и плотности поплавка
по отношению к измеряемой жидкости. Вследствие этого обращен-
ный ротаметр должен быть предварительно откалиброван для каждой
применяемой жидкости. Удобство обращенного ротаметра состоит
в том, что он может быть установлен не только на входе жидкости
в колонку, но и на выходе, что позволяет избежать лишних ком-
муникаций.
3. УСТРОЙСТВО ДЛЯ ОТБОРА И АНАЛИЗА ХРОМАТОГРАФИЧЕСКИХ ПРОБ
Контроль за хроматографическим разделением анализируемых
смесей может осуществляться тремя способами: а) определением
компонентов разделенной смеси непосредственно в слое адсорбента,
2* .35
без вымывания веществ из колонки; б) последовательным анализом
отобранных порций раствора, вытекающего из колонки; в) непре-
рывным определением изменения концентрации анализируемых
веществ в вытекающем растворе теми или иными химическими или
физико-химическими методами.
В жидкостной адсорбционной хроматографии наиболее часто
пользуются вторым способом, так как непрерывные методы анализа
во многих случаях требуют довольно громоздкой и сложной аппара-
туры и нередко бывают трудно осуществимы, а послойный анализ
возможен только в редких специальных случаях.
Фракционный метод анализа. При фракционном методе анализа
возникает необходимость отбора большого числа проб, объем или
масса которых должны быть известны. Эта операция является очень
трудоемкой и требует для осуществления много времени и внима-
ния. Поэтому большое значение имеют разработанные в Институте
физической химии АН СССР [15] специальные автоматические и
полуавтоматические устройства для отбора проб при хроматографи-
ровании жидкостей. Некоторые из этих устройств выпускаются
промышленностью.
Для отбора проб могут быть применены устройства карусельного
типа, состоящие из диска с укрепленными на нем приемниками —
пробирками или колбами. Возможно применение линейных коллек-
торов, более компактных по конструкции, но сложнее по изготов-
лению. В линейных коллекторах приемники располагаются последо-
вательно один за другим. Как в тех, так и в других коллекторах
хроматографическая колонка располагается над одним из прием-
ников. После заполнения приемника до определенного веса или
объема вытекающей жидкостью под колонку автоматически пода-
ется очередной пустой приемник.
Приспособления для отбора фиксированного количества жидкос-
ти могут базироваться на различных принципах. К обычно приме-
няемым устройствам относятся следующие:
1. Каплесчетные устройства. Действие этих устройств может
быть фотоэлектрическим, контактным, пьезоэлектрическим и др.
Электрический импульс возникает при падении капли жидкости.
Специальная электрическая схема прибора позволяет суммировать
заданное число импульсов, после чего она передает сигнал испол-
нительному механизму карусельного или линейного коллектора,
подставляющему под колонку новый пустой приемник.
2. Устройства, дозирующие по объему. В этих устройствах за-
данный объем жидкости определяет специальный дозатор, в кото-
рый поступает жидкость из хроматографической колонки. Дозатор
снабжен, электромагнитным клапаном и уровнемером. Уровнемер
может быть фотоэлектрическим, контактным или иметь другое уст-
ройство. При достижении в дозаторе определенного уровня электро-
магнитный клапан срабатывает и жидкость стекает в приемник.
Одновременно срабатывает механизм коллектора. Можно приме-
36
нить сифонные дозаторы. Дозировка по объему может производиться
также без дозатора, непосредственно в пробирках, собирающих
фильтрат.
3. Устройства, дозирующие по весу. В основу устройства отбора
проб по весу положен принцип гидростатических весов.
4. Карусельный коллектор с часовым механизмом. В этом устрой-
стве диск с приемниками приводится в непрерывное вращение ча-
совым механизмом. Коллектор снабжен специальным устройством,
при помощи которого капли, не попадающие непосредственно в
приемник, стекают в него с наклонной поверхности, которой снаб-
жены все промежутки между отверстиями приемников. Скорость
потока вытекающей из колонки жидкости, а также скорость враще-
ния карусельного коллектора служит мерой объема собираемой в
Рис. 17. Схема работы проточной рефрактометрической установки:
I — источник света; 2 — призма с растворителем; 3 — проточная призма;
4 — фотоэлементы; 5— усилитель; 6— самопишущий гальванометр
приемник жидкости. Разумеется, что скорость потока должна быть
строго постоянной. Основное преимущество устройства состоит в
независимости его работы от наличия источника электрического
питания.
Что касается передвижения коллекторов, то оно может осуществ-
ляться как от электромоторов, так и при помощи пружин. Во всех
случаях сигнал для срабатывания передвигающего механизма по-
дается от дозатора.
Непрерывный метод анализа. Работа проточных устройств с
целью непрерывного определения концентрации анализируемых
компонентов разделяемой смеси может быть основана на различных
принципах. Известны устройства, работающие на принципе измере-
ния показателя преломления, диэлектрической проницаемости,
электрической емкости, электропроводности, радиоактивного излу-
чения и т. д. Во всех случаях используются проточные кюветы спе-
циальных конструкций.
1. Рефрактометрические кюветы. Одна из возможных схем ра-
боты рефрактометрической установки для непрерывного определе-
ния изменения показателя преломления жидкости, вытекающей из
хроматографической колонки, представлена на рис. 17. Луч света
от источника 1 через диафрагму проходит специальное устройство,
состоящее из двух полых прямоугольных призм, из которых приз-
ма 2 не проточная, а призма 3 проточная. Отклонение луча на оп-
ределенный угол зависит от разности показателя преломления жид-
37
костей, заполняющих полые призмы. По выходе из призмы луч
попадает на два .фотоэлемента 4, соединенные посредством усилите-
ля 5 с самопишущим гальванометром 6. Если оба фотоэлемента
освещены одинаково, то в цепи не возникает электрический ток.
При смещении луча в сторону одного из фотоэлементов гальванометр
будет фиксировать наличие фототока, сила которого будет связана
с величиной отклонения луча. Последняя будет зависеть от отноше-
ния показателя преломления жидкости, заполняющей непроточную
кювету 2, к показателю преломления жидкости, протекающей
через проточную кювету 3.
Если в стационарную кювету поместить жидкость, служащую
проявителем, то в случае протекания через проточную кювету чис-
того проявителя луч света не будет преломляться. При появлении
в проявителе веществ, вымываемых с адсорбента и обладающих
иным, чем проявитель, показателем преломления, будет иметь
место преломление света и луч отклонится.
Конструкция и размеры кюветы могут быть подобраны в зави-
симости от условий проведения эксперимента. Для водных раство-
ров кювета может быть изготовлена из органического стекла. Для
органических растворителей необходимо применять металл. В обоих
случаях для изготовления стеклянных окошек на пути луча света
необходимо применять плоскопараллельные пластинки из оптиче-
ского стекла.
Дифференциальное фотореле может быть собрано по мостовой
схеме. В качестве гальванометра может служить стрелочный нуль-
инструмент или самописец типа ЭПП-09.
2. Денситометрические кюветы. При хроматографировании ок-
рашенных веществ можно применять денситометрические установки.
3. Кондуктометрические кюветы. В хроматографическом анализе
неорганических соединений за изменением концентрации можно сле-
дить по изменению электропроводности раствора. Для этой цели
применяются проточные кондуктометрические кюветы, представ-
ляющие собой небольшой стеклянный сосуд с впаянными платино-
выми электродами. Такая кондуктометрическая ячейка включается
в качестве одного из плеч мостовой схемы, в диагональ которой под-
ключается гальванометр или самопишущий потенциометр. Мост
настраивается на сопротивление ячейки при заполнении ее раство-
рителем. Появление в растворителе хроматографируемых веществ,
изменяющих его электропроводность, вызывает разбалансировку
моста, что и фиксируется самописцем. Во избежание смешения двух
разделенных в колонке веществ кювета должна иметь возможно
малый объем.
4. Диэлькометрические кюветы. Если хроматографическому ана-
лизу подвергаются вещества, диэлектрическая постоянная которых
значительно отличается от ее значения для растворителя, то имеет
смысл применять диэлькометрические кюветы и измерять в анали-
зируемом растворе изменение диэлектрической постоянной. Метод
38
обладает высокой степенью точности, а также достаточно высокой
чувствительностью. Большим преимуществом метода является то,
что отпадает необходимость ввода в кювету посторонних материа-
лов, так как обкладки конденсатора помещаются снаружи кю-
веты.
Проточные кюветы могут иметь разнообразные формы. Простей-
шей является кювета в виде стеклянной трубки с укрепленными на
Рис. 18. Оптическая схема интерферометра:
/, 2— кюветы; 3,4 — компенсирующие пластинки;
5, 6, 7 — лннзы; 8 — диафрагма; 9, 10 — микро-
метрические винты
ее наружных стенках кольцеобразными электродами. По указанным
выше причинам кюветы должны иметь как можно меньший объем.
Ячейка может быть вклю-
чена в любую схему, пред-
назначенную для диэлько-
метрического или высоко-
частотного титрования.
5. Интерферометры.
Наблюдение за изменением
концентрации в анализи-
руемых жидкостях по выхо-
де из колонки можно вести
при помощи проточного жидкостного интерферометра типа ИТР-2
или других типов интерферометров. Принципиальная схема интер-
ферометра приведена на рис. 18. Одна кювета интерферометра за-
полняется чистым растворителем, в то время как через другую не-
прерывно протекает анализируемая жидкость. Изменение свойств
последней по сравнению со свойствами чистого растворителя вызы-
вает смещение интерференционных полос в приборе. Поворотом
специального барабана первоначальное положение полос может
быть восстановлено. В этом случае по величине делений на барабане
интерферометра можно судить об изменениях концентрации. Интер-
ферометрический метод весьма чувствителен. К сожалению, наблю-
дение по ходу анализа производится визуально, а запись—вручную,
что значительно осложняет проведение опыта.
. / 4. ПОСЛОЙНЫЙ АНАЛИЗ НЕПОСРЕДСТВЕННО В КОЛОНКАХ
Классический хроматографический анализ по М. С. Цвету поз-
воляет качественно определять состав смеси по окраске зон адсор-
бента после разделения смеси окрашенных веществ. В этом случае
отпадает необходимость вымывания из колонки компонентов раз-
деленной смеси. Однако этот прием послойного анализа смеси ок-
рашенных веществ имеет существенные ограничения, так как для
его осуществления требуется, чтобы вещества разделяемой смеси
были окрашены, адсорбент бесцветен, а колонка прозрачна. Для
проведения количественного анализа в этом случае необходимо ана-
лизировать каждую зону отдельно после разрезания всего столба
адсорбента на отдельные части соответственно окрашенным зонам.
Поэтому в случае количественных определений метод оказывается
39
весьма громоздким и связан каждый раз с уничтожением хромато-
графической колонки.
Существуют, однако, приемы, позволяющие проводить качест-
венный и даже количественный анализ неокрашенных веществ не-
посредственно в колонке. В этих приемах либо используется спо-
собность некоторых веществ люминесцировать при освещении их
ультрафиолетовым излучением, либо в адсорбент вводятся специаль-
ные индикаторы, флюоресцирующие под действием ультрафиоле-
товых лучей в присутствии тех или иных соединений. Наконец, воз-
можно применение цветных индикаторов, окрашивающих зоны
определенных соединений при действии видимого света.
Метод, основанный на свойстве некоторых веществ люминесци-
ровать под действием ультрафиолетового излучения, принято на-
зывать улыпрахроматографическим анализом. Работы в этой области
были начаты еще в 1933—1934 гг. М. А. Константиновой-Шлезин-
гер [16]. Основы метода применительно к анализу нефтей и нефте-
продуктов были разработаны Ф. М. Эфендиевым [17]. Этот метод
весьма чувствителен и требует для анализа небольших количеств
вещества. После хроматографирования колонку облучают ультра-
фиолетовым светом, под действием которого зоны анализируемых
веществ начинают светиться. Длина светящейся зоны пропорцио-
нальна количеству анализируемого вещества. Расчеты ведут по
результатам нескольких измерений.
Ультрахроматографический метод с флюоресцирующими инди-
каторами был предложен А. Л. Конрадом [18]. Метод применяется
в том случае, когда компоненты анализируемой смеси не люминес-
цируют сами под действием ультрафиолетового облучения. Добав-
ляемые в хроматографируемую смесь специально подобранные ин-
дикаторы образуют окрашенную светящуюся зону при ультрафио-
летовом облучении только в присутствии определенных соединений.
Количество добавляемого индикатора обычно составляет 0,001—
0,0005% от взятого для анализа количества смеси. Правильно по-
добранные индикаторы позволяют определять содержание того или
иного компонента с достаточной степенью точности и с высокой чув-
ствительностью.
Хроматографический анализ с цветными индикато-
рами для анализа бензинов и керосинов был впервые применен
А. Л. Ле-Розеном [19]. Общий ход анализа в этом методе остается
таким же, как и в ультрахроматографическом методе. В этом слу-
чае отпадает необходимость применения источника ультрафиолето-
вого излучения, но требуется специальная подготовка адсорб-
ционных колонок. Перед загрузкой адсорбента в колонку на ее
внутренние стенки в виде узких продольных полос наносится выб-
ранный цветной индикатор. Если одновременно требуется несколько
индикаторов, то каждый из них наносится в виде полос, не сопри-
касающихся друг с другом. После этого в колонку засыпается ад-
сорбент и вводится исследуемая смесь. Количественный состав сме-
40
си рассчитывается на основании данных о величине окрашенных
зон, образующихся в результате взаимодействия анализируемых
веществ с индикатором. Во многих случаях метод позволяет полу-
чать вполне надежные результаты.
Хроматографическое разделение смесей радиоактивных
веществ и специально синтезированных соединений с одним или
несколькими «мечеными» радиоактивными атомами в молекуле так-
же позволяет производить послойный анализ компонентов смеси
непосредственно в колонке. В этом случае
можно применить устройство, схема кото-
рого изображена на рис. 19 [15]. Счетчик
у- или достаточно сильного Р-излучения,
помещенный в свинцовую защиту, пере-
двигается вдоль колонки после того, как
произведено хроматографическое разделе-
ние изучаемой смеси. При наличии в ка-
кой-то зоне колонки радиоактивного веще-
ства счетчик передает соответствующий
сигнал на пересчетное или записывающее
устройство. По интенсивности восприни-
маемого счетчиком излучения можно су-
дить о количестве радиоактивного вещества
в зоне.
В отличие от ультрахроматографическо-
го метода, а также метода с флюоресци-
рующими и цветными индикаторами, тре-
бующими прозрачных хроматографических
колонок, в радиометрическом методе могут
I." 1.3
Рис. 19. Схема прибора
для измерения изменения
радиоактивности вдоль
колонки:
I— колонка; 2,3 — штативы;
4— свинцовая защитная ка-
мера; 5 — блок; 6 — проти-
вовес; 7— щель; 8— счетчик
Гейгера
применяться и непрозрачные колонки, на-
пример, алюминиевые. Однако в этом ме-
тоде должна быть обязательно учтена энер-
гия радиоактивного излучения и в соответ-
ствии с ее величиной выбран материал колонки и толщина ее сте-
нок. Общее требование состоит в том, чтобы стенки колонки погло-
щали как можно меньшую часть радиоактивного излучения.
Выбор того или иного метода хроматографирования, а также
последующего анализа веществ зависит от многих причин. Поэтому
дать какие-то общие рекомендации не представляется возможным.
В каждом отдельном случае, прежде чем остановиться на каком-
либо варианте проведения опыта, экспериментатор должен ясно
представить себе цель своего опыта, требуемую степень точности
анализа, а также оценить возможности лаборатории, в которой бу-
дет поставлен опыт. Только после этого следует остановить свой
выбор на том методе, который поможет решить стоящую задачу с
наибольшей точностью и чувствительностью, а также с наименьшей
затратой труда и времени.
41
ПРАКТИЧЕСКИЕ РАБОТЫ
Работа 1
ОПРЕДЕЛЕНИЕ АДСОРБЦИОННОЙ АКТИВНОСТИ
СИЛИКАГЕЛЯ [14, 20]
Приборы « реактивы
1. Хроматографическая стеклянная колонка с краном, длиной 65 см и диа-
метром 10 мм, с рубашкой, позволяющей омывать колонку термостатированной
жидкостью.
2. Штатив химический с двумя лапками.
3. Термостат типа ТС-15М.
4. Резиновые трубки.
5. Рефрактометр типа Аббе (РЛУ).
6. Набор почвенных сит.
7. Ступка фарфоровая.
8. Склянка, с притертой пробкой емкостью 50 мл.
9. Воронка с длинной ножкой.
10. Пробирки градуированные на 10 мл или мерные цилиндры на 10 мл.
11. Силикагель марки МСМ или КСМ.
12. Раствор бензола в н-гептане 10%-ный по объему. Бензол не должен со-
держать тиофена.
Цель работы. Установить адсорбционную активность выбран-
ного образца силикагеля по отношению к бензолу в сравнимых, стан-
дартных условиях.
Сущность работы. Адсорбционную активность силикагеля при-
нято характеризовать количеством бензола, адсорбированного им
из раствора в гептане. При пропускании такого раствора определен-
ной концентрации через слой силикагеля происходит адсорбция
бензола. После насыщения адсорбента бензолом, адсорбирующимся
значительно лучше гептана, из колонки начинает вытекать раствор,
содержащий наряду с гептаном бензол, т. е. наступает проскок наи-
более сильно адсорбирующегося вещества.
По количеству собранного чистого гептана можно вычислить
отвечающее ему количество бензола, адсорбированного определен-
ным количеством силикагеля. Г. С. Ландсберг, Б. А. Казанский и
др. [14] принимают за адсорбционную активность силикагеля ко-
личество бензола, адсорбируемое 100 г силикагеля из 10%-ного по
объему раствора бензола в н-гептане. Эти исследователи вычисляют
адсорбционную активность силикагеля по формуле:
„ Угса 100 ,1П.
а* ~ (100—c6).g Рб’ ( 9)
где аЛ — адсорбционная активность 100 г силикагеля, выраженная
в граммах бензола; — объем чистого гептана, собранного при вы-
текании его из колонки до момента проскока бензола, мл; сб — содер-
жание бензола в исходной смеси, объемн.%; g— количество взятого
силикагеля, г; рб—плотность бензола,
4?
Для определения активности силикагеля применяется фронталь-
ный хроматографический метод.
Выполнение работы. Измельчают силикагель и отбирают его
фракцию, остающуюся на сите № 0,25 и проходящую через сито
№ 0,50. Тщательно отмывают от пыли, декантируют и высушивают
в сушильном шкафу при 150° С в течение 6 ч.
Навеску подготовленного таким образом силикагеля в количест-
ве 10 г всыпают в колонку, непрерывно постукивая по колонке для
наиболее плотной упаковки в ней адсорбента. Колонку закрепляют в
штативе строго вертикально. Термостатную рубашку присоединяют
к насосу термостата, устанавливают температуру воды термостата
20° С и начинают прокачку термостатной воды через рубашку хро-
матографической колонки.
Проверяют содержание бензола в приготовленном растворе.
Для этого определяют показатель преломления раствора строго при
20° С и, пользуясь графиком, находят по показателю преломления
смеси концентрацию в ней бензола. Такой график можно вычертить
по данным табл. 8 [20]. При определении показателя преломления
Таблица 8
Показатели преломления смесей бензола
с «-гептаном при 20° С
Содержание бензола, объемн. % «20 nD Содержание бензола, объемн. % я20 nD
0,0 1,3878 45,0 1,4352
3,0 1,3905 50,0 1,4408
5,0 1,3927 55,0 1,4462
8,3 1,3961 60,0 1,4520
10,0 1,3977 65,0 1,4578
15,0 1,4029 70,0 1,4638
20,0 1,4081 75,0 1,4697
25,0 1,4132 80,0 1,4788
28,6 1,4171 85,0 1,4819
30,0 1,4187 90,0 1,4881
35,0 1,4242 95,0 1,4946
40,0 1,4296 100,0 1,5010
для термостатирования рефрактометра можно пользоваться тем же
термостатом, что и для термостатирования хроматографической
колонки.
После того как колонка приготовлена, осторожно, через воронку
с длинной ножкой по стенке колонки, чтобы не взмутить слой адсор-
бента, приливают 20 мл раствора бензола в н-гептане. Колонку
закрывают пробкой с хлоркальциевой трубкой и вытекающий из
колонки раствор начинают собирать в градуированную пробирку
или мерный цилиндр. Необходимо с возможно большей точностью
установить момент проскока бензола и замерить объем чистого
гептана. Опыт показывает, что момент проскока можно уста-
43
навливать по появлению хорошо видимых в проходящем свете
диффузионных струек жидкости, возникающих при падении капли,
содержащей бензол, в чистый гептан, находящийся в пробирке.
В этот момент следует прекратить отбор пробы и точно замерить
объем гептана в пробирке. Этот объем и будет величиной Vr в фор-
муле (19). Проверку момента появления бензола можно сделать
рефрактометрически, отобрав несколько капель фильтрата непосред-
ственно с кончика колонки и определив в них количественное содер-
жание бензола, пользуясь графиком, или качественно по положи-
тельному результату формалитовой пробы.
Полученные значения величин Уг, с6 и g подставляют в формулу
(19) и вычисляют значение адсорбционной активности силикагеля.
Чтобы сохранить гептан и бензол для последующих определений .
и одновременно регенерировать силикагель, после определения
объема гептана при проскоке в колонку вливают 3—5 мл этилового
спирта, а вытесняемый при этом раствор бензола в «-гептане соби-
рают в отдельную посуду. После поглощения всего количества спир-
та в колонку вливают дистиллированную воду и продолжают вы-
теснение до тех пор, пока желтое пятно, обычно образующееся на
границе бензола и спирта в столбике силикагеля, не передвинется
к концу колонки. Вытесненная смесь и м-гептан могут быть снова
использованы для определения активности силикагеля.
Регенерацию силикагеля производят кипячением его после вы-
грузки из колонки с большим количеством воды с последующей
сушкой при 150° С.
Работа 2
ОПРЕДЕЛЕНИЕ ИЗОТЕРМЫ АДСОРБЦИИ УКСУСНОЙ
КИСЛОТЫ НА АКТИВИРОВАННОМ УГЛЕ ФРОНТАЛЬНЫМ
ХРОМАТОГРАФИЧЕСКИМ МЕТОДОМ
Приборы а реактивы
1. Стеклянные хроматографические колонки с краном, длиной 25 см, диамет-
ром 10 мм — 5 шт.
2. Штативы химические с одной лапкой — 5 шт.
3. Склянки с притертыми пробками емкостью 100—150 мл — 5 шт.
4. Колбы конические для титрования емкостью 100 мл — 5 шт.
5. Мерные цилиндры на 100 мл — 5 шт.
6. Воронки химические диаметром 3—5 см — 5 шт.
7. Активированный березовый уголь БАУ.
8. Растворы уксусной кислоты: 0,005 н.; 0,01 н.; 0,02 н.; 0,05 н.; 0,1 н.
9. Раствор индикатора, индикаторная бумага.
Цель работы.Получить экспериментальные данные о зависимости
величины удельной адсорбции уксусной кислоты на активированном
угле от ее концентрации в водном растворе и по этим данным по-
44
строить изотермы адсорбции в координатах (с, а0) и Про-
верить совпадение опытных данных с теорией.
Сущность работы. Определение изотермы адсорбции произво-
дится фронтальным методом. При пропускании водного раствора
уксусной кислоты через слой угля происходит ее адсорбция, вели-
чина которой зависит от концентрации подаваемого раствора.
В начале опыта из колонки будет вытекать чистый растворитель
(вода). После насыщения всего слоя адсорбента кислотой произойдет
проскок и в вытекающем из колонки растворе можно определить
присутствие кислоты.
Объем вытекающего из колонки чистого растворителя до прос-
кока, т. е. удельный удерживаемый объем Vr, будет зависеть от
концентрации исходного раствора согласно формуле (6):
а° = Кдс,
где а°— величина удельной адсорбции уксусной кислоты на угле,
ас — исходная концентрация уксусной кислоты в растворе.
Определяя величину удельного удерживаемого объема для раст-
воров различных концентраций, можно рассчитать соответствующие
значения удельной адсорбции а° и, пользуясь этими данными, по-
строить изотерму адсорбции.
Чтобы установить применимость уравнения изотермы адсорбций
Лэнгмюра (5) к изучаемому случаю, экспериментальные данные на-
носятся на график в координатах . Если полученные таким
образом экспериментальные точки лягут на прямую, то, следова-
тельно, адсорбция уксусной кислоты на угле описывается уравне-
нием Лэнгмюра и может рассматриваться как мономолекулярная
адсорбция.
Выполнение работы. На технических весах взвешивают пять
порций березового активированного угля БАУ, предварительно
измельченного и просеянного через сита № 0,50 и 0,25, промытого
водой и просушенного в сушильном шкафу при 105—110° С. Вес
требуемой пробы устанавливается предварительно в зависимости
от размеров колонки.
Каждую порцию угля засыпают в стеклянные хроматографичес-
кие колонки с краном, предварительно поместив в нижнюю часть
колонки небольшой ватный тампон для предотвращения высыпа-
ния угля. Постукиванием колонки о поверхность стола утрамбовы-
вают насыпанный уголь, после чего, во избежание взмучивания
верхних слоев при наливании раствора, уголь прижимают толстой
алюминиевой сеткой. Колонки укрепляют в штативах в строго вер-
тикальном положении. Под каждую колонку подставляют мерный
цилиндр для сбора вытекающего раствора. Колонки, мерные ци-
линдры и склянки с растворами уксусной кислоты нумеруют.
45
Открывают краны на колонках и в каждую колонку вливают
раствор уксусной кислоты в строгом соответствии с номерами скля-
нок и колонок. В процессе хроматографирования следят за тем,
чтобы над слоем угля в колонках постоянно находился раствор,
а также за тем, чтобы не было разрывов в столбе адсорбента и
воздушных пробок. Поворотом кранов устанавливают скорость
фильтрования в пределах 1—2 мл/мин.
По мере вытекания раствора из колонок следят за появлением
в нем кислоты, начиная с колонки с наибольшей концентрацией
раствора. Появление кислоты в вытекающем растворе определяют
по изменению окраски индикаторной бумажки под действием кап-
ли, взятой с кончика колонки. Отмечают с точностью до 0,5 мл
объем чистого раствора, вытекшего до появления заметной концен-
трации кислоты, для каждой колонки. При появлении кислоты
заливку раствора в колонку прекращают.
Зная вес адсорбента g в колонке и объем растворителя VR, вы-
текшего из колонки до появления кислоты, рассчитывают величину
удельного удерживаемого объема Vr для каждой колонки по фор-
муле
Зная исходную концентрацию кислоты в растворе с и значение
VR для каждой колонки, по формуле (6) рассчитывают величину
удельной адсорбции а°. По полученным данным строят график в
координатах (с, а°) и второй график в координатах М- , .
Форма записи
Определение изотермы адсорбции уксусной кислоты на активированном угле
Если экспериментальные точки удовлетворительно ложатся на
прямую второго графика, то это означает применимость уравнения
Лэнгмюра для случая адсорбции уксусной кислоты на угле. В про-
46
тивном случае применяют уравнение Фрейндлиха
а°-КфСп', / (20)
где Кф ин' — константы.
Строя по экспериментальным данным график в координатах
1g с и 1g а0, судят о соответствии уравнения (20) этим данным. По ре-
зультатам наиболее соответствующего экспериментальным данным
графика рассчитывают либо константы уравнения (5) Лэнгмюра
(аю, Ь), либо уравнения (20) Фрейндлиха (Кф,п').
Работа 3
ХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ КЕРОСИНА
КОМБИНИРОВАННЫМ МЕТОДОМ
Приборы « реактивы
1. Хроматографическая колонка телескопического типа (см. рис. 9) с при-
способлением для равномерной подачи проявляющей жидкости.
2. Карусельная установка для автоматического отбора проб (может приме-
няться любой тип установки, например, выпускаемый промышленностью тип
ХККВ-1).
3. Пробирки для сбора проб. •.
4. Рефрактометр типа Аббе (РЛУ).
5. Термостат типа ТС-15М.
6. Колба Вюрца на 100—150 мл с холодильником.
7. Баня водяная.
8. Керосин, свободный от сернистых соединений.
9. Пентан, изопентан или петролейный эфир (фракция 35—60° С).
10. Этиловый спирт 96%-ный.
11. Силикагель марки КСМ, МСМ или ШСМ, измельченный до величины зе-
рен 0,5—0,25 мм, просеянный, промытый и высушенный при 150° С.
12. Дистиллированная вода.
Цель работы. Определение содержания ароматических углеводо-
родов в керосине хроматографическим методом.
Сущность работы. Отделение ароматических углеводородов,
содержащихся в керосине, осуществляется комбинированным хро-
матографированием порции керосина на силикагеле. Парафино-
нафтеновые углеводороды поглощаются силикагелем значительно
слабее ароматических. Поэтому, применяя в качестве проявляющего
растворителя один из легких парафиновых углеводородов — пен-
тан, изопентан или петролейный эфир, можно вымыть из колонки
только парафино-нафтеновую часть керосина. Ароматические угле-
водороды остаются при этом на адсорбенте. Их вымывание может
быть осуществлено при помощи какого-либо вытеснителя, например,
этилового спирта. Зная навеску взятого для анализа керосина и
количество десорбированных ароматических углеводородов, можно
рассчитать их содержание в керосине.
47
Разделение смеси парафиновых и нафтеновых углеводородов
на силикагеле невозможно. Оно может быть осуществлено на акти-
вированном угле.
Выполнение работы. Зная активность выбранного образца си-
ликагеля аа (работа 1) и его количество, загруженное в колонку,
можно приблизительно рассчитать количество керосина, необходи-
мое для загрузки, если известно содержание в нем ароматических
углеводородов. Количество керосина Укер в миллилитрах будет
равно:
VKep = ^> (21)
где g — навеска силикагеля, г; аа— активность силикагеля, ;
сар— приблизительное содержание ароматических углеводородов
в керосине, объемн.%; k0— коэффициент использования силикаге-
ля, обычно принимаемый равным единице.
Взвешенное количество силикагеля засыпают в колонку, соблю-
дая правила, указанные в работе 1. Колонку закрепляют в штативе
карусельной установки строго вертикально.
Рассчитанную по формуле (21) навеску керосина растворяют в
равном объеме петролейного эфира и количественно переносят на
адсорбент в верхнюю часть*колонки. После того, как весь влитый в
колонку раствор впитается в адсорбент, на его поверхность насы-
пают дополнительно 1—2 г чистого силикагеля для предотвращения
взмучивания слоя, содержащего адсорбированные вещества. Если
время для проведения опыта ограничено, то на этой стадии опыт сле-
дует прервать.
Затем в приспособление для равномерной подачи жидкости нали-
вают петролейный эфир и осторожно вводят его в верхнюю часть
колонки. Скорость протекания петролейного эфира должна быть
равной 0,25—0,5 мл/мин. Ее можно установить либо при помощи
крана на выходе из колонки, либо путем подбора соответствующего
диаметра капилляра, которым заканчивается колонка. Обращен-
ный ротаметр для измерения скорости потока следует лучше помес-
тить в поток жидкости между напорным резервуаром и колон-
кой.
Отбор проб производят автоматически, задаваясь либо требуе-
мым объемом, либо определенным числом капель, или же (при
постоянной скорости потока) определенным промежутком времени.
Объем каждой пробы не должен превышать 5 мл.
Для каждой пробы определяют показатель преломления. По-
лученные данные записывают в таблицу и наносят на график в ко-
ординатах: объем раствора — показатель преломления. Получают
выходную хроматографическую кривую проявительного анализа.
В начале опыта показатель преломления будет равен показателю
преломления петролейного эфира. Затем, по мере вымывания пара-
48
фино-нафтеновой части керосина, он будет возрастать и, достигнув
максимума, начнет падать до значения, соответствующего показа-
телю преломления исходного петролейного эфира. При этом прек-
ращают подачу петролейного эфира, продолжая, однако, отбор
проб.
После того, как весь петролейный эфир впитается в адсорбент
и верхний слой адсорбента откроется, в колонку вводят этиловый
спирт (30—50 мл) и начинают вытеснительный анализ адсорбиро-
ванных ароматических углеводородов.
Когда адсорбент пропитается залитым количеством спирта, в
колонку подают дистиллированную воду и продолжают вытеснение.
Отбор проб и определение показателя преломления в них продол-
жают, следя одновременно за передвижением темного кольца, об-
разующегося на границе зоны ароматических углеводородов и эти-
лового спирта.
Как только показатель преломления начинает возрастать, его
определение можно прекратить и следить только за передвижением
темного кольца.
Для того чтобы спирт не попал во фракцию ароматических угле-
водородов, последние собирают в пробирки до появления первой
желтой капли из пограничного кольца. Желательно, чтобы эта кап-
ля уже не попала в ароматические углеводороды, так как она, как
правило, содержит в основном смолистые вещества, а не аромати-
ческие углеводороды. Поэтому на первой желтой капле отбор аро-
матической фракции заканчивают. Стекающие спирт и воду соби-
рают в общую емкость и не используют. Для облегчения регенера-
ции силикагеля его промывают достаточно большим количеством
воды (250—300 мл).
Все пробы, соответствующие ароматической фракции, т. е. про-
бы, начиная с той, для которой показатель преломления оказался
выше показателя преломления петролейного эфира, до последней
пробы, взятой перед появлением желтой капли, объединяют, пере-
носят в предварительно взвешенную колбу Вюрца и нагревают на
водяной бане для удаления оставшегося петролейного эфира. После
отгонки дают остатку остыть, затем взвешивают непосредственно в
колбе. Зная навеску взятого керосина и вес ароматической фрак-
ции, рассчитывают содержание ароматических углеводородов в
керосине в вес.%.
Силикагель подвергают регенерации, как указано в рабо-
те 1.
Для получения более надежных результатов опыт следует пов-
торить и вычислить среднее значение содержания ароматических
углеводородов.
Пробы, содержащие парафино-нафтеновые углеводороды, объе-
диняют и передают для анализа на активированном угле (рабо-
та 4).
49
Работа 4
ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ СМЕСИ ПАРАФИНО-
НАФТЕНОВЫХ УГЛЕВОДОРОДОВ, ВЫДЕЛЕННЫХ
ИЗ КЕРОСИНА
Приборы а реактивы
1. Хроматографическая колонка с восходящим потоком жидкости (рис. 11,6)
длиной 1 м, диаметром 10 мм.
2. Карусельная установка для автоматического отбора проб (любого типа).
3. Пробирки для отбора проб.
4. Склянки с притертыми пробками — 2 шт.
5. Рефрактометр типа Аббе (РЛУ).
6. Термостат типа ТС-15М.
7. Колба Вюрца с холодильником.
8. Баня водяная.
9. Активированный березовый уголь марки БАУ с размерами зерен
0,25—0,50 мм, просеянный, промытый водой и высушенный при 105°С.
10. Петролейный эфир (фракция 35—60° С).
Цель работы. Получить раздельно фракции парафиновых и наф-
теновых углеводородов керосина и по результатам данной и преды-
дущей работ определить содержание парафиновых, нафтеновых и
ароматических углеводородов в керосине.
Сущность работы.Хроматографирование керосина на силикаге-
ле дает возможность количественно выделить только ароматические
углеводороды, но не позволяет получить раздельно парафиновые и
Нафтеновые. Поэтому парафино-нафтеновая фракция, выделенная
из керосина в предыдущей работе, подвергается хроматографирова-
нию на активированном угле марки БАУ. Для увеличения четкости
разделения следует применять колонки с восходящим потоком жид-
кости и достаточной высоты. Применяется проявительный метод,
причем проявителем может служить’ петролейный эфир.
Данные хроматографирования керосина на силикагеле и пара-
фино-нафтеновой фракции на угле позволяют получить представле-
ние о групповом составе углеводородов керосина.
Выполнение работы. Взвешенное количество активированного
угля марки БАУ засыпают в колонку с восходящим потоком жид-
кости так, как это указано в работе 2. Во избежание всплывания
угля его верхний слой должен быть прижат тяжелой металличес-
кой сеткой. Вес угля должен быть больше веса парафино-нафтено-
вой фракции (без растворителя) в 50 раз.
После установления колонки с адсорбентом в строго вертикаль-
ном положении в нее вливают всю полученную в работе 3 парафино-
нафтеновую фракцию, растворенную в петролейном эфире. По окон-
чании впитывания фракции в колонку подают непрерывным потоком
и с постоянной скоростью проявитель — петролейный эфир. Ско-
рость потока не должна превышать 0,5 мл/мин. Сбор фракций про-
изводят автоматически при помощи карусельной установки по 5 мл
50
в пробе. В каждой пробе определяют показатель преломления и по
полученным данным строят выходную хроматографическую кривую
в координатах: объем фракций — показатель преломления. При
этом получается ступенчатая кривая.
При четком разделении показатель преломления фракций снача-
ла возрастает, достигает максимума, затем снижается до значения,
соответствующего исходному петролейному эфиру. Эта часть хро-
матограммы соответствует парафиновым углеводородам.
Затем показатель преломления начинает снова возрастать и,
достигая максимума, снижается. Эта часть хроматограммы должна
соответствовать нафтеновым углеводородам.
Опыт заканчивают, как только показатель преломления после
второго снижения достигнет значения показателя преломления пет-
ролейного эфира.
Пробы, соответствующие первой, парафиновой фракции, объеди-
няют, сливают в предварительно взвешенную колбу для перегонки и
отгоняют петролейный эфир на водяной бане. Остаток после остыва-
ния колбы взвешивают. Получают вес парафиновых углеводородов.
С пробами, соответствующими второй, нафтеновой фракции, по-
ступают аналогичным образом. Получают вес нафтеновых углеводо-
родов.
Зная вес исходного керосина (работа 3), рассчитывают содержа-
ние в нем ароматических, нафтеновых и парафиновых углеводоро-
дов в вес.%. Определяют показатель преломления для всех трех
выделенных фракций углеводородов.
По данным работ 3 и 4 подсчитывают баланс опыта.
Форма записи
Баланс опыта по хроматографированию керосина
Фракции керосина Получено Показатель прелом- ления
г %
Ароматические углеводороды Нафтеновые углеводороды Парафиновые углеводороды
Всего Взято Потери 100
Примечание. Расход вещества, пошедшего на определение показателя
преломления, должен учитываться при вычислении веса каждой фракции.. По-
этому для анализа следует брать определенное его количество.
51
Работа 5
ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ СМЕСИ НОРМАЛЬНЫХ
И ИЗОПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
НА МОЛЕКУЛЯРНЫХ СИТАХ
Приборы и реактивы
1. Стеклянная хроматографическая колонка с краном, длиной 180 см, диа-
метром 1 см.
2. Штатив химический с двумя лапками.
3. Колбы плоскодонные на 200 мл — 2 шт.
4. Молекулярные сита типа СаА.
5. Рефрактометр проточного типа или типа Аббе (РЛУ).
6. Ректификационная колонка или колба с елочным дефлегматором.
7. Колбы плоскодонные на 150—250 мл —2 шт.
8. Смесь н-октана с изооктаном (2,2,4-триметилпентан) в соотношении 1 : 1
по объему.
9. Изопентан.
10. н-Пентан.
Цель работы. Пользуясь особым «просеивающими свойством цео-
литов, разделить смесь нормального и изопарафинового углеводоро-
дов на индивидуальные компоненты и определить количественное
содержание каждого из них.
Сущность работы. Экспериментальные данные свидетельствуют
о том, что молекулярные сита с размерами соединяющих «окон»
порядка 5 А сорбируют парафиновые углеводороды нормального
строения, тогда как углеводороды, имеющие разветвленную струк-
туру, не сорбируются. Объяснение этому явлению находят в том,
что наличие разветвленной цепи не позволяет такому углеводороду
проникнуть сквозь «окно» цеолита и, следовательно, сорбция в
этом случае не может произойти.
Пользуясь этим свойством молекулярных сит, можно разделять
смеси нормальных и разветвленных парафиновых углеводородов,
если пропускать их смесь через слой цеолита определенной длины.
На этом и основано разделение смеси октана нормального и изо-
строения. В каждом отдельном случае успех разделения обусловли-
вается прежде всего правильным выбором типа цеолита, т. е. выбо-
ром соответствующих размеров «окон».
Выполнение работы. Хроматографическую колонку заполняют
цеолитом типа СаА (5 А), измельченным и просеянным через сито
№ 0,50 и задержанным на сите № 0,25. Колонку устанавливают
строго вертикально. Осторожно, по стенкам вводят в колонку
10 .мл искусственной смеси н-октана и изооктана (2,2,4-триметилпен-
тан). После пропитки цеолита введенным раствором начинают про-
мывание колонки изопентаном. Последний легко вымывает несор-
бировавшийся изооктан и оставляет на цеолите н-октан.
52
Вытекающий из колонки раствор направляют в проточную приз-
му рефрактометра непрерывного действия [15]. Раствор, выходящий
из призмы, собирают в колбу. В постоянную призму рефрактометра
заливают чистый изооктан. Применение рефрактометра проточного
типа позволяет значительно увеличить чувствительность измерения.
В зависимости от типа применяемого рефрактометра результаты
измерений могут быть записаны автоматически на ленте самописца
или получены непосредственным отсчетом отклонения преломлен-
ного луча. В последнем случае выходная хроматографическая кри-
вая должна быть вычерчена на миллиметровой бумаге.
Как только показатель преломления вытекающего раствора
после некоторого возрастания упадет до значения, соответствующего
показателю преломления чистого изопентана, промывание изопента-
ном заменяют промыванием н-пентаном, который обеспечивает де-
сорбцию н-октана. При этом в постоянную призму рефрактометра
заливают н-пентан, а для сбора раствора подставляют другую колбу.
После того, как показатель преломления раствора достигнет ве-
личины показателя преломления н-пентана, промывание колонки
прекращают.
После окончания хроматографирования каждый из растворов
подвергают ректификации для удаления растворителей. После от-
гонки растворителя остаток взвешивают и рассчитывают содержа-
ние каждого из компонентов смеси в весовых процентах. Определяют
показатель преломления выделенных соединений и сравнивают по-
лученные данные с табличными значениями. По полученным дан-
ным судят о чистоте выделенных веществ.
Работав
ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ СМЕСИ
ТРАНС- И ЦИС-АЗОБЕНЗОЛА НА АКТИВИРОВАННОЙ
ОКИСИ АЛЮМИНИЯ [21]
Приборы и реактивы
1. Хроматографическая стеклянная колонка длиной 200 мм И диаметром
20 мм.
2. Штатив химический с лапкой.
3. Баллон с азотом.
4. Кварцевая лампа.
5. Колбы плоскодонные емкостью 100 и 250 мл — 3 шт.
6. Аппарат Сокслета.
7. Колба Вюрца на 250 мл с холодильником.
8. Кристаллизаторы — 2 шт.
9. Окись алюминия активированная для хроматографии.
10. Азобензол.
53
11. Петролейный эфир (фракция 35—60° С).
12. 1%-ный раствор метилового спирта в петролейном эфире. (Яд!)
13. Сульфат натрия безводный (х. ч.).
Цель работы. Количественное отделение цис-формы от транс-
формы хроматографическим методом.
Сущность работы. Цис-изомер азобензола адсорбируется на окиси
алюминия значительно сильнее, чем его транс-изомер. Поэтому,
применяя метод промывания, можно сравнительно легко вымыть
из колонки транс-азобензол и полностью отделить его от цис-изо-
мера, который остается адсорбированным на окиси алюминия.
Последний может быть удален с адсорбента либо применением более
сильного вымывающего вещества, либо экстракцией его непосред-
ственно из порции адсорбента, извлеченной из колонки. После пере-
кристаллизации из петролейного эфира можно получить чистый
цис-азобензол с т. пл. 71° С.
Выполнение работы. 1 г товарного азобензола (транс-форма с
т. пл. 68° С) растворяют в 50 мл петролейного эфира. Раствор осве-
щают кварцевой лампой, установленной на расстоянии 30—40 см,
в течение 30 мин. (Работать в темных очках!) В ре-
зультате облучения часть транс-азобензола изомеризуется в
ifuc-азобензол, вследствие чего раствор окрашивается в красный
цвет.
Весь раствор вводят в колонку, заполненную активированной
окисью алюминия. После того как весь раствор впитается в адсор-
бент, колонку промывают 100 мл петролейного эфира. Раствор, со-
держащий транс-азобензол, собирают в колбу и затем переносят в
установку для отгонки петролейного эфира. После отгонки раство-
рителя и перекристаллизации получают кристаллы чистого транс-
азобензола, которые взвешивают на технических весах.
Оставшийся на адсорбенте цис-нзомер образует резко очерченную
красную полосу длиной 4—5 см. Для его извлечения колонку слегка
просушивают продуванием через нее азота, а затем извлекают ок-
рашенную часть адсорбента шпателем с загнутым концом и перено-
сят извлеченный адсорбент в аппарат Сокслета. Экстракцию произ-
водят 150 мл 1%-ного раствора метилового спирта в петролейном
эфире. По окончании экстракции отмывают из экстракта водой
метиловый спирт, сушат экстракт безводным сульфатом натрия,
фильтруют и отгоняют петролейный эфир под уменьшенным дав-
лением, так чтобы температура в колбе не превышала 22° С. Выде-
лившиеся оранжево-красные кристаллы цис-азобензола перекрис-
таллизовывают из холодного петролейного эфира, после чего чистые
кристаллы взвешивают и рассчитывают выход обоих изомеров из
взятой навески азобензола.
Все работы с азобензолом (облучение и хроматографирование)
следует проводить в атмосфере азота, для того чтобы предотвратить
возможное окисление азобензола в азоксибензол.
54-
Работа 7
ХРОМАТОГРАФИЧЕСКОЕ ОПРЕДЕЛЕНИЕ АЛКАЛОИДОВ
В ТИНКТУРЕ БЕЛЛАДОННЫ [22]
Приборы и реактивы
1. Хроматографическая стеклянная колонка длиной 15 см, диаметром
1,5 см.
2. Ультрафиолетовый осветитель (например, типа ОУ-1).
3. Штатив химический с лапкой.
4. Колбы конические для титрования емкостью по 100 мл—3 шт.
5. Пипетка иа 2 мл.
6. Бюретка иа 5 или 10 мл с делением до 0,01 мл.
7. Силикагель марки КСМ, ШСМ или МСМ, измельченный до 150—170 меш.
8. Спирто-водный раствор белладонны.
9. Спирт ректификат (96%-иый).
10. Титрованный раствор серной кислоты (0,01 и).
11. Титрованный раствор едкого натра (0,01 и.).
12. Насыщенный водный раствор Хинин-сульфата.
Цель работы. Выделение алкалоидов, содержащихся в тинктуре
белладонны, в чистом виде и количественное их определение.
Сущность работы. Проявление адсорбированных силикагелем
алкалоидов, содержащихся в тинктуре белладонны, водно-спирто-
вым раствором позволяет выделить в чистом виде три алкалоида:
атропин, гиосциамин и скополамин. Их обнаружение возможно
благодаря свойству этих алкалоидов светиться при освещении их
растворов ультрафиолетовым светом. Количественное определение
путем титрования растворов серной кислотой в присутствии серно-
кислого хинина также производится при облучении титруемого
раствора ультрафиолетовыми лучами. Метод обладает высокой точ-
ностью, требует незначительного количества анализируемого ве-
щества, а также затраты небольшого времени для производства
анализа.
Выполнение работы. 1 мл спирто-водного раствора белладонны
наносят на силикагель, которым предварительно заполнена хрома-
тографическая колонка. После того как весь раствор впитается в
адсорбент, начинают промывание 96%-ным этиловым спиртом.
Промывание ведут при непрерывном освещении колонки ультрафио-
летовым светом в затемненном помещении. При этом верхний слой
адсорбента имеет серо-коричневую окраску, несколько ниже видна
узкая серо-зеленая зона, еще ниже — бледная красно-коричневая.
Нижняя часть адсорбента светится бледно-голубым светом.
Раствор собирают в виде трех фракций: первую — светящуюся
бледно-голубым оттенком, вторую — красно-коричневым и третью
— серебристо-зелено-гойубым. Первая фракция соответствует ат-
ропину, вторая — гиосциамину и третья — скополамину.
Количественное содержание'каждого из выделенных алкалоидов
определяют титрованием. С этой целью в каждую колбу с фракциями
55
добавляют по 2 мл титрованного 0,01 н. раствора щелочи и по одной
капле раствора хинин-сульфата. Раствор освещают ультрафиолето-
вом светом и титруют 0,01 н. H2SO4. Вначале растворы не светятся.
При достижении значения pH 4,5 появляется ясно видимое свече-
ние: голубое для атропина и гиосциамина и зеленовато-голубое для
скополамина. Появление свечения считается концом титрования.
Концентрация алкалоида рассчитывается по количеству кислоты,
которое требуется для нейтрализации избытка щелочи, непрореа-
гировавшей с алкалоидом. Титр раствора серной кислоты может
быть установлен как по титрованным растворам чистых алкалои-
дов путем их титрования в ультрафиолетовом свете, так и по титро-
ванному раствору щелочи в присутствии хинин-сульфата.
Работа 8
ЛЮМИНЕСЦЕНТНО-ХРОМАТОГРАФИЧЕСКОЕ ИЗУЧЕНИЕ
СОСТАВА НЕФТИ
Приборы и реактивы
1. Хроматографическая стеклянная колонка длиной 15 см, диаметром
1,5 см.
2. Ультрафиолетовый осветитель типа ОУ-1 или кварцевая лампа со свето-
фильтром.
3. Люминескоп — прибор, позволяющий наблюдать свечение зон под дейст-
вием ультрафиолетового излучения [17, 23].
4. Штатив химический с лапкой.
5. Линейка с делениями.
6. Карандаш по стеклу.
7. Окись алюминия для хроматографии.
8. Нефть для исследования.
Цель работы. Определить люминесцентно-хроматографическим
методом соотношение между основными группами веществ, состав-
ляющих нефть: асфальтенами, смолами, маслами и легкими фрак-
циями (керосинами и бензинами).
Сущность работы. Люминесцентно-хроматографический метод
основан на способности различных групп веществ, составляющих
нефти, люминесцировать под действием ультрафиолетовой радиации
различным свечением. При этом характер люминесцентных хрома-
тограмм закономерно связан с химическим составом веществ, об-
разующих светящиеся зоны. Известно, например, что темно-коричне-
вые зоны соответствуют соединениям типа асфальтенов; коричневые
зоны связаны с наличием смолистых веществ; желтое или желто-
зеленое свечение вызывается масляными фракциями, а зоны голу-
бого или фиолетового свечения относятся к легким фракциям. Было
замечено также, что длина зоны той или иной окраски пропорцио-
нальна количеству данной группы веществ.
56
Поэтому люминесцентно-хроматографический метод позволяет
качественно или полуколичественно за сравнительно короткое вре-
мя определить относительное содержание наиболее важных веществ,
составляющих нефть, и тем самым получить представление о характе-
ре и основных свойствах нефти.
Сочетание люминесцентно-хроматографического метода с проя-
вительным позволяет определить не только качественное, но и ко-
личественное содержание основных групп веществ, составляющих
нефть. Метод может быть применен для анализа не только нефтей,
но и битумов, масел и некоторых других нефтепродуктов.
Выполнение работы. Хроматографическую колонку на длину
140 мм заполняют окисью алюминия для хроматографии. Колонку
устанавливают в штативе строго вертикально, после чего в нее
вливают отвешенное количество нефти (2—3 г). Для ускорения
впитывания нефти в адсорбент можно применить отсос через нижний
конец колонки водоструйным насосом.
После полного впитывания нефти в адсорбент колонку снимают
со штатива и помещают в люминескоп, освещая источником ультра-
фиолетового излучения. Наблюдают отдельные зоны свечения и
отмечают границы этих зон при помощи карандаша для записи по
стеклу. Записывают цвет люминесценции каждой зоны и прекра-
щают облучение.
Колонку вынимают из люминескопа и измеряют длину каждой
зоны. Для нефтей обычно наблюдается 3—4 зоны. Для полуколи-
чественного расчета содержания отдельных зон сумму всех длин зон
принимают за 100 и затем рассчитывают процентное содержание
каждой зоны. Считая плотность каждой зоны одинаковой (что допус-
тимо только для полуколичественных расчетов), можно принять,
что относительная длина зоны будет соответствовать относительному
содержанию тех веществ, которые обусловили то или иное свече-
ние. Анализ повторяют два-три раза и берут средние значения от-
носительного содержания веществ для каждой зоны. Результаты
анализа вносят в таблицу.
Форма записи
Люминесцентно-хроматографический анализ нефти
№ опыта Масса про- бы нефти, г Общая дли- на зон, см Цвет и длина зоны
черно-корич- невая коричневая желтая голубая
см % см 1 % см % см о/ /0
1 2 3
Среднее — — — — — —
57
Работай
ОЧИСТКА ХЛОРБЕНЗОЛА И ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ
В НЕМ ПРИМЕСИ ХЛОРНОГО ЖЕЛЕЗА
ХРОМАТОГРАФИЧЕСКИМ МЕТОДОМ
Приборы и реактивы
1. Хроматографическая колонка длиной 150 см, диаметром 10 мм.
2. Фотоколориметр типа ФЭК-М.
3. Штатив химический с лапкой.
4. Колбы конические для титрования емкостью 150 мл — 2 шт.
5. Хлорбензол с примесью хлорного железа.
6. Силикагель марки МСМ измельчения 0,25—0,5 мм.
7. Раствор роданида аммония (10%-ный).
8. Раствор соли железа (эталонный).
9. Раствор азотной кислоты (1 : 1).
Цель работы. Фронтальным хроматографическим методом на
силикагеле получить хлорбензол, свободный от примеси хлорного
железа и определить содержание этой примеси.
Сущность работы. В процессе хлорирования бензола с целью
получения хлорбензола на железном катализаторе образуется хлор-
ное железо, которое загрязняет продукт хлорирования. Содержание
хлорного железа обычно бывает незначительным, вследствие чего
удаление его обычными методами затруднено.
Принцип хроматографической очистки состоит в том, что если
примесь обладает значительно большим адсорбционным сродством
к выбранному адсорбенту, чем основное вещество, то при длитель-
ном пропускании смеси через слой адсорбента примесь накапли-
вается в колонке до тех пор, пока адсорбент полностью не насы-
тится ею. После этого произойдет проскок примеси и опыт следует
прекратить.
Вымывая затем с адсорбента поглощенную примесь каким-либо
сильно десорбирующим растворителем, можно получить раствор
со значительно более высокой концентрацией примеси и определить
ее количественное содержание обычными аналитическими методами.
Таким образом можно решить сразу три задачи: очистить вещество
от примеси, определить содержание этой примеси и получить ее
в концентрированном виде.
Выполнение работы. Хроматографическую колонку заполняют
силикагелем, предварительно очищенным от примесей железа ки-
пячением с раствором соляной кислоты. Через колонку непрерывно
пропускают хлорбензол, содержащий небольшую примесь хлорного
железа. Время от времени с кончика колонки отбирают в пробирку
несколько капель хлорбензола, добавляют к ним раствор роданида
аммония и несколько капель азотной кислоты. Появление слабо-
розовой окраски роданида железа (III) свидетельствует о начале
проскока хлорного железа. При отрицательной реакции на ион
Fe3+ фильтрование хлорбензола продолжают.
58
При проскоке прекращают подачу хлорбензола, дают ему стечь
с адсорбента и замеряют его количество. Затем в колонку вводят
дистиллированную воду и вытесняют адсорбированное хлорное же-
лезо. При подаче в колонку воды сбор фильтрата начинают в от-
дельную колбу. Промывную воду контролируют на содержание
иона Fe3+. При отрицательной реакции промывание водой прекра-
щают.
Отделяют от водного слоя хлорбензольный и в водном слое опре-
деляют содержание иона Fe3+ фотоколориметрнческим методом с
роданидом аммония.
Эталонный раствор соли железа для получения калибровочной
кривой приготовляют следующим образом. Навеску 0,864 г невы-
ветренных железо-аммонийных квасцов (х. ч.) растворяют в воде,
подкисленной 5 мл конц. H2SO4, и доводят объем раствора водой до
1 л. Приготовленный таким образом раствор содержит 0,1 мг же-
леза в 1 мл.
Зная количество пропущенного через слой адсорбента хлорбен-
зола и содержание железа в водном фильтрате, рассчитывают коли-
чество примеси хлорного железа в хлорбензоле.
ЛИТЕРАТУРА
Использованная литература
1. М. С. Цвет. Хроматографический адсорбционный анализ. Изд-во
АН СССР, М., 1946.
2. R. Kuhn, A. Winterstein, Е. Lederer. Z. physiol. Chem.,
1931, 197, 141
3. A.J.P. Martin, R. L.M. Synge. Biochem. J., 1941,35, 1358.
4. T. Б. Г а и о n, E. H. Гайон, Ф. M. Шемякин. ДАН СССР,
1947 58 595
5. E. H. Г а п о н, Т. Б. Гайон. ДАН СССР, 1948, 60, 401.
6. А. Т. J a m е s, A. J. Р. Martin. Analyst, 1952, 77, 915; Biochem. J.,
1952, 50, 679.
7. А. Кейлеманс. Хроматография газов. ИЛ, М., 1959. стр. 27.
8. А. А. Ж у х о в и ц к и й. Н. М. Т у р к е л ь т а у б, Т. В. Геор-
гиевская. ДАН СССР, 1953, 92, 987.
9. А. А. Жуховицкий, Н. М. Туркельтауб. Зав. лаб,,
1956, № 10, 1252.
10. С. К л а с с о н. Адсорбционный анализ смесей. Госхимиздат, М.—Л.,
1950, стр. 74.
11. Д. П. Тимофеев. Кинетика адсорбции. Изд-во АН СССР, М., 1962.
12. В. Н. 3 р е л о в, Г. И. Кичкин. Хроматография в нефтяной и
нефтехимической промышленности. Гостоптехиздат, М., 1963, стр. 23—28.
13. В. А. Соколов, Н. С. Торочешни ков, Н. В. К е л ь ц е в.
Молекулярные сита и их применение. Изд-во «Химия», М., 1964.
14. Г. С. Л а н д с б е р г и др. Определение индивидуального углеводород-
ного состава бензинов прямой гонки комбинированным методом. Изд-во АН СССР,
М., 1959, стр. 48, 64.
15. К. В. Ч м у т о в, В. Г. А в г у л ь. Автоматические приборы в коло-
ночном хроматографическом анализе. Изд-во АН СССР, М., 1961.
16. М. А. Константинов а-Ш лезингер. Люминесцентный ана-
лиз. Физматгиз, М., 1961,
59
17. Ф. М. Эфендиев. Люминесцентный метод исследования нефтей и
битумов. Гостоптехиздат, Баку, 1953.
18. A. L. С о n г a d. Analyt. Chem., 1948, 20, № 8, 725.
19. A. L. Le Rosen, P. H. M о n a g п а п, C. A. Rivet,
E. D. Smith, H. A. Suter. Analyt. Chem., 1950, 22, № 6, 809.
20. А. Ф. Плата. Краткое руководство к практикуму по химии нефти.
Изд-во МГУ, 1960, стр. 34.
21. Н. А. Ф у к с. В сб. «Реакции и методы исследования органических
соединений», кн. 1, Госхимиздат, 1951, стр. 211.
22. В. С. Краснова. ЖПХ, 1945, 18 , 284.
23. В. Н. Ф л о р о в с к а я, В. Г. Мелков. Введение в люминесцент-
ную битуминологию. Гостоптехиздат, М., 1946.
Рекомендуемая литература
1. «Хроматография». Сб. 1, ИЛ, М., 1949.
2. М. М. С е н я в и н. Усп. хим., 1949, 18, вып. 2, 183.
3. Н. А. Фукс. Усп. хим., 1949, 18, вып. 2, 206.
4. «Исследования в области хроматографии». Изд-во АН СССР, М., 1952.
5. В. В. Рачинский, Т. Б. Гапон. Хроматография в биологии.
Изд-во АН СССР, М„ 1953.
6. Ф. М. Шемякин, Э. С. М и ц е л о в с к и й, Д. В. Романов.
Хроматографический анализ. Введение в теорию и практику. Госхимиздат, М.,
1955.
7. Н. Ф. Ермоленко. Хроматографический адсорбционный анализ
и его развитие. Изд-во АН БССР, 1955.
8. «Состав и свойства нефтей и бензино-керосиновых фракций». Изд-во
АН СССР, М., 1957.
9. «Состав и свойства высокомолекулярной части нефтей». Изд-во АН СССР,
М„ 1958.
10. «Исследования в области промышленного применения сорбентов». Изд-во
АН СССР, М„ 1961.
11. В. В. Рачинский. Введение в общую теорию динамики сорбции
и хроматографии. Изд-во «Наука», М., 1964.
ГЛАВА И
ИОНООБМЕННАЯ ХРОМАТОГРАФИЯ
Ионообменная хроматография приобрела за последние десятиле-
тия первостепенное значение как метод препаративного разделения
и аналитического определения самых различных смесей неоргани-
ческих и органических соединений. В основе ионообменной хрома-
тографии лежит обратимый стехиометрический обмен ионов, содер-
жащихся в хроматографируемом растворе, • на подвижные ионы
веществ, называемых ионитами или ионообменниками. Разделение
смеси содержащихся в растворе ионов основано на неодинаковой
способности их к обмену с ионами ионита.
Между ионообменной хроматографией и адсорбционной моле-
кулярной имеется существенное различие. Если молекулярная ад-
сорбционная хроматография основана на явлении адсорбции, под-
чиняющейся в первом приближении теории Лэнгмюра, то ионообмен-
ная основана на стехиометрическом обмене ионов раствора с ионами
ионита. В соответствии с этим вымывание адсорбированных веществ
в молекулярной хроматографии может производиться чистым раст-
ворителем, тогда как в ионообменной в качестве вымывающего ве-
щества необходимо применять растворы электролитов.
Четкой грани между этими двумя методами, однако, провести
нельзя, так как обычные адсорбенты часто действуют так же, как
иониты, а на ионитах частично имеет место физическая адсорбция.
Несмотря на это, ионообменная хроматография обладает существен-
ными специфическими особенностями и должна рассматриваться
как самостоятельный раздел хроматографического метода.
По знаку заряда обменивающихся ионов иониты разделяются
на катиониты, или катионообменники, и аниониты, или анионооб-
менники. Существуют также амфотерные иониты, способные осу-
ществлять одновременный обмен катионов и анионов. Такие иониты
называются амфолитами.
Можно представить, что ионит состоит из каркаса, связанного
валентными силами или же силами решетки [1]. Каркас обладает
положительным или отрицательным зарядом, который компенсиру-
ется зарядом ионов противоположного знака, так что в целом ионит
нейтрален (рис. 20). Ионы, компенсирующие заряд каркаса, носят
61
название противоионов. Способность ионита к обмену обусловли-
вается тем, что противоионы обладают определенной подвижностью
в пределах каркаса.
Если ионит, содержащий противоионы только одного вида А,
поместить в раствор, в котором находятся ионы другого вида В,
/{-матрица с фиксй- © -противоионы
Р рованными ионами
Рис. 20. Схематическое изобра-
жение структуры синтетической
ионообменной смолы
то ионы А будут покидать ионит и
переходить в раствор, а ионы В бу-
дут в строго эквивалентном коли-
честве переходить в ионит. При
достижении равновесия ионит и
раствор будут содержать ионы А и
В в определенном количественном
соотношении, определяемом кон-
стантой ионообменного равнове-
сия.
В порах ионита содержатся не
только противоионы, но и раство-
ритель и растворенные вещества.
Поэтому наряду с обменом в
ионите происходят и такие про-
цессы, как набухание, вызываемое
поглощением растворителя, и ад-
сорбция растворенных веществ, при поглощении которых вместе
с противоионами в ионит может проникнуть эквивалентное коли-
чество подвижных ионов того же знака, что и заряд каркаса.
Такие ионы носят название коионов. Следовательно, содержание
противоионов в ионите определяется не только величиной заряда
каркаса, но и содержанием коионов.
Описанная модель достаточно полно отражает важнейшие свой-
ства ионитов и объясняет, почему обмен является стехиометричес-
ким процессом. Эта модель показывает, что ионный обмен основы-
вается на статистическом распределении противоионов между
ионитом и раствором, в котором не участвуют ни вещество каркаса,
ни коионы. Поэтому такой ионообменный процесс можно иллюстри-
ровать, например, следующими реакциями:
катионный обмен
RAn-H + +Na + +CrztRAn-Na + -f H + -J-C1-
анионпый обмен
RKt+OH-+№+4-Cl- RKt+Cl-4-Na + +OH-
Здесь R— полимерный радикал, образующий вместе с ионо-
генной группой каркас ионита, а Ап” и Kt+ — ионогенная группа или
фиксированный ион, обусловливающий заряд каркаса.
В связи с тем, что свойства ионита зависят от природы его про-
тивоиона, при характеристике ионита целесообразно указывать,
62
какой именно ион является противоионом. Если, например,
противоионами какого-либо катионита являются ионы Н + , то го-
ворят, что этот катионит находится в водородной форме (Н-форме).
Анионит, для которого противоионом является, например, хлорид-
ион, будет находиться в хлоридной форме (Cl-форме).
Статистическое распределение противоионов различных видов
между ионитом и раствором должно было бы обусловить одинаковые
соотношения между концентрациями этих ионов в обеих фазах после
установления равновесия. В действительности же это условие не
выполняется, вследствие чего имеется возможность путем ионного
обмена практически полностью избавляться от какого-либо иона в
растворе. Важнейшими причинами этого являются следующие.
1. Силы электростатического взаимодействия между заряженным
каркасом и различными противоионами неодинаковы; оказывает
значительное влияние величина заряда ионов.
2. Кроме чисто электростатических сил, проявляются и другие
силы взаимодействия между ионом и окружающей средой. В ионите
и растворе эти силы могут значительно различаться.
3. Противоионы крупных размеров по чисто стерическим при-
чинам не могут проникать в тонкопористые иониты.
Перечисленные причины, вызывающие преимущественное пог-
лощение ионитом противоионов одного вида, определяют селектив-
ность ионита. Последнее свойство, зависящее от природы ионита,
а также условий проведения эксперимента (температура, pH раст-
вора и др.) является важнейшим для ионитов и обусловливает
широкие возможности применения ионообменников для решения
ряда практических задач.
Обменная емкость ионитов. К числу важнейших свойств ионитов
относится их обменная емкость. Полная обменная емкость данного
ионита является постоянной величиной и определяется в первую
очередь числом фиксированных ионов, т. е. ионов, определяющих
заряд каркаса, так как их электрический заряд в каждый данный
момент времени и на любом участке ионита должен компенсиро-
ваться зарядом противоионов. Поэтому в идеальных условиях пол-
ная обменная емкость определенного количества данного ионита
является величиной постоянной, не зависящей от состояния ионита
и от природы противоиона. В реальных условиях она зависит от
ряда факторов, в частности от pH раствора, что усложняет одно-
значное определение этого понятия. Поэтому при определении об-
менной емкости необходимо указывать условия, при которых она
определена.
Обменная емкость, определяемая в статических условиях, может
отличаться от величины, полученной в динамических условиях.
Последняя характеризуется двумя показателями: динамической
обменной емкостью до проскока (ДОЕ) и полной динамической обмен-
ной емкостью (ПДОЕ). ДОЕ представляет собой емкость ионита,
определяемую по появлению данного иона в вытекающем из колонки
63
растворе. ПДОЕ определяется по полному прекращению извлече-
ния данного иона из раствора. Это различие можно пояснить гра-
фически. ДОЕ определяется площадью прямоугольника (рис. 21, а),
основанием которого является объем (в литрах) раствора, вытекаю-
щего из колонки до наступления проскока, а высотой — исходная
концентрация обменивающегося иона (вмг-экв/л). ПДОЕ выражается
площадью над выходной хроматографической кривой (рис. 21,6) и
может быть определена как интеграл
ь
SaMbN~ 5 У dx-
а
Сравнение заштрихованных площадей (см. рис. 21,а и б) показы-
вает, что ПДОЕ больше, чем ДОЕ.
Объем фильтрата У,л
а
Рис. 21. Схема определения динамической обменной
емкости до проскока (а) и полной динамической обменной емкости (б)
Любое определение емкости относится к данному количеству
ионита, либо к его весу (весовая емкость), либо к его объему (объем-
ная емкость). В научной литературе обменную емкость обычно вы-
ражают в миллиграмм-эквивалентах сорбируемого иона на грамм
отмытого от сорбированных веществ сухого ионита, находящегося в
водородной (для катионитов) и хлоридной (для анионитов) форме.
Отнесение обменной емкости к Cl-форме анионита обусловлено тем,
что вес сухого анионита в ОН-форме определить непосредственно
нельзя. В технике распространение получила величина обменной
емкости, выраженная в килограммах СаО на 1 м9 насыпного объема
набухших зерен насыщенного и отмытого ионита.
ИОНООБМЕННОЕ РАВНОВЕСИЕ
Равновесия, устанавливающиеся при ионообменных процессах,
представляют большой практический и теоретический интерес.
В первом приближении ионообменное равновесие может быть
описано законом действия масс.
