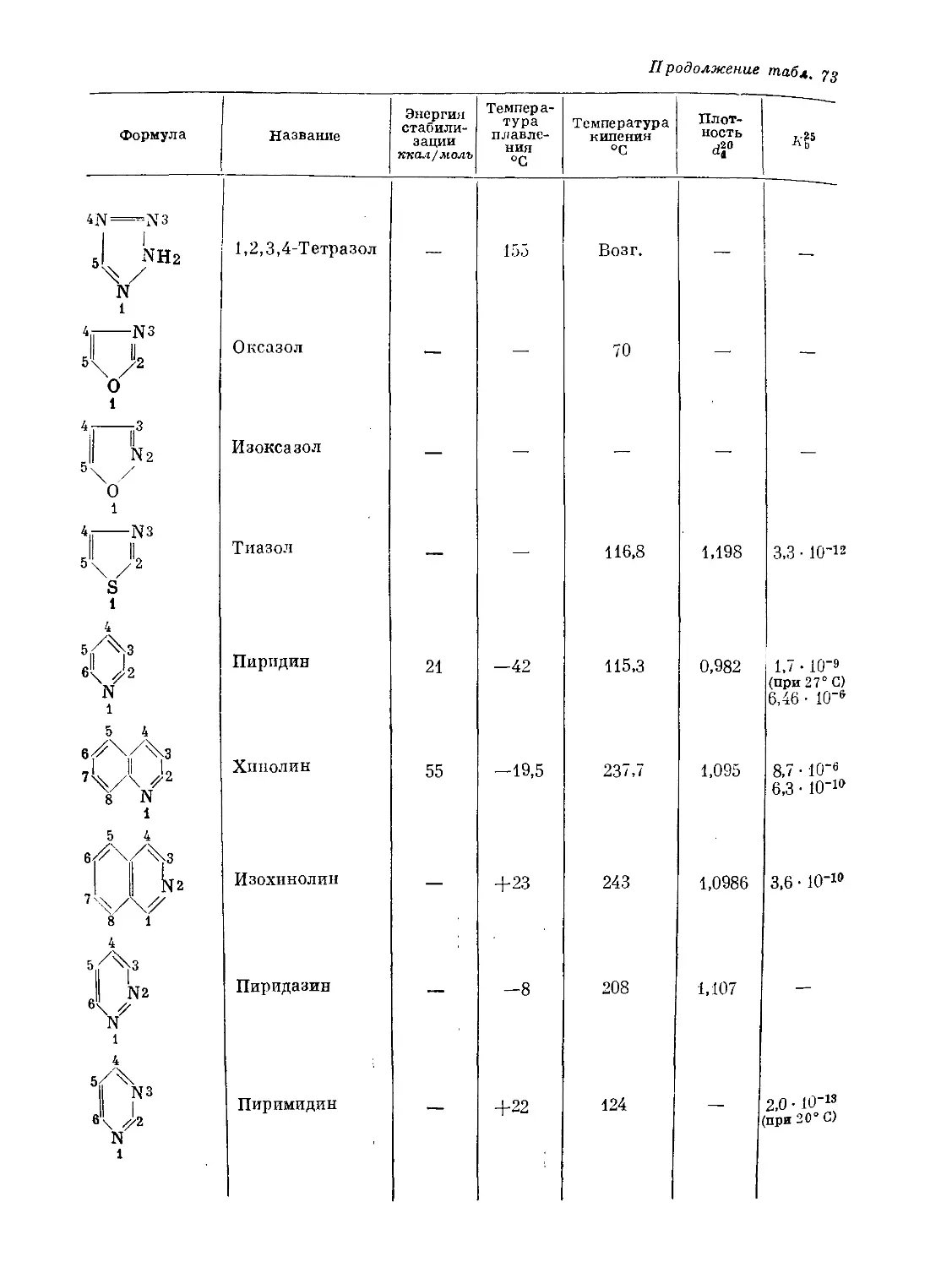
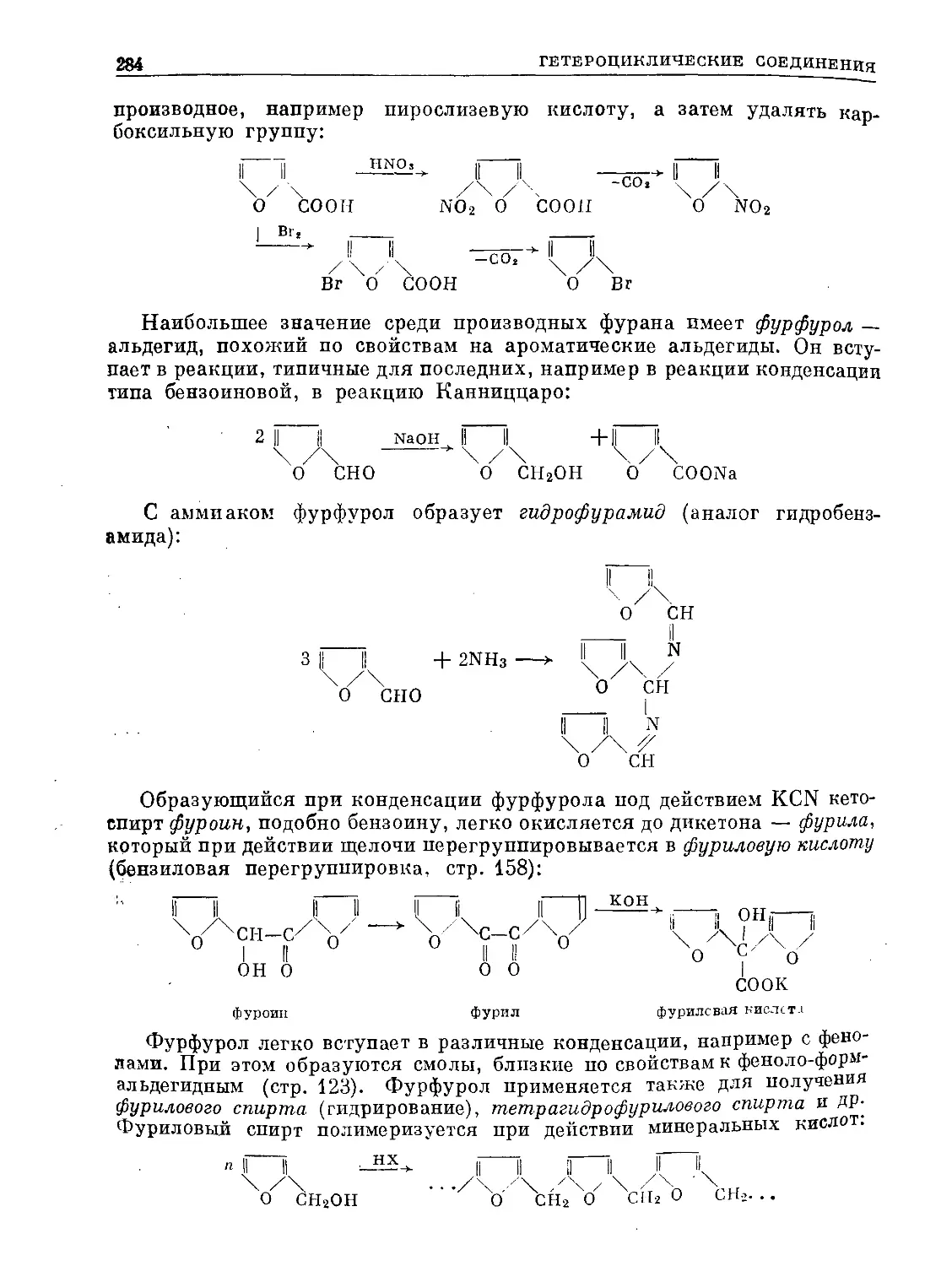
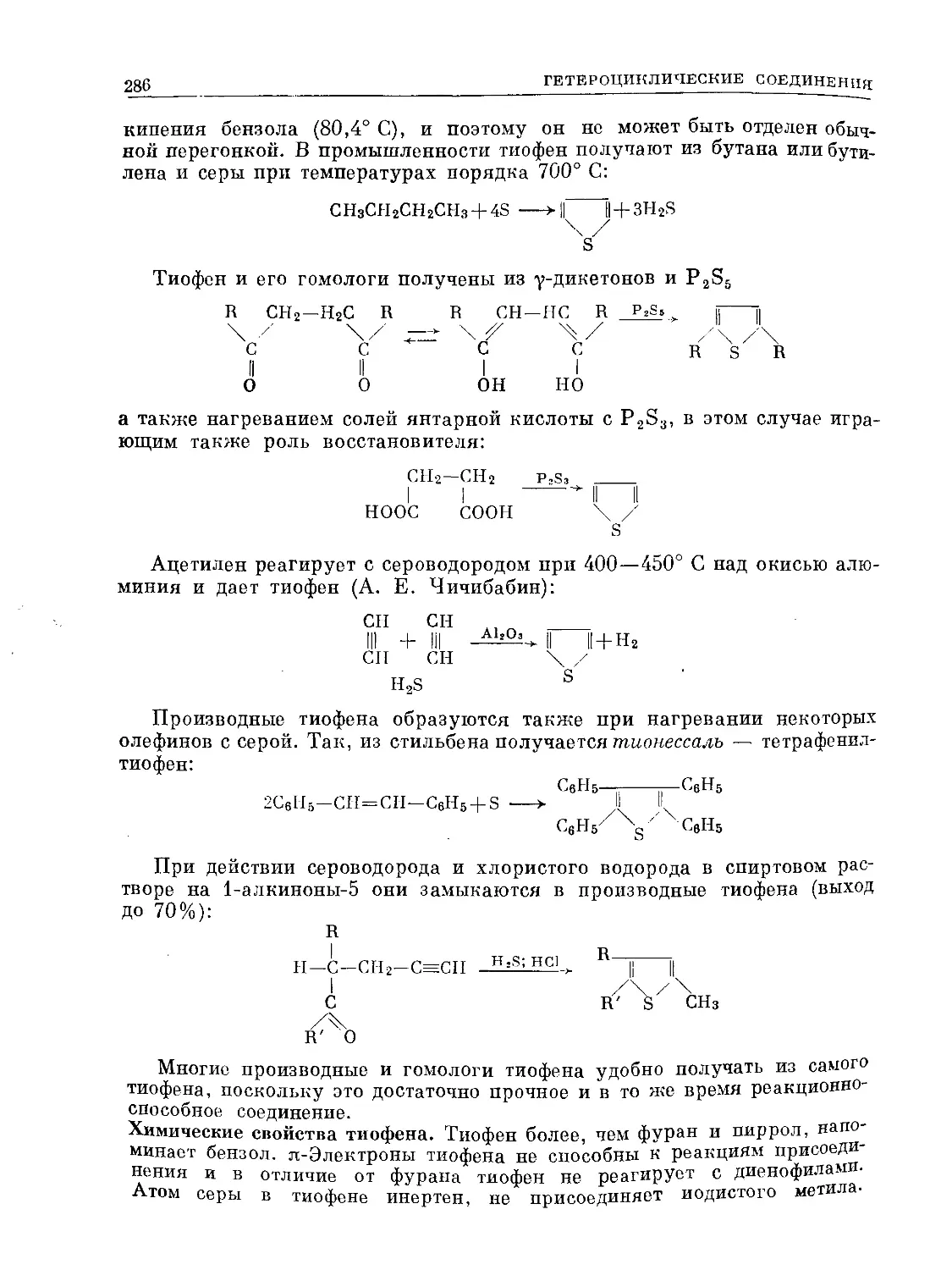
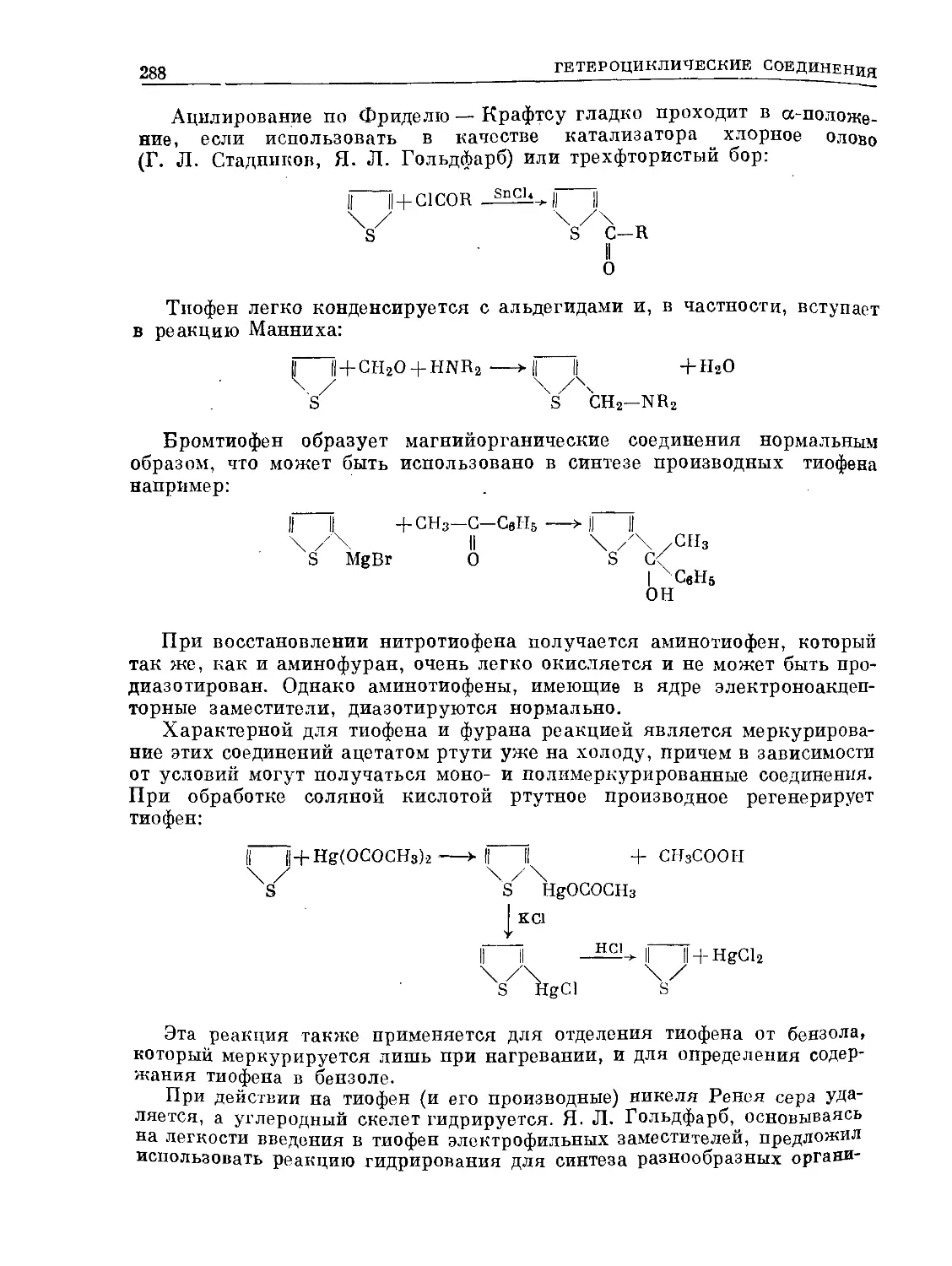
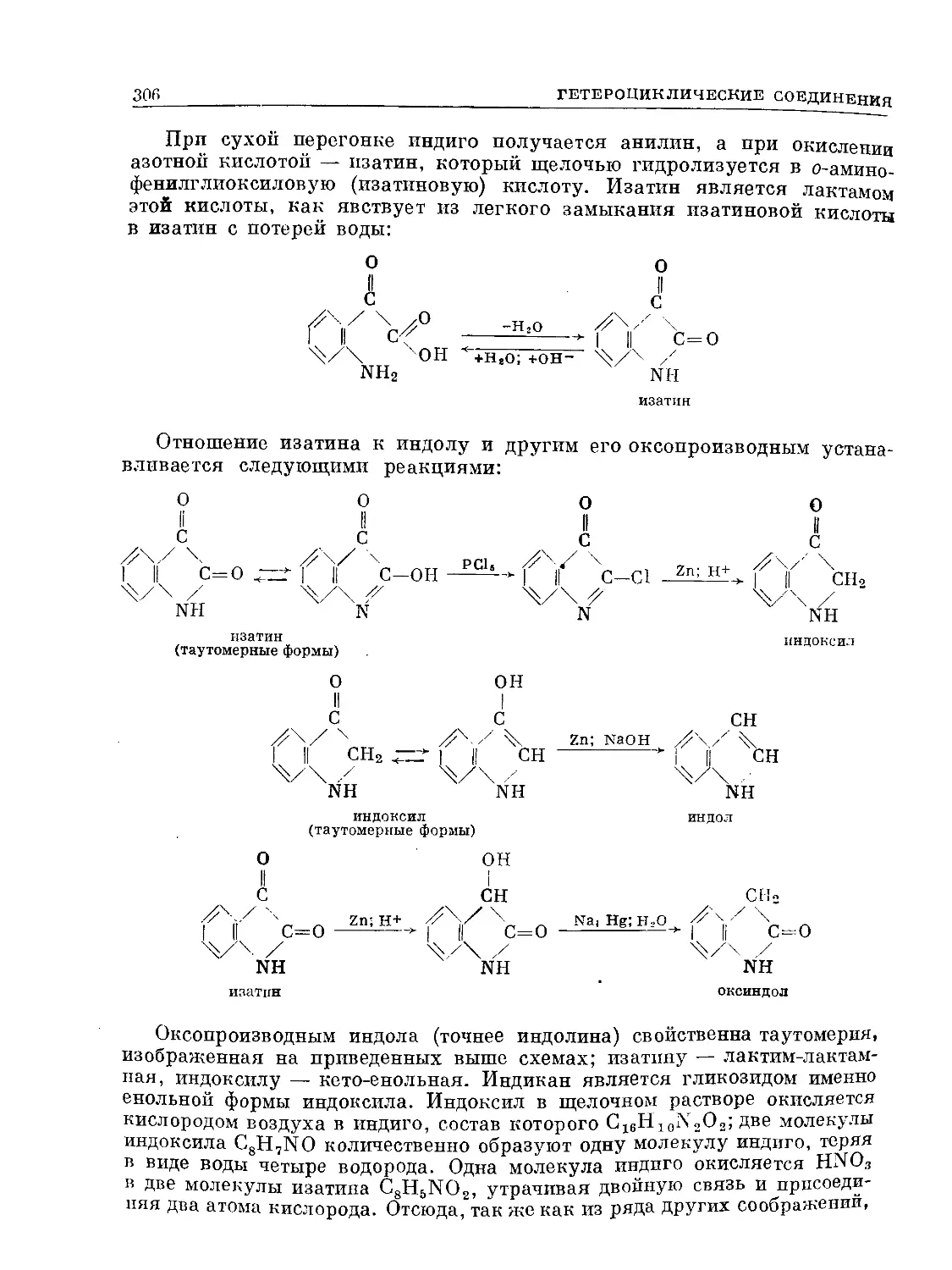
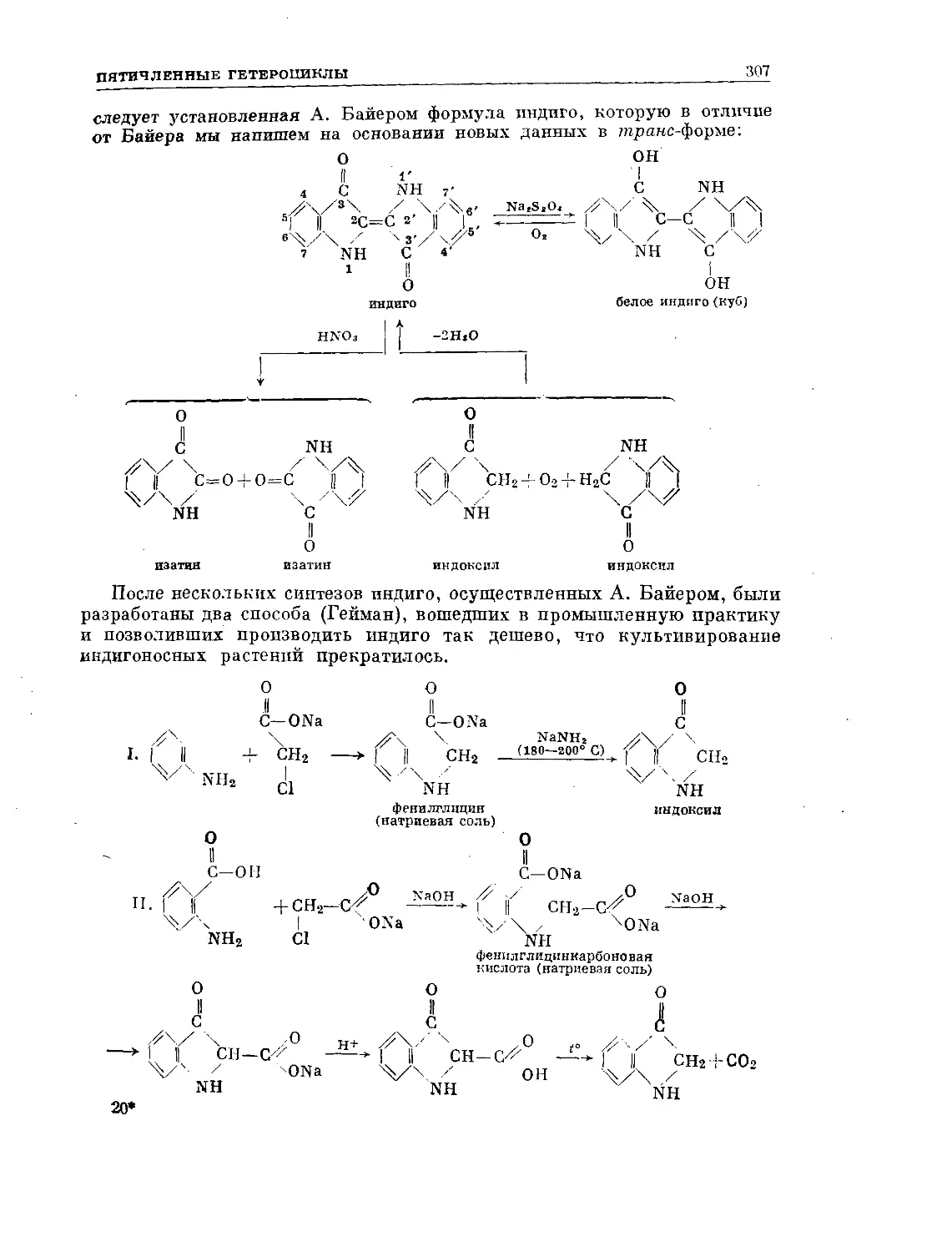
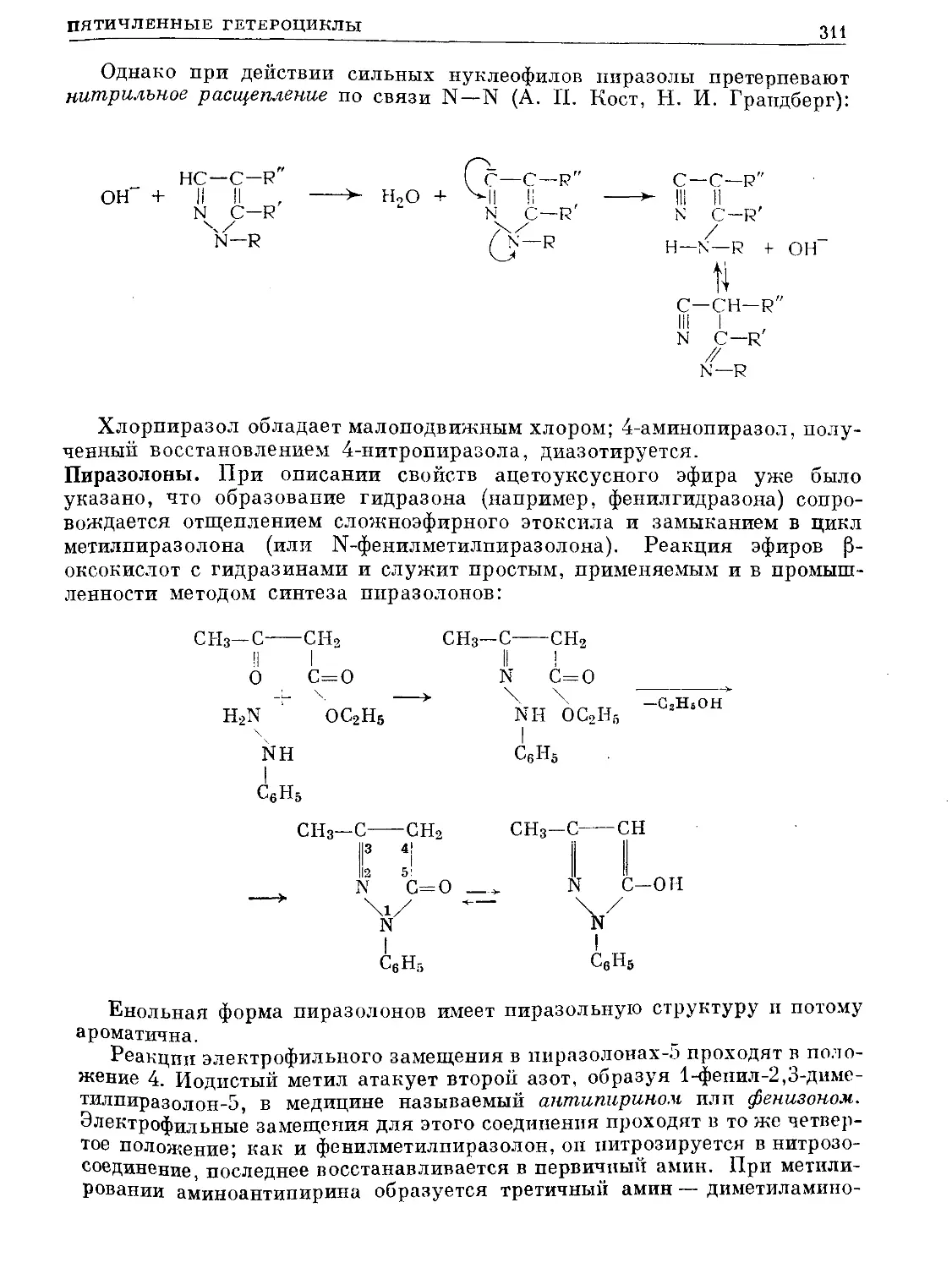
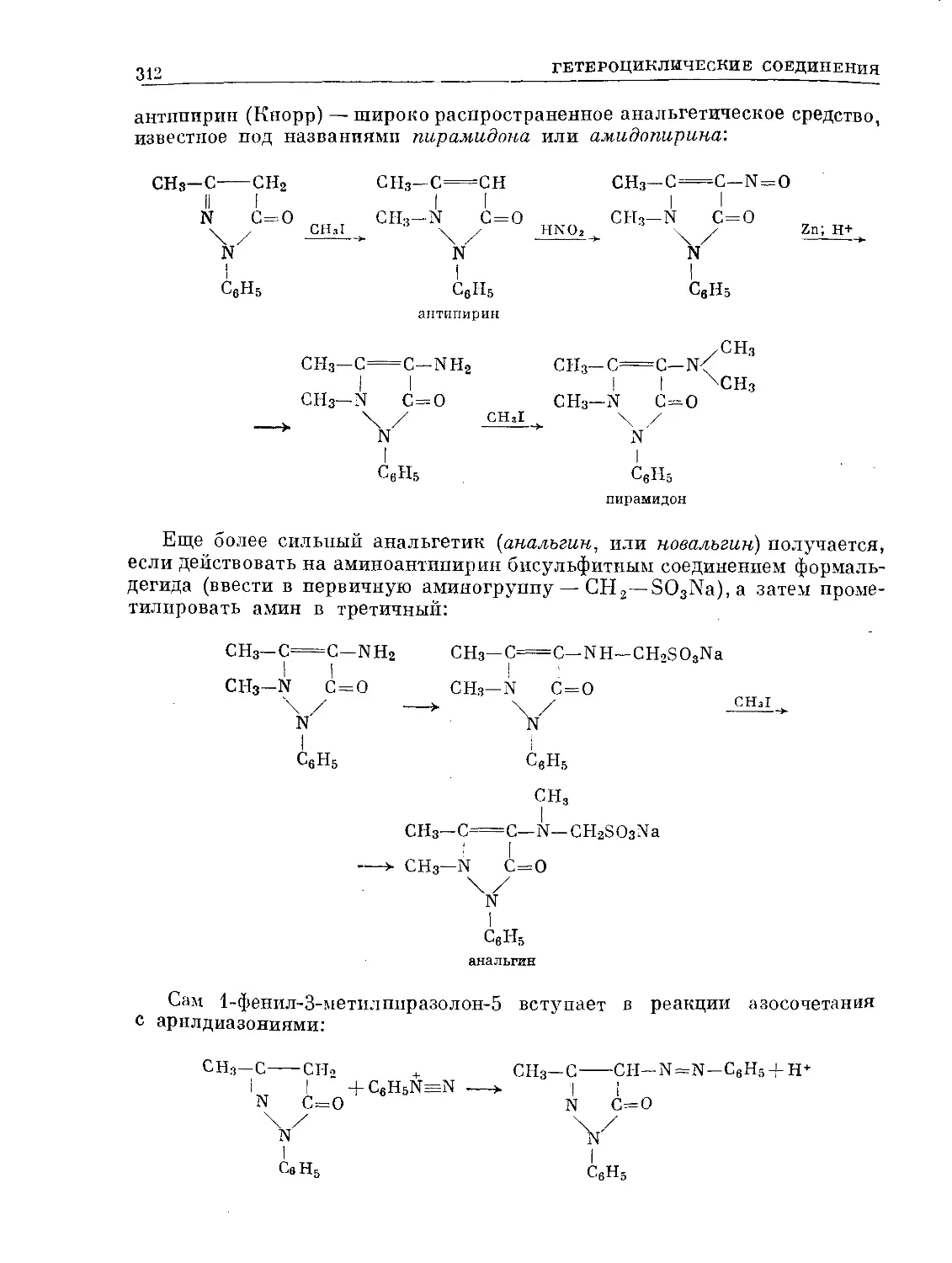
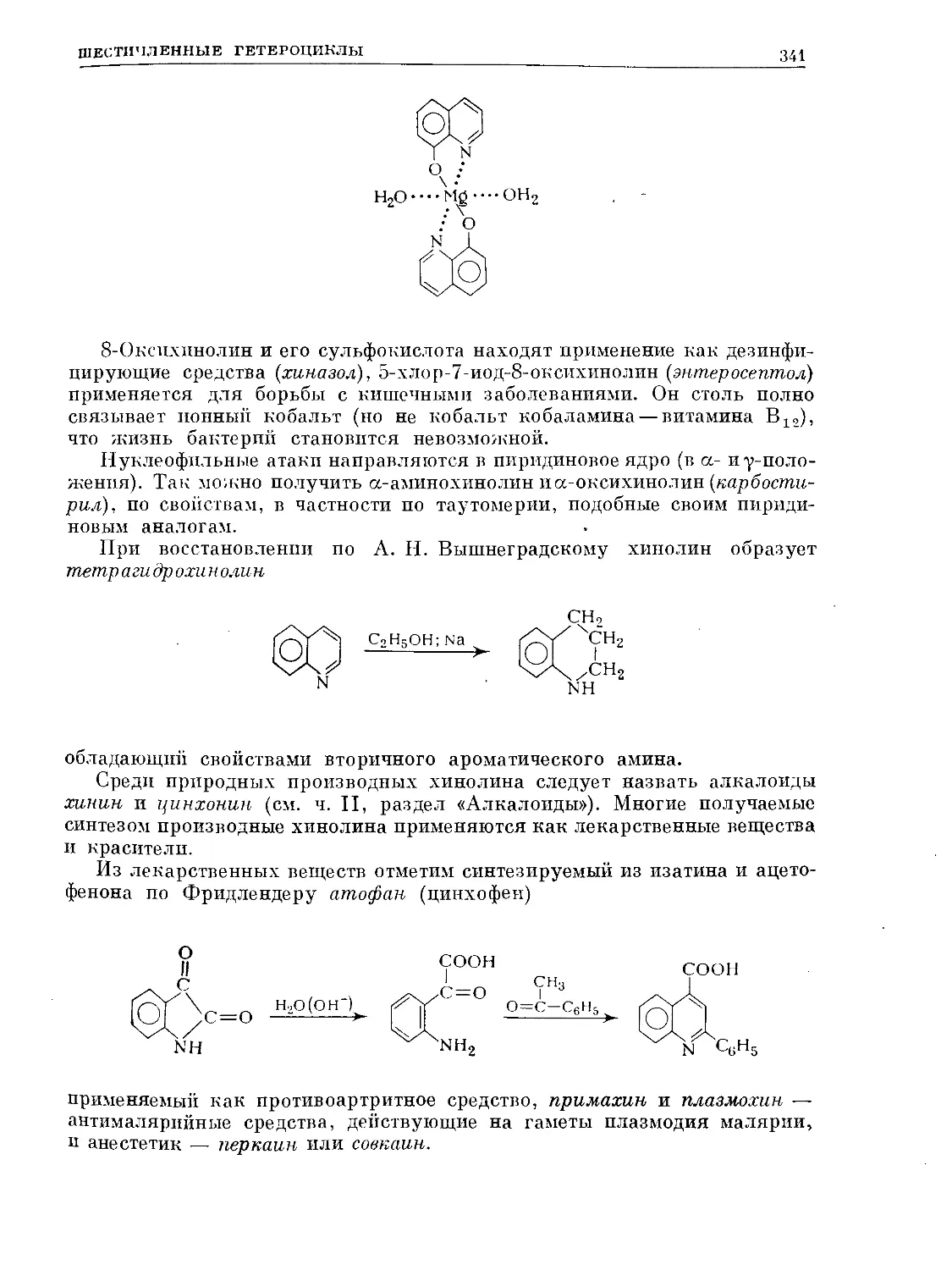
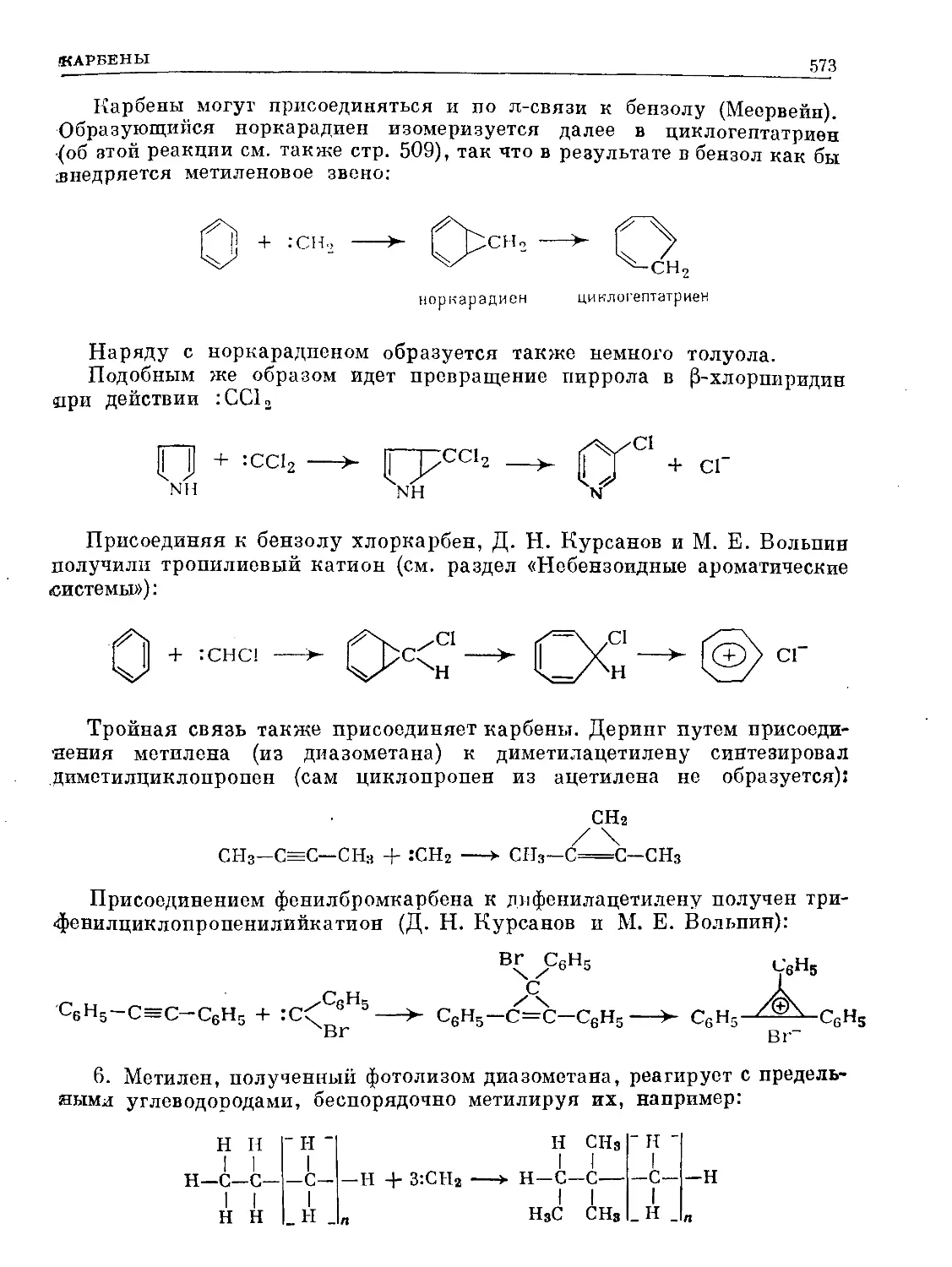
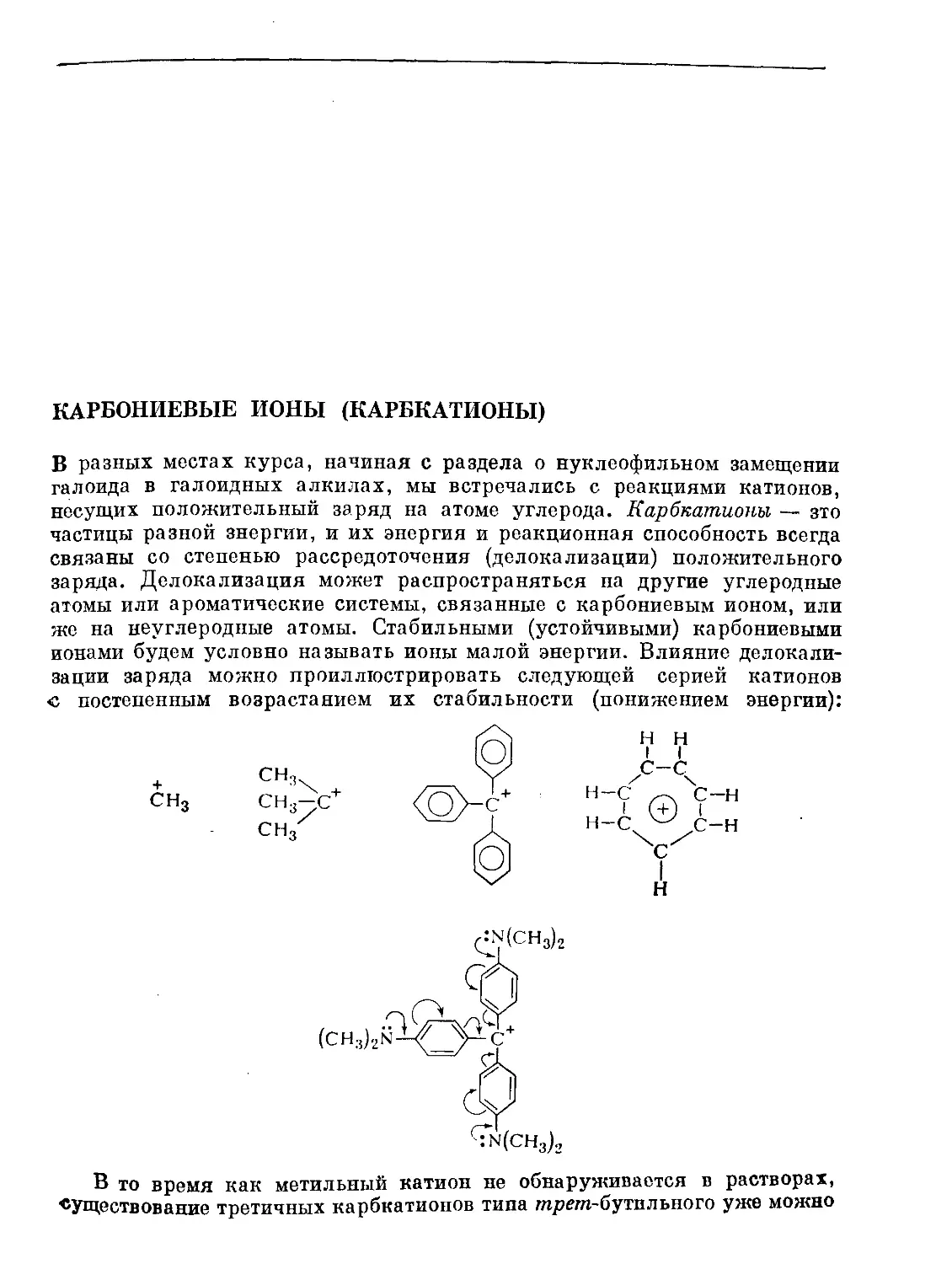
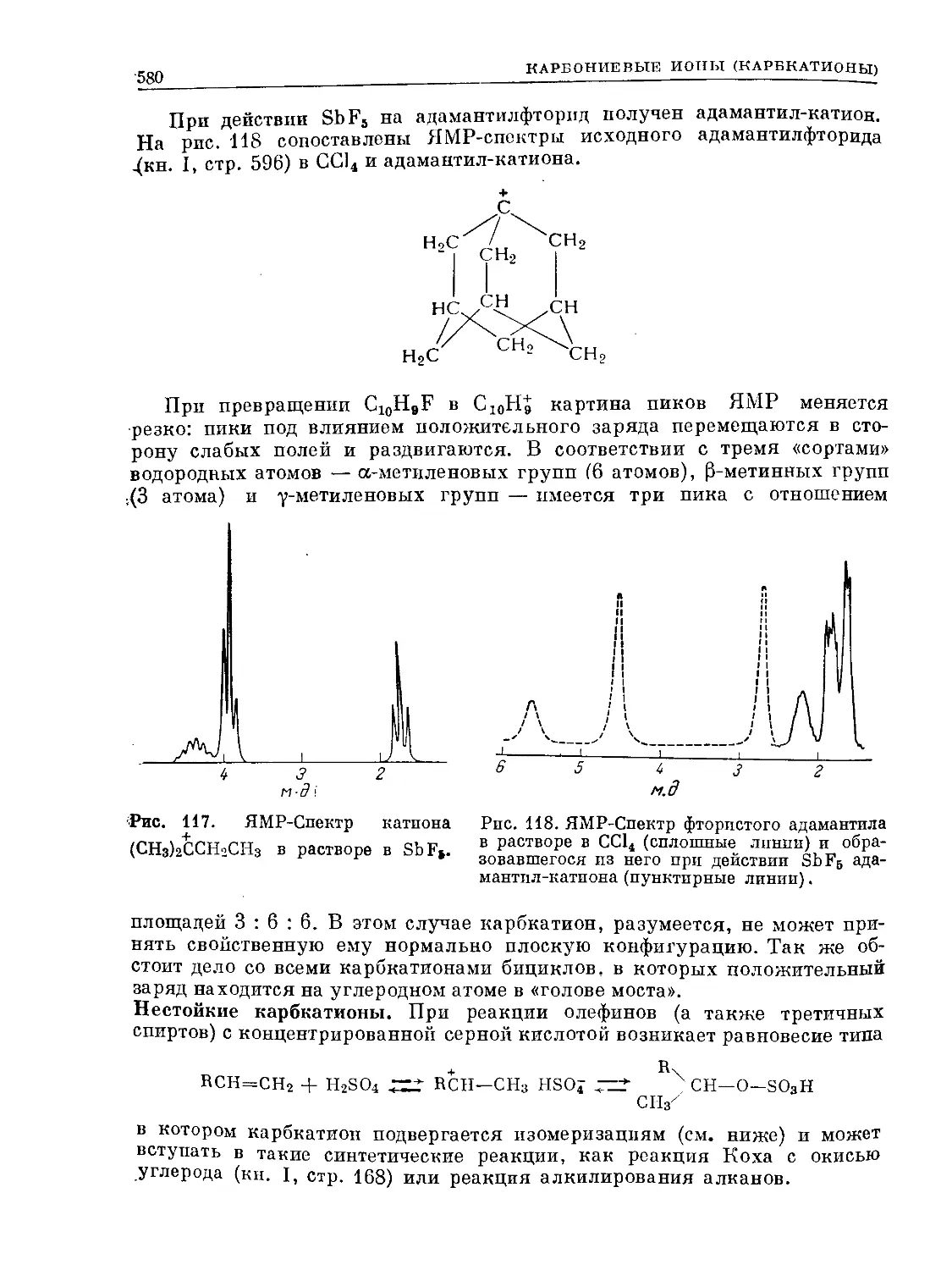
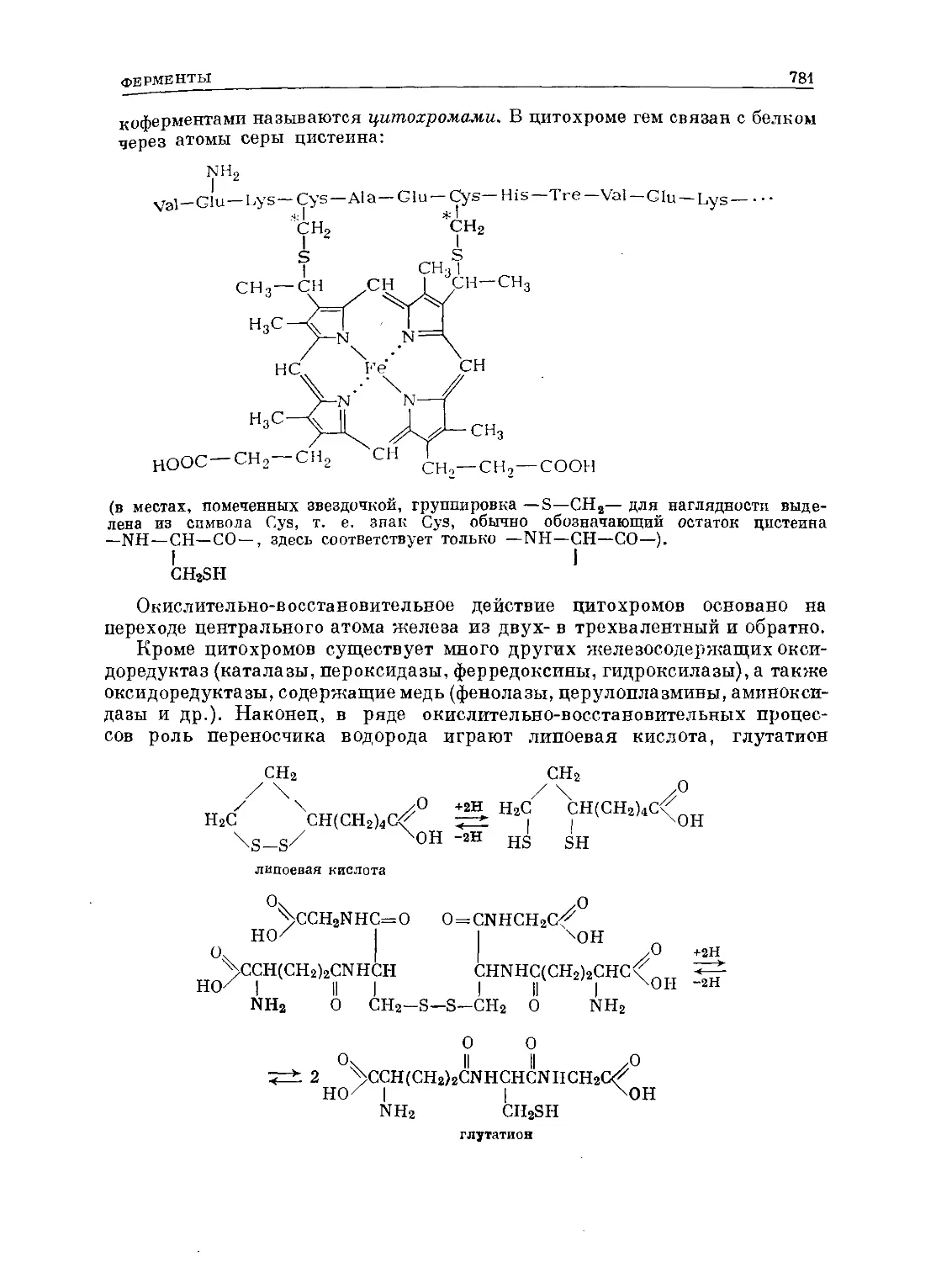
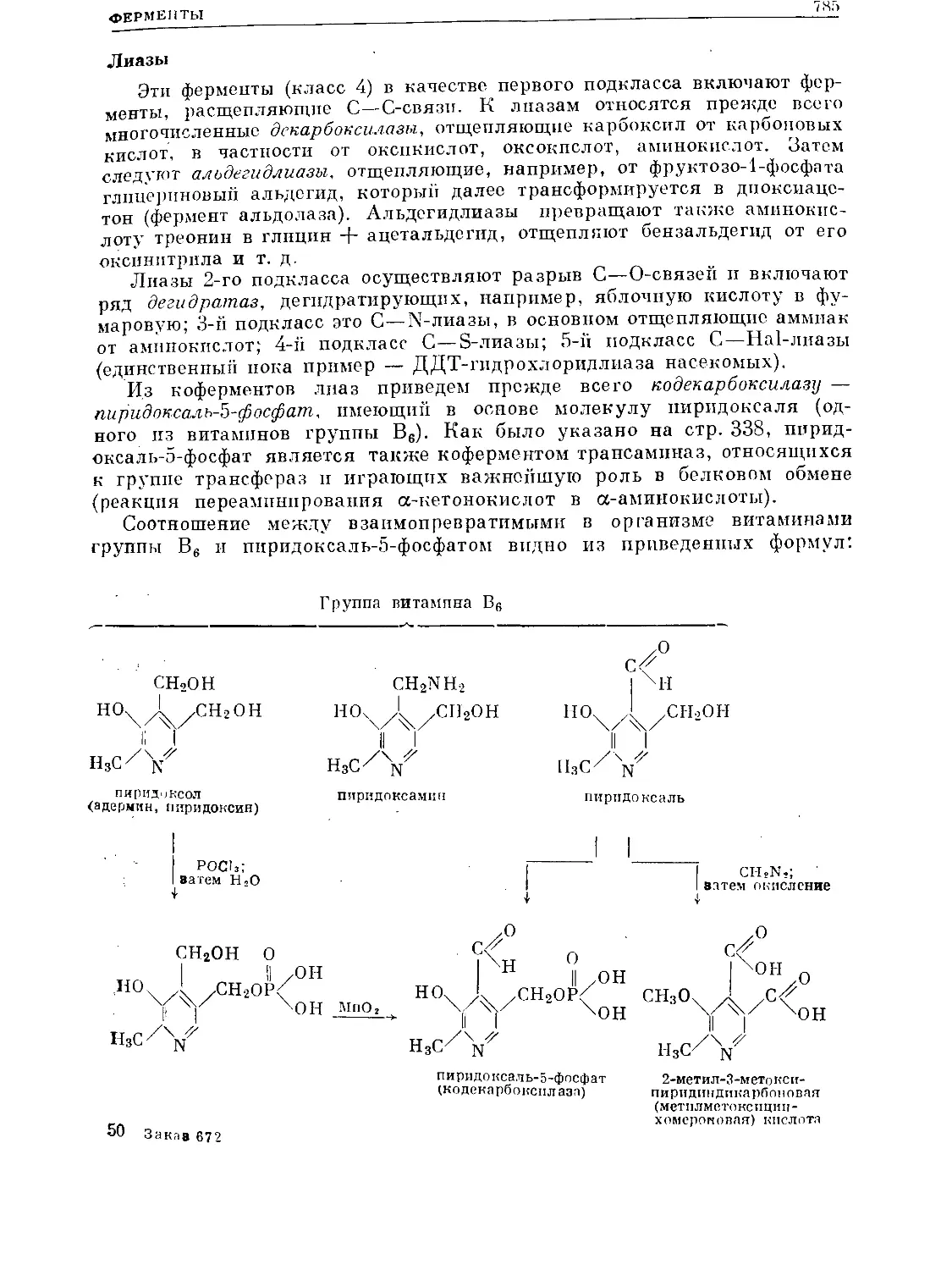
/






































































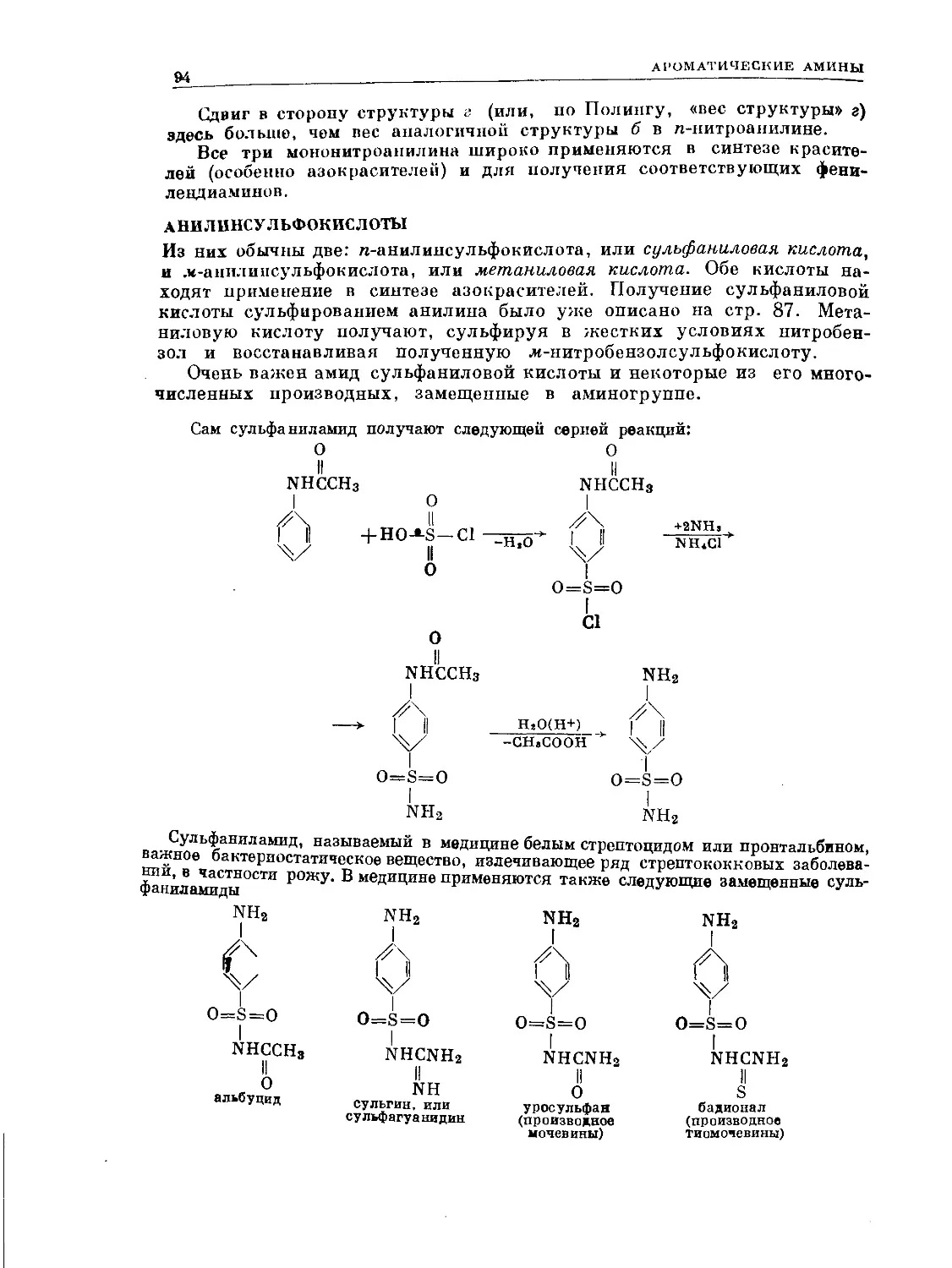







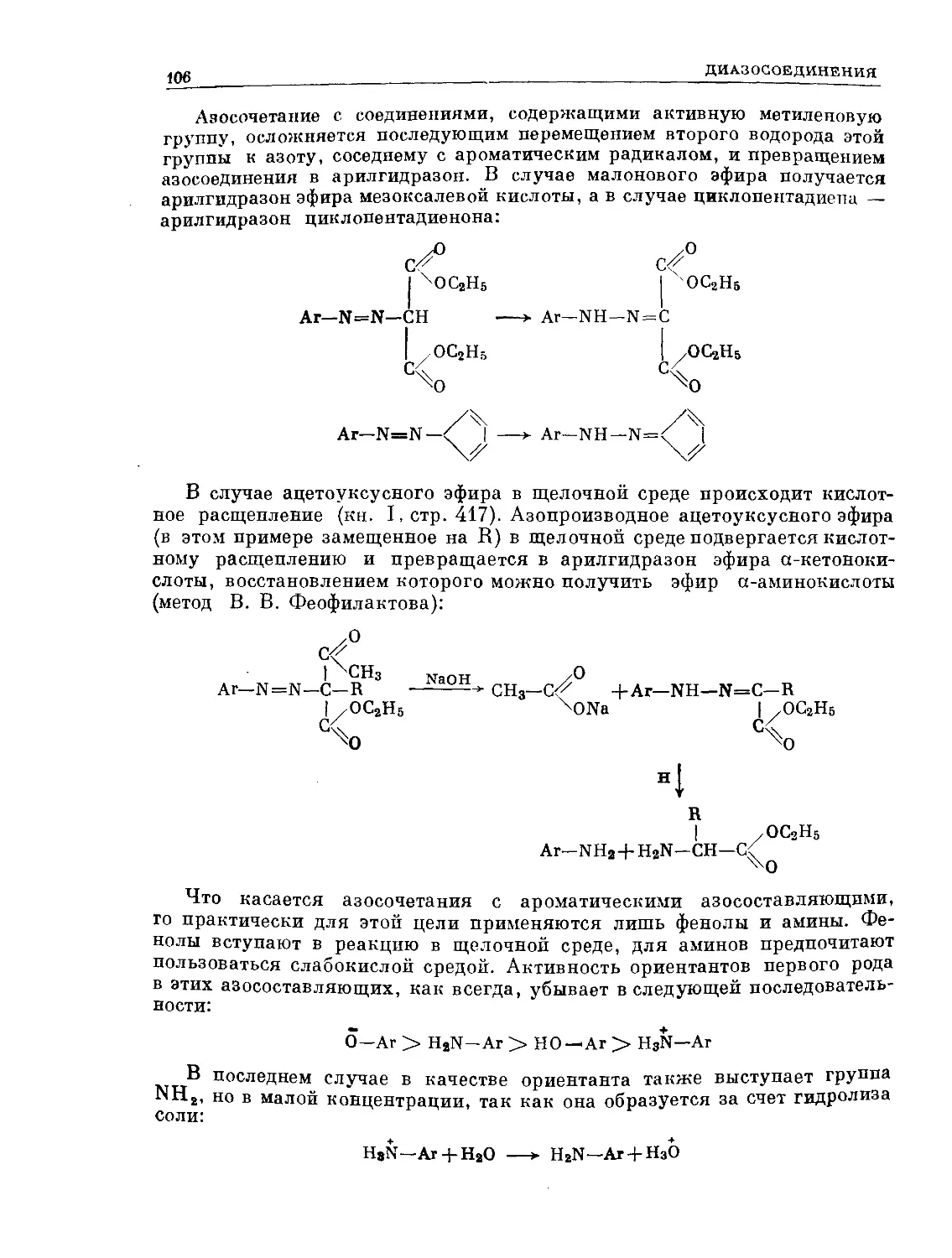
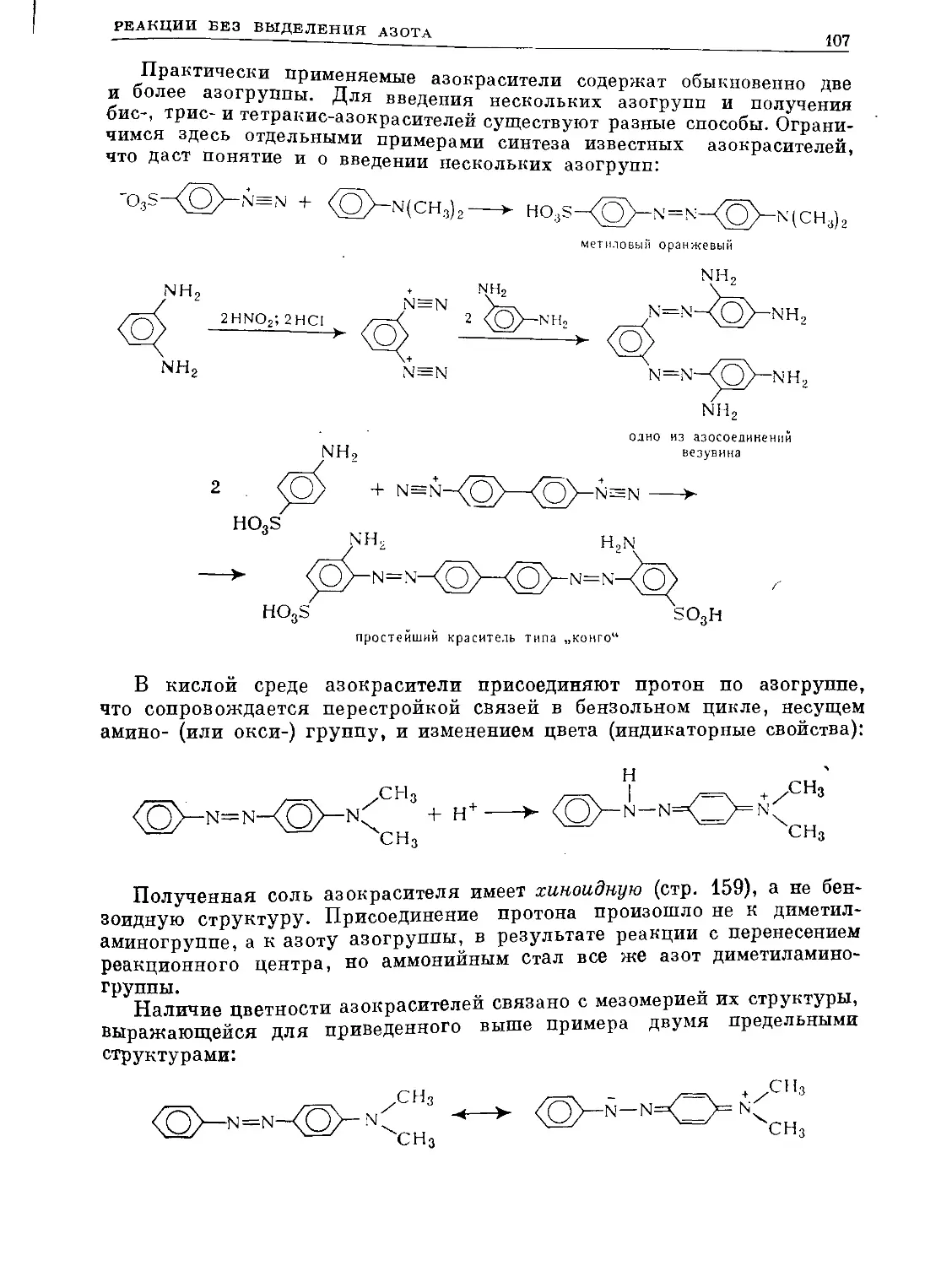





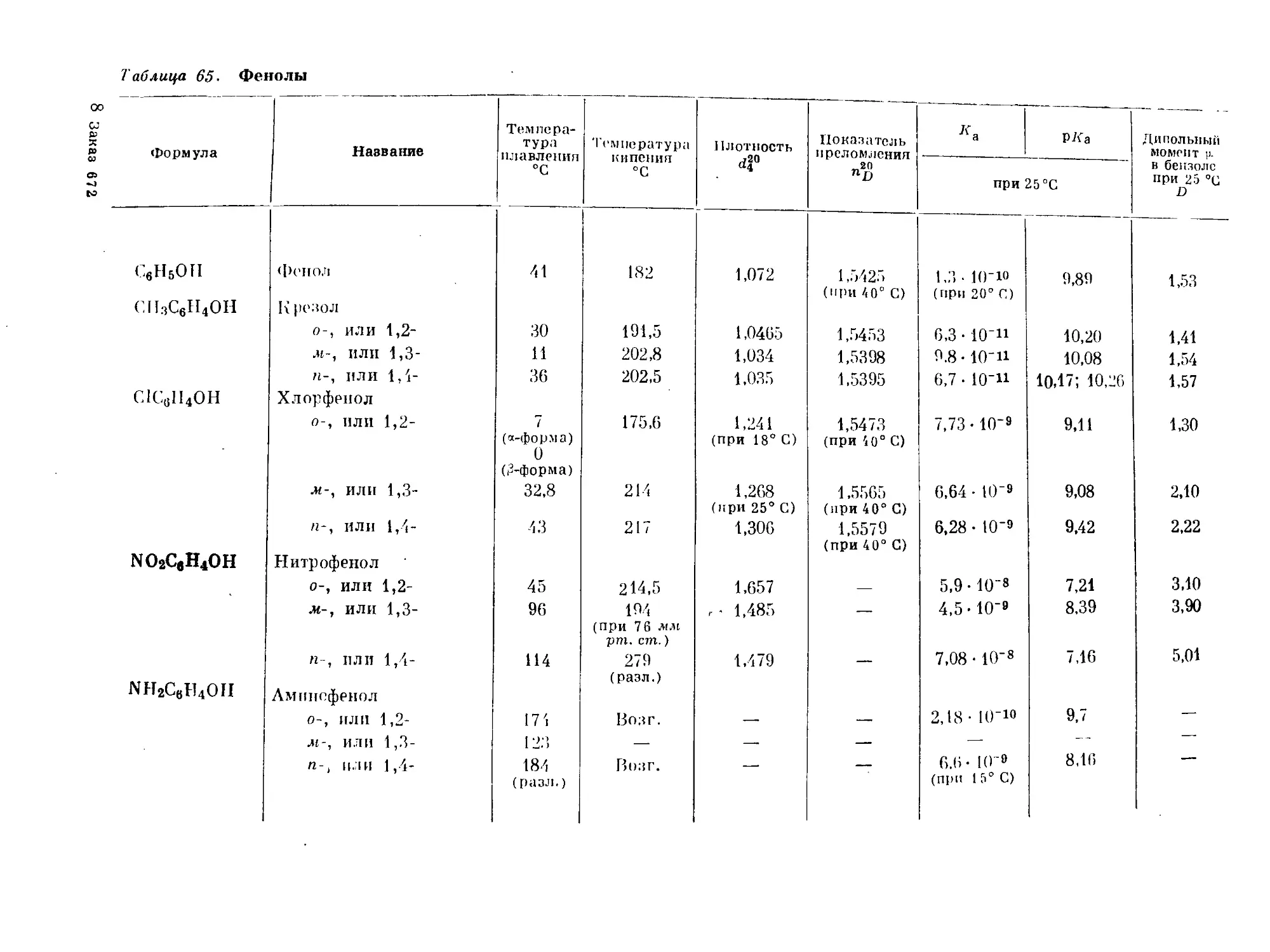



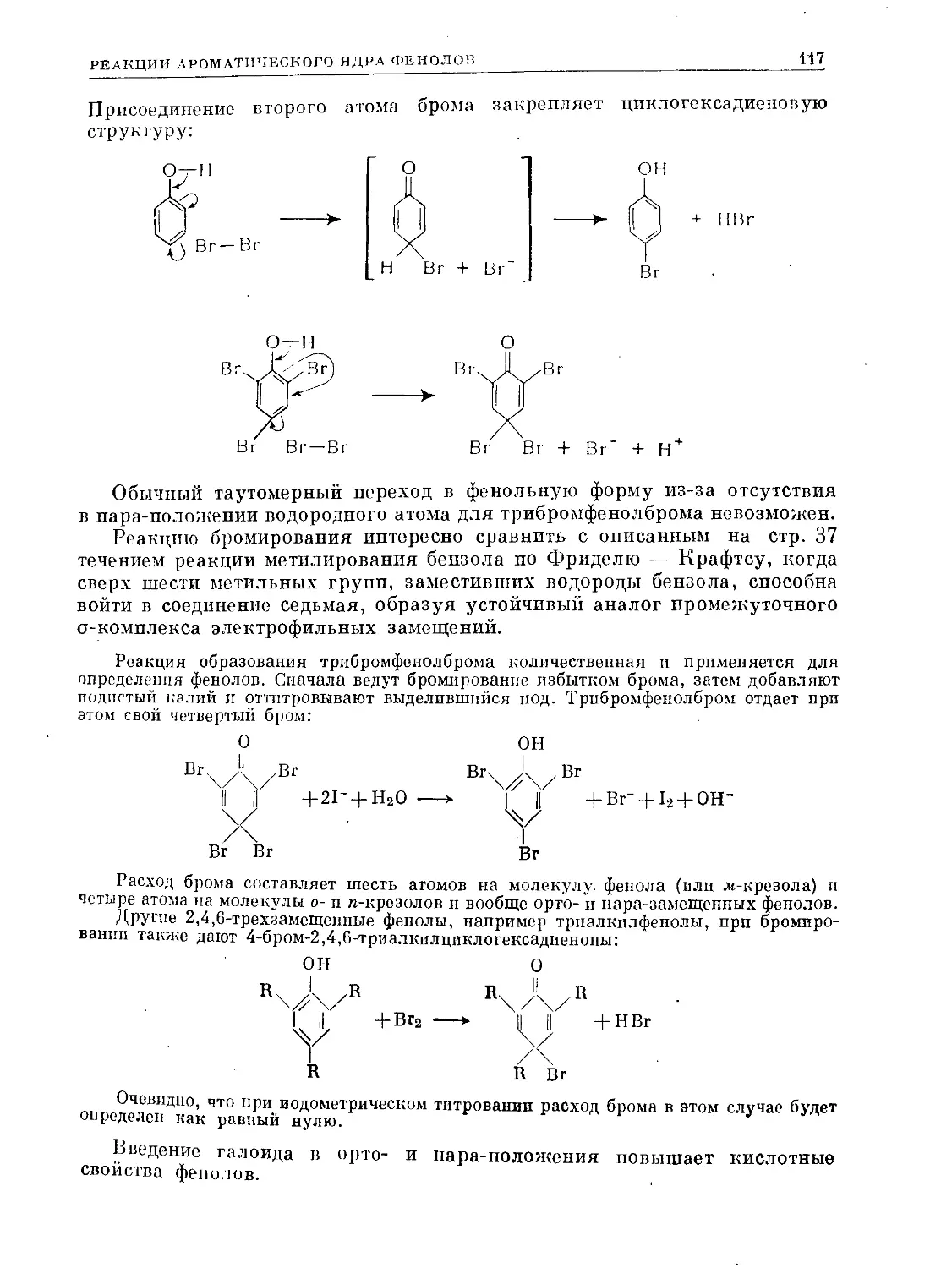






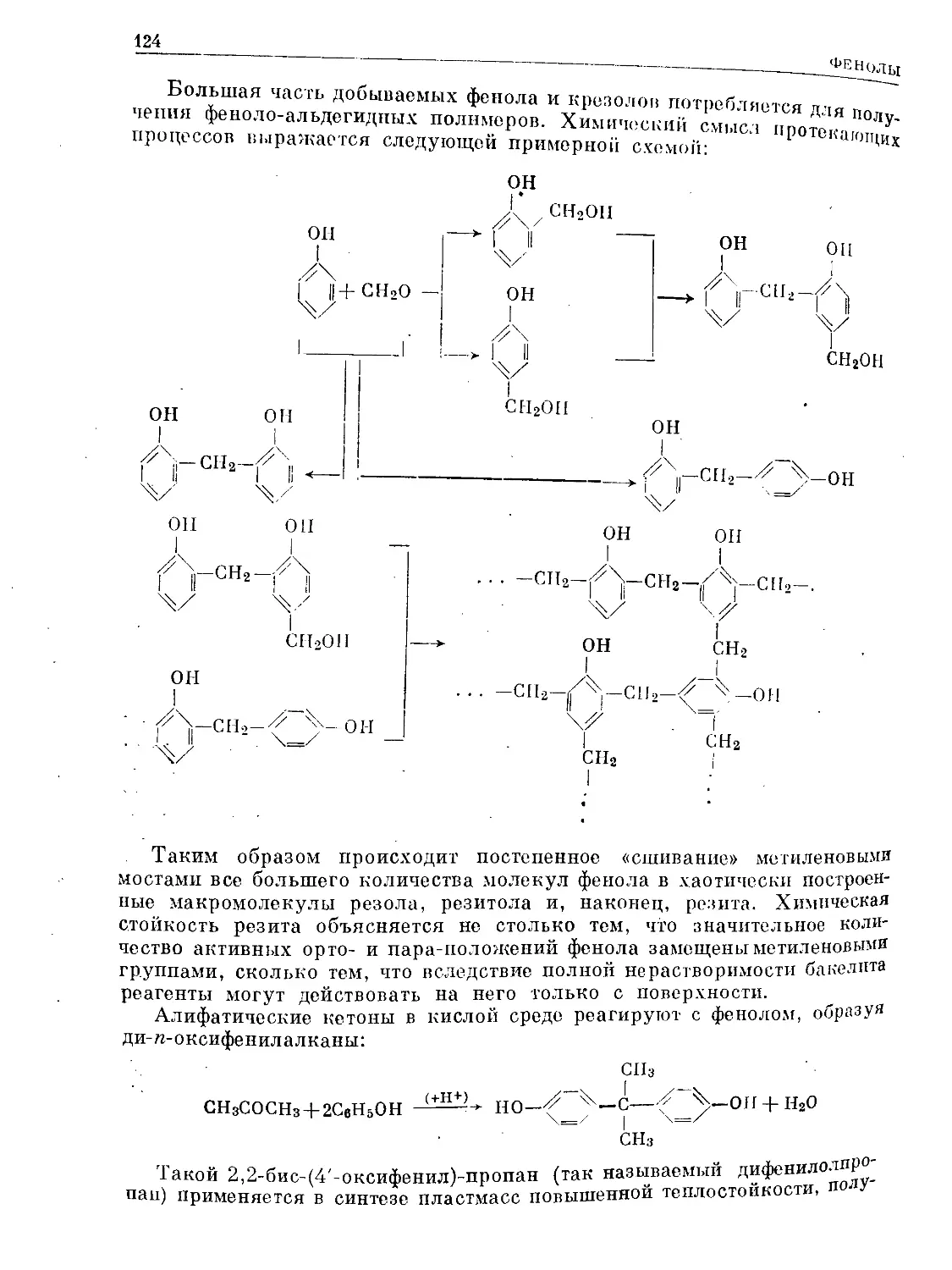


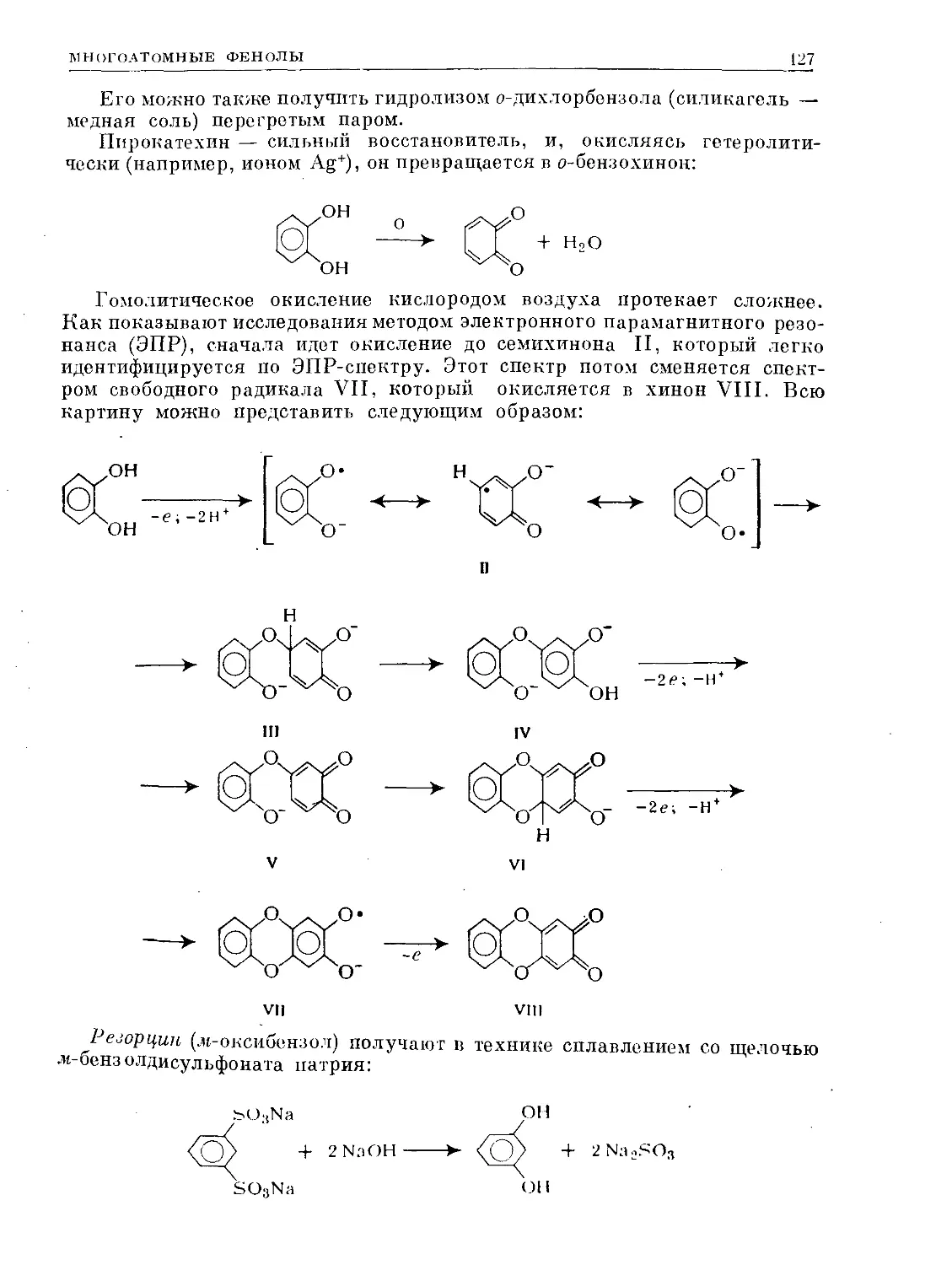
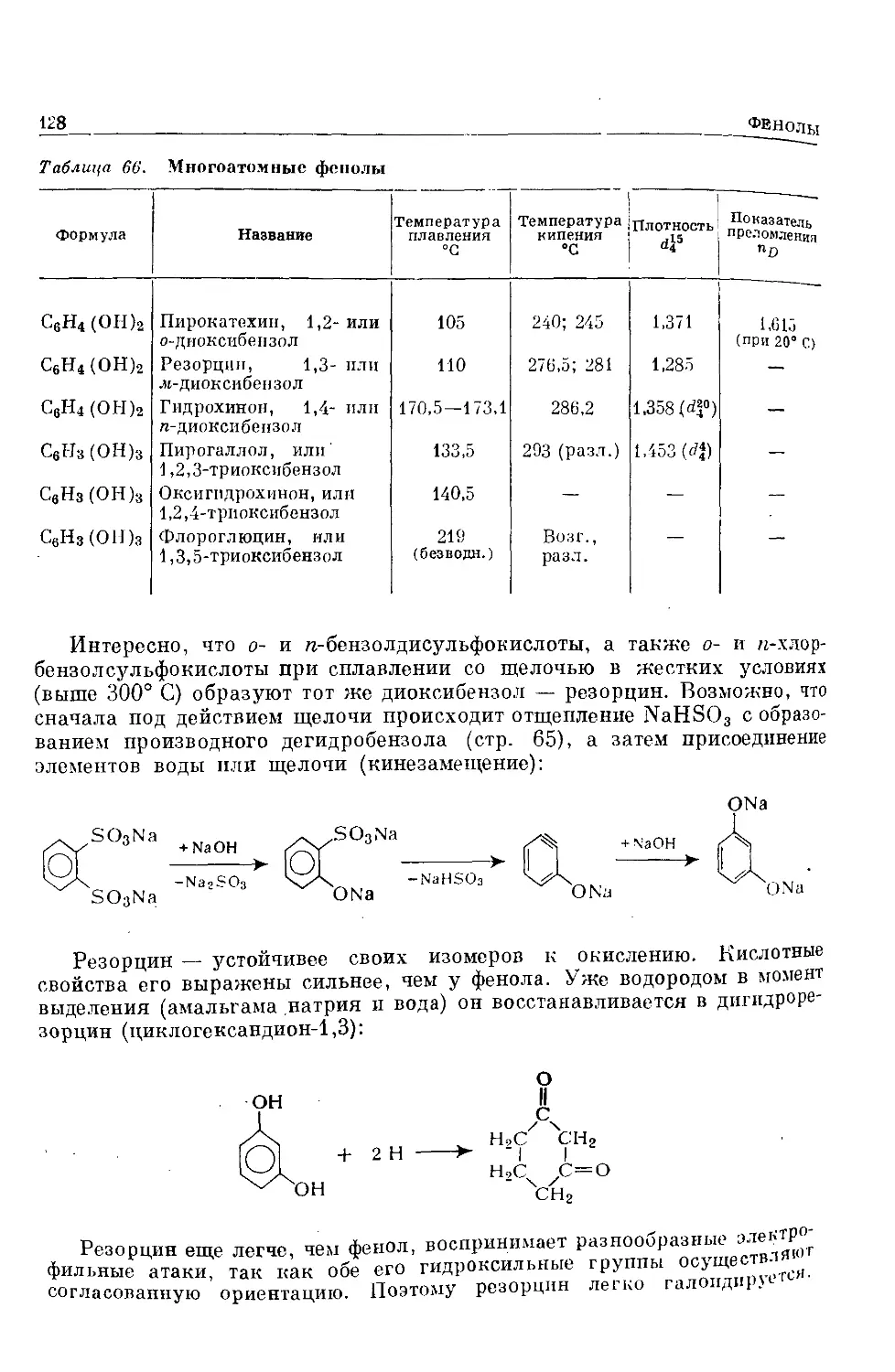

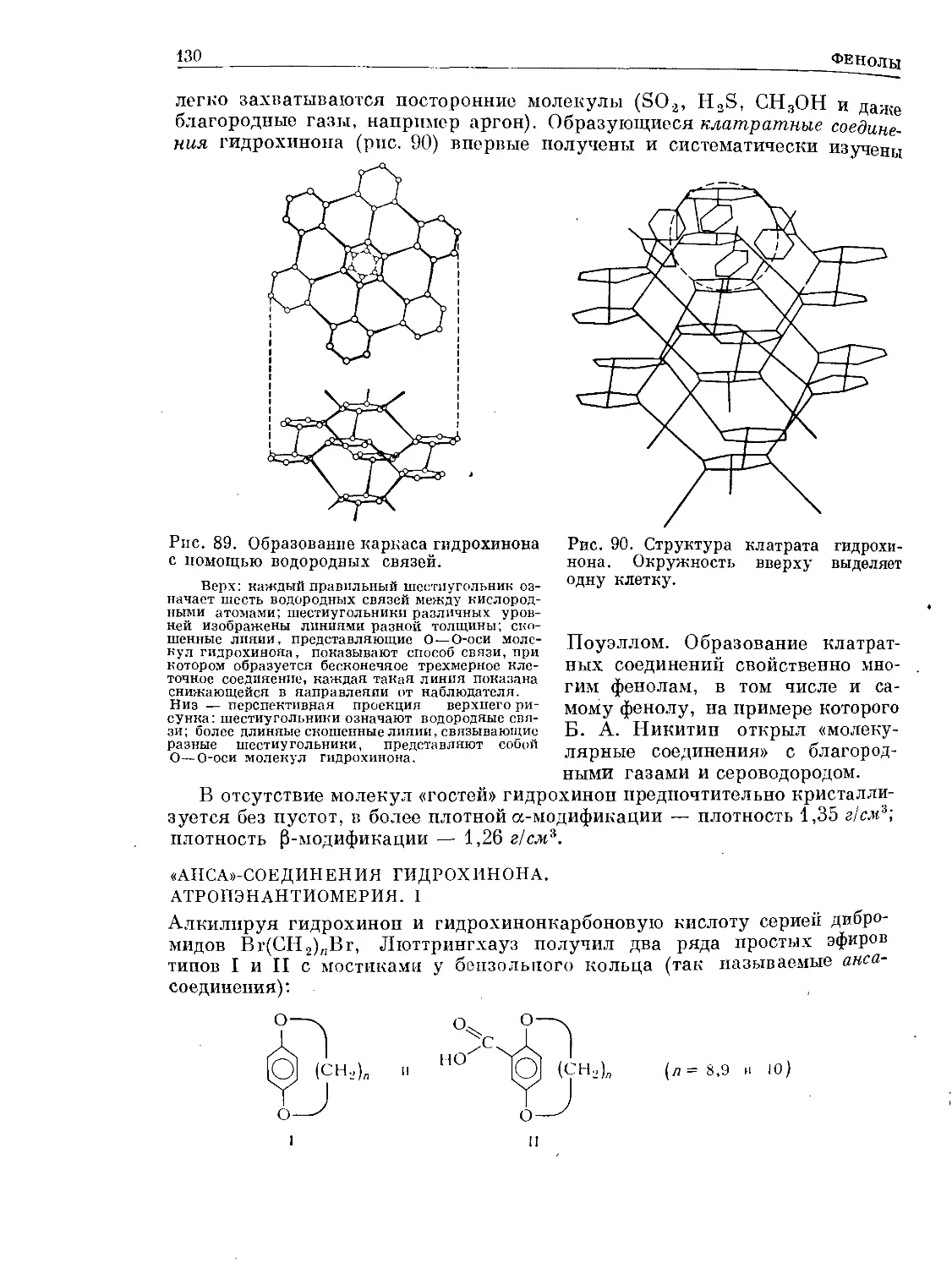


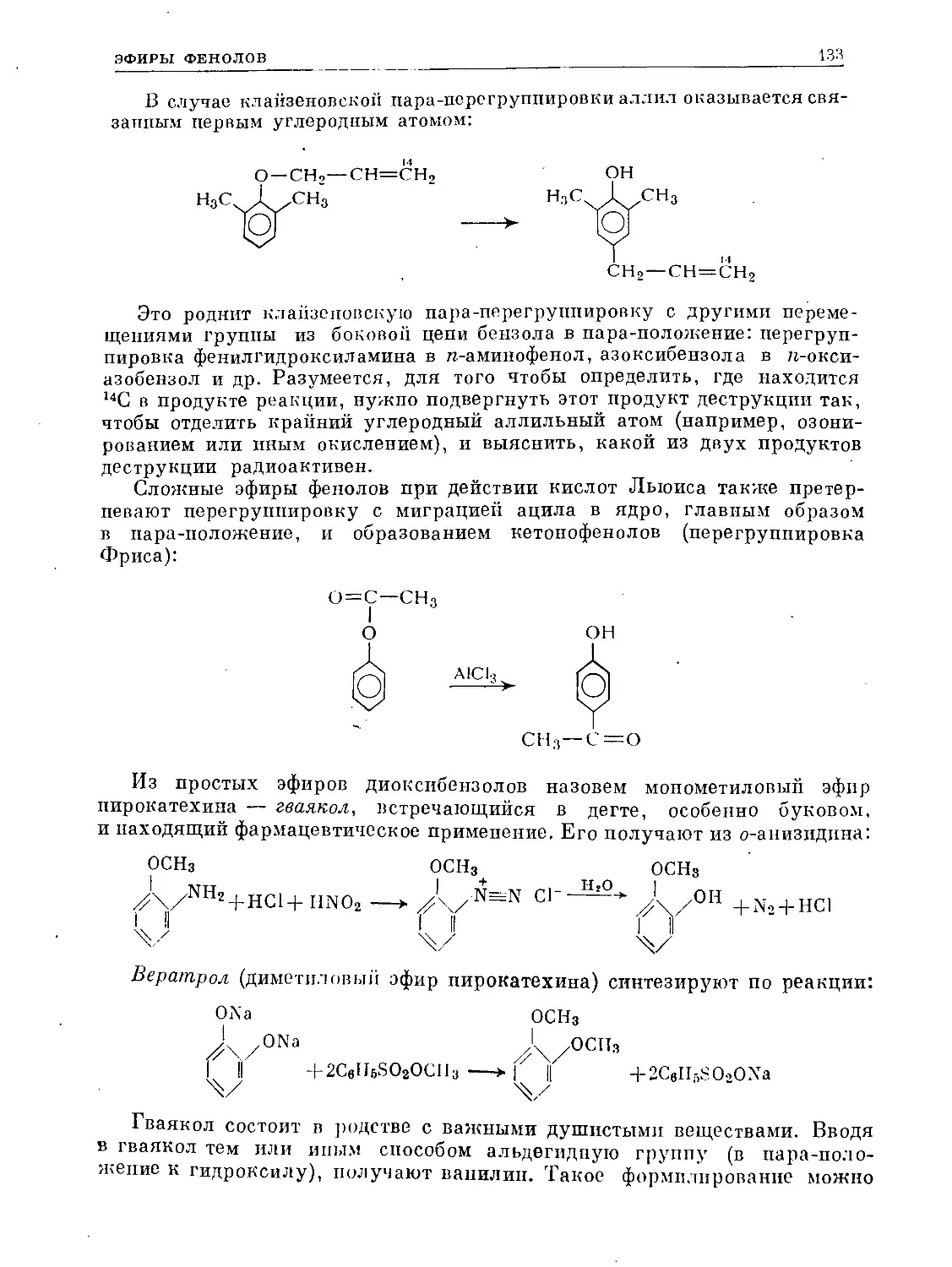



















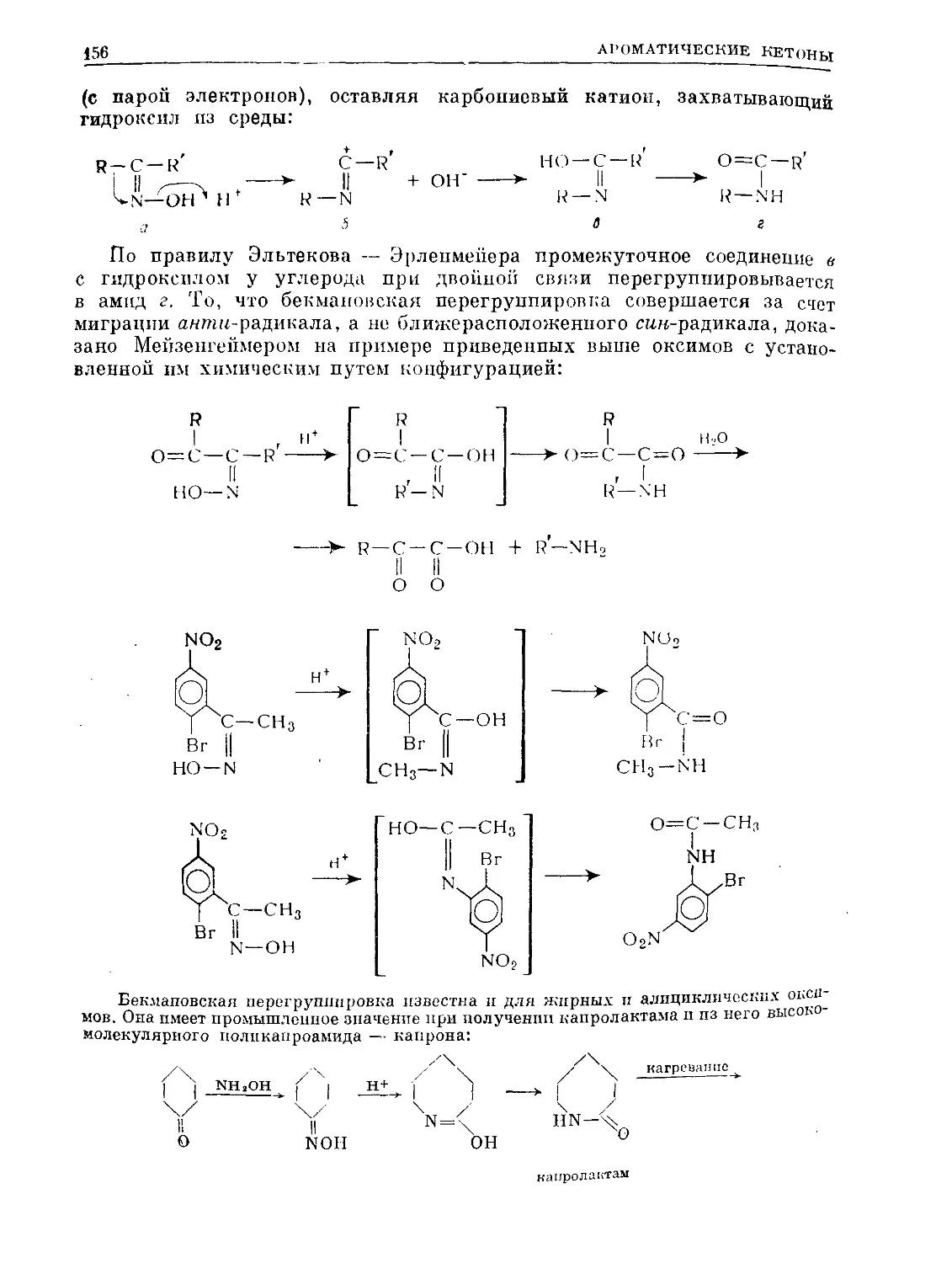


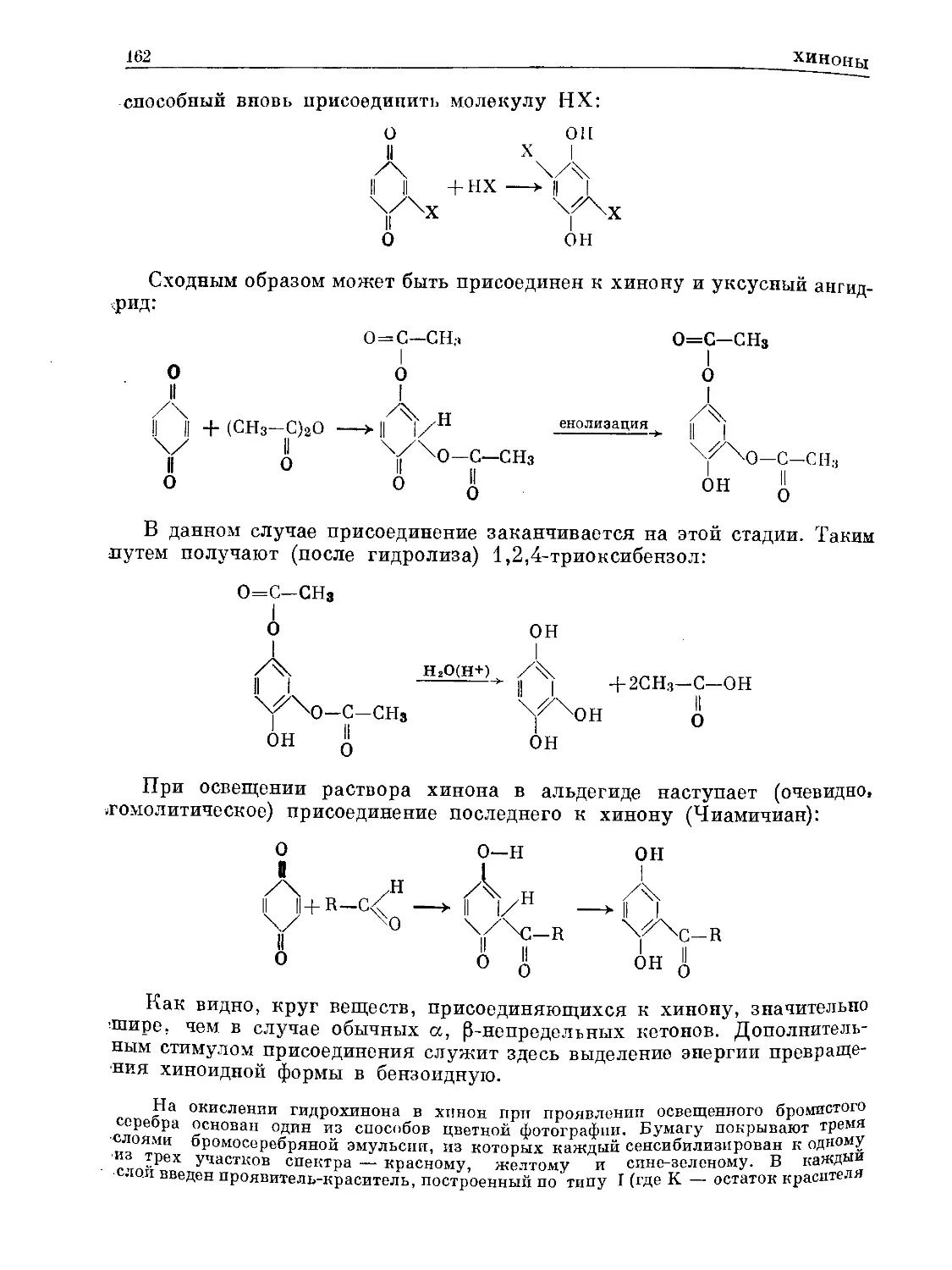


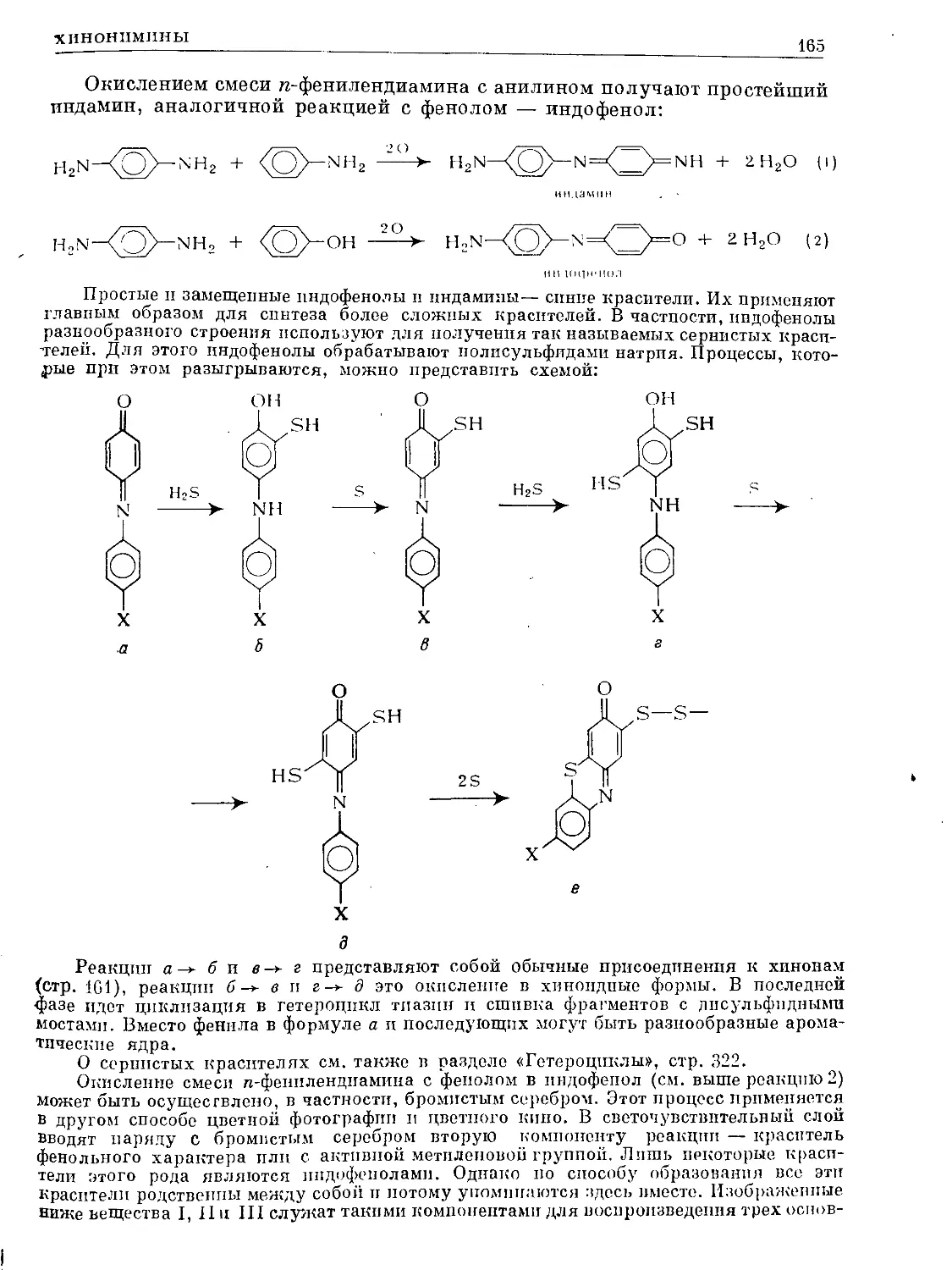
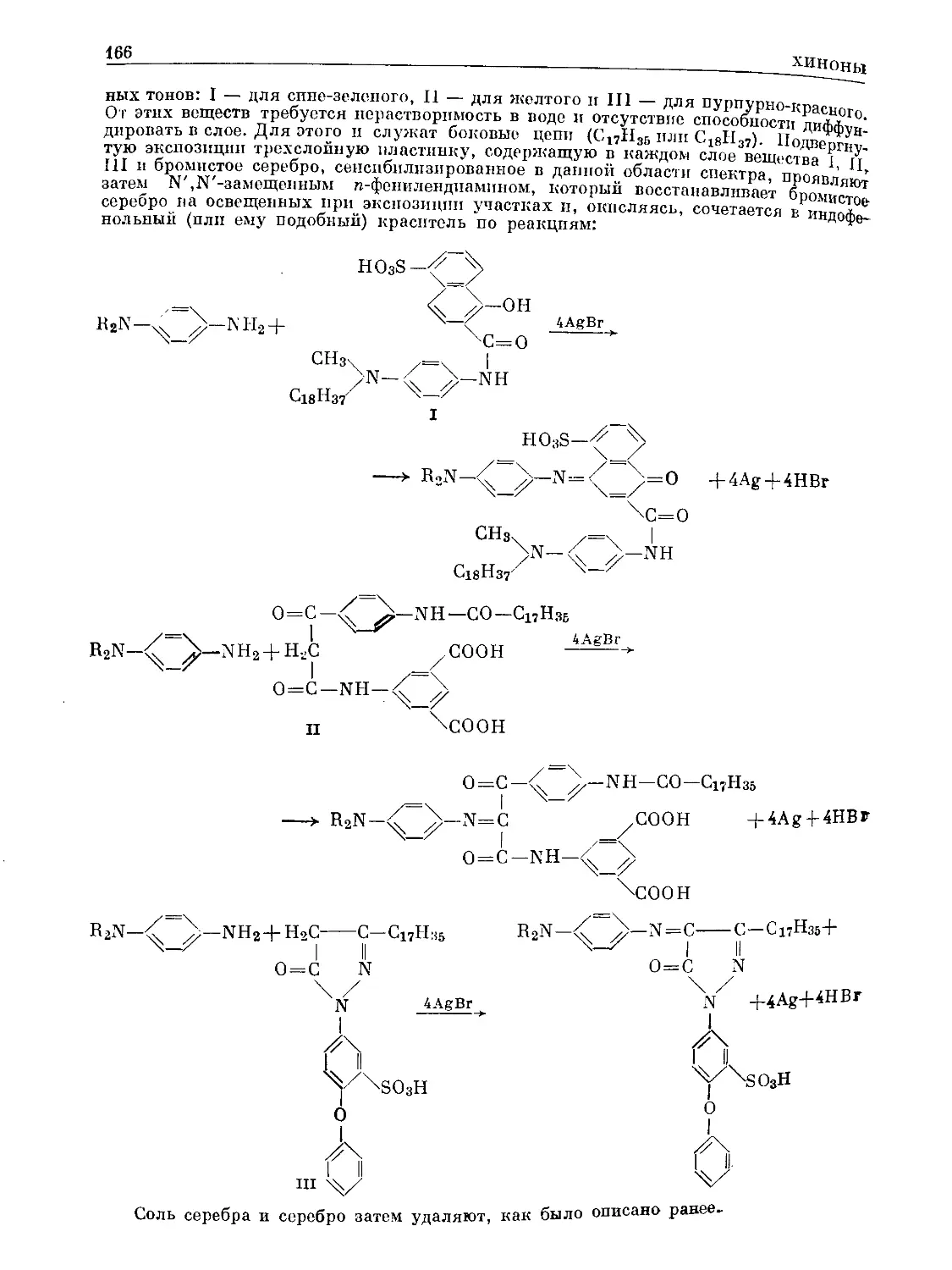



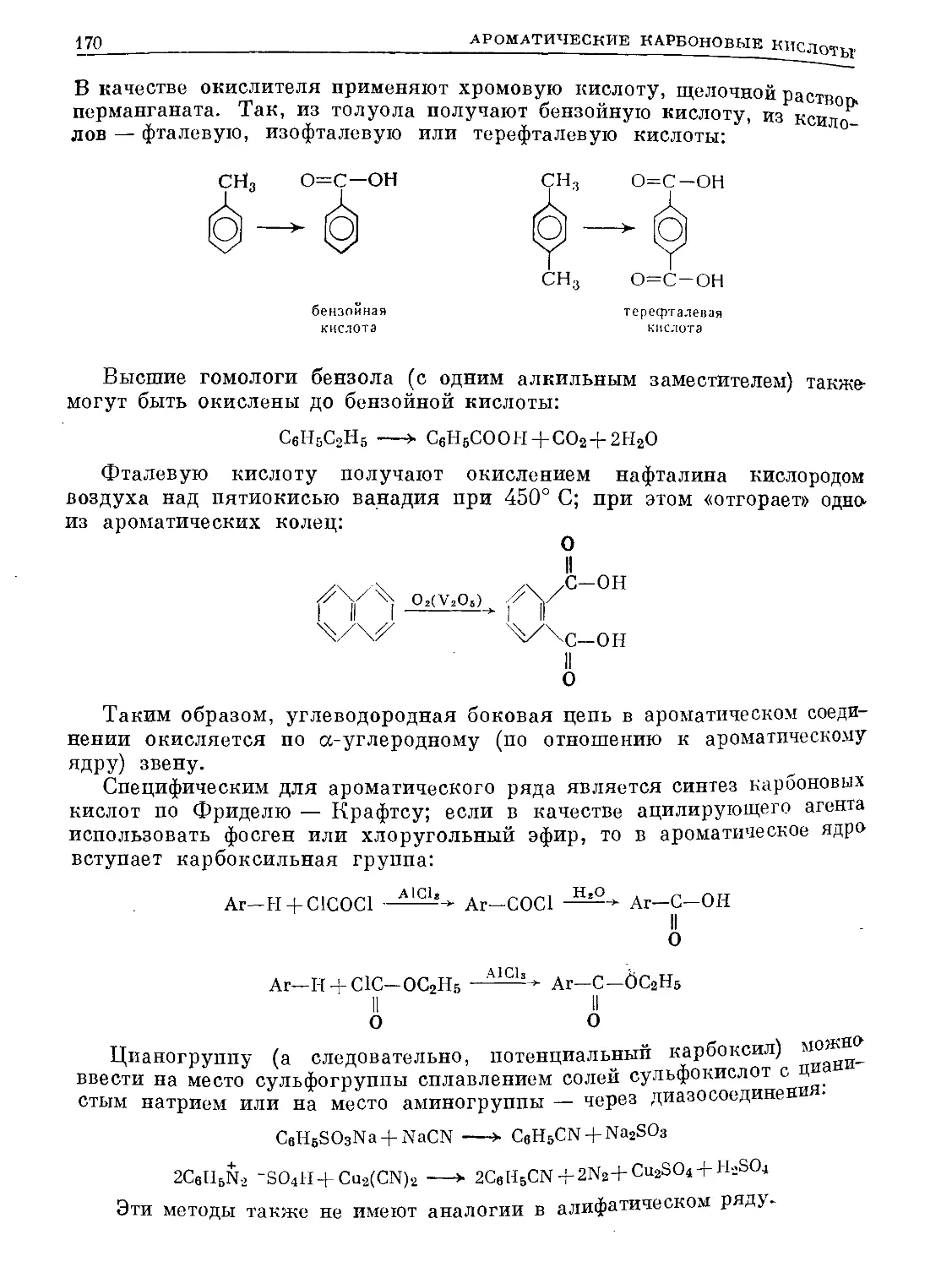



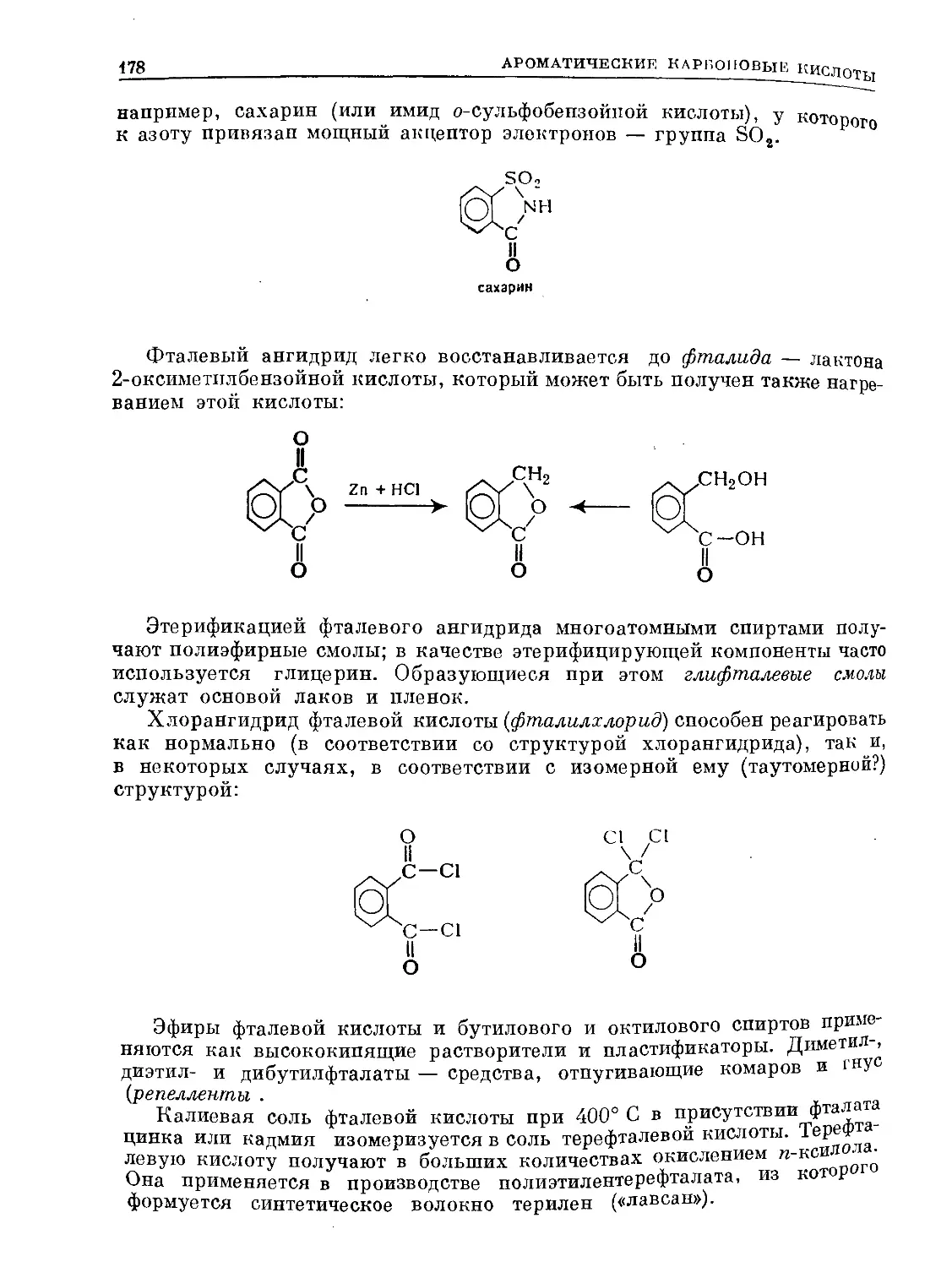
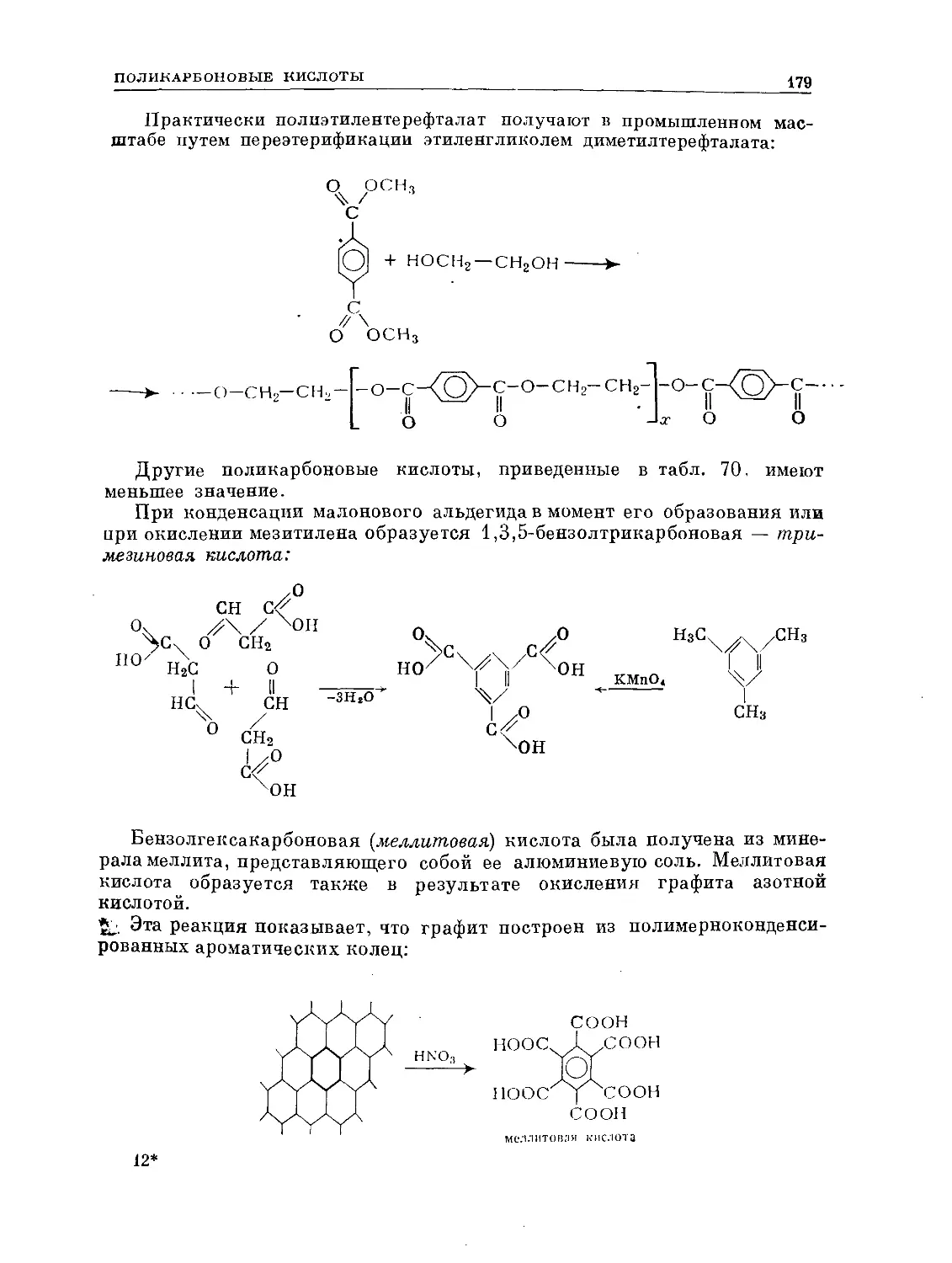



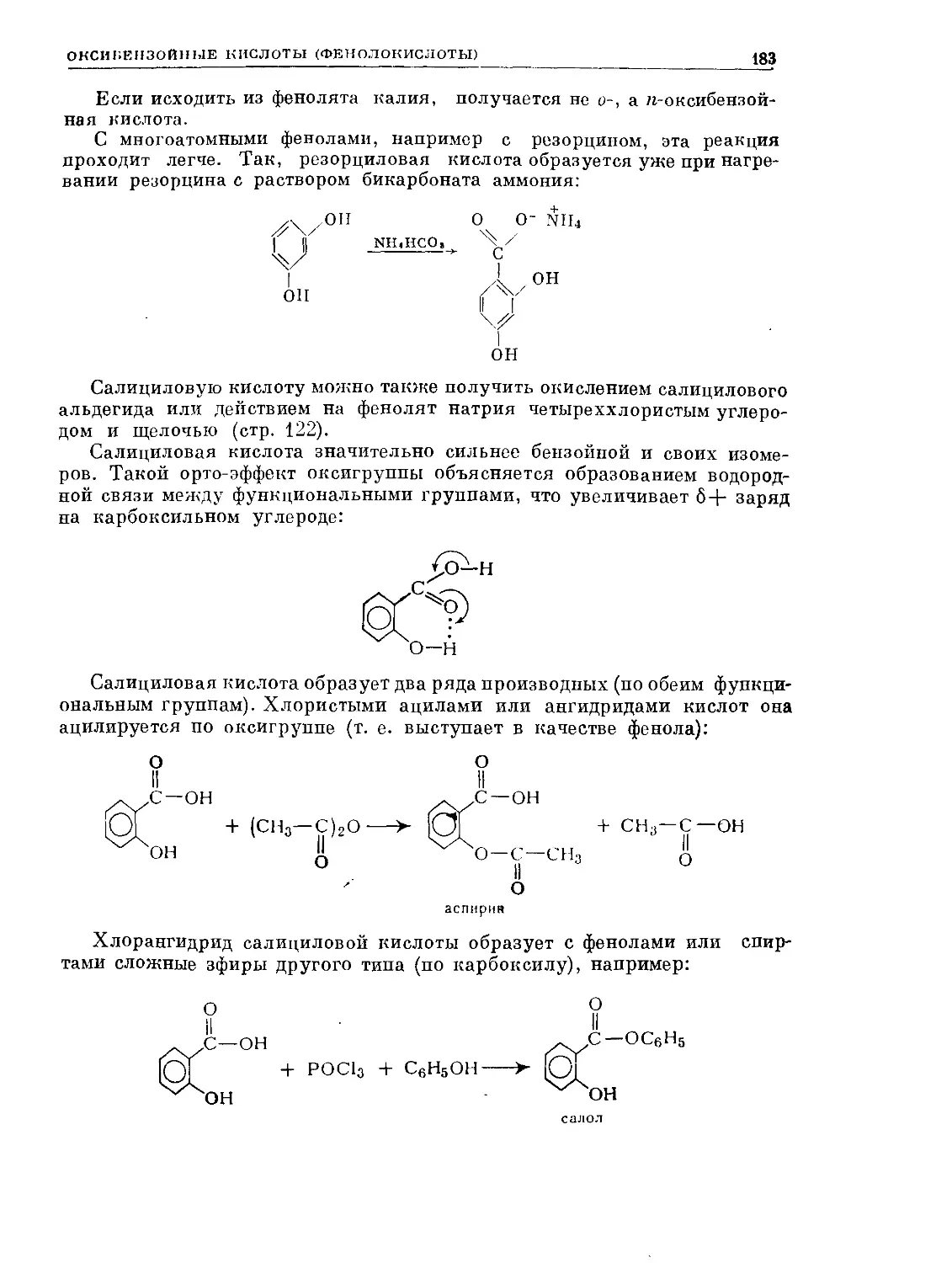
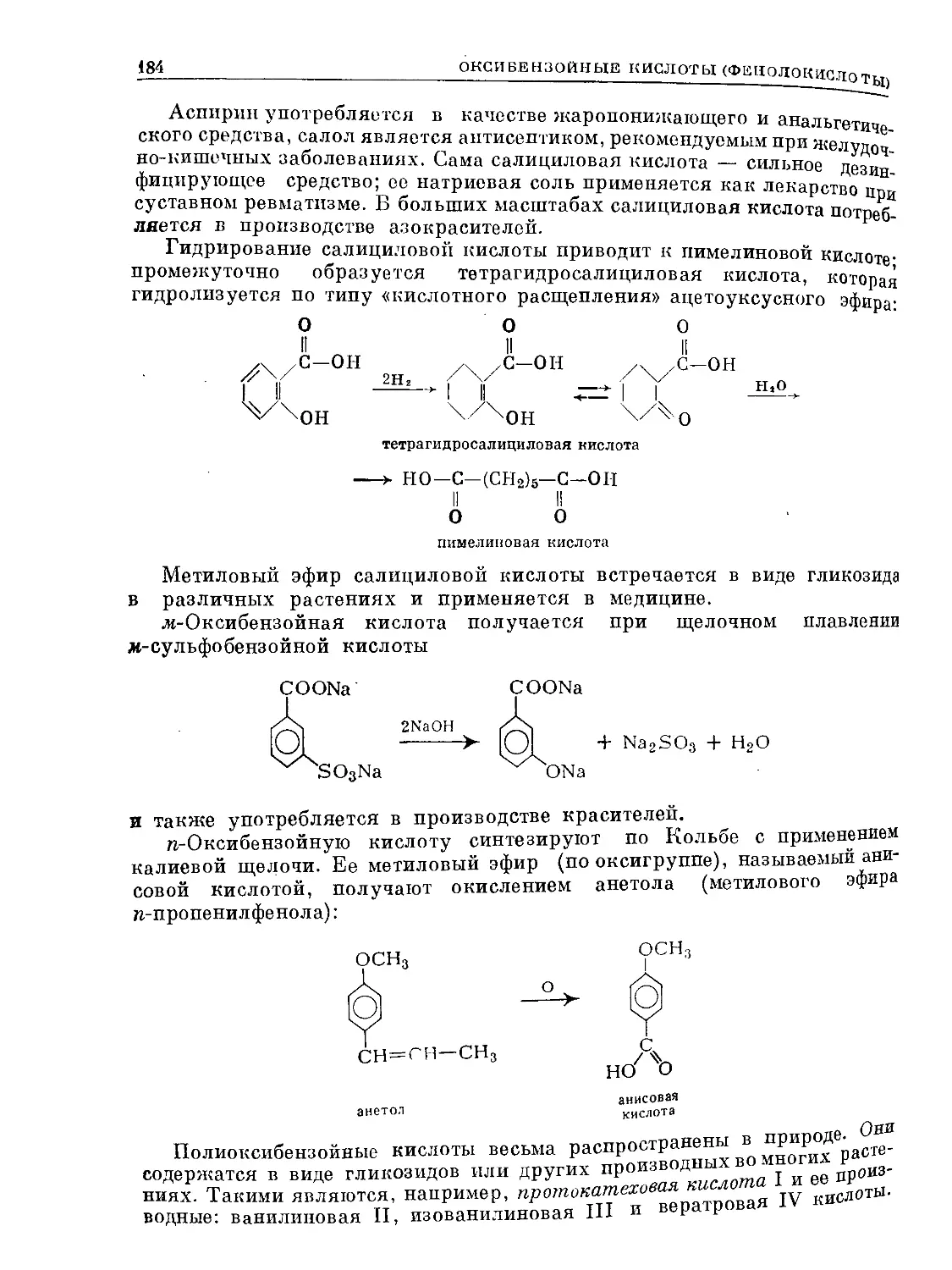
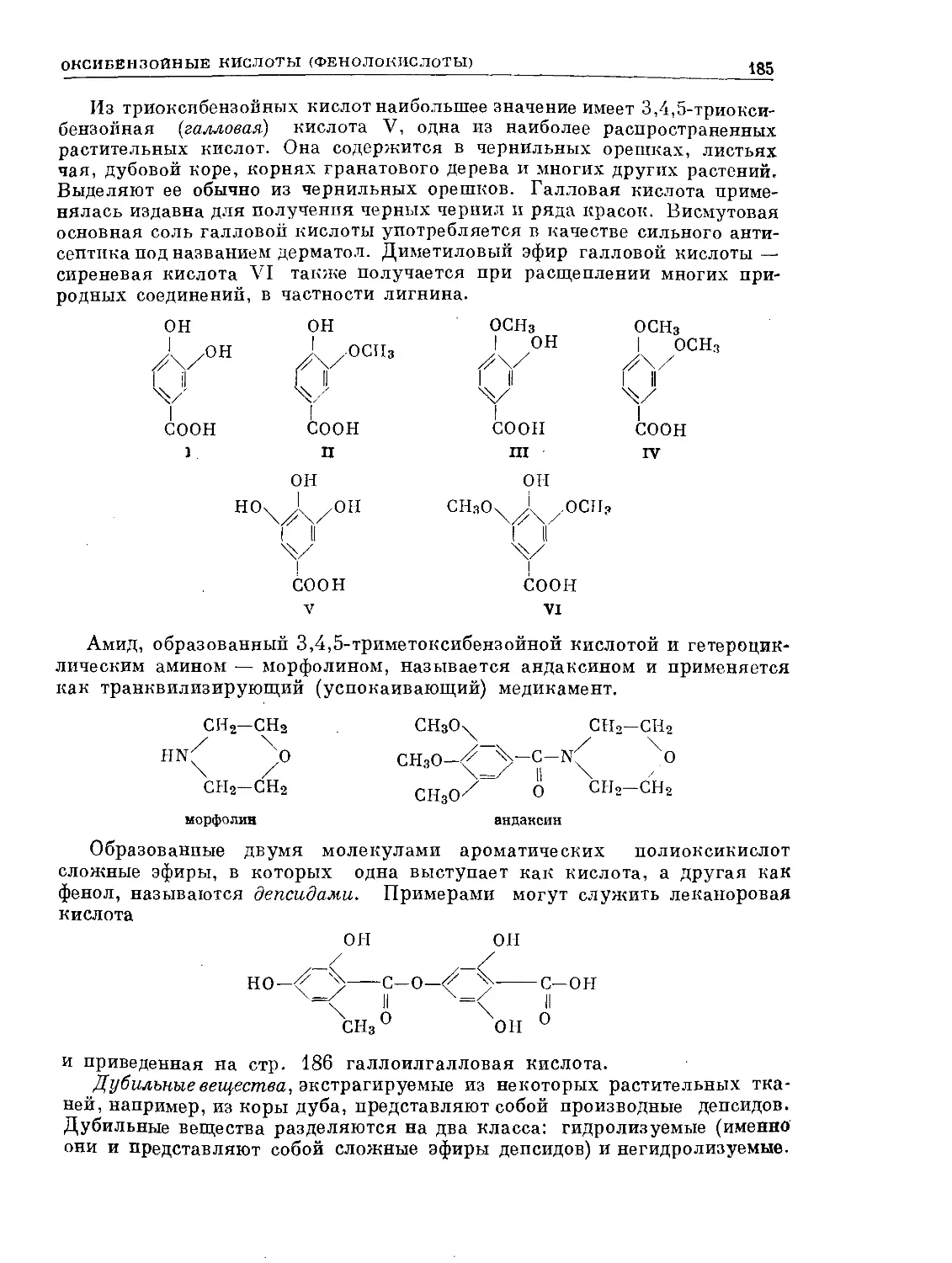
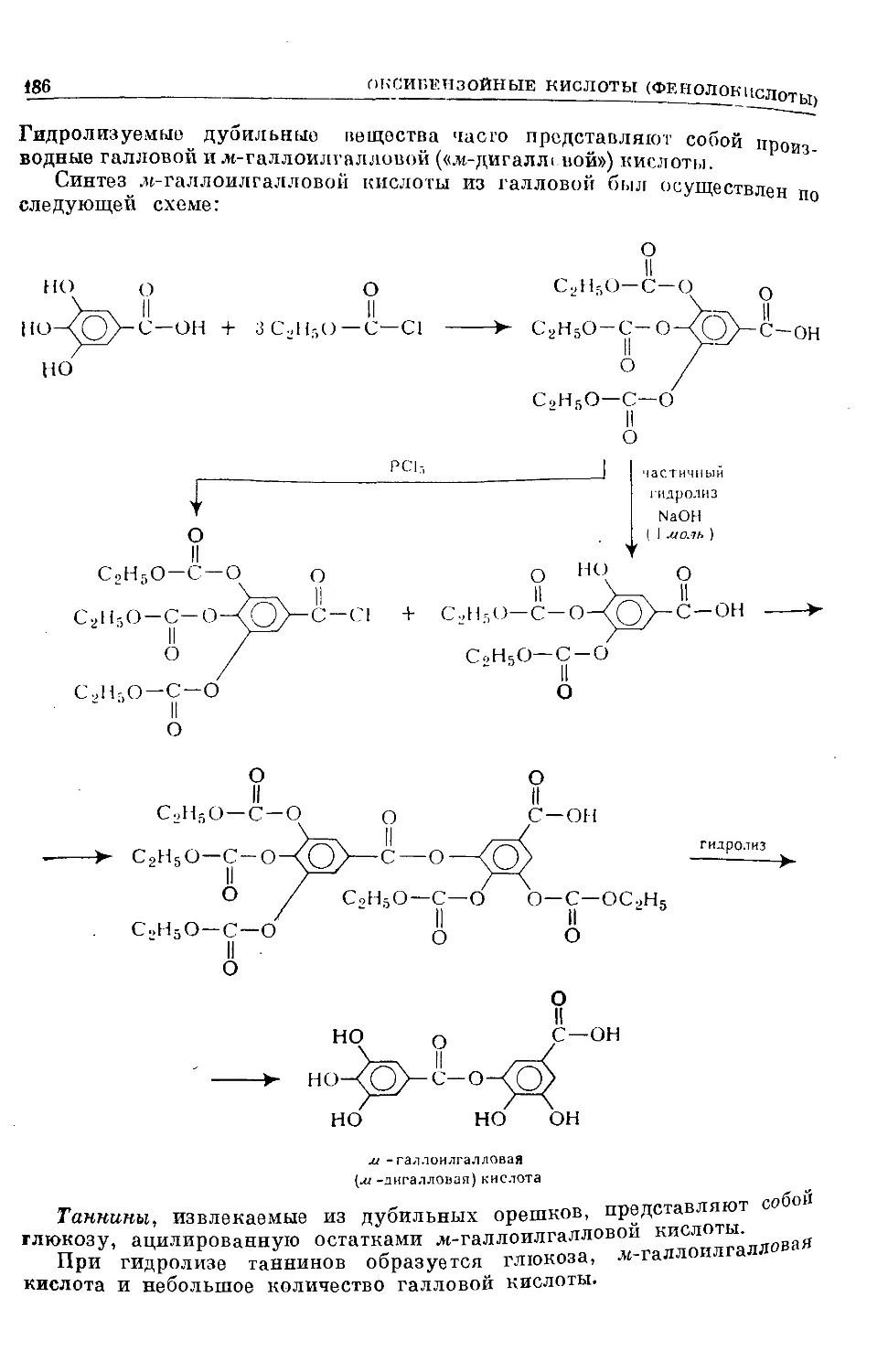



