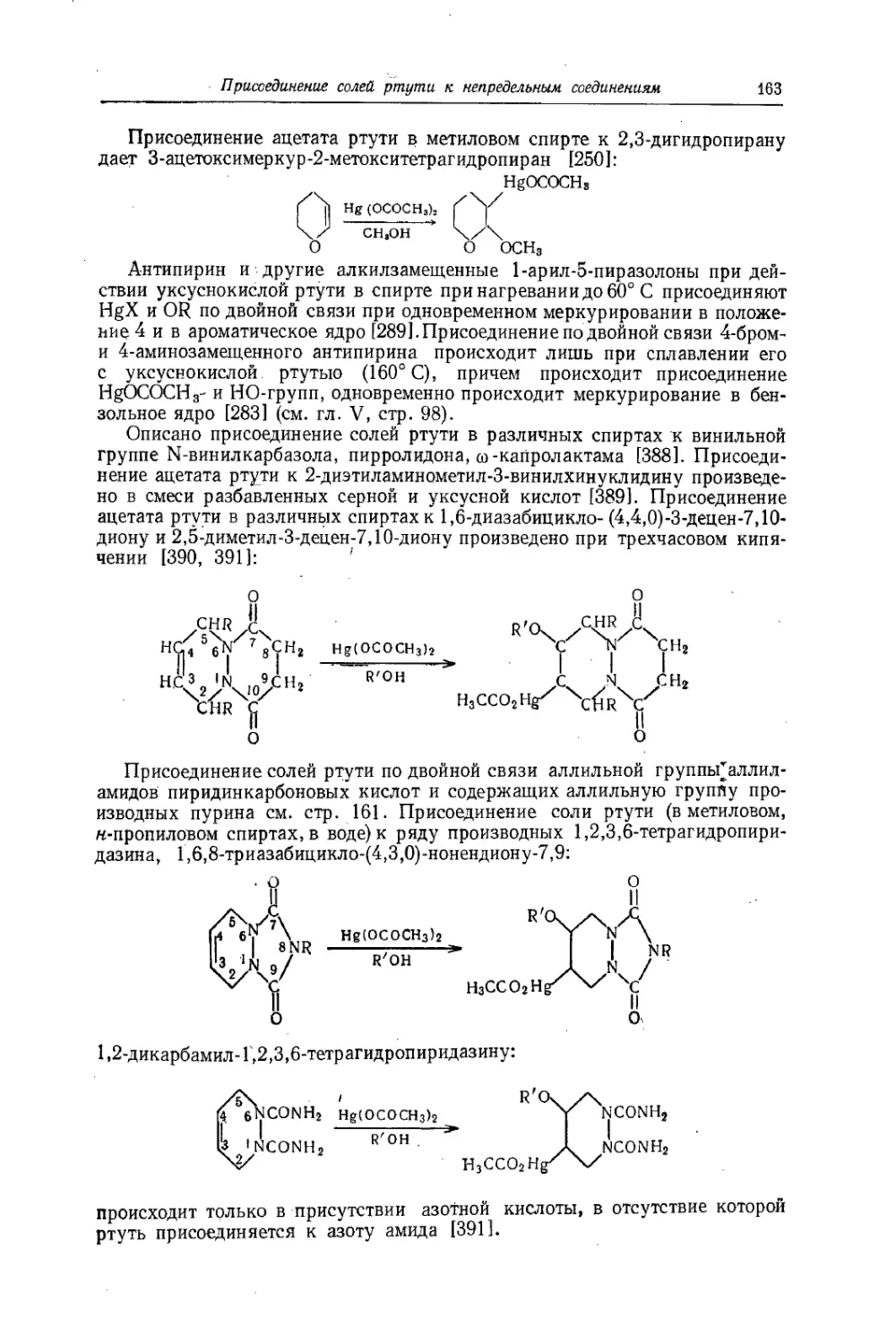
/









































































































































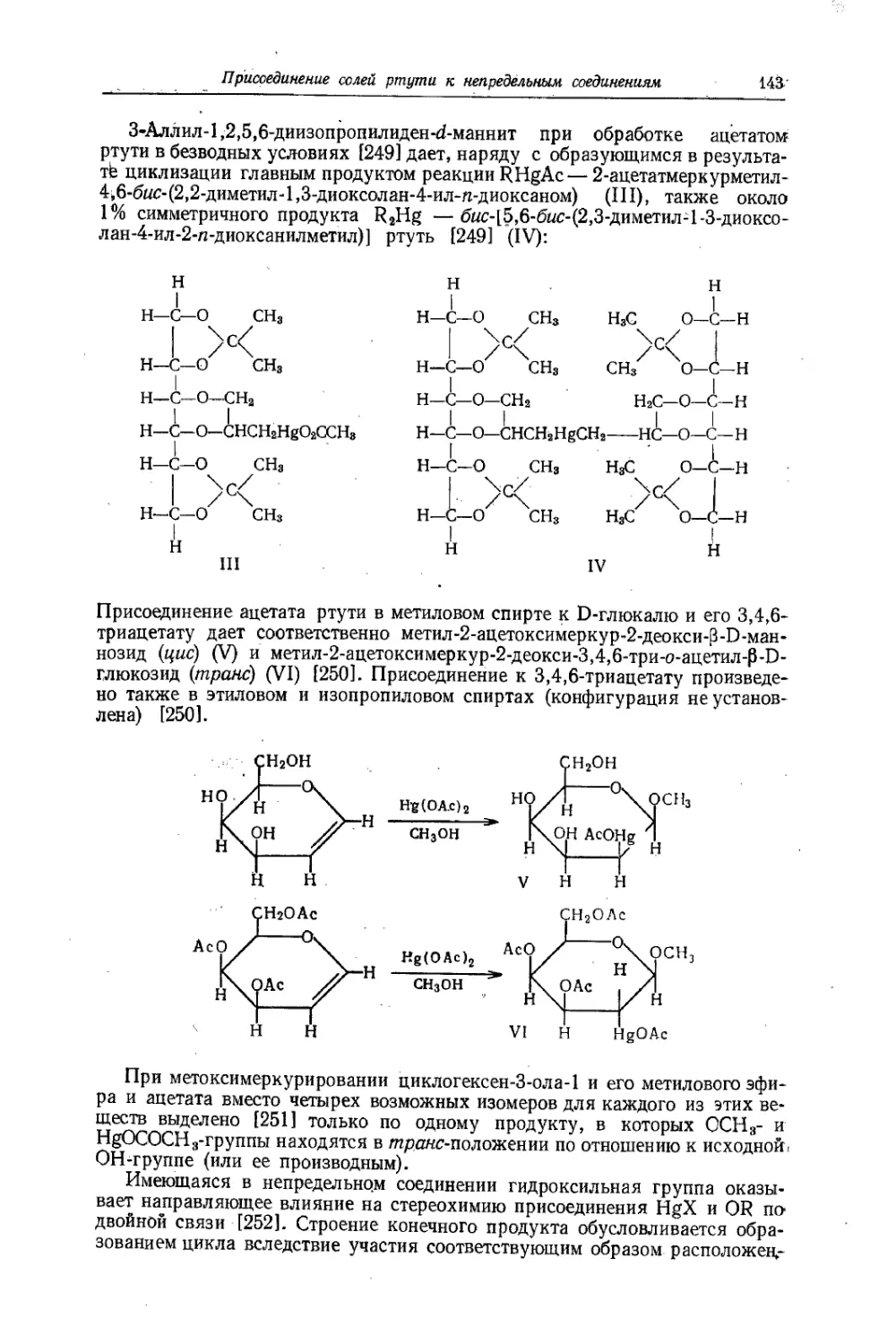
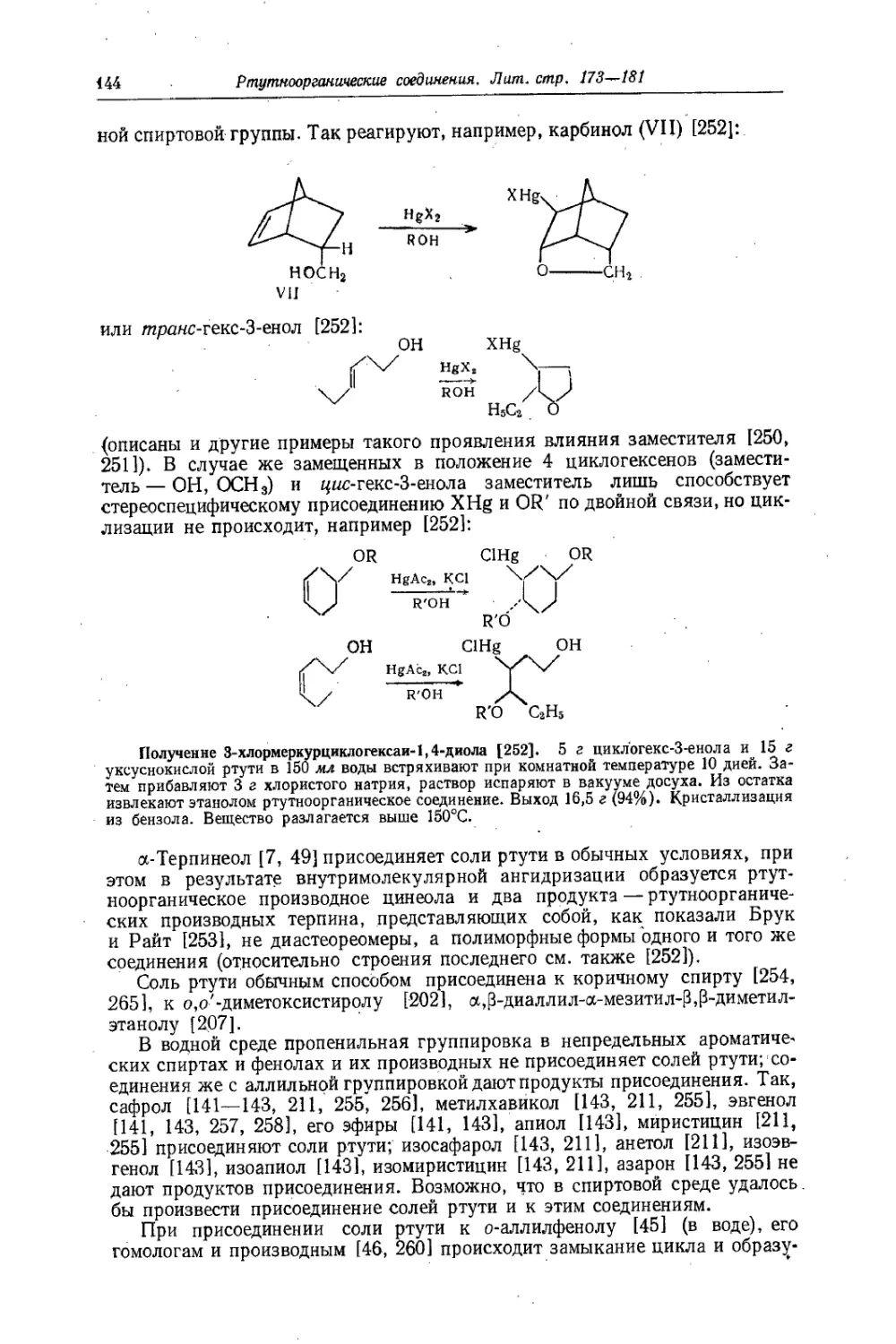











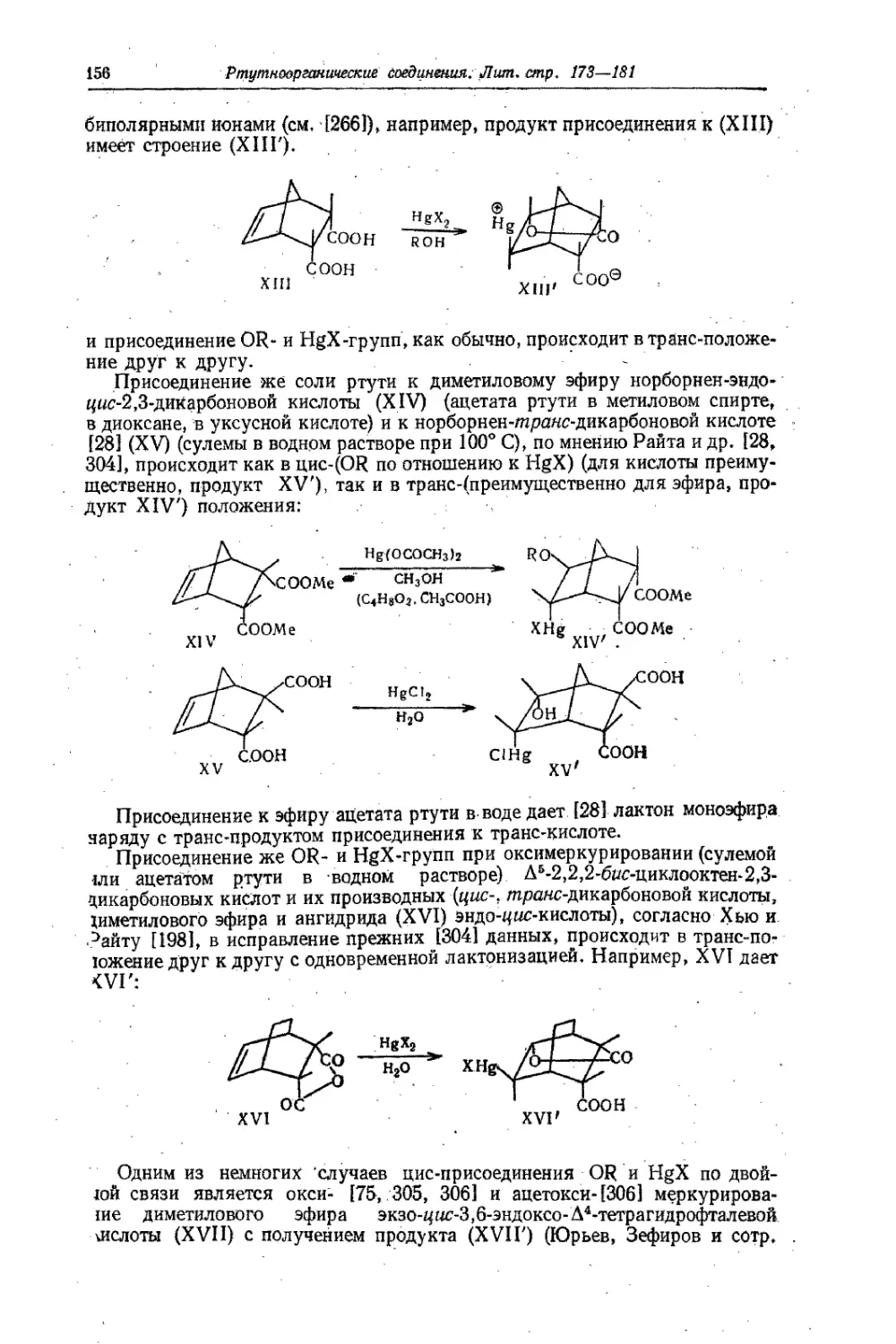
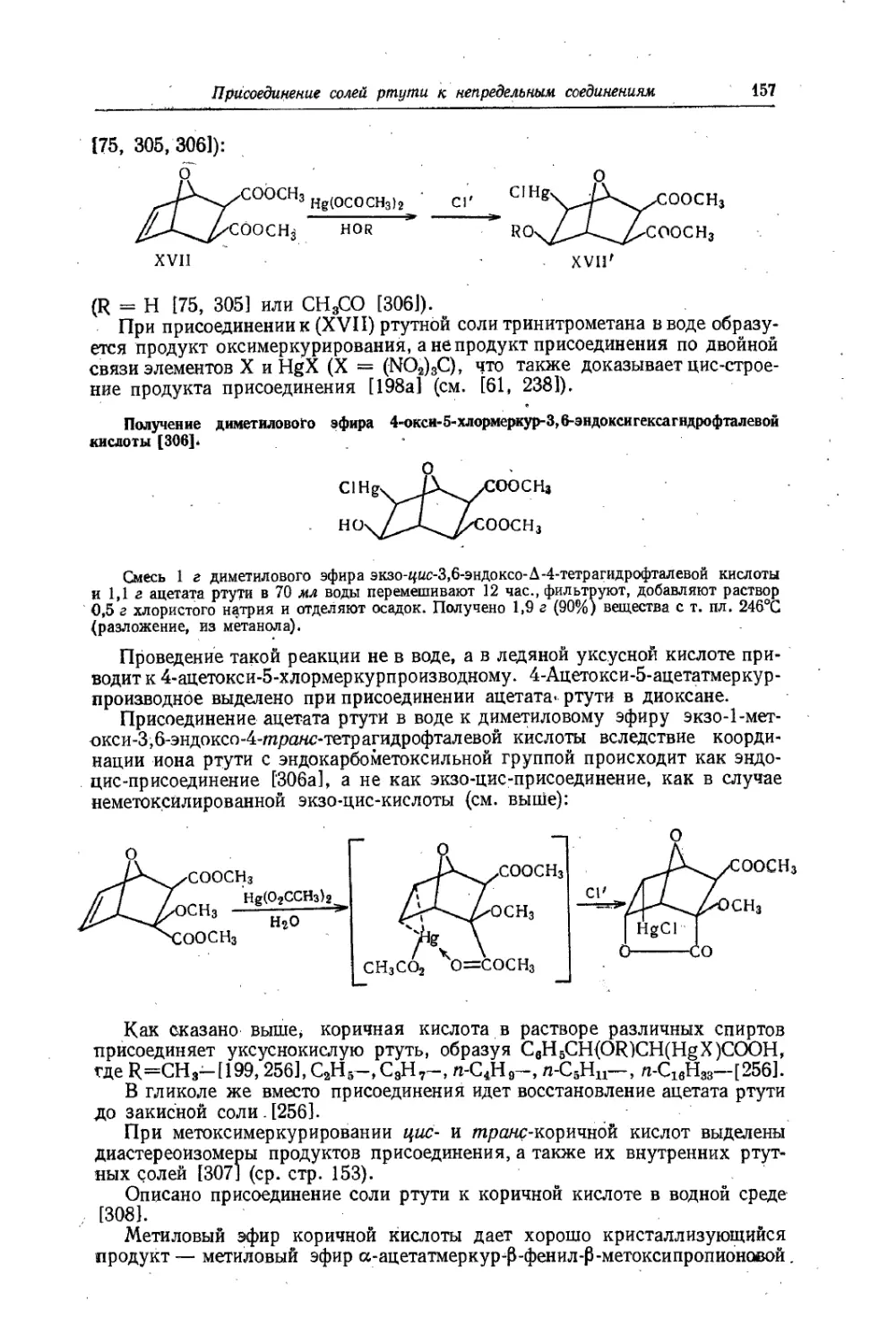




















































































































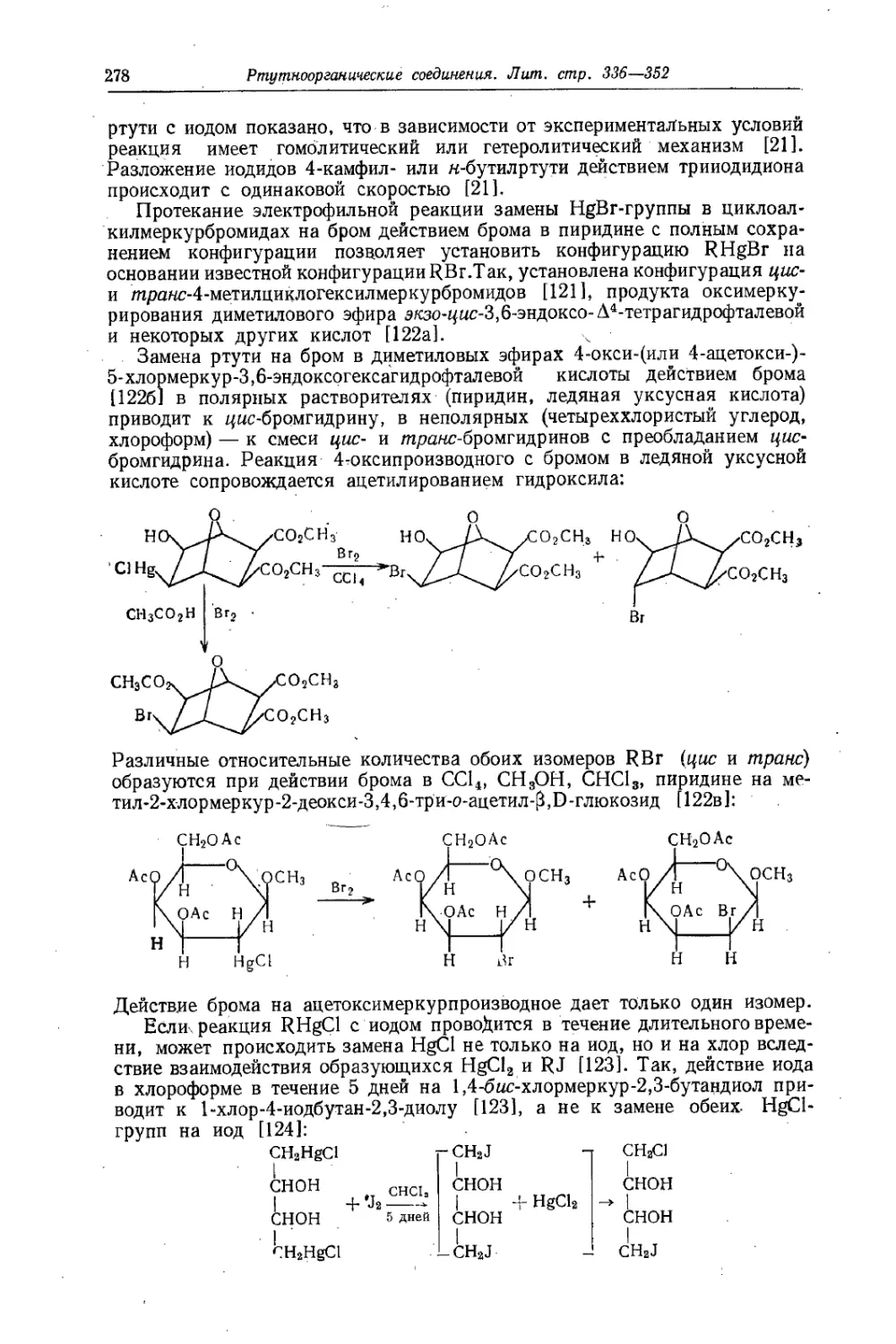

























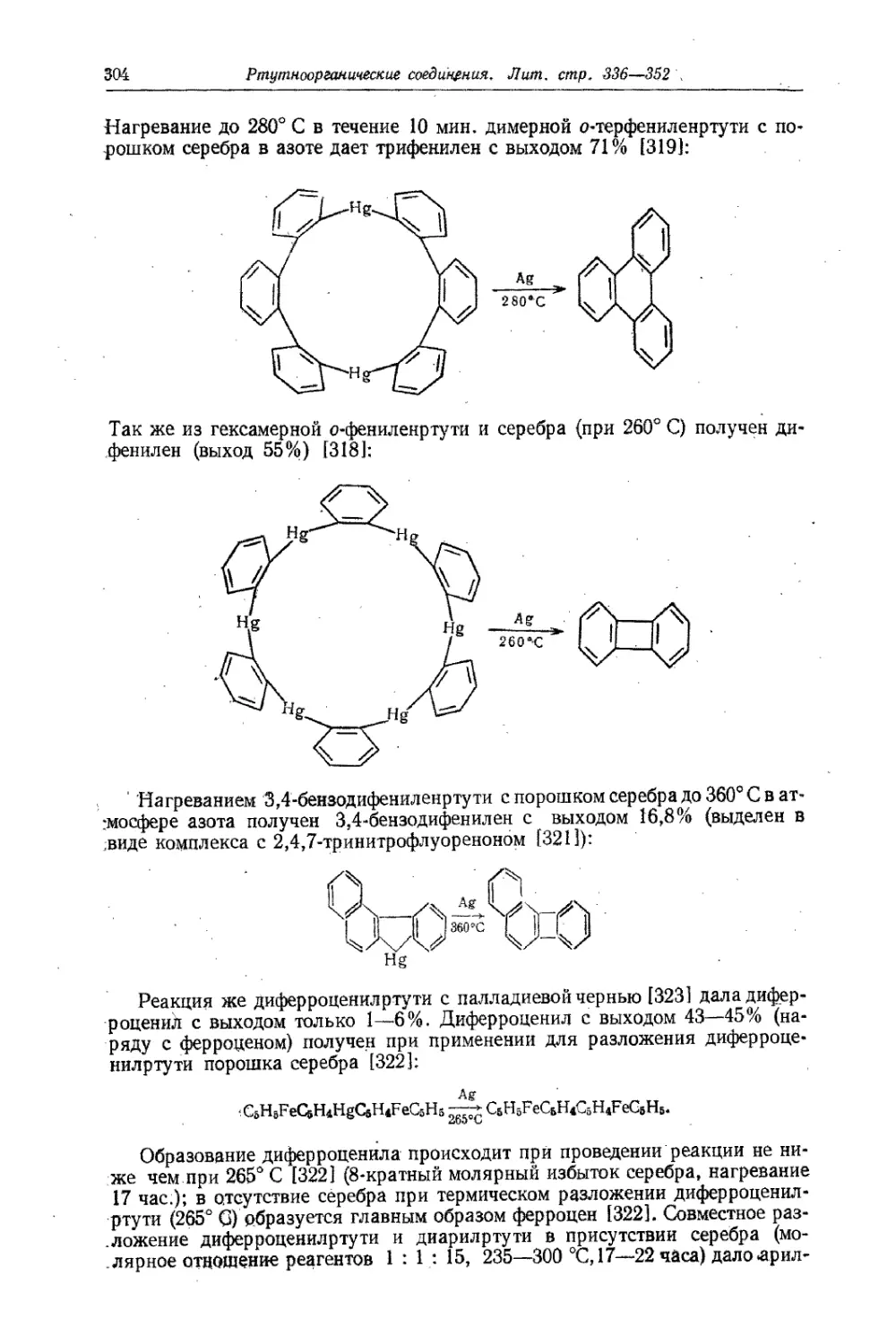












































































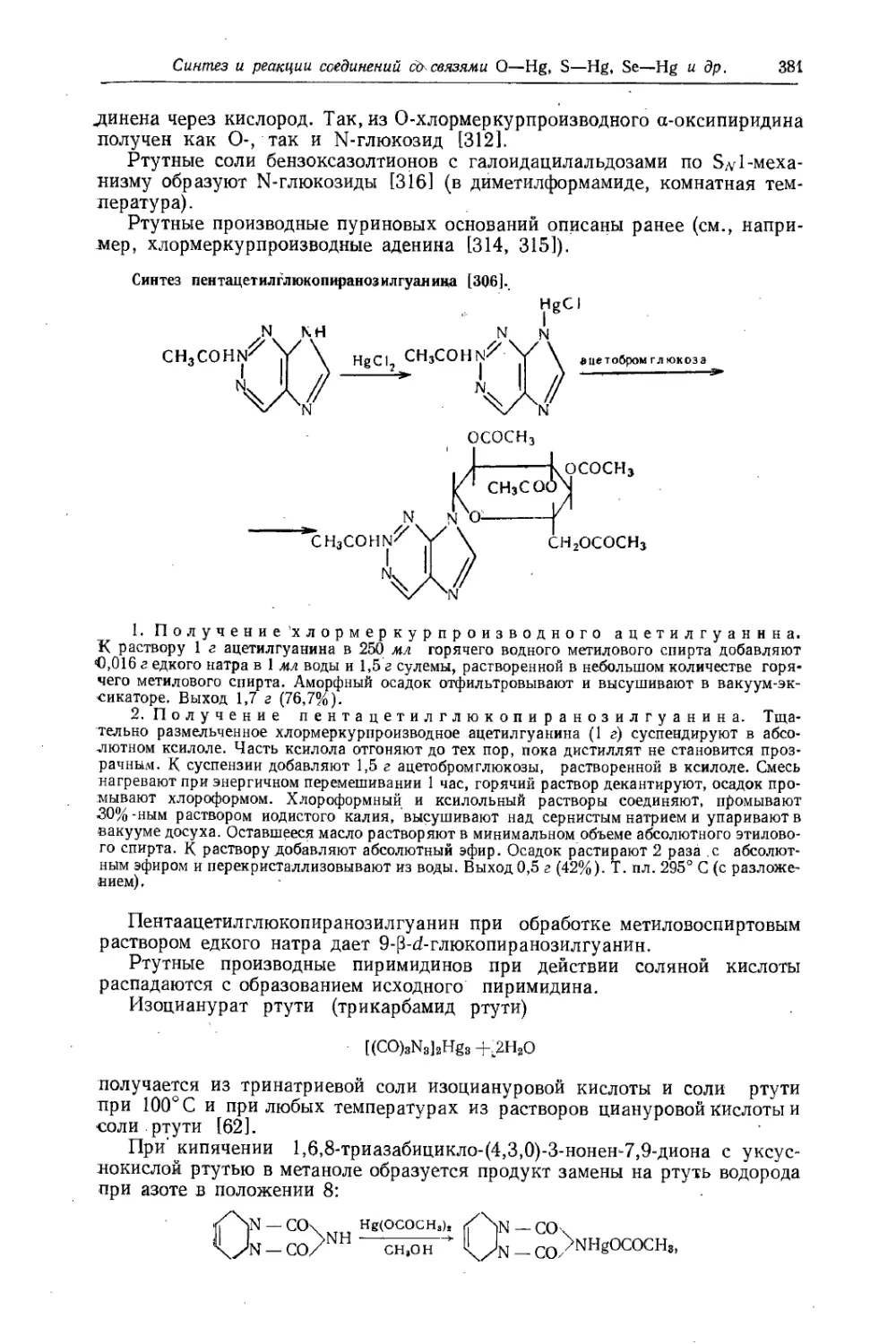






























































Text
АКАДЕМИЯ НАУК СССР
ИНСТИТУТ ЭЛЕМЕНТООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Под общей редакцией
А. Н. НЕСМЕЯНОВА и К. А. КОЧЕШКОВА
МЕТОДЫ
ЭЛЕМЕНТО-
ОРГАНИЧЕСКОЙ
ХИМИИ
ИЗДАТЕЛЬСТВО «НАУКА»
Москва-1965
АКАДЕМИЯ НАУК СССР
ИНСТИТУТ ЭЛЕМЕНТООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
А. Г. МАКАРОВА и А. Н. НЕСМЕЯНОВ
МЕТОДЫ
ЭЛЕМЕНТО-
ОРГАНИЧЕСКОЙ
ХИМИИ
РТУТЬ
ИЗДАТЕЛЬСТВО «НАУКА»
Москва*1965
итветственный редактор
член-корреспондент АН СССР
К. А. КОЧЕШКОВ
ПРЕДИСЛОВИЕ
Настоящая Кнйгй является звеном в серии монографий «Методы элемен.
тоорганической химии», издаваемых под общей редакцией А. Н. Несмея-
нова И К. А. Кочешкова. Вся серия будет посвящена методам синтеза, пре-
образования и использования в синтезе металлоорганических и элементо-
органических соединений. Из этой серии уже вышли в свет в 1963—1964 it.
монографии С. Т. Иоффе и А. Н. Несмеянова (Mg, Be, Са, Sr, Ba), А. Н. Не-
смеянова и Р. А. Соколик (В/А1, Ga, In, Т1), |Н. И. Шевердиной и К. А.Ко-
чешкова (Zn, Cd). Настоящая книга посвящена ртутноорганическим соеди-
нениям, в основу ее положена монография Л. Г. Макаровой и А. Н. Не-
смеянова «Синтетические методы в области металлоорганических соединений
ртути», изданная в 1945 г. За двадцать лет, прошедших с тех пор, хи-
мия металлоорганических соединений развивалась очень интенсивно, это
повлекло к переработке огромного материала и увеличению объема книги
более чем вдвое при сохранении крайне сжатого изложения. В книге исчер-
пывающе отражена литература в области синтетических методов, связанных
с ртутноорганическими соединениями, до 1 января 1964 г. (считая по
РЖХим) и включены важнейшие работы за 1964 г. и отчасти 1965 г.
При работе над книгой в разной мере использованы следующие моно-
графии. .
W h i t m о г е' F.j'C. Organic compounds of mercury. N. Y., Chem. Catalog Co. N. Y.
1921 (Русск. перевод, Госхимиздат, 1938).
C h a 11 J. Chem. Revs., 48, 7 (1951).
Ze iss H, Organometallic Chemistry. N. Y., Reinhold Publ. Corporation, 1960.
R о c ho w 5. G., Hurd D. T., L e w i s R. N. The chemistry of organometallic Compo-
unds, N. Y\„ Willey J., 1957 (Русск. перевод, ИЛ, 1963).
Krause E., Grosse A. Die Chemie der Metalloorganischen Verbindungen. Berlin,
Bontraeger, 1937.
Newton Friend. A Textbook of Inorganic Chemistry, vol. XI; Goddard A. E., God-
dard D. Organometallic compounds, London, Griffin Comp., 1928, p. 1.
Schmidt J. Organometallverbindungen, Th. II. Stuttgart, Wissenschaftliche Verlagge-
sellschaft, 1934. (Русск. перевод, ОНТИ, 1937).
Traite de Chimie organique (sous la direction de V. Grighard, G. Dupont, E. Locquin), N 5,
Paris Masson et Cie, 1937.
H ou b e p J., W e i 1 V. Die Methoden der organischen Chemie, vol. IV. Leipzig, Verlag
3. Thieme, 1924 (S c h 1 e n k W. Organometall Verbindungen).
Предисловие
Abderhalden Е. Handbuch der biologischen Arbeitsmethoden, Abt. I, Theil 2,
2 Halfte, Heft 4 (E. Klarmann-Bloomfield. Darstellung metallorganischer
Verbindungen). Berlin-Wien. Urban & Schwarzenburg, 1929.
G a r z u 1 у R. Organometalle. Sammlung chemischer und Chemisch-technischer Vortrage.
Stuttgart, F. Enke, 1927.
С о a t e s G. E. Organometallic compounds. London, Methuen, 1956.
С о a t e s G. E. Organometallic compounds, 2-nd ed., London, Methuen, 1960.
Metal-organic compounds, 131 Nat. meet., Am. Chem., Soc., Adv. in Chem. Series, vol. 23.
Washington, 1959.
ВВЕДЕНИЕ
Среди истинных металлоорганических соединений непереходных метал-
лов, т. е. соединений, содержащих прямую связь такого металла с углеро-
дом, ртутноорганические соединения относятся к числу наиболее прочных,
уступая в этом отношении только соединениям мышьяка и, может быть,
пятивалентной сурьмы. Хотя устойчивость разных типов ртутноорганиче-
ских соединений варьирует широко, все они отличаются инертностью к
кислороду и окислителям, воде и по крайней мере слабым кислотам, не
реагируют с огромным большинством типов органических кислородных сое-
динений и обычно достаточно инертны также к действию галоидных алкилов.
Все это резко отличает органические соединения ртути от натрий-, литий-,
магний-, цинк- и алюмйнийорганических и им подобных соединений. В свя-
зи с этим и применение ртутноорганических соединений как средств синтеза
ограничено и не идет в сравнение с использованием, например, реактива
Гриньяра.или литийорганических соединений. Наиболее важными типами
реакций ртутноорганических соединений являются два:
1. Обмен ртутноорганических соединений с галогенидами металлов и не-
металлов, приводящий к получению разнообразных металлоорганических
и, шире, элементоорганических соединений. Именно этим путем были, на-
пример, впервые получены арилдихлорарсины и диарилхлорарсины, арил-
дихлор- и диалкилхлорфосфины. Этот способ является одним из наиболее
простых путей синтеза иодониевых солей и т. д.
2. Замещение ртути ртутноорганических соединений при действии сво-
бодного металла на этот металл с получением металлоорганических соеди-
нений последнего. Этот метод оправдал себя в синтезе металлоорганических
соединений некоторых металлов (например, натрия, алюминия и др.) и ис-
пользуется до сих пор.
Таким образом, область применения ртутноорганических соединений
в синтезе — это металлоорганический синтез. Если второй тип применения
специфичен для ртутноорганическйх соединений, то в области первого типа
обмена магнийорганические соединения вследствие своей выдающейся реак-
ционной способности имеют во многих случаях большие преимущества пе-
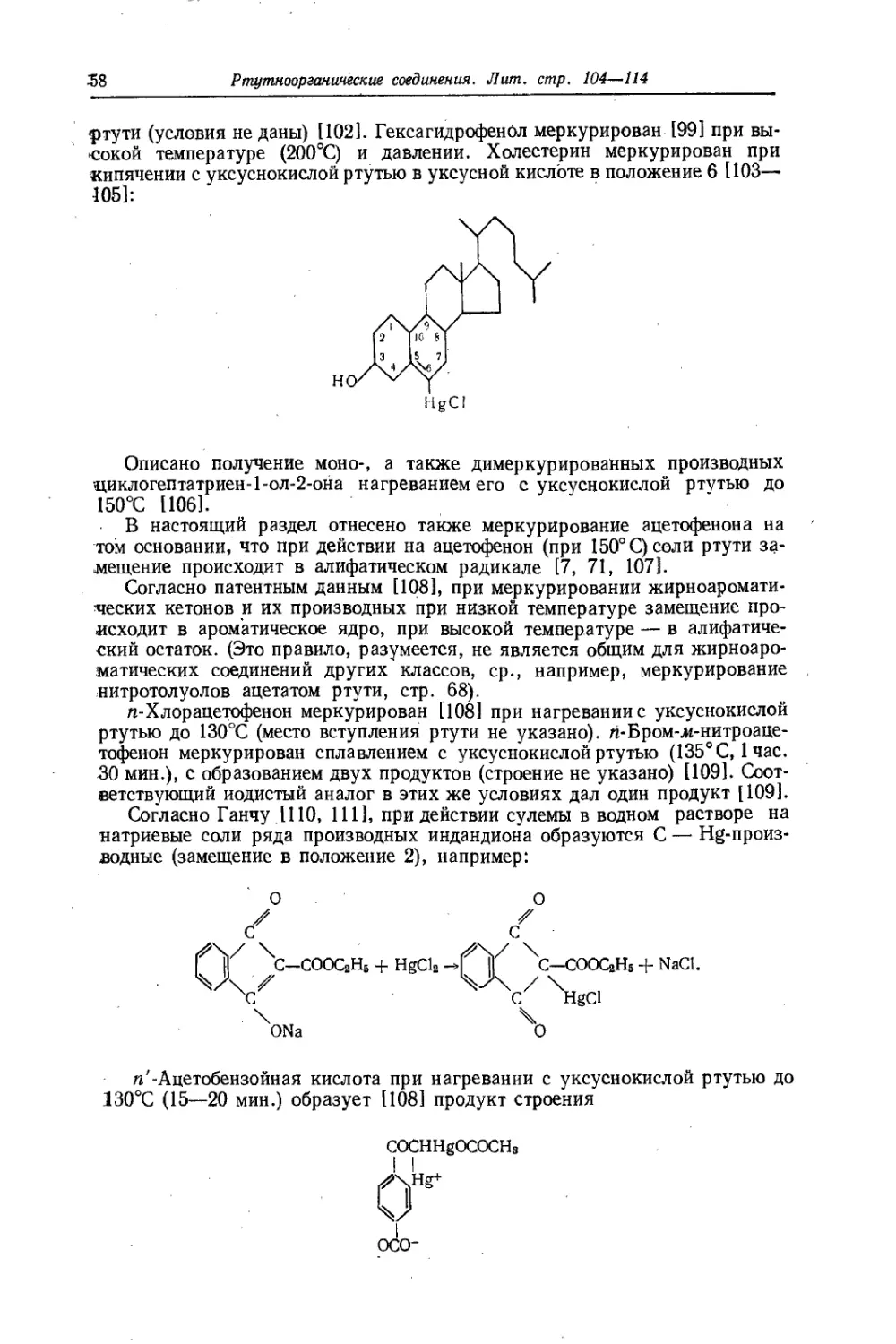
ред ртутными. Эти преимущества сводятся к следующему.
I, Магнийорганические соединения реагируют часто с такими галогени-
дами,\по отношению к которым ртутноорганические инертны.
II. Оперировать с магнийорганическими соединениями вследствие про-
стоты их приготовления и неядовитости удобнее и безопаснее. Однако ртут-
нрорганические соединения не во всех случаях могут быть заменены реак-
тивом Гриньяра по таким причинам:
1) реактив Гриньяра не применим во многих случаях вследствие восста-
новления им галогенида (например JC13 или CeHsJC^, SbCl5);
2) эфир, употребляемый как растворитель в реактиве Гриньяра, часто
связывается в комплекс с получаемым металлоорганическим соединением;
3) применение ртутноорганического соединения позволяет вводить в об-
мен гораздо большее разнообразие радикалов, тогда как посредством реак-
тива Гриньяра можно ввести только алкильные, некоторые алкенильные,
8 соединения. Лит. стр. И—13
аралкильные и арильные, реже гетероциклические радикалы, не содер-
жащие функциональных групп, за исключением немногих, устойчивых
к действию магния или магнийорганического соединения.
Близко примыкает ко второму типу реакций — переходу радикалов от
атома ртути на свободный металл —и переход радикалов на такие восста-
новители, как SnCl2, GeJ2, также приводящий к синтезу металлоорганиче-
ских соединений и, вероятно, далеко недостаточно развитый.
Упомянутая выше пассивность и вялая реакционная способность,
присущая органическим соединениям ртути, обусловливает существование
огромного разнообразия этих соединений. Ртуть комбинируется с самыми
разнообразными органическими молекулами и уживается почти с любыми
функциональными группами, входящими в структуру связанных с ней ра-;
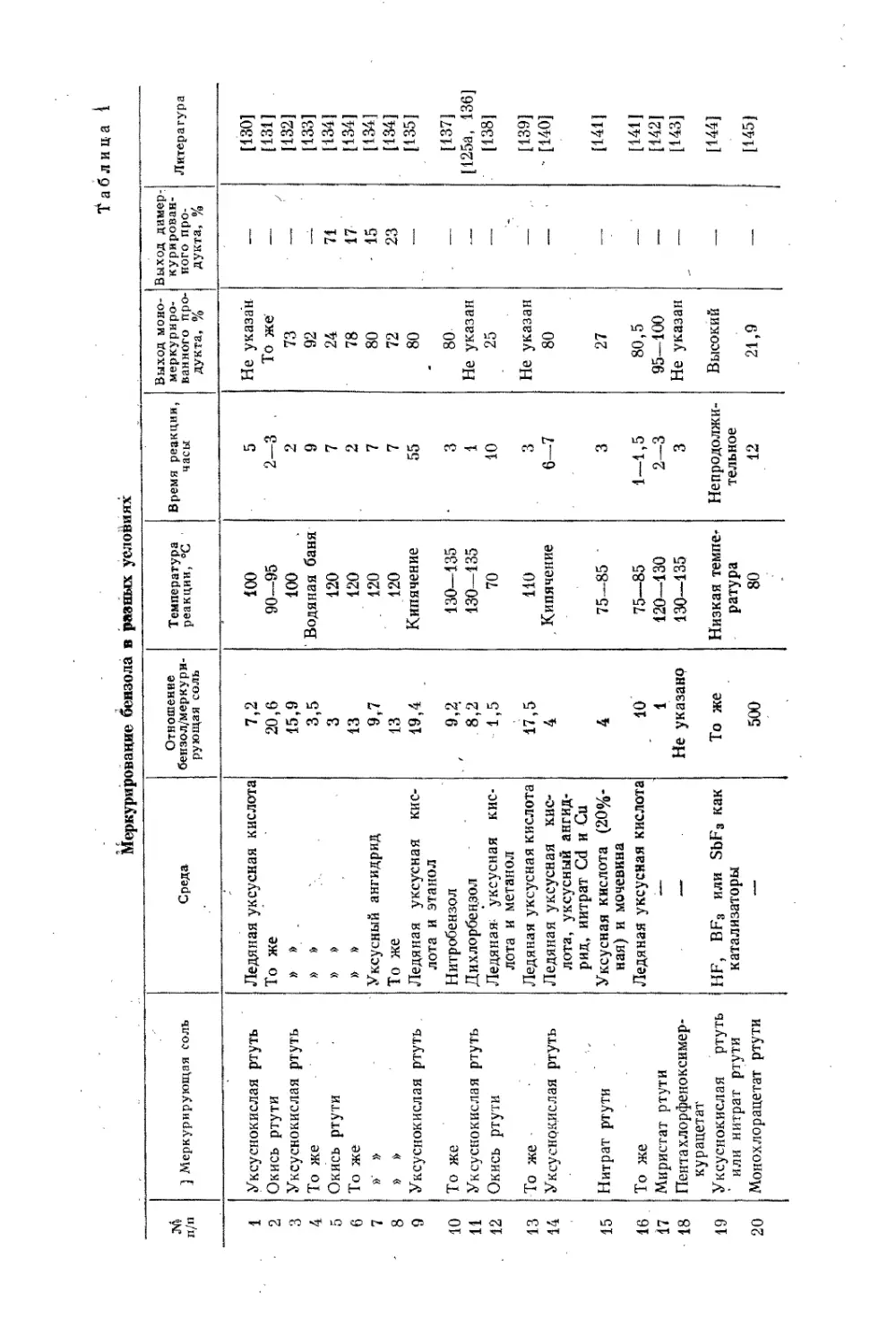
дикалов. Несмотря: на то, что могут существовать всего три основных типа:
ртутноорганических соединений — полное симметричное R2Hg, такое же
несимметричное RHgR' и сметанное (ртутноорганическая соль) RHgX
(относительно существования органических соединений закисной ртути
см. в гл. XI), — большое разнообразие достигается за счет разнохарактер-,
ности радикалов. Остаток R может принадлежать к предельному и непре-
дельному классам, алициклическим, ароматическим, разнообразнейшим
гетероциклическим соединениям и, как сказано выше, может иметь в. своем
составе почти любые группировки атомов. В исходной органической моле-
куле RH на ртуть может быть замещен один или несколько водородных ато-
мов, связанных с углеродом, в пределе — все эти атомы (меркарбиды).
Та же устойчивость ртутноорганических веществ обусловливает и лег-
кость их образования и разнообразие методов введения ртути в органиче-
скую молекулу, что в свою очередь позволяет осуществить почти любые ком-
бинации в молекуле ртутноорганического соединения.
Ниже приведены методы синтеза ртутноорганических соединений. .
1. На первом месте должен быть поставлен значительно превосходящий
все остальные способы по широте применения метод меркуриррвания— по-
лучение ртутноорганических солей заменой водорода на ртуть при дей-
ствии солей или окиси ртути:
RH + HgX2RHgX + НХ.
В жирном ряду он применим главным образом к соединениям, содержащим
подвижный водород. В ароматическом, а также гетероциклическом рядах
область его применения почти безгранична. Неудобством этого метода яв-
ляется одновременное образование смеси изомеров и нередко получение да-
и полизамещенных соединений (гл. V).
2. Взаимодействие органических соединений магния (и лития) с гало-
идной солью ртути. Этим путем с хорошими выходами получаются индави
дуальные как полнозамещенные алифатические, ароматические и некоторы
гетероциклические соединения ртути
2RMgXHgX2-> RHgR-I-2MgX2,
2RLi + HgXs -> R2Hg + 2LiX, ( *
так и ртутноорганическне солн
RMgX + HgXs RHgX + MgX»,
RLi+ HgXaRHgX + LiX. U
Это наиболее удобный метод лабораторного получения ртутноорганических
соединений жирного ряда с простейшими радикалами — гомологами мети-
ла (гл. II).
Введение 9*
3. Сюда же примыкают методы синтеза ртутноорганических соединений че-
рез органические соединения других легких металлов, такие как ныне уже
оставленный метод синтеза через цинкорганические соединения, и имеющий,
вероятно, некоторые перспективы метод синтеза органических соединений
ртути через алюминийорганические соединения (гл. Ш). .
4. Получение ртутноорганических соединений заменой на ртуть кислот-
ных остатков в карбоновых, ар ил борных, сульфиновых кислотах, а также*
JO2 в иодосоединениях и атомов тяжелых металлов в их органических сое-
динениях. Эти реакции приводят к индивидуальным ртутноорганическим;
соединениям типа RHgX (в случае карбоновых и арилборных кислот и
типа R3Hg) ароматического, а для карбоновых, борнЫх и сульфиновых ки-
слот и алифатического рядов. Подробнее об области применения каждого-
из этих методов см. при описании метода (гл. IX).
5. Новым методом синтеза главным образом жирных, а также алицикли-
ческих и ароматических -соединений ртути является инициируемое переки-
сями разложение ртутных солей карбоновых кислот
(RCOO)2Hg + (R'COO)a R'HgOOCR + СО2.
Метод применим для синтеза ртутноорганических соединений, не содер-
жащих заместителей, способных реагировать с перекисями или ингибиро-
вать цепные реакции (гл. X).
6. Диазометод синтеза ароматических ртутноорганических соединений.
ArNaX-HgXa4-2Cu-> ArHgX + N2 + 2CuX, '
2ArN2X*HgX2 + 6Cu + 6nNH3 -* Ar2Hg + 2N2 + 6CuX-/tNH,M- Hg
является наиболее удобным и универсальным методом синтеза индивидуаль-:
ных ароматических ртутноорганических соединений как полных, тай и ртут-
ноорганических солей. Метод не имеет ограничений, свойственных методам.
2, 3, 5, 8 и частично 4 (гл. VII).
7. Аналогичен диазометоду метод синтеза ртутноорганических солей,
через иодониевые соединения, практического значения, однако, не имеющий
(гл. VIII).
8. Действие амальгамы натрия на галоидные производные углеводоро-
дов (а также диалкилсульфаты)
<2RX + NaaHg.-> R2Hg + 2NaX.
Этот самый старый из ныне употребляющихся (все реже) способов получения
полнозамещенных соединений ртути довольно широко применим как в жир-
ном, так и в ароматическом ряду. Область применения этого метода ограни-
чена синтезом соединений, не содержащих заместителей, способных реаги-
ровать, с амальгамой натрия (гл. IV).
9. Присоединение солей ртути по двойной и тройной связям, широко*
применяемо^ в ряду олефинов, ацетиленов и всевозможных их производных,
ведет к своеобразной и интересной области недоступных иными путями про-
дуктов присоединения солей ртути к непредельным веществам — квази-
комплексным соединениям — ₽-замещенным на Hal, ОН, OR и т. д. ртутно-
органическим солям (гл. VI):
RV1^ = CRHIRIV + HgX2 + ROH R^’C (OR)—C (HgX) R,nR,v + HX,
HC=CH + HgCl2 -> ClHC=CHHgCl.
10. Близок к предыдущему метод размыкания трехчленного цикла
солью ртути, приводящий к у-замещенным ртутноорганическим солям (гл^
tO Ртутноорганические соединения. Лшп. стр. 11—13
11. Метод синтеза ртутноорганических солей взаимодействием галоидных
алкилов с металлической ртутью:
RX + Hg-» RHgX,
'(где X = J и в некоторых случаях также Вт). Так, этим методом получены
нерфторалкильные соединения ртути и эфиры а-броммеркуркарбоновых ки-
слот (гл. I).
12. Электролитическое получение полнозамещенных ртутноорганиче-
ских веществ:
a) 2RRTO + Hg + 6Н (RR'CH)aHg + 2НаО,
•путь а приводит к соединениям R2Hg с вторичными радикалами;
б) NaMeinR4 + Hg R2Hg,
путь б приводит к синтезу простейших низших предельных ртутнооргани-
песких соединений (гл. П). Оба метода имеют ограниченное применение.
Переход от соединения типа RHgX к соединениям типа R2Hg осуще-
ствляется методом симметризации (см. гл. XIII); обратный переход—взаи-
модействием RoHg с солями или окисью ртути по уравнению (см. гл.
XIII)
RaHg + HgXa-» 2RHgX.
Из методов синтеза ртутноорганичёских соединений методы 1, 2, 9 и ча-
стично 4 применены для получения соединений RHgR' (см. гл. XII).
В гл. XV дается краткий обзор способов получения и некоторых реакций
таких соединений ртути, в которых ртуть соединена с органическим остат-
ком не через углерод, а через гётероатом Z: RZHgX или (Rn-i Z”)2Hg, где
Z = О, S, Se, N, Ge, Si, а также продуктов замены водорода на ртуть в
Л нал килфосфонатах.
Что касается приемов работы и способов обращения с ртутноорганиче-
•скими соединениями, то, поскольку последние являются вполне устойчи-
выми к воздействию воздуха и влаги, в большинстве случаев твердыми, пре-
красно кристаллизующимися из органических растворителей веществами,
реже жидкостями, способными к перегонке, методы выделения, очистки и
идентификации их являются обычными методами органической химии.
Очень многие, хотя далеко не все, ртутноорганические вещества являются
высокоядовитыми. В этом отношении выдающуюся опасность представляют
только летучие соединения — диметилртуть и ее гомологи, Диаллилртуть,
а также алкилртутные соли, имея дело с которыми, необходимо принимать
все меры предосторожности, работая только под хорошо действующей тя-
гой и обеспечивая полную дегазацию всей посуды и остатков вещества дей-
ствием растворов брома или хлора в четыреххлористом углероде, хлоро-
форме4' или царской водкой. Работа с ароматическими или гетероцик-
.лическими соединениями, а также жирными, содержащими функциональ-
ные группы в органическом радикале, более спокойна. Однако и здесь
необходимо принимать во внимание летучесть при несколько повышенных
температурах таких соединений, как дифенилртуТь. Во всех случаях необ-
ходимо тщательно оградить себя от возможного распыления и рассыпания
ртутноорганических соединений. Некоторые ртутноорганические веще-
ства, например ClCH2HgCl, CH^)2CHgX, ClCH=CHHgCl, а также и ди-
•фенилртуть, раздражают кожу и вызывают ожоги.
Многочисленные работы посвящены исследованию физических свойств
простейших ртутноорганических соединений. Рассмотрение их не входит
ж задачу авторов, отсылающих читателя к оригинальной литературе.
Введение
И
Ниже приводятся литературные ссылки на оригинальные работы по
рефрактометрии и плотностям [1—9], по Спектрам поглощения в ультра-
фиолетовой области [1'0—25], ИК-спектрам [19а, 23, 25—42J, по раман-
епектроскопии [29—49], микроволновой спектроскопии [41,50—52], элек-
тронному [53—55] и ядерному магнитному резонансам [25, 42, 56—62],
масс-спектрам [63—68], рентгеноструктурному анализу [69—876],магнето-
химическим исследованиям [88—92], по измерению диэлектрической по-
стоянной и дипольному моменту [5,32,89—108], по электропроводности
[109—1141, полярографии (см. гл. XVII), по определению парахора Ш5,
116], измерению упругости пара низших алкильных соединений [3, 101,
117—121], электронографическому исследованию молекулярной структуры
[122, 123], измерению теплот сгорания и образования, теплоемкости и сво-
бодной энергии [3 , 22, 116, 121, 124—1361, а также скрытой теплоты .воз-
гонки [117, 137].
Кроме того, исследование тех или иных физических свойств ртутноорга-
нических соединений во многих случаях дается также при соответствующем
ртутноорганическом соединении в оригинальной литературе.
ЛИТЕРАТУРА
1. G h i г a. Atti. Accad. naz. Lincei Rend. Classe Sei. fis. mat. nat. [5], 3, 1, 298 (1894);
Gazz., 24, 1, 311 (1894).
2. Krause E. Ber., 59, 935 (1926).
3. J о n e s W. J., E v a ns D. P., Gul we 1 1 T., G r i f f i t h s D. C. J. Chem.
Soc., 1935, 39.
4. Blitz W., Sapper A. Z. anorg. allg. Chem., 186, 387 (1930).
5. Oes per P. F., Smyth Ch. P. J. Am. Chem. Soc., 64, 173 (1942).
6. W i 1 d e W. K. J. Chem. Soc., 1949, 72.
7. Vogel A. Cresswell W. T., Leicester J. J. Phys. Chem., 58, 174
(1954).
8. Cresswell W. T., Leicester!, Vogel A. I. Chem. and Ind., 1953, 19.
9. Иоффе Б. В., Морачевский А. Г. ЖАХ, 10, 3 (1955).
10. С г у m b 1 е С.- R. J. Chem. Soc., 105, 668 (1914).
11. Pur v i s I, Е., McCleland N. P. J. Chem. Soc., 101, 1514 (1912).
12. Asundi R. K., R aoC. M. B„ S a m ti e 1 R. Proc. Ind. Acad. Sci., 1A, 542 (1935).
13. Thompson H. W., FrewingJ. J. Nature, 135, 507 (1935).
14. T h о m p s о n H. W., Linnet t J. W. Proc. Roy. Soc., A156, 108 (1936).
15. Thompson H. W., Linnet t J. W. Proc. Roy. Soc., A169, 539 (1937).
16. Leandr 1 G„ T u n d о A. J. Chem. Soc., 1954, 3377.
17. -G о w e n To c k B. G., Tro t m a n J. J. Chem. Soc., 1955, 1454.
18. Lean dr i G., T u n d о A. C. A., 47, 4195(1953); Boll. sci. facolta Chim. ind,
Bologna, 10, 160 (1952).
19. G о w e n 1 о c k B. G., T г о t m a n J. Chem. a. Ind., 1954,309.
19a. Словецкий В. И., T а р та к о в с к и й В. А., Новикове. С. Изв.
АН СССР, ОХН, 1962, 1400.
20. L е а и d г i G„ S р i n е 1 1 i D. С. А„ 54, 5530 (1960); Ricera Sci., 29, 541 (1959).
21. T h о m*p son H. W. Proc. Roy. Soc., A150„ 603 (1935).
22. Hor.pwi tz M. G„ К 1 Q t z I. M. J. Am. Chem. Soc., 77, 5011, (1955).
23' . H ec м.ея но в A. H., К'ази цын а Л. А., Луценко И. Ф., Руденко
Г. А. ДАН СССР, 127, 115 (1959).
24. L е a n dr i G., S p i ne 1 1 i D. Ann. chim. (Italy), 50, 1581 (1960).
25. Д в о p я н ц e в а Г. Г., Кочеткова H. С., Материкова P. Б.,
Шейнкер Ю. H. ДАН СССР, 159, 847. (1964).
26. С о s t a G., P u x e d d e n A. C. A., 53, 14689 (1959); Univ. Studi Trieste Fac. Sci.
phys. Chim., 27, 13 (1958).
27. Brodersen K. Ber., 90, 2703 (1957); Z. anorg. allg. Chem., 298, 34 (1958).
28. К a e s z H. D., S t о n e F. G. A. Spectrochim. Acta, 1959, 360.
29. F r i t z U. P. Ber., 92, 780 (1959). ,
30. GutowskiH. S. J. Chem. Phys., 17, 128 (1949).
31. В a u n W. Z. Analyt. Chem., 31, 1308 (1959).
32. S aw a t sky H., W r i g h t G. F. Canad. J. Chem., 36, 1555 (1958).
33. C h a t t J., DunkansonL. A., Venanzi L. M. C. A„ 51, 5559 (1957);
Suomen Kemistilehti, 29B, 75 (1956).
34. А м а к а с у О., СиндоХ., ИтоН. РЖХим., 1961, 18Л 237.
12
Ртутноорганические соединения
35. В оу d D. R. J., Т horn psonH« W., Wi 11 i a ms R. L. Discuss. Faraday
Soc., 1050, N 9 154. .
36. Во рисевичН. А., Петрович П. И., Залесская В. А. ДАН БССР,
4, 510 (1960).
37. Т a k а п о К. С. А., 56, 10071 (1962).
38. Стерлии Р. Н., Д у б о в С. С. Ж. Всесоюзн. хим. общ., 7, 117 (1962).
38а. Downs A. I. J. Chem. Soc., 1963; 5273.
39. Gautier Н., G о u р i 1 R., M a ngeney G., Didelot S., Merlier R.
Chim. analyt., 49, 12 (1962).
40. Margoshes M., F a s s e 1 V. A. Spectrochim. acta, 7, 14 (1955).
41. She line R. K. J. Chem. Phys., 18, 602 (1950).
42. ,H a 1 p e r n J-., К e 11 1 e S. F. A. Chem. and Ind., 1961, 668.
43. P a i N. G Proc. Roy. Soc., A149, 29 (1935).
44. D о n z e 1 о t Pi, Ch a ix M. C. r„ 201, 501 (1935).
45. Koh Ir a u sch K. W. F., Pongratz A., S e к a R. Monatsh., 70, 213 (1933).
46. FehdrF., Kolb W., LeverenzL. Z. Naturforsch., 2a, 454 (1947).
47. Несмеянов A. H., БорисовА. E., Бату евМ. И. Изв. АН СССР,,
ОХН, 1949, 567.
48. G о g g i n Р. L., Woodward!. A. Trans. Faraday Soc., 58, 1495 (1962).
49. N а у a r M. R., SarafJ. R. J. Indian Chem. Soc., 20, 312 (1943).
50. Gordy W., Sheridan J. Phys. Rev., 79, 224 (1950).-
51. Gordy W., She.ridan J. J. Chem. Phys., 22, 92 (1954). _
52. С о x J. T., G a u m a n n T., Thomas W. J. O. Discuss. Faraday Soc., 19,
52(1955).
53. Gow enlock R. G„ Jones P. P., OvenallD. W. J. Chem. Soc., 1958,535.
54. S m a 1 1 e r B., Ma thesonM. S. J. Chem. Phys., 28, 1169 (1958).
55. Gordy W., Me С о r m i с к C. G. J. Am. Chem. Soc., 78, 3243 (1956). >
56. С о у 1 e T. D., S t a f f о r d S. L., S t о n e F. G. A. Spectrochim. acta, 17, 968
(1961).
56a. M о у D., EmersonM., О 1 i v e г J. P. Inorg. Chem., 2, 1261 (1963).
566. W e 11 s P. R., KitchingW. Tetrahedron L., 1963, 1531. .
57. С о t t о n F. A., L e t о J. R. J. Am. Chem. Soc., 80, 4823 (1958).
58. Strohmeier W., Lemmo n R. M. Z. Naturforsch., 14a, 109 (1959).
59. К г e s p a n C. G. J. Org. Chem., 25, 105 (1960).
60. P i p e r T. S., W i 1 к i n s о n G. J. Inorg. Nuclear Chem., 3, 104 (1956),
61. D e s s у R. E„ F 1 a u t t T. J., J a f f ё H. H., R e у n о 1 d s G. F. J. Chem.
. Phys., 30, 1422 (1959).
62. Gu towsky H. S., Раке G. E. J. Chem. Phys., 18, 162 (1950).
63. Gowenlock B. G., На у nes R. M., M а у e r J. R. Trans. Faraday Soc.,
58, 1905(1962).
64. D i b e 1 e г V. H., M о h 1 e r F. L. J. Res. Nat. Bur. Stand., 47, 337 (1951).
65. D i b e 1 e r V. H. J. Res. Nat. Bur. Stand., 49, 235 (1952).
66. I n g о 1 d С. K-, LossingF. P. J. Chem. Phys., 21, 368, 1135 (1953).
67. Hob rock В. C., Kiser R. W. J. Phys. Chem., 66, 155 (1962).
68. Д у б о в С. С., Ч е л о б о в Ф. Н., С т е р л и н Р. Н. Ж. Всесоюзн. хим. общ.,
7, Ns 5, 585 (1.962).
69. G Га с о m е 1 1 о G. С. А., 45, 5019 (1951); Proc. XI Internal. Congr. Pure and Appl.
Chem., 2, 99 (1947).
70. H о w e 1 1 s E. R., P h i 1 1 i p s D. C., Rogers D. Acta Cryst., 3, 210 (1950).
71. Китайгородский А. И. Изв. АН СССР, OXH, 1947, 259.
72. Китайгородский А. И., ГрденичД. Изв. АН СССР, ОХН, 1948, 262.
73. ГрденичД., Китайгородский А. И. ЖФХ, 23, 1161 (1949).
74. Кл тайгородскийА. И., Стручков Ю. Т. Изв. АН СССР, сер. физ.,
15, 147 (1951).
75. К и -Ка й г о р о д с к и й А. И. ДАН СССР, 93, 675 (1953).
•76. М а 1 у J., Киса L. Chem. Listy, 47, 1575 (1953).
77. Е m е 1 ё и s Н. J. Proc. Chem. Soc., 1958, 231.
78. В a и n Wm. L. Analyt. Chem., 33, 308 (1959).
79. G r d e n i c D. Arkiv. Kemi, 22, 14 (1950),
80. G r d e n i ё D. Acta Cryst., 5, 367 (1952).
81. Gr d e n i c D. Ber., 92, 231, (1959).
82. W r i g h t G. F., Brook A. G. Acta Cryst, 4, 50 (1951).
83. A b e г с г о m b i e M. J., R о d g m a n V. A., В h a г и c h a K. R.. Wright
G. F. Canad. J. Chem., 37, 1328 (1959).
84. T г о 11 e г J. Canad. J. Chem., 40, 1218 (1962).
85. С a r I i s 1 e С. H., Palmer R. A. Acta Cryst., 15, 129 (1962).
86. Z i 6 1 k о w s k a B. Roczniki Chem., 36, 1341 (1962).
87. E h r 1 i c h H. W. J. Chem. Soc., 1962, 509.
Введение
13
87а. Пахомов В. И. Кристаллография, 5, 800 (1960); 7, 456 (1962).
876. Пахомов В. И. Ж. структ. хим., 4, 594 (1963); 5, 917 (1964).
88. F i s с h е г Е. О., PiesbergerV. Z. Naturforsch., 11b, 758 (1956).
89. М a t s u n a g a I. С. A., 51, 17283 (1957); Bull. Chem. Soc. Japan, 30, 227 (1957).
90. К a d*o m t z e f f I. Bull. Soc. chim. France, 1940, D 394.
91. Kadom tzef f I. C. r„ 230, 443 (1950).
92. Kadomtzeff I. C. r„ 228, 68141949). .
93. Horning W. C., L a u t e n s c h 1 a e g e r F., W r i g h t G. F. Canad. J.
Chem., 41, 1441 (1963).
94. Ma t thews В. C. Zbl., 1906, 1, 224.
95. В e r g m a n n E., S c h fl t z W, Z. Phys. Chem., B19, 401 (1932).
96. Hamp son G. C. Trans. Roy. Soc., 30, 877 (1934).
97. К e s 1 e r M. Croat. Chem. Acta, 34, 123 (1962).
98. Curran W. J., W e n z k e H. H. J. Am. Chem. Soc., 57,2162 (1935).
99. С о о p I. E., S u t t о n L. E. J. Chem. Soc., 1938, 1269.
100. S m у t h С. P. J. Org. Chem., 6, 421 (1941).
101. Ратман Ф. Г. ЖПХ, 9, 591 (1936).
102. C u r r a n В. C. J. Am. Chem. Soc., 64, 830 (1942).
103. M a 1 a t e s t a L. C. A., 42, 3630 (1948); Rend. ist. Lombardo scl., 78,1 (1944—1945).
104. H e i n F., Schleede A., К a 1 1 m e у e r H. Z. anorg. Chem., 311, 260 (1961).
105. Armstrong R. S., Le Fevre C. G., Le F ё v r e R. J. W. J. Chem. Soc.,
1957, 371.
106. S i p о s J., Sawa tzky H., W r i g h t G. F. J. Am. Chem. Soc., 77, 2759 (1955).
107. C u r r a n В. C. J. Am. Chem. Soc., 63, 1470 (1941).
408. Kesler M. РЖХим., 1964, 20Б 220.
109. J о h n s J. В., P e t e r s о n W. D., H i x о n R. M. J. Phys. Chem., 34,2218 (1930).
110. J о h n s J. В., H i x о n R. M. J. Phys. Chem., 34, 2226 (1930).
111. E v a n s W. V., Pearson R. J. Am. Chem. Soc., 64, 2865 (1942).
112. S t г о h m e i e r W. Z. Elektrochem., 60, 396 (1956).
113. J a n d e r G., Brodersen K. Z. anorg. Chem., 265, 117 (1951).
114. M а у n a r d J. L.,- Howard H. C. J. Chem. Soc., 123, 960 (1923).
115. S u g d e n R. J. Chem. Soc., 1929, 316.
116. G i bl i ng T. W. J. Chem. Soc., 1944, 380. 383.
117. Thompson H. W., L i n n e 11 I. W. Trans. Faraday Soc., 32, 681 (1936).
118. P h i 1 1 i p s G. F„ D i x о n В. E., Li dzey R. G. C. A., 54, 7044 (1960);
J. Sci. Food, Agr., 10, 604 (1959). •
119. C h a r n 1 e у T., SkinnerH. A. J. Chem. Soc., 1951,1921.
120. LongL. H., Cat tanach J. J Inorg. Nuclear Chem., 20, 340 (1961).
121. M о r t i m e г С. T., Pritchard H. O., Skinner H. A. Trans. Faraday
Soc., 48, 220 (1952).
122. В г о c k w a у L. O., J e n k i n s H. O. J. Am. Chem. Soc., 58, 2036 (1936).
123. A 1 1 e n P. W., S u 11 о n L. E. Acta Cryst., 3, 46 (1950).
124. В e r t h e 1 о t M. C. r„ 129,918 (1899).
125. Smith R. H., Andrews D. H. J. Am. Chem. Soc., 53, 3661 (1931).
126. Fairbrother D. M., S k i n n e r H. A. Trans. Faraday Soc., 52, 956 (1956).
Г27. Chernick С. I., S k i n n e r H. A., W a s s б I. Trans. Faraday Soc., 52, 1088
(1956)
128. L a u t s c h W. F. Chem. Tech. Berlin, 10, 419 (1958). :
129. Carson A. S., Carson E. M., Wilmshurst B. R. Nature, 170, 320 (1952).
130. Prices. J. W., Trotman -Dickenson A. F. Trans. Faraday Soc., 53,
939 (1957).
131. S k i n n e r H. A. Rec. trav. chim., 73, 991 (1954).
132. Hartley K-, Pritchard H. O., Skinner H. A. Trans. Faraday Soc.,
- 46, 1019X1950).
133. Hartley K-, P r i t c h a r d H. O., Ski nner H. A. Trans. Faraday Soc.,
47, 254 (1951).
134. L о n g L. H., NorrishG. W. Trans. Roy. Soc., A241, 587 (1949).
135. Hart ley K.( Pri tchar d H. O., S k i n n e r H. A. Trans. Faraday Sdc„
48,220 (1955).
136. Stevenson K. Discuss. Faraday Soc., 10, 35 (1951).
137. C a r s о n A. S., Stranks D. R., W i 1 m s h u r s t B. R. Proc. Roy. Soc , A244^
c 72 (1958).
Г лава I
ВЗАИМОДЕЙСТВИЕ RHal С МЕТАЛЛИЧЕСКОЙ РТУТЬЮ
И СОЛЯМИ РТУТИ
Несмотря на то, что впервые ртутноорганические соединения были по-
лучены действием металлической ртути на галоидное производное углево-
дорода (именно в 1853 г. Франкланд [1] нашел, что CH3J реагирует на сол-
нечном свету с металлической ртутью, давая CH3HgJ), реакция
RXHgRHgX
как метод синтеза ртутноорганических соединений не нашла широкого при-
менения.
До недавнего времени реакция была применена главным образом к йо-
дистым алкилам, так как простейшие хлористые алкилы, как правило, не
реагируют с металлической ртутью, а реакция бромидов проходит труднее»
Взаимодействие алкилиодидов с металлической ртутью идет в большинстве
случаев с трудом и не гладко. Кроме CH3J [1, 21, в эту. реакцию был введен
C2H5J [3, 4, 5], иодистый пропил [2], иодистый аллил [6, 7'1 (в последнем
случае реакция идет легко и для взаимодействия с ртутью не требуется
действия света), иодистый пропаргил [81, иодистый метилен (продукты
JCH2HgJ [2,9, 37] лучше с мелкораздробленной ртутью и при освещении уль-
трафиолетовым светом [9а] и JHgCH2HgJ [9,37]), йодоформ [9], С6Н6С = CJ
[10] (реакцию вели при 100°С), CC13J [11], CHC12J [1], 3-иод-1,Г-дихлорпро-
пен-1 [11] (три последних вещества реагируют с металлической ртутью как
при облучении ультрафиолетовым светом, так и без освещения» нагреванием
до 70—80°С и энергичным перемешиванием реакционной массы), иодистые
аллилы, содержащие группировку СХУ=СНа1 — CH2J (где X и У = Н
или Cl, Hal = Cl или Вг [12]), 3-иод-1,1.-дихлор-2-метилпропен [11, 13],
этиловый эфир иодуксусной кислоты [14]. ArJ при продолжительном облу-
чении и лучше при перемешивании щеточной мешалкой, снимающей осадок
go стенок [14], вступают в реакцию с металлической ртутью, образуя с нез-
начительным выходом ArHgJ [14—16]. Так получены C6H6HgJ [14—161
(выход 3%) и a-C10H7HgJ [14, 16] (выход 6—8%).
Майнард [17], исследовавший реакцию ртути с CH3J, показал, что ка-
тализатором этой реакции является образующаяся под действием прямого
солнечного света Hg2J2.
Синтез иодистой аллилртути [6]. Смесь йодистого аллила и металлической ртути при
встряхивании быстро становится желтой кристаллической массой, из которой горячий
спирт и эфир извлекают иодистую аллилртуть, выкристаллизовывающуюся по охлаж-
дении в виде бесцветных блестящих чешуек. Т. пл. 135° С.
Взаимодействие RHal с Hg и ее солями 1.5»
Данным методом Эмелеусом и Хазсельдином [18—'221 впервые были
получены перфторалкильные соединения ртути. Реакцию ведут или при
продолжительном облучении солнечным светом, или светом ртутной лампы,,
или при продолжительном нагревании в запаянной трубке при высокой тем-
пературе (CF3J, 260—290°С, 12 час. [19], C2F5J, 120°С, 5—6 дней при облу-
чении, n-C3F7J, 220°С, 24 часа [22])* Хороший выход CF3HgJ достигнут про-
ведением реакции в растворителе, каковым здесь явился перфторметил-
циклогексан и при одновременном применении как нагревания, так и облу-
чения [19].
Получение иодистой трифторметилртути[ 19]. Раствор 7,5 г CF8J в 4 дм перфторметил-
циклогексана нагревают в запаянной трубке с 10 мл Hg при 110° С при встряхивании и облу-
чении светом ртутной лампы в течение 36 час. По охлаждении выделяются кристаллы йоди-
стой трифторметилртути. Растворитель удаляют и извлекают эфиром иоднстую трифтор-
метилртуть. Выход 7,5 г (80%, считая на вошедшие в реакцию 4,7 г CFsJ). Белые кристаллы,,
возгоняются при 80°С. Т.пл. 112,5°С [18].
. При реакции полимера из CF3J и этилена с избытком этилена в присут-
ствии ртути образуется CF3CH2CH2HgJ [23].
При реакции RraAsJ3_ra [24] и CeH5SbJ2 [25] с R'J в присутствии Hg
побочно образуется R'HgJ.
При фотолизе бромистого метила в присутствии ртути, возможно, обра-
зуется диметилртуть [26].
При взаимодействии бромтрихлорметана с металлической ртутью оп-
тимальный выход (41%) бромистой трихдорметилртути получен при облу-
чении и одновременном нагревании до 70—80°С энергично перемешиваемой
реакционной смеси в течение 3 час. в присутствии азодинитрила бнс-изо-
масляной кислоты [11].
Бромпроизводные, в молекуле которых связь углерод — бром активиро-
вана сопряжением с кратной связью:
NsC—С—Вг, >С=С—С—Вг или О=С—С—Вг,
I ] I
способны в мягких условиях реагировать с металлической ртутью, давая
рТутнооргайические соединения RHgBr [15, 17, 27]. Бромбензилцианид ре-
агирует с металлической ртутью при комнатной температуре без раствори-
теля или в растворе (в эфире) [27].
Реакция с металлической ртутью этилового [Г4, 28] и/-ментилового эфи-
ров а-бромфенилуксусной кислоты, циннамилбромида [14] не требует об-
лучения и идет при комнатной температуре (выходы RHgBr соответствен-
но 73, 31 и 82%). При встряхивании в течение 4 час. ХС6Н4СНВгСООС2Н6
со ртутью получены XC6H4CH(HgBr)COOC2H5 (X = p-F, p-Br, p-J,o-CH3,
p-(CH3)3C, m-CH3 [29], o-Br, p-NO2, p-C2Hs, p-(i-C3H7), p-CH3[29a]
(см. также [30]). Циннамилбромид настолько энергично реагирует с
ртутью, что во избежание полимеризации и осмоления RBr последний
необходимо разбавлять этиловым спиртом. Реакция бромистого аллила
С металлической ртутью, медленно идущая уж>> *в темноте, ускоряется
при облучении, лучше ультрафиолетовым светом; в последнем случае
во избежании образования ртутноорганических полимеров необходимо
весьма энергичное перемешивание, лучше щеточной мешалкой. Целый
ряд бромистых алкилов не образует в указанных условиях с металли-
ческой ртутью ртутноорганических соединений; броммалоновый эфир и
а-бромфенилуксусный альдегид дали ртутноорганические полимеры [14,16].
46 Ртутноорганические соединения
Получение циннамнлмеркурбромида [14]. 19,7 г циннамилбромида (0,1 моля) смешивают
е равным объемом 96%-ного этилового спирта и с 100,3 г металлической ртути (0,5 г-атома).
После 15-минутного энергичного встряхивания реакционная смесь слегка разогревается и
загустевает. Непрореагировавшую ртуть сливают, в смесь добавляют этиловый спирт до ка-
шеобразного состояния, осадок отфильтровывают. Затем осадок экстрагируют горячим
бензолом. Бензольный раствор упаривают до начала кристаллизации. После окончания
-кристаллизации (2—3 часа) циннамилмеркурбромид отсасывают и промывают холодным
бензином. Вес 32,7 г (выход 82%). После кристаллизации из бензола т. пл. 340—345°С
при быстром нагревании.
Взаимодействие с металлической ртутью галоидных галоидзамещенных
аллилов: СН2=::СС1СС12Вг, СС12=СНСН2Вг (2—3 дня на рассеянном
свёту, 3—4 часа при освещении УФ-светом), СН2=СВгСС13 (только при ос-
вещении УФ-светом) приводит к одному и тому же . продукту,
CCl2=CClCH2HgBr [13].
Дифенилртуть.получена взаимодействием бромбензола и сулемы в при-
сутствии металлического натрия [31]:
2С6Н3Вг + HgCl2 + 4Na (CeH5)2Hg + 2NaCl + 2NaBr.
Вещество с т. пл. 152°С, полученное наряду с бромистой стирилртутью при
действии металлического натрия на смесь бромстирола и сулемы, принятое
Дас-Гупта [32] за дистирил ртуть, оказалось [33] смесью бромистой стирил-
ртути (т. пл. 202°С) и дистирилртути (т. пл. 137°С). Образу1ощийся при этом
малорастворимый высокоплавкий остаток также не представляет собой ин-
дивидуального соединения [33].
Йодистые алкилы с солями закисной ртути образуют соль алкилртути
[34]. При 18-часовом кипячении избытка йодистого метила с сульфатом
закисной ртути иодистая метилртуть получена с выходом 80%, с иодистым
этилом—выход 10—15%; иодистый бутил и иодбензол с Hg2(SO4)2 не дают
RHgJ; иодистый метил с ацетатом или бензоатом закисной ртути при
40—80°С образует соответствующую соль метилртути с выходом 25 и 50%.
При реакции бензоата ртути с избытком RJ (R = СН3, СаН5, л-С4Н9, СеН5)
при 40—80°С в присутствии солей закисной ртути образуются соли ал-
килртути [34].
Синтез иодистой метилртути [34], 0,025 моля сульфата закисной ртути нагревают
.в 50 мл йодистого метила в течение 18 час. при температуре кипения йодида. Из продуктов
реакции выделяют 0,020 моля метилмеркуриодида с т. пл. 143°С. Выход 80% в расчете на
взятую соль.
При разложении бис-(триэтилгермил)ртути в бромбензоле под дейст-
вием УФ-света образуется наряду с триэтилбромгерманом дифенилртуть
435]. ,
Взаимодействие йодацетиленов с иодной или цианистой ртутью [36],
.прямо приводящее к полнозамещенным соединениям ртути
2RC=CJ + HgX2 (RC=C)2Hg + 2XJ,
являете^ особым случаем получения ртутноорганических соединений, не
имеющем аналогий среди реакций других типов галоидных производных
углеводородов.
ЛИТЕ РАТУ РА
1. Frankland Е. Ann., 85, 365 (1853).
2. Пат. США 2914451 (1959): С, А., 54, 5467 (1960),
3. Strecker A. Ann., 82, 75 (1854).
4. Buckton G. Ann., W8, 103 (1858).
-5. J о s e J., E s p i n о z a С. C, A., 42, 2924 (1948); Rev. quim. farm. (Santiago, Chile),
4, N 56, 2 (1947).
Взаимодействие RHal с Hg и ее солями
17
6. Z i п i n N. N. Ann., 96, 363 (1855).
7. Linnemann Е. Ann., 140, 180 (1866).
8. Henry L. Ber., 17, 1132 (1884).
9. S a к u r a i I. J. Chem. Soc., 37, 658 (1880); 39, 485 (1881); 41, 360 (1882).
9a. Simmons H. E., Smith R. D, J. Am. Chem. Soc., 81, 4256 (1959).
10. N e f I. U. Ann., 308, 299 (1898).
11. Несмеянов A. H., Фрейдлнна P. X., ВелнчкоФ. К. Изв. AH
СССР, OXH, 1958; 40; ДАН СССР, 114, 557 (1957).
12. ФрейдлинаР. X., ВелнчкоФ. К- Изв. АН СССР, ОХН, 1961, 55.
13. ФрейдлинаР. X., В е л н ч к о Ф. К. Изв. АН СССР, ОХН, 1959, 1225.
14. Н е с м е я н о в А. Н., Р е у т о в О. А. Изв. АН СССР, ОХН, 1953, 655.
15. Р а з у в а е в Г. А., Шубенкой. А. ЖОХ, 21, 1974 (1951).
16. Реутов О. А., Беспрозванный М. А. ДАН СССР, 80, 765 (1951).
17. М а у п а г d I. L. J. Am. Chem. Soc., 54, 2108 (1932).
18. Banks A. A., E m e 1 e u s H. J., H asze 1 di ne R. N., Kerrigan V.
J. Chem. Soc., 1948, 2188.
19. EmeleusH. J.-, H asze 1 d i ne R. N. J. Chem. Soc., 1949, 2948.
20. EmeleusH. J. Bull. Soc. chim. France, 1953, 909; Fortschr. chem. Forsch., 2,
609.
21. Haszeldine R. N. Angew. Chem., 66, 693 (1954).
22. E m el ё u s H. J., Lagowski J. J. J. Chem. Soc., 1959, 1497.
23. H a s z e 1 d i n e R. N. J. Chem. Soc., 1949, 2856.
24. В a i g M. M., C u 1 1 e n W. R. Canad. J. Chem., 39, 420 (1961).
25. BaigM. M., C u 1 1 e n W. R. Canad.-J. Chem., 40, 161 (1962).
26. Boynton Ch. F., jr.,.T а у 1 о r H. A. J. Chem. Phys., 22, 1929 (1954).
27. К p e т о в A. E., А б p а м о в В. А. ЖОХ, 7, 1572 (1937).
28. Н е с м е я н о в А. Н., Р е у т о в О. А., П о д д у б н а я С. С. Изв. АН СССР,
ОХН, 1953, 649.
29. Белецкая И. П., Реутов О. А.,- Артамкнна Г. А. ЖОХ, 32, 241 (1962).
29а. Белецкая И. П., Артамкнна Г. А., Шевлягина Е. А., Р е у -
то в О. А. ЖОХ, 34, 321 (1964).
30. Р е у т о в О. А., Б е л е ц к а я И. П., А р т а м кина Г. А. ЖОХ, 30, 3220
(1960).
31. М i с h а е 1 i s A., R eese А. Вег., 15, 2876 (1882).
32. D a s G u р t а Н. N. J. Indian Chem. Soc., 14, 400 (1937).
33. Несмеянов А. Н., Кудрявцева Т. А. Уч. зап. МГУ, 151, 57 (1951).
34. Л а т я е в а В. Н., Малышева А. В., Разуваев Г, А. Ж. Всесоюзн.
хим. общ., 7, 594 (1962).
35. ВязанкннН. С., Разуваев Г. А., Гладышев Е. Н. ДАН СССР, 151,
1326 (1963).
36. V a u g h n Т. Н. J. Chem. Soc., 55, 3453 (1933).
37. Blanchard E. P., Blomstrom D. C.', S1 mm оnsH. E. J. Organomet. Chem.,
3, 97 (1965).
Глава 11
ПОЛУЧЕНИЕ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПОСРЕДСТВОМ РЕАКТИВА ГРИНЬЯРА
И ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ ЛИТИЯ
Действие реактива Гриньяра на соли ртути является наиболее употре-
бительным методом синтеза алкил-, циклоалкил- и арилалкилртутных солей
(RHgX), а также соответствующих полных симметричных ртутнооргани-
ческих соединений R2Hg. Однако получение больших количеств вещества
R2Hg удобнее осуществлять взаимодействием алкилгалогенидов с амаль-
гамой натрия в тех случаях, когда этот метод применим. Синтез посредством
реактива Гриньяра имеет применение и для получения простейших арома-
тических соединений ртути, хотя в этом случае проще пользоваться диазо-
методом и иногда прямым меркурированием.
Первые работы по применению реактива Гриньяра для синтеза металло-
органических соединений, в том числе и ртутноорганических, принадлежат
Пфейферу [1]. В дальнейшем наиболее важные работы в этой области опуб-
ликованы Гильпертом и Грютнером [2], Марвелом и Гудом [3], Гильма-
ном и Броуном 14], Слотта и Якоби [51.
При взаимодействии галогенида ртути с магнийорганическими соедине-
ниями проходят следующие реакции:
HgX2 + RMgX RHgX + MgX,,
RHgX + RMgX -> RaHg + MgX2,
RaHg-f-HgXa-^ 2RHgX.
Кроме того, в случае неодинаковости галоидов в соли ртути и реактиве
Гриньяра идет обмен галоидов.
В качестве исходной соли ртути чаще всего используют сулему, которая
и наиболее доступна и лучше других галогенидов растворима в эфире. При
введении ее в реакцию в твердом виде, если нет достаточного размеши-
вания, часто наступает образование твердых комьев, совершенно нарушаю-
щих течение реакции. Поэтому наиболее целесообразно вводить сулему,
помещая ее в гильзу соединенного с реакционной колбой экстрактора и под-
вергая постепенному вымыванию эфиром в реактив Гриньяра. Бромная
ртуть, хотя и немного менее растворима, имеет то преимущество, что, будучи
гораздо более рыхлым порошком, не образует комков (см., например, полу-
чение CeH5CH2HgCl на стр. 22). Иодная ртуть, несмотря на малую раство-
римость, вполне пригодна и находит применение при синтезе ртутноорга-
нических иодидов. Что касается применимости различных галоид-
производных соединений, то пригоден любой из них, способный дать
реактив Гриньяра. При осуществлении первой фазы реакции, т. е. при
синтезе ртутноорганических галогенидов, необходимо употреблять ртутную
соль и реактив Гриньяра, содержащие один и тот же галоид. Нарушение
этого правила ведет к получению подчас очень, трудно разделимых смесей.
Так, при действии бромистого н-бутилмагния на сулему получена пример-
Получение RjHg и RHgX посредством RMgX и RLi
19
но эквимолекулярная смесь хлористой и бромистой н-бутилртути, которую
так и не удалось разделить [3]. Интересно отметить, что температура плав-
ления такой смеси очень мало отличалась от близких точек обоих галоге-
нвдов. Аналогично дело обстояло в случае бромистых «-и изопропилмагния
и сулемы. Подобные же трудности возникают и при реакции алкилмаг-
нийиодидов с сулемой.
По Хинкёлю и Энжелу [6] в результате реакции CH3MgJ с HgCla обра-
зуется молекулярное соединение 2CH3HgJ •CH3HgCl. Поскольку взаимодей-
ствие хлоридов алкилмагния с сулемой с образованием хлористой алкил-
ртути, по Марвелу и Гульду, идет не так гладко, наилучшей комбинацией
для получения ртутноорганической соли Слотта считает бромную ртуть и
бромистый алкилмагний. Йодиды и хлориды он считает более удобным полу-
чать уже из бромидов через гидроксиды (см. гл. XIV, стр. 330). Бромная ртуть
имеет то преимущество, что, будучи растворимой в горячей воде, в отличие от
иодной позволяет легко отделить продукт реакции. Впрочем и иодную ртуть
можно удалить без затруднения осторожным растворением в холодном вод-
ном растворе КД, в котором ртутноорганические иодиды нерастворимы.
Румпф [7] предлагает использование реакции бромистого алкилмагния
с сулемой и превращение полученной таким образом смеси обоих галоидных
алкилртутей в чистую бромистую или хлористую алкилртуть через этоксид
алкилртути, подвергая смесь галогенидов действию раствора алкоголята
натрия в спирте и осаждая фильтрат посредством водной соляной кислоты
или бромистого водорода.
Если имеют в виду получение полного ртутноорганического соединения,
то можно, разумеется, брать реагенты, включающие и разные галоиды.
Осуществление второй стадии реакции имеет свои трудности: малая раство-
римость ртутноорганического галогенида в эфире обусловливает медленное
течение реакции. Поэтому для получения хорошего выхода диалКилртути
необходимо механическое перемешивание, длительное нагревание и
большой избыток (30—70%) реактива Гриньяра [3]. Можно получить еще
лучшие выходы (70—80% вместо 45—65%), если повысить температуру ре-
акции таким путем [4, 8]: после введения всей,сулемы из экстрактора в ре-
акционную колбу, экстрактор заменяют на прямой холодильник и начи-
нают отгонку эфира, постепенно доводя водяную баню до кипения и про-
должая механическое размешивание. При этом способе работы не требуется
избытка реактива Гриньяра и сокращается время взаимодействия. В случае
особо трудной растворимости в эфире промежуточно образующейся соли
RHgX реакцию ведут с добавкой бензола.
Целесообразно ’ также получать диалкилртуть действием реактива
Гриньяра RMgX на ртутноорганический галогенид RHgX. Так получены,
например, дидодецил-, дитетрадецил-, дигексадецил-, диоктадецил- [9],
дибензил- [10, 11], ди-0-фенилэтил- [121, ди-0-метил-0-фенилпропил- [131,
ди-н-вторичнобутилфенил-[10], дифенилртуть [14], ди-у-бЬенилпропилртуть
НО]. \ ‘
Получение диалкилртути с третичными радикалами должно проводиться
вследствие ^Малой термической устойчивости такого рода веществ при ком-
натной температуре. Такие вещества, а также, в меньшей степени, соеди-
нения ртути с вторичными радикалами относительно мало устойчивы, полу-
чаются с значительно более низкими выходами, чем их аналоги с первичными
жирными или же ароматическими радикалами, и гораздо менее изучены.
Во всех случаях реактив Гриньяра должен быть освобожден от остатков
магния декантацией или лучше фильтрованием.
Синтез ртутноорганических соединений при помощи магнийорганических
соединений может проводиться в тетрагидрофур.ане. Как показано [15] на
примере синтеза ди-«-пропилртути и применено к получению дициклопрот
20
Ртутноорганические соединения. Лит. стр. 33—35
пил- [15] и дивинилртути [15, 16], проведение реакции не в эфире, а в тет-
рагидрофуране снижает продолжительность реакции и значительно повы-
шает выход ртутноорганического соединения. Но, разумеется, при выборе
среды реакции следует сообразовываться со свойствами получаемого про-
дукта. Так, низкокипящие вещества, как диметилртуть (т. кип. 92°С),
из-за большого удобства выделения лучше получать в эфире. Проводят ре-
акции и в смеси эфира и тетр а гидрофур ан а.
Ртутноорганические соединения могут быть получены при помощи маг-
нийорганических соединений и в безэфирной среде— в ксилоле [17], в геп-
тане [18] и, как это показано на единичных испытанных примерах, с выхо-
дами не меньшими, чем если реакцию вести в эфире.
Другие ртутные соединения, кроме галоидных солей двухвалентной рту-
ти, практически не применяются как исходный материал для препаративных
целей при-синтезе ртутноорганических соединений посредством реактива
Гриньяра. Однако следует отметить, что некоторые вещества, содержащие
ртуть, связанную с углеродом, при достаточной лабильности этой связи,
вступают в обмен с реактивом Гриньяра по типу.
RHgX 4- R'MgBr -> R'HgX + RMgBr и т. п.
Таковы реакции, бромистой а-нафтилртути с бромистым этилмагнием,
дающие диэтил ртуть [2], аллифенилртути с бромистым фенилмагнием [19]:
CH2=CHCH2HgC6H5 + C6H5MgBr -» CsH5MgCH2CH=CH2 + CeH6HgBr,
галогенмеркурацетофенона с бромистым этил- и фенилмагнием [20]
CGH5COCHaHgX + RMgX CeH5COCH2MgX + RHgX,
наконец, бромистой о-оксифенилртути с бромистым этилмагнием [201:
HOCsHiHgBr + 2C2H5MgBr -> BrMgOC8H4MgBr + С2Н6 + C3H5HgBr.
Но бпс-бромацетиленид ртути с бромистым фенилмагнием образует бром-
ацетиленид магния и дифенилртуть [21]:
(BrC=C)2Hg + 2CeH6MgBr 2BrC=CMgBr + (CeH6)aHg.
Наконец, сюда же относятся реакции продуктов присоединения солей
ртути к олефинам, их производным и ацетиленам. Многие типы этих ве-
ществ реагируют с реактивом Гриньяра как свободная соль ртути [22, 23]:
GsHgOCaHiHgBr + 2C2H5MgBr -» 2С2Н4 + C2He + C2HsHgBr + Mg2OBr2,
ClCH=CHHgCl + QHsMgBr C2H2 + C2H5HgCl + MgClBr,
CeH&CH-CH2HgX + CaH^gX -> CeHsCH=CH2 -ф C2H5HgX ф CHsOMgX.
OCHs
О реакциях обмена радикалами R2Hg с RMgX, R2Hg и RLi см. главу
XIV, стр. 318.
ПОЛУЧЕНИЕ РТУТНООРГАНИЧЕСКИХ ГАЛОГЕНИДОВ
Синтез бромистой метилртути [б]. Бромистый метилмагний получают из 12 г магния
в 300 мл абсолютного эфира и 48 г бромистого метила. Раствор дополнительно нагревают в
течение 0,5 часа на водяной бане и отфильтровывают через стеклянную вату. При легком
потряхивании прибавляют 200 г мелкокристаллической бромной ртути и взвесь кипятят
еще 1 час. По охлаждении смесь разлагают 20 мл воды, зфир отгоняют, разложение зааер-
шают действием избытка воды. Полученную кашу отсасывают и дважды кипятят с 1 % -ной
соляной кислотой. Выход 139 г (94%). Перекристаллизация из этанола. Потеря при этом,
как и при перекристаллизации всех таких галогенидов, составляет 10%. Т. пл. 172°С (см.
также [24]).
Получение RaHg и RHgX посредством RMgX и RLi 21
Синтез иодистой метилртути [3]. К реактиву Гриньяра из 2,4 г магния, 15 г йодисто-
го метила и 50 мл эфира, профильтрованному через стеклянную вату и помещенному в колбу,
снабженную обратным холодильником, капельной воронкой и мешалкой с ртутным затво-
ром, прибавляют при перемешивании 30 г сулемы в 150 мл эфира с такой скоростью, чтобы
эфир спокойно кипел, затем кипятят еще 1 час, добавляют, охладив, немного воды и эфир
отгоняют. К остатку прибавляют 200 мл воды и 5—10 мл конц. соляной кислоты, чтобы
удалить избыток сулемы и соли магния. Йодистую метилртуть отсасывают и кристаллизуют
из спирта. Выход 29—30 г (85—88%), т. пл. 142—143°С.
Однако, по Хинкелю и Энджелу [6], при работе по этой прописи по-
лучается молекулярное соединение 2C-H3HgJCH3HgCl. Чистая йоди-
стая метилртуть получается 16], если вместо сулемы взять эквивалентное
количество иодной ртути.
Синтез бромистой этилртути [5]. Реактив Гриньяра из 12 г магния, 55 г бромистого
этила и 200 мл эфира нагревают в течение 30 мин., отфильтровывают через стеклянную вату
и в него постепенно вводят 200 г HgBra так, чтобы эфир все время слегка кипел. Дальнейшая
обработка тождественна с описанной для бромистой метилртути. Выход 140 (90%). Пере-
кристаллизация из этанола. Т. пл. 198°С (198,5°С [25]).
Описано влияние среды реакции и природы галоида в галогениде ртути
на выход C2H5HgJ при реакции йодистого этилмагния с галогенидом ртути
[17], тоже—при синтезе бромистой этилртути из бромистого этилмагния [17].
Эти соли этилртути получаются с хорошим выходом, если реакцию вести
в ксилоле.
Получение бромистой «-пропилртути [5]. Бромистый н-пропилмагний из 12 г магния,
62 г бромистого пропила 'в 100 мл эфира и 200 г бромной ртути вводятся во взаимодействие,
как это описано выше для бромистой метилртути. Единственным различием является рас-
слоение смеси после добавления 100 г ртутной соли. Выход 140 г (86%). Кристаллизация из
этанола, т. пл. 140° С.
Подобным образом получены [5] также бромистые н-бутилртуть (т. пл.
136°С), н-амилртуть (т. пл. 127°С), н-гексилртуть (т. пл. 127,5° С), «-гептил-
ртуть (т. пл. 118,5°С), н-цетилртуть (т. пл. 101,5°С). Выходы для средних
членов ряда 50%, для цетильного производного—90%.
Из бромистого изопропилмагния и сулемы бромистая изопропилртуть
получена в виде двойной соли с HgCl [26]. См. также стр. 24. ,
Получение бромистой вторичной бутилртути [27]. Реактив Гриньяра из 100 г вторич-
ного бромистого бутила в 300 мл эфира прибавляют к тонко измельченной бромной ртути
(300 г), суспендированной в 250 мл эфира, с такой скоростью, чтобы поддерживалось кипение.
Смесь сильно загустевает и ее энергично перемешивают во время и 2 часа после прибавле-
ния, а затем разлагают холодной 0,5%-ной водной серной кислотой, затем эфирный слой от-
деляют и эфир отгоняют в вакууме. Остающийся осадок дважды кристаллизуют из спирта;
выход 97 г (70%), т. пл. 39°С.
Получение бромистой третичной бутилртути [28]. В эфирный раствор бромистого тре-
тичного бутилмагния на холоду вводят избыток бромной ртути. Через час смесь разлагают
водой, эфирйый слой отделяют и сушат над хлористым кальцием. Эфир выпаривают. Если
продукт нагреХать хотя бы в растворе, он разлагается, выделяя закисную бромистую ртуть.
Его не удается перекристаллизовать. Плавится с разложением при 106°С.
Описан синтез хлористой третичной бутилртути [19].
Получение хлористой иеопеитилртути [29]. Реактив Гриньяра, полученный с 90%-ным
выходом из 105 г (1 моль) неопентилхлорида, 24 г (1 г-атом) магния и 350 мл сухого эфира
с такой скоростью приливают к 270 г (избыток 32 г) измельченной сулемы, суспендирован-
ной в 1 л сухого эфира, чтобы поддерживалось непрерывное умеренное кипение. После это-
го смесь кипятят 3 часа при перемешивании. Эфир отгоняют, остаток перемешивают с 1 л
воды. После кристаллизации из 2 л 95%-ного этилового спирта выход составляет 239 г
(90%) хлористой неопентилртути; т. пл. 117—118°С.
Получение хлористой неопентилртути см. также [30, 31, 42].
Из хлористого триметилсилилметилмагния и сулемы получена хлористая
триметилсилилметилртуть с выходом 80% [25, 32].
22 Ртутноорганические соединения. Лит. стр. 33—35
Из 1 моля хлористого 1-(триметилсилил)этилмагния и 2 молей сулемы
в эфире получен 1-(хлормеркур)этил(триметилсилан) CH3CH(HgCl)Si(CH3)3
[33]; т. пл. 97°С; выход 40%.
Описано получение хлористой 5-оксиамилртути из хлористого триметил-
силилоксиамилмагния и сулемы (с последующим гидролизом Si —О-связи
разбавленной соляной кислотой) [34].
Хлористая н-октилртуть получена через магнийорганическое соедине-
ние в безэфирной среде [18].
Синтез хлористой я-октилртути [18]. К 3 г (0,12 моля) опилок магния добавляют при
перемешивании в токе азота раствор 26,7 г (0,11 моля) йодистого «-октила в 50 мл гептана.
Добавление ведут при слабом кипении растворителя. Реакционную массу нагревают в те-
чение 2 час., затем охлаждают льдом; при перемешивании добавляют малыми порциями
20 г (0,074 моля) сулемы и смесь нагревают в течение 1,5 час. Реакционную массу отфильтро-
вывают, осадок несколько раз промывают на фильтре 30 мл кипящего гептана. При охлаж-
дении фильтрата выпадают белые кристаллы. После обработки раствором едкого кали в ме-
таноле и затем насыщенным раствором хлористого калия получают «-октилмеркурхлорид.
Выход 21 г (81%), т. пл. 115°С.
Получение хлористой 2,2,3,3-тетраметил-1-бутилртути [35]. 6,3 г магния нагревают
с кристалликом иода до удаления последнего, добавляют 2,2 мл бромистого этила, 80 мл
эфира, смесь нагревают до кипения, и в течение 8 час. вводят раствор 33,4 г (0,225 моля)
хлоргексаметилэтана в 150 мл эфира. Кипячение затем продолжают 19 час. Выход магний-
органического средине..ия составляет 70% . Полученный так реактив Гриньяра медленно при
размешивании вливают в раствор 70 г сулемы в 250 мл абс. эфира, смесь кипятят с обратным
холодильником 4 часа, эфир отгоняют, добавляют 300 мл воды и 10 мл соляной кислоты;
массу нагревают на водяной бане. Ртутноорганическое соединение отсасывают, промывают
водой, перекристаллизовывают сначала из водного этилового спирта, затем из метанола и,
наконец, из петролейного эфира. Выход 35%. Окончательная т. пл. 170—171°С.
Получение гидроокиси к-додецилртути [36]. В колбе емкостью 300 мл готовят реактив
Гриньяра из 2,5 г магния и 24,9 г (0,1 моля) бромистого н-додецила (т. кип. 140—142° C/11 мм)
в 150 мл эфира. Затем вводят маленькими порциями эквивалентное количество сухой суле-
мы. Смесь кипятят 3 часа, дважды прерывая кипение и охлаждая, чтобы размешать массу
стеклянным шпателем, затем разлагают ее 75 мл воды, отгоняют эфир, охлаждают, отса-
сывают и продукт (38 г) сушат на воздухе. Его нагревают затем на водяной бане в течение
10 мин. с 160 мл 50% -ной смеси бензола со спиртом и 100 мл 40%-ного раствора едкого кали
в метиловом спирте. Кипящую жидкость фильтруют. Из фильтрата по охлаждении выпадает
гидроокись, которую перекристаллизовывают из пиридина. Т. пл. 80°С. Т. пл. хлорида
111,5°С; Миллс [9]даетдля хлорида т. пл. 114—114,5°С; т. пл. бромида 108° С. Выход 69%,
считая на лаурилбромид [36] (см. также [37]).
Аналогично получены [36] гидроокиси, хлориды и бромиды н-тетраде-
цилртути, н - гекс адеци л ртути и н-октадецилртути. См. также [9].
Получение хлористой бензилртути [2]. При сильном и непрерывном встряхивании (пе-
ремешивании) вводят порциями по 2 г 46,7 а (1,1 моля) тонко измельченной сухой сулемы
в раствор 23,8 г хлористого бензилмагния в 100 мл эфира. Если реакцию предоставить са-
мой себе, даже всего на несколько секунд, то-масса плотно прилипнет ко Дну и едва может
быть отделена. Ускорять реакцию нагреванием нецелесообразно, так как и в этом случае
наступает образование комков. После внесения всей сулемы колбу встряхивают (переме-
шивают реакционную смесь) еще некоторое время и оставляют стоять на 24 часа, после чего
кипятят 2 часа с обратным холодильником. Затем смесь разлагают льдом с серной кислотой,
остаток,трижды нагревают с 1%-ным раствором соляной кислоты до температуры не выше
80°С для удаления избытка сулемы, так как сырой продукт легко плавится. Перекристал-
лизация из смеси равных частей ксилола и спирта. Т. пл 104° С; выход чистого продукта
43 г (84%). См. также [38—40].
Описан синтез бромистой [10, 41] и хлористой [42] |3 -фенил этил ртути
(в последнем случае наряду с RaHg).
При взаимодействии бромистого винилмагния с сулемой [43], бромной
[12, 43] или иодной [43] ртутью (1 моль RMgX : 1 моль HgX2) в тетрагидро-
фуране получена хлористая винилртуть с. выходом 78%, бромистая винил-
ртуть с выходами 64% [12] и 67% [43] и иодистая винилртуть с выходом 83%
[43].
Получение RaHg и RHgX посредством RMgX и RLi 23
Получение бромистой винилртути [12]. В трехгорлую колбу (1 л), снабженную ртутным
затвором с механической мешалкой, капельной воронкой, окруженной рубашкой с сухим
льдом — холодильником длиной в 60 см, наполненным охладительной смесью из ацетона
и сухого льда и закрытым хлор кальциевой трубкой,— помещают 4,86 г (0,2 моля) магния
и кристаллик иода в 50 мл абсолютного тетрагидрофурана. К нагретой на водяной бане при
60—65° С смеси прибавляют при размешивании по каплям раствор 25,5 г (0,23 моля) сухого
бромистого винила в 50 мл абсолютного тетрагидрофурана. Реакция начинается через 20—
30 мин. после прибавления 1—2 мл раствора и сопровождается разогреванием и обесцвечи-
ванием реакционной массы. По мере прибавления раствора магний исчезает, а реакционная
смесь окрашивается в коричневый цвет.
Если по окончании прибавления раствора остался не вошедший в реакцию магний, то
добавляют по каплям еще некоторое количество раствора бромистого винила в абс. тетра-
гидрофуране до полного исчезновения магния. Прибавление продолжают в течение 3—4 час.
К полученному реактиву Гриньяра (при энергичном размешивании) при температуре бани
5—8°С прибавляют по каплям раствор 68,4 г (0,189 моля) сухой бромной ртути в 90 мл аб-
солютного тетрагидрофурана; при этом выпадает белый объемистый осадок. Продолжитель-
ность прибавления 1—1,5 часа. Затем продукты реакции нагревают При 60—65° С в течение
2 час. Реакционную массу разлагают, приливая по каплям в течение 10 мин. 100 мл 3%-ной
соляной кислоты при охлаждении льдом с водой/Полученный раствор выливают в 1 л хо-
лодной воды, выпавший осадок отфильтровывают, промывают водой и сушат над хлористым
кальцием в течение 12 час. Перекристаллизацией из четыреххлористого углерода получают
36,8 г (64%) бромистой вииилртути с т. пл. 169,5—171°С.
Из бромистого бутенилмагния и бромной ртути получена бромистая
транс-кротилртуть [45].
Описано применение ультразвука для получения галогенида пропаргил-
ртути из смеси бромистого пропаргила, магния и сулемы [44].
Из бромистого стирилмагния и бромной ртути в эфире бромистая
стирилртуть получена с выходом 36% [46].
Синтез бромистой циклогексилртути [47]. В раствор 23 г бромистого циклогексилмаг-
ния в 100 мл абс. эфира вносят небольшими порциями 45 г (1,03 моля) очень тонко измель-
ченной бромной ртути при непрерывном энергичном размешивании. Сначала с значитель-
ным выделением тепла происходит растворение. Спустя некоторое время наступает разделе-
ние слоев. При дальнейшем внесении бромная ртуть более не растворяется и при постоянном
умеренном кипении эфира постепенно превращается в хлопья. Смесь нагревают до кипе-
ния еще 2 часа, разлагают льдом, подкисляют бромистоводородной кислотой и весь
эфир удаляют при 60° С в токе воздуха. Отсосанный и промытый холодным абс. спир-
том осадок в течение 30 мин. при перемешивании обрабатывают для удаления избытка бром-
ной ртути 500 мл горячей воды, отсасывают в горячем состоянии, тщательно промывают во-
дой и спиртом и высушивают при 60°С. Выход 35 г (78%).Перекристаллизация из бензола.
Бесцветные блестящие листочки; т. пл. 153°С.
Получение хлористой циклогексилртути [47]. Вещество получают из 20' г хлористого
циклогексилмагния в 100 .ил эфира и 48 г сулемы (аналогично бромистой циклогексилртути).
Выход 30 г. После кристаллизации из спирта или бензола — бесцветные блестящие листоч-
ки. Т. пл. 163— 164°.С.
Из бромистого 4-метилциклогексилмагния и сулемы или бромной ртути
[48] получена с 65%-ным выходом смесь бромистой цис- и транс-4-метил-
циклогексилртути, разделенная на основании разной их растворимости
в бензоле. Менее растворимый транс-изомер перекристаллизован из бензо-
ла. Цис-изомер из маточника очищен хроматографированием на окиси
алюмини'д.
Описанб получение изомеров 2-хлормеркуркамфанов ’ реакцией Гринь-
яра из хлористого d, /-борнила [49, 50].
Из бромной ртути и нортрициклилхлорида или смеси его с дегидронор-
борнил хлоридом через реактив Гриньяра получен нортрициклилмеркур-
бромид:
gC ’ /и g в г
+ HgBr2
+ MgClBr
24
Ртутноорганические соединения. Лит. стр. 33—35
Получение бромистой фенилртути [2]. К 28,7 г бромистого фенилмагния в 100 мл эфира
при сильном встряхивании добавляют малыми порциями 72 a HgBrs (1,25 моля), и смесь
сильно кипятят 4 часа. Затем ее охлаждают, эфир, уже не содержащий CeHsMgBr, сливают,
остаток трижды нагревают до кипения с 1%-ной соляной кислотой, чтобы удалить избы-
точную HgBr2, основательно промывают горячей водой, спиртом и эфиром и высушивают при
100° С. Выход на сырой продукт 56 г (98%). Перекристаллизация из пиридина. Т. пл. 275°С
(см. также [17, 41, 51]).
Хлористая фенилртуть получена из хлористого фенилмагния и сулемы
(17].
Получение бромистой а-нафтилртути [2]. К 22,3 г бромистого а-нафтилмагния в 100 мл
эфира добавляют при сильном встряхивании небольшими порциями 43 г HgBr2 (125% от
теории).
Смесь кипятят 4 часа, затем охлаждают, эфир сливают с остатка, последний трижды
извлекают кипящей 1 % -ной бромистоводородной кислотой,чтобы удалить избыточную HgBr2,
основательно промывают спиртом и эфиром, перекристаллизовывают из пиридина. Выход
чистого продукта 30 г (75%); т. пл. 202° С.
1,4-Диброммеркурбензол с38%-ным выходом выделен при действии су-
лемы (лучше брать HgBra [52]) на реактив Гриньяра, полученный по мето-
дике «в сопровождении» из магния и n-дибромбензола и бромистого этила
[52]. Из-за меньшей растворимости 1,4-диброммеркурбензола он легко от-
делим от одновременно образующихся бромистой этилртути и бромистой
n-бромфенилртути. От последней бромистая этилртуть в свою очередь от-
деляется отгонкой с паром.
Получение 1,4-диброммеркурбензола [52]. К перемешиваемой взвеси 2,40 г маг-
ния в 50 мл сухого эфира прибавляют в течение 30 мин. 2 мл бромистого этила в 50 мл эфира.
Затем в течение 75 мин. прибавляют раствор 4,72 г (0,02 моля) и-дибромбензола и 3,85 мл
(всего 0,06 моля) бромистого этила в 100 мл эфира. Энергичное кипение поддерживают в те-
чение 12 час., затем прибавляют еще 1,2 мл бромистого этила. Когда через 1 час весь магний
оказывается вошедшим в реакцию, в течение 24 час. вводят 27,2 г (0,1 моля) сулемы (поме-
щенной в экстрактор Сокслета).
Твердый продукт отфильтровывают и тщательно промывают водой, кипящим этиловым
спиртом и кипящим бензолом. Осадок — 1,4-Диброммеркурбензол, вес 4,88 г (38%).
От фильтрата отгоняют растворители и остаток подвергают перегонке с паром. Из от-
гона получают 11,9 а бромистой этилртути. В перегонной колбе остается бромистая п-бром-
фенилртуть 1,96 г (23%). После кристаллизации из бензола т. пл. 234—236°С.
Для получения ArHgCNO (Аг — С6НБ, ос-СюН7) использован фульми-
нат ртути [126].
Ниже приводятся краткие данные о получении ртутноорганических
солей реакцией Гриньяра:
RHgCI: R = СН3 [25, 53], С2Н6 [25, 53], п-С3Н7 [25, 53], г-С3Н7 [54,
53], «-С4Н9 [25 , 53], г-С4Н9 [53], /г-С6Нп’ [53], /-С5НП [25, 53],
(СН3)3С(СНа)2 [55], 1-С10Н7СН2 [56], 1-метилциклогексил [54], 3-дибензо-
фурил [57].
RHgBr. R =СН3 [25], СаНБ [25, 58], п-С3Н7 [25], z-C3H7 [41], г-С4Н9
[41], s-C4H9 [41], i-C5Hn [25], f-CsHn [28], n-C6H13 [25], n-C7H15 [25],
(CH3)3CCH2CH2CH2 [59], n-C8H17 [25,J, s-C8H17 [28], л-С9Н19 [41], C12H26
[41], C6H5CH2 [40, 41], CeH5CH2CH2 [41], c-C6Hn [411, C6H5 [41],
o-CH3CeH4 [2,41], m-CH3CeH4 [41], p-CH3C6H4 [2, 41, 60], BrHg(CH2)5HgBr
[2, 26].
RHgBr HgCl: R = и-С3Н7; z'-C3H7, zi-CgHu, 1-СБН4х, н-СвНхз, С«НБСН2,
c-CeHn, o-CH3OCeH5, p-C2H5OCeH4 [26].
RHgJ: R= CH3 [25], C2H5 [25], C3H7 [25, 61], i-C3H, [61], n-C4H9
[25], i-C4H9 [25], i-C5Hu [25], n-CeHu [61].
Получение RaHg и RHgX посредством RMgX и RLi 25
ПОЛУЧЕНИЕ ПОЛНОЗАМЕЩЕННЫХ РТУТНООРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
Получение полнозамещенных ртутноорганических соединений в эфире
(и тетрагидрофуране)
Синтез диметилртути [3]. Следует иметь в виду особую токсичность диметилртути и дру-
гих летучих жирных полнозамещенных соединений ртути, поэтому с этими соединениями
необходимо «работать под хорошо действующей тягой, дегазируя посуду хлорной или бром-
ной водой. Реактив Гриньяра получают из 180 г йодистого метила, 30 г магния в 500 мл абс.
эфира. Его отфильтровывают через стеклянную вату, нагревают до кипения и в него через,
широкий обратный холодильник в течение 45 мин. порциями по 5—10 г вносят 100 г сулемы.
По окончаний прибавлений всей сулемы смесь кипятят 10—12 час., затем охлаждают и раз-
лагают медленным введением через обратный холодильник 250 мл воды; эфирный' слой от-
деляют, водный экстрагируют 100 мл эфира, соединенные эфириые растворы промывают
15—20 мл воды, высушивают хлористым кальцием и тщательно перегоняют на эффективной
колонке. После того, как большая часть эфира была отогнана, остаток переносят в меныпукэ
колбу и продолжают перегонку. Для получения лучшего выхода весь эфир перегоняют еще-
1 раз. Выход 51—56 г (61—66%); т. кип. 89—92*С; т. кип. чистого вещества при 740 мм
92°С, d^2,2 2,9541, п^’2 1,5327. См. также далее (стр. 30) получение диметилртути [4, 81
с отгонкой эфира во время реакции. Описано получение диметилртути, содержащей Hg203
I(CH3)3SiCH2]2Hg получена из (CHa)3SiCH2MgCl и HgCl2 в тетрагидро-
фуране [63]. Выход 48,5%, т. кип. 49—50°С/0,35 мм, fio 1,4869.
Реакцией (CH3)2C6H5SiCH2MgCl с HgCl2 в эфире (извлечение сулемы в
экстракторе Сокслета и затем кипячение в течение 72 час.) получен [64J
[(CH3)2CeH6SiCH2]2Hg с выходом 71%.
Получение диэтилртути* [3]. Синтез проводят в таких же условиях, как и предыдущий,
за исключением того, что применяют механическое размешивание. В реактив Гриньяра из
125 г бромистого этила, 25 г магния и 500 мл эфира введено 97 г сулемы. Отделение диэтил-
ртути от эфира не представляет затруднений. Во избежание разложения перегонку лучше
производить под уменьшенным давлением. Выход диэтилртути, кипящей от 97 до 98° С при
125 мм, составил 55—59 г (60—63%). При атмосферном давлении т. кип. 159°С, d23’2 2,4237,
п23’2 1,5399.
Выход диэтилртути зависит от продолжительности нагревания [65]. Так,
при кипячении на водяной бане после введения сулемы в течение 10—;
14 час. выход диэтилртути составил 50%, при кипячении в течение 20—48 час.
средний выход 70—85%, считая на взятую сулему (условия реакции:
550 а бромистого этила, 120 г металлического магния, 2000 мл абс. эфира,
фильтрование от невошедшего в реакцию магния, постепенное прибавле-
ние сулемы — 395 г; после кипячения разложение 5—7 % -ной соляной кисло-
той, сушка эфирного раствора хлористым кальцием, перегонка диэтил-
ртути в вакууме [65]) (см. также [4, 8, 65—67а]).
Описан синтез диэтилртути с меченой ртутью [68].
При взаимодействии реактива Гриньяра с каломелью по уравнению
[69]
\ 2CaH5MgBr Hg2Cl2 —» (CaHslaHg -(- Hg -f- MgCl2 4- MgBr2
образуется диэтилртуть.
Получение ди-к-пропилртути* [3]. В реактив Гриньяра из 21 г магния, 108 г броми-
стого пропила и 500 мл эфира вносят, как это описано в обоих предыдущих синтезах, 98 г
сулемы. Выделено 47—53 а (45—51%) дипропилртути с т. кип. 82—84°С при 19 мм (при
атмосферном давлении т. кип. 189—191°С [41]), d20 2,0208, 1,5170. См. также [5, 9, 67].
При проведении реакции в тетрагидрофуране [15] (подробной прописи
не дано) выход 75% (см. также [6, 75а]).
* Токсична! Работать под хорошо действующей тягой.
26
Ртутноорганические соединения. Лит. стр. 33—35
Ди-н-пропилртуть наряду с бромистой н-пропилртутью получена из бро-
мистого н-пропилмагния, взятого в избытке, и бромистой ртути [70]. Из
хлористого н-пропилмагния и сулемы выход 65% [71].
Синтез диизопропилртути* [3]. Реактив Гриньяра из 24,5 г магния и 130 г изопропил-
бромида вводят в реакцию с 80 г сулемы в описанных выше условиях. Выход продукта,
кипящего при 119—121° С, составляет 50—51 г (60%); d^° 2,0024, 1,5263 (см. так-
же [67]).
Из хлористого изопропилмагния и сулемы в смеси эфира и тетрагидро-
фурана диизопропилртуть получена [67а] с выходом 87%.
Синтез ди-Я-бутилртути* [3]. Подобно предыдущему синтезу 98 г сулемы введено во
взаимодействие с реактивом Гриньяра, приготовленным из 21 г магния и 134 г н-бутилбро-
мида. Продукт необходимо перегнать дважды для того, чтобы освободиться от бутилмеркур-
галогенидов. Выход 55—56 г (47%), пределы кипения 120—123°С при 23 мм, 1,7779,
п2^ 1,5057. В перегонной колбе осталось около 42 г смеси бутилмеркургалогенидов. Выхо-
ды с отгонкой эфира достигают 67% [4], 80% [8], 92% [72] (см. также [73]).
Получение ди-вторичнобутилртути* [28]. Реактив Гриньяра, полученный из 12 г магние-
вой стружки и 75 г вторичнобутилбромида в 500 мл эфира, вводят в реакцию с 45 г сулемы.
Смесь кипятят с обратным холодильником 8 час. и затем перерабатывают, как описано выше.
Выход ди-вторичнобутилртути с т. кип. 93—96°С при 18 мм составлял 35 г (66%), 1,763,
Пд 1,511 (см. также [10, 73]).
При проведении реакции хлористого вторичнобутилмагния с сулемой
в среде диметоксиэтана в присутствии бензола ди-вторичнобутилртуть по-
лучена с выходом 20% [39].
Получение ди-третичнобутилртути* [28]. В течение 45 мин. в раствор бромистого тре-
тичнобутилмагния, полученный из 6 г магния, 35 г бромида и 500 мл эфира, малыми порци-
ями вводят £0 г бромной ртути. Реакция ведется в двухгорлой колбе, снабженной мешалкой
и пробкой с хлорнокальциевой трубкой, при непрерывном действии мешалки в течение
6—8 час. и при охлаждении водой. После разложения водой эфирный слой отделяют, промы-
вают водой, высушивают хлористым кальцием, после чего эфиру дают свободно испаряться.
По мере концентрации раствора выпадает бромистая третичнобутилртуть, которую отделя-
ют. По удалении всего эфира остаток перегоняют в вакууме. Выход ди-третичнобутилртути
(т. кип. 78—82° С при 5 мм) 2 г (9%). Продукт содержит примесь углеводорода;
1,749, 1,521.
Подобным же образом получены ди-третичноамилртуть [28], выход 21%,
т. кип. 80—84° С при 5 мм, d™ 1,649, 1,492; ди-вторичнооктилртуть [281
(8 г магния, 70 а вторичнооктилбромида, 21 г сулемы, 500 мл эфира), выход
52%, d™ 1,338, пзо 1,334.
Получение ди-н-амилртути [74] (реакцию вели в присутствии декана),
ди-d,/-амил ртути [74, 75], динеопентил ртути [31, 42], ди-н-гексил ртути [74]
(в присутствии додекана) см. приведенные ссылки. Из RMgCl и HgCl2 в
тетрагидрофуране получена (+V(C3H7CH)CH3(CH2)2Hg и соответствующая
рацемическая R2Hg [75а].
Получение ди-я-гептилртути* [76]. Реактив Гриньяра получают из 36 г магния и 270 г
бромистого и-гептила с выходом 60% . В него вводят 108 г сухой сулемы. Перемешивание
и легкое кипение поддерживается в течение 4 дней. Смесь перерабатывают обычным обра-
зом [3], ди-н-гептилртуть перегоняют при 119—122°С при давлении 0,005—0,01 мм (при
этом отогналось немного гептилмер кур галогенида). Выход 144 г (90%), 1,4935, d° 1,474.
Ди-(5-метилгексил-2)-ртуть получена из 5-броммеркур-2-метилгексана и
бромистого 5-метилгексил-2-магния [77].
* Токсична) Работать под хорошо действующей тягой.
Получение Rahg и RHgX посредством. RMgX и RLi
27
Описано получение ди-н-октилртути [71], ди-н-нонил-, ди-н-ундецил-
ртути [78], дидодецил-, дитетрадецил-, дигексадецил-, диоктадецилрту-
ти [9].
Получение дибеизилртути* [11, 79]. В реактив Гриньяра, полученный из 3,8 г магния
и 20 г хлористого бензила, постепенно прибавляют 21,4 г сулемы при энергичном перемеши-
вании, затем смесь осторожно нагревают в течение нескольких часов, затем разлагают водой,
эфирный слой отделяют от воды и сырой хлористой бензилртути. После отгонки эфира оста-
ток состоит главным образом из дибеизилртути. После перекристаллизации из спирта [79]
(или бензола) [60] вес 10 г. Т. пл. 111°С (см. также [79]).
Лучший результат получается при замене сулемы на хлористую бензил-
ртуть [111. Из 3,8 г магния, 20 г хлористого бензила и 35 г хлористой бензил-
ртути получено 40 г дибеизилртути [II].
Можно также дополнительной порцией реактива Гриньяра обраба-
тывать лишь образующуюся одновременно примесь хлористой бензил-
ртути. Работают следующим образом [80].
В реактив Гриньяра из 40 г хлористого бензила, 15 г порошка магния и 500 мл
эфира после отделения от избытка магния вносят при перемешивании и охлаждении
35 г тонко измельченной сухой сулемы; смесь кипятят 5 час. при перемешивании,
разлагают избыток реактива Гриньяра слабой уксусной кислотой, декантируют без
промывания и концентрируют до образования кристаллов. Т. пл. 110—111° С. При
концентрировании маточника из обоазующегося масла получают продукт, содержащий
хлористую бензилртуть, который обрабатывают хлористым бензилмагнием по описан-
ной выше прописи.
Описано получение дибеизилртути [10], а также ди-о-хлорбензилртути
[40], диф-фенилэтилртути [10,42], у-фенилпропилртути [10], б«с-(-ди-0-
метил-Р-фенил эти л (ртути из хлористого неофилмагния и хлористой неофил-
ртути [13].
Дивинилртуть получена из бромистого винилмагния и сулемы. Реакцию
ведут в тетрагидрофуране. Выход 60% [15], 80% [16], 85% [43]. Если вместо
сулемы взять бромную или иодную ртуть, то одновременно образуется и га-
лоидная вини л ртуть [16].
Получение дивинилртути* [43]. Для получения бромистого винилмагния в условиях
реакции Гриньяра холодильник заполняют сухим льдом и реакцию ведут в атмосфере азота.
Для начала реакции прибавляют несколько капель йодистого метила. Средой является тет-
рагидрофуран, перегнанный над литийалюминийгидридом. К полученному в этих условиях
раствору 2 молей винилмагнийбромида в 600 мл тетрагидрофурана при энергичном пере-
мешивании и охлаждении ледяной водой прибавляют раствор 0,77 молей сулемы в 250 мл
тетрагидрофурана. После того как сулема прибавлена, реакционную смесь перемешивают
при 55° С в течение 12 час., затем охлаждают до комнатной температуры; избытфк реактива
Гриньяра гидролизуют приблизительно 250 мл насыщенного раствора хлористого аммония.
Органический слой декантируют в колбу для перегонки, водный раствор солей промывают
несколькими порциями эфира, промывной эфир присоединяют к тетрагидрофурановому
раствору. Большую часть растворителей отгоняют при нормальном давлении. Перегонку
заканчивают в вакууме, прибавив парафинового масла в качестве носителя. Выход ди-
винилртуги 167 г, т. кип, 59,5°С/20 мм.
Получение бпс-перфторвинилртути из йодистого перфторвинилмагния
и сулемы ведут при —10 ч—5°С в эфире [81]; т. кип. 65—66°С/17 мм.
Реакцией бромистого алкинмагния (комплекс Иоцича) с сулемой полу-
чены диацетилениды ртути [82]:
2RC = CMgBr + HgCla -> (RC = C)2Hg + 2MgClBr.
* Токсична! Работать под хорошо действующей тягой.
28
Ртутноорганичежие соединения. Лит. стр. 33—35
Получение ди-^-феиилацетиленида ртутя[82]. Растворб.Оа сулемы в 25мл тетрагидрофу-
рана прибавляют в течение 5 мин. к нагреваемому с обратным холодильником раствору бро-
мистого феиилэтинилмагния(из5>1гфенилацетилена) в 100мл тетрагидрофурана. Через 1 час
смесь обрабатывают водным раствором хлористого аммония, большую часть растворителя от-
гоняют в вакууме и остаток извлекают эфиром. Из эфирного^экстракта получают 6,38 г
(84%) ди-р-фенилацетиленида ртути в виде бесцветных пластинок, т. пл. 123—124°С. Кри-
сталлизация из этанола.
Подобным же образом получена ди-(окт-1-инил)ртуть. Выход 69%.
Бесцветные пластинки, т. пл. 82—83°С. Кристаллизация из легкого петро-
лейного эфира.
Диаллилртуть получена из бромистого аллила и бромной ртути при
проведении реакции в эфирно-тетрагидрофурановом растворе при комнат-
ной температуре [83].
Получение диаллилртути [83]. К эфирному раствору бромистого аллилмагння, приго-
товленному в токе азота из 29,4 а (0,242 моля) бромистого аллила и 5,85 г (0,240 моля) маг-
ния в 150 мл абсолютного эфира, прибавляют по каплям 22 г (0,061 моля) HgBra в 35 мл
абсолютного тетрагидрофурана при 5—7° С. После перемешивания в течение часа при ком-
натной температуре реакционная масса разлагается 150 мл насыщенного водного раствора
хлористого аммония при 5° С. Эфирный раствор высушивают, растворитель отгоняют, оста-
ток перегоняют в вакууме. Собирают фракции: I фракция — т. кип. 61еС/3мм, вес 1,1 г;
II фракция — т. кнп. 71—72,5° С/3 мм, вес 10,5 г; III фракция — т. кип. 74—75Q С/3 мм,
вес 2,1 г.
Всего получено диаллилртути (т. кип. 71—75°С/3 мм) 12,6 г (73%). Повторно перег-
нанный продукт имеет т. кип. 58—58,5° Q/1,5 мм, 1,6309, 2,3180, MR 43,42.
Из хлористого бутен-1-ил-4-магния и сулемы в тетрагидрофуране (реак-
цию ведут при 50° С) получена [83а] дибутенилртуть с выходом 60%.
Дициклопропилртуть получена из бромистого циклопропилмагния и
сулемы в тетрагидрофуране с выходом 64% [15].
Синтез дициклопропилртути* [15]. Реакцию ведут в атмосфере сухого азота.
К 4,6 г (0,19 моля) магния в трехгорлой колбе (250 мл), снабженной мешалкой, холодиль-
ником, термометром и капельной воронкой, приливают тетрагидрофурана (перегнанного
над натрием) столько, чтобы покрыть поверхность магния, и около 3 мл циклопропилбро-
мида. Реакцию начинают осторожным нагреванием раствора до кипения в течение несколь-
ких минут. Остальные 0,19 мл циклопропилбромида, растворенные в 100 мл тетрагидрофу-
рана, прибавляют в течение часа при 15—20° С при охлаждении ледяной водой. Смесь нагре-
вают до 50—60° С в течение часа. К раствору реактива Гриньяра прибавляют по каплям при
перемешивании раствор 20,6 а сулемы (80% от теории) в 50 мл тетрагидрофурана. Слабо на-
гревают в течение ночи. Вязкий осадок отсасывают, фильтрат извлекают эфиром, промывают
несколькими порциями воды, сушат сульфатом магния, отгоняют растворитель на водяной
бане, перегоняют дициклопропилртуть при 110—112°С (18мм); выход 13,7 г (64%).
Синтез дициклогексилртути* [47]. В раствор 35 г бромистого циклогексилмагния в
100 мл абс. эфира вносят при непрерывном энергичном перемешивании небольшими пор-
циями 5,5 г (16,7% от теории) тонко измельченной сулемы. Смесь кипятят 0,5 часа, разла-
гают льдом и разбавленной соляной кислотой; эфириый слой сушат сульфатом натрия
и и спаряют да 1/д. Затем пропусканием тока сухого воздуха охлаждают ниже 0° С, причем
выделяется Небольшое количество кристаллов бромистой циклогексилртути, которые от-
фильтровываю^ от масла. При встряхивании масла с трехкратным объемом эфира оно за-
кристаллизовывается. Кристаллы отсасывают, промывают охлажденным ледяной водой
спиртом, растворяют в горячем спирте, в профильтрованный раствор прикапывают воду, на-
гретую до 60°С, точно до тех пор, пока образующаяся муть при перемешивании не перестанет
растворяться. Охлаждают в охладительной смеси при энергичном перемешивании. Выпа-
дающий осадок отсасывают, промывают 50%-ным спиртом и сушат при 40°С в темном ва-
куум-эксикаторе над пятиокисью фосфора. Выход 3 г вследствие больших потерь при очи-
стке. Дициклогексилртуть плавится при 78—79° С в прозрачную жидкость, которая через
несколько секунд выделяет ртуть. Вещество при хранении в темном вакуум-эксикаторе
уже через 2 часа окрашивается в серый цвет, через 24 часа расплывается в черное масло.
* Токсична! Работать под хорошо действующей тягой.
Получение КгН§ и RHgX посредством RMgX и RLi
29
Получение дифенилртути описано на стр. 30, а также в оригинальной
литературе [1, 2, 10, 14, 52, 84—86].
Условия, способствующие хорошему выходу дифенилртути, как, очёвид-
но, и других полнозамещенных соединений ртути,— избыток реактива
Гриньяра и отсутствие металлического магния [52].
Ди -м -фторфен ил-, ди-ж- и ди-п-хлорфенилртуть получены из
соответствующих галоидфенилмагнийбромидов и галогенидов ртути в смеси
эфира и тетрагидрофурана при нагревании [83]. Получение (p-ClCeH4)2Hg
см. также [88].
Получение д нмфторфен илр тут и [83]'. К реактиву Гриньяра, полученному из 10,9 г
(0,0623 моля) л«-фторбромбензола, 1,52 а (0,0623 моля) магния в 30 мл абсолютного эфира,
прибавляют раствор 7,47 г (0,0207 моля) бромной ртути в 65 мл тетрагидрофурана при 0—
5® С. После нагревания в течение 4 час. 30 мин. реакционную массу разлагают 100 мл насы-
щенного раствора хлористого аммония при 5—10° С. Растворитель отгоняют, а остаток на-
гревают с 50 мл лигроина (т. кип. 100—125° С). Выделено 4,31 а (62,6%) ди-л«-фторфенил-
ртутист. пл. 106—117°С. После повторной кристаллизации из спирта продукт реакции
плавится при 117—119°С.
Из нерастворимого в лигроине остатка получают 2,26 г (29%) бромистой лг-фторфенил-
ртути, т. пл. 234—239° С. После повторной кристаллизации из спирта выделено 1,28 г веще-
ства с т. пл. 240°С.
Реакцией бромистого • пентафторфенилмагния с сулемой получают бис-
пентафторфенилртуть [87].
Получение ffac-пентафторфенилртути [87]. Чистую сухую сулему (6,8 г, 25 ммолей),
Помещенную в гильзу аппарата Сокслета, извлекают в течение часа, кипятя раствор неболь-
шого избытка бромистого пентафторфенилмагния (51 ммоль) в 200 мл эфира (раствор
CeFsMgBr получен обычным образом в присутствии иода с последующим кипячением в те-
чение 5 час.) и затем удаляют эфир под вакуумом;: твердый осадок возгоняют при 130°С/
/10 мм и получают белый возгон бис-пентафторфенилртути (9,8 г, или 73%). После кристал-
лизации из четыреххлористого углерода т. пл. 142,3°С.
Ди-л1-анизилртуть получена реакцией Гриньяра с примесью RHgCI,
вследствие чего для получения ее в чистом виде потребовалась симметри-
зация продукта реакции обработкой иодистым натрием в спирте [89].
Описаны синтезы (p-C6H8OC6H4)2Hg [90] и (p-CH2=CHCeH4)2Hg
[91, 92].
Взаимодействием реактива Гриньяра из /г-бромстирола с бромной ртутью
® тетрагидрофуране получена Hg(p-C6H4CH=CH2)„, где /1 = 2, 3, 4 [93].
Из p-(BrMg)2CeH4 и CeH5HgBr получен (p-C6H5Hg)2CeH4 [94].
Гексамерная о.-фениленртуть получена Виттигом с сотр. [95] из о-фени-
лен-бцс-магнийбромида (в свою очередь полученного из о-дилитийбензола
о бромистого магния) и сулемы с выходом 88%:
30 Ртутноорганические соединения. Лит. стр. 33—35
Получение полнозамещенных ртутноорганических Соединений
с отгонкой эфира
Выход полнозамещенных ртутноорганических соединений повышается,
если их синтез проводится следующим образом [8].
Реактив Гриньяра декантируют с невошедшего в реакцию магния в 2-литровую трех-
горлую колбу, снабженную мешалкой с ртутным затвором и экстрактором Сокслета, сое-
диненным с обратным холодильником. В патрон экстрактора помещают 0,5 моля сулемы*
В колбу доливают эфир до объема 800 ли, и содержимое колбы приводят в состояние умерен-
ного кипения. После того как вся сулема перейдет из гильзы в колбу, экстрактор заменяют
на прямой холодильник, температуру водяной’бани постепенно повышают до кипения и
поддерживают при этой температуре 1 час. Перемешивание продолжается во время от-
гонки и нагревания. Затем баню охлаждают, отогнанный эфир возвращают в колбу и произ-
водят гидролиз посредством воды или раствора хлористого аммония с небольшим количе-
ством аммиака. После отделения эфирного слоя, водный слой дважды извлекают порциями
эфира по 25 мл и соединенный эфирный раствор сушат над 10 г хлористого кальция и пере-
гоняют через подходящую колонку. Легко летучую галоидную н-бутилртуть легко и почти
полно удаляют вымораживанием (вода со льдом) после отгонки эфира. В случае галоидных
метил- и этилртути в этом нет надобности по причине их малой летучести.
Этим путем получены следующие выходы полных ртутноорганических
соединений при применении на 0,5 моля сулемы указанных в скобках мо-
лярных количеств галоидного алкила: (CH3)2Hg— 70% (CH3J — 1,15);
(C2H6)2Hg — 82% (ОДД — 1,15); (n-C4H9)aHg — 80% (n-C4H9Br — 1,15)
[4, 8]; (2-CH3C4H8)2Hg — 80% (2-CH3C4H8Cl — 1,16), [96], а также
[(CH3)(C6H5)CHCH2]2Hg -70% [42].
Получение дифенилртути(14]. Используется только что описанный метод с небольшим из-
менением. По причине малой растворимости фенильных соединений ртути в эфире в броми-
стый фенилмагний еще до добавления сулемы вводят смесь 500 мл бензола и 300 мл эфира.
Дифенил ртуть извлекают из реакционной Смеси хлороформом и дважды перекристаллизо-
вывают из него же или из спирта. Дифенилртуть выделяется в виде мелких иголочек, т. пл.
125°С. Выход 70,6% нз 1,15 моля бромистого фенилмагния и 0,5 моля сулемы.
В аналогичных условиях, но после введения сулемы—кипячение в тече-
ние 3 дней, дифенилртуть получена с выходом 81% [97].
Дифенилртуть получена [98] с выходом 85% при проведении синтеза ее по
Гриньяру в атмосфере азота с последующей.заменой эфира на ксилол.
Получение дифеиилртути [98]. Реактив Гриньяра нз 32 г (1,33 г-атом) стружек магния,
256 г (1,6 моля) свежеприготовленного бромбензола, 400 мл абс. эфира, полученный в ат-
мосфере азота, под давлением азота передавливают через воронку с пористым фильтром в
наполненную азотом колбу, прибзвляют 100 мл абс. эфира и при сильном перемешивании
при кипячении в течение 30 мни. прибавляют 136 г (0,5 моля) сулемы порциями по 5—7 г
в атмосфере азота. Затем добавляют еще 100 мл эфира и смесь нагревают 2 часа иа водяной
бане, часть эфира при этом отгоняется с азотом. Приливают 300 мл абсолютного ксилола,
повышая температуру до 75° С. Через 2 часа прибавляют еще 300 мл абс. ксилола и нагре-
вают еще 4часа до 90° С. В течение всей реакций смесь перемешивают так сильно, чтобы оса-
док не оседал иа дио. После охлаждения ксилольно-эфирный раствор отсасывают от осадка,
который исчерпывающе извлекают ксилолом и затем отбрасывают. Реакционную смесь при
наружном охлаждении разлагают разбавленной соляной кислотой со льдом, органиче-
ский слой отделяют, соединяют с ксилольиым раствором, применявшимся для экстрагиро-
вания, сушат хлористым кальцием, ксилол отгоняют в вакууме. Остаток — дифенилртуть.
Выход 150 г (85%), т. пл. 121—125° С. После кристаллизации из метанола т. пл. 125° С.
ПОЛУЧЕНИЕ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПРИ ПОМОЩИ ЛИТИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Органические соединения лития также применяют для синтеза ртутно-
органических соединений. Условия получения такие же, как при синтезе
посредством магнийорганических соединений.
Получение ИгН§ и RHgX посредством RMgX и RLi
31
Получение бромистой изопропенилртути [99]. а) Эфирный раствор изопропениллития
приготовляют в трехгорлой колбе в атмосфере сухого азота из 2,2 г (0,32 моля) металличе-
ского лития в 275 мл сухого эфира и 20 г (0,16 моля) бромистого изопропенила в 50 мл сухо-
го эфира. Реакцию ведут при 5—7° С до полного перехода металлического лития в раствор.
При более быстром ведении реакции выход продукта значительно снижается.
б) В эфирный раствор изопропениллития медленно в течение 40—45 мин. прибавляют
57 а (0,158 моля) сухой бромной ртути при энергичном перемешивании при»5—8° С. После
2-часового перемешивания реакционную массу разлагают медленным прибавлением 100 мл
4%-ного раствора бромистоводородной кислоты, дважды промывают бромистоводородной
кислотой той же концентрации, затем водой и отделяют образовавшийся осадок. Сухой не-
перекристаллизованный осадок весит 30 а и плавится при 164—166°С. После перекристал-
лизации из ацетона т. пл. 167°С.
Получение дибеизилртути [100]. К 100 мл 0,1 N раствора бёнзиллития в эфире прибав-
ляют 1,2г сулемы и непродолжительное время встряхивают. Через несколько мгновений рас-
твор становится бесцветным. После обработки из эфира получают 0,97 г (51%) дибензил-
ртути, т. пл. 110—111°С.
Получение изомерных алкенилртутных соединений реакцией алкенил-
литиевых соединений с галогенидами ртути во избежание превращения од-
ного изомера в другой при повышенной температуре ведут при температурах
не выше 5—8°С, а в отдельных случаях и при более низких температурах.
Цис- и щранс-пропениллитиевые соединения взаимодействуют с бромной
ртутью в эфире при 5—8°С [101—102], в тетрагидрофуране при 6—8° С с об-
разованием бромистой пропенилртути (выход 80%) той же конфигурации,
что и исходное литиевое соединение (см. также [83а]).
Ци-цис- (выход 85,5%) и ди-транс-пропенилртуть (выход 88%) полу-
чены [103] из соответствующих изомерных RL1 в эфире и сулемы в тетрагид-
рофуранё. Реакцию ведут при температуре не выше 5° С.
Синтез ди-^чс-пропенилртути [103]. В эфирный раствор цис-пропениллития, приго-
товленный при 5° С из 1,146 г (0,1654 моля) лития в 90 мл сухого эфира и 10 г (0,0826 моля)
1<ис-бромистого пропенила в 10 мл эфира, медленно прибавляют 7,84 г (0,0288 моля) сулемы
в 8 мл тетрагидрофурана при 3—5° С- После 2-часового перемешивания при комнатной тем-
пературе реакционную массу разлагают насыщенным раствором хлористого аммония при
0—3° С, промывают холодной водой и высушивают плавленым хлористым кальцием. Из
продукта реакции полностью отгоняют растворитель в вакууме при комнатной температуре.
Остаток — жидкий прозрачный продукт — весит 7 г; 85,5% . Ди-г{цс-пропенилртуть имеет
т. кип. 79—80°С/14 мм, п2Д 1,5628.
Из пгранс-пропениллития и бромистой 1(ис-пропенилртути (в эфире)
получена цш>/пранс-дипропенилртуть с выходом 91% [103].
Галогениды изопропенилртути получены из изопропениллития с бром-
ной ртутью или сулемой в эфирном растворе при 5—8° С [99]. В последнем
случае наряду с хлористой образуется также бромистая изопропенилртуть
за счет иона брома из раствора изопропениллития (который был получен
из изопропенилбромида и лития).
Бромистая а-стирилртуть получена [105] из а-стириллития с выходом
33%. Описано получение со-дистирилртути [46].
Цис- и /пранс-а-стильбениллитиевые соединения превращаются с суле-
мой в эфирном растворе со строгим сохранением конфигурации радикала
в цис-, цис- и транс-, /пранс-ди-а-стильбенилртуть (выходы 50 и 33% соответ-
ственно) [106]. Реакцию ведут в атмосфере азота при температурах —40-4-
—35 и —30 20° С.
1,4-Дилитийтетрафенилбутадиен (полученный из дифенил ацетилен а) при
реакции с сулемой в эфире (получасовое кипячение) образует тетрафенил -
меркурациклопентадиен (выход 34%, считая на дифенилацетилен) и сое-
динение (C28H20)2Hg3 (выход 7%) [107].
Из 4-камфиллития и сулемы 4-хлормеркуркамфан получен [104] с вы-
ходом 57%. Дициклопентенил- и дициклогексенил ртуть получены при про-
ведении реакции в тетрагидрофуране [108].
32
Ртутноорганические соединения. Лит. стр. 33—35
Синтез ди-1-циклопентенилртути [108]. Раствор 103 г (1 моль) свежеперегнанного
1-хлорциклопентена в 100 мл тетрагидрофурана прибавляют по каплям в течение 75 мин.
к 13,9 г (2 г-атом) литиевой проволоки в 500 мл тетрагидрофурана, температуру которого
поддерживают при —10° С. Непрореагировавший литий удаляют и затем прибавляют рас-
твор 68 г (0,25 моля) сулемы в 150 ли тетрагидрофураиа при0°С. Реакционную смесь пере-
мешивают 3 часа при комнатной температуре и затем гидролизуют водой При 0s С. Водный
слой извлекают этиловым эфиром, соединенные органические слои сушат перед отгонкой
растворителей. Остаток после кристаллизации из этанола дал 71 г (85%) дн-1-циклопен-
тенилртути, т. пл. 63,5—64,5°С.
Так же получена [108] ди-1-циклогексенилртуть, представляющая собой
бесцветную вязкую жидкость; т. кип. 122—125° С/0,4 мм, df 1,832, п®
1,5918.
В таких же условиях, как и дибензилртуть (см. выше), из п-толиллития
и сулемы получена ди-л-толилртуть [10, 109].
При получении этим методом ди-л-диметиламинофенилртути после вве-
дения сулемы реакционную смесь кипятят в течение 1 часа. Выход 12% 1109].,
Из эквимолярных количеств n-литийдиметиланилина и сулемы в эфире
получена хлористая и-диметиламинофенилртуть [НО].
бис-(о-,м-к п-Трифторметилфенил)ртуть получены [111, 112] из соответ-
ствующих ArLi и сулемы (2 моля : 1 моль) в эфире. При соотношении ArLi
и HgCla 1 : 1 получены ArHgCl [112].
Гексамерная о-фениленртуть (см. стр. 29) получена [95] из о-дилитий-
бензола и сулемы или металлической ртути в эфире. Выход 72%. в первом
случае и 25% во втором (см. также гл. IV).
Аналогично из о, о'-дилитийдифенил а получена [113] о-дифениленртуть,
имеющая тетрамерное циклическое строение [114]:
Получение тетрамерной о-дифениленртути [113]. При сливании 1,9 г (0,007 моля) суле-
мы в 50 мл абсолютного эфира с раствором 0,0073 моля о,о'-дилитийдифенила в эфире вы-
деляется дифениленртуть. Кристаллизация из нитробензола, т. пл. 335—336°С [НО]. Выход
97%.
При взаимодействии хлористого о-литийдифенила с сулемой получена
[ПО] хлористая о-дифенил ил ртуть.
Получение хлористой о-дифенилнлртутн [НО]. К раствору 0,4 г (1,5 ммоля) сулемы
в 20 мл абс. эфира прибавляют 5 мл свободного от солен эфирного раствора, содержащего
0,9 ммолей о-литийдифенила, эфир отгоняют, основательно промывают осадок водой и кри-
сталлизуют из лигроина. Бесцветные иглы ст. ,пл, 161—162,5° С.
Реакцией 3-бифенилиллития с эквимолекулярным количеством сулемы
в диэтиловом эфире получен [115] 3-хлормеркурбифенил с выходом 70%.
Бромистая инденилртуть получена с небольшим выходом из инденилли-
Получение RaHg и RHgX посредством RMgX и RLi
за
тия «Сулемы (в присутствии иона брома) [116] (среда — эфир, реакцию ве-
дут в дзоте). Попытки получения в этих же условиях диинденил- и дифлу-
оренилртути из инденил- и флуорениллития и сулемы привели к образова-
нию металлической ртути и диинденила и дифлуоренила [1161.
При энергичном встряхивании в течение 12 час. 3-пирениллития и су-
лемы в эфире е 80%-ным выходом получена ди-3-пиренилртуть [117].
Из 1-(2-литийфенил)-2-литийнафталина и сулемы в эфире получена с выхо-
дом 65,9% 3,4-бензодифениленртуть [118J:
бис-а, а-Пиридилртуть получена [119] с небольшим выходом из 2-пири-
диллития и сулемы в эфирном‘растворе при—20° С.
Действием хлористой фенилртутина продукт взаимодействия бензилаце-
тилена с амидом лития в жидком аммиаке получена CeHeHgC==CCHaC6H6
[120]. 1 '
С незначительным выходом образуются ртутноорганические соединения
при взаимодействии литийорганических соединений с металлической ртутью.
Таким путем получены: ди-н-бутилртуть (реакцию вели в циклогексане)
[121], дибензилртуть (15-часовое встряхивание в эфире) [100], дифенил-
ртуть (взаимодействие в течение 94,5 часа в эфире) [122, 123].
При перемешивании 5%-ного эфирного раствора 1,5-дилитийпентана
с избытком ртути (под аргоном) получены циклопентаметиленртуть
(n-CsH11)2Hg и смесь полимерных продуктов [—(СНа)$ — Hg—]« воз-
можно циклического строения [124]. Последние превращаются в димёр при-
продолжительном кипячении в бензоле.
Ртутноорганические соединения получены при аналогичной реакции со
ртутью со, со '-дилитиевых производных бутана, гексана и декана [1241.
Так же, как иногда происходит обмен радикалами между ртутнооргани-
ческими и магнийорганическими соединениями (см. выше),' может проис-
ходить обмен радикалами между ртутноорганическими и лйтййбрганиче-
скими соединениями;
R2Hg + 2R'Li -> 2RLi + R/Hg,
например, при R = р-С1С6Н4, R' = n-C4H9 [125]
ЛИТЕРАТУРА
1. Pfeiff er P., Tr us kier P. Вег., 37, 1125(1904).
2. H i l p er t S„ Gru 11 пег G. Ber., 48, 906 (1915); 46,1675(1913); 47, 177 (1914).
3. M а г V e 1 C. S., Gou I d V. L. J. Am. Chem. Soc., 44, 153 (1922),
4. Gilman H., Brown R. E, J. Am. Chem. Soc., 51, 928 (1929).
5. Slotth К,- H., Jacobi K. R. J. prakt. Chem., 120, 249 (1928).
6. H i n k e 1 L. E., A nge 1 T. H. J, Chem. Soc?, 1927, .1948.
7. R u m p f P. Bull. Soc. chim. France, 9, 535 (1942). '
8. Gilman H., Brown R. E. J. Am. Chem. Soc., 52, 3314 (1930);
9. Meals R. N. J. Org. Chem., 9, 211 (1944).
10. NerdelF., MakowerS. Naturwiss., 45,/490 (1958).
11. Jones L. W., Werner L. J. Am. Chem, Soc., 40, 1266 (1918).
12. Несмеянов A, H., БорисовА. E., Савельева И. С., Голубева
Е. И. Изв. АН СССР, ОХН, 1958, 1490.
13. W i n s t е i п S., Traylor T. G. J. Am. Chem. Soc., 77, 3747 (1955).
14. S t e i и k о p f W., Bielenberg W., A u g e n s t a d - J e n s e n H. Ann.,
430, 41 (1922). .
15. R e y.n о 1 d s G'. F., D e s s у R. E., J a f f ё H. H. J. Org. Chem., 23, 1217 (1958).
34
Ртутноорганические соединения
16. В а г t о с h а В. В,j В г i п с к m а п F. Е., Kaesz Н. D., S t о п е F. G. А.
Proc-. Chem. Soc., 1958, 116.
17. J о s е J., E s p i n о z а С. C. A., 42,2924 (1948); Rev. quim. farm. (Santiago, Chili),
4, N 56, 2 (1947).
18. 3 a x a p к и н Л. И., Охлобыстин О. Ю. ДАН СССР, 149, 1299 (1962);
19. К h а г а з с h М. S., SwartzS. J. Org. Chem., 3, 405 (1938).
20. Несмеян ов А. Н., Печерская К. А. Изв. АН СССР, ОХН, 1941, 67.
2Г. Неем еянов'А. Н., Кочетков Н. К. Изв. АН СССР, ОХН, 1949, 587;
Уч. зап. МГУ, вып. 132, 51 (1950).
22. Wr i gh t G. F. J. Am. Chem. Soc., 57, 1993 (1935).
23. Wright G. F. J. Am. Chem. Soc., 62, 1991 (1940).
24. Erlenmeyer H„ L e о M. Helv. Chem. Acta, 15, 1171 (1932).
25. M a r v e 1 C. S., G a u e r k e C. G„ H i 1 1 E: L. J. Am. Chem. Soc., 47, 3009 (1925).
26. К a k u T., H a g i w a г a Y. C. A., 52, 20005 (1958).
27. Char m an H. B., H u ghes E. D„ I ngo 1 d C. J. Chem. Soc., 1959, 2523.
28. M a r v e 1 C. S., С a 1 v e г у H. O. J. Am. Chem. Soc,, 45, 820 (1923).
29. Whi t mor e F. C., W i 11 1 e E. L., Harriman B. R. J. Am. Chem. Soc.,
61, 1585 (1939). -
30. Whitmore F. C., Fleming G.H. J. Am. Chem. Soc., 55, 4161 (1933).
31. W h i t m о r e F. C., R о h r m а и n E. J. Am. Chem. Soc., 61, 1591 (1939).
32. Whi t mor e F. C., S о m m e r L. H. J. Am. Chem. Soc., 68, 481 (1946).
33. Sommer L. H., W h i t m о r e F. C. J. Am. Chem. Soc., 68, 485 (1946).
34. S p e i e r J. L. J. Am. Chem. Soc., 74, 1003 (1952).
35. Wh i t mor e F, C„ Mar ker R. E., Plambeck L., jr. J. Am. Chem. Soc.,
63, 1626 (1941).
36. R u m p f P. Bull. Soc. chim. France, 9, 538 (1942). ,
37. Massie S. P. C. A., 41, 3044 (1947); Iowa StateColl. J. Sci., 21, 41 (1956).
38. К h a r a s c h M. S., L e g a u 11 R. R., Sp row Is W. R. J. Org. Chem., 3,
409 (1938).
39. С о h e n H. L., W r i g h t G. F. J. Org. Chem., 18, 432 (1953).
40. W a r e F. E., • H i x о n R. M. J. Am. Chem. Soc., 60, 1262 (1938).
41. Hill E. L. J. Am. Chem. Soc., 50, 167 (1928).
42. C r i e g e e R., D i m г о t h P„ Schempi R. Ber., 90, 1337 (1957).
43. Bar to ch a B., S t о n e F. J. A. Z. Naturforsch., 13b, 347 (1958).
44. R e n a u d P. Bull. Soc. chim. France, 1950, 1044.
45. S 1 e e z e r P. D., W i n s t e i n S., Young W. G. J. Am. Chem. Soc., 85,
1890(1963).
46. W r i g h t G. F. J. Org. Chem., 1, 457 (1936).
47. GriittnerG. Ber., 47, 1651 (1914).
48. Jensen F. R., G a 1 e L. H. J. Am. Chem. Soc., 81, 1261 (1959); 82, 145 (I960).
49. P e у т о в О. А., Л у Ц з и и - ч ж у, Б у н д е л ь Ю. Г. Вести. МГУ, сер. мех.,
астр. физ. хим., 13, № 5, 111 (1958).
50. Winstein S., Vogelfanger Е., Pande К. Chem. and Ind., 1962, N 49,2061.
51. Bergstrom P., D e w a r M. M. J. Am. Chem. Soc', 51, 3387 (1929).
52. S a w a t z k у H., W r i g h t G. F. Canad. J. Chem., 36, 1555 (1958).
53. I n d u e Y., S h i n t a n i G. J. Agric. Chem. Soc. Japan, IB, 1141 (1942); C. A., 43,
1138 (1949).
54. Robson I. H., W r i g h t G. F. Canad. J. Chem., 38, 21 (1960).
55. Scott R., jr., G о r d о п M. I. J. Org. Chem., 21, 385 (1956).
56. Grummitt О., В u c k A. C. J. Am. Chem., Soc., 65, 295 (1943).
57. G i 1 m а п H., S m i t h E. W„ О a t f i e 1 d H. J. J. Am. Chem. Soc., 56, 1412
(1934).
58. Rothstein E., S a v i 11 e R. W. J. Chem. Soc., 1952, 2987.
59. Whitmore F. H о m e у e r A. H. J. Am. Chem. Soc., 55, 4555 (1933).
60. P о p e W. J., G i b s о n C. S. J. Chem. Soc., 101, 735 (1912).
61. К г e e\y о у M. M., Hansen R, L. J. Am. Chem. Soc., 83, 626 (1961).
62. Drehman U. J. prakt. Chem., 9, 321 (1959).
63. S e у f e r t h D., Frey er W. J. Org. Chem., 26, 2604 (1961).
64. F r i t z G., В u r d t H. Z. anorg. allg. Chem., 314, 35 (1962).
65. M e л ь и и к о в H. H., РокицкаяМ. С. ЖПХ, 12, 1802 (1939).
66. Японск. пат. 3228 (1952); С. А., 48, 2763 (1954).
67. Р i t z е г К. S., Gu t о wsk у Н„ S. J. Am. Chem. Soc., 68, 2204 (1946).
67а. Cowan D. Q.,.Mosher Н. S. J. Org. Chem., 27, 1 (1962).
68. Нефедов В. Д., Синотова Е. Н. ЖФХ, 32, 2392 (1958).
69.. К i п g Н. S. Zbl., 1931, II, 3425; Proc. Nova Scot. Inst. Sci., 17, 28 (1927).
70. Go d d a r d A. E. J. Chem. Soc., 123, 1161 (1923).
711 W i 1 d e W. K. J. Chem. Soc., 1949, 75.
72. G i 1 m а п H., Z о e 1 1 n e r E. A., Selby W. M.t В о a t h e г С. H. Rec.
trav. chim., 54, 587 (1925).
Получение RaHg и RHgX посредством RMgX и RLi 35
73 F г 'е у F. Е., H e p p H. J. J. Am. Chem. Soc., 55, 3357 (1933).
74. Jones W. J., E van s D. P., Gu Iwell T., Griffiths D. C. J. Chem.
Soc 1935 42
75. Mos her'H. S., Loeffler P. K. J. Am. Chem. Soc., 78, 4959 (1956).
75a. Fray G. I., Robinson R. Tetrahedron L., 18, 261 (1962).
•76. H a g e r F. D., M a r v e 1 C. S. J. Am. Chem. Soc., 48, 2689 (1926).
77. Реутов О. А, У г л о в а Э. В. Изв. АН СССР, ОХН, 1959, 1691.
78. В a h г G., М е i е г G. Z. anorg. allg. Chem., 294, 22 (1958).
79. Wolff P. Ber., 46, 64 (1913).
80. В a nu s A. G. Zbl., 1923, III, 1265; Anales Soc. Espanola Fis. Quim., 20, 667 (1922).
81. С т e p л и н P. H., Ли В e й - г а и, К н у н я н ц И. Л. Изв. АН СССР, ОХН,
1959, 1506.
82. Е g 1 i п g t о n G., Me C r a e W. J. Chem. Soc,, 1963, 2295.
83. Борисов A. E., Савельева й. С., Сер дюк С. P. Изв. АН СССР,
ОХН, 1965 (в печати).
83а. Foster D. J., T-o b 1 e r E. J. Org. Chem., 27, 834 (1962).
84. Letsinger R. L., P о 11 ar t D. F. J. Am. Chem. Soc., 78, 6079 (1956).
85. M а и у л к и н 3. М., ТатаренкоА. И., Юсупов Ф. Сб. ЖОХ, 2, 1308
(1953).
86. Hartmann Н., BeermannC., Czempik Н. Z. anorg. allg. Chem., 287,
261 (1956).
87. Chambers R. D., Coates G. E., Livingstone J. G„ Musgra-
ve W. K. R. J. Chem. Soc., 1963, 4367.
88. D e s s у R. E., Kim J.-Y. J. Am. Chem. Soc., 82, 686 (1960).
89. H i n t о n R. С., M a n n F. G. T о d d D. J. Chem. Soc., 1961, 5454.
90. D a v i e s W. С., M о r'r i s C. J. O. R. J. Chem. Soc., 1932, 2880.
91, К о т о н M. M., Киселева T. M., Флоринский Ф. T. Высокомолекул.
соед., 2, 1639 (1960).
92. К о т о н M. M„ Киселева Т. М. ДАН СССР, 131, 1072 (1960).
93. D г е f a h 1 D., Р 1 о t п е г G., Lorenz D. Angew. Chem., 72, 454 (1960).
94. М а 1 n a^r М., -Grdenic D. J. Chem. Soc., 1959, 3639.
95. Wittig G., В ickelhau p t F. Ber., 91, 883 (1958).
96. Murahashi S., NozakuraS., TakeuchiS. Bull. Chem. Soc. Japan,
33, 658(1960).
97. В a c h m a n W. E. J. Am. Chem. Soc., 55, 2827 (1933).
98. D’ A n s J., Zimmer H., Bergmann H. Ber., 85, 583 (1952).
99. Несмеянов A. H., Б op исов A. E., Новикова H. В. ДАН СССР, 94,
289(1954).
100, Z i e g 1 e г К. Ber., 64, 445 (1931).
101. H e с м e я н о в A. H., БорисовА. E., Новикова H. В. Изв. АН СССР,
ОХН, 1959, 1216.
102. Несмеянов A. H., БорисовА. E., Новикова Н. В. ДАН СССР,
119, 504 (1958).
103. БорисовА. Е. Изв. АН СССР, ОХН, 1961, 1036.
104. W i п s t е i n S., Т г а у 1 о г Т. G. J. Am. Chem. Soc., 78, 2597 (1956).
105. Борисов А. Е., Новикова Н. В. Изв. АН СССР, ОХН, 1957, 1258.
106. Несмеянов.А. Н., БорисовА. Е., ВолькенауН. А. Изв. АН СССР,
1954, 992.
107. В г а у е Е. Н., Hubei W., С а р 1 i е г I. J. Am. Chem. Soc., 83, 4406 (1961).
108. Foster D. J., ToblerE. J. Am. Chem. Soc., 83, 851 (1961).
109. Au st i n P. R. J. Am. Chem. Soc., 54, 3726 (1932).
110. W i t t i g G., H e r w i g W. Ber., 88, 962 (1955).
111. P о w e 1 1 H. В., M a u n g M. T., L agowsk i J. J. J. Chem. Soc., 1963, 2484.
112. M a u n gM. T., LagowskiJ. J. J. Chem. Soc., 1963, 4257.
113. W i t t i g G., Herwig W. Ber., 87, 1511 (1954).
114. W i 11 Lg G., Lehmann G. Ber., 90, 876 (1957).
115. R a u s c h M. J. Inorg. Nuclear Chem., 1, 414 (1962).
116. D’A ns J., Zimmer H., V. BrauchitschM. Ber., 88, 1507 (1955).
117. В e r g A. Acta chem. scand., 3, 655 (1949).
118. C a v a M. P., S t u c k e r J. F. J. Am. Chem. Soc., 77, 6022 (1955).
119. G i 1 m a n H„ G r e g о г у W. A., S p a t z S. M. J. Org. Chem., 16, 1788 (1951).
120. Японск. пат. 2677 (1960); РЖХнм, 1961, 21Л406.
121. Ziegler К., С о 1 о n i u s H. Ann., 479, 149 (1930).
122. Талалаева T. В., Кочешков К. А. ЖОХ, 8, 1831 (1938).
123. Англ. пат. 854776 (1960); РЖХим, 1961, 18Л91.
124. В е i n е г t G., Р а г г о d J. С. г., 255, 1930 (1962).
125. G i 1 m а п Н., Y а 1 е Н. L. J. Am. Chem. Soc., 72, 8 (1950). 1
126. Beck W., S c h u i e r e r E. J. Orgavomet. Chem., 3, 55 (1965).
Глава III
ПОЛУЧЕНИЕ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ЧЕРЕЗ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
ПРОЧИХ ЛЕГКИХ МЕТАЛЛОВ
Органические соединения металлов It II и III групп периодической си-
стемы также были предложены для получения ртутноорганических соеди-
нений. Синтезы при помощи органических соединений натрия и серебра
нетипичны и были применены только для двух особых случаев. В настоя-
щее время почти уже оставлен одйн из наиболее старых способов получе-
ния ртутноорганических соединений — через. органические соединения
цинка. Известные перспективы имеет синтез алифатических ртутнооргани-
ческих соединений при помощи алюминийорганических соединений, пока
испытанный лишь на небольшом числе примеров синтеза как RaHg (R=AIk
или Аг), так и RHgX (R = Alk).
ОБРАЗОВАНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ РТУТИ
ЧЕРЕЗ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ НАТРИЯ И СЕРЕБРА
Дициклопентадиенилртуть получена реакцией циклопентадиенилнатрия
с сулемой [11.
Получение дицнклопентадиенилртути [1]. 3,5 г кусочков натрия в 100 мл тетрагадрофу-
рана обрабатывают 13 мл циклопентадиена. После прекращения выделения водорода раствор
охлаждают до —30° С и, к нему медленно Прибавляют раствор 13,5 г сулемы в 150 мл тетра-
гидрофурана. Затем к раствору прибавляют 1 мл воДы при 0а С, растворитель отгоняют в
вакууме, остаток извлекают эфиром. Эфирный раствор промывают водой и высушивают. Ох-
лаждением до—78° С из‘него выделяют 3 г (20%) днциклОпентадиенилртути. Возмож-
на кристаллизация таким же путем из эфира. Бледно-желтые кристаллы, разлагающиеся
при 83—85’G (начало разложения от 60° С).
При проведении реакций.] бис-(натрийацетиленил)бензола с сулемой
получен полимер [1а].
Обработкой двойной соли ацетиленида серебра с азотнокислым серебром
состава AgCa-6AgNO3 раствором азотнокислой ртути в азотной кйслоте
получают ацетиленид ртути в виде двойной соли с азотнокислым серебром
состава HgC2-3AgNO3 [2, 3].
ПОЛУЧЕНИЕ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ЧЕРЕЗ ’ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ЦИНКА
Взаимодействие солей ртути и ртутноорганических солей с полнозаме-
щенными органическими соединениями цинка может быть выражено сле-
дующими уравнениями:
RaZn 4* HgX2 —> RaHg 4* ZnXz (1)
RaZnR’HgX-> RaHg + R'ZnX. (2)
Синтез R4Hg через органические соединения Na, Ag, Zn и Al 37
Смешанные цинкорганические соединения RZnX для получения ртутнб-
органических соединений не применялись. Реакция (1) была изучена на син-
тезе диэтил- [4] и диметилртути [4, 5]. Реакцией (2) из иодистой аллил-
ртути и диэтилцинка получена диэтилртуть [6, 71. По уравнению (2) Франк-
ланд из иодистой метилртути и диэтилцинка получил диэтилртуть [81; при
попытке получения метилэтилртути из (CH3)2Zn и C2H6HgX он получил
смесь диэтил- и диметилртути [4]. В настоящее время этот метод синтеза
ртутноорганических соединений вытеснен методом синтеза посредством бо-
лее доступных и удобных в обращении магний органических соединений.
Однако Годемаром 19] для получения ряда RHgX (X = Cl, Вг) при
R=CH2=CH—СН2—;CeH6CH2—;СвН5С=С-СН2—иСвН6СН=С=СН—СН2
применены недавно цинкорганические соединения R2Zn в тетрагидрофу-
ране, в случае этих радикалов R, по данным автора, легче получаемые,
чем соответствующие магнийорганические соединения RMgX.
Синтез хлористой аллилртути [9]. Реакцию ведут в токе азота в колбе, снабженной ме-
шалкой, вращающейся со скоростью 200—300 об/мин, холодильником, вводом азота, термо-
метром и капельной воронкой с оттянутым в капилляр носнком. 'Все сообщения с внешней
средой защищены трубками с хлористым кальцием.
В реакцию вводят 16,3 г (0,25 моля) цинковых стружек (полученных обработкой неболь-
шого куска электролитического цинка сначала на токарном станке, что дает опнлкн тол-
щиной 0,1 мм, а затем — в дробильной машине измельченные в стружки длиной около
1 мм), 30 г (0,25 моля) бромистого аллила и 125 мл тетрагидрофурана. Цинк заливают не-
большим количеством растворителя и приливают несколько граммов бромистого аллила. Ре-
акцию начинают подогреванием. После прекращения кипения реакционную смесь охлажда-
ют в бане со льдом; поддерживая температуру 10° С в реакционной смеси в течение 2 час., по
каплям вводят остальной раствор бромистого аллила в тетрагидрофуране. Получают проз-
рачный желтовато-зеленый раствор дналлилцинка. Количество не вступившего в реакцию
цинка не превышает 0,5—0,8 е.
Раствор 15 г (0,055 моля) сулемы в 20 мл тетрагидрофурана вводят при комнатной тем-
пературе в 0,05 моля полученного раствора диаллилцйнка. Смесь перемешивают в течение
1 часа, затем упаривают и вводят по каплям при перемешивании 500 мл 2% -ноГо раствора
серной кислоты. Тетрагндрофуран и неорганические соли переходят в раствор, ртутно-
органическое соединение выпадает на дно. Декантируют верхний слой, осадок обильно про-
мывают водой, быстро холодным спиртом и сушат в вакууме. Вес сырого продукта 11,5 г,
выход 83%. Продукт достаточно чист, т. пл. 110° С. После кристаллизации из спирта т. пл.
Ш°С.
Аналогично получаются бромистая аллилртуть, т. пл. 125°С, выход 69%;
хлористая (т. пл. 110°С, выход 83%) и бромистая (т. пл. 118°С, выход 75%)
бензилртуть; хлористая (т.пл. 108°С, выход77%) ибромистая (т. пл. 114°С,
выход 68%) фенил-З-пропин-2-ил-ртуть; хлористая (т. пл. 122°С, выход бр%)
и бромистая (т. пл. 125°С, выход 53%) фенил-1-бутадиенил-1,2-ил-ртуть.
ПОЛУЧЕНИЕ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПРИ ПОМОЩИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ АЛЮМИНИЯ
Органические соединения ртути могут быть получены действием органи-
ческих соединений алюминия на соли ртути и (для одного известного в лите-
ратуре примера) на металлическую ртуть. Реакцию ведут при нагревании в
инертном растворителе (углеводороды, дихлорэтан, эфир).
Из смешанных алюминийорганических соединений, как RAlCla 110]
или R2A1C1 [10], так и из их смеси [11—13] RA1C12 + R2A1C1 и сулемы по-
лучают с хорошим выходом RHgCl (R = CHS и C2HS, но не высшие алкилы).
Реакцию ведут в бензоле, гексане, дихлорэтане при умеренном нагревании
Или даже при комнатной температуре.
Хлористая этилртуть с выходом- 91% получена из сулемы и
CjHgAlC^.NaCl (побочного продукта при получении катализатора полимери-
зации этилена низкого давления) в бензоле, ксилоле или другом неполярном
растворителе [10, 13, 14]. Преимуществом применения в качестве исходного
38 Ртутноорганические соединения. Лит. стр. 39
соединения этого алюминийорганического комплекса является его невос-
пламеняемость и отсутствие' необходимости вести реакцию в атмосфере
инертного газа.
Получение хлористой этилртути [10]. В трехгорлую колбу, снабженную мешалкой, ка-
пельной воронкой, обратным холодильником и термометром, в условиях исключения влаги
воздуха помещают 112,8 г (0,42 моля) HgCla и 180 мл ксилола. При энергичном перемеши-
вании прибавляют по каплям раствор 76,8 г (0,49 моля) комплексной соли CaHeAlCk.
NaCl в 180 мл сухого ксилола. Температуру смеси поддерживают при 45—50° С. По оконча-
нии введения комплексной соли перемешивают еще в течение 30 мин. и оставляют стоять на
12 час. при комнатной температуре. После этого приливают 300 мл воды при энергичном пе-
ремешивании так, чтобы температура реакционной смёси во время гидролиза не превышала
40° С, для чего внутрь можно вводить лед. Воду следует сначала приливать медленно. При
этом выпадает белый осадок хлористой этилртути, который отсасывают, промывают водой
и небольшим количеством спирта. После сушки получено 100,5 г хлористой этилртути с
т. пл. 192—193° С. Выход 91% .
Из полнозаметенных соединений алюминия, А1к3А1, и соли ртути, в за-
висимости от относительных количеств реагентов и природы аниона в соли
ртути, получают Alk2-Hg [15, 16] или AlkHgX [10, 13, 16]. Для получения
Alk2Hg необходимо брать избыток А1к3А1 и лучше ацетат, а не галогенид ртути.
Если исходить из А1к3А1, то можно получить ртутноорганические сое-
динения и с алкилами высшими, чем этил. Реакцию ведут в гексане, эфире
[16], бензоле [10] при умеренном нагревании или при комнатной темпера-
туре. Из триэтилалюминия и бромной ртути (в гексане) получена диэтил-
ртуть (выход 58,6%). Из эфирата три-н-пропилалюминия и ацетата ртути
в этих же условиях — ди-н-пропилртуть (выход 79%). Из эфирата трипро-
пилалюминия й из триизобутилалюминия и бромной ртути в эфире —- смесь
R2Hg и RHgBr. Из эфирата триизопропилалюминия и бромной ртути в эфи-
ре—бромистая изопропилртуть [16].
• Получение диэтилртути [16]. К суспензии 27,1 г (0,1 моля) измельченной хлорной
ртути в 50 мл гексана добавляют в токе азота при перемешивании 11,4 г (0,1 моля) триэтил-
алюм.иния в 50 мл гексана (40—50° С). Реакционную массу перемешивают при 40° С 2 часа,
после чего ее разлагают смесью льда и бикарбоната натрия. Водный слой экстрагируют эфи-
ром; после высушивания хлористым кальцием и отгонки эфира и гексана получено 15,0 г
диэтилртути (выход 58,5%). Т. кип. 95—96°С/90лш, п22 1,5400, d20 1,4658.
Продукт взаимодействия триэтилалюминия с этиленом при нагревании
в течение часа при 80° С с сулемой в циклогексане дал твердое соединение
H(C2H4)„HgCl (п=4-4-5) [17].
При встряхивании в течение 20 час. трифенилалюминия с металличе^
ской ртутью в ксилоле получена дифенилртуть [18]. На результат реакции
не влияет, ведут ли ее в атмосфере азота или без него.
Описано получение диалкилртути электролизом Me(AlAlk3R) (Me =
= К, Na) на ртутном аноде [19]. Диэтил ртуть так получена с выходом 60%.
При действии карбида алюминия на водный раствор сулемы происходит
метилирование последней [20]. По мнению Гильперта и Дитмара [20], реак-
ция идет через промежуточное образование карбида ртути. При ведении ре-
акцйи в растворе 10%-ной соляной кислоты с избытком сулемы с 30%-ным
выходом получена хлористая метилртуть; в растворе, нейтральная реакция
в котором поддерживается осторожным прибавлением соляной кислоты.,
получена диметил ртуть.
LiAl(CeH5)4 (без выделения его из реакционной среды при его получении),
в эфирном растворе с сулемой образует CeHsHgCl [23].
Получение хлористой метилртути [20]. В раствор 25 г сулемы в 130 г 10%-ной НС1 при
сильном встряхивании маленькими порциями вносят 15 г карбида алгбминия. Происходит
сильное разогревание. Вскоре выделяются листочки хлористой метилртути. Поддерживают
температуру 90° С. Хлористую метилртуть отгоняют с водяным паром. Выход 30%, т. пл.
170° С (см. также [21, 22]).
Синтез RaHg через органические соединения Na, Ag, Zn и Al
39
ЛИТЕРАТУРА
1; W i 1 к i n s о n G., Piper T. S. J. Inorg. a. Nucl. Chem., 2, 32 (1956).
la. Корш а к В. В., Сладков A. M., Л у н e в а Л. К. Изв. АН СССР, ОХН,
1962, 728.
2. Англ. пат. 616318 (1949); С. А., 43, 5175 (1949).
3. Пат. США 2474869 (1949); С. А., 43, 7670 (1949).
.4. Buck ton G. В. Ann., 109, 219 (1959).
5. D i b е 1 е г V. Н. Analyt. Chem., 27, 1958 (1955).
6. Krassowsky H. Вег., 3, 625 (1870).
7. Oppenheim А. Вег., 4, 671 (1871).
8. FranklandE. Ann., 11, 57 (1859).
9. Gaudemar М. Bull. Soc. chim. France, 1962, 974; C. r., 254, 1100 (1962).
10. E с к s t e i n Z., DahlingW., HetnarskiB., P a s у n к i e w i с z-S.
Przem. Chem., 39, 225 (1960).
И. Пат. США,2473434 (1949); С. A„ 43, 7953 (1949).
12. Пат. ФРГ 954878 (1956); С. А., 53, 11226 (1959).
13. Eckstein Z., D a h 1 i n g W., Hetnarski В., P a s у nkiewich S.
Bull. Acad. Polon. Science, VIII, 161 (I960).
14. Swirska А., Kotler-Braj tburg J., Dahling W., Pasynkie-
w i c z S. Przem. Chem., 39, 371 (1960).
15. Англ. пат. 820146 (1959); С. A., 54, 6550 (1960). Пат. ФРГ 1048581 (1959);С. А., 54,
*24401 (1960).
16. Захаркин Л, И., О х л о б ы с т и н О. Ю. ДАН СССР, 116, 236 (1957); Изв.
АН СССР, ОХН, 1959, 1942. Пат. СССР 110947 (1958); С. А., 52, 18217 (1958).
17. Англ. пат. 763826 (1956); С. А., 52, 1203 (1958).
18. Б р и л к н н а Т. Г. У4. зап. ГГУ, 24, 165 (1953).
19. Пат. СССР 132136 (1960); РЖХнм, 1962, 9Л98.
20. Н i 1 р е г t S., D i t t m a r P. Ber„ 46, 3738 (1913).
21; G i Iman H., Jones R. G.. J. Org. Chem., 10, 505 (1945).
22. Нем. пат, 554513 (1931); Zbl., 1932, II, 2229.
23. Traylor T..G. Chem. and Ind., 1959, 1223.
Глава IV
СИНТЕЗ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПРИ ПОМОЩИ АМАЛЬГАМ НАТРИЯ, ЛИТИЯ, КАЛИЯ И КАДМИЯ
В то время как металлическая ртуть реагирует с трудом и лишь с це»|но-
гими галоидными производными углеводородов, амальгама натрия взаимо-
действует с галоидными алкилами уже на холоду, с галоидными арилами при
нагревании по уравнению
2RX -}- NagHg —* RaHg -j- 2NaX.
Этот (один из наиболее старых) открытый еще Франкландом [1] метод
синтеза ртутноорганических соединений используется до настоящего вре-
мени, так как он в отличие от меркурирования, а также методов синтеза за-
меной на ртуть кислых групп непосредственно приводит к полнозамещенным
жирным, алициклическим, некоторым жирноароматическим и ароматиче-
ским ртутноорганическим соединениям. Метод в основном имеет ту же об-
ласть применения, что и синтез соединений R2Hg через магнийорганические
соединения, но иногда дает лучшие, чем последний, выходы. Для получения
веществ R2Hg в большом масштабе метод более удобен, чем синтез их реак-
цией Гриньяра.
В жирном ряду этим методом получены ртутные производные углево-
дородов как с первичными (ряд диметил—диамилртуть, а также диоктил-
ртуть и производные углеводородов изостроения: диизобутил- и диизоамил-
ртуть), так и со вторичными радикалами (диизопропилртуть 12]) с выхода-
ми для соединений с первичными радикалами в отдельных случаях до 80—
85% (диэтилртуть). Вторичные галоидалкилы дают меньшие выходы. По-
лучены также недоступные синтезом через реактив Гриньяра ртутноорга-
нические производные ^-карбоновых [3] (но неа-карбоновых) кислот. Метод
приложим и к алициклическому ряду, где из бромистого циклогексила та-
ким путем получена дициклогексилртуть. Иодистый циклогексил неожи-
данным образом дает с амальгамой натрия не дициклогексилртуть, а иоди-.
стую циклогекбилртуть. Из моногалоидозамещенных жирноароматических
углеводородов дибензилртуть этим методом не получается: при взаимодей-
ствии бромистого бензила с амальгамой натрия проходит только реакция
Вюрца и образуется дибензил. Однако ди-со-стирилртуть с незначитель-
ным выходом была получена этим методом.
В особых случаях — при синтезе перфторалкильных соединений ртути
применяют амальгаму не щелочных металлов, а амальгаму кадмия (хуже —
амальгамы меди и серебра).
Примененный к ароматическому ряду главным Образом в работах Отто
[4, 5] и Михаэлиса [6, 71 метод синтеза через амальгаму широко использо-
ван для получения ртутноорганических производных углеводородов, а так-
Синтез RaHg 'при помощи амальгам Na, Li, К и Cd. 41
же ртутноорганических производных эфиров фенолов и диалкиланили-
нов.
Двугалоидозамещенные углеводороды (ортозамехценные в ароматиче-
ском и жирноароматическом рядах, 1,4-дииод-[8,9] и 1,4-дибромбутан, 1,5-
дйбромпентан и 1,6-дибромгексан 110], но не их низшие гомологи в жир-
ном ряду) при реакции с амальгамой натрия образуют циклические соеди-
нения с гетероатомом — ртутью— полимерного характера в ароматическом
и, возможно, в жирном ряду.
Реакция галоидных производных углеводородов с амальгамой натрия
с образованием ртутноорганического соединения гладко и с удовлетвори-
тельным выходом идет только в присутствии катализатора, каковым здесь
являются главным образом уксусноэтиловый и уксуснометиловый эфиры.
Последние могут быть заменены в некоторых случаях хлоругольным эфиром
и, как показано Льюисом и Чемберленом [11] на примере диизобутилртути,
также, но с худшим эффектом другими карбонилсодержащими соединения-
ми: ацетоном, изовалерьяновым альдегидом, уксусным ангидридом.
Согласно Жиралю [12], замена этилацетата на этилформиат или ацетон
увеличивает выход диэтилртути при синтезе ее из йодистого этила и амаль-
гамы натрия, применение же метил- или этилформиата или ацетона вместо-
Метилацетата при синтезе диметилртути из йодистого метила и амальгамы
натрия уменьшает выход.
Обычно катализатор берут в количестве —х/10 объема галогенида,
но на примере реакции бромистого этила [131 и йодистого изобутила [11]
показано, что наилучшие выходы диалкилртути получаются, если отно-
шение алкилгалогенида и этилацетата равно 1 : 0,5 моля. Снижает выход
R2Hg примесь спирта в этилацетате; небольшая примесь уксусной кислоты
на выход не влияет.
При синтезе ртутноорганических соединений посредством амальгамы
натрия в качестве исходных галогенидов применяются как иодиды, так и
бромиды. Хлориды, хотя и реагируют, но дают незначительные выходы.
Амальгаму натрия берут в избытке, обычно содержащем двойное против-
теории количество натрия. Реакцию галоидных алкилов ведут с низкопро-
центной жидкой амальгамой, для получения диарилртуТи требуется при-
менение более высокопроцентной амальгамы (см. стр. 44). Галоидные ал-
килы реагируют с амальгамой натрия уже на холоду (лучше при охлаж-
дении до0—5°С), галоидные арилы — при нагревании до температуры 140—
150° С. В большинстве случаев реакцию ведут в течение нескольких часов,
хотя в случае йодистого изобутила [11] перемешивание по окончании до-
бавления амальгамы, длившееся 45 мин., не увеличило выхода.
Энергичное встряхивание или перемешивание существенно для полу-
чения хороших выходов.
Общая схема синтеза ртутноорганических соединений посредством
амальгамы натрия представляется, так. Галогенид в смеси с растворите-
лем (или без него) и этилацетатом встряхивают или перемешивают с из-
бытком амальгамы натрия на холоду (R = Aik) или при нагревании (R — Аг)
в круглодонной колбе с обратным холодильником. По окончании реакции
(обычно несколько часов) добавляют воду для растворения галогенида натрия
и выделяют R2Hg (диарилртуть — перекристаллизовывая, диалкилртуть —
перегоняя с паром).
Показана [14] возможность проведения рассматриваемой реакции и без.
предварительного получения амальгамы (см. стр. 47).
Синтез диметилртути [1]. В круглодонной колбе с обратным холодильником попере-
менно встряхивают и охлаждают водой 10 вес. ч. йодистого метила, 1 вес. ч. уксуснометн-
лового эфира [15] и 0,2% амальгамы [16] натрия (1,2 г-атом натрия на 1 моль иодистого-
метила). Конец реакции наступает тогда, когда 'температура падает и прн кипячении н«-
42
Ртутноорганические соединения. Лит. стр. 48—49
скольких капель прозрачной жидкости с азотной кислотой выделяются лишь следы иода.
Если йодистого натрия выпадает так много, что жидкость загустевает и, таким образом, не
достигается достаточно тесного соприкосновения ее с амальгамой, рекомендуется отогнать
наиболее летучую часть содержимого колбы на водяной баие и заставить реагировать со све-
жей амальгамой. По окончании реакции, содержимое колбы смешивают с водой и перего-
няют на масляной бане, температуру которой не следует поднимать выше 160° С. Эфиропо-
добный отгон после отделения водного слоя взбалтывают со спиртовым раствором едкого
кали для удаления уксуснометилового эфира, промывают водой и очищают перегонкой.
Т. кип. 92° С.
Выход диметилртути из йодистого натрия и 0,4%-ной амальгамы натрия
равен 60% [77].
Из бромистого метила и 0,5 %-ной амальгамы при ведении реакции в тече-
ние 3,5—5 час. при —5 ч-—3°С с применением якорной мешалки выход
диметилртути 50—70% [17].
При приготовлении диметил (диэтил) ртути галоидный алкил может быть
заменен диметил(диэтил)сульфатом [18] (см., однако, [12]). В реакцию всту-
пает одна метильная группа диметилсульфата.
Получение диметилртути из диметилсульфата и амальгамы натрия [18]. 2 кг 0,5%-ной
свежеприготовленной амальгамы натрия (1 г-атом натрия) встряхивают в толстостенной
склянке (1 л) для реакций под давлением с 55 г не содержащего кислорода диметилсулцфата
(1 моль) и 10 г метилацетата. Вначале необходимо лишь очень медленно встряхивать, так как
реакция идет быстро с саморазогреванием до 60—70° С. Смесь встряхивают до полного осты-
вания массы, осторожно (небольшое давление) открывают сосуд, растворяют соль прибав-
лением 200 мл воды и отгоняют со слабым током водяного пара, охлаждая приемник ледяной
водой. Выход сырого продукта 38 г (75%). Очистка, как и при синтезе нзйодистого метила
(ср. [12]).
Получение’диэтнлртути из бромистого этила и амальгамы натрия [13]. В сосуд, снаб-
женный мощной механической мешалкой, помещают 0,5%-ную амальгаму натрия, взятую
в 20%-ном избытке, и при перемешивании в течение 1—2 час. в три приема прибавляют
-смесь бромистого этила с уксусноэтиловым эфиром, взятым в отношении 1 : 0,5 моля. Пере-
мешивают при охлаждении льдом до 0° С в течение 5—6 час. По окончании реакции диэтил-
ртуть отгоняется нз реакционной массы с водяным паром. После отделения от воды ее сушат
хлористым кальцием и перегоняют в вакууме. Выход 75—80%; т. кип. 159°С, 2,42346,
п™ 1,5599.
В качестве катализатора этой реакции применен диметилформамид
<0,01—0,05 моля) [19].
СС13Вг с 0,5%-ной амальгамой Na дает неожиданно CCl3HgBr [78].
Из хлористого этила и амальгамы натрия диэтилртуть получается с вьь
ходом не выше 24% [13].
Получениедиэтилртути из йодистого этила и амальгамы натрия [20]. 2900 г (1,25
г-атом натрия) 1%-ной амальгамы натрия перемешивают с 156 г (1 моль) йодистого этила и
10 мл этилацетата в 1 л петролейного эфира в течение 24 час. Затем прибавляют воду к жид-
кости, ставшей черной от выделившейся коллоидной ртути; смесь перемешивают 30 мин.
для разложения амальгамы, слой петролейного эфира отделяют декантацией, промывают,
сушат и перегоняют. Выход 60—70%.
Диэтцлртуть из C2H6J, 0,2%-ной амальгамы натрия в присутствии этил-
ацетата Получена с выходом 67% [21].
Выход диэтилртути увеличивается, если в реакции C2HSJ с амаль-
гамой натрия вместо этилацетата применить формилацетат или ацетон [12]]
Получение диэтилртути см. также [1, 15, 16, 21а—24].
Получение ди-н-пропилртути [17]. В железный аппарат, снабженный якорнопропел-
лерной мешалкой, делающей 400 об!мин, загружено 45 кг 0,5%-ной амальгамы натрия,
986 г бромистого н-пропила и 250 г этилацетата. Реакционную смесь при наружном охлаж-
дении ледяной водой перемешивают в течение 4 час. По окончании реакции дипропилртуть
отгоняют с водяным паром, промывают водой, сушат хлористым кальцием и перегоняют в
вакууме. Т. кип. 89—91°С при 15 мм, 2,0201, 1,5162. Выход чистой ди-н-пропил-
ртути 791 г (69%).
Синтез RaHg при помощи амальгам Na, Li, К и Cd
43
Получение ди-н-пропилртути см. также [2, 25, 26].
Из-фромистого изопропила и 7%-ной амальгамы натрия (в присутствии
этилацетата, 5—6-часовое взаимодействие) получена диизопропилртуть
«‘ выходом 8—10% [2]. Из йодистого н-бутила и амальгамы натрия (про-
центное содержание амальгамы и условия реакции не указаны) полу-
чена ди-н-бутилртуть наряду с иодистой ди:н-бутилртутью [27].
Синтез ди-н-бутилртути из йодистого н-бутила, 0,4 %-ной амальгамы
натрия осуществлен с выходом 64 % [22]. На примере синтеза ди-н-бутилртути
изучена [17] зависимость выхода диалкилртути от природы галоида в гало-
идном алкиле и процентного содержания амальгамы. Наилучший выход
(до 82%) ди-н-бутилртути получен, если исходить из йодистого н-бутила
в условиях синтеза ди-н-пропилртути (см. выше). Если исходить из броми-
стого н-бутила, то оптимальный выход ди-н-бутилртути составляет 70%.
С хлористым бутилом выход не превышает 17%. С более высокопроцентной
амальгамой, чем 0,5%-ная, в значительной степени проходит реакция Вюр-
ца [17].
Получение диизобутилртути из бромистого изобутила [17]. В условиях синтеза ди-н-
пропилртути с применением якорной мешалки (400 об/мин) введено в реакцию 550 г изобу-
тилбромида, 21 кг 0,5%-ной амальгамы натрия и 170 г этилацетата. Перемешивание продол-
жалось 4 часа. По окончании реакции диизобутилртуть отогнана с водяным паром, промыта
водой, высушена хлористым-кальцием и разогнана в вакууме. Т. кип. 108—109°С/25 мм,
1,7670, Пд 1,4969. Выход 393 г (63%). При разгонке диизобутилртути выделено около
45 г 2,5-диметилгексана, т. кип. 107—109°С/752 мм, cP® 0,6990, «р 1,3995. (См. также [11,
22, 28—33].)
Синтез диизобутилртути из йодистого изобутила [11]. В круглодонную колбу (2 л),
снабженную капельной воронкой с хлоркальциевой трубкой, термометром, обратным хо-
лодильником и мешалкой и охлаждаемую льдом, помещают 0,294 моля (31 мл) йодистого
изобутила и 0,147 моля (14,3 мл) этилацетата. Когда смесь примет температуру ледяной
бани, 0,25% -ную амальгаму натрия, приготовленную из 0,591 моля (13,6 г) натрия и 27,2 мо-
ля (5440 г или 400 мл) ртути, прибавляют с такой скоростью, чтобы температура смеси не
поднималась выше 15°С. После того как вся амальгама прибавлена (на что требуется около
45 мин.), прибавляют эфир, избыток натрия разлагают прибавлением воды. Эфирный слой
отделяют, сушат хлористым кальцием и перегоняют. Диизобутилртуть перегоняется в пре
делах 202—206°С, лучше в вакууме (константы см. выше). Выход 56,5%.
Ди-н-(?)-амилртуть получена из н-(?)-амилхлорида и 2%-ной амальгамы
натрия [20].
Получение диизоамилртути [17]. Реакцию проводят с применением якорной мешалки
в условиях синтеза ди-н-пропилртути. Берут 450 г изоамилбромида, 17 кг 0,5%-ной амаль-
гамы натрия, 130 г этилацетата. Реактор охлаждают ледяной водой. Перемешивание реак-
ционной смеси ведут в течение 4 час. По окончании перемешивания продукт реакции отго-
няют с Водяным паром. Образовавшееся масло после высушивания делят в вакууме по
фракциям. Получено]диизоамилртути 258 г (50%) с т. кип. 122—125°Спри 1&мм,^° 1,6381,
Пд 1,4981 и около.100 г 2,7-диметилоктаиа с т. кип. 60°С/15 мм, 0,7282, п& 1,4074.
Получение диизоамилртути см. также [1, 22, 29].
При взаимодействии йодистого гексила с амальгамой натрия Франклан-
ду и Дюппа не удалось получить ртутноорганического соединения [1].
Ди-н-октилртуть получена взаимодействием н-октилиодида с очень раз-
бавленной амальгамой натрия [34].
Циклическое соединение, содержащее 2 атома ртути в цикле, получено
из 1,4-дибромбутана и амальгамы натрия [10].
Получение 1,6-димеркурациклодекана [10]. Смесь 450 г (0,2 моля) 1%-ной амальгамы
натрия, 15,3 г (0,07 моля) 1,4-дибромбутана, 3 мл этилацетата и 25 мл бензола встряхивают
36 час. при 70°С. Охлажденную, смесь встряхивают с 25 мл воды, бензольный слой отделяют,
остаток промывают 25 мл бензола. Из соединенных бензольных растворов испаряют бензол.
Остающееся масло (10,79 г) после промывания холодным эфиром оставляет полутвердый
осадок. После кристаллизации из смеси эфира и бензола т. пл. 44—45,2°С.
кк Ртутноорганические соединения. Лит. стр. 48—49
Соединение, вероятно, аналогичного строения получено из 1,4-дииод-
бутана [8, 9].
При 19-часовом встряхивании 1,4-дииодтетрафенилбутадиена при комнат-
ной температуре с 0,73%-ной амальгамой натрия в толуоле получен [35J
тетрафенилмеркурациклоп ентадиен с выходом 65%, а также соединение
(C28H20)2Hg2.
1,5-Дибромпентан реагирует с 1 % -ной амальгамой натрия с образовани-
ем полимерных и циклических соединений, содержащих ртуть в цикле [36]'
(см. также [10]).
Из 1,6-дибромгексана и 1 % -ной амальгамы натрия (в смеси этилацетата
и бензола, встряхивание в течение 1 дня) получено циклическое соедине-
ние— меркурациклогептан [10].
Получение меркурбис-Р,в'- пропионовой кислоты [3]. В охлажденный снегом с солью
раствор 10 г этилового эфира р-иодпропионовой кислоты в двойном объеме абс. эфира в те-
чейие 30 мин. небольшими порциями вносят 220 г 0,5%-ной амальгамы натрия (1,1 г-атом
натрия). Следует избегать разогревания смеси. Для полноты реакции смесь встряхивают
2 часа при комнатной температуре, затем приливают воды для растворения выделившегося
йодистого натрия и прибавляют эфира. Эфирный слой отделяют. После отгонки эфира оста-
ется некристаллизующееся масло.
Для омыления его сильно встряхивают с 70 мл IN раствора едкого натра до полного
растворения. При 15—20°С на это требуется 7—8 час. Раствор охлаждают до 0вС и прибав-
ляют 16 мл 5 N серной кислоты, при этом медленно выделяются бесцветные кристаллы мер-
кур-б«с-пропионовой кислоты. Их отсасывают после часового хранения в ледяной воде и про-
мывают небольшим количеством холодной воды. Оптимальный выход 2,1 г (27%). После
кристаллизации из 20-кратного количества воды (избегать продолжительного нагревания)
т. пл. 147—148°С.
Аналогично получен [37] метиловый эфир меркур-бпс-пропионовой ки-
слоты и из него меркур-бис-пропионовая кислота (выход 29%).
К а-галоидозамещенным жирным кислотам этот метод не применим [381.
Взаимодействие 1,2,3,4-тетрабромциклобутана с 0,5% ^ной амальгамой
лития (50%-ный избыток) в абсолютном эфире (встряхивание 15—20 час.)
приводит к нестойкому ртутному соединению, не содержащему брома [39].
Амальгама натрия с йодистым циклогексилом даёт иодистую циклогек-
силртуть [40], с бромистым циклогексилом образуется нормальный продукт
реакции — дициклогексилртуть [41].
Из Лментилбромида при 11-часовом встряхивании с 1%-ной амальга-
мой натрия получен 3,3-меркур-бмс-ментан с выходом 35% [42].
Применением нагревания и действием более высокопроцентной амаль-
гамы, чем при синтезе диалкилртути, получены следующие ртутноорганиче-
ские производные ароматических углеводородов: дифенилртуть [5, 6, 43—
45] (также меченная С14) [46], ди-о-толилртуть [5, 47], ди -м-тол ил ртуть (2,7—:
3%-ная амальгама при 180° С) [7], ди-о-ксилил ртуть (катализатор—хлор-
угольный эфир) [481, ди-л«-ксилилртуть (2%-ная амальгама, 12-часовое
нагревание до 140—150° С)'[49], ди-и-ксилюлртуть (110° С, катализатор хлор-
угольный эфир) [501, димезитилртуть [7, 51], дипсевдокумилртуть [71, ди-
/i-пропилфенилртуть (как в присутствии хлоругольного эфира, VaK и дей-
ствием 1%-ной амальгамы натрия в ксилоле в присутствии этилацетата)
[52], дицимилртуть [53, 54], дипентаметилфенилртуть (катализатор —
хлоругольный ‘ эфир) [54], бг/ол-дифениленфенилртуть [71, ди-а-нафтил-
ртуть (4%-ная амальгама натрия, 18-часовое нагревание до 140—150°С)
[4, 55—57], ди-[3-нафтилртуть (4%-ная амальгама, 24-часовое нагревание
до 140° С) [551.
Синтез дифенилртути [43]. В круглодонную колбу (1 л), соединенную с обратным хо-
лодильником, помещают 900 г 3% -ной амальгамы натрия, 180 г бромбензола, 200 мл сухого
толуола или сухого ксилола и 10 мл этилового эфира уксусной кислоты. Смесь кипятят
с обратным холодильником (при частом перемешивании от руки) в течение 12 час. на масля-
ной бане при 130°С.
Cuwitee .RaHg при помощи амальгам Na, Li, К и Cd 45
Смесьпереносят (под тягой) еще горячей на складчатый фильтр, стараясь, чтобы ртуть
осталась в колбе; Дифенилртуть извлекают 600 мл кипящего бензола в течение приблизи-
тельно 10 час. Полученный бензольный раствор упаривают в вакууме на масляной бане, тем-
пературу которой доводят к концу отгонки до 110°С. Твердый остаток после отгонки ра-
створителя промывают охлажденным до 0рС 95%-ным спиртом, пока он не станет бесцвет-
ным. Обычно достаточно четырех промываний порциями по 50 мл. Выход 65—75 г (32—37%).
Т. пл. 125° С.
Улучшенный метод синтеза дифенилртути [58]. Смесь 900 г 3%-ной амальгамы натрия,
180 г бромбензола, 200 мл толуола и 10 мл этилацетата кипятят 12 час. на масляной бане,
температура которой 130° С. Переносят еще горячую массу в большую воронку, по возмож-
ности отделив от ртути. Извлекают по методу, описанному в книге «Синтезы органических
препаратов» [59]. Извлечение заканчивают через 10 час. От раствора, содержащего дифенил-
ртуть, отгоняют растворитель в вакууме прн 20 мм, поднимая температуру к концу от-
гонки до 110°С. Оставшийся осадок после отгонки толуола и бензола промывают холодным
спиртом. Выход 96 г (47%). Т. пл. 122—123°С.
Получение ди-0-нафтилртути [55]. 50 г 0-бромнафталина, 40 г ксилола, 5 г этилацетата
и 300 г 4% -ной амальгамы натрия кипятят с обратным холодильником на масляной баие
в течение 24 час. Во время реакции приблизительно через 12 час. прибавляют еще немного
этйлацетата. После отделения ртути остаток кипячением со спиртом освобождают от орга-
нических примесей, отмывают водой от бромистого натрия и кристаллизуют из амилового
спирта. Т. пл. 238°С. Выход 17 а.
0-Хлорнафталин также дает ди-3-нафтилртуть в этих условиях, но вы-
ход при этом хуже [57]..
Так же получена ди-(а-нафтил)ртуть [55]; т. пл. 249°С.
Амальгама натрия при реакции с бромистым и хлористым бензилом не
дает ртутноорганического соединения, а лишь дибензил [5], При взаимо-
действии (о-бромстирола с 7,5%-ной амальгамой натрия при 140°Св тече-
ние 7 час. с незначительным выходом была получена ди-(со-стирил)ртуть
[60] (см., однако, стр, 16),
Обработка 5-фтор-6-бром-1,2,4-триметилбензола амальгамой лития в эфи-
ре привела к образованию 43% 1,2,4-триметилфениленртути (возможно, по
аналогии с о-фениленртутью, см. ниже, в виде гексамера) и других, не со-
держащих ртути, продуктов [61]. Ортофторбромбензол в. этих условиях
дал 2% о-фениленртути [611 (гексамер).
о-Дибромбензол при нагревании 5 час. с 2%-ной амальгамой натрия
в петролейном эфире в присутствии этилацетата дает вещество, не плавя-
щееся до 300°С, которому Веччиотти [621 приписал строение гетероцикла
с двумя ртутными гетероатомами, но которое, однако, как показал Виттиг
[63], представляет собой гексамер строения (I) (см. гл. II, стр. 29).
Синтез гексамернбй о-фениленртути [63]. В сосуде Шленка из йенского стекла под азо-
том получают амальгаму натрия из 200 г ртути и 2 г (87 г-атом) натрия. К ней при-
бавляют 7,1 г (30 мл) о-дибромбензола в 40 мл абс. эфира. После 3—4-дневного встря-
хивания декантируют верхний слой, разлагают нижний слой сначала холодной, затем
горячей водой, отделяют его от ртути и после сушки извлекают его при кипячении так
долго днметилформамидом, пока последний при охлаждении уже не будет выделять
кристаллов (около трех раз, по 100 мл). Получают 4,3, гл (52%, считая на о-дибромбен-
зол) о-фенилеиртути, т. разл. 326° С.
Определение молекулярного веса и кристаллической структуры [64]
показало, что это гексамер (CeH4Hg)e (см. стр. 29).
Методика выделения продукта реакции видоизменена [65] следующим
образом.
После гидролиза нерастворимый остаток высушивают и много раз извлекают при
перемешивании N-метилпирролндоном (для сырого продукта, полученного из 28,4 г о-ди-
бромбензола, 7 г Na и 680 г Hg требуется около 350 мл N-метилпирролидона). Полу-
ченные мутные экстракты осветляют получасовым центрифугированием при 3500 об/мин.
Из темного раствора-метанолом (половинный объем) гексамерная о-фениленртуть вы-
-саживается в виде белого порошка. Кристаллизация из диметилформамида или из йо-
дистого метила. Выход 13,5 г (41%).
46 Ртутноорганические соединения. Лит. стр. 48—49
При проведении этой реакции в более полярной среде — тетра-
гидрофуране или, лучше, диметиловом эфире этиленгликоля в качестве
главного продукта реакции получена димерная о-терфениленртуть [661
наряду с гексамерной о-фениленртутью (см. гл. II, стр. 29). Вместо амаль-
гамы натрия могут быть применены также амальгама лития и амальгама
калия [66].
Получение димерной о-терфениленртути [66]. 9,5 г о-дибромбензола и амальгаму нат-
рия, приготовленную из 4,6 г натрия и 15 мл ртути, в 100 мл абс. диметилового эфира эти-
ленгликоля встряхивают под азотом 30 мин. н после затухания реакции еще в течение
12 час. После прибавления воды ртуть отделяют в делительной воронке, прибавляют эфир
и отсасывают о-фениленртутъ (гексамер), которую кипятят с небольшим количеством толу-
ола. Соединенные толуольный и эфирный растворы упаривают до начала выпадения кри-
сталлов из горячего раствора. После этого одновременным добавлением толуола и отгонкой
диметилового эфира этиленгликоля заменяют последний на толуол (объем толуольного рас-
твора около 30 мл). При охлаждении выкристаллизовывается слабо окрашенная в корич-
невый цвет о-терфеииленртуть. Кристаллизуют из толуола с активированным углем, т. пл.
292—293°С. Выход 40—42%, считая на о-дибромбензол.
(о,®'-Х1ибром-о-ксилол при 24-часовом нагревании (2%-ная амальгама
натрия, петролейный эфир, этилацетат) дает неплавящееся соединение
предполагаемого строения [67]:
_СН2
jHg
'сн2
Возможно, что это тоже полимер.
При обработке n-дибромдифенила амальгамой натрия в ксилоле Миха-
элис [68] получил желтое нерастворимое вещество, которому он приписал
формулу Hg(CeH4CeH4)2Hg, но которое, вероятно, представляет собой
полимер.
Алкокси- и алкиламинортутноорганические производные ароматиче-
ского ряда получают этим методом, хотя и при таком же продолжительном
нагревании, но действием несколько более низкопроцентной амальгамы,
чем при получении ртутноорганических производных ароматических угле-
водородов.
Получение ди-я-анизилртути [69]. 100 г п-броманизола, 80 г ксилола и 0,1 объема ук-
сусноэтилового эфира кипятят иа масляной бане при 140°С в течение 24 час. с 1,5%-ной
амальгамой натрия, содержащей двойное против теории количество натрия. Колбу часто
встряхивают и через каждые 8час.прибавляют немного уксусноэтилового эфира. Застываю-
щий по охлаждении в твердую массу продукт реакции отделяют от ртути, извлекают бензо-
лом, часть которого затем упаривают. Выкристаллизовавшуюся ди-п-анизилртуть промы-
вают петролейным эфиром. Перекристаллизация из бензола. Т. пл. 202°С. Выход около
40 г (50%).
Видоизменением этой методики без предварительного синтеза амаль-
гамы является следующая [14].
Синтез RaHg при помощи амальгам Na, Li, К и Cd 47
К помещенным в трехгорлую колбу 388 г ртути и 30 г ксилола, нагретым до 160°С, при-
бавлено 0,4 г-атом металлического натрия, затем 37 г (0,2 моля) n-броманизола, растворен-
ного в смеси 6 мл этилацетата и 100 мл ксилола. Смесь кипятят 24 часа при 160°С, гидро-
лизуют водой и сушат. Отгоняют растворитель, остаток кристаллизуют из метанола, т. пл.
ди-п-анизилртути( 202—204°С (выход не указан) [14].
Так же, как ди-«-анизилртуть, получают ди-о-анизилртуть (т. пл.
108°С), ди-о-фенетилртуть (т. пл. 83°С) и ди-п-фенетилртуть (т. пл. 135,5°С)
[55].
Получение бас-га-диметиламииофенилртутн [70]. 100 г п-бромдиметиланилина, 70 г
ксилола и 0,1 объема этилацетата кипятят 24 часа с 1,5%-ной амальгамой натрия, содержа-
щей двойное против теории количество натрия. Продукт реакции извлекают бензолом, после
упаривания которого выкристаллизовывается бис-л-диметиламинофенилртуть. Кристал-
лизация из бензола. Т. пл. 169°С.
Подобным же образом получена ди-(3-диметиламино-4-метилфенил)-
ртуть [71].
5ш>перфторалкильные соединения ртути синтезированы этим методом
из иодперфторалкилов с применением амальгамы не щелочных металлов,
а амальгамы серебра, меди, цинка [72] и, лучше всего, амальгамы кадмия
[72, 73], реакция с которой идет при комнатной температуре (для проведе-
ния реакции с амальгамой меди и серебра требуется нагревание амальгамы
Ag до 80°С, амальгамы Си до 160—180°С, амальгамы Zn — 4 дня при ком-
натной температуре).
Получение бнс-перфторметилртути [72]. 3 г иодтрифторметана конденсируют в трубку
пирекс, содержащую амальгаму кадмия (из 3 г кадмия и 20 мл ртути). После встряхивания
в течение 24,час. прн комнатной температуре жидкая фаза исчезает и образуется твердое ве-
щество. Извлекают эфиром, после испарения которого получают бис-перфторметилртуть,
очищенную возгонкой. Т. пл. 163°С. Выход 35—40%, считая на вошедший в реакцию CF3J
(50-60%).
Так же получена бас-перфторэтилртуть (встряхивание 14 дней при 30°C
C2FbJ и амальгамы кадмия). Выход 60%.
Подобным же образом бис-тридейтерометилртуть получена с 75%-ным
выходом нагреванием .в запаянной трубке (1 день при 160°С или 3 дня прн
125°С) йодистого тридейтерометила с амальгамой кадмия [74].
При взаимодействии бромпентафторбензола с амальгамой лития полу
чена бис- пентафторфени л ртуть [751.
Синтез бпс-пеитафторфенилртути [75]. 5 г бромпентафторбензола в 10 мл эфира при-
бавляют к перемешиваемой взвеси амальгамы лития (0,3 г лития и 200 г ртути) в 30 мл эфи-
ра при 0°С. Происходит энергичная реакция, и вязкая амальгама становится подвижной.
Через 18 час. прибавляют 50 мл воды, эфирный слой отделяют и соединяют с эфирными,
экстрактами (2 X 50 мл) водного слоя, затем высушивают сульфатом магния. Эфир отго-
няют. Коричневый осадок после перегонки с паром и возгонки дает бис-пентафторфенил-
ртуть, бесцветные кристаллы, 2,75 г, т. пл. 117—118° С.
При хранении,вещества т. пл. его возрастает до 136—137°С, при перекристаллиза-
ции из четыреххлористого углерода тотчас после получения т. пл. 138—139°С. Вещест-
во, вероятно, существует в виде нескольких полиморфных разностей.
При кипячении в ксилоле бромбензола со ртутью в присутствии метал-
лического лития получена дифенилртуть [761.
48 'Ртутноорганические соединения
ЛИТЕРАТУРА
1. Frank land Е„ D и р р а В. F. Ann., 130, 104 (1864).
2. Gore t М. Zbl., 1922, III, 1371; Bull. sci’. Pharm., 29, 297(1922).
3. Fischer E. Ber., 40, 386 (1907).
4. О t t о R., M о r i e s G. Ann., 147, 164 (1868).
5. Dreher E., О 11 о R. Ann., 154, 94 (1870).
6. M i c h a e 1 i s A. Ann,, 181, 290 (1876).
7. M i c h a e 1 1 s A. Ber., 28, 588 (1895).
8. В г a и n J. Ber., 46, 1792 (1913).
9. В г a и n J. Ber., 47, 490 (1914).
10. S a w a t z к у H„ W r i g h t G. F. Canad. J' Chem., 36, 1555 (1958).
11, Lewis H.F., Chamberlin Ё. J. Am. Chem. Soc., 51, 291 (1929).
12. G i r a 1 F. C. A., 36, 3149 (1942); Ciencia (тех.), 2, 114 (1941).
13. Мел ь н и ков H. И„ P о к и ц к а я М. С. ЖПХ, 12, 1802 (1939).
14, D е s s у R. Е., Kim J.-Y. J, Am. Chem. Soc., 82, 686 (i960).
15. J о n e s L. W„ Werner L.F. J. Am. Chem. Soc., 40, 1257 (1918)1
16. M fl 1 1 e r A., S а и e r w a 1 d A. Monatsh:, 48, 737 (1927).
17. M e л ь н и к о в Н. И. ЖОХ, 16, 2065 (1946).
18. FuchsK. J. prakt. Chem., .2, 119, 209(1928).
19. Р а з у в а е в Г. А., П е т у х о в Г. Г., X р и п у н о в В, П., Тимошенко
3. Ф„ С е р г ее в а О. М. Пат. СССР 113714 (1958);. С. А., 53, 6082 (1959).
20. М о г t о n А. А., Н е с h е n b 1 е i kn.e г I. J. Am. Chem. Soc., 58, 1024 (1936).
21. V i t t е G., Mesnard P. Bull. Soc. chim. France, 8, 579 (1941).
21a. В i a 1 a s J., Eckstein Z.,Ejmocki Z.,Hetnarski B.,Sobo’tka W.,
Szymaszkiewicz J. Przem. Chem., 40, 567 (1961).
22. W i 1 d e W. K. J. Chem. Soc., 1949, 75.
23. J о s e J., Espinoza C.C. A., 42, 2924 (1948); Rev. chim. farm. (Santiago, Chile),
4, № 56, 2 (1947).
24. Chapman E. L Ann,, 139, 128 (1866).
25. Щер.б a ko-b А. ЖРФХО.12 , 353 (1881).
26, CahoiirsA. Compt. rend., 76, 133, 748 (1873).
27. T i f f e n e a u M. Zbl., 1921, III, 26; Bull. Sci. Pharm., 28, 65 (1921).
28. Muller.A. J. Am. Chem. Soc. 57, 1142 (1935). .
29. M a r q u a r d t A. Ber., 21, 2035 (1888).
30. Chapman E. T„ Smi th M. H. J. Chem. Soc., 22, 163 (1869).
31. C a h о u r s A. C. r„ 77, 1405 (1873); J. prakt. Chem. [27], 8, 397 (1873).
32. Соколове. ЖРФХО, 19, 202 (1887). . - ’
33. P о n z i о G. Gazz., 30, 24 (1900).
34. Eichler E. Ber., 12,1880 (1879).
35. Bray 1 E. H., H ii b e 1 W., Cap Her I. J. Am. Chem. Soc., 83, 4406 (1961),
36. H i 1 p er t S., G r fl t t n er G. Ber., 47, 186 (1914).
37. Л ат я ева В. Н„ ОльдекопЮ. А. Уч.зап.ГГУ, 1958, вып. 32, 187.
38. Schrauth W„ Schoeller W. Вег., 41, 2088 (1908).
39. A u г a m М., Marica E,,NenitzescuC. D. Ber., 92,1088(1959); A u г a m М.,
MaricaE., PoganiJ., NenitzescuC. D. Angew. Chem., 71, 626 (1959).
40. К у p с а н о в H. И. ЖРФХО, 31, 535 (1899).
41. Т i f f е n е a u М., G a и n a g ё Е. С. А., 1921, 2432; Bull. Sci. Pharm., 28, 7 (1921).
42. Р е у т о в О. А., Л у Ц з ин-чжу, Бундель Ю. Г. Вести. МГУ, сер. мех.
мат., астр. физ. хим., 13, № 5, 111 (1958).
43. Синтезы органических препаратов, сб. 1. М., ИЛ, 1949, стр. 204.
44. L a d е и b и г g A. Ann., 173, 151 (1874).
45. Aronheim В. Ann., 194, 148 (1878).
46. Р а з у в а е в Г. А., Митрофанова Е. В.; Петухов Г.Г. ЖОХ, 30, 1996
(1960).
47. Drfeher Е„ Otto R. Ann., 154, 171 (1870).
48. Jacobsen О. Вег., 17, 2372 (1884).
49. W е 1 1 е г J. Вег., 20, 1718 (1887).
50. J а с о b s е п О. Вег., 14, 2112 (1881).
51. Р а з у в а е в Г. А., Ольдекоп Ю. А„ Королевам. Н. ЖОХ, 21, 650
(1951).
52. Р a t е г п о Е„ Colombo Zbl., 1877, 823; Ber., 10, 1749 (1877).
53. Meyer R. J. prakt. Chem., 2, 34, 103 (1886).
54. J a c 6 b s e n O. Ber., 22, 1215 (1889).
55. M i c h a e 1 i s A. Ber., 27, 244 (1894).
56. О t t о R. Ann., 154, 188 (1870).
57. C h a 11 a w a у F. D. J. Chem. Soc., 65, 877 (1894).
58. С a 1 v er у О. H. J. Am. Chem. Soc., 48, 1009 (1926).
59. Синтезы органических препаратов, сб. 1. М., ИЛ, 1949. стр. 262.
Синтез RaHg при помощи амальгам Na, Li, К и Cd
49
60. DasGupta Н. N. Zbl., 1938, I, 597; J. Indian chem. Soc., 14, 400 (1937).
Gl. WittigG., HarleH. Ann., 623, 17 (1959).
62. Ve с c h i о 11 i L. Ber., 63, 2275 (1930).
63. W i t t i g G., В i eke 1 hau p t F. Ber., 91, 883 (1958).
M. Gr deni c D. Ber., 92, 231 (1959).
65. W i 11 i g G„ E b e 1 H. F. Ann., 650, 20 (1961).
66. W i 11 i g G., H a h п E., T о c h t e r m.a n n W. Ber,, 95, 431 (1962).
67. Vecchiott i L. Gazz., 63, 110 (1933).
68. M i c h a e 1 i s A. Ber., 27, 588 (1894).
69. Michaelis A., R a b inerson I. Ber., 23, 2344 (1890).
70; Schenk A., Michaelis A. Ber., 21, 1501 (1888).
71. P e s c i L. Z. anorg, allg. Chem., 17, 280 (1898).
72. Emeleus H. J„ H a s z e 1 d i ri e R. N. J. Chem. Soc., 1949, 2953.
73. В a n u s J., Emeleus H. J., Haszeldine R. N. J. Chem. Soc., 1950, 3041.
74. BevegeE. E., Renaud R., Leitch L.C. Can. J. Chem., 31, 1259 (1953).
75. С о e P. L., S t e p h e n s R., Tatlow J. C. J. Chem.Soc., 1962. 3227.
76. Англ. пат. 854776 (1960); РЖХим, 1961, 18Л91.
77. К о т о н М. М. ЖОХ, 22, 1136 (1952).
78. Несмеянов А. Н., Фрейдлина Р. X., Величко Ф. К. ДАН СССР,
114, 557 (1957).
Глава V
ВВЕДЕНИЕ РТУТИ НА МЕСТО ВОДОРОДНОГО АТОМА
(МЕРКУРИРОВАНИЕ)
Самым важным методом синтеза ртутноорганических соединений являет-
ся непосредственное замещение атома водорода на остаток — HgX (X —
анион) при действии солей ртути по схеме RH -^HgX2 = RHgX -рНХ.
Метод этот, обоснованный работами Песчи, Гофмана и особенно Димрота,
находит применение в некоторых областях алифатического ряда и широ-
кое повсеместное применение в ароматическом и гетероциклическом рядах.
Меркурирование является реакцией электрофильного замещения и про-
исходит по месту наибольшей электронной плотности в меркурируемой мо-
лекуле. В ароматическом ряду это подтверждается огромным числом фактов,,
указывающих на более легкую меркурируемость более нуклеофильных аро-
матических ядер; и из явлений ориентации меркурирования, в основном,
подчиняющейся обычным законам ориентации электрофильного замещения.
Соединение алифатического ряда должно обладать подвижным водород-
ным атомом и способностью давать протонизируемый карбанион для того,
чтобы ртуть могла быть введена в него меркурированием. Особенно легко
меркурируются такие соединения, как малоновый и ацетоуксусный эфиры,
ацетиленовые производные со свободной группой С=СН, циклопентадиен.
Замещение водорода в этих соединениях может происходить и за счет вто-
рой валентности ртути, что, вообще говоря, является исключением. Так, ма-
лоновый эфир с окисью ртути дает (С2НВООС)2СН — Hg —СН(СООС2НБ)2,
ацетилены RC=CH с ртутноорганическими гидроокисями R'HgOH
образуют ацетилениды RC=CHgR', причем для осуществления этих
реакций достаточно проведения их при комнатной температуре.
Более жесткие условия требуются для меркурирования спиртов, кислот,
кетонов и альдегидов. Внедрение ртути происходит здесь, разумеется, в
a-положение, но редко дело ограничивается вступлением в молекулу одно-
го атома ртути, причем одновременно с вступлением ртути часто наступают
функциональные изменения (карбонила), примеры которых видны из даль-
нейшего изложения. Гомологический ряд алкилртутных солей, очевидно,
не может быть получен посредством меркурирования.
Все это очень ограничивает возможности метода в жирном ряду. Наобо-
рот, все ароматические соединения, в том числе и ароматические гетероциклы
и их бензопроизводные, а также ферроцен и некоторые другие ценовые сое-
динения, с большей или меньшей легкостью при нагревании, а иногда и при
комнатной температуре взаимодействуют с меркурирующими агентами,
образуя алкилртутные соли. Ароматическое ядро, активированное по от-
ношению к обычным реакциям электрофильного замещения (нитрованию,
Введение ртути на место водородного атома (меркурирование) 51
сульфированию), активно и к меркурированию, т. е. среди производных беп-
золанаиболее легко (уже в водных растворах, при комнатной температуре)
меркурируются амины, далее идут фенолы. Одноатомные фенолы обычно
меркурируют при повышенной температуре. Меркурирование резорцина
и флороглюцина происходит так легко, что только для первого удается ог-
раничить вступление ртути стадией мономеркурированного продукта, при-
чем реакция идет на холоду в водном растворе.
Толуол реагирует легче, чем бензол, а нафталин еще легче.
В нитробензол и другие производные бензола, содержащие мета-ориен-
танты, атом ртути вступает труднее, чем в бензол: реакция с уксуснокислой
ртутью течет при нагревании без растворителя, до 150°C. Особенно лег-
ко меркурируются суперароматические гетероциклы: пиррол, фуран и тио-
фен. Пиридин, наоборот, меркурируется в жестких условиях. В мягких
условиях меркурируется ферроцен и в сравнительно мягких условиях дру-
гие меркурирующиеся. ценовые соединения.
Вступление ртути в ароматические соединения подчиняется обычным
правилам ориентации электрофильного замещения. Аномальная ориента-
ция, наблюдающаяся при меркурировании уксуснокислой ртутью нитро-
бензола, бензойной кислоты, бензофенона, толуола (преимущественное об-
разование орто-изомера-в первых случаях, образование в значительных ко-
личествах мета-изомера в последнем случае), объясняется низкой степенью
ионизации ацетата ртути в этих неполярных средах и тем, что реакции эти
идут при высоких температурах (до 150°С), при которых меркурирующий
агент обладает г^алой селективностью. Возможно, что в этих условиях ре-
акция в значительной степени имеет гомолитический характер. Как пока-
зали Клаппрот и Уэстхёймер [1], при меркурировании нитробензола и толу-
ола сильно ионизированной солью ртути —перхлоратом ртути в водной
хлорной кислоте, где, очевидно, меркурирующим агентом является ион
ртути в той или иной гидратированной форме, реакция идет в мягких усло-
виях и распределение изомеров подчиняется обычным для электрофильного
замещения ориентирующим влияниям.
Следует учитывать также возможность обратимости реакции мер кури-
рования при высоких температурах и вероятность перехода одного изомера
в другой — более устойчивый — при большой длительности реакции
(см. стр. 91).
Изучение кинетики реакции меркурировании на примере меркуриро-
вания алкилбензолов показало, что реакция имеет второй порядок. Основ-
ным недостатком метода меркурировании в ароматическом ряду, ограничи-
вающим его применение, является образование смеси всех возможных изо-
меров . мономеркуриррванных соединений и одновременное образование
полимеркурированных продуктов. Такие смеси не всегда хорошо раз-
делимы. .
Все же благодаря простоте и дешевизне прямое меркурирование арома-
тических соединений является главным методом синтеза в этом ряду.
Преимущество меркурировании перед синтезами через магнийорганиче-
ские и подобные соединения — возможность] введения ртути в молекулы
с заместителями, не безразличными к реактиву Гриньяра, и т. п. Отсутствие
необходимости готовить исходные соединения является преимуществом пе-
ред методами синтеза через сульфиновые, арилборные кислоты и подобные
соединения. Арилртутные соли без заместителей или с простыми замести-
телями в том случае, когда доступен соответствующий амин, проще и чище
получаются диазометодом. Если, однако, принять во внимание, что мерку-
рированию доступны сложнейшие ароматические молекулы, например
многократно замещенные производные бензола и конденсированные арома-
тические углеводороды, полифенилметановые производные, в том числе кра-
52 Ртутноорганические соединения. Лит. стр. 104—114
сители этого ряда и разные другие красители, гетероциклы, алкалоиды, то
станет ясно, что метод прямого замещения водородного атома на атом ртути
является универсальным.
В качестве мер курирующих агентов применяют как окись ртути, так
и ее соли: ацетат ртути (пригодны также ртутные соли и других карбоновых
кислот), сульфат, перхлорат, нитрат. Уксуснокислая ртуть наряду с окисью
ртути относится к числу наиболее интенсивно меркурирующих реактивов,
так как выделяющаяся слабая уксусная кислота не расщепляет получаю-
щуюся ртутноорганическую соль и при высоких температурах. Однако при •
меркурировании уксуснокислой ртутью ароматических соединений при вы-
соких. температурах ацетат ртути обладает малой селективностью и приме-
нение его влечет за собой образование неупорядоченной смеси изомеров (см. •
выше). В этих случаях меркурирование перхлоратом ртути при низ-
ких температурах приводит к нормальной ориентации. Также, при мерку-
рировании уксуснокислой ртутью, прибавление даже небольших количеств
хлорной кислоты во много раз увеличивает скорость меркурирования, поз-
воляя вести его при комнатной температуре. Ориентация при этом стано-
вится более специфичной (см. при меркурировании толуола).
Нитрат ртути успешно применим для меркурирования жирных кето-
нов по а-углероду, а также для меркурирования ароматических соединений,
в особенности в виде смеси с окисью ртути и безводным сульфатом кальция.
Роль последних ясна из уравнений:
RH + Hg(NO3)2 RHgNO3 + HNO3,
HgO + 2HNOS Hg(NO3)2 + H2O,
C3SO4 —[- 2H2O —> CsSOv2H2O.
Галогениды ртути относятся к самым вяло меркурирующим агентам и
р^еют ограниченное применение. Сулема, по понятной причине, более энер-
гично действует в присутствии ацетата или бикарбоната натрия. Наиболее
слабо рдеркурирует цианистая ртуть, применение которой ограничено полу-
чением некоторых ацетиленидов ртути. Интенсивно меркурирующими
агентами являются меркурацетамид и ртутная соль тринитрометана. По-
следняя не представляет собой, однако, достаточно универсального сред-
ства.
,sv Реакцию меркурирования проводят или в растворе, особенно часто в
водном, или действуя меркурирующим агентом прямо на жидкое или рас-
плавленное твердое соединение при разных температурах, от комнатной до
180;—200°С, в зависимости от большей или меньшей трудности взаимодей-
ствия. Реакцию ведут до исчезновения иона ртути, для чего обычно подще-
лачивают пробу реакционной смеси едким натром и убеждаются в отсут-
ствии желтого осадка окиси ртути. Продукт или выкристаллизовывается
из реакционной смеси сам и тогда отделяется и отмывается от обычно
применяемого избытка меркурируемого вещества или высаживается добав-
лением к реакционной смеси раствора хлористого натрия или бромистого ка-
лия в виде соответствующего галогенида, всегда менее растворимого и более
высокоплавкого, чем, например, первичный продукт меркурирования —
ацетат. Очищают перекристаллизацией из разных органических раствори-
телей, причем полимеркурированные продукты обычно не растворяются,
а орто-изомер часто значительно более растворим, чем пара-изомер, и ухо-
дит в маточнике. В качестве растворителей (в порядке увеличения раство-
ряющей способности) наиболее часто применяют бензол, хлороформ, спирт,
этилацетат, ацетон, диметилформамид, пиридин.