64
Из уравнения реакции обмена двух одновалентных ио-
нов А+ и В +
AZ+B+ BZ+A +
согласно закону действия масс следует
[BZ][A + ] „ [BZ]^ „ [BJ. ,99
[AZ] [В + ] А а,в или [AZ] ДА,в [A-]’
и если твердую фазу обозначать чертой сверху, то
[в ]_г/- [В + ] /по\
[А+] Ка-в[А + ]’ (23)
где [А + ] и [В + ] — концентрация ионов в твердой фазе.
Здесь /Сд^— коэффициент избирательности, или константа
ионного обмена.
Аналогично для реакции обмена двухвалентного иона
на одновалентный
2AZ+B2+BZ2+2A +
применение закона действия масс даст соотношение
[B2+]_^ [B2 + ]
[А + ]2 лА.в [А + ]2 •
(24)
Более точно условие ионообменного равновесия описывается
уравнением Б. П. Никольского [2]:
где ад, «в —активности соответствующих ионов в ионите; ад, ав—
активности ионов в растворе; ?д, £в — заряды обменивающихся
ионов.
Из приведенных уравнений следует, что равновесное отношение
концентраций илй активностей ионов в твердой фазе является ли-
нейной функцией соответствующего отношения в жидкой фазе, ра-
зумеется, с учетом величины зарядов ионов. Если эту зависимость
изображать графически (рис. 22) и по оси абсцисс откладывать рав-
новесные отношения в растворе, а по оси ординат — равновесные
отношения в твердой фазе, то можно получить изотермы ионного
обмена (если равновесие достигнуто и измерения произведены при
постоянной температуре). Прямолинейность изотермы указывает
на соблюдение закона действия масс, если же изотерма представляет
собой кривую, это означает, что имеются отклонения от этого закона.
Для разбавленных растворов обычно имеет место удовлетворитель-
ное выполнение закона действия масс.
3 Б. В. Айвазов
65
Константа ионного обмена равна тангенсу угла наклона прямой,
изображающей изотерму ионного обмена, к оси абсцисс. Физический
смысл константы ионного обмена заключается в том, что она поз-
воляет дать количественную характеристику способности ионита
к обмену с различными ионами из раствора. При этом возможны три
случая.
1) Ла, в > 1 — ион, находящийся в растворе, имеет большее срод-
ство к иониту, чем ион, первоначально соединенный с ионитом. Об-
мен из раствора будет протекать доста-
точно полно.
2)Ла, в < 1 — ион раствора имеет
меньшее сродство, чем ион ионита. Обмен
будет незначительным.
3) Ла, в =1 —в этом случае сродство
обоих ионов одинаково.
Таким образом, различие в величинах
констант ионного обмена является важ-
Рис. 22. Изотерма ионного
обмена:
“aZA „ “aZA
Х= а^В ; У = -Ъ1/гВ
D D
нейшим фактором, обусловливающим
применение ионообменников для разде-
ления смесей ионов.
Применение комплексообразователей.
Большие возможности для улучшения
разделения смесей дает применение комп-
лексообразователей. Так называются ве-
щества, образующие с катионами или
анионами хроматографируемой смеси комплексные соединения раз-
личной прочности.
Применение комплексообразователей позволяет, во-первых, от-
делить одни катионы от других, если выбранный комплексообразо-
ватель образует комплексные соединения только лишь с частью из
них. В этом случае катионы, не прореагировавшие с комплексообра-
зователем, будут обладать константой ионного обмена, существенно
отличающейся от константы ионного обмена образовавшегося ком-
плекса. Возможен и такой случай, когда реагирующие с комплексо-
образователем катионы образуют комплексный анион и поэтому,
не обмениваясь с противоионами катионита, вымываются из колонки
первыми же порциями растворителя.
Во-вторых, именно в комплексных соединениях наиболее зна-
чительно проявляются тонкие различия в величинах ионных радиу-
сов, строении электронных оболочек, величинах констант нестой-
кости и т. п. Поэтому перевод, например, ионов металлов в комплекс-
ные ионы позволяет значительно увеличить различие в константах
ионного обмена и тем самым существенно улучшить разделение сме-
сей близких по свойствам ионов. В. частности, таким путем были
разделены смеси катионов редкоземельных элементов и получены
наиболее чистые препараты их соединений с очень близкими свойст-..
вами.
66
Важно также и то, что величины константы нестойкости очень
многих комплексов значительно зависят от pH среды, что существен-
но облегчает проведение процесса разделения.
Очевидно, что для разделения смеси катионов в том случае, ког-
да образовавшиеся комплексы представляют собой анионы, следует
применять аниониты.
Наряду с галогенидами, сравнительно легко образующими ком-
плексные соединения с ионами многих металлов, в качестве комплек-
сообразователей применяют такие вещества, как лимонную, винную,
молочную кислоты, а также аммиак, глицерин, трилон Б и др.
ДИНАМИКА ИОННОГО ОБМЕНА
Количественная теория хроматографического процесса ионного
обмена основывается на законах динамики сорбции веществ. Под
динамикой сорбции мы понимаем развитие процесса поглощения
вещества в условиях его прохождения через слой сорбента.
Если предположить, что в хроматографическую колонку с по-
стоянным сечением и постоянной плотностью пористого материала
(сорбента) введен определенный объем смеси веществ известной кон-
центрации, то задача будет состоять в том, чтобы найти функцию
распределения каждого вещества по длине колонки. Эта задача ре-
шается на основе уравнения баланса В. В. Рачинского [3].
Допуская, что диффузия вещества вдоль колонки отсутствует,
на основании закона сохранения вещества В. В. Рачинский выводит
уравнение, определяющее количество /-того растворенного вещества,
поступившего в элементарный слой сорбента в колонке толщиной
dx за время А/
/ яг. \ dSf „ дс:
ср„Ы = ( с.-Д- -g^dx ) v„\t-\-dx-5d А/ Д-Vndx-^7-&t. (26)
1 р \ ' 1 ox j Р 1 ot 1 dt '
Здесь х— расстояние от начала слоя сорбента в колонке до данного
сечения, измеряемое условно весом пористого материала (сорбента),
г; t— время, мин\ ир— скорость поступления раствора или раство-
рителя, мл/мин; Vn—удельный объем пор, мл/г\ с(—концентра-
ция г-того вещества в растворе, д«М/л/л; S°— количество г-того
вещества, поглощаемое единицей веса сорбента, мМ1г.
Физический смысл отдельных членов уравнения (26) заключается
в следующем:
(с< + ср — количество вещества, перенесенного из дан-
ного элементарного слоя dx за время А/;
dx-g— — количество вещества, поглощенного в элементар-
ном слое dx за время А/;
дс'
V® dx А/ — количество вещества, оставшегося в порах эле-
ментарного слоя dx за время А/ в растворенном состоянии.
3*
-07
Поделив обе части уравнения (26) на v^tdx, получим уравнение:
дс; V® дс:
___L -I 0—-
дх 1 ир dt
1 dS,9
—=о
Up dt
или, при постоянной скорости течения,
дс; 0 дс, dSQ
______________________L д_ у® —!_ д_L = о
дх ‘ dV rdV и
(27)
(28)
где V—объем поступившего в колонку раствора или растворителя,мл.
Уравнения (26), (27), (28) носят название уравнений баланса.
С учетом кинетики сорбции полная система уравнений, описывающих
хроматографический процесс обмена, будет иметь вид
дс, . дс; д$9
<29)
= fz(S», S®, .... S®, с1( с2, ... , с„), (30)
где 1<г<п (п— число компонентов смеси веществ).
Решая эту систему уравнений, необходимо вывести функции
распределения веществ по длине слоя сорбента, т. е. следует найти
Ci = ft(x, V) I
S? = F,.(x, V) I
(31)
Первая из этих функций должна выражать распределение вещества
в растворе, вторая — в сорбированном состоянии.
Точный расчет ионообменной хроматограммы может быть выпол-
нен при помощи уравнений, полученных интегрированием системы
дифференциальных уравнений, описывающих динамику ионного
обмена [4]. Здесь же мы рассмотрим только приближенный метод
расчета простейшей системы, состоящей из трех одновалентных
ионов.
Взаимное расположение зон каждого иона разделяемой смеси,
а также их перемещение вдоль слоя сорбента при вымывании может
быть рассчитано при помощи метода, разработанного Е. Н. Талоном
и Т. Б. Талон [5J. Рассмотрим кратко основы этого метода на приме-
ре разделения смеси двух одновалентных ионов В+ и С+.
При хроматографическом ионообменном разделении смеси двух
ионов в обмене будут участвовать три иона, включая противоион
ионита А + . Теория допускает, что обмен каждой пары ионов проис-
ходит независимо от присутствия других ионов.
В случае обмена двух ионов будут иметь место следующие обмен-
ные реакции:
AZ+BY BZ+AY
AZ+CY CZ+AY
BZ+CY 5±CZ + BY
68
Соответственно каждому из этих трех уравнений можно написать
три уравнения изотерм ионного обмена:
|вЧ
[А + ]
-КА
[В + ]
[A + J
1С+]_ Кк [С+]
1с+] к
ШЧ=7<В’С
[с+]
[В + ] ’
(32)
где /Сд, в! Ка, с и Ав, с— константы ионного обмена.
Сделаем допущение, что ионообменное равновесие устанавли-
вается практически мгновенно. Это означает, что при прохождении
раствора через слой ионита время установления ионообменного рав-
новесия меньше времени нахождения в данном объеме поступающего
раствора. Следствием этого допущения является независимость
распределения обменивающихся ионов по длине колонки от скорости
прохождения раствора через слой сорбента.
Пусть в колонке находится ионит AZ, насыщенный противоионом
А+. При прохождении через колонку раствора, содержащего ве-
щества BY и CY, ион А+ будет обмениваться на ионы В+ и С+. При
этом раствор будет обогащаться веществом A Y и протекать по колон-
ке вниз. В этом случае в любом бесконечно малом слое сорбента
будет устанавливаться равновесие. Однако оно будет все время
нарушаться вследствие того, что поступающий сверху свежий ра-
створ будет приходить в контакт с ионитом, более богатым ионом
А + , чем это соответствует равновесному состоянию.
При рассмотрении распределения вещества по слою сорбента
колонка, согласно методу, предложенному Е. Н. Гапоном, разби-
вается на i элементарных слоев, причем для каждого такого слоя
по уравнению (32) рассчитывается сорбционное равновесие. Для по-
лучения надежных результатов необходимо брать достаточное ко-
личество слоев.
Для удобства расчета в уравнения [32] введем безразмерные ве-
личины. Обозначим первоначально сорбированное количество иона
через S0; первоначальную концентрацию иона в растворе с°; емкость
сорбента Sm; приращение количества сорбированного иона при
переходе от исходного состояния к равновесному Sp. Тогда, в силу
эквивалентности обмена
5а+5в+5с= Sa + SB + Sc = Sm = const;
ca+cb+cc=ca +cb + cc = 2c° = 2c = const.
Эквивалентные доли веществ в сорбированном состоянии (а) и в
растворе (иг) для иона А+ обозначим
оО
Я А — ,
SA
a a = c-
CA
1Па-2с-
69
Приращение количества сорбированного иона на единицу ем-
кости при переходе от исходного состояния к равновесному будет
равно
Для других ионов можно получить аналогичные выражения.
Кроме этого, введем удельную пористость сорбента Д° = и
Sc® Г 2 ”1
У° “о“ — • Тогда безразмерной величиной, называемой ионным
L^J
отношением колонки, будет величина
Д°у°
gS,„ '
(33)
Оно показывает соотношение между количеством ионов в объеме
раствора V мл и в навеске сорбента g г. Замена этими безразмерными
величинами соответствующих величин в уравнениях (32) приведет
к следующим новым уравнениям:
1 ав ~ас Д’у’/Яд + dB + dc
ав + <*в А’ В Д0?0™^ —dB
1 ав ~ас~^в _£> Д ° у °/п д -J- d в -J- dc
аС~~de А’ С Д0?0™^. — dc
При помощи этих уравнений можно рассчитать значения dB и
dc при заданных других величинах, а также значения равновесных
Рис. 23. Изменение относительной концент-
рации иоиов А+, В+ и С+ по длине слоя
ионита 1:
а — противоионы в иоиите; б — иоиы в растворе,
находящиеся в равновесии с ионитом
долей mt и для любого
слоя сорбента, если осталь-
ные величины заданы.
Рассмотрим результаты
расчета хроматограмм для
одного конкретного случая.
Примем К а, в= ЮО, а
Ка.с = 1- Это означает,
что ионы А+ и С+ облада-
ют одинаковой способ-
ностью к обмену, а ион
В+ обменивается с ионом
А+ в 100 раз сильнее, чем
ион С+.
На рис. 23, а представ-
лено изменение концент-
рации противоионов по длине слоя ионита в колонке после
нанесения на него определенной порции анализируемой смеси
BY и CY, рассчитанное по уравнениям [341 и [35]. На рис. 23, б
70
представлено распределение концентрации ионов в растворе,
находящемся в равновесии с ионитом. По оси абсцисс откладывается
длина слоя ионита в колонке, по оси ординат — относительное
S/
содержание каждого иона на ионите и, = или каждого иона в
с.-
растворе 1П: = .
Верхние слои колонки находятся в равновесии с исходным эк-
вимолекулярным раствором BY и CY. Поэтому они имеют состав
99% BZ и 1% CZ без примеси AZ. В порах верхних слоев ионита
равновесный раствор имеет состав 50% BY и 50% CY, также без
примеси AY. Затем идет узкая переходная смешанная зона, содер-
жащая ионы В+ и С+. В переходной зоне содержание иона В+ резко
падает и затем быстро становится равным нулю; содержание иона
С+ резко возрастает и достигает максимума; содержание иона А +
становится величиной, отличной от нуля, и затем быстро возрастает.
За смешанной зоной, в которой можно обнаружить все три иона,
лежит зона ионов А+ и С+. Ионит в этой зоне содержит AZ и CZ,
причем содержание первого возрастает по длине колонки, а послед-
него падает. В конце колонки содержание AZ становится равным
100%, так как здесь ионит еще не приходит в соприкосновение с раст-
вором. В растворе, находящемся в порах ионита в смешанной зоне,
концентрация ионов В+ падает до нуля, а концентрация ионов С+
резко возрастает, достигая максимума. В порах следующей зоны
находится раствор, содержащий CY и AY. Вещество BY в этой зоне
отсутствует.
Длительное промывание ионита раствором электролита приво-
дит в конце концов к разделению зон и последовательному вымыва-
нию из колонки всех компонентов раствора.
ЭЛЕКТРОХРОМАТОГРАФИЧЕСКИЙ МЕТОД
А. А. Шабанов, В. И. Горшков и Г. М. Панченков [6] совместили
хроматографический метод разделения смеси ионов на ионитах с
электрофорезом. Такой метод был назван ими электрохроматогра-
фическим. В этом методе на слой ионита в колонке накладывается'
электрическое поле, направление которого может совпадать или
быть противоположным направлению движения зон смеси разделяе-
мых ионов. При электрохроматографическом разделении смеси ка-
тионов зоны в колонке, в которой на выходе находится анод, тор-
мозятся под действием электрического поля.
Авторы метода рассчитывают линейную скорость движения каж-
дой зоны сорбированных веществ без наложения электрического поля
нхр по нижеследующей формуле
___ UpC Ср с
(36)
71
где up—линейная скорость движения вымывающего раствора,
см/мин-, vp — объемная скорость движения вымывающего раствора,
мл/мин; с — концентрация вымывающего иона, мг-экв/мл; q —
обменная емкость ионита, мг-экв/мл; S — поперечное сечение колон-
ки, см2; К а, в — константа ионного обмена иона, образующего зону,
на ион вымывающего раствора.
Если связь между объемом раствора VMaKC, требующимся для
того, чтобы вымыть зону данного иона из колонки до появления
максимальной концентрации иона и константой ионного обмена
ЛА, в, выразить уравнением:
_______ К А, В?^
макс___7.
(37)
где I — высота слоя ионита в колонке, то, исходя из уравнения (36),
можно получить, что
и
=
хр V
v макс
(38)
Отсюда следует, что при помощи уравнения (38) можно рассчи-
тывать скорость движения каждой зоны разделяемой смеси ионов
без наложения электрического поля, если известно положение мак-
симумов каждого иона на выходной кривой.
Можно предположить, что скорость движения зоны в случае
наложения электрического поля ихр ,эл будет равна разности двух
величин, действующих в противоположных направлениях: хромато-
графической составляющей скорости ихр, т. е. скорости зоны без на-
ложения электрического поля, и электрофоретической составляющей
иэл, т. е. скорости движения зоны катиона, происходящего только
под действием электрического поля с градиентом потенциала Е
(в/см). При этом иэл равно
«эл = 60^, (39)
где А — эквивалентная электропроводность иона в твердой фазе
ионита, см21г-экв-ом; F— константа Фарадея, к!г-экв.
Тогда
«Хр., ЭЛ «Хр «ЭЛ' (40)
При помощи этого уравнения можно рассчитывать скорости дви-
жения зон ионов разделяемой смеси в электрохроматографическом
методе. Кроме того, из уравнения (40), пользуясь уравнениями
(38) и (39), можно вывести зависимость Кмакс от приложенного
напряжения Е:
---60—, (41)
хр., эл у у р > \ /
vмакс кмакс Г
72
где VMaKC — положение максимума зоны при наложении электри-
ческого поля. Из уравнения (41) следует, что
—— = —--------(42)
V V Flv '
к макс к макс 1 tvp
т. е. 1/Имакс является линейной функцией от Е.
Таким образом, электрохроматографический метод позволяет
изменять условия разделения смеси ионов путем воздействия на
процесс электрическим полем и тем самым дает дополнительную
возможность для разделения смеси даже очень близких по свойствам
ионов.
МЕТОДЫ ИОНООБМЕННОЙ ХРОМАТОГРАФИИ
В ионообменной хроматографии, так же как и в адсорбционной
молекулярной, можно применять как фронтальный и вытеснитель-
ный методы, так и проявительный. Рассмотрим особенности этих
методов.
Фронтальный метод применяется главным образом для очистки
или извлечения примесей. В качестве примера рассмотрим анализ
смеси электролитов BY, CY и DY на ионите, насыщенном противо-
ионами А+. Допустим, что по способности к обмену ионы можно
расположить в ряд: A+<B+<C+<;D + . Тогда ионит будет хуже всего
поглощать ионы В+ и лучше всего ионы D + . Поэтому вначале, при
приливании раствора в колонку с ионитом, ионы В+ будут переме-
щаться быстрее остальных ионов и будут концентрироваться в зоне,
непосредственно следующей за зоной ионов А+. Если колонка дос-
таточно длинна и в смеси содержится достаточное количество ионов
В+, то после полного вытеснения из колонки ионов А+ в вытекающем
растворе прежде всего появляются ионы В + . Ионы D+ удерживают-
ся ионитом в большей степени, чем ионы С+. Поэтому спустя неко-
торое время в хроматографическом растворе на выходе из колонки
наряду с ионами В+ появятся ионы С+.
Верхние слои ионита, через которые непрерывно протекает ра-
створ, содержащий ионы В + ,С+ и D + , приходят в равновесие с ра-
створом. Таким образом, в результате ионного обмена верхние слои
ионита будут пропускать раствор без изменения его состава. Когда
зона, в которой ионит пришел в равновесие с раствором, дойдет до
нижней части колонки, то начнут проскакивать и ионы D + . После
этого вытекающий раствор будет иметь первоначальный состав.
Таким образом, фронтальный анализ позволяет получить только
первую фракцию, содержащую противоион А+, остальные фракции
будут содержать, наряду с ионами А+ , другие ионы анализируемого
раствора соответственно способности ионов к обмену, а затем уже
пойдет исходный раствор, свободный от противоионов А + . Фронталь-
ный анализ на ионитах отличается от фронтального адсорбционного
анализа тем, что при ионном обмене общая концентрация вытекаю-
щего раствора (в эквивалентах) всегда остается постоянной, тогда
73
как при адсорбции она с каждым новым фронтом ступенчато повы-
шается.
Вытеснительный метод в ионообменной хроматографии нашел
себе большее применение, чем в адсорбционной. Анализируемую
смесь ионов подают в колонку в виде отдельной пробы, а затем про-
изводят вытеснение раствором такого электролита, ион которого
обладает наибольшим сродством к выбранному иониту.
Пусть ионит насыщен противоионом А+, а в анализируемом
растворе имеются ионы В + , С+ и D + . В качестве вытеснителя при-
меним раствор, содержащий ион Е+. Ионы расположены по способ-
ности к обмену в ряд: A+<B+<C+<D+<E+. Анализируемый ра-
створ прежде всего приходит в равновесие с верхними слоями иони-
та. После этого ионит промывается раствором, содержащим ион Е + .
Последний вытесняет из верхних слоев все другие ионы и пере-
двигает их перед собой, образуя четко выраженный фронт.
В протекающий раствор наиболее интенсивно проникают ионы
А + , тогда как в ионите наиболее прочно удерживаются ионы D + .
Поэтому зона, в которой находится смесь ионов, при прохождении
через колонку обогащается в нижней своей части ионами А+,
а в верхней —ионами D + . Остальные ионы соответственно способ-
ности к обмену располагаются полосами, следующими непосред-
ственно одна за другой и перемещающимися с одинаковой ско-
ростью. В хроматографическом фильтрате ионы появляются в той
последовательности, в которой они располагаются в вышеприведен-
ном ряду. Естественно, что в реальных условиях всегда образуются
переходные зоны, содержащие два соседних иона; размеры этих
смешанных зон тем меньше, чем правильнее выбраны условия
опыта. Ион вытеснитель Е+ появится в фильтрате лишь после того,
как все ионы в ионите будут заменены на ион Е+.
От адсорбционного вытеснительного анализа метод существенно
не отличается и может применяться главным образом для препара-
тивного разделения смеси ионов в больших количествах.
Проявительный метод является наиболее распространенным
методом ионообменной хроматографии. Для пояснения метода рас-
смотрим разделение смеси трех ионов В + , С+ и D+ на ионите, насы-
щенном противоионами А+. Вымывание проводят раствором, содер-
жащим тот же ион, которым насыщен ионит, т. е. раствором соеди-
нения AY. Допустим, что обменный ряд ионов остается прежним,
т. е. A+<B+<C+<D + . Таким образом, вымывающий противоион
А+ менее прочно связан с ионитом, чем все остальные ионы смеси.
Порцию смеси подают в верхнюю часть колонки и после дости-
жения равновесия проводят вымывание раствором AY. Четко вы-
раженного фронта образоваться в этом случае не может, поскольку
ионит предпочтительнее поглощает ионы В + , С+ и D+ и поэтому
удерживает их прочнее, чем ион А+. В связи с этим ионы А+ опере-
жают ионы смеси. Несмотря на это за счет постоянного поступления
новых порций раствора AY все ионы, поглощенные из раствора иони-
74
том, в конце концов замещаются на ионы А+, причем вначале это
замещение происходит в верхних слоях ионита. В связи с тем, что
ионы В+ занимают в обменном ряду второе место после А+, они
вымываются быстрее остальных ионов. Наиболее трудно вымы-
ваются ионы D+. Эти различия приводят к тому, что смесь ионов
разделяется в колонке на отдельные полосы, каждая из которых
содержит только ионы одного вида. Полосы перемещаются с различ-
ной скоростью. Быстрее будет перемещаться зона, содержащая ион
В+, медленнее зона иона С+ и последней будет зона иона D + . Помимо
ионов В + , С+ и D + , каждая зона содержит вымывающие ионы А+.
В процессе передвижения вдоль слоя ионита расстояние между
зонами увеличивается, вследствие чего, при достаточной длине
колонки, между зонами соответствующих ионов возникают зоны,
в которых содержатся только ионы вымывающего вещества. Обра-
зуется обычная проявительная хроматограмма такого же типа, как
и при адсорбционном разделении молекул веществ.В вытекающем
растворе одна за другой появляются отдельные порции раствора,
содержащие ионы В+, С+ и D+ на фоне ионов А+.
На практике по возможности следует создавать такие условия,
при которых не происходит перекрывания полос. С другой стороны
нужно избегать слишком большого их растягивания и чрезмерно
больших расстояний между ними. Это достигается правильным
выбором условий проведения опыта, т. е. выбором ионита, вымы-
вающего иона, установлением соответствующей длины слоя ионита,
скорости вымывания и т. п. В процессе вымывания можно менять,
скачкообразно или непрерывно, состав вымывающего вещества или
же применять комплексообразователи. Все это позволяет достигать
весьма четкого разделения смеси, состоящей из большого числа
различных ионов.
ИОНИТЫ
Свойствами ионитов обладает большое число различных природ-
ных и синтетических веществ. Важнейшими из них являются синте-
тические смолы, угли и некоторые минеральные иониты. Несмотря
на то, что все иониты построены по одному типу, отдельные их виды
обладают различными свойствами, что обусловливает широкие воз-
можности применения ионитов в самых различных областях практики.
Минеральные иониты. Природные минеральные иониты
являются, как правило, кристаллическими силикатами, жесткая
решетка которых несет избыточный заряд. Наиболее важными пред-
ставителями этой группы ионитов являются цеолиты, способные
к обмену катионов. К ним относятся минералы:
анальцим Na[Si2A10e]-Н2О
шабазит (Са, Na2)[Si2A10(;]2-6H20
гармотом (К2, Ba) [Al.2Si5O14]-5H2O
гейландит Ca[Si3A10s]2-5H20
натролит Na2[Si3Al2O1(l]-2Н2О
75
Они обладают правильной пространственной сетчатой структурой
со сравнительно большими расстояниями между узлами решетки.
Роль противоионов играют ионы щелочных и щелочноземельных
металлов, которые не связаны с какими-либо определенными
местами в решетке.
Некоторые алюмосиликаты, обладающие катионообменными
свойствами, имеют рыхлую слоистую структуру. Их противоионы
находятся в межплоскостных пространствах. К ним отно-
сятся: монтмориллонит Al2[Si4O10(OH)2]-nH2O и бейделлит
А12 [ (ОН) 2 AlSi 3О6ОН ] • 4Н 2О.
Некоторые представители алюмосиликатов, например, монт-
мориллонит, обладают способностью к анионному обмену. Однако
единственным минеральным анионитом, когда-либо применявшим-
ся в качестве анионообменника, является гидроксилапатит
[Са5(РО4)3]ОН.
Цеолиты имеют жесткую по сравнению с другими ионитами
структуру, вследствие чего они слабо набухают, а подвижность их
противоионов оказывается мала. Большие катионы и нейтральные
молекулы из-за своих размеров не могут проникать в ионит. Благо-
даря этому свойству цеолиты могут применяться как ионные или
молекулярные сита.
Синтетические неорганические иониты. Известны две группы
неорганических синтетических ионитов: плавленые пермутиты и
гелеобразные пермутиты. Они представляют собой гидратирован-
ные алюмосиликаты.
Плавленые пермутиты получают сплавлением смеси соды, по-
таша, полевого шпата и каолина. Несмотря на то, что плавленые
пермутиты имеют неправильную структуру, они очень сходны
с цеолитами.
При приготовлении гелеобразного пермутита к сернокислому
раствору сульфата алюминия и жидкого стекла добавляют раствор
едкого натра. Выпавший алюмосиликатный студень высушивают.
При сходном химическом составе с цеолитами такие гели обладают
неправильной структурой геля, подобной структуре органических
синтетических ионитов.
В качестве неорганического ионита может служить специально
активированная окись алюминия. Способ получения окиси алюми-
ния, способной к ионному обмену, разработан Е. Н. Гапоном и
Г. М. Шуваевой [7]. Он состоит в получении окиси алюминия осаж-
дением из раствора алюмината натрия. Получаемому таким образом
катиониту соответствует формула [(А12О3)Х-A1071Na+. Обрабатывая
такой катионит 2 н. раствором азотной кислоты, получают анионит
[(AI2O3)x-A1O+1NO3-.
Кроме пермутитов и окиси алюминия, из синтетических неорга-
нических ионитов следует назвать гели железа и обладающие анио-
нообменными свойствами гели циркония.
Иониты на основе углей. Некоторые сорта каменных углей,
76
мягкие и твердые бурые угли обладают свойствами слабокислых
ионитов и могут применяться даже без специальной обработки.
В них в качестве противоионов действуют главным образом подвиж-
ные карбоксильные группы гуминовых составляющих. Гелеобраз-
ные бурые угли, жирные каменные угли и блестящие бурые после
обработки их растворами едкого натра и соляной кислоты обладают
хорошими катионообменными свойствами, также определяемыми
наличием подвижных карбоксильных групп.
Сульфирование бурых, каменных углей и антрацитов дымящейся
серной кислотой позволяет вводить в угли подвижные сульфо-
группы, а также, после окисления, карбоксильные группы. Суль-
фирование способствует протеканию реакций поликонденсации и
превращает уголь в гель. Благодаря этому иониты на основе суль-
фированных углей (сульфоугли) приближаются по своим свойствам
к синтетическим органическим ионитам. Однако по сравнению
с последними, сульфированные угли обладают менее определен-
ными свойствами, неоднородным составом, а также меньшей хими-
ческой стойкостью, особенно по отношению к щелочам.
Иониты на основе синтетических смол. Наибольшее практиче-
ское значение имеют синтетические органические иониты, получае-
мые на основе полимерных веществ — синтетических смол. Эти
иониты получили всеобщее признание и широкое применение
вследствие их большой химической и механической прочности,
высокой обменной емкости, а также большого разнообразия свойств
и, прежде всего, высокой селективности и избирательности.
Синтетические ионообменные смолы представляют собой типич-
ные гели. Их каркас, так называемая матрица, состоит из непра-
вильной высокополимерной пространственной сетки углеводород-
ных цепочек. В матрице закреплены группы, несущие заряд, так
называемые фиксированные ионы. У катионитов это чаще всего
группы — SOj",—COO-,—РО^-, — AsO^-; у анионитов группы
—NHJ, >NH+, =N + , r-S + .
Матрица ионита гидрофобна. Введение фиксированных ионов
означает введение в гидрофобную матрицу гидрофильных групп,
вследствие чего матрица приобретает способность к набуханию,
а смола превращается в полиэлектролит. Зерно ионита по существу
является гигантской молекулой. Чтобы ее растворить, нужно разор-
вать прочные С—С связи. Поэтому иониты нерастворимы во всех
тех растворителях, которые не разрушают сам ионит.
Таким образом, синтетические ионообменные смолы являются
гелями полиэлектролитов, способными к набуханию. Однако их
набухаемость ограничена благодаря наличию в полимерной моле-
куле поперечных связей.
В противоположность каркасу цеолитов, каркас синтетической
смолы не обладает правильной периодической структурой. Вслед-
ствие этого размеры пор синтетических ионитов неодинаковы и слу-
жить ионными ситами они не могут.
77
Рис. 24. Кривые, характеризующие
иониты (зависимость обменной ем-
кости ионита ОЕ от pH раствора)[1]:
/—I тип ионитов; 2—II тнп нонитов;
3—III тип ионитов; 4—IV тип ионитов
I ТИП — иониты. п
Свойства синтетических ионитов в основном определяются
числом и типом фиксированных ионов, а также строением матрицы,
особенно количеством поперечных связей в ней. Число гидрофиль-
ных групп и количество поперечных связей в матрице определяют
наряду с другими факторами такие важные свойства ионита как
степень набухания, подвижность противоионов, электропровод-
ность и другие кинетические свойства, связанные с движением
ионов [I, 8, 9].
В основу классификации ионообменников Б. П. Никольский
[10] положил их отношение к водородным (для катионитов) и гидро-
ксильным (для анионитов) ионам.
Такая классификация связана с
особенностями поглощения ионита-
ми водородных или гидроксильных
ионов из водных растворов элек-
тролитов. Она в значительной сте-
пени отражает одно из важнейших
свойств ионитов — их обменную
емкость, являющуюся функцией
активности или концентрации (для
разбавленных растворов) катионов
и ионов водорода или анионов и
ионов гидроксила.
Согласно этой классификации
различают четыре типа ионитов
как для катионитов, так и для
анионитов.
оявляющие свойства
сильных кислот или сильных оснований. Ка-
тиониты этого типа характеризуются легкостью вытеснения из них
ионов водорода другими катионами раствора и зависимостью обмен-
ной емкости от pH в очень узкой области (рис. 24, кривая /). Об-
менная емкость такого типа катионитов быстро возрастает с ростом
pH раствора и уже при малых значениях pH, достигая предельной
величины, остается постоянной при дальнейшем возрастании значе-
ний pH. Фиксированными ионами в катионитах I типа чаще всего
являются группы — SO3H, которые легко диссоциируют на остаю-
щийся в каркасе ион — SO3~ и протон, являющийся противо-
ионом.
Аниониты I типа легко обмениваются ионами гидроксила на
анионы из раствора, причем их обменная емкость так же зависит
от рОН раствора, как обменная емкость катионитов от pH. Анио-
ниты этого типа содержат четвертичные амины (==NOH), легко
диссоциирующие на ионы гидроксила и ионы ==N + , входящие
в каркас ионита.
К числу катионитов I типа относятся синтетические смолы КУ-2,
КУ-3, КУ-4, СДВ-2, СДВ-3 и АР.
78
К анионитам I типа относятся синтетические смолы АВ-16, АВ-17.
II тип — иониты, проявляющие свойства
слабых кислот или оснований. Для катионитов
этого типа характерно, что при малых значениях pH раствора боль-
шинство катионов не вытесняет из них ионов водорода. При возра-
стании pH раствора обменная емкость катионитов этого типа резко
возрастает и достигает предельного значения (рис. 24, кривая 2).
Величина pH раствора, при которой начинает резко возрастать
обменная емкость катионита, зависит от концентрации катиона
в растворе и природы ионита. Чем слабее выражены кислотные
свойства катионита, тем более высоким значениям pH соответствует
подъем кривой 2 на рис. 24. Фиксированными группами катионитов
II типа служат группы, характерные для слабых кислот:— СООН,
—SiO3H, —ОН и др.
Свойства анионитов II типа зависят от рОН так же, как зависят
свойства катионитов от pH. Аниониты II типа имеют в качестве
фиксированных ионов группы—NH+, >NH+ или =NH + .
К катионитам II типа относятся катионит силикагель, стекло
ЭС-1, применяемое для изготовления стеклянных электродов, а
также синтетические смолы КБ-4, КБ-4П2, СГ-1.
Анионитами II типа являются синтетические смолы ЭДЭ-10П,
АН-2Ф, АН-1, ММГ-1 и НО.
III тип — иониты смешанного типа, про-
являющие свойства смеси сильной и слабой
кислот или, соответственно, оснований. Иониты
этого типа обладают двумя предельными значениями обменной
емкости в зависимости от pH или рОН раствора (рис. 24, кривая <?).
Для катионитов первая величина предельной обменной емкости
связана с присутствием в них сильнокислотных групп, а вторая обус-
ловлена наличием слабокислотного фиксированного иона.
В качестве примера катионитов, относящихся к III типу, можно
привести синтетические смолы КУ-1, КУ-6, КБУ-1, СНФ, КФ-1,
КФУ, СМ-12, а также сульфоуголь.
К числу анионитов этого типа относится синтетический анионит
ПЭК.
IV тип — иониты, обменная емкость кото-
рых с ростом pH или рОН непрерывно возра-
стает. Иониты, относящиеся к этому типу, ведут себя подобно
смеси многих кислот или оснований различной силы. Зависимость
обменной емкости от pH для катионитов такого типа, изображенная
графически, близка к прямой (рис. 24, кривая 4). Для ионитов
этого типа часто невозможно найти предельное значение обменной
емкости.
Типичным примером ионитов IV типа могут служить почвы,
глины, глауконит.
Чтобы установить тип ионита, по экспериментальным данным
строят график зависимости обменной емкости от pH или рОН рас-
79
твора. Для определения этой зависимости лучше всего пользоваться
методом кривых титрования (стр. 90).
Возможности изготовления ионитов со специфическими свой-
ствами на основе органических полимерных смол практически
неограничены. Современный синтез позволяет в зависимости от
назначения ионита варьировать вид и число фиксированных ионов,
строение матрицы, а также число’ поперечных связей в ней.
В Советском Союзе и за рубежом выпускается большое количе-
ство различного рода ионообменных смол. Краткая характеристика
наиболее важных смол отечественного производства приведена
в табл. 9, смол, выпускаемых иностранными фирмами, — в табл. 10.
В ионообменной хроматографии, особенно в качественном ана-
лизе ионов, в качестве носителя ионогенных групп, способных
к обмену с ионами из раствора, иногда применяют бумагу. В этом
случае фильтровальную бумагу пропитывают каким-либо химиче-
ским реагентом, обладающим группами, способными к обмену, и
чаще всего проводят анализ смеси ионов капельным методом [11].
АППАРАТУРА. ПОДГОТОВКА ИОНИТА
Важнейшим условием успешного решения практических задач
при помощи ионообменной хроматографии является правильный
выбор типа ионообменника, его подготовка, а также определение
условий проведения опыта, особенно размеров колонки. Поэтому
хроматографированию должна предшествовать подготовка ионита,
испытание определенных его свойств и установление на их основе
оптимальных размеров (длины и диаметра) хроматографической
колонки.
Одним из факторов, существенно ухудшающих полноту разде-
ления анализируемой смеси и способствующих размыванию зон
компонентов разделяемой смеси, является так называемый спгеноч-
ный эффект. Для устранения этого эффекта или,по крайней мере,
для сведения его действия к минимуму, необходимо применять
иониты однородного зернения. Соотношение диаметра колонки и
диаметра отдельного зерна не должно быть менее чем 40 : 1. Этим
определяются нижние границы размеров колонок. Можно рекомен-
довать следующие размеры колонок в зависимости от размеров зерен
ионита:
Диаметр
зерна ионита,
мм
1,5
0,5
0,3
Диаметр
колонки, мм
60
20
12
Высота
колонки, мм
300—400
150—250
100—120
В качестве колонок рекомендуется применять стеклянные трубки
с краном внизу. Равномерная подача вымывающего раствора, регу-
лировка его скорости производятся так, как это описано в гл. I.
80
Для отбора проб вытекающего из колонки раствора применяют спе-
циальную аппаратуру [12]. Как правило, в ионообменной хромато-
графии колонку заполняют и промывают сверху. При этом особо
важно соблюдать строгую вертикальность колонки, равномерность
набивки и отсутствие пылевых и воздушных подушек в слое
ионита.
Немаловажное значение имеет степень загрязнения воды, при-
меняемой в качестве растворителя. Особенно это надо иметь в виду
при определении обменной емкости. Наличие посторонних ионов
может нарушить обмен противоионов и исказить общую картину
обмена. Поэтому к воде, применяемой в ионообменной хроматогра-
фии, должны предъявляться повышенные требования в отношении
содержания в ней солей. В особо точных измерениях следует при-
менять бидистиллят, хранящийся в кварцевой или полиэтиленовой
посуде.
Для контроля хроматографических фракций могут применяться
любые аналитические методы, пригодные для качественного и коли-
чественного определения хроматографируемых ионов или комплек-
сов, образуемых ими. Как правило, анализ проводят методом вымы-
вания с последующим определением вымываемого вещества в вы-
текающем из колонки растворе. Реже применяют послойный анализ
без вымывания из колонки, так как он требует наличия окраски у
анализируемых ионов и бесцветного ионита.
Важной стадией работы с ионообменниками является их под-
готовка к проведению эксперимента и регенерации после отработки.
Товарные иониты часто представляют собой смеси, в которых
наряду с основным веществом — высокополимерной смолой — при-
сутствуют различные примеси, особенно ионы железа. Поэтому их
следует предварительно подвергать очистке. Кроме того, ионит
должен быть доведен до набухания, так как только в набухшем
состоянии его следует загружать в колонку.
Подготовка катионитов.Описанные ниже методики предложены
К- М. Ольшановой с сотрудниками [11]. Товарный образец катио-
нита измельчают, просеивают и для подготовки отбирают фракцию
с величиной зерен 0,5—0,25 мм. Порцию этой фракции катионита
помещают в химический стакан и заливают пятикратным по объему
количеством насыщенного раствора хлорида натрия. Залитый раст-
вором катионит оставляют для набухания на 24 ч, после чего раствор
декантируют, а катионит переносят в делительную воронку, в кото-
рой его промывают не менее чем пять раз 5%-ным раствором соля-
ной кислоты. Обьем промывающего раствора должен быть в 30 раз
больше объема катионита. Каждый раз катионит взбалтывают с раст-
вором и оставляют в контакте с ним на 2 ч при периодическом пере-
мешивании. После пятого промывания раствором соляной кислоты
производят промывание дистиллированной водой до нейтральной
реакции по метиловому оранжевому. Обработка катионита раство-
ром соляной кислоты переводит его в Н-форму.
81
Таблица 9
Синтетические ионообменные смолы, выпускаемые или разработанные в СССР [9]
Марка Активные груп- пы Размер частиц, мм Насыпной вес сухого иоиита, г/мл Удельный объем в набухшем со- стоянии, мл/г Обменная емкость, мг-экв/г Исходное сырье
ПОЕ по 0.1 н. NaOH или НС1 Стат. ОЕ по 0,1 н. СаС12
Катиониты
а) сульфояислотные
КУ-2 SO3H 0,25—1,0 0,8 2,5 4,9—5,1 4,3—4,9 Стирол, дивинилбензол
КУ-3 » 0,35—1,5 0,65 2,5—3,0 5,5 5,2 Винилнафталин, дивинилбензол
КУ-4 » 0,65 3,0 5,6 5,4 Аценафтилен, дивинилбензол
КУ-5 0,3—2,0 0,55 5—7 3,0 2,5 Нафталинсульфокислоты, формальде- гид
НСФ 0,3—1,5 0,45 5,0—7,0 3,0 2,4 Нафталин, формальдегид
сдв 0,35—1,5 0,60 •3,2 4,2 4,0 Стирол, дивинилбензол
КУ-21 » 0,55 4,0 5,5 4,5 Нафталинсульфокислоты, формальде- гид
СБС » 0,75 2,0—2,5 3,0 2,3 Стирол, бутадиен
КУ-1 SO3H, он 0,3—2,0 0,74 2,75—3,0 4,5—5,1 1,8—2,2 Фенолсульфокислоты, формальдегид
КУ-7 » 0,3—1,5 0,75 3,0 5,5 3,2 Фенол, бензальдегид-2,4-дисульфокис- лота, формальдегид Фенол, сульфокислоты алифатических альдегидов, формальдегид
КУ-8 » » 0,65 5,0 6,0 4,0
КУ-9 » 0,60 5,0 6,0 3,5 Фенол, сульфокислоты алифатических кетонов, формальдегид
МСФ-3 0,3-2,0 0,65 2,8 4,3 1,8 Хлорбензолсульфокислота, формаль- дегид
сн 0,3-1,5 0,65 3,0 5,2 3,0 Фенольные новолаки
СНФ » 0,65 4,0 5,2 2,8 »
КУ-6 КУ-6Ф SO3H, соон » » » 0,75 0,8 2,7 3,0 5,5 5,6 3,4 3,8 Аценафтен, формальдегид То же и фенол
СМ-12 » 0,35 — 4,5 — Стирол, малеиновый ангидрид, диви- нилбензол
КБУ-1 — б) карб 7,0 оксильные
КБ-1 соон 0,3—1,5 0,6 2,7 10,0 0 Метакриловая кислота, дивинилбен- зол
КБ-2 » 0,3-1,0 0,7 — 10—11 — Метилметакрилат, дивинилбензол
КБ-3 0,3—1,5 0,6 2,8 6,7 0,1 Акрилонитрил, дивинилбензол
КБ-4 » 0,3—0,8 — — 9—10 0,1 Метилметакрилат, дивинилбензол
КБ-ЧП-2 » 0,5—1,2 — — 10,0 — —
КС-1 0,3—1,5 0,7 2,8 10,0 1,0 Малеиновый ангидрид, метилакрилат, дивинилбензол
кн » 0,25—1,5 0,6 — 6,0 0 Акрилонитрил, дивинилбензол
км » 0,35 — м 7,5 0 Метакриловая кислота
кмд » » 0,35 —. 8,5 0 То же
кмг » » 0,35 — 7,7 0 »
КР » » 0,7 —- 7,0 0
кмт » 0,25—0,8 • 0,55 3,5 10,1 0,4 »
СГ-1 0,8—2,0 0,55 —. 8,9 — —
КБ-5 соон, ©н 0,25—1,5 0,6 6,0 7,5 0 Резорцин, монохлоруксусная кислота, формальдегид
КФУ 0,2—0,8 0,6 3,0 6—7 — Феноксиуксусная кислота, фенол, фор- мальдегид
крфу 0,25—1,5 0,45 — 4,0 0 Фенол, резорцин, монохлоруксусная кислота
в) фосфорнокислотныв
КФ-1 РО3Н2 0,3—2,0 0,7 2,7 5,0 0,5 Стирол, дивинилбензол .
КФ-2 » — 0,7 2,7 7,0 1,0 То же
КФ-3 —» 0,65 3,0 3,5 0,5 Винилнафталин, дивинилбензол
КФ-4 » — 0,65 3,0 5,5 1,0 То же
ФВ » 0,4 — 4,3 — Поливиниловый спирт
gg РФ РО3Н2, он — 0,5 — 5,0 — Фенол, резорцин, формальдегид
00
П родолжение табл. 9
Марка Активные груп- пы Размер частиц, мм Насыпной вес сухого ионита, г /м л Удельный объем в набухшем со- стоянии, мл/г Обменная мг-э ПОЕ по 0,1 н. NaOH или НС1 емкость, кв/г Стат. ОЕ по 0,1 н. СаС12 Исходное сырье
Аниониты
а) сильноосновные
АВ-15 АВ-16 N + (CH3)3 NH; N N+—\ZZ/ 0,3—1,0 0,4—2,0 0,59 0,69 2,9 4—5 9,8—10,5 1,62 1,8—2,0 Стирол, дивинилбензол, триметила- мин Пиридин, полиэтиленполиамины, эпи- хлоргидрин
АВ-17 N+ (СН3)3 0,4—1,2 0,74 — 4,3 3,5—4,0 Стирол, дивинилбензол, амины
АВ-18 + Z 0,3—1,0 0,7 3,0 3,0 1,5 Стирол, дивинилбензол, пиридин
АВ-19 N + (CH3)3 Z X » 0,6 3,2 3,0 2,5 Винилнафталин, дивинилбензол, три- метиламин
АВ-20 N+— у » 0,5 4,5 6,0 1,5 Винилпиридин, дивинилбензол
АВ-27 = N + » 0,6 2,6 4,0 3,7 Стирол, дивинилбензол, диметиламин, оксиэтилен
пэк ^N;NH;N+(R)3 » 0,65 3,5 6,0 1,88 Полиэтиленполиамины, эпихлоргид- рин
ЭДЭ-10П NH;-N;N+(R)3 0,4—1,7
АН-2Ф 0,3—2,0
АН-1 » »
АН-9 NH;-N; ОН »
ММГ-1 NH; sN 0,25—2,5
АН-4К » 0,3—1,5
АН-7К » »
АН-10 nh2 »
АН-15 » »
АН-23 N+—
НО NH. N 0,3—2,5
н » 0,3—2,0
мн »
б) слабоосновные
3,4 8,5—9,0 0,9—1,0 Полиэтиленполиамины, эпихлоргид- рин
2,7 10,6 — Полиэтиленполиамины, фенол
— — — Триметилолмеламин
3,0 4,5 — Фенол, формальдегид, аммонийные соли
1,65 3,8 — Мочевина, меламин, гуанидин, фор- мальдегид
4,0 6,5 — Поливинилхлорид, аммиак
3,5 7,4 — Поливинилхлорид, полиэтиленполиа- мины
4,0 14,0 — Аллиламины
2,0 5,5 Стирол, дивинилбензол
5,0 6,5 — Винилпиридин, дивинилбензол.
— 4,1 — Мочевина, меламин, гуанидин
— 4,4 — То же
— 4,1 »
Некоторые наиболее часто встречающиеся иониты иностранных марок
с Марка Основа Активные группы Полная ста- тическая об- менная ем- кость, мг-экв/см3 Макси- мально до- пустимая величина pH Допусти- мая тем- пература, °C Примечание
Катиониты
1 Дуолит СЗ; Леватит PN и KSN; Вофатит F, Р, D; Цеокарб (церолит) 215 и 315 Фенольная смола SO3H 0,6—1,1 9—11 50—100 Сильнокислот- ные
2 Дуолит CS-100; Леватит CNO; Во- фатит CN То же соон 1,2 9,0 40 Слабокислотные
3 Леватит CNS > SO3H соон — 8,5 30—40 Амфотерный
4 CFB; «о»-ионит Алифати- ческий эфир Р03н2 2,3 9,0 90 Среднекислот- ный
5 Амберлит JR-112 и-120; Чемпро С-20; Дауэкс С-50; Дуолит С-25; Ионэк С-240; Леватит S-100; Нальцит HCR; HGR; HDR; Пермутит Q; Пер- мутит RS; Вофатит KPS-200; Цеокарб (церолит) 225 Стироль- ная смола SO3H 1,9—2,2 14 120 Сильнокислот- ные
6 Амберлит JRC-50; Дуолит CS-101; Пермутит Н-70 и С; Вофатит СР-500; Цеокарб (церолит) 226 Смола на основе акри- ловой кис- лоты соон 3,5—4,5 14 100 Слабокислотные
7 CFB «р»-ионит Виниль- ная смола SO3H 1,1 10 40—100 Среднекислот- ный
"Оо
Аниониты
1 Амберлит JRA-400; JRA-401; Деа- цидит FF; Дауэкс-1; Дуолит А-42; А-42 LC; Нальцит SRR; Пермутит ESB и S1 Стироль- ная смола N+ (R)3 N+ (СН3)3 1,0—1,2
2 Амберлит JRA-410; JRA-411; Дау- экс-2; Дуолит А-40, А-40 LC; Ионэк А-550; Нальцит SAR; Пермутит S2 и ES Стироль- ная смола N+ (алкнл)2Х X алкилол 1,0—1,2
3 Амберлит JR-45; Дауэкс-3; Дуолит А-30, А-70; Нальцит WBR; Деаци- дит G То же NH2; NH 1,4—1,8
4
5
Вофатит L-165; Леватит MN
Амберлит JR-4B; Дуолнт А-6; А-7;
Ионэк А-300; Леватнт МЛН; Перму-
тит Е; Вофатит N и MD
Фенольная
смола и дру-
гие продук-
ты конден-
сации
NH2; NH
100
0,7—0,8
2,0-2,5
40—50
S0—60
Сильноосновные.
(Приведены дан-
ные для Cl-фор-
мы)
Сильноосновные.
(Приведены данные
для С1-формы)
Слабоосновные.
(Приведены дан-
ные для ОН-фор-
мы)
Сильноосновные
Слабоосновные
до среднеосновных
(в завнснмости от
продукта) (приве-
дены данные для
ОН-формы).
Отмытый от кислоты катионит переносят на воронку Бюхнера,
отфильтровывают, а затем подсушивают на фильтровальной бумаге
до такого состояния, чтобы зерна свободно отделялись друг от
друга.
Подготовленный таким образом катионит хранят в банке с
притертой пробкой и используют по мере надобности.
В большинстве случаев такой подготовки бывает достаточно,
чтобы при работе с катионитом получить удовлетворительные ре-
зультаты. В специальных случаях требуется более тщательная
подготовка [И].
Подготовка анионитов. Анионит измельчают, просеивают, отби-
рают фракцию с размером зерен 0,5—0,25 мм и обрабатывают на-
сыщенным раствором хлорида натрия так же, как и катионит. Затем
анионит переносят в делительную воронку и промывают 2%-ным
раствором соляной кислоты до полного удаления ионов железа
(проба с роданидом аммония). После этого анионит промывают
десятикратным объемом дистиллированной воды, 5%-ным раствором
щелочи, а затем 10%-ным раствором щелочи до отрицательной
реакции в фильтрате на хлорид-ион (проба с нитратом серебра).
Заканчивают подготовку анионита промыванием дистиллированной
водой, освобожденной кипячением от двуокиси углерода и затем
охлажденной. Промывку прекращают после получения в фильтрате
нейтральной реакции по фенолфталеину.
Подготовленный таким образом анионит хранят под водой в банке
с притертой пробкой, залитой парафином. Только непосредственно
перед загрузкой анионита в колонку его вынимают из банки, отжи-
мают на фильтровальной бумаге и подсушивают в вакуумном
эксикаторе над хлоридом кальция в присутствии кусочков щелочи.
Пбдсушку прекращают, кбгда зерна анионита начинают легко отде-
ляться друг от друга. В таком виде анионит загружают в ко-
лонку.
При подготовке слабоосновного анионита производят замену
раствора щелочи раствором карбоната натрия постепенно увеличи-
вающейся концентрации от 5 до 10%.
Регенерация использованных катионитов и анионитов произво-
дится аналогично описанным методам.
Определение физико-химических свойств ионитов рассматри-
вается при описании практических работ.
ПРАКТИЧЕСКИЕ РАБОТЫ
Работа 1
ОПРЕДЕЛЕНИЕ ПОЛНОЙ ДИНАМИЧЕСКОЙ ОБМЕННОЙ
ЕМКОСТИ КАТИОНИТА [11, 13]
Приборы и реактивы
1. Стеклянная хроматографическая колонка с краном длиной 20 см, диамет-
ром 1,5 см.
2. Склянка Мариотта для равномерной подачи раствора в колонку.
3. Штативы химические с лапками.
4. Мерные цилиндры или градуированные пробирки на 25 мл — 10 шт.
5. Колбы конические для титрования емкостью 100 мл —2 шт.
6. Бюретка для титрования на 25 мл.
7. Микробюретка на 2 мл.
8. Пипетки на 2, 5 и 10 мл.
9. Катионообменная смола КУ-1.
10. Раствор сульфата меди (0,05 и.).
11. Раствор йодистого калия (20%-иый).
12. Титрованный раствор тиосульфата натрия (0,05 н.).
13. Раствор крахмала (1%-ный).
14. Раствор серной кислоты (2 н.).
Цель работы.Получить основную характеристику ионообменной
смолы — ее обменную емкость.
Сущность работы. Основной характеристикой ионообменников
является полная динамическая обменная емкость, выраженная
в мг-экв/г и равная сумме количества всех обменивающихся ионов.
Эта величина не зависит от природы насыщающего иона, размеров
колонки, а также от случайных факторов. Определяется полная
динамическая емкость фронтальным методом по полному поглоще-
нию какого-либо иона из испытуемого раствора данным количест-
вом ионита.
Выполнение работы. Колонку заполняют заранее подготовлен-
ным катионитом КУ-1, строго соблюдая требования равномерной
и плотной упаковки. Для заполнения берут 5 г катионита. Колонку
закрепляют в штативе строго вертикально и катионит смачивают
дистиллированной водой, замеряя требуемый для этой цели объем.
Затем колонку присоединяют к склянке Мариотта (см. рис. 13),
в которую заливают раствор насыщающего иона (раствор сульфата
меди). Подставляют под кран колонки мерный цилиндр, открывают
кран и начинают отбор вытекающего из колонки раствора. По-
воротом крана устанавливают требуемую скорость истечения
(3—4 мл!мин). При проведении опыта необходимо следить, чтобы
в колонке не образовывались воздушные пузыри и чтобы катионит
не всплывал.
Пробы вытекающего из колонки раствора собирают по 25 мл
и в каждой пробе производят определение содержания ионов меди
89
йодометрическим методом. Для этого аликвотную долю раствора
переносят в колбу для титрования, добавляют 4 мл 2 н. раствора
серной кислоты, 10 мл 20%-ного раствора йодистого калия и тит-
руют 0,05 н. раствором тиосульфата натрия в присутствии крахмала
до обесцвечивания синего раствора. Первые порции фильтрата,
когда концентрация ионов меди мала, следует титровать из микро-
бюретки. Пропускание раствора через колонку прекращают после
того, как содержание насыщающего иона в фильтрате сравняется
с его концентрацией в исходном растворе.
По разности титров исходного раствора и фильтратов определяют
количество ионов меди (в мг-экв), поглощенное ионитом из каждой
порции исходного раствора. Эту сумму делят на взятую навеску
катионита в пересчете на абсолютно сухое вещество и таким образом
определяют, общее количество ионов меди, поглощенных катиони-
том (в мг-экв/г), т. е. полную обменную емкость катионита в динами-
ческих условиях. Количество ионов меди, поглощенное ионитом,
рассчитывается исходя из всего объема фильтрата, не содержащего
ионов меди, и исходной концентрации меди в растворе. Для опреде-
ления обменной емкости до проскока это количество ионов меди
делят на взятую навеску катионита.
По данным опыта строят выходную хроматограмму, откладывая
по оси абсцисс объем фильтрата в миллилитрах, а по оси ординат —
количество ионов меди в фильтратах, в мг-экв.
Ра бота 2
ОПРЕДЕЛЕНИЕ ЗАВИСИМОСТИ ОБМЕННОЙ ЕМКОСТИ
КАТИОНИТА ОТ pH РАСТВОРА [13]
Приборы и реактивы
1. Склянки с хорошо притертыми пробками емкостью 150 мл — 8 шт.
2. Аппарат для встряхивания.
3. Бюретки для титрования на 25 и 100 мл.
4. Пипетки иа 50, 10, 5, 2 и 1 мл.
5. Колбы конические для титрования на 250 мл — 2 шт.
6. Ламповый потенциометр (pH-метр) типа ЛП-5; ЛП-58 и др. со стеклянным
электродом.
7. 'Титрованный раствор NaOH (0,1 н.).
8. Раствор хлорида натрия (0,2 н.).
9. Титрованный раствор соляной кислоты (0,1 н.).
10. Катионит КУ-2.
11. Дистиллированная вода.
Цель работы. Определить функциональную зависимость обмен-
ной емкости катионита,, находящегося в Н-форме, от значения pH
раствора.
Сущность работы.Сущность определения зависимости обменной
емкости от pH заключается в построении двух кривых титрования
90
раствора какого-либо вещества (вступающего в обмен с ионами
ионита) раствором щелочи или кислоты в отсутствие и при наличии
ионита. Если, например, катионит обладает кислотными обмени-
в первом случае потре-
Рис. 25. Кривые зависимости
pH раствора щелочи и кисло-
ты от их концентрации в от-
сутствие (/) и при наличии
ионообмениика (2)
вающимися группами, то при титровании раствора в его присут-
ствии расход щелочи будет больше, чем при титровании того же
раствора в отсутствие катионита, так к;
буется дополнительное количество ще-
лочи на нейтрализацию ионов Н+, вы-
тесненных из катионита. Вследствие
этого кривая титрования в присутствии
катионита будет сдвинута в сторону воз-
растания объема щелочи, как это по-
казано на рис. 25. Разность абсцисс
двух точек, лежащих на разных кри-
вых титрования при одном и том же
значении pH, дает возможность вычис-
лить количество ионов Н + , вытесненных
с катионита при данном значении pH.
Зная количество ионов Н+, можно рас-
считать количество катионов а, обменяв-
шихся с ионами Н+ в ионите. Измеряя
эту разность для различных значе-
ний pH, можно найти зависимость ве-
личины а от pH. Расчет значений а про-
водят по формуле:
Здесь — число миллилитров титрованного раствора щелочи,
соответствующее данному значению pH раствора в присутствии
катионита, мл\ Vo — то же без катионита; с — концентрация раст-
вора щелочи, мг-экв!мл\ g — навеска сухого катионита, г. Значение
а выражают в мг-экв!г сухого катионита.
Выполнение работы. Катионит марки КУ-2 в Н-форме высу-
шивают при 105°С в сушильном шкафу и после остывания берут на
технических весах восемь навесок его по 1 а. В каждую склянку
с навеской (предварительно пронумерованную) наливают по 50 мл
0,2 н. раствора NaCI и различные количества 0,1 н. раствора NaOH
(0; 1; 2; 3; 4 мл и т. д.). Затем добавляют воды, так чтобы общий
объем жидкости в каждой склянке составил 100 мл. Благодаря
этому равновесная концентрация NaCI во всех склянках будет оди-
наковой и равной 0,1 н.
. Все сосуды плотно закрывают пробками, встряхивают на аппа-
рате для встряхивания в течение 2—3 ч и оставляют стоять в течение
суток.
Через сутки для каждого раствора определяют pH на ламповом
pH-метре, применяя стеклянный электрод, причем растворы не
фильтруют. Данные опыта наносят на миллиметровую бумагу и
91
строят график, откладывая по оси абсцисс число миллилитров раст-
вора NaOH (взятого в начале опыта), а по оси ординат значения pH,
полученные измерениями на рН-метре.
Для получения кривой в отсутствие катионита берут в восемь
склянок по 50 мл 0,2 н. раствора NaCl, добавляют раствор щелочи,
как в опыте с катионитом, и доводят объем каждого раствора до
100 мл. Для построения кривой в области рН<;7 в две склянки
с раствором NaCl добавляют вместо щелочи 1 и 2 мл 0,1 и. раствора
соляной кислоты. Затем для каждого раствора определяют pH на
рН-метре.
Обе кривые наносят на один график, по которому и определяют
разность абсцисс точек, соответствующих одинаковым значениям pH.
По разностям (Vx—Vo), пользуясь формулой (43), вычисляют значе-
ния а для различных величин pH и полученные результаты сводят
в таблицу. По данным этой таблицы строят график в координатах
pH—а. По форме кривой определяют, к какому типу ионитов отно-
сится изучаемый катионит (см. рис. 24).
Форма записи
Зависимость а от pH растаора
№ склян- ки рн раствора с катио- нитом pH раствора без ка- тионита по кри- вым ти- трования . pH для каждой разности Vi-Vo для каж- дого зна- чения pH а
Работа 3
ОПРЕДЕЛЕНИЕ НЕКОТОРЫХ ФИЗИКО-ХИМИЧЕСКИХ
И ФИЗИКО-МЕХАНИЧЕСКИХ СВОЙСТВ ИОНИТОВ [9]
Приборы и реактивы
1. Цилиндры мерные на 100 мл и на 250 мл.
2. Пикнометры на 10 мл — 2 шт.
3. Эксикатор вакуумный.
4. Водяной термостат типа ТС-15М.
5. Колба круглодонная на 250 мл с обратным холодильником.
6. Штатив химический с лапками.
7. Баня водяная.
8. Воронка для фильтрования с пористым стеклом № 1.
9. Склянка Бунзена.
9?
10. Колба коническая на 200 мл.
11, Бюретка для титрования на 25 мл.
12. Пипетки на 10 и 20 мл.
13. «-Октан или изооктан.
14. Растворы серной кислоты: 5 н.; 1 : 4.
15. Раствор метилового оранжевого.
16. Раствор перманганата калия (0,01 н.).
17. Раствор щавелевой кислоты (0,01 н.).
18. Катионит в Н-форме, измельченный до размеров зерен 0,5—0,25 мм,
например, КУ-2.
Цель работы. Получить представление о разнообразных методах
испытания ионитов и определяемых при этом их свойствах, знание
которых необходимо при решении практических задач применения
ионитов.
Сущность работы. Иониты характеризуются рядом физико-хи-
мических и физико-механических свойств. К первым относятся:
обменная емкость, способность ионита к регенерации, скорость
ионного обмена, химическая стойкость.
К физико-механическим свойствам ионитов относятся: влаж-
ность, фракционный состав в набухшем состоянии, механическая
прочность, насыпной вес, истинная плотность в гидратированном
и негидратированном состоянии, удельный объем, набухаемость и
растворимость.
Определение обменной емкости ионитов проводят статическими
или динамическими методами. К статическим методам относятся
метод потенциометрического титрования, метод определения обмен-
ной емкости по активным группам, метод определения равновесной
обменной емкости.
Динамическую обменную емкость можно характеризовать по
величине полной динамической обменной емкости (ПДОЕ), по вели-
чине динамической обменной емкости до проскока (ДОЕ) и по ско-
рости процесса обмена. Один из методов определения динамической
обменной емкости, а именно, метод определения ПДОЕ, был рас-
смотрен в работе 1. Подробное описание других методов определе-
ния обменной емкости можно найти в [9, 11].
Химическая стойкость ионитов определяется по изменению пол-
ной обменной емкости ионита после воздействия на него какого-либо
реагента, по окисляемости раствора, находившегося в контакте
с ионитом, и по потере веса ионитом. В настоящей работе рассмат-
ривается определение химической стойкости ионита по окисляе-
мости раствора.
Из физико-механических характеристик ионитов в работе пред-
лагается определить насыпной вес, истинную плотность в гидрати-
рованном и негидратированном состоянии, удельный объем и на-
бухаемость ионита.
Выполнение работы. 1. Определение насыпного
веса ионита. В мерный цилиндр на 100 мл насыпают 50 г
товарного ионита, предварительно размельченного и просеянного
да
до величины зерна 0,5—0,25 мм. После уплотнения пробы постуки-
ванием о деревянную крышку стола определяют объем взятого
ионита. Насыпной вес выражают в г/мл и вычисляют по формуле:
= (44)
rfleg — навеска ионита, a; V — объем ионита, мл.
2. Определение истинной плотности ионита
в гидратированном состоянии. Для определения
плотности ионита в гидратированном состоянии навеску 5 г предва-
рительно набухшего ионита (gt) помещают в сухой взвешенный пик-
нометр с известным водным числом и заполняют пикнометр на 3/4
водой. Помещают пикнометр в вакуумный эксикатор для удаления
пузырьков воздуха, затем переносят в термостат, в котором выдер-
живают при 20° С в течение 1 ч. Доводят объем водой до метки,
снова выдерживают при 20° С и взвешивают (g2). После удаления
содержимого пикнометр промывают, заполняют дистиллированной
водой, выдерживают в термостате при 20° С и снова взвешивают
(g3). По полученным данным определяют вес вытесненной воды:
который практически соответствует объему V пробы набухшего
ионита. Отсюда вычисляют истинную плотность ионита (в г/мл)
в гидратированном состоянии по формуле:
p=f. (45)
Для определения плотности ионита в негидратированном состоя-
нии вторую навеску ионита (gj) высушивают до постоянного веса
при 105° С и повторяют с ней определение, заменив воду н-октаном
или изооктаном. Вес вытесненного октана определяют по формуле:
^=/1+^8 — ^
где gj — вес пикнометра, ионита и октана; g'3 — вес пикнометра
и октана; g’t — вес вытесненного ионитом октана. /
Зная плотность октана, вычисляют объем пробы ионита V', а
по нему и истинную плотность ионита в негидратированном
состоянии
Р'=Д. (46)
3. Удельный объем гидратированного
ионита Vo (в мл/г) вычисляют по формуле:
’'• = £’ (47)
94
а удельный объем негидратированного иОнита по формуле:
(48)
4. Определение набухаемости ионита. Под
набухаемостью ионита понимают изменение его удельного объема
при переходе из сухого в набухшее состояние. Абсолютная набухае-
мость (в мл/г) равна разности удельных объемов набухшего и сухого
ионита, т. е. (Vo—V'o).
Относительная набухаемость (в %) определяется как отношение
удельного объема в набухшем состоянии к удельному объему
в сухом.
5. Определение химической стойкости
ионита по окисляемости фильтрата. Берут
навеску катионита в Н-форме, приблизительно 1 г, вносят в кругло-
донную колбу емкостью 250 мл, наливают 100 мл 5 н. раствора сер-
ной кислоты, вставляют в горло колбы пробку с обратным холо-
дильником и содержимое колбы выдерживают в течение 30 мин
на кипящей водяной бане. Колбу вынимают из водяной бани, дают
ей остыть и ионит отделяют от жидкости фильтрованием через
воронку с пористым фильтром № 1. Ионит промывают дистиллиро-
ванной водой до нейтральной реакции по метиловому оранже-
вому.
Фильтрат и промывные воды собирают в один сосуд, измеряют
объем при помощи мерного цилиндра и определяют окисляемость
этого раствора. Для этого 20 мл раствора переносят в коническую
колбу на 200 мл, добавляют 10 мл раствора серной кислоты (1 : 4)
и 20 мл 0,01 н. раствора перманганата калия. Содержимое колбы
кипятят в течение 10 мин на плитке. В случае исчезновения розовой
окраски раствора добавляют еще 20 мл раствора перманганата
калия и кипятят еще 5 мин. Операцию повторяют до тех пор, пока
розовая окраска не перестанет исчезать после 5 мин кипячения раст-
вора. При каждом добавлении раствора перманганата вводят по
10 мл раствора серной кислоты (1 : 4).
После этого в горячий раствор вводят 20 мл 0,01 н. раствора щаве-
левой кислоты и избыток кислоты титруют 0,01 н. раствором пер-
манганата калия до появления заметной розовой окраски раствора.
Титрование следует проводить так, чтобы за время всей операции
температура титруемого раствора снизилась не более чем до 70° С.
Окисляемость в мг О2 на грамм сухого ионита рассчитывают по
формуле:
Окисляемость = (Укмпо4—Vh,c,oJ 5-0,08,
где Укмпо4 — объем прилитого и пошедшего на титрование 0,01 н.
раствора перманганата калия, мл\ Ун2с2о4 — объем прилитого
0,01 н. раствора щавелевой кислоты, мл.
95
Работа 4
ОПРЕДЕЛЕНИЕ ЗАВИСИМОСТИ ИОННОГО ОБМЕНА
ОТ ПОЛЯРНОСТИ РАСТВОРИТЕЛЯ [14]
Приборы и реактивы
1. Колбы плоскодонные на 100 мл с пробками — 30 шт.