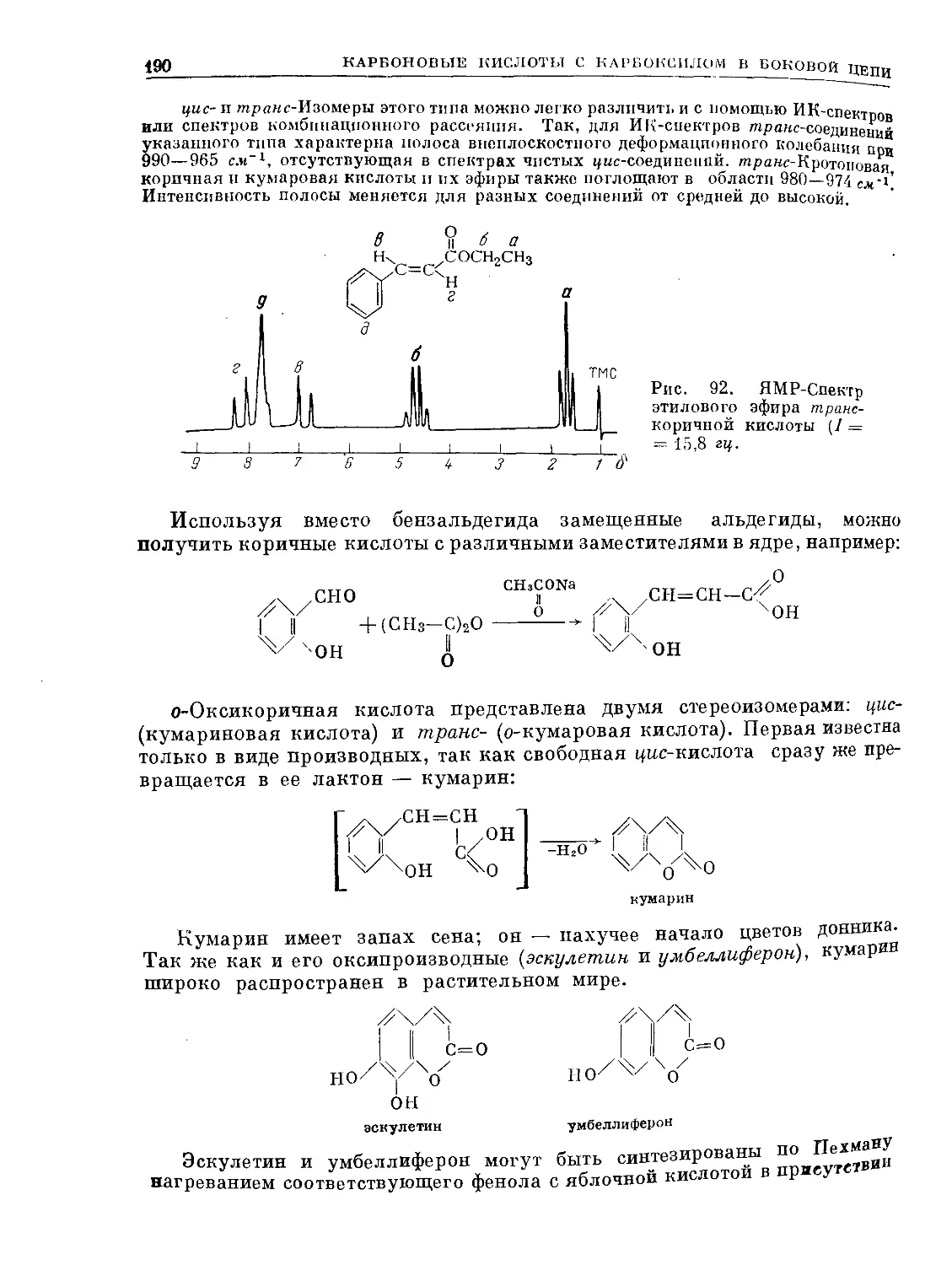
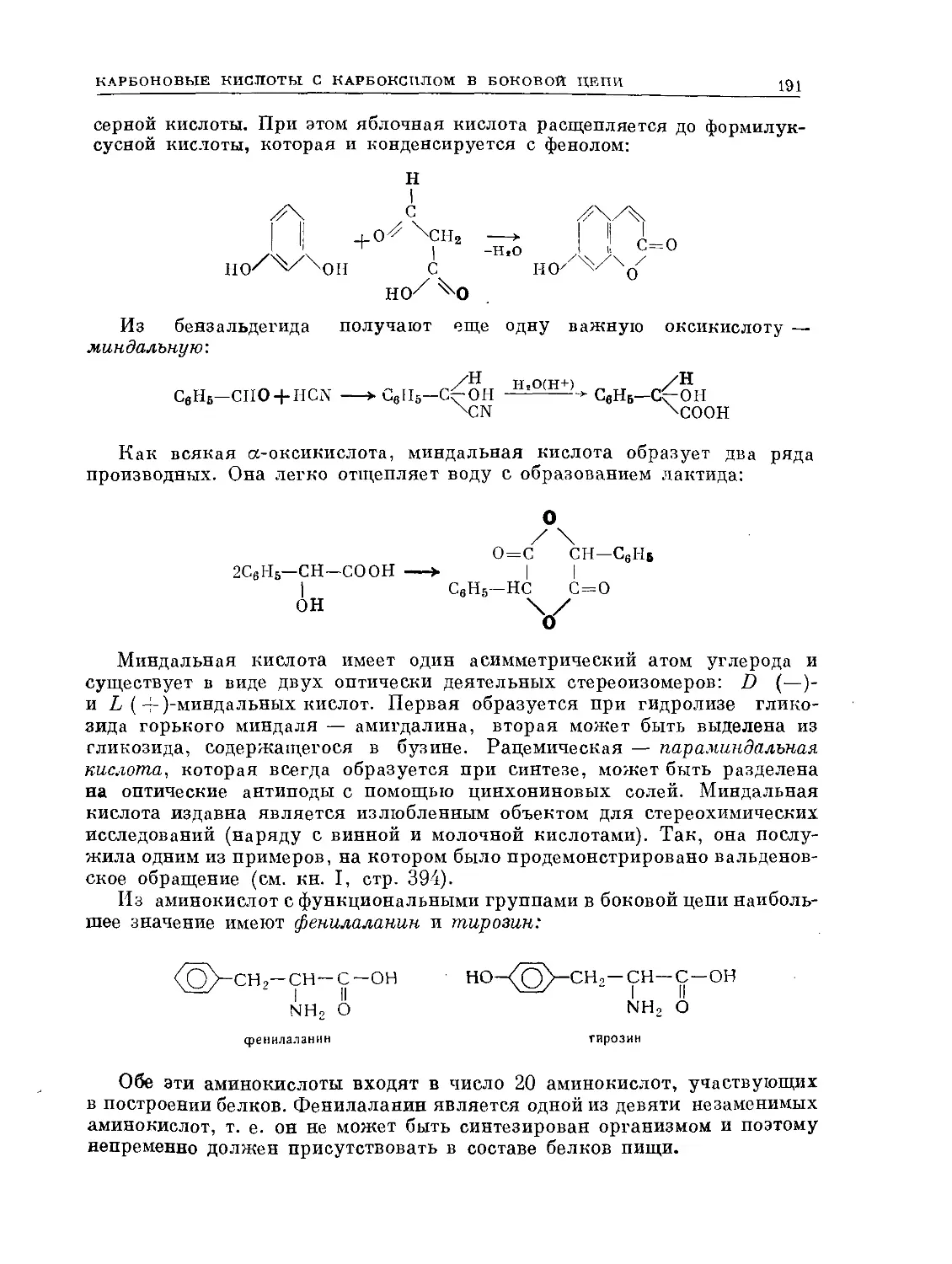








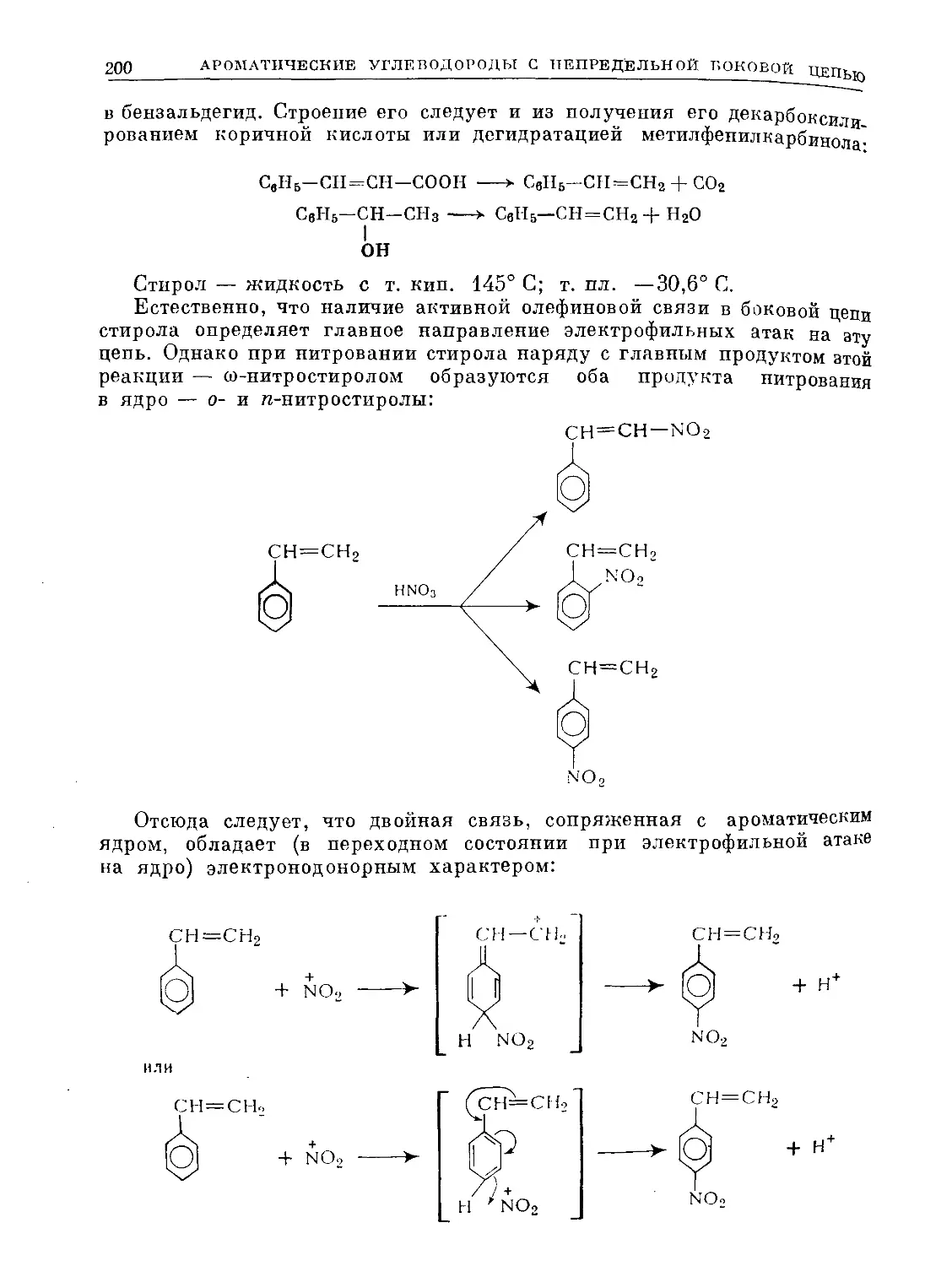











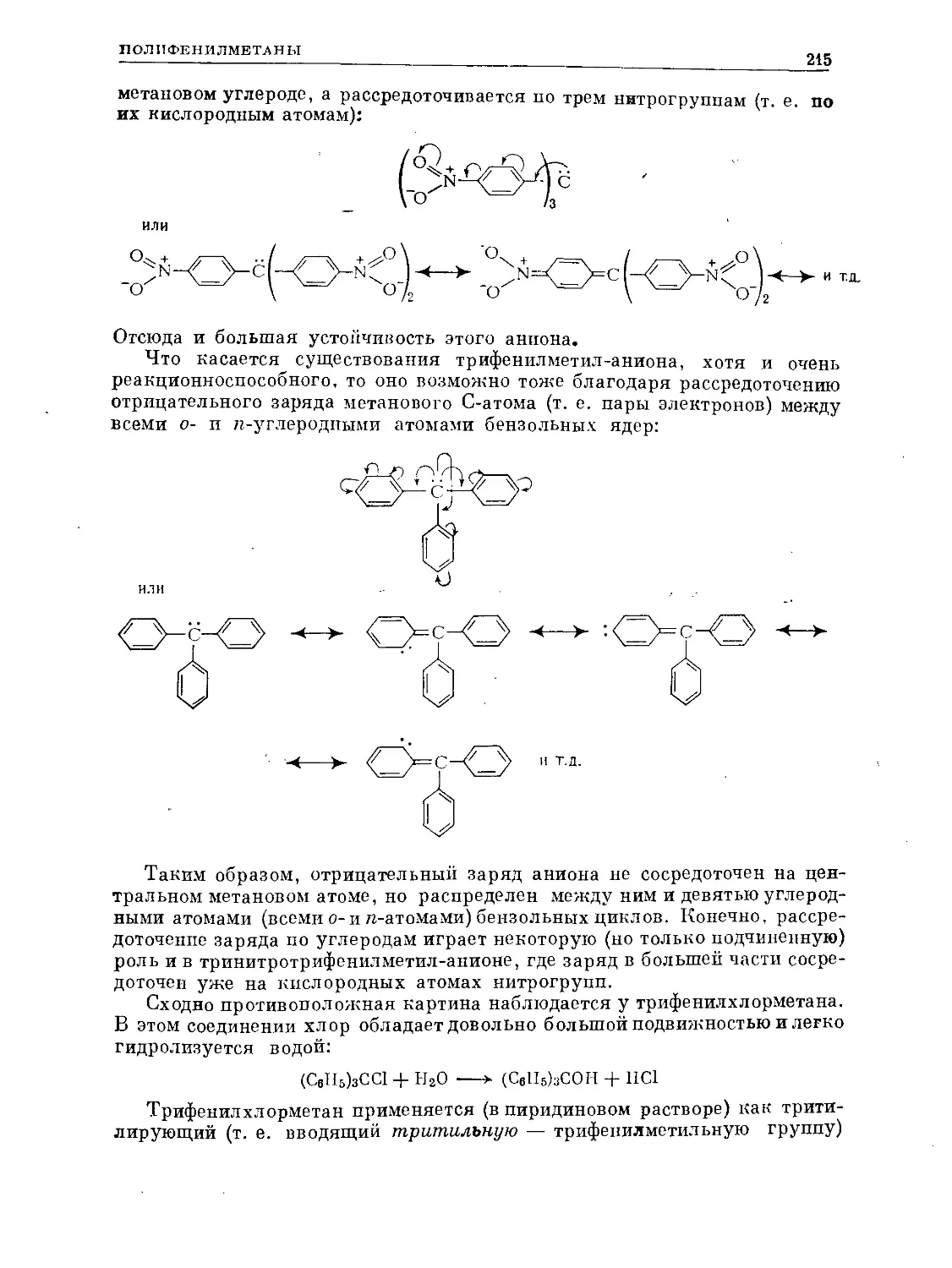
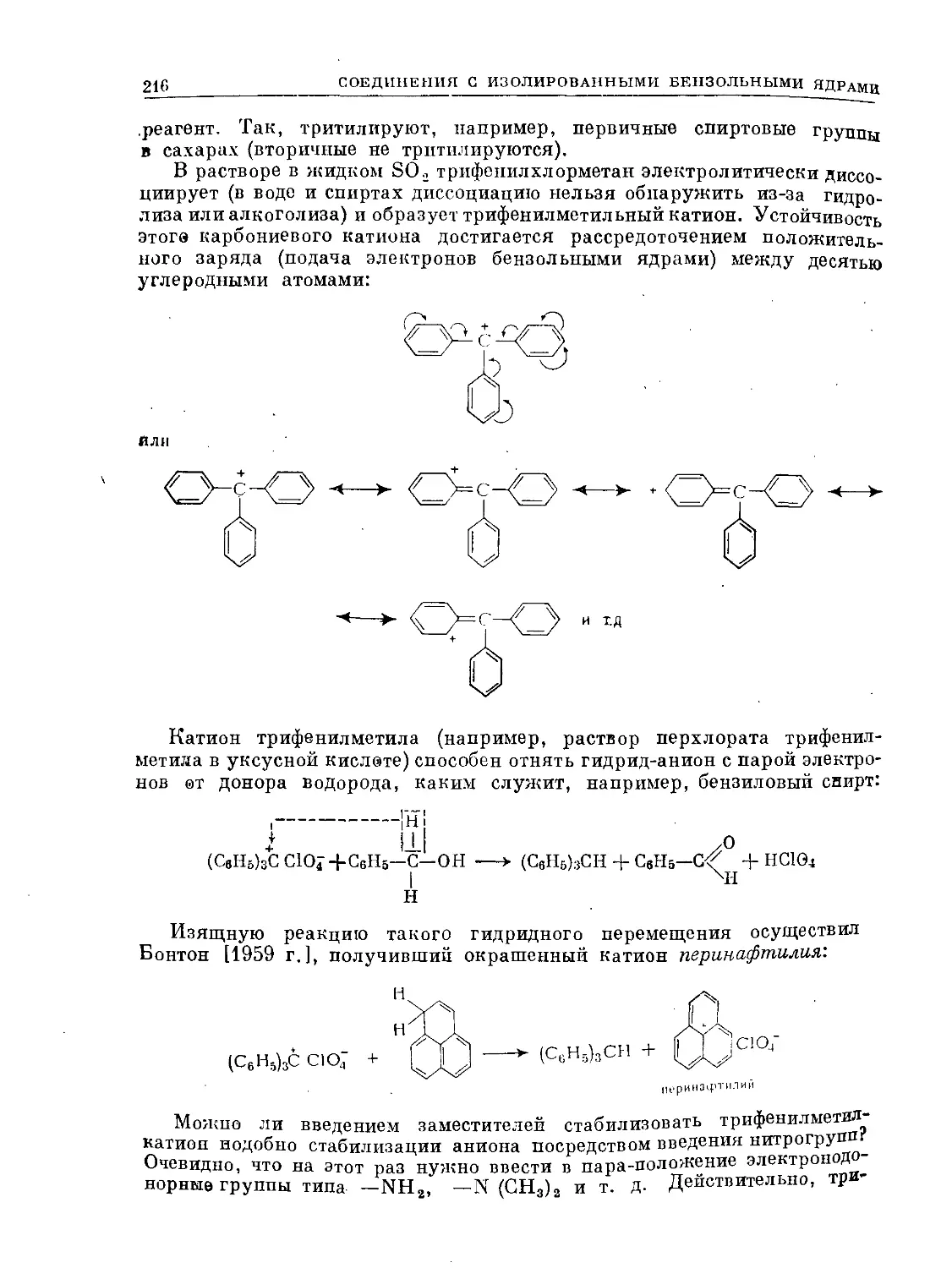
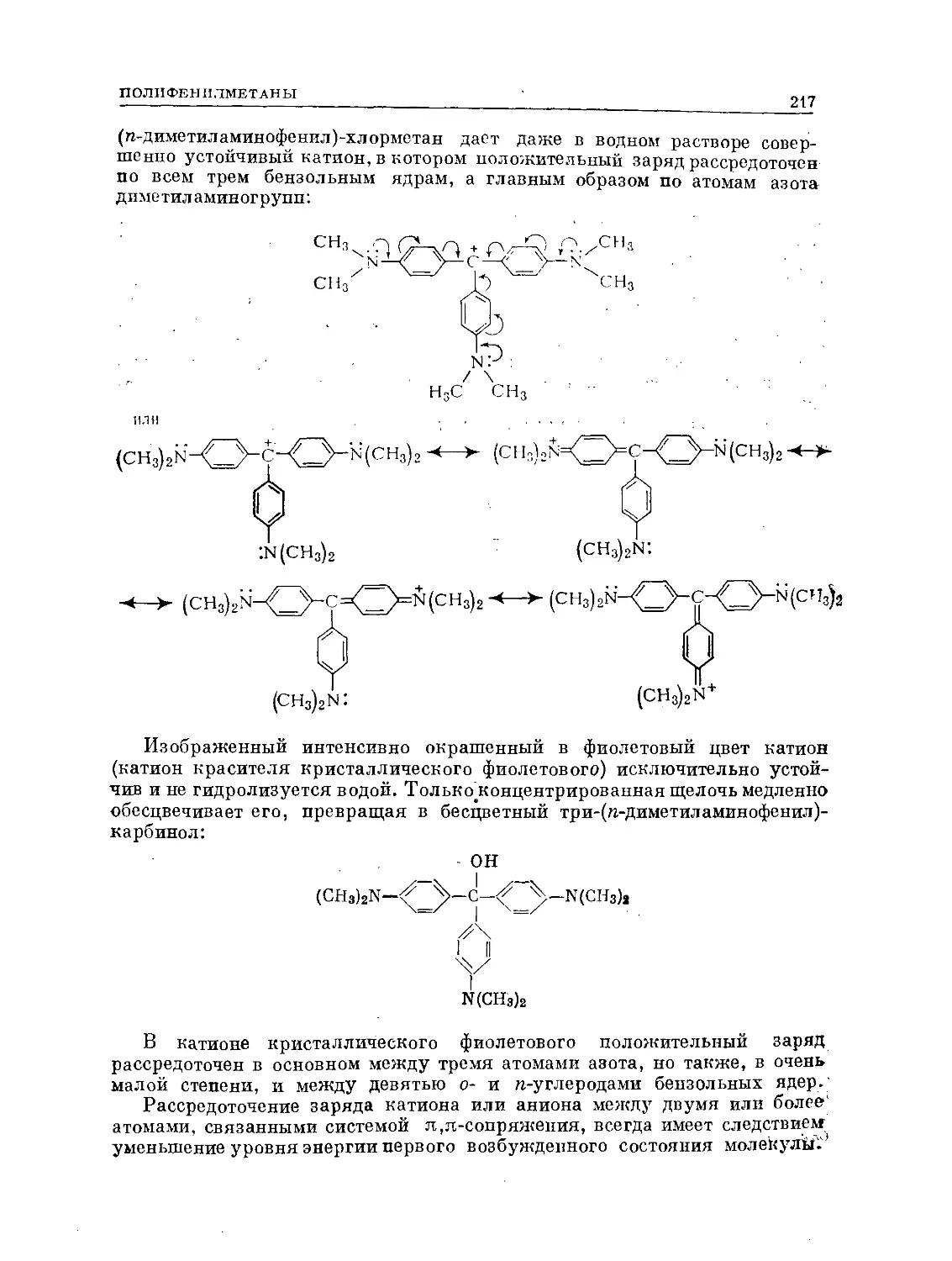

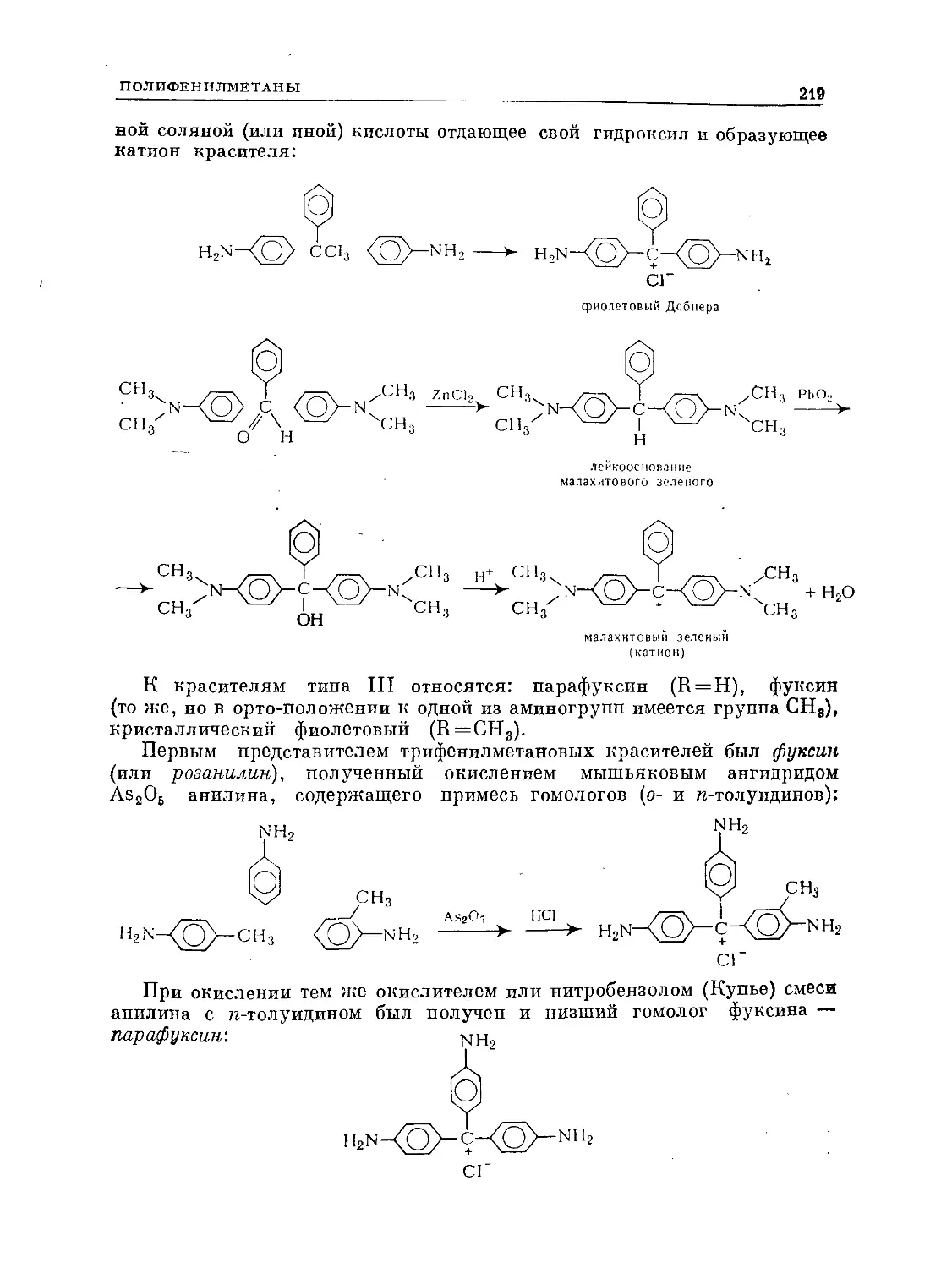
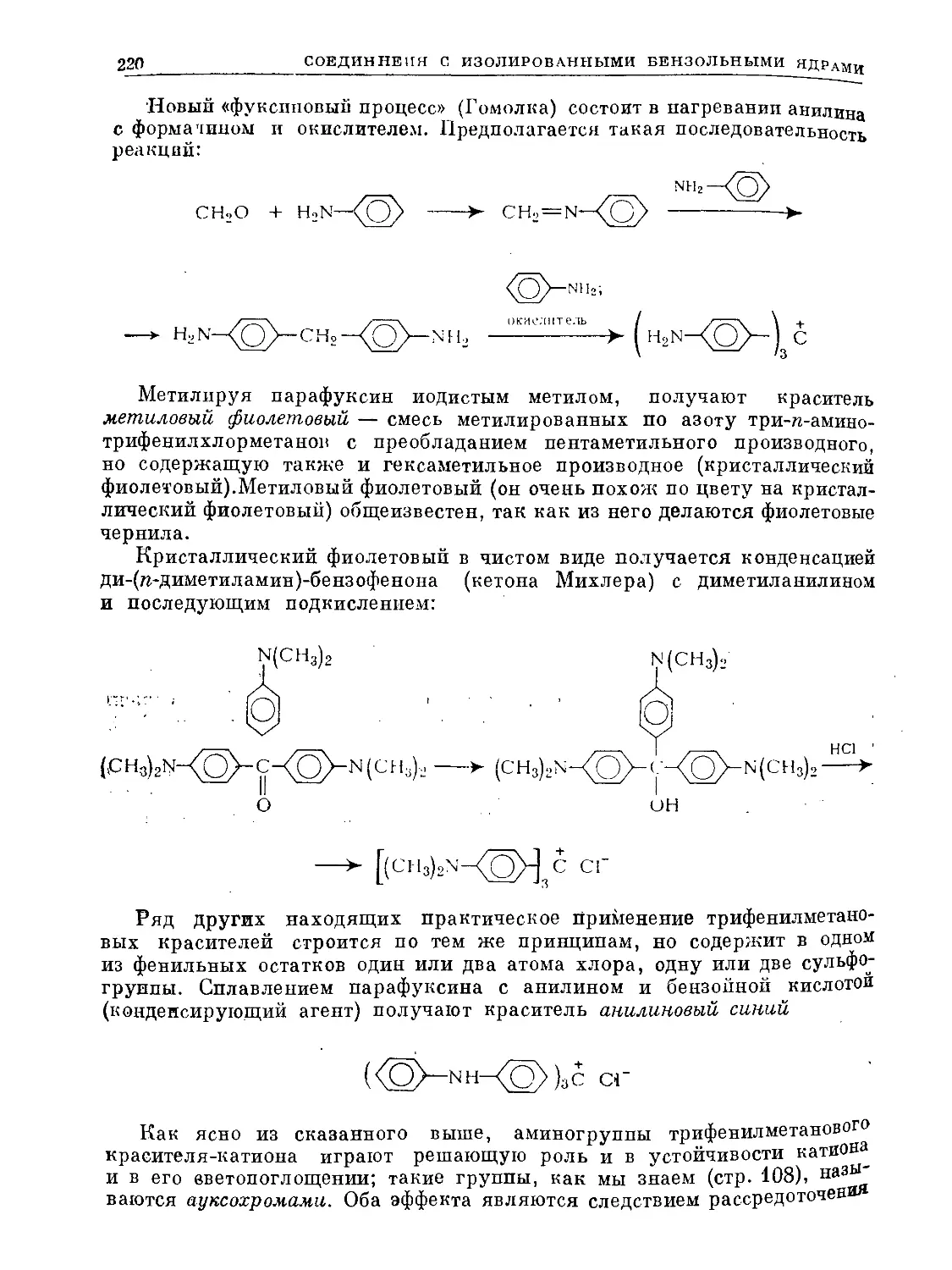


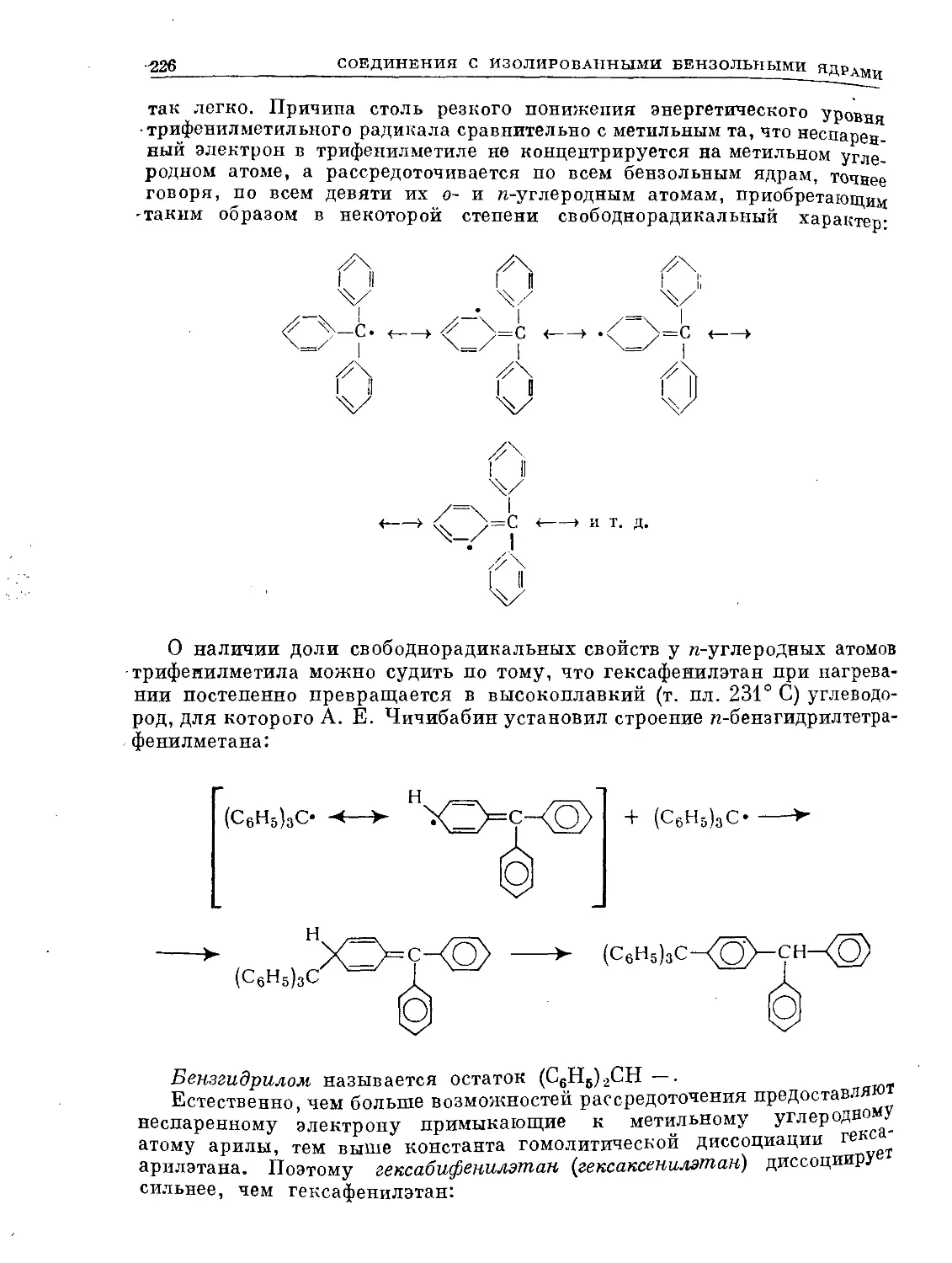





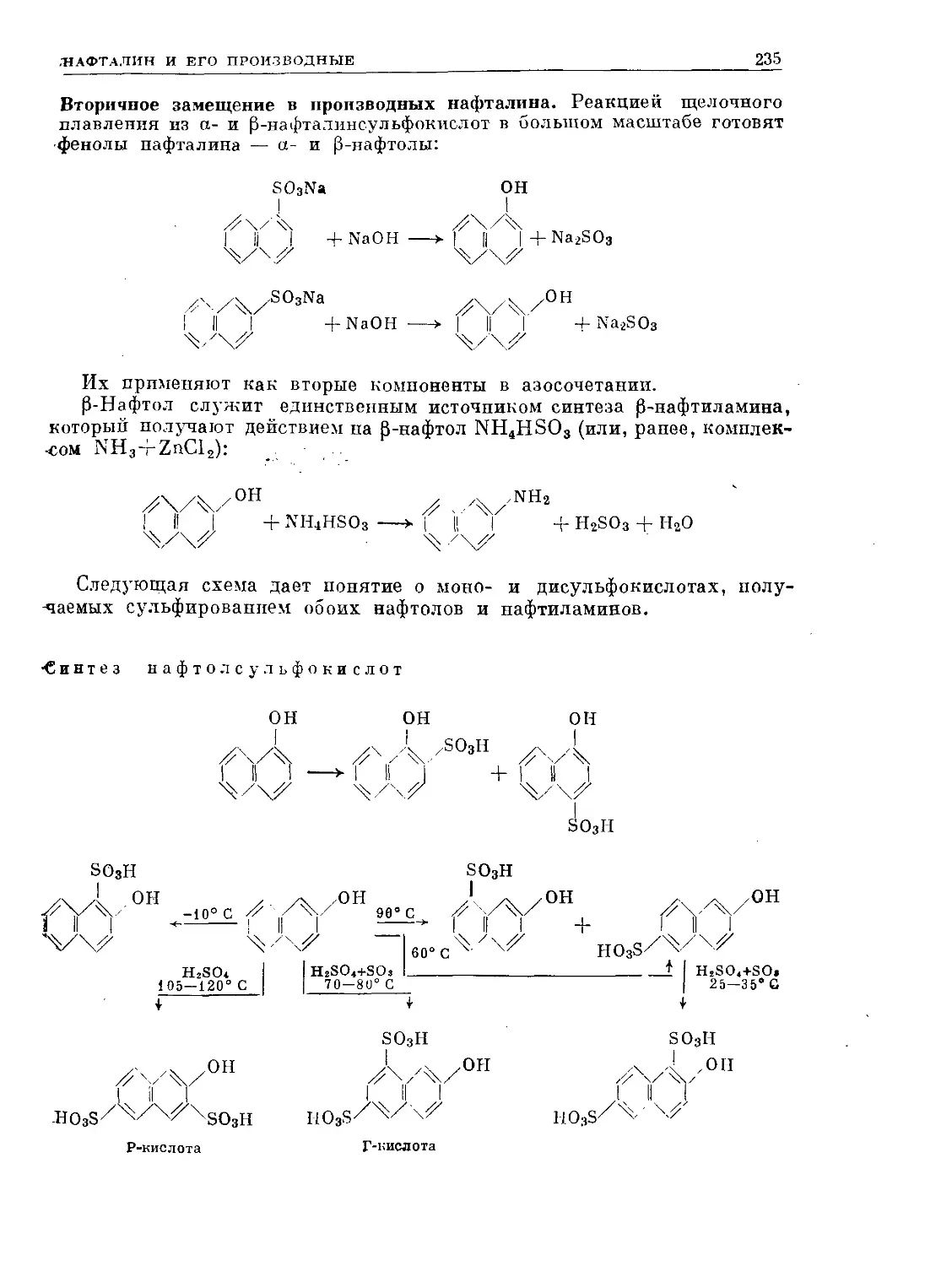
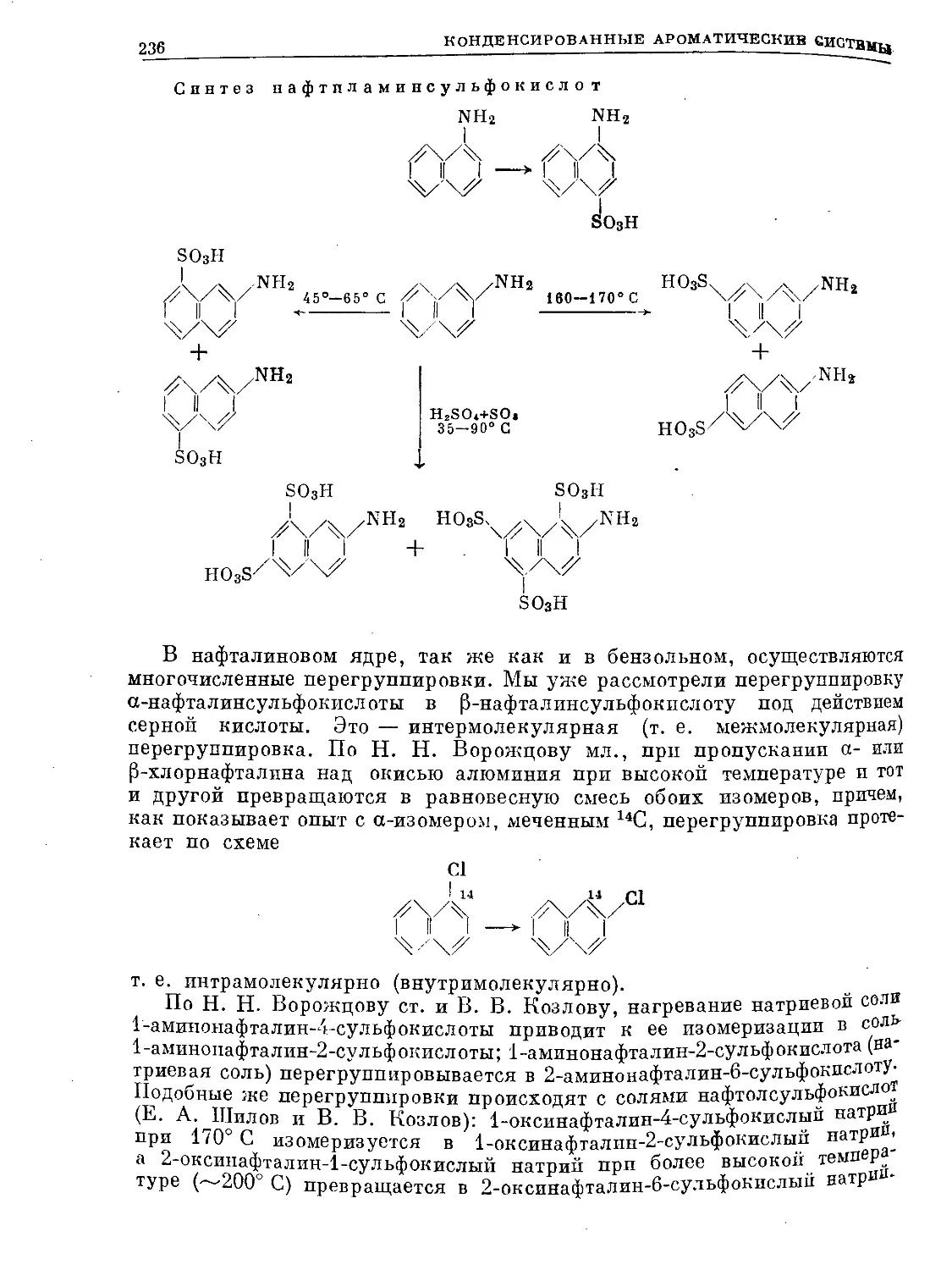
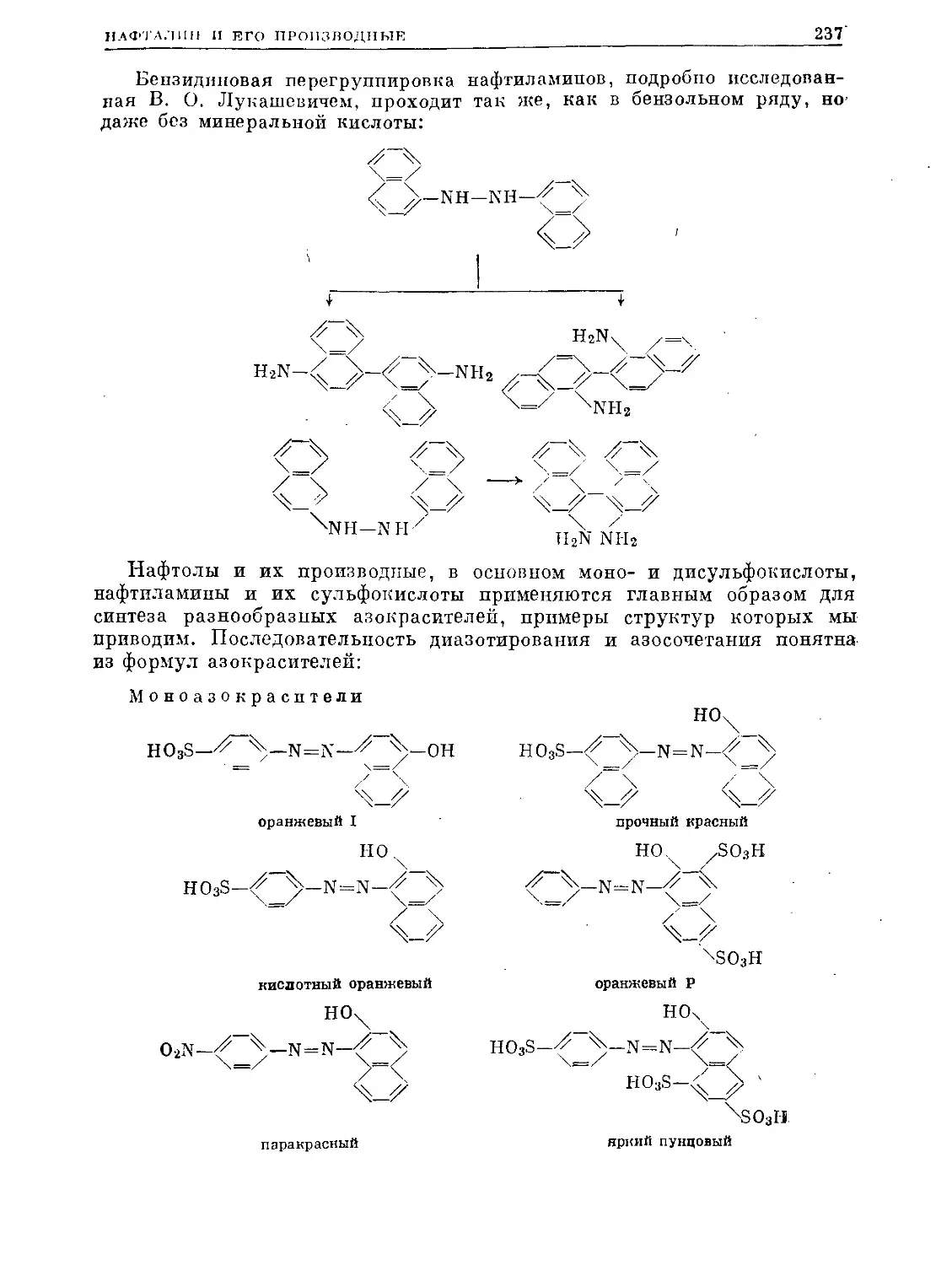
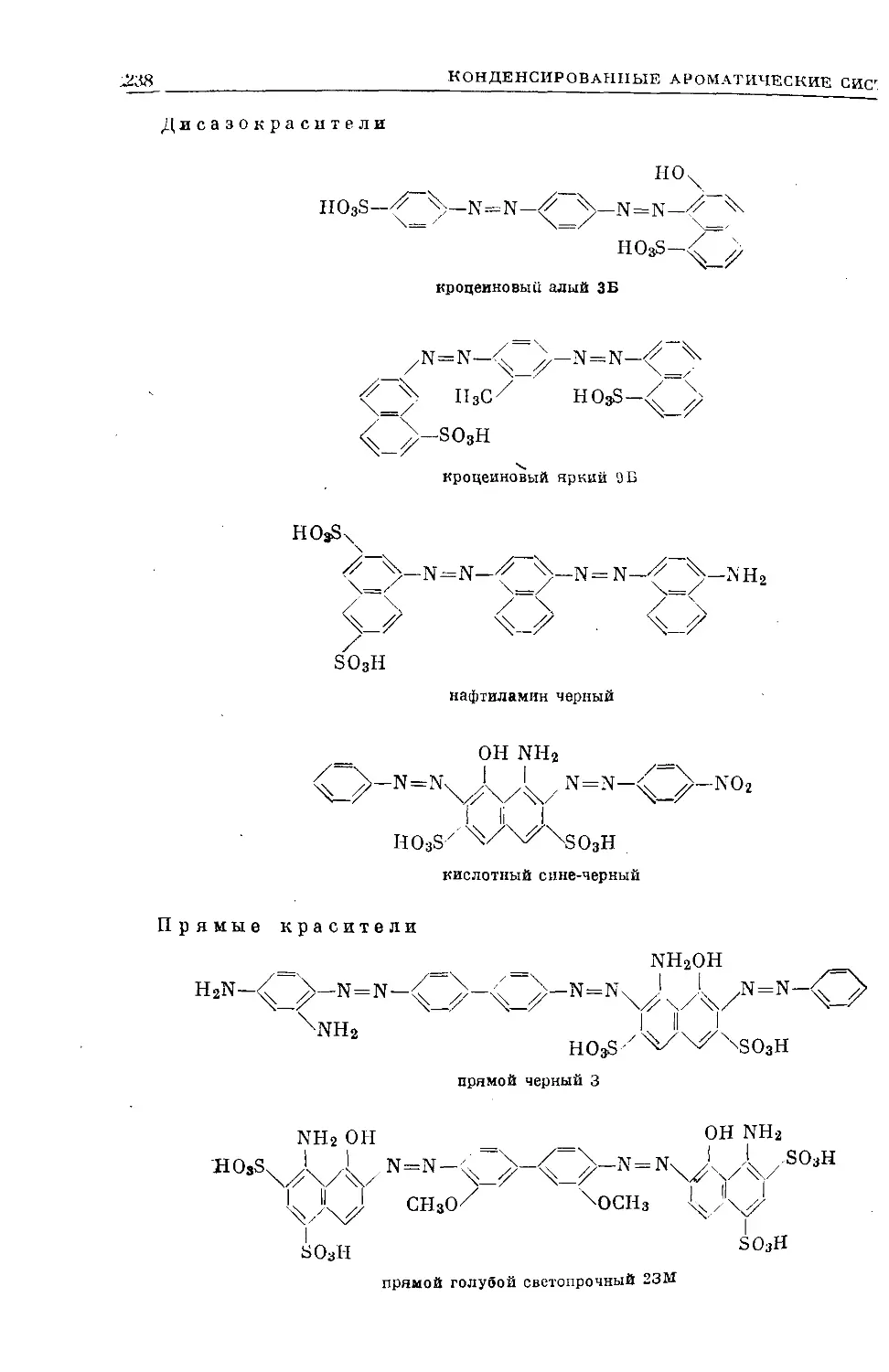

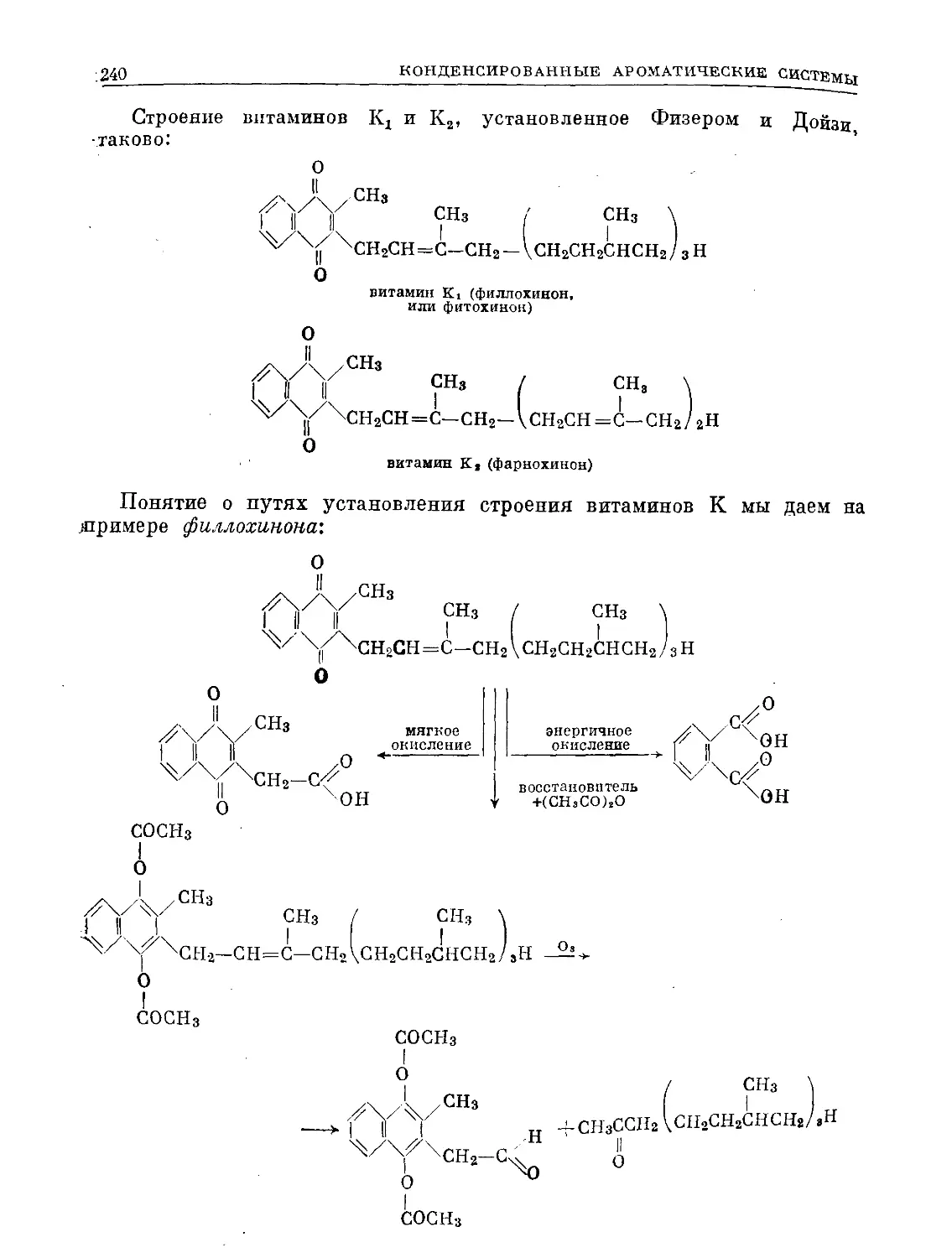
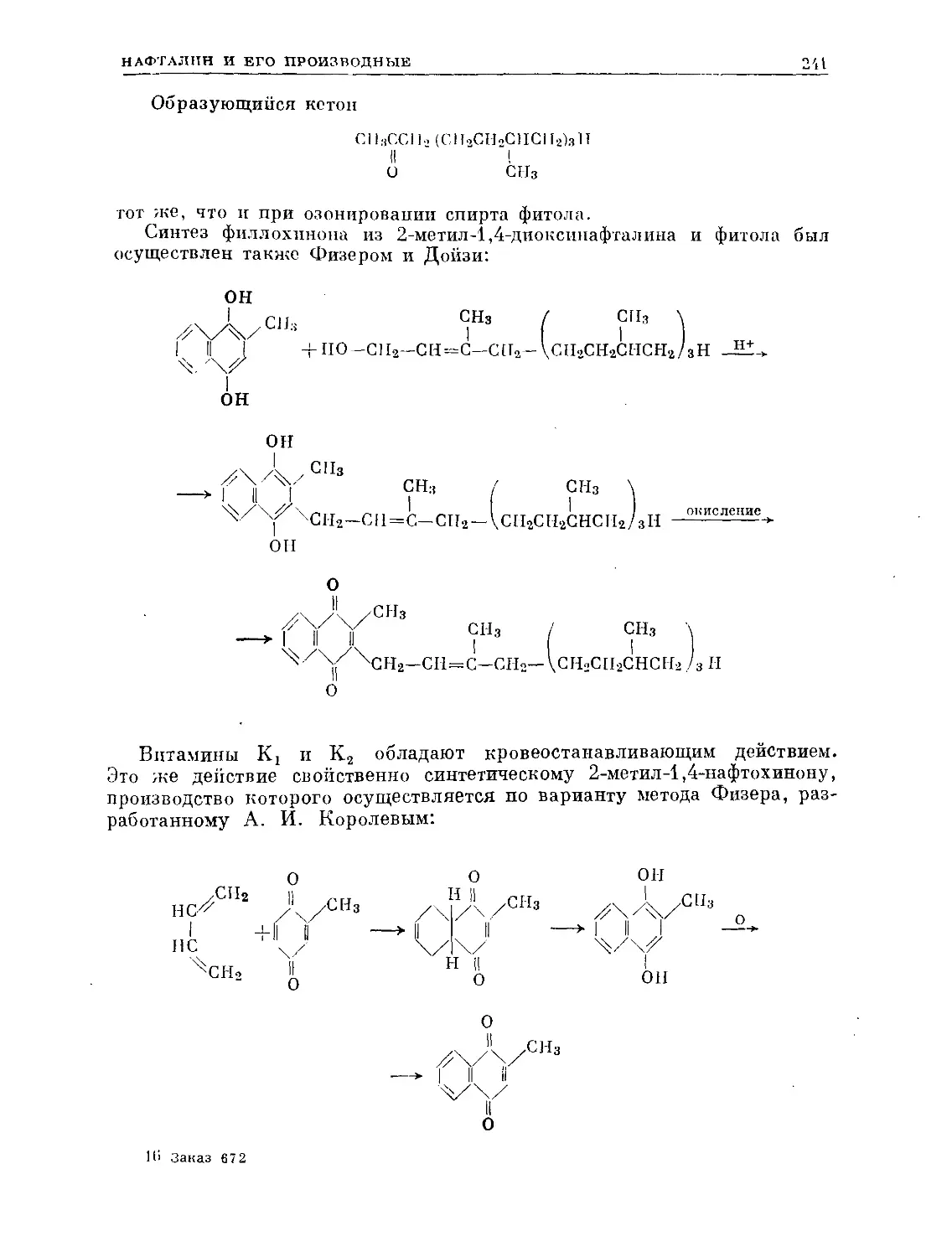

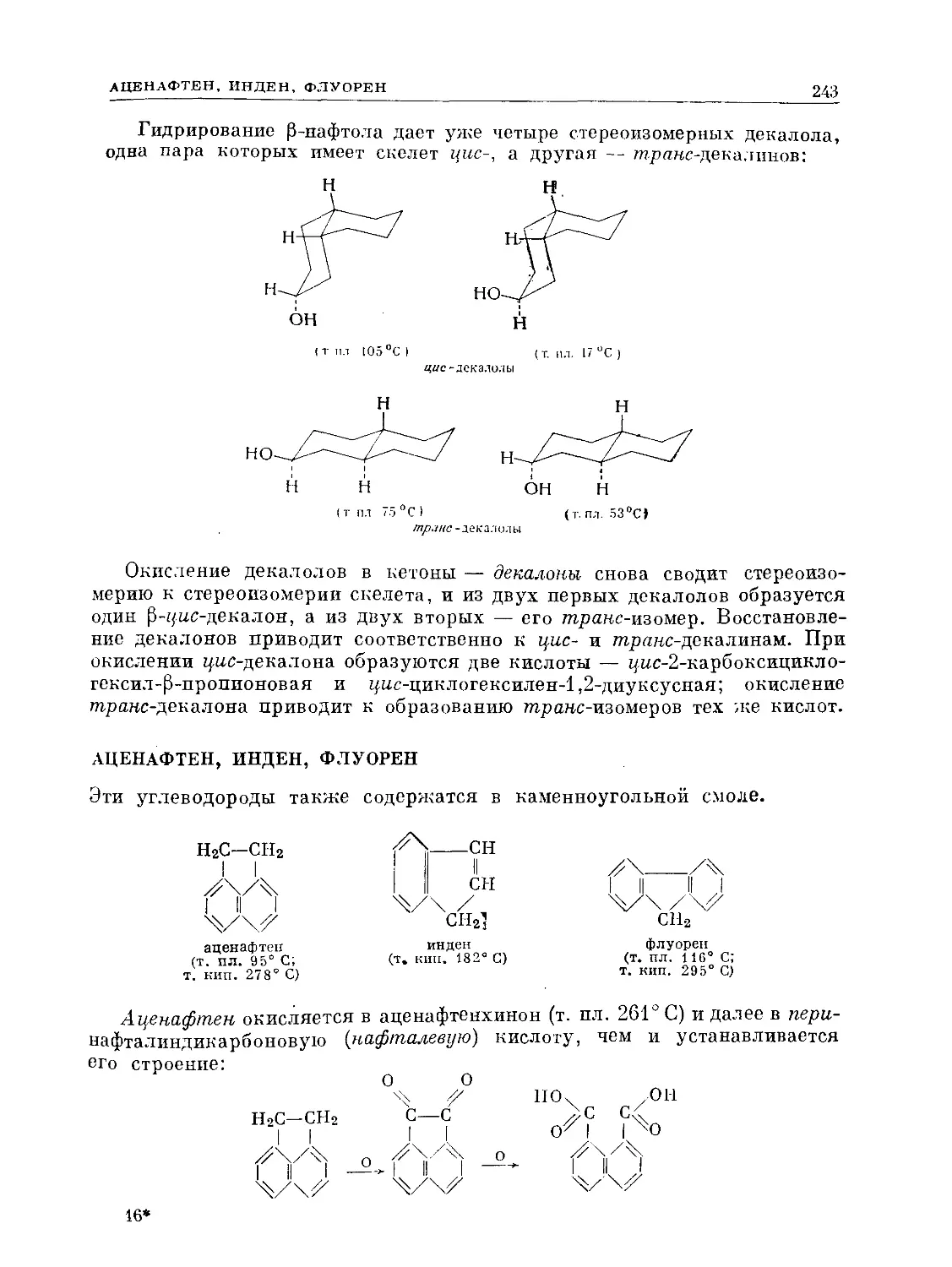
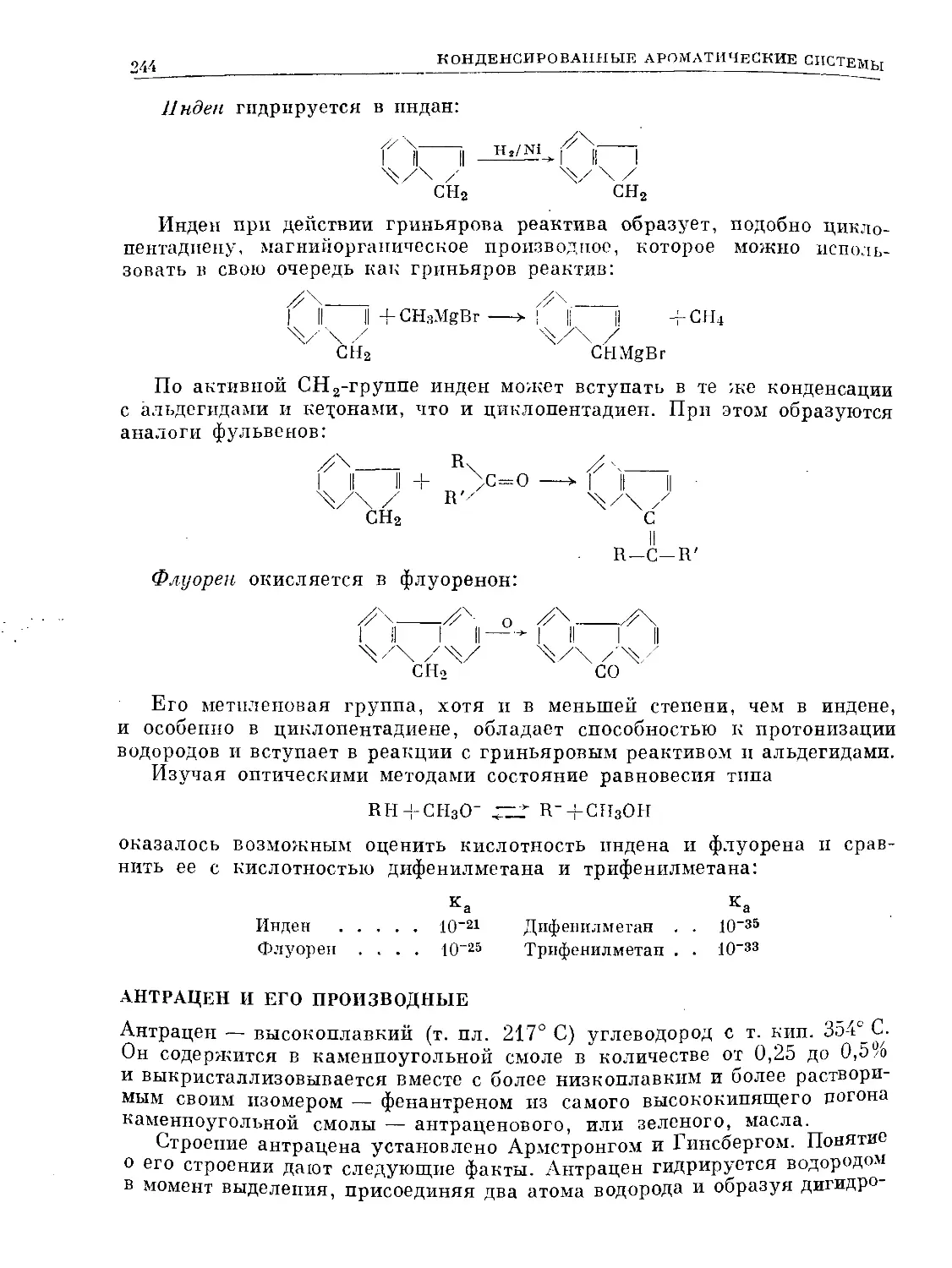

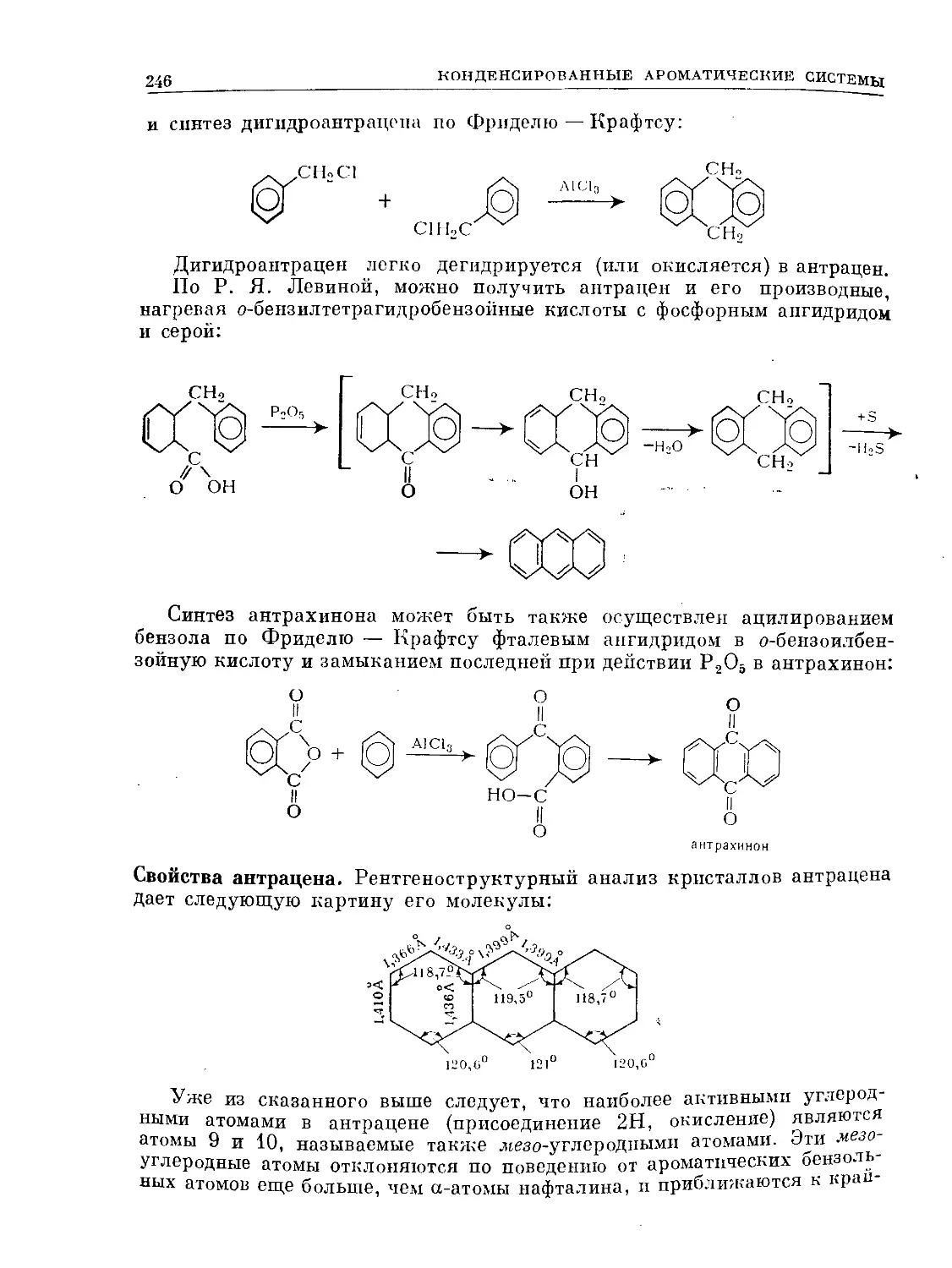
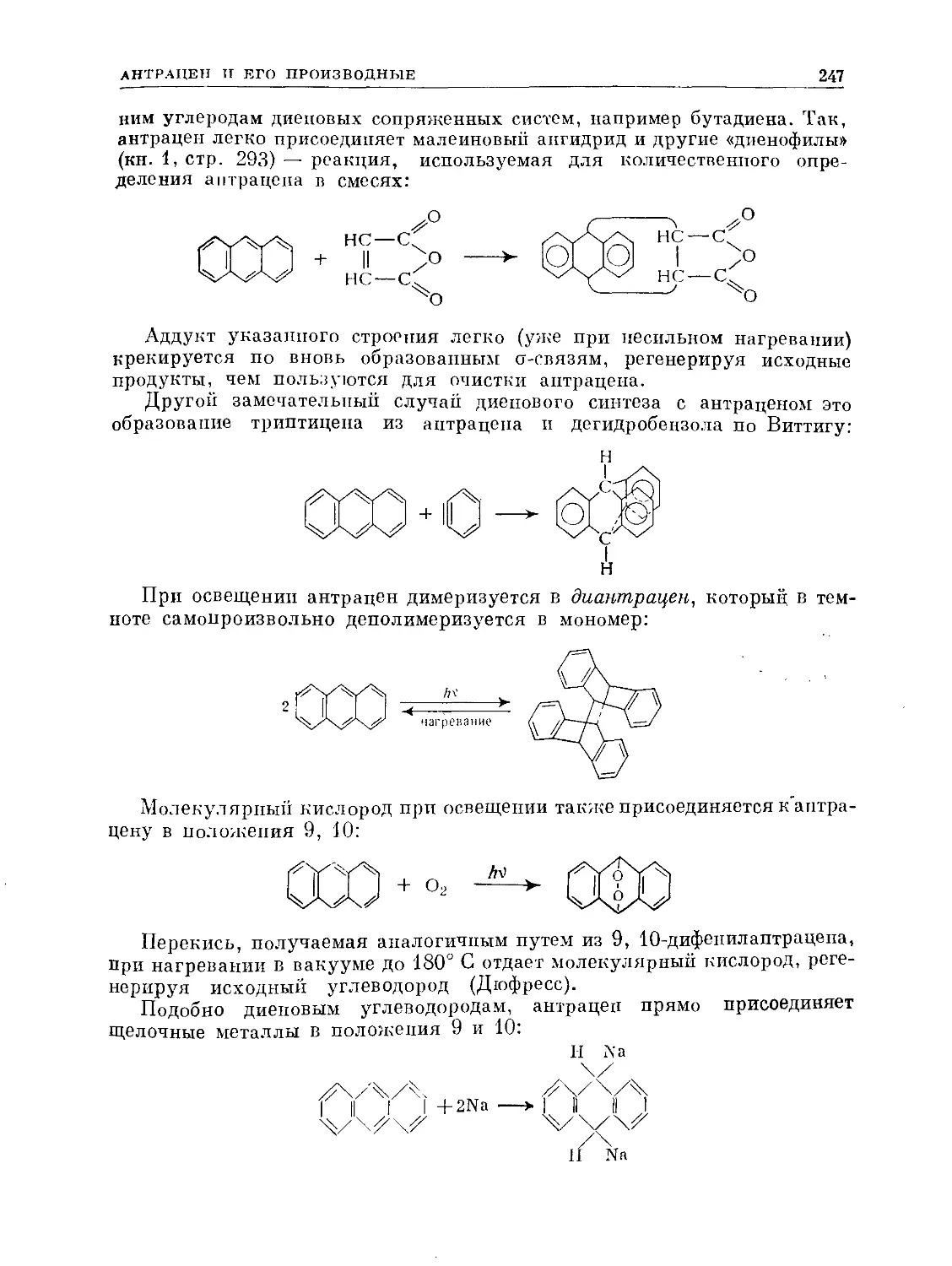


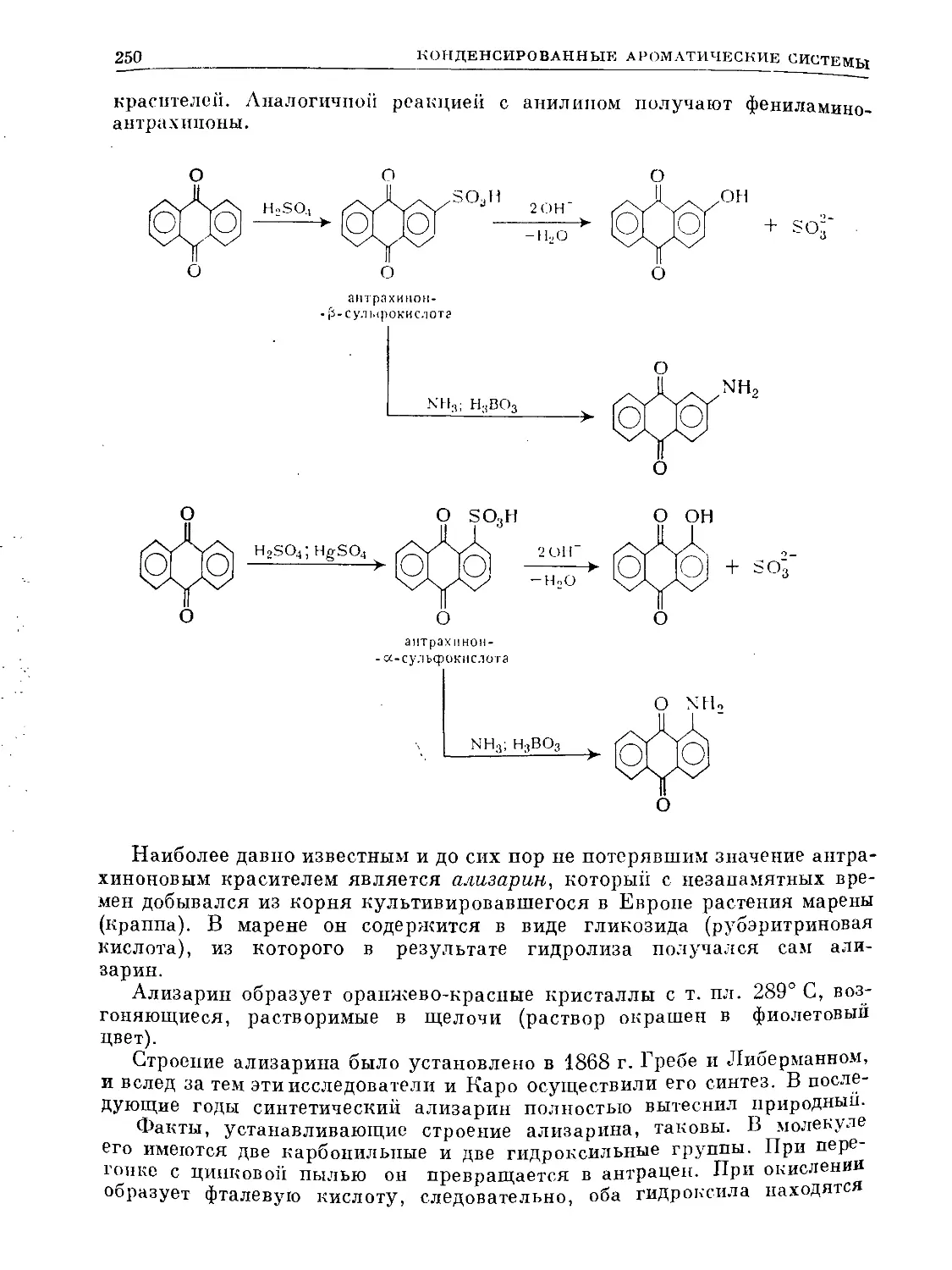
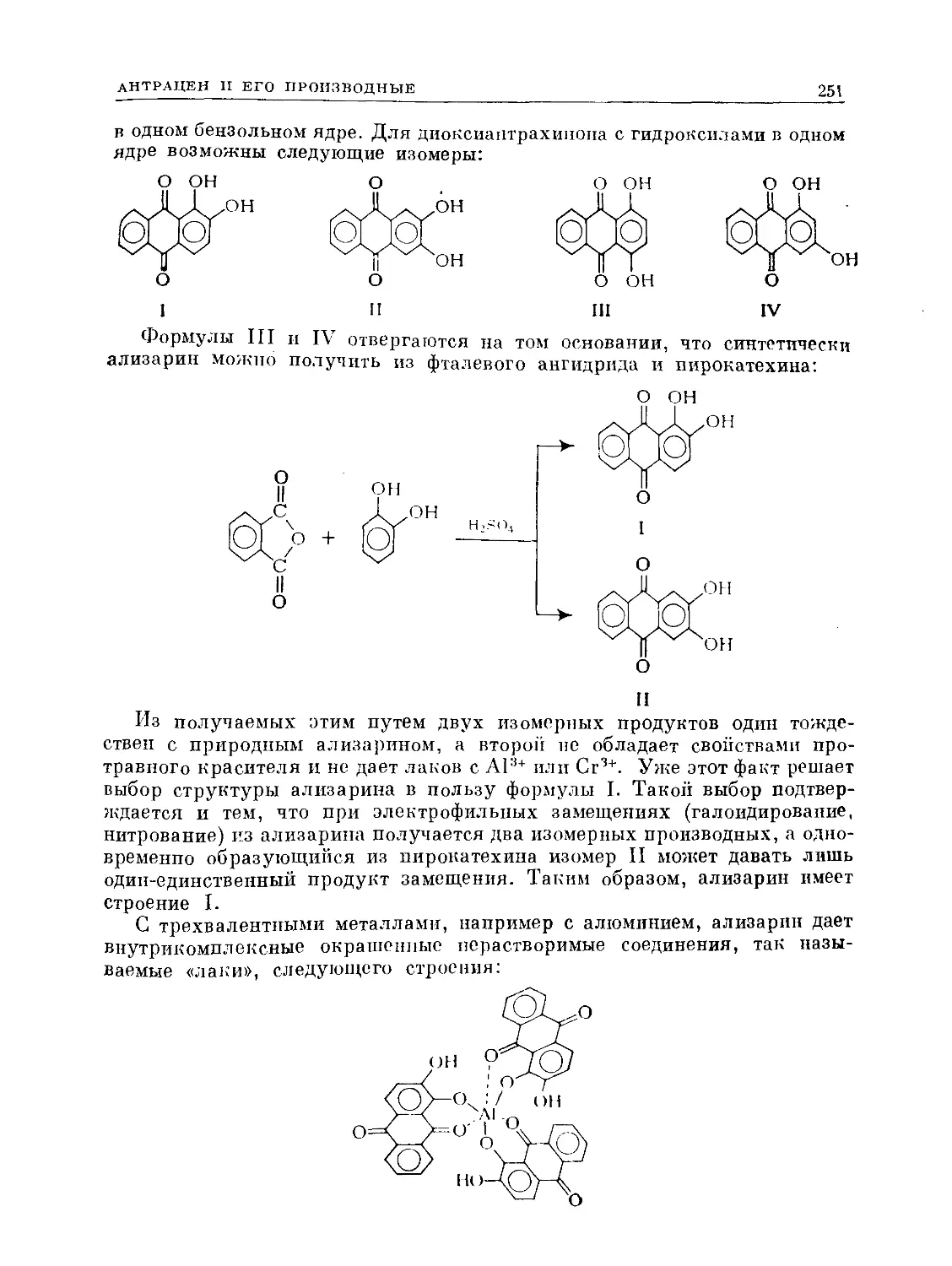

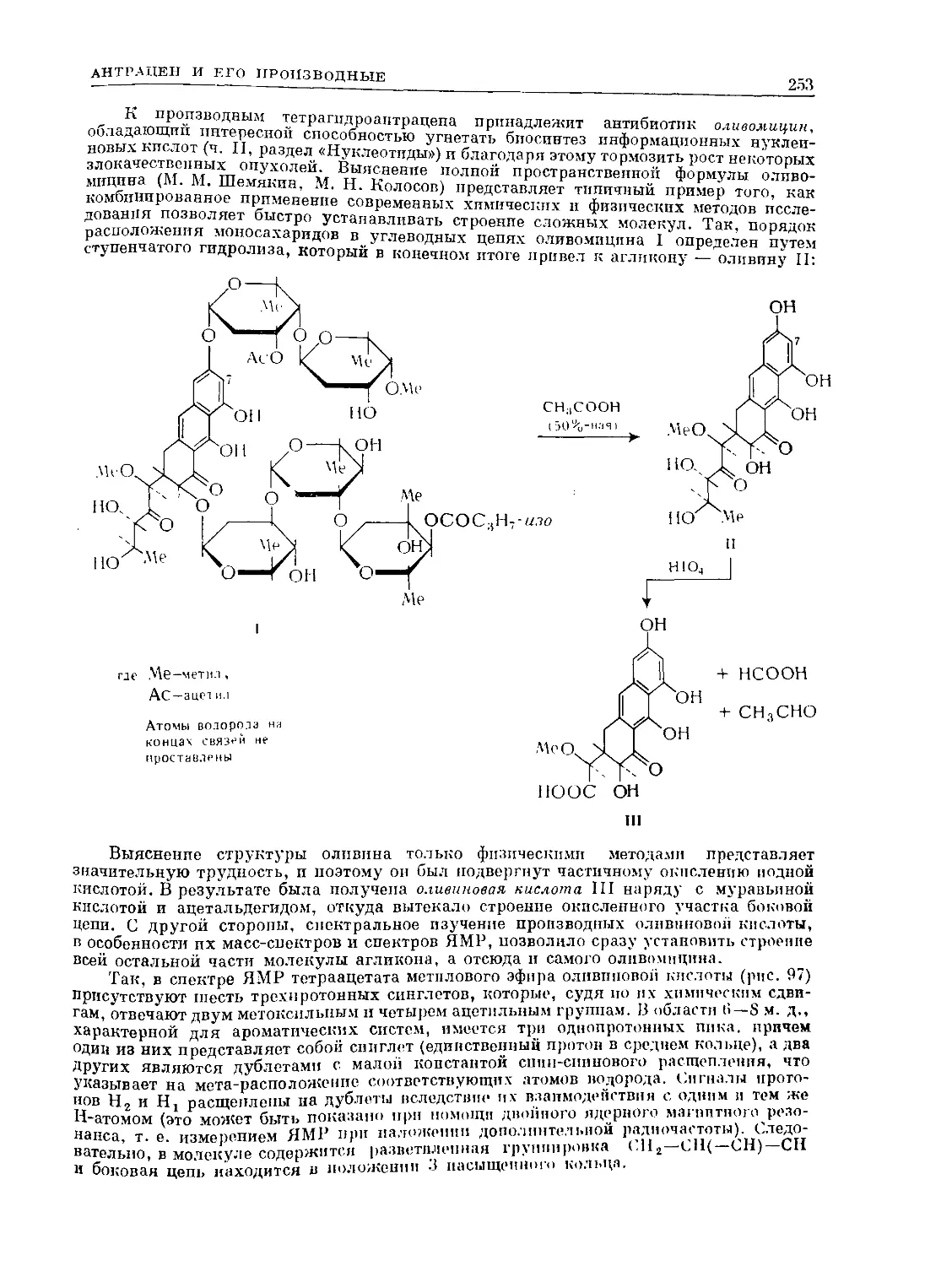
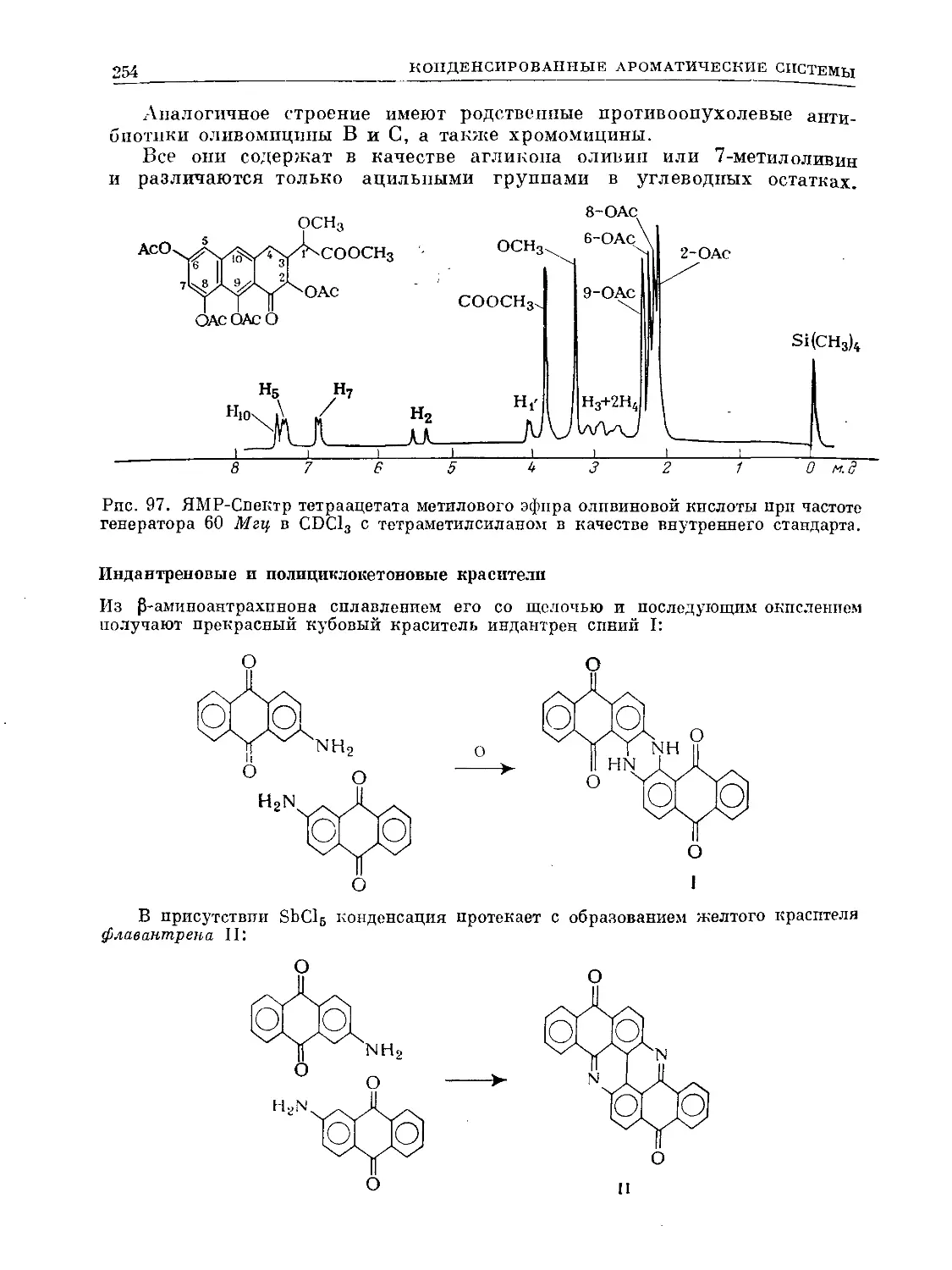
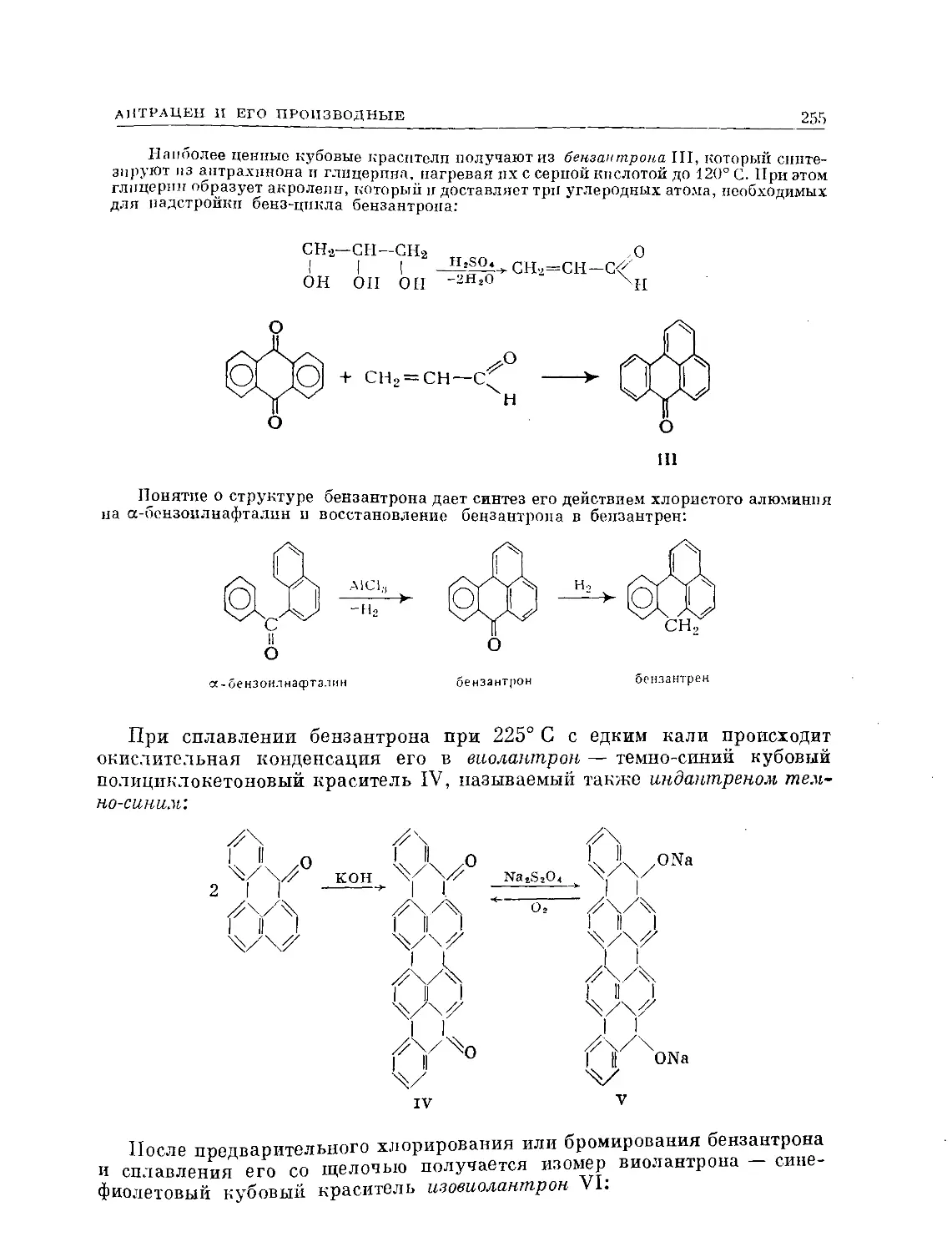

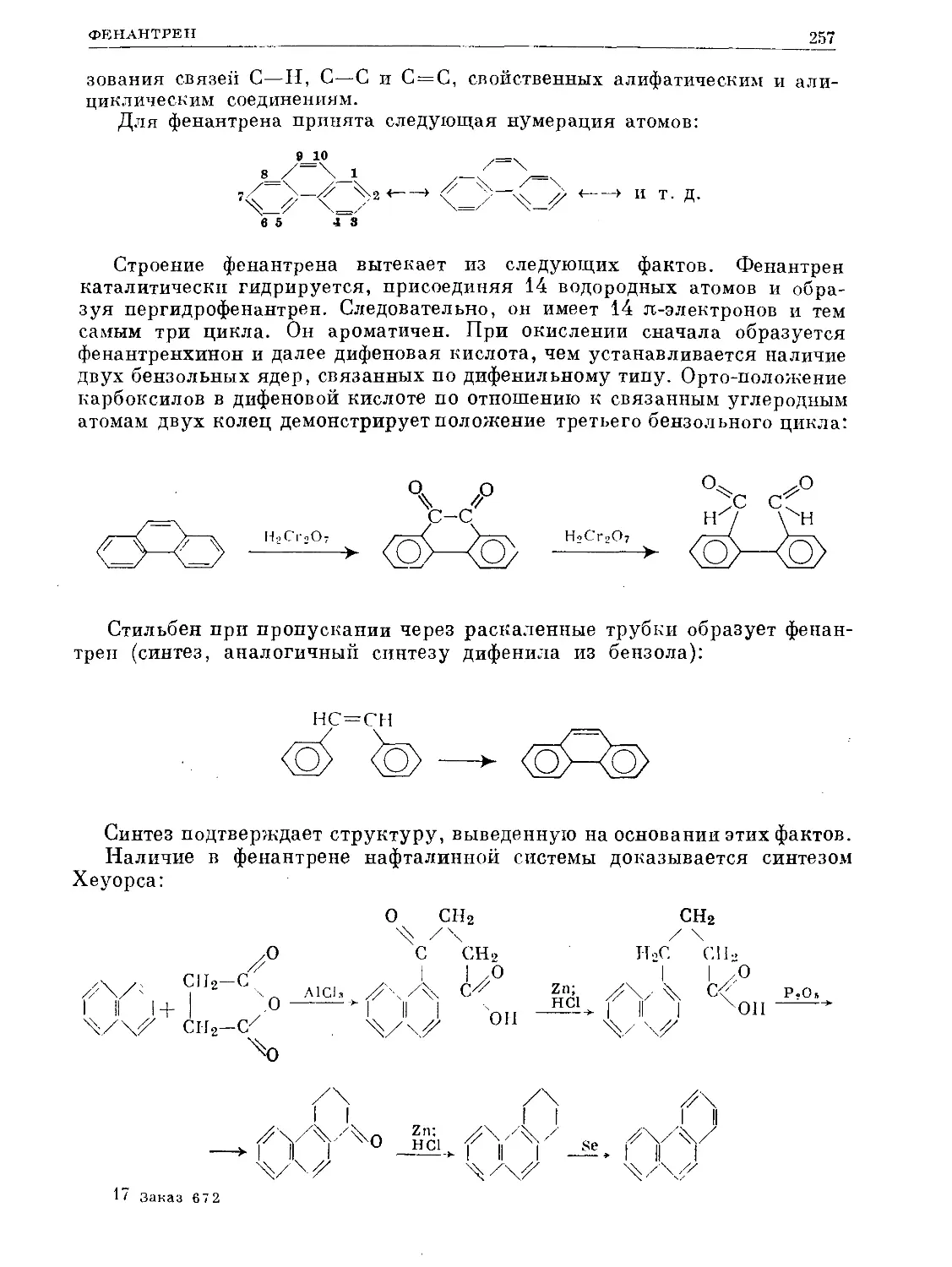



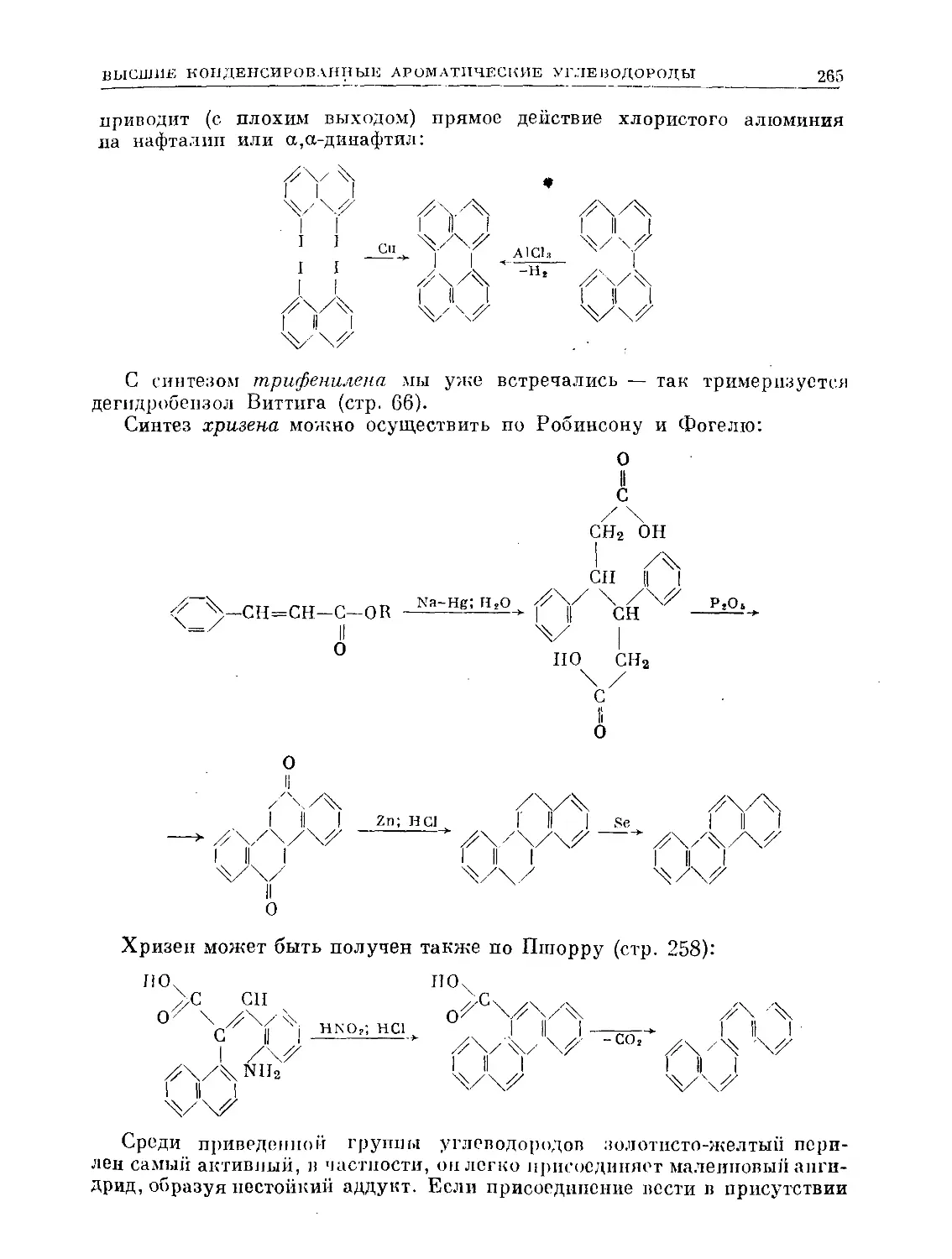
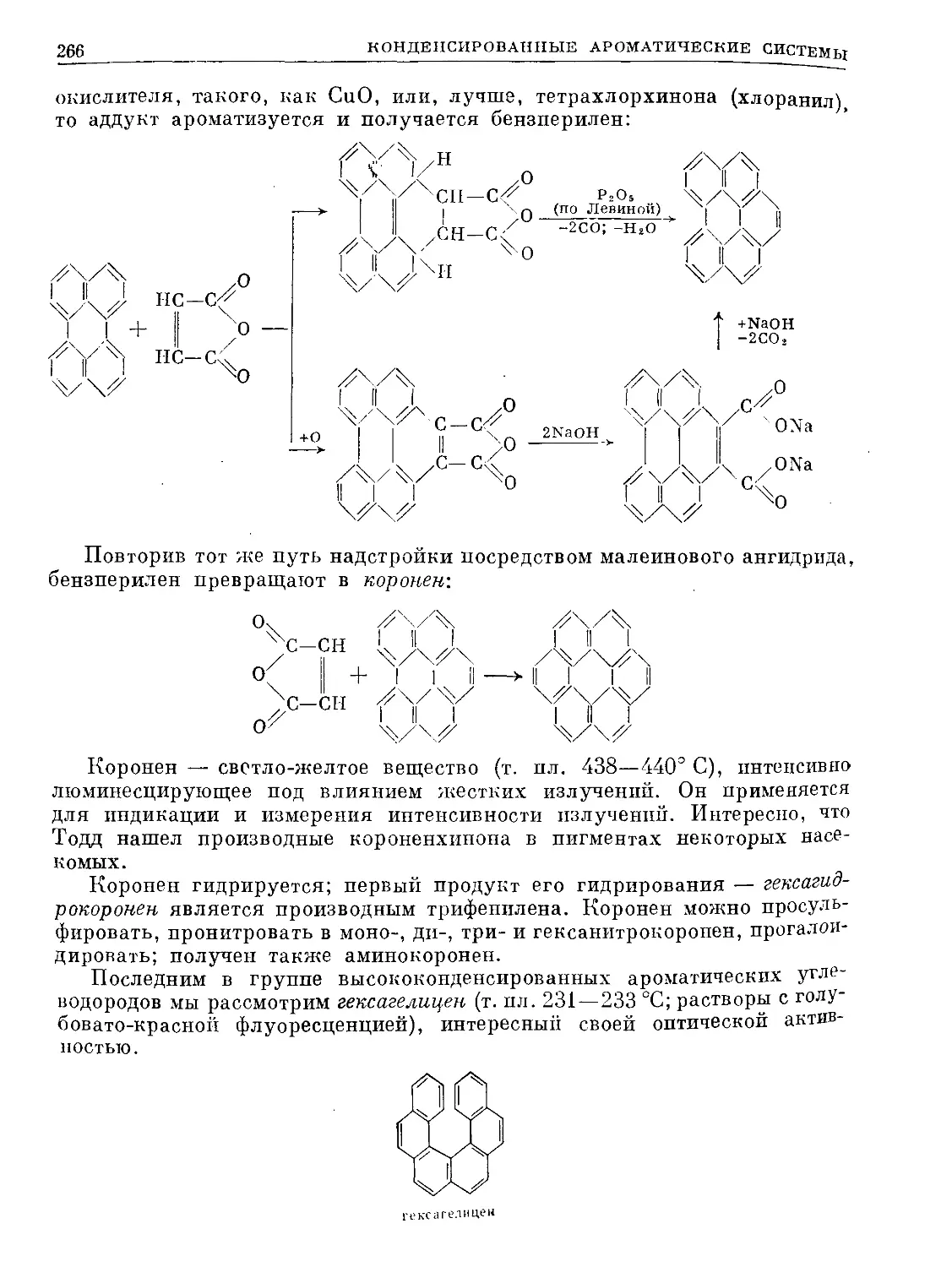
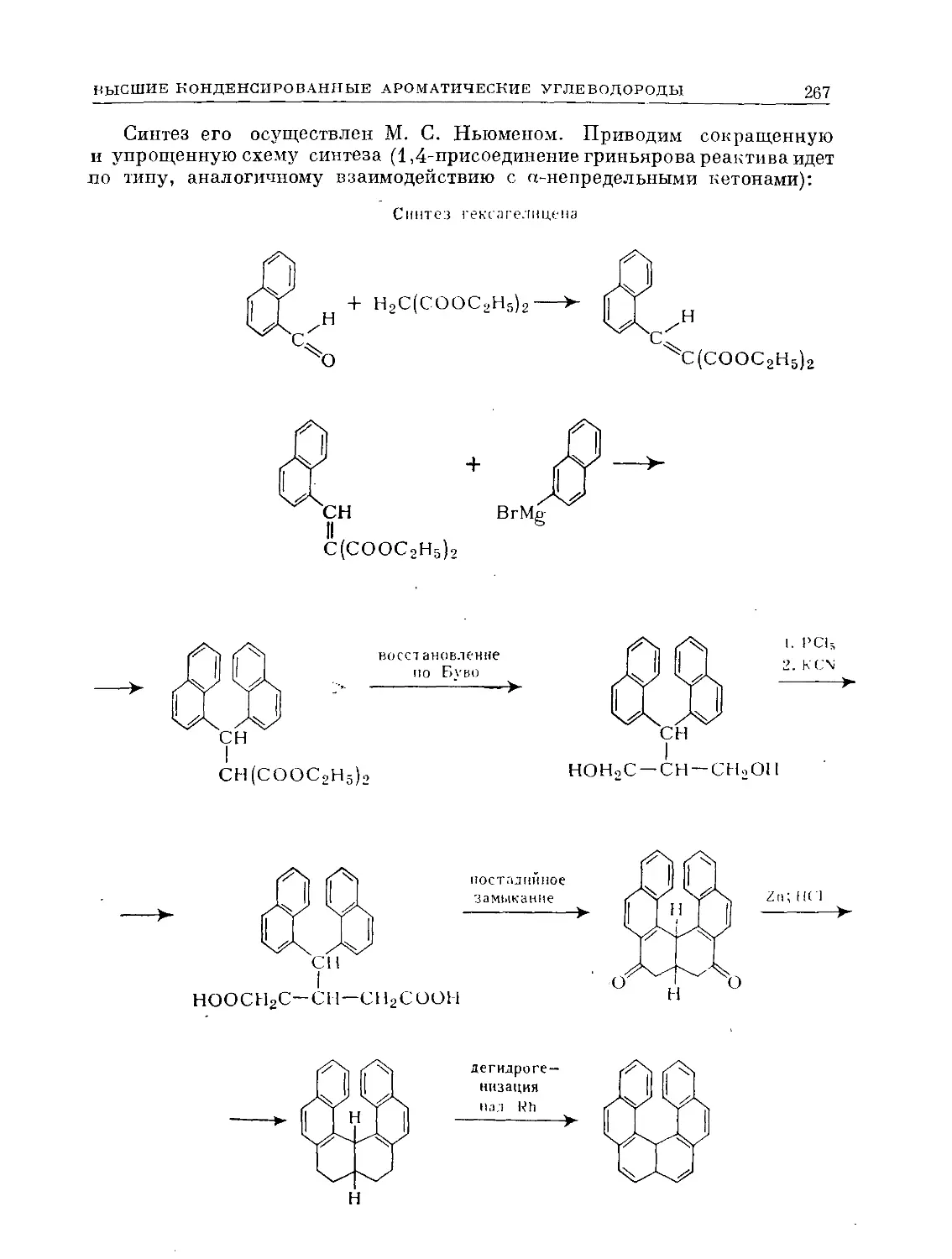





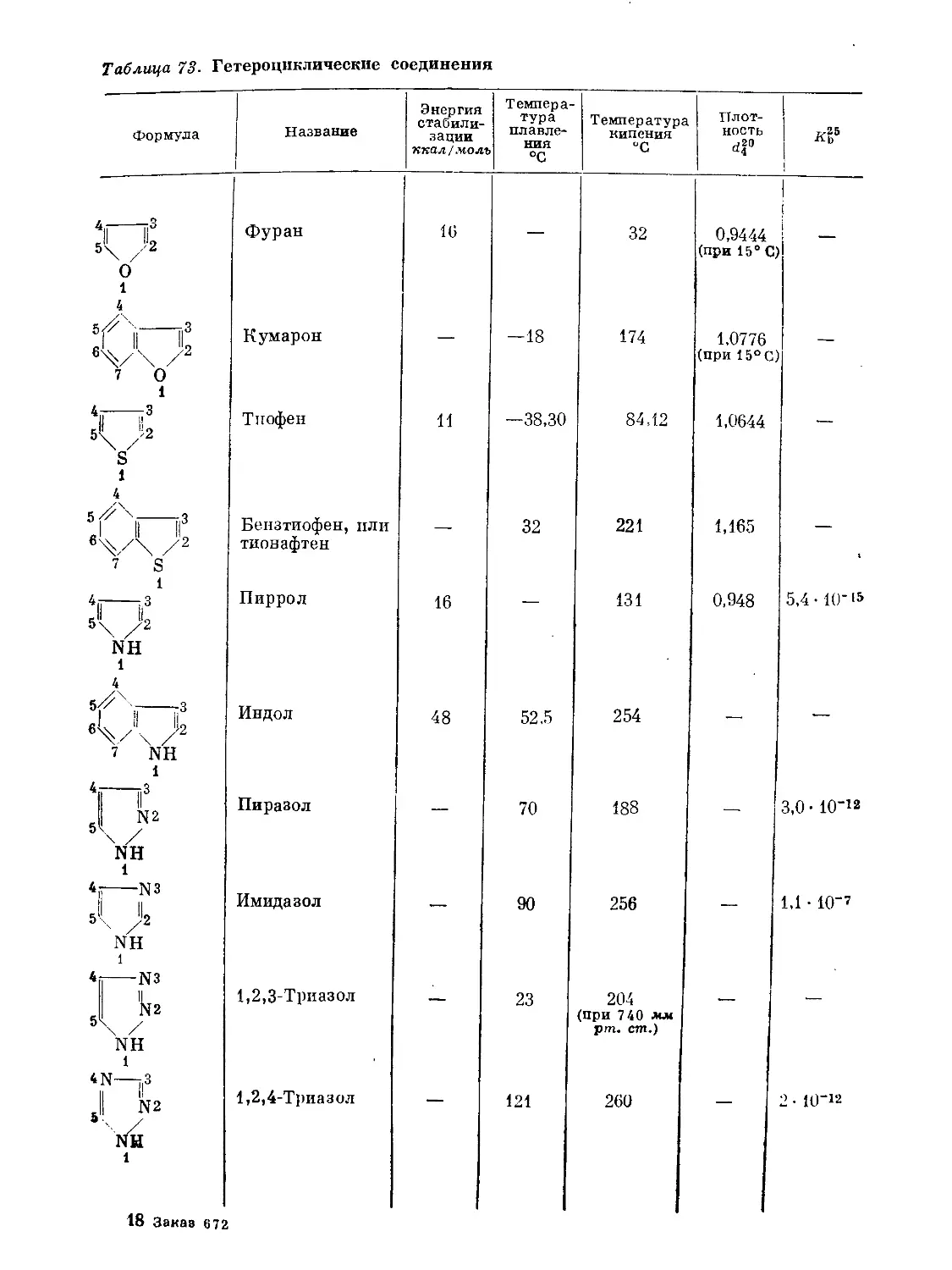












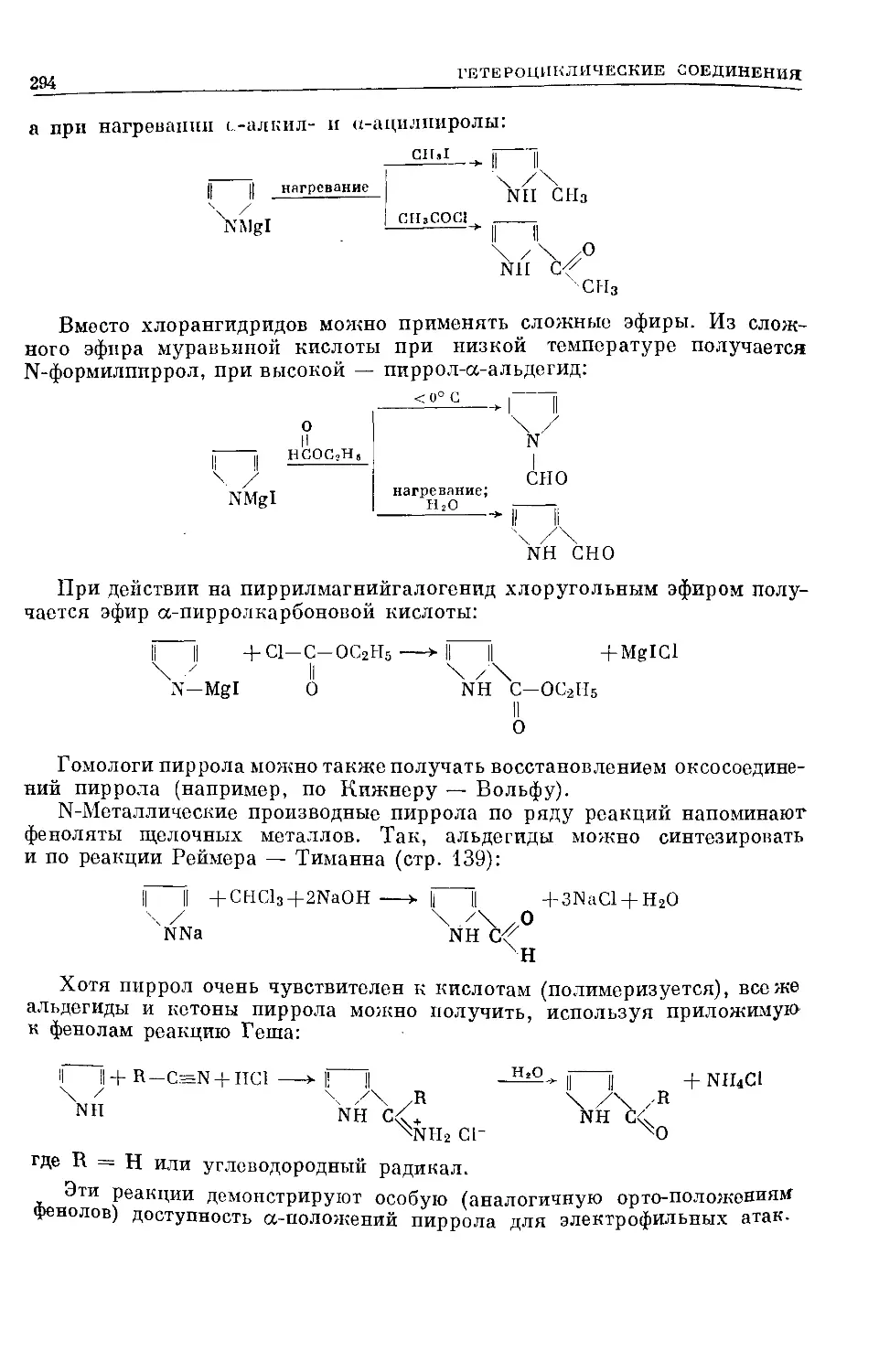


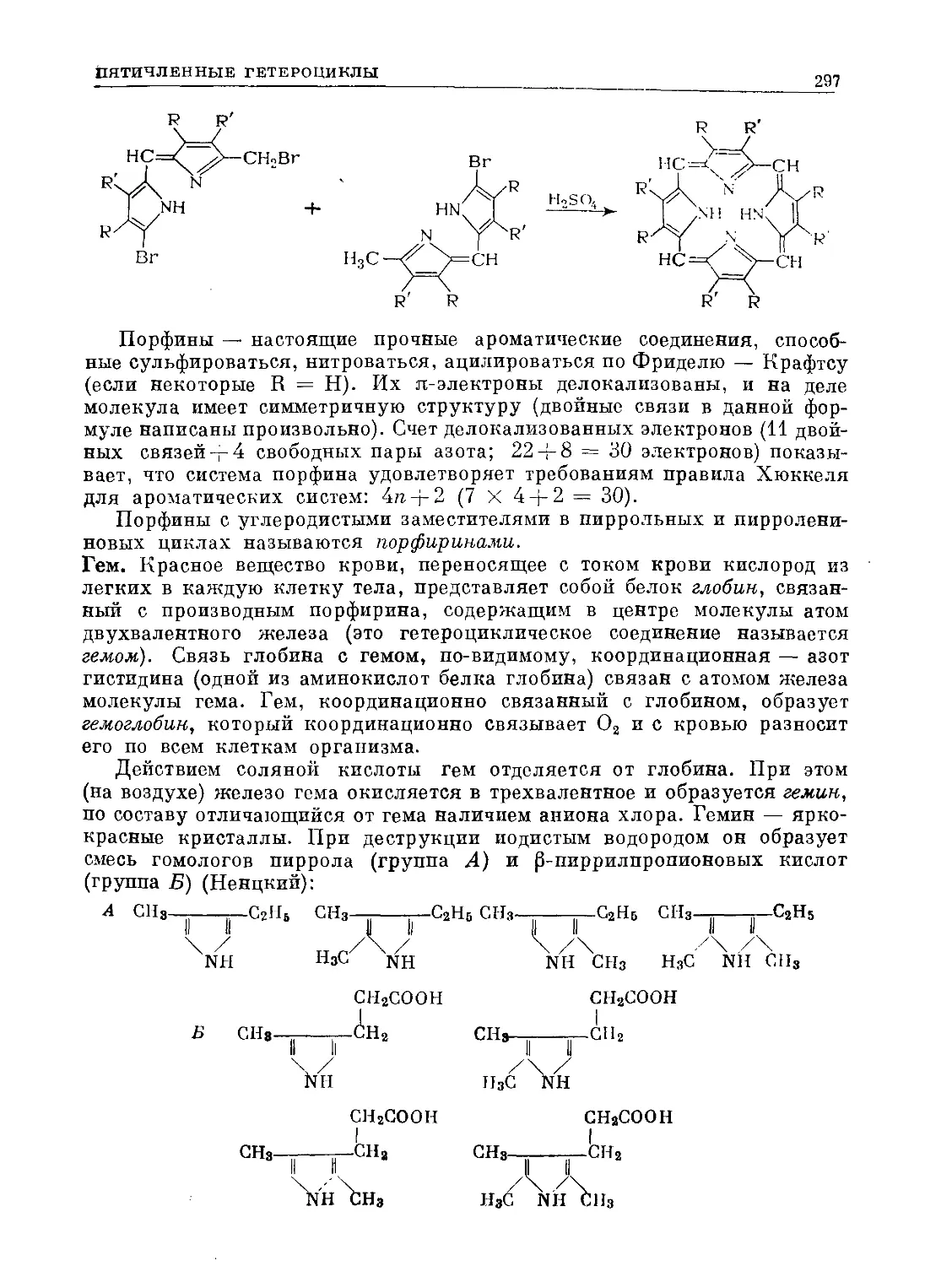

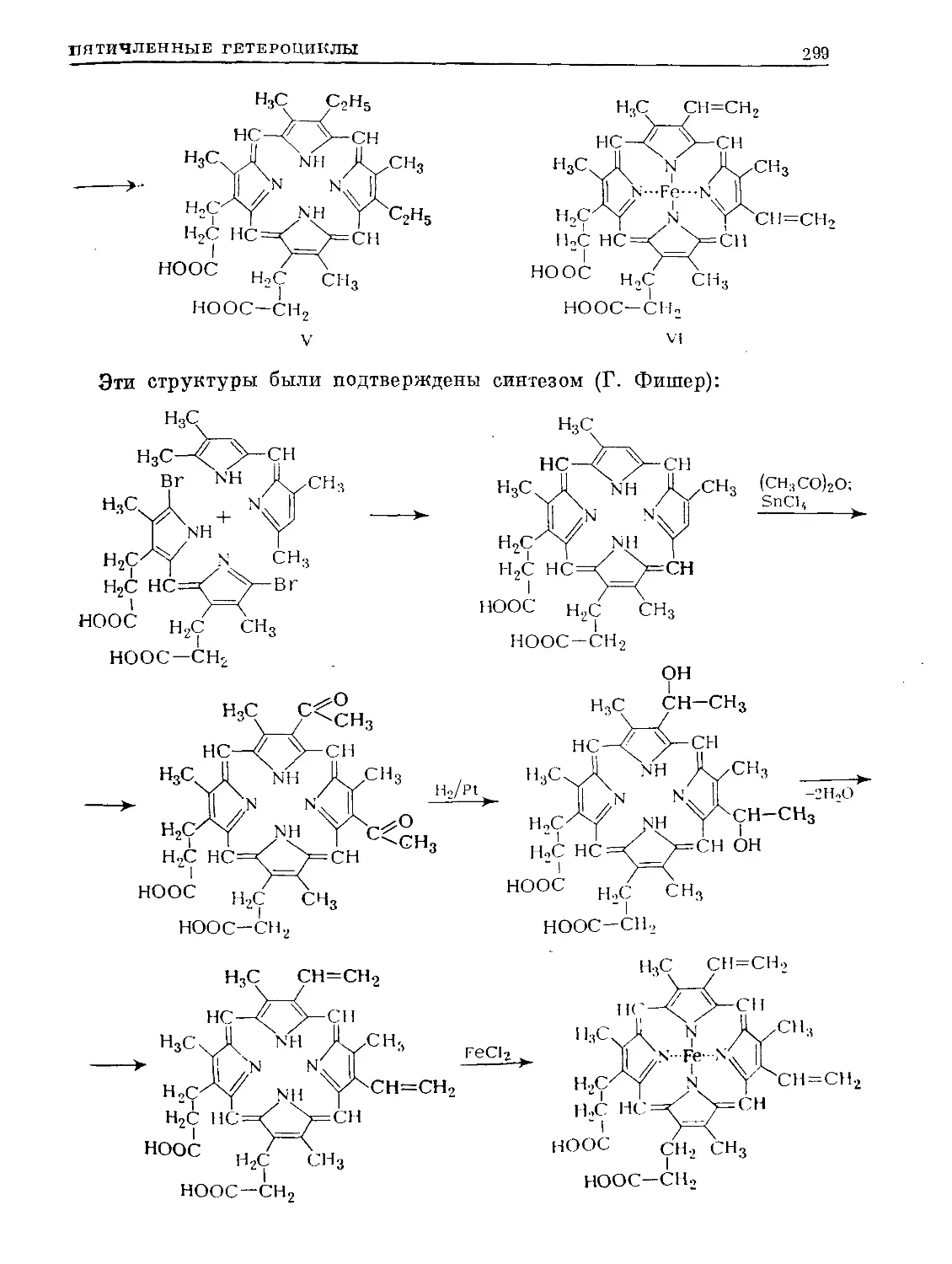
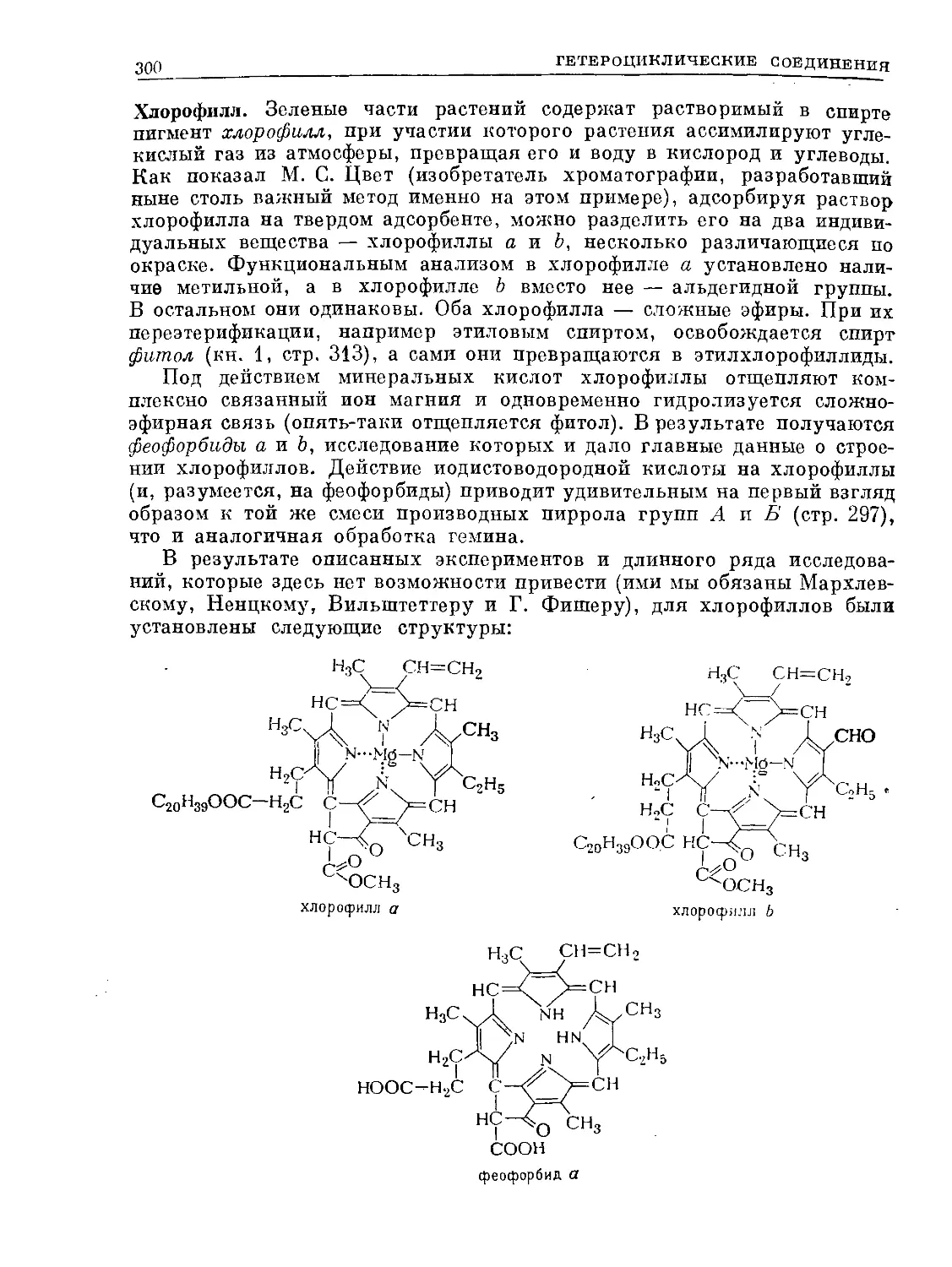
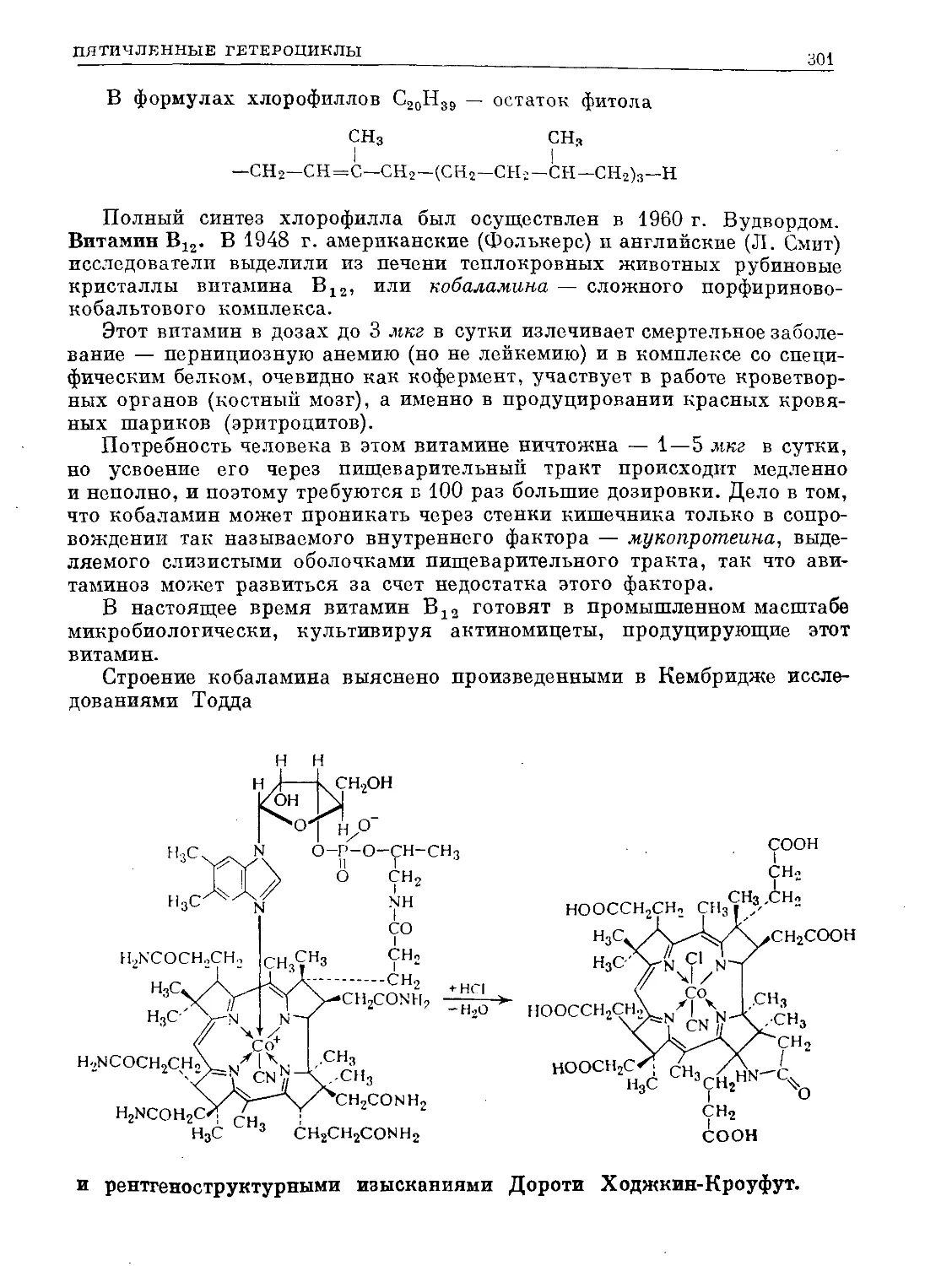