Введение ртути на место .водородного атома (меркурирование)
5 3
МЕРКУРИРОВАНИЕ ЖИРНЫХ И АЛИЦИКЛИЧЕСКИХ СТРУКТУР
Течение реакции меркурировании в алифатическом ряду сравнительно
с меркурированием ароматических и гетероциклических соединений отли-
чается некоторым своеобразием. Действие солей ртути на алифатические
соединения нередко не ограничивается заменой одного атома водорода на
ртуть, но ведет к получению, полимеркурированных продуктов вплоть до
образования сполна меркурйрованных соединений — меркарбидов.
Следует иметь в виду, что в тех случаях, когда были получены поли-
меркурированные продукты (см. далее меркурирование альдегидов, кето-
нов, ряда карбоновых кислот и др.) не всегда можно быть уверенным "в том,
что эти вещества представляли собой индивидуальные соединения, Дак как
полимеркурированные соединения в большинстве случаев нерастворимы
и с трудом поддаются очистке. Приписываемые авторами этим веществам
структурные формулы, основанные часто лишь на данных элементарного
анализа, содержащие двойную связь C=Hg, трех- и четырехчленные циклы
с участием атома ртути, не соответствуют действительности. Несомненно,
что такие вещества, а возможно, что и другие полимеркурированные соеди-
нения представляют собой полимеры.
В некоторых случаях действие меркурирующих агентов, кроме замены
водорода на ртуть, имеет своим следствием изменение функции меркури-
руемого соединения. Однако во многих случаях меркурирование алифати-
ческих соединений, так же как меркурирование ароматических и гетеро-
циклических соединений, не сопровождается побочными реакциями и ско-
рость реакции мер курирования зависит лишь от подвижности замещаемого
водорода. Так, соединения с особенно подвижным водородом: производные
малоновой [2, 3], метилмалоновой [4],. метилен-бис-малоновой [5, 61, аллил-
малоновой [7], циануксусной [8, 9], цианпропионовой [8], нитроуксусной
[10—12], диазоуксусной [13—15], ацетоуксусной [3, 16—18] кислот лег-
ко меркурируются по а-углероду при простом встряхивании на холоду
с раствором соли ртути или с окисью ртути. Меркурирование первых двух
кислот сопровождается отщеплением одной молекулы СО2 (см. гл. IX,
стр. 206). При нагревании малоновой кислоты с окисью ртути в щелочной
среде образуются полимеркурированные продукты [191. Подобные соедине-
ния меркурируются также действием сулемы в присутствии соды и глице-
рина (эфиры [20—21], амиды [20, 21], замещенные амиды [20] и анилиды
[21] ацетоуксусной и малоновой кислот), действием меркурацетамида (эфир
метилмалоновой кислоты [4], цианацетамид и его N-алкил- и N-арилпроиз-
водные [20]), действием ртутной соли тринитрометана [22] (эфиры малоно-
вой, нитроуксусной, ацетоуксусной кислот,, а также ацетил ацетон). Аце-
тоуксусный эфир меркурируется по метилену также ртутными солями дру-
гих карбоновых кислот, кроме уксусной, с образованием димеркурпроиз-
водных 418], при действии окиси ртути при нагревании ацетоуксусный
эфир образует соединение, в котором на два атома ртути приходится три
остатка эфира [23] и продукты более сложного строения [23а].
Меркурирование ацетиленов во избежание присоединения соли ртути
по кратной связи ведут действием щелочного раствора иодной ртути.
Таким путем легко меркурируются с замещением водорода СН-группы
и образованием (HC=C)2Hg ацетилен [24], его однозамещенные произ-
водные [25—27] с образованием (RC=C)aHg, ацетиленовые эфиры
ROC2H4C^CH[26], разнообразные алкил- [27] и арилацетилены [24—25,27—
29]. Так же промеркурированы 7-о- и п-метоксифеноксигептил-1-ины [30],
1-бутилтиобутен-3-ин-1 [31а], фенилмеркаптоацеТилен [31], 1-тридецил-
[32], п-толилмеркаптоацетилены [33], 2-карбэтокси-2-метил-1-этинилцикло-
гексилкарбинол [34], транс-ундец-7-ен-1-ин [35], 1-фенилбутадиинил Е36{,
54 Ртутноорганические соединения. Лит. стр.. 104—114
сполна дейтерированный метилацетилен [37], м-гептадец-1-ин [38], дец-
цис- (и транс-)-З-ен-1 -ины [39], ct-нафтил ацетилен [40], бис-(нафтилэти-
нил)- и бис-{ 1-тиенилэтинйл)ртуть получены также действием на соответ-
ствующие замещенные ацетилены окиси ртути в спирте в присутствии ще-
лочи при кипячении в течение 5 час. [41];1{ис-(и транс-)-пент-2-ен-4-ины
[42], 4-и-метоксифенилбутин-1 [43], 4,4-дифтор-2-бутен-1-ин [44], нона-
диин-1,4 [45], п-нитрофенилацетилен [46], ш-фторалкииы F(CH2)ftC=CH
(п = 3,4, 5, 6) [47], 1,1,1-трифторпропин [48], пентафтор бу тин [48], моно-
и диацетиленовые спирты [49], фенилдиацетилен [50], 3-бром-2,4,6-три-
метилфенилацетилен [25], п-диметиламинофенилацетилен [51], монохлор-
ацетилен [52], замещенные фенилэтинилкарбинолы [53], бутилацетилен
[54], ацетиленилдивинил [55], 1-/пранс-2-бромвинил-2-этинилбензол [56]
(меркурирован в присутствии w-бутиламина; исследована кинетика такой
реакции).
Меркурирование галоидозамещенных этиленов [57,58] также ведут дей-
ствием щелочного растрора иодной или цианистой ртути. Цианистая ртуть
в щелочном растворе пррявляет наряду с меркурирующим действием спо-
собность отщеплять галридоводород, поэтому при действии этого мерку-
рирующего агента на полигалоидозамещенный этан [57] образуются ртут-
ные производные галоидрзамещенных этиленов, например:
2СНВг2СНВг2 + Hg(CN)2 + 2КОН -» (CBr2^CBr)2Hg + 2КВг + 2KCN + 4Н2О,
а из полигалоидозамещенных этиленов — ртутные производные галоидо-
замещенных ацетиленов [57—601 (последние не обладают квазикомплекс-
ными свойствами [611):
С1 С1
. 2 /С=с/ + Hg (CN)2 + 4КОН(СС1=С)2 Hg + 2КС1 + 2KCN + 4Н2О.
Н Н
Согласно Фиц Джибону [58], из дихдорэтиленов так реагирует лишь цис-
изомер; /пранс-дихлорэтилен не отщепляет HCI и продуктом реакции явля-
ется (CHCl=CCl)2Hg. Трихлорэтилен также не отщепляет НС1 и дает
бос-перхлорвинилртуть [62, 63]. Дифтор ацетилен ид ртути получен пропу-
сканием фтор ацетилен а. через водный раствор азотнокислой ртути [64].
Ацетилен может быть промеркурирован даже действием ртутнооргани-
ческих гидроокисей с образованием RHgC=CHgR [64—661 и RHgCssCH [67].
На основании изучения кинетики реакций монозамещенных ацетиленов
с RHgX и Me2HgX4 в присутствии основания — триэтиламйна (реакции
имеют псевдопервый порядок из-за избытка ацетилена и основания) пред-
ложен [68] механизм этих реакций через четырехцентровое переходное со-
стояние.
Соединения с менее подвижным водородом меркурируются в более же-
стких условиях — действием Избытка соли ртути при нагревании, что вле-
чет за собой образование полимеркурированных продуктов. Так, например,
ацетат натрия при кипячении с окисью ртути в конц. щелочи дает соеди-
нение, которому Гофман [69] приписал формулу Hg=C(HgOH)COONa.
Продукт реакции хлорацетата калия с окисью ртути [69] представляет
собой смесь, а не является продуктом меркурирования [69а]. При кипяче-
нии уксуснокислой ртути в уксусной кислоте получается соединение со-
става Ci0H10O10Hg5 [70]. Меркурированная уксусная кислота HgCH2CO2
может быть получена при нагревании уксуснокислой ртути выше ее темпе-
ратуры плавления [71]. Описано меркурирование ацетата и пропиона-
та натрия сплавлением с ацетамидом и окисью ртути [72]. Описана реакция
ацетата ртути с метил-а-элеостеаратом [731.
Введение ртути на место водородного атома (меркурирование)
55
Продукту меркурировзния уксусного ангидрида ацетатом ртути припи-
сана структура тримеркурированной уксусной кислоты [74]:
(CH3COOHg)3C-C=0
I I
Hg+ О-
При нагревании меркурамидов, например меркурацетамида, полу-
чаются вещества строения А [78].
со
СН3/ >NH
Hg
А
Меркурирование ацетамида см. также [75]. О меркурировании ацил-
амидов см. также гл. XVI. В пировиноградной кислоте действием азот-
нокислой ртути в присутствии соли двухвалентной меди заменяются все
три водорода метильной группы с образованием соединения [76]:
Hg
О</ хс—со—соон
Hg HgNO3
Меркурирование сложных эфиров сульфоалифатических дикарбоновых
кислот, как, например, сульфосукцината, произведено [77] действием окиси
ртути при 4—5°C (строение продуктов не приводится).
При многочасовом кипяченииэтиловдго спирта с окисью ртути в присут-
ствии едкого натра образуется полностью меркурированный этан — этан-
оксигексамер карбид, по Гофману имеющий строение, соответствующее фор-
муле Б [79].
HOHg HgOH
Hg-c-c-Hg.
О< / \ >0
\Hg Hg/
Б
Метиловый спирт не дает аналогичных соединений [79]. Этот же меркар-
бид получен при таком же действии окиси ртути на пропиловый, аллиловый,
бутиловый и амиловый спирты, уксусную кислоту, крахмал, тростниковый
сахар, целлюлозу [79].
Высказывались [80] предположения о его полимерной природе. Име-
ются доказательства в пользу строения его как производного метана, а не
этана [81].
Альдегиды, как правило, окисляются действием обычных меркурирую-
щих агентов [82]. Соединение CaH0HgaC103 получено действием хлорно-
ватокислой ртути на ацетальдегид [83]. При реакции ацетальдегида с окисью
ртути в щелочном растворе получено быстро полимеризующееся со-
единение состава (C4H8O5Hg3)^ [79, 84].
По Гофману [79] ацетальдегид при продолжительном кипячении
с окисью ртути и водно-спиртовой щелочью переходит в тр имер курировав-
/ Hg \
ный ацетальдегид HOHg ( / I ССНО, затем в соответствующую кислоту
' Hg >
/ Hg \ г / Hg \
HOHglox^ \|ССО3НИ, наконец, в меркарбид HOHgI о/ />)с~ *
56
Ртутноорганические соединения. Лит. стр, 104—114
Образование этаноксигексамеркарбида из этанола идет таким же путем.
При действии на содержащий ацетальдегид водный раствор уксуснокис-
лой ртути при 0°С едким натром до щелочной реакции образуется аморфное
соединение C2H4O2Hg [82]. Ацетальдегид реагирует с раствором HgO в
Na2SO3 в присутствии щелочи с образованием нерастворимого соединения
C2H2O2Hg2 [85]. При действии подкисленным азотной кислотой раствором
азотнокислой ртути на ацетальдегид образуется соединение C2HO4NHg2
[16].
Описано образование нерастворимых соединений при действии серно-
кислого раствора сернокислой ртути на ацетальдегид и его высшие гомо-
логи [86].
Мономеркурированный ацетальдегид Несмеянов и Луценко получили
присоединением соли ртути к простым виниловым эфирам (см. гл. VI,
стр. 146).
При действии окиси ртути на ацетон (в присутствии едкого барита, на
холоду) [84] и уксуснокислой ртути на ацетон [87] и метил этил кетон [87]
(при нагревании) наряду с меркурированием происходит изменение функ-
ции этих соединений. Продукту, образующемуся из окиси ртути и ацетона,
приписано [84] строение
НН
I . I
HOHg—С—Hg—С—HgOH
I I
Н3С —С—О — С—СНз
он он
Другие способные к энолизации-кетоны дают подобные же соединения,
которые в большинстве случаев, однако, были выделены в виде нераствори-
мых полимеров [84, 88].
Соединению из ацетона (R — Н) или метилэтилкетона (R = СН3) и ук-
суснокислой ртути приписано [87] строение
CH2R CH2R
НО—С—О— C—OH
CH3OCOHgx I
>C
CHsOCOHg/ I
R
| /HgOCGCHs
| ^HgOCOCHa
R
Мономеркурированный ацетон получен косвенным путем через бутилизопро-
пениловый эфир (см. гл. VI).
Мономеркурированный ацетон легко и с хорошим выходом удается
получить, применяя в качестве меркурирующего средства смесь азот-
нокислой ртути, окиси ртути и сульфата кальция [88] (см. стр.
52). Этой мер курирующей смесью удалось промер курировать [89] в а-по-
ложение и другие жирные кетоны, а также ацетофенон (в СН3-группу).
При этом в метилэтилкетоне атакуются оба a-положения с образованием
двух мономеркурированных соединений, в метилизопропил кетоне проис-
ходит замена на ртуть только третичного водорода, но не первичного. Пи-
наколин дает а-меркурированное соединение с выходом 33%. Ртутная соль
тринитрометана также меркурирует ацетон (70 час. в эфире, 50 час. в воде)
и циклопентанон (5 дней в воде), выход количественный в обоих случаях
[22]. Продукт—мономеркурированное соединение.
Введение ртути на место водородного атома (меркурирование)
57
О нерастворимых соединениях, образующихся при действии на ацетон
кислого раствора азотнокислой ртути [16], сернокислого раствора серно-
кислой ртути [3, 86,90,'91], цианистой ртути в щелочном растворе [92, 931
и при действии на ацетон и другие кетоны щелочного раствора иодной рту-
ти [941 см. оригинальную литературу.
Предельные ациклические углеводороды, как правило, не меркуриру-
ются. Из алициклических же углеводородов был промеркурирован цикло-
пентадиен, который меркурируется в метиленовую группу, в весьма мягких
условиях — действием спиртового раствора сулемы [95, 96]. Продукт —
димеркурированное соединение, возможно полимер [96]. В присутствии
ацетата натрия образуется смесь хлористой циклопентадиенилртути и ди-
циклопентадиенилртути [97]. По другим данным [122а], сулема или аце-
тат ртути в присутствии ацетата натрия дает Cfi(HgX)6; в отсутствии
ацетата натрия с сулемой образуется C6H5HgCl, с ацетатом ртути —
C5H2(HgC2H3O2)4.
Смесь CEHEHgJ и (C5H5)2Hg образуется при реакции циклопента диен а
с водным K2HgJ4 [98]. fiuc-циклопентадиенилртуть с т. пл. 83—85°С может
быть выделена из этой смеси извлечением тетрагидрофураном или петролей-
ным эфиром [98а].
При взаимодействии 1-пирролидиноциклопентена с сулемой или бром-
ной ртутью происходит образование иммониевой соли, сопровождающееся.
С-меркурированием [986]:
~ HgX
Энамины же, не содержащие пространственно затрудненного азота, мер ку-
рируются по нему (см. гл. XVI).
Для введения ртути в молекулу декагидронафталина требуется воздей-
ствие уксуснокислой ртути при высоких температурах (200°С) и давлении;
[99].
Описана попытка меркурирования 1,1-пентаметиленбицикло-(0,1,4)-геп-
тана [100].
В настоящем разделе упомянем также о меркурировании производных:
алициклических’ углеводородов: камфоры [101], оба а-водорода которой
подвергаются замещению при действии иодной ртути в растворе едкого нат-
ра или этилата калия.
При действии на камфору щелочного раствора K2HgJ4npn 100°С получе-
но соединение состава (C10H14O)3Hg4.J2 [101], из которого при обработке его-
холодной ледяной уксусной кислотой и разбавлении раствора водой при
высаживании галогенидом щелочного металла получена мономеркуриро-
ванная камфора:
Метиловый эфир камфоркарбоновой кислоты меркурирован [102] при на-
гревании с окисью ртути до 160—180°С (строение продуктов не указано).
Камфоркарбоновая кислота меркурирована действием уксуснокислой
58
Ртутноорганические соединения. Лит. стр. 104—114
ртути (условия не даны) [102]. Гексагидрофенбл меркурирован [99] при вы-
сокой температуре (200°С) и давлении. Холестерин меркурирован при
кипячении с уксуснокислой ртутью в уксусной кислоте в положение 6 [103—
105]:
Описано получение моно-, а также димеркурированных производных
щиклогептатриен-1-ол-2-она нагреванием его с уксуснокислой ртутью до
150°С [106].
В настоящий раздел отнесено также меркурирование ацетофенона на
том основании, что при действии на ацетофенон (при 150° С) соли ртути за-
мещение происходит в алифатическом радикале [7, 71, 107].
Согласно патентным данным [108], при меркурировании жирноаромати-
ческих кетонов и их производных при низкой температуре замещение про-
исходит в ароматическое ядро, при высокой температуре — в алифатиче-
ский остаток. (Это правило, разумеется, не является общим для жирноаро-
матических соединений других классов, ср., например, меркурирование
нитротолуолов ацетатом ртути, стр. 68).
п-Хлорацетофенон меркурирован [1081 при нагревании с уксуснокислой
ртутью до 130°С (место вступления ртути не указано). п-Бром-.м-нитроаце-
тофенон меркурирован сплавлением с уксуснокислой ртутью (135° С, 1 час.
30 мин.), с образованием двух продуктов (строение не указано) [109]. Соот-
ветствующий иодистый аналог в этих же условиях дал один продукт [109].
Согласно Ганчу [ПО, 111], при действии сулемы в водном растворе на
натриевые соли ряда производных индандиона образуются С — Hg-произ-
водные (замещение в положение 2), например:
n'-Ацетобензойная кислота при нагревании с уксуснокислой ртутью до
130°С (15—20 мин.) образует [108] продукт строения
COCHHgOCOCHs
осо-
Введение ртути на место водородного атома (меркурирование)
59
n-Ацетофениларсиновая кислота меркурирована в этих же условиях
в течение 1 часа [108]. Строения продукта не приводится.
О меркурировании n-ацетофениларсиновой и -стибиновой кислот см. при
меркурировании ароматических кетонов.
Об образовании ртутных производных нитробензила и нитробензилиде-
на при кипячении нитротолуолов в.щелочном растворе с окисью ртути см.
при меркурировании ароматических углеводородов.
Меркурирование ацетиленов [27]. Сначала получают щелочной раствор иодной ртути.
Для этого растворяют 66 г (0,486 г-экв) хлорной ртути в растворе 163 г (0,61 г-экв) йодистого
калия в 163 мл воды и прибавляют 125 мл 10%-ного раствора едкого натра (0,31 г-экв).
Затем для получения ацетиленидов ртути (RC^C)2Hg в охлажденный разбавленный раствор
2 экв. щелочной нодной ртути медленно прибавляют по каплям при размешивании мешалкой
раствор 1 экв. замещенного ацетилена в 20 объемах 95%-иого спирта. Тотчас образуется
осадок. Смесь перемешивают 2 — 3 мин., осадок отсасывают, промывают 50%-ным спиртом.
Смесь не следует оставлять долго стоять перед фильтрованием, иначе осадок становится
серым вследствие вторичной реакции, при которой, по-видимому, выделяется металличе-
ская ртуть. Сырой продукт обычно очищают кристаллизацией из спирта или бензола. Мож-
но также растворить его в небольшом количестве бензола или эфира и высадить петролей-
ным эфиром [27].
Т. пл. (RC = C)2Hg: при R = СНз —203—204° С; С2Нв — 162 —163°С [27]; п-СзН7 —
118,8° С; П-С.Н. — 96,4° С; i-C4H9 — 91—92° С [27]; п-С8Нп — 61° С [27]; n-CeHiS —
—80,7° С; п-С7Н1б— 68,5° С; п-С8Н7 — 84° С; n-C9Hi9 — 79°С; СюН21 — 85° С[27]; С9НБСН2 —
— 106,5— 107,5° С [27]; С8НБ — 124,5 — 125°С [27]; р-СНзСеШ — 199—202°С [27];
р-СНзОС6Н4 — 207 — 209°С [27]; p-CaHsCeHi — 142—143°С; р-С1СеН4 — 221 — 222°С;
т-С1СвН4—138—139°С; о-С1С8Н4—212 — 213°С; p-BrCeHt —256—257°С [112]; С1 — 185°С.
Получение RHgC = CHgR [65]. Через прозрачный раствор 8 г RHgX в 800 мл 10%-ного
водного раствора едкого кали (для R—высших алкилов и арилов, лучше брать спиртовый рас-
твор щелочи, так как соответствующие RHgOH мало растворимы в воде [65, б, в]), помещен-
ный в закрытый сосуд, пропускают чистый ацетилен. Раствор тотчас мутнеет, и выпадает
белый осадок. Газ пропускают, время от времени встряхивая сосуд до тех пор, пока рас-
твор больше не будет поглощать ацетилена. Осадок отсасывают, промывают водой и 70%-
ным спиртом и перекристаллизовывают из подходящего растворителя, например ацетона,
бензола, пиридина, метилового и этилового спиртов, четыреххлористого углерода.
Т. пл. R^gC = CHgR: при R= СНз—232оС;СаНБ — 195°С; п-СзН7 — 151°С; QHg —
125°С; СвНн — 91°С; CeHi3 — 104—105°С; х-СзН7 — 110°С; s-C4H9 — 105,5°С; №НЦ—
106°С; С7Н15 —Эб'С; C8Hi7 — Ю8°С; C9Hi8 — 98°С; С1оН21 — Ш°С; СБНБ — 178—
179°С; о-СНзС8Н4 — 205—206° С; р-СНзСеН4 — 190—191°С.
Синтез 2-о-(/тгранс-2'-бромвинил)фенилацетиленида ртути[56]. Раствор 1-траяс-2-бром-
винил-2-этинилбензола (3,96 г) в н-бутиламиие (10 мл) прибавляют к раствору уксуснокис-
лой ртути (3,8 г) в этом же растворителе (15 мл). Через 2 мин. после прекращения выделения
тепла смесь выливают в охлажденную льдом 0,5 N серную кислоту (50 мл) и нейтральную
часть извлекают этилацетатом. Ацетиленид (4,5 а, 78%) кристаллизуется из бензола в бес-
цветные пластинки, т. пл. 187—188°С.
Таким же образом получены ртутные производные фенилацетилена,
окт-1-ина, З-тетрагидропиранилоксипроп-1-ина с выходами 98, 78 и 86%
соответственно.
Получение бас-перхлорвинилртути [63]. В сосуд на 500 мл помещают 500 г (0,2 моля)
Hg(CN)2, 23 г (0,41 моля) КОН и 200 мл воды. К этой смеси прибавляют 80 г (0,61 моля)
трихлорэтилена н реакционную смесь перемешивают при механическом встряхивании.
36 час. Нижний маслянистый слой отделяют, избыток трихлорэтилена удаляют в вакууме.
Белый осадок 30,0 г, т. пл. 70—73°С. Перекристаллизация из пентана дала 28,9 г (86%)
бис-перхлорвинилртути, т. пл. 72—73°С.
60
Ртутноорганические соединения. Лит. стр. 104—114
Получение этаноксигексамеркарбнда [79].
HOHg HgOH
/Hg^c-cZHg.
О< / \ >0
XHg Hg/
10 г металлического натрия растворяют в 150 мл этилового спирта и прибавляют 40 г
тонко измельченной желтой окиси ртути. Смесь умеренно кипятят в круглодонной колбе
с длинным горлом в течение 16 час. на водяной бане; спирт отгоняют и остаток промывают
водой. Серо-желтое вещество освобождают от металлической ртути и избытка окиси ртути
обработкой 8—1,0% -ной азотной кислотой- й получают таким образом нитрат основания в до-
вольно чистом виде. Для освобождения от примеси (небольшие количества коричневатых
смол) его нагревают с 10% -ным раствором едкого натра на водяной бане около 30 мии., про-
мывают и обрабатывают теплой (40° С) 10%-ной азотной кислотой. Таким образом по-
лучается чистый нитрат белого цвета (выход 34 а). Основание получают из него кипячением
с чистым (полученным из металла) едким натром в виде красивого лимонно-желтого, тем-
неющего на свету порошка с высоким удельным весом.
По строению его относят к производным метана, а не этана [81].
Получение тримеркурдиацетондихлррида (2,6-диметил-2,6-дигидрокси-3,5-дихлормер-
кур-1-окса-4-меркурациклогексана) [84].
CIHg—CH—Hg-CH—HgCl
НО(СН3)С — О — С(СН3)ОН(
Полученную из 20 г сулемы сырую окись ртути встряхивают с раствором 6 мл аце-
тона в 100—150 мл воды при постепенном прибавлении баритовой воды до полного раство-
рения. Все высаживают углекислотой, избыток которой удаляют током воздуха. После
отфильтровывания углекислого бария прозрачный раствор выпаривают на водяной бане.
Тримеркурдиацё’гонгидрат остается в виде вязкого сиропа. При-обработке этого соедине-
ния разбавленной соляной кислотой, избытка которой следует избегать, получается три-
меркурдиацетондихлорид. Т. пл. 110°С.
Так же получается бромид, т. пл. 127°С.
Получение тетраацетатмеркурдиацетоигндрата (3,3,3',3'-тетраацетатмеркур-2,2'-днгидр-
оксивторичнобутилоиого эфира) [87].
НО(СН3)С — О —С (CH3)OHj
I I
(H3COCOHg)2CCH3 Н3СС (HgOCOCHsh
25 г сухой уксуснокислой ртути и 50 г ацетона (полученного из бисульфитиого соеди-
нения) нагревают под давлением при 100°С в течение 2 час. После охлаждения и фильтро-
вания испаряют на водяной бане. Остается желтое масло, которое, будучи залито 95% -ным
спиртом, переходит в хлопьевидный осадок. После кристаллизации из спирта т. пл. 157°С.
Получение хлористой фенацилртути [71]. 2 вес. ч. уксуснокислой ртути и 3 вес. ч.
ацетофенона нагревают на масляной бане до 150°С до отсутствия реакции на ион ртути.
Смесь фильтруют горячей и обрабатывают избытком водного раствора NaCl. Осадок промы-
вают эфиром для удаления избытка ацетофенона и перекристаллизовывают нз высококипя-
щего лигроина. Хлористая фенацилртуть получается в виде игл. Т. пл. 145—146°С.
Получение бромистой фенацилртути [107]. Смесь 40 г (0,12 моля) уксуснокислой ртути
и 60 г (0,6 моля) ацетофенона (при перемешивании) быстро нагревают на масляной бане до
150°С. Сначала выпадает желтай окись ртути, которая потом нацело растворяется. Нагре-
вание прозрачного раствора продолжают еще несколько минут до появления мути и пока ре-
акция со щелочью не показала отсутствия неорганической соли ртути. Горячую реакцион-
ную смесь отфильтровывают и выливают в раствор 15 г КВг в 100 мл воды. Продукт реак-
ции — маслянистая жидкость, которая при хранении закристаллизовывается. го-Броммер-
курацетофенон отфильтровывают, промывают петролейным эфиром и дважды кристаллизуют
из спирта. Т. пл. 158—159°С. Выход 25 г (50%).
Получение диацетатмеркурмалоновоэтилового эфира [3]. 20 г слегка влажной уксус-
нокислой ртути растворяют в 120 а воды и встряхивают с 7,5 мл эфира малоновой кислоты.
Жидкость фильтруют, быстро нагревают до 80°С и тотчас охлаждают водой. Получается
кашицеобразная масса, которую отфильтровывают, промывают холодной водой и отсасы-
вают. Выход 11 г. Такой же осадок медленно выделяется при хранении ненагретого раство-
ра [3].
Введение ртути на-место водородного атома {меркурирование)
61
Получение внутренней соли димеркурмалоновой кислоты [3].
-О
+ Hg \о
/с\
+HgZ со
К раствору 1,5 г малоновой кислоты в 8 мл воды приливают 40 мл раствора сернокислой
ртути (содержащего 3 г-атом ртути иа 1 моль малоновой кислоты), слегка нагревают; вы-
деляется белый мелкокристаллический осадок, который отсасывают, промывают водой и
спиртом. Выход 6,5 г.
Получение внутренней соли оксимеркуруксусной кислоты [2].
+Hg—сн2
^>со
°
100 г малоновой кислоты и 120 г едкого натра растворяют в 300—400 мл воды и нагре-
вают до кипения. Затем вносят в раствор небольшими порциями 200 г осажденной окиси
ртути (1 моль). В течение 30 мин. вся окись ртути растворяется, и внутреннюю соль выса-
живают (после разбавления жидкости приблизительно до 1500 мл) прибавлением 950 г
разбавленной 15,9%-ной серной кислоты (около 1,5 моля). Для полного отщепления угле-
кислоты смесь нагревают в течение нескольких часов на водяной бане и промывают затем
теплой водой до исчезновения реакции на серную кислоту. Выход почти теоретический. Ве-
щество на плавится, при нагревании в капилляре чернеет при температуре около 200°С,
разлагается приблизительно при 250°С.
Получение внутренней соли оксимеркурпропионовой кислоты [4]. 1. Получение
меркурацетамида. В расплавленные 60 г ацетамида вносят 80 г желтой окиси рту-
ти. При медленном повышении температуры до 180°С окись ртути растворяется, давая ок-
рашенный в слабо-желтый цвет сплав. Его растворяют в 400 мл воды, фильтруют; фильтрат
испаряют досуха иа водяной бане и перекристаллизовывают из метилового спирта. Выход
около 100 г (80%), т. пл. ~159°С.
2. Получение внутренней соли оксимерку рпропиоио-
вой кислоты. .
СНз—СН-С=О
I I
Hg+ О-
Энергичво встряхивают смесь 5,8 г (1,25 моля) метилового эфира метилмалоновой ки-
слоты и раствора 10 г меркурацетамида (1 моль) в 50 мл воды. К полученной эмульсии при-
бавляют 3 мл 20% -ного раствора соды, нагревают на водяной бане; при этом эфир пере-
ходит в раствор, из которого выделяется немного хлопьевидного белого осадка в том случае,
если взятый метилмалоновый эфир содержал незамещенное соединение. После хранения
в течение 30 мин. раствор фильтруют, охлаждают до —60^-70°С и к нему прибавляют
80 мл (2,5 моля) IN NaOH. Минуты через две разбавляют 200 мл воды и прибавляют 100 мл
1М Нг5О4. При этом выпадает белый хлопьевидный осадок. Его отфильтровывают, суспен-
дируют в воде и кипятят до прекращения выделения СО2,- Отсасывают, промывают, сушат
в вакууМе над серной кислотой. Выход 8 г (93%). Вещество разлагается, ие плавясь.
Меркурирование кетонов азотнокислой ртутью [89]. Общая процедура.
Опыты проводятся в трехгорлой колбе (500 мл), снабженной пропеллерной мешалкой для
высокоскоростного перемешивания, термометром, капельной воронкой, в атмосфере сухого
азота. Меркурируемое соединение является средой, в которой ведут реакцию. Обычно сна-
чала вводят окись ртути и сульфат кальция. После 5—10 мин. энергичного перемешивания
(10 000 об/мин) прибавляют нитрат ртути. Подъем температуры предотвращают охлажде-
нием водой. Подъем температуры наблюдается около часа. После того как исчезнет красный
цвет окиси ртути, температуру поддерживают еще 30 мнн. Охлажденную смесь обычно филь-
труют для удаления неорганических солей. Меркурированный продукт растворим, и его
выделяют или в виде нитрата из сконцентрированного в вакууме фильтрата или чаще посте-
пенным добавлением йодистого калия в виде иодида (избытка йодистого калия, который мо-
жет действовать симметризующе, следует избегать). Полученную смесь перемешивают 15—
30 мии. и фильтруют. Упариванием фильтрата получают ртутноорганический иодид, кото-
рый кристаллизуют из ацетона, спирта, пентана, бензола или их смеси.
62
Ртутноорганические соединения. Лит. стр. 104—114
Меркурирование ацетона. Смесь 396 г (6,83 моля) ацетона, 20 г (0,092 моля) окиси
ртути, 55 г (0,4 моля) сульфата кальция и 20 а (0,092 моля) азотнокислой ртути реагирует
при 56°С. После добавления 25 г йодистого калия получают 55 г иодмеркурацетона в виде
бесцветных пластинок. Его кристаллизуют дважды из ацетона и дважды из бензола.
Выход 78%.. Т. пл. 98—100°С.
Меркурирование метилизопропилкетона. 250 мл кетона, 17 г окиси ртути, 45 г сульфата
кальция и 25 г нитрата ртути реагируют друг с другом при 51°С. Затем добавляют 15 г йо-
дистого калия. Сырой продукт, представляющий собой смесь масла и твердого вещества, вно-
сят в 20 мл ацетона, фильтруют и оставляют при —72°С на 4 дня. Получают 7 г кристалли-
ческого вещества, после кристаллизации из смеси бензола и лигроина т. пл. 72,7—75,5°С.
Получение меркур-бпс-диазоуксусноэтилового эфира [14]. В маленькие колбочки отвеши-
вают по 2 г (1 моль) этилового эфира диазоуксусной кислоты., охлаждают ледяной водой и
постепенно добавляют вычисленное количество (V2 моля) желтой окиси ртути. Первые не-
большие порции окиси ртути в течение 30 мин. полностью растворяются с небольшим разо-
греванием. Выделения газа не происходит. Масса загустевает. Когда вещество начинает
кристаллизоваться, прибавляют немного эфира. К концу реакции иногда происходит сла-
бое выделение азота. Извлекают теплым эфиром, отфильтровывают от невошедшей в реак-
цию окиси ртути и примеси металлической ртути. При быстром испарении эфира на водяной
баней потирании стенок стеклянной палочкой выделяются зеленовато-желтые кристаллы.
Выход около 70% .После кристаллизации из эфира т.пл. 104°C (с разложением). Меркур-бис-
диазоуксуснометиловый эфир получают [14] .аналогичным образом. Он хуже растворим
в Эфире. Т. пл. 123°С (с разложением).
Меркурирование^циклопеитадиена [97]. В охлаждаемую льдом смесь 250 г дигидрата
ацетата натрия, 66 г свежепёрегнанного мономерного циклопентадиена и 500 мл метанола
медленно, при периодическом перемешивании, прибавляют по каплям раствор 272 г сулемы
в 1 л метанола. Через 1—2 дня образовавшийся осадок отсасывают и промывают метанолом.
Получают 270—280 г сырого продукта — смеси хлористой циклопентадиенилртути и ди-
циклопентадиенилртути. Эта смесь разлагается при температуре около 95°С. Быстрым из-
влечением н-октаном Из этой смеси может быть получено бесцветное вещество, по сравнению
с сырым продуктом обогащенное дициклопентадиеиилртутью.(Ср. [122а].)
МЕРКУРИРОВАНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
Реакция меркурирования ароматического ядра открыта Димротом.
Меркурирование ароматических углеводородов происходит с относительной
трудностью — лишь при повышенной температуре, нередко под давлением.
Бейзол меркурируют при многочасовом нагревании до 110° С с уксусно-
кислой ртутью или же в присутствии этилового спирта или ледяной уксус-
ной кислоты на водяной бане, причем образуется почти исключительно одно-
замещенное соединение.
При меркурировании бензола уксуснокислой ртутью в ледяной уксусной,
кислоте в автоклаве при 110° С выход мономеркурированного продукта до-
стигает 92% [113]. Подробное исследование влияния условий реакции
(молярного отношения компонентов — бензола, уксусной кислоты, соли
ртути, длительности и температуры реакции) на выход мономеркурирован-
ного, а также примеси димеркурированного продукта показало [114], что
оптимальными условиями для получения мономеркурированного продукта
(выход 92%) является молярное отношение бензола и соли ртути 30 : U
отношение уксусной кислоты и соли ртути 30 : 1, перемешивание около
3 час. при 110° С. В реакцию можно брать уксусную кислоту, содержащую
до 5 молей воды на 1 моль соли ртути.
Гладко и с хорошим выходом (до 80%) идет меркурирование бензола
действием азотнокислой ртути в присутствии окиси ртути и сульфата каль-
ция и в отсутствии воздуха. Реакцию ведут в атмосфере СО2 во избежание
вторичных реакций, нитрования, взрывов и т. п. [115].
Мономеркурированный бензол, выделенный в виде C6H5HgNO3-CeH5HgOH
[116], является промежуточным продуктом при оксинитровании бензола
в пикриновую кислоту действием азотной кислоты в присутствии азотно-
кислой ртути [116, 1171. Исследование [118] кинетики реакции меркуриро-
Введение ртути на место водородного атома (меркурирование) 6$
вания бензола азотнокислой ртутью в азотной кислоте и ацетатом ртути,
в уксусной кислоте показало, что реакция имеет константу скорости II по-
рядка (и I порядка по отношению к иону ртути и к бензолу) и катали-
зируется кислотами, в частности сильно ускоряется в присутствии НС1О4>
что находится в согласии с предположением об ионном механизме меркури-
рования в этих условиях.
Меркурирующему агенту при меркурировании бензола уксуснокислой
ртутью в. уксусной кислоте в присутствии хлорной кислоты приписывают
электрофильную природу [119].
Некоторые сведения по меркурированию бензола в разных условиях:
приведены в таблице (см. [1201, см. также [121, 122, 123—128]).
Отделение димеркурированного бензола от мономеркурированного см.
[146]. CeDe меркурируется медленнее, чем С6Нв [129].
Меркурирование бензола ртутной солью триннтрометана происходит
в сравнительно мягких условиях (5 час. на кипящей водяной бане) и приво-
дит к мономеркурированному продукту (выход 58,5% [147]). Меркуриро-
вание бензола в водном растворе очень сильно ускоряется прибавлением
нейтральной соли, особенно сильное действие оказывает перхлорат натрия
[148а]. Этот эффект вызван удалением воды от иона ртути. Реакция имеет
II порядок (I по бензолу и соли ртути). К меркурированию бензола дейст-
вием перхлората ртути применено видоизмененное уравнение Гаммета
[148а], Значение р — — 5.
Из димеркурированных производных бензола, при меркурировании
уксуснокислой ртутью, главным изомером является орто-изомер, при мер-
курировании действием перхлората ртути в хлорной кислоте в любых кон-
центрациях последней преобладает мета-изомер, однако в условиях, когда,
пара-изомер нерастворим, его образуется больше, чем мета-изомера [1481.
Это доказывает обратимость реакции меркурирования бензола действием'
перхлората ртути в. хлорной кислоте. Наличие подвижного равновесия
имеет место при образовании всех полимеркурированных продуктов в этих
условиях [1481. При меркурировании бензола наблюдается большой пер-
вичный изотопный эффект водорода [129].
Толуол меркурируется легче бензола при кипячении с уксуснокислой
ртутью [149, 150]. Продукты — все три мономер курированиях изомера,
образующихся в отношениях о : м : п = 43 : 13 : 44 [150, 151], 41:21: 37’
[11 (это отношение определено превращением ТОлилмеркурбромидов дей-
ствием Вг82 в радиоактивные бромтолуолы) и 32 : 15,7 : 51,8 при 90° С (ана-
лиз бромтолуолрв методом ИК-спектроскопии [152]). Ср. [201].
Выход смеси мономер курированных толуолов до 98% при соотношениях:
в продукте о: м: п — 43 : 13 : 44 получен [1511 при введении по каплям обо-
греваемого паром раствора окиси ртути в уксусной кислоте в кипящий то-
луол в течение 30 мин. и последующего кипячения в течение часа. Отноше-
ние компонентов (окиси ртути, уксусной кислоты и толуола) равно соответ-
ственно 1 : 9 : 15.
Более йрко выраженный ориентирующий эффект, свойственный электро-
фильному замещению в толуоле, выявляется при меркурировании его пер-
хлоратом ртути в хлорной кислоте — о:м:п= 19:7:74 (40%-ная НС1ОФ
при 25° С), 27 : 13 : .60 (20%-ная НСЮ4 при 85° С) (анализ бромто-
луолов, по меченому Вг82). Близкие значения процентного распреде-
ления изомеров получены при меркурировании толуола уксуснокислой
ртутью в присутствии хлорной кислоты [152, 1531 (анализ бромтолуолов-
методом ИК-спектроскопии). Скорость меркурирования толуола перхлора-
том ртути, а также ацетатом ртути в присутствии хлорной кислоты (и в-
меньшей степени в присутствии перхлората натрия) значительно выше,
чем ацетатом ртути [152]. Реакция меркурирования толуола и бензола!
Таблица 1
•j г . '< , . . . . «
Меркурирование бензола в разных условиях
п/п ] Меркурирующая соль Среда Отношение бензол/меркури- рующая соль Температура реакции, “С Время реакции, часы Выход моно- меркуриро- ванного про- дукта, % Выход димер- курирован- ного про- дукта, % Литература
1 Уксуснокислая ртуть Ледяная уксусная кислота 7,2 100 5 Не указан — (130]
2 Окись ртути То же 20,6 90—95 2-3 . То же — (131]
3 Уксуснокислая ртуть » » 15,9 100 2 73 ' —« [132]
4 То же » » 3,5 Водяная баня 9 92 [133]
5 Окись ртути » » 3 120 7 24 71 [134]
6 То же » » 13 120 2 78 17 [134]
7 » » Уксусный ангидрид 9,7 120 7 80 - 15 [134]
8 » )> То же 13 120 7 72 23 [134]
9 Уксуснокислая ртуть Ледяная уксусная кис- лота и этанол 19,4 Кипячение 55 80 — [135]
10 То же Нитробензол 9,2 130—135 3 80 — [137]
11 Уксуснокислая ртуть Дихлорбен.зол 8,2 130—135 1 Не указан — [125а, 136]
12 Окись ртути Ледяная уксусная кис- лота и метанол 1,5 70 10 25 —- [138]
13 То же - Ледяная уксусная кислота 17,5 110 3 Не указан —- [139]
14 Уксуснокислая ртуть Ледяная уксусная кис- лота, уксусный ангид- рид, нитрат Cd и Си 4 , Кипячение 6-7 80 — - [140]
15 Нитрат ртути Уксусная кислота (20%- ная) и мочевина 4 75—85 ‘ 3 27 — [141]
16 То же Ледяная уксусная кислота 10 75—85 1—1,5 80,5 — [141]
17 Миристат ртути — 1 120—130 2—3 95—100 J — [142]
18 Пентахлорфеноксимер^ курацетат Не указано 130—135 3 Не указан —. [143]
19 Уксуснокислая ртуть или нитрат ртути HF, BF3 или SbF3 как катализаторы То же Низкая темпе- ратура Непродолжи- тельное Высокий —" [144]
20 Монохлорацетат ртути — 500 80 12 21,9 —* [145]
Введение ртути на место водородного атома {меркурирование)
65
в этих условиях имеет константу скорости II порядка [152] (I порядка по
отношению к каждому углеводороду при мер курировании смеси толуола и
бензола и по отношению к ацетату ртути [117, 118]).
С увеличением продолжительности меркурирования толуола в назван-
ных выше условиях распределение образующихся изомеров приближается
к статистическому (40% орто-, 40% мета-, 20% пара-[152]).
Меркурирование толуола ртутной солью тринитрометана (3 часа при
кипячении) дает смесь мономеркурированных в орто- и пара-положения
продуктов (общий выход 51,5%) [147]. -
Описаны значения изотопного эффекта при меркурировании толуола,
C6H6CD3 и СвН6СТ3 [154]. Действием уксуснокислой ртути в присутствии
перекиси водорода на толуол получен мономеркурированный продукт [1221.
Кипячением толуола с уксуснокислой ртутью в течение 5 час. получено
димеркурированное соединение [155].
При меркурировании толуола пентахлорфеноксимеркурацетатом (130—
135° С, 3 часа) получены о- и n-толилмеркурпентахлорфеноксиды (выходы
не указаны) [143].
f Двенадцатичасовое нагревание толуола (85—87° С), а также ксилолов
(100° С) с избытком уксуснокислой ртути в уксусной кислоте дает полимер-
курированные продукты [156, 157]. м-Ксилол меркурирован [158] ацета-
том ртути в ледяной уксусной кислоте в присутствии небольшого количе-
ства 70 %-ной хлорной кислоты на холоду в положение 4. Замена водорода
ядра на ртуть в молекулах мезитилена [159], псевдокумола [159], дурола
[159, 160], изодурола [159], 1,2,3,4-тетраметилбензола [159], пентаметил-
бензола [159] и п-цимола [161, 162] происходит при длительном кипячении
с уксуснокислой ртутью в спиртовом (полиметилбензолы), спиртово-уксус-
нокислом («-цимол [161] и дурол [160]) или уксуснокислом (п-цимол [162])
растворах. Повышение температуры и продолжительности реакции ведет
к образованию димер кур ированных соединений. Ниже приводятся значе-
ния относительных скоростей меркурирования ряда алкилбензолов, вы-
численных из факторов парциальной скорости [152] и совпадающих
с найденными экспериментально: бензол 1; толуол 5,0; о-ксилол 16,0;
лг-ксилол 34,5; п-ксилол 8,2; гемимеллитол 68; псевдокумол 49; мезитилен
209; пренитол 126; изодурол 257; дурол 30,0; пентаметилбензол 224; этил-
бензол 4,2; изопропилбензол 3,9; третичный бутилбензол 3,2.
Применение уравнения Гаммета к реакциям меркурирования алкилбен-
золов показывает, в частности, что эти реакции зависят от пространст-
венных факторов [163].
Описано меркурирование вторичного бутилбензола в присутствии пере-
киси водорода [1261 (продукт — мономеркурированное соединение).
Меркурирование третичного бутилбензола [119] в ледяной уксусной
кислоте (при 50, 70, 90° С) дает около 70% пара-и 30% мета-изомера; мер-
курированного в орто-положении продукта образуется менее 1%. Бифенил
в этик .же условиях дает 69—75% пара-, 21—23% мета-, 4—7,6% орто-изо-
мера [164].
Кипячение флуорена с уксуснокислой ртутью, в опровержение прежних
данных [165], дает не монозамещенный в положение 4, а димеркурирован-
ный продукт [166]. Этот же продукт, а не смесь двух продуктов, меркури-
рованных в положение 3 и 4 [165], получен [166] при сплавлении флуорена
с уксуснокислой ртутью при 145° С. Однако при изучении факторов пар-
циальной скорости реакции меркурирования флуорена, которое проводили
при взаимодействии избытка флуорена с уксуснокислой ртутью в уксусной
кислоте в присутствии бензола при 25° С [164], главным продуктом реакции
явилось монозамещенное в положение 2 соединение. Во всех опытах продукт
идентифицирован заменой HgX на Вг. Причем скорость меркурирования
66 Ртутноорганические соединения. Лит. стр. 104—114
флуорена значительно выше, чем скорость меркурирования бифенила в тех
же условиях, что может быть объяснено его копланарностью, обуслов-
ленной наличием СН2-мостика [164].
В молекулу нафталина ртуть вступает легче, чем в молекулы бензола и
толуола: уксуснокислая а-нафтилртуть с трудно отделимой примесью бо-
лее высокомеркурированных соединений получена при 20-минутном нагре-
вании до 120° С нафталина и уксуснокислой ртути [1681.
Для введения одного атома ртути в молекулы других углеводородов
с конденсированными ядрами (антрацен, фенантрен, аценафтен) необходимо
применение в течение 4—8 час. [1691 (или более [170]) высокой температуры
(аценафтен 115—120° С, антрацен 130—140° С, фенантрен 120Q С) при по-
вышенном давлении [1691.
Меркурирование аценафтена нагреванием при указанной выше темпе-
ратуре в течение 15 час. с уксуснокислой ртутью в уксусной кислоте в ав-
токлаве дает продукт, замещенный в положение 5 (выход 28%) [1701:
н2с—сн2 . Н2С—сн2
I J 1.1
сн3соон
HgO2CCH3
Меркурирование фенантрена в названных для него выше условиях ук-
суснокислой ртутью в уксусной кислоте (в исправление прежних данных
[169]) дало три мономеркурированных в положения 1, 3 и 9 продукта (об-
щий выход 25%) и два дизамещенных продукта (общий выход 25%) [170].
При меркурировании уксуснокислой ртутью в нитробензоле в присутствии
хлорной кислоты в качестве катализатора (40° С, 3 часа) получены эти же
продукты наряду с, возможно, полимеркурированными соединениями [170].
При меркурировании фенантрена в уксусной кислоте раствором окиси
ртути в хлорной кислоте (60° С, 6 час. или 30° С, 4. часа) получены моно-
и димеркурированные продукты. В отсутствие уксусной кислоты при ком-
натной температуре (150 час.) реакция не идет [170].
При нагревании до 130° С в течение часа 5-кратного (в молях) количества
стильбена с уксуснокислой ртутью в ледяной уксусной кислоте не проис-
ходит присоединения соли ртути по двойной связи, а образуется три моно-
меркурированных в положения 2, 3 и 4 продукта и немного 4,4'-димерку -
рированного соединения [171].
О присоединении соли ртути по этиленовой связи стильбена см. следую-
щую главу.
Полистирол меркурирован 10-часовым кипячением с ацетатом ртути
в уксусной кислоте [172]. Меркурированный полистирол, содержащий
1 атом ртути на одно звено полистирола, получен с почти количественным
выходом при нагревании на водяной бане в течение 20 час. нитробензольных
растворов полистирола и диизобутирата ртути [173]. Полистиролы мерку-
рированы также нагреванием при 130° С в течение около 2 час. с уксусно-
кислой ртутью в уксусной кислоте, а также в присутствии НС1О3. В зави-
симости от типа полистирола и от условий меркурирования полученные
продукты имеют различное содержание ртути [173а].
Нагревание на водяной бане в течение 2 час. в уксусной. кислоте дифе-
нилена и уксуснокислой ртути дает 2-ацетоксимеркурдифенилен [1741.
При кипячении 3,4-бензопирена с избытком уксуснокислой ртути в ук-
сусной кислоте получено димеркурированное производное [175].
Из небензоидных ароматических углеводородов промеркурирован азу-
Введение ртути на место водородного атома (меркурирование) 67
лед 11761, который в реакции меркурировании оказался более реакцион-
носпособным, чем тиофен и фуран и столь же реакционноспособен, как
Пиррол. Уже при действии сулемы в присутствии ацетата натрия в спирто-
вом растворе при комнатной температуре он мгновенно образует димерку-
рированное соединение состава C10HeCl2Hgs. Хотя положение ртути в нем
не определено, но ртуть, очевидно, находится в 5-членном кольце. Соеди-
нение это имеет сначала коричневую окраску, постепенно переходящую
в сине-серую.
В таких же условиях, как и соответствующие углеводороды, меркури-
руют уксуснокислой ртутью их галоидозамещенные.
При 12-часовом кипячении эквимолекулярных количеств фторбензола
и уксуснокислой ртути в ледяной уксусной кислоте выделен лишь мерку-
рированный в орто-положение продукт с выходом 11% [177].
При 2-часовом кипячении получен ацетат n-фторфенилртути [178].
Однако при меркурировании избытка фторбензола уксуснокислой ртутью
при 90° С образуется 59,4% пара-, 6,9% мета- и 33,7% орто-изомеров (раз-
деление газовой хроматографией смеси бромфторбензолов, полученных
из продукта меркурировании) [182]. С понижением температуры выход
орто- и мета-изомеров уменьшается, а пара-изомера увеличивается. Расчет
факторов парциальной скорости указывает на некоторую активацию ато-
мом фтора пара-положения, сильную дезактивацию мета- и небольшую
дезактивацию орто-положения [1821.
При сплавлении хлорбензола с лауринатом ртути (130—140° С, 2 часа)
выход мономеркурированного продукта достигает 95—100% [142]. При
меркурировании избытка хлорбензола уксуснокислой ртутью при 90® С
получено 30% орто-, 21,5% мета-, 48% пара-изомеров [182]. Главным про-
дуктом меркурирования хлорбензола при кипячении с эквимолекулярным
количеством уксуснокислой ртути (140° С; 2,5 часа) является пара-изомер
[172,179] (выход 40 % [179]) (см. также [133,180]). Из продуктов меркуриро-
вания в этих условиях бромбензола выделены пара-изомер (выход 25%),
орто-и мета-изомеры (выходы не указаны) [1791. При проведении реакции
в запаянной трубке (120° С) получен димеркурированный в орто* и пара-
положения продукт [181]. В условиях меркурирования фтор- и .хлор-
бензолов при 90° С бромбензол дает 28% орто-, 26% мета-, 46% пара-
изомеров [182]
При меркурировании хлор/ и бромбензолов дезактивированы все поло-
жения и в особенности мета-и орто-. Для пара-положения порядок реак-
ционной способности: F Н С1 Вг, для мета-положения: Н > С1
~ Вт > F. Для меркурирования монозамещенных производных бензола
(в том числе галоидопроизводных) выполняется четкая линейная коррелят
ция между 1g и <т+ константой электрофильного замещения [1821
(в этих случаях для меркурирования значение р = — 4,0).
Иодбензол при нагревании с уксуснокислой ртутью (140° С, 1,5 часа)
дал только 4% пара-изомера [179]. Остальные продукты не идентифициро-
ваны.
Кипячение п- дихлорбензол а с уксуснокислой ртутью без растворителя
или в уксуснокислом растворе дает смесь мономеркурированного, всех
трех димеркурированных, три-и тетрамеркурированных продуктов [183].
Согласно данным Петровича [184], в уксуснокислом растворе при 125° С
получается мономеркурированный продукт и, при более продолжительном
нагревании, неожиданно получен (2,5-Cl2C6H3)2Hg. Мономеркурирован-
ное соединение может быть получено только в последних условиях при про-
ведении реакции с эквимолярным количеством уксуснокислой ртути и п-
дихлорбензола не до конца или при продолжительном кипячении трехкрат-
ного избытка n-дихлорбензола с ацетатом ртути [183].
68
Ртутноорганические соединения. Лит. стр. 104—114
Трихлорбензол при 5-часовом кипячении с сулемой в изоамиловом спир-
те дает мономеркурированный продукт [185].
гг-Дибромбензол меркурирован нагреванием с ацетатом ртути в запаян-
ной трубке до 120° С в течение 4 час. (образуется димеркурированное в по-
ложения 2 и 5 соединение) [1811.
Р-Бромнафталин при сплавлении с ацетатом ртути дал а-ацетатмеркур-
р-бромнафталин [1861.
В молекулы нитропроизводных углеводородов ртуть вступает с боль-
шим трудом, чем в молекулы самих углеводородов. Нитробензол меркури-
руют при нагревании с уксуснокислой ртутью без растворителя до 150° С
[169]; при этом образуется главным образом орто-изомер, небольшое коли-
чество пара-меркурированного продукта и до 40% мета-производного
[1,187—191]. Как показали КлаппротиУзстхеймер [1], такое неупорядочен-
ное распределение изомеров имеет место при меркурировании уксуснокис-
лой ртутью, при меркурировании же перхлоратом ртути нитробензола в
60%-ной хлорной кислоте (комнатная температура, 8—10 дней) образуется
89% мета-изомера и 11% смеси орто-и пара-изомеров, в40%-ной хлорной
кислоте (95° С, 11 час.)—63% мета-, 37% смеси орто-и пара-изомеров [1]
(см. стр. 51).
Огата и Тсусида [192, 192а] при меркурировании нитробензола
окисью ртути в азотной кислоте (99° С; 7 час.) и уксуснокислой ртутью
(155° С; 2,5 часа) не обнаружили принципиальной разницы в процентном
составе образующихся изомеров. Они нашли, что в первом случае образует-
ся изомеров: 37% орто-, 56,5% мета-, 6,5% пара-; вО втором случае — 27%
орто-, 67% мета-, 5,5% пара-.
Нитротолуолы (орто- и пара-) меркурируются в зависимости от усло-
вий в ядро или в боковую цепь: при кипячении в щелочном растворе с окисью
ртути получаются производные нитробензила [193] и нитробензилидена
[193, 195], при нагревании же до 140—150° С нитротолуолов с уксуснокислой
ртутью без растворителя образуются меркурированные в ядро соединения
[ 194,196,197].Ртутная соль тринитрометана не меркурирует нитросоединения,
с некоторыми из них образуя лишь комплексы состава NO2CeH5Hg [C(NO2)3]2
[198].
Меркурирование бензола [149, 199]. При нагревании свободного от тиофена бензола
в течение многих часов с сухой уксуснокислой ртутью при 110 — 120° С образуется уксусно-
кислая фенилртуть (т. пл. 1493 С) и нерастворимый в бензоле о-диацетатмеркурбензол
CeHjlHgO^CGHs)».
Получение уксуснокислой феннлртути в присутствии этилового спирта [135]. Нагре-
вают смесь 80 мл бензола, 15 а уксуснокислой ртути и 20 мл 95%-ного этилового спирта
на водяной бане с обратным холодильником. Тотчас выпадающий желтый осадок переводят
в раствор прибавлением нескольких миллилитров ледяной уксусной кислоты. Через 5 час.
прибавляют еще 20 мл спирта. Через 55 час. выделяется около 1 г закисной уксуснокислой
ртути, которую отфильтровывают. Фильтрат испаряют досуха на водяной бане и осадок кри-
сталлизуют из 95%-ного спирта. Т. пл. 149°С. Выход 12,6 г (80%).
.. Меркурирование бензола в ледяной уксусной кислоте при 100° С [130]. Раствор 50 г
уксуснокислой ртути в 50 мл ледяной уксусной кислоты в толстостенной склянке смешивают
с 100 мл свободного от тиофена бензола. При этом уксуснокислая ртуть опять выпадает.
Закрытую склянку в течение 5 час. нагревают на кипящей водяной бане. После охлаждения
отфильтровывают от нерастворившегося осадка, многократно промывают бензолом и после
испарения фильтрата получают уксуснокислую фенилртуть. Если хотят получить хлорид,
то после отгонки большей части бензола фильтрат обрабатывают спиртовым раствором
хлористого кальция, осадок отфильтровывают, сушат и промывают горячей водой. Хло-
ристая фенилртуть кристаллизуется из спирта. Т. пл. 252° С*.
Получение тринитрометилфенилртути [147]. 1. Получение ртутной соли
три нитрометан а. К 16 г (0;07 моля) свежеприготовленной окиси ртути в 50 мл
эфира (ртутная соль тринитрометана может быть получена с количественным выходом
в водном нли спиртовом растворах) прибавляют при комнатной температуре небольшими
порциями эфирный раствор (1 : 1 по объему), 20 г (0,13 моля) тринитрометана, что сопро-
* Т. пл. хорошо очищенной CjHjHgCl равна 271° С (ср. [200]).
Введение ртути на место водородного атома (меркурирование)
69
вождается повышением температуры до 30°С. Реакционную смесь перемешивают в течений
15 мин. и отфильтровывают эфирный раствор от невошедшей в реакцию окиси ртути. Эфир
испаряют. Ртутная соль тринитрометана выделяется в виде густого светло-желтого масла,
которое закристаллизовывается в течение 5—6 час. Кристаллы отделяют от масла на пори-
стом фильтре; температура разложения 200—205°С; выход 26,5 г (80%). Ртутная соль
тринитрометана хорошо растворяется в воде, спирте, хлороформе, ацетоне, бензоле,
эфире, этилацетате, уксусной кислоте, не растворяется в петролейном эфире, гексане,
изооктане.
2. Получение тринитрометилфенилртути. 5 г ртутной соли
трннитрометана растворяют в 20 мл бензола и 3 мл эфира (эфир берут для лучшей раствори-
мости ртутной соли тринитрометана). Раствор отфильтровывают от незначительного коли-
чества (50—100 мл} неорганических примесей (продуктов разложения ртутной соли) и на-
гревают в течение 5 час. на водяной бане (температура бани 85—90°С). После окончания
нагревания горячий раствор фильтруют; из фильтрата при охлаждении выделяются бес-
цветные кристаллы тринитрометилфенилртути, их отфильтровывают и промывают несколь-
ко раз водой. Из маточника при отгонке бензола при 250 мм до остатка 5—10 мл удается вы-
делить еще 0,5 г тринитрометилфенилртути. Общий выход 2,5 г (58,5%). После перекристал-
лизации нх четыреххлористого углерода получено 2 г тринитрометилфенилртути с т. пл.
Меркурирование толуола [149]. 1 вес. ч. уксуснокислой ртути и 5 вес. ч. толуола быстро
реагируют друг с другом при кипячении с обратным холодильником. Все полностью пере-
ходит в раствор. Затем охлаждают, при этом выделяется немного труднорастворимого соеди-
нения. Фильтрат высаживают хлористым натрием и не вошедший в реакцию толуол отгоняют
с водяным паром. В остатке — мягковатая масса, затвердевающая при охлаждении. Дроб-
ной кристаллизацией из бензола она с трудом может быть разделена на два соединения.
Более труднорастворимое соединение получается в виде прекрасных кристаллов с т. пл.
230—231°С повторной кристаллизацией из хлороформа или ацетона и представляет собой
хлористую п-толилртуть.
Вещество все же содержит примеси изомеров [201]. Т. пл. чистой хлористой п-толил-
ртути 232—233°С. Остающаяся в бензольном маточнике хлористая о-толилртуть повторной
кристаллизацией из спирта не может быть полностью отделена от изомеров. Т. пл- 140—
142°С вместо 145—146’ С [149]. По данным Коффи [150], прн меркурировании толуола (5—
6-часовое кипячение с уксуснокислой ртутью) получается и мета-меркурированное соеди-
нение. Количество образующихся орто-, мета-, пара-меркурированных соединений отно-
сится, как 43 : 13 : 44.
Получение уксуснокислой 2,4-диметилфенилртути [158]. 32 г (0,1 мол») уксуснокислой
ртути и 3 мл 70%-ной хлорной кислоты прибавляют при перемешивании к раствору 15 а
(0,15 моля) л-ксилола в 200 мл ледяной уксусной кислоты. Раствор перемешивают до начала
образования осадка. Выливают в двойной объем воды и осадок отфильтровывают. Кристал-
лизация из водного спирта, т. пл. 129—130,8°С.
Хлорид плавится при 155—156°С.
Меркурирование нафталина [168]. В 60 г расплавленного нафталина вносят 30 г уксус-
нокислой ртути, и смесь, хорошо перемешивая и разбивая образующиеся комья, нагревают
на масляной бане при 120’Сдо тех пор, пока она не станет прозрачной (~ 20 мни.). Отгоняют
избыток нафталина с водяным паром. Остаток сушат, извлекают лигроином, т. кип. 100—
120’С. Выкристаллизовавшаяся уксуснокислая а-нафтилртуть очищается многократной
повторной кристаллизацией из лигроина и спирта. Т. пл. 154°С. Меркурированный в а-
положение нафталин легче изолировать в виде хлорида. Для этого реакционную смесь перед
перегонкой с паром обрабатывают раствором хлористого натрня, отгоняют нафталин, сушат
остаток, экстрагируют эфиром для окончательного освобождения от нафталина и кристал-
лизуют из двадцатикратного количества амилового спирта. Кипящий раствор (медленно, на
водяной бане) охлаждают до 80°С, выделяющиеся при этом в виде порошка полимеркури-
рованные соединения быстро отсасывают. Остаток, выделившийся при полном охлаждении
фильтрата, извлекают холодным хлороформом н кристаллизуют из бензола. Т. пл. хлори-
стой а-нафтилртути 189°С.
Получение 2-ацетоксимеркурдифенилеиа [174].
/\-/\-н§о2ссн3
0,3 г дифенилена и 0,65 г уксуснокислой ртути в 10 мл уксусной кислоты нагревают на
водяной бане в течение 2 час. Полученный желтый раствор разбавляют водой, не вошедший
в реакцию дифенилен (0,212 а) удаляют быстрой перегонкой с паром.Нелетучие вещества со-
бирают при 0Х н извлекают в экстракторе Сокслета теплым бензолом. Получают 0,187 а
(79%) 2-ацетоксимеркурдифенилена. Кристаллизация нз бензола. Желтый порошок, т. пл.
176—177Х.
70
Ртутноэрганические соединения. Лит. crtip. 104—114
Меркурирование антрацена [169]. 10 г уксуснокислой ртути и 5 г антрацена в 45 мл
уксусной кислоты нагревают в автоклаве до 130—140°С в течение 7—8 час. В течение часа
весь антрацен растворяется с темно-красным окрашиванием. По охлаждении все застывает.
Застывшую массу нагревают с дополнительной порцией уксусной кислоты на водяной бане,
пока большая часть не растворится. Раствор отфильтровывают горячим, фильтрат упари-
вают иа одну треть и тотчас приливают спиртовый раствор хлористого кальция. Тотчас
образуется осадок. Его промывают водой, спиртом и эфиром и сушат в вакуум-эксикаторе.
Выход серовато-желтой хлористой антранилртути 30%, т. пл. 181—183°С. Вещество пло-
хо растворимо в бензоле и нерастворимо в большинстве органических растворителей.
Получение 2-ацетоксимеркур-1,4-дихлорбензола [183]. 6,5 г (3 моля) уксуснокислой рту-
ти и 10 г (10 молей) n-дихлорбензола в 25 г уксусной кислоты кипятят 5 час. По охлаждении
выливают в воду. Выпавший осадок 2-ацетатмеркур-1,4-дихлорбензола весит 2,6 г, после
кристаллизации из спирта т. пл. 175°С. При более продолжительном нагревании (до 11 час.)
вещество симметризуется и при кипячении в течение 13 час. главным продуктом реакции
является бис-(2,5-дихлорфенил)ртуть.
Меркурирование нитробензола [168]. Ацетат ртути (1 вес. ч.) нагревают на масляной бане
до 150°С с 5 вес. ч. нитробензола до тех пор, пока все не перейдет в раствор и от прибавления
разбавленного раствора едкого натра не будет больше выделяться желтая окись ртути.
При охлаждении выделяется немного (0,6% от количества взятого ацетата ртути) вещества,
кристаллизующегося в блестящих листочках. Фильтрат от этого побочного продукта вы-
саживают раствором хлористого натрия и избыток нитробензола отгоняют с водяным паром.
Остается полутвердая, при охлаждении затвердевающая масса. Ее измельчают в порошок
и повторно извлекают кипящим лигроином (т. кип. 100—120°С), при этом почти все раство-
ряется. При охлаждении выделяются слабо-желтые иглы хлористой о-нитрофенилртути.
После кристаллизации из ледяной уксусной кислоты соединение получают чистым. Малень-
кие желтоватые листочки.'Т. пл. 181—182°С.
Меркурирование нитробензола хлорнокислой ртутью [1]. В 250 мл 60%-ной хлорной
кислоты, помещенные в колбу (500 мл), вносят 5,42 г окиси ртути и 10 мл нитробензола. Ре-
акционную смесь встряхивают через каждые несколько часов и оставляют стоять при ком-
натной температуре (23°С) на 8—10 дней. Затем ее выливают в двойной объем ледяной воды
и водным раствором хлористого натрия высаживают смесь нитрофенилмеркурхлоридов. Оса-
док промывают водой, 50%-ным спиртом, эфиром, петролейным эфиром и сушат. Вес 7,9—
8,7 г. Плавится в пределах 215—221°С. Полученную смесь нитрофенилмеркурхлоридов в
растворе хлороформа встряхивают в течение многих часов с раствором брома в бромистом
калии. Извлекают хлороформом, хлороформенные вытяжки сушат, хлороформ испаряют.
В остатке определяют процентное содержание лг-бромнитробензола и суммы о- и п-
броминтробензолов на основании образования только последними аммониевых соединений
с аминами; для этого аликвотную часть остатка нагревают много часов с пиперидином, вы-
ливают воду н водный слой, промытый бензолом, титруют по Фольгарду, что дает содержа-
ние бромионов из орто- и пара-изомеров. Так находят выход 11% суммы орто- и пара-изо-
меров, мета-изомера — по разности 89% (см. также [289]).
Меркурирование о-иитротолуола в щелочном растворе. Получение хлористой о-иитро-
бензилртути и о-нитробеизальдимеркуроксида [193]. Смесь полученной из 297 г сулемы вы-
саживанием едким натром и хорошо промытой окиси ртути (2 моля) и 2,5 л воды, содержа-
щей 44 г (2 моля) NaOH, нагревают иа масляной бане до кипения в колбе, снабженной об-
ратным холодильником и трубкой для ввода воздуха. Последняя применяется для переме-
шивания реакционной смеси; вместо этого может быть применена мешалка. При пропуска-
нии сильного тока воздуха в течение 9 час. небольшими порциями прибавляют 75 г о-нитро-
толуола (1 моль). Кипятят еще 1,5 часа и по охлаждении отделяют осадок от желтовато-
красной жидкости. Продукт желтого цвета с серовато-зеленым оттенком,вызванным примесью
небольшого количества металлической ртути. Вес сухого о-нитробензальдимеркуроксида
291 г, выход 96,4%.
Из маточника избытком соляной кислоты высаживают 5,2 г сырой хлористой о-нитро-
бензилртути. Последнюю очищают, обрабатывая спиртовой щелочью на холоде, разбавляя
водой и высаживая профильтрованный раствор соляной кислоты. Выделившийся хлорид
растворяют при слабом нагревании в большом количестве разбавленного аммиака, раствор
фильтруют при охлаждении, нагревают до кипения и в кипящий раствор приливают соля-
ную кислоту. Этот процесс повторяют несколько раз. Хлористая о-нитробензилртуть по-
лучается в виде бесцветных соединенных в пучки иголок, т. пл. 145—146°С.
Баур с сотр. [202] описал соединения состава MeHg2(SCN)6.-ArH,
где Ar Н— бензол, толуол, ксилол, нафталин.
Введение ртути на место водородного атома (меркурирование')
71
МЕРКУРИРОВАНИЕ АРОМАТИЧЕСКИХ ОКСИСОЕДИНЕНИЙ
Фенолы являются одними из наиболее легко мер курирующихся соеди-
нений. Одноатомные фенолы обычно меркурируются при нагревании (на
кипящей водяной бане) в водном растворе уксуснокислой ртутью или
окисью ртути, нередко в присутствии щелочей. Сам фенол дает при этом
два мономеркурированных в орто- и пара-положения продукта и 2,4-ди-
меркурированное соединение [71, 149, 199, 2031. Не образуется димеркури-
рованного соединения при сплавлении фенола с уксуснокислой ртутью без
растворителя. При сплавлении при 100°С главный . продукт— пара-
изомер [204], при 170° С — орто-изомер [205]. Второй атом ртути может
быть также введен в мономеркурированные фенолы в присутствии щелочи
и цианистого натрия действием сулемы [206]. При вливании избытка
расплавленного фенола в кипящий раствор окиси ртути в уксусной кислоте
образуется мономеркурированное в орто-положение соединение [155].
Тримеркурированный фенол 2,4,6-(CH3COOHg)3C6H2OH-3CH3COOH
с т. пл. 116—120° С (34,5 г) получен при 5-минутном кипячении 65 г HgO,
10 г фенола и 200 мл ледяной уксусной кислоты [155], в аналогичных усло-
виях получены 2,4,6-(XHg)3CeH2OH, с X=NO3 или CN [155].
Скорость меркурирования фенола уксуснокислой ртутью возрастает при
добавлении азотной и,’ в большей степени, хлорной кислоты: добавление
уксусной кислоты уменьшает скорость меркурирования [207]. Реакция
меркурирования фенола в присутствии кислот имеет константу скорости
реакции II порядка [207].
Для меркурирования фенолов с успехом применимы, кроме уксуснокис-
лой ртути и окиси ртути, и другие меркурирующие агенты: меркурацет-
амид, оксимеркурцианид [208] (меркурирование n-крезола и м-ксиленола),
сулема в присутствии бикарбоната натрия и глицерина. Этой смесью мер-
курированыфенол [209], а-, и Р-нафтол, тимол, карвакрол, n-нитрофенол с об-
разованием мономеркурированных соединений. Однако при меркуриро-
вании при помощи этой меркурирующей смеси во многих случаях обра-
зуются соединения с О — Hg-связью.
В таких же условиях, как и фенол, нагреванием'водных или водно-спир-
товых растворов с уксуснокислой ртутью или окисью ртути или же сплавле-
нием с солью ртути без растворителя меркурируют хлорфенолы [182, 210,
211, 253], n-фторфенол (даже на холоду) [178]) и нитрофенолы [182, 210—
215,238]. На примере меркурированияо-нитрофенола видно влияние раствори-
теля на строение образующихся продуктов: в то время как сплавление
о-нитрофенола с уксуснокислой ртутью дает 6-ацетатмеркур-2-нитрофенол,
при проведении реакции в водном растворе получается главным образом
4-ацетатмеркур-2-нитрофенол [214]. В таких же условиях, как фенол, мер-
курируются динитрофенолы [216] и гомологи фенола (крезолы [9,71, 122,181$
208, 210, 217—220, 223], ксиленолы [208], тимол [9, 71, 224, 225], карвакрол
[225], rt-третичнобутилфенол [226], изоамилфенол [226], о- и п-циклогексил-
фенолы 4227], о- и п-бензилфенолы [227], нафтолы).
Из а-нафтола получают, в зависимости от относительного количества
реагентов, димеркурированное в 2 и 4 положения соединение или мономер-
курированный в положение 4 продукт[186,228](при меркурировании сулемой
в присутствии бикарбоната натрия и глицерина [229] получается только
мономеркурированное соединение). Из ^-нафтола действием уксуснокислой
ртути в уксусной кислоте на холоду [230] получают мономеркурированный
в a-положение продукт [228, 230, 231 ]. При меркурировании сулемой в при-
сутствии бикарбоната натрия и глицерина, возможно, в другое положение
[229]. О получении последнего соединения из 2-окси-1-нафтойной кислоты
см. гл. IX.
72 Ртутноорганические соединения. Лит. стр. 104—114
При хранении на холоду водно-спиртовых растворов ацетата ртути
(0,5 моля) и следующих алкилфенолов (1 моль): 1,1-бис-(4'-окси-6-метилфе-
нил)циклогексана, 1,1-бпс-(4'-оксифенил)циклогексана, 4-тетраметилбу-
тилфенола получены их мономеркурированные производные [232]. Димер-
курированные в каждом фенольном кольце производные последних трех
алкилфенолов [232], а также 2,2,5,5-тетра-(4'-оксифенил)гексана [232,
233] получены при кипячении спиртово-уксуснокислых растворов этих фе-
нолов и уксуснокислой ртути (берут 1 моль ацетата ртути на каждое сво-
бодное орто-положение фенола).
Аналогично фенолу или в более жестких условиях меркурируют галоид-
алкилфенолы [234—236], в частности хлоркрезолы [237], галоидксиле-
нолы [208, 222], хлор- [239] и иод- [240] тимол, галоидпроизводные арилфе-
нолов [241] (хлорзамещенный о-оксидифенил нагреванием в течение 2 час.
при 140°С [241] и нитроалкилфенолы [236] (нитрокрезолы [242—245],
нитрозо- и галоиднитрозотимол [239], нитро-[186, 214, 246] и динитро-
[246] нафтолы).
З-Нитро-4-оксидифенил при кипячении в водно-спиртовой среде с ацета-
том ртути дает мономеркурированный продукт с почти количественным
выходом [247].
О действии пол незамещенных ртутноорганических соединений и ртутно-
органических солей на фенол и замещенные фенолы, что, по мнению авто-
ров соответствующих работ, приводит к их меркурированию, см. гл. XII.
Меркурирование фенола в водном растворе [149, 199]. При смешении концентрирован-
ных водных растворов фенола и уксуснокислой ртути прн комнатной температуре медленно
(при нагревании на водяной бане очень быстро) выделяется значительное количество белых,
соединяющихся в шаровидные агрегаты кристаллических. игл окенфенилдимеркурацетата
НОСвНз(НёОСОСН8)2 (т. пл.216—217°С). В маточнике находятся два других соединения, луч-
ше всего изолируемых в виде хлоридов следующим образом: горячий раствор высаживается
хлористым натрием, образовавшийся осадок тотчас отфильтровывают. Он состоит в основном
из хлористой п-оксифенилртути p-HOCeHiHgCl, соответствующее орто-соединение полу-
чается из маточного раствора в виде прекрасных ланцетовидных кристаллов. Пара-соеди-
нение может быть очищено повторной кристаллизацией из ацетона (т. пл. 224—225° С),
орто-соедииение — кристаллизацией из спирта (т, пл. 152,5° С).
Меркурирование фенола без растворителя [204]. 12 г фенола расплавляют нагреванием
на водяной бане и при перемешивании постепенно прибавляют 25 г уксуснокислой ртути.
После того как вся уксуснокислая ртуть растворилась, приливают кипящую воду и кипя-
тят несколько минут. Затем прибавляют горячий водный раствор 5 а NaCl. Тотчас выпадает
хлористая я-оксифеннлртуть. Смесь фильтруют горячей. Раствор по охлаждении выделя-
ет кристаллы хлористой о-окенфенилртути. Выход пара-изомера 18 г, орто-изомера 7 г.
При проведении реакции при 170° С главным продуктом реакции яв-
ляется орто-изомер (см. [2051).
Димеркурированный фенол получен сплавлением фенола с ацетатом
ртути и выделением продукта в виде оксалата; последний получен также
10-часовым кипячением фенола, щавелевой кислоты и окиси ртути в воде
[248].
Получение внутреннего фенолята о-оксимеркурфенола [149]. При введении углекис-
лоты в щелочной раствор хлористой о-оксифенилртути выпадает белый, трудно раствори-
мый в большинстве органических растворителей, растворимый в феноле порошок. Строение
о-
o-CeHj
'нё+
Меркурирование п-крезола [71]. К раствору 21,6 г окиси ртути в 500 мл воды и 18 г
ледяной уксусной кислоты прибавляют раствор 10,8 г n-крезола в небольшом количест-
ве спирта и нагревают на водяной бане. На холоду реакция требует для своего завершения
(отрицательная проба со щёлочью на ион ртути) около 2 дней, при 90°С —30 мин. При про-
ведении реакции при нагревании на водяной бане образуется только днмеркурированное
Введение ртути на место водородного атома (меркурирование}
73
соединение. Отделяют выделившийся в почти чистом виде 3,5-диацетатмеркур-п-крезол (со-
держит 1 молекулу кристаллизационной воды). Он разлагается прн температуре около 200°С
с красным окрашиванием. Горячий маточник обрабатывают водным раствором хлористого
натрия. Выпавший осадок извлекают холодным спиртом. Спиртовый раствор концентрируют,
высаживают водой. Образующийся осадок 2-хлормеркур-п-крезола многократно кристал-
лизуют нз бензола. Т. пл. 166°С.
Получение уксуснокислой 2-окси-1-нафтилртутн [230]. Растворяют 20 г окиси ртути
в 520 мл горячей ледяной уксусной кислоты, по охлаждении раствор отфильтровывают от
небольшого количества нерастворимых блестящих листочков и смешивают с раствором 13,2 а
(3-нафтол а в уксусной кислоте; почти тотчас выделяется тяжелый кристаллический осадок
Р-оксинафтнлмеркурацетата. Количество его при многочасовом хранении увеличивается.
Выход количественный. После однократной кристаллизации из ледяной уксусной кислоты
плавится прн быстром нагревании прн 185°С (с разложением).
Меркурирование Р-нафтола действием сулемы и бикарбоната натрия в присутствии
глицерина [229]. Получение хлористой 2-окси-1-нафтилртути. К раствору [3-нафтола (4,3 г)
в 50%-ном спирте прибавляют 15 мл глицерина н затем раствор 8,1 г сулемы в 50%-ном
спирте до прекращения выделения осадка. К смеси прибавляют раствор 2,5 г бикарбоната
натрия в водном спирте и все энергично перемешивают; при этом появляется желтовато-
белый осадок, происходит обильное выделение СОг. Через несколько часов осадок отфиль-
тровывают н промывают очень разбавленным раствором НС1 и водой. Кристаллизация из
спирта. Т. пл. 180°С (с разложением).
Меркурирование о-нитрофенола [213]. Раствор 22,3 г уксуснокислой ртути в 200 мл
воды и 1 мл ледяной уксусной кислоты прибавляют по каплям к теплому раствору 10 г
о-нитрофенола в 200 мл воды н 10 мл 40%-ного раствора едкого натра. Тотчас выпадает
оранжевый осадок. После'перемешивания в течение 2 час. ставший желтым осадок отфиль-
тровывают и тщательно промывают водой, содержащей небольшое количество уксусной
кислоты. Сырой продукт растворяют по возможности полностью в 5% -ном растворе едкого,
натра, фильтруют и вновь высаживают уксусной кислотой. Осадок отфильтровывают, про-
мывают водой, метиловым спиртом н эфиром до удаления последних следов нитрофенола.
Выход 16,5 г. Осадок представляет собой смесь моно- и днмеркурированных продуктов-
(главным образом 4-ацетатмеркур-, но также и б-ацетатмеркур-2-ннтрофенола и 4,6-диаце-
татмеркур-2-нитрофенола). Четырехкратной кристаллизацией нз ледяной уксусной кисло-
ты можно получить продукт, представляющий собой в основном мономеркурированное со-
единение.
Растворением последнего в разбавленной щелочи, отфильтровыванием от нерастворнв-
шегося осадка и высаживанием фильтрата соляной кислотой получают осадок, который про-
мывают водой от неорганических галогенидов и несколько раз извлекают кипящим метило-
вым спиртом. Нерастворимый осадок представляет собой смесь моно- и днмеркурированных
продуктов. Из метиловоспиртового фильтрата выкристаллизовывается 4-хлормеркур-2-
нитрофенол в виде кремово-желтых гроздьев, т. пл. 205’С [214], 6-хлормеркур-2-нитрофе-
нол - желтовато-коричневые пластинки с т. пл. 185°С [214].
4-Оксимеркур-2-нитрофенолят натрия (меркурофен) может быть получен [213] как
нз хлорида, так и нз ацетата возможно полным растворением в разбавленном растворе едко-
го натра, отфильтровыванием от нерастворившегося осадка и концентрированием фильтрата
в вакууме. По охлаждении выкристаллизовывается красный осадок. Его тщательно промы-
вают охлажденной льдом водой и перекристаллизовывают нз горячей воды.
Еще более облегчает меркурирование накопление нескольких гидро-
ксильных групп в молекуле: резорцин [71, .249—256], салигенин [2571
и флороглюцин [249] меркурируются в водном растворе на холоду (са-
лигенин также в спиртовом растворе при нагревании). При этом толь-
ко для резорцина получен мономеркурированный продукт, салигенин
дает димеркурированное производное, флороглюцин — исключительно
тримерйурированное соединение. Так же меркурирован нитросалигенин,
n-оксифенилкарбинол и его нитропроизводное [257].
Меркурирование резорцина [71]. Полученный прн комнатной температуре раствор
28,3 г (1 моль) тонко измельченной уксуснокислой ртути в 60 мл воды вливают постепенно-
при перемешивании в раствор 29,7 г (3 моля) резорцина в 25 мл воды. После хранения в
течение 15 мин. реакция заканчивается (проба со щелочью не дает осадка окиси ртути).
Более продолжительного хранения следует избегать, так как при этом может выделиться
смесь трудноразделимых ацетатов меркурнрованного резорцина. Раствор быстро отсасы-
вают от небольшого количества выделившихся хлопьев в концентрированный раствор хлори-
стого натрия. Спустя короткое время выделяется смесь хлормеркур- н бис-хлормеркуррезор-
цина. Ее оставляют на льду некоторое время, затем отсасывают и промывают ледяной водой.
Затем растворяют в большом количестве эфира, отделяют от незначительного количества-
серого осадка, эфир отгоняют, под конец — в вакууме, н осадок сушат при комнатной темпе-
ЛЬ Ртутноорганические соединения. Лит. стр. 104—114
ратуре в эксикаторе. Выход 16 а (в водном маточнике остается значительная часть веще-
ства, однако его трудно получить оттуда в чистом'виде). Осадок представляет собой смесь
моно- и димеркурнрованного резорцина, разделяемую обработкой большим количеством
кипящего хлороформа, в котором димеркурированное соединение нерастворимо. Много-
кратной кристаллизацией из хлороформа 4-хлормеркуррезорцин, содержащий хлороформ,
получают в виде призм с т. пл. 105°С. Прн хранении в вакууме над парафином его получают
свободным от хлороформа, т. пл. 123°С, при 170°С окрашивается в кроваво-красный цвет.
Нерастворимый в хлороформе осадок растворяют в небольшом количестве эфира. После
испарения эфира получают 2,5 г 4,6-дихлормеркуррезорцина; порошок, темнеющий прн
температуре около 200°С.
Тримеркурированный резорцин получен действием избытка уксусно-
кислой ртути в спиртовом растворе [256]. Действием в этих же условиях
1 и 2 молями уксуснокислой ртути наэтил- и гексилрезорцин получают ди-
41 мономеркурированные производные [256].
Мономеркурированные 5-метилрезорцины и 4-хлоррезорцины получают
[9] при хранении с эквимолекулярным количеством уксуснокислой ртути
в водно-спиртовом растворе на холоду.
При меркурировании многоядерных фенолов: о,о'-, п,п'-диоксифенила
[258, 259], ди-о-крезола [259] уксуснокислой ртутью в водном или уксус-
нокислом растворе при умеренной температуре (40—50° С) получены моно-
и полимеркурированные продукты.
Однако в случае полиатомных фенолов с орто- или пара-положением
•ОН-группы (пирокатехин [257, 260], гидрохинон, пирогаллол) действие
-солей ртути не ведет к получению меркурированных продуктов, а вызывает
окисление.
Нитрорезорцин меркурирован при 2-часовом нагревании на водяной
‘бане с окисью ртути в водном растворе [213]. При этом получается или мо-
номеркурированное в положение 4 [213] или димеркурированное в поло-
жения 2,4 соединение [261], получающееся и при меркурировании уксусно-
кислой ртутью [261]. Согласно Оно [261], с повышением pH выход умень-
шается. 2,4-Динитрорезорцин с уксуснокислой ртутью при комнатной тем-
пературе при pH 2,5—3,5 образует мономеркурированный в положение
•6 продукт [261]. При pH 4,6—6,0 последний продукт содержит такжё ртуть,
связанную с кислородом [261]. Меркурирование же моно-и динитропро-
изводных гидрохинона и пирокатехина ацетатом или окисью ртути невоз-
можно из-за восстановительного действия первых [261].
Замена водорода гидроксильной группы на алкил (арил) несколько
затрудняет меркурирование, и эфиры фенолов меркурируются с большим
трудом, чем свободные фенолы: при нагревании до 2 час. с уксуснокислой
ртутью без растворителя (анизол [71, 262], нитроанизол [2371, фенетол,
ф-этоксинафталин [1861) или же при многочасовом нагревании в водной
взвеси (анизол, фенетол, метиловый эфир п-крезола [263, 264]), спиртовом
(диметил овый'эфир резорцина [265], метиловые эфиры тимола и карвакрола
[225] или гуксуснокислом растворах (дифениловый эфир и его монобром- и
моноиодзаметценные) [266], причем образуются преимущественно моно-
меркурированные соединения (ср., однако, [225, 263]).
При меркурировании анизола сернокислой ртутью в водном, подкис-
ленном серной кислотой растворе (температура не указана) получен [267]
димеркурированный в положения 2 и 4 продукт. Меркурирование серно-
кислой ртутью амилфенилового эфира приводит к мономеркурированному
•соединению, а дифенилового эфира — к полимеркурированному продукту
1267].
Мономеркурирование в a-положение метилового эфира 0-нафтола азот-
нокислой ртутью происходит при комнатной температуре, действие же кис-
лого сульфата ртути приводит к неидентифицируемым веществам
1268].
Введение ртути на место водородного атома (меркурирование) 75
Измерение скорости меркурирования ацетатом ртути в ледяной уксусной
кислоте при 25° С анизола .(1,85),-п-метил-(0,931), п-метокси-(0,634), п-тре-
тичнобутиланизолов (1,66) и дифенилового эфира (0,267) показало, что реак-
ции эти имеют второй порядок (в скобках даны значения констант скоро-
стей) [269]. Мономеркурированный п-метоксианизол получен при 65°С
[221].
Гваякол при кипячении в течение часа с 2 молями уксуснокислой ртути
в спиртово-уксуснокислом растворе (3 : 1) дал димеркурированный в поло-
жения 4 и 6 продукт [270]
ОН
• I
H3COCOHg—ОСНз
HgOCOCHa
5-Нитрогваякол меркурирован продолжительным кипячением с уксус-
тюкйслой ртутью в разбавленной уксусной кислоте [271, 272] с образова-
нием моно- и димеркурированных продуктов. Арилоксижирные кислоты
(арилоксиуксусная и арилоксипропионовая) меркурируются в ароматиче-
ское ядро при продолжительном нагревании на водяной бане с уксуснокис-
лой ртутью в уксуснокислом [273—275] или спиртово-уксуснокислом
растворе [275а], с окисью ртути в щелочном растворе или сплавлением с ук-
суснокислой ртутью без растворителя [273, 274]. Однако аллильные эфиры
фенола и 0-нафтола меркурируются при непродолжительном (2—3 мин.)
кипячении с уксуснокислой ртутью в уксуснокислом растворе, ари этом
не происходит присоединения ртути по двойной связи аллильной группы
[2761.
Анетол при действии уксуснокислой ртути в водном растворе, по-види-
мому, в значительной степени меркурируется в бензольное ядро [2771.
При действии уксуснокислой ртути в водном растворе на эвгенол (12 час.,
комнатная температура) образуется продукт, возможно, меркурирован-
ный в ядро [278].
Введение "гидроксила и в алкильную группу эфиров фенола облегчает
меркурирование даких эфиров: мономеркурированные в бензольное кольцо
моноарильные эфиры гликолей были получены действием на холоду в спир-
товом растворе 0,5 моля ацетата ртути [279]. Димеркурированные произ-
водные получаются при 2—3-часовом кипячении эквимолекулярных коли-
честв реагентов [2791.
Получение уксуснокислой п-анизнлртутн [71]. Сухую уксуснокислую ртуть (1 моль)
и избыток аннзола (8 молен) нагревают на водяной бане до тех пор, пока проба не будет
более образовывать желтого осадка с раствором едкого натра. Раствор по охлаждении вы-
деляет кристаллы уксуснокислой n-анизилртути, которые могут быть перекристаллизова-
ны из разбавленного спирта. Т. пл. 176,5°С. Маточный раствор содержит уксуснокислую
о-анизилртуть.
Повторение этой реакции в условиях Димрота (48 час., нагревание)
показало [262], что при этом образуется около 65% меркурированного в
пара-положение’продукта, менее 1 % орто-изомера, а также и димеркуриро-
ванноеАоединение. Меркурирование анизола уксуснокислой ртутью в ле-
дяной уксусной кислоте при 25° С, изученное кинетически [119, 269] (кон-
станта скорости М порядка), показало, что при этом образуется 86% пара-и
14% орто-изомера. Найдены также факторы парциальной скорости реакции
меркурирования для этих положений в анизоле [119].
30-Минутное нагревание на водяной бане спиртового раствора анизола
и ртутной соли тринитрометана дает с общим выходом 41 % смесь мономер-
76 Ртутноорганические соединения. Лит. стр. 104—114
курированных в орто- и пара-положения продуктов [147]. Для ряда произ-
водных ароматических алкоксисоединений (см. стр. 75) реакции меркури-
рования уксуснокислой ртутью удовлетворительно подчиняются видоиз-
мененному уравнению Гаммета с константой электрофильного замещения ст*;
величина р = — 4,10 [269].
Получение аллильного эфира я-хлормеркурфенола [276]. 15 г аллильного эфира фенола
(избыток) вливают в смесь 9 мл 80% -ной уксусной кислоты и 37 мл воды; в полученную в
результате встряхивания эмульсию вносят пасту окиси ртути в количестве 6,5 а (в пересче-
те на сухую смесь). По окончании добавления окиси ртути смесь нагревают до кипения и
вливают в кипящий раствор хлористого натрия в 50 мл воды. Все нагревают до кипения в<
течение 2—3 мин. и оставляют охлаждаться. Первоначально выделившееся масло часа че-
рез 2 закристаллизовывается. Перекристаллизовано из хлороформа. Выход 23,5 а (57% ,
считая на окись ртути). Т. пл. чистого вещества 101—101,5°С.
При одновременном присутствии в молекуле наряду с гидроксилом и
других функциональных групп течение меркурирования определяется на-
личием гидроксила. В тех же условиях, что фенол и его гомологи, меркури-
руются прочие ароматические окси соединения. На этом основании мерку-
рирование оксиальдегидов, оксикетонов, оксикарбоновых, оксисульфоно-
вых, оксиарсиновых кислот, фталеинов и других оксипроизводных трифе-
нилметанового ряда рассматривается в настоящем разделе.
В спиртовом или водном растворе на холоду или при нагревании мерку-
рируется салициловый альдегид [280, 281] с образованием 3,5-диацетатмер-
курсалицилового альдегида. Так же меркурированы м- и п-оксибензальде-
гиды, 2,4-диоксибензальдегид и флороглюцинйльдегид с образованием ди-
меркурированных соединений [282], 3-нитро-и 5-нитросалициловые альде-
гиды [280], нитро-ж-оксибензальдегиды с образованием моно- и димеркури-
рованных продуктов [283] и монометиловые эфиры диоксибензальдегидов,
например ванилин [282, 284, 285], который меркурирован также при 10-ча-
совом нагревании с окисью ртути в щелочном растворе [286], изованилин
[282Ь Введение одного атома ртути в молекулу диметилового и тримети-
лового эфиров галлового альдегида требует кипячения в течение 4 час. с ук-
суснокислой ртутью в спирте [287]. Меркурирование диалкил- (алкилы—
СН3 и С3Н7) и моноалкил-(алкилы—(-С4Н9, r-C5Hu) фенолальдегидов
производят сплавлением с уксуснокислой ртутью без растворителя [226].
Оксйарилалкилкетоны, например, оксифенилметилкетон [108, 288, 289],га-
лоид оксиарил а л кил кетоны [109] и семикарбазоны этих соединений [108],
меркурируют при нагревании без растворителя уксуснокислой ртутью
(или другим меркурирующим агентом: цианистой или сернокислой ртутью)
до 70—90° С в течение около 1,5 часа.
Получение 5-оксимеркурваннлнна [280]«
СНО
I
HOHg £ ГСИ»
К pacTBopy*-t36 г сулемы в 400 мл горячей воды прибавляют горячий раствор 100 г ед-
кого натра в 100 мл воды. К полученной смеси при энергичном перемешивании прибавляют
25 г ванилина. Кипятят при перемешивании в течение 10 час. По охлаждении осадок от-
фильтровывают и промывают водой; соединенные фильтрат и промывные воды подкисляют
серной кислотой (1 : 3). Выделившийся плотный белый осадок отфильтровывают, промы-
вают водой, сушат в вакуум-эксикаторе. Выход 40 г. Кристаллизация из воды, метилового
спирта, ацетона или диоксана дает бесцветные иглы 5-оксимеркурванилина, т. пл. 235°С.
Введение ртути на место водородного атома (меркурирование) 77
Меркурированная салициловая кислота получается при кипячении в
воде салициловой кислоты с свежеосажденной окисью ртути [71] или ук-
суснокислой ртутью [291], а также уже при хранении, в особенности в сы-
ром состоянии, салициловокислой ртути при комнатной температуре [291],
быстрее при нагревании до 100—120° С [71, 291, 292]. Паолини рекомен-
дует для этой реакции температуру 170° С [293]. Согласно Руппу [294]
и Бедекеру [295], продукт представляет собой смесь ангидридов 3- и 5-окси-
меркурсалициловых кислот.
В совершенно чистом виде 3-оксимеркурсалициловая кислота получается
при непродолжительном действии сернокислой ртути на салициловую кис-
лоту [294]. Однако, согласно Укай [296], при этом образуется меркуриро-
ванный в положение 5 продукт. При постепенном внесении сернокислого
раствора сульфата ртути в кипящий водный раствор салициловой кисло-
ты, по мнению Сова [267], замещение происходит в положение 4 по отно-
шению к карбоксилу.
Эфиры салициловой кислоты меркурируют действием уксуснокислой
ртути без растворителя [297]: метилсалицилат при кипячении [297], с этил-
•салицилатом реакция количественно проходит лишь при 180° С [297].
Описано меркурирование салициловой кислоты и ее производных [298—
302], салицилсульфоновой кислоты [303], гликолевого эфира салициловой
кислоты [304], ацетилсалициловой кислоты [253, 278, 305—307], нитро-
салициловой кислоты [309, 313], фенацилсалициловой кислоты [290], ме-
тилендисалициловой кислоты' [258] (мономеркурированный продукт).
При кипячении с солью или окисью ртути в воде, спирте или уксусной
кислоте или нагреванием в воде их ртутных солей меркурируют другие
ароматические оксикарбоновые кислоты: лг-оксибензойную кислоту [301,
308, 310], n-оксибензойную кислоту [253, 237, 301, 311], крезотиновую кис-
лоту [312], резорциловую кислоту (меркурирование [J-резорциловой кисло-
ты сернокислой ртутью в водном растворе дает моно-и димеркурированный
продукт [313]), 2-окси-З-нафтойную кислоту [258, 314], 1-окси-2-нафтойную
кислоту [258], 5-алкил-2,4-диоксибензойную кислоту [315]) (алкил—С2Н5,
п-С3Н7, п-С4Н9, п-С5Нп), 5-н-бутил-2,6-диоксибензойную кислоту [315]
и галловую кислоту, два или три гидроксила которой метилированы [287],
а также /г-фенолсульфокислоту [317] и нафтолсульфоновые кислоты:
2-оксинафтил-6-сульфоновую, 1 -оксинафтил-4-сульфоновую, 1 -оксинафтил-
5-сульфоновую [228].
В таких же условиях меркурируют окси- [318—320] и амино-[320] окси-
арил арсиновые кислоты. Меркурирование аминооксиарил арсиновых кислот
описано М^шманом [321].
Меркурирование"салициловой кислоты [71]. Смесь эквимолекулярных количеств све-
жеосажденной окиси ртути и салициловой кислоты в воде продолжительное время нагре-
вают на водяной бане до тех пор, пока проба не будет полностью растворяться в щелочи.
Полученное вещество растворяют в растворе соды и вновь высаживают углекислотой. По-
лученная внутренняя соль о-оксимеркурсалициловой кислоты представляет собой тонкий
белый порошок, разлагающийся при нагревании, не плавясь.
Вещество идентично Hydrargyrum salicylicutn фармакопеи. Согласно
позднейшим данным [293, 294], оно представляет собой смесь ангидридов
3-гидроксимеркур- и 5-гидроксимеркурсалициловых кислот. Разделение
такой смеси см. [295, 322].
Получение метилового эфира 3-хлормеркурсалнциловой кислоты [297]. 15 г уксусноки-
слой ртути, 15 а метилового эфира салициловой кислоты и 2,5 г ледяной уксусной кисло-
ты кипятят 40 мин. с обратным холодильником. После этого проба с едким натром на ион
ртути отрицательная. При охлаждении нз вязкой реакционной смеси выделяются игло-
образные кристаллы. Смесь оставляют стоять на ночь, затем тщательно отсасывают/Вес
78 Ртутноорганические соединения. Лит. стр. 104—114
осадка метилового эфира ацетатмеркурсалициловой кислоты 9 г (52%). Очищают растворе-
нием в кипящем этилацетате, упаривают и высаживают^петролейным эфиром. Т. пл. 202®С
с небольшим предварительным спеканием.
Остающиеся в маточнике около 48% меркурированного продукта легко и полностью
могут быть высажены водным раствором хлористого натрия в виде кристаллов метилового
эфира 3-хлормеркурсалицнловой кислоты. Выход 6 г (36,6%). Общий выход около90%.
Очищают растворением в этилацетате и высаживанием петролейным эфиром.
Меркурирование оксисоединений, например салициловой, 2,3-оксинаф-
тойной кислот, ускоряется в присутствии щелочи [314].
МеркурированиеЗ-окси-2-иафтойион кислоты в щелочном растворе [314].30 г (0,16 моля)1
кислоты в 500 мл воды и 150 мл (0,9 моля) 6 V NaOH нагревают до кипения н при пере-
мешивании постепенно прибавляют 52 г (0,16 моля) уксуснокислой ртути, растворенной в-
300 мл воды и 10 мл (0,15 моля) уксусной кислоты. Тотчас выпадает осадок окиси ртути.
Смесь кипятят несколько минут (под конец с животным углем), отфильтровывают и при-
бавляют, избыток раствора NaCl и разбавленной соляной кислоты до кислой реакции. Оса-
док — желтый неплавкий порошок — представляет собой, по-виднмому, внутреннюю соль-
4-окснмеркур-3-окси-2-нафтойной кислоты.
Точно таким же образом производят меркурирование салициловой кис-
лоты [314]. Из 15 г салициловой кислоты получают 27 г оксимеркурсалици-
ловой кислоты.
Таким же путем из 100 г фенола получают 215 г ангидрооксимеркурфе-
нола [314]. Однако попытка меркурирования а-нафтола в щелочном раство-
ре привела только к окислению его. о-Нитрофенол также не удается мер-
курировать в щелочном растворе [314].
Кумарин и его производные меркурируются в ядро без затрагивания
двойной связи (исключение 7-окси-4-метилкумарин, см. далее) лишь в при-
сутствии щелочи, действием как уксуснокислой ртути [323—327] и окиси
ртути (при кипячении) [323—325], так и меркурацетамида [323]. XHg-
группа вступает в положения 6 и 8. Продукт из кумарина и уксуснокислой
ртути имеет строение
HaCOCOHg-j^Y^
ЧА/СО
I О.
HgOCOCHs
При действии солей и окиси ртути на производные кумарина в отсутствие
щелочи наряду с меркурированием происходит присоединение по двойной
связи (см. гл. VI, стр. 159).
Однако 7-окси-4-метилкумарин при кипячении с уксуснокислой ртутью
в метиловом спирте не присоединяет соли ртути, а лишь меркурируется
в положение 8 [328].
Оксиазосоединения меркурируются уксуснокислой ртутью в водном,,
спиртовом или слабоуксуснокислом растворе при более продолжительном
(до 7 час.) нагревании, чем соответствующее оксисоединение [329—331].
Бензаурин и фенилдиоксидибензопиран образуют при кипячении их
щелочных растворов с водным раствором сулемы моно- и димеркурирован-
ные соединения [3321. :
По Уитмору и Лейку [333], наличие спирта в реакционной среде при
меркурировании уксуснокислой ртутью способствует связыванию образую-
щейся уксусной кислоты и благоприятствует полимеркурированию. Так,
при меркурировании аурина уксуснокислой ртутью в зависимости от рас-
творителя и от относительных количеств аурина и соли ртути получены
соединения с одним (1 моль аурина, 1 моль ацетата ртути, среда — этилаце-
тат и уксусная кислота), двумя (1 моль аурина, 2 моля ацетата ртути, спирт
и уксусная кислота), тремя (1 моль аурина, 4 моля ацетата ртути, спирло-
Введение ртути на место водородного атома (меркурирование)
вый раствор) атомами ртути в молекуле. При действии на аурин шестикрат-
ным, (в молях) количеством уксуснокислой ртути в уксусной кислоте обра-
зуется соединение с шестью атомами ртути в молекуле (I) и одновременно-
вещество, шестикратно меркурированное в ядро и имеющее два ацетокси-
мерку ростатка, связанных с кислородом (И)'.
О
AcHg || HgAc
(Ас = СН3СО2)
Ауринтрикарбоновая кислота при кипячении с уксуснокислой ртутью*
в уксусной кислоте образует тримеркурированное соединение [258] (в каж-
дое бензольное кольцо вступает одна HgX-группа). Так же меркурированя
«нафтохромзеленая» [258].
Фенолфталеин [334—336], его гомологи, как крезолфталеин [335, 337],.
тимолфталеин [335], '3-3-бис-(4'-окси-3'-нитрофенил)фталид [338], флуо-
ресцеин [334—337, 339, 340], этил-[341], гексил- [341] и другие алкилфлуо-
ресцеины [342] (продукты — мономеркурированные' соединения), 4,5-
динитрофлуоресцеины [338], оксифлуоресцеины [337], производные дифе-
нол- и дирезорцинизатина [343, 344] и другие оксисоединения трифенилме-
танового ряда меркурируются в фенольное кольцо в спиртовом, водном,
часто щелочном растворе при кипячении в течение нескольких часов. В боль-
шинстве случаев меркурирование происходит в орто-положение к гидрок-
силу, и в молекулу может быть введено несколько атомов ртути [334].
В сам фенолфталеин действием ацетата ртути в спиртовом или водном рас-
творе или суспензии может быть введено 3 атома ртути вместо 4, кипяче-
нием же с окисью ртути в слабощелочном растворе — только 1 атом ртути.
[334]. Так же меркурированы галоидозамещенные фенолфталеины [345],
2,7-диоксифлуоран [276], ди- и трибромпроизводные 6-оксифлуорана [346].
Меркурирование фенолфталеина уксуснокислой ртутью [334]. Раствор 3 г фенолфталеина*
в 50 мл спирта смешивают с профильтрованным раствором 25 г (8 молей) уксуснокислой-
ртути в 50 мл воды и 50 мл ледяной уксусной кислоты. При хранении в течение ночи при
комнатной температуре меркурирования не происходит, наблюдается лишь выделение-
небольшого количества уксуснокислой закиси ртути. Ее отфильтровывают и фильтрат на-
гревают иа водяной бане. Быстро, в течение 2' час., образуются кристаллы в виде розеток
плотных листочков. Нагревают еще в течение часа и смесь оставляют на ночь. Кристалли-
ческую пасту отфильтровывают, промывают спиртом (для удаления невошедшего в реакцию
фенолфталеина) и, наконец, водой для удаления избытка соли ртути. Кристаллы сушат на
воздухе в течение нескольких дней и затем при 100°С в течение часа. Полученный триаце-
токсимеркурфенолфталеин нерастворим в органических растворителях, растворим в рас-
творе едкого натра с темно-красным окрашиванием.
Меркурирование фенолфталеина окисью ртутн в щелочном растворе [334]. 3 г фенол-
фталеина растворяют в 25 мл 1 N едкого натра, раствор доводят добавлением воды до-
150 мл и кипятят с 3 а желтой окиси ртутн 3 часа. Выделяющийся в небольшом количестве-
серый осадок отделяют отстаиванием в высоком цилиндре в течение нескольких дней. Пос-
ледние следы его отделяют центрифугированием. Из фильтрата углекислотой высаживают
мономеркурированный фенолфталеин в виде слегка красноватого творожистого осадка.
Его отделяют центрифугированием, сушат на водяной баие, промывают спиртом и сушат
при 110°С. Вещество не плавится, нерастворимо в обычных растворителях, растворимо лишь-
в ледяной уксусной кислоте с образованием мутного раствора. Не кристаллизуется.
80
Ртутноорганические соединения. Лит. стр. 104—114
Меркурирование флуоресцеина [334]. 3,3 г флуоресцеина растворяют в 200 мл 0,1 N рас-
твора едкого натра и подкисляют 5 мл ледяной уксусной кислоты. Прибавляют профиль-
трованный раствор 25 а (8 молей) уксуснокислой ртути в 200 мл воды, слегка подкисленной
уксусной кислотой для предотвращения гидролиза. Смесь кипятят 10 час., прибавлением
воды по мере испарения поддерживая объем постоянным. Освобождают от ртути центри-
фугированием, промывают и сушат. При промывании часть вещества теряется в виде тон-
кой суспензии. Выход около 10 г. Вещество представляет собой смесь тетрагндрокси- и
тетраацетоксимеркурфлуоресцеииа с преобладанием первого. Нерастворимо в обычных
растворителях, растворимо в щелочи.
Дибромфлуоресцеин меркурируется уксуснокислой ртутью [334, 336,
347—3501 в уксуснокислом или окисью ртути в щелочном (см., однако, [334])
растворе при нагревании [347, 3481. Количество введенных атомов ртути
и состав образующихся продуктов зависят от продолжительности нагрева-
ния и pH среды [347, 348]. Эозин при нагревании на водяной бане с уксус-
нокислой ртутью в водном растворе дает мономеркурированное производное
[351].
В условиях меркурирования фенолфталеина, флуоресцеина и их произ-
водных меркурируются также и другие галоидированные флуоресцеины
[336, 352, 353], фенолсульфонфталеин [335, 354], его алкилзамещен-
ные [3351, резорцинсульфонфталеин [355, 356], его галоидные производ-
ные, салицилсульфонфталеин [357], резорцинсукцинеины [336], «сахареи-
ны» [336], галоидные производные резорцинсахареина [358, 359] и тому
подобные соединения. Продукты — моно-, ди- иногда и более меркуриро-
ванные в орто-положение к гидроксилу соединения.
Меркурирование днбромфлуоресцеииа [334]. Получение 4-оксимеркур-2,7-дибромфлуо-
ресцеина.
HgOH
49 г 2,7-дибромфлуоресцеина растворяют в растворе 8 г едкого натра в 50 мл воды и
разбавляют до 200 мл. Раствор перемешивают и обрабатывают 12,5 мл ледяной уксусной
кислоты. При энергичном перемешивании образуется вязкий осадок. Предварительно при-
готовленный раствор 22,5 г (теоретическое количество 21,7 г; избыток берется ввиду воз-
можной потери в виде закисной ртути, обычно образующейся при растворении имеющейся
в продаже окиси ртути в уксусной кислоте) окиси ртути в 25 мл ледяной уксусной кислоты
и 50 мл воды,-разбавленный после растворения до 100 мл и профильтрованный, прибавляют
к раствору красителя и все разбавляют до объема около 500 мл. Смесь кипятят до отсутст-
вия реакции иа иои ртути с сульфатом аммония, иа что требуется от 4,5 до 6 час. Во время
кипячения осадок становится темнее и несколько более зернистым. Отфильтровывается он,
однако, с трудом, поэтому его промывают от уксусной кислоты и ацетата натрия цент-
рифугированием и сушат при 110°С. Выход почти количественный.
Оксимеркурдибромфлуоресцеин представляет собой красный порошок, обладающий
ясно выраженными электрическими свойствами. Он нерастворим в обычных растворителях.
Растворим в 2 экв. едкого натра. Натриевая соль 4-оксимеркур-2,7-дибромфлуоресцеина
носит коммерческое название «меркурохром».
Ароматические спирты меркурируются в более жестких условиях, чем
фенолы. Бензиловый спирт—при 6-часовом кипячении с ацетатом ртути
в водном растворе [360]. Так же меркурируется фенилэтиловый спирт
Введение ртути на место -водородного атома (меркурирование)
81
[3611. В обоих случаях образуется два мономеркурированных в положения
2 и 4 продукта, в то время как в этих же условиях ш-фенилпропиловый
спирт [361] также дает два монозамещенных соединения, но в положения
3 и 4.
Меркурирование бензилового спирта [360]. 15 г бензилового спирта и 45 г уксусно-
кислой ртути в 25 мл воды кипятят 6 час., фильтруют, фильтрат по каплям Прибавляют к
насыщенному раствору хлористого натрия. Выпавший осадок весит 46 г. 10 г его извлекают
горячей водой, охлаждают, выпавший осадок после кристаллизации из воды или возгонки
дает 5,6 г 2-С1Н§СвН4СН2ОН; иглы, т. пл. 120—120,5°С. Из маточника или остатка после
извлечения спиртом получают 1,4 г 4-ClHgCeH4OH, т. пл. — 250°С.
Действие сульфата ртути на бензиловый спирт дает мономеркурирован-
ный в орто-положение продукт [267].
МЕРКУРИРОВАНИЕ ПРОИЗВОДНЫХ ТИОФЕНОЛОВ
Сами тиофенолы со свободной сульфгидрильной группой с солями или
окисью ртути образуют устойчивые тиофеноляты ртути (см. гл. XVI).
Вступление атома ртути в тиосоединения происходит менее гладко и с
большим трудом, чем в их кислородные аналоги. Так, метилфенилсульфид
.меркурирован при нагревании с уксуснокислой ртутью на паровой бане
(выход около 40%) [362, 363] или при кипячении с уксуснокислой ртутью
в изоамилацетате [364].
Дифенилсульфид реагирует с уксуснокислой ртутью в кипящем изоамил-
ацетате (141—142° С) [364, 365].
В этих же условиях [364], а также при нагревании компонентов без
растворителя до 170—180° С [366] дифенилсульфон дает мономеркурирован-'
ны.й продукт.
Кипячение спиртовых растворов о, о'- и п, п'-динитродифенилдисульфи-
дов с уксуснокислой ртутью приводит к внутренней соли соответствующих
мер курированиях нитротиофенолов [367].
Дибензиловый эфир димеркаптана
R& [SR]
tH5c2 ССН3
II II
НС CHj
'Xc/Z
(R—СвН5СНа) меркурируется в условиях, близких к условиям меркуриро-
вания тиофена — действием сулемы в присутствии ацетата натрия в спир-
товом растворе при кипячении [368]. Продукт — димеркурированное в
а, а'-положения к тионной группе соединение. Мономеркурированное произ-
водное соответствующего диметилового эфира выделяется уже на холоду
при действии сулемы в растворе метилового спирта [368].
Меркурирование 4-окси-, 2-метил-4-окси- и 4,4'-диоксидифенилсульфида
проходит гладко при получасовом нагревании в водно-спиртовом растворе
с уксуснокислой ртутью [369] с образованием соответственно моно- и ди-
меркурированных продуктов. Спиртовый раствор тиосалициловой кислоты
при 24-часовом хранении на холоду с раствором эквимолекулярного количе-
ства окиси ртути в разбавленной уксусной кислоте образует мономёркури-
рованный продукт [253].
82 Ртутноорганические соединения. Лит. стр. 104—114
Но меркурирование S-метилтиосалициловой кислоты заканчивается
только при 230° С и приводит к образованию неидентифицируемых продук-
тов [365].
Описано меркурирование дитиосалициловой кислоты [293].
Получение уксуснокислой /1-фенилмеркаптофенилртути [365]. 10 г дифенилсульфида
н 20 г уксуснокислой ртути в 50 мл изоамилацетата кипятят в течение 5 час., после чего
проба не дает черного окрашивания с сернистым аммонием. Раствор отфильтровывают го-
рячим от нерастворимого осадка соли закиси ртути (4 г). Из фильтрата после охлаждения по-
лучают 2,5 г смеси ди- и тримеркурированных продуктов. Их также отфильтровывают и
фильтрат упаривают в вакууме; при этом выкристаллизовывается 2,5 г мономеркуриро-
ванного соединения,-фильтрат от которого после высаживания петролейным эфиром дает
масло (кристаллизующееся после обработки бензолом), которое после осаждения
водой из раствора в ледяной уксусной кислоте дает еще 1 г кристаллов. Т. пл.
p-CHgCOaHgCeHiSCeHs после кристаллизации из лигроина 148°С.
Из метилового раствора ацетата высаживанием хлористым кальцием с количествен-
ным выходом получают хлористую п-фенилмеркаптофенилртуть. Т. пл. 181° С.
Получение о-хлормеркурдифенилсульфона [366]. Нагревают 2,5 г дифеиилсульфона с
3,7' г уксуснокислой ртути и несколькими каплями уксусной кислоты до 170—180°С
(до затвердевания). Полученную массу кипятят несколько минут с концентрированным рас-
твором хлористого натрия. Образующийся осадок весит 4,2 г. После кристаллизации из
спирта т. пл. 247—248°С.
МЕРКУРИРОВАНИЕ АРОМАТИЧЕСКИХ АМИНОВ
Амины меркурируются еще легче, чем фенолы. В простейших случаях
реакцию ведут на холоду в водном или водно-спиртовом растворе; обычно
применяют уксуснокислую ртуть. Так меркурируют анилин [9, 71, 169,
370—374] (скорость меркурирования анилина ацетатом ртути уменьшается
от добавления азотной и хлорной кислот [207]), моно- и диалкиланилины,
(алкил—метил [71, 147,374—376] и этил [374,376,377]), бензиланилин [376,
378] (реакцию ведут при 50° С), дифениламин [9, 379, 380] (сплавлением ди-
фениламина с сулемой и последующим кипячением в течение часа в уксус-
ной кислоте получен 2,2',4,4'-тетрахлормеркурдифениламин [380]). Также,
как анилин, меркурируют галоиданилины (см. стр. 87), п-бромдиме-
тиланилин [381], п-[190, 301, 382—386], о-[301, 370, 385—390], ти-толуиди-
ны [301, 385, 387], диметил-п-толуидин [382, 383], М,М'-ди-п-толилформ-
амидин [391] (кипячение с уксуснокислой ртутью в спиртовом растворе);
продукт имеет.вероятное строение
НзС—N—СН
I II
I N
Hg |
СН3
Так же как анилин, меркурируют 2- и 4-аминодифенилы [392], бензидин
[393, 394] (в растворе ледяной уксусной кислоты), толидин [394], а-нафтил-
амин (дает только 2,4-диацетатмеркур-1-нафтиламин [228, 374], а при
рН4,2—монрмеркурированное производное [301]), Д-нафтиламин (продукт
1-ацетатмеркур-2-нафтиламин [228,301]), фенил-Р-нафтиламин (продукт
/С6Н4 х
имеет строение Hg<( ^>NH [395]), Р,Р-динафтиламин (продукт бис-1-
ХЗщНц
Введение ртути на место водородного атома (меркурирование)
83
ацегатмеркурнафтил-2-амйн при нагревании с уксуснокислой ртутью в
уксусной кислоте [186]), 2-аминофлуорен (в положение 3) [396], л-амино-
фейайтриметиламмонийацетат (димеркурированное производное [371]).
Одйако ариламмониевые соли, не имеющие других заместителей в ядре,
Весьма инертны к меркурированию. Триметилфениламмонийацетат не мер-
Курируется ни при хранении в течение 2 месяцев на холоду, ни при 24-ча-
совом кипячении в водном растворе с уксуснокислой ртутью [397]. При
20-часовом же нагревании фенилтриметиламмонийнитрата до 95° С с рас-
твором окиси ртути в разбавленной азотной кислоте образуется продукт
только мета-замещения (выделен в виде продукта замены HgX на бром
[167, 192а]).
Для меркурирования ароматических аминов в таких же мягких усло-
виях могут быть применены также другие меркурирующие агенты, такие
как лактат ртути [398] (меркурирование анилина, нафтиламинов), ртутная
соль тринитрометана [147] (анилин, диметиланилин).
Ртуть вступает в молекулу аминов, согласно общим правилам замещения
в аминах. Так, сам анилин дает смесь двух мономеркурированных в пара-и
орто-положения соединений [71, 370] и, при избытке уксуснокислой ртути,
2,4-димеркурированный продукт [371] и тримеркурированное соединение
при действии сульфата ртути [267].
В условиях меркурирования аминов меркурированы в ароматическое ядро
при азоте аминогруппы многочисленные производные 2-ариламинотиазолов
(4-стирил-2-ариламиноТиазолы [399], 2-ариламино-4-метилтиазолы [400,
401], 2-ариламино-4,5-диметилтиазолы [401—403], 2-ариламино-4-метил-5-
этилтиазолы [404], 2-ариламино-4-фенилтиазолы [401, 405 , 406], 2-арил-
амино-4-бромфенилтиазолы [406], 2-ариламино-4-бромфенил-5-бромтиазолы
[406], 2-ариламино-4-триазолидон и его производные [407—409], 2-арил-
амино-4-метил-5-карбэтокситиазолы [410], 2-ариламино-4-(2-тиенил )тиазо-
лы [411], 2-фениламино-4,5,6,7-тетрагидробензотиазол [412]); также мер-
курированы в ароматическое ядро при азоте в положение 3 S-N-арилтиазо-
лины (2-ариламино-З-фенил-, 4,5-арил- и -алкилтиазолины [413], З-о- [414],
л«-[415], п-[416, 417] тол ил-2,4-тиазол индионы, З-о-толил-5-бензилиден-,
2,4-тиазолиндионы [417], алкилированные, ар ил ирова иные и имеющие дру-
гие заместители в положениях 4 и 5 2-(п-толилимино)-3-(п-толил)тиазолины
[418], 3-арил-2-арилиминотиазолидоны-4 [419] (меркурируются оба аро-
матических ядра), 3-фенилтиазолидин-2,4-дион [420], 2-(и-фторфениламино-
4-тиазолидон [421]).
В условиях меркурирования аминов также меркурируют арилмочевины
[422], арилтиомочевины [422, 423].
Ацетилирование аминогруппы по общим правилам затрудняет меркури-
рование. Так, ацетанилид [424, 425], ацеттолуидиды [391, 393, 4261 мерку-
рируются лишь при температуре не ниже 100° С.
Константа скорости реакции II порядка меркурирования ацетанилида
составляет 0,191 • 105 л/моль-сек [269] в ледяной уксусной кислоте при 25°С.
Уксуснокислад п-ацетаминофенилртуть получается при кипячении ацет-
анилида в водном растворе с уксуснокислой ртутью [424]. При сплавлении
ацетанилида при 115—140° С с избытком уксуснокислой ртути получают
три-[427], тетра-[428] и пентамеркурированные [429—431] соединения.
В этих условиях при температуре не выше 150° С ацетил-а-нафтиламин дает
тетрамеркурированное производное [429, 430, 432]. Однако введение ртутй
в молекулу аминооксисоединений возможно лишь в том случае, если амишя
группа ацилирована (или метилирована); аминооксисоединения со свобод-
ной аминогруппой восстанавливают соли ртути, а не меркурируются. Моно-
и димеркурированные производные N-метилированных, о-ацетилированных
аминофенолов [265], N-ацетилированных аминокрезолов [433] и аминоал^'
84 Ртутноорганические соединения. Лит. стр. 104—114
килфенолов [434, 435]. получены при непродолжительном нагревании или
даже на холоду в' водно-спиртовом или водно-уксуснокислом растворе с
уксуснокислой ртутью. ж-Оксифенилтриметиламмонийацетат при кипя-
чении в спирте с уксуснокислой ртутью прямо дает симметричное соедине-
ние R2Hg [265]. При сплавлении с избытком уксуснокислой ртути я-ацет-
анизидина (125°С) [4361 и я-ацетфенетидина (140°С) [437] получаются три-
и тетрамеркурированные продукты. Кратковременное кипячение избытка
л-анизидина с уксуснокислой ртутью дает монозамещенный продукт
ОСН3
| HgOCOCHs
nh2
Замещение на аминогруппу водорода ядра в молекуле нитропроизводных
углеводородов, кетонов, карбоновых, сульфоновых и арсиновых кислот
облегчает меркурирование, позволяя вести его в более мягких условиях,
нередко приближающихся к условиям меркурирования самих аминов.
Меркурирование о-, м-, я-нитроанилинов [301, 438, 439] и нитротолуи-
динов [440] производят при нагревании в спиртовом или водно-спиртовом
растворе. Так же меркурированы З-нитро-1-нафтиламины [441—443]. Дру-
гие нитронафтиламины меркурируются при кипячении в уксуснокислом
растворе. Описано меркурирование 6-нитро-1-нафтиламина [263, 444].
Мёркурирование других мононитро-1- и -2-нафтиламинов см. в других
работах [442, 443].
!:’ я-Аминоацетофенон меркурируют при кратковременном нагревании
с уксуснокислой ртутью выше 100° С [9, 108] (состав продукта не указан).
Меркурирован и семикарбазон я-аминоацетофенона [108]. я-Бром-ж-ами-
ноацетофенон меркурируется при нагревании в течение 2 час. на водяной
бане с уксуснокислой ртутью в водно-метиловоспиртовом растворе [109].
В ядро антраниловой кислоты ртуть введена при 100° С [445].
При умеренном кратковременном нагревании с уксуснокислой ртутью
в спиртово- или водно-уксуснокислом растворе меркурированы эфиры ан-
траниловой [445] и я-аминобензойной [304, 446] кислот и эфиры моно-
[304, 445] и ди-Ь[-алкил [4451 антраниловой кислоты с образованием, в за-
висимости от относительных количеств реагентов, моно- и димеркурирован-
ных Продуктов. В отсутствие уксусной кислоты в чисто спиртовом растворе
образуются N—Hg-соединения, переходящие в С—Hg-соединения при
добавлении уксусной кислоты. Этиловый эфир я-аминобензойной кислоты
Меркурирован также при многочасовом хранении с уксуснокислой ртутью
в уксуснокислом растворе и при нагревании до 130—160 °C без раствори-
теля [446]. Эфиры анилидожирных кислот меркурированы действием уксус-
нокислой, ртути в водно-спиртовом растворе на холоду [304, 447]. Этиловый
эфир фенилглицина дал при этом с €2%-ным выходом мономеркурирован-
ный продукт, этиловый эфир а-анилидопропионовой кислоты — как моно-,
так и димеркурированное соединение, а высшие гомологи — этиловые эфи-
ры а-анилйдомасляной и изовалериановой кислот и при взаимодействии эк-
вимолекулярных количеств реагентов дают димеркурированные продукты
[447]. В этй соединения и в другие производные аминобензойных кислот
ртуть вступает в орто-положение к аминогруппе [304, 445—447] (ср. [448]).
Согласно данным Шербулье и Мори [448], при меркурировании я-ами-
нобензойной кислоты, производимом при кипячении в водном растйоре,
ртуть вступает в орто-положение к карбоксилу, а не в мета-, как это считает
Шеллер и сотр. [446] для этилового эфира я-аминобензойной кислоты (мер-
Введение ртути на место водородного атома (меркурирование) 85
курирование уксуснокислой ртутью в спиртово-уксуснокислом растворе);
но ни в том, ни в другом случае положение ртути не было определено.
Метиловый эфир ацетилантраниловой кислоты меркурирован при сплав-
лении с уксуснокислой ртутью при 120—130°C [445] (продукт— мономер-
курированное соединение). Л1-Ациламинобензойные кислоты (ацил—ацетил,
бензоил) меркурируются в положение 6 при нагревании без растворителя
с окисью ртути или уксуснокислой ртутью или нагреванием их ртутных
солей до 150—175° С [449].
Метаниловая [450], п-аминооксифенилсульфоновая [4501, Р-нафтилами-
но-6-сульфоновая, а-нафтиламино-4-сульфоновая [228] кислоты меркури-
руются при кратковременном кипячении с солью ртути в водном растворе.
n-Аминобензолсульфамид с уксуснокислой ртутью в разбавленном вод-
но-уксуснокислом растворе на холоду образует мономеркурированное
производное [451], при нагревании на водяной бане— смесь моно-идимер-
курированных соединений [452], образующуюся также при кипячении его
водного или спиртового раствора с раствором окиси ртути в уксусной
или ряде других органических кислот [453]. Димеркурированные соеди-
нения требуют для своего образования более продолжительного нагре-
вания.
3,5-Диацетиламино-4-оксифениларсиновую и З-ацетиламино-4-оксифе-
ниларсиновую кислоты меркурируют в виде натриевых солей в водном
растворе на холоду (последнюю также при нагревании) [321]. В последнем
соединении ртуть вступает в положение 5.
О меркурировании амино-и аминооксиариларсиновых кислот говорится
еще в других работах [318, 320].
Согласно данным Машмана [321], полученные Райциссом и др. [318]
вещества не являются С—Hg-соединениями, так как они слишком быстро
дают реакцию на ртуть с сернистым аммонием.
Реакция меркурирования аминов проходит через промежуточную ста-
дию присоединения к аминогруппе соли ртути, что может быть изображено
схемой:
Н Н
NH2 H3CCX2ONHgOCOCH3 HNHgOCOCH3 NH2
j + Hg (OCOCHS)^Q
I II HgOCOCHs
Изолированы лабильные промежуточные продукты (I) и (II) при мер-
курировании анилина и его гомологов [455], n-аминобензойной кислоты
[448, 456], эфиров о- и п-аминобензойных кислот [445 , 446], амино- и
ацетаминооксикарбоновых и арсиновых кислот [456], а также моно- и
полимеркурированных анилинов [454].
п-Нитродиметил(и диэтил)анилины*'не меркурируются [397], так как,
по мнению Хараша [397], они являются слишком слабыми основаниями
для образования промежуточных продуктов аммониевого типа, тогда как
более сильноосновные изомеры — о- и .и-нитромонометйланилин, п-нитро-
моноэтиланилин, а также n-нитроанилин образуют продукты замещения
водорода ядра на HgO2CCH3. Вероятнее, что в первом случае действуют про-
странственные препятствия.
Трудности меркурирования основных трифенилметановых красителей —
восстановление ртутной соли до металлической ртути и окисление краси-
теля — могут быть обойдены, если меркурировать не сам краситель, а его
бесцветное производное, которое не способно под действием кислот (в»том
86
Ртутноорганические соединения. Лит. стр. 104—114
числе и выделяющихся при меркурировании) превращаться в краситель,
например цианид.
Димеркурированные производные малахитовой зелени получены кипяче-
нием 4,4'-быс-диметиламинотрифенилацетонитрила или карбинола леико-
соединения этого красителя с уксуснокислой ртутью в этилацетате и пос-
ледующим превращением полученных бесцветных меркурированных произ-
водных действием ультрафиолетового света (в случае нитрила) или кислоты
(в случае карбинола) в меркурированный краситель [457 , 458].
О меркурировании малахитовой зелени см. также [332, 333, 459, 460].
Парафуксин дает тримеркурированное соединение [333] при продолжи-
тельном кипячении с эквимолекулярным количеством уксуснокислой ртути
в водном растворе. Описано меркурирование основания фуксина и других
красителей трифенилметанового ряда [332].
Азоксибензол меркурирован [462] нагреванием с уксуснокислой ртутью
в ледяной уксусной кислоте до 135—140 °C с образованием двух мономер-
курированных в орто-положение и пара-положение продуктов. Моногалои-
дозамещенные азоксибензолы меркурированы при 140—160° С [463].
CH3COOHg-rpynna вступает в галоидсодержащее ядро:
+ Hg (О2ССН3)2 -> CH3CO2Hg
N : N (О) —
N-Бензилацётамид меркурируется в мягких условиях (в водном раство-
ре), образуя два монозамещенных в орто- и пара-положение продукта
[361].
Меркурирование анилина [169, 370]. К раствору 31,8 г уксуснокислой ртути в 160 мл
воды прибавляют 18,6 г свежеперегнанного анилина. После непродолжительного хранения
выделяются короткие светло-желтые призмы, количество которых постепенно увеличи-
вается. Они представляют собой чистую уксуснокислую л-аминофенилртуть. При более дол-
гом хранении к ннм примешиваются ясно различимые бородавчатые шаровидные мягкие
агрегаты, состоящие из тонких кристалликов неясной структуры, представляющие собой
орто-соединение. Если предоставить кристаллизацию самой себе и спустя значительное
время (~ 3 час.) отфильтровать призматические кристаллы, затем слить маточник с выде-
ляющихся кристаллов орто-соединения, загрязиеииого примесью небольшого количества
пара-соединеиия, и лишь после этого собрать выделившееся орто-соединение, то обычно мо-
жно достичь полного разделения обоих. Пара-соединения получается в чистом состоянии
40 г, орто-соединенйя — 3 г, Т. пл. уксусной п-амииофенилртути 166—167°С. Т. пл. орто-
изомера 158—160°C.
Другой метод разделения орто- и пара-изомеров основан на разной растворимости хло-
ридов. После того как выделилась главная масса призматических кристаллов уксуснокис-
лой n-амииофенилртутн, к маточнику, содержащему наряду с уксуснокислой о-аминофеиил-
ртутью еще и значительное количество растворенного в уксусной кислоте пара-соедииеиия,
приливают раствор поваренной соли. При этом выпадает смесь аморфного нераствори-
мого пара-хлорида и кристаллического орто-хлорида, легко разделяемых кристаллизацией
из горячего спирта, в котором хлористая о-амииофенилртуть хорошо растворима. Т. пл.
n-хлормеркураннлина 188°С.
Получение 2,4-диацетоксимеркуранилина [372]. К 479 г (7,99 моля) ледяной уксусной
кислоты при комнатной температуре при перемешивании прибавляют 251 г (2,99 моля) би-
карбоната натрия. После того как вспенивание прекратится, приливают осторожно, не да-
вая пениться, 3,2 л воды. pH полученного раствора равен 4,30. Прибавляют 318,6 г (1 моль)
уксуснокислой ртути, от чего pH становится равным 4,25. При непрерывном перемешива-
нии прибавляют 93 г (1 моль) анилина и смесь оставляют стоять на 48 час. в темноте для
полного осаждения продукта. При соблюдении сказанных выше условий кислотности после
обильного промывания осадка несколькими порциями горячей воды и сушки в Вакуум-
эксикаторе над едким натром получают 285,3 г (93,5%) чистого продукта с т. пл. 209°С.
При недостаточно строгом соблюдении значений pH выход снижается, и при рН)>
> 4,25 вещество загрязнено n-ацетоксимеркуранилином, который может быть удален про-
мыванием хлороформом. При pH < 4,25 продукт загрязнен растворимым 2,4-диацетоксн-
меркуранилииийацетатом (см. также [371]).
Введение ртути на место водородного атома (меркурирование) 87
Получение уксуснокислой п-диметиламинофеиилртути [375]. Сливают вместе растворы
60 г уксуснокислой ртути в 400 мл 50% -кого этилового спирта и 50 а диметиланилина в 96% -
ном этиловом спирте. Вначале прозрачная смесь через несколько минут превращается
в густую кашу мелких игл. Кристаллизация из этилового спирта. Т; пл. 165°С.
Получение уксуснокислой 2-амнно-5-хлорфенилртути [387]. К раствору 32 г уксус-
нокислой ртути в смеси 100 мл воды и 200 мл спирта в присутствии небольшого количества
уксусной кислоты прибавляют раствор 13 г n-хлоранилина в спирте. Через 10 час. выдели-
вшийся осадок отделяют и перекристаллизовывают из этилового спирта. Т. пл. 207°С (см.
также [301]).
В аналогичных условиях меркурируют ж- [301, 464, 465] и о-[301, 466]
хлоранилин, 2,2-дихлоранилин [467], п- [468], л/-[469, 470], о-[388, 468]
броманилин, п-[4611, м- [465, 470], о-[4611 иоданилин.
Для введения одного атома ртути в молекулы лг-галоиданилинов, особен-
но склонных к образованию ди- и тримеркурированных соединений, сле-
дует брать избыток галоиданилина.
Получение и-ацетатмеркур-л<-иитроанилииа[438]. Вместе сливают раствор 9,6 г уксус-
нокислой ртути в 40 мл воды и 4,2 г ленитроанилина в 60 мл этилового спирта и нагревают
3 часа на водяной бане. Горячий раствор фильтруют, остаток повторно извлекают этиловым
спиртом, подкисленным уксусной кислотой. Остается темно-красный осадок (т. пл. 225°С),
представляющий собой о.п-диацетатмеркур-лг-нитроанилин. Алкогольный экстракт вы-
паривают и остаток перекристаллизовывают из смеси спирта и уксусной кислоты. п-Аце-
татмеркур-л-нитроанилин— желтые иглы. Т. пл. 183°С.
Получение 2,4-диацетатМеркур-1-нафтнламнна [228]. К раствору 4,2 г а-нафтиламина
в 20 мл спирта и 20 мл ледяной уксусной кислоты прибавляют 50 мл кипящей воды. Полу-
ченный раствор соединяют с горячим раствором 10 г уксуснокислой ртути в 50 мл воды.
Сперва выпадает ярко-желтый осадок, который тотчас же переходит в белые кристаллы.
Их отсасывают. Выход 9,5 г. Сушат в вакууме над серной кислотой.
Получение 2-ацетоксимеркур-3-ннтро-1-нафтиламина [441]. 7,6 а З-нитро-Гнафтилами-
на в 100 мл спирта обрабатывают раствором 12,8 г уксуснокислой ртути в 50 мл воды,
подкисленным 2 мл уксусной кислоты. Смесь кипятят 1 час. После охлаждения и разбавле-
ния водой получают 14 г 2-ацетоксимеркур-3-нитро-1-нафтиламина в виде коричневых
микроскопических игл. Т. пл. около 200°С.
Получение 2,4-б«с-ацетоксимеркур-3-нитро-1-нафтиламина [441]. Из 3,8 г 3-нитро-
нафтиламина, 60 мл спирта, 13,2 г уксуснокислой ртути, 50 мл воды, 2 мл уксусной кис-
лоты при кипячении в течение 3 час. получают 12,5 г 2,4-бис-ацетоксимерку р-3-нитро-
1-нафтиламина.
Получение 4-ацетокснмеркур-6-иитрр-1-нафтиламина [444]. Кипящий раствор 5 г
6-иитро-1-нафтиламина в 30 мл уксусной кислоты прибавляют к раствору 8,6 г уксуснокис-
лой ртути в 15 мл уксусной кислоты и смесь кипятят 5 мин. Полученный осадок 4-ацетокси-
мерку р-6-нитро-1-нафтнламина плавится при температуре выше 400е С.
Получение уксуснокислой и-ацетамннофеннлртути [423]. В кипящий раствор 13,5 г
ацетанилида в 300 мл воды прибавляют небольшими порциями водный раствор 32 г уксус-
нокислой ртути и кипятят до тех пор, пока проба с едкой щелочью не будет давать белый
осадок. По охлаждении уксуснокислая n-ацетаминофеиилртуть выкристаллизовывается в
виде бесцветных пластинок. Перекристаллизация из этилового спирта. Т. пл. 218—220°С.
Получение 3-ацетоксимеркурбензидина [395]. В стакан (1 л), снабженный механиче-
ской мешалкой, помещают раствор 31,8 г (0,1 моля) уксуснокислой ртути в 150 мл ледяной
уксусной кислоты. Мешалку пускают в ход и медленно прибавляют раствор 18,4 г (0,1 мо-
ля) бензидина в 300 мл ледяной уксусной кислоты. Тотчас начинает выделяться осадок.
После смешения растворов смесь перемешивают около 10 мии. и фильтруют. Осадок промы-
вают сначала ледяной уксусной кислотой, затем эфиром и сушат на воздухе в темноте.
Выход 55 г. Т. пл. 144—146°С (с разложением). Вещество нерастворимо в воде, спирте, эфи-
ре и бензоле.
Так же получен [395] ацетоксимеркур-о-толидин, т. пл. 120°C (с разло-
жением).
Получение’этилового эфира 2-ацетатмеркурфеиилглнцина [447]. К раствору этилового
эфира фенилгл'ицииа в 50 мл метилового спирта приливают при перемешивании раствор
20 г (17,8 г равно 1 молю) уксуснокислой ртути в 50 мл воды. Происходит помутнение и вы-
деление желтоватого масла, исчезающего при дальнейшем перемешивании, в случае необ-
ходимости прибавляют немного метилового спирта. Через 10—15 мин. в реакционной смеси
отсутствуют ионы ртути. Затем ее охлаждают в охладительной смеси при потирании стек-
лянной палочкой, при этом выпадает плотный желтовато-белый кристаллический осадок,
количество которого увеличивается при дальнейшем хранении. После 24-часового^хранения
88
Ртутноорганические соединения. Лит. стр. 104—114
в холодильнике выделение большей части осадка закончено. Затем отсасывают, промыва-
ют от вьщелившейся уксусной кислоты холодной водой, сушат в вакууме над серной кис-
лотой. Выход 20 г (82%). Из маточника при дальнейшем хранении выделяется небольшое
количество менее чистого продукта. После двухкратной кристаллизации из хлороформа с
тщательным отсасыванием растворителя и сушкой при 60°С т. пл. 132®С с предварительным
размягчением при 128®С.
К раствору 2 г ацетата в 30 мл спирта прибавляют раствор 0,26 г хлористого натрия в
воде и сильно разбавляют водой. Полученный хлорид кристаллизуют из метилового спир-
та. Т. пл. 152,5 С.
Меркурирование 2-ариламиио-4-метил-5-карбэтокситиазолов [410].
Hg(O2CCH3)j
1 моль 2-ариламино-4-метил-5-карбэтокситиазола в смеси спирта и разбавленной ук-
сусной кислоты обрабатывают водным раствором 1,3 моля уксуснокислой ртути, подкислен-
ным уксусной кислотой. Через некоторое время выпадает осадок. Реакционную смесь ос-
тавляют на ночь. Осадок отфильтровывают и очищают повторным промыванием горячей
водой, спиртом и очень разбавленной уксусной кислотой.
Получение внутренней соли n-оксимеркурантраииловой кислоты [445].
NH2
I со;
сг
Hg+
Во взвесь 10 г антраниловой кислоты и 16 г окиси ртути в 100 мл воды пропускают силь-
ный ток водяного пара. Через 2—3 часа реакционная масса обесцвечивается. Если проба
растворяется в щелочи, реакция закончена. Для очистки ангидрид растворяют в ®/4 моля
1 У щелочи, если нужно, фильтруют, высаживают 6/« моля 1 N серной кислоты и осадок
отмывают водой от SO4". После сушки вес желтого аморфного продукта 23,5 г (96%).
Получение метилового эфира З-ацетатмеркур-2-амииобензойной кислоты [445]. 9 г
метилового эфира антраниловой кислоты приливают к теплому раствору 22,5 г уксусно-
кислой ртутн (1 моль) в 100 мл метилового спирта и 10 мл ледяной уксусной кислоты. На-
гревают 15 мин. до 50 °C, оставляют на ночь на льду. На следующий день осадок отсасывают,
промывают небольшим количеством метилового спирта, эфиром. Выход 22 г (90%^ Осталь-
ное получают из маточника при трехкратном разбавлении водой. Продукт — желтоватые
иглы — кристаллизуют из воды или спирта; вещество плавится с разложением при 180—
182®С.
Получение этилового эфира лг-ацетатмеркур-га-аминобеизойной кислоты [446]. Смесь 3 г
этилового эфира n-аминобензойной кислоты и 6,3 г уксуснокислой ртути нагревают на
глицериновой бане. При 130°С смесь становится жидкой и выделяется уксусная кислота.
При 160°С расплав затвердевает. Затем охлаждают, продукт измельчают и извлекают ме-
тиловым спиртом. Раствор упаривают и разбавляют горячей водой. По охлаждении выкри-
сталлизовываются блестящие иглы. Выход 2,3 г.
В растворе уксусной кислоты реакцию требуется вести 24 часа. Из тех же количеств
исходных веществ получается 3,3 г продукта. Оба метода дают небольшое количество
димеркурированндго производного, которое остается в виде нерастворимого осадка при
извлечении метиловым спиртом. Этиловый эфир л-ацетатмеркур-л-аминобензойной кисло-
ты плавится при 182° С, затем затвердевает и вновь плавится при 228° С.
Получение л-ацетоксимеркурсульфаниламида [451]. Раствор 17 г сульфаниламида в
разбавленной соляной кислоте обрабатывают раствором 40 г уксуснокислой ртути в 400 мл
воды, подкисленной уксусной кислотой. Тотчас образуется осадок. Смесь хорошо пере-
мешивают и оставляют на ночь. Осадок отсасывают, тщательно промывают горячей водой,
спиртом с очень разбавленной уксусной кислотой. Т. пл. выше 350°С. Выход 85%.
Согласно работе Гуха-Сиркар и др. [453], т. пл. моноацетоксимеркурсульфаниламида
(кристаллизация из разбавленной уксусной кислоты) 24б°С.
Введение ртути на место водородного атома (меркурирование)
89
Получение 3-ацетиламино-4-окси-5-оксимеркурфениларсиновой кислоты [321].
NHCOCH3
(HO)2OAs— 0Н
^HgOH
Взвесь 2,75 г З-ацетнламнно-4-оксифениларсниовой кислоты в 30 мл ледяной воды бы-
стро растворяют прибавлением И мл 2 N NaOH и обрабатывают охлажденным льдом рас-
твором 2,2 г уксуснокислой ртути в 20 мл уксусной кислоты и 20 мл воды. После 6—7-
дневного- хранения в темноте желтовато-коричневый осадок промывают водой и сушат.
От примеси непрореагнровавшей арснновой кислоты продукт освобождают встряхиванием
с метиловым спиртом. Выход почти количественный. Вещество не изменяется при нагрева-
нии до 300°С. Продукт получают бесцветным путем растворения его в холодном разбавлен-
ном едком натре, встряхиванием с животным углем и высаживанием разбавленной соля-
ной кислотой.
Если реакцию вести при нагревании на водяной бане, процесс заканчивается в течение
1 часа. Продукт получается окрашенным.
Получение димеркурированиого 4,4'-бис-диметиламинотрифенилацетонитрила [457].
М(СНз)2
I
Q-HgOH
;-C-<^^-N(CH3)2
CN ^HgOCOCHe
Раствор 7,1 г (0,02 моля) 4,4'-диметиламинотрнфенилацетонитрила в 150 мл этилацета-
та обрабатывают 3 мл уксусной кислоты и 12,8 г (0,04 моля) твердого ацетата ртути; смесь
умеренно кипятят с обратным холодильником в течение 70 мин. до растворения ацетата
ртути. Прн этом выделяется немного ацетата одновалентной ртути. Реакционную смесь
охлаждают до комнатной температуры, отфильтровывают от 1,56 г осадка [главным обра-
зом HggfCHsCOaJa], упаривают в вакууме до 30 мл н отфильтровывают от вновь выпавшего
осадка (0,81 г), затем разбавляют 130 мл метилового спирта и сразу приливают 30 мл 4 N
раствора КОН. Быстро образуется осадок слабо-кремового цвета. Его оставляют стоять
4 часа, отсасывают и сушат в эксикаторе. Выход нечистого продукта 8,55 г. Для очистки
4 г продукта растворяют в 100 мл метилового спирта, содержащего 1 мл уксусной кислоты.
Раствор фильтруют и фильтрат обрабатывают 16 мл 2 N раствора едкого кали в метиловом
спирте. Через 24 часа осадок собирают, промывают на фильтре небольшим количеством
метилового спирта и воды и сушат в эксикаторе. Вещество чернеет и разлагается при нагре-
вании выше 200*С.
Фотохимическое превращение меркурированного бис-диметиламииотрифеиилацетонит-
рила в меркурированную малахитовую зелень. Метоксимеркургидрокснмеркур-бпс-
диметиламинотрифеиилацетонитрил растворяют в метиловом спирте, содержащем 1%
уксусной кислоты (2 г в 100 мл). Раствор помещают в кварцевый сосуд, охлаждаемый
током воды для предотвращения кипения спирта и облучают кварцевой лампой. Вначале
бесцветный раствор быстро принимает синюю окраску, которая углубляется по мере уве-
личения продолжительности освещения. При обработке щелочью смесь дает белый осадок,
представляющий собой смесь исходного нитрила и гидроксимеркурцнаномеркур-бис-диме-
тиламинотрифенилкарбинола.
Ы(СНз)2
I HgOH
C-<^^-N(CH3)2
0Н ^HgCN
90
Ртутноорганические соединения. Лит. стр. 104—114
МЕРКУРИРОВАНИЕ АРОМАТИЧЕСКИХ КЕТОНОВ
Ароматические кетоны меркурируются в довольно жестких условиях:
при нагревании с уксуснокислой ртутью без растворителя до температуры не
ниже 150° С или при продолжительном нагревании с ацетатом ртути в ук-
суснокислом растворе.
Из чисто ароматических кетонов, не содержащих других, кроме карбо-
нила, функциональных групп, промеркурированы лишь несколько соеди-
нений. Бензофенон как при сплавлении его с уксуснокислой ртутью [71,
167, 471], так и при нагревании в уксуснокислом растворе с уксуснокислой
ртутью [472] (98°С, 6 час.) дает главным образом мономеркурированный в
орто-положение продукт. Согласно данным Димрота [71] и Гриньяра и Абель-
мана [471], при этом в небольшом количестве образуется полимеркуриро-
ванное соединение. По Огата и Тсусида [167,192а], продукт меркурирования
наряду с орто-изомером содержит в первом случае 5%, а во втором случае
15% мета-изомера .(анализ продукта заменой Н§СО2СН3-группы на Вг).
Флуоренон [167, 192а] как при кипячении с уксуснокислой ртутью без
растворителя (2,5 часа), так и в уксуснокислом растворе (10 час.) дает моно-
меркурированный только в положение 1 продукт (идентифицирован в виде
1-бромфлуоренона):
СО /СО\ /HgOCOCHs
+ дососи»)» -
При 6-часовом кипячении антрона с уксуснокислой ртутью в незначи-
тельном количестве выделена симметричная 9-дигидродиантрахинонилртуть
[473]. При нагревании антрона с сернокислой ртутью в среде моногидрата
серной кислоты при 50—70° С в течение 2 час. образуется также в незначи-
тельном количестве сернокислая 9-дигидроантронилртуть [473].
Бензатрон сплавлением с эквимолекулярным количеством уксуснокис-
лой ртути дает соединение строения [472]
О
HgOCOCHs
В менее жестких условиях — кипячением с уксуснокислой ртутью в
спиртовом или уксуснокислом растворе—промеркурирован ксантон, его
нитро- и аминопроизводные [474].*
Меркурирование антрахинона произведено многочасовым воздейст-
вием раствора окиси ртути в полифосфорной кислоте при 140—145° С
[475] (ср. [167]), а также окисью или основным сульфатом ртути в диметил-
сульфате (150° С) (образуется мономеркурированный в положение 1 про-
дукт, в последних случаях — наряду с а-и fJ-антрахинонсульфокислотами)
[475].
При меркурировании 1-антрахинонсульфокислоты атом ртути вступает
в первую очередь в «-положение несульфированного ядра. Второй атом рту-
ти вступает в то же ядро в пара-положение к первому [475]. Меркурирова-
ние ведут действием окиси ртути в ортофосфорной кислоте (1 час, 120—
125° С).
Аценафтенхинон, по данным Димрота [167, 168], не меркурируется.
Введение ртути на место водородного атома (меркурирование)
91
n-Ацетофенил арсиновая и n-ацетофенилстибиновая кислоты меркури-
рованы [108] в ароматическое кольцо действием окиси ртути без нагрева-
ние Строение продуктов не приводится.
О меркурировании ацетофенона и его производных в метильную группу и
индандиона см. стр. 58, об аминоарилалкилкетонах — стр. 84.
Получение о-хлормеркурбензофенона [71]. Сухую уксуснокислую ртуть нагревают
с 3 молями бензофенона при 150°С до отсутствия реакции на ион ртути. Теплую смесь
выливают в раствор хлористого натрия, охлаждают и взбалтывают с эфиром.После испаре-
ния эфира остается бензофенон и мономеркурированный продукт с незначительной приме-
сью димеркурированного производного. Бензофенон удаляют промыванием горячим лиг-
.ройном. Остаток кристаллизуют из спирта. Т. пл. о-хлормеркурбензофенона 167—168°С.
Выход 40%, считая на уксуснокислую ртуть (ср. [167]).
Получение 1-ацетатмеркурбензантрона [472]. Тонко измельченную и тщательно пере-
мешанную смесь эквимолекулярных количеств уксуснокислой ртути и бензантрона нагре-
вают в широкой пробирке на парафиновой бане при 171°С в течение 30 мин. Полученную
по охлаждении клейкую желтовато-коричневую массу нагревают до кипения в смеси спир-
та и уксусной кислоты, отфильтровывают горячей. После охлаждения выделяется желтое
вещество, которое отфильтровывают, промывают спиртом до тех пор, пока он еще окраши-
вается в желтый цвет, затем эфиром. Вещество, высушенное в вакууме над хлористым каль-
цием, плавится с вспениванием при 141—142°С. Выход около 60%.
МЕРКУРИРОВАНИЕ АРОМАТИЧЕСКИХ КАРБОНОВЫХ, СУЛЬФОНОВЫХ
И АРСИНОВЫХ КИСЛОТ И ИХ ПРОИЗВОДНЫХ
Атом ртути с трудом вступает в ароматическое ядро, содержащее другие
мета-ориентанты: карбоксил, сульфогруппу. Ароматические карбоновые кис-
лоты меркурируют сплавлением с уксуснокислой ртутью без растворителя
или многочасовым кипячением в щелочном растворе с окисью ртути или
уксуснокислой ртутью. При нагревании ртутных солей карбоновых кислот
(бензоата ртути до 170° С [711 или, согласно Песчи, до 130—140° С [476])
происходит замена на ртуть водорода ядра, что также является широко
применяемым методом синтеза ртутнозамещенных карбоновых кислот.
Димрот [71] и Песчи [477] считали, что при сплавлении ртутной соли
бензойной кислоты или при нагревании бензойной кислоты с ацетатом ртути
образуется только орто-изомер. Однако исследование [190] методом ИК-
спектроскопии бромбензойных кислот, полученных заменой HgX на бром
в продуктах меркурирования бензойной кислоты (130° С, 1,5 часа) пока-
зало, что при этом образуются все три изомера в отношении о: м : п = 57:
: 25 : 18. В данном случае нет аномалии ориентации (вступление ртути преи-
мущественно в орто-положение к карбоксильной группе), так как здесь
замещение происходит в соли карбоновой кислоты; карбоксилат же в от-
личие от свободной карбоксильной группы является орто-пара-ориентан-
том.
Согласно же данным Огата и Тсусида [192, 192а] при 10-часовом кипя-
чении в растворе ледяной уксусной кислоты уксуснокислой ртути с избыт-
ком бензойной кислоты образуются мета- и орто-меркурированные продук-
ты в отношении 4 : 1 (идентифицированы продукты замены HgOCOCH3-
группы на бром в виде бромбензамидов).
Не исключено также, что при длительном кипячении может происхо-
дить миграция HgX из одного положения бензольного ядра в другое (см.
стр. 51).
Бензойная кислота'меркурирована в орто-положение действием суль-
фата ртути [289] (условия не указаны).
Любым из перечисленных методов меркурируют о-толуиловую кислоту
[478], алкил- и алкокси- [479] и г алоидзамещенные карбоновые кислоты [478—
480],например, внутренний ангидрид оксимеркур-о-хлорбензойной кислоты
получен нагреванием ртутной соли о-хлорбензойной кислоты до 140—145° С
92
Ртутноорганические соединения. Лит. стр. 104—114
[4801; n-фторбензойную кислоту меркурируют при 12-часовом нагревании
в ледяной уксусной кислоте [178]. Нитробензойные куслоты меркурируют
сплавлением их с HgO, или их солей (например аммониевых) с уксуснокис-
лой ртутью или нагреванием их ртутных солей до 200—250° С [316,481, 482].
По последнему методу в орто-изомере на Hg заменяется' СОО [632].
Из эфиров карбоновых кислот меркурирован бензойнометиловый эфир
4—5-часовым кипячением с уксуснокислой ртутью в присутствии неболь-
шого количества уксусной кислоты [168, 297]. Ртуть вступает в орто-, а,
согласно Огата и Тсусида [167, 192а], также и в мета-положение.
В. 1-нафтойной кислоте при кипячении в течение 44 час. в щелочном рас-
творе с уксуснокислой ртутью ртуть занимает положения 5 и 8 [483].
О реакции фталевокислого натрия с ацетатом ртути и образовании при
этом ангидрида оксимеркурбензойной кислоты см. гл. IX (стр. 206).
Для введения ртути в ядро терефталевой кислоты требуется 240-часо-
вое кипячение терефталата натрия в водном растворе с уксуснокислой
ртутью или сплавление диэтилтерефталата с ацетатом ртути [484].
При нагревании меркурбензамида, меркурфталимида ртуть вступает
в ядро [77]. При меркурировании бензамида уксуснокислой ртутью 6-ча-
совым кипячением в уксуснокислом растворе или без растворителя ртуть
вступает только в орто-положение [167, 192а] (продукт идентифицирован
в виде о-бромбензамида).
Только в пара-положение вступает уксуснокислая ртуть в фенилуксус-
ную кислоту (в уксуснокислом растворе, 98° С, 12 час.) и в ее метиловый
эфир (7-часовое.кипячение компонентов). Продукты реакции идентифици-
рованы в виде п-бромбензойной кислоты, полученной в первом случае после
замены HgOCOCH3-rpynnbi на бром и окисления СНаСООН-группы, во втором
случае — после замены HgOCOCH3-группы на бром, омыления и окисления
[167, 192а]. Продуктом меркурирования бензонитрила уксуснокислой рту-
тью (7-часовое кипячение в ледяной уксусной кислоте) является меркури-
рованная N-бензоил-М-фенилмочевина, p-CH3COOHgC6H4NHCONHCOCeH5
[4851.
Сплавление равных количеств уксуснокислой ртути и бензолсульфоновой
кислоты до 105° С в течение 2 час. [190] дает смесь мономеркурированных
продуктов в отношении о : м : п — 24 : 61 : 15, как показывает исследова-
ние ИК-спектров смеси бромпроизводных, полученных заменой HgX-
группы на бром в смеси меркурированных продуктов.
При меркурировании п-толуолсульфоновой кислоты, осуществленном
кипячением кислоты (в течение часа) или ее натриевой соли (в течение не-
скольких часов) в водном растворе с уксуснокислой ртутью, ртуть вступает
в орто-положение к метилу [486].
Бензолсульфоновая кислота, галоиднитробензолсульфоновые кислоты
в этих условиях дают смесь аморфных нерастворимых и неидентифицируе-
мых веществ [486].
При действии раствора перхлората ртути, содержащего хлорную кис-
лоту, на ионообменную смолу из сульфированного полистирола (Dowex
50W-x8) (при 60, 80—85° С, а также при 25° С) наряду с нормальным обме-
ном Hg++ на Н+ происходит необратимое поглощение ионов ртути, обязан-
ное, возможно, реакции меркурирования молекул сульфокислоты полисти-
рола в ядро [487]
—СН—СН2—
SO3H+
—сн—сн2—
+ Hg++
+ 2Н+
Введение ртути на место водородного атома {меркурирование) 93
При меркурировании 4-карбоксифениларсиновой [318] кислоты (в ус-
ловиях мер курирования бензойной кислоты) ртуть вступает в мета-поло-
жение к карбоксилу.
О меркурировании оксикарбоновых, оксисульфоновых и оксиарсиновых
кислот см. стр. 77, аминокарбоновых, аминосульфоновых и аминоарсино-
вых кислот — стр. 84, л-ацетобензойной кислоты (в метильную группу)—
стр. 58, антрахинонсульфокислоты и л-ацетофениларсиновой и стибиновой
кислот— стр. 90—91, кислот гетероциклического ряда — стр. 96 и сл.
Меркурирование бензойной кислоты [476]. 32 г уксуснокислой ртутн сплавляют с
20 г бензойной кислоты нагреванием до 130—140°С (Димрот [71] рекомендует температуру
до 170°С) до отсутствия реакции на ион ртути со щелочью. Сплав охлаждают, измельчают
и кипятят около 2 час. с 70 г кристаллической соды. По охлаждении опять фильтруют,
пропускают ток СОа через раствор и выделившийся осадок обрабатывают углекислым ам-
монием, пока все не растворится. Из светло-желтого раствора выделяется аммонийная соль
меркурбеизойиой кислоты; при действии уксусной кислоты она образует внутреннюю соль
в виде аморфногб белого порошка. В кристаллическом виде эту внутреннюю соль можно
получить, если аммонийную соль перевести в натриевую и ее раствор обработать углекисло-
той. Продукт в основном представляет собой внутреннюю соль о-оксимеркурбензойной
кислоты [477]. Согласно Огата и сотр., при таком меркурировании бензойной кислоты
образуется и мета-, и п^ра-изомер (см. выше).
Получение метилового эфира о-хлормеркурбензойной кислоты [297]. В круглодонной
колбе растворяют при нагревании 20 г тонко измельченной уксуснокислой ртути в 20 г ме-
тилового эфира бензойной кислоты и 3 а ледяной уксусной кислоты, затем подвергают сла-
бому кипячению с обратным холодильником; через 3—4 часа реакция на ион ртути отрица-
тельная. При охлаждении раствор отфильтровывают от небольшого количества закисной
соли ртути и металлической ртути, от фильтрата отгоняют в вакууме уксусную кислоту и
избыток метнлбензоата. Остаток — 19 г желтоватого (при охлаждении) вязкого масла.
Затем приливают ацетон (около 200 мл) до тех пор, пока количество осадка не перестанет
увеличиваться. Аморфный продукт (5,6 г) представляет собой метиловый эфир диацетат-
меркурбензойной кислоты, который в обычных растворителях, кроме уксусной кислоты,
нерастворим.
В ацетоновом фильтрате находится мономеркурированное соединение, которое после
отгоики ацетона остается в виде светло-желтой некристаллизующейся смолы. Выход 13,3 г.
Растворением этого ацетата в водном спирте и высаживанием водным раствором хло-
ристого натрия переводят его в хлорид. Полученные 9,1 г метилового эфира о-хлормеркур-
бензойиой кислоты очищают растворением в этилацетате и высаживанием петролейным
эфиром. Т. пл. 162°С(с предварительным смоканием от 142°С). По данным Несмеянова
[488], т. пл. 184,5° С (кристаллизация из разбавленного спирта или этилацетата).
Меркурирование 1-нафтойной кислоты [483]. Получение смеси 5-оксимеркур-1-
нафтойной кислоты и ангидрида 8-оксимеркур-1-нафтойной кислоты. Раствор 172 г
(1 моль) 1-нафтойной кислоты в 210 мл 5 N едкого натра и 2 л воды помещают
в колбу, снабженную капельной воронкой, мешалкой с ртутным затвором и обратным
холодильником, соединенным с ловушкой, наполненной взвешенным количеством ед-
кого кали для улавливания выделяющейся углекислоты. При перемешивании смесь
нагревают до кипения. Постепенно прибавляют раствор 318 г (1 моль) уксуснокислой
ртути в 1,5 л воды и 60 мл уксусной кислоты. Тотчас же образуется белый зернистый осадок
и медленно выделяется газ. Смесь кипятят 44 часа. Через систему пропускают воздух, сво-
бодный от углекислоты. Количество поглощенной едким кали углекислоты равно 4,98 г.
Осадок кремового цвета в колбе обрабатывается 280 мл 5 N едкого натра. Часть его рас-
творяется, остаток чернеет из-за наличия в нем соединений закисной ртути. Вес нераство-
римой части 80 г. Щелочной фильтрат насыщают углекислым газом, образовавшийся оса-
док отфильтровывают, а затем вновь растворяют в растворе едкого натра и вновь высажи-
вают углекислотой; вес осадка 240 г. Из фильтрата при подкислении соляной кислотой
получают обратно 34 г 1-нафтойной кислоты. Высаженный углекислотой осадок представ-
ляет собой (как показывает исследование хлорнафтойных кислот, полученных заменой рту-
ти на хлор) смесь меркурированных в положения 5 и 8 продуктов с преобладанием первого.
Меркурирование терефталевой кислоты [484]. Получение внутреннего ангидрида 2-ок-
симеркуртерефталевой кислоты. Раствор 34 г (0,2 моля) терефталевой кислоты в небольшом
избытке раствора едкого натра смешивают с раствором 44,7 г (0,2 моля) окиси ртути в не-
большом избытке уксусной кислоты и 10 мл ацетата натрия и кипятят с обратным холодиль-
ником 240 час. К этому времени смесь не дает реакции на неорганическую ртуть и полно-
стью растворяется в растворе едкого натра. Осадок отфильтровывают, промывают и сушат,
вес его 102 г. Из фильтрата при подкислении получают 10 г терефталевой кислоты. Мер-
курированный продукт перемешивают в течение часа с 1 л конц. раствора аммиака и 500 мл
воды. Нерастворившийся осадок отфильтровывают и еще раз обрабатывают таким же обра-
зом. Оба экстракта подкисляют уксусной кислотой. Белый осадок отфильтровывают, педе-
94
Ртутноорганические соединения. Лит. стр. 104—114
носят в большой стакан и растворяют в очень небольшом избытке горячего насыщенного
раствора карбоната натрия. Раствор отфильтровывают от небольшого количества нерас-
творимого вещества, охлаждают и обрабатывают углекислотой. При этом высаживается
смесь мономеркурированной кислоты и небольшое количество димеркурнрованного про-
дукта. Смесь перемешивают около часа с 500 мл насыщенного при 20Q С раствора
уксуснокислого натрия и 1 л воды. При такой обработке растворяется мОномеркурирован-
иый продукт. Фильтрат подкисляют 40 мл ледяной уксусной кислоты, фильтруют, промы-
вают водой и спиртом и сушат в вакууме над пятиокисью фосфора; выход чистого ангид-
рида 2-оксимеркуртерефталевой кислоты 18 г. Дополнительные количества продукта могут
быть получены из всех остатков и из маточника.
Меркурирование диэтилтерефталата [484]. Смесь 40 г (0,18 моля) диэтилтерефталата,
57,5 г (0,18 моля) уксуснокислой ртути и 2 мл ледяной уксусной кислоты нагревают 70 час.
при 117°С (температура кипения н-бутилового спирта). Проба на неорганическую ртуть от-
рицательная. Вязкую массу подвергают перегонке с паром в течение 24 час. Остаток об-
рабатывают раствором аммиака, отфильтровывают и подкисляют. Меркурированную те-
рефталевую кислоту очищают по описанному выше методу.
Меркурирование n-толуолсульфоновой кислоты [486]. 190/ (1,1 моля) п-толуолсульфоно-
вой кислоты растворяют в 750 мл воды и 35 мл уксусной кислоты, раствор кипятят и филь-
труют. Приготовляют такой же раствор 320 г уксуснокислой ртути. Оба раствора нагре-
вают до кипения, сливают вместе и кипятят в течение часа. К этому времени проба, раз-
бавленная равным объемом воды, с едким натром должна давать прозрачный, раствор. Мер-
курирование закончено. Смесь фильтруют горячей; осадок (81 г) отбрасывают. Фильтрат по
охлаждении выделяет 135 г кристаллического продукта. Его извлекают водой в аппарате
Сокслета. Состав твердого воздушносухого осадка соответствует формуле
СН3 СНз
| HgOH | Hg+
(вероятно:
I I
SO3H.H2O so;
МЕРКУРИРОВАНИЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ
Степень легкости замены водорода на ртуть в гетероциклических соеди-
нениях варьирует в широких пределах. Очень легко — действием смеси су-
лемы и уксуснокислого натрия на холоду в водном растворе, меркурируют-
ся в a-положение суперароматические гетероциклы — фуран, тиофен,
селенофен и их производные, имеющие хотя бы одно свободное а-положение.
Так меркурированы фуран [489], 2-иодфуран [4891, сильван [489, 4901
и другие 2-алкилфураны [491—4931, р-иодсильван [489], фурфуриловый
спирт [490, 494, 4951, 2-метоксифуран [496, 497} и 2-метилмеркаптофуран
[497], 2-метилбензофуран [4981, 4,5,6,7-тетрагидро-3,6-диметил-[4991,
2-фенил-[499, 5001 и 2-этил-[500] бензофуран, 2,4-диалкил-[498, 5001, 2,3,4-
и 2,3,5-триалкилфураны [498, 5001, 2-а-ацилалкилфураны [501], эти-
ловый эфир пирослизевой кислоты [494] (в спиртовом растворе), метиловый
эфир З-метил-4-фур ил карбоновой кислоты [502], фурфурол [5031.
Фурфурол меркурируется также действием сулемы в водном растворе
при кипячении [494]. При меркурировании фурфурола действием уксус-
нокислой ртути в уксусной кислоте при 140—150° С [504] получают моно-
меркурированный в a-положение продукт наряду с другим неидентифици-
рованным веществом [503].
Ряд 2,5-диалкилзамещенных фуранов [500, 505] меркурируют в fJ-no-
ложение действием сулемы и ацетата натрия. Однако введение ртути таким
образом в P-положение при комнатной температуре происходит медленнее,
чем в а-положение [5001, и обычно его проводят при нагревании [500, 5053
или при более долгом стоянии.
Метиловый эфир 5-бром-2-фуранкарбоновой кислоты меркурируют в по-
ложение 4 сплавлением с уксуснокислой ртутью при 160° С [5061.
Введение ртути на место водородного атома (меркурирование) 95
2,5-Диметилфуран гладко дает мономеркурированный продукт при дей-
ствии., сулемы при комнатной температуре в этиловом спирте в присутствии
меньйе обычно применяемого количества уксуснокислого натрия [500].
Мономеркурированный в ^-положение (не содержащий других замести-
телей); фуран может быть получен только косвенным путем — нагреванием
смешанной соли ацетата (а-фуроата) ртути (см. гл. IX, стр 208). При дей-
ствии на фуран и диацетат фурфурола раствором избытка уксуснокислой
ртути на холоду образуется соответственно тетраацетатмеркурфуран
[5071 и тримеркурированное производное диацетата фурфурола, изолиро-
ванное в виде хлорида [508]
ciHg Hgci
X
CIHg ° СН(ОСОСНз),
Тетрамеркурированный фуран получается при кипячении в течение часа
водного раствора эквимолекулярных количеств 5-бензамидометилфуран-2-
карбоновой кислоты и сулемы [509] (см. также стр. 208).
Фуран меркурируют также ртутной солью тринитрометана (в эфире),
выход мономеркурированного в a-положение продукта 20—25% [147].
Фуран и сильван меркурируют цианатом ртути в присутствии ацетата нат-
рия при хранении на холоду в водно-спиртовом растворе [510]. Фуран об-
разует при этом смесь моно- и димеркурированных продуктов, сильван —
только 5-моно'меркурпроизводное [5101.
Тиофен в a-положение может быть промеркурирован даже действием
ртутноорганических солей (см. гл. XII, стр. 233). Меркурирование суле-
мой в присутствии ацетата натрия в водном растворе на холоду, впервые
примененное Фольгардом [511] к тиофену, дает в качестве главного продук-
та а-хлормеркуртиофен [511, 5121 наряду с небольшим количеством 2,5-
дихлормер кур тиофена [512]. При меркурировании тиофена этой же мерку-
рирующей смесью, но при кипячении [513] или уксуснокислой ртутью на
холоду [514] образуется 2,5-димеркурированное соединение. При отделе-
нии тиофена от содержащего его бензола, по Димроту [149], меркурирова-
нием на холоду уксуснокислой ртутью (медленнее сульфатом или нитратом
ртути) тиофен выделяется в виде димеркурированного продукта, которому
Димрот приписал строение
/СА
HOHg S HgOCOCHs
(ср. [514, 515]).
Дёйствие на тиофен сернокислой ртути в водном растворе дает при
встряхивании на холоду тримеркурированный продукт [514] (см. также
[516]).Действие сульфата или нитрата ртути приводит к аналогичным продук-
там, но требует большего времени [149]. Действие на тиофен цианата
ртути в присутствии ацетата натрия (водно-спиртовый раствор на холоду)
дает смесь моно- и димеркурированного продуктов [510].
Для того чтобы ввести ртуть в Р-положение тиофена, необходимы более
жесткие условия. Тетраацетатмеркуртиофен получен из тиофена и 4 мо-
лей уксуснокислой ртути при нагревании на водяной бане (согласно
Паолини [517] —на холоду) в уксуснокислом растворе [518]. Тетрааце-
татмеркуртиофен с меченым атомом серы получен из а-тиофенкарбоновой
кйслоты действием окиси ртути в уксусной кислоте [519].
96 РтутноорганичесНие соединения. Лит. стр. 104—114
Действием сулемы и уксуснокислого натрия на холоду в водном или в
водно-спиртовом растворе меркурируют в a-положение многочисленные
производные тиофена с незамещенным a-положением. Если свободны оба
a-положения, то легко образуются и димеркурированные соединения. Так
меркурированы, например: 2-тиотолен [511, 512, 520—5231, 3-тиотолен
[511-т513], 2-этилтиофен [523—525], 3-этилтиофен [521], 2-пропилтиофен
[523], 3-изопропилтиофен [5111, 2-третичнобутилтиофен [525], 2-изоамил-
тиофен [523], 2-бензилтиофен [523], 2-фенилтиофен [512, 5261, 3,4-тиоксен
[512, 527], 2,3-тиоксен [523], 2,4-тиоксен [523], З-метил-2-этилтиофен [528],
2-метил-З-этилтиофен [528], 3,4-диэталтиофен [529], 2,4-дифенилтиофен
[530—532], 2,3-дифенилтиофен [533], разнообразные монохлор- [534],-бром-
1512, 513, 535—537],-иод-[512,522,538,5391 и полихлор-[5341, -бром-[535—
5361, -иод-[522, 537, 538] замещенные тиофены и гомологи тиофена —
2-метокситиофен [5391, 2-метоксиметилтиофен [5401, 2-тиеиилэтиловый эфир
[5241, 2-тиенилметилкарбинол [541], 2-тиофенкарбинол [542], 2-ацетилами-
нотиофен [543], (монозамещенный продукт, действием ацетата ртути—поли-
меркурированные соединения [544]), 2,2'-дитиенил-[545], 2-тертиенил-[547],
5-метил-1547], 5-бутил- [54612,2'-дитиенилы, 1-метил-а-тертиенил [547],
2-тиенилметил- и 2-тиенилэтилсульфиды [524], 2-тиенилэтиловый эфир
[524], тиофен-2-карбоновая кислота [201].
Известны единичные случаи меркурирования производных тиофена дей-
ствием сулемы в спиртовом растворе и в4 отсутствие ацетата натрия. См.,
например, меркурирование 2-ацетиламино-4-бромтиофена [544], 2-ацетил-
аминотиофена ,[544] (монозамещенный продукт).
Так же в очень мягких условиях (ср. меркурирование полисти-
рола, стр. 66) происходит меркурирование поливинилтиофена. Уже через
1—2 мин. при сливании теплых бензольных растворов поли-а-винилтиофена
и диизобутирата ртути количественно образуется продукт меркурирования,
содержащий атом ртути на каждое звено поли-а-винилтиофена [173].
Некоторые производные тиофена с замещенными обоими а-положениями
могут быть промеркурированы действием сулемы и ацетата натрия, но уже
при более длительной выдержке в водно-спиртовом или спиртовом растворе
или при кипячении (см., например, меркурирование 2,5-диэтилтиофена
Д548], З-метил-2,5-диэтилтиофена [5481, 2-метокси-5-метилтиофена [5401,
5'-метил-[525], 5'-этил-[5491, 2',3', 1,2-циклогептилтиофена, 2-метил-
и 2-пропил-4,5,6,7-тетрагидробензотиофена [5501). а, а'-Дифенилтиофен,
вопреки данным Штейнкопфа [523], ни в названных условиях, ни при ки-
пячении с уксуснокислой ртутью в уксусной кислоте не меркурируется
[5331.
Замещенные в a-положение производные тиофена лучше меркурируются
действием уксуснокислой ртути в спиртовом, уксуснокислом или водном
растворе, что, особенно при наличии электроположительных заместителей,
благоприятствует образованию полимеркурированных продуктов. Так,
например, меркурирование 2,5-тиоксена уксуснокислой ртутью дает ди-
меркурированное производное [513], а меркурирование действием сулемы
и ацетата натрия — мономеркурированный продукт [512].
Действием уксуснокислой ртути меркурированы галоидзамещенные
производные тиофена [512,534,545], 2-метил-5-этилтиофен [551], 5,5'-
диметил-4,4'-диэтил-2,2'-дитиенил [529]. Так же меркурированы нитро-
производные, тиофена 1529, 552, 553].
Действием уксуснокислой ртути на холоду меркурированы 3-метил-
тионафтен [5541, 3-фенилтионафтен [554], 5-метил-З-фенилтионафтен [5541,
2-метил-З-фенилтиофен [555, 556], З-метил-4-фенилтиофен [5561, 2-метил-
4-этил тиофен [5571, 4-метил-2-(1 -оксицикл огексил)тиофен [558], 1,4,4-
триметил-4,5,6,7-тетрагидроизотианафтен [5591, тиофен-2-карбоновая
Введение ртути на -место водородного атома (меркурирование)
97
кислота [553], 2-ацетиламинотиофен [544], о-(2-теноил)бензойная кислота
[560], 2-алкилтиофены [5611; 2-этилтиофен при нагревании на водяной бане
в течение часа с ацетатом ртути в 50 %-ной уксусной кислоте дал монортут-
ное соединение 1561]. 2,4-Дихлор тиофен при нагревании в течение 5 час.
с ацетатом ртути в водной уксусной кислоте дал монортутное соединение
[561а]. Но тионафтен с уксуснокислой ртутью в спиртовом растворе на хо-
лоду дает уксуснокислую [J-тионафтенилртуть [5621, и при нагревании как
с уксуснокислой ртутью, так и с сулемой в присутствии ацетата натрия
образуется димер кур ированное соедйнение [5621. Согласно же другим дан-
ным [563], при нагревании тионафтена с уксуснокислой ртутью в спирто-
вом или уксуснокислом растворе образуется мономеркурированный про-
дукт. Выделение тионафтена из нафталина меркурированием производят
кипячением с ацетатом ртути в метиловом спирте в течение часа [564].
2,3-Дифенилтиофен при 3-часовом кипячении с уксуснокислой ртутью
в уксусной кислоте образует смесь RHgO2CCH3 и RaHg-соединений [5331.
Применяют также сплавление с ацетатом ртути; так меркурированы,
например, 5-бензотиенон, иодзамещенные 5-бензотиеноны [565]. 5-Бензо-
тиенон при кипячении с сулемой и уксуснокислой ртутью в ледяной уксус-.
ной кислоте дает мономеркурированный в положение 2 продукт, при сплав-
лении с уксуснокислой ртутью — смесь моно- и днмеркурированных соеди-
нений, превращаемую в димеркурированный продукт кипячением с ацетатом
ртути в метилцеллозольве [5651.
В условиях меркурирования тиофена (действием сулемы и ацетата нат-
рия) меркурированы в а-положение селенофен [520, 566], 3-метилселенофен
1567], 2,4-диметил селенофен 1567], 2',3,4-триметилселенофен [568], 2,4-
дифенилселенофен [530, 531].
Для монохлормеркурпроизводных селенофена, 2-метилселенофена, 2,5-
диметилселенофена даются температуры плавления [520] без описания
метода синтеза.
Как и в случае тиофена, при меркурировании селенофена уксуснокислой
ртутью в уксусной кислоте или водным раствором основного сульфата ртути
выделено димер кур ированное соединение [514], в последнем случае — двой-
ное соединение димеркурированного продукта и сернокислой ртути.
Для введения ртути в ^-положение и в случае селенофена требуются бо-
лее жесткие условия (см., например, [520]).
Очень легко меркурируется пиррол. При меркурировании пиррола из-
бытком уксуснокислой ртути на холоду образуется тетраацетатмеркурпир-
рол [507] (ср- действие на пиррол сулемы, приводящее к образованию
N—Hg-соединений; гл. XVI, стр. 380).
При меркурировании пиррола цианатом ртути (водно-спиртовый рас-
твор на холоду) образуется димеркурированное производное [510].
Образующиеся при действии сулемы на пиррол и замещенные пирро-
лы ртутные соединения применены для разделения продуктов расщепле-
ния гемина [569].
В мягких условиях уксуснокислой ртутью (на холоду или при нагрева-
нии, в водном или спиртовом растворе) меркурируются в пиррольное кольцо
N-фенилпиррол [5701 (с образованием в зависимости от относительных
количеств реагентов ди- и тетрамеркурированного соединения), индол
[5711 (продукт—2,3-диацетатмеркуриндол), скатол [571], метилкетол [571]
(продукты—мономер кур ир ованные соединения) и другие замещенные (при
азоте и а-углероде) индолы [572], карбазол и тетракарбазол [573] (с об-
разованием R2Hg, с избытком уксуснокислой ртути — тример кур ирован-
ные продукты), N-этилкарбазол [5741.
О меркурировании дифенол- и дирезорцинизатина см. при меркуриро-
вании фенолов.
98 Ртутноорганические соединения. Лит. стр. 104—114
1-Фенил пир азол при 15-минутном нагревании на водяной бане с уксус-
нокислой ртутью в уксусной кислоте образует 4-ацетоксимеркур-1-фенил- .
пиразол [5751; 5,5'-диметил-1,Г-дифенилпиразолил дает 4,4'-бнс-ацеток-
симеркурсоединение в этих же условиях при получасовом нагревании
[576], а 4,4'-бш> ацетоксимер кур-1 ,Г,5,5'-тетрафенил-3,3'-бипиразолил
образуется уже при 5-часовом кипячении реакционной смеси [576].
При обработке антипирина и других алкилзамещенных 1-арил-5-пира-
золонов спиртовым раствором уксуснокислой ртути при комнатной темпера-
туре или при 60° С происходит меркурирование в положение 4 пиразоло-
нового кольца, в бензольное ядро и присоединение HgX и OR по двойной
связи 15771. Например, из 1-фенил-2,3-диметил-5-пиразолона получен про-
дукт строения [5771
/HgOCOCHa
(CH8COOHg)aCeH3—2 g| HgOCOCH3
N-C<
I XCH3
CH3
Если же атом водорода в положение 4 замещен бромом, метилом или ди-
метиламиногруппой, то даже при продолжительном кипячении не проис-
ходит замещения на ртуть во всей системе. В соединения с Вг и NHa в по-
ложении 4 ртуть может быть введена лишь при 160° С. З-Фенил-5-пиразолон
и 1-фенил-3-метил-5-хлорпиразол при нагревании с уксуснокислой ртутью
в метиловом спирте лишь меркурируются, а не присоединяют соли ртути по
двойной связи [577] (см. также гл. VI, стр. 163).
При кипячении антипирина в водном растворе с ClHgNH2 или с уксус-
нокислой ртутью получено соединение с одним HgX-остатком в молекуле,
которому Раньо [5781 приписывает строение меркурированного в положе-
ние 4 продукта. Соединению, содержащему два HgOCOCHg-остатйа
в молекуле, полученному при 15-минутном нагревании антипирина с
уксуснокислой ртутью при 150° С, он приписывает строение меркури-
рованного в пиразольном кольце (в положение 4) и в бензольном ядре
[5781.
Описаны ртутные производные пиразолона с N—Hg-связью [5791, с
О—Hg-связью [580] (см. также гл. XVI, стр. 362, 380).
Тиазол меркурируется многочасовым кипячением с избытком уксусно-
кислой ртути в водной уксусной кислоте [581); продукт—тримеркуриро-
ванное соединение. Мономеркурированный тиазол получен из 2-аминотиа-
зола диазометодом (см. гл. VII, стр. 185).
В более мягких условиях происходит меркурирование в положение 5
2-амино- и 2-амино-4-метилтиазола [582] (действие уксуснокислой ртути
в водном растворе, на холоду), 2-ацетиламинотиазола [583] и 2-ацетил-
амино-4-арилтиазолов. [582] (непродолжительное нагревание на водяной
бане) и 2-ацетиламинотиазола (на холоду) [582]., Последние меркури-
рованы также действием сулемы и ацетата натрия в водно-спиртовом или
спиртовом растворе при непродолжительном кипячении [584,5851. В продук-
тах меркурирования уксуснокислой ртутью на основании их малой раствори-
мости предполагают [582] наличие наряду с С—Hg- также N—Hg-связи.
2-Ацетиламино-4-гексилтиазол при нагревании в течение 30 мин. с аце-
татом натрия и ацетатом ртути в уксусной кислоте до 125° С дает 2-ацет-
амино-4-гексил-5-тиазолилмеркурацетат [586].
О меркурировании ариламинотиазолов в ароматическое ядро см. при
меркурировании аминов.
Введение ртути на место водородного атома (меркурирование) 99
З-Арилсидноны меркурируются в положение 4 действием уксуснокислой
ртути или сулемы и уксуснокислого натрия в водном, водно-метиловоспир-
товом или водно-ацетоновом растворе при кипячении [587] или даже
да холоду [588]. Продукты представляют собой смесь ди-4-(3-арилсвднон)-
ртути и соответствующего RHgX-соединения,
А г—N---С—HgX
. 4-^СО
... Q
Меркурирование 3-пиридилсиднона сулемой в присутствии ацетата нат-
рия в водном спирте приводит в зависимости от температуры и продолжи-
тельности реакции к мономеркурированному в положение 4 продукту,
соответствующему К2Н§-соединению, или к комплексу 3-пиридилсиднбна
с сулемой [589]. Найдены условия взаимного превращения этих продуктов
[589].
Кипячением 2-фенил-а,р-нафтотриазола с уксуснокислой ртутью в ле-
дяной уксусной Кислоте, после отгонки растворителя и последующего на-
гревания до 145°С, получают тримеркурированное производное [590]. Опи-
саны попытки и меркурирования 2,6-диметил-4-пирона действием сулемы
в спиртовом растворе при кипячении [591].
В условиях меркурирования супер ароматических гетероциклов (дей-
ствие раствора сулемы в присутствии ацетата калия на холоду) меркуриро-
ван А2-хромен с образованием 2-хлормеркур-А2-хромена [592].
При кипячении бензо-1,4-дитиациклогексадиена-2,5 с сулемой в спир-
товом растворе получен с хорошим выходом мономеркурированный в поло-
жение 2 продукт [593]
0\3-HgCl
Действие же уксуснокислой ртути приводит к более сложным продук-
там, из которых не выделено чистого вещества [5931.
Из всех гетероциклических соединений пиридин меркурируется с наи-
большим трудом: при нагревании его с уксуснокислой ртутью в запаянной
трубке до 180°С (согласно Свэни и др. [594]—до 155° С) в течение 2,5 час.
Согласно данным Закса и Эберхартингера [595], при этом образуются моно-
и димеркурированные в p-положение соединения; Мак-Клилэнд и Вильсон
[5961 в этих условиях получили лишь p-хлормеркурпиридин (после добав-
ления хлористого натрия). Одновременно образуется двойное соединение
пиридина с хлористой ртутью [597].
Согласно данным Свэни и сотр. [594], оптимальные условия меркури-
рования пиридина уксуснокислой ртутью (выход мономеркурированного
в P-положение продукта 49,5%) следующие: нагревание 2,5 часа до 155°С
в присутствии небольшого количества воды, что предотвращает образова-
ние полимеркурированных продуктов.
Интересно подчеркнуть, что вступление ртути в пиридин подчиняется
обычным законам ориентации электрофильного заместителя, вступающего
в пиридин, в противоположность тому, что имеет место, например, для мер-
курирования нитробензола уксуснокислой ртутью.
З-Пиридилмеркурхлорид получен диазометодом [598]; изучены свой-
ства 3-пиридилмеркурхлорида [599]. 2-Пиридилмеркурхлорид и 6-хлор-
3-пиридилмеркурхлорид получены через соответствующие сульфиновые
кислоты [5991.
100 Ртупгноорганичеекие соединения. Лит. стр. 104—114
а-Пиколин с уксуснокислой ртутью (150° С) дает мономеркурированное
в положение 5(?) соединение (выход 61%) [5941.
y-Пиколин дает мономеркурированный в P-положение продукт при
нагревании в течение 6 час. с водным раствором уксуснокислой ртути до
110° С в запаянной трубке [600].
Пиридиноксид меркурирован нагреванием с уксуснокислой ртутью в при-
сутствии уксусной кислоты в течение 2—3 час. до 100—130° С. Продуктом
реакции является не меркурированное в положение 4 соединение [601], а
смесь моно- и димеркурированных в a-положение продуктов [602, 603].
Для получения только мономеркурированного (в a-положение) продукта
отношение пиридиноксида и ацетата ртути должно быть 6 : 1 [602]. При
меркурировании действием сернокислой ртути в присутствии большого ко-
личества серной кислоты главным продуктом реакции является замещенное
в ^-положение соединение [603].
О меркурировании ксантона см. при меркурировании ароматических
кетонов (стр. 90).
Активирование молекулы введением заместителей первого рода в случае
пиридина сказывается особенно сильно: 2-амино-[604—605] и 2-оксипроиз-
водные пиридина меркурируются в водном или спиртовом растворе на хо-
лоду. Из 2-аминопиридина при этом образуется димеркурированный в
положения 3 и 5 продукт [605]. 2-Ацетиламинопиридин при слабом на-
гревании с уксуснокислой ртутью в метиловом спирте [605] или с окисью
ртути [6061 прямо образует симметричное соединение вероятного
строения.
CH3COHN—NHCOCHs.
N N
2-Аминопиридин[5941, 2- [605,607], 3- [6071 и 4- [607]оксипиридины мерку-
рируются также при кипячении в воде (продукты для оксипиридинов соответ-
ственно : 3,5-диацетатмеркур-2-оксипиридин, 2-ацетатмеркур -3-оксипир идин,
З-ацетатмеркур-4-оксипиридин [607]). Но 4-аминопиридин меркурируется
в положения 3 и 5 при нагревании с окисыб ртути и уксусной кислотой
до 140° С в течение 28 час. [608].
Хинолин [609, 610], изохинолин [609,6111, метилхинолины [609,610,
612] меркурируются также в довольно жестких условиях: при нагревании
с уксуснокислой ртутью до 150° С в течение нескольких часов. Хинолин
дает при этом два мономеркурированных в положения 3 и 8 продукта и ди-
меркурированное соединение. Ртуть в изохинолине вступает в пиридиновое
кольцо в ^-положение к азоту. Но уже наличие в молекуле хинолина не
только гидроксила, но даже и карбоксила и сульфогруппы делает возмож-
ным вести меркурирование в более мягких условиях: в водном растворе на
холоду (8-оксихинолин [9, 613—615] и его бром- и дибромзамещенные 1613])
или при кипячении (хинолин-2-окси-8-карбоновая [615], хинолин-8-окси-
5-сульфоновая [615], о-фенилцинхониновая [615—617], хинолин-8-сульфо-
новая кислоты [615]).
Хинолиноксид [601] и 6-метил-, а также 4-бром- [463] хинолиноксид
при нагревании до 130—140° С в течение 1—4 час. замещаются уксуснокис-
лой ртутью в бензольное кольцо в положение 8. В 8-бромхинолиноксиде
при этих условиях замещение происходит е пиридиновом ядре [463].
Меркурирование 5-нитро-8-оксихинолина уксуснокислой ртутью при
90°С, pH 3 и 5,2 в течение 12 час. приводит к выделению продукта в виде
комплексного фенолята ртути, меркурированного в положение 7 [3091.
Имеются патентные данные о меркурировании акридина [618].
Введение ртути на место водородного атома (меркурирование) 101
Натриевая соль 2,6-диокси-4-аминодигидропиримидинуксусной кисло-
ты очень легко взаимодействует с солями ртути с образованием меркуриро-
ванного по углероду продукта [6191.
Описано меркурирование феносафранина и его гомологов [6201.
Относительно легко, при кипячении в водном растворе с уксуснокислой
ртутью, происходит замена водорода в положении 8 в имидазольном цикле
на ртуть в молекулах пуриновых оснований (метил кофеин а, тетрахлорме-
тилксантина, метилтрихлоркофеина) [6211.
При многочасовом кипячении метиленовой сини с 5 молями уксусно-
кислой ртути в водном, подкисленном уксусной кислотой растворе полу-
чают в незначительном количестве мономеркурированный продукт (строе-
ние не установлено) [6221. Бензольное производное лейкооснования мети-
леновой сини при кипячении в течение 20 мин. с 3 молями уксуснокислой
ртути в водно-спиртовом, подкисленном уксусной кислотой растворе обра-
зует мономеркурированный продукт с выходом 52%. Положение ртути в
нем не определено [6221.
Меркурирование тиофена [511, 512]. В сосуде большого объема сливают вместе 10 г
тиофена, 100 а этилового спирта, 100 г насыщенного на холоду водного раствора сулемы и
200 г 33%-ного водного раствора уксуснокислого натрия и при частом встряхивании остав-
ляют стоять на 4—5 дней. Выделившийся белый кристаллический осадок смеси моно- и ди-
меркурированного соединения кристаллизуют из спирта (лучше нз ацетона [512]). При
этом выкристаллизовывается хлористая а-тиенилртуть. Т. пл. 183°С.
Нерастворимый осадок представляет собой димеркурированное соединение (разлагает-
ся около 275°С, не плавится) [514].
В. этих условиях из 85 г тиофена при 6-дневном хранении получено 217 г моно- и 18 г
димеркурированного продукта [512].
Меркурирование фурана [489]. К раствору 540 г хлорной ртути в 8 л воды прибавляют
раствор 1088 г уксуснокислого натрия в 4 л воды и затем 136 г фураиа в 850 мл спирта. По-
чти тотчас начинает выделяться осадок, образование которого заканчивается через 2 дня,
после чего его отсасывают и экстрагируют кипящим спиртом. Фильтруют горячим. В оса-
дке—2,5-дихлормеркурфуран (164 г). Охлажденный спиртовый фильтрат дает 202 г (33,6%)
хлористой а-фурилртути. Т. пл. после кристаллизации из спирта 151°С.
Совершенно в таких же условиях меркурируются гомологи и производные тиофена н
фураиа (см. стр. 94, 96).
Т. пл. 2-хлормеркур-5-метилфурана 127°С [490] (134° С [489]); 5-метил-3-иод-2-хлор-
меркурфурана 193,5°С [489]; 2-метил-5-хлормеркуртиофена 204°С [512] (в предварительно
нагретой до 190 °C бане, кристаллизация из спирта [512]); 5-хлормеркур-2-хлортиофена
213° С [514]; 2-хлормеркурселенофена 201—202° С (с разложением) [566].
Меркурирование сильвана циановокислой ртутью [510]. а. Получение циано-
вокислой ртути [623]. 27,25 г тонко измельченной сулемы растворяют в 600 мл
сухого эфира и прибавляют 33 г чистого цианата серебра (теорет. количество 30,0 г). В те-
чение I часа встряхивают и оставляют стоять на 1 день. Раствор не содержит хлора.
Осадок, состоящий из AgCl, AgOCN и Hg(OCN)2, исчерпывающе извлекают эфиром.
Получают 27 г чистой Hg(OCN)a. Препарат следует, хранить в сухом месте, лучше в ва-
кууме.
б. Получение циановокислой 5-метилфурил-2-ртути. Рас-
твор 24,9 г циановокислой ртути в 440 мл воды смешивают с раствором 33 г ацетата натрия
в 150 мл воды. В полученный раствор вливают 60,0 мл сильвана. Спустя короткое время
выпадает желтоватый осадок. Сосуд, в котором ведут реакцию, каждый день открывают.
Через 5 дней увеличения давления ие заметно. Через 8 дней осадок промывают водой и
сушат в вакууме. Вес 26,0 г (теорет. количество 28,3 г). Осадок извлекают (200 мл эфира),
при этом остается 2,6 г серовато-коричневого вещества. Эфирный раствор упаривают на
водяной бане до объема 50 мл\ при этом выделяется осадок в виде табличек. По охлаждении
в холодильнике осадок отфильтровывают и сущат в вакууме. Вес циановокислой 5-метнл-
фурил-2-ртути 21,3 г Т. пл. 114—115° С.
Получение 2-метокси-3,5-тиеиил-б'«С-меркурхлорида [539]. Раствор 0,1 мл 2-метокси-
тиофена в 1,0 ли этанола встряхивают 20 час. с 8,5 г насыщенного водного раствора суле-
мы и 1,7 г 33%-иого водного раствора ацетата натрия. Тяжелый белый осадок отфильтро-
вывают, промывают водой и сушат. Вес 0,47 г. После двух кристаллизаций из смеси ди-
метилформамида с этанолом получено 0,38 г (65%) бесцветных иголок. Т. пл. 268—270® С
(с разложением).
102
Ртутноорганические соединения. Лит. стр. 104—114
Из маточника при кристаллизации получено вещество, которое кристаллизуется из
этанола в бесцветных иглах (т. пл. 139—141° С), представляющее собой, возможно, 2-мет-
окси-5-тиенилмеркурхлорид.
Синтез 3,4-ди(ацетоксимеркур)-2-метил-5-этилтиофена[551]. 2 г 2-метил-5-этилтиофена
(т. кип. 158—159° С/761 мм) прибавляют к профильтрованному раствору 5,1 г уксуснокислой
ртути в 50 мл 67% -нон уксусной кислоты. Полученный раствор взбалтывают при 32—40° С
в течение 2 час.; через 10—15 мин. начинает выпадать кристаллический осадок. На следу-
ющий день осадок отфильтровывают, промывают 70%-ной уксусной кислотой, затем горя-
чей водой и спиртом. Выход 4,1 г (40%). После кристаллизации из ледяной уксусной кис-
лоты получено 2 г белоснежных кристаллов, которые промыты ледяной уксусной кислотой,
затем спиртом. При нагревании в капилляре со скоростью 9° С в 1 мин. вещество темнеет
при 244—245° С, сплавляется с появлением черных блестящих точек при 249—250’ С, далее
не изменяется при нагревании до 280° С.
Получение тетраацетатмеркурпиррола [507]. К раствору 15 г уксуснокислой ртути в
60 мл воды прибавляют 1 г пиррола; слегка саморазогревающуюся смесь энергично встря-
хивают в течение нескольких минут. Тотчас выделяется белый кристаллический осадок
тетраацетатмеркурпиррола, количество которого увеличивается при прибавлении неболь-
шого количества аммиака. Тетраацетатмеркурпиррол при нагревании разлагается, не пла-
вясь, и нерастворим в обычных растворителях.
Получение 2,3-днацетатмеркуриидола [571]
HgOCOCHs
HgOCOCHa
К раствору 27,22 г уксуснокислой ртути в 100 мл воды прибавляют 5 г индола и энер-
гично перемешивают в течение I часа. Образуется объемистый белый осадок, который от-
сасывают, промывают водой до отсутствия реакции на ион ртути в промывных водах, затем
эфиром и кристаллизуют из ледяной уксусной кислоты. Белый неплавкий порошок.
Получение 4-хлормеркур-1-фенилпиразола [575]. 8,64 г 1-фенилпиразола и 19,1 г ук-
суснокислой ртути в 75 мл уксусной кислоты нагревают до 90° С в течение 15 мин. Затем
прибавляют 30 мл воды. Раствор оставляют стоять, при этом из него выделяется 1-ацетокси-
меркур-1-фенилпиразол — бесцветные иглы. Т. пл. 191° С.
К 2,2 г вещества в 70 мл 50% -ной уксусной кислоты прибавляют при 90°С раствор 0,33 г
хлористого натрия в 20 мл 50%-ной водной уксусной кислоты. Выпадающий 4-хлормер-
кур-1-фенилпиразол кристаллизован из ксилола. Т. пл. 226° С.
Получение триацетатмеркуртиазола [581]
HeCOCOHg HgOCOCHs
HgOCOCHs
Раствор 0,85 г тиазола, 9,6 г уксуснокислой ртути, 1 мл уксусной кислоты в 30 мл во-
ды нагревают с обратным холодильником на водяной бане. Приблизительно через 1 час
начинает выпадать белый осадок. Реакция заканчивается через 12—15 час. После охлаж-
дения осадок отфильтровывают, промывают водой, спиртом и эфиром. После сушки на воз-
духе вес 8—8,2 г. Промывают дважды холодной уксусной кислотой и остаток кристалли-
зуют из нее же. Вещество слегка темнеет около 280° С; не плавится до 300° С.
Меркурирование пиридина [593]. 1 моль уксуснокислой ртути растворяют в 8 молях
пиридина и прибавляют 8 молей воды. Нагревают под давлением в стеклянном или облицо-
ванном стеклом сосуде 2,5 часа при 155° С. Затем отфильтровывают от некоторого коли-
чества нерастворимых продуктов и отгоняют в вакууме летучие вещества, Остаток кристал-
лизуют из бензола. Уксуснокислая 3-пиридилртуть — шелковистые белые иглы — весьма
растворима в воде. Т. пл. 178° С. Выход 49,2% (на сырой продукт).
Меркурирование хинолина [610]. Избыток хинолина нагревают с уксуснокислой ртутью
в течение 5 час. до 150—160° С. По охлаждении выпадает осадок диацетатмеркурхинолина,
фильтрат от которого после прибавления эфира при продолжительном хранении дает еще
некоторое количество аморфного осадка, представляющего собой это же димеркуриро-
ванное соединение, на стенках выделяется в виде шаровидных агрегатов хинолин-
3-меркурацетат; после кристаллизации из воды плавится при 212° С [605]. Фильтрат после
испарения эфира, прибавления хлористого натрия и отгонки хинолина с водяным паром дает
аморфную хлористую 8-хинолилртуть.
Введение ртути на место водородного атома (меркурирование)
103
Меркурирование пуриновых оснований [621]. 19 г пурина растворяют в колбе (1,5 л)
в 1 л водной уксусной кислоты (5 : 100) и прибавляют раствор 30 г уксуснокислой ртути в
смеси 20 мл воды и 10 мл уксусной кислоты. Смесь кипятят в течение 2 дней, фильтруют и
медленно упаривают до объема приблизительно 300 мл. При охлаждении выкристаллизо-
вывается 8-ацетатмеркурпурин.
МЕРКУРИРОВАНИЕ ЦЕНОВ
Супер ароматическая природа ферроцена проявляется также в том, что
его меркурирование, в отличие от меркурирования бензола, гладко осуще-
ствляется в мягких условиях; действием уксуснокислой ртути при комнат-
ной температуре в эфирно-спиртовом растворе. При этом образуется смесь
ацетатов моно- и димеркурированного (в разные кольца циклопентадиенов)
ферроцена, которые были, выделены в виде хлоридов действием хлористого
калия (общий выход 65%) [624]. Хлористая ферроценилртуть — оран-
жево-желтое кристаллическое вещество, плохо растворимое в эфире,
бензоле, метиловом и этиловом спиртах; кристаллизуется из ксилола и бу-
тилового спирта. Т. пл. 194—196° С (с разложением). Дихлормеркурферро-
цен — светло-желтое порошкообразное вещество; не плавится, слабо рас-
творяется в кипящих бутиловом спирте, дихлорэтане и ксилоле, несколько
лучше — в горячих диоксане, этиленгликоле и циклогексаноле. Димерку-
рированный ферроцен образуется даже при молярном соотношении ферро-
цена к ацетату ртути, равном 2 : 1. Такие же продукты выделены при мерку-
рировании ферроцена ацетатом ртути в уксусной кислоте при кипячении
1625]. Описацо выделение продуктов меркурирования ферроцена в виде
ацетатов [626].
Изменением относительных количеств реагентов можно направлять
реакцию в сторону преимущественного образования моно- или димеркури-
рованного продукта [625]. При относительных молярных количествах фер-
роцена и ацетата ртути (1 : 1) образуется 64% димеркурированного и 19%
мономеркурированного продукта. При молярных количествах ферроцена
и ацетата ртути (5 : 1) получено 50% мономеркурированного и 11% димер-
курированного продукта. n-Нитрофенилферроцен меркурируется с боль-
шим трудом, чем ферроцен: при действии ацетата ртути в условиях мерку-
рирования ферроцена (комнатная температура, эфирно- или бензольно-
спиртовый раствор) образуется ди-(хлормеркур)-п-нитрофенилферроцен с
выходом 15% [627].
Меркурирование рутеноцена проводится в таких же условиях, как мер-
курирование ферроцена [628]. При меркурировании уксуснокислой ртутью
в растворе ледяной уксусной кислоты (кипячение 3 часа) получено 21—
25% моно- и 71—79% димеркурированного производного.
Так же в сравнительно мягких условиях происходит меркурирование
циклопентадиенил- и метилциклопентадиенилтрикарбонилмарганца [629,
630]. Оно оеуществлено действием на них уксуснокислой ртути в спирто-
вом растворе,, а также без растворителя. При молярном соотношении ис-
ходных веществ (1 : 1) в среде спирта образуются моно- и димеркурирован-
ные продукты: часть исходного циклопентадиенилтрикарбонилмарганца
остается непрореагировавшей. При большом избытке последнего (20 : 1)
в отсутствие растворителей идет образование только мономеркурированных
производных.
Реакция циклопентадиенилтрикарбонилмарганца с уксуснокислой ртутью в среде эти-
лового спирта [629]. Смесь 6,12г (0,03 моля) циклопентадиенилтрикарбонилмарганца, 9,56г
(0,03 моля) уксуснокислой ртути и 20 мл абсолютного этилового спирта при перемешива-
нии нагревают при 70° С в течение часа. Ацетат ртути полностью переходит в раствор.
К раствору при комнатной температуре добавляют спиртовый раствор хлористого кальция
(2 г в 20 мл). Растворитель из смеси удаляют в вакууме, остаток экстрагируют ацетоном.
Ацетоновый раствор испаряют, твердый остаток последовательно экстрагируют эфиром,
104
Ртутноорганические соединения
бензолом и ацетоном. Из эфирной вытяжки выделяют 1,2 г (19,6%) исходного соединения,
из бензольной вытяжки — хлормеркурциклопентадиенилтрикарбонилмарганец (3,23 г;
24,5%) (т. пл. 135—136° С), из ацетоновой вытяжки — ди-(хлормеркур)циклопентадиенил-
трикарбонилмаргаиец (2,8 г; 13,9%) (порошок кремового цвета, не плавится, растворяется
в ацетоне, ие растворяется в бензоле, этиловом спирте, хлороформе).
Аналогично действием ацетата ртути в этиловом спирте произведено
меркурирование циклопентадиенилтрикарбонила рения с образованием
моно- и димеркурированных продуктов [6311.
ЛИТЕРАТУРА
1. К 1 а р р г о t h W. J., WestheimerF. Н. J. Am. Chem. Soc., 72, 4461 (1950).
2. Schrau th W., Schoe 1 1 er W. Ber., 41, 2089 (1908).
3. BiilmannE. Ber:, 35, 2571 (1902).
4. Schoeller W., Schrau th W. Ber., 42, 777 (1909).
5. Пат. США 2953588 (1960); РЖХим, 1961, 23Л458.
6. Левина P. Годовиков Н. Н. ЖОХ, 24, 1242 (1954).
7. В i i 1 m а п n Е., Н о f f A. Rec. trav. chim., 36, 306 (1916).
8. Petterson L. J. prakt. Chem., 2, 86, 458 (1912).
9. M ah a p a t r a S., GhoshJ. L., Guha SircarS. S. J. Indian Chem. Soc.,
32, 613 (1955).
10. S c h о 11 R;, N у b e r g B. Ber., 39, 1956 (1906).
11. Prager W. Monatsh., 33, 1285 (1912).
12. Steinkopf W. Ann., 434, 21 (1923).
13. CurtiusTh., Buchner E. J. prakt. Chem., 2, 38, 411 (1888).
14. Buchner E. Ber., 28, 215 (1895).
15. H e с м e я н о в A. H., П о в x Г. С. ЖОХ, 4, 958 (1934); Ber., 67, 971 (1934).
16. Hoffmann K. A. Ber., 31, 2212 (1898).
17. L i p p m а п n E. Zbl., 1896, 29.
18. P a t e 1 K- S., Mankad B. N. Current. Sci., 30, 335 (1961).
19. В i i 1 m a n n E., W i 11 J. Ber., 42, 1067 (1909).
20. N a i k K. G„ S h a h L. D. J. IndianChem. Soc., 7, 655 (1930); 8, 29 (1931).
21. N a i k K- G., P a t e 1 R. P. J. Indian Chem. Soc., 9, 185, 533 (1932).
22. Новикове. С., Годовикова T. И., Тартаковский В. А. Изв.
АН СССР, ОХН, 1960, 669.
23. М i с h а е 1 А. Вег., 38, 2083 (1905).
23а. HellonR., Oppenheim А. ' Вег., 10, 701 (1874).
24. М a nc ho t W., Н a a s I. Ann., 399, 123 (1913).
“24а. N е f J. U. Ann., 308, 299 (1899).
25. A d a m s R., Theobald C. W. J. Am. Chem. Soc., 65, 2383 (1943).
26. M с C u s k e r P. А., К г о e g e r J. W. J. Am. Chem. Soc., 59, 213 (1937).
27. Johnson J. R., McEwen W. L. J. Am. Chem. Soc., 48, 469 (1926).
28. NewmanM. S, К u t n e r A. J. Am. Chem. Soc., 73, 4’199 (1951).
29. С r i s t о 1 S.. J., Norris W. P. J. Am. Chem. Soc., 76, 3005.(1954). '*
30. N in e h a m A. W. J. Chem. Soc., 1953, 2601.
31. P a r h a m W. E., S t r i g h t P. L. J. Am. Chem. Soc., 78, 4783 (1956).
31a. Прилежаева E. H., Цымбал Л. В., Шоста ковскийМ. Ф. ЖОХ,
31, 2487 (1961).
32. L u m b Р. В., S m i t h J. C. J. Chem. Soc., 1952, 5032.
33. Truce W. E., Hill H. E„ Boudaki anM. M. J. Am.Chem. Soc., 78, 2760
(1956).
34. G г о b C. A., R u m p f J. A. Helv. Chim. Acta, 37, 1479 (1954).
35. D о b s о n N. A., R a p h a e 1 R. A. J; Chem. Soc., 1955, 3558.
36. NakagawaM. J. Chem. Soc. Japan, Pure Chem. Sect., 72, 561 (1951)).
37. L e i t c h L. C., R e n a u d R. Can. J. Chem., 30, 79 (1952).
38. E 1 s t e г В. В., P a u 1 P. F. M. J. Chem. Soc., 1951, 893.
39. Crombie L, G r i f f i n В. P. J. Chem. Soc., 1958, 4435.
40. В e r t i n D. C. r„ 229, 660 (1949); Ann. chim. France, 12, 8, 296 (1953).
41. Японок, пат. 11124 (1961); С. A., 56, 4794 (1962).
42. A 1 1 a n J. L. H., W h i t i n g M. C. J. Chem. Soc., 1953, 3314.
43. N e 1 s о n N. A., W о 11 e n s a k J. C. J. Am. Chem. Soc., 80, 6626 (1958).
44. Аигл. пат. 809319 (1959); С. A., 53, 15973 (1959).
45. G e n s 1 e r W. J., Mahadevan A. P„ C a s e 11 e J., jr. J. Am. Chem. Soc.,
78, 163, 167 (1956).
46. C r i s t о 1 S. J., В e gо о n A., N or r i s W. P., R a m e у P. S. J. Am. Chem.
Soc., 76, 4558 (1954).
Введение ртути на место водородного атома (меркурирование) 105
47. Pattison F. L. М., Norman J.'J, J. Am. Chem. Soc., 79, 2311 (1957).
48. Has z e 1 d i n e R. N. J. Chem.'Soc., 1951, 588; 1952, 3484; Nature, 165, 152 (1950).
49. C a d i о t P. Ann. chim. France, 13, 1, 214 (1956).
50. NakagawaM. C. A., 45, 7081 (1951).
fil. Barbieri P. C. r., 231, 57(1950).
52. R о e d i g A. Ann., 569, 161 (1950).
53. Clapperton E. T„ MacGregor W. S. J. Am. Chem. Soc., 71, 3234 (1949).
54. Gens ler W. J., Mahadevan A. P. J. Org. Chem., 21, 180 (1956).
55. Долгопольский И. M., Д о б p о м и л ь с к а я И. М., Подкопаева
К. Н. ЖОХ, 17, 1695 (1947).
56. Е g 1 i n t о n G., Me Crae W. J. Chem. Soc., 1963, 2295.
57. H о f m а и n K. A., Kirmreuther H. Ber., 41, 314 (1908); 42, 4232 (1909).
58. Fitz Gibbon M. J. Chem. Soc., 1938, 1218.
59. В a s h f о r d L. A., E m e 1 e u s H. J„ В r i s с о e H. V. A. J. Chem. Soc., 1938,
1358.
60. T r u c e W. F., BoudakianM. M., Heine R. F., McManimie’R. J.
J. Am. Chem. Soc., 78, 2743 (1956).
61. Несмеянов A. H., Кочетков H. К. Изв. АН СССР, ОХН, 1949, 587;.
Уч. зап. МГУ, вып. 132, 51 (1950).
62. Англ. пат. 427979 (1939); Zbl., 1936, I, 642.
63. S е у f е г t h D., ToweR. H. Inorg. Chem., 1, 185 (1962).
64. M i d d I e t о n W. J., Sharkey W. H. J. Am. Chem. Soc., 81, 803 (1959).
65a . S p a h r R. J., V о g t R. R., N i euwland J. A. J. Am. Chem. Soc.1, 55,
2465 (1933).
656. S p a h r R. J., V 6 g t R. R., NieuwlandJ. A. J. Am. Chem. Soc., 55,3728
(1933).
65b. S p a h r R. J., Vo g t R. R., Nieuwland J. A. J. Am. Chem. Soc., 55,
4206 (1933).
66. Z u p a n c i с B. G. Monatsh., 92, 907 (1961).
67. Пат. США-2251778 (1941); С. A., 35, 7638 (1941).
68. D e s s у R. E., Budde W. L, Woodruff C. J. Am. Chem. Soc., 84, 1172
(1962).
69. Hofmann K. A. Ber., 32, 870 (1899).
69a. W e 111 s P. R., К i t ch i ng W. Austral. Chem. J., 16, 1123 (1963).
70. M a r s h J. E., Jersey Flemings truthers R.J. Chem.Soc., 129, 2658 (1927).
71. DimrothO, BerJ 35, 2853 (1902).
72. Пат. США 2551734 (1951); С. A., 45, 6807 (1951).
73. P 1 a n e k R. W., O’C о n n о r R. T., Goldblatt L. A. J. Am. Oil Chem.
Soc., 33, 350 (1956).
74. Sand J., Singer F. Ber., 36, 3707 (1903).
75. Bro derse n K„ Kunke 1 L. Z. anorg. allg. Chem., 298, 34 (1958).
76. M a r q u a r d R. P., L u с e E. N. Analyt. Chem., 31, 418 (1959).
77. Пат. США 2325411 (1944); С. A., 38, 433 (1944).
78. Пищи му к а П. С. ЖОХ, 10, 305 (1940).
79. Н о f m а п п К. А. Вег., 31, 1904 (1898); 33, 1328 (1900).
80. W е i s s А.,. W е i s s A. Z. anorg. allg. Chem., 282, 324 (1955).
8L Б л e ш н н с к и й С. В., У суб аку нов М. Сборник математической конферен-
ции, посвященной 100-летию акад. Курнакова. Фрунзе, 1963.
82. Lassere A. Zbl., 1905, П, 1125.
83. Hofmann К. А. Вег., 38, 1999 (1905).
84. A u 1 d S. М., Н а п t z s с h A. Ber., 38, 2677 (1905).
85. L е у s A. Bull. Soc. chim. France [3], 33, 1317 (1905).
86. D enig es G. Ann. chim. France, 7, 18, 396 (1899).
87. Sand J., GensslerO. Ber., 36, 3699 (1903).
88. К и t s c h e г о f f M. Ber., 17, 20 (1884).
89. M о r t о n A. A., P e n n e r H. P. J. Am. Chem. Soc., 73, 3300 (1951).
90. Oppenheimer C. Ber., 32, 986 (1899).
91. D e n i g e s G. C. r„ 126, 1,868 (1898); 127, 963 (4898); Ann. chim. France [7], 18, 398
(1899).
92. M a r s h J. E., Jersey Fleming Struthers R. J. Chem. Soc., 87, 1878 (1905).
93. Scott-Wilson H. Zbl., 1911, II, 903.
94. MontignieE. Bull. Soc. Chim. France [4], 43, 115 (1928).
95. Thiele I. Ber., 34, 68 (1901).
96. Hof mann K. A., S e i 1 e г E. Ber., 39, 3187 (1906).
97. I s s 1 e i b K-, В r a c k A. Z. Naturforsch., 11b, 420 (1956).
98. W i 1 k i n s о n G„ P i p e r T. S. J. Inorg. a. Nucl. Chem., 2, 32 (1956).
98a. Fischer E. O., Fritz H. P. Inorg. Chem. Radiochem., 1, 56 (1959).
986. Broderse n!K.. О p i t z G., Breit inger D. Ber., 97, 2046 (1964).
99. Австр. пат. 100723 (1923); Zbl., 1926, II, 1586.
106
Ртутноорганические соединения
100. К о с т А. Н., Г р а н д б е р г И. И. ЖОХ, 25, 2064 (1955).
101. Marsh J. Е., Jersey Fleming Struthers R. J. Chem. Soc., 95, 1777
(1909); Marsh J. E. J. Chem. Soc., 97, 1410 (1910).
102. Нем. пат. 275932 (1912); Zbl., 1914, II, 367.
103. Merz W. Zbl., 1926, II, 1050.
104. L e v i n R. H., SpielmanM. A. J. Am. Chem. Soc., 62, 920 (1940).
105. Пат. США 3034962 (1962); С. A., 57, 12598 (1962). '
106. Пат. США 2675400 (1954); С. А., 49, 1814 (1955).
107. Несмеянов А. Н., Печерская К- А. Изв. АН СССР, ОХН, 1941, 67.
108. Англ. пат. 206507 (1923); Zbl., 1927, I, 950.
109. Австр. пат. 112339 (1923); Zbl., 1929, II, 652.
110. HantzschA., GajewskiF. Ann., 392, 302 (1912).
ill. H a n t z s c h A., L i s t e r J. Ann., 392, 319 (1912).
112. О 11 о M. M. J. Am. Chem. Soc., 56, 1393 (1934).
113. К о b e K. A., L u e t h P. F., jr. Ind. Eng. Chem., 34, 309 (1942).
114. McMahon K. S„ Kobe K. A. Ind. Eng. Chem., 49, 42 (1957).
115. M о r t о n A. A.,M a r s h a 1 1 R. R.,A 1 den R. E.,M a g a t E. E. J. Am. Chem.
Soc., 69, 908(1947).
116. Carmack M., В a i z e r M. M., Handr ick G. R., Kissinger L. W„
S p e c h t E. H. J. Am. Chem. Soc., 69, 785|(1947).
117. Westheimer F. H., Segel E., Schramm R. M. J. Am. Chem. Soc.,
69, 773 (1947).
118. S c h r a m m R. M., Klapproth Wm„- Westheimer F. H. J. Phys, et
Colloid Chem., 55, 843 (1951).
119. Brown H; C., D u b e c k M. j. Am. Chem. Soc., 81,5608(1959).
120. К о b e K- A., D о u m a n i T. F. Ind. Eng. Chem., 33, 170 (1941).
121. Пат. США 2353312 (1944); С. A., 38, 5844 (1944).
122. Англ. пат. 778271 (1957); С. А., 52, 1228 (1958).
122а. W a t t G. W., В a у e L. J. Inorg. Nuci. Chem., 26, 1531 (1964).
123. McGary C. W., jr. Diss. Abs., 15, 977 (1955).
124. Ю Гын-ман. РЖХим, 1958, № 4, 11358.
125. Пат. США 2754241 (1956); С. А., 51, 2859 (1957).
125а. Англ. пат. 325266 (1928); Zbl., 1930, I, 3723,
126. Франц, пат. 37531 (1929); Zbl., 1931, I, 1360.
127. Пат. США 2050018 (1936); Zbl., 1936, II, 3195.
128. Франц, пат. 843092 (1938); Zbl., 1939, II, 4353.
129. К г е s g е A. J., Brennan J. F. Proc. Chem. Soc., 1963, 215.
130. R о e d e r G., В 1 a s i N. Ber., 47, 2748 (1914).
131. Англ. пат. 325846 (1928); Zbl., 1930, II, 306.
132. Gr awe T. В., H a r r i s s S. E., Christiansen W. G. J. Am. Pharm. Ass.,
25, 752 (1936).
133. Nenizescu C. D„ I sacescu D. A., GruescuC. Bull. Soc. chim. Ro-
mania, 20, 127 (1938).
134. Пат. США 2075971 (1935); Zbl.. 1937, II, 288.
135. M a у n a r d J. L. J. Am. Chem. Soc., 46, 1510 (1924).
136. Пат. США 1786094 (1929); Zbl., 1931, I, 1518.
137. Нем пат. 548902 (1928); 1932, II, 1074.
138. Японск. пат. 5333 (1951); С. А., 47, 9357 (1953).
139. Т i с h у V., Т о m a n М. Chem. pram. ysl., 7, 176 (1957).
140. Bergman М. Chem. Techn., 6, 302 (1954).
141. S u п g L. C. A., 54, 359 (1960).
142. Пат. ФРГ 960280 (1957); С. A., 53, 14004 (1959).
143. Японск. пат. 2216 (1959); С. А., 54, 9847 (1960).
144. Пат. США 2502222 (1950); С. А., 44, 6882 (1950).
145. О л ь д е к о п Ю. А., М а й е р Н. А. ЖОХ, 30, 3472 (1960).
146. К о b е К. А., McMahon К. S. С. А., 51, 10838 (1957); Drug Standards, 25, 10
(1957).
147. Новикове. С., Годовикова Т. И., ТартаковскийВ. А. ДАН
СССР, 124, 834 (1959); Изв. АН СССР, ОХН, I960, 505.
148. М а 1 a i у a n d i М., SawatzkyH., Wright G. F. Can. J. Chem., 39, 1827
(1961).
148a. Perrin Ch., Westheimer F. H. J. Am. Chem; Soc., 85, 2773 (1963).
149. D i m г о t h O. Ber., 32, 758 (1899).
150. Coffey S. J. Chem. Soc., 127, 1029 (1925).
151. В a r d u h n A. J., К о b e K. A. Ind. Eng. Chem., 38, 247 (1946).
152. В г о wn H. C„ McG ar у C. W., jr. J .Am. Chem. Soc., 77, 2300, 2306, 2310 (1955).
153. Brown H. C., Le Roi N e 1 s о n К. J. Am. Chem. Soc., 75, 6292 (1953).
Введение ртути на место водородного атома (меркурирование)
107
154. Sw a i п С. G., Knee Т. Е. С., К г е s g е A. J, J. Am. Chem. Soc., 79, 505 (1957).
155. Р о е t h к е W„ F й г s t W. С. А., 56, 1469 (1962); Arch. Pharm., 294, 52 (1961)
156. Англ. пат. 406725 (1932); Zbl., 1934, II, 1981.
157. Пат. США 2506289 (1950); С. А., 44, 6434 (1950).
158. В е п к е s е г R. А., Н о к е D. I., Hickner R. A. J. Am. Chem. Soc., 80,
5294 (1958).
159. S т i t h L. I., Т а у 1 о г F. L. J. Am. Chem. Soc., 57, 2370 (1935).
160. Sawatzky H., Wright G. F. Can. J. Chem., 36, 1555 (1958).
161. Newstrom J. E„ Kobe K. A. J. Am. Chem. Soc., 57, 1640 (1935).
162. D о u m a n i F. F., К о b e K. A. Ind. Eng. Chem., 38, 248 (1946).
163. M c G a г у C. W., jr., Ok a mo to Y., В г о w n H. C. J. Am. Chem. Soc,, 77,
3037 (1955).
164. Brown H. C., Dub eck M., G о 1 d m a n G. J. Am. Chem. Soc., 84, 1229 (1962).
165. M i 11 e r H. F., В a c h m a n G. B. J. Am. Chem. Soc., 57, 2447 (1935).
166. Campbell N„ Stafford W. H. J. Chem. Soc., 1952, 299.
167. Ogata Y., Tsuchida M. J. Org. Chem., 20, 1644 (1955).
168. D i m г о t h O. Ber., 35, 2032 (1902).
169. G о s w a m i M., DasGupta H. N. J. Indian Chem. Soc., 8, 475 (1931); Zbl., 1931,
II, 3477.
170. Carey J. G,, M i 11 а г I. T. J. Chem. Soc., 1962, 3278.
171. D r e f a h I G., L a n g b e i n G. Ber., 90, 145 (1957).
172. Смирнов P. H. ДАН СССР, 119, 508 (1958).
173. Глушкова В, П„ Делинская Е. Д., Кочешков К- А. ДАН СССР,
129, 109 (1959).
173а. Okawara М„ Т а п a k a J.-, I m о t о Е. С. А., 57, 4854 (1962).
174. Baker W., В art on J. W., McOmie J. F. W. J. Chem. Soc., 1958, 2666.
175. D a n s i A., S a 1 v i о n i E. C. A., 38, 4937 (1944); Ricerca sci., 13, 489 (1942).
176. A n d e r s о n A. G., jr., С о w e.e s ЕЛ J., T a z u m a J. J., Nelson J. A.
J. Am. Chem. Soc., 77, 6321 (1955); A n d e r s о n A. G., jr., Nel so n J. A. J. Am,
Chem. Soc., 72, 3824 (1950).
177. D u n к e r M. F. W., St arkey E. B. J. Am. Chem. Soc., 61, 3005 (1939).
17g. I r w i ng H., К 1 w a n A. M. J. Chem. Soc., 1963, 4288.
179. Hanke M. E. J. Am. Chem. Soc., 45, 1321 (1923).
180. Лю Гын-ман, Цой И-рён. РЖХим, 1959, 16, 57175.
181. Kalinowski В. Roczn. Chem., 9, 131 (1929); Zbl., 1929, I, 2301.
182. В г о w п Н. С., Goldman G. J. Am. Chem. Soc., 84, 1650 (1962).
183. Мате 1 i Е, Manne si er-Mame 1 i A., RodighieroG. Chim. e ind.,
32, 471 (1950).
184. П e т p о в и ч П. И. Ж. Всесоюзн. хим. общ., 5, 105 (1960); ЖОХ, 30, 2808 (1960).
185. Японок, пат. 1778 (1958); С. А., 53, 5197 (1959).
186. К г у n s к i J. Roczn. Chem., 8, 71 (1928); Zbl., 1928, II, 2143.
187. H о I 1 e m a n A. F. Rec. trav. chim., 42, 355 (1923).
188. D i m г о t h O. Ann., 446, 148 (1925).
189. Jurgens J. Rec. trav. chim., 45, 61 (1926).
190. Jackson G. R., F г a n t M. S. J. Am. Chem. Soc., 77, 5625 (1955).
191. Kenner J? Nature, 156, 369 (1945).
192. О g a t a Y., T s u c h i d a M. J. Org. Chem., 20, 1637 (1955).
192a. ОгатаИ., ХодзёМ. РЖХим, 1964, 15ж174.
193. Reissert А. Вег., 40, 4209 (1907).
194. CoffeyS. J. Chem. Soc., 1926, 637, 3215.
195. Пат. США 2089006 (1936); Zbl., 1938, I, 727.
196. Burton H., Hammond F., К e n п e г J. J. Chem. Soc., 1926, 1802.
197. I s h i w a t a S., H у e о S. J. Chem. Soc. Japan, 63, 1446 (1942).
198. Новикове. С., Годовикова T. И., Тартаковский В. А. Изв.
АН СССР, ОХН, 1960, 863.
199. D i ш г о t h О. Ber., 31, 2154 (1898).
200. S 1 о t t а К. Н., J а с о b i К. R. J. prakt. Chem., 120, 249 (1929).
201. S t е i п к о р f W. Ann., 413, 310 (1917).
.202, В a u r R., Schellenberg M., Schwarzenbach G. Helv. chim.
acta, 45, F. Ill, 775 (1962).
203. M a m e 1 i E. Gazz., 52, 1, 352 (1922).
204. Whitmore F.C., Middleton E. J. Am. Chem. Soc., 43, 619 (1921).
205. Синтезы органических препаратов, сб. 1. М., ИЛ, 1949, стр. 477.
206. Нем. пат. 683570 (1936); С. А., 36, 4278 (1942).
207. Wronsky М. Roczn. Chem., 34, 947 (1960).
208. В о г d е i а п u С. V. Zbl., 1937, II, 1562; Ann. Sci. Univ. Jassy I, 23, 212 (1937).
209. NeogiP., Chatterjee M. P. J. Indian Chem. Soc., 5, 221 (1928).
210. Нем. пат. 234851 (1910); Zbl., 1911, I, 1769; 497615 (1923); Zbl., 1930, II, 1429.
108
Ртутноорганические соединения
211. Франц, пат. 609478 (1926); Zbl., 1927, I, 347. Пат. США 1890774 (1927); Zbl., 1933, 1„
1501. ’ . • •
212. Hantzsch A., A li I d S. М. Вег., 39, 1105 (1906).
213. R a i z i s s G. W., ProskouriakoffA. J. Am. Chem. Soc., 44, 787 (1922).
214. H о d g s о n H. H. J. Am. Chem. Soc., 49, 2840 (1927); Hodgson H. H., Tur-
ner H. S. J.'Chem. Soc., 1944, 8.
215. N a i к K. G., Mehta B. F. J. Indian Chem. Soc., 12, 783 (1935).
216. О h n о T. C. A., 51, 270 (1957); J. Pharm. Soc. Japan, 76, 718 (1956).
217. Нем. пат. 484995 (1926); Zbl., 1930, I, 1370.
218. M a m e 1 i E. Gazz., 56, 948 (1926).
219. Mameli E., PiaggesiF. Gazz., 62, 158 (1932).
220. Пат. США 2423044 (1947); С. A., 42, 1378 (1948).