2. Колбы конические для титрования на 100 мл — 10 шт.
3. Бюреткн для титрования на 25 мл — 5 шт.
4. Пипетки на 25, 10 и 5 мл — по 5 шт.
5. Ареометры для определения плотности этилового спирта.
6. Спирт этиловый 96%-ный.
7. Титрованный раствор хлорида калия (0,25 н.).
8. Раствор трилона Б (0,1 н.)
9. Катионит — эспатит в Са-форме.
Цель работы. Показать на примере одного катионита, что
обменная емкость ионита зависит от полярности растворителя,
например, от его диэлектрической постоянной.
Сущность работы. Одним из решающих факторов, определяющих
изменение ионного обмена, является диэлектрическая постоянная
растворителя. Зависимость константы ионного обмена Кд. в от
диэлектрической постоянной среды определяется влиянием послед-
ней на отношение коэффициентов активностей ионов равновесного
раствора и, следовательно, при изменении состава растворителя
у]/21
Alg/G. в = А1ё^, (49)
'2
где ух и у2 — коэффициенты активности обменивающихся ионов;
и z2 — заряды обменивающихся ионов.
Для проверки правильности данного положения необходимо
определить значения константы ионного обмена в различных
диэлектрических средах, определить в этих же средах отношения
коэффициентов активностей обменивающихся ионов при равновес-
ных концентрациях и сопоставить их изменения.
Константу ионного обмена можно рассчитать по уравнению
Е. Н. Гапона [15], которое в случае обмена двухзарядных катионов
на однозарядные принимает вид:
а„с,'г г'
TS 2 11;
Ад, в
(50)
Я1С2Т2
где аг и а2— количества адсорбированных ионов; q и с2—моляр-
ные концентрации ионов в равновесии.
Логарифм отношения коэффициентов активностей можно опреде-
лить, согласно теории Дебая и Гюккеля [16], по уравнению:
у/2 _ 1 / е2 e2Pi ( е2 е2р2
П у2 2 \ikTrx zkT (1 + Г1Р1)/ \2efeTr2 2e.kT (1 4-r2p2)
e2Pi
(51)
96
где е — величина элементарного заряда; е — диэлектрическая
постоянная среды; k — константа Больцмана; Т — температура,
° К; t\, гг —радиусы двух- и однозарядного ионов; рх, р2— обрат-
ные значения радиусов ионных сфер двух- и однозарядных ионов.
Для частного случая рассматриваемого обмена ионов калия
с ионами кальция уравнение (51) может быть упрощено:
, Y1 2 е2 / 1 1 \
lg22_ = ^^(----------) (52)
® у2 4,6йГе\Г1 г2' v ’
Изучая ионный обмен на каком-либо катионите из растворов
разной полярности, можно рассчитать зависимость логарифма кон-
станты ионного обмена от диэлектрической постоянной раствори-
1 Yi/2
теля, полученные изменения сопоставить с изменениями 1g--------
и убедиться, что эти изменения равнозначны.
Выполнение работы. В тридцать пронумерованных колб вносят
по 5 а катионита (эспатит в Са-форме), взвешенного с точностью до
0,01 г. В шесть первых колб вливают водные растворы хлорида
калия следующих концентраций: 0,05; 0,075; 0,10; 0,15; 0,20 и
0,25 н. Затем готовят водноспиртовые растворы хлорида калия
с содержанием спирта: 30, 50, 70 и 90 вес. %. Для каждого раствора
данной концентрации спирта приготовляют шесть растворов хло-
рида калия указанных выше концентраций и вливают в колбы с на-
веской катионита.
Для установления равновесия колбы непрерывно встряхивают
в течение одних суток. После этого в аликвотной доле каждого
раствора определяют концентрацию обменивающегося иона кальция
титрованием 0,1 н. раствором трилона Б.
По экспериментальным данным рассчитывают изотерму ионного
обмена, пользуясь уравнением, выведенным из уравнения Е.Н. Та-
лона (50):
(1ООсо aCa) (^макс О /Со\
А а, в =-----Т=-----------' Р'Л
1/ аСа .
V -2-(flCa-0
где пСа — количество ионов Са, вытесненных из 5 а эспатита
в Са-форме, мг-экв/г\ с0 — начальная концентрация ионов калия
(нормальность); пмакс — максимальная емкость эспатита, мг-экв/г-,
Кь. в — константа ионного обмена.
Максимальную емкость эспатита пмакс рассчитывают для каж-
дого раствора с данной диэлектрической постоянной по уравнению
Лэнгмюра [5]. Далее для каждого раствора рассчитывают среднее
значение Ал. в> берут логарифм Ад, в и затем строят один общий
график в координатах: обратная величина диэлектрической постоян-
ной — логарифм константы ионного обмена. При этом обычно
получается прямая линия.
4 Б. В. Айвазов
97
тУ2
По формуле (52) рассчитывают значения 1g-А—, подставляя в
• Тг
формулу значения постоянных
е = 4,803 -10-10; k= 1,38-Ю’16;
Т = 298; гг = гСа = 3,7-Ю'8;
г2 = гк = 2,47-Ю-8.
Для значений диэлектрической постоянной е пользуются следую-
щими величинами: вода — 78,5; 30%-ный раствор этанола — 61,1;
50%-ный—49,0; 70%-ный — 38,0; 90%-ный — 28,0.
У1/2
—— сопоставляют с полученными
у1/2
значениями lg^A. в- Изменения A 1g-А— и A 1g Кд в должны быть
Г2
равнозначны.
Экспериментальные данные вносят в таблицу.
Форма записи
Зависимость константы ионного обмена от полярности растворителя
Расчетные данные для 1g
Работа 5
ОПРЕДЕЛЕНИЕ МЕДИ И ЦИНКА ПРИ ИХ СОВМЕСТНОМ
ПРИСУТСТВИИ НА КАТИОНИТЕ КУ-2 [17]
Приборы и реактивы
1. Хроматографическая колонка длиной 15 см, диаметром 10—12 мм.
2. Штатив химический с лапкой.
3. Колбы конические для титрования на 250 мл — 2 шт.
98
4. Стаканы на 100 и 250 мл — 2 шт.
5. Бюретка на 5 мл.
6. Катионит КУ-2 подготовленный (см. стр. 81).
7. Раствор, содержащий ионы меди и цинка (0,1 н. относительно каждого
иона.
8. Раствор соляной кислоты (3 н.).
9. Раствор оксалата аммония (0,25 н.).
10. Раствор перекиси водорода (6%-иый).
11. Раствор аммиака (25%-ный).
12. Титрованный раствор трилоиа Б (0,1 н.).
13. Мурексид, твердая смесь с хлоридом натрия (1 : 100).
14. Эриохром черный Т, твердая смесь с хлоридом натрия (1 : 100).
15. Хлорид аммония (х. ч.).
16. Раствор метилового оранжевого.
Цель работы. Количественное разделение иопределение содержания
катионов меди и цинка в растворе по принципу комплексообра-
зования.
Сущность работы. Ионы меди и цинка образуют комплексные
соединения с оксалат-ионом, константы нестойкости которых раз-
личны
Kicu (Сго4)2р- =9,1 • 10~9; /<[Zn (С,о4),]‘- = 2,5-10~8.
Если колонку, на катионит которой предварительно нанесен
раствор, содержащий катионы меди и цинка, промыть раствором
оксалата аммония, более стойкий анион комплекса меди перейдет
в раствор, а ионы цинка останутся связанными с катионитом. По-
этому при промывании слоя катионита раствором оксалата аммония
в фильтрате можно обнаружить только медь. После промывания
колонки раствором соляной кислоты, вследствие десорбции, ионы
цинка будут обнаружены во втором, кислом фильтрате.
Для анализа следует применять катиониты, содержащие сульфо-
группы.
Метод может быть рекомендован для определения содержания
меди и цинка в сплавах.
Выполнение работы. Хроматографическую колонку заполняют
заранее подготовленным катионитом КУ-2 в Н-форме, измельчения
0,5—0,25 мм. Затем 4—5 мл раствора, содержащего ионы меди и
цинка, вливают в колонку. Колонку с катионитом промывают водой
до нейтральной реакции по метиловому оранжевому и затем вводят
40—60 мл 0,25 н. раствора оксалата аммония.
Вследствие перехода иона меди в комплекс, весь содержащийся
в катионите ион меди вымывается в фильтрат. Последний обрабаты-
вают 6%-ным раствором перекиси водорода (6—7 капель) при на-
гревании в течение 10 мин. После охлаждения добавляют раствор
аммиака до образования медно-аммиачного комплекса (синее окра-
шивание). Полноту вымывания медного комплекса из колонки
проверяют также обработкой последних порций фильтрата пере-
кисью водорода при нагревании с последующим добавлением после
охлаждения раствора аммиака. Отсутствие синего окрашивания
4
99
указывает на полноту вымывания меди. В случае наличия синего
окрашивания фильтрата его приливают к основной массе фильтрата
и продолжают промывать колонку раствором оксалата аммония до
полного удаления меди.
Весь фильтрат после обработки перекисью водорода и аммиаком
титруют трилоном Б в присутствии мурексида. После добавления
мурексида раствор становится желтым или коричневым, что зави-
сит от количества добавленного мурексида, а также от концентрации
ионов меди. В точке эквивалентности окраска резко переходит
в фиолетовую. По количеству пошедшего на титрование раствора
трилона Б рассчитывают содержание иона меди в исходном растворе.
Оставшийся на катионите цинк удаляют промыванием 3 н. раст-
вором соляной кислоты. Расход кислоты составляет примерно
50—60 мл. К фильтрату добавляют избыток раствора аммиака
(35—40 мл) и 5,5—6,5 г хлорида аммония, с таким расчетом,
чтобы получить буферный раствор, необходимый для определения
цинка. Полученную смесь разбавляют дистиллированной водой до
100 мл и титруют трилоном Б в присутствии индикатора эриохром-
черного Т (0,01—0,05 г) до перехода винно-красной окраски
в синюю. По количеству титрованного раствора трилона Б, пошед-
шего на титрование, рассчитывают содержание ионов цинка
в исходном растворе.
Работа 6
ОПРЕДЕЛЕНИЕ СВИНЦА, МЕДИ И ЦИНКА ПРИ ИХ СОВМЕСТ-
НОМ ПРИСУТСТВИИ НА АНИОНИТЕ ЭДЭ-10П [17]
Приборы и реактивы
1. Хроматографическая колонка длиной 15 см, диаметром 10—12 мм.
2. Штатив химический с лапкой.
3. Колбы конические для титрования на 250 мл — 3 шт.
4. Делительная воронка на 50 мл.
5. Пипетки на 2, 5, 10 и 25 мл — по 1 шт.
6. Колба мерная иа 25 мл.
7. Буферный ацетатный раствор pH» 7.
8. Раствор диэтилдитиокарбамииата натрия (3%-ный).
9. Хлороформ, ч. д. а.
10. Анионит ЭДЭ-10 П, подготовленный и измельченный до 0,5—0,25 мм,
в С1-форме.
И. Растворы соляной кислоты (2 н. и 0,5 н.).
12. Раствор, содержащий ионы свинца, цинка и меди ( 3 мг-экв каждого иона
в 50 мл воды).
Цель работы. Разделение и количественное определение катионов
в. растворе при помощи анионита.
Сущность работы. Разделение смеси ионов свинца, меди и цинка
основано на том, что ионы свинца и цинка образуют с раствором
2 н. НО комплексные анионы [РЬО3]”' и [ZnCl;)Г , в то время как
ионы меди не образуют комплексных анионов.
.100
Если такая смесь вступает в контакт с анионитом, то анионы
[РЬС131 и IZiiCl.J вступят в обменную реакцию с анионами смолы,
а катионы меди нет. Таким образом, при промывании слоя анионита,
содержащего смесь ионов свинца, цинка и меди, 2 н. раствором
соляной кислоты первые два иона будут задержаны на колонке,
а медь вымыта. Понижая затем концентрацию кислоты, применяе-
мой для вымывания, можно последовательно удалить с анионита
сначала цинк, а затем и свинец.
Выполнение работы. Хроматографическую колонку заполняют
заранее подготовленным анионитом ЭДЭ-10 П в С1-форме. 50 мл
исследуемого раствора свинца, цинка и меди наносят на анионит
в хроматографической колонке. Затем слой анионита промывают
150 мл 2 н. соляной кислоты. При этом медь переходит в фильтрат
№ 1, в котором и определяют ее концентрацию одним из известных
методов, например, комплексометрически (работа 5) или иодомет-
рически.
Для вымывания цинка, оставшегося на анионите вместе со свин-
цом в виде хлоридных комплексов, колонку промывают 150—200 мл
0,5 н. раствора соляной кислоты. Цинк переходит в фильтрат №2,
в котором содержание определяют полярографическим [18] или
комплексометрическим методами (работа 5).
Свинец вымывают 200 мл воды, нагретой до 80—90° С, в фильтрат
№ 3 и определяют спектрофотометрически [17] или любым
другим подходящим методом.
Спектрофотометрическое определение свинца в растворе состоит
в следующем. В делительную воронку на 50 мл вливают 5 мл (алик-
вотную долю) фильтрата № 3, содержащего свинец, 5 мл ацетатного
буферного раствора, 5 мл хлороформа и 2 мл 3%-ного раствора
диэтилдитиокарбамината натрия. Содержимое делительной воронки
энергично встряхивают в течение 3—5 мин. Хлороформенный слой
сливают в мерную колбу на 25 мл. К оставшемуся водному слою
снова прибавляют 5 мл хлороформа и экстракцию повторяют вновь.
Слой хлороформа отделяют и сливают вместе с первой его порцией.
Экстракцию повторяют еще раз.
Доводят объем жидкости в мерной колбе чистым хлороформом
до метки, тщательно перемешивают содержимое колбы и в получен-
ном хлороформенном растворе диэтилдитиокарбамината свинца оп-
ределяют оптическую плотность при А = 340 ммк на спектрофото-
метре типа СФ-4. В качестве нулевого раствора применяют чистый
хлороформ, а в качестве стандартного раствора — специально
приготовленный раствор диэтилдитиокарбамината свинца известной
концентрации согласно описанной выше методике.
Вычисление концентрации свинца во взятой пробе сРь произво-
дят по формуле
Cpb=%f-\ (54)
101
где Dx — измеренная оптическая плотность исследуемого раствора;
Оэ — оптическая плотность эталонного раствора; с3 — концентра-
ция эталонного раствора, моль!л.
Далее производят расчет концентрации свинца в фильтрате
№ 3 и в пробе исследуемого раствора.
Работа 7
ОТДЕЛЕНИЕ ФОСФАТ-ИОНОВ ОТ НЕКОТОРЫХ КАТИОНОВ
II И III АНАЛИТИЧЕСКИХ ГРУПП МЕТОДОМ ИОНООБМЕННОЙ
ХРОМАТОГРАФИИ НА АНИОНИТЕ [13]
Приборы и реактивы
1. Хроматографическая колонка длиной 18 см, диаметром 10 мм.
2. Колбы конические для титрования на 100 мл — 2 шт.
3. Штатив химический с лапкой.
4. Анионит типа ЭДЭ-10 П, предварительно подготовленный и измельченный
до 0,5—0,25 мм.
5. Раствор соляной кислоты (2 и.).
6. Кислый раствор, содержащий ионы железа, кальция, магния, хлора и фос-
форную кислоту (0,05 н. относительно каждого иона).
7. Раствор K4Fe(CN)e (5%-иый).
Цель работы. Отделение фосфат-ионов от катионов II и III ана-
литических групп, существенно затрудняющих определение этих
катионов в их присутствии.
Сущность работы. Общеизвестны затруднения, возникающие при
определении ионов II и III аналитических групп в присутствии
фосфат-ионов. Поэтому большое значение имеют методы, позволяю-
щие осуществлять предварительное их удаление. Существенное
преимущество перед другими методами имеет метод ионообменной
хроматографии.
Пользуясь свойством анионитов вступать в обмен только с анио-
нами, можно отделить из смеси с катионами фосфат-ион. Для этого
обычно применяют анионит типа ЭДЭ-10 П в Cl-форме. Разделение
следует осуществлять из солянокислой среды.
Выполнение работы. Колонку заполняют набухшим анионитом,
смачивают дистиллированной водой и наносят 10—15 мл ис-
следуемого раствора. Затем колонку промывают 150—200 мл ди-
стиллированной воды, при этом имеющиеся в исследуемом растворе
катионы вымываются в фильтрат № 1. Промывание прекращают
после получения отрицательной реакции на все катионы, присут-
ствующие в исследуемом растворе. Скорость промывания должна
составлять 2 мл/мин.
Полнота отмывания катионов контролируется в отдельной пробе
по ионам железа раствором K4Fe(CN)6. Отсутствие синего окраши-
102
вания свидетельствует о полноте вымывания всех катионов. Филь-
трат № 1 анализируют на содержание ионов железа, кальция и
магния обычным аналитическим методом в отсутствие фосфат-ионов.
Фосфат-ионы удаляют с анионита промыванием раствором 2 н.
соляной кислоты (приблизительно 100 мл) со скоростью 4—5 мл/ мин.
Определение содержания фосфат-ионов производят обычным ана-
литическим методом с молибденовой жидкостью.
Регенерация анионита осуществляется одновременно с удалением
фосфат-ионов, так как при промывании соляной кислотой анионит
переходит в Cl-форму.
Работа 8
КОНЦЕНТРИРОВАНИЕ ВЕЩЕСТВА ИЗ
РАЗБАВЛЕННЫХ РАСТВОРОВ МЕТОДОМ ИОНООБМЕННОЙ
ХРОМАТОГРАФИИ
Приборы и реактивы
1. Хроматографическая колонка длиной 12 см, диаметром 10 мм, с принуди-
тельным фильтрованием (см. рис. 12).
2. Водоструйный насос.
3. Штатив химический с лапкой.
4. Бюретка для титрования на 25 мл.
5. Колба коническая для титрования на 250 мл.
6. Пермутит измельчения 0,10—0,25 мм.
7. Раствор нитрата меди (0,1 н.).
8. Титрованный раствор тиосульфата натрия (0,1 и.).
9. Раствор соляной кислоты (5%-ный).
10. Раствор крахмала (0,2%-иый).
11. Иодид калия (х. ч.).
Цель работы. Сконцентрировать какое-либо вещество, содержа-
щееся в растворе в небольшой концентрации, определить его коли-
чественное содержание и степень извлечения при концентрировании.
Сущность работы. Если какой-либо ион в растворе обладает
большой константой ионного обмена, то, пропуская этот раствор
через слой ионообменника, можно сконцентрировать его в неболь-
шом объеме сорбента. Вымывая затем этот ион небольшим объемом
какого-либо растворителя, можно значительно повысить его перво-
начальную концентрацию. Если выделение иона не требуется,
а возникает лишь необходимость его количественного определения,
то такой ион можно титровать в присутствии ионообменника. Для
этого нет необходимости вымывать определяемый ион с ионообмен-
ника, а следует ту часть ионита, которая содержит этот ион, пере-
нести в колбу для титрования и титровать его соответствующим
раствором. Такое извлечение ионита можно очень просто осущест-
вить, если он бесцветен, а ион окрашен.
103
Выполнение работы. Колонку заполняют пермутитом (2 г),
добиваясь его уплотнения постукиванием о поверхность стола.
Закрепляют колонку в пробке, которую вставляют в колбу Бун-
зена. Подключают водоструйный насос и пропускают через сорбент
0,001 н. раствор нитрата меди (2 л этого раствора готовят разведе-
нием 0,1 н. раствора).
После того, как 2 л 0,001 н. Cu(NO3)2 будет пропущено через
слой пермутита, прекращают фильтрование и окрашенную зону
пермутита количественно переносят в колбу для титрования.
Содержимое этой колбы заливают затем 40—50 мл 5%-ного раствора ,
НО и добавляют 2 г сухого иодида калия KI, не содержащего
свободного иода.
Раствор, содержащий выделившийся иод, титруют 0,1 н. раст-
вором тиосульфата натрия Na2S2O3 в присутствии крахмала. По
количеству раствора тиосульфата, пошедшего на титрование вы-
делившегося иода, рассчитывают содержание иона Си2+ в пер-
мутите^ Сравнивая эти данные с результатом титрования исходного
0,1 н. раствора нитрата меди и зная количество раствора нитрата
меди, пропущенного через слой пермутита, рассчитывают процент
извлечения меди из раствора.
Работа 9
ПОЛУЧЕНИЕ ВОДЫ ВЫСОКОЙ СТЕПЕНИ ЧИСТОТЫ МЕТОДОМ
ИОНООБМЕННОЙ ХРОМАТОГРАФИИ
Приборы и реактивы
1. Хроматографические колонки длиной 1 м, диаметром 10 жж —2 шт.
2. Штативы химические с лапками — 2 шт.
3. Колбы плоскодонные кварцевые на 200 мл — 2 шт.
4. Мост переменного тока для измерения электропроводности, типа Р-38,
или специально собранная установка с звуковым генератором.
5. Сосуд для измерения электропроводности с платиновыми электродами.
6. Катионит КУ-1.
7. Анионит АН-1.
8. Исследуемая вода.
9. Раствор соляной кислоты (3 н.).
10. Раствор едкого натра (3 н.).
11. Раствор хлорида калия (0,01 н.).
12. Дистиллированная вода.
Цель работы. Получение очень чистой воды, удельная электро-
проводность которой на два порядка ниже, чем у обычной дистилли-
рованной воды.
104
Сущность работы. Пропускание воды, содержащей различные
соли, последовательно через слои катионита в Н-форме и анионита
в ОН-форме дает возможность получить воду, свободную от катионов
и анионов, вследствие чего ее удельная электропроводность должна
быть весьма низкой. Поэтому, если порцию воды профильтровать
сначала через слой катионита в Н-форме, то можно получить
кислую воду. Если же затем эту порцию пропустить через слой
анионита в ОН-форме, то гидроксильные группы анионита будут
обменены на анионы, содержащиеся в воде, и кислая вода станет
нейтральной. Таким путем можно получить очень чистую воду,
в чем нетрудно убедиться, измерив ее удельную электропровод-
ность.
Регенерация отработанных ионитов производится фильтрова-
нием через слой катионита раствора соляной кислоты, а через слой
анионита — щелочи.
Выполнение работы. Одну хроматографическую колонку запол-
няют катионитом КУ-1 в Н-форме, предварительно измельченным до
зерен диаметром 0,10—0,25 мм и набухшим. Другую колонку за-
полняют анионитом АН-1 в ОН-форме также набухшим и с тем же
измельчением. Колонки устанавливают строго вертикально. Пред-
варительно измеряют электропроводность исследуемой воды, уста-
новив константу сосуда по раствору хлорида калия известной кон-
центрации.
Исследуемую воду (250 мл) пропускают через колонку с катио-
нитом. Фильтрат собирают в кварцевую колбу во избежание появ-
ления в воде катионов вследствие выщелачивания стекла. После
того как вся вода стечет с катионита, ее фильтруют через слой
анионита, собирая фильтрат в другую кварцевую колбу.
Фильтрату дают стечь с анионита, после чего измеряют его удель-
ную электропроводность. Сосуд для измерения электропроводности
предварительно несколько раз тщательно споласкивают исследуе-
мым фильтратом.
Вычисление удельной электропроводности очищенной воды х
производят по формуле
К
где К — константа сосуда; /?Нг0 — измеренное сопротивление
воды.
Если опыт проведен тщательно, то полученная вода должна
обладать удельным сопротивлением порядка 5-10-8 ом^-см-1.
Регенерацию ионитов производят не каждый раз, а лишь после
того, как удельное сопротивление получаемой воды начнет возра-
стать. Колонки после регенерации следует тщательно промыть
дистиллированной водой до полного удаления ионов хлора с катио-
нита и ионов натрия с анионита.
105
Работа 10
РАЗДЕЛЕНИЕ СМЕСИ АМИНОКИСЛОТ НА ИОНООБМЕННЫХ
СМОЛАХ [19]
Приборы и реактивы
1. Хроматографическая колонка длиной 12 см, диаметром 10 мм.
2. Штатив химический с лапкой.
3. Колбы конические для титрования на 250 мл — 2 шт.
4. Бюретка для титрования на 25 мл.
5. Анионит АН-2Ф, измельчения 0,5—0,25 мм.
6. Раствор уксусной кислоты (0,05%-ный).
7. Растворы соляной кислоты (1 и. и 0,003 н.) .
8. Раствор нингидрина (готовится непосредственно перед употреблением).
9. Раствор исследуемых аминокислот: глутаминовой и аспарагиновой по
20 мг каждой кислоты в 10 мл воды.
10. Раствор нитрата серебра (1%-иый).
Цель работы. Показать, что ионообменивающие адсорбенты
пригодны для разделения смесей не только неорганических ионов,
но и органических, каковыми, в частности, являются аминокислоты
в водных растворах. Опыт проводится на примере смеси простейших
аминокислот на анионите АН-2Ф в Cl-форме.
Сущность работы. Разделение смеси аминокислот методом ионо-
обменной хроматографии основано на различии в их изоэлектриче-
ских точках. Аминокислоты можно условно разделить на три
группы; основные, нейтральные и кислые. К первым относятся
диаминомонокарбоновые кислоты с изоэлектрической точкой, лежа-
щей при pH 7. Нейтральные аминокислоты представляют собой
моноаминомонокарбоновые кислоты с изоэлектрической точкой,
лежащей в пределах pH = 5,5—6,5. Наконец, кислые аминокислоты
являются моноаминодикарбоновыми кислотами. Их изоэлектриче-
ская точка находится при рН<5.
Таким образом, в зависимости от pH среды поведение различных
групп аминокислот будет различным, их ионы будут обладать раз-
ным знаком и числом зарядов. Это свойство и позволяет применять
ионообменники для разделения смеси аминокислот.
Для разделения пригодны не только синтетические смолы, но и
природные ионообменники: пермутиты, окись алюминия, обрабо-
танная соответствующим образом, активные земли и др.
Успех разделения зависит от правильного выбора pH среды,
а также соответствующей формы ионообменника.
Выполнение работы. Разделение аминокислот на ионообменни-
ках производят на примере смеси кислых дикарбоновых амино-
кислот: глутаминовой и аспарагиновой.
Хроматографическую колонку заполняют набухшим анионитом
АН-2Ф и обрабатывают 1н. раствором соляной кислоты, а затем во-
дой до отрицательной реакции на Cl-ион (проба с нитратом серебра),
106
колонку смачивают 0,003 н. раствором соляной кислоты (рНЗ),
после чего наносят 5 мл раствора исследуемых аминокислот. Нано-
симый раствор также должен иметь рН=3—4, так как при таком
значении pH эти кислоты достаточно хорошо обмениваются с
ионами анионита.
Вытеснение аминокислот производят сначала 0,05%-ным раство-
ром уксусной кислоты, причем вытесняется глутаминовая кислота.
Вымывание прекращают после получения отрицательной реакции
на нингидрин. Индикаторный раствор нингидрина готовят в день
определения смешением 95 объемных частей 0,5%-ного раствора
нингидрина в ацетоне, 1 части ледяной уксусной кислоты и 4 частей
воды. В присутствии аминокислоты раствор дает синюю окраску.
После получения отрицательной реакции на глутаминовую кис-
лоту колонку промывают 1 н. раствором соляной кислоты. При этом
вымывается аспарагиновая кислота. Прекращают вымывание после
отрицательной реакции с нингидрином.
Р а б о т а 11
РАЗДЕЛЕНИЕ СМЕСИ ИОНОВ ЛИТИЯ И НАТРИЯ ПРИ ПОМОЩИ
ИОНООБМЕННОЙ ЭЛЕКТРОХРОМАТОГРАФИИ [20]
Приборы и реактивы
1. Электрохроматографическая колонка специальной конструкции; длина
полезной части 10 см, диаметр 14 мм.
2. Градуированные пробирки илн мерные цилиндры на 10 мл — 20 шт.
3. Колбы конические для титрования на 100 мл — 2 шт.
4. Бюретка для титрования на 25 мл.
5. Источник тока — батарея аккумуляторов, напряжением 600 в.
6. Амперметр постоянного тока на 0,5 а.
7. Вольтметр постоянного тока на 650—1000 в.
8. Реостаты.
9. Пламенный фотометр, позволяющий определять литий и натрий.
10. Катионит КУ-2 в Cd-форме, измельчения 200 меш.
11. Раствор хлорида аммония (0,2 н.).
12. Раствор хлорида кадмия (0,2 и.).
13. Раствор исследуемых ионов (хлориды лнтня и натрия в эквимолекуляр-
ном соотношении, 0,5 и. по каждому иону).
Цель работы. Показать, что применение электрофоретического
эффекта наряду с ионным обменом увеличивает возможности разде-
ления смеси близких по свойствам ионов. Получить электрохромато-
грамму раствора солей лития и натрия.
Сущность работы. В отличие от растворов обычных электроли-
тов,где переносчиками тока являются катионы и анионы, в ионо-
обменной смоле, находящейся в каком-либо полярном растворителе,
В переносе тока участвуют только ионы одного знака — катионы
для катионитов или анионы для анионитов. К- С. Спайглер и
К- Д- Кориэл [21] впервые показали, что при наложении электри-
107
Ческого поля зона того или иного катиона, находящегося в колонке
с катионитом, передвигается в сторону катода. Ими было сделано
предположение, что за счет различия в подвижности ионов электро-
миграция в смоле могла бы служить методом разделения смеси
ионов. В. И. Горшков и др. [6] применили эту идею для разделения
смеси близких по свойствам ионов, на-
Рис. 26. Схема колонки для
электрохроматографического
разделения смеси ионов:
1— колонка; 2 и 3 — вводы для
электродов; 4— капилляр для
вывода фильтрата; 5— пористая
перегородка; 6 — термостат
звав метод электрохроматографическим.
Сущность метода состоит в том, что
на слой ионообменника, в котором про-
изводится разделение смеси ионов в рас-
творе, накладывается электрическое
поле. Направление электрического поля
выбирается так, что оно тормозит дви-
жение зон разделяемой смеси ионов. Это
торможение приводит к увеличению раз-
личия в скоростях движения зон, что,
в свою очередь, увеличивает расстояние
между пиками зон и, следовательно,
улучшает разделение.
Улучшение разделения особенно за-
метно, если подвижность иона, выхо-
дящего первым из колонки без наложе-
ния электрического поля, оказывается
меньше подвижности иона, образующего
следующую зону. Для вытеснения сле-
дует использовать ион с большей под-
вижностью, чем последний ион разделяе-
мой смеси, а катионит насыщать, наобо-
рот, наименее подвижным ионом. Таким
образом, если удачно подобраны ионы
относительно их подвижности, то эффект наложения электрического
поля получается значительным. На полноту разделения оказывает
влияние также и плотность тока, проходящего через систему.
Выполнение работы. Колонку специальной конструкции
(рис. 26) заполняют набухшим катионитом КУ-2. Высота слоя
смолы должна быть равна 10 см.
Катионит переводят в Cd-форму, для чего колонку с катионитом
промывают раствором хлорида кадмия до насыщения им всего слоя
смолы. Контроль за появлением в фильтрате ионов кадмия можно
производить по образованию желтого осадка сульфида кадмия
после приливания раствора сульфида натрия или путем пропуска-
ния через фильтрат сероводорода.
Избыток ионов кадмия и хлора удаляют длительным промыва-
нием колонки дистиллированной водой. Промывание прекращают,
когда качественная реакция на ион кадмия в фильтрате делается
отрицательной.
Остаток воды в колонке сливают и вводят 1 мл исследуемого
108
раствора ионов лития и натрия. После впитывания этого раствора
весь объем колонки заполняют раствором промывающего вещества,
т. е. 0,2 н. раствором хлорида аммония. К электродам подключают
батарею аккумуляторов напряжением 600 в. (Осторожно!
Работать в резиновых перчатках!) При помощи
высокоомных реостатов, включенных по схеме потенциометра,
устанавливают такое напряжение, при котором плотность тока на
электродах была бы равна 0,1 а!см2. Катодом должен быть верхний
электрод, анодом — нижний.
После достижения требуемой плотности тока начинают хромато-
графирование. Устанавливают скорость фильтрации 0,25 мл!мин.
и в колонку приливают раствор промывающего вещества, обеспе-
чивая постоянный уровень его над сорбентом. Хроматографический
фильтрат собирают в градуированные пробирки или мерные
цилиндры по 10 мл и анализируют каждую пробу на пламенном
фотометре. Литий определяют на длине волны 670,8 ммк, натрий —
589,6 ммк.
По полученным данным в координатах «объем фильтрата —
концентрация анализируемых ионов» строят хроматограммы. Для
целей качественного определения по оси концентрации можно
откладывать непосредственные отсчеты величин силы тока, полу-
ченные на пламенном фотометре, так как эти величины в интервале
определяемых концентраций пропорциональны концентрациям со-
ответствующих ионов. В случае количественного определения
необходимо предварительно откалибровать прибор или производить
измерения, пользуясь стандартными растворами, и результаты
измерений пересчитывать на концентрацию.
ЛИТЕРАТУРА
Использованная литература
1. Ф. Гельферих. Иониты. ИЛ, М., 1962, стр. 235.
2. Б. П. Никольский, В. И. Парамонова. Усп. хим., 1939, 8,
1535.
3. В. В. Рачинский, Т. Б. Гапон. Хроматография в биологии.
Изд-во АН СССР, М., 1953, стр. 50.
4. В. В. Рачинский. Введение в общую теорию динамики сорбции и
хроматографии. Изд-во «Наука», М., 1964.
5. Е. Н. Гапон, Т. Б. Г а п о н. ЖПХ, 1948, 21, 937; ДАН СССР,
1948, 59, 921; ДАН СССР, 1948, 60, 817; ЖФХ, 1948, 22, 859, 979; ЖАХ, 1948, 3,
203; Усп. хим., 1948, 17, 452.
6. В. И. Горшков, А. А. Шабанов, Г. М. П а н ч е н к о в.
ЖФХ, 1960, 34, 2530; 1962, 36, 1695.
7. Е. Н. Г а п о н, Г. М. Шуваева, ДАН СССР, 1950, 70, 1007.
8. И. Э. А п е л ь ц и н, В. А. К л я ч к о, Ю. Ю. Лурье,
А. С. Смирнов. Иониты и их применение. Стандартгиз, 1949.
9. К. М. С а л д а д з е, А. Б. Пашков, В. С. Титов. Ионооб-
менные высокомолекулярные соединения. Госхимиздат, М., 1960,
10. «Хроматография». Изд-во ЛГУ, 1956, стр. 5.
109
U.K. M. Ольшанова, М.А. Потапова, В. Д. Копылова,
Н. М. Морозова. Руководство по ионообменной, распределительной и оса-
дочной хроматографии. Изд-во «Химия», М., 1965.
12. К. В. Ч м у т о в, В. Т. А в г у л ь. Автоматические приборы в коло-
ночном хроматографическом анализе. Изд-во АН СССР, М., 1961.
13. А. А. Морозов, И. А. Кисель, Н. Л. Оленович. Прак-
тическое руководство по хроматографическому анализу. Изд-во Одесского универ-
ситета, 1961.
14. «Хроматография, ее теория и применение». Изд-во АН СССР, М., 1960,
стр. 128.
15. Е. Н. Г а п о н. ЖОХ, 1933, 3, 145.
16. В. К. С е м ч е н к о. Физическая теория растворов. Гостехиздат, М.,
1941.
17. В. Б. А л е с к о в с к и й, В. В. Бардин и др. Физико-химические
методы анализа. Практическое руководство. Изд-во «Химия», М., 1964.
18. Е. Н. Виноградова, З.А. Галлай, З.М,Финогенова.
Методы полярографического и амперометрического анализа. Изд-во МГУ, 1960.
19. О. Самуэльсон. Применение ионного обмена в аналитической
химий. ИЛ, М., 1955.
20. «Ионообменная технология». Изд-во «Наука», М., 1965.
21. К. S. S р i е g 1 е г, С. D. Coryell. J. Phys. Chem. 1952, 56,106.
Рекомендуемая литература
1. «Хроматографический метод разделения ионов». ИЛ, М., 1949.
2. «Хроматография». Сб. 1, ИЛ, М., 1949.
3. «Ионный обмен». ИЛ, М., 1951.
4. Р. Кунин, Р. Майерс. Ионообменные смолы. ИЛ, М., 1952.
5. Ионообменивающие вещества. Методы испытания. ГОСТ 5695—53. Стац-
дартгиз, М., 1953.
6. «Теория и практика ионообменных материалов». Изд-во АН СССР, М.,
1955.
7. Ф. М. Шемякин, Э. С. Мицеловский, Д. В. Романов.
Хроматографический анализ. Госхимиздат, М., 1955.
8. «Исследования в области ионообменной хроматографии». Изд-во
АН СССР, М„ 1957.
9. «Исследования в области ионообменной, распределительной и осадочной
хроматографии». Изд-во АН СССР, М., 1959.
10. «Ионный обмен и его применение». Изд-во АН СССР, М., 1959.
11. «Ионообменная технология». Металлургиздат, М., 1959.
12. «Исследования в области промышленного применения сорбентов». Изд-во
АН СССР, М„ 1961.
13. Р. Гр и. с с ба х. Теория и практика ионного обмена. ИЛ, М., 1963.
14. «Ионообменные сорбенты в промышленности». Изд-во АН СССР, М., 1963.
15. «Исследования в области ионообменной и молекулярной хроматографии».
Изд-во Белорусск. гос. ун-та, Минск, 1963.
16. «Ионный обмен и хроматография». Рефераты и краткие сообщения.
Изд-во Воронежск. гос. ун-та, 1965.
ГЛАВА III
ОСАДОЧНАЯ ХРОМАТОГРАФИЯ
Разделение смеси ионов может быть осуществлено не только
методом ионообменной хроматографии, но и методом осадочной
хроматографии, разработанным Е. Н. Гапоном и Т. Б. Гапон [1]
в 1948 г.
Отличие адсорбционных хроматограмм от неадсорбционных было
еще отмечено М. С. Цветом [2], проанализировавшим образование
осадков на фильтровальной бумаге. М. С. Цвет полагал, что наряду
с адсорбционными хроматограммами на бумаге могут возникать
осадочные хроматограммы. Например, при капилляризации во
влажном воздухе спиртового хлорофиллового экстракта спирт,
поднимаясь по бумаге, обогащается водой, вследствие чего раство-
ренные в нем пигменты выпадают в осадок, образуя осадочную
хроматограмму. Многие из описанных в капельном анализе реакций
на бумаге также могут быть отнесены к осадочным хромато-
граммам.
Основным фактором, определяющим разделение смеси веществ
в осадочной хроматографии, является образование труднораство-
римых осадков в определенном порядке. Однако последовательное
выпадение осадков в зависимости от их растворимости служит осно-
вой хорошо известного в аналитической химии метода дробного
осаждения, не являющегося хроматографическим методом. Поэтому
основным отличительным свойством осадочной хроматографии
является не только последовательное образование в колонке осад-
ков, обладающих различной растворимостью, но и многократность
процесса их образования и растворения. Последнее обусловливается
большой поверхностью образующихся в колонке осадков и обра-
тимостью процесса. Многократность элементарных актов образова-
ния и закрепления осадка в определенном месте колонки, а также
его растворения, наряду с различием в растворимости получаю-
щихся осадков, определяют направление процесса.
Таким образом, успех разделения смеси веществ методом оса-
дочной хроматографии в первую очередь определяется различием
растворимости образующихся осадков; во-вторых, закреплением
Ш
осадков в месте их образования, обеспечивающим многократность
процесса. При этом считается, что равновесие между раствором и
веществом, образующим осадки и находящимся в твердой фазе,
устанавливается практически мгновенно. Кроме того, при рассмот-
рении процесса хроматографирования пренебрегают наличием про-
дольной диффузии. Эти допущения вполне обоснованы, так как
практически на процесс разделения смеси веществ методом осадоч-
ной хроматографии эти явления не оказывают влияния.
ОБРАЗОВАНИЕ ОСАДКОВ
Теория осадочной хроматографии была развита К- М. Ольшано-
вой [3,4]. Согласно этой теории, порядок расположения зон обра-
зующихся в колонке осадков зависит от способа получения хромато-
грамм, При этом следует различать два случая:
1) раствор хроматографируемых веществ вносится в колонку,
в твердой фазе которой содержится осадитель;
2) раствор осадителя вводится в колонку, твердая фаза которой
содержит определяемые вещества; этот вариант имеет значительно
меньшее практическое значение.
В первом случае при соприкосновении порции раствора, содер-
жащего анализируемые вещества, например, катионы А"+ и Вт+
с слоем колонки, в котором присутствует осаждающий анион Z*-,
образуется осадок, который остается в месте выпадения. Если пред-
положить, что осадок A^Zn менее растворим, чем осадок BftZm,
то в первую очередь при соприкосновении раствора с осадителем
образуется осадок AjZn, причем в растворе содержание иона А" +
будет уменьшаться до тех пор, пока его концентрация [А"+] не
достигнет величины, определяемой отношением произведений раст-
воримости осадков A/;ZfJ и BftZm. После этого на следующем слое
колонки осадитель начнет реагировать с катионом Вт+ и будет
образовываться осадок B^Z^. Следующая свежая порция раствора
не вызовет никаких изменений при контакте с зоной, занятой осад-
ком A^Zn, и поступит в зону, соответствующую осадку B^Z^.
В связи с тем, что осадок B^Zm более растворим, чем осадок AftZn,
в момент соприкосновения новой порции раствора, богатой катио-
ном Ап + , с осадком B^Zm будет происходить растворение послед-
него и образование осадка AfeZn
BftZm4-fe/nAn+ -ч- mAAZ„+feBm+
Таким образом, длина зоны осадка AtZn увеличится, а порция
раствора с уменьшенным содержанием катионов А"+ и увеличенным
содержанием катионов Вт+ поступит в следующий слой колонки,
где будет образовываться осадок BAZm. При этом длина зоны этого
осадка также возрастет по сравнению с ее длиной в первоначальном
слое. Новое поступление свежего раствора приведет к повторению
картины, причем зона осадка AftZn будет оставаться в верхней части
112
колонки и несколько расширяться, в то время как зона осадка BftZ
будет передвигаться вниз по колонке.
Порядок расположения зон образующихся осадков можно опреде-
лить, исходя из значений произведений их растворимости. При хро-
матографировании раствора, содержащего два указанных катиона,
в колонке будут происходить следующие реакции:
k\n+ + nZk~^AkZn\
Отношение произведений активностей ПА образующихся осадков
равно отношению произведений активностей соответствующих
ионов, согласно уравнению:
В А, у Од п+ • ОуМ —
п/—(56)
где адп+, а^т+, агь- — активности ионов А"+, Вт+ и Zk~, а п,
т, k — валентности соответствующих ионов.
В связи с тем, что активность иона-осадителя az/.~ для каждого
осадка в одной и той же колонке постоянна, ее значение из уравнения
(56) можно исключить. Поэтому, с учетом валентностей пит,
получим:
,57)
(nAB*Zm)" ak£m+
Для осадков с весьма малым значением произведения раство-
римости коэффициентом активности можно пренебречь, тогда урав-
нение (57) перейдет в уравнение
(npAtzn)m [А"+С
Из уравнения (58) следует, что порядок распределения осадков
в колонке можно рассчитать, если известны произведения раствори-
мости образующихся осадков и валентности ионов, участвующих
в их образовании. Верхняя зона всегда будет занята менее раство-
римым осадком, нижняя — наиболее растворимым из всех обра-
зующихся при хроматографировании осадков.
Из уравнения (58) следует также, что концентрация иона-оса-
дителя в колонке в этом случае не оказывает влияния на порядок
расположения зон осадков. Также не будет оказывать влияния на
порядок расположения осадков и соотношение концентраций ионов,
но лишь в том случае, если произведения растворимостей осадков
отличаются не менее чем на 3 порядка. В противном случае увели-
чение концентрации иона, образующего более растворимый осадок,
может привести к изменению порядка расположения зон в колонке.
Иллюстрацией к этому положению может служить следующий
пример. Пусть в колонке находятся ионы серебра, а хроматогра-
113
фируемый раствор содержит карбонат- и хромат-ионы. ПРд£2сго4 =
= 1,1 • 1012, а ПР Ag2co,= 8,2-10~12, т. е. осадок хромата серебра в
восемь раз менее растворим, чем осадок карбоната серебра. Следо-
вательно, при хроматографировании верхнюю зону в колонке дол-
жен занять осадок хромата, а нижнюю — осадок карбоната се-
ребра. Такое положение будет действительно иметь место, если
концентрация хроматографируемых ионов эквимолекулярна. Если
же концентрация хромат-иона будет в десять раз меньше концент-
рации карбонат-иона, например, концентрация хромат-иона равна
0,1 г-ион!л, а карбонат-иона — 1 г-ион!л, то в первую очередь будет
достигнуто произведение растворимости карбоната серебра, вслед-
ствие чего он и займет верхнюю зону в колонке. Сказанное под-
тверждается расчетом концентрации ионов серебра, необходимых
для образования соответствующих осадков. Концентрация ионов
серебра, необходимая для выпадения осадка хромата серебра, будет
[Ag+] = = = 3,4.10-ь а-цон/4.
I J
Концентрация ионов серебра, необходимая для выпадения
осадка карбоната серебра
[Ag+]= j/^4^-2 = 2,9.10-е г.ион/л.
Из этих данных следует, что карбонат серебра действительно
должен выпасть в осадок ранее хромата серебра.
Разделение осадков в хроматографической колонке будет про-
исходить тем лучше, чем значительнее они отличаются по своей
растворимости. Поэтому при хроматографировании каждый эле-
ментарный акт образования и растворения осадка связан с значи-
тельным изменением концентрации осаждаемого иона. Теоретиче-
ски можно считать разделение полным, если в растворе концентрация
иона, образующего осадок, равна или меньше 0,1% его первона-
чальной концентрации.
Рассмотрим, будет ли происходить разделение зон осадков
гидроокисей при хроматографировании растворов ионов Си2+,
Со2+ и Ag+ с концентрацией каждого иона 0,1 г-ион!л. Произведе-
ния растворимости гидроокисей этих ионов соответственно равны:
ПРси (ОН)2 = 5,6.10-20;
ПРсо(он)2-2,0.10-1в;
nPAg он = 2,0 • 10-8.
Для решения вопроса о том, произойдет ли разделение зон
осадков гидроокиси меди и гидроокиси кобальта, рассчитаем кон-
центрацию ионов меди в растворе в момент начала выпадения
114
осадка гидроокиси меди. Этот расчет можно сделать по формуле
km
Г А"+1 = V 59
1 J У (ПРвЛ2и)п •
В данном случае имеем k = 1; п = 2; т = 2, поэтому
2+1 5,6-10-20-10“* п о in —5 ,
[Cu2+J =----2-io-™---= 2,8 • 10 8 г-ион/л,
что равно 0,028% первоначальной концентрации иона меди. Следо-
вательно, теоретически разделение зон осадков гидроокисей меди
и кобальта должно произойти.
При расчете концентрации ионов кобальта по формуле (59)
k = 1; п = 2; т = 1. Поэтому концентрация Со2+ в растворе
в момент начала выпадения осадка гидроокиси кобальта равна
гг, 2+, 2-ю—1в-10—2 к 1П_, .
[Со2 ] = —(2'7Го^)2— =5-10 1 г-ион/л.
Полученная концентрация составляет 5% первоначальной концен-
трации ионов кобальта, поэтому разделение зон должно быть
неполным.
Приведенный расчет показывает, что при хроматографировании
раствора, содержащего ионы меди, кобальта и серебра, в рассматри-
ваемом случае в верхней части колонки должна образоваться зона
осадка гидроокиси меди, затем зона, содержащая два осадка:
гидроокиси кобальта и гидроокиси серебра. Экспериментальные
данные подтверждают теоретические расчеты. Однако следует иметь
в виду, что практически выпадение чистого осадка происходит чрез-
вычайно редко вследствие соосаждения, сорбции посторонних ве-
ществ и по другим причинам. Поэтому теоретические расчеты,
исходящие из предположения, что выпадающий осадок имеет точно
стехиометрический состав, отвечающий его химической формуле,
не всегда могут выполняться с достаточной полнотой.
Для улучшения разделения зон рекомендуется промывание полу-
ченных в колонке осадков водой. Это способствует удалению из
зоны посторонних веществ, в частности, механически задержанных,
а также соосажденных ионов.
ВИДЫ ОСАДОЧНЫХ ХРОМАТОГРАММ
Образование осадков при хроматографировании может происхо-
дить в двух случаях: в жидкой фазе, без прямого участия твердой
фазы, и в результате взаимодействия жидкой фазы с твердой.
В связи с этим можно разделить осадочную хроматографию на два
вида.
К первому виду относятся те случаи, когда формирова-
ние осадка происходит только при участии компонентов, находя-
щихся в жидкой фазе. Оно может обусловливаться различными при-
чинами, например, малой- растворимостью отдельных компонентов
115
смеси, изменениями величины pH раствора в процессе хроматогра-
фирования, химическим взаимодействием компонентов смеси друг
с другом и др. Во всех случаях этого вида осадочной хроматографии
образование осадков происходит без участия специального осади-
теля, находящегося в твердой фазе, а связано с изменениями усло-
вий существования компонентов в растворе. Примером может слу-
жить образование осадочной хроматограммы на колонке, заполнен-
ной окисью алюминия. При соприкосновении раствора определяе-
мых ионов с окисью алюминия, имеющей pH порядка 8—9, происхо-
дит понижение кислотности раствора. В результате, если раствор
содержит, например, ионы Fe8+, Cr8 + , Sb8+ образуются малораство-
римые гидроокиси этих металлов, выпадающие в осадок. Порядок
и расположение осадков зависят от их растворимости.
Вторым примером этого вида хроматограмм могут служить
хроматограммы, образующиеся в колонках, в которых содержатся
малорастворимые в применяемом растворителе вещества, образую-
щие с ионами хроматографируемого раствора труднорастворимые
осадки. К их числу относятся, например, хроматограммы, получае-
мые на 8-оксихинолине или диметилглиоксиме. В таких случаях
часть твердого вещества, находящегося в колонке, переходит
в жидкую фазу и вступает в реакцию с хроматографируемым раст-
вором, а другая часть остается в твердой фазе и служит носителем
образующихся осадков.
К первому виду хроматограмм следует также отнести осадочные
хроматограммы, образуемые ионообменниками, обменивающийся
ион которых способен давать малорастворимые осадки с ионами
хроматографируемого раствора.
Роль носителя в первом виде осадочной хроматографии сводится,
во-первых, к йзменению условий, а следовательно, и свойств раст-
вора, и, во-вторых, к достаточно прочному удерживанию образую-
щихся осадков на своей поверхности.
Ко второму виду осадочной хроматографии могут быть
отнесены те случаи, когда формирование осадков происходит в ко-
лонках, состоящих из двух химически взаимодействующих фаз:
жидкой, содержащей растворенное вещество, и твердой, вещество
которой, вступая в химическое взаимодействие с компонентами раст-
вора, образует нерастворимые осадки. Кроме осадителя, в твердую
фазу должен еще входить носитель. Носителем может быть практи-
чески нерастворимое в применяемом растворителе высокодисперс-
ное вещество, химически инертное ко всем составным частям хро-
матографируемого раствора и хорошо удерживающее на своей
поверхности образующиеся осадки. Образование осадков в таком
случае происходит на поверхности носителя в результате взаимо-
действия адсорбированных молекул осадителя с ионами раствора.
Этот вид хроматографии проводится в колонках, заполненных
механической смесью носителя и осадителя. Роль носителя здесь
сводится к удержанию на своей поверхности как осадителя, так и
116
образующихся осадков. Этот вид осадочной хроматографии является
наиболее распространенным и может выполняться в двух модифи-
кациях: колоночной и бумажной, тогда как первый вид осадочной
хроматографии может быть осуществлен только в виде колоночного
варианта. В колоночном варианте в качестве носителя применяются
сыпучие вещества, в бумажной хроматографии носителем служит
бумага. Механизм образования осадочных хроматограмм как в ко-
лоночной, так и в бумажной хроматографии аналогичен.
ЗАКРЕПЛЕНИЕ ОСАДКОВ
Для получения четкой осадочной хроматограммы необходимо,
чтобы образующиеся осадки достаточно прочно удерживались в месте
их выпадения в колонке. В противном случае осадки под действием
протекающего раствора будут сползать вниз и разделение смеси
анализируемых веществ не будет достигнуто.
Прочность закрепления осадков зависит от природы сил взаимо-
действия вещества, образующего осадок, с веществом, заполняющим
колонку, а также от вида осадочной хроматограммы.
При образовании осадков в жидкой фазе, т. е. в случае пер-
вого вида хроматограмм, закрепление их на твердом
носителе обусловлено главным образом взаимодействием кристаллов
осадка с носителем и может носить как адгезионный характер, так
и чисто механический. В случае образования осадочных хромато-
грамм на колонках, содержащих ионообменники, закрепление
осадков происходит вследствие действия межмолекулярных сил
взаимного притяжения или химического взаимодействия. При про-
мывании таких хроматограмм водой смещения зон, как правило,
не происходит.
При получении осадочных хроматограмм второго
вида закрепление осадков может происходить как вследствие
действия ван-дер-ваальсовских сил взаимного притяжения, так и
вследствие сорбции. Не исключено также и чисто механическое за-
держивание осадков в порах зерен носителя.
Следовательно, можно предположить, что на прочность закреп-
ления осадков в колонках должны влиять природа носителя и сте-
пень его дисперсности. Прежде всего имеет значение сорб-
ционная емкость применяемого носителя. Чрезмерное
увеличение сорбционной емкости носителя может привести к значи-
тельной сорбции хроматографируемых веществ и ухудшит разделе-
ние смеси. Слишком малая сорбционная емкость может обусловить
плохое закрепление осадка и также ухудшит разделение анализи-
руемой смеси.
Существенную роль играет также дисперсность но-
сителя. Увеличение степени дисперсности улучшает закреп-
ление осадков, так как приводит к возрастанию числа пор и умень-
шению их размеров. Однако увеличение степени дисперсности носи-
117
теля приводит к уменьшению скорости протекания хроматографи-
руемого раствора и применяемых растворителей, что влечет за собой
излишнюю затрату времени на анализ. Поэтому наилучшими раз-
мерами зерен применяемых носителей следует считать 0,1—0,02 мм.
Определенное влияние на образование осадочных хроматограмм
оказывает природа и концентрация применяе-
мых осадителей. Осадитель должен удовлетворять двум
основным требованиям, а именно: сорбироваться на носителе и обра-
зовывать с хроматографируемыми веществами труднорастворимые
осадки, растворимость которых должна различаться по крайней
мере на три порядка.
Осадителями в осадочной хроматографии могут служить как
неорганические, так и органические вещества. При этом органиче-
скими осадителями могут быть вещества, образующие нормальные
соли, комплексные соединения, адсорбционные органические соеди-
нения, вещества, участвующие в окислительно-восстановительных
реакциях.
Концентрация осадителя, хотя и не играет роли в определении
порядка расположения зон осадков, оказывает существенное влия-
ние на их формирование, так как она влияет на величину сорбции
осадителя и, следовательно, на плотность образующихся осадков.
На практике следует определить оптимальную концентрацию оса-
дителя на избранном носителе и придерживаться ее при проведении
эксперимента.
Природа применяемых растворителей также оказывает немало-
важное значение на образование и закрепление осадков. Влияние
природы растворителя связано прежде всего с растворимостью в нем
как образующихся осадков, так и применяемых осадителей. Раство-
римость осадков должна быть наименьшей, а осадитель не должен
десорбироваться растворителем. Существенное значение имеет кис-
лотность растворов, применяемых при хроматографировании. Так,
например, уменьшение pH раствора для большинства труднораст-
воримых осадков приводит к увеличению их растворимости, что,
в свою очередь, приводит к ухудшению условий получения осадоч-
ной хроматограммы.
Представляет интерес прбмьШй*йие .осадочных хроматограмм
растворителями, действующими селективно, т. е. растворяющими
одни и не растворяющими другие осадки. Такие растворители могут
служить для вымывания одних осадков, тогда как другие остаются
в колонке. Это в значительной степени может улучшить условия
разделения анализируемой смеси.
ВТОРИЧНЫЕ ЯВЛЕНИЯ В ОСАДОЧНОЙ ХРОМАТОГРАФИИ
Изменения хроматограмм, происходящие во времени, назы-
ваются вторичными явлениями. Наиболее характерными из них
являются выравнивание границ зон, образуемых осадками, увели-
118
чение длины зон и изменение их окраски. Эти явления вызываются
старением осадков, их сползанием вниз по слою носителя, образова-
нием комплексных соединений, а также другими процессами. Вто-
ричные явления часто зависят от условий проведения хроматографи-
ческого опыта. Все это требует, чтобы изучение осадочных хромато-
грамм проводилось в течение короткого времени после их получе-
ния. Рекомендуется, чтобы это время не превышало 3—5 мин.
Изменение условий опыта может уменьшить возникновение вто-
ричных явлений. Так, увеличение концентрации осадителя приводит
к уменьшению изменения длины первоначальной зоны и умень-
шению скорости выравнивания границ зон, что благоприятно сказы-
вается на более длительном сохранении первоначального состояния
осадочной хроматограммы. Увеличение концентрации хромато-
графируемых веществ приводит к противоположному результату.
Такой же эффект оказывает и повышение температуры опыта.
Уменьшение скорости изменения длины зоны и выравнива-
ния ,границ с увеличением концентрации осадителя объясняется
возрастанием плотности образующихся осадков и уменьшением
в связи с этим их способности к перемещению. Обратный эффект,
возникающий с увеличением концентрации хроматографируемого
раствора, может быть объяснен возрастанием количества ионов
в порах носителя, их диффузией вниз по колонке и увеличением
в связи с этим длины зоны образующегося осадка. При этом возра-
стает количество осадка, образующегося за счет ионов, находя-
щихся в порах носителя.
Возникновение вторичных явлений может существенно иска-
зить первоначальную картину распределения осадков, что, без-
условно, должно учитываться при изучении осадочных хромато-
грамм.
Интересные явления возникают в осадочных хроматограммах
иодидов ртути [3].
техника эксперимента
КОЛОНОЧНЫЙ ВАРИАНТ
Как уже указывалось, получение осадочных хроматограмм воз-
можно как в колонках, так и на бумаге. Рассмотрим сначала мето-
дику проведения опыта в колоночном варианте. В этом случае
хроматографирование обычно осуществляется в стеклянных колон-
ках небольшого размера (диаметр 4—5 мм, длина 100—150 мм).
Наиболее ответственной частью подготовки к эксперименту является
приготовление твердой фазы, т. е. носителя, содержащего осади-
тель. Различают два способа приготовления: «сухой» и «мокрый».
При подготовке по «сухому» способу носитель и осадитель рас-
тирают до заданного зернения, просеивают и тщательно смешивают
в определенном соотношении. Набивку колонки производят сухой
смесью, добиваясь тщательного уплотнения носителя. Заполнение
119
колонки рекомендуется вести не отдельными порциями, а сразу
всей навеской, так как послойное уплотнение приводит к образова-
нию слоев различной плотности, что вызывает искажение осадочной
хроматограммы.
При «мокром» способе осадитель растворяют в воде или другом
растворителе и полученным раствором пропитывают носитель, из-
мельченный до зерен определенного размера. Суспензию вносят
в колонку и дают отстояться. Избыток растворителя затем отсасы-
вают и колонку с носителем высушивают в термостате. Дополни-
тельного уплотнения смеси в колонке при этом не производят.
Из двух методов «сухой» метод получил наибольшее распростра-
нение, как более быстрый и удобный.
Приготовив колонку со смесью носителя и осадителя, начинают
фильтрование хроматографируемого раствора, причем образуются
зоны, соответствующие получаемым нерастворимым осадкам. Для
лучшего разделения зон по окончании фильтрования хроматогра-
фируемого раствора колонку промывают чистым растворителем.
Если произведения растворимости осадков существенно разли-
чаются, то при этом можно получить зоны, содержащие осадок
только одного компонента разделяемой смеси.
Как уже указывалось ранее, промывание можно осуществлять
также растворителями, растворяющими осадки селективно. При
этом растворяющиеся осадки полностью вымываются из колонки,
а анализируемое вещество определяется в хроматографическом
фильтрате.
В случаях, когда образующиеся осадки окрашены, а носитель
бесцветен, качественный анализ хроматограммы производится на
основании окраски каждой из зон, а также порядка их располо-
жения.
Количественный анализ может быть произведен путем измере-
ния длин окрашенных зон. Этот метод основан на том, что осадок
в зоне распределен равномерно, вследствие чего имеет место прямая
зависимость между величиной зоны осадка и концентрацией веще-
ства в растворе, образующего осадок. Для проведения количествен-
ного анализа в каждом конкретном случае предварительно строят
калибровочный график зависимости длины зоны осадочной хромато-
граммы от концентрации анализируемого вещества.
Если осадки бесцветны, то в колонку после их формирования
вводят раствор, содержащий вещества, дающие специфические
окраски с осажденными ионами. Во всех остальных случаях анализ
хроматограммы может быть произведен только после последова-
тельного вымывания осадков из колонки.
Наряду с селективными растворителями вымывание осадков
можно проводить также, пропуская через колонку раствор, содер-
жащий вытеснитель, т. е. вещество, образующее с осадителем
осадок, растворимость которого меньше других осадков, находя-
щихся на носителе. В этом случае наблюдается перемещение вниз
120
всей хроматограммы в порядке, соответствующем первоначальному
расположению зон. Анализ удаленных с носителя осадков произво-
дится соответствующими аналитическими методами.
Следует отметить, что в осадочной хроматографии повторное
использование хроматографической колонки, в отличие от молеку-
лярной и ионообменной хроматографии, невозможно. Это объяс-
няется тем, что при образовании осадков осадитель расходуется
необратимо, что требует после каждого опыта обновления смеси
носителя и осадителя.
БУМАЖНЫЙ ВАРИАНТ
В бумажном варианте осадочной хроматографии в качестве носи-
теля применяются полоски или круги фильтровальной бумаги.
Бумага на короткое время погружается в раствор осадителя.
Концентрация раствора подбирается опытным путем в зависимости
от условий опыта. После пропитки бумагу развешивают и сушат на
воздухе.
Раствор анализируемых веществ наносят по каплям на фильтро-
вальную бумагу, предварительно пропитанную раствором осади-
теля и высушенную. При этом каждую следующую каплю наносят
после впитывания предыдущей. Вследствие различной раствори-
мости образующихся осадков они располагаются кольцеобразно
вокруг центра, которым является место нанесения капель анализи-
руемого раствора. Полученную хроматограмму промывают, при-
касаясь к центру хроматограммы капилляром или микропипеткой,
наполненной растворителем. При этом вследствие действия капил-
лярных сил происходит передвижение растворителя от центра к
периферии, что вызывает дополнительное разделение зон осадков.
В случае образования бесцветных осадков проявление можно
осуществлять, опрыскивая бумагу, после хроматографирования и ее
высушивания, раствором таких веществ, которые образуют с осаж-
денными ионами окрашенные соединения.
Качественный анализ по бумажной хроматограмме производится
так же, как и для колоночной хроматограммы, т. е. по окраске и
месту расположения осадков. Количественное определение возможно
по ширине окрашенных зон. При этом количественное определение
хроматографируемых веществ основано, как и в колоночном ва-
рианте, на равномерном распределении осадка по длине зоны и свя-
занной с этим прямой зависимости ширины зоны от концентрации
вещества в хроматографируемом растворе.
Вытеснение зон в бумажном варианте осадочной хроматографии,
по-видимому, возможно; однако в литературе оно не описано.
Качественное и количественное определение веществ на бумаге
методом осадочной хроматографии становится весьма удобным и
быстрым, если хроматографируемый раствор содержит одно или
несколько радиоактивных веществ. В этом случае присутствие
и количество вещества в той или иной зоне может быть установлено
121
Таблица Ц
Условия получения осадочных хроматограмм я окраска зон для наиболее важных катионов
и анионов яз водных растворов
№ п.п. 1 Определяе- мый ион Осадитель Носитель Содержание осадителя Приготовление колонки Получение хроматограммы Окраска зоны
1. Алюми- ний К4 [Fe (CN)e] Фильтроваль- ная бумага — Пропитка вод- ным 4—5%-ным раствором осади- теля На бумагу наносят 1 каплю исследуемо- го раствора, 3—4 кап- ли воды, 3—4 капли раствора ализарина (0,1 г в 100 мл спир- та) и подвергают воз- действию паров NH3 Сушат и действуют 3—4 каплями 1 н. уксусной кислоты Красная
2. Висмут Иоднд натрия Иодид калия Окись алю- миния безвод- ная Фильтроваль- ная бумага 10% Сухая смесь Пропитка вод- ным 4—5%-ным ра- створом осадителя На бумагу наносят 3 капли исследуемого раствора Черная Черная
3. Железо K4[Fe(CN)„] Купферон Окись алю- миния безвод- ная То же 0,1-0,5 . мг-экв на 1 г носителя Осадитель вно- сится в виде раст- вора Из насыщенного спиртового раст- вора Сухая смесь К раствору добав- ляют 1—2 капли 2 н. уксусной кислоты Синяя или голубая Коричне- вая
4. Кальций Гидрофосфат натрия двузаме- щенный Окись алю- миния в анион- ной форме 10% К раствору добав- ляют крупинку инди- катора— мурексида Розовая
5 Кобальт
Рубеановодо-
родная кислота
а-Нитрозо-
Р-нафтол
Силикат нат-
рия
Окись алю-
миния безвод-
ная
То же
Фильтроваль-
ная бумага
6 Медь
7 Мышьяк
Рубеановодо-
родная кислота
К4 [Fe (CN)aJ
Силикат нат-
рия
Нитрат се-
ребра
Окись алю-
миния безвод-
ная
То же
Фильтроваль-
ная бумага
Окись алю-
миния безвод-
ная
8 Никель Рубеаново- Окись алю-
дородная кис- миния безвод-
лота ная
NO со
1% Сухая смесь Коричне- вая
1% Из насыщенного спиртового раст- вора В колонку вносят 0,2 мл исследуемого раствора Красно- бурая
• Пропитка 4— 5%-ным раствором осадителя 1 капля исследуе- мого раствора+ 3—4 капли воды + 2 капли 10%-ного раствора аммиака, затем суш- ка и проявление ра- створом рубеановодо- родной кислоты (0,2г в 100 мл спирта) Желтая
1% Сухая смесь В колонку вносят 0,2 мл исследуемого раствора Черная
0,1 мг-экв на 1 г носителя Осадитель вно- сится в виде раст- вора — Бордовая
— Пропитка 4— 5%-ным водным ра- створом осадителя Так же, как для кобальта Оливково зеленая
1% Сухая смесь В прибор для опре- деления мышьяка по- мещают исследуемый раствор и металличе- ский цинк. Газоот- водной трубкой сое- диняют с хроматогра- фической колонкой Черная
1% То же Фиолето- вая
Е Е Определяе- мый ион Осадитель Носитель
Никель Диметилгли- оксим Окись алю- миния безвод- ная
Силикат нат- рия Фильтроваль- ная бумага
9 Олово 5%-ный вод- ный раствор этилксантогена- та калия То же
10 Ртуть (I) Хромат ка- лия Окись алю- миния в. анион- ной форме
Хлорид нат- рия То же
Продолжение табл. 11
Содержание осадителя Приготовление колонки Получение хроматограммы Окраска зоны
1% Сухая смесь В колонку вносят 0,2 мл исследуемого раствора, а затем 1—2 капли аммиака Ало-розо- вая
— Пропитка 4— 5%-ным водным раствором осади- теля Так же, как для кобальта Синяя
— Пропитка раст- вором осадителя На бумагу наносят 1—2 капли раствора осадителя, 0,04 мл ис- следуемого раствора и снова 1—2 капли раствора осадителя Желтая
0,8 мг-экв на 1 г носителя Осадитель вно- сится в виде ра- створа — Коричне- во-красная
0,5 мг-экв на 1 г носителя То же 3—4 капли раство- ра индикатора днфе- ннлкарбазона вводят в исследуемый ра- створ перед хромато- графированием Фиолето- вая
и Ртуть (11) Иодид нат- рия Окись алю- миния безвод- ная
Иодид калия Фильтроваль- ная бумага
12 Свинец Хромат ка- лия Окись алю- миния в анион- ной форме
Иодид натрия Окись алю- миния безвод- ная
Иодид калия Фильтроваль- ная бумага
13 Серебро Хромат ка- лия Окись алю- миния в анион- ной форме
Иодид натрия Окись алю- миния безвод- ная
Иодид калия Фильтроваль- ная бумага
14 Хлорид Нитрат рту- ти (I) Окись алю- миния в анион- ной форме
10%
Сухая смесь
Красная
Пропитка вод- ным 4—5%-ным раствором осади- теля Наносится 3 капли исследуемого ра- створа Красная
0,5 мг-экв на 1 г носителя Осадитель вно- сится в виде ра- створа — Желтая
10% Сухая смесь — •
— Пропитка вод- ным 4-—5%-ным раствором осади- теля Наносится 3 капли исследуемого раство- ра »
0,8 мг-экв Осадитель вно- В колонку вносят Коричне-
на 1 г носителя сится в виде ра- створа 0,2 мл исследуемого раствора во-красная
10% Сухая смесь — Белая
— Пропитка раст- вором осадителя (4—5%-ный) Наносится 3 капли исследуемого раство- ра Желтая
0,2 мг-экв Осадитель вно- В хроматографи- Сине-фио-
на 1 г носителя сится в виде ра- створа руемый раствор вно- сится спиртовой ра- створ дифенилкарба- зона (1%-ный) летовая
Таблица 12
Ряды растворимости осадков, получаемых в различных условиях |3]
Носитель Осадитель Ряд растворимости для осажденных ионов
Окись алюминия Диметилглиоксим Cu2+ > Ni2 + >Со2 +
То же Родизонат натрия Pb2+ > Fe3+ > Ni2 + > Ва2+
» Рубеановодородная кислота Cu2+ >Ni2+ >Со2 +
» Ксантогенат калия Cu2+ >Sn4+ > Ni2+ = Co2 + > Fe3+
» а-Нитрозо-р-наф- тол Fe3+ > Ni2+ >Co2+
» Купферон Fe3+>Ni2+ = Fe2+ >Co2+
» Нитрат серебра I->Br~>Cl-
» Иодид калия Ag + > Hg2+ > Bi3+ > Pb2+ >Cu2+
Силикагель Силикат натрия Hg2 + >Cu2+ > Ag+ =Co2+
Апатит Едкий натр Bi3+> Fe3+=-. Hg2+= = Ag+ >Co2+ >Cu2+ >Ca2+
Сульфид цинка Сульфид цинка Hg2 + = Ag+ >Sb3+ > Bi3+ >Cd2 + > >Cu3+= Pb2+> Fe3 +
путем измерения радиоактивного излучения соответствующим счет-
чиком или же методом радиоавтографии. Последний метод состоит
в наложении бумажной хроматограммы на светочувствительную
пленку или бумагу с последующим проявлением светочувствитель-
ного материала. По полученным черным пятнам и по интенсивности
их почернения можно судить о месте расположения и о количестве
радиоактивного осадка. Возможно также применение люминесцент-
ного анализа.