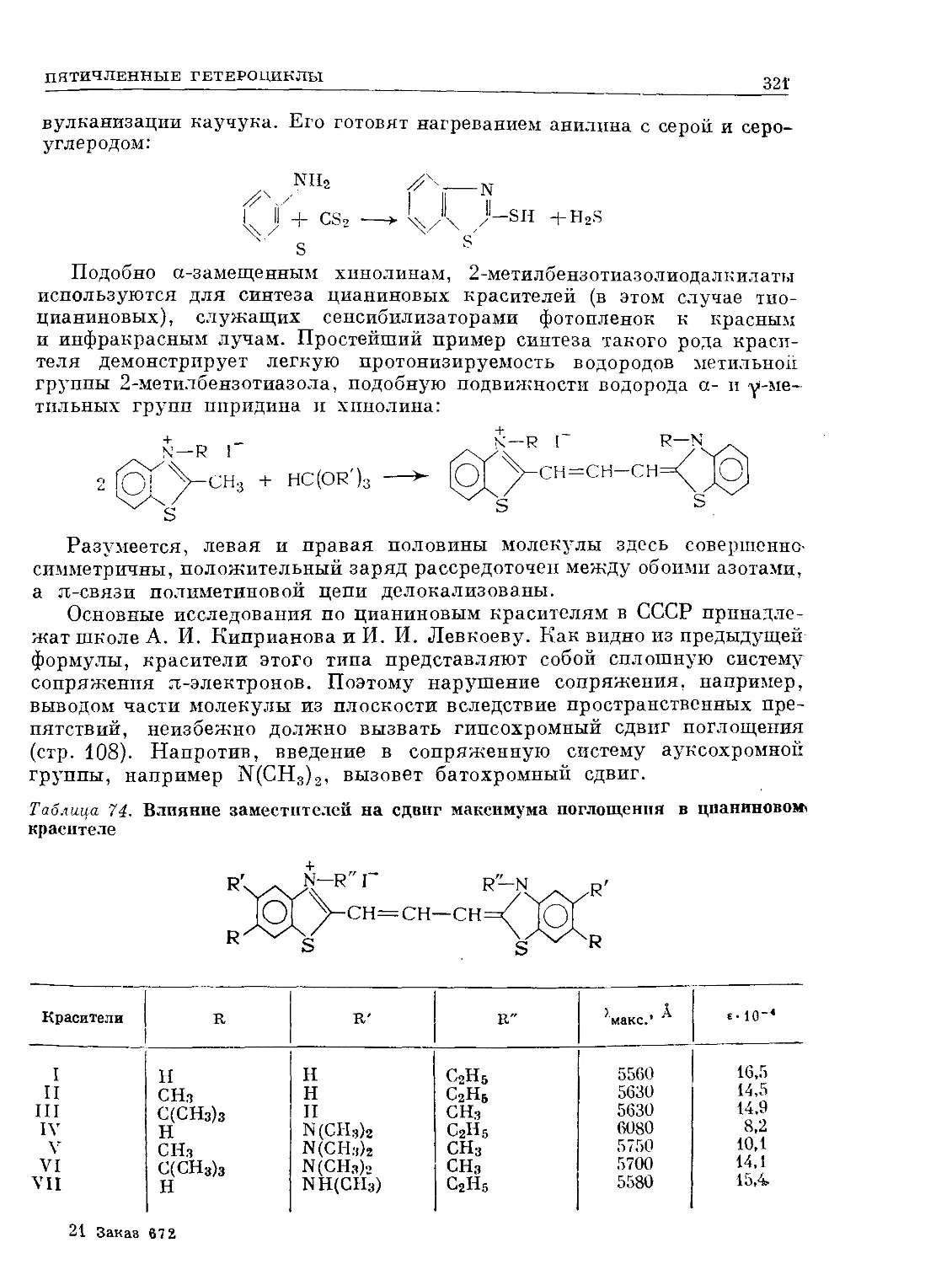










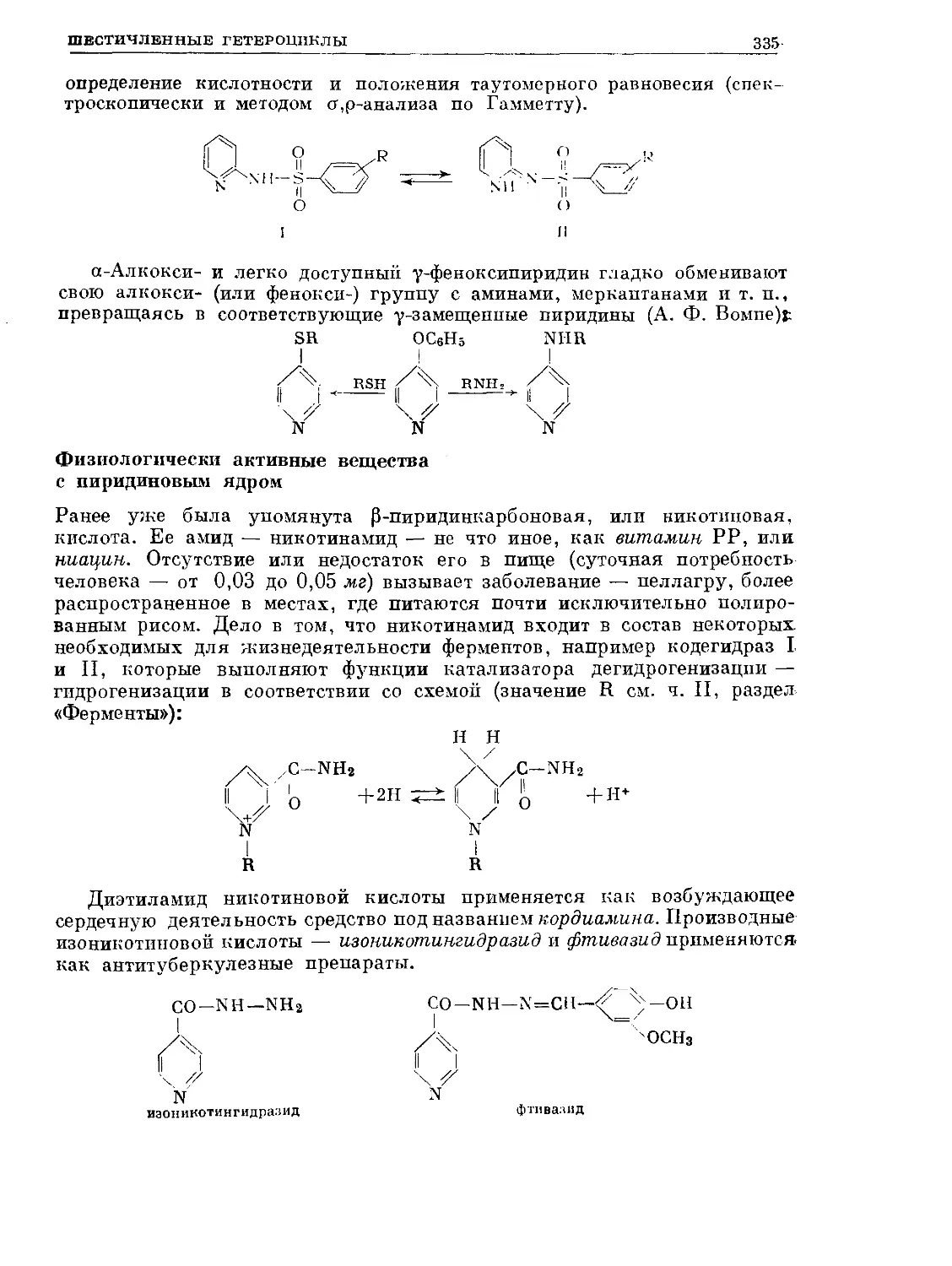

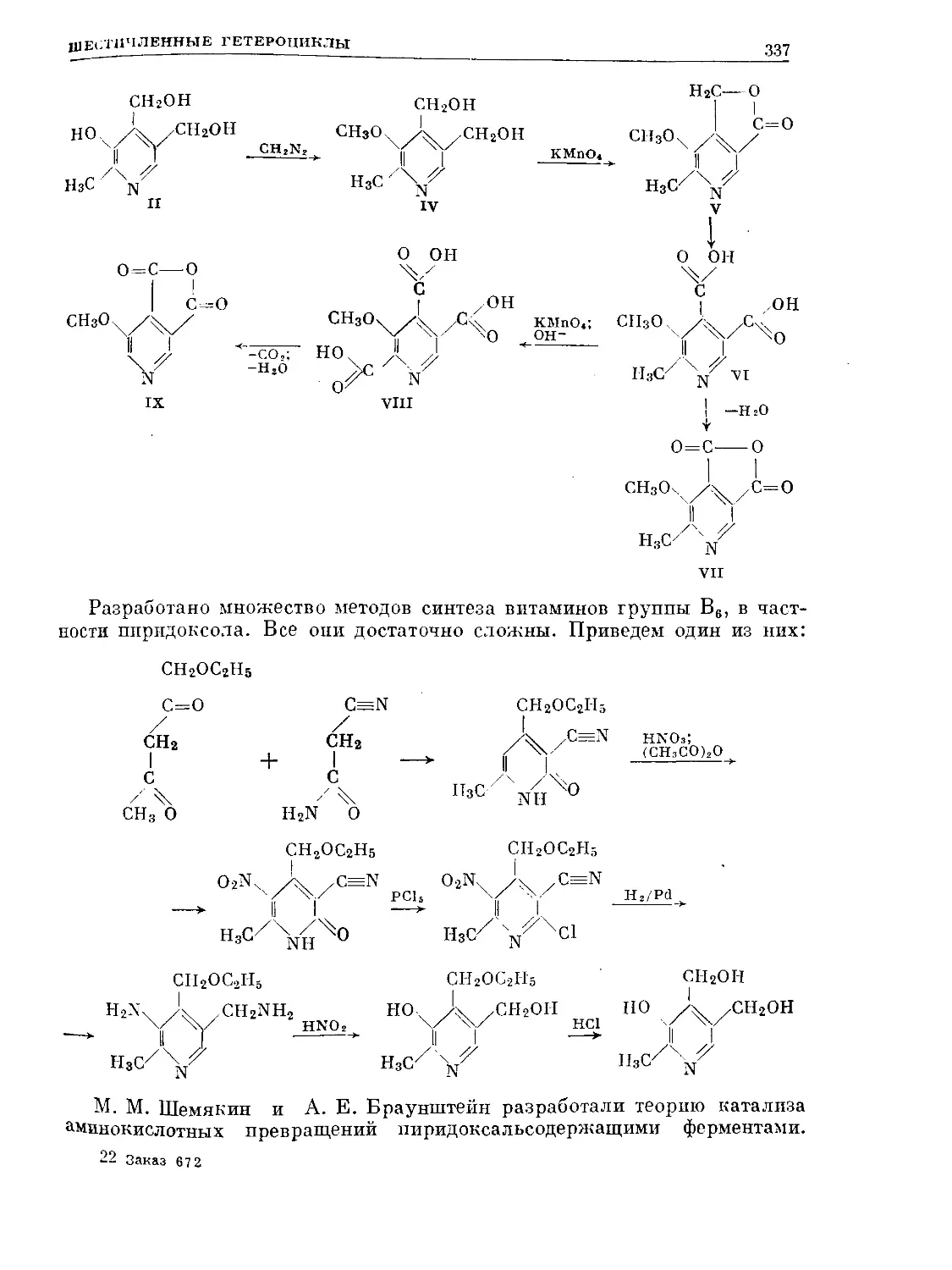
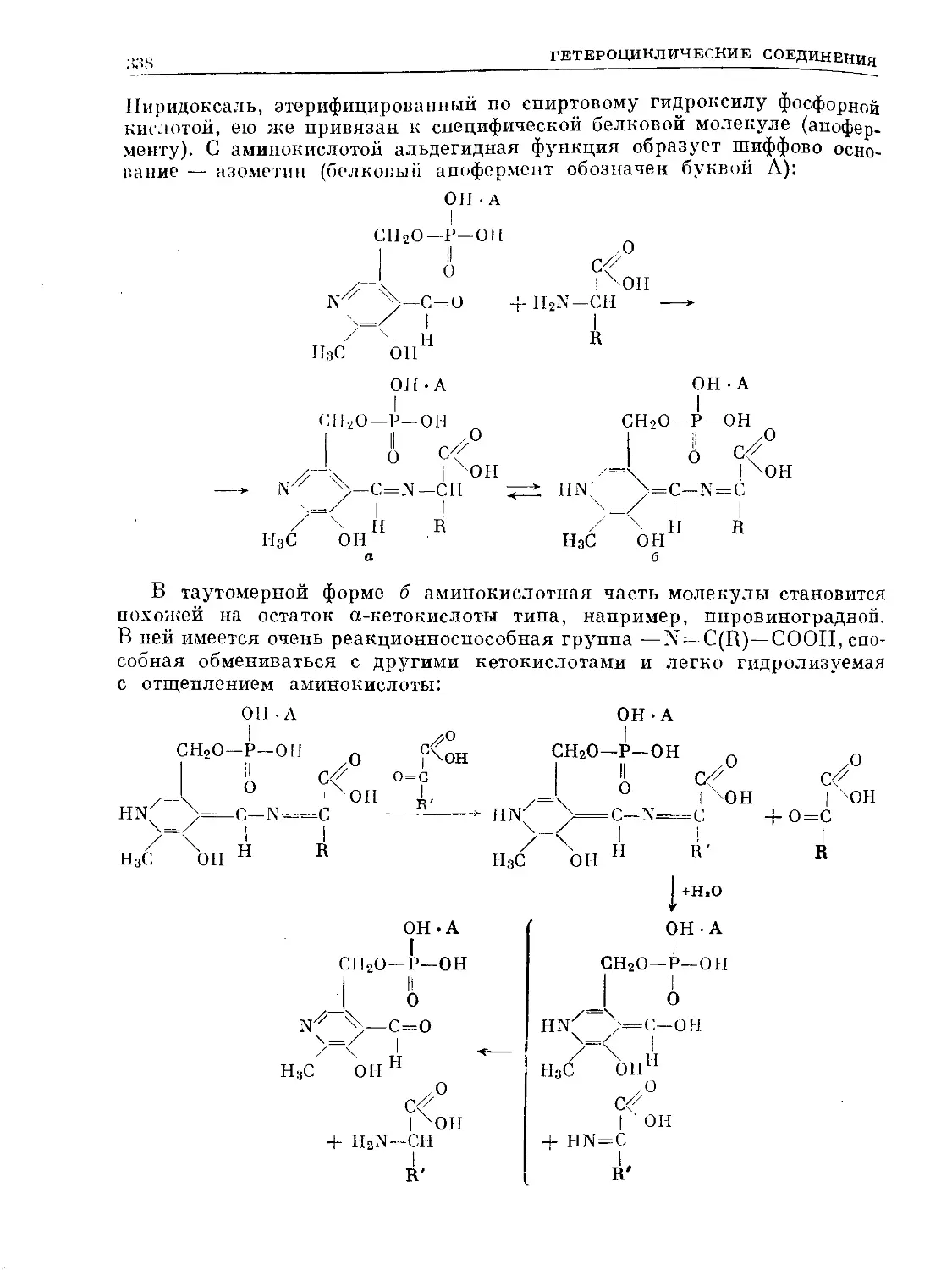

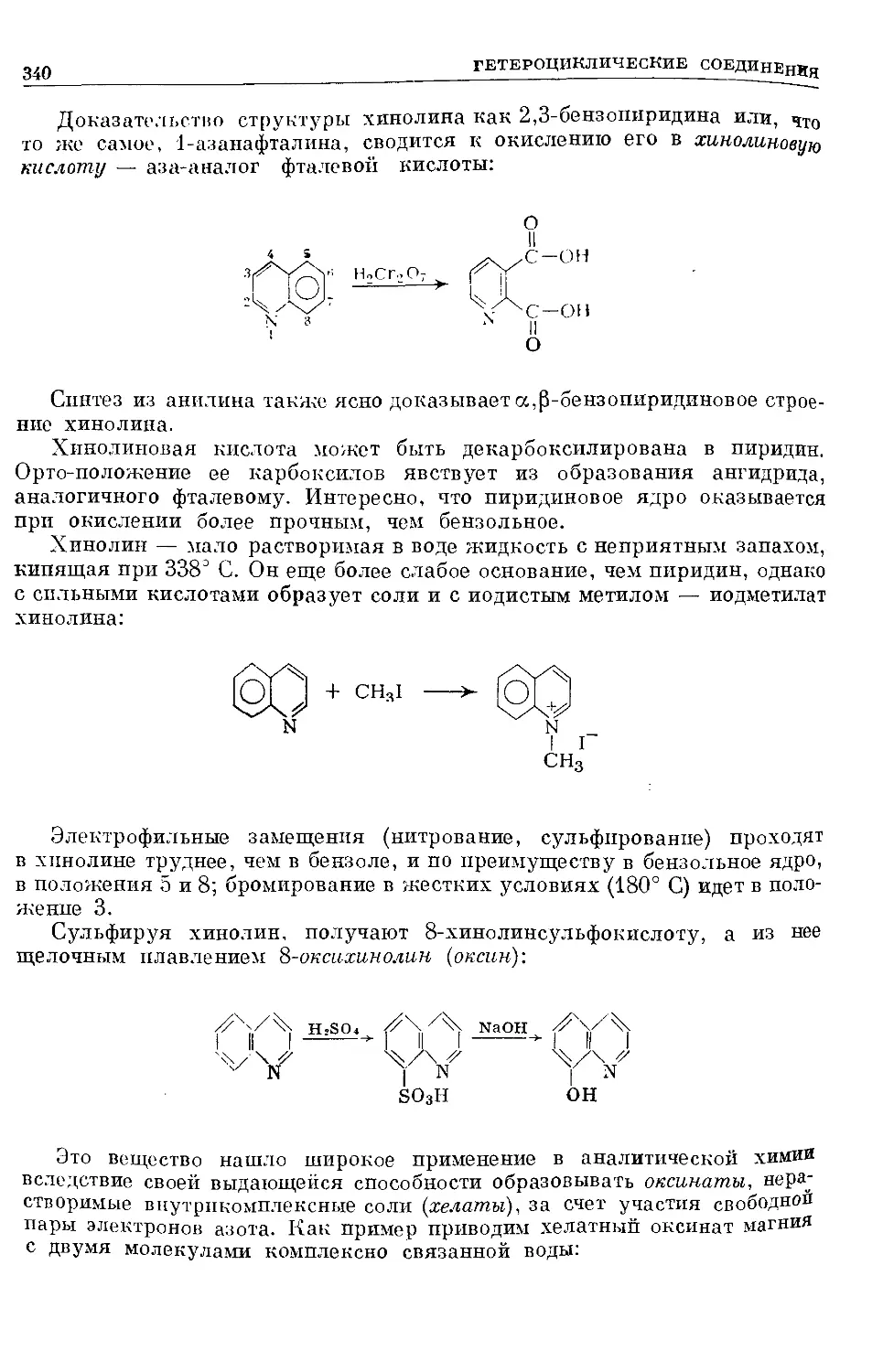
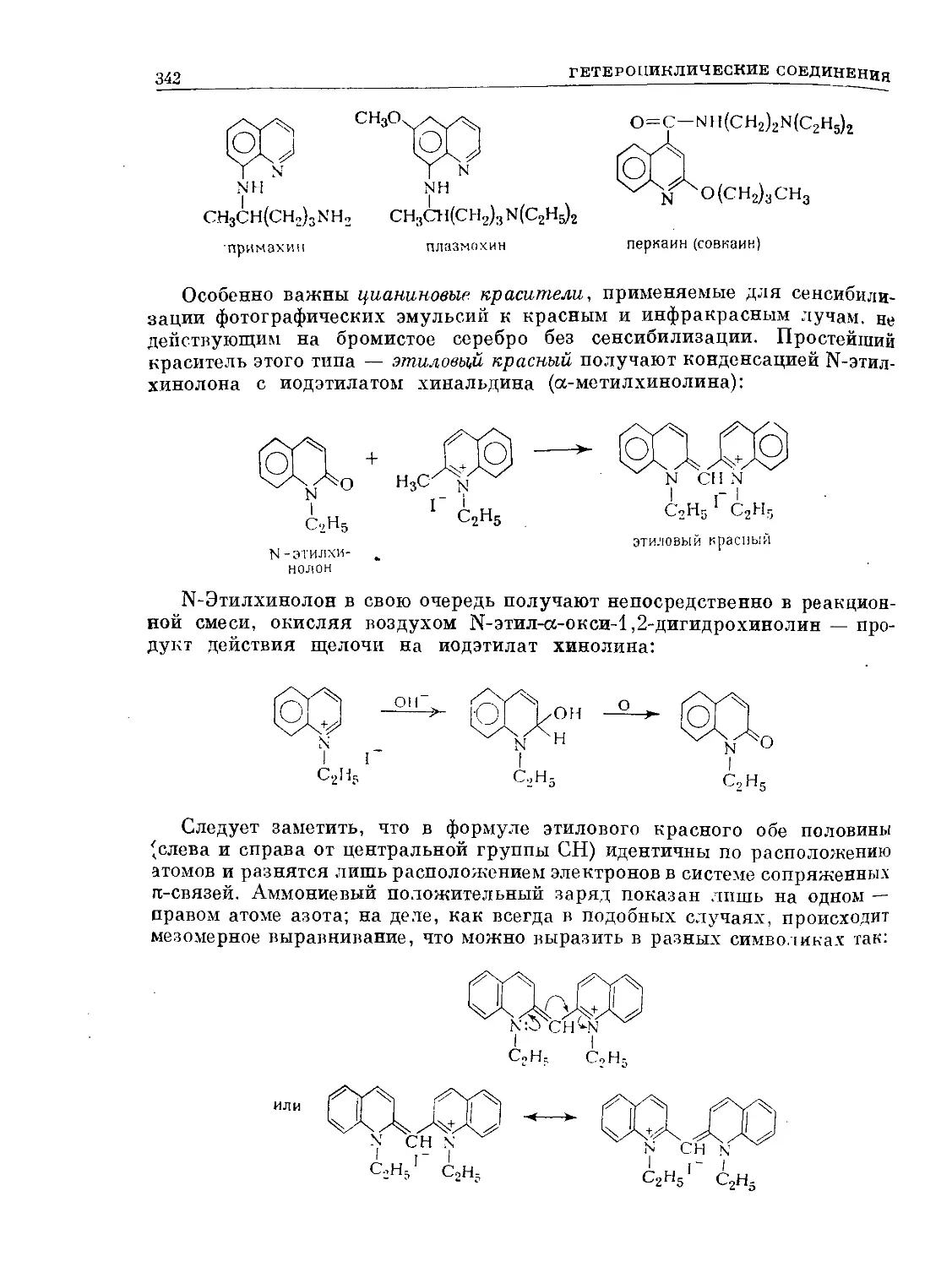

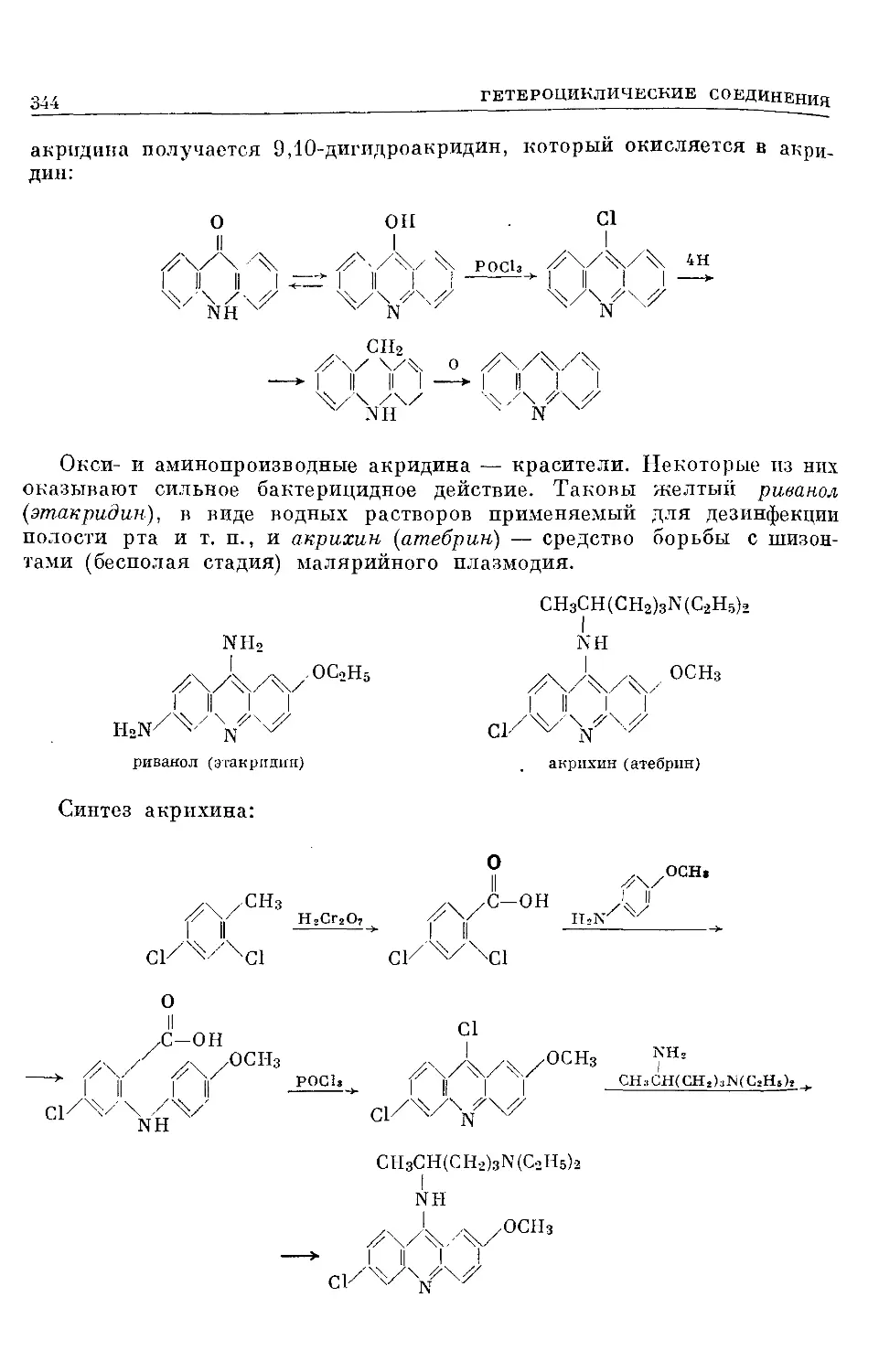
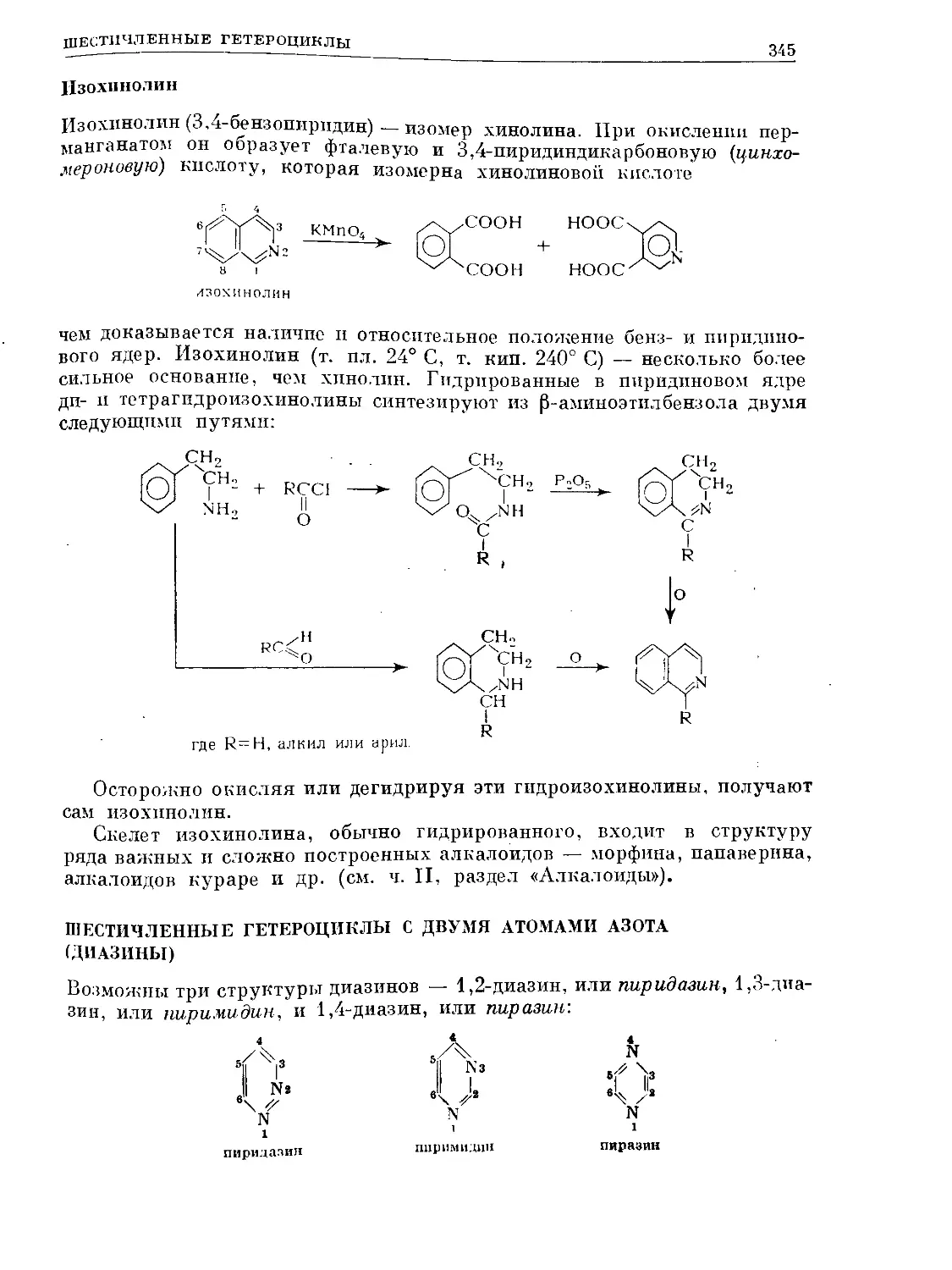

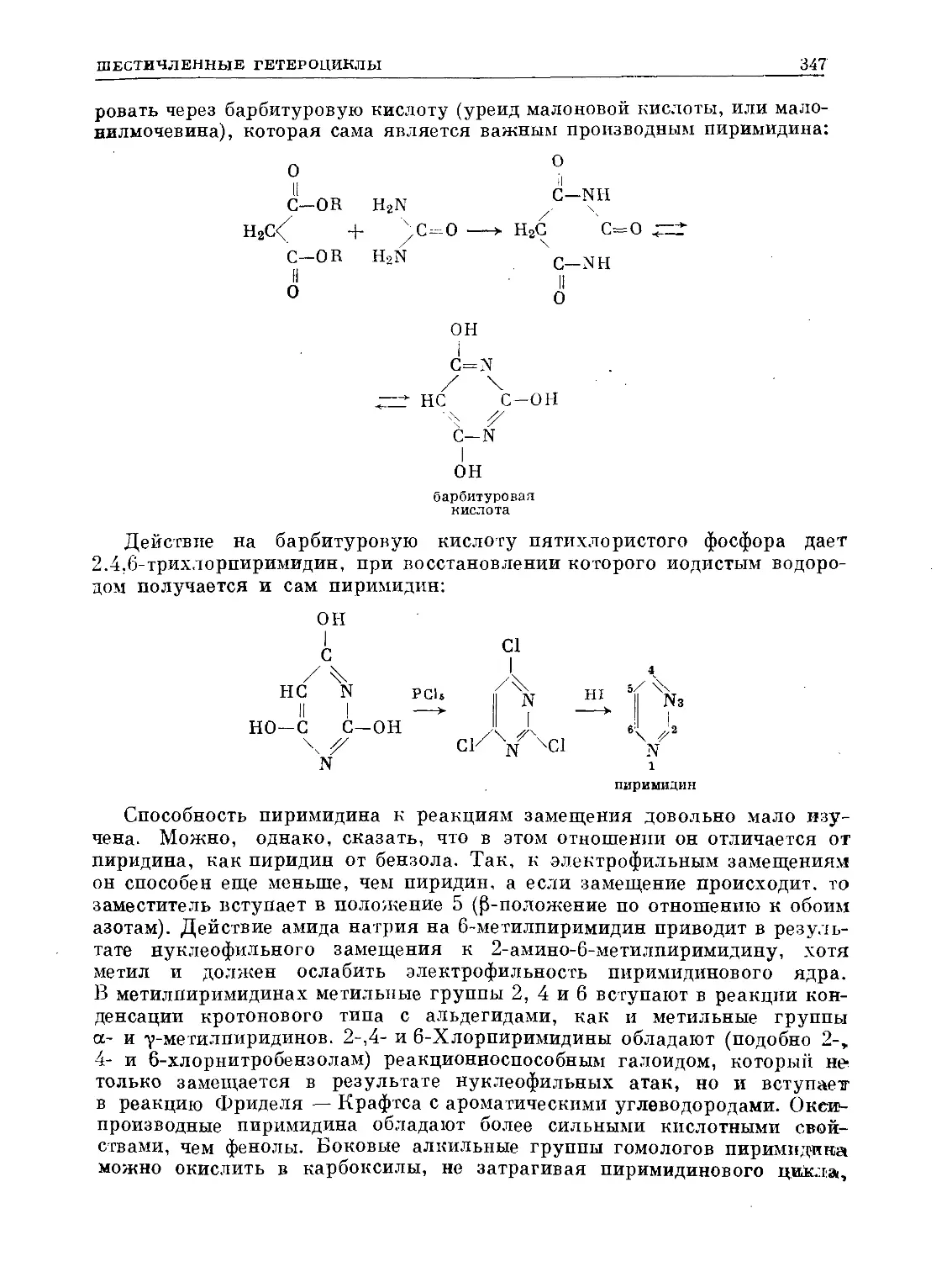

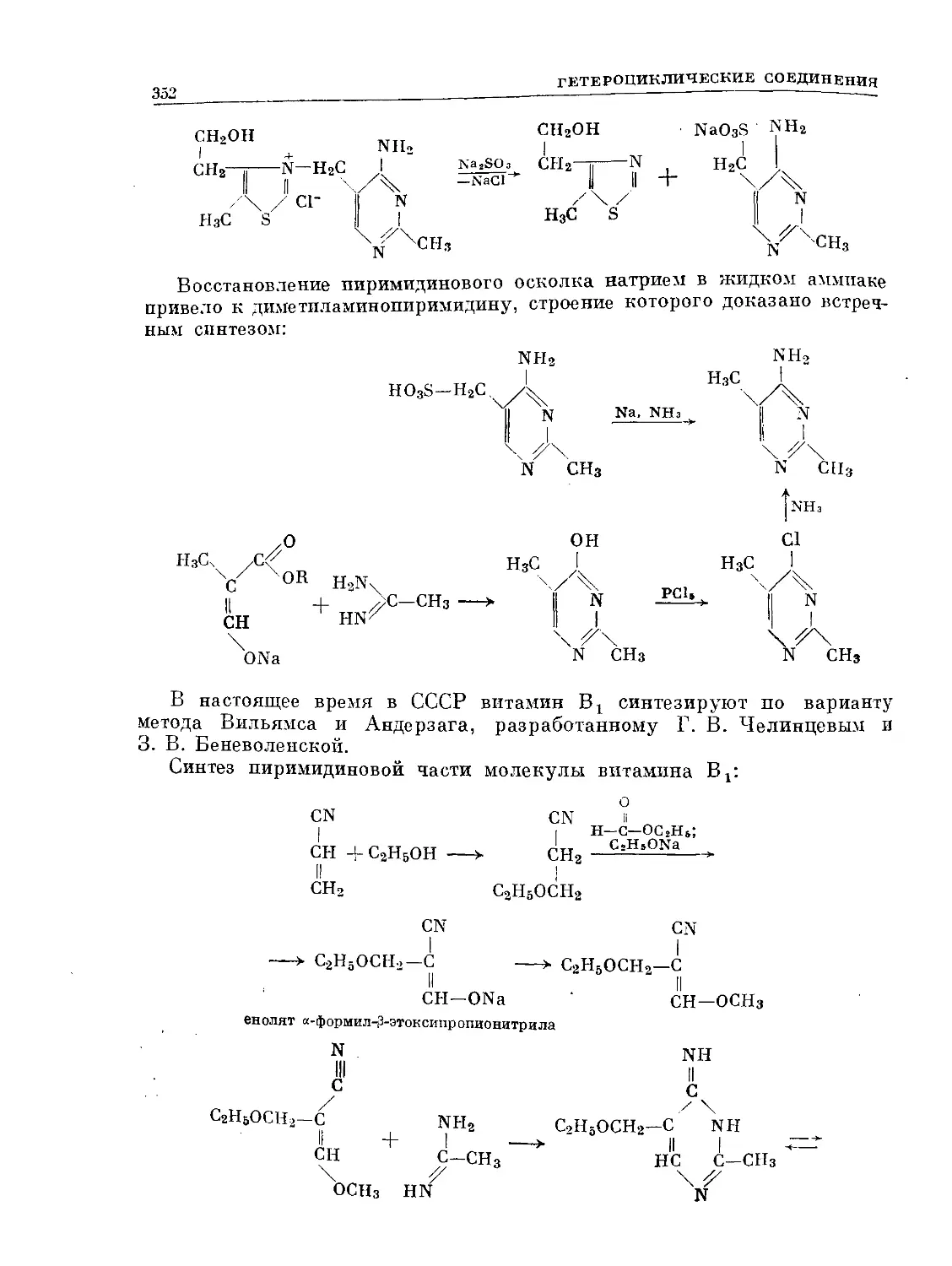
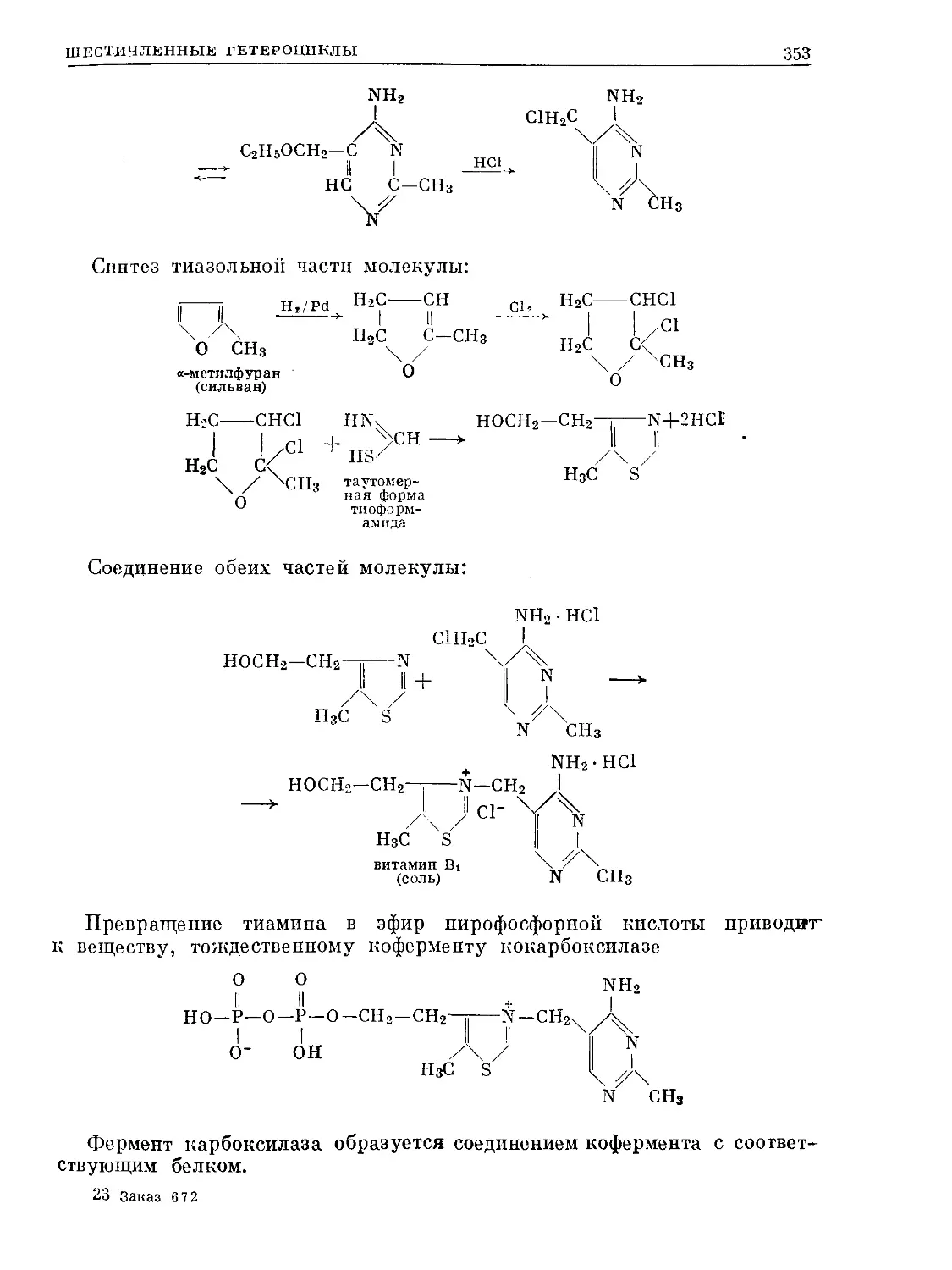
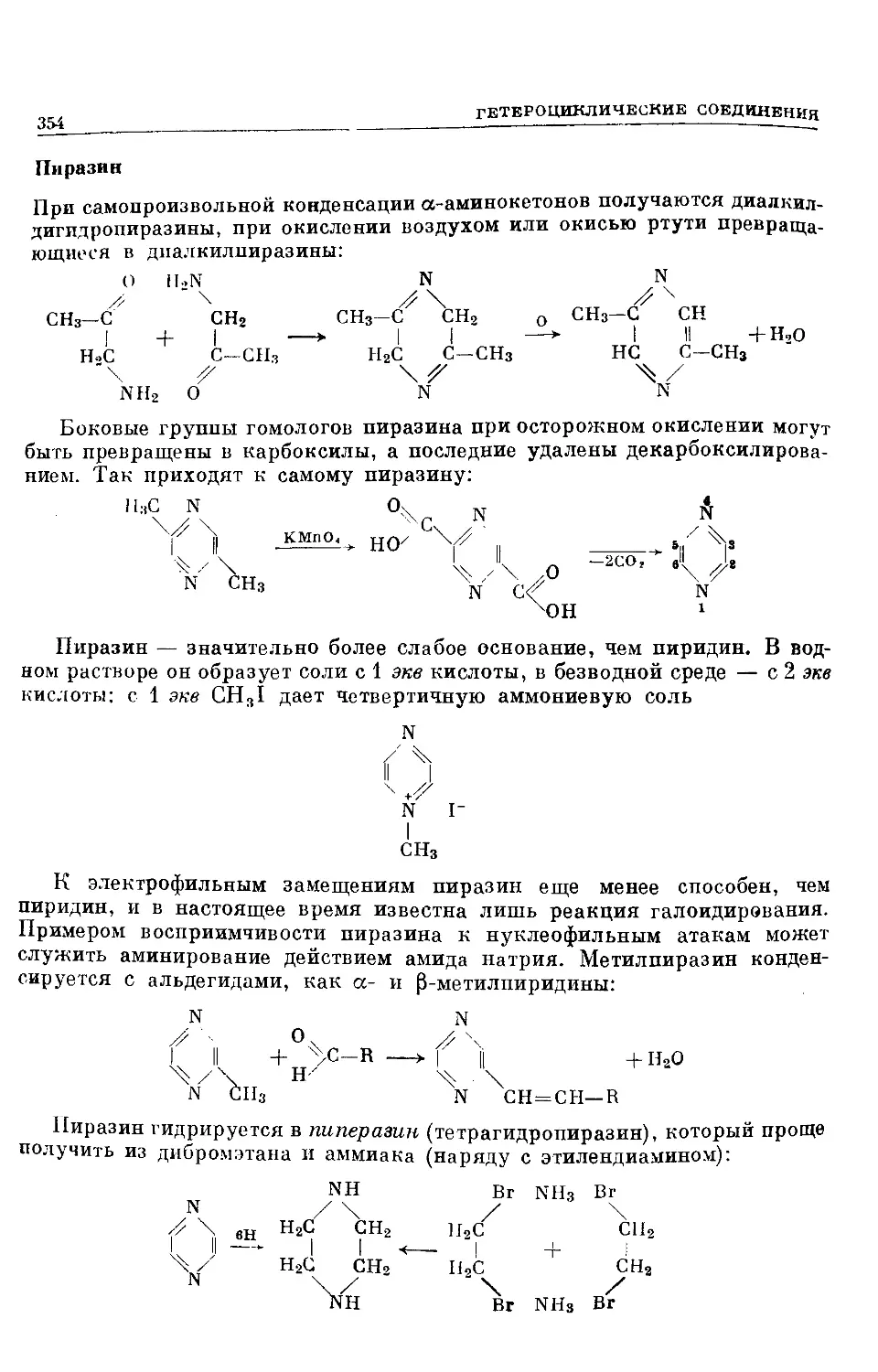
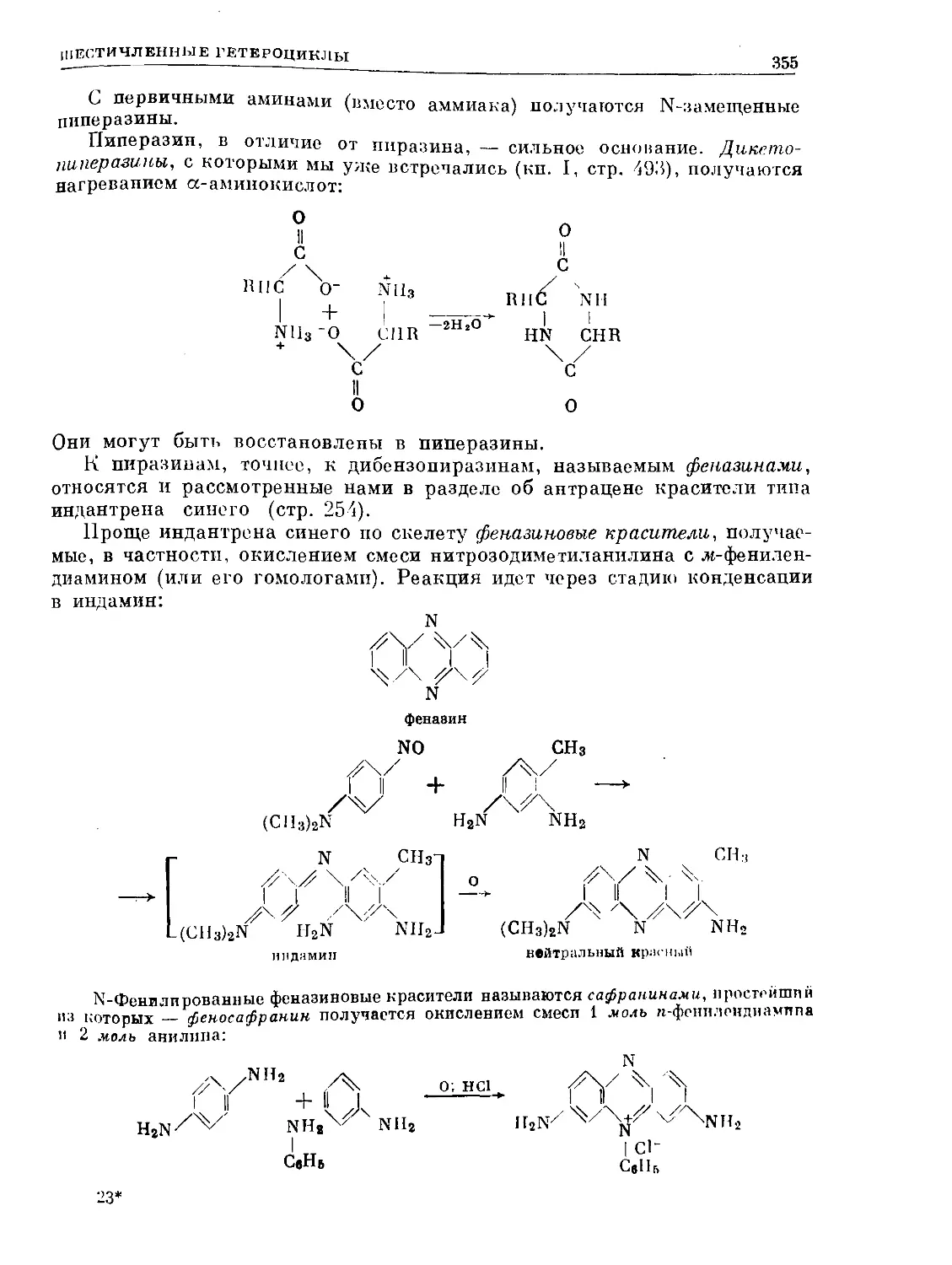

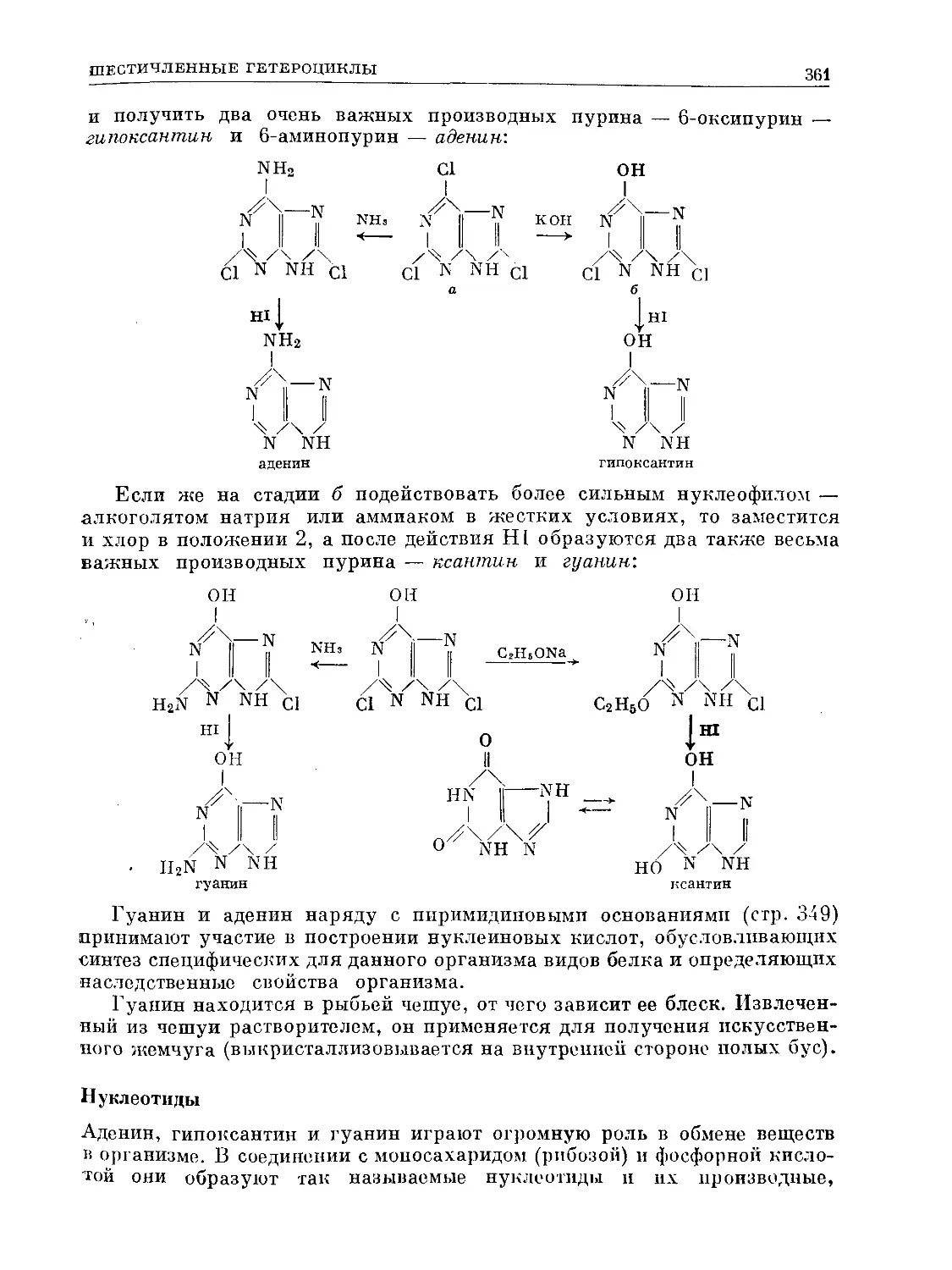
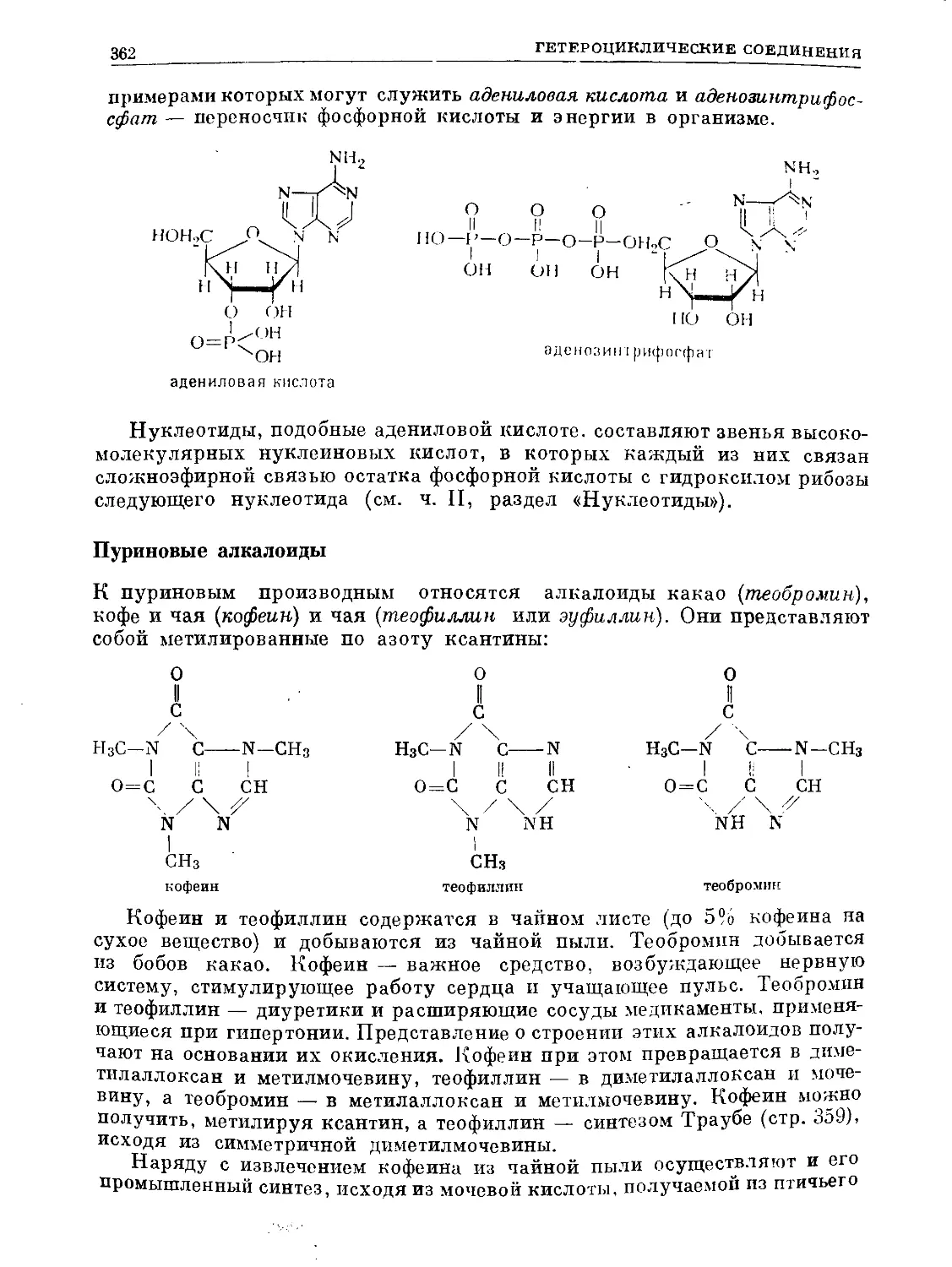
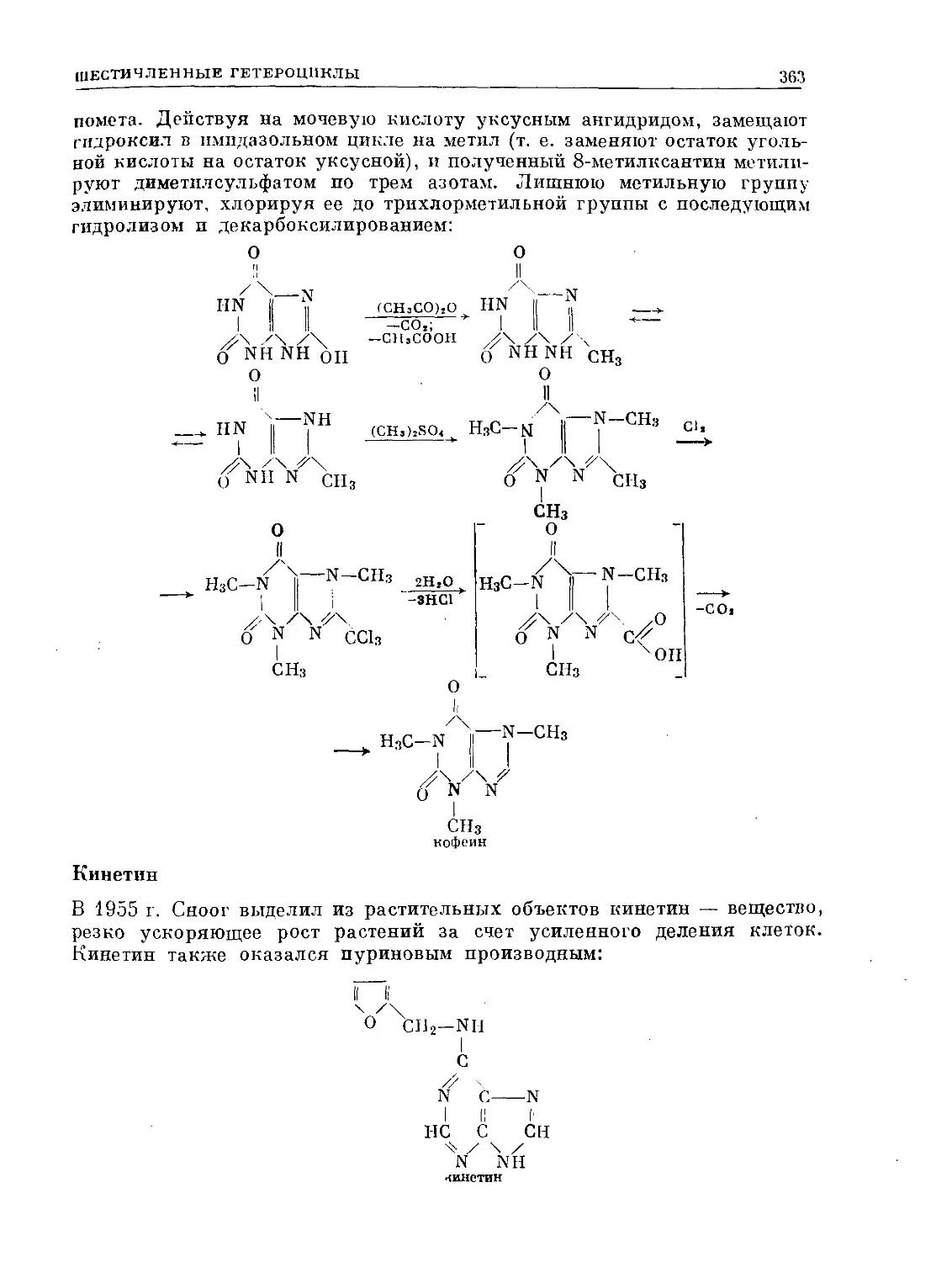


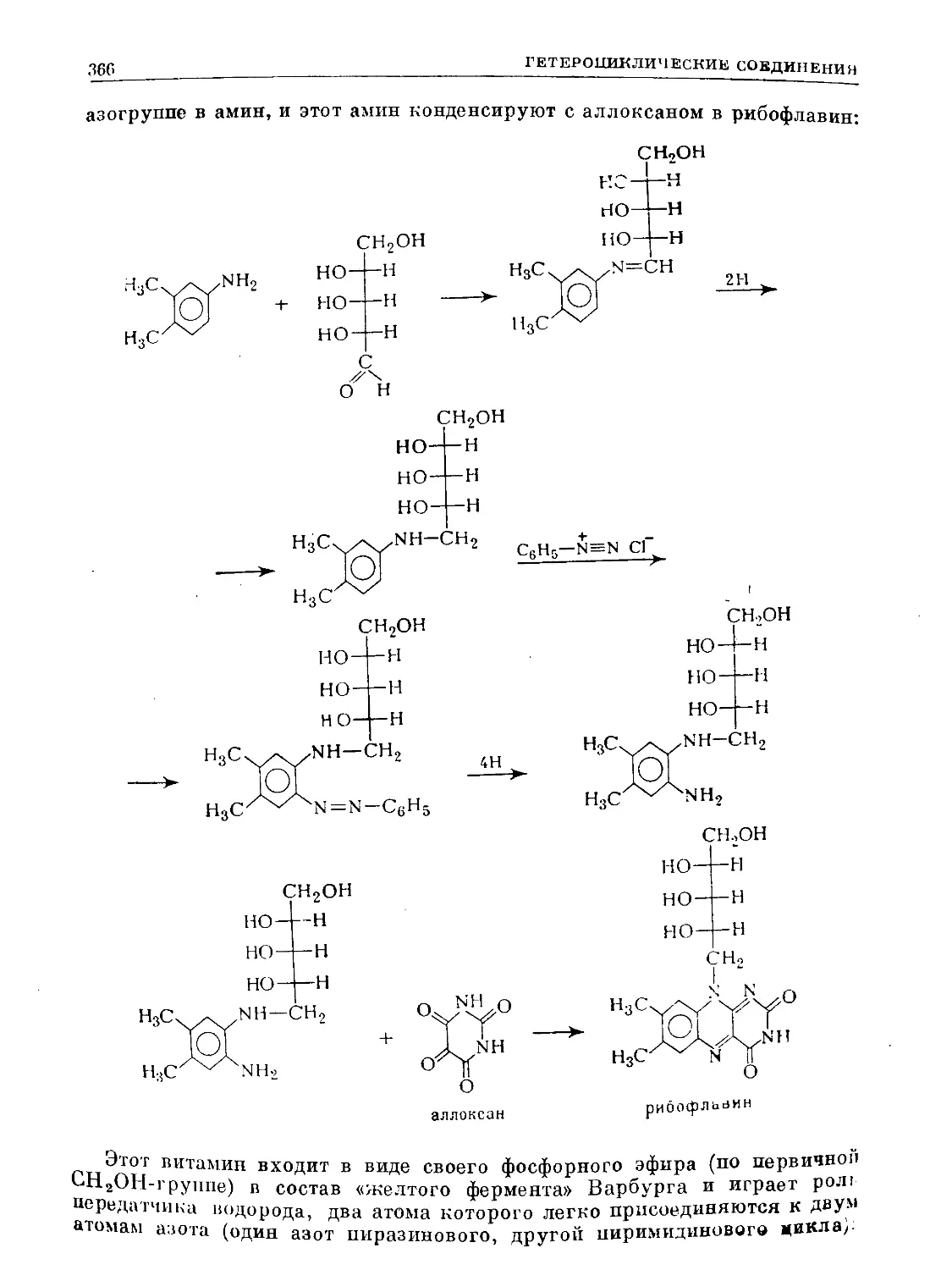
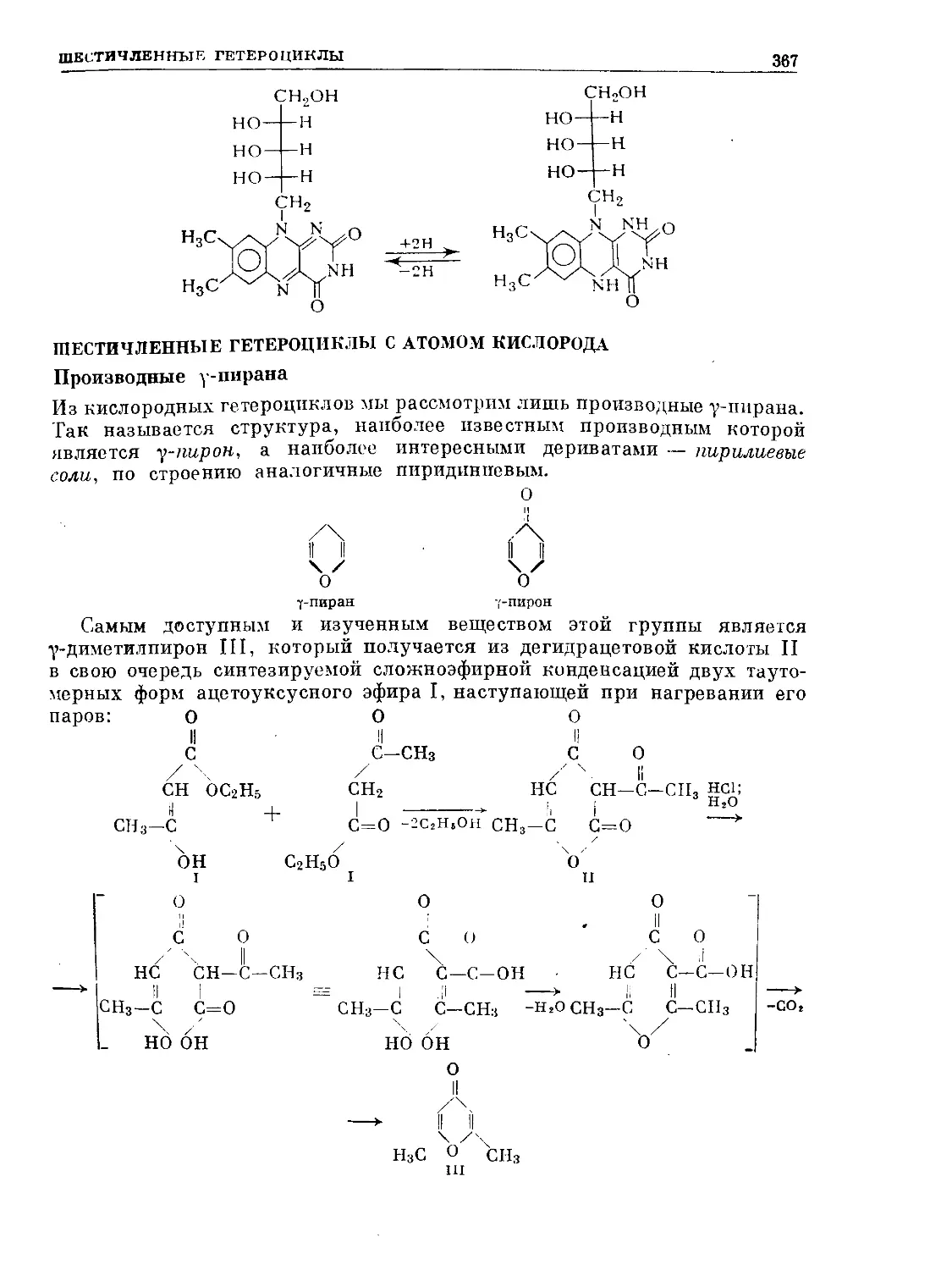
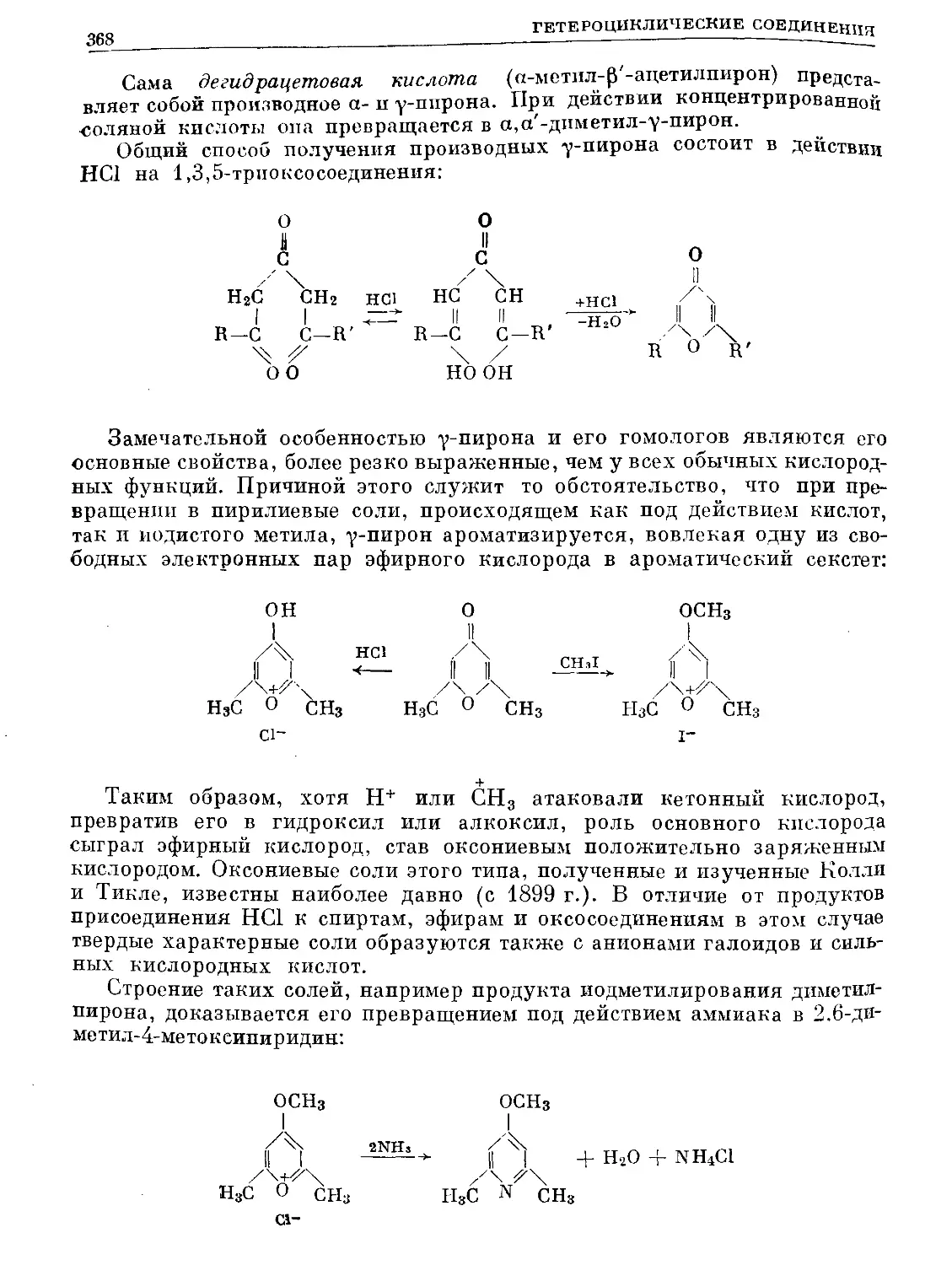
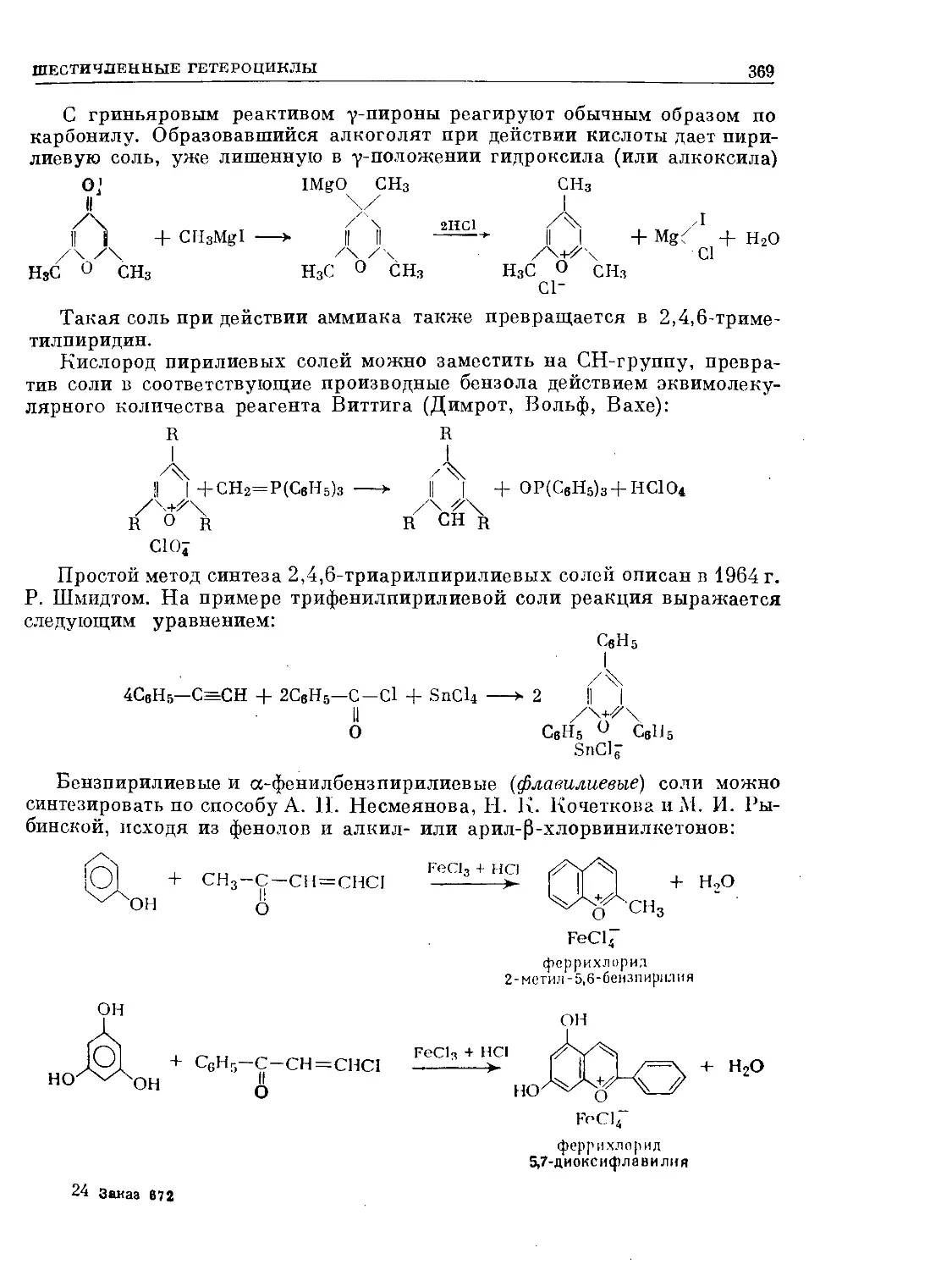

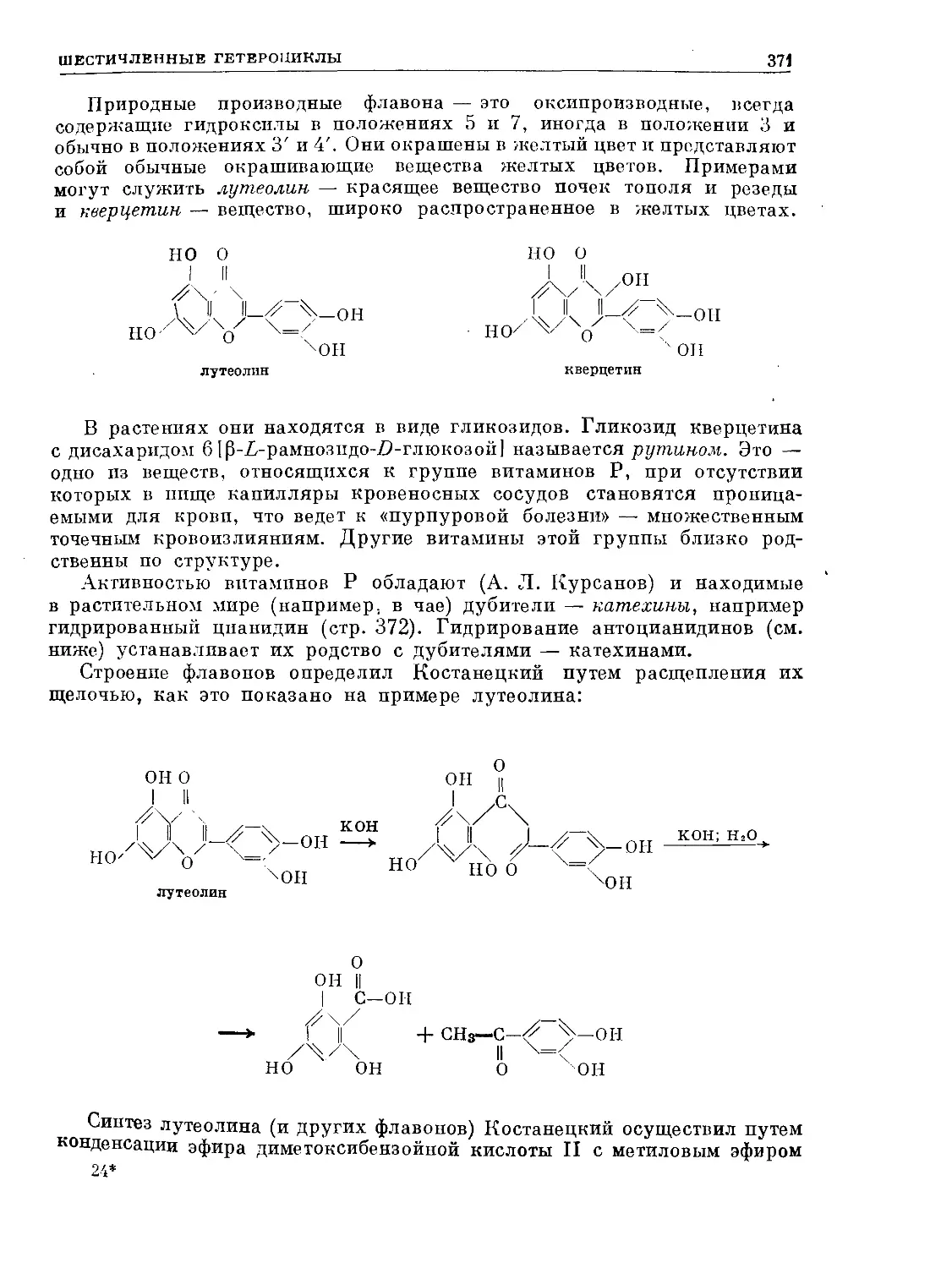
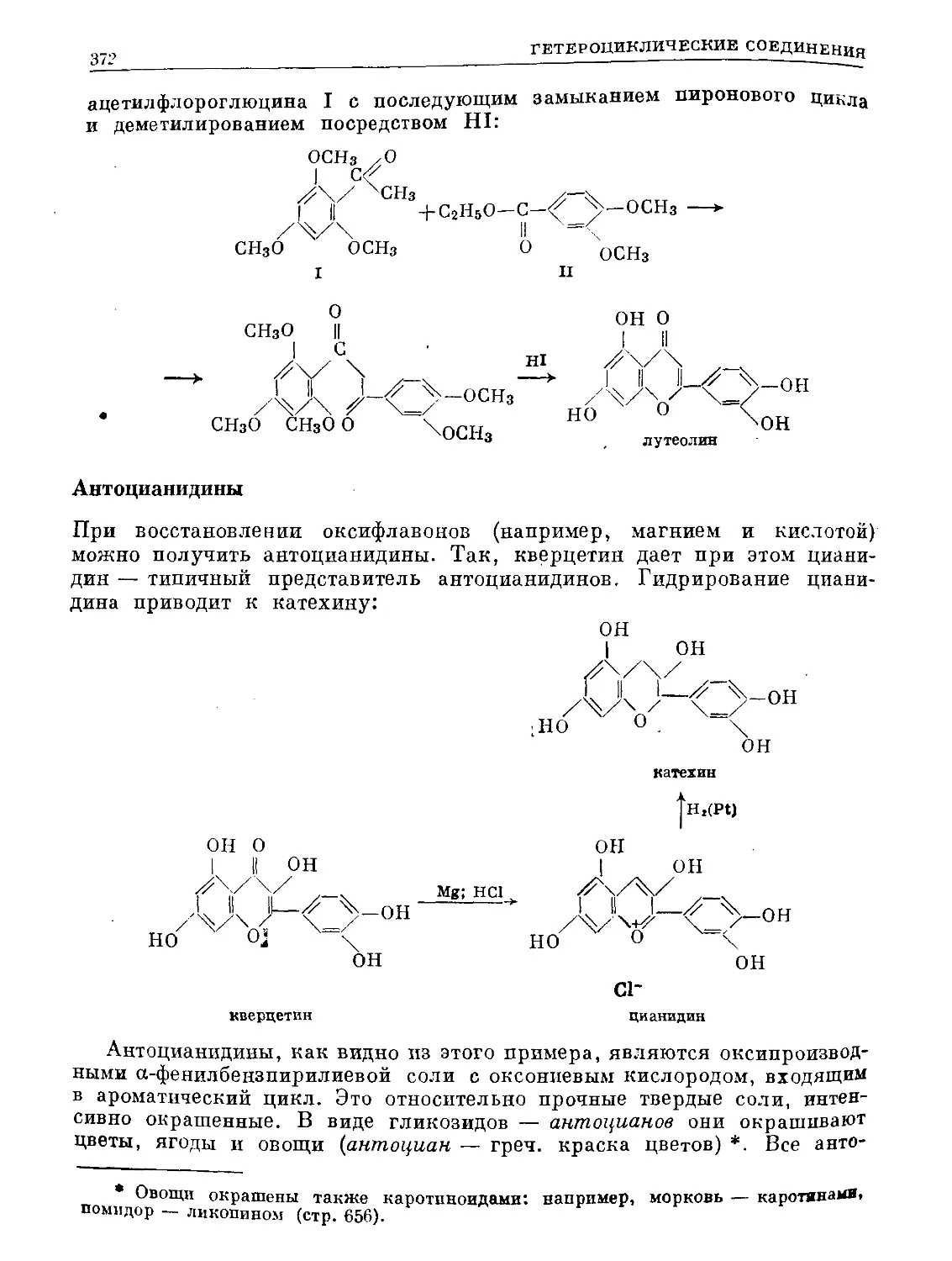
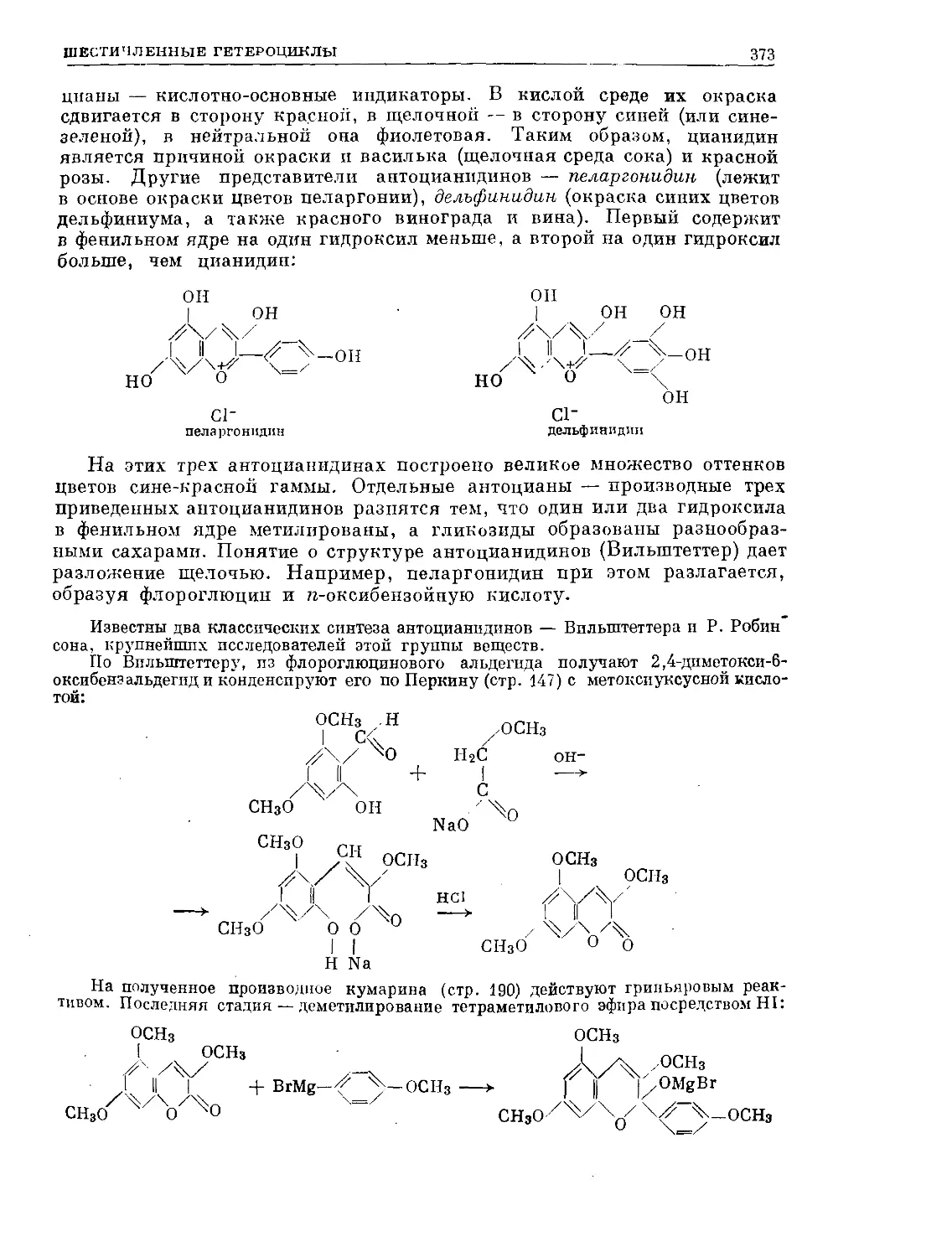
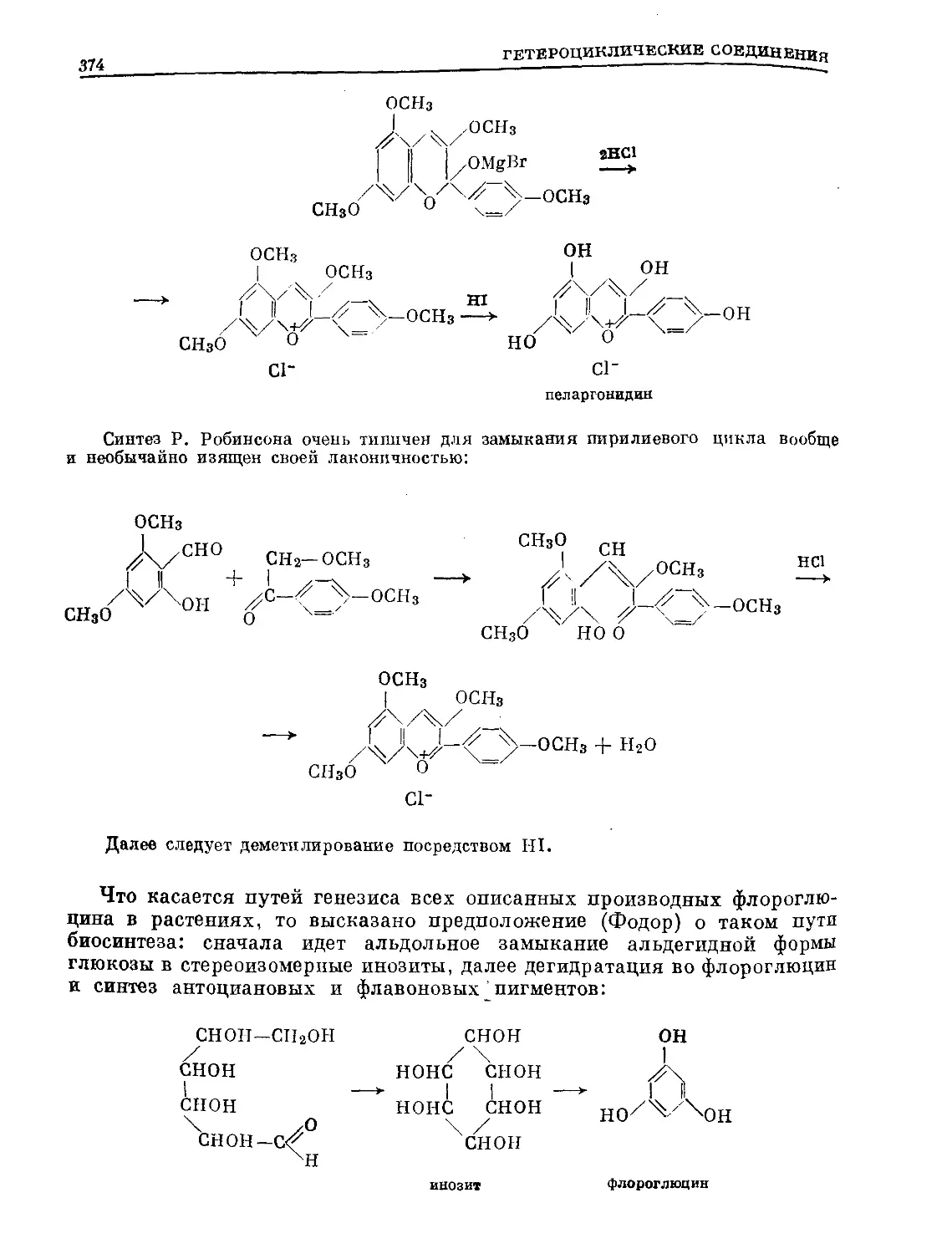
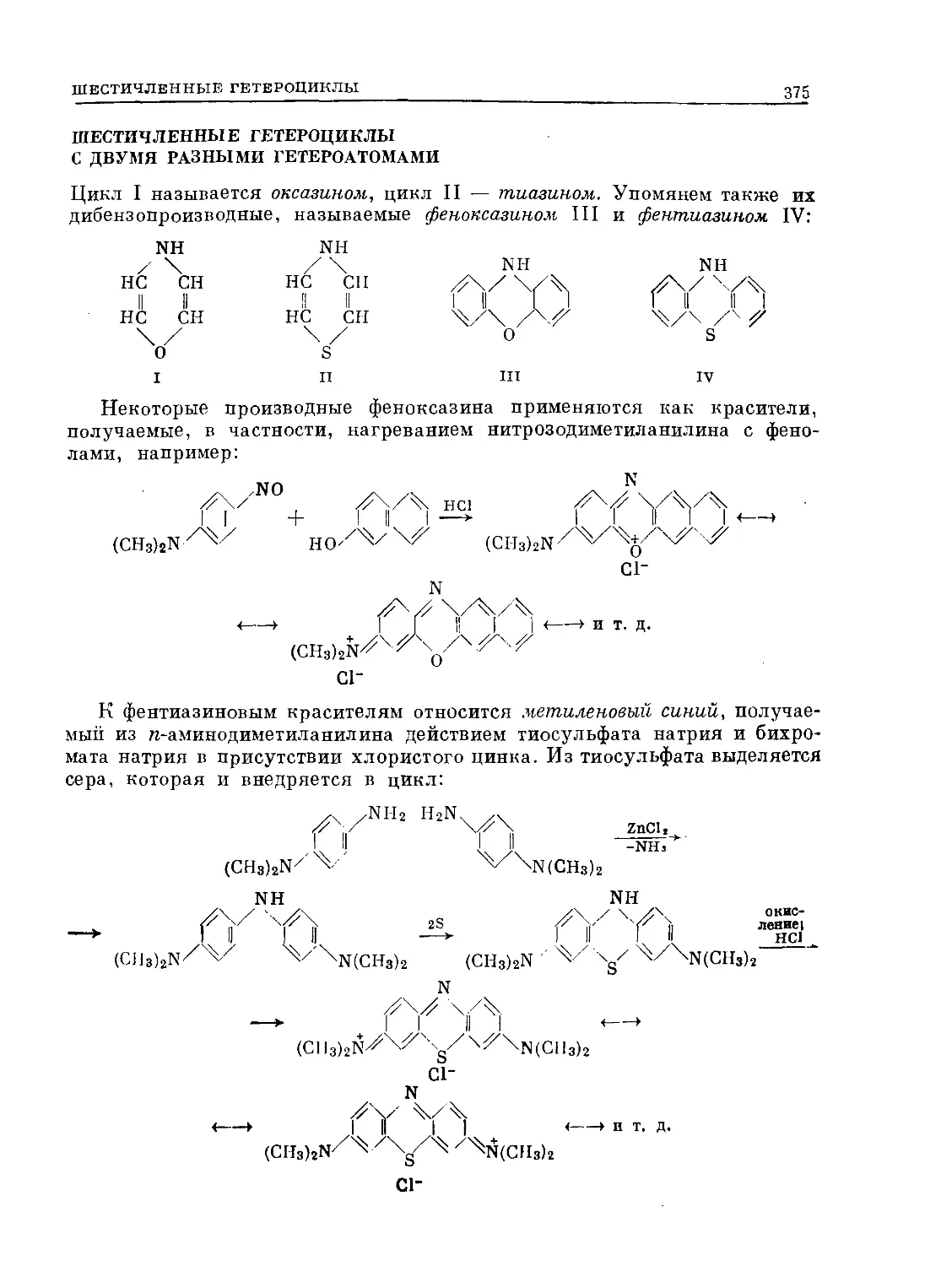
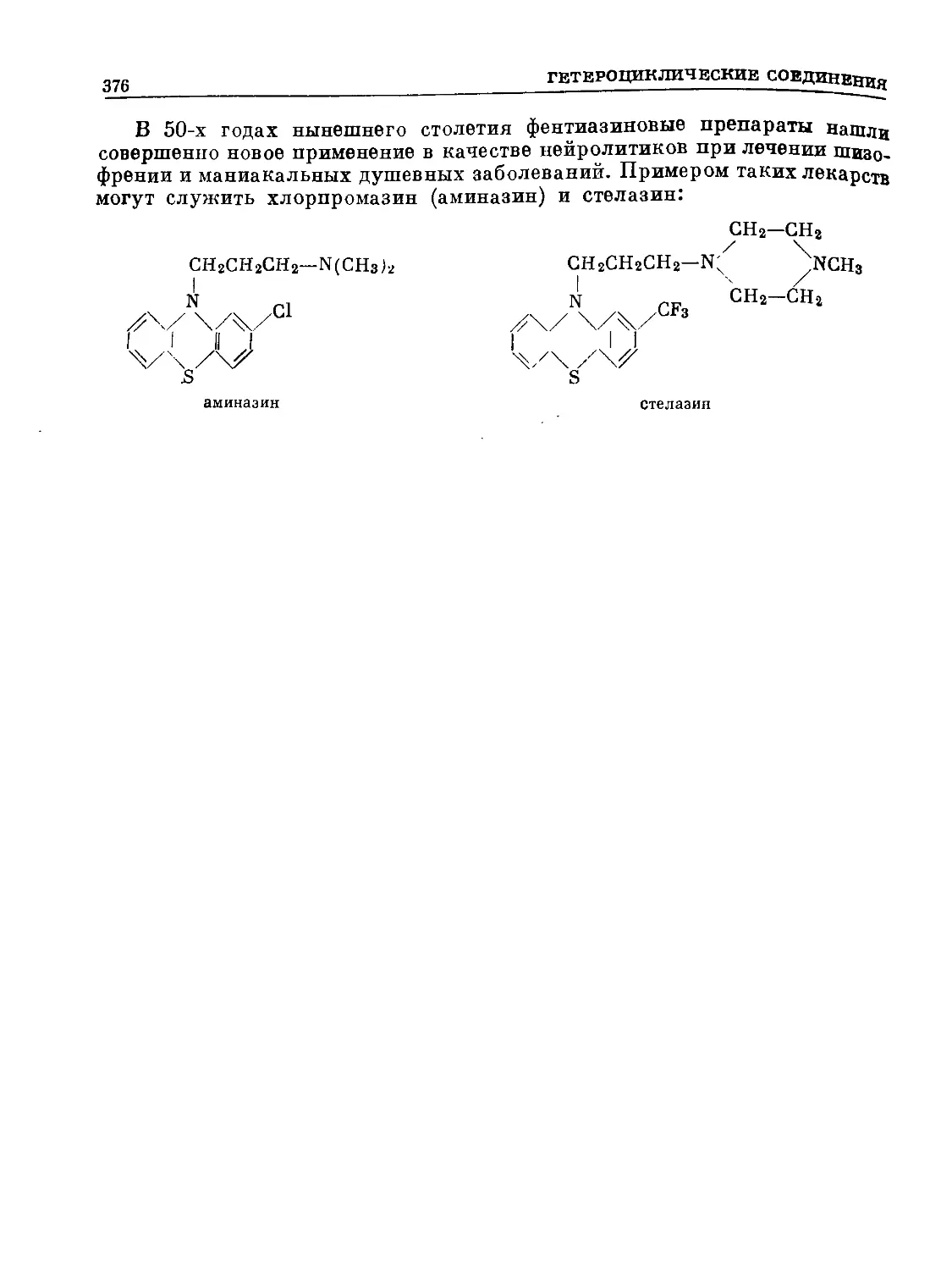
















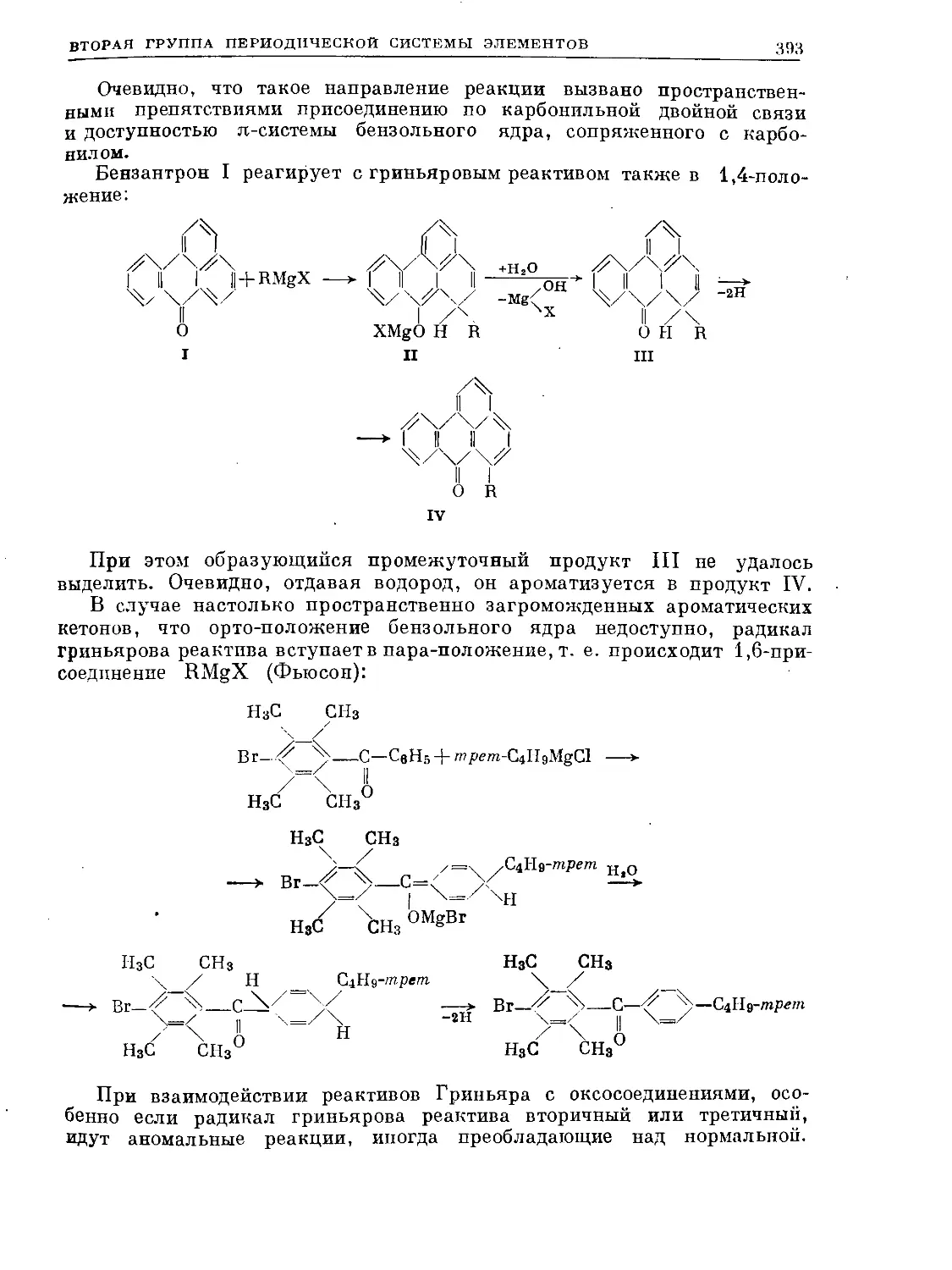

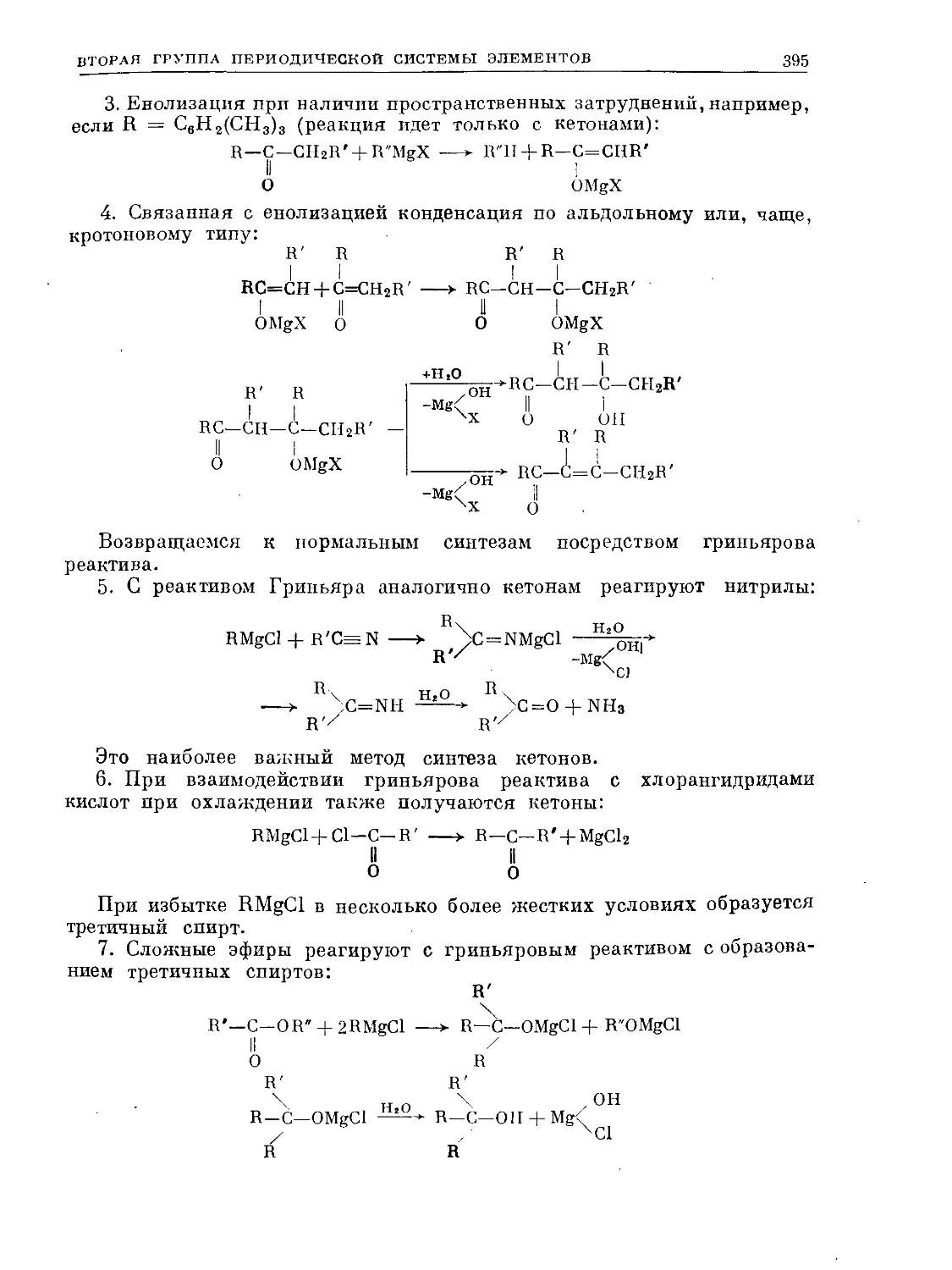
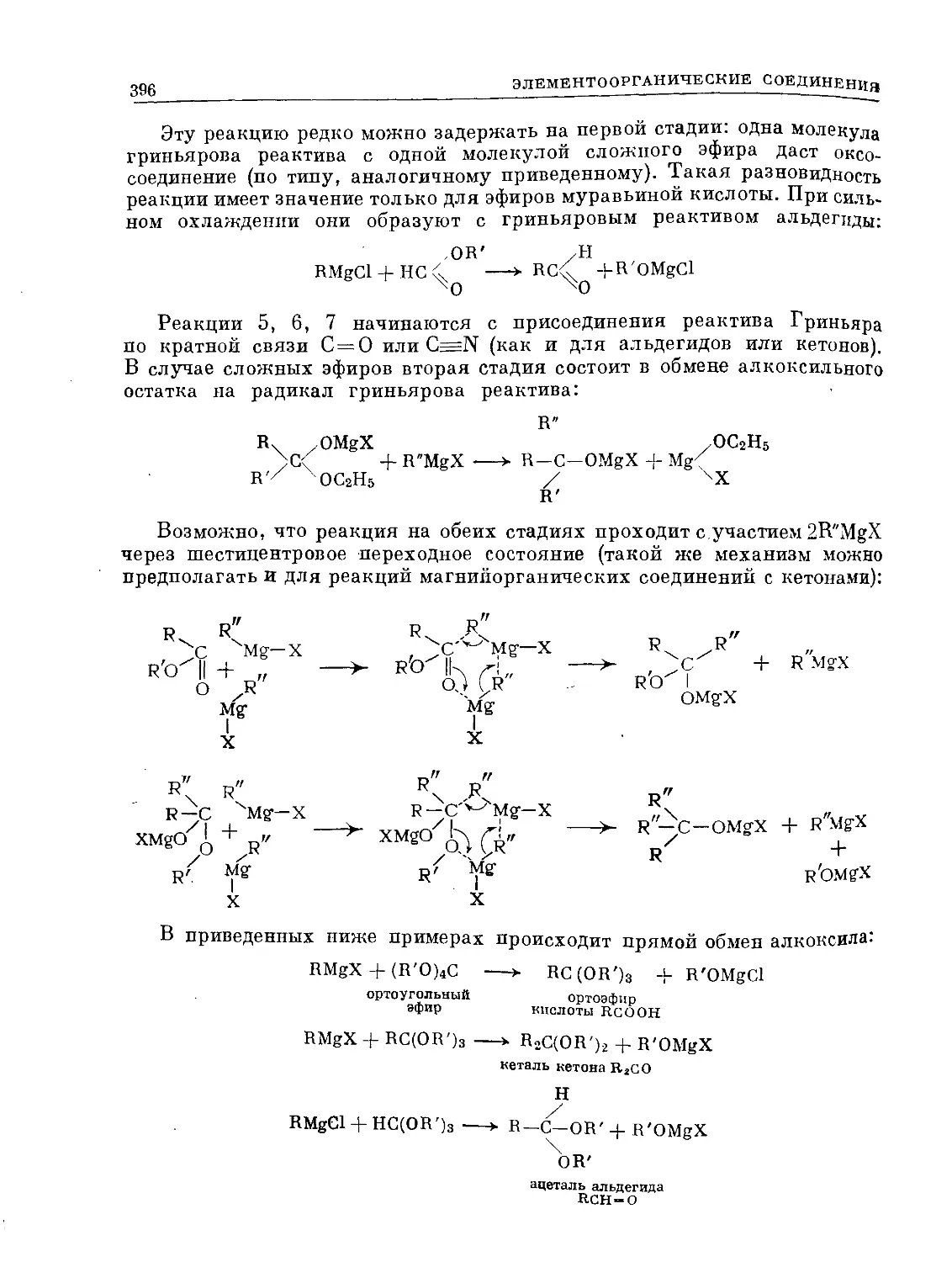