221. Wirth T. H„ Davidson N. J. Am. Chem. Soc., 86, 4322 (1964).
222. Bordeianu С. V. Zbl., 1938, I, 60; Ann. Sci. Univ. Jassy, I, 23, 240 (1937).
223. Mameli E., Mameli-Mannessier A. Zbl., 1932, I, 2758.
224. Mameli E., Mameli-Mannessier A. Gazz., 52, II, 1 (1922).
225. В и r t J. Zbl., 1936, II, 778; J. Amer. Pharm..Assoc., 25, 112 (1936).
226. Henry T. A., S h a r p T. M. J. Chem. Soc., 1926, 2432.
227. Haugh I. W„ Gar lan d С. E., Pray H. H. J. Am. Chem. Soc., 53, 2697 (1931).
228. BriegerR., SchulemannW. J. prakt. Chem., 2, 89, 134 (1914).
229. NeogiP., Mukherjee G. K. J. Indian Chem. Soc., 12, 211 (1935).
230. В amb erger E. Ber., 31, 2626 (1898).
231. К e г к h о f I. G. Rec. trav. chim., 51, 755 (1932).
232. N i e d e r 1 J. B., Shukis A. J. J. Am. Chem. Soc., 66, 844 (1944).
233. N i e d e r 1 J. B., N a g e I R. H. J. Am. Chem. Soc., 63, 1235 (1941).
234. Пат. США 2137236 (1934); Zbl., 1939, I, 5007.
235. Пат. США 2163745 (1934); Zbl., 1939, II, 3148.
236. Пат. США 2217155 (1941); С. А., 35, 1069 (1941).
237. Наг t М. С., A n d е г s е п Н. Р. J. Am. Chem. Soc., 57, 1059 (1935).
238. Оно Т. РЖХим., 1957, 66175.
239. Wayne-RudxiyA., BurtJ. Zbl., 1939, II, 1859; J. Am. Pharm. Ass., 28, 28&
(1939).
240. M a m e 1 i E. Gazz., 52, II, 18 (1922).
241. Пат. США 2240025 (1941); С. A., 35, 4919 (1941).
242. Пат. США 1928436 (1931); Zbl., 1934, I, 248.
243. Пат. США 1630072 (1923); Zbl., 1927, II, 1080.
244. Пат. США 1953263 (1932); Zbl., 1934, II, 2103.
245. Пат. США 1985949 (1931); Zbl., 1935, II, 1581.
246. О h п о Т. С. А., 51, 324 (1957); J. Pharm. Soc. Japan, 76, 722 (1956).
247. Н а г г i s S. Е., С h г i s 11 a n s е n W. G. J. Amer, Pharm. Assoc., 22, 723 (1933).
248. R e i c h e 1 I., Rod L. РЖХим, 1959, № 5, 15460.
249. L e у s A. Zbl., 1905, I, 1531; J. Pharm. Chim., 6, 21, 358 (1905).
250. Lefevre C., DesgrezC. Zbl., 1935, I, 3784; C. r„ 200, 762 (1935).
251. Англ. пат. 312246 (1928); Zbl., 1930, I, 2150.
252. Австр. пат. 114031 (1928); Zbl., 1929 II, 3057.
253. Guha-SircarS. S., Rout M. K- J. Indian Chem. Soc., 29, 769 (1952).
254. F и к a m а и c h i H. C. A., 45, 8472 (1951); J. Pharm. Soc. Japan, 64, 305 (1944).
255. Fukamauchi H. J. Pharm. Soc. Japan, 64, 310 (1942); C. A., 45, 2897 (1951).
256. S a n d i n R. B. J. Am. Chem. Soc., 51, 479 (1929).
257. H a r t M. С., H irschie 1 der A. D. J. Am. Chem. Soc., 42, 2678 (1920).
258. Domini к iewicz M. Roczn. Chem., 12, 79 (1932).
259. В о о n - L о n g N. C. A., 43, 5017 (1949); J. Pharm. Assoc. Siam I, 4, 5 (1948).
260. Англ. пат. 24981; С. A., 6, 1547 (1912).
261. О h n о T. С. А., 51, 270 (1957); J. Pharm. Soc. Japan, 76, 726 (1956).
262. SiposJ.C., Sawatz к у H., W r i g h t G. F. J. Am. Chem. Soc., 77, 2759 (1955)-
263. D i m г о t h O. Ber., 54, 1504 (1921).
264. M a n c h о t W. Ann., 421, 331 (1920).
265. К h a г a s c h M. S., ChalkieyL. J. Am. Chem. Soc., 46, 1216 (1924).
266. Schroeder W. D., Br e w s t e г R. Q. J. Am. Chem. Soc., 60, 751 (1938).
267. Sowa F. J. Пат. США 2607790 (1952); С. A., 47, 6983 (1953).
268. M о r t о n J. W. С. A., 49, 8162 (1955); Iowa State Coll. J. Sci., 28, 367 (1954).
269. В г о w n H. C., D и b e с к M. J. Am. Chem. Soc., 82, 1939 (I960).
270. Mameli E. Gazz., 52, II, 23 (1922).
271. Пат. США 2392811 (1946); С. A., 40, 3234 (1946).
272. D r ake N. L„ H а г г i s H. C., J a e g e r Ch. B. J. Am. Chem. Soc., 70, 168(1948)-
273. Guha-SircarS. S., DattaS. K. J. Indian Chem. Soc., 27, 357 (1950).
274. D a 11 a S. K., Banerjee G. J. Indian Chem. Soc., 30, 568 (1953).
Введение ртути на место водородного атома (меркурирование)
109
275. J о s h i К. С., В a h е 1 S. С. J. Indian Chem. Soc., 37, 365 (I960).
275а. Srivastava К-С. Bull. chim. Soc. Japan, 37, 583 (1964).
276. Несмеянов A. H. Шацкая P. X. ЖОХ, 5, 1268 (1935).
277. Chauveau J. Bull. Soc. Chim. France, 1949, 614.
278. Priester R. Rec. trav. chim., 57, 811 (1938).
279. S h u k i s A. J., T a 1 1 m a n n R. C. J. Am. Chem. Soc,, 66, 1462 (1944).
280. Whitmore F.C., Mi ddleton E. J. Am. Chem. Soc., 45, 1330 (1923).
281. Henry T. A., Sharp T, M, J. Chem. Soc., 121, 1055 (1922).
282. Henr у T. A., Sharp T. M. J. Chem. Soc., 1930, 2279.
283. Henry T. A., S h a r p T. M. J. Chem. Soc., 125, 1049 (1924).
284. Paol in i V. Gazz., 51, II, 188 (1921).
285. Rossi G., R a g n о M. Zbl., 1939, II, 2771; Ann. chim. Applicata, 29, 146 (1939).
286. P e a r 1 J. A. J. Am. Chem. Soc., 70, 2008 (1948).
287. Sharp. T. M. J. Chem. Soc., 1937, 852.
288. Нем. пат. 463517 (1923); Zbl., 1928, II, 1385.
289. Dessy R. E., Kim J.-Y. J. Am. Chem. Soc., 82, 686 (1960).
290. Нем. пат. 486494 (1922); Zbl., 1930, I, 892.
291. В u г о n i G. Gazz., 32, 11, 307 (1902).
292. L i n t n e r C. J. Z. angew. Chem., 13, 708 (1900).
293. P а о 1 i n i V. Gazz., 65, 836 (1935).
294. Rupp E„ G e r s c h H. Zbl., 1927, II, 607; Arch. Pharm., 265, 323 (1927).
295. BoedeckerF., Wunstorf O. Zbl., 1926, 1, 60; Arch. Pharm., 263, 430 (1925).
296. U k a i T., Y a m am a t о Y„ KanatomoS., M a t s u s h i г о К. C. A., 50,
1665 (1956); J. Pharm. Soc. Japan. 75, 280 (1955).
297. S c h о e 1 1 e r W., Sthrau th W., H u e t e r R. Ber., 53, 634 (1920).
298. В r e n a n s P., R a p i 11 у В. C. r., 193, 55 (1931).
299. В r i e g e r R. Zbl., 1912, I, 753.
300. G a d a m e r I. Arch. Pharm., 256, 263 (1919); C. A., 13, 1364 (1919).
301. G u h a - S i r c a r S. S., Rou t M. K. J- Indian Chem. Soc., 29, 779 (1952).
302. Нем. пат. 255030 (1911); Zbl., 1913, I, 353.
303. I s h i h a r a K- Zbl., 1930, I, 47, 48; J. Pharm. Soc. Japan, 49, 134, 140 (1929).
304. Нем. пат. 248291 (1910); Zbl., 1912, II, 211.
305. Ger ngross O., Kersasp H. Ann., 406, 248 (1914).
306. Bucht a 1 a H. Z. Physiol. Chem., 83, 249 (1913).
307. Schmidt H. Pharm. Z„ 60, 724 (1916).
308. Нем пат. 229781 (1909); Zbl., 1911, I, 276.
309. О h n о T. C. A., 51, 270 (1957); J. Pharm. Soc. Japan, 76, 729 (1956).
310. E n g e 1 m a n n G. С. A., 7, 2094 (1913).
311. L a j о u x H. C. A., 9, 2527 (1915).
312. Lespagnol A., Bar D. Bull. Soc. chim. France, 5, 3, 1107 (1936).
313. Fukam auc hi H. J. Pharm. Soc. Japan, 64, 309 (1944); C. A., 45, 8473 (1951).
314. F о x A. L., W h i t m о r e F. C. J. Am. Chem. Soc., 51, 2196 (1929).
315. О v e r b a u g h t S. C., S a n d i n R. B. J. Am. Chem. Soc., 57, 1658 (1935).
316. Whitmore F. C., Middleton E. J. Am. Chem. Soc., 44, 1546 (1922)-
317. Rupp E., Herrmann A. Arch. Pharm., 254, 500 (1916); Zbl., 1917, I, 10.
318. R a i z i s s G._ W., К о 1 m e r I. A., G a v г о n J. L. J. Biol. Chem., 40, 533 (1919).
319. Stieglitz!., К h a r a s c h M. S., H a n k e M. E. J. Am. Chem. Soc., 43,
1185 (1921).
320. Англ. пат. 12472 (1908).
321. Maschmann E. Ber., 59, 216 (1926).
322. Нем. пат. 394363 (1920); Zbl., 1924, II, 1023.
323. Nai k К, G„ Patel A. D. J. Chem. Soc., 1934, 1043.
324. SenR. N., Chakravarti D. J. Indian Chem. Soc., 6, 847 (1929); Zbl., 1930,
I, 2244.
325. SenR. N., Chakravarti D. J. Indian Chem. Soc., 7, 247 (1930).
326. Ahmad S. Z., Desai R. D. Zbl., 1937, II, 229; Proc. Indian Acad. Sci., Sect.,
A5, 277 (1930).
327. A h m a d S. Z., D e s a i R. D. Zbl., 1938, I, 4627; Proc. Indian Acad. Sei., Sect.
A6, 89 (1937).
328. R а о P. S., S a s t r i V. D. N., S e s h a d r i T. R. Zbl., 1939, II, 638.
329. Smith Cl., Mitchell A. D. J. Chem. Soc., 93, 847 (1908).
330. M i t c h e 11 A. D„ S m i t h Cl. J. Chem. Soc., 95,1430 (1909),
331. Proskou r ia kof f A., RaizissG. W. J. Am. Chem. Soc., 47, 1374 (1925).
332. Greenbaum F. R. Zbl., 1929, I, 1690; Arch. J. Pharm., 101, 34 (1929).
333. Whitmore F.C., L e u c k G. J. J. Am. Chem. Soc., 51, 2782 (1929).
334. W h i t e E. C. J. Am. Chem. Soc., 42, 123, 55 (1920).
335. Пат. США 1549942 (1921); Zbl., 1926, I, 728.
110
Ртутноорганические соединения
336. Нем. пат. 308335 (1914); Zbl., 1918, II, 881.
337. R о h a t g i Н. L. РЖХим, 1960, 4, 14576.
338. О h п о Т. С. А., 51, 313 (1957); J. Pharm. Soc. Japan, 76, 733 (1956).
339. Нем пат. 201903 (1906); Zbl., 1908, II, 1307.
340. R о h a t g i H. L. РЖХим, 1959, 18, 65391; Indian J. Appl. Chem., 21, 2, 102 (1958).
341. S a n d i n R. B„ Sutherland J. W. J. Am. Chem. Soc., 51, 1773 (1929).
342. N о v e 1 1 i A. Zbl., 1935, I, 3691; Ann. Pharm. Bioquim., 4, 29 (1933).
343. Harr is S. E., Christiansen W. G. Zbl., 1934, 1,3590; J. Am. Pharm. Soc.,
23, 108 (1934).
344. Пат. США 1954619 (1932); Zbl., 1934, II, 3147.
345. Пат. США 1952166 (1931); Zbl., 1934, II, 2103.
346. RohatgiH. Z. Sharma U. РЖХим, 1960, 18, 73492; Indian J. Appl. Chem.,
22', 128 (1959).
347. N о g a s e Y„ О h n о T. C. A., 49, 254 (1955); J. Pharm. Soc. Japan, 73, 1337 (1953).
348. Nogase Y., О h n о T. C. A., 49, 254 (1955); J. Pharm. Soc. Japan, 73, 1340 (1953).
349. P a n L. C. Zbl., 1937, II, 3745; Chem. Ind (China), 12, 71 (1937).
350. Пат. США 1692237 (1927); Zbl., 1929, I, 1584.
351. C a n d e 1 i A. C. A., 44, 4635 (1950).
352. Пат. США 1860003 (1925); Zbl., 1932, II, 896.
353. Пат. США 1786172 (1927); Zbl., 1931, I, 1947.
354. Пат. США 1863241 (1928); Zbl., 1932, II, 1473.
355. Dunning F., Fari n h old L. H. J. Am. Chem. Soc., 51, 804 (1929).
356. Пат. США 1863268 (1929); Zbl., 1932, II, 1474.
357. Harden W. C. J. Am. Chem. Soc., 49, 3141 (1927).
358. Пат. США 186133 (1930);- Zbl., 1932, II, 896.
359. Пат. США 1757176 (1928); Zbl., 1930, II, 1251.
360. UkaiT., Yamamoto Y., Y о t suz u ka M. C. A., 50, 5665 (1956).
361. UkaiT., Yamamoto Y., Yotsuzuka M., I chimur a F. C. A., 51,
280 (1957); J. Pharm. Soc. Japan, 76, 657 (1956).
362. Webb F. Jowa State Coll. J. Sci., 17, 152 (1942); C. A., 37, 3413 (1943).
363. Gilman Ц., Webb F. J. Am. Chem. Soc., 71, 4062 (1949).
364. Caccia-BavaA. M., Vitali T. Ann. chim. (Roma), 40, 21 (1950).
365. S a c h s G., О 11 M. Ber., 59, 171 (1926).
366. UkaiT., Y a m a m о t о Y., H i r a n о S., Yo ts uzu ka M. C. A., 50, 10033
(1956); Pharm. Bull. (Japan), 3, 105 (1955).
367. Ca. ccia-Bava A. M., Vitali T. An. chim. Applicata, 38, 460 (1948).
368. Arndt F., Tr a verso G. Ber., 89, 124 (1956).
369. Пат. США 1984174 (1933); Zbl., 1935, I, 2407.
370. Piccinini A., Ruspaggiari G. Gazz., 22, II, 604 (1892); Zbl., 1893, I, 564.
371. V e с c h i о t t i L. Gazz., 44, II, 35 (1914).
372. Bru i се T. C. J. Am. Chem. Soc., 72, 1398 (1950).
373. Koehler W. R., Dowb en R. M. Arch. Biochem. Biophys., 93, 501 (1961).
374. Guha-SircarS. S., Mitra J. J. Indian Chem. Soc., 28, 80 (1951).
375. P e s c i L. Gazz., 23, II, 521 (1893); Ber., 27, 128 (1894).
376. P e s c i L. Z. anorg. allg. chem., 15, 208 (1897).
377. Piccinini A. Gazz., 23, II, 534 (1893).
378. Prussia L. ’ Gazz., 27, 14 (1897).
379. P r u s s i a L. Gazz., 28, 129 (1898).
380. KharaschM. S., Piccard I. J. Am. Chem. Soc., 42, 1860 (1920).
381. W h i t m о r e F. C. J. Am. Chem. Soc., 41, 1841 (1919).
382. P e s c i L. Gazz., 28, II, 101 (1898).
383. P e s c i L. Z. anotg. allg. Chem., 17, 276 (1898).
384. ReitzensteinF., Stamm G. J. prakt. Chem., 2, 81, 159 (1 910).
385. Schrauth W., Schueller W., R о t h e r J. Ber., 45, 2808 (1912).
386. V e с c h i о t t i L. Gazz., 51, II, 208 (1921).
387. Vecchi о t t i L. Gazz., 56, 155 (1926); 54, 411 (1924).
388. VecchiottiL., Copertini. Gazz., 59, 525 (1929).
389. Vecchiotti L., A j u t о A. Gazz., 65, 832 (1935).
390. Jacobs W. A., Hei delberger M. J. Biol. Chem., 20, 513 (1915).
391. В ra d 1 e у W„ Wright I. J. Chem. Soc., 1956, 640.
392. Bell F. J. Chem. Soc., 133, 2770 (1928).
393. Bernardi A., Tartarini M. Gazz,, 57, 223 (1927).
394. M с M a h о n E„ M a r v e 1 C. S. J. Am. Chem. Soc., 52, 2528 (1930).
395. Rossi G., С e c h e t t i B. Gazz., 55, 869 (1925).
396. W e i s b u r g e r E. K-, Weisburger J. H., R a у F. E. J. Org. Chem.,
16, 1697 (1951).
397. К h a r a s c h M. S., JacobsohnI. M. J. Am. Chem. Soc., 43, 1894 (1921).
398. Пат. США 2536750 (1951); С. A., 45, 2622 (1951).
Введение ртути на место водородного атома (меркурирование)
Ш
399. R о u t М. К. J. Indian Chem. Soc., 33, 796 (1956).
400. Р u j а г i Н. К., R о u t М. К. J. Indian Chem. Soc., 31, 257 (1954).
401. Bhargava P. N., N a g a b h u s - h a n a m M. J. Indian Chem. Soc., 36, 434 (1959).
402. Das N. K., Mahapair a G. N., R о u t M. K. J. Indian Chem. Soc., 32, 55
(1955).
403. R о u t M. K., Pa dh i B„ Das N. K. Nature, 174, 516 (1954).
404. Maha pa tr a G. N., R о u t M. K- J. Indian Chem. Soc., 31, 933 (1954).
405. Mahapatra G. N.,Roui M. K. J. Sci. Ind. Res. (India), 13 B, 378 (1954).
406. Mahapatra G. N. J. Am. Chem. Soc., 79, 988 (1957).
407. P u j a г i H. K., Rou t M. K. J. Sci. Ind. Res., 14B, 448 (1955).
408. M a h a p a t r a G. N., Rou t M. K. J. Indian Chem. Soc., 32, 715 (1955).
409. Rou t M. K- J. IndianChem. Soc., 33, 690 (1956).
410. R о u t M. К., P u j a r i H. K- J. Am. Chem. Soc., 75, 4057 (1953).
411. Mahapatra G. N., R ou t M. K. J. Indian Chem, Soc., 34,653 (1957).
412. Mahapatra G. N., Rout M. K. J. Sci. Ind. Res., 13B, 407 (1954).
413. Mahapatra G. N. J. Proc. Inst. Chemists, 31, 113 (1959).
414. Bhargava P, N„ R a о R. P. J. Indian Chem. Soc., 32, 463 (1955).
415. В h a r g a v a P. N., P a n t u 1 у A. J., R a о R. P. J. Indian Chem. Soc., 34,
475 (1957).
416. Bhargava P. N., SastryM. S. J. Sci. Research Benares Hindu Univ., 6,196
(1955); C. A., 52, 11821 (1958).
417. В h a r g a v a P, N,, R a о R. P., S a s t r i M. S. J. Indian Chem. Soc., 33, 596
(1956).
418. R a u 1 В. B„ M a h a p a t r a G. N. J. Indian Chem. Soc., 38, 919 (1961).
419. R a о R. P. РЖХим, 1961, 20Ж193; Proc. Nat. Acad. Sci. India, 1960, A29, 101.
420. Rao R. P. J. Indian Chem. Soc., 38, 787 (1961).
421. Joshi К. С., В ah e 1 S. C. J. Indian Chem. Soc., 39, 121 (1962).
422. Guha-Sircar S. S., Patnaik K. K- J- Indian. Chem. Soc., 27, 535 (1950).
423. Mahapatra G. N. J. Indian Chem, Soc,, 33, 923 (1956).
424. P e s c i L. Z. anorg. allg. Chem., 15, 222 (1897); Gazz., 24, II, 449 (1894); Ch. Z., 23,
58 (1899), '
425. P i с c i n i n i A. Gazz., 24, II, 453 (1894).
426. R oss i G„ В о с c h i C. Gazz., 55, 93 (1926).
427. RossJ’G. Gazz., 52, I, 189 (1922).
428. R a f f о M., R о s s i G. Gazz., 44, 109 (1914).
429. Raffo M., Rossi G. Zbl., 1912, II, 2070.
430. Raffo M„ Rossi G. Gazz., 42, II, 623 (1912).
431. Bernardi A. Gazz., 56, 337 (1926).
432. Rossi G., В о e c h i C. Gazz., 56, 817 (1926).
433. ProskouriakoffA., Titherington R. J. J. Am. Chem. Soc., 52, 3978
(1930).
434. R a g n о M. Zbl., 1940, I, 532; Ann. chim. Applicata, 29, 414 (1939).
435. R a g n о M. Zbl., 1940, I, 3247; Ann. chim. Applicata, 30, 72 (1940).
436. R a g п q M. Gazz., 70, 423 (1940).
437. R agno M. Zbl., 1939, П, 2771; Ann. chim. Applicata, 29, 148 (1939).
438. Khar.asch M. S., Lommen F. W, M., Jacobsohnl. M. J. Am. Chem.
Soc., 44, 793 (1922).
439. Пат. США 1787630 (1923); Zbl., 1931, I, 1812.
440. Goddard D. J. Chem. Soc., 1937, 984.
441. H о d g s о n H. H., Hathway D. E. J. Chem. Soc., 1945, 123.
442. Hodgson H. H-, Ha t hway D. E. J. Soc. Dyers and Colourists, 61, 283 (1945).
443. Hodgson H. H., HathwayD. E. J. Soc. Dyers and Colourists, 62,241 (1946).
444. Hodgson H. H., Turner H. S. J. Chem. Soc., 1943, 391.
445. Scho e 11 er W., H u e t e r R. Ber., 47, 1930 (1914).
446. Schneller W„ Schrau th W„ Liese E. Ber., 52, 1777 (1919).
447. S c h о e 1 1 e r W„ S c h r a u t h W., GoldackerP. Ber., 44, 1301 (1911).
448. Cherbuliez E., Mor i M. Helv. chim. acta, 28, 17 (1945).
449. Нем. пат. 264388 (1912); Zbl., 1913, II, 1262.
450. Нем. пат. 281009 (1913); Zbl., 19'15, I, 73.
451. Mahapatra G. N. J. Indian Chem. Soc., 34, 901 (1957).
452. Rodighiero G. Ann. chim. applicata, 39, 621 (1949).
453. Guha - S ircar S. S., A 1 i I. J. Indian Chem. Soc,, 28, 83 (1951).
454. W i г t h T. H., Davidson N. J. Am. Chem. Soc., 86, 4318 (1964).
455. Albert A., Schneider W. Ann., 465, 257 (1928).
456. Maschmann E. Ann., 450, 85 (1926).
457. C h a 1 к 1 e у L. J. Am. Chem. Soc., 63, 981 (1941).
458. C h a 1 к 1 e у L. Zbl., 1940, II, 46; Science (N. S.), 91, 300 (1940).
459. Greenb.aumF R. Zbl., 1929,1, 1960; Am. J. Pharm., 101,34 (192S).
112
Ртутноорганические соединения
460. Пат. США 2325038 (1944); С. А., 38, 117 (1944).
461. Vecchio 11 i L., MicchettiA. Gazz., 56,480(1926).
462. Ukai T., 11 о Y. J. Pharm. Soc. Japan, 73, 821 (1953).
463. U к a i T., Yama mo to Y., I t о Y., Y a n a g i A., YotsuzukaM. C. A.,
. 50, 5664 (1956); J. Pharm. Soc. Japan, 75, 490 (1955).
.464. Vecchiotti L„ Carani N. Gazz., 56, 216(1926).
465. V e с c h i о 11 i L. Gazz., 57, 485 (1927).
466, VecchlottiL., MicchettiA. Gazz., 55, 372 (1925).
467. VecchiottiL., CaraniN. Gazz., 56, 147 (1926).
468. V e с c h i о 11 i L. Gazz., 58, 231 (1928).
469. Vecchiotti L. Gazz., 58, 181 (1928).
470. VecchiottiL., SperanziniG. Gazz., 59, 363 (1929).
471. Grignard V., AbelmannA. Bull. Soc. chim. France, 19, 18 (1916).
472. Bernardi A. Zbl., 1937, II, 2833; Gazz., 67, 380 (1927).
473. К о з л о в В. В. ЖОХ, 18, 757 (1948).
474. Shanmugana t h an Sp. C. A., 50, 7107 (1957).
475. Д о к у н и х и н И. С., Гаева Л. А.'- Ж. Всесоюзн. хим. общ., 6, 112 (1961).
7, 236 (1962).
476. Р е s с i L. Atti Accad. naz. Lincei, Rend. Classe Sci. fis. mat. nat., 5, 9, 1, 225; Zbl.,
1900, I, 1097.
477. P e s c i L. Atti. Accad. naz. Lincei, Rend. Classe Sci. fis. mat. nat., 5, 10, I, 362; Zbl.,
1901, II, 108.
478. Нем. пат. 234914 (1910); Zbl., 1911, II, 112.
479. Нем. пат. 249332 (1911); Zbl., 1912, II, 465.
480. Нем. пат. 229574 (1900); Zbl., 1911, I, 275.
481. Blumental F. Biochem. Z., 32, 59 (1911).
482. Нем. пат. 249725 (1910); Zbl., 1912, II, 777.
483. Whi tmor e F. C„ F о x A. L. J. Am. Chem. Soc., 51, 3363 (1929).
484. W h i t m о r e F. C„ I senhour L. L. J. Am. Chem. Soc., 51, 2785 (1929).
485. Ogata Y:, Tsuchida M. J. Org. Chem., 21, 1324 (1956).
486. Whitmore F.C., Ehrenfeld E. J. Am. Chem. Soc., 48, 789 (1926).
487. W a 1 t о п H. F., Martinez I. M. J. Phys. Chem., 63, 1318 (1959).
488. Несмеянов A. H. ЖРХО, 61, 1393 (1929); Ber., 62, 1010 (1929).
489. G i 1 m a n H., W r i g h t G. F. J. Am. Chem. Soc., 55, 3302 (1933). .
490. Paul R. Bull. Soc. chim. France, 2, 2233 (1935).
491. N о r m a n t H. Ann. chim. (France), 17, 335 (1942).
492. Gilman H., Calloway N. O. J. Am. chem. Soc., 55, 4197 (1933).
493. Paul R, Bull. Soc. chim. France, 2, 2227 (1935).
494. Baroni A., Marini-BettoloG, B. Gazz., 70, 670 (1940).
495. C h u t e W. J., Orcha r d W. M„ W r i g h t G. F. J. Org. Cehm., 6, 157 (1941).
496. M a n 1 у D. G., A m s t u t z E. D. J. Org. Chem., 21, 516 (1956).
497. ГиллерсС. А., Зелмен 3. Я. Изв. АН Латв. ССР, 1958, 11, 97.
498. Morel Th., VerkadeP. Е. Rec. trav. chim., 70, 35 (1951).
499. Eastman R. H. J. Am. Chem. Soc., 72, 5313 (1950).
500. VerkadeP. E., Morel Th., Geritsen H. G.Rec. trav. chim., 74, 763 (1955).
501. F e t i z о n-М., В a r a n g e r P. C. r., 234, 2296 (1952).
502. G i 1 m a n H., В u r t n e r R. J. Am. Chem. Soc., 71, 1213 (1949).
503. I r i e T., W а к a у a m a S. C. A., 45, 4213 (1951); Science (Japan), 19, 231 (1949).
504. Irie T., Wakayama S. C.A., 45, 4213 (1951); Science (Japan), 18, 328 (1948).
505. H u r d Ch. D., W i Ik inson K. J. Am. Chem. Soc., 70, 739 (1948).
506. В e с к W. W„ H a m i I t о n C. S. J. Am. Chem. Soc., 60, 620 (1938).
507. Ci usa R., Gr ilia G. Gazz., 57, 323 (1927).
508. Scheibler H., JeschkeJ., BeiserW. J. prakt. Chem., 136, 232 (1933).
509. C i n n e i d e R. О. C. A., 38, 1230 (1944); Proc. Roy. Irish. Acad., 49B, 143 (1943).
510. So derb ack E. Acta chem. scand., 13, 1221 (1959).
511. V о 1 h a r d J. Ann., 267, 176 (1891).
512. S t e i n к о p f W., В auermeister M. Ann., 403, 50 (1914).
513. SteinkopfW., Ki 11 i ngsta d A. Ann., 532, 288 (1937).
514. В r i s с о e H. V. A., P e e 1 J. B., Y о u n g G. W. J. Chem. Soc., 1929, 2589.
515. Schwalbe C. Ber., 38, 2208 (1905).
516. D e n i g e s G. C. r., 120, 628, 781 (1895); Bull. Soc. chim. France [3], 15, 1004 (1896).
517. P а о 1 i n i V. Gazz., 37, I, 58 (1907).
518. PaoliniO., SilbermannB. Gazz., 45, П, 385 (1915).
519. Buu-Hoi N. P. Bull. Soc. chim. France, 1958, 1407.
520. Арбузов Б. А., К а т a e в E. Г. ДАН СССР, 96, 983 (1954).
521. S t e i n к о p f W. Ann., 428, 123 (1922).
522. Steinkopf W., Hanske W. Ann., 527, 264 (1937).
Введение ртути на место водородного атома (меркурирование) 113
523. SteinkopfW. Ann., 424, 23 (1921).
524. Steinkopf W., Leonhardt P. Ann., 495, 166 (1922).
525. CagniantP., CagniantP. Bull. Soc. chim. France, 1952, 713; 1956, 1152.
526. Horton A. W. J. Org. Chem., 14, 761 (1949).
527. Ю p ь ев Ю. K-, X м e л ь н и ц к и й Л. И. ДАН СССР, 92, 101 (1953).
528. Steinkopf W., М е г с к о 11 A. Ann., 545, 45 (1940).
529. Steinkopf W., Frofflmel Н., L e о J. Ann., 546, 199 (1941).
530. В о g e r t M. T., H e г г e г a P. P. J. Am. Chem. Soc., 45, 238 (1923).
531. В о g e r t M. T., Andersen C. N. J. Am. Chem. Soc., 48, 223 (1926).
532. Parham W. E., De Lai tsch D. M,. J. Am. Chem. Soc., 76, 4960 (1954).
533, Fromm E., FantlP., Lei bsohn E. Ann., 457, 267 (1927).
534. Steinkopf W.( Kohler W. Ann., 532, 250 (1937).
535. Steinkopf W., J acob H., P e n z H. Ann., 512, 136(1934); SteinkopfW.
Ann., 513, 281 (1934).
536. Steinkopf W., R 6 s 1 e r R., Setzer L. Ann., 522, 35 (1936).
537. SteinkopfW., Jacob H. Ann., 515, 273 (1935).
538. Steinkopf W., S e m i 11 H. F., Fiedler H. Ann., 527, 237 (1937); Stein-
kopfW., H a n s к e W. Ann., 532, 236 (1937).
539. Sice J. J. Am. Chem. Soc., 75, 3967 (1953).
540. M a t s u m о t о I., S о n e T. C. A., 57, 85, 28 (1962).
541. KuhnR., Dann 0. Ann., 547, 293 (1941).
542. D u n n F. W., Di ttmer K. J. Am. Chem. Soc., 68, 2561 (1946).
543. D a n n О., M 6 1 1 e r E. F. Ber., 80, 23 (1947).
544. Hurd Ch. D., Priestley H. M. J. Am. Chem. Soc., 69, 859 (1947).
545. S t e i n к о p f W., K-о h 1 e r W. Ann., 522, 17 (1936).
546. Uhlenb rock J. H., BijlooJ. D. Rec. trav. chim., 78, 382 (1959).
547. Steinkopf W., Lei tsmann R., Hofmann К. H. Ann., 546, 180 (1941).
548. CagniantP., CagniantP. Bull. Soc. chim. France, 1953, 713.
549. CagniantP., CagniantP. Bull. Soc. chim. France, 1955, 680.
550. CagniantP., CagniantP. Bull. Soc. chim. France, 1955, 1252.
551. Г о л ь дф a p б Я. Л., Г о p у ш к и н а Г. И., Федоров Б. П. Изв. АН
СССР, ОХН, 1956, 340.
552. S t е i п к о р f W. Ann., 545, 38 (1940).
553. Rhodehamel Н. W., jr., D e d e r i n g E. F. C. A., 36, 7238 (1942); J. Am.
Pharm. Assoc., 31, 281 (1942).
554. DannO., KokorudzM. Ber., 91, 172 (1958).
555. Б p о у и А. С., ВороиковМ. Г. Научи, бюлл. ЛГУ, 20, 6 (1948).
556. В о р о н к о в М. Г., Б р о у н А. С, ЖОХ, 18, 70 (1948).
557. П е р в е е в Ф. Я., Кудряшова Н. И. ЖОХ, 23, 976 (1953).
558. ПервеевФ. Я., Куреньгии а Т. Н. ЖОХ, 25, 1619 (1955).
559. Birch S. F., С u 1 1 и m Т. V., Dean R. A., Redford D. G. Tetrahedron,
7, 311 (1959).
560. W е i n m а у г V. J. Am. Chem. Soc., 74, 4353 (1952).
561. C a m p a i g n e E., DiedrichJ. L. J. Am. Chem. Soc., 73, 5240 (1951).
561a. P г о f f t E„ К и b a t A. Ann., 634, 185 (1960).
562. C h a 1 1 e n g e r F., M i 1 1 e r S. A. J. Chem. Soc., 1939, 1005.
563. В e z d r i k A., Friedlander P., KonigerP. J. Chem. Soc., 41, 232 (1908).
564. Weissgerber R„ KruberO. Ber., 53, 1557 (1920).
565. W e i t k a m p W., Hami 1 ton C. S. J. Am. Chem. Soc., 59, 2699 (1937).
566. Umezawa S. Zbl., 1939, II, 3821; Bull. chem. Soc. Japan, 14, 155 (1939).
567. Юрьев Ю. К., С адовая H. К., Га льберштам M. А. Ж0Х, 28, 620
(1958).
568. Юрьев Ю. K-, Садова я H. К., Га льберштам M. А. ЖОХ, 29, 1970
(1959).
569. Ф н ш'е р Г., О р т Г. Химия пиррола, т. 1. Л., ОНТИ, Хнмтеорет, 1937, стр. 93.
570. Р 1 a n с h е г G., R о s s i G. Zbl., 1925, I, 2076; Gazz., 55, 61 (1925).
571. M i n g о i a Q. ZbL, 1930, П, 2780; Gazz., 60, 509 (1930).
572. Нем. пат. 236893 (1910); Zbl., 1911, II, 404.
573. С e c h e 11 i В., S a r t i U. Gazz., 60, 189 (1930).
574. G i 1 m a n H., К i r b у R. H. J. Org. Chem., 1, 146 (1936).
575. F i n a r I. L„ G о d f г e у К. E. J. Chem. Soc., 1954, 2293.
576. F i n a r I. L. J. Chem. Soc., 1955, 1205.
577. Schrau th W., Bauerschmidt H. Ber., 47, 2736 (1914).
578. R ag no M. Gazz., 68, 741 (1938).
579. Pa deri C. Zbl., 1919, II, 226; Arch, pharm. sperim., 26, 359 (1918).
580. О 1 i ver i-Ma nd a 1 a E. Zbl., 1921, III, 643; Gazz., 51, 1125 (1921).
581. T r a v a g 1 i G. Gazz., 85, 926 (1955).
582. T r a v a g 1 i G. Gazz., 78, 592 (1948).
114
Ртутноорганические соединения
583. Ferreira Р. С. Ciencia (тех.), 10, 139 (1950).
584. D а s В. J. Sci. Ind. Research (India), 15В, 613 (1956); С. Д., 51, 8725 (1957).
585. R о u t М. К. J. Indian Chem. Soc., 33, 741 (1956).
586. Т a s h i к a Y. С. А., 56, 8699 (1962).
587. Яшу некий В. Г., Васильева В. Ф., ШейнкерЮ. Н. ЖОХ, 29,
2712 (1959).
588. N a k a h а г а К., О h t а М. С. А., 53, 5251 (1959);. Nippon Kagaku Zasshi, 77, 1306
(1956).
589. Tien J. M., Hunsberge r J. M. J. Am. Chem. Soc,, 83, 178 (1961).
590. C h i g i E., Pozzo-Balbi T. C. A., 37, 3092 (1943); Ann. chim. farm., 1940, 141.
591. Woods L. L. J. Org. Chem., 22, 341 (1957).
592. M a i 11 e P. Ann. chim. (France), 9, 431 (1954).
593. P a r h a m W, E., S t r i g h t P. Z., H a z e к W. R. J. Org. Chem., 24, 262 (1953).
594. S w a n e у M. W., S к e e t e r s M. J., S h r e v e R. N. Zbl., 1940, II, 235; Ind.
Eng. Chem. ind. Edit., 32, 360 (1940).
. 595. S a c h s G., E b e r h a r t i n g e r R. Ber., 56, 2223 (1923).
596. McC lei a n d N. P., W i 1 s о n S. H. J. Chem. Soc., 1932, 1263.
597. KanvindeC. K., BbrkarR. S., Kothare A. N„ N adkarny V. V.
C. A., 37,2008 (1943).
598. Несмеянов A. H., Луценко И. P. ЖОХ, 2, 382 (1941).
599. H u r d C. D., M о r r i s s e у C. J. Am. Chem. Soc., 77, 4658 (1955).
600. C lemo G. R., Swan G. A. J. Chem. Soc., 1948, 198.
601. UkaiT. Yamamoto Y., Hirano S. C. A., 48, 9946 (1954); J. Pharm. Soc.
Japan, 73, 823 (1953). . '
602. Van A m m e r s M., Den H e r t о g H. J. Rec. trav. chim., 77, 340 (1958).
603. Van A m m e r s M., Den H e r t о g H. J. Rec. trav. chim., 81, 124 (1962).
604. Несмеянов A. H., Макарова Л. Г. Неопубликованные результаты.
605. Австр. пат. 112128 (1925); Zbl., 1929, II, 652.
606. Р i е г о n i A. Zbl., 1927, I, 3003; Atti Accad. naz. Lincei. Rend.classe Sci. fis. nat. nat.
6, 5, 303 (1927).
607. Takahashi T., YonedaF. РЖХим, 1960, 8, 30865; Chem. and Farmacol.
Bull., 6, 611 (1958).
608. P г о f f t E„ О t t о К. H. J. prakt. Chem., 8, 156 (1959).
609. U k a i T., Zbl., 1931, II, 2330; J. pharm. Soc. Japan, 51, 73 (1931).
610. U k a i T. Zbl., 1928, I, 353; J. pharm. Soc. Japan, 47, 119 (1927).
.611, U k a i T. Zbl., 1928, II, 53; J. pharm. Soc. Japan, 48, 75 (1928).
612. UkaiT. Zbl., 1928, II, 1885; J. pharm. Soc. Japan, 48, 116 (1928).
613. S e n R. N„ Mukherjee G. K. J. Indian Chem. Soc., 11, 541 (1934).
614. P i г г о n e F. Zbl., 1940, II, 53; Atti Accad. naz. Lincei Rend. classeSci. fis. mat. nat.,
7, 1, 50 (1939).
615. Нем. пат. 289246 (1913); Zbl., 1916, I, 194.
616. С e c h e t t i B., G о d i E. Zbl., 1929, I, 899; Gazz., 58, 764 (1928).
617. Do m i n i ki ewicz M. Roczn. Chem., 11, 664 (1931).
618. Пат. США 1259517; С. A., 12, 1909 (1918).
619. Нем. пат. 224491 (1909); Zbl., 1910, II, 608.
620. Нем. пат. 286097 (1914); Zbl., 1915, II, 569.
621. С о v е 1 1 о М. 1934, I, 3858; Rend. Accad. Sci. fis. Mat. Napoli j4], 3, (72), 65 (1933).
622. C h a 1 k 1 e у L. J. Am. Chem. Soc., 47, 2055 (1925).
623. Soderback E. Acta chem. Scand., 11, 1622 (1957).
624. Несмеянов A. H., Перевалова Э. -Г., Головня P. В., Несмея-
нов a O. A. ДАН СССР, 97-, 459 (1954).
625. Rausch M., Vogel M„ Rosenberg H. J. Org. Chem., 22, 900 (1957).
626. Пат. США 2835686. (1958); С. A., 52 (1958).
627. H e с м e я н о в A. H., П е р е в а л о в а Э. Г., ГоловняР.В., Симу-
коваН. А., СтаровскийО. В. Изв. АН СССР, ОХН, 1957, 638.
628. R a u s с h М. D., Fischer Е. О., G г u b е г t Н. J. Am. Chem. Soc., 82, 76
(1960). . . . ,
629. Несмеянов А. Н., Анисимов К. Н., Валуева 3. П. Изв. АН СССР,
ОХН, 1962, 1683.
630. F i s с h е г Е. О., Р 1 е s z.k е К. Вег., 91, 2719 (1958).
631. Несмеянов А. Н., Колобова Н. Е., Анисимов К. Н., Барыш-
никова Л. И. Изв. АН СССР, ОХН, 1964, 1135. <
632. MayuranathanP. S. J. Chem. Soc., 1957, 495.
Г лава VI
ПРИСОЕДИНЕНИЕ СОЛЕЙ РТУТИ
К НЕПРЕДЕЛЬНЫМ СОЕДИНЕНИЯМ
И ПРОИЗВОДНЫМ ЦИКЛОПРОПАНА
ВВЕДЕНИЕ
Среди методов синтеза ртутноорганических соединений особое место за-
нимает метод получения их путем присоединения солей ртути к олефинам,
ацетиленам и их производным, кетенам и окиси углерода. Этим методом
синтезируются только вещества, пока лишь в единичных случаях получен-
ные иными способами (см. стр. 119). Вещества эти замечательны тем, что
в своих химических реакциях они ярко проявляют двойственность, в из-
вестном смысле имитирующую различную реакционную способность тауто-
мерных соединений. В одних случаях они .реагируют как истинные метал-
лоорганические вещества, образованные путем присоединения соли ртути
по кратной связи непредельной молекулы, в других же реакциях они ведут
себя подобно л-комплексным аддуктам соли ртути и непредельного соеди-
нения; в реакциях, в которых эти адденды подвергаются замещению. Это
в свое время дало повод к длительной дискуссии об Их строении (см. ниже),
в которой одни исследователи рассматривали эти вещества как настоящие
металлоорганические соединения — продукты присоединения солей ртути
по кратной связи; другие же приписывали им строение комплексных со-
единений (л-комплексов в современной терминологии) (см. стр. 117).
Реакции продуктов присоединения солей ртути
к олефиновым соединениям
Продукты присоединения солей ртути к олефинам, открытые Гофма-
ном и Зандом [1—7], известны в настоящее время для широкого ряда соеди-
нений, содержащих С=С-связи. В зависимости от растворителя,
в котором происходит присоединение соли ртути к олефину и который всег-
да принимает участие в реакции, образуются следующие типы соединений:
\ СН2 — CH2+]HgXa +Н2О-^ HOCHaCH2HgX + НХ [1,5,7] (1)
2CHS = СН3 + HgX2 + HSO -» О (CH2CH2HgX)2 [6] (2)
СНа = СНа + HgXs + AlkOH-> AlkOCHaCHaHgX + НХ [8,9] (3)
СНа = CHa+.HgXa + HOCOR -» RCOOHgCH2 CHaOCOR + HX [10 — 15, 20] (4)
CHa = CHa + HgX2+R2NH.-^R3NCH2CHaHgX + HX [16— 19} (5)
Получение продуктов типа R'CH(Z) — CHR"HgX, где Z = Ar [21—25]
или остаток ацетоуксусного эфира [26, 26al, R', R" = H или Aik, и типа
AlkSCR'H — CHRffHgX [27] описано ниже (стр. 137).
116 Ртутноорганические соединения. Лит. стр. 173—181
Всем этим веществам, несомненно, принадлежит указанное выше строе-
ние, так как они гладко замещают ртуть на водород при восстановлении
амальгамой натрия или амальгамированным алюминием в присутствии воды
или при действии избытка гидразингидрата [28, 29]. Так, алканол ртутные
соли (1 и.2) образуют при этом соответствующий спирт, например, продукт
присоединения к этилену дает этиловый спирт [7]:
HOCH2CH2HgX ^?НОСНйСН8,
продукт присоединения к стиролу C6HBCH(OH)CH2HgOCOCH3 — фенил-
метилкарбинол [30], продукт присоединения к метилстиролу — фенил-
диметилкарбинол [30]:
C8H5C(CHs)OH-CH2HgOCOCHsС8н5 (СНзИ сон,
продукт присоединения к 3-метилциклогексену — 2-экзометил-эндо-цик-
логексанол [31].
Алкоксиалкилртутные соли (3) при действии амальгамы натрия дают
простые эфиры [281:
' AlkOCHaCHjHgX ^^AlkOCgHs;
продукты присоединения солей ртути в среде вторичных аминов (5) — тре-
тичные амины. Также действие иода на продукты присоединения солей рту-
ти к алкенам. (1) приводит к иодгидринам, например [5];
HOCHaCH2HgX i НОСНгСНД
реакция нормальная для ртутноорганических солей. Действие сероводо-
рода на некоторые из этих соединений в определенных условиях ведет
к замене ртути на водород [32] (но сернистые щелочи дают сульфид
(HOCaH4Hg)aS) [331.
Указанное выше строение продуктов присоединения солей ртути к ал-
кенам доказывается также характерными реакциями на функциональную
группу — ацилированием [5, 28] спиртового гидроксила действием галоид-
ных ацилов (например этанолмеркуриодид с хлористым бензоилом дает бен-
зоильное производное С 6H5COOCH2CH2HgJ [5,161 ]), действием фенилизоциа-
ната (пример — образование фенйлуретана C6H5NHCO2CH2CH2HgBr дей-
ствием фенил изоцианата на этанолмеркурбромид [34]) или путем солеобра-
зования за счет третичного азота амииоалкилртутной соли [19]:
RaNCHaCHaHgCl + НС1 (RaHNCH2CH3HgCl)+ С1~.
Возможно провести и окисление спиртовой функции в кислотную или
в кетонную. Так, окисление этанолмеркурбромида действием брома и ще-
лочи приводит к броммеркуруксусной кислоте [1,51:
HOCH2CH2HgBr BrHgCH2COOH.
Окисление перманганатом калия продуктов присоединения солей ртути
к олефинам приводит к мономер кур ированным кетонам [35]. Окисление
2-ацетоксимеркурциклогексанола действием перекиси водорода и соли
двухвалентного железа дает 2-ацетоксимер курциклогексанон [36]. Извест-
ны также примеры отнятия элементов воды от алканолртутных солей, что
приводит к винилртутным соединениям, например [371:
(CsHsh COHCH2HgOCOCH8(СвН8)2 C=CHHgOCOCHs.
Присоединение солей ртути к непредельным соединениям
117
Связанная с углеродом галогенмеркургруппа XHg ведет себя в ряде
реакций так же, как она ведет себя в обычных ртутноорганических соедине-
ниях [38], подобных метилмеркурхлориду CH3HgCl, например заменяет
галоид на арил при действии ароматических оловоорганических и других
металлоорганических соединений [341:
2HOCH2CH2HgBr + (CeH5)2 SnO + 4NaOH -> 2HOCH2CH2HgC6H5 + Na2SnO3 +
+ 2NaBr + 2H2O.
При действии диазометана также происходит в известной мере анало-
гичная реакция замены галоида на бромметильный радикал [39]:
HOCHaCHaHgBr + CH2N2 -» HOCHaCH2HgCH2Br -J- N2.
Однако под действием реагентов, связывающих соли ртути в комплекс
(KJ [401, KCN [11, KCNS и т. д.), и CH3J [1,411 все без исключения упомя-
нутые типы продуктов присоединения солей ртути к олефинам легко при
комнатной температуре элиминируют олефин, например:
ROCaH4HgCl + 4KJ + Н2О -» СаН4 K2HgJ4 + ROH + КОН + КС1.
Алканолмеркурсоли и их эфиры легко и количественно распадаются с от-
щеплением олефина под действием даже разбавленных минеральных кислот:
ROCaH4HgX + НХ ROH + С2Н4 HgX2,
₽-аминоалкилмеркурсоли образуют соли при действии минеральных кислот,
например:
RssNCAHgX + НС1 [R2NHC2H4HgX]+CK
Описана кинетика реакции ₽-окси- и Р-алкоксиалканртутных солей с ки-
слотами (гл. XIV, стр. 273), а также механизм действия кислот (стр. 119).
При действии на соединения типа (3) ₽-галогенмеркурэтилметиловый
эфир — соединения с более активной кратной связью (кетен) также происхо-
дит выделение этилена [41].
Это совершенно исключительная легкость элиминации олефина из ал-
канолртутных солей (и их эфиров), вероятно, самый яркий пример ^-элими-
нации, дала в свое время повод Маншо [42] рассматривать эти вещества
как комплексные, соединения типа C2H4.Hg(OH)X, C2H4.Hg(OAlk)X и т. п.
(л-комплексы в современной терминологии) и, как уже сказано, выз-
вала длительную дискуссию об их алканольном (соответственно эфирном)
[1—7, 33, 43—46] или комплексном (в общем виде R4R2C = CR3R4-
•Hg(OR)X) [42, 47] строении, или таутомерии в смысле схемы [48, 49]:
СН2- /ОН СН2ОН
Il ;-Hg< |
СН/ \Х CHa.HgX
Решающими работами по выяснению структуры обсуждаемых соедине-
ний были следующие. Адамс и сотр.- [45, 46] присоединением сулемы к
о-аллилфенолу получили 1-хлормеркурметил-1,2-дигидробензофуран
----сн2
M\0/CHCH2HgClj
который оказался устойчив к действию соляной кислоты и, что особенно
важно, при действии амальгамы натрия оказался способным симметри-
зоваться. С иодом он дает 1-иодметил-1,2-дигидробензофуран. Вещества
118
Ртутноорганические соединения. Лит. стр, 173—181
с аналогичной структурой и свойствами получаются и из алл ил резорцинов
[501:
ОН
-----СН2 ----сна
V\0/CHCH2HgCl H0/^A0/CHCHaHgCl.
Способность к симметризации не может быть согласована с представле-
нием о комплексной структуре этих веществ. Впрочем, эти соединения под
воздействием ряда других симметризующих агентов размыкаются в исход-
ные аллилфенолы. Осуществленное Несмеяновым и Фрейдлиной [34] уже
упоминавшееся арилирование ароматическими оловоорганическими соедине-
ниями этанолмеркурбромида (и других подобных соединений) привело к
несимметричным соединениям типа
I I
НО—С—С—HgCeH5,
в случае комплексного строения эта реакция должна была бы привести к ди-
фенилртути. Сюда же примыкает алкилирование этанолмеркурбромида
диазометаном, приведшее к несимметричному соединению, но неустойчиво-
му и распадающемуся выше 0° С с выделением этилена:
HOCHaCHaHgCHaBr -» СгН4 ф HOHgCHaBr -» СН2О + Hg + НВг
НВг + HOHgCHaBr BrHgCHaBr + HaO
и структура его может быть изображена с равным правом как комплексная
139].
Важнейшие доказательства «главновалентной» природы продуктов при-
соединения солей ртути к непредельным соединениям (как по двойной,
так и по тройной связи; см. также далее во введении к этой главе, стр. 120)
внесены стереохимическими исследованиями в этой области.
Так, Марвел и сотр. [431 доказали образование двух диастереомеров
I -ментилового эфира а-броммеркурф-фенилф-метоксигидракриловой кисло-
ты при присоединении ртутной соли к /-ментиловому эфиру коричной кис-
лоты (аналогичный результат был получен и с корично-а-борниловым эфи-
ром [44]), что,, по их мнению, должно было доказать появление двух новых
асимметрических углеродных атомов в согласии с уравнением
CSH5CH = СНСООС10С19 + HgXa + СН8ОН -> С6Н5С*Н (ОСН3) — С*Н (HgX) COOCioHi»
и тем подтвердить формулы Гофмана.
Оставалось не ясным, почему не обнаружены два остальных требуемых
теорией диастереомера. Пфейффер [47] указал, что молекулярная струк-
тура
COOCioHie
С8Н6СН А*......Hg(OCH8)X
объясняет факт существования именно двух диастереомеров. Однако отсут-
ствие двух остальных стереоизомеров могло зависеть и от трудностей выде-
ления.
В работах Несмеянова и Борисова даны основы стереохимии продуктов
присоединения солей ртути к ацетиленовым соединениям (см. ниже).
Аналогично Райтом в целом ряде исследований было показано на при-
мере главным образом алициклических алкенов, что присоединение солей
Присоединение солей ртути к непредельным соединениям
119
ртути по двойной связи происходит стереоспецифично, причем из цис- и
транс-изомерных олефинов получаются соответственно различные диасте-
реомеры, не содержащие примеси второго диастереомера (см., например,
(5И). Присоединение соли ртути не сопровождается цис-транс-изомериза-
цией олефина, а «дезоксимеркурирование» приводит к олефину исходной
конфигурации (см. стр. 134, 144, 155). Эти данные также не совместимы с
представлением о комплексной природе этих веществ.
• Наконец, удалось получить вещества, рассматриваемые в настоящей
главе, и встречным синтезом, а не путем присоединения солей ртути По крат-
ной связи. Так, Несмеяновым и Луценко 1-хлормеркурпропанол-2 был по-
лучен восстановлением изопропилатом алюминия хлормеркурацетона, од-
ного из впервые полученных ими а-хлормеркуроксосоединений (подробнее
о работах Несмеянова и Луценко по получению и особенностях поведения
ртутных производных оксосоединений см. ниже).
Спектр ядерного магнитного резонанса продуктов присоединения соли
ртути к этилену в воде и в метаноле подтверждает их строение как нормаль-
ных соединений с a-связью С—Hg, а не л-комплексов [52].
. В настоящее время на основании всей этой серии исследований метал-
лоорганическую структуру продуктов присоединения солей ртути по крат-
ной связи в духе формул Гофмана можно считать установленной.
На основании изучения распределения циклогексена между СС14 и вод-
ным раствором азотнокислой окиси ртути Уинстейн и Люкас с сотр. [53]
пришли к выводу о наличии в реакции присоединения солей ртути к алке-
нам предварительной стадии образования комплексов, выражаемой уравне-
ниями:
C6HiO + Hg++ztC0HiOHg++
CeHio + Hg++ + H20^.[C6HioHg+ + ОН + н+.
(2)
Строение гипотетического иона реакции (1) можно выразить, по мнению
авторов, резонансными формулами:
СН
С4Н8 Hg++,
\н
СН+
С4Н
СН+
Hg+,
СН
С*Н8/| \ig++.
СН
Ион—продукт реакции (2) имеет аналогичное строение с ОН-группой,
связанной со ртутью. Строение обычного типа «гофмановского» продукта,
получаемого дальнейшим превращением этих гипотетических форм, авторы
не обсуждают.
Описано обсуждение электронной структуры комплексов Уинстейна,
Люкаса и сотр. [54].
Образование подобных л-комплексов ртуть — олефин того или иного
С Z
строения типа ц ->Hg+X принимается в настоящее время (см., например,
[55, 56]) в качестве промежуточной стадии присоединения солей ртути по
двойной связи (реакция оксимеркурирование дезоксимеркурирование)
также на основании изучения кинетики как реакции оксимеркурирования
(см., например, [57—591), так и, в особенности, реакции дезоксимеркуриро-
вания под действием кислот (см., например, [60—651); присоединение соли
ртути проходит по реакциям второго порядка.
В качестве одного из возможных примеров схематического изображения
течения реакции присоединения соли ртути по двойной связи с участием
120
Ртутноорганические соединения. Лит. стр., 173—181
л-комплекса может быть предложен следующий:
С
II -> Hg+x + X-
с
и -» HgX
с
+ ROH ROC — С — HgX + Х+
Н+ + Х-^НХ
Образование аналогичных л-комплексов предполагается также в реак-
циях 0-арилалкилацетоацетатов и -спиртов, (3-диарилалканов и этил-а-(2-
ацетоэтил)ацетоацетата при взаимодействии соответственно ароматиче-
ских углеводородов [21—25] и ацетоуксусного эфира [26] с олефинами в
присутствии солей ртути, идущем через промежуточное образование соли
0-арилалкилртути и эфира 1-(а-ацетоацетил)этил-2-ртути.
По механизму, защищаемому главным образом Райтом [27, 51, 66—72],
считающим концепцию «меркуриниевого иона» в смысле представлений
Уинстейна, Люкаса и др. или л-комплекса недостаточно убедительной, пред-
полагается (в особенности для реакций, идущих стереоспецифично) моле-
кулярное присоединение алкокси- (ROHgX) или основной (HOHgX) ртут-
ной соли к олефину по схеме:
HgX2 + ROH ROHgX + HX;
ROHgX + R'CH = CHR' ROCR'HCR'HHgX.
Этот механизм базируется также на кинетических данных, в частности
на большей скорости присоединения в спирте, чем в воде [66] (в случае
ионного присоединения отношения, по мнению Райта, должны были бы
быть обратными). В литературе имеются возражения против этой точки
зрения [60—62, 64].
Не исключено, что реакция окси-(алкокси) меркурирования осуществ-
ляется разными механизмами в зависимости от природы ненасыщенного
соединения, в частности от природы заместителей у одного или у обоих
атомов углерода двойной связи [62].
Присоединение OR- и HgX-групп по двойной связи, как правило, проис-
ходит в транс-положение друг к другу, что также говорит в пользу проме-
жуточного образования л-комплекса [27, 51]. Соколовым и Реутовым [731
транс-присоёдинение к циклогексену доказано на основании расчетов моле-
кулярных вращений цис- и транс-продуктов присоединения. Известны не-
многие случаи цис-присоединения [74, 75 и др.].
Реакции продуктов присоединения солей ртути
по тройной связи
Еще резче выявляется двойственное поведение продуктов присоедине-
ния солей ртути по тройной связи, в особенности на примере простейших
представителей. Ацетилен при действии на него сулемы в солянокислой,
спиртовой [76—79] или водной [79, 80] среде дает продукт состава
НС = CH-HgCl2 (I) (Бигинелли), которому на основании некоторых свойств
Дженкинс [77] приписал структуру ClCH=CHHgCl (см. стр. 166). Этот
продукт способен симметризоваться под влиянием аммиака (см. гл. XIII,
стр. 255), образуя соединение coCTaBa(CH=CH)2HgCl2(II),Ho соответствующее
Присоединение солей ртути к непредельным соединениям 121
по своим реакциям также и структуре (ClCH=CH)2Hg. Свойства обоих ве-
ществ подробно изучены Несмеяновым, Фрейдлиной, Борисовым. Веще-
ства I и II с легкостью отщепляют сулему под влиянием агентов, ее связы-
вающих, например тиосульфата, цианистого калия, йодистого калия, серо-
водорода, трифенилфосфина (образование комплекса [(C6H5)3P]2HgCl2)[80].
Оба вещества арилируются хлористым дифенилоловом, как свободная суле-
ма, в нейтральной среде образуя CeH5HgCl, в щелочной — (C6H6)2Hg (от-
личие от алканолртутных солей [80]). Диазометан так же реагирует с веще-
ством (I), как со свободной сулемой, давая хлорметилмеркурхлорид
ClCH2HgCl [39,80].
Арилазокарбонрвокислые соли с Р-хлорвинилмеркурхлоридом образуют
хлористую арилртуть, а также АгСН=СНС1 [81]. Однако щелочь пре-
вращает ClCH=CHHgCl в гидроокись, а последняя может быть превращена
в ряд солей ClCH=CHHgX, где X — анион брома, циана, уксусной или
бензойной кислоты [82]. Наряду с этим иод от обоих соединений отщепляет
иодхлорэтилен [80], а действие на ClCH=CHHgCl хлористого водорода или
бромистого водорода приводит к образованию хлористого винила [83],
что ясно доказывает хлорвинильную структуру этих ртутных соединений.
Несмеяновым, Борисовым [84] по реакции
(guc-ClCH=CH)3Sb + 3HgCl2 -> 3 qMc-ClCH=CHHgCl + SbCl3,
- азатем изомеризацией транс-ClCH—CHHgCl в цис-изомер действием переки-
си [85] получен цис-изомер Р-хлорвинилмеркурхлорида (вещества Бигинел-
ли), гладко симметризующийся аммиаком в бензольном растворе в жидкую
ди-г<нс-(Р-хлорвинил)ртуть [861, и показано [86], что вещество Бигинелли
(I) и продукт его симметризации (вещество Дженкинса (II)) представляют
собой транс-изомеры. Затем Фрейдлиной и Ногиной [87] цис-хлорвинил-
меркурхлорид получен прямым соединением паров сулемы с газообразным
ацетиленом. Кинетика реакции изучена Смирновым-Замковым и Шиловым
[881. По химическому поведению эти вещества подобны своим транс-изоме-
рам, обнаруживая двойственное химическое поведение: реакции хлорви-
нильных ртутноорганических соединений, с одной стороны, и комплексных
соединений сулемы и ацетилена — с другой. Однако реакции отщепления
ацетилена от цис-изомеров протекают заметно медленнее, чем от веществ,
транс-конфигурации.
Эти вещества являются вполне устойчивыми и. не обнаруживают ни
в какой степени взаимного превращения стереоизомеров, притом не только
при хранении в индивидуальном состоянии или в растворе, но при реакции
симметризации и обратной реакции
(С1СН = CH)2Hg + HgCl2 - 2С1СН = CHHgCl.
Транс-изомер ClCH=CHHgCl переходит в цис-изомер только при дей-
ствии перекисей [851 (подобно тому, как под действием перекиси бензоила
хлористая транс-2-метоксициклогексилртуть переходит в цис-изомер [14]}
и при облучении ультрафиолетовой радиацией [89]. (О переходах одного
геометрического изомера алкенильного соединения ртути в другой см.
гл. XIV.) Далее Несмеяновым и Борисовым изучены реакции передачи
Р-хлорвинильно го радикала от атома ртути на другие металлы, например:
(С1СН = СН)2 Hg + SnCl2 -> (С1СН = СН)2 SnCl2 + Hg,
2 (C1CH = CH)2 Hg + Т1Вг3 - (C1CH = CH)2 TIBr + 2CICH = CHHgBr
и обратные переходы, например:
(С1СН = СН)2 SnBr2 + 2HgBr2 -> 2C1CH = CHHgBr.
122
Ртутноорганические соединения/. Лит. стр. 173—181
СХЕМА
Присоединение солей ртути к непредельным соединениям
123
Для всех переходов, изученных отдельно для транс- и цис-рядов и могу-
щих быть представленными схемой, строго доказано их осуществление
без изменения стереохимической конфигурации передаваемого радикала
(схема, стр. 122).
Эти реакции были первыми, на которых Несмеяновым и Борисовым
190, 91 ] было установлено правило сохранения конфигурации при электро-
фильных и гомолитических замещениях при олефиновом углероде. Это
правило подтверждено также методом обмена атома ртути на радиоактив-
ный изотоп [92].
Такие же передачи алкенильных радикалов, осуществленные этими же
авторами для впервые полученных ими цис- и транс-изомеров продуктов
присоединения солей ртути к диметил- 193, 94] и дифенил-[95] ацетилену,
также происходят без изменения стереохимической конфигурации.
Существование устойчивых цис- и транс-изомеров Р-хлорвинильных со-
единений, ртути не позволяет объяснить двойственное реагирование этих
соединении таутомерией или наличием подвижного равновесия аддукта с
продуктами его диссоциации, но заставляет придать единую структурную
формулу каждому из этих веществ.
Более сложные полимеркурированные продукты, получаемые в резуль-
. тате присоединения солей ртути к ацетилену в водной среде (см. стр. 166),
также обладают способностью отщеплять ацетилен (например при действии
KCN) и, с другой стороны, в отличие от олефиновых производных, при
реакции с кислотами дают нормальную реакцию замещения ртути на водо-
род, приводящую в случае ацетилена, к ацетальдегиду, а в случае аллиле-
на — к ацетону, как это установлено Кучеровым [96, 97]. Бильц и Мумм
198] приписали одному из этих соединений формулу (ClHg)3C—СНО. На
основании исследований Мидльтона и Баррета [99—101] продуктов присо-
единения уксуснокислой ртути к кислотам ацетиленового ряда, где доказана
структура —С=С—HgO2CCH3, вещество (ClHg)3CCHO должно, вероятно,
(JHgOaCCHg
иметь строение (ClHg)2C=CH—OHgCl. Действительно, вещества Мидльтона
и Баррета при действии хлора образуют моногалоидозамещенные оксо-
соединения
/ 4- HCI I / fl
. -с=с/ + С12 ——1 с = с/ -» С-СН
OHgX HgX . Он С1 О С1
тогда как структура —С—С—HgX привела бы к дигалогенкетону
О ^HgX
—С—С—Cl. ПриДействии FeCl3образуется окрашенный энолят С=С—HgX.
О \1 OFeCk
Впрочем, хлорирование «трихлормеркурацетальдегида» ведет к хлоралю
198], что, однако, вряд ли доказывает структуру; Альтернативным для этого
типа соединений должно быть строение молекулярного типа, защищавшееся
______________С
5 Маншо [102]. |]|HgO«HgX2. О строении другого производного ацетилена
. —С
„ C2HO4NHg2 ПОЗ] см. далее (стр. 166).
Так же как и присоединение солей ртути к олефинам, присоединение со-
лей ртути по тройной связи, как это следует из полученных Смирновым-
124
Ртутноорганические соединения. Лит. стр. 173—181
Замковым и Шиловым [88] данных по изучению кинетики реакции ацети-
лена с сулемой в парах (реакция третьего порядка), проходит через образо-
вание л-комплекса.
Реакции продуктов присоединения солей ртути
к окиси углерода
К этому же классу соединений со свойствами, промежуточными между
свойствами комплексных и -истинных металлоорганических веществ, при-
мыкают открытые Шеллером и Шраутом [104] продукты присоединения
окиси углерода к солям ртути, которые получаются при пропускании окиси
углерода под небольшим давлением (2 атм) в раствор уксуснокислой ртути
в метаноле или этаноле. Продукт имеет строение ацетатмеркурмуравьиного
эфира CH3COOHgCOOCH3 или (в этаноле) CH3COOHgCOOC2H6, так как
он с хорошим выходом восстанавливается в соответствующий муравьиный,
эфир (амальгамой алюминия) и с галоидом дает галогенугольный эфир (иден-
тифицированный в виде уретана). Ацетат-ион обменивается на галоид, об-
разуя, например, ClHgCOOCH3. Последнее вещество действием трифенил-
фосфина симметризуется в бис-карбометоксифенилртуть:
2ClHgCOOCHs + 2 (Свн5)з Р (CH8OOC)2 Hg + [(С6Н5)3 Р]2. HgCl2.
Однако продукты присоединения окиси углерода к солям ртути при плав-
лении, действии кислоты или других симметризующих агентов легко
выделяют окись углерода. Поэтому высказывались [105] суждения об
их структуре как молекулярных соединений окиси углерода, анало-
гичных таким комплексам, как PtCl2-2CO, CuCl.CO-2H2O [106] и др.
(см. [107]).
Окись углерода присоединяет сулему также в среде вторичных аминов
[108], в пиперидине образуя продукт состава
CsHioN-C-HgClCsHwNH.
Строение продуктов присоединения солей ртути к окиси углерода как
истинных металлоорганических соединений XHgCOOCH3 подтверждено
ИК-спектрами и спектрами протонного резонанса [109]. Описано примене-
ние полярографических измерений к установлению строения продуктов из.
этилена и окиси углерода [ПО].
Строение продуктов присоединения солей ртути
к олефинам, ацетиленам и окиси углерода
Таким образом, весь этот класс ртутноорганических соединений — про-
дуктов присоединения солей ртути к этилену, ацетилену, их гомологам и
функциональным производным и окиси углерода,— имея структуру настоя-
щих металлоорганических-веществ, по реакционной способности имити-
руют в ряде реакций л-комплексные соединения. На деле это лишь исклю-
чительно легко протекающая р-элиминация. На основании этого такие ве-
щества были названы Несмеяновым [111] квазикомплексными (см. также
[112—115]). Этот класс ртутноорганических соединений расположен в се-
редине ряда, начинающегося такими металлоорганическими соединениями,
как льюизиты C1CH=CHAsC12, (C1CH=CH)2AsC1 и (ClCH=CH)3As, и кон-
чающегося комплексными продуктами присоединения к олефинам солей
Присоединение солей ртути к непредельным соединениям 125
цинка, например (CH3)2C=CH2-2ZnQla-H?O, описанными Кондаковым
[116, 1171, двойными солями типа PtCl2-CaH4, PtCl2-2CO и т. д. [107]
(л-комплексы).
Двойственную - реакционную способность продуктов. присоединения
солей ртути/ к олефинам и ацетиленам — легкую элиминацию олефина и
ацетилена/приближение металла и анионной части-аддукта к ионному со-
стоянию'— Несмеянов объясняет наличием , в этих молекулах о,о-сопря-
женной системы связей [118, 119]:
НО—СН2—СН2—HgCI
Реакционная способность таких систем, как это свойственно сопряжен-
ным системам, характеризуется протеканием реакции не только по связи
1—2, но и, вследствие переноса реакционного центра [118, 119], по связи
1—4:
HgCk + CHsCHsOH
1-2Z
1 а з 4 У
CIHg -СН2-СН2-ОН нНС1<
\1—4
HgCl2 + СНа = СН2 + Н2О
HgCh + CHa = CHCl
1—1/'
CIHg - СН = СН - С1 + НС1/г_4
\gCla + CH==CH.
Так как разрывающиеся связи обе являются о-связями, (о,о-сопряже-
ние), то протекание реакции 1—4 есть не что иное, как реакция элиминиро-
вания исходного олефина или ацетилена. Как показало исследование спек-
тров комбинационного рассеяния p-хлорвинильных соединений металлов
[120], в них отсутствует какое-либо «предсуществование» тройной связи
и, следовательно, не статический, а динамический эффект сопряжения
[118] объясняет квазикомплексные свойства этих соединений. Причиной
особых свойств этих соединений и продуктов присоединения солей ртути
к олефинам является повышенная поляризуемость Hg—С-(сравнительно-
го связью Н—С) и С1—С-связей, находящихся в благоприятном для сопря-
жения положении 1—3. Действительно, открытые и изученные Левиной
с сотр. (см. стр. 171) продукты взаимодействия солей ртути с циклопропано-
выми соединениями, в которых CIHg и С1 находятся в у-положении, совер-
шенно не проявляют квазикомплексных свойств.
Транс-изомеры Р-хлорвинильных соединений ртути (а также других
металлов) в большей степени проявляют квазикомплексные свойства, с боль-
шей легкостью выделяя ацетилен под влиянием агентов, связывающих су-
лему, чем цис-изомеры. Вообще в них связь С—Hg лабильнее, чем в цис-
изомерах. Это особенно наглядно проявляется в реакции цис-транс-ди-Р-
хлорвинилртути [121] и цыс-транс-дипропенилртути [122] с хлористым
таллием:
Н С1 Н С1
2 || || -i-T1C1s—»
н \Hg/ н
126 Ртутноорганические соединения. Лит. стр. 173—181
в которой почти мгновенно происходит обмен транс-хлорвинильной (-про-
пенильной) группы, а цис-хлорвинильный (-пропенильный) радикал остает-
ся связанным со ртутью. Кроме того, транс-изомеры имеют большую моле-
кулярную рефракцию, чем цис-изомеры [123]. По-видимому, это объясняет-
ся большей способностью к сопряжению в транс-расположенных группах
с параллельным расположением осей о-облаков.
Двойственная реакционная способность продуктов присоединения со-
лей ртути к окиси углерода также объясняется наличием б.з-сопряже-
ния.
Реакции и строение а-меркурированных оксосоединений
Продукты присоединения солей ртути к простым и сложным виниловым
эфирам. Изученные Несмеяновым, Луценко и сотр. [124] ртутноорганические
производные оксосоединений — альдегидов и кетонов, получаемые присо-
единением солей ртути к виниловым эфирам и винилацетатам (а также дру-
гими путями, см. стр. 146 этой главы):
OR
CH2=CHOR + HgAc2 -h H2O AcHgCHaCrf .....1 C1'.> ClHgCH2CHO
\ — ROH
OH
CH2=CR'OR + HgAca + H2O AcHgCH2CR'0R -—3L'^ClHgCH2C(O)R-,
| —ROH
OH
хотя и имеют высоколабильную связь С—Hg, не проявляют квазикомплекс-
ных свойств; действие на них таких агентов, как K.CN, KJ в водном раство-
ре, приводит к уксусному альдегиду [125]:
ClHgCH2CHO + 4KJ + Н2О -» K2HgJ4 Ц- CHSCHO + НС1 + КОН.
Однако выделение вин ил алкилового эфира происходит при действии на
меркур-бпс-ацетальдегид (в спирте) соединения с более активной кратной
связью— кетена (Фосс, Луценко, Несмеянов [41]) (одновременно образует-
ся этиловый эфир меркур-бпоуксусной кислоты). Обсуждаемые соединения
имеют строение меркурированных альдегидов и кетонов ClHgCH2CHO и
ClHgCHaCOR. При восстановлении изопропилатом алюминия они дают
квазикомплексные спирты ClHgCH2CH2OH [124], при действии аммиака
симметризуются, образуя Hg(CH2CHO)2 и Hg(CH2COR)2 [126]. Энольные
структуры ClHgCH=CHOH и ClHgCH2=O(OH)R (сами по себе маловероят-
ные) для них исключены, так как они не дают реакции на активный водород
(по Церевитинову — Чугаеву [125]). Также исключаются для этих веществ
и энолятные структуры CH2=CHOHgCl и СН2=С (CH3)OHgCl, хотя бы как
следствие таутомерного равновесия, например:
ClHgCHaCOCH3 СН2=С-СН8
OHgCl
на основании неосаждения окиси ртути при действии щелочи на их водные
растворы и на основании устойчивости хлормеркурацетона к действию на
него перманганата калия в водном растворе на холоду. В то же время эти
вещества содержат очень лабильно связанную ртуть. Они легко, гидроли-
зуются водными растворами гипосульфита натрия, иоднстого калия и по-
добных соединений, способных уводить ртуть в комплексное соединение
(уравнение см. выше). Бромистый фенилмагний с хлормеркурацетальдеги-
Присоединение солей ртути к непредельным соединениям 12?
дом также количественно дает хлористую фенилртуть [125]:
ClHgCH2CHO + CeH5MgCl -» CeH5HgCl + CH2=CHOMgCl.
Однако эти заведомо С-металлические соединения действием ацилирующих
средств ацилируются по кислороду и под действием хлористого ацетила
(и хлористого бензоила) они превращаются [125, 127^130] в соответствую-
щие энолацетаты и энолбензоаты CH2=CHOCOR и CH2=C(CH3)OCOR:
ClHgCH2CHO + RCOC1 CH2=CHOCOR + HgCl2.
По кислороду также ацилируются мер кури рованные альдегиды и кетоны
действием хлорангидридов фосфорной [131], сульфо-[132] и других кислот
[131, 133], а действие на меркурированные альдегиды щелочных металлов,
или хлорного железа дает винилаты соответствующих металлов [134].
Двойственная реакционная способность хлормеркур альдегидов и хлор-
меркуркетонов, типичная для кетоэнолов, объясняется, как уже сказано,
не кетоэнольной таутомерией, а наличием сопряжения, в этих случаях про-
стой и двойной связей (о, л-сопряжение):
Эти вещества способны и к 1—2-реакциям, к которым относятся действие
брома, приводящее к бромзамещенным оксосоединениям [124]:
CIHgCHaCHO + Вг2 -> ВгСН2СНО + HgClBr,'
реакции веществ, уводящих ион ртути (см. выше), реакции алкилирования
трифенилхлор- и трифенилбромметаном [129]:
XHgCH2CHO + (СвН5)8СХ -> (СвН5)зССН2СНО + HgX2.
Действие же три-(л-нитрофенил)хлорметана [129] и особенно реакции
ацилирования, направляющиеся на кислород, являются проявлением 1—4-
реагирования этих систем — результатом переноса реакционного центра
молекулы вследствие сопряжения на кислород:
( |]| (^ II
CiHg—С — С=О 4- CIC—СН3-*HgCl2 + с=с—о—с—сн3
1 ‘ о о
ciHg—с—С—о
HgHal2 + CH2=CHOC(C6H4NO2)3
Продукты присоединения к кетенам. Фоссом, Луценко и Ивановым
[136] присоединением в спиртовой среде окиси ртути к кетену получены
эфиры меркур-бнс-уксусной кислоты:
HgO + ЗСН2=СО + 2ROH -> Hg (CH2COOR)2 + CHgCOOH,
и присоединением ацетата ртути к а-алкоксиакрилонитрилам эфиры моно-
меркурированной уксусной кислоты [135]:
CH2=C.(OR) CN + Hg (ОСОСНз)2 н- Н2О -> CH3COOHgCH2COOR.
128 Ртутноорганические соединения. Лит. стр. 173—181
\ Меркурированная уксусная кислота и ее эфиры получены ими [41]
и другими путями — действием кетена на другие квазикомплексные соеди-
нения: Р-меркурированные оксисоединения и а-меркурирбванные оксосое-
динения [411 (тем самым осуществлен переход от одного типа квазикомп-
лексных соединений к другому). По своим химическим свойствам эфиры мо-
номеркурированной уксусной кислоты аналогичны квазикомплексным со-
единениям.
Эфиры меркурированной уксусной кислоты содержат две системы со-
пряженных связей ртуть — углерод и углерод—кислород <з-л(1—4)
и а~з(1— 4');
при их ацилировании получены продукты реакций по трем направлениям:
--->. R'COCHaCOOR
6 OCOR'
1 2 3 л 1—4 /
-Hg-CH2-C<^ 4, +R'COC1 --- СНа=с/
OR i4, OR
---у R'COOR 4- СНа=С=О.
При действии хлористого ацетила на изобутиловый эфир меркур-бнс-
уксусной кислоты выделен изобутилацетоацетат (реагирование по 1—2-
направлению) и изобутилацетат—продукт 1—4'-реагирования; при дей-
ствии хлористого ацетила на метиловый эфир меркур-бпс-уксусной кислоты
и хлористого изовалерила на изобутиловый эфир меркур-бнс-уксусной
кислоты получены соответственно а-метоксивинилацетат и а-изобутокси-
винилизовалерат — продукты реагирования по 1—4-направлению.
Реакции и строение продуктов присоединения солей ртути
к ацетиленовым спиртам, оксосоединениям и кислотам
А. Н. Несмеяновым и Н. К- Кочетковым получены продукты присоедине-
ния сулемы к ацетиленовым спиртам [138], ацетиленовому кетону [139] и к
ацетиленовым кислотам и их эфирам [140]. Полученные вещества проявляют
свойственную квазикомплексным соединениям двойственную реакционную
•способность.
В продукте присоединения сулемы к ацетиленовому кетону — фенил-
этилметилкетону имеет место одновременное сопряжение Hg—С с С—С1-
и С=Ю-связями:
HgCI
3 /"К 3
С6н5—С=С— с— СНз
fl 2 II
*-С1 о
4 4
И так же, как в хлормеркуральдегидах и хлормеркуркетонах, действие
ацилирующих средств здесь направляется на кислород [139], происходит
О-ацилирование вследствие сопряжения Hg — С- и С=О-связей (реагирова-
ние по связям 1—4 сопряженной системы). В то же время способность су-
лемы к отщеплению в этом соединении резко усилена сравнительно с Р-
хлорвинильными соединениями за счет одновременного сопряжения Hg—С
Присоединение солей ртути к непредельны» соединениям 129
и с С—С1 и со связями С=О: вещество отщепляет сулему не только при дей-
ствии агентов симметризации (KJ, KCN, Na2S2O3), но и (как и продукты
присоединения сулемы к ацетиленовым спиртам и кислотам) под действием
NaCl и даже при перекристаллизации из эфира [1391. При действии кислот
происходит замена HgCl-группы на водород— реакция» обычная для ртут-
ноорганических соединений [1391. Так же более резко выраженные квази-
комплексные свойства, сравнительно с p-хлорвинильными соединениями,
в продуктах присоединения сулемы к ацетиленовым кислотам (отщепление
сулемы не только под действием агентов симметризации, в том числе и ам-
миака, но и под действием NaCl) объясняются повышенной обменоспособ-
ностыо ртутного атома под влиянием сопряжения Hg—С и с С—С1- и с С=О-
группами [140]:
HgCl
rh
R—С=С—С—ОН.
41
Область применения метода синтеза ртутноорганических соединений
посредством присоединения солей ртути к непредельным молекулам доста-
точно широка и своеобразна, так как получаемые этим путем вещества
практически не могут быть получены ни одним другим методом (за исключе-
нием некоторых единичных случаев, см. стр. 119). Первоначально выстав-
лявшиеся ограничения — утверждения о неприсоединении солей ртути
системами сопряженных двойных связей [481, связями, сопряженными с
ароматическим ядром (пропенильными группами) [42, 141—143], некото-
рыми транс-соединениями [72, 144, 145] — постепенно отпадают, так как
найдено (Несмеянов, Луценко [146, 1471), что бутадиен [147, 148],
1,1-дифенилэтилен [147], 1-фенилбутадиен-1,3 [72], изопрен 1147], пиперилен
[147], дипропенил [147] в водной среде с более или менее хорошим выходом
дают обычные продукты присоединения; так же ведут себя стирол и по
крайней мере а-замещенные стиролы (ссылки см. стр. 135);
Наконец, найдены катализаторы [149] (перекиси и BF3), способствую-
щие присоединению солей ртути к некоторым транс-соединениям (стильбен),
которые раньше считались неспособными к присоединению. С другой сто-
роны, прибавление веществ, образующих слабые комплексы с солями рту-
ти, такие как пиридин и нитрилы, замедляет реакцию присоединения солей
ртути к олефинам [62, 149]. Однако приходится принимать во внимание,
что присоединение к веществам с группировкой =СН2 происходит обычно
более гладко, чем к веществам с группировкой =CHR или =CR'R", к
цис-изомерам легче, чем к транс-изомерам [72, 144, 150], и что присоеди-
нение в спиртовой среде проходит значительно более гладко, чем в водной,
где оно в случае последнего типа соединений (=CHR или = CR'R") подчас
сопровождается реакцией (иногда доминирующей) окисления и образовани-
ем закисной соли ртути.
ПРИСОЕДИНЕНИЕ СОЛЕЙ РТУТИ ПО ДВОЙНОЙ СВЯЗИ
Присоединение солей ртути к алкенам,
циклоалкенам и их производным
Присоединение [солей ртути к олефинам и их производным. Хорошо
диссоциирующие соли окисной ртути (нитрат, сульфат и т. п.) и соли
карбоновых кислот, и из них особенно ацетат, присоединяются к угле-
водородам, содержащим этиленовую связь, по реакции Гофмана и Занда
130 Ртутноорганические соединения. Лит. стр. 173—181
[1]. В водном растворе по обе стороны двойной связи с ее разрывом становят-
ся группы ОН и HgX, гдеХ—анион взятой соли ртути. Присоединение идет
в соответствии с правилом Марковникова: атом ртути, как и атом водорода
при присоединении по кратной связи галоидводорода, связывается с более
гидрогенизованным, т. е. более отрицательным атомом углерода (исключе-
ние— присоединение фторной ртути к полифторолефинам). Так полу-
чаются алканолртутные соли. Для этилена, кроме этого типа соедине-
ний HOCH2CH2HgX, изолированы еще производные этилового эфира
XHgCH2CH2OCH2CH2HgX — продукты ангидризации этанолртутных солей.
Строение таких продуктов присоединения к этилену подтверждено изу-
чением [521 спектров протонного резонанса, которые показывают также,
что в щелочном растворе алканолртутная соль (гидроксид) постепенно
превращается в производное эфирного строения.
При впервые осуществленной Шеллером и Шраутом [8, 9] реакции соли
ртути HgX2 с непредельными углеводородами в среде спирта ROH по обе
стороны двойной связи становятся остатки RO и HgX. Порядок присоеди-
нения и в этом случае подчинен тем же закономерностям. Самое присоеди-
нение совершается в этом случае быстрее и еще легче, чем в водной среде,
причем в метиловом спирте легче, чем в этиловом. Единственный продукт
реакции — алкоксиалкилртутные соли ROCR'R"CR"'R""HgX. Так, из
этилена и ацетата ртути в среде спирта получается моноацетатмеркурэти-
ловый эфир CH3COOHgC2H4OC2H5 [104,151]. Так же-получен оксалат [206].
Гораздо менее разработана реакция присоединения уксуснокислой рту-
ти в среде уксусной кислоты (Югель, Ибу [11]): пропускание этилена
в раствор уксуснокислой ртути в ледяной уксусной кислоте дает
CH3COOCH2CH2HgOOCCH3. Применяют также соли ртути других карбо-
новых кислот (см. стр. 136). Присоединение ацилатов (в том числе ацета-
тов) ртути ведут и в инертных растворителях — диметиловом эфире эти-
ленгликоля, диоксане, петролейном эфире и т. п.
Сулема также может быть использована для присоединения по этиленовой
связи, но в этом случае приходится с самого начала реакции вводить не-
много щелочи и постепенно добавлять ее по мере хода процесса. Впрочем,
подобный прием употребляется обычно и в случае применения других солей
ртути с сильными кислотами, и только уксуснокислую ртуть присоединяют
без подщелачивания. Это связано с тем, что накопление в результате реак-
ции сильной кислоты вызывает обратную реакцию, а уксусная кислота в
слабых растворах относится к продуктам присоединения индифферентно.
При взаимодействии'с сулемой в названных условиях олефины очень часто
образуют осадок молекулярного соединения состава: 1 моль алканолмер-
курхлорида, 1 моль сулемы. В частности, этилен дает HOCH2CH2HgCl•
•HgCl2. Для выделения алканолртутной соли в индивидуальном состоянии
в этом случае осадок обрабатывают водным (например 10%-ным) раствором
едкого натра, причем сулема превращается в окись ртути, а алканолртутная
соль в виде алканолмеркургидроксида переходит в водный раствор, из ко-
торого затем в результате подкисления уксусной кислотой (вместо этого
можно нейтрализовать щелочь пропусканием СО2) при одновременном до-
бавлении хлорида или бромида щелочного металла осаждается алканолмер-
кургалогенид.
Более обычно и с большим удобством в качестве исходной ртутной соли
используют ацетат. Реакция проводится пропусканием в водный (соответ-
ственно спиртовый) раствор газообразного или приливанием рассчитанного
количества жидкого олефина при перемешивании, продолжающемся до тех
пор, пока не прекратится выпадение желтого осадка окиси ртути при взаи-
модействии пробы раствора с едкой щелочью, что происходит обычно до-
вольно быстро. Алканолмеркурацетат или прямо выпадает из раствора
Присоединение солей ртути к непредельным соединениям 131
в виде кристаллов или иногда в виде жидкости, или остается в растворе,
так как описываемые ацетаты часто растворимы в воде (и тем более в спир-
те). В первом случае вещество удобно изолировать в виде ацетата. Перекри-
сталлизация (из спиртов, бензола, лигроина и т. д.) не представляет затруд-
ненйй. Во втором и третьем случаях всю массу вещества лучше превратить
й хлорид или бромид, что делается осторожным приливанием при размеши-
вании'водного раствора хлористого натрия или бромистого калия до прекра-
щения выпадения осадка. Избытка галогенида следует избегать, так как он
действует растворяющим образом.
Для алкоксимеркурирования алкенов было рекомендовано {152] при-
менять также смешанные соли ртути типа CH3COOHgX (где X—анион мине-
ральной кислоты), так как при этом в реакционной массе не накапливается
НХ.
Скорость образования продуктов присоединения солей ртути к олефи-
нам наибольшая у олефинов с двойной связью у первого углерода и быстро
уменьшается по мере удаления двойной связи от конца цепи [12]. Так же
олефины с разветвленной цепью реагируют быстрее нормальных, однако в
ряду олефинов с открытой цепью, естественно, оказывает влияние в этом
отношении лишь разветвление, начинающееся близко к двойной связи;
продукты же присоединения соли ртути к метилциклогексенам с более уда-
ленной от двойной связи метильной группой, например продукт присоеди-
нения к 4-метилциклогексену, образуются с большей скоростью, чем продук-
ты присоединения соли ртути к 2- и 3-метилциклогексенам [31].
Получение этано’лмеркургалогенидов [1]. К раствору азотнокислой окиси ртути прибав-
ляется едкое калн до образования неисчезающего белого осадка основной соли. При пропус-
кании этилена раствор через несколько минут опять становится прозрачным. Вновь добав-
ляют едкое кали до образования иенсчезающего белого осадка и опять растворяют его
пропусканием этилена, повторяя это до прекращения поглощения этилена.
К щелочному раствору прибавляют тогда столько галогенида калия (КС1, KBr, KJ),
чтобы на 1 атом ртути приходился 1 атом галоида, и, спустя несколько часов, высаживают
кристаллическую этанолртутную соль углекислотой. Часть, остающаяся в растворе,также
может быть получена в чистом виде после испарения и извлечения затем метиловым спиртом.
Перекристаллизация из метилового спирта. HOCHaCHsHgCl, т. пл. 155° С; HOCHaCHaHgBr,
т. пл. 158® С (см. также [153]). Другие соли см., например, [236].
Получениеа-ацетоксимеркур-Р-метокснэтана [104, 154]. В раствор 20 г уксуснокислой
ртути в 100 мл метилового спирта пропускают свободный от воздуха этилен. Через час
реакция окончена. После отгонки в вакууме спирта и уксусной кислоты остается желтое
масло, затвердевающее при хранении в течение 24 час. в вакууме над серной кислотой.
Выход 14 г (82%).Т.пл. после кристаллизации из петролейного эфира 42°С. Галоидная
соль получается осаждением водного раствора ацетата галогенидом калия [104]. Т. пл-
бромида 58° С (другие соли см. [56, 155, 156]).
Получение ртутных производных этилового эфира [1]. Приготовляют возможно более
нейтральный раствор сернокислой ртути растворением окиси ртути в 30%-ной серной
кислоте и добавлением затем окиси ртути до начала выпадения основной соли, которую
отфильтровывают. Раствор в течение 9 час. насыщают этиленом, образовавшийся осадок
отфильтровывают, промывают и растворяют в 15% -ном растворе едкого кали, к профиль-
трованному раствору прибавляют хлористого калия и смесь насыщают углекислотой. Хло-
{>ид выпадает в виде белых кристаллов. Т. пл. 190° С. Так же получается бромид
т. пл. 200° С), иодид (т. пл. 161*0) [5] и другие соли [56; 153, 155].
Присоединение солей ртути к этилену производят также в разнообраз-
ных гидроксилсодержащих средах [158].
Ртутная соль тринитрометана присоединяется к этилену в воде и нитро-
метане с образованием 1,1,1-тринитро-З-тринитрометилпропилртути (Но-
виков, Тартаковский, Годовикова [159]). В последнем растворителе одновре-
менно образуется также соответствующее симметричное соединение — бис-
тринитропропилртуть, являющаяся единственным продуктом реакции при
проведении присоединения в этиловом спирте. Хлористая тринитрометил-
ртуть также присоединяется к этилену (в воде) с образованием хлористой
132
Ртутноорганические соединения. Лит. стр. 173—181
1,1,1 -тринитропропилртути-3 [159]. 1,1,3,3-Тетранитробутан присоеди-
няется к этилену в метиловом спирте с образованием симметричного соеди-
нения [CH3C(NO2)2CH2C(NO2)2CH2CH2]2Hg [159а].
Приготовление водного раствора ртутной соли тринитрометаиа [159]. К энергично
перемешиваемой взвеси 140 г свежеприготовленной окиси ртути в 1200 ли воды прибавляют
небольшими порциями (по 10—15 мл) 200 г тринитрометана. Окись ртути постепенно рас-
творяется, к концу реакции образуется прозрачный раствор ртутной солн трииитрометана.
Значение pH раствора должно быть не выше 2.
Получение 1,1,1-трииитро-3-(трииитрометилмеркур)пропана [159]. В водный раствор
ртутной соли трииитрометана (10 г в 50 мл воды), помещенный в цилиндр, пропускают при
комнатной температуре ток этилена. Через 15—20 мин. после начала реакции выпадает оса-
док 1,1,1-тринитро-3-(тринитрометилмеркур)пропана. Этилен пропускают в общей слож-
ности 6 час., отфильтровывают осадок через каждые 2 часа. Затем реакционную смесь
оставляют стоять 12 час., в течение которых выпадает еще некоторое количество
продукта. Каждую порцию отфильтрованного осадка промывают 2—3 раза водой и высу-
шивают на воздухе. Общий выход 9,8 г (93%). Т. пл. 167° С (с разложением) при медленной
кристаллизации из воды.
Обработка продукта 6 W соляной кислотой при комнатной температуре приводит к
1,1,1-тринитро-З-хлормеркурпропану. Выход 90%, т. пл. 142° С (из хлороформа).
Получение (йгс-(трииитропропил)ртути [159]. В .раствор 10 г ртутной соли тринитро-
метана в 30 мл этилового спирта пропускают при комнатной температуре этилен в течение
12 час. Через 1—1,5 часа из раствора начинают выделяться кристаллы бпс-(тринитропропил)-
ртути; после окончания реакции их отфильтровывают, промывают небольшим количеством
спирта и затем несколько раз водой. Из фильтрата при разбавлении водой высаживают
еще некоторое количество бис-(тринитропропил)ртути. Общий выход 9,4 г (84%). После
двукратной кристаллизации из хлороформа т. пл. 155° С (с разложением).
Таким же путем получен симметричный продукт из пропилена, плавя-
щийся при 124—125° С (с разложением). Кристаллизация из хлороформа.
Присоединение уксуснокислой ртути к этилену в среде полиметилено-
вых и полиэтиленовых гликолей и моноэтиловых эфиров полиэтиленглико-
лей производят при 70—90° С [160]; если эфир ароматический — присоеди-
нение ведут при температуре, на 5—10° С выше температуры плавления
эфира гликоля, иногда употребляя последний как растворитель или ведя
реакцию в суспензии эфира гликоля в бензоле [160].
Присоединение уксуснокислой ртути к этилену в уксусной кислоте дает
соли Р-ацетоксиэтилртути.
Получение хлористой 0-ацетоксиэтилртути [24]. В смесь 80 г уксуснокислой ртути и
80 г уксусной кислоты пропускают этилен со скоростью 3 л! час при комнатной температуре
до отсутствия реакции на ион ртути со щелочью. Отфильтровывают от небольшой примеси
закисиых солей ртути. К фильтрату прибавляют 600 мл холодной воды и затем 170 мл
10%-ного раствора хлористого калия. Образовавшуюся хлористую Р-ацетоксиэтилртуть
(42 г) отфильтровывают, сушат и кристаллизуют из лигроина. Т. пл. 64—65°С. Также
получена [161] иодистая Р-ацетоксиэтилртуть. Т. пл. 83—85° С.
Действием раствора серной кислоты в ледяной уксусной кислоте
на продукт присоединения ацетата ртути к этилену получен сульфат
(CH3OOXH2CH2Hg)aS04 [162].
Сулема присоединяется к этилену в среде пиперидина [16], а также и
других аминов (см. стр. 137).
Получение М-(Р-хлормеркурэтил)пиперидина [16]. Взвесь 5 г сулемы в 50 мл сухого
пиперидина насыщают этиленом, находящимся под избыточным давлением в 35—40 см рт. ст.,
при механическом взбалтывании до исчезновения реакции на ион ртути. После отделе-
ния от выпавшего осадка CsHioNH-HCl фильтрат упаривают в вакууме.
Остаток сушат в эксикаторе над серной кислотой до исчезновения запаха пиперидина.
После перекристаллизации из петролейного эфира т, пл. 72—73° С. Это вещество отщеп-
ляет этилен с KJ, KCN; устойчиво к кислотам.
Солянокислую соль получают насыщением эфирного раствора 0,2 г CsHioNCHaCHaHgCl
током сухого хлористого водорода. Т. пл. 132° С-
Присоединение солей ртути к непредельным соединениям
133
По двойной связи этилена присоединены полихлорфеноляты ртути
(реакций ведут в инертном растворителе — тетрагидрофуране, возможно,
под давлением) И 63].
Пропилен с уксуснокислой и азотнокислой ртутью в воде обра-
зует пропанолртутные соли, изолированные в виде галогенидов
(Cl, Вг) XHgCH2CHOHCH3, и в качестве побочного продукта — дигалоген-
меркуризопропиловый эфир [XHgCH2(CH3)CH]2O [2, 7, 12, 157, 164—166].
Присоединение в спирте см., например, [169].
Присоединение к пропилену ртутной соли тринитрометана в воде дает
(O2N)3CCH(CH3)CH2HgCl(NO2)3, в спирте— соответствующее симмет-
ричное соединение [159] (см. стр. 132).
Присоединение ацетата ртути к бутену-1 и бутену-2 осуществлено в ме-
таноле [167]. Транс- и, легче, гщс-2-бутены присоединяют уксуснокислую
ртуть как в спиртовом [168], так и в водном [150] растворе.
Изобутилен в тех же условиях дает метил-2-пропан-2-ол-ртутные соли
(CH3)2C(OH)CH2HgX. Существование иодистой изобутенилртути, Описан-
ной Гофманом и Зандом [2], сомнительно. Присоединение соли ртути к ами-
лену произведено в разнообразных гидроксилсодержащих средах [158].
Присоединение соли ртути к пентену-2 в метиловом спирте дает смесь вици-
нальных метоксимеркурхлоридов [169]. Произведено присоединение аце-
тата ртути в воде к 2,3,3-триметилбутену-1 [1701:
(СНз)зСС (СН8) = СН2 + Hg (О2ССН3)2 + НаО (СНз)зСС (СН3) OHCHa.HgO2CCH3.
Соли ртуди-в метиловом спирте присоединены к 2,7-пентадецену, 1-но-
надецену, ундецену [342] и другим олефинам [12, 168].
Триметилсилилэтилен с уксуснокислой ртутью в водном растворе
образует продукт присоединения строения (CH3)gSiCH(HgOCOCH3)CH2OH
[171].
Из полифторалкенов в обычных условиях присоединяет соль ртути три-
фторэтилен и 1,1,1-трифторпропилен. Последний образует продукт присо-
единения с ртутью в положении 2 [172]. Продуктом присоединения ацетата
ртути к 1,1-дифторэтилену [172] в уксусной кислоте в результате реакций
/О /Р
CFa=CHa -ь Hg (СН3СОа)2 -> CHSC<4 CF2CH2HgOC<^
О СН3
Н2О
-2HF
О
II Н20
(СН3С—О—G—СНа—HgOCOCHa) — СН8СООН + HOOCCH2HgOCOCH3
является меркурированная уксусная кислота; в метиловом спирте—ееэфир
CH3OCOCH2HgOCOCH3 [172].
В то время как действием сулемы на олефины нельзя получить продуктов
присоединения элементов сулемы по двойной связи, фторная ртуть присо-
единяется по конечной двойной связи полифторолефинов в среде трехфтори-
стого мышьяка [173] (при 50° С присоединение к CF2=CF2, CF2—CHF,
CH2=CF2, CF2=CFC1) или в среде фтористого водорода (в автоклаве при
85° С, при 100° С, а в некоторых случаях и при более высокой температуре)
происходит присоединение к CF3CF = CF2 [174—176], CHF2CF2CF = CF3
[1751, CF3CH - CF2 [176,177], C1CF2CF2CF = CF2 [175] и H(CF2)8CF=CF2
[175], (CF3)2C=CF2) с образованием симметричных продуктов R2Hg. Ртуть
присоединяется при этом не к первому, а ко второму атому углерода Общая
схема реакции следующая:
2R'R’C=CFa + HgFa (R'R"CCF3)a Hg.
Д34 Ртутноорганические соединения. Лит. стр. 173—181
Попытки присоединения фторной ртути в среде фтористого водорода
,к неконечной двойной связи в таких соединениях, как октафтор-2-бутен
и гексафторциклобутен, были безуспешны [175]. Действие смеси сулемы и
фторной ртути в присутствии фтористого водорода на перфторпропилен дало
(CF3)2CFHgCl [177].'
Лишь следы фтористой перфторэтилртути получены при действии фтор-
ной ртути на перфторэтилен в отсутствие растворителя (автоклав, 100° С,
4 часа), и с умеренным выходом (24%) образовалась CF3CClFHgFH3 HgF2 и
CF2=CC1F в этих условиях (10 час.) [178].
Перечисленные ниже непредельные углеводороды легко образуют продук-
ты присоединения соли ртути при хранении на холоду в течение от несколь-
ких часов до нескольких дней или при слабом нагревании смеси водных или
спиртовых растворов компонентов;- непредельное соединение может быть
также растворено в органическом растворителе, например эфире, и прибав-
лено к водному или спиртовому раствору соли ртути.
Произведено присоединение соли ртути к следующим содержащим двой-
ную связь алициклическим углеводородам: циклопентену [179], дицйк-
допентену [7, 164], циклогексену, 2-метил-, 3-метил- и 4-метилциклогек-
сенам [31, 185] (для 4-метилциклогексена выделены как диастереомеры, таю
и изомеры положения; обнаружена конформация цис- и транс-метилцикло-
гексилмеркурбромидов [188—190]), 3-фенилциклогексену [191], метилен-
циклогексану [169], сантену [192], камфену [143, 193], изолауролену [194,
195], карвоментену [1961 и норборнилену.
Многочисленные работы посвящены окси- (преимущественно алкокси-)
меркурированию циклогексена, на котором были изучены стереохимия и
механизм присоединения солей ртути к непредельным соединениям. Так
было проведено изучение кинетики метоксимеркурирования и разделение
диастереоизомеров [14,61,182—1841 (в частности получены доказательства,
опровергающие прежние данные [14] о присоединении—ОСН3 и HgX в транс-
положения друг к другу 167,73,180,182,250]), изучение кинетики и механиз-
ма присоединения ацетата ртути в этиловом [70], н-пропиловом [70], изо-
пропиловом [13, 27] и в других спиртах [17, 66, 70], присоединение/-лактата
ртути [14, 72], манделата ртути [15] в метиловом спирте, трифтор ацетата
ртути [13], нитрата ртути [68], ацетата ртути в воде [34, 66], в этиловом
спирте [34, 72, 185], в петролейном эфире [66] и других растворителях
[29, 69, 72, 179, 183, 186, 187].
Продукт присоединения к норборнилену ацетата ртути в воде, в метило-
вом спирте или в диоксане [28, 197] или оксиперхлората ртути в воде [74]
представляет собой (после осаждения хлористым натрием), как это доказано
[74] и подтверждено также спектром ядерного магнитного резонанса [180],
не эндо-З-алкокси-экзо-2- [28, 198], а экзо-3-метокси- (или окси)-экзо-2-
норкамфанилмеркурхлорид (1):
что является одним из немногих известных примеров цис-присоединения
OR и HgX подвойной связи (см. также стр. 120). При действии на норборнен
ртутной соли тринитрометана в воде образуется продукт оксимеркуриро-
вания, а не продукт присоединения X и HgX [X = (NO2)3C1, что также до-
казывает цис-конфигурацию продукта присоединения [198а].
Образованию продуктов цис-присоединения при оКсимеркурировании
циклического алкена благоприятствует напряжение в непредельной моле-
Присоединение солей ртути к непредельным соединениям
135
куле [74]. Так, подобно норборнену, цис-продукты оксимеркурирования
получены в случаебензнорборнена (действие смеси окиси ртути и уксуснокис-
лой ртути в воде. 24 часа): . .
и в случае дициклопентадиена (действие смеси окиси ртути и сулемы -в
50%-ном водном ацетоне):
В этом случае не происходит присоединения к менее напряженному пятй-
членному кольцу. Так же менее напряженный, чем норборнилен, бицикло-
(2,2,2)-октен при действии окситозилата ртути в воде дает смесь равных ко-
личеств цис- и транс-продуктов присоединения и смесь этих же продуктов
при действии ацетата ртути в смеси воды и ацетона, тогда как ненапряжен-
ный циклогексен образует продукт только транс-оксимеркурирования
(см. выше). Но при действии ацетата ртути в уксусной кислоте в течение не-
скольких минут бицикло-(2,2,2)-октен образует только продукт цис-присо-
единения элементов уксуснокислой ртути по двойной связи [1986].
К дициклопентадиену произведено присоединение ацетата ртути в воде
[181], в спиртах [223], а также в 50%-ной смеси D2O и CD3COCD3. Про-
дукт присоединения трифтор ацетата ртути в СН3ОН неустойчив.
Ниже перечислены ароматические углеводороды' (а также некоторые гид-
роароматические углеводороды и галоидпроизводные гидроароматических
углеводородов) с двойной связью в боковой цепи, к которым было произведе-
но присоединение солей ртути: стирол [7, 20, 30, 72, 141, 164, 167, 187,
199, 200], а-метилстирол [30, 199], [3-метилстирол [72], а-этилстирол
[201], 2,6-диметоксистирол [202], 1,1 - дифенилэтилен [37, 202], цас-стиль-
бен [29, 72, 149, 204], транс-стильбен (только в присутствии катализаторов:
фтористого бора [203], перекиси бензоила и аскаридола [149 , 203]), а-метил-
стильбен [29, 51/69, 205], аллилциклопентан [147], аллилбензол [147, 206,
207], пропенилбензол [72, 200] (согласно Райту [72] только в среде спирта,
но не в воде), п-изопропенилкумол [208], 2-метил-1-фенилпропен-1 [69, 200,
209], 2-транс-аллилдекалин [147] и дигидронафталин [164, 206].
При этом, согласно Парку и Райту [200], в то время как при метокси-
мер курировании HgX-группа вступает только в р-положение по отношению
к фенилу в стироле и только в а-положение в 2-метил-1-фенилпропене, как
транс-, так и ^uc-пропенилбензол дает смесь обоих изомеров положения.
Присоединение ацетата ртути в метиловом или этиловом спирте к эндо-
и экзо-конформациям хлорированного производного гексагидронафгалина
проходит по схеме [210]:
136
Ртутноорганические соединения. Лит. стр. 173—181
Следует отметить, что весьма часто в комбинации с ароматическим или
алициклическим ядром аллильная группировка гладко присоединяет ацетат
ртути, а пропенильная группировка — CH = СН — СН3 часто не способна
присоединять соли ртути, а претерпевает окисление. Впрочем, это различие,
по-видимому, относится в первую очередь к присоединению в водных, но не
в спиртовых растворах, так как, например пропенилбензол, в воде не при-
соединяющий ацетата ртути [147], в спирте это гладко делает [72]. На этом
различии основан метод разделения аллильных и пропенильных соединений
в эфирных маслах [211]. Разница в способности присоединения солей ртути
аллильной и пропенильной группировок была применена к установлению
строения кариофилленов [192].
Различающиеся друг от друга положением двойной связи пинен (двой-
ная связь в цикле) и нопинен (семициклическая двойная связь) различно от-
носятся к уксуснокислой ртути: в то время как пинен оХисляется ею (в спир-
товом растворе на холоду), нопинен при двухдневном стоянии с уксусно-
кислой ртутью в спирте дает продукт присоединения согласно следующей
схеме [212]
(см. также [213]).
Согласно данным Югеля и Ибу [11], при взаимодействии олефина и ук-
суснокислой ртути в растворе ледяной уксусной кислоты получаются соеди-
нения, строение которых авторы выразили общей формулой:
СНзСОО
СН3СО2 или C„H2reHg (СНзСОО
С„Н2„— этилен; октен-2,3; дуодецен-1,2; тетрадецен-1,2; гексадецен-1,2).
Так же получено соединение C16H32Hg(CH2ClCO2)2 взаимодействием гек-
садецена с раствором хлорацетата ртути в хлоруксусной кислоте [11].
При растворении пальмитата и стеарата ртути в гексадецене получены со-
единения состава CieH32-HgX2 (где Х=С18Н35О2 и Ci8H33O2 [11]). Всем этим
соединениям следует приписать структуру' RCOOCR1R3CR1R2HgOOCR.
Аналогичные продукты присоединения элементов соли ртути X и HgX
по двойной связи общей формулы XCreH2„HgX получены также, например,
действием на этиленовые углеводороды (этилен, пропилен, стирол) ртутных
солей (ацетат, пропионат, а также сульфат) в присутствии безводных карбо-
новых кислот [10], действием уксуснокислой ртути на циклогексен (в петро-
лейном эфире [66] и в третичнобутиловом спирте в присутствии эфирата
фтористого бора [1986]) и на норборнен (в диоксане [28]), ацетата, пропиона-
та, бутирата ртути на стирол [20] (условия не указаны [20]), ртутной соли
тринитрометана (в воде) на этилен, пропилен, стирол, циклопентен и цикло-
гексен (X=C(NO2)3) [159] и ртутной соли 1,1,3,3-тетранитрОбутана к эти-
лену ( в воде) [159а].
Продукты присоединения по двойной связи (децена, циклогексена, сти-
рола) ртутных солей двухосновных кислот, согласно данным Шпенглера^ и
Присоединение солей ртути к непредельным соединениям 137
Вебера [215], имеют циклическое строение. Например, при 5-минутном
нагревании при 80—100° С стирола и терефталевокислой ртути получено со-
единение строения
ос —— со
I I
OHgCH2CH(C6H5)O
В случае децена и циклогексена требуется более длительное нагревание.
Описано присоединение к олефинам ртутных солей оптически активных
карбоновых кислот с целью разделения образующихся изомеров [14, 15].
Осуществлено присоединение солей ртути к олефинам в среде аминов:
присоединение к этилену в среде Пиперидина (см. стр. 132), в среде мезидина
’[17], пиридина [17], к аллиламидам (см. стр.161) в среде пирролидина
[18], морфолина, 1-метилпиперидина [18], ко многим алифатическим олефи-
нам, циклогексену, стиролу в среде метил-и этиланилина и даже в среде ани-
лина [12]. В отличие от обычных продуктов присоединения солей ртути к
олефинам в воде и спиртах продукты присоединения в аминах устойчивы к
действию кислот, в остальном проявляют квазикомплексные свойства.
Продукты присоединения солей ртути к олефинам, содержащие вместо
алкоксигруппы ацил аминогруппу, получены реакцией такого соединения,
как бис-(л-этоксиэтилмеркур)ацетилен и подобных веществ с амидами и
имидами [216] кислот или бензол сульфон амидом [217] при кипячении в
спиртовом растворе. Так получены, например, вещества строения
(H2NCONHCH2CH2HgC)2, (CH3CONHCeH10HgC)2 и т. п.
(О присоединении к этилену полихлорфенолятов ртути см. стр. 133.)
Кроме воды, спиртов, анионов кислот, аминов, ароматические углеводороды
[21—251 ацетоуксусный эфир |26] оказались обладающими достаточными
электрс j-донорными.свойствами, чтобы явиться вторым компонентом в про-
дуктах присоединения солей ртути к алкенам. Такие соединения общей фор-
мулы ArCRiR2CR8R4HgX и CH8COCH(CRjR2CR8R4HgX)COOC2H6 получе-
ны Итикава с сотр. [21—26] при взаимодействии продуктов присоединения
солей ртути к олефинам с ароматическими соединениями (например анизо-
лом) в уксусной кислоте в присутствии катализаторов (кислородных кислот)
(олефины: этилен [21], пропилен, бутилен-2, циклогексен, стирол [22]) и с
ацетоуксусным эфиром (катализатор—эфират фтористого бора) (олефины:
этилен [26, 26а], пропилен, стирол [26]). В большинстве случаев образую-
щиеся вещества не выделялись, а сразу же подвергались разложению с целью
получения 0-арилалкиловых спиртов, ацетатов и а(0-ацетоалкил) ацетоаце-
татов (см. гл. XIV, стр. 272 и 273). Выделены иодистая и уксуснокислая
Р-(п-метоксифенил)этилртуть и а-(2-хлормеркурэтил)ацетоуксусноэтило-
вый эфир CH3COCH(CH2CH2HgCl)COOC2H5.
Получение иодистой Р-(п-метоксифенил)этилртути [21]. Прозрачный раствор продукта
присоединения соли ртути к этилену, полученный из 64 г уксуснокислой ртути, 60 мл ук-
сусной кислоты и этилена, приливают при перемешивании к смесн 100 г анизола н 100 г
98,6%-ной фосфорной кислоты прн 22—23° С. Через 1,5 часа прибавляют 400 мл воды.
Образовавшееся тяжелое масло, смешивают с небольшим количеством воды и 700 мл
5%-ного раствора ноднстого калия при перемешивании. Образующийся белый осадок весит
46 а. После пятикратной кристаллизации нз спирта т. пл. 146—148° С.
Получение хлористой а-(транс- ?)-2-метоксициклопеитнлртутн [179]. Взвесь 89,4 г
(0,28 моля) уксуснокислой ртути в растворе 20 мл (0,226 моля) цнклопентена в 400 мл ме-
тилового спирта перемешивают прн 25° С. Через 2 мин. смесь становится гомогенной. Че-
рез 12 час. реакция заканчивается. Раствор обрабатывают 78 мл (0,195 моля) 10%-ного
водного раствора едкого натра и отфильтровывают от желтой мути. К фильтрату прили-
вают 5%-ный избыток раствора хлористого натрия. Образовавшийся осадок (64 г, выход
88%, т. пл. 76—77° С) после кристаллизации из 195 мл этилового спирта имеет т. пл. 83,3—
83,7° С.
438
Ртутноорганические соединения. Лит. стр, 173—181
Аналогично получен бромид, т. пл. 82,0—82,3° С, и иодид, т. пл. 60,5—61,0° С.
Получение 2-ацеТатмеркурциклогексанола-1 [34]. Сливают вместе О,9 г циклогексена и
3,5 г уксуснокислой ртути в 15 мл воды. Образовавшийся осадок через 5 дней отсасывают
и перекристаллизовывают из этилацетата. Выход количественный. Т. пл. 112,2—113° С.
Получение 1-ацетокси-2-ацетоксимеркурциклогексана [66]. Взвесь 20 г (0,063 моля)
ацетата ртути в 8 мл (0,08 моля) циклогексена и 10 мл петролейного эфира (т. кип. 60—70° С)
энергично перемешивают в течение 7 час. при 35° С. Выделившийся на стенках колбы белый
осадок соскабливают, промывают петролейным эфиром. Вес сухого осадка 23 г (выход
91%), т. пл. 86—89°С. Вещество неустойчиво и уже через 5 мин. начинает обнаруживать
сильный запах циклогексана и уксусной кислоты.
При добавлении 0,003—0,006 молей эфирата фтор исто гобора-или азотной кислоты вре-
мя реакции сокращается до 1 часа. На свойства конечного продукта эти катализаторы не
влияют. Несколько более устойчив 1-ацетокси-2-хлормефкурциклогексан (после кристалли-
зации из ацетона имеющий т. пл. 102—103° С), получаемый сливанием метанольного рас-
твора ацетата и водного раствора хлористого натрия, но он также медленно разлагается
уже при 25° С.’
Получение 2-ацетатмеркур-З-окситетрагидронафталииа [164],
СН2
f^Y4|CHHg°2CCH3
СНг
Эфирный раствор 1,4-дигидронафталина взбалтывают в течение 24 час. с водным рас-
твором уксуснокислой ртути. Выделяются длинные иглы; после перекристаллизации из бен-
зола или лигроина т. пл. 122° С.
Получение ацетатмеркурметилфенилкарбинола [30]. 3,27 г (0,031 моля) стирола зали-
вают раствором 10 г (0,031 моля) уксуснокислой ртути в 40 мл воды. Через сутки раствор
со щелочью не дает осадка окиси ртути. Образовавшийся маслянистый продукт раство-
ряют на холоду в минимальном количестве ацетона. Отфильтрованный раствор длительно
охлаждают смесью снега с солью. Через 3 часа выпадает кристаллический осадок. Выход
9 г, что соответствует 75%. После повторной кристаллизации из бензола т. пл. 77—79° С
(с разложением).
Описано получение его солей с органическими серусодержащимн соединениями мер-
каптидов, ксантогенатов, диалкилтиофосфатов и т. п. [218].
Имеются данные о температурах плавления (разложения) различных
C8H5CH(OR)CH2HgX, где X — различные анионы, a_R — Н алкилы или
ацилы [20].
Получениехлормеркурметилбеизилкарбинола [147]. К 2,55 г (0,025 моля) аллилбензола
(т. кип. 155,5—156° С/738,5 мм, п% 1 ,5171) приливают раствор 8 г ацетата ртути в 40 мл
воды. Через 1,5—2 часа начинается выпадение маслообразного ртутноорганического со-
единения. Спустя 12 час. проба щелочью показывает, что реакция присоединения закончи-
лась. Выпавшее масло, растворенное в несколько приемов в горячей воде, при.прибавлении
раствора 1,8 г хлористого калия, дает 8,4 г (90%) белого осадка хлормеркурметилбензил-
карбинола. После кристаллизации из водного спирта — шелковистые иглы с т. пл. 75° С.
Получение 1,1,1-трифтор-3-этоксипропил-2-меркурнитрата [172]. В трехгорлую колбу
помещают 30 мл этанола и 17 г Hg(NC^)2-0,5HaO и при энергичном перемешивании при
комнатной температуре пропускают 1,5 л 1,1,1-трифторпропилена. Через 1 час весь нитрат
ртути растворяется; реакция на ион ртути со щелочью отрицательная. При хранении выпа-
дает новый осадок. На следующий день осадок отфильтровывают, маточник упаривают в
вакууме досуха. Выход 1,1,1-трифтор-3-этбксипропил-2-меркурнитрата 85%. Вещество
не плавится.' Соответствующий хлорид имеет т. пл. 37—38°С (из водного спирта).
Получеинебис-перфторизопропилртути [175]. 240 г (1,01 моля) фторной ртути, 70 г
безводного фтористого водорода и 300 г (2 моля) гексафторпропилена нагревают в бомбе из
нержавеющей стали при 110° С в течение 12 час. Бомбу охлаждают и открывают вентиль.
Продукт помещают в неплотно закрытую полиэтиленовую бутыль со снятым колпаком,
чтобы испарить избыток фтористого водорода. Последние следы фтористого водорода
удаляют перемешиванием продукта с порошком фтористого натрия или же фтористый во-
дород извлекают водой и продукт затем сушат над пятиокисью фосфора. При перегонке
получено 323 г (66%) биоперфторизопропилртути. Т. кип. 115—116° С, т. пл. 20—21° С,
л о 1,3244, с/25 2,5302. Перед перегонкой вещество должно быть высушено, иначе пере-
гоняется азеотропная смесь (т. кип. 90°С), содержащая около 4,3% воды.
Присоединение солей ртути к непредельным соединениям 139 .
Если исходная фторная ртуть содержит примесь хлор-иона, то в качестве
побочного продукта образуется хлористая перфторизопропилртуть (т. кип.
173т—178° С), получаемая из бнс-перфторизопропилртути при действии тио-
фосгена.
’ В условиях, аналогичных синтезу быс-фторизопропилртути, получены
{175]: б«с-(3-Н-1-трифторметилпентафторпропил)ртуть (т. кип. 172,3—
173° С, выход 73%, т. застывания 0°С), бш>(3-хлор-1-трифторметилпен-
тафторпропил)ртуть (выход -5%, т. кип. 85—93°С/20 мм, т. пл.— 10° G),
бис-1,\-дихлор-2,2,2-трифторэтилртуть (выход 69%, т. пл. 180—185° С).
бис-Перфтор-третичнобутилртуть получена аналогично из фторной ртути
в безводном фтористом водороде и смеси газов, содержащей 17,3% перфтор-
изобутилена, 76,4% перфторциклобутана и 6% перфторметилциклобутана,
при нагреваний в бомбе при 200° С в течение 12 час. Выход 33%, т. пл..65—
*66° С [175].
Синтез бяс-(1,2,2,2-тетрафторэтил)ртути [173]. 23,9 г (0,10 моля) HgF2, 25 г (0,30
моля)- трифторэтилена, 15 мл AsFs нагревают до 50° С в течение 4 час. в закрытой трубке
из нержавеющей стали при встряхивании. После отгонки в вакууме трехфтрриСтого мышья-
ка остающийся твердый осадок возгоняют при 80—90° С при атмосферном давлении. Возо-
тнанное белое вещество весит 26,6 г (66%, считая на HgF2) и плавится при 78—79е С.
Описано количественное определение двойных связей в органических
«соединениях, основанное на присоединении соли ртути 1187, 219—224].
Описано образование под давлением жидких комплексов пропилена и
1- и 2-бутена с уксуснокислой ртутью и пропилена с азотнокислой ртутью, не
являющихся продуктами присоединения солей ртути по двойной связи [225].
Присоединение солей ртути к диеНам. Алифатические углеводороды,
содержащие две двойных связи в молекуле, также присоединяют ацетат
ртути. Так, диаллил при действии водного раствора этой соли (с последую-
щей обработкой раствора КС1) образует циклическое соединение [146]
(ср. [226]) ‘
сн2—сн2
I I •
ClHgCH2CH CHCH2HgCl.
Ч'о'/
Аналогичное циклическое соединение получено при действии на диал-
лил ртутной соли тринитрометана [227].
Вопреки старым данным Занда [48] и углеводороды с сопряженными свя-
зями— бутадиен-1,3 [147, 147а], изопрен, пиперилен, дипропенил [147] —
дают с уксуснокислой ртутью даже в водной среде продукты присоединения
2Hg(OH)X. Однако следует отметить, что углеводороды с конечными
СН2-группами, связанными двойной связью (диаллил [146], бутадиен, изо-
прен), реагируют быстрее и дают гораздо лучший выход продукта, чем, на-
пример, дипропенил (выход ртутноорганических соединений в этом случае
составил всего 13% наряду с большим количеством закисной соли ртути).
Необходимо сопоставить этот факт с разной степенью легкости присоедине-
ния солей ртути аллильной и пропенильной группировками, соединенными
•с ароматической или алициклической системой (см. выше).
Бутадиен-1,3 с уксуснокислой ртутью в метиловом или этиловом спирте
дает мезо- и рацемическую формы 2,3-диалкокси-1,4-диацетоксимеркурбу-
танов [148, 149, 167, 228].
Согласно данным Мак-Нили и Райта [71], бутадиен не Дает продукта
1,4-присоединения. Действием на бутадиен-1,3 азотнокислой ртути в вод-
ном этиленгликоле получен (после обработки иодистым калием) 2,3-бис-
(иодмеркурметил)-1,4-диоксан, из которого заменой группьт HgX на иод
140
РтутноорганИческие соединения. Лит. стр, 173—181
выделены соответствующие цис- и /лранс-иодметилпроизводные диоксана
[229L
Бутадиен, изопрен, гексадиен-1,5 при действии соли ртути в разных гид*
роксилсодержащих средах присоединяют HgX и OR к каждой двойной
связи [167]. Ацетат ртути в метиловом спирте присоединяет также винил*
циклогексен-1 [167].
Для 1-фенилбутадиена-1,3 выделен продукт присоединения как по одной
двойной связи (действием 1/Й ацетата ртути), так и по обеим двойным связям
(действием 27И HgX2 или действием ацетата ртути на мономеркурированный
продукт [72]).
Синтез хлормеркурметокснфеннлбутена [72]. К раствору 3,18 г (0,01 моля) ацетата
ртути в 25 мл метанола прибавляют 1,56 г (0,012 моля) свежеперегианного фенилбутадие-
иа. Через 5 мин. проба ие дает осадка окиси ртути со щелочью (10% NaOH). Через 4 часа
его отфильтровывают от соли закиси ртути и к фильтрату медленно прибавляют 10%-иый
водный хлористый натрий. Выделившееся масло быстро затвердевает, его отфильтровывают,
дважды промывают водой и дважды петролейным эфиром. Выход продукта с т. пл. 89* С
3,35 г (85%). После кристаллизации из 25 мл этанола т. пл. 92° С.
Синтез диацетоксимеркурдиметоксибутана [72]. К раствору 6,36 г (0,02 моля) уксусно-
кислой ртути в 55 мл метанола прибавляют 1,43 г (0,011 моля) феиилбутадиена (т. кип.
86°С/П мм). Через 2 Часа проба не дает осадка окиси ртути с 10%-ным водным раствором
едкого натра. Через 12 час. отгоняют 40 мл метанола, остаток фильтруют и охлаждают до
10°С. Выход кристаллического продукта 7,62 г (35%), т. пл. 142—145°С. Кристаллизация
из смеси 25 мл бензола и 35 мл петролейного эфира (т. кип. 60—70°С) повышает т. пл. до 149®С
с небольшой потерей продукта.
Получение 2-метил-1,4-дихлормеркур-2,3-диоксибутаиа [147]. 1,7 г (0,025 моля)
изопрена заливают равтвором 15 г уксуснокислой ртути в 80 мл воды. Присоединение ртут-
ной соли заканчивается в несколько минут. Часть углеводорода заполнмеризовывалась на
стейках сосуда. К продукту присоединения прибавляют раствор 3 г хлористого калия в
40 мл воды. Выпадает осадок грязно-белого цвета. Проба аммиаком показывает присутствие
солей закисной ртути. Осадок обрабатывают 10% -ной щелочью. К фильтрату прибавляют
10%-ную уксусную кислоту до нейтральной реакции. Выпадает 8,3 г (58%) 2-метил-1,4-
дихлормеркур-2,3-диоксибутана. После двукратной перекристаллизации из этилацетата
т. пл. 147—148°С.
Получение 2,3-ди-(иодмеркурметнл)-1,4-диоксана [229].
JHgCH2CH
I
О
'сНа-СН2
CHCHaHgJ
I
О
Раствор 243 г (1,12 моля) окиси ртути в 300 мл (4,7 моля) конц. азотной кислоты (уд.
вес 1,42) и 150 мл воды соединяют с 100 мл воды и 500 мл свежеперегнаниого этиленгли-
коля. В смесь, охлажденную до 20°С, пропускают 1,3-бутадиен со скоростью 2 пузырька
в 1 сек. Реакция заканчивается через 3 часа (проба с едким натром на присутствие окиси
ртути). Смесь охлаждают до — 5°С, осадок 2,3-ди-(нитратмеркурметил)-1,4-диоксана отса-
сывают и растворяют его затем в 2л 1%-ного едкого натра (слабое выделение ртути). Кеме-
си прибавляют раствор 133 г (0,801 моля) йодистого калия в 500 мл воды. Получено 276 а
(0,359 моля) 2,3-ди-(иодмеркурметил)-1,4-диоксана, т. пл. 196—198°С.
Доказана аналогичность состава продуктов взаимодействия аллена с су-
лемой C6H8Hg6Cl6O3 и сулемы с аллиленом [97, 230].
Соли ртути к 1,2-диметилциклогексадиену-1,4 не присоединяются [231].
Присоединение солей ртути к бициклогептадиену происходит по меха-
низму сопряженного 1,5-присоединения с образованием нортрицикленовых
производных
HgX 2
ROH
xHg\lx^y°R
/ / /
Присоединение солей ртути к непредельным соединениям 141
[R ОССН3 при присоединении сулемы в ледяной уксусной кислоте
[232], В — СН3 при присоединении сулемы в метиловом спирте (комнатная
температура, 3 дня [233]), R — С2Н5 в этиловом спирте (так же и затем кипя-
чение [233])]. Но, согласно патенту [214], присоединение сулемы в теплом
(60° С) этиловом спирте к бицикло-(2,1,1)-гепта-2,5-диену дает с 83%-ным
выходом 2-этоксибицикло-(2,2,1)-гептен-3-меркурхлорид. Согласно этому'
же патенту, аналогично получены диэтоксибицикло-(2,2,1)-гептан-бис-
меркурхлорид, диметоксибицикло-(2,2,1)-гептан-бмс-меркурхлорид.
Получение 1-хлормеркур-5-метоксииортрициклена [233]. К раствору 35 г сулемы в
300 мл не содержащего ацетона метанола прибавляют 12.г бицикло-(2,2,1)-гепта-2,5-диена.
Сосуд закрывают и оставляют стоять 3 дня. За это время в реакционной смеси образуется
•объемистый осадок. Его отфильтровывают, промывают холодным метанолом и сушат в эк-
сикаторе фосфорным, ангидридом. Выход сырого продукта 14 г. После кристаллизации из
метанола получают 5,5 г бесцветных игольчатых кристаллов, т. пл. 121—121,5°С.
Имеются данные о возможном промежуточном образовании продуктов
присоединения солей ртути-при реакции циклооктатетр аена с солями ртути
[234]. '
Описано выделение из смеси углеводородов олефинов и диолефинов на
основании разной скорости присоединения ими солей ртути [235].
О присоединении к дициклопентадиену см. стр. 135.
Присоединение солей ртути к оксисоединениям,содержащим этиленовую связь
Присоединение к оксисоедииениям и простым эфирам. Так же легко,
как к олефиновым углеводородам, происходит присоединение солей ртути
к непредельным спиртам и к содержащим ненасыщенную боковую цепь
ароматическим оксисоединениям и их производным.
Аллиловый спирт дает с солями ртути два рода соединений: производные
пропиленгликоля HOCH2CHOHCH2HgX и производные дипропиленоксида
[3—6, 141, 145, 167, 236, 448, 449]
XHgCH3CH-CH2O
ОСНа—CHCHaHgX
Первые получаются действием аллилового спирта на раствор азотнокис-
лой ртути при одновременной нейтрализации образующейся кислоты добав-
лением щелочи [4]. Ртутные производные дипропиленоксида получаются из
кислого раствора солей ртути[236]. Для них доказано траис-строение и поли-
мерная природа [237].
Описано взаимодействие галогенидов ртути с аллиловым спиртом [3,
226,236]. .
Обсуждался механизм образования алленовых производных при действии
солей ртути на аллиловйй спирт [3, 236].
Производное пропиленгликоля образуется также при присоединении
к аллиловому спирту ртутной соли тринитрометана [238], с из-
бытком аллилового спирта в воде образуется симметричное соединение
Hg [CH2CHC(NO2)3CH2OH]2.
Встряхивание ацетата ртути с аллиловым спиртом приводит к присоеди-
нению ацетата ртути и образованию моноацетата ацетатмеркурпропиленгли-
коля CH2(HgOCOCH8)CH(OCOCH3)CH2OH [169].
Получение броммеркурпропиленгликоля [4]. К раствору 100 г желтой окиси ртути в
20%-ной азотной кислоте прибавляют разбавленный раствор едкого кали до появления
неисчезающего белого осадка основной соли. Прибавляют по каплям при перемешивании и
охлаждении водой время от времени аллиловый спирт до растворения осадка, затем вновь
f42
Ртутноорганические соединения. Лит. стр. 173—181
едкого кали до образования осадка основной соли, которую также растворяют введением
аллилового спирта. Так повторяют до внезапного потемнения осадка и щелочной реакции
жидкости. К раствору прибавляют столько бромистого калия, чтобы на Гатом ртути при-
ходился 1 атом галоида, после хранения в течение 24 час. высаживают бромид пропускани-
ем углекислоты. Т. пл. 84—86°С, разложение при 110°С.
Получение дипропиленоксиддимеркурсульфата [236].
ГНё-СН2-СН-СН2-О ]
I ! . SO4.
L О —СНа—CH-CHa-Hg.
Профильтрованный раствор 75 г окиси ртути в смеси 30 мл воды и 1ф мл разбавленной.-
серной кислоты (1 : 1) перемешивают с 30 мл аллилового спирта. Выделившийся через-
2—4 часа сульфат фильтруют, промывают холодной водой и сушат. Выход 60 г. Так же-
получают нитрат, вместо серной пользуясь 40%-иой азотной кислотой. Галоидные соли
получают действием на нитрат галогенида калия и высаживанием углекислотой. Т. пл.,
бромида’ 251°С. Эти вещества, по-видимому, являются полимерами [237].
Бутен-1-ол-4 с ртутной солью тринйтрометана дает продукт
присоединения элементов этой соли по двойной связи, который с
избытком непредельного (в спирте) образует симметричное соединение
Hg[CH2CHC(NO2)3CH2OH]2 [238].
Пентен-1-ол-5 [146], гексен-1-ол-5 [146], тётраметил-1,1,5,5-пентен-1-ол’
[49] и метил-5-гептен-1-ол-5 [49] присоединяют в водной среде и ацетат и
хлорид ртути, пентен-1-ол-5 также присоединяет ртутную соль тринитро-
метана [238], образуя циклические соединения, например:
СН2-СН2.
СН2=СН—СНа—СН2-СН2—ОН + HgCl2 -> ClHgCH2CH СН2 + НС1
V
Получение 2-хлормеркурметилтетрагидрофурана [146]. 4,3 г (0,05 моля) пентен-1-ола-5-
(т. кип. 136—I38aQ прибавляют к раствору 16 г уксуснокислой ртути в 50 мл воды. При-
соединение ртутиой соли проходит очень быстро; на дне собирается маслообразное ртутно-
органическое соединение, растворяющееся при добавлении воды. При добавлении раствора
хлористого калия выпадает 2-хлормеркурметилтетрагидрофуран в виде масла. Масло-
отделяют и растворяют в эфире. Раствор высушивают над плавленым сернокислым натри-
ем. После удаления эфираостается 12,8 г масла2-хлормеркуртетрагидрофурана. Выход 82%..
Ацетат пентен-1 -ола-5 дает с ртутной солью тринитрометана в воде аци-
клический продукт (NO2)3CHgCH2CHC(NO2)3CH2CH2CH2OCOCH3, в сухом
нитрометане — симметричное соединение Hg[CH2CHC(NO2)3CH2CH2CH2OH]2.
[238].
2,6-Диметилгептен-5-ол-2 при присоединении соли ртути в воде [7, 49,
239] образует как циклический продукт, так и продукт присоединения с от-
крытой цепью [239]. Описано присоединение ацетата ртути в низших спир-
тах к содержащим двойную связь высшим спиртам. [240].
Метоксимеркурирование монометилового эфира винилэтиленгликоля да-
ет нормальный продукт XHgCH2CH(OCH3)CH(OH)CH.(OCH3) с выходом
31% [2411.
ВинилгЛюкоза [242] и аллил ацеторамноза [2431 при умеренном нагре-
вании в течение нескольких часов с уксуснокислой ртутью в уксусной ки-
слоте образуют продукты присоединения элементов уксуснокислой ртути
по двойной связи.
Присоединение уксуснокислой ртути.как в воде, так ив диоксане к
аллил-а-манниту сопровождается ангидризацией и образованием диоксано-
вого цикла и дает смесь изомеров [244].
Имеются литературные данные о присоединении соли ртути к о-аллило-
вым эфирам d-маннита, сорбита и других многоатомных спиртов [245—248L
Присоединение солей ртути к непредельным соединениям
143-
З-Аллил-1,2,5,6-диизопроцилиден -d-маннит при обработке ацетатом
ртути в безводных условиях [249] дает, наряду с образующимся в результа-
тЬ циклизации главным продуктом реакции RHgAc—2-ацетатмеркурметил-
4,6-бпс-(2,2-диметил-1,3-диоксолан-4-ил-п-диоксаном) (III), также около
1% симметричного продукта R2Hg — б«с-[5,6-бас-(2,3-диметил-1-3-диоксо-
лан-4-ил-2-л-диоксанилметил)] ртуть [249] (IV):
Н
I
н—С—О сн3
I /с\
н—с—ох СНз
н—с—о—сна
I I
н—с—О—CHCHaHgOaCCHs
Н—С-0 СНз
I X
Н-С-0 СНз
н
III
н н
I 1
Н-С-0 СНз НзС О-С-Н
I /с\ /с\ I
Н-С—0х СНз СНзХ О-С-Н
I 1
Н—С—О—СНз НзС—О—с—н
II II
Н—С—О—CHCHsHgCHs—нс—о—с—н
I - ' I
Н-С-0 СНз НзС О—с—н
I /с\ /с\ I
н—С— 0Х СНз НзС О—С—н
Присоединение ацетата ртути в метиловом спирте к D-глюкалю и его 3,4,6-
триацетату дает соответственно метил-2-ацетоксимеркур-2-деокси-3-П-ман-
нозид (цис) (V) и метил-2-ацетоксимеркур-2-деокси-3,4,6-три-о-ацетил-Р-Л-
глюкозид (транс) (VI) [250]. Присоединение к 3,4,6-триацетату произведе-
но также в этиловом и изопропиловом спиртах (конфигурация не установ-
лена) [250].
При метоксимеркурировании циклогексен-3-ола-1 и его метилового эфи-
ра и ацетата вместо четырех возможных изомеров для каждого из этих ве-
ществ выделено [251] только по одному продукту, в которых 0СН3- и
Н§ОСОСН3-группы находятся в транс-положении по отношению к исходной,
ОН-группе (или ее производным).
Имеющаяся в непредельном соединении гидроксильная группа оказы-
вает направляющее влияние на стереохимию присоединения HgX и OR по*
двойной связи [252]. Строение конечного продукта обусловливается обра-
зованием цикла вследствие участия соответствующим образом расположен,-
144
Ртутноорганические соединения. Лит. стр. 173—181
ной спиртовой группы. Так реагируют, например, карбинол (VII) [252]:
(описаны и другие примеры такого проявления влияния заместителя [250,
251 ]). В случае же замещенных в положение 4 циклогексенов (замести-
тель— ОН, ОСН3) и £{«с-гекс-3-енола заместитель лишь способствует
стереоспецифическому присоединению XHg и OR' по двойной связи, но цик-
лизации не происходит, например [252]:
HgAc2, КС1
R'OH "*
CIHg OR
HgAc2, KC1
R'OH
Получение 3-хлормеркурциклогексаи-1,4-диола [252]. 5 г циклогекс-3-енола и 15 г
уксуснокислой ртути в 150 мл воды встряхивают при комнатной температуре 10 дней. За-
тем прибавляют 3 г хлористого натрия, раствор испаряют в вакууме досуха. Из остатка
извлекают этанолом ртутноорганическое соединение. Выход 16,5 г (94%). Кристаллизация
из бензола. Вещество разлагается выше 150°С.
а-Терпинеол [7, 49] присоединяет соли ртути в обычных условиях, при
этом в результате внутримолекулярной ангидризации образуется ртут-
ноорганическое производное цинеола и два продукта — ртутноорганиче-
ских производных терпина, представляющих собой, как показали Брук
и Райт [253], не диастеореомеры, а полиморфные формы одного и того же
соединения (относительно строения последнего см. также [252]).
Соль ртути обычным способом присоединена к коричному спирту [254,
265], к о,о'-диметоксистиролу [202], а,Р-диаллил-а-мезитил-Р,Р-диметил-
этанолу [207].
В водной среде пропенильная группировка в непредельных ароматиче-
ских спиртах и фенолах и их производных не присоединяет солей ртути; со-
единения же с аллильной группировкой дают продукты присоединения. Так,
сафрол [141—143, 211, 255, 256], метилхавикол [143, 211, 255], эвгенол
[141, 143, 257, 258], его эфиры [141, 143], апиол [143], миристицин [211,
255] присоединяют соли ртути; изосафарол [143, 211], анетол [211], изоэв-
генол [143], изоапиол [143], изомиристицин [143, 211], азарон [143, 255] не
дают продуктов присоединения. Возможно, что в спиртовой среде удалось
бы произвести присоединение солей ртути и к этим соединениям.
При присоединении соли ртути к о-аллилфенолу [45] (в воде), его
гомологам и производным [46, 260] происходит замыкание цикла и образу-
Присоединение солей ртути к непредельным соединениям
145
ются ртутноорганические производные 1-метил-1,2-дигидробензофурана:
снасн=сна
нао
CHaCHCHaHgX
О CHaHgX
Такое же замыкание цикла происходит при действии солей ртути на 2- и
4-аллилрезорцины [501 и о-аллил-п-третичнобутилфенол [261].
Аналогично 2,2-дифенилпентен-4-ол-1 при продолжительном стоянии
с уксуснокислой ртутью в метиловом спирте дает соответствующее производ-
ное тетрагидрофурана [197].
Получение 1-хлормеркурметил-1,2-дигидробеизофурана [45]. При перемешивании
в течение 3 час. взвеси 25 г о-аллилфеиола в растворе 52,5 а сулемы в 1000 мл воды
выделяется белый осадок, который после кристаллизации из спирта весит 66,5 г.
Т. пл. 137°С.
Получение 1-ацетатмеркурметил-1,2-дигидробеизофурана [45]. Раствор 23,7 г уксус-
нокислой ртути в 100 мл воды при перемешивании медленно прибавляют к взвеси 10 г
о-аллилфенола в 100мл воды.Реакционную массу перемешивают 30 мин., в течение которых
выделяется тяжелое сероватое масло. Слитый с масла водный раствор выделяет к следую-
щему дню кристаллы. Масло постепенно в течение нескольких часов становится твердым
осадком, который смешивают с кристаллами и перекристаллизовывают из воды или спирта.
Т. пл. 80—81°С. Выход почти количественный (см; также стр. 117).
При действии окиси ртути в 80%-ной уксусной кислоте на аллильные
эфиры фенола и 0-нафтола происходит меркурирование в ядро без затраги-
вания двойной связи [262] (см. гл. V, стр. 75).
Предложен метод [263] отделения винильных производных от этильных
из смеси хинных алкалоидов, основанный на способности образования пер-
выми продуктов присоединения солей ртути.
Присоединение ацетата или нитрата ртути к диаллиловому эфиру в водной
среде с последующей обработкой хлористым калием приводит к 2,6-дихлор-
меркурметилдиоксану [146, 167]
/°\ /°\
сна сна сна сна
I I КС1 I ]
СНа = CH CH = СНа + HgXa + НаО 2±i. ClHgCHa-CH CHHCHjHgCl.
\0/
Он представляет собой смесь цис- и транс-изомеров [264] с преобладанием
первогр [55].
Подобное же замыкание цикла с образованием ртутного производного
диоксана происходит при присоединении ацетата ртути в воде к моноалли-
ловому эфиру глицерина [244, 245] и этиленгликоля [244].
К. аллилацетату [187]; коричному эфиру n-нитробензойной кислоты [265]
соли ртути присоединяются в обычных условиях.
Окись ртути присоединена в воде по двойной связи аллильной группы
n-фенолсульфокислой соли 1 -аллил-4-диэтиламиноэтокси-5-метоксибензола,
замещенной n-аллилоксибензойной кислоты, ее эфира, п-аллилоксибен-
золсульфокислоты [2581, уксуснокислая ртуть присоединена в воде к ал-
лилоксибензойной кислоте и ее эфирам [266], аллилсульфонамиду [259].
Присоединение к тиоаналогам оксисоединений. Диаллилсульфид обра-
зует с ацетатом ртути в водном растворе циклический продукт [167, 266а],
146
Ртутноорганические соединения. Лит. стр. 173—181
с сулемой же — продукт присоединения ациклического строения [267]:
А
CH3CO2HgCH2 Х° rHaHgOaCCHs
(ClHgCH2CH (ОН)СН2)а S
Синтез 2,6-ди-(ацетатмеркурметил)-1,4-тиоксана [266а]. К перемешиваемому раствору
0,5 моля ацетата ртути в 500 мл воды быстро прибавляют 0,251 моля аллилсульфида. Через
29 час. реакция на ион ртути отрицательная. Осадок отфильтровывают и высушивают до
постоянного веса. Выход бис-ацетоксимеркурсоединения 81,6 г; т. пл. 166—182°С (с разло-
жением). После нескольких кристаллизаций из ацетона т. пл. 197—199°С (с разложением).
Из фильтрата от бис-ацетоксимеркурпроизводного после подщелачивания иодистым калием
высаживают дииодмёркурпроизводиое. Вес после сушки 62,6 г. Общий выход 82%.
Присоединением ацетата ртути в воде к диаллилсульфону получают соот-
ветствующий циклический сульфон [266а].
Диметаллилсульфид присоединяет ацетат ртути в воде с образованием
2,6-диметил-2,6-б«с-(ацетоксимеркур)-п-оксатиана [266а]:
[CH2=C(CH3)CH8]aS+ Hg(O2CCH3)2+ нао -Л н3с /А сн3
,«ул,
Присоединение к простым и сложным виниловым эфирам. Несмеянов,
ЛуценкО и Верещагина [124] нашли, что при действии уксуснокислой ртути
в водной среде на простой виниловый эфир (винилэтиловый, винилбутиэто-
вый, винилизоамиловый, дивиниловый) происходит обычное присоединение
соли ртути по двойной связи, причем ориентация соответствует правилу
Марковникова. Вслед за этим молекула теряет элементы спирта и обра-
зует раствор ацетатмеркурацетальдегида. Осаждением раствора ионом хлора
или брома легко получить хлормеркур- или броммеркурацетальдегид
— твердые вещества:
OR
СН2=СН—OR+HgAc2 +Н2О -* AcHgCH2 — Сн/ -•> AcHgCH2CHO Я' GlHgCH2CHO.
ОН
Аналогично из изопропенил-н-бутилового эфира получено простейшее
меркурированное производное кетона — хлормеркурацетон [124].
Так же как уксуснокислую ртуть, винилэтиловый эфир присоединяет
ртутную соль тринитрометана [227].
Получение хлормеркурацетальдегида [124]. К профильтрованному раствору 32 г (0,1
моля) уксуснокислой ртути в 150 мл воды постепенно при сильном встряхивании добавля-
ют 10 г (0,1 моля) винилбутилового эфира. Быстро происходит присоединение со слабым
разогреванием. К раствору, отфильтрованному от следов выделившейся ртути, прибавляют
раствор 7,5 г (0,1 моля) хлористого калия в минимальном количестве воды. Сразу же вы-
падают белые кристаллы. Выход 24 г (85%). Хлормеркурацетальдегид перекристаллизован
из горячей воды. Т. пл. 130—13ГС.
Получениехлормеркурацетона [124]. 1,2 г изопропенил-н-бутилового эфира (0,01 моля)
прибавляют к раствору 3,2 г ацетата ртути в 15 мл воды. Виниловый эфир быстро растворя-
ется со слабым разогреванием. Выпадает немного закисных солей ртути, которые отфиль-
тровывают. При добавлении 0,75 г хлористого калия в 3 мл воды раствор мутнеет и через
2—3 мин. начинают выпадать хорошо образованные кристаллы, которые через 15 мин. отса-
сывают и промывают холодной водой. Выход 1,7 г (58%). После перекристаллизации из
метилового спирта т. пл. 104°С. 1
Присоединение солей ртути к непредельным соединениям 1Г475
При кратковременном кипячении р-фенилвинилэтилового эфира с суле-
мбй и окисью ртути в водно-ацетоновом растворе удается выделить хлор-
меркурфенилацетальдегид (выход 88%) [268]
2СвН5СН — CHOCsHs -ф HgCl2 + HgO -ф Н2О -» 2СвН5СНСНО -ф 2С2Н,ОН.
HgCI
Двухчасовое кипячение р-фенилвинилэтилового эфира с избытком уксус-
нокислой ртути в водном растворе приводит к нерастворимой смеси поли-
мерных веществ, содержащих ртуть [102, 268], по-видимому [268], продук-
тов окисления и меркурирования первоначально образовавшегося моно-
меркурированно.го фенилацетальдегида. а-Фениловый эфир (а также а-ацет-
оксистирол) гладко реагируете уксуснокислой ртутью в водном раство-
ре и образует ю-ацетоксимеркурацетофенон [268]:
сн2 = СС6Н5 -|- (СН8СОО)2 Hg -ф Н2О -> CeHeCOCH2HgOCOCH3 -ф CHgCOOH -ф СаНвОН.
ос2н6
Получение хлормеркурфенилацетальдегида [268]. К смеси 1,5 г (0,01 моля) 3-фенил-
винилэтилового эфира, 1,1г (0;005 моля) окиси ртути, 5 мл ацетона и 1- мл воды приливают
раствор 1,35 г (0,005 моля) сулемы и 8 мл ацетона. Реакционную смесь нагревают до ки-
пения н быстро фильтруют. После охлаждения - фильтрата и испарения части ацетона
отделяют кристаллы хлормеркурфенилацетальдегида, которые промывают небольшим ко-
личеством охлажденного ацетона и абсолютным эфиром. Выход 3,2 г (88%), т. пл. 121—
123°С.
Галогенмеркуроксосоединения получены [127] также присоединением
соли ртути в воде при комнатной температуре к энолацетатам оксосоедине-
ний (с последующим добавлением иона галоида):.
С1' •-
RCH = СНОСОСНз -ф Hg (ОСОСН3)2 -ф Н2О —> CIHgCHRCHO -ф 2СНзСООН.
Из винилацетата получен хлормеркурацетальдегид (выход количествен-
ный [127]), из Р-метил- и.р-этилвинилацетатов— соответствующие хлормер-
куралкилальдегиды [269], из изопропенилацетата — хлормеркураце-
тон, из циклопентилацетата — о-хлормеркурциклопентанон (выходы —
70—85% [127]).
Получение хлормеркурацетальдегида из винилацетата [127]. К раствору 16 г (0,05
моля) уксуснокислой ртути в 75 мл воды постепенно при сильном встряхивании добавляют
4,3 г (0,05 моля) свежепёрегнанного вйнилуксусного эфира (т. кип. 72—74°С). Быстро про-
исходит присоединение со слабым разогреванием. В раствор, отфильтрованный от следов
закисных солей ртути, прибавляют раствор 3,8 г (0,005 моля) хлористого калия в 10 мл
воды. Сразу же выпадают белые кристаллы. Выход количественный. Перекристаллизация
из воды, т. пл. 129—130°С (с разложением).
Описано присоединение солей ртути к винил-, а также аллил ацетату [187].
Реакция присоединения ацетата ртути по двойной связи использо-
вана [270] для качественного анализа смесей винилацетата и винилал-
киловых эфиров дикарбоновых кислот при помощи хроматографии на
бумаге.
Проведение реакции присоединения соли ртути к простым и сложным
виниловым эфирам в спиртовой среде в присутствии окиси ртути, связываю-
щей выделяющуюся уксусную кислоту, является методом синтеза ацеталей
меркурированного ацетальдегида [128].
2СН2 = CHOR -ф (CH3CO2)2Hg -ф HgO + 2ROH^ 2CH3COOHgCH,CH(OR)2 -ф
-ф Н2О ClHgCH2CH(OR)2.
148
Ртутноорганические соединения. Лит. стр. 173—181
Получение диэтилового ацеталя хлормеркурацетальдегида из винилэтилового эфира
[128]. К смеси 15,9 г (0,05 моля) уксуснокислой ртути и 20 мл абсолютного этилового
спирта при перемешивании и охлаждении приливают 7,5 г (0,1 моля) винилэтилового эфи-
ра. По окончании присоединения соли ртути к виниловому эфиру в реакционную смесь не-
большими порциями (1 г каждая) вносят 10,8 г (0,05 моля) сухой желтой окиси ртути, до-
бавляя каждую новую порцию после растворения предыдущей. Затем реакционную смесь
выливают в раствор 7,5 г (0,1 моля) хлористого калия в 60 мл воды, отделяют тяжелое мас-
ло, растворяют его в 100 мл сухого серного эфира и сушат хлористым кальцием. После
фильтрования эфир и спирт отгоняют (в вакууме) и остаток сушат в вакуум-эксикаторе
над пятиокисью фосфора. Таким образом получают 26 г (73%) диэтилового ацеталя хлор-
меркурацетальдегида в виде бесцветного тяжелого масла.
Взаимодействие окиси ртути в присутствии уксуснокислой ртути в водно-
спиртовой среде с винилалкиловыми эфирами приводит с. превосходными
выходами прямо к меркур-быс-ацетальдегиду, а с а-замещенными винилал-
киловыми эфирами — к меркургбос-кетонам [2711:
н2о
CH3COOHgOH +CHa=CR'OR-> [HOHgCH2CR'(OR)OCOCH3] сн qr-*
-» Hg (CH1CR'O)2 + ROH + СНзСООН,
R' = H или Aik.
При проведении этой реакции с винилэтиловым эфиром в среде абсолют-
ного спирта получен неполный ацеталь меркур-бнс-ацетальдегида [1281:
OCsHb
(О»Н6О)в CHCH2HgCH2CH
\>н
Получение меркур-б«С-ацетальдегида [271]. К смеси 5,4 г желтой окиси ртути, 0,2 г
уксуснокислой ртути, 1 мл воды и 2 мл спирта прибавляют по каплям при встряхивании
6 г винилбутилового эфира. Окись ртути с разогреванием растворяется. Реакционную
смесь еще теплой отфильтровывают от следов металлической ртути и фильтрат сильно ох-
лаждают. Выпавшие кристаллы отделяют, промывают холодным спиртом, эфиром и высу-
шивают в вакуум-эксикаторе над пятиокисью фосфора. Выход меркур-бас-ацетальдегида
6 г (86%). Т. пл. 93—95°С (из спирта).
В аналогичных условиях меркур-бис-ацетальдегид получают из винил-
этилового, винилизопропилового, винилфенилового эфиров. Выходы соот-
ветственно 87, 90, 81%.
Получение диацетонйлртути [271]. К смеси 2,7’ г желтой окиси ртути, 0,1 г уксусно-
кислой ртути, 3 мл спирта и 0,5 мл воды при встряхивании по каплям прибавляют 3,3 г
изопроценилэтилового эфира. После растворения окиси ртути реакционную смесь отфиль-
тровывают от следов металлической ртути и фильтрат испаряют .в вакууме. Выход ди ацето-
нилртути 3,8 г (98%). Т. пл. 689С (из бензола).
Меркур-быс-ацетальдегид получают действием на дивинилацетали альде-
гидов окиси ртути в присутствии уксуснокислой ртути [2721:
осн = сн2
/ HgfOCOCH,),
RCH 4- HgO----------► RCHO -j- Hg (CH2CHO)2.
\>CH = CH2
Получение меркур-бис-ацетальдегида [272]. К смеси 21,6 г желтой окиси ртути, 0,8 г ук-
суснокислой ртути, 4 мл воды и 8 мл спирта прибавляют по каплям при перемешивании 14,2 г
дивинилбутираля. Окись ртути растворяется с разогреванием. Теплую реакционную смесь
фильтруют й охлаждают ледяной водой. Выпавшие кристаллы отделяют, промывают холод-
ным спиртом и эфирой. Выход сухого меркур-б«с-ацетальдегида 24,8 г (85%). Т. пл. 93—
94°С.
Приссединекие солей ртути к непредельным соединениям 149
Меркур-биоацетальдегид получен 1273] при действии на простой
виниловый эфир гидроокиси арйлртути через очевидное промежуточное об-
разование несимметричного ртутноорганического соединения:
OR
ArHgOH + СНа = CHOR 2^^ ArHgCHjCH
^ОСНз
которое затем распадается:
OR , OR \
гАгНёСЩСН7 ->AraHg-|- Hg] СЬкСН^ |
^ОСНз \ \>CH3A
(см. стр. 234 и 256)
По двойной связи простых виниловых эфиров происходит присоединение
элементов соли ртути и без участия растворителя — при проведении реак-
ции в эфире [274].
Получение ацетата ацетоксимеркурацетальдегидмоноэтилацеталя [274]. К 3,2 г
(0,01 моля) уксуснокислой ртути прибавляют Ю лы абсолютного эфира, к охлажденной
смеси приливают 0,72г винилэтилового эфира. Уксуснокислая ртуть быстро растворяется.
При охлаждении льдом с солью выпадает ацетат ацетоксимеркурацетальдегидмоноэтилаце-
таля. Выход 3,6 г (92,3%). Т. пл. 38—39°С (из эфира).
Об особенностях химического поведения меркурированных оксосоедине-
ний см. стр: 126—128.
О присоединении соли ртути к а-алкоксиакрилонитрилам см. стр. 153.
Взаимодействие винилалкилсульфидов с солями ртути см. гл. XVI,
стр. 367.
Присоединение солей ртути
к аминам, содержащим этиленовую связь
Диаллиламин в водной среде гладко присоединяет ацетат ртути, образуя
производное морфолина, после осаждения изолированное в виде 2,6-дихлор-
меркурметилморфолина [146].
NH NH
СН^ • ^СНа dH2 ^СНз
СН3 = СН СН = СНа + HgXa + НаО -> X HgCHa — СН СН — CHaHgX
W
Получение 2,6-ди-(хлормеркурметил)морфолина [146]. 5 г (0,05 моля) диаллиламина
(т. кип. 108—111°С) прибавляют к раствору 30 г уксуснокислой ртути в 100 мл воды. Амин
растворяется с заметным разогреванием. Через 12 час. отфильтровывают 0,3 г закисной со-
ли ртути. При добавлении к фильтрату раствора 7 г хлористого калня в 50 мл воды выпа-
дает 26 г белого осадка. Последний при действии 10% -ной щелочи растворяется, оставляя
небольшое количество желтой окисн ртути. После усреднения фильтрата выпадает белый
осадок 2,6-.ди-(хлормеркурметил)морфолииа. Соединение нерастворимо в органических
растворителях. При нагревании( разлагается не плавясь.
Соль ртути присоединяется в спиртовой среде к октадецен-9-ил-амину,
бпс-октадецен-9-ил-амину, гендецен-1-ил-амину [275]. п-Кротиламинофенил-
уксусная кислота присоединяет соли ртути в спиртах и гликолях [2661. Осу-
ществлено присоединение соли ртути в метиловом спирте к о- и п-аллилами-
нобензойной кислоте [2761.
Присоединение к аллиламидам кислот см. стр. 161.
150
, Ртуггмоорганические соединения. Лит. стр. 173—181
Присоединение солей ртути
к оксосоединениям, содержащим этиленовые связи,
. и их функциональным производным
Присоединение солей ртути к кетонам олефинового ряда. Насы-
щенные кетоны, как бензальацетофенон Г149, 277], бензальхдораце-
тофенон [277], . дибензальацеТон [277], а также ненасыщенные жирные
кетоны с длинной цепью [2781 и арилалкен- или арилалкадиенкетоны с
длинной цепью алкена или алкадиена [279] дают продукты присоединение
при действии на холоду уксуснокислой ртути в спирте. Реакция бензаль-
ацетофенона сильно ускоряется в присутствии диметилперекиси, перекиси
водорода, эфирата фтористого бора и замедляется примесями, загрязняю-
щими уксуснокислую ртуть: ацетонитрилом, пиридином, диэтилсульфидом,
трпнс-стирилцианидом [149]. . '
' Присоединение к а, р-ненасыщенным оксосоединениям, как всегда к та-
ким системам, происходит против правила Марковникова.
Получение а-ацетоксимеркур-р-метокси-₽-фенилпропиофеиона [277]. Раствор 25 г . бен-
зальацетофенона и 38 г уксуснокислой ртути в 250 мл абс. метилового спирта оставляют
стоять до отсутствия реакции на ион ртути с едким натром. После упаривания раствора
выкристаллизовывается ртутное соединение. Выход 53 г. Т. пл. после кристаллизации из
метилового спирта 115°С.
Арилвинилкетоны образуют с уксуснокислой ртутью в метиловом спир-
те при хранении при комнатной температуре продукты присоединения (вы-
деленные в виде г-алогенидов) АгСОСН (HgHal)CH2OMe [280].
Присоединение солей ртути к кетенам и их производным. "Реакция
кетена с ацетатом или окисью ртути в спиртах дает с хорошим выходом
эфиры меркур-бис-уксусной кислоты [136]. Лучше брать окись ртути
в присутствии небольшого количества уксуснокислой ртути. Реакция
идет по схеме:
HgO + ЗСН, = С = О + 2ROH Hg(OGOS-H3?UHg (CHaCOOR)a + CHSCOOH.
Получение метилового эфира меркур-бис-уксусиой кислоты [136]. В энергично пере-
мешиваемую взвесь 20 г (0,092 моля) окиси ртути и 3 г (0,009 моля) ацетата ртути в 120 мл
абс. метилового спирта пропускают кетен (0,4—0,6 моля) до полного растворения окиси
ртути. Во время пропускания кетена реакционный сосуд охлаждают так, чтобы темпера-
тура смеси не была выше 20—25° С. По окончании реакции раствор фильтруют от неболь-
шого количества выделившейся металлической ртути и растворитель удаляют в вакууме
водоструйного насоса при температуре бани не выше 30°С. Остающиеся кристаллы с т. цл.
96—98°С, представляющие собой метиловый эфир меркур-б«с-уксусной кислоты, загряз-
ненные примесью метилового эфира ацетоксимеркуруксусной кислоты, растворяют
в 100 мл абс. метилового спирта и пропускают еще 0,2 моля кетена. После испарения рас-
творителя в вакууме получают : 35 г (количественный выход) метилового эфира меркур-б«с-
уксусной кислоты, который промывают минимальным количеством эфира. Т. пл. 99—100°С.
Кристаллизация из метанола^
При реакции с кетеном Р-меркурированных оксисоединений и.а-мёрку-
рированных оксосоединений происходит вытеснение из этих квазикомплек-
сов соединением с более активной кратной связью (кетеном) соединений с
менее активными связями (олефина и винилалкилового эфира) и с хорошими
выходами получаются, в зависимости от среды реакции, меркурированная
уксусная кислота и 'ее эфиры (Луценко, Фосс [41, 137]). С Р-галогенмер-
курэтилметиловым эфиром по уравнению
XHgCHaCHtOCH* + СНа = С'= О + ROH-> XHgCHaCOOR + СНа = СНа + СНзОН
Присоединение солей ртути к непредельным соединениям 151'
и с а-галогенмеркуркетонами по схеме
XHgCHaCOR' + СН2 = С = О + ROH -» XHgCH2COOR
(R = Н или Aik) получена галогенмеркуруксусная кислота и ее эфиры,
а с ацетатом ацетоксимеркурацетальдегидмоноэтилацеталя — эфир меркур-
бис-уксусной кислоты.
Получение хлормеркуруксусной кислоты [137]. В раствор 1,5 г (0,005 моля) [3-хлор-
меркурэтилметилового эфира в 75 мл горячен воды пропускают 0,05 моля кетена. При ис-
парении растворителя выпадают крупные кристаллы хлормеркуруксусной кислоты. Вы-
ход 1,35 г (90%). После кристаллизации из метилового спирта т. пл. 196° С (запаянный
капилляр).
Получение метилового эфира хлормеркуруксусной кислоты. В раствор 11 г (0,037 моля)
хлормеркурэтилметилового эфира в 100 мл абс. метанола пропускают шестикратный из-
быток кетена. После испарения растворителя остается 9,2 г (выход 90%) метилового эфира
хлормеркуруксусной кислоты; после перекристаллизации из метанола т. пл. 83°С. Бром-
меркурэтилметиловый эфир реагирует} с избытком кетеиа и в среде бромистого этила с об-
разованием метилового эфира броммеркуруКсусной кислоты с выходом 82%.
Получение метилового эфира броммеркуруКсусной кислоты. В охлажденную до 5° С
взвесь 6,25 г (0,0185 моля) броммеркурацетона в 15 мл абс. метанола пропускают 0,05 мо-
ля кетена. После испарения спирта получают 6,6 г (количественный выхЬд) метилового
эфира броммеркуруКсусной кислоты. Перекристаллизация из смеси бензола и н-октана.
Т. пл. 80—81°С.
Меркур-бис-альдегиды и -кетоны с кетеном в воде и спиртах образуют
меркур-бие-уксусную кислоту и ее эфиры, например [41, 137]:
Hg (СН2СНО)2 + 2Н2О 4- 2СН2 = С = О Hg (СН2СООН)2 4- 2СН3СНО.
Получение третичнобутилового эфира меркур-бйС-уксусной кислоты. Во взвесь 43 г
(0,15 моля) меркур-б«с-ацетальдегида в 100 мл абс. третичнобутилового спирта при охлаж-
дении пропускают 0,6' моля кетена до полного растворения осадка. Спирт испаряют, по-
лучают 64,3 г (количественныйвыход) третичнобутилового эфира меркур-бис-уксусиой кис-
лоты. После перекристаллизации из гексана т. пл. 85—86°С.
При проведении реакции а-галогенмеркур- и меркур-бис-оксосоеди-
нений с кетеном в бензоле получены а, p-непредельные эфиры галоген-
меркур-бис-уксусной кислоты [41, 137]:
О
— HgCH2 — COR 4- СН2 = С = О — HgCH2C^
\)С = СН2
Получение а-пропилвинилового эфира меркур-бие-уксусной кислоты. В раствор 15 г
(0,03 моля) меркур-бис-метилпропилкетона в 50 мл бензола при охлаждении пропускают
0,1 моля кетена.Растворитель испаряют,получают 13,45 г (количественный выход) а-пропил-
винилового эфира меркур-бис-уксусной кислоты. После перекристаллизации из абсолют-
ного метанола т. пл. 70—71°С (см. стр. 127).
Взаимодействие диацеталя кетена с уксуснокислой ртутью в безводном
ацетоне приводит (после высаживания хлористым калием) с 60%-ным выхо-
дом к эфиру три-(хлормеркур)уксусной кислоты в результате реакции при-
соединения по двойной связи и меркурирования [281]:
СН2 = С (OR)2 4- (СН3СО2) Hg
OR
(СНзСО2Н8)з С — С—OR
^ОСОСНз
О О
cl'
(CH3CO2Hg)3 С — С 4- CHsCOaR - (ClHg)3 СС
\)R \>R
152
Ртутноорганические соединения. Лит. стр. 173—181
Получение этилового эфира три-(хлормеркур)уксусной кислоты [281]. К раствору 2 г
ацеталя кетена в 5 мл сухого ацетона быстро (небольшими порциями) прибавляют 1 г аце-
тата ртути. Реакция идет с разогреванием, раствор принимает красноватую окраску вслед-
ствие частичной полимеризации ацеталя кетена. Раствор разбавляют водой до появления
слабого помутнения и к нему приливают по каплям раствор хлористого калия до прек-
ращения выпадения осадка. Выход 5 г (60%, считая на уксуснокислую ртуть). Кристалли-
зация из метилового спирта. Разлагается не плавясь.
Менее реакционноспособные диэтилацетали хлор- и бромкетена с уксус-
нокислой ртутью в водном спирте образуют с небольшим выходом этиловые
эфиры меркур-бис-хлор- (и бром-)уксусной кислоты [2811; выход несколько
повышается, если действовать окисью ртути в присутствии ацетата ртути..
При проведении реакции в эфире в результате’ одновременного присоеди-
нения по двойной связи и меркурирования образуется этиловый эфир ди-
(ацетоксимеркур)хлор- (и бром-)уксусной кислоты [2811
2СНХ— С (ОС2Н5)24- 2Hg (ОСОСН3)2 (CH3COOHg)2CX — СООС2Н5 !- ХСН2СООС2Н5+
-( CHgCOOCgHg,
Получение этилового эфира ди-(ацетоксимеркур)хлоруксусной кислоты [281]. К рас-
твору 6 г ацеталя хлоркетена в 5 мл эфира присыпают! г уксуснокислой ртути. Через сут-
ки осадок отфильтровывают и промывают эфиром. Выход 2,3 г. После испарения раствори-
теля получают еще 1,3 а вещества. Выход 3,6 г (90%, считая на ацетат ртути). Кристалли-
зуют из спирта или дихлорэтана. Т. пл. 159—160° С (разлагается).
Получение этилового эфира ди-(хлормеркур)хлоруксусиой кислоты [281]. К раствору
0,2 г этилового эфира ди-(ацетоксимеркур)хлоруксусной кислоты в 20 мл воды добавляют
по каплям 1 N раствор хлористого калия до прекращения выпадения осадка. Кристалли-
зация из спирта. Выход 0,13 г (65%). Т. пл. 260—262°С (с разложением, в запаянном
капилляре).
О присоединении уксуснокислой ртути к а-алкоксиакрилонитрилам
см. стр. 153.
Действие на диэтилацеталь кетена гидрооксифенилртути дает, через
промежуточное образование полнозамещенного несимметричного ртутно-
органического соединения, дифенилртуть и меркур-бис-этилацетат, выделен-
ный после реакции с сулемой в виде хлормеркурэтилацетата [273] (см. гл.
XII, стр. 234).
Взаимодействие диметил ацеталя диметилкетена с хлористым бутилом,
хлористым цинком и сулемой приводит с 99%-ным выходом к метиловому
эфиру а-хлормеркуризомасляной кислоты [282].
Присоединение соли ртути в спиртах к оксопроизводным некоторых ге-
тероциклов, содержащих двойную связь в цикле, см. стр. 162.
Аллилацетоноксим присоединяет соли ртути на холоду в присутствии ще-
лочи, давая производные дигидроизоксазина [49]:
Н2С = СН — СНа — СН2 — С — СН3+ HgX (ОН) —
II .•
HON
ГН2С — CHCHaCHaCCHs 1
II II
XHg ОН HON J
СНа —СНа
-э- XHgCH2CH ^ССНз
\>-----
Аналогичное соединение дает метилгептеноноксим [49]. О сопровож-
дающемся меркурированием присоединении HgXOAlk к антипирину и дру-
гим алкилзамещенным 1-арил-5-пиразолонам см. стр. 163 (ср. также гл. V,
стр. 98).
В настоящей главе рассматривается присоединение сулемы по двойной
связи карбонильного производного фосфорилида (VIII) (условия не указа-
Присоединение солей ртути к непредельным соединениям 153
ны), которое приводит к образованию меркурированных солей фосфония
(IX) [3621:
(С6Н6)з Р = СНСОСвНб + HgCk
С1-
VIII
HgCI
(С6Н5)8Р+-С-Н
\освн5.
IX
Соль (IX) является квазикомплексным соединением и проявляет двой-
ственную реакционную способность. С бензальдегидом она реагирует с
элиминированием сулемы и образованием транс-бензоилстирола. С другой
стороны, метилат натрия отщепляет от этой соли хлористый водород', давая
фосфорилид, содержащий ртуть:
Г HgCI п HgCI
(Се H 5)3 Р+ - с-н ci- (C8hs)8p = c/
\oCeH5- \oc3hs
При действии разбавленной соляной кислоты последнее соединение, при-
соединяя хлористый водород, превращается в соль (IX). -
Присоединение солей ртути к карбоновым кислотам,
содержащим этиленовые связи,
и их производным
Непредельные кислоты этого типа присоединяют соли и окись ртути в тех
же условиях, что и олефины. В случае а, 0-непредельных кислот атом ртути
становится в «-положение к карбоксилу, а гидроксил (или алкоксил в спир-
товой среде) — в p-положение к карбоксилу. Это было показано, например,
путем переведения продукта присоединения ацетата ртути к кротоновой ки-
слоте в водной среде в р-оксимасляную кислоту действием сероводорода;
реакция — специфическая для именно этих веществ. Согласно Бильману
[145,284], только цис-, но не транс-, конфигурации»,Р-непредельных кислот
способны к присоединению соли ртути. Так, по данным Бильмана [145],
акриловая, кротоновая, малеиновая, итаконовая, цитраконовая, аллоко-
ричная кислоты присоединяют, а коричная, фумаровая и мезаконовая не
присоединяют солей ртути. Однако это относится только к реакциям в вод-
ной среде, тогда.как в растворе различных спиртов эти кислоты присоеди-
няют уксуснокислую ртуть (из перечисленных испытывалась только корич-
ная кислота).
Акриловая кислота[145, 167, 285] и ее эфир [167], Р-циклопентилакрило-
вая [286] кислота в обычных условиях (действием, например, ацетата ртути
в метиловом спирте) образуют продукты присоединения со ртутью в a-поло-
жении к карбоксилу. Изучена кинетика метоксимеркурирования эфиров
акриловрй кислоты (реакция II порядка) [62] и метакриловой кислоты [450].
Если в диацетале кетена двойная связь пассивирована заменой одной OR-
группы на CN, взаимодействие полученных а-алкоксиакрилонитрилов
с уксуснокислой ртутью в водном растворе приводит к эфиру мономеркури-
"рованной уксусной кислоты [135].
OR
СН2 = С// + HgAc2 + Н2О->
\n
ORn
СНзСООН§СН2С—CN
HgACj
CHaCOOHgCHaCOOR 4-
+ Hg (CN)a + 2СНзСООН.
154 Ртутноорганические соединения. Лит. стр. 173—181
В отличие от почти мгновенного присоединения к пропилену реакция
требует для своего проведения нескольких часов'.
На 1 моль непредельного соединения надо брать 2 моля уксуснокислой
ртути; если брать 1 моль уксуснокислой ртути, то выход продукта реакции
снижается с 90—95 до 40—45%.
Получение ацетатмеркурпропилацетата [135]. К охлажденному раствору 63,6 г (0,2
моля) уксуснокислой ртути в 150 мл воды при энергичном встряхивании в несколько при-
емов прибавляют 11,Г г (0,1 моля) а-пропоксиакрилоиитрнла. Выпадает белый, осадок,
который отделяет. Маточный раствор упаривают на холоду и выпавший осадок ацетатмер-
курпропилацетата снова отделяют. Общий выход 34,6 г (95%). Кристаллизация из мети-
лового, спирта. Т. пл. 109—111 °C.
В обычных условиях осуществлено присоединение ацетата ртути в мети-
ловом спирте к изовалериановой кислоте [287].
При присоединении солей ртути в метиловом спирте или в воде к аллил-
уксусной и аллилдифенил уксусной кислотам образуются лактоны [288, 289,
451,452]
ClHgCH3CH-CH2 . •
о
о=с:—cr2
R=H,C6Hb. Произведено присоединение солей, ртути в-спиртах и гликолях
к 2-нонёновой [266], 5-фенил-2-пентеновой [266] кислотам. Описано присо-
единение в обычных условиях ацетата ртути в метиловом спирте к а-пропил-
кротоновой кислоте [290], к 2,7-карбэтокси-2-гептеновой кислоте [2911.
Ацетат ртути в водном растворе обычным образом присоединяется к унде-
циленовой кислоте [2921, продукт взаимодействия ундецил ено вой кислоты
с окисью ртутн в водном растворе (после подкисления уксусной кислотой)
выделен в виде лактона 1293]. Соли ртути в метиловом спирте присоединены
к олеиновой и эруковой кислотам [294].
В обычных условиях произведено присоединение ацетата ртути в присут-
ствии низших алифатических спиртов к нитрилам олеиновой (продукт—
9-ацетоксимеркур-Ю-алкоксипальмитинонитрил), ундециленовой (продукт—
1-ацетоксимеркур-2-метоксиундеканонитрил) и других высших непредель-
ных кислот 1295].
Эфиры ненасыщенных кислот, например олеиновой и линолевой, в част-
ности триолеины, легко дают продукты присоединения при действии ртут-
ных солей в алкогольном растворе, причем образуются эфиры меркурирован-
ных эфирокислот [8, 296, 297]. Эти производные жирных кислот представля-
ют собой масла высокого удельного веса [296].
Так же как в случае других непредельных соединений, присоединение
ацетата ртути в метиловом спирте к эфирам высших непредельных кислот
(олеиновой, элаидиновой, эруковой, брассидиновой) быстрее происходит
к цис-изомерам, чем к транс-изомерам (константа скорости второго порядка
[298]). Так, константы скорости реакции присоединения ацетата ртути
к метиловым эфирам олеиновой и элаидиновой кислот относятся, как
12,5 : 1.
Синтез 9-ацетоксимеркур-10-метоксипальмитииовой кислоты[298]. К раствору 14г ук-
суснокислой ртути в 250 мл метанола, содержащего 2,5 мл воды и 1 мл ледяной уксусной
кислоты, приливают 10 а метилового эфира олеиновой кислоты. После смешения в почти
наполненном доверху сосуде оставляют стоять 24 часа в темноте при комнатной температу-
ре. Отгоняют большую часть метанола иа водяной бане при 90°С; остаток извлекают 200 мл
хлороформа н промывают водой. После сушки и отгонки в вакууме хлороформа остаток
для полного удаления растворителя 5 час. держат в вакууме при 30—40°С и 0,01 атм. По-
сле фильтрации от следов мути через стеклянный фильтр получают 13,2 а(66%)бесцветной
жидкости.
Присоединение солей ртути к непредельным соединениям 155
Разная скорость присоединения солей ртути цис- и транс-изомерами
непредельных кислот и их производных применена для определения состава
смёСи эфиров элаидиновой и олеиновой кислот [299].
В- случае эфиров ненасыщенных жирных кислот, содержащих две и более
двойных связей (линолевой и линоленовой), с разной скоростью происходит
образование моно- и диаддукта (а также триаддукта в случае эфира лино-
леновой кислоты) [300J.
Хаульмугровая кислота и ее этиловый эфир дают продукты присоедине-
ния при действии уксуснокислой ртути на холоду в смеси этилового спирта
и уксусной кислоты [301]. Вероятное строение продукта присоединения
в случае кислоты: _________-
HgCH-CH (СН2)12СС)А
II.
С2Н5ОСН сн2
''сНа
Присоединение уксуснокислой ртути к диаллилуксусной (в этиловом -
спирте) и диаллиламалоновой кислотам (в метиловом спирте) произведено
по обеим аллильным группам [302]. В первом случае получен продукт
(CH2CH(OC2H5)CH2HgBr\
QHsQiCCH ш J °
\:H2CHCH2HgBr / а .
Присоединение ацетата ртути к бициклогептенкарбоновой кислоте и
к 4-окси- (и 4-метокси)циклогексенкарбоновой кислоте и ее нитрилу произ-
ведено при 2—7-часовом хранении в. метиловом спирте [252]. В первом слу-
чае оно сопровождается лактонизацией.
Присоединение ацетата ртути в воде к 1,4-метилен-Д6-циклогексен-2-
карбоновой кислоте [стереоизомер (X)] приводит к двум лактонам: (XI),
образующемуся с выходом 77%, и (XII), образующемуся с выходом. 3%:
что является первым примером образования при оксимеркурировании двух
диастереомеров [303].
Стереоизомерная кислота (X')
также образует два
продукта в отношении 4: 1, которым, однако, не удалось приписать опре-
деленной конфигурации [3031.
. Присоединений солей ртути к циклоолефиновой дикарбоновой кислоте —
норборнен-эндо-^цс-2,3-дикарбоновой кислоте (XIII) (ацетата ртути в мети*
ловом спирте [304],.сулемы в воднбм растворе при 80°С[28]) и к диметило-
воМу эфиру 1,4-экзоэтилен-Дб-циклогексен-эндо-2,3-дикарбоновой кислоты
{304], согласно [28, 198, 304], происходит в эндо-цис-положения, так как
после, соответствующей обработки продукта присоединения дают внутрен
ние ртутные соли карбоновых кислот и лактоны. Однако Кривой и Каунт*
[611 считают более вероятным, что эти внутренние ртутные соли являютст
156 Ртутнооргаяимеские Соединения. Лит. стр. 173—181
биполярными ионами (см. [266}), например, продукт присоединения к (XIII)
имеет строение (XIII').
соон
соон
XIII
Hg*2
ROH
W д°°э
и присоединение OR- и HgX-групп, как обычно, происходит в транс-положе-
ние друг к другу. -
Присоединение же соли ртути к диметиловому эфиру норборнен-эндо-
цис-2,3-дикарбоновой кислоты (XIV) (ацетата ртути в метиловом спирте,
в диоксане, в уксусной кислоте) и к норборнен-трапс-дикарбоновой кислоте
[281 (XV) (сулемы в водном растворе при 100° С), по мнению Райта и др. [28,
304], происходит как в цис-(OR по отношению к HgX) (для кислоты преиму-
щественно, продукт XV'), так и в транс-(преимущественно для эфира, про-
дукт XIV') положения: ,
Присоединение к эфиру ацетата ртути в воде дает [281 лактон моноэфира
наряду с транс-продуктом присоединения к транс-кислоте.
Присоединение же OR- и HgX-групп при оксимеркурировании (сулемой
<ли ацетатом ртути в водном растворе) Д5-2,2,2-бпс-циклооктен-2,3-
цикарбоновых кислот и их производных (цис-, транс-дикарбоновой кислоты,
щметилового эфира и ангидрида (XVI) эндо-^нс-кислоты), согласно Хью и
2айту [1981, в исправление прежних [304] данных, происходит в транс-по-
южение друг к другу с одновременной лактонизацией. Например, XVI дает
KVF:
Одним из немногих случаев цис-присоединения OR и HgX по двой-
кой связи является окси- [75, 305, 3061 и ацетокси-[3061 меркурирова-
ше диметилового эфира экзо-/р/с-3,6-эндоксо-Д4-тетрагидрофталевой
шслоты (XVII) с получением продукта (XVII') (Юрьев, Зефиров и сотр.
Присоединение солей ртути к непредельным соединениям 157
[75, 305, 3061):
О
Cl' CIHg\J^\^COOCH3
"* «ОхАХ^ЛсООСНз
XVII'
(R = Н [75, 305] или СН3СО [306]).
При присоединении к (XVII) ртутной соли тринитрометана в воде образу-
ется продукт оксимеркурирования, а не продукт присоединения по двойной
связи элементов X и HgX (X = (NO2)3C), что также доказывает цис-строе-
ние продукта присоединения [198а] (см. [61, 238]).
Получение диметилового эфира 4-окси-5-хлормеркур-3,6-эндоксигексагндрофталевой
кислоты [306]. *
О
С1Н g\ С Нз
н //СООС Н з
Смесь 1 г диметилового эфира экзо-г|ис-3,6-эндоксо-Д-4-тетрагидрофталевой кислоты
и 1,1 г ацетата ртути в 70 мл воды перемешивают 12 час., фильтруют, добавляют раствор
0,5 г хлористого натрия и отделяют осадок. Получено 1,9 г (90%) вещества с т. пл. 246°С
(разложение, из метанола).
Проведение такой реакции не в воде, а в ледяной уксусной кислоте при-
водит к 4-ацетокси-5-хлормеркурпроизводному. 4-Ацетокси-5-ацетатмеркур-
производное выделено при присоединении ацетата> ртути в диоксане.
Присоединение ацетата ртути в воде к диметиловому эфиру экзо-1-мет-
окси-3,6-эндоксо-4-гтгра«с-тетрагидрофталевой кислоты вследствие коорди-
нации иона ртути с эндокарбометоксильной группой происходит как эндо-
цис-присоединение [306а], а не как экзо-цис-присоединение, как в случае
неметоксйлированной экзо-цис-кислоты (см. выше):
Как сказано выше* коричная кислота в растворе различных спиртов
присоединяет уксуснокислую ртуть, образуя C6H5CH(OR)CH(HgX)COOH,
где R—СН3— [199,256], С2Н5— ,С3Н7—, n-C4H9—, n-C5Hu—, га-С1вН33—[256].
В гликоле же вместо присоединения идет восстановление ацетата ртути
до закисной соли.[256].
При метоксимеркурировании цис- и /пронс-коричной кислот выделены
диастереоизомеры продуктов присоединения, а также их внутренних ртут-
ных солей [307] (ср. стр. 153).
Описано присоединение соли ртути к коричной кислоте в водной среде
[3081.
Метиловый эфир коричной кислоты дает хорошо кристаллизующийся
продукт— метиловый эфир а-ацетатмеркур-Р-фенил-]3-метоксипропионовой.
158. Ртутноорганические соединения. . Лит. стр. 173—181
кислоты' по схеме [8, 72, 296, 450]
С6Н5СН=СНСО2СН3 4- (СН3СОО)2 Hg 4- СНзОН -> СНзСООН 4- CeH5CH—СНСО2СН3
CH3t) hgOOCCHe
Получение метилового эфира а-ацетатмеркур-р-фенил-0-метоксипропионовой кислоты
[8]. К раствору 19,6 г уксуснокислой ртути в 100—120 мл метанола приливают 10 г мети-
лового эфира тракс-коричной кислоты и смесь хорошо перемешивают. Через 4 часа
начинается выделение кристаллов, и через 6 час. проба с едким натром не обнаруживает
присутствия иона ртути.
Спустя два дня количество выделившихся кристаллов достигает 18 г (64—65%). До-
полнительное количество продукта может быть получено нз маточника упариванием в виде
ацетата или же высажено водным раствором поваренной соли в виде хлорида. Ацетат кри-
сталлизуют из уксусноэтилового эфира при добавлении, в случае необходимости, петро-
лейного эфира. Т. пл. 139°С.)
Так же получают ртутные производные эфиров алкоксикислот общей
формулы C6H6CH(OR)CH(HgX)COOAlk, где R = С3Н5 [8, 72], м-С3Н7,
Z-C3H7, п- и /-С4Н9 [9], ведя реакцию в соответствующем спирте.
Найдены температуры плавления этиловых эфиров а-галоген- (и а-ацет-
окси-)меркур-Ргметоксигидрокоричных кислот [20]. Согласно Райту [72], до-
бавление небольшого количества азотной кислоты значительно ускоряет
взаимодействие солей ртути с непредельными кислотами; так, присоединение
уксуснокислой ртути в метаноле к метиловому эфиру транс-коричной кисло-
ты .при добавлении небольшого количества азотной кислоты (0,05 г на 0,01
моля уксуснокислой ртути) происходит в течение 75 мин., в отсутствие же
азотной кислоты реакция заканчивается через 36 час.
Присоединение же к цис-изомеру происходит тотчас же без катализа-
тора [72]..
К аллиловому эфиру^коричной кислоты при действии раствором серно-
кислой ртути в метиловом спирте присоединение происходит по обеим двой-
ным, связям. Продукт (после обработки NaCl) имеет строение [9, 309]:
CeHsCH — СНСООСН2 — СН — сн2
tcH3HgCl dcH3 HgCl
При действии продукта присоединения соли ртути к эфиру коричной ки-
слоты в спирте на полуаллиламид камфорной кислоты получается продукт
присоединения соли ртути к двойной связи аллильной группы этого послед-
него соединения [310].
/-Ментиловый [43] и d-борниловый [44] эфиры коричной кислоты
дают продукты присоединения при действии на холоду в течение не-
скольких дней раствора уксуснокислой ртути в метиловом спирте (см.
стр. 118).
Цис- и транс-стирилцианид реагируют с ацетатом ртути в метиловом
спирте лишь в присутствии катализаторов: HNO3 [144] или эфирата фтори-
стого бора [149], при этом транс-стирилцианид образует и RaHg-соединение
[149].
При 20-часовом кипячении метиловоспиртового раствора п-метоксико-
ричной кислоты с избытком уксуснокислой ртути наряду с присоединением
по двойной связи происходит меркурирование в ядро: продукт — а-3,5-
триацетат меркур-р-4-диметоксидигидрокоричная кислота [311].
о- и лг-Нитрокоричные кислоты дают продукты присоединения при много-
часовом (30 час. для орто-, . 10 час. для мета-) кипячении с уксуснокислой
ртутью в метиловом спирте [311] (ср. [256]).
Глутаконовая кислота при действии уксуснокислой ртути дает смесь
двух соединений: полученного путем замены водорода СН2-группы мерку-
Присоединение солей ртути к непредельным соединениям 159
рированного продукта (XVIII) и продукта присоединения по двойной связи
одновременно меркурированного (XIX) [312]:
O-Hg O-Hg Hg-O-
I I 1111
СО—снсн = снсоон со—снснон—сн—со .
XVIII XIX1
Эвгёнолуксусная[268,361], аллилфеноксиуксусная[314] кислоты присое-
диняют уксуснокислую ртуть в растворе метилового спирта. О присоединений
солей ртути к и-аллилоксибензойной кислоте и к эфиру аллилоксибенз- и
сульфокислоты см. стр. 145.
В обычных условиях, на холоду, уксуснокислая ртуть в Метиловом
спирте присоединяется $ о- и n-аллиламинобензойным кислотам и их эфи-
рам [276].
о- и n-Диаллилацетаминобензойная кислоты и диаллилацетофтальимид
присоединяют соль ртути в метиловом спирте по каждой двойной связи [276].
На присоединении R"OHgX к олефиновым кислотам основан способ син-
теза а-амино-Р-оксикарбоновых кислот из непредельных кислот [287, 315,
316]:
RCH= CHCOOR' HgX> RCH —CHCOOR' Br^
R"OH ir HgX
RCH — CHCOOR' RCH — CHCOOR' CHCOOH
Jr" Br Ar* NH2 HO NHa
Кумарин, кумаровая и кумариновая кислоты и их производные присо-
единяют соли и окись ртути и дают различные продукты в зависимости от
условий реакции (температура, растворители) и от того, действовать ли су-
лемой, окисью или уксуснокислой ртутью. Так, сулема в органических рас-
творителях на холоду присоединяется по двойной связи кумарина (также
7-метил кумар ина), образуя соединения типа
R О
MkzCHgCl
СНС1
R = Н или СН3 [317], которые получаются также при кипячении с сулемой
кумаровой и 4-метилкумаровой кислот [318]. Аналогичное соединение полу-
чается из 5-нитрокумаровой кислоты [318], Кумаровая [254], метилированная
при фенольном гидроксиле кумаровая [254] (см., однако, [319]) и кума-
риновая [254] кислоты обычным образом (присоединение RO и HgX) при-
соединяют соль ртути при действии уксуснокислой ртути в метиловом спирте
на холоду. При нагревании же этих кислот [319, 320], а также 4-метилку-
маровой кислоты [320] и метиловых эфиров кумаровой [319, 320] и 4-метйл-
Кумаровой [320] кислот и кумарина [256, 317] (ср. [254]), умбеллиферрона
[321] (в этом случае при любых соотношениях реагентов), 4,7-диметилку-
марина [321] с уксуснокислой ртутью в метиловом спирте происходит, наря-
ду с присоединением по Двойной связи, меркурирование в ядро перечислен-
ных выше кислот, их эфиров в положения 3 и 5 и кумарина, например:
ОН
|Q]/ + Hg (ОСОСН3)а + СН3ОН -
. • сн=снсоон
160 Ртутноорганические соединения. Лит. стр. 173—181
HgOCOCH3
СН3СООН.+ Jj_.CH(0CH3)CH(Hg0C0CHg) соон
СНоСОО
умбеллиферона в положения 6 и 8; 4, 7-диметилкумарина в положение 6.
5-Нитрокумаровая кислота и ее метиловый эфир в этих условиях дают
соответственно 3,а-диацетатмеркур-5-нитро-|3-метоксидигидрокоридаую кис-
лоту и ее эфир [320]:
ОН
/к JL-CH = CHCOOR + Hg (ОСОСН3)2 + СН3ОН ->
O2N
HgOCOCHs
- СН3СООН + |^J|_CH(0CH3)CH(Hg0C0CH3)C02R
OSN
R H или CH3. '
5-Нитро-2-метоксикоричная и -аллокоричная кислоты лишь присоеди-
няют ацетат ртути в метиловом спирте на холоду, не меркурируясь [319].
При действии окиси ртути в щелочном растворе на кумарин, 7-метилкумарин
и 6-нитрокумарин также происходит лишь присоединение по двойной связи
[322], например:
,ОН
Hg(QH), f у
ОН' kVXCH(OH)CH(HgOH)COOH
Кумариновая кислота и в водном растворе реагирует с тремя молекулами
уксуснокислой ртути на холоду с образованием продукта присоединения
по двойной связи, одновременно меркурированного в ядро в положения
3 и 5 [320].
Синтез а-ацетоксимеркур-$,2-диметоксидигидрокоричной кислоты [319].
ОСН3
. . CH(QCH3)CH(HgOCOCH3)COOH
При смешении эквимолекулярных количеств метилированной при фенольном гидрок-
силе кумариновой кислоты (1,8 г в 10 мл метилового спирта) и уксуснокислой ртути (3,2 г
в 20 мл метилового спирта) через несколько минут начинает выпадать белый кристалличе-
ский осадок. Реакция заканчивается через 4 часа. Осадок отсасывают и тщательно промы-
вают метиловым спиртом. Разлагается при 204°С.
Синтез а,3,5-триацетоксимеркур-р,2-диметоксидигидрокоричной кислоты [319].
HgOCOCH3
I OCH3
XX
CHaCOOHg CH(OCH3)CH(HgOCOCH3)COOH
12,5 г (более чем 3 моля) уксуснокислой ртути и 1,8 г (1 моль) метилового эфира ку-
марииовой кислоты в растворе метилового спирта кипятят в течение 20 час. Образующийся
в течение первого получаса темный осадок отделяют и отбрасывают. Образовавшийся бес-
цветный осадок промывают несколько раз метиловым спиртом. Т. разл. 220—221’С.
Присоединение солей ртути к непредельным соединениям 161
Присоединение соли ртути в метиловом спирте к З-карбокси-8-аллилку-
марину произведено [323] только по двойной связи аллильной группы без
затрагивания двойной связи пиронового кольца. 6-Аллил-7-гидрокси-
'4,8-диметилкумарин тоже присоединяет сулему в метиловом спирте лишь
по двойной связи аллильной группы [324].
О меркурировании производных кумарина без затрагивания двойной
связи, осуществляемом действием меркурирующих агентов в присутствии
щелочи, см. гл. V, стр. 78.
Присоединение уксуснокислой ртути в спирте к аллиловым эфирам ксан-
тогеновых кислот сопровождается заменой тионной серы на кислород, а
также превращением тионной функции в тиольную [325].
О присоединении солей ртути к сложным эфирам, содержащим двойную
связь в спиртовом остатке, см. стр. 147.
Присоединение солей ртути к алкениламидам кислот
Винилфталимид присоединяет уксуснокислую ртуть в метиловом спйрте
по двойной связи винильной группы [326].
.0 присоединении соли ртути в водных или спиртовых растворах к аллил-
амидам кислот, производимом в обычных условиях присоединения солей ртути
по двойной связи (иногда на холоду, часто при кипячении), см. соответствую-
щую литературу: к аллилмочевине [152, 327—335], аллилалкилмочевине
[336], аллилбиурету [332, 337], аллилгидантоинам [338], аллилвероналу
[339], моно- и полиаллилбарбитуровым кислотам [340], пропионил-
аллилмочевине [331], N-аллилпарабановой кислоте [331], Р-карбокси-
пропионилаллилмочевине [341], аллиламиду пропионовой кислоты [331],
N-аллилкарбамил-Р-карбамилпропионовой кислоте [343], аллиламидам
некоторых алифатических оксикарбоновых кислот [331, 334, 345], 4-ал-
лилкарбамоилциклогексанкарбоновой кислоте [347], к аллилсукцинимиду
[348—350], аллиламидам адипиновой [348] и азелаиновой [349] кислот, к ал-
лиламиду гидантоиновой кислоты [1521, к аллиламиду [346,351—353] и со-
держащему, возможно, иную ненасыщенную цепь моноамиду камфарной кис-
лоты [354] (см. также [310]), к диаллиламиду циклогександикарбоновой кис-
лоты [355], к моно алл ил амиду триметилциклопентандикарбоновой кислоты
[356], к N-аллилбензамиду (в среде пиперидина [357]), к N-аллилкарбама-
там о-[266, 332,355, 358, 359], м-[266] и п-феноксиуксусной кислоты, к
аллиламиду n-аминосалициловой кислоты [360], к аллиламиду гип-
пуровой кислоты [152], к диаллиламиду сахариновой кислоты [344],
к аллиламидам замещенных нафтойных кислот [363], к N-аллиламиду
и -полуамиду фталевой кислоты [355, 364], к N-аллилфтальимиду [365—
370], к аллиламиду никотиновой (в среде пиперидина [3311), хинолиновой
[331] и других замещенных пиридинкарбоновых кислот [302, 331, 371—
375], к аллиламидам алкансульфоновых [152], алкансульфамовых [152,
363], кислот, к N-аллилсульфонамидобензойной кислоте [3761, замещенных
урацилкарбоновых кислот [152], к аллиламидам тиофенкарбоновых [152]
и тиофенсульфоновых [152] кислот, к аллиламиду теофиллин-7-уксусной
кислоты [377], к аллиламидам ряда кислот в среде вторичных аминов [18],
357].
Получение М-(3-карбоксипропионил)-М'-(2-метокси-3-галоидмеркурлропил)мочевины
[341]. К горячему раствору 15 г (0,048 моля) уксуснокислой ртути в 200 мл метанола быстро
приливают горячий раствор 1\-ф-карбоксипропионил)-М'-аллилмочевииы (10 г; 0,05 моля) в
150 мл метанола. Тотчас выпадает белый осадок. Полученную взвесь кипятят 3 часа, ох-
лаждают в течение ночи до комнатной температуры, фильтруют и полученный осадок про-
мывают 100 мл метанола. Выход 20 г (91%). Т^пл. сырого продукта 177—178,5°С (,с разло-
жением). После очистки растворением в водном растворе бикарбоната натрия (но не щело-
162 Ртутноорганические соединения. Лит, стр. 173—131
чи) и высаживанием уксусной кислотой, выход 90%, т. пл. 188,5—190,5°С (с разложе-
нием).Галогенвд получают медленным растворением 0,05 моля полученного продукта в 50л«л
воды, содержащей 0,052 моля галогенида калия или натрия. Отфильтровывают от незначи-
тельного осадка, фильтрат подкисляют 3 мл уксусной кислоты. Выпавший осадок промыва-
ют водой и перекристаллизовывают из метанола (50 мл на 1 г для хлорида и 80 и на 1 г
для бромида). Выходы продуктов 98% (сырого) и 92—93%- (после кристаллизации). Т. пл.
хлорида 161—162,5°С, бромида 163—164°С.
Присоединение соли ртути к аллиламидам а-аминокислот (глицина, ала-
нина, /-лейцина, d,/-метионина, d,/-фенилаланина) возможно лишь в том
случае, если аминогруппа в них защищена бензоилированием [3781.
Соли ртути обычным образом присоединяются к аллилу, находящемуся
в боковой цепи соединения пуринового ряда. [379—3851 и в содержащих
аллильную группу уретанах [380, 381, 3861.
Присоединение солей ртути
к гетероциклическим соединениям,
содержащим этиленовые связи в цикле или боковой цепи
При нагревании в течение 5 дней раствора тионафтенсульфона в метило-
вом спирте с окисью ртути в уксусной кислоте образуется- [387] продукт
присоединения по реакции:
Ml ll + HgCCHaCOa^ + CHsOH-^f) I 00113
ЧА/ 4A/~HgOCOCH:
SOa SO2
Произведено присоединение уксуснокислой ртути в воде по двойной свя-
зи аллильной группы сульфата Ы-метилтетрагидрохинолин-8-аллилового
эфира [2581. Присоединение соли ртути к 3-аллилоксисульфолану произ-
ведено действием на 3-аллилоксисульфолан в метаноле раствора ацетата
ртути в ледяной уксусной кислоте с последующим кипячением смеси в тече-
ние — 12 час. [313]. Продукт представляет собой масло.
OCHSCH = CH2
Hg(OCOCHs)2
СН8ОН
OCH2CH(OCH3)CH2HgO2CCH3
Произведено присоединение соли ртути к 2,5-дигидротиофен-5-диоксиду
(сульфолену-3) [3131:
Hg(OCOCH,i)2 CHs0 I 1
СН.ОН * Ч/
оС/2
HgOCOCH3
Присоединение соли ртути к сульфолену-3 [313]. Получение З-ацетоксимеркур-2-меток-
сисульфолана. К раствору 11,8 г сульфолена-3 в абсолютном метаноле прибавляют 2,42 г
перекиси бензоила, затем раствор 31,9 г ацетата ртути и 7,2 г уксусной кислоты в горячем
метаноле. Раствор кипятят в течение 12 час. и затем концентрируют при 50—60°С. Остав-
шееся масло перемешивают с ацетоном и нерастворимый белый осадок отбрасывают. Рас-
творитель испаряют при комнатной температуре, масло растворяют в хлороформе, об-
рабатывают животным углем и извлекают эфиром. Остается бесцветное масло. Через не-
сколько недель масло закристаллизовывается. Т. пл. 105°С. Выход 63%. Хлорид, полу-
ченный из ацетата действием хлористого натрия в водном растворе, при кристаллизации
разлагается.
Присоединение солей ртути к непредельным соединениям 163
Присоединение ацетата ртути в метиловом спирте к 2,3-дигидропирану
дает 3-ацетоксимеркур-2-метокситетрагидропиран [250]:
HgOCOCHs
| |l Hg (ОСОСН3)г Iх
\/ СНаОН \/\
о о осн3
Антипирин и другие алкилзамещенные 1-арил-5-пиразолоны при дей-
ствии уксуснокислой ртути в спирте при нагревании до 60° С присоединяют
HgX и OR по двойной связи при одновременном меркурировании в положе-
ние 4 и в ароматическое ядро [289]. Присоединение по двойной связи 4-бром-
и 4-аминозамещенного антипирина происходит лишь при сплавлении его
с уксуснокислой ртутью (160° С), причем происходит присоединение
HgOCOCH3- и НО-групп, одновременно происходит меркурирование в бен-
зольное ядро [283] (см. гл. V, стр. 98).
Описано присоединение солей ртути в различных спиртах к винильной
группе N-винилкарбазола, пирролидона, со-капролактама [388]. Присоеди-
нение ацетата ртути к 2-диэтиламинометил-З-винилхинуклидину произведе-
но в смеси разбавленных серной и уксусной кислот [389]. Присоединение
ацетата ртути в различных спиртах к 1,6-диазабицикло-(4,4,0)-3-децен-7,10-
диону и 2,5-диметил-3-децен-7,10-диону произведено при трехчасовом кипя-
чении [390, 391]: '
CHR
hc<5^nZ78ch2
НС<2/\'0/СНг
CHR jj
О
Hg(OCOCH3)2
^~R'OH
О
Р'О. /ЧР’Д,
А Л А Н2
H3CCO2Hg<
О
Присоединение солей ртути по двойной связи аллильной группы’аллил-
амидов пиридинкарбоновых кислот и содержащих аллильную группу про-
изводных пурина см. стр. 161. Присоединение соли ртути (в метиловом,
«-пропиловом спиртах, в воде) к ряду производных 1,2,3,6-тетрагидропири-
дазина, 1,6,8-триазабицикло-(4,3,0)-нонендиону-7,9:
Hg(OCOCH3)2
R'OH
1,2-дикарбамил-Г,2,3,6-тетрагидропиридазину:
R'o
у/5\ f
( 6NCONH2 Hg(OCOCH3)2
k 'nconh2 r/°h
V/ H3CCO2H
NCONHjj
NCONH2
происходит только в присутствии азотной кислоты, в отсутствие которой
ртуть присоединяется к азоту амида [391].
164
Ртутноорганические соединения. Лит. стр. 173—181
Получение 4-хлормеркур-5-метокси-1,2-дикарбамилгексагидропиридазина [391]. 8,5 г
(0,05 моля) 1,2-дикарбамил-1,2,3,6-тетрагидропиридазина растворяют в 1 л кипящего
метанола. К полученному раствору прибавляют горячий раствор 15,9 г (0,05 моля) уксус-
нокислой ртути в 125 мл метанола, содержащий 0,5 мл конц. азотной кислоты. Выделяю-
щийся (похожий на мел) белый осадок растворяют вновь нагреванием и прибавлением
12,5 мл ледяной уксусной кислоты. Реакционную смесь оставляют стоять при комнатной
температуре на 18 час. Выделившийся осадок весит 18 г (выход сырого 4-ацетоксимеркур-5-
метокси-1,2-дикарбамилгексагидропиридазина 78%).
0,039 моля осадка суспендируют в растворе, содержащем 1,6 г (0,04 моля) едкого.натра
в 150 мл воды. Нагреванием смеси на паровой бане до 35—40чС осадок растворяют, раствор
фильтруют через диатомовую землю для освобождения от примеси нерастворимых черных
солей закиси ртути. Фильтрат насыщают углекислотой и к полученному раствору 4-гидр-
оксимеркурпроизводного прибавляют раствор, содержащий 2,4 г (0,04 моля) хлористого
натрия в 10 мл воды. Полученный раствор охлаждают и вновь насыщают углекислотой.
Выкристаллизовавшийся продукт дает 11,6 г (0,022 моля) 4-хлормеркур-5-метокси-1,2-ди-
карбамилгексагидропиридазина. Выход 44%; т. пл. 224—229°С. Сырой продукт очищают
растворением в растворе 0,9.г (0,022 моля) едкого натра в 100 мл воды при 3°С; Раствор
фильтруют и фильтрат охлаждают и насыщают углекислотой. Выделившийся осадок про-
мывают абсолютным спиртом и эфиром, сушат в вакууме. Выход 8,3 г (40%). Т. пл. 232—
233° С.
ПРИСОЕДИНЕНИЕ СОЛЕЙ РТУТИ К ОКИСИ УГЛЕРОДА
Эта реакция во многом аналогична присоединению солей ртути к этилену
в спиртовой среде и, как последняя, была найдена также Шеллером и
Шраутом [104]. Реакция протекает при насыщении спиртового раствора
уксуснокислой ртути окисью углерода при избыточном давлении в 1 атм и
встряхивании. Как и в случае олефинов, спирт участвует в образовании ко-
нечного продукта
Hg (ООССН3)а + СО + ROH CH3COOHgCOOR + СН3СООН.
И здесь реакция в метиловом спирте течет быстрее, чем в этиловом, однако
присоединение совершается значительно труднее, чем в случае этилена.
Реакция сопровождается побочной: восстановлением уксуснокислой ртути
до закисной соли.
Доказательство строения основано на реакциях:
NH3
CIHgCOOCHs + J2 -» HgJ2 + CICOOCHs —- H2NCOOCH3,
AlHg+H2O
CH3COOHgCOOC>H3 ----------- HCOOCaHB
и подтверждено ИК-спектрами и спектром протонного резонанса [1091
(см. также стр. 124).
Как всегда, в ртутноорганических соединениях замена ацетат-иона на
галоидный совершается очень легко приливанием раствора галогенида калия
или натрия. Нагревание выше температуры плавления ведет к разложению
с выделением СО. Нагревание со щелочью приводит к выпадению металла и
образованию карбоната. Избыток сероводорода, так же как и все симметри-
зующие агенты (кроме трифенилфосфина — единственного симметризатора
для этого соединения, см. гл. XIII), разрушает соединение с отщеплением
СО, но эквивалентное количество H2S осаждает белый кристаллический
сульфид.
Получение метилового эфира ацетатмеркурмуравьиной кислоты [104]. 26г (85%) ук-
суснокислой ртути в 100 мл метанола помещают в толстостенный сосуд емкостью 300 мл,
снабженный тубусом с пришлифованной к нему трубкой с краном и еще одной трубкой с
краном. Каучуком с узким просветом сосуд соединяют с азотометром, груша которого сое-
динена длинным вакуумным каучуком, позволяющим создать избыточное давление в 1 атм.
Краны смазывают графитом, шлифы заливают парафином, каучуки укрепляют кольцами из
медной проволоки. После вытеснения'воздуха окисью углерода посредством поднятия гру-
ши азотометра создается избыточное давление в 1 атм и сосуд встряхивается на машине.
Приссединение солей ртути к непредельным соединениям 165
Происходит медленное непрерывное поглощение СО, которое заканчивается через сутки.
Всего поглощается 1151 мл СО (приведено к 0°С и 760 мм), из них 21 мл соответствует рас-
творимости газа в реакционной смеси. Рассчитанное количество составляет 1113 мл. Кроме
того, 1,2 г уксуснокислой ртути восстанавливается до закисной соли. По окончании погло-
щения последняя отфильтровывается и от фильтрата в вакууме при температуре бани 30—
35° отгоняются метанол и уксусная кислота. Маслянистый остаток застывает в кристалли-
ческую корку звездообразно сросшихся иголочек. Корку растворяют в 75 мл теплого хло-
роформа, раствор отфильтровывают и высаживают добавлением 5—6-кратного количества
петролейного эфира. Тотчас возникающий осадок после 24-часового хранения в ледяном
шкафу превращается в звездчатые иголочки. Для увеличения выхода все охлаждают еще в
течение 2 час. в охладительной смеси, осадок отсасывают и промывают небольшим коли-
чеством холодного метанола, затем эфиром. Выход 15 г (88%). Т. пл. 110° С (с разложе-
нием). Вещество легко растворимо в метиловом и этиловом спиртах и хлороформе, уме-
ренно — в теплой воде, уксусном эфире и бензоле, трудно — в эфире, петролейном эфире
и лигроине.
Превращение в хлормеркурмуравьинометиловый эфир производится добавлением рас-
твора 0,5 г хлористого натрия в 5 мл воды к раствору 2 г ацетата в 10 мл метанола. После
12 час. хранения во льду и" 2 час. охлаждения в смеси осадок отсасывают. Выход 1,5 г
(80%). Т. пл. 110°С (с разложением).
Получение ацетатмеркурмуравьиноэтилового эфира из 20 г уксуснокислой ртути в 150
мл абсолютного спирта производится совершенно аналогично описанному, но поглощение
СО занимает 3 суток. Переработка тождественна описанной выше. Выход 12,5 г (90%).
При нагревании в капилляре начало размягчения происходит при 65° С, быстрое разложе-
ние— при 125° С. Растворимость подобна таковой метилового эфира. ClHgCOOGHs пла-
вится с разложением при 88° С.
О симметризации продукту присоединения соли ртути к окиси углерода
см. гл. XIII, стр. 258.
Избыток бромистого бензила с карбометоксимеркурацетатом (кипячение
в хлороформе в течение около 23 час.) дает бензил ацетат, метилбензиловый
эфир, карбометоксимеркурбромид, закисные и окисные соли ртути [453].
При реакции бромистого бензила с карбометоксимеркурбромидом (в анало-
гичных условиях) происходит выделение окиси углерода -и образование
метил бензилового эфира и бромной ртути [453]. Фотохимическое разложе-
ние карбометоксимеркуриодида в бензоле (освещение ультрафиолетом
6 час., 6—10° С, атмосфера азота) дает метилбензоат и, возможно, толуол
[453]. Весьма замечательно, что карбоксимеркурацетат с этиленом в мета-
ноле (в автоклаве, 60° С, 5 час.) дают (3-метоксиэтилмер кур ацетат [453].
Это — один из двух известных случаев получения одного типа квазикомп-
лексного соединения из другого.
Окись углерода легко присоединяется к сулеме в среде вторичных ами-
нов [108]. При насыщении раствора сулемы в сухом пиперидине окисью
углерода при комнатной температуре с количественным выходом получен
продукт состава
C5Hio>N—С—HgCI -CsHjoNH
ПРИСОЕДИНЕНИЕ СОЛЕЙ РТУТИ ПО ТРОЙНОЙ'СВДЗИ
Присоединение солей ртути к ацетиленовым углеводородам
Первые работы по присоединению ртутных солей к ацетиленовым угле-
водородам принадлежат Кучерову [96, 97], который на примере аллилена
и ацетилена установил состав полученных им соединений и констатировал
образование ацетона и соответственно ацетальдегида при разложении этих
веществ в условиях кислой среды; эта реакция получила широкое промыш-
ленное использование.
Состав и характер осадков, получающихся при пропускании ацетилена
в водные растворы солей ртути, зависят от условий среды и от того, какая
166 Ртутноорганияеские соединения. Лит. стр. 173—181
соль взята. Действие ацетилена на подкисленный азотной кислотой раствор
азотнокислой окиси ртути приводит к образованию кристаллического осад-
ка, которому приписано строение Hg=C(HgNO3)CHO [103, 392] (см. также
[393, 394]). По-видимому, то же вещество получается при меркурировании
ацетальдегида^
Лучше объясняет свойства этого соединения строение, предложенное для
него Коршаком и Замятиной [395] (см.-стр. 168). Продукт присоединения
азотнокислой ртути к ацетилену в уксусной кислоте имеет состав
C2H2N2O7Hg и вероятное строение (O3NHg)2CH — СНО [395]. Более иссле-
дован продукт взаимодействия ацетилена с водным раствором сулемы [98,
396—398], которому Бильц н Мумм [98] приписали структуру трихлормер-
курацетальдегида C(HgCl)3CHO. Несомненно, что в этих случаях, наряду
с присоединением, происходит и замещение одного из подвижных водород-
ных атомов ацетилена на ртуть. Наряду с последним веществом в условиях
его получения [77, 79, 399], а лучше в абс. спиртовом растворе [76] обра-
зуется собственно продукт присоединения C2H2HgCla (Бигинелли), для кото-
рого установлена структура ClCH=CHHgCl. Освещение вопроса о строе-
нии этого соединения см. стр. 124. Это вещество наиболее просто и с почти
количественными выходами получается [80] насыщением ацетиленом креп-
кого раствора сулемы вводной 15 %-ной соляной кислоте. Оно представляет
собой транс-изомер (см. также стр. 121). Транс-строение этого соединения
подтверждено также рентгеноструктурным исследованием [400, 401].
Впервые полученный Несмеяновым и Борисовым [84] обменной реакцией
(c-ClCH=CH)3Sb-|-HgCla ф/с-р-хлормеркурвинилхлорид получается [87]
также соединением ацетилена и сулемы в парах при 100—110° С (Фрейд-
лина, Ногина [87]).
трпщ>|3-Хлорвинилмеркурхлорид переходит в цис-изомер в присутствии
перекисей [85] (реакция в четыреххлористом или четырехбромистом угле-
роде [402] сопровождается образованием 1,1,1,3-тетрахлор- соответственно
1,1,1-трйбром-З-хлорпропилена) и под действием ультрафиолетового света
(Несмеянов и Борисов [85]).
Роль пгрпнс-хлорвинилхлорида в реакции Кучерова описана Клебанским
и Титовым [403], а также другими авторами.
Получение хлористой игранс-р-хлорвииилртути [80]. Раствор 72 г сулемы в 75 мл
соляной кислоты при сильном перемешивании насыщают на холоду ацетиленом. Через
40 мин. начинает выделяться белый кристаллический осадок. Через 3 часа осадок отфильтро-
вывают, промывают водой и сушат. Вес 25 г. В фильтрате растворяют 23 г сулемы и в тече-
ние 3 час. при перемешивании насыщают ацетиленом. Выделяется еще 28 г продукта, к
фильтрату от которого вновь добавляют 26 г сулемы и через 3 часа перемешивания и на-
сыщения ацетиленом получают еще 31,5 г продукта. Соединенные осадки перекристаллизо-
вывают из бензола. Т. пл. 123—124° С. Из хлорида нетрудно получить соли других кислот
[82].
Получение хлористой цмс-р-хлорвииилртути [87]. В двугорлую колбу помещают 3 г
сулемы, нанесенной на стеклянную вату. Поддерживая внутри колбы температуру85—100°С,
в колбу пропускают ацетилен в течение 3 час. Продукт экстрагируют четыреххлористым
углеродом. Выход 1,7 г (51%); т. пл. сырого продукта 68—70° С. После многократной кри-
сталлизации из петролейного эфира (фракция 80—100° С) т. пл. 77—78° С.
О механизме этой, реакции см, стр. 121.
Описано промежуточное образование хлорвинилмеркурхлорида при
катализируемом сулемой взаимодействии ацетилена с треххлористым мышья-
ком [404].
транс-fi-Хлорвинилхлорид при встряхивании с водным раствором азот-
нокислой ртути образует [395] тот же продукт состава C2HO4NHg2, который
получается при действии ацетилена на водный раствор азотнокислой ртути.
Двухзамещенные ацетилены (диметил- и дифен ил ацетилен) также присо-
единяют соли ртути, но не сулему, а ацетат ртути. Диметилацетилен реаги-
Присоединение солей ртути к непредельным соединениям
167
рует с уксуснокислой ртутью в ледяной уксусной кислоте, образуя три
изомерных ртутноорганических продукта, изолированных в виде хлоридов:
два геометрических изомера а,0-диметил-0-ацетоксивинилмеркурхлорида
(I и II) и структурный изомер — 2-ацетокси-3-хлормеркурбутен-1 (III)
493,94].
I
СН3ООС СНз
/С = С\
СНзХ HgCl
СН3 СНз сн2
^>с —с< СНз—СН—С^
СНзСОО HgCl Hga ОСОСНз
II III
Разработаны условия получения каждого из этих изомеров в отдельно-
сти [941.
Получение хлористой ягра«с-1-метнл-2-ацетокси-1-пропеи-1-ил-ртути [94] (формула 1).
К охлажденному до —18° С раствору 7 г (0,02 моля) уксуснокислой ртути в 25 мл ледяной
.уксусной кислоты приливают 1,2 г (0,02 моля) диметилацетиЛена, охлажденного смесью
льда с солью. Реакционную массу в запаянной стеклянной, ампуле оставляют стоять при
комнатной температуре. Через 24 часа охлажденную смесью льда с солью ампулу вскры-
вают. Осадок одновалентной уксуснокислой ртути (0,2 г) отделяют. Фильтрат обрабатыва-
ют насыщенным водным раствором хлористого калия. Выпавший осадок отделяют и пере-
кристаллизовывают из четыреххлористого углерода. При комнатной температуре выделяют
кристаллы, плавящиеся прн 140°С, выход 5,4 г (70%).
При обработке фильтрата насыщенным водным раствором бромистого натрия получен
соответствующий бромид с т. пл. 162—463° С. Выход 65%.
Получение хлористой цас-1-метил-2-ацетокси-1-пропеи-1-ил-ртути [94] (формула II).
То же количество исходных продуктов после 24 час. хранения нагревают при 56° С в те-
чение 2 час. Выделяется одновалентной уксуснокислой ртути 0,3 г (4,5%). Фильтрат об-
рабатывают аналогично предыдущему опыту. Выделенный при этом осадок перекристалли-
зовывают из смеси лигроина с бензолом (2 : 1). Выпавшие при этом кристаллы отфильтро-
вывают. Дважды перекристаллизованные, они плавятся при 140°С. Выход 0,4 г (4,4%).
Из частично упаренного фильтрата дополнительно выделяют кристаллы в виде игл. Пе-
рекристаллизованные из лигроина (т. кип. ПО—130°С), они плавятся при 95—96° С. Даль-
нейшая перекристаллизация температуры плавления не изменяет. Выход 3,66 г (40,4%).
Получение хлористой 1-метил-2-ацетокси-2-пропен-1-ил-ртути [94] (формула III). К
охлажденному до —18° С раствору 13 г (0,04 моля) уксуснокислой ртути в 39 мл ледяной
уксусной кислоты прибавляют 2,2 г (0,04 моля) диметилацетнлена. В запаянной ампуле
продукты реакции нагревают при 76° С в течение 2,5 час. Выделение продуктов реакции
проводят в условиях опытов, аналогичных описанным выше. Выделено одновалентной
уксуснокислой ртути 1 г (9,5%). После двукратной перекристаллизации хлорида выделяют
продукт с т. пл. 130°С. Выход 2,4 г (17%).
Бромид плавится при 127—128° С.
Присоединение элементов сулемы к 1,4-дихлорбутину-2 осуществлено
действием на него насыщенным раствором сулемы в насыщенном растворе
хлористого натрия [138].
Получение 1,2,4-трихлор-3-хлормеркурбутеиа-2 [138]. 1 г 1,4-дихлорбутина-2 сме-
шивают с 10 мл насыщенного раствора сулемы в насыщенном растворе хлористого натрия.
Смесь оставляют на два дня (при частом встряхивании); через два дня кристаллический
осадок отфильтровывают, промывают водой. Фильтрат насыщают сулемой и оставляют на
сутки. Выделившиеся кристаллы присоединяют к основной порции. После перекристал-
лизации из водного метилового спирта выделяются бесцветные мелкие таблички. Т. пл.
85—86°С. Выход 2,4 г (77%). Вещество хорошо растворимо в ацетоне, спирте, растворимо в
бензоле, хлороформе,' нерастворимо в воде.
При катализируемом ртутью присоединении уксусной кислоты к гекси-
ну-3 предполагается промежуточное присоединение—HgCOOCH3-rpynn
и образование л-комплекса [405].
Дифенилацетилен присоединяет ацетат ртути в ледяной уксусной кисло-
те (2,5 часа при температуре не выше 96° С) с образованием только одного
соединения, по-видимому, цис-изомера [93, 406, 407]. Согласно другим дан-
ным [408], при этом образуется как цис-, так и транс-изомер; последний
168
Ртутноорганические соединения. Лит. стр. 173—181
образуется в результате перегруппировки первоначально образовавшегося
цис-изомера.
Однозамещенные низшие гомологи ацетилена с водными растворами со-
лей ртути образуют нерастворимые аморфные осадки, которые при действии
кислот переходят в кетоны. Из метил ацетилена образуется ацетон [96,97].
Бильц и Мумм [98] осадку из метилацетилена и сулемы приписали строе-
ние трихлормеркурацетона CH3COC(HgCl)3, осадку из ацетилена, при дей-
ствии кислот дающему метилэтилкетон,— строение трихлормеркурметил-
этилкетона (ClHg)3CCOC2Hs [98].
Продукту присоединения азотнокислой ртути к ацетилену в водном рас-
творе, полученному в свое время Гофманом [98, 396, 409], приписано [395]
строение СН= С—HgNO3.
О----Hg
Получение ацетилпропнонилметана нз днметилдиацетилена в присутствии соли ртутн
[410]. Прн нагревании смеси раствора диметнлацетнлена в этиловом спирте со спиртовым
раствором сулемы в запаянной трубке при 100° С в течение 6 Дней получают ацетилпропио-
нилметан. Т. кип. 158°С.
При действии уксуснокислой ртути в ледяной уксусной кислоте при
70—100° С на высшие гомологи ацетилена—гептин-1, октин-1, а также
фенилацетилен происходит (сопровождающееся заменой подвижного водо-
рода при тройной связи на ртуть) присоединение соли ртути согласно схе-
ме [101]:
-С~СН + 3Hg (СН3СО2)2 + Н2О -> — С = с (HgO2CCH3)2 + ЗСНзСО2Н.
OHgO2CCH3
Как показали кинетические исследования, фенилацетилен в воде практи-
чески не присоединяет сулему ни в присутствии L1C1 в метаноле, ни в при-
сутствии NaCl [411]. Реакция фенилацетилена с сулемой в запаянной труб-
ке при 50° С идет со взрывом [411].
При действии уксуснокислой ртути (при 60—70° С) на винилацетилен
СН2=СН—С = СН или на получающийся из него действием K2HgJ4
или уксуснокислой ртути в уксусной кислоте при комнатной температуре
винилацетиленид ртути (СН2=СН—C=C)2Hg образуется соединение
СН2 = CH—C=C(HgO2ccH3)2, которое с соляной кислотой дает метилвинил-
О—HgO2CCH3
кетон; от действия брома оно переходит в трибромметилвинилкетон
СН2=СНСОСВг3 [412].
Бутадиениловые сложные эфиры СН2—CRCH=CHOAc образуются
при взаимодействии кислот в присутствии солей ртути с соединениями
CH2=CR—C=CR', где R и R' = Н, арил или алкил [413]. Ацетилен
и алкилацетилены со спиртами в присутствии солей ртути дают ацетали
[414, 4151. При взаимодействии алкилацетиленов и гликолей в присутствии
солей ртути образуются циклические ацетали [416] (см. также [417]).
Продукт строения C6H5CCl=C(HgCl)—С(ОС2Н5) =СН—СН3 полу-
чен при действии сулемы в водно-спиртовой смеси на фенилметилдиацети-
лен CflH5C=C—С=ССН3 [418].
Ряд других монортутных производных непредельных углеводородов с
ртутью у олефинового углерода: пропенильные [419], изопропенильные
[420], стирильные [421], стильбенильные [95] и винильные [422] соединения
ртути получены Несмеяновым, Борисовым и сотр. через соответствующие
литий- и магнийорганические (см. гл. И), а также таллий- и оловоорганичес-
кие соединения (см. гл. IX).
Присоединение солей ртути к непредельным, соединениям
169
О сохранении геометрической конфигурации изомеров при гомолитичес-
ких и электрофильных реакциях этих ртутноорганических соединений см.
стр. 122, а также гл. XIII и XIV.
Присоединение солей ртути к ацетиленовым спиртам
Ацетиленовые спирты легко образуют аддукты с сулемой [1381. Из-за
лабильности аддуктов для их получения применяются насыщенные раство-
ры сулемы в насыщенном растворе хлористого натрия. При действии такого
раствора на пропаргиловый спирт, диметилэтинилкарбинол, бутиндиол,'
1,4-,2,5-диметилгексин-3-диол-2,5 получены соответственно 2-хлор'меркур-
бутен-2-диол-1,4; 2,5-диметил-3-хлор-4-хлормеркургексен-3-диол-2,5 — пре-
красно кристаллизующиеся вещества, обладающие ярко выраженным ха-
рактером квазикомплексных соединений (см. стр. 128) (Несмеянов и Ко-
четков [138]).
Присоединение сулемы к пропаргиловому спирту [138]. 2,5 г пропаргилового спирта
(в виде моногидрата, т. кип. 97—98°С) смешивают с 20 мл насыщенного раствора сулемы
в насыщенном растворе хлористого натрия. Смесь мутнеет и через несколько минут закри-
сталлизовывается со слабым разогреванием. Через час кристалл отфильтровывают, про-
мывают и высушивают на воздухе. После перекристаллизации из водного метилового спир-
та или бензола выделяются тонкие бесцветные иглы. Т. пл. 104—105°С, выход 2-хлор-З-
хлормеркурпропен-2-ола 6,0 г (55%).
Аддукт сулемы и метилового эфира пропаргилового спирта получен ана-
логично при встряхивании эфира с насыщенным раствором сулемы в насы-
щенном растворе хлористого натрия [4111.
Уксуснокислая ртуть в уксусной кислоте образует с ацетатом бутин-2-
ола-1 продукт присоединения (после высаживания хлористым калием) 1,3-
диацетокси-2-хлормеркурбутен-2 [423].
В настоящем разделе рассматривается присоединение солей ртути к аце-
тиленэтиленгликолям. Действие сулемы (от 1 до 6 час. кипячения в спирте)
на диметилфенилфенилацетиленилэтиленгликоль приводит к циклическому
продукту [424]. Действие на него уксуснокислой ртути (2,5—4 часа при
89° С) дает продукт открытого строения [425]:
СНз СзНб
/С---С\
СНз ОН 0НС=ССвНб
HgCl2/ \Hg (CH3CO?)2
СеНвС = С—HgCl-
НзС. | I ГвН5
)С С<
НзС/ \/ хон
о
С6Н5
СНзч I
>С—С=С—СО—С6Н5
сн/l I
ОН HgOCOCHa
Другие ацетиленилпинаконы также по-разному относятся к сулеме, и
уксуснокислой ртути. Так, дифенилметилфенилацетиленилэтиленгликоль
лишь с ацетатом ртути (3 часа на холоду) дает продукт присоединения откры-
того строения [426], не образуя содержащего ртуть соединения с сулемой.
Вторичнотретичные же гликоли (1,4-дифенил-2-метилацетиленилгликоль
[427], 5,5-диметил-1,2-дифенилгексин-3-диол-1,2 [428], 2-метил-4-фенилбу-
тин-З-диол-1,2 [429], 1,2,4-трифенилбутин-3-диол-1,2 [430] и З-метил-5-фе-
нилпентин-4-диол-2,3 [430]) с сулемой в результате отщепления НС1 и
Н2О и замыкания цикла образуют меркурированные в [3-положение заме-
щенные фураны.
170
Ртутноорганические соединения. Лит. стр. 173—181
Синтез хлористой 2,5-дифеиил-4-метилфурил-3-ртути [427]. Смесь 3 г 1,3-Дифенил-2-
метилацетиленилгликоля и 3,25 г сулемы оставляют стоять 3 дня при комнатной темпе-
ратуре. Осадок промывают несколько раз спиртом, кристаллизуют из бензола. Т. пл. 237—
238°С. Выход 4,9 а (87%).
Присоединение солей ртути к ацетиленовым кетонам
Присоединением сулемы к фенилэтинилметилкетону получен [139] 1-фе-
С1—С = С—С = О
нил-1-хлормеркурбутен-1-он-3, содержащий систему > в ко-
торой связь Hg—С одновременно сопряжена с простой связью С1—С и
двойной связью С=О, что обусловливает повышенную лабильность этого
соединения и его резко выраженные квазикомплексные свойства. Вещество
отщепляет сулему не только при химическом воздействии связывающих ее
реагентов, но в значительной степени даже и при перекристаллизации из
эфира (см. стр. 128).
Присоединение сулемы к фенилэтинилметилкетону [139]. 5 а фенилэтинилметилкетона
и 100 мл насыщенного раствора сулемы в насыщенном растворе хлористого натрия встря-
хивают в течение 4 час. Уже через несколько минут появляется кристаллический продукт
присоединения. Осадок отфильтровывают, промывают водой и высушивают над хлористым
кальцием. Фильтрат насыщают сулемой, добавляют. 5 г фенилэтинилметилкетона и опера-
цию повторяют. Высушенные соединенные осадки обрабатывают дважды небольшим коли-
чеством абсолютного эфира, высушивают в вакууме. .Выход 1-фенил-1-хлор-2-хлормеркур-
бутена 28,0 г (97%). После перекристаллизации из эфира образуются бесцветные мелкие
иглы. Т. пл. 112—113°С (с разложением).