В табл. 11 приведен перечень осадителей, носителей и условия
получения осадочных хроматограмм для наиболее важных катионов
и анионов из водных растворов. Полезными п|>и определении по-
рядка расположения зон могут быть также ряды растворимости
осадков, полученных для разных осадителей и на различных носи-
телях. Эти ряды приведены в табл. 12.
ПРАКТИЧЕСКИЕ РАБОТЫ
Р а б о т а 1
КАЧЕСТВЕННЫЙ АНАЛИЗ СМЕСИ КАТИОНОВ Fe3+ и Си2+
МЕТОДОМ КОЛОНОЧНОЙ ОСАДОЧНОЙ ХРОМАТОГРАФИИ 14]
Приборы и реактивы
1. Хроматографическая стеклянная колонка с краном или оттянутым концом,
длиной 100 мм, диаметром 4 мм.
2. Штатив химический с лапкой.
3. Колба коническая на 100 мл.
4. Пипетка на 1 мл.
5. Окись алюминия в анионной форме, измельчения 0,05—0,1 мм.
126
6. Едкий натр (х. ч.).
7. Раствор сульфата железа (III) и сульфата меди (II) (1 н. для каждого иона).
8. Раствор К4 [Fe (CN)e] (10%-ный).
Цель работы. Установить качественный состав смеси, рассчи-
тать на основании произведений растворимости труднораство-
римых гидроокисей указанных ионов порядок образования осадочной
хроматограммы и проверить полученные расчетным путем данные
на практике.
Сущность работы. Различие в произведении растворимостей
труднорастворимых осадков гидроокисей железа и меди позволяет
получить для каждого осадка раздельную зону, если через слой
осадителя, смешанного с носителем, пропускать раствор указанных
ионов. После проявления образующиеся осадки имеют характерные
окраски: синюю для ионов железа и коричнево-красную для ионов
меди. Все это дает возможность произвести качественное определе-
ние указанных ионов из одной пробы раствора. В качестве осади-
теля рекомендуется едкий натр, носителя — окись алюминия в ани-
онной форме, а проявителя — раствор ферроцианида калия.
Выполнение работы.Окись алюминия в анионной форме должна
быть приготовлена заранее. (См. стр. 76). Затем готовят смесь
окиси алюминия в анионной форме с едким натром. Для этого
едкий натр, служащий осадителем, растворяют в воде из такого
расчета, чтобы содержание ионов ОН- при смешении раствора с но-
сителем составляло 0),дмг-экв на 1 г сухого носителя. Раствор сме-
шивают с окисью алюминия, служащей в данном опыте носителем,
и смесь затем сушат в сушильном шкафу при температуре не выше
50° С. Сушку прекращают при достижении воздушно-сухого со-
стояния.
Полученной смесью заполняют хроматографическую колонку
на половину ее высоты. При заполнении следят за равномерной упа-
ковкой носителя. Колонку укрепляют в штативе строго вертикально
и приступают к хроматографированию.
В колонку вносят 5—6 капель исследуемого раствора сульфатов
железа и меди и промывают небольшим количеством воды. По мере
продвижения раствора вдоль колонки образуются окрашенные
зоны гидроокисей железа (III) и меди (II). Чтобы получить более
четко различающиеся по окраске зоны, в колонку после образо-
вания осадочной хроматограммы вводят 5—6 капель раствора
K4IFe(CN)e]. При контакте образовавшихся осадков с раствором
проявителя появляются окраски: синяя для зоны гидроокиси
железа (III) и коричнево-красная для гидроокиси меди (II).
В соответствии со значениями произведений растворимости
гидроокисей анализируемых ионов (ПРРе(0Н)3 — 3,8-10-38;
ПРСи<0Н). = 5,6- 10-20) верхней зоной должна быть зона гидро-
окиси железа, после проявления она окрашивается в синий цвет,
а нижней — зона гидроокиси меди с коричнево-красной окраской
после проявления.
127
Работа 2
КАЧЕСТВЕННЫЙ АНАЛИЗ СМЕСИ КАТИОНОВ Sb3+ и Sn2 +
МЕТОДОМ КОЛОНОЧНОЙ ОСАДОЧНОЙ ХРОМАТОГРАФИИ [5]
Приборы а реактивы
1. Стеклянная хроматографическая колонка с краном или оттянутым концом
и расширением для пробки № 20, длиной 100 мм, диаметром 5 мм.
2. Штатив химический с лапкой.
3. Аппарат Киппа для получения сероводорода.
4. Окись алюминия безводная, измельчения 0,05—0,1 мм.
5. Соляная кислота (концентрированная).
6. Раствор анализируемых веществ SbCl3 и SnCl2 (0,1 н. для каждого иона).
Цель работы. Установить качественный состав смеси, опре-
делить порядок расположения окрашенных зон осадков и объяснить
его.
Сущность работы.В отличие от предыдущей работы, в которой
осадитель находился в твердом виде на носителе в колонке, а опре-
деляемые ионы — в хроматографируемом растворе, в настоящей
работе получение и разделение по зонам труднорастворимых осад-
ков осуществляется иным образом. Колонка содержит носитель и
определяемые ионы, тогда как осадитель — сероводород — подается
в колонку в процессе хроматографирования. Как и в случае колонки
с осадителем на носителе, в процессе подачи сероводорода обра-
зуются отдельные окрашенные зоны нерастворимых осадков, кото-
рые и можно использовать для качественного анализа неизвестного
раствора.
В процессе хроматографирования образуется две окрашенных
зоны: оранжевая — сульфида сурьмы и буро-коричневая — суль-
фида олова.
Выполнение работы. Хроматографическую колонку заполняют
окисью алюминия, следя за плотностью ее упаковки. Высота слоя
сорбента должна быть 40—50 мм. Приготовленную колонку закреп-
ляют в штативе строго вертикально и в нее вносят раствор анализи-
руемых веществ в количестве 5 капель. Затем колонку промывают
5 каплями концентрированной соляной кислоты. После впитывания
в сорбент соляной кислоты к колонке присоединяют трубку от аппа-
рата Киппа, плотно закрыв колонку пробкой (работать под
т я г о й!). Через колонку пропускают в течение 2—3 мин серово-
дород. При этом очень быстро образуется осадочная хромато-
грамма: оранжевая зона (Sb3+) и буро-коричневая зона (Sn2+). По-
лученные результаты сопоставляют с расчетными, сделанными
по произведениям растворимостей (ПР3ьаза = 3- 10 2Т; ПР3пз =
= МО'28).
128
Работа 3
КАЧЕСТВЕННЫЙ АНАЛИЗ СМЕСИ КАТИОНОВ Hg2 + , Bi3+
И Cu2+ НА ИОНИТЕ В 1-ФОРМЕ [4]
Приборы и реактивы
1. Хроматографическая колонка с краном или оттянутым концом, длиной
100 мм, диаметром 4 мм.
2. Штатив химический с лапкой.
3. Анионит марки ММГ, измельчения 0,05—0,1 мм.
4. Раствор соляной кислоты (5%-ный).
5. Раствор бикарбоната натрия (5%-ный).
6. Раствор иодистоводородной кислоты (0,2 н.).
7. Раствор исследуемых ионов: нитраты ртути (II), висмута и меди (II) по
4 мг-экв/мл для каждого иона.
Цель работы. Определить качественный состав, смеси катионов
и показать, что осадочную хроматограмму можно получить на
анионите.
Сущность работы. При взаимодействии иодид-иона анионита
с катионами раствора либо образуются осадки, выпадающие и за-
крепляющиеся на поверхности анионообменника, либо выделяется
свободный иод, образующий окрашенную в бурый цвет зону. При
наличии значительной разницы в величинах произведений раство-
римости образующихся осадков, их зоны в колонке разделяются,
что позволяет произвести качественное определение состава раст-
вора, содержащего катионы.
Выполнение работы. После измельчения и отбора соответствую-
щей фракции анионита марки ММГ его заливают водой и оставляют
набухать на 24 ч. Набухший анионит помещают в колонку (высота
его слоя должна быть равна 30 мм) и промывают 5%-ным раствором
соляной кислоты, а затем горячей водой до тех пор, пока pH фильт-
рата не станет равным 2,5.
После этого колонку промывают 5%-ным раствором бикарбоната
натрия до полного удаления хлорид-ионов, а избыток бикарбоната
натрия удаляют промыванием водой до значения pH в фильтрате
7,4-8.
Затем анионит промывают 0,2 н. раствором иодистоводородной
кислоты для перевода его в 1-форму. После полного насыщения
анионита иодид-ионом колонку промывают водой до получения
в фильтрате pH = 2,5—3.
В колонку вводят 0,2 мл исследуемого раствора и промывают
водой. При этом образуется осадочная хроматограмма: верхняя,
красная зона — ртуть (II); средняя, черная — висмут и нижняя,
бурая вследствие выделившегося при взаимодействии с ионом меди
свободного иода — медь (II).
По полученным окрашенным зонам определяют качественный
состав катионов раствора.
5 Б. В. Айвазов
129
Работа 4
КАЧЕСТВЕННЫЙ АНАЛИЗ СМЕСИ КАТИОНОВ Си2+, Со2 + И
Ni2+ МЕТОДОМ ОСАДОЧНОЙ ХРОМАТОГРАФИИ НА БУМАГЕ [5]
Приборы и реактивы
1. Фильтровальная бумага марки «синяя лента». Кружок диаметром
70—100 мм.
2. Пульверизатор или мягкая кисточка.
3. Раствор осадителя Na2SiO3 (5%-ный).
4. Раствор проявителя (рубеановодородная кислота) — 0,2 г в 100 мл эти-
лового спирта.
5. Раствор аммиака (10%-ный).
6. Исследуемый раствор сульфатов меди и никеля и хлорида кобальта
(0,1 н. для каждой соли).
Цель работы. Определить качественный состав смеси и устано-
вить порядок расположения зон соответствующих ионов.
Сущность работы. Наряду с колоночной осадочной хроматогра-
фией анализ смеси ионов можно производить методом осадочной
хроматографии на бумаге. В этом случае роль носителя играет
фильтровальная бумага, которая предварительно пропитывается
раствором выбранного осадителя. Неокрашенные осадки можно
проявлять соответствующими реагентами. В остальном принцип
получения осадочной хроматограммы на бумаге не отличается от
ее получения в колонке.
Чаще всего для хроматографирования пользуются кружком
фильтровальной бумаги, в центр которого наносят несколько ка-
пель анализируемого раствора. При этом зоны окрашенных осадков
располагаются в виде концентрических колец.
Преимущество осадочной хроматографии на бумаге перед ее
вариантом в колонке состоит в быстроте получения качественных
результатов и в большом удобстве осуществлять проявление неокра-
шенных осадков.
Выполнение работы. Кружок фильтровальной бумаги марки
«синяя лента» диаметром 70—100 мм пропитывают 5%-ным раство-
ром силиката натрия и высушивают на воздухе. В центр подготов-
ленной таким образом и высушенной бумаги наносят одну каплю
исследуемого раствора. Дав впитаться этой капле, наносят 3—4
капли воды, причем каждая капля должна предварительно впи-
таться. Затем в центр же наносят 2 капли раствора аммиака, после
чего бумагу сушат на воздухе. Наконец, в центр высушенной бумаги
наносят несколько капель раствора проявителя. При этом отчетливо
проявляются концентрически расположенные зоны: ближайшая
к центру оливково-зеленая зона меди, в середине — желтая зона
кобальта и наиболее дальняя — синяя зона никеля.
По окраске зон и порядку их расположения производится каче-
ственный анализ смеси.
130
Работа 5
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ НИКЕЛЯ МЕТОДОМ
ОСАДОЧНОЙ ХРОМАТОГРАФИИ НА БУМАГЕ [6,7]
Приборы и реактивы
1. Фильтровальная бумага марки «синяя лента» или хроматографическая
бумага марки Б Ленинградской бумажной фабрики № 2. Полоса шириной 8 см
и длиной 22 см.
2. Держатель для бумаги.
3. Капилляр стеклянный емкостью 0,002—0,005 мл.
4. Стакан химический на 500 мл.
5. Предметное стекло.
6. Часовое стекло.
7. Чашка Петри.
8. Измерительная линейка.
9. Водный раствор диметилглиоксима (0,12%-ный).
10. Водный раствор глицерина (12%-ный).
11. Эталонные растворы никеля (0,4005 г №С12-6Н2О растворяют в 100 мл
0,1 н. раствора соляной кислоты, выбирая невыветрившиеся кристаллы. Содержа-
ние никеля в 1 мл этого раствора равно 1,0мг. Из этого раствора готовят требуемые
эталонные растворы соответствующим разбавлением).
12. Исследуемый раствор никеля (~ 150 мкг/мл).
Цель работы. Показать, что осадочная хроматография на бумаге
позволяет определять содержание неорганических катионов не
только качественно, но и количественно. Определить количество
ионов никеля в исследуемом растворе.
Сущность работы. Если фильтровальную бумагу пропитать
осадителем, а затем на высушенную бумагу нанести каплю раствора,
содержащего ион, образующий с осадителем нерастворимый осадок,
то в месте нанесения капли раствора образуется окрашенное или
неокрашенное пятно осадка. В случае, если в капле раствора содер-
жится избыток иона, то этот избыток остается на бумаге. При промы-
вании этого пятна чистым растворителем избыточные ионы увле-
каются им, переносятся по бумаге и реагируют с новыми порциями
осадителя. При этом за движущимся по бумаге растворителем
образуется окрашенный или неокрашенный след осадка в виде пика.
Наблюдения показывают, что высота пика связана с количеством
иона в растворе. Последнее свойство может быть положено в основу
количественного определения ионов в растворе методом осадочной
хроматографии на бумаге. С целью получения надежных результа-
тов количественное определение производят путем сравнения высот
пиков, полученных для исследуемого раствора, с высотами пиков,
полученных для стандартных растворов в тех же условиях.
Выполнение работы. Полоску бумаги пропитывают раствором
диметилглиоксима и высушивают на воздухе.
Для нанесения капли анализируемого и стандартного растворов
на бумагу следует подготовить специальный капилляр. Капилляр-
5* 131
Рис. 27. Схема проявле-
ния осадочной хромато-
граммы на бумаге в слу-
чае опредения количест-
венного содержания ве-
щества по высоте конуса
пятна [7]:
/—крышка камеры; 2—фронт
растворителя; 3 — стакан; 4 —
бумага; 5—пятна, образован-
ные осадками; 6 — раствори-
тель
ную трубку диаметром 0,5 мм оттягивают с одного конца на газовой
горелке в виде конуса и обрезают капилляр длиной 25—30 мм.
Нижний торец капилляра отшлифовывают. Затем капилляр тща-
тельно промывают эфиром, этиловым спиртом и дистиллированной
водой. Чистый капилляр заполняется водой мгновенно. Если этого
не происходит, то промывание повторяют. Хранят капилляр в ди-
стиллированной воде.
Для калибровки капилляра его наполняют дистиллированной
водой, наружные стенки вытирают фильтровальной бумагой и
взвешивают на аналитических весах. Затем
удаляют воду из капилляра прикоснове-
нием фильтровальной бумаги и снова взве-
шивают. По разности результатов взвеши-
ваний определяют емкость капилляра.
Капилляр несколько раз промывают
исследуемым раствором и затем на край
полоски бумаги на расстоянии 1,5—2 см
от одного из ее концов наносят каплю ис-
следуемого раствора. На одной линии, на
расстоянии 1,5 см от первой капли наносят
вторую и затем, на таком же расстоянии
от второй, третью каплю. После этого бу-
магу высушивают на воздухе.
В стакан емкостью 500 мл наливают
25 мл проявляющего раствора — 12%-ного
раствора глицерина в воде. Полоску бума-
ги закрепляют в специальном штативе и
устанавливают его в стакане так, чтобы тот
край бумаги, на который нанесены капли
раствора, погрузился в проявляющий рас-
твор, причем пятна от капель должны
находиться выше уровня проявителя. Стакан покрывают чашкой
Петри и оставляют на 20—25 мин (рис. 27).
Проявляющий раствор поднимается по капиллярам бумаги и
захватывает непрореагировавшие ионы никеля, вследствие чего
окрашенные зоны растягиваются и принимают пикообразную
форму.
Через 20—25 мин хроматограмму вынимают из стакана, высуши-
вают и линейкой измеряют высоту пятен от центра до конца
пика. За истинное принимают среднее арифметическое из трех
измерений. Точность метода зависит от тщательности проведения
опыта.
Для получения пиков стандартного раствора наносят на хромато-
графическую бумагу в аналогичных условиях и тем же капилляром
капли стандартного раствора такой концентрации, которая наи-
более близко подходит к концентрации исследуемого раствора
(~ 150 мкг/мл). Хроматограмму проявляют, измеряют высоту
132
пиков и сравнивают результаты, полученные для стандартного
раствора, с результатами исследуемого раствора. Результаты изме-
рений вносят в таблицу.
Форма записи
Количественное определение никеля в растворе
Содержание никеля, мг/мл Высота пиков, мм
в стандартном растворе в исследуемом растворе для стандарт- ного раствора для исследуе- мого раствора
1. 2. 3.
Среднее
По полученным данным вычисляют концентрацию никеля в ис-
следуемом растворе, считая, что если концентрация стандартного
раствора близка к концентрации исследуемого, то высоты пиков про-
порциональны концентрации. Полученные данные о концентрации
раствора сопоставляют с истинным содержанием никеля в растворе
и делают заключение о точности метода.
Работа 6
КАЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ГАЛОГЕНИДОВ МЕТОДОМ
КОЛОНОЧНОЙ ОСАДОЧНОЙ ХРОМАТОГРАФИИ [4]
Приборы и реактивы
1. Стеклянная хроматографическая колонка длиной 150 мм, диаметром 5 мм.
2. Штатив химический с лапкой.
3. Колба коническая на 100 мл.
• 4. Ступка фарфоровая с пестиком.
5. Окись алюминия безводная, измельчения 0,05—0,1 мм.
6. Нитрат серебра (х. ч.).
7. Раствор исследуемых галогенидов: NaCl, NaBr и Nal. Концентрация каж-
дого аниона 0,01 н.
Цель работы.Определить качественный состав анионов, уста-
новить порядок расположения окрашенных зон осадков галогенидов
серебра.
Сущность работы. Подобно катионам, анионы также могут быть
обнаружены и проанализированы методом осадочной хроматогра-
фии. Для этого требуется подобрать в качестве осадителя соответ-
.133
ствующий катион, а хроматографированию подвергнуть раствор,
содержащий анализируемые анионы.
В настоящей работе хроматографированию подвергается раствор
галогенидов натрия, а в качестве осадителя применяется нитрат
серебра. Методика работы не отличается от методики определения
катионов.
Выполнение работы. Безводную окись алюминия тщательно
растирают и перемешивают с нитратом серебра, который служит
осадителем. Соотношение окиси алюминия и нитрата серебра
180 : 1. Приготовленную смесь вводят в хроматографическую ко-
лонку и тщательно уплотняют. Высота слоя носителя должна быть
равна 100 мм.
В колонку вводят 0,2 мл исследуемого раствора и после его
впитывания промывают 4—5 мл дистиллированной воды. Колонку
оставляют на солнечном свету, под действием которого происходит
проявление хроматограммы: верхняя зона, принадлежащая иодиду
серебра, остается желтой, средняя — зона бромида серебра —
становится серо-голубой и нижняя — зона хлорида серебра —
фиолетово-серой.
Пользуясь табличными данными о произведениях растворимости
галогенидов серебра, рассчитывают порядок расположения зон
осадков и сопоставляют полученный экспериментально с расчетным.
Определяют качественный состав исследуемого раствора.
Работа 7
КАЧЕСТВЕННЫЙ АНАЛИЗ СМЕСИ ИОДИД- И БРОМИД-ИОНОВ
ОКСИХРОМАТОГРАФИЧЕСКИМ МЕТОДОМ [8,9]
Приборы и реактивы
1. Стеклянная хроматографическая колонка длиной 100 мм, диаметром 4 мм.
2. Фарфоровая ступка с пестиком.
3. Окись алюминия в анионной форме, измельчения 0,05—0,1 мм.
4. Перйодат калия (окислитель).
5. Растворимый крахмал (индикатор).
6. Раствор серной кислоты (2 н.).
7. Анализируемый раствор иодида и бромида натрия (0,01 н.).
8. Спиртовой раствор флюоресцеина (1%-ный).
Цель работы. Показать возможность качественного определения
смеси анионов в растворе методом оксихроматографии. Произвести
качественный анализ указанной смеси анионов.
Сущность работы. Подобно тому как на колонке, заполненной
осадителем, хроматографируемые ионы, вступая с ним в контакт,
образуют осадки, располагающиеся вдоль слоя осадителя в порядке
возрастания их растворимости, разделение смеси ионов можно полу-
чить и тогда, когда колонка заполнена каким-либо окислителем,
134
а хроматографируемые ионы окисляются, образуя зоны продуктов
окисления в порядке возрастания их окислительно-восстановитель-
ного потенциала. Такой метод был предложен К- М. Ольшановой
с сотрудниками [8] и назван оксихроматографическим методом.
Таким образом, в оксихроматографическом методе разделение ве-
ществ основано на различии их окислительно-восстановительных
потенциалов.
Для успешного разделения смеси.веществ, так же как в осадоч-
ной хроматографии, необходимо, чтобы колонка была заполнена
каким-либо носителем, способным удерживать как окислитель или
восстановитель, так и продукты окисления (восстановления), а
вещества разделяемой смеси должны обладать различными, окис-
лительно-восстановительными потенциалами, причем окисли-
тельно-восстановительный потенциал окислителя должен быть
выше окислительно-восстановительного потенциала всех компонен-
тов разделяемой смеси.
По методике проведения анализа окислительно-восстановитель-
ный метод не отличается от метода осадочной хроматографии.
Выполнение работы. Окись алюминия в анионной форме
тщательно перемешивают и растирают в фарфоровой ступке с перйо-
датом калия и крахмалом, взятыми в соотношении по весу соответ-
ственно 10 : 3 : 1. Полученной смесью плотно заполняют хромато-
графическую колонку до высоты 50 мм. Колонку закрепляют в шта-
тиве строго вертикально.
В колонку предварительно вводят 2 капли 2 н. раствора серной
кислоты-и затем 0,2 мл исследуемого раствора галогенидов натрия.
После впитывания раствора колонку промывают 5 мл спиртового
раствора флюоресцеина. Вдоль колонки образуются следующие
зоны: верхняя, синяя — иода, нижняя, красно-оранжевая — брома.
По окраскам зон устанавливают качественный состав взятого
раствора.
ЛИТЕРАТУРА
Использованная литература
1. Е. Н. Гапон, Т. Б. Г а п о н. ДАН СССР, 1948,60,401.
2. М. С. Ц в е т. Хроматографический адсорбционный анализ. Изд-во АН
СССР, М„ 1946.
3. К. М. О л ь ш а н о в а, В. Д. Копылова, Н. М. Морозова.
Осадочная хроматография. Изд-во АН СССР, М., 1963.
4. К. М. О л ь ш а н о в а, М. А. Потапова, В. Д. Копылова,
Н. М. Морозова. Руководство по ионообменной, распределительной н оса-
дочной хроматографии. Изд-во «Химия», М., 1965.
5. А. А. Морозов, Н. А. Кисель, Н. Л. О л е н о в и ч. Практи-
ческое руководство по хроматографическому анализу. Изд-во Одесск. гос. ун-та,
1961.
6. Е. С. Бойчинова, В. Б. Алесковский. Труды ЛТИ
им. Ленсовета, 1958, вып. 48, 94.
7. В. Б. Алесковский и др. Физико-химические методы анализа.
Изд-во «Химия», М„ 1964.
135
8. К. М. Ольшанова, А. Н. Щ е к о л д и н а. Сб. «Хроматография,
ее теория и применение». Изд-во АН СССР, М., 1960, стр. 383.
9. К. М. Ол ыи а н о в а, А. С. Кон ищев а, Н. М. М о р о з о в а.
Сб. «Ионообменная технология». Изд-во «Наука», М., 1965, стр. 252.
Рекомендуемая литература
1. Ф. М. Ш е м я к и н, Э. С. Мицеловский, Д. В. Романов.
Хроматографический анализ. Госхимиздат, М., 1955.
2. В. В. Рачинский, Т. Б. Гапон. Хроматография в биологии.
Изд-во АН СССР, М., 1958.
3. «Исследования в области ионообменной, распределительной и осадочной
хроматографии». Изд-во АН СССР, М., 1959.
4. «Хроматография, ее теория и применение». Изд-во АН СССР, М., 1960.
5. «Ионообменная технология». Изд-во «Наука», М., 1965.
ГЛАВА VI
АДСОРБЦИОННАЯ ГАЗОВАЯ ХРОМАТОГРАФИЯ-
ХРОМАТЕРМОГРАФИЯ
РАВНОВЕСНАЯ ХРОМАТОГРАФИЯ
Допустим, что в реальных условиях процесс хроматографирова-
ния газовой смеси отвечает условиям, близким к равновесным. Тогда
мы можем облагать, что газ протекает через хроматографическую
колонку с такой скоростью, которая обеспечивает установление термодина- мического равновесия. В этом случае мы должны допустить отсутствие диффузии с — c*dc
dx
газа вдоль потока и внутрь зерен адсор- бента, а также не учитывать кинетику адсорбции и десорбции. Согласно теории равновесной хро- матографии, рассмотренной А. В. Кисе- левым [1], исходя из сделанного допу- х x + tfx Рис. 28. Изменение концен- трации с компонента газовой фазы в поперечном слое хро- матографической колонки [1]
щения, составим уравнение материального баланса для какого-то
слоя в колонке толщиной dx (рис. 28). Если объемная скорость газа,
проходящего через слой dx, равна <о см3!мин, то в слое произойдет
накопление вещества, равное
— (О
(дс \ , 7
моль/мин.
(60)
Здесь — градиент концентрации в слое dx, образовавшийся
в нем ко времени t. Вещество в слое dx распределится между газовой
фазой и адсорбентом так, что его количество, приходящееся на еди-
ницу длины слоя адсорбента, при постоянной концентрации составит
V?c+V°ca, где — объем газовой фазы, a Va — объем газа
в адсорбционном слое, приходящийся на единицу длины сорбента;
са — концентрация вещества в слое адсорбента, ас — в газовой
фазе.
137
Скорость изменения количества вещества в слое dx на расстоя-
нии х от входа в колонку будет равна:
(Vt°c4- VaCa)l dx моль/мин. (61)
X
В связи с тем, что скорость накопления вещества и скорость рас-
пределения между газом и адсорбентом по условию материального
баланса должны быть равны между собой, можно написать:
(62)
—. (63)
,\dt J x \ dt J x ' '
Чтобы перейти от скорости изменения концентрации вещества
в адсорбенте к скорости изменения его концентрации
/ дс\
в газе ( gj 1 , можно написать, что
\ dt J х \ дс Jх \dt Jх • ’
Производная Дает зависимость концентрации вещества
в адсорбенте от его концентрации в газе, т. е. представляет собой
изотерму адсорбции, не зависящую ни от времени t, ни от расстоя-
ния х. Поэтому выражейие (64) принимает вид
дса \ ___dca /5с \
dt Jх dc \dt J х
(65)
Подставив выражение (65) в (63), получим:
(66)
Так как концентрация с в газе является функцией х и t, то ее пол-
ный дифференциал равен:
dc=(^} dx+(^} dt. (67)
\dxjt \dt j x v ’
Поделив уравнение (67) на dt, при постоянной концентрации
с (dc—0), получим уравнение
ldc\ __ (дс\
\dijx~~~ \dxJt k
/ dx\
из которого следует, что ( \ представляет собой линейную ско-
рость перемещения вещества в колонке
/ /Эл* \
<69’
13§
/ дс \
(66) и сокращая на (,
равновесной газовой
f дх\
Заменяя в уравнении (68) на ис, вводя его в уравнение
получим основное уравнение теории
хроматографии
<о
yoiyo
а de
(70)
(71)
которое связывает линейную скорость перемещения газа вдоль
слоя адсорбента ис при данной концентрации с с объемной скоростью
потока газа <о и изотермой адсорбции.
Из уравнения Генри са = Гс, следует, что
____-р
de •
Подставив (71) в (70), получим:
<о
U.== —;----- .
с Г» + У“Г
(72)
Рассматривая уравнение (72), можно прийти к заключению, что
скорость перемещения газа вдоль слоя адсорбента при данной кон-
центрации зависит от коэффициента Генри Г. Она тем больше, чем
меньше Г, т. е. чем хуже адсорбируется газ. Следовательно, хро-
матографические зоны компонентов газовой смеси, обладающих раз-
личными значениями коэффициента Генри, будут передвигаться
вдоль слоя адсорбента с различными скоростями, что и обеспечивает
разделение смеси газов.
Каждой концентрации с в газовой фазе соответствует своя
постоянная скорость перемещения вдоль слоя адсорбента ис.
Поэтому то распределение концентрации данного компонента газа,
которое создалось у входа в колонку в момент впуска газа, остается
при дальнейшем движении газа по слою адсорбента без существен-
ных изменений. Каждый компонент будет передвигаться со свойст-
венной ему, но постоянной на всем протяжении слоя адсорбента
скоростью.
Однако этот случай практически выполняется только тогда,
когда изотерма адсорбции линейна и не имеют место отклонения от
закона Генри. Если же форма изотермы адсорбции нелинейна, то
de
производная в уравнении (70) не постоянна и величина ее
будет изменяться с изменением концентрации. Поэтому и скорость
перемещения зоны адсорбированного компонента не будет
постоянна. Это обстоятельство приведет к тому, что в случае выпук-
лой изотермы адсорбции (описываемой, например, уравнением
de
Лэнгмюра), производная ~ при малых концентрациях будет
больше, чем при больших. Тогда, как это следует из уравнения
139
(70), скорость перемещения газа при малых концентрациях будет
меньше, чем скорость перемещения больших концентраций. След-
ствием этого явится искажение хроматографической зоны, конец
ее сильно растянется, образуются «хвосты», которые могут накла-
дываться на зоны следующих компонентов, что существенно ухуд-
шает разделение смеси газов.
В случае вогнутой изотермы адсорбции будет иметь место обрат-
ная картина, вследствие которой хроматографические зоны будут
растягиваться вперед, что также приводит к ухудшению разделения
смеси.
Вопрос о правильном выборе условий проведения хроматографи-
ческого разделения и анализа той или иной газовой смеси имеет
первостепенное значение для успешного решения практических
задач. Поэтому нам представляется целесообразным кратко изло-
жить причины, чаще всего приводящие к ухудшению разделения
анализируемых газовых смесей, и те теоретические представления,
знание которых позволяет избежать этих затруднений и найти
наиболее правильные условия разделения.
ТЕОРИИ ЭФФЕКТИВНОСТИ ХРОМАТОГРАФИЧЕСКИХ колонок
Теория равновесной газовой хроматографии позволяет сделать
вывод о том, что любые смеси газов могут быть хроматографически
разделены, так как для различных по своей структуре и свойствам
молекул коэффициенты Генри должны быть различны. Однако
в реальных условиях мы наблюдаем очень частые случаи невозмож-
ности более или менее полного разделения смесей даже различных
по своим свойствам газов. Одной из существенных причин, ослож-
няющих картину хроматографического разделения газовых смесей,
является уже рассмотренное нами отклонение реальных изотерм
адсорбции газов от линейности, т. е. от уравнения изотермы адсорб-
ции Генри. Немаловажную роль в возникновении трудностей разде-
ления играют диффузионные и кинетические факторы, приводящие
к искажению распределения концентрации десорбирующихся
веществ и размыванию хроматографических полос. Эти искажения
возникают и в тех случаях, когда линейность изотермы соблюдается,
что вызывает дополнительные осложнения при выборе условий
хроматографирования. Поэтому необходимо рассмотреть и эту
сторону вопроса.
В процессе хроматографирования в потоке газа, кроме движения
его молекул в направлении и со скоростью потока, возникает про-
дольная диффузия навстречу потоку, перенос и диффузия газа
к зернам адсорбента и от них, а также диффузия в поры, т. е. так
называемая внутренняя диффузия. Кроме того, молекулы газа,
находящиеся на адсорбенте, отстают от молекул, находящихся
в газовой фазе, вследствие конечной скорости адсорбции и десорб-
ции, а также наличия поверхностной диффузии.
140
Все эти явления в конечном счете приводят к тому, что разные
участки зоны данного компонента смеси газов начинают передви-
гаться вдоль слоя адсорбента с различными скоростями. Последнее
обстоятельство неизбежно приводит к расширению хроматографи-
ческой полосы и, как следствие этого, к снижению максимальной
концентрации и к ухудшению разделения смеси.
Задача теории состоит в том, чтобы оценить влияние всех этих
искажающих картину разделения факторов, выяснить роль различ-
ных диффузионных и кинетических факторов и хотя бы прибли-
женно указать те пути, посредством которых можно было бы добиться
исключения или ослабления их влияния. К сожалению, однако,
сложность самих процессов, а также значительная неопределенность
геометрии применяемых колонок с набивкой не позволяют в настоя-
щее время дать совершенно четкие рекомендации для устранения
влияния отрицательно действующих факторов. Но даже возможность
приближенной оценки делает теорию весьма полезной.
Наибольшее распространение получили теория эквивалентных
тарелок А. Дж. П. Мартина [2,3], диффузионная теория Дж. Дж.
Ван-Деемтера [4,5] и теория критерия разделения А. А. Жуховиц-
кого и Н. М. Туркельтауба [6,7], учитывающие приближенно диф-
фузионные и кинетические факторы и базирующиеся на полуэмпи-
рических и эмпирических константах.
В теории А. Дж. П. Мартина хроматографическая колонка
мысленно разбивается на ряд последовательных участков — «та-
релок», подобно тому, как это делается в теории дистилляционных
колонок. Считается, что хроматографируемый газ проходит каждую
тарелку прерывными порциями, переносимыми газом-носителем,
причем за каждую порцию газа-носителя между твердой фазой —- ад-
сорбентом — и газовой фазой устанавливается равновесие для всех
компонентов разделяемой смеси. Каждая новая порция газа-носи-
теля, подаваемая на первую тарелку, приводит к распределению
компонентов газа между газообразной и твердой фазами, в резуль-
тате чего часть компонентов газа переходит на вторую тарелку, на
которой также устанавливается равновесное распределение газа
между газообразной и твердой фазами. С каждой новой порцией
газа-носителя концентрация компонентов исследуемого газа на
первых тарелках будет уменьшаться, а на следующих возрастать.
В результате такого передвижения и перераспределения каждый
компонент анализируемого газа окажется на нескольких тарелках,
причем на одних из них — средних — компонент будет находиться
при максимальной концентрации, на других при меньших. Про-
изойдет «размывание» компонента по нескольким тарелкам, вслед-
ствие чего максимальная концентрация окажется ниже исходной
концентрации анализируемого газа.
Такой прием замены реального процесса, протекающего непре-
рывно, многоступенчатым процессом позволяет сравнительно легко
получить уравнение, выражающее форму размывания полосы.
141
Уравнение подобного типа можно получить и из диффузионной
теории, что позволяет связать обе теории и вывести новую функцию
«высоты эквивалентной теоретической тарелки» (ВЭТТ).
Рассмотрим материальный баланс для данного компонента ана-
лизируемого газа на п-й тарелке хроматографической колонки
(рис. 29). Пусть очередной впуск
газа-носителя, равный объемуdV,
принесет на эту тарелку порцию
компонента анализируемого га-
за, равную сп_^ dV молей. Здесь
сп__1 — концентрация газа в га-
зовой фазе на (п—1)-й тарелке
до впуска новой порции газа-
Рис. 29. Схема прохождения газовой носителя.
смеси через серию последовательных В результате этого впуска
ступеней (тарелок) [1] с тарелки перейдет на
(п+1)-ую тарелку тот же объем
газа dV, содержащий cndV молей данного компонента. Вследствие
этого на n-й тарелке данный компонент останется в количестве,
равном (сп„1 — с„) dV молей, которое распределится между газо-
образной и твердой фазами п-й тарелки и вызовет соответству-
ющие изменения концентрации в газовой фазе на величины dcn
и в твердой на dcai „, а также изменения соответствующих коли-
честв компонента на V® dcn и V°dca< п. В результате этого можно на-
писать, что
(c„-i-c„)^ = m„ + Vadca,„. (73)
Если допустить, что каждый раз при поступлении новой порции
газа-носителя равновесие на каждой тарелке успевает установиться
и что система подчиняется уравнению Генри, то
Vardcn + Vldca, „ = (V? + V° Г) dcn = Еэфф dcn, (74)
где Уэфф — эффективный объем тарелки, равный У’+У"Г и пред-
ставляющий собой тот объем газа, который содержал бы весь
de
понент, как в газовой, так и в твердой фазах, а Г = а’—
dcn
уравнение 71).
Тогда уравнение (73) примет вид
(cn-i-cn)^ = V344)dc„,
или
dcn dV
-1 сп) Гэфф
Если для упрощения ввести относительные концентрации уп
и относительные объемы 0, приняв, что с0 — концентрация га-
за на первой тарелке, а V — весь объем га.за, прошедший через
142
ком-
(см.
(75)
(76)
колонку, то
Уп = ?\ *Уп=^> (77)
еО СО
₽ = d₽=I^T> (78)
vэфф vэфф
и уравнение материального баланса примет следующий вид:
dVn = dp. (79)
Уп-1 — Уп ' v '
Решение этого уравнения возможно, если принять в качестве
начальных условий
У1 = У2= • • • = Уп^
(при 0=0 и р0^р).
Здесь Р0=' и , причем Ги— объем исследуемого газа, введен-
* эфф
ный в колонку. Таким образом, объем чистого газа-носителя будет
равен V—V„. При 0>100 и принятых начальных условиях решением
уравнения (79) без больших погрешностей будет уравнение Гаусса:
1 -(₽-п)2/2П
Уп ---------Ё
ап У 2яп
(80)
Уравнение (80) является уравнением хроматографической кри-
вой и представляет собой распределение концентрации данного
компонента газа на n-й тарелке хроматографической колонки. Мак-
симуму на этой кривой отвечает условие
dyn___ 1 < 0— -(₽-П)2/2И
<*Р “ У2лп \ ~ J в
(81)
Отсюда можно установить, что максимальному значению уп
будет соответствовать 0=п. Следовательно,
Уп(макс) = Т7===- ' . 1 (82)
Если относить концентрацию компонента в газе не к его началь-
ной концентрации с0, а к максимальной на хроматографической кри-
вой снакс, то, поделив уравнение (80) на уравнение (82) и переходя
от у к с, можно получить уравнение хроматографической кривой
в довольно простой форме:
С -(3-П)2/2П
-----= е
Смакс
(83)
Можно охарактеризовать хроматографическую кривую, соот-
ветствующую кривой Гаусса, точками перегиба, которые находят,
приравнивая нулю вторую производную уп по 0. Тогда из уравнения
143
(81) получим
/JJ-П\»
лгуп1 ГМ—РУ__________П р \ 2п j —о
d₽2 у'2лп 1\ п 1 п J
откуда > или п—₽=Кп.
Отсюда можно рассчитать абсциссы точек перегиба:
(84)
и р2 = « + ]/«. (85)
При этом ординаты точек перегиба будут соответственно равны:
- -(я ± Уп-П)*/ЪП — —
~^—==е =е2 == 0,607. (86)
^макс
На рис. 30 представлена кривая хроматографической полосы в
коордйнатах 0—с/смакс, с максимумом при 0=п. На высоте, равной
0,607 смакс, кривая имеет точки перегиба. Ширина этой кривой на
данной высоте может быть вычисле-
на из уравнения (85) и будет рав-
на 2 ]/п. Соответственно полуши-
рина р’1/2 = ]/«- Половинная вы-
сота будет иметь ширину полосы,
равную 2,36 ]/п, а полуширину —
соответственно р;'/2 = 1,18JMп. Отре-
зок по оси 0 между точками пере-
сечения абсциссы с касательными,
проведенными к точкам перегиба
кривой, будет равен 4]/п, а по-
луширина =2 ]/rt.
Приведенные выражения очень важны, так как они позволяют
вычислить число эффективных теоретических тарелок хроматогра-
фической колонки, т. е. определить ее разделительную способность
в зависимости от выбранных условий опыта, или, наоборот, рассчи-
тать необходимые условия проведения опыта при заданном числе
эффективных теоретических тарелок.
Для такого расчета необходимо определить положение максиму-
ма хроматографической полосы и характеристику ее размывания,
выраженную через ширину полос на разных высотах.
В соответствии с выражениями (78) получим:
n = 0c =FT или Vt=nVM- (87)
имакс v эфф
Полуширина хроматографической полосы в единицах объема
газа на соответствующих высотах выразится следующими форму-
144-
лами:
У1/2^=УЭфф]/и;^
H/2V= 1,18УЭфф/п;
p-i/2^ = 2УЭфф У п. (88)
Отсюда следует, что
п = у = уц^у = У • (89)
U1/2V> \н1/2т/ U1/2v; 1 7
Таким образом, зная величину удерживаемого объема и полу-
ширину хроматографической полосы, можно рассчитать п — число
эквивалентных теоретических тарелок колонки, необходимое для
обеспечения разделения смеси.
Важной величиной является также высота эквивалентной тео-
ретической тарелки (ВЭТТ) Н, которая получается делением дли-
ны слоя адсорбента на число теоретических тарелок
Я = 4, (90)
(и"' Т V
в частности Н = 1 / • Отсюда следует, что ВЭТТ растет с
увеличением размывания полосы, т. е. размывание приводит к ухуд-
шению разделения.
Из рассмотренного следует, что теория тарелок позволяет рассчи-
тывать одну из важнейших характеристик хроматографических ко-
лонок — высоту эквивалентной теоретической тарелки, или, ины-
ми словами, разделительную способность колонки.
А. А. Жуховицкий и Н. М. Туркельтауб [7] вводят понятие кри-
терия разделения, при помощи которого также возможно характе-
ризовать разделительную способность хроматографической колонки
и рассчитывать оптимальные параметры опыта. >
Основным показателем хроматографического процесса является
выходная хроматографическая кривая. Поэтому ее параметры и
могут лечь в основу характеристики работы хроматографической
колонки, выражаемой, например, посредством критерия разделения.
Как показали А. А. Жуховицкий и Н. М. Туркельтауб, для ли-
нейной изотермы адсорбции выходная хроматографическая кривая
одного компонента определяется тремя величинами: положением
максимума, которое может быть определено по величине удержива-
емого объема VR, концентрацией в максимуме смакс и параметром о,
определяющим форму выходной кривой в соответствии с уравнением
с = смаксе-”2, (91)
где х — отсчитывается от максимальной ординаты.
Ширина полосы в единицах объема
р. = 4ГЗ ’ (92)
145
а расстояние между максимумами двух выходных кривых,
выраженное также в единицах объема, равно
AV₽ = T/S^.
(93)
С
Садх
Стах
Рис. 31. Хроматографическая кри-
вая в случае неполного разделения
компонентов смеси:
с' — максимум первого компонента;
с" — максимум второго компонента
Здесь I — длина слоя адсорбента; S — поперечное сечение ко-
лонки; Г>Эфф — Эффективный коэффициент диффузии; а — линей-
ная скорость га'за-носителя; A Q — разность теплот адсорбции двух
компонентов разделяемой смеси;
R — газовая постоянная; Т — аб-
солютная температура.
Чтобы характеризовать разде-
лительную способность колонки,
вводится два критерия разделения:
к = bVR _ ДУд
1 Ц1 + Ц2 ~ 2ц ’
(94)
X
и
(95)
Смакс । Смакс .
Л2 с
‘'мин
Второй критерий разделения К2
вводится в том случае, когда
происходит неполное разделение двух компонентов и измерить
AVR и р невозможно. Если в случае неполного разделения на вы-
ходной кривой имеется минимум (рис. 31), то в качестве параметров,
определяющих критерий, выбираются величины смин и емакс для двух
компонентов.
Из уравнений (92), (93) и (94) нетрудно найти связь критерия
разделения с параметрами опыта. Приближенно она может быть
выражена уравнением:
К — — 1Л 1а
Если принять приближенно, что
2с
гл ^макс
'2 р ' f
VMHH
(96)
то можно вывести связь между и К2:
(97)
Уравнение (96) может быть выражено несколько иначе, если
вспомнить, что зависимость коэффициента Генри от температуры
выражается уравнением
о
Г = Ае^ <98)
146
и считать, что для двух компонентов разделяемой смеси значение А
одинаково. Тогда
4Г = ГСТ „ (99)
г
Если принять ~=т и в уравнении (99) заменить ДГ на 1\ —Г2,
а Г на Г* Гз, то
К, = -И}-1. т/J«~ (100)
Л1 4(т+1) Г Рэфф • v '
Можно показать, что 2 D3^/a=H. Следовательно,
1/?£. (101)
Л1 4(m + l) г Н ' ’
Отсюда следует, что если требуется получить определенное зна-
чение Klt т. е. заданную полноту разделения, то необходимо иметь
минимальную длину слоя адсорбента /мин.
^мин = 8^(^±])2 Н, (102)
откуда число теоретических тарелок
'=й-8М^Т (103>
При заданных значениях и Н необходимая длина колонки
будет определяться множителем , обратную величину кото-
рого принято называть коэффициентом селективности и обозначать
Кс. Тогда
= (Ю4)
Величина Кс характеризует результат однократного разделе-
ния смеси. Она равна нулю при отсутствии разделения и стремится
к единице при полном разделении. Связь Кс с дается уравнением:
(Ю5)
Выведенная связь критерия разделения с параметрами опыта
позволяет заранее выбирать наиболее подходящие условия прове-
дения опыта, обеспечивающие-лучшее разделение. При этом экспе-
риментатор имеет возможность выбирать адсорбенты, температуру
•опыта, длину и сечение колонки, газ-носитель, его скорость и давле-
ние и, наконец, способ фиксации анализируемых веществ, т. е.
принцип действия и, главным образом, чувствительность детектора.
Практически разделение достигается при значениях 1.
147
Из рассмотрения уравнений (96) и (99) следует, что на величину
критерия разделения Кх прежде всего оказывает влияние природа
адсорбента, так как Кх прямо пропорционально A Q. Отсюда следу-
ет, что выбор адсорбента должен свестись к требованию наибольшей
селективности адсорбции компонентов анализируемой смеси. Эта
селективность должна проявляться в различии коэффициентов
Генри для компонентов разделяемой смеси и в разности теплот ад-
сорбции. Следует отметить, что важна именно достаточная разность
теплот адсорбции A Q, а не абсолютные ее величины Q. Часто слиш-
ком большие значения Q и Г нежелательны, так как это приводит
к затруднениям в десорбции, к ухудшению разделения и затягива-
нию времени опыта.
Расстояние между максимумами выходных кривых, выраженное
разностью удерживаемых объемов, не зависит от скорости газа-но-
сителя, тогда как ширина полосы р, связана с ней. При очень ма-
лых скоростях ширина полосы уменьшается с увеличением скорости.
В области же значительных скоростей, когда начинает играть роль не
продольная диффузия, а вихревая — она не зависит от скорости.
В кинетической области ширина полосы будет возрастать с увеличе-
нием скорости. Поэтому для лучшего разделения следует выбирать
такую скорость газа-носителя, при которой значение функции р=
=f(a) отвечало бы минимуму.
Одной из причин плохого разделения является не увеличение
скорости, а наличие значительного перепада давления в случае
длинных колонок, который приводит к существенному изменению
скорости по длине колонки.
На ширину полосы оказывает влияние степень зернения адсор-
бента. Эта зависимость не одинакова при различных скоростях газа-
носителя, так как она связана с диффузией. При малых скоростях,
при которых имеет место молекулярная диффузия, ширина полосы
практически не зависит от величины зернения. При достижении об-
ласти вихревой диффузии р, пропорционально корню квадратному
из диаметра зерна, в области внешней диффузии — корню квадрат-
ному из куба диаметра, а в области внутренней диффузии — первой
степени диаметра. Большое значение имеет равномерность зернения.
Наконец, на величину критерия разделения оказывает
влияние температура. Зависимость от температуры является слож-
ной, так как она влияет не только на величину адсорбции, но и на
коэффициент Генри и через эти две величины на критерий разде-
ления.
ПРИЧИНЫ РАЗМЫВАНИЯ ХРОМАТОГРАФИЧЕСКИХ ПОЛОС
Как уже было указано, причины размывания хроматографических
полос связаны с процессами диффузии в газе и порах адсорбента, а
также с процессами массообмена между газом и адсорбентом. ДЛя
упрощения этой сложной картины А. В. Киселев [ 1 ] в развитой им
148
теории описывает все эти процессы как единый процесс диффузии,
приписывая и массообмену эквивалентный по результатам процесс
диффузии и вводя эффективный коэффициент диффузии £)эфф.
Это упрощение позволяет представить процесс размывания хро-
матографической полосы как процесс, эквивалентный процессу
диффузии, эффективный коэффициент диффузии которого равен
сумме эффективных коэффициентов диффузии отдельных его стадий.
Введя ОЭфф, можно воспользоваться уравнениями молекулярной
диффузии, заменяя коэффициент молекулярной диффузии в них
суммарным эффективным коэффициентом диффузии.
В процессе хроматографирования молекулы анализируемого
газа, увлекаемые потоком газа-носителя вдоль колонки, одновре-
менно движутся также и хаотически во всех направлениях, причем
движение их вдоль потока вызывает размывание полосы. Пусть
время, за которое вследствие диффузии молекула сместится на рас-
стояние 6, будет t. Тогда, согласно уравнению диффузии Эйнштейна,
можно записать:
6а = 2Г>£, (106)
где D — коэффициент молекулярной диффузии в газе.
Вследствие того, что путь между зернами в хроматографической
колонке с набивкой является извилистым, коэффициент продольной
диффузии Dn отличается от коэффициента свободной молекулярной
диффузии D, что можно учесть, введя коэффициент извилистости
= (Ю7)
где уи может быть равно или больше единицы.
При движении газа через колонку с насадкой, даже при лами-
нарном потоке газа, происходит некоторое завихрение газа вокруг
зерен насадки. Это обстоятельство также приводит к размыванию
хроматографической полосы. Время, в течение которого газ движется
около зерна диаметром d3, равно приблизительно d3/a, где а — ли-
нейная скорость газа. В соответствии с уравнением Эйнштейна
можно написать
d32«HB|-, (108)
где DB — коэффициент вихревой диффузии.
Отсюда
DB^d3a = M3a, ' (109)
где А — коэффициент пропорциональности, не зависящий от ско-
рости газа а. Из уравнения (109) следует, что, в отличие от коэффи-
циента продольной диффузии DB, коэффициент вихревой диффузии
DB зависит от скорости газа.
В реальных процессах адсорбция и десорбция происходят с ко-
нечными скоростями, что также приводит к размыванию. Этот про-
149
цёСС массообмена между газом и адсорбентом в простейшем случае
можно выразить следующим уравнением:
de
(Н°)
где с — концентрация в газовой фазе, t — время, ум— константа
скорости массообмена. Если провести интегрирование от с0 до с и /0
до t,
YM = TInT> (111)
где с0 — начальная концентрация при /=0.
Из уравнения (111) вытекает, что — является временем, в те-
Ум
чение которого концентрация меняется в е раз. За это время мо-
лекулы данного компонента газа переместятся, при линейной ско-
рости потока газа-носителя се, на расстояние
6 = -U. (112)
ГМ
Это смещение полосы можно представить себе в виде диффузион-
ного процесса с коэффициентом диффузии массообмена DM и величи-
ну смещения выразить в соответствии с уравнением Эйнштейна
62 = 2Ом1 (113)
ГМ
Сопоставляя это уравнение с уравнением (112), получаем:
или
(114)
Эффективный коэффициент диффузии является суммой
трех коэффициентов диффузии: продольной Dn, вихревой DB и мас-
сообмена Ом, которыми мы охарактеризовали процессы, вызываю-
щие размывание хроматографической полосы в колонке:
О8фф=Оп + Ов + Ом. (П5)
Подставив в это выражение соответствующие значения коэф-
фициентов диффузии из уравнений (107), (109) и (114), получим
Оэфф = УиО + Н« + 2^«2- (Н6)
Это уравнение устанавливает связь между линейной скоростью
потока газа-носителя а и эффективным коэффициентом диффузии,
характеризующим сложный процесс движения хроматографической
полосы в колонке.
150
Уравнение Фика для молекулярной диффузии имеет вид
(117)
где D — коэффициент молекулярной диффузии. Интегрируя это
уравнение и заменяя D на РЭфф, можно получить уравнение
(Дх)2
= (П8)
^макс
Это уравнение является уравнением Гаусса. Оно позволяет рас-
считать значение смещения полосы Ах относительно среднего зна-
чения х (при с=смакс), т. е. величину полуширины полосы, для ко-
торой определяется значение с, за время I, прошедшее от начала
впуска пробы газа в начале колонки до появления концентрации с
на расстоянии х+Ах (рис. 32).
Если принять, что с/с™х ^стп
нис. oz. ч-'орма хроматографической
Выражая эту полуширину по- полосы по теории диффузии [1]
лосы в единицах объема газа, т. е.
умножая Ах на свободное поперечное сечение колонки SCB, получим:
SCBAx = 2SCB /РЭфф/ = АГ. (120)
Учитывая, что время появления газа с концентрацией с у выхода из
колонки длиной I при скорости движения полосы адсорбированного
газа ис составляет:
/ = -,
ис
а
а также, что ис = у, получим:
/ = £, (121)
и окончательно:
АГ = 2SCB # (122)
Обратим внимание на то, что уравнение (118), характеризующее
размывание хроматографической полосы, идентично уравнению (83),
выведенному на основании теории тарелок. Оба они являются урав-
нениями Гаусса и характеризуют распределение концентрации ана-
151
лизируемого газа по ширине хроматографической полосы. Это об-
стоятельство позволяет связать друг с другом обе теории и на этом
основании выразить основную величину И, применяемую в теории
тарелок, через эффективный коэффициент диффузии Пэфф.
В самом деле, из уравнения (83) следует,что на высоте хромато-
графической кривой, характеризуемой как с=/(Р), равной 0,368,
а
А^ = УэФФ/2^. (123)
Приравнивая (123) к уравнению (122), получаем:
У9ФФГ2Л = 25св]/М^
или
пУэфф = 25св (124)
Умножив обе части уравнения (124) на п и разделив и умножив его
правую часть на I, имея в виду, что SCB/=V; Н= и К/Кэфф=п,
при Р=н, можно получить:
2Рэфф1 _ 1
аН ~
или
И = 2£эФфГ . 025)
Подставив в уравнение (125) значение Пэфф из уравнения (116),
получаем уравнение Ван-Деемтера [4, 5] связывающее величину
Н с а:
Д = 2МЧ + ^££ + £а. (126)
Следовательно, имеется какая-то оптимальная скорость газа-
носителя а, при которой значение Н становится наименьшим, т. е.
эффективность колонки наибольшей. Поэтому следует выбирать
такую скорость газа-носителя, при которой Н будет близко к ми-
нимальному значению и только немного будет увеличиваться с
изменением а.
Таким образом, размывание хроматографических полос, являю-
щееся одной из существенных причин плохого разделения смесей
веществ методом газо-адсорбционной хроматографии, может быть
устранено или же сведено до минимума правильным выбором усло-
вий проведения хроматографического опыта, проведением его в
оптимальных условиях. Однако не всегда существуют реальные воз-
можности осуществления таких условий опыта. Кроме того, в слу-
чае сложных смесей учет всех возможных факторов практически
152
невозможен, что затрудняет успешное применение газо-адсорбцион-
ной хроматографии для разделения и анализа более или менее слож-
ных смесей.
Выход из положения может быть найден двумя путями: либо
изменением свойств адсорбента, что может привести к спрямлению
изотермы адсорбции и уменьшению адсорбционного сродства по от-
ношению к сильно адсорбирующимся компонентам смеси, либо пу-
тем одновременного воздействия на адсорбированное вещество газа-
носителя и движущегося температурного поля. Последнее было пред-
ложено А. А. Жуховицким [8] в виде нового метода хроматографии,
названного им хроматермографией.
X РОМАТ Е РМОГРАФИЯ
Термический фактор в адсорбционной газовой хроматографии
является одним из наиболее действенных. Воздействие температуры
на хроматографическую полосу приводит к выпрямлению изотермы
адсорбции, ускорению внутренней диффузии и, что самое главное,
позволяет изменять адсорбционные свойства в широком диапазоне,
делая многие процессы адсорбции обратимыми.
Однако действие термического фактора имеет и обратный эффект.
Так, повышение температуры вызывает уменьшение расстояния
между максимумами двух хроматографических кривых, что приводит
к уменьшению критерия разделения. Ширина полосы увеличива-
ется в случае продольной диффузии, что также влечет за собой ухуд-
шение разделения. Поэтому применение термического фактора в
хроматографии может дать положительный эффект только в опреде-
ленных случаях. Таким случаем, приводящим к резкому улучшению
хроматографического разделения на адсорбентах, является одновре-
менное воздействие на разделяемую смесь потока газа-носителя и
движущегося во времени и пространстве температурного поля.
Одним из наиболее важных результатов одновременного воздей-
ствия на хроматографическую полосу потока газа-носителя и тем-
пературного поля является сжатие полосы, что и приводит к сущест-
венному улучшению разделения. Такое сжатие может произойти,
если замыкающий край полосы будет двигаться быстрее, чем перед-
ние слои. Чтобы это осуществить, требуется наличие движущегося
температурного поля с градиентом температуры, возрастающей про-
тив направления потока га за-носителя. Если условия действия по-
тока газа-носителя с одновременным действием температурного поля
с температурой, возрастающей в направлении, противоположном
направлению движения газа, соблюдаются, то мы имеем дело со ста-
ционарной Хроматермографией. Теория стационарной хроматермо-
графии разработана А. А. Жуховицким и Н. М. Туркельтаубом [7].
Рассмотрим для простоты движение по слою адсорбента одного
компонента газа, обладающего линейной изотермой адсорбции.
153
Пусть скорость движения полосы адсорбированного газа
= (127)
а коэффициент Генри Г зависит от температуры:
Q
Г = Ае*т.
Здесь Q — теплота адсорбции, А — коэффициент пропорциональ-
ности.
Из уравнения (98) следует, что скорость движения полосы ком-
понента не одинакова в различных местах температурного поля, а
следовательно, и подлине слоя адсорбента. Обозначим скорость дви-
жения температурного поля через w. Если uc>w, то полоса адсор-
бированного вещества будет передвигаться быстрее движения печи.
Поэтому полоса попадет из зоны с высокой температурой в зону с
более низкой, так как температура падает в направлении движения
газа. При этом значение Г возрастет, что приведет к уменьшению
ис. Вследствие этого в некоторой зоне температурного поля, отвеча-
ющей температуре десорбции компонента в данных условиях, будет
соблюдаться равенство uc—w. После того, как полоса займет эту
зону, она будет двигаться со скоростью, равной скорости движения
печи, т. е. температурного поля. Следовательно, и любая другая
полоса любого другого компонента разделяемой смеси газов будет
двигаться с этой же скоростью, но при иной, свойственной другому
компоненту температуре десорбции. Поэтому ту температуру, при
которой происходит десорбция данного компонента и движение его
полосы вдоль слоя адсорбента со скоростью uc=w, называют харак-
теристической температурой данного компонента и обозначают 7\а .
Из уравнений (98) и (127), а также полагая, что
можно вывести зависимость характеристической температуры от
теплоты адсорбции и параметров опыта
• (128)
ха₽ R In Дц ' '
Если T]=const, т. е. условия опыта заданы, то Тхар зависит от
теплоты адсорбции данного компонента на выбранном адсорбенте.
Таким образом, разность характеристических температур двух
компонентов разделяемой смеси газов будет определяться разницей
их теплот адсорбции. Для гомологического ряда парафиновых уг-
леводородов, согласно правилу Траубе, AQ=Qch2, т. е. разность
теплот адсорбции двух соседних членов гомологического ряда яв-
ляется величиной постоянной и равной теплоте адсорбции группы
СН2. Измеряя характеристические температуры компонентов раз-
1 54
деляемой смеси, что не представляет особых трудностей, можно
рассчитать теплоту адсорбции для каждого компонента смеси.
Если для какого-либо опыта Q=const, то Т будет зависеть от
т]. Следовательно, изменяя величину т], можно добиться разделения
смеси. Если, например, увеличивать скорость газа-носителя а,
оставляя постоянной скорость движения печи w, то при этом полоса
компонента будет передвинута в область более низких температур.
Если же увеличивать w, оставляя а постоянной, то полоса будет на-
ходиться в области более высоких температур. Таким образом,
можно подобрать такое значение т], которое обеспечит разделение
смеси, даже если разность Q будет незначительной.
Итак, хроматермография позволяет «расставить» все компоненты
сложной смеси по областям своих характеристических температур,
причем после «расстановки» все компоненты разделенной смеси бу-
дут передвигаться с одной и той же скоростью, равной скорости дви-
жения печи. Это обстоятельство делает возможным применение
хроматермографии для качественного анализа и идентификации ве-
ществ из смеси. В самом деле, если все компоненты строго фиксиро-
ваны по своим характеристическим температурам, то положение ком-
понента на хроматермограмме будет однозначно идентифицировать
его. Отсутствие пика на хроматермограмме при данной температуре
укажет на отсутствие в смеси одного определенного компонента.
Уравнение (128) характеризует также и ширину хроматографи-
ческой полосы. Действительно, из этого уравнения следует, что в
данных условиях вещество может находиться на адсорбенте только
при свойственной ему Следовательно, полоса сжимается,
концентрация данного компонента повышается, происходит обо-
гащение. Это свойство является важнейшей особенностью хроматер-
мографии. В отличие от проявительной и распределительной хро-
матографии, для которых характерно размывание полосы, т. е.
уменьшение концентрации вещества, в хроматермографии проис-
ходит сжатие полосы, наблюдается повышение концентрации. Это
обстоятельство не только облегчает условия разделения смеси
близких по свойствам веществ, но и имеет самостоятельный практи-
ческий интерес. Этим свойством обогащения можно воспользоваться
для повышения концентрации веществ.
Для характеристики этого свойства введем понятие коэффициен-
та обогащения О. Мы будем называть коэффициентом обогащения
отношение максимальной концентрации смакс к начальной концен-
трации с0.
О = £мак_с. (129)
со
Стационарная хроматермография позволяет решать ряд задач
по разделению газовых смесей. Она обладает большими преимущест-
вами перед обычным проявительным методом, к их числу относится
обогащение, возможность определения теплот адсорбции по харак-
155
теристическим температурам, значительное сужение хроматографи-
ческих полос, приводящее к лучшему разделению и облегчающее
десорбцию сильно адсорбирующихся веществ. Однако расстояния
между хроматографическими полосами компонентов разделяемой
смеси в этом случае оказываются меньше, чем в простом прояви-
тельном методе. Это происходит вследствие того, что в стационарной
хроматермографии различные компоненты движутся по слою ад-
сорбента с одинаковыми скоростями, тогда как в проявительном
методе лучше адсорбирующийся компонент движется со скоростью
меньшей, чем хуже адсорбирующийся.
Для увеличения степени разделения в хроматермографии необ-
ходимо, чтобы движение лучше адсорбирующегося вещества про-
исходило при более низкой температуре, чем движение хуже адсор-
бирующегося. Тогда первый будет двигаться медленнее второго. Ре-
шение этого вопроса возможно, если температурное поле будет
иметь градиент температуры со знаком, противоположным направ-
лению потока газа-носителя. При этом различают два возможных
варианта: направление движения печи с обратным градиентом сов-
падает с направлением потока или оно противоположно направлению
потока. В первом варианте на компоненты разделяемой смеси будет
надвигаться поле с понижающейся температурой и движение ком-
понентов будет тормозиться. Этот метод получил название метода
адсорбционного торможения. Однако вследствие того, что метод
требует специальных мер для преодоления возможности необрати-
мой адсорбции, он не получил своего развития.
Осуществление второго варианта приводит к тому, что на ком-
поненты разделяемой смеси будет надвигаться поле с возрастающей
температурой, вследствие чего все компоненты в конце концов бу-
дут десорбированы, причем ускоренно [7].
Метод хроматермографии получил распространение для разде-
ления и анализа не только газовых смесей, но и жидких веществ
[9]. В последнем случае разделение происходит в паровой фазе, а
температурное поле служит одновременно для испарения жидкости.
Существенным преимуществом такого разделения жидких смесей
перед обычным проявительным методом, требующим жидкого проя-
вителя, является то, что в этом методе проявителем служит газ, а
пары компонентов разделяемой смеси по выходе из колонки конден-
сируются и собираются в чистом виде, свободными от проявителя.
ТЕПЛОДИНАМИЧЕКИЙ МЕТОД
Метод хроматермографии позволяет приблизить хроматографи-
ческий метод анализа к непрерывному методу, что имеет очень боль-
шое практическое значение. На основе хроматермографии А. А.
Жуховицким и Н. М. Туркельтаубом [7] был разработан так назы-
ваемый теплодинамический метод, представляющий собой сочетание
непрерывного фронтального метода с движущимся температурным
156
полем. Направление градиента температурного поля при этом про-
тивоположно направлению потока разделяемой смеси газа и движе-
нию печи. Подобно фронтальному методу анализа, в теплодинами-
ческом методе анализируемая смесь газа подается в колонку непре-
рывно. Однако в отличие от фронтального метода, благодаря воз-
действию движущегося температурного поля, одновременно с раз-
делением смеси на отдельные компоненты происходит подготовка
адсорбента к приему следующих порций анализируемого газа. Та-
ким образом, подача газа для анализа происходит непрерывно, а
результаты анализа выдаются периодически.
Рассмотрим наиболее простой случай. Пусть газ-носитель, со-
держащий одно анализируемое вещество с концентрацией с0, не-
прерывно пропускается через колонку с адсорбентом. При условии
постоянства температуры, линейности или выпуклости изотермы ад-
сорбции, а также при отсутствии факторов, вызывающих размыва-
ние, через время t какой-то слой адсорбента полностью отработа-
ется, т. е. будет насыщен анализируемым веществом. При этом за
слоем концентрация изменится скачком до с0. Это время t будет
равно
t = (130)
ас0 ' '
где а0 — начальное количество вещества в единице объема слоя
адсорбента. Резкий фронт этого слоя движется со скоростью
ис = ^. (131)
Если же одновременно на фронт воздействует изменяющееся темпе-
ратурное поле, то скорость его передвижения будет меняться со
временем.
Пусть скорость движения температурного поля (печи) w будет
больше, чем скорость передвижения фронта. Тогда температурное
поле будет надвигаться на слой адсорбента, насыщенного анализи-
руемым веществом, вследствие чего температура в нем будет воз-
растать, что приведет к увеличению скорости передвижения его
фронта. Наконец, скорость движения фронта сравняется со ско-
ростью движения печи:
uc = w.
При этом фронт будет передвигаться при определенной харак-
теристической температуре. Таким образом,
ас0
Щ» = —,
«о
или
Со w
— =— =Т1
а0 а 1
(132)
157
(133)
(134)
(135)
Если соблюдается уравнение Генри, то уравнение (132) совпа-
дает с (127), а характеристическая температура будет определяться
из уравнения (128).