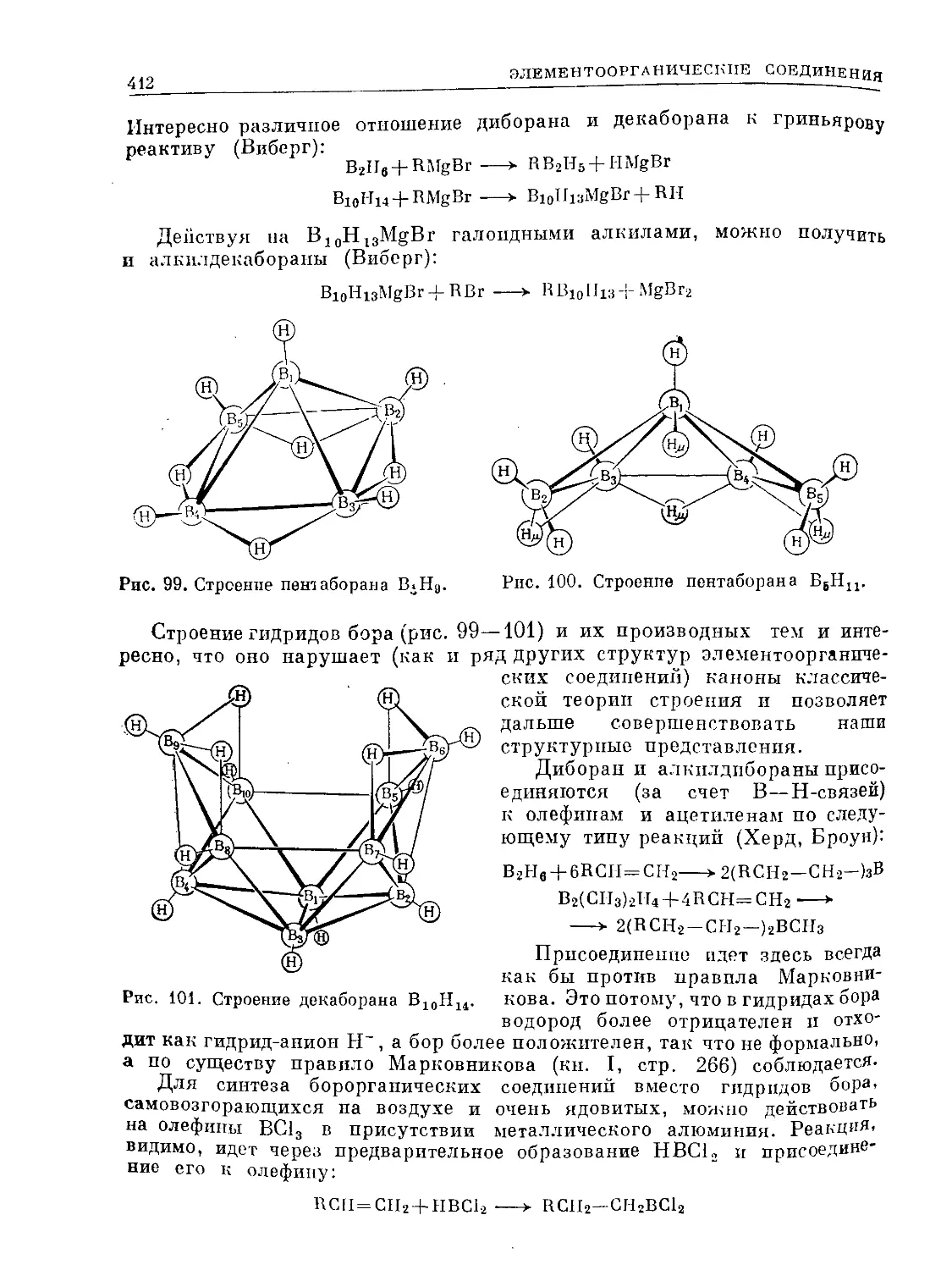



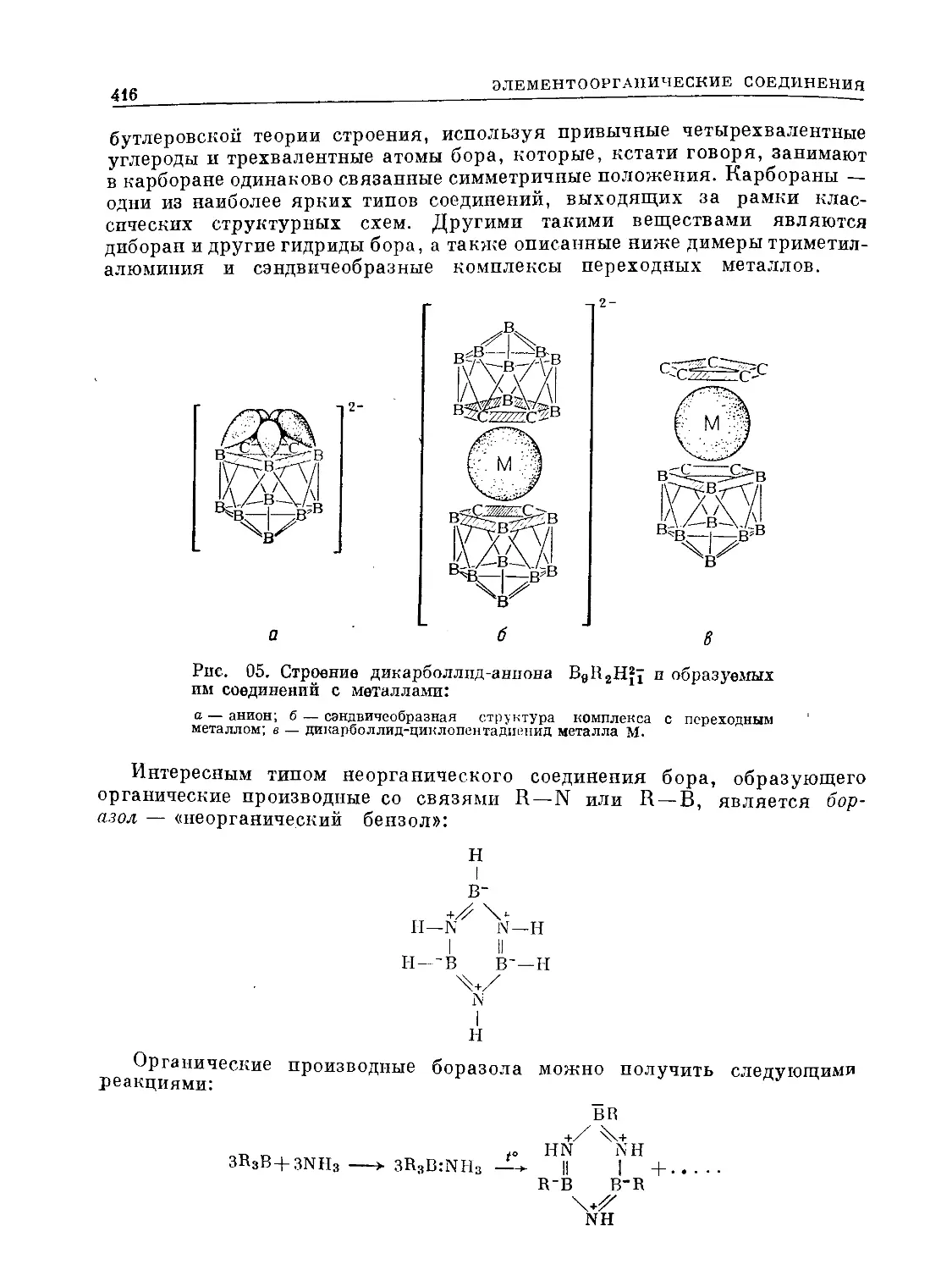

























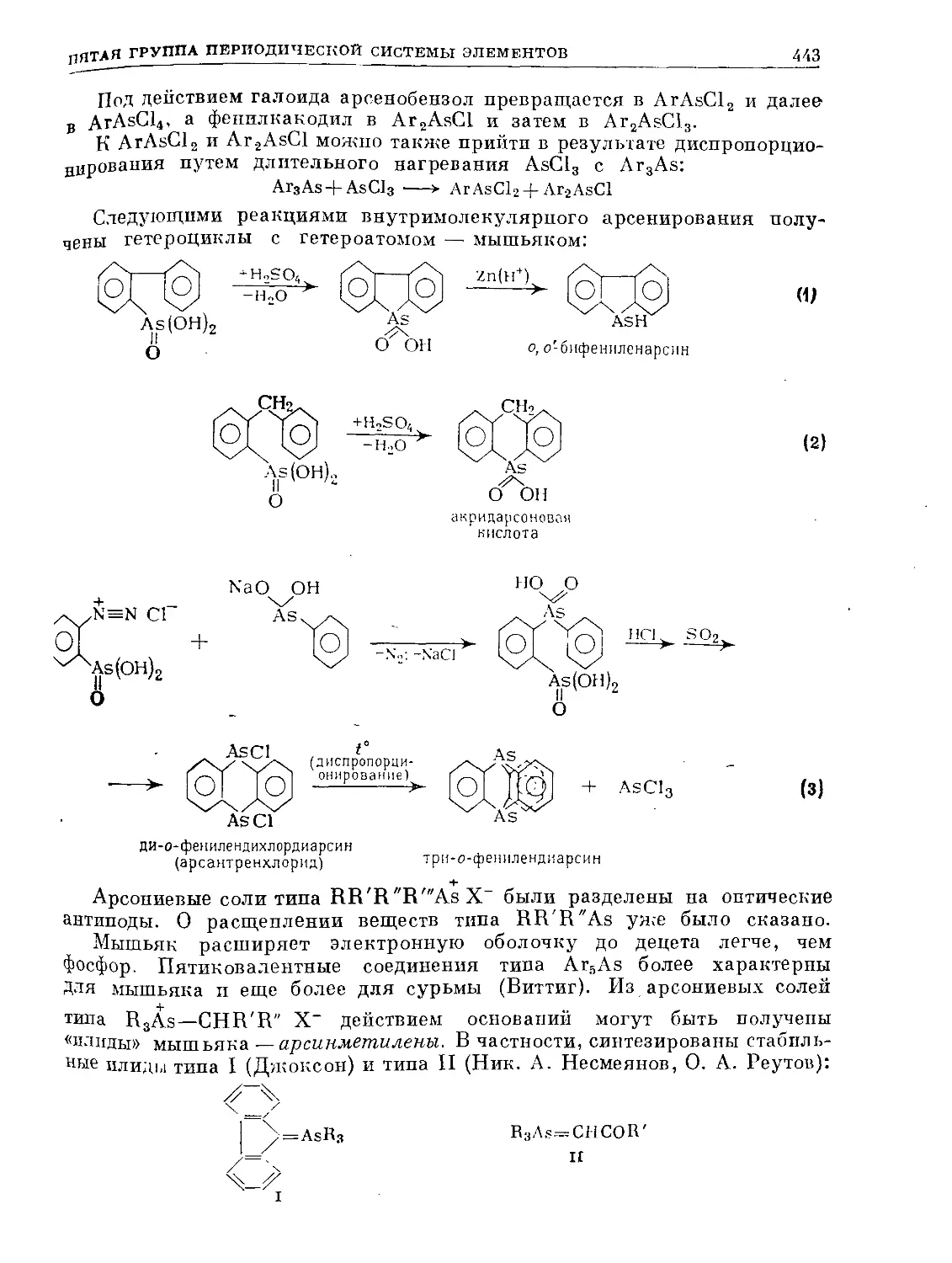













































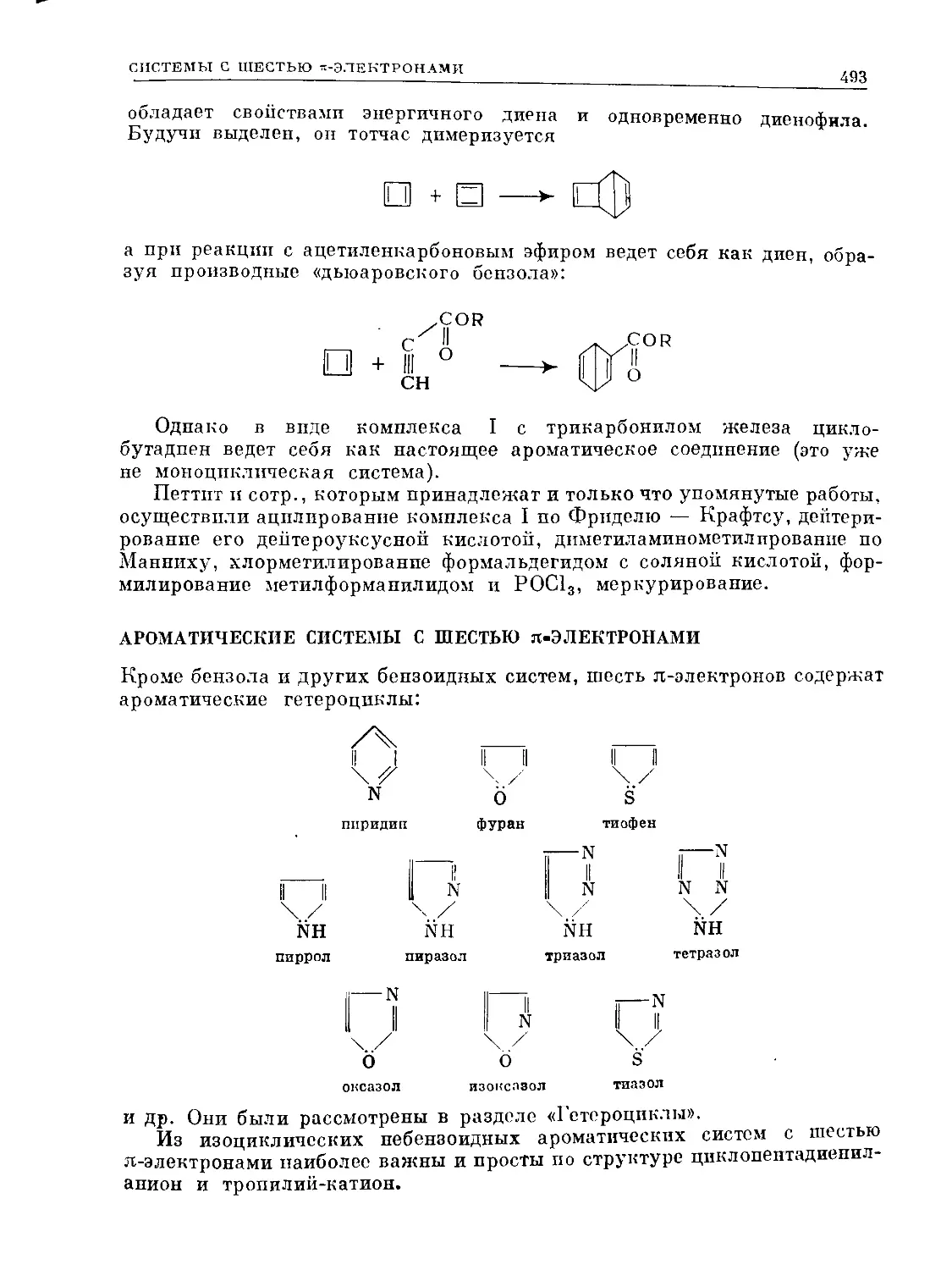

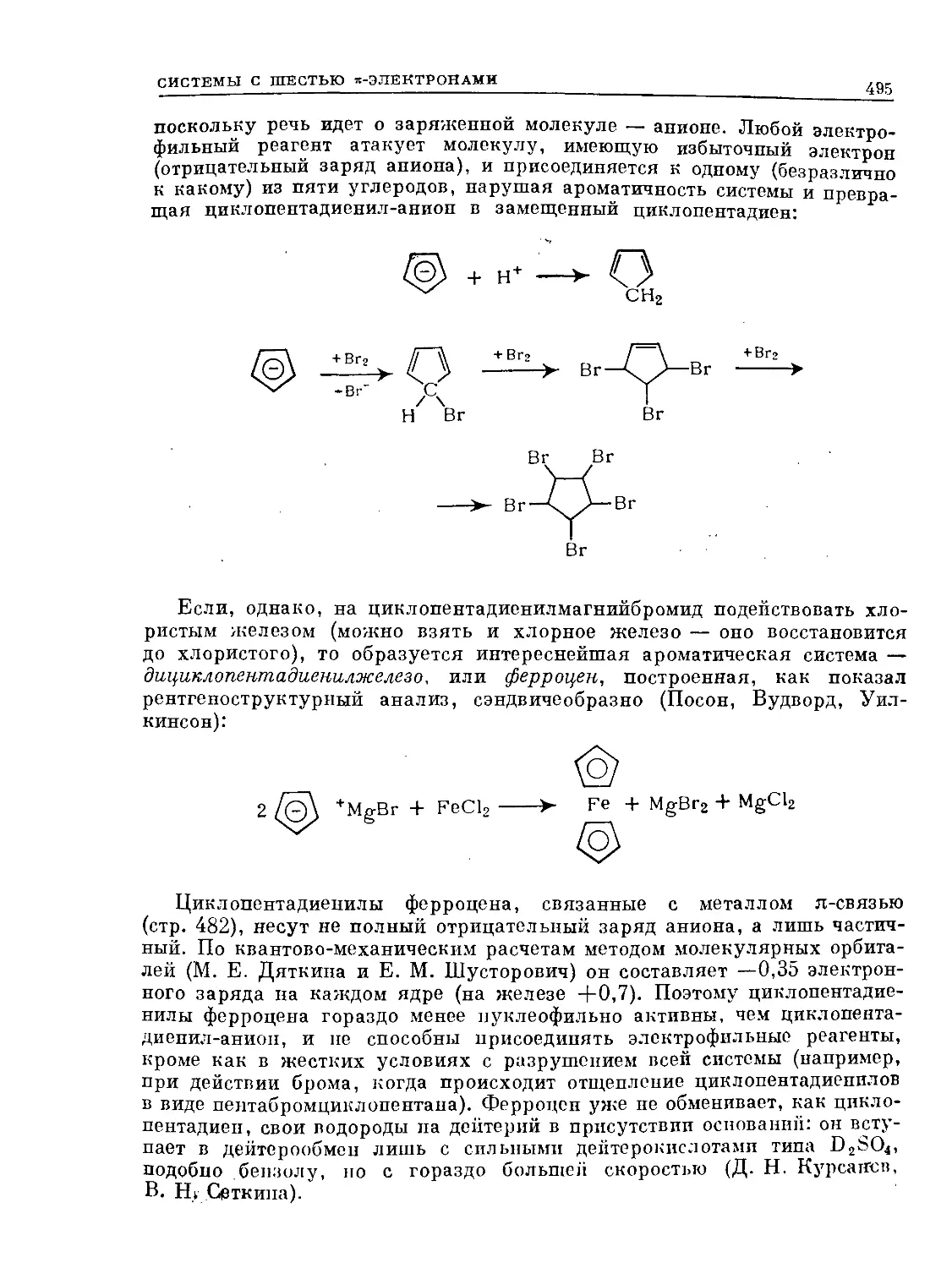

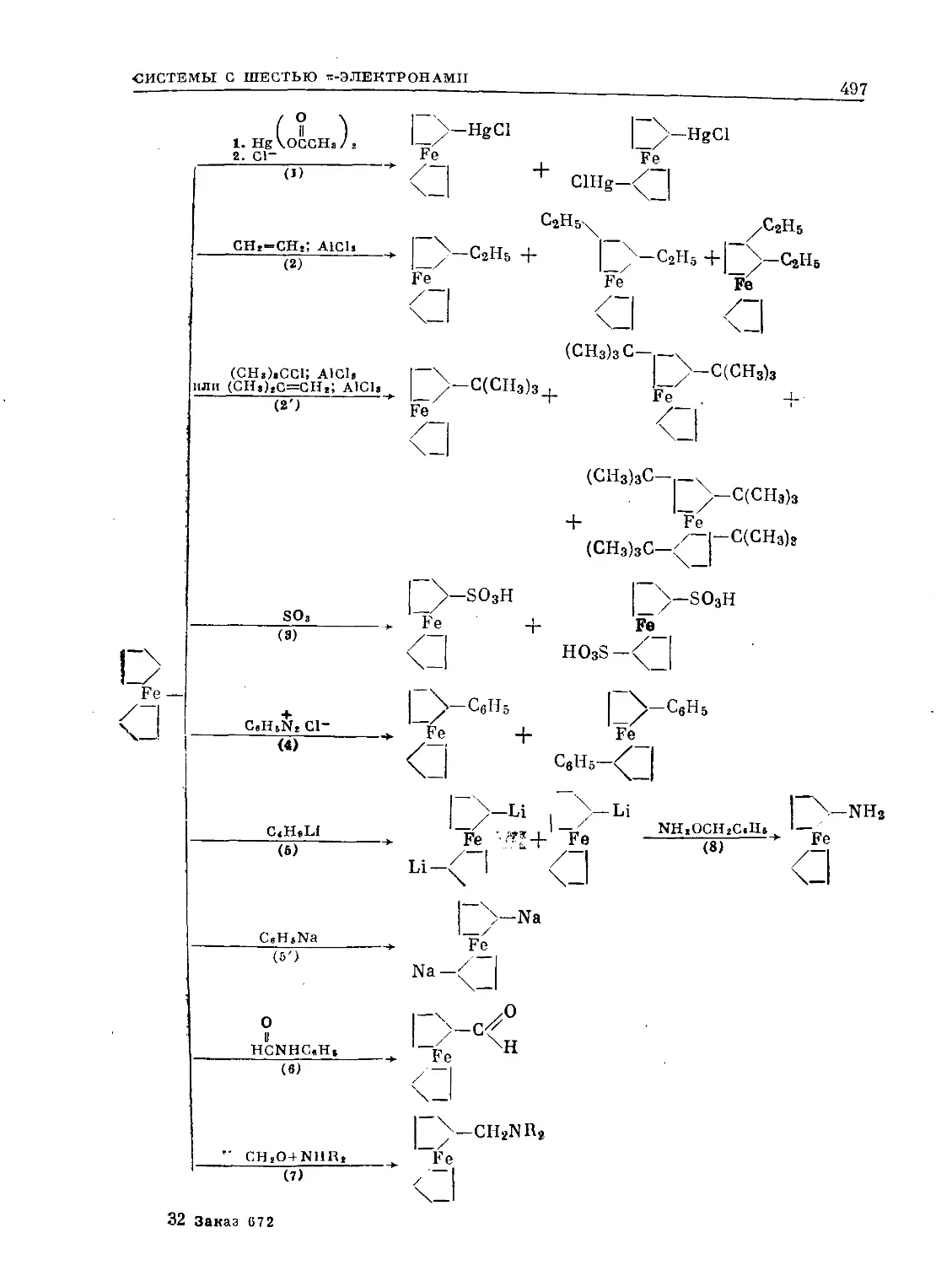

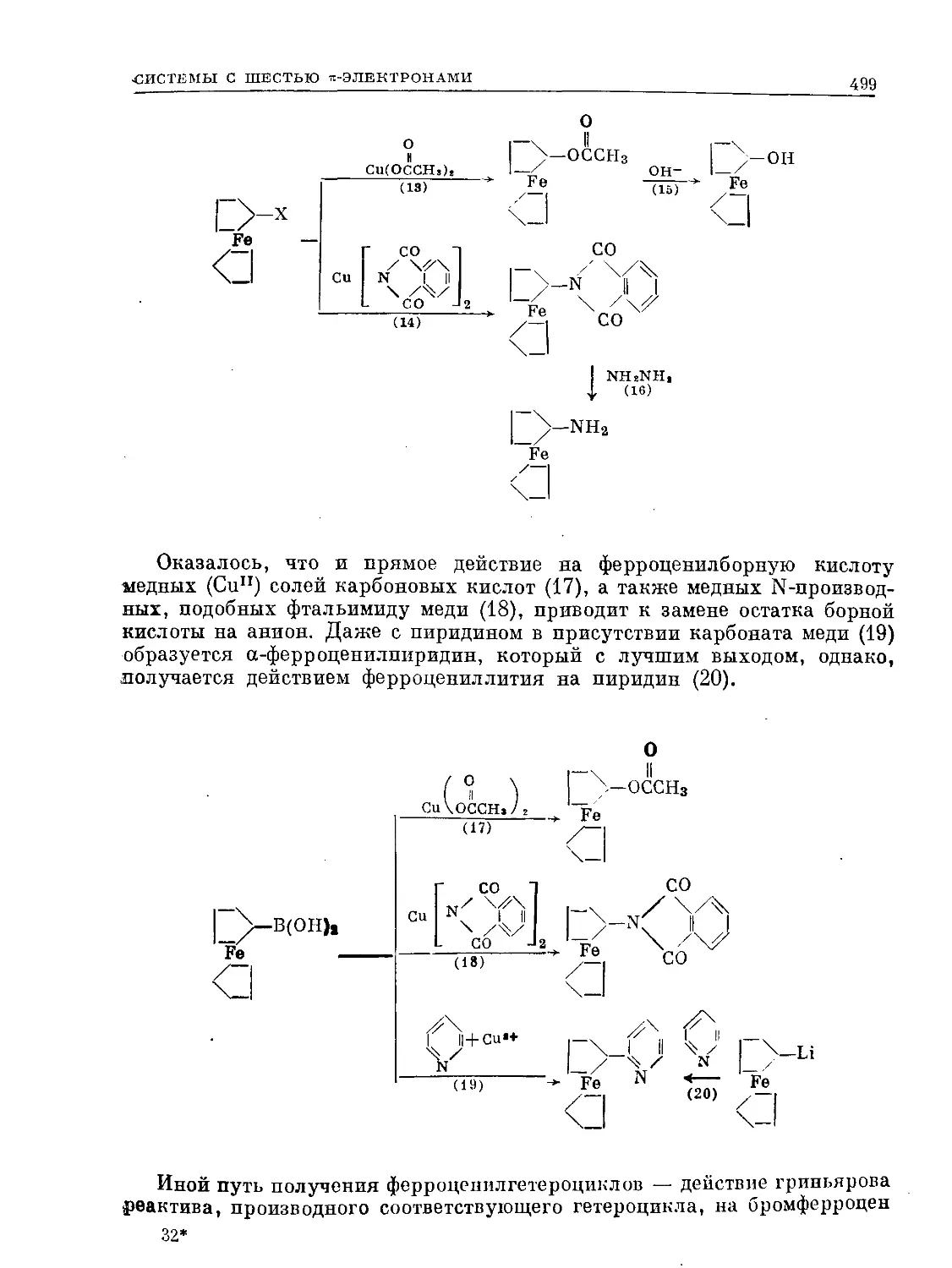
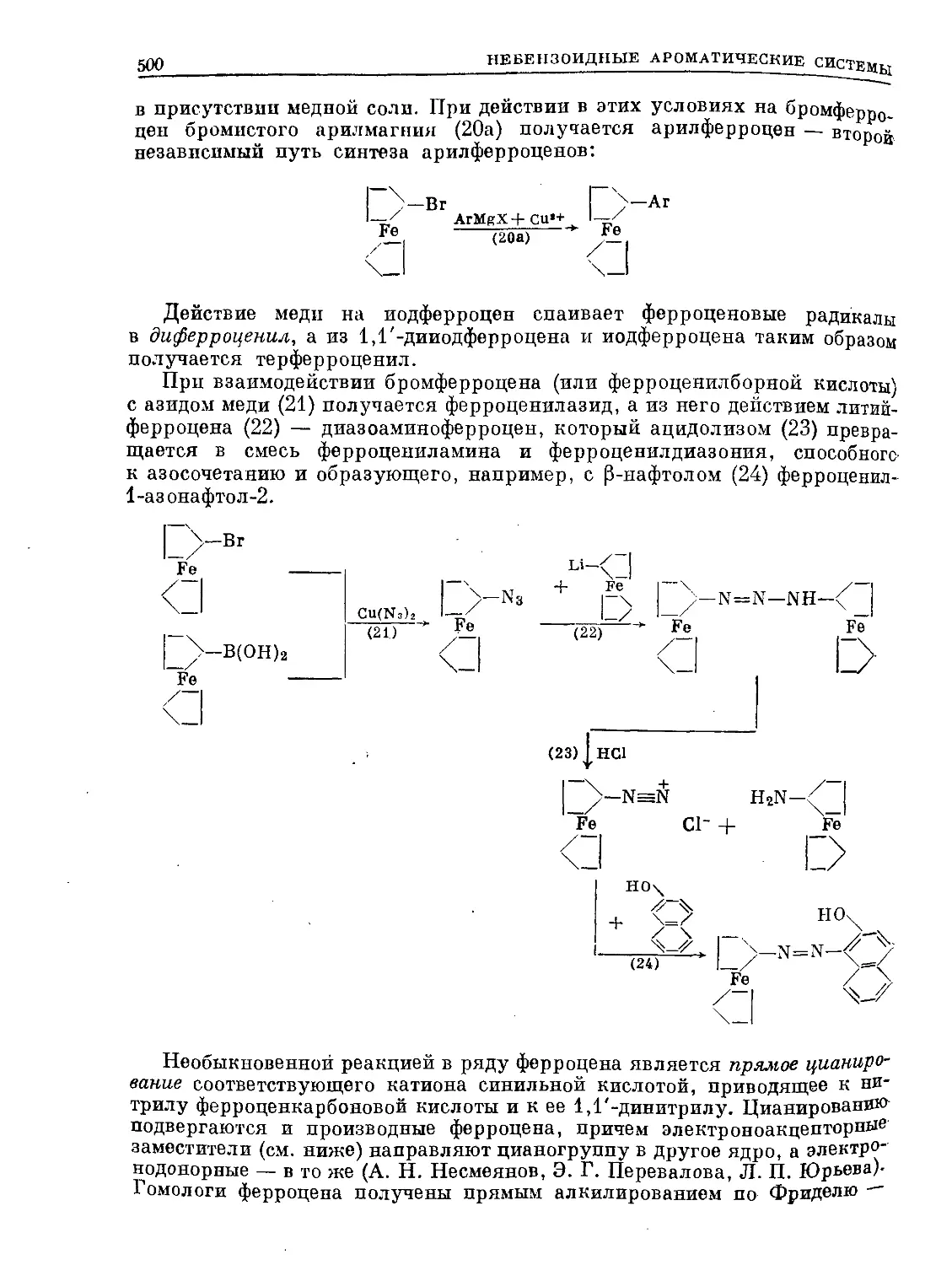


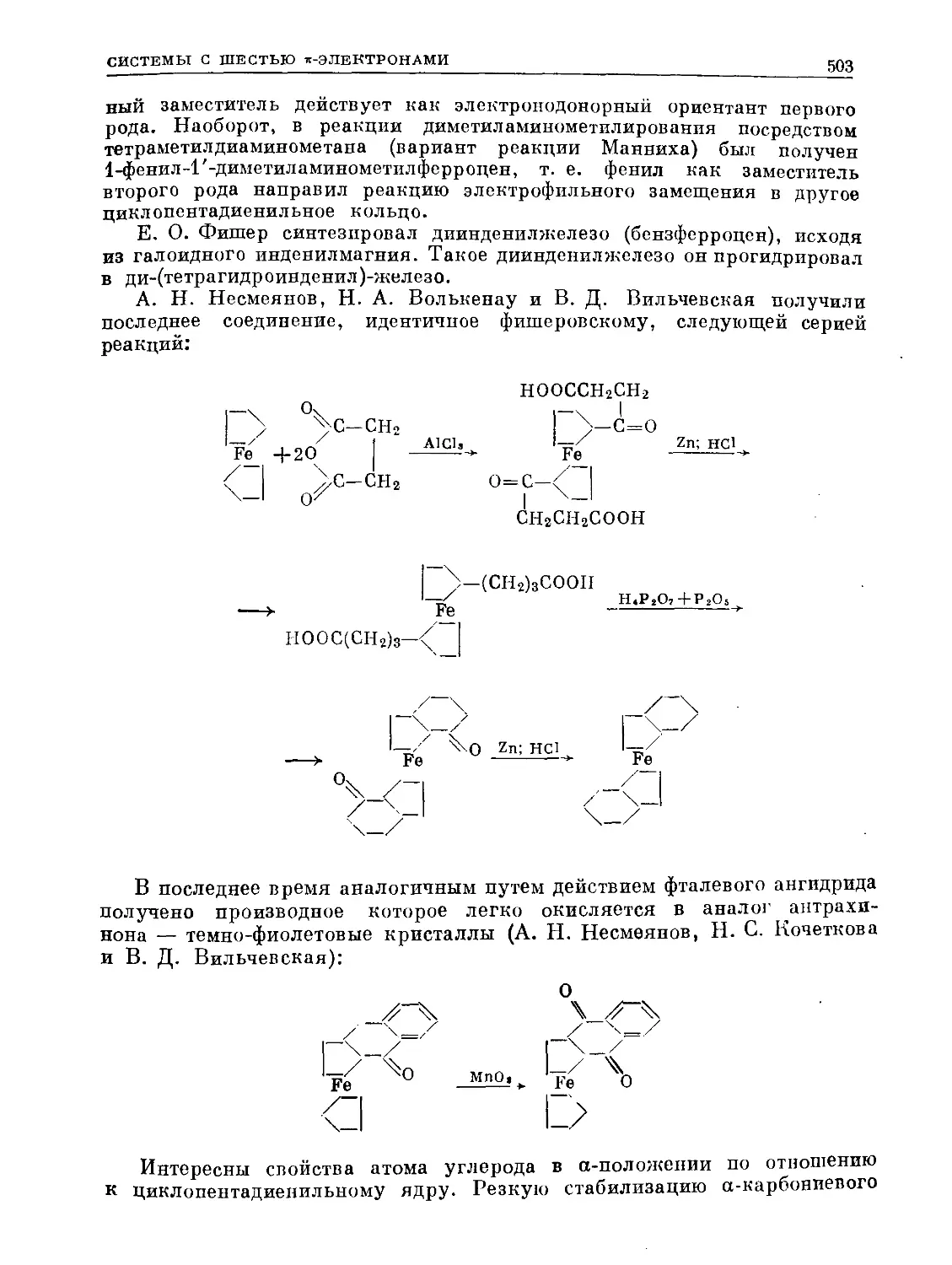


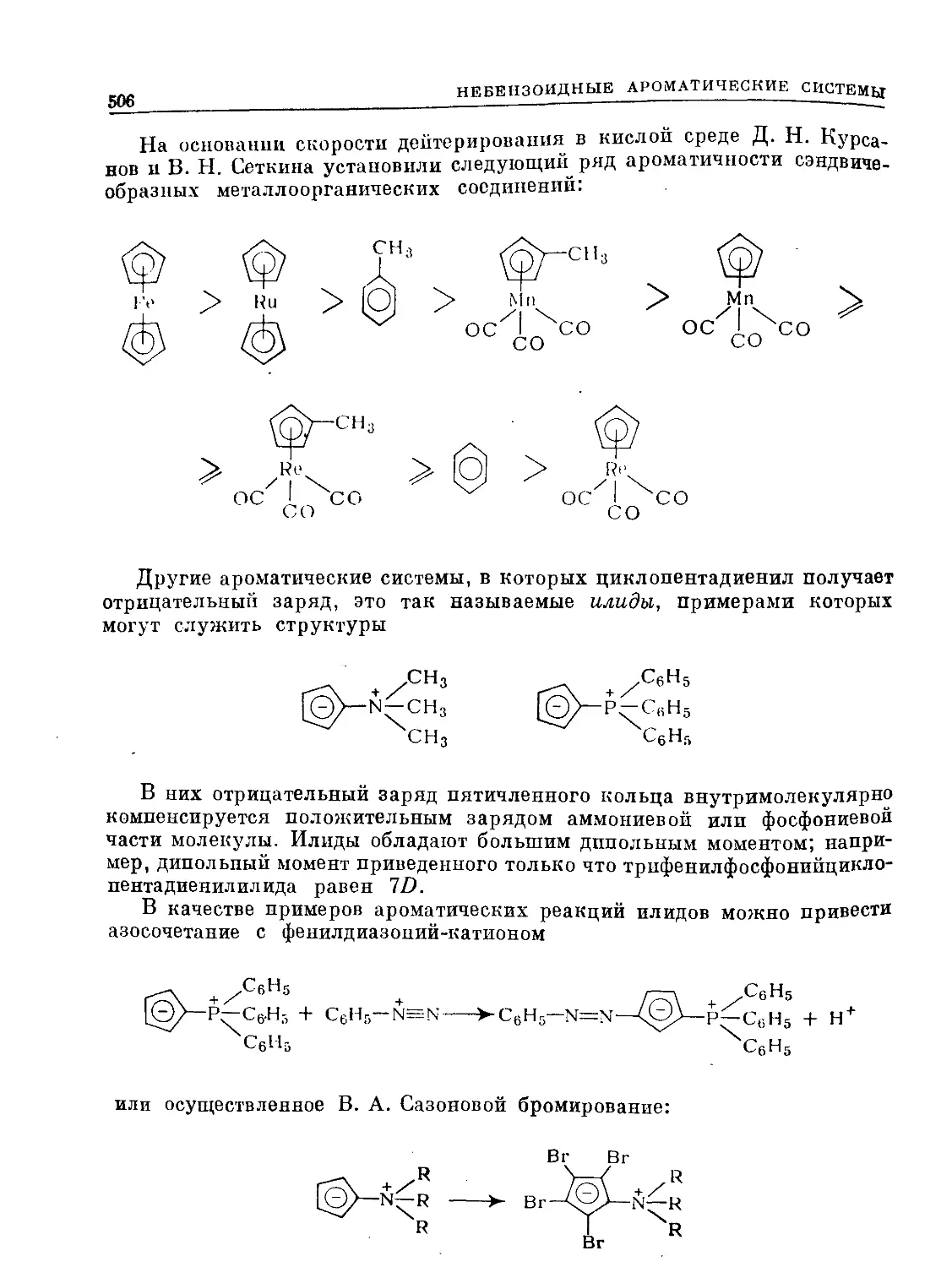
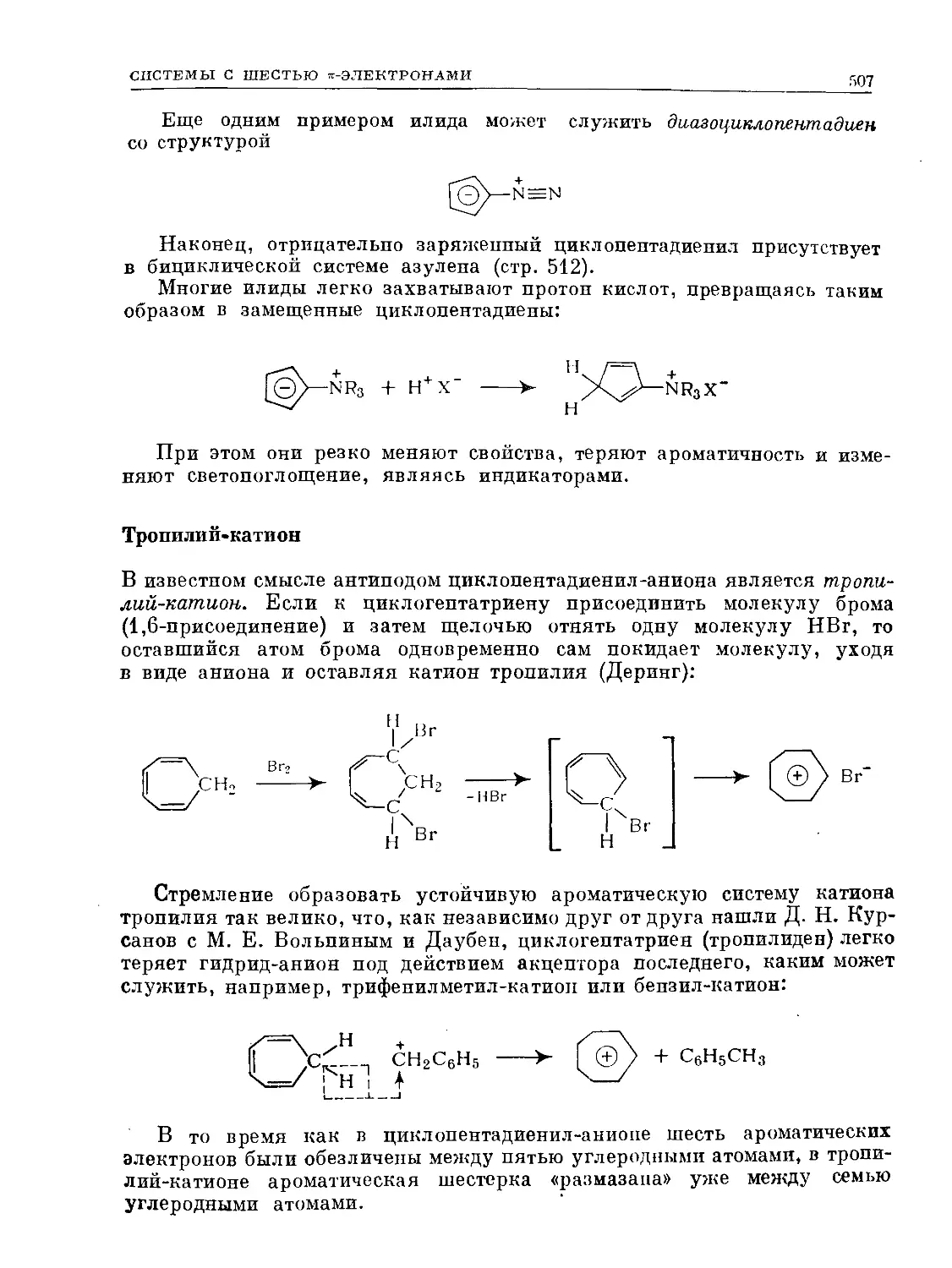

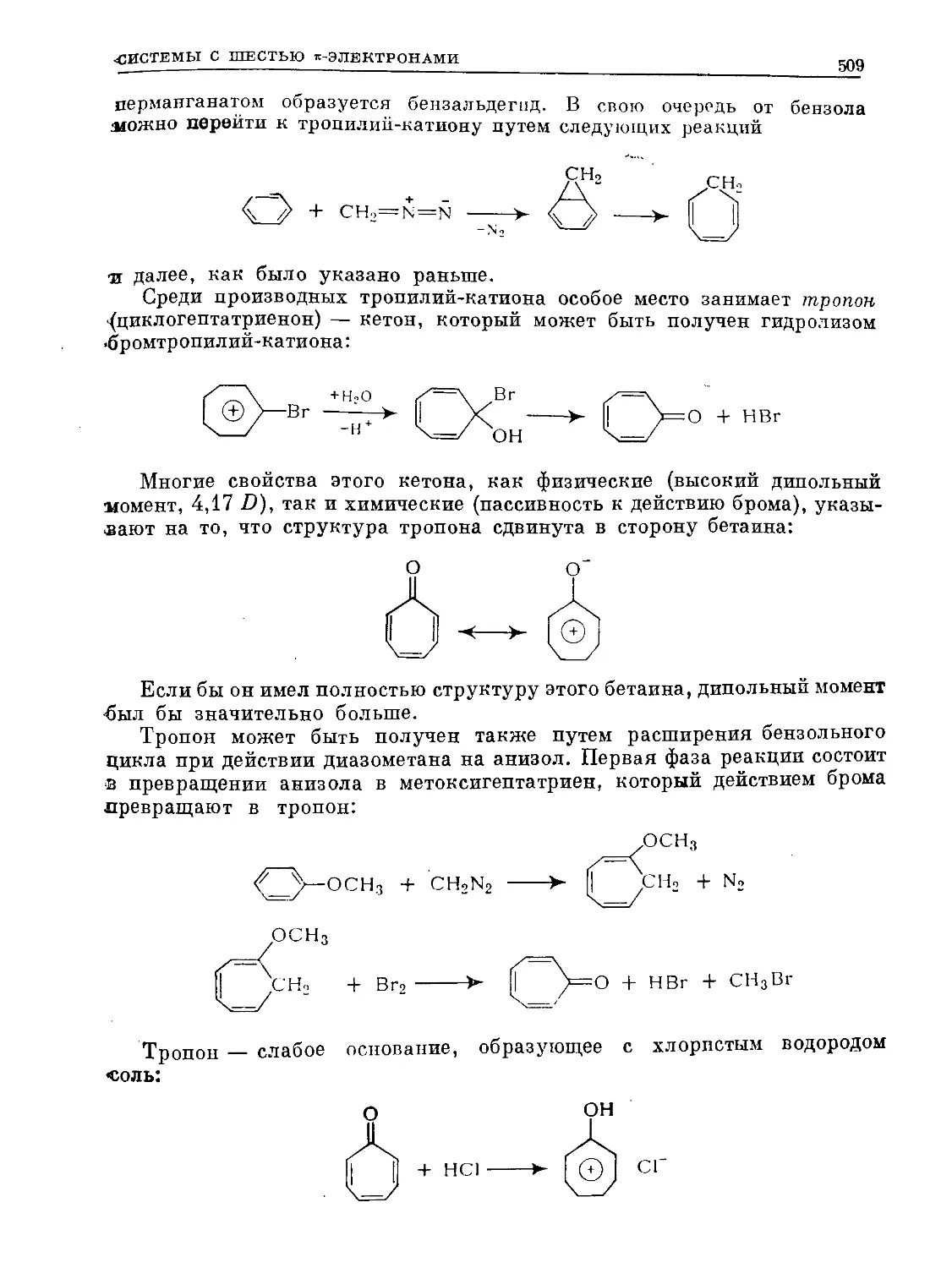
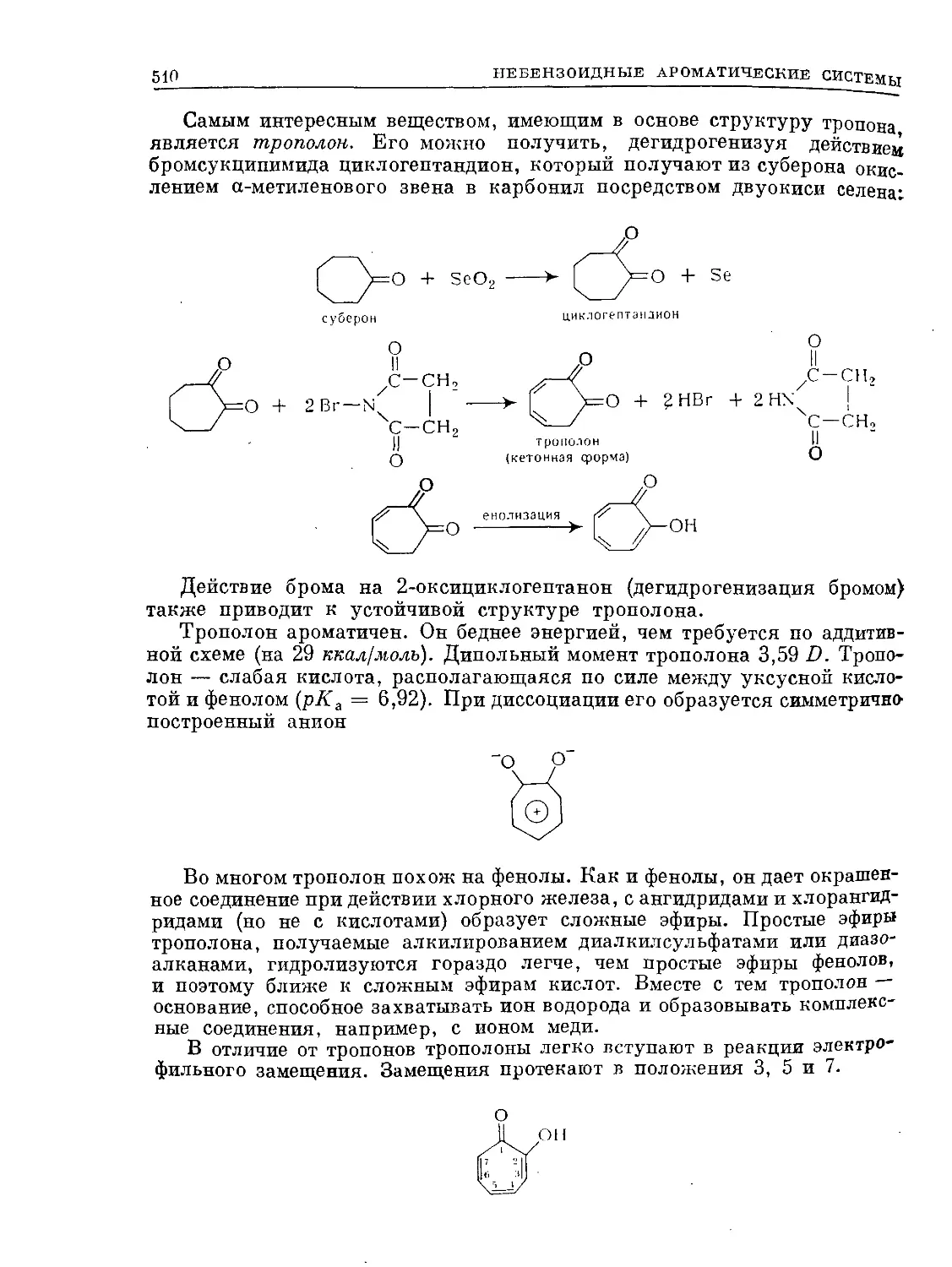
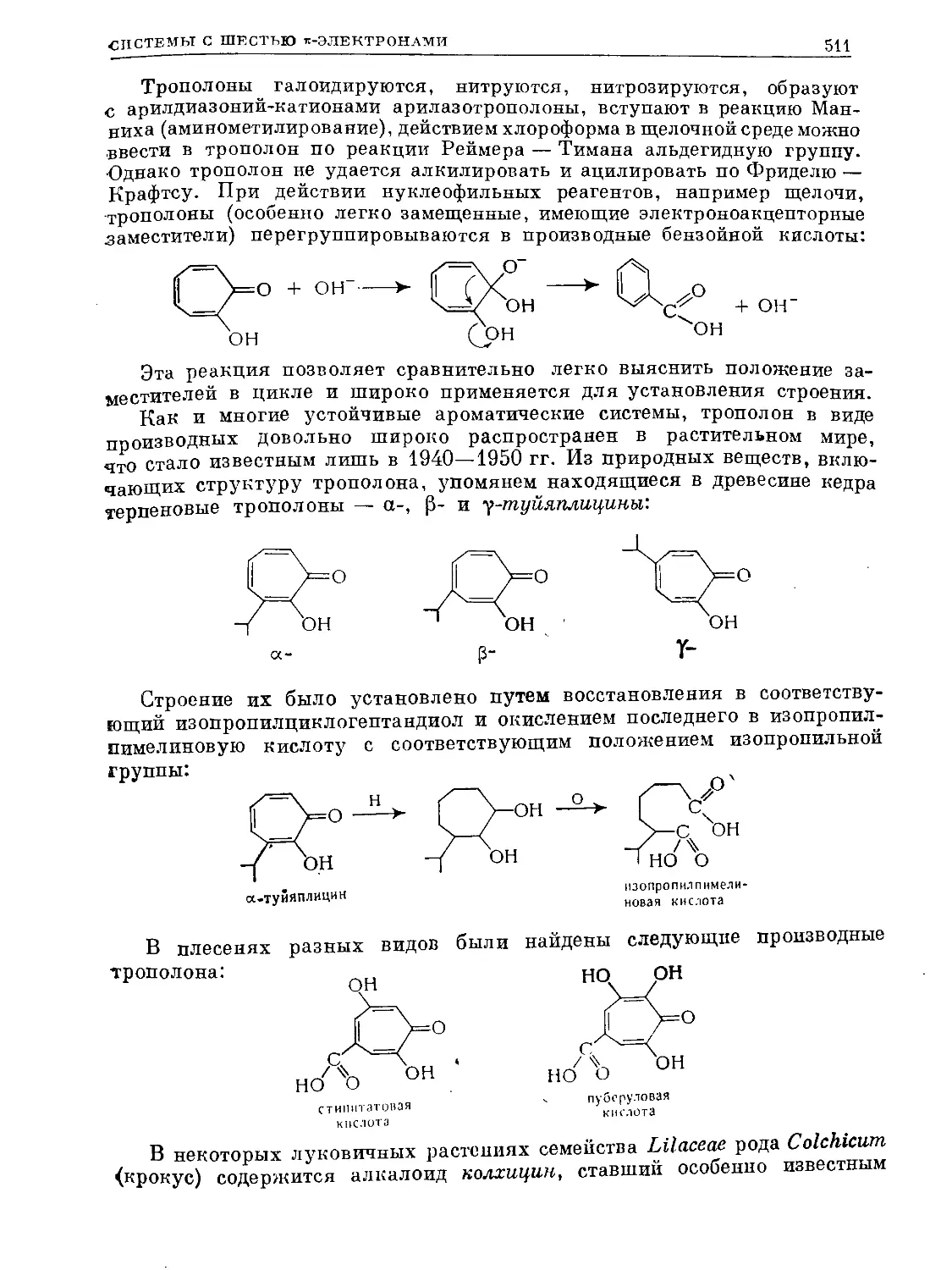

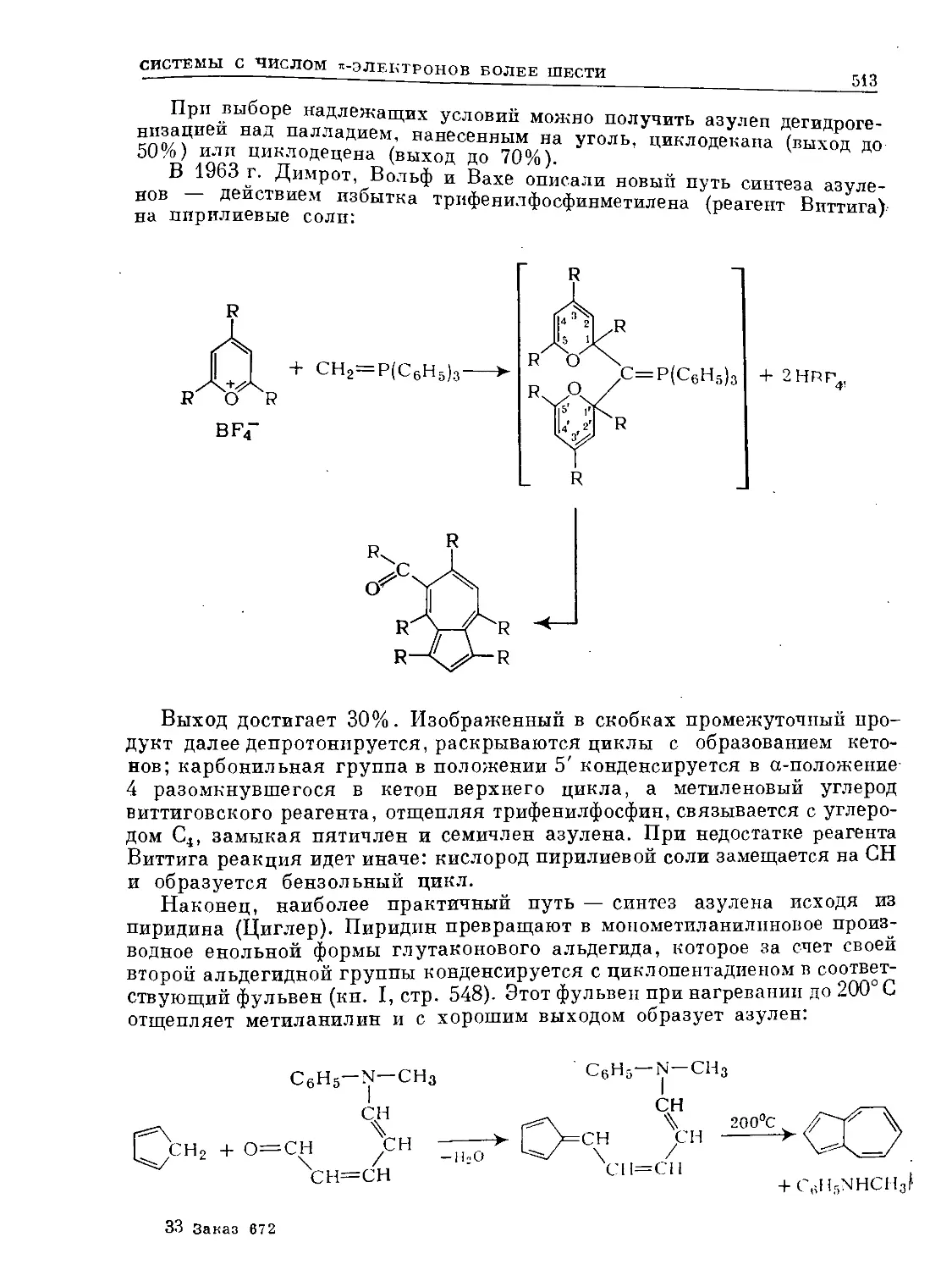
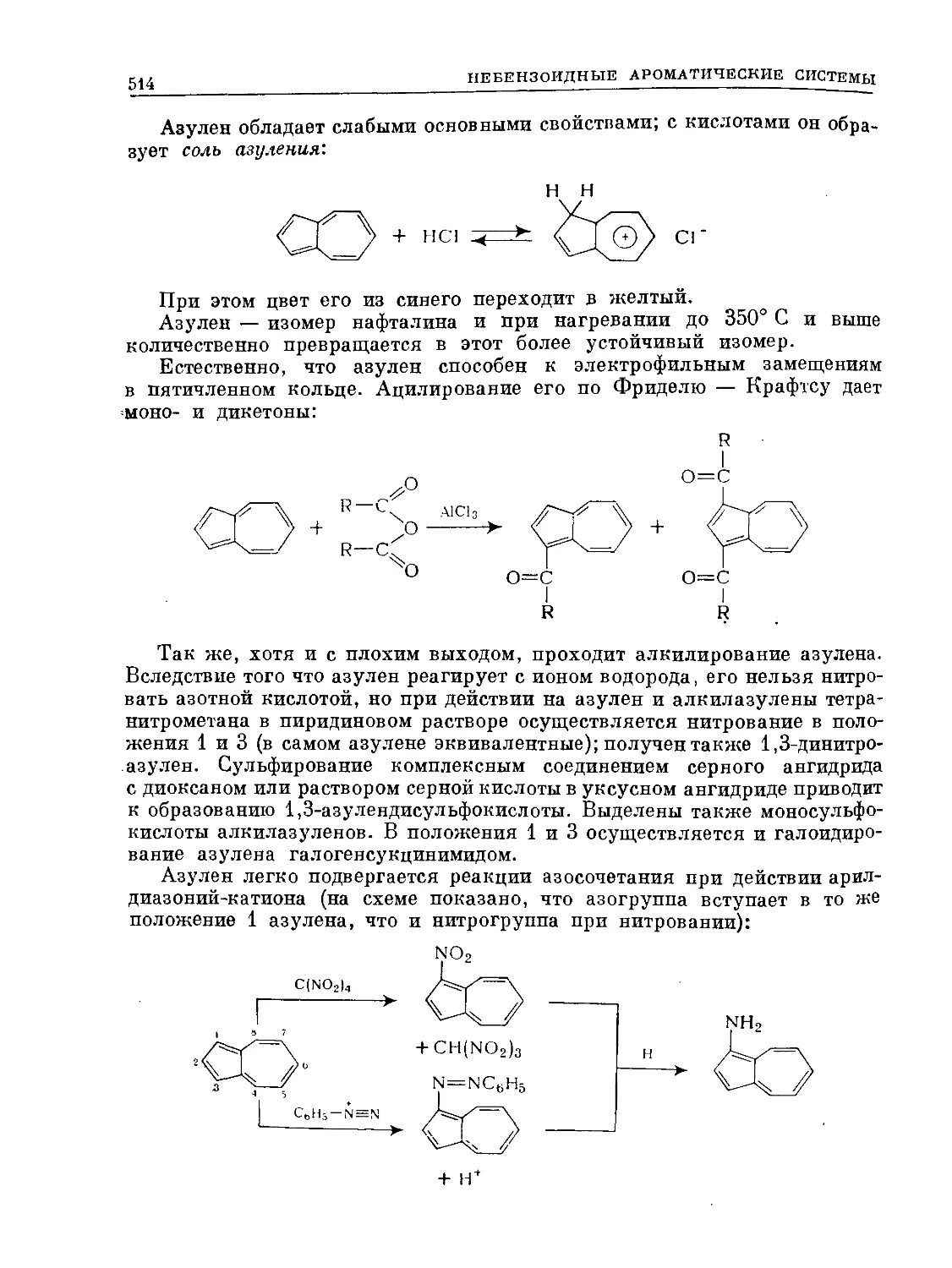















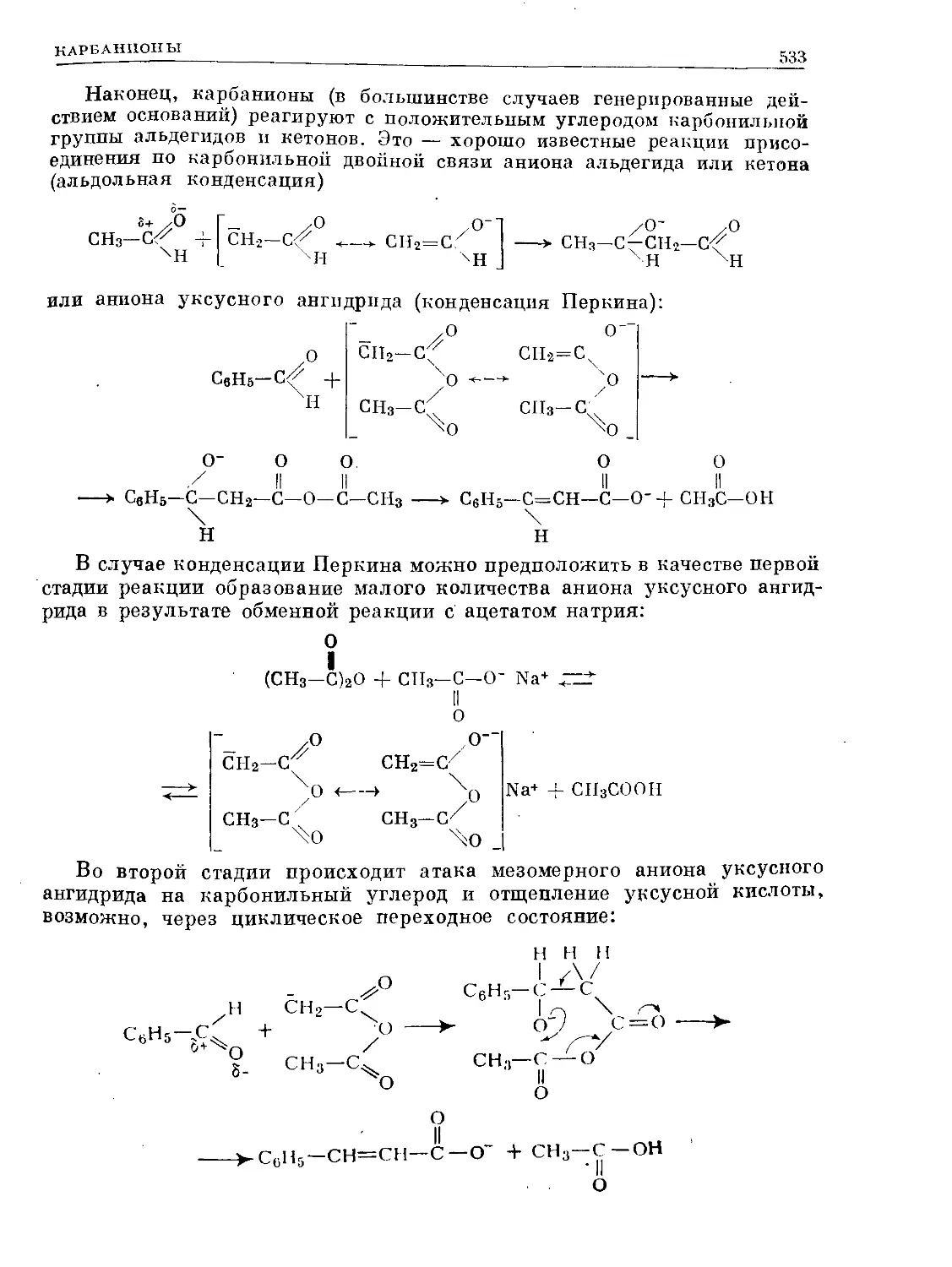

















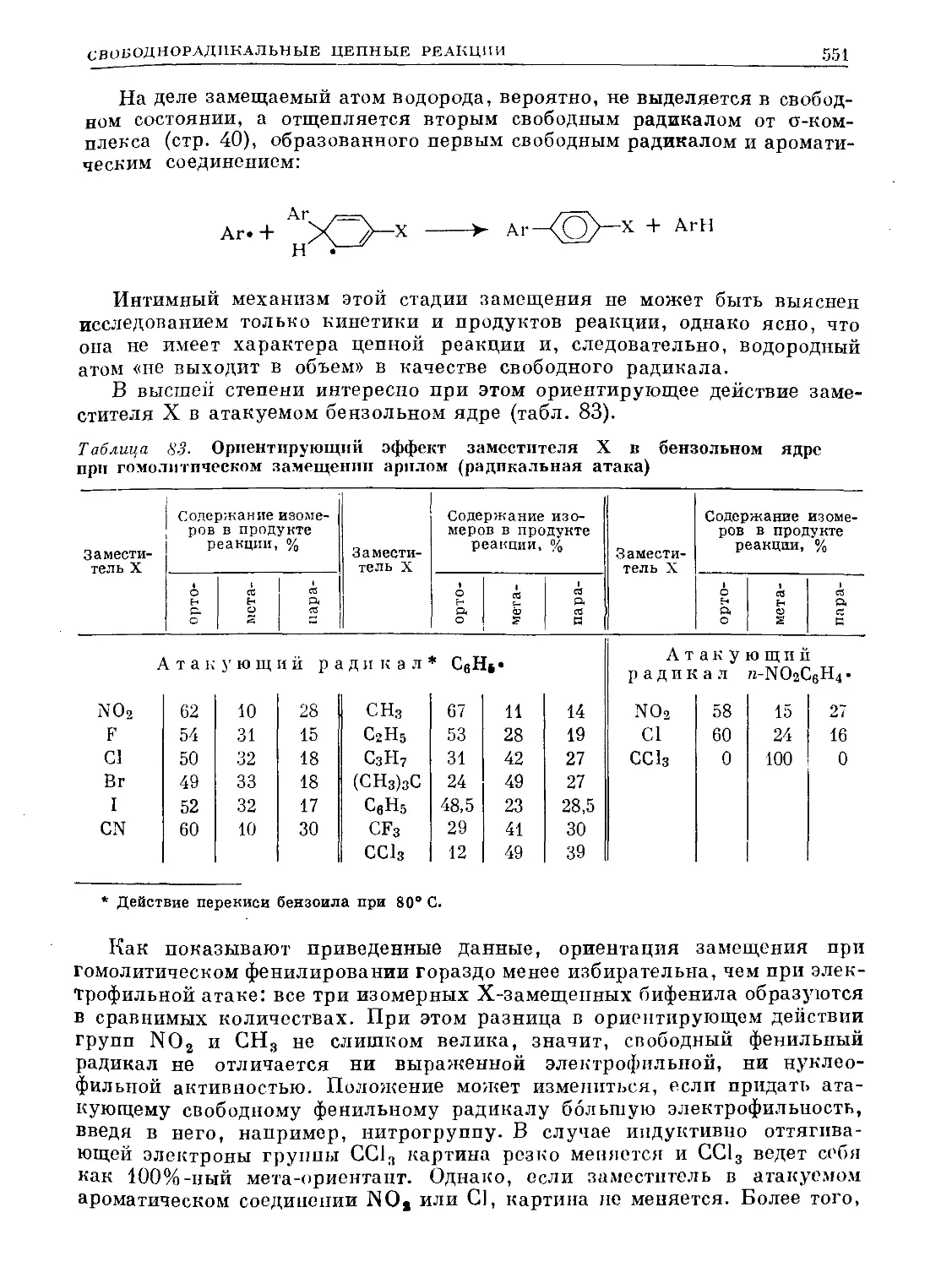
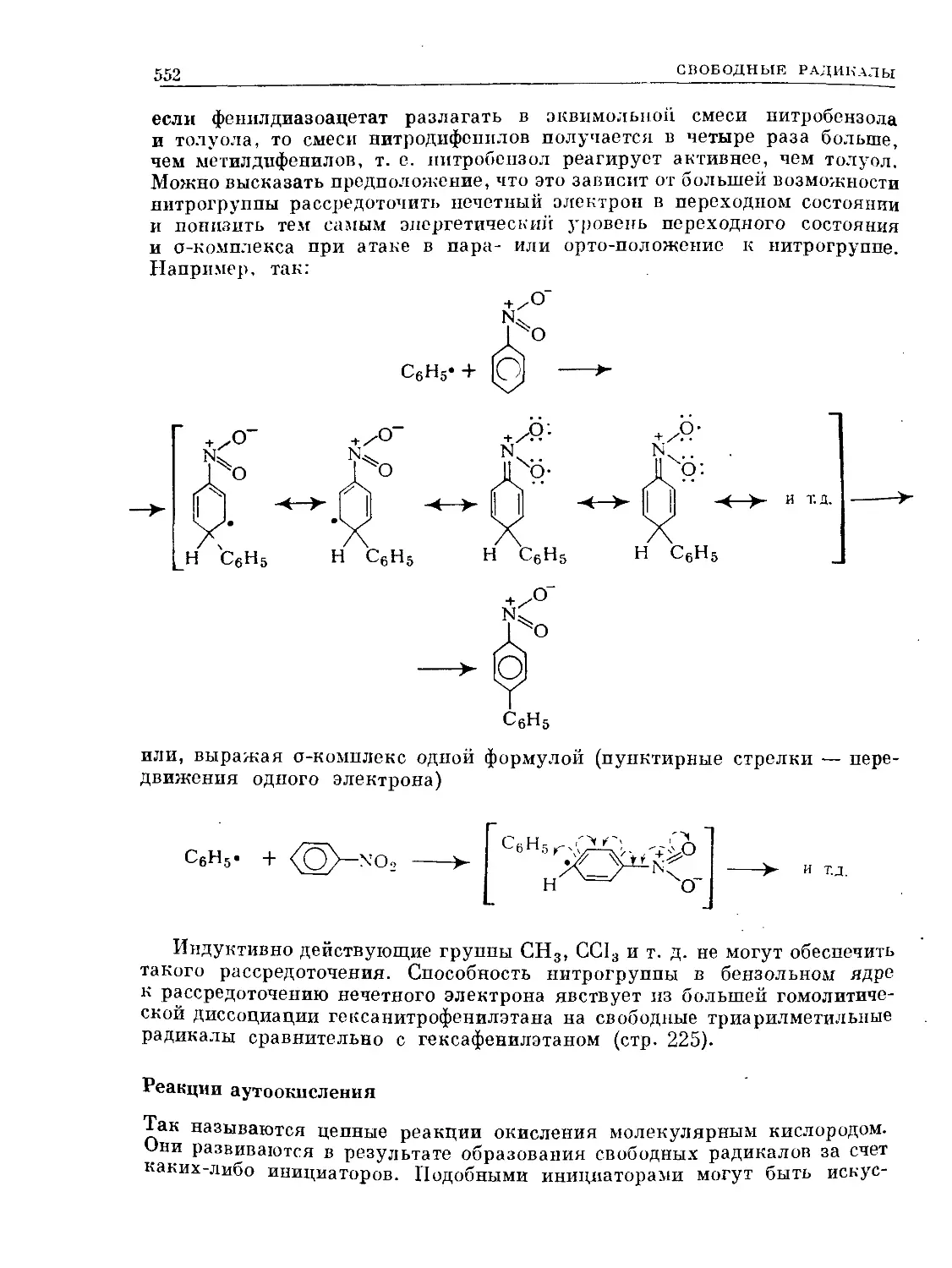


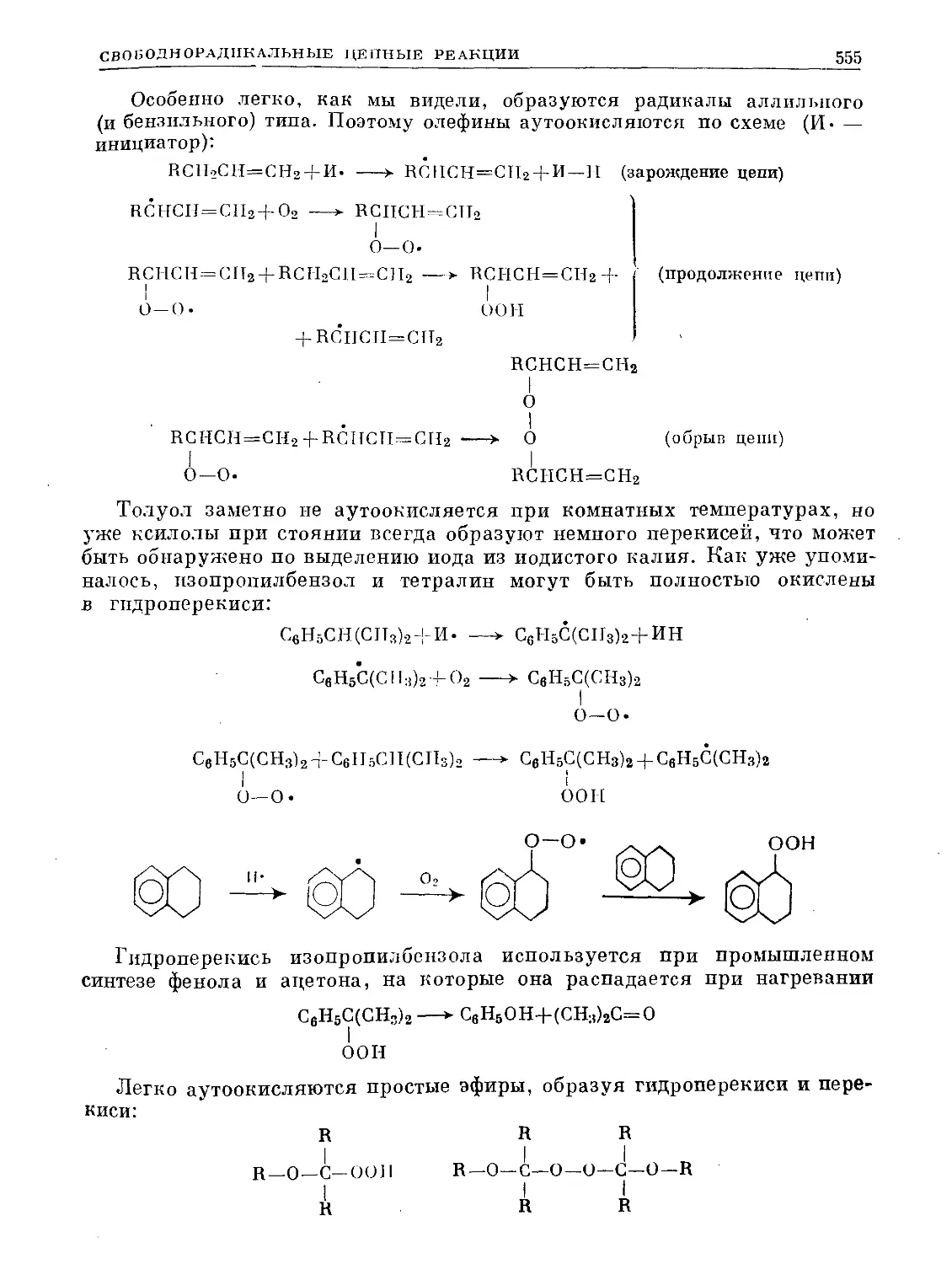























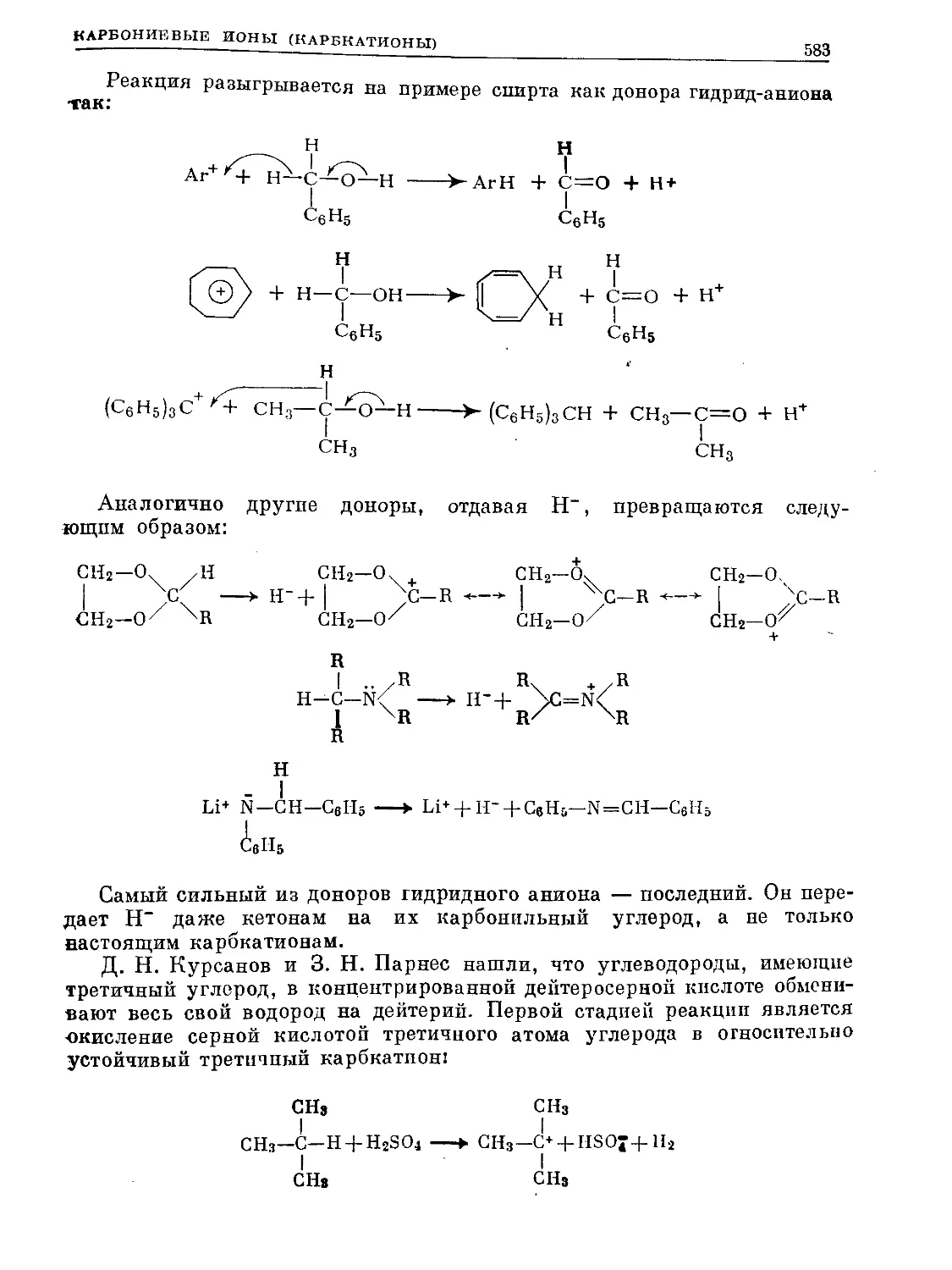

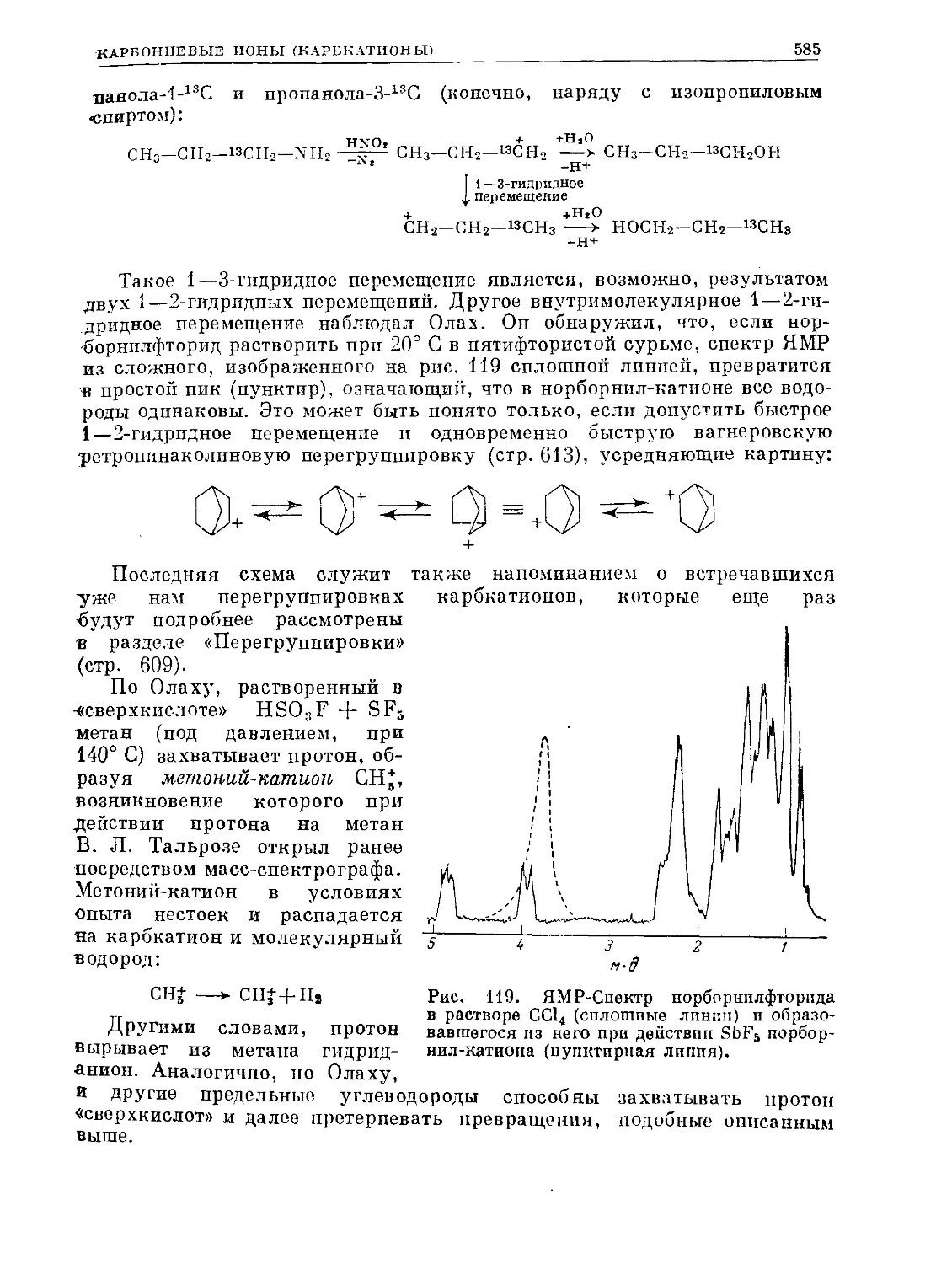








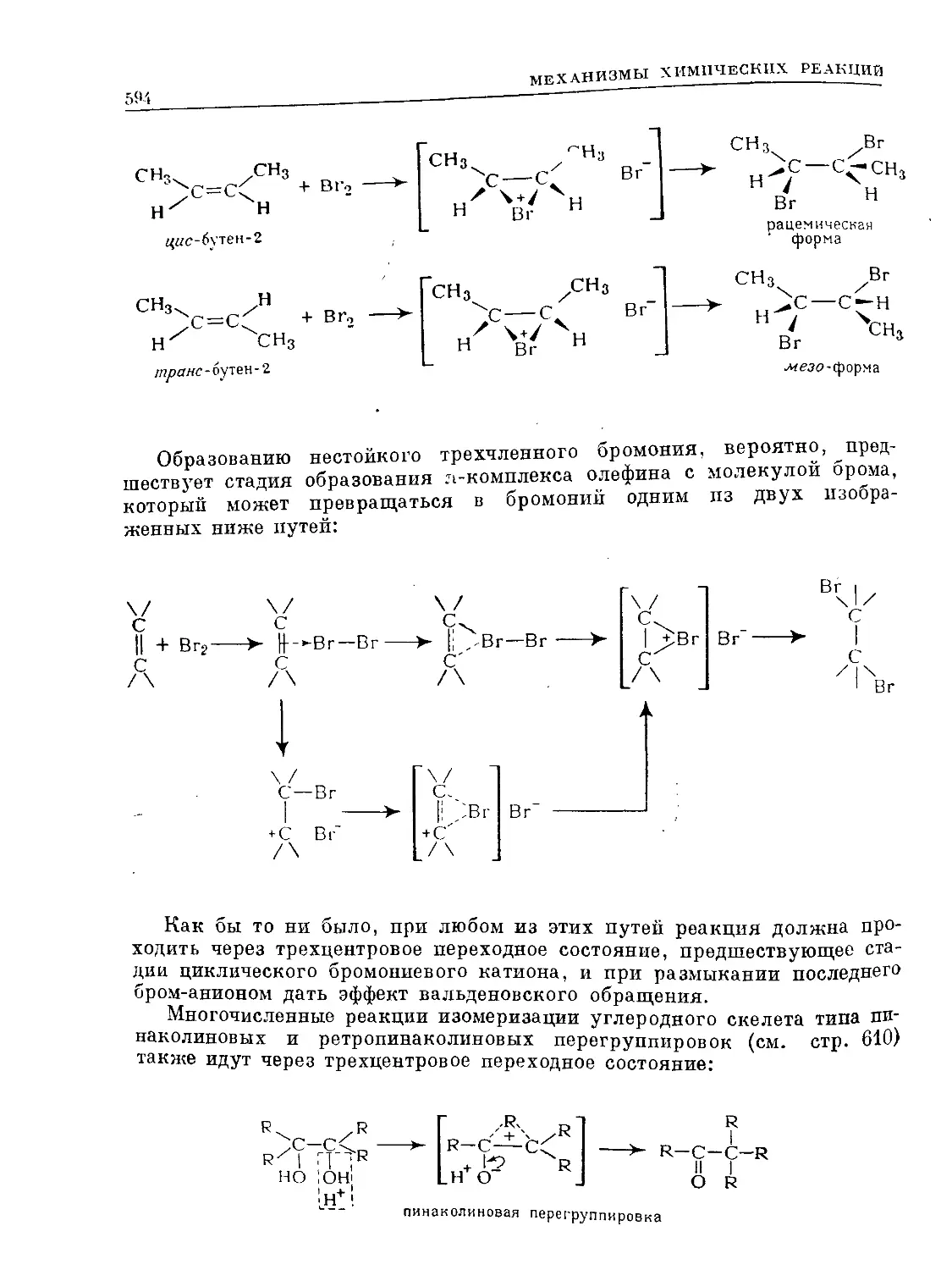
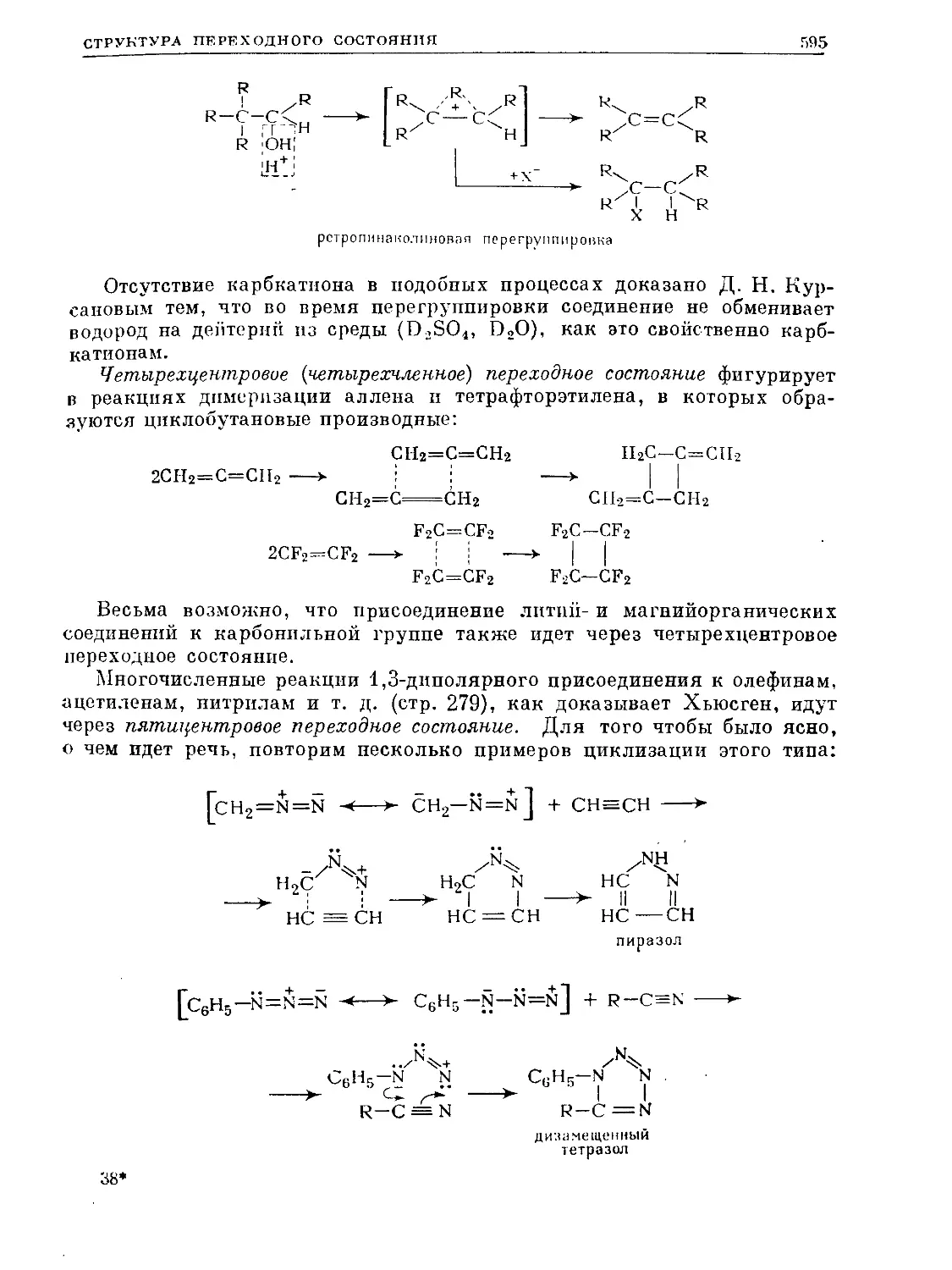








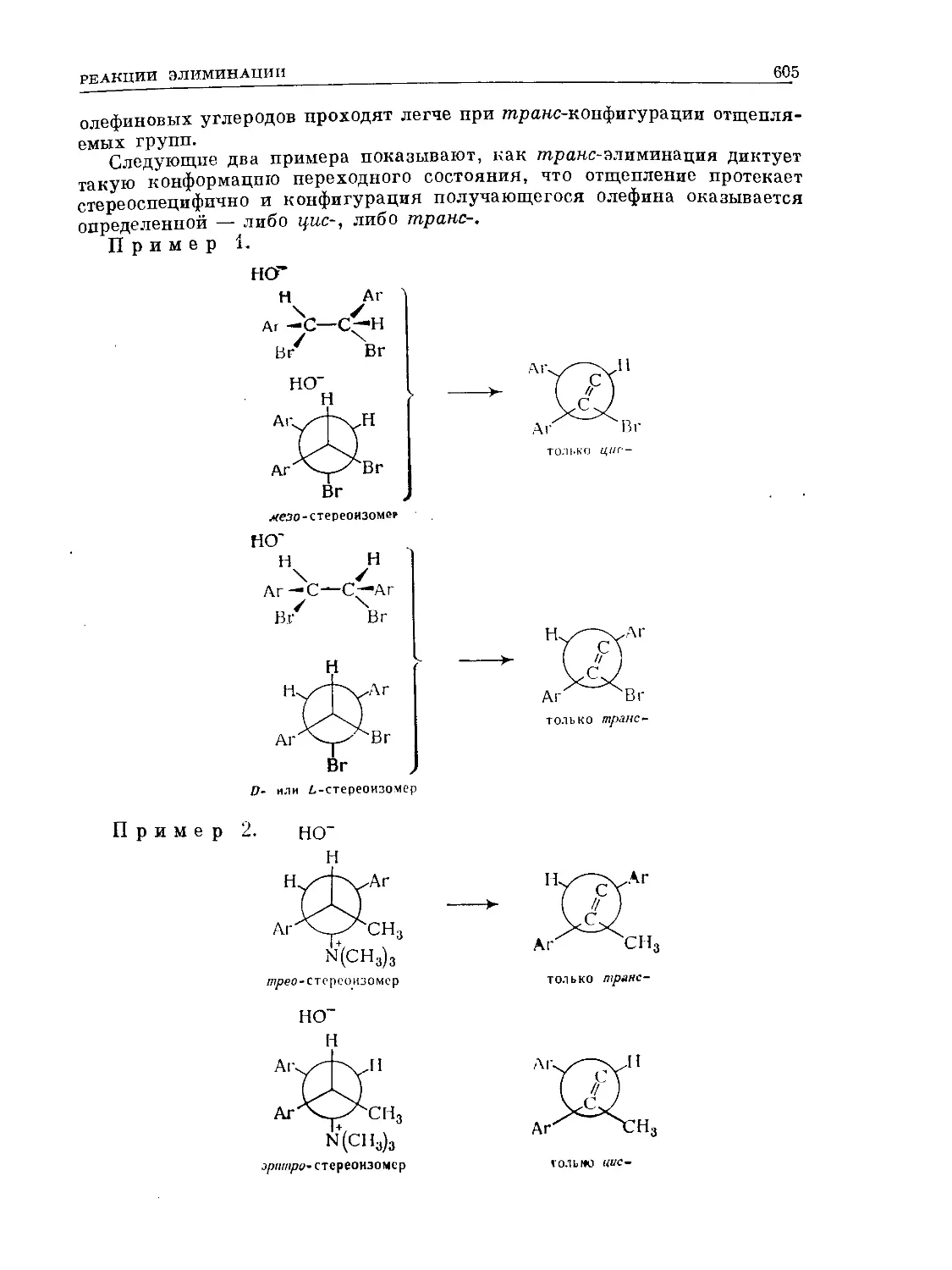






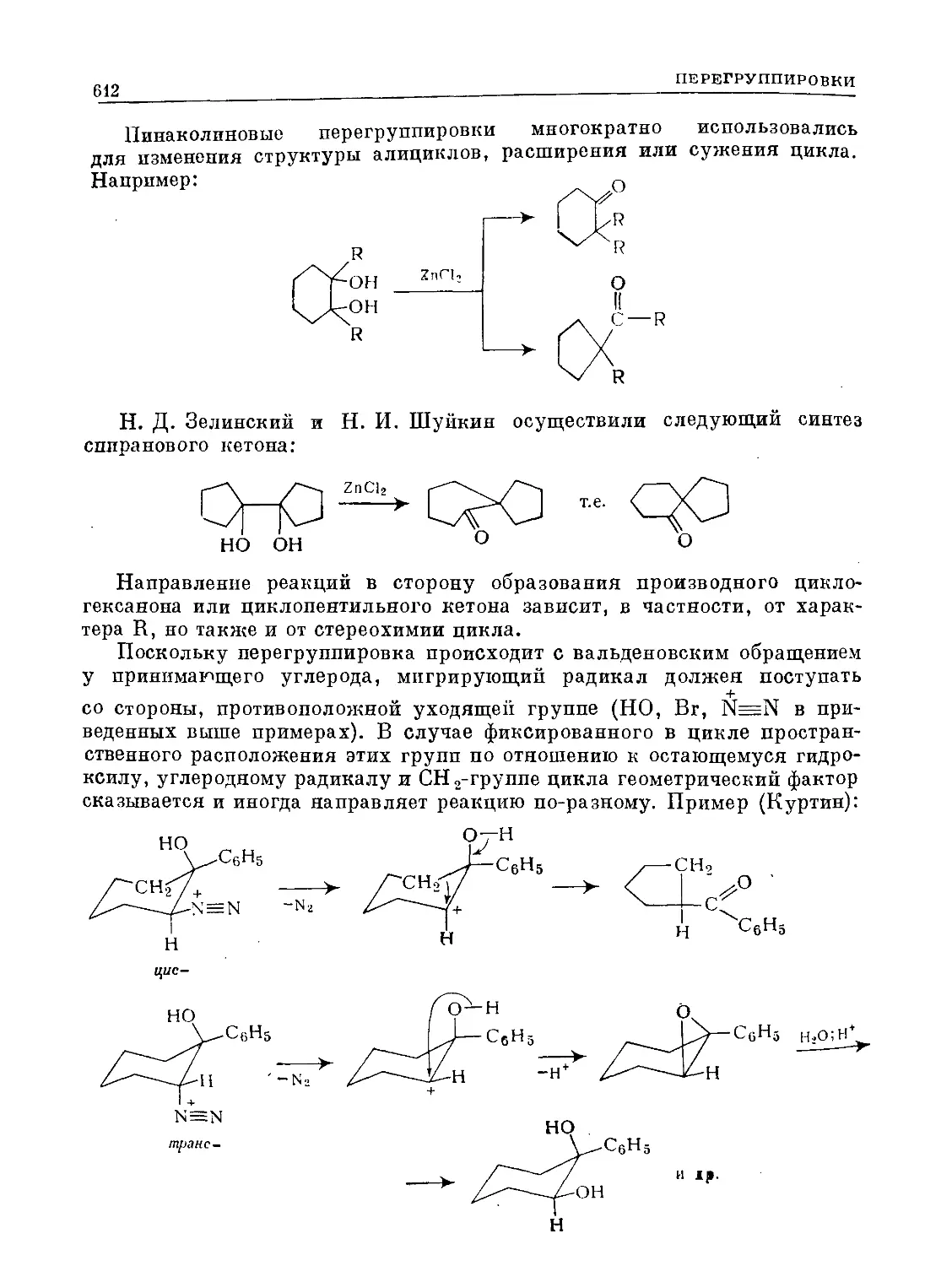

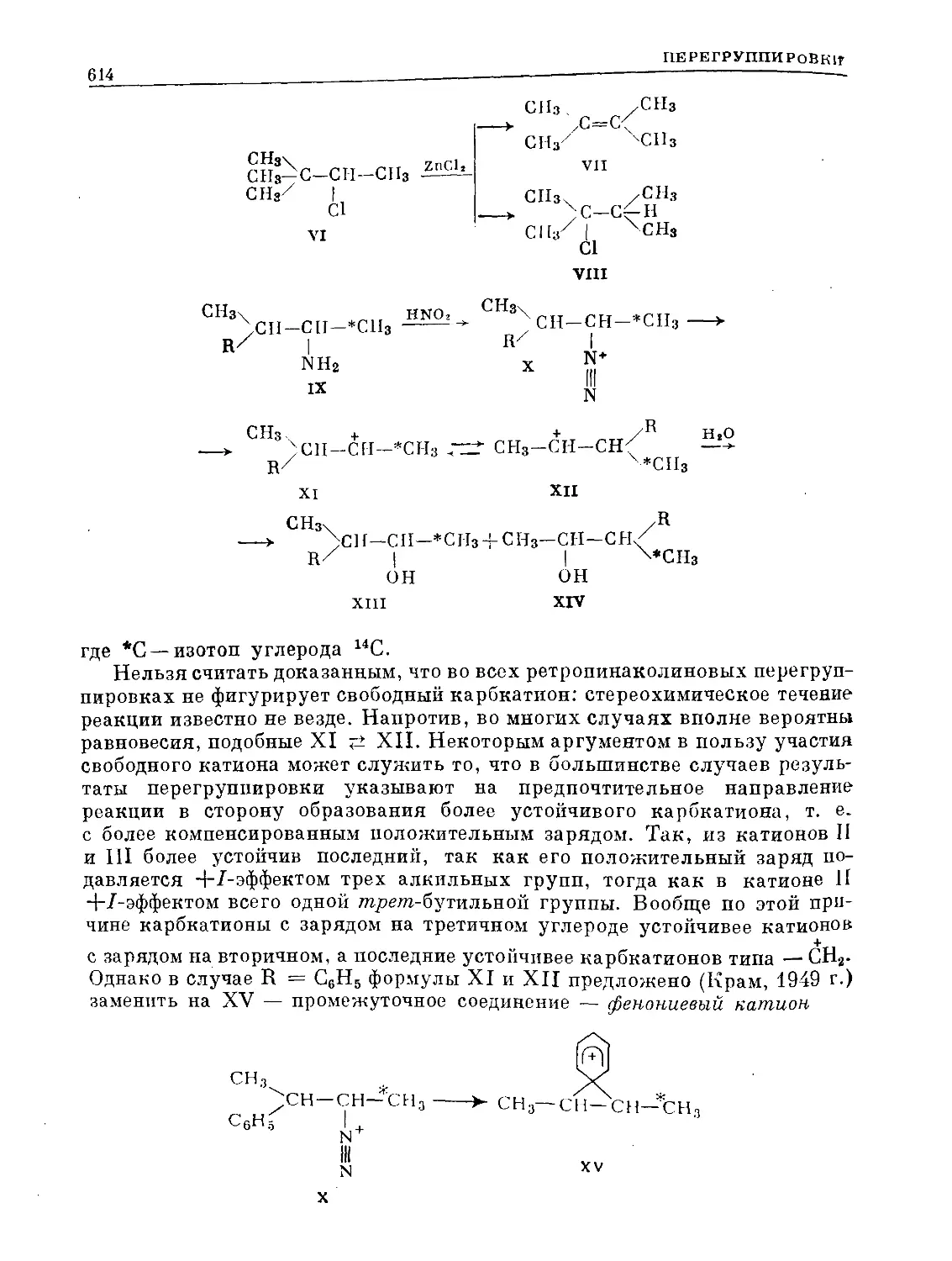
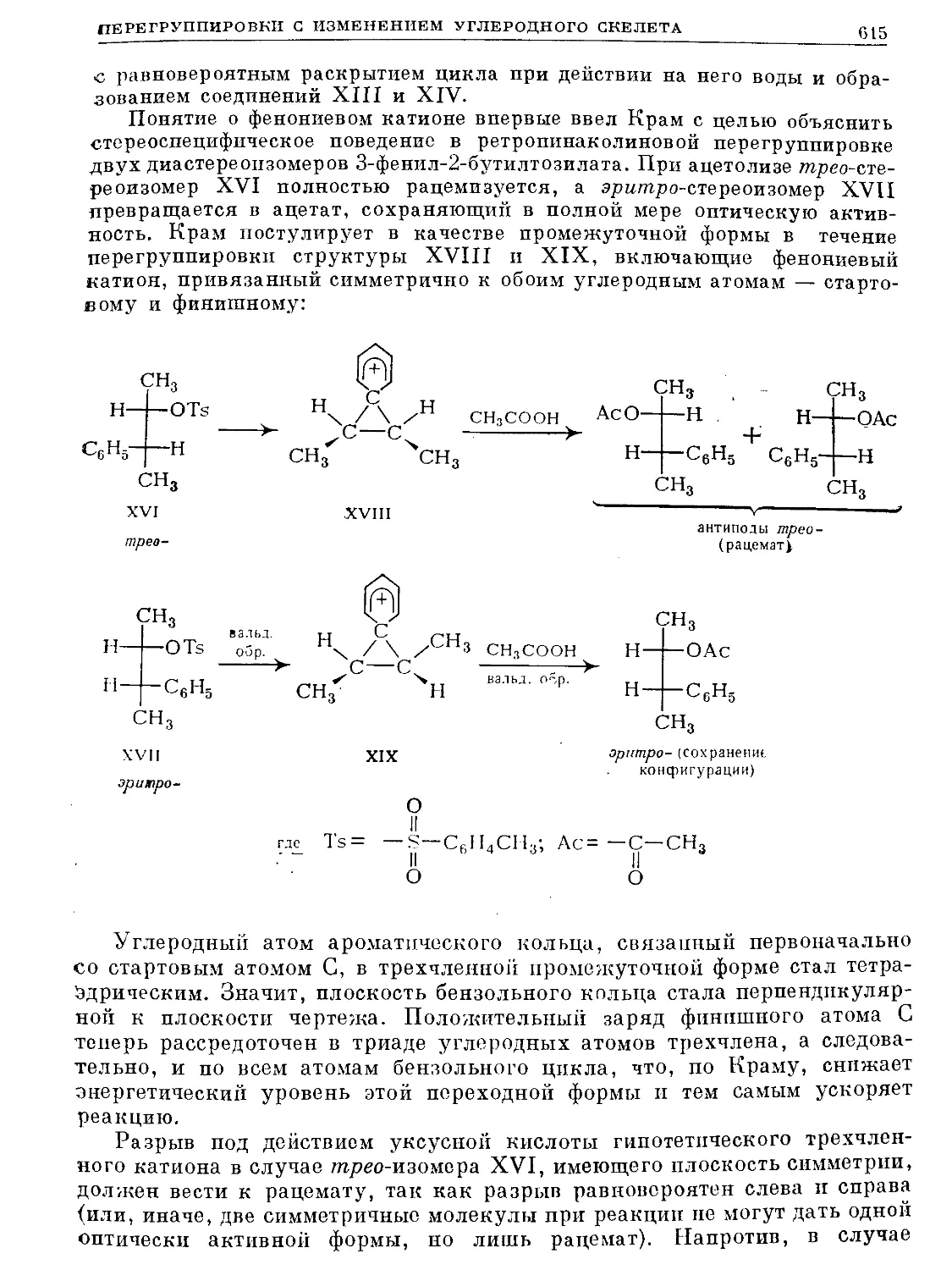

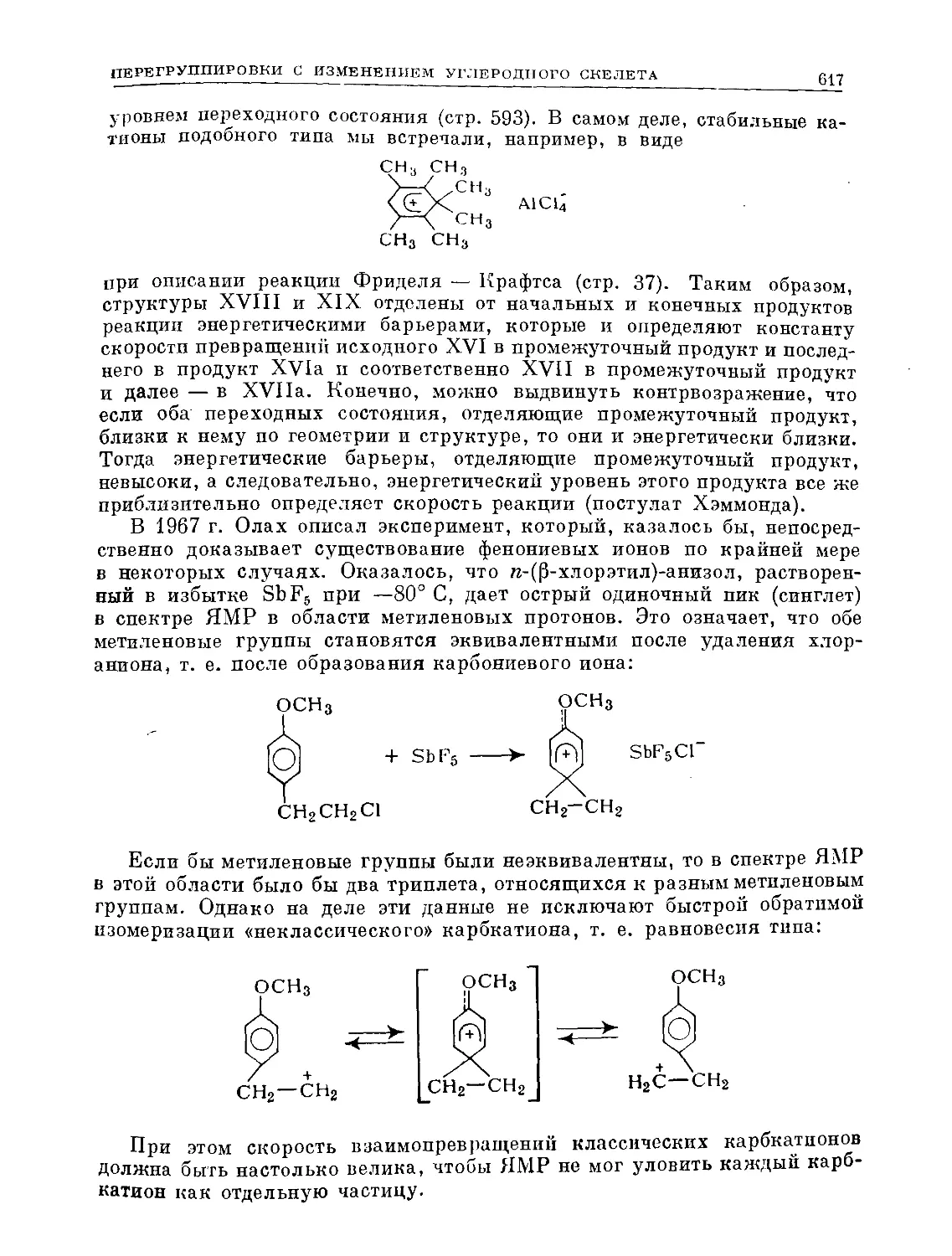
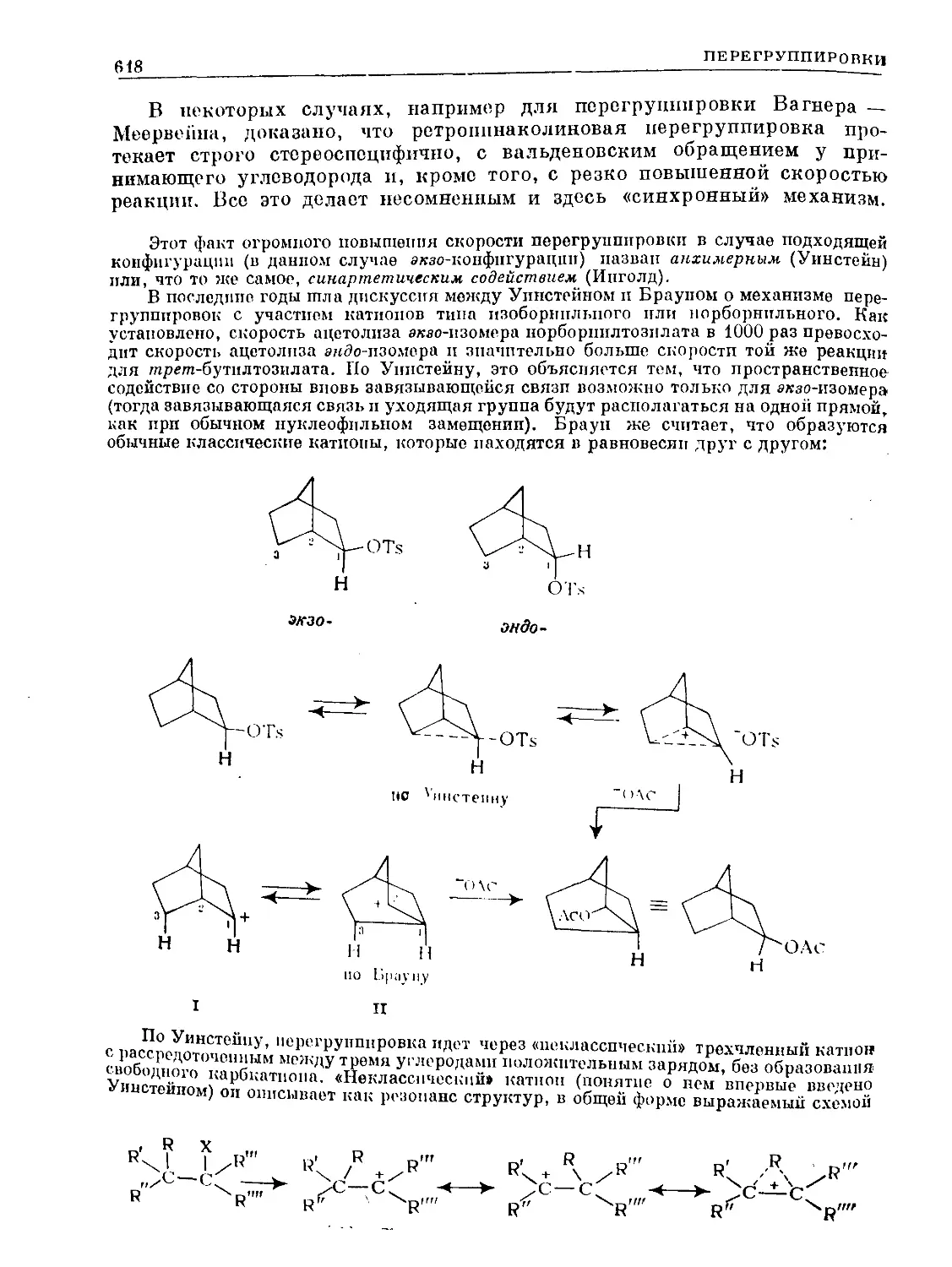


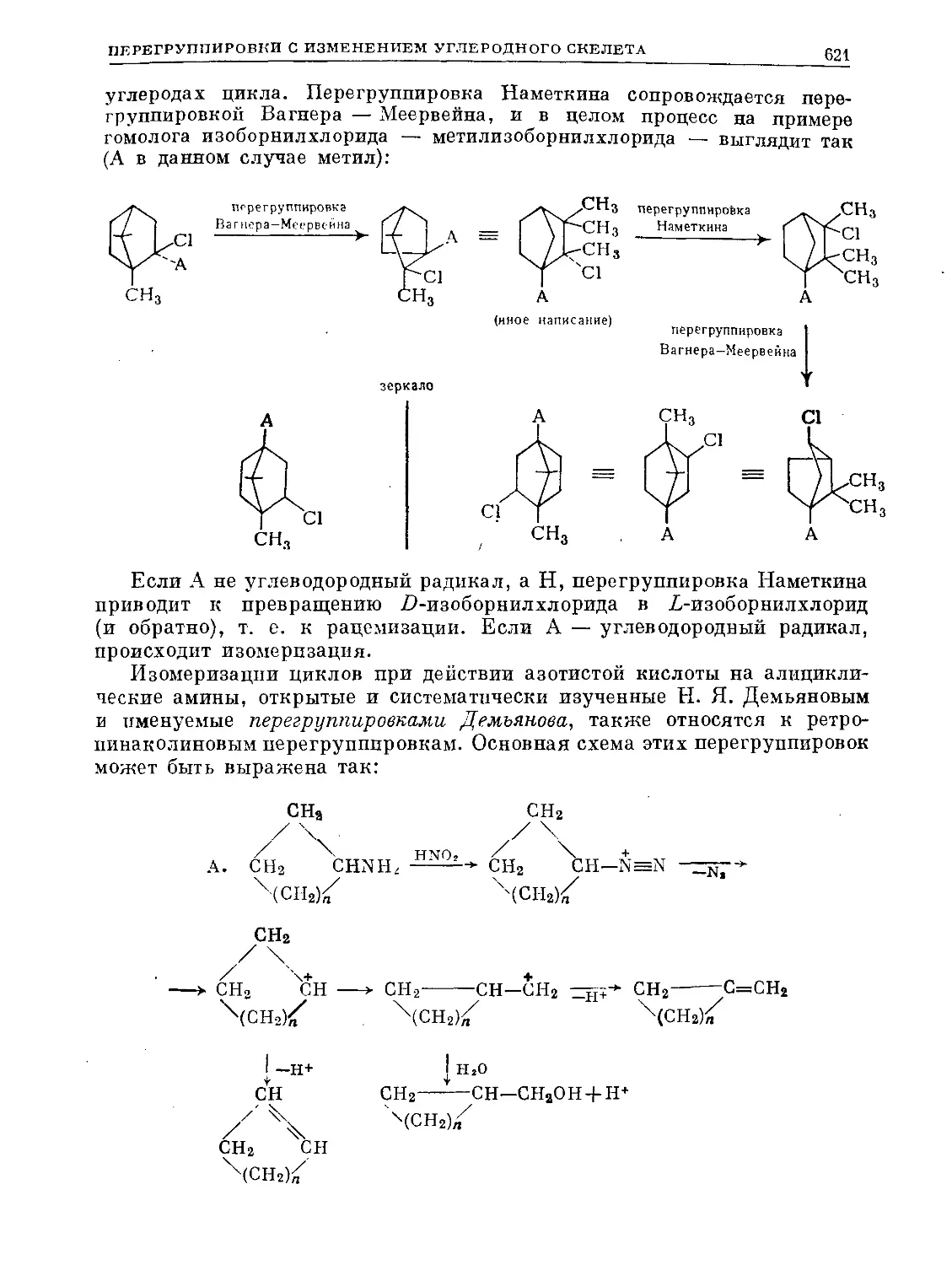















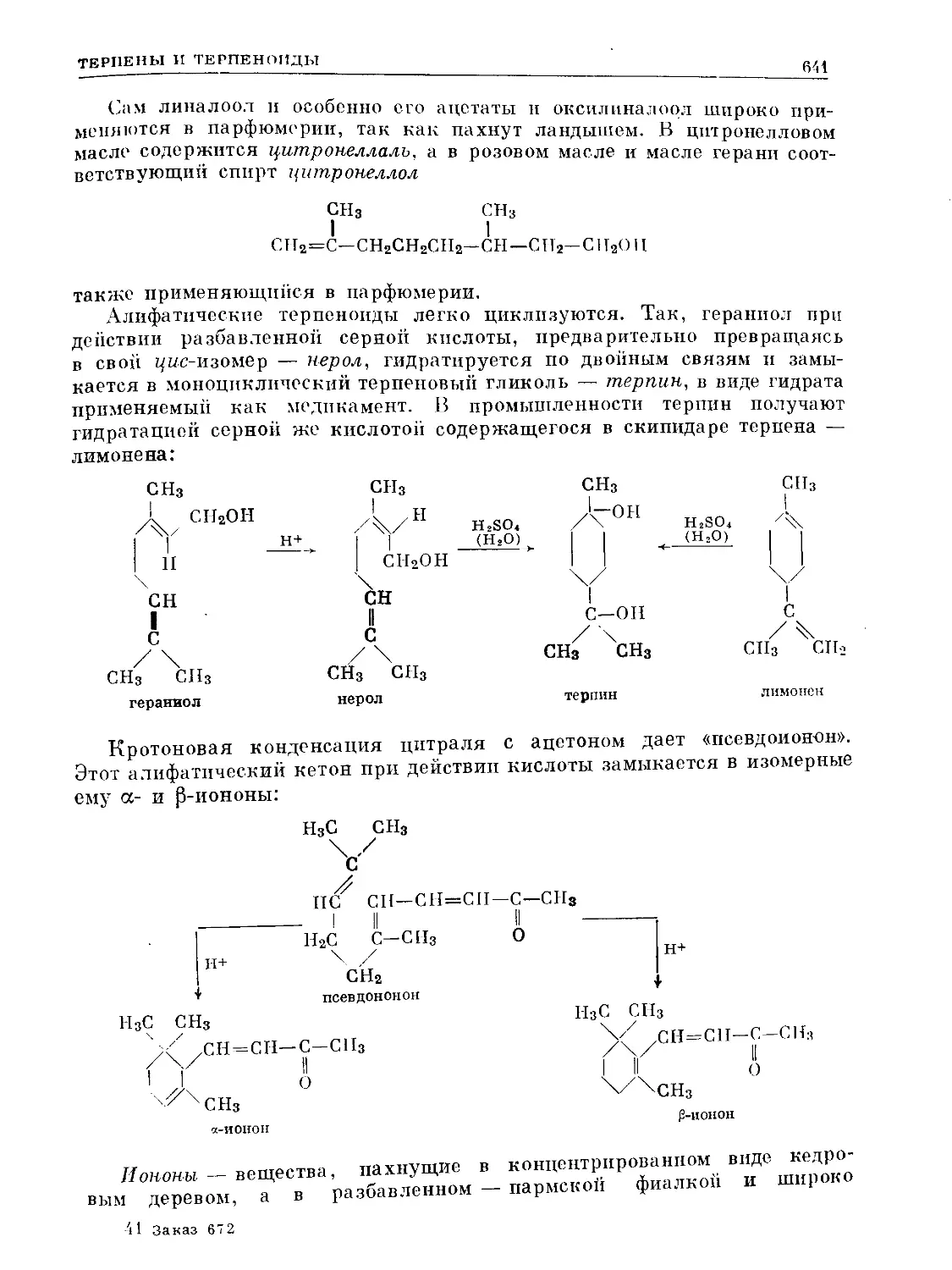
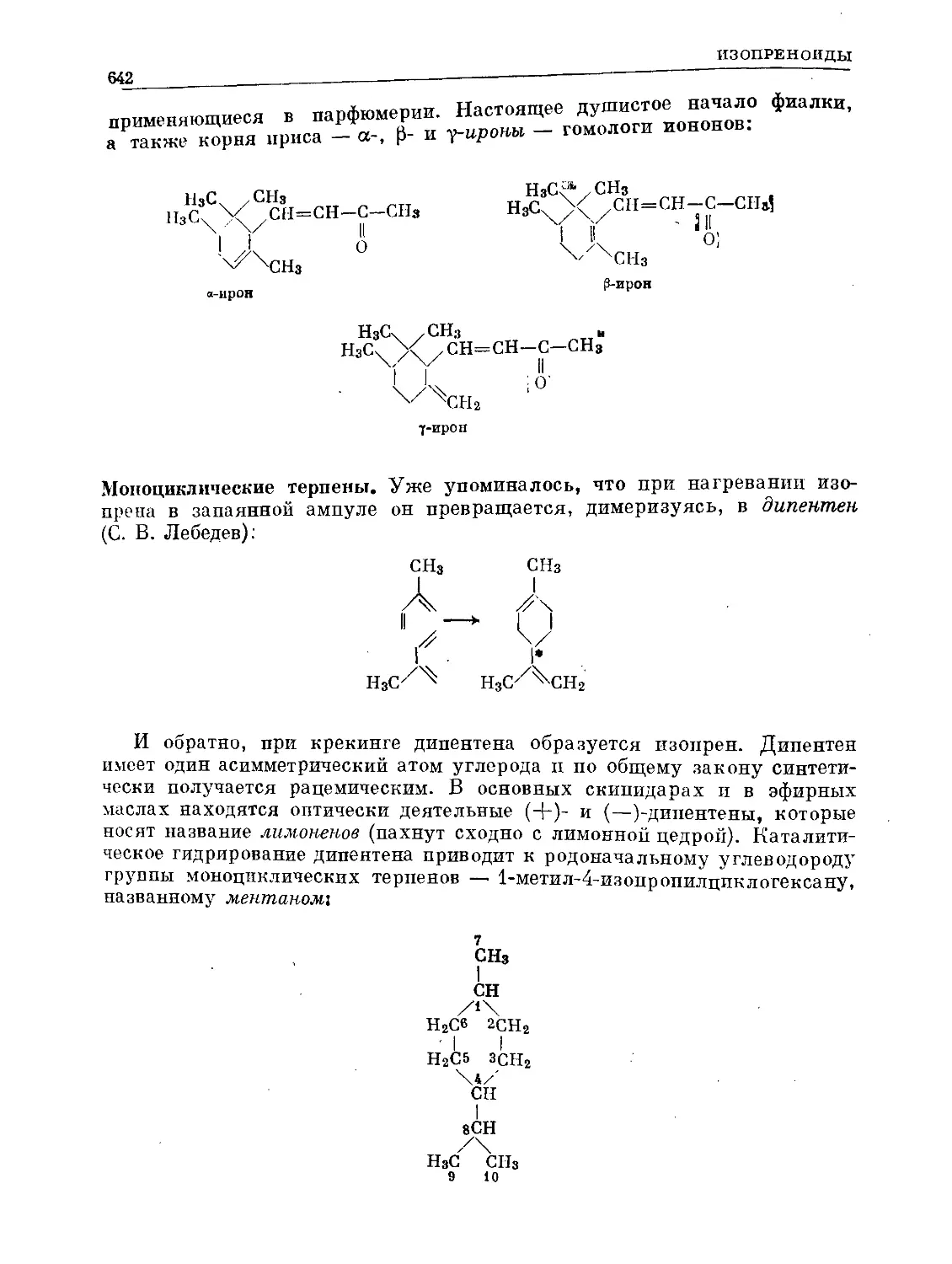
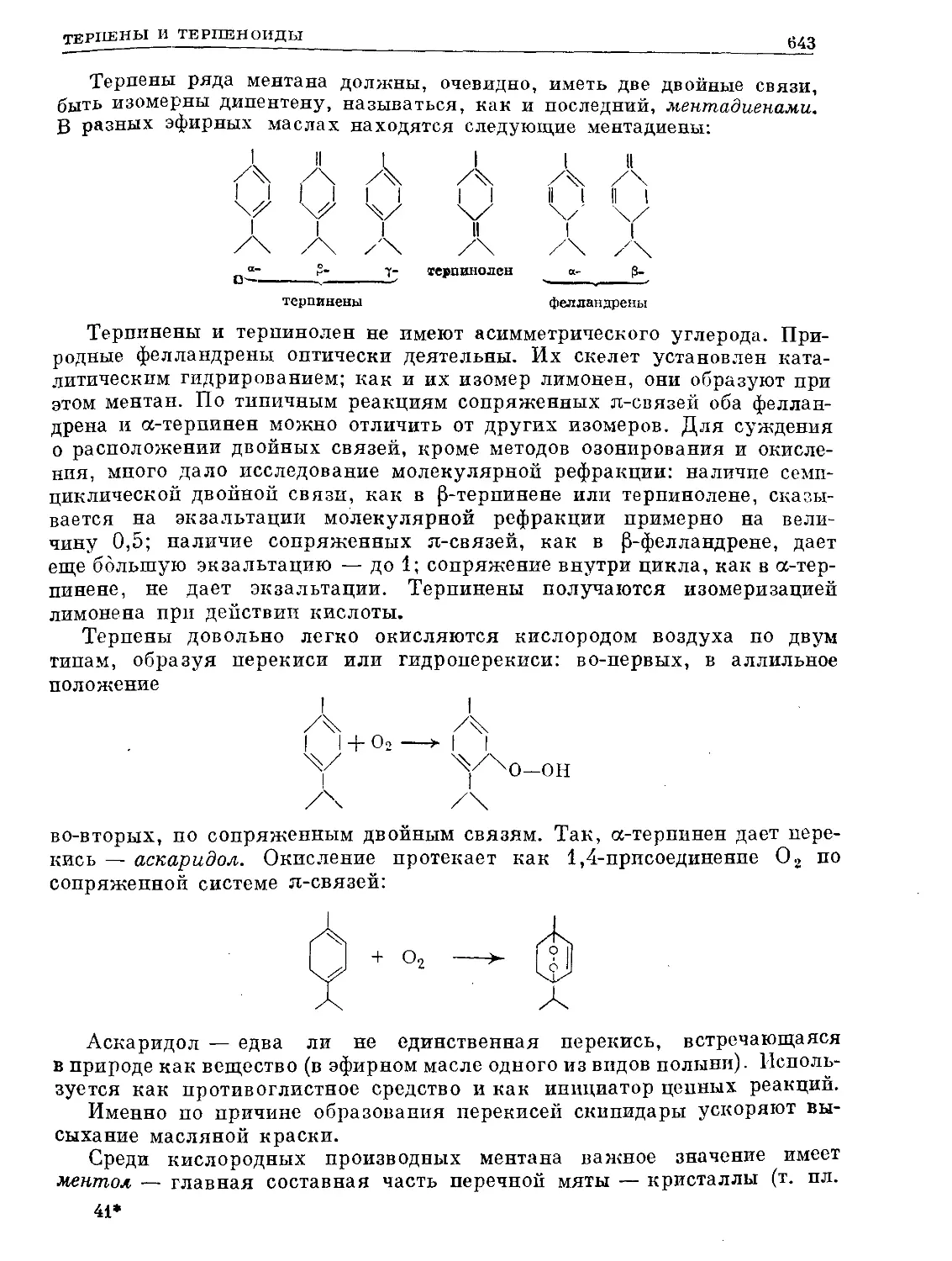
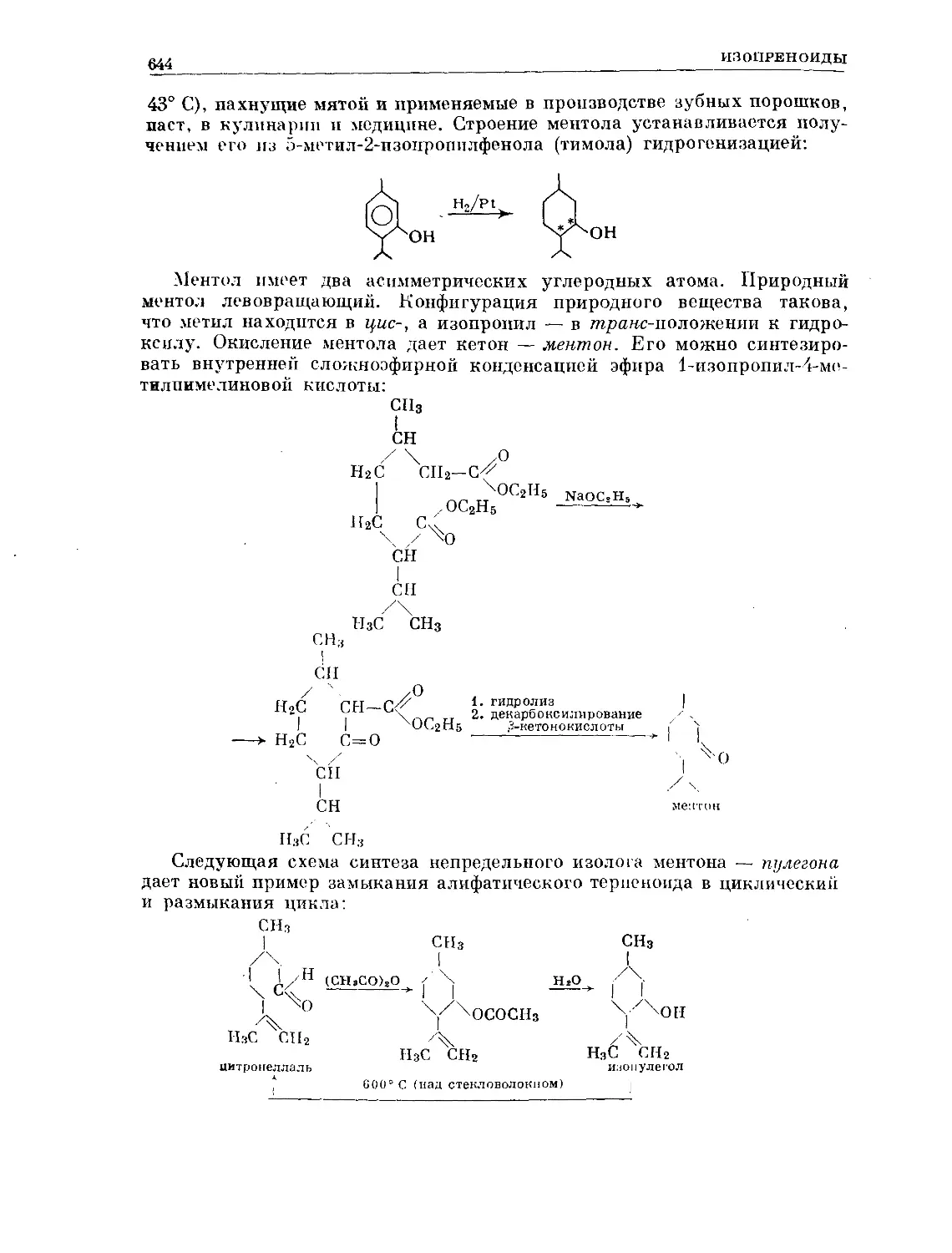