Присоединение солей ртути к ацетиленовым кислотам
Простейшие продукты присоединения соли ртути к низшим ацетилено-
вым кислотам и их эфирам гладко и с хорошими выходами получаются взаи-
модействием соответствующих ацетиленовых кислот и их эфиров с насы-
щенным раствором сулемы в насыщенном водном растворе хлористого натрия
[140]. Для низших гомологов реакция идет очень быстро и заканчивается
в течение 1—2 час. Так получены продукты присоединения сулемы к про-
пиоловой [140] и тетроловой [140, 431] кислотам и их эфирам [140], также
к эфирам фенилпропиоловой и ацетилендикарбоновой [140] кислот. Сулема
становится в a-положение к карбонилу, а галоид — в p-положение, т. е. име-
ет место обычное для ненасыщенных кислот обращение правила Марковни-
кова. Полученные соединения ведут себя как типичные квазикомплексные
соединения, еще более легко отщепляя сулему под действием связывающих
ее агентов, чем Р-хлорвинилмеркурхлорид.
Несколько более устойчивы аддукты сулемы и эфиров ацетиленовых
кислот.
Получение а-хлормеркур-3-хлоракриловой кислоты [140]. 1,00 г пропиоловой кислоты
смешивают с 10 мл насыщенного раствора сулемы в насыщенном растворе хлористого нат-
рия. Через несколько минут смесь после потирания палочкой закристаллизовывается.
Через 2 часа кристаллы отфильтровывают, промывают небольшим количеством воды, вы-
сушивают в эксикаторе над хлористым кальцием. После перекристаллизации из воды полу-
чают мелкие блестящие белые иглы. Т. пл. 178—179°С (с разложением). Выход 4,45 г (91%).
Получение метилового эфира хлормеркурфумаровой кислоты [140]. 5,00 г диметилового
эфира ацетилендикарбоновой кислоты смешивают с 30 мл насыщенного раствора сулемы в
насыщенном растворе хлористого натрия. Смесь несколько раз энергично встряхивается.
Через несколько часов выделяются кристаллы, через 3 дня их отфильтровывают, про-
мывают водой и высушивают на пористой пластинке. Из фильтрата (после насыщения су-
лемой) выделяют дополнительную порцию кристаллов. После перекристаллизации из ме-
тилового спирта получаются бесцветные иглы. Т. пл. 145—145,5°С. Выход 10,72 г (73%).
Продукты присоединения сулемы к метиловым эфирам пропиоловой,
фенилпропиоловой и ацетилендикарбоновой кислот получены также при
Присоединение солей ртути к непредельным соединениям
181
430. Ф а б р и ц ы А., Куба л я И. ЖОХ, 31, 476 (1961).
431. Бабаян А. Г., Г.р и г о р я н А. А. ЖОХ,, 26, 1945 (1956).
432. Двор ко Г. Ф. Укр. хим. ж., 28, 945 (1962).
433. Seher А. С. А., 54, 8623 (1960); Arch. Pharm,, 292, 519 (1959).
434. Л е в и н а Р. Я., Костин В. Н. ДАН СССР, 97, 1027 (1954).
435. Левина Р. Я., Костин В. Н. ЖОХ, 23, 1054 (1953).
436. Левина Р. Я., Костин В. Н., Тартаковский В. А. Вестник МГУ, сер.
физ.-мат. и естеств. наук, № 1, 77 (1956).
437. Ле в и н а Р. Я., К о с т и н В. Н., Тартаковский В. А. ЖОХ, 27, 881
(1957).
438. Левина Р. Я-, Костин В. Н., Ш ан а за ров К- С, ЖОХ, 29, 40 (1959)
439. Левина Р. Я-, Костин В. Н., Тартаковский В. А. ЖОХ, 26, 2998
(1956).
440. Левина Р. Я., Костин В. Н., Г е м б и ц к и41 П. А., В и н о г р а д о в А. Д.
Вести. МГУ, № 1, 67 (1961).
441. Левина Р. Я., Костин В. Н„ Ким Д я й Гир, УстынюкТ. К- ЖОХ,
29, 1956 (1959).
442. Левина Р. Я-, Ш ан а за ров К. С., Костин В. Н., ТрещоваЕ. Г.
ЖОХ, 32, 2637 (1962).
443. Левина Р. Я., Мезенцова Н., Лебедев О. В. ЖОХ, 25, 1097 (1955).
444. Несмеянова О. А., Лукина М. Ю., Казанский Б. А. ДАН СССР,
153,114 (1963)/
445. Несмеянова О. А., Лукина М. Ю., Казанский Б. А. ДАН СССР, 153,
357 (1963).
446. Пахомов В. И. Кристаллография, 5, 800 (1960).
447. Пахомов В. И. Кристаллография, 7, 456 (1962).
448. Edsall J. Т., Maybury R. Н., Simpson R. В., StraessleR.J. Am.
Chem. Soc., 76, 3731 (1964).
449. E 1 d j arn L., Jellutn E. Acta Chem. Scand., 17, 2610 (1963).
450. Chaudhuri A. K-, M a 11 i k К- E., D a s M. N. Tetrahedron, 19, 1981 (1963).
451. Аракелян ’С. В., Рашидян Л. Г., Дангян M. T. Изв. АН Арм. ССР,
сер. хим. наук, 17, 173 (1964).
452. Аракелян С. В., Данг я и М. Т., Аветисян А. А. Изв. АН Арм. ССР,
15, 437 (1962).
453. Fanta G. F. J. Org. Chem., 29, 1610 (1964).
172
Ртутноорганические соединения. Лит. стр. 173—181
(R = Н или А1к) лишь формально аналогична реакции присоединения со-
лей ртути к олефинам. Получаемые вещества совершенно не обладают квази-
комплексными свойствами и представляют собой вполне устойчивые ртутно-
органические соединения (см. стр. 125). Реакция проводится в воде или спир-
те при охлаждении и приводит к образованию у-меркурированных спиртов
или соответственно их простых эфиров. •
Разрыв цикла происходит по наиболее поляризованной связи цикла —
между наиболее замещенными и наименее замещенными атомами углерода.
Присоединение соли ртути идет по правилу Марковникова. Реакция приме-
нена к 1,1,2-триметилциклопропану [434], 1,1,2,2-тетраметилциклопропа-
ну [170, 435], 1,1,2-триметил-2-этилциклопропану [436], фенилциклопро-
пану [437] (к фенилциклопропану присоединена также в метиловом
спирте ртутная соль тринитрометана; этим методом происходит
присоединение —HgC(NO2)3 и—ОСН3[227]) к n-анизил- и и-толилциклопро-
пану [438], к метил- и 1,1-диметилциклопропану [439], 1,1-диметил-2-ал-
килциклопропанам [440] (присоединение к которым происходит в несколько-
более жестких условиях— при продолжительном, 20-часовом для первого и
10—12-часовом для второго, встряхивании на качалке при комнатной темпе-
ратуре), к мостиковым бициклическим углеводородам: 1,2,3-триметил-1441],
1,3,5,5-тетраметил-[442] и 1,2-диметил-3-этил-[441] (0,1,3)-бициклогексануг
к спиро-(2,4)-гептану [4431, к п-циклопропилкумолу [208].
Получение уксуснокислой 3-окси-2,2,3-трнметилбутилртути [435]. К профильтрован-
ному раствору 41,6 г уксуснокислой ртути в 150 мл воды прибавляют 12,8 г тетраметил-
циклопропана. Реакционную емесь оставляют на 6 дней в закрытой склянке при —5°G
при периодическом встряхивании. После окончания реакции (исчезновение углеводород-
ного слоя и отсутствие иона ртути) частично выделяется осадок. От раствора в вакууме
(5 мм) отгоняют 4/s объема воды. Из остатка при охлаждении выделяются белые кристаллы.
Кристаллизация из гептана. Т. пл. 69—70°С. Выход 64%. В спирте реакция проходит за
3 дня.
Несмеяновой, Лукиной и Казанским [444, 445] реакция циклопро-
пановых углеводородов с ацетатом ртути в метиловом спирте исследована
полукинетически. Показано, что электродонорные заместители, в особенности
способные подавать электроны по сопряженному механизму, повышают
реакционную способность циклопропанового кольца по отношению к элек-
трофильному агенту — ацетату ртути, электроакцепторные понижают эту
способность. Так, по уменьшению их реакционной способности по отношению
к ацетату ртути циклопропановые углеводороды могут быть расположены
в ряд (цифры показывают количество прореагировавшего углеводорода
в процентах за 3 часа): фенилциклопропан (71), 1,2-диметилциклопропан
(транс) (48,5), 1-метил-2-этилциклопропан(шранс) (18,5), этилциклопропан
(3,5), изопропилциклопропан (1,5).
Гораздо менее реакционноспособны и мало разнятся по реакционной
способности между собой (цифры — количество прореагировавшего угле-
водорода в процентах за 21 час) транс- 1,2-дифенилциклопропан (3,8), цис-
1,2-дифенцлциклопропан (3,5), 1,1,-дифенилциклопропан (2/5). Метилцикло-
пропилкетон в принятых условиях не реагирует [444].
Более реакционноспособны, чем фенилциклопропан, изображенные-
ниже углеводороды (в скобках — относительная величина скорости реак-
ции) [445]:
<2>5>
(2,2)
(2)
/\-СвН6 (1)
Присоединение солей ртути к непредельным соединениям
173
Произведено рентгенографическое исследование кристаллической струк-
туры продуктов присоединения солей ртути к циклопропановым производ-
ным [446, 447 ].
ЛИТЕРАТУРА
1. Hofmann К. A., Sand J. Вег., 33, 1340 (1900).
2,'Hofmann К. A., Sand J. Вег., 33, 1353 (1900).
3. Nofmann К- A., Sand J. Вег., 33, 1358 (1900).
4. Hofmann К. А„ S a n d J. Вег., 33, 2692 (1900).
5. S a n d J. Вег., 34, 1385 (1901).
6. S a n d J. Вег., 34, 2906 (1901).
7. Sand J., Singer F. Ber., 35, 3170 (1902).
8. Schoeller W., Sc'hrauth W., Struensee R. Ber., 43, 695 (1910).
9. Schoeller W„ Schraut h W„ Struensee R. Ber., 44, 1048 (1911).
10. Нем. пат. 459145 (1928); Zbl., 1928, II, 1615.
U.HiigelG., Hibou J. Chim. et ind., 21, (2 bis), 296 (1929).
12. Spengler G., Frommel H., ShaffR., Faul Ph., L о n s k у P.
Brennstoff-Chemie, 37, 47 (1956).
13. Rodgman A., S h e a r e r D. A., W г I g h t G. F. Can. J. Chem., 35, 1377
(1957).
14. R о m e у n J., W r i g h t G. F. J. Am. Chem. Soc., 69, 697 (1947).
15. В u c k 1 e s R. E„ S m i t h R. J. J. Am. Chem. Soc., 74, 3174 (1952).
16. Фрей длин a P. X., Кочеткова H. С. Изв. АН СССР, ОХН, 1945, 128.
17. Sachs G. J. Chem. Soc., 1949, 733.
18. Wendt G., S h e 11 у В. V., В r u c e W. F. J. Am. Chem. Soc., 81, 4233 (1959).
19. H e с м e я и о в А. Н., Кочеткова Н. С., Ф р ей длин а Р. X. Изв.
АН СССР, ОХН, 1947, 347.
20. Spengler G., Weber A. Brennstoff - Chemie, 40, 22 (1959).
21. Ichikawa К-, Fukushima S., О u c h i H., Tsuchida M. J. Am.
Chem. Soc., 80, 6005 (1958).
22. Ichikawa K., Fukushima S., Ouchi H., TsuchidaM. J Am.
Chem. Soc., 81, 3401 (1959).
23. Ichikawa K, F u j i t a K-, О u c h i H. J. Am. Chem. Soc., 81, 5316 (1955).
24, Ichikawa К, О u c h i H., J. Am. Chem. Soc., 82, 3876 (1960). #
25. Японск. пат. 13015 (1960); РЖХим, 1962, 16Л93; 1961, 21529; РЖХим, 1964, 4Н83.
26. Ichikawa К., Ouchi Н., Fukushima S. J. Org. Chem., 24, 1129 (1959).
26a. Ichikawa K-, Fukushima T., Ouchi H. РЖХим, 1964, 17H124.
27. В e r g О. W., Lay W. P„ R о d g m a n A., W r i g h t G. F. Can. J. Chem.,
36, 358 (1958).
28. AbercrombieM. J., Rodgman A., Bharucha K. R., Wright G. F.
Can. J. Chem., 37, 1328 (1959).
29. W r i g h t G. F. Can. J. Chem., 30, 268 (1952).
30. Несмеянов A. H., Ф p e й д л и н а Р. X. ЖОХ, 7, 2748 (1937).
31. Р а г k W. R. R„ W г i g h t G. F. Can. J. Chem., 35, 1088 (1957).
32. В i i 1 m a n E. Ber., 43, 568 (1910).
33. Hofmann K- A„ Leschewski K- Ber., 86, 123 (1923).
34. Несмеянов A. H., Фрейдлииа P. X. ЖОХ, 7, 43 (1937); Ber., 69, 1631
(1936).
35. Луценко И. Ф., Сивкова Р. И. ЖОХ, 29, 1182 (1959).
36. Robson J. Н., Wright G. F. Can. J. Chem., 38, 1 (1960).
37. PataiS., Harnik М., Hoffmann Е. J. Am. Chem. Soc., 72, 923 (1950).
38. ФрейдлинаР. X., Несмеянов A. H., Кочешков К. А. ЖОХ, 5,
1171 (1935); Ber., 68, 565 (1935).
39. Несмеянов A. H., Фрейдлииа P. X., Токарева Ф. А. ЖОХ, 7,
262 (1937); Ber., 69,2019 (1936).
40. Laubie H. C. A., 51, 15889 (1957); Bull. soc. pharm. Bordeaux, 96, 65 (1957).
41. Фосс В. Л., Ж а д и и a M. А., Луценко И. Ф., Несмеянов А. Н.
ЖОХ, 33, 1927 (1963).
42. М а п с h о t W., К 1 u g A. Ann., 420, 170 (1920).
43. Sandborn L. Т., Marvel С. S. J. Am. Chem. Soc., 48, 1409 (1926).
44. Griffith E., Marvel C. S. J. Am. Chem. Soc., 53, 789 (1931).
45. Adams R., Roman F. L., Sperry W. N. J. Am. Chem. Soc., 44, 1781 (1922).
46. M i 1 I s L., A d a m s R. J. Am. Chem. Soc., 45, 1842 (1923).
47. Pfeiffer P. Naturwiss., 14, 1100 (1926).
48. S a n d J. Ann., 329, 135 (1903).
49. S a n d J., S i n g e r F. Ann., 329, 166 (1903).
174
Ртутноорганические соединения
50. Несмеянов А. Н„ 3 а р е в и ч Т. С. ЖОХ, 6, 140 (1936); Вег., 68, 1476 (1935).
51. W г i g h t G. F. Ann. New. York Acad. Sci., 65, 436 (1957).
52. С о t t о n F., Leto J. R. J. Am. Chem. Soc., 80, 4823 (1958).
53. Lucas H. J., H epner F. R., Winst ei n S. J. Am. Chem. Soc., 61, 3102
(1939).
54. Dewar M. S. Ball. Soc. chim. France, 18, C71 (1951).
55. Summer b el I R, K., Lest ina G„ Wai t e H. J. Am. Chem. Soc., 79, 234
(1957).
56. Gomez Aranda V., Sanz PastorO. Combustible, 10, 183 (1950).
57. В r a n d R. B., Plum O. Acta Chem. scand., 7, 97 (1953).,
58. A 1 1 e n E. R., С а г 11 i d g e J., Tay lor M. M., T i p p e r C. F. H. J. Phys.
Chem., 63, 1437 (1959).
59. Allen E. R. J. Phys. Chem., 63, 1442 (1959).
60. Кг ее voy M. M. J: Am. Chem. Soc., 81, 1099 (1959).
61. Кг ее voy M. M., К о w i 11 F. R. J. Am. Chem. Soc., 82, 739 (1960).
62. M a 1 1 i к K. L., D a s M. N. J. Am. Chem. Soc., 82, 4269 (1960).
63. Kreevoy M. M., DitschL. T. J. Org. Chem., 25, 134 (1960).
64. Kreevoy M. M., D i t s c h L. T. J. Am. Chem. Soc., 82, 6127 (1960).
65. Kreeyoy M. M., G i 1 j e J. W., Ditsch L. T., Batorewicz W.,
Turner M. A. J. Org. Chem., 27, 726 (1962).
66. В г о о к A. G., W r i g h t G. F. Can. J. Research, 28B, 623 (1950).
67. W r i g h t C. F. Chem. in Canada, 2, No 9, 29 (1950).
68. В г о о к A. G., W r i g h t G. F. Can. J. Chem., 29, 308 (1951).
69. S h e a r e r D. A., Wr i g h t G. F. Can. J. Chem., 33, 1002 (1955).
.70 . Ro dgman A., Wright G. F. J. Org. Chem., 18, 1617 (1953).
71. M c N e 11 у К- H„ W r i g h t G. F. J. Am. Chem. Soc., 77, 2553 (1955).
72. Wr i g h t G. F. J. Am. Chem. Soc., 57, 1993 (1935).
73. С о к о л о в В. И., Р е у т о в О. А. ДАН СССР, 148, 867 (1963).
74. Т г а у 1 о г Т. G,, В а к е г A. W. Т etrahedron L., 19,14 (1959); J. Am. Chem. Soc.,
85, 2746 (1963).
75. Ю p ь e в Ю. К., 3 е ф и р о в Н. С., ПриказчиковаЛ. П. ЖОХ, 32,
2744(1962).
76. С h а р m a n Е. Т., J е n k i n s W. J. Zbl., 1920, I, 153; J. Chem. Soc., 115, 847
(1919).
77. Jenkins W. J. Zbl., 1921, HI, 608; J. Chem., Soc., 119, 747 (1921).
78. F e r b e r E., Romer E. J. prakt. Chem., 139, 277 (1934).
79. В i g i n e 1 1 i P. Zbl., 1898, I, 925; Ann. di Pharmacoterap. e chim., 1898, 16.
80. Несмеянов A. H., Фрей длин a P. X. ДАН СССР, 24, 59 (1940).
81. Реутов О. А., Феднева Е. И. ЖОХ, 27, 2506 (1957).
82. ФрейдлинаР. X. Изв. АН СССР, ОХН, 1942, 14.
83. В а р ш а в с к и й С. Л. ДАН СССР, 29, 315 (1940).
84. Несмеянов А. Н., Борисов А. Е. Изв. АН СССР, ОХН, 1945, 146.
85. Несмеянов А. Н., БорисовА. Е., Вильчевская В. Д. Изв. АН
СССР, ОХН, 1949, 578; Уч. зап. МГУ, 132, 33 (1950).
й. Несмеянов А. Н., БорисовА. Е., Гуськова А. Н. Изв. АН СССР,
ОХН, 1945, 639.
87. Ф р е й д л и н а Р. X., Ногина О. В, Изв. АН СССР, ОХН, 1947, 99.
88. С м и р н о в - 3 а м к о в И. В., Шилов Е. А. ДАН СССР, 73, 723 (1950).
89. Несме я нов А. Н., БорисовА. Е., Абрамова А. Н. Изв. АН СССР,
ОХН, 1947, 289.
90. N е s m е у а п о v A. N., В о г i s s о v А. Е. Tetrahedron, 1, 158 (1957).
91. Несмеянов А. Н., БорисовА. Е. ДАН СССР, 60, 67 (1948).
92. Несмеянов А. Н., РеутовО. А., КнолльП. Г. ДАН СССР, 118,
99(1958).
93. БорисовА. Е., Ви льчевская В. Д., Несмеянов А. Н. ДАН СССР,
90, 383 (1953).
94. Несмеянов А. Н„ БорисовА, Е., Вильчевская В. Д. Изв.
АН СССР, ОХН, 1954,1008.
95. Н е с м е я н о в А. Н., БорисовА. Е., В о л ь к е н а у Н. А. Изв. АН СССР,
ОХН, 1954, 992.
96. К у ч е р о в М. Г. ЖРФХО, 13, 542 (1881); Вег., 14, 1532, 1540 (1881).
97. KutscheroffM. Вег., 17, 13 (1884).
98. В i 1 t z Н., М u m m О. Вег., 37, 4417 (1904).
99. Myddleton W. W., Barrett A. W. J. Am. Chem. Soc., 49, 2258 (1927).
100. M v d d 1 e t 6 n W. W., В e r c h e m R. G., Barr ett A. W. J. Am. Chem.
Soc'. 49. 2264 (1927).
111. Myddleton W. W., Barrett A. W., SeagerJ. H. J. Am. Chem. Soc.,
52, 4405 (1930).
Присоединение солей ртути к непредельным соединениям 175
102. М a n с h о f W, Ann., 417, 93 (1918).
103. Hofmann К- А. Вег., 31, 2212 (1898).!
104. Schoell er W., S с h г a u t h W., Essers W. Ber., 46, 2864 (1913).
105. Mancho t W. Ber., 53, 984 (1920).
106. M a n c h о t W., F r i e n d J. N. Ann., 359, 100 (1908).
107. Pfeiffer P. Organische Molekiilverbindungen. Stuttgart, Verlag F. Enke, 1927.
108. Фрейдлина P. X„ Как Э. И. Изв. АН СССР, ОХН, 1948, 548.
109. Н а 1 р е г n J., К е t t 1 е S. F. A. Chem. and Ind,, 1961, 668.
ПО. S u 1 1 a m С. Zbl,, 1939, I, 93; Atti R. st. Veneto Sci. mat. natur, 97, 221 (1937—1938).
111. Несмеянов A. H., Фрейдлина P. X., Борисов A. E. Юбилейный сбор-
ник, посвященный 30-летню Великой Октябрьской социалистической революции,
часть 1. М., Изд-во АН СССР, 1947, стр. 658.
112. Ш и л о в Е. А. ДАН СССР, 18, 643 (1938).
113. Н е с м е я н о в А. Н. ЖОХ, 21, 1733 (1951).
114. Чел ни пев Г. В. Изв. АН СССР, ОХН, 1949, 410.
115. Несмеянов А. Н. Изв. АН СССР, ОХН, 1950, 541.
116. Kondakov I. Rec, trav. chim., 25, 864 (1892).
117. Kondakov I. Rec. trav. chim., 26, 1012 (1893).
118. Несмеянов A. H. Уч. зап. МГУ, 132, 5 (1950).
119. H e с м e я н о в A. H., К а б а ч и и к М. И. ЖОХ, 25, 41 (1955).
120. Несмеянов А. Н., Батуев М. И., Б о р и с о в А. Е. Изв. АН СССР, ОХН,
1949, 457.
121. Н е с м е я н о в А. Н., Борисов А. Е„ Ш е п е л е в а Р. И. Изв. АН СССР,
ОХН, 1949, 582.
122. Б о р и с о в А. Е. Изв. АН СССР, ОХН, 1961, 1036.
123. Несмеянов А. Н„ Б о р и с о в А. Е. Изв. АН СССР, ОХН, 1945, 252.
124. Н е с м е я н о в А. Н., Луценко И. Ф., Верещагина Н. И. Изв. АН
СССР, ОХН, 1947, 63.
125. Несмеянов А. Н., Л у ц е и к о И. Ф. ДАН СССР, 59, 707 (1948).
126. Несмеянов А. Н., Луценко И. Ф., Хомутов Р. М. ДАН,СССР, 83,
837 (1953).
127. Несмеянов А. Н.,Луценко И. Ф., Туманова 3. М. Изв. АН СССР,
ОХН, 1949, 601.
128. Несмеянов А. Н., Луценко И. Ф., Хомуто в[Р. М. Изв. АН СССР,
ОХН 1957 942.
129. Несмеянов А. Н., Перевалова Э. Г. Изв. АН СССР, ОХН, 1954, 1002.
130. Якубович А. Я., Разумовский В. В., Вострух ин а 3. Н., Розен-
штейн С. М. ЖОХ, 28, 1930 (1958).
131. Лу цен ко И. Ф., К р а й ц 3. С. ДАН СССР, 135, 860 (1960).
132. Н е с м е я н о в А. Н., ЛуценкоИ. Ф., Хомутов Р. М., Дубовиц-
кий В. А. ЖОХ, 29, 2817 (1959).
133. Несмеянов А. Н., Луценко И. Ф., К р а й ц 3. С., Б о к о в о й А. П. ДАН
СССР, 124, 1251 (1958).
134. Несмеянов А. Н., Луценко И. Ф., Хомутов Р. М. ДАН СССР, 120.
1049 (1958).
135. Л у цен к о И. Ф, Б а де в к о в а Л. П„ Ф о с с В. Л. ЖОХ, 27, 3261 (1957).
136. Л у цен ко И. Ф„ Фосс В. Л., И в а н о в а Н. Л. ДАН СССР, 141, 1107(1961).
137. ЛуценкоИ. Ф., Фосс В. Л., ЖадинаМ. А. ЖОХ, 35 (1965) (в печати).
138. Несмеянов А. Н., Кочетков Н. К- Изв. АН СССР, ОХН, 1949, 76.
139. Несмеянов А. Н., Кочетков Н. К. Изв. АН СССР, ОХН, 1949, 305.
140. Несмеянов А. Н., Кочетков Н. К-, Дашунин В. М. Изв. АН СССР,
ОХН, 1950, 77.
141. М а п с h о t W. Ann., 421, 316 (1920).
142. В а 1 b i а п о L., Р а о 11 n i V. Ber., 35, 2994 (1902).
143. В а 1 b i а п о L., Р а о 1 i n i V. Ber., 36, 3575 (1903).
144. Brown W. H., W r i g h t G. F. J. Am. Chem. Soc., 62, 1991 (1940).
145. В i ilman E. Ber„ 35, 2571 (1902).
146. H e с м e я н о в А. Н., ЛуценкоИ. Ф. Изв. АН СССР, ОХН, 1943, 296.
147. Несмеянов А. Н., Л у ц е н к о И. Ф. Изв. АН СССР, ОХН, 1942, 366.
147а. Summerbell R. К., ForresterS. R. J. Org. Chem,, 26, 4834 (1961).
148. Johnson J. R., JoblingW. H., Bodamer G. W. J. Am. Chem. Soc., 63,
131 (1941).
149. В i r k s A. M„ W r i g h t G. F. J. Am. Chem. Soc., 62, 2412 (1940).
150. T h о m a s M. H., Wetmore F. E. W. J. Am. Chem. Soc., 63, 136 (1941).
151. Hope. пат. 60693 (1939); Zbl., 1939, II, 4280.
152. Whitehead C. W., T r a v e r s о J. J. J. Am. Chem. Soc., 80, 2182 (1958).
153. Gomez Aranda V., Sanz Pastor O., Martinez Gordon L. '7„.
РЖХнм, 1957, 23070; Combustibles, 15, No 78-79, 102 (1956).
176
Ртутноорганические соединения
154. Spahr R. J., Vogt R. R., Ni euwl a n d J, A. J. Am. Chem. Soc., 55, 3731
(1933).
155. Sanz PastorO. C. A„ 47, 2688 (1953).
156. Пат. ФРГ 967765 (1957); С. A., 53, 13057 (1959).
157. Sand J. Ber., 34, 2910 (1901).
158. Нем. пат. 704409 (1941); С. A., 36, 2087 (1942).
159. Тартаковский В, А., Новикове. С., Годовикова Т. И. Изв. АН
СССР, ОХН, 1961, 1043.
159а. Тартаковский В. А., Савостьянова И. А., Новиков С. С. Изв.
АН СССР, сер. хим., 1963, 1330.
160. ShukisA. J., TallmannR. С. J. Am. Chem. Soc., 65, 2365 (1943); 66, 1462
(1944).
161. Kreevoy M. M., Bodem G. В. J. Org. Chem., 27, 4539 (1962).
162. Пат. США 3031484 (1962); С. A., 57, 13803 (1962).
163. Пат. ФРГ 912452 (1954); С. А., 52, 15579 (1958).
164. SandJ., GensslerO. Ber., 36, 3699 (1903).
165. Martinez Gordon J. L., Gomez Aranda V. РЖХим, 1959, 19513; Com-
bustibles, 16, No 86, 214 (1956); 16, No 87, 290 (1956); 17, No 89, 12, 83, 196 (1957).
166. Англ. пат. 908744 (1962); РЖХим, 1963, 20H49.
167. Goodman L., Ross L. O., Gre.ene M. O., GreenbergJ., BakerB.R.
РЖХим, 1961, 24Ж186; J. Med. and Pharm. Chem., 3, No 1, 65 (1961).
168. Tausz J„ Peter N. Zbl., 1919, II, 125; Petroleum, 13, 649 (1918).
169. Robson I. H., Wright G. F. Can. J. Chem., 38, 21 (1960).
170. Левина P. Я., Гл ад штейн Б. М. ДАН СССР, 71, 65 (1950).
171. S е у f е г t h D., К a h 1 е n N. Naturforsch., 14b, 137 (1959).
172. Кнунянц И. Л., ПервоваЕ. Я-, Тюленева В. В. Изв. АН СССР,
ОХН, 1956, 843; 1961, 88.
173. Krespan С. G. J. Organ. Chem., 25, 105 (1960); Пат. США 2844614 (1958); С. А.,
53, 3061 (1959)..
174. Middiet on W. J., Howard Е. G., Sharkey W. Н. J. Am, Chem. Soc.,
83, 2589 (1961).
175. A 1 d r i c h P. E„ H о w a r d E. G., Linn W. J., Middleton W. J.,Shai-
key W. H. J. Org. Chem., 28, 184 (1963).
176. M i 1 1 e r W. T., jr., Freedman M. B. J. Am. Chem. Soc., 85, 180 (1963).
177. M i 1 1 e r W. T., jr., Freedman M, B., Friend J. H., Koch H. F. J. Am,
Chem. Soc., 83, 4105 (1961).
178. Goldwhite H., Haszeldine R. N., Mukherjee R.N. J. Chem. Soc.,
1961, 3825.
179. Brook A. G., D о n о v a n R., W r i g h t G. F. Can. J. Chem., 31, 536 (1953).
180. Anderson M. M., Henry P. M. Chem. and Ind., 50, 2053 (I960.
181. Gina M., Polla G. Chimica e industria (Milan), 35, 888 (1953).
182. В г о о k A. G., W r i g h t G. F. Acta cryst., 4, 50 (1951).
183. В г о о k A. G„ W r i g h t G. F. J. Am. Chem. Soc., 72, 3821 (1950).
184. В Г о о k A. G., W a 1 f e S. J. Am. Chem. Soc., 79, 1431 (1957L
185. Англ. пат. 459.462 (1937); Zbl., 1937, I, 3518.
186. Raliant J., Wittig G. Bull. Soc. Chim. France. 1957, 798.
187. D a s M. N. Anal: Chem., 26, 1086 (1954).
188. Jensen F. R., Gale L. H. J. Am. Chem. Soc., 81, 1261 (1959).
189. Jensen F. R., Ga le L. H. J. Am. Chem. Soc., 81, 6337 (1959).
190. J ensen F. R., G a 1 e L. H. J. Am. Chem. Soc., 82, 145 (1960).
191. Schaeffer H. J., С о I 1 i n s C. J. J. Am. Chem. Soc., 78, 124 (1956).
192. Deussen E. J. prakt. Chem., 114, 63 (1926).
193. BalbianoL., PaoliniV. Gazz., 36, I, 270 (1906).
194. Zelinsky N., Lepeschkin N. Ann., 319, 308 (1901).
195. Grossley A. W., Renouf N. J. Chem. Soc., 1906, 42.
196. Henbest H. B., McElhi nney R. S. J. Chem. Soc., 1959, 1834.
197. R о w 1 a n d R. L. J. Am. Chem. Soc., 73, 2381 (1951).
198. Chiu D. D. K., Wright G. F. Can. J. Chem., 37, 1425 (1959).
198a. Зефиров H. С., ПриказчиковаА. П., Юрьев Ю. К. ДАН СССР, 152,
869 (1963).
1986. Traylor Т. G. J. Am. Chem. Soc., 86, 244 (1964).
199. Levy М. С. А., 54, 10934 (1960).
200. Park W. R. R„ Wright G. F. J. Org. Chem., 19, 1435 (1954).
201. Несмеянов A. H., Печерская К- А. Изв. АН СССР, ОХН, 1941, 67.
202. Ш а мш у р и н А. А. ЖОХ, 16, 99 (1946).
203. Rodgman A., Wright G. F. Can. J. Research. Sect./ В38, 623 (1956).
204. Downing D. С., Wright G. F. J. Am. Chem. Soc., 68, 141 (1946).
205. Rodgman A., Wright G. F. J. Am. Chem. Soc., 76, 1382 (1954).
Присоединение солей ртути к непредельным соединениям
177
206. Англ. пат. 411507 (1934); Zbl., 1934, И, 2286.
207. Geissman Т. А., Horowitz R.M. J. Am. Chem. Soc., 73, 5759 (1951).
208. Левина P. Я-, Костин В. H., ГембицкийП. А., Шостаковский
С. М„ Трещав а Е. Г. ЖОХ, 31, 1185 (1961).
209. Ветшал L., Н а 1 1 R. Н., Р у k е R. Q., W г 1 g h t G. F. Can. J. Chem., 30,
541 (1952).
210. Япоиск. пат. 9431 (1961); С. A., 56, 5855 (1962).
211. В a 1 b i a n о L. Ber., 42, 1502 (1909).
212. Gasopou las I. Ber., 59, 2184 (1924).
213. KergomardA. Ann. chim., 8, 153 (1953).
214. Японск. пат. 14714 (1961); С. A., 56, 12945 (1962).
215. S p e n g 1 e r G., W e b e r A. Brennstoff-Chemie, 43, 234 (1962).
216. Пат. США 2369339 (1945); С. A., 39, 3618 (1945).
217. Пат. США 2329883 (1944); С. А., 38, 1604 (1944).
218. Spengler G., Weber A. Brennstoff-Chemie, 40,-55 (1959).
219. Budesinsky В. Coll. Czechoslov. Chem. Communication, 22, 1147 (1957); Chem.
Listy, 51, 259 (1957).
220. M a r q u a r d R. P„ L u с e E. N. Analyt. Chem., 20, 751 (1948); 21, 1194 (1949).
221. M a r t i n R. W. Analyt. Chem., 21, 921 (1949).
222. .Johnson J. B., F 1 e t c h e r J. P. Analyt. Chem., 31, 1563 (1959).
223. Hofmann K- A., S e i 1 e r E. Ber., 39, 3187 (1906).
224. H о r n s t e i n I. Analyt. Chem., 23, 1329 (1951).
225. Francis A. F. J. Am. Chem. Soc., 73, 3709 (1951).
226. КучеровM. Г. ЖРФХО, 24, 330 (1892).
227. H о в и к о в G. С., Т а р т а к о в с к и й В. А., Г о д о в'и к о в а Т. И., Г р и-
бовБ. Г. Изв. АН СССР, ОХН, 1962, 276.
228. Пат. США 2400436 (1946); С. А., 40, 4484 (1946).
229. Summerbell R. К-, Lestina G. J. J. Am. Chem. Soc., 79, 3878 (1957).
230. Lessen W., DornoK. Ann., 342, 185 (1905).
231. HiickelW. Ber., 88, 338 (1955).
232. Wins te i n S., ShatavskyM. Chem. and Ind., 1956, 56.
233. A 1 e x a n d e r R. A., Baenziger N. C., CarpenterC., DoyleJ.R.
J. Am. Chem. Soc., 82, 535 (1960).
234. Cop e A. C., N e 1 son N. A., Smith D. S. J. Am. Chemi Soc., 76, 1100 (1954).
235. Пат. США 2395954 (1946); С. A., 40, 3122 (1946).
236. В i i I m a n n E. Ber., 33, 1641 (1900).
237. S u rri.m erbell R. K., Stephens J. R. J. Am. Chem. Soc., 77, 6080 (1955).
238. Новикове. С., Тартаковский В. А., Годовикова T. И., Гри-
бов Б. Г. Изв. АН СССР, ОХН, 1962, 272.
239. Ro dgman A., W г i g h t G. F. J. Org. Chem., 17, 988 (1952),
240. Пат. США 2284067 (1942); С. A., 36, 6302 (1942).
241. BlickeF. F„ В i e 1 J. H. J. Am. Chem. Soc., 76, 3163 (1954).
242. Жданов Ю. А., КорольченкоГ. А., К у б а с с к а я Л. А., Криво-
ручко P. M. ДАН СССР, 129, 1049 (1959).
243. Жданов Ю. А., КорольченкоГ. А., КубасскаяЛ. А. ДАН СССР,
128, 1185 (1959).
244. W е г n е г L. Н„ S с h о 1 z С. R. J. Am. Chem. Soc., 76, 2701 (1954).
245. Пат. ФРГ 946539 (1956); С. А1, 53, 12208 (1959).
246. Пат. США 2790812 (1957); С. А., 51, 14833 (1957).
247. Англ. пат. 791125 (1958); С. А., 52, 16238 (1958).
248. Пат. США 2790816, 2790817 (1957); С. А., 51, 16527, 16528 (1957),
249. R о s е n W. Е., Z i е g 1 е г J. В. J. Am. Chem. Soc., 77, 762 (1955).
250. Ma nolopoulosT. P., Mednik M„ L i c h t i n N. N. J. Am. Chem. Soc.,
84, 2203 (1962).
251. Hen best H. B., Nicholls B. Proc. Chem. Soc., 1957, 61.
252. Henbest H. B., Nicholls B. J. Chem. Soc., 1959, 227.
253. В г о о k A. G.,W r i g h t G. F. J. Org. Chem., 22, 1314 (1957).
254. Biilmann E. Ann., 388, 272 (1912).
255. В a 1 b i a n о L. Gazz., 36, I, 244 (1906).
256. Matej ka K. Ber., 69, 274 (1936).
257. UkaiT. Zbl., 1937, I, 3804; J. Pharm. Soc. Japan, 57, 6 (1937).
258. Австр. пат. 99678 (1925); Zbl., 1926, I, 493.
259. Пат. ФРГ 923788 (1955); С. A., 51, 12973 (1957).
260. Bartz Q. R„ Mi Her R. F„ Adams R. J. Am. Chem. Soc., 57, 371 (1955)
261. S e n A. B., Rostogi R. P. J. Indian Chem. Soc., 30, 355 (1953).
262. Несмеянов A. H., Шацкая P. X. ЖОХ, 5, 1268 (1935).
263. Thro nH., Dirscherl W. Ann., 515, 252 (1935).
264. Summerbell R. K-, Stephens J. R. J. Am. Chem. Soc., 76, 731 (1954)?
178 Ртутноорганические соединения
265. Levy М. Докл. Болг. АН, 13, 427 (1960).
266. W h i t е h e a d С. W. J. Am. Chem. Soc., 80, 2178 (1958).
266a. Summerbell R. К., P о 1 k a c k i E, S. J. Org. Chem., 27, 2074 (1962).
267. A s h A. S. F„ Challenger F., Greenwood D. J. Chem. Soc., 1951, 1877.
.268 . Несмеянов A. H., Луценко И. Ф., Хомутов P. M. Изв. АН СССР,
ОХН, 1960, 217.
269. С u г t i n D. Y., Н и г w i t z M. J. J. Am. Chem. Soc., 74, 5381 (1952).
270. Женод a p о в а С. M., АдамоваМ. Н., Л я п о т а Л. А. ЖАХ, 18, 285
(1963).
271. Луценко И. Ф., Хомутов Р. М. ДАН СССР, 102, 97 (1955).
272. ВеретеноваТ. И., Балин А. Н. ЖОХ, 33, 2079 (1963); пат. СССР 143397
(1963); С. А., 57, 9881 (1962).
273. Луценко И. Ф., Юркова Е. И. Изв. АН СССР, ОХН, 1956, 27.
274. Луценко И. Ф., Хомутов Р. М., Елисеева Л. В. Изв. АН СССР, ОХН,
1956, 173.
275. Пат. США 2356884 (1944); С. А., 39, 1246 (1945).
276. Dominikiewicz М„ К i j е w s k a M. Zbl., 1940, II, 1573; Arch. Chem. Far-
macji, 3, 8 (1939).
277. Middleton E. B. J. Am. Chem. Soc., 45, 2763 (1923).
278. Пат. США 2262430 (1942); С. A., 36, 1741 (1942).
279. Пат. США 2286226 (1942); С. А., 36, 7244 (1942).
280. Као Y. S., Pan Р. С., L о h S. Н., С h е n С. Н., Hsu Н. Y. G А., 53, 7088,
13097 (1952).
281. Л у ц е н к о И. Ф., Ф о с с В. Л. ДАН СССР, 98, 407 (1954).
282. M e Е 1 v a i n S. М.,’ А 1 d г i d g е С. L. J. Am. Chem. Soc., 75, 3987 (1953).
283. Schrauth W., Bauerschmidt H. Ber., 47, 2736 (1914).
284. В i i 1 m a n n E. Ber., 41, 4340 (1908).
285. I zu mi j ya.N. C. A., 45, 7011 (1951).
286. Roth R., Erlenmeyer H. Helv. chim. acta, 38, 1276 (1955).
287. Tatsuok- aS., Murakani M., Ueno T., Mor i mo to A. C. A., 46, 2498
(1952); Ann. Repts. Takeda Research Lab., 9, 7 (I960).
288. Rowla nd R. L., P e г г у W. L., F r i e d m a n H. L. J. Am. Chem. Soc., 73,
1040 (1951).
.289. R о w 1 a n d R. L., Kluehesky E. F. J. Am. Chem. Soc., 73, 5490 (1951).
290. D г у L. J. C. A., 49, 6825 (1955).
291. Пат. США 2428955 (1947); С. A., 42, 1317 (1948).
.292. Iritani.N., Misumi M., N a g i w a r a Y. C. A., 52, 2743 (1958).
-293. Японск. пат. 4222 (1955); С. A., 51, 16520 (1957).
294. Нем. пат. 271820 (1914); Zbl., 1914, I, 1384.
295. Пат. США 2289590 (1943); С. А., 37, 498 (1943).
296. R а 1 s t о n А. М., Christensen С. W., J о s h G. Zbl., 1937, I, 3393; Oil and
Soap, 14, 5 (1937).
297. Нем. пат. 228877 (1910); Zbl., 1911, I, 102.
298. JantzenE., Andreas H. Angew. Chem., 70, 656 (1958); Ber., 92, 1427 (1959).
299. Connor T„ W r i g h t G. F. J. Am. Chem. Soc,, 68, 256 (1946).
300. Jantzen. E., Andreas H. Ber., 94, 628 (1961).
301. Dean A. L„ Wrenshall R„ Fu j i mo to G. J. Am. Chem. Soc., 47,403 (1925).
302. Австр. пат. 112624 (1929); Zbl., 1929, II, 602.
303. M a 1 a i у a n d i M., Wright G. F. Can. J. Chem., 41, 1493 (1963).
304. M c N e 1 1 у К. H., R о dgriian A., Wr i gh t G. F. J. Org. Chem 20, 714 (1955).
305. Юрьев Ю. К., Зефиров H. С., Пр н к азчнкова Л. П. ОХ, 33, 1793
(1963).
306. Ю р ь е в Ю. К-, Зефиров Н. С., Шайдерова Л. П. ЖОХ, 33, 818 (1963).
306а. Зефиров Н. С., Приказчнкова Л. П., Бондарева М. А., Юрьев
Ю. К- ЖОХ, 33, 4026 (1963).
307. Р а г k W. R„ W г i g h t G. F. J. Org. Chem., 19, 1325 (1954).
308. Negoro К. C. A., 47, 9929 (1953).
309. S a h P. P. T„ T s e u C.-Z. Zbl., 1937, II, 1789; J. Chin. Chem. Soc., 5, 134 (1937).
310. Австр. пат. 115016 (1929); Zbl., 1930, I, 1331.
311. R a n g a s w a m i S., R а о V. S., S es h a d r i T. R. Zbl., 1938, II, 3078; Proc. Indian
Acad. Sci. Sect., A7, 296 (1938).
312. VerkadeP. E. Rec. trav. chim., 45, 475 (1926).
313. L о e v B. J. Org. Chem., 26, 4394 (1961).
314. Пат. США 234-3547 (1944); С. A., 38, 3093 (1944).
315. S c h r a u t h W., G e 1 1 e r H. Ber., 55, 2783 (1922).
316. AbderhaldenE., Heyns K. Ber.^ 67, 530 (1934).
317. Seshadri T, R., R а о P. S. Zbl., 1937, I, 2370; Proc. Indian. Acad. Sci., A4
112 (1936).
Присоединение солей ртути к непредельным соединениям
179
318. S е s h a d г i Т. R., R а о Р. S. Zbl., 1937, I, 4621; Proc. Indian Acad. Sci., 4, 157
(1936).
319. R angaswam i S., R а о V. S., SeshadriT. R. Zbl., 1939, I, 104; Proc.
Indian Acad. Sci. Sect., A7, 312 (1938).
320. R а о P. S., S e s h a d r i T. R. Zbl., 1937, H, 3457; Proc. Indian Acad. Sci. Sect.,
A4, 630 (1936).
321. R а о P. S., S a s t r i V. D. N., S e s h a d r i T. R. Zbl., 1939, II, 638; Proc. In-
dian Acad. Sci. Sect., A9, 22 (1939).
322. SeshadriT. R„ RaoP.S. Zbl., 1937, I, 2757; Proc. Indian Acad. Sci. Sect.,
A3, 293 (1936).
323. Пат. США 2667442 (1954); С. A., 49, 1818 (1955).
324. SastriV. D. N., Narasimhachari N., R a j agop a 1 an P., Seshad-
riT. R., ThiruvengadamT. R. C. A., 48, 8227 (1954); Proc. Indian Acad.
Sci. Sect., A37, 681 (1953).
325. ФрейдлинаР. X., Чуковская E. Ц. Изв. АН СССР, ОХН, 1957, 187.
326. Kato К- С. А., 56, 493 (1962).
327. Rowland R. L., Perry W, L.,- F о r e m a n E. L., F r i e d m a n. H. L.
J. Am. Chem. Soc., 72, 3595 (1950).
328. Rowland R. L., Perry W. L., Gerstein S. J. Am. Chem. Soc., 73, 3691
(1951).
329. Датск. пат. 84183 (1958); С. A., 53, 11238 (1959).
330. Англ. пат. 730628 (1954); С. А., 49, 12533 (1955).
331. Werner L. Н„ S с h о 1 z С. R. J. Am. Chem. Soc., 76, 2453 (1954).
332. Y а 1 е Н. L. J. Am. Chem. Soc., 79, 4762 (1957).
333. Англ. пат. 792992 (1958); С. А., 52, 18222 (1958).
334. Пат. США 2800471 (1957); С. А., 52, 16381 (1958).
335. Пат. ФРГ 959907 (1957); С. А., 53, 18865 (1959).
336. Пат. США 2635982 (1953); С. А., 48, 6460 (1954).
337. Англ. пат. 790906 (1958); С. А., 52, 16213 (1958).
338. G u b i t z F: W„ М с К е о n W. В., jr. РЖХим, 1962, 24Ж423; J. Med. Pharmac,
Chem., 5, 168 (1962).
339. Н а 1 р е г n A., J о n е s J. W„ G г о s s Е. G. С. А., 43, 2169 (1949).
340. N a d k а г n i М. V., J о n е s J. W. С. A„ 44, 6525 (1950). '
341. Pe ar so n D. E„ Si ga 1 M. V., jr. J. Org. Chem4 15, 1055 (1950).
342. As in ger F., Fell B., Hadik G., Stefan G. W., 97, 1568 (1964).
343. Пат. США 2208941 (1941); С. A., 35, 281 (1941).
344. Пат. США 2163296 (1939); Zbl., 1939, II, 2815.
345. R о w 1 a n d R. L.,Perr у W. L., G e г s t e i n S. J. Am. Chem. Soc., 73, 91 (1951k
346. Пат. США 2117901 (1938); С. A., 32, 5584 (1938).
347. Пат. США 2749361 (1956); С. А., 51, 465 (1957).
348. Англ. пат. 692953 (1953); С. А., 48, 10056 (1954).
349. S z е k е г е s L. Gazz., 79, 56 (1949).
350. Англ. пат. 782050 (1957); С. А., 52, 2100 (1958).
351. Англ. пат. 447877 (1936); Zbl., 1936, II, 1763.
352. Франц, пат. 799381 (1936); Zbl., 1937, I, 1278.
353. W е п d t G.,.B г и с е W. F..J. Org. Chem., 23, 1448 (1958).
354. Канад, пат. 401561 (1941); С. А., 36, 1736 (1942).
355. Пат. США 2672472 (1954); С. А., 49, 3253 (1955).
356. Пат. США 2834795 (1958); G. А., 53, 2118 (1959).
357. Пат. США 2922789 (I960); С. А., 54, 9973 (I960).
358. Пат. США 2685594 (1954); С. А., 49, 11014 (1955).
359. Пат. США 2698858 (1955); С. А., 49, 14032 (1955).
360. Пат. США 2695305 (1954); С. А., 49, 15969 (1955).
361 М i n g о i a Q. С. А., 38, 4999 (1944).
362. Несмеянов Ник. А., Н о в и к о в В., Р е у т о в О. А. Изв. АН СССР, ОХН,
1964, 772.
363. F о у е W. О., К о t a k Н. М., К е f f е г е m J. J. С. А., 47, 5922 (1953).
364. Англ. пат. 553232 (1943); С. А., 38, 5047 (1944).
365. Carrara G., Mori Е. Gazz., 73, 113 (1943).
366. С а г г а г a G. Gazz., 79, 201 (1949).
367. Szekeres L. Gazz., 79, 832 (1949).
368. C a r r a r a G. Gazz., 79, 834 (1949).
369. Pearson D. E., S i g a 1 M. V., jr., Krug R. H. J. Org. Chem., 15, 1048 (1950).
370. В а к e r B. R., Q u e г г у M. V., S c h a u b R. E„ W i 1 1 i a m s J. H. J. Ore.
Chem., 17, 58 (1952).
371. Швейц, пат. 186461, 183121 (1936); Zbl., 1937, I, 4128.
372. Нем. пат. 642582 (1937); Zbl., 1937, I, 4920. •
373. Швейц, пат. 188235 (1937); Zbl., 1937, II, 106.
180 Ртутноорганические соединения
374. Швейц, пат. 176757 (1934); Zbl., 1936, I, 812. 188799 (1935); Zbl., 1937, II, 123
375. Schimmel С. Zbl., 1938, I, 1826. Pharm. Monatsh., 18, No 11, 65 (1937).
376. Пат.. США 2704767 (1955); С. A., 50, 15586 (1956).
377. Eckstein M., Gorczyca M., К Q c w a A., Z e j с A, C. A., 51, 16487 (1957)
Dissert. Pharm., 9, 83 (1957).
378. Foye W. O„ Mode R. A. C. A., 49, 7194 (1955); J. Am. Pharm. Assoc., 44, 76
(1955)
379. Ahivl пат. 365211 (1932); Chem. Zbl., 1932, II, 2207,
380. Англ. пат. 447405 (1936); Chem. Zbl., 1937, I, 131.
381. Нем. пат. 637188 (1936); Chem. Zbl., 1937, I, 2216.
382. UkaiT., Hayashi M., Abe. H. Zbl., I, 1937, 3803.
383. Англ. пат. 802881 (1958); С. A„ 53, 12317 (1959).
384. Пат. ФРГ 826295 (1952); С. А., 47, 1728 (1953).
385. Пат. США 2576855 (1951); С. А., 46, 6149 (1952).
386. Нем. пат. 154905 (1938); Zbl., 1939, I, 2248.
387. С h а 11 е n g e.r F., С 1 а р h a m Р. J. Chem. Soc., 1948, 1615.
388. Пат. ФРГ 1005964 (1957); С. А., 53, 1.8058 (1959).
389. Фуршта това В.Я..М их л ина Е. Е., Рубцов М. В. ЖОХ, 29, 477 (1959).
390. Пат. США 2921068 (1960); С. А.* 54, 8867 (1960).
391. G u b i t z F. W„ С I a г к e R. L. J. Org. Chem., 26, 559 (1961).
392. Hofmann K. A. Ber., 31, 2785 (1898).
393. Erdman H., KothnerP. Z. anorg. allg. Chem., 18, 48 (1898).
394. Kot hner P. Ber., 31, 2475 (1898). ,
395. Коршак В. В., Замятина В. А. Изв. АН СССР, ОХН, 1946, 111.
396. Hof m a nn К. A. Ber., 32, 874 (1899).
397. В i 1 t z H„ Reinkob er K. Ann., 404, 219 (1914).
398. Keiser E. H. Am. Chem. J., 15, 537 (1893).
399. В r a m e J. S. J. Chem. Soc., 87, 427 (1905).
400. Китайгородский А. И. Изв. АН СССР, ОХН, 1945, 170.
401. Китайгородский А. И. Изв. АН СССР, ОХН, 1947, 259.
402. Б о р и с о в А. Е. Изв. АН СССР, ОХН, 1951, 524.
403. Клебанский А. Л,, Т и т о в В. Д. ЖПХ, 20, 1005 (1947).
404. Jones W. Е,, R о s s е г R. J., Woodward F. N. J. Soc. Chem. Ind., 68,
258 (1949).
405. Lemaire H., L u c a s H. J. J. Am. Chem. Soc., 77, 939 (1955).
406. D r e f a h 1 G., H e u b 1 e i n G., W i n t z e r A. Angew. Chem., 70, 166 (1958).
407. Несмеянов A. H., Борнео в A. E.,С а в e л ь e в а И.С., О с и п о в aM. A.
Изв. АН СССР, ОХН, 1961, 1249.
408. DrefahlG., SchaafS. Ber., 30, 148 (1957).
409. Н о f m a n п К- A. Ber., 38, 1999 (1905).
410. G г i n е г G. Ann. chim. [6], 26, 363 (1892).
411. Д в о p к о Г. Ф., Ш и л о в E. А. Укр. хим. ж.,’28, 833 (1962).
412. Carothers W. Н., Jacobson R. A., BerchetG. J. J. Am. Chem. Soc.,
55, 4665 (1933).
413. Пат. США 1963108 (1934); Zbl., 1936, I, 2439.
414. Hennion'G. F., N i e u w 1 a n d J. A. J. Am. Chem. Soc., 57, 2006 (1935).
415. С о n t a r d i A., C i о с с a B. Zbl., 1937, I, 3872; Ric. Sci. progr. techn. [2J, 7, II,
610 (1936); итал. пат. 340195 (1936); Zbl., 1937, I, 4557.
416. К i 1 1 i a n i D. В., Hennion G. F., N ieuwland J. A. J. Am. Chem. Soc.,
58, 1658 (1936).
417. Губен И. Методы органической химии, т. III, вып. 1. М.—Л., Госхимтехиздат,
1934, стр. 239.
418. Р г е v о s t Ch. Ann., chim. [10], 10, 377 (1928).
419. Несмеянов A. H., Борисов А. Е., Новикова Н. В. Изв. АН СССР,
ОХН, 1959, 1216.
420. Несмеянов А. Н., БорисовА. Е., Новикова Н. В. ДАН СССР, 94,
289 (1954).
421. БорисовА. Е., Новикова Н. В. Изв. АН СССР, ОХН, 1957, 1258.
422. Несмеянов А. Н., БорисовА. Е., Савельева И. С., Голубе-
ва Е. И. Изв. АН СССР, ОХН, 1958, 1490.
423. МацоянС. Г„ ЧухаджянГ. А., Вартанян С. А. ЖОХ, 30, 1206
(1960).
424. Венус-Данилова Э. Д., Фабрицы А. ЖОХ, 26, 1609 (1956).
425. Венус-Данилова Э. Д., Фабрицы.А. ЖОХ, 26, 1901 (1956).
426. Венус-Данилова Э. Д., Ф абрн цы А. ЖОХ, 26, 2458 (1956).
427. Фабрицы А., Венус-Данилова Э. Д. ЖОХ, 28, 3227 (1958).
428. Фабрицы А., Куб ал я И. ЖОХ, 30, 3604 (1960).
429. Фабрицы А., ГощинскийС. ЖОХ, 29, 81 (1959).
Присоединение солей ртути к непредельным, соединениям
181
430. Ф абрицы А., К у б а л я И. ЖОХ, 31, 476 (1961).
431. Б а б а я н А. Г., Г.р н г о р я н А. А. ЖОХ,„ 26, 1945 (1956).
432. Д в о р к о Г. Ф. Укр. хим. ж., 28, 945 (1962).
433. S е h е г А. С. А., 54, 8623 (1960); Arch. Pharm., 292, 519 (1959).
434. Л е в и н а Р. Я., Костин В. Н. ДАН СССР, 97, 1027 (1954).
435. Левин а Р. Я., Костин В. Н. ЖОХ, 23, 1054 (1953).
436. Левина Р. Я., К о с т и и В. Н., Тартаковский В. А. Вестник МГУ, сер.
физ.-мат. и естеств. наук, К® 1, 77 (1956).
437. Левина Р. Я., Костин В. Н., Тартаковский В. А. ЖОХ, 27, 881
(1957).
438. Левина Р, Я-, Костин В. Н., Ш а на з а р о в К. С. ЖОХ, 29, 40 (1959)
439. Л е в н н а Р. Я-, Костин В. Н., Тартаковский В. А. ЖОХ, 26, 2998
(1956).
440. Левина Р. Я-, Костин В. Н., Г ембицкий П. А., Виноградов А. Д.
Вестн. МГУ, № 1, 67 (1961).
441. Левина Р. Я., Костив В. Н., Ким Д я й Гир, УстынюкТ. К- ЖОХ,
29, 1956 (1959).
442. Левина Р. Я.,' Шанаэаров К. С., Костин В. Н., Трещова Е. Г.
ЖОХ, 32, 2637 (1962).
443. Левина Р. Я., Мез ен нова Н., Лебедев О. В. ЖОХ, 25, 1097 (1955).
444. Несмеянова О. А., Лукина М. Ю„ Казанский Б. А. ДАН СССР,
153,114 (1963).
445. Несмеянова О. А., Лукина М. Ю., Казанский Б. А. ДАН СССР, 153,
357 (1963).
446. Пахомов В. И: Кристаллография, 5, 800 (1960),
447. Пахомов В. И. Кристаллография, 7, 456 (1962).
448. Edsall J. Т., Maybury R. Н., Simpson R. В., Straessle R. J. Am.
Chem. Soc., 76, 3731 (1964).
449. E 1 d j a r n L., Jell urn E. Acta Chem. Scand., 17, 2610 (1963).
450. C h a u d h u r i А. К., M a 1 1 i k K. L., Das M. N. Tetrahedron, 19, 1981 (1963),
451. Аракелян С. В., Рашидян Л. Г., Данг ян M. T. Изв. АН Арм. ССР,
сер. хим. наук, 17, 173 (1964).
452. Аракелян С. В., Дангян М. Т., Аветисян А. А. Изв. АН Арм. ССР,
15, 437 (1962).
453. Fanta G. F. J. Org. Chem., 29, 1610 (1964).
Глава VII
ДИАЗОМЕТОД СИНТЕЗА
РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Удобным и универсальным методом синтеза ароматических соединений
ртути является предложенный Несмеяновым [1,2] диазометод: разложение
двойных солей галогенида арилдиазония и галоидной соли ртути порошком
меди [можно также применять «медную бронзу» (Naturkupfer С.)]. Реакция
проводится в ацетоне, этилацетате или спирте, а также в воде и дает ртутно-
органические соли [1]
ArN2X- HgX2 + 2Cu ArHgX + 2CuX + Na,
При ведении реакции с избытком меди и добавлении водного аммиака
получаются полнозамещенные ртутноорганические соединения [2]
2ArN2X-HgX2 + 6Cu + 6NH3—» Ar2Hg + 6CuX-NHa + 2N2,
В том и другом случае обычно получают выходы 45—75% (от теорет.), в от-
дельных случаях — и выше.
Для получения этим методом соединений с сильно отрицательными
заместителями (нитрогруппа, карбоксил, сульфогруппа, два атома хлора)
реакцию нужно вести при сильном охлаждении и особенно тщательно забо-
титься. о перемешивании [3].
Отличием диазометода от методов синтеза ртутноорганических соедине-
ний посредством магнийорганических соединений и через амальгаму натрия
является возможность получения веществ с такими заместителями, как
NO2, СООН, SO3H, СООСН3, не индиферентными к реактиву Гриньяра или
натрию. В отличие от меркурирования диазометодом получаются соедине-
ния, свободные от изомеров и замещенные в выбранное положение.
Синтез ртутноорганических (и вообще металлоорганических) соединений
диазометодом, как при разложении двойных диазониевых солей с галогени-
дами ртути действием постороннего восстановителя, так и в других вариан-
тах диазометода (см. ниже), по-видиМому, осуществляется гомолитическим
механизмом. Свободный металл (металл-восстановитель в случае разложе-
ния двойных диазониевых солей) действует как восстановитель на катион
диазония, переводя его в диазоформу, распадающуюся затем гомолитически
с образованием радикала арила, который и арилирует металл:
Аг—N+=N:+Me-> Ar:N = N:Me+-> N2+Ar.+ Me+
Такое воздействие металлов на разложение диазониевых солей под-
тверждается образованием при этом обычных продуктов гомолитического
Диазометод синтеза ртутноорганических соединений
183
распада. Так, при разложении борофторидов арилдиазониев в нитробензо-
ле и эфире бензойной кислоты в присутствии порошков меди [41, кадмия,
цинка или серебра [51 образуется смесь главным образом о- и п-нитроби-
арилов и о- и n-дифенилкарбоновых кислот (после омыления) соответственно:
COOAlk
CeH6NaBF4 +
Си (Cd, Zn, Ag)
В отсутствие же металлов борофториды арилдиазониев распадаются
гетеролитически с образованием арилкатиона, вступающего в ароматическое
ядро, содержащее мета-ориентанты — в нитробензол и алкилбензоат —
в мета-положение к мета-ориентанту, с образованием ти-нитробиарилов
[6, 7, 8] и эфиров л!-дифенилкарбоновой кислоты и переэтерифицируя
[6, 8, 9] эти эфиры (Несмеянов, Макарова)
CeH5N2BF4 +
COOAlk
6
CeHsN2BF4 ц-
COOAlk
4* CeHgCOOCeHs
(см. также [10]).
Приписывая гетеролитический механизм реакции
p-XCeH4SbCl4-CeH5N2Cl 4- Fe -* p-XCeH4 (CeH5) SbCl3 + N2 + FeCl2
на основании увеличения ее скорости с увеличением электронодонорных
свойств заместителя X, Реутов [11] предполагает, что и синтез других метал-
лоорганических соединений по реакции Несмеянова совершается гетероли-
тическим механизмом.
О механизме получения ртутноорганических соединений диазометодом
см. также [12—14].
Изучены спектры двойных диазониевых солей с сулемой в видимой
ультрафиолетовой [15] и инфракрасной [16] областях.
Синтез ртутноорганических соединений посредством диазометода совер-
шается в два приема:
а) получение двойной соли сулемы (или иного галогенида окисной ртути)
и хлористого (или другого галогенида) арилдиазония;
б) разложение двойной соли медью.
184
Ртутноорганические соединения. Лит. стр. 193—194
ПОЛУЧЕНИЕ ГАЛОГЕНИДА АРИЛРТУТИ
Получение двойной соли хлористого арилдиазония
и .сулемы
Эту операцию производят по одному из следующих способов:
1) 1 моль амина (если амин твердый, но низкоплавкий, лучше его пред-
варительно расплавить) или его хлористоводородной соли заливают 300 мл
крепкой соляной кислоты и, если надо, греют до полного переведения в соль.
После охлаждения в смесь вводят 300 г мелко измельченного льда и диазо-
тируют при энергичном перемешивании и иногда наружном охлаждении
быстрым вливанием насыщенного раствора (70 г) нитрита натрия. Получен-
ный диазораствор сразу вводят при перемешивании в раствор 270 г сулемы
в 270 мл крепкой соляной кислоты, к которому добавлено 300 г льда. Образо-
вавшийся осадок двойной диазониевой соли отсасывают, промывают водой,
спиртом и эфиром и сушат на воздухе. Этот способ работы рекомендуется
в наиболее обычных случаях. Он дает очень чистую диазониевую соль с вы-
ходом 80—90 %.
2) В раствор 270 г сулемы в 300 мл конц. предварительно охлажденной
до —10-;---15° С соляной кислоты прибавляют 300 г мелко измельченного
льда и при энергичном перемешивании и наружном охлаждении снегом
с солью приливают насыщенный раствор нитрита натрия, взятый с избытком
—20%. В полученный таким образом раствор сразу вливают предваритель-
но охлажденный раствор 1 моля амина в эфире (минимум тройное количест-
во) при энергичном встряхивании и наружном охлаждении. Встряхивание
и растирание продолжают в течение нескольких минут после сливания
растворов. Выпавший осадок растирают, отсасывают и промывают, как
сказано при описании первого способа. Так работают в случае относительно
растворимых или низкоплавких двойных солей (например, карбалкоксиза-
мещенных). В этом случае первый способ дает относительно низкие выходы
иногда жидкой двойной соли.
3) Солянокислую соль амина растворяют в минимальном количестве
95%-ного спирта и, добавив 2—3 капли конц. соляной кислоты, диазотиру-
ют небольшим избытком амил’нитрита. Диазораствор вливают в раствор эк-
вимолярного количества сулемы в объеме эфира, превышающем раза в 3—4
объем употребленного спирта. Выпавшую отсосанную двойную соль промы-
вают спиртом и эфиром. Этот метод дает количественный выход двойной
соли, но требует затраты большего количества растворителей и работы с
большими объемами. Применяется в случае дорогих аминов.
Диазониевые соли следует по возможности применять в день синтеза.
В случае необходимости хранения их не следует оставлять в закрытой посу-
де и рекомендуется рассыпать тонким слоем. При соблюдении этих условий
можно безопасно работать с сотнями граммов двойных солей.
Приготовление порошка меди
Порошок меди готовят, просеивая 60 г цинковой пыли в холодный рас-
твор 250 г медного купороса в 1,5—2 л воды. Затем взвесь, тщательно пере-
мешивая, отсасывают и тщательно промывают осадок водой, спиртом и
эфиром и высушивают его на воздухе. Медь должна иметь чистый цвет
(не темноватый).
Из потемневшей меди чистая медь может быть регенерирована промыва-
нием разбавленной соляной кислотой (и затем водой, спиртом и эфиром,
как сказано выше).
Диазометод синтеза ртутноорганических соединений
185
Вместо меди можно при разложении двойных солей применять иные вос-
становители (Hg, Zn, SnCl3 и т. д. [17—22]). Однако меДь дает лучшие ре-
зультаты.
Разложение двойной диазониевой соли
в органических растворителях
Двойную соль вносят небольшими порциями в охлаждаемую снегом
с солью, хорошо механически перемешиваемую взвесь порошка меди (50%
избытка) в растворителе (большей частью — в ацетоне) (30—50 мл на каждые
10 г соли). Температуру во время разложения лучше держать ниже —5° С.
По окончании разложения растворитель выпаривают, твердый осадок про-
мывают небольшим количеством эфира и извлекают из него ртутнооргани-
ческую соль подходящим растворителем (обычно бензол, ксилол или ацетон),
которую перекристаллизовывают из одного из перечисленных растворите-
лей или из хлороформа или спирта. Этим методом с хорошими выходами
были получены ртутноорганическ^е производные углеводородов: бензола
[1, 17, 23—27], его гомологов (о-[1, 28, 29], м-[28—30], п -[1, 26, 28] толил-,
2,5-ксилил-[31] меркургалогениды), нафталина (а- [1, 28], 0-11, 32] нафтил-
меркурхлориды), бифенила [33, 34], трифенилметана и трифенилкарбинола
[35], галоидзамещенных бензолов (о- [36], п-[1,27, 37] хлорфенил-, 2,5-ди-
хлорфенил [1, 3], о- [27], п- [1, 271 бромфенил-, м- [361, п-иодфенил-[ 1 ]
меркурхлориды и о-иодфенилмеркуриодид 1381), нитрозамещенных бензо-
лов (о- [3], ж-[31, л-[3, 26] нитрофенилмер кур хлор иды и 2-хлормер кул-4-нит-
ротолуол [39]), .ж-трифторметилбензола [29, 40], эфиров фенолов (о-[1, 41,
42], п-[41,42] анизил-, о-[41], п-[1, 41] фенетилмеркургалогениды, 2-и 4-
хлормеркурдифениловый эфир [41], 2-хлормеркур-4-метилдифениловый
эфир [43]), фенолов (м- [44] ил-[1] оксифенилмеркурхлориды), карбоновых
кислот (о-хлормеркурбензойная кислота [3]) и их эфиров (метиловый
[1, 30], этиловый[30] эфиры о-[1,30, 36], .и-[30] и /г-[30] хлормеркурбензой-
ных кислот [30]), бензолсульфоновой кислоты [3] и ее производных [45],
антрахинона (монохлормеркурпроизводное из а-аминоантрахинона [46,471
и бис-хлормер кур производное из 1,5-диаминоантрахинона [48], стильбена
[49], тиоанизола [50], производных нафтооксазина [51] и нафтотиазина
[52], бензофенона [63], тиазола [87]. Худшие результаты дают ₽-ами-
нопиридин и продукты его замещения [53].
Разложение двойной диазониевой соли можно проводить и в воде, приме-
няя энергичное перемешивание [36]. При этом выходы ртутноорганических
соединений не хуже, чем при разложении в ацетоне, за исключением нитро-
и ему подобных замещенных, требующих глубокого охлаждения, недости-
жимого в воде.
Синтез хлористой фенилртути [1]. При смешивании 23 г анилина, 100 мл конц. соля-
ной кислоты, 300 мл воды, 18 г нитрита натрия и 68,5 а сулемы в 70 мл криц, соляной кисло-
ты с добавлением 70 г льда при работе по первому способу (стр. 184) получают 75 г двойной
соли хлористого фенилдиазония и сулемы, io г этой соли и 3 г меди в 40 мл ацетона, дают
(экстракция ацетоном) 3,9 г (51% от теорет.) хлористой фенилртути. Т. пл. 258°С. Если
разложение вести при охлаждении ниже —10°С, выход 77% [12].
Синтез хлористой я-этоксифенилртути [1]. При смешивании 4,28 г солянокислого
п-фенетидина, 20мл соляной кислоты, 80 мл воды, 2,2г нитрита натрия и 8,1 г сулемы в 15 мл
конц. соляной кислоты с добавлением 15 г льда получают при работе по первому способу
5,2 г двойной солн хлористого n-этоксифенилдиазония и сулемы, выпадающей через 5 мин.
после сливания растворов.
3,5 г этой соли и 1 ? меди в 10 мл спирта дают (извлечение хлороформом) 2,1 г (77%)
хлористой л-этоксифенилртути. Т. пл. 249—250°С.
Синтез хлористой /г-толилртути [1]. При смешивании 32,1 г и-толуидина, 150 мл конц.
соляной кислоты, 150 г снега, 22 г нитрита натрия и 81 г сулемы в 80 мл конц. соляной
кислоты и 80 г льда при работе по первому способу получают 75 г двойной соли хлористо-
го п-толилдиазония и сулемы.
186
Ртутноорганические соединения. Лит. стр. 193—194
6,5 г этой соли, 1,8 г меди, 30 мл ацетона дают (экстракция ацетоном) 3,3 г (66%) хло-
ристой л-толилртути. Т. пл. 238—239°С.
Синтез хлористой о-толилртути [1]. При смешивании 10,7 г о-толуидииа в 40 мл конц.
соляной кислоты, 40 г льда с 7 г нитрита натрия и 27 г сулемы в 30 мл конц. соляной кис-
лоты и 30 г льда получают по первому способу 23 г двойной соли хлористого о-толилдиазо-
иия и сулемы.
4,25 г этой соли, 1,2 г меди в 20 мл спйрта дают (экстракция ацетоном) 2,15 г (66%)
хлористой о-толилртути. Т. пл. 143°С.
Синтез хлористой га-хлорфенилртути [1]. При смешивании 10 г n-хлоранилина в 40 мл
эфира и 21,3 г сулемы в 30 мл конц. соляной кислоты с добавлением 30 г льда и 7 г нитри-
та натрия получают при работе по второму способу (стр. 184) 28,7 г двойной соли хлористо-
го л-хлорфенилдиазоиия и сулемы. 16г.этой соли и 4,3 г меди в 50 мл ацетона дают (экстрак-
ция ацетоном, перекристаллизация из бензола) 5,6 г (46%) хлористой п-хлорфенилртути.
Т. пл. 240° С.
Синтез хлористой а-нафтилртутн [1]. При смешивании 14,3г ot-нафтиламина, 10 мл конц.
соляной кислоты, 100 г льда, 9 г нитрита натрия и 27 г сулемы в 40 мл конц. соляной кис-
лоты и 40 г льда получают 31 г двойной соли хлористого а-иафтилдиазония и сулемы свет-
ло-желтого цвета.
4,6 г соли и 1,6 г меди в 30 мл ацетона дают (экстракция ацетоном) 2,1 г хлористой
сс-нафтилртути (58%). Т. пл. 191°С. Описан синтез [3-изомера [32].
Синтез хлористой п-оксифеиилртути [1]. При смешивании 1,45 г солянокислого
n-аминофенола в 20 мл спирта с добавлением 1,5 г амилнитрита и 2,7 г сулемы в 60 мл эфира
при работе по третьему способу (стр. 184) получают 4,0 г двойной соли хлористого л-окси-
фенилдиазония и сулемы.
3 г соли и 1 г меди в 10 мл ацетона дают (экстракция ацетоном) 1,3 г (56%) хлористой
л-оксифенилртути. Т. пл. 226—227°С.
Синтез хлористой п-карбэтокснфеиилртути [30]. При смешивании 45 г этилового
эфира n-аминобеизойной кислоты, 170 мл соляной кислоты (1 : 5), 18 г нитрита натрия и
68 г сулемы в 68 мл конц. соляной кислоты и 70 г льда получают 101 г двойной соли
л-карбэтоксифенилдиазоиия и сулемы.
20 г соли, 5 г меди в 50 м охлажденного до —10°С ацетона дают (экстракция этилаце-
татом) 7 г (44,7%) хлористой n-карбэтоксифенилртути. Кристаллизация из бензола. Т. пл.
223’ С. •
При синтезе ртутноорганических солей с сильно отрицательными заме-
стителями в ядре (нитрогруппа, сульфогруппа, карбоксил, два атома хлора)
для получения хороших выходов необходимо при разложении медью двой-
ной соли применять крайне энергичное перемешивание (мешалка Витта)
и разложение вести в ацетоне или этилацетате (последний растворитель
в случае хлористой о- и zz-нитрофенилртути) при температуре не выше
—10° С. Лишь для получения о-хлормеркурбензойной кислоты необходимо
применять более глубокое’охлаждение —70° С [3].
При соблюдении этих условий получены вполне удовлетворительные вы-
ходы ртутноорганических соединений: хлористой о-нитрофенилртути 42%,
п- 56%, м- 60%, о-хлормеркурбензойной кислоты 39%, п-хлормеркурбензол-
сульфоновой кислоты 30%; хлористой|3,5-дихлорфенилртути 32% [3].
В случае карбокси- и сульфосоединений, не дающих двойных солей нуж-
ного типа, разложение производят, внося во взвесь меди в растворителе
эквимолекулярную смесь диазония и сулемы в растворителе в твердом виде.
В последнем случае необходимо' соблюдать крайнюю осторожность, так как
такие смеси весьма взрывчаты.
Получение хлористой о-(илн л-)нитрофеинлртути [3]. 60 г о-нитранилина растворяют
в смеси 150 мл конц. соляной кислоты и 75 мл воды, нагретой до 90°С. По охлаждении до
4-25° С вводят 250 г снега, причем температура падает до —20°С. При энергичном размеши-
вании мешалкой Витта внутрь жидкости сразу вливают раствор. 36,5 г нитрита натрия в
72 мл воды. Осадок, выпавший при охлаждении, немедленно растворяется. Через 1—2 мин.
в полученный диазораствор вводят сразу раствор 136 г сулемы в 80 мл конц. соляной кис-
лоты. Скоро начинается выпадение осадка и вся жидкость застывает в кашеобразную мас-
су. Осадок отсасывают, промывают минимальным количеством воды, затем спиртом и эфи-
ром. Выход 190 г.
В' охлажденную до —20’С энергично перемешиваемую мешалкой Витта взвесь 8,4 г
порошка меди в 100 мл этилацетата небольшими порциями вносят 20 г двойной соли
Диазометод синтеза ртутноорганических соединений
187
с такой скоростью, чтобы температура не поднималась выше —15°С. По окончании внесе-
ния смесь размешивают еще 30 мин. Осадок отсасывают и извлекают горячим ацетоном.
Ацетон и этилацетат отгоняют досуха. Соединенные осадки промывают эфиром. Кристал-
лизация из ацетона. Выход 6,5 г (41%). Т. пл. 185°С.
В этих же условиях получена хлористая n-нитрофенилртуть (из 69 г п-нитроанилина
получают 144г двойной соли хлористого n-нитрофенилдиазония и сулемы). 20 г двойной
соли после разложения в этилацетате дают 8,6 г (55%) хлористой n-нитрофенилртути. Кри-
сталлизация из нитробензола. Т. пл. 265° С.
Получение хлористой лг-нитрофенилртути[3]. 69 г л-нитроаиилина растворяют в емеси
150 мл конц. соляной кислоты и 150 мл воды, нагретой до 90° С. Затем раствор смешивают
с 500 мл воды и охлаждают до 4-7°С. При энергичном размешивании вливают раствор 35 г
нитрита натрия в 70 мл воды. Бывшая первоначально в осадке соль амина переходит в
раствор. Сразу вливают раствор 136 г сулемы в 136 мл конц. соляной кислоты. Все засты-
вает в кашу. Осадок отсасывают, промывают небольшим количеством воды, спирта, эфира.
Вес 210 г. Разложение ведут в тех же условиях, что и о- и n-изомеров, но вместо этилаце-
тата берут ацетон. Выход из 20 г двойной соли 9,2 г (59%). Т. пл. 235°С [3].
Получеииея-хлормеркуртрифеиилкарбииола [35]. 13,75 г мелко растертого п-аминотри-
фенилкарбинола размешивают в смеси 40 мл воды и 5 мл соляной кислоты и добавляют еще
20 мл крепкой соляной кислоты. Выпадает красный мелкий осадок соли, который диазо-
тируют раствором 5 г нитрита натрия в 15мл воды. Отфильтрованный диазораствор вливают
в раствор 25 г сулемы в 25 мл крепкой соляной кислоты. Выпавшую соль отсасывают, про-
мывают трижды эфиром и высушивают на воздухе. Выход 21 г (91%) соли состава
[(CeHsbCXOHJCeHaNaClbHgCla. К 20 г соли прибавляют 100 мл охлажденного этилаце-
тата, содержащего 5,5 г сулемы, и прн хорошем перемешивании и охлаждении добавляют
5 г порошка меди. Через 1 сутки растворитель отгоняют, остаток хорошо промывают
теплым петролейным эфиром и извлекают горячим уксусноэтиловым эфиром. Раствор
концентрируют и выделяют 10 г (40%), считая на амин, белых кристаллов. Перекристал-
лизация. из бензола. Т. пл. 158°С (соединение с 1 молем СвНв). Бензол удаляют при 140рС,
после чего т. пл. 93°С.
Получение иодистой о-иодфенилртути [38]. 27,4 г(0,125 моля) о-иоданилнна растворяют
при нагревании в 220 ли 10%-ной серной кислоты. Смесь охлаждают до 0°С, а затем к ней
медленно, при энергичном перемешивании, добавляют 8,7 г (0,125 моля) нитрита натрия.
Полученный диазораствор оставляют стоять во льду и К нему приливают насыщенный рас-
твор иодной ртути в иодистом натрии. Выпавший осадок двойной дназониевой солн промы-
вают водой, спиртом и многократно эфиром. Выход сырого продукта 75 г (75%). После пе-
реосаждения абс. эфиром из ацетона и нитрометана соль* плавится при 69—69,5°С (с раз-
ложением).-
44 г двойной соли, 33 г меди в 250 мл ацетона (не выше — 5°С). Экстракция ацетоном
(1 л) дает 10,26 г (35%) иодистой о-иодфеннлртути. После двукратной кристаллизации из
смеси нитрометана и бензола (1 : 1) т. пл. 154°С.
Разложение двойной дназониевой соли в воде [36]
Двойную соль хлористого арилдиазония и сулемы вносят небольшими
порциями в охлаждаемую до 0°С (льдом с водой) | хорошо механически
перемешиваемую взвесь порошка меди (50% избытка) в воде (50—60 мл на
каждые 10 г соли). Температуру во время разложения лучше держать около
0° С. По окончании разложения (через 4—5 час.) осадок отсасывают, промы-
вают небольшим количеством спирта, эфира и извлекают из него ртутноорга-
ническую соль (бензолом, ацетоном, или этил ацетатом). Перекристаллизо-
вывают из одного из перечисленных растворителей или из хлороформа или
спирта.
Разложением двойной дназониевой соли в воде с хорошими выходами,
практически с такими же, как и при разложении в ацетоне, а в ряде случаев
и с более высокими'выходами, были получены ртутноорганические производ-
ные углеводородов: бензола, его гомологов, нафталина, галоидозамещенных
бензолов, эфиров карбоновых кислот. Хлористая л-нитрофенилртуть полу-
чена с выходом 27%, что объясняется невозможностью применения глубоко-
го охлаждения [36] (ср. [3]).
Синтез хлористой феиилртути. 25 г двойной дназониевой солн, 7,5 г меди в 100 мл
воды дают (экстракция ацетоном) 10 г (52,8%) хлористой фенилртути. Т. пл. 251 °C.
Синтез хлористой /г-толилртути. 12,5 г двойной дназониевой соли, 3,5 г меди в 60 мл
воды дают (экстракция ацетоном) 6,47 г (68%) хлористой /г-толилртути. Т. пл. 237°С. *
188 Ртутноорганические соединения. Лит. стр. 193—194
Синтез хлористой а-нафтилртути. 14,1 г. двойной диазониевой соли, 4,95 г меди в 100 мл
воды дают (экстракция ацетоном) 7,4 г (66,8%) хлористой а-нафтилртути. Т. пл. 190—
191 °C.
Синтез хлористой лг-бромфенилртути. 13 г двойной диазониевой соли, 3,5 г меди и
90 мл воды дают (экстракция бензолом) 4,5 г (43,4%) хлористой м-бромфенилртути. Т. пл.
198°С.
Синтез хлористой и-карбэтоксифеиилртути. 22,2 г двойной диазониевой соли, 5,8 г
меди в ПО мл воды дают (экстракция этнлацетатом) 10 г (56%) хлористой л-карбэтоксифе-
нилртути. Т. пл. 220°С.
ПОЛУЧЕНИЕ ДИАРИЛРТУТИ
10 г двойной соли хлористого арилдиазония и сулемы смешивают с 8 г
порошка меди и заливают 50 мл ацетона, предварительно охлажденного. Ми-
нут через 5 после того, как реакция затихает, приливают равный объем
25%-ного водного аммиака, тщательно перемешивают в течение нескольких
минут и оставляют реакционную смесь на 1 сутки при комнатной температу-
ре. Затем ее греют 1—2 часа с обратным холодильником, приливают большой
избыток воды, чтобы высадить все ртутноорганическое соединение, осадок
отсасывают, промывают водой, затем небольшим количеством эфира и из-
влекают полученное диарилртутное соединение подходящим растворителем
(спирт, бензол, хлороформ). Перекристаллизация обычно из них же. Так
получены полнозамещенные ртутноорганические производные углеводоро-
дов (дифенил-[2, 54], ди-о-[2], ди-д{-[55], ди-и-[2, 56]толил-, дипсевдокумил-
[551, ди-а-[2], ди-0-нафтил-[32] ртуть), их галоидозамещенных (ди-и-фторфе-
нил-[58], ди-о-, ди-л{-[59], ди-п-160] хлорфенил-, бис-2,5-дихлорфенил-, ди-п-
бромфенил-[2, 38, 61], ди-о-[38], ди-п-иодфенил-[2] ртуть), их нитрозамещен-
ных (ди-и-нитрофенилртуть [2], бпс-(5-нитро-2-метилфенил)ртуть [39]), эфи-
ров фенолов (ди-о-[2,41], ди-ж-[57], ди-и- [41, 55] анизил-, ди-о- [55],
ди-п-[55]фенетил-, б«с-феноксифенил-[41]ртуть), галоидозамещенных эфи-
ров фенолов (бис-З-иод-4-метоксифенилртуть [62]), эфиров бензойных
кислот (меркур-бис-о- и -л-бензойнометиловые эфиры [30]), бифенила [41],
антрахинона [46] и пиридина [64].
Получение дифенилртути [2]. При смешивании 4,65 г анилина в 20,0 мл конц. соля-
ной кислоты, 20,0 г льда,3,5 г нитрита натрия в 7,0 мл воды, 13,5 г сулемы в 15,0 мл конц.
соляной кислоты с добавлением 15,0 г льда получают 20,35 г двойной соли хлористого фе-
нилдиазония и сулемы.
10 г этой соли, 8 г порошка меди, 40 мл ацетона и затем 40 мл 25% -ного аммиака дают
после извлечения спиртом 3,0 г (68%) дифенилртути. Т. пл. 125°С (перекристаллизация
из спирта).
Получение ди-о-толилртути [2]. При смешивании 10,7 г о-толуидина, 40 мл соляной
кислоты и 40 г льда, 7,0 г нитрита натрия в 14 мл воды, 27 г сулемы в 30 мл соляной кисло-
ты и 30 г льда получают 16,4 г двойной соли хлористого о-толилдиазоиия и сулемы.
10 г этой соли, 9 г медн, 50 мл ацетона и 50 мл аммиака после извлечения хлорофор-
мом дают 5,3 г ди-о-толилртути. Т. пл. 107°С.
Получение ди-а-иафтилртути [2]. При смешивании 4,78 г а-нафтил амина, 15,0 г конц.
соляной кислоты, 15,0 г льда, 2,3 г нитрита натрия в 7 мл воды, 9,09 г сулемы в 10 мл со-
ляной кислоты и 10 г льда получают 13,9 г двойной соли, хлор истого а-нафтилдиазония и
сулемы.
10 г этой соли, 8 г меди, 50 мл ацетона и50мл 25%-ного аммиака дают 2,6 г ди-а-наф-
тилртути. Т. пл. 249°С. Выход53%. Аналогично получена диф-нафтилртуть [65].
Получение ди-л-бромфеиилртути [2]. При смешивании 30 г n-броманилина в 75 мл
эфира, 47,1 г сулемы в 50 мл конц. соляной кислоты и 100 г льда и 20 г нитрита натрия
получают по второму способу 31 г двойной соли хлористого n-бромднфенилазония и сулемы
(с содержанием азота, несколько отличающимся от вычисленного).
1.0 г двойной соли, 8 г меди, 50 мл ацетона, 50 мл 25%-ного аммиака дают (экстракция
хлороформом, кристаллизация из ацетона), 3,0 г ди-п-бромфенилртути. Т. пл. 243—244°С.
Получение ди-й-иодфенилртути [2]. При смешивании 22 г n-иоданилина в 100 мл эфи-
ра, 30 а сулемы в 30 мл конц. соляной кислоты и 30 г льда и 10 г нитрита натрия получают
по второму способу 45 г двойной солн хлористого n-иодфенилдиазония и сулемы.
10 г этой соли, 8 г меди, 50 мл ацетона и 50 мл 25%-ного аммиака дают (экстракция
Диазометод синтеза ртутноорганических соединений
189
хлороформом) 3,9 г ди-п-иодфенилртути. Выход 70%. Кристаллизация из пиридина или
ксилола. Т. пл. 270—272°С.
Получение меркур-бмс-л<-бензойнометилового эфира [30]. При смешивании 21,7 г ме-
тилового эфира м-аминобензойной кислоты в 100 мл эфира и 39 г сулемы в 40 мл соляной
кислоты с добавлением 40 г льда и насыщенного водного раствора 15 г нитрита натрия
получают 46 г двойной соли хлористого л-карбометоксифенилдиазония и сулемы.
10 г этой соли, 2,5 г меди, 25 мл ацетона (при —10°С) и еще 5,5 г меди и 30 мл 25% -ного
водного аммиака дают 1,4 г меркур-бмс-л-бензойнометилового эфира. Перекристалли-
зация из уксусноэтилового эфира. Т. пл. 129°С.
При нагревании в течение 1 часа 30 мин. при 180° С смеси диазотирован-
ного в моногидрате серной кислоты а-аминоантрахинона с взвесью серно-
кислой ртути в этой же кислоте образуется прямо с почти количественным
выходом а,а'-диантрахинонилртуть [46]. Аналогично получают р,Р'-изомер
[46] (выход 94,7%). Эти случаи являются единственными примерами прямо-
го получения полнозамещенных соединений ртути из диазосоединения без
участия восстановителя и симметризатора.
Получение а,а'-диантрахиноннлртути [46]. 1,0 г а-аминоантрахинона растворяют в 30 мл
моногидрата серной кислоты при 603С в течение 3 час. и добавляют 7,5 г сухого нитрита
натрия. Полученный раствор диазония тонкой струей вносят в течение 15 мин. в тонкую
взвесь 0,73 г сернокислой окиси ртути в 100 мл моногидрата серной кислоты. Смесь нагре-
вают до 180°С и выдерживают'при этой температуре 1,5 часа. Массу охлаждают и выливают
в 300 мл воды. Образовавшийся осадок серого цвета отфильтровывают, промывают 3%-ной
серной кислотой до полного исчезновения реакции на ионную ртуть в фильтрате, затем
водой и кипящим спиртом для удаления возможной примеси в видеа-оксиантрахинона
Промытый осадок высушивают в шкафу при 110°С. Выход 1,35 г (98,5%). Кристаллизация
из уксусной кислоты. Т. пл. 275—276°С.
Хлористая арилртуть может быть получена действием металлической
ртути на соль арилдиазония в воде и других растворителях [12, 14, 18, 19,
66], однако только при применении энергично действующей мешалки осо-
бой конструкции соль арилртути образуется с выходом от 20 до 72% [66].
Способ Мак-Клюра и Лоуи, предложенный ими в 1931 г., не имеет, однако,
никаких преимуществ перед описанным выше методом Несмеянова, опубли-
кованным в 1929 г.
Получение хлористой феиилртути из хлористого фенилдиазония и металлической ртути
[66]. 5 г перегнанного анилина растворяют в 30 мл коиц. соляной кислоты и 50 мл воды.
Раствор охлаждают до 0°С и диазотируют обычным образом. Диазораствор дистиллирован-
ной водой доводят до объема 200 мл. В стакан емкостью 600 мл помещают слой ртути в
1,5см и сюда вливают диазораствор. Стакан помещают в ледяную баню, и температура в нем
поддерживается ниже 5°С в течение всего эксперимента. Ртуть и раствор перемешиваются
в течение 4 час. со скоростью 1000 об/мнн мешалкой, сделанной из стеклянной трубочки
диаметром 4 мм, имеющей форму якоря с отверстием на дне и многими капиллярными от-
верстиями по нижним сторонам полукруглых лопастей якоря. Осадок отсасывают, кипятят
в течение нескольких минут со 100 мл воды, что заставляет металлическую ртуть собраться
в комки и позволяет затем декантацией отделить ют нее осадок,который отсасывают и кри-
сталлизуют из ацетона. Т. пл. 251°С. Выход 45%.
Реакций арилазокарбоновокислого калия с сулемой приводит [671 к об-
разованию арилмеркурхлоридов с выходом до 54% (арил —фенил, л-толил,
и-бромфенил, п-нитрофенил, n-сульфоксифенил, а-нафтил). Судя по ха-
рактеру побочных продуктов, реакция идет по гомолитическому механизму.
Синтез хлористой феннлртутн [67]. 2 г калиевой соли фенилазокарбоновой кислоты
в течение 30 мин. присыпают при комнатной температуре к раствору 8,7 г сулемы (тройной
избыток) в 100 мл ацетона при механическом перемешивании. Перемешивание продолжа-
ют еще в течение 30 мин. Растворитель отгоняют, сухой отстаток отмывают от су-
лемы раствором хлористого натрия, промывают водой. Экстракция ацетоном дает 1,78 г
хлористой фенилртути (54%). Т. пл. 251—252°С. Вес каломели 1,03 г. Реакция калиевой
соли фенил азокарбоновой кислоты с бромиой ртутью дает бромистую фенилртуть Выход
40%. Т. пл. 275°С.
190 Ртутноорганические соединения. Лит. стр. 193—194
При действии фенилазокарбоновокислого калия на хлористую фенил-
ртуть получена [671 дифенилртуть с выходом 94%.
О реакции арилазокарбоновокислого калия с p-хлорвинилмеркурхлори-
дами, приводящей к хлористой арилртути, см. гл. VI, стр. 121.
При продолжительном перемешивании в четыреххлористом углероде
с металлической ртутью фенилазотрифенилметана [20, 21] и бензиламино-
диазобензола 168] образуется с небольшим выходом (15% в последнем случае
[68]) хлористая фенилртуть.
Образующаяся при разложении нитрозоацетанилида”в среде меченого
бензола (в присутствии металлической ртути) уксуснокислая фенилртуть
не активна [22].
СИНТЕЗ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ РТУТИ
ПРИ ПОМОЩИ ЖИРНЫХ ДИАЗОСОЕДИНЕНИЙ
Лиячометаи ЛРЯгиПТ’РТ г П'т.гпй. полной ISO] и /уэпмигэй J7OJ prynw (Г&п-
лерман) без участия восстановителя, образуя галоидную галоидметилртуть
по уравнению:
СТШ + HgX2 XCH2HgX 4- N2.
Реакцию ведут в эфире.
Дифенилдиазометан с сулемой реагирует аналогичным образом 169]:
(C6H5)2CN2 + HgCl2 N2 + (CeH6)2C (Cl) HgCI.
Будучи взят в избытке, диазометан с солями ртути по уравнению
XHgX + 2CH2N2—> 2NH- XCH2HgCH2X
дает дигалоидметилртуть [69].
быс-Иодметилртуть может быть получена, по Геллерману’ и' Ньюмену,
с выходом 75%, если в качестве растворителя вместо эфира взять
диоксан [71],
При действии же диазометана в эфирном растворе на ацетат ртути не
происходит выделения азота, но образуется в качестве главного продукта
реакции взрывчатое полимерное соединение (—HgCNN—) наряду с неболь-
шим количеством бмс-диазометилртути Hg(CHN2)2 [71а].
При взаимодействии диазометана с ртутноорганическими солями RHgCl
(R = С6Н5, СН3С6Н4, С6Н5СН2) образуются два полнозамещенных ртутно-
органических соединения [69]:
2RHgCl + 2CH2N2 2N2 + (2RHgCH2Cl) -> R2Hg + (ClCHs)2Hg.
Промежуточный продукт RHgCH2Cl устойчив в случае хлористой бензил-
ртути. При действии диазометана на арилртутные соли карбоновых кислот
также образуются достаточно устойчивые соединения типа RCO2CH2HgAr
[72, 73]. Бромистая трихлорметилртуть с диазометанрм образует трихлор-
Метилмонобромметилртуть [74] (см. также гл. XII).
Получение хлористой хлорметилртути [69]. В раствор 13,6 г сулемы в 200 мл эфира
при размешивании пропускают эквимолекулярное количество диазометана (2,1 г), полу-
чаемого медленным прибавлением 12 г нитрозометилуретана к смеси 60 мл эфира, и
18 мл 25%-ного раствора едкого кали в метиловом спирте с такой скоростью, чтобы реак-
ционная смесь была бесцветна. Быстро происходит реакция с выделением газа, под
конец выделяются кристаллы. После испарения эфира остается хлористая хлорметил-
ртуть. Выход количественный. Кристаллизация из спирта. Т. пл. 131°С. .
Диазометод синтеза ртутноорганических соединений
191
Дихлорметилртуть (ClCH2)2Hg получается также при введении 2 молей
дназометана в эфирный раствор 1 моля сулемы. По окончании введения рас-
твор должен быть желтым от.избытка диазометана. Раствор фильтруют от
слёдов каломели и быстро испаряют в токе сухого воздуха. Остается слегка
окрашенное масло, закристаллизовывающееся через несколько дней. Пе-
рекристаллизация из эфира. Т. пл. 37—40° С.
Оба вещества очень ядовиты и дают ожоги кожи.
Диазотирование солянокислого метиламина амилнитритом в присут-
ствии сулемы и меди дает с незначительным выходом хлористую хлорметил-
ртуть [751.
При взаимодействии сулемы и диазоуксусного эфира [75] по уравнению
4CHN2COOC2H5 + 3HgCl2 -^QCHaClCOOQHs + Hg (ClHgCClCOOC2H4)2 + 4Na
образуется продукт замены диазогруппы на HgCl и С1, одновременно проис-
ходит меркурирование а-водородного атома.
Руководствуясь формальной аналогией электронной структуры трифенил-
фосфинметилена (СвН5)3Р—СН2 с таковой диазометана, Сейферт и Грим
[76, 771 ввели трифенилфосфинметилен в реакцию с бромной ртутью в эфире
при комнатной температуре. Продукт реакции высажен в виде бис-трибром-
меркуроата [(C6H5)3P+CH2l2Hg(HgBr3)2_, бпс-тетрафенилбората или рей-
неката.
СИНТЕЗ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ДЕЙСТВИЕМ АРИЛГИДРАЗИНОВ НА СОЛИ И ОКИСЬ РТУТИ
Арилгидразины окисляются окисью и солями ртути, арилируя их
(Э. Фишер). Так, фенилгидразин реагирует с окисью ртути, образуя с зна-
чительным выходом дифенилртуть [78, 79].
Получение дифенилртути из фенилгидразина. К эфирному раствору 10 г фенилгидра-
зина прибавляют избыток желтой окисн ртути. Тотчас выделяется азот. После испарения
ефира остается 4 г дифенилртути. Т. пл. 125° С.
В этой реакции исследована также возможность применения и других
растворителей [80].
При действии фенилгидразина на водный раствор меркурацетамида обра-
зуется наряду с другими продуктами дифенилртуть [81].
При действии арйлгидразинов на уксуснокислую ртуть в присутствии
уксуснокислой меди (очевидно, через промежуточное образование диазосо-
единения [82]) с хорошим выходом получается уксуснокислая ар ил ртуть
[83]. Так получена уксуснокислая фенил-, n-нитрофенил- и п-бромфенил-
ртуть. Уксуснокислая фенилртуть действием фенилгидразина переводится
в дифенилртуть.
Получение уксуснокислой фенилртути [83]. К подкисленному 3 мл конц. уксусной кис-
лоты раствору 31,8 г уксуснокислой ртути в 200 мл воды прибавляют концентрированный
раствор 1 г уксуснокислой меди. Раствор нагревают до 90° С и при хорошем перемешивании
постепенно прибавляют к нему 5,5 г фенилгидразина. Мгновенно наступающая реакция
сопровождается выделением азота и металлической ртути. Раствор принимает лилово-крас-
ную окраску, исчезающую только после прибавления всего фенилгидразина. Для оконча-
ния реакции смесь нагревают; после охлаждения Образовавшийся осадок отсасывают и
кристаллизуют из 5%-иой уксусной кислоты в присутствии животного угля. Выход 11,8 а
(70%). Т.пл. 146-147°С.
192
Ртутноорганические соединения. Лит. стр. 193—194
СИНТЕЗ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ЧЕРЕЗ ГИДРАЗОНЫ
Как показали Несмеянов, Реутов и Лосева [84—86], гидразоны альде-
гидов и кетонов легко реагируют с уксуснокислой ртутью при умеренной
температуре (на холоду или при 70—90° С) в среде воды, метанола или аб-
солютного бензола с выделением азота, закисных солей ртути и образова-
нием ртутноорганических соединений, в которых ртуть находится при вто-
ричном или третичном атоме углерода. Реакция, очевидно, проходит через
промежуточное образование диазосоединений, после выделения азота, из
которых происходит «сопряженное присоединение» ацетоксимеркургруппы
по освобождающимся валентностям и, в зависимости от рода растворителя,
соответствующего электроотрицательного остатка: ацетоксигруппы в слу-
чае бензола, алкоксигруппы в случае спирта, гидроксила в воде. В послед-
нем случае образуются, однако, не а-оксиалкильные производные ртути, а
производные простых эфиров:
AlkOH yHgOCOCH3 < РР'Г/ -LHcrrt (ОСНГНоК J_ мл
RR'C=NNH2+Hg(OCOCH3)2 — xOAlk н2о Г /HgOCOCH3
RR'C=NNHa + Hg (ОСОСНз) X J2 в бензоле HgOCOCHs 2 > N2 + Hg2 (OCOCH3)2 + RR'C/ OCOCHs
2
В большинстве случаев происходит образование димеркурированных
соединений независимо от того, берется ли в реакцию эквимолекулярное или
меньшее количество ртути за счет меркурирования по соседнему углероду:
R"OH
RC=NNH2 + Hg(OCOCH3)2------► RC— HgOCOCHa
R'-CH2 P^OR"
R'—CH— HgOCOCHa
R" = H, Aik или CH3CO.
Реакция применена к гидразонам альдегидов и кетонов алифатического
(ацетальдегида, ацетона, метилэтилкетона, бутирона), алициклического
(циклопентанона, циклогексанона, 4-метилциклогексанона, камфоры) и
ароматического рядов (о-нитробензальдегида, бензофецона). Выходы в боль-
шинстве случаев хорошие, в отдельных случаях достигают 96—98%.
Реакция гидразонов, циклогексанона и 4-метилциклогексанона с уксус-
нокислой ртутью в водной среде приводит к образованию мономеркуриро-
ванного циклогексена-1 и 4-метилциклогексена-1.
Синтез а.а'фф'-тетрахлормеркурдиизопропилового эфира [85].
ClHgCH2 HgCK
С |О.
СНз А
К нагретому до 70° С раствору 44 г (0,14 моля) уксуснокислой ртути в 250 мл воды
прибавляют по каплям при интенсивном механическом перемешивании 5 г (С .97 моля)
свежеперегнанного гидразона ацетона. Мгновенно начинающаяся реакция сопровождается
разогреванием, выделением азота и закисной уксуснокислой ртути. После прибавления
всего гидразона закисная уксуснокислая ртуть полностью восстанавливается до метал-
лической ртути. Реакционнук} смесь перемешивают еще 5 мин., затем отфильтровывают.
Диазометод синтеза ртутноорганических соединений
193
от металлической ртути. При обработке фильтрата 10%-ным раствором хлористого калия
выпадает белый объемистый осадок а,а',р,р -тетрахлормеркурдиизопропилового эфира,
вес 17 г. Вещество (после двукратного переосаждения водой из ацетонового раствора)
разлагается выше 150° С, не плавясь.
Синтез 1-(ацетоксимеркур)циклогексена-1 и 1-(хлормеркур)циклогексена-1 [86]. К на-
гретому до 90°С раствору 28,5 г (0,09 моля) уксуснокислой ртути и 1 г (0,005 моля) уксус-
нокислой меди в 250 мл воды прибавляют по каплям при интенсивном перемешивании 5 г
(0,045 моля) гидразона циклогексанона. Реакция сопровождается разогреванием, выделе-
нием азота и закисной уксуснокислой ртути. Реакционную смесь отфильтровывают и
фильтрат охлаждают. Выпадают белые кристаллы 1-(ацетоксимеркур)циклогексена-1,
116 ^Нб^С^’ СЧИТЭЯ На гиДРазон- Т. пл. (после перекристаллизации из метанола)
Водный фильтрат, оставшийся после выделения 1-(ацетоксимеркур)циклогексена-1,
обрабатывают 10%-ным раствором хлористого калия. Выпадает белый хлопьевидный
осадок 1-(хлормеркур)циклогексена-1; вес 5 г. Т. пл. (после перекристаллизация из то-
луола) 191—192°С.
ЛИТЕРАТУРА
1. Несмеянов А. Н. ЖРФХО, 61, 1393 (1929); Вег., 62, 1010 (1929).
2. Несмеянов А. Н., К а н Э. И. ЖРФХО, 61, 1407 (1920); Вег., 62, 1018 (1929).
3. Несмеянов А. Н., Глушнев Н. Ф., Епифанский П. Ф., Ф л е г он-
то в А. М. ЖОХ, 4, 713 (1934); Вег., 67, 130 (1934).
4. Макарова Л. Г., Матвеева М. К. Изв. АН СССР, ОХН, 1960, 1974.
5. Н е с ме я н о в А. Н., Макарова Л. Г., Матвеева М. К- Изв. АН СССР,
ОХН, 1961, 1898.
6. Н е с м е я н о в А. Н., М а к а р о в а Л. Г. Изв. АН СССР, ОХН, 1947, 213.
7. Макарова Л. Г., Матвеева М. К., ГрибченкоЕ. А. Изв. АН СССР,
ОХН, 1958, 1452.
8. N esmeya по w A. N., М а к а г о w a L. G., Tolstaya Т. R. Tetrahedron,
1, 145 (1957).
9. Макарова Л. Г., ГрибченкоЕ. А. Изв. АН СССР, ОХН, 1958, 693.
10. М а к а р о в а Л. Г. Сб.: Реакции и методы исследования органических соединений,
вып. 3. М., Госхимиздат, 1954, стр. 75.
И. Реутов О. А. Изв. АН СССР, ОХН, 1956, 943.
12. Н е с м е я н о в А. Н. Уч. зап. МГУ, 3, 291 (1934).
13. Waters W. A. J. Chem. Soc., 1937, 2007.
14. М a k i п F. В., W a t е г s W. A. J. Chem. Soc., 1938, 843.
15. Anderson L. С., Steedly J. W., jr. J. Am. Chem. Soc., 76, 5144 (1954).
16. К а з и ц ы н а Л. А., РеутовО. А., КикотьБ.С. ЖОХ, 33, 1564 (1963).
17. Австр. пат. 172320 (1952); С. А., 47, 3343 (1953).
18. D u п k е г М. F. W., Starkey Е. В., J е п к i п s G. L. J. Am. Chem. Soc., 58,
2308 (1936).
19. D u n к e r M. F. W., Starkey E. B. J. Am. Chem. Soc., 61, 3005 (1939).
20. Разуваев Г. А., Федотова E. И. ЖОХ, 21, 1118 (1951).
21. Федотова E. И. Уч. зап. ГГУ, 23, 127 (1952).
22. Разуваев Г. А., П е т у х о в Г. Г., 3 а т е е в Б. Г. ДАН СССР, 127, 803 (1959).
23. Англ. пат. 638565 (1950); С. А., 44, 9478 (1950).
24. Пат. США 2650238 (1953); С. А., 48, 10765 (1954).
25. Е t t е 1 V., N о s е к J. Chem, Listy, 47, 1413 (1953).
26. Пат. США 2593114 (1957); С. А., 47, 1188 (1953).
27. KharaschM. S., Pines Н„ L е v i n е J. Н. J. Org. Chem., 3, 347 (1938).
28. Wilde W. K- J. Chem. Soc., 1949, 75.
29. N e r d e 1 F., M а к о w e г S. Naturwiss., 45, 490 (1958),
30. Нес м e яновА. H., Макарова Л. Г. ЖОХ, 1, 598 (1931).
31. П e т p о в и ч П. И. ЖОХ, 29, 2387 (1959).
32. Синтезы органических препаратов, сб. 3. М., ОНТИ, 1935, стр. 92; сб. 2, М„ ИЛ, 1949,
стр. 557.
33. С о u г t о t С., В a s t a n i М. G. С. г., 203, 197 (1936).
34. Н u 1 1 F. В. J. Am. Chem. Soc., 60, 321 (1938).
35, Н е с м е я н о в А. Н., Эскин И. Т, Изв. АН СССР, ОХН, 1942, 116.
36. Несмеянов А, Н., Макарова Л. Г., ПоловянюкИ.В. ЖОХ, 1965, № 4
(в печати).
37. Gilman Н., Y а 1 е Н. L. J. Am. Chem. Soc., 72, 8 (1950).
38. Н е с м е я н о в А. Н., И с а е в а Л. С. ЖОХ, 1965 (в печати).
39. Петрович П. И. ЖОХ, 30, 2808 (1960); Ж. Всесоюзн. хим. общ., 5, 106 (1960).
40. Пат. США 2050075 (1936); Zbl., 1937, I, 1192.
41. L a m d a n S. С. А., 43, 4236 (1949); Rev. asoc. bioquim. Argentina, 14, 295 (1947).
194
Ртутноорганические соединения
42. С h а 1 1 е л g е г F., М i 1 1 е г S. A. J. Chem. Soc., 1938, 894.
43. С a m р b е 1 1 I. G. М., Turner Е. Е. J. Chem. Soc., 1938, 37.
44. Несмеянов А- Н., Торопова Е. М. ЖОХ, 4, 664 (1934).
45. Е 11 е 1 V., Nosek J. Coll. Czechoslov. Chem. Commun., 14, 74 (1949).
46. Ко з л о в В. В. ЖОХ, 18, 1376 (1948).
47. Козлов В. В., Б е л о в Б. И. ЖОХ, 25, 410 (1955).
48. К о з л о в В. В., Смолин Д. Д. ЖОХ, 19, 745 (1949).
49. D г е f a h LG., Stange G. J. prakt. Chem., 9, N 6, 311 (1959).
50. Эскин И. T. Изв. АН СССР, ОХН, 1947, 405.
51. Conti L., Leandri G. Boll. Sci. fac. chim. ind. Bologna, 15, 33 (1957).
52. Conti L., L e a n d r i G. Boll. Sci. fac. chim. ind. Bologna, 14, 60 (1956).
53. Несмеянов A. H., ЛуценкоИ. Ф. ЖОХ, 11, 382 (1941).
54. Gilman H., Marp le К. E. Rec. Trav. chim., 55, 133 (1936).
55. Несмеянов A. H., Новикова H. H. Изв. АН СССР, ОХН, 1942, 372.
56. Krause E., D i 11 m a r P. Ber., 63, 2401 (1930).
57. H i n t о n R. C.., Mann F, G., Todd D. J. Chem. Soc., 1961, 5454.
58. Б о p и с о в A. E. Савельева И. С., Сердюк С. P. Изв. АН СССР, ОХН,
1965 (в печати).
59. Я г у п о л ь с к и й Л. М., Ю ф а П. А. ЖОХ, 28, 2853 (1958).
60. Н е с м е я н о в А. Н., Кочешков К-А. ЖОХ, 1, 219 (1931); Вег., 64, 628 (1931).
61. Gilman Н., J о n е s R. G. J. Am. Chem. Soc., 63, 2482 (1941).
62. Gilman H., Moore F. W„ Jones R. G. J. Am. Chem. Sk>c., 63, 2482 (1941).
63. Э с к и н И. T. Изв. АН СССР, ОХН, 1942, 297.
64. Н u г d С. D., М о г i s s е у С. J. J. Am. Chem. Soc., 77, 4658 (1958).
65. Синтезы органических препаратов, сб. 2. М., ИЛ, 1949, стр. 222.
66. McClure R. Е., Lowy A. J. Am. Chem. Soc., 53, 319 (1931).
67. Н е с м е я н о в А. Н„ Р е у т о в О. А. Изв. АН СССР, ОХН, 1948, 316.
68. Р а з у в а е в Г. А., Овчинникова А. Г. ЖОХ, 23, 435 (1953).
69. Н е 1 1 е г m a n L., Newman М. D. J. Am. Chem. Soc., 54, 2859 (1932).
70. ФрейдлинаР. X., Несмеянов А. Н., Токарева Ф. А. ЖОХ, 7, 262
(1937); Вег.,’ 69,. 2019 (1936).
71. Wittig G., Schwarzenb ach К- Ann., 650, 1 (1961).
71а. W г i g h t A. N., Kramer A. W., S t e e 1 G. Nature, 193, N 4896, 903 (1963).
72. P f e i f f e r P., J a g e r H. Ber., 80, 1 (1947).
73. Pfeiffer P., Schulz e-B entrop R., La Roche К- H. Ber., 85, 232 (1952).
74. Фрейдлина P. X., Величко Ф. К- Изв. АН СССР, ОХН, 1959, 1225.
75. Н е с м е я н о в А. Н., П о в х Г. С. ЖОХ, 4, 958 (1934); Вег., 67, 971 (1934).
76. S е у f е г t h D., G г i т S. О. J. Am, Chem. Soc., 83, 1610 (1961).
77. G r i m S. O., Seyferth D. Chem. and Ind., 1959, 849.
78. Fischer E. Ann., 190, 99 (1877).
79. Fischer E. Ann., 199, 332 (1879).
80. Hardie R. L., Thomson R. H. J. Chem. Soc., 1957, 2512.
81. Forster. J. Chem. Soc., 73, 790 (1898).
82. Б p у к e p А. Б., Левин Б. В. ЖОХ, 28, 2725 (1958).
83. S e i d e O. A., S c h e r 1 i n S. M., В r a z G. I. J. prakt. Chem., 138, 55 (1933).
84. Несмеянов A. H., P e у т о в О. А., Л о с е в а А. С. ДАН СССР, 111, 835 (1956).
85. Несмеянов А. Н., Реутов О. А., Л о с е в а А. С., X о р л и н а М. Я. Изв.
АН СССР, ОХН, 1958, 1315.
86. Н е с м е я н о в А. Н., Р е у т о в О. А., Л о с е в а А. С., X о р л и н а М. Я. Изв,
АН СССР, ОХН, 1959, 50.
87. Travagli G. Gazz., 85, 926 (1955).
Глава VIII
СИНТЕЗ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ЧЕРЕЗ ГАЛОГЕНОНИЕВЫЕ СОЕДИНЕНИЯ
Ароматические ртутноорганические соединения могут быть получены
через галогенониевые (иодониевые, бромониевые, хлорониевые) соединения
действием на них металлической ртути (в случае иодониевых соединений при
повышенной температуре) или разложением двойных солей галогенидов
диарилгалогенониев с галогенидом ртути действием восстановителя.
Впервые Сэндин, Мак-Клюр, Ирвин [1] при кипячении хлористого
диарилиодония с металлической ртутью в воде или «-пропиловом спирте
получили хлористую арилртуть
Ar2JCl + Hg -» ArHgCl -|~ ArJ.
Так же получены хлористая фенил- и п-толилртуть.
Синтез хлористой фенилртути [1]. 3 г хлористого дифенил иодрни я в 150 мл «-пропи-
лового спирта кипятят с 2 г ртути в течение 5 час., затем фильтруют. После охлаждения
из фильтрата выкристаллизовывается 1,25 г (40%) хлористой фенилртути. Т. пл. 251°С [2J.
Макарова и Несмеянов [2, 3] в этих же условиях дают выход 50%. Бен-
зол и особенно ацетон оказались еще лучшей средой для этой реакции [4].
В последнем растворителе при кипячении в течение 3 час. из соответствую-
щих хлористых диарилиодониев получены ArHgCl, где Аг (в скобках—выхо-
ды): C6HS (76%), р-СН3С6Н4 (55%), р-СН3ОС6Н4 (53%), р-С1СвН4
(77%), р-ВгС6Н4 (75%), m-O2NC6H4 (40%), р-С2Н5ОСОС6Н4 (47%).
При разложении смешанных хлоридов диарилиодониев на ртуть пе-
реходит более электроположительный радикал; так, разложение соединения
C6H6(p-CH3OC6H4)JCl дает C6HBHgCl (выход 70%); разложение соединения
(p-CH3OC6H4)(p-C2H5O2CC6H2)JCl приводит к p-C2H5OCOC6H4HgCl (56%).
В случае небольшой разницы в электроотрицательности обоих радикалов
образуются оба галогенида арилртути:
CeH6 (p-JCeH<) JC1 + Hg -» C,H£HgCl 4- p.JC,H4HgCl.
Разложением йодистого дифенилиодония при 24-часовом взбалтывании
с металлической ртутью при 80° С в среде меченого бейзола (С614Н6) получе-
на с 15%-ным выходом неактивная иодистая фенилртуть [5].
Бромониевые и хлорониевые соли — галогениды (иодиды, бромиды и
хлориды) дифенилбромония и дифенилхлорония при взбалтывании с метал-
лической ртутью в изопропиловом спирте при комнатной температуре (иоди-
стый дифенилбромоний даже в воде при кипячении) арилируют металличе-
скую ртуть, образуя соответствующий галогенид фенилртути с выходом*до
196
Ртутноорганические соединения
45% (бромистая фенилртуть из бромистого дифенилхлорония получена с вы-
ходом 66%; Несмеянов, Толстая, Исаева [6, 7]).
В то же время в отличие от борофторидов арилдиазониев борфториды ди-
арилгалогенониев не образуют с металлической ртутью ртутноорганических
соединений [2, 3, 6], хотя они в состоянии арилировать менее благородные
(более нуклеофильные) металлы — таллий, олово, свинец, висмут,— спо-
собные превратить катион галогенония в форму, образующую ковалентную
связь с металлом.
Аналогично двойным солям галогенидов арилдиазониев с галоидной
ртутью также и двойные соли галогенидов диарилгалогенониев с галогени-
дом ртути при разложении их действием металлов-восстановителей в соот-
ветствующем растворителе образуют органические соединения ртути. Раз-
ложением двойных солей хлористого дифенилиодония и сулемы действием
ртути, порошков цинка или меди в ацетоне или спирте (для цинка и меди),
а также и в воде (для ртути) получена хлористая фенилртуть с выходом до
34%; при разложении в присутствии ртути в воде выходы АгН§С1.для Аг =
= С6Н6 — 70%, р-С1С6Н4 — 45%, р-СН3ОС6Н4 — 28%. При разложении
(C6Hs)2Cl-HgCl2 действием порошка железа в ацетоне или спирте главным
продуктом реакции (выход 39 и 21 %) является дифенилртуть [4]:
2 (СвНв)2СЬНбС12 + 2Fe (C6H3)2Hg + 2CeH3 + 2FeCl2 + Hg.
При разложении двойных солей йодистого дифенилхлорония и йодистого
дифен ил бр омон и я с иодной ртутью порошком меди в ацетоне на холоду об-
разуется фенилмеркуриодид с выходом 22 и 38,5% соответственно [8].
Разложение двойной солн йодистого дифенилхлорония и иодиой ртути. Получение
иодистой фенилртути [8]. 3,8 г двойной соли йодистого дифенилхлорония и иодиой ртутн
и 1 г меди в 100 мл сухого ацетона встряхивают при комнатной температуре 7 час. Полу-
чают 0,39 г иодистой фенилртути. Выход 22%. Т. пл. 270°С.
Разложение двойной соли йодистого дифенилбромонии и иодиой ртути. Получение
иодистой фенилртути [8]. 3,3 г двойной соли йодистого дифенилбромония и иодной ртути и 1 г
меди в 90 мл сухого ацетона встряхивают при комнатной температуре 4 часа. Получают
0,63 г иодистой фенилртути с т. пл. 270°С. Выход 38,5%.
Разложение хлористого дифенилбромония металлической ртутью. Получение хлористой
фенилртути [7, 8]. 0,4 г хлористого дифенилбромония, 2 мл ртути, 4 мл изопропилового
спирта встряхивают при комнатной температуре в течение часа. Получают 0,22 г.(47%)
хлористой фенилртути. Т. пл. 260°С.
Разложение бромистого дифенилхлорония металлической рутью. Получение бромистой
фенилртути. Раствор 0,2г борофторида дифенилхлорония в минимальном количестве воды,
насыщенной бромистым натрием, 1,5 мл ртути, 3 мл изопропионового спирта встряхивают
при комнатной температуре 30 мин.и Получают 0,17 г (66%) бромистой фенилртути.
Т. пл. 280°С.
ЛИТЕРАТУРА
1. S а п d i n R. В., М с С 1 u г е F. Т., Irwin F. J. Am. Chem. Soc., 61, 2944 (1939).
2. Макарова Л. Г., Несмеянов А. Н. Изв. АН СССР, ОХН, 1945, 617.
3. Несмеянов А. Н., Макарова Л. Г. Уч. зап. МГУ, 132, кн. 7, 109 (1950).
4. РеутовО. А., Пти цын а О. А., Ху X у н - в е н. ДАН СССР, 122, 825 (1958).
5. Р а з у в а е в Г. А., П е т у х о в Г. Г., 3 а т е е в Б. Г. ДАНСССР, 127, 803 (1959).
6. Несмеянов А. Н., Толстая Т. П., И с а е в а Л. С. ДАН СССР, 117, 996
(1957).
7. Н е с м е я н о в А. Н., Тол стая Т. П., И с а е в а Л. С. ДАН СССР, 125, 330
(1959).
8. Несмеянов А. Н., Р е у т о в О. А., Т о л с т а я Т. П., П т и ц ы и а О. А.,
Исаева Л. С., ТурчинскийМ. Ф., Бочкарева Г. П. ДАН СССР, 125,
1265 (1959).
Глава IX
СИНТЕЗЫ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ РТУТИ
ЗАМЕНОЙ НА РТУТЬ КИСЛОТНЫХ ОСТАТКОВ,
АТОМОВ ТЯЖЕЛЫХ МЕТАЛЛОВ
И НЕКОТОРЫХ МЕТАЛЛОИДОВ
В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Высокая степень электрофильности ртути выражается в том, что в ор-
ганических соединениях способен замещаться на ртуть не только водород
(собственно меркурирование), но и атомы металлоидов в некоторых груп-
пировках (меркурирование в широком смысле) [11.
Важной группой методов синтеза ароматических соединений ртути яв-
ляется замена на ртуть остатков кислот: борной в арил(алкил)борных
кислотах, сернистой в сульфиновых кислотах, йодноватой в иодосоедине-
ниях и карбоксила в карбоновых кислотах. Реакции эти, имеющие глав-
ную область применения в ароматическом ряду, в случае остатков СООН,
В(ОН)2, SO2H применимы и к синтезу алифатических соединений ртути.
Замена на ртуть атомов тяжелых металлов — олова, свинца, висмута,
таллия, кадмия, кремния, трехвалентных сурьмы и мышьяка — в их ариль-
ных (частью в алкильных и алкенильных) соединениях также может
служить для целей синтеза ароматических и жирных (предельных и не-
предельных) соединений ртути.
В отдельных, частных случаях были заменены на ртуть некоторые метал-
лоиды — бор (в иных, чем арил(алкил)борные кислоты, его органических
соединениях), кремний, фосфор, а также титан и хром в их органических
соединениях, что также рассматривается в настоящей главе.
ЗАМЕНА НА РТУТЬ ОСТАТКА БОРНОЙ КИСЛОТЫ,
А ТАКЖЕ БОРА
В ДРУГИХ ЕГО ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
При сливании кипящих водных растворов арил(алкил)-и диарилбор-
ных кислот и сулемы, бромной или уксуснокислой ртути образуются ртут-
ноорганические соли [21. Реакция эта течет количественно и крайне бы-
стро с любыми арилборными (в частности, диарилборными) кислотами:
RB (ОН)2 + HgX2 + Н2О -VRHgX + В (ОН)з +НХ,
R2BOH + 2HgX2 + Н2О -> 2RHgX + В (ОН)3 + 2НХ.
Из соответствующих арилборных кислот этой реакцией получены сле-
дующие ртутноорганические соединения: хлористая фенилртуть [2, 3],
о-толилртуть [4, 51, м-толилртуть [5, 6], п-толилртуть [5, 7], о-хлор-
фенилртуть [51, п-бромфенилртуть [5, 7, 14], м-[51 и п-[4,51 анизилртуть,
о-карбоксифенилртуть [5], jw-карбоксифенилртуть [5], н-карбоксифенил-
ртуть [5, 9], де-оксифенилртуть [7], а-[51 и ₽-[3,4] нафтилртуть, о-нитро-
фенилртуть [10], 4-метил-З-нитрофенилртуть [7], 4-метил-З-оксифенил-
198
Ртутноорганические соединения. Лит. стр. 213—216
ртуть [7], а также хлористая бензилртуть [6J, уксуснокислая п-бромфе-
нилртуть [5], о-, м- и п-анизилртуть [5], о-, м-, п-толилртуть [5], а-[5]
и Р-151 нафтилртуть, jw-карбоксифенилртуть, бромистая фенилртуть [3,11]
(см. также [121), 2-оксиметилфенилртуть [3], 2-формилфенилртуть [3],
2-метил-3,5-динитрофенилртуть [3], 2-нитро-4-аминофенилртуть [31, 3-нит-
ро-5-аминофенилртуть [3], а-фурилртуть [13], а-тиенилртуть [13].
Синтез хлористой лг-оксифенилртути [7]. К раствору 0,3 г л-оксифенилборной кислоты
в 100 мл воды при 50°С по каплям прибавляют насыщенный раствор сулемы до тех пор, пока
не прекратится образование осадка. Через 10 мин. осадок отсасывают и промывают горя-
чей водой до удаления избытка сулемы. Выход практически количественный. Кристалли-
зация нз 30%-ного спирта. Т. пл. 240—241,5°С.
Немногие арилборные кислоты, такие как З-нитро-5-карбометоксифе-
нилборная кислота, в водном растворе с трудом или совсем не реагирую-
щие с галоидной ртутью, с высоким выходом арилируют соль ртути, если
реакцию вести в уксуснокислом растворе в присутствии каталитического
количества хлорной кислоты [3].
Получение З-нитро-5-карбометоксифенилмеркурхлорида [3]. 0,1 г З-нитро-5-карбомет*
оксифенилборной кислоты растворяют в 3 мл ледяной уксусной кислоты при 80—90°С,
прибавляют 0,16 г уксуснокислой ртути и затем 0,05 мл 60%-иой хлорной кислоты.
После выдержки в течение 2 мин. при этой температуре раствор вливают в 15 мл воды,
содержащей 0,2 г хлористого калия. Тотчас образуется белый осадок. Выход 92%. Кри-
сталлизация из этилового спирта. Т. пл. 247—249°С. Так же получен [3] 2-нитро-4-
карбометоксифенилмеркурхлорид. Выход 97%; т. пл. 172—173° С.
Взаимодействие н-пропилборной кислоты с сулемой проходит в жестких
условиях: при нагревании вводе до 140—150°Св течение 20 час. образуется
хлористая н-пропилртуть с 55%-ным выходом [14]. Одновременно проходит
вторичная реакция:
QHjHgG + HgCI2 -> Hg2Cl2 + C3H7Cl.
При кипячении экзо- или эндо-5-норборнен-2-борной кислоты с экви-
молекулярным количеством сулемы в 83 %-ном водном ацетоне до почти
полного испарения ацетона получена с 80—83%-ным выходом хлористая
нортрициклилртуть [14а].
Экзо-изомер реагирует в сотни раз быстрее, чем эндо-изомер. Возмож-
но, что реакция идет через образование л-комплекса с последующим транс-
аннулярным вытеснением борной кислоты:
Хлористая со-стирилртуть с количественным выходом получена из стирил-
борной кислоты и сулемы при непродолжительном нагревании их в ацето-
не [15], то же — хлористая транс-р-хлорвинилртуть [8].
Замена на ртуть кислотных остатков и атомов тяжелых металлов 199
Другие моноалкилборные кислоты в водном растворе (30-минутное ки-
пячение) не реагируют с галогенидами ртути [3]. Однако реакция легко
проходит в ледяной уксусной кислоте при нагревании [3].
Получение я-октилмеркурхлорида [3]. 0,10 г «-октилборной кислоты растворяют в
2 мл холодной ледяной уксусной кислоты, прибавляют 0,26 г ацетата ртути и нагревают
до 90—100° С в течение 4—5 мин,, затем выливают в раствор 0,1 г хлористого натрия в
15 мл воды, Тотчас выпадает осадок к-октилмеркурхлорида. Выход 0,18 г (81%). Т. пл.
115—116° G.
Изобутилмеркурхлорид получен [3] таким же путем. Выход 50%.
Т. пл. 54—55° С.
Получение хлористой ферроцеиилртути [16]. К горячему раствору 0,16 г ферроценил-
борной кислоты в 20 мл воды добавляют водно-ацетоновый раствор 0,19 г сулемы. Тотчас
выпадает желтый осадок. Смесь нагревают несколько минут. Осадок отфильтровывают,
промывают водой, высушивают в вакуум-эксикаторе над пятиокисью фосфора. Получают
0,22 г хлористой ферроцеиилртути. Выход 7694. Т, разл. 190—192° С. После кристаллиза-
ции из ксилола т. разл. 192—194° С.
Хлористая 1-(Г-хлорферроценил)ртуть и бромистая 1-(Г-бромфер-
роценил)ртуть получены [17] аналогично из 1-(Г-галоидферроценил)бор-
ной кислоты (водно-спиртовый раствор) и водно-ацетонового раствора (со-
ответственно) сулемы или бромной ртути с выходом свыше 80%.
Если при синтезе ртутноорганических соединений через арил(алкил)-
борные кислоты вместо соли ртути взять окись ртути, получается с хоро-
шим выходом.полнозамещенное ртутноорганическое соединение [14].
Изучение [18] кинетики этой реакции (реакция бимолекулярная, пер-
вого порядка по каждому из компонентов) позволяет предполагать, что
реакция образования Ar2Hg проходит через промежуточное образование
ArHgOH:
АгВ (ОН)а + HgO + Н2О ArHgOH + В (ОН)3,
ArHgOH + АгВ (ОН)2 -* Ar2Hg + В (ОН)3.
Получение ди-я-толилртути. 2,72 г (2 моля) n-толилборной кислоты, 2,16 г (1 моль)
желтой окиси ртути в 50 мл воды при 100° С дают 2,7 г ди-и-толилртути. Т. пл. (после кри-
сталлизации из бензола) 236° С [14].
Из фенилборной кислоты и азотнокислой окиси ртути в присутствии
ацетата натрия количественно получена дифенилртуть [19], в присут-
ствии же йодистого калия, нитрата аммония и аммиака также с количест-
венным выходом получена иодистая фенилртуть [19].
м-Бромфенилборная кислота и азотнокислая окись ртути при 15-ми-
нутном кипячении в водном растворе, содержащем 1 % едкого натра, также
дают с количественным выходом ди-п-бромфенилртуть [19а]. Аналогично
получена ди-п-бромфенилртуть, содержащая Hg202 [19а].
В настоящее время метод синтеза ароматических ртутноорганических
соединений через борные кислоты применяется главным образом для иден-
тификации арил(диарил)борных кислот и вряд ли может иметь широкое
применение вследствие необходимости готовить исходные арилборные
кислоты (обычно реакцией Гриньяра). Возможно, что в применении к али-
фатическим соединениям метод получил новые перспективы, поскольку
широкий круг предельных и непредельных борорганических соединений
стал доступен за счет реакций присоединения к олефинам и ацетиленам
боргидридов и галогенидов.
О применении" арилборных кислот среди других арилирующих средств
для синтеза несимметричных полнозамещенных ртутноорганических
соединений см. гл. XII, стр. 231.
200
Ртутноорганические соединения. Лит. стр. 213—216
В некоторых случаях бор и в других его органических соединениях мо-
жет быть заменен на ртуть.
В мягких условиях триарил-[20, 21] и триалкил-[22, 231 бор отдает
сулеме все три радикала. Триэтилбор в водном растворе с сулемой [22],
окисью ртути (в щелочном растворе [24]), с ацетатом ртути (в растворе
диметилового эфира этиленгликоля [23, 24], в воде при комнатной тем-
пературе [251) дает диэтилртуть. Описана реакция триаллилбора с су-
лемой [261. Реакция триарилбора с сулемой в водноспиртовом растворе
проходит количественно с образованием хлористой арилртути (арил —
фенил, н-диметиламинофенил).
С HgO в воде для получения Alk2Hg введены в реакцию и другие
А1к3В (А1к— октил, винил, 1-гексенил, 1-октадеценил, октадецил) [22].
Тетраарилдибороксиды (арил — фенил [27], п-толил [28]) при взаимо-
действии в водно-спиртовом растворе на холоду с сулемой количественно
отдают ей все четыре арила:
Аг2ВОВАга + 4HgCl2 + 5НаО 4ArHgCl + 4НС1 + Н3ВО3.
Количественно переходят на ртуть все соединенные с бором арилы тет-
раарилборметаллов при взаимодействии с сулемой (в водном [20, 21, 291,
метанольном [20, 21, 29] или ацетоновом [30] растворах на холоду):
Me ВАг4 + 4HgCl2 + ЗН2О -> 4ArHgCl + MeCl + ЗНС1 + В(ОН)3
Me—литий, натрий, калий, аммоний). Так же ведут себя арилы триарил-
цианоборнатрия. Тетрафенилборнатрий с гидроокисью ртути в ацетоне
дает дифенилртуть и гидроокись фенилртути [29, 31], с ацетатом ртути в
водном щелочном растворе — дифенилртуть [32].
Синтез диэтилртути из триэтилбора и окиси ртути [24]. Нагретую до 75° С взвесь
0,05 моля окиси ртути в воде, содержащую 0,15 моля едкого натра, обрабатывают 0,05 моля
триэтнлбора при перемешивании мешалкой. Конец реакции заметен через 10 мин. после
растворения окиси ртути и выпадения на дно тяжелого масла при прекращении переме-
шивания. Перегонка масла дает с 95%-ным выходом диэтилртуть. Т. кип. 67—68° С (19 мм).
Реакция триэтилбора с ацетатом ртути в 1,2-диметоксиэтане (2-ча-
совое кипячение) дает диэтилртуть с выходом 66% [24].
Синтез хлористой фенилртути из тетрафенилборкалия и сулемы [30]. 0,1224 а тетрафе-
ннлборкалия растворяют в ацетоне, добавляют немного воды и затем ацетонового раство-
ра 0,5 г сулемы. Тотчас выпадает осадок хлористой фенилртути. Для полного разрушения
борорганического соединения смесь нагревают на водяной бане и ацетон отгоняют. Хло-
ристую фенилртуть отфильтровывают и промывают водой. Т. пл. 252° С. Выход 0,41 г
(95%).
Тетраэтилборнатрий с сульфатом или ацетатом ртути в водном щелоч-
ном растворе при нагревании дает диэтилртуть с выходом 80% [32].
бис-2,2 '-Дифенил енборат тетраметил аммония после разрушения его
кипячением в течение часа в ацетоновом растворе с 2 N серной кислотой
(реагирует, очевидно, образовавшаяся 2-дифенилборная кислота) и по-
следующего взаимодействия с сулемой в метиловом спирте образует хлори-
стую о-дифенилртуть [21].
кг»//—»тт \ HgCI2
.Nt(CH3)4-------> o-CeHsCeHaHgCl;
Замена на ртуть кислотных остатков и атомов тяжелых металлов
201
При проведении этого опыта в присутствии йодистого натрия образуется
£21] ди-о-дифен ил ртуть (o-CeH6CeH4)2Hg.
При обработке же бис-2,2'-дифениленбората тетраметиламмония щело-
чью в ацетоновом растворе и последующего кипячения с сулемой получен
с 51%-ным выходом о,о'-бпс-хлормеркурдифенил [21]
О'СЗ
\gC^ HgCl
а 3-часовое кипячение в спиртовом растворе в присутствии йодистого нат-
рия приводит с 90%-ным выходом к о,о'-дифениленртути [21], представля-
ющей собой тетрамер [33] (см. гл. II, стр. 32). ,
С другой стороны, при взаимодействии с металлической ртутью (при
продолжительном 70—80-часовом встряхивании в хлороформе лучше при
температуре — 10 ---15°С) тетраарилборметаллы (катион — литий,
калий, аммоний) отдают ртути только один арил, давая диарилртуть; три-
арилбор с металлической ртутью не реагирует [34]. В случаеМе[В(С6Н6)3Аг]
(Me—литий [35], калий [36], аммоний [36]) и 1л(ВС6Н5Аг3) [35],
(Аг — толил, а-нафтил) образуется Ar2Hg. Двойная соль Li [В(С6Н6)4] • 2ЫВг
[36] с металлической ртутью в хлороформе (70—80-часовое встряхивание}
образует хлористую фен ил ртуть.
ЗАМЕНА НА РТУТЬ ОСТАТКА СЕРНИСТОЙ КИСЛОТЫ
Открытая Петерсом [371 реакция взаимодействия с сулемой сульфино-
вых кислот
RSOaH + HgCIa RHgCl ф- SO2 + HC1
в свое время была довольно широко использована для синтеза ртутноорга-
нических солей, так как до открытия диазометода она была наиболее про-
стым способом синтеза тех изомеров ртутноорганических соединений с за-
местителем в ядре, которые не получаются прямым меркурирова-
нием. Реакция вначале была разработана для синтеза ароматических
соединений ртути, ныне же применяется й к синтезу алифатических, гете-
роциклических соединений и ртутных производных ценов. В некоторых
случаях реакция проходит на холоду, обычно же она идет с большим тру-
дом, чем взаимодействие с солями ртути арил(диарил)борных кислот (лишь
при кипячении в большинстве случаев не менее 1 часа).
Реакцией Петерса получены простейшие алкильные соединения ртути
[38]. \ .
Получение хлористой метилртути [38]. 50 г метилсульфината натрия в 200 мл этилового
спирта смешивают с 135 г сулемы в 600 мл спирта. Тотчас образуется белый осадок, из
которого хлорную ртуть отгоняют с паром. Т. пл. 170° С. Из фильтрата добавочная порция
соли метилртути может быть высажена иодистым калием в виде иодистой метилртути.
Т. пл. 152°С.
Таким же образом получены галогениды этил- и бутилртути [38]. Из
1-додецилсульфината натрия хлористая 1-додецилртуть получена с вы-
ходом 49,3% [39]. Рассматриваемой реакцией получены ртутноорганиче-
ские производные камфоры [40].
202
Ртутноорганические соединения. Лит. стр. 213—216
Синтез камфор- 10-меркурхлорида [40].
CH2HgCl
Камфор-10-сульфиновую кислоту (1 моль) кипятят 4—5 час. со спиртовым раствором
хлорной ртути (2 моля). Продукт переносят в воду; спустя 12 час. осадок собирают, про-
мывают спиртом и извлекают горячим хлороформом. После двукратной кристаллизации
из спирта, к которому прибавлено немного хлороформа, т. пл. 166° С.
З-Хлор- и 3-6ромкамфор-10-сульфиновые кислоты дают хорошие выхо-
ды ртутных производных по реакции Петерса лишь в пиридине в качестве
реакционной срёды, или же (значительно хуже), если взяты щелочные суль-
финаты вместо свободной кислоты [41].
Получение 3-хлоркамфор-10-меркурхлорида [41]. 9 г З-хлоркамфор-10-сульфиновой кис-
лоты кипятят с 18 г хлорной ртути в 30 мл пиридина в течение 3 час. Охлажденный рас-
твор, отделенный от металлической ртути, осторожно прибавляют к избытку разбавленной
соляной кислоты. Осадок собирают, промывают (под конец спиртом), сушат и извлекают
в аппарате Сокслета хлороформом, из которого по охлаждении выделяется чистое веще-
ство в виде длинных игл. Т. пл. 218—219° С. Выход 40—50%.
З-Хлор и 3-бромкамфор-л-сульфиновые кислоты с солями ртути по
реакции Петерса не реагируют [41].
Получение хлористой фенилртути из беизолсульфиновой кислоты [37]. Растворяют
3 г (1 моль) беизолсульфиновой кислоты в водном спирте и 6 г сулемы (более 1 моля) в во-
де. Кипящие растворы сливают вместе. С выделением сернистого газа образуется белый
осадок. Затем кипятят до исчезновения запаха сернистого газа, отсасывают, растворе-
нием в пиридине освобождают от металлической ртути, высаживают водой, промывают
спиртом и растворяют в горячем эфире. Концентрированный эфирный раствор по охлаж
дении выделяет хлористую фенилртуть. Т. пл. 251° С. Выход не указан.
При проведении реакции в воде в присутствии серной кислоты выход
хлористой фенилртути 73,6% [42] (см. также [43]).
В некоторых случаях лучшие выходы получаются, если не допускать
образования при реакции соляной кислоты, применяя вместо свободных
сульфиновых кислот их натриевые соли или ведя реакцию в пиридине.
Получение хлористой л-нитрофенилртути [44]. 8 а м-нитробензолсульфиновокислого
натрия растворяют в 40 мл воды и приливают раствор 21 г сулемы в 100 мл спирта. Затем
прибавляют ледяную уксусную кислоту в количестве, несколько большем, чем нужно
для выделения сульфнновой кислоты (2,3 мл), и смесь кипятят 20 час. Из образовавшегося
осадка извлекают хлористую м-нитрофеннлртуть ацетоном. Кристаллизация из спирта.
Т. пл. 236—237° С. Выход 42% (см. также [165]).
Так же получены хлористые о- и н-нитрофенилртуть [44].
Метод получения ароматических соединений ртути через сульфиновые
кислоты как нельзя лучше применим к галоидным производным бензола.
При кипячении соответствующих галоидбензосульфиновых кислот с тремя
эквивалентами уксуснокислой ртути в ледяной уксусной кислоте (после
отфильтрования солей закиси ртути) от прибавления воды или щелочи из
растворов выпадают уксуснокислые соли галоидфенилртути. Так получены
уксуснокислые о-, м-, n-хлор-, о-, м-, n-бром- и и-иодфенилртуть [45],
хлористая 2,4-дихлорфенил-, 2,5-дихлорфенил-, 2,4,6-трихлорфенилртуть
Замена на ртуть кислотных остатков и атомов тяжелых металлов 203
146]. Соответствующие хлористые соли получают прибавлением хлорис-
того натрия к водным или спиртовым растворам ацетатов.
Получение уксуснокислой я-хлорфенилртути [45]. К раствору 11,3 г свежеприготов-
ленной, еще сырой п-хлорфенилсульфиновой кислоты в 50 мл ледяной уксусной кислоты
приливают раствор 50 г уксуснокислой ртути (не менее 3 моль-зкв) в 100 мл ледяной уксус-
ной кислоты. Тотчас образуется белый осадок сульфината ртути, который кипятят в те-
чение 15 мин.; при этом ртутная соль полностью превращается в уксуснокислую п-хлорфе-
нилртуть. Ее отфильтровывают от одновременно образовавшихся солей закисной ртути и
высаживают уксуснокислую и-хлорфенилртуть прибавлением щелочи или воды. Выход
14,0 г. Т. пл. 193° С. Кристаллизация из спирта.
Маточный раствор от получения сульфнновой кислоты обрабатывают 100 мл 10%-иого
водного раствора уксуснокислой ртути. Выпавший сульфинат ртути (4,7 г) отсасывают,
смешивают с 40 мл ледяной уксусной кислоты и 4 г уксуснокислой ртути, нагревают до
кипения, уксуснокислую n-хлорфенилртуть изолируют, как описано выше. Добавочно
получено 1,5 г. Общий выход 15,5 г (70%).
Метод с успехом применим и для получения хлористой [37, 47—49]
(и других солец [501) п-толилртути, 5- и 4-хлормеркур-2-нитротолуола [511,
2-хлормеркур-4-нитротолуола [52] (с лучшим выходом, однако, получаемого
диазометодом [52]), ряда изомеров хлористой хлорнафтилртути [53], хло-
ристой этилфенилртути [54], хлористой о-бензилфенилртути [55), хлори-
стой 2,5-дихлорфенилртути [52], хлористой о-(1-нафтил)фенилртути [56],
6-окси-5-хлормеркуркумарина [57], 2-хлормеркур-3,4',5-триметилдифе-
нилметана из натриевой соли 2-(4-метилбензил)-4,6-диметилбензол-
сульфиновой кислоты [58], 4-стильбенилмеркуриодида [59].
Из ж-бензолдисульфиновой кислоты и сулемы (20-минутное кипячение
в щелочном растворе) лг-дихлормеркурбензол получен с 82%-ным выходом
[60].
Получение 1,3-дихлормеркурбензола [60]. Взвесь 4,70 г (0,0238 моля) .ч-беизолдисуль-
финовой кислоты в 10 мл воды, подщелоченную прибавлением 50% -иого раствора едкого
иатра, прибавляют к кипящему раствору 16,35 г (0,06 моля) сулемы в 50 мл воды. Через
0,5 мин. появляется творожистый осадок, и на водяной бане все загустевает в пасту. Через
20 мин. сильно кислый раствор разбавляют 50 мл воды и нагревают иа водяной бане 6 час.
Осадок отфильтровывают и промывают горячей водой до удаления солей ртути (0,015 моля
которых вымывается), затем кипящим спиртом и кипящим бензолом. Остаток, представ-
ляющий собой 1,3-бпс-хлормеркурбензол, весит 70,7 г. Выход 82%. Вещество ие плавится.
Кипячением в течение 2 час. лг-бензолдисульфината калия с уксусно-
кислой ртутью в ледяной уксусной кислоте получен [61] л1-ди ацетоксимер -
курбензол с выходом около 70%.
Лри 3-часовом кипячении щелочного раствора 1,8-антрахинондисуль-
финовой кислоты с водным раствором сулемы получен [62] 1,8-бис-хлор-
меркурантрахинон с выходом 85%.
Этим методом получены 2-хлормеркур-и 2,8-дихлормеркурдибензофуран
(63].
Получение 2-хлормеркурдибензофурана [63]. Горячий раствор 12,7 г (0,05 моля) ди-
беизофурансульфината-2-натрия в 150 мл воды быстро при перемешивании прибавляют
к 27,2 г (0,1 моля) сулемы в 200 мл кипящей воды. Получают 2-хлормеркурдибензофураи
с 82%-ным выходом. Кристаллизация из ацетона. Т. пл. 236,5—237°С.
Хлористая 2-пиридилртуть и 6-хлор-З-пиридилмеркурхлорид синтези-
рованы [64] из соответствующих сульфиновых кислот в водном растворе
кипячением в течение 15 мин. с сулемой и отстаиванием смеси в течение
2 час.
Реакция Петерса применима также и к синтезу ртутных производных
ценов. Так, хлормеркурферроцен и биохлормеркурферроцен легко могут
быть получены из соответствующих ферроценсульфиновых кислот [65]*
204
Ртутноорганические соединения. Лит. стр. 213—216
Получение хлормеркурферроцена [65]. К раствору 0,31 г (0,001 моля) натриевой соли
ферроценсульфиновой кислоты добавляют спиртовый раствор 0,27 г (0,001 моля) сулемы.
Сразу выпадает осадок хлормеркурферроцена. После высушивания вес 0,41 г. Выход ко-
личественный. Т. пл. 194—196° С.
Получение днхлормеркурферроцена [65]. При нагревании 0,32 г (0,0078 моля) спирто-
вого раствора натриевой соли ферроцендисульфиновой кислоты и 0,55 г (0,002 моля) су-
лемы выделяется в виде желтого осадка дихлормеркурферроцен. Выход 0,41 г (80%).
Из натриевой соли марганецтрикарбонилциклопентадиенилсульфино-
вой кислоты при 45-минутном кипячении с сулемой в водно-спиртовом рас-
творе получен с 80%-ным выходом хлормеркурциклопентадиенилмарга-
нецтрикарбонил [66].
Лоудон показал, что реакция Петерса не применима для установления
строения продуктов присоединения солей ртути к непредельным соеди-
нениям [67].
ЗАМЕНА НА РТУТЬ ОСТАТКА ЙОДНОВАТОЙ КИСЛОТЫ
Несмеяновым и Макаровой [68] найден метод синтеза ртутноорганиче-
ских гидроокисей арилированием окиси ртути ароматическими иодосоеди-
нениями: »
ArJO2.-{- HgO + AgOH -> ArHgOH -j- AgJOs.
Эта реакция аналогична синтезу иодониевых оснований по В. Мейеру:
ArJO2 4- ArJO 4- AgOH -> Ar2JOH 4- AgJO3.
Метод был приложен к синтезу ртутноорганических соединений углеводо-
родов, их галоид- и нитропроизводных. Реакцию ведут при кипячении в во-
де взвеси окиси ртути с ароматическим иодосоединением в присутствии
щелочи (лучше всего окиси серебра). Образовавшуюся ртутноорганиче-
скую гидроокись выделяют в виде солей добавлением соответствующей
кислоты или соли щелочного металла при подкислении.
Окись ртути приготовляют действием избытка едкого натра на горячий
водный раствор сулемы с последующей декантацией до нейтральной реак-
ции и отсасыванием. Применяют в виде влажной пасты.
Получение бромистой фенилртути [68]. 1,18 г иодобензола, 2 г пасты окиси ртути,
2 г пасты окнси серебра кипятят со 100 мл воды в течение 4 час. Осадок отфильтровывают,-
вновь заливают 100 мл воды и опять кипятят в течение часа; так повторяют до прекраще-
ния образования осадка с бромистым калием в фильтрате, что достигается после трехкрат-
ной смены воды. Полученная высаживанием соединенных фильтратов водным раствором
бромистого калия при подкислении уксусной кислотой бромистая фенилртуть весит 1,66 г,
после перекристаллизации из ацетона— 1,56 г (88%). Т. пл. 275° С. Без повторного ки-
пячения выход снижается до 70—80%.
Если вместо добавления к фильтрату бромистого калия отогнать до
малого объема в вакууме без доступа углекислоты воздуха воду, то выкри-
сталлизовывается в виде призматических игл гидроокись фенилртути.
Аналогично бромистой фенилртути получены бромистая и-толилртуть,
хлористые о-, м-, n-хлорфенилртуть, о- и ж-нитрофенилртуть [68].
В случае плохой растворимости гидроокиси следующим образом изме-
няют процесс выделения ртутноорганического соединения: осадок после
кипячения смешивают с щелочной солью соответствующей кислоты, сма-
чивают небольшим количеством слабой кислоты и из смеси извлекают ртут-
ноорганическую соль подходящим растворителем.
Получение хлористой я-иитрофенилртути [68]. 1,4 г л-нитроиодобензола, 2 г пасты
окиси ртути, 2 г пасты окиси серебра кипятят 12 час. со 100 мл воды. Осадок отсасывают,
растирают с избытком хлористого натрия и 5 мл 2 N уксусной кислоты, отфильтровывают
и извлекают ацетоном. Выход хлористой л-нитрофенилртути 0,95 г (54%). Т. пл. 265 °C.
Замена на ртуть кислотных остатков и атомов тяжелых металлов
205
Этот метод дает возможность получать сразу ряд солей одной и той же
ртутноорганической гидроокиси путем осаждения частей фильтрата раз-
личными анионами.
ЗАМЕНА НА РТУТЬ КАРБОКСИЛА
в карбоновых кислотах
Вступление ртути вместо карбоксила в органическое соединение возмо-
жно в ряде случаев как нагреванием ртутных солей карбоновых кислот,
так и нагреванием карбоновых кислот с солями ртути. В последнем случае
ртутноорганические соли образуются промежуточно.
Выделение карбоксила и вступление ртути на его место при нагревании
ртутных солей жирных кислот происходит легко в том случае, если один
из атомов водорода при а-углеродном атоме замещен таким радикалом,
как СН3СО, С6Н5СО [69].
Реакция с бензоилуксусной кислотой проходит различно в зависимости
от условий:
С2Н5ОН, нагревание
(C6H5COCHaCOO)2Hg —
СНС13, нагревание
2СО2 + (СвН5СОСН2)2 Hg
С6Н5СОСНСО + СвН5СОСН2СООН
I I
Hg-o
Нагреванием в вакууме до 85—90°С ртутных солей а,а-диалкилацето-
уксусных кислот Харашем и Стэвли [69] получены меркур-бис-ацетодиал-
килметаны, где алкил — СН3, С2НБ.
Несмеяновым, Луценко и Ананченко [70] значительно улучшен этот
метод синтеза а-меркурирования оксосоединений декарбоксилированием
ртутных солей р-кетокислот. Синтез конечных продуктов — а-хлормеркур-
кетонов осуществляется при этом не в три, как у Хараша и Стэвли, -а в
одну стадию, и выход а-хлормер кур кетонов достигает 50—70% (у Хараша
и Стэвли выход был 5—7%). Реакцию проводят 5-минутным нагреванием
водного раствора -кетокислоты с ацетатом ртути.
Получение метил-а-хлормеркуризопропилкетона [70]. 48 г (0,37 моля) диметилацето-
уксусной кислоты прибавляют по каплям к раствору 115 г ацетата ртути в 300 мл воды.
При этом выпадает осадок ртутной соли диметилацетоуксусной кислоты. Декарбоксилиро-
вание проводят при нагревании водного раствора этой ртутной соли. Окончание реакции
отмечается по исчезновению иона ртути в растворе. Раствор становится прозрачным. Го-
рячий раствор отфильтровывают и к еще теплому раствору по каплям прибавляют раствор
26,7 г хлористого калия в 150 мл воды. Выпадает белый осадок, который вначале быстро
растворяется, но по мере прибавления хлористого калия выпадает вновь в виде белых
кристаллов. Осадок отсасывают, сушат в эксикаторе над хлористым кальцием. Выход
89 г (77%). После кристаллизации из этилового спирта т. пл. 124°С.
Получение З-метил-З-хлормеркурпентанона-2 [70]. 43 г метилэтилацетоуксусной кис-
лоты прибавляют к раствору 80 г ацетата ртути в 275 мл воды. Водный раствор ртутной
соли декарбоксилируют при нагревании на водяной бане до 45—50° С. По окончании де-
карбоксилирования к раствору прибавляют раствор 18 г хлористого калия в 50 мл воды,
причем выпадает вязкий осадок. Из обычных органических растворителей вещество выпа-
дает в виде масла. Его высушивают в эксикаторе над пятиокисыо фосфора. Выход 72 г
(72,9%).
Аналогично получены [70]: З-этил-З-хлормеркурпентанон-2 (выход
58%, после кристаллизации из метилового спирта т. пл. 77° С); 2-метил-
2-хлормеркурциклопентанон (при 125° С разлагается, не плавясь); 2-метил-2-
хлормеркурциклогексанон (т. пл. 128°С).
При взаимодействии трихлор ацетата натрия и сулемы или бромной
ртути при соотношении реагентов 1 : 1 образуется CCl3HgHal, из трихлор-
ацетата натрия и сулемы или ацетата ртути при соотношении реагентов
206
Ртутноорганические соединения. Лит. стр. 213—216
2 : 1 образуется (CCl3)2Hg [71 ].4По мнению Логана [71], в этих реакциях про-
исходит декарбоксилирование промежуточно образующегося CCl3COOHg X,
а не внедрение дихлоркарбена по связи Hg—X.
Синтез б'аг-трихлорметилртути [71]. Раствор 27,2 г (0,1 моля) сулемы и 37,0 а (0,2 мо-
ля) трихлорацетата натрия в 150 мл монометилового эфира этиленгликоля кипятят 1 час,
вливают в воду, эфиром извлекают б«с-трихлорметилртуть. Экстракт промывают водой,
сушат сульфатом магния. Эфир отгоняют, остаток кристаллизуют из хлороформа. Выход
31,2 а (71,5%). Т. пл. 140—141° С.
При применении ацетата ртути выход 54%. С HgBr2 образуется только
CClgHgBr; (CCl3)2Hg из трихлорацетата натрия и бромной ртути полу-
чить не удалось [71].
Согласно патентным данным [71а], ртутные соли поли- и перфторалкил-
карбоновых кислот декарбоксилируются при нагревании до высокой тем-
пературы с образованием поли-(пер-)фторалкилкарбоната поли-(пер-)-
фторалкилртути. Так, нагреванием трифтор ацетата ртути до 300° С перего-
няется трифторацетат трифторметилртути; т. кип. 250—260°С, т. пл. 83—
92°С. Аналогично получены пентафторпропионат пентафторэтилртути,
т. кип. 113° С при 9 мм, т. пл. 49—51°С; перфторгексаонат перфторамил-
ртути, т. пл. 69,5—70° С, ЗН-тетрафторпропионат 2Н-тетрафторэтилртути,
т. кип. 165°С при 13 мм, т. пл. 58—58,5° С.
При кипячении в воде или бензоле хлормеркуркамфоркарбоната
С10Н15ОСО2 HgCl медленно выделяется СО2 и образуется хлормеркуркамфора
C10H16OHgCl [716]. Различные ртутные производные камфоры получены
выделением углекислоты из ртутных солей камфоркарбоновых кислот
[716].
О сопровождающемся отщеплении 1 молекулы СО2 меркурирование
малоновой и метилмалоновой кислот см. гл. V, стр. 53.
Декарбоксилирование как метод синтеза ртутноорганического соеди-
нения впервые применил Песчи [72]. Сплавлением фталевой кислоты с ук-
суснокислой ртутью он получил внутреннюю соль о-гидроксимеркурбензой-
ной кислоты (см. также [75]):
СО
Л-™» + Hg (СНзСОО)2 [Y \ о 4- СО2 4- 2СНзСООН.
Hg/
В ароматическом ряду, как было показано Харашем [73], ртутноорга-
нические соединения при повышении температуры образуют ртутные соли
таких карбоновых кислот, карбоксил которых легко элиминируется при
нагревании.
Метод этот имеет, следовательно, узкую область применения, но в ее
пределах сохраняет свое значение, так как ни прямое меркурирование, ни
диазометод, ни синтез через реактив Гриньяра к получению такого рода
ртутноорганических соединений часто не применимы.
Получение 2,2',4,4',6,6'-гексанитродифенилртути [73]. Ртутную соль 2,4,6-тринит-
робензойной кислоты, помещенную в пробирку, нагревают до 210° С в бане с серной кис-
лотой и держат при этой температуре до прекращения выделения углекислоты. Потеря
в весе точно соответствует двум молекулам СО2 на каждую молекулу ртутной соли. Со-
держимое пробирки извлекают ацетоном, после упаривания которого получают осадок
2,2',4,4',6,6'-гексанитрофенилртути. Кристаллизация из ацетона и спирта. Т. пл. 272°С.
Получение внутренней соли 2-оксимеркуризофталевой кислоты [74]. К раствору 40 г
(0,2 моля) гемимелитового ангидрида в 110 мл 6 N едкого натра и 170 мл воды приливают
раствор 43,3 г (0,2 моля) окиси ртути в 40 мл ледяной уксусной кислоты и 70 мл воды;
смесь нагревают 16 час., после чего углекислота больше не выделяется; образовавшийся
осадок растворим в едком натре. Его отфильтровывают, промывают и сушат до постоян-
ного веса при 105° С. Выход 70 г (96%) [74].
Замена на ртуть кислотных остатков и атомов тяжелых металлов
207
При продолжительном кипячении 3-нитро-[781,4-нитро-[76,77],3-хлор-,
3-бром-[78], 4-хлор-, 4-бром- [77,79] фталевых кислоте уксуснокислой ртутью
выделяется одна молекула углекислоты и ртуть замещает место карбок-
сила в положении 2.
При 5—6-часовом нагревании до 185—190° С ртутных солей мономети-
ловых эфиров (как а-, так и p-эфира) 3-нитрофталевой кислоты происходит
с количественным выделением углекислоты, замена ее на ртуть и образо-
вание из обоих эфиров одного и того же продукта [2-(COOCH3)-6-(NO2)-
•C6H3]2Hg [80].
При нагревании ртутных солей о-(180°С, 4 часа),.и-и п-(190°С, 6 час.)
нитробензойных кислот [761 только из первой происходит выделение угле-
кислоты и замена ее на ртуть с образованием с 16%-ным выходом ди-о-
нитрофенилртути, для мета- и пара-изомеров имеет место меркурирование в
ядро в положение 2 без выделения углекислоты (см. гл. V, стр. 92).
При нагревании ртутной соли нафталевой кислоты [81] или при
98-часовом кипячении водно-щелочного раствора нафталевой кислоты с рас-
твором окиси ртути в уксусной кислоте [82] образуется (в последнем слу-
чае количественно) внутренняя соль 8-меркур-1-нафтойной кислоты.
При нагревании ртутной соли антрахинон-о-дикарбоновых кислот до
прекращения выделения углекислоты происходит замена на ртуть одного
из карбоксилов, например [831
О COO—Hg
О Hg—О
сбУ°
Р-Окси-а-нафтойная кислота при хранении с раствором уксуснокислой
ртути в ледяной уксусной кислоте на холоду заменяет свой карбоксил на
ртуть [84]. На способности ртути заменять карбоксил в карбоновых кисло-
тах основан предложенный Родионовым и Федоровой [85] метод синтеза
вератровой кислоты из гемипиновой, могущий быть представленным следую-
щей схемой:
^\-СООН
СНэО-l^JI—соон
ОСНз
Hg(CHaCO2)2
натревание
A“C0\n Hcl
cH3o-^J_Hg/° —
I
ОСНз
Синтез вератровой кислоты из гемипиновой [85]. 1. Раствор 35 г гемипиновой кисло-
ты в 50 мл 25%-ного раствора едкого натра и 150 мл воды кипятят с раствором 62 г уксус-
нокислой ртути в 300 мл воды и 50 мл уксусной кислоты в течение приблизительно
60 час. (до исчезновения реакции на ион ртути с едким натром). Полученный осадок
меркурированной вератровой кислоты промывают водой, растворяют в едком натре,
высаживают соляной кислотой.
2. 30 г меркурированной вератровой кислоты после 5 час. кипячения с 120 мл конц.
соляной кислоты дают 13,2 г вератровой кислоты.
3-Часовое нагревание на паровой бане водного раствора триметилизо-
галлофлавина с сулемой в присутствии бикарбоната калия приводит к за-
мене СО2 на HgCl [861.
Очень легко замещается карбоксил в ct-фуранкарбоновых кислотах
[871. При кипячении а-фуранкарбоновокислого натрия с водным раствором
сулемы гладко получается соответствующая хлористая фурилртуть:
QHsOCOaNa + HgCl2 C4HaOHgCl + СО2 + NaCl.
208
Ртутноорганические соединения. Лит. стр. 213—216
Реакция была проведена с натриевыми солями а-фуранкарбоновой кис-
лоты, ее 5-бром-, 5-иод-, метил- [87] и 3-метил-4-карбометокси-[88] заме-
щенными.
При кипячении 5-бензамидометилфуран-2-карбоновой кислоты в вод-
ном растворе с сулемой наряду с меркурированием происходит замена на
ртуть не только карбоксильной, но и бензамидометильной группы [89].
Продуктом реакции является тетрахлормеркурфуран [89].
С 3-фуранкарбоновыми кислотами реакция замены карбоксила на ртуть
не идет.
Если же вместо натриевой соли взять свободную кислоту, то происхо-
дит только декарбоксилирование, ртутного соединения не получается, по-
видимому, вследствие отщепляющего действия образующейся соляной
кислоты.
При нагревании смешанного ацетата фуроата ртути, получающегося дей-
ствием водного раствора уксуснокислой ртути на а-фуранкарбоновую кис-
лоту, неожиданно образуется меркурированный в p-положение фуран [87].
Получение хлористой 5-бром-2-фурилртути [87]. Водный раствор 1 моля 5-бром-2-
фуроата натрия (полученного смешением точно эквивалентных количеств кислоты и едкого
натра) прибавляют к раствору 270 г (1моль) сулемы в 5 л воды' После хранения в течение
часа смесь фильтруют и кипятят до прекращения выделения углекислоты. По охлаждении
ртутное соединение отфильтровывают и кристаллизуют из спирта. Выход 76%. Т. пл.
177° С.
______Получение хлористой 3-фурилртути [87]. Смешанный ацетатфуроат ртути формулы
jl^Jj_. получают прибавлением раствора 336 г (3 моля) а-фуранкарбо-
0 'cOOHgOOCCHa
новой кислоты в 3 л воды к раствору 477 г (1,5 моля) уксуснокислой ртути в 7,5 л
воды. Образовавшийся осадок отсасывают. Вес 490 г (88,3%).
840 г (2 моля) ацетоксимтркурфуроата в сосуде, снабженном мешалкой, термометром
и присоединенном через скляику с 30%-ным едким натром прямым холодильником с при-
емником для собирания фураиа, нагревают на масляной бане, температуру которой под-
нимают до 210° С, в течение 10 час. Температура внутри сосуда 110° С и лишь к концу реак-
ции достигает 135° С. Через 10 час. выделение газа заканчивается. Вес осадка 547 г. Оса-
док растворяют в 900 мл 95%-ной уксусной кислоты; полученный раствор растворяют в
8 л воды, фильтруют и к фильтрату прибавляют хлористого натрия до прекращения обра-
зования осадка, который отсасывают, промывают водой, сушат. Вес 270 г. Этот осадок
помещают в аппарат Сокслета и извлекают эфиром. После испарения эфира и перекристал-
лизации остатка из спирта получено 87,5 г хлористой (3-фурилртути. Выход 12%, считая
на уксуснокислую ртуть. Т. пл. 184,5° С.
В ряду тиофена также легко происходит замена карбоксила на ртуть
в сс-тиофенкарбоновых кислотах. Реакция идет при действии уксусно-
кислой ртути на холоду. В некоторых случаях она сопровождается мер-
курированием. Так, действием окиси ртути в уксусной кислоте на а-тио-
•фенкарбоновую кислоту (содержащую меченый атом серы) получен тет-
раацетатмеркуртиофен [90]. 2,3-Дихлор-2-тиофенкарбоновая кислота при
3-часовом хранении с раствором окиси ртути в уксусной кислоте обра-
зует 2,3-дихлор-4,5-дихлормеркуртиофен, но 2,3-дибром-5-тиофенкарбоно-
вая кислота в этих же условиях не меркурируется, а только заменяет
карбоксил на ртуть, образуя 2,3-дибром-5-хлормеркуртиофен [91]. В 4,5-
дииод-З-тиотолен-2-карбоновой кислоте карбоксил заменяется на ртуть
при кипячении с сулемой в растворе уксусной кислоты, в ее 5-моноиод-
аналоге — при действии ацетата ртути в уксусной кислоте (нагревание
на водяной бане) [92].
Получение 2,3-дихлор-4,5-дихлормеркуртиофеиа [92]. 40 г 2,3-дихлор-5-тиофеикарбо-
яовой кислоты в 200 мл ледяной уксусной кислоты-прибавляют к горячему раствору 130 г
уксуснокислой ртути в 240 мл ледяной уксусной кислоты. Почти тотчас начинает выде-
ляться углекислота. При энергичном перемешивании поддерживают легкое кипение. Ми-
Замена на ртуть кислотных остатков и атомов тяжелых металлов
209
нут через 15 начинает выделяться белый осадок 2,3-дихлор-4,5-диацетоксимеркуртиофена.
Через 3 часа отсасывают, высаживают водой из фильтрата дополнительное количество осад-
ка и соединенные осадки кипячением с избытком хлористого натрия превращают в 2,3-
дихлор-4,5-дихлормеркуртиофен. Вещество не плавится.
О методе синтеза несимметричных соединений ртути, протекающего по
схеме [93, 941
RCOOAg + CIHgR' -> AgCl + RCOOHgR' CO2 + RHgR',
см. гл. XII.
ЗАМЕНА НА РТУТЬ АТОМОВ ТЯЖЕЛЫХ МЕТАЛЛОВ,
А ТАКЖЕ НЕКОТОРЫХ МЕТАЛЛОИДОВ
Как полные, так и смешанные ароматические и алифатические ме-
таллоорганические соединения олова (ароматические [95—98, 114], ал-
кильные [99, 100], алкенильные [101—1101), свинца (ароматические [95,
III—114, 116], алифатические [114—120]), трехвалентной сурьмы (аромати-
ческие [95, 121, 122], хлорвинильные [103,104, 123], в частности этой реак-
цией была впервые получена хлористая цис-f}-хлорвинилртуть [123]),
висмута (ароматические [124—126], алифатические [127, 128]), а также ди-
фенилкадмий [129] и соли моноарил-[14, 130—132] и диарил-[132] таллия
уже на холоду и очень быстро при кипячении в органическом растворите-
ле, лучше всего спирте, иногда ацетоне (для дифенилкадмия, перфторви-
нилтрибутил-[102а], триэтил-[110] иди- и трибутилвинилолова [102] в эфире,
а для тетраэтилсвинца — в уксусной кислоте [133]) с галогенидом ртути
образуют галогенид арил(алкил)ртути, частично, а при достаточном коли-
честве соли ртути — сполна, отдавая ртути свой радикал:
RmM„Hal„_m +mHgHal2 -> RHgHal +
Так же идет реакция при взаимодействии гетероциклического соедине-
ния мышьяка — трифур ил арсин а с сулемой в водно-спиртовой среде [134].
Двойные соединения Ar3Sb-HgCl2 разлагаются при кипячении в спирте с
образованием ArHgCl (Аг = м- и n-толил [121], n-анизил и п-фенетил
[122]). В случае Аг — о-толил ArHgCl в этих условиях не образуется [121].
Ароматические арсиноксиды арилируют уксуснокислую ртуть при
продолжительном кипячении в уксусной кислоте [135]. Но при действии
(CF3)3As на HgJ2 (2 дня при 105°Сили 1 день при 175°С), CF3AsC12 на Hg и
НС1 (CF3)2AsHal (Hal = Cl или J) на Hg+ трифторметильных соединений
ртути не образуется [135а].
Реакция окиси ртути с ароматическими металлоорганическими соеди-
нениями олова [95, 96] (также алкенильными соединениями олова [106,
107]), свинца [95], трехвалентной сурьмы [95], трехвалентного мышьяка
[95, 136, 137], висмута [124], проводимая при кипячении в водно-спирто-
вой щелочйой среде, приводит неизменно к диарил(диалкенил)ртути.
Реакция в тех же условиях, с теми же соединениями алкил- или арилмер-
кургидроксида (практически применяют хлористую алкил- или арилртуть
в присутствии щелочи) ведет к несимметричным ртутноорганическим сое-
динениям (см. гл. XII, стр. 231).
При взаимодействии R4Pb[137а] или R4Sn [138] с Hg2(NO3)2 в метило-
вом спирте при 25°С получена R2Hg (R = СН3, С2Н5).
Тетраэтилсвинец реагирует с пентахлорфенолятом [118] ртути (в ша-
ровой мельнице, в присутствии инертного носителя—талька и спирта,
многочасовое перемешивание) с образованием соответствующей соли
этилртути.
210 ' Ртутноорганические соединения. Лит. стр. 213—216
Реакции олефиновых металлоорганических (в частности квазикомплекс-
ных) соединений с солью ртути или с металлической ртутью (см. стр. 211)
протекают особенно легко и так же, как обратные реакции, проходят с сохра-
нением стереохимической конфигурации передаваемого радикала (Несмеянов,
Борисов) (см. гл. VI).
Для получения ртутноорганических соединений взаимодействием гало-
генида ртути с солью моноарилталлия удачным явилось найденное Глушковой
и Кочешковым [130—1321 Применение диизобутирата арилталлия.
Метод полезен для установления строения арилталлийдиацилатов, полу-
чаемых таллированием ароматических соединений методом этих авторов.
Разработанное Пановым и Кочешковым [1121 взаимодействие экви-
молекулярных количеств диацилата диарилсвинца с ацилатом (ацил—аце-
тат, изобутират) ртути, проводимое в одноименной кислоте при комнатной
температуре, явилось методом синтеза ранее недоступных соединений свин-
ца типа АгРЬХ3. Одновременно образуется соль арилртути [1121.
За исключением упомянутых выше патентных методик, все перечислен-
ные операции проводятся однотипно и могут быть иллюстрированы сле-
дующими примерами.
Синтез хлористой метилртути [100]. К раствору 9 г (0,033 моля) сулемы в 40 мл абсо-
лютного спирта прибавляют 3 г (0,0.16 моля) тетраметилолова. Происходит разогревание,
выпадает чешуйчатый осадок. По окончании реакции осадок отфильтровывают (т. пл.
170° С). Из фильтрата дробной кристаллизацией получены: из первой и второй фракций —
хлористая метилртуть с т. пл. 170° С, из третьей фракции — кристаллы с т. пл. 120—
ПО'5 С — смесь хлористой метилртути и дихлордиметилстаннана; из четвертой фракции
(остаток после полного испарения спирта и кристаллизации из лигроина)—дихлордиметил-
станнан с т. пл. 107° С. Получено хлористой метилртути 7,4 г (89%), диметилдихлорстан-
нана 3,2 г (87%). Третья фракция (0,22 а) не учтена.
Синтез хлористой ц«с-₽-хлорвииилртути [123]. К раствору 13,3 г (0,0483 моля) сулемы
в 40 мл абсолютного спирта приливают 5 г (0,0163 моля) цис-триф-хлорвинилстибина (по-
тучение его'см. [166]) в 10 мл абсолютного спирта. После перемешивания в течение часа
при комнатной температуре реакционную массу выливают в 50' мл 10%-ной соляной кис-
лоты, осадок отфильтровывают и высушивают. Сырого продукта получено 14 г (96%).
После трехкратной.кристаллизации из петролейного эфира (т.кип.60—90°С) т. пл. 78,5—79° С.
Синтез хлористой трансф-хлорвинилртути [101]. К 0,35 г (0,00128 моля) сулемы, рас-
творенной в 2 ал абсолютного спирта, прибавляют 0,2 г (0,00064 моля) хлористого трачс-
диф-хлорвинилолова с т. пл. 77,5—78,5° С. После получасового нагревания при 50° С
и стояния в течение часа при комнатной температуре нз реакционной массы выделяются
белые кристаллы в виде игл. Отсосанные и высушенные, они весят 0,2 г. Из маточного раст-
вора дополнительно выделяют 0,15 а того же вещества. Перекристаллизованное из бензола
оио плавится при 123°С (с разложением). Выход 0,35 г (92%).
Синтез хлористой фенилртути -[95]. 1,72 г (0,005 моля) хлористого дифенилолова
растворяют в 10 мл спирта, раствор нагревают до кипения и в него быстро вливают кипя-
щий раствор 2,71 г (0,01 моля) сулемы в 10 мл спирта. Тотчас выпадает осадок хлористой
фенилртути, который отсасывают, промывают спиртом н высушивают. Вес 2,75 г (90%).
Т. пл. 252°С.
Получение перфторвииилмеркурхлорида [НО]. Раствор, содержащий 6,0 г (0,021 моля)
триэтилперфторвинилолова и 57 г (0,021 моля) сулемы в 25 мл эфира, кипятят 8час.От-
гоняют 2/з эфира, к остатку прибавляют 75 мл пентана. Выделившийся осадок отфильтро-
вывают (4,0 г, т. пл. 93—97° С) и затем возгоняют при 70°С/10 мм. Выход хлористой пер-
фторвинйлртути 3,4 г (51%). Т. пл. 106°С.
Получение хлористой я-анизилртути [130]. К раствору 1 г диизобутирата п-анизилтал-
лия в 15 мл спирта приливают раствор 1 г сулемы в 15 мл спирта. После нагревания в те-
чение 15 мин. на водяной бане (бурый осадок) прибавляют по каплям соляную кислоту
до обесцвечивания; осадок отсасывают, промывают спиртом, абс. эфиром и перекристал-
лизовывают из спирта. Т. пл. 239—240° С.
Аналогично получают бромистую n-анизилртуть (реакцию ведут в
ацетоне). Т. пл. 250-250,5°С [130].
При реакции циклопентадиенилталлия с сулемой (в тетрагидрофуране,
при —30 н—-40° С) с количественным выходом получена [139] дицикло-
пентадиенилртуть без примеси хлористой циклопентадиенилртути. Этот
метод получения дициклопентадиенилртути является наилучшим.
Замена на ртуть кислотных остатков и атомов тяжелых металлов 211
Получение дициклопеитадиенилртути [139]. Смесь 6,0 г циклопентадиенилталлия (по-
лучен накануне и высушен над хлористым кальцием в течение ночи) и 15 мл сухого тетра-
гидрофурана помещают в колбу, снабженную мешалкой, капельной воронкой и хлоркаль-
циевой трубкой. При температуре —40 ---50° С и эффективном перемешивании по каплям
за 2,5 часа прибавляют раствор 2,35 г сулемы в 30 мл тетрагидрофурана. После выдержки
в течение часа при —30 -=—-40° С проба’ капли раствора со щелочью на присутствие окиси
ртути отрицательна. Ойадок (вес 5,17 г) быстро отфильтровывают от раствора, промывают
иа фильтре дважды (по 7 мл) тетрагидрофураном и высушивают на воздухе. Из осадка путем
возгонки в вакууме возвращают 0,26 г циклопентадиенилталлия. Раствор немедленно упа-
ривают досуха без нагревания в вакууме водоструйного насоса за 20—25 мин. Остаток
быстро обрабатывают абсолютным эфиром (20 + 20+10 мл); выпавшие при охлаждении
до —40° С лимонно-желтые иглй дициклопентадиенилртутн .отделяют декантацией, промы-
вают 5—7 мл абсолютного эфира, высушивают в тонком слое на фильтровальной бумаге
за 5 мин.; вес 2,21 г. Испарением эфирного раствора получено еще 0,64 г дициклопентади-
енилртути. Выход 97,5% на взятую сулему. После перекристаллизаций из абсолютного .
эфира т. пл. 82—83,5° С (с разложением). Дициклопентадиенилртуть устойчива только
при температуре около —30-=—40° С.
Синтез бромистой «-тиеиилртути из диизобутирата а-тиенилталлия и бромной ртути [131J.
К 0,3 г диизобутиратаа-тиенилталлия, растворенного в 2 мл спирта, прибавляют 0,4 г
бромной ртути в 4 мл спирта. При нагревании в течение 15 мин. образуется коричнево-
бурый осадок. По охлаждении прибавляют по каплям бромистоводородную кислоту до
посветления осадка. Осадок отсасывают и перекристаллизовывают из ацетона, Т. пл.
168° С.
Синтез дифеиилртути [95[. К 0,005 моля (1,72 г) хлористого дифенилолова в 10 мл
спирта добавляют 30 мл кипящего водного 5 iV раствора едкого натра. Выпадает белый
осадок окиси дифенилолова. Прн продолжающемся кипении вливают горячий раствор
0,005 моля (1,35 г) сулемы в 8 мл спирта. Тотчас появляется желтый осадок окиси ртути,
в течение 5—6 сек. исчезающий и заменяющийся белым осадком. Через 1 мин. смесь охлаж-
дают, добавляют 30 мл воды, осадок отсасывают. Это чистая дифенилртуть с т. пл. Г25° С.
Вес 1,7 г [1].
Реакция диацётата дифенилсвинца с уксуснокислой ртутью. Получение триацетата
фенилсвинца и ацетата фенилртути [112]. 1,92 г (1 моль) уксуснокислой ртути растворяют
при слабом нагревании в 40 мл ледяной уксусной кислоты и сюда после охлаждения до
комнатной температуры прибавляют 2,88 г (1 моль) днацетата дифенилсвинца, который
легко растворяется. Через сутки, когда реакция практически закончена, к реакционной
массе прибавляют по каплям из микробюретки 1,28 г 4,7 N раствора соляной кислоты в
спирте (что отвечает 1 молю) для превращения образовавшейся уксуснокислой фенилртути
в хлористую фенилртуть. Выпавший осадок отфильтровывают и промывают спиртом. Т. пл.
после кристаллизации из ксилола 258° С. Выход 1,68 г (80%). Фильтрат оставляют в ва-
куум-экснкаторе над едким кали до испарения уксусной кислоты, твердый осадок (3,63 г)
растворяют в 15 мл горячего сухого свежеперегнанногр.этилацетата и отфильтровывают.
Фильтрат охлаждают в смеси льда и соли; выделившиеся кристаллы отсасывают, промы-
вают гексаном. Выход триацетата фенилсвинца с т. пл. 101—102° С составляет 2,19 г (79%).
Кристаллизация из этилацетата.
Синтез ди-л-хлорфенилртути [95]. К слитым на холоду растворам 0,27 г (0,001 моля)
сулемы в 3 мл спирта и .0,6 г (0,002 моля) двухлористой n-хлорфенилсурьмы в 4 мл спирта
добавлено 5 мл 5 7V кипящего водного раствора едкого натра. Выпадает сероватый осадок.
Затем добавляют 25 мл воды. Осадок отсасывают и извлекают горячим ацетоном. Получено
0,34 г ди-п-хлорфенилртутн (80%). Т. пл. nocjje перекристаллизации из ацетона 242° С.
Синтез хлористой а-фурилртути [134]. Сливают вместе раствор 1 г трифуриларсииа
в 20 мл этилового спирта и раствор 3,2 г сулемы в 100 мл воды со спиртом и нагревают до
кипения. По охлаждении выкристаллизовывается хлористая а-фурилртуть. Затем прибав-
ляют воду, осадок отсасывают. Выход 1,6 г (60%). Т. пл. 151°С.
Взаимодействие с металлической ртутью ди-р-хлорвинильных [140] и .
диарильных [140] соединений свинца и алкенильных [103, 104, 141—147] ’
соединений таллия R2T1X (в спиртовом или ацетоновом растворе на холо-
ду или при нагревании), а также триметилталлия [1.48], (в эфире, комнат-
ная температура), трифенилталлия [149] (кипячение в бензоле) и бромисто-
го дифенилталлия [149] (в пиридине при кипячении) приводит к полноза-
мещенному соединению ртути.
Синтез ди-^мс-р-хлорвинилртути [141]. 0,1 г хлористого ди-цис-р-хлорвинилталлия
и 0,4 г металлической ртути в 10 мл метилового спирта встряхивают при комнатной темпе'
ратуре в течение 90 час. Осадок и непрореагировавшую ртуть отфильтровывают, однохло-
ристый таллий удаляют декантацией с ртути. Вес металлической- ртути 0,35 г. Вес
212
Ртутноор'ганические соединения. Лит. стр. 213—216
выделившегося хлористого таллия 0,06 г (теоретический выход).Спиртовый фильтрат упари-
вают в вакууме досуха, остаток эстрагируют петролейным эфиром. После отгонки раство-
рителя на водяной бане под водоструйным насосом остается жидкость. Это ди-^ис-р-хлор-
винилртуть (Ид 1,6152). Вес 0,09 г.
Синтез ди-(1-метил-2-ацетокси-1-пропен-1-ил)-ртути [143]. В двухгорлую цилиндриче-
скую колбу, снабженную мешалкой и обратным холодильником, помещают 0,25 г хлори-
стого ди-(1-метил-2-ацетокси-1-пропен-1-ил)-таллия в 40 мл ацетона н 2,3 г металлической
ртути. Реакционная масса при энергичном перемешивании нагревается до 60° С в течение '
10 час. Раствор отделяют от хлористого таллия и непрореагировавшей металлической рту-
ти. Фильтрат упаривают досуха, остаток экстрагируют бензолом. После отгонки бензола
выделяются кристаллы, которые плавятся при 107—111° С. Дважды перекристаллизован-
ные из петроленного эфира (т. кип. 40—60° С) плавятся при 112—113° С. Выход перекри-
сталлизованного продукта 0,09 г (40,9%).
При 2-часовом нагревании до 140—150° С без растворителя тетраэтил-
кремния с сулемой удалось получить с 15%-ным выходом хлористую этил-
ртуть [1001. Арилтриметилкремний также арилирует уксуснокислую ртуть
(25° С, ледяная уксусная кислота) с образованием арилмеркурацетатов [150,
151], [(C6H5)3SiO]2Hg при нагревании до 110°С перегруппировывается в
(CeH5)3SiOHgC6H5 [1521.
Реакция триметилфенилеилайа с уксуснокислой ртутью. Получение хлористой феиил-
ртути [150]. Раствор 15 г (0,1 моля) триметнлфенилсилана и 3,6 г (0Д11 моля) уксуснокис-
лой ртути в 200 мл ледяной уксусной кислоты оставляют стоять при комнатной температу-
ре до завершения реакции (проба на отсутствие реакции на ион ртути), после чего выли-
вают в раствор избытка хлористого натрия. Осадок отфильтровывают и сушат. Выход
хлористой фенилртути 3,25 г (92%). Т. пл. 249,5—250,5° С.
Так же получены хлористая о- и я-толилртуть, 3- и 4-хлормеркур-о-
ксилол, хлормеркур-п-ксилол, 2-, 4- и 5-хлормеркур-лг-ксилолы [150].
При помощи органических соединений германия соли ртути до настоя-
щего времени не были проалкилированы.
Ьпс-циклопентадиенилфенилтитан, фенилирует как сулему, так и ме-
таллическую ртуть 'с образованием хлористой фенилртути [153]. Реакции
эти ведут в хлороформе (в первом случае—также в бензоле, выход хлористой
фенилртути 90%).
Перфтор триметил фосфин при нагревании в течение 36 час. до 100°С
с окисью ртути в запаянной трубке образует диперфторметилртуть с вы-
ходом 96% [154].
Пентафенилсурьма [155] и пентафенцлфосфор [156, 157] арилируют
металлическую ртуть. При встряхивании их в течение многих дней при
комнатной температуре с металлической ртутью в. хлороформе, наряду с
хлористым тетрафенилстибонием (соответственно хлористым тетрафенил-
фосфонием) и другими продуктами, с количественным выходом получена
хлористая фенилртуть.
Трифенилвисмут (2 г) и металлическая ртуть (14 г) при нагревании (250° С,'
10 мин.) дает’смесь состава; 74% трифенилвисмута, 24/4 дифенилртути, 2%
дифенила [158].
Комплексы трифенилхрома: (СвН5)3Сг-(ТГФ)3 и (СеН5)3Сг-(ТГФ)3-
ЗМ§С1Вг-(ТГФ)3 с сулемой в тетрагидрофуране быстро количественно дают
хлористую фенилртуть [159, 160]. Катион пентаквобензилхрома в водном
растворе с сулемой образует с небольшим выходом хлористую бензилртуть
С6Н5СН2Сг (H2O)S++ + HgCU + Н2О CeH5CH2HgCl + Сг(Н2О)в++ + а-.
Но катион пентакводихлорметилхрома С12СНСг(Н2О++)6 не реагирует с
сулемой [1621.
Замена на ртуть кислотных остатков и атомов тяжелых металлов 213
Наличие ст-связи С—Zr в циклопентадиенил ьных производных цирко-
ния, содержащих арильные группы, (C6H6)aArZr2O (Аг=СвН5, р-СН3СвН4),
доказано образованием с небольшим выходом ArHgCl при реакции их с
сулемой (тетрагидрофуран, 60—65°С, перемешивание2 часа) [163]. C8H5HgCl
образуется с хорошим выходом при взаимодействии n-C5H6Fe(CO)2C6H5-6
с HgCl2 [164].
ЛИТЕРАТУРА
1. Несмеянов А. Н., Корешков К-А. ЖОХ, 4, 1102 (1934); Вег., 67, 317 (1934).
2. Michaelis A., Becker Р. Вег., 15, 180 (1882).
3. Т о г s е 1 1 К. Acta chem. scand., 13, 115 (1959).
4. Michaelis A. Ber., 27, 244 (1894).
5. К 6 n i g W., Scharrnbeck W. J. prakt. Chem., 128, 153 (1930).
6. Khotinsky E„ Melamed M. Ber., 42, 3090 (1909).
7. Bean F. R., Johnson J. R. J. Am. Chem. Soc., 54, 4415 (1932).
8. Б о p и с о в A. E. Изв. АН СССР, ОХН, 1951, 402.
9. Michaelis A. Ann., 315, 19 (1901).
10. Seaman W., J о h n s о n J. R. J. Am. Chem. Soc., 53, 711 (1931).
11. A i n 1 e у A. D.,C ha I le nger F. J. Chem. Soc., 1930, 2171.
12. T о rs e 1 I K- Arkiv Kemi, 10, 473 (1957).
13. Johnson J. R„ Van CampenM. G., Grummitt O. J. Am. Chem. Soc., 60,
111 (1938).
14. Challenger F:, R i c h a r d s О. V. J. Chem. Soc,, 1934, 405.
14a. M a t t e s о n D. S., W a 1 d b i 1 1 i g J. O. J. Am. Chem. Soc., 85, 1019 (1963).
15. С а з о н о в а В. А., Кронрод H. Я. ЖОХ, 26, 1876 (1956).
16. Несмеянов А. И., Сазонова В. А., Дрозд В. H. ДАН СССР, 126, 1004
(1959).
17. Несмеянов А. Н., Сазонова В. А., Дрозд В. Н., Ни коно ва Л. А.
ДАН СССР, 131, 1088 (1960)
18. К u i v i 1 а Н. G., М u 1 1 е г Т. С. J. Am. Chem. Soc., 84, 377 (1962).
19. Н о 1 z b е с h е г Z. Chem. Listy, 46, 17 (1952).
19а, Horning W. С., Lautenschlaeger F., Wright G. F. Can. J. Chem.,
41, 1441 (1963).
20. W i t t i g G., Keicher G., Rffckert A., R a f t P. Ann., 563, 110 (1949).
21. W i t t i g G., H e r w i g W. Ber., 88, 962 (1955).
’22. Пат. США 2950305, 2950303 (I960); РЖХим, 1961, 23Л88; С. А., 55, 3434 (1961).
23. Пат. США 2969381 (1961); РЖХим, 1962, 12Л67.
24. Н о п е у с u t t J. В., R i d d 1 е J. М. J. Am. Cheth. Soc., 81, 2593 (1959).
25. Пат. США 2950304 (1960); РЖХим, 1961, 22Л69.
26. 3 a x a p к и н Л. И., С т а н к о В. И. Изв. АН СССР, ОХН, 1960, 1896.
27. Neu R. Вег., 87, 802 (1954). '
28. Neu R. Вег., 88, 1761 (1955).
29. Montequi R., DoadrioA., Serrano С. РЖХим, 1957, 65996; An. Real,
soc. esp. fis. у quim., B52, Nil, 595 (1956).
30. H e с м e я н о в A. H., С а з о н о в а В. А. Изв. АН СССР, ОХН, 1955, 48.
31. Montequi R., DoadrioA., Serrano С. С. А., 51, 11906 (1957); Pubis,
insv. guim. «Alonso Barba» (Madrid), 10, 183 (1956).
32. Пат. США 2950302 (1960); РЖХим, 1961, 22Л71.
33. Wittig G., Lehmann G. Ber., 90, 875 (1957).
34. Разуваев Г. А., БрилкинаТ. Г. ДАН СССР, 85, 815 (1952).
35. P а з у в a e в Г. А., Б p и.л к и н a T. Г. ДАН СССР, 91, 861 (1953).
36. Разуваев Г. А. Уч. зап. ГГУ, сер. хим., 32, 169 (1958).
37. Рче t е г s W. Вег., 38, 2567 (1905).
38. Англ. пат. 805156 (1958); С. А., 53, 12177 (1959).
39. Marvel С. S., Adams С. Е., Johnson R. S. J. Am. Chem. Soc., 68, 2735
(1946).
40. L о u d о n J. D. J. Chem. Soc., 1933, 823.
41. Lou don J. D. J. Chem. Soc., 1935, 535.
42. Японск. пат. 178876 (1949); С. A., 46, 529 (1952).
43. W.i 11 e n b e r g:D., W u F. G., G i 1 m a n H. J. Org. Chem., 23, 1898 (1958).
44. К h a r a s c h M. S., C h a 1 k 1 e у L. J. Am. Chem. Soc., 43, 611 (1921).
45. H a n k e M. E. J, Am. Chem. Soc., 45, 1321 (1923).
46. KharaschM. S., Legault R. R., SprowlsW. R. J. Org. - Chem., 3,
409 (1938).
47. W h i t m о r e F. С., Hamilton?. H.; Thurman N. J. Am. Chem. Soc.,
45, 1066 (1923).
214 Ртутноорганические соединения
48. Т г u с е W. Е., McMani m f е R. J. J. Am. Chem. Soc,, 76, 5745 (1954).
49. Haley Т. J., Е d w а г t s L. D., J о h n s о n С. H. C. A., 40, 6446 (1946); J. Am.
Pharm. Assoc., 35, 179 (1946).
50. Poethke W., Furst W. РЖХим, 1962, 13Ж284; Arch. Pharmazie., 296/66, 524
(1961).
51. Coffey S. J. Chem. Soc., 128, 639 (1926).
52. Петр о ан ч П. И. Ж0Х, 30, 2808 (1960).
53. В e a t t i e R. W., Wh i t mo r e F. C. J. Am. Chem. Soc., 55, 1567 (1933).
54. Whitmore F. C., So b'atzki R. I. J. Am. Chem. Soc., 55, 1128 (1933).
55. T r u c e W. E., R a у W. J., jr. J. Am. Chem. Soc., 81, 481 (1959).
56. В о r d w e 1 1 F. G., Me Re 11 i n W. H., Babcock D. J. Am. Chem. Soc., 73,
5566 (1951).
57. Usgaonkar R. N., J a d h a v G. V. J. Indian Chem. Soc., 36, 346 (1959).
58. T r u c e W. E., R a у W. J., jr., Norman O. L., Eickemeier D. B.J. Am.
Chem. Soc., 80, 3625 (1958).
59. Dref ahi G^, S t a n g e G. J. prakt. Chem., 9, 311 (1959).
60. M a 1 a i у a n d i M., S a w a t z к у H., W r i g h t G. F. Can. J. Chem., 39, 1827
(1961).
61. Carmack M., Baizer M. M., Handrick G. K-, Kissinger Z. W.,
S p e c h t E. H. J. Am. Chem. Soc., 69, 785 (1947).
62. Козлов В. В., С м о л и н Д. Д. ЖОХ, 19, 863 (1949).
63. Gilman A., S m i t h E. W., О a t f i e 1 d H. J. J. Am. Chem. Soc., 56, 1412
(1934).
64. H u r d C. D., Morrissey С. I. J. Am. Chem. Soc., 77, 4658 (1955).
65. П e p e в а л о в а Э. Г.-, Несмеянова О. А., Лукьянова И. Г. ДАН
СССР, 132, 853 (1960).
66. Cais М., Kozikowski J. J. Am. Chem. Soc., 82, 5667 (1960).
67. L о u d о n J. D., ShulmanN. J. Chem. Soc., 1939, 1066.
68. H e с м e я н о в A. H„ M а к a p о в а Л. Г. ЖОХ, 3, 257 (1933); Ber., 66, 199 (1933).
69. К h a r a s c h.M. S., S.t a v e 1 e у F. W. J. Am. Chem. Soc., 45, 2961 (1923).
70. H e с м e я н о в A. H., Луценко И. Ф., Ананченко С. H. Уч. зап. МГУ,
132, № 7, 136 (1950).
71. Logan T. J. J. Org. Chem., 28, 1129 (1963).
71a. Пат. США. 3043859. (1962); С. A., 57, 14944 (1962).
716. P i с о n M. Zbl., 1930, II, 1221; C. r., 190, 1430 (1930).
72. P e s c i L. Atti RAL (5), 10, 1, 362 (1901-).
73. Kharasch M. S. J. Am. Chem. Soc., 43, 2238 (1921).
74. Whitmore F. C„ P e r k i n s R. P. J. Am. Chem. Soc., 51, 3352 (1929).-
75. Bruno M., В e 1 1 u с о U. C. A., 52, 16939 (1958); Ricerca sci., 26, 2384 (1956).
76. M a у u r a n a t h a n P. S. J. Chem. Soc., 1957, 495.
77. Chaudhari M. B„ Nargund K. S. C. A., 45, 1975 (1951).
78. W h i t m о r e F. С., C u 1 h a n e P. I. J. Am. Chem. Soc., 51, 602 (1929).
79. Синтезы органических препаратов, сб. 1. М., ИЛ, 1949, стр. 41. ..
80. Mayurana t han Р. S. J. Chem. Soc., 1957, 493.
81. С о r b e 1 1 i n i A., F о s s a t i V. Zbl., 1936, II, 4008; R. 1st. lomardo Scilettere,
Rend. (2), 69, 258 (1936).
82. Leuck G. J'., P e r k i n s R. P., W h i t m о r e F. C. J. Am. Chem. Soc., 51,
1831 (1929).
83. Whitmore F. C., Carnahan F. L. J. Am. Chem. Soc., 51, 856 (1929).
84. Kerkhof I. G. Rec. trav. chim., 51, 755 (1932).
85. R.o d io no w W. M., Fedorowa A. M. Zbl., 1933, II, 373; Arch. Phar. Ber.
Dtcsh. pharm. Ges., 271, 282 (1933). -
86. GrimshawJ., Haworth R. D. J. Chem. Soc., 1956, 418.
87. G i Im an H„ W r i g h t G. F. J. Am. Chem. Soc., 55, 3302 (1933).
88. Gilman H., Burtner R. J. Am. Chem. Soc., 71, 1213 (1949).
89. C i n n e i d e R. О. C. A., 38, 1230 (1944); Proc. Roy. Irish. Acad., 49B, 143 (1943).
90. В u u - H о i N. P. Bull. Soc. chim. France, 1958, 1407.
91. St einkopfW.,. Kohler W. Ann., 532, 250 (1937).
92. Steinkopf W., H a n s k e W. Ann., 532, 236 (1937).
93. К h a r a s h M. S., G r a f f 1 i n M. W. J. Am. Chem. Soc., 47, 1948 (1925).
94. К о t e n J. A., A d a m s R. J. Am. Chem. Soc., 46, 276 (1924).
95. Несмеянов A. H„ К о ч e ш к о в К- А. ЖОХ, 4, 1102 (1934); Вег., .67, 317
(1934).
96. Кочешков К- А., Н а д ь М. М. ЖОХ, 5, 1158 (1935); 4, 1434 (1934); Вег., 67,
717 (1934).
97. Кочешков К. А., Т а л а л а е в а Т. В., П и к и н а Е. И. ЖОХ, 8, 1844 (1938).
98. Bah г G., G е 1 i u s R. Вег., 91, 812 (1958).
. 99. S е у f е г t h D. J. Am. Chem. Soc., 79, 5881 (1957).
Замена на ртуть кислотных остатков и атомов тяжелых металлов 215
100. М а и у л к и н 3. М. ЖОХ,. t Гб, 235.(1946).
101. Н е с м е я н о в А. Н., Борисов А. Е., А б р а м о в а А. Н. Изв. АН СССР,
ОХН, 1946, 647.
102. S е у f е г t h D. J. Org. Chem., 22, 478 (1957).
102a. SeyferthD., R a a b G., В raende K. A. J. Org. Chem., 26, 2934 (1961).
103. Несмеянов A. H., Борисов A. E. ДАН СССР, 60, 67 (1948).
104. Несмеянов A. H., БорисовА. E. Tetrahedron, 1, 158 (1957).
105. Несмеянов A. H., БорисовА. Е., Новикова Н. В. Изв. АН СССР,
ОХН, 1959, 259,
106. Несмеянов А. Н., Борисов А. Е., Новикова Н. В. Изв. АН СССР,
ОХН, 1959, 1216.
107. Несмеянов А. Н., Фрейдлина Р. X., БорисовА. Е. Юбилейный
сборник к 30-летию Великой Октябрьской социалистической революции. М., Изд-во
АН СССР, 1947, стр. 658.
108. Н е с м е я н о в А. Н., Кудрявцева Т. А, Уч. зап. МГУ, 151, 57 (1951).
109. Несмеянов А. Н., БорисовА. Е., ВолькенауН. А. Изв. АН СССР,
1956, 162.
110. S е у f е г t h D., Towe R. H. Inorg. Chem., I, 185 (1962).
111. К г a u s e E„ S c h m i t z M. Ber., 52, 2150 (1919).
112. Кочешков К- А.,П ановЕ.М. Изв. АН СССР, ОХН, 1955, 711; П а и о в Е. М_,
Кочешков К- А. ДАН СССР, 85, 1037 (1952),
113. Ср. Goddard А. Е., Ashley J. N., Evans R. В. J. Chem. Soc., 1922, 978.
114. Англ, пат 331494; Zbl., 1930, И, 2688.
115. Пат. США 2344872 (1944); С. А., 38, 3776 (1944).
116. Пат. США 1987685 (1935); Zbl., 1935, II, 437.
117. W h е 1 е n М. S. Advances in Chem., Ser. 23, 82 (1959).
118. Пат. США 2430287 (1947); С. А., 42, 1014 (1948).
1'19. Пат. США 2673869 (1954); С. А., 49, 5512 (1955).
120. A i n 1 е у A. D., Е 1 s о n L. A., S е х t о n W. A. J. Chem. Soc., 1946, 776.
121. М i с h а е 1 i s A., GenzkenU. Ann., 242, 164 (1887).
122. L 6 1 о f f С. Вег., 30, 2834 *(1897).
123. Несмеянов А. Н., БорисовА. Е., Гуськова А. Н. .Изв. АН СССР
ОХН, 1945, 639.
124. Макарова Л. Г. ЖОХ, 7, . 143 (1937).
125. Gillmeister А. Вег., 30, 2843-(1897).
126. Challenger F., Ridgway L. R. J. Chem. Soc., 121, 104 (1922).
127. Dilnhaupt D. F. Ann., 92, 378 (1854).
128. FranklandE. J. Chem. Soc., 13, 177 (1861).
129. Несмеянов A. H., Макарова Л. Г. ЖОХ, 7, 2651 (1957).
130. Глушкова В. П., Кочешков К. А. ДАН СССР, 103, 615 (1955).
131. Глушкова В. П., Кочешков К. А. Изв. АН СССР, ОХН, 1957, 1186.
132. Глушкова В. П., Кочешков К- А. Изв. АН СССР, ОХН, 1957, 1193.
133. Пат. США 2452595 (1948); С. А., 43, 1805 (1949).
134. Lowe W. G., Н a m i11 о п С. S. J. Am. Chem. Soc., 57, 1081 (1935).
135. BinkleyS. В., H a m i 1 t о n C. S. J. Am. Chem. Soc., 59, 1716 (1937).
135a. C u 1 1 e n W. R. Can. J. Chem., 41, 317 (1963).
136. Нем. пат. 272289 (1914); Zbl., 1914, I, 1469.
137. Пат. США 2391452 (1945); С. A., 40, 4482 (1946).
137a. Tagliavini G., Belluco U. C. A., 57, 13785 (1962).
138- Tagliavini G., CattaliniL., Belluco U. РЖХим, 1963, 20 Ж248.
139. Несмеянов A. H., Матернкова P, Б., Кочеткова H. С. Изв. AH
СССР, ОХН, 1963, 1334.
140. Несмеянов A. H., ФрейдлинаР. X., Кочетков А. К- Изв. АН СССР,
ОХН, 1948, 127; 1950,203. Уч. зап. МГУ, 132, № 7, 144 (1950).’
141. Н е с м е я н о в А. Н., Б о р и с о в А. Е., Шепелева Р. И. Изв. АН СССР,
ОХН, 1949, 582.
142. Несмеянов А. Н., ФрейдлинаР. X., Кочетков А. К- Изв. АН
СССР, ОХН, 1948, 445.
143. Несмеянов А. Н„ БорисовА. Е., ВнльчевскаяВ. Д. Изв. АН
СССР, ОХН, 1954, 1008.
144. Несмеянов А. Н., БорисовА. Е., Новикова Н. В. ДАН СССР, 94,
289 (1954).
145. Нес м е я н о в А. Н., БорисовА. Е., Савельева И. С., Голубева
Е. Н. Изв. АН СССР, ОХН, 1958, 1490.
146. БорисовА. Е., Новикова Н. В. Изв. АН СССР, ОХН, 1957, 1258.
147. Несмеянов А. Н., БорисовА. Е., Савельева И. С., Осипова М. А.
Изв. АН СССР, ОХН, 1961, 1249.
<48. G i 1 m a n Н., J о n е s R. G. J. Am. Chem. Soc., 68, 517 (1946).
216
Ртутноорганические соединения
149. Gilman Н., J о n е s R. G. J. Am. Chem. Soc., 61, 1513 (1939).
150. Benkeser R. A., Hoke D. I., Hickner R. A. J. Am. Chem. Soc., 80,
5294 (1958).
151. В e n к e s e r R. A., Liston T. V., S t a n t о n G. M. Tetrahedron L., 1960,
N 15, 1.
152. Ghosh A. K-, Hansing С. E., Stutz A. I., MacDiarmid A. G.
J. Chem. Soc., 1962, 403.
153. Разуваев Г. А., Латяева В. H., В ы ш и н с к а я Л. И. ДАН СССР, 134,
612.(1960); ЖОХ, 31, 2667 (1961).
154. Griffiths!. Е., В u г g А. В. J. Am. Chem. Soc., 82, 5759 (I960).
155. Разуваев Г. А., Осанова Н. А., Ч у л а е в Н. И., Цигин Б, М. ЖОХ,
30, 3234 (1960).
156. Разуваев Г. А., Осанова Н. А. ДАН СССР, 104, 552 (1955).
157. Разуваев Г. А., Осанова Н. А. ЖОХ, 26, 2531 (1956).
158. HilpertS., GriittnerG. Ber., 46, 1675 (1913).
159. Н е г w i g W., Zeiss H. H. J. Am. Chem. Soc., 79, 6561 (1957).
160. Herwig W., Z e i s s H. H. J. Am. Chem. Soc., 81, 4798 (1959).
161. A n e t F. A. L., Leblanc E. J. Am. Chem. Soc., 79, 2649 (1957).
162. Anet F. A. L. Can. J. Chem., 37, 58 (1959).
163. БрайнинаЭ. M., Дворянцева Г. Г., Фрейдлина Р. X. ДАН СССР,
156, 1375 (1964).
164. Несмеянов А. Н., Чаповский Ю. А., Макарова Л. Г. Изв. АН СССР,
серия химия., 1965.
165. Newell М. Р., Argus М. F., R а у F. Е. J. Am. Chem. Soc., 78, 6122 (1956).
166. Несмеянов А. Н., Борисов А. Е. Изв АН СССР, ОХН, 1945 , 251.
Глава X
СИНТЕЗ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ДЕЙСТВИЕМ СВОБОДНЫХ РАДИКАЛОВ
(ИЗ ПЕРЕКИСЕЙ И ДРУГИХ ИСТОЧНИКОВ)
НА РТУТНЫЕ СОЛИ КАРБОНОВЫХ (И ДРУГИХ) КИСЛОТ
И НА МЕТАЛЛИЧЕСКУЮ РТУТЬ
Разуваевым и Ольдекопом предложен [1,2] новый метод синтеза алкиль-
ных (а также арильных) соединений ртути, состоящий в разложении ртут-
ных солей карбоновых кислот, инициируемом свободными радикалами из
перекисей ацилов. В общем виде реакция может быть выражена схемой:
(RCOO)2Hg + (R'COO)2 R'HgOOCR + RHgOOCR + 2COS.
Метод заслуживает более широкого применения, которого он еще не ус-
пел найти вследствие своей новизны. Этим методом с хорошими выходами из
доступных веществ легко могут быть получены соединения типа RHgX глав-
ным образом жирного, а также алициклического и ароматического рядов,
но, разумеется, не содержащие заместителей-ингибиторов, реагирующих с
перекисями, таких как амино-группа, гидроксил и т. п. В случае синтеза
жирных соединений ртути преимуществом этого метода перед наиболее
обычным методом получения алифатических ртутноорганическйх солей —
через магний- и литий-органические соединения — является возможность
получения жирных солей ртути с таким заместителем, как галоид, отсут-
ствие необходимости работать с абсолютными растворителями и с таким
растворителем, как эфир.
В случае синтеза ароматических соединений ртути (в отличие от мерку-
рирования) этим методом, так же как и диазометодом, получаются свобод-
ные от изомеров индивидуальные арилртутные соли; однако в отличие от
диазометода им не могут быть получены соединения, содержащие заме-
стители-восстановители или ингибиторы цепных реакций. В этом ряду
метод был применен пока к синтезу только соли фенилртути.
В алициклическом ряду этот метод был испытан пока только на одном
примере.
РАЗЛОЖЕНИЕ ПЕРЕКИСЕЙ
В ПРИСУТСТВИИ СОЛЕЙ РТУТИ *
Реакция идет по цепному механизму. Схема цепи:
(RCO2)2 -> 2RCO2- (1)
RCO2 • —> R • -j- CO2 (2)
* Выходы даны, считая на соль ртути, за исключением тех случаев, где это специально
оговорено.
218
Ртутноорганические соединения. Лит. стр. 222—223
R
j,
(R'CO2)2Hg + R- (R'CO2HgO2CR') RHgO2CR' + R'CO2. (3)
R'CO2--> R'- +C02 (4)
(R'COab Hg — R'-—> R'HgO2CR'+ R'COr (5)
R'CO2- ^ R'--CO, (6)
Таким образом, в случае различных R и R' образуется смесь ртутноор-
ганических соединений, что является недостатком метода.
Разложение сОли двухвалентной ртути проводят при нагревании в бен-
золе или другом растворителе, лучше всего в ледяной уксусной кислоте
(или для алкоилперекисей — в одноименной с перекисью кислоте) в при-
сутствии небольшого количества перекиси (около 0,05—0,1 моля на 1 моль
соли ртути).
В случае разложения ацетата ртути перекисью ацетила в ледяной ук-
сусной кислоте выход соли метилртути достигает 99%, считая на соль рту-
ти (выше 100%, считая на взятую перекись, что указывает на цепной
механизм реакции).
Для инициирования разложения ацетата ртути применяют, кроме пере-
киси ацетила [1, 2, 19], также дибензоилперекись [1—4] (при 98°С в
уксусной кислоте выход соли метилртути 95,5% [4], при 12-часовом кипяче-
нии в бензоле выход соли метилртути 54% [2] (70% [4]), соли фенилртути
41,7% (на перекись бензоила) (11,7% [4]), ди-л4-нитробензоилперекись [5]
(в уксусной кислоте выход соли метилртути около 70%), дитретичнобутил-
перекись [6] (19 час. кипячения в уксусной кислоте дало соль метилртути с
выходом 50%), 1,1-диацетилдигидроперекись циклогексила [4] (нагревание
до 80 и 98° С в уксусной кислоте или кипячение в бензоле; выходы соли
метилртути 95,4, 98 и 96,5% соответственно), дициклогексилпероксидикар-
бонат [3,-7] (нагревание до 98° С в уксусной кислоте; выход ацетата метил-
ртути 83%), 1,1-бис-ацетоксиперокси-циклогексанон и -дициклогексилпе-
рекись в бензоле и уксусной кислоте [31].
Перекись третичного бутила при своем распаде генерирует метилради-
калы; при разложении триметилацетата ртути дитретичнобутилперекисью
24-часовым кипячением в хлорбензоле получена соль метилртути • с выхо-
дом 35,7% [8].' Из бензоата ртути и перекиси третичного бутила в хлорбен-
золе (20-часовое кипячение) также получена соль метилртути [61 (выход
20%). Уксуснокислая метилртуть получена [9] с выходом 90% действием
перекиси ацетила, растворенной в диэтилфталате, на раствор окиси ртути
в смеси уксусной кислоты и уксусного ангидрида при 90° С.
Изучено влияние условий реакции на количества образующихся про-
дуктов при декарбоксилировании ацетата ртути под действием свободных
радикалов из 1,1-дигидробензоилперекиси и перекиси бензоила [9а].
Разложение проводили в. уксусной кислоте при 80 и 97—98° С и в бензоле
при 80° С. При разложении ацетата ртути в уксусной кислоте действием
1,1 -дигидробензоилперекиси выход ацетата метилртути при 80° С — 94—95 %,
при 98° С—98%. Повышение температуры сокращает продолжительность
реакции с 3—3,5 часа до 40—60 мин. При разложении ацетата ртути дей-
ствием 1,1-дигидробензоилперекиси в бензоле добавление уксусной кисло-
ты увеличивает выход.
При разложении ацетата ртути действием перекиси бензоила в уксус-
ной кислоте при 80°С выход соли метилртути равен 87%, а соли .фенил-
ртути— 5,3%. Проведение разложения при 97—98°С и увеличение
количества перекиси бензоила увеличивает количество соли фенилртути
Синтез RHgX действием радикалов на Hg и H-g-соли кислот 219
до 60% и уменьшает количество соли метилртути до -39%. Разложение
при 97—98°С сокращает продолжительность реакции с,6—7 час. (80°С)
до 1,5—2 час.
При проведении разложения ацетата ртути действием перекиси бензои-
ла в бензоле добавление небольших количеств уксусной кислоты значи-
тельно ускоряет реакцию и при проведении реакции при 80°С увеличивает
выход и соли метилртути, и соли фенилртути. При этой температуре в от-
сутствие уксусной кислоты 34% ацетата ртути остается непрореагировав-
шими.
Разложение пропионата ртути в пропионовой кислоте под действием
перекиси пропионила дает соль этилртути с выходом 89% 13, 10], Разло-
жением пропионата ртути перекисью бензоила в пропионовой кислоте (при
97—98°С) получена соль этилртути с выходом 54% и соль фенилртути с
выходом 16%; при проведении этой реакции в бензоле при 80°С главным
продуктом реакции является .соль фенилртути (выход 50%), соль этилрту-
ти образуется с выходом 24% [10] (34% [2]). Для инициирования разло-
жения пропионата ртути применена перекись водорода [10]; разложение
проведено в смеси пропионовой кислоты и пропионового ангидрида при
температуре около 100°С. Выход соли этилртути 76%. При разложении
монохлорацетата ртути кипячением с перекисью бензоила или перекисью
м’онохлорацетила в бензоле получена соль фенилртути с выходом до 60%
и 45% соответственно; в последнем случае образуется также монохлор аце-
тат хлорметилртути [3, 11]. Аналогичная картина наблюдается при раз-
ложении н-каприната ртути перекисью бензоила или перекисью н-капри-
ноила в бензоле: выход соли фенилртути 88,5 и 16,5% соответственно;
выход соли нонилртути в.последнем случае 46,5% [3, 12].
Соль фенилртути при разложении перекисью бензоила в этих и анало-
гичных случаях обязана своим возникновением как замене алкилрадика-
ла фенилрадикалом из перекиси, так и меркурированию бензола ацетатом
ртути (см. гл. V).
Нагревание перекиси камфеноноила с ртутной солью камфеноновой
кислоты без растворителя в расплаве при 70—80°С оказалось [13] един-
ственным путем получения а-хлормеркуркамфенилона (после осаждения
хлористым калием). -
Выход соли фенилртути из бензоата ртути и перекиси бензоила в ук-
сусной кислоте (облучение ультрафиолетовым светом) невелик [5]. При ки-
пячении триметил ацетата ртути с перекисью бензоила в бензоле в течение
10 час. соль фенилртути выделена [81 с выходом 94,8% (на перекись бен-
зоила).
Описано образование соли фенилртути, содержащей меченый углерод,
при инициируемом перекисью бензоила распаде ацетата ртути в среде
бензола, содержащего С14 [14].
Получение соли метилртути из ацетата окисиой ртути и перекиси ацетила в уксусной
кислоте [2]. 58,8 г ацетата ртути, 230 мл ледяной уксусной кислоты, 3,0 мл уксусного ан-
гидрида н 2,0 г перекиси ацетила при перемешивании мешалкой нагревают на кипящей
водяной бане в трехгорлой колбе с обратным холодильником в течение 1 часа. По оконча-
нии нагревания разбавляют водой (в 2—3 раза), иодистым натрием осаждают иодистую
метилртуть. Выход 62,8 <(99%. считая на ацетат ртути). Из уксуснокислого раствора осто-
рожным выпариванием реакционной смеси можно получить ацетат метилртути; после воз-
гонки т. пл. 128° С.
Реакция пропионата ртути с перекисью пропионила в пропионовой кислоте. Получение,
солей этилртути [10]. а. Приготовление раствора Перекиси про-
пионила в пропион о-в ой кислоте. 10 мл 27%-ной перекиси водорода при-
ливают к смеси 52 г пропионового ангидрида и 2 г едкого натра при непрерывном охлажде-
нии водой со снегом и перемешивании. Содержание перекиси в растворе определяют иодо-
метрическим титрованием.
б. Реакция пропионата ртути с перекисью пропионила
п р и 9 7—9 8° С. К нагретой смеси 10,4 г пропионата ртутн (0,03 моля) и 100мл пропиоио-
220
Ртутноорганические соединения. Лит. стр. 222-—223
вой кислоты порциями, по 5 мл, приливают в течение 2 час. 44,0 мл раствора перекиси про-
пионила в пропионовой кислоте, содержащего 8,95 а (0,06 моля) перекиси. Нагревание и
перемешивание продолжают 3,5 часа. От реакционной массы отгоняют этилацетат и в ва-
кууме — пропионовую кислоту. Остаток растворяют в воде, из раствора хлористым ка-
лием высаживают 4,89 е хлористой этилртути. После кристаллизации из спирта и эфира
т. пл. 194° С. Выход 61,5%. Из маточного раствора действием избытка йодистого калия
получают 2,82 г иодистой этилртути. Т. пл. 181° С (после возгонки). Общий выход солей
этилртути 88,8%.
Реакция пропионата ртути с перекисью водорода в смеси пропионовой кислоты и про-
пионового ангидрида. Синтез соли этилртути [10J. К смеси 10,4 г пропионата
ртути (0,03 моля), 100 мл пропионовой кислоты и 40 мл пропионового ангидрида,* поме-
щенной в четырехгорлую колбу, снабженную мешалкой с ртутным затвором, обратным
холодильником, капельной воронкой и термометром, прибавляют в течение 23 мин. при
перемешивании (при комнатной температуре) 10 мл 42%-ной перекиси водорода. Смесь
перемешивают еще 25 мин., затем температуру поднимают до 97—98° С. При этой темпера-
туре массу перемешивают 2 часа. Выделение газа, вначале бурное, заканчивается за
1,5 часа. От реакционной массы, отделенной от металлической ртути, отгоняют в вакууме
растворитель, остаток растворяют в воде; из раствора действием хлористого калия осаж-
дают хлористую этилртуть (5,57 г; 70,0%). Из маточного раствора действием избытка йоди-
стого калия получают 0,68 г (6,35%) иодистой этилртути. Общий выход солей этилртути
76,35%.
Разложение перекисями солей закисной ртути карбоновых кислот так-
же приводит к получению ртутноорганических солей, но выходы при этом
хуже, чем при применении солей окисной ртути.
• Так, при продолжительном кипячении в. бензоле уксуснокислой за-
киси ртути с перекисью ацетила получена [2] соль метилртути (выход
49%), считая на взятую перекись, с перекисью бензоила соль фенилртути
(выход 29% на перекись бензоила) [2], с перекисью третичного бутила в
уксусной кислоте — соль метилртути (выход 36%) [6] (при кипячении в
хлорбензоле выход соли метилртути 18%).
При разложении перекиси ацетила в уксусной кислоте в присутствии
солей ртути неорганических кислот HgSO4, HgJ2, Hg2SO4, Hg2Cl2 соль ме-
тилртути образуется с незначительным выходом и ее не образуется при про-
ведении разложения в присутствии сулемы [15].
РАЗЛОЖЕНИЕ СОЛЕЙ РТУТИ, ИНИЦИИРУЕМОЕ РАДИКАЛАМИ,
ОБРАЗУЮЩИМИСЯ ПОД ДЕЙСТВИЕМ УФ-СВЕТА .
Источниками алкилрадикалов, инициирующих декарбоксилирование
соли ртути и ее- алкилирование, могут являться не только перекиси, но и
продукты распада под действием ультрафиолетового света [16, 17] самих
солей ртути карбоновых кислот:
ftv
(RCO2)2Hg -► RCOOHg' + RCCV
RCO2‘ R' -J- CO2
R’ 4- ’HgCOOR -> RHgCOOR
При облучении ацетата окисной и закисной ртути в ледяной уксусной
кислоте, пропионата ртути как в пропионовой кислоте [17, 18], так и в бен-
золе выходы солей метил- и этилртути достигают 70% [16, 17, 19, 201. При
облучении ацетата ртути в бензоле выход ацетата метилртути 30—34 % [14,
19, 20]. Аналогично ведет себя «-бутират ртути в бензоле [20]. Однако об-
лучение триметилацетата ртути в бензоле не дал'о ртутноорганического сое-
динения, а привело к выделению металлической ртути [9].
Получение иодистой метилртути при облучении ацетата ртути [16]. Опыт проводят
в кварцевой колбе, снабженной обратным холодильником. В колбу по горизонтали впаяно
Синтез RHgX действием радикалов на Hg w Hg-соли кислот
221
кварцевая трубка, в которую помещают ртутную лампу FIPR-4. Уровень реакционной
смеси выше впаянной трубки. Теплота, выделяемая лампой при ее горении, достаточна
для поддержания растворителя в состоянии кипения. 25,0 г ацетата ртути в 135 мл ледя-
ной уксусной кислоты облучают в течение 10 час. К реакционной массе, отделенной от
ртути, добавляют 250 мл воды и 15 г йодистого натрия. Выход иодистой метилртути 19,0 г
(71%).
РАЗЛОЖЕНИЕ СОЛЕЙ РТУТИ, ИНИЦИИРУЕМОЕ РАДИКАЛАМИ,
ОБРАЗУЮЩИМИСЯ ИЗ ДРУГИХ ИСТОЧНИКОВ
Метилрадикалы, образующиеся при разложении тетраацетата свинца,
также могут инициировать декарбоксилирование соли ртути [21]. И в этом
случае лучшей средой является ледяная уксусная кислота. Разложение
ведут при кипячении с избытком ацетата ртути до прекращения выделения
газа. Выход соли метилртути выше 80%, считая на тетраацетат свинца
(3,6%, считая на ацетат ртути) [21].
Фенилиодозоацетат, подобно перекисям, но гораздо слабее иниции-
рует декарбоксилирование ацетата ртути при кипячении в уксусной кисло-
те, анизоле или кумоле с образованием соли метилртути [22]. При кипяче-
нии фенилиодозоацетата с металлической ртутью в бензоле или уксусной
кислоте в результате реакции радикала СН3 (продукт термического
распада фенилиодозоацетата) с образующимся ацетатом закисной ртути
получена уксуснокислая метилртуть (в последнем случае с выходом 2,2%);
при проведении этой реакции на холоду соединений метилртути не обра-
зуется.
РАЗЛОЖЕНИЕ ПЕРЕКИСЕЙ
В ПРИСУТСТВИИ металлической РТУТИ *
Ртутноорганическйе соединения получаются также разложением пере-
кисей в присутствии металлической ртути [23, 24]. Алкилирование (ари-
лирование) ртути происходит лишь при проведении реакции при кипяче-
нии [251. Реакцию ведут в бензоле, уксусной кислоте; при применении че-
тыреххлористого углерода выходы хуже. Соль метилртути (после добав-
ления соответствующего аниона) получена при разложении в присутствии
металлической ртути перекиси ацетила [23, 24] (при 20-часовом кипячении
в бензоле выход соли метилртути равен 64,7%), диацетилированной
1,1-дигидроперекиси циклогексила [26] (при 6—7-часовом кипячении в бензо-
ле выход — 32,5%, в уксусной кислоте — 63,7%, в четыреххлористом
углероде соль метилртути образуется с незначительным выходом), перекиси
третичного бутила [6] (при 20-часовом кипячении в уксусной кислоте соль
метилртути получена с незначительным выходом).
Также из перекиси бензоила получена соль фенилртути [23] (при
20-часовом кипячении в бензоле выходЗ! ,5% [23], при нагревании в четырех-
хлористрм углероде выход незначительный [27]). Полученная с незначитель-
ным выходом при разложении перекиси бензоила в С|4Н8 в присутствии ме-
таллической ртути хлористая фенилртуть содержала 3% фенильных групп
растворителя [28].
При разложении в присутствии металлической ртути ацетилбензоил-
перекиси (кипячение в бензоле в течение 4,5 часа) получено (после осажде-
ния хлористым натрием и иодистым калием) 13,8% хлористой фенилртути
и 31,3% иодистой метилртути [29]. Разложение м-нитробензоилбензоил-
нерекиси и n-нитробензоилбензоилперекиси (кипячение в бензоле 11 и
* Выходы даны, считая на взятую перекись.
222
Ртутноорганические соединения
20 час. соответственно) в присутствии металлической ртути дает с небольшим
выходом только хлористую фенилртуть 129].
Образующиеся при термическом разложении бензола и толуола радика-
лы фиксируются на ртути [30].
Реакция между перекисью ацетила и ртутью в бензоле [23]. а. Получение рас-
твора п е.р екиси ацетила. В широкогорлую открытую колбу или стакан по-
мещают 10 г перекиси калия и 100 мл бензола. Реакционную’смесь при быстро работающей
мешалке, препятствующей замерзанию бензола, охлаждают снегом с солью до—10° С
и к ней по каплям прибавляют уксусный ангидрид. После прибавления всего уксу-
сного ангидрида перемешивание продолжают еще около 1 часа. После этого добавляют
лед сначала очень небольшими порциями, затем больше, до полного разложения пере-
киси калия. Слой бензола отделяют от водного слоя, , промывают водой и сушат серно-
кислым натрием; количество получившейся перекиси определяют йодометрическим
титрованием.
б. Получение хлористой метилртути [23}. В трехгорлую колбу,
снабженную механической мешалкой с ртутным затвором, капельной воронкой и обратным
холодильником, помещают металлическую ртуть и над ней Небольшой слой бензола. Бен-
зол доводят до кипения и при одновременном перемешивании в колбу из капельной ворон-
ки, конец которой погружен в бензол, прибавляют раствор 9,9 г перекиси ацетила в 200 мл
бензола. Прибавление продолжают медленно и заканчивают за 8 час. при нагревании реак-
ционной массы на водяной бане. Затем бензольный слой нагревают еще 12 час. По оконча-
нии реакции бензольный слой отделяют от ртути и многократно отмывают водой.
При прибавлении к этой водё насыщенного раствора йодистого калия выпадает бе-
лый осадок иодистой метилртути. Выход 18,6г (64,7%, считая на взятую перекись).
Т. пл. 144—145° С.
ЛИТЕРАТУРА’’
1. Разуваев Г. А.,Ольдекоп Ю. А.,Майер Н. А. ДАН СССР, 98, 613
(1954).
2. Разуваев Г. А., Ольдекоп Ю. А.> Майер Н. А. ЖОХ, 25, 697
(1955).
3. Майер Н. А., Ольдекоп Ю. А. РЖХим, 1961, 22 Ж27; Сборник научных
работ Института физико-органической химии АН БССР, вып. 8, 113 (I960).
4. Ольдекоп-Ю. А., Майер Н’. А. ЖОХ, 30, 275 (1960).
5. Разуваев Г. А., Ольдекоп Ю. А. Тр. по химии и хим. технологии, № 1,
178 (1958).
6. Ольдекой Ю. А., Азановская М. М. ЖОХ, 30, 2291 (I960).,
7. Р а з у в а е в Г. А., Т е р м а н Л. М. Ж- Всесоюзн, хим. общ., 6, 473 (1961); ЖОХ,
31, 3132 (1961).
8. Ольдекоп Ю. А., Василевская Т. В. ЖОХ, 28, 3008 (1958).
9. Пат. Чехосл. 98129 (1959); С. А., 56, 7356 (1962).
9а. Ольдекоп Ю. А., М а й е р Н. А. Вести. АН БССР, сер. физ.-техн, наук,
№ 2, 37 (1960). •
10. Ольдекоп Ю.А., М а й е р Н. А. ЖОХ, 30, 619 (1960).
11. О л ь д е к о п Ю. А., М а й е р Н. А. ЖОХ, 30, 3472 (1960).
12. О л ь д е к о п Ю. А., М а й е р Н. А. ЖОХ, 30, 3017 (1960).
13. Несмеянов А. Н„ К р и ц к а я И. И. ДАН СССР, 121, 477 (1958).
14. Р а з у в а е в Г. А., Петухов Г. Г., Затеев Б. Г. ДАН СССР, 127, 348
(1959).
15. О л ь д е х о п Ю. А., М а Й е р Н. А. ЖОХ, 30, 299 (1960).
16. Р а з у в а'е в Г. А., О л ь д е к о п Ю. А. ДАН СССР, 105, 738 (1955).
17. О л ь д е к о п Ю. А., М а й е р Н. А. ДАН БССР, 4, 288 (1960).
18. О л ь д е к о п Ю. А., М а й е р Н. А. ДАН СССР, 131, 1096 (1960).
19. О л ь д е к о п Ю. А., М а й е р Н. А. ЖОХ, 30, 303 (i960).
20. Оль де коп Ю. А., Майер Н. А., Гессельберг В. И. РЖХим,
1961, 22Ж26; Сборник научных работ Института физико-органической химии АН
БССР, вып. 8, 37 (1960). .
21. Разуваев Г. А., Ольдекоп Ю. А., Сорокин Ю. А., Т в е р д о в а В. И.
ЖОХ, 26, 1683 (1956).
22. Ольдекоп Ю. А.,Майер Н. А., Б ыстрая А. В. ДАН СССР, 6, 503 (1962).
23. Р а з у в а е в Г. А., Ольдекоп Ю, А., Г р о’б о.в Л. Н. ДАН СССР, 88, 77
(1953); ЖОХ, 23, 589 (1953).
24. Р а з у в а е в Г. А. Сб. Вопросы химической кинетики, катализа и реакционной спо-
собности. М., Изд-во АН СССР, 1955, стр. 790.