Понятие характеристической температуры при теплодинами-
ческом методе применимо также и при выпуклой изотерме адсорб-
ции. Однако в этом случае характеристическая температура будет
зависеть от концентрации. Пусть изотерма адсорбции изучаемого
вещества описывается уравнением Лэнгмюра
Ьс
а-ахТ+Ьс'
где и b — постоянные. Тогда, в соответствии с уравнением (132)
1 Ьс0
п = - < ,
00
откуда постоянная в уравнении Лэнгмюра
Ь =---!— .
W — со
Учитывая, что b зависит от температуры
Q
b = boeRr,
получим выражение для характеристической температуры в тепло-
динамическом методе:
<136>
Таким образом, фронт вещества будет двигаться при своей ха-
рактеристической температуре со скоростью печи. Отличие от ста-
ционарной хроматермографии состоит в том, что в слой вещества
все время поступают новые порции смеси. Кроме того, надвигаю-
щееся температурное поле десорбирует вещества, поток газа уносит
их вперед, где они адсорбируются на свежем адсорбенте. Однако
адсорбция будет происходить вблизи зоны характеристической тем-
пературы, вследствие чего как ранее десорбированные, так и вновь
поступающие вещества будут накопляться вблизи этой зоны. Если
смесь содержит несколько компонентов, то все они будут накоплять-
ся вблизи зон своих характеристических температур.
Рассмотрим наиболее интересный вариант теплодинамического
метода. Согласно этому варианту, анализируемая смесь непрерывно
подается в колонку с адсорбентом, на слой которого надвигается
печь. Длина печи значительно меньше длины слоя адсорбента в
колонке. Печь, достигнув нижнего края колонки, возвращается
в исходное положение и снова продолжает двигаться по слою ад-
сорбента. При таком варианте осуществляется теплодинамическое
обогащение компонентов и происходит разделение их смеси. Зоны
158
компонентов передвигаются вместе с движением печи. Освобождаю-
щаяся за печью область адсорбента охлаждается и начинает играть
роль своеобразного фильтра, поглощающего все компоненты смеси.
Благодаря этому в ту часть колонки, где продолжается движение
печи, поступление смеси прекращается и она продолжает работать
в условиях стационарной хроматермографии. Одновременно в хо-
лодной части печи происходит накопление анализируемой смеси.
Таким образом, новый цикл анализа начинается с того слоя адсор-
бента, на котором уже адсорбировано некоторое количество смеси,
что еще больше способствует обогащению смеси анализируемым
компонентом.
Экспериментальное выполнение такого анализа, сочетающего
обогащение с разделением, возможно в три стадии: фронтальное
накопление вещества, промежуточная ступень, позволяющая уве-
личить концентрацию и разделить компоненты смеси, т. е. тепло-
динамический процесс, и термическая десорбция.
Очевидно, что действие температуры на стадии накопления ве-
щества излишне, поскольку необходимо сохранить на адсорбенте
все количество накопленного вещества. Кроме того, без применения
температурного поля будет накапливаться больше вещества. В са-
мом деле, при фронтальном анализе передний фронт адсорбирован-
ного вещества будет двигаться по адсорбенту медленнее, чем при
теплодинамическом методе. Поэтому при одинаковых скоростях
подачи газа при фронтальном анализе накопится больше вещества,
чем при теплодинамическом.
Пусть х — расстояние от края полосы фронта до конца слоя,
на котором совершается первая стадия — накопление.
__ а/ _ I
Время второй стадии — теплодинамического процесса, т. е. сжатия
полосы, определяется значением скорости движения печи w. Тогда
время второй стадии tz
(137)
Отсюда можно определить время первой стадии
/Г
Или, если обозначить — = — время полного насыщения, то
G = U-?2, (139)
т. е. время полного насыщения должно быть уменьшено на время
прохождения печи.
На второй стадии количество адсорбированного вещества не пре-
вышает того количества, которое было адсорбировано при стадии
159
фронтального накопления, но концентрация вещества за счет сжа-
тия полосы резко возрастает. При этом некоторая малая доля слоя
после окончания второй стадии должна быть свободна от адсорби-
рованного вещества, чтобы можно было осуществить третью ста-
дию — воздействие термического фактора, десорбцию. В третьей
стадии а=0, так как весь подаваемый газ задерживается в первой
стадии и не поступает в зону термической десорбции.
Для лучшего разделения адсорбент выбирают так, чтобы он
хорошо адсорбировал вещество, подлежащее концентрированию,
и плохо адсорбировал газ-носитель.
РАСЧЕТ ХРОМАТОГРАММ
В газовой хроматографии, как правило, анализ проводится не
путем отбора отдельных порций анализируемого газа и не послойно,
как это часто производится в хроматографическом анализе жидко-
стей, а непрерывно, непосредственно на выходе из колонки. Для
этой цели применяются различного рода детекторы. В большинстве
случаев применяют дифференциальные детекторы, позволяющие
получать хроматограммы, состоящие из ряда пиков. Успех хрома-
тографического анализа, а также его точность в значительной сте-
пени зависят от метода расчета полученных хроматографических
пиков.
Для количественного определения состава анализируемой смеси
по хроматографическим кривым, получаемым при помощи диффе-
ренциальных детекторов, можно измерять высоту максимумов
пиков h, площадь пиков П или произведение удерживаемого объема
на высоту пика VRh.
Концентрация в максимуме смакс пропорциональна высоте пика.
В тоже время смакс пропорциональна количеству нанесенного ве-
щества <?;.
макс 2Г У л1Н ’
Отсюда следует, что высота пика h пропорциональна количеству
нанесенного вещества.
Площадь пика П также характеризует количество нанесенного
вещества где
+ со
q~S § aCjdt. (141)
— со
Здесь cz — концентрация i'-того компонента в смеси; а — линейная
скорость газа; S — площадь поперечного сечения колонки.
Ширина полосы р выражается соотношением
160
Удерживаемый объем VR пропорционален длине и сечению колонки,
а также коэффициенту Генри
Vfi=rS/. (142)
Отсюда следует, что
= {143)
4 Г Ыэфф V Ь’эфф
где В — постоянная величина.
Так как площадь пика П = ^-,то
__ VRh = 2B-^=. (144)
г Ъ'эфф
Следовательно, VRh пропорционально количеству нанесенного ве-
щества.
Следовательно, как высота пика h, так и его площадь П, а также
произведение VRh могут служить для определения количества ана-
лизируемого вещества.
Площадь пиков определяют планиметром, взвешиванием на
аналитических весах вырезанного из бумаги пика и сравнением
веса куска той же бумаги известной площади, либо умножением
половины высоты пика на его ширину. В этом случае высота и ши-
рина пика определяются так, как это показано на рис. 2. Удержи-
ваемый объем определяют по оси объемов от момента впуска порции
анализируемой смеси до максимума пика.
Все три параметра могут применяться для расчета количества
анализируемого вещества по полученной хроматограмме одним из
следующих методов: нормировкой, внутренней стандартизацией,
нормировкой с введением калибровочных коэффициентов и абсо-
лютной калибровкой [7]. Рассмотрим каждый из этих методов.
Метод нормировки. Методом нормировки определяют соотно-
шение между количеством анализируемых компонентов и высотами
пик или другими параметрами хроматографических кривых. При
этом сумма одного из параметров, например, высоты, для всех ком-
понентов смеси принимается равной 100%. Тогда высота пика од-
ного из компонентов, выраженная в процентах, будет давать про-
центное содержание этого компонента в смеси.
Метод применим только в том случае, когда измеряемый пара-
метр для каждого компонента одинаково зависит от концентрации.
В наиболее часто применяемых детекторах, измеряющих разность
теплопроводностей анализируемого компонента и газа-носителя,
такая зависимость будет почти всегда неодинаковой. Это обстоя-
тельство следует учитывать при применении метода нормировки.
Метод внутренней стандартизации. При использовании метода
внутренней стандартизации к анализируемой смеси добавляют опре-
деленное количество известного вещества. Это вещество может
6 Б. Б. Айвазов 161
отсутствовать или присутствовать в анализируемой смеси. По полу-
ченной хроматограмме сравнивают значение одного из трех пара-
метров для введенного и анализируемых веществ. Так как любой
из параметров пропорционален количеству вещества, то таким путем,
зная содержание введенного вещества, можно определить содержа-
ние искомого компонента и состав всей смеси. При этом следует
иметь в виду, что условия, необходимые для метода нормировки,
остаются справедливыми и обязательными и в методе внутренней
стандартизации. Поэтому лучше в качестве стандартного вещества
применять один из присутствующих в смеси компонентов. Еще
лучше проводить стандартизацию для каждого из анализируемых
компонентов. Следует также иметь в виду, что коэффициент стан-
дартизации будет постоянным лишь в том случае, если линейная
зависимость между концентрацией вещества и показаниями детек-
тора сохраняется во всем диапазоне исследуемых концентраций.
Метод нормировки с введением калибровочного коэффициента.
В этом методе также принимают сумму одного из параметров за
100%, однако предварительно проводят калибровку для всех ана-
лизируемых компонентов, после чего для каждого из них находят
калибровочный коэффициент. Калибровку проводят так, что один
из постоянно присутствующих и преобладающих компонентов смеси
принимают за сравнительный, а калибровочный коэффициент для
этого компонента приравнивают единице. Тогда калибровочные
коэффициенты для других компонентов рассчитывают по формулам:
zz _V, . ь' . ь' С^7?Л)сС1 /IK'i
где /гс, сс относятся к веществу, принятому за сравнительное, a hi
н с; — к искомым веществам.
Метод абсолютной калибровки. Наиболее точным методом яв-
ляется метод абсолютной калибровки. В этом методе по хромато-
граммам известных веществ, взятых в разных, но точно измеренных
количествах, строят график зависимости высоты пиков или значе-
ний других параметров от количества нанесенного на адсорбент
вещества. При хроматографировании анализируемой смеси, содер-
жащей те же компоненты, пользуются полученными калибровоч-
ными графиками. Анализ смеси, состоящей из i компонентов, тре-
бует построения i графиков. Для анализа малых концентраций
веществ этот метод является единственно надежным.
Экспериментальные данные свидетельствуют о том, что, кроме
метода абсолютной калибровки, наиболее точные данные позволяет
получить метод нормировки с введением калибровочных коэффи-
циентов. Относительная ошибка этого метода составляет не более
1—2%. Важно и то, что при этом методе нет необходимости знать
точное количество пробы. Ошибки, возникающие в связи с коле-
баниями параметров опыта, учитываются калибровочными коэф-
фициентами.
162
Для точности измерений имеет значение форма выходных кри-
вых. В случае острых пиков (удерживаемый объем VR мал) измере-
ние площадей может привести к существенным ошибкам вследствие
недостаточной точности измерения ширины пиков.- Большие зна-
чения VR приводят к сильному размазыванию полосы и снижению
концентрации в максимуме пика. В этих случаях расчеты лучше
вести по площади. Однако на точности измерения площадей сказы-
ваются колебания в скорости газа-носителя, в скорости движения
ленты самописца, а также форма выходной кривой. При измерении
площади планиметром форма кривой не играет роли. Метод взве-
шивания также дает возможность избежать влияния ее формы.
Однако в этом случае ошибка возникает вследствие неоднородности
бумаги по плотности, а также при взвешивании, особенно, если
площади малы. Расчет площади по результатам измерений высот
и ширины пиков дает ошибку, величина которой связана с характе-
ристикой кривой. Этот метод оказывается совершенно непригодным
в случае кривых несимметричной формы.
Расчет по произведению удерживаемого объ-
ема на высоту пика имеет то преимущество, что он дает
возможность рассчитывать концентрацию анализируемых веществ
в том случае, когда отсутствует полное разделение двух соседних
компонентов. Однако ошибки при этом методе расчета сильно воз-
растают, если значение Г^>1.
Метод расчета по высотам пиков имеет преимущества
перед расчетом по площадям в том отношении, что он предъявляет
менее строгие требования к полноте разделения. Он особенно при-
годен в тех случаях, когда пики узки и высоки. Поэтому этим
методом следует пользоваться в хроматермографии, теплодинамиче-
ском методе, а также в капиллярной хроматографии. При абсолют-
ной калибровке, особенно при расчете по методу высот пиков, сле-
дует обращать внимание на то, чтобы вся аппаратура работала в
одинаковых условиях.
Окончательный выбор способа расчета и метода калибровки сле-
дует сделать только после учета всех возможных ошибок и оценки
их величины.
ОПРЕДЕЛЕНИЕ НЕКОТОРЫХ ФИЗИКО-ХИМИЧЕСКИХ ВЕЛИЧИН
Наряду с чисто аналитическими задачами, газовая хроматогра-
фия позволяет также проводить определение ряда физико-химиче-
ских характеристик, касающихся как анализируемых веществ, так
и адсорбентов [7]. Это новое направление хроматографии получило
значительное развитие за последние годы.
Определение изотермы адсорбции. Одной из важнейших харак-
теристик хроматографического метода является удерживаемый
объем. В случае газо-адсорбционной хроматографии из величины
удерживаемого объема можно рассчитать коэффициент Генри. Если
6*
163
изотерма адсорбции линейна, удерживаемый объем не зависит от
концентрации хроматографируемого вещества. В случае нелиней-
ной изотермы он зависит от концентрации. Поэтому, как впервые
показал Е. Дж. Глюкауф [10], из величин удерживаемых объемов
можно рассчитать изотерму адсорбции. При этом необходимо, од-
нако, чтобы размывание хроматографической полосы, вызванное
иными, чем криволинейность изотермы, причинами, было мало.
При криволинейной изотерме и равновесной хроматографии
скорость движения полосы при данной концентрации может быть
выражена следующим уравнением:
или
и<: da/dc’
at
da/de
(146)
(147)
где at представляет собой объем газа-носителя (V/j(c)), который
должен быть пропущен до момента появления концентрации с. От-
сюда
= (148)
de х ' '
Для вычисления величины адсорбции а необходимо проинтегри-
ровать уравнение (148). Так как а=0, при с=0, поэтому
а с
Sda=T (c)dc-
о о
(149)
Для определения интеграла в этом выражении необходимо изме-
рить всю выходную кривую до с=0, что невыполнимо. Выход из
затруднения может быть найден, если интеграл выразить через
величину, измеряемую непосредственно, т. е.
с 6макс смакс
^VR(c)dc= VR(c)dc— VR (с) de. (150)
0 0 с
Первый член правой части уравнения (150) равен количеству
нанесенного вещества qR Отсюда количество адсорбированного
вещества
смакс
<7;— $ VR(c)dc
а =-------~х------. (151)
Таким образом, для измерения величины а необходимо опреде-
лить заштрихованную площадь на хроматографической кривой, как
это показано на рис. 33.
161
Недостатком этого метода является необходимость проведения
опыта в условиях равновесной хроматографии. А. А. Жуховицким
с соавторами [11] разработан метод, позволяющий проводить ис-
следование в неравновесных условиях. Он основан на применении
хроматермографии.
Необходимо выяснить область выходной кривой, в которой вы-
полняются условия равновесия. Такой областью является область
концентраций, лежащая вблизи ма-
ксимальной концентрации на выход-
ной кривой. Если уравнение кинети-
ки адсорбции
g = B(c-i/), (152)
то при стационарном процессе а=f(y),
а у~х— wt. Здесь х — расстояние,
t — время, w — скорость движения
адсорбционной волны, равная скоро-
сти движения печи. Тогда
da du
4t = ~wtf
(153)
Отсюда следует, что
Рис. 33. Кривая к расчету изо-
термы адсорбции из хромато-
граммы [11]
da В , ч
— т- = — (с — у).
dy w ' -ji .
(154)
В максимуме величины адсорбции а, в котором ^ = 0,
ствляется равенство с=у и наступает адсорбционное равновесие.
Для определения величины а следует обратиться к уравнению
осуще-
ас—D ~г = wa,
dy .
откуда
с D de,
а —---------т-.
П w dy
de
Если в максимуме выходной кривой = 0, то
—- ^макс
П
(155)
(156)
(157)
Таким образом, величину адсорбции а легко рассчитать по мак-
симуму выходной кривой.
При отсутствии продольной диффузии (0=0),
и максимумы с и а совпадают. Следовательно, если влияние про-
дольной диффузии мало, можно определить связь а с с, меняя на-
чальные концентрации изучаемого газа и определяя смакс по вы-
ходным кривым.
165
Если £)=Н=0, то рассчитывать а следует по уравнению (156),
определив предварительно коэффициент диффузии D. Последнее
бывает необходимым в сравнительно редких случаях, главным об-
разом при высоких температурах.
Другой метод, позволяющий определять изотерму адсорбции,
проводя опыт в неравновесных условиях, основан на применении
фронтального анализа. Этот метод, как и только что рассмотренный,
разработан А. А. Жуховицким и Н. М. Туркельтаубом [7].
Как было подробно разобрано в первой главе, фронтальный ме-
тод позволяет определить изотерму адсорбции, измеряя удерживае-
мый объем и вычисляя величину адсорбции по уравнению (6)
a = VRc.
Однако это условие требует соблюдения адсорбционного равновесия
и отсутствия продольной диффузии.
Предлагаемый метод позволяет рассчитать изотерму адсорбции
без соблюдения этих условий. Для этого на выходной кривой фрон-
тального анализа необходимо найти точку, которая передвигалась
-бы по слою адсорбента со скоростью, выражаемой уравнением (127)
Такой точкой является точка, в которой концентрация равна по-
ловине начальной, т. е.
Для получения изотермы адсорбции опыт проводят при различных
начальных концентрациях и по выходным кривым определяют удер-
живаемый объем, отвечающий концентрации, равной ~, т. е.
Уд, 1/2. Рассчитывают удельный удерживаемый объем и вычисляют
величину удельной адсорбции а0 по формуле
а° = с0^,1/а. (158)
Определение поверхности адсорбента.Вторая задача, решае-
мая при помощи хроматографического метода, это определение по-
верхности адсорбента. Методика измерения, предложенная Е. Кре-
мер [12], основывается на предположении, что постоянная А в урав-
нении (98) пропорциональна числу центров поверхности адсорбен-
та, на которых может происходить адсорбция. Поэтому
Q
VR^k'SaAce^, (159)
где 5аяс — поверхность адсорбента, a k' — постоянная, зависящая
от изменения энтропии при адсорбции. Е. Кремер полагает, что
величины k' и Q мало изменяются при адсорбции, если адсорбенты
отличаются друг от друга величиной поверхности, но имеют близ-
кий химический состав. В таком случае упрощается определение
166
поверхности адсорбента. Формула (159) позволяет вычислить по-
верхность адсорбента, если известны Q и k’ и измерены удерживае-
мые объемы. Для измерений берут близкие по химическому составу
адсорбенты, но обладающие различной поверхностью.
Определение теплоты адсорбции. К числу следующих задач по
определению физико-химических констант,, решаемых хроматогра-
фическим методом, следует отнести определение теплоты адсорбции.
Последнее может быть легко выполнено, если измерить характери-
стические температуры для изучаемых веществ на данном адсор-
бенте. Пользуясь уравнением (128), можно по измеренным Тхар
рассчитать теплоту адсорбции.
Идентификация веществ. Хроматографический метод позволяет
также идентифицировать вещества из их смеси. Распространение по-
лучили методы идентификации, основанные на характеристиках
хроматографических кривых, главной из которых является удер-
живаемый объем. Экспериментальные данные показывают, что удер-
живаемый объем связан с числом атомов углерода в молекулах ор-
ганических веществ, принадлежащих одному гомологическому ряду.
Известен ряд других зависимостей.
Если допустить, что теплота адсорбции пропорциональна числу
п атомов углерода в молекуле, а изменение энтропии не зависит от
него, то можно получить связь удерживаемого объема с числом п:
]gVR = Мп + В. (160)
Определив удерживаемые объемы для каждого из компонентов
смеси и построив по полученным данным для веществ с известным
числом п график в координатах п—IgV^, можно идентифицировать
неизвестное вещество смеси, если оно принадлежит к тому же гомо-
логическому ряду.
Известна также связь удерживаемого объема с температурой ки-
пения компонентов смеси, если они принадлежат к одному гомоло-
гическому ряду:
lgVa=W + i. (161)
Пользуясь этим уравнением, можно идентифицировать неизвест-
ный компонент смеси гомологического ряда или же определить тем-
пературу кипения одного из членов гомологического ряда.
По ширине полосы можно также определить величины физиче-
ских характеристик, определяющих размывание полос (£)п, Z)B, D
и др.).
Все эти методы нашли широкое применение в хроматографиче-
ской практике.
АППАРАТУРА
Любой прибор для газовой хроматографии должен состоять из
следующих основных узлов, обеспечивающих разделение и анализ
смеси:
167
1) источник постоянного потока газа-носителя;
2) дозирующее устройство для количественного ввода пробы
анализируемого газа;
3) хроматографическая колонка, работающая в изотермических
условиях или в условиях непрерывного изменения температурного
поля вдоль слоя сорбента;
4) устройство, фиксирующее компоненты разделенной смеси по
выходе их из колонки (детектор);
5) устройство, преобразующее возникающий в детекторе импульс
в легко воспринимаемый сигнал, например, в запись на бумажной
ленте;
6) в отдельных случаях необходимо приспособление для улавли-
вания компонентов смеси после ее разделения.
Рис. 34. Стабилиза-
тор потока газа в
хроматографиче-
ской колонке:
1 — пористая перего-
родка; 2 — уравни-
тельный сосуд с
ртутью
ГАЗ-НОСИТЕЛЬ
Поток газа-носителя должен подаваться в хроматографическую
колонку непрерывно с постоянной и определенной скоростью, при-
чем должен быть обеспечен требуемый перепад
давления газа-носителя на входе и выходе из
колонки. Как правило, газ-носитель подается
из соответствующего газового баллона через
редуктор. По выходе из редуктора газ обычно
обладает постоянным давлением и скоростью.
Однако для обеспечения лучшей стабилизации
давления можно рекомендовать специальные
стабилизаторы, например стабилизатор, изобра-
женный на" рис. 34. Этот стабилизатор состоит
из отростка, в котором имеется боковое отвер-
стие с впаянной в него перегородкой 1 из пори-
стого стекла. К отростку, кроме того, присое-
динен уравнительный сосуд 2, заполненный
ртутью. Во время работы уравнительный сосуд
устанавливают так, чтобы большая часть пори-
стой перегородки была закрыта ртутью. При по-
нижении давления в системе ртуть перекрывает
перегородку, при повышении — открывает. Уста-
навливая давление и сопротивление системы
постоянными, можно поддерживать постоянной и скорость потока
газа-носителя.
Измерение скорости потока можно производить любой системой
ротаметров при обязательном условии нерастворимости газа-носи-
теля в жидкости, которой заполнен ротаметр. Предложен пенный
ротаметр, оказавшийся весьма удобным в работе [5]. Устанавливать
ротаметры удобнее на выходе газа из системы, т. е. после детектора.
Пропускаемые через колонку газы должны быть предварительно
осушены и освобождены от примесей, мешающих четкой работе
хроматографической установки. Для осушки можно применять
168
поглотительные трубки, заполненные безводным хлоратом магния,
пягиэкисью фосфора, хлоридом кальция, силикагелем и т. д.
В качестве таза-носителя наиболее часто применяют водород,
гелий, аргон, азот, воздух, реже двуокись углерода. Все они долж-
ны быть очищены от мешающих примесей. Особенно это относится
к двуокиси углерода, которая часто содержит довольно много по-
сторонних примесей, в частности, воздух.
ДОЗИРУЮЩИЕ УСТРОЙСТВА
Знание точного количества введенного в колонку газа является
одним из главных требований успешного выполнения количествен-
ного определения компонентов разделяемой смеси. Учитывая, что
количественная дозировка газа значительно труднее дозировки
жидкости, выбору дозатора, соответствующего поставленной задаче,
должно быть уделено серьезное внимание. Особенно важен выбор
дозатора при анализе небольших количеств газа.
К дозирующим устройствам предъявляются следующие требова-
ния: они должны иметь минимальный объем, в них должно отсут-
ствовать мертвое пространство, материал дозатора не должен ад-
сорбировать анализируемую смесь или химически с ней реагировать,
введение пробы не должно прерывать поток газа-носителя или
нарушать иным образом режим работы колонки. Дозировка и введе-
ние пробы является Одной из важнейших операций газо-хромато-
графического опыта. Поэтому несоблюдение вышеуказанных требо-
ваний может привести к ухудшению разделения и потери точности.
Количество наносимой пробы газа в газо-адсорбционной хрома-
тографии обычно составляет 1—100 мл. Поэтому объемы дозаторов
должны быть также в этих пределах.
Достаточно высокую точность анализа, а также соблюдение ука-
занных выше требований обеспечивает применение кранов-дозато-
ров, позволяющих отсекать известный объем газа и затем вводить
его в колонку. Недостатком этого типа дозаторов является необхо-
димость применения смазки. Поэтому их не рекомендуется приме-
нять для дозировки газов, адсорбирующихся на смазке.
Для введения проб газа большого объема можно пользоваться
различного рода сосудами (типа петли) точно известного объема.
В перечисленных типах дозаторов проба газа сначала отсекается,
а затем вводится в колонку продуванием объема дозатора потоком
газа-носителя. Такая система позволяет, не прекращая потока га-
за-носителя, полностью переносить весь объем анализируемого га-
за в колонку (рис. 35).
Дозаторы другого типа основаны на выдавливании измеренного
объема анализируемого газа в поток газа-носителя. К этому типу
дозаторов относятся различной конструкции газовые бюретки, по-
зволяющие выдавливать газ какой-либо запирающей жидкостью.
Запирающие жидкости не должны растворять исследуемый газ и
169
Рис. 35. Схема дозирующего устройства
(а — заполнение; б — дозировка):
/— ввод пробы; 2— поток газа-носнтеля; 3— дози-
рующий объем; 4— в хроматографическую колонку
колонки. Последние удобны тем, что
реагировать с ним, В частности, можно применять насыщенный вод-
ный раствор хлорида натрия или ртуть.
В лабораторной практике в качестве дозатора часто применяют
медицинские шприцы. Проба газа в этом случае отбирается в шприц,
а затем вводится непосредственно на адсорбент через самоуплот-
няющийся резиновый колпачок. При этом поток газа-носителя не
прекращается. Этот метод очень прост, однако он не обеспечивает
полной герметичности и поэтому не может дать высокой точности.
Во всех случаях введения
пробы путем выдавливания
следует создавать в выдав-
ливающем устройстве дав-
ление, несколько превы-
шающее давление газа-но-
сителя.
ХРОМАТОГРАФИЧЕСКИЕ
КОЛОНКИ
В газовой хроматогра-
фии применяют прямые,
U-образные и спиральные
они компактны и поэтому
их легче термостатировать. Внутренний диаметр колонок варь-
ируется от 2 до 12—15 мм. Значительно реже применяют ко-
лонки больших размеров. Длина колонки устанавливается в зави-
симости от поставленной задачи. Чаще применяют колонки от 2 до
20 м. Материалом для изготовления колонок может служить стекло,
медь, латунь, сталь и др.
Очень большое значение имеет равномерная и достаточно плот-
ная упаковка сорбента в колонке. При использовании спиральных
колонок этого добиться довольно трудно. Поэтому в ряде случаев
сорбент вносят в прямую колонку, которую затем сгибают в спираль.
После набивки колонку с сорбентом продувают газом-носителем,
чтобы освободить сорбент от сорбированных посторонних веществ.
В газо-адсорбционной хроматографии очень важно обеспечить
постоянство температуры в течение всего опыта. Это требование свя-
зано с тем, что влияние температуры на величину адсорбции газов
очень велико. Поэтому колонки, а также и детекторы, помещае-
мые непосредственно на выходе из колонки, должны термостати-
роваться. Колебания температуры не должны превышать ±0,5° С,
а в отдельных случаях требуется еще большее постоянство темпе-
ратуры.
В качестве термостатов применяют как воздушные, так и жид-
костные термостаты, выпускаемые промышленностью, или само-
дельные. К ним предъявляются следующие требования: они долж-
ны обеспечивать поддержание температуры на протяжении всего
опыта на заданном уровне и с требуемой точностью. Кроме того,
170
термостаты должны иметь перемешивающее устройство, обеспечи-
вающее равномерный нагрев по всей длине колонки. Для спираль-
ных колонок можно рекомендовать жидкостные термостаты типа
ТС-15М и ТС-24. В качестве термостатной жидкости при температу-
рах ниже 99° С можно пользоваться водой, а при более высоких
температурах — трансформаторным маслом или другими высоко-
кипящими и неагрессивными жидкостями.
В хроматермографии необходимо обеспечить изменение темпера-
туры колонки во времени и по ее длине. Это достигается либо при-
менением движущихся печей с определенным градиентом темпера-
туры, либо специальным ступенчатым обогревом самой колонки с
шаговым искателем. Изменение температурного поля при этом мо-
жет быть как линейным, так и нелинейным. Возможно примене-
ние устройств для программированного изменения температуры.
Разделение смеси низкокипящих газов требует применения таких
специальных термостатов, которые позволили бы проводить экспе-
римент при температурах много ниже 0° С, вплоть до температур,
близких к температуре жидкого воздуха.
ДЕТЕКТОРЫ
Важнейшим узлом газо-хроматографической установки является
детектор, который должен реагировать на изменение состава газа
по его выходе из колонки и немедленно передавать эти изменения
фиксирующему прибору. Характеристикой детектора в значитель-
ной степени определяется точность и чувствительность всей хрома-
тографической установки, а также размер вводимой пробы, время
анализа и другие условия.
Детектор должен возможно меньше реагировать на изменения
внешних условий, а также на случайные изменения. Он должен
быть также малоинерционным. Для улучшения работы детектора и
повышения его чувствительности объем пространства между концом
елся сорбента и детектором, а также собственный его объем должны
быть минимальными.
Существует несколько принципов классификации детекторов.
Согласно наиболее распространенной классификации, детекторы
делятся на дифференциальные и интегральные. Первые передают
мгновенное изменение какой-то характеристики, вторые суммиру-
ют изменение ее за определенный отрезок времени. В свою очередь
дифференциальные детекторы подразделяют на концентрационные,
показывающие изменение концентрации на выходе из колонки, и
потоковые, дающие произведение концентрации на скорость, т. е.
поток вещества.
Можно классифицировать детекторы по характеру протекающего
в нем процесса. По этой классификации различают детекторы хи-
мические, физические и детекторы, показания которых зависят
или не зависят от природы газа-носителя.
17]
В общем случае зависимость сигнала I от концентрации газа
для любого детектора можно записать в виде уравнения
I = Gv‘c. (162)
Если г=0, то детектор измеряет концентрацию; при i=l он изме-
ряет произведение концентрации на скорость, т. е. величину потока
вещества. Строго говоря, большинство применяемых детекторов не
принадлежит ни к одной из этих групп. Однако в тех областях ско-
ростей, с которыми обычно приходится работать, показания детек-
торов с достаточной достоверностью могут передавать величину
концентраций или величину потока.
Вопрос о том, какой из типов детекторов целесообразно приме-
нить в том или ином случае решения практических задач, необходи-
мо рассматривать в каждом конкретном случае. В общем случае
следует учитывать следующее:
1. Так как хроматограмма регистрируется непосредственно в ко-
ординатах t—с, а количество вещества равно величине площади
в координатах V—с (где У=^цг//), то
' с 0 г е
<?,= ) cdV= j cvdt = v j cdt. (163)
Отсюда следует, что количество вещества можно найти суммирова-
нием потока сц. Поэтому потоковые детекторы дают выходные хро-
матографические кривые, количество вещества по которым следует
определять, измеряя площади пиков.
2. В ряде случаев необходимо применять концентрационные де-
текторы и определять состав смеси по величине h или VRh. Если же
скорость движения потока вещества непостоянна, как это имеет
место, например, в хроматермографии, то применение концентра-
ционного детектора может привести к серьезным ошибкам.
3. Определение физико-химических характеристик хроматографи-
ческим методом следует проводить при помощи концентрационных
детекторов.
4. Интегральные детекторы применяют при вычислении количе-
ства нанесенного вещества 7Z.
Рассмотрим наиболее распространенные типы детекторов [5, 7,
13, 14, 15].
ДИФФЕРЕНЦИАЛЬНЫЕ ДЕТЕКТОРЫ
Катарометр. Наиболее распространенным детектором этого типа
является термокондуктометрическая ячейка, или катарометр.
Принцип действия катарометра состоит в том, что количество тепла,
отводимого от нагретой нити, зависит от теплопроводности газа,
омывающего эту нить. Поэтому, если меняется состав газа, то изме-
няется его теплопроводность, с изменением которой будет изменяться
сопротивление нагретой нити. Если сопротивление нити уравно-
вешено и включено в плечо моста, то нарушение баланса моста вслед-
ствие изменившегося сопротивления нити приведет к отклонению
172
пера записывающего устройства и вместо прямой нулевой линии
будут записаны кривые, показывающие эти отклонения.
На этом принципе можно изготовить термокондуктометриче-
скую ячейку, поместив ее камеру непосредственно на выходе газа
из хроматографической колонки. Схема устройства такой ячейки
следующая. По оси стеклянной или металлической трубки натяги-
вается платиновая или вольфрамовая нить, нагреваемая электри-
ческим током до определенной температуры. Газ-носитель омывает
эту нить и отводит часть выделяемого ею тепла. Нить включается
в мост Уитстона, который уравновешивается. Подключенный к
соответствующим точкам моста записывающий прибор, например
самопишущий потенциометр, в этом случае вычерчивает нулевую
линию. При изменении состава газа-носителя вследствие появления
в нем компонентов анализируемой смеси температура нити изме-
няется и нарушается баланс моста, что и фиксируется записываю-
щим прибором.
При изготовлении катарометра следует учитывать, что его чув-
ствительность зависит от длины и диаметра нити, а также от объема
ячейки, в которую помещена нить. Чувствительность возрастает
при увеличении длины нити и уменьшении объема ячейки. С уве-
личением диаметра нити возрастает отвод тепла, однако резко умень-
шается сопротивление нити, что уменьшает чувствительность. По-
этому рекомендуется применять нить с диаметром 0,02—0,04 мм.
Оптимальным диаметром ячейки можно считать 3—4 мм.
В качестве материала для изготовления нитей катарометра
следует применять металлы с большим удельным сопротивлением и
достаточно большим температурным коэффициентом. Поэтому чаще
всего применяют платиновые или вольфрамовые нити. Еще лучше
изготовлять сопротивление из полупроводниковых материалов,
т. е. заменять нитевые сопротивления термисторами.
На чувствительность катарометра оказывает влияние разность
температур нити и стенок ячейки: чем больше эта разность, тем выше
чувствительность. Обычно повышение температуры нити ограничи-
вается тем, что многие компоненты газа начинают так или иначе ка-
талитически реагировать на нагретой нити. Наиболее часто вели-
чина тока накала нити составляет 150—200 ма.
В анализе газовых смесей решающим является существенное от-
личие теплопроводности компонентов анализируемой смеси от теп-
лопроводности газа-носителя. Поэтому наибольшей чувствительно-
сти катарометра можно достичь при применении в качестве газа-но-
сителя водорода, теплопроводность которого значительно отли-
чается от теплопроводности большинства газов и паров многих
органических веществ. Теплопроводность некоторых газов, наибо-
лее часто встречающихся в практике хроматографического анализа,
приведена в табл. 13.
Конструкция ячейки катарометра может быть различной. Про-
точная ячейка (рис. 36, а) не обладает инерцией, но чувствительна
173
каналами. о этом случае в одном из
Рис. 36. Типы ячеек по теплопроводности
(катарометров):
а — проточные; б — полудиффузионные; в —
диффузионные
к колебаниям скорости потока газа. В отличие от нее, диффузион-
ная ячейка (рис. 36, в) не чувствительна к изменению скорости по-
тока газа, но обладает значительной инерцией. Поэтому лучше
применять полудиффузионную ячейку (рис. 36, б), обладающую
сравнительно небольшой инерцией и достаточной чувствительно-
стью.
Чтобы уменьшить влияние колебаний температуры окружающей
среды, ячейки обычно изготовляют из массивного куска металла.
Такие ячейки не чувствительны к колебаниям температуры в 2—3°
и поэтому могут не термостатироваться.
Наиболее целесообразной конструкцией является ячейка с двумя
каналов помещается сопротив-
ление, служащее измеритель-
ным плечом моста, а в дру-
гом находится сопротивление
сравнения. Тогда через срав-
нительную ячейку проходит
чистый газ-носитель, а через
измерительную — анализируе-
мая смесь. Если на выходе
из колонки газ-носитель не
содержит компонентов анали-
зируемой смеси, то обе ячейки
находятся в одинаковых ус-
ловиях и мост уравновешен.
При появлении в измери-
тельной ячейке одного из ком-
понентов анализируемой смеси вследствие иной его теплопроводно-
сти нарушается баланс моста, что и отмечается соответствующим
прибором. Техническое оформление такой схемы может осуще-
ствляться одним из трех вариантов:
1) газ-носитель сначала проходит через камеру сравнения, а
затем поступает в колонку, по выходе из которой проходит измери-
тельную камеру;
2) перед входом в колонку газ-носитель разветвляется на два
потока, один из которых проходит сравнительную камеру, а дру-
гой колонку и измерительную камеру;
3) по выходе из колонки газ-носитель проходит измерительную
камеру, а затем поступает в холодильник, в котором конденсируют-
ся анализируемые компоненты. Очищенный таким образом газ-но-
ситель поступает в камеру сравнения.
Последний вариант пригоден только для анализа легко конден-
сирующихся веществ и применяется редко.
Термохимический детектор. Кроме катарометров, широкое при-
менение в практике хроматографического анализа нашел прибор,
основанный на измерении теплового эффекта сгорания горючих
газов — термохимический детектор. В детекторах этого типа со-
174
Таблица 13
Теплопроводность и температурный коэффициент
теплопроводности некоторых газов и паров
Вещества Теплопроводность кал/сМ'Сек-град Коэффициент теплопро- водности от 0 до 100° С
10s при 0° С воздуха X у/л у водорода при 0° С
при 0° С при 100° С
Воздух 5,83 1 ,со 1,00 0,140 0,03265
Водород 41,60 7,14 7,10 1,030 0,03261
Гелий 34,80 5,97 5,53 0,837 0,03193
Метан 7,21 1,25 1,45 0,173 0,03655
Кислород 5,90 1,012 1,032 0,142 0,00303
Азот 5,81 0,996 0,996 0,140 0,03264
Окись углерода . . . 5,59 0,959 0,924 0,134 0,00262
Аммиак 5,22 0,897 1,04 0,125 0,0058
Ацетилен 4,53 0,777 0,903 0,109 0,0048
Этан 4,36 0,750 0,970 0,105 0,00583
Этилен 4,19 0,720 0,980 0,101 0,00763
Аргон 4,07 0,645 — 0,0983 0,00311
Пропан 3,58 0,615 . 0,832 0,0861 —
Двуокись углерода . . 3,52 0,605 0,700 0,0846 0,00495
Метанол 3,45 0,592 0,727 0,0829 0,0054
н-Бутан 3,22 0,552 0,741 0,0774 —
н-Пентан 3,12 0,535 0,702 0,0750 —
изо-Пентан 3,03 0,515 0,718 0,0722 —
н-Гексан 2,96 0,508 0,662 0,0712 —
Ацетон 2,37 0,406 0,557 0,0570 0,0372
Хлористый метил . . 2,20 0,377 0,530 0,0529 —
Бензол 2,16 0,370 0,583 0,0519 0,0098
Водяной пар — — 0,775 — 0,00725
Циклогексан — — 0,576 — —
Этанол — — 0,7С0 — 0,0063
Диэтиловый эфир . . — — 0,747 — —
Хлороформ 1,58 0,269 0,328 0,0380 —
Четыреххлористый уг-
лерод — — 0,228 — 0,0053
держащиеся в анализируемой смеси горючие газы сжигаются по
выходе из колонки на тонкой платиновой проволоке, нагретой до
800—900° С. Платиновая проволока в этом приборе является одно-
временно катализатором и плечевым сопротивлением в мостовой
схеме. При сгорании на проволоке какого-либо вещества температу-
ра ее повышается, вследствие чего сопротивление изменяется, что
приводит к нарушению баланса моста. Поэтому прибор может фик-
сировать наличие горючих газов в анализируемой смеси.
В литературе описан ряд конструкций термохимических детек-
торов. Наиболее распространенным у нас в СССР детектором этого
типа является газоанализатор ПГФ-1.
175
Пламенный детектор. Близким по принципу действия к термо-
химическому детектору является пламенный детектор. Принцип
его действия заключается в том, что при наличии органического ве-
щества в анализируемой смеси водородное пламя горелки, в кото-
рое поступает анализируемая смесь, удлиняется, а температура его
изменяется. Вследствие этого помещенная над пламенем термопара
подвергается воздействию изменяющейся температуры и передает
эти изменения самопищущему прибору или другому индика-
тору.
Пламенный детектор обладает малой инерцией, а также важным
преимуществом перед другими типами детекторов в том отношении,
что позволяет обходиться без предварительной калибровки для всех
компонентов анализируемой смеси, так как между теплотой сгора-
ния и площадью пика существует линейная зависимость. К недо-
статкам его следует отнести необходимость подачи двух газов: во-
дорода и воздуха или кислорода, высокие требования к чистоте
г аза-носителя, а также необходимость тщательной регулировки ве-
личины потока.
Ионизационные детекторы. Весьма чувствительными являются
ионизационные детекторы. К этой группе относятся детекторы, в
которых ионизация молекул может осуществляться под действием
электрического разряда в вакууме, либо в пламени при наличии
электрического поля, или же под действием радиоактивного излу-
чения.
Принцип работы ионизационного детектора первого типа состоит
в том, что при давлении 0,02—0,1 мм рт. ст. ионизация инертных
газов, например гелия, затруднена, тогда как другие газы ионизи-
руются значительно легче. Поэтому при наложении напряжения,
меньшего, чем ионизационный потенциал гелия, ионизация будет
возникать лишь в присутствии каких-либо газов или паров. В каче-
стве разрядной трубки применяют неоновую лампу, причем для
обеспечения требуемого вакуума в детектор подают только часть
выходящих из колонки газов.
—Работа пламенно-ионизационного детектора основана на том,
что пламя чистого водорода почти не содержит ионов и обладает
поэтому очень малой электропроводностью. При наличии других
веществ электропроводность пламени возрастает, что и может слу-
жить индикатором на присутствие в газе-носителе анализируемых
веществ.
Детекторы такого типа благодаря своей высокой чувствительно-
сти нашли широкое применение, особенно в капиллярной хромато-
графии, и монтируются в большинстве современных хроматогра-
фов, выпускаемых промышленностью.
Конструкция пламенно-ионизационных детекторов может быть
различной, однако основными узлами любой из них является во-
дородная горелка, в пламя которой помещают два электрода из
платиновой проволоки, на которые от анодной батареи подается по-
176
стоянное напряжение. Чувствительность детектора определяется
расстоянием между электродами, подаваемым на них напряжением,
а также скоростью потока водорода. Для каждой конструкции уста-
навливаются оптимальные значения этих параметров.
В случае появления в водороде газов или паров анализируемых
веществ (за исключением СО, СО2, COS, CS2, H2S, О2, инертных
газов) происходит ионизация пламени, причем, вероятно, возника-
ют радикалы и электроны. Последние удаляются из водородного
пламени вследствие наличия положительного напряжения на одном
из электродов. Количество возникающих электронов и радикалов
пропорционально концентрации анализируемого вещества в газе-
носителе, поступающем в горелку. Следовательно, и ток ионизации
пропорционален концентрации анализируемого вещества. В связи
с тем, однако, что сила тока ионизации весьма мала, для преобразо-
вания его в сигнал, передаваемый самописцу, следует применять
соответствующие усилители.
Чувствительность пламенно-ионизационного детектора весьма
высока, что является одним из его достоинств. Так, например, наи-
меньшая концентрация пропана, определяемая с достаточной уве-
ренностью, равна (4—8)-10“9 мг!мл. Другим важным достоинством
пламенно-ионизационного детектора является относительная про-
стота его конструкции, малая инерционность и независимость его
показаний от колебаний температуры. Все это делает пламенно-
ионизационный детектор одним из наиболее распространенных
детекторов, особенно в капиллярной хроматографии.
К недостаткам пламенно-ионизационных детекторов следует от-
нести необходимость применения одновременно двух газов: водо-
рода и азота высокой степени чистоты.
В качестве источника ионизации можно применять радиоактив-
ное излучение, в частности p-излучение. Однако такого типа детек-
торы, так же как и детекторы с электрическим разрядом, являются
мало чувствительными и поэтому не получили широкого распро-
странения. Исключение составляет аргоновый детектор Дж. Е. Ло-
велока [16], оказавшийся самым чувствительным из всех известных
в настоящее время детекторов. Дело в том, что атомы аргона обла-
дают одним из самых высоких потенциалов ионизации (11,6 эв).
При воздействии на них радиоактивного излучения, например р-из-
лучения стронция-90 или прометия-147, возникают возбужденные
метастабильные атомы, которые при столкновении с молекулами дру-
гих веществ могут передать свою избыточную энергию электронам
этих молекул. Если при этом окажется, что потенциал их иониза-
ции будет ниже энергии возбуждения атомов аргона, то произойдет
ионизация молекул. В результате возникнет ток ионизации, кото-
рый может оказаться значительно больше тока, вызванного иони-
зацией самого аргона. Это свойство дает возможность определять
весьма малые примеси паров почти всех органических веществ,
а также газов, кроме N2, СО2, О2, СН4 и паров воды.
177
К настоящему времени предложено несколько различных кон-
струкций аргоновых детекторов. Разрез одной из наиболее приня-
тых из них приведен на рис. 37. Основной частью детектора служит
латунный электрод 1, через который поступает газ из колонки. Вто-
Из кошки
Рис. 37. Схема аргонового иони-
зационного детектора:
1 — латунный электрод; 2 — тефло-
новая изолирующая прокладка;
3 — латунная камера 4 — фольга со
слоем радиоактивного вещества (Sreo)
лая чувствительность к
рым электродом являются стенки ка-
меры 3, изготовляемые из толстой
латуни для защиты от радиоактив-
ного излучения. Изоляцией между
электродами служит тефлоновая про-
кладка 2. В качестве источника ра-
диоактивного излучения применяют
фольгу 4, покрытую слоем радио-
активного стронция-90 или проме-
тия-147. На электроды подается на-
пряжение 400—500 в.
Аргоновые детекторы оказались
весьма чувствительными. С их по-
мощью можно определять до 5-10'9 г
вещества, что позволяет уменьшить
наносимую пробу вещества до 0,2 мкг.
Кроме того, важным свойством де-
текторов этого типа является линей-
ная зависимость между концентра-
цией вещества в аргоне и величи-
ной ионизационного тока, что зна-
чительно облегчает калибровку. Ма-
изменениям температуры и давления
также относится к положительным качествам этих детекторов.
ИНТЕГРАЛЬНЫЕ ДЕТЕКТОРЫ
К интегральным методам детектирования относятся: 1) автома-
тическое титрование; 2) измерение объема или давления газа и
3) измерение электропроводности раствора.
Автоматический титратор. Принцип действия автоматического
титратора состоит в том, что анализируемый газ по выходе из ко-
лонки поглощается каким-либо реагентом, избыток которого затем
титруется. Объем раствора измеряется и автоматически регистри-
руется. Этим методом можно определять содержание кислых или
основных газов, а также меркаптанов, альдегидов, кетонов и других
соединений, для поглощения которых можно подобрать соответст-
вующие растворы и индикаторы. Достигаемая точность определения
достаточно высока.
Объемно-хроматографический метод. Этот метод определения со-
держания газов был разработан одновременно в СССР Д. А. Вяхи-
ревым [17] и в Чехословакии Я. Янаком [18]. Принцип метода со-
стоит в том, что газ-носитель на выходе из колонки поглощается
каким-либо раствором, а анализируемые газы им не поглощаются и
178
собираются в сосуд, в котором их объем может быть измерен. Обыч-
но в качестве поглощающего раствора применяют 45—48%-ный
раствор едкого кали, а в качестве газа-носителя — двуокись угле-
рода. Во избежание больших ошибок двуокись углерода должна
быть совершенно свободной от примесей непоглощающихся в щелочи
газов, в частности, воздуха. Кроме того, этим методом нельзя ана-
лизировать газы, заметно растворяющиеся в щелочах или конден-
сирующиеся при температуре анализа. Все это накладывает су-
щественные ограничения на применение метода.
Количество газа, накопившегося над раствором щелочи, опре-
деляют в азотометре с точностью до 0,01 мл, причем измерения ведут
при постоянном давлении путем перемещения уравнительного со-
суда до выравнивания уровня раствора щелочи в сосуде с уровнем
в азотометре. Измерения можно автоматизировать с помощью фото-
элемента, помещаемого на линии мениска раствора в азотометре, и
самописца. Метод измерения электропроводности аналогичен ме-
тоду титрования.
В заключение следует отметить, что универсальных детекторов
нет. Каждый из рассмотренных типов имеет свою область примене-
ния и для решения той или иной конкретной задачи является наи-
лучшим. Поэтому экспериментатору прежде всего следует четко
поставить задачу, а затем, в соответствии с требованиями, выдви-
гаемыми этой задачей, сделать выбор наиболее оптимального типа
детектора. В табл. 14 приведены наиболее важные характеристики
детекторов различных типов.
РЕГИСТРИРУЮЩИЕ ПРИБОРЫ
Для измерения или записи импульса, передаваемого детекто-
ром, необходимо применять какой-либо регистрирующий прибор или
устройство. В большинстве случаев, если импульс, подаваемый де-
тектором, является электрическим сигналом, применяют чувстви-
тельные показывающие или записывающие милливольтметры и
потенциометры. Наиболее распространенным и подходящим для
этой цели прибором является уравновешенный регистрирующий
потенциометр типа ЭПП-09 со временем пробега каретки не более
2 сек. Эти приборы позволяют производить непрерывную автомати-
ческую запись хроматографической кривой.
ХРОМАТОГРАФИЧЕСКИЕ ПРИБОРЫ
Хроматографы можно разделить на две группы соответственно
назначению: лабораторные и промышленные. Это деление оправдано
тем, что требования, предъявляемые к лабораторным и промышлен-
ным хроматографам, обычно различны. В приборах лабораторного
типа необходимо обеспечить наиболее полное разделение очень
сложных по составу смесей. При этом время, потребное для прове-
дения анализа, не является определяющим. Кроме того, лабора-
торный прибор должен быть возможно более универсальным и чув-
179
Сравнительная характеристика
№ п.п. Название детектора Принцип действия Что измеряется записывающим прибором
Дифференциаль
1 Катарометр Изменение теплопро- водности газа Изменение сопротив- ления
2 Термохимиче- ский Теплота сгорания То же
3 Пламенный Форма и температура пламени Термоэлектродвижу- щая сила
4 Пламенно-иони- зационный Ионизация в пламени Ток ионизации
5 Аргоновый ио- низационный Ионизация Р-излуча- телем Ток ионизации Интегральные
6 Титрометр Обратное титрование Объем титрующего раствора
7 Объемный Поглощение газа-но- сителя раствором Объем анализируемо- го газа
8
Кондуктометр
Изменение электро-
проводимости вследст-
вие химической реак-
ции
Изменение электро-
проводности раствора
* +— сильное влияние; ± — слабое влияние;-отсутствие влияния
ствительным. К промышленным приборам не предъявляется требо-
ваний широкой универсальности, но зато приборы должны обес-
печивать проведение анализа в возможно более короткие сроки
с максимальной точностью при наибольшей автоматизации.
’ Существующее деление, однако, не исключает возможности при-
менения приборов лабораторного типа для решения задач промыш-
ленности и наоборот. При этом следует иметь в виду, что для реше-
ния простых задач не следует прибегать к очень сложным и дорого-
180
Таблица 14
детекторов различных типов
Граничная чувствитель- ность Постоян- ная времени, сек Область линейности Зависимость* показаний прибора от Область применения
темпера- туры давления скорости
ные детекторы 10“3—10“5 мг/см3 1—2 Большая + ± i Заполненные ко-
10“3— 10"6лг/сл3 1 Малая ± лонки Заполненные ко-
10-4 мг/сек 1 Средняя лонки Анализ только го- рючих веществ Заполненные ко-
10-9—10~10мг/сек 10“3 Большая — лонки Капиллярная хро-
4-Ю"8— 3—2-10-8 Большая + . матография. Анализ примесей Заполненные и ка-
2-10-11 мг/сек детекторы IO"3 мг Большая пиллярные колонки. Анализ примесей Анализ химически
10”1 мг Большая + активных газов и паров Заполненные ко-
10-6 мг Большая лонки Непоглощаемые щелочью газы. Газ- носитель СО2 Анализ химически
активных газов и паров
стоящим приборам, обращение с которыми требует более высокой
квалификации.
В табл. 15 приведены наиболее важные характеристики хрома-
тографов лабораторного и промышленного типа, выпускаемых оте-
чественной промышленностью (см. также [19—26]). Описание хро-
матографов иностранных фирм можно найти в соответствующих
проспектах, либо в книге А. А. Жуховицкого и Н. М. Туркельта-
уба [7].
181
Сведения о хроматографах
I №№ пп 1 Название и мар- ка прибора Изготови- тель Назначение прибора Детектор Дозатор
Лабораторные
1 Титрометриче- ский газоанали- затор внииягг Определение ма- лых количеств угле- водородов и окиси углерода в воздухе Сжигание на платино- вой спирали н поглощение полученной СО2 раство- ром Ва(ОН)2; обратное тн- трова ине НС1 Бюретка
2 Хромвтермохи- мический газоана- лизатор ХТХГ-1 внигни Определение водо- рода, окиси углеро- да и предельных уг- леводородов до С6 включительно Термохи- мический с самописцем ЭПП-09 Микробю- ретка или га- зовая петля
3 Переносный хроматограф ПХ-1 внигни Анализ Н2, О2, N2, СО, предельных и непредельных угле- водородов до С8 включительно Катаро- метр Кран
4 Хроматермо- граф № 4 внигни Анализ СО, О2, N2 предельных и непре- дельных углеводо- родов Азотометр Бюретка
5 Хроматермо- граф № 8 внигни То же Катаро- метр или термохими- ческий Бюретка или газовая петля
6 Универсальный хроматермогра- фический газо- анализатор УХТГ-1 внигни То же Катаро- метр н тер- мохимиче- ский, соеди- ненные по- следователь- но Микробю- ретка
1
1
182 .
Таблица 15
выпускаемых в СССР
Тип и размеры колонки Газ- носитель Адсор- бент Температур- ные условия Г раничиая чувствитель- ность, объеми. % ( ® а У я л ° А О.Н 5 Ч ь о 3 о о 2 н в к Примеча - иие
хроматографы Стеклянная Воздух Активи- рованный уголь КАД До 40 °C 10~‘ ±4
1-я с углем для Н8, СО и СН4, 2-я газожид- костная Воздух Уголь АГ или СКТ. Но- ситель с жидкой фазой До 40 °C 0,01 ±5
То же Гелий или азот То же До 40 °C 0,01 ±5
1 -я с силикаге- лем, 2-я с углем Дву- окись уг- лерода 1-я с си- ликаге- лем, 2-я с углем АГ Движуща- яся электро- печь с гра- диентом тем- пературы 0,1—0,05 ±5
То же Водо- род, ге- лий, азот, воздух То же То же 0,1—0,001 ±5
1-я в виде баранки, с си- ликагелем, 2-я газо- жидкостная, 3-я с углем, 2-я и 3-я спиральные Азот или воз- дух Сили - кагель, диатомит с жидкой фазой, уголь Вдоль 1-й колонки не- прерывно движется печь с гра- диентом зем- перзтуры 0,1—0,0001 ±5
183
С с Название и марка прибора Изготови- тель Назначение прибора Детектор Дозатор
7 Лабораторный хроматограф ЛХМ-7А СКВ иох им. Зелин- ского АН СССР Анализ смесей ор- ганических соедине- ний, кипящих до 450° С Катарометр и пламеиио- ионизапиои- ныЙ, Само- писец ЭПП- 09 Шприц и микрошприц
8 Хромат ограф лабораторный ХЛ-2 СКВ.АНН завод «КИП» Анализ газов, не реагирующих со ще- лочью Азотометр Бюретка
9 Хроматограф лабораторный хл-з СКБ АНН завод «КИП» Анализ любой сме- си газов Катарометр с термисто- рами. Само- писец ЭПП-09 Шестихо- довой край или шприц
10 Хроматограф лабораторный ХЛ-4 Завод «Моснефте- кип» Анализ паров жид- костей с высокой температурой кипе- ния Катарометр с термисто- рами. Само- писец ЭПП- 17-М2 Шприц
11 Хроматограф лабораторный ХЛ-5 СКБ АНН То же То же Шприц
12 Хромат ограф ГСТЛ Завод «Моснефте- кип» Анализ предель- ных и непредельных углеводородов Термохими- ческий или катарометр. Отсчет визу- альный по стрелочному прибору Газовая петля
13 Универсальный хроматограф УХ-1 Таллин- ский завод измеритель- ных ’ прибо- ров Прибор универ- сальный. Позволяет определить любую смесь газов и паров Катаро- метр с тер- мисторами. Самописец ЭПП-17М Шприц
184
Продолжение табл. 15
Тип и размеры колонки Г аз- носитель Адсор- бент Температур- ные условия Граничная чувстви- тельность, объеми. % Относи- тельная погреш- ность, % Примечание
Наполненная, длиной до 16 м, с диаметром от 2 до 8 мм. Капил- лярная, длиной 200 м, диаметр 0,25 мм Любой Любой Изотерми- ческий ре- жим до 300° С. Про- грамнрован - ный линей- ный от 50 до 300° С —
U-образные сменные, метал- лические Дву- окись уг- лерода » До 40“ С 0,1 i 1 0
Спиральная металлическая Водо- род, ге- лий, азот > До 140° С ю-3 —
U-образные металлические, длиной до 12 л Водо- род, ге- лий, азот » До 150е С 0,05- — 0,005 ±2
То же То же > То же 0,05— 0,005 ±2 Разработан, но ие выпу- скается
Четыре после- довательно сое- диненных метал- лических колон- ки Воздух » Нагрев ко- лонки проис- ходит во вре- мени до 1 00° С 0,1 — 0,001 ±5 Возможно подключение самописца
Две металли- ческие колонки с переключе- нием иа парал- лельную и пос- ледовательную работу. Длина 1 0 м Любой Любой До 220° С 0,0! ±2
185
Е Е 5? Название и марка прибора Изготови- тель Назначение прибора Детектор Дозатор
14 Лабораторный газовый хрома- тограф <Цвет>. Модель 1-64 ОКБА Дзержин- ский филиал Анализ любых смесей газов н жид- костей, кипящих до 400° С Катарометр и пламенно- ионизацион- ный Край-доза- тор, микро- шприц и до- затор-испа- ритель
15 То же. Модель 2-65 То же То же Пламен ко- лонизацион- ный диффе- ренциальный То же
16 То же. Модель 3-66 То же То же. Возможен попеременный ана- лиз иа различных сорбентах Катарометр четырехпле- чевой и пла- меино-иони- зациоиный дифференци- альный То же
17 Радиохромато- граф ХГ2301 СКВ АП АН СССР Химический и ра- диохимический ана- лиз смесей, кипящих до 300° С Ионизаци- онный по се- чению иони- зации, арго- новый и про- точный счет- чик радиоиз- лучений 'шприц
18 Универсальный хроматограф ХГ1302 То же Анализ любых смесей газов и жид- костей, кипящих до 300° С Катаро- метр и четы- ре ионизаци- онных Кран-доза- тор и шприц
18&
Продолжение табл. 15
Тип и размеры колонки Газ- носигель Адсор- бент Температур- ные услов ИЯ Г раничиая чувстви- тельность, объеми. % От носи- тельная погреш- ность, % Примечание
Наполненные из нержавеющей стали, длиной до 6 м; диаметр 4 мм; капилляр- ная, длиной 50 м; диаметр 0,25 мм Водород Любой Изотерми- ческий режим до 300°С Катаро- метр 5-10 — » объемн. % ДИП 5-10“’ объеми. % ±2
То же плюс микронабивиые от 1 до 3 м То же То же Изотерми- ческий ре- жим до 400°С программи- рованный ли- нейный до 400° С ю-« объемн. % ±2
То же; кроме того, предусмот- рены стеклян- ные наполнен- ные колонки То же То же То же 10-’ объемн. % ±2
Из нержавею- щей стали, дли- ной 6 м; диа- метр 6 мм Аргон Любой Изотерми- ческий ре- жим до 200° С —10“4 объеми. %; Для счетчика 10~3 ми- крокюри ±4
Набор колонок, общая длина 4 м; диаметр 2,4 н 6 мм Аргон Любой Изотерми- ческий режим до 300 °C; программиро- ванный ли- нейный со скоростью от 4 до 10° С в мин Высшая 5-10-1’ моль/сек ±2
187
Е Е Название и Изготови- Назначение
марка прибора тель прибора Детектор Дозатор
Промышленные
19 Хроматермо- граф промышлен- ный ХТП-2 ВНИИКА Нефтегаз Анализ пирогаза Катаро- мет р Автомати- ческий
20 Хроматермог- раф геохимиче- ский ХТГ-3 ВНИИКА Нефтегаз Анализ газов при геохимических ис- следованиях Три ката- рометра с тремя само- п исцами Дозатор с пневматиче- ским управ- лением
21 Хроматермог- раф теплодина- мический ХТД ВНИИКА Нефтегаз Анализ предель- ных н непредельных углеводородов в по- токе Катаро- метр само- писцы ЭПП-09 и ЭМД-247 Непрерыв- ная подача
22 Хроматограф промышленный автомат ический ХПА-2 СКВ АНН завод «КИП» Анализ предель- ных и непредельных углеводородов до C.s вкл. Катаро- метр с терми- сторами. Са- мописец ЭПП-09 Автомати- ческий
23 Хроматограф промышленный автоматический ХПА-3 Завод «Моснефтекип» Непрерывный кон- троль потоков жид- ких веществ нефте- перерабатывающих и нефтехимических установок Два смен- ных катаро- метра на тер- мисторах. Самописец ЭПП-09М2Х Автомати- ческий
188
Продолжение табл. 15
Тип и размеры колонки Газ- носитель Адсор- бент Температур- ные условия Граничная чувстви- тельность, объемн. % Относи- тельная погреш- ность, % Примечание
хроматографы
Металличе- ская трубка длиной 3,5 м Водород Окись алюми- ния А-57 До 2 50°С — —
Три взанмно- пе реключ ающие- ся колонки Любой Любой Изотермиче- ский режим до 300°С; про- грамироваи- ный линейный со скоростью от 4 до 10°С в мин —
Колонка в ви- де баранки Нет > Непрерыв- но движущая- ся электро- печь с гради- ентом темпе- ратуры 0,004 ±5 Отбор про- бы осущест- вляется не- прерывно
Спиральная металлическая Любой > До 40° С 0,01 ±5
Три металли- ческих колонки, длиной 3, 6 н 9 м Водо- род, ге- лий, азот Твер- дый но- ситель с жидкой фазой До 120° С — —
189
№Ws nn 1 Название и марка прибора Изготови- тель Назначение прибора Детектор Дозатор
24 Хроматограф промышленный автоматический ХПА-4 Завод «Моснефте- кип» Непрерывный кон- троль газовых по- токов нефтеперера- батывающих и неф- техимических уста- новок Катаро- метр с тер- мисторами. Самописец ЭПП-09М2Х Автомат»- ческий
25 Хроматограф с выходом на систему АУС СКВ АНН Непрерывный кон- троль и управле- ние нефтеперераба- тывающими и неф- техимическими ус- тановками по соста- ву газа Катарометр с термисто- рами Автомати- ческий
26 Промышленный автоматический газовый хрома- тограф типа РХ-1 ОКБА, Дзержин- ский филиал Определение одно- го или нескольких компонентов в сло- жных смесях орга- нических веществ, а также выдача сигна- ла для автоматиче- ского управления процессом по одно- му компоненту Пламенно- иочнзацион- иый . Автомати- ческий мем- бранного ти- па, только для газов
27 То же. Тип РХ-5 То же То же То же То же; для газов и жид- костей
J 90
Продолжение табл. 15
Тип и размеры колонки Газ- иоситель Адсор- бент Температур- ные условия Граничная чувстви- тельность, объемн. % , Относи- тельная погреш- ность, % Примечание
U-образная ме- таллическая; длиной 6 м Водо- род, ге- лий, азот, воздух Любой До 80° С 0,05— 0,005 ±4 —
U-образные металлические; длиной от 1 до 6 м Азот, гелий, воздух Любой До 80° С 0,05— 0,005 — Разработан, но не выпу- скается
U-образные, наполненные, длиной от 1 до 6 м; . диаметр 4 мм Азот, воздух Любой Изотерми- ческий ре- жим до 80® С 10“4 объемн. % 2,5—6
То же. Длина от 0,5 до 6 м То же То же До 180°С То же 2,5—10
191
ПРАКТИЧЕСКИЕ РАБОТЫ
Работа 1
ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ СМЕСИ АЗОТА
И КИСЛОРОДА (ВОЗДУХ) НА МОЛЕКУЛЯРНЫХ СИТАХ
Приборы и реактивы
1. Хроматограф типа ПХ-1 или ГСТЛ с самописцем.
2. Хроматографическая колонка стеклянная U-образная, длиной 32 см,
внутренним диаметром 3 мм.
3. Молекулярные сита типа СаА. Измельчение 0,5—0,25 мм.
4. Баллон с гелием.
Цель работы. Показать возможность разделения смеси низко-
кипящих газов методом газовой хроматографии.
Сущность работы. Проба воздуха, наносимая на сорбент, может
быть затем разделена на азот и кислород, если выбранный сорбент
обладает различным сорбционным сродством к компонентам воз-
духа. Таким избирательным сорбентом могут служить молекуляр-
ные сита типа СаА. Газом-носителем в этом случае должен служить
гелий. Результат анализа может быть зафиксирован каким-либо
детектором и записан на самописце.
Выполнение работы. Специально изготовленную хроматографи-
ческую колонку тщательно заполняют сорбентом, предварительно
высушенным в токе сухого воздуха при 400° С. При заполнении
колонки следят за тем, чтобы плотность набивки сорбента была до-
статочной и равномерной по всей ее длине. Заполненную колонку
присоединяют к хроматографу типа ПХ-1 или ГСТЛ вместо имею-
щейся первой колонки прибора. При работе с ГСТЛ присоединяют
самописец типа ЭПП-09.
Включают ток газа-носителя и тщательно продувают им всю
систему до полного удаления из нее воздуха. Добиваются установ-
ления нулевой линии на ленте самописца, после чего вводят пробу
воздуха для анализа путем продувки газом-носителем крана-доза-
тора или газовой петли, в которых до продувания должен находить-
ся воздух, подлежащий анализу.
При правильном проведении анализа на ленте самописца поя-
вится два пика: первый будет относиться к кислороду, второй —
к азоту. Пик кислорода появляется на четвертой, а азота на седьмой
минуте от момента ввода пробы для анализа.
После окончания анализа, что устанавливают по возвращению
пера самописца на исходную нулевую линию, прекращают ток
газа-носителя и движение ленты самописца. Измеряют высоты пиков
кислорода и азота и определяют их относительное содержание в
объемных процентах. Полученные результаты сравнивают с таб-
личными данными.
192
Работа 2
ХРОМАТОГРАФИЧЕСКОЕ ОПРЕДЕЛЕНИЕ СМЕСИ НИЗКО-
КИПЯЩИХ ГОРЮЧИХ ГАЗОВ НА УГЛЕ И МОЛЕКУЛЯРНЫХ
СИТАХ [7]
Приборы, и реактивы
1. Хроматограф типа ПХ-1 или ГСТЛ с самописцем.
2. Хроматографические колонки стеклянные U-образные, длиной 32 см,
внутренним диаметром 3 мм.
3. Молекулярные сита типа СаА. Измельчение 0,5—0,25 мм.
4. Уголь марки СК.Т. Измельчение то же.
5. Баллон со сжатым воздухом или гелием.
6. Газометр с анализируемой смесью водорода, окиси углерода и метана.
Цель работы. Показать возможность разделения и анализа смеси
низкокипящих горючих газов, а также изменение порядка их выделе-
ния в зависимости от природы сорбента.
Сущность работы. Если две одинаковых пробы анализируемого
газа нанести на разные сорбенты, которыми заполнены две одина-
ковые хроматографические колонки, то по виду хроматограммы
можно заметить различие в адсорбционных свойствах этих сорбен-
тов. Один сорбент может полностью разделить анализируемую смесь,
другой разделить лишь частично или не разделить совсем. В другом
случае разделение может быть полным как на одном, так и на дру-
гом сорбенте. Однако порядок расположения вымываемых компо-
нентов смеси будет различным. В настоящей работе состав смеси и
сорбенты выбраны так, что разделение компонентов смеси достига-
ется на обоих сорбентах, однако изменяется порядок их выхода из
колонки.
Выполнение работы. Одну колонку заполняют молекулярными
ситами так, как это описано в предыдущей работе. Другую за-
полняют углем марки СК.Т, соблюдая те же условия заполнения.