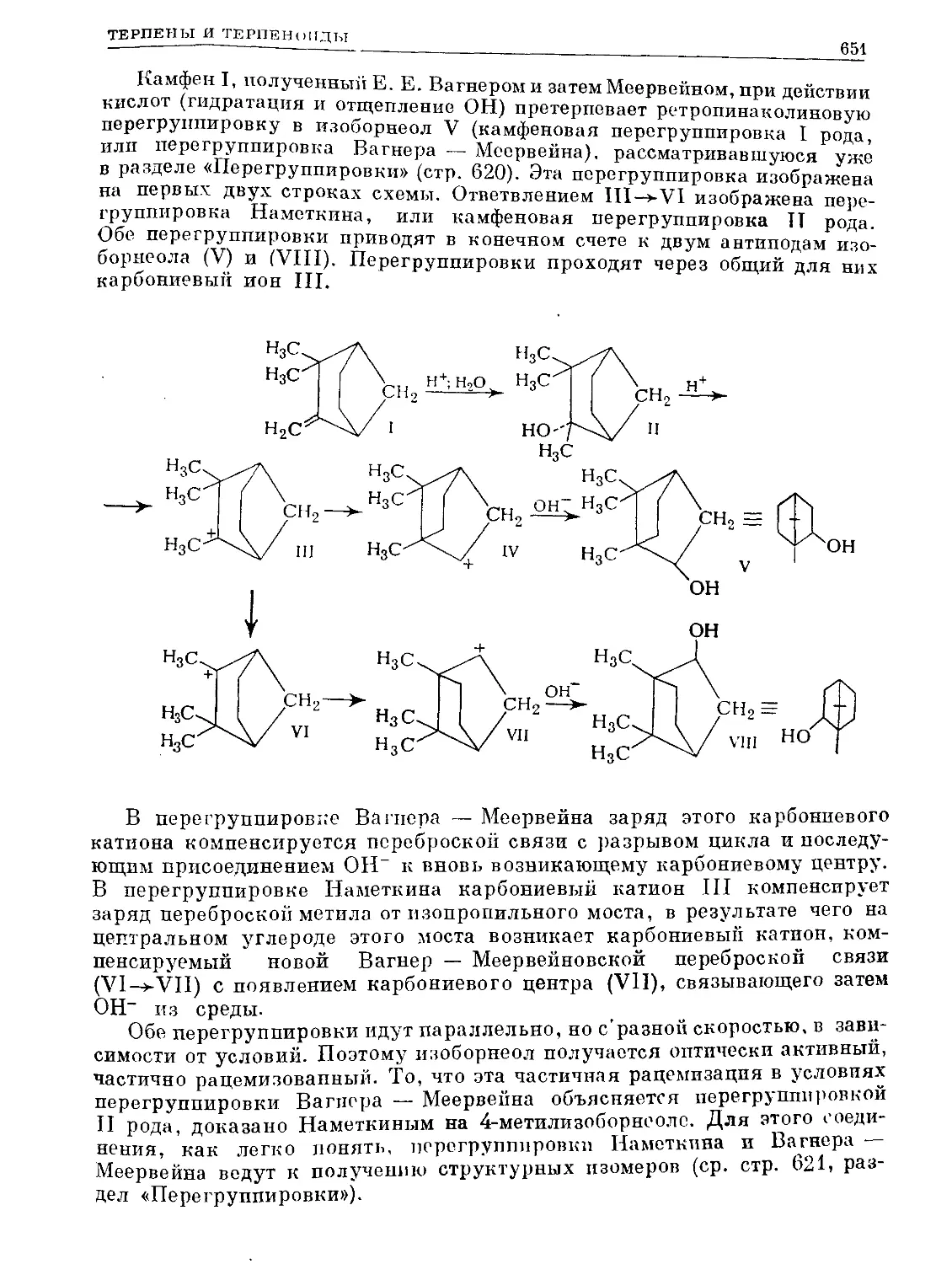
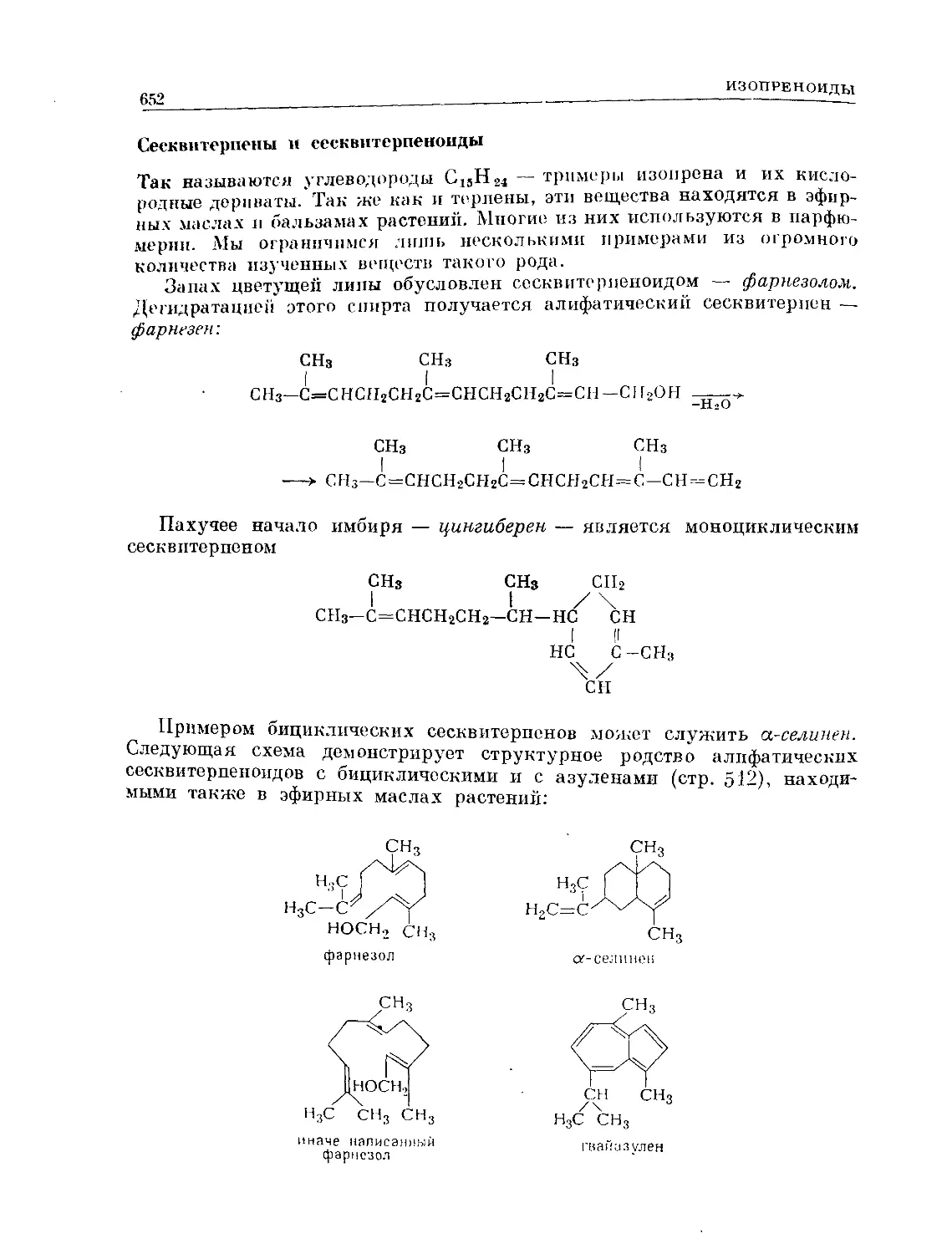

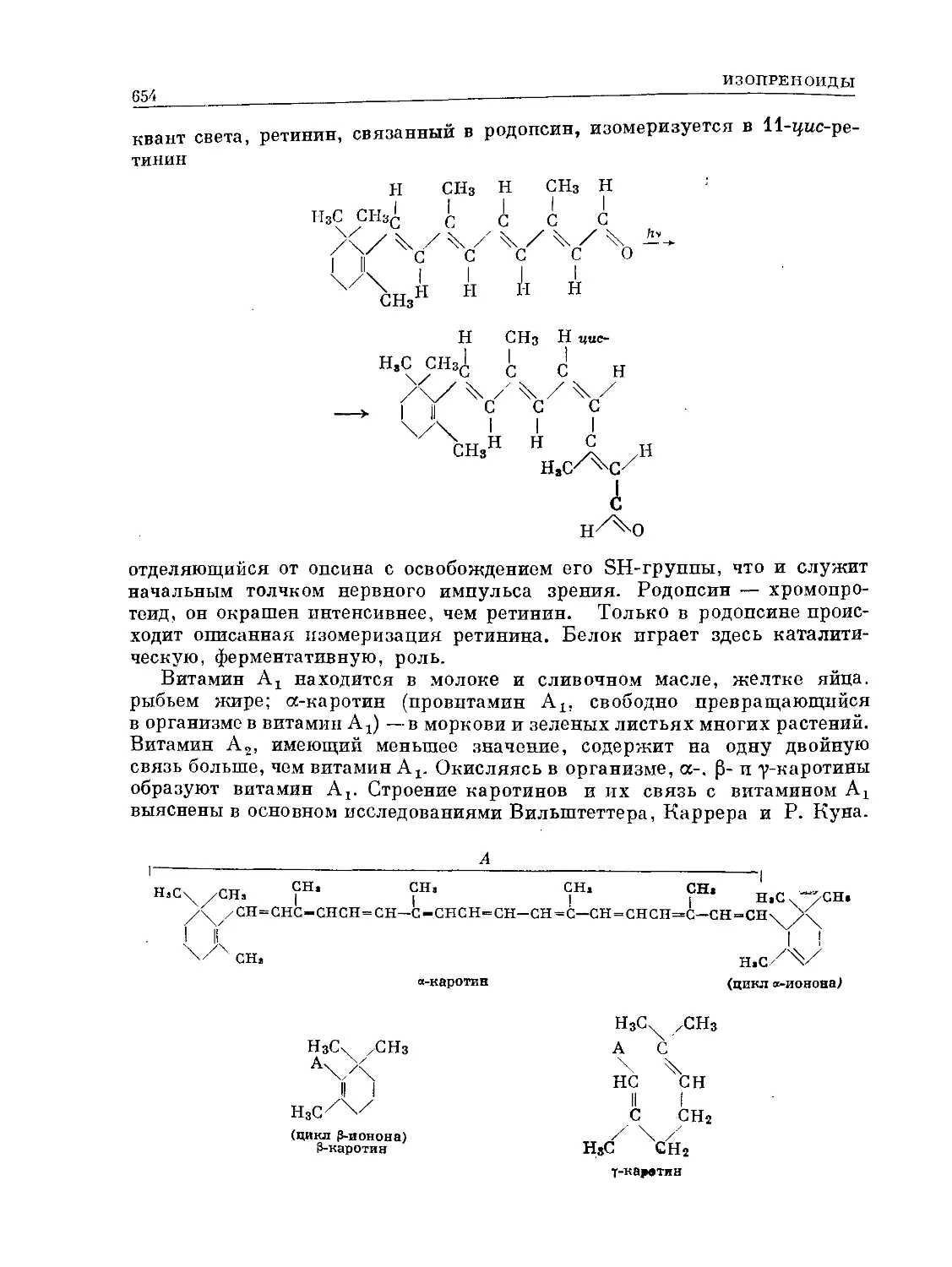
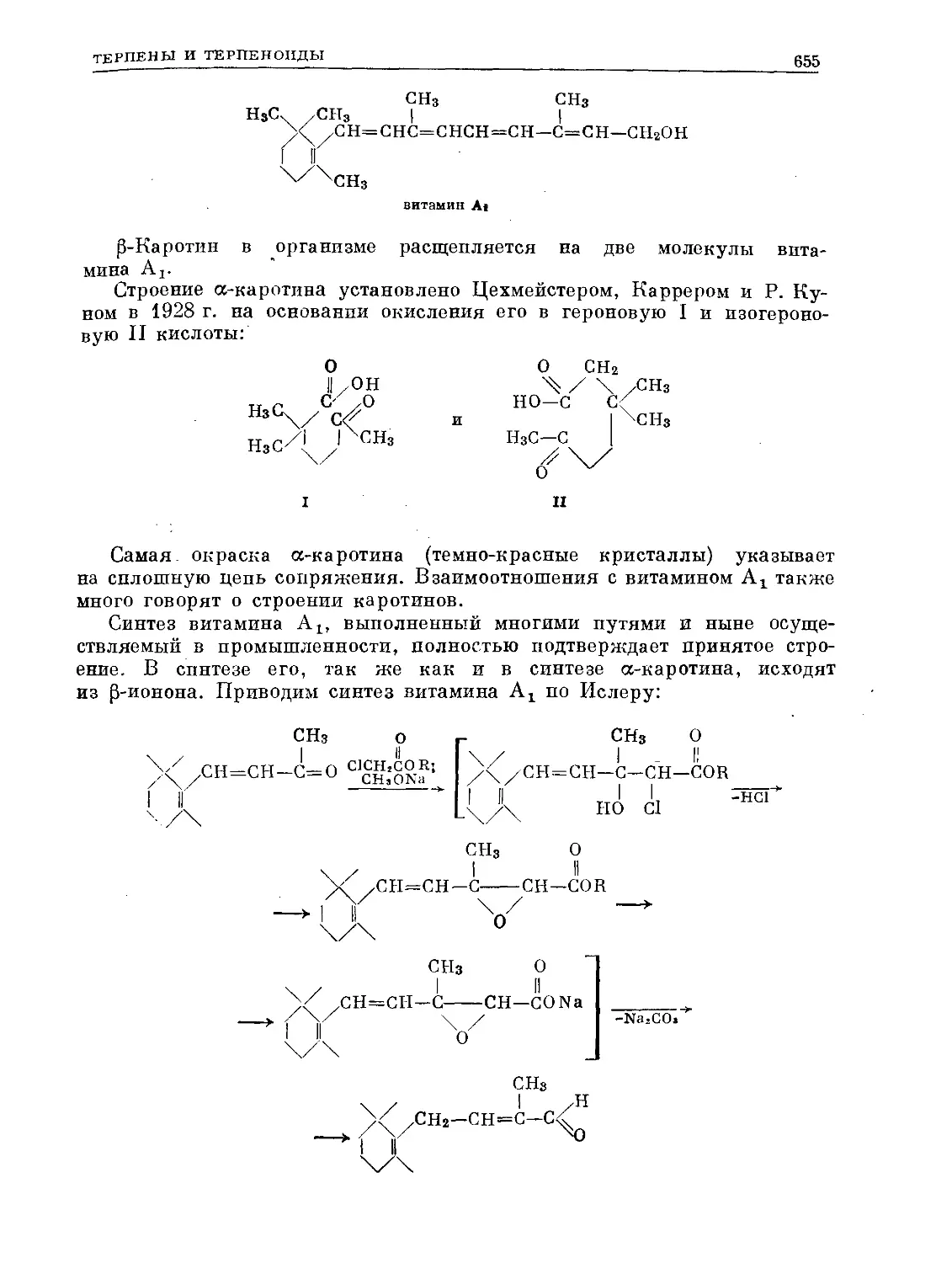










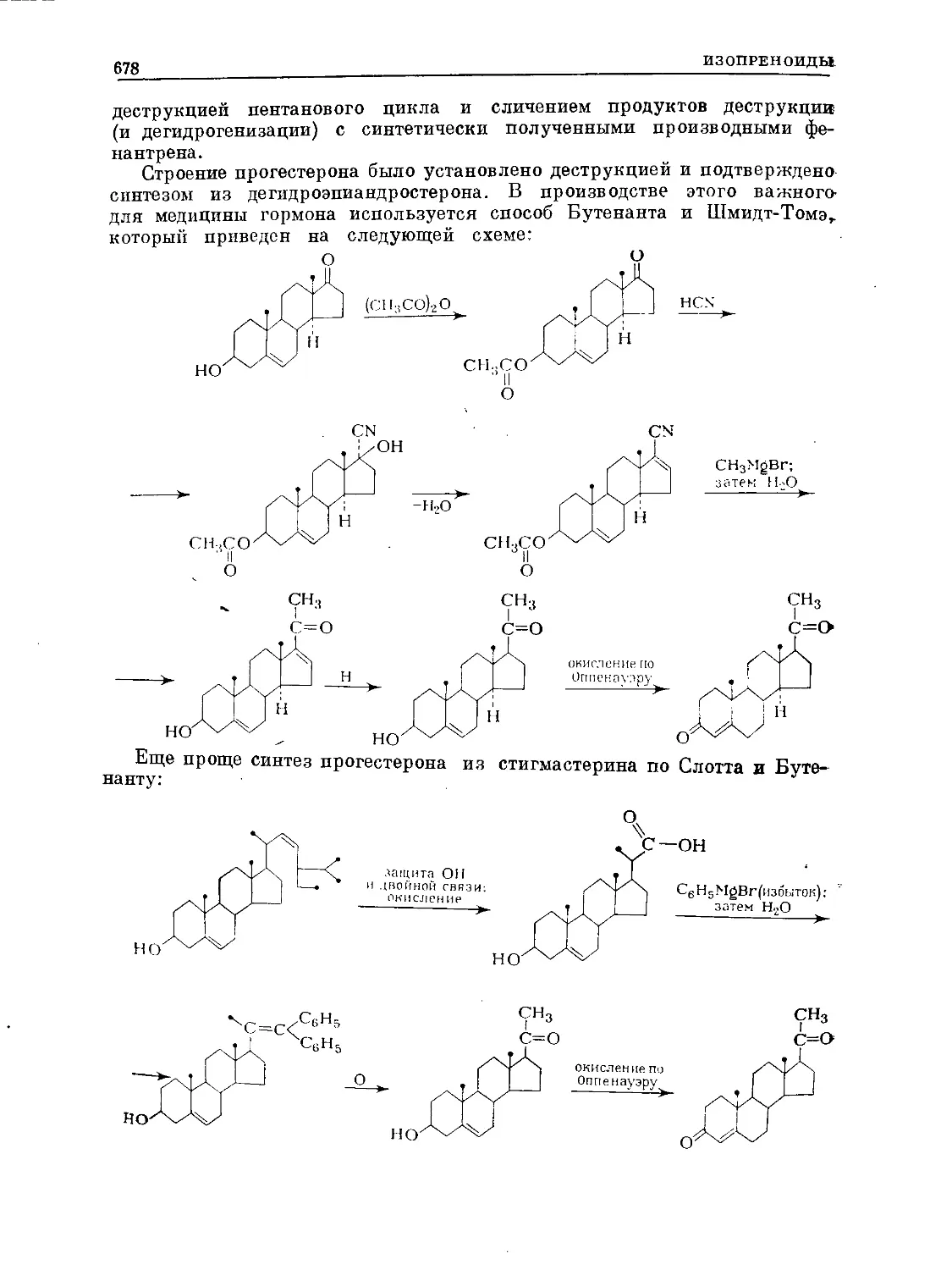
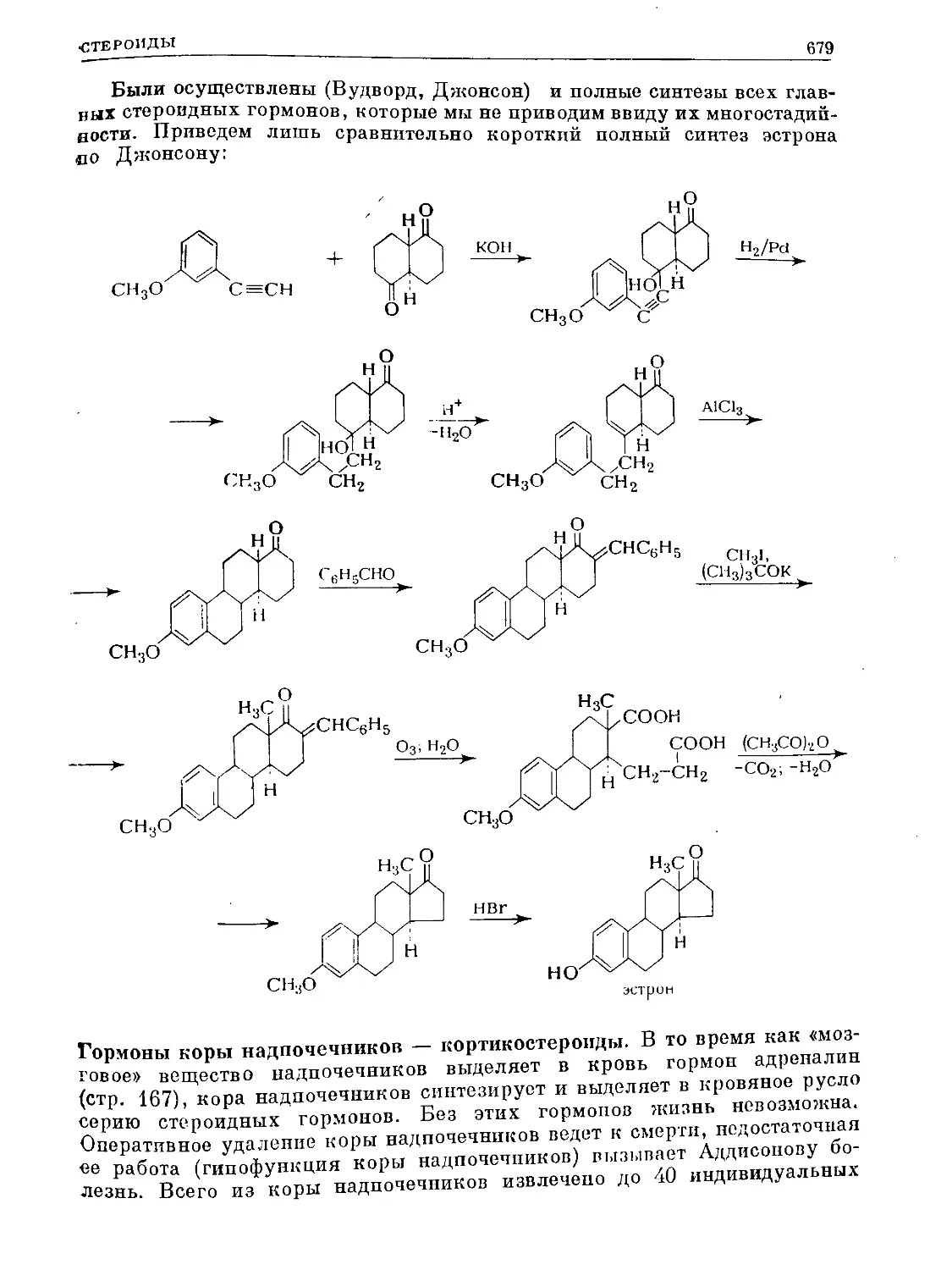
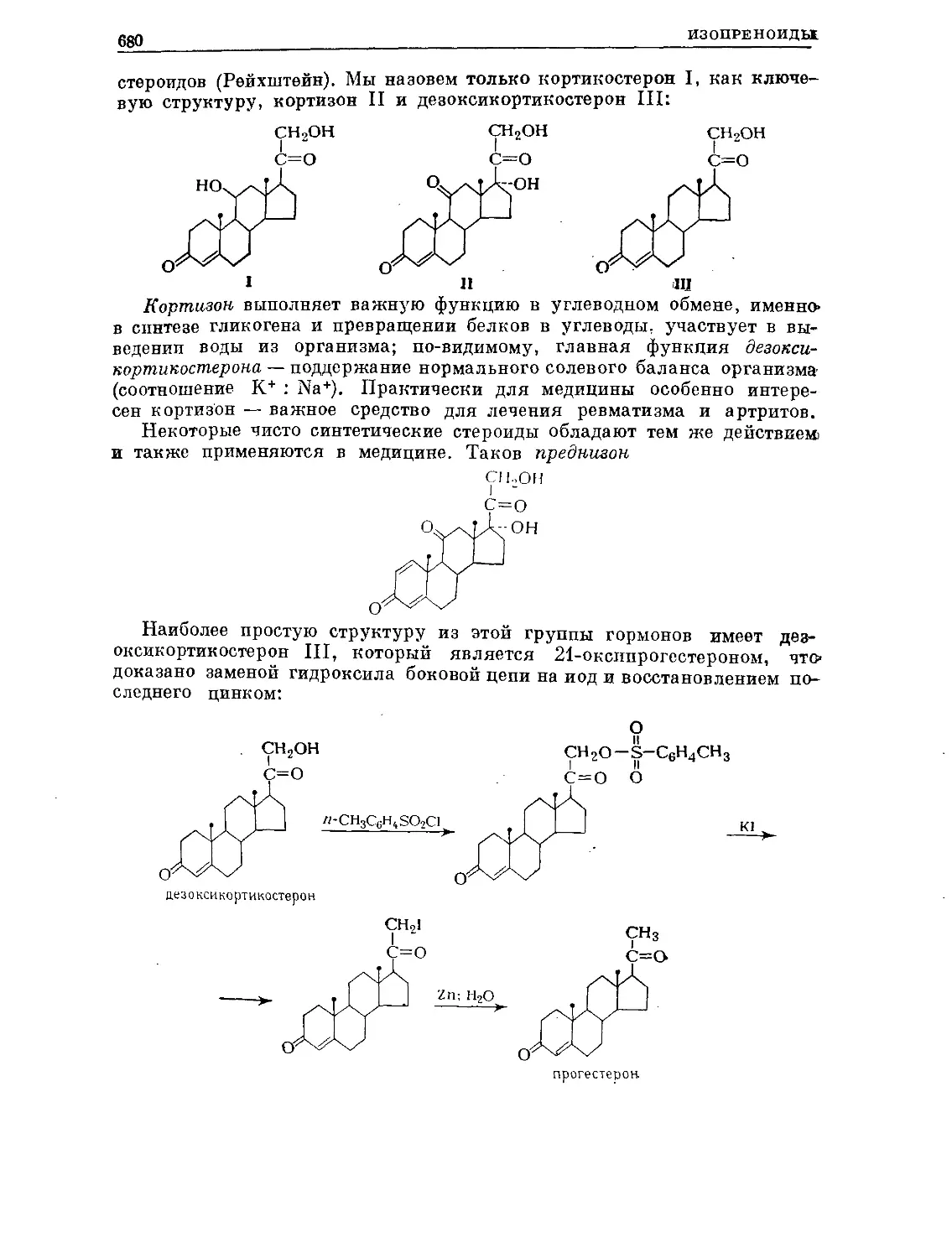
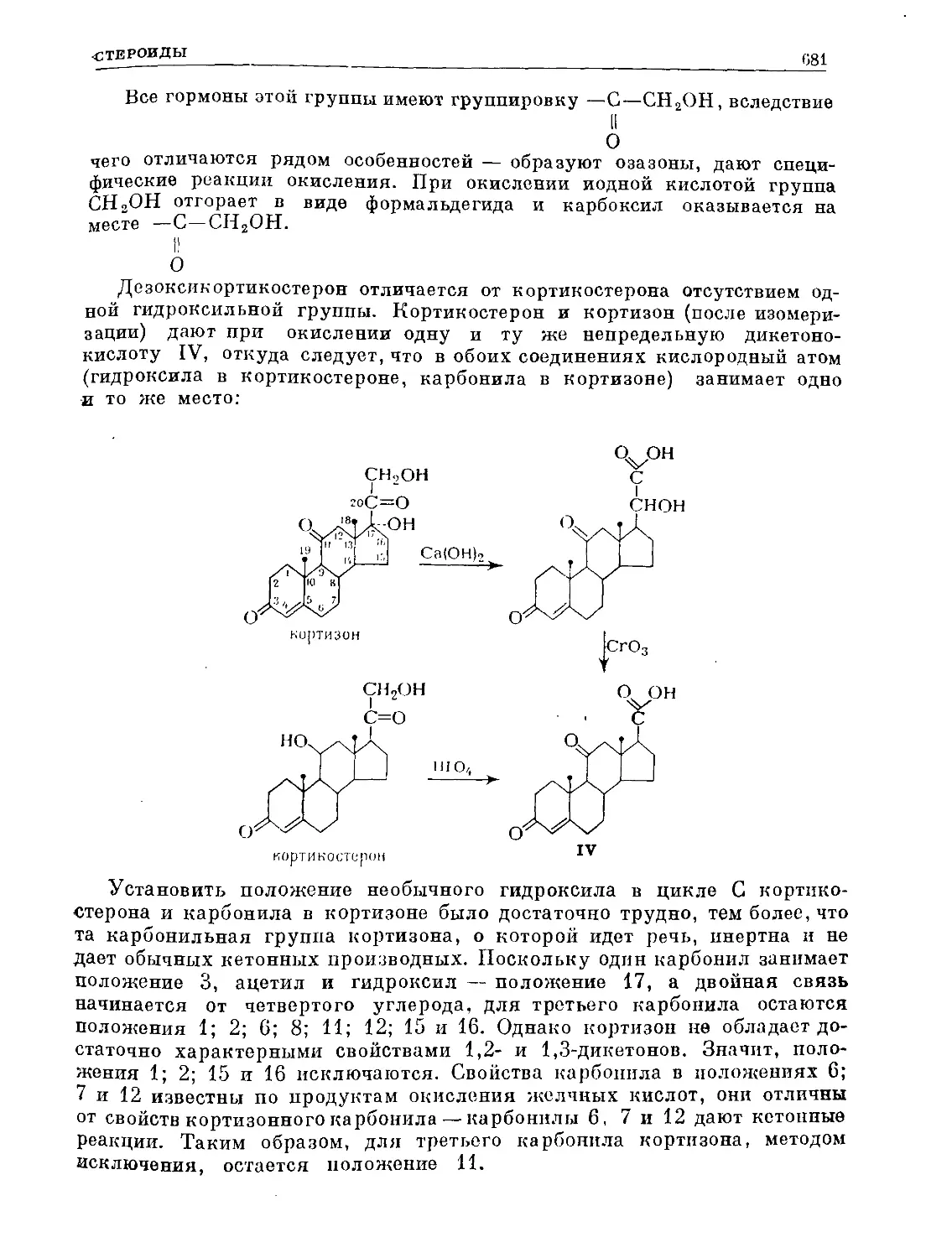
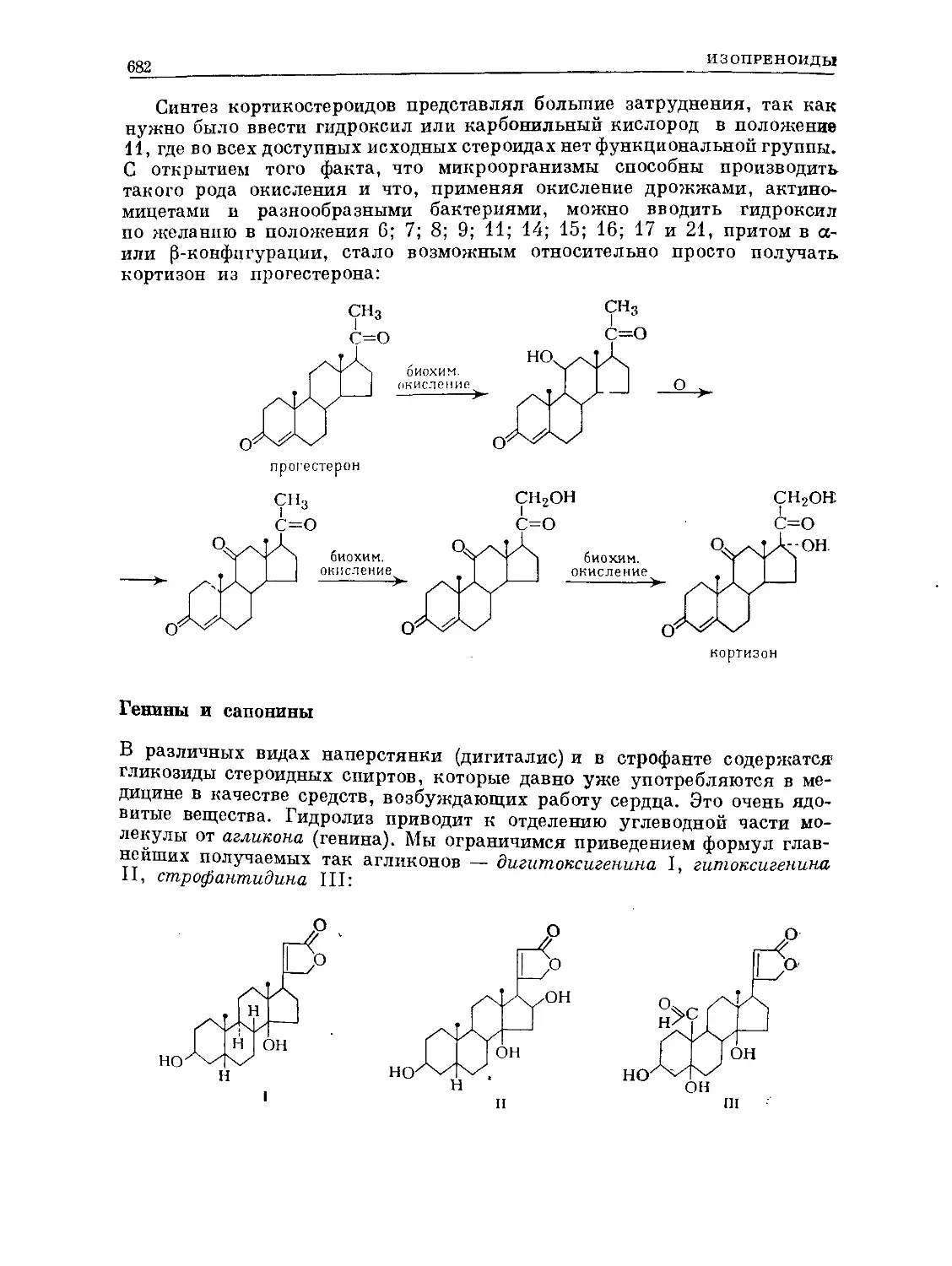




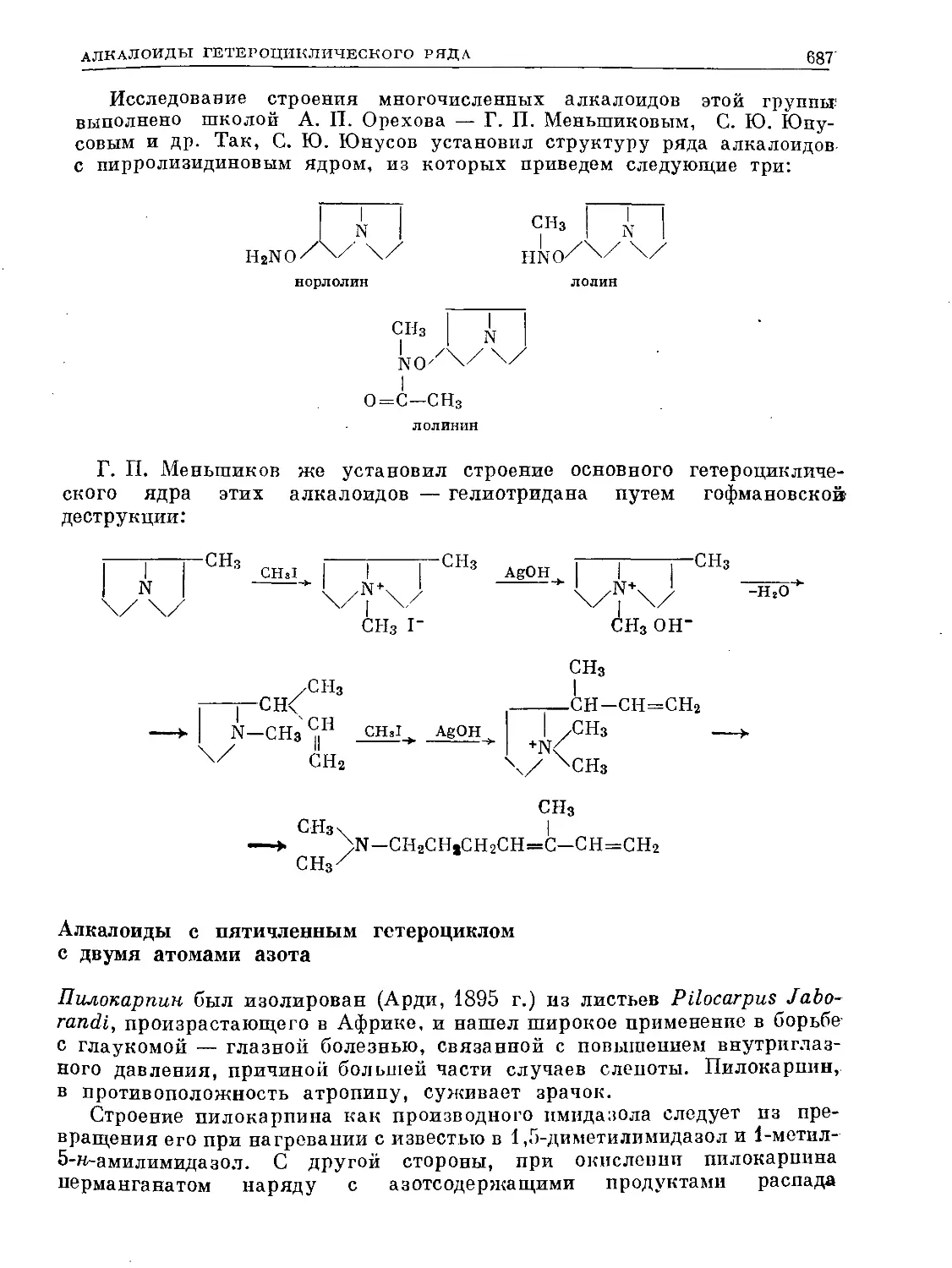
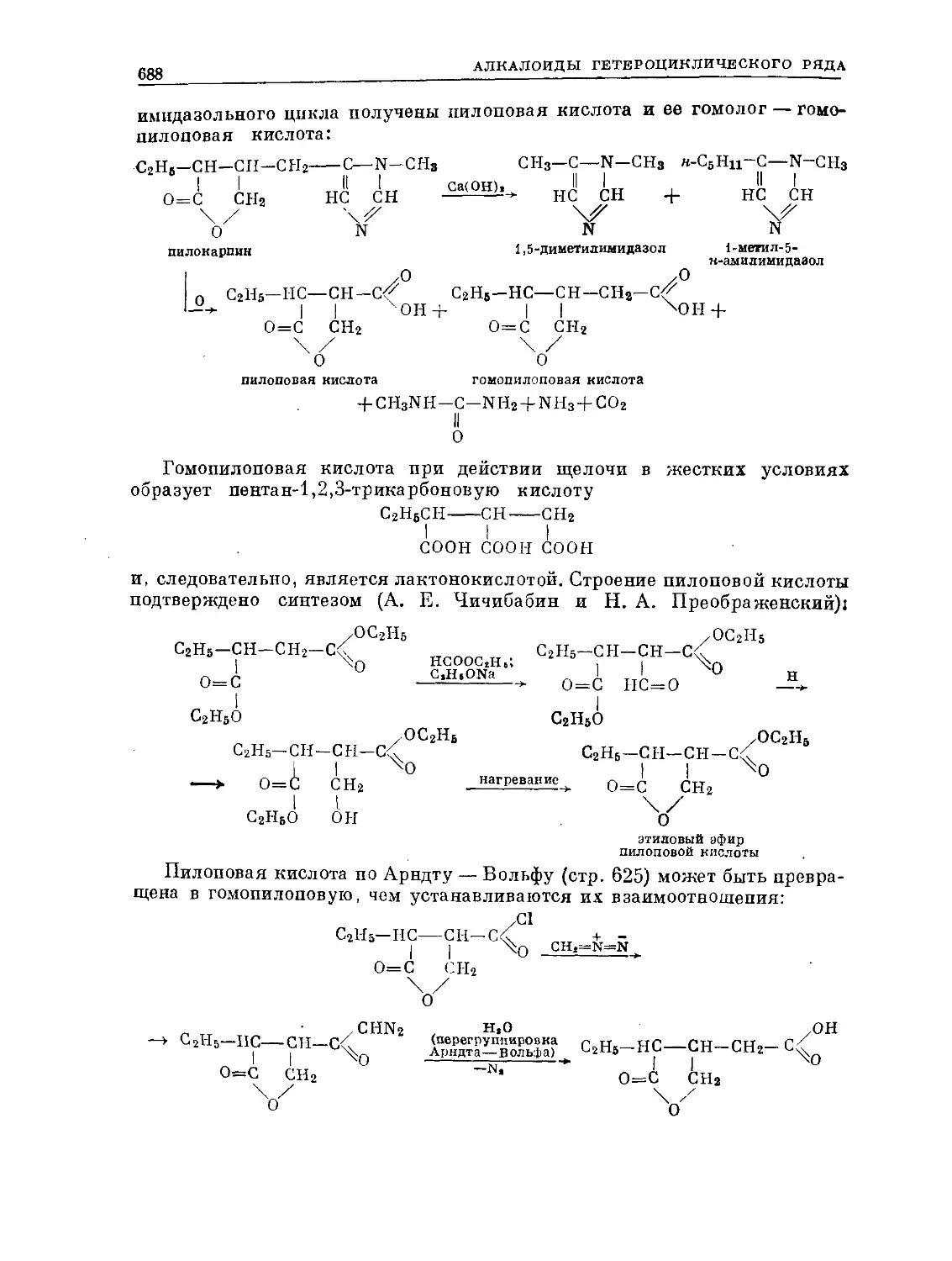
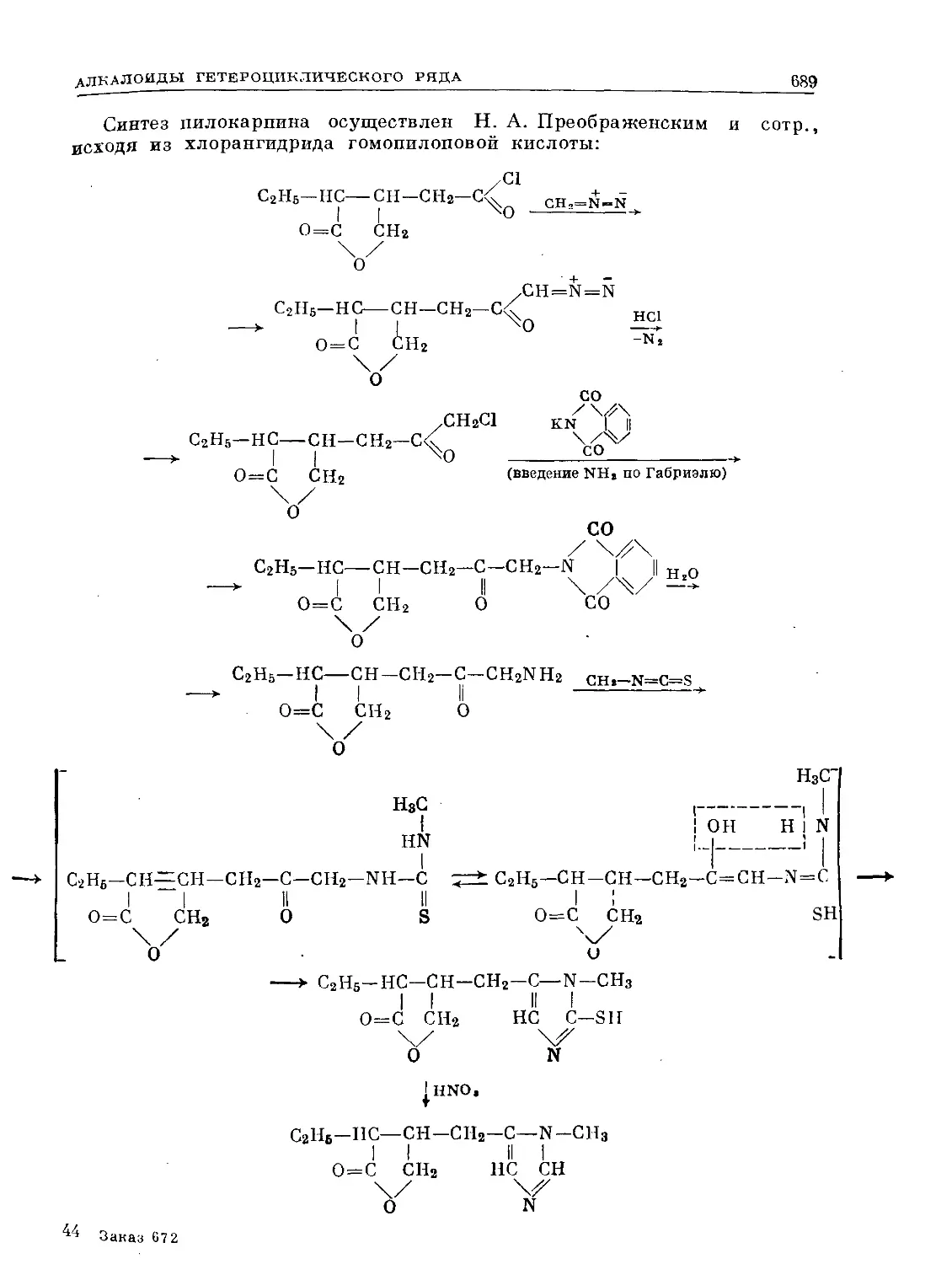

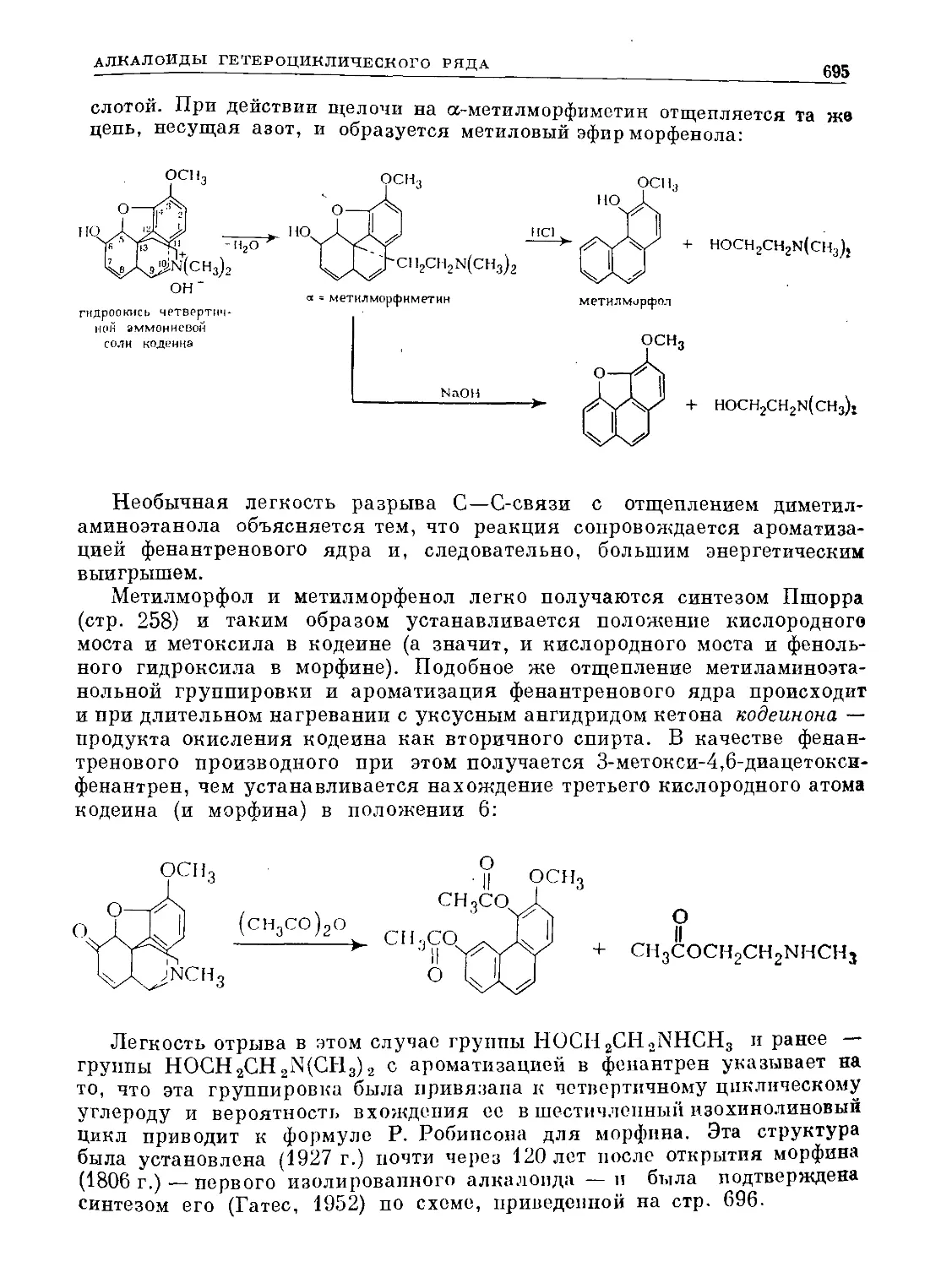
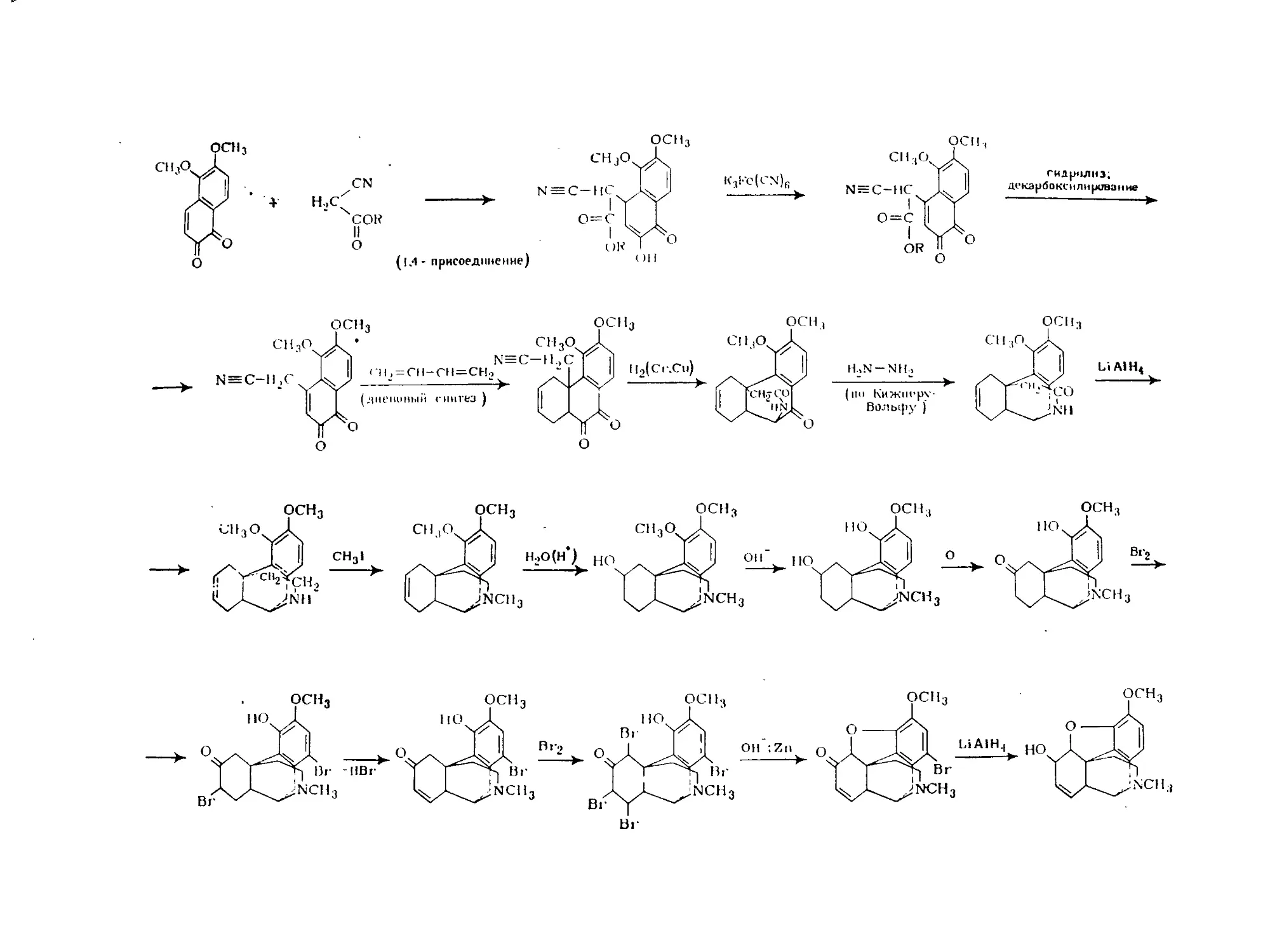

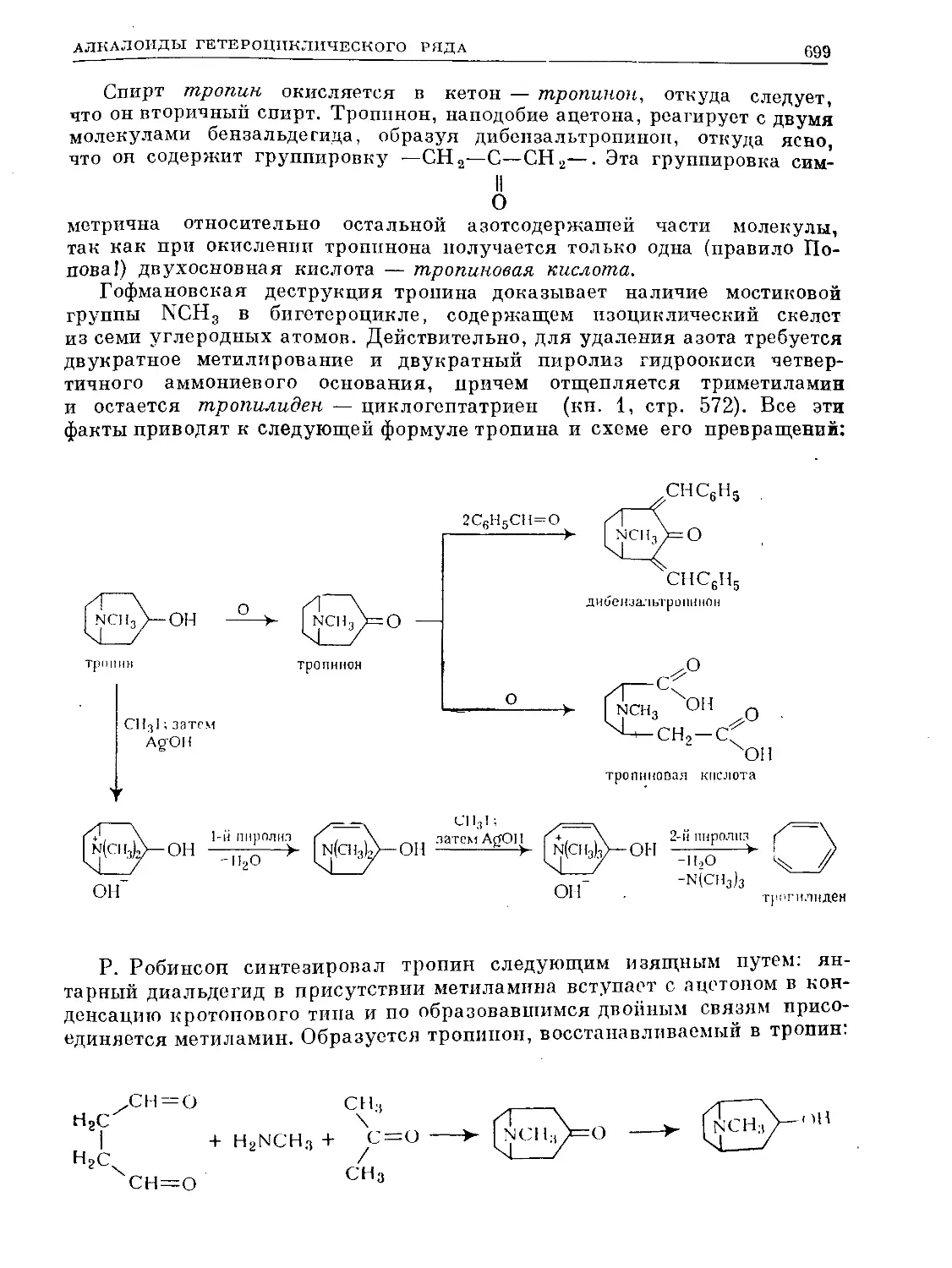



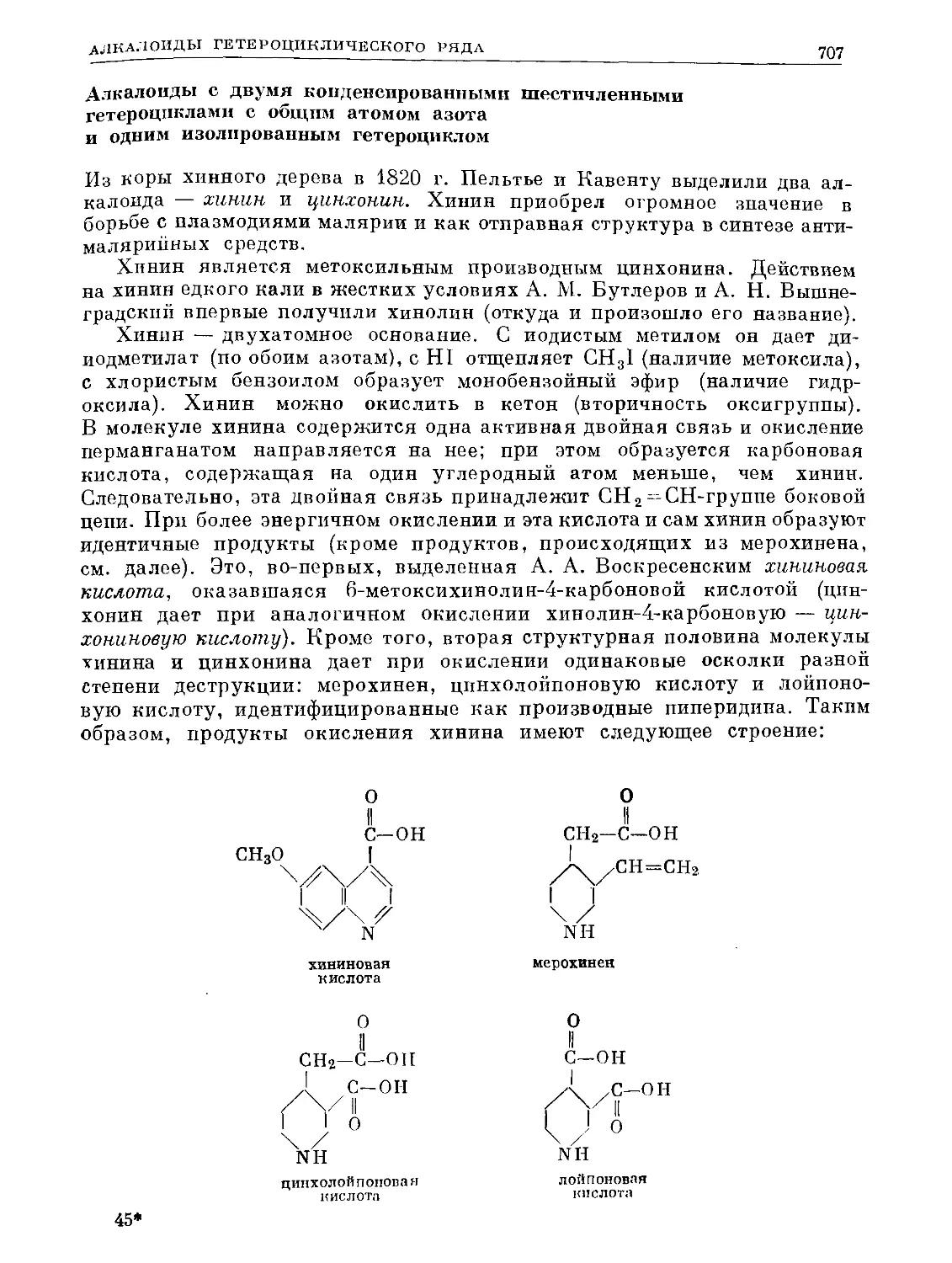
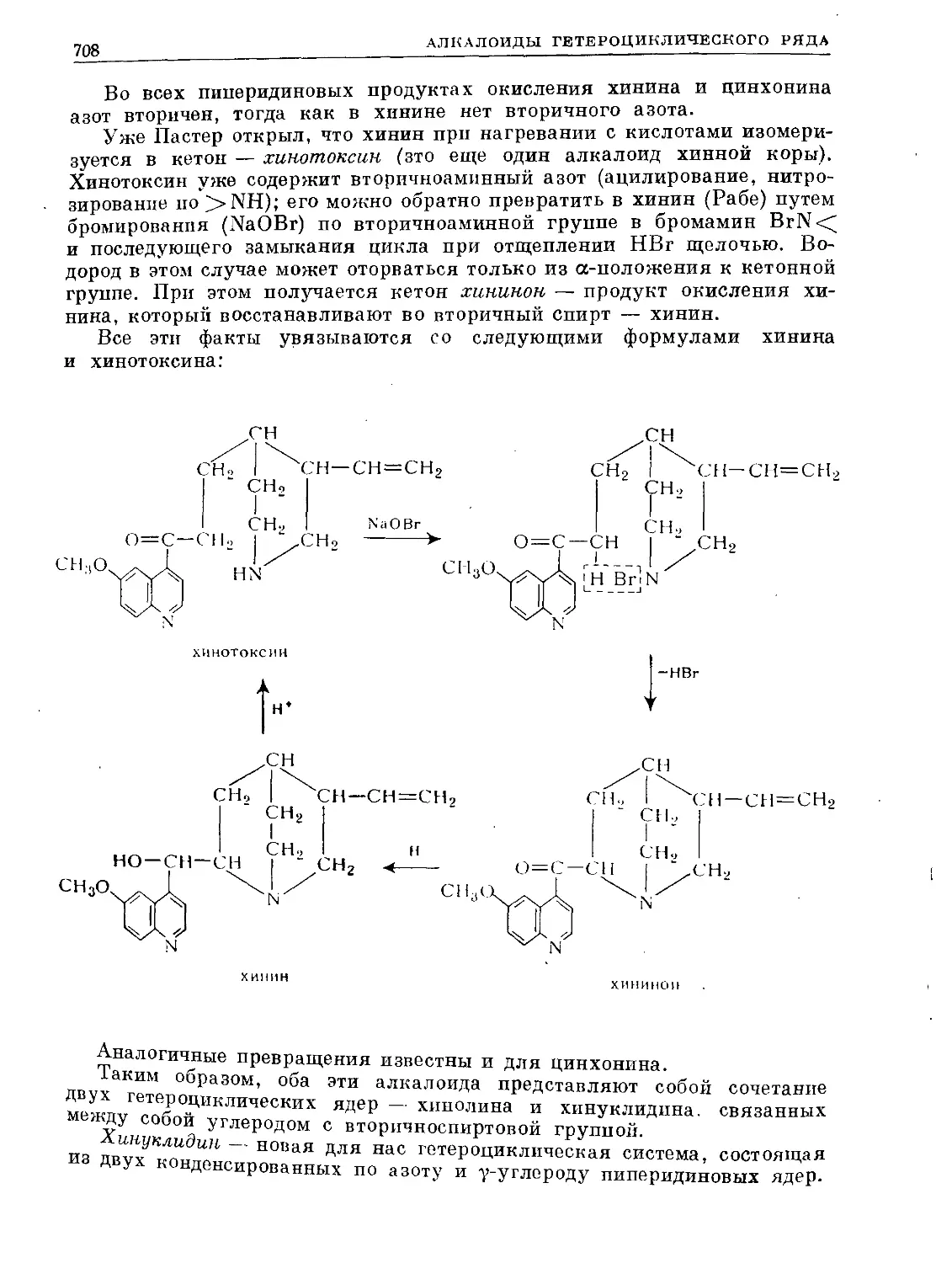
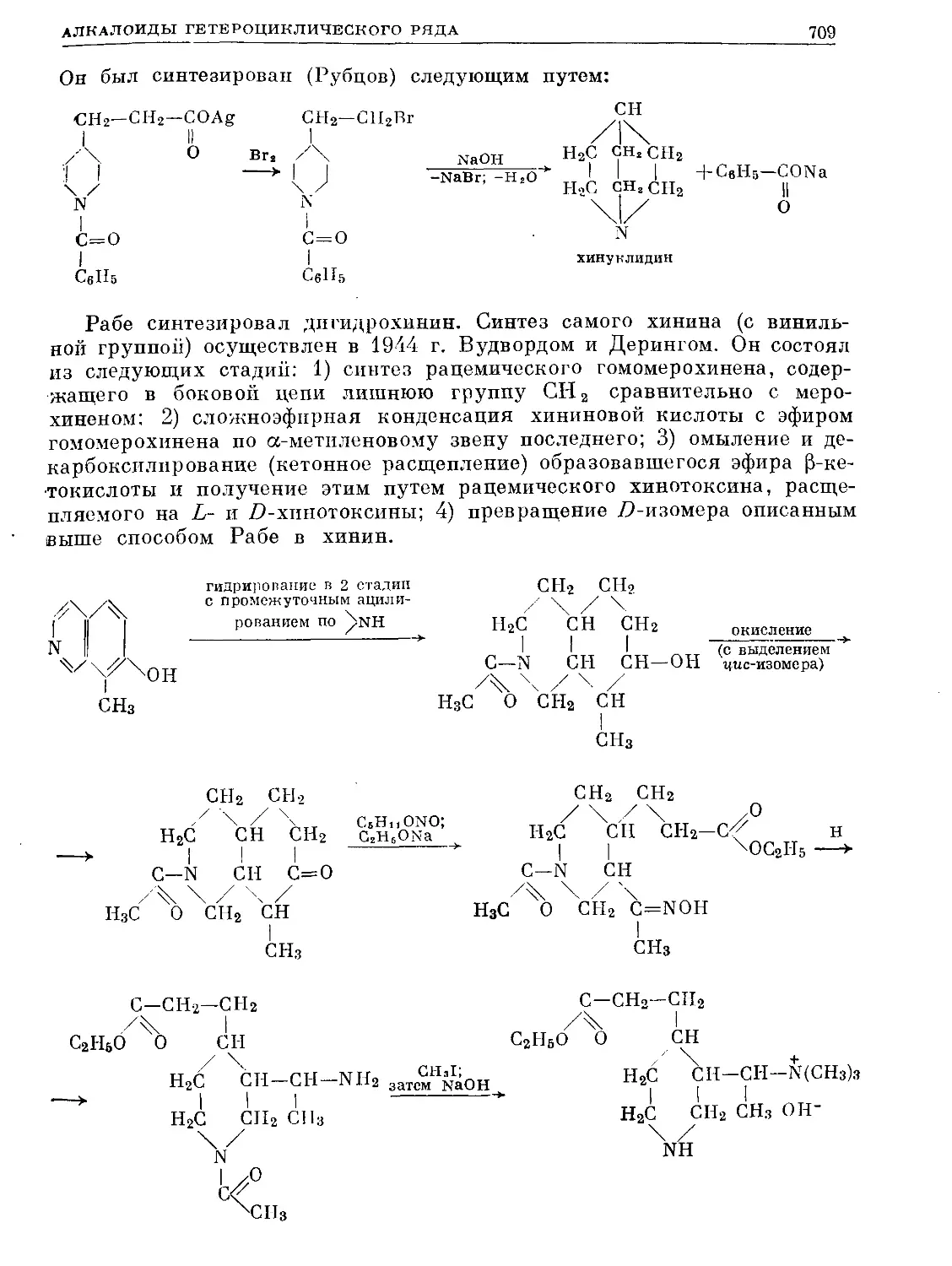
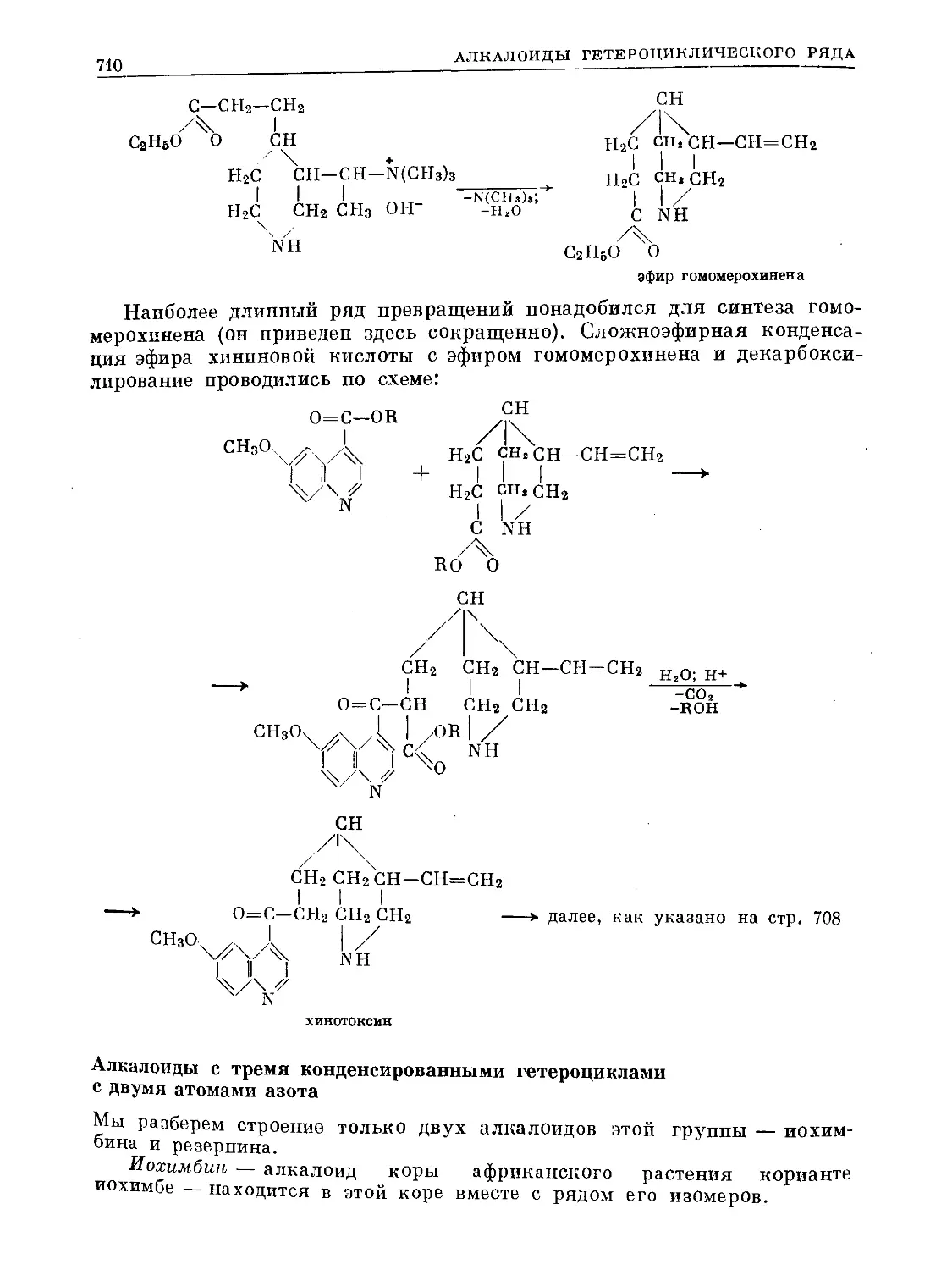
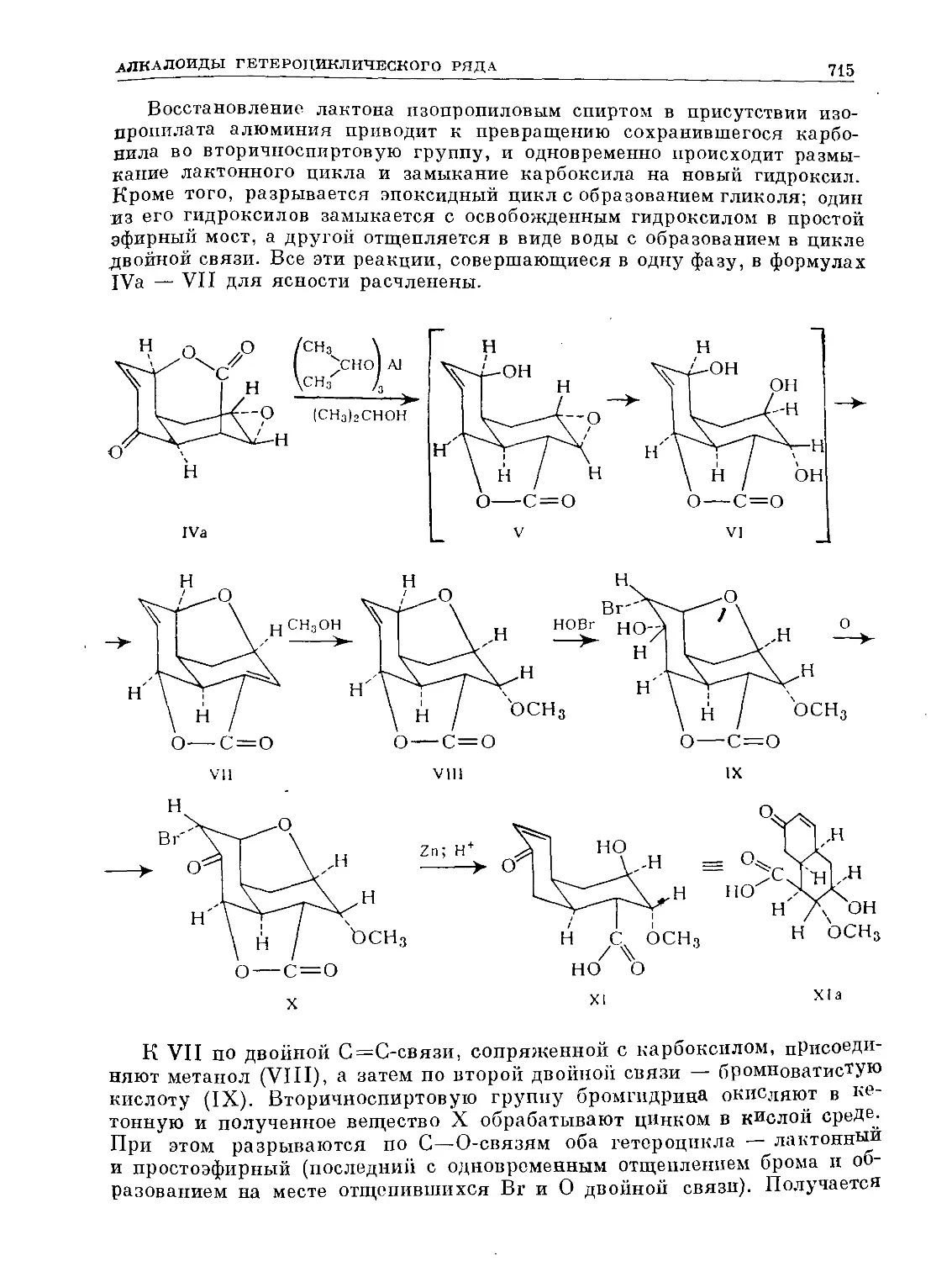
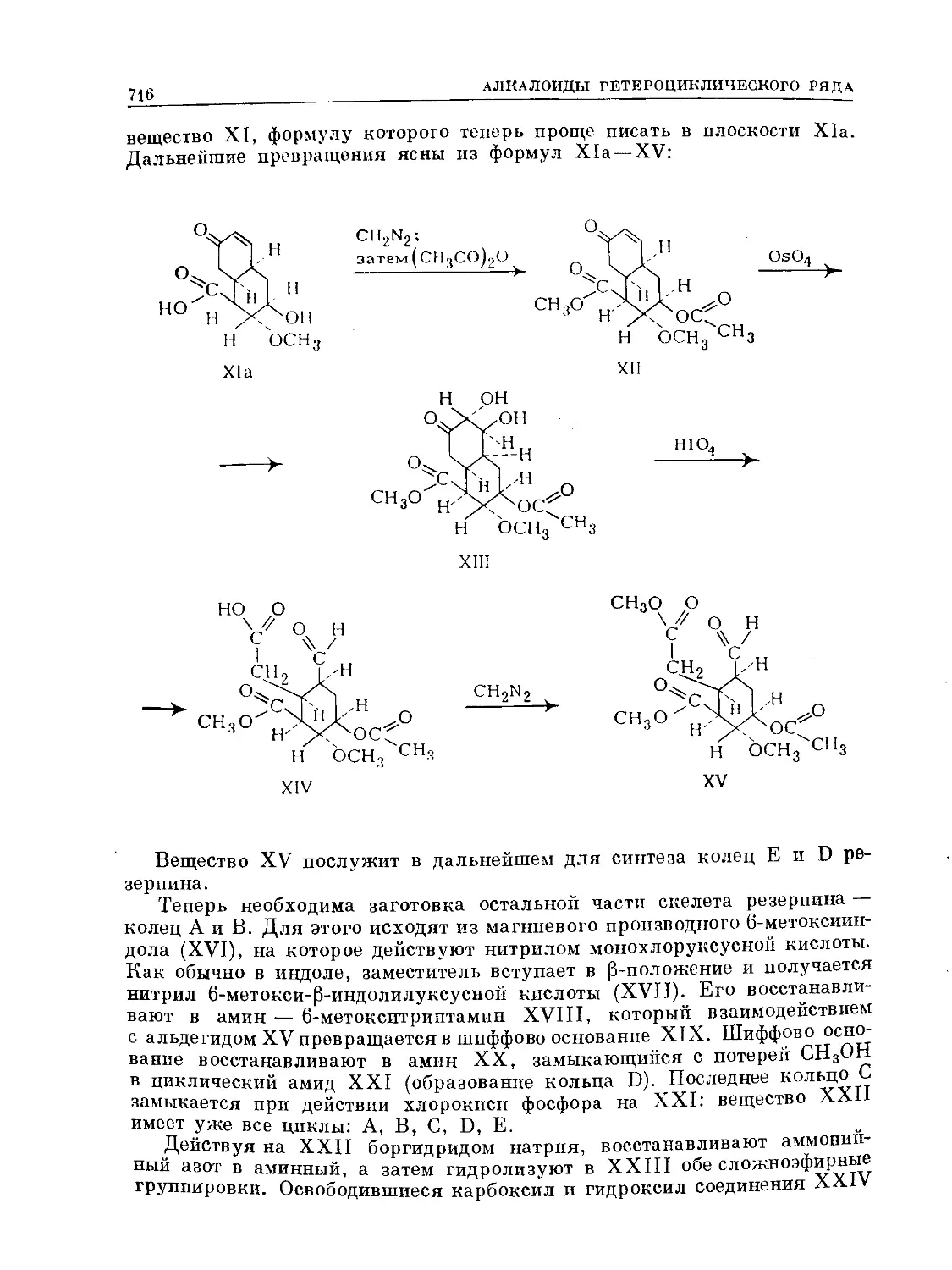
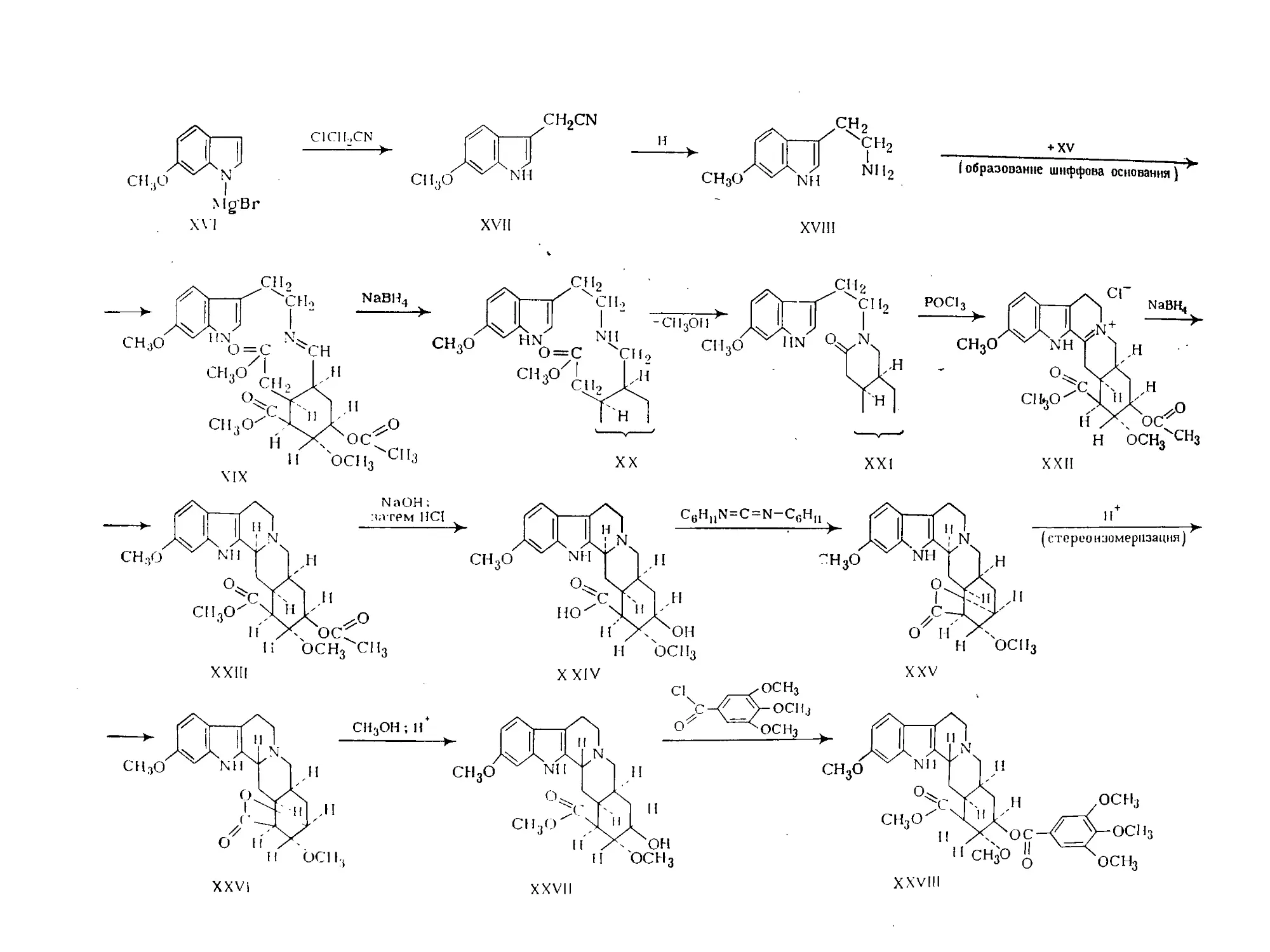

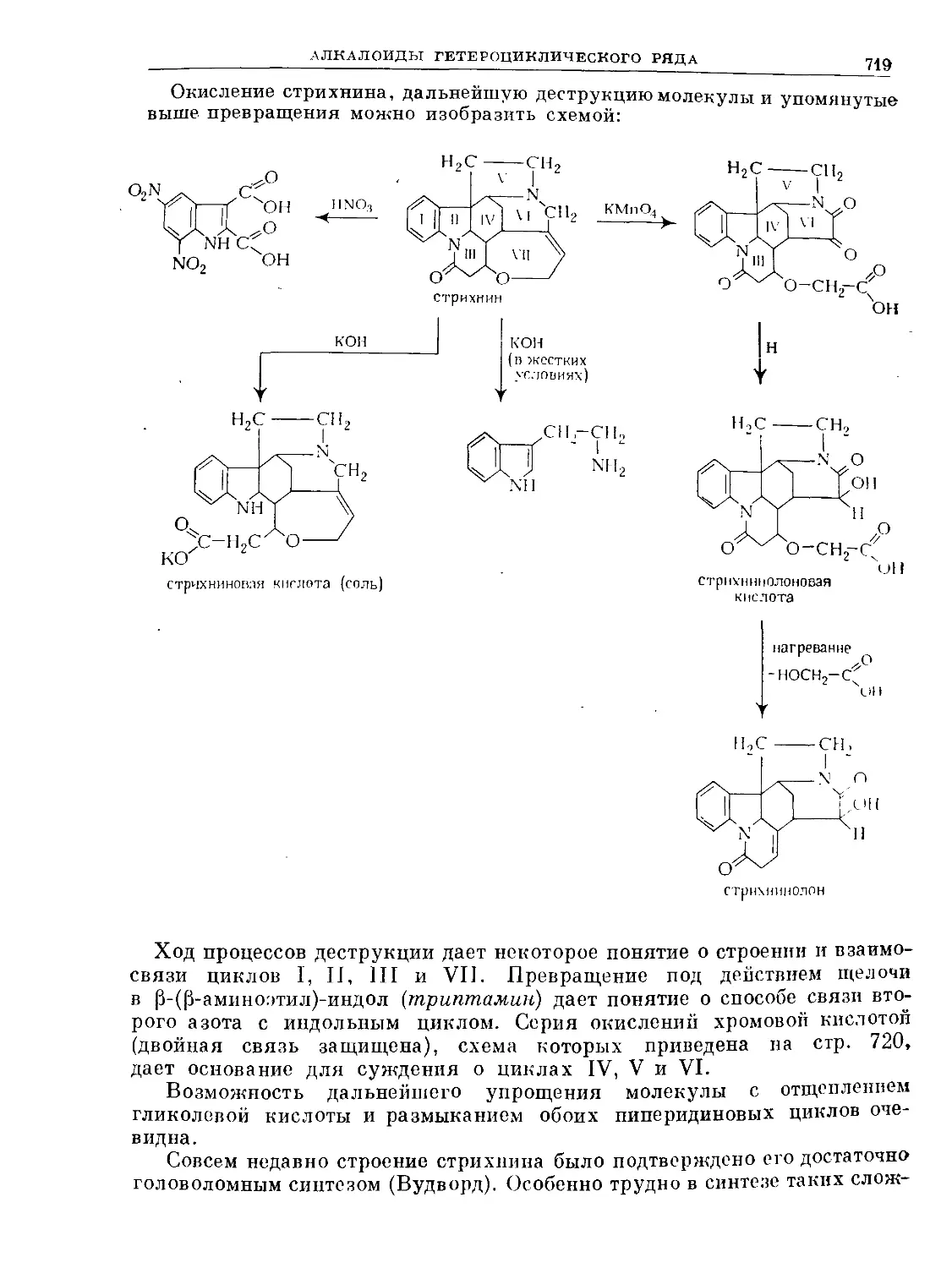
















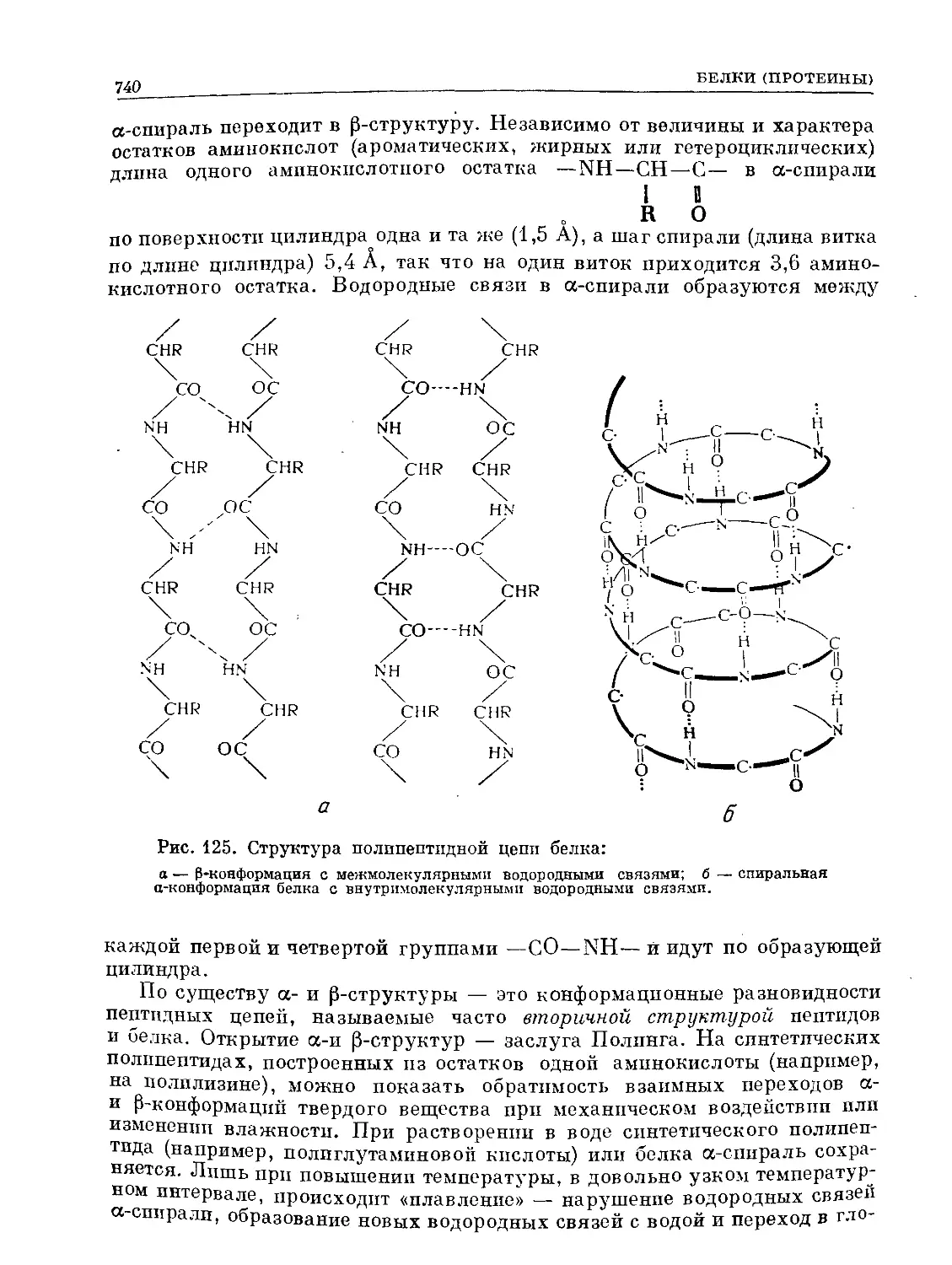

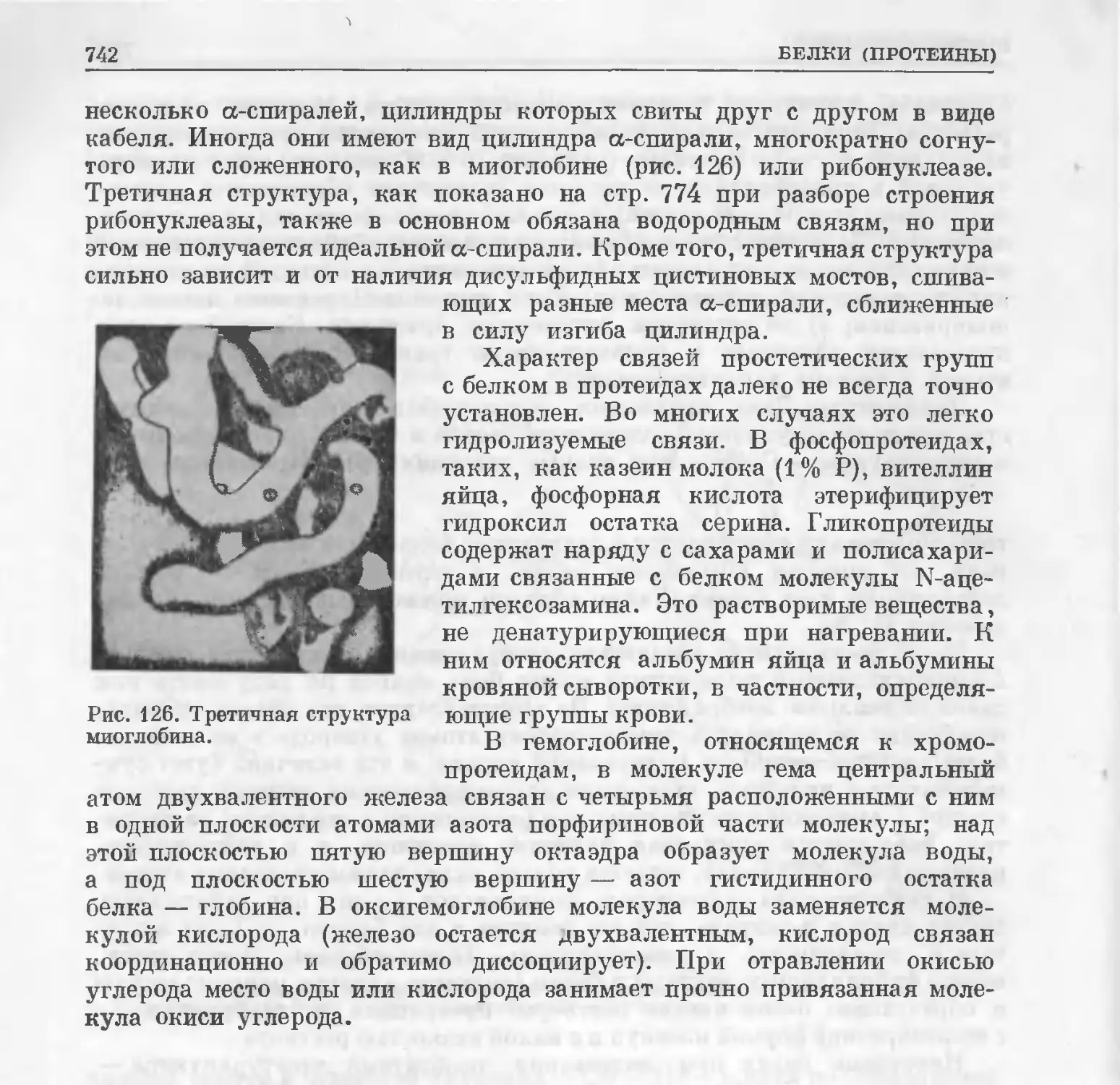


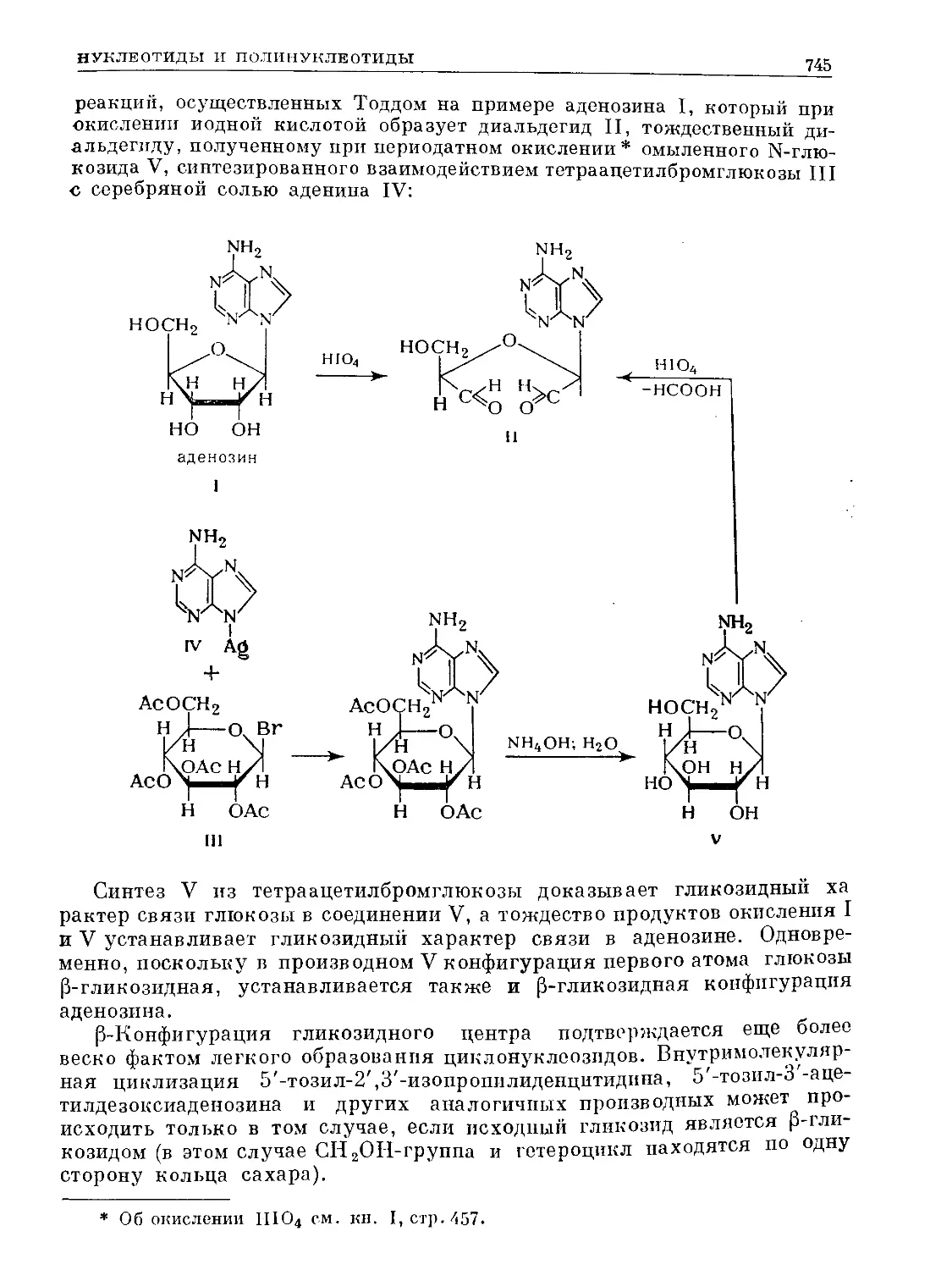
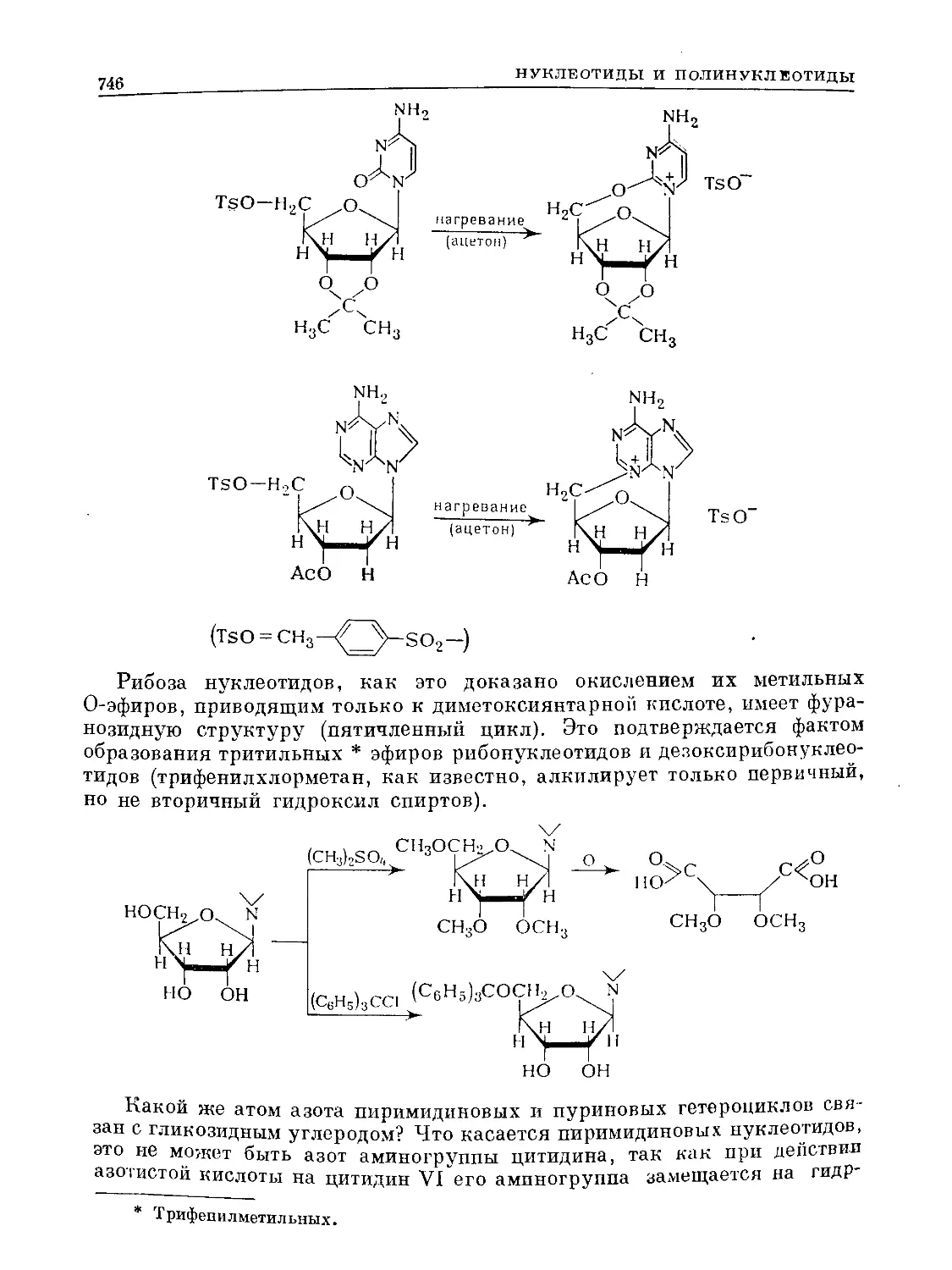
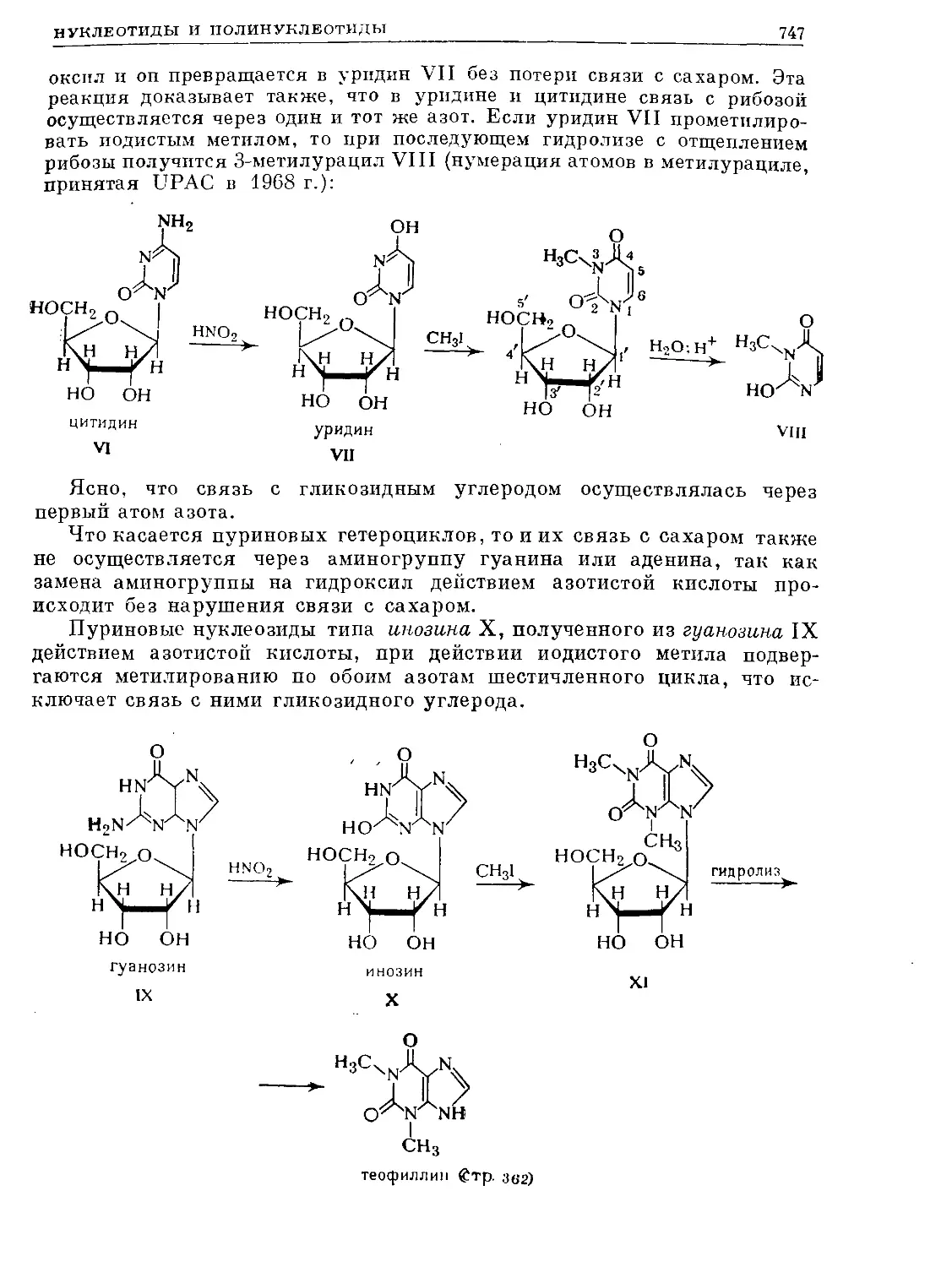
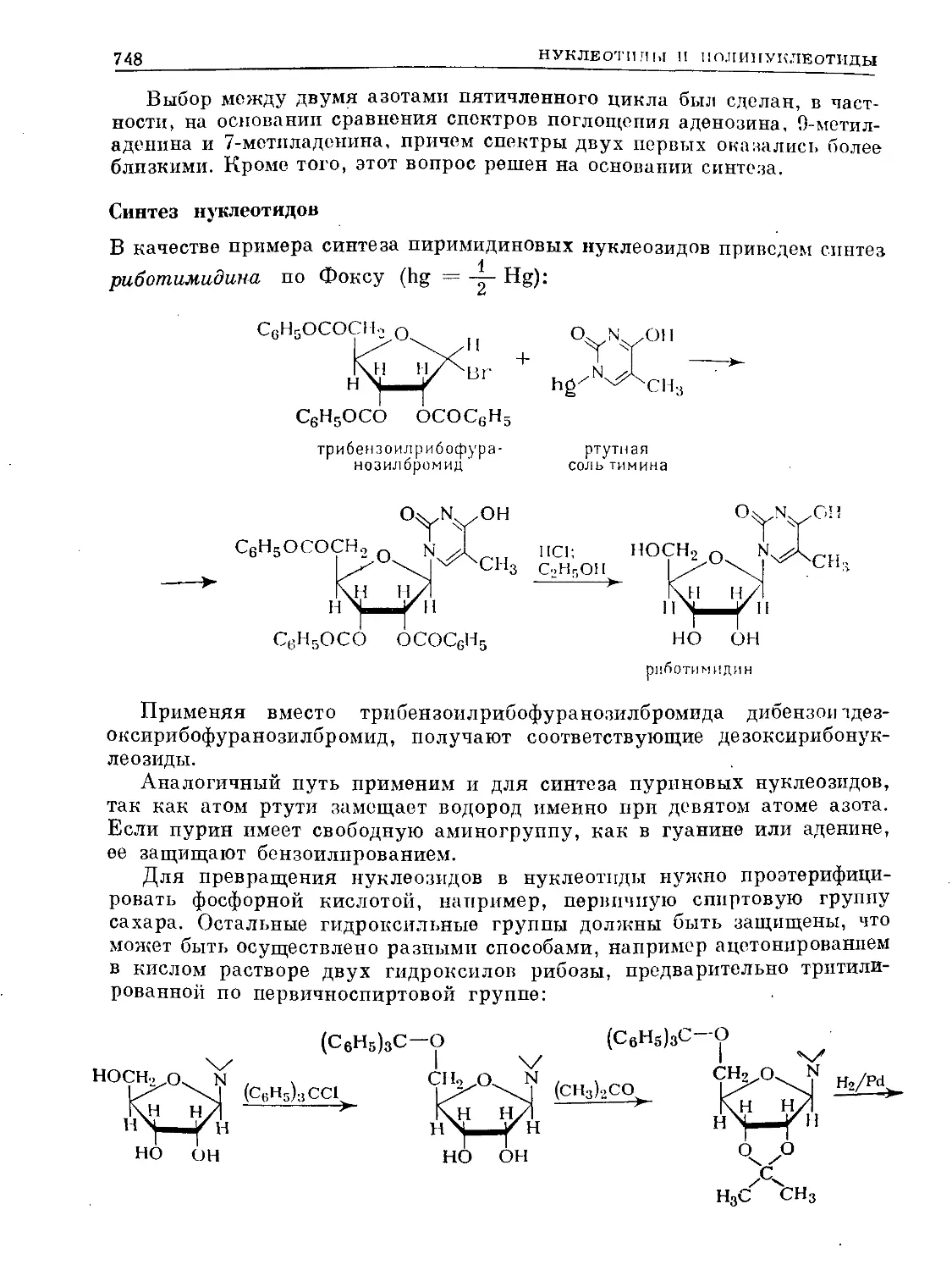
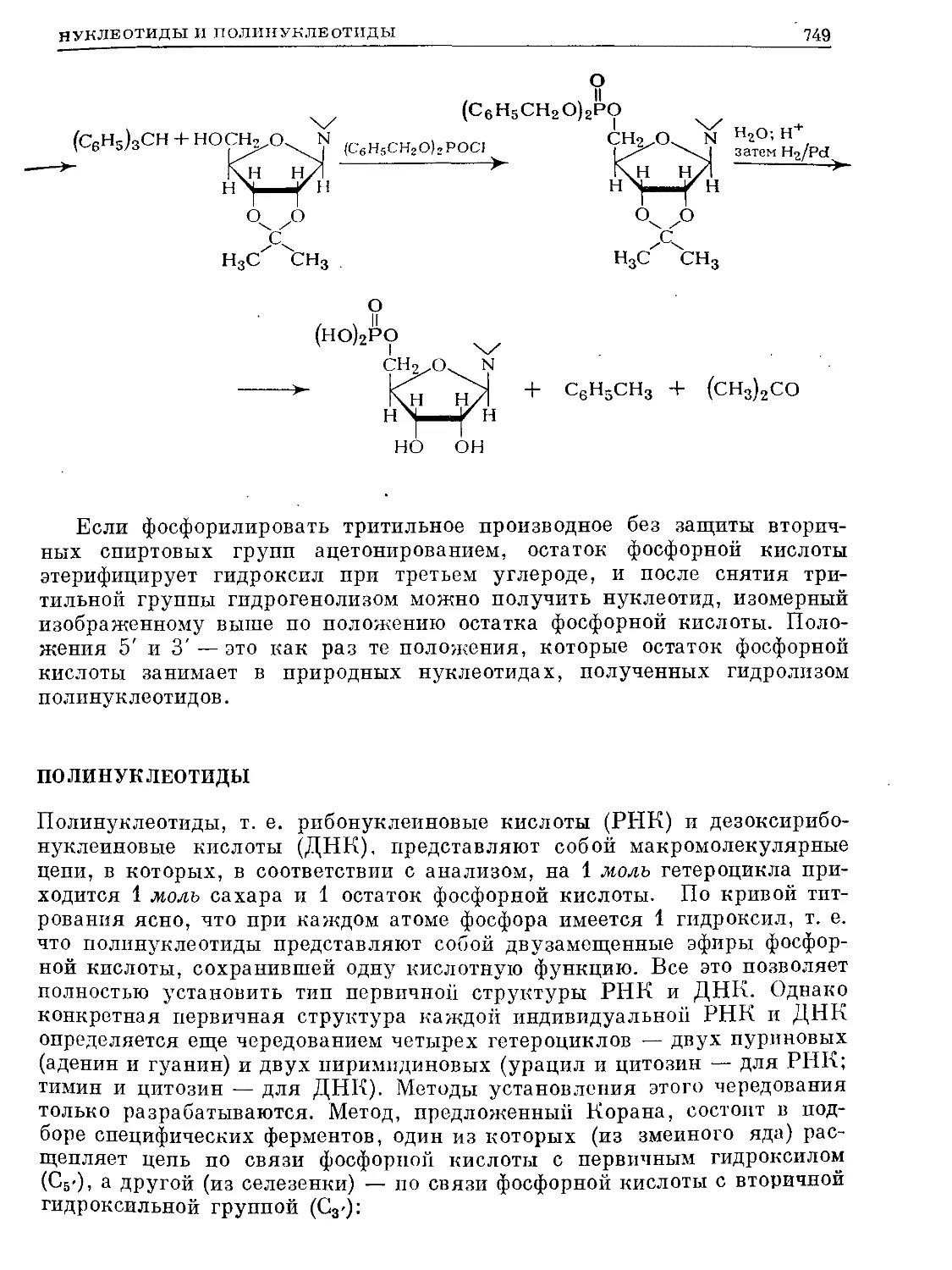




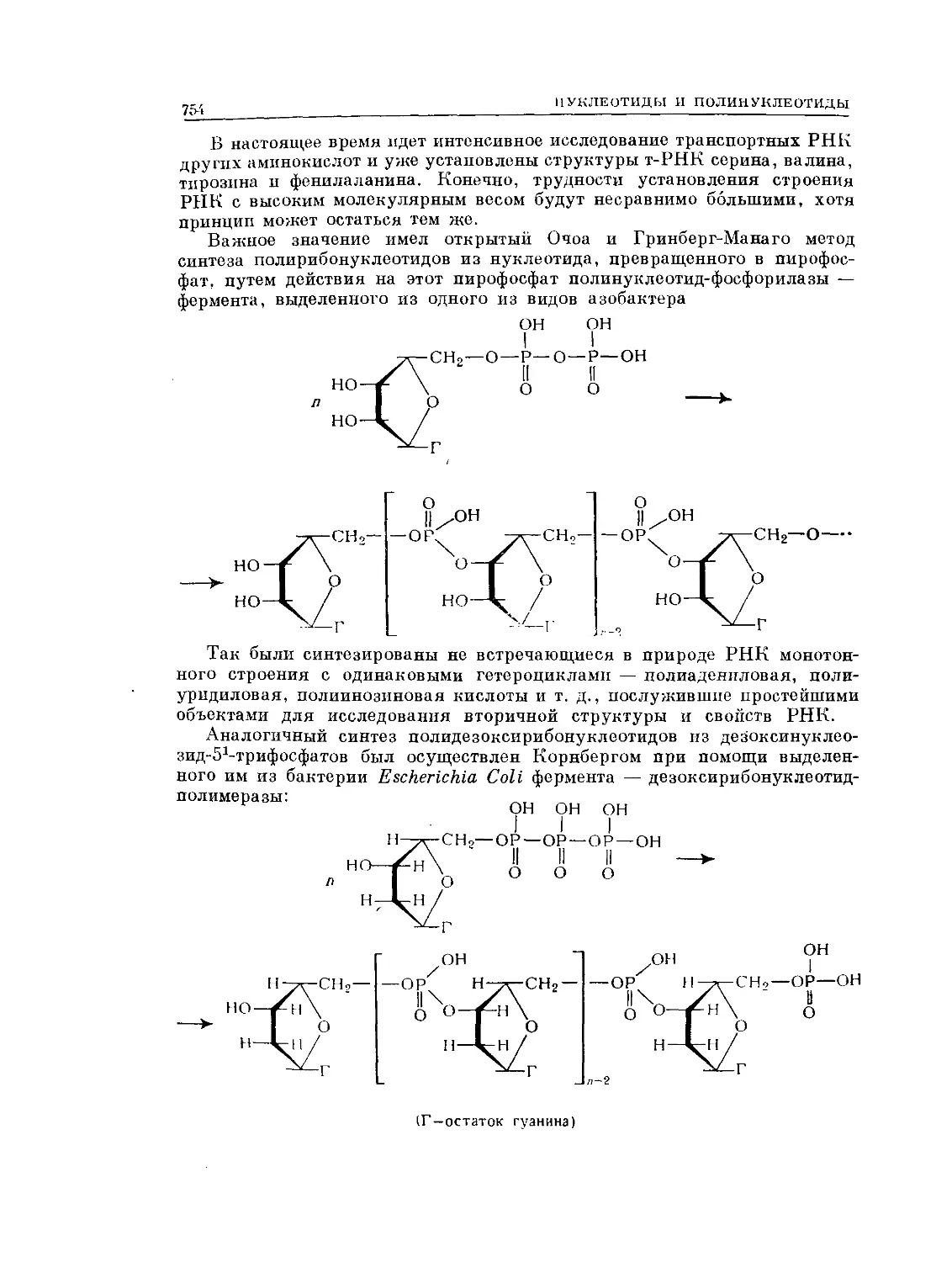
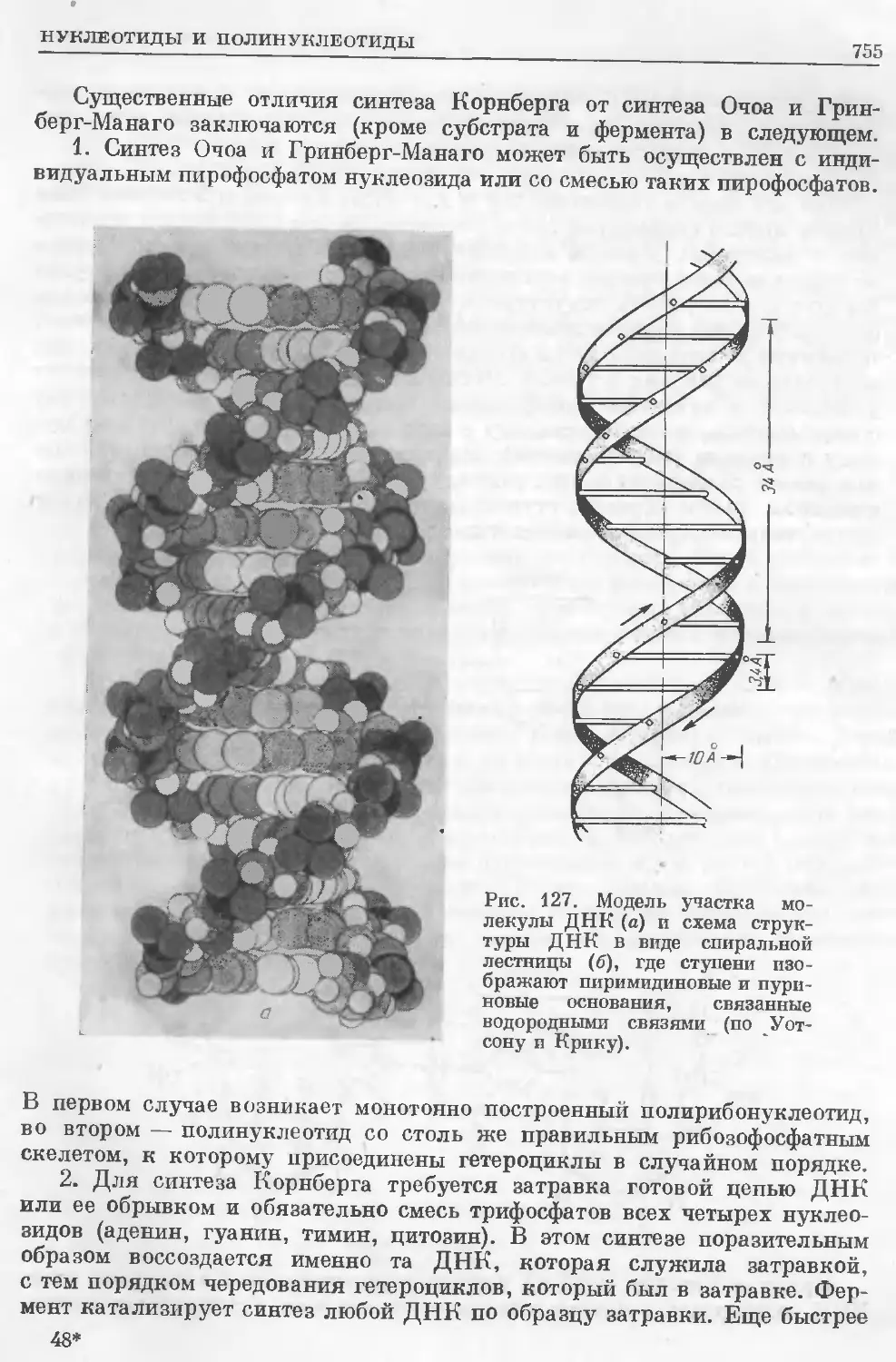
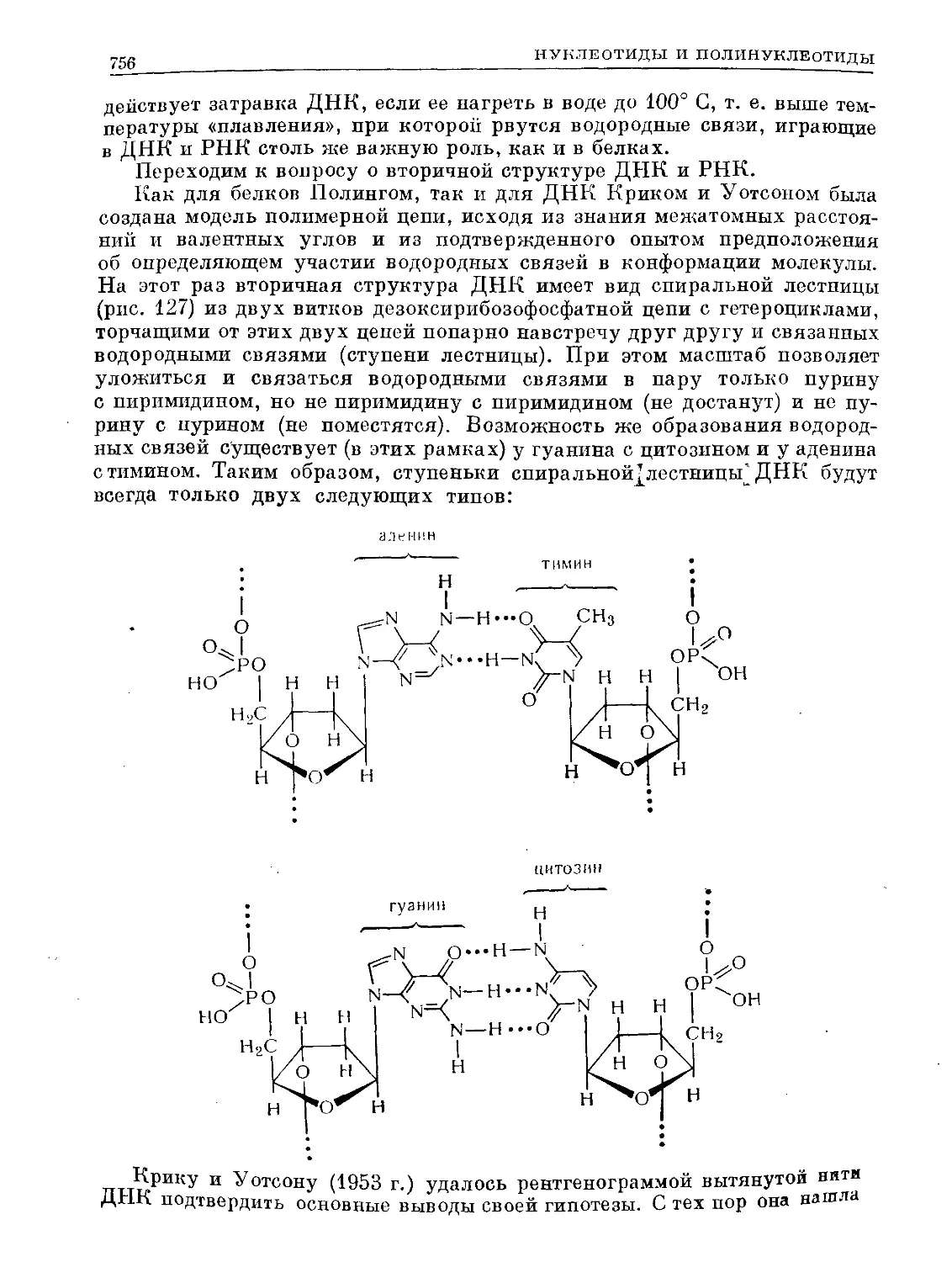