Синтез RHgX действием радикалов на Hg и Hg-соли кислот 223
25. Разуваев. Г. А., Ольдекоп Ю. А. ЖОХ, 27, 196 (1957).
26. О л ь д е к о п Ю. А., Чижевская И. И. Сборник научных работ Института
физико-органической химии АН БССР, вьш. 7, 68 (1959).
27. Р а з у в а е в Г. А. Уч. зап.. ГГУ, 15, 81 (1949).
28. Л а т я е в а В. Н. РЖХим,. 1961, 21Ж2; Сб. научных работ Института физико-орга-
нической химии АН БССР, вып. 8, 58 (1960).
29. Разуваев Г. А., Ольдекоп Ю. А., Л атяев’а В. Н. ЖОХ, 26, 1110 (1956).
30. Н е i и F., М е з ё е Н. J. Вег., 76, 430 (1943).
31. ОльдекопЮ. А., Майер Н. А., Пшеничный В. Н. ЖОХ, 34, 317 (1964).
Глааа XI
ПОЛУЧЕНИЕ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ЭЛЕКТРОЛИЗОМ
Имеется несколько методов электролитического синтеза ртутнооргани-
ческих соединений, не получивших, однако, до настоящего времени широ-
кого применения.
1. Получение симметричных ртутноорганических соединений при элек-
тролитическом восстановлении кетонов на ртутном аноде [1]:
;2RR'CO + Hg + 6ГГ -> (RR'CH)aHg + 2Н2О.
Эта реакция приводит к полным ртутным производным углеводородов с
вторичными радикалами. (Случай сохранения кетонной функцйи и образова-
ния первичного ртутноорганического соединения описан ниже, см. стр.
226.)
2. Получение полнозамещенных жирных соединений ртути электро-
лизом NaBAlk4 или NaAlAlk3OR на ртутном аноде:
NaMeAlk4 -р Hg AlkaHg -f-MeAlks.
3. Получение соединений состава (RHg)re электролизом ртутноорганиче-
ских солей в жидком аммиаке.
4. Электролитическая симметризация/ртутноорганических солей (см.
гл. XIII).
При электролитическом восстановлении кетонов на ртутном катоде на-
ряду с получением соответствующего спирта происходит образование полно-
замещенных органических соединений ртути, могущее, при соблюдении
необходимых условий (главным образом температуры, а также концентра-
ции, силы и плотности тока и длительности процесса), стать преобладающей
реакцией.
Так, при электролизе ацетона получены диизопропилртуть и изопропи-
ловый спирт 12]. При электролизе метил этил кетона в приведенных ниже
условиях с выходом до 30% получена дивторичнобутилртуть [1]. В послед-
нем случае образуется также какое-т,о другое, содержащее ртуть органиче-
ское соединение, разлагающееся при нагревании с выделением металличе-
ской ртути [вероятно, за счет примеси соединения типа (RHg)„L
При электролизе на ртутном катоде кетонов С6Н5СН2СОСН3,
СН3СОСОСеНа, циклопентанона, циклогексанона и 2-, 3- [3]и4- [3, 4] метил-
циклогексанонов получены соответственно (CeH6CH2CHCH3)aHg, дицик-
лопентилртуть, дициклогексилртуть и диметилциклогексилртуть.
Получение ртутноорганических соединений электролизом
225
Электролиз ведут в 5 %-ной серной кислоте, в которой суспендирован
кетон. Анод—слой ртути в 15 см2, катодом служит пористый свинцовый
цилиндр. Для разных кетонов продолжительность электролиза варьирует-
ся от 4 до 8 час., сила тока 2—4а, температура 55—60°С (в некоторых слу-
чаях комнатная) [3]. Выходы 25—30%.
С6Н5СН2СОСН3 восстанавливается с образованием ртутного соединения
в 5%-ной серной кислоте только при 55° С, а также в 30 %-ной серной кис-
лоте или в смеси ледяной уксусной и 18%-ной соляной кислот. Аналогич-
но ведет себя циклогексанон. Циклопентанон образует ртутноорганическое
соединение легко [3]. Кетоны с СО-группой, соседней с бензольным ядром,
не восстанавливаются с образованием ртутноорганических соединений.
В этих условиях не удалось получить ртутноорганического соединения
при электролизе ментона и дибензилкетона.
Диментилртуть получена [5] из ментона в электролите — конц. сер-
ной кислоте на ртутном катоде при 77—80° С.
Получение дивторичнобутилртути [1J. На дно стеклянного стакана шириной 11,5 см,
высотой 16 см, служащего в качестве катодного пространства и помещенного в баню при
40° С, наливают слоем в 1 см ртуть, в которую вводят защищенный стеклом медный про-
вод, оканчивающийся платиновым концом. В качестве анодного пространства служит
пористый фарфоровый сосуд (внешний диаметр 7 см), который подвешивают посредством
прочного резинового кольца и парафинированной деревянной крышки в стакане таким
образом, чтобы дно его находилось на расстоянии 2—2,5 см от поверхности ртути. Анод —
полый цилиндр нз свинца, внутри которого циркулирует вода и который посредством па-
рафинированной деревянной ручки свободно подвешивается в фарфоровом сосуде. Анодной
жидкостью служит 20%-ная серная кислота. В катодное пространство помещают 30 г ме-
тилэтилкетона и 300 мл 30%-ной серной кислоты. Прн силе тока 25 а и 45—50° С (оптималь-
ная температура для реакции) напряжение на клеммах от 7,8 до 8,4 в. Уже спустя несколь-
ко минут после включения тока начинается выделение янтарно-желтых капель диалкилрту-
тн. Через 2 часа количество образовавшегося масла диалкилртути достигает 41,5 г. Его
отделяют, фильтруют, сушат сернокислым натрием и перегоняют из нагретой не выше 80° С
водяной бани в вакууме 0,12—0,32 мм. Большая часть перегоняется при 46° С и представ-
ляет собой чистую дивторичнобутилртуть. Т. кип. 91—93°С/15 мм. Остающееся после пе-
регонки желтое масло (иногда до 30% от сырого веса продукта) бурно разлагается при тем-
пературе бани 90° С с выделением металлической ртути и не может быть перегнано без
разложения даже в высоком вакууме.
Получение днизопропилртути [2]. Прн электролизе на ртутном катоде 40%-ного раст-
вора серной кислоты, содержащего Ч& моля ацетона в течение 30 а-час при плотности тока
2,8—5,5 а/см2 получены динзопропнлртуть и изопропиловый спирт.
Электролизом NaB(C2H5)4 с ртутными электродами получена диэтил-
ртуть с выходом 81%. Одновременно образуется триэтилбор [6].
Получение диэтилртути электролизом NaB(C2Hs)4 на ртутном аноде [6]. Электролиз
ведут в цилиндрическом стеклянном сосуде емкостью около 400 мл. Сосуд внизу снаб-
жен краном для отделения продуктов реакции. Верхний край сосуда плоско отшлифо-
ван, что дает возможность плотно закрывать его плоской алюминиевой крышкой толщи-
ной 16 мм. Ртуть, служащую в качестве катода, наливают в широкий стеклянный ста-
кан (например емкостью 250 мл), который посредством трех припаянных ножек устанав-
ливают на дне сосуда. В этот стакан посредине помещают приклеенный при помощи
подходящего клея стаканчик емкостью 30 мл, в который наливают ртуть, служащую в
качестве анода, соединенную с источником тока посредством изолированного (посколь-
ку он смачивается электролитом) вращающегося стержня. К стержню в пространстве,
заполняемом электролитом, прикрепляется лопастная мешалка из изолирующего мате-
риала или покрытая изолирующим материалом. При помощи этой мешалки поверхность
ртути освобождается от продуктов реакции.
Оба стакана наполняют ртутью до краев. Края внешнего и внутреннего стаканов
должны быть одинаковой высоты. Катод, таким образом, имеет кольцеобразную форму,
анод расположен в нем концентрически. Провод, соединенный с катодом, также изоли-
рован и проходит через отверстия в крышке, кроме того, имеются отверстия для входа
и выхода инертного газа, для термометра и посредине—для стержня, подводящего ток
к аноду.
226
Ртутноорганические соединения. Лит. стр. 227
NaB(C2H6)i растворяют в воде, тщательно освобожденной от воздуха при помощи
инертного газа (зота или, лучше, аргона), так как продуктом реакции, кроме (CaHs^Hg
и углеводородов, является триэтилбор. Во время электролиза в самый сосуд инертный газ
не пропускают, но им при помощи Т-образного ответвления «обвевают» сосуд, в котором
ведут электролиз. Во время электролиза сосуд охлаждают орошением водой или при по-
мощи небольшого настольного вентилятора. Температура во время электролиза держит-
ся около 20°С. Электролизу подвергают (13 а/дм2 на аноде, 6,5 в) 31,6 г (0,211 моля)
NaB(CaH5)j. Вскоре образуются бесцветные капельки смеси диэтилртути и триэтилбора,
собирающиеся на дне; После прохождения 14,450 а-сек (0,150 ф) получают 15,7 г (0,122
моля, 81%) диэтилртути (т. кип. 52°С/10 мм) и 13,0 а (0,133 моля, 87%) триэтилбора
(т. кип. 94—95°С).
Описано получение диалкилртути электролизом Me[Al(Alk)3R)
(Me—Na, К) на ртутном аноде [7].
Электролизом Me[Al(C2H5)3OR] на ртутном аноде (катод — медный)
получена [8] диэтилртуть с выходом 82%.
При электровосстановлении СН3СОСН=СН2 на ртутных электродах, при
катодном потенциале от 1,05 до 1,30 в, pH 5, образуется соединение
Hg(CH2CH2COCH3)2, выделенное в виде семикарбазона и 2,4-динитрофенил-
гидразона [9].
При электролизе RHgX (R = СН3 [10—13], С2Н5 [10, 12], п-С3Н7 [10,
12], г-С3Н7[11, 13], п-С4Н9 [11, 13], s-C4H9 [14], n-C5Hn [14], z-C5Hu [14],
n-C6H13 [14], n-C7H15 [14],n-C8H17 [10], c-C6Hn[14], C6H5[10, 12], атакже
p-CH3C6H4 [14]; X=Cl[10, 12, 14], Br[13], J [6, 14], NO3 [11]) в жидком ам-
миаке на катоде выделяются соединения состава RHg, как это впервые было
показано Краусом [10] (в случае C6H5CH2HgCl и транс-ClCH^CHHgCl
таких соединений выделить не удалось [12]). CH3Hg получен и при элек-
тролизе водного или спиртового раствора хлористой метилртути [10].
Эти соединения представляют собой в большинстве случаев черные, вы-
сокой электропроводности порошки, разлагающиеся при комнатной темпера-
туре на R2Hg и металлическую ртуть. Соединения с вторичными алкильны-
ми группами менее устойчивы, чем содержащие алкилы нормального стро-
ения. Многие из них начинают разлагаться уже при—50°С.
Вещества эти рассматриваются как «органические металлы» с делокали-
зованными свободными электронами. Структурной ячейкой в них, по Кот-
су [15], является RHg+ с ковалентной связью ртути с алкилом. Возможно
[14], что такая структура стабильна в пределах температурной устойчивости
этих веществ — с повышением же температуры электроны оттягиваются
к атому ртути и все связи R с Hg в кристаллической решетке выравни-
ваются [12, 13]. С этим представлением согласуется, в частности, потеря
оптической активности при электролизе в жидком аммиаке оптически ак-
тивной бромистой вторичнобутилртути и проведении цикла [14]:
Н£Вг8
R°HgBr (RHg)re R2Hg + Hg--------------> RHgBr,
где R° — оптически активный радикал.
Электролиз продуктов присоединения солей ртути к этилену и окиси
углерода в целях определения их строения описан в гл. VI.
Получение ртутноорганических соединений электролизом
227
ЛИТЕРАТУРА
1. Та tel J. Вег., 39, 3626 (1906).
2. Haggerty С. Zbl., 1930, I, 338; Trans. Am. Electrochem. Soc., 56, 5 (1929).
3. A r a i T. Bull. Chem. Soc. Japan, 32, 184 (1959).
4. J e n s e n F. R., G a 1 e L. H. J. Am. Chem, Soc., 82, 145 (1960).
5. Schall C., Kirst W. Z. Electrochem., 29, 537 (1923).
6. Ziegler K-, SteudelO. W. Ann., 652, 1 (1962).
7. Пат. СССР 132136(1960); РЖХим, 1962, 9Л98.
8. Пат. ФРГ 1127900 (1962); С. А., 57, 11235 (1962).
9. HolleckL., MarquardingD. Naturwiss., 49, 468 (1962).
10. К г a u s С. A. J. Am. Chem. Soc., 35, 1732 (1913).
11. R i с е F. О., Evening В. L. J. Am. Chem. Soc., 56, 2105 (1934).
12. G о w e n 1 о с к B. G., Trotman J. J. Chem. Soc., 1957, 2114.
13. Go wen 1 ос к В, G., Prit.char d - Jo nes P., О v e n a 1 1 D. W. J. Chem. Soc.,
1958 535 '
14. В i 1 1 i n g e В. H. M., G о w e n 1 о с к В. G. J. Chem. Soc., 1962, 1201.
15. Coates G. E. Quart. Revs., 4, 226 (1950).
Глава XII
МЕТОДЫ СИНТЕЗА
НЕСИММЕТРИЧНЫХ ПОЛНОЗАМЕЩЕННЫХ1
РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ RHgR'
СИНТЕЗ RHgR’
ЧЕРЕЗ МАГНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Полнозамещенные несимметричные ртутноорганические соединения
RHgR' были впервые получены в относительно чистом виде Гильпертом
и Грюттнером [II (ср. [2]) действием магнийорганических соединений на
арил(алкил)ртутные соли:
' RMgX + R'HgX R'HgR + MgX2.
Так получены бензилфенил-[ 1 ], бензилэтил-[3], этилфенил-П1, бензил-
о-толилртуть [11. При синтезе этих соединений необходимо избегать повы-
шения температуры, так как некоторые несимметричные соединения неус-
тойчивы и даже при обыкновенной температуре (легче при нагревании)
склонны распадаться на смесь двух симметричных соединений :
2RHgR'-> RaHg + R^Hg.
По данным Гильперта и Грюттнера, несимметричные соединения могут
быть получены через магнийорганические соединения только одним путем,
например: C6H6CH2HgC6H5— действием CeH5MgBr на C6H5CH2HgBr;
если же действовать CeH5CH2MgBr на CeH5HgBr, то фенилбензилртуть
не образуется. Хараш [4, 5] показал, однако, что, избегая избытка ре-
актива Гриньяра и тщательно следя за температурой, можно получать
RHgR' как действием RMgX на R'HgX, так и действием R'MgX на RHgX.
Харашем с сотр. так получены:
CeH5HgCH8,CeH5HgCeHu, CeH5HgC4H9-n, CH3HgCH2CeH5, C6H5HgC10H7,
C2H5HgC10H7, CH3HgC4H9-n, CeH5HgCeH4OCH3-o, CH8HgCeHn,
C2H5HgC6Hn, C6H5Hg(CH3)3C6H2-l,3,5, C3H7HgC4H8-n, CeH6HgCeH4CH3-p,
n-C4H9HgCsHu-i, CH3Hg(CH3)3C6H2-l ,3,5 [41, CeH5HgC2H4C6H5,
€6H5CH2HgC2H4C6H5, CeH5HgC4H3S [2], C2H5HgC7Hltf, C4H9HgC7Hls,
i-C4H9HgC4H9, z-C3H7HgC8H„ CeH6HgC6H4Cl-p, CeH5HgC6H4Cl-0,
CeH5HgCeH4Cl-m, o-ClCfiH4HgCeH4Cl-p, C6H5HgCeH4CH3-o,
o-CH3C6H4HgC6H4CH3-p, o-CH3CeH4HgCeH4Cl-p, o-CH3CeH4HgC6H4CH3-p,
o-CH3C6H4HgCeH4Cl-p, C6HsHgC6H4CH3-p, C6H5HgC6H4CH3-m,
m-CH3C6H4HgCeH4CH3-p, m-ClCeH4HgC6H4Cl-o, m-CH3C6H4HgCeH4Cl-p,
o-CH3C6H4HgCeH4OCH3-o, CH3HgC6H4Cl-m, p-CH3OCeH4HgCeH4OCH3-o,
a-C10H7HgCeH4OCH3-p, a-C10H7HgCeH4OCH3-o [5], C6H6HgC6H4F-m,
C6H5HgC6H4F-p, CeHsHgCeH4Br-o, C6HsHgCeH4Br-m, CeHsHgC6H4Br-p,
Синтез несимметричных ртутноорганических соединений RHgR'
229
C6H5HgCeH4CF3-m , m-ClC6H4HgC6H4CF3-m, C6H5CH2HgCH2C6H4Cl-o,
C6H5CH2HgCH2C6H4Cl-m, C6H5CH2HgCH2C6H4Cl-p,
m-ClC6H4CH2HgCH2C6H4Cl-o, p-ClC6H4CH2HgCH2C6H4Cl-o, [6],
C6H5CH2HgC4H9-Z, .C8H5CH2HgC3H5, C6H5HgC8H5 [7], C6H5HgC6H3Cl2-2,4,
o-ClC6H4HgC6H3Cl2-2,4, m-ClC6H4HgC6H3Cl2-2,4, C6H5CH2HgC6H3Cl2-2,4,
C6H5HgC3H3Cl2-2,5, m-ClC6H4HgC6H3Cl2-2,5, CHsHgC6H3Cl2-2,5,
m-ClC6H4HgCH2C6H5 [81, CH2—CHHgR (R=C2H5, СЙН6) [77].
Аналогично получены соединение (CH3)3SiCH2HgR (R = CH3, C6H13,
С6Н5) 19], p-CH2=CHC6H4HgC6H5 [10, 11], C4H9Hg(CH2)5HgC4H9 (из 1
моля BrHg(CH2)5HgBr и 2 молей C4H9MgBr [12]), (—)вторичнобутил(+)-
вторичнобутилртуть [13], C4H9HgC6H4OCH3-o [14], CH2=CHCH2HgC2H5 [15]
(из CH2=CHCH2MgBr и C2H5HgBr).C3H5HgBr n^HsMgJ не даютRHgR',
а образуют Hg, (C2H5)2HgJ и C2H5HgJ; также C3H5HgBr и (C2H6)2Zn дают
(C2H5)2Hg, диаллилртуть и металлическую ртуть [15].
Из ди-1,4-хлормеркурбензола и 1,4-дихлормеркурдурола и CH3MgX (X—
J или С1) получены ди(метилмеркур)бензол и ди-(1,4-метилмеркур)ду-
рол соответственно [16]. Описано получение г(г/с-2-метоксициллогексил-
неофилртути из хлористой г{г/с-2-метоксициклогексилртути и ’хлористого
2-метил-2-фенилпропилмагния [17] .
Несмеяновым с сотр. [18] получены CeH5CH2HgCeHn, C2H5HgC4He-n,
a-C10H7HgC6H2(CH3)3, n-C4H9HgCH2C6H6, C6H5HgCH2C6H5, n-C4H9HgC6Hu.
цис, транс-Дипропенилртуть синтезирована из бромистой цг/с-пропе-
нилртути и т/хшс-пропениллития [19]. Котон с сотр. синтезировал п-сти-
рилфенилртуть [201. Синтезированы также CF2=CF—HgR (R — СН2=СН,
С2Н5, С6Н5) [211.
n-бис-Фенилмеркурбензол получен с выходом 16% из п-бг/с-броммагний-
бензола и бромистой фенилртути (10 час. перемешивания в эфире) [22].
Получение этилбутилртути [18]. В раствор Гриньяра, приготовленный из 10,9 г (0,1
моля) бромистого этила и 2,4 г (0,1 моля) магния в 45 мл сухого эфира, вводят небольшими
порциями 16,88 г (0,05 моля) измельченной бромистой бутилртути при 2—10° С и энергич-
ном перемешивании. Спустя 2 часа реакционную массу разлагают 100 мл 1%-ного раствора
серной кислоты при 5° С. После высушивания эфирного слоя хлористым кальцием раство-
ритель отгоняют под водоструйным насосом. Остаток 13 г (90%) представляет собой под-
вижную жидкость; Пр’5 1,5114.
Получение бензилфенилртути [1]. К 32 г бромистого фенилмагния (4 моля) в абс. эфи-
ре при перемешивании небольшими порциями прибавляют 10,5 г тонко измельченной су-
хой хлористой бензилртути. При умеренном кипении эфира происходит растворение. По-
сле получасового хранения (более продолжительного, а тем более кипения, следует избе-
гать) разлагают 1%-ной серной кислотой, эфир отделяют, сушат и испаряют в вакууме
при 40° С. Остающееся окрашенное в слабо-желтый цвет масло встряхивают в делительной
воронке с 5-кратным объемом холодного абс. спирта, затем вливают при перемешивании в
200 мл кипящего абс. спирта; раствор тотчас фильтруют и масло выделяют вновь сильным
охлаждением. Эту операцию повторяют дважды и затем слегка еще окрашенное масло в
эксикаторе над пятиокисью фосфора при 35° С доводят до постоянного веса [1] (см. так-
же [18]).
\
При взаимодействии бромистой трихлорметилртути с C8H6MgBr наряду
с фенилтрихлорметилртутью получена бромистая фенилртуть [231.
Несимметричные соединения типа AlkHgC=CH (Aik = СН3, С2Н6,
п-С3Н7 и ЛС4Н9) получены реакцией галоидной алкилртути с бромистым
этинилмагнием [24]. £
Синтез этилэтинилртути [24]. Синтез’ведут при постоянном пропускании через аппарат
сухого азота. К охлаждаемому водой со льдом свежеприготовленному раствору бромистого
этинилмагния [25] из 2,07 г (0,086 г-атом) магния в 125 мл тетрагидрофурана при переме-
шивании магнитной мешалкой прибавляют по каплям в течение 50 мин. раствор 8,75 г (0,0247
230 Ртутноорганические соединения. Лит. стр. 236—237
моля) иодистой этилртути в 50 мл тетрагидрофурана. По окончании прибавления реакцион-
ную смесь перемешивают 17 час. при комнатной температуре. Затем ее охлаждают до +5° С
и медленно прибавляют при перемешивании и охлаждении льдом смесь 450 мл 6,6%-ного
водного раствора нитрата аммония и 190 мл эфира. Нижний водный слой отделяют и дваж-
ды извлекают эфиром порциями по 100 мл. Соединенные эфирные экстракты промывают
двумя порциями воды по 200 мл и сушат безводным сульфатом магния. После испарения
растворителя в вакууме остается 5,37 г желтого кристаллического продукта с характер-
ным неприятным запахом. Возгонка вещества при комнатной температуре при давлении
1 мм дает 4,6 г белого кристаллического сублимата. Т. пл. 71—72° С. Выход 73,4%.
Аналогично получены и другие AlkHgC^CH. CH8Hg=CH плавится
при 117—118°С. Высшие члены ряда представляют собой малолетучие жид-
кости.
Получение га-толилфенилртути [4]. К 2 молям бромистого фенилмагния в абс. эфире
прибавляют 1 моль тонкоизмельченной хлористой n-толилртути небольшими порциями
при непрерывном перемешивании. Температуру поддерживают около 5° С, чтобы избежать
разложения продукта на две симметричные молекулы. Экспериментально найдено, что
разложение происходит почти количественно, если смесь держать при температуре кипе-
ния эфира в течение 30 мин.
Смесь затем встряхивают 30 мин. или до тех пор, пока не растворится вся хлористая
толилртуть. Избыток реактива Гриньяра разлагают затем 0,1%-ным раствором серной
кислоты. При этом сосуд погружают в ледяную баню и температуре не дают подняться
выше 10° С. Несимметричное ртутиоорганическое соединение извлекают эфиром, эфириый
экстракт сушат безводным сульфатом натрия, фильтруют, эфир отгоняют в вакууме без
доступа влаги. Полученный продукт тщательно промывают 5 раз спиртом для удаления
сулемы и симметричного продукта и опять сушат в вакууме. Т. разл. —-120°С.
Синтез перфторвинилвниилртути CF2=CFHgCH—СН2 [21]. К эфирному раствору
йодистого перфторвинилмагиия или к раствору бромистого перфторвинилмагния в тетра-
гидрофураие (0,1 a RMgBr в 100 мл растворителя) при сильном перемешивании приливают
эквимолекулярный раствор бромистой винилртути в тех же растворителях при —10° С.
После прибавления RHgBr перемешивание продолжают при 0° С в течение 12 час. Раство-
ритель отгоняют в вакууме, остаток подвергают перегонке. Т. кип. перфторвииилвинил-
ртути 45°С/2 мм. Выход 35%, 1,5220, 2,9510.
Также получена перфторвинилэтилртуть (выход 30%, т. кип. 53° С/
/12 мм, п.% 1,4711, df 2,7805).
СИНТЕЗ RHgR'
ДЕКАРБОКСИЛИРОВАНИЕМ RCOOHgR'
Хараш предложил свой метод синтеза ртутноорганических соединений
декарбоксилированием ртутных солей карбоновых кислот (см. гл. IX) для
синтеза несимметричных ртутноорганических соединений [26]. При нагре-
вании в высоком вакууме ртутноорганических солей карбоновых кислот,
получаемых действием хлористой арил(алкил)ртути на серебряные соли
карбоновых кислот, выделяется углекислота и образуется несимметричное
соединение ртути:
RCOOAg + CIHgR' -> AgCl + RCOOHgR' -» CO2 + RHgR'.
Ртутноорганические соли карбоновых кислот могут быть получены так-
же действием карбоновой кислоты на гидроокись ар ил (алкил) ртути
[27]. Метод применим к кислотам с легко отщепляемым карбоксилом,
особенно в тех случаях, когда R представляет собой радикал с резко
отрицательными заместителями. Этого типа несимметричные соединения
устойчивы и не склонны превращаться в смесь двух симметричных со-
единений.
Получение фенил-2,4,6-тринитрофенилртути [26]. К взвеси 3 г серебряной соли три-
нитробензойной кислоты в 30 мл бензола прибавляют при встряхивании 2,4 г хлористой
фенилртути Выпадает плотный белый осадок. Встряхивают 30 мин., затем заливают 20 мл
Синтез несимметричных ртутноорганических соединений RHgR' 231
бензола и извлекают в течение нескольких часов в экстракционном аппарате. Экстракт
охлаждают, выделившуюся ртутную соль отфильтровывают и сушат в вакууме над серной
кислотой. Выход 3,1 г, т. пл. 228° С.
Сосуд с 2 г фенилмеркуртринитробензоата присоединяют к высоковакуумному насо-
су и нагревают на бане до 222°С. При этой температуре держат в течение 5 мин., причем
содержимое сосуда темнеет и сжимается. Держат еще в течение 5 мин. при 226° С и затем
поднимают температуру до 228° С. Как только вещество начнет плавиться, баню удаляют
и насос отключают. Сероватый продукт промывают на фильтре 15—20 мл эфира. Выход
1,73 г. После кристаллизации из бензола и промывания эфиром т. пл. 227,5 ° С.
Подобным же образом получены и-толил-2,4,6-тринитрофенилртуть
{26, 27] и этил-2,4,6-тринитрофенилртуть 127]. Синтез CCl3HgCeH6 взаимо-
действием CCl3COONa с CeH5HgCl, по мнению Логана [27а], является реак-
цией декарбоксилирования промежуточно образующегося CCl3COOHgCeH5,
а не внедрением дигалокарбена по связи Hg—С.
СИНТЕЗ RHgAr' АРИЛИРОВАНИЕМ RHgOH
АРОМАТИЧЕСКИМИ СОЕДИНЕНИЯМИ ОЛОВА, СУРЬМЫ(Ш) И БОРА
Не имеющим ограничений, свойственных синтезу несимметричных сое-
динений ртути через магнийорганические соединения (невозможность полу-
чения соединений с реакционноспособными заместителями) и декарбокси-
лированию ртутноорганических солей карбоновых кислот (приложимому
лишь к легко отдающим СО2 кислотам), является метод синтеза RHgR'
арилированием в щелочной среде ртутноорганических гидроокисей арома-
тическими соединениями олова, трехвалентной сурьмы и бора (Несмеянов,
Фрейдлина, Кочещков) [28]:
2RHgOH + RgSnO 2RHgR' + H3SnO3,
RHgOH + R'B(OH)>-> RHgR'4- HSBOS,
2RHgOH + 2R'SbO -» 2RHgR' + Sb2Os + HaO.,
При сливании вместе кипящих растворов (или взвеси) ртутноорганиче-
ской соли и арилирующего соединения в водно-спиртовой щелочи, не-
продолжительном нагревании смеси до температуры не выше 80° С и ох-
лаждении выкристаллизовывается несимметричное соединение ртути.
В случае жидкого несимметричного соединения его извлекают из реакцион-
ной смеси эфиром. При необходимости реакцию можно вести и при комнат-
ной температуре.
Синтез я-китрофенилфенилртути (28]. Кипящий раствор 1,9 г (0,005 моля) хлористой
л-нитрофенилртути в 120 мл спирта смешивают с 5 мл 20%-ного водного раствора едкого
натра и с раствором 1 г (0,003 моля) хлористого дифенилолова в 20 мл спирта. Смесь
фильтруют горячей и фильтрат разбавляют 100 мл воды. Полученную взвесь после
оседания (15—20 мин.) отсасывают. Осадок n-нитрофенилфенилртути весит 2,1 г (коли-
чественный выход). После двухкратной кристаллизации из бензола т. пл. 144—145° С
(с разложением).
Синтез метилфенилртути [28]. Сливают вместе кипящие растворы 3,4 г (0,01 моля)
иодистой метидртути в спирте и 1,2 г (0,01 моля) фенилборной кислоты в 20 мл спирта и
5 мл 20%-ного едкого иатра. Смесь разбавляют двухкратным объемом воды, извлекают
эфиром; эфирный слой промывают 25%-ным едким натром, водой и сушат хлористым каль-
цием. После отгонкн эфира продукт реакции весит 1,85 г (81%).
Так же из хлористой ц-хлорфенилртути и фенилборной кислоты полу-
чена с 80%-ным выходом n-хлорфенилфенилртуть [29].
Синтез га-хлорфеиилбензилртути [28]. Сливают вместе раствор 2 г (0,08 моля) п-хлор-
фенилстибиноксида в 200 мл кипящего спирта, 5 мл 20%-ного едкого натра и 2,6 г (0,08
моля) кипящего спиртового раствора хлористой бензилртути; горячую смесь отфильтро-
вывают, фильтрат разбавляют равным объемом воды. Через 2 часа образовавшийся осадок
отсасывают и сушат. Выход 56%. После двухкратной кристаллизации из лигроина т. пл.
104—108° С.
232 ' Ртутноорганические соединения. Лит. стр. 236—237
Продукты присоединения солей ртути к двойной связи ненасыщенных сое-
динений также могут быть арилированы в щелочной среде ароматическими
соединениями оловаи давать своеобразные несимметричные ртутнооргани-
ческие соединения [301.
2HOCH3CH3HgX‘+ Ar3SnX3 + 4NaOH 2HOCH3CH3HgAr + H3SnO3 + 4NaX + H3O.
(см. также гл. VI, стр. 117).
Синтез Р-оксиэтил-га-толилртути [30}. Горячий раствор 3,02 г (0,007 моля) хлористого
ди-я-тол идолов а в 10 мл этилового спирта вливают в кипящий раствор 3,25 г (0,01 моля)
бромистой p-оксиэтилртути в смеси 70 мл. спирта и 5 мл 20%-кого раствора едкого кали.
Кипятят 3 мин. на водяной бане, разбавляют 150 мл холодной воды и извлекают эфиром.
Эфирный слой промывают водой, сушат сульфатом натрия и эфир отгоняют в вакууме.
Выход Р-оксиэтил-п-толилртути 1,85 г (55%). После двухкратной кристаллизации из пет-
ролейного эфира получены бесцветные кристаллы. Т. пл. 52,5—53,5° С.
Взаимодействие бромистой трихлорметилртути с дифенилдихлор стан-
наном протекает по разным направлениям в зависимости от количества
взятой в реакцию щелочи [23]. При стехиометрическом отношении реаген-
тов в соответствии с уравнением
2CClsHgBr + (CeH5)3SnCt3 + 6NaOH -> 2CCl3HgCeH6 + Na3SnOs + 2NaCl + 2NaBr + 3HaO
получена фенилтрихлорметилртуть с выходом 49%. С большим количест-
вом щелочи реакция идет с образованием дифенилртути(29%). В обоих слу-
чаях образуется значительное количество неплавких и нерастворимых
осадков, содержащих ртуть.
Синтез феиил-о-оксициклогексилртути [30]. Сливают вместе горячие растворы 1,8 г
(0,005 моля) уксуснокислой о-оксициклогексилртути в 40 мл этилового спирта, 0,9 г (0,025
моля) хлористого дифенилолова в 10 мл спирта и 5 ли 25%-ного раствора едкого натра и
нагревают-3 мин. на водяной бане. Высаженный водой осадок (1,6 г; 85%) дважды перекри-
сталлизовывают из петролейного эфира. Т. пл. 101—102° С.
Действие фенилборной кислоты (в водно-спиртовой щелочи, при ки-
пячении) на CHCl2HgCl приводит к C6H5HgCHCla [39а].
СИНТЕЗ RHgR' ДЕЙСТВИЕМ ДИАЗОМЕТАНА НА RHgCl
Особым случаем получения несимметричных ртутноорганических сое-
динений является действие диазометана на RHgCl (R=CeH5, CH3CeH4,
С6Н5СН2) [31], на CCl8HgBr [32] и на ArHgO2CR [33, 34], например;
RHgCl -Ь CH3N3 RHgCH3Cl + Ns.
Из полученных несимметричных соединений устойчивы лишь хлорме-
тилбензилртуть CCl3HgCH2Br и ArHgCH2OCOR, остальные тотчас разла-
гаются на смесь дихлорметилртути и диарилртути.
Получение беизилхлорметилртути [31]. В хорошо перемешиваемую охлажденную до
0°С взвесь 8,18 г хлористой бензилртути в 200 мл эфира прибавляют 1,05 г (5%-иый из-
быток) диазометана в эфирном растворе. Реакция идет медленно с выделением азота, ко-
торый прекращает выделяться после того, как прибавлен 1 эквивалент диазометана; в это
время вся взвесь растворяется. После испарения эфира и избытка диазометана в сухом
воздухе остается незакристаллизовывающееся масло CeHgCHgHgCHsCl, которое сушат
до постоянного веса в вакуум-эксикаторе над парафином и пятиокисью фосфора.
Крайне неустойчивы и существуют только при низких температурах
такого типа несимметричные соединения, полученные из этанолмеркурсо-
лей, HOCH2CH2HgCH2X [35].
Синтез несимметричных ртутноорганических соединений RHgR'
233
СИНТЕЗ RHgR' ПРИ ПОМОЩИ ДИГАЛОКАРБЕНОВ
Дигалокарбен (дихлоркарбен, получаемый разложением CCl3COONa
[36, ср. 27а] или этилтрихлорацетата [37], дихлор*[38] или дибром-[38]
карбены, получаемые действием третичнобутилата калия на галоформ) спо-
собен внедряться по связи Hg—Hal с образованием трихлорметильных,
в том числе несимметричных соединений ртути[(Разуваев и сотр. [36], Реутов
и Ловцова [38, 39а]):
[Cl HgCl — -CCl3HgCI,
ArHgCl ArHgCCig.
Последнюю реакцию ведут в бензоле при комнатной температуре [38—
39а]. Аналогично получены ArHgCHHal2, если взять СН2На12 вместо
СННа13 в реакцию с третичнобутилатом калия и ArHgHal [39а].
fArHgHal - СННа1^ ArHgCHHah.
По мнению Сейферта [40], в случае применения третичнобутилата калия
и галоформа имеет место простое вытеснение галоиданиона СС13-анионом:
ArHgBr 4-СС1"-> ArHgCCIa 4-Вг“.
Получение феннлтрихлорметилртути [38]. К суспензии 4,9 г (0,016 моля) фенилмеркур-
хлорида в 100 мл абс. бензола добавляют 5 мг (0,062 моля) абс. хлороформа и постепенно-
при перемешивании и охлаждении ледяной водой—третичнобутилаткалия из 1,2 а
(0,03 г-атом) калия. После 30 мин. перемешивания почерневшую реакционную смесь выли-
вают в воду, осадок отфильтровывают, промывают бензолом. После испарения бензола по-
лучают 1,7 г CeHaHgCCh, т. пл. 114° С (из спирта). Осадок, не растворившийся в бензоле,
экстрагируют горячим ацетоном, после испарения которого выделяют смесь фенилмеркур-
хлорида и феннлтрихлорметилртути (2,32 а). Из нее холодным бензолом вымывают 1,55 г
CeHsHgCCla с т. пл. 112—114Q С. В остатке 0,55 г непрореагировавшего фенилмеркурхло-
рида с т. пл. 256° С. Общий выход феннлтрихлорметилртути 3,25 г (52%).
СС12 спосооен внедряться в одну С—Н-связь некоторых симметричных
ртутноорганических соединений с ртутью, связанной с вторичным угле-
родом. Эти соединения при термической обработке распадаются не на
смесь двух симметричных ртутноорганических соединений, а на ртутно-
органическую соль и непредельное соединение. Внедрение CHHal по HgX-
связи ртутноорганических соединений произведено по схеме [76]:
RHgX +ГСН2Х2 + (СНз)зСОК-> RHgCHX2 + КХ + (СН3)8СОН.
Реакцию ведут в абсолютном бензоле при охлаждении ледяной водой.
CHHTE^RHgR' «СОСИММЕТРИЗАЦИЕЙ» RHgX и R HgX
Несимметричные соединения RHgR' получаются при «сосимметризации»
RHgX и R'HgX, где Rt4=R', осуществляемой действием аммиака в хлоро-
форме и других органических растворителях. Так, «сосимметризованы»
p-XC6H4CH(HgBr)COOC2H5 и p-X'C6H4CH(HgBr)COOC2H5 [41, 42], этило-
вые эфиры а-броммеркурарилуксусных кислот с бромистой бензил-
ртутью [43].
Механизм этой реакции описан в начале гл. XIII.
234
Ртутноорганические соединения. Лит. стр. 236—237
СИНТЕЗ RHgR' ДЕЙСТВИЕМ RHgX (и R2Hg)
НА СОЕДИНЕНИЯ С ПОДВИЖНЫМ ВОДОРОДОМ
Соединения с подвижным водородом: ацетилен [44], фенилацетилен
[45], флуорен [45], циклопентадиен [45], инден [45], трифенилметан [45],
пропионовая [45], малоновая [45], циануксусная [45] кислоты, а также
трихлорэтилен [45] и наряду с этим супер ароматические гетероциклы, как
тиофен [46] и, неожиданно, акридин [46], 2,6-диметилпиридин [46], хи-
нальдин [46] — меркурируются ртутноорганическими основаниями RHgOH,
давая несимметричные соединения по схеме:
RH + R'HgOH RHgR' + Н2О.
(см. гл. V, стр. 54).
Синтез трихлорэтиленциклогексилртути [45]. 30 г гидроокиси циклогексилртути в
200 мл бензола обрабатывают 15 г трихлорэтилена. После отделения растворителя остается
кристаллический осадок. Т. пл. 44я С.
Синтез 2,4-дифенилтиенилфенилртути [46].
СвН5
. АА
CeHsHg S СбН5
Из смеси спиртовых растворов 11,96 г гидроокиси фенилртути и 4,72 г 2,4-дифенил-
тиофеиа выпадает продукт с т. пл. 114° С.
Неустойчивые несимметричные продукты присоединения ароматических
ртутноорганических оснований к непредельным соединениям, например:
OR
ArHgOH + СН2 = CHOR ArHgCHaCH^
\)СН,
тотчас распадаются с образованием двух симметричных ртутноорганических
соединений [47] (см. гл. XIII).
Гидроокись арилртути мер курирует также p-дикарбонильные соедине-
ния, образуя моно- и димер кур ирова иные С-производные, которые в боль-
шинстве случаев неустойчивы и легко симметризуются [48]. Выделено ди-
арилртутное производное дибензоилметана [48]. Устойчивы продукты
меркурирования» гидроокисью арилртути р-дисульфонильных соединений
[48].,
В мягких условиях (в спиртовом растворе, при комнатной температуре
или при слабом нагревании) и гидроокись [48] и ацетат [49] арилртути
образуют с фенолами Hg—О-производные, неспособные превращаться в
меркурированные фенолы.
Hg—О-продукт взаимодействия дивинилртути с фенолом легко распа-
дается с выделением металлической ртути и винилового эфира фенола [49].
Диарилртуть [50] с пентахлорфенолом и фенилмер кур ацетат с полихлор-
фенолами [51], гидроокись и ацетат фенилртути с полигидрофенолами [52]
также образуют HgO-производные.
Согласно же Котону и сотр., гидроокись [53] и ацетат [54] фенил-
ртути, полнозамещенные симметричные ртутноорганические соединения,
такие как Alk2Hg(Alk = СН3 [55], С2Н5 [56], п-С5Ни [57]) и Ar2Hg (Аг =
=С6Н5 [54, 58, 59], о-[60] и р- [60] СН3С6Н4, р-СН3ОС6Н4 159. 61, 62],
о-НОС6Н4 [62], о-[62, 63], т-[62, 63] и р-[591 NO2GeH4, а-С10Н7 [58],
p-NH2C6H4 [59, 62, 64]), а также полнозамещенные несимметричные соедине-
Синтез несимметричных ртутноорганических соединений RHgR' 235
ния ртути RHgR' [651 (CH3HgC6H5, CH3HgC10H7-a, C6H5HgC10H7-a) меркури-
руют фенолы с образованием несимметричных ртутноорганических соеди-
нений. По мнению авторов, при этом образуются моно-, ди- и тримерку-
рированные соединения в зависимости от характера заместителей в феноле.
Реакции эти проводили при 130—140° С в запаянных ампулах без раствори-
теля (в единичных случаях в спирте, в случае диарилртути также в кипя-
щем ксилоле) в течение нескольких часов.
Такое нагревание R2Hg и RHgX (в том числе RHgHal [66]) с полифе-
нолами и а- и ^-нафтолом приводит к разрушению ртутноорганического сое-
динения с выделением металлической ртути и в случае большей части
ArHgHal—Hg2Hal2. Дибензилртуть [67], так же реагирует со всеми фено-
лами с выделением металлической ртути.
Дифенилртуть, согласно этим же авторам, меркурирует (при 150°С,
3 часа) салициловый альдегид и следующие кетоны: ацетофенон, бензальаце-
тон, бенз аль ацетофенон с образованием мономеркурированных соединений
168].
Ди-л-тол ил ртуть меркурирует в этих же условиях уксусный, пропионо-
вый, масляный альдегиды, бензальдегид, о- и ж-оксибензальдегиды [69]. Ак-
ролеин в этих же условиях дает димеркурированный продукт (при 180° С —
монозамещенный продукт), а паральдегид (120—150° С) образует ди-п-
толилмеркуркротоновый альдегид, в присутствии воды (3 часа, 150°С)
образуется металлическая ртуть и /z-толилмеркуркротоновый альде-
гид. Введены в реакцию в этих же условиях с ди-п-аминофенилртутью
пропионовый и масляный альдегиды, акролеин, бензальдегид, о-оксибен-
зальдегид с ’ образованием п-аминофенилмеркуралкил(арил)бензальде-
гида [69].
Однако в свете работ Несмеянова и Кравцова [48], доказавших строение
продуктов взаимодействия ArHgOH с фенолами, нитрозофенолами и т. п.
как ArHgO-соединений (см. выше, а также гл. XIV, стр. 336), строение по-
лученных Котоном и сотр. продуктов взаимодействия ртутноорганических
соединений с фенолами как ArHgC-соединений следует поставить под сом-
нение.
В соответствии с высокой термической устойчивостью перфторированных
соединений ртути несимметричное ртутноорганическое соединение CH3HgC6F5
отличается особой устойчивостью — оно не разлагается при нагрева-
нии до 200°С и получается нагреванием диметилртути и бис-пентафторфе-
нилртути [70].
Получение метилпентафторфенилртути [70]. 0,5 г бос-пентафторфенилртути и избыток
диметилртути помещают в эвакуированную трубку Кариуса и нагревают до 60Q С. Твер-
дое вещество тотчас растворяется, но вновь выкристаллизовывается при охлаждении.
После того как смесь нагревается до 60° С, в течение 12 час. н по охлаждении осадок уже не
выкристаллизовывается, трубку вскрывают и отгоняют в вакууме избыток диметилртути.
Остаток с т. пл. 34° С представляет собой метилпентафторфенилртуть.
СИНТЕЗ RHgR' РЕАКЦИЕЙ ПЕРЕРАСПРЕДЕЛЕНИЯ РАДИКАЛОВ
В РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Несимметричные ртутноорганические соединения получены [71] реак-
цией перераспределения радикалов в ртутноорганических соединениях по
схеме:
RaHg 4- R'HgX RHgX + RHgR'.
Так, при реакции хлористой карбометоксиртути с R2Hg (R — С6Н5,
/>-СН3ОС6Н4, п-С4Н9, СО2СН3) получены RHgCO2CH3.
236
Ртутноорганические соединения
Синтез фенилкарбометокснртути [71]. Раствор дифенилртути (10,5 г, 0,030 моля) в
теплом (55° С) бензоле (100 мл) прибавляют к раствору хлористой карбометоксиртути
(10 г, 0,033 моля) в теплом бензоле (100 мл). Тотчас выпадает осадок хлористой фенилрту-
ти. Смесь охлаждают, отфильтровывают хлористую фенилртуть. Фильтрат испаряют до-
суха в вакууме при температуре ниже 50° С. Кристаллизуют из эфира при охлаждении.
Выход 8 г (70%). Т. пл. 68—69,5е С.
Карбометокси-п-метоксифенилртуть с т. пл. 89°С и н-бутилкарбомет-
оксиртуть, представляющая собой масло, получены подобным же образом;
в последнем случае реакцию ведут в спирте.
Калингерт с сотр. [72—74] сообщил об образовании RHgR' при вза-
имодействии R2Hg с R2Hg или R4Pb в присутствии катализатора
А1С13.
При взаимодействии R3Bi с R2Hg обнаружен обмен радикалами [75].
ЛИ ТЕРАТУРА
1. HilpertS., GriittnerG. Ber., 48, 906 (1915).
2. Steinkopf W., Bielenberg W., Augenstad-Jensen H. Ann., 430,
41 (1923).
3. Go ddard D. J. Chem. Soc., 123, 1169 (1923).
4. Kharasch M. S., Marker R. J. Am. Chem. Soc., 48, 3130 (1926).
5. Kharasch M. S., F 1 e n n e r A. L. J. Am. Chem. Soc., 54, 674 (1932).
6. Kharasch M. S„ Pines H., L e v i n e J. H. J. Org. Chem., 3, 347 (1938).
7. Kharasch M. S., SwartzS. J. Org. Chem., 3, 405 (1938).
8. Kharasch M. S., Legau 1 t R. R., SprowlsW. R. J. Org. Chem., 3, 409
(1938).
9. Whitmore F. C., S о m m e r L. H. J. Am. Chem. Soc., 68, 481 (1946).
10. К о т о и M. M., Киселева T. M., ’ Флоринский Ф. С. Изв. АН СССР,
ОХН, 1959, 948.
11. Котой M. M., Киселева Т.М. ДАН СССР, 131, 1072 (1960).
12. В a h г G., М е i е г G. Z. anorg. allg. Chem., 294, 22 (1958).
13. J е п s е n F. R. J. Am. Chem. Soc., 82, 2469 (1960).
14. Б о p и с о в A. E., Осипова M. А. Изв. АН СССР, ОХН, 1961, 1039.
15. R о t h s t e i n E., S a v i 1 1 e R. W. J. Chem. Soc., 1952, 2987.
16. W r i g h t G. F„ Sawat zky H. Can. J. Chem., 36, 1555 (1958).
17. W i n s t e i n S., T г а у 1 о r T. G., Garner C. S. J. Am. Chem. Soc., 77, 3741
(1955).
18. Несмеянов A. H., Б о p и с о в E. А., Г о л у б e в а Е. И., Ковредов А.Н.
Изв. АН СССР, ОХН, 1961, 1582.
19. Б о р и с о в4 А. Е. Изв. АН СССР, ОХН, 1961, 1036.
20. К о т о н М. М., Киселева Т. М., Флоринский Ф. Т. Высокомолекул. соед.,
2, 1639 (1960). »
21. Стерлии Р. Н., Ли Вей-ган, Кнунянц И. Л. ДАН СССР, 140, 137 (1961).
22. М а 1 n а г М., G г d е n 1 с D. J. Chem. Soc., 1959, 3639.
23. Несмеянов А. Н., Фрейдлина Р. X., Величко Ф. К- ДАН СССР, 114,
557 (1957).
24. К г a u t М., L е i t с h L. С. Can. J. Chem., 43, 549 (1963).
25. Organic Syntheses, N. Y., John Wiley Sons, 1959, vol. 39, p. 56.
26. К h a r a s c h M. S., G r a f f 1 i n M. W. J. Am. Chem. Soc., 47, 1948 (1925).
27. К о t e n J. A., A d a m s R. J. Am. Chem. Soc., 46, 2768 (1924).
27a. Logan T. Y. J.,Org. Chem., 28, 1129 (1963).
28. Фрейдлина P. X., H e с м e я н о в А. Н., К о ч е ш к о в К- А. ЖОХ, 5, 1171
(1935); .Вег., 68, 565 (1935).
29. G i 1 m а n Н., Y а 1 е Н. L. J. Am. Chem. Soc., 72, 8 (1950).
30. Несмеяно в А. Н., Фрейдлниа Р. X. ЖОХ, 7 (1937); Вег., 69, 1631 (1936).
31. Н е 1 1 е г т a n L., NewmanM. D. J. Am. Chem. Soc., 54, 2859 (1932).
32. Ф р е й д л и и а Р. X., В е л и ч к о Ф. К. Изв. АН СССР, ОХН, 1959, 1225.
33. Р f е i f f е г Р., J a g е г Н. Вег., 80, 1 (1947).
34. Pfeiffer Р., Schulze-BentropR., La Roche К- Н. Вег., 85, 232
(1952).
35. Фрейдлина Р. X., НесмеяновА. Н., Токарева Ф. А. ЖОХ, 7, 262
(1937); Вег., 69, 2019 (1936).
36. Разуваев Г. А., Василейская Н. С., Никитин а Л. А. Тр. по химии
и хим. технологии (Горький), 1960, № 3, 638.
37. S с h w е i z е г Е. Е„ О’ N е i 1 1 G. J. J. Org. Chem., 28, 851 (1963).
Синтез несимметрических ртутноорганических соединений RHgR'
237
38. РеутовО. А., Л о в ц о в а А. Н. Изв. АН СССР, ОХН, 1960, 1716; ДАН СССР,
139, 622 (1961).
39. Seyferth D., BurlitchJ. М., HeerenJ. К. J. Org. Chem., 27, 1491 (1962).
39a. Реутов О. А., ЛовцоваА. H. ДАН СССР, 154, 166 (1964).
40. S е у f е г t h D., В u г 1 i t с h J. М. J. Am. Chem. Soc., 84, 1757 (1962).
41. РеутовО. А., Белецкая И. П. ДАН СССР, 131, 853 (1960).
42, Б ел ец к а я И. П., А р т а м к и н а Г. А., Р е у т о в О. А. ДАН СССР, 149, 90
(1963).
43. Б е л е ц к а я И, П., Артамкина Г. А., Р е у т о в О. А. Изв, АН СССР, ОХН,
1963, 765.
44. S р a h г R. J., V о g t R. R., N ieuwland J. A. J. Am. Chem. Soc., 55, 2465,
3728 (1933).
45. Франц, пат. 812603 (1936); Zbl., 1937, II, 1895.
46. Пат. США 2085065 (1938); Zbl., 1938, I, 374.
47. Луценко И. Ф., Юркова E. И. Изв. АН СССР, ОХН, 1956, 27.
48. Несмеянов А. Н., Кравцов Д. Н. Изв. АН СССР, ОХН, 1962, 431.
49. F о s t е г D. J., Т о b 1 е г Е. J. Am. Chem. Soc., 83, 851 (1961).
50. L е а п d г i G., S р i п е 1 1 i D., S а 1 v е т i п i A. Ann. chim., 50, 1046 (1960).
51. Японок, пат. 6547 (1960); РЖХим, 1961, 21 Л 228.
52. Франц, пат. 827299 (1938); С. А., 32, 7923 (1938). Франц, пат. 49782; С. А., 36, 2566
(1942).
53. Лаб у зов С. М. ЖОХ, 22, 93 (1952).
54. Котой М. М., ЖОХ, 20, 2096 (1950).
55. КотонМ. М. ЖОХ, 22, 1136 (1952).
56. КотонМ. М. ЖОХ, 19, 730 (1949).
57. КотонМ. М. ЖОХ, 19, 734 (1949).
58. Котов М. М., Зор ин а Т. М. ЖОХ, 17, 1220 (1947).
59. Мартынова В. Ф. ЖОХ, 26, 894 (1956).
60. КотонМ. М., Ч е р н о в И. А. ЖОХ, 19, 2104 (1949).
61. КотонМ. М., Мартынова В. Ф. ЖОХ, 25, 705 (1955).
62. К о т о н М..М., Мартынова В. Ф. Изв. АН СССР, ОХН, 1955, 1063.
63. М а р т ы н о в а В. Ф. ЖОХ, 27, 1056 (1957).
64. КотонМ. М., Мартынова В. Ф. ЖОХ, 25, 594 (1955).
65. К о т о н М. М., Большакова А. А. ЖОХ, 23, 2023 (1953).
66. Ко т о н М. М„ 3 о р и н а Т. М. .ЖОХ, 17, 1303 (1947).
67. К о т о н М. М., Большакова А. А. ЖОХ, 18, 1290 (1948).
68. К о т о н М. М., 3 о р и н а Т. М. ЖОХ, 19, 1137 (1949).
69. М а р т ы н о в а В. Ф. ЖОХ, 32, 962 (1962).
70. С h а m b е г s R. D., С о a t е s G. Е., Livingstone J. G., Musgrave
W. К. R. J. Chem. Soc., 1963, 4367.
71. P a u I i k F. E., D easy R. E. Chem. and Ind., 37, 1650 (1962).
72. Calingaert G., В e a 11 у H. A., N e a 1 H. R. J. Am. Chem. Soc., 61, 2755(1939).
73. Calingaert G., S о г о о s H., H n i z d a V. J. Am. Chem. Soc., 62, 1107 (1940),
74. Ca lingaer t G., Soroos H„ Sh a p iro H. J. Am. Chem. Soc., 63, 947 (1941).
75. Challenger F., Ridgway L. R.J. Chem. Soc., 121, 104 (1922).
76. Land grebe J. A., Mathis R. D. J, Am. Chem. Soc., 86, 254 (1964).
77. В ar to ch a B., Stone F. G. A. Z. Naturforsch., 13b, 347 (1958).
Глава XIII
СИММЕТРИЗАЦИЯ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
И ОБРАТНАЯ ЕЙ РЕАКЦИЯ
Большинство методов синтеза органических соединений ртути при-
водит к соединениям типа RHgX, поэтому одной из важнейших реакций
ртутноорганических соединений является реакция симметризации — пе-
реход ртутноорганических солей RHgX к полнозамещенным ртутнооргани-
ческим соединениям RaHg. Схематически можно представить, что ртутно-
органическая соль RHgX находится в равновесии с продуктами «симмет-
ризации»:
2RHgX RaHg + HgX2.
Для того чтобы реакция прошла до конца в сторону образования R2Hg,
необходимо присутствие реактива, полностью уводящего ион ртути в виде
или нерастворимой соли (например, HgS, HgFe(CO)4), комплекса (напри-
мер K2HgJ4), или металлической ртути и тем смещающего равновесие вправо.
Таким образом, симметризующими агентами являются восстановители:
металлы (Na, амальгама натрия и др. металлов, Си, Zn), щелочной раствор
станнита натрия, гипосульфит, гидразин и его производные и комплек-
сообразователи: K(Na)J, KCN, KCNS, Na2S2O3, NH8 (сюда же в некото-
рых случаях могут быть отнесены K2S, NaOH, трифенилфосфин). Получе-
ние полнозамещенных, ртутноорганических соединений при электролизе
некоторых ртутноорганических солей является также восстановительной
симметризацией. В тех случаях, когда анион ртутноорганической соли
способен образовать с ионом ртути нерастворимое соединение, симметри-
зация иногда происходит в отсутствие других симметризующих агентов;
такова симметризация ртутноорганических сульфидов и тиосульфатов,
идущая лишь при умеренном нагревании, например:
(RHg)2S ^R2Hg + HgS.
Диспропорционирование несимметричных ртутноорганических соеди-
нений с образованием смеси двух симметричных соединений
2RHgR' R2Hg + R'Hg
лежит в основе симметризации действием непредельных соединений (см.
стр. 256) и некоторых элементоорганических соединений (бутиллития;
см. стр. 257). Немногие, являющиеся исключениями, случаи симметриза-
ции в отсутствие восстановителей или комплексообразователей описаны
ниже (стр. 258),
Симметризация ртутноорганических соединений
23»
Исследованы кинетика, стереохимия и механизм реакции симметриза-
ции ртутноорганических соединений и обратной реакции. Несмеяновым,
Реутовым и сотр. найдено, что симметризация оптически активных ртут-
ноорганических соединений—диастереомеров Z-ментиловых [1—4] и эти-
лового [4] эфиров а-броммеркурфенилуксусной кислоты под действием ам-
миака — является реакцией второго порядка (как по RHgX, так и по NH3
[5]) (S£ 2-механизм) и протекает с сохранением конфигурации у асим-
метрического атома углерода, затрагиваемого в ходе реакции, на основа-
нии чего ими высказано [1—3] правило о сохранении конфигурации при
реакциях электрофильного замещения у насыщенного углеродного атома
(ср. правило о сохранении конфигурации у ненасыщенного атома углерода
при реакциях электрофильного и гомолитического замещения, выведенное
на основании изучения поведения квазикомплексных ртутноорганических
соединений; см. гл. VI).
Величина бимолекулярйой константы скорости симметризации под дей-
ствием аммиака для C6H5CH(HgBr)COOR зависит от природы R [6] и для
p-XC6H4CH(HgBr)COOC2H5 — от природы X [7]. Влияние заместителей
X удовлетворяет уравнению Гаммета [81. Скорость реакции симметризации
p-XC6H4CH(HgBr)COOC2H5 повышается, если в реакцию вводить неодина-
ковые молекулы этих ртутноорганических солей, половина которых со-
держит в качестве X электронодонорный и половина — электроноакцеп-
торный заместители, что облегчает разрыв связей С—Hg и Hg—Вг в пе-
реходном состоянии [9, 10].
При помощи меченой ртути показано, что при такой «сосимметризации»
в образующееся RHgR' переходит преимущественно атом ртути из мо-
лекулы ртутноорганической соли, содержащей менее электроотрицательный
заместитель X, что, по мнению Реутова и др., согласуется с представле-
нием о механизме реакции, как идущей через четырехцентровое пере-
ходное состояние (ср., однако, работы Дженсена [44])..
В реакцию «сосимметризации» с этиловыми эфирами а-броммеркурарил-
уксусных кислот вступает бензилмеркурбромид, который сам не симмет-
ризуется аммиаком. При этом разрыв связи С—Hg в четырехцентровом
переходном состоянии осуществляется в молекуле бромистой бензилртути,
как это доказано [11] применением бромистой бензилртути, меченной Hg203.
Ниже описана реакция, обратная симметризации, в случае R^=R'(стр.
259).
Симметризация устойчивых диастереоизомеров 3-броммеркур4-камфо-
ры и З-броммеркур-Й-камфоры при действии тиосульфата натрия протекает
с сохранением конфигурации [12], и, следовательно, эта реакция также яв-
ляется реакцией электрофильного замещения.
С другой стороны, на примере симметризации иодистой 2-метоксицикло-
гексилртути [13] и бромистой вторичнобутилртути [14] под действием
станнита натрия показано, что симметризация, протекающая по радикаль-
ному механизму, происходит с обращением конфигурации. В последнем
случае обе алкильные группы рацемизуются.
Естественно, однако, что обращения конфигурации не происходит даже
при участии симметризатора, действующего по радикальному механизму —
станнита натрия — в том случае, если атом ртути жестко закреплен у го-
ловы моста, как это имеет место в случае 4-хлормеркуркамфана [15].
Факт обращения конфигурации при симметризации 3-броммеркуркам-
1 форы [16, 17] продуктов присоединения солей ртути к непредельным сое-
‘ динениям [19] под действием гидразингидрата показывает, что симметри-
; зация под действием этого реагента также является гомолитической [16,17].
Стереоизомерные алкилртутные соли (бромистые цис- и трцнс-4-метил-
1 циклогексилртуть, бромистая £-(—)вторичнобутилртуть) симметризуются
240
Ртутноорганические соединения. Лит. стр. 260—263
действием магния стереоспецифйчно с 85—97%-ным сохранением конфи-
гурации [20]. Предложен механизм этой реакции с участием органического
соединения одновалентной ртути [20].
Протекающая по гомолитическому механизму симметризация электро-
лизом происходит с рацемизацией [21].
Райтом [22] показано, что реакция, обратная симметризации, также про-
текает с сохранением конфигурации. Это же показано Несмеяновым и
Реутовым [23, 24] в частичное исправление их прежних данных [1—3].
Реутов И сотр. считают, что ту же кинетику и стереохимию, что
и реакция симметризации (под действием аммиака [1—10], тиосульфата
натрия [12])
2RsCHgX -» (RsC)2Hg + HgX, (1)
и реакция, обратная симметризации [23, 24]
(RsC)2Hg+ HgXa 2R3CHgX, (2)
имеют и другие реакции электрофильного замещения у насыщенного атома
углерода — реакции изотопного обмена ртутноорганических соединений, с
соединениями, содержащими меченую ртуть:
RsCHgX + Hg203xa R3CHg20SX + HgX2 [25-37], (3)
RsCHgX + RHg®osX R<tCHg203X + RHgX [38], (4)
(RsC)aHg + RsCHg203X (RsC)2Hg®03 RsCHgX [39]. (5)
Реакции (3—5) бимолекулярные, имеют первый порядок по каждому из
компонентов и протекают с сохранением конфигурации. На этом основа-
нии Реутовым [40] предложен сходный механизм для этих реакций (5^2),
который он изображает общей схемой с одним и тем же для этих реакций
переходным состоянием;
X
X Y Hg^ X
RsC— HgZ + ^HgstRsC'' \ Щ RsCHg + HgZ
R'Z Hg'' R' \
R'
(реакция (1) : R3C — R', Y=X—галоид; реакция (2): X — R3C, R'=Y—
галоид; реакция (3); Y= X—галоид; реакция (4): R3C= X, R = Y—галоид;
реакция (5): R3C = X — R', Y=галоид) (ср., однако, [41]). Однако Джен-
сеном и Рикборном [44] показано, что кинетические данные Реутова,
Белецкой и др. [ 1—10] по реакции симметризации аммиаком и 5£2-механизм,
предлагаемый ими для этой реакции, неверны. Они не согласуются, напри-
мер, с фактом мгновенного образования нерастворимого комплекса HgBr2-
•NH3. Следовательно, кинетика и механизм этой реакции подлежат пере-
смотру.
Иные, шести- и четырехзвенные переходные состояния предложены Дес-
си [42] для реакции
RHgR' + HgHal/jt RHgHal + R'HgHal.
Аналогичные взгляды развиваются Хьюзом, Ингольдом и сотр. Так,
Чаменом, Хьюзом и Ингольдом [43] на основании изучения кинетики
реакции обратной симметризации дивторичнобутилртути с бромной,
уксуснокислой и азотнокислой ртутью и с бромной ртутью в присутствии
Симметризация ртутноорганических соединений
241
бромистого лития подтвержден механизм равновесной реакции симметри-
зации
2RHgX RaHg + HgX3
и течение этой реакции в обоих направлениях с сохранением конфигурации.
Тот же 5г2-механизм принят ими для некатализируемых реакций (3)
[45, 46] и (5) [47] на основании кинетических данных и сохранения стерео-
химической конфигурации атома углерода, соединенного с обменивающейся
ртутью (см. гл. XIV, стр. 320).
При реакции дивторичнобутилртути [141 и (—)-вторичнобутил, (ф)-вто-
ричнобутилртути [14, 481 с бромной ртутью конфигурация сохраняется.
Подтвержден [491 также второй порядок и 5£2-механизм реакции об-
ратной симметризации R2Hg (R — разнообразные алкилы и арилы) с HgX2
(X = Cl, Br, J). На основании наличия зависимости величины константы
скорости от полярности заместителя X и полярности связи Hg—Hal пред-
ложен [49] четырехцентровый механизм переходного состояния этой реак-
ции с участием в нем ионной пары XHg+ и X".
Зависимость скорости реакции
RHgR -[-HgJ2-> RHgJ + R'HgJ
от природы R и R' и природы растворителя является следствием изменения
энтропии активации этой реакции и поляризуемости С—Hg-связи [421.
При помощи изотопного метода изучалась реакция, обратная симметри-
зации
RHgR' + HgX2 RHgX + R'HgX
в случае R=^R' (см. также гл. XIV, стр. 320). Определена относительная
прочность связи в RHgR' по распределению Hg203 между RHgR' и
R 'HgBr в реакции RHgR' с Hg203Br2 [50,51]. Ник. А. Несмеянов и О. А. Реутов
нашли, что в фенилэтилртути, в исправление ошибочных данных Десси [42],
происходит разрыв связи фенила со ртутью [51].
Взаимодействие ц.ас-2-метоксициклогексилнеофилртути с сулемой,
меченной радиоактивной ртутью, протекает с сохранением конфигурации
затрагиваемого углеродного атома циклогексанового кольца [41]. При
этом происходит статистическое распределение меченой ртути между обо-
ими радикалами.
Предположения о возможности возникновения в качестве промежуточной
стадии симметричного продукта в реакциях:
RHgX + Hg*Xa RHg*X + HgXa
RHgX + Hg* - RHg*X + Hg
экспериментально не подтверждаются [29].
Действие симметризаторов-комплексообразователей] (KJ, KCNS, Na2S2O3,
KC1 и др.) на a-мер курированные альдегиды, кетоны, эфиры карбоновых
кислот вследствие ярко выраженного о-л-сопряжения в этих соединениях
приводит не к симметризации их, а к гидролизу Hg—С-связи и образованию
энолятов, например [521:
CIHgCHaCHO + 4NaJ Na2HgJ* + NaCl + CHa=CHONa.
Ни один из симметризующих агентов не применим с одинаковым успе-
хом ко всем классам ртутноорганических соединений. Одни из них с боль-
шим удобством и успехом применимы в одной, другие — в другой области.
Одним из наиболее универсальных способов является симметризация
станнитом натрия, который широко применим для симметризации алифати-
ческих предельных и непредельных, жирноароматических, алициклических,
242 Ртутноорганические соединения. Лит. стр. 260—263
ароматических, гетероциклических соединений ртути. Почти универсаль-
ным действием обладает медь в присутствии аммиака или других азотистых
оснований, применявшаяся и для симметризации продуктов присоединения
солей ртути к непредельным соединениям и а-меркуриррванных оксосоеди-
Нений.
В последнее время все шире применяется аммиак в индифферентном рас-
творителе (бензол, хлороформ и т. п.), который является и в отсутствие меди
прекрасным, пригодным для симметризации самых разнообразных соеди-
нений, очень мягко действующим средством. Он симметризует и такие ве-
щества с крайне лабильной связью С—Hg, как а-галогенмеркуроксо- и
а-галогенмеркуркарбоксисоединения, которые в водных растворах (а также
в спиртах) большинством обычных агентов симметризации — комплексо-
образователей (KJ, NaCN, Na2S2O3) гидролизуются по уравнению типа
RHgX 4- 4J' + Н2О RH + Hgj" + ОН' + X'
(см. также стр. 126, 241).
Таким образом, применение симметризации аммиаком следует особенно
рекомендовать.
Йодистый калий широко применяется в особенности для симметризации
ароматических, гетероциклических ртутноорганических солей и ртутных
производных ценов. Он, как и другие комплексообразователи (KCN, KCNS,
Na2S2O3), непригоден для симметризации квазикомплексных соединений.
Об ограничениях, свойственных этому симметризатору, см. также стр. 250.
Одним из наиболее часто употребляющихся симметризаторов является
водный раствор тиосульфата натрия — один из мягких симметризующих
агентов, применимый там, где ртутный атом связан лабильно, однако ус-
тупающий в этом отношении аммиаку в индифферентной среде.
Для симметризации простейших алифатических соединений ртути
применяют цианистый натрий, станнит натрия, медь в присутствии азоти-
стых оснований. Особенно успешна в этих случаях симметризация электро-
лизом.
Специфическим симметризатором низших перфтор алкильных соеди-
нений ртути является амальгама кадмия. Действие на перфтор алкиль-
ные соединения ртути других симметризующих агентов, таких как нат-
рий, щелочной раствор станнита натрия, иодистый или цианистый калий в
водном или ацетоновом растворе, не приводит к образованию симметричных
соединений [78]. При действии этих (кроме Na) симметризаторов на
CF3HgJ образуется фтороформ [78].
Продукты присоединения сблей ртутй по двойной связи ациклических
алкенов не симметризуются; они разрушаются под действием симметриза-
торов-комплексообразователей (KJ, KCN, KCNS, Na2S2O3) с выделением
олефина, что является одним из проявлений их квазикомплексных свойств
(см. гл. VI); симметризатор.ы-восстановители, например амальгама нат-
рия, заменяют ртуть в них на водород. Исключением является симметри-
зация продукта присоединения соли ртути к окиси углерода действием
особого симметризующего средства — трифенилфосфйна.
Особый случай представляет собой также симметризация продукта
присоединения сулемы к аллилфенолу действием амальгамы натрия.
Продукты же присоединения солей ртути к алициклическим алкенам
симметризованы действием аммиака, гидр аз ин гидрата, иногда и дейст-
вием других симметризаторов. а-Галогенмеркуроксосоединения симметри-
зуются действием аммиака в бензоле или хлороформе, а в одном случае —
действием меди в присутствии пиридина. Квазикомплексные продукты при-
соединения солей ртути по тройной связи, выделяя ацетилен под действием
всех обычных агентов симметризации — комплексообразователей, прекрас-
Симметризация ртутноорганических соединений
243
но симметризуются действием аммиака в бензоле или хлороформе. Продукт
присоединения ацетата ртути к дифенилацетилену симметризуется дей-
ствием йодистого калия в ацетоне.
Но такие чрезвычайно лабильные квазикомплексные соединения, как
продукты присоединения сулемы к ацетиленовым спиртам, кетонам и кис-
лотам, не симметризуются, а разрушаются при действии всех симметри-
зующих агентов.
Другие алкенильные ртутноорганические соединения, не являющиеся
квазикомплексными соединениями, могут быть в ряде случаев симметризо-
ваны действием, например, йодистого калия в ацетоне, цианистого калия,
амальгамы натрия, станнита натрия, бутиллития.
Симметризация ароматических и гетероциклических монортутных со-
лей может быть произведена разнообразными симметризаторами в зависи-
мости от лабильности связи С—Hg.
Полимеркурированные соединения симметризуются в жестких условиях
действием, например, меди в пиридине, амальгамы натрия и пр.
Об области применения разнообразных других симметризующих средств
см. ниже, при каждом из них. Следует иметь в виду, что применявшиеся
пока лишь в отдельных случаях специфические симметризаторы могут ока-
заться пригодными и для более широкого круга веществ.
РЕАКЦИИ СИММЕТРИЗАЦИИ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Симметризация действием металлов
Натрий, амальгама натрия и сплав олово — натрий.
2RHgX + 2Na R2Hg + Hg + 2NaX,
2RHgX + Na2Hg-> R2Hg + 2NaX + 2Hg
Известны лишь единичные случаи симметризации посредством металли-
ческого натрия. Таково получение производных диф-тиенилртути, которое
осуществлено при нагревании хлористой [J-тиенилртути и ее производных с
натрием в ксилоле [53].
Известны случаи симметризации таким путем и меркурированных в
a-положение тиофенов [54], которые, однако, чаще симметризуют более
мягко действующими средствами, такими как иодиды, иногда роданиды ще-
лочных металлов, станнит натрия.
Получение 2,5,2',5'-тетраметил-3,3'-дитненилртутн [53].
К кипящему раствору 5,6 г 3-хлормеркур-2,5-тиоксена в 30 мг сухого ксилола при-
бавляют 1,5 г натрия и 30 мин. нагревают с обратным холодильником при сильном встря-
хивании. Затем отфильтровывают от темного остатка, коричневый фильтрат очень сильно
упаривают, выпавший коричневато-белый осадок сушат. Дробной кристаллизацией из
бензина (т. кип. 70—120° С) получают два продукта: плавящийся при 152—155° С пред-
ставляет собой исходное вещество и полученный продукт, плавящийся при 144—145° С. Вы-
ход 0,3—0,4 г.
Действие натрия, диспергированного в смеси нонана и бензола, на хлор-
мер курферроцен дает диферроценилртуть [55].
Амальгама натрия в индифферентном растворителе или в спирте при-
менялась более широко для симметризации главным образом ртутноорг
ганических производных углеводородов. Посредством амальгамы натрия
244
Ртутноорганические соединения. Лит. стр. 260—263
симметризованы уксуснокислая и иодистая фенилртуть [561, иодистая
а-нафтилртуть [571, броммеркурпентаметилен, дающий при этом гетероцик-
лическое соединение — цикломеркурпентаметилен [58, 59] (меркурацикло-
гексан)
СН2СН2
\lg.
При помощи амальгамы натрия Адамсом, Романом и Сперри [18] было
осуществлено превращение 1-хлормеркурметил-1,2-дигидробензофурана в
соответствующее симметричное соединение:
—СН2 Na/Hg, /—.СН2 \
\JcHCH2HgCl \\JkJcHCHay2Hg'
Получение бис-{\-метил-1,2-дигидробензофурнл)ртути [18]. К взвеси 15 г 1-хлормер-
курметнл-1,2-дигидробензофураиа в 100 мл абсолютного спирта, помещенной в кругло-
донную колбу, соединенную с обратным холодильником, прибавляют небольшими порциями
через обратный холодильник-3%-иую амальгаму, содержащую 1,5 г натрия. Слегка подо-
гревают, чтобы реакция пошла, и продолжают осторожно нагревать до разложения амаль-
гамы (20 мин.), после чего кипятят 30 мин.. Под конец кристаллы 1-хлормеркурметил-1,2-
дигидробензофурана исчезают. Образуется серый осадок. После охлаждения осадок,
состоящий из 2,5 г хлористого натрия и органических загрязнений, отфильтровывают,
фильтрат испаряют. Выделившийся осадок перекристаллизовывают из эфира. Выход 6 г
чистого вещества. Из маточника может быть получено около 1 г загрязненного продукта,
что составляет 50% выхода. Т. пл. 93° С.
Следует иметь в виду, что действие амальгамы натрия (особенно в воде)
во многих случаях вызывает не симметризацию ртутноорганических соеди-
нений, а замену группы HgX в них на водород.
При взаимодействии со сплавом олово — натрий (15% Na) ч«с-(3-часовЬе
кипячение в бензоле) и транс-(3-часовое кипячение в ксилоле) а-хлормеркур-
стияьбенов не образуется оловоорганических соединений, а имеет место
симметризация этих ртутноорганических соединений [60]. Выход транс-
а-меркур-быс-стильбена 77%, цис-изомера — 97%. В случае транс-изомера
при проведении реакции в бензоле для достижения 70%-ного выхода не-
обходимо восьмичасовое кипячение.
Медь. Медь сама по себе не является особенно удобным симметризующим
средством, так как требует длительного нагревания с раствором ртутноорга-
нической соли и была примерена в немногих случаях (см., например, [61—
64]). Метод же симметризации медью в присутствии органического [65, 66]
или неорганического [66, 67] азотистого основания NA, предложенный Гей-
ном и Ваглером 165]:.
2RHgX4-2Cu + NA-> R2Hg+2CuX-NA + Hg,
является одним из наиболее удобных и универсальных. Он дал превосход-
ные результаты в применении к жирным [65] (а-меркурированное оксосое-
динение —эфир а-хлормеркуруксусной кислоты образует при этом симмет-
ричный продукт с небольшим выходом [68]), алициклическим [69] и к сле-
дующим классам ароматических соединений: меркурированным углеводо-
родам [67, 701, их галоид- и нитрозамещенным [65, 67, 71], фенолам [72]
и тиофенолам [183], аминам и их производным [65, 73], кетонам [62],
кислотам [74], сложным эфирам [74, 75]. В качестве основания NA при-
меняют аммиак, амины, пиридин и его гомологи.
Симметризация ртутнсорганических соединений
245
Однако симметризация без аммиака или пиридина предпочтительнее в
тех случаях, когда аммиак не индифферентен к симметризуемому веществу.
Симметризация без аммиака дает в этом случае более высокие выходы
(ср. примеры симметризации хлормеркурбензойных эфиров).
Получение метилового эфира меркур-бис-уксусной кислоты [68]. К раствору 6,2 г (0,02
моля) метилового эфира хлормеркуруксусной кислоты в 20 мл абс. пиридина прибавляют
4 г меди. Наблюдается разогревание и позеленение раствора. Через неделю жидкость от-
фильтровывают от осадка, пиридин удаляют в вакууме и остаток дважды экстрагируют
5 мл горячего бензола. После упаривания части бензола добавляют равный объем гексана.
Выпадает темный осадок (в основном соли меди). Его отфильтровывают и к фильтрату
приливают эфир до помутнения; при охлаждении до —30° С выпадают кристаллы, слегка
окрашенные в светло-зеленый цвет. После кристаллизации их из смеси бензола fc эфиром
выход 0,8 г. Т. пл. 96—98° С. Повторная кристаллизация из смеси метанола с эфиром дает
0,5 г (14%) метилового эфира меркур-бис-уксусной кислоты с т. пл. 99—100° С.
Получение дифенилртути [65]. Раствор 0,5 а бромистой фенилртути в 4 мл пиридина в
сосуде Шленка, наполненном азотом (или углекислотой), обрабатывают 0,8 г меди (в виде
мелко нарезанных кусочков проволоки). Жидкость в течение нескольких минут окраши-
вается в интенсивный желто-зеленый цвет, и происходит выделение ртути частично в виде
амальгамы. При встряхивании сосуда в течение недели также не наступает больше види-
мых изменений. Пиридин отгоняют в вакууме при температуре бани ие выше 40° С. Оста-
ток отмывают от неорганических солей ртути холодным водным раствором аммиака. Кри-
сталлизация из спирта. Т. пл. 125° С.
Получение дибензилртути [65]. Около 4 г хлористой бензилртути, обработанные, как
описано выше, медью в пиридине, дают 7,5 г дибензилртути. Т. пл. 111° С. Выход 75%.
Получение ди-п-хлорфенилртути [65]. Из 5 г хлористой ге-хлорфеннлртутн, 6,5 г меди н
35 мл пиридина в условиях предыдущего опыта получено 2,5 г ди-п-хлорфенилртути. Вы-
ход 81%. Кристаллизация из ацетона. Т. пл. 242—243° С.
Получение бмс-о-нитрофенилртути [65]. 4,5 г хлористой о-нитрофенилртути раство-
ряют в 30 мл пиридина, прибавляют 5,3 г комочков медной проволоки и оставляют стоять
в сосуде, наполненном углекислотой, в течение 1 дня. Пиридин отгоняют в вакууме при
температуре не выше 40° С, остаток смачивают спиртом и отмывают от солей меди водным
раствором аммиака. Выход бис-о-нитрофенилртути 2,3 а (92%). После кристаллизации из
спирта или ацетона получают темноватые иголочки. Т. пл. 206—207° С.
Получение ди-я-диметиламииофенилртути [65]. 2 а хлористой п-диметиламинофенилрту-
тн, 10 мл пиридина и 1 г меди дали по предыдущей прописи 1,0 а (81%) ди-п-диметилами*
нофенилртути. Т, пл. 167—168° С.
Так же получена ди-а-нафтилртуть (т. пл. 243°С), ди-л-бромфенилртуть
(т. пл. 245°С). Этим методом симметризованы также иодистая метилртуть и
хлористая этилртуть [65].
Получение меркур-бас-л-беизойиоэтилового эфира действием меди без аммиака [64].
3,84 а (0,01 моля) n-хлормеркурбензойноэтилового эфира и 3 а меди в 150 мл бензола ки-
пятят в течение 10 час. Получено 2,4 а (96%) вещества с т. пл. 192° С. Перекристаллиза-
ция из спирта.
Получение меркур-бас-я-бензойиометилового эфира действием меди и аммиака [74].
6,6 а метилового эфира л-хлормеркурбензойной кислоты смешивают с 4а порошка меди,
заливают 60 мл ацетона и тщательно размешивают. Затем при взбалтывании вливают
30 мл 25%-ного водного аммиака, смесь энергично встряхивают 10 мин., после чего вы-
ливают в избыток воды (300 мл) и осадок отсасывают. После просушивания при 100° С его
извлекают горячим уксусноэтиловым эфиром. Выход 2,85 а (68%). Перекристаллизация из
хлороформа или ацетона. Т. пл. 264—265° С. Для труднее омыляемого этилового эфира в
этих же условиях выход 88,5% .
Прибавление порошка меди и 25 % -ного аммиака при получении ртутно-
органических соединений разложением двойных солей хлорной ртути и
хлористого арилдиазония прямо дает диарилртуть из двойной соли без
необходимости выделения хлористой арилртути (см. гл. VII).
Цинк. Цинк также может явиться симметризующим средством. Он был
применен для этой цели в единичных случаях. Так, л-хлормеркурбензойно-
этиловый эфир при продолжительном нагревании с цинковой пылью’в толу-
оле дал с 58%-ным выходом ди-л-карбэтоксифенилртуть [64].
246
Ртутноорганические соединения. Лит. стр. 260—263
Нагревание у-меркурированных спиртов, содержащих вторичную и
третичную спиртовые группы, с цинковой пылью через промежуточное
образование соответствующих симметричных ртутноорганических соединений
приводит к отщеплению металлической ртути [76].
Под действием цинковой пыли в щелочном растворе этил-а-(2-хлор-
меркурэтил)ацетоацетат претерпевает кетонное расщепление и образует
бис-у-ацетопропил ртуть [771.
Магний. Действием магния (условия не указаны) симметризованы броми-
стые цис- и тпранс-4-метилциклогексилртуть и бромистая L-(—)вторично-
бутилртуть [201.
Амальгамы кадмия, меди и серебра. Специфическими симметризаторами
перфтор алкильных соединений ртути являются амальгамы кадмия, меди
и серебра [78].
Получение бкс-трифторметилртути [78]. 2,0 г иодистой трифторметилртути нагревают
с амальгамой серебра (10 г серебра, 25 мл ртути) в запаянной трубке при 140® С во вра-
щающейся печи в течение 20 час. Затем избыток амальгамы отделяют, твердый осадок
извлекают эфиром. После испарения эфира получено белое твердое вещество, после воз-
гонки при 70° С (при атмосферном давлении) т. пл. (в запаянном капилляре) 163® С. Вы-
ход 80%.
С амальгамой кадмия (120—130°С, 10 час.) выход дифторметилртути
80—90%, с амальгамой меди (120°С, 12 час.) выход 20%, в ацетоне выход
50-60%.
Симметризация действием гидросульфита натрия
Действием гидросульфита натрия симметризована соль о-нитрофенил-
ртути [78а].
Симметризация действием гидрата закиси железа
Гидрат закиси железа применен для симметризации, например [184],
меркурированных нитробензойных кислот, с одновременным их восстанов-
лением.
Симметризация действием станнита натрия
Прекрасным и очень общим является метод симметризации действием
щелочного раствора станнита натрия
2RHgX + Na2SnO2 + НаО^ R2Hg + Hg + 2NaX + H2SnOs.
Он пригоден для симметризации разнообразных классов как аромати-
ческих, так и жирных ртутноорганических соединений. Им симметризованы
ртутноррганические соединения жирных углеводородов [14] (в том числе
алкенильных углеводородов [13, 79,181 ]); в частности, так получена дивинил-
ртуть [80], диаллилртуть [81, 82] (лучшим симметризатором для иодистой
аллилртути является, однако, цианистый калий [83]). Станнитом натрия
симметризованы также соли цис- и /пранс-стильбенилртути [84], 3-алкано-
лы [85, 861, некоторые продукты присоединения солей ртути к непредель-
ным соединениям, например продуктметоксимеркурирования стирола [90а],
производные терпенов: цинеола [87], камфана [88, 89] и камфоры [901
(также с CiHg-группой у головы моста [15]), ртутноорганические про-
Симметризация ртутноорганических соединений
247
изводные жирноароматических [91] и ароматических (см., например, [92,
95]) углеводородов, эфиров фенолов [94, 951, ароматических кислот [96],
гетероциклов [97, 98], ферроцена [55] и др.
В редких случаях при действии щелочного раствора станнита нат-
рия на некоторые, особые, ртутноорганические соли происходит не сим-
метризация, а восстановление ртутноорганического соединения. Так, при
действии щелочного 0,5 N раствора станнита натрия на CF3CClFHgF
это соединение не симметризуется, а ртуть в нем заменяется на водо-
род [99].
Алкилы, содержащие две HalHg-группы на концах цепи, симметризуясь
под действием станнита натрия, образуют циклические соединения, в ко-
торых ртуть является членом цикла. Так получен, например, 1,6-димер-
курациклодекан из 1,4-дихлормеркурбутана [100]. Аналогично ртутное
производное этилового эфира O(CH2CH2HgBr)2 при действии щелочного
раствора станнита натрия образует соединение, которому Занд [101] припи-
сал строение 1-окса-4-меркурациклогексана, но которое, однако, согласно
рентгенографическим исследованиям [102], представляет собой димер—1,7-
диокса-4,10-димер курациклододекан:
CH2CH2HgCH2CH2
cf \)
^CHsCHaHgCHaCH^
Получение 1,7-диокса-4,10-димеркурациклододекана [101]. Растворяют диброммеркур-
диэтиловый эфир в 10%-ном едком кали и в этот раствор приливают отфильтрованный
щелочной раствор станнита до прекращения выпадения осадка. Последний полно отмыва-
ется декантацией и высушивается над H2SO4. Серо-фнолетовый осадок в количестве 5 г
заливают бензолом, запаивают в трубке и греют до 140° С в течение нескольких часов.
После охлаждения вскрывают. Над выделившейся ртутью жидкость заполнена бесцветны-
ми тонкими длинными призмами. Перекристаллизация нз бензола или из хлороформа.
Т. пл. 145° С. Выход 60%.
Для продукта, полученного действием станнита натрия на имеющее
транс-строение [103] ртутное производное дипропиленоксида [101], до-
казана [103] полимерная природа:
XHgH2C
CH2HgX
Получение дибеизилртути [91]. К взвеси 15 г иодистой бензилртути в 10 мл спирта
и 225 мл воды прибавляют щелочной раствор станнита натрия из 25 г едкого иатра в 125 мл
воды и 10 е,дигидрата хлористого олова в 125 мл воды. Перемешивают мешалкой в течение
часа. После этого осадок отфильтровывают, промывают водой, сушат и экстрагируют аце-
тоном. Загрязняющую фильтрат мелкораздробленную ртуть удаляют прибавлением не-
большого количества цинковой пыли, затем фильтруют. К фильтрату прибавляют воду
до появления мути, охлаждают до —15° С. Выделяются игольчатые кристаллы днбензил-
ртути. Выход 64 г (93,2%). Т. пл. 111° С.
Получение дивинилртути [80]. К раствору 10 г (0,0326 моля) бромистой вннилртути
в 120 мл ацетона прибавляют раствор станнита натрия, приготовленный из 4,02 г (0,017
моля) дигидрата хлористого олова в 36 мл воды и 8,27 г едкого натра, растворенного в
30 мл воды. Спустя 30 мии. нз реакционной массы отгоняют фракцию с т, кип, 58—87° С
(приемник охлаждают до —25ч—30° С), к которой затем приливают насыщенный водный
раствор хлористого натрия. Происходит отделение маслообразного продукта; он высуши-
вается и перегоняется при 90—91° С/80ки; 1,5978; 2,7713. Выход 53%.
248
Ртутноорганические соединения. Лит. стр. 260—263
Получение 10,10-меркур-бис-камфоры [90J.
1 г хлористой камфор-10-ртути растворяют в горячем ацетоне, выливают в 150 мл
воды и полученную суспензию обрабатывают щелочным раствором станнита натрия (из
10 г NaOH, 8 г SnCla-2H2O, 200 мл воды). Перемешивают 2 часа. Осадок отфильтровы-
вают и извлекают кипящим ацетоном. После осторожного разбавления водой и охлаждения
бис-камфор-10-ртуть выделяется в виде кристаллов. Т. пл. 255—256° С.
Получение дифенилртути из уксуснокислой фенилртути [93]. К раствору 30 а уксусно-
кислой фенилртути в 300 мл воды при энергичном механическом перемешивании прибавля-
ют раствор, приготовленный смешением 125 мл 40%-ного NaOH и 50 г SnCk^HsO в 25 мл
воды. Перемешивание продолжают в течение часа. Образуется серовато-черный осадок,
который отсасывают, промывают водой и извлекают ацетоном. Если через фильтр проходит
тонкоизмельченная ртуть, то добавляют немного цинка. К фильтрату добавляют воду до по-
явления легкой неисчезающей мути. Вместо ацетона можно извлекать бензолом. Выделив-
шиеся после охлаждения кристаллы отсасывают и сушат. Вес 13,2а (95,6%). Т. пл. 125° С.
Получение дифенилртути из хлористой фенилртути [97]. В раствор 2,5 а хлористого
олова в 5 мл воды в 30%-ном водном растворе едкого натра вносят раствор 6,2 г хлористой
фенилртути в 40 мл пиридина. Через несколько часов осадок отфильтровывают, сушат и
извлекают из него дифенилртуть 95%-ным спиртом. После кристаллизации из спирта вы-
ход 3,6 г (90%). Т. пл. 124—125° С-
Получение диферроцеиилртути [55]. К взвеси 2,1 г (0,005 моля) хлористой ферроце-
нилртути в 20 мл 95%-ного этанола и 50 мл воды прибавляют раствор станнита натрия,
полученный из 5,0 г едкого натра в 25 мл воды и 1,8 г дигидрата хлористого олова в 25 мл
воды. Желтовато-оранжевый цвет тотчас исчезает и выделяется серовато-черный осадок.
После перемешивания в течение 3 час. осадок отсасывают, промывают водой, сушат и из-
влекают кипящим ксилолом. После охлаждения получают 1,0 г (70%) диферроцеиилртути
в виде желтовато-оранжевых кристаллов. После кристаллизации из ксилола т. пл.
234—235° С (с разложением).
Симметризация действием гидразина и родственных соединений
Гидразингидрат, соли гидразина, фенилгидразин (последний также в
присутствии меди [105]), семикарбазид, а также гидроксиламин дают хоро-
шие выходы диарилртутных соединений из ртутноорганических производ-
ных углеводородов. Были получены дифенил-[104], ди-п-толил-[104], ди-
п-хлорфенил-[106], дипентафторфенилртуть [106а],ди-а-фурилртуть [104].
Метод был применен и к жирному ряду на примере хлористой н-бутилрту-
ти, давшей ди-н-бутилртуть с 40%-ным выходом [104].
Гидразингидрат применен для симметризации оптически активных
ртутноорганических соединений, в частности 3-броммеркуркамфоры [16,
17], 2-хлормеркуркамфана [88] (смесь диастереомеров), а также продук-
тов присоединения солей ртути к непредельным соединениям [18, 19].
Однако при его применении происходит обращение конфигурации этих сое-
динений [17—19]. Следует иметь в виду, что действие избытка гидразина
на продукты присоединения солей ртути к олефинам вызывает отщепление
ртути и замену ее на водород [22], иногда сопровождающееся отщеплением
OR (но не ОН)-группы [107] и регенерацией двойной связи.
Замена ртути на водород действием гидразина описана в гл. XIV, стр. 301.
Действием гидразингидрата симметризованы также разнообразные дру-
гие RHgCl (R=Ar, а- и 3-нафтил, а-тиенил) [108].
Симметризация смеси диастереомеров З-броммеркур/камфоры гидразиигидратом [17].
В раствор 0,6 г гидразингидрата в 15 мл диоксана вносят при перемешивании 2 г 3-бром-
меркур-1-камфоры. Через 30 мин. осадок выпавшей металлической ртути отфильтровывают,
а фильтрат разбавляют водой до 150 мл. Выпавший осадок 3,3-меркур-бис-/-камфоры от-
фильтровывают и промывают водой. Вес 0,6 г (50%). После перекристаллизации из 70% -
ного водного диоксана т. разл. 200—205° С.
Симметризация ртутноорганических соединений
249
Симметризация же экзо-^1/с-3-гидрокси-2-норборнилмеркурхлорида дей-
ствием гидразина в метанольном растворе в присутствии щелочи проис-
ходит с сохранением конфигурации [106а]:
n2h4
HgCl СН3ОН
Hg.
NaOH
Синтез ди-экзо-З-окси-экзо-2-норборнилртути [106а]. К раствору 5,6 г (0,016 моля)
экзо-^пс-З-гидрокси-2-норборнилмеркурхлорида в 72 мл абсолютного метанола прибавля-
ют раствор 1,28 г едкого натра в 4 мл воды и 1,09 г 95% -ного гидразина. Тотчас выделяется
ртуть. Раствор кипятят 6 час. и сливают со ртути в 300 мл эфира. Ртуть последовательно'
промывают метанолом, водой, ацетоном, затем сушат и взвешивают. Выход ртути 1,814 г
(56,2%), Эфирный раствор промывают двумя порциями дистиллированной воды по 200 мл,
сушат сульфатом натрия, упаривают до объема около 20 мл и охлаждают. Выкристалли-
зовывается осадок; вес 1,243 г, т, пл. 152—152,5° С. Из фильтрата может быть выделен
также экзо-норборнеол (0,6685 г, 37%). Т. пл. 115—127° С.
Симметризация действием алкоголя та натрия (калия)
4RHgX + R'CH2ONa + 4NaOH 2R2Hg + R'COONa + 4NaX +'2Hg + 2H2O
Метилат и этилат натрия применяли в единичных случаях с переменным
успехом в качестве симметризующих агентов.
При 6-часовом нагреваний хлористой о-нитрофенилртути в метиловом
спирте с метилатом натрия была получена ди-о-нитрофенилртуть с 80%-
ным выходом [78а]. В применении же к меркурированным хлорнафтали-
нам метод дал плохие результаты [109].
Симметризация электролизом
При электролизе растворов ртутноорганических солей в результате
восстановительной симметризации получаются с выходами от 70 до 100%
полнозамещенные соединения ртути. В случае уксуснокислой метилртути
необходимая электропроводность водного раствора достигается добавлением
пиридина. Симметризацию сернокислых и янтарнокислых солей алкилрту-
ти (алкил — СН3, С2Н5, С3Н7, С4Н9, j-C5Hu) ведут в присутствии сернокис-
лого натрия.
При электролизе хлористой 0-хлорвинилртути происходит ее симметри-
зация [ПО].
Симметризация уксуснокислой метилртути [111]. Сосудом для электролиза служит
сужающийся книзу стеклянный цилиндр, снабженный внизу краном, с анодом из графита,
помещенным в пористую глиняную камеру, и катодом—платиновой жестью, располагаемой
на расстоянии нескольких миллиметров концентрически вокруг глиняной камеры. Сосуд
окружен водяной рубашкой, посредством которой в нем поддерживают температуру 30—
40° С. В него помещают 25%-ный водный раствор уксуснокислой метилртути, к которой
добавлен пйридин (2 моля на 1 моль уксуснокислой метилртути). При силе тока 2,5 а,
начальном напряжении 30—35 в, плотности тока 1,8 а! дм2 катода электролиз длится 2 часа
10 мин. Выход диметилртути 92%. Выход по току 90%.
Симметризация сернокислой метилртути [112]. Электролиз ведут без диафрагмы в
цилиндрическом сосуде диаметром 18 мм, на дне которого помещен катод, анод — спираль
из платиновой проволоки диаметром 0,5 мм. Сосуд погружен в водяную баню. Источник
тока — батарея свинцовых аккумуляторов напряжением 6,2 в.
Электролизу подвергают (при 20° С, плотности тока 0,02 а/с.и2) раствор 3% -ной сер-
нокислой этилртути и 0,1 г сернокислого натрия в 15 мл воды. Через 3—4 часа выделение
диэтилртути заканчивается. Ее отделяют в микроделительной воронке от ртути и раствора,
сушат хлористым кальцием. Выход 1,2 г (около 90%). Т. кип. 159° С.
В этих же условиях были симметризованы сернокислая метил-, пропил-, бутил- (пер-
вые 15 мин. температура 50е С, затем 20® С), изоамил- (температура 56° С, плотность тока
0,04 а/см2) ртуть [112].
250
Ртутноорганические соединения. Лит. стр. 260—263
Симметризация янтарнокислой пропилртути [112]. Раствор 1 г янтарнокислой про-
пилртути и 0,1 а сернокислого натрия в 15 мл воды подвергают электролизу при 80—90° С
и плотности тока 0,03 а'см2. По окончании электролиза и охлаждении электролита дипро-
пилртуть отделяют от ртути и воды и сушат хлористым кальцием. Выход 70%. Т. кип.
186—187° С.
(Г 1 Так же при электролизе янтарнокислой этилртути (при плотности тока
0,04 а/сж2) получена диэтилртуть [112].
Симметризация действием йодистого калия (натрия)
Иодистый калий (натрий) является превосходным симметризующим
средством для многих классов ртутноорганических соединений. Реакция
симметризации посредством йодистого калия изучена Уитмором н Собац-
ким [113], которые нашли, что эта реакция обратима
2RHgJ + 2К J RaHg + K2HgJ4,
и если R2Hg значительно более растворима в реакционной среде, чем RHgX
(как это имеет место для RС6Н5), то вследствие большой скорости обрат-
ного процесса реакция практически не идет и KJ неприменим для симмет-
ризации таких соединений. Вследствие обратимости этой реакции для ее
количественного прохождения слева направо необходимо брать K(Na)J
в большом избытке сравнительно с RHgX, в, отношении приблизительно
10:1.
Этим методом симметризованы в ряде случаев алкенильные (но не ква-
зикомплексные) соединения ртути [60, 180]. Особенно легко симметризуются
действием Na(K)J (а также действием K.CNS, см. стр. 252) пергалоидви-
нилртутные соли. Даже действие на них эквимолекулярных количеств этих
реагентов ведет к симметризации, тогда как для симметризации остальных
ртутноорганических солей требуется избыток Na(K)J [114]. Принимая во
внимание указанное выше ограничение, этим методом гладко и с хорошими
выходами можно симметризовать ртутноорганические производные аромати-
ческих углеводородов [ Г1£>], бифенил а [ 116,117, 1246], хлорнафталинов [109],
галоидалкиламинов [118], эфиров нитробензойных кислот [119], гетероцик-
лов (меркурированные в а-положение тиофен [53,54,97,120, 121] и селено-
фен [122] и их производные, меркурированный в а-положение тионафтен
[123]). Меркурированные эфиры фенолов и фенолы с ацилированной гид-
роксильной группой также могут быть симметризованы этим методом. Фе-
нолы же со свободным гидроксилом под действием йодистого калия разла-
гаются [124]:
HOCeH4HgJ + 3KJ + Н2о ->• CeHsOH + K2HgJ4 + КОН.
Разложение под действием йодистого калия (как и других комплексо-
образователей) квазикомплексных соединений, в том числе а-меркуроксо-
соединений, описано в начале этой главы и в гл. VI.
При обработке 2,2'-дихлормеркурдифенила раствором йодистого натрия
в этиловом спирте получена циклическая тетрамерная о-дифениленртуть
[124а]. Вероятно, полимерное (возможно циклическое) строение имеют про-
дукты симметризации [в частности при действии иодистых калия (натрия)]
и других днмеркурированных соединений, например дихлормеркуртиофена
[97], дихлор мер курферроцена [1246].
Симметризация ртутноорганических соединений
251
Так, 1,Г-дихлормеркурферроцен при действии этанольного раствора
йодистого натрия образует соединение состава (C10H8FeHg)x И строения,
показанного ниже:
Он, вероятно, представляет собой смесь линейных и, возможно, цикли-
ческих полимеров с невысоким значением х.
Получение играяс-а-меркур-бас-стильбена [60].
Hg
К раствору 2 г (0,0021 моля) транс-а-броммеркурстильбена с т. пл. 157—158° С в 150 мл
ацетона и 10 мл воды прибавляют раствор 3,35 г (0,02 моля) йодистого калия в 54 мл аце-
тона и 6,5 мл воды. Смесь стоит при комнатной температуре сутки. Затем осадок отфильтро-
вывают и высушивают.. Получают 1,2 г (99%) траяс-а-меркур-бис-стильбеиа с т. пл.
239—241s С после перекристаллизации из диоксаиа.
При симметризации в этих условиях цш>а-броммеркурстильбена с т. пл.
118—120° С, вследствие значительной растворимости цмс-а-меркур-бис-
стильбена в водном ацетоне, по окончании реакции продукт высаживают
из реакционной смеси добавлением воды. Выход 53%. Т. пл. 145—146° С
после перекристаллизации из бензола со спиртом. Одновременно вернулось
7,5% не вошедшего в реакцию RHgBr. цыс-а-Броммеркурстильбен дает
цпс-а-меркур-бис-стильбен с выходом 94% при симметризации станнитом
натрия и с выходом 88% при симметризации аммиаком.
Получение ди-и-толилртути описано в «Синтезах органических препа-
ратов» [115].
Получение дн-о-бифеиилилртутн [124а]. К раствору 16 г сулемы в 480 мл абсолютного
эфира прибавляют по частям 160 мл эфирного раствора (57 ммолей) о-литийбифеинла
(обычным образом приготовленного из о-иодбифенила и лития в эфире), кипятят в течение
часа (отсутствие пробы Гилмана на активное металлоорганическое соединение). После
отгонки эфира и взбалтывания остатка с 400 мл горячей воды отсасывают, промывают
водой и осадок извлекают кипящим этиловым спиртом (700 мл). К спиртовому экстракту,
выделяющему Фри охлаждении кристаллы, прибавляют 30 г йодистого натрия и кипятят
8 час. для полного превращения хлористой о-бифенилилртути в. ди-о-бифеинлилртуть.
При охлаждении до 0° С последняя выкристаллизовывается. Осадок отсасывают и кристал-
лизуют из лигроииз. Образуются иглы ст. пл. 161—162° С. Смешанная проба с хлористой
о-бифенилилртутью дает депрессию температуры плавления. Выход 84%.
Получение ди-(3-бифеиилил)ртути [117]. 4,58 г 3-хлормеркурбифенила кипятят 20 час.
с 7,50 г йодистого натрия в 250 мл этилового спирта. Реакционную смесь фильтруют го-
рячей. Ди-(3-бифеиилил)ртуть (т. пл. 187—189° С) выкристаллизовывается из фильтрата.
Выход 56%. После кристаллизации из н-гептана т. пл. 188,5—189* С.
Получение дн-а-тиеинлртутн [53]. К раствору 10 г хлористой а-тиеиилртути в 175 мл
ацетона прибавляют раствор 10 г NaJ в 75 мл ацетона. Получившийся белый осадок спустя
несколько часов отсасывают, фильтрат сильно разбавляют водой, выпавший прн этом
осадок присоединяют к первому и промывают водой. Кристаллизация из бензола. Выход
5,3 г (92,6%). Т. пл. 198—199° С.
252 Ртутноррганические соединения. Лит. стр. 260-^263
Получение 2,5,2',5'-дитиенилдиртути [97].
Hg
IZys\ / \_l
“ Hg “
К раствору 1г 2,5-дихлормеркуртиофеиа в 100 мл пиридина при энергичном переме-
шивании приливают в течение 6 час. раствор 0,75 г (2 моля) йодистого натрия в 80 мл пи-
ридина. После этого перемешивают еще 45 мин. Образовавшийся осадок отсасывают. Для
полного удаления пиридина, который прочно удерживается, продолжительное время
обрабатывают водяным паром, отсасывают и затем промывают горячей водой, спиртом и
эфиром. Выход после сушки в вакууме при 60° С 0,37 г (72%). Вещество не плавится. Воз-
можно, оно представляет собой полимер
\ S / п
Симметризация действием цианистого калия (натрия)
2RHgX + 4KCN - RaHg + 2КХ + K2Hg (CN)*
Цианистый калий был первым симметризующим средством, применен-
ным еще Бектоном для получения диметил- и диэтилртути [125]. В арома-
тическом ряду он применялся очень мало, хотя дает достаточно хорошие
выходы. Цианистый натрий является прекрасным симметризующим аген-
том для превращения хлористой p-нафтилртути в д и-(р-нафтил) ртуть [108].
Цианистый калий в концентрированном растворе является лучшим сим-
метризующим средством для иодистой аллилртути. В отличие от других
симметризующих ее агентов он превращает ее в диаллилртуть количествен-
но и на холоду [83]. Для симметризации квазикомплексных соединений
он непригоден.
Симметризация действием роданистого калия (натрия)
2RHgX + 4KCNSR2Hg + 2КХ + K2Hg (CNS)<
Роданистый калий (натрий) является симметризующим средством той
же области применения, что иодистый калий (натрий), давая, однако, иног-
да несколько худшие выходы. Им симметризованы, например, эфиры фе-
нолов [125], производные тиофена [97, 126].
Но KSCN, наряду с Na(K)J и Р(СеН5)3, является симметризатором par
excellence для пергалоидвинилртутных солей. Действие на последние уже
одного эквивалента KCNS приводит с выходом до 90% к соответствующим
R2Hg [114]. Как и другие комплексообразователи, KCNS непригоден для
симметризации квазикомплексных соединений.
Получение б«с-перхлорвиннлртутн [114]. 1,36 г (0,014 моля) тиоцианата калия в
25 мл безводного ацетона прибавляют по каплям при быстром перемешивании к 5,0 г (0,014
моля) хлористой перхлорвинилртути в 25 мл ацетона. Образовавшийся белый осадок
(0,71 а) отфильтровывают; после испарения фильтрата досуха получают 6,3 г (т. пл. 149—
154° С) другого белого осадка. Его промывают 5 раз горячим пентаном порциями по 10 мл.
Оставшиеся белые кристаллы 2,4 г (т. пл. 140—150° С) после кристаллизации из воды дали
2,0 г (91%) тиоцианата ртути. Т. пл. 175° G. После испарения пентана получена бис-пер-
хлорвинилртуть с выходом 2,8 г (88%). После кристаллизации из пентана т. пл. 72—73р С.
Симметризация действием хлористого кальция
Известны два случая симметризации действием хлористого кальция —
уксуснокислой а-тионафтенилртути [124] и продукта присоединения аце-
тата ртути к диметилфенилфенилацетиленилэтиленгликолю [127].
Симметризация ртутноорганических соединений 253
Симметризация действием серноватистокислого натрия
2RHgX Ц- ЗМазЗгОз —> RgHg -f- 2NaX -j- Nag [Hg (8203)2)2
Серноватистокислый натрий является лучшим средством для симметри-
зации соединений с легко отщепляемой ртутью — меркурированных ами-
нов {92, 128—131], фенолов [72, 92], а также ртутных производных кам-
форы [12, 161, некоторых гетероциклов: меркурированного в а-(но и в
Р)-положение фурана [132],фурилового спирта [133]. Им симметризованы
также меркурированный пиридин [75] и хинолин [134] и ртутные произ-
водные ценов [1246, 135—137]. При симметризации 3-броммеркуркамфоры
употреблен прием [16], предотвращающий течение обратной реакции взаи-
модействия симметричного ртутноорганического соединения с солью рту-
ти: реакцию ведут в гетерофазной среде бензол — вода; более раствори-
мое полнозамещенное ртутноорганическое соединение растворяется в
бензоле и тем самым выводится из сферы реакции.
Разложение квазикомплексных ртутноорганических соединений под дей-
ствием Na3S2O3 описано на стр. 242 и в гл. VI.
Получение ди-п-амииофеиилртути [129, 130]. К взвеси 20 г уксуснокислой п-амино-
фенилртути в 75 мл воды, нагретой до 45—50° С, прибавляют при перемешивании нагре-
тый до такой же температуры раствор 16 г гипосульфита натрия в 200 мл воды. Уксус-
нокислая фенилртуть при этом растворяется и образуется небольшой осадок сернистой
ртути. Смесь нагревают до температуры не выше 65—70° С, профильтровывают и к фильт-
рату, имеющему температуру не выше 55° С, прибавляют раствор 90 г гипосульфита
натрия в 70 мл воды. Выпавший, осадок ди-п-аминофеиилртути после 2-часового стоя-
ния отсасывают, промывают водой н сушат. Выход 8 г (73%). После кристаллиза-
ции из диоксана т. пл. 174° С (капилляр вносят в предварительно нагретую до 165° С
баню). Так же симметризованы [129] соединения, содержащие диалкиламиногруппу. Вы-
ходы 70—75%.
Получение ди-п-амино-о-бромфенилртути [131]. 4-Ацетатмеркур-2-бромаиилин переме-
шивают в кашу с избытком 50%-кого водного раствора тиосульфата натрия и оставляют
стоять на 48 час., после чего осадок отсасывают. п-Амино-о-бромфеиилртуть — белые че-
шуйки. Т. пл. 125° С.
Получение ди-лг-оксифенилртути [72]. 8 г хлористой л-оксифенилртути растворяют
в растворе 17 г кристаллического гипосульфита натрия в 110 мл воды. Вскоре после раство-
рения начинается помутнение и затем выпадение осадка. Через сутки выпавший осадок
отсасывают и промывают. Вес 4,6 г (выход количественный). Кристаллизация из спирта.
Вещество не плавится, разлагается выше 265° С.
Получение ди-а-фурилртутн [132]. К 50 г тиосульфата натрия в 200 мл воды быстро
прибавляют 0,1 моля хлористой а-фурил'ртути. Смесь сильно встряхивают в течение не-
скольких минут. Вскоре начинает выпадать осадок, который отсасывают через 8 час. Вы-
ход 95%. Кристаллизация из ацетоно-водного раствора или из спирта. Т. пл. 114° С.
Гладко симметризуется насыщенным водным раствором тиосульфата нат-
рия хлористая ферроценилртуть, образуя диферроценилртуть [137]. Хлор-
меркурцикЛопентадиенилтрикарбонил марганца при действии тиосульфата
натрия образует меркур-бг/с-циклопентадиенилтрикарбонилмарганец с вы-
ходом 78% [135].
Получение меркур-бпс-циклопентадиеиилтрикарбоиилмаргаица [135]. Раствор 2,2 г
(0,005 моля) хлормеркурциклопентадиенилтрикарбонил марганца в 2 мл ацетона прили-
вают к раствору 8 г тиосульфата натрия в 30 мл воды. Смесь при перемешивании выдержи-
вают при комнатной температуре в течение 2 час. и оставляют на ночь. Осадок отфильтро-
вывают, промывают водой, высушивают над хлористым кальцием, экстрагируют бензолом.
Из раствора бензол испаряют; остаток, который весит 1,19 г (78,3%),— это светло-желтое
кристаллическое соединение, растворяется в бензоле, этиловом спирте. Т. пл. 174,5° С
(с разложением). После двухкратного высаживания из бензола или четыреххлористого
углерода т. пл. 178—179° С [138].
254
Ртутноорганические соединения. Лит. стр. 260—263
Симметризация действием сернистых щелочей
2RHgX + K2S R2Hg + 2КХ +HgS
В настоящее время сравнительно редко применяют мало удобный метод
симметризации сернистыми металлами — сернистым калием или натрием
или сульфгидратом бария. В качестве промежуточного продукта получа-
ются ртутноорганические сульфиды, разлагающиеся при нагревании на
R2Hg и HgS.
Единственным способом симметризации соли а-меркуркамфенилона яви-
лось нагревание сульфида до 214—220° С, приведшее к ди-а-камфенилонил-
ртути с 80%-ным выходом [139]. Действием сернистых щелочей симметри-
зованы ртутноорганические производные ароматических углеводородов
[56], нитросоединений [140], кислот [141], аминов [142], фенолов [125,
144], гетероциклов [124].
Однако Мак-Котченом и Кобе [145] разработаны условия превращения
этим методом уксуснокислой фенилртути в дифенилртуть с выходом выше
90%.
Получение дифенилртути [ 145 ]. а. Разложение без растворителя.
75 г уксуснокислой фенилртути растворяют в 1 л дистиллированной воды, 100 г уксусно-
кислого аммония, 25 мл конц. раствора аммиака в 2-литровой эрленмейеровской колбе.
В раствор пропускают сероводород до полного осаждения. Осадок отфильтровывают,
промывают дистиллированной водой и отсасывают по. возможности досуха, затем подсу-
шивают в печи при температуре около 120° С в течение 20 мин. и еще раз просасывают на
воронке. Осадок разлагают нагреванием в печи в течение часа. Смесь охлаждают, кускй
измельчают для большей легкости извлечения. Дифенилртуть извлекают обработкой 600 мл
ацетона, осадок сернистой ртути отфильтровывают и промывают ацетоном. Из раствора вы-
езживают дифенилртуть 3,5 л воды, отсасывают и сушат на воздухе в течение ночи. Выход
33,7 г (85,4%).
б. Разложение в толуоле. Осадок фенилмеркурсульфида (полученный из
37 г уксуснокислой фенилртути, 1 л воды, 25 мл аммиака, 30 г уксуснокислого ам-
мония, как описано выше) промывают кипящей водой и отжатый иа фильтре нагревают
в круглодонной колбе с обратным холодильником и ловушкой для воды с 500 мл
толуола в течение 35 мин. так, чтобы удалить за это время всю воду. Затем отсасы-
вают воду, осадок промывают горячим толуолом, который затем отгоняют до объема
75 мл. Дифенилртуть выкристаллизовывается по охлаждении. Толуол отгоняют со спир-
том. Из спиртового раствора дополнительное количество кристаллов высаживают пяти-
кратным объемом воды. Выход 19,0 г (97,5%).
Получение ди-(3-иитро-4-толил)ртути [140].
NO2
(НзС-<О>-)аНб
3,7 г 4-хлормеркур-2-нитротолуола, помещенные в водный раствор 1,2 г кристалличе-
ского Na2S, постепенно превращаются в бесцветный сульфид. Смесь нагревают на водяной
бане 2 часа, черный продукт фильтруют, промывают, сушат, извлекают ацетоном. Темный
экстракт фильтруют холодным; черная коллоидная HgS проходит через фильтр, и остается
белый кристаллический осадок. Так повторяют несколько раз. Кристаллизация из толуо-
ла, т. пл. 291° С.
При смешении при комнатной температуре гидроокиси фенилртути с
сероуглеродом смесь вскипает. Продуктом реакции является выделенная
с 30%-ным выходом дифенилртуть [146]. Реакция идет через вероятное
промежуточное образование фенилсульфида ртути:
2C6H5HgOH + 2CS2 - (CeH6Hg)2S + 2COS + H2S, ’
(CeH5Hg)2 S HgS + (CeH5)2 Hg
(см. также гл. XIV, стр. 334).
Симметризация ртутноорганических соединений
255
Симметризация действием щелочей
2RHgX + 2КОН R2Hg + 2КХ + Н2О + HgO
Едкие щелочи также могут действовать в качестве симметризующих
средств, но так как ртуть выделяется при этом в виде несколько раствори-
мой окиси ртути, которая может реагировать с R2Hg с образованием RHgOH,
то симметризация этим методом идет менее полно, чем при действии обыч-
ных симметризующих агентов [147].
Симметризация действием аммиака
2RHgX + 2NH3'^ RaHg + HgX2.(NH3)2
Аммиак является мягко действующим хорошим агентом симметризации
продуктов присоединения солей ртути к непредельным алифатическим и
ароматическим соединениям и широко применяется для этих соеди-
нецйй. Реакции симметризации аммиаком к тому же наиболее изучены с
точки зрения механизма реакции и стереохимического течения. При его
применении не происходит обращения конфигурации стереоизомеров*. Его
-все шире успешно применяют для симметризации ртутноорганических сое-
динений тогда, когда желательно избегать применения более жестко дей-
ствующих средств (см., например, [148, 149]) и в случае а-меркурза-
мещенных жирных оксосоединений и кислот.
Действием аммиака симметризована хлористая циклопентадиенилртуть
[143].
Обычно реакцию ведут в растворе хлороформа, дихлорэтана, иногда в
бензоле. Оба продукта присоединения соли ртути к ацетилену строения
ClHgCH=CHCl, как транс- [150], так и цис- [151], симметризуются дей-
ствием аммиака, образуя ди-ф-хлорвинил)ртуть.
Получение транс,/прамс-ди Р-хлорвиннлртути [150]. При пропускании аммиака через
хлороформенный раствор хлористой трансф-хлорвинилртути выпадает белый осадок пре-
-ципитата ртути, по отделении которого и испарении растворителя из фильтрата получают
соединение (G1CH = CH)2Hg. Т. пл. 60° С.
Получение ц«е,цис-ди-0хлорвинилртути [151]. В раствор 2,6 г хлористой цис-р-хлор-
винилртути (т. пл. 73 °C) в 35 мл сухого бензола пропускают аммиак в течение 6 мин. со
скоростью 80—100 пузырьков в минуту. Выпавший осадок отфильтровывают, из фильтрата
медленно выпаривают растворитель при 30—35° С, а остаток его удаляют в вакуум-эксика-
торе при 10 мм в течение нескольких дней. Вес полученной бесцветной жидкости 1 г (70,9%
выхода), ijuc,tfuc-Диф-хлорвинилртуть не затвердевает при охлаждении до —70° С, 4°
2,8090, 4° 1,6308.
Аммиак является также наилучшим средством симметризации а-гало-
генмёркуркетонов [152], которые гидролизуются всеми водными раствора-
ми других симметризаторов. Для предотвращения вредного действия из-
бытка аммиака, снижающего выход меркур-бш?-кетона, раствор аммиака
в дихлорэтане или хлороформе приливают к раствору галогенмеркуркетона.
до прекращения образования преципитата. Реакцию ведут при комнатной
температуре; повышение температуры снижает выход [152].
Получение диацетоиилртути [152]. К раствору 9 г (0,03 моля) хлормеркурацетона в
250 мл дихлорэтана прибавляют раствор аммиака в дихлорэтане до прекращения выпаде-
ния преципитата. Осадок профильтровывают, промывают дихлорэтаном, растворы соеди-
няют и дихлорэтан испаряют на холоду. Выход диацетоиилртути 4,5 г (количественный).
После перекристаллизации из смеси бензола и гептана т. пл. 69° С.
* В случае других симметризаторов эта сторона реакции осталась почти неизученной.
256 Ртутноорганические соединения. Лит. стр. 260—263
Но продукты присоединения сулемы к ацетиленовым спиртам [1531,
к ацетиленовым кислотам и их эфирам [1541 под действием аммиака (а
также других агентов симметризации) не симметризуются, а разлагаются
на исходные компоненты. Не симметризуются аммиаком, а образуют с ним
комплекс эфиры хлормеркуруксусной кислоты [68]. Эфиры же а-галоид-
меркурарилуксусных кислот гладко симметризуются действием аммиака
в хлороформе и других растворителях [6—11].
Механизм симметризации аммиаком и строение переходного состояния
при этой реакции описаны в начале главы. Там же и в гл. XII приводятся
данные о реакции «сосймметризации» аммиаком RHgX и R'HgX и образо-
вании RHgR'.
Симметризация действием пиридина
2RHgCl + C3H5N RaHg + C5H5N HgCl2
RHgCl (где R — производные индандиона), представляющие собой,, со-
гласно Ганчу [155], С — Hg-соединения, симметризуются при действии
пиридина на холоду.
Симметризация действием триэтаноламина
Уксуснокислая фенилртуть симметризована [156] действием триэтанол-
амина. Реакцию ведут в присутствии молочной и лимонной кислот. Обра-
зующееся сначала двойное соединение при нагревании разлагается,
давая дифенилртуть:
CsHsHgOCOCHs + N(CH2CH2OH)3-> СбН5^(СНзСО2)Н(СН2СНгОН)3-»(СбН5)2 Hg +
+ CH3CO2Hg(CH3CO2)N(CH2CH2OH)3 + N(CH2CH2OH)3.
Симметризация действием непредельных соединений
При присоединении ртутноорганических оснований к непредельным со-
единениям образуются неустойчивые несимметричные ртутноорганические
соединения [157], например:
OR
сн,он /
ArHgOH + CH2=CHOR-----> ArHgCH2CH
\сН3
тотчас распадающиеся на смесь двух симметричных ртутноорганических
соединений:
ОСНз ОСН8
2ArHgCH2CH'/ - Ar2Hg + Hg ( СНг-СН^ j .
\* \ \ /2
OR OR
Реакция может быть использована [157] для симметризации ртутноорга-
нических солей в очень мягких условиях; для этого нет необходимости вы-
делять исходное ртутноорганическое основание ArHgQH, получаемое при
кипячении ртутноорганической соли ArHgX с СН3ОН — КОН.
В качестве активных непредельных соединений применены простые ви-
ниловые эфиры, диацеталь кетена. Образующиеся одновременно меркур-
Симметризация ртутноорганических соединений
257
бис-ацетальдегиды и меркур-бис-кетоны (или производные тех и других)
не выделяют как таковые, но после отделения Ar2Hg, прибавлением к филь-
трату соли ртути, изолируют в виде ртутноорганических солей [157].
Получение дифенилртути [157]. К раствору 5 г хлористой фенилртути в 100 мл мета-
иола прибавляют 26,5 мл 40% -ного метанольного раствора едкого кали и кипятят 30 мин.
После охлаждения фильтруют и к фильтрату приливают 1,5—2 г винилэтилового эфира..
На следующий день отгоняют */« растворителя и отсасывают выделившуюся дифенилртуть.
Выход 2,6 г (92%). Т. пл. 119—120° С.
В аналогичных условиях из 5 г хлористой n-толилртути и 2г винилэтилового эфи-
ра получено 2,7 г ди-и-толилртути (95,5%). Т. пл. 237—238°С.
Симметризация действием бутиллития
Реакция симметризации ртутноорганических соединений может проис-
ходить под действием бутиллития. В частности, так симметризованы сте-
реоизомерные ртутные производные стильбена. Симметризация действием
бутиллития, вероятно, идет через стадию образования нестойкого, несим-
метричного полнозамещенного соединения ртути [158]:
2СвН6СН=С (HgCl) СвН6 + 2C<HeLi [2CeH5CH=C (HgQHg) CeH5 + LiCl]
-> (CeH6CH=CCeH6)2Hg + (C4He)2 Hg.
Получение /пра«с-а-меркур-бпс-стильбеиа[158]. 1 г транс-а-хлормеркурстильбена раст-
воряют в смеси 40 мл бензола и 60 мл эфира и к раствору при —40 н 30° С в атмосфере
азота прибавляют эквимолекулярное количество титрованного эфирного раствора бутил-
лития. Сразу начинает выпадать осадок. Смесь стоит при той же температуре 40 мин.,
затем ее разлагают 50 мл 3%-ной соляной кислоты; осадок транс-а-меркур-бис-стильбена
отфильтровывают и слои разделяют. Получено 0,5 г (83%) тра«с-а-меркур-бис-стильбена
с т. пл. 239—241° С. Из эфирно-бензольного слоя выделено незначительное количество
транс-а-хлормеркурстильбена.
Аналогично из 1 г /р/с-а-хлормеркурстильбена получено 0,4 г (66%) quc-а-меркур-
бис-стильбена [158], выделившегося после отгонки в вакууме растворителей из высушен-
ного хлористым кальцием эфирно-бензольного слоя. Т. пл. после перекристаллизации из
спирта с бензолом 145—147° С. Реакция проведена в атмосфере азота.
При попытке действием бутиллития трансметаллировать хлормеркурцик-
лопентадиенилмарганецтрикарбонил произошла симметризация последнего
и образование ди-(циклопентадиенилмарганецтрикарбонил)ртути [138].
Симметризация действием дифенилртути
Дифенилртуть применена [159] в качестве симметризующего агента.
Она соединяется с образующимся галогенидом ртути в малорастворимую
галоидную фенилртуть]1 которая легко может быть отделена от более рас-
творимого симметричного соединения. Реакция идет через промежуточное
образование несимметричного соединения RHgC6H5, что подтверждено
спектром ЯМР [182].
Действием дифенилртути симметризованы соединения с «подвижным»
атомом водорода: этиловые эфиры а-броммеркур- и л-бром-а-броммеркур-
фенилуксусной кислоты, 3-броммеркуркамфора, 3-бензилкамфора, транс-[1-
хлорвинилмеркурхлорид.
Получение этилового эфира а-меркур-бис-фенилуксусной кислоты [159]. Раствор 4,5 г
(0,01 моля) этилового эфира а-броммеркурфенилуксусной кислоты в 30 мл хлороформа
сливают с раствором 1,8 г (0,005 моля) дифенилртути в 15 мл хлороформа. Осадок броми-
стой фенилртути отфильтровывают, промывают небольшим количеством хлороформа. Из
фильтрата после испарения хлороформа получено 2,28 г (85%) этилового эфира а-меркур-
бпс-феиилуксусной кислоты. Т. пл. 104—105° С.
258 Ртутноорган ические соединения. Лит. стр. 260—263
Симметризация действием третичных фосфинов
2RHgX + 2RgP -> RaHg + (R^P), HgXs:
Комплексообразователи—третичные фосфины являются симметризатора-
ми избирательного действия. Характер их реакции с RHgX зависит от
природы как R, так и R' и от среды реакции. Так, бромистая метилртуть
симметризуется действием триэтилфосфина в бензоле, в эфире же образует
фосфониевую соль_[СН3Н§Р(С2Н5)3]Вг [160]. Обычно для симметризации
применяют трифенилфосфин. С CH3HgBr [148] в эфире и с цис- 184] и
транс-[ Ц4] p-ClCH=GHHgClB спирте'он образует лишь комплекс [(CeHs) SPJ2 •
• HgHal2, но с хлористой перхлорцинилртутью в эфире или в этаноле наря-
ду с этим комплексом получен с выходом до 88% и продукт симметризации
(CCla=CCl)2Hg [114]. Продукт метоксимеркурирования окиси углерода —
хлористая карбометоксиртуть с трифенилфосфином в бензоле дает симмет-
ричное соединение— бис-карбометоксиртуть [161]. Хлористая фенилртуть
с трифенилфосфином образует комплекс 1 : 1, но не симметризуется [1611
Но при нагревании в течение 30 мин. в бензоле уксуснокислой фенил-
ртути с трибутилфосфинбм происходит реакция [162а]:
2CeHsHgOCOCH3 + (n-C4Hg)3P -> Hg ф (С6Н6)2 Hg ф (СНаСО)3 Hg ф (й-С*Н8)» РО.
Получение б’ие-перхлорвииилртути. К 0,5 г (0,014 моля) хлористой перхлорйинилрту-
ти в 50 мл этанола прибавляют 3,7 г (0,014 моля) трифенилфосфина в 350 мл Этанола. Тот-
час образуется белый осадок, который отсасывают и промывают Спиртом. Вес 4,8 г (86%)
(GeHebPHgCla, т. пл. 274—276е С. Из соединенных этанольных растворов после концент-
рирования их до ОД первоначального объема получено 2,8 г (88%) бис-перхлорвинилртути.
Т. пл. 72—73е С. 1
Получение бис-карбометоксиртути [161]. При вливании раствора 23 г (0;08 моля) хло-
ристой карбометоксиртути в 250 мл бензола в раствор 20,8 е (0,08 моля) трифенилфос-
фнна в 250 мл бензола тотчас выпадает комплекс трифенилфосфина и сулемы. От филь-
трата отгоняют растворитель в вакууме при температуре не выше 50° С. Перекристаллиза-
ция из эфира. Т. пл. 84,5° С.
Симметризация действием пентакарбонила железа
и его производных
HgFe(CO)4 очевидно, наряду с R2Hg является конечным продук-
том действия пентакарбонила железа на гидроокись фенилртути в метило-
вом спирте [164] и реакции, происходящей при прибавлении раствора же-
лезокарбонилгидрида к раствору гидроокиси метилртути и других ртутно-
органических оснований [164].
При действии на RHgOH (R = CeH5, С2На) CaFe(CO)4 образуется
(RHg)2Fe(CO)4, тотчас диспропорционирующееся на R2Hg и HgFe(CO)4.
В случае R = СН3 соответствующее (RHg)2Fe(CO)4 устойчиво, но и оно
медленно диспропорционируется с образованием (CH3)2Hg и HgFe(CO)4
[163]. .
\ Симметризация действием окиси алюминия
Йодистая о-иодфенилртуть симметризуется при хроматографировании на
окиси алюминия (растворители бензол—циклогексан). Выход R2Hg 94%
[165].
Симметризация без участия симметризующих агентов
В редких случаях некоторые ртутноорганические соли сймметризуются
при нагревании; так, меркурированный бензантрон симметризуется при
нагревании его Др температуры плавления [166]. При нагревании до 200° С
в вакууме (10 мм) фтористой фенилртути получается дифенилртуть [167].
Уксуснокислая 2,5-дихлорфенилртуть симметризуется [712 при нагрева-
Симметризация ртутноорганических соединений
259
нии в л-ксилоле или мезитилене до 142—143° С. JCH2HgJ при 2,5-часо-
вом кипячении в тетрагидрофуране симметризуется в (JCH2)2Hg [186].
. При нагревании иодистой о-иодфенилртути образуется б«с-о-иодфенил-
ртуть [165].
(C6H6)3SiOHgC6H5 при нагревании в течение 75 мин. при 145° С в атмо-
сфере азота переходит в дифенилртуть [168] с образованием полимерных
фенилсилоксанов.
Получение бис-2,5-(дихлорфеиил)ртути [71]. Раствор 0,5 г ацетоксимеркур-и-дихлорбен-
зола в 17 мл и-ксилола (или мезитилена) нагревают в течение 10 час. при 142—143° С. По-
сле отгонки большей части растворителя получено 0,21 г бис-(2,5-дихлорфенил)ртути.
Кристаллизация из смеси ацетона и хлороформа. Т. пл. 237° С. В продуктах реакции име-
ется металлическая ртуть.
РЕАКЦИЯ, ОБРАТНАЯ СИММЕТРИЗАЦИИ
Важной с препаративной точки зрения является реакция, обратная
симметризации,— получение ртутноорганических солей при взаимодейст-
вии полнозамещенных ртутноорганических соединений с солями и окисью
ртути
RaHg + HgX2-* 2RHgX.
Обычно реакция идет очень легко. В большинстве случаев для ее осу-
ществления достаточно нагревания эквимолекулярных количеств реаген-
тов в спиртовом или ацетоновом, тетрагидрофурановом [80] или даже эфир-
ном [169] растворе; иногда, в особенности с солями ртути органических
И двухосновных кислот [170, 171], реакцию ведут без растворителя: сплав-
лением, а если один из «компонентов: жидкость, то в некоторых случаях
даже и без нагревания; с трехосновными кислотами реакцию ведут без
растворителя в присутствии небольшого количества воды [172]; с ртутны-
ми солями органических кислот и в среде одноименной кислоты [80].
При образовании ртутноорганической соли реакцией как транс, транс-,
так и цис, цис-симметричных продуктов присоединения солей ртути по
тройной связи с солями ртути конфигурация стереомеров строго сохраня-
ется (Несмеянов, Борисов [151, 173 и другие работы этих авторов]).
Также сохраняется конфигурация при взаимодействии с солями ртути
оптически активных симметричных ртутноорганических соединений.
О реакции Hg203Cl2 с RHgR', протекающей с сохранением конфигура-
ции и сопровождающейся изотопным обменом, см. гл. XIV, стр. 320.
Полученная взаимодействием оптически активных симметричных ртут-
ноорганических соединений (дивторичнобутилртути и бис-2-метилгексил-5-
ртути) с солью ртути оптически активной кислоты (ртутной солью моноэти-
лового эфира d-винной кислоты) смесь двух (для каждого случая) диасте-
реомеров разделена кристаллизацией на оптические антиподы, которые об-
работкой бромистым кальцием выделены в виде устойчивых бромидов [174].
Произведено расщепление на оптические антиподы солей вторичнобу-
Тилртути—тартрата и Z-фенилгликолята [43].
Циклические ртутноорганические соединения, содержащие ртуть в ка-
честве члена цикла, реагируют с галогенидом ртути с раскрытием цикла,
образуя галогенид алкил(арил)ртути [100, 175]. Так, гексамерная о-фени-
ленртуть при 8-часовом кипячении в ацетоновом растворе с сулемой дает
о-бис-хлормеркурбензол [175].
Получение RHgNO3 (R — арил [138, 176, 177], аралкил или алкил
[138, 176]) реакцией R2Hg в ацетоне с водной азотнокислой ртутью ведут в
присутствии азотной кислоты во избежание образования основного нитрата.
Подобно другим RHgX реакцией R2Hg с Hg(CNO)2 получены фуль-
минаты арилртути [185].
260
Ртутноорганические соединения
В некоторых случаях — с частично ионным характером связи С—Hg в
RJHg — реакция, обратная симметризации, не происходит. Так, попытки
приготовить C6H5C=CHgJ из (C6H5C=C)2Hg и HgJ2 не удались [162].
(CF3CFCF3)2Hg также не реагирует с сулемой (100° С, 24 часа) [178].
Получение метилового эфира .и-броммеркурбензойной кислоты [74]. 0,12 г меркур-
биг-.и-бензойнометилового эфира, 0,09 г бромной ртути в 2 мл этилового спирта нагрева-
ют в запаянной пробирке в течение 2 час. на водяной бане. После охлаждения пробирку
вскрывают, слегка упаривают спирт. Выход 0,22 г (90%) метилового эфира л-броммеркур-
бензойной кислоты. Перекристаллизация из бензола. Т. пл. 204° С.
Получение хлористой 0-нафтйлртути [170]. Кипятят раствор в амиловом спирте ди-
0-нафтилртутй и сулемы. После охлаждения выкристаллизовывается хлористая 0-нафтил-
ртуть. Перекристаллизация из этилового спирта. Т. пл. 271° С.
Получение янтарнокислой этилртути [171]. 2,6 г диэтилртути нагревают* на водяной
бане до 100° С с 3,2 г янтарнокислой ртути. Продукт нацело растворяется в спирте (янтар-
нокислая ртуть в спирте почти не растворима). Перекристаллизация из спирта. Выход чи-
стого вещества 4,7 г (81%). Т. пл. 157—158° С.
Получение хлористой ци^Рхлорвинилртути [151]. К раствору 0,15 г (0,00046 моля)
грс-ди-(0-хлорвинил)ртути в 8 мл сухого эфира прибавляют 0,125 а (0,00046 моля) сулемы
в 8 мл сухого эфира. Немедленно взятая проба не показывает со щелочью реакции на ион-
ную ртуть. После испарения растворителя оставшийся белый кристаллический осадок
плавится при 77—78° С, весит 0,26 г (96%). После перекристаллизации из смеси бензола и
петролейного эфира (1 : 2) т. пл. 78,5—79° С.
Получение хлористой этилртути см. [179].
ЛИТЕРАТУРА
1. Несмеянов А. Н., РеутовО. А., Поддубная С. С. Изв. АН СССР,
ОХН, 1953, 850.
2. Несмеянов А. Н., РеутовО. А., Поддубная С. С. ДАН СССР, 88,
479 (1953).
3. Н е с м е я н о в А. Н., РеутовО. А. Изв. АН СССР, ОХН, 1955, 739; Вести.
МГУ, № 4-5, 133 (1955)’.
4. Несмеянов А. Н., РеутовО. А., Белецкая И. П., М а р д ал ей-
швилиР. Е. ДАН СССР, 116, 617 (1957).
5. Реутов О. А., Белецкая И. П., Мардалейшвили Р. Е. ЖФХ, 33,
152, 1962 (1959).
6. Р е у т о в О. А., Белецкая И. П. Сб. Кинетика и катализ. М., Изд-во АН СССР,
1960, стр. 48.
7. РеутовО. А. Белецкая И. П., Артамкнна Г. А. Сб. Кинетика и ката-
лиз. М., Изд-во'АН СССР, 1960, стр. 55.
8. Реутов О. А., Б е ле ц к а я И. П., Артамкнна Г. А. ЖФХ, 36, 2582 (1962).
9. Р е у т о в О. А., Белецкая И. П. ДАН СССР, 131, 853 (1960).
10. Б е л е ц к а я И. П., Артамкнна Г. А., Р е у т о в О. А. ДАН СССР, 149,
90 (1963).
11. Белецкая И. П., Артамкнна Г. А., РеутовО. А. Изв. АН СССР,
ОХН, 1963, 765.
12. Р е у т о в О. А., Л у Ц з и н - ч ж у. ЖОХ, 29, 182 (1959).
13. Т г а у 1 о г Т. G., W i n s t е i п S. J. Org. Chem., 23, 1796 (1958).
14. J е и s е n F. R., W h i р р 1 е L. D., Wedegaert ner D. K., Landgre-
b e J. A.- J. Am. Chem. Soc., 81, 1262 (1959).
15. Wi ns tei n S., Traylor T. G. J. Am. Chem. Soc., 78, 2597 (1956).
16. P e у т о в О. А., Л у Цзи н-чжу. ДАН СССР, ПО, 575 (1956).
17. Р е у т о в О. А., Л у Ц з и н - ч ж у. ЖОХ, 29, 1207 (1959).
18. Adams R., R о m a n F. L., Sperry W. N.J. Am. Chem. Soc., 44, 1789 (1922).
19. В г о о k е A. G., DonovanR„ W г i g h t G. F. Can. J. Chem., 31, 536 (1953).
20. J e n s e n F. R., L a n d g r e b e I. A. J. Am. Chem. Soc., 82, 1004 (1960).
21. В i 1 1 i n g e В. H. M., Gowenlock B. G. J. Chem. Soc., 1962, 120].
22. W г i g h t G. F. Can. J. Chem., 30, 268 (1952).
23. Несмеянов A. H., Реутов О. А., У Я н - цей, Лу Цзин-чжу. Изв.
АН СССР, ОХН, 1958, 1327.
24. Реутов О. А., У г л о в а Э. В. Изв. АН СССР, ОХН, 1959, 1691.
25. Р е у т о в О. А., К н о л л ь П. Г., У Я н - цей. ДАН СССР, 120, 1052 (1958).
26. Н е ф е д о в В. Д., Синотова Е. Н., Фролов Н. Я- ЖФХ, 30, 2356 (1956).
27. Нефедов В. Д., Синотова Е.Н. Сборник работ по радиохимии. Изд-во ЛГУ,
1955, стр. 113.
Симметризация ртутноорганических соединений 261
28. С и н о т о в а Е. Н. ЖНХ, 2, 1205 (1957).
£9 Реутов О. А., Смол ина Т. А., Калявин В. А. ЖФХ, 36, 119 (1962).
30. Р е у т о в О. Аб С о к о л о в В. И., Белецкая И. П. ДАН СССР, 136, 631
(1961). '
31 Р е у т о в О. А., Б е л е ц к а я И. П., У Я н - ц е й. Сб. Кинетика и катализ. М.,
Изд-во АН СССР, 1960, стр. 43.
32. Р е у т о в ,0. А., С м о л и н а Т. А., К а л я в и н В. А.- ДАН СССР, 139, 389
(1961).
33. С г о s s J. М., Р i n a j i a n J. J. С. А., 45, 3694 (1951).
34. A n s а 1 о n i А., В е 1 m о n d i G., С г о a t t о V. С. А., 51, 5517 (1957); Ricerca
Sci., 26, 3315 (1956).
35. Н е ф е д о в В. Д., С и н о т о в а Е. Н. Сборник работ по радиохимии. Изд-во
ЛГУ, 1955, стр. ПО.
36 Cerfontain Н., Van A k е n G. М. F. J. Am.- Chem. Soc., 78, 2094 (1956).
37. РеутовО. А.’, Соколов В. И., Белецкая И. П. Изв. АН СССР, ОХН,
1961, 1213.
38. РеутовО. А., Ху Хун-ве и, Белецкая И. П., Смолина Т. А. ЖФХ,
35, 2424 (1961).
39. Р е у т о в О. А., Кар по в Г. П., У г л о в а Э. В., М а л я н о в В.. А. Изв. АН
СССР, ОХН, 1960, 1311; ДАН СССР, 134, 360 (1960); Tetrahedron L., 19, 6 (1960).
40. ReutowO. A. Acta Chim, Acad.-Sci. Hung., 18, 439 (1959); Angew. Chem., 72,
198 (1960); Records of chem. Progress,, № 1, 1 (1961).
41. Winstein S., T г а у Го r T. G., G а г n е г С. S. J. Am. Chem. Soc., 77, 3741
(1955).
42. Dessy R. E., Lee У. K., Kim J.-Y. J. Am. Chem. Soc., 83, 1163 (1961).
43. C h a r m a n H, В., H u g h e s E. D., I n g о 1 d C. J. Chem. Soc., 1959, 2523,2530.
44. Jensen F. R-, Rickborn B. J. Am. Chem. Soc., 86 , 3784 (1964).
45. Hughes E, D., Ingold C., Thorpe F. G., V о 1 g e r H. C. J. Chem. Soc,,
1961, 1133.
46. H u g h e s E. D.,'Vo 1 ger H. C. J. Chem. Soc., 1961, 2359.
47. Charman H. В., H u g h e s E. D„ Ingol d C.,T hor pe F. G. J. Chem. Soc.,
1961, 1121.
48. J e n s e n F. R. J. Am. Chem. Soc., 82, 2469 (1960).
49. Dessy R. E., Lee I. K- J. Am. Chem. Soc., 82, 689 (1960).
50. Brodersen K., Schlenker U. Ber., 94, 3304 (1961).
51. Несмеянов H. А., Реутов О. А. ДАН СССР, 144, 126 (1962).
52. H e с м e я н о в А. Н., Луценко И. Ф., Хомутов Р. М. ДАН СССР, 120,
1049 (1958).
53. S t е i n к о р f W. Ann., 413, 310 (1917).
54. Steinkopf W., BauermeisterM. Ann., 403, 50 (1914).
55. Rausch M., Vogel M., Rosenberg H. J. Org. Chem., 22, 900 (1957).
56. D r e h e r E., О t t о R. Ann., 154, 93 (1870).
57. Ot'to R. Ann., 154, 190 (1870).
58. H i 1 p e r t S., GriittnerG. Ber., 47, 186 (1914).
59. В r a u n J. Ber.,-47, 490 (1914).
60. H e с м e я н о в A. H., Борисов A. E„ Волькенау H. А. Изв. АН СССР,
ОХН, 1956, 162.
61. С о n t a r d i A., C i о с с a B. Ann. Chim. applicata, 23, 362 (1933).
62. Э с к и н И. T. Изв. АН СССР, ОХН, 1942, 297.
63. Э с к и н И. T., Несмеянов А. Н. Изв. АН СССР, ОХН, 1942, 116.
64. Э с к и и И. Т. Изв. АН СССР, ОХН, 1942, 302.
65. Н е i n F., W a g 1 е г К- Вег., 58, 1499 (1925).
66. Не i n F„ W ag 1 е г К. D. Нем. пат. 444666; Zbl., 1927, II, 740.
67. Несмеянов А, Н., Каи Э. И. ЖРХО, 61, 1407 (1929); Вег., 62, 1018 (1929).
68. Ф о с с В. Л., Луценко И. Ф., Несмеянов А. Н. ЖОХ, 35 (1965). .
69. Lou don J. D. J. Chem. Soc,, 1935, 535.
70. Dref a h I G., S t a n g e G. J. prakt. Chem., 9, 311 (1959).
71. Петрович П. И. Ж- Всесоюзн. хим. общ. 5, 106 (1960).
72. Н е с м е я н о в А. Н., Торопова Е. М. ЖОХ, 4, 667 (1943). .
73. R о d i g h i e г о G. Ann. chim. applicata, 39, 621 (1949).
74. Несмеянов A. H., Макарова Л. Г. ЖОХ, 1, 598 (1931).
75. H u r d C. D., MorisseyC. J. J. Am. Chem. Soc., 77, 4658 (1955).
76. Л e в и н a P., Я., Костин В. H. ЖОХ, 28 3307 (1958).
77. Ichikawa K-, О u c h i H., Fukushima S. J. Org. Chem., 24, 1129 (1959).
78. Emeleus H. J., H a s z e 1 d i n e R. N. J. Chem. Soc., 1949, 2953.
78a. Sachs G., Furst K. Monatsh., 53/54, 550 (1929).
79. БорисовА- E., Новикова H. В. Изв. АН СССР, ОХН, 1957, 1258.
262 Ртутноорганические соединения
80. Н е с м е я н о в А. Н., Борисов А. Е., С а в е л ь е в а . И. С., Голубе-
в а Е. И. Изв. АН СССР, ОХН, 1958, 1490.
81. Vijayaraghavan К. V. С. А., Зв, 4477 (1942); J. Indian Chem. Soc., 17, 589
(1940).
82. К i г г m а п А., К 1 е i n е - Р е t е г М. Bull. Soc. chim. France,; 19.57, 894.
83. Vijayaraghavan К- V. C. A., 38, 2006 (1944); J. Indian Chem. Soc., 20, 318
(1943). .
84. H есмеяно в A. H., БорисовА. E. Изв. АН СССР, ОХН, 1945, 146.
85. Левина P. Я., Костин В. H. ЖОХ, 23, 1054 (1953).
86. Л е в и н а Р. Я., Ко ст ин В. Н., Тартаковский В. А. ЖОХ, 27, 881
(1957). '
87. S a n d J., S i n g e r F. Ber.-, 35, 3171 (1902),
88. P e утов О. А., ЛуЦзин-чжу, БундельЮ. Г. Вести. МГУ, 13, № 5, 111
(1958).
89. Реутов О. А., Л у Цзин-чжу. ЖОХ, 29, 1617 (1959).
90. L о u d о n J. D. J. Chem. Soc., 1933, 825.
90а. S р е n g 1 е г G., W е b ё г A. Brennst.- Chem., 43, 234 (1962).
91. М а у n а г d I. L. J. Am. Chem. Soc., 54, 2118 (1932).
92. D i m г о t h О. Ber., 35, 2032 (1902).
93. Maynard I. L. J. Am. Chem. Soc., 46, 1510 (1924).
94. ,H e с м e я и о в A. H., Шацкая P. X; ЖОХ, 5, 1268 (1935).
95. D i m г о t h O. Ber., 35, 2853 (1902).
96. Whitmore F. C., W о о d w a r d G. J. Am. Chem. Soc., 48, 333 (1926).
97. S t e i n k о p f W., Bielenberg W., Augenst ad-Jensen H. Ann., 430,
41 (1923).
98. P г о f f t E., О t t о К. H. J. prakt. Chem., 8, 156 (1959).
99. G о 1 d w h i t e H., H a s z e 1 d i n e R. N., MuknerjeeR. N. J. Chem. Soc.,
1961, 3825.
100. Sawatzky H„ Wright G. F. Can. J. Chem., 36, 1555 (1958).
101. Sand J. Ber., 34, 2910 (1901).
102. Grdenic D. Acta Cryst., 5, 367 (1952).
103. Summerbell R. K., Stephens J. R. J. Am. Chem. Soc., 77, 6080 (1955).
104. G i Im a n H., В a r n e 11 M. M. Rec. trav. chim., 55, 563 (1936).
105. S e i d e O. A., S c h e r 1 i n S. M., В r a z G. I. J. prakt. Chem., 138 (1), 55 (1933).
106. Gilman H„ Y a 1 e H. L. J. Am. Chem.' Soc., 72, 8 (1950).
106a. Traylor T. G., Baker A. W. J. Am. Chem. Soc., 85, 2746 (1963).
107. Henbest H. B., Nicholls B. J. Chem. Soc., 1959, 227.
108. Spinelli D., Salvemini A. Ann. chim. (Italy,), 50, 1423 (1960).
109. Beattie R. W., W h i t m о r e F. C. J. Am. Chem. Soc., 55, 1567 (1933).
110. Gowenlock B. G., T г о t m a n J. J. Chem. Soc., 1957, 2114.
111. M a у n a r d I. L., H о w a r d H. C. J. Chem. Soc., 123, 960 (1923).
112. Мел ьников H. H., P о к и ц к а я М. С. ЖОХ, 7, 2596 (1937).
113. Whitmore F. С., Sobatzki R. J. J. Am. Chem. Soc., 55, 1128 (1933).
114. Seyferth D„ ToweR. H. J. Inorg. a. Nucl. Chem., 1, 185 (1962).
115. Синтезы органических препаратов, сб. 1. М., ИЛ, 1949, стр. 201.
116. L a m d а п S.. Rev. assoc, bioquim. argentina, 114, 295 (1947); С. A., 43, 4236 (1949).
117. R a u s c h M. D. J. Inorg. a. Nucl. Chem., 1, 414 (1962).
118. Whitmore F.C. J. Am. Chem. Soc., 41, 1841 (1915).
119. W h i t m о r e F. С., Middleton E. J. Am. Chem. Soc., 44, 1546 (1922).
120. S t e i n k о p f W. Ann., 424, 40 (1921).
121. S t e i n k о p f W., Leonhardt P. Ann., 495, 166 (1932).
122. Bogert M. T., Herrera P. P. J. Am. Chem. Soc., 45, 328 (1923); 48, 223 (1926).
123. C h a 1 1 e n g e r F„ M i 11 e r S. A. J. Chem. Soc., 1939, 1005.
124. Whitmore F. C., Middleton E. J. Am. Chem. Soc., 45, 1753 (1923).
124a. W i4 t i g G., Herwig W. Ber., 88, 962 (1955).
1246. Rausch M. D. J. Org. Chem., 28, 3337 (1963).
125. В u c k t о n G. B. Ann., 108, 105 (1858).
126. S t e i n k о p f W. Ann., 428, 123 (1922).
127. Венус-Данилова Э. Д. Tp. Ленингр. технол. ин-та им. Ленсовета, 60, 32
(1960).
128. Schoeller W., SchrauthW. Ber., 53, 634 (1920).
129. Чалов В. П. ЖОХ, 18, 608 (1948).
130. БорисовА. Е. (в печати).
131. V е с с h i о 11 i L. Gazz., 58, 243 (1928).
132. Gilman H. J. Am. Chem. Soc., 55, 3302 (1933).
133. Chu t e W. J., Orchar d W. M„ Wr igh t G. F. J. Org. Chem., 6, 157 (1941).
134. UkaiT. Zbl., 1929, I, 1108.
135. Несмеянов A. H., Анисимов К. H., Валуева 3. П. Изв. АН СССР,
ОХН 1962, 1683.
Симметризация ртутноорганических соединений
263
136. Несмеянов А. Н., Сазонова В. А., Дрозд В. Н., Никонова Л. А.
ДАН СССР, 131, 1088 (1960).
137. Несмеянов А. Н., Перевалова Э. Г., Головня Р. В., Несмеяно-
ва О. А. ДАН СССР, 97, 459 (1954).
138. Cais М., Kozikows к у J. J. Am. Chem. Soc., 82, 5667 (1960).
139. Несмеянов А. Н., Кр и цк ая И. И. ДАН СССР, 121, 477 (1958).
140. CoffeyS. J. Chem. Soc., 128, 639 (1926).
141. S с h о е 1 1 е г W., Schrauth W., Hueter R. Ber., 53, 637 (1920).
142. Pesci L. Gazz., 23, II, 529 (1893); 28, II, 446 (1898).
143. Несмеянов A. H., Дворянцева Г. Г. и др. ДАН СССР, 159, 847 (1964).
144. Fourneau Е., Vila A. J. pharm. Chim., 6 (VII), 433 (1912).
145. McCutchanR. Т., Kobe К. A. Ind. Eng. Chem., 46, 675 (1954). Пат. CHIA
2628241 (1953); С. A., 48, 718 (1954).
146. R e i c h 1 e W. T. Inorg. Chem., 1, 650 (1962).
147. Whitmore F. С., H a n s о n E. R., C a r n a h a n F. L. J. Am. Chem. Soc.,
51, 894 (1929).
148. Реутов О. А., БеспрозванныйМ. А. ДАН СССР, 80, 765 (1951).
149. Несмеянов А. Н., Р е у т о в О. А. Изв. АН СССР, ОХН, 1953, 655.
150. J enk i ns W. J. Zbl., 1921, III, 608; J. Chem. Soc., 119, 747 (1921).
151. H e с м e я н о в A. H., БорисовА. E., Гуськова A. H. Изв. АН СССР,
ОХН, 1945, 639.
152. Несмеянов A. H., Луценко И. Ф., Хомутов Р. М. ДАН CCCR, 88,
837 (1953).
153. Несмеянов А. Н., Кочетков Н. К. Изв. АН СССР, ОХН, 1949, 76.
154. Несмеянов А. Н., Ко.четко в Н. К-, Д а ш у н н н В. М. Изв. АН СССР,
ОХН, 1950, 77.
155. На n tzsch A., G a j е w s k i F. Ann., 392, 302 (1912).
156. Пат. США 2423261 (1947); С. A., 42, 1376 (1948). Англ. пат. 605442 (1948); С. А., 43,
5528 (1949).
157. ЛуценкоИ. Ф., Ю р к о в а Е. И. Изв. АН СССР, ОХН, 1956, 27.
158. Несмеянов А‘. Н., БорисовА. Е., В о л ь к е н а у Н. А. Изв. АН СССР,
ОХН, 1954 , 992.
159. Реутов О. А., Белецкая И. П., Филиппенко Л. Р. Научные доклады
высшей школы. Химия и хим. технология, 1958, 754.
160. Cross R. J., Lauder А., С о a t е s G. Е. Chem. and Ind., 47, 2013 (1962).
161. Paulik F. E., Dessy R. E. Chem. and Ind., 37, 1650 (1962).
162. D e s s у R. E„ Budde W. L., W о о d r u f f C. J. Am. Chem. Soc., 84, 1172
(1962).
162a. M u k a j a m a T., К u w a j i m a J., S u z u k i Z. J. Org. Chem., 28,2024 (1963).
163. H e i n F„ H e u s e r E. C. A., 37, 3685 (1943); Z. anorg. allg. Chem., 249, 293 (1942).
164. H e i n F„ P о b 1 о t h H. C. A., 37, 26764 (1943); Z. anorg. allg. Chem., 248, 84 (1941).
165. Wittig G„ Ebel H. F. Ann., 650, 20 (1961); Angew. Chem., 72, 564 (1960).
166. Bernardi A. Gazz., 67, 380 (1937); Zbl., 1937, II, 2833.
167. Wright G. F. J. Am. Chem. Soc., 58, 2653.(1936).
168. G h 6 s h А. К.. H ansing С. E., S t u t z A. I., Mac D i а г m i d A. G. J. Chem.
Soc., 1962, 403.
169. M e a 1 s R. N. J. -Org. Chem., 9, 211 (1944).
170. M i c h a e 1 i s A. Ber., 27, 244 (1894).
171. Мельников H. H., Рокицкая M. С. ЖОХ, 7, 2518 (1937).
172. Мельников H. H., Рокицкая M. С. ЖОХ, 11, 592 (1941); ЖПХ, 14, 446
(1941).
173. Несмеянов A. H., Борисов A. E.,C а в e л ь e в а И. С., О с и n о в aM. A.
Изв. АН СССР, ОХН, 1961, 1249.
174. Р е у то в О. А., У г л о в а Э. В. Изв. АН СССР, ОХН, 1959, 757.
175. W i t t i g G., Bickelhaupt F. Ber., 91, 883 (1958).
176. Пат. США 2366683 (1945); С. A., 39, 2079 (1945).
177. Канад, пат. 424236 (1944); С. А., 39, 710 (1945).
178. Miller W. Т., jr., F г е е d m а п М. В. J. Am. Chem. Soc., 85, 180 (1963).
179. VitteG., MesnardP. Bull. Soc. Chim. France, 8, 350 (1941).
180. Несмеянов A. H., БорисовА. E., Вильчевская В. Д. ДАН СССР,
90, 383 (1953).
181. Н е с м е я н о в А. Н., БорисовА. Е., Новикова Н. В. Изв. АН СССР,
ОХН, 1959, 1216.
482. Jensen F. R., Miller J. J. Am Chem. Soc., 86, 4735 (1964).
183. Эскин И. T. Изв. АН СССР, ОХН, 1947, 405.
484. Нем. пат. 249729; Zbl., 1912, II, 777.
185. Beck W., Schuierer E. J. Organomet. Chem., 3,55 (1965).
186. Blanchard E. P., jr., Blomstrom D. C., Simmons H. E. J. Organomet.
Chem., 3, 97 (19651.
Глава XIV
РЕАКЦИИ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Органические соединения ртути по сравнению с другими металлоорга-
ническими соединениями отличаются незначительной реакционной способ-
ностью. Так, они не реагируют связью С—Hg с подвижным водородом
воды, спиртов и аминов и лишь подвержены ацидолизу минеральными кис-
лотами с разной скоростью для разных типов ртутноорганических соеди-
нений. Будучи совершенно инертны по отношению к кислородным соедине-
ниям, в частности к карбонильной группе, а также к двойным связям C=N,
C=S, N=O и с трудом вступая в реакцию с органическими галогенида-
ми в отсутствие инициирующего действия ультрафиолетового света и пере-
кисей (о фотохимических реакциях ртутноорганических соединений см.
стр. 325), ртутноорганические соединения находят ограниченное применение
в качестве средств синтеза главным образом для получения других метал-
лоорганических соединений при своем взаимодействии с металлами и -неор-
ганическими и металлоорганическими галогенидами.
Действию кислот, галоидов и других реактивов, разрывающих связь
С—Hg, легче поддаются ароматические соединения, чем жирные. Полно-
замещенные соединения R2Hg значительно реакционноспособнее ртутно-
органических солей RHgX.
ДЕЙСТВИЕ[КИСЛОРОДА
Ртутноорга'нические соединения устойчивы к действию кислорода воз-
духа; с ними работают без применения мер предосторожности в этом от-
ношении. Некоторые ртутноорганические соединения (в частности с ртутью,
соединенной с вторичными и третичными атомами углерода алифатических
соединений) при длительном хранении выделяют металлическую ртуть,
по-видимому, в результате окислительного действия кислорода воздуха.
Так ведет себя диизопропилртуть [1], дициклогексилртуть [2]. Одновре-
менно образуются продукты окисления органической части молекулы —
кетон и спирт.
Длительное воздействие кислорода на диизопропил- и дициклогек-
сил- [2а], дитре’Гичноамил-[26], дибензил ртуть [26] изучено в некоторых рас-
творителях. Диизопропилртуть (в запаянной трубке, в изопропиловом спирте
при встряхивании) с кислородом реагирует па уравнению
(»-СзН7)2Нё + О2 [(£ - C3H7)2Hg.O2] - Hg + (СНз)2СО +(СН8)2СНОН
(для обнаружения изопропилового спирта как продукта реакции бралась
меченная С14 диизопропилртуть).
Реакции ртутноорганических соединений
265
При проведении реакции с СС14 (17—20° С, 20 час.) образуются те же
продукты, а также хлористая изопропилртуть и хлористый изопропил;
при увеличении относительного количества диизопропилртути и проведе-
нии реакции при 44° С в течение 18 час. не образуется металлической рту-
ти и изопропилового спирта и меняются относительные количества других
продуктов реакции. Продуктами реакции в хлороформе являются металл и-'
ческая ртуть, хлористая изопропилртуть, ацетон, изопропиловый спирт.
При 40-часовом пропускании при 60° С сухого кислорода в раствор ди-
циклогексилртути в изопропиловом спирте образуются металлическая
ртуть (64%), циклогексанол и циклогексанон; в хлороформе (25°C, 25 час.),
кроме металлической ртути (20%), цикЛогексанола и циклогексанона, об-
разуются также в небольших количествах циклогексан и циклогексен,
а также хлористая циклогексилртуть (70% от исходной дициклогексилртути);
в четыреххлористом углероде (30 час. при комнатной температуре) образу-
ются в меньших количествах те же продукты и 76% хлор истой , циклогек-
силртути. Аналогичные продукты получены при окислении дитретично-
амилртути в СС14, СНС13, бензоле, циклогексане. Но и дибензилртуть
при действии кислорода дает бензоальдегид.
Скорость реакции расщепления действием хлорной кислоты в воде
иодистой изопропил- и третйчнобутилртути значительно увеличивается и
порядок ее изменяется при проведении ее в присутствии кислорода [3],
что приписано его окислительному действию на эти ртутноорганические
соединения.
В присутствии кислорода воздуха возрастает скорость термического
разложения дифенилртути в бензоле на ртуть, дифенил и обмен фенильны-
ми радикалами между дифенилртутью и меченым бензолом [4], а также
увеличивается степень фоторазложения дифенилртути в бензоле на ртуть
и дифенил [4].
Действие озона на алкенильные ртутноорганические соединения опи-
сано в этой главе (стр. 275) и гл. XV.
ДЕЙСТВИЕ КИСЛОТ
Замена ртути на водород и другие реакции
с кислотами ртутноорганических соединений, не являющихся продуктами
присоединения солей ртути к олефинам
Связь Hg — С легче разрывается действием галоидоводородных кислот
и несколько более устойчива по отношению к кислородным кислотам.
Полнозамещенные соединения ртути легко разлагаются минеральными и
несколько труднее карбоновыми кислотами [5—18, 21, 22] до ртутноорга-
нических солей
R2Hg + НХ -» RHgX + RH. (1)
На ртутноорганические соли карбоновые кислоты обычно не действуют
(о действии карбоновых кислот в жестких условиях см. дальше), минераль-
ные кислоты разлагают, их особенно легко при повышенной температуре:
RHgX-j-НХ-» RH + HgX2. (2}
Прочность различных ртутноорганических соединений по отношению
к кислотам варьирует в широких пределах. Наиболее легко кислоты отщеп-
ляют ацетилены от. ацетиленидов типа RC=-CHgR. Наоборот, продукты
присоединения ацетилена к сулеме (ClCH=CHHgCl) устойчивы к не слиш-
ком крепкой соляной кислоте.
266 Ртутноорганические соединения. Лшп. стр. 336—352
В продуктах присоединения сулемы к ацетиленовым кетонам и ацети-
леновым кислотам под действием кислот происходит обычная для ртутно-
органических соединений.замена HgX. на водород (см. гл. VI, стр. 129).
Ртуть, занимающая место наиболее «подвижного» водорода, отщепля-
ется кислотами наиболее легко. Арилы всегда отщепляются легче, чем ал-
килы. Однако перфторарилртутные соединения отличаются устойчивостью
к кислотам. Так, бис-пентафторфенилртуть может быть перекристаллизо-
вана из конц. серной кислоты [44]. Так же устойчивы к действию кислот
перфторалкильные соединения ртути. Среди жирных ртутных соединений
особой прочностью , отличаются меркарбиды. Этаноксигексамеркарбид при
действии холодной соляной кислоты переходит в хлорид, не отщепляя
ртути [45]:
HOHg HgOH
/Hg^C-C^Hg\0 НС1 (clHghC_c (HgCI)8> •
\Hg Hg/
(ClHg)2CH—CH(HgCl)2, получаемый при действии разбавленной НС1
на цианид, имеющий, по Гофману, состав (NCHg)HgC—CHg(HgCN), дей-
ствием кипящей концентрированной соляной кислоты превращается в
ClHgCH2—CH2HgCl, далее неизменяющийся [46].
Блешинский и Усубакунов [47] нашли, что конечным продуктом аци-
долиза меркарбида действием НС1 является не 1,2-дихлормеркурэтан, а
хлористая метилртуть, что является одним из приводимых ими доказательств
того, что меркар.бид Гофмана представляет собой Производное метана, а не
этана.
бис-Пер хлор винилртуть не реагирует с 10%-ной соляной кислотой
[47а], но с бромистоводородной кислотой и с раствором бромистого водоро-
да в хлороформе образует бромную ртуть и трихлорэтилен с выходом 83%
[476].
С трудом разлагаются кислотами некоторые Р- и даже а-меркурдикарбо-
новые кислоты; так, а-меркурди-Р-фенилангидрогидракриловая кис-
лота
СвН5СН-СН—СООН
О Hg
СбН5СН-СН-СООН
разлагается лишь при длительном кипячении с концентрированной соля-
ной, уксусной или азотной кислотой [47в]. Не реагирует с соляной кисло-
той даже при кипячении меркур-бис-л-дифенил (m - CsH6C8H4)2Hg.
Легкость распада Alk2Hg пОд действием соляной кислоты [43] падает
в ряду Aik: /-С4Н9 > i-C4H9 и-С4Н9 и j-C3H7 > п-С3Н7.
Исследована кинетика ацидолиза R2Hg под действием различных кис-
лот: муравьиной (R = СвН5 [10, 22]), уксусной (R =/г-С4Н9 [17],
s-C4H9 [17], СН2=СН-СНа [53], СвН5С(СН3)2СН2 [17], 4-камфил [21],
<%Н5 [10, 17, 22]), трихлоруксусиой (R = СвНв [23]), соляной (R = СН3
[24], СаН5 [23, 24, 25], п-С3Н7 [24, 25], z-C3H7 (24, 25], СвН5СН2
[18, 26], С6Н5СН3СН3 [18], С6Н5СН2СН2СН2 [18], СН2=СН [24,^7,
28], С6Й5СН=СН [28J-, цис- и транс-С1СН=СЙ [27, 28], цис- и mpakc-
СН3СН-СН [27, 28], цис- и трдас-СН3ОСОС(СН3)=С(СН;3) [27, 28],
цис- и mpuuc-C6H5CH = С(С6Н5) [28], ^цс-СН8ОСОС(СеН5) =С(С6На) [27],
ZC6H4C=C 123] (где Z=H, 3-СН3, 2,4-(СН3)а, 2,6-(СН3)а, 4/-C4H9, 4-F,
3-G1, 4-С1), с-С3Н5 [24], СеН5 [18, 24, 25, 27-29], о-СН3С6Н4 [18, 27, 281,
т-СН3С6Н4 [18, 27, 28], р-СН3СвН4 [18, 24, 27, 28], p-s-C4H9CeH4 [18],
о-СН3ОС8Н4 [27, 28], р - СН3ОС6Н4 [25, 27, 28], p-FC6H4 [27, 28], о-С1С9Н4
[27], т-С1С6Н4 [27], р-С1С6Н4 [25,271, а-С10Н, [27, 281, Р-С1ОН7[23],
Реакции ртутноорганических соединений
267
л-С4Й8С==С [231, m-NO2CsH4[25], р-СвН5С6Н4 [25], о-фенилен [23]), броми-
стоводородной (R = С6Н5 123, 29]), -хлорной (R =п-С4Н9 [17], s-C4H9
[17], СеН6С(СН3)2СН2 [17], С6Н5 [10, 17, 22, 23]), «-толилсульфоновой
(R — С6Н6 [23]). См. также ниже.
Реакция ацидолиза имеет второй порядок (5в2-механизм). В большом
избытке кислоты при ацидолизе действием уксусной, муравьиной кислот
реакция имеет кажущийся первый порядок.
Относительная легкость отщепления радикала кислотой определяется
его положением в ряду электроотрицательности, выведенном Харашем
[30—36] на основании реакции
RHgR' 4- НС1 RHgCl + R'H,
где радикал R' обозначается как более электроотрицательный; R' распо-
лагаются в ряд: CN 4>р-С6Н4ОСН34> о-С6Н4ОСН3> а-С10Н7^> о-С6Н4СН3^>
J> p-CeH4CH3 > /п-С6Н4СН3 >С6Н6>- p-CsH4Cl > О’СвН4С1 >пг-С6Н4СГ>
СН3^> С2Н6> С3НС> С4Н9^> СвН5СН2> С(СН3)3; а-тиенил и а-фурил,
д также «-оксифенил, «-аминофенил расположены в самом начале ряда.
Ряд Хараша подвергнут обсуждению 137—39].
Уинстейн и Трэйлор, Десен, Несмеянов и Борисов и др. дополнили ряд
Хараша другими радикалами на основании изучения кинетики реакции
соляной кислоты с Alk2Hg[ 17, 23,24,286], с Ar2Hg[18, 23—25,27,28], с сим-
метричными квазикомплексными и другими алкенильными соединениями
ртути [27, 28], с соединениями типа (XC6H4C~C)2Hg [23]. Вот некоторые
из этих дополнений: р-СН3ОС6Н4> р-С6Н5С6Н4 > С6Н5 > p-FC6H4 >
р-С1С6Н4> m-NO2C6H4 [25], р-СН3С6Н4>о-СН3С6Н4> m-CH3C6H4 [184,
Т-С6Н5С3Н6 > С6Н5СН2СН2 > С6Н5СН2 [18].
Положение разветвленных алифатических групп в ряду электроотрица-
тельности на основании разложения RHgR' спиртовым’ раствором НС1
видно из следующего ряда [28а]:
СвН5 > СНз > С2Н5 > я-СзН, >
я-С4Нэ; (СН3)зССН2СН2 А „ ,
n-C6Hls; (СН8)3ССН2СН2СН21>СНзСНа^Н > (СН’)з ССН (CHs) ’
СНз
. СвН6СН2> [(СНз)з С; (СН3)зССН2].
Группы, расположенные в скобках, не различаются по этому методу,
т. е. дают смеси R'HgCl и RHgCl.
Несмеяновым, Борисовым и Савельевой [27, 28, 286] на основании изу-
чения скорости ацидолиза R2Hg действием соляной кислоты в 90 %-ном
водном диоксане получен следующий ряд для R:
1,3,5-(СН3)3С6Н2 > тра«с-СН3СН=СН > р-СаНвОСвН4 > ^мс-СН8СН = СН
> p-CH3OCeH4 > CeH5CH = CH > тра«с-СН3ОСОС(СН3) = С(СН3) >
> 1Щс-СН3ОСОС(СН3)=С(СН3) "> а-тиенил ^>о-СН ^>С6Н4 4> р-С2Н5С6Н4 >
>р-СН3С6Н4>ц«с-СвН5СН=С(С6Нв)>о-СН3ОС6Н4>а-С10Н7>пг-СН3СвН4>
>СН2=СН > C6HB>p-FCeH4>«t-CH3OCeH4 > /пра«с-С6Н5СН =С(СвН5)>
>р-С1С6Н4> p-BrCeH4> m-FC6H4> т-С1СвН4>СН3ОСОС(С6Н5)С(С6Н5)> .
> о-СН3ОСОС6Н4>тра«с-С1СН=СН>С2Н5> р-СН3ООССвН4>ццс-С1СН=
=СН > о-СН3ООСС6Н4> «-С4Н9 > о-С1С6Н4 > С6Н5СН2.
Как видно из этих данных, в зависимости от природы и положения
заместителей в бензольном ядре скорость ацидолиза Ar2Hg падает в рядах:
р-СН3ОС6Н4 > о-СН3ОС6Н4 > С6Н5 > /п-СН3ОСвН4; о-СН3С6Н4>
> р-СН3С6Н4 > /п-СН3СеН4 [28]; р-С2Н5С6Н4 > р-СН3С6Н4 [281.
268
Ртутноорганические соединение. Лит. стр. 336—352
Накопление метильных групп в орто- и пар а* положениях вызывает
значительное ускорение реакции ацидолиза под действием соляной кисло-
ты, и димезитилртуть имеет наибольшую константу скорости этой реакции.
Пассивируют бензольное ядро к электрофильной атаке, как всегда, галои-
ды в орто-, пара- и мета-положениях, ацетокси- и карбометоксигруппы в
орто- и пара-положениях. p-Хлорвинильные соединения также реагируют
медленнее, чем дивинилртуть.
Для галоидозамещенных бензолов скорость реакции падает в ряду:
р-С1С6Н4 > /п-С1СвН4 > о-С1СвН4 и p-FC6H4 > р-С1С6Н4 > p-BrCeHt
(то же в ряду мета-производных).
На основании скорости образования RH в реакции RHgCHCl2 со спир-
товым раствором НС1 радикалы R располагаются в ряд СС13^> С6Н5
>СНС12 [39а].
Предельные алифатические соединения реагируют значительно медлен-
нее большинства ароматических и непредельных алифатических соедине-
ний. Транс-изомеры непредельных ртутноорганических соединений, кроме
co-меркур-бнс-стильбена, имеют большую константу скорости, чем цис-
изомеры. Скорость реакции диаллилртути с соляной кислотой наибольшая
из всех изученных R2Hg [286].
Скорость разложения соединений RHgR соляной кислотой в смеси ди-
метилсульфоксида и диоксана зависит от гибридизации атакуемого атома
углерода. Эти скорости возрастают в ряду sp3, sp2 и sp при приближенных
факторах 1, 100 и 1000 [23].
По убывающему значению констант скорости (уравнение псевдопервого-
порядка) реакции R2Hg с хлористым водородом в водном тетрагидрофуране
(15% Н2О) R могут быть также расположены в ряд: СН2=СН > CF2=CF>
>С2Н5 [40]. При аналогичном разложении перфторвинилвинил-, перфторви-
нилэтил-, перфторвинилфенилртути образуется трифторэтилен [40].
Ряд, полученный Десен и др. [24] (действием на R2Hg соляной кислоты
в смеси диметилсульфоксид — диоксан, 10:1): с-С3Н5> Н2С=СН >
>С6Н5 C2HS > z-C3H7^> zz-C3H7>CH3, несколько не совпадает с рядом
Хараша и др. [28].
Десси с сотр. [24, 24а] предложен четырехцентровЫй механизм:
атаки ртутноорганического срединения кислотой:
R — Hg—R R—Hg—R RH
t — X
H —Cl H—Cl RHgCl
в котором предполагается электрофильная, молекулярная атака или атака
ионной парой НС1. Это подтверждается тем, чт* при разложении замещен-
ных ароматических соединений ртути хлористым водородом заместители,
притягивающие электроны, понижают скорость реакции, а группы, являю-
щиеся донорами электронов, увеличивают скорость реакции.
На основании сходства значений энергии активации й энтропии актива-
ции реакции разрыва С6Н5—Hg-связи под действием соляной кислоты на
CeHsHgC2Hi и на (CeHs)2Hg Десси [23] уточняет, что разрыв RHgR' дей-
ствием соляной кислоты является мерой доступности электронов у атакуе-
мого углерода, а не электроотрицательности отщепляющейся группы. Одна-
ко относительно возможности передачи электрических влияний через атом
ртути данные противоречивы [23, 24]. Роль аниона кислоты HHal в ста-
дии, определяющей скорость реакции ацидолиза,, проявляется в значи-
тельном изменении значений £* и AS* при переходе от НС1 к НВг [23].
Линейная зависимость 1g К от функции кислотности Н различных си-
Реакции ртутноорганических соединений 269
стем растворителей указывает на атаку ионными парами; отклонения,
проявляемые системой НС1—диоксан, являются результатом атаки молеку-
лярной НС1. Между диэлектрической постоянной растворителя и скоро-
стью реакции ацидолиза достаточно ярко выраженной зависимости не на-
блюдается.
Описано применение уравнения Гаммета к реакции ацидолиза Ar2Hg
действием соляной кислоты [23, 25, 28], в частности для (ZCeH4C=C)aHg[23L
Для ацидолиза мета- и пара-замещенных ароматических ртутнооргани-
ческих соединений наблюдалась [28] линейная зависимость 1g К от о Гам-
мета, <т+ и ° + —. Наилучшая корреляция наблюдается при применении
СЛ
[23, 25, 28]. Таким образом, атака НС1 на Ar2Hg протекает хотя в
общем и подобно электрофильным замещениям в ароматическом ядре (кор-
реляция с сг+), но все же несколько отклоняется (в сторону корреляции со).
Для непредельных ртутноорганических соединений применимо[28а] урав-
нение Тафта с полярными константами заместителей о* (для всего радика-
ла, связанного со ртутью). Наблюдающаяся [286] корреляция 1g К7Ко от
Ор Гаммета, сг£ и °. + для заместителей R непредельных ртутноорганиче-
ских соединений свидетельствует о большем сопряжении ^-заместителей
непредельной системы с реакционным центром по сравнению с ароматиче-
ской системой. Значение р Гаммета для арилацетиленовых соединений
(ZC6H4C=C)2Hg показывает, что реакция их с соляной кислотой менее
чувствительна к 'заместителям в кольце по сравнению с ароматическими
соединениями [231-
Ацидолиз AlkHgJ действием хлорной и серной кислот идет как реак-
ция псевдопервого порядка из-за избытка кислоты [41, 42]. По относитель-
ной реакционной способности в этой реакции радикалы располагаются в
ряд: с-С3Н4 >» СН3 >• C2HS)> а-С3Н7)> z-C3H7> n-C6Hu> /-С4Н9 [42].
Кинетика этой реакции (для Aik = i-C3H7 и ЛС4Н9) зависит от при-
сутствия кислорода [3].
Действие DC1 на тетрахлормеркуртиофен приводит к дейтеротио-
фену [48].
Действием ТС1 на тетрахлормеркуртиофен (также содержащий меченую
серу) получен тетратритилировангдяй тиофен [49].
При ацидолизе ди-А-(—)вторйчнобутйлртути и ди-г{ис- и ди-транс-
4-метилциклогексилртути действием дейтерокислот по схеме
RaHg + DX---> RD + RHgX
имеет место частичное сохранение конфигурации R в первом случае (дей-
ствие CH3COOD и DC1 в диоксане) и высокая степень сохранения конфигу-
рации (расщерлСние действием DC1 в диоксане) в двух последних случаях
[50]. В этих случаях другие кислоты (CH3COOD, D2SO4) в диоксане дают
RD преимущественно транс-конфигурации [50].
Описано действие серной кислоты и других сульфирующих агентов
на ртутные производные антрахинона [59—62], а также действие на ртут-
ноорганические соединения азотной кислоты, содержащей окислы азота
(стр. 317).
Тиофенолы реагируют'с дифенилртутью и RHgX (R=ArnAlk) [63, 64]
при 130° С как кислоты с образованием 'тиофенолята ртути или метал-
лической ртути. При 6-часовом кипячении в бензоле Ar2Hg с тиофенолами
образуются тиофеноляты, ArHgAr' отщепляют радикалы в соответствии
с рядом электроотрицательности Хараша [64].
270
Ртутноорганические соединениям Лит. стр. 336—352
. Действие фосфорной кислоты на бис-этилмеркурацетилен приводит к
кислым моно- и диэтилмеркурфосфатам [651.
Сложные эфиры органических кислот при высокой температуре (150° С,
6—9 час.), согласно Котону [161, реагируют с дифенилртутью по схеме
(CeH^Hg + RCOOR' -» RCOgHgCeHs + CeH6R'.
B(OC4H9)S не реагирует с (C6H5)2Hg при 140°С [661 и с ArHgOH и
ArHgOCOCH3 при 150°С [67].
Хлоргидрат диМетиланилина реагирует с Ar2Hg при 130—150° С, по-
добно свободной соляной кислоте, образуя АгН и HgCl2 [68]. Хлоргидрат
триметиламина с Ar2Hg в спирте и бензоле при 100° С образует ArHgCl,
при 130° С в воде и без растворителя (запаянная трубка) вызывает распад
Ar2Hg и ArHgCl до Hg и HgCl2 [69].
Подобно кислотам, реагируют с полнозамещенными ароматическими
ртутноорганическими соединениями амиды [70] и имиды [70—72] кислот:
протон амидов и имидов соединяется с радикалом, RHg-остаток образует
RHgN-производное.
Фенилтригалоидметилртуть реагирует с хлористым водородом (в толуо-
ле при 80° С) по двум -направлениям:
I—> СвН9 CClaBrHgCl
C6HsHgCCl2Br + НС1 —
U CeHsHgBr + CHCls
C6H5HgCBr3 с HC1 не образует бромоформа, главным продуктом реакции
является бензол [50al. C6H5HgCCls с метанольным раствором НС1 дает
хлороформ и C6H5HgCl [538].
Аналогично с карбоновыми кислотами (в бензоле при 60—80° С) и спир-
тами (в этилбензоле при 80—85° С) образуются соответственно сложные
RCOOCHC12 и смешанные простые дихлорметиловые эфиры ROCHC12 [50а].
Также основным, продуктом реакции перекиси бензоила с (C2H5)2Hg
или с (CeH5)2Hg в бензольном растворе является RHg-бензоат [72а].
Реакции ртутноорганических соединений с кислотами,
приводящие к выделению металлической ртути
(реакция демеркурирования)
Монозамещенные ртутноорганические соединения R2Hg и RHgX при
нагревании их с уксусной кислотой до высокой температуры в запаян-
ной трубке полностью разлагаются с выделением металлической ртути
[51]. Диметйлртуть дает при этом (250° С) металлическую ртуть,
этилен, метан и водород [51]; диэтилртуть (160—190° С, несколько часов)—
ртуть, этилацетат, этилен, этан и водород [51]; диизоамилртуть (190—
200° С, 16 час.) — ртуть, амилацетат, изопентан [51]; уксуснокислая бен-
зилртуть (170° С, 7 час.)—ртуть и бензилацетат; дибензилртуть (170° С,
2 часа [51,52])—кроме двух последних продуктов, также толуол и дибен-
зил; уксуснокислая фенилртуть и дифенилртуть (220—230° С) — бензол,
дифенил и смолу -[511 но ср. и ([53].)
Ацетаты алкенилртути при нагревании их выше 100° С в уксусной кис-
лоте разлагаются с образоваием алкенилацетатов и выделением металли-
ческой ртути [53]. Ацетаты метил-, фенил- и циклопропил ртути в этих
условиях не разлагаются [53].
И при умеренных температурах при разложении кислотами достаточно
широкого круга ртутноорганических солей может происходить более да-
леко идущий сольволиз, при котором ртутноорганическая соль разлдга-
Реакции ртутноорганических соединений 271 ’
етсЯ с выделением металлической ртути, с образованием продуктов взаимо-
действия органического остатка с кислотным остатком (например, сложного
Эфира взятой в реакцию кислоты) и в ряде случаев с образованием олефина
по схеме:
RHgZ 4-НХHZ Н-HgRXолефин.
Реакция наблюдалась в качестве побочной при расщеплении R2Hg хлор-
ной кислотой в уксусной кислоте (для. R — неофил [17]), .дейтероуксусной
и другими дейтерокислотами (для R—L-{—)вторичнобутил и 4-метил-
циклогексил [50]).
Дивинилртуть с карбоновыми кислотами (при умеренном нагревании
без растворителя или в спирте, бензоле, толуоле, гептане) образует вини-
ловые эфиры их с выделением металлической ртути через промежуточное
образование RCOOHgCH=CH2 [&4].
Дивинилртуть реагирует с фенолами (нагревание на водяной бане в
течение 1—3 час.), тиофенолами (мгновенная экзотермическая реакция при
комнатной температуре), алкилтиолами (115° С, 3 часа) подобно тому, как
она реагирует с карбоновыми кислотами: с выделением металлической
ртути и образованием винилариловых эфиров, виниларил- и винилалкил-
сульфидов [54]. Так же рёагируют с тиофенолами другие R2Hg (R—
алкенил) [53].
Взаимодействие /г-толуолсульфиновой кислоты с дивинилртутью [54]
(в этаноле, водяная баня) и дициклопропилртутью [64а] приводит к обра-
зованию одних и те?, же продуктов — этилена, винил-/г-толилсульфона, и
выделению металлической ртути, тогда как с органическими карбоновыми
кислотами дивинилртуть образует их виниловые эфиры [54]. (См. выше.)
Кинетическое изучение [19] расщепления уксуснокислой циклогексил-
ртути уксусной кислотой как в отсутствие, так и в присутствий хлорной
кислоты при 25° С показало, что эта реакция имеет первый порядок. Такое
же изучение сольволиза перхлоратов алкилртути (алкил—СН3, С2Н8,
z-C3H7, n-C4H9, s-C4H9, АС4Н9[20]), перхлоратов и других солей циклогек-
силртути [19] в воде и уксусной кислоте позволяет предположить, что- в
переходном состоянии в реакции участвует ион карбония [19, 20]
RHgX RHg+ + Х-,
RHg+^R++Hg,
R+ -> продукты реакции.
Скорость формолиза и ацетолиза C2H5HgC104 сильно возрастает при
введении арила (арил—фенил, м- и zz-толил) и особенно фенила и одновре-
менно двух метильных групп в положение 2|Д54а]. К этой реакции
применено уравнение Гаммета[54а].
Соли фенилртути, в особенности перхлорат и тозилат в уксусной кисло-
те и даже в трлуоле, в присутствии хлорной кислоты тоже претерпевают
сольволиз такого типа (до фенилмеркуркатиона), на что указывает образо-
вание феррицинийкатиона при проведении реакции в присутствии фер-
роцена [546]. Сольволиз солей кротилртути действием кислот происходит
с гораздо большей'скоростью, чем сольволиз предельных ртутноорганиче-
ских соединений и сопровождается аллильной перегруппировкой радикала
[54в]. Так, при действии соляной кислоты в этилацетате или хлорной кис-
лоты в уксусной кислоте кротилмеркургалогенид, а также кротилмеркур-
ацетат (только при действии соляной кислоты) образуют олефин — бутен-1.
Аналогично циннамилацетат при действии соляной кислоты образует
аллилбензол [54в1. Сольволиз кротилмеркурацетата действием ук-
сусной кислоты при 50° С дает почти исключительно вторичнобутенилацетат,
•272
Ртутноорганические соединения. Лит. стр. 336—352
в присутствии хлорной кислоты — 71% этого же продукта и 29%' смеси
транс- и цис-кротилацетатов:
н
АсОН
СН3СНСН=СН2
СНз
сн2
ОАс
99,5 %
Ас°-_ 25°с .
50°С
HgOAc нею,
СН3СН=СНСН2ОАс
транс цис
0,5 %
28,5% 0,5%
С
н
Первый порядок имеет реакция демер курирования уксуснокислых бензил-
ртути, (С(л-метоксифенил)Этилртути и нортрициклилртути [55а] в водной
уксусной кислоте (последняя также в уксусном ангидриде) в присутствии
хлорной кислоты, идущая с образованием соответствующего алкилацетата
и ртути. В этих же условиях реакция уксуснокислой бензилртути с ацета-
том ртути, проходящая по схеме [55]
ArCH2HgAc + HgAca АгСН2Ас + Hg2Ac2’
имеет второй порядок. См. также стр. 275.
Константы скорости реакций первого порядка демеркурирования дей-
ствием хлорной кислоты в уксусной кислоте уксуснокислых экзонорбор-
нилртути, циклогексилртути и эндонорборнилртути относятся, как 5-Ю3:
: 43 : 1 [55а, б].
При взаимодёйствии (в бензоле, при кипячении) дифенилртути и трибу-
тилфосфина в присутствии кислот (карбоновых, моноэфира фосфорной кис-
лоты, /г-толуолсульфокислоты) образуется ангидрид кислоты, трибутил-
фосфиноксид и выделяется металлическая ртуть, например [55в]:
(C6HS)2 Hg + 2(п-С*Н9)3 Р + 2RCOOH Hg + (RCO)2 О + (и-С4Н9)3 РО + СвНв.
Аналогично через промежуточное образование дифенилртути, идет реак-
ция фенилмеркурацетата с трибутилфосфином в присутствии кислоты [55в]
2C6H5HgOC0CH3 + (я-С4Нв)3 Р + 2RCOOH 2Hgj+ (СН3СО)2 О + (RCO)2 О +
+ 2 (п-С4Н,)3 РО + 2C6H6.‘f
Из эквимолекулярных количеств дифенилртути, трибутилфосфина^
бензойной кислоты и этанола (кипячение в течение 8 час.) получается с
76%-ным выходом этилбензоат, металлическая ртуть (85%) и трибутил-
фосфиноксид (85%). Если вместо спирта применен анилин, то получается
с хорошим выходом анилид взятой кислоты [55в].
Действие кислот на продукты присоединения солей ртути
к олефинам и их производным
Иным типом реакции является действие кислот на продукты присоеди-
нения олефинов и их производных к основной соли ртути, протекающее с
регенерацией олефина (реакция деоксимеркурирования), например:
HOCH2CH2HgX + НХ|-—> С2Н4'+ Н2О + HgX2.
Эта реакция элиминации олефина идет легко на холоду при действии
минеральных кислот и характерна для этого типа веществ [76] (см. также
гл. VI). Согласно Занду [77], бензоилирование гидроксила в таких соеди-
нениях в ряде случаев несколько повышает их устойчивость к действию
Реакции ртутноорганических соединений
273
кислот, однако полученные соединения претерпевают иного типа сольволиз
по схеме:
2RCOOCH2CH2HgJ RCOO" + СН2=СН2 + HgJa + COOCH2CHaHg+.
Такой сольволиз легко происходит и под действием уксусной кислоты, а
также в нейтральной среде в смеси воды со спиртом или диоксаном [78].
Изучена кинетика разложения соляной кислотой продуктов присоеди-
нения солей ртути к этилену [79, 80], цис- и транс-стильбену [81]; соля-
ной, трифторуксусной [81, 82], хлорной [83, 86] кислотами — продукта
присоединения к циклогексену; хлорной кислотой — продуктов присоеди-
нения солей ртути к этилену [84—86] и его тетрадейтероаналогу [84], к
пропилену [87], к циклопентену [85, 86], бицикло-(2,2,1)-гептену [85, 86],
акрилонитрилу [87а], стиролу [876], также дейтерированному в положе-
ния 1 и 2 [88J; соляной, трифторуксусной и уксусной кислотами — про-
дукта присоединения к окиси углерода [89а].
При регенерации алкенов действием кислот на продукты присоедине-
ния к алкенам солей ртути геометрическая конфигурация алкенов сохраня-
ется (например цис- и транс-стильбен [89]).
Под действием кислот элементы XHg и OR легче отщепляются от транс-
изомеров — продуктов присоединения к циклоалкенам, чем от их цис-
изомеров, легче от продуктов присоединения соли ртути к олефинам в воде
(соли (З-оксиалкилртути), чем от продуктов присоединения соли ртути в
спирте (соли Р-алкоксиалкилртути) [85а].
Карбоновые кислоты при умеренной температуре, за'редкими исключе-
ниями, на рассматриваемые в этом разделе вещества не действуют, но раз-
ложение продукта присоединения соли ртути к окиси углерода действием
трифторуксусной и уксусной кислот в диметилсульфоксиде ускоряется в
присутствии целого ряда веществ—ускорителей реакций, связывающих
соль ртути [89а]. Эти вещества по их ускоряющему действию могут быть
расположены в ряд [89а]:
C6H5SH > J- > Br- > Cl" > (СвНб)3 РО > (С6Н5)3 Р, СвН5ОН.
О механизме деоксимеркурирования под действием кислот продуктов
присоединения солей ртути по двойной связи см. гл. VI, стр. 119, а также,
например, [90, 91].
При разложении при температуре 40° С в присутствии сильных кислот
(H2SO4, Н3РО4, НС1О4, ArSO3H) ртутноорганических солей (без выделения
их), образующихся при реакции ароматических соединений с продуктами
присоединения ацетата ртути к олефинам (этилену [56], пропилену, 2-бу-
тену, стиролу, циклогексену [57]), происходит их демеркурирование с вы-
делением металлической ртути (иногда сопровождающимся образованием
закисной ртути) и образованием р-арилалкилацетатов и р-диарилалканов.
Реакции эти имеют второй порядок [57а].
Ртутноорганические соединения, образующиеся при реакции ацетоук-
сусного эфира с продуктами присоединения солей ртути к олефинам (эти-
лену, пропилену, стиролу), демеркурируются при умеренном нагревании в
уксусной кислоте в присутствии эфирата фтористого бора с образованием
металлической ртути иа-(2-ацетоалкил)ацетоуксусногоэфира [58], например:
BF, О
снзсосн (CHaCH2HgococH3) coocsHs сн>сооц>
Hg + СНэСОСН (СНаСНаС0СН3) СООСаНе.
В случае стирола выделен продукт циклизации — производное фурана.
274 Ртутноорганические соединения. Лит. стр. 336—352
ДЕЙСТВИЕ ЩЕЛОЧЕЙ
Щелочи, как правило, не разрывают С—Hg-связь. Исключением явля-
ется образование NaOCeH4HgOH из ди-о-оксифенилртути при действии
щелочи [92], а также полный распад с выделением металлической ртути
меркур-бис-хлормеркуруксусноэтилового эфира [93], бромметилмеркурбро-
мида [94] при нагревании со слабыми растворами щелочи.
Продукт реакции монохлоруксусной кислоты и окиси ртути, которому
Гофман приписал структуру комплекса хлористого калия с солью хлормер-
курхлоруксусной кислоты, при действии щелочи выделяет окись ртути с
одновременным образованием соли гликолевой кислоты [95].
Продукт присоединения ацетата ртути к ацеталю хлоркетена — этило-
вый эфир ди-(ацетоксимеркур)хлоруксусной кислоты—разрушается дейст-
вием 20%-ного водного раствора едкого кали [96].
При обработке щелочью происходит отщепление соли ртути, присоединен-
ной по двойной связи производных кумарина [97,98] (см. гл. VI, стр. 159).
Однако обычные продукты присоединения солей, ртути к непредельным
соединениям, в том числе и к кумарину [62, 99], устойчивы к щелочам.
В гл. VI описано действие оснований, в том числе щелочей,на р-хлорвиниль-
ные соединения ртути.,
ДЕЙСТВИЕ СЕРОВОДОРОДА
И СУЛЬФИДОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ
За редкими исключениями полные ртутноорганические соединения не
реагируют с сероводородом или сернистыми щелочами. Ртутноорганические
соли обычно образуют бесцветные сульфиды (RHg)3S, часто имеющие тен-
денцию при нагревании или хранении отщеплять (особенно в ароматиче-
ском ряду и в случае наиболее электроотрицательных радикалов) сернистую
ртуть по схеме
(RHg)2S^R2Hg + HgS,
чем пользуются иногда для симметризации ртутноорганических солей
(гл. XIII).
Сравнительно легко заменяется ртуть на водород действием сероводорода
на пентафторфенилртутные соединения [99а].
Продукты присоединения солей ртути к олефинам обычно также дают
бесцветные сульфиды, довольно быстро разлагающиеся. В отличие от реак-
ции их с кислотами продукты присоединения солей ртути к а,р-непредель-
ным кис-лотам при действии сернистой щелочи не регенерируют непре-
дельное соединение, а, замещая атом ртути на водород, с хорошим выходом
образуют [1-оксикислоты [100], например:
СвНвСН—СН—СООН 4- Na2S—> СвН5СН—СН2—СООН.
I I I
ОН HgCI ОН
Сульфиды щелочных металлов, в том числе сернистый аммоний, в неко-
торых случаях отщепляют атом ртути от ртутноорганических соединений.
Так, атом ртути легко отщепляется от солей меркурированных уксусной и
пропионовой к ислот при действии сернистого аммония [101, 102].
ДЕЙСТВИЕ НЕКОТОРЫХ ОКИСЛИТЕЛЕЙ,
В ЧАСТНОСТИ СОЛЕЙ РТУТИ
Некоторые ртутноорганические соли, особенно со ртутью, соединенной
с вторичным углеродом, при умеренной температуре окисляются солями
окисной ртути, восстанавливая их до одновалентной ртути или до металли-
Реакции ртутноорганических соединений
275
ческой ртути и образуя (в зависимости от среды реакции) в воде — спирты
и продукты их окисления, в спиртах — простые эфиры. При окислении
RHgNO3 действием азотнокислой окиси ртути образуются также сложные
эфиры азотной кислоты. В настоящем разделе не рассматриваются те слу-
. чаи окисления органических соединений действием солей ртути, где можно
предположить лишь промежуточное образование ртутноорганических со-
единений.
Азотнокислой ртутью произведено окисление (комнатная темпе-
ратура) в метаноле азотнокислых бензилртути [103], циклогексилртути
[104], ее же в воде [1041; окисление циклогексилртути в смеси воды и мети-
лового спирта перазотной кислотой дает те же продукты, что окисление
.азотнокислой ртутью в метиловом спирте [104]. v
Окисление ртутноорганических ацетатов солями ртути протекает мед-
леннее, чем нитратов, но дает продукты того же типа [104]. При окислении
уксуснокислой циклогексилртути надуксусной кислотой в метиловом спир-
те (7-часовое кипячение в смеси спирта и хлороформа) выделен циклогекса-
нон (за счет окисления первично образующегося циклогексанола надуксус-
ной кислотой и Hg++) и Hg+.
Окисление ртутноорганических нитратов и ацетатов является реакцией
второго порядка. Авторы [104] считают, что реакция имеет гомолитиче-
ский характер.
Hg+ и соответствующий простой эфир получены при окислении уксусно-
кислой 1-фенил-2-метокси-2-метилпропилртути уксуснокислой ртутью в при-
сутствии эфирата фтористого бора (в СН3ОН, 50° С, 24 часа). При окис-
лении уксуснокислой 4-оксициклогексилртути реактивом Фентона в воде
получен формилциклопентан и 2-хлормеркурциклогексанон (после добав-
ления хлористого натрия [104]).
Циклогексилртутные соли (трифторметилацетат и нитрат без катали-
затора, ацетат— лишь в присутствии эфирата фтористого бора), 2-пропил-
меркурацетат, 3-амилмеркурацетат претерпевают в растворе метилового
спирта при умеренной температуре (50° С) гетерополяркое мономолекуляр-
ное аутоокисление с образованием ртути и олефина [105]. Образование соот-
ветствующего олефина имеет место и в случае 1-метил-1-циклогексилмеркур-
ацетата [105].
Действие озона на алкенильные ртутноорганические соединения, про-
водимое в обычных условиях действия его на органические соединения,
помимо озонирования органической части молекулы, приводит к разрыву
связи С—Hg [106] (см. также гл. XV).
ДЕЙСТВИЕ КАРБОНИЛОВ МЕТАЛЛОВ
При действии на RHgX и R2Hg пентакарбонила железа при нагревании
образуется HgFe(CO)4 [107]. Mn(CO)6H с RHgOH образует [Mn(CO)s]2Hg
[108].
ДЕЙСТВИЕ ГАЛОИДОВ
Свободные галоиды разлагают ртутноорганические соединения в две
стадии:
R2Hg + X2^RHgX + RX, (1)
RHgX 4- Х2^ RX + HgX2. (2)
. Полнозамещенные соединения реагируют энергичнее ртутноорганиче-
ских солей.
276 Ртутноорганические соединения. Лит. стр. 336—352
Диметил- и диэтилртуть самовоспламеняются в сухом хлоре, будучи
внесены в него по каплям. С иодом и бромом они реагируют без воспламе-
нения по уравнению (1). Реакция проходит более спокойно в растворителе
и в этих условиях широко применяется для определения положения ртути
в молекуле (обычно употребляют бром или иод в растворителях — водных
KBr, KJ, в спирте, сероуглероде, хлороформе, четыреххлористом углеро-
де, уксусной кислоте), а также для получения трудно доступных иными
путями галоидопроизводных.
В качестве мягко действующего средства может быть применен диоксан -
дибромид [114].
Изучена кинетика и стереохимия реакции s-C4H9HgBr с Вг2вСС14 [109а],
а также кинетика реакции
RaHg -|- Ja ~* RHgJ -|- RJ
в четыреххлористом углероде [1091. По возрастающему значению величин
энергии активации этой реакции радикалы могут быть расположены в ряд:
р-СН3ОС6Н4 < о-СН3ОС6Н4 < ₽-С10Н7 <а-С10Н7 < р-СН3СвН4<
о-СН3С6Н6 <С6Н5 < p-C2H5O2CCeH4< p-FCeH4 <a-C4H3S <р-С1С6Н4 <
< СН3< а-С2Н5О2СС2Н4<^ С2Н6<^ ц-С4Н9< rt-C6Hu < r-C5Hu< /-С3Н7 <
<С6Н3СН2, в основном совпадающий с рядом Хараша.
CeH6HgSCeH6 с бромом дает CeH6HgBr и (C6H5)2S [291].
Реакции эфира a-броммеркурфенилуксусной кислоты и бромистой бензил-
ртути с иодом в присутствии CdJ2(e водном диоксане)—это реакции второго по-
рядка, вотсутствиеС(Н2—это реакции первого порядка, идущие по радикаль-
ному механизму [110]. Второй порядок, имеет также электрофильная реак-
ция хлористой транс- и tfuc-^-хлорвинилртути с J2 в присутствии CdJ2
в водном диоксане [111а], в метаноле [1116], диметилформамиде [111в], про-
текающая с сохранением конфигурации радикала[111в]; в отсутствие CdJ2
в бензоле, четыреххлористом углероде, диметилсульфоксиде реакция имеет
суммарный первый порядок [111г].
Реакция хлористой бензилртути с иодом в присутствии избытка йоди-
стого кадмия имеет суммарный второй порядок (растворители — диметил-
формамид, метиловый и этиловый спирты [112а], 70%-ный водный диоксан
[112а, 1126]). Реакция хлористой бензилртути с иодом в четыреххлористом
углероде при освещении лампой накаливания 150 вт имеет первый порядок по
отношению к иоду и нулевой по отношению к ртутному соединению [112в,
1116]. В диметилформамиде кинетика реакции более сложная: в этом рас-
творителе в темноте и без нагревания реакция не идет [112в]. Реакция бен-
зилмеркурхлорида с бромом (и NH4Br) имеет второй порядок в диметил-
формамиде, метиловом спирте и 70%-ном водном диоксане [113а]. Кинетика
реакции бензилмеркурхлорида с бромом (в СС14 первый порядок по отно-
шению к брому и нулевой порядок по отношению к бромистой бензилрту-
ти) показывает, что реакция идет через образование свободных радикалов
[1136]. Добавление кислородсодержащих веществ изменяет порядок на вто-
рой. Скорость реакции уменьшается при проведении ее в растворителях:
СН3ОН > С2Н5ОН > (C2HS)2O > i-C3H7OH > У-С4Н9ОН [1136]. Предпо-
ложено, что бром поляризуется образованием комплексов с этими соеди-
нениями и реакция с хлористой бензил ртутью проходит, как бимолекуляр-
ное электрофильное замещение [1136].
Действие брома в бензольном растворе [114] на тиранс-а-меркур-бис-
стильбен приводит к замене ртути на бром, при действии же диоксандибро-
мида (в диоксане) можно" изолировать промежуточную ступень—транс-а-
•броммеркурстильбен. tfuc-Изомер R2Hg, как и RHgBr, и при действии диок-
сандибромида при комнатной температуре прямо дает RBr [114]. Реакции
эти проходят стереохимически чисто, с образованием только одного геомет-
Реакции ртутноорганических соединений
277
рического изомера:;
н свн5
Вг2 с,н.
с6н5 С6Н6
>С=с/
BrHg н
С4Н«О2- Вгг
CjHfcOj1 Вг8
В гл. VI описано действие галоидов на продукты присоединения солей
ртути к непредельным соединениям.
Бромистые цис- и mpawc-4-метилциклогексилртуть [115] и оптически
активный 2-броммеркурбутан [116а] отщепляют HgBr-группу действием
брома в условиях, благоприятствующих радикальному механизму (в СС14,
сероуглероде; для 2-броммеркурбутана — в сероуглероде) с полной ра-
цемизацией образующихся RBr. В неполярных растворителях,-, содержа-
щих. небольшие количества спирта, имеет место частичное, сохранение
конфигурации. В полярных средах (пиридин, 0° С) отщепление HgBr от пер-
вых двух веществ происходит с сохранением конфигурации, а для 2-бром-
меркурбутана, в исправление прежних данных [1166], конфигурация со-
храняется лишь частично. Стереоспецифическому течению реакции для
последнего вещества благоприятствует низкая температура (—65° С), а для
первых двух веществ — проведение реакции не в азоте, а в присутствии
кислорода воздуха.
Действием иода в водном хлороформе на 2,6-бис-(ацетоксимеркурметил)-
n-оксатиан получены цис- и транс-изомеры дииодида [117]:
S
. н>ан
CH3CO2HgCH2 О CH2HgO2CCH3
S S
Н /X Н Н /X CH2J
i >U< + >О<
JCH2 О CH2J JCH3 о н
V цис-, т. пл. 73° С ’ транс-, т. пл. 64—68° С
Диметил-2,6-бис-(ацетоксимеркурметил)-п-оксатиан при действии иода в
этих же условиях также дает оба изомера RJ [117]. Соответствующие суль-
фоцы дают только один продукт, более высокоплавкий (цис-) [117].
Действие брома [118, 119] на чистые диастереомеры 2-броммеркур-З-
фенилпропионовых кислот дает смесь двух диастереомерных 2-бром-З-фе-
нилпропионовых кислот [118]. Относительные количества образующихся
диастереомеров зависят от температуры, растворителя и других эксперимен-
тальных условий [118]. Хлористая неопентилртуть превращается галоида-
ми в соответствующие бромид или иодид без изменения конфигурации
[120]. На основании кинетического изучения реакции иодистой 4-камфил-
278
Ртутноорганические соединения. Лит. стр. 336—352
ртути с иодом показано, что в зависимости от экспериментальных условий
реакция имеет гомолитический или гетеролитический механизм [21].
Разложение иодидов 4-камфил- или «-бутилртути действием трииодидиона
происходит с одинаковой скоростью [21].
Протекание электрофильной реакции замены HgBr-группы в циклоал-
килмеркурбромидах на бром действием брома в пиридине с полным сохра-
нением конфигурации позволяет установить конфигурацию RHgBr на
основании известной конфигурации ДВг.Так, установлена конфигурация цис-
и /пранс-4-метилциклогексилмеркурбромидов [121], продукта оксимерку-
рирования диметилового эфира экзо-г{ис-3,6-эндоксо-А4-тетрагидрофталевой
и некоторых других кислот [122а].
Замена ртути на бром в диметиловых эфирах 4-окси-(или 4-ацетокси-)-
5-хлормеркур-3,6-эндоксогексагидрофталевой кислоты действием брома
[1226] в полярных растворителях (пиридин, ледяная уксусная кислота)
приводит к ifuc-бромгидрину, в неполярных (четыреххлористый углерод,
хлороформ) — к смеси цис- и /пра«с-бромгидринов с преобладанием цис-
бромгидрина. Реакция 4-оксипроизводного с бромом в ледяной уксусной
кислоте сопровождается ацетилированием гидроксила:
Различные относительные количества обоих изомеров RBr (цис и транс)
образуются при действии брома в СС14, СН3ОН, СНС13, пиридине на ме-
тил-2-хлормеркур-2-деокси-3,4,6-три-о-ацетил-^,В-глюкозид [122в]:
Действие брома на ацетоксимеркурпроизводное дает только один изомер.
Если\ реакция RHgCl с иодом проводится в течение длительного време-
ни, может происходить замена HgCl не только на иод, но и на хлор вслед-
ствие взаимодействия образующихся HgCl2 и RJ [123]. Так, действие иода
в хлороформе в течение 5 дней на 1,4-бис-хлормеркур-2,3-бутандиол при-
водит к 1-хлор-4-иодбутан-2,3-диолу [123], а не к замене обеих. HgCl-
групп на иод [124]:
CH2HgCl - CH2J и СН2С1
СНОН снсц 1 СНОН 1 СНОН
| +'J2 : 1 + HgCl2 —> 1
СНОН 5 дней СНОН СНОН
CH2HgCl 1 -CH2J 1 CH2J
Реакции ртутноорганических соединений
279
Возможно произвести замену HgX на хлор действием хлора, образую-
щегося при реакции хлорноватой кислоты с соляной кислотой. Так, 2-ацет-
оксимеркурдифенилен превращен в 2-хлордифенилен действием смеси со-
ляной кислоты и бертолетовой соли [125].
Получение 2-хлордифенилена [125]. К перемешиваемой смеси 0,180 г 2-ацетоксимер-
курдифенилена, 50 мл конц. соляной кислоты и 25 мл хлороформа по каплям прибавляют
насыщенный водный раствор 0,020 г КСЮз. Через 30 мин. таким же образом прибавля-
ют еще 0,005 г КСЮз и перемешивание продолжают в течение 4 час. Хлороформенный слой
промывают водным раствором соды. Продукт возгоняют. Выход сырого продукта 0,056 г,
т. пл. 56—60° С. Его обрабатывают спиртово-уксуснокислым раствором 2,4,7-тринит-
рофлуоренона (продукт — т. пл. 133—134° С) и вновь выделяют из этого комплекса, про-
пуская бензольный раствор его через А!2О3. Чистый 2-хлордифенилен — бледно-желтые
пластинки; т. пл. 67,5—68,5’ С.
2-Ацетоксимеркурдифенилен превращен в 2-бромдифенилен действием
оаствора брома в хлороформе, в 2-иоддифенилен — действием раствора
иода в иодистом калии.
Реакция замены атома ртути на галоид в диацетиленидах ртути явля-
ется удобным методом синтеза галоидацетиленов. При работе в отсутствие
избытка галоида не происходит присоединения галоида к двойной связи[312].
Получение 1-траяс-2'-бромвйнил-2,2'-бромэтинилбензола [311].
—СН=СН Вг-транс
К перемешиваемому раствору 3,4 г бпс-(о-транс-2'-бромвинилфенилэтинил)ртути в 150 мл
бензола прикапывают раствор 1,81 г брома в 20 мл бензола. Красный цвет не исчезает пос-
ле прибавления около 80% брома. После окончания прибавления смесь перемешивают
еще 10 мин. и растворитель затем отгоняют в вакууме при 60° С. Остаток извлекают не-
сколько раз легким петролейным эфиром и экстракт освобождают от растворенной бромной
ртути пропусканием через короткую колонку с 5 г животного угля, содержащего 0,1 вес. ч.
10% -ной смеси палладия на угле. Испарением элюата получают 2,45 г бледио-желтого мас-
ла (79%) 1-траяс-2'-бромвинил-2,2'-бромэтинилбензола; т. пл. около—10° С, т кип. 60Q С
при 10~4 мм.
1-Бром-окт-1-ин и 1-бромфенил ацетилен получены подобным же обра-
зом с выходом 75 и 87%, соответственно, с применением в качестве раствори-
теля четыреххлористого углерода вместо бензола [311].
Действие на транс, транс- и цис, ^цс-ди-([3-хлорвинил)ртуть эквимолеку-
лярного количества .хлора (в четыреххлористом углероде, при комнатной
температуре в первом случае, при 0° С во втором) дает хлорвинилмеркур-
хлорид той же конфигурации, что и исходное симметричное соединение
[126].
Действие хлора на цас,цас-ди-(Р-хлорвинил)ртуть. Получение ц«сф-хлорвииилмеркур-
хлорида [126].
Н ИНН
4 ( \с=с/ ) Н8 + С12 + RCl
'Cl <’ Cl HgCI
К раствору 0,15 г (0,000464 моля) ццс-ди-(Р-хлорвинил)ртути в 5 мл четыреххлористого
углерода приливают при перемешивании 0,032 г (0,000927 г-атом) хлора, растворенного
в 5 ли охлажденного до 0° С четыреххлористого углерода. После получасового хранения
при комнатной температуре выделившийся осадок отфильтровывают и высушивают, он
плавится при 78,5° С. Выход 0,1 г (72,4%). Перекристаллизованный из петролейного эфи-
ра, он плавится так же.
Реакция цис-р-хлорвинилмеркурхлорида [127] с эквивалентным коли-
чеством иода в эфирном растворе приводит к 1,2-хлориодэтилену той же
конфигурации.
280 Ртутноорганические соединения. Лит. стр. 336—352
Реакция цис[1-хлорвинилмеркурхлорида с иодом [127]. Получение Цис-1,2-хлориодэти-
лена. К раствору 1,5 г цис-|3-хлорвинилмеркурхлорида в 25 мл сухого эфира приливают
1,2 г иода, растворенного в сухом эфире. После двухдневного стояния при комнатной
температуре отфильтровывают Образовавшийся осадок. Фильтрат обесцвечивают несколь-
кими каплями водного раствора тиосульфата и высушивают безводным сернокислым нат-
рием. После отгонки эфира остаток перегоняют на масляной бане при температуре 109—
113° С, 1,5805. Выход 0,62 г (67%).
быс-Перхлорвинилртуть обычным образом реагирует с хлором только
при сильном освещении [47а], реакции ее с бромом (в СС14) или с иодом
(в кипящем ксилоле) не могли быть остановлены на стадии получения
RHgX, даже когда были взяты эквимолекулярные количества реагентов
[476].
Реакция диэтилового ацеталя хлормеркурацетальдегида с бромом. Получение диэти-
лового ацеталя бромацетальдегида [128].
ClHgCH2CH(OC2H6)2 + Bra - ВгСН2СН(ОС2Н5)2 + HgBrCl
К раствору 26 г диэтилового ацеталя хлормеркурацетальдегида в 50 мл сухого хлоро-
форма при охлаждении и перемешивании прибавляют по каплям раствор 12 г брома в 30 ли
хлороформа. Через несколько минут после прибавления брома ртутную соль отсасывают,
осадок промывают хлороформом н от соединенных фильтратов отгоняют растворитель. При
перегонке остатка получают 11,2 а (80%) дйэтилового ацеталя бромацетальдегида. Т. кип.
45—46° С/10 мм\ п2° 1,4405; d20 1,2853.
Реакция этилового эфира меркур-бис-уксусной кислоты с бромом [129].- К раствору
11,5 а (0,031 моля) этилового эфира меркур-бяс-уксусной кислоты в 15 мл четыреххлори-
стого углерода прибавляют по каплям при взбалтывании раствор 10 г (0,062 моля) брома
в 10 мл четыреххлористого углерода (вначале при охлаждении, а под конец при нагрева-
нии на водяной бане до 50° С). После охлаждения отделяют бромную ртуть (10 г, 91%) и
дважды фракционируют фильтрат; собирают фракцию с т. кип. 165—168° С. Выход этило-
вого эфира бромуксусной кислоты 6,9 г (69%); «р 1,4510; d4° 1,503.
В столь инертных соединениях, какими являются перфтор алкильные
соединения ртути, замену ртути на галоид производят действием галоида
без растворителя в автоклаве при высокой температуре [130].
Получение гептафтор-2-иодпропана [130]. 540 г (1 моль) бмс-перфторизоиропилртути
и 510 г (2 моля) иода нагревают в автоклаве при 200° С в течение 8 час. Автоклав погру-
жают в ледяную баню и открывают вентиль. Охлажденный продукт отфильтровывают;
применением ловушки с твердой углекислотой предотвращают потери при отсасывании.
Получено 454 г сырого продукта, который при перегонке дает 439 г (74%) бледно-розового
гептафтор-2-иодпропана. Т. кип. 40° С.
При действии брома (в четыреххлористом углероде, при —15° С) на
фенилвинилацетиленил- и фенилаллилртуть [131] образуется бромистая
фенилртуть. Но при действии брома в хлороформе на CeHBHgCHCla
происходит разрыв связи между ртутью и ароматическим кольцом [39а].
Действие брома и гипобромита калия на циклопентадиенилртуть, осу-
ществленное Несмеяновым, Кочетковой и Материковой [132], приводит
не только к замене металла на галоид, но и сопровождается бромирова-
нием органической части молекулы. Ввиду термической неустойчивости
дициклопентадиенилртути выше —15° С реакции с бромом вели при тем-
пературе—17н--------20° С.
Реакция дициклопентадиенилртути с бромом [132]. а. Получение 1,2, 3-три-
бромциклопентена-4. В раствор 0,33 г дициклопентадиенилртути в 20 мл
я-гептана и 3 мл бензола, охлажденного до —20°С, по каплям прибавляют 4 мл гептанового
раствора брома, содержащего 0,127 г брома в 1 мл раствора, причем Температура бани
поддерживается в пределах —17 н 204 С. Поглощение брома происходит очень быстро.
При этом по стенкам колбы образуется кремовый мелкокристаллический осадок (дальней-
шее прибавление брома дает иеисчезающую окраску в растворе). Полученный бесцветный
Реакции ртутноорганических соединений ' 281
раствор сливают, осадок промывают 2 раза я-гептаном (по 10 мл), который присоединяют
к основному раствору. Вес воздушно-сухого осадка бромной ртути 0,35 г. Гептан выпари-
вают. Вес кристаллического кремового осадка 0,57 г. На свету быстро темнеет (!), имеет
специфический запах непредельных бромидов. Т. пл. 47—55° С. После хроматографирова-
ния на силикагеле (d = 12 мм, h = 300 мм) из смеси гептан — бензол (8 : 2), затем геп-
тан— бензол (1 : 1) при защите от света) получено.0,41 г прозрачных бесцветных, сильно
преломляющих свет кристаллов с т. пл. 63—64° С. Выход трибромциклопентена 93%.
б. Получение изомерных пентабромциклопентанов. При
температуре опыта—2-;—5° С поглощается 6 мл раствора брома; после хроматографиро-
вания на силикагеле (d = 12 мм, h ~ 300 мм, из гептана) получены изомеры пентабром-
циклопентана. Т. пл. 96—101°С и 105—107° С. Выход 49%.
Реакция дициклопентадиеиилртути с гипобромитом калия. Получение гексабромцикло-
пентадиена [132]. Раствор 0,34 г дициклопентадиеиилртути в 20 мл бензола прибавляют
к охлажденному до -)-2° С реактиву, приготовленному из 20 мл воды, 4,5 г едкого кали и
0,65 мл брома. При 0-г-4-2° С реакционную смесь выдерживают 2 часа с эффективным ме-
ханическим перемешиванием, затем отфильтровывают 0,2 г HgO. Бензольный слой отде-
ляют, водный 2 раза промывают бензолом. Объединенный бензольный раствор высушивают
NaaSO4. Испарением бензола получают 0,68 а желтых кристаллов, после хроматографии
(силикагель, d = 12 мм, h = 100 мм, из н-гептана) получено 0,60 г бромида с т. пл. 70—
72° С. Повторной хроматографией получен CsBr6 с т. пл. 86—86,5° С.
Получение 4-бромфлуорена из хлористой 4-флуорилртути [134]. 40 г хлористой 4-флуо-
рилртути, взвешенной в 200 мл ледяной уксусной кислоты, перемешивают с 14,5 г брома
в 25 мл ледяной уксусной кислоты в течение 8 час. Смесь нагревают до кипения и фильтру-
ют горячей. В теплый фильтрат пропускают сероводород до удаления ртути и сернистую
ртуть отфильтровывают. По охлаждении фильтрата выкристаллизовывается осадок 4-бром-
флуорена, после перекристаллизации из уксусной кислоты т. пл. 165° С. Выход 16,1 г
(66%).
Получение о-иодфенола см. [132а], о-иодкрезола [133].
При нагревании. (шс-пентафторфенилртути с иодом в течение 15 час.
при 150—155° С в запаянной трубке получено небольшое количество пен-
тафториодбензола [134а]. Замена HgCl-группы в продукте меркурирования
полистирола на иод 3-часовым кипячением с иодом в хлороформе дает ко-
личественно поли-(п-иодстирол) [13461.
Только путем замены ртути на галоид впервые удалось получить [135,
136] галоидные производные ферроцена. Иод- и бромферроцены получены
действием иода и брома на хлористую ферроцен ил ртуть, дииод- и дибром-
ферроцены—действием соответствующих галоидов на 1,Г-дихлормеркурфер-
роцен [135] (дииодферроцен — также действием иода в ксилоле на продукт
его симметризации [136а]), Г-хлор-1-иод- й Г-бром-1-иодферроцены—дей-
ствием горячего раствора иода в ксидоле на хлористую 1-(1'-хлорферроце-
нил)ртуть и бромистую 1-(1 '-бромферроценил)ртуть [136].
Получение иодферроцена [135]. При постепенном приливании к горячему ксилольному
раствору хлористой ферроценилртути раствора иода в ксилоле выпадает светлый серо-зе-
леный осадок, являющийся комплексом хлористой ферроценилртути с иодом, легко пере-
ходящим под действием водного раствора тиосульфата натрия в диферроценилртуть. При
прибавлении большого избытка иода в комплексе происходит замена ртути на иод. Свет-
лый серо-зеленый осадок превращается в черный. По охлаждении раствора осадок, промы-
тый спиртом, тщательно измельчают и перемешивают в течение 1,5 час. с водным раство-
ром тиосульфата натрия. Эту операцию повторяют 2 раза, после чего осадок несколько
раз промывают эфиром. Эфирные вытяжки соединяют. Эфир испаряют. Выход иодферро-
цена 64%. Кристаллизация из метилового спирта при охлаждении насыщенного раствора
до —10° С. Т. пл. 44—45° С.
Хлормеркурциклопентадиенилтрикарбонил марганца легко реагирует с
иодом или хлористым иодом с образованием иодциклопентадиенилтрикарбо-
нила марганца [137].
Реакция хлормеркурциклопентадиеиилтрикарбонила марганца с иодом [137]. К суспен-
зии 2,2 г (0,005 моля) хлормеркурциклопентадиенилтрйкарбонила марганца в 25 мл абсо-
лютного четыреххлористого углерода при температуре кипения растворителя и перемеши-
вании приливают по каплям раствор 2,5 г (0,01 моля) иода в 100 мл абсолютного четырех-
хлористого углерода. После прибавления половинного количества иода окраска иода пе-
рестает исчезать. Охлажденную смесь отфильтровывают, фильтрат обрабатывают водным
282 Ртутноорганические соединения. Лит. стр. 336—352
раствором тиосульфата натрия, промывают водой, подсушивают над MgSO^. Из раствора
растворитель удаляют в .вакууме, остаток — чистый иодциклопентадиенилтрикарбонил
марганца.. Т. пл. 33—34° С. Выход 1,52 г (92,1%).
Свободный родан действует на'ртутноорганическое соединение подобно
галоидам [139].
RHgX + (CNS)2 -+ RCNSJ+ HgXCNS
Реакция диферроценилртути с роданом, приводящая к замене ртути
на серу, описана в настоящей главе, стр. 293.
Хлористый иод реагирует и с полнозамещёнными соединениями и с
ртутноорганическими солями по уравнениям [140]:
RaHg + 2JC1 2RJ + HgCl2,
RHgX + JClRJ + HgXCl.
Действие ЯС13на ртутноорганические соединения см. стр. 295.
С полнозамещенными ароматическими соединениями ртути реагируют,
подобно кислотам, N-бромамиды [73], N-бромимиды [70, 74, 751 (с диви-
нилртутью [64а]) и гипобромиты [70] кислот. Положительный атом брома
перечисленных производных кислот соединяется с радикалом, RHg-оста-
ток в случае N—Вг- производного образует RHgN-производное. Например:
RHgR' + BrN (СОСНа)а -> RBr -f- R'HgN (СОСН2)2.
В случае несимметричных RHgR'(R =f= R') бром соединяется с более элект-
роотрицательным радикалом ряда Хараша [74, 75]. Реакцию ведут при
умеренном нагревании, кислотное производное в некоторых случаях играет
роль растворителя.
При облучении УФ-светом реакция идет по радикальному механизму:
бром присоединяется уже7 не к радикалу, а к атому ртути (см. стр. 328).
Действие хлористого нитрозила на полизамещенные полифторхлор&л-
кильные соединения ртути сопровождается заменой ртути на хлор [140а].
Основной реакцией является замена ртути на NO (см. стр. 316).
РЕАКЦИЯ С ГАЛОГЕНИДАМИ
И ДРУГИМИ СОЛЯМИ ЭЛЕМЕНТОВ,
А ТАКЖЕ ИХ АЛКИЛ(АРИЛ)ГАЛОГЕНИДАМИ И ГИДРИДАМИ
С препаративной точки зрения важными являются реакции нормаль-
ного обмена галогенидов элементов с полными ртутноорганическими соеди-
нениями или ртутноорганическими солями, протекающие по уравнениям:
R2Hg + M(n»X„ -> RHgX 4- RM(n) X„_t
R2Hg + 2M<'1>X^HgX2 + 2RM^’X„_1>
RHgX + -+ HgX2 + RM^X^.
•
По сравнению с реактивом Гриньяра применение в подобных обменных
реакциях ртутноорганических соединений позволяет синтезировать более
узкий круг металлоорганических соединений. Зато метод позволяет ввести
в связь с данным элементом весьма разнообразные радикалы, в том числе
неиндифферентные к реактиву Гриньяра.
Галоидные соли, щелочных и щелочноземельных металлов не вступают
с ртутноорганическими соединениями в реакцию рассматриваемого типа.
Реакции ртутноорганических соединений
283
Для целей синтеза металлоорганических соединений путем использования
данной реакции не были применены и галогениды Be, Mg, Zn и Cd. Про-
стейший пример названных выше реакций — взаимодействие ртутноорга-
нических соединений с солью меченой ртути Hg*X2 описан в этой главе
(стр. 320). Посредством описанного обмена нельзя также получить метал-
лоорганических соединений элементов нечетных рядов периодической си-
стемы (от К ДО Ni, от Rb до Pd и т. д.). Об инициирующем распад ртутно-
органических соединений действии галогенидов Си, Мп, Fe, Со, Ni см.
стр. 327. Обмен с галоидпроизводными С, N и О также не дает нужного
результата. Галогениды В, In, Tl, Si, Ge, Sn,.P, As, Sb, Bi, S, Se, Те,
J вступают в реакцию обсуждаемого типа и использованы для получения
металлоорганических соединений этих элементов (Ладенбург, Фридель и
особенно Михаэлис).
Наиболее легко вступает в реакцию, подобно самим свободным галоидам,
хлористый и треххлористый иод.
Реакции с ароматическими (а с JC13 также и с p-хлорвинильными) со-
единениями протекают при комнатной температуре в водной взвеси, с JC1
образуя арилиодид (см. предыдущий раздел, стр. 282), а с JC13—хлористый
,диарил-(ди-р-хлорвинил)иодоний и арил-р-хлорвинилиодидхлорид. При
комнатной температуре идет, реакция с BC1S, AsCl3 и более вяло с РС13.
Однако обычно приходится применять нагревание, особенно для заверше-
ния реакции, и подчас довольно высокое (200—300° С). Условия реакции
необходимо выбирать более жесткими в соответствии с желанием прийти к
более высокоалкилированному (соответственно арилированному) прбдукту
и более полно (до галоидной ртути) деалкилировать ртутноорганическое
соединение. Выбор условий зависит и от радикала, переходящего от ртути
к другому атому. Более трудно обмениваются предельные жирные радика-
лы, легче — фенил, еще легче — фенилы, содержащие в орто- и пара-по-
ложениях ориентанты первого рода, например анизил, а также фурил,
тиенил и другие суперароматические 5-членные гетероциклы.’ Подобно
фенильным радикалам, легко обмениваются и винильные.
Следует отметить, что рассматриваемый метод синтеза элементооргани-
ческих соединений использован мало, хотя в некоторых случаях (например
при использовании в качестве исходных полиртутных соединений) он за-
ключает возможности, отсутствующие у других методов. Отчасти это зави-
сит от того, что в случае простейших радикалов синтез посредством реак-
тива Грцньяра может быть использован с еще большим удобством, а для
получения металлоорганических производных сложных радикалов в слу-
чае наиболее интересовавших исследователей элементов (например As,
Sb) существуют и иные более удобные пути, например диазометод. Все же
данная группа синтетических методов, заключающая много неиспользо-
ванных возможностей, имеет богатые перспективы развития.
Синтез органических соединений элементов
III группы периодической системы
Синтез борорганических соединений. Взаимодействием галогенидов
бора с органическими соединениями ртути получены алкенильные и аро-
матические галогениды бора.
Первые получают на холоду или при умеренном нагревании. Так по-
лучены двуфтористый винилбор (из BF3 и дивинилртути) [141], двуброми-
стый винилбор [142], двухлористый p-хлорвинилбор (реакцией RHgCl с
ВС13 при 50° С) [143], хлористый ди-3-хлорвинилбор [143], двухлористый
перфторвинилбор [144] (из R3Hg и ВС13, условия не указаны).
284 Ртутноорганические соединения. Лит. стр. 336—352
Получение двубромистого винилбора [142]. 15,0 г (0,06 моля) трехбромистого бора
конденсируют на 12,5 г (0,05 моля) дивинилртути, смесь оставляют нагреваться до комнат-
ной температуры и оставляют стоять на некоторое время. Продукт реакции очищают пере-
гонкой в вакууме. Т. кип. 35° С/50 мм. Выход 8,1 г (82%).
При взаимодействии треххлористого бора с диарилртутью, начинаю
щемся уже на холоду, образуется хлористый арилбор [66, 145—147]:
Ar2Hg.+ 2ВС13 -» 2АгВС12 + HgCl2. (1)
В свою очередь последний с избытком диарилртути дает хлористый
диарилбор [147]:
ArBCh + Ar2Hg -> Аг2ВС1 + ArHgCl. (2)
Если Аг—углеводородный остаток, реакцию (1) Михаэлис проводил лри
180—200° С в запаянной трубке; взаимодействие с дианизил-, дифенил-
ртутью идет при комнатной температуре.