Сначала к прибору присоединяют одну из колонок и анализируют
газ на одном сорбенте. Кран-дозатор или газовую петлю заполняют
из газометра анализируемым газом. Предварительно газ тщательно
осушают. Включают ток газа-носителя, которым в данном случае
может служить воздух, подаваемый из баллона или от воздуходув-
ки. Продувают всю систему (кроме крана-дозатора или газовой
петли) газом-носителем и добиваются постоянства нулевой линии
самописца. Вводят пробу анализируемого газа продуванием крана-
дозатора газом-носителем и наблюдают запись результатов анализа
на самописце.
В случае применения в качестве первого сорбента угля, на хро-
матограмме первым записывается пик водорода, затем окиси угле-
рода и последним — пик метана. Водород выходит на первой минуте
от момента впуска газа, окись углерода — на второй, а метан —
на четвертой.
7 Б. В. Айвазов
193
По окончании анализа колонку с углем отсоединяют и вместо нее
присоединяют колонку с молекулярными ситами. Процедуру ана-
лиза повторяют. На хроматограмме получают также три пика. Од-
нако второй пик в этом случае будет относиться к метану, а третий
к окиси углерода. Водород будет также выходить на первой ми-
нуте, метан — на второй, а окись углерода — на пятой.
После окончания анализа хроматограммы вынимают из само-
писца и по ним рассчитывают содержание каждого компонента смеси
в процентах. Расчет производят по методу абсолютной калибровки,
измеряя высоты пик. (Калибровочные графики выдает преподава-
тель.) Полученные результаты анализа для двух сорбентов сравни-
вают между собой и с истинным составом смеси. Расхождения не
должны превышать допустимых отклонений для выбранных при-
боров.
Р а б о т а 3
ХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ СМЕСИ ГАЗОВ,
СОДЕРЖАЩЕЙ ОКИСЛЫ АЗОТА
Приборы и реактивы
1. Хроматограф типа ПХ-1 или ГСТЛ с самописцем.
2. Хроматографическая стеклянная U-образная колонка длиной 180 см и
диаметром 6 мм.
3. Силикагель марки МСМ. Измельчение 0,5—0,25 мм.
4. Криостат, содержащий смесь сухого.льда и ацетона (в качестве криостата
может служить цилиндрический стеклянный сосуд Дьюара).
5. Термометр низкотемпературный.
6. Баллон с гелием.
7. Баллон с жидкой углекислотой.
8. Ацетон.
9. Смесь газов, состоящая из кислорода, азота, двуокиси азота, закиси азота,
окиси углерода и двуокиси углерода. (Хранить над водой иельз я!)
Цель работы. Определение состава шестикомпонентной смеси,
содержащей различные по своей природе и свойствам газы.
Сущность работы. Разделение многокомпонентной смеси мето-
дом адсорбционной хроматографии из одной пробы связано с труд-
ностями вследствие большого различия в адсорбционных свойствах
Отдельных компонентов разделяемой смеси. Лучшее разделение
может быть достигнуто, если в процессе хроматографирования де-
сорбцию различных компонентов смеси производить при разных
температурах. Этот принцип и применен в настоящей работе. Ад-
сорбция всех компонентов смеси на силикагеле производится при
низкой температуре. При этой же температуре происходит десорбция
кислорода, азота, двуокиси азота и окиси углерода. Наиболее трудно
десорбируемые газы: закись азота и двуокись углерода десорбиру-
ются при комнатной температуре. Таким путем удается полностью
разделить смесь, состоящую из шести компонентов.
194
Выполнение работы. Колонку заполняют предварительно вы-
сушенным в токе воздуха при 400° С силикагелем, после чего ее
помещают в криостат с холодильной смесью и присоединяют к хро-
матографу. Устанавливают температуру в криостате не выше
—70° С и начинают продувку всей системы газом-носителем со ско-
ростью 30 смНмин. После установления самописца на нулевой линии
на адсорбент вводят порцию газа, продувая кран-дозатор газом-но-
сителем. При температуре криостата происходит десорбция сначала
кислорода, который выходит из колонки на четвертой минуте,
затем азота (на пятой минуте), двуокиси азота (на одиннадцатой
минуте) и окиси углерода (на четырнадцатой минуте). После десорб-
ции окиси углерода колонку освобождают от криостата и нагревают
до комнатной температуры. При этом первой проявляется закись
азота и последней — двуокись углерода.
По окнчании анализа хроматограмму расшифровывают, опреде-
ляя состав смеси в объемных процентах.
Р а б о т а 4
ХРОМАТОГРАФИЧЕСКОЕ ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ
ГАЗООБРАЗНЫХ УГЛЕВОДОРОДОВ В ВОЗДУХЕ
Приборы и реактивы
1. Титрометрический газоанализатор ВНИИЯГГ.
2. Силикагель марки МСМ или ШСМ, измельчения 0,5—0,25 мм.
3. Баллон со сжатым воздухом или воздуходувка.
4. Газометр с воздухом, содержащим примесь метана, этана и пропана.
5. Титрованный раствор гидроокиси бария (0,01 н.).
6. Титрованный раствор соляной кислоты (0,01 н.).
7. Раствор метилового оранжевого (индикатор).
Цель работы. Определить содержание примеси газообразных
углеводородов в воздухе при помощи титрометрического газоанали-
затора.
Сущность работы. Смесь газообразных углеводородов можно
разделить при помощи адсорбционной хроматографии в том случае,
если в качестве адсорбента применить силикагель, а сама смесь
не содержит непредельных компонентов. Определение содержания
каждого компонента после разделения смеси можно произвести путем
сжигания углеводорода на платиновой спирали с последующим по-
глощением образующейся двуокиси углерода раствором гидроокиси
бария и титрованием оставшегося избытка этого раствора титрован-
ным раствором соляной кислоты.
Таким образом, содержание простейших углеводородов (метана,
этана и пропана) в воздухе можно определить на титрометрическом
газоанализаторе ВНИИЯГГ. При отсутствии готового прибора его
можно изготовить собственными силами в лаборатории.
7*
195
Выполнение работы. Воздух, содержащий примесь метана, этана
и пропана, подается из газометра в титрометрический газоанализатор,
схема которого приведена на рис. 38. Газ поступает через трубку I,
проходит очистительную систему 2, 3, 4 и при помощи трехходового
крана 6 разделяется на два равных потока. В одном из них в ко-
лонке 13 производится сожжение углеводородов до двуокиси угле-
рода, по количеству которой определяют суммарное содержание
Рис. 38. Схема хроматографического титрометрического газоанализа-
тора:
1, 12, 13 — колонки для сожжения; 2,3, 24, 28 — барботеры с раствором едкого
калн; 4 — осушитель с хлоридом кальция; 5— ртутный затвор; 6, 7, 16, 17, 18',
19, 26, 27 — краиы; 8, 9 — аспираторы; 10, 11 — уравнительные сосуды; 14,
15 — поглотительные камеры ; 20, 21 — мнкробюретки; 22—сливная склянка;
23 — кран, к которому присоединяется резиновая Груша для создания вакуума;
25 — колонка с адсорбентом.
Линин: I — ввод анализируемого газа; II — ввод промывающего воздуха;
III— к склянке с соляной кислотой; IV — к склянке с гидроокисью барня;
V — к резиновой груше
углеводородов в воздухе. Поглощение двуокиси углерода произво-
дится в сосуде 15, заполненном раствором гидроокиси бария. Дру-
гой поток газа поступает в хроматографическую колонку 25, за-
полненную силикагелем. Количество газа, пропущенного через си-
стему, определяется в аспираторах 8 и 9. Требуемый для анализа
объем газа может колебаться в зависимости от концентрации угле-
водородов от 50 до 400 мл.
После адсорбции в колонку 25 через трубку 16 подается газ-но-
ситель — воздух. По выходе из колонки воздух, содержащий де-
сорбированные углеродороды, поступает в колонку с платиновой
спиралью 12, на которой происходит сгорание углеводорода до
19(э
двуокиси углерода. Последняя поглощается в сосуде 14 раствором
гидроокиси бария. Избыток раствора титруют раствором соляной
кислоты в присутствии метилового оранжевого. По расходу соляной
кислоты рассчитывают количество содержащегося в воздухе угле-
водорода. Титрование избытка гидроокиси бария производят не-
посредственно в поглотительных сосудах 14 и 15. Весь анализ за-
нимает 3V2 часа.
Работа 5
ОПРЕДЕЛЕНИЕ СОСТАВА СМЕСИ ГАЗООБРАЗНЫХ
УГЛЕВОДОРОДОВ МЕТОДОМ ХРОМАТЕРМОГРАФИИ
Приборы и реактивы
1. Универсальный хроматермографический газоанализатор УХТГ-1.
2. Силикагель марки АСК, измельчения 0,25—0,5 мм.
3. Активированный уголь марки АГ, измельчения 0,25—0,5 мм.
4. Баллон с сжатым воздухом или азотом.
5. Газометр, содержащий смесь углеводородов (метан, этан, пропан, бутан).
Цель работы. Проанализировать сложную смесь углеводородных
газов и показать преимущество хроматермографии перед обычной
адсорбционной хроматографией.
Сущность работы. Как уже говорилось выше, хроматермогра-
фический метод обладает тем преимуществом, что применение дви-
жущегося температурного поля приводит к сжатию полос, исклю-
чающему или существенно уменьшающему размывание. Кроме того,
значительно сокращается время анализа. Поэтому в настоящей ра-
боте предлагается при помощи хроматермографического метода
проанализировать сравнительно сложную смесь углеводородов.
Анализируемая смесь проходит две последовательно включенные
адсорбционные колонки, первая из которых служит для разделения
смеси сильно адсорбирующихся компонентов, а вторая — для раз-
деления смеси слабо адсорбирующихся. Десорбция компонентов
смеси в первой колонке осуществляется хроматермографическим
методом при помощи движущейся вдоль колонки печи с градиентом
температуры. Анализ осуществляется на универсальном хроматер*
мографическом газоанализаторе УХТГ-1 (рис. 39).
Выполнение работы. Анализируемую смесь из газометра подают
в бюретку 16 (см. рис. 39) и отмеряют объем, равный 5 мл, с точ-
ностью до 0,05 мл. Отмеренный объем газа выдавливают в колонку 15,
заполненную силикагелем. Газ-носитель, которым может служить
воздух или азот, подается со скоростью 15 см?1мин. Одновременно
с подачей газа-носителя включают движение печи, которая должна
быть предварительно нагрета до температуры 120—130° С. Скорость
движения печи должна быть равна 2,5 см/мин.
497
В процессе анализа метан и этан движутся в колонке с силикаге-
лем быстрее печи, а остальные углеводороды со скоростью, равной
скорости движения печи. По выходе из колонки поток газа, содер-
жащий неразделенные метан и этан, направляется в колонку 12,
заполненную углем. Колонка 13 отключается. На угле происходит
адсорбция и разделение смеси метана и этана, что фиксируется на
ленте самописца. После того, как самописец зафиксирует выход
Рис. 39. Схема универсального хроматермографа УХТГ-1:
/— реометр; 2— осушитель с твердым едким кали; 3 — осушитель с хло-
ридом кальция; 4, 5, 7, 8, 11 — краны; 6 — дополнительное сопротивление;
9, 10 — детекторы; 12— колонка с углем; 13— колонка для газо-жидкостной
хроматографии; 14— движущаяся электропечь; 15— колонка для хроматер-
мографии; 16—бюретка для ввода газа в установку
этана, колонку 12 с углем отключают и газы, выходящие из колонки
15, направляют непосредственно в детектор. В процессе хроматер-
мографирования из колонки с силикагелем последовательно десор-
бируются пропан и бутан. При этом самописец фиксирует два хоро-
шо разделенных и резких пика.
Анализ заканчивают расчетом состава газа по полученной хро-
матограмме и сравнением полноты разделения, времени, потребного
для производства анализа, и точности результатов анализа, полу-
ченных в этой работе, с результатами предыдущей работы, т. е.
сравнивают хроматермографический метод с методом обычной ад-
сорбционной хроматографии.
Работа 6
ОПРЕДЕЛЕНИЕ СОСТАВА СМЕСИ ГАЗООБРАЗНЫХ УГЛЕВО-
ДОРОДОВ ТЕПЛОДИНАМИЧЕСКИМ МЕТОДОМ
Приборы и реактивы
1. Универсальный хроматермографический газоанализатор УХТГ-1 или
горизонтальная теплодинамическая установка ТДУ-1,
195
2. Система склянок для непрерывной подачи газа в колонку £ постоянной
скоростью.
3. Силикагель марки АСК, измельчения 0,25—0,5 мм.
4. Газометр с анализируемым газом.
Цель работы. Непрерывное определение состава газа хроматогра-
фическим методом.
Сущность работы.Теплодинамический метод представляет собой
сочетаниефронтальыого и хроматермографического методов анализа.
Из всех известных вариантов хроматографии он наиболее близок
к непрерывному процессу. Сущность его состоит в том, что анали-
зируемая смесь газов непрерывно подается на колонку, заполненную
адсорбентом. При этом плохо адсорбирующиеся компоненты про-
ходят слой адсорбента, не задерживаясь, и фиксируются детектором,
а сильно адсорбирующиеся накапливаются в холодной части ад-
сорбента и затем последовательно вымываются вследствие воздейст-
вия движущегося температурного поля. В то время как с одно-
го конца колонки происходит вымывание десорбирующихся
компонентов смеси, на другом ее конце — холодном, идет накоп-
ление свежей порции анализируемого газа. В результате на лен-
те самописца периодически появляется запись состава анализи-
руемого газа. Каждая запись соответствует одному циклу работы
установки.
Выполнение работы. В случае работы на установке УХТГ-1
колонки 12 и 13 (см. рис. 39) отключаются и анализируемый газ
по выходе из колонки 15 поступает непосредственно в детектор.
Колонка 15 заполняется силикагелем АСК. Печь нагревается до
температуры 120—130° С в максимуме.
Анализируемую смесь, состоящую из пропилена, пропана, «-бу-
тилена и бутана, непрерывно подают в колонку со скоростью
10 см31мин. Для подачи газа с постоянной скоростью его выдавли-
вают из склянки насыщенным раствором хлорида натрия, вытекаю-
щим из напорного сосуда Мариотта. Выдавливаемый из склянки газ
проходит осушительную систему из трубок с хлоридом кальция и
пяти окисью фосфора. После осушки на пути газа устанавливают
капилляр, устраняющий возможные пульсации потока газа. Затем
газ поступает в баранкообразную колонку установки. Одновременно
с подачей газа включают движение печи, которая должна быть пред-
варительно нагрета до требуемой температуры. Скорость движения
печи должна быть равна 2,5 см!мин.
В процессе анализа газ подается непрерывно, в то время как
печь проходит каждый данный слой адсорбента периодически, со-
вершая после первого оборота второй и т. д. При этом на ленте само-
писца периодически производится запись состава анализируемого
газа. В качестве детектора используют катарометр.
Получив на ленте самописца три-четыре хорошо воспроизве-
денных записи состава газа, опыт прекращают, для чего выключают
199
подачу газа и останавливают печь. Одновременно выключают на-
грев печи и всю установку. По полученным данным вычисляют сред-
ний состав газа *.
Работа 7
РАЗДЕЛЕНИЕ СМЕСИ КСИЛОЛОВ МЕТОДОМ
ХРОМАТЕРМОГРАФИИ В ПАРОВОЙ ФАЗЕ [9]
Приборы и реактивы
1. Хроматермографическая установка, состоящая из: а) стеклянной колонки
длиной 1 м 25 см, диаметром 10 мм, с капилляром на конце длиной 0,75 м и диа-
метром 2 мм; б) движущейся вдоль колонки цилиндрической печи с градиентом
температуры от 25 до 150° С (снизу вверх), внутренним диаметром 12 мм и длиной
0,5 м; в) приспособления для равномерного движения печи сверху вниз (могут
быть использованы часы-ходики или часовой механизм, или синхронный электро-
мотор Уоррена).
2. Силикагель марки АСК, измельчения 0,25—0,5 мм.
3, Пробирки для сбора конденсата — 10 шт.
4. Рефрактометр типа Аббе (РЛУ).
5. Баллон с сжатым воздухом или воздуходувка.
6. Смесь о- и л-ксилолов,-
Цель работы. Разделить смесь жидких углеводородов (ксилолов)
и показать на ее примере применимость хроматермографии для
разделения жидких веществ.
Сущность работы. Метод хроматермографии позволяет успешно
разделить и проанализировать смесь не только газообразных веществ,
но и жидких, но жидкости должны быть предварительно превра-
щены в пар, так как разделение происходит в паровой фазе.
Преимущество хроматермографии состоит в том, что испарение
жидкости, нанесенной на адсорбент, происходит под воздействием
той же движущейся печи, которая служит для создания движущего-
ся температурного поля с градиентом температуры. Одновременное
воздействие потока газа-носителя облегчает испарение, которое
благодаря этому происходит при температурах более низких, чем
температуры кипения этих жидкостей.
Таким образом, нанося определенную порцию смеси жидких
веществ на колонку с адсорбентом, продувая ее потоком газа-носи-
теля и прстепенно прогревая слой адсорбента движущейся печью
с градиейтом температуры, можно испарить нанесенную жидкость
и разделить ее на составляющие компоненты. По выходе из колонки
пары могут быть сконденсированы и собраны в чистом виде. После
окончания анализа адсорбент оказывается пригодным для следую-
щего анализа.
* Метод расчета устанавливается преподавателем.
200
Метод может служить для препаративных целей, так как он по-
зволяет получить компоненты разделенной смеси без примеси вы-
мывающих веществ.
Выполнение работы. Хроматографическую колонку заполняют
силикагелем. Слой силикагеля должен составлять 0,75 м. Затем
колонку подвешивают строго вертикально и устанавливают печь
в верхней части колонки так, чтобы она закрывала свободную от
адсорбента часть и была направлена своим холодным концом вниз.
Печь нагревают до температуры в ее верхнем конце 120—130° С.
Взвешенную порцию смеси о- и п-ксилолов (1,5 г) вливают в колон-
ку и после того как вся жидкость впитается в адсорбент, к верхней
части колонки присоединяют резиновую трубку, по которой подают
сжатый воздух от воздуходувки или баллона. Включают поток га-
за-носителя (воздуха) и движение печи. Скорость газа-носителя
должна быть около 160 см3/мин, а движение печи сверху вниз —
2,5 см/мин.
По мере продвижения печи вдоль колонки происходит испарение,
адсорбция и десорбция компонентов смеси с одновременным ее
разделением. Первым начинает выходить из колонки о-ксилол. Его
пары, попадая в капилляр, конденсируются и стекают в пробирку.
Во время опыта пробирки сменяют после того, как объем жидкости
в них составит 0,2—0,3 мл. Жидкость в каждой пробирке взвешива-
ют, после чего определяют ее показатель преломления. По получен-
ным данным строят хроматограмму в координатах: масса — пока-
затель преломления. Данные хроматограммы должны указать на
полноту разделения смеси и на количество каждого компонента.
По этим данным вычисляют состав смеси, или, если ее состав был
известен, выход каждого из компонентов.
Опыт прекращают после того, как верхний край печи достигнет
нижнего конца хроматографической колонки. После окончания опы-
та отключают поток газа, двигатель печи и ее нагрев. Адсорбент
может быть использован для следующего опыта без регенерации.
Работа 8
ГРАДУИРОВКА ХРОМАТОГРАФА
Приборы и реактивы
1. Хроматограф типа ХЛ-3.
2. Силикагель марки МСМ, измельчения 0,5—0,25 мм.
3. Чистые газы: метан, этан, пропан, бутан (смесь).
4. Баллон со сжатым водородом.
Цель работы. Получить данные, позволяющие производить коли-
чественный расчет состава анализируемой смеси по хроматограм-
мам.
201
Сущность работы. Чтобы иметь возможность определять не толь-
ко качественный, но и количественный состав анализируемой смеси,
надо знать зависимость параметров, определяемых по хроматограм-
мам, т. е. зависимость высот или площадей пиков от концентрации
или количества вещества, обусловливающего появление пика на
хроматограмме. Для дифференциальных хроматографов существу-
ет четыре метода установления такой зависимости: нормировка,
внутренняя стандартизация, нормировка с введением калибровоч-
ных коэффициентов и абсолютная калибровка. В работе предлага-
ется произвести калибровку одного хроматографа с катарометром
в качестве детектора всеми четырьмя методами и сравнить их между
собой. Калибровка производится для смеси газообразных углево-
дородов: метана, этана, пропана и бутана.
Выполнение работы*. I. Метод нормировки. Запол-
няют колонку силикагелем марки МСМ и производят анализ смеси
газа, вводя его порцию посредством шестиходового крана илн шпри-
цем. В качестве газа-носителя применяют водород. (Баллон
с водородом должен находиться вне поме-
щения для анализа, выход газа из прибора
направлять в атмосферу вне помещения
лаборатории!)
Продувают всю систему водородом до тех пор, пока положение
нулевой линии на самописце не станет устойчивым (прекратится
дрейф). Вводят порцию анализируемого газа. Через 4—5 мин само-
писец вычертит четыре пика. Опыт повторяют три раза, после чего
прибор останавливают, снимают диаграмму и приступают к ее рас-
шифровке. Для этого измеряют циркулем высоту каждого пика в
миллиметрах, все высоты складывают и их сумму принимают за
100%. По полученным высотам рассчитывают содержание каждого
компонента в процентах.
Измеряют площади полученных пиков одним из методов: пла-
ниметром, взвешиванием пика, вырезанного из хроматограммы, на
аналитических весах, или умножением половины высоты пика на
ширину его основания, считая пик за правильный треугольник.
Сумму площадей принимают за 100%, откуда рассчитывают содер-
жание каждого компонента в смеси и сравнивают с данными, полу-
ченными для расчета по высотам. Результаты обеих измерений сво-
дят в таблицу.
2. Метод внутренней стандартизации. Под-
готавливают прибор так же, как в предыдущем определении, и
после достижения постоянной нулевой линии вводят строго опре-
деленную порцию анализируемого газа. Получив хроматограмму,
вводят такой же объем той же смеси, добавив в нее предварительно
* Работа должна выполняться одновременно несколькими студентами, так
чтобы каждый студент произвел градуировку одним методом. После окончания
работы преподаватель и студенты сопоставляют полученные результаты,
202
строго определенное количество одного из компонентов в чистом
виде, например бутана.
После получения записи результатов хроматографирования опыт
прекращают и рассчитывают хроматограмму. Для этого измеряют
высоту и площадь пика, относящегося к бутану, как на хромато-
грамме, полученной для анализируемого газа, так и на хромато-
грамме после введения известной добавки бутана. Если количество
введенного в смесь чистого бутана известно и равно а6, высота пика
для бутана в смеси до введения его добавки в чистом виде h, а вы-
сота пика после введения добавки чистого бутана h-J-A/i, то коли-
чество бутана в анализируемой смеси можно рассчитать из со-
отношения;
аб+^6_хб
h-f-Ah h ’
откуда
Чтобы рассчитать концентрацию бутана в исходной смеси, необхо-
димо знать массу взятой пробы, или, если концентрацию рассчиты-
вать в объемных процентах, следует знать объем введенной пробы
и объем добавленного бутана *.
3. Метод нормировки с введением калибро-
вочных коэффициентов. Составляют смесь из чистых
индивидуальных компонентов или берут смесь строго известного
состава и после подготовки прибора наносят ее порцию на адсор-
бент. Опыт повторяют три раза, в результате чего получают три
хроматограммы. Для их расшифровки поступают следующим обра-
зом: измеряют высоты и площади пиков. Вводят для расчета кали-
бровочные коэффициенты, причем для одного из компонентов смеси,
например метана, коэффициент приравнивают единице. Остальные
коэффициенты рассчитывают по формулам (145):
где Kh— калибровочный коэффициент для случая измерения вы-
сот; Кп — то же для случая измерения площадей; сс и с;— концен-
трации сравнительного и определяемого z-того компонента; Лс, ht,
Пс, П(— соответственно высоты и площади пиков на хроматограмме
этих компонентов.
* Если позволяет время, опыт повторяют для каждого компонента, произ-
ведя затем расчет содержания каждого из иих аналогичным образом. Полученные
данные сводят в таблицу, аналогичную таблице, приведенной на стр. 204, и срав-
нивают с результатами, рассчитанными по методу нормировки.
203
Форма записи
Результаты измерений концентрации компонентов
анализируемого газа методом нормировки по высотам
и площадям пиков
Рассчитав калибровочные коэффициенты для каждого компонен-
та анализируемой смеси, умножают высоты или площади, полу-
ченные для каждого компонента, на соответствующие коэффициен-
ты и полученные произведения складывают. Приняв их сумму за
100%, рассчитывают содержание каждого компонента в процентах,
Результаты расчета вносят в таблицу, подобную таблице, приве-
денной выше, и сравнивают с данными, рассчитанными по ме-
тоду нормировки без калибровочных коэффициентов. Из сравнения
можно убедиться, что результаты, полученные двумя методами,
оказываются различными. При этом чем больше теплопроводность
анализируемого компонента отличается от теплопроводности ве-
щества, принятого за сравнительное, тем большее различие между
результатами опыта будет иметь место.
4. Метод абсолютной калибровки. При ана-
лизе смесей, содержащих вещества в малых концентрациях, наи-
более целесообразно применять метод абсолютной калибровки.
В этом случае составляют смесь воздуха и какого-либо компонента,
например бутана, в строго определенной концентрации (можно
взять 2% бутана по объему). После подготовки прибора и получе-
ния постоянства нулевой линии определенную порцию этой смеси
вводят в колонку. Смесь разбавляют воздухом вдвое и вводят в
колонку точно такой же ее объем. Разбавление и ввод пробы повто-
ряют несколько раз. Таким путем получают хроматограмму, на ко-
торой вычерчены пики бутана для его смесей с воздухом при кон-
центрациях 2; 1; 0,5; 0,25; 0,125 и 0,0625%. После этого измеряют
высоты и площади всех пиков и по полученным данным строят два
калибровочных графика в координатах: высота пиков — концент-
рация в объемных процентах и площадь пиков — концентрация в
объемных процентах.
Градуировку повторяют для каждого компонента анализируе-
мой смеси и строят соответствующие калибровочные графики.
204
Работа 9
ОПРЕДЕЛЕНИЕ ТЕПЛОТЫ АДСОРБЦИИ Я-БУТАНА
НА СИЛИКАГЕЛЕ ХРОМАТЕРМОГРАФИЧЕСКИМ МЕТОДОМ
Приборы и реактивы
1. Хроматермограф № 5.
2. Потенциометр типа ПП и спаренная термопара медь — константан или хро-
мель — копель.
3. Силикагель марки МСМ, измельчения 0,5—0,25 мм.
4. Баллон с сжатым водородом.
5. Чистый н-бутан.
Цель работы. Определить один из важнейших физико-химических
параметров адсорбции — теплоту адсорбции — новым хроматер-
мографическим методом.
Сущность работы. Из теории хроматермографии следует, что
характеристическая температура десорбции компонента смеси свя-
зана с теплотой его адсорбции Q на адсорбенте и отношением ско-
рости движения температурного поля к скорости потока газа-но-
сителя т] уравнением (128), которое может быть переписано в сле-
дующем йиде:
— 1gП = 2,3RTxap + • О64)
Поэтому, меняя величину т] и измеряя характеристическую темпе-
ратуру десорбции какого-либо газа, можно по величине наклона
прямой, построенной по экспериментальным данным в координа-
тах -------Ig-q, рассчитать теплоту адсорбции данного компонента
хар
на выбранном адсорбенте (Q).
Выполнение работы. Подготавливают прибор к работе, нагре-
вают печь хроматермографа №5 до температуры в максимуме 100°С
и устанавливают ее в исходном верхнем положении. На выходе газа
из колонки, т. е. в самом нижнем слое адсорбента, устанавливают
спаренную медь-константановую или хромель-копелевую термопару
и присоединяют ее холодные спаи к чувствительному гальваномет-
ру или к потенциометру типа ПП. Устанавливают определенную
скорость потока газа-носителя и наносят порцию чистого н-бутана
или его смеси с воздухом. Устанавливают заданную скорость дви-
жения печи и включают механизм, опускающий ее вниз. Следят за
температурой и выходом н-бутана. В момент достижения максимума
пика н-бутана измеряют температуру, при которой происходит его
десорбция, и полученное значение записывают.
По окончании опыта печь возвращают в исходное положение и
повторяют опыт с такой же порцией н-бутана, но при другом зна-
чении тр Производят 4—5 опытов с различными значениями т|, на-
пример с т] равным 0,028; 0,056; 0,080; 0,10; 0,18.
205
Полученные данные вносят в таблицу и по ним строят график в
координатах =Д--103—1g ц. Из наклона прямой рассчитывают зна-
1 хар
чение Q в кал!моль. Сопоставляют рассчитанную величину с име-
ющимися табличными данными.
Работаю
ПОСТРОЕНИЕ ИЗОТЕРМЫ АДСОРБЦИИ ПРОПАНА
ПО ХРОМАТОГРАФИЧЕСКИМ ДАННЫМ [27]
Приборы и реактивы
1. Хроматограф типа XЛ-3 со стеклянной U-образной колонкой длиной 50 см,
диаметром 6 мм.
2. Силикагель марки МСМ, измельчения 0,5—0,25 мм.
3. Баллон с сжатым водородом.
4. Чистый пропан.
Цель работы. Пользуясь экспериментальной выходной хромато-
графической кривой индивидуального вещества (пропана), рассчи-
тать и построить изотерму его адсорбции.
Сущность работы. В теоретической части настоящей главы были
рассмотрены некоторые методы построения изотермы адсорбции
по хроматографическим данным. В этой работе предлагается еще
одйн метод, позволяющий построить изотерму адсорбции по дан-
ным проявительного анализа.
Вследствие наличия зависимости хроматографической выходной
кривой десорбированного вещества от формы его изотермы адсорб-
ции, последнюю можно рассчитать из хроматографических данных.
Для этого следует рассчитать количество вещества а®, адсорбирован-
ного единицей массы адсорбента, как функцию его концентрации
с. В свою очередь, концентрация, соответствующая данной точке
хроматографической кривой, пропорциональна ее высоте h
c = k"h. (165)
Для определения коэффициента пропорциональности проводят
калибровочный опыт с тем же веществом и определяют площадь
выходной хроматографической кривой П в см2. Тогда количество
адсорбированного вещества а° определится из уравнения
= (166)
где со — объемная скорость потока газа-носителя, см3/мин;
и — скорость движения ленты самописца, см/мин.
Отсюда рассчитывают k”
*''S- (167)
206
Для расчета величины о0 как функции с можно воспользоваться
уравнением
С
a° = f(c)=| ^(V-Ve)dc, (168)
О
где g — вес адсорбента, г; V — объем газа-носителя, прошедшего
через колонку длиной / от впуска газа до появления концентрации с
на выходе из колонки, слс’; Vn— свободный объем колонки, см3.
Уравнение (168) позволяет рассчитать изотерму адсорбции по
экспериментальной хроматографической кривой проявительного
анализа. При этом значение с выражают через величины, связанные
с показателями детектора, а именно:
c = k''h^~h. (169)
toll ' r
Объем V можно вычислить из соотношения
' <17°)
где L — расстояние на ленте самописца от момента впуска ве-
щества, см. Подставляя в (168) значения с и V из (169) и (170), по-
лучим:
а° = f <с) = V Пл “ j dh'’ <I71>
0
ft
HA=\(L-L0)dh.
0
Интегрируя по всему удерживаемому объему, соответствующему де-
сорбционной ветви выходной хроматографической кривой, полу-
чают значения ПЛ при разных h и по полученным данным строят
график изотермы адсорбции.
Выполнение работы. Подготавливают прибор и добиваются по-
стоянства нулевой линии. Колонку с силикагелем термостатируют
при температуре 40° С. Газ-носитель (водород) пропускают с по-
стоянной скоростью, равной 46 см3! мин.
После установления постоянной нулевой линии в колонку шпри-
цем вводят 10 мл 1%-ной по объему смеси воздуха с парами,про-
пана. Смесь должна быть тщательно составлена, так как необхо-
димо знать точное количество введенного пропана. Получают хро-
матографическую кривую пропана и снова добиваются постоянства
нулевой линии.
Вводят новую точно такую же порцию смеси воздуха и паров
пропана. По получении новой выходной кривой опыт прекраща-
ют, ленту вынимают из самописца и приступают к обработке хро-
матограмм, . :
207
Для первой хроматограммы определяют площадь выходной кри-
вой в см2 одним из методов, указанных в работе 8, например путем
взвешивания. Одновременно определяют высоту выходной кривой.
По полученным данным рассчитывают ко-
Рис. 40. К расчету изо-
термы адсорбции по хро-
матограмме:
заштрихованный участок
указывает, как следует ве-
сти интегрирование для по-
лучения кривой изотермы
адсорбции
эффициент пропорциональности k” цо (167)
и концентрацию с по (165).
Выходную кривую второй хроматограм-
мы подвергают графическому интегриро-
ванию. Для этого всю хроматограмму
делят на пять-шесть участков, проводя
прямые, параллельные оси времени, как
это показано на рис. 40. Для каждого
участка измеряют высоту h и рассчиты-
вают по формуле (169) соответствующую
концентрацию с, а по (171) площадь, за-
штрихованную на рис. 40. По значе-
ниям ПА для каждого h рассчитывают
величину а0. Для расчета последней не-
обходимо знание массы адсорбента g.
Поэтому адсорбент до его засыпки в ко-
лонку взвешивают.
Рассчитанные величины вписывают в
таблицу, по данным которой строят график
изотермы адсорбции в координатах с—а0.
Форма записи
Данные для построения изотермы адсорбции пропана
№ п.п. h, см П&, с.«2 а®, мг г с, мг см3 kn = и = 11 II 3 ъо
ЛИТЕРАТУРА
Использованная литература
1. Я. И. Герасимов и др. Курс физической химии, т. 1. Госхим-
издат, М.,: 1963.
2. A. J. Р. М а г t i n, R. L. М. S у n g е. Biochem. J. 35, 1358, 1941.
3. Э. Байер. Хроматография газов. ИЛ, М., 1961.
4. J. J. van Deemter, F. J. Zul derweg, A. Klinkenberg.
Chem. Eng. Sci., 5, 271, 1956.
5. А; Кейле мане. Хроматография газов. ИЛ, M., 1959.
6. А. А. Жуховицкий, Н. М. Т у р к е л ь т а у б. Усп. хим., 26,
. 992, 1957.
7. А. А. Жуховицкий, Н. М. Т у р к е л ь т а у б. Газовая хро-
матография. Гостоптехиздат, М., 1962.
208
8. А. А. Жуховицкий, О. В. Золотарева, В. А. Соко-
лов, Н. М. Туркельтауб. ДАН СССР, 77, 435, 1951.
9. Б. В. Айвазов, «Хроматография, ее теория и применение». Изд-во
АН СССР, М„ 1960, стр. 438.
10. Е. J. Glueckauf. J. Chem. Soc., 1302, 1321, 1947.
11. Н. М. Туркельтауб, В. П, Шварцман, В.В. Нау-
мова, А. А. Жуховицкий. ЖФХ, 30, 417, 1956.
12. Е. Kremer. Angew. Chem., 71, 512, 1959.
13. М. Ш и н г л я р. Газовая хроматография в практике. Изд-во «Химия»,
М., 1964.
14. Г, Ш а й. Теоретические основы хроматографии газов. ИЛ, М., 1963.
15. Е. П. Фесенко, А. А. Д а ц к е в и ч, В. Р. А н д е р с. «Газовая
хроматография». Труды 1-й Всес. конф. Изд-во АН СССР, М., 1960.
16. J. Е. L о v е 1 о с k J. Chromat., 1, 35, 1958.
17. Д. А. Вяхирев, А. И. Брук, С. А. Г у г л и н а. ДАН СССР,
90 577 1953
18. ’j. J а п a k. Collection., 18, 798, 1953; Chem. listy, 47, 817, 1953.
19. «Хроматография, ее теория и применение». Изд-во АН СССР, М., 1960.
20. «Газовая хроматография». Труды 1-й Всес. конф. Изд-во АН СССР.,
1960.
21. Н. М. Туркельтауб. Хроматографические газоанализаторы.
ГИНТИ, М., 1959.
22. Хроматографические методы анализа газов. Каталог. ГосИНТИ, М., 1960.
23. Хроматограф ХЛ-3. Моснефтекип. Каталог. Гостоптехиздат, М., 1962.
24. Хроматографы РХ. Автоматический контроль и регулировка. Каталог.
Дзержинск, 1964.
25. Газовые хроматографы «Цвет». Модели 1-64; 2-65; 3-66. Каталог. Дзер-
жинск, 1966.
26. «Газовая хроматография». Труды 3-й Всес. конф. Изд. Дзержинского
филиала ОКБА, Дзержинск, 1966.
27. Практические работы по физической химии. Под ред. Я. И. Герасимова
и др., ч. 4. Изд-во МГУ, 1962.
Рекомендуемая литература
1. К. Филлипс. Хроматография газов. ИЛ, М., 1958.
2. С. Б р у н а у э р. Адсорбция газов и паров. Т. 1, ИЛ, М., 1948.
3. «Успехи и достижения газовой хроматографии». Гостоптехиздат, М., 1961.
4. «Газовая хроматография». Труды симпозиума в Амстердаме. ИЛ, М., 1961.
5. Газовая хроматография. Библиографический указатель отечественной
и зарубежной литературы. (1952—1960 гг.). Изд-во АН СССР, М., 1962.
6. Д. П. Тимофеев. Кинетика адсорбции. Изд-во АН СССР, М., 1962.
7. «Разделение и анализ углеводородных газов». Изд-во АН СССР, М., 1963.
8. В. Н. Зрелое, Г. И. К и ч к и и. Хроматография в нефтяной н
нефтехимической промышленности. Гостоптехиздат, М., 1963.
9. «Газовая хроматография». Труды 2-й Всес. конф. Изд-во «Наука», М.,
1964.
10. «Газовая хроматография». Труды симпозиума в Эдинбурге. Изд-во «Мир»,
М„ 1964.
11. «Газовая хроматография». Вып. 1. НИИТЭХим, М., 1964.
12. Молекулярная хроматография. Изд-во «Наука», М., 1964.
13. В. В. Рачинский. Введение в общую теорию динамики сорбции
и хроматографии. Изд-во «Наука», М., 1964.
14. В. А. Соколов, Н. С. Торочешников, Н. В. Кельцев.
Молекулярные сита и их применение. Изд-во «Химия», М., 1964.
15. Г. Б е р ч ф и л д, Э. Сторрс. Газовая хроматография в биохимии.
Изд-во «Мир», М„ 1964.
16. К- А. Г о л ь б е р т, М. С. В иг дер га уз. Курс газовой хрома-
тографии. Изд-во «Химия», М. 1967.
209
ГЛАВА V
ГАЗО-ЖИДКОСТНАЯ ХРОМАТОГРАФИЯ
Существенным этапом развития хроматографии явилось откры-
тие А. Дж. П. Мартином и А. Т. Джеймсом [1] в 1952 г. метода газо-
жидкостной хроматографии. Наличие прямолинейной изотермы
распределения, возможность большого выбора неподвижных жид-
ких фаз,- а также применимость метода для анализа и разделения
смеси веществ в теоретически неограниченной области их выки-
пания обусловили стремительное развитие метода и широкое его
внедрение в химию, биологию, промышленный анализ и т. д.
В отличие от адсорбционной хроматографии, в газо-жидкостной
имеет место распределение компонентов разделяемой смеси между
газообразной и жидкой фазами» причем последняя является не-
подвижной. Жидкая фаза наносится на твердый инертный носитель»
задача которого состоит в локализации жидкости в пространстве и
в таком состоянии, при котором обеспечивается наилучшая массо-
передача. Успех газо-жидкостной хроматографии в основном опре-
деляется выбором и правильным нанесением жидкой фазы. При этом
должна быть обеспечена максимальная селективность жидкой фазы.
Отношение концентрации анализируемого вещества в жидкой
неподвижной фазе к его концентрации в газовой фазе играет перво-
степенную роль в разделении смеси веществ. Оно называется ко-
эффициентом распределения. Если коэффициент распределения не
зависит от концентрации растворяющегося вещества, то изотерма
распределения линейна и тогда хроматографические пики оказы-
ваются симметричными, если, конечно, не действуют другие раз-
мывающие факторы.
Приведенные в предыдущей главе основные закономерности,
имеющие место в газовой адсорбционной хроматографии, остаются
справедливыми и для газо-жидкостной хроматографии, с той лишь
разницей, что в последнем случае мы должны рассматривать не
процесс адсорбции и десорбции газа или пара на поверхности твер-
дого вещества — адсорбента, а процесс растворения и выделения газа
или пара в жидкой пленке, удерживаемой твердым инертным но-
сителем.
210
Таким образом, основное уравнение равновесной газовой хрома-
тографии (72), выведенное в предыдущей главе, остается справед-
ливым и для случая газо-жидкостной хроматографии. Его рассмот-
рение позволяет сделать вывод о том, что скорость перемещения
газа вдоль слоя жидкой неподвижной фазы при данной концентра-
ции зависит от коэффициента Генри Г. Она тем больше, чем меньше
Г, т. е. чем труднее растворяется газ или пар в выбранной жидко-
сти. Отсюда следует, что хроматографические зоны компонентов
разделяемой смеси, обладающих различными значениями коэффи-
циента Генри, будут передвигаться вдоль слоя с разными скоро-
стями, что и обеспечит разделение смеси.
Значительное преимущество газо-жидкостной хроматографии
перед газо-адсорбционной состоит в том, что, как правило, распре-
деление подчиняется линейной изотерме, тогда как адсорбция про-
исходит по нелинейной изотерме. Уравнение (72), как мы уже от-
мечали в гл. IV, будет справедливым только в случае линейной
изотермы. Второе преимущество заключается в том, что коэффици-
ент Генри в случае растворения значительно больше изменяется
при переходе от одного вещества к другому, чем это имеет место при
адсорбции. Все это и обеспечивает большие возможности газо-жид-
костной хроматографии в разделении сложных смесей.
В условиях газо-жидкостной хроматографии газ-носитель не
должен растворяться в неподвижной фазе и адсорбироваться твер-
дым носителем. Поэтому для газа-носителя Г=0, и, следовательно,
исходя из уравнения (72), скорость перемещения газа-носителя а
будет равна
(172)
Таким образом, линейная скорость перемещения газа-носителя
определяется его объемной скоростью и занимаемым им объемом
в колонке.
Линейная скорость перемещения компонентов разделяемой
смеси не поддается измерению. Поэтому в газо-жидкостной хрома-
тографии пользуются относительной скоростью перемещения ком-
понента
(173)
выражает отношение объема неподвижной фазы к
объему газа, находящегося в колонке.
В связи с тем, что обычно Г^>1, а следовательно, и 2Г,'<Л, то
величина Поэтому уравнение (173) может быть упрощено
(174)
211
Так как для различных веществ, даже относящихся к одному гомо-
логическому ряду, значения констант Генри обязательно разли-
чаются, то теория равновесной хроматографии утверждает возмож-
ность разделения любых по сложности смесей на их составляющие.
Встречающиеся отклонения объясняются тем, что либо имеет место
криволинейность изотермы распределения, либо действуют диффу-
зионные и кинетические факторы, приводящие к размыванию полос.
Константа RF имеет большое значение для распределительной
хроматографии, так как ее значение является характерным для
каждого компонента смеси. Н. А. Фукс [21 предложил называть
коэффициент Rr подвижностью.
Другой важной константой газо-жидкостной хроматографии яв-
ляется коэффициент распределения Кр.
Успех разделения смеси веществ методом газо-жидкостной хро-
матографии обеспечивается в основном правильным выбором не-
подвижной фазы, причем она должна обладать наибольшей селек-
тивностью. Кроме того, следует учитывать ряд других требований,
предъявляемых к неподвижным фазам. Все это заставляет нас преж-
де всего рассмотреть необходимые условия выбора неподвижных
жидких фаз, а интересующихся теорией газо-жидкостной хромато-
графии отослать к соответствующим монографиям [3—5].
ВЫБОР ЖИДКОЙ ФАЗЫ
Одним из основных требований к жидкостям, применяемым в
качестве неподвижных фаз, является их полная химическая инерт-
ность как по отношению к компонентам разделяемой смеси, так и
по отношению к твердому носителю. Не менее важным являются
требования малой вязкости, незначительной летучести, высокой се-
лективности. Последнее определяет значение коэффициента распре-
деления. Необходимо также, чтобы неподвижная фаза прочно
удерживалась на поверхности выбранного твердого носителя и
была бы достаточно термически устойчивой. Несмотря на довольно
жесткие требования, предъявляемые к неподвижным жидким фа-
зам, в литературе описано очень большое число жидкостей, приме-
няемых в качестве неподвижных фаз. В табл. 16 приведен список
и основные свойства только наиболее широко применяемых жидких
фаз. В ней же указаны вещества, смеси которых могут быть разде-
лены на данных жидких фазах.
Чтобы наиболее эффективно использовать жидкую фазу, сле-
дует правильно выбрать ее количество, наносимое на твердый но-
ситель. Оно зависит от характера поставленной задачи и обычно
колеблется от 0,1 до 30—40% от веса твердого носителя. Количе-
ство жидкой фазы вызывает изменение величины коэффициента
селективности Кс, в частности, при увеличении толщины пленки
жидкой фазы 60, а следовательно, и при увеличении количества на-
носимой жидкой фазы, Кс возрастает. А. А. Жуховицкий и Н. М.Тур-
,212
Таблица 16
Растворители, наиболее часто применяемые
в качестве неподвижных жидких фаз
в газо-жидкостной хроматографии
растворитель Максималь- ная рабочая температура, 0 С Разделяемые смеси веществ
Вазелиновое масло (смесь Гомологические ряды углеводо-
жидких парафинов высокой 130 родов и других жидких оргаии-
чистоты) ческих соединений
Сквалан (2, 6, 10, 15, 19, 23- 160 Гомологические ряды углеводо-
гексаметнлтетракозан) родов, спиртов, кетонов, эфиров
Октадекан 150 Гомологические ряды углеводо-
Гексатриаконтан 160 родов То же
Апиезон (высоковакуумная 280 Гомологические ряды высококи-
смазка) пящих соединений различных
Высококипящее авиационное классов
масло с 00 Сераорганические соединения
Фталаты: 100 120 Смеси углеводородов, спиртов,
дибутил фенолов, кетонов, альдегидов, га-
диизоамил лоидопроизводных, сложных эфи-
диоктил IbU 170 180 170 ров
динонил
дидецил диизодецил Дио ктилсебацинат Смеси углеводородом
Тритолилфосфат 100 Гомологические ряды мерка-
Т рикрезилфосфат 180 птанов и сульфидов Смеси углеводородов, спиртов,
Бензилдифенил 150 сложных эфиров, сераорганйче- ских соединений, сероводорода с воздухом и углеводородами Смеси ароматических углеводо-
Силиконовое масло (ДС-200 240 родов, галоидопроизводных, спир- тов, аминов Смеси высококипящих веществ
и ДС-703) различных классов
Диметилформамид 20 Смеси иизкокипящих изомеров
Диметилсульфолан (2,4-ди- углеводородов То же
метилтиофандиоксид) 40
Дифенилформамид 100 Смеси средиекипящих изомеров
Р, Р'-Оксидипропиоиитрил 70 углеводородов (пентаны — октаны) Смеси углеводородов и других
Р, Р'-Тиодипропионитрил 70 классов соединений различной структуры Смеси газообразных углеводоро-
дов и сернистых соединений
213
Продолжение табл. 16
Растворитель Максималь- ная рабочая температура, ° С Разделяемые смеси веществ
Р, Р'-Иминодипропионитрил 70 Сераорганические соединения
Дигл вдерни 150 Смеси спиртов, аминов, кетонов
Полиэтиленгликоль-200 100 Смеси углеводородов, спиртов,
» 400 120 кетонов, альдегидов, галоидопро-
» 1000 150 изводных, сераорганических сое-
» 1500 200 динений
Адипат полиэтиленгликоля 150 Сераорганические соединения
Растворы азотнокислого се- ребра в этиленгликоле 40 Смеси низкокипящих углеводо- родов, принадлежащих различным
Алкилариловый эфир с по- лиоксиэтиленовой цепочкой (Тритон X) Полиэфир адипиновой кисло- ты и пропиленгликоля (Рео- 150 150 гомологическим рядам Сераорганические соединения Гомологический ряд органиче- ских дисульфидов и полисульфи-
плекс-400) Эвтектическая смесь NaNO3: KNO3: LiNO3= 18,2:54,5:27,3 — ДОВ Смесь алифатических и цикли- ческих сульфидов
кельтауб [5], исходя из предположения о наложении вихревой и
внутренней диффузии в газо-жидкостной хроматографической ко-
лонке, выводят формулу, устанавливающую связь между /<с и 60
Ле = 3 (1 ~ г 6°АГ , (175)
где % — доля свободного сечения колонки, г — радиус зерен твер-
дого носителя.
Несмотря на то, что увеличение количества наносимого для ана-
лиза вещества приводит к уменьшению числа теоретических таре-
лок и, следовательно, к ухудшению разделения, возрастание селек-
тивности при увеличении количества жидкой фазы позволяет одно-
временно увеличивать и количество наносимой для разделения смеси.
Очевидно, что оптимальное количество жидкой фазы должно уста-
навливаться в каждом отдельном случае в зависимости от кон-
кретных условий опыта и поставленной задачи.
Процесс нанесения жидкой фазы должен обеспечить удержа-
ние определенного ее количества на твердом носителе, а также рав-
номерное покрытие зерен носителя ее пленкой. Эти условия могут
быть выполнены несколькими приемами, из которых наиболее
часто применяется следующий. Рассчитывают требуемое количест-
во жидкой фазы и определенную навеску растворяют в таком раст-
214
ворителе, который был бы достаточно летуч и одновременно хорошо
растворял бы выбранную жидкую фазу. Затем подготовленный,
т. е. просушенный и измельченный твердый носитель тщательно
пропитывается полученный раствором. После этого растворитель
испаряют, нагревая всю массу на водяной бане при перемешива-
нии. Для окончательного удаления растворителя пропитанный
жидкой фазой носитель рассыпают на фильтровальной бумаге и-
оставляют на воздухе до полного высушивания. Полученный таким
образом носитель, содержащий жидкую фазу, внешне не отличается
от носителя без нее. Поэтому его следует хранить в склянках с со-
ответствующей надписью.
Маловязкие, а также сравнительно летучие фазы следует на-
носить непосредственной пропиткой ими твердого носителя, т. е.
без применения летучего растворителя. При этом необходимо до-
биваться, чтобы все взятое количество носителя было пропитано
жидкостью, а также особенно тщательно следить за равномерной
его пропиткой. Последнее может быть достигнуто, если брать из-
быток жидкой фазы и после полной пропитки удалять избыток
жидкости путем фильтрования всей массы под вакуумом на воронке
Бюхнера. Количество нанесенной жидкой фазы определяется в
этом случае по привесу твердой фазы после полного удаления из-
бытка жидкости.
При выборе наиболее подходящей жидкой фазы часто исполь-
зуют качественные представления о природе связи между молеку-
лами веществ, подлежащих анализу, и молекулами растворителя.
С этой точки зрения целесообразно обычно применяемые жидкие
фазы разделять на две группы: полярные и неполярные.
Рассмотрим задачу разделения смеси веществ, принадлежащих
к одному гомологическому ряду. Если взять соСедние члены такого
ряда, то изменение изохорно-изотермического потенциала ДЕ,
отнесенное к компонентам смеси, отличающимся иа группу СН2,
будет равно
ДГ=ДЯ-7’Д5.
Здесь Д/7 — изменение энтальпии — отличается от величины теп-
лоты сорбции группы СН2 только знаком и не зависит от концент-
рации вещества. AS — изменение энтропии — относится к стандарт-
ным состояниям рассматриваемых растворов.
Для сравнения разделительной способности выбранного раство-
рителя целесообразно сопоставлять относительные удерживаемые
объекты веществ разделяемой смеси VR> отн, т. е. удерживаемые
объемы, отнесенные к удерживаемому объему какого-либо компо-
нента смеси. Часто их относят к удерживаемому объему первого
члена гомологического ряда.
Относительный удерживаемый объем является величиной, Ха-
рактерной для выбранного растворителя, определяющей его селек-
тивность,
215
Связь удерживаемых объемов с коэффициентом селективности
может быть выражена следующим уравнением:
k- vW-vR,i Z17R4
Vr, 2+ ¥*,’$> ( 76)
Зная, чтоУД!0тп=УЛ12/УД11, и полагая, что для двух соседних членов
гомологического ряда VSil^VSt2, получим
Яс^уО^.от»-1). (177)
Отсюда следует, что коэффициент селективности может быть оп-
ределен из величины относительного удерживаемого объема.
В свою очередь, относительный удерживаемый объем должен
быть связан с разницей изменения изохорно-изотермических потен-
циалов процессов растворения компонентов анализируемой смеси
ДГ2>1 = ДГ2 —ДГр
Тогда
Vr 2 КР 2 х-
= = e RT'
Если принять, что АЕ21 не зависит от длины цепи молекул
веществ гомологического ряда, то уравнение (178) будет.подтвер-
ждать известное правило Траубе, согласно которому коэффициент
распределения для членов данного ряда изменяется по закону гео-
метрической прогрессии. Если же принять также, что стандартные
значения энтропий растворения AS для соседних членов ряда оди-
наковы, то
VR,^ = e-Q^/RT, (179)
где Qch2— теплота растворения группы СН2. Таким образом, отно-
сительный удерживаемый объем при переходе от одного члена ряда
к соседнему будет изменяться на определенную постоянную вели-
чину, связанную с теплотой растворения группы СН2.
При появлении в молекуле новой группы СН2 выигрывается
работа взаимодействия и затрачивается работа раздвижения мо-
лекул. Последняя будет зависеть от полярности молекул раствори-
теля. Поэтому значение величины Qch2 для полярных растворите-
лей меньше, чем для неполярных. Отсюда следует, что разделение
смеси веществ одного гомологического ряда будет более успешным
на неполярной жидкой фазе.
Для разделения смеси неполярных и полярных соединений сле-
дует применять полярную жидкую фазу, так как в этом случае
удельный удерживаемый объем полярного соединения будет боль-
ше, чем на неполярной фазе, что приведет к увеличению расстояния
между зонами полярного и неполярного соединений.
Разделение смеси полярных веществ также следует проводить
на полярных жидких фазах.
216
Выбор наиболее подходящей жидкой фазы облегчается рядом
опытных закономерностей. Установлено, что величины удерживае-
мых объемов на данной жидкой фазе изменяются в определенной
последовательности при переходе в органическом соединении от од-
ного заместителя к другому. Например, на вазелиновом масле удер-
живаемые объемы для соединения С5НПХ, где X — различные за-
местители, возрастают при переходе от одного заместителя к друго-
му в следующем порядке:
—ОСН3<—Вг<—С1<—1<—NO2<—СССН3<—NH2<—CN<—ОН
На другой фазе порядок увеличения удерживаемых объемов мо-
жет быть иным.
Компоненты разделяемых смесей могут образовывать с вещест-
вом жидкой фазы комплексные соединения. При этом важная роль
принадлежит водородной связи. Последняя растет с ростом величи-
ны дипольного момента связи. Эти свойства также могут служить
ориентиром в выборе жидкой фазы.
Правильно выбранная жидкая фаза может обеспечить разделе-
ние достаточно сложных смесей. Однако это удается не всегда, осо-
бенно для очень сложных систем. В этих случаях рекомендуется
применять либо смеси нескольких жидких фаз, нанесенные на один
твердый носитель, либо соединять последовательно несколько ко-
лонок, заполненных твердым носителем с различными жидкими
фазами. Изменяя количественное соотношение растворителей или
последовательность расположения колонок, можно варьировать
условия разделения в широком диапазоне.
В табл. 17 приведены значения относительных удерживаемых
объемов для нескольких гомологических рядов углеводородов на
двух неподвижных фазах [5].
ВЫБОР ТВЕРДОГО НОСИТЕЛЯ
Задача твердого носителя состоит в локализации жидкой фазы
на значительной и достаточно доступной поверхности. Поэтому в
качестве материала для твердых носителей предпочтительно приме-
нять вещества с развитой макропористостью и достаточно малой
микропористостью. Последнее необходимо для того, чтобы исклю-
чить адсорбцию анализируемых соединений поверхностью твердого
носителя. Кроме этого, носитель не должен обладать каталитиче-
ской активностью. Нецелесообразно также применение гидрофиль-
ных материалов, так как на них трудно получить воспроизводимые
результаты. Что касается величины зернения носителя, то здесь
предъявляются те же требования, что и в газо-адсорбционной хро-
матографии, т. е. размеры зерен должны обеспечить достаточно раз-
витую поверхность, хороший доступ газа-носителя и минимальное
сопротивление его потоку.
217
Таблица 17
Относительные удерживаемые объемы некоторых углеводородов
на полярных и неполярных жидких фазах (при 25° С)
У глеводороды Температура кипения, °C Жидкая фаза
Сквалан Трикре- зилфос- фат
н-А л к а н ы
Этан - 88,6 0,03 0,03
Пропан — 42,1 0,09 0,10
Бутан — 0,5 0,31 0,32
Пентан 36,1 1,00 1,00
Гексан 68,7 3,20 2,92
Гептан 98,4 9,83 8,07
н-А лкены-1
Этилен —103,7 >0,02 0,03
Пропилен — 47,7 0,08 0,13
Бутен-1 — 6 3 0,25 0,41
Пентен-1 30,0 0,81 1,20
Гексен-1 63,5 2,66 3,61
Гептен-1 93,6 8,62 —•
Циклены
Циклопеятан 49,3 2,12 3,43
Метилциклопентан 71,« 4,07 5,02
Циклогексан 80,7 5,80 7,34
1,1-Диметилциклопентан 87,8 6,84 —
1,3-Диметилциклопентан (цис) 91,7 8,55
1,2 Диметилциклопентан (транс) 91,9 8,55 —.
Ц2-Диметилциклопентан (цис) 99,5 И,2 «
Метилциклогексан 100,9 11,8 ——
Этилциклопентан 103,5 13,0 —
1, 1, З-Триметилциклопентан 104,9 12,4 —
Перечисленные выше требования к твердому носителю позволя-
ют рекомендовать довольно широкий ассортимент материалов, удов-
летворительно выполняющих функцию носителя. Поэтому в лите-
ратуре можно найти сведения о применении в качестве носителей
многих материалов. Наибольшее распространение получили веще-
ства, приведенные в табл. 18.
Рассмотренные в гл. IV положения, касающиеся выбора газа-
носителя, детектора и т. д., а также методики проведения опыта
остаются в силе и для газо-жидкостной хроматографии. Однако
следует иметь в виду, что метод газо-жидкостной хроматографии
позволяет анализировать не только газообразные, но и жидкие ве-
щества. Поэтому в газо-жидкостной хроматографии для анализа
жидких смесей могут применяться только те приборы, в которых
имеются приспособления для испарения введенных в колонку жид-
218
Таблица 18
Вещества, наиболее часто применяемые в качестве
твердых носителей
Носитель
Удельная
поверхность
носителя, Л12/г
Инзенский кирпич ИНЗ-600 (СССР)
Дмитровский кирпич (СССР) . . .
Апрелевский кирпич (СССР) . . .
Диатомит (СССР)...............
Каолин (СССР) ................
Трепел (СССР).................
Ресорб (ЧССР).................
Светлофильтр (ЧССР) ......
Инфузорная земля (ЧССР) . , . .
Тефлон (США)..................
Хромосорб (США)...............
Целит (США) ..................
Огнеупорный кирпич (США) . . .
Стерхамол (ФРГ)...............
4—8
14
42
6
6—12
5—8
7,5
14—35
0,2
1,4-2,8
1,7
3
5—10
костей, а также имеется возможность поддерживать на протяжении
всего опыта температуру колонки и детектора на уровне, исключаю-
щем конденсацию паров жидких компонентов анализируемой смеси.
Не менее важным является также требование высокой точности
дозировки проб жидких веществ, вводимых в колонку, так как при
анализе жидкостей количество вводимого вещества по объему долж-
но быть значительно меньше объема вводимого газа. Очевидно, что
в этих случаях наиболее целесообразно для ввода проб применять
микрошприцы или же дозаторы специальной конструкции.
Калибровка приборов и расчеты количества анализируемых ве-
ществ по хроматограммам производятся так же, как и в адсорбцион-
ной газовой хроматографии.
ПРАКТИЧЕСКИЕ РАБОТЫ
Работа 1
ХРОМАТОГРАФИЧЕСКОЕ ОПРЕДЕЛЕНИЕ СМЕСИ
ГАЗООБРАЗНЫХ УГЛЕВОДОРОДОВ Сх—С6
Приборы и реактивы
1. Хроматограф УХ-1 или ХЛ-4. Колонка длиной 6 м, диаметром 6 мм.
2. Твердый носитель — диатомит, измельчения 0,5—0,25 мм, пропитанный
днизоамилфталатом. Содержание жидкой фазы 15 вес. % от твердой фазы.
219
3. Микрошприц иа 0,5 мл.
4. Баллон с сжатым водородом.
5. Газообразная смесь углеводородов Сх—С6. Состав смеси может быть раз-
личным*.
Цель работы. Проанализировать сложную смесь, определить ко-
личественный состав смеси и убедиться в преимуществе газо-жидко-
стной хроматографии перед адсорбционной.
Сущность работы. Вследствие высокой селективности выбранной
жидкой фазы нанесенная на колонку смесь газообразных угле-
водородов разделяется, причем каждый компонент выходит после-
довательно один за другим. По выходе из колонки газ-носитель,
содержащий анализируемые компоненты, проходит детектор, при-
чем возникающий в нем импульс • преобразуется и записывается
на ленте самописца в виде хроматографического пика. По высотам
или площадям пиков одним из разобранных в гл. IV методов опре-
деляют количественное содержание каждого компонента смеси.
Выполнение работы. Колонку заполняют диатомитом, пропитан-
ным 15% диизоамилфталата; При заполнении колонки тщательно
слёдят за равномерным уплотнением твердой фазы. Колонку уста-
навливают в хроматограф,, проверяют герметичность всей системы
и включают ток газа-носителя. (Водород из колонки не
выпускать в помещение!) Включают в электрическую
сеть прибор и начинают подготовку прибора к анализу. Температу-
ру термостата колонки устанавливают 50° С, давление газа-носи-
теля на входе в колонку 1,5 ати, скорость газа-носителя 3 л/час.
Чувствительность самописца следует выбрать так, чтобы пик ве-
щества, содержащегося в большем количестве, уложился на шкале.
Когда дрейф нулевой линии самописца прекратится и нулевая
линия будет вычерчиваться в виде прямой, параллельной оси вре-
мени, прибор считается подготовленным к анализу. После этого
через головку колонки, закрытую самоуплотняющимся резиновым
колпачком, шприцем вводят 0,3 мл анализируемой газообразной
смёси. Момент впуска автоматически отмечается на нулевой линии
самописца.
Если смесь состоит из метановых и олефиновых углеводородов
состава Сг—С5, то порядок выхода компонентов на выбранной жид-
кой фазе будет следующим: метан, этан, этилен, пропан, пропилен,
изобутан, н-бутан, изобутилен вместе с бутиленом-1, /пранс-бутен-2,
цпс-бутен-2, изопентан, 3-метил-бутен-1, н-пентан, пентен-1,
2-метилбутен-1, пентен-2, 2-метилбутен-2. Вся операция хромато-
графирования продолжается 20—25 мин.
По окончании опыта отключают ток газа-носителя, выключают
из сети прибор, вынимают из самописца ленту и приступают к ее
расшифровке, получив у преподавателя указание о порядке распо-
* Смесь составляется преподавателем.
220
ложения компонентов и о методе расчета хроматограммы. Опреде-
ляют количественный состав смеси. Результаты анализа сводят
в таблицу.
Работа 2
ОПРЕДЕЛЕНИЕ ЗАВИСИМОСТИ ВЕЛИЧИНЫ
УДЕРЖИВАЕМОГО ОБЪЕМА ОТ ЧИСЛА АТОМОВ УГЛЕРОДА
В МОЛЕКУЛЕ АЛИФАТИЧЕСКИХ СПИРТОВ [5]
Приборы и реактивы
1. Хроматограф ХЛ-4 с выносной стеклянной колонкой длиной 180 см, диа-
метром 4 мм.
2. Твердый носитель целит, измельчения 0,5—0,25 мм, пропитанный три-
крезилфосфатом (20% от веса носителя).
3. Микрошприц иа 0,5 мл.
4. Баллон с сжатым азотом.
5. Смесь спиртов: метилового, этилового, н-пропилового, н-бутилового и
н-амилового.
Цель работы. Установить математическую зависимость между
величинами удерживаемых объемов компонентов хроматографируе-
мой смеси веществ, принадлежащих к одному гомологическому ряду,
и числом атомов углерода в их молекулах.
Сущность работы. Для идентификации компонентов разделенной
смеси, особенно если они относятся к одному гомологическому ряду,
можно воспользоваться линейной зависимостью логарифма удержи-
ваемого объема данного члена гомологического ряда от числа ато-
мов углерода в его молекуле. В самом деле, если принять,.что теп-
лота растворения при переходе от одного члена гомологического
ряда к другому изменяется пропорционально числу атомов углеро-
да п в молекуле, а изменение энтропии не зависит от этого числа, то,
в соответствии с формулой (160), можно представить себе, что
lgV^=X«+B.
Хроматографируя смесь веществ, принадлежащих к одному го-
мологическому ряду, например, спиртов, и определяя величины
удельных удерживаемых объемов для каждого члена ряда, строят
график в координатах п—IgV#, откуда определяют значения коэф-
фициентов А и В. Пользуясь выведенным уравнением и измеряя
удельный удерживаемый объем какого-либо неизвестного члена
этого же гомологического ряда, находят для него значение п и
идентифицируют искомое вещество.
Выполнение работы. Колонку U-образной формы заполняют
предварительно приготовленным твердым носителем, содержащим
20% трикрезилфосфата. Колонку присоединяют к хроматографу
ХЛ-4 вместо имеющейся в нем металлической колонки. Продувают
221
газом-носителем всю систему со скоростью 50 см3/мин, включают
прибор и добиваются постоянства нулевой линии.
После выведения системы на режим в колонку через .резиновый
колпачок вводят 0,05 мл жидкой смеси спиртов. Получают хрома-
тограмму, на которой первым пиком должен быть пик метилового
спирта (на 3-й минуте), вторым — пик этилового спирта (на 4-й ми-
нуте), третий пик будет относиться к н-пропиловому спирту (на
7-й минуте), четвертый к н-бутиловому (на 13-й минуте) и пятый
к н-амиловому спирту (на 22-й минуте). В случае отсутствия в смеси
того илй иного компонента на хроматограмме будет отсутствовать
соответствующий ему пик.
После окончания анализа, т. е. после выхода последнего, компо-
нента и возвращения пера самописца на нулевую линию, установку
выключают, диаграмму вынимают и приступают к измерению удер-
живаемых объемов. Для этого из максимумов высот пиков опускают
на нулевую линию перпендикуляры и измеряют расстояние между
перпендикуляром на нулевой линии и отметкой впуска смеси на
диаграмме. Зная это расстояние (в см), скорость движения ленты
самописца (в см/мин) и скорость газа-носителя (в см3/мин), рассчи-
тывают величину удерживаемого объема для каждого компонента
смеси. Для определения величины удельного удерживаемого объе-
ма V# полученные значения делят на массу загруженного в колонку
твердого носителя с жидкой фазой. Вычисляют логарифм удельных
удерживаемых объемов и по полученным данным строят график в
координатах п—1g Уд. По наклону прямой определяют значение
коэффициента А и вычисляют коэффициент В*.
Все измеренные и расчетные данные должны быть сведены в
таблицу.
Работа 3
ХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ СМЕСИ УГЛЕВОДОРОДОВ
С6 НА КОЛОНКАХ, ЗАПОЛНЕННЫХ НОСИТЕЛЕМ
С РАЗЛИЧНЫМИ ЖИДКИМИ ФАЗАМИ
Приборы и реактивы
1. Хроматограф ХЛ-3 с выносной колонкой.
2. Стеклянные U-образные колонки длиной 1м, диаметром 4 мм — 2 шт.
3. Целит, пропитанный трикрезилфосфатом (25%). Измельчение 0,5—0,25 мм.
* По желанию преподавателя может быть дана новая смесь неизвестного
состава, после хроматографирования которой при помощи полученных коэффи-
циентов уравнения (160) должна быть произведена идентификация неизвестных
веществ.
222
4. Инзенский кирпич, пропитанный гексадеканом (25%). Измельчение
0,5—0,25 мм.
5. Мнкрошприц .на 0,5 мл.
6. Баллон с сжатым водородом.
7. Жидкая смесь пентанов и пентенов. Можно взять н-пентан, изопентан,
пентен-1 и 3-метилбутен.
Цель работы. Показать, что применение последовательно сое-
диненных колонок, заполненных носителями, содержащими различ-
ные жидкие фазы, позволяет осуществлять значительно лучшее раз-
деление сложной смеси, чем в случае разделения на колонках с
одной жидкой фазой.