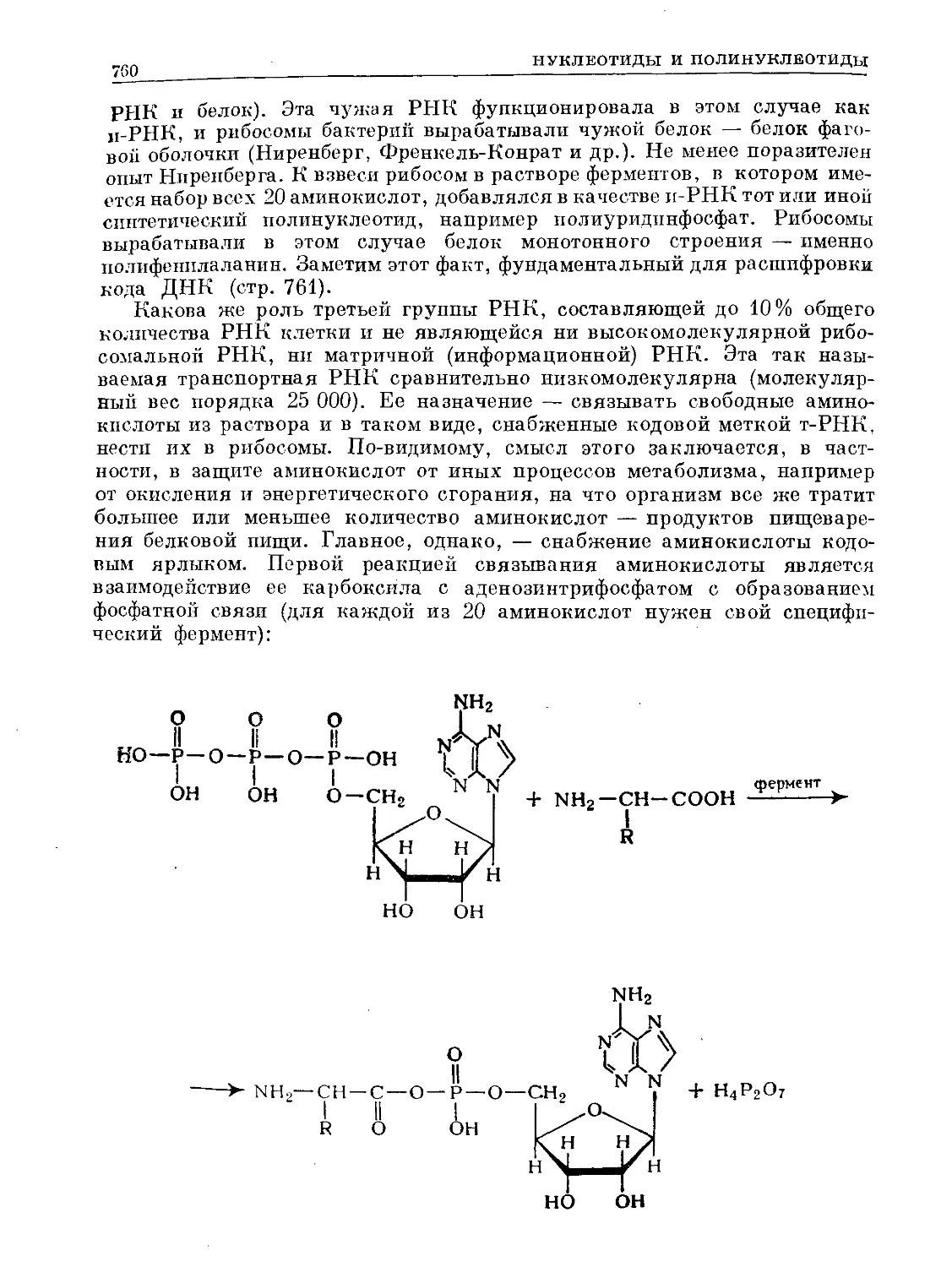

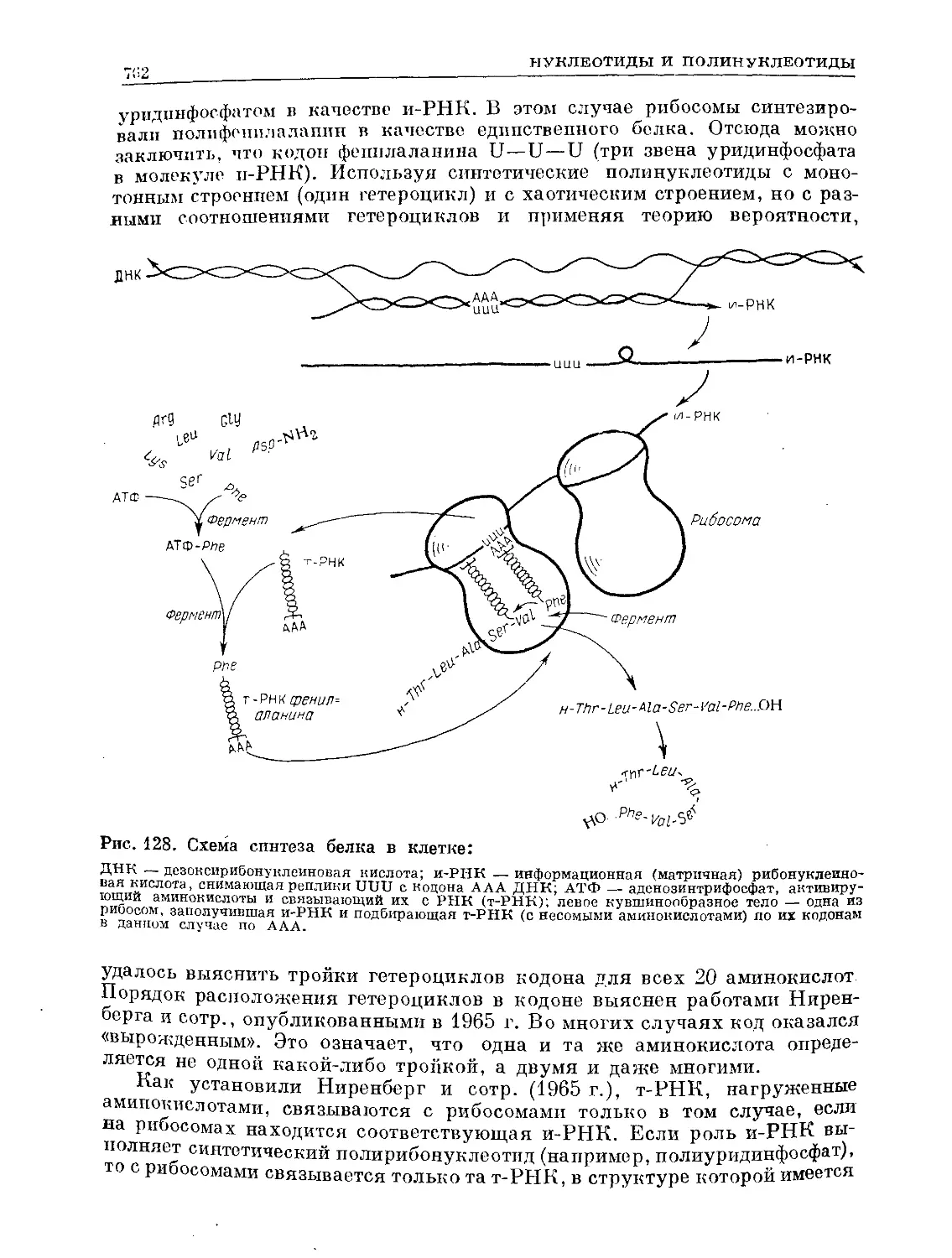





























































Text
Памяти Николая Васильевича Несмеянова,
отца и деда
НАЧАЛА
ОРГАНИЧЕСКОЙ ХИМИИ
В ДВУХ КНИГАХ
ИЗДАТЕЛЬСТВО «ХИМИЯ»
А.Н. НЕСМЕЯНОВ, Н.А.НЕСМЕЯНОВ
НАЧАЛА
ОРГАНИЧЕСКОЙ ХИМИИ
КН И ГА ВТОРАЯ
МОСКВА 1970
УДК 547 (075.8)
НГ55
Несмеянов Л. II., Несмеянов Н. А.
Начала органической химии
Руководство предназначено для первоначального систематического
изучения органической химии. Его объем несколько шире программы
химических специальностей университетов. Первая часть руководства
построена по «классической схеме»; вторая часть, скорее, предназначена
для внимательного прочтения, чем для глубокого изучения. В книгу I
вошли разделы первой части — введение, алифатический ряд п алпцпклы,
в книгу II — ароматический ряд, гетероциклы п вся вторая часть. Элемен-
тарные сведения по тем разделам, которые во второй части развиты в це-
лые главы, имеются п в первой части книги, в которую включены также
сведения о методах физического исследования п понятия о химической
связи.
Материал изложен в порядке нарастающей трудности, вначале доста-
точно просто и подробно, затем все более лаконично.
«Начала органической химии» могут служить как для самостоятель-
ного знакомства с предметом, так п для изучения его в университетах
и химических вузах. Книга будет несомненно интересна и для аспиран-
тов, преподавателей, молодых ученых п инженеров, работающих в области
органической химии.
АЛЕКСАНДР НИКОЛАЕВИЧ НЕСМЕЯНОВ
НИКОЛАЙ АЛЕКСАНДРОВИЧ НЕСМЕЯНОВ
НАЧАЛА ОРГАНИЧЕСКОЙ ХИМИИ
2-5-3
B3-34-69-33
Книга вторая
Издательство «Химия», М., 1970 г. 82 4 с.
Редакторы Ф. Б. Рабинович, Р. С. Ромм. Техн, редактор Е. Г. Шпак
Художник К. М. Егоров. Корректоры Л. П. Курьянова, Е. П. Павлов
Т0ч614. Подписано к печати 11 /Н1 1970 г. Формат бумаги 70 X 100‘/>в. Печ. л. о1,5.
л‘ '>»>.95. Уч.-изд. л. 59.4. Типогр. бум. AS 1. Тираж зО ООО ЭКЗ.
ьз л,. 34—19G9 г.—.V8 зз. Цена 2 р. 26 к., в суперобложке 2 р. 3 0 к. Зак. Л« 6 /--
Ленинградская типография .41 14 «Красный Печатник» Главполиграфпрома
комитета по печати при Совете Министров СССР,
московский проспект, дом 91.
КНИГА 1
ЧАСТЬ ПЕРВАЯ
Введение
Ациклические соединения
Предельные соединения
Непредельные соединения
Гетерофункциональные соединения
Алициклические соединения
Алициклические углеводороды и их производные
КНИГА 2
Ароматические соединения
Бензол и его производные
Многоядерные ароматические соединения
Гетероциклические соединения
ЧАСТЬ ВТОРАЯ
Элементоорганические соединения
Небензоидные ароматические системы
Карбанионы
Свободные радикалы
Карбены
Карбоплевые ионы (карбкатионы)
Механизмы химических реакций
Реакции элиминации
Перегруппировки
Изопреноиды
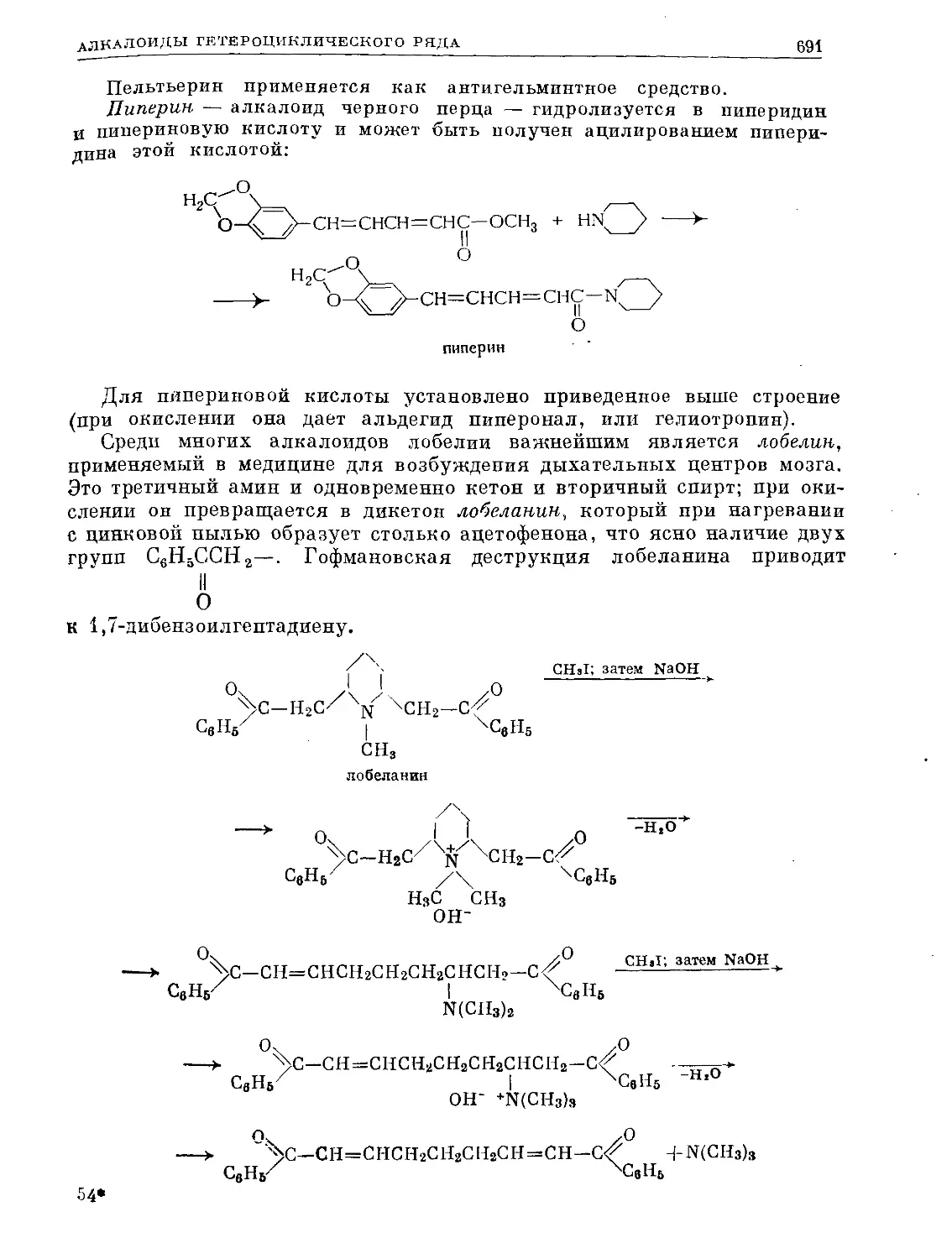
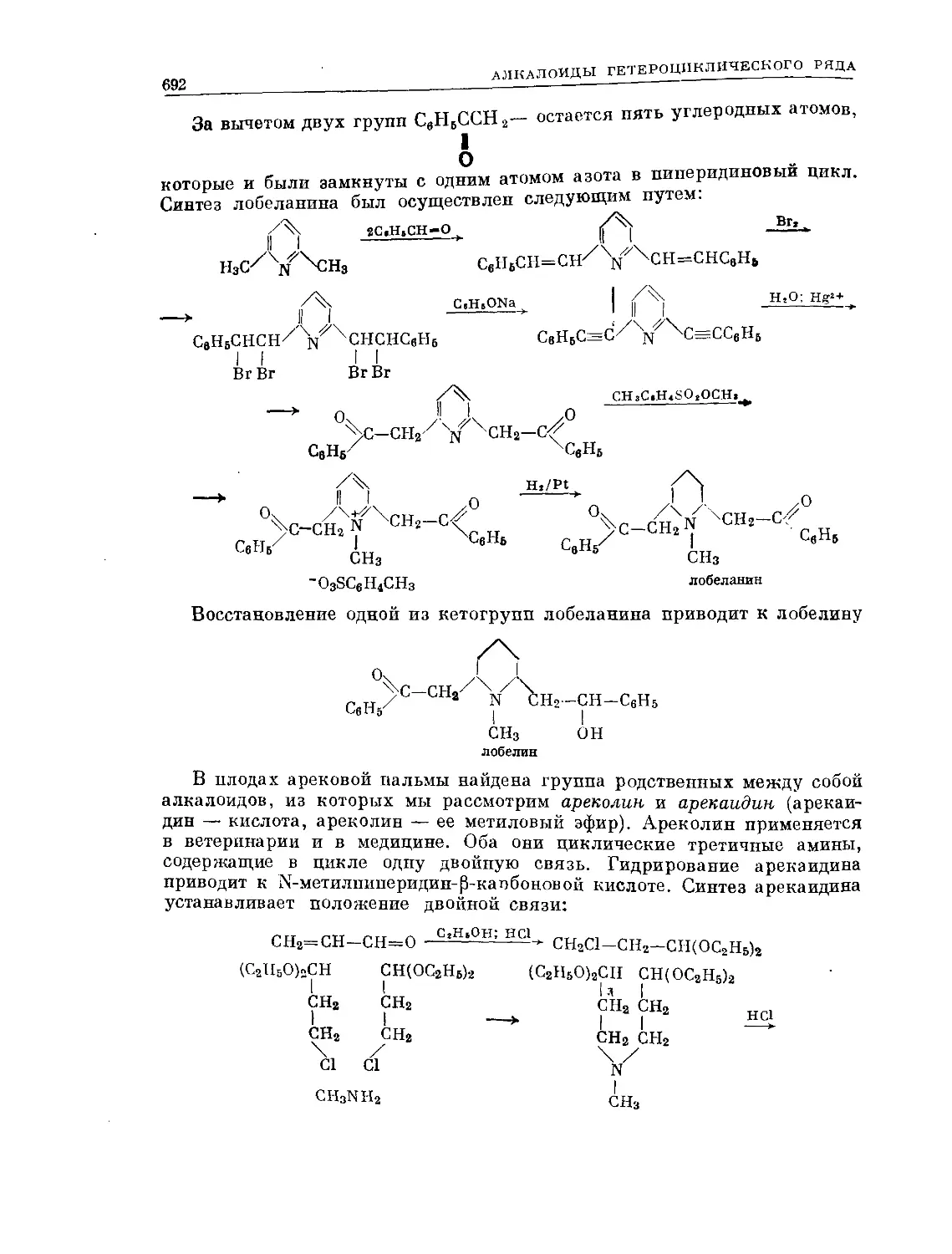
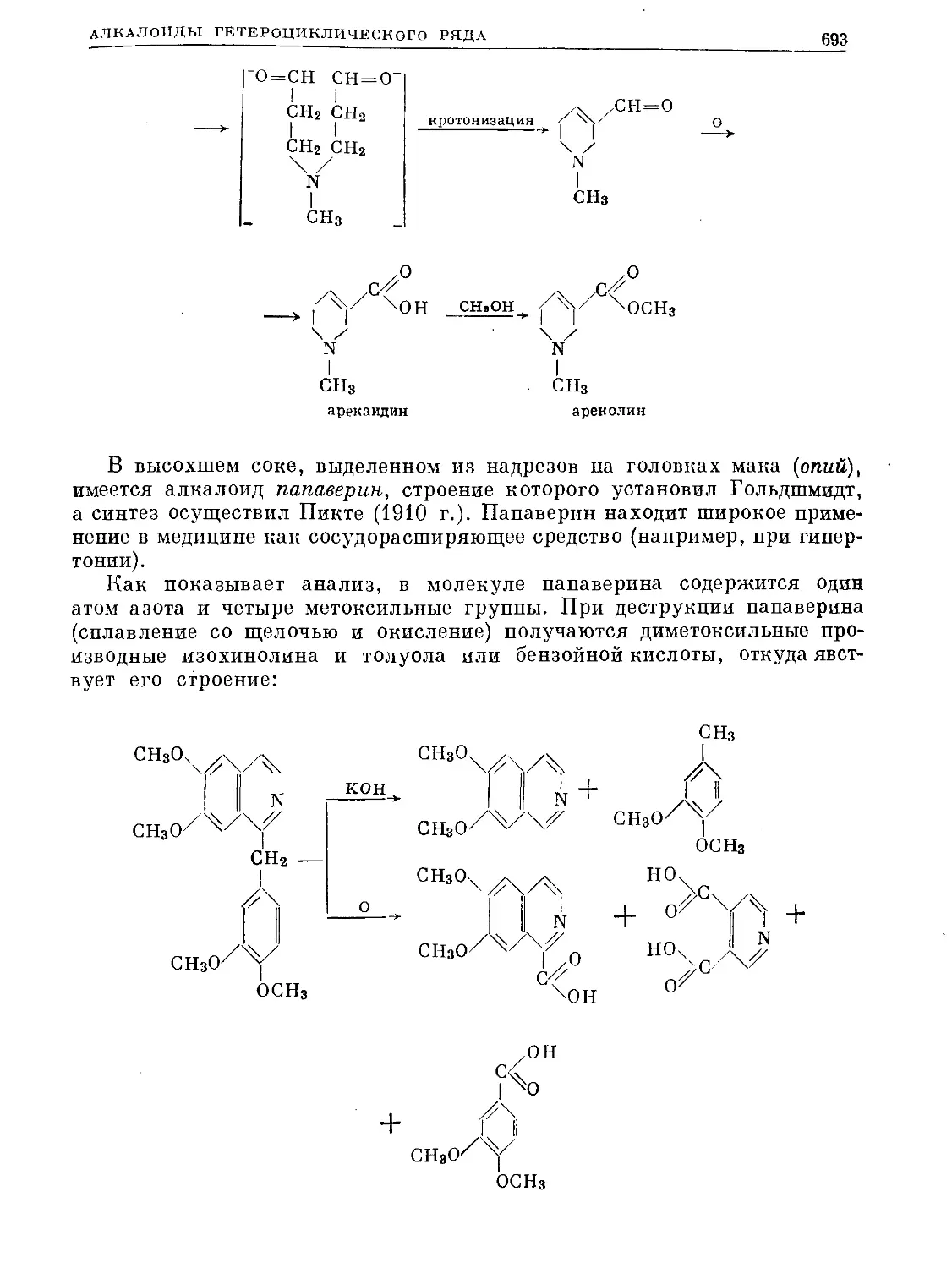
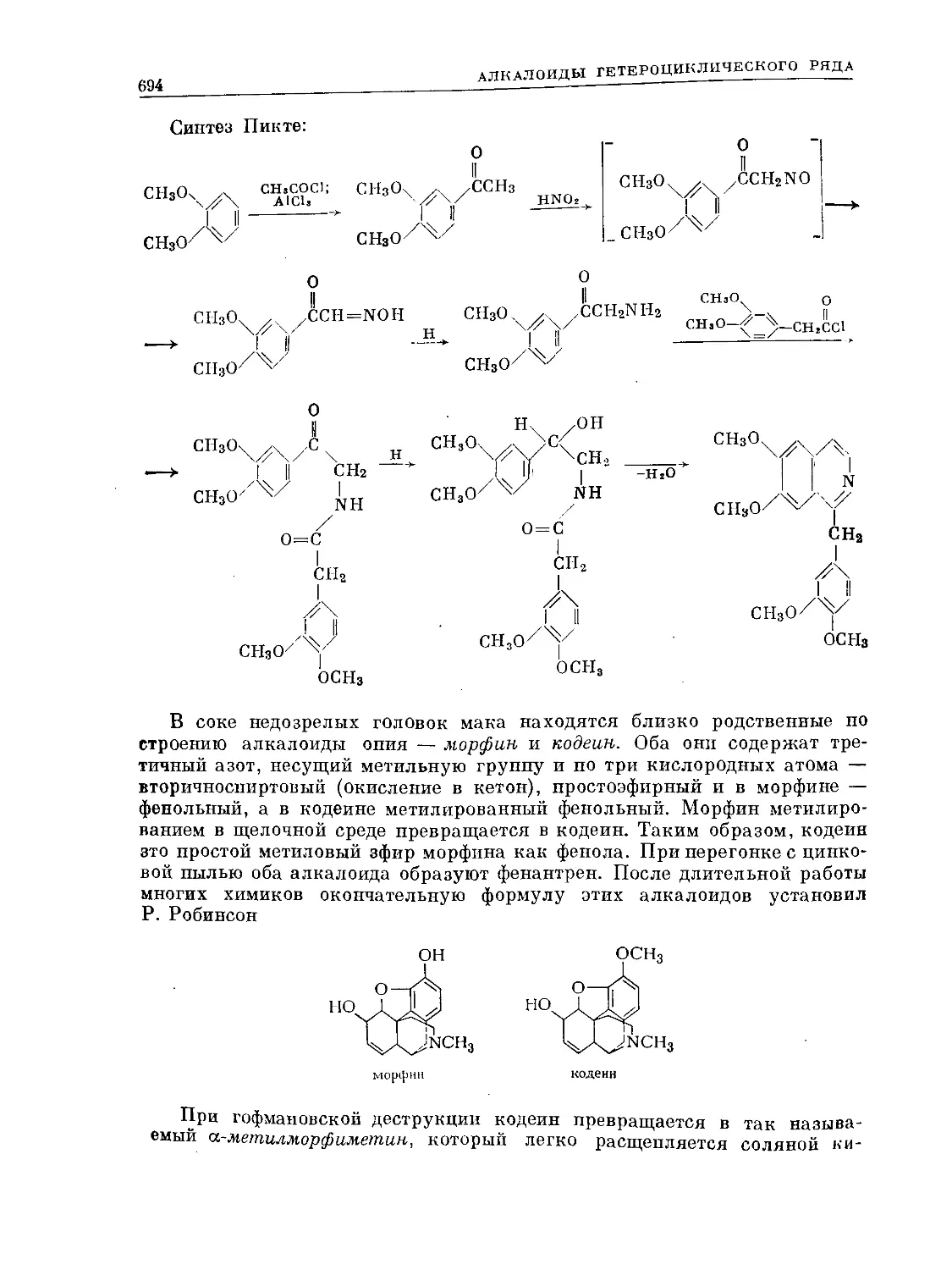
Алкалоиды гетероциклического ряда
Белки (протеины)
Нуклеотиды и полинуклеотиды. Синтез белка в
организме
Ферменты
АРОМАТИЧЕСКИЕ
СОЕДИНЕНИЯ
БЕНЗОЛ
И ЕГО ПРОИЗВОДНЫЕ
БЕНЗОЛ И ЕГО СТРОЕНИЕ
В начале этого курса, во «Введении», уже было сказано о чисто случайном
происхождении термина «ароматические соединения», присвоенного в на-
чале XIX столетия бензолу, его производным и соединениям, в основе
которых лежат углеводороды с конденсированными бензольными циклами
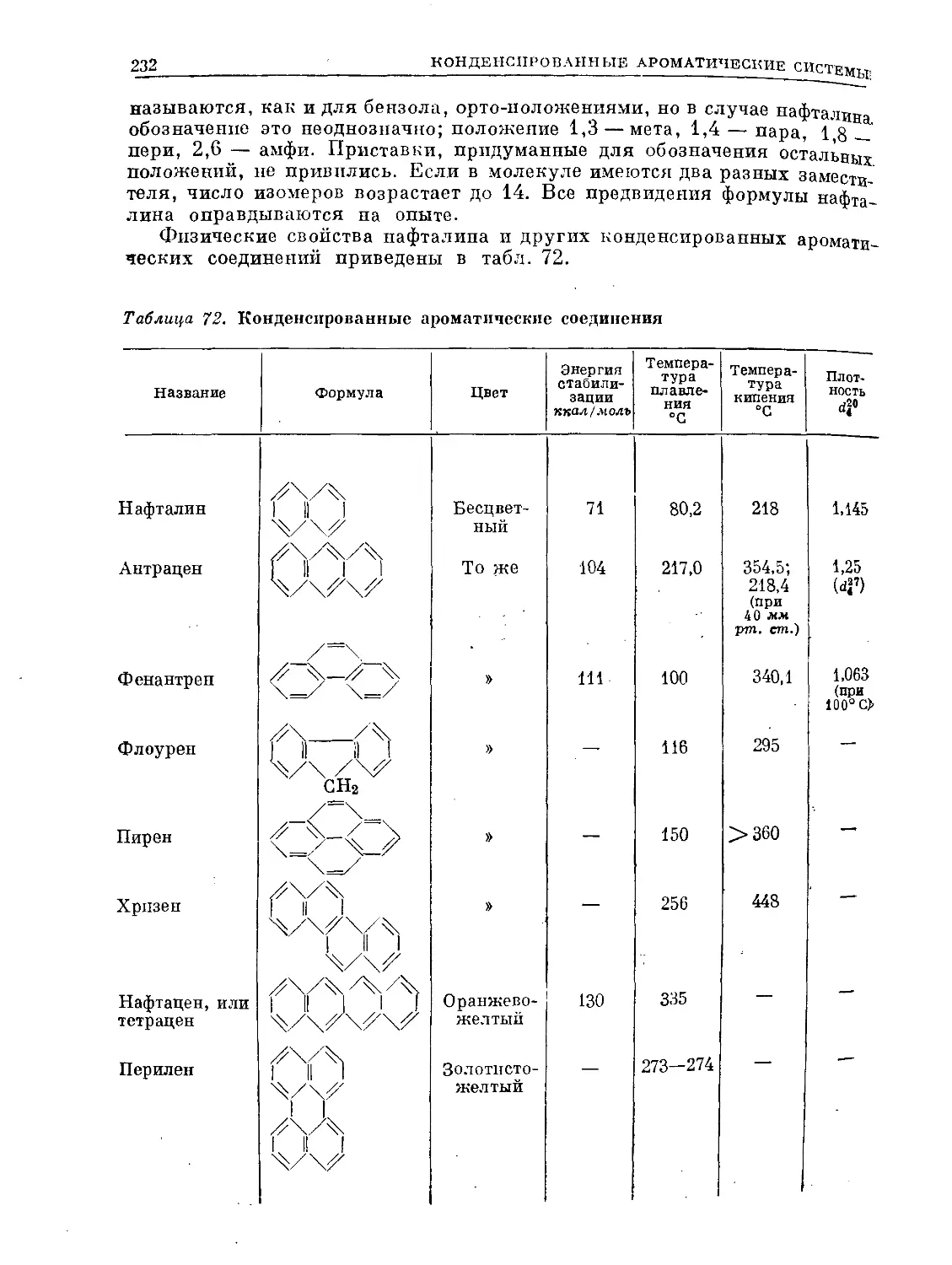
(нафталин, антрацен и др.). Первыми известными химикам веществами этого
ряда (бензойный альдегид, бензойная кислота, толуол и т. д.) были или
хорошо пахнущие вещества, или вещества, выделенные из ароматных баль-
замов (толуол из толуанского бальзама) или других благовонных экзоти-
ческих продуктов (бензойная кислота из росного ладана). Сам родоначаль-
ный углеводород ароматического ряда — бензол открыт в 1825 г. Фарадеем
в светильном газе, из которого он выкристаллизовывался при охлажде-
нии. В главных чертах строение бензола было установлено Кекуле уже
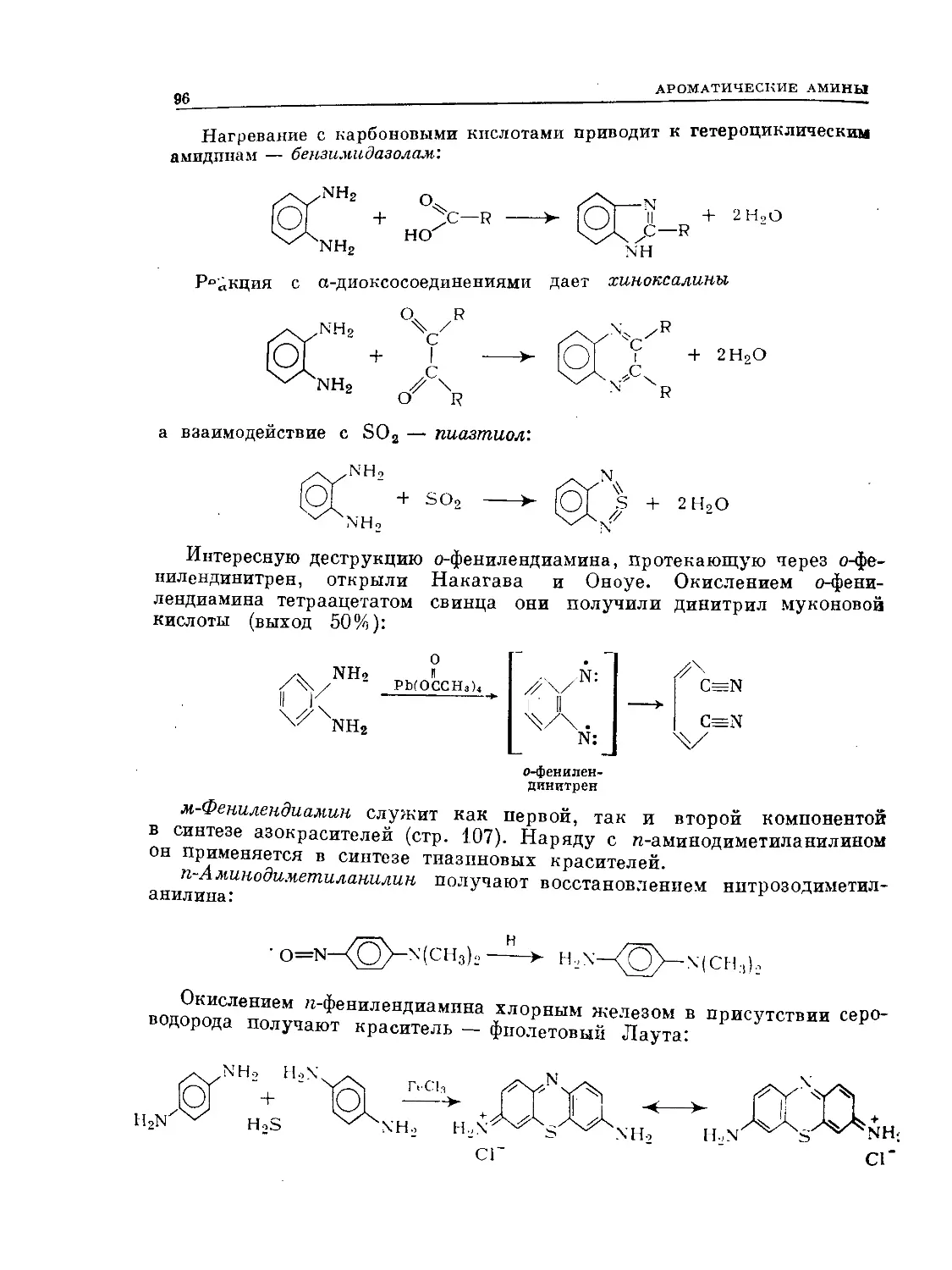
в 1865 г. — через пять лет после создания Бутлеровым теории химического
строения. Несмотря на это в строении бензола и его производных остава-
лась загадка, к которой химики возвращались па протяжении всей после-
дующей истории и которая разъяснилась лишь сравнительно недавно.
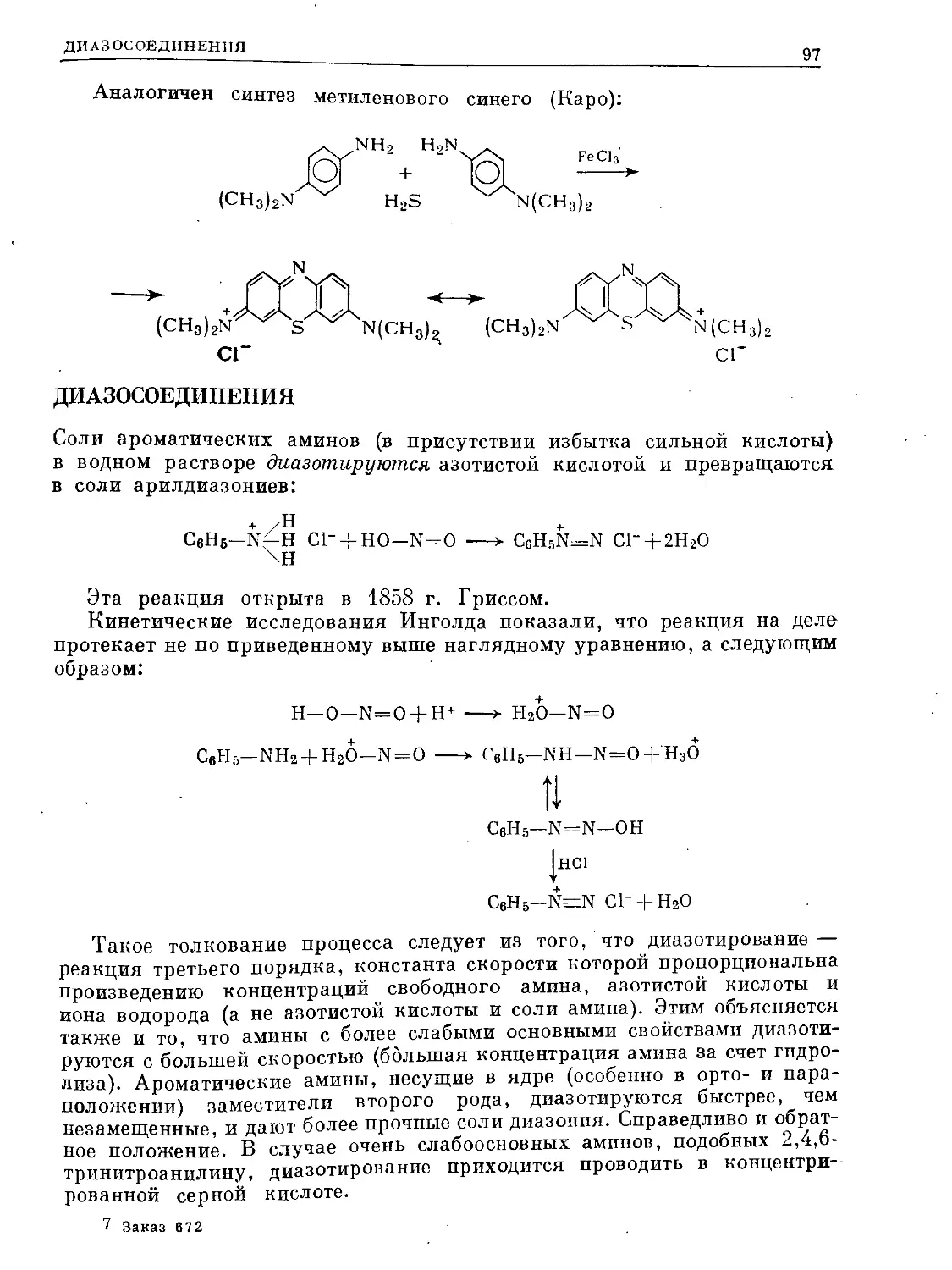
Известные Кекуле факты, послужившие основой для предложенной им
«формулы Кекуле» бензола, были следующие. Бензол
сн
НС сн
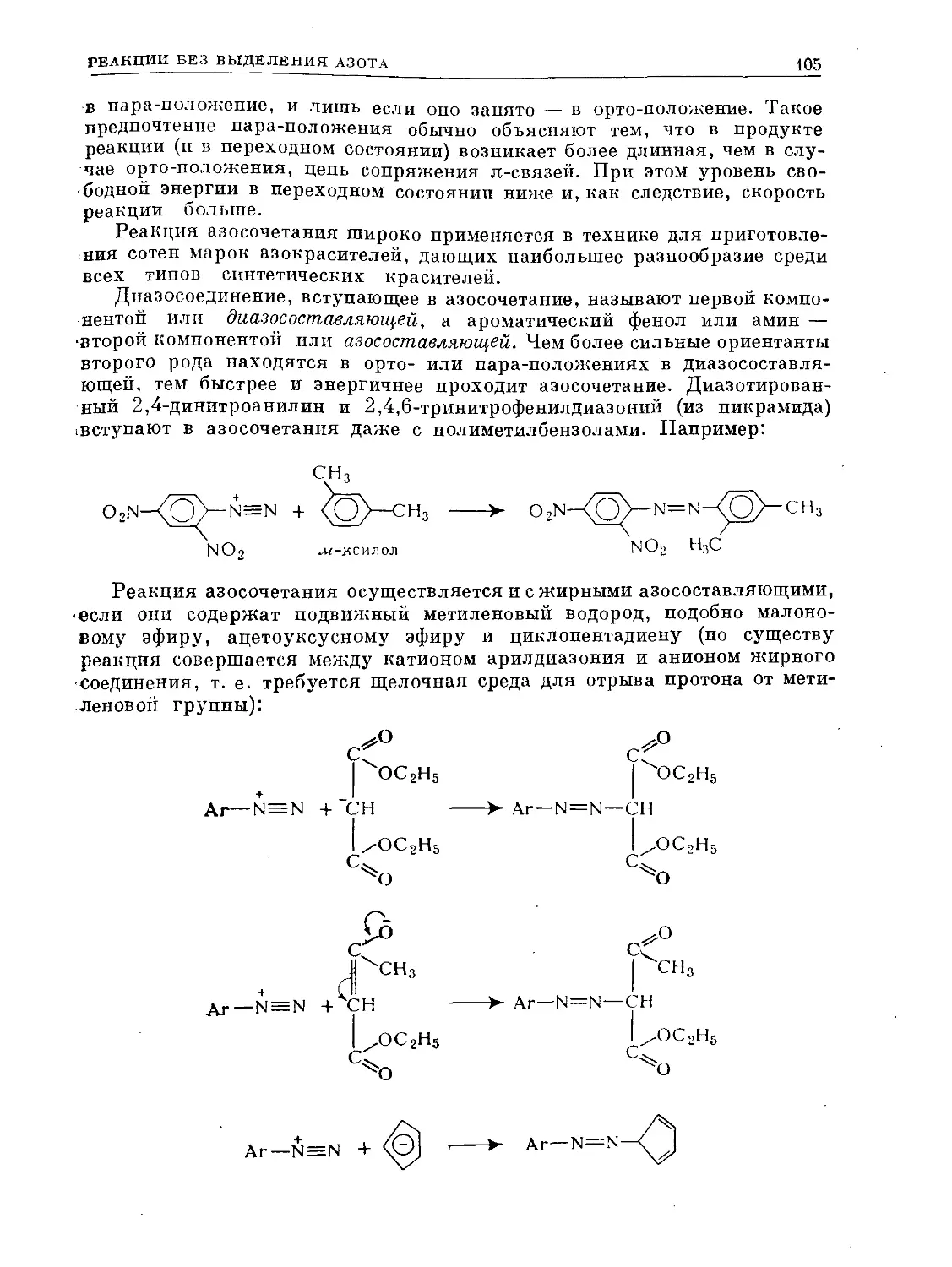
II I
НС сн
сн
и его гомологи удовлетворяют формуле С„Н2л_в, что при очевидной для
бепзола моиоциклической структуре должно обозначать наличие в нем
трех двойных связей (или одной тройной и одной двойной).
В бензоле равноценны все углеродные атомы, так же как и все водород-
ные, что совместимо только с предположением о циклической структуре.
Таким образом, замещение любого водородного атома бензола приводит
к одному и тому же мопозамещенному продукту. С другой стороны,.
10
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫ^
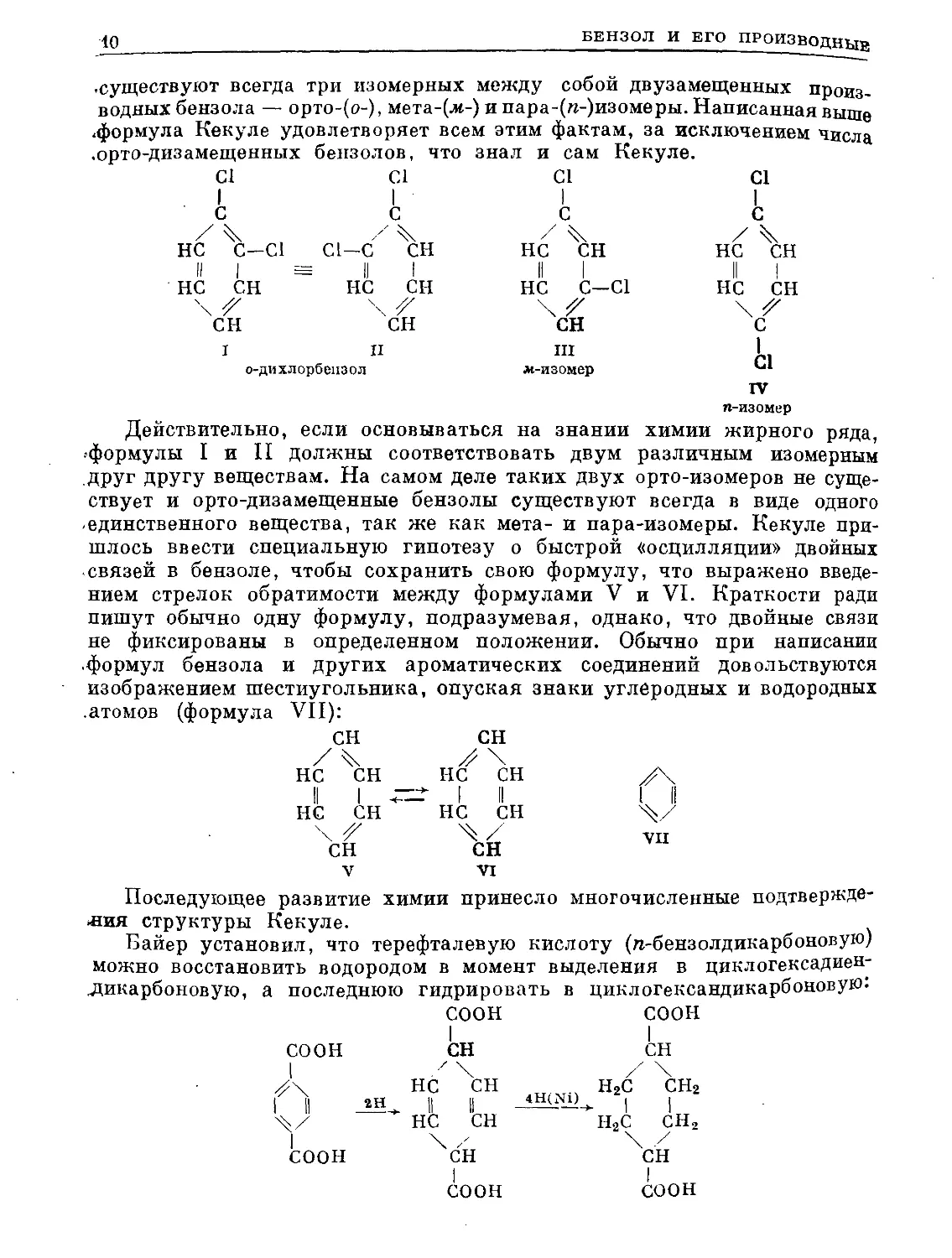
•существуют всегда три изомерных между собой двузамещенных произ-
водных бензола — орто-(о-), мета-(ле-) и пара-(п-)изомеры. Написанная выше
•формула Кекуле удовлетворяет всем этим фактам, за исключением числа
•орто-дизамещенных
C1
бензолов, что знал и сам Кекуле.
С1
С1
С1
НС
нс
с
с
С
С
C-CI
С1—с
сн
НС
сн
НС
сн
сн
нс
сн
нс
С—С1
нс
сн
СН
I
СН
и
о-ди хлорбензол
СН
III
м-изомер
с
С1
иг
п-изомер
химии жирного ряда
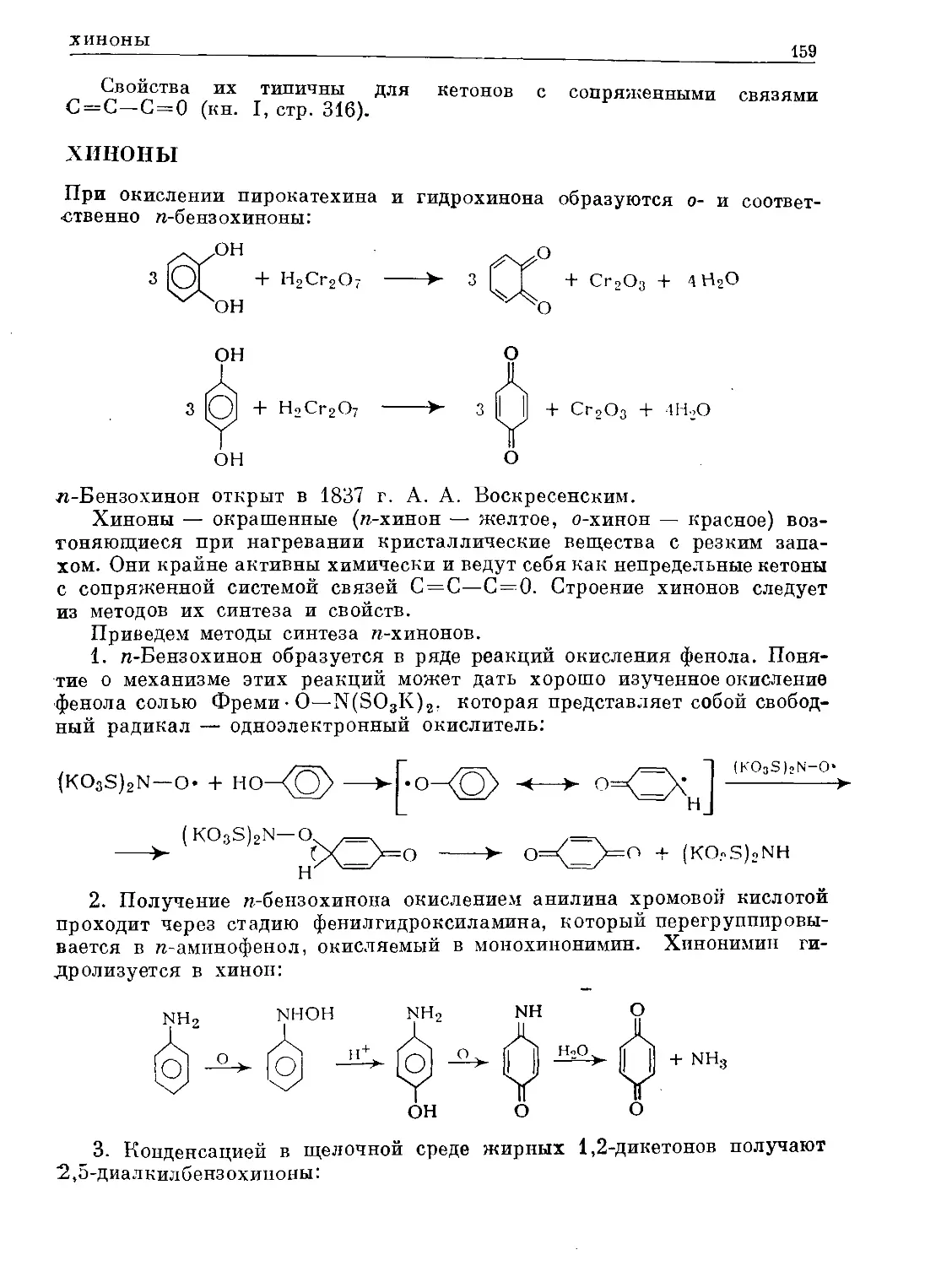
Действительно, если основываться ___________
•формулы I и II должны соответствовать двум различным изомерным
друг другу веществам. На самом деле таких двух орто-изомеров не суще-
ствует и орто-дизамещенные бензолы существуют всегда в виде одного
•единственного вещества, так же как мета- и пара-изомеры. Кекуле при-
шлось ввести специальную гипотезу о быстрой «осцилляции» двойных
связей в бензоле, чтобы сохранить свою формулу, что выражено введе-
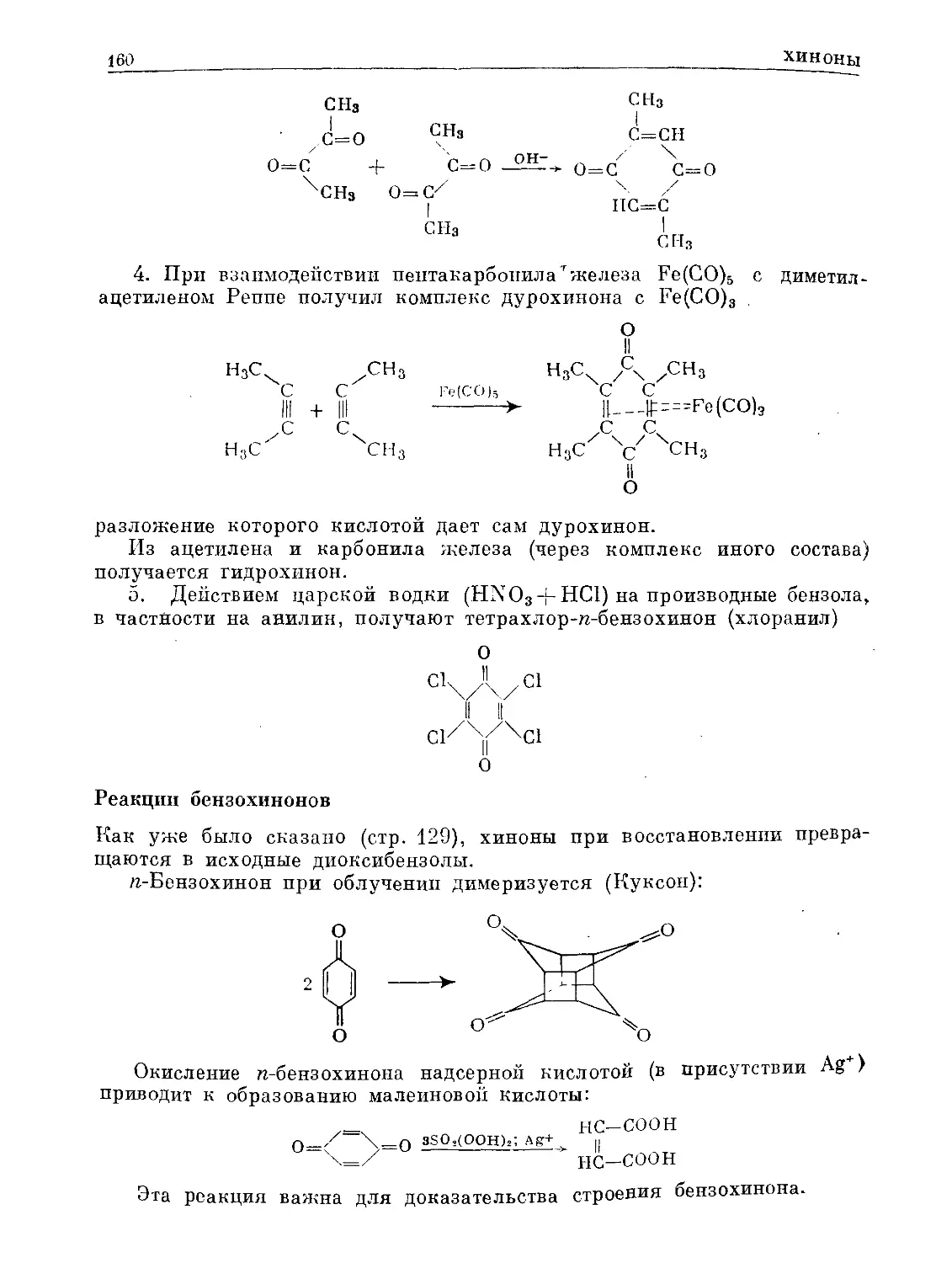
нием стрелок обратимости между формулами V и VI. Краткости ради
пишут обычно одну формулу, подразумевая, однако, что двойные связи
не фиксированы в определенном положении. Обычно при написании
•формул бензола и других ароматических соединений довольствуются
изображением шестиугольника, опуская знаки углеродных и водородных
.атомов (формула VII):
СН
на знании
СН
НС
СН
НС
СН
НС
СН
НС
СН
VII
СН
VI
Последующее развитие химии принесло многочисленные подтвержде-
ния структуры Кекуле.
Байер установил, что терефталевую кислоту (п-бензолдикарбоновую)
можно восстановить водородом в момент выделения в циклогексадиен-
Ликарбоновую, а последнюю гидрировать в циклогександикарбоновую.
СООН
I
СН
нс \н
II II
НС сн
^СН
СН
СООН
СООН
СООН
2Н
4H(Ni)
Н2С
I
Н2С
СООН
СН
СН2
сн,
сн
I
СООН
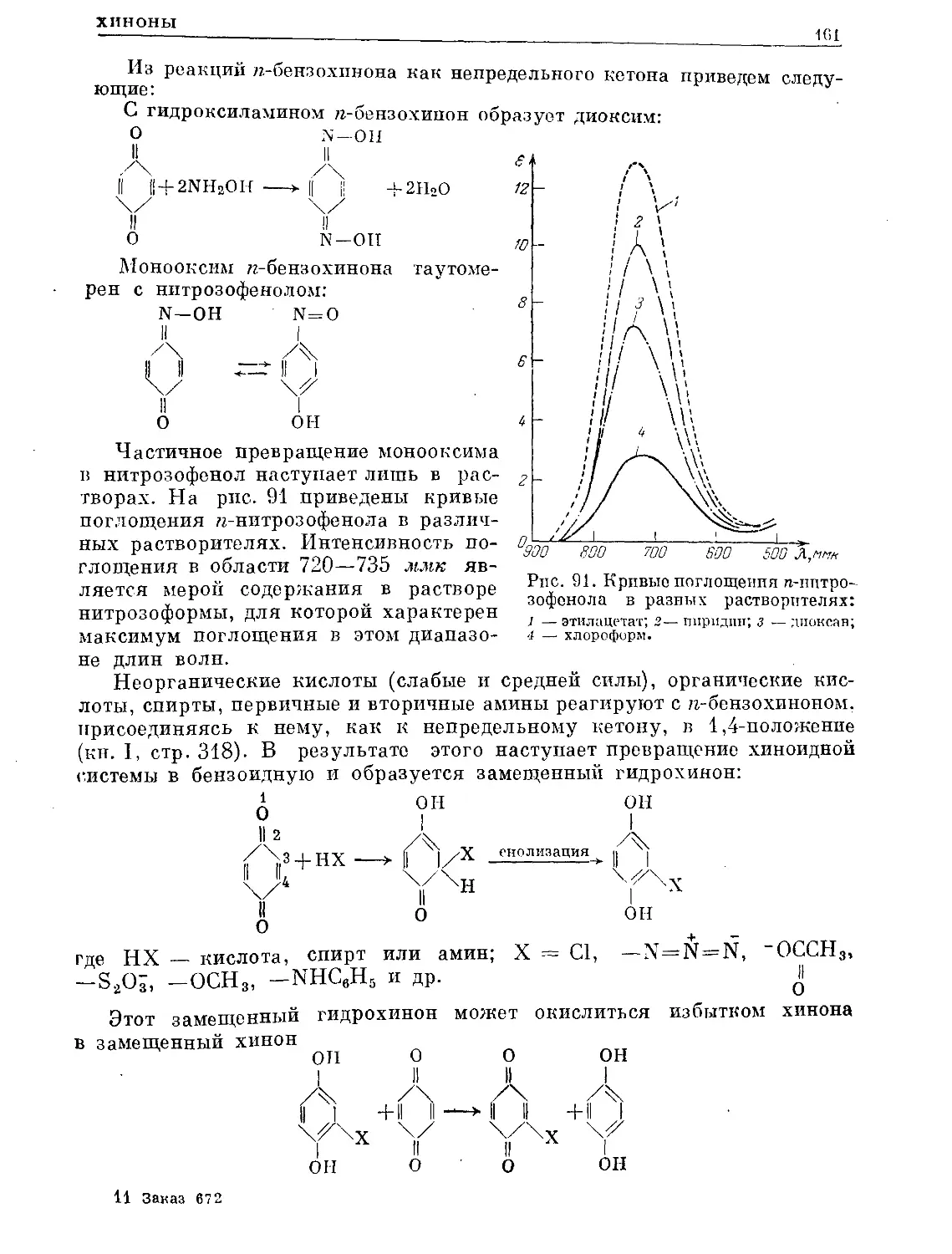
БЕНЗОЛ И ЕГО СТРОЕНИЕ
11
Таким образом, были впервые связаны ряды бензола и циклогексана
и воочию доказано наличие в бензоле шестичленного цикла.
Около 1900 г. Сабатье, используя найденный им метод гидрирования
непредельных соединений молекулярным водородом над никелем, показал,
что те же отношения существуют для самого бензола и циклогексана:
I II aH«(Ni) г
сн2
н2с \н2
I I
Н2С сн2
сн2
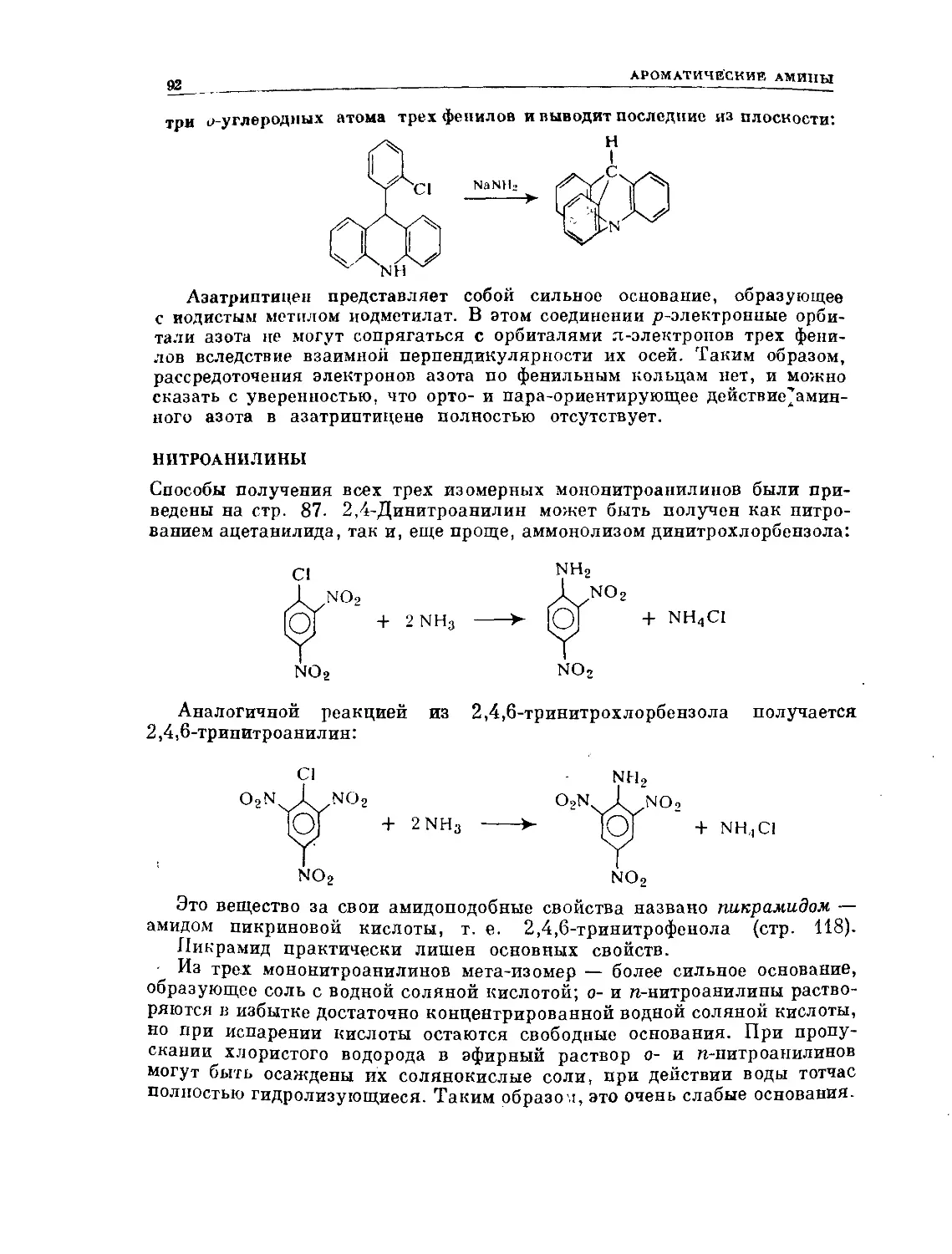
В 1912 г. Н. Д. Зелинский осуществил обратную реакцию — аромати-
зацию циклогексана в бензол над платиновым или палладиевым катали-
затором:
СН2
н2с сн2
I I II 1 + ЗН2
Н2С сн2
сн2
В 1904 г. Гарриес проозонировал бензол, причем озонирование про-
ходило по типу озонирования олефинов. Это доказывает, что все шесть
углеродных атомов бензола по валентному состоянию олефиновые; озонид
бензола после гидролиза превращается в глиоксаль и перекись водорода:
/СН
нсх ЧСН
II I
НС\ /;Н
ХСН
о
Х°
Н( о |
о/1 сн
I О I
° I /сн
НС <3 I
НС\
с>
ЗН2О2
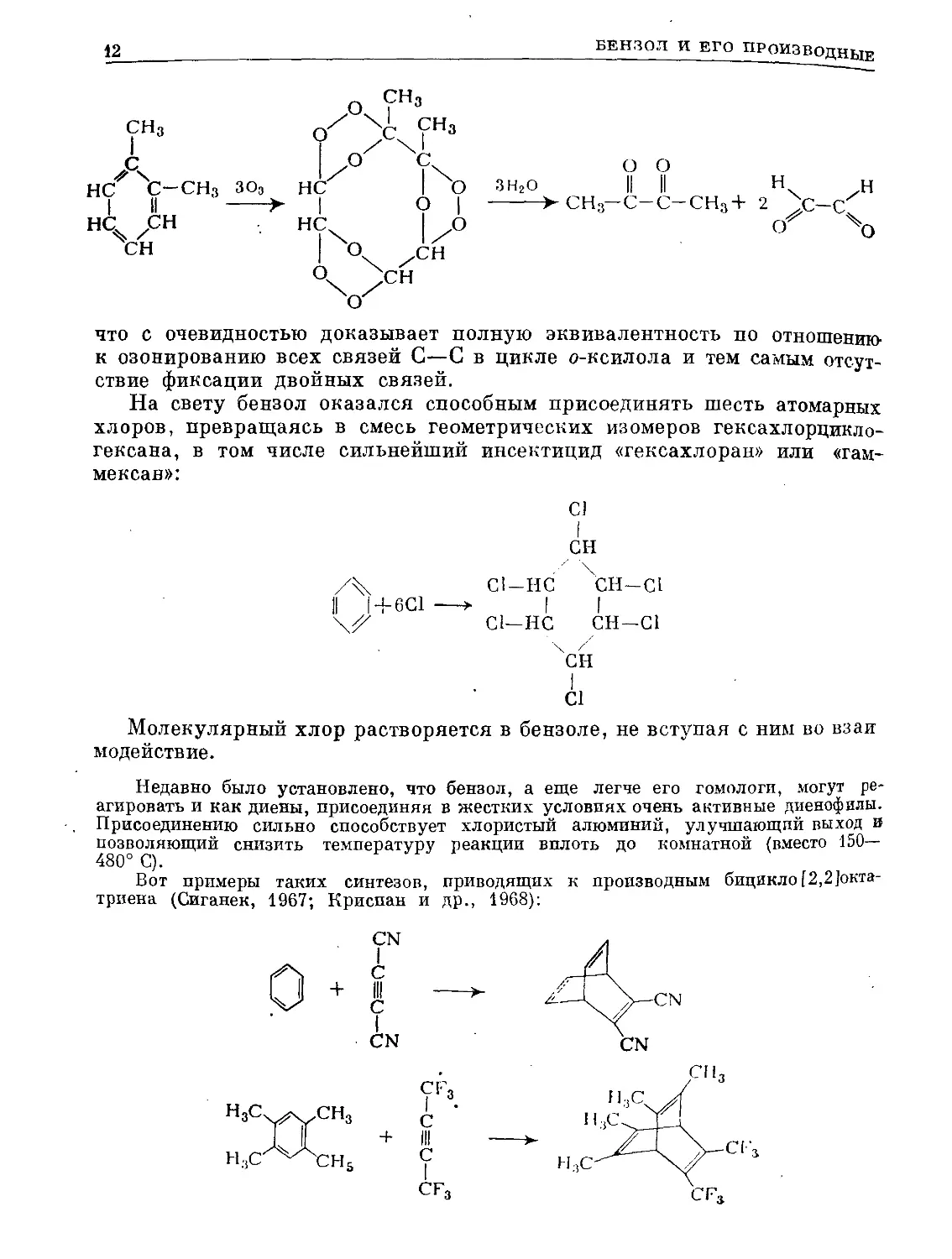
Озонирование о-ксилола (о-диметилбензола) приводит к глиоксалю,
метилглиоксалю и диацетилу в молекулярном соотношении 3:2:1
(Вибо, 1941 г.)
I
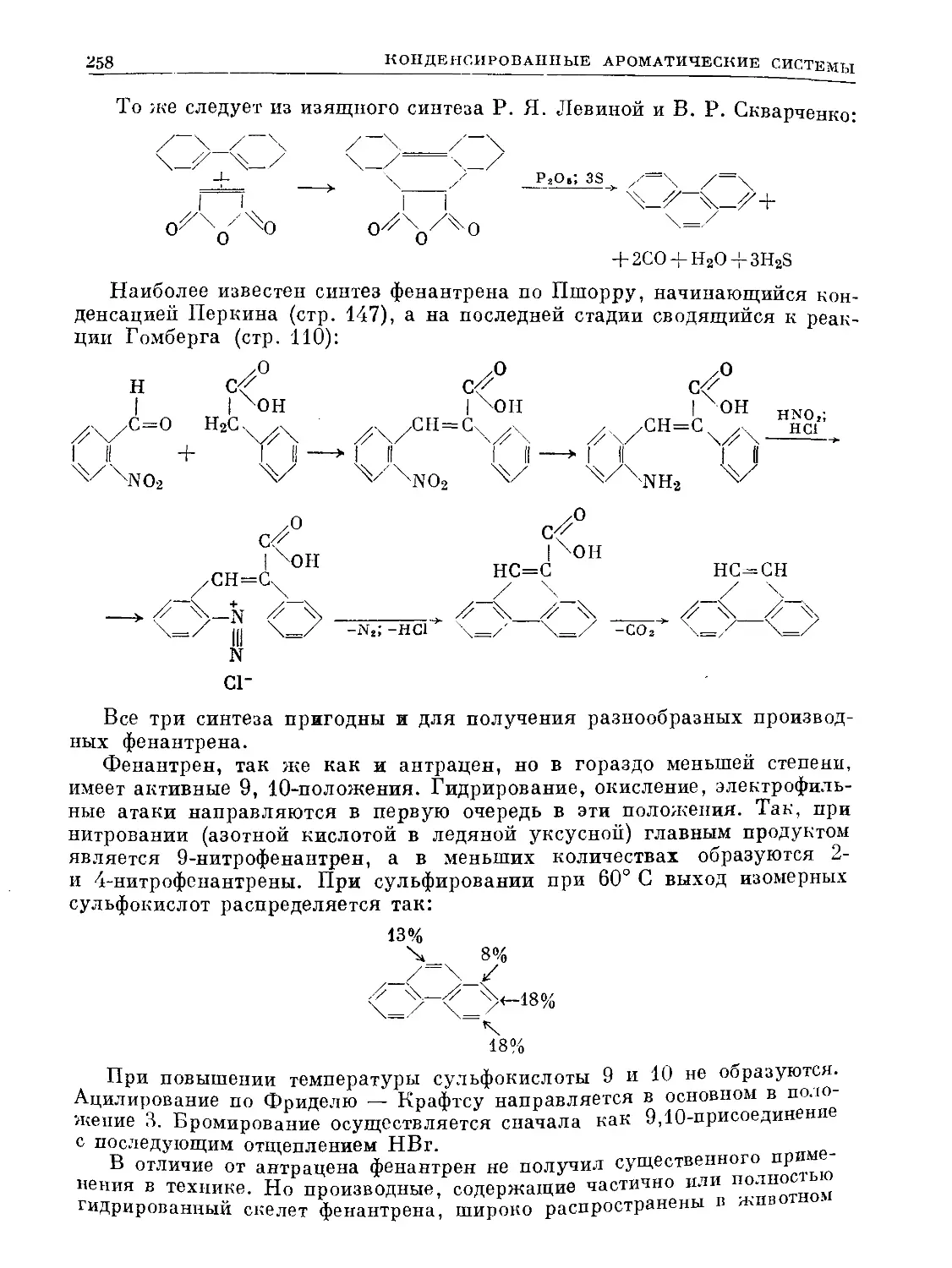
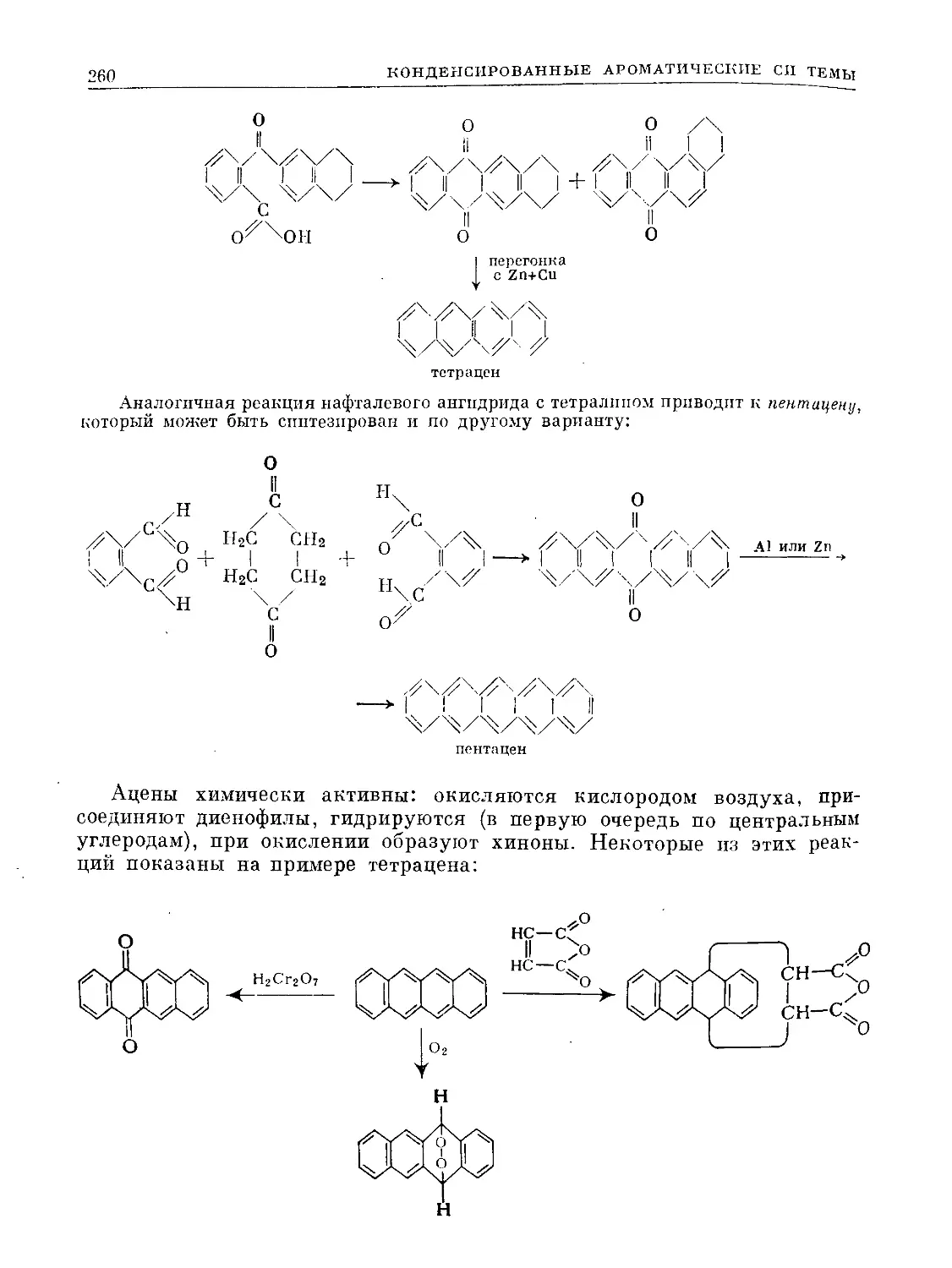
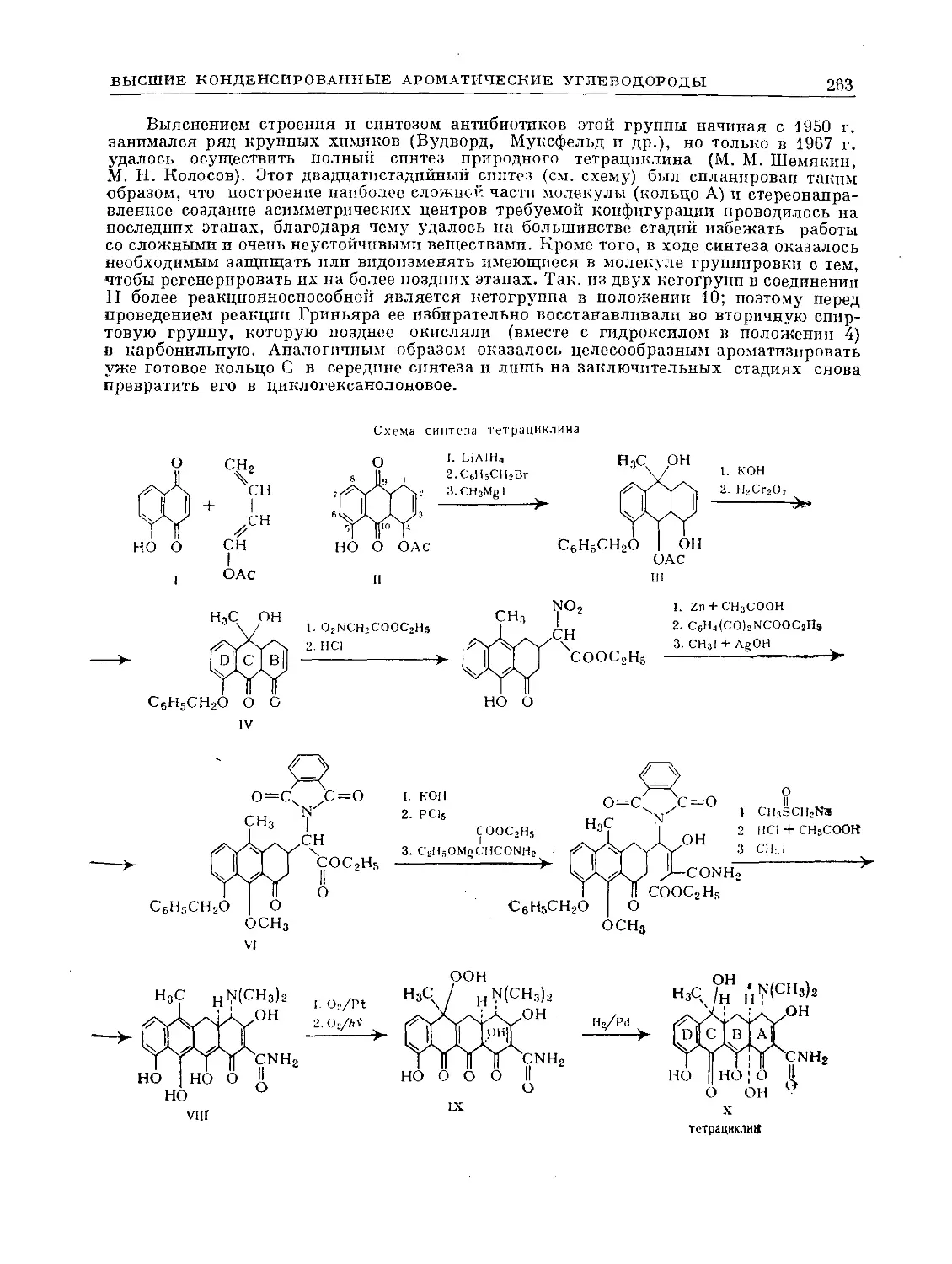
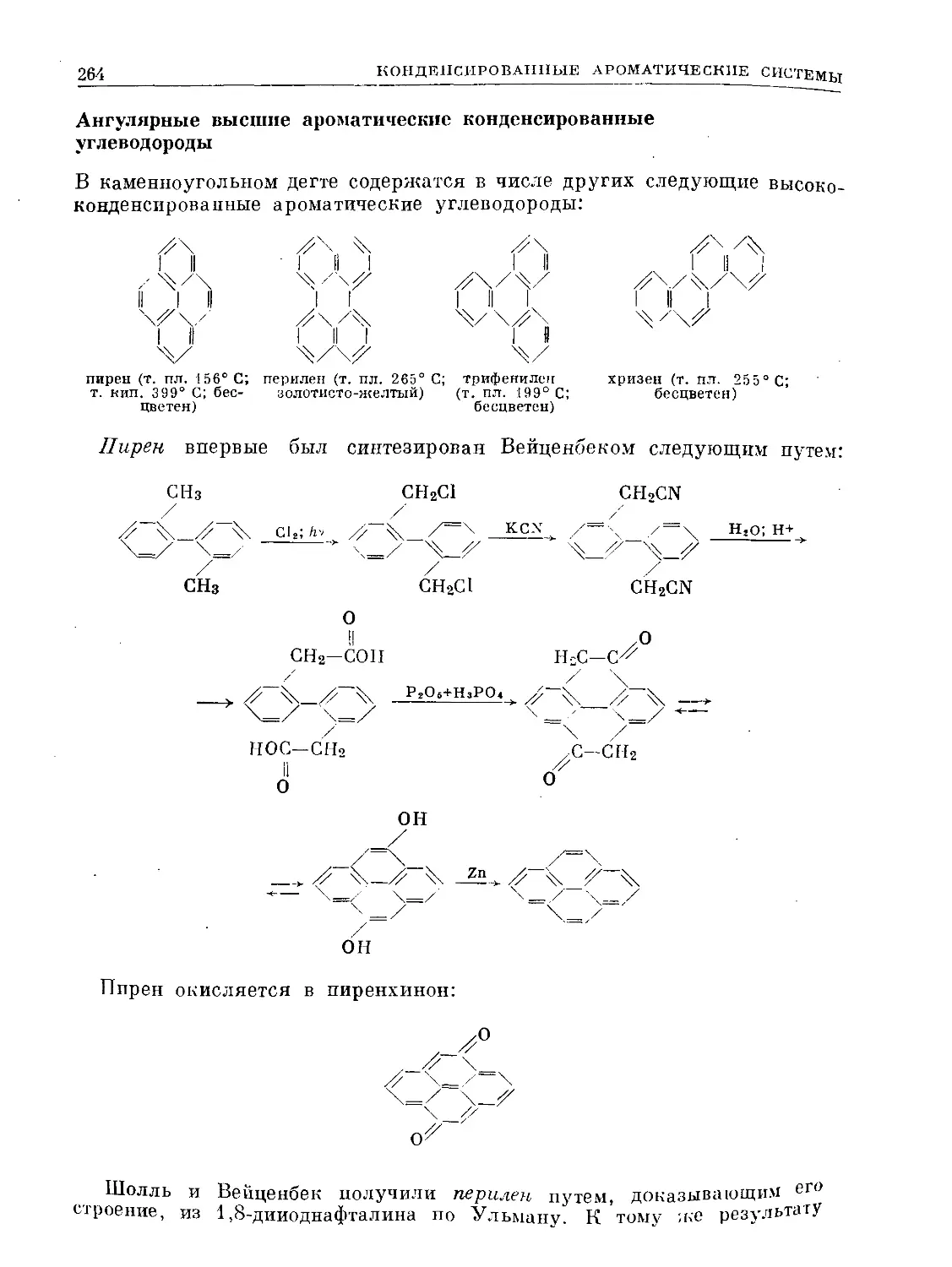
ХО
С НС О |
нсх %с—СН3 3°з ' С-СНз
II I 3—> I о I
НС ^СН о. | сн
чсн НС СУ |
НС\ <3
° .1
3ICO II
----> 2СН3-с-С
Ч)
12
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
<у ?н’
СИ3 0/ V сн,
А 1/° ?\
НС С-СНз ЗОэ нс I о
I » —>- 1 9 >
нс СН НС о
IX /СН
о ^сн
хс>
о о
зн2о II II
----> сн3—с—с—сн3+
что с очевидностью доказывает полную эквивалентность по отношению
к озонированию всех связей С—С в цикле о-ксилола и тем самым отсут-
ствие фиксации двойных связей.
На свету бензол оказался способным присоединять шесть атомарных
хлоров, превращаясь в смесь геометрических изомеров гексахлорцикло-
гексана, в том числе сильнейший инсектицид «гексахлоран» или «гам-
мексан»:
CJ
I
СН
CI-HC ХСН-С1
II I + 6C1 > ( |
С1—НС СН—Cl
\н
С1
Молекулярный хлор растворяется в бензоле, не вступая с ним во взаи
модействие.
Недавно было установлено, что бензол, а еще легче его гомологи, могут ре-
агировать и как диены, присоединяя в жестких условиях очень активные диенофилы.
Присоединению сильно способствует хлористый алюминий, улучшающий выход и
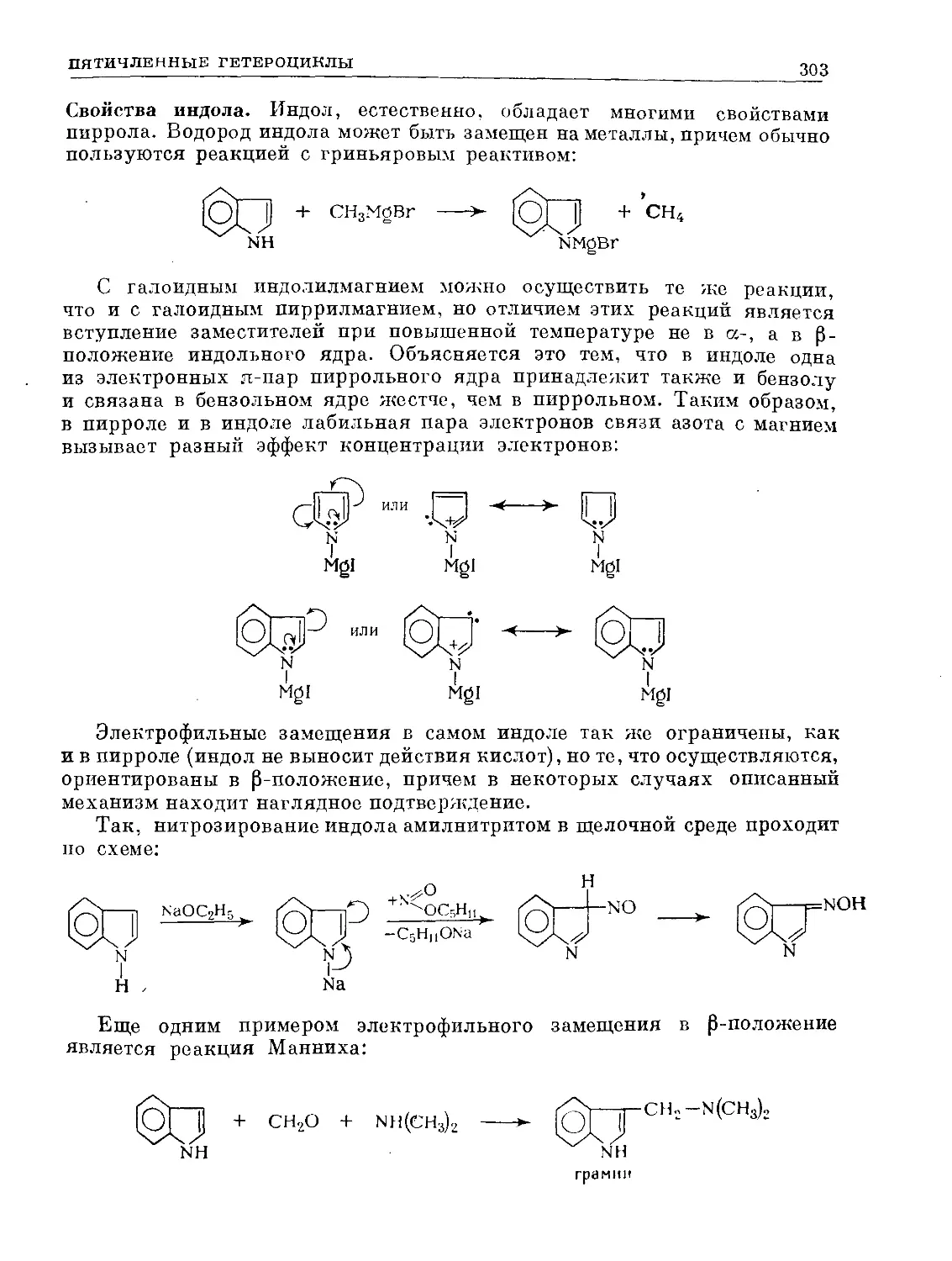
позволяющий снизить температуру реакции вплоть до комнатной (вместо 150—
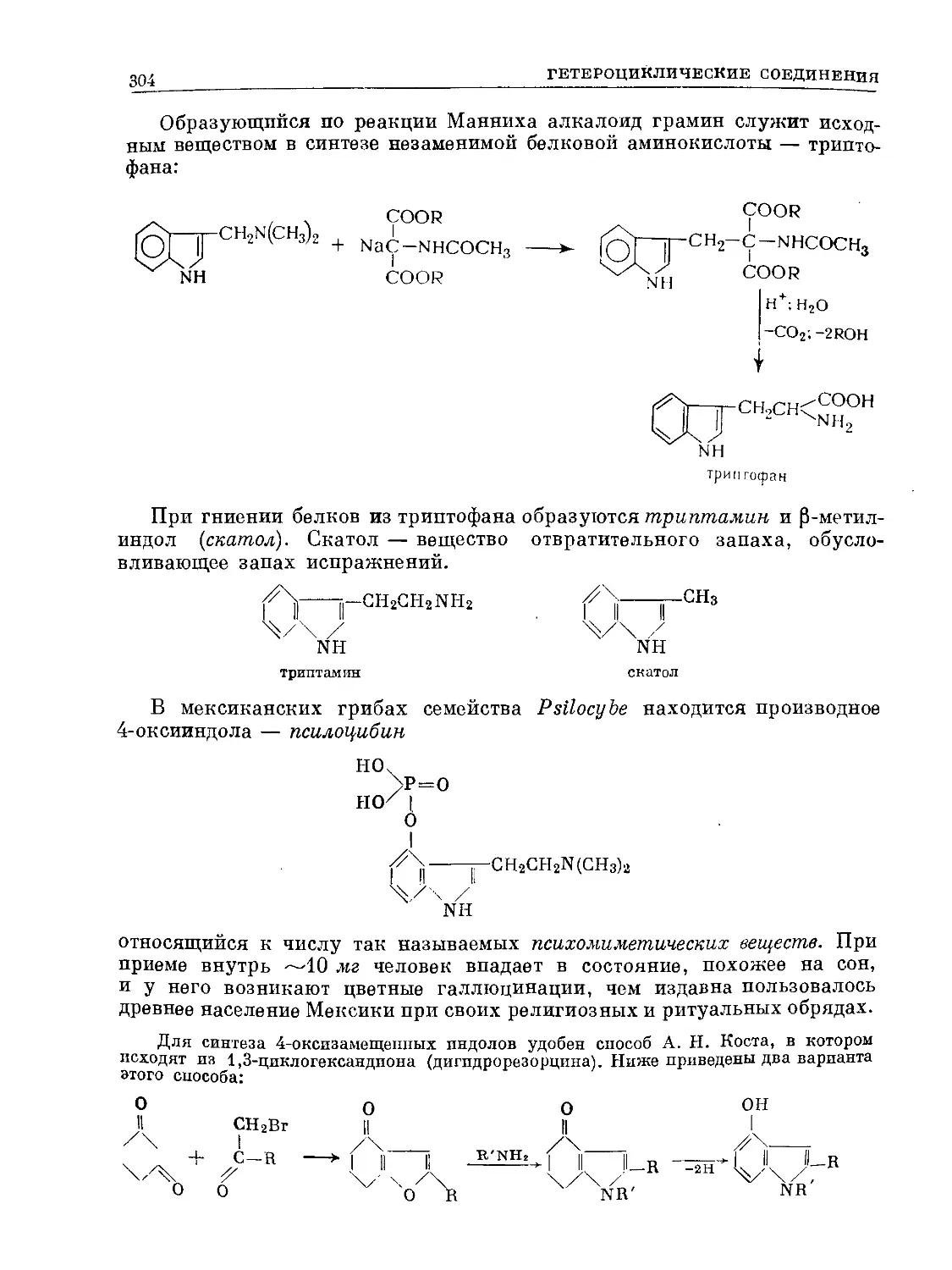
480° С).
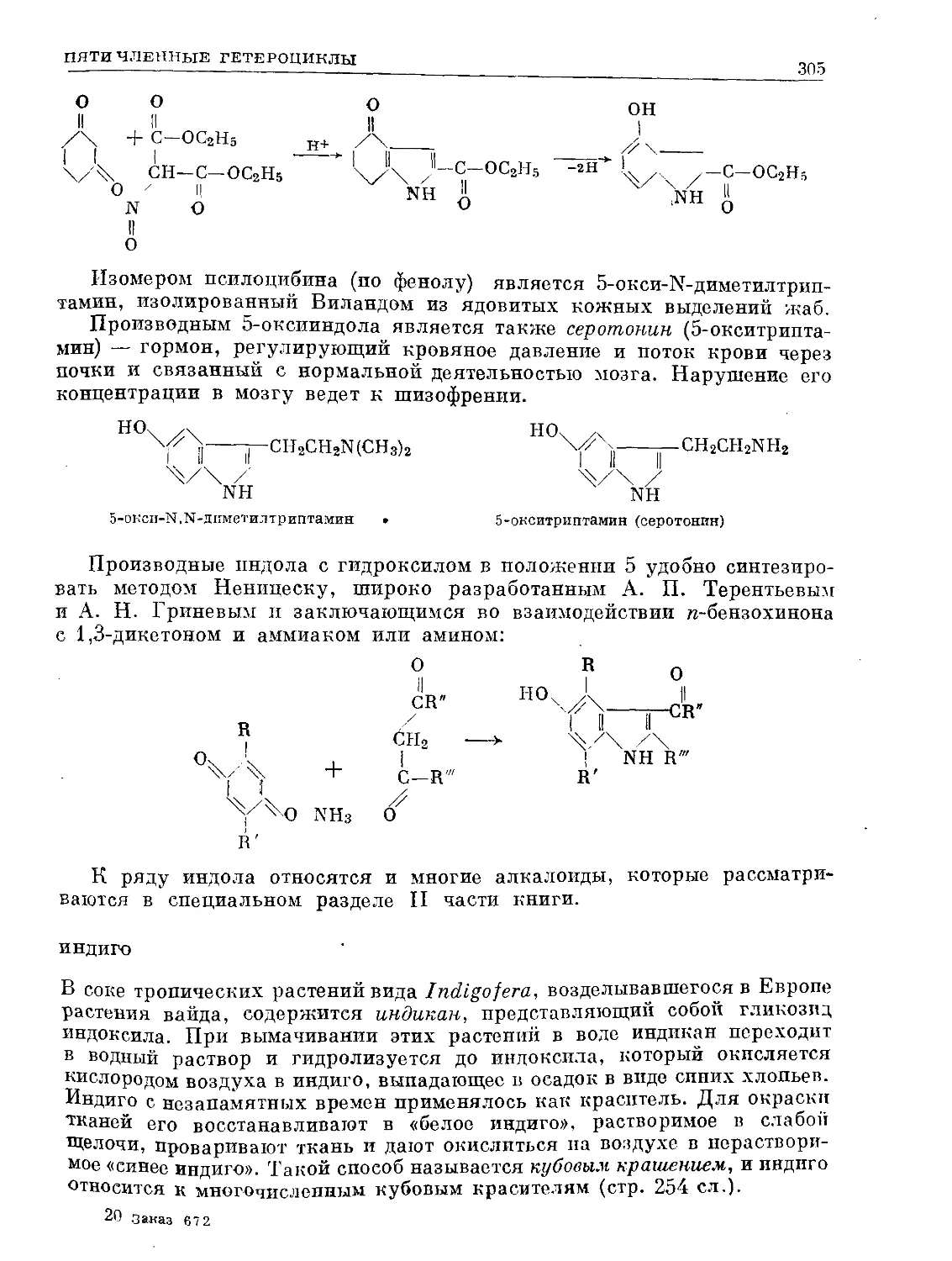
Вот примеры таких синтезов, приводящих к производным бицикло [2,2]окта-
триена (Сиганек, 1967; Криспан и др., 1968):
БЕНЗО л II ЕГО СТРОЕНИЕ_________________________________________12
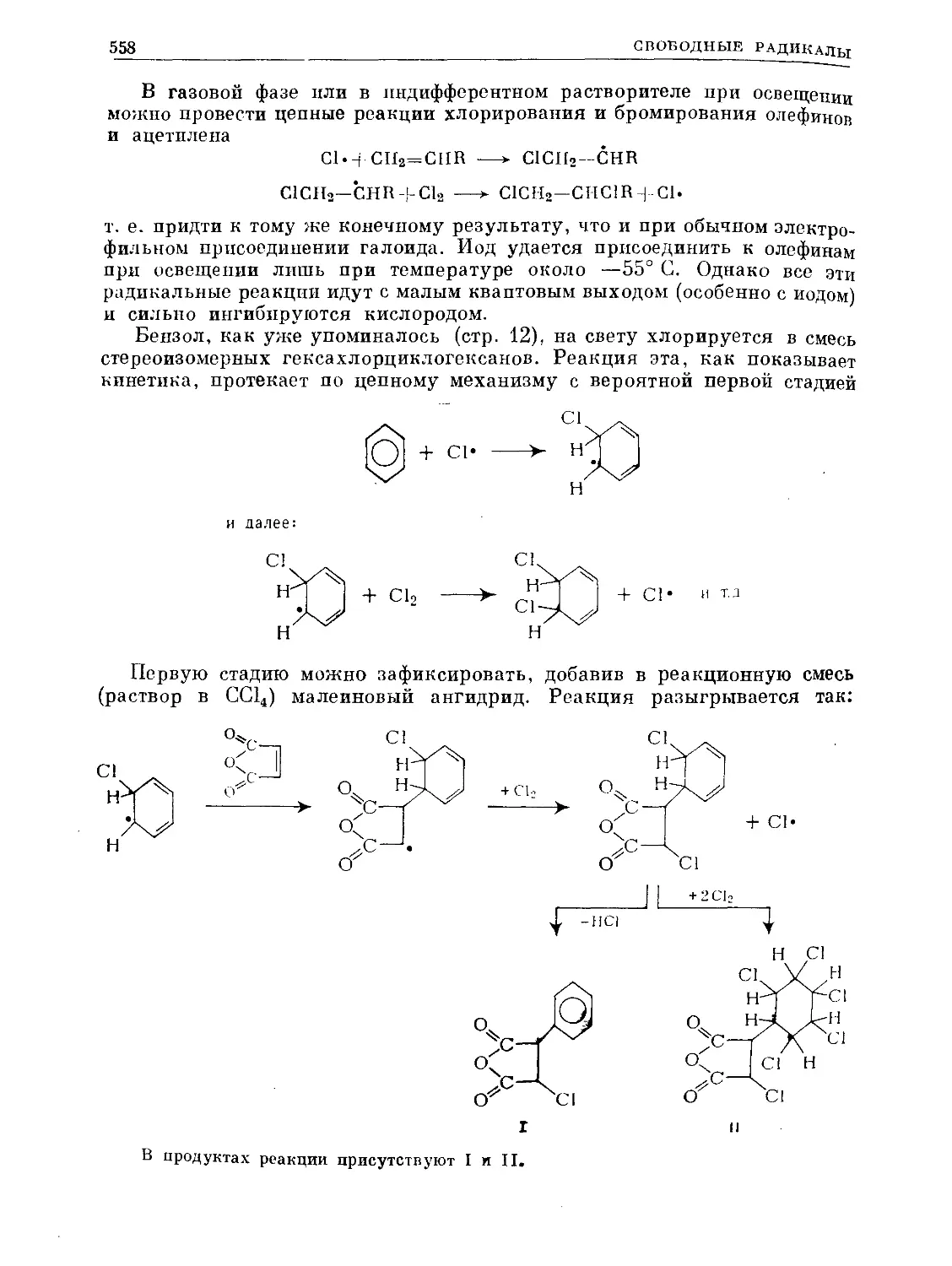
При облучении реакционной смеси и малеиновый ангидрид присоеди-
няется к бензолу как к диену, образуя
Предложенное Кекуле строение бензола подтверждается, многими
синтезами бензола, его гомологов и производных из соединений жирного
ряда, которые будут приведены на стр. 19.
Несмотря на такое, казалось бы, блестящее согласие формулы с фак-
тами, многие крупные химики возвращались к пересмотру структуры бен-
зола и предлагали иные формулы (Клаус, Ладенбург, Байер), предста-
вляющие яыне лишь исторический интерес. Эта неудовлетворенность
формулой Кекуле основывалась на том, что бензол (п все ароматические
соединения) в реакциях присоединения несравненно инертнее олефинов.
Реализуются лишь редкие реакции присоединения (3H2/Ni; О3; 6С1),
а многие из реагентов, которые присоединяются к олефинам, в аромати-
ческом ряду приводят к замещению водородов. Особенно характерна
стойкость бензола к окислителям (КМпО4, Н2Сг2О7).
Большая инертность бензола сравнительно с олефинами (на примере
циклоолефинов, наиболее близких к бензолу) явствует из следующего
энергетического сопоставления:
Pt
---►
СН2
Н2С \н2
| | +28,8 ккал
н2с сн2
ХСН2
? |!+ ЗН2
сн2
Н2с ^СНг
I I
н2с сн2
+ 49,8 ккал
Если бы каждая двойная связь бензола энергетически была равноценна
двойной связи циклогексена, то при гидрировании бензола в циклогексан
выделилось бы 28,8 X 3 = 86,4 ккал/моль, т. е. на 36,6 ккал/молъ больше,
чем в действительности. На эту величину бензол энергетически беднее
(и, значит, устойчивее), чем был бы циклоолефин с фиксированной струк-
турой Кекуле — циклогексатриен.
Любое нарушение «бензоидиости» ароматического соединения, напри-
мер путем присоединения хотя бы пары атомов за счет изъятия из секстета
двух электронов, вело бы к дополнительной потере «энергии резонанса»
14
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
(36,6 ккал/моль), что и является одной из причин редкости подобных
реакций.
сн
нс^хсн2
I I
нс^ хсн2
сн
2С1
СН
НС^ХСН-С1
НСХ/СН—С1
хсн
Из приведенных ранее (см. метод МО, кн. I, стр. 241 сл.) молекуляр-
ных диаграмм видно, что в случае бутадиена можно еще приближенно>
говорить о двойных связях между углеродами 1—2 и 3—4, хотя л-связи
между углеродами 1—2 и 3—4, хотя л-связи
локализованы в бутадиене не в такой сте-
пени, как в этилене. На это указывает
существенно отличный от двух (~ 1,45)
порядок связи между атомами углерода
2—3, отличные от нуля,
большие, значения энергии
(в методе МО) или энергии
языке теории резонанса) и,
сколько сокращенная длина
углеродами 2—3 (1,48 А вместо 1,54 А для
ординарной связи С—С). Таким образом,
представление бутадиена структурной фор-
мулой СН2=СН—СН=СН2 неоднозначно,
локализованных двухцентровых л-связях уже
е
хотя и не-
делокализации
резонанса (на
наконец, не-
связи между
Рис. 80. Схема уровней орби-
тальных энергий для моле-
кулы бензола.
В бензоле же говорить о
ХтХТов углеппиГ * М°ЛеКуле ^зола одинаковы, как и все
ных данных- мптгр ’ ЧТ° совеРшенно ясно вытекает из эксперименталь-
оавна 1 zn А кУла копланарна, длина каждой из шести связей
гексагональная ВСро ВаЛеНТНЫе углы Равны 120°, симметрия молекулы
в согласии е чкгп ЗУЛЬТЙТЫ квантов°-™ических расчетов находятся
рЛ с ЭКСПеРИментальными данными.
исходит6 и°°шестт?У М° ЛКАО в л-электронном приближении Хюккеля
Уровней энрпгип w атомных Рг~°рбиталей углерода. Качественно схема
показаны на рис ^спРСДелспие по ним электронов в основном состоянии
Рг’Орбитали^углеиопа Ь^ЭЯ ЛИНИя означает энергию электрона на атомной
горизонтальные сплошиыр ппЗУЮ Зй начало отсчета на шкале орбитальных энергий;
женным ниже пунктипяЛЛ^п™ ~ УР°ВНИ орбитальной энергии. Уровням, располо-
тали, а лежащим вы inf С00тве'гствУК>т связывающие молекулярные орби-
тивоположным ciiiinm.i разрыхляющие; стрелками обозначены л-электроны (с про-
ижным спином) на связывающих орбиталях.
В осяп^Л^УЛЯ₽НЫе °Р^итали относятся к л-типу (рис. 81).
кую орбиталь1,сос.тоянии молекулы два электрона занимают самую глубо-
\a2u)j принадлежащую нижнему невырожденному значению
БЕНЗОЛ И ЕГО СТРОЕНИЕ
15-
орбитальной энергии. Остальные че-
тыре электрона заполняют две
связывающие молекулярные орбита-
ли (elg), принадлежащие следующему
в порядке возрастания двухкратно
вырожденному уровню энергии. Три
разрыхляющие (или антисвязыва-
ющие) молекулярные орбитали (л[,
л*, nJ) расположены симметрично
связывающим относительно начала
отсчета на шкале орбитальных энер-
гий (см. рис. 80). На эти орбитали
я-электроны бензола переходят при
возбуждении поглощением в ближней
ультрафиолетовой области (л —► л*-
переход). Первый максимум погло-
щения в электронном спектре бен-
зола соответствует наименьшему
кванту света и, следовательно, наи-
большей длине волны X 250 ммк
(2500 А).
На рис. 81 показаны граничные
поверхности молекулярных л-орбита-
лей бензола, а на рис. 82 дано при-
близительное изображение граничной
поверхности трехмерной л-электрон-
ной плотности в молекуле бензола.
Молекулярная диаграмма бензола,
находимая в л-электронном прибли-
жении Хюккеля, имеет следующий
вид:
Эффективные заряды атомов рав-
ны нулю; симметрия позволяет огра-
ничиться указанием порядка одной
связи 1,66 и индекса свободной
валентности 0,40 одного атома; по-
рядки о-связей приняты равными
единице (кн. I, стр. 249).
Диаграмма хорошо отражает
выровненность связей и равноцен-
ность атомов.
Энергия делокализации по сравнению с системой трех локали-
зованных двухцентровых л-связей формулы Кекуле в единицах 6 рав-
на 2 (~36 ккал/молъ).
Рис. 81. Граничные поверхности моле-
кулярных л-орбиталей бензола (вид'
сверху):
z,, тг2, к, — связывающие орбитали (соответ-
ствснио а2и, e,g, e,g); т*, “а - “з—разрых-
ляющие орбитали; пунктирныз линии 0 —
узловые плоскости.
Рис. 82. Граничная поверхность трех-
мерной л-электронпой плотности в мо-
лекуле бепзола (приблизительное изоб-
ражение).
16
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
Э. Хюккель в 1931 г. сформулировал и обосновал в рамках своего л-элек-
тронного приближения в методе МО ЛКАО так называемое (4п + 2)-цра_
вило.
Согласно этому правилу, плоские моноциклические системы, состоя-
щие из атомов, вносящих в л-электронную систему молекулы 4п 2
р-электронов, являются, подобно бензолу, ароматическими циклами
с замкнутыми электронными оболочками (все связывающие и только
связывающие орбитали заполнены электронами), стабилизованными
в силу значительной величины энергии делокализации.
Хюккель предвидел, однако, возможность дестабилизующего влия-
ния напряжения о-скелета.
Такого рода системы будут рассмотрены и ниже в разделе «Небензоид-
ные ароматические системы».
Интересно, что циклооктатетраен, не удовлетворяющий правилу Хюк-
келя, совершенно лишен ароматического характера*. Существуют и гете-
роциклические ароматические системы (см. «Гетероциклы») и в их числе —
•-«неорганический бензол», боразол'.
вн вн вн
/ +/^ \ +
HN NH HN NH HN NH
I I — II I — I II
НВ ВН НВ- -вн НВ- -вн
\ Z \+z ^+/
NH NH NH
Переходя к описанию л-электронной структуры бензола с точки
зрения резонанса, мы представляем ее как резонанс пяти так называемых
«канонических» структур — Двух структур Кекуле (а и а') и трех струк-
тур Дьюара (б) **
Напомним, что понятие о резонансе структур исходит из стремления
использовать по возможности привычные в классической химии спосооы
изображения строения молекул. Резонансные структуры не предполага-
ются существующими в отдельности даже в качестве возбужденных
состояний молекул.
* Однако при действии щелочного металла циклооктатетраен захватывает два
электрона и превращается в ароматический двузарядный анион С8Н|_, удовлетворя-
ющий правилу Хюккеля (4 -2 + 2 = 10).
** Такое обозначение структур Дьюара законно, поскольку л-связи между это
мами углерода в пара-положении имеют формальный характер и существенной являет-
ся, с одной стороны, антипараллельность спинов р-электронов на атомных рг-оро
талях этих атомов, а с другой — полнота набора канонических структур. Следуе ,
конечно, иметь в виду, что стрелки не подразумевают указания на абсолютную орпе
бензол и его строение
17
В резоиаисе пяти канонических структур вес * структуры а или а
гораздо больше, чем вес какой-либо из структур б. Вес одной структуры
Дьюара б и вес одной структуры Кекуле а относятся как 0,19 : 1; в про-
центах вес одной структуры Кекуле составляет 39%, тогда как вес одной
структуры Дьюара — 7%. Энергетически резонанс пяти канонических
структур беднее структуры Кекуле **, на величину энергии резонанса,
а именно на 36,6 ккал/молъ, а эта последняя значительно беднее струк-
туры Дьюара.
Несмотря на отсутствие в бензоле локализованных двойных связей,
его структуру обычно и сейчас изображают формулой Кекуле. Наряду
с ней применяют и другие обозначения, стремящиеся выразить большую
симметричность молекулы. В дальнейшем мы будем пользоваться первой
и последней из приведенных формул:
Структуры Дьюара б, вносящие малый вклад в резонанс структур,
лучше изображать как I, а не так, как ее изображал сам Дьюар (фор-
мула II) во избежание употребления одной формулы для обозначения
разных соединений.
о о
I II
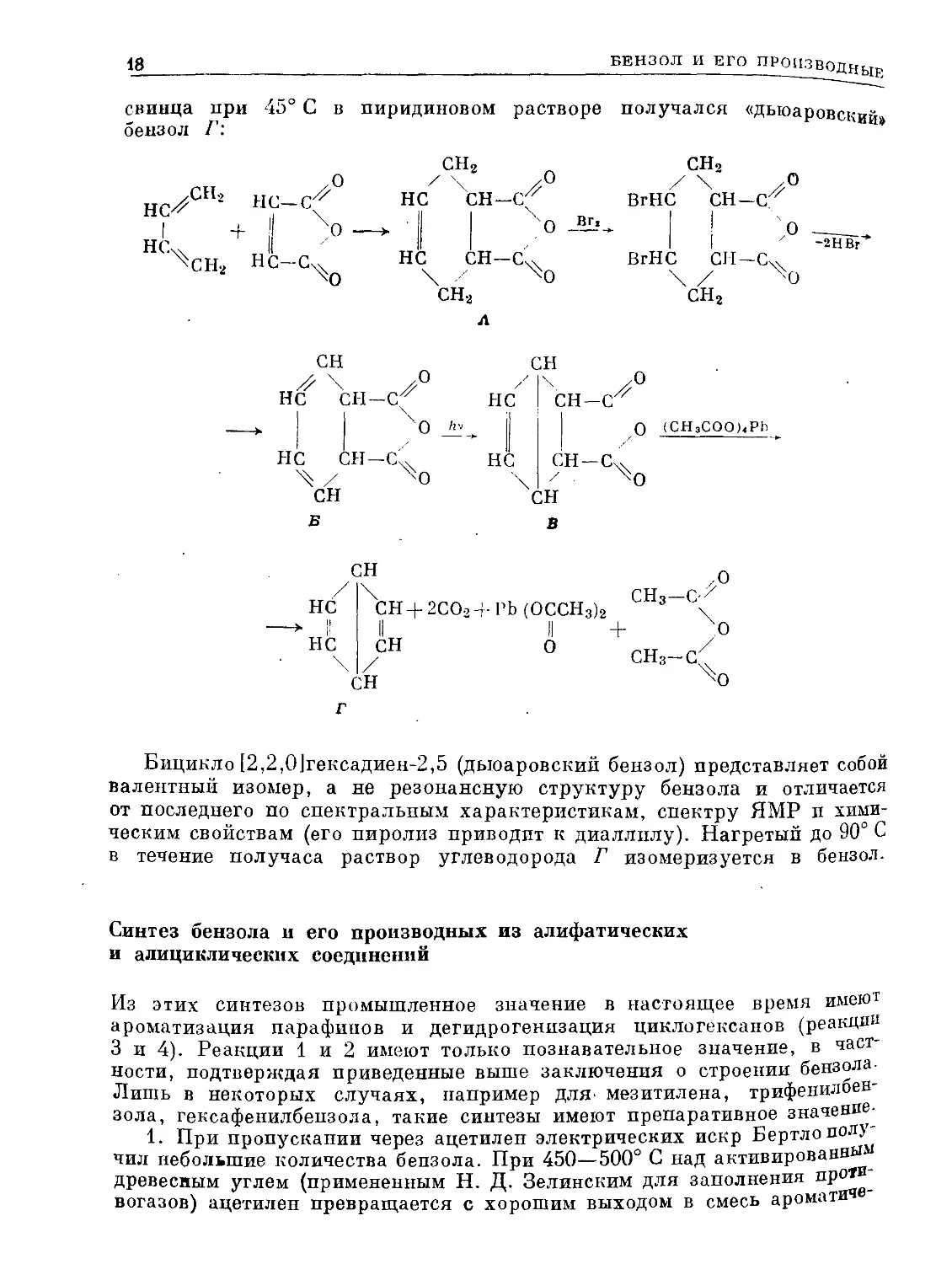
Дело в том, что недавно получен как химический индивид так называ-
емый дъюаровскмй бензол II, в молекуле которого имеется только четыре
л-электрона, геометрия ядерной конфигурации иная, чем в бензоле,
и связь между атомами 1 и 4 не является л-связью. Этот дьюаровский
бензол, или бицикло [2,2,0]гексадиен-2,5, получен в 1963 г. Ван Тамеле-
ном и Паппасом следующим путем: аддукт бутадиена-1,3 и малеинового ан-
гидрида (А) превращали в ангидрид циклогексадиен-3,5-дикарбоновоп-1,2
кислоты (Б), который под действием ультрафиолетового облучения пере-
группировывался в продукт В; при декарбоксилировании В тетраацетатом
* Весом резонансной структуры называется квадрат модуля коэффициента при
соответствующей структуре в линейной комбинации, представляющей резонанс.
В случае действительных волновых функций и коэффициентов это — квадрат коэффи-
циента, Чем больше вес структуры, тем опа в некотором точно математически опреде-
лимом смысле ближе к линейной комбинации, представляющей резонанс.
** Энергию структуры Кекуле рассчитывают по аддитивной схеме из термохими-
ческих данных.
18
БЕНЗОЛ И ЕГО
Л£^ОДНЫЕ
свинца при 45° С
беизол Г:
в пиридиновом растворе
получался «дыоаровский»
СН2
р„ /° / \ /°
нс^ 2 нс-с-^ НС CH-Cz/
| | \q —* [ | Хо _В£‘
Н(\ч II '' II I
^СН2 НС—нс СН—С\Х
\ / ^0
сн2
сн2
/ \ /О
ВгНС СН-С^
I I >______
I I -2НВГ
ВгНС сн—схч
\ / хо
сн2
// \ /)
нс сн—с^
сн
сн-с(х
чо
сн
НСХ
сн—с
(СНзСООПРЬ
сн
нс7" ^сн+гсоз+вь
II II
нс сн
сн
(ОССН3)2
II +
о
/О
СН3-С-^
^0
СНз-С^
^0
Бицикло [2,2,0]гексадиен-2,5 (дыоаровский бензол) представляет собой
валентный изомер, а не резонансную структуру бензола и отличается
от последнего по спектральным характеристикам, спектру ЯМР и хими-
ческим свойствам (его пиролиз приводит к диаллилу). Нагретый до 90° С
в течение получаса раствор углеводорода Г изомеризуется в бензол.
Синтез бензола и его производных из алифатических
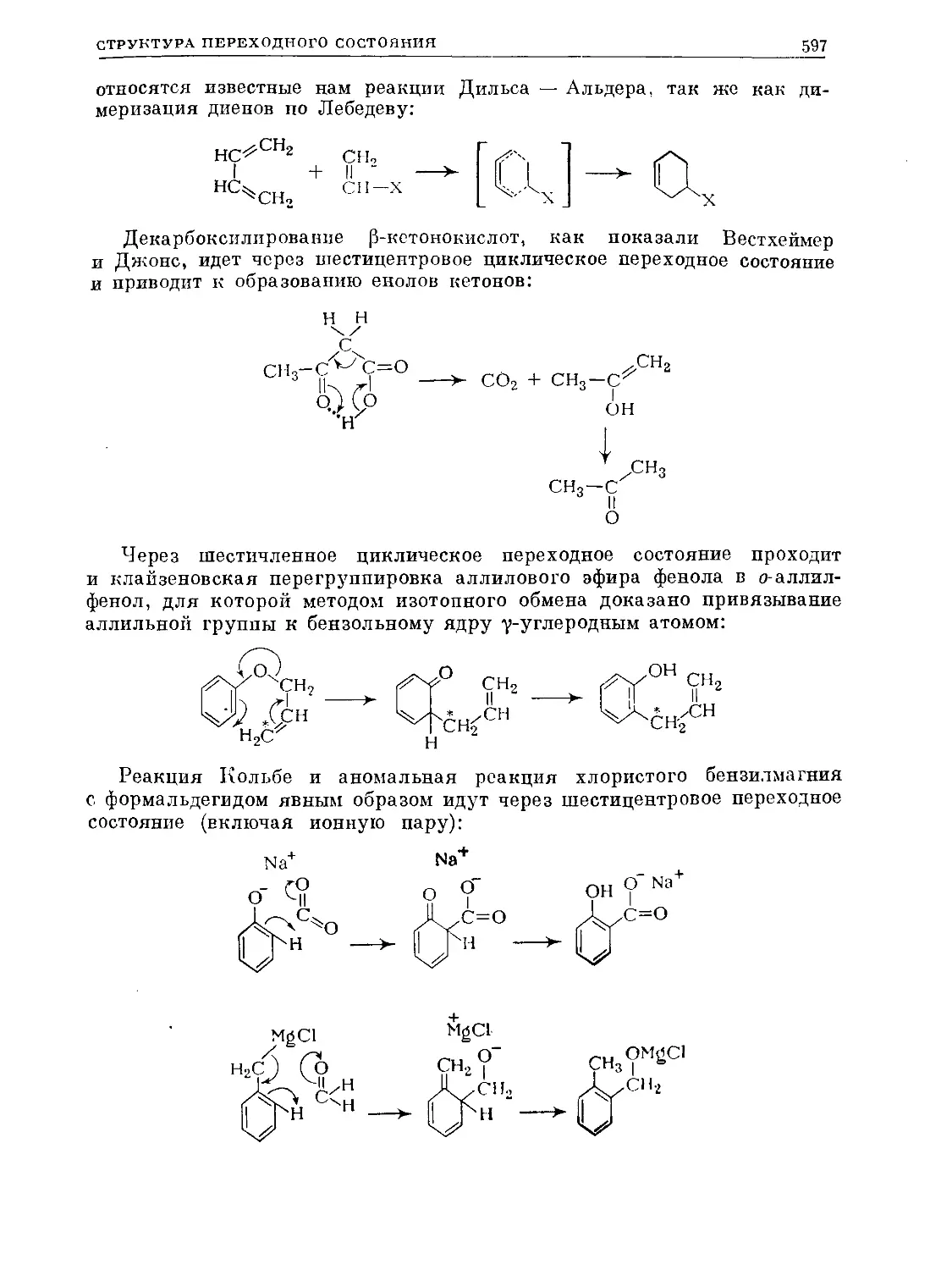
и алициклических соединений
Из этих синтезов промышленное значение в настоящее время имеют
ароматизация парафинов и дегидрогенизация циклогексанов (реакции
о и 4). Реакции 1 и 2 имеют только познавательное значение, в част-
Лишь в некоторых ВЫШе заклК)чения о строении бензола,
зола, гексафенилбрпчлЛ Я ’ папРимеР Для мезитилена, трифенилбен-
1. При HnonvcKaтти « такие синтезы имеют препаративное значение-
чил небольшие количествl?3 ацети2®н электрических искр Бертло полу-
Древесным углем (примененным н’ 450~500° С Над активированным
вогазов) ацетилрп ппйвпа ШМ ^елинским Для заполнения дроти-
} ацетилен превращается с хорошим выходом в смесь ароматиче-
БЕНЗОЛ И ЕГО СТРОЕНИЕ
ских углеводородов, содержащую бензол (Н. Д. Зелинский, Б. А. Казан-
ский, 1922 г.):
НС
Ч
нс сн \
нс + сн *
Ту же реакцию можно осуществить по Реппе (1948 г.) «мокрым путем»
под действием дикарбонил-дитрифенилфосфинникеля l(CeH&)3P]2Ni(CO)2,
получаемого из трифенилфосфина и тетракарбонила никеля. Тот же ката-
лизатор трпмеризует монозамещепные ацетилены в 1,3,5-замещенные бен-
золы (см. ч. II, раздел «Элементоорганические соединения») и в изомерные
им 1,3,4-производные бензола. Со времени открытия Реппе выяснено,
что то же действие оказывает ряд комплексных катализаторов. Так, по
М. Е. Вольпину, [(С2Н5О)3Р]4СоС1 также катализирует тримеризацию
ацетиленов в бензолы.
Катализаторы Циглера [Al(C2H5)3+TiCl4, см. кп. I, стр. 398], вызываю-
щие полимеризацию этилена и его гомологов, тримеризуют как моно-, так
и дизамещенные ацетилены соответственно в 1,3,5-тризамещенные и гекса-
замещенные бензолы (Француз; Лютц). По данным В. О. Репхсфельда
и К. Л. Маковецкого, лучше всего осуществлять эту тримеризацию смесью
триизобутилалюминия с четыреххлористым титаном в соотношении
Al : Ti = 3 : 1.
Дифенилацетилен превращается в гексафенилбензол под действием
многих катализаторов, в частности под действием СгС13 в момент его
восстановления гриньяровым реактивом (Цейсс).
2. Ацетон при действии концентрированной серной кислоты образует
1,3,5-триметилбензол (мезитилен). Реакция представляет собой случай
циклической кротоновой конденсации трех молекул кетона:
СН3
С
0^ \ЗН3
н+
СН3 + О
i II
СН3—С С-СНз
\ /
О СНз
СНз
I
С
нс хсн +зн,о
I II
СНз—С С—СНз
/
сн
Подобным образом ведут себя все кетоны типа В—СО—СН3. Так,
из ацетофенона СвН5СОСН3 можно получить 1,3,5-трифенилбепзол.
Этот тип реакций циклизации вообще довольно распространен, а при
наличии в исходном соединении альдегидной группы, связанной с очень
подвижной метиленовой группой, протекает самопроизвольно, часто
с количественным выходом. Так, реакции, в которых должен был бы
получиться полуальдегид малоновой кислоты НООС—СН2—СНО,
9*
20
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
на деле приводят к кротоновой конденсации последнего в тримезиновую
кислоту:
СООН
I
сы2
/ СООН
ОНС СНО |
L + —>
Н2С СН2 II |
/ /\ /\^\
МООС ОНС СООН НООС СООН
3. Замечательная реакция ароматизации парафинов открыта Н. Д. Зе-
линским, Б. А. Казанским и А. Ф. Плата. При пропускании н-гексана,
«-гептана и других парафинов, имеющих цепь не менее чем из шести
углеродных атомов, над платиновым катализатором при ~300с С обра-
зуются бензол (в первом случае), толуол (во втором) или их гомологи
и выделяется водород:
СН3-СН2—СН2-СН2—СН2-СН3
СН3
СН3-СН2-СН2-СН2-СН2-СН2-СН3 -ZU Q +4Н2
Было выяснено, что того же эффекта можно добиться, применяя в каче-
стве катализатора окись хрома, осажденную на окиси алюминия
(Б. Л. Молдавский), при более высокой температуре (> 400° С). Эти
реакции широко используются при ароматизации погонов нефти.
4. Как уже отмечалось, пропуская парообразный циклогексан (или
его гомологи) над платиной или палладием при —300° С, можно количе-
ственно превратить его в бензол (или гомологи) (Н. Д. Зелинский, 1912).
Позднее было найдено, что подобная дегидрогенизация циклогексанов
может быть осуществлена при нагревании их с серой до —400° С (Ружичка)
или с селеном (—500—600° С), причем водород уходит в виде сероводорода
или селеноводорода.
5. Циклогексен, циклогексадиен и их гомологи при действии платины
или палладия диспропорционируются в бензол и циклогексан (или гомо-
логи) уже при’комнатной температуре (реакция «необратимого катализа»
Зелинского):
БЕНЗОЛ И ЕГО СТРОЕНИЕ
Превращения бензола в алифатические
и алициклические соединения
Как ни прочен бензол, однако, энергичные воздействия разрушают его
ароматическую систему. Сюда прежде всего относится энергичное окисле-
ние воздухом над V2OS в качестве катализатора, приводящее к малеино-
вому ангидриду:
нс/\
! Og: || О + 2СОг + 2Н2О
у нс\/
Процесс используется в промышленности.
В организме (кроликов) бензол окисляется в муконовую кислоту
и таким образом выводится из организма. При протекании этой реакции
сохраняется углеродный скелет из шести атомов п две двойные связи
бензола:
О /\соон
U —“ VC00H
Реакция гидрирования бензола в циклогексан уже рассматрива-
лась (см. стр. 11).
Восстановление бензола посредством HI проходит с изомеризацией
и приводит к метилциклопентану (Н. М. Кижнер).
Действуя ультразвуком на бензол, Цейхмейстер (1957) получал аце-
тилен, изолированный им в виде ацетиленида меди; действием ультра-
фиолетового облучения он изомеризовал бензол в фульвен:
СН2
В 1968г. Уорд и Уишнок облучением бензолав жидком состоянии даль-
ним ультрафиолетовым светом (1650—2000 А) получили валентный изомер
бензола — дыоаровский бензол (бицикло [2,2,0]гексадпеп-2,5) наряду
99
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
с двумя другими валентными изомерами бензола бензваленом и фуль-
веном:
би цикл о гекс алией-2,5
бензвален
сн2
фульвен
Источники ароматических углеводородов
Бензол и его ближайшие гомологи, нафталин и другие конденсированные
ароматические углеводороды в виде многочисленных своих производных
находят разнообразное применение в промышленности — в производстве
пластмасс, синтетических каучуков, красителей, как лекарственные
вещества, растворители, полупродукты для разных отраслей органической
промышленности. Их мировое потребление измеряется многими миллио-
нами тонн. Одним из главных, а до недавнего прошлого единственным
источником их была каменноугольная смола, получающаяся при коксова-
нии угля для нужд металлургии (ранее с целью производства светильного
газа). При нагревании коксующегося угля в закрытом пространстве
вплоть.до 1000° С происходит пиролиз, в результате которого остается
кокс, отходят коксовые газы и отгоняется каменноугольная смола —
тяжелая густая черная масса (выход ~3% от веса угля).
Выход и состав каменноугольной смолы сильно зависят от темпера-
туры и скорости коксования. Чем они выше, тем меньше выход каменно-
угольной смолы и тем она беднее ценными фенолами. В этом смысле
интересы металлургической промышленности и химической не совпадают.
Часть летучих углеводородов содержится в коксовом газе, выделя-
ющемся в количестве 250 м3 на 1 т угля. В этом газе имеется (в 1 л )
около 40 г бензола и толуола — соотношение бензола и толуола в смеси
от 3 : 1 до 5 : 1. Углеводороды выделяют из газа поглощением их более
тяжелыми фракциями каменноугольной смолы. Остальную часть этих
углеводородов выделяют из каменноугольной смолы, которую сначала
подвергают разгонке на грубые фракции (см. стр. 23).
После фракционирования получают сырую бензольно-толуольную
Фракцию (~1% от количества смолы), нафталин (—6%), от которого
отделяют фенол и крезолы (в сумме 1,5%), пользуясь их растворимостью
в водной щелочи; выделяют также —1%. антрацена.
Все возрастающим по важности источником ароматических углеводо-
родов является нефтепереработка. Из нефти получается более половин0
всей ароматики.
ИЗОМЕРИЯ ЗАМЕЩЕННЫХ БЕНЗОЛОВ
23
Фракции каменноугольной смолы
Легкое масло (фракция 80—170° С; выход 3—5%) Содержит циклопептадиеп и ароматические углево- дороды—бензол, толуол, о-, м- и re-ксилолы, не- много полиметилбензолов и этилбензола, а также азотистые и сернистые гетероциклы
Среднее масло (фракция 170—240° С; выход ~Ю%) Содержит главным образом нафталин, оба метил- пафталпиа, фенол, о-, м- и п-крезолы
Тяжелое масло (фракция 210—270° С; выход Ю-15%) Нафталин, крезолы, гомологи и фенолы нафталина, дифенил, аценафтен, частично хпиолнн
Антраценовое или зеленое масло (фракция до 360° С; выход — 20%) Антрацен, фенантрен, флуорен, индол, карбазол
Пек (остаток от перегонки; выход ДО 60%) Уголь, высшие конденсированные ароматические углеводороды
Некоторые виды нефти (например, майкопская, румынская, нефть
острова Борнео) в заметном количестве содержат бензол и его гомологи.
Из соответствующих фракций можно извлечь ароматические’углеводороды.
пользуясь селективными растворителями, например жидким сернистым
ангидридом и фурфуролом, или адсорбцией на силикагеле, при помощи
которой отделяются легко адсорбирующиеся ароматические углеводороды.
Однако так добывается лишь малая часть ароматических углеводо-
родов. Большая их часть получается пиролизом или каталитической аро-
матизацией углеводородов нефти. Пиролиз, проводимый при —800° С
в трубчатках, превращает алициклические и жирные углеводороды нефти
в ароматические углеводороды, осколочные олефины и низшие алканы.
Основной путь получения ароматики — каталитическая ароматизация,
основанная на реакциях, описанных на стр. 20. В этом процессе приме-
няется и платиновый катализатор, работающий при более низких темпе-
ратурах («платформинг») и окись хрома, отложенная На окиси алюминия,
и другие окислы металлов (Mo, V) при 450—500° С. При переработке
нефти соотношение бензола, толуола и ксилолов составляет 1:4:5,
тогда как в каменноугольной смоле преобладает бензол: его в пять раз
больше, чем толуола, и в 15 раз больше, чем ксилолов.
Изомерия замещенных бензолов
Мы уже упоминали, что монозамещеппые бензолы не имеют изомеров,
например существует только один монометилбензол — толуол. Специаль-
ной серией реакций было доказано, что если в монозамещенном бензоле
24
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
замещать последовательно каждый из остальных водородов и элиминиро-
вать (т. е. замещать на водород) первый заместитель, то будет получаться
все один и тот же монозамещениый бензол.
Дизамещенные бензолы с одинаковыми заместителями существуют
в трех и только трех изомерных формах (изомеры положения):
орто-.или
1,2-j и-Х-бензол
пара -,или
М -Ди-Х-бензоЛ
Для указания положений заместителей в бензольном ядре обычно
пользуются цифровыми обозначениями атомов углерода бензола. Обозначе-
ния орто-, мета- и пара- введены Кернером (сокращенно пишут о-, м-, п-).
Тризамещенные (с одинаковыми заместителями) бензолы также суще-
ствуют в виде трех изомеров:
симметрический, или
1,3,5-три-Х-бензол
смежный, или
1,2,3-три-Х-бензол
1,3,4-три-Х-бензол
Тетразамещеиных бензолов, очевидно, тоже три (в них оставшиеся
незамещенными два водорода могут находиться в орто-, пара- или мета-
положении друг к другу):
X-бензол
Наконец, пентазамещенный бензол (опять-таки при одинаковых заме-
стителях) бывает один.
Гораздо больше комбинаций получается при разных заместителях-
Гак, при трех разных заместителях возможно 10 изомеров положения.
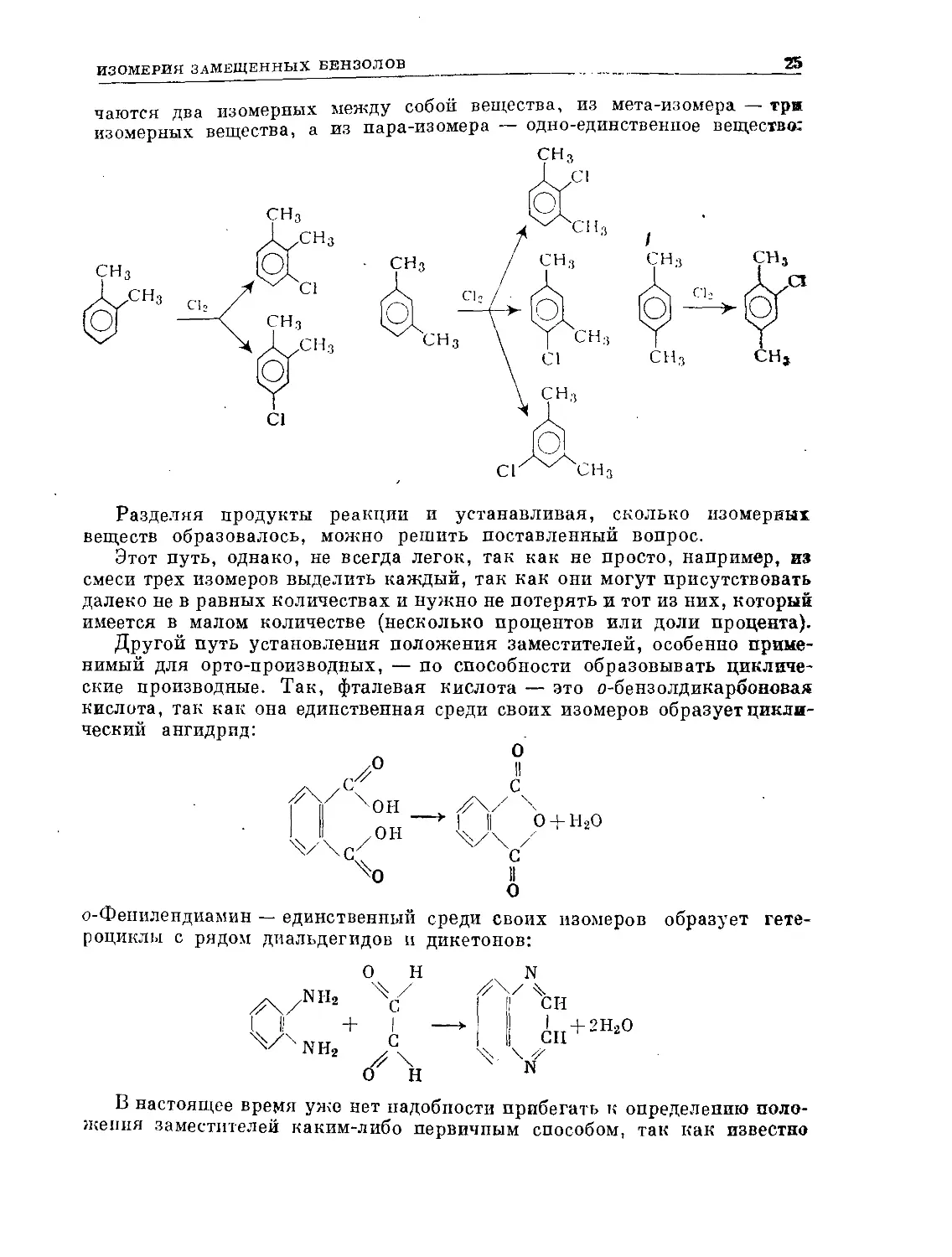
Для того чтобы выяснить, какое из трех изомерных веществ является
2₽то-, какое мета- и какое пара-изомером, было использовано правило
Кернера: при введении нового заместителя прямой реакцией замещения
водорода на галоид, нитрогруппу и т. п. (см. ниже) из орто-изомера полу-
ИЗОМЕРИЯ ЗАМЕЩЕННЫХ БЕНЗОЛОВ 25
чаются два изомерных между собой вещества, из мета-изомера — три
изомерных вещества, а из пара-изомера — одно-единственное вещество:
СНЯ
Разделяя продукты реакции и устанавливая, сколько изомерных
веществ образовалось, можно решить поставленный вопрос.
Этот путь, однако, не всегда легок, так как не просто, например, из
смеси трех изомеров выделить каждый, так как они могут присутствовать
далеко не в равных количествах и нужно не потерять и тот из них, который
имеется в малом количестве (несколько процентов или доли процента).
Другой путь установления положения заместителей, особенно приме-
нимый для орто-производных, — по способности образовывать цикличе-
ские производные. Так, фталевая кислота — это о-бензолдикарбоновая
кислота, так как она единственная среди своих изомеров образует цикли-
ческий ангидрид:
о-Фенилендиамин — единственный среди своих изомеров образует гете-
роциклы с рядом диальдегидов и дикетонов:
О Н
+ 2Н2О
В настоящее время уже нет надобности прибегать к определению поло-
жения заместителей каким-либо первичным способом, так как известно
26
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
положение заместителей в множестве производных, к которым можно
свести любое новое вещество рядом подходящих реакций. Например,
зная, что во фталевой кислоте карбоксилы находятся в орто-положений
друг к другу, можно установить соответствие положений во фталевой
кислоте и о-ксилоле, окисляя последний хромовой кислотой во фталевую
кислоту .
или действуя бромом на фталат серебра и замещая таким образом
карбоксилы на бром, получить о-дибромбензол:
ФИЗИЧЕСКИЕ МЕТОДЫ УСТАНОВЛЕНИЯ
СТРОЕНИЯ ОРГАНИЧЕСКИХ МОЛЕКУЛ*. 1И
Разработанный Дебаем в 1912 г. способ измерения дипольных моментов
молекул дал в руки химиков абсолютный метод установления расположе-
ния заместителей в бензольном ядре. Однако он появился тогда, когда
в этом его применении практически уже почти миновала необходимость.
Если известны дипольные моменты двух монозамещенных бензолов щ
и р.2, то по формуле векторного сложения можно вычислить дипольный
момент р. двузамещенного бензола с двумя такими заместителями:
И = /И? + Ра+ 2р.Щ2 cos а
Здесь а — угол между векторами дипольных моментов двух заместите-
лей в одном бензольном ядре. Для орто-положения а = 60°, cos а = /г»
для мета-положения а — 120°, cos а = 1; для пара-положения а = 180 ,
соз а = —1.
При одинаковых заместителях, электрические моменты которых
направлены по диагоналям бензольного шестиугольника, дипольные
моменты дизамещенпых бензолов по сравнению с дипольным моментом
* См. также ки. I, стр. 341 и .г8(’.
ФИЗИЧЕСКИЕ МЕТОДЫ УСТАНОВЛЕНИЯ СТРОЕНИЯ. III
27
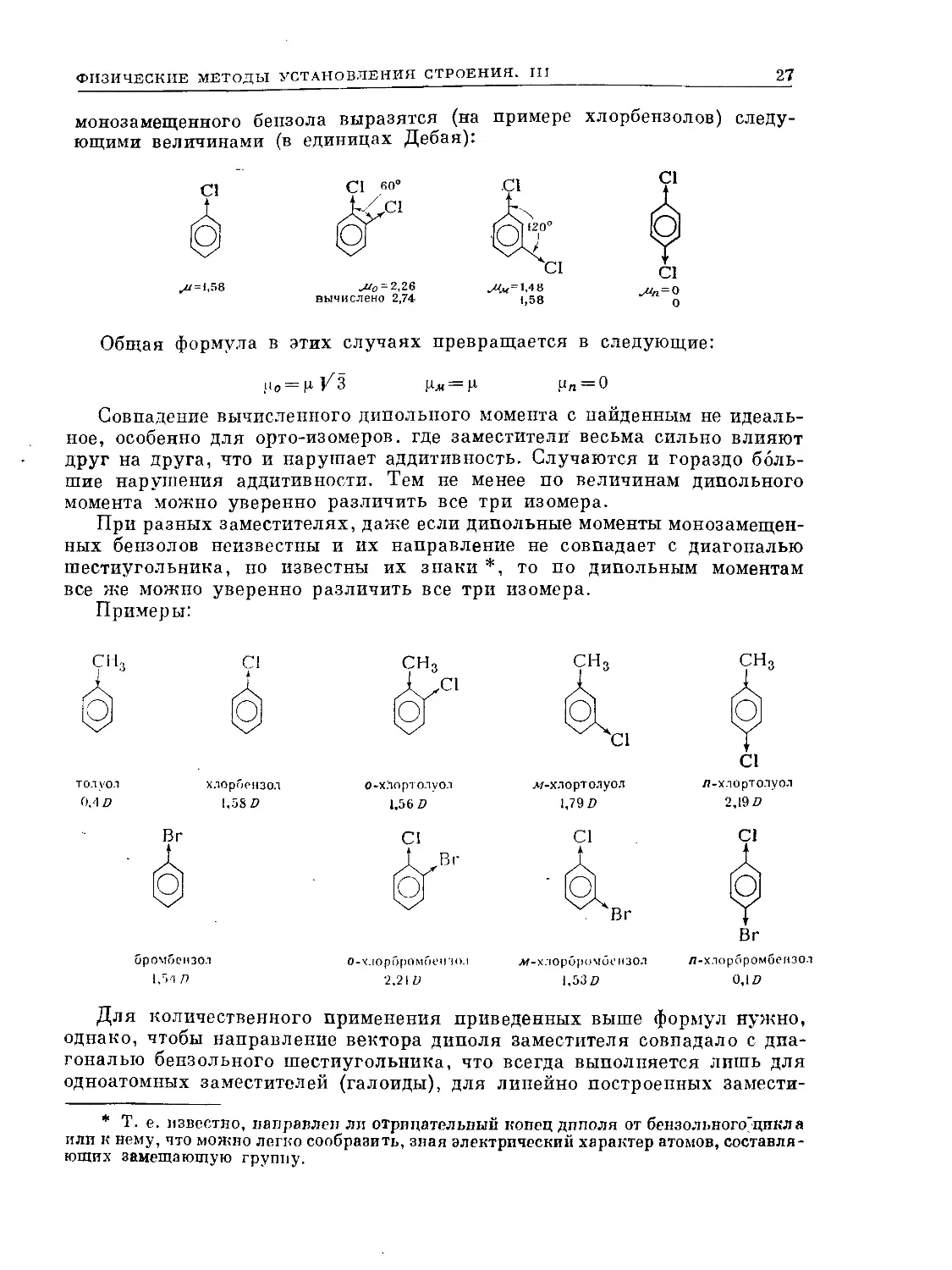
монозамещенного бензола выразятся (на примере хлорбензолов) следу-
ющими величинами (в единицах Дебая):
58
^и0 - 2,26
вычислено 2,74-
Общая формула в этих случаях превращается в следующие:
!*о — Ц Um— И Рп — О
Совпадение вычисленного дипольного момента с найденным не идеаль-
ное, особенно для орто-изомеров, где заместители весьма сильно влияют
друг на друга, что и нарушает аддитивность. Случаются и гораздо боль-
шие нарушения аддитивности. Тем не менее по величинам дипольного
момента можно уверенно различить все три изомера.
При разных заместителях, даже если дипольные моменты монозамещен-
ных бензолов неизвестны и их направление не совпадает с диагональю
шестиугольника, но известны их знаки *, то по дипольным моментам
все же можно уверенно различить все три изомера.
Примеры:
толуол хлорбензол
бромбензол
1.5-1 D
Для количественного применения приведенных выше формул нужно,
однако, чтобы направление вектора диполя заместителя совпадало с диа-
гональю бензольного шестиугольника, что всегда выполняется лишь для
одноатомных заместителей (галоиды), для линейно построенных замести-
* Т. е. известно, направлен ли отрицательный конец диполя от бензольного'цикла
или к нему, что можно легко сообразить, зная электрический характер атомов, составля-
ющих замещающую группу.
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
телей (C=N, —Се=С—В) и для заместителей с симметричным расположе-
нием атомов, т. е. таких, в которых «центр тяжести» заряженных атомов
ложится па диагональ шестиугольника бензола (СН3, СС13, NO2). Уже
для карбоксилов это условие не выполняется, и, например, этиловый
эфир терефталевой кислоты имеет ц = 2,3 D.
В табл. 59 приведен ряд примеров соблюдения аддитивности диполь-
ных моментов бензолов с простейшими заместителями.
Как видно, аддитивность для пара- и мета-производных бензола
выполняется лучше, чем для орто-замещенных. Отклонения последнего
рода объясняются геометрическим искажением молекулы — выводом
заместителей из плоскости кольца, отклонением угла бензольного шести-
угольника от 120°. Нарушение аддитивности совершенно другого происхо-
ждения бывает в случае, если замещающие группы (это яснее всего для
пара-расположенных заместителей) обладают противоположным мезомер-
ным эффектом (+М- и —М-) и, значит, находясь в противоположных поло-
жениях бензольного кольца, действуют согласованно. Увеличение ц
по сравнению с рассчитанным является следствием сопряжения (резо-
нанса). Это особенно явно в случае n-нитрофенола (см. табл. 59), строение
которого может быть выражено следующими структурами:
*=0 Н или
’О
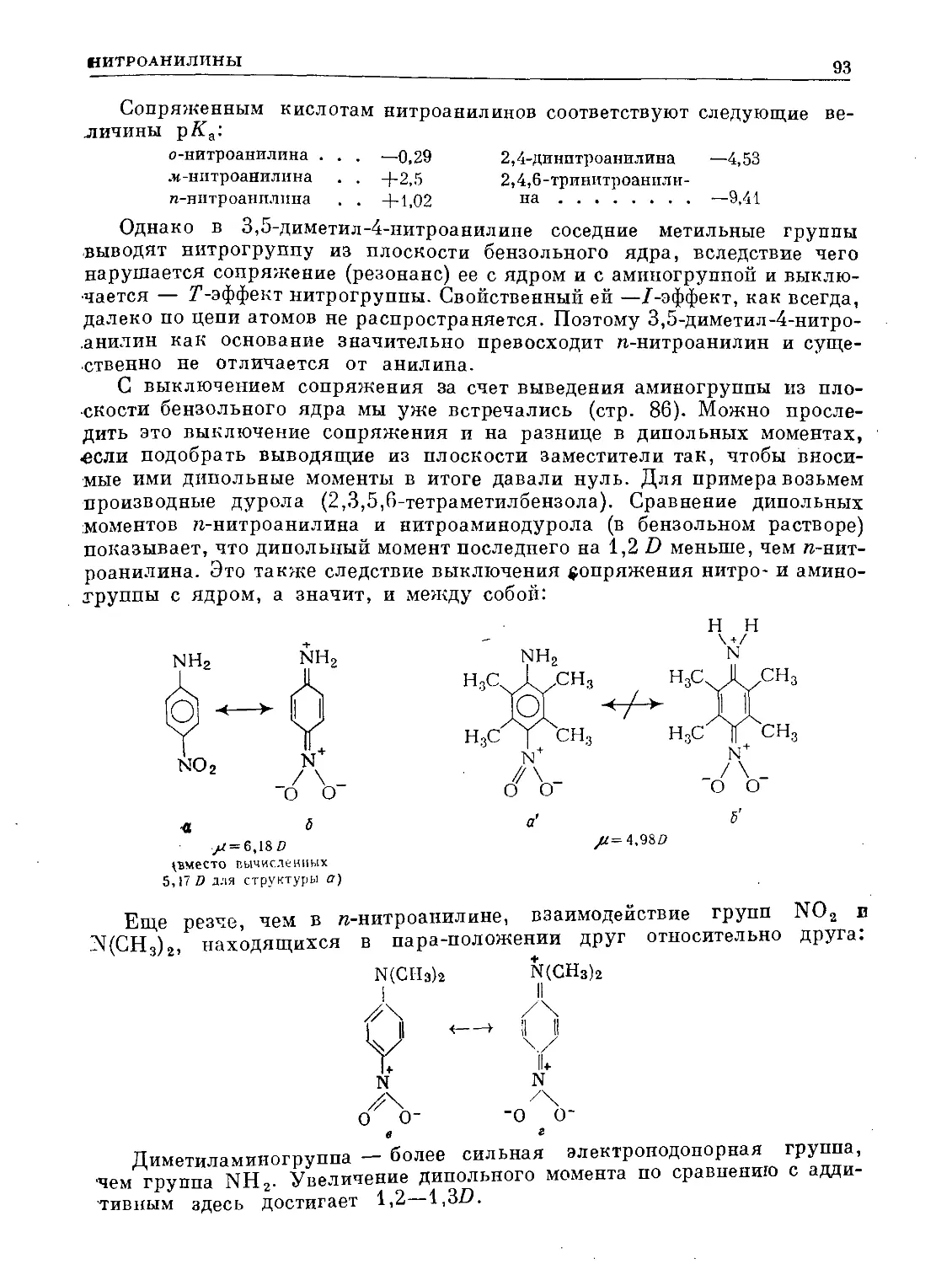
Такой эффект связан с углублением окраски соединения (стр.108)
и впервые на этом основании был констатирован В. А. Измаильским еще
в 1913 г. (явление названо им мезотропией). Привился предложенный
иозднее Инголдом, широко исследовавшим этот эффект, термин мезомерия',
эще позднее Полингом был введен термин резонанс. По признаку неадди-
тивности дипольных моментов эффект мезомерии можно проследить
иа n-нитроанилипе (стр. 93) и п-диметиламинонитроанилине.
Явление того же типа прослеживается и на монозамещенных бензолах
(стр. 27). Как уже указывалось, дипольный момент галоидбензолов
заметно меньше, чем дипольный момент соответствующих галоидалканов.
Это объясняется мезомерией такого типа:
ВОПРОСЫ НОМЕНКЛАТУРЫ
29
Таблица 59. Аддитивность дипольных моментов (в 1)) замещенных бензолов
Монозамещеняый бензол Дизамещенные бензолы
замести- тели и заместители орто- мета- пара-
^набл. ^выч. ^набл. ^выч. ^набл. ’^ВЫЧ.
4
С1 1,58 С1, С1 2,27 2,74 1,48 1,58 0 0
Вг 1,54 Вг, Вг 2,1 2,67 1,46 1,54 0 0
no2 3,98 no2, no2 6,00 6,90 3,89 3,98 0 0
nh2 1,53 nh2, nh2 1,45 — 1,79 1,80 1,5 —
он 1,6 no2, Cl 4,1 4,97 3,4 3,47 2,50 2,40
no2, nh2 4,24 3,66 4,94 4,72 6,2 5,17
Cl, nh2 1,77 1,71 — — 2,27 2,30
no2, oh — — — — 5,07 4,34
При наличии электроноакцепторных заместителей дипольный момент
ароматических соединений больше, чем жирных:
//= 4,03 27
+
СН, —
3 ^о-
Д-=3,15 27
^ = 3,9422
сн3—C=N
yz=3,51O
Во всех случаях сказывается обычная «податливость» бензольного
ядра к «требованиям» замещающей группы.
Таким образом, дипольные моменты говорят многое не только о распо-
ложении замещающих групп в бензольном ядре, но и об их взаимодействии
с бензольным ядром и через бензольное ядро друг с другом.
Вопросы номенклатуры бензольных производных
Для углеводородов бензольного ряда и их производных применяются
тривиальные, «радикальные» и женевские названия. Тривиальные назва-
ния не требуют пояснений. В основе «радикальных» названий (имеющих
совершенно частное значение) лежат наименования радикалов:
СвН.5 ..........Фенил (от первого старинного
названия бензола — «фен»)
С6Н4 ...........Фенилен (о-, м- или п-)
СНзСвЩ .... Толил (о-, м- или п-)
CeHsCH2 .... Бензил
СвНьСН..........Бензилиден
Галоидпроизводные по этой номенклатуре называются так: СвН5С1 —
хлористый фенил (неупотребительно); С1С6Н4СН3 — хлористый толил
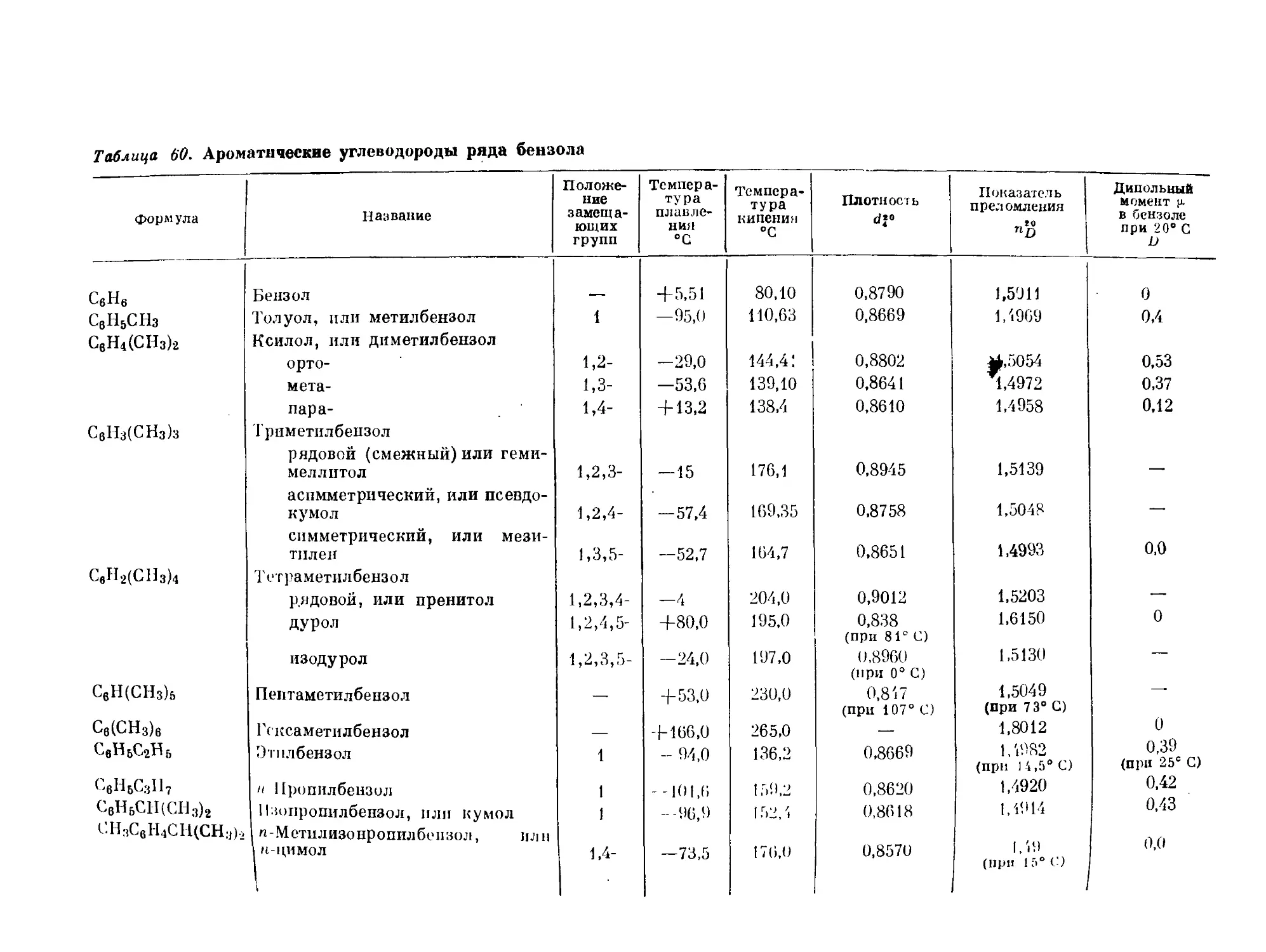
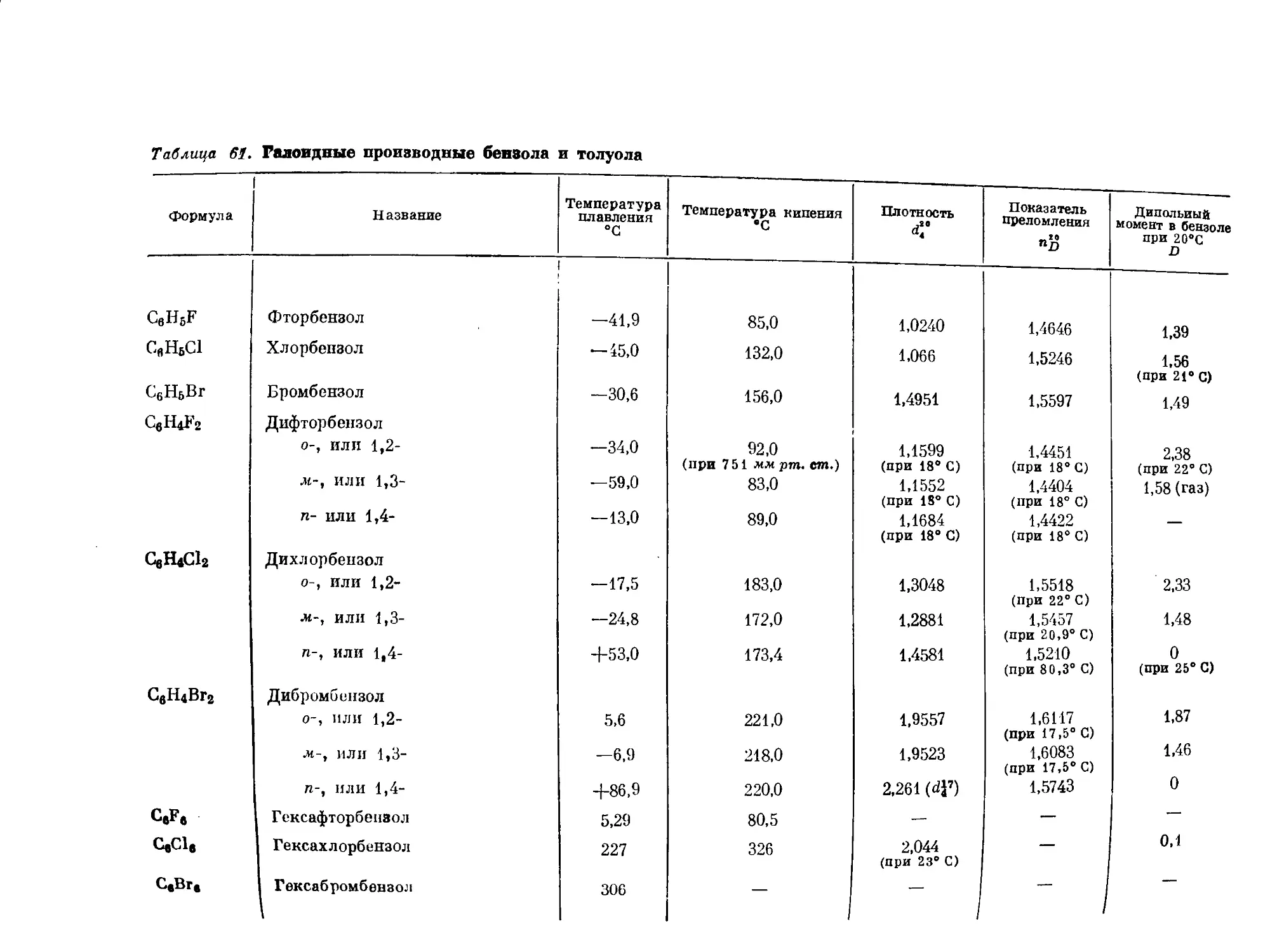
Таблица 60. Ароматические углеводороды ряда бензола
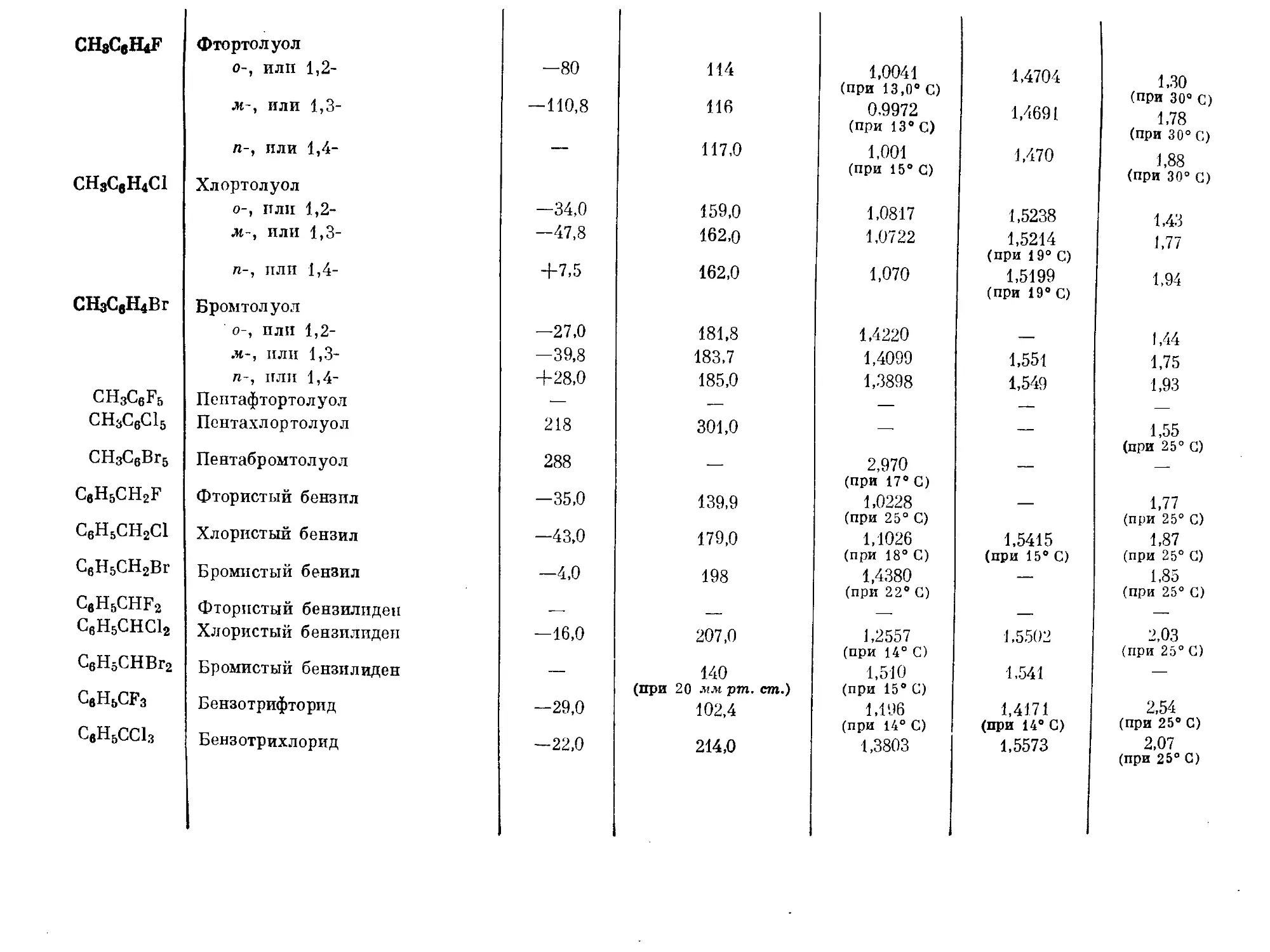
Форм ула Название Положе- ние замеща- ющих групп
с8н8 С8Н5СНз С6Н4(СНз)г С6Нз(СН3)з С.Н2(СН3)4 Бензол Толуол, или метилбензол Ксилол, или диметилбензол орто- мета- пара- Триметилбепзол рядовой (смежный) или геми- меллитол асимметрический, или псевдо- кумол симметрический, или мези- тплен Тетраметилбензол рядовой, или пренитол ДУРол 1 1,2- 1,3- 1,4- 1,2,3- 1,2,4- 1,3,5- 1,2,3,4- 1,2,4,5-
изодурол 1,2,3,5-
С6Н(СН3)6 Пептаметилбензол —
С8(СН3)6 СвЩСгНь Г<ксаметилбензол Этилбензол 1
СвН6С3П7 С8Н6СН(СН3)г СН3СвИ4С1-1(СНа).> " Иропилбензол Изопропилбензол, или кумол п-Метилизонропилбензол, или | н-цимол 1 1 1,4-
Темпера- тура плав ле- пил °C Темпера- тура кипения °C Плоти ОС г Ь djo Показатель преломления nD Дипольный момент и. в бензоле при 20° С 1J
+5,51 80,10 0,8790 1,5911 0
—95,0 110,63 0,8669 1,1969 0,4
-29,0 144,4! 0,8802 >,5054 0,53
—53,6 139,10 0,8641 1,4972 0,37
+ 13,2 138,4 0,8610 1,4958 0,12
— 15 176,1 0,8945 1,5139 —
—57,4 169,35 0,8758 1,5048 —
—52,7 164,7 0,8651 1,4993 0.0
—4 204,0 0,9012 1,5203 —
+80,0 195,0 0,838 (при 81° С) 1,6150 0
—24,0 197,0 0,8960 (при 0° С) 1,5130 —
+ 53,0 230,0 0,817 (при 107° С) 1,5049 (при 73° С) —
+ 166,0 265,0 .— 1,8012 0
- 94,0 136,2 0,8669 1,1982 (при 14,5° С) 0,39 (при 25° С)
— 101,6 159,2 0,8620 1,4920 0,42
- 96,9 152,1 0,8618 1,1914 0,43
—73,5 176,0 0,8570 1,19 (при 15°С) 1 0,0
РЕАКЦИИ ЭЛЕКТРОФИЛЬНОГО ЗАМЕЩЕНИЯ
31
(также неупотребительно), СвН5СН2С1 — хлористый бензил; С3Н6СНС12 —
хлористый бензилиден.
По женевской номенклатуре перед названием углеводорода ставятся
названия замещающих групп с номерами углеводородных атомов, несущих
заместители, например:
С1
1,2-хлор-
1-МРТОКСИ-
I
4-иол-л/-ксило.г,
или 4-иод-1,3-яи-
метилбензол
нитробснзол 4-хлорбензол
2,6-срторбромтолуол,
или 2,6-срторбром-
1-мотилиензол
В табл. 60 приведены названия и физические свойства углеводородов
бензольного ряда.
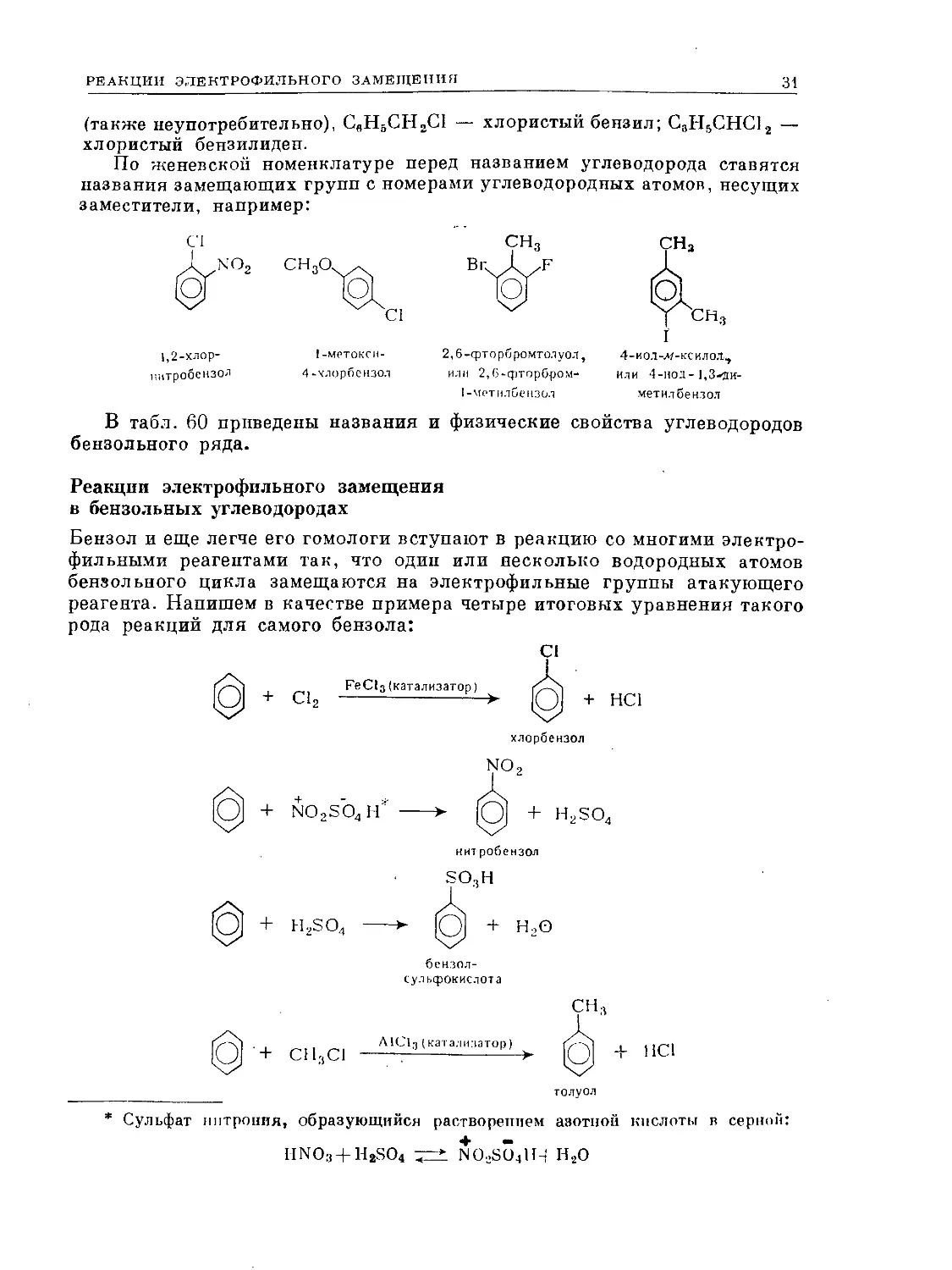
Реакции электрофильного замещения
в бензольных углеводородах
Бензол и еще легче его гомологи вступают в реакцию со многими электро-
фильными реагентами так, что один или несколько водородных атомов
бензольного цикла замещаются на электрофильные группы атакующего
реагента. Напишем в качестве примера четыре итоговых уравнения такого
рода реакций для самого бензола:
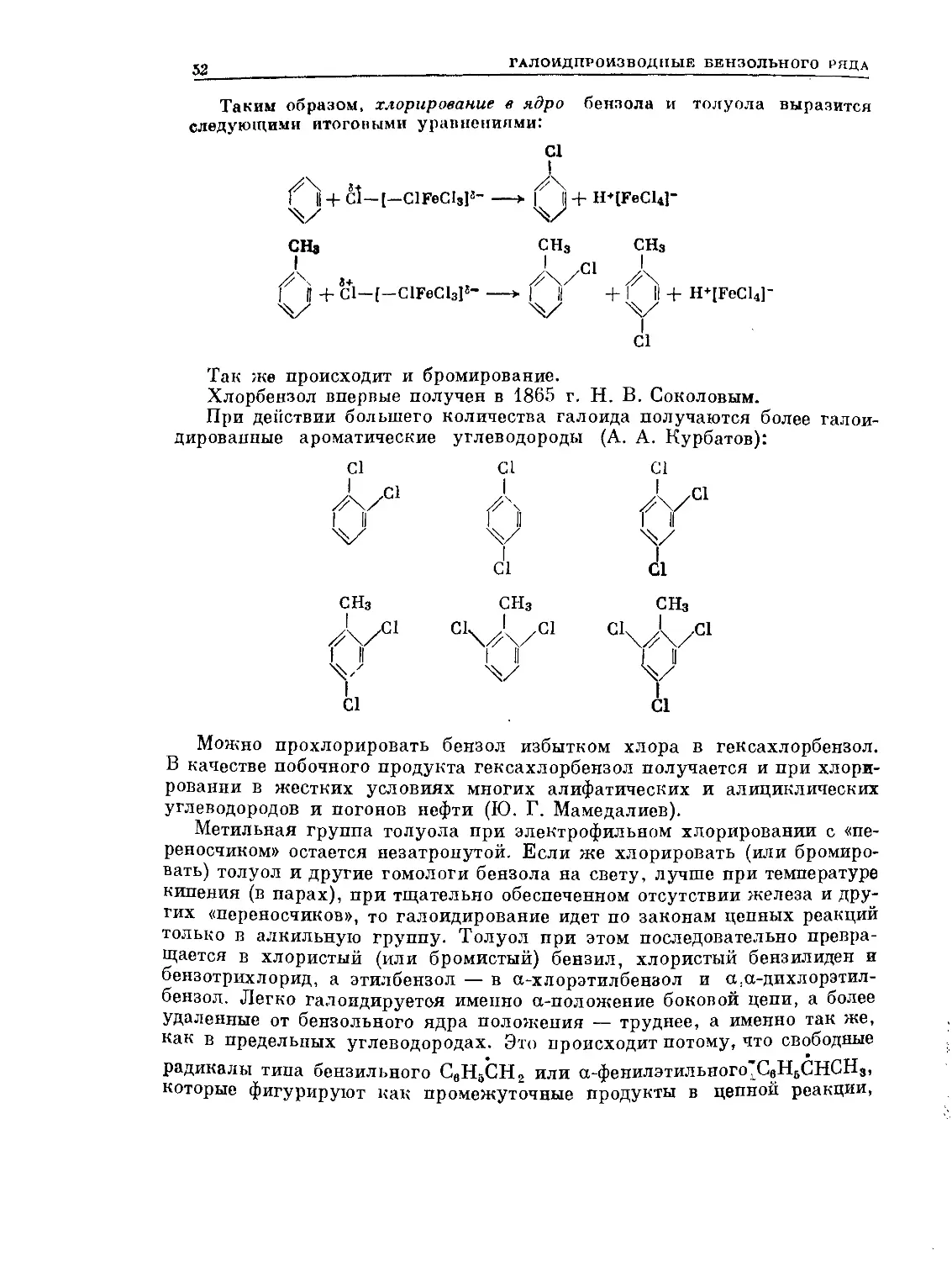
CI
ГеС13(катализатор) Z-S
[OJ + С12 -------------------> [QJ + НС1
хлорбензол
no2
(С>) + no2so4h*—> [О| + h2so4
нитробензол
SO3H
[0] + H2so4 —► (Q) + Н2©
бензол-
сульсрокислота
толуол
* Сульфат нитрония, образующийся растворением азотной кислоты в серной:
HNO3 + H2SO4 NO..SO4IH Н2О
32 БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЙ
Вс© эти реакции будут подробно разобраны в соответствующих разде-
лах. Сейчас для пас важно отметить, что такого рода легко идущие при
комнатной температуре замещения являются наиболее характерным
свойством ароматических соединений, резко отличающим их от предель-
ных соединений. Что касается олефинов, то под влиянием тех же реаген-
тов они претерпевают в обычных условиях иные реакции — электрофиль-
ное присоединение по двойной связи или полимеризацию, так что возмож-
ность реакции замещения остается непроявленной.
По всей вероятности, первой фазой атаки электрофильного реагента
является образование л-комплекса (Дьюар), в котором электрофильная
частица, в наших примерах Cl+, NO2, SO3H, бна, связывается с молеку-
лой бензола за счет всех его л-электронов (как мы увидим дальше, иногда
могут образоваться и прочные изолируемые л-комилексы). Затем электро-
фильная частица вытягивает из бензольного цикла (из ароматической ше-
стерки) пару электронов связи, причем остальные четыре л-электрона
располагаются в две сопряженные двойные связи
или, с использованием формулы Кекуле:
а
Конечно, зти л-электроны обобщены, как мы видели на примере бута-
диена. Но, кроме того, соседний с местом атаки (формула в) положитель-
ный центр (карбкатион) не остается безучастным к соседней системе
л-электронов, и фактически л-электроны растекаются по всем пяти угле-
родным атомам, оставшимся во втором валентном состоянии, а атакован
пый атом становится тетраэдрическим углеродом (в первом валентно
состоянии). Поэтому вместо формулы в часто пишут формулу г (о-комплеь
с положительным зарядом, рассредоточенным по всем пяти углеродн
атомам.
РЕАКЦИИ ЭЛЕКТРОФИЛЬНОГО ЗАМЕЩЕНИЯ
33
По сути дела этот промежуточный комплекс еще правильнее изобра-
жать как резонансный гибрид структур:
Из изображения ясно, что распределение положительного заряда неравно-
мерное и максимально в положениях 2, 4 и 6 по отношению к углероду,
принимающему атаку. Затем из атакованного, ставшего тетраэдрическим
углеродного атома отделяется протон, и привязывавшая последний элек-
тронная пара вливается в состав вновь образующейся ароматической
шестерки л-электронов (в' -> д).
В дальнейшем мы будем пользоваться как формулами типа в, так и фор-
мулами типа г, не делая между ними различия. Структуры типа в или г,
в которые переходит образовавшийся вначале л-комплекс, по аналогии
называются O'-комплексами. Для крат-
кости мы иногда не совсем правильно
будем называть эти o'-комплексы реак-
ционными комплексами или переход-
ными состояниями, а сейчас объясним
разницу между истинным переходным
состоянием и о-комплексом.
Переходным состоянием называется
высшая энергетическая точка на пути
реакции (кн. I, стр. 94), энергетический
перевал. Реакция может проходить и че-
рез несколько таких перевалов. Схема-
тически это показано на рис. 83. Пер-
вый невысокий пик — переходное со-
стояние, ведущее к малоустойчивому
л-комплексу, первый минимум — л-ком-
плекс. Второй пик — переходное со-
Рис. 83. Энергетическая диаграмма
реакции электрофильного замещения
в бензольном ядре.
стояние к o'-комплексу, а следующий за ним неглубокий минимум —
сг-комплекс. Наконец, третий, последний небольшой пик — переход-
ное состояние от o'-комплекса к продуктам реакции. Энергетически вто-
рой пик, неглубокий минимум о-комплекса и третий пик разнятся
незначительно — их ординаты почти равны. По постулату Хэммонда
(см. ч. II, раздел «Механизмы химических реакций») при этом условии
и по геометрии O'-комплекс близок к обоим соседним переходным состоя-
ниям, что дает нам право указанного упрощения.
1. Протонирование и дейтерирование бензола. Самой простой и потому
поучительной реакцией электрофильного замещения в бензоле и вообще
в ароматическом ряду является протонирование и дейтерирование.Бензол
растворим в крепких безводных кислотах, а в дейтерокислотах обменивает
один за другим все свои водороды на дейтерий *. С галоидоводородом
* Конечно, аналогично происходит и обмен водородов бензола на водороды
кислоты.
3 Заказ 67 2
бензол и его производные
34
в присутствии галоидного алюминия (т е. с кислотой HA1C1J бензол
и его гомологи образуют твердые при низкой температуре комплексы.
Аналогичные комплексы образуются с НВ1'4 (например, комплекс толу-
ола, т. пл. —65° С). Как показывает подробное спектроскопическое,иссле-
дование этих веществ, проведенное Перкампусом, они г”—-
имеют следующее
строение:
BF4"
протонированный
толуол
И Н
в г;
протонированный
бензол
Действительно, Олах синтезировал
такой комплекс следующим путем
//
СН2—С
+ | ‘ J^NBr
СН,—
бромирование
в аллильное
положение
------------->
+ AgBF4
- AgBr
bf4~
причем продукт реакции оказался идентичным продукту прямого протони-
рования толуола. С такого рода структурами, постулированными Ингол-
дом как промежуточные при электрофильных замещениях в ароматиче-
ских циклах, мы уже встречались на стр. 32, 33.
Макор, Гофстра и Ван-дер-Ваальс непосредственно определили основность ряда
ароматических углеводородов, в том числе бензола и его гомологов, измеряя пх распре-
деление между гексаном и безводным фтористым водородом. Константа основности
Kb равна в этом случае:
„ = [АгН*] [F~1 -
Ь ИгН] /АгН
где в квадратных скобках концентрации соответствующих ионов или молекулы;
/ — коэффициенты активности ионов (/+ и /_) и молекулы.
Ниже приведены константы основности ароматических углеводородов:
te-b'b >gA'b
Бензол..............—9,4 л1-Ксилол............—3,2
Толуол................—6,3 Мезитилен............—0,4
л-Ксилол............—5,7 Гексаметплбензол +1,4
Как видно, бензол очень слабое основание, захватывающее протон
указанным выше механизмом. По мере алкилирования сила ароматиче-
ских углеводородов как оснований повышается на много порядков.
еакции захвата электрона. Все ароматические углеводороды при
действии сильных доноров электронов способны захватить один электрон
и ооразовать радикал-анион, дающий сигнал ЭПР (ч. II, стр. 538). Бензол
захватывает электрон труднее, чем другие ароматические углеводороды,
он за ирает электрон лишь от калия, но не от натрия, и образует недиссо-
Циированную ионную пару СвН6К. Электрон занимает низшую из
незанятых орбиталей бензола.
РЕАКЦИИ ЭЛЕКТРОФИЛЬНОГО ЗАМЕЩЕНИЯ
35
3. Реакция металлирования. Как открыл П. П. Шорыгин, бензол всту-
пает в следующую реакцию обмена с натрий- и калийалкилами:
ядре п, веро-
водород бен-
©NH3
• СН
Рис. 84. Схематичное
изображение клат-
ратного комплекса
CeHe-Ni(CN)2NH3.
C6He4-CH3Na --> C6H3Na + CH4
Эта реакция вряд ли имеет аналогии среди реакций электрофильного
и нуклеофильного (см. дальше) замещений в ароматическом
ятно, должна рассматриваться как атака алкиланионом на
зола. Она аналогична действию гриньярова реактива
на ацетилен.
4. Клатратные соединения бензола. К. А. Гофман
открыл «молекулярное соединение» бензола
С8Н8 • Ni(CN)2NH3, долгое время остававшееся загад-
кой. Пауэлл и Рейнер получили ряд аналогичных
соединений для производных бензола и ароматичес-
ких гетероциклов (тиофен и др.). При помощи рент-
геноструктурных исследований была выявлена клат-
ратная (стр. 129) природа этих веществ: бензол «за-
стревает» в пустотах решетки, образуемой неоргани-
ческими ингредиентами соединения (рис. 84).
5. Синтез гомологов бензола. Гомологи бензола
можно синтезировать, действуя натрием на смесь
галоидного алкила и галоидбензола (реакция Вюр-
ца — Фиттига), например:
C6H5Br+C2H5Br+2Na —> C6H5C2H5 + 2NaBr
Однако эта реакция лишь частично проходит в
нужном направлении. Наряду с целевыми продук-
тами образуются диарил (в данном примере дифенил С8Н5С8Н5) и про-
дукты взаимодействия двух жирных радикалов 2С2Н8-,т. е. алкан,
алкен и диалкил (в данном случае С2Н8, С2Н4 и С4Н1П). Поэтому гораздо
шире используется алкилирование бензола галоидными алкилами или
олефинами по реакции Фриделя — Крафтса, т. е. с применением безвод-
ного хлористого алюминия в качестве катализатора *. Реакция про-
ходит уже при комнатной температуре:
С8Н84-С1СНз —— - С6Н5СНз+ НС1
С8Н8 + пС1СНз С8Н8_„(СНз)„+пНС1
CeHe + GCJCHs Св(СН3)в + 6НС1
С6Н6+С1СН2СНз^^*С8Н5СНгСНз+НС1
С8Н8 + СН2=СН2 СвН5СН2СНз
С8Н8 + СН2-СНСНз С8Н5СН(СНз)2
СН3
С8Н8Ч CH2=CZ -—С8Н5С(СНз)з
______________ 'СНз
* Применением в органической химии этого замечательного катализатора наука
обязана Г. Г. Густавсопу (1878 г.), профессору Лесной академии (ныне Тимирязевской
сельскохозяйственной академии).
3*
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
36
Взаимодействие очень активных олефинов с бензолом можно вызвать
не только действием хлористого алюминия, но и кислот (концентрирован-
ном серной, плавиковой, фосфорной). Как видно из приведенных примеров,
несимметрично построенные олефины вводят в бензол вторичные или тре-
тичные алкильные радикалы. Первичные радикалы, кроме метила и этила,
таким путем ввести нельзя. По существу вариант реакции Фриделя —
Крафтса с олефинами можно рассматривать как присоединение бензола
к олефинам, проходящее в соответствии с правилом Марковникова (водо-
род бензола присоединяется к «более гидрогенизированному» олефиновому
углероду, а фенильный радикал — к его партнеру). Вполне возможно при-
менение бромистого алюминия и бромистых алкилов. Кроме галоидного
алюминия катализаторами реакции Фриделя — Крафтса могут служить
в порядке ослабевающего действия BF3 (только для фтористых алкилов),
GaCl3, SnCl4, FeCl3. С[их помощью алкилируются только ароматические
соединения, более нуклеофильные, чем бензол. Для алкилирования олефи-
нами предпочитают применять в качестве катализатора фосфорную кислоту.
Реакции Фриделя — Крафтса это типичные реакции электрофильного
замещения в бензольном цикле. Смысл действия катализатора Фриделя —
Крафтса (это всегда льюисова кислота) на галоидный алкил и олефин
выражается следующими схемами:
s+ ГС1д1С11°_
RCl-f-AlCl3 -> R [С1А1С1]
Активирование хлористым алюминием может быть ограничено просто
поляризацией галоидного алкила RC1. . .А1С13 без полной его ионизации:
+ С1
R'CH=CH2+A1C13 ---> R'CH—CH,:-A1CI
Cl
Образующиеся карбониевые катионы или комплексы с 6+ на углероде
уже являются сильными электрофильными реагентами, которые и атакуют
своим положительным углеродом ароматический цикл. Однако галоидный
алюминий не индифферентен к бензольному ядру и активирует его. Это
подтверждается тем, что в присутствии хлористого алюминия бензол
образует сэндвичеобразные л-комплексы с металлами (стр. 416, 458 сл.),
которые без хлористого алюминия не образуются. Однако попытки
Б. Н. Меншуткина доказать методом физико-химического анализа обра-
зование соединений хлористого алюминия с бензолом привели к отрица-
тельному результату, тогда как бромистый алюминий (тоже катализатор
реакции Фриделя — Крафтса) такие соединения образует.
По В. В. Коршаку и Н. Н. Лебедеву, реакция галоидного алкила
с ароматическим углеводородом осуществляется так, что в комплексе
RC1-A1C13 синхронно разрывается связь С—С1 и образуется связь С—С.
В нитробензоле, обычной среде для этих реакций (сам нитробензол не
вступает в реакции Фриделя — Крафтса), сначала образуется комплекс,
катионная часть которого и связывается с галоидным алкилом, поляризуя
его и катализируя реакцию:
' C6H5NO2, /С1
Ч/А1/
_C6H5NO2Z ХС1
2C6H5NO2 + А12С16 ->
[АЮКГ
к+
РЕАКЦИИ ЭЛЕКТРОФИЛЬНОГО ЗАМЕЩЕНИЯ
37
C6H5NO2. /С11 +
Ж
C6H5NO2z чС1.
+ RC1 --->
C6H5NO24 |
zA1
CeH5NO2''Z I
г+
Cl—R
В среде с меньшей диэлектрической проницаемостью комплекс дей-
ствует уже как ионная пара АК и слабее поляризует RC1. Поэтому здесь
требуется активация и бензола, так что вся реакция выражается схемой:
_ । 8+ —4- -I_
АК- . .Cl—R + НАг. . .АК-> ArR + HCl + 2KA
В высшей степени интересно, что алкилирование бензола галоидным
метилом не заканчивается образованием гексаметилбензола. Далее обра-
зуется сг-комплекс такого строения и состава (конечно, с противостоящим
анионом А1С17)
Н3С СН3
Н3сЛ+сн;!
А1С14'
НзС'уХНр
сн3
который является прототипом сг-комплексов при электрофильном замеще-
нии в бензольном ядре и подобен сг-комплексам бензола и толуола с прото-
ном или дейтроном, приведенным на стр. 34.
Особенностью синтезов с хлористым алюминием является то, что он
катализирует не только алкилирование, но и дезалкилирование гомологов
бензола, так как обладает способностью разрывать углерод-углеродные
связи. Кроме того, в более жестких условиях он способствует изомериза-
ции цепи углеродных атомов галоидного алкила или олефина. Таким обра-
зом, в реакциях алкилирования с применением А1С13 могут встретиться
осложнения.
И. П. Цукерваник разработал реакцию алкилирования ароматических
углеводородов спиртами в присутствии хлористого алюминия и показал,
что в этом случае спирт ROH превращается в алкилирующий агент
ROA1G12. Подобное алкилирование спиртами идет также в присутствии
трехфтористого бора. Олефины алкилируют ароматические соединения
и в присутствии кислот — серной, фосфорной и др. Промышленные
процессы такого рода, например получение этилбензола и изопропилбен-
зола, разработали в СССР М. А. Далин и Ю. Г. Мамедалиев.
Гомологи бензола можно синтезировать и путем ацилирования по
Фриделю — Крафтсу с последующим восстановлением ацилбензола
(кетона) в алкилбензол по Клемменсену. Ацилирование бензола идет только
до стадии моноацилбензола и не сопровождается изомеризацией:
\ 8+
||+ R—С—G1
О:А1С13
С-К CHiR
+ О:Л1С1з +«
6. Синтез диарил- и триарилметанов. Альдегиды в сильнокислой среде
энергично реагируют с ароматическими углеводородами и даже с более
пассивными их галоидпроизводными уже при низкой температуре. Конеч-
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
38
ные продукты этой реакции - - диарилалкапы или, в случае ароматиче-
ских альдегидов, триарилметаны. Реакция протекает, конечно, через
стадию образования вторичного спирта (который обычно не изолируют)
по схеме: п
Н । /И
R_C - II* —> R-С.* ’ -к-с
I Ч)-н
он
Таким образом, реакция проходит как электрофильное замещение
в ароматическом ядре при атаке карбкатиона.
В растворе фтористого водорода альдегиды аналогично реагируют
с толуолом уже при —70° С. Хлораль при комнатной температуре в кон-
центрированной серной кислоте вступает в описанную реакцию с хлор-
бензолом, образуя известный инсектицид ДДТ:
Н
CClsCHO + 2С6Н5С1 —Cl
X=Z ] =/
СС13
О такого рода конденсациях см. также стр. 213, 219.
ОРИЕНТАЦИЯ ЗАМЕЩЕНИЯ
Реакции электрофильного замещения в ряду бензола подчиняются следу-
ющим правильностям, установленным эмпирически (Голлеман).
1. Место вступления электрофильного*" заместителя определяется
(в основном) характером уже присутствующих одного или нескольких
заместителей в бензольном цикле.
2. Заместители (ориентанты) делятся на две группы:
а) заместители первого рода, или орто-пара-ориентанты, направля-
ющие новый заместитель в орто- и пара-положение по отношению к себе
(здесь и ниже ориентанты даются в порядке убывающей ориентирующей
способности; R — означает алкил): о о
—О, — NR2, —NHR, —NH2, —ОН, —OR, — NH—С—R, —О—С—R,
—СНз и другие алкилы, алкенилы, I, Вг, Cl, F
б) заместители второго рода, или мета-ориентанты, направляющие
вступающий заместитель в мета-положение:
* * z° ,Н R
—NR3, —-CN, —SO3H, —CF3, —CCI3, —c^0> — c\x0’
/0 ,0
—CZ , -c^
OH \0R
ОРИЕНТАЦИЯ ЗАМЕЩЕНИЯ
39
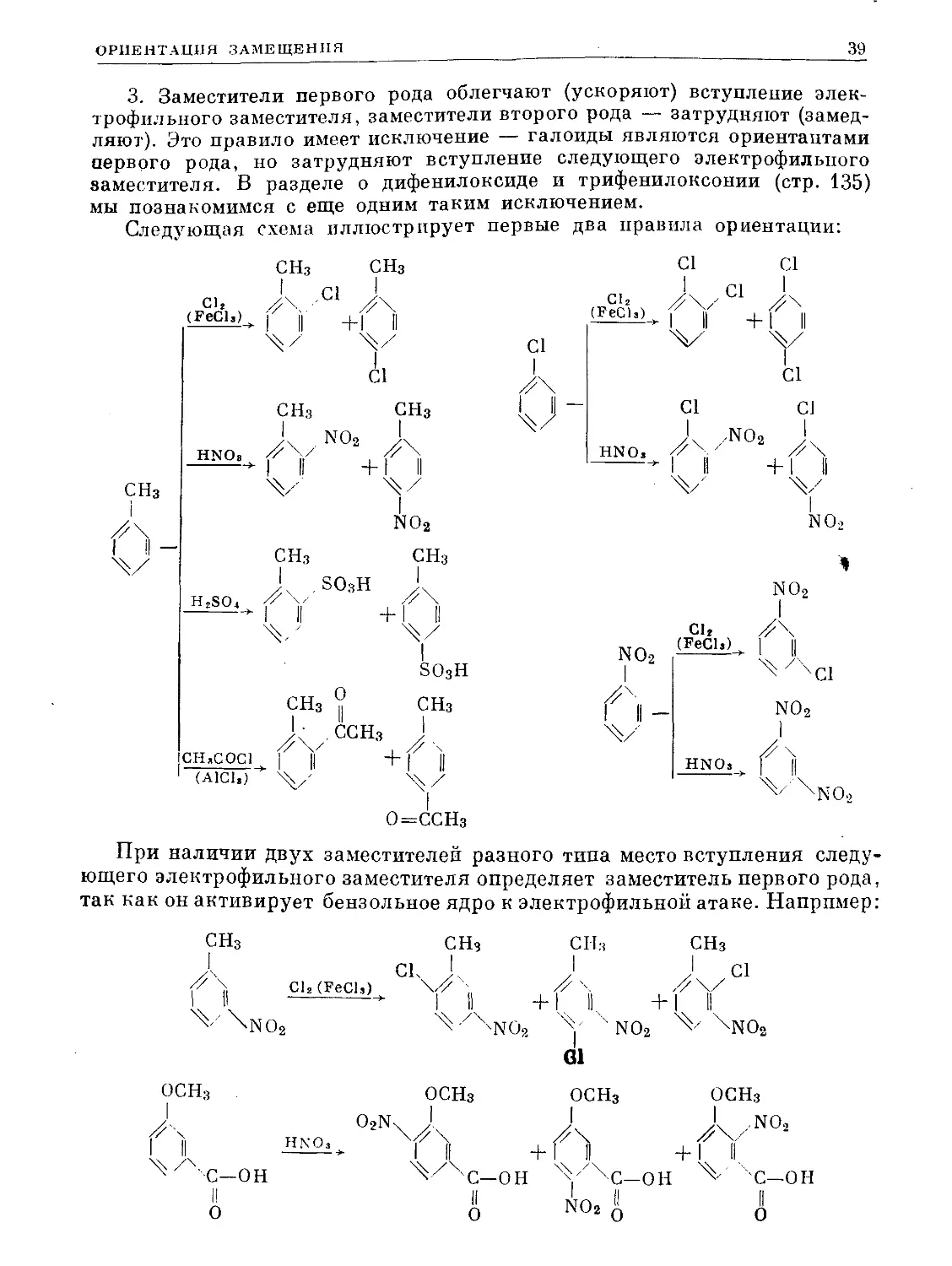
3. Заместители первого рода облегчают (ускоряют) вступление элек-
трофильного заместителя, заместители второго рода — затрудняют (замед-
ляют). Это правило имеет исключение — галоиды являются ориентантами
первого рода, но затрудняют вступление следующего электрофильного
заместителя. В разделе о дифенилоксиде и трифенилоксонии (стр. 135)
мы познакомимся с еще одним таким исключением.
Следующая схема иллюстрирует первые два правила ориентации:
СНз СН3
сн
(FeCla)
,С1
+1
С1
HNOa
СНз
no2
СНз
N02
H2SO4
CHaCOCl
(А1С1,)
СНз
СНз
SO3H
SO3H
СНз
СНз
. ССН3
сн
(FeCh).
HNOa .
О=ССНз
NO2
о
При наличии двух заместителей разного типа место вступления следу-
ющего электрофильного заместителя определяет заместитель первого рода,
так как он активирует бензольное ядро к электрофильной атаке. Например:
40
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
Если же оба ориентанта одного рода, то место вступления определяет
более сильный, а при не слишком разнящейся ориентирующей способности
получаются все изомеры, требуемые как одним, так и другим ориентантом.
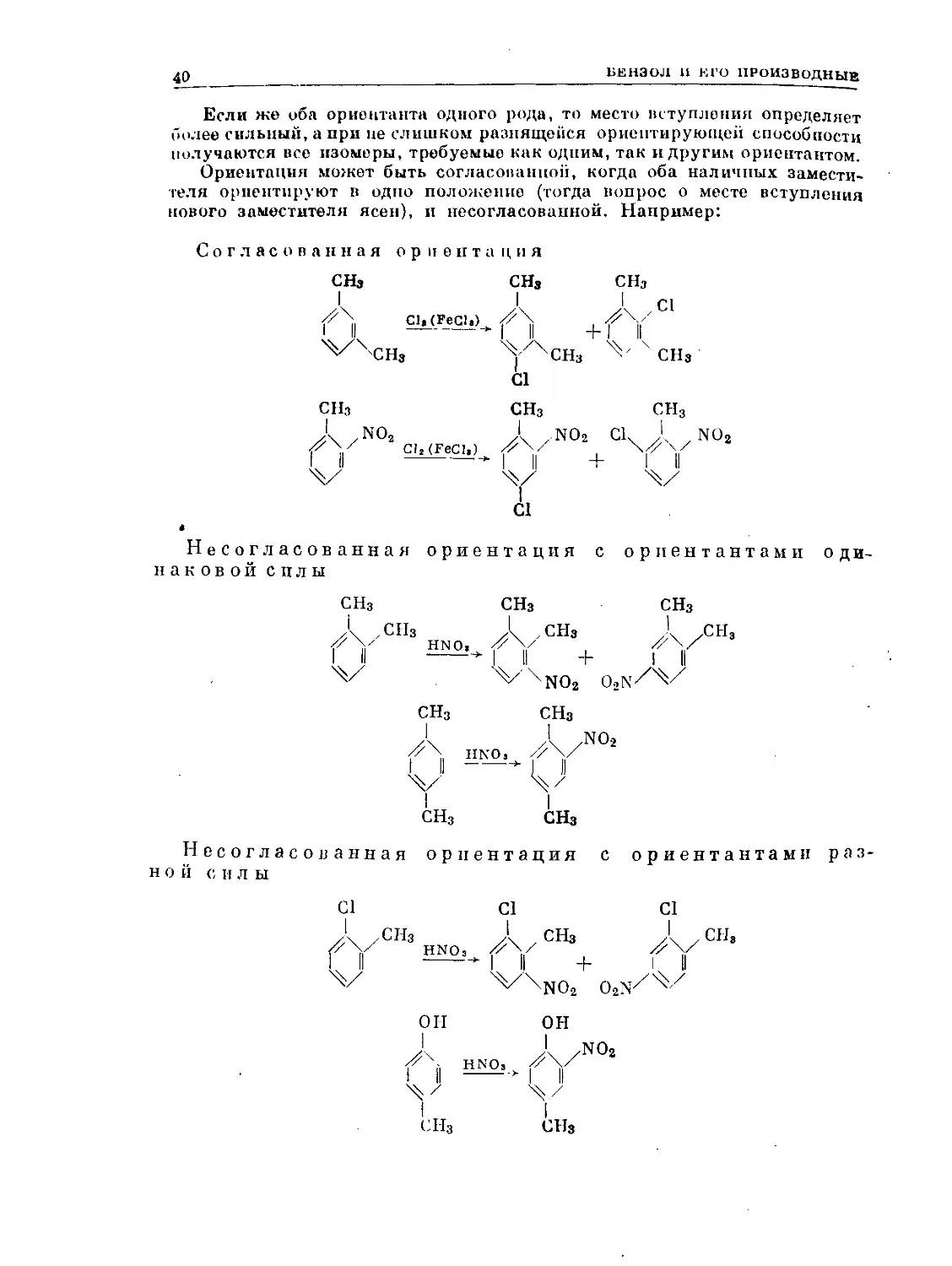
Ориентация может быть согласованной, когда оба наличных замести-
теля ориентируют в одно положение (тогда вопрос о месте вступления
нового заместителя ясен), и несогласованной. Например:
Согласованная ориентация
Несогласованная ориентация
наковой силы
с ориентантами
о ди-
СНз
HN0,
СНз
СН3
А/СНз
। II
СНз СНз
/\ /\ ZN°2
и
СНз СНз
Несогласованная ориентация с ориентантами раз-
ной с н л ы
С1 С1 С1
ОН
СНз
ПРИЧИНА ЯВЛЕНИИ ОРИЕНТАЦИИ
41
Возникает вопрос, как увязать все только что сказанное с изложенным
на стр. 24 правилом Кернера об установлении орто-, пара- пли мета-
расположения групп в двузамещенных бензолах? Правило Кернера исхо-
дит из образования всех возможных изомеров при вступлении третьего
заместителя, а правила ориентации ограничивают число возможных изо-
меров. Дело в том, что правила ориентации определяют главные направле-
ния реакции, пренебрегая теми изомерами, которых получается очень
мало (доли процента или немногие проценты), а способ Кернера основы-
вается на учете всех продуктов реакции, в том числе и образующихся
в очень малых количествах. В этом причина кропотливости способа.
ПРИЧИНА ЯВЛЕНИИ ОРИЕНТАЦИИ
Действие ориентанта заключается в изменении свободной энергии акти-
вации G* переходного состояния сравнительно с незамещенным бензолом
(см. также стр. 33). Ориентанты
первого рода понижают эту вели-
чину для переходного состояния
вступления нового электрофиль-
ного заместителя, особенно в орто-
и пара-положения (рис. 85). В ре-
зультате в соответствии с выраже-
нием для константы скорости реак-
ции (кн. 1, стр. 95)
до*
. ~ ит
к = —— е
h
резко (в показателе!) повышается
константа скорости реакции заме-
щенного бензола сравнительно с
незамещенным бензолом. Пониже-
Рорды переходное^ состояния- .
к 6-комплексу
Рис. 85. Энергетическая диаграмма заме-
щения на электрофил (N02) в бензоле
и в мета- и пара-положения толуола.
ние G* происходит благодаря
подаче электронов ориентантом
первого рода к углероду, с которым этот ориентант соединен и на котором
по преимуществу возникает в переходном состоянии положительный за-
ряд. Примеры:
Атака в орто-положение
бензол и его производные
42
Атака в пара-положение
Атака в пара-положение
NO2
Атака в орто-положение
Из приведенных схем видно,
бензольное кольцо, вытягивает из
нов * и привязывается этой парой
в переходном состоянии и в
что электрофильный реагент, атакуя
ароматического секстета пару электро-
к атакуемому углероду, сохраняющему
ст-комплексе связь и с водородным
атомом.
Атакованный углеродный атом становится в о-комплексе тетраэдри-
ческим углеродом, а оставшиеся четыре л-электрона располагаются в виде
Двух сопряженных двойных связей в кольце. Вследствие оттяжки пары
электронов, образовавшей связь с атакующей группой, на соседнем с атаку-
емым атоме углерода возникает положительный заряд. Этот положитель-
ный заряд может быть частично нейтрализован (компенсирован), если он
ходится на углероде, несущем орто-пара-ориентант, т. е. электроно-
нор, при условии, что атаке подвергается углеродный атом
Рто"или пара-положении по отношению к углероду, несущему
ориентант.
мостиХаНИЗМ компенсайии положительного заряда различен в зависи-
несет °Т хаРактеРа орто-пара-ориентанта. Если ориентант первого рода
на ключевом атоме свободную (неподеленную) пару электронов
* Предварительную стадию образования л комплекса мы
поэтому не изображаем.
здесь не учитываем и
ПРИЧИНА ЯВЛЕНИЙ ОРИЕНТАЦИИ
(—NH,, —ОН, —ОСН3, —С1: и др.), то он передает эту пару в совместное
обладание связанному с ним углероду бензольного кольца, устанавливая
с ним двойную связь и переходя в результате в положительно заряжен-
ное — аммониевое, оксониевое и т. п. — состояние. Это знакомый нам
4~/’-эффект (ср. кн. I, стр. 333).
Такая система гораздо беднее энергетически, чем система положи-
тельного карбониевого углерода. Поэтому свободная энергия акти-
вации переходного состояния ниже, чем в отсутствие ориентирующей
группы. Если орто-пара-ориентант, например алкил, не имеет свободной
пары электронов, то он частично компенсирует возникший положитель-
ный заряд связанного с ним карбониевого углерода путем сдвига пары
электронов связи от алкила к бензольному карбониевому углероду в соот-
ветствии с 4-7-эффектом, присущим алкильным группам (нужно помнить,
что понятие об 4-1-эффекте возникает только при сравнении эффекта,
оказываемого алкилом, со стандартом — водородным атомом). Сравним,
например, бензол и толуол:
При атаке нитрогруппой (нитроний-катионом) возникший в орто-
положении к месту атаки положительный'заряд лучше нейтрализуется
соседней СН3-группой (толуол), чем водородом (бензол), так как электрон-
ная пара связи Н3С : С обслуживает бензольный углерод в большей
степени, чем электронная пара связи Н : С.
В случае электрофильной атаки в мета-положение компенсации заряда
за счет ориентанта не происходит:
Поэтому, например, нитрование толуола в мета-положение должно
было бы происходить примерно с той же скоростью, что и незамещенного
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
44
бензола. На деле ориентанты первого рода, резко ускоряющие процесс
электрофильного замещения в орто- и пара-положения, все же несколько
ускоряют и замещение в мета-положение. Так, толуол нитруется в орто-
и пара-положения примерно в 40 раз быстрее, чем бензол, а в мета-положе-
ние тоже быстрее, по лишь в два-три раза. Эту передачу влияния в мета-
положение объясняют обычно индуктивным эффектом (+/-эффектом),
обозначаемым прямой стрелкой, символизирующей смещение электрон-
ной пары простой (о-) связи сравнительно со стандартом:
СН3
NO2
Поскольку /-эффекты быстро затухают по цепи атомов, они очень слабы
сравнительно с /’-эффектами и повышение скорости замещения в мета-
положение невелико.
ГИПЕРКОНЪЮГАЦИЯ И ОРТО-ПАРА-ОРИЕНТАЦИЯ
Все алкилы во много раз увеличивают скорость электрофильного замеще-
ния в бензольном ядре. Однако Бэкер и Натан отметили неожиданную
последовательность алкилов как орто-пара-ориентаптов при их располо-
жении в ряд по ориентирующему действию, в частности по скорости хлори-
рования и бромирования. Относительные скорости галоидирования при
24° С в 15%-ной водной уксусной кислоте изменяются следующим
образом:
СНз СН2СН3 СН(СНз)2 С(СН3)з
I I I I '
. й /Л //\ //\ //^ //\
Алкилбензолы........I II I II I II | II | II
Скорость
хлорирования . . . 0,29 100 94 51 32
бромирования ... — 110 76 44 23
Эта последовательность прямо противоположна ряду тех же алкилов
в порядке убывающего +/-эффекта:
~-С(СН3)з > — СН(СНз)2 > -СН2СН3 > -СН3
Если сопоставить в той же последовательности дипольные моменты
алкилбензолов, то окажется, что толуол имеет в этом ряду наименьший
дипольный момент, а mpem-бутил — наибольший, т. е. что метил (в нере-
агирующеи молекуле) передает ядру меньший отрицательный заряд,
-S/1 ^-бУ™льная группа. Это согласуется с понятием индуктивного
' эффекта (кн. I, стр. 192) как эффекта подачи ковалентной пары электро-
ов связи атомом X атому Y в комбинации X—>У сравнительно с
ОРИЕНТИРУЮЩИЙ ЭФФЕКТ ГАЛОИДНОГО ЗАМЕСТИТЕЛЯ
45
Ясно, что чем больше алкильных групп несет ключевой (связанный
с бензолом) атом углерода, тем больше его -(-/-эффект:
Алкплбензолы
СНз
СН3->С->СбН5
ch3Z
СНз
Н-»С-»СвН5
СНз
СНз-»СН2->С6Н5 СНз->С6Н5
Дипольный момент, D ... 0,7
0,65 0,58 0,37
Описываемый парадокс, получивший название эффекта Натана —
Бэкера, был объяснен большей поляризуемостью Н—С-связи сравни-
тельно с С—С-связыо и возможностью благодаря этому тем большей подачи
электронов алкильной группы уже за счет + 7'-эффекта, чем с большим
числом водородов связан ее ключевой атом (гиперконъюгация):
fi о н
Cl-Cl —> Cl+н++сг
H
На языке резонанса гиперконъюгацию в молекуле толуола изобра-