Однако, согласно Гильману и Муру [66], и в случае, если в реакции (1)
Аг — углеводородный остаток, нет необходимости в применении высокой
температуры и давления. Так, получасовое взаимодействие при комнатной
температуре дифенилртути с треххлористым бором (в хлорбензоле) дает
после гидролиза, фенил борную кислоту с выходом 52—70% [66].
Реакция Ar2Hg с ВВг3 (получение АгВВг2) более удобна, чем с ВС13,
ее ведут не в запаянной трубке, а при кипячении с обратным холодильни-
ком в бензоле [1481. Описан синтез хлористого [146] (т. пл. 0°С, т. кип.
175° С) и бромистого [148] фенилбора, хлористого о-толилбора [147], бро-
мистого и-толилбора [146, 148], хлористого n-толилбора (т. пл. 27° С)
[146], о-(т. кип. 212° С), м- (т. кип. 218° С) [148], п- (т. кип. 206° С) [146]
ксилилбора, бромистого псевдокумилбора [146, 148], хлористых а-(т. кип.
124° С/25 мм) и р-(т. пл. 116° С) нафтилбора [147], о- и и- (т. кип. 182° СУ
/170 мм, т. пл. 300° С) анизилбора [147], о- и п- (т. кип. 400° С/220 мм,
т. пл. 2° С) фенетилбора [147].
Получение фенилборной кислоты [66]. Взвесь 0,05 моля дифенилртути в 500 мл хлор-
бензола помещают в трехгорлую круглодонную колбу, снабженную мешалкой. Колба
соединена с приемником, охлаждаемым сухим льдом в ацетоне и снабженным выходным
ртутным затвором и трубкой для ввода газа, опущенной ниже уровня жидкости. Вводная
трубка соединена с баллоном с треххлористым бором через ртутный затвор, служащий для
удаления хлора, могущего присутствовать в треххлористом боре, и для наблюдения за
скоростью пропускания последнего. В реакционную смесь пропускают 100%-ный избыток
треххлористого бора (количество введенного треххлористого бора определяют по умень-
шению веса баллона с треххлористым бором). По окончании введения треххлористого бора
перемешивают в течение 30 мин. Смесь фильтруют для отделения хлористой фенилртути и
фильтрат гидролизуют медленным прибавлением льда. Образовавшуюся кислоту извле-
кают четырьмя порциями 10%-него раствора едкого кали. Общий объем 10%-кого КОН
250 мл. Водный слой промывают 100 мл эфира и затем подкисляют. Выделяющийся слегка
желтоватый осадок кристаллизуют из воды. Выход 70,5%.
Реакцию (2) ведут при нагревании до 300—320° С в запаянной трубке.
n-Хлормеркурфенол, n-хлормеркурбензойная кислота, метил-п-карбок-
сифенилртуть не дают при многочасовом нагревании с треххлорйетым бо-
ром в хлорбензоле борорганических соединений [147]. При реакции в раз-
ных условиях дифенилртути с три-я-бутоксибором также не получено бор-
органического соединения [66].
Образование адюминийорганических соединений. Описано получение
из галогенидов алюминия и винилртутных соединений (в бензоле или
четыреххлористом углероде) винилалюминийгалогенидов в виде неустой-
чивых, легко полимеризующихся веществ [149].
Реакции ртутноорганических соединений
285
Синтез органических соединений индия. Согласно Годдару [150], при
кипячении в ксилоле в течение 37 час. дифенилртути и безводного трех-
хлористого индия образуется хлористый дифенилиндий.
, Получение хлористого дифенилиндия [150]. Безводный треххлористый индий (из
1,16 г металлического индия) и 10,77 г дифенилртути (3 моля) кипятят в 50 мл ксилола
-37 час. Отсосанный осадок для удаления ртутноорганических соединений извлекают в
экстракторе сухим бензолом. Остаток в гильзе (0,647 г) представляет собой хлористый
дифенилиндий — нерастворимый, неплавкий кристаллический порошок кремового цвета.
Синтез таллийорганических соединений. Из ди-н-пропилртути и хло-
ристого таллия образуется хлористый ди-н-пропилталлий и хлористая
я-пропилртуть [151 ]. При взаимодействии хлористого таллия в этих же усло-
виях с диизоамил-, дибензил-, бензилэтилртутью, а также иодистой 2-тие-
нилртутью не образуется органических соединений таллия [151].
Полнозамещенные ртутноорганические соединения с ртутью у олефи-
нового атома углерода: 3-хлорвинильные [152, 153], пропенильные [154,
155], изопропенильные [156, 157], бутенильные [106], винильные [158],
а- [159] и и- [160] стирильные соединения ртути реагируют в мягких усло-
виях (в эфирном растворе) с треххлористым или трехбромистым таллием с
образованием соответствующих галогенидов диалкенилталлия. При соотноше-
нии 1 моля R2Hg к 2 молям Т1С13 ди-^zzc, цис- и дд-транс, транс-1-метпл-2-
ацетокси-1-пропен-1-ил-ртуть также с высоким выходом дали [106] соот-
ветствующие цис- и /пранс-КТ1С12-соединения. Транс-изомеры перечислен-
ных ртутноорганических соединений реагируют легче цис-изомеров. Так,
при сливании эфирных растворов цис, транс-дихлорвинилртути и треххло-
ристого таллия тотчас выпадает осадок хлористого ди-транс-3-хлорвинил-
таллия и лишь через 4 часа начинает образовываться осадок хлористого
ди-г{«с-р-хлорвинилталлия [152]. цис, транс-Дипропенилртуть с треххло-
ристым таллием в этих условиях образует хлористый ди-транс-лропенилтал-
лий [155]. Все эти реакции проходят стереохимически чисто, с сохранением
конфигурации передаваемого радикала (см. гл. VI).
Реакция транс,гя/>анс-дипропенилртутнс трехбромистым таллием. Получение бромис-
того транс, /ге/ихнс-днпропенилталлия [154].
/СНз Н
( \с=с/ ) Т1Вг
\ HZ 4
К 1 г (0,0035 моля) свежеприготовленной, но не перегнанной транс, транс-детроаег
иилртути в 3 мл эфира прибавляют 0,8 г (0,0018 моля) трехбромистого таллия; при этом
наблюдается моментальное выделение обильного осадка. После 1 часа выдержки осадок
отфильтровывают и промывают ацетоном. Сухого продукта получают 0,61 г (94%). После
двухкратной перекристаллизации из пиридина выделяют кристаллический продукт, желтею-
щий при нагревании до 360°С, не плавясь. Из эфирного фильтрата выделяют 0,96 г (84%)
бромистой транс-пропенилртути с т. пл. 118,5—119,5° С.
Получение двухлористого ц«с-1-метил-2-ацетокси-1-пропен-1-ил-таллия [106].
СН3 СНз
>=с<
; СНзСОО Т1С13
К эфирному раствору 1,5 г (0,003 моля) ди-(1-метил-2-ацетокси-1-пропен-1-ил)-ртути
(т. пл. 101° С) приливают эфирный раствор 2,16 г (0,006 моля) треххлористого таллия.прн
комнатной температуре. Выделение кристаллического осадка было замечено только через
10 мин. цосле соединения реагентов. После 40-минутного стояния при комнатной темпе-
286 Ртутноорганические соединения. Лит. стр. 336—352
ратуре осадок отделяют, промывают эфиром и перекристаллизовывают из воды. Темпера-
тура разложения 135° С. Выход 80% . Повторно перекристаллизованное из спирта вещество
разлагается (не плавясь) при 135° С; оно хорошо растворимо при нагревании в спирте,
ацетоне, воде, плохо — в бензоле и эфире.
Получение хлористого, дифенилталлия [161]. К раствору 2,8 г дифенилртути в 50 -г
сухого эфира добавляют 2,5 г треххлористого таллия В том же растворителе, причем сразу
же выпадает белый осадок. Через несколько часов последний отсасывают и извлекают для
удаления хлористой фенилртути бензолом. Остаток — хлористый дифенилталлий. Т. пл.
250° С.
Бромистый ди-о-ацетоксифенилталлий получен с небольшим выходом
при 3-часовом кипячении спиртового раствора R2Hg (1 моль) иТ1Вг3-4Н2О
(—2 моля) [162].
Гораздо лучшие результаты дает разработанный Глушковой и Кочеш-
ковым [163, 164] метод синтеза RT1X2, а также R2T1X взаимодействием
диарилртути с солью трехвалентного таллия органической кислоты — три-
изобутиратом таллия. В обоих случаях выходы хорошие. Реакции эти про-
ходят быстро в мягких условиях (при кратковременном нагревании в хло-
роформе). Метод применен к синтезу ароматических и тиенильных соедине-
ний таллия и заслуживает широкого применения.
При взаимодействии эквимолекулярных количеств R2Hg и Т1Х3 полу-
чаются соли моноарилталлия:
R3Hg + Т1 (OCOQH8-j)3 -* RT1 (ОСОСШНЬ + RHg (OCOC4H»-i)
(R=фенил, а- и [J-нафтил, n-хлор- и п-бромфенил, n-анизил, а-тиенил).
Синтез триизобутирата таллия [163]. При нагревании в течение 5 мин, до растворения
2 г ТкОз (0,045 моля) в 10 мл кипящей изомасляной кислоты и последующем охлаждении
выделяется белый кристаллический осадок, который отсасывают н промывают петролей-
ным эфиром. Получают 1,8 г вещества (выход 44%) с т. пл. 174—174,5° С- После пере-
кристаллизации из бензола т. пл. 179—179,5° С.
Получение динзобутирата га-хлорфенилталлия [164]. 4,65 г (0,01 моля) триизобутирата
таллия в 15 мл хлороформа приливают к 4,25 г (0,01 моля) взвеси ди-«-хлорфенилртути в
15 мл того же растворителя (при сливании растворов ди-я-хлорфеиилртуть начинает пере-
ходить в раствор, оставшийся небольшой. осадок растворяется при нагревании). После
некоторого испарения хлороформа выпавший осадок отсасывают и промывают петролей-
иым эфиром. После перекристаллизации из дихлорэтана получают 3 г вещества ст. пл.
229° С (с разложением) и 0,6 а вещества с т. пл. 224—225° С. Выход продукта 73,5%.
Получение диизобутирата га-анизилталлия [163]. К раствору 4,65 г (0,01 моля) триизо-
бутирата таллия в 10 мл хлороформа (нагревание) приливают взвесь 3,82 г (0,01 моля)
ди-л-аиизилртути в 25 мл хлороформа. После нагревания смеси иа водяной бане и фильт-
рования из фильтрата, после охлаждения выделяют 3,15 г (выход 65%) динзобутирата
n-анизилталлня, после кристаллизации из дихлорэтана т. пл. 196° С.
Взаимодействие 2 молей диарилртути с 1 молем триизобутирата таллия
(в тех же условиях)
2RaHg + Т1 (OCOQHe-i)s -» R2TI (OCOQHo-i)
является'методом синтеза солей диарилталлия [164]. Так получены с хо-
рошими .выходами соли дифенил- [164], ди-п-толил- [164], ди-п-анизил-
[164] таллия.
Получение изобутирата ди-п-аннзилталлня [164]. 2,35 е (0,005 моля) триизобутирата
таллия в 10 мл хлороформа приливают к суспензии 4,15 а (0,01 моля) ди-л-анизилртути в
10 мл того же растворителя и нагревают до полного растворения осадка. После некоторого
испарения растворителя иа холоду выкристаллизовавшийся осадок отсасывают, промыва-
ют петролейным эфиром, перекристаллизовывают из дихлорэтана. Получено 2 г (66%) ве-
щества с т» пл. 246° С. После повторной кристаллизации из того же растворителя т. пл.
252° С.
По относительной легкости отрыва радикала от несимметричных ртут-
ноорганических соединений под действием треххлористого таллия (в эфире
Реакции ртутноорганических соединений
287
или смеси эфира с толуолом) по уравнению
RHgR' + TICI3 -» R2TICI + RHgCl
(RHgR'= C8H5HgC2H5, a-C10H7HgC2H5, a-C10H7HgC8H6, а также
o-CH3OCeH4HgC10H7‘a, a-C10H7HgC8H2(CH3)3) радикалы R могут быть рас-
положены в ряд, в основном совпадающий с рядом радикалов Хараша,
полученным на основании расщепления RHgR' под действием кислот [1651.
C4H9HgCH2C8H5 с 'треххлористым таллием дал лишь RHgCl, R'HgCl
и хлористый таллий [155].
Не имеющей аналогий в химии ртутноорганических соединений с лока-
лизованными сг-связями Hg — С является реакция дициклопентадиенил-
ртути с гидроокисью Таллия (I), приведшая к образованию циклопентадие-
нилталлия с выходом 30% [166]. Реакцию ведут в водно-метанОльном рас-
творе при +2° С.
Синтез органических соединений элементов
IV группы периодической системы
Дихлор-(и дибром-)карбен, образующийся при разложении фенил-
тригалоидметилртути, внедряется по связи СН метиновых и метиленовых
групп углеводородов с образованием дигалопроизводных углеводородов.
Реакцию ведут в избытке углеводорода [167]. Таким путем получены [167]
(в скобках выходы) реакцией с C6HgCH2CH3 — С8Н5СН(СН3)СС12Н (35%),
с С8Н5СН(СН3)2 —С8Н5(СН3)2ССС12Н (58%), с сг/с1о-С8Н12— срс/о-СдНцСС^Н
(32%), с С8Н5СН2СН3 — С6Н6СН(СН3)СВг2Н (6,5%).
Реакция фенилтригалоидметилртути — C8H5HgCCl2Br, C8H5HgCClBr2,
C8H6HgCBr3—с олефинами предложена как метод синтеза г&и-дигалоидцик-
лопропанов [168, 406а]; Вг в СНа13-группе облегчает реакцию [340]
Hal Hal
RHgCHals + >С=СО Y +RHgHal.
^>C—
Синтез гексахлорциклопропана [168]. 0,1 моля фенил(бромтрихлорметил)ртути в
1 моле тетрахлорэтилена нагревают при 90° С в течение часа. Выпадает бромистая фенил-
ртуть (выход 94%) и образуется гексахлорциклопропан. Выход 74%, т. пл. 103—104° С.
Аналогично из тетрахлорэтилена и С8Н5СС1Вг2 получен бромпентахлор-
циклопропан (выход 48%), т. пл. 106,4—107,6°С; из тетрахлорэтилена и
CeH6HgCBr3— 1,1-дибромтетрахлорциклопропан; выход 26%, т. пл. 114—
115°С [168].
Из этилена и C6H5HgCBr3 (автоклав, в бензоле, 24 часа) получен 1,1-
дибромциклопропан (выход 53%), с CeH5HgCCl2Br (хлорбензол) — Г,1-ди-
хлорциклопропан (выход 65%), из транс-стильбена и C8H5HgOCl2Br
(в бензоле, 2,25 часа, 85° Q получен 1,1 -дихлор-2,3-дифенилциклопропан с
выходом 90% [168].
Метиленовая группа, генерируемая при разложении бис-галоидметил-
ртути или галогенида галоидметилртути, присоединяется по двойной связи
циклогексена с образованием норкарана (реакция ведется при кипячении
в бензоле в течение 8 дней, в последнем случае в присутствии дифенилртути),
например [166а]:
JCH2HgJ -HCeH^Hg + Г Г р +2CeH5HgJ,
288
Ртутноорганические соединения. Лит. стр. 336—352
Синтез кремнийорганических соединений. Из этой области осуществлен
синтез арилтрихлор силанов, требующий нагревания до высокой темпе-
ратуры.
' Ar2Hg + 2SiCU-^ 2ArSiCl3 + HgCh.
Получение треххлори^того фенилкремния [169]. Нагревают 100 г дифенилртути и
50 г четыреххлористого кремния в запаянной трубке до 300° С в течение многих часов.
Продукт перегоняют, получают 30 а фракции с т. кип. 197—198° С, представляющей со-
бой треххлорнстый фенилкремний.
Аналогично получен треххлористый л-толилкремний [169].
Реакция же четыреххлористого кремния, а также хлор алкил (ар ил и
алкокси)сйланов с ртутными производными оксосоединений производится
в кипящем изопентане и приводит к полной замене галоидов на винил окси-
группу и образованию с хорошим выходом соответственно тетравинилокси-
силанов или винилокси-алкил-, арил- или -алкоксисиланов [170].
«Hg(CH2CHO)2 + Щ-п SiC1n R4-«Si (ОСН=СН2)ге +ClHgCH2CHO,
R = СНз, С3Н5, СвНз, С1СН2, С2Н5О; п = 1, 2, 3, 4.
Получение триметилвинилоксисилана [170]. К 32 г (0,11 моля) меркур-бпс-ацетальде-
гида в 70 мл изопентана приливают при размешивании раствор 11 г (0,11 моля) трнметил-
хлорсилана в 20 мл изопентана. Смесь кипятят в течение 2 час., осадок отфильтровывают,
дважды промывают изопентаном, изопентан отгоняют. При разгонке получено 8,4 а (72%)
триметилвйиилоксисилана с т. кип. 74—75° С, Пр 1,3885, d|° 0,7720; (C^5)sSiOCH=CHa
с т. кип. 53—54° С/20 лои, ftp 1,4275; Si(OCH=CH2)4 с т. кип. 79—80° С/2 мм, 1,4896.
Йодиды триалкилкремния легко реагируют с эфирами меркур-бнс-ук-
сусной кислоты по схеме:
2Alk3SiJ + Hg (CH?COOR)2-> 2Alk3SiCH2COOR -f- HgJa.
Так, из йодистого триэтилкремния и метилового эфира меркур-бгл>уксус-
ной кислоты получен метиловый эфир триэтилсилилуксусной кислоты с
выходом 52% [171].
Дигалокарбен, образующийся при разложении фенилтригалоидметил-
ртути, внедряется и по связи Si—Н арилсиланов с образованием (с вы-
ходами 77—90%) дигалоидпроизводных кремнийорганических соеди-
нений [167]. Реакция применена к R3SiH, (C8H5)2SiH2, растворитель —
бензол.
При кипячении C8H5HgCCl2Br или CeH8HgCBr3 и триметилвинилсилана
(30-часовое кипячение в бензоле) получен 1,1-дихлор- и 1,1-дибром-2-три-
метил силилциклопропан соответственно [168]. Выход 67 и 57%.
Синтез германийорганических соединений. Эфиры меркур-бмс-уксусной
кислоты взаимодействуют в инертном растворителе с триалкилгерманами
(в тетрагидрофуране) или лучше с иодистыми триалкилгерманами (в пет-
ролейном эфире) с образованием с хорошим выходом эфиров триалкилгер-
манилуксусной кислоты [171]:
/ 0 \ 0
2R3GeJ + Hg | СНаС^ j -> R3GeCHaC^ + HgJa.
\ \)R/a ^OR
Реакции ртутноорганических соединений
289
Реакцию проводят в присутствии пиридина, связывающего образую-
. щуюся иодную ртуть в комплекс HgJ2-2C5H5N, легко отделяемый фильтро-
ванием.
Получение метилового эфира трипропилгерманилуксусной кислоты [171]. К раствору
17,5 г метилового эфира меркур-бис-уксусной кислоты и 6 мл пиридина в петролейном
эфире прибавляют по каплям 33 г йодистого трипропилгермания, затем кипятят 30 мин.
и фильтруют. Фильтрат разгоняют. Выход 19,5 г (71%), т. кип. 81,5—84°С/1,5 мм,
1,4580, 1,0518. Так же получен этиловый (т. кип. 105—108° С/5 мм, 1,4562,
1,0418), метиловый и пропиловый эфиры трибутилгерманилуксусной кислоты.
Реакции эфиров меркур-б«с-уксусной кислоты с галоидными триамил-
германиями ведут в тетрагидрофуране при кипячении в течение часа.
При нагревании (80° С) дивинилртути с четыреххлористым германием
по реакции •
.GeCU -f-. (CH2=CH)aHg CHa=CHGeCl3 + CH2=CHHgCl
с 70%-ным выходом получен винилтрихлоргерманий [172].
Взаимодействие диарилртути с четыреххлористым германием проведено
в автоклаве при 180° С и с небольшим выходом привело к ArGeCl3 [173].
Гораздо лучшие результаты дает реакция диарилртути с галогенидами
двухвалентного германия (см. стр. 311.).
Дигалокарбен, образующийся при разложении фенилтригалоидметил-
ртути, способен присоединяться и по связи Ge — Н. Как и с кремнийорга-
ническими соединениями, реакцию ведут в бензоле [167]. Таким путем
из (C6HB)3GeH получен (C6HB)3GeCCl2H. Выход 88%.
В общем виде реакция может быть изображена уравнением:
CeH5HgCX2Br + ^М—Н -> 2м—СХ2Н + C6H5HgBr
М = С, Si, Ge; X = Cl или Br.
Синтез оловоорганических соединений. При нагревании смеси дифенил-
ртути и хлорного олова образуется [174] смесь фенильных оловоорганиче-
ских соединений, из которой можно выделить хлористое дифенилолово.
Однако гораздо лучшие результаты дает реакция восстановления дифе-
нилртути хлористым оловом (см. стр. 313).
При взаимодействии фенил(бромдихлорметил)ртути с гидридом три-
н-бутилолова происходит реакция [413а]:
(n-CiH9)3SnH + C6H5HgCCl2Br - (n-C4H9)3SnBr + С6Н5НбСС12Н.
Синтез свинцовоорганических соединений. В области синтеза свинцо-
воорганических соединений жирного и ароматического рядов реакция об-
мена этого типа, предложенная Кочешковым и Надь [176], дает широкие
препаративные возможности. Вследствие малой устойчивости тетрагалоге-
нидов свинца в качестве исходной соли свинца используют тетраацетат.
Реакция течет по уравнению:
R2Hg + Pb (ОСОСН3)4 R2Pb (ОСОСН3)а + Hg (ОСОСН3)2
и ведется в хлороформе при комнатной температуре- Выходы превосходны.
При других соотношениях реагентов взаимодействие диарилртути й
тетраацилата (ацетата, изобутирата) свинца является методом синтеза мо-
290
Ртутноорганические соединения. Лит. стр. 336—352
ноарильных соединений свинца [177, 178]:
AraHg + Pb (OCOR)4 -> ArPb (OCOR)3 + ArHgOCOR.
Реакция также проводится в хлороформе [177,178], с ббльшим трудом—
в бензоле [179].
Естественно, диарилртуть может арилировать моноарильное соединение
свинца. Так получен диацетат диарилсвинца [178а].
Реакции с галогенидами титана
Реакция с четыреххлорйстым титаном идет по уравнению [180]:
2 (CeHs)2Hg + TiCl4-> 2CeHBHgCl + Ti2Cle + C6H5CeH3.
См. также [181]. Но (C6H5)2TiCl2 с (C8H6)2Hg дает (C5H6)2Ti(CeH5)2 [138].
Синтез органических соединений элементов
V группы периодической системы
Синтез фосфорорганических соединений. Реакции ртутноорганических
соединений с треххлористым фосфором (может быть взят также трехбро-
мистый фосфор) протекают по уравнениям:
R2Hg + РС13 RPCk + RHgCl,
RHgCl + РС13 RPCU + HgCl2,
RaHg + RPCl2 -» RaPCl + RHgCl
(1>
(2)
(3)
и являются важным методом синтеза алкил(арил)дихлорфосфинов и ди-
арилхлорфосфинов.
Реакция треххлористого фосфора с ртутноорганическими соединениями
[182—1841 протекает гораздо легче' чем взаимодействие SiCl4, но более
вяло, чем AsCl3. Обычно эти реакции ведут при 180—250° С. Лучшие вы-
ходы получаются, если РХ8 или RPX2 брать в избытке.
Как всегда,- реакция алкил(арил)дихлорфосфинов с диарилртутью
RPCla + RaHg R2PC1 + RHgCl
происходит значительно труднее, чем реакция R2Hg с РС13, а реакция
А1кРС12 с диалкилртутью (алкил — остаток предельного углеводорода) во-
обще не была использована. Этой реакцией получают и смешанные диарил-
хлорфосфины RR'PCl. Для ее проведения можно использовать не только
диарилр'Гуть, но и галоидную арилртуть, особенно в случае легко обмени-
вающегося арила, т. е. арила, более электроотрицательного (по Харашу,
см. стр. 267), чем фенил, например толила.
Получение алкилдихлорфосфниов [183]. 20 г совершенно сухой диалкилртути и 60 г
свободного от соляной кислоты треххлористого фосфора нагревают в запаянных трубках до
250° С в течение 6 час. Такое нагревание имеет целью разрушить хлористую алкил ртуть,
распадающуюся с образованием олефина и каломели, так как иначе она мешает, частично
перегоняясь с фосфином. При получении этилдихлорфосфина исходят из 15 г диэтилртути и
45 г треххлористого фосфора. После вскрытия трубок жидкость сливают с кристаллов и
тщательно разгоняют без доступа влаги. Выход низок.
Этилхлорфосфин имеет т. кип. 114—117° С, н-пропилхлорфосфин — 140—ПЗ^С, изо-
пропилхлорфосфин — 135—138° С, изобутилдихлорфосфин— 155—157° С, изоамилди.
хлорфосфин— 180—183° С.
Реакции ртутноорганических соединений
291
Реакцию дивинилртути с галогенидами трехвалентного фосфора ведут
в более мягких условиях. Получены винилдихлор- [141, 185] и винилди-
бромфосфины [142, 185].
Получение винилдибромфосфина [142]. 13,5 г (0,05 моля) трехбромистого фосфора и
и 12, 7 г (0,05 моля) дивинилртути при перемешивании нагревают при 80° С в течение
12 час. От реакционной смеси отгоняют жидкость, дымящуюся на воздухе. При повторной
перегонке получают 8,5 г (78%) винилдибромфосфина. Т. кип. 60°С/20 мм.
[(CH3)3SiCH2]2Hg не реагирует с РС13 при 76° С и при 12-часовом кипя-
чении в бензоле [185а].
Получение фенилдихлорфосфнна [184]. 10 г дифенилртути нагревают с 34 г хлористого
фосфора в запаянной трубке до 180° С. После охлаждения жидкость сливают с образовав-
шейся в большом количестве хлористой фенилртути и подвергают перегонке. После того как
отогнался избыток хлористого фосфора, между 216 и 220° С перегоняется слегка мутная жид-
кость, при хранении выделяющая немного металлической ртути. Жидкость сливают со ртути'
и после повторной перегонки получают совершенно чистой; для CeHsPCU разные авторы
дают отличающиеся друг от друга температуры кипения (225, 221—222, 222° С); d|° 1,319.
Описано получение о- [187, 188], м- [188] и п-толилдихлорфосфина [188],
л-хлорфенилдихлорфосфина [189], а-нафтилдихлорфосфина [190], п-ди-
метиламинофенилдихлорфосфина [186].
При получении дифенилхлорфосфина [191] действием дифенилртути на
фенилдихлорфосфин-, несмотря на большой избыток последнего и высокую
температуру (222° С), реакция идет только до стадии хлористой фенил-
ртути.
Улучшенный метод синтеза дифенилхлорфосфина [192]. В круглодонной колбе, снаб-
женной обратным холодильником и хлоркальциевой трубкой, в токе углекислого газа на-
гревают до 230—240° С 1,5 часа 55 г дифенилртути и 125 г фенилдихлорфосфнна. Продукт
реакции извлекают 4 раза бензолом. Бензол и фенилдйхлорфосфин отгоняют при нормальном
давлении до 260° С; остаток фракционируют в вакууме. Первая фракция 178—189° С/16 мм
(56 г) дает при повторной перегонке 50 г чистого дифенилхлорфосфина с т. кип. 179—
180° С/16 мм ; вторая фракция 188—230° С/16 мм (20 г) представляет собой главным обра-
зом окись дифенилхлорфосфина.
С избытком дифенилртути выход дифенилхлорфосфина [193] состав-
ляет 55%.
Аналогично можно синтезировать и несимметричные диарилхлорфосфи-
ны RR'PCl. Толильный радикал переходит легче, чем фенил.
Получение фенил-га-толилхлорфосфина [194]. 78 г фенилдихлорфосфнна и 60 а бромистой
(очевидно, с тем же успехом можно взять эквивалентное количество хлористой) п-толилрту-
ти нагревают до 270° С 2—3 часа в колбе с обратным холодильником в токе углекислого газа.
Реакция вначале идет бурно. Смесь извлекают бензолом, растворитель отгоняют после фильт-
рации, остаток фракционируют в токе углекислого газа под уменьшенным давлением. Со-
бирают при 100 jtAt фракцию 230—240°С (30 а, 63%) и возвращают, кроме того, 24 a CeHsPClz.
Подобный же результат получают, исходя из толилдихлорфосфина и
дифенилртути [148, 195]. Аналогично получен фенилпсевдокумилхлор-
фосфин [195]; л-анизилфенилхлорфосфин синтезирован из ди-лг-анизилрту-
ти и фенилдихлорфосфнна (215—220° С, 1 час, под азотом) [195а].
Синтез мышьяковоорганических соединений. Рассматриваемый метод •
основан на группе реакций
R2Hg + AsClg-» RHgCl + RAsCh,
RHgCl + AsCls HgCl2 + RAsCla,
292
Ртутноорганические соединения. Лит. стр. 336—352
R2Hg -|- 2AsCl3 —> HgCl2 -J- 2RAsCl2,
R2Hg + R'AsCla -» RHgCl + RR'AsCl,
RHgCl + R'AsCla HgCl2 + RR'AsCl,
RaHg AsCl3 —> HgCl2 4" RaAsCl,
2RHgCl + AsCl3 -* HgCl2 + RaAsCl,
RaHg + R'AsS HgS + R'RaAs
и использован шире, чем любой другой метод данного раздела.
Применив его, Михаэлис впервые получил представителей столь важ-
ных классов мышьяковых соединений, как арилдихлорарсины и диарил-
хлорарсины, в частности фенилдихлор арсин и дифенил хлорарсин. До на-
стоящего времени метод находит применение особенно в тех случаях, когда
арил- или алкиларсиновые кислоты, проходя через которые, теперь обычно
получают хлорарсины, нельзя синтезировать по Барту (через диазосоеди-
нения) или по Мейеру (из алкилгалогенидов). Таков, например, случай
производных фурана, тиофена и т. д.
Реакция AsCl3 с ртутноорганическими соединениями относится к типу
легко протекающих, даже диэтилртуть уже при комнатной температуре с
разогреванием вступает в реакцию с AsCl3, деалкилируясь, впрочем, толь-
ко до стадии C2H5HgCl. Особенно легко реагируют, как всегда, ртутные
производные радикалов электроотрицательной части ряда Хараша, в ча-
стности фурила, тиенила и т. д.
Труднее проходит замещение второго атома хлора AsCl3 с ртутноорга-
ническими соединениями. Точно так же требует более жестких условий
использование радикала ртутноорганической соли RHgCl. С целью дове-
дения реакции до конца (до состояния равновесия) ее завершают обычно
при большем или меньшем, но не слишком длительном и высоком нагре-
вании.
Имеется целый ряд работ, положивших основы данного метода [191—
196, 197—207]. Описано также получение несимметричных диарилхлорар-
синов [199], синтез жирных хлорарсинов [203, 2081: р-хлорвинилдихлор-
арсина (люизит I) (из AsCl3 и хлористой р-хлорвинилртути [209]), пер-
фторвинилдихлорарсина[40, 144].
При взаимодействии CF2=CFHgC2H5 и AsCl3 получены CF2=CFAsCla
> и C2H5HgCl [40]: Синтезированы анизил- [201,210] и фенетилдихлор арсины
[210], феноксифенилдихлорарсин [211], о- и п-толилдихлорарсины [200,
210], ди-я-толилхлорарсин [203], о-, м- и п-ксилилдихлорарсины [212],
псевдокумилдихлорарсин [212], а-нафтилдихлорарсин [190, 208, 210, 213],
Р-нафтилдихлорарсин [212], первичные [207, 214—216], вторичные [214] и
третичные [214] арсины ряда тиофена и фурана [204, 205].
Синтез сурьмяноорганических соединений. Диметйлртуть энергично
реагирует с треххлористой сурьмой с образованием соединения хлористой
метилртути с хлористой триметил сурьмой и выделением металлической
ртути [217].
При взаимодействии дифенилртути с треххлористой сурьмой (в ксило-
ле, запаянная трубка, 130° С) получается хлористая трифенилсурьма и
треххлористая дифенилсурьма [218].
В более мягких условиях (при кипячении в бензоле) единственным
сурьмяноорганическим продуктом реакции является хлористая дифенил-
сурьма [219]. Последняя не образуется при реакции с треххлористой сурь-
мой бромистой фенилртути (условия не указаны) [220].
Реакции ртутноорганических соединений
293
Синтез висмуторгамических соединений. Получение висмуторганиче-
ских соединений реакцией BiX3 с ртутноорганическими соединениями
изучено мало и только для ароматического ряда. Оно не всегда идет
гладко. Так, действие треххлористого висмута на дифенилртуть (5 час., в
кипящем хлороформе) привело не к висмуторганическому соединению, а
лишь к образованию C6H6HgCl [222, 223].- Но взаимодействием дифенил-
ртути с трехбромистым висмутом в эфире в течение 20 час. с количест-
венным выходом получен трифенил висмут [224].
В случае ди-о-карбоксифенилртути обмен в эфирной среде проходит с
трудом и реакцию удается провести лишь в отсутствие растворителя [221].
При реакции дифенилртути и (a-CiQH7)BiBr2 получена [224] главным
образом бромистая фенилртуть, некоторое количество (a-C10H7)2Hg и
(C6Hs)3Bi и весьма незначительное количество, возможно, a-Ci0H7Bi(C6H5)a.
При реакции (C6H5)2Hg с (а-С10Н7)3В1Вг2 неожиданно получено не соеди-
нение Ar3BiAr2', а смесь органических соединений трехвалентного висмута
[(a-CiOH7)3Bi, (C6H5)3Bi, a-C10H7Bi(C6H5)2] и органических соединений
ртути [C6H5HgBr, (a-C10H7)2Hg] [224].
Образование ванадийорганических соединений. При взаимодействии
дифенилртути с VOC13 и VC14 в растворе циклогексана при комнатной
температуре образуются неустойчивые ванадийорганические соединения;
C6H5VOC13 иС6Н5УС12 [225].
Синтез органических соединений элементов
VI группы периодической системы
Действие хлористой серы испытано на меркарбидах. Из них хлорид
C2Hg6Cl6 не реагирует с хлористой серой при 120° С; основание C2Hg6O2(OH)a
с раствором S2C12 в бензоле, отщепляя часть ртути, дает соединение [225а]:
(CiHg)jC-C(HgCl)2
При реакции тионилхлорида с R2Hg разрываются одна (R = С6Н6
[226], C6H4N(CH3)2 [227]) или обе (R = р-С10Н7 [226]) С — Hg-связи.
В первом случае получены соответствующие RHgCl, продукты второй реак-
ции— р-С10Н7С1 и металлическая ртуть.
Галоидангидриды ар ил сульфокислот, например бензол- и толуолсуль-
фохлориды, с диарилртутью с незначительным выходом дают диарилсуль-
фо ны [228]:
2RSO2C1 + R'2Hg 2RSO2R'.
Хлормеркурацетальдегид с серным ангидридом в дихлорэтане (12-часо-
вое нагревание на водяной бане) образует с 41%-ным выходом сульфоук-
сусную кислоту (выделена в виде бариевой соли) [229].
Описано получение сульфопроизводных антрахинона действием на его
ртутные производные сульфирующих средств [59—62].
Диарилртуть (RC6H4)2Hg с арилсульфенхлоридами R'C6H4SC1 в хло-
роформе при комнатной температуре в темноте при 48-часовом взаимодей-
ствии образует несимметричные ди ар ил сульфиды RC6H4SC6H4R' [230].
Диферроценилртуть с роданом образует комплекс, переходящий при
обработке тиосульфатом натрия в диферроценилдисульфид с выходом 15%
[231].
При взаимодействии диарилртути с четырехбромистым селеном в серо-
углероде образуются диарилселениды
SeBr4 -I- 3R2Hg R2Se RBr SRHgBr.
294
Ртутноорганические соединения. Лит. стр. 336—352
В присутствии избытка SeBr4 происходит полное деарилирование ди-
арилртути 1232]:
2SeBr4 + 3RsHg 3HgBr2 + 2R2Se + 2RBr.
Из диферроценилртути и SeBr4 (растворитель — хлороформ, кипячение
в течение часа, последующая обработка тиосульфатом натрия) получен
Диферроценилселен с выходом 21% [231].
Диалкилртуть при осторожном нагревании с двуокисью селена дает
диалкил селен [233].
Галогениды диарилселена с диарилртутью реагируют по уравнениям
[234, 2351:
R2SeCl2 -f- R2Hg —> R2Se -f- RC1 -f- RHgCl
(в сероуглероде, ацетоне или четыреххлористом углероде на холоду),
RHgCl + RaSeCla^- RaSeCl-HgCh
(при нагревании в ацетоне или без растворителя).
Для разных арилов у соединений ртути и селена также получены сме-
шанные соединения селена ArSeAr' [236].
Действие хлористого теллура на дифенилртуть (при многочасовом на-
гревании до 200° С в запаянной трубке) неожиданно дает хлорбензол и
амальгаму теллура [237]
R2Hg + ТеС12 2RC1 + HgTe.
При кипячении RHgCl строения
оОх
с ТеС14 в СНС13 (для X = СН3) или в ацетонитриле (X = СООС2Н5)
получен соответствующий треххлористый арилтеллур [2381:
RHgCl + ТеС14 RTeCls + HgCl2.
Симметричные' (в случае Аг = Аг') и смешанные диарилтеллуриды по-
лучены (кипячение в диоксане в отсутствие влаги) по реакции [2391:
АгТеС13 + QHgAr' -» HgCl2 + АгАг'ТеС12 -> АгТеАг'.
Попытка получения органических соединений урана при нагревании
диэтилртути с четырехфтористым ураном (150—170° С) [240] была неу-
дачной.
Синтез органических соединений элементов
VII группы периодической системы
Получение иодониевых солей и иодидхлоридов. Весьма плодотворна
реакция Вильгеродта [241, 2421 — получение иодониевых солей при взаи-
модействии арилиодидхлоридов с диарилртутью (или ртутноорганической
солью):
C6H4JC12 + (CeH5)2Hg (CeH5)2JCl + CeHBHgCl,
C6H6JC12 + C6H5HgCl (CeHsWCl + HgCk.
(1)
(2)
Реакции ртутноорганических соединений
295
Применяют обычно реакцию (1); она идет гладко в мягких условиях
(при комнатной температуре, в водной взвеси или в растворе, в органическом
растворителе).
Получение хлористого дифенилиодония [241, 242]. 5 г дифенилртути растворяют с
5 г фенилиодидхлорида в небольшом количестве воды, заливают водой, встряхивают
12 час. и затем фильтруют. После упаривания фильтрата выкристаллизовывается хлористый
дафенилиодоний в виде игл. Т. пл. 230° С. Твердый отфильтрованный остаток содержит,
кроме CeHsHgCl.enxe двойную соль (CeH5)aJCl-HgCla. Кипячением с водой последнюю рас-
творяют, из фильтрата высаживают сероводородом ртуть; из упаренного фильтрата получают
новое количество хлористого дифенилиодония.
Так же легко происходит реакция обмена ртутноорганических аромати-
ческих соединений (обоих типов: ArHgX и Ar2Hg) с треххлористым иодом
12431: '
2ArHgCi 4- JC13 АгаJC1 + 2HgCla,
AraHg -+ JC13 AraJCl + HgCla.
Подобным же образом проходят реакции^ винильными ртутноорганиче-
скими соединениями. Эти реакции представляют собой простой метод син-
теза иодониевЫх солей. При избытке треххлористого иода и постепенном
введении в реакцию диарилртути можно осуществить синтез арилнодид-
хлорида:
AraHg + JC13 ArJCla + ArHgCl.
Эта реакция имеет препаративный смысл в случае винильного типа ртут-
ных соединений.
Получение двойной соли хлористого дифенилиодония и сулемы [243]. 2,5 г (0,007 моля)
дифенилртути заливают раствором 2 г (0,008 моля) треххлористого иода в разбавленной со»
ляной кислоте (1 : 10) при энергичном перемешивании (необходимо охлаждать). Через
15—20 мин. реакция заканчивается; осадок отсасывают, промывают спиртом и эфиром и пе
рекристаллизовывают из воды. Т.пл. 168—170° С; выход 2 г (50%); состав соли (C«H»)aJCI*
•HgCk
* Хлористая Р-хлорвинилртуть, как и ди-ф-хлорвинил)ртуть, при взаи-
модействии с JC18 или хлорвинилиодидхлоридом образует двойную соль
сулемы и ди-р-хлорвиниЛиодония [244].
Получение хлористого ди-р-хлорвииилиодоиия [244]. 26 а.(0,15 моля) транс-Р-хлорви-
нилмеркурхлорида заливают 30 мл 3%-ной соляной кислоты, сюда же постепенно прибавля-
ют раствор 17,5 г (0,075 моля) JC13 в 30 мл 3%-ной соляной кислоты. Реакция заканчивается
через 20—30мин., при этом появляются маслянистый слой и кристаллический осадок. Оса-
док тщательно отмывают горячим хлороформом от непрореагировавшего остатка С1СН =
=CHHgCl. Перекристаллизованный из воды продукт плавится при 149—150° С и имеет со-
став (ClCH=CH)2JCl-2HgCl. Выход 3,9 г. 2 г этого продукта растворяют в 300 мл воды,
раствор подкисляют соляной кислотой и насыщают сероводородом в течение 40 мин. Серни-
стую ртуть отфильтровывают, фильтрат упаривают в вакууме при температуре 30—40° С.
Вес кристаллического остатка 0,5 г (72%). Хлористый ди-р-хлорвинилиодоний, перекристал-
лизованный изводы, плавится с разложением при НО—112°С; образует пикрат; т. пл. 140° С.
С эквимолекулярными количествами хлористой хлорвинилртути и трех-
хлористого иода образуется [З-хлорвинилиодидхлорид.
Синтез транс-Р-хлорвииилиодидхлорида [245]. 1,32а (0,004 моля) транс-ClCH^CHHgCl
с т. пл. 124°С заливают 5 мл 16%-ной соляной кислоты, сюда при охлаждении добавляют
раствор 1 а (0,004 моля) JCI3 в 5 мл 15%-ной соляной кислоты. Смесь охлаждаю^ и встряхива-
ют 30 мин. Полученный иодидхлорид промывают 15%-ной соляной кислотой, спиртом и два
раза петролейным эфиром. Т. пл. 71° С. Выход 64%.
296 Ртутноорганические соединения. Лит. стр. 336—352
РЕАКЦИЯ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ОРГАНИЧЕСКИМИ ГАЛОГЕНИДАМИ
Взаимодействие ртутноорганических соединений с органическими гало-
генидами лишь в немногих случаях (для ртутноорганических соединений,
в которых С — Hg-связь не активирована сопряжением) и большей частью
в жестких условиях проходит подобно взаимодействию галоидных алкилов
с органическими соединениями активных металлов (лития, натрия,, иногда
магния) — с образованием углерод-углеродной связи по уравнениям:
R2Hg + 2R'X 2RR' + HgX2,
R2Hg + R'X-* RR'-т» RHgX,
RHgX + R'XRR'+ HgX2.
*
Так проходит реакция в тех случаях, когда под действием ртутноорга-
нического соединения от галоидуглеводорода не может отщепиться НХ.
Таковы реакции дифенилбромметана [2461 с дибутил-, дифенил-, ди-п-
толилртутью, идущие в ксилоле при температуре от 200° С (ди-ц-толил-
ртуть) до 340° С (дибутилртуть) и дающие R2R 'СН; реакция дифенилртути с
хлористым бензилиденом (при 150° С), дающая трифенилметан [2471; реакция
дифенилртути с бромистым'этиленом (запаянная трубка, 180—200°C,6— 10час.),
идущая с образованием бромистой фенилртути и р-фенилэтилбромида [2481;.
реакции n-хлормеркурдифенилового эфира с хлористым бензилом [249], хло-
ристой а-фурилртути са-хлорметилфураном[250] и реакции п-хлор мер ку рди-
фенилового эфира [249], хлористой фенил- [251], а-фурил- [250], а-тиенил-
[252], а-селенилртути [253], уксуснокислой а-тионафтенилртути [254],
диэтил- [255], дифенил- [251, 256, 257], дитолил- [257], ди-а-тиенил- [252,
258, 259] ртути с хлор ангидридами кислот, дающие кетоны. В присутствии
хлористого алюминия эти реакции протекают в более мягких условиях
[260, 2611.
Реакция хлористой фенилртути с хлористым бензоилом в присутствии хлористого алю-
миния. Получение бензофенона [261]. К смеси 3,2 г (0,01 моля) хлористой фенилртути и
1,7 г (0,01 моля и 20% избытка) хлористого бензоила при размешивании и охлаждении в те-
чение 30 мин. прибавляют 1,3 г (0,01 моля) безводного хлористого алюминия. Реакционную
смесь нагревают в течение 2 час. на водяной бане прн 50° С, затем разлагают льдом и перего-
няют с водяным паром. Получают 1,04 г бензофенона (выход 59%).
При проведении реакции в сероуглероде или в нитробензоле выходы ниже.
В некоторых же случаях при реакции с хлорангидридами кислот обра-
зования кетонов не происходит; так, при кипячении фтористой фенилртути
с хлористым ацетилом происходит выделение фтористого ацетила и с 90 %-‘
ным выходом образуется хлористая фенилртуть [262].
Диферроценилртуть при 4-часовом кипячении в бензоле с трифенилхлор-
метаном дает с 18%-ным выходом трифенилметилферроцен [231]; с гало-
идангидридами карбоновых и сульфоновых кислот реакция идет труднее,
при еще более продолжительном кипячении в бензоле — с образованием
(с незначительным выходом) ацетилферроцена и фенилферроценилсульфона
[2311.
При действии на меркурированный в a-положение фурфуриловый спирт
трифенилхлорметана в пиридине происходит разрыв С — Hg-связи и обра-
зуется трифенилметиловый эфир фурфурилового спирта [263].
В мягких условиях реагируют с галоидуглеводородами с образованием
С — С-связи лишь соединения, содержащие подвижную ртуть; так, дейст-
вие на ртутные производные оксосоединения трифенилгалоидметана на хо-
лоду приводит к трифенилметилацетальдегиду и трифенилметилацетону
[264—266].
Реакции ртутноорганических соединений
297
Получение трифенилметилацетальдегида [265]. Раствор 3 г трифенилбромметана в
25 мл абсолютного бензола прибавляют к 3 г сухого йеперекристаллизованного тщательно1
растертого броммеркурацетальдегида. Смесь нагревают 2 часа при 50—60° С и 4 часа кипя-
тят. Затем осадок отфильтровывают, бензол отгоняют. Получают 2,4 г сырого трифенил-
метилацетальдегида, плавящегося при 85—95° С. После многократной кристаллизации из
лигроина и затем из метилового спирта т. пл. 99—100,5° С.
n-Нитрофенилгидразон трифенилметилацетальдегида имеет (из толуола) т. пл. 245—
246° С.
Действие же на ртутные производные оксосоединений три-(п-нитрофенил)-
бромметана [2651 и хлорангидридов карбоновых [128, 264—267] (сульфоно-
вых [268], фосфорной [269], фосфористой [270], фосфиновых [269]) кислот-
направляется на кислород и приводит к виниловым эфирам соответствую-
щих кислот.
Взаимодействие хлормеркурацетальдегида с хлористым ацетилом (реакция с переносом;
реакционного центра). Получение винилацетата [266].
ClHgCH2CHO + СНзСОС!-------> СНзСОСН=СНа + HgCU
К 28 а (0,1 моля) хлормеркурацетальдегида в 15 лм сухого ксилола приливают раствор
7,8 г (0,1 моля) хлористого ацетила в 10 мл ксилола. Через 1 мин. реакционная смесь замет-
но разогревается. Через 1 час ксилольный раствор отделяют и твердую часть промывают
ксилолом. Из соединенных ксилольных растворов с 30-сантиметровым дефлегматором Вигрэ1
отгоняют фракцию 72—74° С. Выход 4,1 г (48%). Очистку винилацетата от последних следов
хлористого ацетила производят взбалтыванием с водной суспензией гидроокиси цинка. Вы-
сушенный и вторично перегнанный винилацетат имеет следующие константы: т. кип. 72—
74°С; 1,3960; d*° 0,9292.
Реакция хлормеркурацетальдегида с хлористым бензоилом идет много
медленнее, чем с хлористым ацетилом (6—8-часовое нагревание в ксилоле
при 50° С).
Взаимодействие хлормеркурацетоиа с хлористым ацетилом (реакция с переносом^
реакционного центра). Получение изопропеиилацетата [266].
CIHgCHaCOCHs + CHsCOCl-------► СН3СООС(СН3)=СН2 + HgCl2
К 16 г (0,05 моля) хлормеркурацетоиа приливают раствор 3,9 г (0,05 моля) хлористого-
ацетила в 30 мл сухого ксилола. Через 1 мин. заметно разогревание. Через час для связыва-
ния остатка хлористого ацетила прибавляют 2 мл хинолина. Твердую часть отсасывают и
дважды промывают ксилолом. Из соединенных ксилольных растворов с 30-саитиметровым
дефлегматором Вигрэ отгоняют фракцию 95—97° С. Вес 3,3 г (66%). При второй перегонке
изопропенилацетат получен со следующими константами: т. кип. 96—97° С; 1,4003;.
0,9127.
4
Так же по кислороду ацилируется продукт присоединения соли ртут»
к фенилэтинилметилкетону [271].
Эфиры меркур-бис-уксусной кислоты действием хлористых ацилов аци-
лируются как по углероду, так и по-обоим атомам кислорода [129]:
О
1 2 зУ
—Hg—СН2—С + R'COCl
\ 4'
OR
R'COCH2COOR
OCOR'
OR
R'COOR + CH2=C=O
I направление
II направление
III направление
Продукты того или иного направления реакции получены в зависимости
от природы R и R' [129].
Реакция метилового эфира меркур-бпс-уксусной кислоты с хлористым ацетилом [129f
(II направление) ^реакция с переносом реакционного центра). Получение а-метоксивииил-
ацетата. К раствору 34 г (0,1 моля) метилового эфира меркур-бис-уксусиой кислоты в 50 мл
298
Ртутноорганические соединения. Лит. стр. 336—352
•сухого хлороформа прибавляют в течение 30 мин. при перемешивании раствор 7,5 мл
(0,1 моля) хлористого ацетила в 10 мл хлороформа. Наблюдается незначительное саморазо-
гревание. Смесь нагревают при 50—55° С в течение 3 час. и оставляют на ночь. Отфиль-
тровывают выделившийся метиловый эфир хлормеркуруксусной кислоты, имеющий
т. пл.82—83° С; к фильтрату добавляют изопентан для более полного отделения ртутиоорга-
яического вещества. После фильтрования раствора и отгонки изопентана и хлороформа оста-
ток перегоняют в вакууме. Собирают 3,8 г (35%) а-метоксивинилацетата с т. кип. 79—85° С;
п2® 1,4121; d20 1,045.
Изобутиловый эфир меркур-бпс-ук.сусной кислоты дал продукт ацили-
рования по углероду — изобутил ацетоацетат, наряду с изобутил ацетатом—
продуктом ацилирования по эфирному кислороду [129] (см. также гл. VI).
При действии хлористого ацетила на этиловый эфир а-броммеркурфе-
нилуксусной кислоты образуется а-фенилацетоуксусный эфир (наряду с
О-ацильным производным фенилуксусного эфира, а также этиловым эфи-
ром фенилуксусной кислоты [272]).
При реакции арилхлорсульфидов с полнозамещенными соединениями
ртути один арил в последних заменяется на хлор [230]. Арилмеркурарил-
сульфиды [230] реагируют с арилхлорсульфидами (в четыреххлористом
углероде при комнатной температуре) по уравнению
ArHgSAr' + Ar'SCl - Ar'SSAr" + ArHgCl.
Действие на ArHgSAr' хлористого ацетила (в бензоле, при кипячении)
или р-СН3С6Н45СЦ также приводит к разрыву S — Hg, а не С — Hg-
связи и образованию соответственно ArHgCl и ArHgJ [230]. п-Толуолсуль-
фохлорид не реагирует с арилмеркурарилсульфидами даже прн кипячении.
Диэтилртуть с йодоформом (при 120° С) образует этилен и ацетилен
[273] наряду с иодистой этилртутью и иодистым этилом [273].
В то же время дифенилртуть, согласно Котону и сотр. [274], не реаги-
рует при 130° С с хлороформом и бромоформом; с йодоформом через проме-
жуточное образование несимметричного соединения C6H5HgCJ3 реагирует
по схеме:
(С6Н6)гН§ + CHJ3 -» СвН6 + C6H5HgCJ3 2C6HsHgJ + QJ4.
С хлороформом и четыреххлористым углеродом дифенил- [275] и дицик-
логексилртуть реагируют при температуре выше 260° С, но в присутствии
треххлористого висмута дифенилртуть при 5-часовом кипячении в хлоро-
форме дает хлористую фенилртуть, бензол и дифенил [276].
Реакция дифенилртути (и других R2Hg) с четыреххлористым углеро-
дом, инициируемая перекисями, течет иначе, чем эта же реакция, иниции-
руемая светом, а именно: в то время как при инициирующем действии све-
та продуктами реакции являются хлористая фенилртуть, хлорбензол и
гексахлорэтан [277, 278], продуктами реакции в присутствии перекисей
являются хлористая фенилртуть и бензотрихлорид [279]. При этой реак-
ции так же констатировано образование гексахлорэтана [279] (см. стр.
327, 328).
Взаимодействие дифенилртути с четыреххлористым углеродом в присутствии перекиси
ацетила [279]. 5 г (0,014 моля) дифенилртути, 15 г (0,097 моля) четыреххдористого углерода
н 0,1 г перекиси ацетила в запаянной трубке нагревают на масляной бане при 120—125° С в
течение 3 час. Охлажденную трубку вскрывают, в реакционную массу вводят еще 0,1 г пере-
киси ацетила, запаивают и вновь греют при той же температуре в течение 2 час. При вскры-
тии трубки наблюдалось давление. Продукты реакции нагревают до 30° С, образовавшийся
осадок феиилмеркурхлорида отфильтровывают и дважды перекристаллизовывают из ацетона.
Вес 2,2 г (54%), т. пл. 257° С. После охлаждения маточного раствора выпадает невошедшая
в реакцию дифенилртуть. После кристаллизации из ацетона т. пл. 124° С, вес 2 г, что соот-
ветствует 40% от взятого количества.
Освобожденный от хлористой фенилртути и дифенилртути продукт реакции подвергают
«фракционной перегонке из колбы Арбузова. После отгоики четыреххлористого углерода
Реакции ртутноорганических соединений 299
В приемник перешла бесцветная жидкость с т. кип. 86—90°С/18мм; 1,5565. Эта бесцветная
жидкость пронитрована. Выделенная м-нитробензойная кислота после кристаллизации из
воды плавится при 141°С, вес 0,25 г, что соответствует 18,7 %, считая на прореагировав-
шую дифенилртуть. Вместе с бесцветной жидкостью перегоняется гексахлорэтан, который
кристаллизуется в холодильнике; после возгонки плавится в запаянном капилляре при
185° С; вес 0,1 г.
Применение Несмеяновым, Борисовым и corp. [279а] этой реакции к
несимметричным ртутноорганическим соединениям
(С,НеСОО)2
RHgR' + CCU-------► RHgCl + R'CCU
позволило вывести ряд радикалов, построенный в порядке убывающего
сродства к свободному радикалу СС13: 2,4,6-(СН3)3С6Н2, а-С10Н7, р-СН3С6Н4,
о-СН3С6Н4, т-СН3С6Н4, С6Н6, С2Н5, С4Н9, С6Н5СН2, С6НИ, совпадающий
с рядом радикалов Хараша, построенным на основании убывающего срод-
ства их к протону в гетеролитической реакции с соляной кислотой.
Диизопропилртуть при 130° С в течение 10 час. в отсутствие кислорода
реагирует с CDC13, СНС13 и СС14, отрывая от них хлор и образуя изопро-
пилмеркурхлорид [281а] (ср. стр. 325: Aik, генерируемый из Alk2Hg,
отрывает от СНС13 водород), одновременно образуется ряд других продуктов
(ср. также стр. 325 этой главы).
Но описанный выше распад R2Hg(R = C2Ha) перекиси ацилов не ини-
циируют. Перекись бензоила с диэтилртутью (в азоте, 12 час., 70—95° С,
без растворителя) .образует C2H5HgOCOR (R =С6Н5, С2Н3С6Н4, СН3),
С2Н6, С2Н4 и очень небольшие количества Hg, СО2, /г-С4Н10, СвН6СООС2Н5
[280].
При нагревании диферроценилртути в абсолютном СС14 выделен хлор-
меркурферроцен (57%), ферроцен (22%) и смола, содержащая ртуть и
хлор (кроме С, Н и Fe) [281]. В присутствии гидрохинона или перекиси
бензоила не образуется ни смолы, ни хлормеркурферроцена. Предложен
цепной механизм этой реакции [28Г].
Йодистый аллил реагирует с диэтилртутью при 120° С с образованием
диаллила, иодистой этилртути и йодистого этила [274], но дифенилртуть
не реагирует с иодистым аллилом при кипячении в ксилоле в течение
300 час. 1246].
Особняком стоит легкое отщепление галоидной ртути при действии
бромистого аллила при —40° С на хлористую о-оксифенилртуть в пиридин-
ном растворе [282].
При действии галоидциана на ртутноорганическое соединение также
не образуется С — С-связи; иодциан в эфирном растворе с диметйлртутью
при 50° С дает цианистую ртуть, при 110°С—-иодную ртуть и метилизонит-
рил [283].
Бромистый циан не разрывает С — Hg-связи, а заменяет X в RHgX
на бром, как это показано на примере а-ацетатмеркур-0-метокси-р-фенил-
этана [284].
В немногих случаях ртутнооргайическое соединение реагирует с органи-
ческим галогенидом с отнятием галоидоводорода и образованием непредель-
ного соединения:
2RCH—CXR2 + R'2Hg -> 2R2C=CRa + HgX2 + 2R'H.
Так реагирует третичный бромистый бутил [246], третичный иодистый амил
[246] с дифенил- и ди-п-толилртутью; 1-бром-1,1,2-трикарбэтоксиэтан [246],
бромистый и иодистый неопентил [285] с ди-/г-толилртутью. Реакции идут
в незначительной степени и в жестких условиях: кипячение в течение 340 час.
300
Ртутноорганические соединения. Лит. стр. 336—352
в толуоле [246], многочасовое нагревание в запаянной трубке при 200° С и
выше [285].
9-Бромфлуорен реагирует по схеме
2RCHBr + 2R'2Hg R2C=CR2 + 2R'H + 2R'HgBr
с образованием бмс-дифениленэтилена [246].
При 340-часовом кипячении в толуоле ди-п-толилртути с стильбендибро-
мидом происходит отщепление галоида и образование непредельного соеди-
нения [246]
СвН6СНВгСНВгС6Н5 + RaHg CeH6CH=CHCeH5 + 2RBr + 2RHgBr.
В большинстве же случаев органические галогениды вовсе не реагируют
с ртутноорганическими соединениями [246] (18 из 32 испытанных RX) даже
при 300—350-часовом кипячении в толуоле.
Действие галоидных алкилов (йодистого метила) на продукты присоеди-
нения солей ртути к этилену характерно для этого рода веществ — оно при-
водит к выделению этилена [286, 287]. Действие галоидных ацилов на гало-
идную р-оксиалкил- [288] и 3-алкоксиалкил- [289] ртуть также ведет к выде-
лению непредельного соединения.
О реакции с галоидалкилами продуктов присоединения солей ртути к
олефинам см. также гл. VI.
Разуваев с сотр. [290] описал реакцию четыреххлористого углерода и
метилового спирта в присутствии дифенилртути.
О фотохимических реакциях ртутноорганических соединений с органи-
ческими галогенидами см. стр. 325.
Реакция дивинилртути с иодистым метиленом в присутствии цинка
или меди приводит к дициклопропилртути [64а].
ДЕЙСТВИЕ ВОССТАНОВИТЕЛЕЙ
НА РТУТНООРГАНИЧЕСКИЕТСОЕДИНЕНИЯ
(В ЧАСТНОСТИ РЕАКЦИЯ С МЕТАЛЛАМИ)
.Цикл реакций, выражаемый схемой:
у RaHg + М" у Hg + R„Mn- (1)
где М—атом металла, n-валентного в своих металлоорганических соединени-
ях, широко использован для получения металлоорганических соединений
разных металлов и для некоторых является главным, а иногда и единствен-
ным путем синтеза этих веществ.
Один из простейших случаев реакции (1) изотопный обмен R2Hg с мече-
ной ртутью рассматривается на стр. 320—323).
Взаимодействием диалкил (соответственно диарил)ртути с металлами
Li, Na, Be, Mg, Zn, Cd, Al, Ga, In, Sn, Bi, Те могут быть получены полные
металлоорганические соединения последних, а реакцией циклопентадиенил fa-
ных соединений ртути с Fe — ферроцен.
Для наиболее энергично реагирующих металлов (Li, Na) реакцию ведут
при комнатной или слегка (например нижеЮ0°С) повышенной температуре,
для других —при температурах 120—150—230° С, в расплаве, или, чаще и
удобнее, используя растворитель, например ксилол. Ароматические со-
единения ртути и в эту реакцию вступают легче, чем жирные, и последние ис-
пользованы гораздо уже. Реакция при нагревании осложняется распадом
RaHg Ra + Hg,
Реакции ртутноорганических соединений
301
настолько резко выраженным в случае дибензилртути и аналогичных соеди-
нений, что последнюю удается использовать для получения металлооргани-
ческих соединений тяжелых металлов только в редких случаях (Sn). Бен-
зилиллитий и бензилнатрий получаются нормально. Существен выбор
растворителя, который, с одной стороны, не должен взаимодействовать в
условиях реакции с ртутными соединениями, с другой —должен быть ин-
дифферентен к полученному металлоорганическому соединению,не говоря уже
о взятом металле. С точки зрения первого условия следует помнить, что
при высокой температуре ряд способных дегидрироваться веществ, какспирты
[292—294] (также содержащие D [299]), гидрохинон [292], тетралин [294],
гидразобензол [274], отдают R2Hg водород, превращая ее в RH и ртуть.
Источником водорода, восстанавливающим образующиеся радикалы, мо-
гут являться вторичные и третичные спиртовые группы, имеющиеся в орга-
нической части молекул ртутноорганического соединения [295]. Ртутноор-
ганические соли реагируютсо спиртами так же[296—298]—с образованием RH
и металлической ртути. Водород берется при этом не из гидроксильной груп-
пы спирта [299].
Кроме того, до RH и металлической ртути восстанавливает RHgCl и
R2Hg такжетриэтилсилан [300], LiAlH4 [21,99а, 301, 303], NaBH4 [302],
NaBH(OCH3)3 [105] (в случае трех последних восстановителей через проме-
жуточное образование LiAlR4 [303] или LiAlH2R2 и NaBR4 соответственно,
которые затем гидролизуют разбавленной кислотой). Реакции последнего
типа могут служить для элиминации ртути там, где это почему-либо нельзя
сделать при помощи-НС1.
Восстановление дифенилртути лнтийалюминийгидридом [301]. 3,5 г дифенилртути в
эфире обрабатывают избытком литийалюминийгидрида. Тотчас выделяется ртуть. Кипятят
30 мин. и ртуть отфильтровывают, вес ее 1,93 г (теоретич. выход 1,95 г). Эфирный раствор
промывают разбавленной серной кислотой, водой и сушат сульфатом натрия. Эфир отго-
няют через колонку Вигрэ, Остаток, закристаллизовывающийся при замораживании, пред-
ставляет собой бензол, плавится при +2° С.
Получение лактона 2,а-оксибицикло-(2,2,1)-гептан-6, а-карбоновой кислоты [302].
17 г лактона 5,0-хлормеркур-6, а-оксибицикло-(2,2,1)-гептан-2, а-карбоновой кислоты
в 50 мл смеси эфира с метанолом (1 : 3) и 3,8 г боргидрида натрия нагревают с обратным холо-
дильником 3 часа при перемешивании; после прибавления соляной кислоты продукт извле-
кают эфиром. Кристаллизация из легкого петролейного эфира. Выход 5,8 г (80%) Т. пл
154—155° С»
XHg-группа восстановительно замещается на Н в RHgX, в частности в
продуктах прцсоединения солей ртути к непредельным соединениям дейст-
вием таких восстановителей, как амальгама натрия в щелочном растворе,
гидразингидрат, иногда H2S, а также электролитически (см. гл. XI).
Получение циклогексан-Щ,4а-диола [302]»
CiHg ОН он
К кипящему раствору 10,7 г 3|3-хлормеркурциклогексан-1р,4а-диола в 85 мл раствора
едкого натра прибавляют 5,5 мл гидразингидрата и смесь кипятят 16 час. Продукт извле-
кают этилацетатом и кристаллизуют из ацетона. Выход 2,95 г (90%). Т. пл. 138,5—140° С.
Для дибензоата т. пл. 150—151° С.
302
Ртутноорганические соединения. Лит. стр. 336—352
Действие гидразина на меркарбид (при 4-часовом кипячении) приводит к
полной замене ртути на водород и образованию, согласно Гофману [46], эта-
на, согласно же Блешинскому и Усубакунову [47] — метана.
Несколько иного рода реакцией является замена на водород ртути в ди-
ферроценилртути. Так, при нагревании диферроценилртути в течение
15 час. в кипящем бензоле с металлическим натрием был выделен ферроцен
(выход 10%) [281]. Большая часть диферроценилртути в реакцию не всту-
пила.
При взаимодействии диферроценилртути с хлористым оловом в петро-
лейном эфире (15 час.) был выделен также ферроцен с выходом 15% и обна-
ружена металлическая ртуть. Большая часть ртути и в этом случае не про-
реагировала [281]. Как сказано выше, растворитель, в котором проводится
реакция ртутноорганического соединения с металлом, естественно должен
быть инертен к металлу и получаемому металлоорганическому соединению.
Это следует иметь в виду при синтезе металлоорганических соединений, реа-
гирующих с веществами, содержащими активный водород (группы ОН,
NH2), т. е. прежде всего при синтезе органических, соединений щелочных ме-
таллов.
Натрийалкилы, по данным Шорыгина [304, 305], реагируют, кроме того,
с ароматическими углеводородами (и гетероциклами типа фурана и тиофена
[306]) по схемам:
RNa + С6Нв CeHsNa + RH,
RNa + CeHsCH3 CeH5CH2Na + RH,
RNa + l^JI-*!^^ +RH.
S S Na
Эти реакции, ограничивающие выбор растворителей при синтезе алки-
лов щелочных металлов, с другой стороны, дают возможность синтеза ариль-
ных, аралкильных и гетероциклических производных щелочных металлов,
исходя из натрийалкилов, в свою очередь полученных при помощи ртутно-
органических соединений.
К реакциям, рассматриваемым в настоящем разделе, относятся найден-
ные Надь и Кочешковым [307] реакции хлористой арилртути с металличе-
ским оловом (метод синтеза тетраарилолова), а также реакция со сплавом
висмут—натрий [308], давшая дифенилртуть и трифенилвисмут.
В заключение следует упомянуть о том, что неметаллические элементы —
водород (при высоком давлении и температуре [292]), сера [237, 309, 310], се-
лен [237, 310, 311], теллур [237, 310, 421] и, наконец, галоиды — вступают
в такого же типа реакцию, забирая у ртутноорганического соединения его
радикал, с tqA, однако, разницей, что в случае серы, селена, теллура и га-
лоида ртуть связывается избытком этих элементов:
\ RaHg + 2Э-» R23 + Hg3
(где Э == S, Se, Те),
RaHg + Х2 RHgX + RX-----» RX + HgX2.
Гораздо менее изученным является переход радикалов из ртутноорганиче-
ских соединений в состав молекул восстановителей неметаллов. Сюда отно-
сятся предложенные Несмеяновым и Кочешковым [423] реакции хлористого
и бромистого олова с R2Hg и RHgX, являющиеся очень удобным методом
синтеза оловоорганических соединений типа R2SnX2, а также реакции ве-
ществ R2Sn и R3Sn — SnR3 с диарилртутью, сопровождающиеся переходом
радикалов на атом олова и образованием полных несимметричных оловоор-
ганических соединений R2SnR2 и R3SnR' [314].
Реакции ртутноорганических соединений
303;
К этому же типу относятся реакции двуиодистого германия с диарилрту-
тью, приводящие к ди-, три- и моноарилированным соединениям германия.
Подробности об области применения и условиях проведения этих реакций
см. ниже (в соответствующих разделах).
Почти не представляет сомнения, что принцип, положенный в основу
последних приемов синтеза, должен иметь более общее значение и что веще-
ства, подобные СгС12 или VC12, и другие достаточно сильные восстановители
также должны отрывать радикалы ртутноорганического соединения. Неиз-
бежной трудностью здесь явится, однако, выбор среды реакции, так как вода
и спирты, будучи способными отдавать свой водород под влиянием восста-
новителя, вероятно, обусловили бы отрыв радикалов R ртутноорганического
соединения в виде RH (ср. раздел о реакции ртутноорганических соедине-
ний с SnCl2).
Сюда же относится взаимодействие арсенобензола с диэтилртутью, дав-
шее фенилдиэтиларсин и ртуть [315, 316].
Действие металлов
без образования металлоорганических соединений
Металлы Си, Ag, Аи, редкие земли, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, U,.
Mn, Ma, Re, Co, Ni, Pa, Rh, Pd, Os, Ir, Pt и, по всей видимости, Ca, Sr, Ba,
Ra, Sc, Y не способны образовывать металлоорганические соединения при
помощи ртутноорганических соединений. Нагревание полнозамещенных
соединений ртути с некоторыми из них (Pt, Pd [294, 317], Ag [318—322})‘.
лишь катализирует распад
R2Hg^R2 — Hg,
иногда (Pd) резко понижая необходимую для него температуру.
Разложение дифенилртути металлическим палладием [317]. Смесь 1 г дифенилртути и
0,5 г металлического палладия, полученного высаживанием из его соли формалином, в за-
паянной пробирке нагревают в течение 12 час. при 100° С. После этого извлекают сначала
эфиром, затем бензолом. После испарения эфирных вытяжек получено 0,37 г (86%) дифени-
ла, после возгонки т, пл. 69° С. В бензольных вытяжках после испарения содержались
следы дифенилртути. Вес промытой эфиром и бензолом ртути 0,5322 г, распад состав-
ляет 94,2%.
Нагревание тетрамерной о-дифениленртути с порошком серебра до 300° С
дает дифенилен [320] наряду с трифениленом (9,5%) [318]:
304
Ртутноорганические соединения. Лит. стр. 336—352 \
Нагревание до 280° С в течение 10 мин. димерной о-терфениленртути с по-
рошком серебра в азоте дает трифенилен с выходом 71% [319J:
Так же из гексамер ной о-фениленртути и серебра (при 260° С) получен ди-
фенилен (выход 55%) [3181:
Нагреванием 3,4-бензодифениленртути с порошком серебра до 360° Св ат-
гмосфере азота получен 3,4-бензодифенилен с выходом 16,8% (выделен в
виде комплекса с 2,4,7-тринитрофлуореноном [321]):
Реакция же диферроценилртути с палладиевой чернью [3231 даладифер-
роценил с выходом только 1—6%. Диферроценил с выходом 43—45% (на-
ряду с ферроценом) получен при применении для разложения диферроце-
нилртути порошка серебра [322]:
Ag
1C5H5FeQiH4HgCsH<FeC5H5 —+ C5H6FeCsH<C5H4FeC6H5.
Образование диферроценила происходит при проведении реакции не ни-
же чем при 265° С [322] (8-кратный молярный избыток серебра, нагревание
17 час.); в отсутствие серебра при термическом разложении диферроценил-
ртути (265° G) образуется главным образом ферроцен [322]. Совместное раз-
.ложение диферроценилртути и диарилртути в присутствии серебра (мо-
. лярное отношение реагентов 1:1: 15, 235—300 °C, 17—22 часа) дало арил-
Реакции ртутноорганических соединений
305
ферроцены с выходами для Аг—фенил 45%, 2-бифенилил—6% ,3-бифенилил—>
22%, 4-бифенилил —20% [322]. ' :
Нагревание полимерного соединения (—CsH4FeCBH4Hg—стр. 251),
полученного симметризацией 1,Г-дихлормеркурферроцена, с порошком
серебра, до 300° С приводит к полиферроценилену-. наряду с очень. неболь-
шими количествами ферроцена и биферроценила [327];.
Получение дифенилеиа[^|| [320]. В приборе для возгонки нагревают на ме-
таллической бане до 296—300° С смесь 2,7 г.тетрамерной дифениленртути и 6 г порошка се-
ребра. В течение часа на охлаждаемом «пальце» выделяются кристаллы дифенилена, которые
время от времени удаляют. Выход 54%. Т. пл. 107—109° С. Хроматографирование раствора
вещества в петролейном эфире через колонку с окисью алюминия повышает т.-пл. до 109,5'—
1110 С. Из°спиртового раствора маточника с пикриновой кислотой получен пикрат.
При действии сплава никель—алюминий ацетат фенилртути уже в водном
щелочном растворе образует дифенил [324].
Синтез органических соединений лития и натрия
через ртутноорганические соединения
Полнозамещенные ртутноорганические производные жирных и аромати-
ческих углеводородов реагируют с Li и Na с образованием Li- и Na-алкилов
и арилов [325]:
• R2Hg + 2Li (Na) 2RLi (Na) + Hg. (1)
Реакции обычно ведут в лигроине, газолине, иногда в эфире. Синтез арот
матических и жирноароматических соединений ведут и в бензолу.
Вследствие крайней чувствительности к кислороду, влаге и углекислоте
простейших литий- и натрийалкилов реакцию (1) и (2) ведут в атмосфере
абсолютно свободного от кислорода и влаги чистейшего азота в специаль-
ной аппаратуре.
Для лития получены, например, этил- [325], я-пропил- [325], я-бутил-,
изоамил-, 3-я-гептил- [326] литий, виниллитий (реакции ведут в пентане [142]
или в эфире [158]), бутил ацетиленид лития из дибутил ацетиленида ртути и
лития в диоксане [328].
Описано получение фениллития в бензоле [325, 329] и эфире [359], о-, м-,
я-толиллития [330] (в эфире); о-дилитийбензол получен при многодневном
встряхивании гексамерной о-фениленртути и лития в эфире [318]; получен
также о-литийдифенил из тетрамерной о-дифенил ил ртути и лития [331];
2,2'-дилитий-о-терфенил из о-терфенилртути и лития [319].
В присутствии небольшого количества AlkLi происходит обменная реак-
ция [332] между органическим соединением ртути и органическим галоге-
нидом:
ч AlkLi
RaHg + 2R'J ZZ R'aHg + 2RJ,
через промежуточные стадии образования органического соединения лития
RaHg + 2R'Li R'aHg -j- 2RLi,j
2R'J + 2RLi 2RJ + 2R'Li,
выделенного в случае, например, R—бензил [354].
Для натрия получены метил-[325], этил-[333, 335, 337], Ятпропил-[325,
336,338] натрий, вторичнобутилнатрий (выделен после карбонизации в виде
306
Ртутноорганические соединения. Лит. стр. 336—352
метилэтилуксусной и метилэтилмалоновой кислот [339], амилнатрий [334],
октил- [325], бензил натрий [325].
При получении бензилнатрия и бутилкалия исходят из дитолилртути
(любой изомер); при нагревании первоначально образующегося толилнат-
рия (калия) происходит переход натрия (калия) в боковую цепь [338],
Винилнатрий и винилкалий получены из дивинилртути и соответствую-
щего металла в пентане [142]. Получен фенилнатрий [325, 341] (ср. [342]),
фурилнатрий [306]. При встряхивании R2Hg с RLi и щелочным металлом
(Na, Cs) в эфире, через промежуточное образование органического соедине-
ния щелочного металла, получены комплексы (R2Li)Me [330, 343] (R = Aik
и Аг). Для получения таких комплексов с калием требуется встряхивание со
сплавом KNa, но не с металлическим калием, который вытесняет литий из
его металлоорганических соединений [330].
Так как диметилртуть лишь медленно реагирует с металлическим Li с
образованием нерастворимого метиллития, выделяющегося по уравнению
(1) и загрязненного металлом, то метиллитий удобнее получать реакцией двой-
ного обмена растворимого алкиллития с диметилртутью
(CHs)2Hg + 2AlkLi 2CH3Li + Alk2Hg. (2)
раств. раств. нераств. раств.
При действии на ртутные производные альдегидов и кетонов растворами
лития и натрия в жидком аммиаке образуются винилаты этих щелочных ме-
таллов [344]: 7
Hg (СН2СНО)2 4- Me/NH3 Me/Hg + СН2 = СНОМе
Me '= Li, Na.
Фениллитий может быть получен как действием лития на дифенилртуть,
так и (экспериментально проще) взаимодействием дифенилртути с этилли-
тием.
Реакцией трансметаллирования хлормеркурферроцена этил-[345], я-бу-
тил- [322, 3451 и фениллитием [345] и диферроценилртути н-бутиллитием
[345] получен ферроцениллитий с выходом 90, 50, 58 и 43% соответственно.
Реакции ведут при 0° С в эфире.
Трансметаллирование 1,Г-б«с-(хлормеркур)ферроцена этиллитием в
эфире при комнатной температуре дало 1,Г-ферроценилдилитий с выходом
44% [345] (выходы рассчитаны по образованию карбоновых кислот после
карбоксилирования).
При проведении ряда синтезов при помощи натрийррганических соедине-
ний нет необходимости изолировать их из реакционной смеси. Так, Шоры-
гин [346], вводя вторую компоненту реакции в смесь диалкил ртути и нат-
рия, осуществил следующие синтезы:
1) (CaH&Hg + Na + СвН5СОСвН5 (CeHs)3C2HsCOH;
у 2) (C2Hs)2Hg + Na + CSHSCOOC,H5 - CeH5 (CiH5)2COH;
3) (CjH8)2Hg + Na + CeH5CHO -+ CeH5C3H5CHOH;
4) (CjHi&Hg + Na 4- COa -> CH3CH2COOH;
5) (CH3)2Hg 4- Na 4- CO2 -> CH3COOH;
6) (i - C5H11)2Hg 4- Na + CO2 -> i - QHeCHsCOOH.
Реакции (1—6) проводились в эфире.
При действии углекислоты на смесь натрия и диалкилртути (алкил—этил
и амил) в пентане или лигроине наряду с монокарбоновой кислотой получа-
ется алкилмалоновая кислота [334].
Реакции ртутноорганических соединений
307
Если со смесью диалкилртути и натрия работать в ароматическом угле-
водороде, то в реакции принимает участие растворитель и при действии угле-
кислоты при комнатной температуре образуется арил карбоновая [304], при
низких температурах также и дикарбоновая [334] кислота (в бензоле смесь
изо- и терефталевой кислот [334]). При работе с гомологами бензола угле-
кислота вступает в боковую цепь: так, толуол дает фенилуксусную [304],
при низких температурах также и фенилмалоновую [334], л-ксилол —
л-толилуксусную [304], этилбензол—гидратроповую {304], о-ксилол—о-то-
лилуксусную [305], л-ксилол — п-толилуксусную [305], мезитилен — 3,5-
диметил фенил уксусную [305], дифенилметан — дифенилу ксусную [305],
л-цимол — л-гомокуминовую [305] кислоты.
При ведении реакции в тиофене, фуране, а-метилфуране, а-метилтиофене
с большим выходом, чем в случае изоциклических углеводородов, образует-
ся а-тиофенкарбоновая [305], сс-фуранкарбоновая [306], 2,5-метилфуранкар-
боновая и 2,5-метилтиофенкарбоновая [306] кислоты.
При действии натрия на диэтилртуть в простых эфирах последние раз-
лагаются образующимся этилнатрием, давая алкоголяты натрия [3471, на-
пример:
CaHgNa -f- (С2Н5)2О —> C2HsONa -4~ GaHe С3Н4,
Синтез органических соединений металлов
II группы периодической системы
Бериллий, магний; цинк и кадмий реагируют с полнозамещенными сое-
динениями ртути по уравнению
R2Hg + Me R2M.e 4- Hg
с образованием полнозамещенных металлоорганических соединений. Это яв-
ляется наиболее удобным способом получения в индивидуальном состоянии
полнозамещенных алифатических соединений магния, ароматических соеди-
нений'бериллия, магйия и цинка. Реакции следует вести в атмосфере инерт-
ного газа (азота, аргона, а при получении органических соединений цинка —
можно и в атмосфере углекислоты). При нагревании диалкилртути с метал-
лическим бериллием в запаянной трубке при 130° С получены диметил-
[349 — 353], в том числе содержащий Be7 [351], диэтил- [355, 356] (ср. [357]),^
ди-н-пропил-, ди-я-бутилбериллий [355, 356].
Описана аппаратура для синтеза диметилбериллия (не в запаянной труб-
ке) [351]. Для получения диарилбериллия требуется нагревание до более
высокой температуры и добавление катализатора: дифенил- и ди-п-толил-
бериллий получены нагреванием диарилртути с эквивалентным количеством
бериллия в присутствии следов сулемы в запаянной трубке до 225° С в те-
чение 6 час. [357], дифенилбериллий, кроме того, в присутствии ВеВг2
(170° С, 72 часа) [358] и нагреванием дифенилртути и бериллия в ксилоле
при 150° С в течение 72 час. [359, 360].
Получение днфеннлбернллня (359, 360]. 9 г дифенилртути, 0,8 г бериллия и 25 мл су-
хого ксилола нагревают в запаянной трубке (все манипуляции под азотом) 72 часа при 150° С.
Ксилол отгоняют в вакууме, растворяют дифенилбериллий в 50 мл абсолютного эфира, от-
фильтровывают от амальгамированного бериллия. При сгущении и охлаждении эфирного
раствора выпадают кубические кристаллы, плавящиеся при 28—32° С в желтоватую жид-;
кость. Препарат теряет эфир только при 130° С в вакууме и тогда плавится при 160—165° С
с разложением. Выход 86% 13601. ‘
308' Ртутноорганические соединения. Лит. стр. 336—352
, При нагревании диалкилртути с магнией в запаянной трубке в течение
нескольких часов получены диметил-(120—130° С, 36 час. (3611; 95—105° С,
24 часа., трижды возогнанный Загний [361а]),диэтил- [3611 (согласно Гил-
ману [3621 лишь в присутствии сулемы), ди-н-бутил- (нагревание в течение
М) час. [3631), ди-(2-метилбутил)-[3641 магний. Реакцией R2Hg с Mg (24 часа,
115—120° С, запаянная трубка, в атмосфере азота) получены без выделения
их из эфирных растворов (C2H5)2Mg с выходом 81 %, (C3H7)2Mg с выходом
89%, (f-C3H7)2Mg с непостоянным выходом [348]. При нагревании дифенил-
ртути с магнием в течение 5—6 час. до 200° С [342, 365, 3661 (в присутствии
амилацетата до 180—185° С), лучше в присутствии небольшого количества
сулемы как катализатора [3671, получен дифенил магний. Диэтил и дифенйл-
магний получаются также при продолжительном встряхивании эфирного
раствора R2Hg с магнием [368].
Нагреванием Диалкилртути с металлическим цинком в запаянной труб-
ке до 120—130° С в течение нескольких часов или при продолжительном
нагревании на водяной бане получены следующие полнозамещенные цинк-
органические производные простейших алифатических углеводородов: ди-
метилцинк (нагревание в течение 24 час. до 120° С; также, при кипячении
диметилртути с гранулированным цинком) [3681, диэтилцинк (100° С,
36 час.) [369], ди*-н-пролилцинк [370, 371], диизобутилцинк (120—130° С
[372, 373], 36 час. [373], на водяной бане 60 час.) [374, 375], диизоамилцинк
(130° С, 36 час.) [369].
Простым и удобным методом синтеза полнозамещенных ароматиче-
ских соединений цинка, позволяющим, в частности, получать цинкарилы с
заместителями в бензольном ядре, является кипячение диарилртути в кси-
лольном растворе с металлическим цинком (Кочешков, Несмеянов, Потро-
сов [376]). Так получены дифенил-, ди-/1-фторфенил-, ди-п-хлорфенил?,
ди-о-толил-, ди-п-диметиламинофенил-, динафтилцинк. С дифенил-, ди-п-
хлорфенил-иди-п-диметиламинофенилцинком реакция заканчивается за 2—3
часа. Ди-р-толилцинк требует для своего получения 5—6-часового нагрева-
ния. Совершенно не реагирует с цинком даже при нагревании до 210° С в.
запаянной трубке ди-л-карбэтоксифенилртуть. Ди-и-бромфенил- и ди-п-
иодфенилртуть, не реагируя с цинком при кипячении в ксилольном раство-
ре, при нагревании с цинком в ксилоле до 220—2301’ С в запаянной трубке
разлагаются, не давая цинкорганических соединений. При нагревании ди-
бензилртути с цинком как в ксилоле, так и без растворителя до 110° С об-
разуется дибензил.
Получение дифенилцинка описано также Гилманом [377] и Хилпертом
1342].
. Диэтилртуть с металлическим кадмием (100—130° С) дает смесь диэтил-
кадмия и диэтилртути [369]. Лёру [361] не удалось выделить (200° С, за-
паянная трубка) при нагревании диметил- и диэтилртути с металлическим
кадмием кадмийорганических соединений. Образующийся при взаимо-
действие дифенилртути и- металлического кадмия дифеннлкадмий не смог
быть выделен из изоморфной смеси его с дифенилртутью [342]..
О получении дйфенилкадмия см. [378].
Синтез органических соединений металлов
III группы периодической системы
Полнозамещенные соединения ртути реагируют с алюминием, галлием и
индием по уравнению
3R2Hg ф 2Ме -^ 2RsMe + 3Hg,
что является методом синтеза металлоорганических соединений этих метал-
лов. Впервые получены этим путем индивидуальные полнозамещенные али-
Реакции ртутноорганических соединений
309
фактические соединения алюминия, галлия и-индия и ароматические соеди-
нения алюминия.
При нагревании диалкилртути с алюминиевыми стружками в запаянной,
наполненной азотом или эвакуированной трубке до 100—120° С в течение
многих часов получены следующие алифатические соединения алюминия:
триметилалюминий [379—383], триэтилалюминий [379, 381^385] (также
при кипячении диметилртути с А1 |*368а]), три-н-пропилалюминий [5, 356,
370, 382, 384], триизопропилалюминий [384] (это вещество в действительно-
сти является [386] смесью нормального и изотрипропилалюминия.так как вто-
ричные А1к3А1 при нагревании перегруппировываются в первичные А1к3А1),
смесь тривторично- и три-н-бутилалюминця [386], исходя из дивторичнобу-
тилртути [386], три-н-бутилалюминий [356], триизоамилалюминий [382,
3871. . .
(-]-)- [C3H7CH(CH^CH2]2Hg и соответствующая рацемическая R2Hg при
нагревании в течение 3 дней с. алюминием в изооктане дает (+)-R3Al и ра-
цемический R3A1 соответственно [353].
Из гранулированного металлического алюминия и дивинилртути в пен-
тане получен свободный от растворителя Тривинилалюминий [149, 388].
Реакция литийалюминийгидрида с избытком дивинилртути дает продукт
LiAl(C2Hg)4 [149, 388].
Действие LiAlH4 на 4-камфилмеркурхлорид в абс. эфире (5 час. при
0°С, затем 12 час. при 25° С) после разложения водой дало камфан (выход
19%) [21].
Реакция A1C13-N(CH3)3 (1 моль) в эфире с дивинилртутью (1,75 моля)
дает (СН2 — CH)3A1-N(CH3)3 [388]. Из 1 моля (CH2=±=CH)2Hg и 2 молей
A1H3.N(CH3)3 получен (СН2 —CH)A1H2-N(CH3)3 [388].
При взаимодействии же RHgCl (R = CsHn, 6 час.; $ -С4Н9, 15 час.) с
хлористым алюминием в бензоле проходит реакция Фриделя—Крафтса и про-
дуктом реакции является алкилбензол (выход 75%) и металлическая ртуть
(в первом случае также Hg2Cl2). [105].
Взаимодействие диперфторвинилртути с алюминийгидридтриэтиламином в
эфире даеттрпс-(перфторвинил)алюминийтриметиламин(СЕ2=СР)3А1Ь1(СН3)3
[389].
Получение литийалюминийтетравинилата [388]. На 7,6 г (0,02 моля) LiAlH* (100 меш,
кристаллический) в 100 мл сухого эфира конденсируют 11,4 г (0,045 моля) дивинилртути.
Реакционную смесь оставляют медленно нагреваться до комнатной температуры, перемеши-
вают 16 час., оставляют на ночь и затем фильтруют в атмосфере азота. За это время выдели-
лось 0,0399 молей водорода. Эфир отгоняют в вакууме и остающийся растворимый мягкий
осадок извлекают несколькими порциями бензола. По охлаждении выделяется белый оса-
док, его отфильтровывают и сушат в вакууме.
Перекристаллизация из бензола, тонкие белые иглы с т. разл. 180° С. Вещество пиро-
форно и горит на воздухе, разбрасывая искры.
Из ароматических полнозамещенных соединений алюминия были
получены при взаимодействии диарилртути с алюминием трифенилалю-
миний [377, 390—393] (также содержащий С14 [394]) и три-п-толилалюми-
ний [395].
Кипячение диарилртути с металлическим алюминием в ксилоле или дру-
гом индиферентном растворителе, например толуоле, октане, в некоторых
случаях декалине, является методом синтеза ароматических соединений алю-
миния (Несмеянов, Новикова [396]). Так получены трифенил-[396, 397],
три-о-толил-, три-л-толил-, трипсевдокумил-, три-о-анизил-, три-и-фтор-
фгнил-, три-п-хлорфенил-, три-а-нафтилалюминий, три-п-бифенилалю-
миний [396]. При кипячении ди-о-фенетил- и ди-р-нафтилртути с метал-
лическим алюминием в октаново-нонановой фракции синтина образуются
вязкие коллоидные растворы, из которых алюминийорганические соединения
310 Ртутноорганические соединения. Лит,- стр. 336—352
не могли быть выделены., Ди-о-хлорфенил- и ди-п-диметиламинофенил-
ртуть при кипячении с металлическим алюминием как в ксилоле, так
и в нонане разлагаются, не давая алюминийорганических соединений. Со-
вершенно не реагируют с металлическим алюминием при кипячении не толь-
ко в ксилоле, но и в декалинеди-п-анизил-, ди-и-фенетил-, ди-и-карбэтокси-
фенил-, ди-п-карбметоксифенилртуть [3961.
Переход радикалов от ртути к алюминию совершается также при взаимо-
действии литийалюминийгидрида при комнатной температуре в эфирном
растворе с дифенилртутью, приводящем к ЫА1Н2(С6НВ)2 и LiAl(CeH5)4,
идентифицированным по образованию с сулемой хлористой фенилртути [303].
Разложение реакционной смеси «кислотой дает бензол [301].
Из металлического галлия и диметилртути (в присутствии металлической
ртути) при нагревании до 110° С в течение 91 часа в запаянном сосуде поду-
чен с хорошим выходом триметилгаллий [398].
Эту же реакцию, в присутствии следов сулемы, проводили [399] и не в
запаянной трубке кипячением в течение нескольких часов.
При продолжительном нагревании диэтилртути с избытком металличе-
ского галлия до 165° С в запаянном сосуде в атмосфере азота получено в
индивидуальном состоянии первое полное металлоорганическое соединение
галлия — триэтилгаллий [4001.
В то же время тривинилгаллий получен из дивинилртути и избытка ме-
таллического галлия при 24-часовом взаимодействии в атмосфере азота при
комнатной температуре. [401].
Из дифенилртути и металлического галлия в условиях синтеза триметил-
и триэтилгаллия (запаянная трубка, 170° С, 50 и лучше 75 час. в атмосфере
азота) получен [393, 4021 трифенилгаллий. Т. пл. 166° С [402].
Взаимодействием металлического индия с диметилртутью в присутствии
небольшого количества сулемы и, лучше, сулемы й следов йода и магния
[403] при 100° С в атмосфере углекислоты получено полное органическое
соединение индия — триметилиндий [404].
Нагреванием дифенилртути и металлического индия (130° С, 50 час.)
в запаянном сосуде в атмосфере СО2[405] или, лучше, атмосфере азота [393,
406] получен с выходом до 80% трифенилиндий. Т. пл. 208° С [406].
Нагреванием 1,5 г металлического индия и 2,25 г дифенилртути до 270° С
в атмосфере азота при давлении 17,2 мм в течение 30 мин. после охлаждения,
извлечения эфиром и испарения последнего было получено масло, затвер-
девающее при прибавлении петролейного эфира. После растворения в эфире
и высаживания петролейным эфиром получено вещество, не плавящееся до
290° С-, представляющее собой соединение окиси фенилиндия и трехокиси
индия СвНв1пО-1п2О8 [150].
Синтез органических соединений элементов
IV группы периодической системы
Получение органических соединений кремния. При взаимодействии
(CeHs)3SiLi с Ar2Hg (Ar= GeHB и р-СН3С3Н4) и C6HsHgBr (в тетрагидрофу-
ране при комнатной температуре) образуются (C6HB)3SiAr, [(C6H5)3SiI2,
(CeHB)3SiOH и выделяется металлическая ртуть [407].
Получение органических соединений германия. При взаимодействии GeJ2
с (CjH^jjHg и (n-C4H9)2Hg выделяется металлическая ртуть, в случае ди-
и-бутилртути выделено германийорганическое соединение, которому при-
писано строение [(C4H8)2Ge]2 [408].
Взаимодействие диарилртути с двуиодистым германием является мето-
дом синтеза ароматических соединений германия [409, 410]. Реакция про-
Реакции ртутноорганических соединений
311
ходит при непродолжительном кипячении эквимолекулярных количеств
GeJ2 и Ar2Hg в толуоле. Основными продуктами реакции являются диарили-
рованные соединения германия, порученные с выходом от 40 до 70% и яв-
ляющиеся единственными продуктами в случае орто-замещенных арилов.
В некоторых случаях образуются также ArgGeJ и ArGeJ3. Реакция осу-
ществлена для Аг—фенил, м-,п- и о-толил, о-,м-, n-хлорфенил, о- и п-бром-
фенил, n-анизил, о- и n-фенетил, а-нафтил,|
Взаимодействие дифенилртути с двуиоднстым германием [410]. Смесь 3,55 г (0,01 моля)
(CeHs)2Hg с 3,26 г (0,01 моля) GeJ2 кипятят 15 мин. с 30 мл высушенного над натрием толуо-
ла в колбе с обратным холодильником, снабженным хлоркальциевой трубкой. Выделение
серо-черного осадка мелкораздробленной металлической ртути наблюдается еще до закипа-
ния толуола, в то же время золотисто-желтый GeJ2 исчезает. Постепенно выделяются зеле-
новато-желтые хлопья. После охлаждения реакционной массы осадок отфильтровывают,
промывают 2 раза небольшими порциями толуола. При обработке осадка раствором KJ зе-
леновато-желтое вещество быстро исчезает. В фильтрате реакцией с аммиаком обнаружен
ион Hg4-*. После промывания остатка водой из него ацетоном извлекают 0,07 г CeHsHgJ.
Т. пл. 260—262° С. Затем 10%-ным .раствором едкого натра извлекают GeO2, который из под-
кисленного концентрированной соляной кислотой фильтрата высаживают сероводородом
в виде GeS2. На фильтре остаются капельки металлической ртути. Толуольный фильтрат
для разрушения ртутноорганнческнх соединений и извлечения неорганических нодидов ртути
взбалтывают в делительной воронке сначала с окрашенной иодом Ш-кислотой (уд. в. 1,7), до
прекращения обесцвечивания Кислоты, затем с бесцветной кислотой для извлечения раство-
ренного в:толуоле иода. После этого толуол отгоняют в небольшом вакууме при нагревании
на водяной бане. В остатке—светло-желтое масло, из которого при охлаждении выделяют
0,37 г бесцветного вещества с т. пл. 137—157° С. Перекристаллизовано из нитрометана, за-
тем из н-гептана. Т. пл. 155—157° С. Жидкую часть остатка после отделения (CeHaJeGeJ
разделяют перегонкой-в вакууме при 2 мм на две фракции: первая — т. кип. 152— 155° С,
0,9 г (светло-желтая жидкость); вторая — т. кип. 160—161,5° С, 1,55 г (светло-оранжевая
жидкость).
Первая фракция не закристаллизовывается при охлаждении; гидролизуется нагревани-
ем с 20%-ным едким натром (добавляют предварительно 15—20 мл спирта). Получают 0,16 г
нерастворимых в органических растворителях бесцветных оксидов (CeHg)2GeO. Т. пл. 283—
293° С. Из щелочного фильтрата при подкислении соляной кислотой получают 0,05 г жел-
товатых хлопьев. Вещество очищают повторным растворением в щелочи. Так выделяют
0,02 г ангидрида фенилгермаииевой кислоты. Вторая фракция при охлаждении закристалли-
зовывается. Получено 1,1 г желтоватого вещества; т. пл. 56—61° С. Перекристаллизация
из нитрометана (2 раза) и из н-гептана дает бесцветные кристаллы (C«H5)2GeJ2 с т. пл. 63—
65° С. Выходы полученных соединений в расчете на процент использованного фенила:
(CeHshGeJa — 39% (0,004 моля); (CeHs)3GeJ — 37% (0,0016 моля); CeHsGeJ» -г 0,5%.
Получение органических соединений олова. Металлическое олово вза-
имодействует с хлористой арил- и аралкилртутью в кипящем ксилоле по
уравнениям:
6ArHgClJ+[3Sn 3Ar2SnCl2 + 6Н, (1)
3Ar2SnCl2^ 2Ar3SnCl + SnCk, (2)
SnCl4 + Sn -» 2SnCl2 (3)
с образованием хлористого триарилолова [307]. Так, с выходами до 70% по-
лучены: хлористое трифенил-, три-п-толил-, три-п-карбэтоксифенил-, три-
бензилолово. При взаимодействий хлористой арилртути со сплавом олово—
натрий (15% натрия) в кипящем ксилоле в результате реакций (4—6)
наряду с гексафенилоловом получается тетраарилолово:
6ArHgCl + 3Na2Sn -+ 3Ar2Sn+ 6NaCl + 6Hg, (4)
3Ar2Sn ->• Ar3Sn —SnAr3 + Sn, (5)
ArsSn—SnAr3~->Ar<SnAr2Sn. (6)
Так получены тетрафенил-, тетра-п-толил-, тетра-п-хлорфенилолово [307].
312
Ртутноорганические соединения. Лит. стр. 336—352
Дифенилртуть с металлическим оловом в кипящем ксилоле с 28,6%-ным
выходом дает тетрафенилолово [307]; при проведении реакции без раство-
рителя в запаянной трубке при 200° С количественно образуется тетрафе-
нилолово [411] (ср. [342]). При нагревании дифенилртути в ксилольном рас-
творе со сплавом олово-натрий выход невелик [307].,
Аналогично стереоизомерные ртутноорганические соединения этилено-
вого ряда — хлористые цис-. и тра«с-(3-хлорвинил)ртуть — реагируют со
сплавом олово — натрий (в бензоле, 45—50° С) по уравнению:
4CHCl=CHHgCl + NaSn-* (С1СН=СН)зЗпС1 + СН=СН + 4NaCl + 4Hg
с образованием из цас-ртутного изомера хлористого три-1{ис-3-хлорвинилоло-
ва, а нзтранс-ртутного изомера — только хлористого три-транс-В-хлор-
винилолова [412]. puc-Изомер требует более продолжительного нагрева-
ния, чем транс-изомер. Взаимодействие ди-транс-(Р-хлорвинил)ртути с
металлическим оловом дает хлористое три-транс-3-хлорвинилолово наряду с
ди-тро«с-₽-хлорвинилдихлороловом и треххлористым 3-хлорвинилоловом
[412]. Образованию хлористого три-траис-Р-хлорвинилолова способствует,
как всегда, большая продолжительность течения реакции.
Оловоорганический-продукт взаимодействия 4^-Ди-(3-хлорвинил)ртути
с металлическим оловом не был выделен в индивидуальном состоянии, но
его рис-конфигурация доказана [412]. Взаимодействие цис- и транс-а-
хлормеркурстильбенов со сплавом олово —натрий влечет за собой лишь сим-
метризацию ртутных соединений; оловоорганических соединений при этом
не образуется [2281 (см. также гл. VI и ХШ).
Триэтилстаннан легко взаимодействует с эфирами меркур-бис-уксусной
кислоты (Луценко, Бауков, Хасапов [413]):
. О . . О О
(CaHslsSnH + Hg( СН2С^ I (C2H5)3SnCH2C^ 4-Hg + CHsC^
' OR'2 OR OR
(R = CHS, 7i-C3H7).
Реакция проходит при нагревании компонентов до 70° С (в азоте) и конт-
ролируется по выделению металлической ртути.
Гидрид триэтилолова реагирует с диэтилртутью (без растворителя,
5 час., 100°С) при любых соотношениях реагентов по уравнению [175]
(CaHsJaHg + г^Н^зБпН (С2Н5)б5п2 + Hg + гСаНв.
Но ср. стр. 289.
Получение пропилового эфира триэтилстаннилуксусиой кислоты [413]. 24,3 а пропилово-
го эфира меркур-бис-уксусной кислоты й 12,5 а триэтилстаннана нагревают в атмосфере азо-
та при температуре 75° С в течение 90 мин. Затем декантируют с осадка металлической рту-
ти (11,5 а;. 96%). При разгонке выделяют 3,1 а пропилового эфира уксусной кислоты (51%)
с т. кип. 99—101° C/743 almн 13,2 а (73% ) пропилового эфира триэтилстаннилуксусиой кисло-
ты с т. кип. 102—105°С/3,5 лии; n^l.4779; 1,2226.
Аналогично получен метиловый эфир триэтилстаннилуксусиой кислоты.
Выход 68%.
Несмеянов и Кочешков [423, 424] нашли, что взаимодействие диарилрту-
ти с SnCl2 и SnBr2 проходит по уравнению
R2Hg SnX2 -* R2SnX2 4- Hg (7)
Реакции ртутноорганических соединений
313
и может служить методом синтеза диарилдихлоролова. Реакцию ведут с без-
- водными SnCl2 и SnBr2 в среде сухого ацетона при температуре кипения по-
следнего. Процесс заканчивается в течение 0,5—1 часа. От слитого с ртути
- раствора отгоняют ацетон, остаток извлекают петролейным эфиром и из него
выкристаллизовывают искомое вещество. Так получены [423—425] дифенил-,
ди-п-толцл-, ди-а-нафтил-, ди-£-нафтил-, ди-п-хлорфенил-, ди-п-бромфенил-,
дй-п-иодфенил-, ди-м-карбэтоксифенилдихлоролово (соответственно дибром-
олово). В среде спирта только дифенилртуть и диарильные соединения рту-
ти, содержащие более электроположительный, чем фенил, радикал (в ряде
Хараша), реагируют нормально по уравнению (7). Остальные вступают в.
реакцию с заменой ртути на водород:
RgHg-f-SnX2 + 2CaHjOH-+2RHHg + (C2HsO)2SnX2. (8)
Соединения, включающие радикалы, еще более электроотрицательные,,
например n-аминофениЛ-, в среде ацетона отщепляют их в виде RH, что-
ограничивает применение метода [423]. Хлористая арилртуть также спо-
собна реагировать по двум основным направлениям:
2RHgCl -|- 2SnCl2 -* R2SnCl2 -f- 2Hg -|- SnCl<, (9
RHgCl + SnCl2;+C2H5OH RH+[Hg + GHsOSnCls (10>
Преобладание того или другого из двух основных направлений реакции
и в этом случае зависит от тех же приведенных выше условий, В то время как
дибензил ртуть достаточно гладко превращается в дибензилдихлоролово^
чисто жирных ртутноорганические соединения мало пригодны для превраще-
ния этим путем в оловоорганические, так как реакция идет крайне медленно
и неполно и сопровождается образованием вследствие побочной реакции
соли RHgCl, в свою очередь, почти вполне инертной к SnCl2.
Однако как транс, транс-, так и цис, цис-изомеры ди-(£-хлорвинил)рту-
ти уже на холоду гладко и быстро реагируют [426] с хлористым оловом в
спиртовом растворе, образуя два стереоизомера хлористого ди-(3-хлорви-
нилолова: твердый, имеющий т. пл. 77,5—78,5°С, и жидкий с т. кип. 101—
102° С/3 мм, $ 1,7494; 4° 1,5675.
Реакция транс,трс«£-ди-(0-хлорвивил)ртути с хлористым оловом.^ Получение
хлористого транс, /мраис-ди-0-хлорвинилолова [426].
С1 Н
[ V = С/ ) SnCl2
\hZ /а
Те (0,0063 моля) транс,щранс-ди-(0-Хлорвинил)ртути в 5 мл абсолютного спирта, слег-
ка подкисленного сухим хлористым водородом, и 1,2 г (0,0063 моля) безводного хлористого-
олова в 5 мл абсолютного спирта сливают вместе. Реакция начинается уже на холоду. Пр»
нагревании в течение 10 мин. при 45—50° С и стоянии в течение часа при комнатной темпе-
ратуре происходит почти количественное выделение металлической ртути (0,5 г; 97%).
После прекращения реакции прозрачный реакционный раствор декантируют с металличе-
ской ртути. Спирт отгоняют возможно полно при 50—60° С (водоструйный насос), затем при-
бавляют 10 мл петролейного эфира, с которым отгоняют остатки спирта. Остаток в колбе,
дважды перекристаллизованный из петролейного эфира, весит 1,05 г (58%). Хлористое
транс, транс-ди-0-хлорвннилолово — белое кристаллическое вещество с т. пл. 77,5—78,5° С,
при 135° С в капилляре желТеет.
Так же с сохранением конфигурации происходит образование бромисто-
го цис- и ягронс-ди-пропенилолова из бромистой цис- и /праяс-пропенилртути
и бромистого олова [427].
В то же время из стереоизомерных ртутных производных стильбена
только ijuc-a-меркур-бис-стильбен с SnCl2 в ацетоне образует двухлористое
ди-а-стильбенилолово наряду с треххлористым а-стильбенилоловом и цис-
314 Ртутноорганические соединения. Лит.. стр. 336—352
стильбеном. Транс-а-меркур-бцс-стильбен не реагирует с SnCl2 в спирте и
без растворителя в расплаве при 240—260° С [313]. деранс-а-Хлормеркур-
стильбен в спирте симметризуется под действием SnCl2, давая транс-а-
меркур-бг/с-стильбен [3131 . Интересно отметить, что ароматические ртутно-
органические бромиды реагируют с SnBr2 иначе, чем хлориды с SnCl2,
а именно [4231:
2RHgBr + SnBra -* R2SnBra + 2HgBr. (11)
Побочным процессом является здесь
RHgBr 4- SnBr2 -> RSnBr3 -4- Hg . (12)
Синтез хлористого дибензилолова [423]. 5,7 г (0,015 моля) свежеприготовленной
дибензилртути н 4,5 г (0,0225 моля) SnCl2 нагревают в 25 мл абс. спирта до кипения.
Реакция идет очень быстро и основная масса ртути выделяется уже в течение первых
минут. Для завершения ее реакционную смесь нагревают до кипения в течение 30 мин.
Выделяется 2,92 г металлической ртути. После отгонки спирта и обработки остатка
большим количеством петролейного эфира получают, после отгонки последнего, 5,56 г
вещества. Т. пл. 160° С. Выход близок-к теоретическому.
Границы применимости реакции ртутноорганических соединений с со-
лями двухвалентного олова расширены проведением этой реакции в инерт-
ных растворителях. Так, в то время как при применении в качестве раство-
рителя спирта или ацетона при проведении реакции цис- и /п/адяс-пропениль-
ных производных ртути с галогенидами олова не удается получить пропениль-
ных соединений олова, последние могут быть получены-с выходом 70—75%
при проведении реакции в петролейном эфире (и лигроине) [154]. При вза-
имодействии диизопропенилртути с бромистым оловом в петролейном эфире
при комнатной температуре и при нагревании до 65° С выделены двуброми-
•стое диизопропенилолово и тетраизопропенилолово [428]. Геометрические'
изомеры дипропенилртути реагируют с бромистым оловом с образованием
только двубромистого дипропенилолова [154]. Этим вариантом метода уда-
ется получить и жирные оловоорганические соединения [428]. В продуктах
реакции диэтилртути с хлористым оловом найдены двухлористое диэтил-
юлово и этилмеркурхлорид [428]. В реакции дифенилртути с хлористым оло-
вом в петролейном эфире при различных температурах получено неизменно
только хлористое трифенилолово [428], которое образуется, очевидно, про-
ходя через стадию (C6H5)2SnCl2, по схеме:
2|('CeH5)2SnCl2 + Hg (СвН5)2 2 (CeH6)3SnCl + HgCU.
Аналогично проходит реакция с бромистым оловом, но с меньшим выходом
бромистого трифенилолова [428]. Ди-n-, ди-о-толил- и ди-а-нафтилртуть
реагирует с двухлористым оловом с образованием только двухлористого
диарилолова [428]. Попытки синтеза ароматических соединений олова с та-
кими заместителями в бензольном ядре, как ОН и N(CH3)2, успеха не имели.
В случае реакции ди-о-оксифенилртути с хлористым оловом был идентифи-
цирован в небольшом количестве фенол, а с хлористой м-диметиламинофе-
нилртутью и дибензилртутью реакция не прошла [428].
Наряду с двугалоидным оловом для получения оловоорганических сое-
динений путем восстановления ртутноорганических могут применяться, по
Несмеянову, Кочешкову и Пузыревой[314], также оловоорганические соеди-
нения типа R3Sn— SnR3 и R2Sn; реакция.их с диарилртутью выражается
уравнениями:
R3Sn~SnR3+R'2Hg^ 2R'SnR3+ Hg, (13)
R2Sn -|- R2Hg —* R2SnR2 -|-Hg. (14)
Реакции ртутноорганических соединений
315
В реакцию (13) введено и жирное соединение ртути—метиловый эфир
меркур-блс-уксусной кислоты [413].
Получение гексаэтилдистаннана [314]. 40 г. триэтилхлоролова (полученного по Кочеш-
кову [430]) нагревают прн 130° С в течение 5 час. с 5 г порошкообразного металлического
натрия в 50 ли изоамилового эфира. Продукт выделяют фракционированием в токе азота.
Т. кип. 160°С/23 мм. Выход 70%.
Получение метилового эфира трнэтилстанннлуксусной кислоты [413].
О
(C2H5)3SnCH2C<^
ОСН3
10,5 г гексаэтилдистаннана, 8,7 г метилового эфира меркур-бис-уксусной кислоты.нагре-
вают в течение часа при 160° С в атмосфере азота. Затем декантируют с металлической ртути
3,4 г (68%). При разгонке выделяют 5,6 г (41%) метилового эфира триэтилстаннилуксусиой
кислоты с т. кип. 69—71°С/1,8 мм. 1,4835; d2® 1,2967.
Получение диэтилднфенилолова [314]. 8,5 г (0,05 моля) диэтилолова нагревают в течение
30,мин. с 17 г дифенилртути (0,05 моля) на масляной баие до 150° С. Реакционную массу пос-
ле охлаждения обрабатывают петролейным эфиром, раствор отделяют от ртути, раствори-
тель отгоняют и остаток фракционируют при 4 мм. Главная фракция 154—156 ° С является
диэтил дифенил оловом, выход 9 г (60%).
Синтез органических соединений металлов
V группы периодической системы
При нагревании металлической сурьмы с диметилртутью и следами суле-
мы в запаянной трубке при 150° С в течение 160 час. получена триметил-
-сурьма [414].
Опыты по взаимодействию диэтилртути с порошком висмута при 120—
140° С, проводившиеся еще Франкландом и Дюпца в 1864,г. [369],привели к
образованию триэтилвисмута.
Реакция дифенилртути с металлическим висмутом не проходит до конца:
при нагревании до 250° С в токе водорода дифенилртути и избытка висмута
получена смесь, содержащая 41% трифенилвисмута, 57% дифенилртути и
:2% дифенила [342].
При взаимодействии хлористой фенилртути и сплава висмут — натрий
(10% натрия) в кипящем ксилоле образуется дифенилртуть и ничтожное
количество трифенилвисмута [308].
Синтез органических соединений элементов
VI группы периодической системы
При нагревании диарил(алкил)ртути с серой, селеном и теллуром до вы-
сокой температуры (190—235° С) происходит реакция
Ar2Hg + 2Э - Аг2Э + Hg3
(Э= S, Se,Te).
Так получены диперфторэтил- [415], дифенил- [416, 417], ди-о-толил-
-(нагревание до 225—230° С) [310], ди-л-толил- [310] сульфиды.
Дибензилртуть с серой (при 100° С) образует не R2S, а лишь дибензил
Реакция вторичноперфторалкилртутных соединений с кипящей серой
является методом синтеза перфтортиокетонов, например [419]:
s ’
(CF3)2CF—HgCF(CF3)2 CF3-C (S) -CF3.
316 Ртутноорганические соединения. Лит. стр. 336—352
Подсобным же образом из бис-(1 -хлор-1,2,2,2-тетрафторэтил)ртути и се-
ры получен трифтортиоацетилфторид [4201 с выходом 80%.
Аналогично диарил сульфидам получены: дифенил- [237],ди-о-толил-(220°С>
[3101, ди-п-толил- (225° С) [3101, ди-а-нафтилселен (190ь С) [3111,
дифенил-(230° С) [2371, ди-о-толил-(225—235° С) [3101, Ди-п-толил-(225—
230° С) [3101, ди-а-нафтилтеллур [421].
Получение ди-о-толилсульфида [310]. 5 г ди-о-толилртути и 0,8 г тонко измельченной
серы нагревают в запаянной, наполненной углекислотой трубке в течение 12 час. до 225—
230° С (последние 4 часа до 235° С). Из реакционной смеси извлекают ди-о-толилсульфид.
эфиром, многократно перегоняют в вакууме, причем он закристаллизовывается й, наконец,
кристаллизуют из спирта. Т. пл. 64° С, т. кип. 175° С/16 мм.
Получение дифеиилселеиа [237]. 4 г дифенилртути и 1,79 г селена нагревают в течение-
4 час. при 220° С в запаянной, наполненной углекислотой трубке. Образуется слегка окра-
шенное масло. Выход 2,2 г (85%). После перегонки при 160—162° С/12 мм продукт бесцвет-
ный.
Получение ди-а-нафтилтеллура [421]. 4,5 г ди-а-нафтилртутн и 2,5 г порошка теллура,,
помещенные в небольшую колбу Аншютца, соединенную с водоструйным насосом, нагревают-
до 190—198° С/16,5 мм в течение около 8 час, После охлаждения затвердевшую массу пере-
гоняют с паром для освобождения от нафталина, собирающегося в верхней части колбы.
Остаток извлекают эфиром; эфирный раствор быстро фильтруют и испаряют. Остаток кри-
сталлизуют из спирта. Образуются блестящие коричневато-желтые листочки. Т. пл. 126,5° С.
Выход сырого продукта 53%. •
Описана неудачная попытка получения органических соединений урана
при нагревании диэтилртути с ураном (150—160° С) [240] (ср. стр. 294).
Синтез органических соединений элементов
VIII группы периодической системы
Продукт меркурирования циклопентадиена, представляющий собой,
смесь хлористой циклопентадиенил ртути и дициклопентадиенилртути, лег-
ко реагирует с порошком железа в тетрагидрофуране, образуя ферроцен
[422]. В противоположность обычным методам получения циклопентадие-
нильных соединений металлов при этом методе нет необходимости применять
безводные растворители.
Получение ферроцена [422]. 150 г продукта меркурирования циклопентадиена смешива-
ют с 30 г порошка железа и при энергичном перемешивании заливают 150 мл тетрагидрофу-
рана. Реакция наступает через несколько мгновений. Ее регулируют охлаждением холодной
водой (не охлаждающей смесью). После уменьшения самопроизвольного взаимодействия*
смесь кипятят 20 мин. на водяной бане. Затем выливают в 500 мл воды, в которой растворе-
но 5 г дитионита натрия, и извлекают петролейным эфиром (т. кип. 65—75° С). После отгон-
ки растворителей остается 12—15 г сырого ферроцена. Кристаллизация из этилового спирта.
Выход 24—30%, считая на циклопентадиен.
РЕАКЦИИ С ОКИСЛАМИ АЗОТА
Реакцией бромистого нитрозила с диарилртутью еще Байером [430а J
по уравнению
Ar2Hg 2NOBr -» 2 ArNO + HgBr2
были впервые получены нитрозосоединения.
Реакция полифторалкильных симметричных ртутноорганических соеди-
нений с хлористым нитрозилом приводит к полифторалкилнитрозосоедине-
ниям [140а]. В некоторых случаях она сопровождается заменой ртути на>
хлор, а при наличии атома водорода в полифторалкильной группе — заме-
ной водорода на хлор [140а]. Среда реакции — диметилформамид.
Ртутноорганические соединения не реагируют с NO.
Реакции ртутноорганических соединений
317
При действии,двуокиси азота на диарилртуть [431—433] или соль арил*
ртути [433] образуется нитрозобензол:
Ar2Hg + N2O4 ArNO + ArHgNO3,
ArHgX + N2O4 ArNO + HgXNO3,
а при действии на ртутноорганические соединения N2O4 в органических рас-
творителях образуются нитросоединения [434].
При взаимодействии диарилртути с трехокисью азота с 50 %-ным выхо-
дом образуется соль арилдиазония по уравнению [431, 432]:
Ar2Hg + N2Os -* ArHgNO3 + ArNaNO3.
От ртутноорганической соли чистая трехокись азота уже не отрывает
ртути.
При одновременном же действии трехокиси и окиси азота азотнокислый
фенилдиазоний образуется с 85%-ным выходом [435], считая по уравнению:
(CeH5)2Hg|+:4N2O3 2CeH5N2O3 + Hg (NO3)2.
В соответствий с этим смесь трехокиси и окиси азота количественно пре-
вращает соль фенилртути в .фенилдиазониевую соль [435].
При действии трехокиси азота на диметилртуть Бамбергер вместо ожида-
емого диазометана получил имидодигидроксамовую кислоту [4371:
HON = С—NH—С = NOH
НО . ОН
Получение азотнокислого феиилдиазоиия из дифенилртути [435]. В охлажденный до
15° С раствор 1,1г дифенилртути в 25 мл СНС1з пропускают ток окиси азота и при перемеши-
вании быстро приливают около 10 мл 10%-ного раствора N2O3 в СНС1з. Перемешивают и
пропускают окись азота в течение 30 мин., после чего раствор в четыре приема встряхивают
с 100 мл воды. Соединенные водные вытяжки содержат всю диа'зониевую соль. Выход фенил-
диазония, определенный после сочетания с р-нафтолом, по весу образовавшегося беизолазо-
3-нафтола, равен 85%.
В то время как не содержащая окислов азота азотная кислота, подобно
другим кислотам, заменяет ртуть в ртутноорганических соединениях на во-
дород [438], действие содержащей окиси азота дзотной кислоты на аромати-
ческие соединения ртути приводит к замене ртути на нитрозогруппу и обра-
зованию нитрозосоединений и продуктов их дальнейших превращенш
(в зависимости от концентрации азотной кислоты): диазосоединений (глав
ным образом в разбавленной азотной кислоте), нитросоединений [434, 436,
438—441], полинитросоединений [434, 436, 438, 4401, нитрофенолов [436,
438] и т. п.
Взаимодействие я-диацетоксимеркурбензола с разбавленной (около 40%)
азотной кислотой при 45° С в течение 1 часа 30 мин. дает в качестве единст-
венного продукта 2,4-динитрофенол [436]. Азотнокислые ос- и 0-нафтил-
ртуть при действии конц. азотной кислоты в присутствии окислов азота
образуют 2,4-динитро-а-нафтол. Первая образует также небольшое количе-
ство а-нитронафталина [442].
Действие азотной кислоты и окислов азота (N2O3) на хлористую бензил-
ртуть приводит в зависимости от условий к фенилнитрозо- и фенилнитроме-
тану или к дифенилфуроксану [443]. Действие нахлористую бензилртуть или
на дибензил ртуть NO2 и N2Q4 дает бензил нитрит и бензил нитрат [443].
Ди-н-бутилртуть и N2O4 при —5° С в хлороформе образует нитрозобутан
[443]. При действии на ароматические сульфо- и карбоновые кислоты и нитро-
соединения нитрозилсерной кислоты в присутствии солей ртути, по мнению
318 Ртутноорганические соединения. Лит. стр. 336—*352
авторов [444], через промежуточное образование ртутноорганических сое-
динений пр реакции Бамбергера образуются диазониевые соли.
Но ртутноорганические соединения, имеющие в a-положении к атому рту-
ти СО-группу, реагирую.т в мягких условиях, в средах с высокой диэлект-
рической постоянной с арилдиазонийборофторидами, с заменой атома рту-
ти на арилазогруппу [449].
РЕАКЦИЯ С КЕТЕНАМИ
Описано образование кетонов при взаимодействии кетена с R2Hg и RHgX
[445—447].
Реакция с кетеном квази комплексных соединений ртути — меркуриро-
ванных оксисоединений и меркурированных оксосоединений — приводит
к образованию с хорошими выходами (в зависимости от среды реакции) мер-
курированной уксусной кислоты или ее эфиров (см. об этом в гл. VI).
ВЗАИМОДЕЙСТВИЕ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С РЕАКТИВОМ ГРИНЬЯРА И ОРГАНИЧЕСКИМИ СОЕДИНЕНИЯМИ
ЛИТИЯ, НАТРИЯ, ЦИНКА, АЛЮМИНИЯ и ДРУГИХ МЕТАЛЛОВ *
При взаимодействии R2Hg (R = С6НВ, р-СвН4СН3, р-ВгСеН4, р-С1С3Н4
[448], СвН5СН2, З-иод-4-метоксифенил) с R'Li в эфире или смеси бензола с
петролейным эфиром происходит обмен радикалами в этих металлооргани-
ческих соединениях с образованием R2Hg и RLi [450, 451].
При реакции реактива Гриньяра с ртутноорганическим соединением типа
RHgX (при условии достаточно подвижного атома ртути, например стоящего
в a-положений к карбонилу) происходит замещение этого атома на галоген-
магнезильный остаток аналогично реакции Церевитинова — Чугаева за-
мены подвижного водорода на галогенмагнезил:
RHgX + CH3MgX -> RMgX + CH3HgX,
RH + CH3MgX -+ RMgX + CH4.
Так ведет себя, например, галогенмеркурацетофенон [452]:
CeH5CCH2HgX ч+ RMgX RHgX + C6H5CCH2MgX
(таутомерно с С6Н5С=СН2), который, вопреки более ранним данным Гринья-
I ।
OMgX
ра и Абельмана [453, 454], не обнаруживает реакций карбонильной группы.
Аналогична реакция с реактивом Гриньяра о-галогенмеркурфенола [455];
ОН
+ 2C2H5MgBr
HgCl
OMgBr
+C3He + C2H8HgCl.
MgBr
Менее подвижный атом ртути не затрагивается, например:
—С— + C2H5MgBr С—
О HgBr • н6с2 ОН^Вг
Ср. гл. II н XII.
Реакции ртутноорганических соединений
31»
Таково же действие бромистого фенилмагния на аллилфенилртуть[138]:
CH2=CHCH2HgCeH5 + CeHsMgBr CeH5MgCH2CH=CH2 + CeHsHgBr.
При реакции ди-(бромацетиленида) ртути с бромистым фенилмагнием
(вэфире) образуетсядифенилртуть и бромистый p-бромацетиленид магния, что
является одним из доказательств отсутствия квазикомплексных свойств в
ди-(бромацетилениде) ртути [456] (ср. действие бромистого фенилмагния на
хлористую /пранс-р-хлорвинилртуть, приводящее к образованию дифенил-
ртути и выделению ацетилена). В подобных реакциях передачи оптиче-
ски активного радикала (в дибутиловом эфире при 80° С):
R2*Hg + R'MgBr R‘R'Hg + R‘MgBr,
[гдеЫ' -R* = (+)C2H5C*H(CH3)CH2CH(CH3) и (CH3)2CHCH2CH2C*H(CH3)h
а также в реакции оптически активной диизогептилртути с рацемическим
изогептиллитием частично сохраняется конфигурация радикала [429].
В согласии с высказанным Кнунянцем, Стерлиным и сотр. [457] положе-
нием о том, что обмен радикалами в металлоорганических соединениях дол-
жен происходить тем легче, чем больше разница в электроотрицательности
обменивающихся радикалов, показана [144] значительная степень обмена
радикалами в реакциях:
(CF3)2Hg + CaH5MgBr—-(QiH^Hg (60%),
ТГФ
(CF3)2Hg + CF2=CFMgBr---- (CF2=CF)2Hg,
ТГФ
(CF2=CF)2Hg + CH2=CHMgBr—»(CH2=CH)2Hg (43%),
ТГФ
(CF3=CF)2Hg + CeII5MgBr-► (CeHe&Hg + CF3=CFMgBr.
Также еще Франкландом [459] при попытке получения несимметричных
полнозамещенных ртутноорганических соединений из диэтилцинка и иоди-
стой метилртути выделена лишь диэтилртуть и из диметилцинка и хлори-
стой этилртути — диметил- и диэтилртуть. Аналогично Гильпертом и Грют-
нером [460] из бромистого бензилмагния и бромистой фенилртути или
бромистой этилртути получена лишь дибензилртуть и из бромистого этил-
магния и бромистой фенилртути или бромистой нафтилртути—диэтилртуть.
Реакция
RaHg -|- R*Mg R*Hg + R2Mg
(в тетрагидрофуране, 25° С) протекает тем быстрее, чем больше разница в
стабильности R и R'. Однако различные диалкилмагниевые соединения реа-
гируют с (C6Hs)2Hg с одинаковой скоростью. По способности образовывать
связь с магнием (по убывающей стабильности) радикалы R располагаются в
ряд: С6Н5С=С>С6Н5>СН3>С2Н6>СН(СН3)2.
(/-С4Н 9)2Mg не вступает в обмен с (z-C3H 7)2Hg и (CeH5)2Hg в течение 5 час.
при — 20° С [458а].
Изучен обмен [99а]:
(C6Fs)2Hg + 2CH3Li 2GeF5Li+ (CH3)2Hg.
Обмен радикалами между диферроценилртутью и н-бутиллитием (эфир-
но-бензольный раствор, 25%) с образованием монолитийферроцена й ди-н-
бутилртути заканчивается за 10 мин. Избыток w-бутиллития, больший чем
320 Ртутноорганические соединения. Лит. стр. 336—352
4 : 1, не увеличивает степени обмена (73%) 1322]. Монолитийферроцен полу-
чен также из хлористой ферроцеиилртути и н-бутиллития [461].
Методом изотопного обмена показано [462, 463], что полнозамещенные
соединения ртути обменивают свои радикалы с органическими соединениями
натрия, магния, цинка, аЛюминия, кремния и олова. Так, диэтилртуть
[462] реагирует са следующими этильными соединениями металлов, содер-
жащими этильную гр у пПу, меченную С14 (в скобках условия реакции и про-
цент обмена): C2HsNa (комн, температура: 2,5 месяца, 9,5%;100°С: 5 час.,
19,9%), (С2НВ)3АГ (100° С; 5 час., 59,5.%). Диэтилртуть, содержащая С14,
тоже реагирует с диэтилцинком (65° С; 20 час.; 27%). (CD3)2Hg обменивается
радикалами с (CH3)2Mg (в тетрагидрофуране, 28° С, 8 дней) и с броми-
стым этилмагнием [361а]. Но между (CD3)2Hg и (CH3)2Cd или (CH3)2Zn,
[361а], а также между (CH3)2Hg и (CH3)2Cd или (CH3)2Zn, как показано
[368а] методом ядерного магнитного резонанса, обмена радикалами
не происходит.
С тетраэтилсвинцом при 100° С в течение 20 час. обмена нет. Не происхо-
дит обмена [463] радикалами также и у дифенилртути с трифенилалюми-
нием, меченным С14; не удалось выделить дифенилртути из реакционной сме-
си взаимодействия дифенилртути и фенилнатрия, меченного С14. В соответст-
вии с инертностью связи С — Si и относительной лабильностью связи
С — Sn обмен (C6H6)2Hg,. содержащей меченый С14, с тетрафенилкремнием
происходит на 0,06% (100° С, 20 час.); в этих же условиях тетрафенилолово
обменивается на 88% [463].
ИЗОТОПНЫЙ ОБМЕН РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С РАДИОАКТИВНОЙ РТУТЬЮ
И СОЕДИНЕНИЯМИ, ЕЕ СОДЕРЖАЩИМИ
Реутов и сотр. нашли, что как полнозамещенные, так и некоторые сме-
шанные ртутноорганические соединения вступают в реакцию обмена с
радиоактивной ртутью (Hg203):
RHgR + Hg* г* RHg*R + Hg, (1)
RHgX + Hg* # RHg*X + Hg, (2)
и с галоидной ртутью, меченной Hg203:
RaHg + Hg*X2 RaHg* + HgX2, (3)
RHgX + Hg*X2 RHg^X + HgX2. (4)
Изотопный обмен имеет место также между двумя полнозамещенными
ртутноорганическими соединениями, одно из которых имеет меченую ртуть:
RaHg + R2Hg* RaHg*-J-R'Hg. (5)
Он происходит между полнозамещенными и смешанными органическими со-
единениями ртути:
RaHg + R'Hg*X R2Hg*+RHgX, (6)
-а также между двумя смешанными ртутноорганическими соединениями, одно
.из которых имеет меченую ртуть:
R'HgX+ RHg*X^.RHg*X+R'HgX. (7)
Реакции ртутноорганических соединений
321
Из смешанных ртутноорганических соединений жирного и ациклическо-
го рядов в мягких условиях (на холоду, в бензоле или ацетоне) реагируют с
Hg203 только а-меркурированные оксосоединения [464, 465]. Для RHgX
реакционная способность к металлической ртути уменьшается в последова-
тельности: а-броммеркурциклогексанон > этиловый эфир а-броммеркурфе-
нилуксусной кислоты /-ментиловый эфир а-броммеркурфенил уксусной
кислоты > фенилмеркурбромид > З-броммеркур-З-бензилкамфора > 3-бром-
меркуркамфора. Проходящий на 100% за 15 час. обмен с Hg203 /-менти-
лового эфира а-бромфенилуксусной кислоты протекает без изменения
стереохимической конфигурации [465]. Хлористая цис- и тронс-р-хлорвинил-
ртуть, вступая в реакцию изотопного обмена с радиоактивной ртутью в аце-
тоне на холоду, также не меняют при этом своей стереохимической конфигу-
рации [466].
Ди -транс- и ди-ццс-хлорвинилртуть легко реагируют в ацетоне на холо-
ду с мелкораздробленной металлической ртутью. Изотопное равновесие
устанавливается за несколько часов [466]. Стереохимическая конфигурация
хлорвинильной группы при этом сохраняется в соответствии с установлен-
ным Несмеяновым и Борисовым правилом (ссылки см. стр. 123, гл. VI).
Ароматические полнозамещенные ртутноорганические соединения всту-
пают в реакцию изотопного обмена с металлической ртутью в мягких усло-
виях:
х—Hg—х + Hg* x-<Q-Hr-^>-x + Hg.
/
Скорость реакции существенно зависит от характера заместителя X, воз-
растая в ряду
NOs,С1<Н<СН8<ОСН3
(ряд получен при проведении реакции в пиридине при 60° С) [467].
Ди-м-анизилртуть реагирует с металлической ртутью уже на холоду в
бензоле: равновесие устанавливается за 16 час. [467]. Для дифенилртути
изотопное равновесие достигается в диоксане при 60° С за 2,75 часа, в кси-
лоле при 140° С — за 30 мин. [467].
Арилртутные соли реагируют с металлической ртутью быстрее, чем ди-
арилртутные соединения [468]. В этом случае электроположительные заме-
стители ускоряют, а электроотрицательные замедляют реакцию изотопно-
го обмена.
В реакции
RHgR' + Hg^Cla RHgCl + R'HgCl,
согласно Уинстейну, Трэйлору и др. [469], происходит статистическое рас-
пределение меченой ртути между двумя радикалами, отщепляющимися от
RHgR'. Однако эти работы, выполненные до установления обменов типа (5),
естественно, не учитывали этого осложнения и подлежат уточнению.
Реакция изотопного обмена между щгс-2-метоксициклогексилнеофил-
ртутью и Hg203Cla также происходит с сохранением конфигурации радикала
[469] (см. также гл. XIII, стр. 240). По распределению Hg203 между RHgBr
и R'HgBr в реакции Hg2o8Br2 с RHgR' (в эфире при —20° С для алифатиче-
ских соединений; в тетрагидрофуране — для ароматических) определена
[470] относительная полярность связи С—Hg в последних (HgBr2 атакует
более полярную связь С—Hg). Для алифатических радикалов прочность
этой связи увеличивается с длиной R(CSH7 ]> СаН5 ]> СН3), для соединения
ArHgAlk прочность связи со ртутью арила, могущего участвовать в сопря-
жении, больше, чем алкила [470]. В соединениях CeH5HgAr (Аг =
322
Ртутноорганические соединения. Лит. стр. 336—352
р-ВгС6Н4 и р-СН3ОСвН4) происходит расщепление связи со ртутью фенила
[4701. •
Данные Десси [471] о том, что в реакции фениЛэтилртути ,с Hg203Cl2
Hg203 в одинаковой степени распределяется между обоими радикалами, осно-
ваны, как показали Ник. Несмеянов и Реутов [4721, на эксперименталь-
ной ошибке; в действительности же происходит разрыв связи со ртутью
фенила.
Ртутноорганические соли, являющиеся а-меркурированными оксосоеди-
нениями, а также хлористые цис- и /прцнс-р-хлорвинилртуть — реагируют
с галоидной ртутью, меченной Hg203 (реакция (4)), в мягких условиях:
а-меркурированные оксосоединения — в бензоле, диоксане, этиловом спирте
при комнатной температуре или при 50° С [473], [3-хлорвинильные соли рту-
ти — в ацетоне на холоду. Реакционная способность а-меркурированных ок-
сосоединений уменьшается в ряду: а-броммеркурциклогексанон 3-бром-
мер кур-3-бензил камфора >• этиловый эфир а-броммеркурфенилуксусной
кислоты > Z-ментиловый эфир а-броммеркурфенилуксусной кислоты >
3-броммер кур камфора. Взаимодействие цис- и транс-8-хлорвинилмеркурхло-
рида [466] и цис- и шрйнс-2-метоксициклогексилмеркурхлорида [465] с
HgCi2 протекает с сохранением стереохимической конфигурации. В послед-
них случаях лучшей средой является диоксан и оптимальная температура
120—130° С [465]. Реакция (4) имеет второй порядок. Измерения проводи-
лись для R = СН3 (в спирте, при 100° С для X =С1 [474, 475], J [476];
при 60° С для X = Вг, СН3СОа [476], в смеси спирта, HNOS и следов воды
при 0°С для X = NO3 [476]), (в спирте, 100° С [475]); С3Н7(тоже
[475, 477]); s-C4H9 (X = Вг, в спирте при 60° С [476]); (СН3)3ССН2 (то же,
130° С [476]); CSH6CH2 (в хинолине [478]); С6Н6СНСООС2Н6 [479, 4801,
р-ХСвН4СН2 (в хинолине при 70° С [481]); С6Н5 (в ледяной уксусной кис-
лоте, 50 %-ном спирте, воде, бензоле [482], в водном бензоле [483]; в пиридине
изучение кинетики затруднительно из-за слишком большой скорости обмена
[475, 489]); р-НООСС6Н5 [485].
В случае R — s-C4H9 реакция (4) протекает с сохранением конфигурации
алкила. Сохранение конфигурации и характер кинетических зависимостей в
реакции (4), в частности, увеличение скорости обмена в ряду X =Br<^J <<
СН3СО2<< NO3 [4761, характер солевого эффекта [476] и незначительное
влияние стерических факторов, проявляющихся для R в порядке СН3 <
<первичный алкил < вторичный алкил [486], говорят в пользу механизма
S£2 для этой реакции. Однако механизм и порядок реакции HgM3Br2 с
а-броммеркурфенилуксуснойкислотой зависят от среды реакции [466]: в пири-
дине [487], диметйлформамиде [487а], водном спирте [487а, 488], бензоле
[487а] реакция имеет суммарный второй порядок (механизм 5^2); в диметил-
сульфоксиде [488а], диоксане [4876] — первый порядок (в последнем случае
механизм S,?l в исправление прежних данных [480]). Реакции, идущиешо
механизму ЗеЬ также проходят с сохранением конфигурации.
Скорбеть реакции HgZ03Br2 с замещенным в пара-положении бензил-
меркурбромидом зависит от природы заместителя, уменьшаясь в ряду
(СНд)2СН >• СН3 .> Н'> Cl > F [481 ]. Реакция протекает значительно быст-
рее в диметилсульфоксиде, чем в хинолине, катализируется добавкой КВг
[481а]. В этом последнем случае влияние заместителей в пара-положении на
скорость реакции иное [481а]. В (CH3)2SO она наибольшая для п-нитробен-
зилмеркурбромида [4886].
Скорость реакции изотопного обмена, например для реакции этилового
эфира а-броммеркурфенилуксусной кислоты с Hg203Br2, также зависит от
полярности растворителя: при 75° С в бензоле за 9 час. обменивается 52%
.сложного эфира, а в 70%-ном водном диоксане за 1,5 часа обмен достигает
89% 1480, 489].
Реакции ртутноорганических соединений
323
Изотопный обмен ртутноорганических солей с галогенидами радиоак-
тивной ртути ускоряется кислотами [482] и основаниями (диэтиЬамином.
пиридином) [473, 480].
Для реакции (4) установлено два типа катализа металла галогенидами
(LiX): одно- и двуханионный, в зависимости от числа участвующих в пере-
ходном состоянии ионов X, привносимых LiX [490]. При обоих типах ката-
лиза стереохимическая конфигурация оптически активных групп R (R =
s-C4H9) сохраняется [490]. Механизм катализируемой реакции (4)
В реакции (5) в случае R' = С6НБ и R = р-С1С6Н4 в пиридине при
60° С время полуобмена составляет 3 часа. R' = р-С1С6Н4 не вступает в реак-
цию изотопного обмена с R2Hg (R = СН2СНО) при 20° С в ацетоновом раство-
ре. Однако при взаимодействии (в случае R' = С6Н9 или р-СНдОСвН^
с R2Hg (в случае R ~ СН2СНО) изотопное равновесие устанавливается в
ацетоне при 20° С практически мгновенно [491].
Изотопный обмен в реакции (6) (для R' =СНа, СН(СН2)2, СН(СНа)2,
R — тот же оптически активный радикал, X = Вг, в этиловом спирте
при 60° С [492] и для R = s-C4H9, R' — оптически активный s-C4HB,
X = Вг в этиловом спирте при 35° С, X = СН3СОО и NO3 при 0° С [493])
сопровождается распределением оптической активности между обоими со-
единениями ртути и проходит с сохранением стереохимической конфигурации.
Кинетика этой реакции, изученная [493] в случае R = s-C4H9 (реакция би-
молекулярная), показала зависимость скорости реакции от полярности анио-
на (скорость реакци.и возрастает в ряду X = Вт < СНаСОО < NO3) и
наличия солевого эффекта. Сохранение стереохимической конфигурации и
кинетические данные указывают на механизм S е 2 для последней реакции
[493]. Второй порядок (первый по каждому из компонентов) имеет реакция в
случае R—изогептил [483а].
В системе C6H5HgBr — (CeH5)2Hg как в пиридине (при 100° С), так и без
растворителя в расплаве имеет место быстрый изотопный обмен [484].
Реакция (7) изотопного обмена между двумя ртутноорганическими соля-
ми — бромистой фенилртутью, содержащей Hg203, и этиловым эфиром
а-броммеркурфенилуксусной кислоты в пиридине, имеет суммарный второй
порядок (первый по каждому из компонентов) [494].
Весьма быстро устанавливается изотопное равновесие между аллилмер-
курбромидом и p-(CH3)2NC6H4Hg203Br [473а].
ПИРОЛИЗ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
При нагревании при высокой температуре ()> 300° С, в отдельных слу-
чаях ниже) ртутноорганические соединения разлагаются с выделением ме-
таллической ртути (и соли ртути, в случае разложения RHgX), полнозаме-
щенные соединения, согласно Приччарду [495], Рэсселу и Бернстайну
[496],— в одну стадию
RaHg 2R- + Hg
с переходом ртути из двухвалентного sp-состояния в состояние s2 нулевой ва-
лентности, или, что менее вероятно, по стадиям:
R2Hg —> RHg R ,
RHg-R’+ Hg.
Возможно, что тот или иной путь распада зависит от температуры [497].
Образующиеся радикалы R’ ведут себя так, как это свойственно радика-
324 Ртутноорганические соединения. Лит. стр. 336—352
лам в газовой фазе: димеризуясь, диспропорционируясь или реагируя с
осколками ртутноорганического соединения или с другими присутствующи-
ми в реакционном сосуде веществами, в частности с зеркалом металла 1498,
499]. Рассмотрение дальнейшего поведения этих образующихся радикалов
не входит в задачу настоящей книги. См. об этом соответствующую лите-
ратуру: пиролиз диметилртути [495, 496, 498—513, 515—5201, диэтилртути
[495,500,520—523], ди-«-пропилртути[495,524,525,529], диизопропилртути.
4495,497,526—529],диперфторизопропилртути [130],ди-н-бутилртути[529—
531], дивторичнобутилртути [531], (при 110° С [43]), дитретичнобутилртути
(при 40° С) [43], дивинилртути [524], цпс-ди-Р-хлорвинилртути [126], дици-
клогексилртути (в бензоле) [2а], дибензилртути [194, 532], дифенилртути
[248, 418, 495, 523,533], 2,2-дифёниленртути [580], хлористой я-пропилртути
[523], ди-(3-окси-3-фенилпропил)ртути (120—130° С, в вакууме) [534],
ди-(3-гидрокси-3-метилбутил)ртути (100—110°С) [534], хлористой цис-З-хлор-
винилртути [127], хлористой [523], бромистой [523], иодистой [535] и
л-винилбензоата [553] фенилртути. Качественно показано, что устойчивость
(XCfiH4)2Hg при 130—150° С зависит от природы заместителя X [536].
При происходящем пиролизе ксантогената (115—125° С/25 мм) и тиоциа-
ната (100—120° С/10 мм) винилртути получены виниловые эфиры соответст-
вующих кислот (в последнем случае смесь винилтиоцианата и вин'илизотио-
цианата) [514].
г Пиролиз бмс-о-иодфенилртути при 230° С (3,5 часа) дает о-дииодбензол;
трифенилен при 600° С — о-дииодбензол, бифенилен (25—64%) наряду с
HgJ3 и Hg2J2 [537].
бис-Пентафторфенилртуть не изменяется при нагревании до 250° С в
запаянной трубке в течение 5 час. [44].
Дибензилртути разложена в нитробензоле [532а] и нитрозобензоле
[5326].
Термический распад) дифенилртути в циклогексане [539] (260—280° С, в
ампулах за 150 час.) и C6De [539а] проходит на — 9% и дает частично иные
продукты, чем ее фотораспад, а, именно, в первом случае — ртуть, бензол,
циклогексен, фенилциклогексан и следы дифенила, и полимерное соедине-
ние (—С6Н4 — Hg—)п, в последнем случае при термораспаде преобладает
немеченый дифенил. Но термо- (280—300° С) и фотораспад дифенилртути в
толуоле происходит одинаково с образованием смолы, бензола (причем от-
рыв водорода фенилрадикалом происходит не из СН3-группы, а из аромати-
ческого ядра толуола), ртути и смеси метилдифенилов, содержащей орто-,
мета- и пара-изомеры [5396]. Разложение диферроценилртути (265° С) дает
ферроцен (68%) и диферроценил (13%) [322]. О термическом разложении
диферроценилртути в присутствии металлов см. стр. 304.
Изучено ускорение полимеризации винилацетата продуктами распада
дифенил- и дибензилртути [540].
Термический распад цкг-ди-^-хлорвинилртути [126]. 0,1178 г ццс-ди-р-хлорвинилртути
нагревают с количественным улавливанием выделяющегося ацетилена аммиачным раствором
хлористой меди, приготовленным по Илосваю. Выделение ацетилена начинается при темпе-
ратуре 100—105° С. Нагревание продолжают в течение часа. Выдувание ацетилена водоро-
дом из реакционной колбы продолжается в течение 40 мин. Титрование раствора образовав-
шейся ацетиленистой меди соответствует 99,12% ацетилена, содержавшегося во взятой
навеске. '
CeH6HgOCOCH3 и C6H6HgNO3 при нагревании с расплавленным бензои-
ном до 150—120° С вызывает пиролитическое образование уксусной кислоты
и азотистой кислоты соответственно [458[.
Реакции ртутноорганических соединений
325
ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Реакции в газовой фазе
R2Hg под действием ультрафиолетового света (широкого диапазона длин
волн) распадаются на радикалы:
RHgR -» RHg* + R и далее
RHg*-» R* + Hg.
В задачу настоящей книги не входит рассмотрение реакций органиче-
ских радикалов R’, образующихся при фотолизе ртутноорганических сое-
динений в газовой фазе. См. об этом соответствующую литературу: фотолиз
диметилртути [542—571], диэтилртути [572—575], ди-к-пропилртути [576],
ди-«-бутилртути [577, 578], диперфторэтилртути [579], дифенилртути
[580], дивинилртути [580а].
О фотолизе хлористой метилртути при 200° С под действием фотовов-
бужденной ртути см., например, [581].
Под действием ультрафиолетового света происходит переход одного гео-
метрического изомера ртутнрорганического соединения этиленового ряда в
другой (см. стр. 329).
Реакции в растворе
Исследования Разуваева и его сотрудников показали, что ртутноорга-
нические соединения в растворе при инициирующем действии ультрафиоле-
тового света утрачивают свойственную им в обычных условиях инертность по
отношению к органическим галогенидам, к веществам, способным дегидри-
роваться (спирты, некоторые простые эфиры и т. п.), и к ароматическим угле-
водородам.
Под действием ультрафиолетового света ртутноорганические соединения
как R2Hg, так и RHgX (X = ОН) в растворе, так же как и в газовой фазе,
распадаются на радикалы:
RaHg -» RHg- + R‘, (1)
RHgOH-» RHg* + ОН. (?)
Образующиеся радикалы RHg’ или реагируют с растворителем, или
(с большей или меньшей легкостью в зависимости от природы R) распадаются
далее:
RHg’-» R’ + Hg, (3)
или реагируют между собой:
2RHg*-» RaHg + R* (4)
(в настоящем разделе не рассматриваются особенности поведения несодер-
жащих ртуть органических радикалов R', образующихся при фотолизе
R2Hg и RHgX).
При фотолизе R2Hg (R — метил’ [278, 582], этил [583], 2-карбометокси-
этил [584], циклогексил [585], фенил [277, 278, 585—597], о- [278, 598],
п- [599] толил, п-хлорфенил [598], и-анизил [600], карбокси- и карбомет-
оксифенил [601], а- [277, 602] и [3-нафтил [603]) в галоидсодержащих рас-
творителях (хлороформе [277 , 572, 582—585, 587, 598—603], бромоформе
[587, 601], четыреххлористом углероде [277 , 278, 582 , 585 ' 590, 595, 598—
603], иодистом метиле [582—584, 593, 602], трихлорэтилене [587], бромистом
[595, 602] и иодистом [602] этиле, дихлорэтане [598, 603], тетрахлорэтане
[586, 602], гексахлорэтане [586], бромистом изобутиле [588], третичнобутил-
хлориде [587], хлорексе [588], бромистом изопентане [602], децилбромиде
326
Ртутноорганические соединения. Лит. стр. 336—352
(589], хлористом [278, 592] и иодистом [592] бензиле, бромбензоле [588]
и иодбензоле [591, 592], о-нитроиодбензоле [594], о-иоданизоле [594],
о-иодацетофеноне [594]) образующийся радикал RHg’ отрывает галоид от
растворителя, давая RHgX. В хлороформе частично идет выделение ртути
за счет реакций (3) и (4).
Так же ведет себя в этих галоидсодержащих растворителях радикал
RHg*, образующийся при фотолизе RHgOH (R = фенил [604, 605], мезитил
[606], п-диметил аминофенил [606]).
При фотореакции дифенилртути с хлорбензолом и ди-га-анизилртути с
четыреххлористым углеродом [600] образуется каломель и смола (в послед-
нем случае также анизол).
В растворителях, не содержащих галоидов (спирты [277, 585—587, 601],
ацетон (277, 587], этилцеллозольв [277, 587, 599], диоксан [601, 607]гдиэти-
довый эфир [587], этилформиат [277], 2,2,4-триметилпентан [277, 587],
изопропил бензол [608, 609], морфолин [610], гексан [611] и др.), разложе-
ние R2Hg (R = метил [611], этил [583], «-бутил [609], фенил [277,585—
587,589, 607, 608, 610, 612], о-толил [277, 598], п-толил [599], п-хлорфенил
[589], п-анизил [600], карбокси- и карбометоксифенил [601], а- [277], fl-
нафтил [603]) и RHgOH ([R = фенил [604, 605, 613] (в бензоле) [614], о- и
т-толил [606], п-нитрофенил [606, 615], n-диметил аминофен ил [613]) не
останавливается на стадиях (1) и (2), но проходит и по (3) с выделением
металлической ртути; образующийся R дегидрирует растворитель, иногда
димеризуется и диспропорционйруется.
При фотолизе R2Hg [4,72а], RHgOH [613] и RHgX [616] в ароматиче-
ских соединениях проходит реакция ароматического замещения:
RaHg + ArH -» RAr +[Hg,
RHgOH + ArH RAr + Hg + H2O.
Для дифенилртути это подтверждено и разложением в меченом бензоле
[4]. При разложении дифенилртути, меченной С14 в положения 1,1, в бен-
юле не происходит изомеризации радикала [532в].
При фотолизе иодистой метилртути в изопропилбензоле образуются метан,
2,3-диметил-2,3-дифенилбутан и иодистая ртуть, при ' фотолизе же иоди-
стой метилртути в бензоле и хлорбензоле метилрадикалы метилируют рас-
творитель, давая соответственно толуол и смесь о-, м- и и-хлортолуолов [616].
От димезитил- [617], ди-и-фенилэтил^- [618], дициклогексил- [585], ча-
стично, диэтил [583] ртути и меркурди-Р-пропионовой кислоты [584] при
фотолизе отрываются оба радикала, образуя в галоидсодержащих раство-
рителях RC1 в СС14 и RH в СНС13, а также Hg2Cl2, в безгалоидных раствори-
телях, образуя RH. Иначе ведут себя при фотолизе также дибензилртуть
[75, 619], гидроокись [620] и сукцинимид [621] бензилртути, во всех раствори-
телях образуя дибензил и ртуть(в СНС1а образуетсяЬ^Ц. Отсутствие взаимо-
действия дибензилртути с растворителем при облучении подтверждено разло-
жением в дейтерированном растворителе [622]. Дифенил, образующийся при
распаде под действием ультрафиолетового света дифенилртути в Сб4Нв, со-
держит 50% [4], а при распаде в CeD6 до 88% меченых бензольных ядер; в
последнем случае образуется в незначительном количестве СвН5С6НБ и
CeD5C6D5 [539а], а также С6Н6 и, содержащий водород дейтеробензол, а
при фотораспаде (CeDs)2Hg в СвНв возникают молекулы C6D5H, CeD4H2,
CeDsH3 и дифенила всех трех типов [539а].
Обмен С—С14 при облучении бромистой фенилртути и С6Н6 и бромистой
фенилртути и СвНвВг только в первом случае идет уже без добавок; с бром-
бензолом он достигает заметной величины только в присутствии добавок —
солей кобальта и алюминия и металлического серебра [623].
Реакции ртутноорганических соединений 327
При фотолизе дифенилртути и дициклогексилртути в средах, содержащих
С14 и D, происходит обмен радикалами [585, 612] (см. выше).
При помощи меченых атомов подтвержден различный механизм фото-
реакций для дифенилртути и для гидроокиси фенилртути [624, 625].
Радикалы, образующиеся при фотораспадедифенилртути в смеси галоид-
содержащего и не содержащего галоида растворителя, реагируют таким об-
разом, что радикал C6H5Hg отрывает галоид от галоидсодержащего компо-
нента смеси, образуя хлористую фенилртуть, а фенилрадикал отрывает во-
дород, образуя бензол [626]. Реакция индуцируется перекисями [627].
В смеси хлор- и бромсодержащего растворителя радикал CeH6Hg’ отрывает
бром предпочтительнее, чем хлор [628]. Если же бром соединен с арилом,
а хлор с алкилом, то сначала отрывается хлор [628].
Сходно с действием ультрафиолетового света действие на ртутнооргани-
ческие соединения галогенидов металлов, не образующих устойчивых ме-
таллоорганических соединений. Они также инициируют распад R2Hg на
радикалы RHg’ и R’ [629]. RHg’ отрывает галоид от галогенида металла, об-
разуя RHgCl; R‘ в случае действия СоС12, FeCl3 [630], NiCl2, МпС12 [629] от-
рывает водород от растворителя, в случае СиС12 отрывает С1 от СиС12 и обра-
зует RC1. Галогениды металлов, взятые из расчета на отщепление двух ра-
дикалов, полностью деарил и руют дифенилртуть, образуя HgCl2 и бензол
(в водородсодержащем растворителе) и хлорбензол—в случае, действия СиС12
[629]. Также полностью деарилируется дифенилртуть с образованием бен-
зола под действием FeCl3, СоС12 и СиС12 в морфолине (СоС12 образует неболь-
шое количество хлористой фенилртути) [630]..
Дибензилртуть разлагается под действием FeCl3 и СоС12 в этилцелло-
зольве до металлической ртути с образованием этиловобензилового эфира
этиленгликоля. Под действием СиС12 в этилцеллозольве и диоксане обра-
зуются сулема и тот же эфир и в диоксане— хлористый бензил [631 ]. С хлор-
ным железом в диоксане образуются хлористая бензилртуть и хлористый
бензил [631 ]. При реакции хлорной меди с диферроценилртутью в диоксане
образуется смесь хлорферроцена и ферроцена [281]:
СиС1г
(CBHBFeCBH4)2Hg—-* CsHsFeCsH^Cl + (CsH6)2Fe.
Фото- и термораспад ртутноорганических соединений в растворителях
не всегда протекает аналогично. Иногда они проходят одинаково. Такова,
например, реакция дифенилртути с метиловым спиртом [292]:
(CoH5)2Hg + СНзОН — 2СвНв -F Hg + СНаО.
Термо- и фотораспад дифенилртути в толуоле дают одни и те же продукты в
приблизительно одинаковых количествах. Об этом также см. выше (стр. 324).
Фотораспад же дифенилртути в циклогексане (100—150 час.) дает бен-
зол (70—80%), дициклогексил, циклогексен, фенилциклогексан, дифенил
[539]. О термораспаде дифенилртути см. стр. 324.
Фотораспад дифенилртути в циклогексене дает бензол и дициклогексе-
нил; образование бензола за счет отрыва фенилрадикалом водорода от рас-
творителя подтверждено применением меченой (Cj4H5)2Hg [539]. Выше
уже описано различное течение фото- и термораспада дифенилртути в
меченом бензоле.
Различно проходит распад дифенилртути в четыреххлористом углероде
под действием света и перекисей:
hv
’ (CeH5)2Hg + 2СС1* - CeH5HgCl + ОСЬ + CeHBCl [277, 278], (1)
(RCOO),
(CeHB)2Hg + CCl4—--► CeHBHgCJ + CeHBCC!3 [279]. (2)
328
Ртутноорганические соединения. Лит. стр. 336—352
Так же как и в последнем случае, проходит реакция Р-хлорвинильных
соединений ртути с четыреххлористым углеродом под действием перекисей
ацетила, бензоила и др. [279].
Реакция дифенилртути с четыреххлористым углеродом под действием ультрафиолетового
света [278]. Раствор 5 г дифенилртути в 75 мл четыреххлористого углерода помещают в
кварцевую колбу, снабженную обратным холодильником, так как раствор от лампы нагре-
вается до слабого кипения. Облучение производят кварцевой ртутной лампой ПРК-2 в те-
чение 50 час. Раствор после облучения окрашивается в коричневый цвет. Из раствора выде-
ляются листочки хлористой фенилртути (3,0 а). После кристаллизации из ацетона т. пл.
257° С.
Раствор четыреххлористого углерода перегоняют с водяным паром. Перешедший с во-
дяным паром четыреххлористый углерод при отгоике дает в остатке гексахлорэтаи, который
после возгонки плавится в запаянном капилляре при 185° С.
Отгон четыреххлористого углерода нитруют и выделяют n-хлорнитробеизол, после кри-
сталлизации из ацетона и метилового спирта т. пл. 83° С. После отгонки с водяным паром
получают 1,75 г густой коричневой массы, которую обрабатывают горячим ацетоном. Из
ацетонового раствора выделяется при охлаждении еще 0,5 г хлористой фенилртути с т. пл.
257° С, а из маточника выделяют 0,8 г иепрореагировавшей дифенилртути. Т. пл. после пере-
кристаллизации 125° С.
Полученный Разуваевым и сотр. ряд: о-анизил>фенил>толил>п-бром-
фенил [633] >сс-нафтил > метил [75], в который располагаются радика-
лы на основании легкости отщепления от ртути в RHgR' в гомолитических
реакциях
RHgR' + CCU RHgCl + R'Cl + (СС13)а,
RHgR' + СНСЬ - RHgCl + R'H + (CCls)2,
в значительной части не совпадает с рядом Хараша, в котором радикалы рас-
полагаются на основании реакции, протекающей по иному, гетеролитическо-
му механизму, но и не совпадает с рядом, полученным Несмеяновым, Бори-
совым и сотр. [279а] на основании индуцируемого перекисями распада
RHgR' в СС14 (см. стр. 299).
Фотолиз дифениленртути, так же как и ее пиролиз, дает дифенилен [580].
При фотолизе иодистой о-иодфенилртути в бензоле в продуктах реак-
ции найдены бифенил-(42%), о-терфенил-(36%), бме-о-дифенилйлртуть (21%)
и неорганические соли ртути [537]. Продуктами фотолиза иодистой о-иод-
фенилртути в циклогексане являются бензол и иодная ртуть [537].
При облучении ультрафиолетовым светом в течение 8 час. иодистой
о-иодфенилртути в.бензоле в присутствии тетрациклона выделен 1,2,3,4-тет-
рафенилнафталин (выход 25%) [6341:
Аналогично протекает фотолиз бме-о-иодфенилртути [634]. Полученные
продукты указывают на течение реакции через промежуточное образование
дегидробензола [537, 634].
Реакция бромсукцинимида и диарилртути при облучении ультрафиолето-
вым светом переходит на радикальный механизм [75] (ср. стр. 282).
Реакции ртутноорганических соединений 329
цис-транс-ПРЕВРАЩЕНИЯ
НЕПРЕДЕЛЬНЫХ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Превращение одного геометрического изомера ртутноорганического со-
единения этиленового ряда в другой может совершаться под действием ульт-
рафиолетового Света, под действием перекисей, иногда под действием нагре-
вания при умеренной температуре (Несмеянов, Борисов и сотр.). Под дей-
ствием ультрафиолетового света д и-транс-p-хлорвинил ртуть гладко прев-
ращается в ди-цос-р-хлорвинилртуть, также хлористая шранс-Р-хлорвинил-
ртуть в абсолютном спирте, толуоле или, лучше всего, в бензоле переходит
в хлористую цпс-р-хлорвинилртуть с выходом 23,7% [6351. Но для ртутного
производного стильбена под действием ультрафиолетового света цис-изомер
превращается в транс-изомер [541].
Превращение тра«е,траяс-ди-(Р-хлорвинил)ртути в ее цис,цис-изомер [635]. 10 г транс,
тра«с-ди-(Р-хлорвинил)ртути, плавящейся при 71—72° С, вносят в кварцевую пробирку,
расплавляют и с расстояния 2—3 см облучают кварцевой лампой ПРК-4 в течение 17 час.; при
этом расплавленное вещество при охлаждении до комнатной температуры не закристалли-
зовывается. После облучения жидкое, немного пожелтевшее вещество подвергают перегонке
в вакууме, в преемник переходит бесцветная жидкость, кипящая при 56° С (6,4-Ю-3 мм).
Получено 4,5 г вещества (45%).
При увеличении продолжительности облучения до 76 час. выход достигает 65,5%. Оста-
ток в перегонной колбе растворяют в. 15 мл бензола, обрабатывают активированным углем и
фильтруют; при охлаждении из бесцветного фильтрата выделяются кристаллы хлористой
тращс-|3-хлорвинилртути. Вес 3,8 г. Т. пл. 122—123° С.
Превращение хлористой т/?аис-р-хлорвииилртути в ее цис-изомер [635]. 4 г хлористой
тралс-р-хлорвинилртути в 25 мл сухого бензола подвергают облучению кварцевой ртутной
лампой ПРК-4 в кварцевом сосуде в течение 50 час. при комнатной температуре с расстояния
2—3 см. Образовавшаяся при этом муть удаляется при помощи активированного угля. Из
прозрачного раствора при охлаждении выделяют 2,4 а кристаллов. Т. пл. 123—124° С
(с разложением). Из фильтрата удаляют растворитель слабым нагреванием при действии во-
доструйного насоса, остаток подвергают дробной кристаллизации из смеси бензола и петро-
лейного эфира (1 : 4). Кристаллы — мелкие лепешечки (исходное вещество — иглы). После
повторной перекристаллизации т. пл. 78° С. Выход 0,95 г (24%).
Превращение цас-а-меркур-бас-стильбена в траис-а-меркур-бас-стильбен [541]. 1 г
цас-а-меркур-бис-стильбена в 50 мл хлороформа облучают светом кварцевой лампы ПРК-4 в
течение 3 час. Уже через 30 мин. начинает выпадать осадок в виде мелких игл. Осадок от-
фильтровывают. Получают 0,50 г (50%) транс-а-меркур-бос-стильбена; т. пл. без перекри-
сталлизации 243—244° С. После испарения хлороформа выделено 0,3 г (30% взятого коли-
чества) исходного цос-а-меркур-бис-стильбена.
Превратить транс-изомеры ртутных производных стильбена в цис-изо-
меры не удалось. Длительное облучение ультрафиолетовым светом разруша-
ет их, но не превращает в цис-модификации.
Хлористая mpaw-p-хлорвинилртуть превращается в цис-изомер в при-
сутствии перекиси [632] в содержащем перекиси ксилоле, в присутствии пе-
рекиси бензоила, ацетила или перекиси натрия в ксилоле, толуоле, диокса-
не, четыреххлористом углероде при нагревании до 95—100° С (в СС14 в за-
паянных ампулах). Гидрохинон предотвращает изомеризацию.
Продукты присоединения солей ртути к непредельным углеводородам
при цагреваний выше температуры плавления обычно разлагаются с выде-
лением исходного непредельного соединения [127]. В некоторых случаях
под воздействием не слишком высоких температур, наряду с термическим
разложением, происходит переход одного геометрического изомера ртутно-
органического соединения этиленового ряда в другой. Так, tjz/c-a-меркур-
быс-стильбен при нагревании переходит в транс-изомер [5.41].
Переходцас-а-меркур-бас-стильбена в транс-а-меркур-бис-супльбе» [541]. 0,5 г цис-
а-меркур-б«с-стильбена нагревают в запаянной трубке при 140—160° С в течение 4 час.,
при этом на дне трубки выделяется металлическая ртуть. По окончании нагревания содер-
жимое трубки последовательно обрабатывают бензолом и диоксаном. Из диоксана выделяют
0,25 г (50%) /праяс-а-меркур-бис-стильбена с т. пл. 242—244°С. Из бензола выделяют 0,1 г
mpawc-стильбена с т. пл. 121—122° С.
330 Ртутноорганические соединения. Лит. стр. 336—352
ОБМЕН АНИОНОВ В РТУТНООРГАНИЧЕСКИХ СОЛЯХ
Ртутноорганический галогенид из растворимой соли RHgX (X — ОН,
СН3СО2, NO3) получают прямо прибавлением к раствору RHgX галогенида
Калия, натрия или кальция. RHgHal выпадает в осадок [636—644]. RHgOH
из RHgHal получают нагреванием RHgHal с эквимолекулярным количест-
вом окиси серебра [620, 645—647] в воде или спирте или с метиловоспирто-
вым раствором едкой щелочи [620, 648, 655]. После отфильтровывания
Ag(K)Hal из фильтрата (после упаривания) получают RHgOH. О
строении метилмеркургидроксида как триметилмеркуроксонийгидроксида
[(CH3Hg)3O]+OH_ см. ниже.
Нейтрализуя раствор соответствующей кислотой, можем осуществить
переход от одной ртутноорганической соли к другой [649]. Нитраты, ацета-
ты, сульфаты и перхлораты из галогенидов получают нагреванием послед-
них в водном или спиртовом растворе с азотнокислым, уксуснокислым или
хлорнокислым серебром [372, 442, 650—653]. После отделения AgHal фильт-
рат упаривают. -
Получение а-нафтилмеркурнитрата [442]. Смешивают эквимолекулярные количества
а-нафтилмеркурхлорида, растворенного вгорячем ацетоне, и азотнокислого серебра в спирте.
После фильтрования и упаривания нитрат выделяется в виде мелких иголочек с т. пл. 137—
138° С.
Фенилмеркурнитрат получен также из фенилмеркурацилатов и азотной
кислоты [654]. Некоторые гидроксиды из ацетатов получают действием вод-
ного [655, 656] или спиртового [657] раствора едкой щелочи или же действуя
водным раствором едкой щелочи на раствор ацилата в органическом раство-
рителе, например бензоле [658].
C6H5CH2HgCNO получено из RHgCl и фульмината калия в метаноле
при охлаждении [659].
Для замены анионов в ртутноорганических солях применяют также и
обычные способы получения солей: взаимодействие ртутноорганической
соли с неорганической солью металла, образующего с анионом ртутноорга-
нической соли растворимую соль, которая затем вымывается [660], или не-
растворимую соль, которая отфильтровывается [661], или нагревание ртут-
ноорганической соли летучей кислоты с нелетучей кислотой [6621.
Сульфиды алкилртути (RHg)2S и смешанные сульфиды RHgSHgR' по-
лучают в чистом виде из галогенидов и водного сульфида натрия в спирте
[663]. Взаимодействие ртутноорганических солей разных классов с гипосуль-
фитом и сернистым аммонием также приводит к (RHg)aS [663а].
Во многих случаях предпочтительно пользоваться симметризацией соли
и последующим нагреванием полученного симметричного соединения с соот-
ветствующей солью окиси ртути в растворе или без растворителя по схеме
2RHgX-> R2Hg; R2Hg + HgX2 2RHgX.
Получение гидроокиси метилртути [648] (ср. [664, 665]). 30 г бромистой метилртути
смачивают метанолом, заливают 100 мл метиловоспиртового раствора едкого кали и нагре-
вают 30 мину на водяной бане. Затем отфильтровывают горячим, добавляют 20 мл воды и
упаривают в вакууме (17 мм) при 60° С. К кашеобразному остатку прибавляют немного воды
и отсасывают. Осадок гидроокиси метилртути растворяют в 40 Мл пиридина при 80° С и
сливают с вновь выделившейся щелочи. Из пиридинового раствора после отфильтровывания
выкристаллизовываются иглы гидроокиси метилртути. Их отсасывают, многократно промы-
вают абсолютным эфиром и сушат над хлористым кальцием. Выход 16 г (68%). Т. пл. 137° С.
Рекомендуется хранить в сосуде с пришлифованной пробкой. Также получены другие
RHgOH (R = С2Н5 (т. пл. 37° С), п-СзН7 (т. пл. 78° С), п-С4Н9(т. пл. 68°С), и-С5Ни
(т. пл. 50° С), п-СвНи (т. пл. 54,5°С), п-С7Н1б (т. пл. 51° С), п-СмНзз (т. пл. 78° С), СвНз
(т. разл. около 200° С), а-СшН7 (т. разд, около 280° С) [648]).
Алкилмеркургидроксиды представляют собой в действительности смесь
алкилмеркуроксида (AlkHg)2O и три(алкилмеркур)оксонийгидроксида
[(AlkHg)8O+jOH_ [664 , 665]. При дегидратировании кипячением в растворе
Реакции ртутноорганических соединений 331
толуола или в вакууме эта смесь переходит в алкилмеркуроксид [т. пл. для
(AlkHg)2O, где Aik = СН3, 139° С, С2Н5—, 47° С, п-С3Н7—, 98° С;
/-СаН7—, 135° С, С4Н9—, 74° С (Грденич)].
Соли (AlkHg)3O+X_ могут быть получены несколькими путями:
а) осторожным действием кислоты на (AlkHg)2O (в метаноле или ацето-
не); так получены для (CHaHg)3OX борофторйд [664], карбонат [665] (про-
пусканием СО2 в метанольный раствор (CH3Hg)2O), перхлорат [665], нитрат
[6651, трихлорацетат [665], бихромат (действие 10%-ного водного раствора
СгОа);
б) прибавлением RHgX к (RHg)2O,(B метаноле); так получен (CHaHg)3OX,
где X — сульфат, карбонат;
в) реакцией двойного обмена (CH3Hg)aOX + КХ', X' = СЮ4, BF4 (в ме-
таноле, в случае действия КМпО4—в ацетоне); так получен также ацетат,
гексафторфосфат, рейнекат;
г) (CH3Hg)3O+NO3~ получен также из (CH3Hg)2O и AgNO3.
Описано также образование дитизонатов триалкилмеркуроксония из
его солей и дитизона [729].
Получение перхлората триметилмеркуроксония [665]. Раствор 5 г диметилмеркур-
оксида в 20 мл метилового спирта нейтрализуют прибавлением по каплям 70%-ной хлорной
кислоты, растворенной в тройном объеме метилового спирта. Прибавлением эфира высажи-
вают кристаллический осадок перхлората. Т. пл. 148° С.
Основание тримеркурметилоксонийгидроксида получено действием рас-
твора едкого кали в метиловом спирте на перхлорат [665].
Аналогично тримётилмеркуроксониевым солям получены [666] триметил-
меркурсульфониевые соли — встряхиванием бензольного раствора диметил-
меркурсульфида (получение его см. ниже) с водным раствором СгО3; полу-
ченный двойным разложением с азотнокислым свинцом бихромат (растира-
ние в ступке с небольшим количеством этанола) переводят в нитрат
(CH3Hg)3S+NO3-.
Течение реакции бромистой алкилртути с третичными фосфинами зави-.
сит от ряда условий, в том числе от природы алкила, фосфина и от среды
реакции. Бромистая этилртуть присоединяется к фенилДиметилфосфину и к
триэтилфосфину в метаноле с образованием соответствующего этилмеркур-
триалкилфосфонийбромида (EtHgPMe2Ph)Br и (EtHgPEt3)Br. Про-
дукт реакции бромистой метилртути с триэтилфосфином в эфире [CH3HgP-
• (С2Н5)а1Вг выделен в кристаллическом виде [667]. О реакциях ацетата
фенилртути с третичными фосфинами см. также [55в]. При взаимодействии
ацетата фенилртути с триэтилфосфитом среди других продуктов получен
диалкилфосфит фенилртути [55в] (ср. гл. XIII, стр. 258).
Получениебцс-метилмеркурсульфида [663]. Теплый раствор 0,5 г Na2S-9H2O (отжатого
на фильтровальной бумаге) в 10 мл спирта смешивают с насыщенным раствором 1 г хлористой
метилртути в 10 мл спирта и реакционную смесь охлаждают током воды. Выделяются широ-
кие листочки метилмеркурсульфида. Выход 0,7 г (90%). Кристаллизация из спирта или хло-
роформа. Т. пл. 445° С.
Получение метилмеркурэтнлмеркурсульфида [663]. Теплый раствор 5,0 г Na2S*9H2O
в 50 мл спирта прибавляют к раствору 5,0 г бромистой метилртути в 50 мл спирта. К смеси
прибавляют теплый насыщенный спиртовый раствор бромистой этилртути (5,0 г- приблизи- •
тельно в 50 мл) и реакционную смесь нагревают; затем быстро фильтруют горячей, фильтрат
охлаждают. Выделившийся сероватый осадок (широкие листочки нз хлороформа) весит
7,0 г (89%). Т. пл. 73,5° С.
Аналогично получен метилмеркур-н-пропилмеркурсульфид. Т. пл. 51° С.
Синтез бис-этилмеркурсульфида [663]. Теплый спиртовый раствор 1,6 г Na2S-9H2O
постепенно прибавляют к теплому раствору 4,0 г бромистой этилртути в спирте. Реакцион-
ную смесь охлаждают током воды. Выпадает серый осадок (3,0 г, выход 94%). После двух
кристаллизаций из бензола получены широкие листочки с т. пл. 105° С. Аналогично получе-
332 Ртутноорганические соединения. Лит. стр. 336—352
ны бкс-я-пропилмеркурсульфид с т. пл. 70° С (кристаллизация из спирта), бмс-изопропил-
меркурсульфид с т. пл. 59,5° С и бис-н-бутнлмеркурсульфид с т. пл. 59° С.
Получение азотнокислой метилртути [668]. К раствору 9 г иодистой этилртути (или эк-
вивалентное количество бромида или хлорида) в 300 мл спирта добавляют раствор 4,25 г
азотнокислого серебра в тепловатой воде и кипятят 2 часа с обратным холодильником на во-
дяной бане. Затем добавляют 200 мл воды, отгоняют спирт, отгоняют воду в вакууме досу-
ха и полученный сероватый нитрат этилртути очищают переосаждением петролейным эфи-
ром из раствора в минимальном количестве метилового спирта. Т. пл. 87° С.
Получение азотнокислой бензилртути [669]. 11,35 г хлористой бензилртути в эфире
смешивают с раствором 5,89 г AgNOg в этиловом спирте. После 1 часа встряхивания AgCl
отфильтровывают. Фильтрат выпаривают при возможно более низкой температуре в ваку-
уме. Остаток перекристаллизовывают из эфира; выделяются белые иглы; т. пл. 90—91 °C (с
разложением).
Синтез гидроокиси бензилртути из бензилмеркурхлорида и влажной окиси серебра [620].
5,00 г хлористой бензилртути растворяют в 100 мл горячего метилового или этилового спир-
та, добавляют влажной окиси серебра, полученной осаждением 3,25 г AgNOs (с учетом 20% -
ного избытка). После 4—5-часового нагревания на водяной бане при 80° С отфильтровыва-
ют осадок хлористого серебра, затем отгоняют избыток спирта. При охлаждении выкристал-
лизовывается гидроокись бензилртути. Выход 2,46 г. (58,4%). Перекристаллизовывают из
спирта. Т. пл. 125—127° С.
Синтез гидроокиси бензилртути из бензилмеркурхлорида и едкой щелочи [620].
5,00 г хлористой бензилртути растворяют в 100 мл горячего метилового или этилового спир-
та, добавляют 0,62 г едкого натра, растворенного в спирте. Смесь нагревают 2 часа на водя-
ной бане при 80° С, отфильтровывают осадок NaCl и отгоняют около 2/3 спирта. Выход гид-
роокиси бензилртути 4,38 г (92,8%).
Синтез гидроокиси фенилртути [657]. В нагретый на водяной бане раствор 12 г уксусно-
кислой фенилртути в 1 л изопропилового спирта постепенно приливают 10 мл 30%-ного рас-
твора едкого кали. Нагревание продолжают еще 1 час, после чего раствор разбавляют рав-
ным объемом воды. Растворитель отгоняют от профильтрованного раствора примерно до
остаточного объема 200 мл. При охлаждении выделяются кристаллы гидроокиси фенилрту-
ти, которую перекристаллизовывают из горячей воды. Получают 7,9 г гидроокиси фенил-
ртути (75%). Т. пл. 200° С, с разложением (см. также [670]).
Синтез гидроокиси фенилртути [658]. К раствору 33,6 г уксуснокислой фенилртути в
горячем бензоле прибавляют при перемешивании 0,15 моля 5—10%-ного едкого натра и че-
рез 20 мин. растворитель отгоняют. Из охлажденного раствора выделяют гидроокись фенил-
ртути. Кристаллизация из воды. Выход 75—79%.
Подобным же образом получены гидроокиси толил-, мезитил- и нитрофе-
нил ртути.
Описано получение гидроокиси фенилртути из основного нитрата
PhHgNO3-PhHgOH [671].
Гидроокись фенилртути получена [670] с выходом 94,6% при кипяче-
нии в течение 1 часа в водном растворе едкого натра с (C6H6Hg)2NH2OCOCH3,
образующейся при сливании кипящего раствора NH4OH с C6H6HgOCOCH3.
Реакция ацетата фенилртути с едкой щелочью, по утверждению автора,
менее удобна [670].
Получение фтористой фенилртути [672]. Раствор 40,6' г (0,17 моля) свежеосажденной
окиси ртути и 14 г (0,35 моля) 50%-ной плавиковой кислотыв400 мл воды встряхивают 5 час.
с 78 г (0,25 моля) хлористой фенилртути, предварительно смоченной этанолом. Осадок
отфильтровывают и извлекают 400 мл кипящего этанола. При охлаждении образуется не-
большое количество осадка. Раствор декантируют и концентрируют в вакууме. Остаток из-
влекают кипящим этанолом и фильтруют. Охлажденный раствор дает 32 г фтористой фенил-
ртути. Т. пл. 170° С. Выход 43%. После кристаллизации из хлороформа т. пл. 171° С. Ве-
роятно, выход может быть повышен при увеличении длительности реакции.
При действии на RHgOH CaFe(CO)4 [673] или железокарбонилгидридов
[107] образуются своеобразные, нестойкие ртутноорганические соли
(RHg)2Fe(CO)4 (см. гл. XIII).
Основной нитрат метилртути получен или обработкой оксида или гидр-
оксида метилртути азотной кислотой (недостаток) [674] или действием оки-
си или гидроокиси ртути на средний нитрат метилртути [675]. Описано
также получение основного нитрата фенилртути [676].
Действием СО2 на RHgOH в спирте получают RHgHCO3, из послед-
них и CS2 в отсутствие спирта получены RHgSH [739].
Реакции ртутноорганических соединений
333
Способами, перечисленными в начале этого раздела, осуществлено полу-
чение фосфата этилртути из гидроокиси этилртути и фосфорной кислоты
[668], диарил- и диалкилфосфатов [681], диалкилтиофосфатов алкил-
[678 —680], арил- [677, 679—682] ртути, метабората 3-пиридилртути [683];
получение фенолятов, в том числе полихлорфенолятов алкил(арил)ртути
[64, 684—697], алкоголятов алкил(арил)ртути [698],нафтолятовалкилрту-
ти[619],8-оксихинолятаалкилртутй[686, 688, 699а],-фенилртути [699,699а],
алкилртутных солей некоторых карбоновых кислот [700, 701], оксалатов в
меркурированных фенолов [702], фенилмеркурсульфатов, -сульфанилатов,
частности соединения C6H5HgO3SCH2CH2(CH3)NOC(CH2)7CH=CH(CH2)7CH3
[703], алкилмеркурсульфонатов [704], фенилмеркурдинафтилметандисуль-
фоната (из фенилмеркурацетата и соответствующей кислоты в спиртовом
растворе [705], соединений типа CeHfiHgNR3OR' (R — алкил, R' — поли-
галоидарил [706]), алкилмеркурсульфидов [679, 707—709], алкилмеркур-
пертиоцианатов [710, 711].
Легко получаются разнообразные меркаптиды и тиофеноляты алкил-
(арил)ртути RHgSR' [712, 713] взаимодействием обычно в водном, иногда
спиртовом «растворе на холоду ацетата или гидроксида алкил(арил)ртути
с R'SII или R'SNa. Так пол учены. RHgSR' (где R — алкил или арил, R' —
алкил [714], CH3CONH [715, 716]), полиалкоксиалкилы [717], алкилы, содер-
жащие диалкиламиногруппу [718, 719, 721—724] (например’(СН3)2ЫСН2СН2
[719,721—724]), замещенный пиримидил, пиридазил [715], бензтиазолил
[725], триазины [726, 727], тропонил [728, 759], тиодиазолил [720].
Дитизонат метилртути CH3HgSC(N=NC6H5)NNHC6Hs получен из
бромистой метилртути и дитизона [729]. RHgSR' получены подобным же
образом [730] из меркаптанов и алкил(арил)меркурациламидов или -суль-
фонамидов, например:
N-HgR
/О RHgSR' 4^CeH5SO2N(HgR)CeH4OCH3-p.
Известны также примеры замены аниона у ртути в продуктах присо-
единения солей ртути к олефинам на SR-группу; так, действием меркаптанов
или тиофенолов на продукты присоединения ацетата ртути в воде к 1-де-
ценкарбоновой-10 кислоте и ее эфиру произведена замена ацетоксигруппы
на SR-остаток [731].
Известен пример, когда заменяемым анионом является остаток теофил-
лина [732]. Произведена также замена аниона хлора в у-хлормеркур-Р-окси-
и метокси)пропилтеобромине на остатки барбитуровых кислот [733].
Согласно Шпенглеру и Веберу [734], продукты метоксимеркурирования
алкенов с аммиаком и аминами образуют аммониевые соли RHgNR'H2X
(R' = H или Aik; R = С6Н5СН(ОСН3)СН2, СН3(СН2)7СН(ОСН3)СН2,
2-метоксициклогексил, СН3(СН2) 12СН(ОСН3)СН2).
Другие примеры замены аниона в продуктах присоединения солей ртути
к непредельным см. гл. VI.
Диоктилсульфосукцинаты алкил(арил)ртути получены кратковременным
кипячением ацетата алкил(арил)ртути с диоктилсульфосукцинатом [735].
Алкил(арил)ртутные производные циангуанидина получены из сереб-
ряных производных последнего и RHgHal [736], или из гуанидина и
RHgOH [737]. В RHgBr (R = CH3, C2HS, n-C3H7, п-С4Н9) атом брома обме-
нивается на меченый атом брома реакцией с HgBr2, изотопное равновесие
устанавливается за 10 мин. [738]. Действием сероуглерода на спиртовый
раствор RHgOH получают ртутноорганические ксантогенаты RHgSSCOR'
[679, 739]. При смешении при комнатной температуре гидроокиси фенилртути
334 Ртутноорганические соединения. Лит. стр. 336—352
с сероуглеродом в отсутствие спирта продуктом реакции является выделенная
с 30 %-ным выходом дифенилртуть [7401. Реакция идет, вероятно, через про-
межуточное образование фенилсульфида ртути (см, гл. XIII, стр. 254, ср.
также [739]).
Винилмеркурксантогенат (й -тиоцианат) получен обменной реакцией
винилмеркурацетата с ксантогенатом (и тиоцианатом) щелочного метал-
ла [514].
Получение этилмеркур-Ы-алкилдитиокарбоксилатов и дитиоКарбаматов
из натриевых солей соответствующих дитиокислот и этилмеркурхлорида
в присутствии триэтаноламина и ацетата аммония произведено в воде на
холоду [740—7421.
Взаимодействием б«с-(этилмеркур)фосфата с Ы-алкил-М-фенилдитиокар-
баматом в водном 'растворе на холоду или этилмеркурхлорида с метилани-
лином и сероуглеродом в щелочном растворе получены этилмеркур-Ы-алкил-
N-фенилдитиокарбаматы [743].
Фенилмеркурацетат превращается в 8-фенилмеркуроксихинолин поли-
хлорфенолят при кипячении в бензоле как с полихлорфенолятом 8-оксихи-
нолина, так и со смесью полихлорфенола и 8-оксихинолина.
Особенно важны для осуществления практической цели получения вод-
норастворимых ртутноорганических соединений такие превращения ртут-
ноорганических солей RHgX, в которых анион X заменяется на остаток,
довольно прочно связывающийся с атомами ртути через азот или серу и
содержащий группировку атомов, способствующую растворению в воде.
Конденсацией ртутноорганических солей или гидроокисей с меркаптокар-
боновыми [744—755, 757, 758, 760, 762, 763,765,766,780], сульфоновыми
[761, 763], арсиновыми [745] кислотами по уравнению
RHgX + HSR' -> RHgSR' НХ
(R' — радикал, содержащий СООН-, SO3H- или AsO3H2-группу) получают
соединения, растворимые в водных растворах щелочей; например, из фос-
фата алкилртути действием ш-меркаптоундеКановой кислоты получены
соли RHSR' (R'=СИН23СООН) [767].
ClCH2HgSR получен реакцией ClCH2HgCl с RSH кипячением в метиловом
спирте (RSH—тиол-яблочная кислота или ее эфиры) [768]. Эфиры алкилмер-
кур-а-сульфгидрйл-«-алкановых кислот общей формулы R"CH(SHgR') COOR
получены взаимодействием R'HgCl и соответствующей меркаптокис-
лоты в спирте в присутствии щелочи на холоду [769]. В патентной ли-
тературе [770] описано получение эфиров алкилмеркуртиосалициловой ки-
слоты.
Получение этилмеркуртиосалициловой кислоты [746]. К взвеси 1 моля тиосалициловой
кислоты‘И I моля бромистой этилртути в 750 мл воды прибавляют по каплям раствор 2,5 мо-
лей бикарбоната натрия в 250 мл воды. Смесь нагревают до 50° С, охлаждают, фильтруют с
углем для удаления избытка бромистой этилртути, подкисляют 162 мл уксусной кислоты;
затем осадок отделяют, промывают 600 мл воды, перемешивают с 500 мл охлаждаемого льдом
абсолютного этилового спирта, вновь промывают холодным спиртом, сушат при 50° С. Вы-
ход 75—80%. Т. пл. Ш°С.
Получение фейилмеркуртиогликолевой кислоты [746]. Взвесь 10 г CeH6HgOH в 80 ч.
этилового спирта обрабатывают 10 г тиогликолевой кислоты. При разбавлении водой выпа-
дают тонкие иглы, Т. пл. 114° С.
Взаимодействием ртутноорганических солей или оснований с имидами [755,
756, 764, 771—781] (например сукцинимидом, фталимидом и др.), иминами
[776, 782] (например хинонимином), амидами [783—789] (например ацет-
амидом, арилсульфонамидом) [787, 788, 790] получают воднорастворимые
ртутноорганические производные этих азотистых соединений с ртутью, со-
единенной непосредственно с азотом.
Действием на 8-(2'-метокси-3-оксимеркурпропил)кумарин-3-карбоно-
вую кислоту теофиллина и пирролидинацето-2,6-диметиланилида в роде
Реакции ртутноорганических соединений
335
получено 7-теофиллинмеркурпроизводное соответствующей аммониевой
солй кислоты [791].
JirHg-производные теофиллина получены реакцией ацетата или хлорида
ариЛртути с теофиллином в этиловом спирте при 5-часовом кипячении [791 —
793].
Предложено получение N-винилмеркуримидов реакцией винилртут-
ных солей с имидами в присутствии оснований [514].
Из AlkHgOH и алкилмеркаптобензимидазола получено соединение [793]:
N<^HgAlk
Аналогичное соединение получено из 2-меркаптобензоксазода [756].
Получение о-оксифенилмеркурсукцинимнда [774]. 3,3 г сукцинимида в 18,7 мл воды при-
бавляют к 10,95 г хлористой о-оксифенилртути в 25 мл теплой воды. Охлаждают насколько
возможно без того, чтобы образовался осадок, н прибавляют 18,7 мл 10%-ного едкого кали.
После встряхивания н охлаждения получают сырой продукт. Кристаллизация из 200 мл
этилового спирта и 100 мл воды. Выход 72,3%. Т. пл. 232—235° С.
С другой стороны, замена SAr'-группы в ArHgSAr'. на галоид может
быть произведена действием хлористого бензоила [794, 795], арилсульфо-
хлоридов[794, 795] и -иодидов [794,795], на N-сукцинимидогруппу—действи-
ем бромсукцинимида [795]. Возможна также замена R' в RHgSR' на R"
действием R"SSR", -(R"S)2Hg, HSR" [794]. Так, например, произведен
обмен этиЛмеркуртиосалицилаТа и ж-толилмёркуртиогликолята с дитизоном
[796]
C2HsHgSCeH4COO- + DzH DzHgC2H5 + HSC6H4COO.
(Dz = C6H6N=NC(S_)=NNHC6H5) при встряхивании при комнатной
температуре бензольного раствора дитизона со слабощелочным водным
раствором этих ртутных соединений.
o-C2H6HgSCeH4COONa в водном растворе при продолжительном
хранении (быстрее на прямом солнечном свету) самопроизвольно превращает-
ся в o-C2H5HgSC6H4COOHgC2H5 [797]. ArHgSAr' превращается, в
ArHgOCOR действием RCOOH [798].
Соли фенилртути с анионами, способными полимеризоваться (п-винил-
бензоат [553], акрилат и метакрилат), получены нейтрализацией фенил-
мер кур гидроксида соответствующими кислотами в спиртовом растворе
[553, 676, 799].
Для продуктов взаимодействия в водном или спиртовом растворе при
комнатной температуре гидроокисей арилртути с нитроанилинами и нитронаф-
тиламинами при помощи ИК-спектррв Несмеяновыми Кравцовым доказано
их строение как HgN-производных [800]; для арилртутных производ-
ных нитрозофенолов (полученных в тех же условиях) доказано их хинон-
оксимное строение в твердом состоянии и наличие металлотропного тау-
томерного равновесия в смысле схемы
OHgR [CI-
NO NO-
NOHgR
в растворе [801], в то время как арилртутные производные нитрозоанилинов
обладают бензоидным строением [802]. Для п-диметиламинофенилртутных
336 Ртутноорганические соединения
производных оксиазосоединений доказано [803] их бензоидное строение
RHgO -<^^-N=NCeH5,
а для производных фенил гидразонов антрахинона, аценафтена и бензила —
хинонгидразонная структура [803], например:
СвНБСОССвН5
NN<fHgAr
ХС6Н5
При взаимодействии гидроокиси арилртути с фенолами в мягких усло-
виях (в спиртовом растворе, при комнатной температуре и при нагревании
[804]) образуются феноляты арилртути, не способные превращаться в не-
симметричные оксиарилртутные соединения (ср. действие на фенолы гидро-
окиси арилртути в жестких условиях, что, по мнению авторов, приводит к
продуктам арилмеркурирования фенолов, ср. стр. 234).
CF3-rpynna в Hg(CF3)2 в соответствии со своими галогеноподобными
свойствами при реакции бис-трифторметиЛртути с KHal входит в состав
комплексного аниона К2[Н§(СРз)2На1а] [805, 806]. Однако криоскопичес-
кие измерения водных растворов опровергают эту точку зрения на их строе-
ние и говорят в пользу того, что такие соединения являются 1,1-аддуктами
Hg(CF3)2 и НаГ [714а]. При взаимодействии диэтилртути с триэтилсиланом
[795а] и пентаэтилдисиланом [7956] образуются соединения C2HbHgSi(C2H5)3
и C2H5HgSi(C2H5)2Si(C2H5)3 наряду с другими продуктами (см. гл. XVI,
стр. 374). C6HsHgSi(C6Hg)3, получаемое действием дифенилртути или броми-
стой фенилртути на (C6H5)3SiLi, неустойчиво и тотчас диспропорциони-
руется на Hg и (C6Hb)4Si [795в].
Описано образование клатратных соединений диэтилртути с три-о-тимо-
тидом [807].
Изучена ассоциация RHg+ с сульфгидрильными и другими функциональ-
ными группами белка [808—818].
ЛИТЕРАТУРА
1. Разуваев Г. А..Петухов Г. Г., Жильцов С. Ф., Кудрявцев Л. Ф,
ДАН СССР, 141, 107 (1961).
2. Разуваев Г. А..Петухов Г. Г., Жильцов С. Ф., Кудрявцев Л. Ф.
ДАН СССР, 135, 87 (1960).
2а. Разуваев Г. А., П е т у х о в Г. Г., Жн льцов С. Ф., Кудрявцев Л. Ф.
ДАН СССР, 144, 810 (1962).
26. Р а 'з у в а е в Г. А., Ж и л ь ц о в С. Ф., Д р у ж к о в О. Н., Петухов Г. Г.
ДАН СССР, 152, 633 (1963).
3. Kreevo у М. М., Hansen R. L. J. Phys. Chem., 65, 1055 (1961).
4. Разуваев Г. А., П е т у х о в Г. Г., К а п л и и Ю. А. ДАН СССР, 135, 342
(1960).
5. С a h о u г s А. С. г., 76, 748 (1873); Zbl., 1873, 275.
6. Otto R. Вег., 2, 542 (1869).
7. Gilman Н., F b г d G. М. Zbl., 1939, II, 3062; Jowa State Coll. J. Sci., 13, 135
(1939).
8. К о т о н M. M. ЖОХ, 9, 912 (1939).
9. К о т о н M. M., 3 о p н н a T. M. ЖОХ, 17, 1303 (1947).
10. Corwin A. H., N ay lor M. A. J. Am. Chem. Soc., 69, 1004 (1947).
11. Большакова А. А. ЖОХ, 24, 266 (1954).
12. P а з у в a e в Г. А. ЖОХ, 24, 1693 (1954).
13. Ч e p н о в И. А. ЖОХ, 20, 325 (1950).
14. Ч e p н о в И. А. ЖОХ, 22, 97 (1952).
15. Котов М. М. ЖОХ, 22, 1136 (1952).
16. Котой М. М., Киселева Т. М. ЖОХ, 22, 1139 (1952).
17, WinsteinS., Traylor Т. G. J. Am. Chem. Soc., 77, 3747 (1955).
18. N e r d e 1 F., Makower S. Naturwiss., 45, 490 (1958).
Реакции ртутноорганических соединений
337
19. Jensen Т. R., Q u е 1 1 е 11 е R. J. J. Am. Chem. Soc., 83, 4477 (1961).
20. Jensen T. R., Quellette R. J. J. Am. Chem. Soc., 83, 4478 (1961).
21. Winstein S., Tr ay lor T. G. J. Am. Chem. Soc., 78, 2597 (1956).
22. Kaufman F., Corwin A. H. J. Am. Chem. Soc., 77, 6280 (1955).
23. Dessy R. E., Kim J.-Y. J. Am. Chem. Soc., 83, 1167 (1961).
24. Dessy R. E., R ey no 1 ds G. F., Kim J.-Y. J. Am. Chem. Soc., 81, 2683 (1959),
24a, К i m J.-Y. Diss. Abstr., 21, 32, 71 (1961).
25. D e s s у R. E., Kim J.-Y. J. Am. Chem. Soc., 82, 686 (1960).
26. P e у т о в О. А., Б e л e ц к а я И. П., А л e й н и к о в a M. Я. ЖФХ, 36, 489
(1962).
27. Несмеянов А. Н., БорисовА. Е., Савельева И. С., Круглова
Н. В. Изв. АН СССР, ОХН, 1961, 726.
28. Несмеянов А. Н., БорисовА. Е., С а в е л ь е в а И. С. Изв. АН СССР,
ОХН, 1961, 2241.
28а . Whitmore F. С., Bernstein Н. J. Am. Chem. Soc., 60, 2626 (1938).
286. Несмеянов А. Н., Борисов А. Е., Савельева И. С. ДАН СССР, 155,
603 (1964).
. 29. Z i т т е г Н., Makower S. Naturwiss., 41, 551 (1954).
30. Kharasch М. S., G г a f f 1 i n М. W. J, Am. Chem. Soc., 47, 1948 (1925).
31. Kharasch М. S., М а г k е г R. J. Am. Chem. Soc., 48, 3130 (1926).
32. К h а г a s с h М. S., F i е n n е г A. L. J. Am. Chem. Soc., 54, 674 (1932).
33. К h а г a s с h М. S., Р i n е s Н., L е v i n е J. Н. J. Org. Chem., 3, 347 (1938).
34. Kharasch М. S., S w а г t z S. J. Org. Chem., 3, 405 (1938).
35. Kharasch M. S., Legault R. R., SprowlsW. R. J. Org. Chem., 3, 409
(1938).
36. Kharasch M. S., Weinhouse S. J. Org. Chem., 1, 209 (1936).
37. H e с м e я н о в A. H., Кочешков К- А. Уч. зап. МГУ, 3, 283 (1934).
,38. Б об ат ин ск а я Ч. С., Кочешков К- А. ЖОХ, 8, 1850 (1938).
39. Whitmore F. С., Bernstein Н. J. Am. Chem. Soc., 60, 2626 (1938).
39а. Реутов О. А., Ловцов а А. Н. ДАН СССР, 154, 166 (1964).
40. С т е р л и н Р. Н., Ли Вей-ган, Кнунянц И. Л. ДАН СССР, 140, 137 (1961).
41. К г е е v о у М. М. J. Am. Chem. Soc., 79, 5927 (1957).
42. К г е е v о у М. М., Н a n s е n R. L. J. Am. Chem. Soc., 83, 626 (1961).
43. М а г v е I С. S., С а 1 v е г у Н. О. J. Am. Chem. Soc., 45, 820 (1926).
44. С h а т b е г s R. D., С о a t е s G. Е., L i v i n g s t о n J.G.,Musgr aveW.K.
J. Chem. Soc., 1962, 4367.
45. Hofmann K. A. Ber., 31, 1904 (1898).
46. H о f m a nn K- A. Ber., 33, 1328 (1900).
47. БлешинскийС. В., УсубакуновМ. Сборник математической конферен-
ции, посвященный 100-летию акад. Курнакова. Фрунзе, 1963.
47а . Hofmann К- A., Kirmreuther Н. Вег., 42, 4232 (1909).
476. Seyferth D., Towe R. Н. Inorg. Chem., 1, 185 (1962).
47в. StruenseeR., Sc ho e 1 1 er W., Schrauth W. Ber., 44, 1057 (1911).
48. S t e i n k о p f L., В о e t i u s. M. Ann., 546, 208 (1941).
49. В u u - H о j N. P. Bull. Soc, chim. France, 1958, 1407.
50. G a 1 e L. H., Jensen F, R., Landgrebe J. A. Chem. and Ind., 1960, 118.
50a. S e у f e r.t h D., Mui J. Y.-P., Todd L. J. J. Am. Chem. Soc., 86, 2961
(1964) . ч:.' :
, 51. Jones L. W., Werner L. J. Am. Chem. Soc., 40, 1266 (1918).
5 2. Wolff P. Ber., 46, 64 (1913).
53. Foster D. J., T о b 1 e r E. J. Org. Chem., 27, 834 (1962)1
54. Foster D. J., T о b 1 e r E. J. J. Am. Chem. Soc., 83, 851 (1961).
54a. Jensen F. R., QuelletteR. J. J. Am. Chem. Soc., 85, 363, 367 (1963).
546. W a n g С. H. J. Am. Chem. Soc., 85, 2339 (1963).
54b. Sleezer.P. D., W i n s t e i n S., Y о u n g W. G. J. Aih. Chem. Soc., 85, 1890
(1963).
55. Ichikawa K-, О u c h i H. J. Am. Chem. Soc., 82, 3876 (1960).
»55a. WinsteinS.-, Vogelf anger E., Pan de K- Chem. and Ind.,1 49, 2061
(1962). ,
556. Winstein S., Yogelfanger E., Pande K- C.,Ebel H.F. J. Am. Chem.
Soc., 84, 4994 (1962).
55в. M i k a i j a m a. T., К u w a j i m a I., S u z u k i Z. J. Org. Chem., 28, 2024. (1963).
56. I ch i k'awa K', Fukushima S., Ou chi H., T s u ch i d a M. J. Am. Chem.
Soc., 80, 6005 (1058). ' ; :
57. Ichikawa K-, F u k u s h rm a S., О u c h i H.,' T s u c h i d a M. J.Art. Chem.
Soc., 81, 3401 (1959). ' ;i .
57a. Ichikawa ;K-, Fuj.it а К., I t 0 h O. J, Am. 'Chem. SoC.,: 84, 2632 (1962).
58. Ichikawa K.., Fuku shim a S. J. Org. Ghent,24, H129 -(1959).
338
Ртутноорганические соединения
59. К о з л о в В. В., Б е л о в Б. И. ЖОХ, 25, 410 (1955).
60. К о з л о в В. В., Б е л о в Б. И. ЖОХ, 25, 565 (1955).
61. Козлов В. В. ЖОХ, 18, 2094 (1948).
62. Доку нихин И. С., Гаева Л. А. Ж- Всесоюзн. хим. общ., 6, 112 (1961).
63. КотонМ. М. ЖОХ, 22, 643 (1952).
64. L е а п d г i G., SpinelliD: Ann. Chimica, 49, 1885 (1959).
64а. Т о b 1 е г Е., Foster D. J. Z.'Naturforsch., 176, 135 (1962).
65. Z u р а п с i с В. G. Monatsh., 92, 907 (1961).
66. Gilman Н., Mo o г е L. О. J. Am. Chem. Soc., 80, 3609 (1958).
67. Snyder H. R., Weaver C. J. Am. Chem. Soc., 70, 232 (1948).
68. КотонМ. M., Мартынова В. Ф. ЖОХ, 19, 1141 (1949).
69. К от он М. М. ЖОХ, 18, 936 (1948).
70. Р а з у в а е в Г. А., В я з а н к и н Н. С. ЖОХ, 22, 640 (1952).
71. Разуваев Г. А., Ольдекоп Ю. А., ВязанкинН. С. ЖОХ, 21, 1283
(1951).
72. Р а з у в а е в Г. А., В я з а н к и н Н. С. Синтезы органических соединений, сб. 2.
М„ ИОХ АН СССР, 1952, стр. 136.
72а. Разуваев Г. А., Вязанкин Н. С., Митрофанова Е. В. ЖОХ, 34,
675 (1964).
73. Р а з у в а е в Г. А., В а с и л е й с к а я Н. С. ДАН СССР, 74, 279 (1950).
74. Разуваев Г. А., ВасилейскаяН. С. ДАН СССР, 67, 851 (1949).
75. Р а з у в а е в Г. А., Б у г а е в а 3. И. Уч. зап. ГГУ, 24, 143 (1953).
76. Hofman пК- A., Sand J. Вег., 33, 1340 (1900).
77. Sand J. Вег., 34, 2906 (1901).
78. К г е е v о у М. М., В о dem G. В. J. Org. Chem., 27, 4539 (1962).
79. S a n d J.-, В r e e s t F. Z. physik. Chem., 59, 424 (1907).
80. Ichikawa K-, О u c h i H., A r a k i S. J. Am. Chem. Soc., 82, 3880 (1960).
81. В e r g O. W., Lay W. P., Rodgman A., Wright G. F. Can. J. Chem., 36,
358 (1958).
82. R о dgm an- A., Shearer D. A., Wright G, F. Can. J. Chem., 35,
1377 (1957).
83. Kreevoy M. M., К о w i t t F. R. J. Am. Chem. Soc., 82, 739 (1960).
84. Kreevoy M. M., D i t s c h L. T. J. Am. Chem. Soc., 82, 6127 (I960).
85. К r e e v о у M. M., G i 1 j e J. W., fcretchmer R. A. J. Am. Chem. Soc., 83,
4205 (1961).
85a. Kreevoy M. M., S t о k k e r G., Kretchtner R. A., Ahmed A. K.
J. Org. Chem.,28, 3184 (1963).
86. Kreevoy M. M., G i 1 j e J. W., D i t s c h L. T., Batorewi.cz W., T u r-
n e r M. A. J. Org. Chem., 27, 726 (1962).
87. Kreevoy M. M. J, Am. Chem. Soc., 81, 1099 (1959).
87a. К r e e v о у M. M., T u r n e r M. A. J. Org. Chem., 29, 1639 (1964).
88. К r e e v о у M. M., E i s e n В. M. J. Org. Chem., 28, 2104 (1963).
89. W r i g h t G. F. Can. J. Chem., 30, 268 (1952).
89a. Dessy R. E., P a u 1 i k F. E. J. Am. Chem. Soc., 85, 1812 (1963).
90. Whitmore F. Chem. Ind. News, 26, 668 (1948).
91. S c h a 1 e g e r L. L., T u r n e r M. А. и др. J. Org. Chem., 27, 3421 (1962).
92. W h i t m о r e F. С., M i d p 1 e t о n E. J. Am. Chem. Soc., 43, 621 (1921).
93. H e с м e я н о в A. H„ П о в x Г. С. Ж0Х, 4, 958 (1934); Ber., 67, 971 (1934).
94. Несмеянов A. H., Фрейдлина P. X., Токарева Ф. А. ЖОХ, 7, 262
(1937); Ber., 69, 2019 (1937).
95. Hofmann K. A. Ber., 32, 870 (1899).
96. H e с м e я н о в A. H., Луценко И. Ф., Ф о с с В. Л. ДАН СССР, 98, 407
(1954).
97. Sen R. N., Chakravarti D. Zbl., 1930, I, 2244.
98. R а о P. S., S e s h a d r i T. R. Zbl., 1937, II, 3457.
99. В i i 1 m a n E. Ann., 388, 259 (1912).
99a. В u r d о n J., С о e P. L., Fulton M., T a t 1 о w J. C. J. Chem. Soc., 1964,
2673.
100, Bi i Iman E, Ber., 43, 568 (1910).
lOl. Schrauth W., Schoeller W. Ber., 42, 782 (1909).
102. F i s c h e r E. Ber., 40, 386 (1907).
103. Shearer D. A-, Wright G. F. Can. J. Chem., 33, 1002 (1955).
104. R о b s о n J. H., W r i g h t G. F. Can. J. Chem., 38, 1 (1960).
105. Robson J. H., Wright G. F. Can. J. Chem., 38, 21 (1960).
106. БорисовА. E., Вильчевская В. Д., Несмеянов A. H. Изв. AH
СССР, ОХН, 1954, 1008.
107. Hein F., Poblot h H. Z. anorg. allg. Chem., 248, 84 (1941).
108. H i e b e r W., Schropp W. Ber., 93,. 455 (1960).
Реакции ртутноорганических соединений
339
109. Разуваев Г. А., Савицкий А. В. ДАН СССР, 85, 575 (1952).
109а. Реутов О. А., Углова Э. В., Белецкая И. П., Светланова Т. Б.
Изв. АН СССР, ОХН, 1964, 1383.
ПО. РеутовО. А., Белецкая И. П. Изв. АН СССР, ОХН, 1960, 1716.
111а . Белецкая И. П., Р е у т о в О. А., Карпов В, И. Изв. АН СССР, ОХН,
1961, 1961.
1116. Б е л е ц к а я И. П., Р е у т о в О. А., Карпов В. И. Изв. АН СССР, ОХН,
1961, 2125.
Шв. Б е л е ц к а я И. П., Р е у т о в О. А., Карпов В. И. Изв. АН СССР, ОХН,
1961, 2129.
111г. Б е л е ц к а я И. П., Р е у т о в О. А., К а р п о в В. И. РЖХим, 1963, 22Ж24.
112а. Белецкая И. П., РеутовО. А., Гурьянова Т. П. Изв. АН СССР,
ОХН, 1961, 1997.
1126. Белецкая И. П., РеутовО. А., Гурьянова Т. П. Изв. АН СССР,
ОХН, 1961, 1589.
112в. Белецкая И. П., РеутовО. А., Гурьянова Т. П. Изв. АН СССР,
ОХН, 1961, 2178.
113а. Б е л е ц к а я И. П., А з и з я н Т. А., Р е у т о в О. А. Изв. АН СССР, ОХН,
1962, 424.
1136. Б е л е ц к а я И. П., Азизяи Т. А., Р е у т о в О. А. Изв. АН СССР, ОХН,
1962, 223.
114. Н е с м е я н о в А. Н., БорисовА. Е., ВолькенауН. А. Изв. АН СССР,
ОХН, 1956, 162.
115. J е n s е п F. R., G а 1 е L. Н. J. Am. Chem. Soc., 82, 148 (1960).
116а. Jensen F. R., Whipple L. D., WedegaertnerD. K-, L a n d g r e-
be J. A. J. Am. Chem. Soc., 82, 2466 (1960).
1166. Jensen F. R„ Whipple L. D., WedegaertnerD. K.. Landgre-
be J. A. J. Am. Chem. Soc., 81, 1262 (1959).
117. S u m m e r b e 1 1 R. К., Polkacki E. S. J. Org. Chem., 27, 2074 (1962).
118. Van Loon E. J.,• C a r t e r H. E. J. Am. Chem. Soc., 59, 2555 (1937).
119. Sc hr au th W., G e 11 e r H. Ber., 55, 2783 (1922).
120. W h i t m о r e F. C., W h i t t 1 e E. Z., H a r r i m a n B. R. J. Am.Chem. Soc.,
61, 1585 (1939).
121. J e n s e n F. R., G a 1 e L. H., J. Am. Chem. Soc., 81, 1261 (1959).
122a. Юрьев Ю. К., Зефиров H. С., ПриказчиковаЛ. П. ЖОХ, 32, 2744
(1962).
1226. Юрьев Ю. К-, Зефиров Н. С., ПриказчиковаЛ. П. ЖОХ, 33, 1793
(1963).
122в. ManolopoulosT. Р., Mednik М., L i с h t i п N. N. J. Am. Chem. Soc.,
84, 2203 (1962).
123. Summerbell R. K., Forrester S. R. J. Org. Chem., 26, 4834 (1961).
124. Несмеянов A. H., Луценко И. Ф. Изв. АН СССР, ОХН, 1942, 366.
125. В a k е г W., В а г t о n J. W., Мс О т i е J. F. W. J. Chem. Soc., 1958, 2666.
126. Несмеянов А. Н., Борисов А. Е., Гуськова А. Н. Изв. АН СССР,
ОХН, 1945, 639.
127. Несмеянов А. Н., БорисовА. Е. Изв. АН СССР, ОХН, 1945, 146.
128. Несмеянов А. Н., Луценко И. Ф., Хомутов Р. М. Изв. АН СССР,
ОХН, 1957, 942.
129. Луценко И. Ф., Фосс В. Л., Несмеянов А. Н. Изв. АН СССР, ОХН,
1965 (в печати).
130. Aldrich Р. Е., Howard Е. G., Linn W. J., М i d d 1 е t о n W. J., Shar-
key W. H. J. Org. Chem., 28, 184 (1963).
131. Ралль К. Б., Петров А. А. ЖОХ, 32, 1095 (1962).
132. Несмеянов A. H., Кочеткова H. С.,Материкова P. Б. ДАН СССР,
147, 113 (1962).
132a. Синтезы органических соединений, сб. 1. М., ИЛ, 1943, стр. 224.
133. D i m г о t h О. Вег., 35, 2853 (1902).
134. М i 11 e r Н; F., В а с h m a n n G. В. J. Am. Chem. Soc., 57, 2447 (1935).
134а. СоеР. L., Stephens R., Т a t 1 о w J. С. J. Chem. Soc., 1962, 3227.
1346. О k a w а г а М., Tana.kaJ., Imo to Е. С. А., 57, 4854 (1962).
135. Несмеянов А. Н., Перевалова Э. Г., Несмеянова О. А. ДАН
СССР, 100, 1099 (1955).
136. Несмеянов А. Н., Сазонова В. А., Дрозд В. Н., Никонова Л. А.
ДАН СССР, 131, 1088 (I960).
136а. R a u s с h М. D. J. Org. Chem., 28, 3337 (1963).
137. Несмеянов А. Н., Анисимов К- Н., Валуева 3. П. Изв. АН СССР»
ОХН, 1962, 1683.
138. Разуваев Г. А., Латяева В. И., Вышинская Л.И.ЖОХ, 31,2667(1961).
340
Ртутноорганические соединения
139. S б d е г b а с к Е. Дпп., 419, 266 (1919).
140. W h i t m о г е F. С., Thorpe М. A. J. Am. Chem. Soc., 55, 782 (1933).
140а. Tarrant Р., О’С о n п о г D. Е. J. Org. Chem., 29, 2012 (1964).
141. В а г t о с h а В., Brinkman F. Е., Kaesz Н. D., Stone F. G. А.
Proc. Chem. Soc., 1958, 116.
142. Bartocha В., Douglas С. M., G г а у М. I. Z. Naturforsch., 14В, 809 (1959).
143. БорисовА. Е. Изв. АН СССР, ОХН, 1951, 402.
144. Стерлин Р. Н., Ли Вей-ган, Кнунянц И. Л. Ж. Всесоюзн. хим. общ.,
6, 108 (1961).
145. Michaelis A., Becker Р. Вег., 13, 58 (1880).
146*. Michaelis A., Becker Р. Вег., 15, 180 (1882).
147. Michaelis А. Вег., 27, 244 (1894).
148. М i с h а е 1 i s A. Ann., 315, 19 (1901).
149. В а г t о с h а В., В i 1 b о А. I., В и b 1 i t z D. E., G г а у M. I. Angew. Chem.,
72, 36 (I960).
150. God d ar a A. E., Go d d ar d D. Organometallic compounds. Newton Friends Text
Book of Inorganic Chemistry, vol. XI. London, 1928, part 1, p. 235. .
151. G о d d a r d A. E. J. Chem. Soc., 123, 1161 (1923).
152. Несмеянов A. H., БорисовА. E., Шепелева P. И. Изв. АН СССР,
1949, 582.
153. Несмеянов A. H., Фрейдлина P. X., Кочетков А. К- Изв. AH
« СССР, ОХН, 1948, 445.
154. Несмеянов A. H., БорисовА. E., Новикова Н. В. Изв. АН СССР,
ОХН, 1959, 1216.
155. Борисов А. Е. Изв. АН СССР, ОХН, 1961, 1036.
156. Несмеянов А. Н., БорисовА. Е., Новикова Н. В. ДАН СССР, 94,
289 (1954),
157. Несмеянов А. Н., БорисовА. Е., Новикова Н. В. Изв. АН СССР,
ОХН, 1959, 259.
158. Несмеян о-в А. Н., Б о р и с о в А. Е., Савельева И. С., Голубе-
ва Е. И. Изв. АН СССР, ОХН, 1958, 1490.
J59. БорисовА. Е„ Новикова Н. В. Изв. АН СССР, ОХН, 1957, 1258.
160. Несмеянов А. Н„ Кудрявцева Т. А. Уч. зап. МГУ, 151, 57 (1951).
161. G о d d а г d А. Е. J. Chem. Soc., 121, 36 (1922).
162. Мель н и ков Н. Н., РокицкаяМ. С. ЖОХ, 7, 1472 (1937).-
163. Глушкова В. П., Кочешков К- А. ДАН СССР, 103, 615 (1955).
164. Глушкова В. П., Кочешков К- А. ДАН СССР, 116, 233 (1957); Изв. АН
СССР, ОХН, 1957, 1193.
J65. Б о р и с о в.А. Е., Осипова М. А. Изв. АН СССР, ОХН, 1961, 1039.
166. Н е с м е я н о в А. Н., Д в о р, я н ц е в а Г. Г., Ко ч е т к о в а Н. С., Мате-
ри ков а Р. Б., Шей н кер Ю. Н. ДАН СССР, 159, 847 (1964).
166а. Seyferth Ь., Е i s е г t М. А., Т о d d L. J. J. Am. Chem. Soc., 86, 121 (1964).
167. Seyferth D., BurlitchJ.M. J. Am. Chem. Soc., 85, 2667 (1963).
J68. Seyferth D., Minasz R. J., Treibe'r A. J. H., В u r 1 i t c h J. M.,
DowdS. R. J. Org. Chem., 28, 1163 (1963).
169. LadenburgA. Ann., 173, 152 (1873); Ber., 6, 379 (1873); F r i e d e 1 C., L a den-
fa u r g A. Ann., 159, 259 (1871); LadenburgA. Ber., 7, 803 (1874).
170. Несмеянов A. H., Луценко И. Ф., Братцев В. А. ДАН СССР, 128,
551 (1959).
171. Б а у к о в Ю. И., Л у ц е н к о И. Ф. ЖОХ, 32, 2746 (1962).
172. В г i n k m a n F. Е„ S t о n е F. G. A. J. Inorg. a. Nucl. Chem., 11, 24 (1959).
173. О г n d о г f f W. К., Т a b е г n D. L., D е n n i s L. М. J. Am. Chem. Soc., 49,
2512 (1922).
174. A г о n h e i m B. Ann., 194,145 (1878).
175. Вязанкин H. С., Разуваев Г. А., Корнева С. П. Ж0Х, 33, 1041 (1933).
176. H а д ь M. M., Кочешков К. А. ЖОХ, 12, 409 (1942).
177. Панов E. M., Лодочникова E. И., Кочешков К- А. ДАН СССР, 111,
1042 (1956).
178. Лодочникова E. И., Панов Е.. М., Кочешков К- А. Изв. АН СССР,
ОХН, 1957, 1484.
178 а. Ло дочникова Е. И., Панов Е. М., Кочешков К- А. ЖОХ, 29,
2253 (1959).
,f9-. С riegeeR., DimrothP., SchempfR. Ber,, 90, 1337 (1957) /
* 80. Разуваев Г. А., Богданов И. Ф. ЖОХ, 3, 367 (1933).
* 81. Levy L. Zbl., 1892, I, 735.
|82; Michaelis A. Ber., 13, 2174 (1880).
;83. Guichard F. Ber., 32, 1572 (1899).
• 84. M i c ha e 1 i s A, Ann., 181, 288 (1876).
Реакции ртутноорганических соединений
341
185. К а е s z Н., Stone F. J. Org. Chem., 24, 635 (1959).
185а. SeyferthD., FreyerW. J. Org. Chem., 26, 260 (1961),
186. Michaelis A., Schenk A. Ann., 260, 7 (1890).
187. Michaelis A., Pane с к C. Ann., 212, 213 (1882).
188. M i c h a e 1 i s A. Ann., 293, 261 (1896); 294, 1 (1896).
189. Ягупольский Л. M., Ю ф а П. А. Ж0Х, 28, 2853 (1958).
190. К e 1 b e W. Ber., 11, 1499 (1878).
191. M i c h a e 1 i s A., L i n к A. Ann., 207, 193 (1881).
192. M e i s e n h e i m e r J., Casper J., HoringM., LauterW., L i c h t e n-
stadtL., Samuel W. Ann., 449, 213 (1926).
193. Hartman H., BeermannC., CzempikH. Z. anorg. allg. Chem., 287,261
(1956).
194. P о p e W„ G i b s о n C. S. J. Chem. Soc., 101, 735 (1912).
195. Wedekind E. Ber., 45, 2935 (1912).
195a. NenitzescuC. D., Isacescu D. A., Gruescu C. Zbl., 1940, I, 532.
196. La С о s t e W., Michaelis A. Ann., 201, 184 (1880).
197. M i c h a e 1 i s A. Ber., 8, 1316 (1875).
198. M i c ha e 1 is A. Ber., 9, 1566 (1876).
195. M i c h a e 1 i s A. Ann., 321, 155 (1902).
200. La С о s t e W., Michaelis A. Ber., 11, 1883 (1878).
201. Roeder G., В 1 a s i N. Ber., 47, 12748 (1914).
202. Michaelis A„ Schulte Q. Ber., 15, 1953 (1882).
203. La С о s t e W. Ann., 208, 3 (1881).
204. В e с к W. W., Hami 1 ton C. S. J. Am. Chem. Soc., 60, 620 (1938).
205. Lowe W. G., H a m i 1 t о n C. S. J. Am. Chem. Soc., 57, 1080 (1935).
206. Lowe W. G, Ham 11 ton C. S. J. Am. Chem. Soc., 57, 2374 (1935).
207. W e i t к a m p W., Hamilton C. S. J. Am. Chem. Soc., 59, 2699 (1937).
208. Steinkopf W„ M i e g W. Ber., 53, 1014 (1920).
209. Jones W. E., Rosser R. J., Woodward F. N. J. Soc. Chem. Ind., 68,
258 (1949).
210. В 1 i ck e F. F„ Sm i th F. D. J. Am. Chem. Soc., 51, 3479 (1929).
211. D a v i e s W. С., О t h e n C. W. J. Chem. Soc., 1936, 1236.
212. M i c h a e 1 i s A. Ann., 320, 271 (1901).
213. M a t s u m i j a K. Zbl., 1926, I, 652.
214. S t e i n к о p f W. Ann., 413, 310 (1917).
215. F i n z i C. Gazz., 45, 280 (1915).
216. F i n z i C. Gazz., 55, 824 (1925).
217. BucktonG. B. J. Chem. Soc., 16, 22 (1863).
218. Hasenbaumer.I. Ber., 31, 2911 (1898).
219. Michaelis A., GiintnerA. Ber., 44, 2316 (1911).
220. Woo ds L. W. C. A., 39, 693 (1945); Iowa State Coll. J. Sci., 19, 611 (1944).
221. КозминскаяТ. K-, Кочешков К. А. Бюлл. ВХО, № 9, 21 (1940).
222. Ману л кин 3. М., Татаренко А. Н., ЮсуповФ. Сборник статей
по общей химии, 2, 1308 (1953).
223. Ман ул кин 3. М. Узб. хим. ж., 2, 41 (1959).
224. Challenger F.,. Allpress С. F. J. Chem. Soc., 1921, 923.
225; С а г г i с k W. L., R е i с h 1 е W. Т., P e n e 1 1 a F., S m i t h J. J. J. Am.
Chem. Soc., 82, 3887 (1960).
225a. H о f m а п n K. A., F e i g e 1 H. Ber., 38, 3655 (1905).
226. Heu man n K., KbchlinP. Ber., 16, 1625 (1883).
227. Michaelis A., Gode hau x E. Ber., 24, 758 (1891).
228. О 11 о R. Ber., 18, 246 (1885).
229. Терентьев А. П., К о с т А. Н., Юркевич А. М., X а с к и н а Е. Е.,
Обреимова Л. И. Вестник МГУ, сер. физ., мат. и естеств. наук, № 4, 121 (1953).
230. Spinelli D., S а 1 v е m i п i A. Ann. chimica, 51, 389, 1296 (1961).
231. Несмеянов А. Н., Перевалова Э. Г., Несмеянов аО. А. ДАН
СССР, 119, 288 (1958).
232. L е i с е s t е г Н. М. J. Am. Chem. Soc., 60, 619 (1938).
233. Мельников Н. Н., Рокицкая М. С. ЖОХ, 8, 834 (1938).
234. Leicester Н. М. J. Am. Chem. Soc., 57, 1901 (1935).
235. Leicester Н. М. J. Am. Chem. Soc., 53, 4428 (1931).
236. С а т р b е 1 1 Т. W„ М с С u 1 1 о и g h J. D. J. Am. Chem. Soc., 67, 1965 (1945).
237. К r a f f t F., L у о n s R. E. Ber., 27, 1768 (1894).
238. Campbell LG. M., T и r n e r E. E. J. Chem. Soc., 1938, 37.
239. Rheinboldt H., VicentiniG. Ber., 89, 624 (1956).
240. A 1 a n E. C. A., 51, 17557 (1957).
241. W i 1 1 g e г о d t C. Ber., 30, 56 (1897).
242. W i 1 1 g e г о d t C. Ber., 31, 915 (1898).
342
Ртутноорганические соединения
243. Фрейдлииа Р. X., Несмеянов А. Н. ДАН СССР, 29, 566 (1940).
244. Фрейдлииа Р. X., БрайнинаЭ. М., Несмеянов А. Н. Изв. АН
СССР ОХН 1945 647
245. Несмеянов А. Н., Фрейдлииа Р. X. Изв. АН СССР, ОХН, 1943, 152.
246. Whitmore F. С., Thur mann N. J. Am. Chem. Soc., 51, 1491 (1929).
247. KekuleA., Franc hi mon t A. Ber,, 5, 907 (1872).
248. D r e h e r E., О t t о R. Ann., 154, 93 (1870).
249. Sc h r a e d e r W. X., Brewster R. Q. J. Am. Chem. Soc., 60, 751 (1938).
250. G i 1 m a n H., W r i g h t G. F. J. Am. Chem. Soc., 55, 3302 (1933).
251. О t t о R. Ber., 3, 197 (1870).
252. V о 1 h a r d J. Ann., 267, 172 (1892).
253. Umesawa S. Zbl., 1939, II, 3821; Bull. Chem. Soc. Japan, 14, 155 (1939).
254. C h a 1 1 e n g e r F., M i 1 1 e r S. A. J. Chem. Soc., 1939, 1005.
255. Мельников H. H., P о к и ц к а я М. С. ЖОХ, 7, 464 (1937).
256. Calvery Н. О. J. Am. Chem. Soc., 48, 1009 (1926).
257. G i 1 т а п Н., N е 1 s о п J. F. J. Am. Chem. Soc., 61, 743 (1939).
258. Steinkopf W., В auermeis ter M. Ann., 403, 50 (1914).
259. Steinkopf W., KiHingstadA. Ann., 532, 288 (1937).
260. Малиновский M. С. Tp. Горьк. гос. пед. ин-та, 1940, № 5, 39; С. A., 37, 3070
(1943).
261. Сколдинов А. П., Кочешков К. А. ЖОХ, 12, 398 (1942).
262. W г i g h t G. F. J. Am. Chem. Soc., 58, 2653 (1936).
263. C h u t e W. J., О r c h a r d W. M., W r i g h t G. F. J. Org. Chem., 6, 157 (1941).
264. Несмеянов A.. H., Луценко И. Ф. ДАН СССР, 59, 707 (1948).
265. Несмеянов А. Н., Перевалова Э. Г. Изв. АН СССР, ОХН, 1954, 1002.
266. Несмеянов А. Н., Луценко И. Ф., Туманова 3. М. Изв. АН СССР,
ОХН, 1949, 601.
267. Якубович А. Я., Разумовский В. В., ВострухинаЗ. Н., Р о-
зеиштейнС. М. ЖОХ, 28 1930 (1958).
268. Несмеянов А. Н., Луценко И. Ф., Хомутов Р. М., Дубовиц-
к и й В. А. ЖОХ, 29, 2817 (1959).
269. Л у ц е н к о И. Ф., К р а й ц 3. С. ДАН СССР, 135, 860 (1960).
270. Н е с м е я н о в А. Н., Л у ц е н к о И. Ф., К р а й ц 3. С., Б о к о в о й А. П.
ДАН СССР, 124, 1251 (1958).
271. Несмеянов А. Н., Кочетков Н. К. Изв. АН СССР, ОХН, 1949, 305.
272. Несмеянов А. Н., РеутовО. А. Изв. АН СССР, ОХН, 1953, 655.
273. S u i d a W. С. Zbl., 1880, 585.
274. К о т о н М. М., 3 о р и н а Т. М., О с б е р г Э. Г. ЖОХ, 17, 59 (1947).
275. Разуваев Г. А. Уч. зап. ГГУ, 15, 81 (1949).
276. Манулкин 3. М. РЖХим, 1959, 15244; Узб. хим. ж., 1958, № 2, 41.
277. Разуваев Г. А., О л ь декой Ю. А. ДАН СССР, 64, 77 (1949).
278. Разуваев Г. А., Ольдекоп Ю. А. Уч. зап. ГГУ, 15, 85 (1949).
279. БорисовА. Е. Изв. АН СССР, ОХН, 1951, 524.
279а. Несмеянов А. Н., Борисов А. Е., Голубева Е. И., К о в ре-
до в А. И. Изв. АН СССР, ОХН, 1961, 1582; Tetrahedron, 18, 683 (1962).
280. Разуваев Г. А., Митрофанова Е. В., Вязанкин Н. С. ДАН СССР,
144, 132 (1962).
281. Несмеянов А. Н., Перевалова Э. Г., Несмеянова О. А. Изв.
АН СССР, ОХН, 1962, 47.
281а. Разуваев Г. А., Жильцов С. Ф., Дружков О. Н., Петухов Г. Г.
ДАН СССР, 156, 393.(1964).
282. Несмеянов А. Н„ Шацкая Р. X. ЖОХ, 5, 1268 (1935).
283. С а 1 m е 1 s G. С. г., 99, 240 (1884).
284. Wright I. F. J. Am. Chem. Soc., 57, 1993 (1935).
285. Wh i t more F. C., R о h r m a n n E. J. Am. Chem. Soc., 61, 1591 (1939).
286. Hofmann K. A., San d J. Ber., 33, 1348 (1900).
287. M а и c h о t W., К 1 fl g A. Ann.; 420, 170 (1920).
288. W r i g h t G. F. J. Am. Chem. Soc., 57, 1993 (1935).
289. S a n d J. Ber., 34, 1385 (1901).
290. Разуваев T. А., ВасилейскаяН. С. ДАН СССР, 80, 69 (1951).