Сущность работы.Анализ сложной смеси, содержащей вещества
с различными свойствами, но с близкими температурами кипения,
часто встречает затруднения в связи со сложностью подбора подхо-
дящей жидкой фазы. Задача может быть значительно облегчена,
если разделение такой смеси производить на двух или более после-
довательно соединенных колонках, содержащих различные жидкие
фазы. В этом случае может быть не только улучшен эффект раз-
деления, но и изменен порядок выхода отдельных компонентов
смеси.
На примере смеси углеводородов, содержащих пять атомов уг-
лерода в молекуле, т. е. смеси пентанов и пентенов, можно убедить-
ся в том, что полное разделение на колонке как с одной, так и с дру-
гой жидкой фазой не достигается. В то же время соединение
этих колонок последовательно обеспечивает полное разделение
смеси.
Выполнение работы. Одну колонку заполняют целитом, пропи-
танным трикрезилфосфатом, т. е. полярной фазой, другую — Ин-
зенским кирпичом, пропитанным гексадеканом, т. е. неполярной
фазой. Первую колонку присоединяют к хроматографу ХЛ-3 вме-
сто металлической колонки. Включают установку, продувают си-
стему водородом и после установления нулевой линии наносят пор-
цию исследуемой смеси в количестве 0,05 мл. Вследствие плохого
разделения исследуемой смеси на полярных жидких фазах на хро-
матограмме вместо четырех пиков получается три. Смесь — 3-ме-
тилбутилена-1 и изопентана не разделяется.
После окончания этого опыта первую колонку заменяют другой,
заполненной носителем с неполярной фазой. Повторяют опыт с
той же смесью. При этом опять получают вместо четырех три пика.
Однако в этом случае не разделяется смесь пентана и пентена-1.
По окончании второго опыта колонку отсоединяют и приступа-
ют к проведению третьего опыта. Первую и вторую колонки соеди-
няют друг с другом последовательно и к хроматографу присоединяют
верхнюю часть первой колонки. Анализируемую смесь наносят
на первую колонку, причем в процессе хроматографирования смесь
проходит и первую, и вторую колонки. В результате анализа по-
лучают хроматограмму, состоящую из четырех пиков.
223
Таким образом, на двух последовательно соединенных колонках
с разными жидкими фазами получают полное разделение смеси.
Результаты трех опытов записывают в журнал.
Р а б о т а 4
ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ СМЕСИ ИЗОМЕРОВ
КСИЛОЛА [6]
Приборы и реактивы
1. Хроматограф УХ-1. Колонка длиной 6 м, диаметром 6 мм.
2. Инзенский кирпич, измельчения 0,5—0,25 льи, пропитанный бензилдифени-
лом (15% от веса кирпича).
3. Микрошприц на 0,1 мл.
4. Баллон с сжатым водородом.
5. Смесь п-, м- и о-ксилолов.
Цель работы. Разделить и проанализировать количественно
наиболее трудно разделяемую смесь изомеров ксилола.
Сущность работы. Правильно выбранная жидкая фаза, а также
достаточная, длина колонки позволяют осуществить разделение
таких смесей, компоненты которых близки по структуре, темпера-
турам кипения и другим физико-химическим свойствам. В частнос-
ти такой смесью является смесь трех изомеров ксилола, знание
состава которой в настоящее время имеет большое значение в связи
с применением n-ксилола для синтеза лавсана. Литературные дан-
ные свидетельствуют о том, что разделение смеси трех изомеров кси-
лола может быть достигнуто на некоторых полярных высокоселек-
тивных жидкостях. В настоящей работе разделение смеси изомеров
ксилола производится на бензилдифениле, которым пропитан Ин-
зенский кирпич.
Выполнение работы. Имеющуюся в хроматографе УХ-1 метал-
лическую колонку заполняют Инзенским кирпичом, предварительно
пропитанным бензилдифенилом, и помещают ее в термостат хрома-
тографа. Температуру термостата доводят до 100° С и выдерживают
ее в течение всего опыта. После продувки системы водородом и вы-
вода установки на заданный режим в колонку вводят 0,05 мл смеси
изомеров ксилола. Опыт повторяют и после окончания приступают
к расшифровке хроматограмм, определяя количественный состав
смеси методом нормировки.
Для определения качественного состава смеси можно проделать
опыт с чистыми изомерами ксилола: в колонку последовательно
вводят каждый из изомеров в чистом виде и определяют величину
удерживаемого объема для каждого изомера. Сравнивая получен-
ные данные с удерживаемыми объемами компонентов смеси, уста-
навливают принадлежность каждого пика тому или иному изоме-
ру. Результаты опыта и расчеты записывают в журнал.
224
Работа 5
ВЫБОР ЖИДКОЙ ФАЗЫ ДЛЯ РАЗДЕЛЕНИЯ СМЕСИ ВЕЩЕСТВ
С БЛИЗКИМИ ТЕМПЕРАТУРАМИ КИПЕНИЯ
Приборы и реактивы
1. Хроматограф типа ХЛ-4 или УХ-1 с двумя одинаковыми выносными стек-
лянными колонками длиной 3,8 м, диаметром 4 мм каждая.
2. Инзенский кирпич, измельчения 0,5—0,25 мм, пропитанный полярной
жидкой фазой — бутиратом диэтиленгликоля (20% от веса кирпича).
3. Инзенский кирпич того же измельчения, пропитанный неполярной жидкой
фазой вазелиновым маслом (20% от веса кирпича).
4. Шприц на 1 мл.
5. С1црсь паров диметилового эфира (т. кип.—23,7° С) и хлористого метила
(т. кип.— 24,0°С); 1 : 1 по объему.
6. Баллон с сжатым водородом.
Цель работы. Показать, что от правильного выбора неподвиж-
ной жидкой фазы зависит успех разделения смеси веществ.
Сущность работы. Хак уже говорилось, разделение смеси ве-
ществ, обладающих различными физико-химическими свойствами и
структурой, но одинаковыми или близкими температурами кипения,
может быть осуществлено на сильно полярных жидких фазах. Не-
полярные фазы затрудняют разделение.
В настоящей работе предлагается проверить это положение путем
хроматографирования смеси двух полярных веществ, кипящих
практически при одинаковой температуре, на полярной и неполяр-
ной жидких фазах.
Выполнение работы. Одну колонку заполняют инзенским кир-
пичом, пропитанным бутиратом диэтиленгликоля, а другую таким
же кирпичом, пропитанным вазелиновым маслом. Сначала к хрома-
тографу присоединяют первую колонку. Устанавливают скорость
газа-носителя (водорода) 25 см?/мин, добиваются постоянства нуле-
вой линии и затем шприцем вводят 0,3—0,5 мл смеси паров димети-
лового эфира и хлористого метила. Температура колонки комнатная.
Первым выходит эфир, затем хлористый метил, причем разделение
смеси происходит очень четко.
После окончания первой части опыта к хроматографу вместо
первой присоединяют вторую колонку, заполненную кирпичом с
вазелиновым маслом. Устанавливают режим работы хроматографа
и вводят 0,3—0,5 мл смеси паров эфира и хлористого метила. Полу-
чают один общий пик, так как разделения не происходит. Результа-
ты записывают в тетрадь и сравнивают между собой.
8 Б. В. Айвазов
225
Работа 6
ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ СМЕСИ
ЖИДКИХ ХЛОРМЕТАНОВ [7]
Приборы и реактивы
1. Хроматограф любой марки с детектором по теплопроводности и термо-
статом.
2. Колонка стеклянная U-образная, длиной 3 м, диаметром 6 мм.
3. Диатомит, измельчения 0,5—0,25 мм, пропитанный этиленгликолем (30%
от веса твердого носителя).
4. Микрошприц на 0,1 мл.
5. Баллон с сжатым водородом.
6. Смесь жидких хлорметанов.
Цель работы. Разделение и количественный анализ смеси жидких
хлорметанов, образующихся при хлорировании углеводородов.
Сущность работы. Требованию хорошего разделения смеси хлор-
метанов вполне отвечает этиленгликоль и его полимеры. На этих
жидких неподвижных фазах можно достичь полного разделения сме-
си всех хлорированных метанов. В настоящей работе производится
разделение смеси жидких ди-, три- и тетрахлорметанов на этилен-
гликоле в качестве неподвижной жидкой фазы. Первым из компонен-
тов этой смеси элюируется четыреххлористый углерод, затем хло-
ристый метилен и последним хлороформ. В качестве детектора может
служить катарометр.
Выполнение работы. Колонку заполняют диатомитом, содержа-
щим 30% этиленгликоля, и помещают в термостат. Устанавливают
температуру термостата 40° С, скорость газа-носителя 45 см3/мин.
После вывода системы на режим в колонку вводят 0,02 мл жидкой
смеси хлорметанов. На хроматограмме получают три пика. Расчет
количественного содержания каждого из компонентов смеси произ-
водят по площадям пиков методом нормировки. Результаты расчета
сводят в таблицу.
Работа 7
ХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ ФРЕОНОВ [8]
Приборы и реактивы
1. Хроматограф ХЛ-4. Колонка длиной 6 м, диаметром 6 мм.
2. Диатомит, измельчения 0,5—0,25 мм, пропитанный трибутилфосфатом
(16% от веса кирпича).
3. Микрошприц на 0,1 мл.
4. Баллон с сжатым азотом.
5. Технический фреон.
226
Цель работы. Определение состава одной из технически важных
жидкостей — фреона, применяемой в холодильных установках.
Сущность работы. Разделение и анализ технического фреона про-
водится как на полярных, так и неполярных жидких фазах. Наилуч-
шие результаты достигаются на трибутилфосфате или на диизоа-
миловом эфире метилфосфоновой кислоты, нанесенных на диатомит.
В оптимальных условиях разделения в техническом фреоне удается
обнаружить фреоны И, 12, 13, 21, 22, а также четыреххлористый
углерод. Анализ проводится на стандартном хроматографе типа
ХЛ-4.
Выполнение работы. Колонку заполняют диатомитом, пропи-
танным трибутилфосфатом, включают установку, устанавливают
температуру термостата 80° С и скорость газа-носителя (азота)
2 л!час. После выведения установки на режим вводят 0,05 м,л тех-
нического фреона и производят хроматографирование.
Последовательность выхода фреонов такова: первым выходит
фреон 13, затем 12, 22, 11 и 21. Последним элюируется четыреххло-
ристый углерод. Время элюирования примерно 1,5 ч.
Количественный анализ производят методом нормировки по
площадям пиков. Результаты анализа записывают в журнал.
Работа 8
ХРОМАТОГРАФИЧЕСКОЕ ОПРЕДЕЛЕНИЕ МИКРОПРИМЕСИ
АЦЕТИЛЕНА В ЭТИЛЕНЕ
Приборы и реактивы
1. Хроматограф любой конструкции с колонкой длиной 3 м, диаметром 4 мм
и с приставным пламенно-ионизационным детектором типа ДИП-1. Может быть
применен любой хроматограф с пламенно-ионизационным детектором, например,
хроматограф типа «Цвет», хроматографы чешской фирмы «Хром-1» или «Хром-2»,
фирмы «Перкин-Эльмер» (США), а также с аргоновым детектором англий-
ской фирмы «Пай» и др.
2. Инзенский кирпич, измельчения 0,5—0,25 мм, пропитанный н-гептадека-
ном (20% отвеса кирпича).
3. Шприц на 10 мл или газовая петля.
4. Баллоны с сжатыми водородом и азотом.
5. Этилен, содержащий примесь ацетилена'порядка 10~3—10~4% по объему.
Цель работы. Показать возможность применения хроматографи-
ческого метода для определения микропримесей при помощи высоко-
чувствительных детекторов. Определить содержание микропримеси
ацетилена в этилене.
Сущность работы. Широкое применение различных газов в
качестве сырья для получения полимерных материалов предъявляет
повышенные требования к чистоте этих газов. Одним из эффектив-
ных методов определения чистоты газов является хроматографичес-
кий метод определения содержания в них примесей посторонних
8*
227
веществ в микроколичествах. Определение микропримесей может
осуществляться двумя путями: либо путем предварительного обо-
гащения анализируемой смеси искомым компонентом, либо приме-
нением высокочувствительного детектора. В настоящей работе
предлагается второй путь, т. е. хроматографирование без обогаще-
ния и применение высокочувствительного пламенно-ионизацион-
ного детектора. В качестве неподвижной жидкой фазы применяется
н-гептадекан. Чувствительность метода 5-10“5 объемн.%.
Выполнение работы. Колонку заполняют инзенским кирпичом,
содержащим 20% н-гептадекана. Устанавливают температуру тер-
мостата 25° С, проверяют работу пламенно-ионизационного детек-
тора и добиваются постоянства нулевой линии. Работу проводят в
режиме самой высокой чувствительности самописца. С этой целью
перед началом опыта указатель на шкале «чувствительность» уста-
навливают на положении 1:1.
В качестве газа-носителя применяют азот. Скорость его подачи
устанавливается равной 3 л!час.
После вывода прибора на режим вводят 10 мл исследуемого
газа. Вследствие большого содержания этилена его пик выходит
за пределы шкалы. После возвращения пера самописца на нулевую
линию появляется небольшой пик ацетилена. Для количественного
определения примеси ацетилена измеряют высоту его пика и по ка-
либровочному графику (берется у преподавателя) устанавливают
его содержание в этилене.
Работа 9
ХРОМАТОГРАФИЧЕСКОЕ ОПРЕДЕЛЕНИЕ МИКРОПРИМЕСИ
БУТАНОЛА В СТОЧНЫХ ВОДАХ [9]
Приборы и реактивы
1. Хроматограф типа «Хром-1» или «Хром-2» или другой тип хроматографа
с пламенно-ионизационным детектором (например, типа «Цвет»).
2. Инзенский кирпич, измельчения 0,5—0,25 мм, пропитанный трикрезил-
фосфатом (20% от веса кирпича).
3. Шприц на 1 мл.
4. Баллоны с сжатыми водородом и азотом.
5, Вода, содержащая растворенный бутиловый спирт (концентрация порядка
0,05%).
Цель работы. Количественное определение примеси бутилового
спирта в воде. Применение хроматографического метода для анализа
водных растворов органических веществ.
Сущность работы. Для определения микропримеси бутилового
спирта в воде берется жидкая фаза (трикрезилфосфат), на которой
спирт хорошо отделяется от воды, а в качестве детектора — пла-
менно-ионизационный детектор, являющийся нечувствительным к
228
воде. Последнее обстоятельство позволяет исключить наложение
пика основного компонента и легко определить пик анализируемого
вещества. Применение трикрезилфосфата позволяет проводить од-
новременное определение микропримесей в сточных водах метило-
вого, этилового и бутилового спиртов, а также бензола и толуола.
Выполнение работы. Колонку заполняют инзенским кирпичом,
пропитанным трикрезилфосфатом. Производят подготовку всей
хроматографической установки, причем работу ведут в режиме выс-
шей чувствительности (см. работу 8). После установления нулевой
линии в колонку вводят 0,1 мл воды, содержащей примесь бутано-
ла. В качестве газа-носителя применяют азот. Его скорость уста-
навливают равной 3 л!час. Температура термостата 25° С.
После получения пика бутанола определяют его высоту, по ко-
торой, пользуясь калибровочным графиком, определяют содержание
бутанола в воде.
Результаты анализа записывают в журнал. Продолжительность
анализа 20 мин.
Работа 10
ХРОМАТОГРАФИРОВАНИЕ АНТРАЦЕНА [10]
Приборы и реактивы
1. Хроматограф любого типа с катарометром, например, УХ-1. Колонка
стеклянная U-образная, длиной 2 м, диаметром '4 мм; специальный испаритель
на температуру до 400° С.
2. Стеклянные микробусы с диаметром бусинок около 200 мк, покрытые сили-
коновым маслом (0,05% от веса стекла).
3. Специальное устройство для мгновенного испарения жидкости.
4. Микрошприц на 0,1 мл.
5. Баллон с сжатым азотом.
6. Раствор антрацена в диэтиловом эфире (1%-ный).
Цель работы. Показать возможность хроматографирования
твердых веществ, обладающих температурой плавления выше тем-
пературы работы колонки.
Сущность работы. Применение газо-жидкостной хроматографии
для разделения и анализа твердых органических веществ встречает
серьезные препятствия из-за трудностей в подборе достаточно тер-
мически устойчивых и нелетучих жидких фаз. Кроме того, затруд-
нения могут возникнуть вследствие термической неустойчивости
анализируемого вещества.
Выход из положения может быть найден, если снизить количест-
во неподвижной жидкой фазы с обычно применяемых 20—30% от
веса твердого носителя до 0,05—0,2%. При этом значительно сни-
жается время удерживания, а также требуемое для анализа коли-
чество вещества. Последнее обстоятельство позволяет вести анализ
с таким количеством паров твердого вещества, которое образуется
при сравнительно низких температурах.
8* Б. В. Айвазов
229
Однако малые количества жидкой фазы вследствие сильной по-
верхностной адсорбции нельзя наносить на носители с развитой
макроповерхностью. Поэтому в данном случае в качестве твердой
фазы применяют стекло, например, стеклянные микробусы диамет-
ром 200 мк. Количество наносимого анализируемого вещества не
должно превышать нескольких десятых миллиграмма.
В настоящей работе предлагается получить хроматограмму ан-
трацена (темп. пл. 216° С, темп. кип. 354° С) при рабочей темпера-
туре колонки 100° С. В качестве жидкой фазы применяется силико-
новое масло.
Выполнение работы. Колонку заполняют стеклянными микро-
бусами с силиконовым маслом (0,05%) и готовят установку к ра-
боте. Газ-носитель — азот, скорость 2 л!ч. Температура термостата
100° С. Температура испарителя 375°С. Чувствительность высшая,
г. е. положение указателя на шкале «чувствительность» на 1:1.
После вывода хроматографа на установленный режим в специа-
льный испаритель вводят пробу раствора антрацена в эфире в ко-
личестве 0,05 мл. После пика растворителя появляется пик антра-
цена. Для того, чтобы убедиться в принадлежности данного пика
антрацену, опыт повторяют с чистым эфиром. В этом случае второй
пик должен отсутствовать. Этот опыт доказывает возможность ана-
лиза таким путем твердых органических веществ, плавящихся при
температуре выше температуры работы колонки.
Работа 11
РАСЧЕТ СОСТАВА БИНАРНОЙ СМЕСИ ПО ПЛОЩАДИ ПИКОВ
МЕТОДОМ ДЖ- К. БАРЛЕТА И Д. М. СМИТА [11, 12]
Приборы и реактивы
1. Хроматограф типа СКВ ИОХ АН СССР с катарометром с вольфрамовыми
нитями. Колонка длиной 4 м, диаметром 4 мм.
2. Диатомит, измельчения 0,5—0,25 мм, пропитанный бутиратом триэтилен-
гликоля (10% от веса кирпича).
3. Микрошприц на 0,1 мл.
4. Компаратор ИЗА-2.
5. Измерительная линейка.
6. Баллон с сжатым гелием.
7. Смесь бензола и н-гексана 1 : 1 (по весу).
Цель работы. Знакомство с новым методом расчета состава ана-
лизируемой смеси по площади пиков и сравнение полученных результа-
тов с расчетом по площадям обычным методом.
Сущность работы. Обычно площадь пика на хроматограмме оп-
ределяют как площадь треугольника с основанием, полученным на
нулевой линии между касательными, проведенными в точках пере-
гиба пика. Исходя из того, что форма пика соответствует кривой
230
распределения Гаусса, Дж. К. Барлет и Д. М. Смит предложили
рассчитывать площади пика по формуле:
П,- = 2,507/г,ф;., (180)
где П, — площадь пика i-того компонента, смг; hi — высота пика,
см; ф(. — стандартное отклонение, см.
Величина ф связана с шириной пика и следующим образом:
ф на расстоянии 0,882/г равно ширине пика ц; на 0,607/г ф = -£
и на 0,324/г ф = у.
Определив значения ф на названных высотах, можно проверить
соответствие формы пика гауссовской кривой распределения.
В случае соответствия, значения ф для данного компонента должны
быть одинаковыми. В противном случае метод не может быть ис-
пользован.
В настоящей работе предлагается определить состав бинарной
смеси, рассчитав его двумя методами; по площади пиков, приняв их
за треугольники, и по площади пиков методом Дж. К. Барлета и
Д. М. Смита, определив соответствующие значения ф. В качестве
объекта исследования берется смесь гексана и бензола в отношении
1:1 по весу.
Выполнение работы. Колонку заполняют диатомитовым кирпи-
чом, пропитанным бутиратом триэтиленгликоля. Включают прибор,
устанавливают температуру термостата, равную 60°С, и скорость
газа-носителя — гелия, равную 40 см*1мин. Добиваются посто-
янства нулевой линии, после чего вводят в колонку микрошприцем
0,05 м,л жидкой смеси. Опыт хроматографирования повторяют три
раза. Затем установку отключают и приступают к измерению хрома-
тограмм.
1. Измеряют масштабной линейкой площадь каждого из полу-
ченных пиков. Для этого к точкам перегиба проводят касательные,
измеряют расстояние между точками их пересечения с нулевой
линией, измеряют высоту треугольника и рассчитывают его площадь,
умножад половину высоты на основание. Получают по три измере-
ния для каждого компонента, из которых рассчитывают средние
арифметические значения.
Среднее значение площади пика гексана складывают со средним
значением площади пика бензола, сумму приравнивают 100% и рас-
считывают содержание гексана и бензола в весовых процентах, при-
няв, что отклонения пера самописца одинаковы для обоих компонен-
тов. Полученные данные сравнивают с истинным составом смеси.
2. Измеряют на компараторе с точностью до 0,02 мм значения
ширины пика ц на расстояниях от нулевой линии, равных 0,882/г,
0,607/г и 0,324/г, и из полученных данных рассчитывают ф. Сравни-
вают их значения между собой и, если они оказываются постоян-
ными в пределах ошибки опыта и измерения, берут среднее арифме-
8** 231
тическое из них и подставляют в формулу (180). Зная среднее зна-
чение ф и Л для каждого компонента, рассчитывают площади пиков.
Значение площади пика гексана складывают со значением площади
пика бензола и сумму принимают за 100%. Отсюда рассчитывают
состав смеси в весовых процентах и сравнивают результат с расче-
том, полученным по площади треугольника, а также с истинным
составом смеси. Все данные сводят в таблицу (см. форму записи).
Форма записи
Работа 12
ИДЕНТИФИКАЦИЯ НЕИЗВЕСТНОГО ВЕЩЕСТВА
ПО ОТНОСИТЕЛЬНЫМ УДЕРЖИВАЕМЫМ ОБЪЕМАМ
Приборы и реактивы
1. Хроматограф типа УХ-1, ХЛ-3 или ХЛ-4 с колонкой длиной 6 м,
диаметром 6 мм.
2. Инзенский кирпич, измельчения 0,5—0,25 мм, пропитанный скваланом
(15% от веса кирпича).
3. То же, пропитанный три крезилфосфатом (15% от веса кирпича).
4. Шприц на 0,1 мл.
5. Баллон с сжатым водородом.
6. Жидкие углеводороды: пентан, гексан, гептан, пентен-1, гексен-1 и геп-
тен-1.
Цель работы. Показать на примере простейшей смеси углеводо-
родов возможность идентификации неизвестного вещества хромато-
графическим методом.
Сущность работы. Зависимость логарифма относительных удер-
живаемых объемов членов одного гомологического ряда, получен-
232
ных на каком-либо растворителе, от логарифма относительных удер-
живаемых объемов этих же соединений, но полученных на другой
жидкой фазе, оказывается линейной. Для другого гомологического
ряда эта зависимость будет также линейна, однако уравнение пря-
мой в этом случае будет иметь иные коэффициенты. Пользуясь этим
свойством, можно экспериментально определить относительные удер-
живаемые объемы членов нескольких гомологических рядов на двух
различных жидких фазах, рассчитать логарифмы их относительных
удерживаемых объемов и построить график в координатах 1g VRi отн
на одной фазе— lg^,oTH на Другой фазе. Затем, определяя отно-
сительные удерживаемые объемы неизвестного вещества на тех же
жидких фазах, при помощи полученного графика можно идентифи-
цировать это вещество.
В данной работе применяется смесь нормальных парафиновых
и олефиновых углеводородов и две жидкие фазы: сквалан и трикре-
зилфосфат.
Выполнение работы. Одну колонку заполняют инзенским кир-
пичом, пропитанным скваланом, другую — кирпичом с трикрезил-
фосфатом. Сначала к хроматографу присоединяют первую колонку,
продувают газом-носителем (водородом), добиваются постоянства
нулевой линии и вводят 0,05 мл жидкой смеси углеводородов из-
вестного состава, т. е. смесь жидких пентана, пентена, гексана, гек-
сена, гептана и гептена, взятых в эквимолекулярном соотношении.
Полученную хроматограмму расшифровывают, т. е. определяют
принадлежность каждого пика, и измеряют удерживаемые объемы
каждого вещества в произвольных единицах. Затем удерживаемый
объем пентана принимают равным единице, а остальные объемы от-
носят к этому удерживаемому объему. Полученные данные сравни-
вают с данными табл. 17.
Первую колонку убирают и присоединяют вторую. Опыт и расче-
ты повторяют для второй жидкой фазы.
Рассчитанные данные относительных удерживаемых объемов
логарифмируют и полученные значения логарифмов откладывают
на графике в координатах lgV^,0TH для сквалана — lgVw,0TH Для
трикрезилфосфата. Точки, относящиеся к ряду н-алканов, соединя-
ют одной прямой, а точки, полученные для ряда алкенов,— другой.
Получают от преподавателя неизвестное вещество и хромато-
графируют его на каждой колонке. Рассчитывают для него относи-
тельные удерживаемые объемы, принимая за единицу удерживаемые
объемы пентана из опытов, проделанных для градуировки, вы-
числяют логарифмы относительных удерживаемых объемов и, поль-
зуясь графиком, устанавливают принадлежность неизвестного ве-
щества к одному из гомологических рядов, а затем идентифицируют
его.
Примечание. Для ускорения работы хроматографирование неизвест-
ного вещества можно проводить непосредственно за хроматографированием смеси
для каждой жидкой фазы.
233
ЛИТЕРАТУРА
Использованная литература
1. А. Т. James, A. J. Р. М а г t i n. Biochem. J ., 50, 679, 1952; Analyst.,
77 915 1952.
2. H. А. Фукс. Усп. хим., 17,45, 1948.
3. А. К e й л e м а н с. Хроматография газов. ИЛ, М., 1959.
4. Г, ПТ а й. Теоретические основы хроматографии газов. ИЛ, М., 1963.
5. А. А. Жуховицкий, И. М. Туркельтауб. Газовая хромато-
графия. Гостоптехиздат, М., 1962.
6. A. Z 1 a t k i s, L. О’В г i е n, Р. R. S с h о 1 к у. Nature, 181, 1794,
1958.
7. Д. А. Вяхирев, Л. Е. Р е ш е т н и к о в а. В сб. «Разделение и
анализ углеводородных газов» Изд-во АН СССР, М., 1963.
8. Т. Н. Ч и ч у г о в а, Г. В. Р а б о в с к и й, В. Н. Залесский.
В сб. «Газовая хроматография». Вып. 1, НИИТЭХим, М., 1964.
9. Б. М. Л у с к и н а, С. В. Сявцилло, В. Д. Меркулов.
III Всесоюзная конференция по газовой хроматографии. Тезисы докладов. М.,
.1964.
10. Ch. Hishta, J. Р. Messerly, R. F. Reschke, D. H. Fre-
dericks, W. D. Cooke. Anal. Chem., 32, № 7, 880, 1960; «Газовая хрома-
тография». Вып. 11, НИИТЭХим. М., 1963, стр. 230.
И . J С. В а г 1 е t, D. М. Smith. Canad. J. Chem., 38 , 2057, 1960.
12. Молекулярная хроматография. Изд-во «Наука», М., 1964.
Рекомендуемая литература
1. Э. Байер. Хроматография газов. ИЛ, М., 1961.
2. К. Филлипс. Хроматография газов. ИЛ, М., 1958.
3. «Хроматография, ее теория и применение». Изд-во АН СССР, М., 1960.
4. «Газовая хроматография». Труды 1-й Всес. конф. Изд-во АН СССР, М.,
1960.
5. «Успехи и достижения газовой хроматографии». Гостоптехиздат М.,
1961.
6. «Газовая хроматография». Труды симпозиума в Амстердаме. ИЛ, М., 1961.
7. Газовая хроматография. Библиографический указатель отечественной и
зарубежной литературы (1952—1960 гг.). Изд-во АН СССР, М., 1962.
8. Практические работы по физической химии. Под ред. Я. И. Герасимова
и др. Ч. IV. Изд-во МГУ, 1962.
9. В. Н. Зрелое, Г. И. Кичкин. Хроматография в нефтяной и
нефтехимической промышленности. Гостоптехиздат, М., 1963.
10. «Газовая хроматография». Труды 2-й Всес. конф. Изд-во «Наука», М.,
1964.
11. «Газовая хроматография». Труды симпозиума в Эдинбурге. Изд-во «Мир»,
М„ 1964.
12. «Газовая хроматография». Вып. 1, НИИТЭХим, М., 1964.
13. «Газовая хроматография». Вып. II. НИИТЭХим, М., 1965.
14. М Шингляр. Газовая хроматография в практике. Изд-во «Химия»,
М„ 1964.
15. Г. Берчфилд, Э. Сторрс. Газовая хроматография в биохимии.
Изд-во «Мир», М., 1964.
. 16. «Газовая хроматография». Вып. III, НИИТЭХим, 1965.
.17. «Газовая хроматография». Вып. IV, НИИТЭХим, М., 1966.
18. «Газовая хроматография». Труды 3-й Всесоюзной конференции по газо-
вой хроматографии. Изд. Дзержинского филиала ОКБА, Дзержинск, 1966.
19. С. Д. Н огаре, Р. С. Д ж у в е т. Газожидкостная хроматография.
Теория и практика. Изд-во «Недра», Л., 1966.
ГЛАВА VI
НОВЫЕ ВАРИАНТЫ ГАЗО-ЖИДКОСТНОЙ
ХРОМАТОГРАФИИ
А. КАПИЛЛЯРНАЯ ХРОМАТОГРАФИЯ
М. Дж. Е. Голеем [1] в 1957 г. был разработан вариант газо-
жидкостной хроматографии, существенно повышающий эффектив-
ность разделения и получивший название капиллярной хроматогра-
фии.
Отличие капиллярной хроматографии от обычно применяемой
хроматографии с колонками, заполненными зерненой твердой фа-
зой, состоит в том, что здесь жидкая фаза наносится непосредствен-
но на стенки тонкого капилляра, служащего хроматографической
колонкой. Это обстоятельство приводит к устранению вредного влия-
ния вихревой диффузии на размывание хроматографических по-
лос, существенно уменьшает сопротивление потоку газа-носителя и
обеспечивает условия стабильности тонких пленок. Все это позво-
ляет значительно увеличивать длину колонок, что улучшает про-
цесс разделения, применять весьма малые дозы анализируемого
вещества, т. е. осуществлять микроанализ, а также существенно
сокращать время анализа, приближая его к экспрессному.
Капиллярная хроматография обладает рядом специфических
особенностей, рассматриваемых теорией [1—3].
Коэффициент Генри в капиллярной хроматографии равен
Г^х + хЛр. (181)
26 /s ч
где Xj = —, (о — толщина пленки; гк— радиус капилляра).
Так как обычно 6«5-10“5 см, а гк«0,01 см, то Xj^lO"2. Учиты-
вая, что
1 1 26
X = 1 — X, = 1---,
1 г ’
к
можно с достаточной точностью считать, что
г=1+^кр. (182)
'к
235
Таким образом, если Др=10, то Г да 1., 1, т. е. коэффициент
Генри имеет малое значение, что невыгодно для разделения, так
как приводит к малому значению коэффициента селективно-
сти Дс.
Расчет показывает, что в капиллярной хроматографии коэффи-
циент селективности может оказаться в десятки раз меньшим, чем
в газо-жидкостной с заполненными колонками. Поэтому одним из
первых условий успешного разделения смесей методом капиллярной
хроматографии должно быть высокое значение коэффициента рас-
пределения /Ср. Он должен быть много более 100, т. е. хорошее разде-
ление смеси методом капиллярной хроматографии может быть дос-
тигнуто для сильно сорбирующихся веществ.
Вторым условием,. определяющим разделение смеси веществ,
является высота эквивалентной теоретической тарелки. Она
должна быть наименьшей. Это условие достигается в капиллярной
хроматографии значительно лучше, чем в хроматографии с запол-
ненными колонками.
Рассмотрим влияние различных факторов на величину размыва-
ния в капиллярной хроматографии.
1. Влияние толщины пленки. Для плохо сорби-
рующихся веществ основную роль в размывании должна играть
внутренняя диффузия, тогда как для сильно сорбирующихся —
внешняя. Поэтому для хорошо сорбирующихся веществ размыва-
ние, а следовательно, и высота эквивалентной теоретической тарел-
ки не должны зависеть от толщины пленки. Для слабо сорбирую-
щихся веществ должно иметь место резкое возрастание величины Н
с ростом б. Практика подтверждает этот вывод.
2. Влияние диаметра капилляра. Влияние раз-
меров диаметра капилляра также связано с природой сорбирую-
щихся веществ. Для плохо сорбирующихся соединений изменение
величины Н при изменении размеров диаметра капилляра почти не
наблюдается, тогда как для веществ сильно сорбирующихся, для
которых размывание определяется главным образом внешней диф-
фузией, имеет место резкое возрастание Н с ростом диаметра и,
следовательно, значительное ухудшение разделения.
3. Влияние природы газа -носителя. Переход
от более тяжелого газа-носителя (аргона) к более легкому (водоро-
ду) вследствие увеличения коэффициента диффузии приводит к
увеличению высоты тарелки Н и, следовательно, к ухудшению раз-
деления. Это влияние природы газа-носителя особенно заметно
для хорошо сорбирующихся веществ, для которых основную роль
играет внешняя диффузия.
4. Влияние температуры. Для сильно сорбирующих-
ся веществ, у которых Др^>1, размывание связано в основном с
внешнедиффузионной массопередачей. Поэтому для них величина
Др мало зависит от температуры. Для плохо сорбирующихся ве-
ществ размывание определяется главным образом внутренней диф-
236
фузией, следовательно, уменьшается с ростом температуры. Эти
выводы подтверждаются экспериментально.
5. Допустимое количество наносимого
вещества. Для капиллярной хроматографии особое значение
приобретает объем наносимой пробы. Как полагает А. Кейлеманс
[3], максимально допустимое количество наносимого вещества q- макс
определяется уравнением
<?Z, макс=0,02^, (183)
где п — число теоретических тарелок, а коэффициент 0,02, введен-
ный А. Кейлемансом произвольно, по данным многих исследовате-
лей является завышенным по крайней мере в 10 раз. Несмотря на
это, формула (183) сохраняет свое значение, причем из нее следует
зависимость величины пробы от всех параметров опыта. В самом
деле
<7,,макс«Г$]/Ш. (184)
В связи с тем, что для капиллярной хроматографии наименьшее зна-
чение высоты теоретической тарелки Н численно близко к размеру
диаметра капилляра 2гк, то
?,-,макс~(2Гк)6/2-
Таким образом, уменьшение диаметра капилляра будет приводить
к существенному уменьшению величины наносимой пробы, что,
в свою очередь, ухудшит условия детектирования. Все это не позво-
ляет уменьшать диаметр без предела.
Из уравнения (184) следует также, что величина допустимой
пробы растет с ростом коэффициента Генри Г. Таким образом, и
это обстоятельство подтверждает целесообразность применения ка-
пиллярной хроматографии для хорошо сорбирующихся веществ.
В общем случае допустимый объем наносимой пробы в капилляр-
ной хроматографии значительно меньше, чем в газо-жидкостной с
заполненными колонками, и не должен превышать 0,2—0,5 мкг.
Дальнейшее увеличение пробы приводит к резкому ухудшению
разделения. Поэтому дозировка пробы, достаточно точное измерение
ее количества имеют в капиллярной хроматографии первостепенное
значение и одновременно вызывают серьезные трудности в приме-
нении этого метода.
Развитие метода капиллярной хроматографии потребовало
также решения ряда других задач, из которых наиболее важной и
сложной оказалось приготовление самих капиллярных колонок.
Прежде всего важно, чтобы материал колонок удовлетворял следу-
ющим требованиям. Он не должен адсорбировать ни неподвижную
фазу, ни определяемые вещества или оказывать на них каталити-
ческое воздействие. Поверхность капилляра должна хорошо сма-
чиваться неподвижной фазой и в то же время должна быть очень
237
гладкой. Наличие на внутренней поверхности микропор, микротре-
щин, окисленного слоя металла совершенно недопустимо, так как
затрудняет получение равномерной пленки жидкой фазы. Кроме
этого, материал, применяемый для изготовления капилляров, дол-
жен позволять вытягивать его при соблюдении строгого постоянст-
ва диаметра на значительной длине. Уже небольшие сужения или
расширения капилляра резко ухудшают разделение, так как не по-
зволяют осуществлять оптимальные условия по всей длине колонки.
Важна также температурная устойчивость материала.
В литературе высказывается мнение, что наилучшим материалом
для капиллярных колонок следует признать стекло и нержавеющую
сталь. Однако имеются ссылки на то, что в качестве материала для
капиллярных колонок может служить медь, алюминий, найлон,
а также стекло, обработанное модификаторами, например, триме-
тилхлорсиланом. В США применяются в основном капилляры из
нержавеющей стали.
Огромное значение для разделения методом капиллярной хро-
матографии имеет способ нанесения тонкой пленки на внутренние
стенк’и капилляра. Задача состоит в том, чтобы обеспечить равно-
мерность толщины пленки на всем протяжении колонки и исключить
возможность образования непокрытых пленкой мест. В настоящее
время применяют в основном два способа нанесения пленки жидкой
фазы: способ продавливания и способ испарения. Оба способа тре-
буют предварительного растворения наносимого вещества в летучем
растворителе.
По первому 5—10%-ный раствор жидкой фазы в летучем раство-
рителе вводится в капилляр и движется в виде «пробки» по капил-
ляру, смачивая его стенки раствором. Движение осуществляется
током инертного газа. Толщина остающейся на стенках капилляра
пленки зависит от концентрации раствора, его вязкости, смачива-
емости стенок капилляра, а также от скорости движения «пробки».
Поэтому для обеспечения постоянной толщины пленки по всей
длине капилляра важно соблюдение постоянства скорости продви-
жения «пробки» смачивающего раствора.
Во втором способе применяют более разбавленные растворы
жидкой фазы в летучем растворителе (1—2%-ные) и таким раство-
ром заполняют весь объем капилляра, закрыв один из его концов.
После заполнения капилляр медленно продвигают вдоль нагретой
печи. При этом происходит испарение растворителя и оседание раст-
воренной жидкой фазы на стенках капилляра.
Получаемые как по первому, так и по второму способу пленки
обычно имеют толщину в пределах от 0,1 до 2 мк.
В практике чаще пользуются первым способом нанесения пленки.
Длина применяемых капилляров измеряется десятками, а иног-
да и сотнями метров, диаметр от 0,1 до 0,5 мм.
Капиллярная хроматография наряду с ее большими достоин-
ствами, позволяющими разделять весьма сложные по составу смеси
238
с применением малых доз анализируемых веществ, обладает, одна-
ко, рядом недостатков, ограничивающих ее применение.
Эти недостатки в основном сводятся к следующему. Если вы-
бранная фаза обладает малой селективностью по отношению к ве-
ществам анализируемой смеси, то это обстоятельство сводит на нет
преимущества высокой эффективности капиллярной колонки и
разделение не может быть осуществлено. Капиллярная хроматог-
рафия методически требует решения более сложных задач. В част-
ности, требуется очень точная дозировка вещества при малых его
количествах, а также высокая чувствительность детекторов, вслед-
ствие весьма малых порций анализируемой смеси. Поэтому в тех
случаях, когда задача может быть решена при помощи газо-жидкост-
ной хроматографии с заполненными колонками, не следует прибе-
гать к методу капиллярной хроматографии.
В капиллярной хроматографии, вследствие малых значений ко-
эффициентов Генри, существенно ограничены возможности обога-
щения. Поэтому метод не может применяться для производственно-
го контроля, особенно для автоматического регулирования контро-
лируемого процесса.
Эти недостатки не умаляют, однако достоинства метода капилляр-
ной хроматографии, который является достаточно эффективным и
тонким методом анализа сложных многокомпонентных смесей. Бе-
зусловно, что метод капиллярной хроматографии должен находить
себе широкое применение, главным образом для решения задач на-
учно-исследовательского характера.
ПРАКТИЧЕСКИЕ РАБОТЫ
»
Работа 1
АНАЛИЗ СМЕСИ ИЗОМЕРОВ КСИЛОЛА МЕТОДОМ
КАПИЛЛЯРНОЙ ХРОМАТОГРАФИИ
Приборы и реактивы
1. Хроматограф с капиллярной колонкой и высокочувствительным детекто-
ром. Может быть рекомендован хроматограф чешской фирмы типа «Хром-2» или
типа «Цвет».
2. Специальное распределительное устройство для ввода микродозы анали-
зируемой смеси.
3. Микрошприц на 1 мк.л.
4. Дннонилфталат.
5. Диэтиловый эфир.
6. Ацетон.
7. Баллоны с сжатыми водородом, воздухом и азотом.
Цель работы. Сравнение метода капиллярной хроматографии с
м£тодом хроматографии с заполненными колонками.
239
Сущность работы. В предыдущей главе в работе 4 был рассмот-
рен метод разделения смеси изомеров ксилола на одной из высоко-
полярных фаз методом газо-жидкостной хроматографии с запол-
ненной колонкой.
В настоящей работе предлагается проанализировать эту же смесь
методом капиллярной хроматографии с применением менее поляр-
Рис. 41. Принципиальная схема рас-
пределительного устройства для ввода
проб в капиллярную колонку [4]:
1, 5, 10 — игольчатые вентили; 2 — мано-
метр; 7— делитель; 11—реометр; 9—капил-
лярная колонка; 8 — тефлоновый саль-
ник; 13 — термостат; 12 — крышка термо-
стата; 3 — резиновый колпачок для ввода
пробы; 4 — латунное тело устройства;
6— канал крана
ной жидкой фазы и сравнить
преимущества и недостатки каж-
дого из методов.
Выполнение работы. В ка-
пиллярную колонку из нержа-
веющей стали длиной 30 м и диа-
метром 0,2 мм предварительно
вводят промывающие жидкости:
ацетон и диэтиловый эфир. Рас-
творители проходят трубку ка-
пилляра под давлением азота.
После окончания промывания
капилляр сушат в токе азота и
затем заполняют неподвижной
жидкой фазой. С этой целью го-
товят 10%-ный раствор динонил-
фталата в диэтиловом эфире и
порцию этого раствора вводят в
капилляр. Продвижение «проб-
ки» раствора осуществляют рав-
номерной продувкой капилляра
током азота под избыточным
давлением. После выхода ос-
татка раствора из колонки ее
некоторое время продувают азо-
том до полного испарения рас-
творителя. Колонку присоединяют к хроматографу.
Хроматограф включают в сеть, устанавливают ток газа-носителя
азота, включают пламенно-ионизационный детектор, для чего по-
дают из баллонов ток водорода и воздуха. Устанавливают темпера-
туру термостата 75° С. Указатель шкалы «Чувствительность» уста-
навливают в положении 1:1 и добиваются постоянства нулевой ли-
нии. После этого вводят пробу анализируемой смеси ксилолов.
Ввод пробы осуществляется следующим образом. В колонке на
ее входе подключают специальное распределительное устройство,
разработанное В. В. Бражниковым и Ф. Н. Фроловым [4] (рис. 41).
Оно представляет собой четырехходовой кран, изготовленный из
латуни и тефлоновых прокладок.
Поток газа-носителя, поступающий из баллона через игольчатый
вентиль 1 и манометр 2, делится в делителе 7 на две части. Одна
из них, большая, сбрасывается через игольчатый вентиль 10 и рео-
240
метр 11 в атмосферу, а другая поступает в капиллярную колонку 9.
Присоединение к колонке можно осуществить при помощи тефлоно-
вого сальника 8. Устройство помещается в термостат, температура
которого должна обеспечивать быстрое испарение вводимой пробы.
В данном случае распределительное устройство может быть помеще-
но в тот же термостат, в котором находится колонка, т. е. при темпе-
ратуре 75° С.
Для определения соотношения потоков сбрасываемого газа и
поступающего в колонку, предварительно, до введения анализиру-
емой смеси, производят калибровку устройства. С этой целью полу-
чают данные для построения калибровочного графика реометра И,
установленного на линии сброса, и графика зависимости скорости
газа-носителя на выходе из колонки от его давления на входе в нее.
Зная из полученных градуировок соотношение между объемом сбра-
сываемого газа и его количеством, поступающим в капиллярную
колонку, рассчитывают объем анализируемой смеси, поступившей в
колонку.
Требуемое соотношение устанавливают следующим образом:
после сброса давления вентилем 5 в камеру через резиновый само-
уплотняющийся колпачок 3 вводят пробуЛанализируемой смеси
ксилолов в количестве 1 мкл. При этом положение каналов крана
должно соответствовать показанному на рис. 41 пунктиром. Пово-
ротом крана в положение, соответствующее показанному сплошной
линией, испарившаяся проба вводится непосредственно в делитель.
Отсюда смесь пробы и газа-носителя поступает в колонку в таком со-
отношении, которое устанавливается предварительно. В данном
случае следует установить соотношение между количеством смеси,
поступающей в колонку и сбрасываемой, равное 1:500. Тогда, если
в смеситель была нанесена проба в количестве 1 мкл, в колонку
поступит 2-10-3 мкл. Этого оказывается достаточно для того, чтобы
обнаружить на хроматограмме три четко выраженных пика. Первым
выходит л-ксилол, затем — n-ксилол и последним о-ксилол. По
высотам пиков методом нормировки рассчитывают относительное
содержание в смеси каждого из изомеров. Полученные данные за-
носят в тетрадь.
Работа 2
АНАЛИЗ БЫТОВОГО ГАЗА МЕТОДОМ КАПИЛЛЯРНОЙ
ХРОМАТОГРАФИИ
Приборы и реактивы
1. Самодельная капиллярная колонка из нержавеющей стали длиной
45—50 м, диаметром 0,3 мм, присоединенная к ионизационно-пламенному детек-
тору типа ДИП-1.
2. Микрошприц на 1 мкл.
3. Гексадекан.
4. Диэтиловый эфир.
5. Баллоны с сжатыми водородом, воздухом и азотом.
241
Цель работы. Быстрое определение состава сложной смеси.
Сущность работы. Небольшая проба газа вводится в капилляр-
ную колонку, причем через 60—75 сек самописец записывает полный
состав семикомпонентной смеси.
Выполнение работы. Капиллярную колонку заполняют 10%-ным
раствором гексадекана в диэтиловом эфире так, как это описано в
работе 1. Сушку колонки производят так же.
После вывода установки на режим и установления постоянства
нулевой линии в колонку микрошприцем вводят 0,5 мкл исследуе-
мого газа. Через 45 сек появляется пик метана, затем выходят этан,
пропан, изобутан, бутан, изопентан и пентан. Последний выходит
на 65—70 сек. Таким образом, весь анализ заканчивается за время,
немногим превышающее одну минуту.
Примечание-. Два последних компонента редко присутствуют в бытовом
газе. Поэтому их пары можно добавлять искусственно.
Б. СТУПЕНЧАТАЯ ХРОМАТОГРАФИЯ
Как уже было сказано, капиллярная хроматография не позво-
ляет осуществлять контроль производственных смесей и тем более —
совмещать контроль с автоматизацией управления процессом.
Стремление улучшить условия разделения многокомпонентных сме-
сей приводит к тому, что на колонку стараются наносить как можно
меньшее количество пробы анализируемых веществ. Это стремле-
ние во многих случаях оправдывается, хотя и приводит к уменьше-
нию концентрации в максимуме полосы по сравнению с исходной по
крайней мере в десятки, а иногда и в сотни раз, что существенно
затрудняет применение хроматографического метода анализа для
решения одновременной задачи автоматизации управления процес-
сом.
Последняя задача для своего разрешения требует, чтобы исход-
ная концентрация при хроматографировании была бы по возможно-
сти сохранена и чтобы получаемый от детектора сигнал в виде прика-
за какому-либо регулирующему процесс устройству был бы доста-
точно устойчивым и независимым от изменения параметров опыта.
Эти требования могут быть выполнены в варианте проявитель-
ной хроматографии, названном его авторами А. А. Жуховицким и
Н. М. Туркельтаубом [3,5] ступенчатой хроматографией.
В ступенчатой хроматографии, в противоположность капил-
лярной, объем пробы анализируемого вещества должен быть дос-
таточно большим. Он должен обеспечивать, даже за счет ухудшения
разделения, сохранение в максимуме хроматографического пика
исходной концентрации вещества. В таком случае возникает устой-
чивый сигнал, который и может быть использован для передачи
приказа об изменении условий протекания процесса.
242
Такая постановка задачи оправдывается тем, что в практике
автоматизации управления процессом очень часто требуется конт-
ролировать процесс не по изменению концентрации всех компонен-
тов смеси, а по одному-двум компонентам. Поэтому задача упро-
щается.
Чтобы устранить понижение концентрации, вызываемое размы-
ванием хроматографической полосы, нужно наносить достаточно
большой объем пробы Уи. Однако это приводит к ухудшению разде-
ления.
Рассмотрим условия появления на хроматограмме двух разде-
ленных полос, высоты которых соответствуют исходной концент-
рации анализируемых веществ. Условия получения такой ступень-
ки требуют, чтобы
VH>2v0, (185)
как это следует из рис. 42. Здесь v0 — объем, в котором произошло
размывание полосы.
Рис. 42. Схема ступенчатой хроматограммы
Решение уравнения диффузии для задачи размывания границы
приводит к тому, что
у0 = 1,6Г]/Ж (186)
Следовательно, условием сохранения ступеньки будет неравен-
ство
УИ>3,2Г VHI,
(187)
а условием разделения двух ступенек
V„ </АГ - 0,72Г УН1.
(188)
Здесь АГ — разность удерживаемых объемов, рассчитанных на
единицу площади сечения слоя.
Таким образом, искомые условия
3,2Г]/Я/<УИ < /АГ — 0.72Г ]/777.
(189)
Из условия совместимости этих неравенств можно получить зна-
чение наименьшей длины слоя /min, на которой возможно получе-
243
ние двух разделенных ступенек. При этом
= (190)
где Кс — коэффициент селективности.
Таким образом, для получения разделенных ступенек следует,
чтобы длина слоя В этом случае возможен диапазон значений
проб анализируемых веществ Уи, позволяющий при данном значении
осуществить разделение смеси двух веществ и получить для
каждого из них ступеньку. Подставив значение /min из уравнения
(190) в любую сторону неравенства (189), получим значение харак-
теристического объема У0>х, который должен быть нанесен на ко-
лонку, чтобы обеспечить одновременно и разделение и образование
ступеньки
V0.x = ^. (191)
Экспериментальные данные подтверждают правильность выве-
денных соотношений.
Определим условия получения на хроматограмме п разделенных
ступенек. Если между первым и последним компонентом помеща-
ются все компоненты смеси, тогда
/(r-rjXn-OV,. (192)
Это условие является необходимым, но недостаточным, так как
компоненты могут поместиться на данном участке, но не разделить-
ся. Кроме того, мы пренебрегли членом 0,72 описывающим
размывание ступеньки.
Поставим условие сохранения ступеньки для компонента, сор-
бирующегося сильнее остальных. Тогда для него
Va > 3,2Г„ VTTl. (193)
Из неравенств (192) и (193) следует, что
1+0,36 -]>). (194)
/ г \
Множитель 1—-=+ близок к 1, если Гп Гг Поэтому разделе-
\ * п/
ние многокомпонентной смеси с образованием отдельных ступенек
возможно, если Гп+1. Однако в том случае, если Г близко к 1,
капиллярная хроматография также не дает удовлетворительного
эффекта разделения.
Для многих практически встречающихся случаев применение
ступенчатой хроматографии для автоматизации управления про-
цессом может дать положительный эффект. Экспериментальные дан-
ные подтверждают теоретические предпосылки.
244
ПРАКТИЧЕСКИЕ РАБОТЫ
Работа 3
ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ ТРИМЕТИЛХЛОРСИЛАНА
И МЕТИЛТРИХЛОРСИЛАНА В ДИМЕТИЛДИХЛОРСИЛАНЕ
МЕТОДОМ СТУПЕНЧАТОЙ ХРОМАТОГРАФИИ [6]
Приборы и реактивы.
1, Хроматограф с катарометром с вольфрамовыми нитями или термисторами
любой системы, например, ХЛ-3 или ХЛ-4. Колонка выносная стеклянная длиной
3,5 м, диаметром 4 мм.
2. Инзенский кирпич, измельчения 0,5—0,25 мм, содержание дибутилфта-
лата 15% отвеса кирпича.
3. Шприц на 1 мл.
4. Баллон с сжатым азотом.
5. Исходное сырье (диметилдихлорсилаи с примесью триметилхлорсилана
и метилтрихлорсилана).
Цель работы. Показать возможность получения ступенек при
введении определенной порции смеси веществ и проанализиро-
вать ее.
Сущность работы. Диметилдихлорсилаи применяется в произ-
водстве термостойкого синтетического каучука. Исходное сырье
может содержать примесь триметилхлорсилана и метилтрихлор-
силана, количество которых в основном сырье необходимо знать.
Так как концентрацию указанных примесей следует определять
по ходу получения диметилдихлорсилана и по содержанию приме-
сей регулировать процесс синтеза, контроль за их содержанием це-
лесообразно проводить по методу ступенчатой хроматографии.
Для правильного проведения анализа рассчитывают по урав-
нению (189) величину наносимой пробы анализируемой смеси и
установленную пробу вводят в колонку. Каждый компонент приме-
сей, а также основное вещество выходят из колонки, образуя от-
дельные ступеньки. Пользуясь предварительно полученными калиб-
ровочными графиками, по высоте ступеньки определяют концентра-
цию компонентов примеси. В качестве неподвижной жидкой фазы
служит дибутилфталат.
Выполнение работы. В готовую хроматографическую колонку,
заполненную инзенским кирпичом, пропитанным дибутилфталатом,
и нагретую в термостате до 40° С, после получения нулевой линии
вводят пробу анализируемой смеси. С этой целью в ловушку, при-
соединенную на входе в хроматографическую колонку и помещенную
в тот же, что и колонка, термостат, вводят шприцем 0,5 мл анализи-
руемой жидкости и продувают ловушку газом-носителем, в качест-
ве которого берут азот. После продувки ловушку отключают, при-
чем подачу газа-носителя со скоростью 30 см?/мин не прекращают.
245
На ленте самописца записываются отдельные ступени, первая
из которых соответствует триметилхлорсилану, вторая — метил-
трихлорсилану, а последняя — основному веществу, диметилди-
хлорсилану. Измеряют высоту каждой из ступенек примеси и, поль-
зуясь калибровочным графиком, полученным у преподавателя, вы-
числяют концентрацию примесей. Результаты измерений и расчета
заносят в журнал.
Ошибка определения не должна превышать ±5% относительных.
В. В АКАНТОХРОМ АТОГРАФИЯ
А. А. Жуховицкий и Н. М. Туркельтауб [3,7] предложили в
1962 г. принципиально новый вариант газо-жидкостной хроматог-
рафии, в котором движение полосы сорбированного вещества заме-
нено движением полосы газа-носителя, свободной от анализируе-
мого компонента. Таким образом, вместо полосы компонента смеси
вдоль колонки движется свободная от него зона — вакансия. Поэ-
тому предложенный метод был назван вакантохроматографическим.
Рассмотрим кратко теорию метода. Пусть через слой сорбента
в колонке непрерывно проходит газ-носитель, содержащий один
компонент примеси. В таком случае через некоторое время по всей
длине слоя устанавливается сорбционное равновесие и на выходе
из колонки состав газа оказывается одинаковым с его составом на
входе. В некоторый момент времени мы введем порцию чистого га-
за-носителя. Тогда равновесие будет нарушено и через слой будет
продвигаться область, свободная от компонента примеси.
Докажем, что поведение такой вакансии совпадает с поведением
полосы компонента в обычной проявительной хроматографии. Для
этого напишем уравнение динамики для линейной изотермы и с уче-
том внешней диффузии:
дс , г, д2с дс . да
+ + (195)
Здесь = —у-) , а граничными условиями будут с(0,/)=0;
с(0, х)=6 (х), где 6 (х) — дираковская функция, передающая очень
тонкую полосу.
В случае движения вакансии дифференциальные уравнения не
должны изменяться, так как не изменяются физические явления
процесса. Будут меняться только начальные и граничные условия,
а именно: с (0, t)=c0', с (0, х)=с0—6 (х).
Если ввести новую переменную с1=с0—с, для которой граничные и
начальные условия будут идентичными с условиями, взятыми при
решении уравнения (195), то
сх (0, 0 = 0 и сх (0, х) = б (х).
246
Если ввести вместо а новую функцию а1=а0—а, где а0=Гс0,
то уравнение (195) примет вид
+ = + (196)
а
<197>
т. е. мы видим, что дифференциальные уравнения для вакансии и
для полосы сорбированного вещества совпадают. Следовательно,
все условия движения вакансии идентичны условиям движения по-
лосы. Поэтому по вакансии можно судить о самом компоненте. В
частности, по величине площади пика вакансии можно, следователь-
но, определить количество компонента в смеси.
Хроматографирование методом вакантохроматографии осущест-
вляется следующим образом. Через слой сорбента в колонке непре-
рывно пропускается анализируемая смесь. После того как на сор-
бенте будет достигнуто сорбционное равновесие, в колонку вводится
порция чистого газа-носителя, который в этом случае можно назвать
газом-дозатором. При этом передвижение вакансий для каждого
компонента будет происходить независимо друг от друга и детектор
зафиксирует столько вакансий, сколько содержится компонентов в
смеси. В каждой вакансии будут находиться все компоненты анали-
зируемой смеси, кроме одного, регистрируемого детектором. Если
через сравнительную камеру детектора пропускать анализируемую
смесь, то детектор после введения порции газа-дозатора будет фик-
сировать вакансии каждого из компонентов, причем параметры
пика будут пропорциональны концентрации компонентов анализи-
руемой смеси.
Таким образом, метод вакантохроматографии позволяет анали-
зировать смеси, хотя разделения их на составляющие при этом не
происходит. Метод введения пробы газащозатора, калибровка
прибора и расшифровка хроматограмм остаются идентичными обыч-
ной газо-жидкостной проявительной хроматографии.
Вакантохроматография обладает специфическими особенностя-
ми, обусловливающими ряд преимуществ этого метода.
1. В отличие от проявительной хроматографии в вакантохро-
матографии анализируемая смесь пропускается непрерывно, а в
нужный момент вводится проба газа-дозатора. Это обстоятельство
существенно упрощает анализ, так как, во-первых, не требует специ-
альных устройств дозаторов в случае, например, анализа агрессив-
ных веществ. Во-вторых, при этом резко сокращается расход доро-
гостоящего газа-носителя (например, гелия). В-третьих, отпадает
необходимость пользоваться большими количествами взрывоопас-
ных газов (водород). Наконец, в качестве газа-дозатора в ваканто-
хроматографии можно применять любой из компонентов анализи-
руемой смеси в чистом виде.
247
2. В вакантохроматографии размывание полос, связанное с
нелинейностью изотермы сорбции, должно быть меньше, чем в про-
явительной. В самом деле, скорость движения полосы данной кон-
центрации определяется величиной daldc. Если рассмотреть уравне-
ние Лэнгмюра
_______Г,- с,-__
' 1 + Г,-с,- + 2ГjCj ’
г __L+££Z2__
dCi !'(l + riC1 + 2r/C7)2’
(198)
(199)
то из этого уравнения следует, что наличие других компонентов
смеси резко уменьшает зависимость да^дс^ от cz. Поэтому иногда
для улучшения разделения следует добавлять к анализируемой
смеси один из сильно сорбирующихся компонентов.
3. Вакантохроматографии позволяет определять изотерму сорб-
ции смеси, что принципиально невозможно в проявительной хрома-
тографии.
4. Вакантохроматографии дает возможность определения неболь-
ших объемов газа новым методом. При отсутствии значительного
размывания площадь пика вакантохроматограммы, выраженная в
единицах объема, непосредственно равна объему введенного газа-
дозатора, а высота пика пропорциональна его объему. Отсюда воз-
никает новая возможность определения малых объемов газа хромато-
графическим методом. В случае применения высокочувствительных
детекторов можно свободно определять объемы порядка 0,01 см3.
К числу недостатков вакантохроматографического метода следу-
ет отнести возникающий при изменении концентрации анализиру-
емой смеси дрейф нулевой линии. При непрерывном изменении
концентрации нулевая линия будет отклоняться, причем величина
отклонения будет определяться разницей концентраций в сравни-
тельной и измерительной камерах детектора. Этого недостатка
можно, однако, либо избежать, либо использовать его для непре-
рывного анализа.
Если в камеру сравнения детектора направлять поток анализи-
руемого газа не непосредственно, а пропустив его предварительно
через такую же колонку с сорбентом, как и газ, поступающий в
измерительную камеру, то тогда все изменения концентрации в ана-
лизируемом газе будут одинаково отражаться как на работе камеры
сравнения, так и на индикаторной камере. Для осуществления этого
следует монтировать хроматографическую установку из двух парал-
лельно работающих, одинаковых по своим размерам колонок, за-
полненных одинаковым количеством сорбента. Регулировка нуле-
вой линии в этом случае может производиться путем изменения ско-
рости подачи газовой смеси в одну из колонок, а также обычными
методами. Впуск газа-дозатора производится в ту колонку, выход
газа из которой направляется в измерительную камеру детектора.
248
Такой прием позволяет исключить влияние изменения концентра-
ции на положение нулевой линии.
Другой метод, предложенный А. А. Жуховицким, Л. М. Лапки-
ным и А. А. Дацкевичем [8] в 1965 г., состоит в том, что дрейф нуле-
вой линии, имеющий место вследствие изменения концентрации
анализируемой смеси, используется для непосредственного измере-
ния концентрации. В этом случае расстояние между точками на
нулевых линиях, соответствующих постоянной и переменной кон-
центрациям в данный момент времени, линейно зависит от скорости
изменения концентрации в смеси, а площадь между этими нулевыми
линиями за определенный промежуток времени равна сумме произ-
ведений времени удерживания на величину показаний детектора, от-
вечающих соответствующим концентрациям. Это обстоятельство
позволяет применять вакантохроматографию в качестве непрерыв-
ного метода анализа и выгодно отличает этот вариант хроматогра-
фического метода от других его видоизменений. Кроме того, в отли-
чие от проявительного метода, вакантохроматография позволяет
определять не только концентрации, но и скорость их изменения
во времени.
Экспериментальные данные хорошо подтверждают теоретиче-
ские представления и указывают на перспективность метода.
ПРАКТИЧЕСКИЕ РАБОТЫ
Работа 4
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ДВУОКИСИ УГЛЕРОДА
В ВОЗДУХЕ МЕТОДОМ ВАКАНТОХРОМАТОГРАФИИ
Приборы и реактивы
1. Хроматограф типа ХЛ-3.
2. Система напорных бутылей емкостью 20—30 л для осуществления непре-
рывной подачи воздуха, содержащего 1% двуокиси углерода.
3. Инзенский кирпич, пропитанный вазелиновым маслом (25% от веса кир-
пича).
4. Шприц на 5 мл.
5. Емкость с сухим воздухом, не содержащим двуокиси углерода.
Цель работы. Показать возможности и преимущества ваканто-
хроматографического метода; определить содержание двуокиси уг-
лерода в воздухе.
Сущность работы. Если воздух, содержащий двуокись углерода,
непрерывно пропускать через колонку, заполненную твердым но-
сителем с какой-либо жидкой фазой, то после установления равно-
весия самописец прибора будет вычерчивать постоянную нулевую
линию. При впуске порции газа-дозатора, которым может служить
воздух, освобожденный от двуокиси углерода, самописец вычертит
9 Б. В. Айвазов
249
пик, соответствующий вакансии двуокиси углерода. Площадь это-
го пика будет пропорциональна содержанию СО2 в воздухе.
Выполнение работы. Хроматографическую колонку заполняют
инзенским кирпичом, пропитанным вазелиновым маслом. Устанав-
ливают систему бутылей, обеспечивающую непрерывную подачу
воздуха с постоянной скоростью, равной 50 см31мин. Поступающий в
колонку воздух предварительно осушают в трубках, наполненных
пятиокисью фосфора. Ожидают, пока нулевая линия самописца
установится на постоянном уровне. Вводят шприцем 5 мл сухого
воздуха, не содержащего двуокиси углерода. При этом самописец
должен вычертить пик вакансии, соответствующей содержанию
двуокиси углерода. Опыт прекращают и по площади полученного
пика рассчитывают содержание двуокиси углерода в воздухе, поль-
зуясь калибровочным графиком, полученным у преподавателя.
Работа 5
ОПРЕДЕЛЕНИЕ МАЛЫХ ОБЪЕМОВ ГАЗА МЕТОДОМ
ВАКАНТОХРОМАТОГРАФИИ
Приборы и реактивы
1. Хроматограф с пламенно-ионизационным детектором, например, хромато-
граф типа «Цвет». Можно применить самодельный хроматограф и детектор типа
ДИП-1. Колонка стеклянная длиной 2 л, диаметром 4 мм.
2. Инзенский кирпич, пропитанный динонилфталатом (25% от веса кирпича),
измельчения 0,5—0,25 мм.
3. Микрошприц до 0,1 мл.
4. Система бутылей, обеспечивающая непрерывную подачу газа с постоянной
скоростью.
5. Емкость с чистым азотом.
Цель работы. Показать, что метод вакантохроматографии
может служить для определения малых объемов газа.
Сущность работы. Площадь пика вакансии пропорциональна
объему нанесенной пробы газа-дозатора. Эту зависимость можно
использовать для определения объема газа. С этой целью предвари-
тельно строят график зависимости площади пика от объема пробы
газа, затем вводят неизвестный объем газа, определяют площадь
полученного пика и по графику рассчитывают объем.
Выполнение работы. Колонку заполняют инзенским кирпичом,
пропитанным динонилфталатом, устанавливают постоянный ток
смеси азота с бутаном, подаваемой из системы напорных бутылей, и
после достижения сорбционного равновесия, наступление которого
определяют по постоянству нулевой линии, вводят пробы газа-до-
затора. Скорость подачи смеси газа 50 см3!мин. Газом-дозатором
служит чистый азот. Объемы вводимых проб: 0,01; 0,02; 0,04; 0,06;
0,08 и 0,1 см3.
250
Объем вводимой пробы должен быть точно известен. Для полу-
ченных вакансий определяют площади пиков и по ним строят гра-
фик в координатах: объем вводимой пробы в см3 — площадь пика
в мм2. При отсутствии размывания должна получиться прямая
линия, проходящая через начало координат.
После построения графика получают от преподавателя пробу
газа (азота) неизвестного объема и вводят ее в колонку. Определя-
ют площадь полученной вакансии и по графику рассчитывают
объем газа. Все данные записывают в журнал.
ЛИТЕРАТУРА
Использованная литература
1. «Газовая хроматография». Труды симпозиума в Амстердаме. ИЛ, М., 1961.
2. Г. Ш а й. Теоретические основы хроматографии газов. ИЛ, М., 1963.
3. А. А. Жуховицкий, И. М. Туркельтауб. Газовая хромато-
графия. Гостоптехиздат, М., 1962.
4. В. В. Бражников, Ф, Я. Ф р о л о в. В сб. «Газовая хроматогра-
фия». Вып. I, НИИТЭХим, М., 1964.
5. А. А. Жуховицкий, Н. М. Туркельтауб. ДАН СССР, 144,
4, 1962.
6. Н. А. П а л а м а р ч у к. В сб. «Газовая хроматография». Вып. I,
НИИТЭХим, М., 1964.
7. А. А. Жуховицкий, Н. М. Т у р к е л ь т а у б. ДАН СССР,143,
№ 3, 646, 1962.
8. А. А. Жуховицкий, Л. М. Л а п к и и, А. А. Д а ц к е в и ч.
ДАН СССР, 162, № 5, 1089, 1965.
Рекомендуемая литература
1. М. Шиигляр. Газовая хроматография в практике. Изд-во «Химия»,
М., 1964.
2. «Газовая хроматография». Труды 3-й Всес. конф. Изд-во «Наука», М., 1964.
3. «Газовая хроматография». Труды 3-й Всес. коиф. Изд. Дзерж. фнл.
ОКБА, 1966.
4. «Успехи и достижения газовой хроматографии». Гостоптехиздат, М., 1961.
5. «Газовая хроматография». Труды симпозиума в Эдинбурге. Изд-во «Мир»,
М., 1964.
9
ГЛАВА VII
РАСПРЕДЕЛИТЕЛЬНАЯ ХРОМАТОГРАФИЯ
НА БУМАГЕ
Различие в коэффициентах распределения компонентов разделя-
емой смеси между двумя несмешивающимися жидкостями может быть
использовано'для хроматографического разделения и анализа,так
же как и различие в коэффициентах распределения между жидкос-
тью и газом. Такой вид хроматографии назван жидкостно-жид-
костной распределительной хроматографией. В зависимости от при-
роды твердого носителя жидкой неподвижной фазы и способа про-
ведения эксперимента жидкостно-жидкостная распределительная
хроматография делится на колоночную и бумажную.