жают так:
Понятие гиперконъюгации не ограничивается только что рассмотрен-
ной областью или явлениями взаимодействия алкилов с ароматическим
ядром, но имеет и общее значение. Это — частный, но важнейший случай
о, л-сопряжения (ср. кн. I, стр. 425), которое касается уже не только
взаимодействия систем Н—С—С= или Н—С — но и систем с не-
водородными атомами.
ОРИЕНТИРУЮЩИЙ ЭФФЕКТ ГАЛОИДНОГО ЗАМЕСТИТЕЛЯ
Особым случаем является ориентирующее действие галопда. Галоиды —
это самые слабые из обычных ориентантов первого рода. Но в отли-
чие от всех других ориентантов первого рода они не ускоряют, а замедляют
реакции электрофильного замещения. Объясняется это противоположным
знаком их —I- и Ч-Т-эффектов. Галоиды оказывают на бензольное ядро
сильный —/-эффект (оттягивание электронов из ядра сравнительно
со стандартной связью Н—С) и слабый -f-71-эффект (способность в момент
ЦЕНЗОМ II ЕГО ПРОИЗВОДНЫЕ
Ну
атаки в орто- и пара-положения подать пару электронов и нейтрализовать
таким образом положительный заряд, возникающий на первом углеродном
атоме цикла). Это выражают следующей схемой:
Понимать это следует таким образом: прямая стрелка показывает,
что бензольное ядро хлорбензола обеднено электронами сравнительно
с бензолом. Однако в случае электрофильной атаки в орто- или пара-
положения все же возникает переходное состояние обычного для орто-
пара-ориентации типа. Правда, уровень свободной энергии этого переход-
ного состояния выше, чем в случае незамещенного бензола, поскольку
компенсация положительного заряда первого углерода за счет неспарен-
ных электронов галоида менее полная, чем для любого другого обычного
ориентанта первого рода. Не компенсируется полностью даже та оттяжка
электронов (сравнительно с бензолом), которая осуществилась за счет
сильного индуктивного^эффекта хлора.
предельное
(недостижимое)
состояние
н no2 ( 11 NO2 J
писанного действия + /’-эффекта было бы достаточно, чтобы объяс-
нить орто-пара-ориентацищ электрофильного замещения галоидом,
а деле, как это часто бывает, -\-Т-эффект сопровождается +ЛГ-эффектом,
т. е. мезомерным эффектом, являющимся постоянной составляющей
(а не только в момент реакции) таутомерного эффекта. Кривые стрелки,
изогоне °Т ХЛ0Ра к бензольному ядру — в орто- и пара-положения, —
Ра'ка10т взаимодействие галогена с бензольным ядром и вне реакции,
то взаимодействие слабо. Лучше всего это можно понять, вспомнив
кн. , стр. 3-±9) разницу дипольных моментов хлорбензола (1,56 D) и хлор-
тпоаНа,ЦНаПРИМеР хл0РЭтана (2,00 D). Хлор в хлорбензоле подает элек-
с^Г ензольяомУ ядру, если сравнивать его с хлором хлорэтана, но
* ьно оттягивает электроны из бензола, если в качестве стандарта взять
МЕТА-ОРИЕНТАЦПЯ
47
сам беызол (т. е. вести сравнение хлора с водородом). О такой сравнитель-
ности оценок электронных влияний не следует забывать.
Другой пример дезактивирующего действия орто-пара-ориентанта см.
при трифенилоксонии (стр. 135). Так же дезактивируют орто-пара-ориен-
танты —СН2С1 и —СН=СН—NO2.
МЕТА-ОРИЕНТАЦИЯ
Мета-ориентанты бывают двух родов — действующие по механизму
—/-эффекта и действующие —/-эффектом. Первые имеют л-связи у клю-
чевого атома:
-S^O
\он
и т. п.
Вторые действуют только благодаря наличию положительного заряда
на ключевом атоме:
СНз
-N^-СНз
/F
О + / -уч
-C~F
\F
\ci
и т. п.
В обоих случаях это действие за счет оттяжки электронов имеет резуль-
татом создание положительного заряда на том углероде бензольного
кольца, который несет мета-ориентант. Мы видели, что атака электрофиль-
ного реагента в орто- и пара-положения по отношению к любому замести-
телю также создает положительный заряд на том же самом углеродном
атоме. Поэтому соответствующее переходное состояние и . сг-комплекс
менее выгодны (энергетически выше), чем у незамещенного бензола.
н no2
При атаке же в мета-положения столь невыгодной комбинации не возни-
кает, и эта атака оказывается единственно возможной. Так как осуще-
ствляется и общая оттяжка электронов из бензольного кольца, в част-
ности из мета-положений (—/-эффект), то и в мета-положения атака
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
48
совершается труднее, чем в незамещенном бензоле, и бензольное кольцо,
замещенное мета-ориентантом, оказывается в целом более пассивным
к электрофильным замещениям.
Когда идет электрофильная атака производного бензола, содержащего
мета-ориентирующие группы, несущие активные свободные пары элек-
тронов, например
то наряду с вступлением электрофильного заместителя преимущественно
в мета-положение, замещение направляется в довольно значительной
степени и в орто-положение. Поэтому в данных примерах при нитровании
получаются кроме мета- также и орто-изомеры!
НО—С=О
2
По-видимому, это объясняется первичным связыванием атакующего
электрофила р-парами или л-электронами ориентанта с последующим
перемещением электрофила в ближайшее орто-положение:
Н+
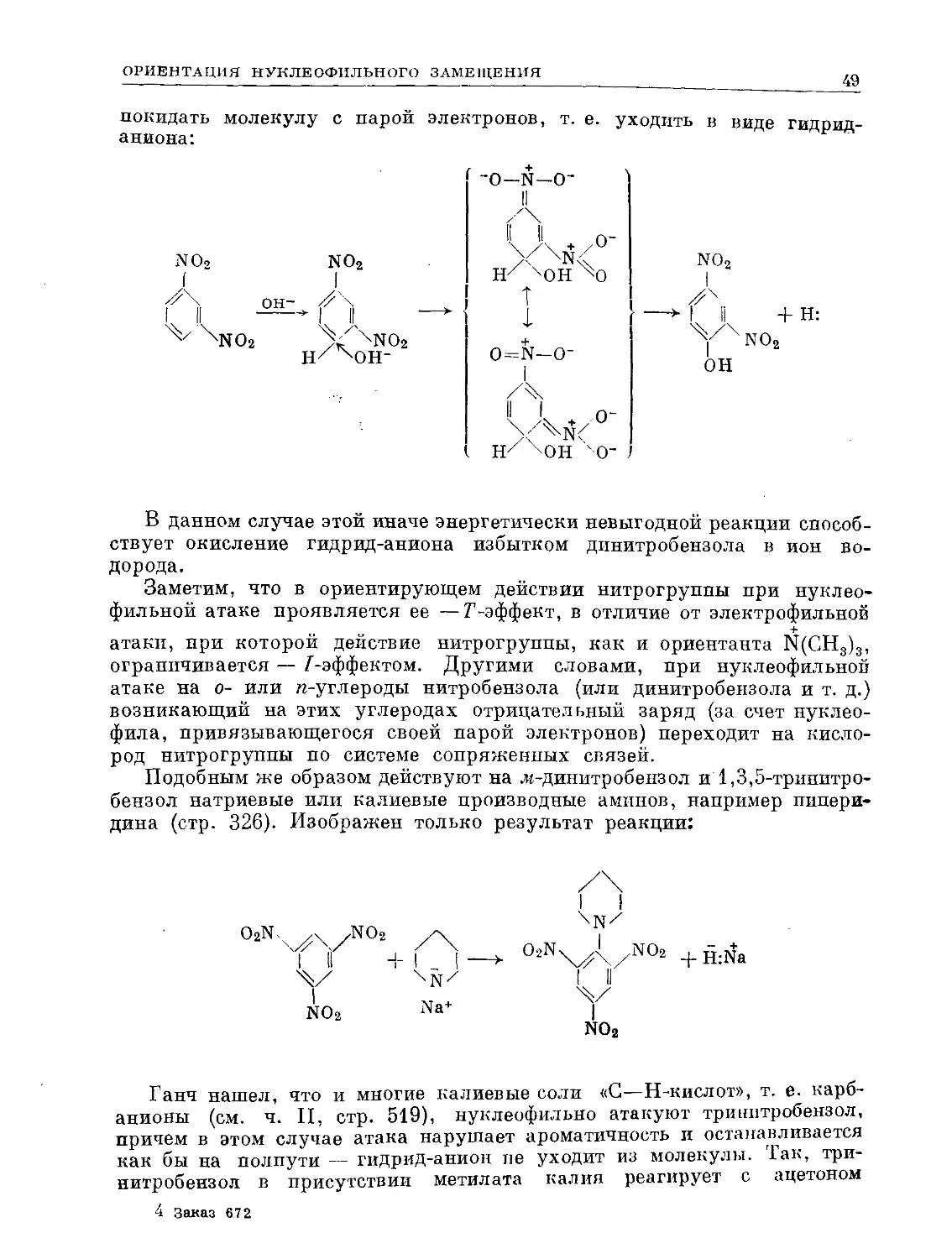
ОРИЕНТАЦИЯ НУКЛЕОФИЛЬНОГО ЗАМЕЩЕНИЯ
До сих пор мы рассматривали только электрофильные атаки на бензоль-
ное ядро. Естественно предположить, что наличие сильных мета-ориентан-
тов, оттягивающих электроны и обнажающих положительные заряды
на углероде, должно способствовать успеху нуклеофильных атак. Преды-
дущие рассуждения показывают, что в бензоле, несущем мета-ориентиру-
ющий заместитель, наиболее подвержены нуклеофильным атакам должны
быть орто- и пара-положения. Действительно, л£-динитробензол реагирует
со щелочью и с амидом натрия, замещая один из орто-или пара-водород-
ных атомов па ОН или, соответственно, на NH2- Этот водород должен
ОРИЕНТАЦИЯ НУКЛЕОФИЛЬНОГО ЗАМЕЩЕНИЯ
49
покидать молекулу с парой электронов, т. е. уходить в виде гидрид-
аниона:
-о—N—0-
no2 no2
f I
//\ он-
I II —* I II
ЧЧО2 Э/ xNO2
HZ Х)Н-
0=N—0-
В данном случае этой иначе энергетически невыгодной реакции способ-
ствует окисление гидрид-аниона избытком динитробензола в ион во-
дорода.
Заметим, что в ориентирующем действии нитрогруппы при нуклео-
фильной атаке проявляется ее —Г-эффект, в отличие от электрофильной
+
атаки, при которой действие нитрогруппы, как и ориентанта N(CH3)3,
ограничивается — /-эффектом. Другими словами, при нуклеофильной
атаке на о- или и-углероды нитробензола (или динитробензола и т. д.)
возникающий на этих углеродах отрицательный заряд (за счет нуклео-
фила, привязывающегося своей парой электронов) переходит на кисло-
род нитрогруппы по системе сопряженных связей.
Подобным же образом действуют на .и-динитробензол и 1,3,5-тринитро-
бензол натриевые или калиевые производные аминов, например пипери-
дина (стр. 326). Изображен только результат реакции:
Ганч нашел, что и многие калиевые соли «С—Н-кислот», т. е. карб-
анионы (см. ч. II, стр. 519), нуклеофильно атакуют тршштробензол,
причем в этом случае атака нарушает ароматичность и останавливается
как бы на полпути — гидрид-анион не уходит из молекулы. Гак, три-
нитробензол в присутствии метилата калия реагирует с ацетоном
4 Заказ 672
БЕНЗОЛ И ЕГО ПРОИЗВОДНЫЕ
50
следующим образом, давая черио-фиолетовый продукт присоединения —
о-комплеке:
/NOg
OgN-^ +
^NOg
О О-
♦ _ II + I
К(СН2—С—СНз) <----► K(CHg=C—СНз)
-о /NO2
+ и =< ZH о
к n=< и
I х=< \СН2-С—СНз
-о no2
Если покидать молекулу продукта приходится не гидрид-аниону,
а, например, аниону хлора, дело происходит гораздо легче. В этом случае
нет необходимости в согласованном действии на место атаки двух мощных
мета-ориентантов, а достаточно и одного. Так, в молекулах о- и п-нитро-
хлорбензолов и других аналогичных соединений имеется, как принято
выражаться, «подвижный» хлор (открытие П. А. Лачинова, 1870 г.),
чем эти молекулы отличаются от хлорбензола, в котором хлор лишь в жест-
ких условиях и медленно вступает в реакции обмена. Этот подвижный
хлор легко обменивается на пуклеофильные группы (ОН, NH2, SH и т. п.).
Причина такой подвижности хлора заключается в электрофильности
связанного с ним углерода, который становится восприимчивым к нуклео-
фильным атакам благодаря способности мета-ориентанта компенсировать
привнесенный атакующим нуклеофилом отрицательный заряд:
+ СГ
ОН
При атаке в мета-положение такой компенсации не происходит,
поэтому хлор в .и-нитрохлорбензоле «неподвижен».
Действие, подобное питрогруппе, оказывают и другие мета-ориентанты.
Это явление — лишь иное проявление законов ориентации замещения
в бензольном ядре. Дело, следовательно, не в «подвижности» галоида,
а в возросшей электрофильности связанного с ним углерода. Разумеется,
электроноакцепторные заместители в орто- и пара-положении облегчают
замещение на нуклеофилы не только галоида (и гидрид-аниона), но и
Других групп, способных покидать молекулу в виде аниона (NH2, ОН
и Др.).
ГАЛОИДПРОИЗВОДНЫЕ БЕНЗОЛЬНОГО РЯДА
51
При накоплении электроноакцепторных заместителей нуклеофильная
атака может остановиться на стадии ст-комплекса, который в этом случае
достаточно устойчив. Мы уже видели это на примере взаимодействия
тринитробензола с анионом ацетона (Ганч, стр. 49). Мейзенгеймер,
действуя этилатом щелочного металла на 2,4,6-тринитроанизол (ани-
зол — метоксибензол С6Н5ОСН3), получил продукт присоединения II,
который лишь в более жестких условиях отщепляет алкоксил, образуя
пР0ДУкт замещения метоксила на этоксил (Ш):
+ СН3О" Na+
Ш
Легко видеть, что о-комплекс II аналогичен, но противоположен по
знаку о-комплексам, образующимся при электрофильной атаке, среди
которых тоже встречаются индивидуализируемые соединения (стр. 34, 37).
ГАЛОИДПРОИЗВОДНЫЕ БЕНЗОЛЬНОГО РЯДА
Из сказанного в предыдущем разделе следует, что бензол, его гомологи
и их производные легко подвергаются замещению на хлор или бром, но
только в присутствии катализаторов, так называемых переносчиков галоида
(FeCl3, А1С13, 1С13 и другие галогениды). Общее свойство этих галогенидов
состоит в способности образовывать с анионом галоида более или менее
прочные анионы типа [FeCl4]“, [А1С14]—, [IC14]~, [FeBrJ-и т. д.
По силе действия переносчики брома располагаются в следующий ряд
(Б. В. Тронов):
FeBr3 > А1Вгз > ZnBr2 > 1Вг3 > SbBrs > CuBr2 > S2Br2 > РВг3
При действии нейтральной молекулы галоида (только хлора или брома)
эта молекула галоида поляризуется в силу захвата (полного или в совмест-
ное обладание) одного из двух атомов молекулы «переносчиком галоида»,
причем второй атом галоида получает 6 + -заряд и действует как элек-
трофильный реагент: g
I |
—Cl-Fe-Cl
Cl
С12 + FeCl3----> Cl-
4*
ГАЛОИДПРОИЗВОДПЫЕ БЕНЗОЛЬНОГО РЯДА
52
Таким образом, хлорирование в ядро бензола и толуола выразится
следующими итоговыми уравнениями:
С1
I
+ cl-f-ClFeClsp-----> + H+[FeCl4]-
СН3
I
.+
1 U + Cl—[—ClFeClsP” >
CH3
I
+ О + H+[FeCl4]-
I
Cl
Так же происходит и бромирование.
Хлорбензол впервые получен в 1865 г. Н. В. Соколовым.
При действии большего количества галоида получаются более галои-
дированные ароматические углеводороды (А. А. Курбатов):
СНз
СН3
СНз
Можно прохлорировать бензол избытком хлора в гексахлорбензол.
В качестве побочного продукта гексахлорбензол получается и при хлори-
ровании в жестких условиях многих алифатических и алициклических
углеводородов и погонов нефти (Ю. Г. Мамедалиев).
Метильная группа толуола при электрофильном хлорировании с «пе-
реносчиком» остается незатронутой. Если же хлорировать (или бромиро
вать) толуол и другие гомологи бензола на свету, лучше при температуре
кипения (в парах), при тщательно обеспеченном отсутствии железа и ДРУ_
гих «переносчиков», то галоидирование идет по законам цепных реакции
только в алкильную группу. Толуол при этом последовательно превра
щается в хлористый (или бромистый) бензил, хлористый бензилиден
бензотрихлорид, а этилбензол — в а-хлорэтилбензол и а,а-дихлорэти
бензол. Легко галоидируется именно а-положение боковой цепи, a 0
удаленные от бензольного ядра положения — труднее, а именно та ’
как в предельных углеводородах. Это происходит потому, что сво д
радикалы типа бензильного СвН5СНг или а-фенилэтильного^СвН6СНСЩ,
которые фигурируют как промежуточные продукты в цепной ре
ГАЛОНДПРОИЗВОДНЫЕ БЕНЗОЛЬНОГО РЯДА
53
более устойчивы и, значит, образуются с меньшей энергией активации
(а следовательно, и с большей скоростью), чем изомерные им радикалы,
например СвН5СН2СН2:
Cl2 2СЬ (инициирование цепи)
CeH5CH3-f-С1 • —> СвЩСНг+НС! (звено цепи)
С6Н5СН2 4- С12 --> С6Н5СН2С1 + С1- и т. д.
Таким же путем (пишем лишь итоговое уравнение) образуются хло-
ристый бензилиден и бензотрихлорид:
С6Н5СН3 + 2С12 -> С6Н5СНС12 + 2НС1
С6Н5СН3 + ЗС12 -> С6Н5СС13 + ЗНС1
Реакцией электрофильного замещения можно ввести в ароматические
углеводороды группу СН2С1 (хлорметилирование). Для этого на арома-
тический углеводород действуют формальдегидом (в виде формалина или
параформа) в присутствии хлористого цинка, пропуская одновременно
хлористый водород (Блан):
СН2О + Н+ —> СН2ОН <----> СН2=О—Н
ArH +СН2ОНгй+^ АгСН2ОН - HC1 АгСН2С1
Интересную реакцию обмена толуола с четыреххлористым углеродом
(225—235° С) описал Ю. А. Ольдекоп:
С6Н5СН3 + СС14 -> С6Н5СН2С1 + СНС13
Иодирование в боковую цепь не идет. Иодированные и фторирован-
ные в цепь продукты получают путем обмена соответствующих хлоридов
с AgF, SbF3, Nal.
Иодирование не требует «переносчика галоида», однако оно происходит
только при наличии достаточно активного ориентанта первого рода.
Ни бензол, ни алкилбензолы иодом непосредственно не иодируются, но аро-
матические амины, ациламины и фенолы иодируются очень легко. Можно,
однако, непосредственно проиодировать и бензол, и его гомологи, если
одновременно действовать окислителем на выделяющийся иодистый водо-
род. Реакция иодирования обратима, иодистый водород восстанавли-
вает полученный иодбензол (в случае бензола) в бензол, а окисление йодис-
того водорода купирует обратную реакцию. Окисление часто произво-
дят йодноватой кислотой. Очень удобно иодировать по Тронову, действуя
на ароматическое соединение иодом в нитрующей смеси (т. е. смеси
концентрированных азотной и серной кислот).
Фторировать ароматические углеводороды неразбавленным элемен-
тарным фтором нельзя, так как реакция заходит дальше — до образования
четырехфтористого углерода и фтористого водорода. Поэтому аромати-
ческие фторпроизводные получают через диазосоедипения по реакции
Шимана (стр. 110). Введение нескольких атомов фтора непосредственно
фторированием бензола при помощи, например, CoF3 или MnF3 приводит
Таблица 61. Галоидные производные бензола и толуола
Формула Название Температура плавления °C
CeH6F Фторбензол —41,9
СЯН6С1 Хлорбензол — 45,0
С6Н6Вг Бромбензол —30,6
Дифторбензол о-, или 1,2- —34,0
л-, или 1,3- —59,0
п- или 1,4- —13,0
СвН4С12 Дихлорбензол о-, или 1,2- —17,5
м-, или 1,3- —24,8
п-, или 1,4- 4-53,0
С^НдВгз Дибромбензол о-, или 1,2- 5,6
•и-, или 1,3- —6,9
п-, или 1,4- 4-86,9
CeFe Гексафторбеизол 5,29
СеС1в Гексахлорбензол 227
СаВга Гексабромбензол 306
Температура кипения •С Плотность Показатель преломления nD Дипольный момент в бензоле при 20°С D
85,0 1,0240 1,4646 1,39
132,0 1,066 1,5246 1,56 (при 21° С)
156,0 1,4951 1,5597 1,49
92,0 1,1599 1,4451 2,38
(при 751 мм рт. ст.) (при 18° С) (при 18° С) (при 22° С)
83,0 1,1552 (при 18° С) 1,4404 (при 18° С) 1,58 (газ)
89,0 1,1684 (при 18° С) 1,4422 (при 18° С) —
183,0 1,3048 1,5518 (при 22° С) 2,33
172,0 1,2881 1,5457 (при 20,9° С) 1,48
173,4 1,4581 1,5210 (при 80,3° С) 0 (при 25° С)
221,0 1,9557 1,6117 (при 17,5° С) 1,87
218,0 1,9523 1,6083 (при 17,5° С) 1,46
220,0 2,261 (dj7) 1,5743 0
80,5 — — —
326 2,044 (при 23° С) — 0,1
CHgCeHiF фтортолуол
о-, или 1,2- —80
m~, или 1,3- —110,8
n-, или 1,4- —
CHsCeH4Cl Хлортолуол о-, пли 1,2- —34,0
jh-, или 1,3- —47,8
п-, пли 1,4- 4-7,5
СНзСвЩВг Бромтолуол о-, пли 1,2- —27,0
.м-, пли 1,3- —39,8
п-, или 1,4- 4*28,0
ch3c6f5 Пептафтортолуол —
CH3C6C15 Пснтахлортолуол 218
CH3C6Br5 Пентабромтолуол 288
CeH5CH2F Фтористый бензил —35,0
C6H5CH2C1 Хлористый бензил —43,0
C6H5CH2Br Бромистый бензил —4,0
CeH5CHF3 Фтористый бензилиден —
C6H5CHC12 Хлористый бензилиден —16,0
C6H5CHBr2 Бромистый бензилиден —
CeHbCF3 Бензотрифторид —29,0
ceH5cci3 Бензотрихлорид —22,0
114 1,0041 1,4704 1,30
(при 13,0° С) (при 30° С)
116 0,9972 1,4691 1,78
(при 13° С) (при зо° С)
117,0 1,001 1,470 1,88
(при 15° С) (при 30° G)
159,0 1,0817 1,5238 1,43
162,0 1,0722 1,5214 1,77
(при 19° С)
162,0 1,070 1,5199 1,94
(при 19° С)
181,8 1,4220 — 1,44
183,7 1,4099 1,551 1,75
185,0 1,3898 1,549 1,93
-— — —
301,0 — — 1,55
(при 25° С)
— 2,970 — —-
(при 17° С)
139,9 1,0228 — 1,77
(при 25° С) (при 25° С)
179,0 1,1026 1,5415 1,87
(при 18° С) (при 15° С) (при 25° С)
198 1,4380 — 1,85
(при 22° С) (при 25° G)
207,0 1,2557 1,5502 2,03
(при 14° С) (при 25° G)
140 1,510 1,541 —
(при 20 ли» pm. cm.) (при 15° С)
102,4 1,196 1,4171 2,54
(при 14° С) (при 14° С) (при 25° С)
214,0 1,3803 1,5573 2,07 (при 25° С)
56
1’АЛОИДИРОИЗВОДНЫЕ БЕНЗОЛЬНОГО РЯДА
к полифторциклогексанам, которые нужно ароматизировать дегидро-
фторированием, чтобы получить смесь полпфторбеизолов.
Оказывается, пиролиз при 630—640° С GBr3F, а также фреонов
(например, GF2G12) приводит к гексафторбепзолу (Дезираи). По Н. Н. Во-
рожцову мл. гексафторбензол можно получить нагреванием гексахлор-
бензола с безводным фтористым калием до 530° G, причем получаются
также тетрафтордпхлорбеизолы и другие полигалогепбензолы. То же может
быть достигнуто в более мягких условиях — нагреванием при 380—430° С
гексабромбензола с фтористым калием.
Гексафторбензол — жидкость с т. кип. 79,5—80° G. В жестких усло-
виях фтор в гексафторбензоле обменивается нуклеофильно. Так, напри-
мер, действием 30%-пого водного раствора аммиака при 150° G в течение
Зч Н. Н. Ворожцов мл. и сотр. получили из гексафторбеизола пентафтор-
анилин (85%) с примесью тетрафтор-лс-фепилендиамина. Пентафтор-
бромбензол образует гриньяров реактив. Исходя их этих и других
первичных соединений, Н. Н. Ворожцов мл. развил целую область
химии перфторароматических соединений.
В табл. 61 приведены физические свойства галоидбензолов и галоид-
толуолов.
Обращает на себя внимание то обстоятельство, что замена водорода
на более тяжелый фтор (в 20 раз тяжелее) практически не меняет темпера-
туру кипения вещества. Это зависит от исключительно малой поляризу-
емости фтора (ср. низкие показатели преломления фторпроизводных сдан-
ными для других галоидпроизводных) и отсюда — малости дисперсион-
ных сил (сил Ван-дер-Ваальса). По той же причине изделия из фторопласта
(политетрафторэтилена) не смачиваются никакими жидкостями.
Химические свойства галоидпроизводных ряда бензола
Галоидные атомы, непосредственно связанные с углеродами бензольного
цикла, «неподвижны» и мало способны вступать в реакции нуклеофильного
замещения. Наиболее способен к этому типу реакций иод, наименее —
фтор.
1. Ароматические галоидпроизводные (кроме фторпроизводных) всту-
пают во взаимодействие с литием, натрием и магнием. Иодбензол и бром-
бензол в эфирном растворе реагируют с магнием лишь немногим более
вяло, чем галоидные алкилы, и образуют галоидный фенилмагний. Так же
реагируют и гомологи этих галоидбензолов. С хлорбензолом подобная
реакция идет при более высокой температуре (~-150° С), поэтому его можно
превратить в хлористый фенилмагний в автоклаве без эфира (П. П. Шо-
рыгин) или в тетрагидрофуране в обычных условиях. В лаборатории,
конечно, предпочтительнее применять для гриньярова синтеза бромиды
и иодиды. Таким же образом в эфире или в углеводородном растворе из
металлического лития получают растворы фениллития, применимые для
тех же реакций, что и растворы галоидного фенилмагния. Обе реакции
идут через стадию образования свободного радикала:
СвН5Вг + Mg---> CeH5- + -MgBr •—> CeHsMgBr
СяН5Вг + Li --► CftH5*-f-LiBr — - CeH5Li
ГАЛОИДПРОИЗВОДНЫЕ БЕНЗОЛЬНОГО РЯДА 57
Это ясно из того факта, что в качестве побочного продукта образуется
довольно много дифенила:
2С6Н5* —>• СвН5-С6Н5
(бромбензол и иодбензол индифферентны по отношению к гриньярову
реактиву).
Реакцию галоидбензолов с натрием можно задержать на стадии обра-
зования натрийорганического соединения
С6Н5Вг 4- Na --> NaBr + C6H5« — * C6H5Na
но фенилнатрий реагирует с бромбензолом
C6H5Br + C6H5Na--> NaBr + С6Н5—С6Н5
так что реакцию можно сразу вести как реакцию Вюрца:
2С6Н5Вг + 2Na -> С6Н5-С6Н5 4- 2NaBr
Можно исходить и из двух разных галоидных арилов или брать галоид-
ный арил и галоидный алкил:
СеЩВг 4~ С2Н5ВГ 4* 2Na -> CgHs—С9Н5 4~ 2NaBr
2. Галоидные арилы, реагируя с медью, тоже образуют диарил (реак-
ция Ульманна):
2С6Н51 4- 2Си -> С6Н5—С6Н5 4- 2CuI
3. Реакции нуклеофильного замещения галоида (но не фтора) в галоид-
ных арилах катализируются металлической медью (Ульманы), иногда
закисью меди и солями двухвалентной меди, так что в довольно жестких
условиях осуществимы такие типы реакций:
СвН5С1 4- NaOH — * С6Н5ОН 4- NaCl
CeH5I 4- Na—О—С—СНз — * С6Н5О—С—СН3 4- Nal
II II
О О
С6Н5Вг 4- NaCN — -> CeH5CN 4- NaBr
В отсутствие меди хлорбензол также можно прогидролизовать ще-
лочью, но для этого требуются очень жесткие условия, например про-
должительное действие 20—40?4-ной щелочи при 300° С и 300 ат. Срав-
нительно недавно найдено, что пары хлорбензола реагируют с парами
воды при атмосферном давлении и —500° С на силикагеле, промотиро-
ванном солью двухвалентной меди (способ Рашига). Этот процесс был
положен в основу одного из способов промышленного получения фенола,
впрочем, не имеющего большой перспективы.
Причина инертности ароматического галоида та же, что инертности
галоида в галоидном виниле (книга I, стр. 305), и связана с меньшим
дипольным моментом связи С — Hal в ароматических и винильпых гало-
генидах сравнительно с предельными. Атомы галоида, обладая свободными
парами электронов, взаимодействуют с л-связями випильной группы и
58
ГАЛОИДПРОИЗВОДНЫЕ БЕНЗОЛЬНОГО РЯДА
с л-системон связей бензольного ядра. Это мезомерный эффект (+ М-эф-
фект, кн. I, стр. 185 и 251), который очень невелик, так как активность р-
электронов хлора мала. Напротив, индуктивный эффект хлора (— /-эффект
т. е. оттяжка электронной пары связи к хлору как очень электроотрица-
тельному элементу) всегда велик, и он наводит положительный заряд на
связанный с этим хлором углерод и далее на орто- и пара-углероды бензола
или на 0-углерод хлористого винила (он соответствует орто-углероду
бензола). Однако эти б+-заряды в орто-, пара- и, соответственно, [J-поло-
жениях оказываются меньше, чем они были бы, если бы не мезомерный
эффект хлора, имеющий обратное направление (+Л/-зффект, подающий
электроны) и уменьшающий упомянутые б+-заряды. В справедливости
этого утверждения можно убедиться, сопоставив дипольные моменты
ряда галогенидов (см. стр. 59). Вследствие уменьшения б+ (на орто-, пара-
или 0-углеродных атомах хлорбензола и соответственно хлористого винила
и их бромистых аналогов) дипольные моменты этих галогенидов значи-
тельно ниже, чем у предельных галоидных алкилов, обладающих только
—1-, но не +Д/-эффектом галоида. Выше уже объяснялось, как наличие
— /-эффекта сказывается на замедлении электрофильных атак на угле-
роды хлорбензола — они обеднены электронами, оттянутыми хлором.
Подобным образом и хлористый винил, несмотря на слабый +Л/-эффект,
благодаря сильному —/-эффекту в целом (конечно, его углероды), обед-
нен электронами. Поэтому присоединение кислот или галоидоводородов
(электрофильная атака) к хлористому винилу идет медленнее, чем к эти-
лену, и тем более к пропилену. Однако электрофильные атаки совер-
шаются и в ароматическом (в орто- и пара-положения) и в этиленовом
галогениде (в ^-положение).
В отношенип галоидбензола это было объяснено раньше (стр. 45).
Аналогично объясняется ориентация присоединения протона (электро-
фильная атака) в |3-п сложение хлористого винила:
. сг
[нсн2—СН—СГ-] -----> снз-снсь
н+ + сн2=сн—СГ. а С]_
[сн2—сн2—ci:] X > сн2—сн2
б о С1
Промежуточный катион а явно энергетически беднее катиона б, потому
что хлор легко компенсирует положительный заряд, появляющийся на
соседнем с ним углероде в результате присоединения протона к крайнему
углероду. Это может быть выражено следующим образом:
Н+ ^CH^CH^Qj:
что символизирует уже не присущий покоящейся молекуле малый
мерный + М-сдвиг электронов, отраженный в уменьшенном дипо
Г-ЛЛОИДПРОИЗВОДНЫЕ БЕНЗОЛЬНОГО РЯДА
59
моменте, а полный таутомерный + 71 (или, что то же самое, электромер-
ный) сдвиг с передачей пары электронов из свиты одного атома в свиту
другого.
Совершенно аналогично дело обстоит с хлорбензолом с той только
разницей, что хлор здесь в состоянии за счет своего 4-У-эффекта хотя бы
частично скомпенсировать положительный заряд, внесенный атакующим
электрофилом, например NO2, при атаке в орто- и пара-положения
(о-комплексы виг), но не в мета-положение (о-комплекс д):
Подача электронов посредством + /'-эффекта хлора в ядро, обед-
ненное его —/-эффектом, доходит только в орто- и пара-положения, где
и происходит (хотя и замедленное) замещение водорода электрофилом:
Как указывалось на стр. 45, все четыре галоида — орто-пара-ориен-
танты, замедляющие, однако, электрофильные замещения в бензольном
ядре. 4- Л/-Эффект фтора больше, чем у других галоидов. Скорости нит-
рования фтор-, хлор- и бромбензолов относятся как 5 : 1,1 : 1 (Инголд).
Однако при ориентации в орто-положение преимущество имеет хлор. Так,
по данным Н. Н. Ворожцова мл., в случае конкурирующей ориентации
хлором и фтором место вступления NO2 определяет хлор. Например,
3,5-дифтор-2,6-дихлорбензол нитруется в орто-положение по отношению
к обоим хлорам и в мета-положение по отношению к фторам.
Естественно, что уменьшенная полярность связи С — X в галоид-
бензолах и галоидном виниле (+Л/-эффект, налагающийся на —/-эффект)
снижает обменоспособность галоида. Например, нуклеофильные атаки
ОН-, NH“ и CN- на углерод, несущий галоид, здесь гораздо менее эф-
фективны и требуют более жестких условий, чем в галоидных алкилах.
Как видно из приведенных ниже значений дипольных^моментов, у га-
лоидных винилов и галоидбензолов они действительно значительно ниже,
чем у соответствующих предельных галогенидов:
С2Н5С1 ........2,0 С2Н5Вг.........2,09
СН2=СНС1 . . . 1,44 СН2=СНВг . . . 1,41
СвН6С1 ........1,56 С6Н5Вг.........1,54
60
ГАЛОИДПРОИЗВОДНЫЕ БЕНЗОЛЬНОГО РЯДА
Углерод, несущий галоид в ароматических и винильных галогенидах,
менее положителен и менее подвержден поэтому нуклеофильным атакам.
Стоит увеличить его положительность, введя в орто- или пара-положение
оттягивающую от него электроны электропоакцепторную группу —NO,,
/° \
— SO3H, —С<^ , ^>С—О, —Сс' , как углерод делается способным
\н \он/
воспринимать нуклеофильные атаки (см. стр. 48) и, значит, галоид ста-
новится подвижным, легко вступающим в обмен с ОН-, NH2, CN-,
„О
и т, п.
Интересно, что фтор в тг-нитрофторбензоле еще значительно легче
замещается на нуклеофилы, чем хлор в тг-нитрохлорбензоле. Поэтому
нитрофторбензолы, особенно 2,4-динитрофторбензол, широко исполь-
зуются для введения 2,4-дипитрофеиилы1ого остатка, например, в кон-
цевые аминогруппы белков (см. кп. I, раздел «Аминокислоты» и ч. II,
раздел «Белки»). Напомним, что в предельных соединениях, наоборот,
хлор подвижнее фтора. Такое обращение подвижности легко объясняется
тем, что хотя фтор и подает связанному с ним углероду свои свобод-
ные электроны, по его способность компенсировать наведенный на этот
углерод нитрогрупной положительный заряд гораздо меньше, чем у
хлора.
Что касается галоидбензолов, не содержащих электронооттягивающих
трупп, то часто, если не всегда, замещение галоида на нуклеофилы про-
ходит не прямо, а через предварительное отщепление галоидоводорода
с образованием нестойкого, в высшей степени реакционноспособного
бензина, или дегидробензола (см. стр. 65), содержащего одну тройную
связь в цикле и способного присоединять воду, аммиак и другие нуклео-
фильные молекулы. Такой вывод (Робертс) был сделан на основании
того, что при действии щелочи на n-хлортолуол образуются два изомера —
п- и .и-крезолы:
Окончательно этот механизм был доказан получением анилина дейст-
вием аммиака на хлорбензол, в котором хлор был связан с радиоактивным
изотопом углерода 14С:
9
+ NH3
-------->
-NH4CI
NH3
NH2
ГАЛОИДПРОИЗВОДНЫЕ БЕНЗОЛЬНОГО РЯДА
61
В 50% анилина аминогруппа оказалась связанной с радиоактивным
углеродом, а в 50% — с соседним с ним углеродным атомом. Такое не-
прямое замещение называется кинезамещением.
Совершенно иначе обстоит дело с а-галоидами боковой цепи. В то
время как более удаленные галоиды боковой цепи обладают нормальной
подвижностью галоида в предельных соединениях, а-галоид более по-
движен. Хлористый бензил — слезоточивое соединение; раздражение
слизистых оболочек носа и глаз — следствие реакционной способности
соединения. Еще большим слезоточивым действием обладает иодистый
бензил — «полицейский слезоточивый газ».
Галоидные бензилы гидролизуются и вообще вступают в обмен с нуклео-
филами в спиртовом растворе по реакции первого порядка, что означает,
что они сначала электролитически диссоциируют (медленная стадия
реакции, определяющая первый порядок всего процесса), затем бензил-
катион очень быстро связывается с нуклеофилом. Поскольку концентрация
нуклеофила никак не влияет на скорость электролитической диссоциации
галоидного бензила, скорость реакции пропорциональна только концен-
трации последнего (реакция первого порядка):
С6Н5СН2С1 —> С6Н5СН2 + С1-
СвН5СН2 + Н2О ---> свн5сн2он + н+
Таково течение реакции в хорошо сольватирующих средах с высокой
диэлектрической проницаемостью.
Легкость электролитической диссоциации галоидного бензила (срав-
нительно с галоидными алкилами) объясняется большей стабильностью
(более низким энергетическим уровнем) бензильного катиона сравнительно
с алкильными катионами. Менее понятно, что в ряде растворителей
хлористый бензил, гидролизуясь по механизму SN2, реагирует быстрее,
чем галоидные алкилы.
Хлористый бензил широко используется для бензилирования — введе-
ния бензильной группы.
Как всегда, скопление двух и более атомов хлора у одного углерода
делает соединение более стабильным и менее подверженным нуклеофиль-
ным атакам. Все же хлористый бензилиден при нагревании гидроли-
зуется в бензальдегид
С6Н5СНС12 + Н2О -> С6Н5СНО + 2НС1
а бензотрихлорид при высокой температуре в присутствии серной кислоты
гидролизуется водой (1 моль) в хлористый бензоил — хлорангидрпд бен-
зойной кислоты:
СвН5СС13 + Н2О --> C6H5C-f + 2НС1
\С1
Это промышленный процесс.
При избытке воды бензотрихлорид гидролизуется в бензойную кислоту:
/О
СвН5СС13 + 2Н2О -► С6Н5С< + ЗНС1
Ч)Н
ГАЛОИДПРОИЗВОДНЫЕ БЕНЗОЛЬНОГО РЯДА
62
Действием трехфтористой сурьмы бензотрихлорид легко превращается
в бензотрифторид:
СвПБСС1.з + SbF3 -> CeH5CF3 + SbCl3
Это соединение исключительно устойчиво, совершенно не гидроли-
зуется и не склонно вступать в реакции обмена. Группы СС13, и особенно
CFS, — сильные мета-ориентанты, пассивирующие бензольное ядро к
электрофильным атакам.
СОЕДИНЕНИЯ С МНОГОВАЛЕНТНЫМИ ГАЛОИДАМИ
Раньше всех (90-годы XIX столетия) стали известны органические соеди-
нения многовалентного иода, открытые В. Мейером. Разработанный им
синтез органических соединений трех- и пятивалентного иода таков:
С1
Свн61 +С12 ---> CeH6I <f -2Ag‘o+H‘°-> CeH6I=0
ХС1
финилиодидхлорид иодозобензол
При нагревании иодозобепзола до 100° С он диспропорционируется,
давая иодбензол и иодобензол:
перегонка q
свн61=о -°-паром свН61 + свн6-к^
иодбензол иодобензол ®
Иодобензол — своеобразное фенилирующее средство, обменивающее
группу С8Н“ на О2-. чем и объясняется течение столь необычной реакции:
zO СвНБч +
СвНв1 = О + СвНБ1^ -I- AgOH ---► >1 ОН“-|-AgIO3
X) CeHZ
гидроокись
дифенилиодония
СБНБх свнБч
>1* ОН- + НС1 ----► Л-С1 + Н2О
СвНБ СБНБ'
дифенилиодоний-
хлорид
Однако путь синтеза этих веществ по В. Мейеру относительно сложен.
Может быть наиболее нагляден путь синтеза некоторых типов орга-
нических соединений многовалентного иода, осуществленный одним из
авторов этой книги и Р. X. Фрейдлиной. При действии на треххлористый
иод дифепилртути хлор замещается па фепил, и в зависимости от взятых
соотношений реагентов можно осуществить две реакции:
1С13+ (CeII5)2Hg--> CeH6ICl2+ CeH5HgCl
фспилиодид-
хлорид
1С13 + 2(CeII5)2Hg ->• (CeH5)2ICl + 2CeH5HgCl
дифенилиодо-
нийхлорид
СОЕДИНЕНИЯ С МНОГОВАЛЕНТНЫМИ ГАЛОИДАМИ 63
Вторая реакция, возможно, проходит в две стадии: образующийся
пОрПервой реакции фенилиодидхлорид затем претерпевает реакцию Виль-
CeH5ICl2 + Hg(CeH5)2 —> (CeH5)2ICl + CeH5HgCl
Фенилиодидхлорид СвН51С12, который можно получить и прямым дейст-
вием хлора на иодбензол, растворенный, например, в хлороформе, —
желтое кристаллическое вещество, постепенно разлагающееся на хло-
ристый водород и n-иодхлорбензол. При действии на СвН51С12 щелочью
получается иодозобензол:
CeH5ICl2 + 2NaOH —> CeHsI=O + 2NaCl + H2O
Иодозобензол — слабое основание и с некоторыми кислотами образует
солеобразные производные:
О
II
/О /ОС—СНз
СвН51 = О -г 2СНзС<^ --> СвН51/ +Н2О
х ОН ХОС-СНз
И иодозобензол и иодбензол при действии кислоты Каро окисляются
в иодобензол:
НО—О. /О /О
СвН51 4- 2 ZS< -------> С6Н51^ -|- 2H2SO4
HOZ Ч) X)
иодобензол
Иодосоединения и, в частности, иодобензол — кристаллические бес-
цветные взрывчатые вещества. И иодобензол и иодозобензол восстана-
вливаются (например, H2SO3 или HI) в иодбензол.
Дифенилиодонийхлорид (С6Н5)21С1 образует растворимые в воде крис-
таллы; в растворе он электролитически диссоциирует на катион дифенил-
иодония (С6Н5)21+ и анион С1~, который легко обменять на любой другой
анион. При действии AgOH получается раствор гидроокиси дифенилиодо-
ния (С6Н5)21 ОН-— основания, окрашивающего лакмус в синий цвет.
По своим аналитическим характеристикам катион дифенилиодопия напоми-
нает катион одновалентного таллия (образует нерастворимые иодид,
хромат, сульфид, растворимую гидроокись). При нагревании до темпе-
ратуры плавления галоидные соли дифенилиодония распадаются:
(СвН5)21Х —>• СвН51 + СвН6Х
Дифенилиодонийиодид (СвН5)2П — димер иодбензола, превращаю-
щийся в него при плавлении. Распад дифенилиодониевых солей в водном
ГАЛОИДПРОИЗВОДНЫЕ БЕНЗОЛЬНОГО РЯДА
64
растворе идет гетеролитически, а в полонизирующих средах гомолити-
чески, что ясно из следующих типичных реакций:
(Cells); I I + Но-->- CcHjHgl + С6Н51
через стадию
Се н 5 -11L f •
с^у.у.-
> с6н51 + С6Н5* + I-
(СеН5),1С1 + НО ---->С6Н5ОН + С0Н51 + сг
через стадию
cgh5:
:ci:
>- CGH51 + С6н5+ + СГ
1
Проще всего получать соли иодония прямым действием раствора йод-
новатой кислоты в серной кислоте на ароматический углеводород (Мэс-
сон):
/О СвИ5х + _
2СвН, 4- НО- 1<г 4- H2SO4 ->- >1 SO4H 4- 2Н2О +0
^0 CeH5Z
Мэссон открыл, а Берипджер подробнее исследовал новый тип диарил-
иодозиловых солей, получаемых конденсацией иодосоединений с бензолом
или его производными в щелочной среде с последующим подкислением:
CeHsIOa + Celle —> Cells—I—СвНь СвН5-1-СвНв
II II
О 01Г О СГ
В 1950—1960 гг. одним из авторов совместно с Т. П. Толстой полу-
чены и изучены хлорный и бромный аналоги дифепнлподопиевых солей
(CeHB)2Cl+ X- и (СвН5)2Вг+Х“. Способ их получения через диазониевые
соединения см. стр. 111. Смысл его состоит в том, что генерированный из
фенилдиазопиевой соли фепил-катион СВН* атакует свободную пару
электронов хлор- или бромбензола, и образуется катион галогенония —
дифенилхлорония или дифенилбромопня:
с0н5ч +
CfjITg—Cl: СвП* —>
Cells
Если X“ —прочный комплексный анион, такой, как ВЕ^пли Hgl,,
соли обоих Дифенилгалогепониов вполне устойчивы и разлагаются лишь —
(СвНь)2Вг.ВЕ., при 120° С и (CeIL,)„CI • В Г4 ври 110° С. Галоидные соли
гораздо менее прочны; так, (С0115).,ВН разлагается при 81° С, а (СвИ5)3СП -
при об С.
Соли дифенилбромопня и дифенилхлорония по химическим свойствам
похожи на соли дифенилиодопия, по еще легче вступают в реакции
ДЕГИДРОБЕНЗОЛ (ЦИКЛОГЕКС АДИЕНИН, «БЕНЗИН») 65
с нуклеофилами: уже на холоду реагируют с водным раствором щелочи по
схеме:
(CeH5)2Cl X- + ОН- ► CeH5OH + C0I-I5CI + X-
Они способны к гомолитическому распаду и реагируют с металлами
подобно солям дифенилиодония:
(CeH5)2Cl—I + Hg —> CeH5HgI + С0Н5С1
В реакциях синтеза солей дифенилбромония п дифенилхлорония сво-
бодные пары электронов галоида исходного галоидбензола обусловливают
проявление галоидом нуклеофильных свойств. Это редкие реакции, где
ковалентно связанный галоид реагирует подобно трехвалентному азоту
и двухвалентному кислороду, в которых, однако, свободные (неподелен-
ные) пары электронов гораздо активнее.
Соли дифенилбромония и дифенилхлорония чувствительны к нуклео-
фильным реагентам и ими разлагаются, но к действию электрофильных
реагентов они устойчивы. Например, борфторид дифенилбромония можно
в жестких условиях пронитровать нитрующей смесью и получить бор-
фторид ди-(.м-нитрофенил)-бромония. Таким образом, положительно заря-
женный галоидный атом бромония — резко выраженный мета-ориентант.
В 1958 г. Остерлинг и сотр. описали перхлорильные производные бен-
//°
зола и его производных, содержащие группу —С1=О. Перхлорил-
^0
бензол (жидкость с т. пл. —3° С, т. кип. 232° С, = 1,185) получается
при пропускании газообразного фторангидрида хлорной кислоты FC103
в теплый бензол в присутствии хлористого алюминия:
О
С6Н6 + FC103 СвН6-С1=0 + HF
II
О
Он выдерживает нитрование нитрующей смесью (в мета-положение)
и сульфирование при температуре до 280° С, но при кипячении со ще-
лочью постепенно гидролизуется до фенола. Получены перхлорильные
производные и для гомологов бензола.
Перхлорильные соединения взрывчаты.
ДЕГИДРОБЕНЗОЛ (ЦИКЛОГЕКСАДИЕНИН, «БЕНЗИН»)
Виттиг открыл замечательную реакцию образования дегидробензола при
действии амальгамы лития на о-фторбромбензол:
5 Заказ 6 72
66
СУЛЬФОКИСЛОТЫ И ДРУГИЕ СЕРНИСТЫЕ СОЕДИНЕНИЯ
Наличие в продуктах реакции дегидробензола констатируют по его
реакциям, так как вещество неустойчиво и по было выделено в свободном
состоянии. Если нет подходящего реагента, оно димеризуется в дифепилен
и тримеризуется в трифенилен:
В присутствии диена, выдерживающего условия реакции (например,
не полимеризующегося), проходит диеновый синтез, причем дегидробеп-
аол играет роль диенофила (стр. 288). Подходящим диеном является
фурап:
н он
н
СУЛЬФОКИСЛОТЫ И ДРУГИЕ АРОМАТИЧЕСКИЕ
СЕРНИСТЫЕ СОЕДИНЕНИЯ
Реакция сульфирования в ряду бензола
Бензол медленно сульфируется при приливании его при встряхивании
(уже при комнатной температуре) в 100%-ную серную кислоту (так
называемый моногидрат) и быстрее — при действии олеума (серная
кислота, содержащая растворенный серный ангидрид):
О
II
Cel-Ie + H2SO4 -> Свн5—s—ОН +Н2О
II
О
бензол сульфо-
кислота
Выделяющаяся вода разбавляет серную кислоту, и скорость суль-
фирования быстро падает, поэтому необходимо брать избыток серной
кислоты. Если порядок введения реагентов в реакцию обратный, то в из-
оытке находится углеводород, что приводит к получению дифенилсульфона
в качестве побочного продукта:
О
2СвНв + H2SO4 --► CeH5-S—СвНБ + 2Н2О
II
О
дифенилсульфон
С> ЛЬФОЬИСЛОТЫ И ДРУГИЕ АРОМАТИЧЕСКИЕ СЕРНИСТЫЕ СОЕДИНЕНИЯ
Бензолсульфокислоту отделяют от серной кислоты одним из двух спо-
собов.
1. Выливают реакционный раствор в концентрированный раствор
поваренной соли. При этом оседает бензолсульфокислый натрий, который,
как и большинство натриевых солей сульфокислот, трудно растворим в воде
и еще хуже в растворах поваренной соли.
2. Осаждают серную кислоту баритовой водой. Тогда бензолсульфо-
кислый барий остается в растворе и свободную сульфокислоту можно
получить, добавляя точно эквивалентное количество серной кислоты
(до прекращения выпадения BaSO4) и выпаривая досуха фильтрат.
С кальциевыми солями отделение менее полное вследствие некоторой
растворимости гипса. Соли сульфокислот со щелочноземельными метал-
лами все растворимы в воде.
Сульфокислоты — сильные кислоты, порядка серной кислоты.
Толуол сульфируется легче, чем бензол, образуя смесь о- и п-толуол-
сульфокислот. В целом реакция сульфирования — типичная реакция
электрофильного замещения и подчиняется правильностям, изложенным
на стр. 38. Поскольку сама сульфогруппа мета-ориентант, вторая суль-
фогруппа вступает в бензол в мета-положение к первой, а полученная
так (действием олеума) лг-бензолдисульфокислота уже настолько пассиви-
рована наличием двух сильных мета-ориентантов, что третью сульфо-
группу ввести сульфированием в бензол не удается.
Доказательством прямой связи серы с углеродом (а не через кислород,
как в эфирах серной кислоты) в сульфокислотах служит их генетическая
связь с меркаптанами. При восстановлении самыми сильными восстано-
вителями сульфокислоты превращаются в меркаптаны, а при окислении
меркаптанов азотной кислотой образуются сульфокислоты:
CeH5-SO3H CeH5-SH
CeH5SO3H CeH5SO2Cl Хткие11^ CeH5-SH
условия)
CeH6-SH C6H5-SO3H
Функциональные производные сульфокислот
Сульфокислоты ароматического ряда, как и вообще сульфокислоты, обра-
зуют следующие функциональные производные, получаемые приведен-
ными ниже реакциями.
Хлорангидриды сульфокислот можно синтезировать, действуя пяти-
хлористым фосфором на сульфокислоту или на ее соль
° °
CeH5_S—ONa + PCI5 > С6Н6—S—Cl + POCI3+ NaCl
11 О
О и
5*
СУЛЬФОКИСЛОТЫ И ДРУГИЕ СЕРНИСТЫЕ СОЕДИНЕНИЯ
68
или сульфируя углеводород хлорсульфоповой кислотой, взятой в из-
бытке:
о
II
СвНв + CISO3II —> CeH5-S—Cl + Н2О
II
О
Сложные эфиры сульфокислот получают реакцией этерификации
О О
II II
Ar—S—ОН + ROH ” Ar—S—OR + Н2О
II II
О О
или действием хлорангидрида сульфокислоты на спирты:
О О
II II
Ar—S—Cl + ROII > Ar—S-OR + НС1
II II
о lo
Сложные эфиры сульфокислот, как и сложные эфиры серной кислоты,—
алкилирующие агенты, находящие практическое применение:
О О
II II
Ar-S-ОСНз + ROH -----> Ar—S—ОН + ROCH3
II II
О о
Здесь ROH может быть спиртом, фенолом, карбоновой кислотой;
можно алкилировать и амины.
Амиды сульфокислот получают реакцией аммиака (а замещенные ами-
ды — аминов) с хлорангидридом сульфокислоты:
О О
II II
Ar-S-Cl + 2NH3-----> Аг-S—NHo + NH4CI
II II
О О
Амиды сульфокислот — нейтральные вещества.
При хлорировании амидов сульфокислот один или оба водорода ами-
догруппы замещаются па хлор и получаются так называемые хлор-
амины, гидролизующиеся с образованием НОС1 и исходного сульф-
амида и обладающие сильным дезинфицирующим действием.
Ai
А
।
o==s=o
I
NHC1
хлорамин Б
СНз СНз
I I ।
NC12 NHC1 NClo
дихлорамин Б хлорамин Т дихлорамин Т
СУЛЬФОКИСЛОТЫ И ДРУГИЕ АРОМАТИЧЕСКИЕ СЕРНИСТЫЕ СОЕДИНЕНИЯ 69
Реакции сульфокислот с замещением сульфогруппы
Из таких реакций важнейшей является реакция «щелочного плавления»
(Кекуле), при помощи которой до сих пор еще получают синтетический
фенол:
О
CeH5—S—ONa + 2NaOH > C5H5ONa + Na2SO3+H2O
О
Здесь примечательно восстановление серы до четырехвалентной за
счет окисления атома углерода, получившего в результате реакции
связь с кислородом.
Аналогично проходит не имеющая существенного препаративного
значения реакция с солью синильной кислоты (М. Г. Кучеров):
О
II
CeH5—S—ONa + NaCN —> CeH5—C=N + Na2SO3
II
О
Другие сернистые функции ароматического ряда
Восстановление хлорангидридов сульфокислот цинком с кислотой
приводит к не слишком устойчивым слабым сулъфиновым кислотам
О
II /О
Ar—S—Cl Zn+HCl^ Ar—S<^
II он
О
которые легко окислить в сульфокислоты или восстановить в меркап-
таны.
Хлорангидриды кислот реагируют с ароматическими углеводородами
в присутствии хлористого алюминия, образуя сульфоны:
О [°
II II
Ar—S—Cl + CeHe AlCla h Ar—S—C6H5 + HC1
Сульфоны очень прочные соединения. Сульфоннаягруппа - сильный
мета-ориеитант, пассивирующий бензольные циклы к электрофильным
замещениям Сульфоны очень трудно восстанавливаются в сулъфидн.
замещениям. * „„„тяется в две стадии: рассчитанным количеством
Дифенилсудьфид окисляется .д. * ф*ксид (C,H,),S^O, а из-
окислителя (например, г /г и \ чп
«ытком сильного окислителя в дифенилсульфо,. (С,Н,),ЬО2.
НИТРОСОЕДИНЕНИЯ РЯДА БЕНЗОЛА
70
Меркаптаны слабыми окислителями превращаются в дисульфиды,
2CeHbSH — - СвНь—S—S—СвН5 + 2Н1
а при действии сильных окислителей дают сульфокислоты.
Как и жирные меркаптаны, ArSH образуют характерные прочные
бесцветные нерастворимые меркаптиды с Hg2+, Pb2+ и катионами других
тяжелых металлов.
НИТРОСОЕДПНЕНИЯ РЯДА БЕНЗОЛА
О строении нитрогруппы см. кн. I, стр. 217. Обычным нитрующим сред-
ством служит нитрующая смесь, т. е. раствор концентрированной азот-
ной кислоты (60—100%-ной) в концентрированной серной кислоте. В та-
ком растворе азотная кислота ведет себя как основание:
HONO2 + H2SO4 NO2 SO3H--I-H2O
Нитроний-катион и является электрофильным нитрующим агентом.
Сама азотная кислота нитрует гораздо более медленно, а так как она
сильный окислитель, то сказываются или* преобладают окислительные
реакции.
Реакции последовательного нитрования бензола изображаются так:
На второй стадии нитрования получаются также небольшие коли-
чества о-динитробензола.
Тринитробензол (идеальное бризантное взрывчатое вещество) . обра-
зуется лишь в жестких условиях и с плохим выходом. Несмотря на уси-
лия химиков многих стран, во время второй мировой войны осуществить
эту реакцию с приемлемым выходом не удалось.
НИТРОСОЕДИНЕНИЯ РЯДА БЕНЗОЛА
71
Толуол, как все алкилбензолы, на всех стадиях
чем бензол. Вот стадии его нитрования:
нитруется легче,
Таким путем получается 2,4,6-трш1итротолуол (тол, или тротил} —
бризантное взрывчатое вещество, служащее для начинки снарядов. В от-
личие от 1,3,5-тринитробензола тротил получается с хорошим выходом.
Нитрование .м-тпретп-бутилтолуола до тринптропродукта приводит к образованию
«искусственного мускуса», имеющего запах, подобный запаху естественного мускуса
(кн. 1, стр. 577). Формулы ряда искусственных мускусов приведены ниже:
СНз
O2N^z^/ZNO2
^|/\C(CH3)3
no2
толуольный
мускус
СНз
°2N\J\/NO2
1 ¥
СНз©/ у
С(СН3)з
амбровый
мускус
СНз
°2N\A/C0CH3
I li
(СНз)3с/^/\сН3 .
N02
кетонный
мускус
Они находят применение в парфюмерии в качестве душистых веществ и фиксато-
ров запаха.
При действии па толуол разбавленной азотной кислоты при высокой
температуре (100—150° С) электрофильное нитрование в ядро подавлено
разбавлением азотной кислоты, и происходит гомолитическое нитрование
толуола по цепному механизму в метильную группу (М. И. Коновалов):
СвН.СНз+• NO2 ----> CeH6CH2. + HNO2 инициирование реакции
6 (одна из возможностей)
CeH5CH2. + HONO2 —> CeH5ClI2NO2 + HO. j
С6Н5СНз+ -О11 —* СвНБСН2.+Н2О )
в a-положение в этих условиях нитруются и гомологи бензола
с более длинной боковой цепью.
Тот же а-нитротолуол можно получить, действуя хлористым бензилом
.на нитрит натрия (см. также кн. I, стр. 216).
CelI5CIl2Cl + NaNOa —► CeH8CH2NO2 + NaCI
ПИТРОСОЕДИНЕНИЯ РЯДА ВЕНЗОЛЛ
72
Таблица 62. Ароматические нитросоединения
Формула Название Температура плавления, °с
CeH5NO2 Нитробензол 5,7
C.H4(NO2)> Динитробензол
о-, или 1,2- 118
м-, или 1,3- 89,57
п-, или 1,4- 173,5
CeH,(NO2)3 Тринитробензол
1,3,5- 61,0
1,2,3- 127,5
1,2,4- 61,0
CH3CeHiNO2 Нитротолуол
о-, пли 1,2- —4,1 (3-форма) —10,6 («-форма)
м-, или 1,3- +15,5
п-, или 1,4- 51,3
СНзСвН3(ПО2)2 2,4-Динитротолуол 70,5
CH3CeH2(NO2)3 2,4,6-Тринитротолуол 80,7
ClCeH4NO2 Нитрохлорбепзол
о-, или 1,2- 32,5
м-, или 1,3- 44,4
и-, или 1,4- 83,5
BrCeH4NO2 Нитробромбензол
о-, или 1,2- 42,0
м-, или 1,3- 56,0
п-, или 1,4- 127,0
ICeH4NO2 Нитроиодбензол
о-. или 1,2- 49,4
м-, или 1,3- 36,0
п-, или 1,4- 171,5
Нитротолуол — одно из немногих нитросоединений, для которого
можно изолировать непрочную сильнокислую ai/u-форму (см. кн. I,
стр. 219), таутомерную с сс-нитротолуолом:
о-
CeH5CH2NO2 + NaOH >
CeH5-CH=N
О’J
свН5—СН=2у/0'
аЧи-форМа
О ~"| jjCl +
Na* —--------Cells—CH=NZ
-Nad NOH
♦ /О-
C9H5-CHt-N^
НИТРОСОЕДИНЕНИЯ РЯДА БЕНЗОЛА
Температура кипения, °с Плотность d4° Показатель прелом- ОП леяия nfi Дипольный момент p- в бензоле при 20° С, D
210,9 1,2030 1,5529 3,97
319 (при 773 лыи рт. ст.) 1,565 (при 17° С) — 5,98
302,8 (при 770 '«.и рт. ст.) 1,571 (dj) — 3,78
299 1,625 0,00
(При 777 мм рт. ст.)
Разл. 1,688 0,8
— — — —-
— 1,73 (d}«)
222,3 1,1630 1,5474 3,69; 4,22
231,0 1,1571 1,5475 4,17
238,0 1,1286 1,5346 4,44
(при 62,5° C)
300 1,521 1,662 4,33
(разл.) (при 15° G) (при 25° С)
240 1,654 1,15
(взрыв.) (при 25° С)
245,7 1,368 (d242) — 4,33
235,5 1,534 3,40
242 1,520 (d|8) 2,57
261 1,6245 (c?so) 4,20
256,5 1,7036 1,5979 3,41
256,0 1,934 (d22) — 2,65
290,0 1,810 (dje) 3,92 (при 22° С)
280,0 1,804 (dje) 3,43 (при 22° С)
288,1 1,809 (dp’») 3,04 (при 22° С)
Мононитропродукты, выпускаемые промышленностью в огромных
масштабах, применяются главным образом для дальнейшего
восстановления в амины (см. раздел «Ароматические амины)». Эта реакция
(Н Н Зинин) служит доказательством того, что азот нитро-
группы соединен с углеродом прямо, а не через посредство кислорода.
Физические свойства ряда ароматических нитросоединении приведены
в табл. 62.
74
ФУНКЦИИ, ОБРАЗУЕМЫЕ ВОССТАНОВЛЕНИЕМ НИТРОСОЕДИНЕНИЙ
ФУНКЦИИ, ОБРАЗУЕМЫЕ НЕПОЛНЫМ
ВОССТАНОВЛЕНИЕМ НИТРОСОЕДИНЕНИЙ
Электрохимическое восстановление нитросоединений в зависимости от
среды (кислой или щелочной) и плотности тока приводит к серии продуктов,
содержащих новые для нас функциональные группы, включающие азот:
кислая среда CellsNO? щелочная среда
нитробензол
СаН5—N=0
питрозобспзол
СаН5-N =N-СаН5
о-
азокснбензол
СаНб—NH-0H
фенилгидрокенламип
CaH5-N=N—СаН5
азобензол
СвНБ—NH—NH-CaH5
гидразобензол
> CaH6NH2
анилин
Каждый из этих продуктов может быть получен также восстановлением
нитросоединения посредством подходящего реагента. Конечным продуктом
серии восстановлений и в кислой и в щелочной среде является пер-
вичный амин — анилин.
Таблица 63. Продукты неполного восстановления нитробензола
Формула Название Темпера- тура плавле- ния, °C Температура кипения, °C Плотность dr
СаН5—N=0 Нитрозобепзол 68 59 (при 18 л«л1 pm. cm.) 293 Разл. —
CaH5-N=N-CaH5 СаНь—N=N-CaH6 X 0 CaH5-NH-NH-CaH5 СаНв—NHOH Азобензол Азоксибепзол 68 36 1,203 1,246
Гидразобензол, или дифе- ннлгидразип Фенилгидроксиламин 126; 131 82 Разд. 1,158 (<4‘)
ФУНКЦИИ, ОБРАЗУЕМЫЕ ВОССТАНОВЛЕНИЕМ НИТРОСОЕДИНЕНИЙ
75
В табл. 63 приведены физические свойства продуктов неполного вос-
становления нитробензола.
Нитрозобензол
Восстановление нитробензола в кислой среде трудно задержать на стадии
образования нитрозобензола. Последний легче получить окислением сле-
дующего продукта восстановления нитробензола — фенилгидроксиламина
(Бамбергер). Это и есть обычный путь синтеза нитрозобензола:
ЗСвН5—NHOH + HsCr2O74-6H+ —> ЗСвН5—N==O-|-7H2O + 2Cr3+
В последние годы опубликован и прямой метод восстановления нитро-
соединений в нитрозосоедипения пентакарбонилом железа Fe(GO)5.
Ароматические нитрозосоединения — бесцветные кристаллические
вещества, димерные в твердом состоянии. В растворе, расплаве и в форме
пара они диссоциируют на две молекулы мономерного нитрозосоединения,
имеющего изумрудно-зелено-голубую окраску, причем устанавливается
равновесие:
2С6Н5-NO [CeH5NO]a
Димер нитрозосоединения имеет строение, аналогичное разбираемым
далее азоксисоединениям *:
CeH5-N=N-CeH5
X X
о о
димер нитровобензола
CeH5-N=N-CeH5
X
о
азоксибензол
Возможно, этим объясняется легкое восстановление нитрозосоедине-
ний в азокси- и далее в азосоединения.
Доводом в пользу такой структуры димера является открытое недавно
существование геометрической изомерии димеров нитрозосоединений
в жирном ряду. Что касается ароматических нитрозосоединеппй, то для
них также существуют цис- и транс-изомеры (или, что то же, син- и анти-
изомеры). Однако сам димерный нитрозобензол известен только
* Напоминаем, что знаки
—N=
X
о
и — N=
о-
эквпваленты и обозначают семиполярную связь — ковалентную пару электронов,
связывающую атомы, несущие полный ионный положительный и отрицательный заряд.
76
ФУНКЦИИ, ОБРАЗУЕМЫЕ ВОССТАНОВЛЕНИЕМ НИТРОСОЕДИНЕНИЙ
в цис-форме, как показывает аналогия его ИК- и раман-спектров с за-
ведомой t/uc-формоп о,о'-нитрозобифенила (он бесцветен, но мономерен)
Для обоих веществ получаются две полосы поглощения между 1389
и 1397 cat-1 и полоса при 1409 cat-1; для ароматических транс-димеров
наблюдается единственная полоса поглощения между 1253 и 1299 cat-1.
Другие пути образования ароматических нитрозосоединений: действие
N2O3 или C1NO на металлоорганические соединения, например на ди-
фенилртуть; бензольное ядро, содержащее сильные ориентанты первого
рода [—ОН, —N(CH3)2], индифферентные к азотистой кислоте, может
быть прямо пронитрозировано в водном растворе действием азотистой
кислоты:
|\О/>—он + но—N=O --> O=N-^O)>—он
(Q^-N(CH3)2 + но-N = O -->- O=N-<g>-N(CH3)2
Нитрозирование проходит на холоду в кислом водном растворе и идет
практически нацело в пара-положение.
Нитрозофенол — особый случай нитрозосоединения. В его растворах
наблюдается таутомерия нитрозофенольной и хиноноксимной форм (ср.
с таутомерией нитрофенолов):
O=N—\О/~ОН ч- >1 Н —О —N=/ 4=0
Положение таутомерного равновесия зависит от применяемого раство-
рителя.
Нитрозодиметиланилин также отличается от обычных нитрозосоеди-
нений. Вследствие согласованного взаимодействия электронодонорнои
диметил аминогруппы и электроноакцепторной нитрозогруппы его струк-
тура заметно смещена в сторону хиноидной формы, и он, как образно го-
ворится, представляет собой резонансный гибрид:
O=N—Ч—N(CH3)2 < > "О—N=<^_>=N(CH3)2
Это обнаруживается по увеличенному дипольному моменту, батоном
ному сдвигу светопоглощения и неспособности образовать димер.
ФУНКЦИИ, ОБРАЗУЕМЫЕ ВОССТАНОВЛЕНИЕМ НИТРОСОЕДИНЕНИЙ
77
взаимодействие двух нитрозогрупп наблюдается в о-динитрозобензоле,
который на деле является гетероциклом — бензфуразаном
Нитрозосоединения способны к многочисленным реакциям конден-
сации. Они реагируют с выделением воды с соединениями, содержащими
группировку H2N—X (будут разобраны далее в этом разделе), и с соеди-
нениями, имеющими подвижную метиленовую группу. Например:
О R
О R
CeH5-N=O + Н2С
O^R
С6Н6-N=C
+н20
СдНб—N — О -|-
анил трикетона
I II C=NC6H5 +н2о
\/\ Z
NH
анил изатина *
Вторая группа реакций — присоединения к нитрозогруппе с обра-
зованием четырехчленного цикла:
(СвН6)2С-СН2
CeH5-N=O + (CeH5)2C=CH2 —> | |
CeH5—N—О
СвН5—N=O + R—N=CH2
CeH5—N—О
I I
H2C-N—R
Наконец, с диенами идет диеновый синтез, в котором нитрозогруппа
выступает в роли диенофила (Ю. А. Арбузов):
СН2
сн
I
сн
^сн2
/СвН5
N
+ 11
О
СН2
НС ^N—СвНб
II I
НС о
сн2
* Анилами (стр. 145) называются продукты взаимодействия ароматических
аминов с оксосоединениями, например:
О.
CeHsNHjj-J- \С—С6Н5 —► СвНб—N—СН—СвНб + Н2О
нх
ФУНКЦИИ, ОБРАЗУЕМЫЕ ВОССТАНОВЛЕНИЕМ НИТРОСОЕДИНЕНИЙ
78
Фенилгидроксиламин
Фенплгпдрокспламин получают восстановлением нитробензола (в виде
взвеси в водном растворе хлористого аммония) посредством цинковой
пыли. Из водного раствора фенилгидроксиламин высаливают поваренной
солью. Это — бесцветное кристаллическое вещество с запахом, раздра-
жающим слизистые оболочки носа. Фенилгидроксиламин очень легко
окисляется и темнеет на воздухе, а при более энергичном окислении пре-
вращается в нитробензол. С нитрозобензолом в щелочной среде он обра-
зует азоксибензол
С6Н5-NHOH + O=N—С6Н5 ► CeH5-N=N-C6H5 + H2O
О
чем п объясняется появление азоксисоединения при восстановлении нит-
робензола в щелочной среде.
Фенплгпдроксплампн — очень слабое основание. Замечательным
(и используемым) свойством фенилгидроксиламина является перегруппи-
ровка (изомеризация) его при нагревании с кислотой в более сильное
основание — д-аминофенол:
—•* НО—/ NHj
п-Ампнофенол — известный фотопроявитель. Проявителями служат и его произ-
водные:
NH—СНз
NH—СН2СООН
NH2
ОН
метод
ОН
глицпн
^^NHj
ОН
амидол
Промышленный процесс получения n-аминофенола осуществляют
в одной ванне, восстанавливая нитробензол и перегруппировывая фенил-
гид р о к сил а мин.
Азоксибензол
Азоксибензол, полученный впервые Н. Н. Зининым, можно синтезиро-
вать восстановлением нитробензола метилатом натрия:
4C6H5-NO2 + 3CH3ONa —> 2C6H6-N=N-CeH5 + 3HCOONa + 3H2O
X
О
В свою очередь азоксибензол восстанавливается (посредством Zn, Fe)
в азобензол. Несимметричное положение кислорода в азоксибензоле сле-
дует из существования изомерии типа (Анжели):
Аг—N=N—Аг' п Аг—N=N—Аг'
ФУНКЦИИ, ОБРАЗУЕМЫЕ ВОССТАНОВЛЕНИЕМ НИТРОСОЕДИНЕНИЙ
79
При окислении Ar—N=N —Аг' получаются оба изомера. То же про-
исходит и при реакции:
Аг—NHOH + O==N—Аг’ —> [аг—NH—N—Аг'] Аг—N—N—Аг'] ТндГ’
НО о- но он
—> Аг—N=N—Аг' + Аг—N=N—Аг'
Нагревание азоксисоединений в кислой среде ведет к их перегруп-
пировке в n-оксиазосоединения (перегруппировка Валлаха):
н+
С6Н5—N=N-C6H6-----* НО—С6Н4—N=N—С6Н5
X
По М. М. Шемякину, азоксибензол передает кислород в пара-поло-
жения обоих фенилов, что доказывается опытом с азоксибензолом, со-
держащим один меченый атом азота (изотоп 15N). О том, что промежу-
точно образуется симметрично построенная молекула, свидетельствует
восстановление в смесь анилина и n-аминофенола с эквивалентным распре-
делением между ними 15N.
Азобензол
Азобензол получается восстановлением нитробензола станнитом натрия
и многими другими восстановителями в щелочной среде:
2CeH5—NO2 + 4Sn(ONa)2
СвН5—N=N—CeH5 + 4Na2SnO3
Его можно получить и реакцией нитрозобензола с анилином:
С6Н5-N=O + H2N-С6Н5 —> CeH5-N=N-C6H5-f-H2O
Все эти реакции приводят к азобензолу с т. пл. 68° С. При облучении
ультрафиолетовым светом этот азобензол превращается в свой малоустой-
чивый геометрический изомер, энергетически более богатый (примерно
на 11 ккал/молъ) и имеющий т. пл. 71° С. Это ^ис-тпранс-изомерия, которая
Для случаев геометрической изомерии с участием азота обычно обозна-
чается как син-анти-изомерия:
обычный транс-, или
анти-азобензол
(т. пл. 68° С)
цис-, или син-
азобензол
(т. пл. 7 1° С)
ФУНКЦИИ, ОБРАЗУЕМЫЕ ВОССТАНОВЛЕНИЕМ НИТРОСОЕДИНЕНИЙ
80
син-Азобензол — неплоское соединение, так как при плоском располо-
жении бензольные остатки наложились бы друг на друга. Вопрос о конфи-
гурации обоих соединений был решен рентгеноструктурным исследованием.
С геометрической изомерией, обусловленной расположением замести-
телей вокруг атомов азота при двойной связи, мы уже встречались при
рассмотрении нитрозосоединений. В дальнейшем мы еще встретимся с этим
явлением, особенно хорошо изученным на примере альдоксимов и кет-
оксимов. Дело в том, что двойная связь N=N (а также связь G=N
в оксимах) не допускает, как и С=С связь, свободного вращения, а угол
N отличен от 180° (он ближе к 120°). Последнее обстоятельство свя-
с/ 4n
зано с тем, что неподеленная (свободная) р-пара электронов азота тре-
бует места.
Азобензол — основание, еще более слабое, чем фенилгидроксиламин. Он
растворим в концентрированной серной кислоте, но высаживается из нее
водой вследствие гидролиза образовавшейся соли. Такая соль в среде
концентрированной серной кислоты легко отнимает у множества типов
органических соединений водородный атом с двумя электронами (гидрид-
анион) и, превращаясь в гидразобензол (стр. 81), перегруппировывается
в бензидин (А. Н. Несмеянов, Р. В. Головня).
Азогруппа представляет собой одну из важнейших функций аромати-
ческого ряда, и многочисленные азосоединения в виде азокрасителей
в очень больших количествах готовятся синтетически. Однако способ
создания азогруппы в синтезе азокрасителей иной (через диазосоединения)
и будет изложен после рассмотрения диазосоединений.
Азобензол представляет собой легкоплавкие, растворимые в орга-
нических растворителях ярко-оранжевые кристаллы. Однако его окраска
далека по интенсивности от окраски азокрасителей, и он не способен
закрепляться на волокне.
Азосоединения при кипячении с раствором хлористого олова или при
действии треххлористого титана количественно восстанавливаются в пер-
вичные амины. Восстановлением азобензола цинковой пылью в щелочи
получают бесцветный кристаллический гидразобензол
CeH5—N=N—СвНэ CeH5-NH-NH-CeH5
который уже кислородом воздуха постепенно окисляется обратно в азо-
бензол. Самым замечательным свойством гидразобензола является коли-
чественно протекающая при кипячении в водных растворах сильных кис-
лот перегруппировка в бензидин (n,ri-диаминодифенил):
Бензидиновая перегруппировка, открытая Н. Н. Зининым, изучалась
многочисленными исследователями. Выяснено, что наряду с п,п'-пере-
ФУНКПИИ, ОБРАЗУЕМЫЕ ВОССТАНОВЛЕНИЕМ НИТРОСОЕДИНЕНИЙ
81
группировкой происходит и «^-перегруппировка и получается, правда
в меньшем количестве, дифенилин (о, и-диаминодифенил)
NH2
Если одно из пара-положений гидразобензола занято заместителем,
происходит семидиновая перегруппировка, приводящая к первично-вторич-
ному диамину ряда бензола (а не бифенила) — семидину, являющемуся
n-замещенным производным и-аминодифениламина:
Н
NH—NH
>
Перегруппировка происходит интрамолекулярно, т. е. внутри одной
молекулы, а не интермолекулярно, т. е. не путем взаимодействия двух
(или более) молекул. Это доказывается тем, что из смеси двух разных
гидразосоединений получаются два и только два разных бензидина,
например:
не получается вовсе
Наиболее вероятной гипотезой механизма реакции, очевидно, сле-
дует считать вариант гипотезы Р. Робинзона, предложенный чешским
ученым Вечержем. Первая стадия — фиксация протонов обоими атомами
азота:
2Н+
н н н н
Такое состояние с двумя полными положительными зарядами на со-
седних атомах азота неустойчиво. Поскольку каждый азот приобретает
тетраэдрическую конфигурацию, противоположные (пара-) концы бен-
зольных ядер сближаются друг с другом. Для гомолитического разрыва
•связи N — N требуется по одному дополнительному электрону на каждый
атом азота. Эти электроны доставляются от пара-углеродных атомов,
которые в свою очередь приобретают свободнорадикальный характер.
6 Заказ 672
АРОМАТИЧЕСКИЕ АМИНЫ
82
Бензольные ядра спаиваются своими пара-углеродными свободноради-
кальными атомами в систему дифенила *:
При действии на азобензол серной кислоты и донора гидридного водо-
рода, например первичного или вторичного спирта, также происходит
бензидиновая перегруппировка. Сначала образуется гидразобензол
который далее перегруппировывается, как показано на стр. 81.
АРОМАТИЧЕСКИЕ АМИНЫ
Простейший ароматический амин — анилин C6H5NH2. Впервые он был
получен сухой перегонкой природного индиго (Фрицше, 1826 г.), откуда
и его название (anil — по-испански, «индиго»). Следы анилина найдены
также в каменноугольной смоле. Промышленное значение анилин при-
обрел после получения его Н. Н. Зининым реакцией восстановления
нитробензола сероводородом:
CeH5NO2-|-3H2S —> CeH5NH2-|-2H2O-|-3S
До недавнего прошлого главным способом промышленного получения
анилина и ряда других ароматических аминов был вариант зининской
реакции — восстановление нитросоединений чугунной стружкой в при-
сутствии небольшого количества соляной кислоты (Бешам):
4CeH5NO2 + 9Fe +4Н2О > 4СвН5ТШ2 + ЗГезО4
По мере увеличения производства анилина на первый план в промыш-
ленности выдвигается каталитическое восстановление нитросоединений:
CeH5NO2 + 3H2 CeH5NHa+2H2O
Пунктирные стрелки обозначают перемещение одного электрона.
ароматические амины
83
В лаборатории излюбленным восстановителем служит металлическое
олово с крепкой соляной кислотой:
2C6H5NO2 + 3Sn+ 14НС1 —-> 2C6H5NH2 • HCl + 3SnCl4
Восстановлением соответствующих нитросоединений получают гомо-
логи анилина — о-, м- и и-толуидины CH3C6H4NH2, а также замещенные
анилины — о-, м- и и-хлора нилины C1C6H4NH2, о- и и-анизидины
CH3OCeH4NH2, о- и и-фенетидины C2H5OC6H4NH2 и т. д.
Восстановление .и-динитробензола (чугунной стружкой или катали-
тически) дает л-фенилендиамин
а при восстановлении полинитросоединений сернистыми металлами вос-
станавливается лишь одна нитрогруппа и из л4-динитробензола образуется
.и-нитроанилин.
Восстановлением о- и n-нитроанилина получаются о- и’п-фениленди-
амины, например:
H2N—CeH4—NO2-o Fe (НЧ:) > H2N-C6H4—NH2-o
Другие способы получения ароматических аминов имеют подчиненное
значение. Назовем некоторые из них.
1. Восстановление (например, при помощи олова и соляной кислоты)
любого из промежуточных продуктов восстановления нитросоединений,
в частности азосоединений (стр. 80).
2. Действие азотистоводородной кислоты на ароматические углево-
дороды (Шмидт):
CeHe + H- N=N=N —► C6H5NH2 + N2
3. Действие амида натрия в жестких условиях на галоидбензолы.
Проведение реакции с галоидбензолом, в котором меченый углерод
(например, радиоактивный 14С) связан с галоидом, показало (Робертс),
что аминогруппа связывается лишь на 50% с этим углеродом, а на 50%
с орто-углеродным атомом. Это объясняется предварительным отщепле-
нием сильноосновным NaNH2 галоидоводорода и образованием дегидро-
бензола, затем уже присоединяющего NH3 к тройной связи (кинезамеще-
ние, см. также стр. 60 и 61):
4. Действие на ароматические соединения гидроксиламин-0-суль-
фокислоты в присутствии А1С13 при нагревании (Ковачик, Беннет).
Преобладает орто-пара-замещение:
СН3С6Нб + NH2-O-SO3H > CH3CeH4NH2 + H2SO4
6*
АРОМАТИЧЕСКИЕ АМИНЫ
84
Таблица 64. Амины
Формула Название । Температура плавления °C Температура кипения °C
c6h5nh2 Анилин —6,2 184,4
CH3ceH4NH2 Толуидин
о-, или 1,2- —24,4 (а-форма) —16,3 (3-форма) 199,8
м-, или 1,3- —31,5 203,3
п-, или 1,4- +45 . 200,3
NO2C6H4NH2 Нитроанилин
о-, или 1,2- 71.5 284,1
м-, или 1,3- 111,8 306,3
ClCeH4NH2 п-, или 1,4- 147,5 331,7
Хлоранилин
о-, пли 1,2- —14
(а-форма) —3,5 (3-форма) 208,8
м-, или 1,3- —10,4 229,8
HSO3C6H4NH2 п- или 1,4- Аминобензолсульфокпслота +70 231
о-, или 1,2- (ортаниловая) — —
м-, или 1,3- (метаниловая) — —
CeH5NHCH3 п-, или 1,4-(сульфаниловая) 288 (разл.) —
Метиланилип —57 195,7
СбН5К(СНз)2 Диметилаиплин +2,5 193,5
(CeH5)2WH Дифениламин 53 302
(C6H5)3N Трифениламин 126,5 365
к т^е °и/РЬ^е $ИЗИЧеские свойства ароматических
гптоп ‘ ; омологи анилина называются о-, м- и
руина сн3) и ксилидинами (две группы СН3).
аминов сопоставлены
n-толуидинами (одна
Основные свойства ароматических аминов
Так*11^ основание гораздо более слабое, чем аммиак и жирные амины,
жен’аой KHcXTlO™»!5'^’-’ Э ДЛЯ ави™на'3,5.10-и (К. для сопря-
в воде да Ы ° ' Действительно, анилин, слабо растворимый
О.ЛКТ? "» окрашивающие ни лакмус, ни фенолфталеин.
Р ует солей с такими слабыми кислотами, как угольная, синиль-
ОСНОВНЫЕ СВОЙСТВА
85
Плотность Показатель преломления „20 nD л'ь Дипольный момент р. в бензоле при 20° С, О
п ри 25° С
1,022 1,5863 — 3,82-10'1° 1,53
1,004 1,5728 — 2,47-10'Ю 1,60
0,989 1,5711 (при 22° С) — 4,92-10'Ю 1,45
1,046 1,5532 (при 5 9° С) — 1,18-10'й 1,31
1,442 — — 6-Ю'4 (при 0° С) 4,96
1,430 — — 2,7-10'5 (при 0° С) 4,85
1,424 •— — 6,32 (при 25° С)
1,213 1,5895 1,77
1,216 1,5942 ММ — 2,68
1,427 — — 8,51-10'5 2,97
— — 3,3-Ю-з
— — 1,85-Ю'4 — —
— 5,9-Ю’4 — —
0,986 1,5702 (при 21е С) — 5-10'Ю 1,64
0,955 1,5582 — 1,15-10-э 1,56
1,159 — — 7,6-10'14 (при 15° С) 0,95
0,774 (при о°С) 1,353 (при 16° С) — — 0,52
ная кислота или сероводород, но с сильными кислотами образует твердые
соли, например
н н
С6Н5-N<f +НС1 —> СвНб-N^H С1-
Н ХН
в водном растворе сильно гидролизованные.
Ослабление основных свойств трехвалентного азота в анилине вызвано
мезомерным эффектом аминогруппы, или, иначе говоря, сопряжением
свободной пары электронов азота с л-электронами ароматического цикла
(р,л-сопряжение). Это значит, что орбиталь р-электронов азота (сво-