291. L e a n d r i G„ S p i n e 1 1 i D. C. A., 54, 5530 (1960).
292. P а з у в a e в Г. A„ К о т о н М. М. ЖОХ, 1, 864 (1931); Вег., 66, 854 (1932).
293. Котон М. М. ЖОХ, 8, 1791 (1938).
294. Разу ваев Г. А., Котон М. М. ЖОХ, 4, 647 (1934); Вег., 66, 1210 (1933).
295. Левина Р. Я., Костин В. Н., ТартаковскийВ. А. ЖОХ, 27, 2049
(1947).
296. Котон М. М. ЖОХ, 9, 1622 (1939).
297. Котон М. М. ЖОХ, 9, 2196 (1939).
Реакции ртутноорганических соединений
343
298. К о тон М. М. ЖОХ, 11, 179 (1941).
299. Разуваев Г. А., Петухов Г. Г., Калинина Р. В. ЖОХ, 26, 1685 (1956).
300. Л у кевиц 3., Гил л ер С. А. Изв. АН Латв. ССР, 1961, № 4, 99.
301. Barton D. Н. R., RosenfelderW. J. J. Chem. Soc., 1951, 2381.
302. Henbest H. H., N i с о 1 1 s R. J. Chem. Soc., 1959, 2277.
303. T г а у 1 о r T. G. Chem. and Ind., 1959, 1223; J. Am. Chem. Soc., 86, 244 (1964).
304. Schorygin P. P. Ber., 41, 2723 (1908).
305. Schorygin P. P. Ber., 43, 1938 (1910).
306. Gilman H., Breuer F. J. Am. Chem. Soc., 56, 1123 (1934).
307. H а д ь M. M„ К о ч e ш к о в К- А. ЖОХ, 8, 42 (1938).
308. Житкова Л. А., ШевердинаН. И., Кочешков К- А. ЖОХ, 8, 839
(1938).
309. D г е h е г Е., О 11 о R. Вег.,.2, 542 (1869).
310. Z е i s е г Н. Вег., 28, 1670 (1895).
311. Е g 1 i n t о n G., McC r a e W. J. Chem. Soc., 1963, 2295.
312. Carothers W. H., JacobsonR. A., BerchetG. J. J. Am. Chem.
Soc., 55, 4665 (1933).
313. Несмеянов A. H., БорисовА. E., Волькенау H. А. Изв. АН СССР,
ОХН, 1956, 162.
314. Несмеянов A. H., Кочешков К. А., Пузыр ева В. П. ЖОХ, 7,
118 (1937).
315. Michaelis A., Schulte С. Вег., 14, 1952 (1881).
316. М i с h а е 1 i s А. Вег., 15, 1956 (1882).
317. Р а з у в а е в Г. А., К о т о н М. М. ЖОХ, 3, 792 (1933).
318. Wittig G., Bickel ha u р t F. Ber., 91, 883 (1958).
319. Wittig G., Hahn E., TochtermannW. Ber., 95, 431 (1962).
320. W i 11 i g G., H e r w i g W. Ber., 87, 1511 (1954).
321. Cava M. P., S t u c k e r J. F. J. Am. Chem. Soc., 77, 6022 (1955).
322. R a u s c h M. D. Inorg. Chem., 1, 414 (1962).
323. Несмеянова О. А., Перевалова Э. Г. ДАН СССР, 126, 1007 (1959).
324. Schwenke Е.,' Papa D:, Whitman В., Ginsberg Н. J. Org. Chem.,
9, 1 (1944).
325. Schlenk W„ H о 1 t z J. Ber., 50, 262 (1917).
326. H ager F. D„ M a r v e 1 C. S. J. Am. Chem. Soc., 48, 2689 (1926); 49, 2323 (1927).
327. R a u s c h M. D. J. Org. Chem., 28, 3337 (1963).
328. GenslerW. J., Mahadevan A. P. J. Org. Chem., 21, 180 (1956).
329. Талалаева T. В., Кочешков К. А. ЖОХ, 8, 1831 (1938).
330. W i 11 i g G., В i c k e 1 h a u p t F. Ber., 91, 865 (1958).
331. W i t t i g G., Herwig W. Ber., 88, 962 (1955).
332. Gilman H., J о n e s R. G. J. Am. Chem. Soc., 63, 1443 (1941).
333. В u c k t о n'G. B. Ann., 112, 222 (1859).
334. Morton A. A., Hechenb le ikner I. J. Am. Chem. Soc., 58, 1024 (1936).
335. Carothers W. H., С о f f m a n n D. D. J. Am. Chem. Soc., 51, 588 (1929).
336. W h i t m о r e F. C., Z о о k H. D. J. Am. Chem. Soc;, 64, 1783 (1942).
337. Gilman H., Y о u n g R. V. J. Org. Chem., 1, 315 (1936).
338. Gilman H., PacevitzH. A., Baine O. J. Am. Chem. Soc., 62, 1514 (1940).
339. L a n e J., U 1 r i c h S. E. J. Am. Chem. Soc., 73, 5470 (1951).
340. Sey fer fh D., Burlitc.h J. M. J. Am. Chem. Soc., 84, 1757 (1962).
341. A c re e S. F. Am. Chem. J., 29, 588 (1903).
342. HilpertS., GriittnerG. Ber., 46, 1675 (1913).
343. Wittig G,, Benz E. Ber., 91, 873 (1958).
344. Несмеянов A. H., Луценко И. Ф., Хомутов Р. М. ДАН СССР, 120,
.1049 (1958).
345. SeyferthD., Hofmann Н. Р., В ur ton R., Helling J. F. Inorg.
Chem., 1, № 2, 227 (1962).
346. Schorygin P. P. Ber., 43, 1913 (1910).
347. Schorygin P. P. Ber., 43, 1931 (1910).
348. Cowan D. Q., Mosher H. S. J. Org. Chem., 27, 1 (1962).
349. Лавров В. ЖРХО, 16, 93 (1884).
350. Burg A. В., S с h 1 е s i n g е г Н. I. J. Am. Chem. Soc., 62, 3425 (1940).
351. М и s а г t R., MelletR., Jaworski R. Bull. Soc. Chim. France, 1956, 445.
352. R a b i d e a и S. W„ A 1 e i M. jr., H о 1 1 e у С. E. C. A„ 50, 3992 (1956); Atomic
Energy Comm., LA 1687, 11 (1954).
353. Fray G. I., RobinsonR. Tetrahedron, 18, 261 (1962).
354. HeinF., Petzchner F., W a g 1 e г K-, S e g i t z F. A. Z. anorg. Chem.,
141, 161 (1924).
355. FranklandE., DuppaB.F. J. Chem. Soc., 13, 181, 194 (1861).
356. C a h о и r s A. Jahresb., 1873, 520; C. r., 76, 1383 (1873).
344
Ртутноорганические соединения
357. Gilman Н., Schulze F. J. Chem. Soc., 1927, 2663k
358. Dessy R. E. J. Am. Chem. Soc., 82, 1580 (I960).
359. W i t t i g G., Meyer F. J„ Lange G.. Ann., 571, 167 (1951).
360. W i t t i g G., H о r n b e r g e r H. Ann., 577, 11 (1952).
361. Lohr P. Ann., 261, 72 (1891).
361a. Dessy R. E., Кар lan F., Coe G. R., Salinger R. ,M. J. Am. Chem.
Soc., 85, 1191 (1963).
362. Gilman H., Schulze F. J. Am. chem. Soc., 49, 2328 (1927).
363. Gilman H., Brown R. E. Rec. trav. chim., 49, 724 (1930).
364. Mos h er H. S., L о e f f 1 e r P. K. J- Am. Chem. Soc., 78, 4959 (1956).
365. Fleck H. P. Ann., 276, 138 (1893).
366. Gilman H., Brown R. E. Rec. trav. chim., 49, 202 (1930).
367. W a g a F. Ann., 282, 320 (1894).
368. Schlenk, Ws, jr. Ber., 64, 736 (1931).
368a. McCoyC. R., A 1 1 r e d A. L. J. Am. Chem. Soc., 84, 912 (1962).
369. Frankland E., D u p p a B. F. Ann., 130, 117 (1864); J. Chem. Soc., 17, 29
(1864).
370. C a h о u r s A. Zbl., 1873, 200; C. r., 76, 133 (1873).
371. Щ e p б а к о в А. ЖРФХО, 13, 350 (1881).
372. C a h o.u r.s A. J. pract. Chem, (2), 8, 398 (1873).
373. Mar qu ar d t A. Ber., 21, 2035 (1888).
374. Соколове. ЖРФХО, 19, 202 (1887).
375. P о n z i o G. Zbl., 1900, II, 624; Gazz., 30, II, 25 (1900).
376. Кочешков К. А., Несмеянов A. H., Потросов В. И. Уч. зап. МГУ,
3, 305 (1934); Вег., 67, 1138 (1934).
377. Gilman Н, К i г b у R. Н. J. Am. Chem. Soc., 63, 2046 (1941).
378. Несмеянов А. Н., Макарова Л. Г. ЖОХ, 7, 2649 (1937).
379. В и с k t о n G. В., О d 1 i n g W. Ann, Chem, (Suppl.), 4, 109 (1865).
380. Wartick T., Schlesinger H. J. J. Am. Chem. Soc,, 75, 835 (1953).
381. Laubengayer A. W., Gilliam W. F. J. Am. Chem. Soc., 63, 477 (1941).
382. R о и x E„ L о и i s e E. Bull. Soc. Chim. France (2), 50,497 (1888).
383. В amfor d С. H., L e v i D. L., N e w i t t D. M. J. Chem. Soc., 1946, 468.
384. Pitzer K. S., Gutowsky H. S. J. Am. Chem. Soc., 68, 2204 (1946).
385. M ii 11 e r A., S a и e r w a 1 d A. Monatsh., 48, 737 (1927).
386. Захаркин Л. И„ Охлобыстин О. Ю. Изв. АН СССР, ОХН, 1958, 1278.
387. MurahashiS., NozakuraS., TakeushiS. РЖХим, 1961, 5Ж 246.
388. В а г t о с h а В., В i 1 Ь о A. J., В u b 1 i t z D. E., G г а у M. I. Z. Naturforsch.,
16B, 357 (1961).
389. Bartocha B., Bilbo A. J. J. Am. Chem. Soc., 83, 2202 (1961).
390. F r i e d e 1 С., К r a f f t s H. Ann. chim. (6), 14, 457 (1888).
391. H i 1 p e r t S., G rii t t n er G. Ber., 45, 2828 (1912).
392. Gilman H., Marple К- E. Rec. trav. chim., 55, 133 (1936).
393. S trohmeier W., H u m p f n e r K- Ber., 90, 2339 (1957).
394. Разуваев Г. A., Митрофанова E. В., Петухов Г. Г. ЖОХ, 30,
1996 (I960).
395. KrauseE., DittmarP. Ber., 63, 2401 (1930).
396. НесмеяновА. H., H о в и к о в а Н. Н. Изв. АН СССР, ОХН, 1942, 373
397. Menzel W. Z. anorg. allg. Chem., 269, 59 (1952).
398. Wiberg E., Johannsen T., StecherO. Z. anorg. allg. Chem., 251, 114
(1943).
399. С о a t e s G. E. J. Chem. Soc., 1951, 2003.
400. Dennis L. M„ Patnode W. J. Am. Chem. Soc., 54, 182 (1932).
401. О 1 i v.e r J. P., Stevens L. G. J. Inorg. Nucl. Chem., 19, 378 (1961).
402. Gilman H., J о n e s R. G. J. Am. Chem. Soc., 62, 980 (1940).
403. Van der Kelen G. P. Bull. Soc. chim. Belg., 65, 343 (1956).
404. D e n n i s L. M., W о r k R. W., R о c h о w E. G., C h a m о t E. M. J. Am.
Chem. Soc., 56, 1047 (1934).
405. S c h u m b N. С., Crane H. I. J. Am. Chem. Soc., 60, 306 (1938).
406. Gilman H., JonesR. G. J. Am. Chem. Soc., 62, 2353 (1940).
406a. Seyferth D., Burlitch J. M., Heiren J. K. J. Org. Chem., 27, 1491
(1962).
407. George M. V„ Lichtenwalter G. D., Gilman H. J. Am. Chem.
Soc., 81, 978 (1959).
408. J а с о b s G. C. r., 238, 1825 (1954).
409. Емельянова Л. И., Макарова Л. Г. Изв. АН СССР, ОХН, 1960, 1710.
410. Емельянова Л. И., Виноградова В. Н., МакароваЛ. Г., Нес-
меяновА. Н. Изв. АН СССР, ОХН, 1962, 53.
411. S h и г о w А. Е., R a s и w a j е w G. А. Вег., 65, 1507 (1932).
Реакции ртутноорганических соединений
345
412. Несмеянов А. Н., БорисовА. Е., Абрамов а А. Н. Уч. зап. МГУ*
132, 73 (1950); Изв. АН СССР, ОХН, 1949, 570.
413. Луценко И. Ф., Б а у к о в Ю. И., ХасаповБ. Н. ЖОХ, 33, 2724 (1963).
413а. S е у f е г t h D., S i m m о n s, jr. H. D. T о d d L. J. J. Organomet. Chem., 2,
282 (1964).
414. Long L. H., Sackman J. F. C. A., 50, 9081 (1956).
415. Пат. США 2844614 (1958); С. A., 53, 3061 (1959).
416. Dreher E„ Otto R. Ber., 2, 542 (1869).
417. Dreher E., О 11 о R. Ann., 154, 37 (1870).
418. P а з у в a e в Г. А., Котон M. M. Ж0Х, 5, 361 (1935).
419. M i d d 1 e t о n W. J., Howar d E. G., S h a r k e у W. H. J. Am. Chem. Soc.*
83,. 2589 (1961).
420. К r e s p a n C. G. J. Org. Chem., 25, 105 (1960).
421. Lyons R. E., Bush G. C. J. Am. Chem. Soc., 30, 834 (1908).
422. I s s 1 e i b К., В r a c k A. Z. Naturforsch., 118, 420 (1956).
423. Нес м e яновА. H., Кочешков К- А. ЖРХО, 62, 1795 (1930); Ber., 63,
2496 (1930).
424. Кочешков К- А., Несмеянов A. H. ЖОХ, 1, 219 (1931); Ber., 64, 628
(1931).
425. Эскин И. T., Несмеянов А. Н., Кочешков К-А- ЖОХ, 8, 35 (1938).
426. Несмеянов А. Н., БорисовА. Е., Абрамова А. Н. Изв. АН СССР,
ОХН, 1946, 647.
427. Несмеянов А. Н., Борисов А. Е., Н о в и к о в а Н. В. ДАН СССР,
119, 504 (1958).
428. Несмеянов А. Н., Б о р и с о в А. Е., Н о в и к о в а Н. В., Оси ло-
ва М. А. Изв. АН СССР,- ОХН, 1959, .263.
429. ReytovO. A. Bull. Soc. chim. France, 1963, 1383.
430. Kozeschkow К- A. Ber., 66, 1661 (1933).
430a. В а у e r A. Ber., 7, 1638 (1874).
431. Bamberger E. Ber., 30, 506 (1897).
432. Kunz I. Ber., ЗГ, 1528 (1898).
433. S m i t h L. I., T а у 1 о r F. L. J. Am. Chem. Soc., 57, 2460 (1935).
434. T и т о в А. И., Л а п т e в H. Г. ЖОХ, 19, 267 (1949).
435. Макарова Л. Г., Несмеянов А. Н. ЖОХ, 9, 771 (1939).
436. Carmack М., В a i г е г М. М., HandrickG. К-, Kissinger L. W.t
S p e c h t E. H. J. Am. Chem. Soc., 69, 785 (1947).
437. Bamberger E. Ber., 32, 3546 (1899).
438. T и т о в А, И., Б a p ы ш н и к о в a A. H. ЖОХ, 17, 829 (1947).
439. Ogata I., Tsuchida М. J. Org. Chem., 21, 1065 (1956).
440. Петрович П. И. ЖОХ, 30, 2808 (1960).
441. П е т р о в и ч П. И. ЖОХ, 29, 2387 (1959).
442. Т и т о в А. И., Л а п т е в Н. Г. ДАН СССР, 66, 1101 (1949).
443. Т и т о в А. И., Р у с а н о в Д. Е. ДАН СССР, 82, 65 (1952).
444. Tedder J. М„ The ak er G. J. Chem. Soc., 1957, 4008.
445. Gilman H., WooleyB. L., Wright G. F. J. Am. Chem. Soc., 55, 2609s
(1933).
446. C hu t e W. J„ О r c h a r d W. M„ W r i g h t G. F. J. Org. Chem., 6, 157 (1941).
447. Gilman H., N e 1 s о n J. Rec. trav. chim., 55, 518 (1936).
448. Gilman H., Y a 1 e H. L. J. Am. Chem., Soc., 72, 8 (1950).
449. Белецкая И. П., Бутин К. П., Реутов О. А. Изв. АН СССР, ОХН*
1964, 1711.
450. Gilman Н., Moore F. W., Jones R. G. J. Am. Chem. Soc., 63, 2482 (1941)*
451. G i 1 m a n H., J о n e s R. G. J. Am. Chem. Soc., 63, 1439, 1443 (1941).
452. Несмеянов A. H., Печерская К- А. Изв. АН СССР, ОХН, 1941, 67.
453. Gr i gnar d V„ Abe 1 man n A. Bull. Soc. chim. France (14), 19, 18 (1916).
454. Abe Imann A. Ber., 47, 293 (1914).
455. Несмеянов A. H., Печерская К- А. Изв. АН СССР, ОХН, 1943, 317..
456. Несмеянов A. H., Кочетков Н. К- Изв. АН СССР, ОХН, 1949, 587;
Уч. зап. МГУ, 132, 1 (1950).
457. Стерлин Р. Н., Пипкина Л. П., НезговоровЛ. Ф., Кнунянц.
И. Л. Хим. наука и промышленность, 6, 809 (1959).
458. F е i g 1 F. Angew. Chem., 73, 656 (1961).
458a, Salinger R. M., Dessy R. E. Tetrahedron L., 1963, 729.
459. FranklandE. Ann., Ill, 57 (1859).
460. HilpertS., GruttnerG. Ber., 48, 906 (1915).
461. Seyferth D., H e 1 1 i n g J. F. Chem. and Ind., 1961, 1568.
462. Назарова Л. M. ЖОХ, 29, 2671 (1959).
463. H а з apo в а Л. M. ЖОХ, 31, 1119 (1961).
346
Ртутноорганические соединения
464. Реуто в О. А., У Я н - ц е й. ДАН СССР, 117, 1003 (1957).
465. Р е у т о в О. А., К н о л л ь П. Г., У Я н - ц е й. ДАН СССР, 120, 1052 (1958).
466. Несмеянов А. Н., Р е у т о в О. А., К и о л л ь П. Г. ДАН СССР, 118, 99
(1958).
467. РеутовО. А., Остапчук Г. М. ДАН СССР, 117, 826 (1957).
468. РеутовО. А., Остапчук Г. А. ЖОХ, 29, 1614 (1959).
469. WinsteinS., Traylor T.G., Garner С. S. J. Am. Chem. Soc,, 77, 3741
(1955).
470. Bro der sen K., Sch lenker U. Ber., 94, 3304 (1961).
•471. Dessy R. E., Lee I. K-, К i m J.-Y. J. Am. Chem. Soc., 83, 1163 (1961).
472. H e с м e я н о в H. A., P e у т о в О. А. ДАН СССР, 144, 126 (1962).
473. РеутовО. А., Смолина Т. А., У Я н - цей, Бубнов Ю. И, Науч-
ные доклады высшей школы. Хим. и хим. технол., 1958, № 2, 324.
473а. С м о л и н а Т. А., Кал яви н В. А., Р е у т о в О. А. Изв. АН СССР, ОХН,
1963 2235
474. Нефедов В. Д., С ин ото ва Е. Н., Ф р о л о в Н. Я- ЖФХ, 30, 2356
(1956).
475. Нефедов В. Д., СинотоваЕ. Н. Сборник работ по радиохимии. Изд-во
ЛГУ, 1955, стр. 113.
476. Hughes Е. D., Ingold С., Thorpe F. G., VolgerH. С. J. Chem.
Soc., 1961, 1133.
477. СинотоваЕ. Н. ЖНХ, 2, 1205 (1957).
478. Р е у т о в О. А., С м о л и н а Т. А., К а л я в и н В. А. ЖФХ, 36, 119' (1962).
479. РеутовО. А., Соколов В. И., Белецкая И. П. ДАН СССР, 136,
631 (1961).
480. Р е у т о в О. А., Белецкая И. П., У Я н - ц е й. Кинетика и катализ.
М., Изд-во АН СССР, 1960, стр. 43.
481. Р е у т о в О. А., Смолина Т. А., КалявинВ. А, ДАН СССР, 139, 389
(1961).
481а. Кал я вин В. А., Смол ин а Т.. А., Р е у т о в О. А. ДАН СССР, 155, 596
(1964).
482. С г о s s J. М., Р i n a j a m J. J. С. А., 45, 3694 (1951).
483. А п s а I о п i А., В е I m о n d i G., С г о a t t о U. С. А., 51, 5517 (1957); Ricerce
Sci., 26, 3315 (1956).
483а. Карпов Т. П., М а л я н о в В. А., У г л о в а В. Э., Р е у т о в О. А. Изв.
АН СССР, ОХН, 1964, 1580.
484. Нефедов В. Д., СинотоваЕ. Н. Сборник работ по радиохимии. Изд-во
ЛГУ, 1955, стр. 110.
485. Cerfontain Н., Van A k е п G. М. F. J. Am. Chem. Soc., 78, 2094 (1956).
486. Hughes Е. D., Vo Iger H. C. J. Chem. Soc., 1961, 2359.
487. РеутовО. А., Соколов В. И., Белецкая И. П. Изв. АН СССР,
ОХН, 1961, 1213.
487а. РеутовО. А., Соколов В. И., Белецкая И. П. Тр. Конференции по
проблеме применения корреляционных уравнений в органической химии, т. 1. Тарту,
1962, стр. 342; Изв. АН СССР, ОХН, 1961, 1561.
4876. РеутовО. А., Соколов В. И., Б е л е ц к а я И. II. Изв. АН СССР,
ОХН, 1961, 1217.
488. РеутовО. А., Соколов В. И., Белецкая И. П., Рябокобыл-
к о Ю. С. Изв. АН СССР, ОХН, 1963, 965.
488а. Реутов О. А., ПрайснерБ., Белецкая И. П„ Соколов В. И.
Изв. АН СССР, ОХН, 1963 , 970.
4886. К ал я в и н В. А., С м о л и н а Т. А., Р е у т 0 в О. А. ДАН СССР, 156, 94
(1964).
489. Реутов О. А., Б е л е ц к а я И. П. ДАН СССР, 131, 853 (1960).
490. Charman Н. В., Hughes Е. D., I n g о 1 d С., V о 1 g е г Н. С. J. Chem.
Soc., 1961, 1142,
491. Реутов О. А., Смолина Т. А., Ху Хун-вен. Изв. АН СССР, ОХН,
1959, 559.
492. Реутов О. А., Карпов Г. П., У г л о в а Э. В., М а л я н о в В. А. Изв.
АН СССР, ОХН, 1960, 1311; ДАН СССР, 134, 360 (i960); Tetrahedron L., 19, 6 (1960).
493. Charman Н. В., Hughes Е. D., Thorpe F. G. J. Am. Chem. Soc., 1961,
1121.
494. РеутовО. А., Ху Хун-вен, Белецкая И. П., Смолина Т. А.
ЖФХ, 35, 2424 (1961).
495. Р г i t с h а г d Н. О. J. Chem. Phys., 25, 267 (1956).
496. R u s s е 1 I М. Е., Ber nstei n R. В. J. Chem. Phys., 30, 607, 613 (1959).
497. В i 1 1 i n g e H. M., Gowen lock B. G. Proc. Chem. Soc., 1962, 24.
498. H a r r i s G. M„ Tickuer A. W. J. Chem. Phys., 15, 686 (1947),
Реакции ртутноорганических соединений
347
499. Н а г г i s G. М., Т i с к п е г A. W. Сап. J. Research, 26В, 343 (1948).
500. G о w е п 1 о с к В. G., Polanyi I. С., Warhurst F. Е. Proc. Roy Soc.,
А218, 269 (1953).
500а. Р а д и ч Л., Кравчук И. IL, Мардалейшвили Р. Е. ДАН СССР, 136,
657 (1961).
501. Ingold К. Н., L о s s i п g F. Р. J. Chem. Phys., 21, 368, 1135 (1953).
502. L о s s i n g F. P., T i с к n e r A. W. J. Chem. Phys., 20, 907 (1952).
503. L о s s i n g F. P., T а п а к a I. J. Chem. Phys., 25, 1031 (1956).
504. К r e c h M., PriceS. J. Can. J. Chem., 41, 224 (1963).
505. Muller К. H., Walters W. D. J. Am. Chem. Soc., 73, 1458 (1951).
506. Y e d d a n a p a 1 1 i L. M., S r i n i v a s a n R., P a u 1 V. J. J. Sci. Ind. Research
(India), 13B, 232 (1954); C. A., 49, 4387 (1955).
507. Cunningham J. P„ TaylorH. S. J. Chem. Phys., 6, 359 (1938).
508. L о n g L. H. Trans. Farad. Soc., 51, 673 (1955).
509. L о n g f i e 1 d J. E., W a 11 e r s W. D. J. Am. Chem. Soc., 77, 6098 (1955).
510. Ganesan R. Z. Phys. Chem., 31, 328 (1962).
511. Y e d d a n a p a 1 1 i L. M., Schubert С. C. J. Chem. Phys., 14, 1 (1949).
512. L a u r i e С. M., Long L. H. Trans. Farad. Soc., 51, 665 (1955).
513. Ganesan R. J. Sci. Industr. Res. (India), 20B, 228 (1961).
514. T о b 1 e r E., F о s t e r D. J. Z. Naturforsch., 176, 136 (1962).
515. Saunders K- W., T а у 1 о r H. S. J. Chem. Phys., 9, 616 (1941).
516. PriceS. I. W., Tro tman - D i ck enson A. F. Trans. Farad. Soc., 53, 939
(1957).
517. CattanachJ., Long L. H. Trans. Farad. Soc., 56, 1286 (1960).
518. L о n g L. H. J. Chem. Soc., 1956, 3410.
519. Srinivasan R. J. Chem. Phys., 28, 895 (1958).
520. T а у 1 о r H. S., J о n e s W. H. J. Am. Chem. Soc., 52, 1111 (1930).
521. Pasf ield W. H. Diss. Abs„ 15, 1325 (1955).
522. Armstrong D. A., Winkler C. A. Canad. J. Chem., 34, 885 (1951).
523. C a r t e r H. V„ Chappell E. I„ Warhurst E. J. Chem. Soc., 1956, 106.
524. Tro t m an - D i ckenso n A. F„ VerbekeG. J. Q. J. Chem. Soc., 1961,
2580.
525. C h i 1 t о n H. T. Y., Gowenlo ck B. G. Trans. Farad. Soc., 50, 824 (1954).
526. Chilton H. T. Y., Gowenlock B. G. J. Chem. Soc., 1953, 3232.
527. Chilton H. T. Y., Gowenlock B. G. Nature, 172, 73 (1953).
528. C h i 11 о n H. T. Y., С о w e п 1 о с к B. G. Trans. Farad. Soc., 49, 1451 (1953).
529. В i 1 1 i n g e В. H. M., Gowenlock B. G. J. Chem. Soc., 1962, 3252; Trans.
Farad. Soc., 59, 690 (1963).
530. Chilton H. T. Y., Gowenlock B. G. J. Chem. Soc., 1954, 3174.
531. F г e у F. E., H e p p H. J. J. Am. Chem. Soc., 55, 3357 (1933).
532. W о 1 I f P. Ber., 46, 64 (1913).
532a. G о b 1 e A. G., L i n d s t о n e A. G., P a u w e 1 s P. I. S. Chem. and Ind.,
1959, 1489.
5326. Bass K- S. Chem. and Ind., 1964, 1712.
532в. Разуваев Г. A., 3 а т e e в Б. Г., M я к о в В. H. ДАН СССР, 154, 164
(1964).
533. JaquissM. Т., SzwarcM. Nature, 170, 317 (1952).
534. Левина Р. Я., Костин В. Н., Тартаковский В. А. ЖОХ, 27,
2049 (1957).
535. Cowperthwaite М., W a rhurst Е. J. Chem. Soc., 1958, 2429.
536. К о т о н М. М , Мартынова В. Ф. ЖОХ, 24, 2177 (1954).
537. W i t t.i g G., E b e 1 H. F. Ann., 650, 20 (1961).
538. Несмеянов A. H., ФрейдлинаР. X., Величко Ф. К. ДАН СССР,
114, 557 (1^57).
539. Р а з у в а е в Г. А., П е т у х о в Г. Г., КаплинЮ. А., Кудрявцев Л. Ф.
ДАН СССР, 141, 371 (1961).
539а. Разуваев Г. А., Петухов Г. Г., КаплинЮ. А., Дружков О. Н.
ДАН СССР, 152, 1122 (1963).
5396. Разуваев Г. А., Петухов Г. Г., Кудрявцев Л. Ф., Шубенко
М. А. ЖОХ, 33 2764 (1963).
540. К о т о н М. М. ДАН СССР, 88, 991 (1953).
541. Н е с м е я н о в А. Н., БорисовА. Е., Волькенау Н. А. Изв. АН
СССР, ОХН, 1954, 992.
542. Trotman-DickensonA. F., Steacie Е. W. R. J. Phys. Chem., 55, 908
(1951).
543. Р h i b b s M. K-, D a r w e n t B. de B. Trans Farad. Soc., 45, 541 (1949).
544. Har’risG. M., S t e a c i e E. W. R. J. Chem. Phys., 13, 559 (1945).
545. Darwent B. de В., P h i b b s M. K. J. Chem. Phys., 22, 110 (1954).
348
Ртутноорганичёские соединения
546. Rebbert R. Е., Steacie Е. W. R. Can. J. Chem., 31,. 631 (1953).
547. Rebbert R. E., Steacie E. W. R. J. Chem. Phys.; 21, 1723 (1953).
548. Ho Iroy d R., N о у e s jr. W. A. J. Am. Chem. Soc., 76, 1583 (1954).
549. Muller К. H., W a 1 t e r s W. D. J. Am. Chem. Soc., 76, 330 (1954).
550. GunningH.E., S t e a c i e E. W. R. J. Chem. Phys., 14, 534 (1946).
551. M i 1 1 e r D. M-, S t e a c i e E. W. R. J. Chem. Phys., 19, 73 (1951).
552. Gomer R., Ki st i akowsk i G. B. J. Chem. Phys., 19, 85 (1951).
553. Котон M. M„ Киселева T. M, Изв. АН СССР, ОХН, 1961, 1783,
554. Marcus R. A., Steacie E. W. R. Z. Naturforsch., 4a, 332 (1949).
555. G о m e r R., N о у e s W. A. J. Am. Chem. Soc., 71, 3390 (1949); 72, 101 (1950).
556. Gomer R. J. Chem. Phys., 18, 998 (1950).
557. Rebbert R. E., Steacie E. W. R. Can. J. Chem., 32, 113 (1954)..
558. Rebbert R. E„ S t e a c i e E. W. R. Can. J. Chem., 32, 40 (1954).
559. Gomer R. J. Am. Chem. Soc., 72, 201 (1950).
560. LinnettJ. W., Thompson H. W. Trans. Farad. Soc., 33, 501 (1937).
561. T h о m p s o n H. W., LinnettJ. W. Trans. Farad. Soc., 33, 874 (1937).
562. Masson C. R., S t e a c i e E. W. R. J. Chem. Phys., 19, 183 (1951).
563. Phi bbs M. K., Darwent B. de B. Can J. Res., B28, 395 (1950).
564. Marcus R. A. J. Chem. Phys., 20, 364 (1952).
565. Kistiakowski G. B., Roberts E. K. J. Chem. Phys., 21, 1637 (1953)L
566. Durham R. W., Steacie E. W. R. J. Chem. Phys., 20, 582 (1952).
567. SzwarcM., Roberts J. S. Trans. Farad. Soc., 46, 625 (1950).
568. E v a n s M. G., SzwarcM. Trans. Farad. Soc., 45, 940 (1949).
569. Steacie E. W. R., D a r w e n t B. de B. Discuss. Trans. Farad. Soc., 2, 80 (1947).
570. Allen A. О., В awn С. E. H. Trans. Farad. Soc., 34, 463 (1938).
571. Martin R. B., Noyes, jr. W. A. J. Am. Chem. Soc., 75, 4183 (1953).
572. I v i n K. J., S t e a c i e E. W. R. Proc. Roy. Soc., A208, 25 (1951).
573. В r a d 1 e у J. N., M e 1 v i 11 e H. W., R о b b J. C. Proc. Roy. Soc., 236, 318
(1956).
573a..Graham W. A. G., Gatti A. R. C. A., 57, 5756 (1962).
574. thrush B. A. Proc. Roy. Soc., A243, 555 (1958). .
575. M о о r e W. J., T а у 1 о r H. S. J. Chem. Phys., 8, 396 (1940).
576. C a u 1 e E. J., S t e a c i e E. W. R. Can J. Chem., 29, 103 (1951).
577. M e 1 v i 1 1 e H. W., R о b b J. C. Proc. Roy. Soc., A196, 479 (1949).
578. M о о r e W. J., Wall L. A. J. Chem. Phys., 17, 1325 (1949).
579. BanusJ., Emeleus H. J., HaszeldineR. N. J. Chem. Soc., 1950,
3041.
580. Fonken G. J. Chem. and Ind., 1961, 716.
580a. Erhard K. Naturwiss., 49, 417 (1962).
581. HirataT.,Sujishi S.,Gunning H. E. J. Am. Chem. Soc., 82, 5045 (1960).
582. Разуваев Г. А., Ольдекоп Ю. А., МанчиноваЗ. H. ЖОХ, 22,
480 (1952).
583. Разуваев Г. А., Ольдекоп Ю. А. Сборник статей по общей химии, 2, 981
(1953).
584. Разуваев Г. А., Ольдекоп Ю. А., ЛатяеваВ. Н. ЖОХ, 24, 260
(1954).
585. Разуваев Г. А., Петухов Г. Г. Тр. хим. и хим. технол. (Горький), 1961,
вып. 1, 150; РЖХим, 1962, 4Ж50.
586. Разуваев Г. А., Ольдекоп Ю. А. ЖОХ, 19, 736 (1949).
587. Разуваев Г. А., Ольдекоп Ю. А. ЖОХ, 19, 1483 (1949).
588. Разуваев Г. А., Ольдекоп Ю. А. ЖОХ, 21, 1122 (1951).
589. Р а з у в а е в Г. А., О л ь д е к о п Ю. А. Сборник статей по общей химии, 1,
275 (1953).
590. Ольдекоп Ю.. А. ЖОХ, 23, 2020 (1953).
591. Разуваев Г. А., ШубенкоМ. А. ДАН СССР, 67, 1049 (1949).
592. Разуваев Г. А., ШубенкоМ. А. ЖОХ, 20, 175 (1950).
593. Разуваев Г. А., ШубенкоМ. А. ЖОХ, 21, 1974 (1951).
594. Разуваев Г. А., ШубенкоМ. А. Сборник статей по общей химии, 2, 1043
(1953).
595. Разуваев Г. А., Ольдекоп Ю. А. ЖОХ, 23, 587 (1953).
596. Разуваев Г. А., Ольдекоп Ю. А. Уч. зап. ГГУ, 17, 171 (1951).
597. Разуваев Г. А. Сб.: Проблемы механизма органических реакций. Киев, Изд-во
АН УССР, 1958, стр. 78.
598. Разуваев Г. А., Ольдекоп Ю. А. ЖОХ, 20, 181 (1950).
599. Разуваев Г. А., Ольдекоп Ю. А., НазарковаН. П. Уч. зап. .ГГУ,
23, 117 (1952).
600. О л ь д е к о п Ю. А. ЖОХ, 22, 478 (1952).
601. Ольдекоп Ю. А., ИдельчикЗ. Б. ЖОХ, 30, 2564 (1960).
Реакции ртутноорганических соединений
349
602. Разуваев Г. А., Ольдекоп Ю. А. ЖОХ, 20, 506 (1950).
603. Ольдекоп Ю. А. ЖОХ, 22, 2063 (1952).
604. Разуваев Г. А., Петухов Г. Г. ЖОХ, 21, 646 (1951).
605. Разуваев Г. А., Петухов Г. Г. Уч. зап. ГГУ, 17, 183 (1951).
606. Петухов Г. Г. Уч. зап. ГГУ, 24, 155 (1953).
607. Разуваев Г. А., Новаторов Н. Ф. Уч. зап. ГГУ, 17, 197 (1951).
608. McD onald Blair J., Bryce-Smyth D., PengillyB. W. J. Chem.
Soc., 1959, 3174.
609. Bryce-Smith D. J. Chem. Soc., 1955, 1712.
610. Разуваев Г. Г., Нов а то ров Н. Ф. Уч. зап. ГГУ, 23, 121 (1952).
611. Derbyshire D. Н., Steacie Е. W. R. Can. J. Chem., 32, 457 (1954).
612. Разуваев Г. А., Петухов Г. Г., ЗатеевБ. Г. ДАН СССР, 127, 348
(1959).
613. Разуваев Г. А., Петухов Г. Г. ЖОХ, 23, 37 (1953).
614. Sow den R. G., D a v i d s о п N. J. Am. Chem. Soc., 78, 1291 (1956).
615. Петухов Г. Г. Сборник статей по общей химии, 2, 989 (1953).
616. С о г b е t t G. Е., W i 1 1 i a m s G. Н. Proc. Chem. Soc., 1961, 240.
617. Разуваев Г. А., Ольдекоп Ю. А., Королевам. H. ЖОХ, 21, 650
(1951).
618. Разуваев Г. А., Субботина А. И. Уч. зап. ГГУ, 23, 149 (1952).
619. P а з у в a e в Г. А., О л ь д e к о п Ю. А. ЖОХ, 19, 1487 (1949).
620. Разуваев Г. А., Субботина А. И. Уч. зап. ГГУ, 17, 187 (1951).
621. Разуваев Г. А., 3 а п е в а л о в а Г. Н. Уч. зап; ГГУ, 23, 123 (1952).
622. Разуваев Г. А., - Петухов Г. Г., Шубенко М. А., Войтович В. А.
Укр. хим. ж., 22, 45 (1956).
623. Коршунов И. А., - Аменицкая Р. В., Орлова А. А., Бата-
лов А. П. ЖОХ, 29, 1992 (1959).
624. Разуваев Г. А., Петухов Г. Г., Рекаш е в а А. Ф., Миклухин
Г. П. ДАН СССР, 90, 569 (1953).
625. Коршунов И. А., Орлова А. А. ЖОХ, 28, 45 (1958).
626. Разуваев Г. А., Ольдекоп Ю. А. ЖОХ, 21, 2197 (1951).
627. Ол ь д екоп Ю. А. Уч. зап. ГГУ, 24, 175 (1953).
628. Ольдекоп Ю. А. Уч. зап. ГГУ, 24, 147 (1953).
629. Федотов М. С. Сборник статей по общей химии, 2, 984 (1953).
630. Разуваев Г. А., Федотов М. С. ЖОХ, 22, 484 (1952).
631. Разуваев Г. А., Федотов М. С. Сборник статей по общей химии, 2, 1517
(1953).
632. Несмеянов А. Н., БорисовА. Е., ВильчевскаяВ. Д. Изв. АН
СССР, ОХН, 1949, 578; Уч. зап. МГУ, 132, 33 (1950).
633. В а с и л е й с к а я Н. С. Уч. зап. ГГУ, 23, 133 (1952).
634. Wittig G., Е b е 1 Н. F. Angew. Chem., 72 , 564 (1960).
635. Несмеянов А. Н., БорисовА. Е., Абрамова А. Н. Изв. АН
СССР, ОХН, 1947, 289.
636. С a j u s I. F., W a d i a J. Н. J. Indian Chem. Soc., 6, 613 (1929).
637. D i mr о th O. Ber'., 35, 2036 (1902).
638. Rei tzenstein F., Stamm G. J. Prakt. Chem. (2), 8, 154 (1910).
639. Pesci L. Z. anorg. allg. Chem., 32, 231 (1902).
640. S c h о e 1 1 e r W., Schrauth W., Goldacker P. Ber., 44, 1305 (1911).
641. W h i t m о r e F. C., L e u c k G. I. J. J. Am. Chem. Soc., 51, 2782 (1928).
642. Пат. США 2118033 (1936); Zbl., 1939, I, 1410.
643. G u h a - S i r c a r S. S., R ou t M. K. J. Indian Chem. Soc., 30, 359 (1953).
644. Несмеянов A. H., Макарова Л. Г. ЖОХ, 3, 257 (1933); Ber., 66, 199
(1933).
645. DunhauptD. J. Prakt. Chem. (1), 61, 415 (1854); Ann., 92, 379 (1854).
646. О t t о R. J. Prakt. Chem. (2). 29, 136 (1884).
647. Z i n i n N. Ann., 96, 363 (1929).
648. S 1 о t t а К. H., Jacobi K. R.’J. Prakt. Chem., 120, :282 (1928).
649. A u 1 d S. M., H ant zsch A. Ber., 58, 2678 (1905).
650. S t r e c k e r A. Ann., 92, 79 (1854).
651. Sand J. Ber., 34, 1385 (1901).
652, RumpiP. Bull. Soc. Chim. France, 9, 535 (1942).
653. Rumpf P. Bull. Soc. Chim. France, 9, 538 (1942).
654, Японск. пат. 3369 (1960); РЖХим, 1961, 22Л70.
655. Prussia L. Gazz., 28, II, 1'29 (1898).
656. M a m e 1 i A., Marne li-Ma’nnes|-er A. Gazz., 52, II, 1 (1922).
657. Пету xob Г. Г. Уч. зап.. ГГУ, 17, Г83 (1951).
350
Ртутноорганические соединения
658. Петухов Г. Г. Синтезы органических соединений, сб. 2. М., ИОХ АН СССР,
1952, стр. 72.
659. Beck W., SchuiererE. J. Organomet Chem., 3, 55 (1965).
660. Пат. США 2131008 (1933); Zbl., 1939, I, 1858.
661. Пат. СССР 57163 (1940); С. А., 38, 4961 (1944).
662. Канад, пат. 375312 (1935); Zbl., 1939, I, 1601.
663. D a d i с М., G г d е п i с D. Croat. Chem. Acta, 32 , 39 (1960).
663а. Японск. пат. 1074 (1959); С. А., 54, 8585 (1960).
664. GrdenicD., ZadoF. Croat. Chem. Acta, 29, 425 (1957).
665. Grdenic D., Z a d о F. J. Chem. Soc., 1962, 521.
666. Grdenic D., Markujic B. J. Chem. Soc., 1958, 2434.
667. Cross R. J.,- Lauder А., С о a t e s G. E. Chem. and Ind., 47, 2013 (1962).
668. R u m p f P. Bull. Soc. chim. France, 11, 550 (1944).
669. J о h n s J. B., Peterson W. D., HixonR.M. J. Am. Chem. Soc., 52, 2820
(1930).
670. Zupancic B. Monatsh., 93, 1298 (1962).
671. Schramm R. M. J. Am. Chem. Soc., 69, 1831 (1947).
672. W r i g h t G. F. J. Am. Chem. Soc., 58, 2653 (1936).
673. H e i n F., H e u s e r E. Z. Anorg. Chem., 249, 293 (1942).
674. Нем. пат. 703897 (1941); С. A., 36, 1048 (1942).
675. Пат. США 2214278 (1941); С. А., 35, 860 (1941).
676. В i s w a s Н. G., Das G u р t a S. J. С. А., 45, 819 (1951); Indian J. Pharm., 12, 9
(1950).
677. Spinelli D., Dell’Erb а С. РЖХим, 1962, 11Ж315.
678. Hu P. F.,Chen W. Y. C. A., 52, 7186 (1958).
679. Spengler G., Weber A. Brennst.-Chem., 40, 55 (1959).
680. AlemanyA., В a 1 u j a M. G;, С о r r a 1 C., Lora-Tama jo M. РЖХим,. .
1961, 17Ж 262.
681. В a 1 u j a M. G., L о r a - T a m a j о M. РЖХим, 1960, 7, 26682.
682. Малиновский M. С., ЮркоД. Т., ТульчинскнйВ. Б. ЖОХ, 30„
2170 (1960).
683. Швейц, пат. 236878 (1945); С. А., 44, 4642 (1950).
684. Пат. США 2331268 (1944); С. А., 38, 1340 (1944).
685. Японск. пат. 2979 (1955); С. А., 51, 16551 (1957).
686. Англ. пат. 736399 (1955); С. А., 50, 7877 (1956).
687. LeandriG., Spinelli D., Salvemini A. Ann. chimica, 50, 1046 (I960).
688. Пат. США 2844508 (1958); С. А., 52, 19001 (1958).
689. Пат. США 2429086 (1947); С. А., 42, 329 (1948).
690. Пат: ФРГ 968815 (1957); С. А., 54, 7669 (1960).
691. Японск. пат. 9131 (1958); С. А., 54, 4496 (1960).
692. Японск. пат. 7371 (1958); С. А., 54, 2257 (1960).
693. Японск. пат. 8966 (1958); С. А., 54, 4496 (I960).
694. Англ. пат. 819130, 820440 (1959); С. А., 54, 9193 , 6649 (1960).
695. Голл. пат. 90773 (1959); С. А., 54, 12469 (1960).
696. Франц, пат. 827299 (1938); 49782 (1939); С. А., 32, 7923 (1938); 36, 2566 (1942).
697. Японск. пат. 168 (1960); РЖХим, 1961, 5JI334.
698. Пат. США 2437450 (1948); С. А., 42, 4608 (1948).
699. Пат. ФРГ 943730 (1956); С. А., 52, 11963 (1958).
699а. F а г a g 1 i a G., "RoncucciL., Barbieri R. Gazz., 93, 1413 (1963).
700. Англ. пат. 737753 (1955); С. А., 50, 15577 (1956).
701. Пат. США 2692204 (1954); С. А., 50, 2668 (1956).
702. R е i с he 1 1., Р о d L. РЖХим, 1959. 15460.
703. Пат. США 2216842 (1941); С. А., 35, 1940 (1941).
704. Пат. США 2794038 (1957); С. А., 51, 16538 (1957).
705. I t о А., Н a m a n a k a W., I nami К. РЖХим, 1959, № 20, 71529.
706. Японск. пат. 3072, 3073 (1960); РЖХим, 1961, 16Л331.
707. Японск. пат. 4566 (1957); С. А., 52, 8183 (1958).
708. Японск. пат. 2611 (1959); С. А., 53, 19316 (1959).
709. Японск. пат. 5918 (1957); С. А., 52, 15566 (1958).
710. Пат. США 2526356 (1950); С. А., 45, 2501 (1951).
711. Пат. США 2521720 (1950); С. А., 45, 1162 (1951).
712. Японск. пат. 17063 (1961); С. А., 56, 4697 (1962).
713. Англ. пат. 628525 (1949); С. А., 46, 4568 (1952).
714. Японск. пат. 8711 (1959); С. А., 54, 6550 (1960).
714а. D о w n s A. J. J. Inorg. a. Nucl. Chem., 26, 41 (1964).
Реакции ртутноорганических соединений
351
715. S a d о Т., М i j a tn о t о F., I а п о К., S а к о I., О s е Н., W a t а п а 1 е J.
Agric. and Biol. Chem., 25, 77 (1961); РЖХим, 1962; 6Ж290.
716. Японск. пат. 10873 (I960); РЖХим, 1962 7ЛЗЗЗ.
717. Пат. США 2883407 (1959); С. А., 53, 15464 (1959).
.718. Швейц, пат. 257397 (1949); С. А., 43, 9355 (1949).
719. Пат. США 2471621 (1949); С. А., 43 , 6648 (1949).
720. Пат. ФРГ 951869 (1956); С. А., 53, 4306 (1959).
721. Англ. пат. 603102 (1948); С. А., 42, 9066 (1948).
722. Англ. пат. 514831 (1939); С. А., 35, 4544 (1941).
723. Швейц, пат. 265758 (1950); С. А., 47, 3869 (1953).
724. Пат. США 2471622 (1949); С. А., 43, 6648 (1949).
725. Hetnarski В„ Eckstein Z., Urbanski Т. Przemysl. Chem., 13, 291;
(1957).
726. Пат. США 2766234 (1956); С. А., 51, 5131 (1957).
727. Англ. пат. 789432 (1958); С. А., 52, 6417 (1958).
728. N о z о е Т., S a t о М., М a t s u i К- С. А., 49 , 6851 (1955).
729. Z a d о F. Croat, chem. acta, 34, 89 (1962).
730. Hetnarski В., H е t narska К. Roczn. Chem., 34, 457 (1960).
731. Японск. пат. 18360 (1960); С. А., 56, 2472 (1962).
732. L е h m а п J. F., Barrack L. Р„ L е h m а и R. A. Science, 113, 410 (1951);.
• С. А., 45, 6300 (1951).
733. Укай, Ямамото, Канатом о, Каваками. РЖХим, 1957, 37687.
734. Spengler G-, Weber А. Вrennst.-Chemie, 43, 234 (1962).
735. Японск. пат. 13625 (1961); С. А., 56, 12945 (1962).
736. Hetnarski В. Roczn. Chem., 35, 1333 (1961).
737. Hetnarski В. РЖХим, 1961, 22Ж215; Bull. Acad. Polon. Chim., 8, 481 (1960).
738. Нефедов В. Д., Синотова Е. Н. ЖНХ, 2, 1162 (1957).
739. К о t е n J. A., A d a m s R. J. Am. Chem. Soc., 46, 2764 (1924).
740. R e i c h 1 e W. T. Inorg. Chem., 1, 650 (1962).
741. Пат. ФРГ 1011221 (1957); С. A., 54, 11965 (1960).
742. Франц, пат. 1072312 (1954); С. А., 53, 6157 (1959).
743. Англ. пат. 897854 (1962); С. А., 57, 13803 (1962).
743а. И и да Т. Japan J. Pharmacy and Chem., 34, 237 (1962); РЖХим, 1963, 17Ж237.
7436. Матяш Л. Ф., Степанов В. М. Изв. АН СССР, ОХН, 1964, 111.
744. Пат. США 1589599 (1924); Zbl., 1926, II, 1692; 1672615 (1927); Zbl., 1929; I, 1045.
745. Нем. пат. 586352 (1932); Zbl., 1934, I, 84.
746. Нем пат. 540609 (1929); Zbl., 1932, I, 1575; 399904 (1924); Zbl., 1925, I, 598.
747. Walter L. A., Fosbinder R. J. Zbl., 1940, II, 1581.
748. Пат. США 2782194 (1957); С. A., 52, 2100 (1958).
749. Sawal., MaedaS., HishimuraH., N a k a j i ma K-, ShimaokaN-
C. A., 51, 10008 (1957).
750. Пат. США 2618645 (1952); С. A., 47, 8775 (1953).
751. Пат. США 2728780 (1955)>С. А., 50, 14000 (1956).
752. Японск. пат. 1131 (1950); С. А., 47, 2216 (1953).
753. Англ. пат. 723433 (1955); С. А., 50, 7131 (1956).
754. Пат. ФРГ 940896 (1956); С. А., 53, 6253 (1959).
755. Англ. пат. 619515 (1949); С. А., 43, 9084 (1949).
756. Eckstein Z., Hetnarski В., Urbanski Т. Przemysl. Chem., 37, 44, 160*
(1958).
757. Пат. США 2790800 (1957); С. А., 51, 14833 (1957).
758. TakagiS., Tsukat ani Н., Tanaka Н. С. А., 47, 111 (1953).
759. N о z о е Т., S a t о М., М a t s u i К. С. А., 48, 5172 (1954).
760. Benesch R-, BeneschR- E.J. Am. Chem. Soc., 78, 1597 (1956).
761. Швейц, пат. 265757 (1950); С. A., 45, 652 (1951).
762. T r i k о j us V. M. Nature, 158, 472 (1946).
763. Пат. США 2096721—4 (1937); Zbl., 1938, I, 1407; 2555114 (1951); C. A., 46, 9127 (1952)..
764, О k u d a T., T aka nashi Y., Tsuruoka M. C. A., 56, 7147 (1962).
765. Чехосл. пат. 86087 (1957); С. A., 52, 2917 (1958).
766. W a 1 do J. H. J. Am. Chem. Soc., 53, 992 (1931); Waldo J.H., ShonleH.S.,
P о w e 1 1 H. N. J. Bakt., 21, 323 (1931).
767. Японск. пат. 519 (1961); С. A., 56, 8570 (1962).
768. Японск. пат. 421 (1961); С. А., 56, 6000 (1962).
769. Японск. пат. 7955 (1962); РЖХим, 1964, 5Н74.
770. Японск. пат. 4117 (1957); С. А., 52, 8194 (1958); 7627 (1958); С. А., 54, 2271 (1960).
771. Англ. пат. 432689 (1935); С. А., 30, 576 (1936).
772. Югослав, пат. 12703 (1936); Zbl., 1937, I, 2437.
773. Пат. США 3039920 (1961); С. А., 57, 15153 (1962).
774. Andersen Н. Р., Н а г t М. J. Am. Chem. Soc., 59, 1115 (1937).
352
Ртутноорганические соединения
775. Австр. пат. 151661 (1935); Zbl., 1938, I, 1855.
776. Пат. США 2087959 (1934); 2087960 (1934); Zbl., 1938, I, 1406.
777. Англ. пат. 512682 (1939); С. А., 35, 857 (1941).
778. Пат. США 2697101 (1954); С. А., 49, 15974 (1955).
779, Англ. пат. 709582 (1954); С. А., 49, 10363 (1955).
780. Rumpf Р. Bull Soc. chim. France, 9, 661 (1942).
781. Bi a las J.,Eckstein Z., Ejmocki Z.,Hetnarski B., Sobotka W.,
Szymaszkiewicz J. Przemysl. Chem., 40, 567 (1961).
782. Пат. США 2118133 (1934); Zbl., 1938, II, 3717.
783. Пат. США 2085066 (1935); Zbl., 1938, I, 374.
784. Нем. пат. 641285 (1934); Zbl., 1937, I, 3518.
785. Пат. США 213553 (1937); Zbl., 1939, I, 2248.
786. Пат. США 3035073 (1962); С. А., 57, 12535 (1962).
787. Пат. США 2452595 (1948); С. А., 43, 1805 (1949).
788. Пат. США 2336359 (1943); С. А., 38, 2972 (1944). Англ. пат. 561849 (1944); С. А., 40,
1174 (1946).
789. Японск. пат. 4565 (1957); С. А., 52, 5758 (1958).
790. Пат. ФРГ 1009420 (1957); С. А., 54, 12067 (1960).
791. Пат. США 2813861 (1957); С: А., 52, 10222 (1958).
792. Т о у о s h i К., Murakami Н. С. А., 56, 7342 (1962).
793. NakajimaS., Tanaka I., SekiT., Апшо Т., Komatsu М. РЖХим,
1961, 5Ж239; J. Pharm. Soc. Japan, 79, 1113 (1959).
794. L е а п d г i С., Spinelli D., S а 1 v е m i п i A. Ric. Sci., 29, 2618 (1959).
795. Spinelli D., S a 1 v e m i n i A. Ann. chim. Ital., 51, 1296 (1961).
795a. ВязанкинН. С., Разуваев Г. А., Гладышев E. H. ДАН СССР, 155,
830 (1964).
7956. ВязанкинН. С., Разуваев Г. А., ГладышевЕ. Н., Гурико-
в а Т. Г. ДАН СССР, 155, 1108 (1964).
795в. George М. V., LichtenwalterG, D., Gilman Н. J. Am. Chem. Soc.,
81, 978 (1959) .
796. W e b b J. L. A., В h a t i a I. S., Cor w i n A. S., S h a r p A. G. J. Am.
Chem. Soc., 72, 91 (1950).
797. Tanaka F., M i t s u n о M. C. A., 47, 4860 (1953).
798. L e a n d r i G., Spinelli D., Monaco G. Ann. chimica, 50, 156 (1960).
799. Котон M. M., Киселева T. M., Флоринский Ф. T. Высокомол. соеД.,
2, 1639 (1960).
800. Кравцов Д. H., К ачку р о в а Их Я. ДАН СССР, 130, 329 (1960).
801. Несмеянов А. Н., Кравцов Д. Н. ДАН СССР, 135, 331 (1960).
802. Несмеянов А. Н., Кравцов Д. Н. ДАН СССР, 137, 614 (1961).
803. Несмеянов А. Н., Кравцов Д. Н. ДАН СССР, 140, 1334 (1961).
804. Несмеянов А. Н., Кравцов Д. Н. Изв. АН СССР, ОХН, 1962, 431..
805. Е m е 1 ё ц s Н. I., Lagowski I. I. Proc. Chem. Soc., 1958, 231.
806. EmeleusH. I., Lagowski I. I. J. Chem, Soc., 1959, 1497.
807. Lawton D., PowellH. M. J. Chem. Soc., 1958, 2339.
808. Simp so n R. B. J. Am. Chem. Soc,, 83, 4711 (1961).
809. Singer S. Y., FothergillJ. E., ShainoffJ. R. J. Am. Chem. Soc.,
82 , 565 (1960).
"810. McD о n n e 1 1 L. R., S i 1 v a R. B., Feeney-R. E. Arch. Biochem. Biophys.,
32, 288 (1951).
811. H e 1 1 e r m a n L. Physiol. Revs., 17, 454 (1937); C. A., 35, 6985 (1941); Cold. Spring
Harbor Symp. Quant. Biol., 7, 165 (1939).
812. H e 11 e r m a n L., C h i n a r d F. P., D e i t z V. R. C. A., 37, 2393 (1943);
J. Biol. Chem., 147, 443 (1943). ’ ,L.
813. В о у e г P. D. J. Am. Chem. Soc., 79, 4331 (1954).
814. S w e n s о n A. D., Boy er P. D. J. Am. Chem. Soc., 79, 2174 (1957).
815. Пол яновский О. Л. Биохимия, 27, 734 (1962). '
• 816. S z а b о 1 с s i G., B i s z k ii E. Biochim. Biophys. Acta, 48, 229 (1961).
817. FedjarnL., JellumE. Acta Chem. Scand., 17, 2610 (1963)..
818. E d.elhoch H., Katchalski E., May bury R. H., Hughes W. L.,
jr., E d s a 1 1 J. T. J. Am. Chem. Soc., 75, 5058 (1953).
Глава XV
ИЗМЕНЕНИЯ В ОРГАНИЧЕСКОЙ ЧАСТИ МОЛЕКУЛЫ
РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Изменения в органической части молекулы ртутноорганических сое-
динений в большинстве случаев производятся обычными методами орга-
нической химии’ и не специфичны для ртутноорганических соединений.
Реакции органической части молекул ртутноорганических соединений, не
сопровождаемые разрывом связи С—Hg, не слишком многочисленны.
В жирном ряду они сводятся к ацилированию гидроксила посредством гало-
идных ацилов [1,2] (в том числе в ртутных производных алкендиолов [3]),
ангидридов кислот [1, 4, 5] и изоциановых эфиров [6], к окислению пер-
вичной спиртовой группы в карбоксильную [4, 7] действием, брома и щело-
чи [7] или щелочным раствором перманганата [4] (и в первом [8], и во вто-
ром случаях выход невелик) и вторично спиртовой группы в кетонную [9, 9а]
(выход 50—60% [9]), к восстановлению альдегидной функции в спиртовую
действием, например, изопропилата алюминия [10], к солеобразованию
меркурированными кислотами, к этерификации кислот действием диазомета-
на [11], к замене алкоксигруппы действием амидов карбоновых [12—14]
и сульфоновых [12] кислот на ацил- или сульфиниминогруппу, к вступле-
нию бис-алкилмеркурацетиленидов [15] и p-хлорвинилмеркурхлорида [16]
в диеновый синтез с циклонами. Ацилали меркурированных оксосоединений
гидролизуют сильно разбавленным раствором серной кислоты в 50 %-ном
спирте [17].
Синтез 2-иодмеркурэтилбензоата [1]. 10 г иодистой £-оксиэтилртути растворяют в 10 г
10%-ного раствора йодистого калия, охлаждают до 0°С и встряхивают раствор с 10 г хло-
ристого бензоила, следя за тем, чтобы температура реакции не поднималась выше 0°С [2].
Выпавший белый осадок очищают [2] промыванием его хлороформенного раствора трижды
10%-ным раствором йодистого натрия и перекристаллизацией из смеси хлороформа и петро-
лейного эфира. Выход после очистки 16%. Т. пл. 115—118° С.
Реакцию необходимо вести при 0° С. При несоблюдении этой меры предосторожности
или если продукт кристаллизовать, как это рекомендует Занд [1], из Этанольного раствора
йодистого натрия, происходит энергичное деоксимеркурирование и конечный продукт пред-
ставляет собой главным образом бензойную кислоту [2].
Восстаиовлеиие хлормеркурацетальдегида в этанолмеркурхлорид [10].
с1н§сн2сно _1£~Сз”7°)з-аД ciHgCH2cH20H
1. Приготовление изопропилата алюминия [18]. Алюминиевые
стружки обрабатывают раствором едкого натра до сильного выделения водорода, затем их
промывают водой до нейтральной реакции и обрабатывают 1%-ным раствором сулемы, про-
мывают водой, спиртом, эфиром и высушивают. В круглодонную колбу, снабженную обрат-
ным холодильником, помещают 1 г алюминиевых стружек, 30 мл абсолютного изопропило-
вого спирта и несколько кристаллов иода. Реакция начинается уже при комнатной темпера-
туре. Колбу нагревают на водяной бане до растворения металла. Раствор отфильтровывают.
2. Восстановление хлормеркурацетальдегида. К7г (0,04 мо-
ля) хлормеркурацетальдегида приливают 12 мл раствора изопропилата алюминия. Раствор
оставляют на 3 дня. Осадок слеживается и становится тягучим. Затем приливают 10%-ный
раствор едкого натра до растворения осадка. В профильтрованный раствор пропускают
354
Ртутноорганическиё соединения. Лит. стр. 356—357
углекислый газ для осаждения этанолмеркурхлорида, который отсасывают и перекристалли-
зовывают из метилового спирта. Т. пл. 151° С.
Получение хлормеркурацетоиа из пропилена [9].
СНз - сн = СН2 4- Hg (СН3СОО)2 СНзСН (ОСОСНз) CH2HgOCOCH3Ki--nO‘’ка
-> CH3COCH2HgCl
В раствор 16 г !уксуснокислой ртути в 150 мл воды пропускают пропилен до исчезнове-
ния реакции на ион ртути; к профильтрованному раствору прибавляют 6 мл уксусной кисло-
ты и затем (небольшими порциями) при размешивании прибавляют 5,3 г перманганата калия.
После окончания реакции для осаждения коллоидальной перекиси марганца раствор нагре-
вают 15 мин. на водяной бане. Осадок отфильтровывают и промывают теплой водой. К фильт-
рату прибавляют раствор 3,7 г хлористого калия. Воду удаляют в вакууме при комнатной
температуре. Выход 7,75 г (53%). Т. пл. 102—102,5° С (из метилового спирта). Т. пл. чисто-
го вещества 103—104° С.
Действием диметил сульфата произведено метилирование окиси ртути
в продуктах оксимер курирования циклоалкенов (см., например, [19]).
Метилирование а-1-хлормеркур-2-оксициклогексаиа. Получение а-1-хлормеркур-2-мет-
оксициклогексана [19].
ОН
В видоизмененную колбу Кляйзена помещают 80 мл не содержащего серы толуола и от-
гоняют 15 мл его для освобождения от следов серы. К остатку под азотом прибавляют 0,59 г
(0,015 г-атом) калия и 10 мл сухого Третичного бутилового спирта; после того как после
перемешивания и кипячения калий растворится, избыток спирта отгоняют. Остающуюся
суспензию охлаждают, прибавляют 1,68 г (0,005 моля) а-1-хлормеркур-2-оксициклогексана
и перемешивают до тех пор, пока через 20 мин. ртутное соединение не растворится. Одновре-
менно через 15 мин. отгоняют в вакууме (ширбкая трубка для отгонки, через которую про-
пускаюх азот) 15 мл дистиллята. К остатку прибавляют 1,89 г (0,015 моля) диметилсульфата
и энергично перемешивают в течение 90 мин.; затем прибавляют 20 мл 10%-ного едкого натра
и после 10-минутного перемешивания толуол отгоняют в вакууме. Остаток отфильтро-
вывают, разбавляют90 мл 1,5%-ного водного раствора хлористого натрия и насыщают угле-
кислотой. 1-Хлормеркур-2-метоксициклогексан (1,57 г, 90%) плавится при 109—112° С.
После кристаллизации из 8 мл метанола остается 1,1 г; т. пл. 113—115,5° С.
Действие йодистого метилена на дивинилртуть в присутствии цинка
в тетрагидрофуране при 40° С приводит к дициклопропилртути [20]. О Дру-
гих изменениях в органической части молекулы в продуктах присоединения
солей ртути к непредельным соединениям см. гл. VI (стр. 116).
Нуклеофильная атака J- в сухом и водном ацетоне на C2H5HgCH2Cl,
идущая по схеме
C2HsHgCH2C! CaHbHgCH^Cl- C2H5HgCH2J + КС1,
и сольволиз n-C4H 9HgCH2Cl в 80%-ном водном этаноле являются реак-
циями второго порядка [20а].
Осуществлена реакция [206]
(CFJ = СН)2 Hg + Вг2 —(CFJBrCHBr)2 Hg,
в то время как несодержащие фтора алкенильные соединения ртути, в том
числе квазикомплексные, под действием галоидов отщепляют HgHal2 (см.
гл. VI и XIV). При действии триметиламина на б«с-иодметилртуть образует-
ся смесь аммониевых солей I и II [21]:
(JCH2)2 Hg [(СНз)з NCH2HgCH2J]+, J-, (I)
[(СНв)з NCH2HgCH2N (СНз)з]2+ 2J~. (II)
Изменения в органической части молекулы RzHg
355
В ароматическом ряду известно, кроме того, нитрование меркурирован-
ного бензола [22], дающее до 50% мета-нитропроизводного, функциональные
изменения аминогруппы, гидроксила и альдегидной группы, а именно,
ацилирование [23—26], алкилирование [23, 27—29] и другие изменения
[29] аминогруппы (в том числе получение четвертичных аммониевых солей
[30]).
Описана также конденсация меркурированных аминов с альдегидами
в меркурированные Шиффовы основания [31], конденсация с хлористым
динитрофенилпиридинием [32], приводящая к обычному под действием ами-
нов разрыву пиридинного кольца, и с ацеталями [27]. В ряде работ исследо-
вано ацилирование [33—35] (в частности действием диалкилхлорфосфата
[36, 37] и алкилирование [38, 39] (в том числе аллилирование [401) гидро-
ксила; (аллилирование о-хлормеркурфенолята натрия действием хлористого
аллила в ацетоне дает только симметричный продукт [40] R2Hg).
Ацетилирование фенольного гидроксила. Получен не ЛГ-ацетоксифеиилмеркурхлорида [35].
ОН ОСОСНз
I I
(СН3СО)2О
.LjJ-HgCl c5hsn~T LHgCl
3 г jw-оксифенилмеркурхлорида растворяют в 6 г пиридина и в этот раствор вливают 1,2 г
уксусного ангидрида в 2,5 г пиридина. Смесь стоит в течение суток, затем ее нагревают 1 час
на водяной бане и выливают в воду, подкисленную соляной кислотой. Выпавший осадок от-
сасывают и перекристаллизовывают из спирта. Т. пл. 199—200° С. Выход количественный.
В литературе имеются данные о диазотировании аминогруппы в мерку-
рированных аминах [31, 41—47] и реакциях, полученных таким образом
диазониев, о конденсации меркурированных альдегидов с аминами в Шиф-
фовы основания [48], об этерификации меркурированных кислот [49]. От-
метим еще нитрозирование [50] и азосочетание меркурированных фенолов
[25, 27, 33, 41, 51—53] и аминов [41], превращение меркурированного нит-
розофенола в нитрозоанилин действием солей аммония [50], превращение
карбоновых кислот в хлорангидриды [49, 53а]. Гладко проходит окисление
метильной [54, 55] и метилольной [561 групп в карбоксильную посредством
щелочного перманганата. .Так получены, например, п- [54] и л«-[55] хлормер-
курбензойная кислота, ангидро-2-оксимеркур-4-нитробензойная кислота
[57].
Окисление лг-хлормеркуртолуола по Уитмору [54]. Получение л<-хлормеркурбеизойной
кислоты [55].
СН3 СООН
I I
КМпО.
^JJ-HgCl ^QjL-HgCl
10 г .м-хлормеркуртолуола, истертого в тонкий порошок, вносят небольшими порциями
в раствор 15 г перманганата калия и 24 г едкого натра в 360 мл воды и растирают там. Смесь
нагревают иа водяной бане до 95° С в течение 15 мин., избыток перманганата разрушают
спиртом, раствор отфильтровывают от перекиси марганца и фильтрат подкисляют соляной
кислотой. Выпавшую л«-хлормеркурбензойную кислоту отсасывают, тщательно промывают
водой и высушивают. Выход 6,42 г. м-Хлормеркурбензойная кислота — бесцветное кристал-
лическое малорастворимое вещество. Т. пл. 273° С.
Меркурированные нитробензойные кислоты восстанавливаются в мер-
курированные аминобензойные кислоты действием щелочных или нейтраль-
ных (но не кислых) восстановителей [58].
356
Ртутноорганические соединения
В системе Ph*HgBr — Ph*Hg (Ph*— фенил, содержащий меченый атом
углерода) в пиридине и диоксане происходит быстрый обмен фенилами [59].
В гетероциклическом ряду в производных фурана первичная спиртовая
группа окислена в альдегид [60], алкилпиридиниевые соли (где Aik =
= R3SnOC(CH2)„X), меркурированные в пара-положение пиридина, полу-
чены нагреванием его в течение нескольких минут с AlkHal [30].
Озонирование ртутных производных алкенов (в обычных условиях
озонирования, в хлороформе) хлористой цис- и транс-1-метил-2-ацетокси-1-
пропен-1-ил-ртути (СН3)(Н3ССОО)С = C(CH3)HgCl и 1-метил-2-ацетокси-
2-пропен-I-ил-ртути CH2=C(OOCCH3)CH(HgCl)CH3[61 ], продукта при-
соединения лактата ртути к циклогексену [62J сопровождается разрывом
связи С—Hg. Возможны реакции и в анионной части молекулы ртутноор-
ганической соли, например нагревацием в вакууме в этиловом спирте три-
этаноламина, метилтриэтаноламина и др.; с этилмеркуртиосалицилатом и
с этилмер кур солями других карбоновых кислот получены этаноламидные
производные этилмеркуртиосалицилата и других карбоновых кислот [63].
8-Фенилмеркуроксихинолинполихлорфеноляты получены (при кипяче-
нии в бензоле) как из 8-фенилмеркуроксихинолината и полихлорфенола,
так и из фенилмеркурполихлорфенолята и 8-оксихинолина [64]. Легко
вступает в реакцию с нуклеофильными реагентами с заменой фтора бис-
пентафторфенилртуть:
(CsF5)a Hg Д (р - XC6F4)2 Hg,
с КОН в третичнобутиловом спирте (1 час, 100° С), с NaOCH3 в метиловом
спирте (кипячение 24 часа). Выходы хорошие. Реакция с гидразингидратом
идет при кипячении в спирте в течение 6 час. и сопровождается выделением
ртути, пентафторбензола, /г-тетрафторфенилгидразона [65].
ЛИТЕРАТУРА
1. Sand J. Вег., 34, 1385 (1901).
2. Kreevoy М. М., Bodem G. В. J. Org. Chem., 27, 4539 (1962),
3. Несмеянов А. Н„ Кочетков Н. К- Изв. АН СССР, ОХН, 1949, 76.
4. S a n d J., S i п g е г F. Ann., 329, 166 (1903).
5. Abercrombie М. J., Rodgman А., В h а г u с h а К. R-, W г i g h t G. F.
Can. J. Chem., 37, 1328 (1959).
6. Несмеянов A. H., Фрейдлииа P. X. ЖОХ, 7, 2748 (1937).
7. Hofmann K. A., S a n d J. Ber., 33, 1340 (1900).
8. San d J., Ge'nssler O. Ber., 36, 3699 (1903).
9. Луценко И. Ф., Сивкова P. И. ЖОХ, 29, 1182 (1959).
9a. T г а у 1 о r T. G., В a k е г A. W. J. Am. Chem. Soc., 85, 2746 (1963).
10. Н е с м е я н о в А. Н., Луценко И. Ф., Верещагина Н. И. Изв. АН
СССР, ОХН, 1947, 63.
ll. ChiuD. D., Wr i gh t G. F. Can. J. Chem., 37, 1425 (1959).
12. Пат. США 2329883 (1944); С. A., 38, 1604 (1944).
13. Пат. США 2369339; С. А., 37, 3618 (1943).
14. U k ad Т. Yamamoto I., К-a n a t о m a S., Kawakami А. С. А., 50,
10737 (1956).
15. А б р а м о в В. С., Ш а и ш и н с к а я Л. А. ДАН СССР, 59, 1291 (1948).
16. Абр амов В. С. Изв. АН СССР, ОХН, 1945, 330.
17. Н е с м е я и о в А. Н., Луценко И. Ф., Хомутов Р. М. Изв. АН СССР,
ОХН, 1957, 942.
18. Lun d Н. Вег., 70, 1520 (1937).
19. Р а г k W. R. R., W г i g h t G. F. Can. J. Chem., 35, 1088 (1957).
20. T о b 1 e r E., F о s t e r D. J. Z. Naturforsch., 176, 135 (1962).
20a. L e d w i t h A., P h i 1 1 i p s L. J. Chem. Soc., 1962, 3796.
206. Смирнов К- M., Г и н с б у p г В. А., Якубович А. Я. Ж. Всесоюзн.
хим. общ,, 8, 231 (1963).
21. W 1 t t i g G., Sc hwarzenb ach К Ann., 650, 1 (1961).
22. C h a 1 1 e n g e r F., R о t h s t e i n E. J. Chem. Soc., 1934, 1258.
Изменения в органической части молекулы RaHg
357
23. D i m г о t h О. Вег., 35, 2032 (1902).
24. Kharasch M.S.,Lommen F. W. М., J а с о b s о h n I. М. J. Am. Chem. Soc.,
44, 793 (1922).
25. Vecchiotti L„ M i c h e t-t i A. Gazz., 55,372 (1925).
HathwayD. E. J. Chem. Soc., 1945, 123.
26. Hodgson U. H., H a t hway D. E. J. Chem. Soc., 1945, 123.
27. ReitzensteinF., Boni tsch G. J. prakt. Chem. (2) 86, 73 (1912).
28. Швейц, пат. 265755 (1950); С. A., 45, 652 (1951).
29. Пат. США 2471622 (1949); С. А., 43, 6648 (1949); 2415555 (1947); С. А., 41, 3133 (1947).
30. М i 11 е г V. L., С h a n J. К- J. Org. Chem., 28, 1938 (1963).
31. Jacobs W. A., H e i de Iberger M. J. Biol. Chem., 20, 513 (1915).
32. ReitzensteinF., Stamm G. J. prakt. Chem. (2) 81, 154 (1910).
33. Dimro th O. Ber., 35, 2853 (1902).
34. Whi (more F. C„ M i d d 1 e t о n E. J. Am. Chem. Soc., 43, 619 (1921).
35. Несмеянов A. H„ Торопова E. М.ЖОХ, 4, 664 (1934).
36. Малиновский M. С., ЮркоД. Г., ТульчинскийВ. Б. ЖОХ, 30,
2170 (1960).
37. Малиновский М. С., Ю’р к о Д. Г., М а ш т а к Н. И. Изв. высших учеб-
ных заведений. Химия и хим. технол., 4, 514 (1961).
38. D i m г о t h О. Вег., 31, 2155 (1898).
39. D i m г о t h О. Вег., 32, 758 (1899). _
40. Н е с м е я н о в А. Н.
41. G u h
42. G u h
43. G u h
44. G u h
45. В r a k e r W., C h r
a
a
a
a
-Si
-Si
- S i
- S i
rear
rear
rear
rear
S.
S.
S.
s.
Шацкая P. X. ЖОХ, 5, 1268 (1935).
R о u t M. K. J. Indian Chem. Soc., 30, 361 (1953).
Rout M. K. J. Indian Chem. Soc., 30, 364 (1953).
Rout M. K. J. Indian Chem. Soc., 30, 366 (1953).
S., Rout M- K- J- Indian Chem. Soc., 30, 435 (1953).
i s t i a n s e n W. G., Zbl., 1937, I, 851.
s.
s.
s.
46. WillstaedtH., Borggar d M., Myrback K. Arkiv Kemi, 1, 331 (1949).
47. McM a h о n E., M a r v e 1 C. S. J. Am. Chem. Soc., 52, 2528 (1930).
48. W h i t m о r e F. С., M i d d 1 e t о n E. J. Am. Chem. Soc., 45, 1330 (1923).
49. W h i t m о r e F. G., Isenhour L. L. J. Am. Chem. Soc., 51, 2785 (1929).
49a. Степанов В. M., M а т я ш Л. Ф. ЖОХ, 33, 316 (1963).
50. К h а г a s с h М. S., Р i с с а г d J. F. J. Am. Chem. Soc., 42, 1855 (1920).
51. Bamberger E. Ber., 31, 2624 (1898).
52. H о d g s о n H. H., T'urner H. S. J. Chem. Soc., 1943, 391.
53. Whitmore F. С., H a m p s о и E. R., L e u c k G. J. J. Am. Chem. Soc., 48,
1013 (1926).
53a. M а т я ш Л. Ф., Степанов В. M. Изв. АН СССР, ОХН, 1964, 111.
54. W h i t m о г е F. С., Woodward G. Е. J. Am. Chem. Soc., 48, 533 (1926).
55. Несмеянов А. Н-, М а к а р о в а Л. Г. ЖОХ, 1, 598 (1931).
56. U k a i Т., Y a m a m о t о Y., Y о t s u z u к a M. C. A., 50, 5665 (1956).
57. Петро ви ч П. И. Ж. Всесоюзн. хим. общ., 5, 106 (1960).
58. Нем. пат. 249725 (1912); Zbl., 1912, II, 777.
59. СинотоваЕ. Н., ВобецкийМ. Ф., Логинов Ю. Н., Евтихеев
Л. Н. Радиохимия, 1, № 6, 687 (1959).
60. С h u t е W. J., Orchard W. М., Wright G. J. Org. Chem., 6, 157 (1941).
61. Несмеянов A. H., БорисовА. E., Вильчевская В. Д. Изв. AH
СССР, ОХН, 1954, 1008; ДАН СССР, 90, 383 (1953).
62. W г i g h t G. F. J. Am. Chem. Soc., 57, 1993 (1935).
63. Японск. пат. 21197 (1961); С. A., 57, 16655 (1962).
64. U nga T. Japan J. Pharmacy and Chem., 34, 237 (1962).
65. В u r d о n J., С о e P. L., Fulton M., Tat low J. L. J. Chem. Soc., 1964,
2673.
Глава XVi
СИНТЕЗ И РЕАКЦИИ СОЕДИНЕНИЙ,
В КОТОРЫХ РТУТЬ СВЯЗАНА
С ОРГАНИЧЕСКИМ ОСТАТКОМ НЕ ЧЕРЕЗ УГЛЕРОД
В соответствии со своей высокой электрофильностью ртуть весьма склон-
на вступать в соединение с электроотрицательными атомами (О, S, Se, N
и в некоторых случаях с Р), присутствующими в органических, молекулах,
и образовывать ртутноорганические производные, не являющиеся истинны-
ми металлоорганическими соединениями, т. е. соединениями со связью угле-
род — металл, но представляющими собой вещества, в которых ртуть соеди-
нена с органическим остатком через гетероатом.
Среди таких веществ наименее устойчивы и сравнительно трудно обра-
зуются соединения с О—Hg-связью. Особенной прочностью и легкостью
образования отличаются соединения с S—Hg-связью и менее широко изу-
ченные органические соединения с Se—Hg-связью. Ртуть соединяется с
атомом азота разнообразных органических азотсодержащих соединений, об-
разуя соединения с N—Hg-связью, прочность которой варьирует в широ-
ких пределах, не превышая, однако, прочности S—Hg-связи в ртутных
производных органических сернистых соединений.
Органические соединения с О—Hg- и N—Hg-связями часто являются
первыми промежуточными продуктами при меркурировании кислород- и азот-
содержащих органических соединений и переходят в соединения, мерку-
рированные по углероду при интенсификации процесса, например, при по-
вышении температуры или при уменьшении pH среды реакции.
Литература по ртутноорганическим соединениям, содержащим ртуть,
соединенную с органическим радикалом через О, S, Se, N, приведена в’на-
стоящей книге не исчерпывающе и выбор (а также и расположение) рассмат-
риваемого в этой главе материала сделан несколько произвольно. Так,
сюда не включены ртутные соли карбоновых, сульфоновых и других кислот,
хелатные и другие подобные соединения ртути с ионизированной или сла-
бой ковалентной связью. Однако в главе кратко рассматриваются ртутные
соли циановой, гремучей, роданистоводородной кислот и т. п. соединений.
В отличие от порядка, принятого в остальной части книги, где реакциям
ртутноорганических соединений посвящена отдельная глава (гл. XIV),
в данной главе свойства и реакции некоторых соединений излагаются при
данном соединении или при группе сходных соединений.
О получении соединений RHgZR', где R — органический радикал, сое-
диненный с ртутью через углерод, R' — органический радикал, соединен-
ный с ртутью через гетероатом Z, см. гл. XIV.
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др. 359
СОЕДИНЕНИЯ с О—Hg-СВЯЗЬЮ
О — Hg-Производные спиртов и фенолов
О—Hg-Производные спиртов (алкоголяты ртути). Метилат и этилат
ртути образуются при действии сулемы на раствор соответствующего алко-
голята натрия в метиловом или этиловом спирте 111. Соединения эти не-
устойчивы.
Получение метилата ртути 11]. При действии на раствор метилата натрия в абсолютном
метиловом спирте раствором эквимолекулярного количества сулемы в метиловом спирте вы-
падает оранжево-красный осадок метилата ртути.
Алкоголяты высших спиртов не получены.
Двойные соединения алкоголятов ртути нисших спиртов и пентакарбо-
нила железа состава (RO)2Hg-2Fe(CO)5 (R = СН3, С2Н5, С3Н7) получены
действием пентакарбонила железа на раствор ацетата ртути (в случае
R = СН3 — также на раствор пропионата ртути) в соответствующем спир-
те [1]. Эти двойные соединения разлагаются действием конц. азотной и
соляной кислот, постепенно разлагаются при кипячении в воде[1]. При дей-
ствии на них сулемы в воднОт ацетоновом растворе происходит реакция, на-
пример, для метилата ртути:
2Fe (CO)6-Hg (ОСНз)2 + 3HgCl2 + 2НаО -> 2Fe (CO)4-Hg2Cl2 + 2СН8ОН + 2НС1.
Продукт присоединения меркурамидхлорида к глицерину получен выса-
живанием аммиаком раствора сулемы в глицерине или действием амидхлори-
да ртути на глицерин [21.
О—Hg-Производные фенолов (феноляты ртути). Ртуть не образует
устойчивых монофенолятов (ArO)2Hg и ArOHgX, где Аг — фенил или оста-
ток гомологов бензола. Неустойчивые феноляты ртути, первоначально
образующиеся при взаимодействии солей ртути с фенолятами щелоч-
ных металлов, довольно быстро перегруппировываются в меркуриро-
ванные в ядро фенолы, растворимые в щелочах без выделения окиси ртути.
Маншо описано [3] образование продуктов присоединения солей ртути
по кислороду меркурированием фенолов и их эфиров. Выделены лишь фе-
ноляты ртути фенолов, содержащих в ядре несколько атомов галоида, нит-
рогруппы и т. д. Сюда относятся, например, такие соединения, как трибром-
фенолят pTyTH(C6H2OBr3)2Hg, получаемый [4, 5] из 2,4,6-трибромфено-
ла и ацетата ртути в разбавленном спирте и представляющий собой желто-
красные чешуйки, довольно легко растворимые в спирте. 2,4,6-Трибром-
и трииодфеноляты закисной ртути выпадают в виде темно-желтых осадков
при прибавлении слегка подкисленного раствора азотнокислой закиси ртути
к спиртовому раствору тригалоидфенола [6].
Пентахлорфенолят [7] ртути (C6Cl5O)2Hg -]- 2Н2О теряет воду при! 10° С,
превращаясь ф (C6Cl5O)2Hg; это соединение образуется в виде аморфного не-
растворимого в обычных растворителях желтого порошка при действии стро-
го нейтрального раствора пентахлорфенолята натрия или калия на водный
раствор соли ртути при нагревании.
Получение пентахлорфенолята ртути [7]. 1 моль пентахлорфенола растворяют в водном
растворе едкого кали или натра, содержащем несколько больше 1 моля КОН (или NaOH).
К полученному раствору при перемешивании прибавляют по каплям сильно разбавленную
азотную кислоту до тех пор, пока не образуется небольшой осадок нерастворимого пен-
тахлорфенола, который отфильтровывают. К полученному прозрачному раствору пента-
хлорфенолята, не содержащему избытка щелочи, прибавляют раствор 1 моля нейтральной
соли ртути. Выпавший осадок пентахлорфенолята ртути отсасывают.
Пентахлорфенолят ртути легко разлагается даже слабыми кислотами.
360
Ртутноорганические соединения. Лит. стр. <583—389
Пикрат ртути [C6Ha(NO2)3O]2Hg-2H2O [5,8? 9] получают при нагревании
в воде не выше 80° С пикриновой кислоты и свежеосажденной окиси ртути.
Зильберрад и др. [9] сообщают, что при всех условиях это соединение по-
лучается только в виде двойной соли [C6H2(NO2)3O]2Hg-Hg(OH)2.6Н2О,
которая взрывает. При кипячении в воде или при действии разбавленной
соляной кислоты оно распадается с образованием продукта меркурирова-
ния пикриновой кислоты в ядро [5]. Этим последним соединением является ве-
щество, полученное Либихом [10] и принятое им за пикрат закисной ртути [5].
Согласно патентным данным действием на (P-C10H7)2Hg уксусной ки-
слоты получается p-C10H7OHgOCOCH3 [11].
4-Нитро-1-нафтол-0-меркурацетат получен при сливании водных раство-
ров уксуснокислой ртути и 4-нитронафтола-1 [12]. В уксуснокислом раство-
ре происходит меркурирование в ядро (см. гл. V).
В то время как фенол и нафтолы при действии сулемы и бикарбоната нат-
рия в присутствии глицерина меркурируются в ядро (см. гл. V, стр. 71),
пирокатехин, резорцин, гидрохинон, орсин, гваякол, флороглюцин, соглас-
но Неоги и Чаттерджи [13], образуют соединения, содержащие группи-
ровку OHgCl, например бис- хлормеркурпроизводное пирокатехина
o-CeH4(OHgCl)2, полученное при действии на водный раствор пирокатехина
сулемы, бикарбоната натрйя и глицерина и представляющее собой зеленый
осадок, разлагающийся около 150° С. С повышением pH среды увели-
чивается содержание O-Hg-продукта [14].
При действии раствора сулемы на натриевую соль 4-фенилазо-1-нафтола
образуется красный осадок соединения, содержащего О—Hg-связь [15].
O-Hg-Производные алкилнитраминов
Ртутные производные алкилнитраминов (RNNO2)2Hg, представляющие
собой, по-видимому, Hg—О-соединения, получают обработкой водного
раствора алкилнитрамина свежеосажденной окисью ртути. Ртутное произ-
водное метиднитрамина — бесцветные иглы, труднорастворимые в холодной
водей спирте [16]; этилнитрамина — листочки или пластинки (взры-
вает) [17].
O-Hg-Соединения кислородсодержащих производных циана
Ртутное производное циановой кислоты, получаемое действием соли ртути
на цианат калия в виде двойной соли с солью калия, представляет собой,
как считают Биркенбах и Кольб [18, 19] на основании анализа раман-спект-
ров, Hg—О-соединение, однако строение как самих цианатов ртути, так
и их двойных солей Hg—О- или Hg—N-соединений еще не достаточно
выяснено [20].
Получение двойной соли циаиата ртути и хлористого калия [18]. В раствор 20 г цианата
калия в 150 мл воды (нейтрализованной добавлением разбавленной соляной кислоты в при-
сутствии метилового красного) при 15—20° С прибавляют при перемешивании по частям
28 г измельченной в порошок сулемы. Тотчас выпадает осадок, и раствор застывает в кашу.
Смесь отсасывают, нромывают многократно 50% -ным, затем чистым этиловым спиртом и эфи-
ром. Выход 29г (80%). После кристаллизации из 100мл воды (40°С), с отфильтровыванием
слабой мути, получают 15 г двойной соли (41,5%) в виде бесцветного тонко кристаллического
порошка.
Гремучую ртуть, фульминат ртути (CNO)2Hg, получают действием раство-
ра азотнокислой ртути в азотной кислоте на этиловый спирт [21—25].
Схема этой реакции следующая [30]: \
СН3СНаОН + HNO3 СН3СНО 4- HNO2 + Н2О -* ONCH2CHO HON = CH - CHO (2?
(°> HON = CHCOOH —t HON = C (NO2)COOH HON = CHNOa ->
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др.
36t
Н—О—N=C 4- HNO2
2HONC + Hg (NO3)2-» Hg (ONC)2 + 2HNO8.
Ее промышленное получение основано на этой реакции.
Этиловый спирт может быть заменен ацетальдегидом, диметилацеталем
или малоновой кислотой [26]. Гремучая ртуть может быть получена также
прибавлением натриевой соли ацинитрометана к водному раствору сулемы
при 0° С [27—29] и последующем кипячении с разбавленной соляной кисло-
той .
2CH2NO2Na + HgCl2 (CH2NOO)2 Hg -»(ONC)2 Hg + 2H2O + 2NaCI.
Одновременно образуется желтая основная соль
cno2
Hg,
2
которая является единственным продуктом, образующимся при приливании
раствора сулемы к раствору натриевой соли нитрометана. Эта соль также
весьма взрывчата.
При кипячении метилнитроловой кислоты HON=CHNO2 в разбавлен-
ной азотной кислоте в присутствии солей ртути также образуется гремучая
ртуть [30]., Ничтожная величина электропроводности фульмината ртути
[161 указывает, возможно, на наличие в ней не Hg—О-, a Hg—С-связи [31].
Получение гремучей ртути [24, 25]. 50 г ртути растворяют в 600 г азотной кислоты
(уд. в. 1,4) и еще теплый раствор (~ 70° С) постепенно при перемешивании вливают
в 550 г этилового спирта (98,5%); по окончании выделения белых паров реакционную смесь
заливают 1 л воды. Выделившуюся гремучую ртуть хранят под водой.
Гремучая ртуть представляет собой кристаллическое вещество белого или
серого цвета. При кристаллизации из воды она содержит х/2 молекулы воды,,
при кристаллизации из спирта или при высаживании из водного аммиака
азотной кислотой получается безводной (ср. [32, 33]).
Гремучая ртуть нерастворима в большинстве органических растворите-
лей, плохо растворима в воде и бензоле, несколько лучше — в теплом ацето-
не, спирте, аммиаке, пиридине, растворе цианистого калия. Она может быть
очищена растворением в водном растворе цианистого калия, пиридине или
водном аммиаке и высаживанием разбавленной кислотой. Гремучая ртуть
при действии соляной кислоты на холоду дает хлороформальдоксим [28, 34],
при нагревании — гйдроксиламин и муравьиную кислоту [35]. При дейст-
вии хлора или брома на гремучую ртуть образуется дигалоидфуроксан. Из-
быток хлора разрушает фульминат ртути с образованием сулемы, хлорциана
и хлорпикрина [36] (ср. [37]). Водный аммиак дает мочевину и гуанидин
[38]. Действие аммиака и -аминов см. также [38—40]. При кипячении
фульмината ртути в воде в присутствии меди или цинка образуются фуль-
минаты этих металлов и выделяется металлическая ртуть [35]. Действием
на фульминат ртути в метиловом спирте в атмосфере водорода амальгамами
натрия [35], калия, щелочноземельных металлов, кадмия, таллия или
марганца образуются их фульминаты [41—44].
Сероводород разрушает гремучую ртуть (при действии на ее водную
суспензию) 'с образованием роданистого аммония, сернистой ртути и угле-
кислоты [36], согласно Камби [45], с образованием тиоформгидроксамовой
кислоты (см. также [38, 46]).
При действии на гремучую ртуть йодистого'этила образуется этилизоциа-
нат и этилцианураты [28], при действии хлористого ацетила образуется
ацетилизоцианат, CH3CON=CO, сулема, немного HCN,.ацетил-и диацетил-
мочевина [47], при действии хлористого бензоила — дибензоилмочевина
362
Ртутноорганические соединения. Лит. стр. 383—389
[48]. При действии на гремучую ртуть теплого концентрированного рас-
твора тиомочевины образуется сернистая ртуть, роданистая ртуть и
мочевина [49]. При смешении диэтилртути с гремучей ртутью взаимодействия
не заметно, при действии на такую смесь хлористого водорода выделена хло-
ристая этилртуть и комплекс состава: 5HgCl2-2NH2OH-HCl-2NH4C1-
•2(СН3)2СО 1501.
Шоллем и сотр. показано, что при взаимодействии гремучей ртути с аро-
матическими углеводородами в присутствии А1С13, А1С13 + 6Н2О и А1(ОН)3
в зависимости от порядка введения реагентов образуются нитрилы '[521
или впервые полученные ими ароматические альдоксимы [51, 53—55].
Взаимодействие гремучей ртути (в виде ее двойной соли с цианистым ка-
лием) с алкиларилпиразолонами приводит к образованию 4-цианалкиларил-
пиразолонов 156].
Литература, посвященная гремучей ртути — одному из важнейших де-
тонаторов — инициирующих взрывчатых веществ, — обширна. Техничес-
кое получение гремучей ртути см., например, [57—61].
Ртутные .производные циануровой кислоты, полученные действием соли
ртути (хлорида и ацетата) при 0° С на щелочный раствор циануровой кисло-
ты, представляют собой, по Ганчу [62], О—Hg-соединения.
О — Hg-Производные некоторых гетероциклов]
О ртутном производном изатина, которое может быть рассматриваемо и
как Hg—О-соединение, и как Hg— N-соединение, см. ниже, при Hg—N-
соединениях.
Изатин-3-гидразон, соответственно, его десмотропная форма при нагре-
вании с окисью ртути в бензоле образует, по Курциусу [631, ртутную соль
3-диазооксиндола
N2 .
/ /С\ \
I с6н/ С — о I Hg.
\ N А
Описаны ртутные производные антипирина с Hg—0-связью [64].
СОЕДИНЕНИЯ с S—Hg-СВЯЗЬЮ
Алкилмеркаптиды ртути
Получение из меркаптанов и некоторых простых сульфидов. Некоторые
реакции. В то время как лишь для некоторых случаев известны алкоголяты и
феноляты ртути, образование прочных соединений (RS)2Hg прохоДит легко и
характерно для меркаптанов (на основании чего они получили [65] свое на-
звание— corpus шегсцгшш captans). Меркаптиды ртути получают действи-
ем RSH на HgO или Hg(CN)2 большей частью в водном растворе или в спир-
те. Реакция образования меркаптидов сопровождается значительным вы-
делением тепла.
Получение этилмеркаитвда ртути [66]. При встряхивании водного раствора цианистой
ртути с этилмеркаптаном меркаптид ртути выпадает в виде белой массы перепутанных
тонких, гибких игл. После кристаллизации из спирта — листочки с т. пл. 76—77 С
[67—69] (78° С [23]).
Аналогично получен [70] метнлмеркаптид ртути (CHslaHg с т. пл. (с разложением)
175° С [70, 71], т. кип. 215-220° С/О.Гжл [72].
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др.
363
Брэдли и corp. [72Г доказывают полимерное строение последнего.
Пропилмеркаптид ртути (т. пл. 71° С) получен из пропилмеркаптана и
окиси ртути [73] или цианида ртути [74]. Вертхейм приводит температуры
плавления ряда алкилмеркаптидов ртути [74]: (/-C3H7S)2Hg— 62—63° С
[ср. 75] (n-C4H4S)2Hg — 85—86° С, (z-C4H9S)2Hg — 94-95° С,
(C5HnS)2Hg - 74—75° С, (f-C8HnS)2Hg - 100° С, (C7HlgS)2Hg - 76-
77° С.
Образование бензилмеркаптида ртути происходит с сильным разогрева-
нием, даже если взаимодействие меркаптана и окиси ртути вести в сильно
разбавленном спиртовом растворе [76, 77].
Получение бензилмеркаптида ртути [76]. В спиртовый раствор бензилмеркаптана вносят
тонкоизмельченную красную окись ртути. Образующуюся белую массу отфильтровывают н
несколько раз извлекают горячим спиртом. Кристаллизуют из этилового спирта. Бензилмер-
каптид ртути представляет собой тонкие нитевидные иглы. Т. пл. 121,5° С [76а].
Взаимодействием RSH с HgX2 (X = Hal, NO3 [77 — 82] и др.) в мяг-
ких условиях, например в спиртовом растворе, действием НХ (X = С1 [70],
NO3 [82]) на (RS)2Hg, нагреванием (RS)2Hg с RX (X = Вг, J) получают
RSHgX [82, 83]. В последнем случае при X = J в зависимости от условий
могут быть получены как RSHgJ [84], так и R3SJ-HgJ2, (R3SJ)2HgJ2 [83,85],
а также R2S- HgJ2 [85]. С2Н5Вг реагирует с большим трудом, чем C2H5J [83].
RSHgX могут быть получены также реакцией двойного обмена:
RSHgX' + КХ RSHgX + КХ'
(см., например, [86]).
C2H5SHgNO3 получен действием паров окислов азота на (C2H5S)2Hg [83].
Возможно, что иодиды построены ковалентно RSHgJ, а нитраты ионизирова-
ны (RSHg)+NO3 [85]. AlkSHgX в отличие от (AlkS)2Hg образуют комплексы
с аммиаком [83]. Галоиды в мягких условиях действуют на (RS)2Hg с образо-
ванием RSHgHal или RSHal [85а—85в].
C2H5SHgO2CCH3 получен также действием на этилтиоацетат уксусно-
кислой ртути в спирте при нагревании или в растворе в уксусном ангидриде
при комнатной температуре [87]:
СНзС — S — C2HS + Hg (О2ССН3)2 -> C2H5SHgO2CCH3.
II
о
Согласно данным Смиса и Симона [79], соли RSHgX (X — галогенид,
ацетат, перхлорат и др.) имеют удвоенный молекулярный вес.
Продукт реакции RSHgX с R'X имеет состав R2S2-HgX2-R'X [89].
Хлористый бензил меркаптид ртути получен действием сулемы на бен-
зилтиосульфат натрия [90], а действием на него цианистой ртути (в водном
растворе на холоде, затем нагревание в течение часа на водяной бане) полу-
чен бензилмеркаптид ртути [76а]. Аналогично, получен этилмеркаптид
ртути [76а]. \
Описаны ртутные производные циклических триэтиленполисульфидов
191].
2-Алкоксиэтилмеркаптаны ROCH2CH2SH при действии сулемы в спирте
образуют белые осадки высокоплавких труднорастворимых в органических
растворителях меркаптомеркурхлоридов ROCH2CH2SHgCl [88, 92—94]
(R = С2Н5, С4Н9 [92, 931). Образованием этих солей они отличаются от
изомерных 1-алкоксиэтантиолов CH3CH(OR)SH.
Получение (C2H5SHgO2CCH3)2 [79]. При перемешивании 16,14 г этилмеркаптида ртути
с 15,94 г уксуснокислой ртути в 60 мл теплой воды образуется прозрачный раствор, из ко-
торого при медленном испарении выделяется чистая соль в ваде крупных кристаллов,
хорошо растворимых в воде, спирте, четыреххлористом углероде.
364
Ртутноорганические соединения. Лит. стр.^383—389
Подробно исследован интересный меркаптид ртути — трифторметилмер-
каптид ртути (CF3S)2Hg, впервые полученный Брандтом, Эмелеусом и
Хассельдином [95, 96] при освещении ультрафиолетовым светом трифторме-
тилдисульфида и ртути, а также взаимодействием сероуглерода и фторной
ртути при 250° С [97, 98].
Получение трифторметилмеркаптида ртути из трифторметилдисульфида и ртути [96].
5,0 г трифторметилдисульфида и 80 г ртути в кварцевой трубке подвергают освещению1
ультрафиолетовым светом при энергичном встряхивании в течение 4 дней. Избыток трифтор-
метилдисульфида удаляют и твердый осадок извлекают эфиром. После испарения при ком-
натной температуре в сухой атмосфере высушенного над пятиокисью фосфора эфирного эк-
стракта получают с 90% -ным выходом трифторметилмеркаптида ртути. Т. пл. 37—38° С.
Получение трифторметилмеркаптида ртути из сероуглерода и фторной ртути [98]. 715 г
(3 моля) фторной ртути и 453 г (около 6 молей) сероуглерода нагревают 4 часа при 250° С в
автоклаве емкостью 1 л, облицованном нержавеющей сталью. После охлаждения до ком-
натной температуры отфильтровывают сернистую ртуть. Из фильтрата испаряют сероугле-
род и низкоплавкий остаток перегоняют при 80—81° С/21 мм. Выход трифторметилмеркап-
тдда ртути от 250 до 350 а (средний выход 72%).
Взаимодействие трифторметилмеркаптида ртути с сулемой дает хлори-
стый трифторметилсульфид ртути [99], обменными реакциями которого
с соответствующими солями серебра (в воде, при комнатной температуре)*
могут быть получены нитрат и ацетат трифторметилмеркаптида ртути [99].
Получение хлористого трифторметилсульфида ртути [99]. 0,301 г трифторметилмеркап-
тида ртути в 10 мл эфира и 0,204 г сулемы в 10 мл эфира встряхивают при комнатной тем-
пературе. Продукт после испарения эфира возгоняют в вакууме при 40—50° С. Выход 0,45 г.
Реакция трифторметилмеркаптида ртути с бромной ртутью в этих усло-
виях не идет [99].
Как показывает спектроскопическое изучение (ИК- и раман-спектры)
реакций трифторметилмеркаптида ртути с ртутными солями галоидоводород-
ных и кислородных кислот, в водных или метиловоспиртовых растворах,
имеет место равновесие
(CFsS)a Hg -j- HgX2 2CFsSHgX
(X = Cl, Br, J [101], OCOCH3, OCOCF3, NO3 [100]).
С серебряными солями этих кислородных кислот происходит обратимое
образование комплексов [100]:
AgX + (CF3S)2 Hg X- AgX (CFsS)a Hg.
С иодистым калием, хлористым и иодистым тетраметил аммонием в ацетоне
трифторметилмеркаптид ртути, как показывает кондуктометрическое титро-
вание, образует комплексы, в которых он входит в состав аниона [102], на-
пример
(CH8)4N+[Hg(SCF3)2Cl]“.
Трифторметилмеркаптид ртути реагирует с треххлористым фосфором уже
при комнатной температуре (запаянная трубка) [99] (ср. [98]). С эквимоле-
кулярными количествами реагентов реакция идет по уравнению
J(CF3S)2 Hg + PC1S -» CF3SPC12 + CF3SHgCl,
с большим количеством трифторметилмеркаптида ртути образуется
(СРзЗ)2РС1 и (CF3S)3P (для получения последнего соединения необходима
нагревание).
Так же идет реакция с треххлористым мышьяком. В этих реакциях три-
фторметилмеркаптид ртути подобен полнозамещенным жирным и аромати-
ческим соединениям ртути Alk2Hg и Ar2Hg, но не бце-трифторметилртути^
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др.
365
которая не реагирует с РС13 и AsCl3 даже при 100° С. С медью при нагре-
вании трифторметил меркаптид ртути образует (CF3S)a:Cu [98].
Реакция трифторметилмеркаптида ртути с медью [98]. При нагревании смеси 6 г тонко
измельченной меди и 9 г трифторметилмеркаптида ртути при'80—100° С имеет место экзо-
термическая реакция с появлением яркой оранжевой окраски. Нагревание продолжают
.30 мин. при 150° С. После охлаждения твердый осадок извлекают 500 мл эфира. Испарение
экстракта дает бледно-зеленый осадок Cu(SCF;0x, не полностью свободный от эфира.
Другие же металлы, как Pb, Т1, образуют с трифторметилмеркаптидом
ртути фтористые соли MeF„ [98]. Арил- и алкилгалогениды, сульфенилгало-
гениды, ацил галогениды образуют с трифторметилмеркаптидом ртути три-
•фторметиларил(алкил)сульфиды,-тиоацилаты, -дисульфиды [98], например:
СН2 = CHCH2SCF3, C2H6CSCF3, CCI3SSCF3.
При взаимодействии трифторметилмеркаптида ртути с хлористым водо-
родом в запаянной трубке при комнатной температуре в течение 48 час.
количественно образуется трифторметилмеркаптан [96].
Легко образуется S—Hg-соединение за счет тиольной группы, присут-
ствующей в органических соединениях и других классов. Например, S —
— Hg-производные типа (RS)2Hg анилидов тиогликолевой, тиомолочной, тио-
масляной кислот тотчас выпадают при сливании вместе спиртовых раство-
ров сулемы и избытка анилида меркапто кислоты [103].
Согласно Рэю [324], тиоамиды реагируют с нитритом ртути в таутомер-
ной форме иминомеркаптанов с образованием продуктов RC(NH)SHgNO2,
неустойчивых и тотчас распадающихся с образованием альдегида, кислоты,
аммиака и неорганической соли ртути.
Тиомочевина реагирует с нитритом ртути, возможно, тоже с первоначаль-
ным образованием H2NC(SHgNO2)=NH, превращающегося далее в более
сложные продукты.
Действием едкого натра и сулемы на водный раствор сульфида
HO2CCH2CH2SC(NH)NH2 получен p-меркурметилмеркаптопропионат натрия
1105].
Соль двухвалентной ртути роданистоводородной кислоты — тиоцианат
или роданид ртути Hg(SCN)a — получается обычными способами: из окиси
ртути и роданистоводородной кислоты [106—108] в водном растворе, из ро-
данидов щелочных металлов и нитрата [106, 107, 109], ацетата [НО] (ср.
[111] (коллоид [112]), хлорида [113].Об образовании роданистой ртути из
роданистоводородной кислоты и гремучей ртути см. выше. Роданистая
ртуть представляет собой иголочки или листочки, трудно растворимые в
холодной, легче—в кипящей воде [106]; лучше, чем в воде, она растворяется
в спирте и несколько растворима в эфире. Разлагается при температуре
около 165° С.с сильным вспучиванием («фараоновы змеи»); весьма склонна
к образованию, с другими роданидами, галогенидами и цианидами комплекс-
ных солей Me[Hg(SCN)3] и Me2[Hg(SCN)4], в которых SCN-группа мо-
жет быть заменена на CN и галоид [Ill] и Н2О [111а] (ср. [114]). Три-
роданиды, как правило, трудно растворимы в воде, тетрароданиды же
легко растворимы [115].
Изучено влияние природы растворителя на образование комплексов
тиоцианата ртути [115а].
Исследование ИК-спектров роданида ртути и комплексов MeHg(SCN)4 и
MeHg(SCN)6-C6H6 (Me = Со, Ni, Cd) заставляет авторов [1156] предполо-
жить в последних наличие своего рода л-о-связи. Роданид ртути весьма
склонен также к образованию двойных солей.
366 Ртутноорганические соединения. Лит. стр, 333—38,9
Монороданиды ртути NCSHgX [где X = С1 [115, 116] (ср. [117]), Вг
[115], J [118], С1О4 [119], СН3СО2 [120, 121], HS [118]] получают
взаимодействием роданида ртути с избытком соответствующей соли ртути в
горячей воде.
В реакцию с R2Hg в диоксане тиоцианат ртути вступает с еще большей
легкостью, чем иодид и цианид ртути [122].
Тиоцианат закисной ртути образуется при сливании очень разбавлен-
ных водных растворов [107, 123, 124] азотнокислой закиси ртути и роданида
щелочного металла и при действии родана на металлическую ртуть [125].
Получающийся белый осадок разлагается при нагревании, но не столь ха-
рактерно, как роданид окисной ртути. Он растворяется в растворе родани-
дов щелочных металлов с образованием M2[Hg(SCN)4] и выделением метал-
лической ртути [107].
Моно тиогликоли [88,126,326] и дитиогликоли [89] и моно-[326] и дитиогли-
церин [326] также образуют соответствующие S—Hg-соединения в услови-
ях, аналогичных получению меркаптидов ртути из меркаптанов. Напри-
мер, при действии нитрита ртути на дитиоэтиленгликоль [89] получены
соединения O2NHgSC2H4SHgNO2 и S2(C2H4SHgNO2)2.
Получение реакцией с солями ртути тиоацеталей, винилсульфидов и
т. п. соединений. Обычные диалкилсульфиды образуют с сулемой комп-
лексные соли. Сульфиды же, являющиеся производными альдегидов —мо-
нотиоацетали, тиоацетали, винилсульфиды, а также некоторых кетонов, не
образуя с сулемой в воде или спирте устойчивых комплексных солей, рас-
щепляются под действием сулемы или окиси ртути с образованием соответ-
ствующего карбонильного соединения (при проведении реакции в воде) или
его ацеталя (при проведении реакции в спирте) и с выделением хлористо-
го водорода и хлормеркурмеркаптида. С тиоацеталями реакцию можно про-
водить в одну или две стадии:
SR
RCH(SR)2 4- HgCl2 + R'OH RCH + RSHgCl + HC1,
\>R'
SR
RCH + HgCl2 + R'OH — RCH (OR')a + RSHgCl + HC1.
\>R'.
При обработке тиоацеталей окисью ртути в воде происходит отрыв одной
SR-группы. При действии сулемы в присутствии карбоната кадмия отрыва-
ются обе SR-группы.
При кипячении соответствующих диэтилмер каптал ей с водным раствором
избытка сулемы получены акролеин [133] и кротоновый альдегид [134].
Реакция была применена для регенерации £-цистеина из S-бензилтиоме-
тил-Д-цистеина действием на раствор последнего в \N соляной кислоте
теплого водного раствора сулемы [135]. При этом выпадает тиобензил-
ртуть.
Легкость отрыва тиольных групп от меркапталей альдегидов действием
сулемы дает возможность, защитив альдегидные группы путем меркаптали-
зации, проводить различные реакции сложных альдегидов и затем регенериро-
вать альдегиды, вводя меркаптали в реакцию с сулемой, напр‘имер:
. HgCls
RCOCH (SR')2 RCH (ОН) CH (SR')2 RCH (ОН) СНО + R'SHgCl.
HjO
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др.
367
Синтез миндального альдегида расщеплением его диэтилмеркапталя действием сулемы и
карбоната кадмия [136}. Раствор 10 г диэтилмеркапталя миндального альдегида в 120 мл аце-
тона и 15 мл воды обрабатывают 50 г карбоната кадмия и 45 г сулемы в 50 мл ацетона и 10 мл
воды. После перемешивания в течение 2 дней при комнатной температуре отфильтровывают.
Фильтрат обрабатывают 10 г сулемы и 10 г карбоната кадмия и нагревают 30 мин. с обрат-
ным холодильником. Соли отсасывают и промывают ацетоном. Фильтрат испаряют в ваку-
уме, остаток извлекают хлороформом, раствор встряхивают с раствором йодистого калия и
отмывают водой от галоида. Сушат сульфатом натрия, хлороформ отгоняют в вакууме. Оста-
ется масло, через некоторое время начинающее выделять кристаллы, которые после обра-
ботки 2—3 раза эфиром и петролейным эфиром кристаллизуют из смеси абсолютного тетра-
гидрофурана и петролейного эфира. Выход 2,5 г (44,5%). Т. пл. 134—137° С (с предваритель-
ным разложением).
Действием сулемы в присутствии веществ, нейтрализующих одновремен-
но выделяющийся хлористый водород (карбоната натрия или кадмия) при
перемешивании, иногда в течение нескольких часов, проводят регенерацию
карбонильных соединений из циклических меркапталей, полученных дей-
ствием 1,2-этан- [137] или 1,3-[138—140] пропандитиолов на карбонильное
соединение.
Так, при действии сулемы в присутствии бикарбоната натрия на 2-фе-
нил-1,3-дитиан-5-сульфонат натрия происходит реакция [140]:
S — СН2 S — СНа
С6Н5-СН ^CHSOsNa HgZ \н5О3На + С6Н5СНО.
\ / - NaHCO, \ /
S — СН2 S —СНа
Получение 1,3-меркурдимеркаптопропан-2-сульфоната натрия [140]. К раствору 401?
сырого 2-фенил-1,3-дитиан-5-сульфоната натрия в 400 мл воды при размешивании н нагре-
вании (температура бани 60—65° С) в течение 1 часа частями прибавляют 35 г сулемы и 12 г
двууглекислого натрия. Затем раствор нагревают еще 1 час, после чего в нем иодометрически
определяют 44,8 г (81%) меркурдимеркаптопропансульфоната натрия. Соль высажена из
раствора спиртом (500 мл) в виде технического продукта 72%-ной чистоты.
В то время как простые виниловые эфиры присоединяют соли ртути по
двойной связи (см. гл. VI), действие сулемы на винилалкилсульфиды в
спиртовом и водном растворах направляется на второй реакционный центр
молекулы — атом серы и приводит к образованию хлормеркуралкилмеркап-
тида [141]:
СН2 = CHSC2H6 4- HgCl2 + 2С2Н5ОН C2H5SHgCl + НС1 + СНзСН (ОС2Н5)2,
СН2 = CHSC2H6 + HgCl2 + Н2О C2H6SHgCl + НС1 + СНзСНО.
Реакция в этиловом спирте проходит количественно и может применять-
ся для титрометрического определения тиовиниловых эфиров [142]. В эфир-
ном растворе реакция винилэтилсульфида с сулемой приводит к об-
разованию неустойчивой комплексной соли приблизительного состава
3CH2=CHSC2H5-4HgCl2, быстро разрушающейся при действии спирта и
воды с образованием ацетальдегида, хлористого водорода и этилмеркапто-
меркурхлорида [141].
Взаимодействие винилэтилсульфида со спиртовым раствором сулемы. Получение этил-
меркаптомеркурхлорида [141]. 0,13 г винилэтилсульфида обрабатывают 4,5 мл 20%-ного
раствора сулемы в этаноле и оставляют на ночь. Выпавший осадок отфильтровывают, про-
мывают спиртом, эфиром и высушивают в вакууме до постоянного веса. Вес осадка 0,37 г
(выход 96,2%). Этилмеркаптомеркурхлорид перекристаллизовывают из кипящего ксилола,
промывают спиртом, эфиром и высушивают в вакууме; при нагревании до 200° С он не пла-
вится, а медленно разлагается при 300—350° С.
Так же проходит реакция 3-ал коксиэтилвинил сульфидов с сулемой в
растворе этанола по уравнению
ROCH2CH2SCH = СН2 + HgCl2 + 2С2Н5ОН -» ROCH2CH2SHgCl + НС1 + СН3СН(ОС2Н5)а
368
Ртутноорганические соединения. Лит. стр. 383—389
с количественным выделением Р-алкоксиэтилтиомеркурхлорида и хлори-
стого водорода [142] (ср. [143]). При проведении реакции в водном рас-
творе образуется ацетальдегид [142].
Как показали Прилежаева, Шостаковский и сотр. [92, 144], из трех ти-
пов диалкоксидиэтилсульфидов:
ROCH2CH2
КОСНгСНа^
3,3'
ROCH2CH2
RO—бНСНз
а, 3
CH3CHOR
S
I
CH3CHOR
а, а'
р,3'-сульфиды при реакции с сулемой образуют типичные для обычных
сульфидов устойчивые комплексные соли. Комплексы с сулемой а,£- и а,а'-
производных тотчас распадаются. <х,Р-Производные образуют {З-алкокси-
этилтиомеркурхлориды и ацетали и выделяют одну молекулу хлористого
водорода:
OR OR
СНзбН + HgCU + R'OH СНзСН + ROCH2CH2SHgCl + НС1.
^SCHaCHaOR ' ^OR'
Комплексные соли а,а'-изомеров распадаются с образованием желтых осад-
ков и выделением двух молекул хлористого водорода.
Действие сулемы на а,р-дибутоксиднэтилсульфид. Получение хлористой 0-бутоксиэтил-
тиортути [144].
ОС4Н9 ос4н9
СНдСН^ + HgCla + С2Н5ОН -> СНзСН7" -f C4H»OCHaCHaSHgCl + НС1
\сН2СН2ОС4Н9 \)С2Н5
К 2,5 г а,Р-дибутоксидиэтилсульфида добавляют 3,5 г сулемы в 15 мл этанола. Немед-
ленно выпадает белый Творожистый осадок. После хранения в течение ночи его отфильтровы-
вают и промывают холодным растворителем. При упаривании фильтрата выпадает новая не-
значительная порция кристаллов. Общий вес осадка 3,5 а (87,2%, считая на взятый сульфид),
После перекристаллизации из большого количества кипящего спирта выпадают белые бле-
стящие таблички с т. пл. 137,5—138 °C, практически нерастворимые в холодном спирте,
эфире, ацетоне.
Аналогично получена хлористая 3-этоксиэтилтиортуть. Т. пл. 155,5—156° С (из спирта).
бнс-р-Алкоксиэтилмеркапто-1,1-этаны при действии сулемы в спирте
образуют соответствующие меркаптидмеркурхлориды и выделяют хлори-
-стый водород [144]:
CH3CH(SCH2CHaOR)2 + 2HgCl2 + 2R'OH --------> CH3CH(OR')2 + 2ROCH2CHaSHgCl +
-J-2HC1.
При нагревании с избытком сулемы в воде выделяется ацетальдегид.
Отделение меркаптидных остатков идет в две стадии.
Действие сулемы на биг-(0-этоксиэтилмеркапто)-1,1-этан. Получение хлористой р-этокси-
этилтиортути [144].
СН3СН (СН2СН2ОС2Н5)2 + 2HgCla + 2С2Н5ОН -> СНзСН (ОСаН5)2 +
+ 2C2H5OCH2CHaSHgCl 4- 2НС1
К 0,2 г p-этоксиэтилмеркапталя ацетальдегида добавляют 0,3 г сулемы, растворенной в
этаноле. Вес немедленно выпавшего белого осадка p-этоксиэтилмеркаптомеркурхлорида по-
сле сушки в вакуум-эксидаторе 0,60 г. После повторной перекристаллизации из кипящего
этанола т. пл. 155,5—156° С.
Таким же путем из 0,2 г [З-бутоксиэтилмеркапталя ацетальдегида получено 0,5 г (теория
—0,49 г) 3-бутоксиэтилмеркаптомеркурхлорида с т. пл. 137,5—138° С.
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др. 369
Реакция с сулемой предложена Хольмбергом [145—1491 для количе-
ственного определения в ряду меркапталей и меркаптолов тиоуксуснОй,
тиогликолевой, тиомолочной, тиогидракриловой кислот (титрование выде-
ляющегося хлористого водорода щелочью или йодометрическое определе-
ние сулемы). В спиртовой среде она разработана Прилежаевой и сотр. для
количественного определения монотиоацеталей [92, 144, 189], меркапта-
лей [141, 1421, винилсульфидов [94 142, 144, 150—157] и ими и другими
авторами для определения разнообразных других.сульфидов, производных
альдегидов и кетонов (например, [158]). В случае нахождения в 3-поло-
жении к SR-группе бензоат-радикала расщепление действием сулемы не
проходит количественно [159].
Продукты присоединения диалкилдитиофосфатов к виниловым [159а]
и тиовиниловым [159а, 160] эфирам ведут себя по отношению к сулеме в
спиртовой среде подобно меркапталям, выделяя два эквивалента хлористо-
го водорода и распадаясь по уравнению
SR'
СНзСН + HgCl2 + 2С2Н4ОН -► СНзСН (ОС2Н6)2 + 2НС1 + R'SHgCl +
\pS(OR)a
+ ROP(S)SHgCl. (1)
Смешанная ртутная соль диалкилдитиофосфорной и соляной кислот
(1) при перекристаллизации диспропорционировалась:
2 (GaH5O)2 PSSHgCl HgCl2 + [(С2Н5О)2 PSS]2 Hg.
Реакция 0,0-диалкил-8-(с-алкилмеркапто)этилдитиофосфатов с сулемой [160]. По-
лучение хлористой тиоэтилртути. К 1 г (0,0036 моля) О,О-диэтил-5-(а-этилмеркапто)-
этилдитиофосфата добавляют 2,2 г (0,0081 моля) сулемы, растворенной в 11 мл 96%-нрго
этилового спирта. Немедленно выпавший белый осадок отфильтровывают через 3 часа и
промывают холодным спиртом. Спирт соединяют с фильтратом, а осадок высушивают в ва-
кууме; вес осадка 1,8 г. При хранении в течение 5 дней из фильтрата выпадает еще 0,15 г
кристаллов. При титровании части фильтрата стандартным раствором едкого натра найдено
соляной кислоты 97,7% (от теорет.). Фильтрат упаривают досуха в вакууме (10 мм)', отгон
количественно собирают в ловушке, охлажденной Сухим льдом с ацетоном. Определение
диэтилацеталя гидроксиламиновым методом (в части отгона) дало 92,6%.
К остальной части добавляют 0,45 г динитрофенилгидразина и 0,5 мл конц. соляной кис-
лоты. После кипячения в течение 30 мин. и охлаждения выпадают золотистые иглы ацеталь-
дегиддинитрофенилгидразона; после перекристаллизации т. пл. 163—164° С. Вес сухого
остатка после разгонки составляет 0,6 г, а после обработки водой для удаления непрореаги-
ровавшей сулемы—0,3 г. Остаток дальше не обрабатывается, так как быстро чернеет. Общий
вес выделенных ртутных солей составляет 2,25 г (по теории 2,6 г).
Соли, выпавшие из раствора (1,96 г), экстрагируются несколько раз кипящим спиртом
(общее количество 75 мл). Вес нерастворимого, в спирте осадка 0,8 г. После перекристалли-
зации из кипящего ксилола выпадают перламутровые листочки хлористой тиоэтилртути,
не плавящиеся, но постепенно разлагающиеся выше 250° С.
Из охлажденного спирта выпадает 0,7 г мелких кристаллов, которые для полного дис-
пропорционирования нагреваются в течение часа с кипящим бензолом. Температура раз-
ложения выпавший из бензола кристаллов диэтилдитиофосфата ртути 121—121,5° С.
Реакция с сулемой (в присутствии углекислого кадмия в спиртовом
растворе, перемешивание в течение 24 час.) с последующим действием
аммиаком применена для превращения 2-(2-бромпропионамидо)пропальде-
гиддиэтилмеркапталя в 2-гидрокси-3,6-диметилпиразин [161 ]:
Вг
(C2H5S)2CH СНСНз
I I
НзССН СО
\nh/
HgCI2
NH,
CdCO,
C2HSOH
N
n-GH-
ил-О-он
N
370
Ртутноорганические соединения. Лшп. стр. 383—389
Расщепление меркапталей сахаров под действием сулемы, впервые
наблюдавшееся Фишером [162], применяется для синтеза монотиоглюко-
зидов и алкилглюкозидов (при действии на меркаптали сахаров одного или
двух эквивалентов сулемы в абсолютном спирте) [163—170]:
SR
—С
|\r
СНОН '
| Hgcl,
О СНОН с2нб0н
СНОН •
—СН
СН2ОН
SR
—o'
I ХОС2Н6
СНОН
О СНОН
СНОН
—сн
СНгОН
ОС2Н6
---
HgCb
С2Н5ОН^ 0
I ХОС2Н5
СНОН
I
СНОН
СНОН
—СН
СНаОН
и для получения моноз в альдегидной форме (действие на меркаптали из-
бытка сулемы в водном спирте) [163, 164, 171—173]:
Синтез глюкозидов моноз из их бензилмеркапталей. Превращение бензилмеркапталей
Z-арабинозы, {-рамнозы н а-галактозы в р-метил-{-арабииозид, а-метил-7-рамнозид и
а-метил-Й-галактозид [169]. Горячий концентрированный раствор 1 моля меркапталя в ме-
тиловом спирте Кальбаума быстро обрабатывают также горячим метиловоспиртовым раст-
вором избытка сулемы (3 моля) и осторожно нагревают на водяной бане в течение 5—10 мин.
Раствор отфильтровывают горячим от осадка бензилтиомеркурхлорида и затем так долго
попеременно нагревают и фильтруют, пока раствор при дальнейшем нагревании не будет
совершенно прозрачным, что обычно достигается в течение 1 часа. Затем удаляют избыток су-
лемы сероводородом и кислый (вследствие выделения хлористого водорода) раствор нейтра-
лизуют углекислым серебром. Фильтрат для удаления небольших количеств растворенной
серебряной соли вновь обрабатывают сероводородом и жидкость, ставшую черно-корич-
невой от выделившегося коллоидального сернистого серебра, упаривают в фарфоровой чаш-
ке при добавлении небольшого количества животного угля до объема 20—30 мл. Отфильтро-
ванный прозрачный нейтральный раствор упаривают в вакууме до консистенции густого
сиропа. р-Метил-{-арабинозид имеет т. пл. 169—170° С (из спирта), выход 90% ;а-метил-й!-га-
лактозид— т. пл. Ш°С (из спирта), выход 80%; а-метил-{-рамнозид—сироп, идентифициро-
ван после ацетилирования в виде триацетата, т. пл. 86—87° С.
Демеркаптализация диэтилмеркапталя диметилизопропилиденксилозы [173]. Раствор
2 г сулемы в 50 мл абсолютного метанола прибавляют в течение 30 мин. к смеси 9 г диэтил-
меркапталя диметилизопропилиденксилозы, 18 г окнси ртути, 5 г безводного сульфата нат-
рия в 80 мл абсолютного метанола. После перемешивания в течение 6 час. при 45° С раствор
отфильтровывают, остаток извлекают-теплым спиртом и соединенные фильтраты обрабаты-
вают 5 мл пиридина и оставляют при 0° С на 2 часа. После фильтрования комплекса пи-
ридина с сулемой фильтрат испаряют в вакууме до консистенции сиропа, который освобо-
ждают от неорганических веществ извлечением бензолом. Сироп представляет собой диме-
тилизопропилиденальдёгидкснлозу; п™ 1,4535.
Защита карбонильной группы сахаров путем превращения в меркапта-
ли широко применяется для проведения разнообразных реакций также и
в этом ряду, с последующей регенерацией сахаров действием на меркапта-
ли сулемы (см., например, многочисленные работы Циннера и сотр. [174—
199]).
Синтез 6-бензоил-2,3,4,5-тетраацетил-а{-/)-глюкозы [187]. 2,9 г (0,005 моля) 6-бензоил-
2,3,4,5-тетраацетил-П-глюкозы ди-«-пропилмеркапталя в 40 мл ацетона и Змл воды пере-
мешивают с 5 г желтой окиси ртути и 5 г сулемы 6 час. при 20° С и 30 мин. при 50° С. Затем
фильтруют, осадок промывают ацетоном и фильтрат в присутствии окиси ртути испаряют в
вакууме. Осадок извлекают хлороформом 4 раза по 25 мл, промывают экстракт 1 N иодистым
калием, водой, сушат сульфатом натрия и испаряют в вакууме. Получают аморфный поро-
шок, который сушат, выдерживают над парафином при 35° С и 15 Торр. Выход 1,58 г
(70%); [а]^ -ф 18,8° (с—2,91 в хлороформе).
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др.
371-
Арилмеркаптиды (тиофеноляты) ртути
Тиофеноляты ртути получают взаимодействием тиофенола' и окиси ртути
в водном или спиртовом растворе при нагревании [76, 127], иногда даже
без растворителя [128]. Лучше реакцию вести в растворе пиридина [129].
Получение тиофеиолята ртути [129]. Встряхивают раствор 5 г тиофенола в 60 мл пири-
дина с 6 г желтой окиси ртути, при этом окись ртути постепенно разлагается с разогрева-
нием.Затем отфильтровывают от не вошедшей в реакцию окиси ртути, осадок промывают пи-
ридином и добавлением к раствору двойного объема воды высаживают фенилмеркаптид рту-
ти. Выход сырого продукта 9,11 г. После кристаллизации из бензола образуются бесцвет-
ные иглы. Т. пл. 152,5—153,5° С.
Описано получение (o-CH3C6H4S)2Hg, (p-CH3C6H4S)2Hg [77], [1,3-
(CH3)2C6H3S]2Hg [130], (a-C10H7S)2Hg [128, 131].
Меркаптид ртути (p-BrCH2CH2C6H4S)2Hg получен прибавлением избытка
раствора ацетата ртути в разбавленной уксусной кислоте к n-бромэтилтио-
фенолу [131а].
Получение а-иафтилсульфида ртути [128]. К измельченной в порошок окиси ртути при-
бавляют по каплям а-тионафтол. При этом происходит бурная реакция с сильным выделе-
нием тепла. Кристаллизация из спирта. а-Тионафтолят ртути представляет собой бледно-
желтый порошок.
Некоторые RHgSC6H5 превращаются в (C6H5S)2Hg'[327J.
При действии хлористого нитрозила на меркаптиды ртути образуются
соответствующие дисульфиды [132].'
Несимметричные дисульфиды получены [85а] по реакции
(P-CioH7S)a Hg + 2 (СНз)з CSJ 2 ₽-CwH,SSC (CHS)3 + HgJ2.
Меркаптиды некоторых гетероциклических соединений
Тиофен-, тианафтенмеркаптиды ртути получены взаимодействием ацетата
ртути с тиофентиолом в теплом спирте [103а].
2-Меркапттиазолин с азотнокислой ртутью в спиртовом растворе образует
[89] меркаптид ртути
/ СН2 — S4 \
| \c-S)Hg,
' СНа—N '2'
образующий с C2H5J комплексное соединение дисульфида с HgJ2 и C2H6J.
Получение меркаптидов ртути действием солей ртути
на некоторые другие S-содержащие соединения
В ряде случаев соли ртути в мягких условиях способны разрывать
С—S-связь некоторых других серусодержащих соединений (ацетилсульфидов,
дитиокарбоновых кислот, сульфоксидов) с образованием соединений с
S—Hg-связью.
Так, например, после нагревания водного раствора двойной солис нат-
рием и ртутью, полученной из этилового эфира дитиокарбаминоуксу-
сной кислоты состава C2H5CO2N(CH3)CSSNH2(CH3)CH2CO2C2H5 спиртом
высаживается в виде зеленого порошка следующее соединение:
NaCO2CH2N(CH3)CSSHgSCH3CO2Na. При выпаривании раствора досуха
получен натриевортутный тиогликолят [104].
. Фенилацетйлсульфид с уксуснокислой ртутью в зависимости от отно-
сительных количеств реагентов образует C6H5SHgX n.nif (CfiH5S)2Hg [200:]?
372
Ртутноорганические соединения. Лит. стр. 383—389
Получение хлормеркурфенилсульфида [200]. 1,1 г уксуснокислой ртути, растворенной в
спирте с добавлением воды и уксусной кислоты, обрабатывают при кипячении 0,5 г
CeHsSCOCHs- Как только раствор начинает мутнеть, нагревание прекращают, фильтруют и
добавляют водный раствор хлористого натрия, Выпадает 0,8 г хлормеркурфенилсульфида.
Кристаллизация из бензола.
Из 1,5 г уксуснокислой ртути и 1 г фенилтиоацетата в спирте получен
(C6HsS)2Hg [200L
При действии сулемы на ArSC(CeHa)3 в спиртовом растворе при комнатной
температуре образуются (ArS)2Hg, с избытком сулемы — ArSHgCl (Аг =
= СвНа, о-, т-, р-СН3С6Н4) [200а].
При действии сулемы (в спиртовом растворе, в присутствии соляной
кислоты) на кумаронилсульфоксидуксусную кислоту образуется хлормер-
куркумаронилмеркаптид [201].
SHgCl
S — СН2СООН с,н„он --------------/
Ч/\/ О + HgC12 HC1
о о
Фотолиз и пиролиз меркаптидов ртути
Постепенное почернение меркаптидов ртути при хранении отмечено еще
Меркером [77К Фотолиз меркаптидов ртути протекает различно, в зависи-
мости от природы органического радикала. Так, в то время как некоторые
меркаптиды ртути (как бензилмеркаптид ртути, циклопентилмеркаптид
ртути) разлагаются при фотолизе с выделением как металлической ртути,
так и сернистой ртути и органических сернистых соединений, другие (нап-
ример, тиофенолят ртути, этилмеркаптид ртути) выделяют при фотолизе
лишь металлическую ртуть (но не сернистую ртуть) [202]. По их возрас-
тающей устойчивости к фотолизу R в (RS)2Hg могут быть расположены в
ряд: R—бензил н-пропил изопропил < циклопентил третичнобу-
тил < фенил [202].
Пиролиз этилмеркаптида [68] и тиофенолята ртути [127, 129] дает ди- ,
этилдисульфид и дифенилдисульфид (соответственно) и металлическую
ртуть (но не сернистую ртуть) [129]. Пиролиз б«с-трифторметилтиортути (в
автоклаве, 350°С, 5 час.) и анализ ПК-спектров летучих продуктов показал
присутствие (CF3)2Sh (CF3)2S2[98]. Продуктами же пиролиза л-толилмеркап-
тида и бензилмеркаптида ртути являются сернистая ртуть и соответству-
ющие сульфиды или продукты их дальнейшего распада [127].
СОЕДИНЕНИЯ с Se —Hg-СВЯЗЬЮ
Алкилселениды ртути (RSe)2Hg получают подобно меркаптидам из алкил-
селенолов и окиси ртути или цианистой ртути [203]. (CH3Se)2Hg получен
приливанием CH3SeH в Hg(CN)3 при охлаждении [203, 204]. Его этильный
аналог—взаимодействием C2HsSeH с HgO [203, 204]. Как и в случае алкил-
меркаптанов, реакции эти экзотермичны.
При пропускании диалкилдиселенидов в водный раствор как сулемы
(подкисленной соляной кислотой), так и цианистой ртути происходит раз-
рыв Se—Se-связи ди алкилдиселенидов (отличие от диалкилдисульфидов)
и образуются RSeHgCl наряду с двойной солью RSeHgCl HgCl2 и, возмож-
но, RSeHgCN (R = СН3) и (RSe)2Hg (R = С2Н5) [205].
Реакция диметилдиселенида с сулемой [205]. Прибавление диметилдиселенида (можно
также его раствора в спирте) к раствору 10 г сулемы в 80 жл воды и 20 мл соляной кислоты
вызывает образование желтоватых игл CHsSeHgCl -HgCh. Т. пл. 123Q С. Вещество, возможно,
содержит немного CHsSeHgCL
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др. 373
Так же получают соли и других алкилселенидов ртути.
Диперфторметилдиселенид не образует двойных солей с сулемой и иод-
ной ртутью; при простом встряхивании с металлической ртутью дает бис-
перфторметилдиселенид ртути (CF3Se)2Hg [206], образующийся также с
количественным выходом при взаимодействии CF3SeCl с Hg.
Получение бяс-трифторметилселенида ртути [206]. 0,618 г (CF3)2Se2 запаивают в квар-
цевой трубке Кариуса с 2 мл ртути и встряхивают 20 час. на расстоянии 4 см от ртутной лам-
пы. Образующиеся летучие продукты представляют собой (CF3)2Se2 и, возможно, (CF3)2Se.
Остаток извлекают эфиром, после испарения которого остаток возгоняют в вакууме при 50°С.
Получают бис-трифторметилселенид ртути 0,385 а в виде желтых игл. После повторной воз-
гонки т. пл. 5° С. Продукт получают с выходом 96% при более энергичном встряхивании и
при непосредственной возгонке, не извлекая его эфиром.
(CF3Se)2Hg с хлором реагирует по уравнению:
(CF3Se)2 Hg + Cl2 -» 2CF3SeCl 4- HgCh,
с вероятным промежуточным образованием CF3SeHgCl. Так же реагирует
бнс-трифторметилселенид ртути с бромом.
(CF3Se)2Hg с НС1 при комнатной температуре реагирует в весьма нез-
начительной степени, при 50—100°С, давая с небольшим выходом CF3SeH,
наряду с другими продуктами [206]. CF3SeHgCl (т. пл. 185—190°С) получен
реакцией (CF3Se)2Hg с HgCl2 в эфире [206].
Ar2Hg с ArSeH в бензоле дают (ArSe)2Hg [328]. (a-C10H7Se)2Hg по-
лучен при обработке спиртового раствора селенонафтола раствором аце-
тата ртути [207].-
Диарилдиселениды с сулемой (в водном спирте) образуют лишь двойные
соединения Ar2Se2• 2HgCl2 [208].
СОЕДИНЕНИЯ с Ge —Hg-СВЯЗЬЮ
При взаимодействии диэтилртути с триэтилгерманийгидридом (моляр-
ное соотношение 1 : 2) в отсутствие кислорода воздуха при 100—120°C об-
разуется этан и бис-(триэтилгермил)ртуть (выход 66,5%) [320]:
2 (C2H5)3GeH + (С2Н5)2 Hg 2CaHe + [(С2Н5)3 Ge]2 Hg.
5нс-(триэтилгермил)ртуты представляет собой термически устойчивое,
но весьма реакционноспособное соединение; так, она легко окисляется
кислородом воздуха с образованием окиси триэтилгермания и выделением
ртути:
2 [(С2Н5)3 Ge]2 Hg + О2 2 [(С2НБ)3 Ge]2 О + 2Hg.
При облучении ультрафиолетовым светом бнс-(триэтилгермил)ртуть коли-
чественно распадается на ртуть и гексаэтилди германий, так же идет эта
реакция и в присутствии бензола. При фотолизе бис-(триэтилгермил)ртути
в четыреххлористом углероде образуется металлическая ртуть, триэтил-
хлоргерманий и гексахлорэтан, в бромбензоле — неожиданно, триэтилбром-
германий, дифенилртуть и металлическая ртуть. Реакция с перекисью
бензоила (в бензоле, 20°С, 3—5 мин.) идет по схеме:
[(С2НВ)3 Ge]« Hg + (CeH6COO)2 Hg + 2 (C2H5)3JGeOCOCeH5.
Так же идет реакция с перекисью циклогексила [322]. Реакция с RBr
идет по уравнению [321]
[(СаН5)з Geh Hg + 2RBr 2 (СзН^з GeBr + R2Hg.
Получение бяе-(триэтилгермил)ртути [320]. Реакцию ведут в атмосфере азота. 6,83 г
триэтилгерманийгидрида и 5,5 г диэтилртути нагревают до 100—120° С в сосуде, снабжен-
ном обратным холодильником, соединенным с газовой бюреткой. Собирают 920 мл (96,8%)
374
Ртутноорганические соединения. Лит. стр. 383—389
этана, идентифицированного хроматографическим методом. Смесь фракционируют в вакуу-
ме в токе азота. Приемником служат ампулы, которые отпаивают по мере отбора фракций,
не прекращая перегонки. Получают 7,34 г (66,5%) бис-(триэтилгермил)ртути. Т. кнп. 118—
120° С/1,5 мм, п® 1,5696. Жидкость лимонно-желтого цвета, отлагающая ртуть при контак-
те с воздухом.
СОЕДИНЕНИЯ с Si —Hg- и Si — Hg — Ge-СВЯЗЯМИ
Взаимодействие диэтилртути с триэтилсиланом протекает более медлен-
но (при 130—140° С) и более сложно, чем реакция ее с триэтилгерманом.
Продуктами реакции являются C2H5HgSi(C2H5)3, [(C2H5)3Si]2 Hg и ме-
таллическая ртуть [321].
бпс-Триэтилсилилртуть образуется также при реакции диспропорциони-
рования [322]:
170°С
2C2H5HgSi(C2H6)3 —(С2Н5)2 Hg + [(С2Н5)з Si]2 Hg.
[(C2H5)3Si]2Hg под действием света разлагается с образованием металли-
ческой ртути и (C2H5)6Si2 [321]. Легко проходит окисление [321]:
[(№>3 Si]2 Hg + Ог [(СаНв)з Si]a О + Hg.
Реакция пентаэтилдисилана и диэтилртути (155—160°С, 19час.) проходит
с образованием, наряду с другими продуктами, соединения со связью [332]:
—SiSiHgSiSi—
/ I I \
(CaHslsSiSi (CaHsh Н (C2Hg)2 Hg —> СгН® -ф- C2H5HgSi (С2Нб)2 Si (С2Нз)3 Ц-
+ [(С3Н5)3 SiSi (С2Н5)а]2 Hg + Hg..
Элементоорганические соединения с Si—Hg—Ge- и Si—Si—Hg—Ge-свя-
зями синтезированы по схеме
(C2H5)3 GeH + C2H5HgR C2He + (С2Н5)3 GeHgR,
где R = (C2HJ3Si и (C2H5)3SiSi(C2H5)2.
Реакции эти идут в мягких условиях—в течение 1—2 час. при 100° С. Пер-
вое из этих соединений получено с выходом 26%, это — жидкость лимонно-
желтого цвета, т. кип. 130—131°Спри1,5 мм, второе получено с выходом
50,4%, т. кип. 159—163°Спри 1 мм. Оба вещества очень чувствительны к
кислороду. Под действием ультрафиолетового света происходит реакция
(С2Н6)3 GeHgSi (С2Н3)з Hg + (С2Н6)3 GeSi (С2Н5)3.
При взаимодействии (C6H5)3SiLi с HgCl2 получен (C6H5)3SiHgCl [323].
СОЕДИНЕНИЯ с N—Hg-СВЯЗЬЮ
N — Hg-Производные аминов
Первичные алифатические амины в мягких условиях (в водном раство-
ре, на холоду или при умеренном нагревании) образуют с солями ртути в
зависимости от относительных количеств реагентов и условий реакций
соединения типа RNHHgX, RN(HgX)2 или [RN(HgX)]2Hg.
Так, при постепенном приливании к концентрированному раствору
метиламина слабо подкисленного раствора сулемы или азотнокислой рту-
ти средней концентрации образуется мелкокристаллический порошок
CH3NH(HgX) (X = Cl или NO3), не растворимый вводе, спирте и эфире [209].
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др. 375
Получение азотнокислой метиламинортути [209]. При добавлении водного раствора
азотнокислой ртути при перемешивании к концентрированному водному раствору небольшо-
го избытка метиламина образуется объемистый белый осадок, который после умеренного на-
гревания на водяной бане превращается в белый порошок. Вещество нерастворимо в обыч-
ных органических растворителях, при нагревании разлагается, не плавясь. Так же получе-
ны нитраты этил- и амиламинортути. .
При приливании раствора метиламина (при перемешивании) к концент-'
рированному раствору сулемы образуется продукт CH3N(HgCl)2 [210].
Взаимодействие сулемы с избытком водного раствора жирных аминов
при нагревании приводит к соединениям, согласно Стромхольму [211],
имеющим состав [RN(HgCl)2l2Hg (R = СН3, С2Н5, л-С3Н7) и строение
R R
\j_Hg—rZ
CIHg7 \gCl
В то время как N-бензилацетамид меркурируется' в бензольное ядро
(см. гл. V), бензиламин, согласно Песни [212], при действии солей ртути обра-
зует только N-ртутные производные. N—Hg-Производное бензиламина
C6H5CH2NHHgCl получено обработкой 4 молей бензиламина в кипящем
водном растворе 1 молем сулемы и кипячением осадка со спиртом [212, 213].
•Соответствующая сернокислая соль получается высаживанием раствора
двойного соединения бензиламина и ацетата ртути сульфатом натрия.
Из сульфата действием гидроокиси бария получено основание состава
CeH5CH2NHHgOH [213].
бцс-(Дитрйфторметиламин)ртуть [(CF3)2N]2Hg получается [214] присое-
динением фторной ртути по двойной связи перфтор- 2-азапропена (авто-
клав, 100° С):
2CF3N = CF2 + HgF2 -+ (CF3)2N - Hg - N (CF3)2
или взаимодействием бромдитрифторметиламина с металлической ртутью
при комнатной температуре (15 час., запаянная трубка):
(CF3)2 NBr + Hg -+ [(CF3)a N]2 Hg.
. Синтез <>ае-(дитрифторметиламин)ртути [214]. 52 г (0,4 моля) перфтор-2-азапропена и
50 а фторной ртути нагребают в автоклаве из нержавеющей стали в течение 15 час. при 100° С.
При открывании автоклава в вакууме получают обратно 5 г перфтор-2-азапропена. При пе-
регонке остатка получают 70 г бис-(дитрифторметиламин)ртути: т. кип. 127° С, т. пл. 17,5Р С.
Выход 79%, Выход, вероятно, выше; несмотря на проведение операций с ртутным соедине-
нием в тщательно высушенной атмосфере, происходят значительные потери при обработке.
бис-(Дитрифторметиламин)ртуть очень чувствительна к малейшим сле-
дам влаги, тотчас разлагаясь с образованием твердого желтого вещества.
При нагревании до 150° С диссоциирует, по-видимому, на HgF2 и исходный
перфторазапсщен, вновь регенерируясь при охлаждении. При действии
брома на быо(дитрифторметиламин)ртути в тщательно высушенном четы1
реххлористом углероде при комнатной температуре атом ртути заменяется
на бром и образуется с 90 %-ным выходом бромди(трифторметил)амин [2141.
[(CF3)2N]2Hg с NOC1 дает (CF3)2NNO, с NO2C1 — (CF3)2NNO2 [214а].
Действием на [(CH3)2N]2Hg ацилхлоридов (запаянная трубка, комнат-
ная температура) ртуть заменяется на ацил [2146]:
[(CF3)2 N]2 Hg + RCOC1 RCON(CF3)2.
Ртутное производное гликоколя представляет собой, возможно,
Hg—N-соединение [16].
376 Ртутноорганические соединения. Лит. стр. 383—389
Третичные энамины с сулемой или бромной ртутью в эфирном растворе
под азотом реагируют по схеме:
R2N - СН = CR2 + 2HgX2 [R2NCH = СН2]+ (HgX3)-.
HgX
Образующиеся аморфные осадки в случае, если X = С1,_ устойчивей, чем
если X = Вг [215а].
При меркурировании ароматических аминов действием обычных мерку-
рирующих средств в качестве первой стадии реакции образуются лабильные
промежуточные вещества, в которых ртуть соединена с органическим
остатком через азот. Благоприятствует образованию таких соединений
недостаток кислоты в реагирующей среде.
N-Меркуранилин C6H5NHHgNHC6H5 получен из анилина, сулемы и
щелочи [212, 215, 216—219].
Нерастворимый в обычных растворителях продукт действия сулемы на
анилин в спиртовом растворе C6H5NHgCl [220, 221], по мнению Песчи
[212] и Димрота [222], представляет собой полимер. Его очищают экстрак-
цией из кипящего спирта [2231. Двойное соединение его с сулемой имеет
состав'5CeH6NHHgCl +2HgCla [213].
Из хлоргидрата анилина и меркурацетамида в воде получен [224, 225}
HgCl
продукт строения CeHsN ,
^HgOH
Описаны N—Hg-соединения нафтиламинов и замещенных нафТилами-
нов [12, 229—232].
Действием и на другие ароматические амины (анилин, метиланилин)
сулемы (также в присутствии соды) образуется ArNHHgCl [13]. Диметил-
анилин дает при этом,продукт Песчи, содержащий N—Hg-связь и одно-
временно меркурированный в ядро (см. гл. V).
Получение аиилинмеркурхлорида [13]. При действии на спиртовый раствор 3 г анилина
раствором 3 г NaHCO3 и горячим водным раствором 9 г сулемы образуется желтый осадок
аиилинмеркурхлорида.
Ртутная соль тринитрометана, меркурируя ароматические углеводороды
и ряд их производных, с анилином, (в спирте, при 20° С) образует N—Hg-
соединение CeH5NHHgC(NO2)3 [226].
При действии азотнокислой ртути на нитроанилии в присутствии щело-
чи Джексон и Пикс [227] получили красные или коричневые осадки,
которым эти авторы приписали строение N-производных нитроаиилинов.
Хараш [228] приписывает ярко окрашенным соединениям, образующимся
при реакции ацетата ртути с о- и n-нитроанилином, строение хинонимино-
ацйнитрбсоединений.
При взаимодействии CF3CONHNHCOCF3 с ацетатом ртути в подкислен-
ном уксусной кислотой водном растворе получена [2146] [CF3CON]2Hg.
Вещество ни в чем нерастворимо и представляет собой, вероятно, полимер.
Взаимодействием (CF3CON)2Hg с CF3COC1 в фторуглеводороде при 50° С
получен (CF3CO)4N2 [2146]. '
Получение тетракис-(трифторацетил)гидразииа [2146]. 38 г (0,29 моля) трифтораце-
тилхлорида и 50 г фторуглеводородного растворителя, кипящего около 50° С, конденсиру-
ют во вращающийся автоклав, содержащий 57 г (0,14 моля) тщательно высушенной
(CFsCON)2Hg. Реакцию ведут 14 час. при 50° С. Летучие продукты удаляют отсасыванием
и остаток фракционируют. Т. кип. 108° С/760 леи. Вес 53 г.
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др. 377
N-Hg-Производные амидов, имидов и гидразидов кислот
Ртуть замещает водород, соединенный с азотом . амидов, легче, чем
с азотом аминов, и еще легче — водород, соединенный с азотом имидов,
кислот.
При растворении окиси ртути в расплавах амидов или в растворах ами-
дов кислот- образуются меркурациламиды (RCONH)2Hg, содержащие
N—Hg-связь, не расщепляющуюся щелочами, разрывающуюся действием
кислот, сульфидов, иона иода. Меркурформамид существует только в раст-
воре [233]. При взаимодействии окиси ртути и формамида в спирте обра-
зуется аморфный осадок HCONHHgOH, с НС1 дающий соединения
HCONHHgCl-HCl, 2HCONHHgCl-3HCl. Меркурацетамид (CH3CONH)2Hg
получен [16, 31, 234—238] сплавлением окиси ртути и ацетамида при 180° С
(рецептуру см. в гл. V, стр. 61). Так же получают другие меркурациламиды.
Описанб получение (ClCH2CONH)Hg [239, 2401, (NO2CH2CONH)2Hg [241],
(CH3CH2CONH)2Hg [242, 243], (C3H7CONH)2Hg [244, 245], меркуртартра-
мида [246], меркуроксамида [244, 247], меркурсукцинамида [248], меркур-
сукцинимида [244, 245, 248, 249], меркурфумарамида [241,245] и др. Описа-
но строение меркурацетамидов [16].
При действии уксуснокислой ртути на сукцинимид в водном растворе
щелочи при охлаждении льдом меркурсукцинимид получается недостаточ-
но чистым [250].
Реакцией ацетата ртути с гексимидом (I) получен N-меркургексимид (Г).
[258]:
СН3 О СНз О .
2 СН2 -/ <>NH Hg(°±C--b^ ( <^Х“--СН2 Hg’
/ \ОН \ \ / \ ОН L
СНз О О 4 СНз о О
1 I'
Ртутные производные амидов .перфторкарбоновых кислот (RCONH)2Hg,
где R = CF3, C2F5, C3F7, получены [251 ] с количественным выходом наг-
реванием амидов этих кислоте избытком красной окиси ртути (150—170° С,
5 мин.) с последующим извлечением продукта реакции кипящим спиртом.
Т. пл. соответственно 219, 244, 286° С.
О некоторых N—Hg-производных барбитуровой кислоты и т. п. сое-
динений см. ниже (стр. 380).
Многочисленные соединения мочевины с окисью и разнообразными соля-
ми Hg (также в виде двойных солей с мочевиной и иногда НХ) CO(NHHgX)2,
где X = ОН [212, 245, 252], С1 [253, 254] (т. пл. 125—128°С) [253],
NO3[252, 254], SO4[254], СН3СО2 [254], получены взаимодействием соли
ртути и мочевины в воде (иногда при слабом нагревании) или в метиловом
или этиловом спиртах. Глассманом с сотр. [255] описано строение нитратов,
меркурмочевины. Изучено титрование мочевины раствором солей ртути
по методу Пфейфера [256, 257].
О ртутных соединениях циклических производных мочевины см. в нас-
тоящей главе на стр. 380.
При действии меркурацетамида в водном или спиртовом растворе на
ацетилуретан AcNHCO2Et и его замещенные амиды AcNHCONHR обра-
зуются соединения общей формулы Hg(NAcCONHR)2. Действие на них
гидразина или фенилгидразина приводит к выделению металлической
ртути [259].
Согласно данным Вильямса и др. [260], ртутные производные алифати-
ческих амидов лучше получаются сплавлением амидов с окисью ртути
[245] и вымыванием из реакционной смеси холодным спиртом, в котором
378 Ртутноорганические соединения. Лит. стр. 383—389
они хорошо растворимы. Ртутные же производные амидов ароматических
кислот: бензамида, n-хлорбензамида, бромбензамидов, толуамидов, о-анис-
амида, салициламида, растворимые в горячем и нерастворимые в холодном
спирте, удобнее получать следующим образом.
Получение ртутных производных амидов ароматических кислот [260]. 5 г желтой окисн
-ртути, 4 г амида прибавляют к 50 мл 95% -ного спирта, смесь кипятят в течение 4 часа, затем
фильтруют горячим через стеклянный пористый фильтр, .охлаждают посредством ледяной
бани;-кристаллы отсасывают и промывают холодным спиртом или эфиром. Т. пл. ртутного
производного бензамида 222° С, п-толуамида 260° С, n-хлорбензамида 258°С, л-бромбензами-
да 266° С, салициламида 290° С (280° С).
Меркурбензамид (C7H6ON)2Hg получен [245, 261, 262] также растворе-
нием окиси ртути.в водном растворе бензамида. Это соединение очень устой-
чиво и может быть перекристаллизовано также из теплого водного рас-
твора едкого кали.
Меркуркуминамид [263] [(CH3)2CHC6H5CONH]2Hg-]-1/2H2O кристалли-
зуется из разбавленного спирта. Т. пл. 190—191 °C. Он нерастворим в
воде, растворим в спирте. Меркурфталимид — порошкообразный осадок
<[264].
Меркурациламиды (RCONH)2Hg восстанавливаются алкилфосфитами с
выделением металлической ртути и,образованием амида и нитрила кислоты
RCN и эфира фосфорной кислоты [265].
Меркурациламиды, в частности меркурацетамид, разрушаются дейст-
вием аммониевых солей, солей закиси ртути с выделением неорганических
соединений ртути [224], действием гидразина, гидразобензола — с выде-
лением металлической ртути [266].
N-Ртутное производное бензоилцианамида получено [267] действием
ацетата ртути на водный раствор его щелочной соли.
Ртутные производные имидов кислот более устойчивы, например, к дей-
ствию соляной кислоты, чем таковые амидов кислот [244].
О получении ртутных производных амидов кислот см. также [268, 269]
(а также гл. V, стр. 53).
Ацильные производные ариламинов также дают с солями или окисью
ртути N—Hg-соединения.
Форманилид с бромной ртутью и этилатом натрия дает N-меркур-бис-
формамид (C6H5NCHO)2Hg. Из форманилида, сулемы и этилата натрия
образуется N-хлор'меркурформанилид C6H5N(HgCl)CHO [270].
Ацетанилид при сплавлении с окисью ртути дает соединение состава
Hg[N(COCH3)C6H5]2 [236, 271]. Это же соединение получается, если к
водному раствору 2 молей- ацетанилида и 1 моля сулемы прибавить соду
до сильно щелочной реакции [272].
Аналогично получаются N-ртутные производные и других ариламидов
кислот, как, например, N-ацилнафтиламинов [229—232] и др.
Фенилмочевина взаимодействует с сулемой в кипящей воде [продукт —
<C6H5NHCONHHgCl)3(HgCl2)2 не дает реакции на ион ртути] или в ацетоне
при нагревании на водяной бане (продукты: Hg[<3C(NH2)NHC6H5]„Cl2, где
п = 1 или 5 [273]).
C6H5(C2H5)NCONH2, (C6H5)2NCONHC2H5, C6H5(C2H5)NCONHC6H5 с
водным раствором сулемы на холоду образуют комплексные соединения
Hg [OC(NHR)NRR']C12 [273]. Ряд замещенных мочевин не взаимодействует
с водным раствором HgCl2 [273].
Дифенил карбазид с нитратами закисной и окисной ртути (в присутствии
буферного раствора — буры и борной кислоты) в водном спирте образует
•соединения с N— Hg-связью; в случае окисной соли ртути происходит окис-
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др.
379
ление дифенилкарбазида до дифенилкарбазона, и продукт реакции пред-
ставляет собой N—Hg-соединение дифенилкарбазона [274].
Как N—Hg-производные ациламидов, так и N—Hg-производные анили-
дов кислот при действии йодистого калия, гипосульфита, а также бромисто-
го аммония распадаются с образованием анилида и соли ртути [229].
При обработке сульфаниламидов уксуснокислой ртутью в 98 %-ной
уксусной кислоте образуются соединения с N—Hg-связью, в горячей
воде — соединения как с N—Hg-, так и с С—Hg-связью, в горячей же
40 %-ной уксусной кислоте образуются соединения только с С — Hg-
связью [275].
Триамид 1,3,5-бензолтрисульфокислоты СбНз(5О2МН2)3 дает при ки-
пячении 1 ч. его с окисью ртути (полученной из 1,3 ч. сулемы) осадок
состава Н£з('С6Н6О6Нз5з)2 [276]. При кипячении 1 ч. раствора амида с
окисью ртути, полученной из 2,6 ч. сулемы, получается осадок
(HOHg)3C6H6O6N3S3 [276].
N—Hg-Производное диацетилгидразида и одновалентной ртути
Hg2N2(COOH3)2 получается сливанием подкисленного водного раствора
азотнокислой закиси ртути и М,ЬГ-диацетилгидразида в виде оранжево-
красного порошка [277]. Его строение подтверждено рентгенографически
и при помощи ИК-спектров. При сливании водных растворов сулемы
(1 моль) и диацетилгидразида (1 моль) N—Hg-производное двухвалент-
ной ртути состава Hg(NHCOCH3)2 получается в более чистом виде,
если эту реакцию, в отличие от рекомендации Штолле [278], проводят в
отсутствие этилата натрия [269]. При нагревании меркурацетамида до
240° С происходит (в результате частичного распада его на металличе-
скую ртуть и диацетилгидразид) меркурирование последнего как по
азоту, так и по углероду и образование продукта Hg2NC2H2O2 [269].
N — Hg-Производные диазоаминосоединений
N—Hg-Производные диазоаминосоединений (ArN=N—N—Ar)2Hg удоб-
нее всего могут быть получены сливанием метиловоспиртовых растворов
ацетата ртути и метиловоспиртового или бензольного раствора диазоами-
носоединения [279—281]. Они могут быть получены также действием окиси
ртути на раствор диазоаминобензола в хлороформе [282]. Ртутное производ-
ное диазоаминобензола образуется также при взаимодействии ацетата рту-
ти и фенилгидразина в водном растворе на холоду [283] (ср. синтез дифе-
нилртути по Фишеру при взаимодействии ацетата ртути и фенилгидразина
в эфирном растворе, гл. VII, стр. 191).
Ртутные (а также и других металлов) производные диазоаминосоедине-
ний существуют в одной форме [284]. Образование же двух более и менее
ярко окрашенных форм этих производных обязано загрязнениям. Таким
образом, основанное на существовании этих двух форм подтверждение фу-
мароидно-маленоидной теории цвета диазосоединений неверно [284].
Получение N-ртутной соли диазоаминобензола [279]. К раствору 0,78 г уксуснокислой
ртути в 20 мл метилового спирта, подкисленному 4—5 каплями разбавленной уксусной кис-
лоты, приливают раствор 0,90 г диазоаминобензола в 10 мл метилового спирта. Тотчас
выпадает хлопьевидный желтый осадок. Выход количественный. Продукт кристалли-
зуют из бензола или лучше из толуола. Образуются желтовато-золотистые иглы.
Т. пл. 232° С.
380
Ртутноорганические соединения. Лит. стр. 383—389
N — Hg-Производные циановой и изоциановой кислот
Ртутное производное циановой кислоты, получаемое действием сулемы
в “метиловом спирте на цианат серебра, представляет собой, согласно дан-
ным раман-спектроскопии [18, 19], Hg—N-производное (OCN)2Hg. Луч-
ше реакции вести в эфире [126, 256]. Методика получения дана в гл. V.
Цианат ртути получен также нейтрализацией окиси ртути циановой кис-
лотой [286] в эфире, а также нагреванием в воде цианата закисной ртути
с цианатом натрия в водном, подкисленном азотной кислотой растворе-
при охлаждении льдом.
Строение цианатов ртути как Hg—О- или Hg—N-соединений еще не-
достаточно выяснено.
N — Hg-Производные некоторых гетероциклов
Обычно при действии солей ртути на азотсодержащие соединения недо-
статок кислоты в реакционной среде благоприятствует образованию
N—Hg-связи, но не С—Hg-связи ([285] и гл., V и VI).
С другой стороны, во многих случаях, в то время как при действии на
азотсодержащие органические соединения ацетата ртути образуются
С—Hg-производные, при взаимодействии их с сулемой образуются соедине-
ния с N—Hg-связью.
Так, при действии на пиррол и его производные уксуснокислой или
циановокислой ртути происходит меркурирование и образуются С—Hg-
производные (см. гл. V, стр. 97).
Действие сулемы на пиррол [286], 2,3-диметил- и 2,4-диметил-З-карб-
оксипиррол [2861, 2-метил-5-изопропилпиррол [287] приводит к образо-
ванию лишь соединений с N—Hg-связью и их двойных солей с сулемой
[288].
Ртутное производное изатина Hg(C8H4O2N)2 (могущее быть рассматри-
ваемо и какН—Hg- и как О—Hg-производное), получаемое с наилучшим вы-
ходом (95%) действием конц. раствора ацетата ртути на кипящий спирто-
вый раствор изатина, представляет собой темно-красные кристаллы [289],
растворимые в разбавленном растворе едкого кали с желтым окрашива-
нием; при подкислении раствора разбавленной серной кислотой дает сое-
динение Hg(NHCeH4COCOOH)2 + 2Н2О.
При действии на антипирин окиси ртути или каломели в присутствии
щелочи, достаточном, чтобы связать весь С1', образуются [64] соедине-
ния типа (R2N)2Hg, при недостатке щелочи—типа RNHgCl [290]. Из раст-
вора имидазола и перхлората ртути при pH >4,5 высаживается соеди-
нение C3H3N2HgC104-H20, возможно полимерной природы [325].
Барбитуровая [267], диэТил- и фенилэтилбарбитуровая [2911 и вио-
луровая [267] кислоты образуют N—Hg-соединения действием ацетата рту-
ти на водные растворы их щелочных солей. Так же получены ртутные про-
изводные метнлурацила [267] и мочевой кислоты [267].
N—Hg-соединения пиримидина и его производных, в том числе пури-
нов, как типа (RN)2Hg, так и THnaRN—HgHal, получают взаимодействием
на холоду или при нагревании галогенида (хлорида или бромида) ртути &
спиртовом растворе со слабощелочным раствором соответствующего азоти-
стого гетероцикла. Конденсация полученных ртутных производных с аци-
лированными галоидпентозами или -гексозами является важным методом
синтеза нуклеозидов (так называемый ртутный метод синтеза нуклеозидов}
[285, 292—311, 313]. Исходным азотистым основанием может являться так-
же кислородсодержащее производное пиридина, в котором ртуть присое-
Синтез и реакции соединений ей связями О—Hg, S—Hg, Se—Hg и др.
381
динена через кислород. Так, из 0-хлормеркурпроизводного а-оксипиридина
получен как О-, так и N-глюкозид [312].
Ртутные соли бензоксазолтионов с галоидацилальдозами по S^l-меха-
низму образуют N-глюкозиды [316] (в диметилформамиде, комнатная тем-
пература).
Ртутные производные пуриновых оснований описаны ранее (см., напри-
мер, хлормеркурпроизводные аденина [314, 315]).
Синтез пентацетилглюкопираноз ил гуанина [306].
1. Получениехлормеркурпроизводного ацетилгуаннна.
К раствору 1 г ацетилгуаннна в 250 мл горячего водного метилового спирта добавляют
€,016 г едкого натра в 1 мл воды и 1,5 г сулемы, растворенной в небольшом количестве горя-
чего метилового спирта. Аморфный осадок отфильтровывают и высушивают в вакуум-эк-
•сикаторе. Выход 1,7 г (76,7%).
2. Получение пентацетилглюкопиранозилгуанина. Тща-
тельно размельченное хлормеркурпроизводное ацетилгуаннна (1 г) суспендируют в абсо-
лютном ксилоле. Часть ксилола отгоняют до тех пор, пока дистиллят не становится проз-
рачным. К суспензии добавляют 1,5 г ацетобромглюкозы, растворенной в ксилоле. Смесь
нагревают при энергичном перемешивании 1 час, горячий раствор декантируют, осадок про-
мывают хлороформом. Хлороформный и ксилольный растворы соединяют, промывают
30%-ным раствором йодистого калия, высушивают над сернистым натрием и упаривают в
вакууме досуха. Оставшееся масло растворяют в минимальном объеме абсолютного этилово-
го спирта. К раствору добавляют абсолютный эфир. Осадок растирают 2 раза с абсолют-
ным эфиром и перекристаллизовывают из воды. Выход 0,5 г (42%). Т. пл. 295° С (с разложе-
нием).
Пентаацетилглюкопиранозилгуанин при обработке метиловоспиртовым
раствором едкого натра дает 9-[3-£/-глюкопиранозилгуанин.
Ртутные производные пиримидинов при действии соляной кислоты
распадаются с образованием исходного пиримидина.
Изоцианурат ртути (трикарбамид ртути)
[(CO)3N3]2Hg3 -|-ДН2О
получается из тринатриевой соли изоциануровой кислоты и соли ртути
при 100° С и при любых температурах из растворов циануровой кислоты и
•соли ртути [62].
При кипячении 1,6,8-триазабицикло-(4,3,0)-3-нонен-7,9-диона с уксус-
нокислой ртутью в метаноле образуется продукт замены на ртуть водорода
при азоте в положении 8:
Z\n — С(Х т„ Hg<ococH3), Z\n — СО .
Vn-CO/NH сн.он^ VN_CO/NHgOCOCH8,
382
Ртутноорганические соединения. Лит. стр. 383—389
тотчас превращающийся в продукт присоединения элементов HgOaCCH^.
и ОСН3 по С=С-связи при добавлении к реагирующей смеси азотной кис-
лоты [317].
СОЕДИНЕНИЯ с Р—Hg-СВЯЗЬЮ
Как показали Фокс и Венецкий [318], диалкилфосфонаты меркуриру-
ются уксуснокислой ртутью или смесью окиси ртути и соли ртути (ацетата,,
галогенида) с образованием соединений с Hg—Р-связью — диалкоксифос-
финилртутных солей:
(RO)2P(O)H + Hg(O2CCH3)2 (RO)a Р (О) HgO2CCH3 + СН3СООН, (1>
(RO)2P(O)H + HgO + HgX2 2 (RO)a Р (О) HgX + Н2О. (2)
Реакция (1) идет в мягких условиях: при комнатной температуре, в рас-
творителе — толуоле (для R — этил) или этилбензоле, н-октане (для
R — другие Aik). Реакцию (2) ведут в кипящем бензоле. Образующуюся по
реакции (1) уксусную кислоту и по реакции (2) воду удаляют азеотропной
отгонкой. Выходы 78—92%. Реакция с уксуснокислой ртутью может быть
проведена также без растворителя. Она идет с значительным выделением
тепла и дает несколько менее чистый продукт, чем при проведении реак-
ции в растворе.
При взаимодействии 2 молей диалкилфосфоната и 1 моля окиси ртути в.
бензоле или эфире получают бяс-(диалкоксифосфинил)ртуть [319]:
2 (RO)2 Р (О) Н + HgO [(RO)2 Р (О)]2 Hg + Н2О.
Взаимодействие бяс-диалкоксифосфинилртути с сулемой в кипящем:
бензоле дает алкоксифосфинилмеркурхлорид:
[(RO)a Р (О)]2 Hg + HgCl2 2 (RO)a Р (О) HgCl.
Получение диэтилфосфинилмеркурацетата [318]. К энергично перемешиваемой взвеси
350,5 г уксуснокислой ртути (1,1 моля) в 500 мл сухого толуола при комнатной температуре-
прибавляют 138 г (1,0 моль) диэтилфосфоната. Смесь перемешивают 2,5 часа, поддерживая
температуру при 25° С при помощи бани со льдом. Отфильтровывают от нерастворимого ве-
щества (3 а). Образующуюся уксусную кислоту удаляют в виде азеотропной смеси с толуо-
лом перегонкой при. 60 мм до тех пор, пока температура не поднимется до 36° С и объем
дистиллята не достигнет 175 мл. (Перегонка при более высокой температуре ведет к
образованию значительных количеств металлической ртути.) После фильтрования испа-
рение фильтрата дает 330 г (83,3% выхода) сырого продукта. Перекристаллизация из
толуола или смеси бензола с эфиром дает чистый диэтоксифосфинилмеркурацетат. Т. пл..
106,8—107,6° С.
Другие диалкоксифосфинилмеркурацетаты получены таким же путем
(алкил—н- и изопропил, н- и изобутил, я-амил, я-гексил, я-гептил). Реакцию
ведут в этилбензоле или лучше в я-октане. При этом необходимо полностью*
отгонять уксусную кислоту, так как сырые ацетаты в ней очень хорошо-
раствор имы.
Хлориды и бромиды диалкоксифосфинилртути могут быть получены
как из ацетатов действием галогенида калия или натрия в водном растворе,,
так и непосредственно из диалкилфосфонатов, как выше сказано, меркури-
рованием их смесью сулемы (или бромной ртути) и окиси ртути в бензоле
при кипячении. Йодиды диалкоксифосфинилртути получены только пос-
ледним способом.
Диалкоксифосфинилмеркурхлориды получены также реакцией бис-
(диалкоксифосфинил)ртути с сулемой в бензоле.
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др. .383’
Получение ди-н-пропоксифосфииилмеркурбромида [318]. Смесь 8,3 г (0,05 моля) ди-
н-пропилфосфоната, 5,4 г (0,025 моля) окисн ртути и 9,0 г (0,025 моля) бромной ртути в
50 мл сухого бензола кипятят 3 часа, (при непрерывной отгонке воды с одновременным ее
улавливанием).’Вскоре после начала "кипения оранжевый цвет окиси ртути переходит в бе-
лый. После охлаждения смесь отфильтровывают от неорганических веществ (2,6 г). Испаре-
ние фильтрата дает сироп, который быстро закристаллизовывается при прибавлении петро-
лейного эфира. Выход 14,8 г (66,4%). Т. пл.-67—68,5° С. После двукратной кристаллиза-
ции из горячего гексана получен чистый ди-н-пропоксйфосфинилбромид с т. пл. 70,0—
70,4° С.
• Получение ди-н-пропоксифосфинилмеркуриодида («-C3H7O)2P(O)HgJ [318]. Смесь
8,3 г (0,05 моля) ди-н-пропилфосфоната, 5,4 г (0,025 моля) окиси ртути и 11,3 г (0,025 моля)-
иодиой ртути в 50 мл бензола кипятят 5 час. Так как происходит сильное образование пены,
то образующуюся воду не собирают. Красный цвет иодида вскоре исчезает, но образую-
щийся осадок имеет окраску окиси ртути. После отфильтровывания от неорганических солей
фильтрат высушивают драйеритом. Растворитель испаряют, при этом выделяется значи-
тельное количество неидентифицируемого желтого аморфного осадка. Его извлекают кипя-
щим гексаном. Экстракт профильтровывают и концентрируют. Получено 12,1 г (49,1%) сы-
рого продукта с т. пл. 74,6—75,6° С; перекристаллизация из теплого гексана дала чистый
ди-н-пропоксифосфинилмеркуриодид с т. пл. 75,2—76,0° С. Подобным же образом получены
диэтокси- и ди-н-бутоксифосфинилмеркуриодиды. Выход 61,0 и 41,9%- соответственно..
Т. пл. 102,0—102,5° С для первого и 56—57,2 0 С для второго.
Получение бйс-(диметоксифосфинил)ртути [319]. К энергично перемешиваемому
раствору 50 г (0,45 моля) диметилфосфоната в 75 мл сухого бензола при комнатной
температуре прибавляют 49 г (0;23 моля) окиси ртути. После того как закончится присоеди-
нение окиси ртути, прибавляют еще 50 мл бензола. Давление уменьшают до 200 мм и смесь,
кипятят при 40—45° С. Образующуюся при реакции воду удаляют в виде азеотропной
смеси с бензолом и улавливают в ловушке. Хотя некоторое количество непрореагировавшей
окиси ртути остается, реакцию можно считать оконченной после того, как в ловушкё больше-
не собирается вода. Продукт выпадает из бензольного раствора при охлаждении. После
однократной кристаллизации из бензола получают 78,9 г (83%) бис-(диметоксифосфинил)-
ртути. После трехкратной кристаллизации из бензола она плавится при -121,6—423° С. «-Бу-
тиловый, изобутиловый и «-пропиловый эфиры, полученные этим методом, очищены хрома-
тографированием реакционной смеси через колонку с окисью алюминия (заполненную
влажным способом). Полученные сырые продукты подвергнуты дополнительной очистке
двукратным хроматографированием их растворов с н-гексаном. .
Получение бис-(диэтоксифосфинил)ртути [319]. К раствору технического диэтил-
фосфоната 138,1 г (1,0 моль) в 150 мл абсолютного эфира прибавляют небольшими
порциями 108,3 г (0,50 моля) окиси ртути. После того как половина окиси ртути прибавлена,,
раствор нагревают, чтобы он хорошо кипел, при этом оранжевая окраска тотчас исчезает.
Когда реакция затихнет, прибавляют остальную порцию окиси ртути и смесь кипятят 6 час.
Образовавшуюся мутную серую жидкость оставляют отстаиваться на ночь; затем отфильт-
ровывают от нерастворимого осадка (1,6г), представляющего собой смесь ртути и не вошед-
шей в реакцию окиси ртути; из фильтрата в вакууме отгоняют растворитель. В охлаждае-
мой ловушке собирается 6,7 г (0,37 моля) воды. Серые пластинки, образующиеся после уда-
ления низкокипящих продуктов, трижды кристаллизуют из петролейного эфира. Выход
б«с-(диэток<;ифосфинил)ртути 216,1 г (91,2%). Т. пл. 56,8—58,2° С.
Реакция бис-(диалкоксифосфинил)ртути с сулемой [319]. Синтез диэтоксифосфинилмер-
курхлорида (CaH5O)2P(O)HgCl. К раствору 4,7 г (0,01 моля) бис-(диэтоксифосфинил)ртути
в 20 мл сухого бензола прибавляют 2,7 г (0,01 моля) сулемы. Смесь кипятят 15 мин. и ох-
лаждают до комнатной температуры. Отфильтровывают от небольшого осадка (0,5 г) и из.
фильтрата получают 5,9 г (78,6%) сырого диэтоксифосфинилмеркурхлорида. После кри-
сталлизации из четыреххлористого углерода и затем из воды т. пл. 103—104° С.
ЛИТЕРАТУРА
l. HockH., StuhlmannH. Вег., 62, 2690 (1929).
2. Нем. пат. 293692 (1916>; Zbl., 1916, II, 439.
3. Manchot W. Ann., 421, 331 (1920).
4. Rupp E., Hermann-A. Arch. Pharm., 254, 497 (1916).
5. H a at z sch A., Auld S. M. Ber., 39, 1105 (1906).
6. Torrey H. A., H u n t e r W. H. J. Am Chem. Soc., 33, 203 (1911).
7. J a m b о n L. Bull. Soc. Chim. France [3], 23, 825 (1900)
8. C h e v r e u 1 M. E. Ann. chim. phys. (1), 32, 113 (1809).
9. Silberrad О., P h i 1 1 i p s H. A. J. Chem. Soc., 93, 474 (1908).
10. L i e b i g J. Ann. Chim. phys., 35, 72 (1827); 37, 286 (1828).
11. Англ. пат. 24981 (1918); С. A., 6, 1547 (1918).
12. К г у n s k i J. Roczn. Ch., 8-, 71 (1928); Zbl., 1928, II, 2143.
384
Ртутноорганические соединения
13. N е о g i Р., Chatterjee М. Р. J. Indian Chem. Soc., 5, 221 (1925).
14. О h и о T. С. А., 51, 270 (1957); J. Pharm. Soc. Japan, 76, 726 (1956).
15. Z i и с к е Т., Bindewaid Н. Вег., 17, 3026 (1884).
16. Ley Н., Kis.se ГН. Вег., 32, 1357 (1899). •
17. U m g г о v е Н., Franchimont А. Р. N. Rec. trav. chim., 16, 385 (1897).
18. BirckenbachL., Ko lb H. Ber., 66, 1571 (1933).
19. Birckenbach L., Kolb H. Ber., 68, 855 (1935).
20. S 6 d e r b а с к E. Acta chem. scand., 11, 1622 (1957).
21. L i e b i g J. Ann. Chini. phys. [11], 24, 294 (1823).
22. Howard, Philos. Transact, of the Roy. Soc., 1800, 204.
23. Woh ler L. Ber., 38, 1345 (1905).
24. Beckmann E. Ber., 19, 993 (1886).
25. L о b г у de В r u y'n C. A. Ber., 19, 1370 (1886).
26. A n g e 1 i с о F. Atti Accad. Lincei [5], 10, 476 (1901).
27. J о n e s L. W. Am. Chem. J., 20, 1 (1898).
28. Nef I. U. Ann., 280, 263 (1894).
29. WohlerL. Ber., 38, 1351 (1905).
30. W i e 1 a n d H. Ber., 43, 3362 (1910).
31. Si dgwick N. W. The chemical elements and their compounds. Oxford, Clarendon
Press 1951
32. К a s t H.* S e 1 1 e H. Ber., 59, 1958 (1926).
33. Schischko.ff L. Ann., 97, 54 (1856).
34. Scholl R. Ber., 27, 2816 (1894).
35. E h r e n b e r g A. J. prakt. Chem. [2], 32, 232 (1885).
36. Keku le A. Ann., 101, 206(1857).
37. W i e 1 a n d H. Ann., 358, 56’ (1908); Ber., 42, 2192 (1909).
38. Steiner A. Ber., 8, 512 (1875)..
39. Langhans Z. Schiesz-Sprengst., 15, 219 (1920).
40. Divers E., KawakitaM. J. Chem. Soc., 45, 13 (1884).
41. W о h 1 e r L., Martin F. Z. angew. Chem., 27, 335 (1914).
42. Wohler L„ Martin F. Ber., 50, 586 (1917).
43. W 6 h 1 e r L., W e b e r A. L. Ber., 62, 3742 (1929).
44. Carstanjen H., Ehrenberg A. J. prakt. Chem., 25, 232 (1882).
•45. C a m b i.L. Gazz., 41, I, 168 (1911).
46. S t e i n e r A. Ber., 9, 779 (1876); Ber., 16, 1484, 2419 (1893).
.47. Scholl R. Ber., 23, 3509 (1890).
48. H о 1 1 e m a n A. F. Rec. trav. Chim., 10, 70 (1891).
49. S c h о 1 v i e n L. J. prakt. Chem. [2], 32, 486 (1885).
50. Григоровичи. ЖРФХО, 37, 1113 (1905).
51. Scholl R. Ber., 32, 3492 (1899).
52. Scholl R. Ber., 36, 10 (1903).
53. Sch о 1 1 R., Kacer F. Ber., 36, 322 (1903).
54. Scholl R., Kr emp er A. Ber., 36, 650 (1903).
55. Scholl R., Bertsch E. Ber., 34, 1441 (1901).
56. L о s с о G. Gazz., 68, 474 (1938).
57. Ullmann F Enzyklopadie der technischen chemie, 2 Aufl. Bd. IV. Berlin—Wien,
1929, S 749.
58. Davis T. L. The Chemistry of powder and Explosives. London—N. Y., 1943, p. 405.
59. К i г к R. E., О t h m e r D. F. Encyclopedia of Chemical Technology, vol. 6. N.Y.
Intern. Science Encyclopedia Inc., 1951, p. 10.
60. Б у б и о в П. Ф. Инициирующие взрывчатые вещества и средства инициирования,
ч. 1. М., Оборонгиз. 1960.
61. Г d р с т А. Г., Яременко Н. Е., Светлов Б. Я- Пороха и взрывчатые
вещества. М.,' Оборониздат, 1957, стр. 108; Теория и технология промышленных взрыв-
чатых веществ. М., ГИЗ Стройматер., 1957, стр. 203.
62. Н а п t z s с h А. Вег., 35, 2717 (1902).
63. С и г t i u s Th., Lang H. J. prakt. Chem. [2], 44, 544 (1891).
64. Paderi C. Zbl., 1919, III, 226; Arch, pharm. Sperim., 26, 359 (1918).
65. Z e i s e W. C. J. prakt. Chem., 1, 257, 345, 396 (1834).
66. К 1 a s о n P. I. J. prakt. Chem. [2], 15, 193 (1877).
67. О t t о R. Ber., 15, 121 (1882).
68. О 11 о R. Ber., 13, 1289 (1880).
69. Liebig J. Ann., 11, 17 (1834).
70. К 1 a s о п P. Ber., 20, 3410 (1887).
71. Nakamura M. Biochem. Z., 164, 32 (1925).
72. В r a d 1 e у D. C., Kunchur N- R- Chem. and Ind., 27, 1241 (1962).
73. Roemer H. Ber., 6, 784 (1873).
74. Wertheim E. J. Am. Chem. Soc., 51, 3661 (1929).
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др.
385
75. В i г с h S. F., N о г г i s W. S. G. Р. J. Chem. Soc., 127, 898 (1925).
76. Vogt K. Ann., 119, 142 (1861).
76a. Gutmann A. Ber., 49, 949 (1916).
77. MarkerC. Ann., 136, 76 (1865).
78. D e b u s H. Ann., 72, 18 (1849).
79. Sm ithG. Mc.P., Semon W. L. J. Am. Chem. Soc., 46, 1325 (1924).
80?JacksonC. L., О p p e n h e i m A. Ber., 8, 1032 (1875).
81. R а у P. C. J. Chem. Soc., 109, 131 (1916).
82. Holman K. A., Rabe W. O. Z. anorg. allg. Chem., 17, 26 (1898).
83. H о f m a n K- A., R a b e W. O. Z. anorg. allg. Chem., 14, 293 (1897).
84. Loir A. Ann., 107, 234 (1858).
85. S m i 1 e s S. J. Chem. Soc., 77, 160 (1900).
85a. Rheinboldt H., Motzkus E. Ber., 72, 657 (1939).
856. Rheinboldt H., Mott F. Ber., 72, 668 (1939).
85b. Rheinboldt H. C. A., 32, 484 (1938); Rev. brasil. Chim. (SaoPaulo), 4, 169
(1937).
86. Sachs G. Z. anorg. allg. Chem., 135, 277 (1924).
87. Sachs G. Ber., 54, 1849 (1921).
88. S w a 1 1 e n L. С., Boor d С. E. J. Am. Chem. Soc., 52, 651 (1930).
89. R а у P. C. J. Chem. Soc., 109, 603 (1916).
90. Purgotti A. Gazz., 20, 24 (1891).
91. R,a у P. C. J. Chem. Soc., 121, 1280 (1922).
92. Прилежаева E.H., Шапиро Э. С., ШостаковскийМ. Ф. Изв.
АН СССР, ОХН, 1951, 438.
93. ШостаковскийМ. Ф.,’ Прилежаева Е. Н., Шапиро Э. С. Изв.
АН СССР, ОХН, 1954, 292.
94. ШостаковскийМ. Ф., Прилежаева Е.Н., Шапиро Э. С. Изв.
АН СССР, ОХН, 1954, 303.
95. В г а n d t G. A. R., Е m е 1 ё u s Н. J., Н a s z е 1 d i n е R. N. J. Chem. Soc.,
1952, 2198.
96. H a s z e 1 d i n e R. N., К i d d J. M. J. Chem. Soc., 1953, 3219.
97. Пат. США 2729663 (1956); С. A., 50, 11362 (1956).
98. Man E. H., Coffman D. D., Muettert ies E. L. J. Am. Chem. Soc.,
81, 3575 (1959).
99. Emeleus H. J., P u g h H. J. Chem. Soc., 1960, 1108.
100. Downs A. J„ E b s w о r t h E. A. V., Emeleus H. J. J. Chem. Soc., 1962,
1254.
101. Downs A. J., Ebsworth E. A. V., Em eleu s H. J. J. Chem. Soc., 1961,
3187.
102. J el 1 i nek F., Lagowskil. I. J. Chem. Soc., 1960, 810.
103. Beckurts H., F r e r i c h s G., H arwig L. J. prakt. Chem. [2], 74, 25 (1906).
103a. ShuetzR. D., Fredericks W. J. J. Org. Chem., 27, 1301 (1962).
104. Нем. пат. 235356 (1911); Zbl., 1911, II, 170.
105. Пат. США 2509198 (1950); С. А., 44, 9478 (1950).
106. Н ermes О. J. prakt. Chem. fl], 97, 465 (1866).
107. Р h i 1 i р р J. Ann. d. Phys, und Chem., 131, 89 (1867).
108. К 1 a s о n P. J. prakt. Chem. [2], 35, 402 (1887).
109. Kar aoglanov Z. Z. anal. Chem., 125, 336 (1943).
110. Peters W. Ber., 41, 3175 (1908).
111. G г о s s m a n n H. Z. anorg. allg. Chem., 37, 411 (1903); Z. Elektr. Chem., 9,736
(1903).
Illa. A g r a w a 1 A. K-, M e h г о t r a R. C. Z. anorg. allg. Chem., 312, 225 (1961).
112. Нем. пат. 475533 (1925); С. A., 23, 3781 (1929).
113. Cohn R. Ber., 34, 3502 (1901).
114. I n о u e T. Japan J. Chem., 3, 147 (1928).
115. R о s e n h e i m А., С о h n R. Ber., 33, 1112 (1900).
115a. Голуб A. M., Романенко Л. И. ЖНХ, 51085 (1960).
1156. Fritz H. P., ManchotJ. Ber., 96, 1891 (1963).
116. Murtry G. C. J. Chem. Soc., 55, 50 (1889).
117. H e r t у С. H., S m i t h J. G. J. Am. Chem. Soc., 18, 906 (1896).
118. R a о S. W. R., W a t s о n H. E. J. Phys. Chem., 32, 1357 (1928). -
119. В о r e 1 1 i V. Gazz., 38, II, 421, 465 (1908).
120. Byk S. J. prakt. Chem. [2], 20, 331 (1879).
121. Rosenheim A., Cohn R. Z. anorg. allg. Chem., 27, 280 (1901).
122. Dessy R. E., Lee I. K- J. Am. Chem. Soc., 82, 689 (1960).
123. Wohler F. Gilberts Ann. Phys, und Chem., 69, 272 (1899).
124. С 1 a u s C. J. prakt. Chem. [1], 15, 406 (1838).
125. So derback E. Ann., 419, 248 (1919).
386
Ртутноорганические соединения
126. В е n n е 11 G. М. J. Chem. Soc., 121, 2139 (1922).
127. Dreher E„ О 11 о "R. Ann., 154, 178 (1870).
128. S c h e r t e 1 A. Ann., 132, 92 (1864).
129. Lecher H. Ber., 48, 1425 (1915).
130. Friedmann W. Petroleum, 11, 978; Zbl., 1916, II, 485.
131. Maikop ar B. Zbl., 1869, 711.
131a. S p i n n e r I. H., A n n о p о u 1 о s J. V., Metanovsky W. Can. J. Chem.,
oq 9^90 riQfill
132. Rheinbo Id t H. Ber., 59, 1321 (1926).
133. Rothstein E. J. Chem. Soc., 1940, 1550.
134. H a 11 R. H., Howe В. K. J. Chem. Soc., 1949, 2723.
135. P i m lo 11 P. J. E., Y о u ng G. T. Proc. Chem. Soc., 1958, 257.
136. WeygandF., Bergmann H. J., Ziemann H., К 1 i! e g e r E. Ber., 91,
1043 (1958).
137. H a c h V. Chem. Listy, 47, 227 (1953).
138. Cram D. J., CordoinM. J. Am. Chem. Soc., 77, 1810 (1955).
139. Cram D. J., A n t a r M. F. J. Am. Chem. Soc., 80, 3109 (1958).
140. ПетрунькинВ. E., Лысенко H. И. ЖОХ, 29, 309 (1959).
141. Ill о с т а к о в с к и й М. Ф., Прилежаева Е. Н., Уварова Н. И. Изв.
АН СССР, ОХН, 1954, 526.
142. Шоста ковский М. Ф., Прилежаева Е. Н. Изв. АН СССР, ОХН,
1954, 517.
143. D a v i е s J. S. Н., О х 1 о г d А. Е. J. Chem. Soc., 1931, 224.
144. П р и л еж'а е в а Е. Н., ШапироЭ.С., Ш о с т а к о в с к и й М. Ф. Изв.
АН СССР, ОХН, .1951, 560.
145. Н о 1 m b е г g В. J. prakt. Chem., 135, 57 (1932).
146. Holmberg В. J. prakt. Chem. [2], 141, 103 (1934).
147. H о 1 m b e r g В. Arch. f. kern. Min. Geol., 12B, N 471, 31 (1938).
148. H о 1 m b er g В. C. A., 38, 2945 (1944); Arch. f. kern. Min. Geol., A15, № 22, 11
(1942). ,
149. Holmberg B. Ark. Kemi, 2, 567 (1950).
150. Ш о с т а к о в с к и й М. Ф., Прилежаева Е. Н., Шапиро Э. С. Изв.
АН СССР, ОХН, 1953, 357.
151. Прилежаева Е. Н., Шапиро Э. С., ШостаковскийМ. Ф. Изв.
АН СССР, ОХН, 1952, 478.
152. Ш о с т а к о в с к и й М. Ф., Богданова А. В., Плотникова Г. И.
ДАН СССР, 124, 107 (1959).
153 . III о с т а к о в с к и й М. Ф., Богданова А. В., Плотникова Г. И.,
Андреев Н. С. Изв. АН СССР, ОХН, 1960, 1279.
154. Ш о с т а к о в с к и й М. Ф., Богданова А. В., Плотникова Г. И.
Изв. АН СССР, ОХН, 1960, 1514.
155. Ш о с т а к о в с к и й М. Ф., Грачева Е. П., Лаба В. И., Якутки-
н а Л. М. ЖОХ, 32, 709 (1962).
156. ШостаковскийМ. Ф., Грачева Е. П., К у л ь б о в с к а я Н. К- ЖОХ,
30, 383 (1960).
157. Ш о с т а к о в с к и й М. Ф., Богданова А. В., Плотникова Г. А.
ДАН СССР, 120, 301 (1958).
158. Михайлов Б. М., Тер-Саркисян Г. С. Изв. АН СССР, ОХН, 1960,
1888.
159. Ш о с т а к о в с к и й М. Ф., Прилежаева Е, Н., Герштейн Н. А.,
Караваева В. А. Изв. АН СССР, ОХН, 1959, 904.
159а. П р и л еж а е в а Е. Н., ЦымбалЛ. В., Шостаковскнй М. Ф^ Изв,
АН СССР, ОХН, 1962, 1679.
160. Мас’грюковаТ. А., Прилежаева Е. Н., УвароваН. И., Шо-
стаковскийМ. Ф., Кабачник М. И. Изв. АН СССР, ОХН, 1956, 443.
161. В aster R. A., Newbold G. Т., S р г i п g F. S. J. Chem. Soc., 1947, 370.,
162. Fischer E. Ber., 27, 673 (1894).
163. Schnei der W., Sep p J. Ber., 49, 2054 (1916).
164. Schneider W., Sepp J., StiehlerO. Ber., 51, 220 (1918).
165. Pacsu E. Ber., 57, 849 (1924).
166. Pacsu E. Ber., 58, 509 (1925).
167. Pacsu E. Ber., 58, 1455 (1925).
168. Pacsu E., Kar у Ch. Ber., 62, 2811 (1929).
169. Pacsu E., T icharich N- Ber., 62, 3008 (1929).
170. P a c s u E., Green J. W. J. Am. Chem. Soc., 58, 1823 (1936),
171, Levene P, A., MeyerC. M. J. Biol. Chem., 69, 175 (1926).
172. L e v e n e P. A., M e у e г С. M. J. Biol. Chem., 74, 695 (1927).
173. D a 11 у О. T., Me I 1 г о у R. J. J. Chem. Soc., 1949, 555.
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др. 387
174. Z i n n е г Н., BrandnerH., R e m b a r z G. Ber., 89, 800 (1956).
175. Zinner H., BrandnerH. Ber., 89, 1507 (1956).
176. Zinner H., Wi ttenburg E. Ber., 94, 2072 (1961).
177. ZinnerH., Wessely K-, BockW., RieckhoffK., St r an d t F.,
Nimmich W. Ber., 90, 500 (1957).
178. ZinnerH., Wessely К- Ber., 90, 516 (1957).
179. Zinner H., RembarzG., Linke H. W., UlbrichtG. Ber., 90, 1751
(1957).
180. ZinnerH., RembarzG., KlocklingH. P. Ber., 90, 2688 (1957).
181. ZinnerH., N i m z H,, V e n n e r H. Ber., 90, 2696 (1957).
182. ZinnerH., NimzH., Ven пег H. Ber., 91, 148 (1958).
183. Zinner H., Dassler C. G., RembarzG. Ber., 91, 427 (1958).
184. ZinnerH., NimzH., VennerH. Ber., 91, 638 (1958).
185. ZinnerH., ThielebeuleW., RembarzG. Ber., 91, 1006 (1958).
186. ZinnerH., NimzH. Ber., 91, 1657 (1958).
187. ZinnerH., BockW., KlocklingH. P. Ber., 92, 1313 (1959).
188. Z i n n e r H., W i g e r t H. Ber., 92, 2893 (1959).
189. ZinnerH., Brandhoff H., S c h m a n d k e H., К r i s t e n H., H a u n R.
lUC1. I 11 IX С 1 Xlt) U 1 4 1* LI 11 LJ 1 1 kJ V 11 Ill CL XI LI JX С XI
Ber 92 3151 (1959)
190. ZinnerH., D a s s 1 e r C. G. Ber,, 93, 1597 (1960).
191. ZinnerH., T hielebeule W. Ber., 93, 2791 (1960).
192. ZinnerH., Wittenburg E. Ber., 94, 1298 (1961).
193. Zinner H., Schmandke H. Ber., 94, 1304 (1961).
194. ZinnerH., P f e i f f e r M- Ber., 94, 2792 (1961).
195. Zinner H., Ernst B., KreienbringF. Ber., 95, 821 (1962).
196. ZinnerH., Stark К-H., MichalzikE., К r i s t e n H. Ber., 95, 1391
(1962).
197. Zinner H., Rehder W., Schmandke H. Ber., 95, 1805 (1962).
198. ZinnerH., Schneider F. Ber., 95, 2295 (1962).
199. ZinnerH., Schnei der F. Ber., 95, 2769 (1962).
200. S a c h s G., О t t M. Ber., 59, 171 (1926).
200a. Gregg D. C., IddlesH. A., Stearns P. W., jr. J. Org. Chem., 16, 246
(1951).
201. H о 1 m b e r g B. Ber., 89, 278 (1956).
202. Kern R. J. J. Am. Chem. Soc., 75, 1865 (1953)..
203. S i e m e n s C. Ann., 61, 360 (1847).
204. Baroni A. Atti Accad. Lincei, 12, 234 (1930).
205. В i r d M. L, Challenger F. J. Chem. Soc., 1942, 570.
206. D a 1 e J. W., Emeleus H. J., H aszel di ne R. N. J. Chem. Soc., 1958^.
2939
207. L о e v e n i c h J., F r e m d 1 i ng H., Fohr M. Ber., 62, 2856 (1929).
208. В г a d t W. E., Green J. F, J. Org. chem., 1, 540 (1936).
209. Raffo M., Scar el la A. Gazz., 45, 1, 127 (1915).
210. R а у P. C., D h a r N. J. Chem. Soc., 103, 3 (1901).
211. S t r 6 m h о 1 m D. Z. anorg. allg. chem., 57, 72 (1908).
212. P esc i L. Z. anorg! allg. chem., 15, 208 (1897); Gazz., 26, II, 54 (1896).
213. Andre G. C. r., 112, 996 (1891).
214. YoungJ. A., TsoukalasS. N., DresdnerR. D. J. Am. Chem. Soe. _
80, 3604 (1958).
214a. Макарове. П., Шпанский В. А., Гинсбург В. А., Щекоти-
хинА.И., ФилатовА.С., Мартынова Л. Л., П а в л о в с к а я И. В.,
Го лованева А. Ф., Якубович А. Я- ДАН СССР, 142, 596 (1962).
2146. Y о u п g J. A., D и г г е 1 1 W. S., D г е s d n е г R. D. J. Am. Chem. Soc., 84.
2105 (1962b
215. Pesci L. Gazz., 27, I, 568 (1897).
215a. BrodersenK-.Opitz G.,Breitinger D.,Menzel D. Ber., 97, 1155 (1964).
216. Pesci L. Atti Accad. naz. Lincei. Rend., 1, 312 (1892).
2177 Pesci L. Gazz., 22, I, 373 (1892).
218. Pesci L. Gazz., 23, II, 529 (1893).
219. Pesci L. Gazz., 28, II, 442 (1898).
220. Forster C. Ann., 175, 29 (1874).
221. Forster C. Ber., 7, 294 (1874).
222. DimrothO., OttoR. Ber., 35, 2037 (1902).
223. H о f m a n n A. W. Ann., 47, 62 (1843).
224. F u r t h O. Monatsh., 23, 1155 (1902).
225. F r a n k 1 i n E. C. Am. Chem. J., 47, 361 (1912).
226. Новикове. С., Годовикова T. И., Тартаковский В. А. ДАН
СССР, 124, 4 , 834 (1959).
"388
Ртутноорганические соединения
227. Jackson С. L., Peaks R. W. Am. Chem. J., 39, 567 (1908).
228. Kharasch M. S., Lomnien F. W. M., JacobsohnI, M. J. Am. Chem.
Члс 44 793 119921
229. P r u s s i a L. Gazz., 28, 123 (1898).
230. Zinin N. N. J. prakt. Chem. (1), 27, 148 (1842). -
231. К 1 e i n O. Ber., 11, 1743 (1878).
232. Pesci L. Gazz., 26, II, 54 (1896).
233. Fischer B., GriitznerB. Arch, pharm., 232, 329 (1893).
234. S t г e с к e r A. Ann., 103, 324 (1857).
235. Markownikoff W. Z. Chem., [1], 6, 535 (1865).
236. О p p e n h e i m A., P f a f f S. Ber., 7, 623 (1874).
237. A n d г ё G. C. r., 102, 116 (1886).
238. Schoeller W., SchrauthW. Ber., 42, 784 (1899).
239. MenschutkinN., Ermolaef f M. Z. Chem., [2], 7, 5 (1871).
240. Francesconi L., de Plato G. Gazz., 33, I, 228 (1903).
241. Prager W. Monatsch., 33, 1285 (1912).
242. Ley H., Fischer W. Z, anorg. allg. Chem., 82, 329 (1913).
243. S e s t i n i F. Z. Chem. [2], 7, 35 (1871).
244. L e у H., Schaefer K. Ber., 35, 1309 (1902).
245. Desaignes V. Ann.,'82, 231 (1852).
246. Grot e K. Ann., 130, 202 (1864).
247. Schutz, Marsh, Gen t her. Z Chem. [2], 4, 303 (1868).
248. MenschutkinN. A. Ann., 162, 168 (1872).
249. LandsbergM. Ann., 215, 209 (1882).
250. Lyons E. J, Am. Chem. Soc., 47, 830 (1925).
251. Patton R. H., Simons J. H. J. Org. Chem., 21, 1199 (1956).
252. L i e b i g J. Ann., 85, 289 (1853).
253. Wert h er. J. prakt. Chem. flj, 35, 63 (1887).
254. Ruspaggiari G. Gazz., 27, I, 6 (1897).
255. Glassma nn B., Skundtna S. Hoppe-Seyler F. A. Z, physiol. Chem., 160,
77 (1926); C. A., 21, 751 (1927).
256. Novak J. Zb!., 1873, 154.
:257. P f 1 ii ge r E. Zbl., 1888, 559.
258. О к u d a T., TakanashiY., T s u г u о к a M. C. A., 56, 7147 (1962).
259. S h a h L. D. J. Indian Chem. Soc., 15, 149 (1938).
260. William sJ. W., Rainey W. T., jr., Leopo 1 d R. S. J. Am. Chem. Soc.,
64, 1738 (1942).
261. Oppenheim A., C zar nomsky. Ber., 6, 1392 (1873).
-262. Ostrogovich A. Ann., 291, 377 (1896).
263. Field F. Ann., 65, 49 (1848).
264. LandsbergM. Ann., 215, 172 (1882).
265. Луценко И. Ф., Тюленева В. В. ЖОХ, 27, 497 (1957).
266. F о г s t е г М. О. J. Chem. Soc., 73, 783 (1898).
267. A u 1 d S. J. М. J. Chem. Soc., 91, 1045 (1907).
-268. KaufmanHs P., S к i b a K. J. C. A., 52, 19183 (1958); Fette, Seife, Anstrich
smittel, 59, 340, 498 (1957).
269. В rodersen K., Kunkel L. Z. anorg. allg. Chem., 298, 34 (1959).
270. W h e e 1 e г H. V., McFarland B. W. Am. Chem. J., 18, 542 (1894).
-271. Piccinini A. Gazz., 24, II, 457 (1894).
272. Piccinini A. Gazz., 24, II, 453 (1894).
273. Gizycki F. V., R e p p e 1 L. РЖХнм, 1956, 71750; Arch. Pharmazie, 289, 61,
№ I> 33 (1956).
'274. К о p e н м а н И. M., Дьячковская А. С. Уч. зап. ГГУ, 1953, № 24, 139.
275. Rodighiero G. Ann. chim. applicata, 39, 261 (1949).
276. J acksonC. L, W i n g J. F. Am. Chem. J., 9, 3251 (1887).
277. Bro dersen K-, К u n k e 1 L. Ber., 91, 2698 (1958):
278. S t о I 1 e R. Ber., 45, 284 (1912).
279. Mangini A., Dejudicibusl. Gazz., 63, 601 (1933).
280. Mangini A. Gazz., 65, 298 (1935).
281. M a n g i n i A. Gazz., 67, 384 (1937).
-282. C i u s a R. Atti Accad. naz. Lincei. Rend. 20, 578 (1911).
-283. Vecchiotti L., Cap 0 d a c qu a A. Gazz., 55, 369 (1929).
284. Dwyer F. P. C. A., 34, 733 (1940); AustralianChem. Inst. J. Proc., 6, 348, 362 (1939).
285. Пат. США 2852506 (1958); С. A., 53, 8174 (1959).
286. Fischer H., Mil 11 er R. Z. physiol. Chem., 148, 155 (1925).
287. Ч у г a e в Л. А., Ш л e з и н г e p H. А. ЖРФХО, 36, 1261 (1904).
288. Wil 1st at ter R., A s a h i n a Y. Ann., 385, 195 (1911).
-289. Pet e r s W. Ber., 40 235 (1907).
Синтез и реакции соединений со связями О—Hg, S—Hg, Se—Hg и др.
389»
290. Oliveri-Mandala Е. ZbL, 1921, III, 643; Gazz., 51, I, 125 (1921).
291. L am i P. C. A., 10, 2613 (1916).
292. Fox J. J., Yung N., Davoll J., В r own G. B. J. Am. Chem. Soc., 78,
2117 (1956).
293. F о x J. J., Yung N., W e m p e n I., D о e г r I. L. J. Am. Chem. Soc., 79,.
5060 (1957).
294. Fox J. J., Y u ng N. Praag. Fed. Proc., 16, 182 (1957).
295. D a v о 1 1 J., Lowy B. A. J. Am. Chem. Soc., 73, 1650 (1951).
296. Michelson A. M., T о d d A. R. J. Chem. Soc., 1955 , 816.
297. Fox J. J., Co di ngton J. E., Yung N. C., Kaplan Z., Lampen I. O.,
J. Am. Chem. Soc., 80, 5155 (1958).
298. KissmannH. M., Weiss M. J. J. Am. Chem. Soc., 80, 2575 (1958).
298a. R esti vo A. R., D о n d z i 1 a F. A. J. Org. Chem., 27, 2281 (1962).
2986. Англ. пат. 875971 (1960); С. A., 57, 15221 (1962).
299. Baker В. R., Joseph J. P., Schaub R. E., WilliamsJ. H. J. Org..
Chem., 19, 1786 (1954).
300. Baker B. R., Schaub R. E. J. Am. Chem. Soc., 77, 2396 (1955).
301. В а к e r B. R., Schaub R. E. J. Am. Chem. Soc., 77, 5900 (1955).
302. Bristow N. W., L у t h о e B. J. Chem. Soc., 1949, 2306.
303. Baker B. R., Hewson K., Thomas H. J., Johnson, J. A. jr. J., Org,
Chem., 22, 954 (1957).
304. Schaeffer H. J., Thomas H. J. J. Am. Chem. Soc., 80, 4896 (1958).
305. Schaeffer H. J., Thomas H. J. J. Am. Chem. Soc., 80, 3738 (1958).
306. Шабарова 3. А., Полякова 3. П., Прокофьев M. А. ЖОХ, 29, 215.
(1959).
307. Baker B. R., Hewson K. J. Org. Chem., 22, 959 (1957).
308. Пат. США 2885396 (1959); С. A., 53, 17158 (1959).
308a. Англ. пат. 877318 (1960); С. А., 56, 8724 (1962).
309. CodingtonJ. Е. J. Am. Chem. Soc., 80, 5164 (1958).
310. Zinner H., WittenburgE. Ber., 95, 1866 (1962).
311. Hoffer M., DiischinskyR., Fox J. J., Yung N. J. Am. Chem. Soc.,.
81, 4112 (1959).
312. Wagner G., P i s c h e 1 H. Naturwiss., 48, 454 (1961).
313. H о f f e r M. Ber., 93, 2777 (1960).
314. Bruns G. Z. physiol. Chem., 14, 533 (1890).
315. Kruger M. Z. physiol. Chem., 18, 423 (1894).
316. Zinner H., P f e i f f er M. Ber., 96, 432 (1963).
317. Gubitz F. M., Clarke R. L. J. Org. Chem., 26, 559 (1961).
318. F о x R. В., V e n e z k у D. L. J. Am. Chem. Soc., 75, 3967 (1953).
319. Venezky D. L., FoxR. B. J. Am. Chem. Soc., 78, 1664 (1956).
320. ВязанкинН. С., Разуваев Г. А., Глады me в E. H. ДАН СССР,
151, 1326 (1963).
321. ВязанкинН. С., Разуваев Г. А., Гладышев Е. Н. ДАН СССР, 155,
830 (1964).
322. ВязанкинН. С., Разуваев Г. А., Гладышев Е. Н., Тур ико-
ва Т. Г. ДАН СССР, 155, 1108 (1964).
323. George М. V., Lichtenwalter G. D., Gi Iman Н. J. Am. Chem. Soc.,
81, 978 (1959).
324. Ray P. C. J. Chem. Soc., Ill, 101 (1916).
325. Brooks P., Davidson N. J. Am. Chem. Soc., 82, 2118 (1960).
326. Carius L. Ann., 124, 221, 257 (1862).
327. Foster D. J., To bier E. J. Org. Chem., 27, 834 (1962).
328. Spinelli D., Dell’Erb а С. РЖХим., 1962, 11Ж 315.
Глава XVII
АНАЛИЗ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
качественное определение ртути
В РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Присутствие ртути в органическом соединении легко определить каче-
ственной пробой.
Вещество прокаливают с окисью кальция или с перманганатом се-
ребра [2] в стеклянной трубке (диаметром 0,—0,5 см, длиной 20—25 см)
и обнаруживают конденсирующуюся на стенках ртуть визуально или пос-
редством дитизона. Или же вещество прокаливают с окисью меди, выделяю-
щиеся пары ртути дают на бумаге, смоченной раствором хлористого палла-
дия, темное пятно восстановленного палладия [31, которое можно сделать
'более контрастным, если поместить в пары аммиака. Если вещество выде-
ляет при своем разрушении непредельные соединения, тоже восстанавли-
вающие хлористый палладий, то пары ртути улавливают сначала на ли-
сточки золота или на стенки трубки, откуда уже, после разрушения
органического вещества, перегоняют на бумагу, смоченную хлористым пал-
ладием 14].
В немногих случаях ртутноорганические соединения могут быть обна-
ружены без предварительного разрушения; например, чувствительной яв-
ляется цветная реакция определения фенилмеркуриона при помощи дити-
зона [4а].
Уксуснокислая фенилртуть и этилмеркурфосфат определены спектро-
скопически при помощи 2-(1,3-диоксоиндан-2-ил-имипо)-1,3-индандиона
[46]. Имеются микрохимические реакции ртутноорганических солей с раз-
ными анионами [51.
КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
\ Элементарный анализ ртутноорганических соединений
на С, Н, Hg и другие элементы
Макрометод. В настоящее время практически не применяются макро-
методы определения ртути совместно с С и Н. При элементарном анализе
ртутноорганических соединений по Либиху ртуть улавливали в изогнутом
конце длинной трубки для сожжения (метод А. В. Гофмана [6] и Франклан-
да и Дюппа [7]) или же, поместив в конце трубки листочки золота, ко-
торые ртуть амальгамирует [1, 8, 9].
При элементарном анализе по Деннштедту (методика разработана
Фильковым и Райциссом [10]) ртуть поглощают посеребренным асбестом.
Анализ ртутноорганических соединений
391
Микрометод. Разработанный Коршун, Шевелевой и Гельман [11] ско-
ростной метод микроопределения углерода, водорода, галоида и ртути из
одной навески органического вещества основан на пиролитическом сож-
жении вещества [12—16] и весовом определении этих элементов.
Углерод и водород определяют, как обычно [12—15], а галоид и ртуть
удерживают в трубке для сожжения в специальных гильзах из прозрач-
ного кварца, наполненных соответственно серебром и золотом (в виде сет-
ки, фольги или проволоки) и определяют по привесу гильз. Отклонения
Рис. 1; Трубка для определения углерода,
водорода и ртути
полученных значений от вычисленных не превышают для углерода и водо-
рода 0,3%, для галоида и ртути — 0,5%. Таким же путем можно опреде-
лять ртуть одновременно с углеродом, водородом и серой [16]. Этот метод
позволяет определять ртуть в веществах, содержащих азот, а также тяжелые
металлы. Если в веществе присутствует и тяжелый металл, и галоид, то
ртуть можно определять только в тех случаях, когда металл не образует
летучих галогенидов при сожжении.
Рис. 2. Трубка для определения углерода, водорода, ртути
и галоида (или серы)
В соединениях, содержащих С, Н, F и Hg, можно определять С, Н, F,
удерживая ртуть золотом в охлаждаемой зоне.
Одновременное определение углерода, водо-
рода, галоида и ртути [11]. Аппаратура. Применяется та же
аппаратура, что и для одновременного определения С, Н и галоида в гиль-
зе [16]. Расположение гильзы с золотом, навески и окислительной зоны
показано на рис. 1. Если вещество содержит галоид, то в трубку для
сожжения помещают также гильзу с серебром, как показано на рис. 2.
В этом случае длина широкой части трубки для сожжения должна быть
не меньше 30 см. Для быстрого охлаждения поглотительных аппаратов и
гильз пользуются металлическими блоками с выемками, соответствую-
щими размерам аппарата Прэгля и гйльзы.
Реактивы. Для удерживания ртути применяется тонкая золотая про-
волока или фольга.. Галоид поглощается металлическим серебром, применя-
392
Ртутнооргаяические соединения. Лит. стр. 395—396
емым в виде сетки, фольги или проволоки весом около 3 г. Работать с посе-
ребренной пемзой в этом случае нельзя, так как она удерживает часть рту-
ти и результаты анализа для ртути и галоида получаются искаженными.
Остальные реактивы те же, что и обычно применяемые для определения
углерода и водорода [12—16].
Выполнение анализа. 1Аъ\ остановимся только на приемах, связанных с
определением ртути и помещением гильзы и пробирки в трубку для сож-
жения, так как в части, относящейся к определению углерода, водорода й
галоида, ход анализа таков же, как и для несодержащих ртуть
веществ [16].
Перед употреблением гильзу кипятят в соляной кислоте (1 : 1) в тече-
ние 10 мин., моют, сушат и прокаливают в трубке для сожжения. В про-
каленную гильзу (в ее узкую часть) помещают слой золота длиной 4 см.
Золото должно лежать неплотно, так, чтобы оно не оказывало сопротивле-
ния току газа.
Гильзу с золотом тарируют, пользуясь тар ильной склянкой с кусочка-
ми стекла. Перед началом работы проводят холостой опыт, во время кото-
рого гильзу помещают в трубку для сожжения. Если гильза уже содержит
ртуть, поглощенную во время предыдущих опытов, то при холостом опы-
те, как и при сожжении, слой золота необходимо охлаждать льдом, чтобы
не было уноса мельчайших частиц ртути за пределы гильзы струей теплого
кислорода, идущей из нагретой трубки для сожжения. При сожжении ртут-
ноорганических соединений гильзу с вставленной в нее пробиркой с навес-
кой (5—8 мг) вдвигают в трубку на расстояние не менее 5 см от окислитель-
ной зоны, устанавливают скорость кислорода 30—35 мл!мин и начинают
сожжение вещества газовой горелкой или электропечью. По окончании
сожжения прокаливают горелкой широкую часть гильзы. Узкую часть на-
гревать не следует во избежание испарения скапливающейся там ртути.
Надо отметить, что ртуть лишь частично попадает на золото. Часть ее осе-
дает на холодных стенках узкой части гильзы в виде тончайшего серого
налета. Когда сожжение окончено, гасят горелку, снимают охлаждение
и продолжают в течение 10 мин. пропускать кислород через трубку для
сожжения. В это время берут навеску для следующего определения. Затем
отсоединяют поглотительные аппараты и трубку сейчас же закрывают
затычкой. Вынимают гильзу, удаляют из нее пробирку, переносят гиль-
зу в весовую комнату, помещают на металлический блок, находящийся
около весов, и взвешивают через 25 мин. после удаления из труб-
ки [16].
Если в анализируемом веществе присутствует галоид, то необходимо
ввести в трубку для сожжения еще одну кварцевую гильзу, наполненную
в узкой части металлическим серебром. Если широкая часть трубки для сож-
жения имеет длину 30 см, то обе гильзы свободно размещаются и сожжение
идет достаточно далеко от резиновой пробки. Нужно внимательно следить,
чтобы весь слой серебра был нагрет до 575°С и ртуть не могла задержаться
на более холодных концах слоя в виде амальгамы серебра. Сожжение про-
водят, как описано выше. По окончании сожжения гасят горелку, снимают
печь с серебра и пропускают кислород еще в течение Юмин.; первыебмин.—
не снимая охлаждение и еще 5 мин. после удаления льда. Гильзы с се-
ребром и золотом взвешивают соответственно через 25 и 30 мин. после уда-
ления их из трубки для сожжения.
В конце каждого рабочего дня рекомендуется регенерировать золото.
Для этого гильзу помещают в чистую кварцевую трубку с оттянутым кон-
цом, соединяют трубку через небольшую промывалку, наполненную азот-
ной кислотой 1:1, со склянкой Мариотта или с водоструйным насосом и
просасывают воздух через трубку, одновременно нагревая золото горелкой
Анализ ртутноорганичёских соединений
393"-
или печью. Когда вся ртуть будет вытеснена с поверхности золота и соответ-
ственно из гильзы, гильзу вынимают, а трубку моют азотной кислотой.
Ртуть может [16а] улавливаться серебром, а не золотом, как обычно,
при одновременном микроопределении С, Н и ртути в веществах, не со-
держащих галоида, сожжением в кислороде при 900—950° С и затем пропу-
сканием паров над Со3О4 (650—680° С) на корунде.
Определение углерода и водорода
в ртутноорганических соединениях
Макрометод. Для определения только углерода и водорода в ртутноорга-
нических соединениях пользуются длинной трубкой для сожжения, ко-
нец которой выдвигают из печи на 15—20 см; на нем осаждается ртуть
и часть воды. По окончании сожжения воду осторожным нагреванием пере-
водят в поглотительный прибор. Можно улавливать ртуть также на золото,
как в микройетоде.
Микрометод. При обычном микроопределении С и Н рекомендуется при-
менять металлическое золото в качестве поглотителя ртути. Золото вкла-
дывают в виде тампона в носик трубки для сожжения. Это позволяет
вытеснять ртуть простым нагреванием носика или менять золото по мере*
его амальгамирования.
Определение ртути и других элементов, кроме С и Н,
в ртутноорганических соединениях
Из предложенных разнообразных «влажных способов» разрушения ртут-
ноорганических соединений передопределением в них ртути, например раз-
ложение нагреванием с серной и азотной кислотами [17], сррной кислотой и
перекисью водорода[18—21], серной кислотой и перманганатом [22—25J
и др., наиболее надежным и универсальным является разложение
по Кариусу дымящей азотной кислотой в запаянной трубке. Минерализо-
ванную этими способами ртуть определяют затем обычными аналитически-
ми методами, как титрование тиоцианатом, осаждение в виде сернистой рту-
ти, электролитически, в том числе микроэлектролитически по данным Вер-
дино [26—28], колориметрически и т. п.
Возможно также разложение ртутноорганического соединения таким
образом, чтобы ртуть была выделена в виде металлической ртути или амаль-
гамы. Для этого производят, например, кипячение вещества с цинковой
пылью в нейтральном [29,30] или щелочном [31,32] растворе (см. также [33]);
образовавшуюся амальгаму растворяют в азотной кислоте и затем обычным
образом определяют ртуть. В некоторых случаях предлагали восстанов-
ление металлическим алюминием в нейтральной или слабощелочной среде
[34], хлорисдым оловом [35], моноэтаноламином и металлическим натрием
в диоксане [36] и др. Однако перечисленные способы менее надежны и уни-
версальны, чем разложение ртутноорганических соединений по Кариусу
или определение ртути пиролитическим сожжением (см. дальше).
Анализ на ртуть ртутноорганических соединений. Ртутноорганическое
соединение разлагают по Кариусу в запаянной трубке дымящей азот-
ной кислотой (уд. в. 1,52). После вскрытия трубки и разбав-
ления водой, в случае если ртутноорганическое соединение не содер-
жало галоида, титруют 0,1 N раствором роданистого аммония в при-
сутствии железных квасцов до слабо-коричневого окрашивания: 1 мл
0,1 N NH4CNS идет на 0,010015 г Hg. Титрование роданистым аммонием
394 Ртутноорганические соединения. Лит. стр. 395—396
неприменимо к соединениям, содержащим галоид, в которых ртуть после
разложения и разбавления водой определяют весовым путем в виде серни-
стой ртути аналогично данному ниже описанию.
После разложения удобно также определять ртуть (и в веществах, со-
держащих галоид) электролитически, соответственно микроэлектролити-
чески.
В соединениях с легко отщепляемой ртутью, например в продуктах
присоединения солей ртути к непредельным соединениям или в арома-
тических соединениях ртути, последнюю удобнее и проще определять ме-
тодом Адамса.
Анализ на ртуть ртутноорганических соединений по методу Адамса [37].
'Около 0,5 г ртутноорганического соединения помещают в круглодонную
колбу (емкостью 200 мл), закрытую резиновой пробкой с двумя отверстия-
ми, в одно из которых вставлена капельная воронка, в другое — трубка
Пелиго, наполненная водой. Через воронку приливают 5 мл конц. соля-
ной кислоты и смесь нагревают до образования прозрачного ^раствора (ми-
нут 10—15). Воду из трубки Пелиго смывают в реакционную колбу и до-
бавляют воды до 10 мл. Через раствор пропускают сероводород до полного
осаждения. HgS фильтруют через взвешенный фарфоровый или стеклянный
тигель с фильтрующим дном, промывают водой, спиртом, сероуглеродом,
сушат при 110° С и взвешивают.
Оба эти метода приложимы также для микроаналитического определе-
ния ртути.
Определение ртути сожжением ртутноорганических соединений старыми
методами (с окисью кальция) [1,38] непригодны для анализа веществ, со-
держащих азот или галоид, в особенности иод или бром. Поэтому предла-
гались две раздельные методики определения ртути сожжением. Одна для
анализа веществ, не содержащих азота: по Боэтиусу [39] — сожжение в
кислороде; сера, хлор и бром удерживаются нагретой окисью свинца, иод —
слоем глиняных черепков, покрытых серебром, или по Юречеку [40] (полу-
микрометод): сожжение в кислороде с платиновым контактом; хлор и бром
поглощают безводным углекислым натрием, иод удерживают серебром,
диспергированным на окиси магния, ртуть — золотом. Для определения
ртути в веществах, содержащих азот, была предложена уже другая ме-
тодика; сожжение в токе углекислоты в трубке, наполненной хроматом
свинца, медью и посеребренными черепками (Боэтиус [39]), или сжига-
ние вещества в токе кислорода, вытеснение последнего углекислым газом,
пропускание ртути, загрязненной нитратом ртути, через раскаленную медь
и улавливание чистой ртути на золото [41].
Коршун и Лавровской [42] разработан универсальный метод определе-
ния ртути (независимо от присутствия других элементов) сожжением веще-
ства в токе азота и пропусканием продуктов разложения через накаленный
до 750° С, полностью удерживающий кислые пары наполнитель, такой же,
как во второй методике Боэтиуса. Пары ртути, конденсирующиеся в конце
сожигаФельной трубки, перегоняют на слой золотой фольги.
Однако эта методика устарела. Ей, а также перечисленным методикам
следует предпочесть более простой универсальный метод пиролитического
сожжения вещества, разработанный Коршун, Шевелевой и Гельман [И],
позволяющий определять и одну ртуть.
Этот же метод позволяет определять одновременно ртуть, галоид и серу,
что удобнее, чем, например, одновременное определение этих элементов
разложением ртутноорганического соединения в бомбе металлическим ка-
лием [43].
По Хернлеру [44] возможно одновременное определение ртути и азота
сожжением по Дюма и улавливанием ртути на золоте.
Анализ ртутноорганических соединений
395
Имеется целый ряд обзоров и книг по органическому анализу, в которых
упоминается определение ртути [27, 45—47].
Описано колориметрическое [48, 49] и спектрографическое определение
ртутноорганических соединений (уксуснокислой фенилртути) [50], а также
полярографическое определение некоторых классов ртутноорганических
соединений, например, алкилртутных соединений [51—56, 58], продуктов
присоединения солей ртути к непредельным [57, 59—61], ар ил ртутных сое-
динений [56, 62—71], ртутных производных гетероциклов [72].
Кимура и Миллер сообщили о разделении паров летучих ртутноорга-
яических соединений и металлической ртути [73].
Изучено значение величины Rf в бумажной хроматографии некоторых
ртутноорганических соединений [74].
Произведено разделение смесей RHgBr(R —Вг, СН3, С2Н5, С3Н7, С4Н9,
р-СН3ОС6Н5, р-ВгСвН4) хроматографией на бумаге с растворителем, содер-
жащим NH4OH [75] (см. также [76]), и при помощи газовой хроматографии
на силиконовом масле при 190—220° С с носителем—водородом [75].
Комплексы дитизона с солями" ртути, а также с CeHgHgNO3 и
C6H5HgOCOCH3 могут быть разделены хроматографией на бумаге и про-
явлены бромистым калием [77].
ЛИТЕРАТУРА
1.
2.
3.
Erdmann О. L., М а г с h а и d R. F. J. prakt. Chem. [1], 31, 385 (1844).
К б г b 1 J., Р f i b i I R. Chem. Listy, 50, 236 (1956).
F e i g 1 E, Qualitative Analysis by spot tests. Elsevier Publ. Co. Inc. N. Y., 1947,
p. 274.
S a c h s G. Analyst, 78, 185 (1953).
4. Sachs G. Analyst, 78, 185 (1953).
4a. В о 11 A., W e n g e r P. Helv. Chim. Acta, 30, 538 (1947).
46. Ozawa К., E g a s h i r a S. C. A., 57, 6590 (1962).
КоренманИ. M., Максакова T. П. Tp. Комиссии по аналит. химии АН
СССР, 3, 200 (1951).
Н о f m a n n A. W. Ann., 47, 63 (1843).
Frankland E., Duppa B. F. Ann., 130, 107 (1864).
К 6 n i g W. J. prakt. Chem. [1], 70, 64 (1857).
Abelmann A. Ber., 47, 2935 (1914).
F a 1 k о v M.,
Коршун
99 (1960).
Коршун
Коршун
К о p ш у н M.
К о р ш у н М.
М.—Л., Госхимиздат, 1949.
5.
6.
'7.
8.
9.
10.
11.
М.
R а
О.,
1 z i ss G. W. J. Am. Chem. Soc., 45, 998 (1923).
Ш е в е л е в а Н. С., Г е л ь м а н Н. Э. ЖАХ, 15, вып. 1
12.
13.
14.
15.
М.
М.
о.,
о.,
о.,
о.,
277 (1947).
176 (1948).
292 (1949).
Климова В. А. ЖАХ, 2,
Климова В. А. ЖАХ, 3,
Климова В. А. ЖАХ, 4,
Гельман Н. Э, Новые методы элементарного микроанализа.
16. К о р ш у н М. О., Гельман Н. Э., Шевелева Н. С. ЖАХ, 13, 695 (1958).
16а. Лебедева А. И., Крамер К- Щ. Изв. АН СССР, ОХН, 1962, 1305.
17. Rupp Е., N б 11 Ph. Zbl., 1905, I, 959; Arch. Pharm., 243,1 (1905); 244, 300, 536 (1905).
18. W 6 b e r A. Z. angew. Chem., 33, 63 (1920).
19. В a u e r H. Ber., 54, 2078 (1921).
20. Tabern D. L., S h e 1 1 b e r g E. F. Ind. Eng. Chem., Anal. Ed., 4, 401 (1932).
21. P f i b i J R., KorosE., BarczaL. Acta Pharm. Hung.; 27, 243 (1957).
22. BordelanuC. V. Ann. Sci. Univ. Jassy, 20, 129 (1935).
23. H i r a i МД Hagatsu R. C. A., 45, 3757 (1951).
24. Kamen K. Chem. Listy, 47, 100 (1953).
25. Rupp E., Kropat K. Zbl., 1912, II, 151.
26. V e r d i и о A. Mikrochem., 6, 5 (1928).
27. Прегл ь Ф. Количественный органический микроанализ. М.— Л., Госхимтехиздат,
1934. Р г eg 1 F. Quantative Organische Mikroanalyse, 7 Aull. Springer Verlag. Wien,
1958.
28. Her nler F., PfennigbergerR. Mikrochem., 21, 116 (1936).
29. P i er c e J. С. C. A., 37, 3019 (1945); Quart. J. Pharm. Pharmacol., 15, 367 (1942).
SO. Chamberg W. P., С г о p p e r F. R., С г о s s 1 e у H. C. A., 50, 4724 (1956);
J. Sci. Food, Agr., 7, 17 (1956).
31. G a u t i e r J. A., P e 1 1 e r i n F. C. A., 52, 15338 (1958); Prods. Pharm., 13, 149
(1958),
396
Ртутноорганические соединения
32. Sanz Р a s t о г О. С. А., 46, 1388 (1952).
33. Chambers W. Р. С. А., 37, 5155 (1943); Quart. J. Pharm. Pharmacol., 16, 6 (1943)_
34. Eckert Н. W. Ind. Eng. Chem., Anal. Ed., 15, 406 (1943).
35. Bartlett J. N., McNabb W. M. Ind. Eng. Chem., Anal. Ed., 19, 484 (1947).
36. R a s c h e r W. H. Ind. Eng. Chem., Anal. Ed., 10, 331 (1938). y
37. Adams R., Ro man F. L, Sper.ry W. N. J. Am. Chem. Soc., 44, 1781 (1922).
38. Meixner A., Krocker F. Mikrochem., 5, 131 (1927).
39. В 6 e t i u s M. J. Prakt. Chem., 151, 239 (1938).
40. J u r ec ek M- Coll. Czech. Chem. Comm., 12, 455 (1947).
41. Jurecek M., VeceraM. Chem. Listy, 45, 445 (1951).
42. Коршун M. О., Лавровская E. В. ЖАХ, 3, 322 (1948).
43. К о p ш у н M. О., Чумаченко M- H. ДАН СССР, 99, 769 (1954).