В колоночной хроматографии в качестве твердого носителя не-
подвижной жидкой фазы может быть применен любой твердый но-
ситель, удовлетворяющий трем основным требованиям: он должен
прочно удерживать неподвижную фазу, легко пропускать подвиж-
ную жидкую фазу и не вызывать побочных явлений (адсорбции ве-
ществ смеси, каталитического воздействия на компоненты смеси
и т. п.). Экспериментальное оформление колоночной жидкостно-
жидкостной распределительной хроматографии практически ничем
не отличается от колоночной адсорбционной хроматографии жидких
веществ (см. гл. I).
В качестве носителя неподвижной жидкой фазы в бумажной хро-
матографии применяется специальная бумага, способная удерживать
в своих порах значительные количества жидкости, являющейся не-
подвижной фазой. В этом случае движение зон компонентов разделя-
емой смеси происходит вдоль тонкого слоя бумаги. Эксперименталь-
ное осуществление этого метода существенно отличается от метода
колоночной хроматографии.
В связи с тем, что из указанных двух вариантов жидкостно-жид-
костной распределительной хроматографии наибольшее распростра-
нение получил второй вариант с применением бумаги в качестве но-
сителя неподвижной фазы, а также в связи со спецификой этого
метода мы рассмотрим теорию и практику только бумажной хрома-
тографии.
252
Теория распределительной колоночной хроматографии, разра-
ботанная А. Дж. П. Мартином, Р. Л. М. Синджем [1] и Н. А. Фуксом
[2], может быть распространена на вариант бумажной хроматогра-
фии.
В распределительной колоночной хроматографии движение
зон компонентов разделяемой смеси может быть количественно
охарактеризовано величиной RF, названной Н. А. Фуксом под-
вижностью
p __ s
Sn + 7<pSH’
где S — поперечное сечение колонки; Sn — поперечное сечение
подвижной жидкой фазы; SH — поперечное сечение неподвижной
Жидкой фазы; — коэффициент распределения.
Так как в случае бумажной хроматографии величину RF изме-
рить не представляется возможным, то для характеристики поведе-
ния зон на бумаге вводят величину Rf
— _ скорость движения зоны одного компонента ____
'/ скорость движения фронта подвижной жидкой фазы
_ ___ " Sn
S Sn + KpSH ’
Величину Rj можно также выразить через смещение зоны при
промывании чистым растворителем. В самом деле, величина смеще-
ния зоны х при промывании колонки чистым растворителем, объем
которого равен V, может быть определена из уравнения
V
X^sn+Kps«’
(200)
(201)
(202)
откуда
R/- v и x— s .
(203)
Если величину смещения фронта растворителя обозначить xf,
то xf определится равенством
(204)
тогда
/?/ = £, (205)
т. е. Rf равно отношению смещения зоны к смещению фронта рас-
творителя.
На рис. 43 дано пояснение этого отношения. Здесь х± и х2 — путь;
пройденный соответственно первым и вторым компонентами разде-
ляемой смеси от начального положения; xf — путь, пройденный
фронтом растворителя. Если хг не равен х2, то и R'f не будет равно
/?)', следовательно, зоны компонентов смеси разделятся. Поэтому
253
Рис. 43. Схема определе-
ния /?j компонентов сме-
си по результатам хрома-
тографирования на бу-
маге:
х2, Xf—путь, пройденный
соответственно первым и вто-
рым компонентами смеси и
растворителем
величина Rf может характеризовать способность двух несмешиваю-
щихся жидкостей к разделению смеси веществ.
В идеальном случае Rf характеризует скорость перемещения
зоны компонента по бумаге и не зависит от присутствия других
веществ, а зависит только от природы выбранных жидких подвиж-
ной и неподвижной фаз. Следовательно, в идеальном случае Rf
определяется только коэффициентом рас-
пределения и параметрами бумаги. На прак-
тике, однако, вследствие взаимодействия
веществ смеси с носителем, отклонения про-
цесса от равновесия и по другим причинам,
Rf зависит от природы носителя, техники
эксперимента и других факторов.
Величины R{, а также коэффициенты
распределения могут быть определены
экспериментально из хроматографических
данных. Для этого следует измерить иа
хроматограмме величины смещения зон ве-
ществ и фронта растворителя. Коэффици-
ент распределения может быть вычислен
из нижеследующего уравнения,
вытекает из уравнения (201)
которое
^р’ s'
(206)
отноше-
5
При этом отношение , равное
нию объемов жидких фаз, а также Rf
можно легко определить из опытных дан-
ных. Измеренные таким путем коэффициенты распределения для
многих веществ хорошо согласуются с экспериментальными дан-
ными, полученными прямым определением.
Величины Rf для компонентов разделяемой смеси должны быть
не очень большими, но и не слишком малыми. В первом случае
смесь не сможет разделиться, а во втором — процесс разделения
будет протекать слишком медленно.
МЕТОДИКИ ПОЛУЧЕНИЯ РАСПРЕДЕЛИТЕЛЬНЫХ
ХРОМАТОГРАММ НА БУМАГЕ [2—9]
В настоящее время получили развитие следующие виды хромато-
графии на бумаге: одномерная, двумерная, круговая и электро-
форетическая. Одномерная и двумерная могут выполняться в двух
вариантах: восходящим и нисходящим потоком растворителя. Все
указанные виды обладают своими преимуществами и недостатками.
Поэтому выбор наиболее подходящего способа хроматографирова-
254
Рис. 44. Камера
для получения ни-
сходящей хромато-
граммы:
1 — цилиндр; 2—кор-
ковая пробка; 3—ван-
ночка с подвижным
р аствор ителем; 4 —
Г-образная стеклян-
ная палочка; 5—по-
лоска бумаги; 6 —
груз; 7 — бюкс с не-
подвижным раствори-
телем
тем лучше, чем
не более 0,005—
ния должен быть сделан самим экспериментатором в зависимости от
конкретных условий.
Получение одномерной восходящей хроматограммы. Простейшим
вариантом является получение одномерной восходящей хрома-
тограммы. В этом случае капля исследуемого раствора нано-
сится на нижнюю часть полоски бумаги на некотором расстоянии
от края. Если неподвижной фазой служит вода,
то бумагу не подвергают специальной пропитке,
так как воздушно-сухая бумага содержит доста-
точное количество влаги (до 20—22%). Подвиж-
ная фаза, насыщенная неподвижной, наливается
на дно сосуда для хроматографирования, кото-
рым может служить цилиндр или пробирка.
Полоска бумаги своим нижним краем опускается
в жидкость, служащую подвижной фазой. Верх-
ний край бумажной полоски закрепляется так,
чтобы бумага свободно свисала вниз. Сосуд,
в котором происходит хроматографирование,
плотно закрывается и помещается в термостат
на все время опыта. Колебания температуры на
протяжении всего опыта не должны превышать
±1,5° С. Вследствие капиллярных сил подвиж-
ная жидкость поднимается вверх по бумаге и
разделяет компоненты смеси на зоны, которые
при наличии различных значений движутся
по слою бумаги с неодинаковыми скоростями.
Опыт считается законченным, когда фронт под-
вижной фазы достигнет верхнего края полоски
бумаги. После этого бумагу вынимают из сосу-
да, высушивают и проявляют.
Нанесение капли раствора на бумагу произ-
водится капилляром или микропипеткой. Вслед-
ствие того, что четкость разделения зависит от
количества нанесенного вещества, причем она
меньше его нанесено, рекомендуется наносить
—0,001 мл раствора.
Ширина полоски бумаги в случае нанесения одной капли обычно
составляет 2—5 см. Длина определяется условиями разделения.
При одновременном хроматографировании ряда растворов берут
широкую полосу бумаги и вдоль ее нижнего края, на так называе-
мую стартовую линию наносят капли исследуемых растворов с
интервалом в 2—3 см.
Получение одномерной нисходящей хроматограммы. Для полу-
чения нисходящей одномерной хроматограммы применяют стеклян-
ный цилиндр емкостью 100—500 мл (рис. 44,/). В корковой пробке
2 укрепляют небольшую ванночку 3, в которую наливают подвиж-
ный растворитель, насыщенный неподвижным. В эту же пробку
255
монтируют Г-образную стеклянную палочку 4, через которую пере-
кидывают полоску бумаги 5 так, чтобы ее верхний конец был опущен
в ванночку с растворителем. На дно сосуда помещают бюкс с непо-
движным растворителем, насыщенным подвижным, для создания в ци-
5
Рис. 45. Кювета для одновре-
менного получения нисходящих
хроматограмм на нескольких
листах бумаги (а — вид сверху;
б — поперечный разрез):
1— кювета для подвижного раство-
рителя; 2 — стеклянные палочки;
3 — места нанесения капель раствора
на бумагу; 4— листы бумаги
линдре атмосферы насыщенных паров,
предотвращающей испарение раство-
рителя с бумаги. На полоску бумаги на
расстоянии 5 см от края наносят
каплю исследуемого раствора и этот
край полоски погружают в ванночку
с подвижной фазой. Для утяжеления
полоски бумаги к ее нижнему концу
прикрепляют стеклянную палочку 6,
вставляя ее в небольшой продольный
разрез. Пробку с ванночкой и бума-
гой вставляют в цилиндр и плотно
его закрывают.
Растворитель из ванночки стекает
вследствие действия капиллярных сил
и сил тяжести вниз по бумаге и обес-
печивает движение зон компонентов
разделяемой смеси. Опыт считается
оконченным, когда фронт раствори-
теля достигнет 3—5 см от нижнего
края бумаги.
Для получения нисходящей хрома-
тограммы нескольких растворов можно
применять специальную кювету, вид сверху и разрез которой
приведены на рис. 45. Такая кювета закрепляется в верхней части
цилиндра или другого сосуда соответствующего размера. Диаметр
кюветы 3—4 см, ее длина определяется диаметром выбранного
сосуда. Пользуясь такой кюветой, можно одновременно под-
вергать хроматографированию несколько анализируемых смесей,
помещая в кювету ряд широких полос бумаги, как это показано
на рис. 45,6.
Получение круговой хроматограммы. Круговая хроматограмма
позволяет получить вместо пятен концентрически расположенные
кольца. Для получения круговой хроматограммы вырезают круг
из соответствующей бумаги и в его центр наносят каплю исследуе-
мого раствора. В качестве сосуда для хроматографирования удобно
в данном случае использовать эксикатор небольшого размера. Диа-
метр бумажного круга должен быть на 2—3 см больше, чем диаметр
нижней узкой части эксикатора. Бумажный круг укладывается над
узкой частью эксикатора (рис. 46). Для подачи подвижного раство-
рителя на круге вырезают полоску — «фитиль» шириной 2 мм так,
как это показано на рис. 47. Фитиль отгибают и опускают в раство-
ритель. Насыщенный неподвижной фазой растворитель наливают
256
на дно эксикатора. Скорость поступления растворителя к центру
круга можно регулировать изменением ширины фитиля.
После того как растворитель дойдет почти до краев бумажного
круга, круг вынимают из эксикатора, высушивают и проявляют.
Хроматографирование, также как и на полосках бумаги, произ-
водят при герметически закрытом эксикаторе и при постоянной тем-
пературе.
Получение двумерной хроматограммы. Если при помощи одного
растворителя разделить сложную смесь не удается, то следует после-
довательно применять два растворителя, обладающих различными
коэффициентами распределения. Такая хроматограмма называется
двумерной.
Рис. 46. Прибор
для получения
круговой хромато-
граммы:
1—бумажный фильтр;
2 — фитиль; 3 — рас-
творитель
Рис. 47. Форма бумаги для
получения круговой хромато-
граммы (Д — место нанесе-
ния капли раствора):
1 — бумажный кружок; 2 — фи-
тиль для подачи растворителя
Для получения двумерной хроматограммы применяют квадрат-
ные листы бумаги размером 20x20, 30x30 или 40x40 см. В начале
опыта каплю исследуемого раствора наносят на бумагу в ее левом
углу на расстоянии 5 см от краев (рис. 48, а). После высушивания
образовавшегося пятна бумагу помещают в сосуд для хроматогра-
фирования, опускают нижний край бумаги в один из выбранных
растворителей и производят хроматографирование по восходящему
методу. После того как фронт подвижной фазы достигнет верхнего
края бумаги, хроматографирование прекращают, бумагу высушивают
и поворачивают ее на 90° против часовой стрелки. При этом место
нанесения капли исследуемого раствора окажется справа, а линия,
по которой происходил подъем зон анализируемых веществ, внизу
(рис. 48, б). В таком положении бумагу помещают в новый сосуд
для хроматографирования, опускают ее нижний конец во второй
растворитель и хроматографируют по восходящему методу. После
достижения фронтом новой подвижной фазы верхнего края бумаги
хроматографирование прекращают, бумагу высушивают, а образо-
вавшиеся зоны анализируемых веществ проявляют заранее выбран-
ным раствором. Получают двумерную хроматограмму такого типа,
как это изображено на рис. 48 (в).
257
Получение электрофоретической хроматограммы. Успех хрома-
тографии на бумаге обусловлен не только широким, ассортиментом
жидких фаз с различными коэффициентами распределения и други-
ми ее достоинствами, но также и возможностью применять одно-
временное или последовательное воздействие на разделяемую смесь
электрического поля. Такой вид хроматографии на бумаге получил
название электрофоретической хроматографии.
Рис. 48. Схема получения двумерной хроматограммы (Д — место нанесения
капли исследуемого раствора):
а — хроматограмма, полученная после хроматографирования первым раствори-
телем, смесь, состоящая из шести компонентов, не разделилась; б — та же хрома-
тограмма перед опусканием бумаги во второй растворитель; в — хроматограмма,
полученная после хроматографирования во втором растворителе и проявления,
смесь разделилась.
Стрелкой указано направление движения фронта подвижной фазы
При одновременном проведении хроматографирования и электро-
фореза бумажный лист пропитывается раствором электролита и за-
крепляется между разноименными электродами. Одновременно с
подачей напряжения на электроды от источника постоянного тоКа
обеспечивают движение вдоль бумаги подвижного растворителя.
Такой процесс значительно сокращает время разделения, однако
выполнение его связано с некоторыми техническими трудностями.
Поэтому более целесообразно проводить оба процесса последова-
тельно.
Электрофорез с последующим хроматографированием осуществ-
ляется следующим образом. Хроматографическую бумагу пропиты-
вают раствором какого-либо летучего электролита, например, ук-
сусной кислотой, наносят каплю анализируемой смеси и проводят
электрофорез, подключая бумагу через электроды к источнику пос-
тоянного тока. После окончания электрофореза бумагу вынимают
из прибора, сушат и переносят в камеру для хроматографирования
по методу восходящей или нисходящей хроматографии. Обработка
хроматограммы после окончания хроматографирования ничем не
отличается от обычной. Метод раздельного электрофореза и хрома-
тографирования позволяет производить эти две операции при раз-
личных значениях pH, что существенно увеличивает возможности
разделения.
?5§
Бумага для хроматографирования
В распределительной хроматографии к бумаге предъявляются
определенные требования: она должна быть химически чистой,
химически и адсорбционно нейтральной, однородной по плотнос-
ти, обеспечивать определенную скорость движения растворителя.
Имеет существенное значение структура и ориентация волокон бу-
маги. Без соблюдения этих требований успех хроматографического
анализа не может быть обеспечен.
Для получения химически чистой бумаги товарную бумагу обра-
батывают различными реагентами, например, аминоуксусной кис-
лотой, трилоном Б, 8-оксихинолином и др., образующими раство-
римые комплексные соединения с присутствующими в бумаге неор-
ганическими ионами. Получающиеся соединения вымываются затем
растворителями, вследствие чего бумага освобождается от неорга-
нических примесей.
В качестве хроматографической бумаги можно применять не
только специально выпускаемую для этой цели бумагу, но и плотные
сорта фильтровальной бумаги, ватмановскую бумагу и др. В Совет-
ском Союзе выпускается четыре сорта хроматографической бумаги:
№1,2,3 и 4. Она отличается по плотности, а следовательно, и по
скорости движения растворителя. Бумага № 1 и 2 менее плотная
и потому может называться «быстрой». Бумага № 3 и 4 может
быть названа «медленной», так как она более плотна.
В качестве характеристики бумаги принимают массу 1 м2 в грам-
мах, толщину листа в миллиметрах, высоту подъема воды за едини-
цу времени (в см!час) и высоту подъема смеси м-бутилового спирта,
уксусной кислоты и воды, взятых в соотношении 4 : 1 : 5 по объему,
за единицу времени.
Для получения четкого разделения необходимо учитывать на-
правление волокон бумаги, причем оно должно совпадать с направ-
лением движения растворителя.
В. любом случае хроматографирования бумага должна содержать
достаточное количество неподвижной фазы, на которой будет произ-
ведено разделение смеси.
Обычные сорта бумаги гидрофильны. Поэтому в случае примене-
ния воды в качестве неподвижной фазы никакого специального
увлажнения бумаги не требуется. Как уже указывалось, воздушно-
сухая -бумага содержит до 20—22% влаги, что вполне достаточно для
проведения опыта.
Для разделения смесей нерастворимых в воде органических
соединений необходимо гидрофильную бумагу превратить в гидро-
фобную. Существуют следующие способы гидрофобизации бумаги:
пропитка различными гидрофобными веществами и ацетилирование.
Пропитку бумаги производят 1%-ным раствором парафина в
нетролейном эфире, 0,5%-ным раствором каучука в бензо-
ле, либо 1—2%-ным раствором очищенного растительного масла
259
в диэтиловом эфире. Проверка гидрофобности бумаги после оконча-
ния ее пропитки одним из указанных растворов и испарения раство-
рителя производится каплей воды. Если капля воды стекает с по-
лоски бумаги, не смачивая ее, то бумага считается гидрофобной.
Хранят такую бумагу в герметичных сосудах в атмосфере паров того
растворителя, который затем будет применяться с этой бумагой в
качестве неподвижной фазы.
Ацетилирование бумаги производят обработкой ее ацетилирую-
щей смесью, которую готовят смешением 90 мл уксусного ангид-
.рида, 10 мл петролейного эфира и 8—10 капель концентрированной
серной кислоты. Бумагу помещают в ацетилирующую смесь на
45 мин, затем вынимают и тщательно промывают в проточной воде
в течение 15 мин, оставляют на 10—15 мин в дистиллированной воде,
а затем промывают и высушивают. Высушенную бумагу проверяют
на гидрофобность.
Метод получения хроматограмм на гидрофобной бумаге, приме-
няющийся для анализа водонерастворимых веществ, получил наз-
вание метода «.обращенных фаз». В этом случае неподвижной фазой
служит неполярный растворитель (углеводород), а подвижной —
полярный (водные растворы спиртов, органических кислот и т. п.).
РАСТВОРИТЕЛИ, ПРИМЕНЯЕМЫЕ В ХРОМАТОГРАФИИ НА БУМАГЕ
Первостепенное значение для успешного решения задачи разде-
ления смеси веществ методом хроматографии на бумаге имеет пра-
вильный выбор подвижной и неподвижной, фаз. Это значение жид-
ких фаз обусловлено тем, что разрешающая способность распреде-
лительной хроматографии зависит от различий в коэффициентах рас-
пределения компонентов исследуемой смеси между двумя жидкими
фазами. Поэтому жидкие фазы для хроматографии на бумаге долж-
ны удовлетворять следующим основным требованиям.
L Обе жидкости не должны смешиваться друг с другом.
2. Подвижной фазой должен быть выбран такой растворитель,
в котором компоненты разделяемой смеси имеют меньшую раство-
римость, чем в растворителе, применяемом в качестве неподвиж-
ной фазы. При этом растворимость в подвижном растворителе не
должна быть очень большой и слишком малой. В первом случае
вещества смеси будут двигаться со скоростью движения фронта под-
вижного растворителя. Во втором — они будут оставаться на месте
их нанесения.
3. Растворимость и, следовательно, коэффициент распределения
компонентов хроматографируемой смеси должны быть различными
для выбранной пары растворителей. В противном случае разделе-
ния смеси веществ не произойдет.
4. Состав растворителя в процессе хроматографирования не дол-
жен изменяться.
260
5. Растворители должны легко удаляться с бумаги, быть без-
вредными для человека и недефицитными.
Для водорастворимых веществ в качестве подвижных фаз приме-
няются органические растворители, насыщенные водой, которая слу-
жит неподвижной фазой. Нерастворимые в воде вещества должны
хроматографироваться водными растворами органических веществ,
а неподвижной фазой в этом случае должны быть неполярные орга-
нические соединения. В настоящее время индивидуальные вещества
используются в качестве жидких фаз сравнительно редко. Большей
частью применяют смеси. Наиболее часто применяемые в хромато-
графии на бумаге смеси растворителей для водорастворимых ве-
ществ приведены в табл. 19.
МЕТОДИКА АНАЛИЗА
Методика качественного анализа. В большинстве случаев хрома-
тограмма на бумаге после проведения опыта по разделению смеси
веществ и испарения подвижной фазы оказывается бесцветной и по-
этому не позволяет непосредственно не только идентифицировать
вещество, но и судить о наличии разделения их смеси. Поэтому по-
лученные хроматограммы должны быть проявлены. Для этой цели
могут служить растворы различных веществ, при взаимодействии
которых с компонентами анализируемой смеси образуются окра-
шенные соединения. В проявленной хроматограмме по окраске пят-
на, образованного тем или иным веществом смеси и проявителем,
можно производить идентификацию вещества. Если проявитель об-
разует со всеми веществами разделенной смеси одинаково окра-
шенные пятна, то идентификация веществ должна производиться
по месту расположения пятна на бумаге. Качественное обнару-
жение веществ в проявленной хроматограмме возможно не только
по окраске пятен, видимой, при дневном освещении, но и по
люминесценции в ультрафиолетовом свете.
Качественный метод обнаружения веществ на хроматограм-
мах с применением специфически окрашивающих реактивов очень
прост по выполнению, но ограничен вследствие сравнительно
небольшого ассортимента известных и достаточно подходящих
проявителей. Возможности метода несколько расширяет примене-
ние люминесценции в ультрафиолетовом свете, а также радиоак-
тивных изотопов.
Применение метода люминесценции очень облегчается, если для
открытия на хроматограммах люминесцирующих веществ использо-
вать специальный ультрахемископ Е. М. Брумберга [10]. Прибор
состоит из ртутно-кварцевой лампы низкого давления, светофиль-
тра и люминесцирующего экрана, компактен и удобен в обращении.
Применение радиоактивных изотопов, т. е. хроматографирова-
ние смеси, один или несколько компонентов которой являются радио-
активными, облегчает задачу качественного анализа. В этом случае
261
Таблица 19
Подвижные фазы, наиболее часто применяемые
в бумажной хроматографии для разделения смесей
водорастворимы х веществ (неподвижная фаза — вода)
Растворители Соотношение растворителей н способ приготовления Смеси каких веществ разделяются
н-Бутиловый спирт, 4:1:5 по объему. После Аминокислоты, уг-
уксусная кислота, вода расслаивания смеси приме- няется ее верхний слой леводы и другие ор- ганические вещества
н-Бутиловый спирт, муравьиная кислота, (20%-ный раствор), вода 5:1:1 по объему Аминокислоты
н-Бутиловый спирт, н-пропиловый спирт, во- да 12:5:3 по объему Аминокислоты
«-Амиловый спирт, пи- ридин 9:1 по весу а-Аминокислоты
Феиол, вода 100 мл дистиллированной воды при легком нагрева- нии растворяют в 4С0 мл фенола Аминокислоты, уг- леводы
Этилацетат, пиридин, вода 2:1:2 по объему Углеводы
Этилацетат, уксусная кислота, вода 3:1:3 по объему Углеводы
Водонасыщеииый н-бу- Концентрация уксусной Органические окси-
тиловый спирт, уксусная кислота кислоты 1—2 моль/л кислоты
н-Бутиловый спирт, насыщенный аммиаком Равные объемы н-бутило- вого спирта и 1,5 и. водно- го раствора аммиака Жирные кислоты
Этиловый спирт, кон- центрированный водный раствор аммиака 99:1 по весу Первые восемь го- мологов жирных кис- лот
Четыреххлористый уг- лерод, уксусная кисло- та, вода 5:1:1 по объему Жирные кислоты и их натриевые соли
н-Бутиловый спирт, Спирт насыщают 1 и. или Иоиы Pb2 + , Hg2+, Bi3+, Cu2+, Cd2 +
соляная кислота 3 и. раствором соляной кис- лоты
Ацетон, соляная кис- лота (1,19 г/см3), вода 87:8:5 по объему Иоиы Zn2+, Mn2+, Co2+, Ni2+ Ионы Cu2+, Fe3+, Mn2 + , Coa + , Ni2 +
Метилэтилкетон, кон- центрированная соляная кислота 92:8 по объему
Метилэтилкетон, соля- ная кислота (30%-иый раствор) 1:1 по объему Ионы металлов пла- тиновой группы
- н-Бутиловый спирт, 5 г феиилуксусиой кис- Большинство кати-
феиилуксусная кислота, азотная кислота лоты растворяют в 50 мл н-бутилового спирта и встряхивают с 50 мл 0,1 н. раствора азотной кислоты. онов
262
П родолжение табл. 19
Растворители Соотношение растворителей и способ приготовления Смеси каких веществ разделяются
Ацетилацетон, концеи- После расслоения применя- ют верхний слой Ацетилацетон насыщают As3+, Sb3+, Sn2+
трированная соляная кислота, ацетон н-Бутиловый спирт, водой. Компоненты смеши- вают в отношении 50:1:50 по объему Смешивают в отношении Анноны
пиридин, 1,5 и. раствор аммиака н-Бутиловый спирт, 2:1:2 по объему 96:2:2 по объему. п-То- Стрептомицин
пиперидин, п-толуол- сульфоновая кислота Этиловый спирт (40%- луолсульфоновая кислота, образуя со стрептомицином соль, сообщает ему гидро- фобные свойства, в резуль- тате чего улучшается раз- деление В 40%-ном этиловом Ni2 + , Cu2 + , Co2 +
ный), трилон Б, кон- центрированная серная кислота спирте приготовляют 0,01 н. раствор трилона Б. На 100 мл этого раствора при- бавляют 4 капли серной кислоты
пятно на хроматограмме, содержащее радиоактивный компонент,
может быть обнаружено либо по счетчику Гейгера — Мюллера,
к которому должна быть поднесена бумажная хроматограмма, либо
при помощи метода радиоавтографии. Последний состоит в том, что
бумажная хроматограмма, содержащая радиоактивный изотоп, кон-
тактирует некоторое время со светочувствительной пленкой или
бумагой. После проявления пленки или бумаги наличие радиоизо-
топа легко обнаруживается по почернению того места, которое со-
ответствовало положению пятна радиоизотопа на первичной хрома-
тограмме. Метод радиоавтографии удобен тем, что с его помощью
можно обнаруживать любое неорганическое или органическое ве-
щество, молекула которого содержит радиоизотоп. ,.
В том случае, когда нет возможности подобрать реактивы, образу-
ющие разноцветные пятна компонентов разделяемой смеси, или ис-
пользовать методы люминесценции и радиоавтографии, применяют
метод «свидетелей». Этот Метод основан на том, что коэффициент
Rf не зависит от присутствия посторонних веществ. Поэтому, если
параллельно с каплей анализируемой смеси на полоску хроматогра-
фической бумаги нанести каплю смеси, содержащей известные ве-
щества, то после проявления хроматограммы можно, сравнивая
положение пятен компонентов анализируемой смеси с положением
пятен известных соединений, идентифицировать неизвестные ве-
щества,
263
Недостатком метода является его громоздкость, необходимость
каждый раз наносить и хроматографировать искусственную смесь.
Поэтому лучшим методом является метод измерения коэффициента
Rf. Сущность этого метода состоит в том, что все анализы прово-
дятся в строго определенных и одинаковых условиях. Тогда необ-
ходимость в нанесении каждый раз искусственной смеси отпадает.
Такая смесь хроматографируется однажды, а затем в тех же усло-
виях проводится хроматографирование анализируемых образцов.
Идентификация производится по значениям Rf. Если два вещества
из анализируемой и контрольной смеси обладают одинаковыми зна-
чениями Rp то, следовательно, они являются идентичными. Не-
достатком метода является тот факт, что величина Rf зависит от
ряда факторов, в том числе от формы и размеров камеры, Ь которой
производится хроматографирование. Поэтому соблюдение строгой
идентичности условий проведения опыта является совершенно обя-
зательным.
Методика количественного анализа. Методы количественного
хроматографического анализа на бумаге могут быть разделены на две
группы: методы, не требующие удаления анализируемого вещества
с бумаги, и методы, основанные на вымывании анализируемых ве-
ществ. Методы, относящиеся к первой группе, основаны на том, что
площадь пятна и интенсивность его окраски зависят от количества
хроматографируемого вещества. Так, например, известно, что если на
хроматографическую бумагу наносить постоянный объем анализиру-
емого раствора, то площади пятен П6ум, получающихся при хрома-
тографировании и проявлении, пропорциональны логарифму концен-
трации анализируемых веществ с [11], т. е.
П6ум = Л1§с + В. (207)
Здесь А и В — эмпирические константы.
Для количественного анализа компонентов смеси на бумагу пред-
варительно наносят равные количества растворов, содержащих оп-
ределенные концентрации веществ, подлежащих затем определению.
После хроматографирования и проявления полученные пятна акку-
ратно очерчивают карандашом и измеряют их площадь планиметром
или путем взвешивания. Проводя опыты с растворами разных кон-
центраций и измеряя для каждой концентрации и каждого вещества
площади полученных пятен, строят калибровочные графики в коор-
динатах 1g с — П6ум. Полученными графиками пользуются при коли-
чественном анализе смесей неизвестных веществ.
Следует иметь в виду, что площадь пятна может зависеть не толь-
ко от концентрации, но и от размера капли, сорта бумаги, качества
растворителя, температуры и других условий опыта. Поэтому толь-
ко при соблюдении одинаковых условий хроматографирования при
калибровке и анализе можно добиться правильных результатов.
Считается, что при соблюдении указанных условий погрешность
метода составляет +5—40% относительных.
264
Более точным является метод измерения интенсивности окраски
пятна. Он основан на том, что концентрация вещества в пятне, а
следовательно, и в исследуемом растворе, и интенсивность окраски
пятна связаны между собой линейно. Поэтому, измеряя интенсив-
ность окраски всего пятна или его максимальной плотности, можно
судить о концентрации вещества в растворе. Количественный ана-
лиз проводят по предварительно полученному калибровочному гра-
фику. Измерения интенсивности окраски пятна производят при по-
мощи специально приспособленного к измерению плотности окраски
денситометра, работающего по принципу фотометрирования прохо-
дящего через хроматограмму светового потока [12, 13].
Ко второй группе методов количественного анализа относится
метод вымывания. Его сущность состоит в том, что полученную хро-
матограмму разрезают на части так, чтобы в каждой находилось
лишь одно пятно. Затем вещество экстрагируют из бумаги и в эк-
стракте измеряют его количество любым из доступных методов.
Для предварительного определения положения каждого пятна на
хроматограмме производят параллельное хроматографирование двух
капель одного и того же раствора. После хроматографирования
одну хроматограмму проявляют, а другую нет. Непроявленную
хроматограмму разрезают в соответствии с результатами проявления
соседней хроматограммы.
Для увеличения количества вещества, получаемого экстракцией
из пятна, производят многократное хроматографирование одной
и той же смеси, вырезают соответствующие места бумаги и подвер-
гают экстракции все пятна данного компонента, полученные во всех
хроматограммах. Таким путем удается собрать количество вещества,
достаточное для всестороннего его изучения. Этот метод является
весьма трудоемким, хотя и более точным, чем рассмотренные выше.
Поэтому его следует применять лишь тогда, когда требуемая точ-
ность не может быть достигнута другими методами, а также в тех
случаях, когда выделенное вещество требуется для дальнейшего изу-
чения его свойств и структуры.
ПРАКТИЧЕСКИЕ РАБОТЫ
Работа 1
ОПРЕДЕЛЕНИЕ КОЭФФИЦИЕНТОВ РАСПРЕДЕЛЕНИЯ
МЕТОДОМ ХРОМАТОГРАФИИ НА БУМАГЕ [9]
Приборы и реактивы
1. Цилиндр с пробкой и крючком для закрепления бумажной полоски.
2. Хроматографическая бумага № 1 или № 2. Полоска длиной 20 см, шириной
2,5 см.
3- . Эксикатор с водой для хранения бумаги.
265
4. Бюкс.
5. Штатив химический с лапкой.
6. Пульверизатор.
7. Раствор н-бутилового спирта, уксусной кислоты и воды, взятых в отноше-
нии 4 : 1 : 5 по объему.
8. Водный раствор исследуемых аминокислот: гистидина, валина и лейцииа
(0,01 эквимолярный раствор).
9. 0,2%-иый раствор нингидрина в ацетоне [9] (хранить в темной склянке).
Цель работы. Определить коэффициенты распределения, трех
аминокислот для системы вода — смесь н-бутилового спирта, уксус-
ной кислоты и воды.
Сущность работы. Для определения коэффициента распределе-
ния вещества между двумя несмешивающимися жидкостями можно
применить уравнение (206), в котором отношение следует за-
менить отношением ~ , т. е. отношением веса подвижной фазы
тя к весу неподвижной фазы ти. В свою очередь, величину тя
можно определить, если знать вес бумажной полоски до хромато-
графирования т6 и содержание в ней влаги в процентах а:
та^. (208)
Величину тп можно определить, если знать вес бумажной полоски
после насыщения ее подвижным растворителем т^ :
тП = тб—тб. (209)
Величину Rf определяют графически, как это показано на рис. 43.
Таким образом, после проведения хроматографического опыта
все величины оказываются экспериментально определенными и рас-
чет величины коэффициента распределения Кр нетрудно сделать по
формуле
<210>
Выполнение работы.Бумажную полоску тщательно высушивают
в сушильном шкафу при 105° С и после высушивания взвешивают в
закрытом бюксе на аналитических весах. Затем полоску бумаги по-
мещают в эксикатор, на дне которого налита вода. Бумагу выдер-
живают в течение суток и после этого взвешивают на аналитических
весах в закрытом бюксе. Из полученной разности в весе бумаги до
и после насыщения ее влагой рассчитывают процентное содержание
влаги а.
Хроматографирование проводят методом восходящей хромато-
графии. Капилляром аккуратно на край полоски бумаги наносят
каплю исследуемого раствора аминокислот, подсушивают ее, нали-
вают на дно цилиндра 10—12 мл растворителя и помещают в цилиндр
бумажную полоску так, чтобы ее нижний край касался раствори-
теля, но капля исследуемого раствора не была бы в него погружена.
Полоску закрепляют в цилиндре, который закрывают пробкой,
266
После прохождения растворителя вдоль полоски бумаги до ее
конца (не доходя 0,5 см до верхнего края) бумагу вынимают, быстро
помещают в бюкс и, закрыв его, взвешивают. Затем полоску сушат
на воздухе, опрыскивают раствором проявителя (раствором нингид-
рина), снова сушат и определяют значения величины Rj для каж-
дого пятна.
Аминокислоты располагаются в следующем порядке: нижнее пят-
но — гистидин, среднее — валин, верхнее — лейцин.
Величины тн и тп определяют по формулам (208) и (209). Вели-
чину а рассчитывают из результатов взвешивания сухой и влажной
бумаги; т6 и тб — по результатам взвешивания влажной бумаги и
бумаги, насыщенной растворителем. Все измеренные и рассчитанные
данные заносят в таблицу и по ним вычисляют значение коэффициен-
та распределения /(р.
Работа 2
КАЧЕСТВЕННЫЙ АНАЛИЗ СМЕСИ КАТИОНОВ
МЕТОДОМ ХРОМАТОГРАФИИ НА БУМАГЕ [14]
Приборы и реактивы
1. Пробирка для хроматографирования с пробкой.
2. Хроматографическая бумага № 3 или 4. Полоска шириной 1,5 см,
длиной 15 см.
3. Штатив для пробирок.
4. Капиллярная пипетка.
5. Пинцет.
6. Пульверизатор.
7. Раствор, содержащий ионы железа и кобальта, по 2 мг/мл каждого иона.
8. Растворитель: смесь н-бутилового спирта (80% по объему) и концентриро-
ванной соляной кислоты (20%).
9, Проявитель: спиртовой раствор роданида аммония (1%-ный).
Цель работы. Качественное определение смеси катионов.
Сущность работы. Смесь двух-трех катионов может быть раз-
делена при помощи одномерной хроматографии на бумаге. Для раз-
деления следует подобрать подходящую подвижную фазу, а для
проявления —- соответствующий проявитель. Для разделения ка-
тионов обычно применяют смесн органических растворителей и
воды, для проявления--водные или спиртовые растворы неорга-
нических и органических соединений, образующих окрашенные
вещества с катионами. По цвету окраски каждого пятна, полу-
ченного после хроматографирования и проявления, устанавливают
качественный состав катионов.
Выполнение работы. Каплю исследуемого раствора наносят пи-
петкой на полоску бумаги на расстоянии 6 мм от края и высушивают.*
В пробирку для хроматографирования вливают 0,5 мл растворите-:
ля, вводят в нее полоску бумаги, которую укрепляют так, чтобы ниж-
267
ний ее конец касался растворителя, а края полоски не касались
стенок пробирки. Верхний конец полоски закрепляют на пробке,
которой закрывают пробирку. Анализ продолжается 2 — 3 ч. Ког-
да фронт растворителя достигнет верхнего края полоски бумаги
(не дойдет на 5 мм до верхнего края), опыт прекращают, полоску
бумаги вынимают и высушивают в сушильном шкафу при 110° С.
Высушенную бумагу опрыскивают из, пульверизатора раствором
роданида аммония и снова высушивают. В случае присутствия ио-
нов Fe3+ и Со2+ после опрыскивания появляются окрашенные пят-
на: красное соответствует иону железа, голубое — иону кобальта.
Работа 3
КАЧЕСТВЕННЫЙ АНАЛИЗ СЛОЖНОЙ СМЕСИ КАТИОНОВ
МЕТОДОМ ДВУМЕРНОЙ ХРОМАТОГРАФИИ НА БУМАГЕ [9]
Приборы и реактивы
1. Сосуд для хроматографирования (батарейный стакан, вегетационный
сосуд) с плотно закрывающейся крышкой.
2. Приспособление для закрепления бумаги.
3. Хроматографическая бумага № 3 или № 4, размером 25 X 25 см.
4. Капиллярная пипетка.
5. Пульверизатор.
6. Растворители: 1) ацетон, содержащий 5% воды и 8% концентрированной
соляной кислоты; 2) н-бутиловый спирт, насыщенный 0,1 н. раствором соляной
кислоты.
7. Проявитель: насыщенный спиртовой раствор 8-оксихииолина.
8. Раствор аммиака (25%-ный).
9. Исследуемый водный раствор ионов Ni2 + , Со2+ , Fe3+, Cu2 + , Cd2+ и Hg3 +
(10 мг/мл каждого иона) (для приготовления берутся хлориды).
Цель работы. Показать, что применение двумерной хрома-
тографии позволяет анализировать сложные смеси, не разделяе-
мые одномерной хроматографией.
Сущность работы. Если хроматографирование сложной смеси
проводить при помощи одной подвижной фазы, то полного раз-
деления достичь бывает трудно. Поэтому . такие смеси обычно
разделяют при помощи двумерной хроматографии, сущность ко-
торой состоит в том, . что сначала смесь хроматографируют
одним растворителем, а затем лист бумаги поворачивают на 90°
и хроматографирование производят другим растворителем. Раз-
деление смеси в этом случае оказывается значительно лучшим.
Выполнение работы. Хроматографическую бумагу предваритель-
но выдерживают в эксикаторе в парах воды. Затем в левый нижний
угол квадратного листа бумаги наносят каплю исследуемого раство-
ра и подсушивают ее. Бумагу помещают в держатель, который встав-
ляют в сосуд для хроматографирования. На дно сосуда наливают
первый растворитель. Бумагу погружают в растворитель на глуби-
ну не более 1 см, а каплю раствора наносят на расстоянии 5 см как
от левого, так и от нижнего краев бумаги. Сосуд плотно закрывают
268
и оставляют стоять до тех пор, пока растворитель не поднимется на
высоту 20—22 см (в зависимости от сорта бумаги процесс продол-
жается 2—4 ч). Хроматограмму вынимают, высушивают и повора-
чивают на 90° так, чтобы ее левый край оказался внизу, закрепляют
в держателе и помещают в другой сосуд, на дно которого наливают
второй растворитель. Сосуд плотно закрывают и хроматографиро-
вание повторяют (процесс также протекает 2—4 ч). Затем хромато-
грамму извлекают из камеры, высушивают и проявляют, опрыски-
вая спиртовым раствором 8-оксихинолина. После опрыскивания бу-
магу выдерживают некоторое время над парами аммиака. При этом
возникают окрашенные пятна, по цвету и расположению которых
Проводят качественный анализ катионов.
Расположение зон катионов должно быть следующим: вдоль
линии подъема первого растворителя располагаются зоны катионов
(снизу вверх): никеля, кобальта, меди, железа. При повторном хро-
матографировании в направлении, перпендикулярном первому,
образуются зоны катионов (слева направо): кадмия (над зоной желе-
за) и ртути (в верхнем правом углу).
Работа 4
КАЧЕСТВЕННЫЙ АНАЛИЗ ПРОСТЕЙШИХ АМИНОКИСЛОТ
НА КРУГОВОЙ ХРОМАТОГРАММЕ
Приборы и реактивы
1. Эксикатор с приспособлением для хроматографирования на бумажных
кружках.
2. Хроматографическая бумага № 1. Кружок диаметром 10—12 см.
3. Капиллярная пипетка.
4. Пинцет.
5. Пульверизатор.
6. Растворитель: составляют смесь из 4 объемов н-бутилового спирта, 1 объема
уксусной кислоты и 5 объемов воды. Смесь тщательно взбалтывают и после рас-
слоения применяют верхний слой.
7. Проявитель: раствор нингидрина в водонасыщенном н-бутиловом спирте
(0,25%-ный).
8. Исследуемый раствор аминокислот (а-аланин и аспарагиновая кислота
в концентрации 0,01 моль/л каждой).
Цель работы. Качественное определение аминокислот в смеси.
Освоение одного из методов хроматографии на бумаге.
Сущность работы. Исследуемая смесь наносится в центре бумаж-
ного круга, а движение растворителя и зон компонентов разделя-
емой смеси происходит от центра к периферии. После проявления
такой хроматограммы образуются окрашенные концентрические
кольца.
Выполнение работы. Перед хроматографированием следует под-
готовить бумагу. С этой целью вырезанный кружок бумаги диамет-
ром 10—12 см обрабатывают раствором 8-оксихинолина или раст-
вором трилона Б для удаления следов катионов металлов. Раствор
8-оксихинолина готовят растворением его в смеси, состоящей из 4
269
частей «-бутилового спирта,. 1 части ледяной уксусной кислоты И
1 части воды по объему. Концентрация 8-оксихинолина должна быть
равна 0,1 вес. %. Раствор трилона Б должен быть 1%-ным.
Для удаления катионов бумагу опускают на 1—2 мин в раствор
8-оксихинолина, затем ее подсушивают и помещают в хроматогра-
фическую камеру, пропуская растворитель до полного удаления тем-
ноокрашенных соединений 8-оксихинолина с катионами металлов.
Промытую бумагу сушат на воздухе. Подготовка бумаги требует
36—40 ч.
В подготовленном кружке бумаги вырезают полоску, как это пока-
зано на рис. 45, отгибают ее и на расстоянии 0,5 см от центра круж-
ка наносят каплю исследуемой смеси. Бумагу помещают в эксика-
тор, на дно которого наливают растворитель, причем отогнутую
полоску бумаги опускают в растворитель, чтобы он поступал к цент-
ру кружка. Эксикатор плотно закрывают. По окончании хромато-
графирования бумагу вынимают, высушивают и опрыскивают
проявителем. Получают два концентрических окрашенных кольца:
первое из них относится к аспарагиновой кислоте, а второе к
а-аланину. После проявления производят качественный анализ.
Р а б о т а 5
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ СМЕСИ ЛИМОННОЙ
И ЯБЛОЧНОЙ КИСЛОТ МЕТОДОМ ВЫМЫВАНИЯ ПЯТЕН [9]
Приборы и реактивы
1. Прибор для хроматографирования методом нисходящей хроматографии.
2. Хроматографическая бумага № 2. Полоска шириной 12 см, длиной 25 см.
3. Выпарительные чашки — 2 шт.
4. Пинцет.
5. Капиллярная пипетка и микробюретка.
6. Пульверизатор.
7. Растворитель: смесь н-бутилового спирта, муравьиной кислоты и воды
в отношении 9 : 1 : 4 по объему.
8. Проявитель: 0,04% -ный спиртовой раствор бромфенолового синего.
9. Водный раствор исследуемых кислот (5 мг/мл каждой кислоты).
10. Фенолфталеин.
11. Карбонат натрия.
12. Спирт этиловый (96%-иый).
13. Раствор щавелевой кислоты (2%-ный).
Цель работы. Показать возможности хроматографии на бумаге
для количественного определения состава смесей. Количественно про-
анализировать простую смесь кислот.
Сущность работы. Одним из методов количественного определе-
ния компонентов смеси после ее хроматографического разделения
является метод вымывания, сущность которого состоит в том, что
сначала смесь разделяют на бумаге, а затем вырезают пятна, соот-
ветствующие компонентам анализируемой смеси, и' их содержимое
экстрагируют. После экстракции количество вещества определяют
любым химическим, физико-химическим или физическим методом.
270
Выполнение работы. Прежде всего готовят бумагу для хромато-
графирования. Для этого вырезанную полосу бумаги № 2 отмывают
1%-ным водным раствором щавелевой кислоты, промывают водой
и сушат на воздухе. На высушенную бумагу на расстоянии 5 см
от верхнего края наносят в ряд 5 капель исследуемого раствора ли-
монной и яблочной кислот. Расстояние между каждой каплей должно
быть равно 2 см, причем все капли должны быть расположены на
одинаковом расстоянии от края полосы, с тем чтобы зоны веществ
могли двигаться параллельно друг другу. Хроматографирование
производят по способу нисходящей хроматографии и прекращают
его, когда фронт проявителя пройдет полосу бумаги (он не должен
доходить 1—2 см до нижнего края полосы).
После окончания хроматографирования полосу бумаги вынима-
ют, высушивают на воздухе и проявляют следующим образом:
полосу раскладывают на стекле, покрывают ее листом плотной бу-
маги так, чтобы открытым оставался один ряд пятен, например,
правый. В таком положении бумагу опрыскивают раствором проя-
вителя, причем на синем фоне проявляются желтые пятна кислот.
Порядок расположения кислот (сверху вниз) следующий: лимонная,
яблочная.
После высушивания проявленной хроматограммы из непрояв-
ленной ее части вырезают места, соответствующие лимонной и яб-
лочной кислотам, отдельно для каждой кислоты, и приступают к
экстракции кислот.
Для этого вырезанные полосы бумаги разрезают на мелкие части
и три раза экстрагируют порциями горячей воды по 10—15 мл.
Экстракт лимонной кислоты собирают в одну выпарительную чаш-
ку, а яблочной — в другую. Растворы упаривают на водяной бане
до объема около 1 мл в каждом. Полученные вытяжки оттитровыва-
ют из микробюретки 0,001 н. раствором фенолфталеината натрия.
Из пошедшего на титрование объема фенолфталеината натрия необ-
ходимо вычесть объем раствора, израсходованного на контрольное
определение. Контрольное определение проводят следующим обра-
зом: из непроявленной и. не содержащей кислот части бумаги выре-
зают полосу, равную по площади полосам, взятым для экстракции
каждой из кислот. Из этих полос извлекают горячей водой присут-
ствующие в бумаге другие кислоты, экстракт упаривают и титруют,
как указано выше. По полученным данным рассчитывают содержа-
ние лимонной и яблочной кислот в мг!мл.
Для получения фенолфталеината натрия растворяют при кипя-
чении 5 г чистого фенолфталеина в 100 мл 20%-ного раствора карбо-
ната натрия. Полученный темно-красный раствор упаривают на во-
дяной бане досуха и остаток растворяют небольшими порциями
96%-ного этилового спирта при нагревании. Извлечение повторяют
до тех пор, пока новые порции спирта не перестанут окрашиваться
в розовый цвет. Красный остаток растирают в порошок и хранят в
склянке с притертой пробкой. Для приготовления титрующего раст-
вора берут 0,35 г этого остатка и растворяют в 1 л воды. Титр раст-
вора устанавливают по серной кислоте.
Конец титрования определяется по появлению неисчезающей
окраски самого раствора фенолфталеината натрия [151.
Работа 6
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ГЛЮКОЗЫ В РАСТВОРЕ
МЕТОДОМ ИЗМЕРЕНИЯ ПЛОЩАДИ ПЯТНА НА БУМАГЕ [9]
Приборы и реактивы
1. Установка для хроматографирования нисходящим способом (см. рис. 44).
2. Хроматографическая бумага № 4, длина полосы 25 см, ширина 10—12 см.
3. Сушильный шкаф.
4. Пульверизатор.
5. Пинцет.
6. Растворитель: смесь н-бутилового спирта, уксусной кислоты и воды в соот-
ношении 4 : 1 : 5 по объему.
7. Проявитель: спиртовой 1%-ный раствор резорцина и водный 0,2 н. раствор
соляной кислоты. Перед употреблением оба раствора смешивают в отношении
1:1 по объему.
8. Растворы глюкозы: 20; 15; 10 и 5 вес. %.
Цель работы. Получить калибровочный график зависимости пло-
щади пятна на хроматограмме от концентрации глюкозы в растворе
и определить ее содержание в исследуемом растворе.
Сущность работы. В связи с тем, что площадь пятна на хрома-
тограмме, образуемого определяемым компонентом при хроматогра-
фировании раствора, линейно связана с логарифмом концентрации
этого компонента (уравнение 207), возникает принципиальная воз-
можность количественного определения концентрации растворен-
ного вещества. Для этой цели следует предварительно подвергнуть
хроматографированию ряд растворов с разными, но известными кон-
центрациями компонента, измерить площадь полученных на бумаге
пятен и построить калибровочный график в координатах: площадь
пятна — логарифм концентрации раствора. Пользуясь этим гра-
фиком, определяют концентрацию исследуемого .раствора.
Выполнение работы. На хроматографическую бумагу по одной
линии наносят капли растворов глюкозы известных концентраций
на расстоянии от края и друг от друга по 2 см. Объем капли должен
составлять приблизительно 5 мкл, причем объемы всех капель дол-
жны быть строго одинаковыми.
После подсушивания бумаги ее помещают в установку для хро-
матографирования методом нисходящей хроматографии. В связи
с тем, что сахара имеют небольшую скорость передвижения, раство-
ритель пропускают через бумагу в течение 24 ч. Затем бумагу выни-
мают из камеры, высушивают и проявляют. После проявления пов-
торное высушивание производят в сушильном шкафу при 80—90° С.
При этом на бледно-розовом фоне появляются цветные пятна, выз-
272
ванные присутствием глюкозы. Площадь каждого пятна тщательно
измеряют. Измерение площади можно производить либо планимет-
ром, либо методом взвешивания. Полученные данные наносят на
график в координатах: площадь пятна — логарифм концентрации
растворов. Через нанесенные точки проводят прямую линию.
Получают от преподавателя раствор глюкозы неизвестной кон-
центрации и опыт повторяют. После измерения площади получен-
ного пятна, пользуясь калибровочным графиком, определяют кон-
центрацию изучаемого раствора.
Следует иметь в виду, что метод позволяет получать удовлетво-
рительные результаты только при условии очень тщательного вы-
полнения всех операций, особенно измерения площади пятен.
Работа 7
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ СМОЛ В НЕФТЕПРОДУКТАХ
МЕТОДОМ ЛЮМИНЕСЦЕНТНОЙ ХРОМАТОГРАФИИ НА БУМАГЕ
Приборы и реактивы
1. Ультрахемископ Брумберга.
2. Стакан на 50 мл.
3. Аппарат Сокслетта на 100 мл.
4. Хроматографическая бумага № 4, длина полосы 15 см, ширина 1 см.
5. Штатив химический с лапкой.
6. Смесь спирта и бензола в соотношении 1: 1 по объему.
7. Исследуемые смолы (0,001%-ный раствор в спиртобензольной смеси).
Цель работы. Качественное определение содержания смол в мас-
лах; знакомство с люминесцентным хроматографическим методом.
Сущность работы. Применение ультрафиолетовой люминесцен-
ции расширяет возможности качественного определения веществ
методом бумажной хроматографии. Если компоненты разделенной
смеси обладают способностью люминесцировать под действием уль-
трафиолетового облучения, проявление бумажной хроматограммы
осуществляется при освещении высушенной после хроматографиро-
вания бумажной полоски ультрафиолетовым светом. Освещение
производится ртутно-кварцевой лампой через специальный фильтр,
задерживающий видимые лучи. По цвету люминесценции можно
судить о природе веществ, подвергшихся хроматографированию.
Этот метод нашел применение в анализе тяжелых нефтепродуктов и
битумов.
Весьма удобным для получения люминесценции на бумаге яв-
ляется ультрахемископ Брумберга [10].
Выполнение работы. Подготавливают хроматографическую бу-
магу (освобождают ее от возможных люминесцирующих примесей).
Для этого полоски бумаги помещают в аппарат Сокслетта и в тече-
ние 6 ч экстрагируют смесью спирта и бензола (1 : 1 по объему).
Подготовленную и пропитанную спиртобензольной смесью полоску
273
бумаги опускают в 0,001 %-ный раствор исследуемой смолы в этой
же смеси, которую наливают на дно стаканчика. Полоску бумаги
закрепляют в штативе так, чтобы она находилась в вертикальном
положении и нижним концом касалась дна стаканчика. Хромато-
графирование производят в течение 12—14 ч. За это время лишний
растворитель и растворитель с бумаги улетучиваются. Сухую по-
лоску бумаги помещают на стекло ультрахемископа, включают при-
бор в сеть и наблюдают свечение.
Смолы, богатые парафино-нафтеновыми углеводородами, напри-
мер, извлеченные из светлых масел, светятся голубым свечением,
чистые смолы, а также смолы с примесью ароматических углеводо-
родов — желтым свечением.
Для анализа рекомендуется брать смолы, извлеченные из нефти
или нефтяных масел хроматографическим методом на силикагеле.
ЛИТ ЕРАТ УРА
Использованная литература
1. A. J. Р. Martin, R. L. М. Synge. Biochem. J., 35, 1358, 1941.
2. Н. А. Фукс. Усп. хим,, 17, 45, 1948.
3. S. Datta, С. Dent, Н. Н а г г i s. Sei., 112, 621, 1950.
4. L. Butter. Analyst., 75, 37, 1950.
5. Г. Д. Елисеева. Изв. высш. уч. заведений. «Хим. и хим. техн.»,
№ 5, 35, 1958.
6. Е. D urrum. J. Am. Chem. Soc., 72, 2943, 1950.
7. H. Strain. Anal. Chem., 23, 25, 1951.
8. M. Лед ер ер. Введение в электрофорез. ИЛ, М., 1955.
9. К. М. О л ь ш а н о в а, М. А. Потапова, В. Д. Копы-
лова, Н. М. Морозова. Руководство по ионообменной, распределитель-
ной и осадочной хроматографии. Изд-во «Химия», М., 1965.
10. Е. М. Б р у м б е р г. ДАН СССР, 72, 885, 1950.
11. R. Fischer, D. Parsons, G. Morrison. Nature, 161, 764,
1948; R. Fischer, D. Parsons, R. Holmes. Nature, 164, 183, 1949.
12. R. Block. Science, 108, 608, 1948.
13. H« Bull, J. Hahn, V. Baptist. J. Am. Chem. Soc., 71, 550,
1948.
14. А. А. Морозов, H. А. К и с e л ь, H. Л. О л e н о в и ч. Практи-
ческое руководство по хроматографическому анализу. Изд-во Одесск. ун-та, 1961.
15. И. М. К о р е н м а н. Количественный микрохимический анализ. Гос-
химиздат, М., 1949.
Рекомендуемая литература
1. «Хроматография». Сб. 1, ИЛ, М., 1949.
2. В. В. Рачинский. Усп. хим., 19, № 4, 445, 1950.
3. В. В. Рачинский, Т. Б. Гапон. Хроматография в биологии.
Изд-во АН СССР, М„ 1953.
4. В. С. Асатиани. Усп. хим., 22, № 3, 289, 1953.
5. Р. Блок, Р. Л е с т р а н ж, Г. Цвейг. Хроматография на бумаге.
ИЛ, М„ 1954.
6. «Исследования в области ионообменной, распределительной и осадочной
хроматографии». Изд-во АН СССР, М., 1959.
7. «Хроматография иа бумаге». Под ред. И. М. X а й с а и К- М а ц е к а.
ИЛ, М„ 1962.
274
СПИСОК ПРИНЯТЫХ ОБОЗНАЧЕНИЙ
а — количество сорбированного вещества, соответствующее равновесному
состоянию при заданной концентрации
а° — то же в расчете на 1 г сорбента
— предельное количество сорбированного вещества
с — концентрация
с0 — начальная концентрация
са — концентрация вещества в слое сорбента
сд — концентрация вытеснителя
Смаке — концентрация, соответствующая максимуму на хроматографической
кривой
смин — концентрация, соответствующая минимуму на хроматографической кри-
вой при неполном разделении двух компонентов
d — диаметр колонки
d^ — диаметр зерна сорбента
dg — насыпной вес сорбента, г/см3
D — коэффициент молекулярной диффузии
D — коэффициент диффузии в ионите
— эффективный коэффициент диффузии
Е — напряжение электрического поля
F — константа Фарадея
АЕ — изменение изохорно-изотермического потенциала при растворении
g — вес сорбента
h — высота хроматографического пика
Н — высота теоретической тарелки
АН — изменение энтальпии при растворении
k — константа Больцмана
Ki — первый критерий разделения
К2 — второй критерий разделения
Аа в — константа ионного обмена для ионов А и В
Кс — коэффициент селективности
Ар — коэффициент распределения
Kh — калибровочный коэффициент по высоте хроматографического пика
КГ[ — то же по площади хроматографического пика
Kvgh— то же по произведению удерживаемого объема на высоту хроматографи-
ческого пика
I — длина колонки или слоя сорбента
/МИн — минимальная длина слоя сорбента, необходимая для разделения смеси
О — коэффициент обогащения
<7; — количество наносимого на сорбент вещества
Q — теплота сорбции
г — радиус зерна сорбента
гк — радиус капилляра
А — газовая постоянная
275
R/.- — относительная скорость перемещения компонентов смеси в газо-жид-
костной хроматографии
—отношение скорости движения зоны компонента к скорости движения
жидкой подвижной фазы
S — поперечное сечение колонки
5адс — поверхность сорбента
SCB — свободное поперечное сечение колонки
Sm — емкость ионита
Sp — приращение количества сорбированного иона при переходе от исход-
ного к равновесному состоянию
Sn — поперечное сечение подвижной жидкой фазы
S„ — то же неподвижной жидкой фазы
AS — изменение энтропии при растворении
t — время
Т — абсолютная температура
7\ар — характеристическая температура
ис — линейная скорость перемещения вещества вдоль слоя сорбента
fp — объемная скорость поступления раствора или растворителя
V — объем газа или раствора
V" — объем газа или раствора, приходящийся на единицу длины сорбента
Vo — свободный объем колонки
— объем газа в сорбционном слое, приходящийся на единицу длины
сорбента
V„ — объем раствора или газа, введенного в колонку для анализа
Vr — удерживаемый объем
— удельный удерживаемый объем
V® — удельный объем пор сорбента
Гэфф — эффективный объем тарелки
w — скорость изменения температурного поля (линейная скорость движения
печи вдоль слоя сорбента)
— смещение фронта растворителя
а — линейная скорость газа-иосителя
у — коэффициент активности раствора
Г — коэффициент Генри
А0 — удельная пористость сорбента
е — диэлектрическая постоянная
х — доля свободного сечения колонки
Л — эквивалентная электропроводность
ц — ширина полосы иа хроматограмме
— полуширина полосы на хроматограмме
тд — время диффузии
tr — время удерживания
о) — объемная скорость газа, см^/мин
ОГЛАВЛЕНИЕ
Стр.
Предисловие.......................................................... 3
Введение......................................................... 5
Глава 1. Молекулярная адсорбционная хроматография жидких
веществ .......................................................... 12
Практические работы.............................................. 42;
Работа 1. Определение адсорбционной активности силикагеля . . ; 42
Работа 2. Определение изотермы адсорбции уксусной кислоты на
активированном угле фронтальным хроматографическим методом . . 44
Р а бота 3. Хроматографический анализ керосина комбинированным
методом......................................................... 47
Ра б о т а 4. Хроматографическое разделение смеси парафино-нафте-.
невых. углеводородов, выделенных из керосина .................. 50
Работа 5. Хроматографическое разделение смеси нормальных и
изопарафиновых углеводородов на молекулярных ситах.............. 52
Работа 6. Хроматографическое разделение смеси транс- и цис-
азобензола на активированной окиси алюминия..................... 53
Работа 7. Хроматографическое определение алкалоидов в тинк-
туре белладонны................................................. 55
Работа 8. Люминесцентно-хроматографическое изучение состава
нефти....................................................... • 56
Работа 9. Очистка хлорбензола и определение содержания в нем
примеси хлорного железа хроматографическим методом.............. 58
Литература............. . . ..................................... 59
Глава II. Ионообменная хроматография........................... . > 61
Практические работы............................................. . 89
Работа 1. Определение полной динамической обменной емкости
катионита...................................................... 89
Ра бота 2. Определение зависимости обменной емкости катионита
от pH раствора.................................................. 90
Работа 3. Определение некоторых физико-химических н физико-
механических свойств ионитов. .................................. 92
Работа 4. Определение зависимости ионного обмена от полярности
растворителя................................................... 96
Работа 5. Определение меди и цинка при их совместном присут-
ствии на катионите КУ-2......................................... 98
Работа 6. Определение свинца, меди и цинка при их совместном
присутствии на анионите ЭДЭ-10П................................ 100
Работа 7. Отделение фосфат-ионов от некоторых катионов II и III
аналитических групп методом ионообменной хроматографии иа анио-
Работа 8. Концентрирование вещества из разбавленных растворов
методом ионообменной хроматографии.............................. ЮЗ
Работа 9. Получение воды высокой степени чистоты методом
ионообменной хроматографии.................................... Ю4
277
Работа 10. Разделение смеси аминокислот на ионообменных
смолах.......................................................... 106
Работа 11. Разделение смеси ионов лития и натрия при помощи
ионообменной электрохроматографии............................... 107
Литература.......................................................... 109
Глава III. Осадочная хроматография.................................. 111
Практические работы................................................. 126
Работа 1. Качественный анализ смеси катионов Fe34 и Си2 + ме-
тодом колоночной осадочной хроматографии ....................... 126
Работа 2. Качественный анализсмеси катионов Sb3* и Sn2 + мето-
дом колоночной осадочной хроматографии . . . . : . . . . 128
Работа 3. Качественный анализсмеси катионов Hg2 + , Bi3+ и Cu2 +
на ионите в 1-форме. ;...........................................129
Р а б b Т а' 4. 'Качественный анализ смеси катионов Си2+,’Со2 и Ni2 +
методом осадочной хроматографии на бумаге....................... 130
Работа 5. Количественное определение никеля методом осадочной
хроматографии на бумаге........................................ 131
Работа 6. Качественное определение галогенидов методом коло-
ночной осадочной хроматографии................................. 133
Работа 7. Качественный анализ смеси иодид- и бромид-ионов окси-
хроматографическим методом...................................... 134
Литература.......................................................... 135
Г л а в a I V. Адсорбционная газовая хроматография. Хроматермо-
графия ..............................................» •............ 137
Практические работы................................................ 192
Работа 1. Хроматографическое разделение смеси азота и кисло-
рода (воздух) на молекулярных ситах ............................ 192
Работа 2. Хроматографическое определение смеси низкокипящих
горючих газов на угле и молекулярных ситах..................... 193
Работа 3. Хроматографический анализ смеси газов, содержащей
окислы азота................................................... 194
Работа 4. Хроматографическое опредёлёнйе содержания газооб-
разных углеводородов в воздухе.................................. 195
Р.а б о т а- 5. Определение состава смеси газообразных углеводородов
методом хроматермографии........................................ 197
Работа 6. Определение состава смеси газообразных углеводородов
теплодинамическим методом..................... . .........-. 198
Работа 7. Разделение смеси ксилолов методом хроматермографии
в паровой фазе ............................................... 200
Работа 8. Градуировка хроматографа,- ........................... 201
Работа 9. Определение теплоты адсорбции н-бутана на силикагеле
хроматермографическим методом.• . . . . ;....................... 205
Работа 10. Построение изотермы адсорбции пропана по хромато-
графическим даииым ............................................. 206
Литература.......................................................... 208
Глава V. Газо-жидкостиая хроматография............................ 210
Практические работы................................................. 219
Работа 1. Хроматографическое определение смеси газообразных
углеводородов С]—С5............. 219
Работа 2. Определение зависимости величины удерживаемого
объема от числа атомов углерода в молекуле алифатических спиртов 221
Работа 3. Хроматографический анализ смеси углеводородов С5
на колонках, заполненных носителем о различными жидкими фазами , 222
• 278
Работа 4. Хроматографическое разделение смеси изомеров кси-
лола .......................................................... 224
Работа 5. Выбор жидкой фазы для разделения смеси веществ
с близкими температурами кипения............................. 225
Работа 6. Хроматографическое разделение смеси жидких хлормета-
нов ........................................................... 226
Работа 7. Хроматографический анализ фреонов ....... 226
Работа 8. Хроматографическое определение микропримеси ацети-
лена в этилене................................................ 227
Работа 9. Хроматографическое определение микропримеси бута-
нола в сточных водах........................................... 228
Работа 10. Хроматографирование антрацена.................. 229
Работа 11. Расчет состава бинарной смеси по площади пиков ме-
тодом Дж. К. Барлета и Д. М. Смита............................. 230
Работа 12. Идентификация неизвестного вещества по относитель-
ным удерживаемым объемам....................................... 232
Литература......................................................... 234
Глава VI. Новые варианты газо-жидкостной хроматографии . . . 235
А. Капиллярная хроматография...................................... 235
Практические работы................................................ 239
Работа 1. Анализ смеси изомеров ксилола методом капиллярной
хроматографии.................................................. 239
Работа 2. Анализ бытового газа методом капиллярной хроматогра-
фии............................................................ 241
Б; Ступенчатая хроматография....................................... 242
Практические работы................................................ 245
Работа 3. Определение примесей триметилхлорсилана и метил-
трихлорсилана в диметилдихлорсилане методом ступенчатой хромато-
графии......................................................... 245
В. Вакантохроматография........................................... 246
Практические работы................................................ 249
Работа 4. Определение содержания двуокиси углерода в воздухе
методом вакантохроматографии.................................. 249
Работа 5. Определение малых объемов газа методом вакантохрома-
тографии....................................................... 250
Литература......................................................... 251
Глава VII. Распределительная хроматография на бумаге .... 252
Практические работы................................................ 265
Работа 1. Определение коэффициентов распределения мето-
дом хроматографии иа бумаге ................................... 265
Работа 2. Качественный анализ смеси катионов методом хромато-
графии на бумаге............................................... 267
Работа 3. Качественный анализ сложной смеси катионов методом
двумерной хроматографии на бумаге ............................. 268
Работа 4. Качественный анализ простейших аминокислот на
круговой хроматограмме......................................... 269
Работа 5. Количественное определение смеси лимонной и яблоч-
ной кислот методом вымывания пятен............................. 270
Работа 6. Количественное определение глюкозы в растворе мето-
дом измерения площади пятна на бумаге.......................... 272
Работа 7. Определение содержания смол в нефтепродуктах мето-
дом люминесцентной хроматографии на бумаге..................... 273
Литература. . ..................................................... 274
Список принятых обозначений........................................ 275
279
Айвазов Борис Викторович
ПРАКТИЧЕСКОЕ РУКОВОДСТВО ПО ХРОМАТОГРАФИИ
Редактор А. В. Бородина
Художественный редактор Т. М. Скворцова
Технический редактор Л. М. Матюшина
Корректор Е. К. Штурм
Т-10354. Сдано в набор 14/IV 1967 г. Подп. к печати
26, IX 1967 г. Формат 60х90’/»в- Объем 17,5 печ. л.
Уч.-изд. л. 16,89. Изд. № Хим. 311. Тираж 20 000 =>кз.
Цена 74 коп.
Тематический план издательства «Высшая школа»
(вузы и техникумы) на 1968 г. Позиция №43
Москва, К-51, Неглинная ул., д. 29/14,
Издательство «Высшая школа»
Ордена Трудового Красного Знамени
Первая Образцовая типография имени А. А. Жданова
Главполиграфпромв Комитета по печати
при Совете Министров СССР. Москва, Ж-54, Валовая, 28
Заказ № 1561
Отсканировал Семенсченко Владимир
chem_vova@mail. u n iv. kiev. u a: vova2002@mail. ru