44. H e r nJ e r F. Zbl., 1930, I, 1186.
45. Jurecek M. Organicka Analysa II, Nacladatelstvi CAV. Praha, 1957, стр. 216.
46. Belcher P., Gibbons D., Sykes A. Mikrochim. Acta, 40, 76 (1952).
47. S у k e s A. Mikrochim. Acta, 1956, 1155.
48. Polley D., Gould C. J. Analyt. Chem., 23, 1286 (1951).
49. IguchiM-, Nishiy ama A., Nagasel. J. Pharm. Soc. Japan, 80, 1437"
(1960).
50. Eldridge A., Sweet T. R. Analyt. Chem., 28, 1268 (1956).
51. Ma л ы г и н a H. И., Крешнякова 3. В. ЖАХ, 13, 250 (1958).
52. С о s t a G. Ann. chim. applicata, 38, 655 (1948).
53. С о s t a G. Ann. chim. (Italy) 1, 207 (1951).
54. U n a b e N., Y a s u k б c h i К. C. A., 53, 9856 (1959); Kumamoto Pharm. Bull.,,
1958, N 3, 94.
55. Kern D. M. H. J. Am-. Chem. Soc., 81, 1563 (1959).
56. Коршу нов И. A., M а л ы г и н a H. И., ВертилинаЛ. К- Тр. по химии,
и хим. технологии, 1, 521 (1958).
57. R о w 1 а п d R. L., К 1 u с h е s к у Е. F. J. Am. Chem. Soc., 73, 5490 (1951).
58. Okamoto К. J. Chem. Soc. Japan, 81, 125 (1960).
59. H a m a m о t q I., Kot a ketnori M. J. Agric. Chem. Soc. Japan, 35, 250 (1961),
60. Usami S. C. A., 51, 17608 (1957); Bunseki Kagaku, 5, 499 (1956).
61. KirrmannA., Kleine-Peter M. Bull. Soc. chim. France, 1957, 894.
62. L a w г e п c e S. M., К г e к e C. W. C. A., 53, 12891 (1959); J. Am. Pharm. Assoc.r
48, 208 (1959).
63. S a t о H. C. A., 52, 16088 (1958); Bunseki Kagaku, 6, 166 (1957).
64. К a j i m и r a T., Y a m a m о t о S. C. A., 50, 10613 (1956); Japan Analyst, 4, 152
(1953).
65. V о j i f V. Chem. Listy, 46, 129 (1952).
66. W и g g a t z e r W. Z., С г о s s J. M. J. Am. Pharm. Assoc., 41, 80 (1952).
67. Vo j i f V. Collect. Czech. Chem. Communs., 16, 488 (1951).
68. Benesch R., BeneschR. E. Arch. Biochem. Biophys., .38, 425 (1952).
69. В e n e s c h R., В e n e s c h R. E. J. Am. Chem. Soc., 73, 3391 (1951).
70. Benesch R., Benesch R. E. J. Phys. Chem., 56 , 648 (1952).
71. Nagasel., Iguchi M., I g и c h i I. C. A., 52, 1151 (1958); Yakugaku Zasshi,
77, 837 (1957).
72. C a s e у P. S., С a г г о 1 e J. J., Stab ica N. R. C. A., 53, 5005 (1959); Proc.
Penna Acad. Sci., 32, 63 (1958).
73. Kimura I., M i 1 1 e г V. L. Analyt. Chem., 32 , 420 (1960).
74. Kanazawa J., Koyama K., Aya M., S a t 6 R. C. A., 52, 12649 (1958).
75. BrodersenK., SchlenkerU. Z. anal. Chem., 182, 421 (1961).
76. В a r t 1 e t t J. N., Curt is G. W. Analyt. Chem., 34, 80 (1962).
77. Becker A., E h i n g e r F. Z. anal. Chem., 187, 110 (1962).
авторский указатель
Абрамова А. Н. 209 , 210 (101), 312(412),
313 (426), 329 (635)
Абрамов В. А. 15 (27)
Абрамов В. С. 353 (15,16)
Аветисян А. А. 154 (452)
Адамова М. Н. 147 (270)
Азановская М. М. 218, 220, 221 (6)
Азизян Т. А. 276 (113а, 1136)
Алейникова М. А. 266 (26)
Амакасу О. 11 (34)
Аменицкая Р. В. 326 (623)
Ананченко С. Н. 205 (70)
Андреев Н. С. 369 (157)
Анисимов К. Н. 103 (629), 104 (631), 253
(135), 281 (137)
Аракелян С. В. 154 (451, 452)
Арбузов Б. А. 96, 97 (520)
Артамкина Г. А. 15 (29, 30), 233 (42, 43),
239 (7, 8, 10, 11), 240 (7, 8, 10), 256 (7, 8,
Ю, П)
Бабаян А. Г. 170 (431)
Баденкова Л. П. 127, 153, 154 (135)
Балин А. Н. 148 (272)
Барышникова А. Н. 317 (438)
Барышникова Л. И. 104 (631)
Баталов А. П. 326 (623)
Батуев М. И. 11 (47), 125 (120)
Бауков Ю. И. 288, 289 (171), 312, 315 (413)
Белецкая И. П. 15 (29—30), 233 (41—43),
239 (4—11), 240 (4—10), 244 (30), 256 (6—
11), 257 (159), 266 (26), 276 (111а—1136),
318 (449), 322 (480, 487а — 488а, 489),
323 (480, 494)
Белов Б. И. 185 (47), 269, 293 (59, 60)
Беспрозванный М. А. 14, 15 (16), 255, 258
(148)
Блешинский С. В. 55, 60 (81), 266 , 302 (47)
Бобашинская Ч. С. 267 (38)
Богданова А. В. 290 (180), 369 (152—154,
157)
Боковой А. IL 127 (133), 297 (270)
Большакова А.\А. 235 (65, 67), 265 (11)
Бондарева М. А. 157 (306а)
Борисевич Н. А. 11 (36)
Борисов А. Е. 11 (47), 19, 22, 23 (12), 28,
29 (83), 31 (99), 101—103, 105, 106,
121 (84—86), 123 (90, 91, 93—95), 124
(111), 125 (120—122), 126 (123), 166 (84,
85, 402), 167 (93, 94, 407), 168 (95, 419—
422), 188 (58), 197, 198 (8), 209 (101, ЮЗ-
107, 109, 123), 210 (101, 123, 166), 211
(ЮЗ, 104, 141, 143—147), 212 (143),
229 (14, 18, 19), 244 (60), 246 (79 , 80),
247 (80), 250, 251 (60), 253 (130), 255 (151),
257 (158), 258(84), 259 (80, 151, 173),
260 (151), 266 (27, 28), 267 (27, 28, 286),
268 , 269 (28), 275 (106), 276 (114), 279
(126, 127), 280 (127), 283 (143), 285 (106,
152, 154—156, 158, 159), 287 (155, 165),
298 (279), 299 (279а), 305 (158), 312 (412),
313 (426, 427), 314 (154, 313, 428), 324
(126, 127), 328 (279, 279а), 329(127,
541, 632, 635), 356 (61)
Бочкарева Г. П. 196 (8)
Браз Г. И. 248 (105)
Брайнина Э. М. 213 (163), 295 (244)
Братцев В. А. 288 (170)
Брилкина Т. Г. 38 (18), 201 (34, 35)
Броун А. С. 96 (555, 556)
Брукер А. Б. 191 (82)
Бубнов Ю. И. 322, 323 (473), 362 (60)
Бугаева 3. И. 282, 326, 328 (75)
Бундель Ю. Г. 23 (49), 44 (42), 221 (22),
246 (88)
Бутин К- П. 318 (449)
Валуева 3. П. 103 (629), 253 (135), 281 (137)
Вартанян С. А. 169 (423)
Варшавский С. Л. 121 (83)
Василевская Т. В. 218, 219 (8)
Василейская Н. С. 233 (36), 282.(73 , 74),
300 (290), 328 (633)
Васильева В. Ф. 99 (587)
Величко Ф. К. 14 (11—13), 15 (11), 16 (13),
190 (74), 229 (23), 232 (23, 32), 270 (538)
Венус-Данилова Э. Д. 169 (424—427),
170 (427), 252 (127)
Веретенова Т. И. 148 (272)
Верещагина Н. И. 126, 127 (124), 353(10)
Вертилина Л. К- 395 (56)
Вильчевская В. Д. 121 (85), 123 (93, 94),
166 (85), 167 (93 , 94), 211, 212 (143),
275, 285 (106), 329 (632), 356 (61)
Виноградова А. Д. 172 (440), 310, 311 (410)
Вобецкий М. Ф. 356 (59)
Войтович В. А. 326 (622)
Волькенау Н. А. 31 (106), 123, 168 (95),
209 (109), 244, 250 (60), 257 (158), 276 (114)
314 (313), 329 (541)
ВоронковМ. Г. 96 (555, 556)
Вострухина 3. Н. 127 (130), 297 (267)
Вышинская Л. И. 212 (153), 290 (138)
Вязанкин Н. С. 16 (35), 270 (70, 72а),
* Цифры без скобок обозначают страницы, где даются ссылки на работы авторов,
цифры в скобках — порядковые номера ссылок, приводимые в конце соответствующих
глав.
398
Авторский указатель
282 (70), 289 (175), 299 (280), 326 (72а),
336 (395а, 7956), 373 (320—322), 374
(321, 322)
Гаева Л. А. 269, 274, 293 (62)
Гальберштам М. А. 97 (567, 568)
Гельман Н. Э. 391 (И, 15, 16), 392 (15, 16),
394 (11)
Гембицкий П. А, 135 (208)., 172 (208, 440)
Герштейн Н. А. 369 (159)
Гиллер С. А. 94 (497), 301 (300)
Гинсбург В. А. 354 (206), 375 (214а)
Гладштейн Б. М. 133 (170), 172 (170)
Гладышев Е. Н. 16 (35), 336 (7956), 373
(320, 322), 374 (322)
Глушкова В. П. 66, 96 (173), 209 (130—132),
210 (130—132), 211 (131), 286 (163, 164)
Глушнев Н. Ф. 182, 185—187 (3)
Годовиков Н. Н. 53 (6)
Годовикова Т. И. 53, 56 (22), 63, 65 (147),
68 (147, 198), 76, 82, 83, 95 (147), 131 —
133, 136 (159), 139 (227), 141,142 (238), 146
(227),* 157 (238), 172 (227), 376 (226)
Голованева А. Ф. 375 (214а)
Головин Р. В. 103 (624, 627), 253 (137)
Голуб А. Н. 365 (115а)
Голубева Е. И. 19, 22, 23 (12), 168 (422),
211 (145), 229 (18), 246, 247, 259 (80),
285 (158), 299 (279а), 305 (158), 328 (279а)
Гольдфарб Я. Л. 96, 102'(551)
Гощииский С. 169 (429)
Горст А. Г. 362 (61)
Горушкииа Г. И. 96, 102 (551)
Грандберг И. И. 57 (100)
Грачева Е. П. 369 (155, 156)
Грденич Д. И (72, 73)
Грибов Б. Г. .139 (227), 141, 142 (238), 146
(227), 157 (238), 172 (227)
Грибченко Е. А. 183 (7, 9)
Григорович П. 362 (50)
ГригорянА. А. 170 (431)
Гробов Л. Н. 221, 222 (23)
Губен И. 168 (417)
Гурикова Т. Г. 336 (7956)
Гурьянова Т. П. 276 (112в)
Гуськова А. Н. 121 (86), 209, 210 (123),
255, 259, 260 (151), 279, 324 (126)
Дангян М. Т. 154 (451, 452)
Дашунии В. М. 128, 129, 170 (140), 256
(154)
Дворко Г. Ф. 168, 169 (411), 171 (411, 432)
Дворянцева Г. Г. 11 (25), 213 (163), 255
(143), 287 (166)
Делинская Е. Д. 66, 96 (173)
Добромильская И. М. 54 (55)
Докунихин И. С. 269, 274 , 293 (62)
Долгопольский И. М. 54 (55)
Дрозд В. Н. 199 (16, 17), 253, 281 (136)
Дружков О. Н. 264 (26), 299 (281а), 324.
326 (539а)
Дубовицкий В. А. 127 (132), 297 (268)
Дубов С. С. 11 (38, 68)
Дьячковская А. С. 379 (274)
Евтихеев Л. Н. 356 (59)
Елисеева Л. В. 149 (274)
Емельянова Л. И. 310 (409, 410), 311 (410)
Е молаев М. 377 (239)
Епифанский П. Ф. 182, 185—187 (3)
Жадина М. А. 117,126, 128 (41), 150, 151 (41„
137)
Жданов Ю. А. 142 (242, 243)
Женодарова С. М. 147 (270)
Жильцов С. Ф. 264(1—26), 299 (281а),
324 (2а)
Житкова Л. А. 302, 315 (308)
Залесская В. А. 11 (36)
Замятина В. А. 166, 168 (395)
Запевалова Г. Н. 326 (621)
Заревич Т. С. 118, 145 (50)
Затеев Б. Г. 185, 190 (22), 195 (5), 219,.
221 (14), 326 (532в, 612), 327 (612)
Захаркин Л. И. 20, 22 (18), 38 (16), 200 (26),.
309 (386)
Зелинский Н. Д. 134 (194)
Зелмеи 3. Я- 94 (497)
Зефиров Н. С. 120 (75), 134 (198а), 156 (75,.
305, 306), 157 (75, 198а, 305—ЗО6а)„
278 (122а, 1226)
Зинин Н. Н. 14 (6), 330 (647)
Зорина Т. М. 234 (58), 235 (66, 68), 265 (9),
298, 299, 301 (274)
Иванова Н. Л. 127, 150 (136)
Идельчик 3. Г. 325 , 326 (601)
Иоффе Б. В. 11 (9)
Исаева Л. С. 187, 188 (38), 196 (6—8)
Ито И. 11 (34)
КабачникМ. И. 125 (119), 369 (160)
Каваками 333 (733)
Казанский Б. А. 172 (444, 445)
Казицына Л. А. 11 (23), 183 (16)
Калинин Р. В. 301 (299)
Калявин В. А. 240 (29, 32), 241 (29), 322
(478, 481, 481а)
Кан Э. И. 124, 165 (108), 182,188(2), 244 (67)
Канатомо 333 (733)
Каплин Ю. А. 265, 311, 322 (4), 324 (539,.
539а), 326 (4, 539а), 327 ь(539)
Караваева В. А. 369 (159)
Карпов Г. П. 240 (39), 276 (1116, 111г),
323 (483а, 492)
Катаев Е. Г. 96, 97 (520)
Качкурова И. Я- 335 (800)
Кикоть Б. С. 183 (16)
Ким Дяй Гир 172 (441)
Киселева Т. М. 29 (91,92), 229 (10, 11, 20),.
265, 270 (16), 324, 325 (553), 335 (799),
336 (553)
Китайгородский А. И. 11 (71—75), 166
(400, 401)
Клебанский А. Л. 166 (403)
Климова В. А. 391,392 (12—14)
Кнолль П. Г. 123 (92), 240 (25), 321, 322'
. (465, 466)
Кнунянц И. Л. 27 (81), 133, 138 (172), 229,
230 (21), 268 (40), 283 (144), 292 (40, 144),
319 (144 , 457)
Ковредов А. Н. 229 (18), 299, 328 (279а>
Козлов В. В. 90 (473), 185 (46—48), 188_
189 (46), 203 (62), 269, 293 (59—61)
Козминская Т. К. 293 (221)
Колобова И. Е. 104 (631)
Коренман И. М. 379 (274), 390 (5)
Кориева С. П. 289 (175)
Авторский указатель
399
Королевам. Н. 44 (51), 326 (617)
Корольченко Г. А. 142 (242, 243)
Коршак В. В. 36 (1а), 166, 168 (395)
Коршун М. О. 391 (11—16), 392 (12—16),
394 (11,42,43)
Коршунов И. А. 326 (623), 327 (625), 395 (56)
Кост А. Н. 57 (100), 293 (229)
Костин В. Н. 135 (208), 172 (208, 434—442),
246 (76, 85, 86), 301 (295), 324 (534)
Котон М. М. 29 (91, 92), 42 (77), 229 (10, 11,
20), 234 (54—58, 60—64), 235 (65—68),
265 (8, 9, 15, 16), 269 (63), 270 (16, 68,
69), 298, 299 (274), 301 (274, 292—294,
296—298), 302 (292), 303 (294, 317), 315
(418), 324 (418, 536, 540, 553), 325 (553),
327 (292), 335 (799), 336 (553)
Кочетков А. К. 2П (140, 142), 285 (153)
Кочетков Н. К. 20 (21), 54 (61), 128 (138—
140), 129 (139, 140), 167, 169 (138), 170
(139, 140), 256 (153, 154), 297 (271), 319
(456), 353, 355 (3)
Кочеткова Н. С. 11 (25), 115(16, 19), 116
(19), 132 (16), 210 (139), 211 (139), 280,
281 (132), 287 (166)
Кочешков К. А. 33 (122), 66, 96 (173), 117
(38), 188 (60), 197 (1), 209 (95—97, 112,
130—132), 210 (95, 112, 130—132), 211 (1,
95, 112, 131), 231 (28), 267 (37, 38), 286,
(163, 164), 289 (176), 290 (177—178а),
293 (221), 296 (261), 302 (307, 308, 314,
423), 305 (329), 308 (376), 311 (307),
312 (307,423 , 424), 313 (423—425), 314
(314, 423), 315 (308 , 314, 430)
Кравцов Д. Н. 234, 235 (48), 335 (800—803)
Крайц 3. С. 127 (131, 133), 297 (269, 270)
Крамер К. Щ. 393 (16а)
Кретов А. Е. 15 (27)
.Крешникова 3. В. 395 (51)
Криворучко Р. М. 142 (242)
Крицкая И. И. 219 (13), 254 (139)
КронродН. Я- 198 (15)
Круглова Н. В. 266, 267 (27)
Кубаля И. 169 (428 , 430)
Кубасская Л. А. 142 (242, 243)
Кудрявцев Л. Ф. 264 (1—2а), 324 (2а, 539,
5396), 327 (539)
Кудрявцева Т. А. 16 (33), 209 (108), 285
(160)
Кудряшова Н. И. 96 (557)
Кульбовская Н. К. 369 (156)
Куреньгина Т. Н. 96 (558)
Курсанов Н. И. 44 (40)
Кучеров М, Г. 123 (96), 139, 141 (226), 165,
168 (96) \
Лаба В. И. 369 (155)
Лабузов С. М. 234 (53)
Лавров В. 307 (349)
Лавровская Е. В. 394 (42)
Лаптев Н. Г. 317 (434, 442), 330 (442)
Латяева В. Н. 16 (34), 44 (37), 212 (153),
221 (28, 29), 290 (138), 325, 326 (584)
Лебедев О. В. 172 (443)
Лебедева А. И. 393 (16а)
Левин Б. В. 191 (82)
Левина Р. Я- 53 (6), 133 (170), 135 (208),
172 (170, 208 , 434—443), 246 (76, 85, 86),
301 (295), 324 (534)
Ли Вей-ган 27 (81), 229 , 230 (21), 268 (40k
283 (144), 292 (40, 144), 319 (144)
Ловцова А. НТ 232 (39а), 233 (38, 39а), 268,
280 (39а)
Логинов Ю. Н. 356 (59)
Лодочникова Е. И. 290 (177—178а)
Лосева А. С. 192 (84—86), 193 (86)
Лукевиц 3. 301 (300)
Лукина М. Ю. 172 (444, 445)
Лукьянова И. Г. 203, 204 (65)
Лунева Л. К- 36 (1а)
Луценко И. Ф. 11 (23), 99 (598), 116 (35),
117 (41), 126 (41, 124—126), 127 (124,
125, 127, 128, 131—136), 128 (41), 129
(146, 147), 135, 138 (147), 139 (146, 147),
140 (147), 145 (146), 146 (124), 147 (127,
128, 268), 148 (128, 271), 149 (146, 273, 274),
150 (41, 136, 137), 151 (41, 137, 281), 152
(273, 281), 153, 154 (135), 159 (268),
185 (53), 205 70), 234 (47), 241 (52), 244
(68), 245 (68), 255 (152), 256 (68,
157), 257 (157), 274 (96), 278 (124), 280-
(128, 129), 288 (170, 171), 289 (171),
296 (264, 266), 297 (128, 129, 264, 266,
268—270), 298 (129), 306, 312, 315 (413),
353 (9, 10, 17), 354 (9), 378 (265)
Лу Цзин-чжу 23 (49), 44 (42), 239 (12, 16,
17), 240 (12, 23), 246 (88, 89), 248 (16, 17,
88), 253 (12, 16)
Лысенко Н. И. 367 (140)
Лю Гын-ман 67 (180)
Ляпота Л. А. 147 (270)
Майер Н. А. 217(1, 2), 218 (1—4, 9а, 19),
219 (2, 3, 10—12), 220 (2, 10, 15, 17—20),
221 (22)
Макаров С. П. 375 (214а)
Макарова Л. Г. 100 (604), 183 (4—7, 9, 10),
185 (30, 36), 186 (30), 187 (36), 188, 189
(30), 195, 196 (2, 3), 204 (68), 209 (124,
129), 213 (164), 244, 245, 260 (74), 308-
(378), 310 (409, 410), 311 (410), 317 (435),
330 (644), 355 (55)
Максакова Т. П. 390 (5)
Малиновский М. С. 296 (260), 333 (682),
355 (36, 37)
Малыгина Н. И. 395 (51,56)
Малышева А. В. 16 (34)
Малянов В. А. 240 (39), 323 (483а, 492)
Манулкин 3. М. 29 (85), 209, 210, 212 (100),
293 (222, 223), 298 (276)
Манчинова 3. Н. 325 (582)
Мардалейшвили Р. Е. 239 , 240 (4, 5)
Мартынова В. Ф. 234 (59, 61—64), 235 (69),
324(536), 270 (68)
Мартынова Л. Л. 375 (214а)
Мастрюкова Т. А. 369(160)
Матвеева М. К. 183 (4, 5, 7)
Материкова Р. Б. 11 (25), 210, 211 (139),
280, 281 (132), 287 (166)
Матяш Л. Ф. 355 (53а)
Маштак Н. И. 355 (37)
Мацоян С. Г. 169 (423)
Мезенцова Н. 172 (443)
Мельников Н. Н. 25 (65), 41 (13), 42 (13,
17), 43 (17), 249, 250 (112), 259 (171, 172),
260 (171), 286(162), 294 (233), 296 (255)
Меншуткин Н. А. 377 (239, 248)
Миклухин Г. П. 327 (624)
400
Авторский указатель
Митрофанова Е. В. 44 (46), 270 (72а), 299
(280), 309 (394), 326 (72а)
Михайлов Б. М. 369 (158)
Морачевский А. Г. 11 (9)
Мяков В. Н. 326 (532в)
Надь М. М. 209 (96), 289 (76), 302, 311,
312 (307)
Назаркова Н. П. 325, 326 (599)
Назарова Л. М. 320 (462, 463)
Незговоров Л. Ф. 319 (457)
Несмеянов А. Н. 11 (23, 47), 14 (11, 14),
15 (11, 14, 28), 16 (14, 33), 19 (12), 20 (20,
21), 22, 23 (12), 31 (99, 101, 102, 105, 106),
53 (15), 54 (61), 58, 60 (107), 75 , 76, 79
(276), 93 (488), 99 (598), 100 (604), 103
(624, 627,629), 104(631), 115 (19), 116 (19,
30, 34), 117 (34, 38, 39, 41), 118 (34, 39, 50),
120 (80), 121 (39, 80 , 84—86), 123 (90—
95), 124 (111, 113, 115); 125(118—121),
126 (41, 123—126), 127 (124, 125, 127—
129, 132—134), 128 (41, 138—140),
129 (139, 140, 146, 147), 134 (34), 135 (30,
147, 201), 138 (30, 34, 147), 139 (146, 147),
140 (147), 145 (50, 146, 262), 146 (124),
147 (127,128 , 268), 159 (268), 148 (128),
149 (146), 150 (41), 153 (362), 166 (80,
.84, 85), 167 (93 , 94, 138, 407), 168 (95,
419, 420, 422), 169 (138), 170 (139, 140),
172 (444, 445), 182 (1—3), 183 (5, 6, 8,
12), 185 (1, 3, 12, 30, 35,36,44,53),
186 (1, 3, 30), 187 (3, 35, 36, 38), 188 (2,
.30, 38, 55, 60, 67), 189( 12,30), 190(67,
70), 191 (75), 192 (84—86), 193 (86), 195
(2,3), 196 (2, 3, 6—8), 197 (1), 1)9 (16, 17),
200 (30), 203 (65), 204 (65 , 68), 205 (70),
209 (95, 101, 103—109, 123, 129), 210 (95,
101, 123, 139, 166), 211 (1, 95,103, 104,
139—145, 147), 212 (143), 213 (164),
219 (13), 229 (18, 23), 231 (28), 232' (23,
30, 35), 234, 235 (48), 239 (1—4), 240
(1—4, 23), 241 (52), 244 (60 , 63 , 67 , 68,
72, 74), 245 (68, 74), 246 (80, 84, 94),
247 (80), 250, 251 (60), 253 (72, 135—137),
254 (139), 255 (143, 149, 151—154), 256
(68, 152), 257 (158), 258 (84), 259 (80,
151, 173), 260 (74, 151), 266 (27, 28), 267
(27, 28, 286, 37), 268, 269 (28), 270 (538),
274 (93, 94, 96), 275 (106), 276 (114), 278
(1241 279 (126, 127), 280 (127—129, 132),
281 (131, 135—137), 285 (106, 152—154,
156—158, 160), 288 (170), 293 , 294 (231),
295 (243—245), 296 (231, 264—266),
297 (128, 129, 264—266, 268, 270, 271),
298 (129, 272), 299 (279а, 281, 282), 302
(281,314,423), 305 (158), 306 (344), 308
(376 , 378), 309 (396), 310 (396, 410), 311
(410), 312 (412, 423, 424), 313(423—427),
314 (154, 313,314,423,428), 315 (314),
317 (435), 318 (452, 455), 319 (456), 321
(466), 322 (466, 472), 324 (126, 127),
327(281), 328(279а, 282), 329 (127), 541,
632, 635), 330(644), 335 (801, 802), 336
(803, 804), .353 (3, 6, 10, 17), 355(3, 35,
40, 55), 356 (61)
Несмеянов Н. А. 241(51), 153(362)
Несмеянова О. А. 103 (624), 253 (137)
281 (135), 293, 294, 296 (231), 299, 302 (281),
304, 305 (323), 327 (281)
Нефедов В. Д. 25 (68), 240 (26, 27, 35),
322 (474, 475), 323 (484), 333 (738)
Никитина Л. А. 233 (36)
Никонова Л. А. 199 (17), 253, 281 (136)
Новаторов Н. Ф. 326 (607, 610)
Новиков В. М. 153 (362)
Новиков С. С. И (19а), 53, 56 (22), 63,
65 (147), 68 (147, 198), 76, 82, 83, 95 (147),
131 (159), 132 (159, 159а), 133 (159), 136
(159, 159а), 139(227), 141, 142(238), 146
(227), 157 (238), 172 (227), 376 (226)
Новикова Н. В. 31 (99, 101, 102), 211 (144,
146), 168 (419—421), 209 (105, 106), 246
(79), 285 (154, 156, 157, 159), 313 (427),
314 (154, 428)
Новикова Н. Н. 188 (55), 309, 310 (396)
Ногина О. В. 121, 166 (87)
Обреимова Л. И. 293 (229)
Овчинникова А. Г. 190 (68)
Огата И. 68, 83, 90 (192а), 91 (192, 192а),
92 (192а)
Ольдекоп Ю. А. 44 (37, 51), 217 (1, 2),
218 (1—6, 8, 9а, 19), 219(2, 3, 5, 8, 10—
12), 220 (2, 6, 10, 15—20), 221 (6, 21—23,
25, 29), 222 (23),270 (71), 298 (277, 278), 325
(277, 278, 582—584, 586—590, 595, 596,
598—603), 326 (277, 278, 583, 584, 586—
601, 617, 619), 327 (626—628), 332 (627),
333 (619)
Орт Г. 97 (569)
Орлова А. А. 326 (623), 327 (625)
Осанова Н. А. 212 (155—157)
Осберг Э. Г. 298, 299, 301 (274)
Осипова М. А. 167 (407), 211 (147), 229 (14),
259 (173), 287 (165), 314 (428)
Остапчук Г. М. 321 (467, 468)
Охлобыстин О. Ю. 20, 22 (18), 38 (16),
309 (386)
Павловская И. В. 375 (214а)
Панов Е. М. 209—211 (112), 290 (177—178а)
Пахомов В. И. 11 (87а, 876), 173 (446, 447)
Первеев Ф. Я. 96 (557, 558)
Первова Е. Я- 133, 138 (172)
Перевалова Э. Г. 103 (624, 627), 127 (129),
203, 204 (65), 253 (137), 281 (135), 293,
294(231), 296 (231, 265), 297 (265), 299,
302 (281), 304 (323), 327 (281)
Петров А. А. 280 (131)
Петрович П. И. 11 (36), 67 (184), 185 (31,
39), 188(39), 203(52), 244, 259(71), 317
(440, 441), 355 (57)
Петрунькин В. Е. 367 (140)
Петухов Г. Г. 42 (19), 44 (46), 185, 190 (22),
195 (5), 219, 221 (14), 264 (1—26), 265
(281а), 301 (299), 309 (394), 311, 322 (4),
324 (2а, 539—5396), 325 (585), 326 (4,
539а, 585, 604—606, 612, 613, 615, 622),
327 (281а, 539, 585, 612, 624), 330, 332
(657, 658)
Печерская К- А. 20 (20), 58, 60 (107), 135
(201), 318 (452, 455)
Пикина Е. И. 209 (97)
Пинкина Л. П. 319 (457)
Пищнмука П. С. 55 (78)
Плотникова Г. И. 369 (152—154, 157)
Повх Г. С. 53 (15), 191 (75), 274 (93),
Поддубная С. С. 15 (28), 239, 240 (1, 2)
Авторский указатель
401
Подкопаева К. Н. 54 (55)
Половянюк И. В. 135, 187 (36)
Полякова 3. П. 380 (306)
Поляновский О. Л. 336 (815)
Потросов В. И. 308 (376)
Прайснер Б. 322 (488а)
Прегль Ф. 393, 395 (27)
Приказчикова Л. П. 120 (75), 134 (198а),
156 (75, 305), 157 (75, 198а, 305, 306а),
278 (122а, 1226)
Прилежаева Е. Н. 53 (31а), 363 (92—94),
367 (141, 142), 368 (92, 142, 144), 369 (92,
94, 141, 142, 144, 150, 151, 159—160)
Прокофьев М. А. 380 (306)
Птицына О. А. 195 (4), 196 (4, 8)
ПузыреваВ. П. 302, 314, 315 (314)
Разуваев Г. А. 14 (15), 16 (34, 35), 42 (19),
44 (46, 51), 185 (20, 22), 190 (20, 22,
68), 195(5), 201 (34—36), 212 (153, 155—
157), 217 (1, 2), 218 (1, 2, 5, 7),
219 (2, 5, 14), 220 (2, 16), 221 (14, 21—24,
25, 27, 29), 222 (23), 233 (36), 264 (1—
26), 265 (4, 12), 270 (70—72а), 276 (109),
282 (70, 73—75), 289 (175), 290 (138, 180),
298 (275, 277, 278), 299 (280, 281а), 300
(290), 301 (292, 294, 299), 302 (292), 303
(294, 317), 309 (394), 311 (4), 315 (418),
322 (4), 324 (2а, 418, 539—5396), 325 (277,
278, 582—589, 591—599, 602), 326 (4, 75,
278, 5326, 539а, 583—Й89, 591, 592, 594,
598, 599, 604, 605, 607, 610, 612, 613,
617—622), 326 (72а, 277), 327 (292, 539,
585, 612, 624, 626, 630, 631), 328 (75),
330, 332 (620), 333 (619), 336 (795а, 7976),
373 (320—322), 374 (321, 322)
Разумовский В. В. 127(130), 297 (267)
Ралль К. Б. 280 (131)
Ратман Ф. Г. 11 (101)
Рашидян Л. Г. 154 (451) ,
Рекашева А. Ф. 327 (624)
Реутов О. А. 14 (14, 16), 15 (16, 28—30),
16 (14), 23 (49), 26 (77), 44 (42), 120 (73),
121 (81), 123 (92), 134 (73), 153 (362), 183
(11, 16), 188, 190 (67), 192 (84-86), 193
(86), 195 (4), 196 (4, 8), 232 (39а), 233 (38,
39а, 41—43), 239 (1—12; 16, 17), 240 (1 —
10, 12, 23—25, 29—32, 37—40), 241 (29,
51), 244 (30), 246(88, 89), 248 (16, 17,
88), 253 (12, 16), 255 (148, 149), 256 (6—
11), 257 (159), 258 (148), 259 (174), 266
(26), 268 (39а), 276 (109а, 110, 1116, 111г,
112в, 113а,. 1136), 280 (39а), 298 (272),
321 (464—468), 318 (449), 319 (429), 322
(465, 466, 472, 473, 478—481а, 487а—
488, 489), 322 (487а), 323 (473, 480, 483а,
491, 492, 494)
Родионов В. М. 207 (85)
Розенштейн С. М. 127 (130), 297 (267)
Рокицкая М. С. 25 (65), 41, 42 (13), 249,
250 (112), 259 (171, 172), 260 (171), 286
(162), 294 (233), 296 (255)
Романенко Л. Й. 365 (115а)
Руденко Г. А. 11 (23)
Русанов Д. Е. 317 (443)
Рябокобылко Ю. С. 322 (488)
Савельева И. С. 19, 22, 23 (12), 28, 29 (83),
167 (407), 168 (422), 211 (145, 147), 246,
247 (80), 259 (80, 173), 266 (27, 28),
267 (27, 28, 286), 268, 269 (28), 285, 305
(158)
Савицкий А. В. 276 (109)
Савостьянова И. А. 132, 136 (159а)
Садовая Н. К- 97 (567, 568)
Сазонова В. А. 198 (15), 199 (16, 17), 200
(30), 253, 281 (136)
Светланова Т. Б. 276 (109а)
Светлов Б. Я- 362 (61)
Сергеева О. М. 42 (19)
Сердюк С. Р. 28, 29 (83)
Сивкова Р. И. 116 (35), 353, 354 (9)
Симукова Н. Д. 103 (627)
Синдо X. 11 (34)
Синотова Е. Н. 25 (68), 240 (26—28, 35),
322 (474, 475, 477), 323 (484), 333 (738),
356 (59)
Сколдинов А. П. 296 (261)
Сладков А. М. 36 (1а)
Словецкий В. И. 11 (19а)
Смирнов К- М. 354 (.206)
Смирнов Р. Н. 66, 67 (172)
Смирнов-Замков И. В. 121, 124 (88)
Смолин Д. Д. 185 (48), 203 (62)
Смолина Т. А. 240 (29, 32, 38), 241 (29),
322 (473, 478, 481, 481а), 323 (473, 491,
494)
Соколов В. И. 120, 134 (73), 240 (30, 37),
244 (30), 322 (479, 487а, 488)
Соколов Е. 43 (32), 308 (374)
Сорокин Ю. А. 221 (21)
Станко В. И. 200 (26)
Старовский О. В. 103 (627)
Степанов В. М. 355 (53а)
Стёрлин Р. Н. П (38, 68), 27 (81), 229,
230 (21), 268 (40), 283 (144), 292 (40, 144),
319 (144, 457)
Стручков Ю. Т. 11 (74)
Субботина А. И. 326 (618, 620), 330, 332 (620)
Талалаева Т. В. 33 (122), 209 (97), 305 (329)
Тартаковский В. А. 11(192), 53, 56 (22),
63, 65 (147), 68 (147, 198), 76, 82, 83,
95 (147), 131 (159), 132 (159, 159а), 133
(159), 136 (159, 159а), 139‘(227), 141,
142 (238), 146 (227), 157 (238), 172 (227,
436, 437, 439), 246 (86), 301 (295), 324
(534), 376 (226)
Татаренко А. Н. 29 (85), 293 (222)
Твердова В. И. 221 (21)
Терентьев А. П. 293 (229)
Терман Л. М. 218 (7)
Тер-Саркисян Г. С. 369 (158)
Тимошенко 3. Ф. 42 (19)
Титов А. И. 317/(434, 438, 442, 443), 330(442)
Титов В. Д. 166 (403)
Токарева Ф. А. 117, 118, 121 (39), 190 (70),
232 (35), 274 (94)
Толстая Т. П. 183 (8), 196 (6—8)
Торопова Е. М. 185 (44), 244, 253 (72),
355 (35)
Трещова Е. Г. 172 (208, 442)
Тудьчинский В. Б. 333 (682), 355 (36)
Туманова 3. М. 127, 147 (127), 296, 297 (266)
Турикова Т. Г. 373, 374 (322)
Турчинский М. Ф. 196 (8)
Тюленева В. В. 133, 138 (172), 378 (265)
402
Авторский указатель
Уварова Н. И. 367 (141), 369 (141, 160)
Углова Э. В. 26 (77), 240 (24, 39), 259 (174),
276 (109а), 323 (483а, 492)
Укай 333(733)
Устынюк Т. К. 172(441)
Усубакунов М. 55, 60 (81), 266, 302 (47)
У Ян-цей 240 (23, 25, 31), 321 (464, 465),
322 (465, 473, 480), 323(473, 480)
Фабрицы А. 169 (424—430), 170 (427)
Феднева Е. И. 121 (81)
Федоров Б. П. 96, 102 (551)
Федорова А. М. 207 (85)
Федотов М. С. 327 (629—631)
Федотова Е. И. 185, 190(20, 21)
Филатов А. С. 375 (214а)
Филиппенко Л. Р. 257 (159)
Фишер Г. 97 (569)
Флегонтов А. М. 132, 185—187 (3
Флоринский Ф. Т. 29 (91), 229 (20), 335 j* 5j
Фосс В. Л. 117, 126 (41), 127 (135, 136),
128 (41), 150 (41, 136, 137), 151 (41, 137,
281), 152 (281), 153 (135), 154 (135), 244,
245, 256 (68), 274 (96), 280, 297, 298 (129)
Фрейдлина Р. X. 14(11—13), 15(11), 16
(13), 115 (16, 19), 116 (19, 30, 34), 117 (34,
38, 39), 118 (34, 39), 120 (80), 121 (39, 80,
82, 87), 124 (108, 111), 132 (16), 134 (34),
135 (30), 138 (30 , 34), 161 (325), 165 (108),
166 (80, 82, 87), 190 (70, 74), 209 (107),
211 (140, 142), 213 (163), 229 (23), 231 (28),
232 (23, 30, 32, 35), 270 (538), 274 (94),
285 (153), 295 (243—245), 353 (6)
Фролов Н. Я. 240 (26), 322 (474)
Хасапов Б. Н. 312, 315(413)
Хаскина Е. Е. 293 (229)
Хмельницкий Л. И. 96 (527)
Ходзе М. 68, 83, 90—92 (192а)
Хомутов Р. М. 126 (126), 127 (128, 132, 134),
147 (128, 268), 148 (128, 271), 149 (274),
159 (268), 241 (52), 255, 256 (152), 280
(128), 297 (128, 268), 306 (344), 353 (17)
Хорлина М. Я- 192 (85, 86), 193 (86)
Хрипунов В. П. 42 (19)
Ху Хун-вен 195, 196 (4), 240 (38), 323 (491,
494)
Цигин Б. М. 212(155)
Цой И-рен 67 (180)
Цымбал Л. В. 53 (31а), 369 (159а)
Чалов В. П. 253 (129)
Чаповский Ю. А. 213(164)
Челинцев Г. В. 124(114)
Челобов\Ф. М. 11 (68)
Чернов И. А. 234 (60), 265 (13, 14)
Чугаев Л. Н. 380 (287)
Чуковская Е. Ц. 161 (325)
Чулаев Н. П. 212(155)
Чумачейко М. Н. 394 (43)
Чуходжян Г. А. 169 (423)
Шабарова 3. А. 380 (306)
Шайдерова Л. П. 156, 157.(306)
Шамшурин А. А. 135, 144 (202)
Шаназаров К- С. 172 (438 , 442)
Шапиро Э. С. 363 (92—94), 368 (92, 144),
369 (92, 94, 144, 150, 151)
Шапшинская Л. А. 353 (15)
Шацкая Р. X. 75, 76, 79(276), 145(262),
246 (94), 299 (282), 328 (282), 355(40)
Шевелева Н. С. 391 (11, 16), 392 (16),
394 (11)
Шевердина Н. И. 302, 315 (308)
Шевлягина Е. А. 15 (29а)
Шейнкер Ю. Н. 11 (25), 99(587), 287(166)
Шепелева Р. И. 125 (121), 211 (141), 285 (152)
Шилов Е. А. 121 (88), 124 (88, 112), 168,
169, 171(411)
Шишков Л. 361 (33)
Шлезингер Н. А. 380 (287)
Шостаковский М. Ф. 53 (31а), 363 (92—94),
367 (141, 142), 368 (92, 142, 144), 369 (92,
94, .141, 142, 144, 150—157, 159—160)
Шостаковский С. М. 135, 172 (208)
Шпанский В. А. 375 (214а)
Шубенко М. А. 14, 15 (15), 324 (5396), 325
(591—594), 326 (591, 592, 594, 622)
Щекотихин А. И. 375 (214а)
Щербаков А. 43 (25), 308 (371)
Эскин И. Т. 185 (35, 50, 63), 187 (35),
188 (63), 244 (62—64, 183), 245 (64), 313
(425)
Ю Гын-ман 63 (124)
Юркевич А. М. 293 (229)
Юрко Д. Г. 333 (682), 355 (36, 37)
Юркова Е. И. 149, 152 (273), 234 (47), 256,
257 (157)
Кфьев Ю. К. 96 (527), 97 (567, 568), 120 (75),
134 (198а), 156 (75, 305, 306), 157 (75,
305—306а), 278 (122а, 1226)
Юсупов Ф. 29 (85), 293 (222)
Юфа П. А. 188 (59), 291- (189)
Ягупольский Л. М. 188 (59), 291 (189)
Якубович А. Я. 127 (130), 297 (267), 354
(206), 375 (214а)
Якушкина Л. М. 369 (155)
Ямамото 333 (733)
Яременко Н. Е. 362 (61)
Яшунский В. Г. 99 (587)
Abderhalden Е. 159 (316)
Abe Н. 162 (382)
Abelmann А. 90 (471), 159 (316)
Abercrombie М. J. 11 (83), 116, 134, 135,
155, 156 (28), 353 (5)
Acree S. F. 306 (341)
Adams С. Е. 201 (39)
Adams R. J. 144 (45,46), 53 (25), 117,
145 (45), 209 (94), 230, 231 (27), 241,
244, 248 (18), 334 (739)
Agrawal А. К. 365 (111а)
Ahmad S. Z., 78 (326, 327)
Ahmed А. К. 273 (85а)
Ainley A. D. 198 (11), 209 (120)
Ajuto А. 82 (389)
Alan Е. 294 (340), 316 (240)
Albert А. 85 (455)
Alden R. Е. 62 (115)
Aldrich Р. Е. 133, 134, 138, 139 (175),
324 (130)
Aldridge С. L. 152 (282)
Alei М. 307, 316 (352),
Alexander R. А. 141 (233)
Ali I. J. 85, 88 (453)
Авторский указатель .
403*
Allan J, L. H. 54 (42)
Allen A. О. 325 (570)
Allen E. R. 119 (58, 59)
Allen P. W. 11 (123)
Allpress C. F. 293 (224)
Allred A. L. 309, 320 (368a)
Almany A. 333 (680)
Amstutz E. D. 94 (496)
Andersen C. N. 96, 97 (531)
Andersen H. P. 74 (237), 334, 335 (774)
Anderson A. G. 67 (176)
Anderson L. G. 183 (15)
Anderson M. M. 134 (180)
Andre C. 375, 376 (213)
Andreas H. 154 (298), 155 (300)
Andrews D. H. 11 (125)
Anet F. A. 212(161, 162)
Angel T. H. 19, 21, 25(6)
Angelico F. 361 (26)
Anmo T. 335 (793)
Annopoulos J. V. 366 (131a)
Ansoloni A. 240 (34), 322 (483)
Antar M. F. 367 (139)
Arai T. 224, 225 (3)
Araki S. 273(80)
Argus M. 202 (165) 1
Armstrong D. A. 324 (522)
Armstrong R. S. 11 (105)
Arndt F. 81 (368)
Aronhein B. 289 (174)
Asahina J. 380(288)
Ash A. S. F. 146 (267)
Ashley J. N. 209 (113)
Asinger F. 133 (342)
Asundi R. K. 11 (12)
Augenstad-Jensen H. 19, 30 (14), 228 (2),
246, 248, 250, 252 (97)
Auld S. J. M. 380 (267)
Auld S. M. 55, 56, 60 (84), 359, 360 (5),
378 (267)
Auram M. 44 (39)
Austin P. R. 32 (109)
Aya M, 395 (74)
Babcock D. 203 (56)
Bachman G. B. 65 (165), 281 (134)
Bachman W. E. 30 (97) •
Baenziger N. C. 141 (233)
Bahel S. C. 75 (275), 83 (421)
Bahr G. 27 (78), 209 (98), 229(12)
Baig M. M. 15(24,-25)
Baine O, 305 (338)
Baizer M. M. 62 (116), 203 (61)
Baker A. W.'120, 134, 135(74), 248, 249
(106a), 353 (9a)
Baker B. R. 133, 135, 139—141, 145, 153
(167), 161 (370), 380 (299—301, 303, 307)
Baker W. 66, 69(174), 279 (125)
Balbiano L. 129 (142, 143), 134 (143, 193),
136 (211), 144 (142, 143, 211, 255)
Baluja M. G. 333(680, 681)
Bamberger E. 71, 73 (230), 317 (431, 437),
355 (51)
Bamford С. H. 309 (383)
Banerjee A. 75 (274)
Banks A. A. 15(18)
Bantis A. G. 27 (80)
Banus J. 47 (73), 325 (579)
Bar D. 77(312)
Baranger P. C. 94 (501)
Barbieri P. 54 (51)
Barbieri R. 333 (699a)
Barcza L. 393 (21)
Barduhn A. J. 63(151)
Barnett M. M. 248 (104)
Baroni A. 94 (494), 372 (204)
Barrack L. P. 333 (732)
Barrett A. W. 123 (99—101), 168 (101),,
171 (100, 101)
Bartlett J. N. 393 (35), 395 (76)
Bartocha B. 20 (16), 22 (43), 27 (16, 43),
229 (77), 283(141, 142), 284 (142, 149),
291 (141, 142), 305, 306(142), 309(149,
388, 389)
Barton D. H. R. 301, 310(301)
Barton J. W.-66, 69(174), 279 (125)
Bartz Q. R. 144 (260)
Bashford L. A. 54 (59) 1
Bass K- S. 324 (5326)
Bastani M. G. 185(33)
Baster R. A. 369 (161)
Batorewicz W. 119 (65), 273 (86)
Bauer H. 393 (19)
Bauermeister M. 95, 96, 101 (512), 243,
250(54), 296 (258)
Bauerschmidt H. 98 (577), 153 (283)
Baun W. L. 11 (31, 78)
Baur R. 70 (202)
Bawn С. E. H. 325 (570)
Baye L. 57 (122a)
Bayer A. 316 (430a)
Bean F. R. 197, 198 (7)
Beattie R. W. 203 (53), 249, 250 (109)
Beatty H. A. 236 (72)
Beck W. W. 24 (126), 94 (506), 259 (185),
292 (204)
Becker A. 395 (77)
Becker P. 197 (2), 284 (145, 146)
Beckmann E. 360, 361 (24)
Beckurts H. 365 (103)
Beermann C. 29(86), 291, 292 (193)
Begoon A. 54 (46)
Beinert G. 33 (124)
Beiser W. 95 (508)
Belcher P. 395(46)
Bell F. J. 82 (392)
Belluco U. 206 (75), 209 (137a, 138)
Belmondi G. 322 (483)
Benesch R. 334 (760), 395 (68—70)
Benesch R. E. 334 (760), 395 (68—70)
Benkeser R. A. 65(158), 212 (150, 151)
Bennett G. M. 366, 380 (126)
Berchem R. G. 123, 171 (100)
Berchet G. J. 168 (412), 279 (312)
Berg A. 33(117)
Berg O, W. 115, 120 (27), 273 (81)
Bergmann E. 11 (95)
Bergmann H. 30 (98), 367 (136)
Berman L. 135 (209)
Bernardi A. 82 (393), 83 (393, 431), 90,
91 (472), 258 (156)
Bernstein H. 267 (28a, 39) ,
Berstein R. B. 323, 324 (496)
Berthelot M. 11 (124)
Bertin D. 54 (40)
Bertsch E. 362 (55)
Bevege E. E. 47 (74)
Bezdrik A. 97 (563)
404
Авторский указатель
Bhargava Р. N. 83 (401, 414—417)
Bharucha К. К. 11= (83), 116, 134, 135, 155,
156(28), 353(5)
Bhatia I. S. 335(796)
Bialas J. 42 (21a), 334(781)
Bickelhaupt F. 29, 32 (95), 45 (63), 259 (175),-
303, 304 (318), 305 (318, 330), 306 (330)
Biel J. H. 142 (241)
Bielenberg W. 19, 29, 30 (14), 228(2), 246,
248, 250, 252(97)
Biginelli P. 120, 166 (79)
Biilmann E. 53 (3, 7, 19), 57 (3), 58 (7), 60,
61 (3), 116 (32), 129(145), 131 (236),
135(32), 141 (145, 236), 142 (236), 144
(254), 153 (145, 284), 159 (254), 274
(99, 100)
Bijloo J. D. 96(546)
Bilbo A. I. 284(149), 309(149, 388, 389)
Billinge В. H. M. 226 (14), 240 (21), 323,
324 (497)
Biltz H. 123 (98), 166 (98, 397), 168 (98)
Biltz W. 11 (4)
Bindewaid H. 360 (15)
Binkley S. B. 209(135)
Bird M. L. 372 (205)
Birch S. F. 96 (559), 363 (75)
Birckenbach L. 360, 380(18, 19)
Birks A. M. 129, 135, 139, 150, 158 (149)
Biswas H. G. 332, • 335 (676)
Biszku E, 336 (816)
Blanchard E. P. 14 (37), 259 (186)
Blasi N. 68(130), 292(201)
Blicke F, F. 142 (241), 292 (210)
BlomstrOm D. C. 14 (37), 259 (186)
Elemental F. 92 (481)
Boather С. H. 26 (72)
Bock W. 370(177, 187)
Bocchi C. 83 (426, 432)
Bodamer G. W. 129, 139 (148)
Bodem G. B. 116, 132 (161), 273 (78), 353 (2)
Boedecker F. 77(295)
Boetius M. 394 (39)
Bogert M. T. 96, 97 (530, 531), 250 (122)
Boll A. 390 (4a)
Bonitsch G. 355(27)
Boon-Long N. 74 (259)
Boord C. 363, 366 (88)
Bordeianu С. V. 71 (208, 222), 72 (208),
393 (22)
Bordwell F. G. 203(56)
Borelli V. 365 (119)
Borggard M. 355 (46)
Bergstrom P. 24 (51)
BorisSov A. E. 123 (90)
Borkar 4?. S. 99(597)
Boudakian M. M. 53 (33), 54 (60)
Boyd D. R. J. 11 (35)
Boyer P. D. 336 (813, 814)
Boynton Ch. F. 15 (26)
Brack A. 57, 62 (97), 316 (422) .
Bradley D. C. 362 (72)
Bradley J. N. 325(573)
Bradley W. 82. 83, 87(391)
Bradt W. E. 373 (208)
Braende K. A. 209 (102a)
Braker W. 355 (45)
Brame J. S. 166(399)
Brand R. B, 119(57)
Brandhoff H. 399, 370 (189)
Brandner H. 370(174, 175)
Brandt G. A. R. 363(95)
Brauchitsch M. 33(116)
Braun J. 41, 44 (8, 9), 244 (59)
Braz G. J. 191 (83), 248. (105)
Brayel E. H. 31 (107), 44 (35)
Breest F. 273 (79)
Breitinger D. 53(986), 376 (215a)
Brenans P. 77 (298)
Brennan J. F. 63 (129)
Breuer F. 302 , 306, 307 (306)
Brewster R. Q. 74 (266), 296 (249)
Brieger R. 71 (228), 77 (228, 299), 82, 87 (228)
Brinckman F. E. 20, 27 (16), 283 (141),
289 (172), 291 (141)
Briscoe H. V. A. 54 (59), 95, 97, 101 (514)
Bristow N. W. 380(302)
Brockway L. O. 11 (122)
Brodersen K. 11 (27, 113), 55 (75), 57 (986),
241 (50), 321, 322 (470), 376 (215a), 378
(269), 379 (269, 277), 395 (75)
Brook A. G. 11 (82), 120 (66, 68), 134 (66,
68, 179, 182—184), 136(66), 137(179),
138 (66), 144 (253), -239, 248 (19)
Brooks P. 380 (325)
Brown G. B. 380(292)
Brown H. C. 63(119, 152), 64 (152, 153),
65(119, 152, 163, 164), 66 (164), 67, 71
(182), 75 (119, 269), 76 (269), 77(119),
83 (269)
Brown R, E. 18 (4), 19, 25, 26 (4, 8), 29 (8),
30(4, 8), 308 (363, 366)
Brown W. H. 129, 158 (144)
Bruce W. F. 115, 161 (18), (353)
Bruce-Smyth D. 326(608, 609)
Bruice T. C. 82, 86 (372)
Bruno M. 206 (75)
Bruns G. 381 (314)
Bublitz D. E. 284 (149),. 309 (149, 388)
Buchner E. 53 (13, 14), 62 (14)
Buchtala H. 77 (306)
Buck A. C. 24 (56)
Buckles R. E. 115, 134, 137(15)
Buckton G. B. 14, 37 (4), 252, 254 (125),
292 (217), .305 (333), 309 (379)
Budde W. L. 54 (68), 260 (162)
Budesinsky B. 139 (219)
Burden J. 274, 301, 319 (99a), 356(65).
Burdt H. 25 (64)
Burg A. B. 212 (154), 307(350)
Burlitch J. M. 233 (39, 40), 287 (167, 168,
406a), 288 (168), 289 (167), 306 (340)
Buroni G. 77 (291)
Burt J. 71 (225), 72 (239), 74 (225)
Burtner R. 94 (502), 208 (88)
Burton H. 68 (196)
Burton R. 306 (345)
Busch- G. C. 302, 316 (421)
Buu-HoT N. P. 95 (519), 208 (90), 269 (49)
Byk S. 366 (120) .
Caccia-Bava A. M. 81 (364, 367)
Cadiot P. 54 (49)
Cagniant P. 96 (525, 548—550)
Cahours A. 43 (26, 31), 265 (5), 307^356),
308 (370, 372), 309 (5, 356, 370), 311 (5),
330 (372)
Cais M. 204 (66), 253, 257, 259 (138)
Cajus I. F. 330(636)
Авторский указатель
405
Calingaert G. 236 (72—75)
Calloway N. О. 94 (492)
Calmels G. C. 299(283)
Calvery H. O. 21, 24, 26 (28), 45 (58), 266
(43), 296 (256), 324 (43)
Cambi L. 361 (45)
Campaigns E. 97 (561)
Campbell I. G. M. 185 (43), 294 (238)
Campbell N. 65(166)
Campbell T. W, 294 (236)
Candeli A. 83(151)
Caplier I. 31 (107), 44 (35)
Capodacqua A. 379 (283)
Carani N. 87 (464, 467)
Carey I. 66(170)
Carius 366 (326)
Carlisle С. H. 11 (85)
Carmack M. 62 (116), 203 (61), 317 (436)
Carnahan F. L. 207 (83), 255 (147)
Carothers W. H. 168 (412), 279 (312)
Carpenter C. 141 (233)
Carrara G. 161 (365, 366, 368)
Carrick W. L. 293 (225)
Carrole J. J. 395 (72)
Carson A. S. 11 (129, 137)
Carson E. M. 11 (129)
Carstanjen H. 361 (44)
Carter H. E. 277 (118)
Carter H. V. 324 (523)
Cartlidge J. 119(58)
Caselle J. 54(45)
Casey P. S. 395(72)
Casper J. 291, 292 (192)
Cattalini L. 209(138)
Cattanach J. 11 (120), 324 (517)
Caule E. J. 325 (576)
Cava M. P. 33, 303, 304 (321)
Cechetti B. 82, 87 (395), 97 (573), 100 (616)
Cerfontain H. 240 (36), 322 (485)
Chaix M. 11 (44)
Chakravarti D. 78 (324, 325), 274 (97)
Chalkley L. 74 , 83, 84 (265), 86 (457, 458),
89 (457), 101 (622)
Challenger F. 97 (562), 146 (?"’), 162 (387),
185(42), 197 (14), 198 (11, Л), 199(14),
209 (14, 126), 236 (75), 250 (123), 293 (224),
296 (254), 355(22), 372 (205)
Chamberlin E. 41, 43 (11)
Chambers R. D. 29 (87), 235 (70), 266, 324
(44), 393 (30 , 33)
Chamot E. M. 310 (404)
Chan J. K. 355, 356 (30)
Chapman E. T. 42 (24), 43 (30), 120, 166 (76)
Chappell E. t 324 (523)
Charman H. B.\21 (27), 240 (43), 241 (47),
259 (43), 323 (490, 493)
Charnley T. 11 (119)
Chatt J. 11 (33)
Chattaway F. D. 44, 45 (57)
Chatterjee M. P. 71 (209), 360, 376 (13)
Chaudhari M. B. 207 (77)
Chaudhuri A. K. 153, 158 (450)
Chauveau J. 75(277)
Chen С. H. 150(28)
Chen W. Y. 333 (678)
Cherbuliez E. 84, 85 (448)
Chernick С. I. 11 (127)
Chevreul M. E. 360 (8)
Chigi E. 99(590)
Chilton H. T. Y. 324(525—528, 530)
Chinard F. P. 336(812)
Chiu D. D. 134 (198), 353 (11)
Chote W. J. 94(495)
Christensen C. W. 154, 158 (296)
Christiansen W, G. 72 (247), 79 (343), 355 (45)
Chute W. J. 253 (133), 296(263), 318(446),
356 (60)
Cinneide R. O. 95(509), 208 (89)
Ciocca B. 168(415), 244 (51)
Ciusa R. 95, 97, 102 (507), 379 (282)
Clapham P. 162 (387)
Clapperton E. T. 54(53)
Clarke R. L. 163, 164 (391)
Claus C. 366 (124)
Clemo G. R. 100(600)
Coates G. E. 29 (87), 226 (15), 235(70),
258 (160), 266 (44), 310 (399), 324 (44),
331 (667)
Cochen H. L. 26 (39)
Codington J. E. 380(297, 309)
Coe G. R. 308, 320 (361a)
Coe P. L. 47 (75), 274 (99a), 281 (134a), 301,
319 (99a), 356 (65)
Coffey S. 63 (150), 68 (194), 69 (150), 203
(51), 254(140)
Coffman D. D. 361, 365, 372(98)
Cohn R. 365 (113, 115, 121)
Collin C. J. 134(191)
Colombo 44 (52)
Colonius H. 33 (121)
Connor T. 155(299)
Contardi A. 168 (415), 244 (61)
Conti L. 185 (51, 52)
Coop I. E. 11 (99)
Cope A. C. 141 (234)
Copertini 82, 87 (388)
Corbellini A. 207 (81)
Corbett G. E. 326 (616)
Cordoin M. 367 (138)
Corral G. 333 (680)
Corwin A. H. 265—267 (10, 22), 335(796)
Costa G. 11 (26), 395(52, 53)
Cotton F. A. 11 (57), 119, 130 (52)
Courtot C. 185(33)
Covello M. 101, 103 (621)
Cowan D. Q. 25, 26 (67a), 308 (348)
Cowees E. J, 67 (176)
Cowperthwaite M. 324 (535)
Cox J. T. 11 (52)
Coyle T. D. 11(56)
Cram D. J, 367 (138, 139)
Crane H. I. 310 (405)
Cresswell W. T. 11 (7, 8)
Criegee R.21,22, 26, 27, 30 (42), 290 (279)
Cristol S. J. 53(29), 54(46)
Croatto 240 (34), 322 (483)
Crombie L. 54 (39)
Cropper F. R. 393 (30)
Cross J. M.240 (33), 322, 323 (482), 395 (66)
Cross R. J. 258 (160), 331 (667)
Crossley H. 393 (30)
Crymble C. R. 11 (10)
Culhane P. I. 207 (78)
Cullen W. R. 15(24, 25), 209 (135a)
Cullum T, V. 96 (559)
Cunningham J. P. 324 (507)
Curran В. C. 11 (102, 107)
Curran W. J. 11 (98)
408
Авторский указатель
Curtin D. Y. 147 (269)
Curtis G. W. 395(76)
Curtius Th. 53 (13), . 362 (63)
Czarnomsky 378 (261)
Czempik H. 29 (86), 291, 292 (193)
Padic M. 330 , 331(663)
Dahling W. 37 (10, 13, 14), 38 (10, 13)
Dale J. W. 373 (206)
Dally О. T. 370 (173)
Dann O. 96 (541, 543 , 554)
D’Ans J. 30 (98), 33 (116)
Dansi A. 66 (175)
Darwent B. 325 (543, 545, 565, 569)
Das B. 98 (584)
Das Gupta H. N. 16 (32), 45 (60), 66, 68,
70, 82, 86(169)
Das Gupta S. J. 332, 335 (676)
Das M. N. 119, 120, 129 (62), 134, 135, 139,
143, 145 (187), 153 (62, 450), 158 (450)
Das N. K. 83 (402 , 403) .
Dassler C. G. 370 (183, 190)
Datta S. K. 75 (273, 274)
Davidson N. 74 (221), 85(454), 326 (614),
. 38Q(325)
Davies J. S. H. 368 (143)
Davies W. C. 29(90), 292(211)
Davis T. L. 362 (58)
Davoll J. 380 (292, 295)
Dean A. L. 155(301)
Dean R. A. 96 (559)
Dedering E. F. 96, 97 (553)
Deitz V. R. 336(812)
Dejudicibus I. 379 (279)
De Laitsch D. M. 96 (532)
Dell’Erba C. 333 (677), 373 (328)
Den Hertog H. 100 (602, 603)
Deniges G. 56 (86), 57 (91), 95 (516)
Dennis L. M. 289 (173), 310 (400 , 404)
Derbyshire D. H. 326 (611) ...
Desai R. D. 78 (326, 327)
Desaignes V. 377, 378 (245)
Desgrez C. 73 (250)
Dessy R. E. 11 (61), 19, 20, 25, 27, 28 (15),
29 (88), 41, 46, 47 (14), 54 (68), 70 (289),
235, 236 (71), 240 (42), 241 (40, 42), 258
(161), 260 (162), 266, 267 (23—25), 268 (23,
24), 269 (23, 25), 273 (89a), 307 (358),
308 (361a), 319 (458a), 320 (361a), 322
(471), 366 (122)
Deussen E. 134, 136 (192)
Dewar M. M. 24 (51)
Dewar M. S. 119(54)
Dhar N. 375(210)
Dibeler VSH. 11 (64, 65), 37 (5)
Didelot S. 11 (39)
Diedrich J. L. 97 (561)
Dimroth O. 54, 58, 60 (71), 63 (149), 66
(168), 68 (188, 199), 69 (149, 168), 70 (168),
. 71, 72 (149, 199), 73 (71), 74,(71, 263),
75, 77, 82, 83 (71), 84 (263), 90 (71, 168),
91 (71), 92 (168), 93 (71), 95 (148), 246 (92,
95), 253 (92), 281 (1-33), 290(179), 330
(637), 355 (23, 33 , 38, 39), 376 (222)
Dimroth P. 21, 22, 26, 27, 30(42)
Dirscherl W. 145 (263)
Ditsch L. T. 119 (63—65), 120 (64), 273 (84,
86)
Dittmar P. 38 (20), 188 (56), 309 (395)
Dittmer K. 96 (542)
Divers E. 361 (40)
Dixon В. E. 11 (118)
Doadrio A. 200 (29, 31)
Dobson N. A. 53 (35)
Doerr L. 380 (293)
Dominikiewiez M. 74, 77, 79 (258), 100 (617),
149, 159 (276)
Dondzila F. A. 380 (298a)
Donovan R. 134, 137 (179), 239, 248 (19)
Donzelot P. 11 (44)
Dorno K- 140 (230)
Douben R. M. 82 (373)
Douglas С. M. 283, 284, 291, 305, 306 (142)
Doumani F. F. 63 (120), 65 (162)
Dowd S. D. 287, 288 (168)
Downing D. C. 135 (204)
Downs A. J. 11 (38a), 336 (714a), 364 (100,
101)
Doyle J. R. 141 (233)
Drake N. L. 75 (272)
Drefahl G. 29 (93), 66 (171), 167 (406, 408),
185 (49), 203 (59), 244 (70)
Dreher E. 40 (5), 44 (5, 47), 45(5), 243,
254 (56), 293 (248), 302 (309), 315 (416,
417), 324 (248), 371, 372 (127)
Drehman U. J. 25(62)
Dresdner R. D. 375 (214, 2146), 376 (2146)
Dry L. J. 154 (290)
Dubeck M. 63 (119), 65(119, 164), 66 (164),
75(119, 269), 76 (269), 77 (119), 83 (269)
Diinhaupt D. 209 (127), 330 (645) .
Dunkanson L. A. 11 (33)
Dunker M. F. W. 67 (177), 185, 189 (18, 19)
Dunn F. W. 96 (542)
Dunning F. 80 (335)
Duppa B. F. 40—43 (1), 307 (355), 308,
315(369), 390(7)
Durham R. W. 325 (566)
Durrell W. S. 375, 376 (2146)
Duschinsky R. 380 (311)
Dwyer F. P. 379 (284)
Eastman R. H. 94 (499)
Ebel H. F. 45 (65), 258, 259(165), 324
(537), 328 (537, 634)
Eberhartinger R. 99 (595)
Ebsworth E. A. V. 364 (100, 101)
Eckert H. W. 393(34)
Eckstein M. 161 (377)
Eckstein Z. 37, 38 (10, 13), 42 (21a), 333
(725), 334 (756, 781), 335 (756)
Edelhoch H. 336 (818)
Edsall 141 (448), 336 (818)
Edwarts L. D. 203 (49)
Egashira S. 390 (46)
Eglinton G. 27,28 (82), 54, 59 (56), 279,
302 (311)
Ehinger F. 395 (77)
Ehrenberg A. 361 (35, 44)
Ehrenfeld E. 92, 94 (486)
Eichler E. 43 (34)
Eickemeier D. B. 203 (58)
Eisen В. M. 273 (876, 88)
Eisert M. A. 287 (166a)
Ejmocki Z. 42 (21a), 334 (781)
Eldjarn L. 141 (449)
Eldridge A. 395 (50)
Elson L. A. 209 (120)
Авторский указатель
407
Elster В. В. 54 (38)
Emeleus Н. J. 11 (77), 15(18—20, 22),
47 (72, 73), 54 (59), 246 (78), 325 (579),
336 (805, 806), 364 (95, 99—101), 373 (206)
Emerson М. 11 (56а)
Engelmann G. 77 (310)
Erdmann О. L. 166 (393), 390, 394 (1)
Erhard К. 325 (580а)
Erlenmeyer Н. 20 (24), 153 (286)
Ernst В. 370 (195)
Espinoza С. 14 (5), 20, 21, 24 (17), 42 (23)
Essers W. 124, 130, 131, 163 (104)
Ettel V. 185 (25, 45)
Evans D. P. 11 (3), 26 (74)
Evans M. G. 325 (568)
Evans R. B. 209 (113)
Evans W. V. 11 (И1)
Evering B. L. 226 (11)
Fairbrother D. M. 11 (126)
Falkov M. 390 (10)
Fanta G. F. 165 (453)
Fantl P. 96, 97 (533)
Faraglia G. 333 (699a)
Farinhold L. H. 80 (355)
Fassel V. A. 11 (40)
Faul Ph. 115, 131, 133, 137 (12)
Fedjarn L. 336 (817)
Feeney R. E. 336 (810)
Feher F. 11 (46)
Feigel H. 293 (225a) •
Feigl E. 390 (3)
Feigl F. 324 (458)
Fell B. 133 (342)
Ferber E. 120 (78)
Ferreira P. C. 98 (583)
Fetizon M. 94 (501)
Fiedler H. 96 (538)
Field F. 378 (263)
Finzi C. 292 (215, 216), 324 (216)
Fischer B. 377 (233)
Fischer E. 11 (88), 40, 44 (3), 57 (98a), 103
(628, 630), 191 (78, 79), 274 (102), 370 (162)
Fischer H. 380 (286)
Fischer W. 377 (242)
Fitz Gibbon M. 54 (58)
Flautt T. J. 11 (61)
Fleck H. P. 308 (365)
Fleming G. H. 21 ,(30)
Fleming-Struthers R. 54 (70), 57 (92, 101)
Flenner A. L. 228 (5)
Fletcher J. P. 139 (222) .
Fohr M. 373 (207)
Fonken G. 324, 325, 328 (580).
Ford G. M. 265(7)
Foreman E. t>. 161 (327)
Forrester S. R. 139 (147a), 278 (123)
Forster C. 376 (220, 221)
Forster M. O, 191 (81), 378 (266)
Fosbinder R. J. 334 (747)
Fossati V. 207 (81)
Foster D. J. 28 (83a), 31 (83a,.108), 32 (108),
234 (49), 266, 270 (53), 271 (53, 54), 282,
300 (64a), 324, 335 (514), 354 (20),
37 (327)
Fothergill J. E. 336 (809) .
Fourneau E. 254 (144)
Fox A. L. 77, 78 (314), 92, 93 (483)
Fox J. J. 380 (292—294, 297, 311)
Fox R. B. 382 (19), 383 (318, 319)
Foye W, O. 161 (363), 162 (378)
Franchimont A. 296 (247), 360 (17)
Francesconi L. 377 (240)
Francis A. F. 139 (225)
Frankland E. 14(1), 37 (8), 40—43 (1), 209
(128), 307 (355), 308, 315 (369), 319 (459),
390 (7)
Franklin E. C. 376 (225)
Frant M. S. 82 , 92 (190)
Fray G. I. 25, 26 (75a), 307, 309 (353)
Fredericks W. J. 365, 371 (103a)
Freedman M. B. 133 (176, 177), 134 (177),
260 (178)
Fremdling H. 373 (207)
Frerichs G. 365 (103)
Frewing J. J. 11 (13)
Frey F. E. 26 (73), 324 (531)
Freyer W. 25 (63), 291 (185a)
Friedel C. 309(390)
Friedlander P. 97 (563)
Friedman H. L. 154 (288), 161 (327)
Friedmann W. 371 (130)
Friend J. H. 133, 134 (177)
Friend J. N. 124 (106)
Fritz G. 25(64)
Fritz H. P. 11 (29), 57 (98a), 365 (1156)
Fromm E. 96, 97 (533)
Frommel H. 96 (529), 115, 131,133, 137 (12)
Fuchs K. 42(18)
Fujimoto G. 155 (301)
Fijita K. 115, 120, 137 (23)
Fukamauchi H. 73 (254, 255), 77 (343)
Fukushima S. 115 (21, 22, 26, 26a), Г20 (21,
22, 26), 137 (21, 22, 26, 26a), 246(77),
273 (56—58)
Fulton M. 274 , 301, 319 (99a), 356 (65)
Furst K. 246, 249 (78a)
Furst W. 65, 71 (155), 203 (50) .
Furth O. 376, 378 (224)
Gadamer I. 77(300)
Gajewski F. 58(110)
Gale L. H. 23 (48), 134 (188—190), 224 (4),
269, 271 (50), 277 (115), 278 (121)
Ganesan R. 324 (510, 513)
Gannage E. 44.(41)
Garjand С. E. 71 (227)
Garner C. S. 229 (17), 240, 241 (41), 321 (469)
Gasopoulos I. 136 (212)
Gaudemar M. 37 (9)
Gauerke C. G. 22, 24 (25)
Gaumann T. 11 (52)
Gautier H. 11 (39)
Gautier J. A. 393 (31)
Gavron J. L. 77 (818), 85, 93 (318)
Geissman T. A. 135, 144 (207)
Gelius R. 209 (98)
Geller H. 159 (315), 277 (119) .
Gensler W. J. 54 (45, 54), 305 (328)
Genssler O. 56, 60 (87), 133—135 (164),
353 (8)
Genzken U. 209 (121)
George M. V. 310 (407), 336 (795b), 374 (323)
Gerissen H. G. 94, 95 (500)
Gerngross O, 77 (305)
Gersch H. 77 (294)
Gerstein S. 161 (328, 345)
Geuther 377 (247)
408
Авторский указатель
Ghira 11 (1)
Ghosh А. К. 212(152), 259 (168)
Ghosh J. L. 53, 71, 74, 82, 84, 100 (9)
Giacomello G. 11 (69)
Gibbons D. 395 (46)
Gibling T. W. 11 (116)
Gibson C. S. 24, 27 (60), 291, 292 , 324 (194)
Gilje J. 119 (65), 273 (85, 86)
Gilliam W. F. 309 (381)
Gillmeister A. 209 (125)
Gilman H. 18 (4), 19 (4, 8), 24 (57), 25 (4,
8), 26 (4, 8, 72), 30 (4, 8), 33 (119, 125),
38 (21), 81 (363), 94 (489, 492, 502), 97
(574), 101 (489), 185(37), 188 (54, 61, 62),
202 (43), 203 (63), 207 (87), 208 (87, 88),
211 (148, 149), 231 (29), 248 (104, 106),
253 (132), 265 (7), 284 (66), 296 (250, 257),
302 (306), 305 (332, 337, 338), 306 (306),
307 (306, 357), 308 (362, 363, 366, 377),
309 (377, 392), 310 (402, 406, 407), 318
(447, 448 , 450, 451), 336 (7956), 374 (323)
Gina M. 134, 135(181)
Ginsberg H. 305 (324)
Giral F. 41, 42 (12)
Gizycki F. V. 378 (273)
Glassmann B. 377 (255)
Goble A. G. 324 (532a)
Godchaux E. 293 (227)
Goddard A. E. 26 (70), 209 (113), 286 (161),
310 (150)
Goddard D. 84 (440), 228 (3), 310 (150)
Godfrey К. E. 98, 102 (575)
Godi E. 100 (616)
Goggin P. L. 11 (48)
GoTdacker P. 84, 87 (447), 330 (640)
Goldblatt L. A. 54(73)
Goldman G. 65, 66 (164), 67, 71 (182)
Goldwhite H. 134 (178), 247 (99)
Gomer R. 325 (552, 555, 559)
Gomez Aranda V. 119, 131 (56), 133 (165)
Goodman L. 133, 135, 139—141, 145, 153
(167)
Gorczyca M. 161 (377)
Gordon M. I. 24 (55)
Gordy W. 11 (50, 51, 55)
Goret M. 40, 43 (2)
Goswami M. 66, 68, 70, 82, 86 (169)
Gould C. 395 (48) ,
Gould V. L. 18, 19, 21, 25—27 (3)
Goupil R. 11 (39)
Govenlock B. G. 11 (17, 19, 53, 63), 226 (12—
14), 240 (21), 249 (110), 323 (497), 324 (497,
500, 525—529, 530)
Grafflin M. W. 209 (93), 230, 231 (26),
.267 (30) v
Gray M. I. 283 (142), 284 (142, 149), 291,
305, 306 (142), 309 (149, 388)
Grdenic D. 11 (79—81), 29(94), 45(64),
229 (22), 247 (102), 330 (663—665), 331
(663—666)
Green J. F. 373 (208)
Green J. W. 370 (170)
Greenbaum F. R. 78 (332), 86 (332, 459)
Greenberg J. 133, 135, 139—141, 145, 153
(167)
Greene M. O. 133 (167), 135, 139—141, 145,
153 (167)
Greenwood D. 146 (267)
Gregg D. C. 372 (200a)
Gregory W. A. 33(119)
Griffin В. P. 54(39) ,
Griffith E. 117, 118, 158 (44)
Griffiths D. C. 11 (3), 26 (74)
Griffiths J. E. 212 (154)
Grignard V. 90 (471)
Grilla G. 95, 97, 102 (507)
Grim S. O. 191 (76, 77)
Grimshaw J. 207 (86)
Griner G. 168 (410)
Grob C. A. 53 (34)
Gross E. G. 161 (339)
Grossley A. W. 134 (195)
Grossmann H. 365 (111)
Grote K. 37.7, 378 (246)
Grubert H. 103 (628)
Gruescu C. 64 (133), 292 (195a)
Grummitt O. 24 (56), 198 (13)
Gruttner G. 18, 20, 22 (2), 23 (47), 24 (2),
28 (47), 29 (2), 44 (36), 212 (158), 228,
229 (1), 244 (58), 306, 308'(342), 309 (391),
312, 315 (342), 319(460)
Griitzner B. 377 (233)
Gubitz F. W. 161 (338), 163, 164 (391)
Guha Sircar S. S. 53 (9), 71 (9, 253), 73 (253),
74 (9), 75 (273), 77 (253, 301), 81 (253),
82 (9, 301, 374), 83 (422), 84 (9, 301),
85 (453), 87 (301), 88 (453), 100 (9), 330
(643), 355 (41—44)
Guichard F. 290 (183)
Gulwell T. 11 (3), 26 (74)
Gunning H. E. 325.(550, 581)
Gfintner A. 292 (219)
Gutmann A. 363 (76a)
Gutowsky H. S. 11 (30, 62), 25, 26 (67),
309 (384)
Haas I. 53 (24)
Hach V. 367 (137)
Hadik G. 133 (342)
Hagatsu R. 393 (23)
Hager F. D. 26 (76), 305 (326)
Haggerty C. 224, 225 (2)
Hagiwara Y. 21, 24 (26)
Hahn E. 45(66), 303—305(319)
Haley T. J. 203 (49)
Hall R. H. 135(209), 366 (134)
Halpern A. 161 (339)
Halpern J. 11 (42), 124, 164 (109)
Hamamoto I. 395 (59)
Hamanaka W. 333 (705)
Hammond F, 68 (196)
Hampson E. R. 355 (53)
Hampson G. C. 11 (96)
Hamilton C. S. 94 (506), 97 (565), 209 (134,
135), 211 (134), 292 (204—207)
Hamilton F. H. 203 (47)
Handrick G. K. 62 (116), 203 (61), 317 (436)
Hanke M. E. 67 (179), 77 (319), 96 (522, 538),
202, 203 (45)
Hansen R. L. 24, 27 (61), 265 (3), 269 (3, 42)
Hansing С. E. 212 (152), 259 (168)
Hanske W. 208 (92)
Hanson E. R. 255 (147)
Hantzsch A. 58(110, 111), 60 (84), 71 (212),
359, 360 (5), 362, 381 (62)
Harden W. C. 80 (357)
Hardie R. L. 191 (80)
Harle H. 45(61)
Авторский указатель
409
Harnik М. 116, 135 (37)
Harriman В. JR. 21 (29), 277 (120)
Harris S. E. 72 (247), 75(272), 79 (343),
324 (498, 499), 325 (544)
Hart M. 72 (237), 73 (257), 74 (237), 334,
335 (774)
Hartley К. 11 (132, 133, 135)
Hartman H. 29 (86), 291, 292 (193)
Harwig L. 365 (103)
Hasenbaumer I. 292 (218)
Haszeldine R. N. 15 (18, 19, 21, 23), 47 (72,
73), 54 (48), 134 (178), 246 (78), 247 (99),
325 (579), 364 (95, 96), 365 (96), 373 (206)
Hathway D. E. 84 (441—443), 87 (441),
355 (26)
Haworth R. 207 (86)
Haugh I. W. 71 (227)
Haun R. 369, 370 (189)
Hayashi M. 162 (382)
Haynes R. M. 11 (63)
Hazek W. R. 99, 102 (593)
Hechenbleikner I. 42 , 43 (20), 306, 307 (334)
Heeren J. K. 233 (39)
Heidelberger M. J. 82 (390), 355(31)
Hein F. 11 (104), 222 (30), 245 (65), 258 (163,
164), 275 (107), 305, 307 (354), 332 (107,
673)
Heine R. F. 54 (60)
Hellerman L. 190 (69), 232 (31), 336 (811,
812)
Helling J. F. 306 (345)
Helion R. 53 (23a)
Henbest H. B. 134 (196), 143, 144 (251, 252),
155 (252), 248 (107), 301 (302)
Hennion G. F. 168 (414, 416)
Henry L. 14 (8)
Henry P. M. 134 (180)
Henry T. A. 71 (226), 76 (226, 281—283)
Hepner F. R. 119 (53)
Hepp H. J. 26 (73), 324 (531)
Hermes O. 365 (106)
Herrera P. P. 96, 97 (530), 250 (122)
Hermann A. 77 (317), 359 (4)
Hernler F. 393 (28), 394 (44)
Herty С. H. 365 (117)
Herwig W, 32 (110, 113), 200, 201 (21), 212
(159, 160), 250 (124a), 30'3 (320), 305 (320,
331)
Hetnarska K. 333 (730)
Hetnarski B. 37 (13), 38 (10, 13), 42 (21a),
333 (725, 730, 736, 737), 334 (756, 781),
335 (756)
Heublein G, 167 (406)
Heuser E. 258 (163), 332 (673)
Heumann K. 293, (296)
Hewson K. 380 (303, 307)
Hey ns K. 159 (316)
Hibou J. 115, 130, 136 (11)
Hickner R. A. 65 (158), 212 (150)
Hieber W. 275 (108)
Hill E. L. 25 (41), 22 (25, 41), 24 (25, 41)
Hill H. E. 53 (33)
Hilpert S. 18, 20, 22, 24, 29 (2), 38 (20),
44 (36), 212 (158), 228, 229 (1), 244 (58),
308, 306 (342), 309 (391), 312, 315 (342),
319 (460)
Hinkel L. E. 19, 21, 25 (6)
Hinton R. C. 29 (89), 188 (57)
Hirai M. 393 (23)
Hirano S. 81, 82 (366), 100 (601)
Hirata T. 325 (581)
Hirschfelder A. D. 73 (257)
Hishimura H. 334 (749)
Hixon R. M. 11 (109, 110), 24, 27 (40), 332
(669)
Hnizda V. 236 (73)
Hobrock В. C. 11 (67)
Hock H. 359 (1)
Hodgson H. H. 71—73 '(214), 53, 84 (441—
444), 87 (441, 444), 355 (26, 52)
Hoffer M. 380 (311, 313)
Hoffmann E. 116, 135 (37)
Hofmann A. W. 376 (223), 390 (6)
Hofmann H. P. 306 (345)
Hofmann K. A. 53 (16), 54 (57, 69), 55
(79, 83), 56 (16), 57 (16, 96), 60 (79),
96 (547), 115 (1—4), 116(133), 117 (1—4,
33), 123 (103), 130, 131 (1), 133 (2),
135 (223), 138 (2), 141 (3, 4), 166 (103, 392,
396), 168 (396, 409), 266 (45, 46, 47a) 272
(76), 274 (95), 280 (47a), 293 (225a), 300
(286), 302 (46), 353 (7), 363 (82, 83)
Hoke D. I. 65 (158, 212 (150)
Holleck L. 226 (9)
Holleman A. F. 68 (187), 361 (48)
Holley С. E. 307 , 316 (352)
Holmberg B. 369 (145—149), 372 (201)
Holroyd R. 325 (548)
Holtz J. 305, 306 (325)
Holzbecher Z. 199 (19)
Homeyer A. H. 24 (59)
Honeycutt JI B. 200 (24)
Horing M. 291, 292 (192)
Hornberger H. 307 (360)
Horning W. C. 11 (93), 199 (19a)
Hornstein I. 139 (224)
Horowitz M. G. 11 (22)
Horowitz R. M. 135, 144 (207)
Horton A. W. 96 (526)
Howard E. G. 133 (174, 175), 134, 138, 139
(175), 315 (419), 324 (130), 360 (22)
Howard H. C. 11 (114), 249 (111)
Howe В. K. 366 (134)
Howells E. R. 11 (70)
Hsfl H. Y. 150 (280)
Hu P.-F. 333 (678)
Hflbel W. 31 (107), 44 (35)
Hiickel W. 140 (231)
Hueter R. 77 (297), 84, 85, 88 (445), 92 (297),
254 (141)
Hugel G. 115, 130, 136 (11)
Hughes E. D. 21 (27), 240 (43), 241 (45—
47), 259 (43), 322 (476, 486), 323 (490, 493)
Hull F. B. 185 (34)
Hiimpfner K. 309, 310 (393)
Hunsberger J. M. 99 (589)
Hunter W. H. 359 (6)
Hurd C. D. 94 (505), 96, 97 (544), 99, 100
(599), 188, 203 (64), 244, 253 (75)
Hurwitz M. 147 (269)
Hyeo S. 68 (197)
lano K. 333 (715)
Ichikawa K. 115 (21—26, 26ak 132 (24),
137 (21—24, 26a), 246 (77), 272 (55), 273
(56—58, 80)
Ichimura F. 81 (361)
Iddles H. A. 372 (200a)
410
Авторский указатель
Iguchi М. 395 (49, 71)
Imoto Е, 66 (173а), 281 (1346)
Inami К. 333 (705)
Ingold С. 11 (66), 21 (27), 240 (43), 241 (45,
47), 259 (43), 322 (476), 323 (490), 324 (501)
Inoue Т. 24 (53), 365 (114)
Irie Т. 94 (503, 504)
Iritani N. 154 (292)
Irwin F. 195 (1)
Irwing H. 67, 71, 92 (178)
Isacescu D. A. 64 (133), 292 (195a)
Isenhour L. L. 92, 94 (484), 355 (49)
Ishihara K. 77 (303)
Ishiwata S. 68 (197)
Issleib K. 57, 62 (97), 316 (422)
Ito A. 333 (705)
Ito Y. 86 (462, 463), 100 (463)
Ivin K. J. 325 (572)
Izumijya N. 153 (285)
Jackson C. L. 376 (227), 379 (276)
Jackson G. R. 82, 91—93 (190)
Jacob H. 96 (535, 537)
Jacobi K. R. 18, 20, 21 (5), 330 (648)
Jacobs G. 310 (408)
Jacobs W. A, 82 (390), 355 (31)
Jacobsen O. 44 (48, 50, 54)
Jacobsohn I. M. 84 (438), 85 (397), 87 (438),
355 (24), 376 (228)
Jacobson R. A. 168.(412), 279 (312)
Jadhav G. V. 203 (57)
Jaeger С. B. 75 (272)
Jaffe H. H. 11 (61), 19, 20, 25, 27, 28 (15)
Jager H. 190 (72), 232 (33)
Jambon L. 359 (7)
Jander G. 11 (113)
Jantzen E. 154 (298), 155 (300)
Jaquiss M. T. 324 (533)
Jaworsky R. 307 (351)
Jellinek F, 364 (102)
Jellum E. 141 (449), 336 (817)
Jenkins G. L. 185, 189 (18)
Jenkins H. O. 11 (122)
Jenkins W. J. 120 (76, 77), 255 (150)
Jensen F. R. 23 (48), 134 (188—190), 224 (4),
229 (13), 239 (14), 240 (20, 44), 241 (14,
48), 246 (14, 20), 257 (182), 269 (50), 271
(19, 20, 50, 54a), 273 (54a), 277 (115—
1166), 278 (121)
Jeschke J. 95 (508)
jobling W. H. 129, 139 (148)
Johannsen T. 310 (398)
Johns J. B. 11 (109, 110), 332 (669)
Johnson G. H. 203 (49)
Johnson J.\A. jr. 380 (303)
Johnson J. B. 139 (222)
Johnson J. R. 53 , 59 (27), 129, 139 (148),
197 (7, 10), 198 (7„ 13)
Johnson R. S. 201 (39)
Jones J. W. 161 (339, 340)
Jones L. W. 19, 27 (11), 41, 42 (15), 270 (51),
361 (27)
Jones P. P. 11 (53)
Jones R. G. 38 (21), 188 (61), 211 (148, 149),
305 (332), 310 (402, 406) 318 (450, 451)
Jones W, E. 166 (404), 292 (209)
Jones W. H. 324 (520)
Jones W. J. 11 (3), 26 (74)
Jose I. 14 (5), 20, 21, 24 (17), 42 (23)
Joseph J. P. 380 (299)
Josh G. 154, 158 (296)
Joshi К. C. 75 (275), 83 (421)
Jurecek M. 394 (40, 41), 395 (45)
Jiirgens J. 68 (189)
Kacer F. 362 (53)
Kadomtzeff I. 11 (90—92)
Kaesz H. D. 11 (28), 20, 27 (16), 283 (141),
291 (141, 185)
Kahlen M. 133 (171)
Kajimura T. 395 (64)
Kaku T. 21, 24 (26)
Kalinowski B. 67, 68 (181)
Kalltneyer H. 11 (104)
Kamen K. 393 (24)
Kanatoma S. 77 (296), 353 (14)
Kanazawa J. 395 (74)
Kanvinde С. K. 99 (597)
Kao Y. S. 150 (280)
Kaplan Z. 308, 320 (361a), 380 (297)
Karaoglanov Z. Z. 365 (109)
Kary Ch. 370 (168)
Kast H. 361 (32), 362 (58)
Katchalski E. 336 (818)
Kato A. 161 (326)
Kaufman F. 265—267 (22)
Kaufman H. P. 378 (268)
Kawakami A. 353 (14)
Kawakita M. 361 (40)
Kefferem J. J. 161 (363)
Keicher G. 200 (20)
Keiser E. H. 166 (398)
Kekule A. 296 (247), 361 (36)
Kelbe W. 291, 292 (190)
Kenner J. 68 (191, 196)
Kergomard A. 136 (213)
Kerkhof L G. 71 (231), 207 (84)
Kern D. M. H. 395 (55)
Kern R. J. 372 (202)
Kerrigan V. 15 (18)
Kersasp H. 77 (305)
Kesler M. 11 (97, 108)
Kettle S. F. A. 11 (42), 124, 164 (109)
Kharasch M. S. 20, 21, (19), 74 (265), 77
(319), 82 (380), 83 (265, 397), 84 (265, 438),
, 85 (397), 87 (438), 185 (27), 202 (44), 203
(46), 205 (69), 206 (73), 209 (93), 228 (4, 5),
229 (6—8), 230 (4, 26), 231 (26), 267
, (30—36), 330 (32), 355 (24. 50), 376 (228)
Khotinsky E. 197, 198 (6)
Kidd J. M. 364, 365 (96)
Kijewska M. 149, 159 (276)
Kiliiani D. B. 168 (416)
Killingstad A. 95, 96 (513), 296 (259)
Kim J.-Y. 29 (88), 41, 43, 46, 47 (14), 70
(289), 266 (23, 25), 240, 241 (42), 267 (23—
25), 268 (23—24a), 269 (23, 25) 322(471)
Kimura I. 395 (73)
King H. S. 25 (69)
Kirby R. H 97 (574), 308, 309 (377)
Kirk R. E. 362 (59)
Kirmreuther H. 54 (57), 266, 280 (47a)
Kirrman A. 246 (82), 395 (61)
Kirst W. 225 (5)
Kiser R. W. 11 (67)
Kissel H 360, 361, 375, 377 (16)
Kissinger L. W. 62 (116), 203 (61). 317 (436>
Kissmann H. M. 380 (298)
Авторский указатель
411
Kistftkowski G. В. 325 (552, 565)
Kitching W. 11 (566). 54 (69a)
Kiwan A. M. 67, 71, 9? (178}
Klapproth W. J. 62 (118), 63 (1), 65 (118),
68, 70 '1)
Klason P. 362 (66, 70), 365 (108)
Klein 0 . 376, 378 (231)
Kleine-Peter Mi 246 (82), 395 (61)
Klieger E. 367 (136)
Klocking H. P. 370 (180, 187)
Klotz I. M. 11 (22)
Kluchesky E. F. 154, 163 (289), 395 (57)
Klug A. 117, 129 (42), 300 (287)
Knee T. E.< C.< 65 (154)
Kobe K. A. 62 (113, 114), 63 (120, 146, 151),
65 (161, 162), 254 (145)
Koch H. F. 133, 134 (177)
Kocwa A. 161 (377)
Kochlin P. 293 (226)
Koehler R; 82 (373)
Kohler W. 96 (534, 545), 208 (91)
Kohlrausch K. W. F. 11 (45)
Kokorudz M. 96 (554)
Kolb H, 360, 380 (18, 19)
Kolb W. 11.(46)
Kolmer I. A. 77, 85, 93 (318)
Komatsu M. 335 (793)
Kondakov J. 125 (116, 117)
Konig W. 197, 198 (5), 390 (8)
Koniger P. 97 (563)
Korbl J. 390 (2)
Kotos E. 393 (21)
Kotak H. M. 161 (363)
Kotakemori M, 395 (59)
Koten J. A. 209 (94), 230, 231 (27), 334
(739)
Kothare A. W. 99 (597)
Kothner P. 166 (393, 394)
Kotler-Brajtburg J. 37 (14)
Kowitt F. R. 119, 120, 134, 155, 157 (61),
273 (83)
Koyama K. 395 (74)
Koziknwski J. 204 (66), 253 , 257, 259 (138)
Krafft F. 294, 302, 316 (237)
Kraffts H. 309 (390)
Krassowsky H. 37 (6)
Kraus C. A. 226 (10)
Krause E. 11 (2), 188 (56), 209 (111), 309 (395)
Kraut M. 229 (24)
Krech M. 324 (504)
Kreevoy M. M. 24, 27 (61), 116 (161), 119 (60,
61, 63—65), 120 (60, 61, 64}, 132 (161),
134, 155, 157 (61), 265 (3), 269 (3, 41, 42),
273 (78, 84-v88), 353 (2)
Kreienbring F. 371 (195)
Kreke C. W. 395 (62)
Kremper A. 362 (54)
Kresge A. J. 63 (129). 65 (154)
Krespan C. G. 11 (59), 133, 139 (173), 315
(420)
Kretchmer R. A. 273 (85, 85a)
Kristen H. 369 (189), 370 (189, 196)
Krocker F. 394 (38)
Kroeger J. W. 53 (26)
Kropat K. 393 (25)
Krug R. H. 161 (369)
Kriiber O. 97 (564)
Kriiger M. 381 (315)
Krynski J. 68, 71, 72 , 74, 83 (186), 360 , 376
(12)
Kubat A. 97 (561a)
Kuca L. 11 (76)
Kuhn R. 96 (541)
Kuivila H. G. 199 (18)
, Kunchur N. R. 362 (72)
Kunkel L. 55 (75), 378 (269), 379 (269, 277)
Kunz I. 317 (432)
Kutner A. 53 (28)
Kutscheroff M. 56 (88), 123, 140, 164, 168
(97)
Kuwajima I. 272 (55в), 258 (162a)
La Coste W. 292 (196, 200, 203)
Ladenburg A. 44 (44), 288 (169)
Lado F. 831 (665), 333 (729)
Lagowski J. J. 15 (22), 32 (111, 112), 336
(805, 806), 364 (102)
Lajoux H. 77 (311)
Lamdan S. 185, 188 (41), 250 (116)
Lami P. 380 (291)
Lampen I. O. 380 (297)
Landgrebe J. A. 233 (76), 239 (14), 240 (20),
241 (14), 246 (14, 20), 269, 271 (50), 277
(116a, 116)
Landsberg M. 377 (249)
Lane J. 306 (339)
Lang H. 362 (63)
Langbein G. 66 (171)
Lange G. 305, 307 (359)
Langhans 361 (39)
La Roche К. H. 190 (73), 232 (34)
Lassere A. 55 (82), 56 (82)
Laubengaver A. W. 309 (381)
Lauder A. 258 (160), 331 (667)
Laurie С. M. 324 (512)
Lautenschlaeger F. 11 (93), 199 (19a)
Lauter W. 291, 292 (192)
Lautsch W. F. 11 (128)
Lawrence S. M. 395 (62)
Lawton D. 336 (807)
Lay W. P. 115. 120 (27), 273 (81)
Leandri G. 11 (16, 18, 20, 24), 185 (51. 52).
269 (64), 276 (291), 333 (64), 335 (794. 798)
Leblanc E, 212 (161)
Lecher H. 371, 372 (129)
Ledwith A. 354 (20a)
Lee Y. K- 240 (42), 241 (42, 49), 322 (471),
366 (122)
Lefevre C. 73 (250)
Le Fevre R. J. W. 11 (105)
Legault R. R. 203 (46), 229 (8), 267 (35)
Lehmann G. 32 (114), 201 (33)
Lehman J. F. 333 (732)
Lehman R. A. 333 (732)
Leibsohn E. 96, 97 (533)
Leicester H. M. 294 (232, 234, 235)
Leicester J. 11 (7, 8)
Leitch L. C. 47 (74), 54 (37), 229 (24)
Leitsmann R. 96 (547)
Lemaire H. 167 (405)
Lemmon R. M, 11 (58)
Leo J. 96 (529)
Leo M. 20 (24)
Leonhardt P. 96 (524), 250 (121)
Leopold R. S. 377 (260)
Lepeschkin N. 134 (194)
412
Авторский указатель
Le Roi Nelson К. 64 (153>
Leschewski К. 116, 117 (33)
Lespagnol A. 77 (312)
Lestina G. 119 (55), 140 (229), 145 (55)
Leto J. R. 11 (57) 119, 130 (52)
Letsinger R. 29 (84)
Leurk G. J. 78, 86 (333), 207 (82), 330 (641),
355 (53)
Levene P. A. 370 (171, 172)
Leverenz L. 11 (46)
Levi D. L. 309 (383)
Levin R. H. 58 (104)
Levine J. H. 185 (27), 229 (6), 267 (33)
Levy L. 290 (181)
Levy M. 135 (199), 144, 145 (265)
Lewis H. F. 41, 43 (11)
Ley H. 360, 361, 375 (16), 377 (16, 242, 244),
378 (244)
Leys A. 56 (85), 73 (249)
Lichtenstadt L. 291, 292 (192)
Lichtenwalter G. D. 310 (407), 336 (795b),
374 (323)
Lichtin N. N. 134, 143, 144, 163 (250), 276,
278 (122b)
Lidzey R. G. 11 (118)
Liebig J. 360 (10 , 21), 362 (69), 377 (252)
Liese E. 84, 88 (446)
Lindstone A. G. 324 (532a)
Link A. 291, 292 (191)
Linke H. W. 370 (179).
Linn W. J. 133, 134. 138, 139 (175), 324 (130)
Linnemann E. 14 (7)
Linnett J. W. 11 (14, 15, 117), 325 (560, 561)
Lintner C. 77 (292)
Lippmann E. 53 (17)
Lister J. 58 (111)
Liston T. V. 212 (151)
Livingstone J. G. 29 (87), 235 (70), 266, 324
(44)
Lobry de Bruyn 360, 361 (25)
Loeffler P. K- 26 (75), 308 (364)
Loev B. 162 (313)
Loevenich J. 373 (207)
Logan T, J. 206 (71), 231, 233 (27a)
Loh S. H. 150 (280)
Lohr P. 308 (361)
Loir A. 363 (84)
Loloff C. 209 (122)
Lommen F. W. M. 84, 87 (438), 355 (24),
376 (228)
Long L. H. 11 (120, 134), 315 (414), 324
(508, 512, 517, 518)
Longfield J. E. 324 (509)
Lonsky P. 115, 131, 133, 137 (12)
Lora-Tamajo M. 333 (680, 681)
Lorenz D. 29 (93)
Losco G. 362 (56)
Lossen W. 140 (230)
Lossing F. P. 11 (66), 324 (501—503)
Loudon J, D. 201 (40), 202 (40 , 41), 204
(67), 244 (69), 246. 248 (90)
Louise E, 309 (382)
Lowe W. G. 209, 211 (134), 292 (205, 206)
Lowy A. 189 (66)
Lowy B. A. 380 (295)
Lucas H. J. 119 (53), 167 (405)
Luce E. N. 55 (76)
Lueth P. F. 62 (113)
Lumb P. B. 53 (32) '
Lund H. 353 (18) ,
Lyons E. 377 (250)
Lyons R. E. 294 (237), 302, 316 (237, 421)
Lythoe B. 380 (302)
Mac-Dianmd A G. 212 (152), 259 (168)
Mac Gregor 54 (53)
Maeda S. 334 (749)
Magat E. E. 62 (115)
Mahadevan A. P. 54 (45, 54), 305 (328)
Mahapatra G. N 83 (402, 404—406, 408,
411-413, 418, 423), 85 (451), 87 (423),
88 (451)
Mahapatra S. 53 (9), 71, 74, 82, 84, 100 (9)
Maikop ar 371 (131)
Maitte P. 99 (592)
Makin F. B. 183 (14)
Mallik K. L. 153, 158 (450)
Malaiyandi M. 63 (148)
Manolopoulos T. P. 134, 143, 144, 163 (250)
Mameli E. 67, 70 (183), 71 (203, 218, 2-19,
223, 224), 72 (240)
Mameli-Mannessier A. 67,70 (183),71 (223,224)
Manchot W. 53 (24), 74 (264), 117 (42), 123
(102), 124 (106), 129 (42), 300 (287)
Marchand R. F. 390, 394 (1)
Marcus R. A. 325 (554, 564)
Margcches M. 11 (40)
Marica E. 44 (39)
Marini-Bettolo G. B. 94 (494)
Marker C. 363, 371, 372 (77)
Marker R. 228, 230 (4), 267 (31)
Marker R. E. 22 (35)
Markusic B. 331 (666)
Marple К. E. 188 (54), 309 (392)
Marquard R. P. 55 (76), 139 (220)
Marquarding D. 226 (9)
Marquardt A. 43 (29), 308 (373)
Marschall R. R. 62 (115)
Marsh J. E. 54 (70), 57 (92, 101), 377 (247)
Martin F. 361 (41, 42)
Martin R. B. 325 (571)
Martin R. W. 139 (221)
Martinez I. M. 92 (487)
Martinez Gordon L. 2. 131 (153), 133 (165)
Marvel C.S. 18, 19(3), 21 (3, 28), 22 (25), 24
(25, 28), 25 (3), 26 (3, 28, 76), 27 (3),
82(394), 117, 118, 158 (43 , 44), 201 (39).
266 (43), 305 (326), 324 (43), 355 (47)
Maschmann E. 77 (321), 85 (321, 456),
89 (321)
Massie 8. P. 22 (37)
Masson C. R. 325 (562)
Matejka K. 144, 157—159 (256)
Matheson M. S. 11 (54)
Mathis R. D. 233 (76)
Matsui K. 333 (728 , 759). 334 (759)
Matsumija K. 292 (213)
Matsumoto 1. 96 (540)
Matsunga I. 11 (89)
Matsushiro K. 77 (296)
Matteson D. S. 198 (14a)
Matthews В. C. 11 (94)
Maung M. T. 32(111. 112)
Maybury R. H. 141 (448). 336 (818)
Mayer J. R. 11 (63)
Maynard J. L. 11 (114), 14, 15 (17), 68 (135),
246, 247 (91), 248 (93), 249 (Шк
Mayuranathan P. S. 92 (632), 207 (76, 80)
Авторский указатель
413
Me Cleland N. Р. 11 (И), 99(596)
Me Clure F. Т. 195 (1)
Me Clure R. Е. 189 (66)
Me Cormick С. G. 11 (55)
Me Coy С. R. 309, 320 (368а)
Me Сгае W. 27, 28 (82), 54, 59 (56), 279,
302 (311)
Me Cullough J. D. 294 (236)
.Me Cusker P. A. 53 (26)
Me Cutchan R. T. 254 (145)
Me Donald Blair J. 326 (608)
Me Donnell L. R. 336(810)
Me Elhinney R. S. 134 (196)
Me Elvain S. M. 152 (2821
Me Ewen W. L. 53, 59 (27)
Me Farland B. W. 378 (270)
Me Gary C. W.63 (123), 64 (152), 65 (152, 163)
Me Gregor W. S. 54 (53) .
Me Jlroy R. 370 (173)
Me Kellin W. H. 203 (56)
Me Keon W. D. 161 (338)
Me Mahon E. 82 (394), 355 (47)
Me Mahon K. S. 62 (114), 63 (146)
Me Manimie R. J. 54 (60), 203 (48)
Me Nabb W. B. 393 (35)
Me Nelly К. H. 120, 139 fl), 155, 156 (304)
Me Omie J. F. 66, 69 (174)
Meals R. N. 19, 22, 25, 27 (9), 259(169)
Mednik M. 134, 143, 144, 163 (250), 276,
278 (122b)
Mehrotra R. C. 365 (1-1 la)
Mehta B. F. 71 (215)
Meier G. 27 (78), 229(12)
Meisenheimer J, 291, 292 (192)
Meixner A. 394 (38)
Melamed M. 197, 198.(6)
Mellet R. 307 (351)
Melville H. W. 325 (573, 577)
Menzel D. 376 (215a)
Menzel W. 309(397)
Merckoll A. 96 (528)
Merlier R. 11 (39)
Merz W. 58 (103)
Mesee H. 222 (30)
Mesnard P. 42 (21) 260 (179)
Metanovskv W. 366 (131a)
Meyer С. M. 370 (171,. 172)
Meyer F. J. 305, 307 (359)
Mever R. 44 (53)
Micchetti A. 86 (461), 87 (461, 466), 355 (25)
Michael A. 53 (23)
Michaelis A. 16 (31), 40 (6, 7), 44 (6,7 55),
45 (55), 46 (68, 69), 47 (55, 70), 197 (2,
4. 9), 209 (121), 259, 260 (170), 284 (145—
148), 290 (182, 184), 291 (148, 184, 186—
188, 191), 292 (191, 196—198, 199, 200,
202, 212, 219), 293 (227), 303 (315, 316)
Michalzik E. 370 (196)
Michelson A. M. 380 (296)
Middleton E. 72 (204), 76 (280), 92 (316),
150 (277), 250 (119, 124), 252, 254 (124),
274 (92), 355 (34, 48)
Middleton W. J. 54 (64), 133 (174,175),
134, 138, 139 (175), 315 (419), 324 (130)
Mieg W. 292 (208)
Mijamoto F. 333 (715) ,
Mikaijama T. 272 (55b)
Millar I. T. 66 (170)
Miller D. M. 325(551)
Miller H. F. 65 (165), 281 (134)
Miller J, 257 (182)
Miller R. F. 144 (260)
Miller S. A. 97 (562), 185 (42), 250(123),
296 (254)
Miller V. L. 355, 356 (30), 395 (73)
Miller W. T. 133 (176, 177), 134 (177),
260 (178)
Mills L. E. 117, 144 (46)
Minasz R. J. 287, 288 (168)
Mingoia Q. 97, 102 (571), 159, 161 (361)
Misumi M. 154 (292)
Mitchell A. D. 78 (329, 330)
Mitra J. 82 (374)
Mitsuno M, 335 (797)
Mode R. A. 162 (378)
Mohler F. L. 11 (64)
Moller E. F. 96 (543)
Monaco G. 335 (798)
Montequi R. 200 (29, 31)
Montignie E. 57 (94)
Moore F. W. 188 (62), 318 (450)
Moore L. O. 284 (66)
Moore W. J. 325 (575, 578)
Morel T. 94 (498, 500), 95 (500)
Mori E. 161 (365)
Mori M. 84, 85 (448)
Mories G. 40, 44 (4)
Morimoto A. 154, 159(287)
Morris C. J. O. R. 29 (90)
Morrissey C. 99, 100 (599), 188, 203 (64),
244, 253 (75)
Mortimer С. T. 11 (121)
Morton A. A. 42 , 43 (20), 56, 61 (89), 62
(115),306, 307 (334)
Morton J. W. 74 (268)
Mosher H. S. 25 (67a), 26 (75), 308 (348 , 364)
Mott F. 363, 366 (856)
Motzkus E. 363 , 371 (85a)
Moy D. 11 (56a)
Muetterties E. L. 364, 365, 372 (98)
Mui J. Y.-P. 270 (50a)
Mukai jama T. 258 (162a)
Mukherjee G. K- 71, 73 (229), 100 (613)
Mukherjee R. N. 134 (178), 247 (99)
Muller A. 41, 42 (16), 43 (28), 309 (385)
Muller К. H. 324 (505), 325 (549)
Muller R. 380 (286)
Muller T. C. 199 (18)
Mumm O. 123, 166, 168 (98)
Murahashi S. 30 (96), 309 (387)
Murakani M. 154, 159(287)
Murtry G. C. 365 (116)
Musart R. 307 (351)
Musgrave W. K. 235 (70), 266, 324 (44)
Myddleton W. W. 123 (99—101), 168 (101),
171 (99—101)
Myrback K. 355 (46)
Nadkarni M. V. 161 (340)
Nadkarny V. V. 99 (597)
Nagabhus-hanam M. 83 (401)
Nagase I. 395 (49, 71)
Nagel R. H. 72 (233)
Nagiwara Y. 154 (292)
Naik K. G. 53 (20, 21), 71 (215), 78 (323)
Nakagawa M. 53 (36), 54 (50)
Nakahara K- 99 (588)
Nakajima K. 334 (749)
Nakajima S. 335 (793)
414
Авторский указатель
Nakamura М. 362 (71)
Narasimhachari N. 161 (324)
Nargund К. S. 207 (77)
Nayar M. R. 11 (49)
Naylor M. A. 265—267 (10)
Neal H. R. 236 (72)
Nef I. U. 14 (10), 53 (24a), 361 (28)
Negoro K. 15/ (308)
Nelson J. A. 67 (176)
Nelson J. F. 296 (257), 318 (447)
Nelson N. A. 54 (43), 141 (234)
Nenitzescu C. D. 64 (133), 292 (195a)
Neogi P. 71 (209, 229), 73 (229), 360, 376 (13)
Nerdel F. 19, 22, 26, 27, 29, 32 (10), 185 (29),
265—267 (18)
Newbold G. T. 369 (161)
Newell M. P. 202 (165)
Newitt D. M. 309 (383)
Newman M. D. 190 (69), 232 (31)
Newman M. S. 53 (28)
Newstrom J. E. 65 (161)
Neu R. 200 (27, 28)
Nicholls B. 143, 144 (251, 252), 155 (252),
248 (107), 301 (302)
Niederl J. B. 72 (232, 233)
Nieuwland J. A. 54 (65a), 59 (656, 65b),
131 (154), 168 (414, 416), 234 (44)
Nimmich W. 370 (177)
Nimz H. 370 (181, 182, 184, 186)
Nineham A. W. 53 (30)
Nishiyama A. 395 (49)
Nogase Y. 80 (347, 348)
Noll Ph. 391, 393 (17)
Norman J. J. 54 (47)
Norman O. L. 203 (58)
Normant H. 94 (491)
Norris W. P. 53 (29), 54 (46)
Norris W. S. G. P. 363 (75)
Norrish G. W. 11 (134)
Nosek J. 185 (25, 45)
Novak J. 377, 380 (256)
Novel li A. 79 (342)
Noyes W. A. 325 (548, 555, 571)
Nozakura S. 30 (96), 309(387)
Nozoe T. 333 (728, 759), 334 (759)
Nyberg B. 53 (10)
Oatfield H. J. 24 (57), 203 (63)
O’Connor D. E. 282, 316 (140a)
O’Connor R. T. 54 (73)
Odling W. 309 (379)
Oesper P. F. 11 (5)
Ogata Y. 68(192), 83 , 90, 91 (167), 92 (167,
485), 317 (439)
Ohno T. 71 (216, 238), 72 (246), 74 (261),
77 (309), 79(338), 80 (347, 348), 100
(309), 360 (14)
Ohta M. 99 (588)
Okamoto K. 395 (58)
Okamoto Y. 65 (163)
Okawara M. 66 (173a), 281 (1346)
Okuda T. 334 (764), 377 (258)
Oliver J. P. 11 (56a), 310 (401)
Oliveri-Mandala E. 98 (580), 380 (290)
O’Neill G. J. 233 (37)
Opitz G. 53 (986), 376 (215a)
Oppenheim A. 37 (7), 53 (23a), 378 (236, 261)
Oppenheimer C. 57 (90)
Orchard W. M. 94 (495), 253 (133), 296 (263),
318 (446), 356 (60)
Orndorff W. K. 286 (173)
Ose H. 333 (715)
Ostrogovich A. 378 (262)
Othen C. W. 292 (211)
Othmer D. F. 362 (59)
Ott M. 81, 82 (365), 371, 372 (200)
Otto К. H. 100 (608), 246 (98)
Otto M. M. 59(112)
Otto R. 40 (4,5), 44(4,5 47,56), 45(5),
244 (56,57), 254 (56), 265 (6), 293 (228),
296 (248,251), 302 (309), 311 (6), 312 (228),
315 (416, 417), 324 (248), 330 (646),
362 (67, 68), 371 (127), 372 (68, 127),
376 (222)
Ovenall D. W. 11 (53), 226 (13)
Overbaught S. C. 77 (315)
Ouchi H. 115(21—24, 26, 26a), 120(21 —
24, 26), 132 (24), 137 (21—24, 26, 26a)
246 (77), 272 (55), 273 (56, 57, 80)
Oxford A. E. 368 (143)
Ozawa K. 390 (46)
Pacevitz H. A. 305 (338)
Pacsu E. 370 (165-170), 371 (169)
Paderi C. 98 (579), 362, 380 (64)
Padhi B. 83(403)
Pai N. G. 11 (43)
Раке G. E. 11 (62)
Palmer R. A. 11 (85)
Pan L. C. 80 (349)
Pan P.-C. 150 (280)
Pande K. 23 (50), 272 (55a)
Paneck C. 291 (187)
Pantuly A. J. 83 (415)
Paolini O. 95 (518)
Paolini V. 76 (284), 77, 82 (293), 95 (517),
129 (142, 143), 134 (143,193), 144 (142, 143)
Papa D. 305 (324)
Parham W. E. 53 (31), 96 (532), 99,402 (593)
Park W. R. R. 116, 131, 134 (31), 135 (200),
157 (307), 354 (19)
Parrod J. C. 33 (124)
Pasfield W. H. 324 (521)
Pasynkiewicz S. 37 (10, 13,14), 38 (10, 13)
Patai S. 116, 135 (37)
Patel A. D. 78 (323)
Patel K. S. 53 (18)
Patel R. P. 53 (21)
Paterno E. 44 (52)
Patnaik К. K. 83 (422)
Patnode W, 310 (400)
Pattison F. L. M. 54 (47)
Patton R. H. 377 (251)
Paul P. F. M. 54 (38)
Paul R. 94 (490, 493), 101 (490)
Paulik F. E, 235, 236 (71), 258 (161), 273 (89a)
Pauwels P. J. S. 324 (532a)
Peaks R. W. 376 (227)
Pearl J. A. 76 (286)
Pearson D. E. 161 (341, 369)
Pearson R. 11 (111)
Peel J. B. 95, 97, 101 (514)
Pellerin F. 393 (31)
Penella F. 293 (225)
Pengilly B. W. 326 (608)
Penner H. P. 56, 61 (89)
Penz H. 96 (535)
Авторский указатель
415
Perkins R. Р. 206 (74), 207 (82)
Perrin С. 63 (148а)
Perry W. L. 154 (288), 161(327, 328, 345)
Pesci L. 47 (71), 82 (375, 376, 382, 383),
83 (424), 87 (375), 91, 93 (476, 477), 206(72),
254 (142), 330 (639), 375 (212), 376 (212,
215—219, 232), 377 (212), 378 (232)
Peter N. 133 (168)
Peters W. 201—203 (37), 365 (110), 380 (289)
Peterson W. D. 11 (109), 332 (669)
Petterson L. 53 (8)
Petzchner 305, 307 (354)
Pfaff S. 378 (236)
Pfeiffer M. 370 (194), 381 (316)
Pfeiffer P. 18, 26, 27, 29(1), 117, 118 (47),
190 (72, 73), 124, 125 (107), 232 (33, 34)
Pfennigberger R. 393 (28)
Pflfiger E. 377 (257)
Phibbs M. K. 325 (543 , 545, 563)
Philipp J. 365, 366 (107)
Phillips D. C. 11 (70)
Phillips G. F. 11 (118)
Phillips H. A. 360 (9)
Phillips L. J. 354 (20a)
Piaggesi F. 71 (219)
Pierce J. C. 393 (29)
Pieroni A. 100 (606)
Piesberger V. 11 (88)
Piccard I. 82 (380), 355(50)
Piccinini A. 82 (370, 377), 83 (370, 425),
86 (370), 378 (271, 272)
Picon M. 206 (716)
Pixnlott P. J. E. 366 (135)
Pinajian J. J. 240 (33), 322, 323(482)
Pines H. 185 (27), 229 (6), 267 (33)
Piper T. S. 11 (60), 36 (1), 57 (98)
Pirrone F. 100 (614)
Pischel H. 381 (312)
Pitzer K. S. 25, 26 (67), 309 (384)
Plambeck L. 22 (35)
Plancher G. 97 (570)
Planek R. W. 54(73)
Plato G. 377 (240)
Pleszke K. 103 (630)
Plotner G. 29 (93)
Plum O. 119 (57)
Pobloth H. 258 (164), 275, 332 (107)
Pod L. 333 (702)
Poethke W. 65, 71 (155), 203 (50)
Polanyi I. C. 324 (500)
Polkacki E. S. 145, 146 (266a), 277 (117)
Polla G. 134, 135, (181)
Pollart D. F. 29 (84)
Polley D. 395 (48)
Pongratz A. 11 (45)
Ponzio G. 43 (33), 308 (375)
Pope W. J. 24, 27 (60), 291, 292, 324 (194)
Powell H. B. 32(111)
Powell H. M, 336 (807)
Powell H. N. 334 (766)
Pozzo-Balbi T. 99 (590)
Prager W. 53 (11), 377 (241)
Pray H. H. 71 (227)
Pregl F. 393, 395 (27)
Prevost C. 168 (418)
Pfibil R. 390(2), 393 (21)
Price S. 11 (130), 324 (504, 516)
Priester R. 75, 77 (278)
Priestley H. M. 96, 97 (544)
Pritchard H. O. 11 (121, 132, 133, 135),
323, 324 (495)
Pritchard-Jones P. 226 (13)
Profft E. 97 (561a), 100 (608), 246 (98)
Proskouriakoff A. 71, 73, 74 (213), 78 (331),
83 (433)
Prussia L. 82 (378, 379), 330 (655), 376, 378,
379 (229)
Pugh H. J. 364 (99)
Pujari H. K. 83 (400, 407, 410), 88 (410)
Purgotti A. 393 (90)
Purvis I. E. 11 (11)
Puxedden A. 11 (26)
Pyke R. G. 135 (209)
Quellette R. J. 271 (19, 20, 54a), 273 (54a)
Querry M. V. 161 (370)
Raabe G. 209 (102a)
Rabe W. O. 363 (82, 83)
Rabideau S. W. 307, 316 (352)
Rabinerson G. 46 (69)
Raffo M. 83 (428—430), 374,„ 375 (209)
Raft P. 200 (20)
Ragno M. 76 (285), 84 (434—437), 98 (578)
Rainey W. T. 377 (260)
Raiziss G. W. 71, 73, 74 (213), 77 (318),
78 (331), 85, 93 (318), 390 (10)
Rajagopalan P. 161 (324)
Raliant J. 134 (186)
Ralston A. M. 154, 158 (296)
Ramey P. S. 54 (46)
Rangaswami S. 158 (311), 159, 160 (319)
Rao С. M. B. 11 (12)
Rao P. S. 78 (328), 159 (317, 318, 320, 321),
160 (320, 322), 274 (98)
Rao R. P. 83 (414, 415, 417, 419, 420)
Rao S. W. R. 365(118)
Rao V. S. 158 (311), 159, 160 (319)
Raphael R, A. 53 (35)
Rapilly B. 77 (298)
Rascher W. H. 393 (36)
Rasuwajew G. A. 312 (711)
Raul В. В. 83 (418)
Rausch M. D. 32 (115), 103 (625, 628), 243,
246, 248 (55), 250 (117, 1246), 251 (117),
281 (136a), 303, 304 (322), 305 (322, 327),
306, 320, 324 (322)
Ray F. E. 83 (396), 202 (165)
Ray P. C. 363 (89, 91), 365 (324), 366 (89),
375 (210), 393 (81)
Ray W. J. 203 (55, 58)
Rebbert R. E. 325 (546, 547, 557, 558)
Redford D. G. 96 (559)
Reese A. 16 (31)
Rehder W. 370 (197)
Reichel I. 72 (268), 333 (702)
Reichle W. T. 254 (146), 293 (225), 334 (740)
Reikober K. 166 (397)
Reissert A. 68, 70 (193)
Reitzenstein F. 82 (384), 330(648), 355(27,
32)
Rembarz G. 370 (174, 179, 180, 183, 185)
Renaud P. 23 (44)
Renaud R. 47 (74); 54 (37)
Renouf N. 134 (195)
Reppel L. 378 (273)
Reynolds G. F. 11 (61), 19, 20, 25, 27, 28 (15),
266—268 (24)
416
Авторский указатель
Rheinboldt Н. 294(239), 363 (85а — 85а),
371 (85а, 132)
Rhodehamel Н. W. 96, 97 (553)
Rice F. О. 226 (11)
Richards О. V. 198, 199, 209 (14)
Rickborn В. 240 (44)
Riddle J. М. 200 (24)
Ridgway L. R. 209 (126), 236 (75)
Rieckhoff К. 370 (177)
Robb J. С. 325 (573, 577)
Roberts Е. К. 325(565)
Roberts J. S. 325 (567)
Robinson R. 25, 26 (75a), 307, 309 (353)
Robson I. H. 24 (54), 116 (36), 133, 134,
163(169), 275 (104, 105), 301. 309 (105)
Rochow E. G. 310 (404)
Rod L. 72 (248)
Rodgman A. 11 (83), 115 (13, 27), 116 (28),
120 (27, 70), 134 (13, 28, 70), 135 (28,
203, 205), 142 (239), 155, 156 (28, 304),
273 (81, 82), 353 (5)
Rodighiero G. 67, 70 (183), 85 (452), 244 (73),
379 (275)
Roeder G. 68 (130), 292 (201)
Roedig A. 54 (52)
Roemer H. 363 (73)
Rogers D. 11 (70)
Rohatgi H. L. 79 (337, 340, 346)
Rohrmann E. 21 (31), 299, 300 (285)
Roman F. L. 117, 144, 145 (45), 244, 248 (18)
Romer E. 120 (78)
Romeyn J. 115, 121, 134, 137 (14)
Roncucci L. 333 (699a)
Rosen 143 (249)
Rosenberg H. 103 (625), 243, 246, 248 (55)
Rosenfelder W. J. 301, 310(301)
Rosenheim A. 365 (115, 121)
Rosier R. 96 (536)
Ross L. O. 133 (167), 135, 139, 140, 141, 145,
153 (167)
Rosser R. J. 166 (404), 292 (209)
Rossi G. 76 (285), 82 (395), 83 (426—430,
432), 87 (395), 207 (570)
Rostogi R. P. 145 (261)
Rother J. 82 (385)
Rothstein E. 24 (58), 229 (15), 355 (22),
366 (133) >
Rout M. K. 71, 73 (253), 77 (253, 301),
81 (253), 82 (301), 83 (399, 400 , 402, 403,
405, 407—412), 84, 87 (301), 88 (310), 330
(643), 98 (585), 355(41—44)
Roux E. 309(382)
Rowland R. L. 134, 145 (197), 153 (289), 154
(288, 289), 161 (327, 328, 345), 395 (57)
Ruckert A. 200 (20)
Rumpf J. A. 53 (34)
Rumpf P. 19 (7), 22 (36), 330 (652, 653),
332, 333 (668), 334 (653, 780)
Rupp E. 77 (294, 317), 359 (4), 393 (17, 25)
Ruspaggiari G. 82, 83, 86 (370), 377 (254)
Russel .M. E. 323, 324 (496)
Sachs G. 81, 82(365), 99 (595), 115, 134,
137(17), 246, 249 (78a), 363 (86, 87), 371,
372 (200), 390 (4)
Sackman J. F. 315(414)
Sado T. 333 (715)
Sah P. P. T; 158 (309)
Sako I. 333 (715)
Sakurai I. 14(9)
Salinger R. M. 308 (361a), 319 (458a), 320
(361a)
Salvemini A. 234 (50), 248, 252 (108), 293,
298 (230), 335(794, 795)
Salvioni E. 66 (175)
Samuel R. 11 (12)
Samuel W. 291, 292 (192)
Sand J. 55 (74), 56, 60 (87), 115 (1—7), 116
(1,5,7), 117(1—7, 48,49), 129(48), 130(1),
131 (5), 133 (2, 7, 157, 164), 134,
135(7, 164), 138 (2), 139 (48), 141 (3—6),
142, 144 (7, 49), 152 (49), 246 (87),
247 (101), 272(76, 77), 273(79), 300 (286,
289), 330 (651), 353 (1, 4, 7, 8)
Sandborn L. T. 117, 118, 158(43)
Sandin R. B. 73, 74 (256), 77 (315), 79 (341).
195 (1)
Sanz-Pastor O. 119 (56), 131 (56, 153, 155).
393 (32)
Sapper A. 11 (4)
Saraf J. R. 11 (49)
Sarti U. 97 (573)
Sastri V. D. N. 78 (328), 83 (416, 417),
159 (321), 161 (324)
Sato H. 395 (63)
Sato M. 333 (728, 759), 334 (759)
Sato R. 395 (74)
Sauerwald A. 41, 42(16). 309(385)
Saunders K. W. 324 (515)
Saville R. W. 24 (58), 229 (15)
Sawa J. 334 (749)
Sawatsky H 11 (32, 106), 24, 29 (52), 41, 43,
44 (10), 63 (148), 65 (160), 74, 75 (26?),
203 (60), 229 (16). 247, 259 (100)
Scarella A. 374, 375 (209)
Schaaf S. 167 (408)
Schaefer K. 377, 378 (244)
Schaeffer H. J. 134 (191), 380 (304, 305)
Schaleger L. L. 273 (91)
Schall C. 225 (5)
Scharp T. M. 71 (226), 76 (226,281,282, 287)
Scharrnbeck W. 197, 198 (5)
Schaub R. E. 161 (370), 380 (299—301)
Scheibler H. 95 (508)
Schellenberg M. 70 (202)
Schempf R. 21, 22, 26, 27, 30 (42), 290 (179)
Schenk A. 47 (70), 291 (186)
Scherlin S. M. 191 (83), 248 (105)
Schertel A. 371 (128)
Schimmel C. 161 (375)
Schleede A. 11 (104)
Schlenk W. 305, 306 (325), 308 (368)
Schlenker O. 241 (50), 321, 322 (470), 395(75)
Schlesinger H. 1. 307(350), 309(380)
Schmandke H. 369 (189), 370 (189, 193, 197)
Schmidt H. 77 (307)
Schmitz M. 209 (111)
Schneider F. 370 (198, 199)
Schneider W. 85 (455), 370 , 371 (163, 164)
Schoeller W. 44 (38), 53, 61 (2, 4), 77 (297),
82 (385), 84 (445—447), 85 (445), 87 (447),
88 (446, 448), 92(297), 115(8,9), 124
(104). 130 (8, 9, 104), 131 (104). 154 (8),
158 (8, 9), 164 (104), 166 (9),. 253 (128),
254 (141), 266 (476), 274 (101), 330 (640),
377 (238)
Scholl R. 53 (10), 361 (34, 47), 362 (51—55)
Scholvien L. 362 (49)
Авторский указатель
417
Scholz С. R. 145 (244), 161 (331)
Schorygtn Р. Р. 302 (304, 305), 306 (346),
307 (304, 305, 347)
Schraeder W. X. 296 (249)
Schramm R. М. 62, 65 (117, 118), 332 (671)
Schrauth W. 44 (38), 53, 61 (2, 4), 77 (297),
82 (385), 84 (446, 447), 87 (447), 88 (446),
92 (297), 98 (577), 115 (8, 9), 124 (104),
130 (8, 9, 104), 131 (104), 154 (8),
158 (8, 9), 159 (315), 163 (283), 164 (104),
166 (9), 253 (128), 254 (141), 266 (476),
274 (101), 277 (119), 330 (640), 377 (238)
Schroeder W. D. 74 (266)
Schropp W. 275 (108)
Schubert С. C. 324 (511)
Schuierer E. 24 (126), 259 (185)
Schulemann W. 71, 77 , 82 , 85, 87 (228)
Schulte C. 292 (202), 303 (315)
Schulze F. 307 (357), 308 (362)
Schulze-Bentrop R. 190 (73), 232 (34)
Schumb N. C. 310 (405)
Schutz W. 11 (95), 377 (247)
Schwalbe C, 95 (515)
Schwarze.nbach G. 70 (202)
Schwarzenbach K. 190 (71), 354 (21)
Schweizer E. E. 233 (37)
Schwenke E. 305 (324)
Scott R. 24 (55)
Scott-Wilson H. 57 (93)
Seager J. H. 123, 168, 171 (101)
Seaman W. 197 (10)
SegelE, 62, 65 (117)
Segitz 305. 307 (354)
Seher A. 171 (433)
Seide O. A. 191 (83), 248 (105)
Seiler E. 57 (96), 135 (223)
Seka R. 11 (45)
Seki T. 335 (793)
Selle H. 361 (32)
Selby W. M. 26 (72)
Semitt H. F. 96 (538)
Semon W. L. 363 (79)
Sen R. N. 78 (324, 325), 100 (613), 145 (261),
274 (97)
Sepp J. 370, 371 (163, 164)
Serrano C. 200 (29, 31)
Seshadri T. R. 78 (328), 158 (311), 159 (317—
321), 160 (319, 320, 322), 161 (324), 274 (98)
Sestini F. 377 (243)
Setzer L. 96 (536)
Sexton W. A. 209 (120)
Seyferth D. 25, 54, 59 (63), 133 (171), 191 (76,
77), 209 (99, 102, 102a, 110), 210(110),
233 (39, 40), 250, 252, 258 (114), 266 (476),
270 (50a), 280 (476), 287 (166a, 167, 168,
406a), 288 (168), 289 (167, 413a), 291
(185a), 306 (340, 345)
Shainoff J.-R. 336 (809)
Shaft R. 115, 131, 133, 137 (12)
Shah L. D. 53 (20), 377 (259)
Shanmuganathan S. 90 (474)
Shapiro H. 236 (74) ,
Sharkey W. H. 54 (64), 133 (174, 175), 134,
138, 139 (175), 315 (419)
Sharma U. 79 (346)
Sharp A. G. J. 335 (796)
Sharp T. M. 76 (283, 287)
Shatavsky M. 141 (232)
Shearer D. A. 115 (13), 120 (69), 134 (13, 69),
135 (69), 273 (82), 275 (103)
Sheline R. K- 11 (41)
Shellberg E. F. 393 (20)
Sheridan J. 11 (50, 51)
Shetty B.V. 115, 137 (18), 150 (280), 161 (18)
Shimaoka N. 334 (749)
Shintani G. 24 (53)
Shoule H. S. 334 (766)
Shreve R. N. 99, 100 (594)
Shuetz R. D. 365, 371 (103a)
Shukis A. J. 72 (232), 75 (279), 132 (160)
Shulman N. 204 (67)
Shurow A. E. 312 (411)
Sice J. 96, 101 (539)
Sidgwick N. W. 361, 377 (31)
Siemens C. 372 (203)
Sigal M. V. 161 (341, 369)
Silbermann B. 95 (518)
Silberrad O. 360 (9)
Silman H. 318 (445)
Silva R. B. 336 (810)
Simmons H. E. 14 (9a, 37), 259 (186)
Simmons J. H. 377 (251)
Simpson R. B. 141 (448), 336 (808)
Singer F. 55(74), 115, 116 (7), 117 (7,49),
133, 134, 135 (7), 142, 144 (7, 49), 152 (49),
246 (87), 353 (4)
Singer S. Y. 336 (809)
Sipos J. C. 11 (106), 74, 75 (262)
Skeeters M. J. 99, 100 (594)
Skiba K. J. 378 (268)
Skinner H. A. 11 (119, 121, 126, 131—133,
135)
Skundina S. 377 (255)
Sleezer P. D. 23 (45), 271 (54b)
Slotta К. H. 18, 20, 21 (5), 330 (648)
Smaller B. 11 (54)
Smiles S. 363 (85)
Smith Cl. 78 (329, 330)
Smith D. S. 141 (234)
Smith E. W. 24 (57). 203 (63)
Smith F. D. 292 (210)
Smith G. M. С. P. 363 (79)
Smith J. C. 53 (32)
Smith J. G. 365 (117)
Smith J. J. 293 (225)
Smith L. I. 65, 78 (159), 317 (433)
Smith M. H. 43 (30)
Smith R. D. 14 (9a)
Smith R. H. 11(125)
Smith R. J. 115, 134, 137 (15)
Smyth С. P. 11 (5)
Snyder H. R. 270 (67)
Sobatzki R. I. 203 (54), 250 (113)
Sobotka W. 42 (21a), 334 (781)
Soderback E. 95, 97 (510), 101 (510, 623),
282 (139), 360 (20), 366 (125)
Sommer L. H. C. 21 (32), 22 (33), 229 (9)
Sone T, 96 (540)
Soroos H. 236 (73 , 74)
Sowden R. J. 326(614)
Spahr R. J. 54 (65a), 59^(656, 65b), 131 (154),
234 (44)
Spatz S. M. 33 (119)
Specht E. H. 62 (116), 203 (61), 217 (436)
Speter J. L. 22 (34)
Spengler J 115 (12, 20), 131, 133 (12), 135
(20), 137 (12, 215), 138 (20, 218), 158 (20),
246 (90a), 333 (679, 734), 334 (679) ,
418
Авторский указатель
Speranzini G. 87 (470)
Sperry W. N. 117, 144, 145 (45), 244, 248 (18)
Spielman M. A. 58 (104)
^Spinelli D. 11 (20, 24), 234(50), 248, 252
(108). 269(64), 276 (291), 293, 298(230),
333 (64,677), 335 (794, 795, 798), 373
(328)
Spinner I. H. 366 (131a)
Spring F. S. 369 (161)
Sprowels W. R. 203(46), 229(8), 267 (35)
Srinivasan R. 324 (506, 519)
Srivastava K. 75 (275a)
Stabica N. R. 395 (72)
Stafford S. L. 11 (56)
Stafford W. H. 65(166)
Stamm G. 82 (384), 330 (638), 355 (32)
Stange G. 185 (49), 203 (59), 244 (70)
Stanton G. M. 212 (151)
Stark K.-H. 370 (196)
Starkey E, B. 67 (177), 185, 189(18, 19)
Staveley F. W. 205 (69)
Steacie E. W. R. 325 (542, 544, 546, 547,
550, 551, 554, 557, 558, 562, 566, 569,
572, 576), 326 (611)
Stearns 372 (200a)
Stecher O. 310 (398)
Steedly J. W. 183(15)
Stefan G. 133 (342)
Steiner A. 361 (38, 46)
Steinkopf W. 19, 29, 30(14), 53 (12), 63,
«69 (201), 95.(512, 513), 96(201, 512, 513,
* 321—524, 528, 529, 534—538, 545, 547,
-552), 101 (512), 208 (91, 92), 228 (2), 243(53,
34), 246, 248 (97), 250(53, 54, 97, 120,
121), 251 (53), 252 (97, 126), 269 (48),
292(208, 214), 296(258, 259)
Stephens J. R. 141, 142 (237), 145 (264),
247 (103)
Stephens R. 47 (75), 281 (134a)
Steudel O. 225, 226 (6)
Stevens L. G. 310 (401)
Stevenson К. 11 (136)
Stieglitz I. 77(319)
Stiehler O. 370, 371 (164)
Stokker G. 273 (85a)
Stolle R. 379 (278)
Stone F. J. A. 11 (28, 56), 20 (16), 22 (43),
27(16, 43), 229(77), 283 (141), 289(172),
291 (141, 185)
Straessle R. 141 (448)
Strand t F. 370 (177)
Stranks D. R. 11 (137)
Strecker A. 14 (3), 330 (650), 377 (234)
Stright P. L. 53 (31), 99, 102 (593)
Strohmeier W. 11 (58, 112), 309, 310 (393)
Stromholm D. 375(211)
Stacker J. F. 33 (118), 303, 304 (321)
Struensee R. 115, 130 (8,9), 154(8), 158
(8, 9), 166 (9), 266 (476)
Stuhlmann H. 359 (1)
Stutz A. I. 212 (152), 259 (168)
Suce E, N. 139 (220)
Sugden S. 11 (115)
Suida W, C, 298 (273)
Sujishi S. 325 (581)
Sullam C. 124 (110)
Summerbell R. K. 119 (55), 139 (147a),
140(229), 141, 142(237), 145(55, 264,
266a), 146 (266a), 247 (103), 277 (117),
• 278(123)
Sung L. K. 64 (141)
Sutherland J. W. 79 (341)
Sutton L. E. 11 (99, 123)
Suzuki Z. 258 (162a), 272 (55 в)
Swain C. G. 65 (154)
Swallen L. C. 363, 366 (88)
Swan G. A. 100 (600)
Swaney M. W. 99, 100 (594)
Swartz S, 20, 21, (19), 28(138), 229 (7),
267 (34)
Sweet T. 395 (50)
Swenson A. D, 336 (814)
Swirska A. 37 (14)
Sykes A. 395(46, 47)
Szabolesi G. 336 (816)
Szekeres L. 161 (349, 367)
Szwarc M. 324 (533), 325 (567, 568)
Szymaszkiewicz J. 42 (21a), 334 (781)
Tabern D. L. 289 (173), 393 (20)
Tafel J. 224, 225(1)
Tagliavini G. 209 (137a, 138)
Takagi S. 334 (7.48)
Takanashi Y. 100 (607), 334 (764), 377 (258)
Takano K. 11 (37)
Takeuchi S. 30 (96), 309 (387)
Tallmann R. C. 75 (279), 132 (160)
Tanaka J. 66 (173a), 281 (1346), 324 (503),
334(758), 335(793, 797)
Tarrant P. 282, 316 (140a)
Tartarini M. 82, 83 (393)
Tashika J. 98 (586)
Tatlow J. C. 47(75), 274 (99a), 281 (134a),
301, 319 (99a), 356 (65)
Tatsuoka S. 154, 159 (287)
Tausz J. 133 (168)
Taylor F. L. 65, 78 (159), 317 (433)
Taylor H. A. 15(26)
Taylor H. S. 324 (507, 515, 520), 325 (575)
Tazuma J. J. 57 (176)
Tedder J. M. 318(444)
Theaker G. I. 318(444)
Theobald C. W. 53 (25)
Thiele I. 57 (95)
Thielebeule W. 370 (185, 191)
Thiruvengadam T. R. 161 (324)
Thomas H. J. 380(303—305)
Thomas M. H. 129, 133 (150)
Thomas W. J. O. 11 (52)
Thompson H. W. 11 (13—21, 35, 117), 325
(560, 561)
Thorpe F. G. 241 (45, 47), 322 (476), 323 (493)
Thorpe M. A. 282 (140)
Thron H. 145 (263)
Thrush B. A. 325 (574)
Thurmann N. 203 (47), 296, 299, 300 (246)
Ticharich N. 370, 371 (169)
Tickner A. W. 324 (498, 499, 502)
Tien J. M. 99(589)
Tiffeneau M. 43 (27), 44 (40)
Tipper C. F. 119 (58)
Titherington R. J. 83 (433)
Tobler E. 28 (83a), 31, 32 (108), 234 (49)
266, 270 (53), 271 (53, 54), 282, 300 (64a)
324, 335 (514), 354 (20), 371 (327)
Tochtermann W. 46 (66), 303-305(319
Todd A. R. 380 (296)
Авторский указатель
41»
Todd D. 29 (89), 188 (57)
Todd L. J. 270 (50a), 289 (413a)
Tomson R. H. 191 (80)
Torrey H. A. 359 (6)
Torsell K. 197(3), 198(3, 12), 199(3)
Towe R. H. 54, 59 (63), 209, 210 (110), 250,
252, 258(114), 266, 280(476)
Travagli G. 98 (581, 582), 102 (581), 185 (87)
Traverso G. 81 (368)
Traverso J. J. 131, 161 (152)
Traylor M. M. 119(58)
Traylor T. G. 19 (13), 31 (104), 38 (23), 120,
134 (74), 135 (74, 1986), 136 (1986), 229(17),
239 (13, 15), 240, 241 (41), 246 (15),
248, 249 (106a), 265, 266 (17, 21), 267,
271 (17), 278 (21), 301 (21, 303), 309 (21),
310 (303), 321 (469), 353 (9a)
Treiber A. J. H. 287, 288 (168)
Trikojus V. M, 334 (762)
Trotman J. 11 (17, 19), 226 (12), 249(110)
Trotman-Dickenson A. F. 11 (130), 324 (516,
524), 325 (542)
Trotter J. J. 11 (84)
Truce W. E. 53(33), 54(60), 203 (48,55,58)
203 (58)
Truskier P, 18, 26, 27, 29 (1)
Tseu C. Z. 158 (309)
Tsoukalas S. N. 375 (214)
Tsuchida M. 68(192), 83, 90 (167), 91 (167),
92 (167, 485), 115 (21, 22), 120 (2,22),
137 (21, 22), 273 (56, 57), 317 (439)
Tsukatani H. 334 (758)
Tsuruoka M. 334 (764), 377 (258)
Tundo A. 11 (16, 18)
Turner E. E. 185 (43), 294 (238)
Turner H. S. 72 (214), 84, 87 (444) 355 (52)
Turner M. A. 119(65), 273 (86, 87a, 91)
Ueno T. 154, 159 (287)
Uhlenbrock J. H. 96 (546)
Ukai T. 77 (296), 81 (360, 361, 366), 82 (366),
86 (462, 463), 100(463, 601, 609—612),
102 (610), 144 (257), 162 (382), 253 (134),
353 (14), 355 (56)
Ulbricht G. 370 (179)
Ullmann F. 362 (57)
Ulrich S. E. 306 (339)
Umezawa S. 97, 101 (566), 296 (253)
Umgrove L. 360 (17)
Unabe N. 395 (54)
Unga T. 356 (64)
Urbanski T. 333 (725), 334, 335 (756)
Usami S. 395(60)
Usgaonkar R. N. 203(57)
Van Aken G. M. F> 240 (36), 322 (485)
Van Ammers M. 100 (602, 603)
Van Campen M. G. 198 (13)
Van Der Kelen G. P. 310 (403)
Van Loon E. J. 277 (118)
Vaughn T. H. 16(36)
Vecchiotti L. 45 (62), 46 (67), 82 (371, 386—
389), 83 (371), 86 (371, 461), 87 (387,
388, 461, 464—470), 253 (131), 355 (25),
379 (283)
Vecera M. 394 (41) .
Venanzi L. M. 11 (33)
Venezky D, L. 382, 383 (318, 319)
Venner H. 370(181, 182, 184)
Verbeke G. J. Q. 324 (524)
Verdino A. 393 (26)
Verkade P. E. 94(498, 500), 95(500), 159(312)
Vicentini G. 294. (239)
Vijayaraghavan К. V. 246 (81, 83), 252 (83)
Vila A. 254 (144)
Vitali T. 81 (364, 367)
Vitte G. 42 (21), 260 (179)
Vogel M. 11 (7, 8), 103 (625), 243, 246,
248 (55)
Vogelfanger E. 23 (50), 272 (55a)
Vogt K. 363, 371 (76)
Vogt R. R. 54 (65a), 59 (656, 65b), 131 (154),
234 (44)
Vojir V. 395 (65, 67)
Volger H. C. 241 (45, 46), 322 (476, 486).
323 (490)
Volhard J. 95,96, 101 (511), 296 (252)
Wadia J. H. 330 (636)
Waga F. 308(367)
Wagler K. 245 (65), 305, 307 (354)
Wagner G. 381 (312)
Wait H. 119, 145(55)
Wakayama S. 94(503, 504)
Waldbillig J. O. 198 (14a)
Waldo J. H. 334 (766)
Walfe S. 134 (184)
Wall L. A. 325 (578)
Walter L. A. 334 (747)
Walters W. D. 324 (505 , 509), 325 (549) ’
Walton H. F. 92(487)
Wang С. H. 271 (546)
Ware F. E. 24, 27 (40)
Warhurst E. I. 324 (523, 535)
Wartick T. 309 (380)
Wasso I. 11 (127)
Watanale J. 333 (715)
Waters W. A. 183 (13, 14), 189 (14)
Watson H. E. 365 (118)
Watt G. W. 57 (122a)
Wayne-Ruddy A. 72 (239) . .
Weaver C. 270 (67)
Webb F. J. 81 (362, 363)
Webb J. L. A. 335 (796)
Weber A. 115, 135(20), 137 (215), 138 (2Cf,
218), 158(20), 246 (90a), 333 (679, 734),
334 (679), 361 (43) ;
Wedegaertner D. K. 239, 241, 246, 277 (116а»
1166)
Wedekind E. 291, 292 (195)
Weinhouse S. 267 (36)
Weinmayr V. 97 (560)
Weisburger E. K. 83 (396)
Weisburger J, H. 83 (396)
Weiss A. 55 (80)
Weiss A. 55 (80)
Weiss M. J. 380 (298).
Weissgerber R. 97 (564)
Weitkamp W. 97 (565), 292 (207)'
Weller J. 44 (49)
Wells P. R. 11 (566), 54 (69a)
Wempen I. 380(293)
Wendt G. 115, 137 (18), 161 (18, 358)
Wenger P. 390 (4a)
Wenzke H. H. 11(98)
Werner L. F. 19, 27(11), 41, 42 (15W
145 (244), 161 (331), 270 (51) '
Wertheim E. J. 363 (74)
420
Авторский указатель
Werther 377 (253)
Wessely К. 370 (177, 178)
Westheimer F. H. 62 (117, 118), 63 (1, 148a),
65 (117, 118), 68, 70 (1)
Wetmore F. E. W. 129, 133 (150)
Weygand F. 367 (136)
Wheeler H. V. 378 (270)
Whelen M. S. 209 (117)
Whipple L. D 239, 241, 246 (14), 277 (116a,
1166)
White E. C. 79, 80 (334)
Whitehead C. W. 131 (152), 145, 149, 154,
156 (266), 161 (152, 266)
Whiting M. C. 54 (42)
Whitman B. 305 (324)
Whitmore F. C. 21 (29-32), 22 (33, 35),
24 (59), 72 (204), 76 (280), 77 (314), 78
(314, 333), 82 (381), 86 (333), 92 (316, 483,
484, 486), 93 (483, 484), 94 (484, 486),
203 (47, 53, 54), 206 (74), 207 (78, 82, 93),
229(9), 249(109), 250 (109, 113, 118, 119,
124), 252, 254 (124), 255 (147), 267 (28a,
39), 273 (90), 274 (92), 277 (120), 282 (140),
296 (246), 299, 300 (246 , 285), 305 (336),
330 (641), 355 (34, 48, 49, 53, 54)
Whittle E. Z. 277 (120)
Wiberg E. 310 (398)
Wieland H. 360 (30), 361 (30, 37)
Wigert H. 370 (188)
Wilde W. К. И (6), 26, 27 (71), 42 , 43 (22),
185 (28)
Wilkinson G. 11 (60), 36 (1), 57 (98)
Wilkinson K. 94 (505)
Willgerodt C. 294, 295 (241, 242)
Williams G. H. 326 (616)
Williams J. H. 161 (370), 381 (299)
Williams J. W. 377 (260)
Williams R. L. 11 (35)
Willstaedt H. 355 (46)
Willstatter R. 380 (288)
Wilmshurst B. 11 (129, 137)
Wilson S. H. 99 (596)
Wing J. F. 379 (276)
Winkler C. A. 324 (522)
Winstein S. 19 (13), 23 (45, 50), 31 (104),
119(53), 141 (232), 229 (17), 239(13, 15),
240, 241 (41), 246 (13, 15), 265, 266 (17,
21), 267 (17), 271 (17, 54b), 272 (55a), 278,
’301,309 (21), 321 (469)
Wintzer A. 167 (406)
Wirth T. H. 74 (221), 85 (454)
Wittenburg E. 370 (176, 192), 380 (310)
Witt J. 53 (19)
Wittenberg D. 202 (43)
Wittig G. 29 (95), 32 (95, 110, 113, 114), 45
(61, 63, 65), 46 (66), 134 (186), 190 (71),
200 (20, 21), 201 (21, 33), 250, 251 (124a),
258 (165), 259 (165, 175), 303 (318—320),
304 (318,319), 305 (318—320, 330, 331,
359), 306 (330), 307 (359, 360), 324 (537),
328 (537, 634), 354 (21)
Wittie E. 21 (29)
Wober A. 393 (18)
Wohler L'. 360 (23), 361 (29, 41—43), 362 (23),
366 (123)
W'olff P. 27 (79), 270 (52), 324 (532)
Wollensak J. C. 54 (43)
Woodruff C. J. 54 (68), 260 (162)
Woods L. L. 99 (591)
Woods L. W. 292 (220)
Woodward F. N. 166 (404), 292 (209)
Woodward G. E. 355 (54)
Woodward L. A. 11 (48)
Wooley B. L. 318 (445)
Work R. W. 310(404)
Wrenshale R. 155 (301)
Wright G. F. 11 (32, 82, 83, 93, 106), 20 (22,
23), 23 (46), 24 (52, 51), 26 (39), 29 (52),
31 (46), 41, 43, 44 (10), 63 (148), 65(160),
74, 75 (262), 82, 83 (391), 94 (489, 495),
101 (489), 115 (13, 14, 27), 116 (28, 29,
31, 36), 119 (51), 120 (27, 51, 66—72), 121
(14), 129 (72, 144, 149), 131 (31), 133 (169),
134 (13, 14, 28, 29, 31, 66—70, 72, 169,
179, 182, 183, 198), 135(28, 29, 51, 69,
72, 149, 200, 203—205, 209), 136 (66, 72),
137 (14, 179), 138 (66), 139 (71, 149),
140(72), 142 (239), 144 (253), 150(149),
155 (28, 299, 303, 304), 156 (28, 304),
157(307), 158 (72, 144, 149), 199 (19a),
203 (60), 207, 208 (87), 229 (16), 239 (19),
240 (22), 247 (100), 248 (19, 22), 253 (133),
259 (100, 167), 273 (81, 82, 89), 275 (ЮЗ-
105), 296 (250, 262, 263), 299 (284), 300
(288), 301, 309 (105), 318 (445, 446), 353
(5, 11), 354 (19), 356 (60, 62)
Wright I. 87 (391)
Wronsky M. 71, 82 (207)
Wu F. G. 202 (43)
Wuggatzer W. Z. 395 (66)
Wunstorf O. 77 (295)
Yale H. L. 33 (125), 161 (332), 185(37),
231 (29), 248 (106), 318 (448)
Yamamoto I. 353 (14)
Yamamoto S. 395 (64)
Yamamoto Y. 31 (366), 81 (360, 361), 82
(366), 86 (463), 100 (463, 601), 355 (56)
Yanagi A. 86, 100 (463)
Yasukochi K. 395 (54)
Yeddanapalli R. 324 (506, 511)
Yoneda F. 100(607)
Yotsuzuka M. 81(360, 361, 366), 82(366),
86, 100 (463), 355 (56)
Young G. T. 366 (135)
Young G. W. 95, 97, 101 (514)
Young J. A. 375 (214, 2146)
Young R. V. 305(337)
Young W. G. 23 (45), 271 (54b)
Yung N. 380 (292—294, 297, 311)
Zado F. 330 (664, 665), 331 (664, 729)
Zeise W. C, 362 (65)
Zeiser H. 302, 316 (310)
Zeiss H. H. 212 (159, 160)
Zejc A. 161 (377)
Ziegler J. B. 143 (249)
Ziegler K. 31 (100), 33 (100, 121), 225, 226 (6)
Ziemann H. 367 (136)
Zimmer H. 30 (98), 33 (116), 266, 267 (29),
369, 371 (189)
Zincke T. 360 (15)
Zinin N. N. 14 (6), 330 (647)
Zinner H. 370 (174—199), 380 (310), 381
(316)
Ziolkowska B. 11 (86)
Zoellner E. A. 26 (72)
Zook H. D. 305 (336)
Zupancic B. G. 54 (66), 270 (65), 332(670)
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ*
8-Ацетатмеркурпурин 103
Ацетатмеркурсоединения см. также Аце-
токсимеркурсоединения
2-Ацетатмеркурфенилглицин, этиловый
эфир 87
а-Ацетатмеркур-З-фенил-Р-метоксипропио-
новая кислота, метиловый эфир 158
2-Ацетатмеркур циклогексанол 138
3-Ацетиламино-4-окси-5-оксимеркурфенил-
арсиновая кислота 89
Ацетилгуанин, хлормеркурпроизводное 381
Ацетилпропионилметан 168
Ацетилферроцен 296
1 - Ацетокси-2-ацетоксимер курциклогексан
138 '
Ацетоксимер курацетальдегидмоноэтилаце-
таль, ацетат 149
З-Ацетоксимеркурбензидин 87
а-Ацетоксимер кур-3,2-диметоксидигидро-
коричная кислота 160
2-Ацетоксимеркурдифенилен 67, 69
реакция с хлором 279
2-Ацетоксимеркур-1,4-дихлорбензол 70
симметризация 259
2,6-бис-(Ацетоксимеркурметил)-п-оксатиан
277
9-Ацетоксимеркур-10г метоксипальмитино-
вая кислота 154
З-Ацетоксимеркур-2-метоксисульфолан 162
а-Ацетоксимеркур-З-метокси-р-фенилпро-
пиофенон 150
а-Ацетоксимеркур-Р- метоксиэта н 131
2-Ацетоксимеркур-3-нитро-1-нафтиламин 87
4-Ацетоксимеркур-6-нитро- 1-нафтиламин 87
2,4-бмс-Ацетоксимеркур-3-нитро-1-нафтил-
амин 87
Ацетоксимеркурсоединения см. также
Ацетатмеркурсоединения '
.и-Ацетоксимеркурсульфаниламид 88
Ацетоксимеркур-о-толидин 87
1-(Ацетоксимеркур)циклогексен-1 193
л-Ацетоксифенилмеркурхлорид 355
З-Ацетоксиэтилртуть 132
Ацетон, электролиз 224
бис-Т-Ацетопропилртуть 245
Бензиламин, реакция с сулемой 375
Бензилмеркаптали моноз, расщепление
сулемой 371
Бензилмеркаптид ртути 363
Бензилнатрий 306
Бензилртуть
азотнокислая 332
окисление нитратом ртути 275
указывающая страницу, на которой приведена
Алканолртутные соли, Р-элиминирование
олефинов 117
Алкилдихлорфосфины 290
Ал кил меркаптиды ртути 362 сл.
реакции с аммиаком, галоидами, окисла-
ми азота 363
Алкилртуть ©-меркаптоундеканат 334
АлкиЛселениды ртути 372
Аллилртуть
иодистая 14, 37, 252
хлористая 37
Аллилфенилртуть 20, 319
Алюминия изопропилат 353
Амиды ароматических кислот, ртутные про-,
изводные 378
Амилнатрий 306
«-Амилртуть бромистая 21, 24, 25
о-Аминофенилртуть хлористая 86
п-Аминофенилртуть уксуснокислая 86
симметризация 253 ’
2-Амино-5-хлорфенилртуть уксуснокислая
87
л-Анизилмеркурбромид, двойная соль с су-
лемой 25
п-Анизилртуть уксуснокислая 75, 210
л-Анизилталлий диизобутират 286
реакция с сулемой 210
Анилин, р₽акция с сулемой 376
Анилинмеркурхлорид 376
Арилборные кислоты 197 сл.
Арилгидразины, реакции с солями ртути 191
сл.
Арилдиазоний хлористый, двойная соль с
сулемой 184
Арилдихлпрфосфины 291
Арилмеркаптиды ртути. 362 сл.
Арилселениды ртути 373
Арилтеллуриды треххлористые 294
л-Ацетаминофенилртуть уксуснокислая 83,
87
Ацетанилид, реакция с сулемой 378
л£-Ацетатмеркур-л-аминобензойная кисло-
та, этиловый- эфир 88
1-Ацетатмеркурбензантрон 91
4-Ацетатмерку^.-2-броманилин 253
1 -Ацетатмеркур метил-1,2-дигидробензофу-
ран 145
Ацетатмеркурмуравьиная кислота, эфиры
метиловый 164
этиловый 124, 165
=бис-1-Ацетатмеркурнафтил-2-амин 83
л-Ацетатмеркур-л«-нитроанилин 87
4-Ацетатмеркур-2-нитрофенол 71, 73
•б-Ацетатмеркур-2-нитрофенол 71, 73
Ацетатмеркурпропилацетат 154
* Жирным шрифтом набрана цифра,
пропись получения данного соединения.
422
Предметный указатель
бромистая 37
двойная соль с сулемой 25
меченная Hg208 239
реакция с иодистым кадмием и иодом
276
гидроокись 332
иодистая, симметризация 247
хлористая 22
демеркурирование 272
реакция с трехокисью азота 317
симметризация медью 245
Бензилфенилртуть 229
Бензилхлорметилртуть 232
3,4-Бензодифениленртуть 33
6-Бензоил-2, 3, 4, 5-тетраацетил-а/-О-глю-
коза 371
Бензол
кинетика меркурирования 63
оксинитрование 63
Бензофенон 296
Бигинелли комплекс см. игранс-р-Хлорви-
нилмеркурхлорид
Бромацетальдегид, диэтилацеталь 280
б^с-Б рома цетил енид ртути 20
1-/пранс-2'-Бромвинил-2,2'-бромэтинилбен-
зол 279
2-о-(тра«с-2'-Бромвинил)фенилацетиленид
ртути 59
бмс-( 1-транс-2'-Бромвинил-2-этинил-2)-
ртуть 279
Броммеркурацетальдегид, реакция с три-
фенилбромметаном 297
Броммеркурацетон, реакция с кетеном 151
З-Броммеркур-З-бензилкамфора, обмен с
Hg203 322
jf-Броммеркурбензойная кислота, метило-
вый эфир 260
З-Броммеркуркамфора 321
обмен с Hg2U3 322
симметризация 239, 248, 257
Броммеркурпентаметилен 243
Броммеркурпропиленгликоль 141
а-Броммеркурстильбен, симметризация 251
Броммеркуруксусиая кислота, метило-
вый эфир 151
а-Броммеркур-Б-фенил-р-метоксигидракри-
ловая кислота, /-ментиловый эфир 118
2-Броммеркур-З-фенилпропионовая кисло-
та 277
а-Броммеркурфенилуксусная кислота
реакции с
иодистым кадмием и иодом 276
хлористым ацетилом 298
этиловый эфир, симметризация 239, 257
а-Броммеркурциклогексанои, обмен с Hg203
322
Бромметилмеркурбромид, реакция со ще-
лочами 274
1-Бромокт-1-ин 279
Бромпентахлорциклопропан 287
Бромуксусная кислота, этиловый эфир 280
1-Бромфенилацетилен 279
.и-Бромфенилртуть хлористая 188
Бромферроцен 281
1-(1'-Бромферроценил)ртуть бромистая 199
4-Бромфлуорен 281
5-Бром-2-фурилртуть хлористая 208
w-Бутил карбометоксиртуть 235
н-Бутил ртуть
бромистая 21
иодистая. 25
хлористая 24
симметризация 248
Вератровая кислота 207
Вильгеродта реакция 294 сл.
Винилацетат 297
Винилбор двубромнстый 284
Винилдибромфосфин 291
Винилкалий 306
Виниллитий 305
N-Винилмеркуримиды 335
Винилмеркурксантогеиат 334
Винилмеркуртиоцианат 334
Винилртуть бромистая 22, 23
симметризация 247
Винилртуть
иодистая 22
хлористая 22
Винилтрихлоргерманий 289
Винилэтилсульфид, реакция с сулемой 368
Вторичнобутилнатрий 305
Вторичнобутилртуть
бромистая 21, 239
симметризация 239, 246
электролиз 226
тартрат и /-фенилгликолят, расщепление
на оптические антиподы 259
Вторичнооктилртуть бромистая 24
Гаммета уравнение 63, 65, 67, 76, 239, 269
Гексабромциклопентадиен 281
н-Гексадецилртуть хлористая 22
2,2',4,4',6,6'-Гексанитродифенилртуть 206
Гексахлорциклопропан 287
Гексаэтилдистаннаи 315
«-Гексилртуть бромистая 21, 25
Гептафтор-2-иодпропан 280
н-Гептилртуть 21
о-Гидроксимеркурбензойная кислота 206
3-Г идроксимерку реал ициловая кислота,
ангидрид 77
экзо-1(«с-3-Гидрокси-2-норборнилмеркур-
хлорид, симметризация 249
Демеркурирование 270 сл.
Деоксимеркурирование 119, 273
Диазоаминобензол, N-ртутная соль 379
Диазометан, реакция с солями ртути 190
Диалкилбериллий 307
Диалкилмагний 307
2,3-Диал кокси-1,4-диацетоксимеркурбутан
139
Диаллилртуть 28, 246
Ди-«-амил ртуть 26, 43
Ди-л.-амино-о-бромфенилртуть 253
Ди-п-аминофенилртуть 253
Дианизилртуть 29, 46, 47, 321
Ди-п-анизилталлий, изобутират 285
а, а'-Диантрахинонилртуть 189
Диарилдихлоролово 313
Диарилртуть, реакции с
бромистым иитрозилом 316
бромсукцииимидом 328
окислами азота 317
Диарилсвинец, диацетат 290
Диарилселениды 293
Предметный указатель
423
о-Диацетатмеркурбензол 68
2,3-Диацетатмеркуриндол 192
9,10-Диацетатмеркуркетоундеценовая кис-
лота, этиловый эфир 171
Диацетатмеркурмалоновоэтиловый эфир 60
2,6-Ди(ацетатмеркурметил)-1,4-тиоксан 146
2,4-Диацетатмеркур-1-нафтиламин 87
4,6-Диацетатмеркур-2-нитрофенол 73
2,4-Диацетоксимеркуранилин 86
.м-Диацетоксимеркурбензол 317
Диацетоксимеркурдиметоксибутан 140
3,4-Ди(ацетоксимеркур)-2-метил-5-этилтио-
фен 102
Диацетоксймеркурхлоруксусная кислота,
реакции со щелочами 274
Ди-о-ацетоксифенилталлий бромистый 286
1,3-Диацетокси-2-хлормеркуррутеи-2 169
Диацетоксимеркурхлоруксусноэтиловый
эфир 152
Диацетонилртуть 148, 255
Дибензилолово хлористое 314
Дибензилртуть 19, 27, 31, 33, 245, 247
окисление 264
пиролиз 324
реакции с
окислами азота 318
серой 316
хлоридами железа, кобальта, меди 327
хлористым оловом 314
З-Дибензофурилртуть хлористая 24
Ди-о-бифенилилртуть 251
Дибромацетиленид ртути 319
1,4-Диброммеркурбензол 24
Диброммеркурдиэтиловый эфир 247
1, 1-Дибромтетрахлорциклопропан 287
Ди-п-бромфенил ртуть 188, 245
1, Г-Дибромферроцен 281
2,3-Дибром-5-хлормеркуртиофен 208
1,1-Дибромциклопропан 287
Дибутенилртуть 28
Ди-н-бутилртуть 26, 30, 33, 43, 248
реакции с
иодистым германием и иодом 311
окислами азота 318
фотолиз 325
«, [3-Дибутоксидиэтилсульфид, реакция с
сулемой 369
Дивинилртуть 20, 27, 246, 247
пиролиз 324
реакции с
галлием 310
иодистым метиленом 354
литийалюминийгидридом 309
трех бромистым бором 284
трехфтористым бором 283
фотолиз 325 сл.
Дивторичнобутилртуть 26, 224, 225
Ди-н-вторичнобутилфенилртуть 19
Дивторичнооктилртуть 26
Дигалокарбены, внедрение по связи угле-
род — ртуть 233, 287
Дигексадецилртуть 19
Ди-н-гексилртуть 26
Ди-н-гептилртуть 26
Ди-экзо-З-окси-экзо-2-норборнилртуть 249
Ди-(3-диметиламино-4-метилфенил)ртуть 47
Ди-п-диметиламинофенилртуть 32, 245
Ди-о-дифенил ртуть 201
Диизбамилртуть 30, 43 .
Диизобутилртуть 43
Диизогептилртуть 319
Диизопропилртуть 26, 38, 40, 43, 224, 225
окисление 264
пиролиз 324
реакции с хлороформом и четыреххлори-
стым углеродом 299
2,3-Ди(иодмеркурметил)-1,4-диоксан 140
Ди-п-иодфенилртуть 188
1, Г-Дииодферроцен 281
Ди-а-камфенилонилртуть 254
Ди-л-ксилилртуть 44
Ди-п-ксилилртуть 44
о-Дилитийбензол 305
Димезитилртуть 44
Диментилртуть 225
Димеркурмалоновая кислота, внутренняя
соль 61
1,6-Димеркурциклодекан 43, 247
n-Диметиламинофенил ртуть
уксуснокислая 87
хлористая 32
симметризация 245
бис-n-Диметил аминофенил ртуть 47
Ди-( 1 -метил-2-ацето кси-1 -пропен-1 -ил)-
ртуть 212, 285
Диметилберрилий 307
Ди-5-метилгексил-2-ртуть 26
Диметилдиселенид 372
Дйметилртуть 25, 30, 37, 38, 41, 42, 249
пиролиз 324
реакции с
алкиллитием 306
алюминием 309
бериллием 307
галлием 310
диметилмагнием 320
индием 310
иодцианом 299
кадмием 308
магнием 308
окислами азота 317
сурьмой 315
треххлористой сурьмой 292
цинком 308
фотолиз 325
Ди-р-метил-р-фенилпропилртуть 19
2,4-Диметилфенилртуть 69
бис-(Ди-р-метил-р-фенилэтил)ртуть 27
Диметилциклогексилртуть 224
б«с-(Диметоксифосфинил)ртуть 383
Ди-а-нафтилртуть 44, 45, 188, 245
реакция с теллуром 316
Ди-Р-нафтилртуть 44, 45
реакция с сулемой 260
Ди-а-нафтилтеллур 316
Динеопентилртуть 26
Ди-З-нитро-4-толилртуть 254
Ди-о-нитрофенил ртуть 249
Ди-я-нонил ртуть 27
1,7-Диокса-4,10-димеркурациклододекан
247
Ди-л£-оксифенилртуть 253
Диоктадецилртуть 19
Ди-я-октилртуть 27, 43
Ди-окт-1-инил-ртуть 28
Дипентаметилфенил ртуть 44
Дипентафторфенилртуть 248
Диперфторизопропилртуть 324
424
Предметный указатель
Диперфторметилдиселенид 373
Диперфторэтилртуть 325
Диперфторэтилсульфид 316
Ди-З-пиренилртуть 33
Дипропенил ртуть 31, 125, 229
транс-/пранс-Дипропеиилртуть, реакция с
бромистым таллием 285
транс, транс-Дипропенилталлий бромистый
285
Дипропиленоксиддимеркурсульфат 142
Ди-м-пропилртуть 19, 25, 38, 42, 250
пиролиз 324
реакция с
алюминием 309
бериллием 307
хлористым таллием 285
цинком 308
фотолиз 325
Ди-п-пропилфенилртуть 44
Ди-н-пропилфосфоиат' 383
Ди-н-пропоксифосфииилмеркургалогениды
383
Дипсевдокумилртуть 44
Ди-а-стильбенилртуть 31
Дистирилртуть 16, 29
Ди-со-стирилртуть 31
Дитетрадецилртуть 19
Ди-а-тиеиилртуть 251
Ди-п-толилмеркуркротоновый альдегид 235
Дитолилртуть 32, 44, 199, 248, 251, 257
Ди-о-тол ил ртуть 44, 188
реакция с серой 316
Дитретичноамилртуть 26
окисление 264
Дитретичнобутилртуть 26
Ди-(триметилсилил)метилртуть 25
бис-(Дитрифторметиламин)ртуть 375
Ди-м-ундецйлртуть 27
Дифеиетилртуть 47
Ди-р-фенилацетиленид ртути 28
Дифенилбериллий 307
Дифенилдиазометан,-реакция с сулемой 190
Дифеиилен 303, 304, 305
о-Дифениленртуть, тетрамер 32, 250
пиролиз 328
реакция с серебром 303
фотолиз 328
бис-тИ-Дифениленфенилртуть 44
бис-Дифениленэтилен 300
о-Дифенил ил ртуть хлористая 32
Дифенилиндий хлористый 285
Дифенилиодоиий хлористый 195, 295
Дифенилкадмий 308
1,3-Д ифе н ил - 2 - мет ил а цети ле ии л гл икол ь,
реакция с сулемой 170
2,5-Дифенил-4-;метилфурил-3-ртуть 170
Ди-т-фенилпропилртуть 19, 27
Дифенилртуть
обмен с Hg203 321
пиролиз 324
получение 16, 44, 47, 243, 245, 248, 254,
257
из фенилгидразина 191
из фенилдиазония 188
через органические производные'
алюминия 38
лития 33
магния 19, 29, 30
олова 211
реакции с
алюминием 309
беррилием 307
висмутом 315
галлием 310
индием 310
иодистым германием 311
йодоформом 298
кадмием 308
литийалюмииийгидридом 301, 310
окислами азота 317
оловом 311, 315, 320
палладием 303
селеном 316
серой 316
сероуглеродом 334
трехбромистым висмутом 293
треххлористой сурьмой 292
треххлористым бором 284
треххлористым таллием 286
хлоридами металлов 293, 294, 313, 327
цинком 308
.четыреххлористым углеродом 298, 328
этиллитием 306
фотолиз 325сл.
Дифеиилсвинец, диацетат 211
Дифеиилселен 316
Дифеиилсурьма хлористая 292
Дцфенилталлий хлористый 286
2,4-Дифеиилтиенилфеиилртуть 234
Дифеиилхлорарсин 292
Дифенилхлорфосфин 291
Дифенилцинк 308
Ди-З-фенилэтилртуть .19, 27
Ди-п-феноксифенилртуть 29
Диферроценил 304
Диоерроценилдисульфид 293
Диферроценилртуть 243, 248
пиролиз 324
реакции с
бутиллитием 306, 320
палладием 304
трифенилхлорметаном 296
хлористой медью 327
хлористым оловом 302
четырехбромистым селеном 293
четыреххлористым углеродом 299
Диферроценилселен 294
Дифторацетиленид ртути 54
Ди-ж-фторфенил ртуть 29
Ди-а-фурилртуть 248, 253
Ди-о-хлорбензилртуть 27
Ди-З-хлорвинилбор хлористый 283
Ди-З-хлорвинилиодоний хлористый 295
транс, транс-Ди-З-хлорвинилолово 313
реакция с сулемой 210
Ди-З-хлорвинилртуть 211, 255
обмен с Hg293 322
пиролиз 324
реакции с
сулемой 260
хлористым оловом 313
хлористым таллием 125
хлором 279
транс, /пранс-Ди-З-хлорвииилртуть, прев-
ращение в ^ис-изомер 329
Ди-1{ис-3-хлорвинилталлий 211
2,3-Дихлор-4,5-дихлормеркуртиофен 208
Предметный указатель
425
1,3-Дихлормеркурбензол 203
1,4-Дихлормеркурбутан 247
2,2'-Дихлормеркурдифенил 250
2,6-Дихлормеркурметилдиоксан 145
2,6-Дихлормеркурметилморфолин 149
4,6-Дихлормеркуррезорцин 74
2,5-Дихлормеркуртиофен 250, 252
1, Г-Дихлормеркурферроцен 204, 250
Дихлормеркуруксусная кислота, этило-
вый эфир 152
Дихлормеркурциклопентадиенилтрикарт
бонилмарганец 103
Дихлорметилртуть 191
1,1 '-Дихлор-2-триметилсилилциклопропан
288
Ди-л-хлорфенилртуть 29, 211, 245, 248
бис-2,5-Дихлорфенилртуть 70, 259
1,1-Дихлорциклопропан 287
Дициклогексенилртуть 31, 32
Дициклогексилртуть 28, 44, 224
окисление 264
пиролиз 324
фотолиз 326
Дициклопентадиенилртуть 36, 57, 62, 211
реакция с бромом 280
Дициклопентенилртуть 31, 32, 224
Дициклопропилртуть 20, 28, 300, 354
Дицимилртуть 44
Диэтилдифенилолово 315
Диэтилмагний 308
Диэтиловый эфир, ртутные производные
Диэтилртуть
получение 42
через органические производные
алюминия 38
бора 200, 225
магния 25, 30
цинка 37
электролизом 38
реакции с
алюминием 309
арсенобензолом 303‘
бериллием 307
висмутом 315
галлием 310
иодистым аллилом 299
йодоформом 298
кадмием 308
литием 305
магнием 308
натрием 307
пентаэтилдисиланом 336, 374
перекисью бензоила 299
три-о-тимотидом 336
триэтилгерманийгидридом 373
триэтилсиланом 336, 374
ураном 316
хлористым мышьяком 292
янтарнокислой ртутью 260
фотолиз 325
Диэтилфосфинилмеркурацетат 382
Диэтилцинк 308
Диэтоксифосфинилмеркурхлорид 383
бис-(Диэтоксифосфинил) ртуть 383
1-Додецилртуть хлористая 201
н-Додецилртуть, гидроокись 22
Изатин, ртутное производное 380
Изатин-[3-гидразон 362
Изоамилртуть хлористая 24, 25
Изобутил ацетоацетат 298
Изобутилртуть хлористая 24
Изопропенилацетат 297
Изопропенилртуть бромистая 31
Изопропилртуть бромистая 24, 25, 38
Изоцианурат ртути 381
Имидодигидроксамовая кислота 317
Инденилртуть бромистая 32
Иодмеркурацетон 62
2,3-бис-Иодмеркурметил-1,4-диоксан 140
2-Иодмеркурэтилбензоат 353
бис-Иодметилртуть 190, 354
Иодониевые соединения 195 сл.
о-Иодфенилртуть иодистая 187, 258, 328
бис-о-Иодфенилртуть 259, 324, 328
Иодферроцен 281
Иодциклопентадиенилтрикарбонилмарга-
нец 281
4-Камфил ртуть иодистая 277
Камфора мономеркурированная 57
Камфор-10-ртуть хлористая 202, 248
Ь1-(£-Карбоксипропионил)-М'-(2-метокси-3-
галоидмеркур)мочевина 161
Карбометокси-п-метоксифенилртуть 235
Карбометоксиртуть хлористая 235, 258
бис-Кар бметокси ртуть 258
л-Карбэтоксифенилртуть хлористая 186,
188
Кетоны, электролитическое восстановле-
ние на ртутном аноде 224
Кротилмеркурацетат, сольволиз 271
транс-Кротилртуть бромистая 23
о-Литийдифенил 305
Люизит I (3-хлорвинилдихлорарсин) 292
Малахитовая зелень меркурированная 89
Медь, порошок, приготовление для диазо-
метода 184
Меркаптали сахаров, расщепление сулемой
371
Меркаптиды ртути 372
Меркаптомеркурхлорид 363
Меркарбид, реакция с полухлористой серой
293
Меркур-бис-ацетальдегид 148, 307
Меркурацетамид 61, 377
Меркур-бис-ацетодиалкилметан 205
Меркурациклогексан 244
Меркурациклогептан 44
Меркурациламиды 377
Меркурбензамид 92, 378
Меркур-бис-л-бензойнометиловый эфир 189,
245, 260
Меркур-бис-диазоуксусноэтиловый эфир
62
1,3-Меркурдимеркаптопропан-2-сульфонат
натрия 368
Меркур-бис-л-дифенил 266
а-Меркурди-З-фенилангидрогидракриловая
кислота, действие кислот 266
Меркурирование
азоксибеизола 86
азулена 67
акридина 100, 234
алкиланилинов 82
426
Предметный указатель
Меркурирование
алкилацетиленов 53
алкйлбензолов 65
5-алкил-2,4-Диоксибензойных кислот 77
алкилселенофенов 97
алкилтиофенов 96
алкилфенолов 72
алкилфлуоресцеинов 79
2-алкилфуранов 94
аллилмалоновой кислоты 53
аллилового спирта 55
аллилфенилового эфира 74
п-аминоацетофенона 84
п-аминобензойной кислоты этилового
эфира 88
п-аминобензосульфамида 85
аминов, промежуточные продукты 85
2-аминодифеннлов 82
аминооксиариларсиновых кислот 77
n-аминоокисифенилсульфоновой кисло-
ты 85
2-аминопиридина 100
л-аминофенилтриметиламмонийацетата 83
аминофенолов 83
2-аминофлуорена 83
анетола 75
анизола 74, 75
а-анилидоизовалериановой кислоты эти-
лового эфира 84
анилина 82, 86
антипирина 98
антраниловой кислоты 84, 88
антрахинона 90
1-антрахинонсульфркислоты 90
антрацена 66, 70
антрона 90
2-арнл амино-4-метил-5-карбоэтокситиа-
зола 88
2-ариламинотиазолов 83
арилацетиленов 53
арилмочевины 83
ариловых эфиров гликолей 75
арилоксипропионовой кислоты 75
арилоксиуксусной кислоты 75
1-арил-5-пиразолонов 98
3-арилсиднонов 99
арилтиомочевины 83
ароматических спиртов 81
ароматическое
влияние заместителей 51
селективность 51
арсиновых кислот, ароматических 91 сл.
ауринтрикарбоновой кислоты 79
аценафтена 66
ацетальдегида 55
ацетамида 55
л-ацетанизидина 84
ацетанилида 87
2-ацетиламинопиридина 100
ацетилантраниловой кислоты метилово-
го эфира 85
ацетил ацетона 53
ацетиленилдивииила 54
ацетиленов 53, 59 сл., 234
ацетиленовых эфиров 53
ацетил-а-нафтиламина 83
ацетилсалициловой кислоты 77
n-ацетобензойной кислоты 58
ацетона 56, 60, 62
ацетоуксусной кислоты 53
n-ацетофениларсиновой кислоты 58
л-ацетофенилстибиновой кислоты 58
ацетофенона 56, 58, 60, 234
л-ацетфенетидина 84
л-ациламинобензойных кислот 85
бензальацетона 234
бензальацетофенона 235
бензальдегида 235
бензантрона 90, 91
бензаурина 78
бензидина 82, 87
бензиланилина 82
N-бензилацетамнда 86
бензилового спирта 81
бенз илфено ло в 71
бензо-1,4-дитиацнклогексадиена-2,5 99
бензойной кислоты 91, 92, 93
бензола 62, 64, 68, 69
бензолсульфокислоты 92
бензонитрила 92
бензофенона 90, 91
бензофуранов 94
бифенила 65
л-бром-ж-аминоацетофенона 84
бромбензола 67
1-транс-2-бромвинил-2-этинилбензола 54,
59
л-бромдиметиланилина 82
бромдифенилового эфира 74
1-бром-Г-иодферроцена 281
3-бромнафталина 68
л-бром-л-нитроацетофенона 58
З-бром-2, 4,6-триметилфенилацетилена 54
5-бром-2-фуранкарбоновой кислоты, ме-
тилового эфира 94
бутилацетилена 54
5-н-бутил-2,6-диоксибензойной кислоты
77
бутилового спирта 55
ванилина 76
вторичнобутилбедзола 65
галлового альдегида 76
галловой кислоты, метиловых эфиров
77
галоиданилинов 87
галоидксиленолов 72
галоидоксиарилалкилкетонов 76
галоидтиофенолов 96
гваякола 75
гексагидрофенола 57
гексилрезорцина 74
н-гептадиен-1-ина 53
декагидронафталнна 54
дец-транс-З-ен-1-ина 54
диазоуксусной кислоты 53, 62
3,5-диацетиламино-4-оксифениларсиио-
вой кислоты 85
диацетиленового спирта .54
дибензоилметана 234
п-дибромбензола 68
2,7-дибромфлуоресцеина 80
ди-о-крезола 74
димеркаптандибензилового эфира 81
4,4'-бис-диметил амннотрифенилацето-
нитрила 89
п-диметиламинофенилацетилена 54
диметиланилина 87
2,6-диметилпиридина 234
Предметный указатель
427
Меркурирование
диметил-н-толуидина 82
2,5-диметилфурана 95
Р, р-динафтиламина 82
динитронафтолов 72
2,4;Динитрорезорцина 74
динитрофенолов 71
2,4-диоксибензальдегида 76
о, о', п, п'-диоксидифенила 74
4,4'-диоксидифенилсульфида 81
2,7-диоксифлуорана 79
дитиосалициловой кислоты 82
N, N'-ди-п-толилформамидина 82
дифениламина 82 ,
дифеиилена 67, 70
дифенилового эфира 74
2,4-дифенилселенофена 97
дифенил сульфида 81, 82
дифенилсульфона 81, 82
2,3-дифенилтиофена 97
4,4-дифтор-2-бутен-1-ина 54
п-дихлорбензола 67, 70
ц«с-дихлорэтилена 54
диэтилтерефталата 94
2,5-диэтилтиофена 96
дурола 65
изоамилфенола 71
изованилина 76
изодурола 65
изохинолина 100
индандиона, натриевой соли 58
индена 234
индола 97, 102
иодбензола 67
иоддифенилового эфира 74
л-иод-лг-нитроацетофенона 58
Р-иодсильвана 94
иодтимола 72
2-иодфурана 94
камфоркарбоновой кислоты 57
камфоры 57
карбазола 97
4-карбоксифениларсиновой кислоты 93
карбоновых кислот, ароматических
91 сл.
2-карбэтокси-2-метил-.1-этииилциклогек-
силкарбинола 53
карвакрола 71, 74
кетонов
алифатических 56
ароматических 90 сл.
жирноароматических 58
крахмала 55
л-крезола 72, 74
крезотиновойхкислоты 77
ксантона 90
ксиленолов 71
.«-ксилола 65, 69
кумарина 78
лейкооснования метиленовой сини 101•
малахитовой зелени 86
малоновой кислоты 61, 234
этилового эфира 53, 60
масляного альдегида 235
мезитилена 65
метаниловой кислоты 85
метилен-бис-малоновой кислоты 53
метилацетилена, сполна дейтерированного
53
2-метилбензофурана 94
метилендисалициловой кислоты 77
метил'изопропилкетона 56, 62
метилкетола 97
метил мало новой кислоты 53
2-метилмеркаптофурана 94
2-метил-4-оксидифенилсульфида 81
5-метилрезорцина 74
S-метилтиосалициловой кислоты 82
метилфенилсульфида 81
метилхинолинов 100 ;
метилциклопентадиенилтрикарбонил мар-
ганца 103
мегилэтилкетона 56
2-метил-5-этилтиофена 102
2-метокси-3,5-тиенил-б«с-меркурхлорида
101
2-метокситиофена 101
4-л£-метоксифенилбутина-1 54
7-о-метоксифеноксигептил-1-ина 53
2-метоксифурана 94
моноацетиленового спирта 54
монохлорацетилена 54
нафталина 69
а-нафтил амина 82, 87
а-нафтиламино-4-сульфоновой кислоты
85
Р-нафтиламино-6-сульфоновой кислоты
85
а-нафтилацетилена 54
1-нафтойной кислоты 93
а-нафтола 71, 78
Р-нафтола 71, 73, 75
нафтолсульфокислоты 77
нитроанизола 74
ж-нитроанилина 87
нитробензойных кислот 92
нитробензола 68, 70
5-нитрогваякола 75
нитрозотимола 72
нитрокрезола 72
нитронафтиламинов 84, 87
нитронафтолов 72
нитро-лг-оксибензальдегида 76
З-нитро-4-оксидифенила 72
5-нитро-8-оксихинолина 100
нитрорезорцина 74
нитросалициловых альдегидов 76
нитротиофенолов 81, 96
нитротолуидинов 84
нитротолуолов 68, 70
нитроуксусной кислоты 53
/г-нитрофенилацетилена 54
п-иитрофенилферроцена 103
нитрофенолов 71, 73
нонадиина-1,4 54
оксиарилалкилкетонов 76
оксиариларсиновых кислот 77
оксибензальдегидов 76
оксибензойных кислот 77
оксидифенилсульфида 81
оксикарбоновых кислот, ароматических
77
оксимеркуруксусной кислоты 61
7-окси-4-метилкумарина 78
1,1-бнс-(4'-окси-6-метилфенил)циклотек-
сана 72
1-окси-2-нафтойной кислоты 77
428
Предметный указатель
Меркурирование
оксифенилметилкетона 76
1,1-бис-(4'-оксифенил)циклогексана 72
окт-1-нна 59
парафуксииа 86
пентаметилбензола 65
4,1 -пентаметиленбицикло-(0,1,4)-гептаиа
57
пеитафторбутина 54
г(«с-пеит-2-еи-4-ииа 54
а-пиколйна 100
пинаколина 56
3-пиридилсидиона 99
пиридина 102
пировиноградной кислоты 55
пиррола 97, 102
1-пирролидиициклопентена 57
полистирола 66
пропилового спирта 55
пропионата натрия 54
пропионового альдегида 235
пропионовой кислоты 234
псевдокумола 65
пуриновых оснований 101, 103
резорциловой кислоты 77
резорцина 73, 74
резорциисульфоифталеина 80
. рутеноцеиа 103
салигенина 73
салициловой кислоты 76, 77, 78, 234
салицилсульфоновой кислоты 77
салицилсульфоифталеина 80
сахара тростникового 55
селенофена 97
сильвана 94, 101
скатола 97
сульфаниламида 88
сульфоновых кислот, ароматических
91 сл.
сульфосукцинага 55
терефталевой кислоты 93
3-тетрагидропиранилоксипроп-1 -ина 59
1,2,3,4-тетраметнлбензола 65
4-тетраметилбутилфенола 72
2,2,5,5-тетра-(4-оксифенил)гексана 72
тиазола 98, 102
тимола 71, 74
тиоиафтеиа 97
тиосалициловой кислоты 81
тиофена 95, 101, 234
тиофенолов 81 сл.
толидина 82
л-толилмеркаптоацетилена 53
толуидинов 82
о-толуиловой кислоты 91
толуола 63, 65, 69
«-толуолсульфокислоты 92, 93
третичнобутилбензола 65
п-третичиобутилфеиола 71
трйалкилфуранов 94
Гтридецилацетилеиа 53
• трифенилметана 234
1,1,1-трифторпропина 54
трихлорбензола 68
трихлорэтилена 54, 59, 234
уксусного альдегида 235
уксусного ангидрида 55
уксусной кислоты 55
«гранс-уидец-7-еи-1-ина 53
фенантрена 66
фенетола 74
фенилацетилена 234
фенилглицинэтилового эфира 84, 87
феиилдиацетилеиа 54
фенилдиоксидибензопирана 78
феиилмеркаптоацетилена 53
феиил-3-нафтиламииа 82
2-феиил-а,3-нафтопиразола 99
1-феиилпиразола 98, 102
З-фенил-5-пиразолона 98
N-фенилпиррола 97
<в-фенилпропилового спирта 81
феиилсалициловой кислоты 77
фенил уксусной кислоты 92
феиилэтинилкарбинолов 54
фенола 71, 72, 74, 75, 76, 78, 234
феиолальдегидов 76
п-фенолсульфокислоты 77
фенолсульфонфталеина 80
феносафранина 101
ферроцена 103
флороглюцина 73
флороглюцинальдегида 76
флуорена 65, 66, 234
флуоренона 90
флуоресцеина 79, 80
фталеинов 79
£о-фторалкинов 54
фторацетилена 54(
п-фторбензойиой кислоты 92
фторбензола 67
«-фторфенола 71
фуксина, основания 86
фурана 94, 101
фурфурилового спирта 94
фурфурола 94
хинальдина 234
хинолина 100, 102
«-хлоранилина 87
«-хлорацетофенона 58
о-хлорбензойной кислоты 91
хлорбензола 67
хлоркрезолов 72
4-хлоррезорцина 74
хлортимола 72
хлорфенолов 71
холестерина 54
Д2-хромена 99
целлюлозы 55
цианацегамида 53
цианпропионовой кислоты 53
циануксусной кислоты 234
циклогексилфенолов 71
циклогептатриен-1-ол-2-она 58
циклопентадиена 57, 62, 233
циклопентадиенилтрикарбонилмаргаица
103
циклопентадиенилтрикарбонилреиия 103
циклопентаноиа 56
«-цимола 65
эвгенола 75
а-элеостеарата 54
эозина 80
этилового спирта 55, 60
этилрезорцииа 74
а-этоксинафталииа 74
Меркурирующие агенты 52 сл
диизобутират ртути 66
Предметный указатель
429
ди-н-толилртуть 235
дифенилртуть 235
карбоновых кислот, ртутные соли 53
лауринат ртути 67
меркурацетамид 71, 52, 53
миристат ртути 64
монохлорацетат ртути 65
нитрат ртути 52, 54, 56, 61, 62, 64
окись ртутн 54, 64
оксимеркурцианид 73
пентахлорфеноксимеркурацетат 65
перхлорат ртути 52, 63
сулема 52, 53, 71
сульфат ртути 52
тринитрометан, ртутная соль 63, 65, 75,
132
цианистая ртуть 52, 54
10,10-Меркур-бис-камфора 248
3,3-Меркур-бас-ментан 44
Меркур-б«с-метилпропилкетон, реакция с
кетенами 151
Меркурмочевина 377
8-Меркур-1-нафтойная кислота, внутренняя
соль 207
Меркурофен-(4-оксимеркур-2-нитрофенолят
натрия) 73
Меркур-бас-пропионовая кислота, метило-
вый эфир 44
Меркур-бцс-3,|3'-пропионовая кислота 44
а-Меркур-бас-стильбен 244, 251, 257
реакции с
бромом 276
хлористым оловом 314
^цс-а-Меркур-бас-стильбен, превращение в
транс-изомер 329
Меркурсукцинимид 377
Меркур-бас-уксусная кислота
бутиловый эфир, реакции с хлористым
ацетилом 298
метиловый эфир 150, 244
реакции с
гексаэтилдистаиианом 315
хлористым ацетилом 128, 297
а-пропилвиниловый эфир 151
третичнобутиловый эфир 151
этиловый эфир, реакция с бромом 280
а-Меркур-бас-фенилуксусная кислота, эти-
( ловый эфир 257
' Меркурфталимид 92
Меркур-бас-хлормеркуруксусноэтиловый
эфир 274
Меркур-бир-циклопентадиенилтрикарбонил-
марганецJJ53
Метиламинортуть, азотнокислая 375
1-Метил-2-ацетбкси-1-пропен-1-ил-ртуть хло-
ристая 167, 356
цис-1 -Метил-2-ацетокси-1 -пропен-1 -ил-тал-
лий двуххлористый 285
бис-1 -Метил -1,2 -дигидробе нзофу р ил ртуть 244
Метиллитий 306
Метилмеркур-н-проп илмеркурсульфид 331
Метилмеркурсульфид 331
Метилмеркурэтилмеркурсульфид 331
Метилнатрий 305
4-Метил-З-нитрофенилртуть хлористая 197
Метилпентафторфенилртуть 235
Метилртуть
ацетат 16, 218, 249, 332
бензоат 16
бромистая 20, 24
гидроокись 330
иодистая 16, 21, 25, 220, 245
хлористая 24, 38, 201, 210, 222, 331
Метил фенил ртуть 231
Метил-2-хлормеркур-2-деокси-3,4,6-три-о-
ацетил-р,D-глюкозид, реакция с бромом
278
Метил-а-хлормеркуризопропилкетон 205
З-Метил-З-хлормеркурпентанон-2 205
1-Метил-1 -циклогексил меркурацетат, окис-
ление 275
4-Метилциклогексилртуть 23, 24, 239, 246,
277, 278
Метилэтилкетон, электролиз 225
а-Метоксивинилацетат 297
2-Метокси-5-тиенилмерку рхлорид 102
(З-(л-Метоксифенил)этилртуть иодистая 137»
272
2-Метоксициклогексилмеркурхлорид 322
1{ис-2-Метоксициклогексилнеофилртуть 229,
321
2-Метоксициклогексил ртуть 239
а-2-Метоксициклопентил ртуть хлористая
137
[З-Метоксиэтилмеркурацетат 165
Миндальный альдегид, диэтилмеркапталь
367
а-Нафтилмеркурнитрат 330
а-Нафтил ртуть
бромистая 24
иодистая 243
хлористая 69, 186, 187
ot-Нафтилртуть хлористая 252, 260
а-Нафтил сульфид ртути 366
бцс-Нафтилэтинилртуть 54
Неопентил ртуть хлористая 21, 277
о-Нитробензальдимеркуроксид 70
о-Нитробензилртуть 70
Нитробензойные кислоты 246
Нитрозометилуретан 190
З-Нитро-5-карбометоксифенилмеркурхло-
рид 198
4-Нитро-1-нафтол-о-меркурацетат 360
бис-о-Нитрофенилртуть 245
о-Нитрофенилртуть хлористая 70, 186, 245,
249
ж-Нитрофенилртуть хлористая 187, 202
n-Нитрофенилртуть хлористая 186, 202,
204
п-Нитрофенилфенилртуть 231
н-Нонилртуть бромистая 24
Норборнилртуть уксуснокислая 272
Нортрицикл ил ртуть хлористая 23, 198, 272
Окись углерода, реакция с солями ртути
124, 164 сл.
5-Оксиамилртуть хлористая 22
2а-Оксибицикло-(2,2,1)-гептан-6, а-карбо-
новая кислота, лактон 301
n-Оксимеркурантраниловая кислота, внут-
ренняя соль 88
о-Оксимеркурбензойная кислота, внутрен-
няя соль 93.
5-Оксимеркурванилин 76
4-Оксимеркур-2,7-дибромфлуоресцеин 80
430
Предметный указатель
2-Оксимеркуризофталевая кислота, внут-
ренняя соль 206
Оксимеркурирование 134 сл,
8-Оксимеркур-1-нафтойная кислота 93
4-Оксимеркур-3-окси-2-нафтойная кислота,
внутренняя соль 78
Оксимеркурпропионовая кислота, внут-
ренняя соль 61
о-Оксимеркурсалициловая кислота 77
2-Оксимеркуртерефталевая кислота, внут-
ренний ангидрид 93
о-Оксимеркурфенол, внутренний фенолят
14
2-Окси-1-нафтилртуть уксуснокислая 73
3-Окси-2,2,3-триметилбутилртуть 172
о-Оксифенилмеркурсукцинимид 335
л<-Оксифенилмеркурхлорид 198, 253, 355
n-Оксифенилртуть хлористая 72, 186
4-Окси-5-хлормеркур-3,6-эндоксогексагид-
рофталевая кислота, диметиловый эфир
157, 278
4-Оксициклогексил ртуть 275
З-Оксиэтилртуть иодистая, бензоилирова-
ние 353
З-Оксиэтил-п-толилртуть 232
экзо,^ис-3,6-энд-Оксо-А4-тетрагидрофтале-
вая кислота, диметиловый эфир 278
н-Октадецилртуть хлористая 22
н-Октилмеркурхлорид 22, 199
Палладий металлический 303
Пентафенилсурьма, реакция с металличе-
ской ртутью 212
Пентафенилфосфор, реакция с сулемой 212
Пентафториодбензол 281
бцс-Пентафторфенилртуть 29, 47
реакция с
иодом 281
конц. серной кислотой 266
Пентацетилглюкопиранозилгуанин 381
Перфтор-2-азапропен 375
Перфторвинилбор двухлористый 283
Перфторвинилдихлорарсин 292
Перфторвинилмеркурхлорид 210, 230, 258
бцс-Перфторвинилртуть 27
б«с-Перхлорвинилртуть 252
Пиррол, реакция с сулемой 380
Поли-(п-иодстирол) 281
Пол ифторал кил нитро зосоед инен ия 317
Пропаргилртуть бромистая 23
Пропилмеркаптид ртути 363
л-Пропилртуть
бромистая 21, 24, 25
янтарнокислая 250
\
Ртутноорганические соединения
ацидолиз, кинетика 266 сл.
бумажная хроматография 395
газовая хроматография 395
меры предосторожности прн работе 10
несимметричные, диспропорционирова-
ние 238
озонирование 356
окисление 275
реакции с
азотной кислотой 317
арилхлорсульфидами 298
N-бромамидами 282
N-бромимидами 282
бромом 276 сл.
бутиллитием 318
галоидами 275 сл.
галоидуглеводородами 296
диазометаном 232
диоксандибромидом 276
дихлоркарбеном 218
иодом 276 сл.
иодом хлористым 272
карбонилами металлов 275
кислородом 264 сл.
кислотами 265 сл.
кетенами 318
литием 305
литийалюмогидридом 301
натрием 305
натрийборгидридом 301
нитрозилом хлористым 282
реактивом Фентона 275
роданом 282
сероводородом 274
тетраацетатом свинца 290
тионилхлоридом 293
треххлористым фосфором 290 сл
фтальимидом 334
хлорангидридами кислот 290 сл.
хлором 276 сл.
щелочами 274
симметризация 238 сл.
симметризующие агенты
алкоголяты калия и натрия 249
алюминия окись 258
амальгама
кадмия 242, 246
меди 246
натрия 41, 242
аммиак 242, 255 сл.
бутиллитий 257
винилэтиловый эфир 257
гидразин 248 сл.
гидроксиламин 248
ди-п-аминофенилртуть 235
дифенилртуть 257 сл.
железа пентакарбонил 258
железо, гидрат закиси 246
калий
иодистый 242, 250 сл.
роданистый 252
цианистый 252
кальций хлористый 252
магний 246
медь 242, 244 сл.
натрий 243
гидросульфит 246
иодистый 250 сл.
станнит 241, 246 сл.
тиосульфат 242, 253
цианистый 242, 252
семикарбазид 248
серебро 246
сероуглерод 254
сплав олово — натрий 244
третичные фосфины 258
триэтаноламин 256
фенилгидразин 248
цинк 245 сл.
щелочи 254, 255
токсичность 10
Предметный указатель
431
Ртуть
ацетилениды 16, 36, 53, 59
гремучая 361, 362
двойная соль с циановой и соляной кисло-
тами 360
закисная, соли 16, 359
метилат 359
пентахлорфенолят 359
пикрат 360
соли, присоединение к
акриловой кислоте
а-алкоксиакрилоннтрилам 127
аллену 140
аллиламидам-а-амннокнслот 161, 162
о-аллиламинобензойной кислоте 149
алл ил ацетату 145
аллилацетоноксиму 152
аллилацеторамнозе 142
аллнлбензолу 135, 138
аллнлбнурету 161
аллнлвероналу 161
2-транс- аллилдекалнну 135
3-аллнл-1,2,5,6-днизопропнлнден-с1-ман-
ннту 143
аллнлдифенилуксусной кислоте 155
аллил-d-манниту 142
аллилмочевине 161
аллиловому спирту 141, 142
3-аллилокснсульфолану 162
аллнлрезорцннам 145
аллнлсукциннмнду. 161
аллнлсульфонамнду 145
о-аллнл-п-третнчнобутнлфенолу 145
аллилуксусной кислоте 154
аллилфеноксиуксусной кислоте 159
о-аллилфенолу 117, 144, 145
аллилциклопентану 135
аллокоричной кислоте 153
антипирину 163
арнлазокарбоновокислому калию 189
1-арнл-5-пиразолону 163
ацетатмеркурметилфенилкарбинолу 138
2-ацетатмеркур-З-окситетрагидронафта-
лину 138
ацетилендикарбоновой кислоте, днме-
тиловому эфиру 170
ацетиленовым кетонам 128, 129, 170
ацетиленовым кислотам 170 сл.
ацетиленовым спиртам 128, 129
ацетнленпинаконам 169
ацетилену 120, 166
бензальацетофенону 150
бензнорборйену 135
бициклогептадиену 140, 141
бициклдгептенкарбоновой кислоте 155
бицикло-(2,2,2)-октену 135
бутадиену 139, 140
бутенам 133
бутен-1-олу-4 142
винилацетату 147
винилацетилену 168
виннлбутнловому эфиру 146, 148
виннлглюкозе 142
N-виннлкарбазолу 163
виниловым эфирам 126 сл.
винилфталимиду 161
винилциклогексену 140
винилэтиленгликоля монометиловому
эфиру 142
виннлэтнловому эфиру 146, 147
гексадиену-1,5 140
гексафторпропилену 138
гексен-1-олу-5 142
гептину-1 168
глутаконовой кислоте 158
1,6-диазобицикло-(4, 4, 0)-3-децен-7, 10-
диолу 163
диаллнламину 149
дналлнлмалоновой кислоте 155
дналлиловому эфиру 145
дналлилсульфиду 145, 146
диаллнлсульфону 146
диаллилу 139
диаллилуксусной кислоте 155
дибензальацетону 150
дивннилбутнралю 148
дивиниловому эфиру 146
днгндронафталину 135, 138
2,3-днгидропнрану 163
днметаллнлсульфиду 146
диметилацетилену 167
2,6-днметилгептен-5-олу-2 142
2,5-днметил-3-децен-7,10-днону 163
днметнлфенилфенилацетиленилэти-
ленгликолю 169
днметилфосфонату 383
1,1-днметнлциклопропану 172
дипропеннлу 139
днпропиленокснду 141
дифенилацетилену 167
1,1-днфенилциклопропану 172
1,2-днфенилцнклопропану 172
1,1-днфеннлэтилену 135
1,4-дихлорбутину-2 167
дициклопентадиеиу 135
2-диэтнламинометил-З-вннилхннукли-
дину 163
изобутилену 133
изолауролену 134
изопрену 139, 140
изопропеннлацетату 147
изопропенил-«-бутиловому эфиру
146
изопропнлэтиловому эфиру 148
итаконовой кислоте 153
камфену 134
М-(3-карбоксипропнл)-М'-алл ил мочеви-
не 161
2,7-карбоэтокси-2-гептеновой кислоте
154
карвоментену 134
кетену 127, 150
коричной кислоты, аллиловым эфирам
118, 158
коричному спирту 144
n-кротиламииофенилуксусной кислоте
149
кротоновой кислоте 153
кумариновой кислоте 159
кумарину 159
кумаровой кислоте 159
малеиновой кислоте 153
метакриловой кислоте 153
метилацетилену 168
1-метил-2-ацетокси-2-пропен-1-ил-рту-
ти хлористой 167
метил-5-гептен-1-олу-5 142
432
Предметный указатель
соли, присоединение к
метилгептеноноксйму 152
2-метил-1,4-дихлормеркур-2,3-диокси-
бутану 140
1,4-метилен- А 5-циклоГексен-2-карбо-
новой кислоте 155
. 3-метилциклогексену 116
нитрокоричным кислотам 158
2-ноненовой кислоте 154
нопинену 136
норборнен-эндо-^мс-2,3-дикарбоновой
кислоте 155
норборнилену 134
окиси углерода 164 сл.
октадецен-9-ил-амину 149
октину-1 168
экзо, «с-3,6-эндо-оксо-Д-4-тетрагид-
рофталевой кислоты диметиловому
эфиру 157
олеиновой кислоте 154
2,7-пентадецену 133
пентен-1-олу-5 142
пинену 136
пиперилену 139
полифторалкенам 133
пропаргиловому спирту 169
пропенилбензолу 135
пропилену 133
а-пропилкротоновой кислоте 154
пропиоловой кислоте 170
а-пропоксиакрилонитрилу 154
сорбиту, о-аллиловому эфиру 142
стильбену 135
стиролу 135, 138
сульфол ену-3 162
а-терпинеолу 144
1,2,3,6-тетрагидропиридазину 163
тетраметил-1,1,5,5-пентен- 1-олу 142
тетраметилциклопропану 172
тионафтенсульфону 162
2,3,3-триметилбутену-1 133
триметилсилилэтилену 133
1,1,2-триметилциклопропану 172
1,1,1-трифторпропилену 138
трифторэтилену 139
ундецену 133
ундециловой кислоте' 154, 171
фенилацетилену 168
фенилбутадиену 140
фенилвинилэтиловому эфиру 147
фенилметилдиацетилену 168
5-фенил-2-пентеновой кислоте 154
фенилциклопропану 172
фенилэтинилметилкетону 170
хаульмугровой кислоте 155
циклогексен-3-олу-1 143
циклогексену 134, 138
p-циклопентилакриловой кислоте
153
циклопентену 134, 137
цитраконовой кислоте 153
эвгенолуксусной кислоте 159
эруковой кислоте 154
этилену 130, 131
этилциклопропану 172
стеарат 136
феноляты 359
этилат 359
этилтиоацетат 363
а-Стирилртуть бромистая 23, 31
ю-Стирилртуть бромистая' 16, 198
п-Стирилфенилртуть 229
Сукцинимид 334
Сулема 18, 130, 169 сл., 361
Сульфоуксусная кислота 293
о-Терфениленртуть 46, 304
Тетраацетатмеркурдиацетонгидрат 60
Тетраацетатмеркурпиррол 102
Тетравинилат литийалюминия 309
Тетрагидрокарбазол 97
н-Тетрадецилртуть хлористая 22
Тетракис(трифторацетил)гидразин 376
2,2,3,3-Тетраметил-1 -бутилртуть 22
2,5,2.5'-Тетраметил-3,3'-дитиенилртуть
243
Тетраметилолово 210
Тетрафенилмеркурациклопентадиен 31, 44
Тетрафенилолово 312
бис-(1,2,2,2-Тетрафторэтил)ртуть 139
а,а/3,3'-Тетрахлормеркурдиизопропиловый
эфир 192
2,2',4,4'-Тетрахлормеркурдифениламин 82
Тетраэтилборнатрий, электролиз 225
Тетраэтилкремний 212
а-Тиенил ртуть
бромистая 211
хлористая 251
а-Тиенилталлий, диизобутират 211
бис-1-Тиенилэтинилртуть 54
а-Тионафтенилртуть уксуснокислая 252
Тионафтенмеркаптид ртути 366
Тиофенмеркаптид ртути 366
Тиоэтилртутъ хлористая 340
Тол ил меркурпентахлорфеиоксид ртути
65
Толилртуть
гидроокись 332
хлористая 69, 185, 186, 187, 257
/г-Толил-2,4,6-трииитрофенилртуть 231
/г-Толилтрихлорсилан 288 ’
/г-Толилфенилртуть 230
Третичноамилртуть бромистая 24
Третичнобутил ртуть бромистая 21, 24,
25
Триацетатмеркуртиазол 102
а,3,5-Триацетоксимеркур-3,2-диметоксиди-
гидрокоричная кислота 160
1,2,3-Трибромциклопентен 280
бис-Тридейтерометилртуть 47
Триизобутират таллия 286
Тримеркурдиацетонгидрат 60
Триметилвинилоксисилан 288
Триметилмеркуроксоний, перхлорат 331
Триметилсилан 212
Триметилсилилметилртуть хлористая 21
1,2,4-Триметилфениленртуть 45
Тринитрометилфенилртуть 68
бис-Тринитропропилртуть 132
1,1,1-Тринитро-3-(тринитрометилмеркур)-
пропан 132
1,1,1 -Трииитро-З-хлормеркурпропан 132
Трипропилгерманилуксусная кислота, ме-
тиловый эфир 289
Трифенилметилацетальдегид 297
Трифенилметилферроцен 296
Трифторацетат ртути 206
Трифтормеркаптид ртути 365
Предметный указатель
433
Трифторметилмеркаптид ртути 364
Т р ифтор метил ртуть
иодистая 15, 246
трифторацетат 206
бис-Трифторметилртуть 32, 246
Трифторметилсульфид ртути хлористый 364
бис-(м-Трифторметилфенил)ртуть 32
1,1,1-Трифтор-3-этоксипропил-2-меркурни-
трат 138
Трифуриларсин 2J1
^ис-Три-З-хлорвинилстибин 210
Трихлормеркуруксусная кислота, этило-
вый эфир 152
бис-Трихлорметилртуть 206
1,2,4-Трихлор-3-хлормеркурбутен-2 167
Трихлорэтиленциклогексилртуть 234
Триэтилперфторвинилолово 210
Триэтилстаннилуксусной кислоты эфиры
312, 315
Фенацилртуть бромистая 60
Фенилаллилртуть 280
Фенилацетиленид ртути 59
Фенилацетилсульфид, реакция с сулемой
372
Фенилбориая кислота 284
Фенил-1 -бутадиенил-1,2-ил-ртуть хлори-
стая 37
Фенилвинилацетиленилртуть 280
Фенилдиазоний азотнокислый 317
Фенилдихлорфосфин 291
о-Фениленртуть, гексамер 29, 32, 45
Фенилкарбометоксиртуть 236
n-Фенилмеркаптофенилртуть хлористая 8
бис-Фенилмеркурбензол 229
п-Фенилмеркурбромид 22, 24
Фенилмеркуртиогликолевая кислота 334
1-Фенил-2-метокси-2-метилпропилртуть ук-
суснокислая 275
Фенил-о-оксициклогексилртуть 232
Фенил-З-пропин-2-ил-ртуть хлористая 37
Фенилртуть
акрилат 335
бромистая 24, 196, 204, 245, 319
n-винил бензоат 335
гидроокись 332
диалкилфосфит 331
метакрилат 335
уксуснокислая 68, 69, 191, 211, 248, 254,
256
фенолят 336
фтористая 259, 332
хлористая 24, 60, 185, 187, 189, 195, 200,
210, 222, 257
Фенилсвинец, триацетат 211
Фенил-я-толилхлорфосфин 291
Фенил-2,4,6-тр инитрофенилртуть 230
Фенилтрихлорметилртуть 229, 233
Фенилтрихлорсилан 288
Фенилферроценилсульфон 296
1-Фенил-1-хлормеркурбутен-1-он-3 170
Ферроцен 316
Ферроцениллитий 306
Ферроценилртуть хлористая 199, 248, 253,
281, 320
4-Флуорилртуть хлористая 281
2-Формилфенилртуть 198
л-Фторфенил ртуть ацетат 67
Фурилртуть хлористая 207, 208, 211, 253
Хараша ряд 267 сл.
8-Хинолилртуть 102
Хинолин-З-меркурацетат 102
лгранс-З-Хлорвинилиодидхлорид 295
/праис-З-Хлорвинилмеркурхлорид 166,
198, 210, 257, 255, 295, 329
^uc-3-Хлорвинилмеркурхлорид 166, 210,
255, 260, 279, 280
2-Хлордифенилен 279
1-Хлор-1'-иодферроцен 281
1,2-Хлориодэтилен 279, 280
3-Хлоркамфор-10-меркурхлорид-202
1,8-бис-Хлормеркурантрахинон 203
Хлормеркурацетальдегид 146, 147, 280, 293,
297, 353
Хлормеркурацетон 111, 146, 255, 297, 354
л-Хлормеркурбензойная кислота 355
п-Хлормеркурбензойной кислоты эфиры 245
о-Хлормеркурбензойнометиловый эфир 93
о-бис-Хлормеркурбензол 259
о-Хлормеркурбензофенон 91
З-Хлормеркурбифенил 32, 251
1,4-бис-Хлормеркур-2,3-бутандиол 278
2-Хлормеркурдибензофуран 203
о,о'-бис-Хлормеркурдифенил 201
о-Хлормеркурдифенилсульфон 82
а-Хлормеркуризомасляная кислота, мети-
ловый эфир 152
2-Хлормеркуркамфан 23
а-Хлормеркуркамфенилон 254
2-Хлормеркур-п-крезол 73
Хлормеркуркумаронилмеркаптид 372
Хлормеркурметилбензилкарбинол 138
1-Хлормеркурметил-1,2-дигидробензофу-
ран 145, 244
2-Хлормеркурметилтетрагидрофуран 142
4-Хлормеркур-5-метокси-1,2-дикарбамил-
гексагидропиридазин 164
1-Хлормеркур-5-метокситрициклен 141
Хлормеркурметоксифенилбутен 140
а-1 - X ло р меркур-2- метоксициклогекса н 354
4-Хлормеркур-2-нитротолуол 254
а-1-Хлормеркур-2-оксициклогексан 354
1-Хлормеркурпропанол 119
4-Хлормеркуррезорцин 74
З-Хлормеркурсалициловая кислота, мети-
ловый эфир 77
а-Хлормеркурстильбен 244
3-Хлормеркур-2,5-тиоксен 243
лг-Хлормеркуртолуол 355
п-Хлормеркуртрифенилкарбинол 187
Хлормеркуруксусная кислота 151
метиловый эфир 244
Хлормеркурфенилацетальдегид 147
4-Хлормеркур-1-фенилпиразол 102
Хлормеркурфеиилсульфид 372
Хлормеркурферроцен 203, 204, 243, 306
Хлормеркурфумаровая кислота, метиловый
эфир 170
а-Хлормеркур-З-хлоракриловая кислота 170
З-Хлормеркурциклогексан-1,4-диол 144
Хлормеркурциклопентадиенилмарганец-
трикарбонил 103, 204, 253, 281
М-(3-Хлормеркурэтил)пиперидин 132
1-Хлормеркурэтилтриметилсилан 22
Хлорметилртуть хлористая 190
бис-(1-Хлор-1,2,2,2-тетрафторэтил)ртуть 316
п-Хлорфенилбензилртуть 231
n-Хлорфенилртуть хлористая 186, 203, 245
434
Предметный указатель
n-Хлорфенилталлий диизобутират 286
л-Хлорфенилфенилртуть 231
1-(Г-Хлорферроценил)ртуть хлористая
199
2-Хлор-3-хлормеркурпропен-2-ол 169
н-Цетилртуть 21
Циклогексан-13,4 “-диол 301
Циклогексилртуть
азотнокислая 275
бромистая 23, 24, 25
иодистая 44
уксуснокислая 271, 272
хлористая 23
Цикломеркурпентаметилен 243
Циклопентадиенилртуть хлористая 57, 255,
316
бис-Циклопентадиенилртуть, реакция с ма-
леиновым ангидридом 57
Циклопентадиенилталлий 211
Циклопентаметиленртуть 33
Циннамилмеркурбромид 16
Этаноксигексамеркарбид 60
Этанолмеркурбромид 116, 117, 131
Этанолмеркурхлорид 353
Этиламинортуть азотнокислая 375
Этилбутилртуть 229
Этилмеркаптид ртути 362
Этилмеркаптомеркурхлорид 368
бис-Этилмеркурсульфид 331
Этилмеркуртиосалициловая кислота 334
Этилртуть
алкоголяты 333
бромистая 21
Этилртуть
иодистая 25, 220
меркаптиды 333
тиофеноляты 333
феноляты 333
фосфат 333
хлористая 24, 37, 38, 220, 245
янтарнокислая 250, 260
Этил-2,4,6-тринитрофенилртуть 231
З-Этил-З-хлормеркурпентанон-2 205
Этилэтинилртуть 229
л-Этоксифенилмеркурбромид 25
/г-Этоксифенилртуть хлористая 185
В-Этоксиэтилтиортуть хлористая 369
ОГЛАВЛЕНИЕ
/
Предисловие..........................................-. ....................... 3
Введение...................................................................... 7
Глава I. Взаимодействие RHal с металлической ртутью и солями ртути ... 14
Глава II. Получение ртутноорганических соединений посредством реактива
Гриньяра и органических соединений лития...................(............. 18
Получение ртутноорганических галогенидов.................................. 20
Получение полнозамещенных ртутноорганических соединений................ 25
Получение полнозамещенных ртутноорганических соединений в эфире (и тет-
рагидрофуране) ................................................... 25
Получение полнозамещенных ртутноорганических соединений с отгонкой
эфира........................................................... 30
Получение ртутноорганических соединений при помощи литийорганических
соединений........................................................... 30
Глава III. Получение ртутноорганических соединений через органические сое-
динения прочих легких металлов........................................ 36
Образование органических соединений ртути через органические соединения
натрия и серебра..................................................... 36
Получение ртутноорганических соединений через органические соединения цинка 36
Получение ртутноорганических соединений при помощи органических соединений
алюминия ................................................................ 37
Глава IV. Синтез ртутноорганических соединений при помощи амальгам натрия,
лития, калия и кадмия............................................... 40
Г л а в а V. Введение ртути на место водородного атома (меркурирование) .... 50
Меркурирование жирных и алициклических структур........................ 53
Меркурирование ароматических углеводородов............................. 62
Меркурирование ароматических оксисоединений............................ 71
Меркурирование производных тиофенолов................................... 81
Меркурирование ароматических аминов.................................... 82
Меркурирование ароматических кетонов................................... 90
Меркурирование ароматических карбоновых, сульфоновых и арсиновых
кислот и их производных.............................................. 91
Меркурирование гетероциклических соединений............................... 94
Меркурирование ценов .................................................... 103
Глава VI. Присоединение солей ртути к непредельным соединениям и произ-
водным циклопропана ................................................. 115
Введение................................•................................ 115
Реакции продуктов присоединения солей ртути к олефиновым соединениям 115
Реакции продуктов присоединения солей ртути по тройной связи .... 120
Реакции продуктов присоединения солей ртути к окиси углерода .... 124
Строение продуктов присоединения солей ртути к олефинам, ацетиленам и
окиси углерода.................................................... 124
Реакции и строение а-меркурированных оксосоединений.................. 126
Реакции и строение продуктов присоединения солей ртути к ацетиленовым
спиртам, оксосоединениям и кислотам.................................. 128
Присоединение солей ртути подвойной связи............................... 129
Присоединение солей ртути к алкенам, циклоалкенам и их производным . 129
Присоединение солей ртути к оксисоединениям, содержащим этиленовую
связь ............................................................... 141
Присоединение солей ртути к аминам, содержащим этиленовую связь .... 146
Присоединение солей ртути к оксосоединениям, содержащим этиленовые связи, 149
и их функциональным производным................................... 150
436
Оглавление
Присоединение солей ртути к карбоновым кислотам, содержащим этиленовые
связи, и их производным................................................. 153 -
Присоединение солей ртути к алкениламидам кислот....................... 161
Присоединение солей ртути к гетероциклическим соединениям, содержащим
этиленовые связи в цикле или боковой цепи.......................... 162
Присоединение солей ртути к окиси углерода............................... 164
Присоединение солей ртути по тройной связи............................... 165
Присоединение солей ртути к ацетиленовым углеводородам............... 165
Присоединение солей ртути к ацетиленовым спиртам..................... 165
Присоединение солей ртути к ацетиленовым кетонам...................... 169
Присоединение солей ртути к ацетиленовым кислотам................... 170’
Присоединение: солей ртути к производным циклопропана.................... 171
Глава VII. Диазометод синтеза ртутноорганических соединений................. 182'
Получение галогенида арилртути ......................................... 184
Получение двойной соли хлористого арилдиазония и сулемы................ 184
Приготовление порошка меди............................................. 184
Разложение двойной диазониевой соли в органических растворителях....... 185
Разложение двойной диазониевой соли в воде ...........*................ 187
Получение диарилртути.................................................. 188
Синтез органических соединений ртути при помощи жирных диазосоединений . . 190
Синтез ртутноорганических соединений действием арилгидразинов на соли
и окись ртути ....................................................... 191
Синтез ртутноорганических соединений через гидразоны................... 192'
Глава VIII. Синтез ртутноорганических соединений через галогенониевые сое-
динения . ............................................................... 195
Глава IX. Синтезы ртутноорганических соединений ртути. заменой на ртуть
кислотных остатков, атомов тяжелых металлов и некоторых металлоидов в
органических соединениях................................................. . 197
Замена на ртуть остатка борной кислоты, а также бора в других его органи-
ческих соединениях................................................... 197
Замена на ртуть остатка сернистой кислоты................................ 201
Замена на ртуть остатка йодноватой кислоты............................... 204
Замена на ртуть карбоксила в карбоновых кислотах.............’........... 205
Замена на ртуть атомов тяжелых металлов, а также некоторых металлоидов 209
Глава X. Синтез ртутноорганических соединений действием свободных радика-
лов (из перекисей и других источников) на ртутные соли карбоновых (и дру-
гих) кислот и на металлическую ртуть......................................... 217
Разложение перекисей в присутствии солей ртути........................... 217
Разложение солей ртути, инициируемое радикалами, образующимися под
действием УФ-света .................................................. 229
Разложение солей ртути, инициируемое радикалами, образующимися из
других источников...................................................... 221
Разложение перекисей в присутствии металлической ртути.................. 221
Глава XI. Получение ртутноорганических соединений электролизом ..... 224
Глава XII. Методы синтеза несимметричных полнозамещенных ртутноорганйче-
ских соединений RHgR'........................................................ 228
Синтез RHgR' через магнийорганические соединения......................... 228
Синтез RHgR' декарбоксилированием RCOOHgR'............................... 230
Синтез RHgAr' арилированием RHgOH ароматическими соединениями олова,
сурьмы (III) и бора ................................................ 231
Синтез RHgR' действием диазометаиа на RHgCl.............................. 232
Синтез RHgR' при помощи дигалокарбенов................................... 233
Синтез RHgR' «сосимметризацией» RHgX и R'HgX............................. 233
Синтез RHgR' действием RHgX (и RaHg) на соединения с подвижным водородом 234
Синтез RHgR' реакцией перераспределения радикалов в ртутноорганических
соединениях......................................................... 235
Глава XIII. Симметризация ртутноорганических соединений и обратная ей реак-
ция ....................................................................... 238
Реакции симметризации ртутноорганических соединений................... 243
Симметризация действием металлов...................................... 243
Симметризация действием гидросульфита натрия........................ 246
Симметризация действием гидрата закиси железа......................... 246
Симметризация действием станнита натрия............................... 246
Симметризация действием гидразина и родственных соединений........... 248
Оглавление
437
Симметризация действием алкоголята натрия (калия)...................... 249
Симметризация электролизом.............................................. 249
Симметризация действием йодистого калия (натрия)....................... 250
Симметризация действием цианистого калия (натрия) ..................... 252
Симметризация действием роданистого калия (натрия)..................... 252
Симметризация действием хлористого кальция............................. 252
Симметризация действием серноватистокислого натрия..................... 253
Симметризация действием сернистых щелочей............................. 254
Симметризация действием щелочей....................................... 255
Симметризация действием аммиака....................................... 255
Симметризация действием пиридина....................................... 256
Симметризация действием триэтаноламина . . . ........................ 256
Симметризация действием непредельных соединений....................... 256
Симметризация действием бутиллития.................................. . 257
Симметризация Действием дифенилртути.................................. 257
Симметризация действием третичных фосфинов............................ 258
Симметризация действием пеитакарбонила железа и его производных . . . 258
Симметризация действием окиси алюминия................................ 258
Симметризация без участия симметризующих агентов....................... 258
Реакция, обратная симметризации .................................... 259
Глава XIV. Реакции ртутноорганических соединений.......................... . 264
Действие кислорода........................................................ 254
Действие кислот........................................................... 265
Замена ртути иа водород и другие реакции с кислотами ртутноорганических
соединений, не являющихся продуктами присоединения солей ртути
к олефинам....................................................... 265
Реакции ртутноорганических соединений с кислотами, приводящие к выде-
лению металлической ртути (реакция демеркурирования).............. 270
Действие кислот иа продукты присоединения солей ртути к олефинам и их
производным....................................................... 272
Действие щелочей......................................................... 274
Действие сероводорода и сульфидов щелочных металлов...................... 274
Действие некоторых окислителей, в частности солей ртути................... 274
Действие карбонилов металлов............................................. 275
Действие галоидов...................................................... 275
Реакция с галогенидами и другими солями элементов, а также их алкил(арил)-
галогенидами и гидридами ............................................ 282
Синтез органических соединений элементов III группы периодической систе-
мы ................................. . ....................... 283
Синтез органических соединений элементов IV группы периодической систе-
мы ............................................................... 287
Синтез органических соединений элементов V группы периодической системы 290
Синтез органических соединений элементов VI группы периодической системы 293
Синтез органических соединений элементов VII группы периодической си-
стемы ............................................................. 294
Реакция ртутноорганических соединений с органическими галогенидами . . 296
Действие восстановителей на ртутноорганические соединения (в частности ре-
акция с металлами)................................................... 300
Действие металлов без образования металлоорганических соединений . . . 303
Синтез органических соединений лития и натрия через ртутнооргаиические
соединения........................................................ 305
Синтез органических соединений металлов II группы периодической системы 307
Синтез органических соединений металлов III группы периодической системы 308
Синтез органических соединений элементов IV группы периодической системы 310
Синтез органических соединений металлов V группы периодической системы . 315
Синтез органических соединений элементов VI группы периодической системы 315
Синтез органических соединений элементов VIII группы периодической системы 316
Реакции с окислами азота................................................. 316
Реакция с кетенами ...................................................... 318
Взаимодействие ртутноорганических соединений с реактивом Гриньяра и органи-
ческими соединениями лития, натрия, цинка, алюминия и других металлов 318
Изотопный обмен ртутноорганических соединений с радиоактивной ртутью
и соединениями, ее содержащими....................................... 320
Пиролиз ртутноорганических соединений ................................... 323
Фотохимические реакции ртутноорганических соединений..................... 325
Реакции в газовой фазе.................................................. 325
Реакции в растворе...................................................... 325
438
Оглавление
Чме-транс-Превращения непредельных ртутноорганических соединений . 329
Обмен анионов в ртутноорганических солях............................ 330
Гл а в а XV. Изменения в органической части молекулы ртутноорганических сое-
динений ............................................................... 353
Глава' XVI. Синтез и реакции соединений, в которых ртуть связана с органиче-
ским остатком не через углерод......................................... 358
Соединения с О—Hg-связью............................................. 359
О—Hg-производные спиртов и фенолов................................. 359
О—Hg-Производные алкилнитраминов................................... 360
О—Hg-Соединеиия кислородсодержащих производных циана .............. 360
О—Hg-Производные некоторых гетероциклов............................ 362
Соединения с S—Hg-связью............................................. 362
Алкилмеркаптиды ртути ......................................... 362
Арилмеркаптиды (тиофеноляты) ртути............................. 371
Меркаптиды некоторых гетероциклических соединений................ 371
Получение меркаптидов ртути действием солей ртути на некоторые дру-
гие S-содержащие соединения...................................... 371
Фотолиз и пиролиз меркаптидов ртути.............................. 372
Соединения с Se—Hg-связью........................................... 372
Соединения с Ge—Hg-связью....................................... . 373
Соединения с Si—Hg- и Si—Hg—Ge-связями............................... 374
Соединения с N—Hg-связью............................................ 374
N—Hg-Производные аминов........................................ 374
N—Hg-Производные амидов, имидов и гидразидов кислот.............. 377
N—Hg-Производные диазоаминосоединений............................ 379
N—Hg-Производные циановой и изоциановой кислот................... 380
N—Hg-Производные некоторых гетероциклов.......................... 380
Соединения с Р—Hg-связью............................................. 382
Глава XVII. Анализ ртутноорганических соединений........................ 390
Качественное определение ртути в ртутноорганических соединениях..... 390
Количественный анализ ртутноорганических соединений................ 390*
Элементарный анализ ртутноорганических соединений на С, Н, Hg и другие
элементы . ....................................................... 390‘
Определение углерода и водорода в ртутноорганических соединениях . . . 393
Определение ртути и других элементов, кроме С и Н, в ртутноорганических
соединениях . .................................................... 393
Авторский указатель................................................... 397
Предметный указатель............................................ . . . . 421
Любовь Геннадиевна Макарова,
Александр Николаевич Несмеянов
Методы элементоорганической химии.
Ртуть
Утверждено к печати
Институтом элементоорганических соединений
Академии наук СССР
Редактор А. И. Родионов
Технические-редакторы Ф. М. Хенох, И.И. Дорохина
Сдано в набор 40/111 1964 г. Подписано к печати 23/IV 1965 г.
Формат 70xl08’/i«. Печ. л. 27 75. Усл. л. 38,01. Уч.-изд. л. 39,8
Тираж 2800 экз. Т-06703. Изд. Ns.3204/04. Тип. зак. № 1546
Цена 2 р. 95 к.
Издательство «Наука». Москва, К-62, Подсосенский пер., 21
2-я типография издательства «Наука». Москва, Г-99,
Шубинский пер., 10
ИСПРАВЛЕНИЯ И ОПЕЧАТКИ
Стр. Строка Напечатано Должно быть
35 12 СН. J. Inorg. Nuclear. Chem. Inorg. Chem.
36 10 сн. AgC2- AgaCa*
70 22 сн. воду в воду
91 14 сн. [192, [167,
106 4 св. 1410 2410
229 13 св. C3H5HgBr C3H5HgI
233 16 сн. C —H C-Hg
333 6 сн. , анидина циангуанидина
334 18 сн. [746] . [765]
353 19 св. и Р-хлорвинилмер- курхлорида [16] (ср. Р-хлорвинилмер- курхлорид [16])
365 17 сн. [ИЗ] [ИЗ] ртути
337 25 сн. (1901) (1913)
424 31 сн. 47 47, 188
429 32 сн. а-Нафтилртуть Р-Н афтил ртуть.
Л. Г. Макарова, А. Н. Несмеянов