бодная пара) перекрывается с л-орбиталями бензольного цикла. Такое
взаимодействие возможно, очевидно, лишь в том случае, если оси
АРОМАТИЧЕСКИЕ АМИНЫ
86 -----------——--------—------------------------------------
n-электронных орбиталей азота приблизительно параллельны осям
р-электронных орбиталей углеродов цикла. Действительно, выведение
аминогруппы из (условно говоря) плоскости цикла тотчас увеличивает ее
основные свойства и уменьшает дипольный момент молекулы. Такое нару-
шение копланарности возможно при наличии двух достаточно громозд-
ких орто-заместителей, и его влияние видно, например, из сравнения
дипольных моментов анилина и 2,3,5,6-тетраметил-1-аминобен.зола (амино-
дурола):
//=1,532)
NHo
//=1,392)
Метильные группы, вообще сообщающие лишь малый дипольный
момент ароматическому ядру, в данном случае расположены симметрично,
так что их собственное итоговое влияние на дипольный момент равно
нулю. К сожалению, нельзя сравнивать основные свойства аминогруппы
в обоих соединениях, так как при орто-расположении метилов поля их
водородов отталкивают атакующий ион водорода, что ослабляет основные
свойства и скрадывает интересующий нас эффект. Разница дипольных
моментов двух ароматических аминов обусловлена только наличием
мезомерного эффекта в случае анилина и его отсутствием у аминодурола.
Реакции электрофильного замещения
в бензольном ядре аминов
Взаимодействие аминогруппы с бензольным ядром особенно сильно про-
является в момент электрофильной атаки в орто- и пара-положения,
например, при галоидировании. сг-Комплексы (см. стр. 33) в этих слу-
чаях имеют следующее строение:
РЕАКЦИИ ЭЛЕКТРОФИЛЬНОГО ЗАМЕЩЕНИЯ
87
Способность азота легко превращаться в положительно заряженный
аммонийный азот и брать на себя (в промежуточном о-комплексе) поло-
жительный заряд, возникающий на связанном с ним углероде цикла
в момент атаки электрофила на о- и и-углеродные атомы, и является при-
чиной снижения уровня энергии переходного состояния и большой
скорости (стр. 33) прохождения электрофильных замещений. Таким
образом, слабые основные свойства ароматической аминогруппы и ее
сильное орто- и пара-ориентирующее действие имеют одну и ту же
причину.
Ориентирующее действие аминогруппы в анилине и его гомологах
настолько сильно, что прямым воздействием иода при комнатной тем-
пературе анилин иодируется в 2,4,6-трииоданилин. Еще более энер-
гично он реагирует с бромом и хлором, образуя 2,4,6-тригалоид-
анилины.
При сульфировании анилина требуется уже повышенная температура,
так как сначала образуется соль, в которой азот, став аммонийным,
перестает быть орто- и пара-ориентантом. Кроме того, в данном случае
сульфирование сначала проходит по аминогруппе, и лишь затем перво-
начально образовавшаяся фенилсульфаминовая I кислота превращается
в и-аминофенилсульфокислоту II (сульфаниловую кислоту) в результате
интрамолекулярной перегруппировки в пара-положение: скорость пере-
группировки пропорциональна концентрации фенилсульфаниловой ки-
слоты и функции кислотности (Врба, Аллан, 1968). Эти авторы дают
следующий предположительный механизм перегруппировки:
Нитровать прямо анилин нельзя — реакция слишком энергична,
а выделяющиеся вследствие окисления анилина окислы азота диазоти-
руют оставшийся неокисленный анилин. Поэтому анилин предварительно
ацетилируют (или формилируют), а образовавшееся ацильное производное
(анилид) нитруют, получая смесь о- и n-питроацетанилидов (или о- и
АРОМАТИЧЕСКИЕ АМИНЫ
88 -----------------------------------
n-аитроформанилидов). Гидролизуя защитную ацильную группу,
чают смесь о- и п-пптроааилинов, которые разделяют.
полу-
Здесь аминогруппа защищена от диазотирующего действия окислов
азота, а бензольное ядро вследствие уменьшения ориентирующей силы
группы NH2 (группа — NH — G — СН3 более слабый орто- пара-ориен-
II
О
тант) становится менее доступным для окисления и избыточного нитро-
вания.
Замещение в аминогруппе
Анилиды. Мы уже встретились с реакцией ацилирования анилина. Аро-
матические амины можно ацилировать хлорангидридами или ангидридами
кислот или сильным нагреванием аминов с кислотами вплоть до отщеп-
ления воды:
CeH5NH2 +R—С—С1 > С6Н5—NH—С—R4-HC1
ii ||
о о
CeH5NH2+R—С—О—С—R ----► ceH5NH—С—R + R—С—ОН
II II II
о 0 0
CeH6NH2+ R—с-он —> CeH5NH3 OOC-R ► CeH5NH—C-R + Н20
п К
0 О
Для формилирования пригоден только последний способ.
О
ЗАМЕЩЕНИЯ В АМИНОГРУППЕ
<89
Ацилированные ароматические амины носят общее название анилидов.
Анилиды толуидинов называются толуидидами, анилины анизидинов —
анизидидами и т. д.
Анилиды можно рассматривать также как фенилированные амиды
кислот:
СНз—С—NH2
СНз—с—nhc6h5
ацетамид
фенилацетамид,
или ацетанилид
Как все амиды, они гидролизуются при нагревании и в кислой, и
в щелочной среде:
C6H5NH—С—R + H-jO —CeHBNH2 +НО—С—R
II II
О о
Анилиды лишены основных свойств.
Некоторые анилиды применяются как лекарственные седативные
и жаропонижающие вещества. Таковы сам ацетанилид (антифебрин),
n-ацетилфенетидин С2Н5О — С6Н4 — NH — С — СН3 (фенацетин).
II
О
Алкиланилины. При нагревании сернокислого анилина с метанолом под
давлением один или два водорода аминогруппы замещаются на метил
и образуются метил- и диметиланилин (преобладание того или другого
вещества зависит от условий реакции). Той же цели можно достигнуть,
пропуская пары анилина и метанола над дегидратирующим катализа-
тором (А12О3):
C6H5NH2 + CH3OH С6Н5Ш1СНз + Н2О
метиланилин
А1 ?О ч
C6H5NH2 + 2CH3OH CeH5N(CH3)2 + H2O
диметиланилин
Метил- и диметиланилин готовятся в больших масштабах, такгкак
моноалкиланилины применяются в некоторых странах как антидетона-
торы моторных топлив. Они же (особенно диалкиланилины) используются
в промышленности синтетических красителей (азокрасители и трифенил-
метановые красители). Кроме того, при нитровании диметиланилина,
наряду с 2,4,6-замещением в ядро, происходит замещение на нитро-
группу одного метила, и получается тетрил — сильное бризантное
взрывчатое вещество:
СН3—N—СН3
+ no2 hso4 —>
СН3—N—NO2
o2n 1 \()2
+ Продукты окисления СН3_
no2
АРОМАТИЧЕСКИЕ АМИНЫ
90
Монометиланилпн, как и вообще вторичные амины, при действии
азотистой кислоты образует нитрозамин, при нагревании перегруппиро-
вывающийся в лг-нитрозометиланилин:
- н2о
+ HONO
Этим монометиланилин отличается от самого анилина, который при
действии азотистой кислоты диазотируется (стр. 97)- Что касается диме-
тиланплпна, то он при действии азотистой кислоты прямо нитрозируется
в пара-положение, образуя тг-нитрозодиметиланилин:
СН3—^т—СН3
+ HONO
------->
- Н2О
N=O
Для нитрозированпя приходится применять разбавленный водный
раствор азотистой кислоты. Уже поэтому данная реакция электрофиль-
ного замещения в ароматическом ядре осуществима лишь с самыми актив-
ными по отношению к электрофильным атакам ароматическими ядрами,
например содержащими такие заместители, как —N(CH3)2 или —ОН.
Нптрозогруппа как заместитель (в растворах, где нитрозосоединение
мономерно, см. стр. 75) благодаря своему—У-эффекту является сильным
электроноакпептором и оттягивает электроны от орто- и пара-углеродных
атомов. В результате пара-углеродный атом, несущий диметиламиногруп-
пу, делается уязвимым для нуклеофильных атак. Поэтому нитрозоди-
метиланплин при кипячении со щелочью отщепляет диметиламин, пре-
вращаясь в нитрозофенол:
АРОМАТИЧЕСКИЕ ВТОРИЧНЫЕ II ТРЕТИЧНЫЕ АМИНЫ
Нагреванием хлористоводородной соли анилина получают вторичный
чистоароматический ампн - дифениламин:
2CeH5NH3 СВ —> CeHjXHCeHs-r-NHiCl-bHCl
ВТОРИЧНЫЕ И ТРЕТИЧНЫЕ АМИНЫ
Дифениламин является антиоксидантом. Многие аналогично пост-
роенные ароматические вторичные амины, например
₽-(нафтил)-фениламин
NH— NH— <\_^>
п (N. М'-дифспил)-фепилепдиамин
служат для защиты пластмасс от окислительного разрушения. Сам ди-
фениламин применяется как стабилизатор пироксилиновых порохов
(связывание окислов азота).
Фенилируя его натриевое производное по Ульману действием иод-
бензола в присутствии порошка меди можно получить и третичный амин —
трифенила мин'.
СвН5ч _ + с
>NNa + CeH5I — -> (CeH5)3N + NaI
CeHs'
Уже дифениламин практически почти лишен основных свойств, все
же он растворим в концентрированной серной кислоте (в таком растворе
Ка его сопряженной кислоты равно 10"1); из такого раствора вода (более
сильное основание) высаживает дифениламин. С окислителями, в част-
ности с азотной кислотой, дифениламин в сернокислом растворе дает
синее окрашивание — чувствительная реакция на ион NO". Реакция
состоит в превращении дифениламина в окрашенную иммониевую соль
следующего строения:
CeH5-N
HSOJ
Трифенил амин в отличие от других третичных аминов не метилируется
иодистым метилом и не образует четвертичных аммониевых солей. Он
индифферентен даже к концентрированной серной кислоте. С самой
сильной — хлорной кислотой он все же образует соль — перхлорат.
В трифениламипе достигается настолько полное перекрывание р-ор-
битали свободной пары электронов азота с л-орбиталями всех трех бен-
зольных ядер и рассредоточение этой пары, что опа практически себя
не проявляет (кроме реакции с хлорной кислотой). 1 рпфешглампп, в част-
ности, не способен к комплексообразованию, не реагирует с иодистым
Метилом.
Молекула трифепиламииа плоская, иначе невозможно было бы
р, л-сопряжение. При нарушении плоскостной структуры основность
азота возрастает. Виттигу удалось, отнимая НС1 от 9-(о-хлорфеппл)-9,10-
Дигидроакридина посредством NaNH2 в жидком аммиаке, замкнуть
пространственный гетероцикл — азатриптицен. Последний представ-
ляет собой как бы трифениламин, в котором группа НС связывает
АРОМАТИЧЕСКИЙ АМИНЫ
три а-углеродных атома трех фенилов и выводит последние из плоскости:
Азатриптицеп представляет собой сильное основание, образующее
с иодистым метилом иодметилат. В этом соединении р-электронные орби-
тали азота не могут сопрягаться с орбиталями л-электропов трех фени-
лов вследствие взаимной перпендикулярности их осей. Таким образом,
рассредоточения электронов азота по фенильным кольцам нет, и можно
сказать с уверенностью, что орто- и пара-ориентирующее действие}амин-
ного азота в азатриптицене полностью отсутствует.
НИТРОАНИЛИНЫ
Способы получения всех трех изомерных мононитроанилинов были при-
ведены на стр. 87. 2,4-Динитроанилин может быть получен как нитро-
ванием ацетанилида, так и, еще проще, аммонолизом динитрохлорбензола:
+ NH4C1
Аналогичной реакцией из 2,4,6-тринитрохлорбензола получается
2,4,6-тринитроанилин:
Это вещество за свои амидоподобные свойства названо гшкрамидом —
амидом пикриновой кислоты, т. е. 2,4,6-тринитрофенола (стр. 118).
Пикрамид практически лишен основных свойств.
Из трех мононитроанилинов мета-изомер — более сильное основание,
образующее соль с водной соляной кислотой; о- и п-нитроанилипы раство-
ряются в избытке достаточно концентрированной водной соляной кислоты,
но при испарении кислоты остаются свободные основания. При пропу-
скании хлористого водорода в эфирный раствор о- и п-питроанилинов
могут быть осаждены их солянокислые соли, при действии воды тотчас
полностью гидролизующиеся. Таким образом, это очень слабые основания.
НИТРОАНИЛИНЫ
93
Сопряженным кислотам
личины р/Га:
о-нитроанилина . . , —0,29
.м-нитроанилина . . 4-2,5
п-нитроанплпна . . 4-1,02
нитроанилинов соответствуют следующие ве-
2,4-динитроанилина —4,53
2,4,6-тринитроанили-
на..................—9,41
Однако в 3,5-диметил-4-нитроанилине соседние метильные группы
выводят нитрогруппу из плоскости бензольного ядра, вследствие чего
нарушается сопряжение (резонанс) ее с ядром и с аминогруппой и выклю-
чается — Т-эффект нитрогруппы. Свойственный ей —Z-эффект, как всегда,
далеко по цепи атомов не распространяется. Поэтому 3,5-диметил-4-нитро-
.анилин как основание значительно превосходит n-нитроанилин и суще-
ственно не отличается от анилина.
С выключением сопряжения за счет выведения аминогруппы из пло-
скости бензольного ядра мы уже встречались (стр. 86). Можно просле-
дить это выключение сопряжения и на разнице в дипольных моментах,
«ели подобрать выводящие из плоскости заместители так, чтобы вноси-
мые ими дипольные моменты в итоге давали нуль. Для примера возьмем
производные дурола (2,3,5,6-тетраметилбензола). Сравнение дипольных
моментов /г-нитроанилина и нитроаминодурола (в бензольном растворе)
показывает, что дипольный момент последнего на 1,2 D меньше, чем п-нит-
роанилина. Это также следствие выключения сопряжения нитро- и амино-
группы с ядром, а значит, и между собой:
.^=6,180 /г-4,980
^вместо вычисленных
5,17 0 для структуры а)
Еще резче, чем в n-нитроанилине, взаимодействие групп NO2 в
УТ(СН3)2, находящихся в пара-положении друг относительно друга:
N(CH3)2 N(CH3)2
9 - 2
N N
</Хо- -c/V
Диметиламиногруппа — более сильная электронодонорная группа,
чем группа NH2. Увеличение дипольного момента по сравнению с адди-
тивным здесь достигает 1,2 1,30.
АРОМАТИЧЕСКИЕ АМИНЫ
Сдвиг в сторону структуры г (или, по Полингу, «пес структуры» г)
здесь больше, чем пес аналогичной структуры б в п-нитроанилине.
Все три мононитроанилина широко применяются в синтезе красите-
лей (особенно азокрасителей) и для получения соответствующих фени-
лендиаминов.
АНИЛИНСУЛЬФОКИСЛОТЫ
Из них обычны две: n-анилинсульфокислота, или сульфаниловая кислота,
и .н-анилипсульфокислота, или метаниловая кислота. Обе кислоты на-
ходят применение в синтезе азокрасителей. Получение сульфаниловой
кислоты сульфированием анилина было уже описано на стр. 87. Мета-
ниловую кислоту получают, сульфируя в жестких условиях нитробен-
зол и восстанавливая полученную .и-нитробензолсульфокислоту.
Очень важен амид сульфаниловой кислоты и некоторые из его много-
численных производных, замещенные в аминогруппе.
Сам сульфаниламид получают следующей серией реакций:
О
II
NHCCH3
О
II
4-HO-tS—С1
о
о
II
NHCCH3
o=s=o
I
Cl
+2NH, ,
NHiGl’>
I
nh2
Н8О(Н+)
-СН.СООН
I
nh2
no ь «аНИЛамид’ называемыи в медицине белым стрептоцидом или пронтальбином,
тйТ» ЬактеРпостатическое вещество, излечивающее ряд стрептококковых заболева-
(ЬанилямаСТН°СТИ РОЖУ‘ " медицине применяются также следующие замещенные суль-
nh2 1 nh2 nh2 1 nh2 1
с 1 0 1 0
o=s=o 1 1 o=s=o 1 o=s=o 1 o=s=o
NHCCH3 II О альбуцид 1 nhcnh2 II NH сульгин, или сульфагуанидин 1 nhcnh2 II О уросульфан (производное мочевины) 1 nhcnh2 II s бадионал (производное тиомочевины)
АРОМАТИЧЕСКИЕ диамины
95
а также ряд веществ типа NH2— Ч—SO2—NHR, где R — тот или иной гетеро-
цикл — пиридин (в сульфидине), пиримидин (в сульфадимезине), тиазол (в сульфазоле).
Эти сульфамидные препараты вызвали в период 1930—1940 гг. подлинный перево-
рот в терапии ряда инфекционных бактериальных заболеваний. Благодаря им стали
легко излечиваться прежде опасные или смертельные болезни — крупозное воспале-
ние легких, заражение крови, стрептококковые ангины и др. Значение сульфамидных
препаратов несколько померкло после открытия в период 1940—1950 гг. ряда антибио-
тиков во главе с пенициллином, которые еще шире по спектру излечиваемых бактери-
альных заболеваний.
Действие сульфамидных препаратов основывается на совершенно новом принципе—
применении «антиметаболпта». Дело в том, что почти все организмы, в том числе
и бактериальные, нуждаются в фолевой кислоте, существенной для их жизнедеятельно-
сти. В молекулу фолевой кислоты входит п-аминобензойная кислота (витамин Hj),
которая является «фактором роста» бактерий. Амид сульфаниловой кислоты по физиче-
ским и геометрическим параметрам молекулы близок к п-аминобензойной кислоте и
потребляется бактерией вместо нее, не создавая веществ, обладающих энзиматической
активностью. Бактерии перестают размножаться и быстро побеждаются организмом-
хозяином. Таким образом, в случае сульфамидных препаратов речь идет о чем-то
принципиально ином, чем ядовитое действие, убивающее бактерии. Непосредственно
сульфамидные препараты не ядовиты для бактерий. Их действие основано па подмене
естественного метаболита (продукта обмена веществ организма). Действие сульф-
амидных препаратов сказывается при достаточной их концентрации, далеко превосхо-
дящей концентрацию природной н-аминобензойной кислоты, п может быть снято
добавлением последней.
АРОМАТИЧЕСКИЕ ДИАМИНЫ
Способы синтеза трех диаминов —о-, м- и п-фенилендиаминов — из
.м-динитробензола и о- и n-нитроанилинов были приведены на стр. 83.
тг-Фенилендиамин можно также легко получить из аминоазобензола
(стр. 104) восстановлением его посредством SnCl2 или TiCI3 в водном
растворе:
N — Х-ЧОУ-NHo + 2SnCl, + 4НС1
п -фенилендпамин
Фенилендиамины — твердые вещества, быстро окисляющиеся и темне-
ющие на воздухе, довольно хорошо растворимые в воде.
о-Фенилендиамин отличается способностью легко образовывать гете-
роциклы. Так, при диазотировании он дает внутреннее диазоаминосо-
единение — азимидобензол'.
z^NH2
4x/K'NH2- hci
+ hno2 ——
АРОМАТИЧЕСКИЕ АМИНЫ
96
Нагревание с карбоновыми кислотами приводит к гетероциклическим
амидинам — бензимидазолам’.
/С—R
НО
Р°'йкция с а-диоксосоединениями дает хиноксалины
а взаимодействие с S02 — пиазтиол'.
+ 2 Н2О
Интересную деструкцию о-фенилендиамина, протекающую через о-фе-
нилендинитрен, открыли Накагава и Оноуе. Окислением о-фени-
лендиамина тетраацетатом свинца они получили динитрил муконовой
кислоты (выход 50%):
о-фенилен-
динитрен
м-Фенилендиамин служит как первой, так и второй компонентой
в синтезе азокрасителей (стр. 107). Наряду с п-аминодиметиланилином
он применяется в синтезе тиазиновых красителей.
п-Аминодиметиланилин получают восстановлением нитрозодиметил-
анилина:
/ /—\\ п
O=N-\O/-><(сН3)о----> Н, \(СН3)2
Окислением n-фенилендиамина хлорным железом в присутствии серо-
водорода получают краситель — фиолетовый Лаута:
СГ
ДИАЗОСОЕДИНЕНИЯ
97
Аналогичен синтез метиленового синего (Каро):
СГ
ДИАЗОСОЕДИНЕНИЯ
Соли ароматических аминов (в присутствии избытка сильной кислоты)
в водном растворе диазотируются азотистой кислотой и превращаются
в соли арилдиазониев:
+ /И
СвНБ—N^H Cl’ + HO—N=O > C6H5N=N C1" + 2H2O
Эта реакция открыта в 1858 г. Гриссом.
Кинетические исследования Инголда показали, что реакция на деле
протекает не по приведенному выше наглядному уравнению, а следующим
образом:
H-O-N=O + H+ —>
Н26-N=0
CeH5-NH2 + H26-N=O -> C6H5-NH—N=O + H3O
c6h5—n=n—oh
lHGI
CeH5-NsN СГ + Н2О
Такое толкование процесса следует из того, что диазотирование —
реакция третьего порядка, константа скорости которой пропорциональна
произведению концентраций свободного амина, азотистой кислоты и
иона водорода (а не азотистой кислоты и соли амина). Этим объясняется
также и то, что амины с более слабыми основными свойствами диазоти-
руются с большей скоростью (большая концентрация амина за счет гидро-
лиза). Ароматические амины, несущие в ядре (особенно в орто- и пара-
положении) заместители второго рода, диазотируются быстрее, чем
незамещенные, и дают более прочные соли диазопия. Справедливо и обрат-
ное положение. В случае очень слабоосновных аминов, подобных 2,4,6-
тринитроанилину, диазотирование приходится проводить в концентри--
рованной серной кислоте.
7 Заказ 672
ДИАЗОСОЕДИНЕНИЯ
98
Соли арилдиазониев с простыми анионами малопрочны, и их не вы-
деляют из водного раствора, а непосредственно подвергают дальнейшим
превращениям. Однако из водного раствора можно осадить (добавлением
соответствующей соли или кислоты) комплексные («двойные») соли,
например:
С«Н5-N=N HgClg (CeH5-N=N)2 SnClJ"
СвЩ—N=N BFj (CeH6-N=N)2 SiFJ"
CeH5—N=N SbClj- CeH6—N==N SbClj-
CeH5—N=N ZnClj C8H5-teN FeClJ
Для получения хлористого арилдиазония в твердом виде соляноки-
слую соль амина растворяют в спирте и диазотируют сложным эфиром
азотистой кислоты; образовавшийся хлористый арилдиазоний высажи-
вают эфиром.
Кроме главного пути синтеза диазониевых солей — диазотирования
аминов, соли диазония можно получить по Бамбергеру двумя путями —
действуя на нитрозосоединения гидроксиламином или NO:
С6Н5—N = O4-H2NOH —> CeH6—n=noh+h2o
CeH5—N = O+2NO ► C6H6-N=N NO^
а так как окислы азота (N2O4 и N2O3) нитрозируют многие металло-
органические соединения, можно получать соли диазония, действуя
этой смесью окислов азота на следующие металлоорганические соеди-
нения (А. Н. Несмеянов, Л. Г. Макарова):
(CeH5)2Hg (C6H5)2Cd (CeH5)4Sn (CeH5)3Bi
Недавно Ботт описал новый путь синтеза ароматических диазониевых
солей действием^на арилсульфинилимиды (нерассмотренный нами класс
соединений) нитрозилперхлоратом (в скобках — гипотетический проме
жуточный продукт реакции):
Ar—N=S=O-J-NO* С1О4 —>
N=O
I СЮ;
Ar—N=S = O
+
--> ArN=N ClOj + SOo
Арилдиазониевые соли — ионно построенные соединения и их вод-
ные растворы — электролиты. Оли хорошо растворимы в воде, хуже
в спирте, нерастворимы в эфире и углеводородах. Фенилдиазонийхло-
риды, сульфаты и нитраты взрывают при нагревании, очень неустойчивы
(ури комнатной температуре могут храниться обычно не более суток).
Боли с комплексными анионами менее растворимы и более устойчивы.
Наиболее прочны соли типа ArN2 BFj, которые можно хранить месяцами.
Строение диазониевых солей, аналогичных во многом аммониевым
солям, установил уже в 1869 г. Бломстранд. Их можно рассматривать
как ониевые соединения молекулы азота, сопоставляя с аммониевыми,
оксониевыми, сульфониевыми и прочими соединениями, являющимися
ониевыми производными атомов N, О, S (ср. кн. I, стр. 23). Реакции с анио-
нами слабых кислот проходят, однако, для диазониевых солей иначе,
ДИАЗОСОЕДИНЕНИЯ
99
чем для других ониевых солей. Обычные ониевые соли могут^только
электрофильно арилировать (или соответственно алкилировать) анион
с передачей последнему ониевого катиона без пары электронов связи:
АгзО + ОН_ > АтОН-рАг—О—Аг
Диазониевые катионы при этом образуют с анионами слабых кислот
(CN“, SO3H~, ОН ) ковалентно построенные диазосоединения:
Аг— N=N:+OH- > Аг—N=N—ОН
Аг—N=N:+CN- > Аг—N=N—C=N
Аг—N=N:+SO3H- —> Аг—N=N—SO3H
Или то же в электронной интерпретации:
Ar—n=n:+':о: н-----► дг—n=n:o:h
Различие обусловлено участием второго атома азота, который вслед-
ствие частичного оттягивания от него пары электронов связи приобре-
тает заряд 6 +
+П 8+
Ar— №N:
а следовательно, и способность воспринимать нуклеофильные атаки.
Особенно подробно изучалось взаимодействие катионов арилдиазо-
ния с гидроксильным анионом. Первой довольно медленно протекающей
реакцией является уже описанное образование диазогидрата
Аг—N=N:-f-OH- > Аг—N=N—ОН (1)
который быстро реагирует со щелочью как кислота, образуя диазотат-
анион:
Аг—N=N—ОН + ОН- > Аг—N=N—О' + НгО (2)
При сливании эквивалентных количеств диазониевой соли и щелочи
благодаря быстрому прохождению реакции (2) лишь половина диазония
успевает превратиться в диазогидрат и далее (быстро) в диазотат-анион,
а половина остается в форме диазоний-катиона:
2Аг—N=N:+20H“ ----> Аг—N=N: + Аг—N=N— О“+Н2О
Диазотат (натрия или калия), образующийся при действии избытка
щелочи, по старым наблюдениям Ганча сначала активен в реакции азосо-
четания с фенолами и третичными аминами (стр. 104), затем самопро-
извольно превращается в изомерную неактивную форму. Первая назы-
вается нормальным диазотатом (и ей соответствует нормальный диазо-
гидрат), вторая — изодиазотатом (ей соответствует изодиазогидрат).
7*
100
ДИАЗОСОЕДИНЕНИЯ
Один из крупнейших исследователей диазосоединений прошлого
века Бамбергер приписывал изодиазогидрату на основании реакций
•его ацилирования структуру первичного нитрозамипа:
Н О О-=С—Н
Ar—N—N—O-|-R—С—Cl > Аг-N—N=O + HC1
Другой исследователь, Ганч, считал, и это принятая и сейчас точка
зрения, что изодиазогидрат^и первичный нитрозамид находятся в отно-
шениях таутомерии:
Ar—N=N—ОН Аг—N—N=O
Н
Как было указано, образование изодиазогидрата по реакции
Аг—N==N:+OH- > Аг—N=N—ОН
протекает медленно. Добавляя к соли диазония щелочь на холоду, Ганч
выделял в осадок вещество, которое в отличие от изодиазогидрата реаги-
ровало с фенолами, образуя азокрасители (стр. 107), и давало иной спектр
поглощения. Это якобы более реакционноспособное вещество было на-
звано им нормальным диазогидратом. Ганч предположил, что выделенный
им «нормальный диазогидрат» находится с изодиазогидратом в отношениях
геометрической снн-анттш-изомерии
Аг—N Аг—N
II II
НО—N N—ОН
ctm-диазогидрат анти-диазогидрат
подобной изученной им же стереоизомерии оксимов (кн. I, стр. 140)
Аг—С—Н Аг—С—Н
II II
N-OH HO-N
син-оксим анти-оксим
Бамбергер считал наличие стереоизомерии диазогидратов не дока-
занным и приписывал им структурную изомерию:
Н
Аг— N=N—ОН Ar—N— N=O
нормальный диазогидрат ' изодиазогидрат
(по Бамбергеру) (по Бамбергеру)
Б. А. Порай-Кошиц потенциометрическим и спектроскопическим ис-
следованием смесей ряда арилдиазониев со щелочью заставил усомниться
в индивидуальности ганчевских «нормальных диазогидратов», приведя
аргументы в пользу того, что это смесь веществ: соли диазония, щелочи
а изодиазогидрата (и его соли) с пеустановившимся равновесием.
На рис. 86 приведены кривые Порай-Кошица, полученные потенцио-
метрическим титрованием соли п-нитрофенилдиазония щелочью (пунктир^-
•ные кривые), и кривые обратного титрования раствора кислотой с разной
скоростью, демонстрирующие медленное приближение к равновесию.
ДИАЗОСОЕ ДПНЕНИЯ
101
, Дв,’ее Вривые светопогл°Щения ряда диазосоединений в кислой среде
(pH ), Д Диазосоединение существует только как катион диазония,
Рис. 86^ Титрование п-нитрофенплдиазония щелочью и
кислотой при 10° С с разной скоростью:
1 — кривые прямого титрования щелочью; 2 — кривые обратного
титрования кислотой; 3 — равновесная кривая.
показывают, что этот катион характеризуется двумя максимумами погло-
щения (рис. 87). В щелочной среде (pH 11 —12), когда диазоний практиче-
ски нацело превратился в изодиазогидрат, на кривой поглощения обнару-
живается характерный для изодиазогидрата единственный максимум
Рис. 87. Кривые поглощения водных рас-
творов п-нитродиазобепзола при раз-
ных значениях pH:
1 — рН4; 2 — рН7; 3 — рН7.38; 4 — рН7,67;
5 —рН7,80; е — рН7,98; 7—рНИ.О.
Рис. 88. Кривые поглощения водных рас-
творов дпазосоединений при рНИ—12:
j __п-нитрофснилдиазоний; 2 — п-диазобензол-
сульфокислота; з — феннлдиазоний; 4 — о-мето-
ксифснилдиазоний; 5 — о-нитро-п-хлорфенилди-
азоний.
<рис. 88). В промежуточной области (pH ~ 7-8) кривые ясно демонстри-
руют наличие в растворе одновременно катиона диазония и изодиазогид-
рата (рис. 87) и отсутствие какой-либо третьей формы.
102
ДИАЗОСОЕДИНЕНИЯ
Е. Мюллер, впервые получивший неустойчивые первичные нитроз-
амины, действуя при —70° С хлористым нитрозилом на первичные аро-
матические амины, превратил этин итрозамипы действием этилата калия
в твердые диазотаты калия:
ArNHa 4- C1N=O ---> ArNIIN = O + HCl
ArNHNO + C2H5OK ----> ArN = NOK
Он установил разную в случае разных Аг способность таких диазо-
татов к азосочетанию, а именно меньшую для NO2CeH4—N = NOH (I),
чем для C6H5N = NOH (II) и поэтому считал, что диазотат (I) это «нор-
мальный» ганчевский, а диазотат (II) — изодиазотат. Однако воочию две
стереоизомерные формы структурно одного и того же диазогидрата или
диазотата металла никогда не были констатированы.
Таким образом, приходится прийти к выводу, что стереоизомерия
арилдиазосоединений на примере диазогидратов и их солей твердо не уста-
новлена и что в данное время нет оснований приписывать единственной
известной форме диазогидратов или диазотатов ту илу иную конфигу-
рацию.
Ганч приписал стереоизомерию диазосоединениям, связанным не только с ОН“,
но и с другими анионами слабых кислот, например CN*. Эти арилдиазоцианпды изу-
чались многими исследователями. По-видимому, здесь не существует стереоизомерии,
а два ряда диазоцианидов структурно изомерны между собой. Последнее установлено
О. А. Реутовым и Л. А. Казицыной путем ИК-спектроскопического исследования двух
рядов изомерных арилдпазоцианидов. При сливании на холоду (—15° С) растворов
арилдиазониевой соли и цианистого калия выделялись «лабильные» арилдиазоцианпды
с частотами ИК-спектра, близкими к частотам изонитрилов (-—2150 см'1). При хранении
эти «лабильные» диазоцианиды превращались в более высокоплавкие «стабильные»
продукты с частотами, близкими к частотам связи C^N (на 40 с.н-1 выше, чем
у «лабильных» продуктов). На этом основании О. А. Реутов п Л. А.Казицына заклю-
чили, что «лабильные цианиды» являются изоцпанидами и что обоим рядам веществ
принадлежат следующие изомерные структуры:
Ar—N=N-C=N: Аг—N=N—NsC:
стабильные лабильные цианиды
цианиды (изоцианиды)
Частоты ИК-спектров связи C=N в нитрилах обычно действительно выше, чем
в изонитрилах. Правда, разница в частотах больше найденной (40 см'1) и равна при-
мерно 100 см*1, а частота стабильных арилдиазоцианпдов несколько ниже, чем
обычно у нитрилов, но это может быть объяснено сопряжением, уменьшающим «крат-
ность» связи C=N.
Неясности в отношении строения диазосоединений, существовавшие
более 100 лет, были вызваны малой устойчивостью этих соединений,
вынуждавшей работать с растворами веществ часто при неустановившемся
равновесии, и невозможностью нормальной индивидуализации твердых
пР°Дуктов вследствие их быстрого разложения. Более устойчивы в твер-
дом состоянии лишь диазониевые соли, структура которых не вызывает
сомнений, и только что^упомянутые цианиды.
реакции без выделения азота
103
Ароматические диазосоединения широко применяются в лабораторных
и промышленных синтезах, основанных на их богатой реакционной спо-
собности.
Различают два типа химических превращений арилдиазониев и арил-
диазосоединений: без выделения азота и с выделением азота.
Реакции диазосоединений без выделения азота
1. При восстановлении диазосоединений солями сернистой кислоты перво-
начально образующийся арилдиазосульфонат восстанавливается в арил-
гидразинсульфонат, который гидролизуется в кислой среде в соль фенил-
гидразина:
О
СвНб—N=N + 'SO3Na —> С6Н6—N=N—S—ONa - H,!h,so?° ”
О
О
-—> С6Н6—NH—NH—S—ONa - - ,S°4i н,° -» CeH5—NH—NH3 -SO4H + NaHSO4
II
О
Этим путем фенилгидразин получают в заводском масштабе.
2. При действии брома на раствор соли арилдиазония оседает пер-
бромид, который с аммиаком дает фенилазид:
C3H5-N=N Вг- + Вг2 -> C6H5-N=N Вгд +4NH-3+ CeH5-N=N=N4-3NH4Br
3. При взаимодействии арилдиазониевых солей с первичными аро-
матическими аминами получаются диазоаминосоединения, называемые
также тприазенами:
АГ—N=N + H2N—Аг' >• Ar—N=N—NH—Аг'+ Н+
Триазенам свойственна трехазотная таутомерия:
Ar—N=N—NH—Ar' Ar—NH—N=N—Аг'
Равновесие смещено в сторону того соединения, в котором протон
связан с более основным азотом.
Диазоаминосоединения образуются также при действии галоидного
арилмагния на арилазиды:
Ar—N=N=N4-Ar'MgBr ► Ас— N=N— N—Аг'
MgBr
—MgBrOH * N—N NH Аг
на
Этой
При
соль
реакцией могут быть получены и алифатические триазены.
действии сильных кислот диазоамипосоединения расщепляются
диазония и первичный амин:
Ar-N=N-NII-Ar' + HCl —> Ar—N=N Cl- + Ar'-NH2
104 ______________________________________ДИАЗОСОЕДИНЕН И Я
Практически вследствие таутомерии ацидолиз идет в обоих возможных
направлениях и образуется также ArNH2 и Ar'N N СГ.
Эта реакция позволяет получать соли диазопия в тех случаях, когда
диазотирование невозможно или амин недоступен. Так, одним из авторов,
В. А. Сазоновой и В. Н. Дроздом были получены диазосоединения фер-
роцена (о ферроцене см. ч. II, стр. 481), который не выдерживает
действия азотистой кислоты. Реакция идет по схеме (Fc — ферроцен):
FcBr + NaN3 Fc-N=N=N ->
—>• Fc-N-N—N-Cells Fc—N=N—NH-CeHs
MgBr
--> Fc—N=N Cl- + CeH5NH3 Cl-
При нагревании с солью амина диазоаминосоединения претерпевают'
перегруппировку в аминоазосоединения. Например, диазоаминобензол
при нагревании с раствором солянокислого анилина превращается в
п-аминоазобензол:
CeH5-N=N-NH-CeHB -C,H,N-P,C1~-+ ceII5-N=N-CeH4-NH8
В отличие от бензидиновой и других интрамолекулярных перегруп-
пировок превращение диазоаминобензола в аминоазобензол — интер-
молекулярная перегруппировка. Она протекает с ацидолизом диазоамино-
соединения на диазоний и амин, которые и вступают в реакцию азосоче-
тания (см. ниже):
CeH5-N=N-NHCeH5 + CeH6NH3 Cl" --> CeH5-N=N Cl- + 2CeH5NH2
CeH5NH2 + CeH5—N=N Cl’ > CeH5—N=N-CeH4NH2-ra
«-Аминоазобензол, содержащий в качестве одной из функциональных
групп уже знакомую нам азогруппу, — простейший азокраситель. Это
слабое основание: pJfa сопряженной его кислоты 4-2,82, рАГа анилина
4-4,6. Таким образом, введение в пара-положение анилина азогруппы
понижает силу основания на два порядка.
4. При действии солей арилдиазониев на ароматические соединения,,
содержащие очень сильные ориентанты первого рода (NH2, NHR, NR2>.
ОН), происходит реакция азосочетания и образуется оксиазо- или амино-
азосоединение:
Аг—N=N
ОН + н
+
Ar-N=N +
----> Al N=N-A^O^>—N(CH3)2 + H +
Азосочетание — новый пример электрофильного замещения в бензоль-
ном ядре (фенола или амина). Замещение происходит по преимуществу
РЕАКЦИИ БЕЗ ВЫДЕЛЕНИЯ АЗОТА
405
в пара-положение, и лишь если оно занято — в орто-положение. Такое
предпочтение пара-положения обычно объясняют тем, что в продукте
реакции (и в переходном состоянии) возникает более длинная, чем в слу-
чае орто-положения, цепь сопряжения л-связей. При этом уровень сво-
бодной энергии в переходном состоянии ниже и, как следствие, скорость
реакции больше.
Реакция азосочетания широко применяется в технике для приготовле-
ния сотен марок азокрасителей, дающих наибольшее разнообразие среди
всех типов синтетических красителей.
Диазосоединение, вступающее в азосочетание, называют первой компо-
нентой или диазосоставляющей, а ароматический фенол или амин —
второй компонентой или азосоставляющей. Чем более сильные ориентанты
второго рода находятся в орто- или пара-положениях в диазосоставля-
ющей, тем быстрее и энергичнее проходит азосочетание. Диазотирован-
ный 2,4-динитроанилин и 2,4,6-тринитрофенилдиазоний (из пикрамида)
вступают в азосочетания даже с полиметилбензолами. Например:
Реакция азосочетания осуществляется и с жирными азосоставляющими,
если они содержат подвижный метиленовый водород, подобно малоно-
вому эфиру, ацетоуксусному эфиру и циклопентадиену (по существу
реакция совершается между катионом арилдиазония и анионом жирного
соединения, т. е. требуется щелочная среда для отрыва протона от мети-
леновой группы):
| ОС2Н5 А1—N = N +"СН - 1 /ОС2Н5 (X хо Аг—N = N + СН 1 /ОС2Н5 cf чо Ar—N=N + (OJ ' .О | ОС2Н5 — >- Ar—N = N—СН 1 ,ос.>н5 ^0 | СН3 — > Ar—N=N— СН 1 /ОСоН5 С. — >- А1—N=N—
106
ДИАЗОСОЕДИНЕНИЯ
Азосочетание с соединениями, содержащими активную метиленовую
группу, осложняется последующим перемещением второго водорода этой
группы к азоту, соседнему с ароматическим радикалом, и превращением
азосоединения в арилгидразон. В случае малонового эфира получается
арилгидразон эфира мезоксалевой кислоты, а в случае циклопентадиепа —
арилгидразон циклопентадиенона:
qz/ С"
I ^ОСгНй |ХОС2Н5
Аг—N=N—СН — -> Аг—NH—N=C
1 /OC,Hs % 1 /ОС2Н5 Ч
Аг—N=N
Ar—NH—N=
В случае ацетоуксусного эфира в щелочной среде происходит кислот-
ное расщепление (кн. I, стр. 417). Азопроизводное ацетоуксусного эфира
(в этом примере замещенное на R) в щелочной среде подвергается кислот-
ному расщеплению и превращается в арилгидразон эфира а-кетоноки-
слоты, восстановлением которого можно получить эфир а-аминокислоты
(метод В. В. Феофилактова):
О
I ХСН3
Аг—N=N—С—R
I /ОС2Н5
С<0
СН3—С<^° +Аг—NH—N=C—R
xONa | .ОС2Н5
ч
ч
R
I /ОСзНй
Аг—NH2+H2N—СН—С<
Что касается азосочетания с ароматическими азосоставляющими,
то практически для этой цели применяются лишь фенолы и амины. Фе-
нолы вступают в реакцию в щелочной среде, для аминов предпочитают
пользоваться слабокислой средой. Активность ориентантов первого рода
в этих азосоставляющих, как всегда, убывает в следующей последователь-
ности:
б—Аг > HaN—Аг > НО—«Аг > HgN—Аг
В последнем случае в качестве ориентанта также выступает группа
NH2, но в малой концентрации, так как она образуется за счет гидролиза
соли:
HSN—Аг+НаО —> HaN—Аг + Н3б
РЕАКЦИИ БЕЗ ВЫДЕЛЕНИЯ АЗОТА
Практически применяемые азокрасители содержат обыкновенно две
и более азогруппы. Для введения нескольких Uorpynn и получения
бис-, трис- и тетракис-азокрасителей существуют разные способы. Ограни- '
чимся здесь отдельными примерами синтеза известных азокрасителей,
что даст понятие и о введении нескольких азогрупп:
-N + <<Э>-ГДСНз)2
2
3
> HO3S-\O>-N=N_^(
метиловый оранжевый
простейший краситель типа „конто11
В кислой среде азокрасители присоединяют протон по азогруппе,
что сопровождается перестройкой связей в бензольном цикле, несущем
амино- (или окси-) группу, и изменением цвета (индикаторные свойства):
Полученная соль азокрасителя имеет хиноидную (стр. 159), а не бен-
зоидную структуру. Присоединение протона произошло не к диметил-
аминогруппе, а к азоту азогруппы, в результате реакции с перенесением
реакционного центра, но аммонийным Стал все же азот диметиламино-
₽УНаличие цветности азокрасителей связано с мезомерией их структуры,
выражающейся для приведенного выше примера двумя предельными
структурами:
108
ДИАЗОСОЕДИНЕНИя
Вес первой структуры больше, но, как видно из направления атак
протона, вторая тоже дает себя ясно знать. и
С наличием сопряжения связано сближение основного энергетич
ского уровня азокрасителя и его возбужденного уровня. Поэтому о”
поглощает в более длинноволновой области спектра не только по сравнен
нию с бензолом, но и сравнительно с азобензолом. Этот батохромный
сдвиг, т. е. сдвиг поглощения в длинноволновую область спектра сравни-
тельно с азобензолом, обязан наличию в пара- (или могло бы быть и в
орто-) положении ориентанта первого рода, способного нести положитель-
ный или отрицательный заряд (в данном случае положительный). Такой
заместитель называется ауксохромом. Этот ауксохром «работает» и в смысле
передачи основных свойств (в данном случае отрицательного заряда)
удаленному атому азота, и в смысле батохромного сдвига поглощения
только при наличии сопряженной системы, реализующейся при плоскост-
ном расположении молекулы. Только при этом условии (и в меру плос-
костности) проявляется сопряжение (мезомерия) и его эффекты.
Так, по А. И. Киприанову и сотр. при переходе от красител с А к кра-
сителю Б
А
наблюдается гипсохромный сдвиг (сдвиг поглощения в более коротко-
волновую область спектра) вследствие того, что о-метильнып замести-
тель заставляет диметиламиногруппу вывернуться и тем (частично)
выключиться из системы сопряжения (ср. стр. 86).
Если, однако, диалкиламиногруппу связать в цикл, прочно удержи-
вающий ее в (почти) плоском положении
то, напротив, сдвиг поглощения сравнительно с А будет батохромньь
Сказанное здесь имеет совершенно общий характер для крас
Реакции диазосоединений с выделением азота
Соли диазония и диазосоединения * непрочны и способны расп д
гетер олитически
СвН5—N=N -----> C6H5 + N2
' е. ияк обобщающее для диазо-
* Название «диазосоединения» мы будем употреблят
ниевых солей и собственно диазосоединений.
РЕАКЦИИ С ВЫДЕЛЕНИЕМ АЗОТА
109*
и гомолитически
CeH5-N=N CI’ ---> CeH5-N=N-Cl —► CeH5- + N2 + Cl.
Направление распада зависит от аниона соли диазония и раствори-
теля. Гомолитическому распаду (гомолизу) способствуют в основном
анионы слабых кислот, но в неводных растворах так распадаются даже
хлориды. Растворители с высокой диэлектрической проницаемостыо-
способствуют гетеролитическому распаду. Борфториды арилдиазония рас-
падаются только гетеролитически. Далеко не во всех типах реакций заме-
щения диазогруппы выяснен характер распада. Из приведенных далее
примеров, следует, что, вероятно, он гетеролитический в реакциях 1,2,
4, 6, 11а, 12 и гомолитический в реакциях 3, 5, 7, 8, 9, 10, 11.
Диазосоединения широко используются (особенно в лабораторной
практике) для синтезов, состоящих в замене диазогруппы на различные
атомы и группы. При этом диазогруппа в результате того или другого-
типа распада удаляется в виде N2. В качестве примеров приведены следу-
ющие типы реакций.
1. Замена диазогруппы на гидроксил нагреванием водного раствора
соли диазония:
Аг—N=N + H2O —> ArOH + N2-|-H+
2. Замена диазогруппы на алкоксил нагреванием диазосоединения со
спиртами:
Аг—N=N + ROH — ArOR + N2 + H+
Реакция имеет ограниченное применение, так как сопровождается
замещением диазогруппы на водород по реакции 3, особенно для диазо-
ниев с мета-ориентантами в ядре. Хорошо идет для неокисляющихся
спиртов.
3. Замена диазогруппы на водород осуществляется многими восстано-
вителями и, в частности, первичными спиртами (в этом случае сопернича-
ющей является реакция 2):
О
+ Н+
н
Поэтому лучше элиминировать (т. е. замещать на водород) диазо-
группу, действуя Н3РО2, муравьиной кислотой или станнитом натрия:
ArN=N +Sn(ONa)2 + H2O ► ArII + N2-|-2Na+-|-HSnO5-
Кроме того, гомолитический распад диазосоединений в среде веществ,
содержащих предельный алифатический водород (даже в алканах, кето-
нах, кислотах), ведет к захвату радикалом Аг атома водорода и к образо-
ванию АгН.
4. Замещение диазогруппы на иод непосредственно действием йоди-
стого калия на водный раствор соли диазония (иодиды арилдиазониев
неустойчивы):
ArN=N + CHs—СН2ОН —> ArH + N2 + CH3-C<^
ArN=N4-I- —► Arl + N2
110 -______________________________________ диазосоединения
5. Замещение диазогруппы на хлор, бром, циан и родан, осуществля
емое по реакции Зандмейера. Соли закисной меди и этих анионов разл '
гают соли диазония, причем место диазогруппы занимает анион (У — гч~
Вг, CN или SCN): ~ U’
2ArN=N SO4H- + 2CuX —> 2АгХ 4-2N24-Cu2SO44-H2SO4
Приходится исходить из сернокислого диазония или заботиться о том
чтобы анион галоида в соли диазония совпадал с анионом медной соли'
6. Замещение диазогруппы на фтор разложением при нагревании сухого
комплексного борфторида диазония (Бальц — Шиманн):
ArN=N BFj —> ArF4-N24-BFs
7. Замещение диазогруппы на остаток сульфиновой кислоты дей-
ствием SO2 в присутствии катализатора — порошка меди (Гаттерман):
+ „ /О
ArN=N + SO2 — - Ar-S<f 4-N2
ХО"
8. Замещение диазогруппы на нитрогруппу действием нитрита натрия
в присутствии порошка меди (Гаттерман):
+ п + /О’
ArN=N4-NOj ——Ar-N<^ +N2
9. Замещение диазогруппы на металлы (А. Н. Несмеянов, 3. И. Кан,
К. А. Кочешков, О. А. Реутов, Л. Г. Макарова) восстановлением метал-
лом двойных диазониевых солей с галогенидом металла или диазониевои
соли с анионом BF4:
CeH5N2Cl • HgCl2 + 2Cu —> C6H5HgCl-|-N2 + 2CuCl
2C6H6N2C1 • HgCl24-6Cu ацет*^+ (CeH5)2Hg + 2N2 + 6CuCl + Hg • Аммиак
2C9H5N2 • BF4 + T1 > (CeH5)2TlF-|-2N2-|-2BF3-|-TlF
(CeH5N2Cl)2 • SnCl4+ Zn —> (C6H5)2SnC12 + 2N2 + ZnCl2
2C6H5N2C1 • SbCl3 + Zn —> (CeH5)2SbCl3 + 2N2 + ZnCl2 + SbC13
3CeH5N2 • BF4 + 2Bi -► (C6H5)3Bi + 3N2 + 3BF3 + BiF3
10. Замещение диазогруппы на остаток мышьяковой кислоты (Б р )
CeH6-N^N 4- AsO|“ —> C6H5-As/Q_ 4- ^2
О
11. Реакция Гомберга. При действии диазониевых ®°оС"бдЫе к азо-
ствии ацетата натрия) на ароматические соединени , свяЗЬ1вается УгЛе"
сочетанию, ароматический радикал диазосоедине
РЕАКЦИИ С ВЫДЕЛЕНИЕМ АЗОТА
114
род-углеродной связью с ароматическим ядром второго реагента, замещая
в нем водород. Так образуются производные дифенила (стр. 203).
__ .ОН
Ar—N=N —О—С—СН3
II
О
В схеме приведено лишь замещение в пара-положение, которое дей-
ствительно всегда преобладает независимо от характера ориентанта X,
так как в реакции Гомберга реагентом является свободный радикал
арил Аг. и замещение это не электрофильное, а гомолитическое. Хотя
пара-замещение и преобладает (даже если X — мета-ориентант, подобный
группе NO2), но получаются и орто-и мета-замещенные бифенилы:
О
no2
11а. Если в нитробензоле проводится разложение борфторида фенил-
диазония, промежуточным продуктом реакции является не свободный
радикал арил, а катион арил, и атака на нитробензол электрофильная:
CeH5-N=N BF7 ---> CeHJ+Na+BFj
В качестве преобладающего продукта замещения в ядре нитробензола
образуется л-нитродифенил (А. Н. Несмеянов, Л. Г. Макарова):
NO2
12. При разложении борфторида фенилдиазония в хлорбензоле, бром-
бензоле, иодбензоле и простом дифениловом эфире наряду с реакцией
Гомберга происходит присоединение образующегося фенил-катиона по
свободной паре электронов галоида или кислорода и образуются соли
(борфториды) ониевых катионов — дифенилхлорония, дифенилбромония,
дифенилиодония и трифенилоксония (А. Н. Несмеянов, Т. П. Толстая):
СеН5—N=N BFj + CellsCl —► св115—Cl—Cells BFj-f-N2
CeHs—N=N BFj+CeH6Br ► Cells—Br—Cells BFj-|-N2
CeH6—N=N BFj + CeHs-O-Cells ---->• CeH6—6-CeII5 BFj + N2
CeH5
112
фЕНОЛЫ
Полученные соли опиевых катионов растворимы в воде и таким обпа
зом могут быть извлечены из реакционной массы и выделены, хотя они
получаются с невысоким выходом (—6%).
13. Разложение диазосоединений в а, ^-непредельных кислотах, кето-
нах и т. д. ведет к присоединению арила и аниона диазосоединения ц0
двойной связи (Меервейн — Кельш) с нарушением правила ориентации
электрофильного присоединения (кн. I, стр. 395), так как реакция течет
гомолитпчески:
Аг—N=N Cl’ > Аг—N=N—Cl > Аг« 4- N2 +С1-
/ОС2Н5 /ОС2Н5
Аг« 4-СН3—СН=СН—Cg +С1. > СНз—СН—СН-С4
^0 I I %
С6Н5 С1
Разложение же борфторида арилдиазония приводит к обратному
лорядку присоединения (А. Н. Несмеянов, Л. Г. Макарова):
♦ /ОС2Н5
Ar—N=N BF7-4-CH3—СН=СН-С<
^0
/ОС2Н5
--> СНз— CH-CH—cd +n2 + bf3
I I ^0
F C6H5
В этом случае реакция осложняется последующим отщеплением HF:
/ОС2Н5 /ОС2Н5
СНз-СН—СН-СС ---------> СН3-СН=С-СЧ\ +HF
I I X) |^0
F С6Н5 СвН5
14. Естественно, что сама диазониевая группировка — сильный элек-
-троноакцептор, оттягивающий электроны из ядра и активирующий о
и n-углероды по отношению к нуклеофильным атакам. Описаны реакции
такого типа:
Вг
<+ С2Н5ОН
NHN СГ -------->
Вг
Вг
,_(
CI—(О/ + СН3—+ N2 + НВГ
--\ н
Вг
Замещение Вг на G1 — результат присутствия группы —-V — N, и совер-
шается оно до замещения последней на водород.
ФЕНОЛЫ
Термин «фенолы» происходит от старинного названия бензола «фен»,
введенного Лораном (1837 г.), и обозначает арОхматическое вещество^
-содержащее гидроксил, связанный непосредственно с углеродом арома
ческого ядра.
Физические свойства фенолов и некоторых их производных приведе
в табл. 65.
8 Заказ 672
Таблица 65- Фенолы
Формула Название Темпера- тура плавлении °C Температура кипения °C
СвН5ОН СН.зС6Н4ОН фенол К резол 41 182
о-, или 1,2- 30 191,5
или 1,3- И 202,8
С1С0П4ОН п-, или Хлорфенол 1,1- 36 202,5
о-, или 1,2- 7 (а-форма) 0 (3-форма) 175,6
Л£-, ИЛИ 1,3- 32,8 214
NO2CeH4OH п-, или Нитрофенол 1,4- 43 217
о-, или 1,2- 45 214,5
М-, или 1,3- 96 194 (при 76 Л1Л1 рт. ст.)
nh2c6h4oii п-, или Амннсфенол 1,4- 114 279 (разл.)
о~, пли 1,2- 171 Возг.
Л1-, или 1,3- 123 —
п-> или 1,4- 184 (разл.) Возг.
Плотность Показатель преломления 20 nD *а РКа Дипольный момент |>- в бензоле при 25 °с D
при 25 °C
1,072 1,5425 (при 40° С) 1,3- 10-W (при 20° С) 9,89 1,53
1,0465 1,5453 6,3 • 10-11 10,20 1,41
1,034 1,5398 9.8-10-Ц 10,08 1,54
1,035 1,5395 6,7 • 10-И 10,17; 10,26 1,57
1,241 (при 18° С) 1,5473 (при '10° С) 7,73-10-9 9,11 1,30
1,268 (при 25° С) 1,5565 (при 40° С) 6,64 - 10"9 9,08 2,10
1,306 1,5579 (при 40° С) 6,28 • 10-9 9,42 2,22
1,657 — 5,9-10-8 7,21 3,10
г- 1,485 — 4,5-10-9 8,39 3,90
1,479 — 7,08 • IO'8 7,16 5,01
,— — 2,18 • 1()-ю 9,7 —
— —- — —
— — 6.6- IO’9 (при 15° С) 8,16
ФЕНОЛы
114 __________________________________________________
Как уже говорилось, каменноугольная смола содержит простейший
из фенолов — оксибензол (собственно, фенол) и его гомологи: о-, м- и п_
крезолы в тем большем количестве, чем ниже температура коксования
угля. Дополнительные количества фенола, мировое потребление которого
достигает миллионов тонн, получаются из бензола. Для этого исполь-
зуется (все в меньших масштабах) старый метод щелочного плавления соли
бензосульфокислоты:
C6H5SO3Na + NaOH —> CeH5OH + Na2SO3
Некоторое количество фенола получают гидролизом хлорбензола пере-
гретым паром (450—500° С) над катализатором — силикагелем, промоти-
рованным ионами Сн2+ (Рашиг):
С«Н.С1 + Н2О <”*-> С.Н5ОН + НС1
Наибольшие перспективы развития имеет разложение перекиси кумола
(изопропилбензола) разбавленными кислотами. Процесс состоит в следу-
ющем:
н+
+ сн3—сн=сн2---►
ООН
I
сн3—с—СНз
II
о
Механизм разложения перекиси приведен в разделе «Перегруппи-
ровки», ч. II, стр. 624.
Путь введения гидроксила на место диазогруппы
+ No + Н +
неэкономичный для получения самого фенола, используется для синтеза
чистых о-, м- и n-крезолов, гваякола (см. далее) и других фенолов. Техни-
ческий крезол (смесь трех изомеров) получают из каменноугольной сМ0Л^;
Фенол — слабая кислота с константой диссоциации при комнатн
температуре в водном растворе 1,3-10~10 (константы диссоциации дрУ1
фенолов приведены в табл. 65). ке
Таким образом, он на несколько порядков кислее воды, не_говоряУ'^
о жирных спиртах, но гораздо слабее уксусной кислоты (1,8-10 )• ,
умеренно растворим в воде (8% при 15° С). Вода растворяется в <pe
с образованием жидкого при комнатной температуре раствора.
РЕАКЦИИ ГИДРОКСИЛА ФЕНОЛОВ 11 •'
фенол — бесцветное легкоплавкое (+zil° С) кристаллическое вещество,
вследствие окисления розовеющее на воздухе. Крезолы менее, чем фенол,
растворимы в воде, подобно фенолу хорошо растворимы в эфире, спиртах,
хлороформе, бензоле.
Фенолы хорошо растворяются в водных растворах щелочей в резуль-
тате образования фенолятов щелочных металлов:
АгОП + NaOH ArO“ Na+-|-TI2O
Гидролиз фенолята (обратная реакция) вследствие слабости кислот-
ных свойств фенола заходит далеко, и требуется избыток щелочи, чтобы
сместить равновесие вправо. Уже двуокись углерода выделяет фенол из
раствора фенолята.
Кислотные свойства фенольного гидроксила вызваны мезомерным
взаимодействием с ароматическим ядром, что выражается символами:
Валентные электроны атома кислорода (в том числе и связывающие
водород с кислородом) оказываются частично рассредоточенными в орто-
и пара-положения бензольного ядра, а водородный атом гидроксила —
протонизированным. Таким образом, большая кислотность фенола (срав-
нительно со спиртами) — это другая сторона сильного орто-пара-ориен-
тирующего действия гидроксила в реакциях электрофильного замещения.
Отношения здесь вполне похожи на отношения в енолах, более кислых,
чем спирты, и в енолятах с их б--зарядом на втором углеродном атоме
(соответствует 6—заряду на о-углеродах фенолята).
Реакции гидроксила фенолов
1. Образование фенолятов (см. выше).
2. Образование простых эфиров фенолов алкилированием фенолятов
ArONa-|-RI ------------------> ArOR-j-Nal
ArONa + (CH3O)2SO2 —> ArOCH3 + CH30-S02ONa
3. Образование сложных эфиров фенолов (в отличие от сложных эфи-
ров спиртов) не может быть достигнуто взаимодействием их с кислотами,
а только ацилированием фенолов (лучше в щелочной среде) галоидангидри-
дами или ангидридами кислот:
ArONa + Cl—C-R —> АгО-С—R-1-NaCl
II II
о О
о
'"C-R
ArONa + O -—> ArO-C—R-l-R—C-ONa
8*
116
ФЕНОЛЫ
4. Замещение гидроксила на хлор при действии РС15, протекает
гораздо труднее, чем для спиртов, и с плохим выходом. В этом случае
происходит главным образом хлорирование в ядро, причем РС15 превра-
щается в РС13. С РС13 в малой степени идет замещение гидроксила на
хлор, а в большей степени — образование трифенилфосфита (эфира фос-
фористой кислоты). С хлорокисью фосфора РОС13 образуется фениловый
эфир фосфорной кислоты.
5. При перегонке с цинковой пылью фенолы превращаются в угле-
водороды:
АгОН + Zn -► АгН + ZnO
Реакции ароматического ядра фенолов
Гидроксил — один из сильнейших, а в щелочном растворе сильнейший
орто-пара-ориентант. В соответствии с этим для фенолов легко проходят
реакции электрофильного замещения.
Галоидирование фенолов. В неводной среде галоидирование фенолов
при соответствующем соотношении реагентов приводит к смеси о- и п-
галоидфенолов, далее к 2,4-дигалоидфенолам и, наконец, к 2,4,6-трига-
лоидфенолам (их лучше получать в водной щелочной среде). В случае
орто- и пара-замещенных фенолов, например крезолов, занятые замести-
телем (например, метилом) места галоидированием не затрагиваются.
Бромирование фенола избытком бромной воды проходит по схеме:
ОН ОН
Вг
Поучительно образование «трибромфенолброма»
циклогексадиенона, который долго считали эфиром
лоты:
— 2,4,4,6-тетрабром-
бромноватистой кис-
ОВг
Вг
Ориентирующая сила гидроксила, т. е. сообщение гидроксилом o;J
фильной активности n-углеродному атому, такова, что этот у
и после замещения связанного с ним водородного атома сп брома,
принять электрофильную атаку электроположительного ат
РЕАКЦИИ АРОМАТИЧЕСКОГО ЯДРА ФЕНОЛОВ
117
Присоединение второго атома брома закрепляет циклогексадиеновую
структуру:
+ НВг
О
Обычный таутомерный переход в фенольную форму из-за отсутствия
в пара-положении водородного атома для трибромфенолброма невозможен.
Реакцию бромирования интересно сравнить с описанным на стр. 37
течением реакции метилирования бензола по Фриделю — Крафтсу, когда
сверх шести метильных групп, заместивших водороды бензола, способна
войти в соединение седьмая, образуя устойчивый аналог промежуточного
о-комплекса электрофильных замещений.
Реакция образования трибромфенолброма количественная и применяется для
определения фенолов. Сначала ведут бромирование избытком брома, затем добавляют
иодистый калий и оттитровывают выделившийся под. Трпбромфенолбром отдает при
этом свой четвертый бром:
ОН
ВгААвг вг\А/Вг
+2Г + На0 —> +Вг- + 12 + ОН-
Вг^ Вг Вг
Расход брома составляет шесть атомов на молекулу, фенола (или л-крезола) и
четыре атома на молекулы о- и n-крезолов п вообще орто- и нара-замещенных фенолов.
Другие 2,4,6-трехзамещенные фенолы, например триалкилфенолы, при бромиро-
вании также дают 4-бром-2,4,6-триалкилциклогексадненоны:
ОН О
r\A/r r\A/r
I^JI, +Вг2 ----► || || +НВг
I /\
R R Вг
Очевидно, что при йодометрическом титровании расход брома в этом случае будет
определен как равный нулю. J
Введение галоида is орто- и пара-положения повышает кислотные
свойства фенолов.
______________-___________________________________________ФЕ П олц
о- и п-Хлорфенолы (см. табл. (>5) легкоплавкие вещества с очен
навязчивым, интенсивнььм карболовым запахом. Они обладают еще более
сильными, чем фенол, дезинфицирующими свойствами. Висмутовая соль
2,4,6-трибромфенола в виде мазей пли присыпок употребляется как наруж-
ное бактерицидное средство.
Простой эфир 2,4-дихлорфенола и гликолевой кислоты, получаемый
действием на 2,4-дихлорфенолят натрия монохлоруксусной кислотой
о—сн..
+ С1СН,—С —ONa
“ II
О
С — ONa
применяется как гербицид при борьбе с сорняками (2,4-дпхлорфенокси-
уксусная кислота, или 2,4-D).
Сульфирование фенолов. Сульфирование фенола при комнатной темпе-
ратуре дает в основном о-фенолсульфокислоту, при 100° С получается
n-изомер, а в более жестких условиях — 2,4-фенолсульфокислота.
Нитрование фенолов. Для получения мононптрофенолов приходится
нитровать фенолы на холоду разбавленной азотной кислотой (—30%-ной),
лучше всего получаемой смешением водного раствора селитры с серной
кислотой (чтобы избежать присутствия окислов азота). Образуется смесь
о- и n-нитрофенолов, из которой о-нитрофенол удаляют отгонкой с водя-
ным паром, а и-изомер выделяют кристаллизацией. .ч-Изомер приходится
готовить обходным путем, например из ж-нитроанилина через at-нитро-
фенилдиазоний. 2,4-Динитрофенол проще всего получить гидролизом
2,4-динитрохлор бензол а.
Тринитрофенол, называемый пикриновой кислотой, производят в про-
мышленном масштабе, нитруя крепкой нитрующей смесью 2,4-фенолди-
сульфокислоту, получаемую сульфированием фенола, без выделения ее
из сульфурационной массы. При этом нитруется не только свободное
шестое положение, но и сульфогруппы замещаются на нитрогруппы —
явление, наблюдаемое и в ряде других случаев. Наличие в феноле сульфо-
групп защищает его и от окисления и от действия окислов азота. Пикри-
новую кислоту можно получить и прямо из бензола, если нитровать его
в присутствии соли ртути — катализирующей окисление промежуточно
образующегося ж-динитробензола до 2,4-динитрофенола, который далее
превращается в пикриновую кислоту.
Свойства нитрофенолов. Введение нитрогруппы в орто- и пара-положетп
значительно повышает константу диссоциации фенола; меньшип апид^Ф^
цирующий эффект оказывает мета-нитрогруппа.
в шесть раз более сильная кислота, '
очень сильная кислота:
Ла
Фенол.........1,3-10"10
о-Ннтрофенол . . 5,9-10"8
и-Нптрсфопол . . 7,08-10-8
чем уксусная, <
А'а
4,8 • 10“9
. 1-Ю"4
1,6- 10"!
м- Нитрофенол . .
2,4-Динптрофенол .
Трннптрофенол .
РЕАКЦИИ АРОМАТИЧЕСКОГО ЯДРА ФЕНОЛОВ
119
Согласованное действие на бензольное ядро обоих ориентантов (ОН
и NO2), находящихся в орто- или пара-положениях друг к другу, выра-
жается схемой:
Щелочь связывает протон гидроксила и позволяет осуществиться
сдвигу электронов в тенденции, обозначенной стрелками. Поэтому нитро-
феноляты натрия по существу уже не имеют строения фенолятов: заряд
аниона в них сосредоточен на нитрогруппе, а не на кислороде бывшей
гидроксильной группы, и ядро имеет хиноидное расположение двойных
связей:
в твердом виде
красный
желтый
Такое перемещение связей ведет к резкому изменению области свето-
поглощения и обусловливает свойства нитрофенолов (а также динитрофе-
нолов) как индикаторов концентрации водородных ионов.
В какой-то малой степени переход протона от гидроксила к нитро-
группе, сопровождаемый хиноидным перемещением связей, происходит
и в самих нитрофеполах, особенно в растворителях, способных, подобно
воде, связывать протон, т. е. речь идет о таутомерии:
ОН
120
фЕНОЛЫ
Для о-нитрофенола получены изомерные эфиры:
осн..
no2
О"
окрашенный
осн3
бесцветный
о-Нитрофенол довольно резко отличается от своего пара-изомера и от
самого фенола тем, что он не ассоциирован в углеводородных растворах.
Фенол, как и все гидроксильные соединения, ассоциирован за счет водо-
родных связей гидроксильных групи, а п~ нитрофено л, кроме того, еще за
счет водородных связей между гидроксилом одной молекулы и нитро-
группой другой:
В о-нитрофеноле водородная связь осуществляется внутри молекулы,
о
/ х
Н
К
*0
поэтому гидроксил уже не способен к образованию водород
ных связей «на стороне». Такое особое строение о-нитрофе-
нола обусловливает и его лучшую растворимость, более низ-
кую температуру плавления, летучесть с водяным паром
(молекулы химически не связаны друг с другом), а также
окраску.
Пикриновая кислота — сильная кислота. С множеством даже слабых
оснований (например, алкалоидов) она дает хорошо образованные кри-
сталлические соли, что широко используется при выделении, характери-
стике и анализе оснований. Этим дело, однако, не ограничивается, так как
и с конденсированными ароматическими углеводородами, такими, как
нафталин и антрацен, пикриновая кислота также образует кристалличе-
ские «молекулярные» соединения (Фрицше) в отношении 1:1. Это свойство
уже не связано с кислотностью пикриновой кислоты, так как подобные
соединения образуют все тринитропроизводные, например тринитробен-
зол с нафталином. В таких молекулярных соединениях одна молекула
является донором электронов, другая акцептором (см. ниже о соедине-
ниях с переносом заряда).
Пикриновая кислота ранее применялась как желтый краситель. Как
все ароматические тринитросоединения, она и ранее (впервые в русско-
японскую войну 1904 г.) находила применение в качестве бризантного
взрывчатого вещества под названиями меленит, лидит, шимоза. Впослед-
ствии она была вытеснена тротилом, так как ее кислотные свойства соз-
дают ряд неудобств — коррозия металла, образование солей, крайне чув-
ствительных к удару. Некоторое применение как взрывчатое вещество
находит также соль пикриновой кислоты — пикрат аммония.
МОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ С ПЕРЕНОСОМ ЗАРЯДА
121
МОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ С ПЕРЕНОСОМ ЗАРЯДА
Существуют соединения 2,4,6-тринитробензола с нафталином и другими
конденсированными ароматическими углеводородами, а также с мезити-
леном (1,3,5-триметилбензолом) и вообще с высокоалкилироваппыми бен-
золами, подобные только что упомянутым пикратам ароматических угле-
водородов. Все они имеют молекулярный состав 1 : 1 и отличаются более
глубокой и интенсивной окраской, чем исходные соединения. Обычно
в этих молекулярных соединениях плоскости обеих ароматических соста-
вляющих молекул расположены одна над другой. В настоящее время
установлено, что способ связи двух молекул между собой таков, что одна
из них (конденсированное ароматическое соединение или алкилбензол)
передает па орбиталь другой (питросоедипения) в полное или совместное
обладание один из своих л-электронов. Такие молекулярные соединения
называются соединениями, с переносом заряда (charge transfer compounds).
В некоторых случаях они образуются только в растворе при смешении
компонентов, и за процессом можно количественно проследить спектро-
фотометрически вплоть до установления равновесия. В других случаях
они образуют прекрасные кристаллы. Способность вступать в такие соеди-
нения не является привилегией только ароматических молекул, но свой-
ственна и другим, имеющим л-связи веществам, если партнерами будут
одна высокодонорная, а другая высокоакцепторная молекула. В качестве
примера высокоакцепторной молекулы в жирном ряду можно назвать
тетрацпапэтплен, который при смешении уже с бензолом образует ярко-
оранжевый комплекс. В свое время И. И. Остромысленский предложил
те.транитрометап как реактив на двойную связь, так как при смешении его
с непредельными соединениями получается желтый раствор, по-видимому,
вследствие подобного же явления.
Наконец, описанные в ч. II (стр. 471) л-комплексы металлов, начиная
с л-комплексов катиона серебра с олефинами
А '
^с==с^
также в известной степени представляются аналогами соединений с.пере-
носом заряда. В них донором является л-связь, акцептором — катион
металла. Такие же аналоги — нестойкие промежуточные л-комплексы
олефинов или ароматических соединений с атакующими электрофильными
частицами, о которых шла речь при рассмотрении реакций олефинов
(ки. I, стр. 265) и в начале раздела, посвященного реакциям аромати-
ческих соединений (см. стр. 32). Принципиальное отличие описыва-
емых здесь соединений заключается в том, что донорами и акцепторами
заряда служат здесь две органические молекулы.
Питрозированис фенолов. При действии водного раствора азотистой
кислоты фенол нптрозируется в пара-положение:
НО^О) +
но — N=O
но
N=O
122
ФЕ НОДЫ
Ннтрозофенол таутомерсн монооксиму /г-бензохинона (о хинонах
стр. 159): ' UI
Наличие нитрозофенол — хиноноксимной таутомерии и константу
таутомерного равновесия в разных растворителях можно выяснить из
спектров поглощения растворов нитрозофенола. Для нитрозофенольной
формы характерен максимум поглощения в области 730 ммк. По величине
коэффициента экстинкции этого максимума судят о содержании нитрозо-
фенольной формы.
Азосочетанпе фенолов как реакция электрофильного замещения уже
была разобрана. Она протекает в слабощелочной среде по схеме:
Ar—N = N + Ar—N=N—ОН
Если в пара-положении имеется заместитель, азогруппа вступает
в орто-положение.
Электрофильные замещения в фенолах с образованием углерод-углеродной
связи. Таких реакций известно много. Поскольку они приводят к полу-
чению бифункциональных соединений, которым будет уделено особое
внимание, реакции эти рассмотрены в разделе ароматических феноло-
кислот, фенолоальдегидов и фенолоспиртов. Здесь мы лишь коротко
упомянем некоторые из них.
При нагревании фенолята натрия в токе СО2 образуется салицилово-
кислый натрий (реакция Кольбе):
При действии на фенолят натрия (избыток щелочи) четыреххлористого
углерода также образуется салициловокислый натрий, а при действий
хлороформа — салициловый альдегид (реакция Реймера Тимана, под
робнее на стр. 139):
ON а /Ч. Na( OJ + ссц ONa A f [О] + СНС13 он ON а + NaCI + Н2° он /Н ,1аОН > [Q^ + NaCI + Н2о
МОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ С ПЕРЕНОСОМ .ЗАРЯД*
123
Действием олефинов на фенолы в присутствии льгоисовых кислот полу-
чают n-алкилфенолы (частный случай реакции Фриделя — Крафтса);
он он
(^] + rch=ch2 ZnC12 > ю)
. RCH—СН3
С синильной кислотой (или нитрилами) в присутствии хлористого
водорода фенолы дают иминоальдегидофенолы или иминокетонофенолы
(реакция Геша), а после гидролиза иминогруппы получаются сами окси-
оксосоединения:
ОН
(Х=Н,арил или алкил)
Наиболее важная реакция этого рода — реакция фенолов с формаль-
дегидом, которая протекает в присутствии как кислот, так и щелочей.
При нагревании фенола (избытка) с формалином и серной кислотой проис-
ходит бурная реакция и образуется растворимый в спиртах, ацетоне
п сложных эфирах полимер линейного строения — «новолак». При щелоч-
ной конденсации фенола с избытком формалина сначала образуется легко-
плавкий сравнительно низкомолекулярный полимер «резол», подобно
новолаку растворимый в органических растворителях. Это — так назы-
ваемый термореактивный полимер: при нагревании происходит дальней-
шая конденсация свободных оксиметиленовых групп с образованием
метиленовых мостов, и полимер приобретает сетчатую структуру. Полу-
чаемый «резитол» нерастворим в органических растворителях, но сохра-
няет некоторую пластичность. При нагревании до 150 С конденсация
идет дальше и получается химически очень устойчивый, неплавкий и нера-
створимый полимер — «резит», который можно нагревать до температуры
—300° С. Таковы три стадии процесса конденсации, объединяемые назва-
нием «бакелитизация» (по имени изобретателя бакелита -- Бакеланда).
Обычно резол перед последующей стадией конденсации смешивают с напол-
нителем (минеральным типа асбеста или органическим типа древесины,
лигнина, целлюлозы) или пропитывают им древесину пли волокнистые
материалы и затем подвергают дальнейшей баке.тнтнзацнп. Этот открытый
в 1909 г. тип феполо-формальдегидных пластмасс и в настоящее время
сохранил свое значение.
124
----------------------------------------- ФЕНоды
Большая часть добываемых фенола и крезолов потребляется для
чепия феноло-альдегидпых полимеров. Химический смысл
процессов выражается следующей примерной схемой:
ПОЛу-
„ротекаю1цИх
Таким образом происходит постепенное «сшивание» метиленовыми
мостами все большего количества молекул фенола в хаотически построен
ные макромолекулы резола, резитола и, наконец, резита. Химическая
стойкость резита объясняется не столько тем, что значительное коЛИ
чество активных орто- и пара-положений фенола замещены метиленовым
группами, сколько тем, что вследствие полной нерастворимости бакелит
реагенты могут действовать на него только с поверхности.
Алифатические кетоны в кислой среде реагируют с фенолом, оора
ди-п-оксифенилалканы:
СПз
СН3СОСНз + 2СвН5ОН НО—С \J/~011 + Н2°
СНз
Такой 2,2-бис-(4'-оксифенил)-пропан (так называемый Дифенилолпр^
пан) применяется в синтезе пластмасс повышенной те ло
АРОКСИЛЫ
125
чаемых путем этерификации фенольных гидроксилов ароматическими
двухосновными кислотами типа терефталевой.
АРОКСИЛЫ
Фенолы, алкилированные в оба орто-положения и в пара-положение, осо-
бенно третичными алкилами, могут быть окислены окисью серебра или
феррицианидом калия в ароксилы — вещества, обладающие свойствами
свободных радикалов (Е. Мюллер):
С(СН3)3 С(СН3)3
(CH3)3C^qVoh + K3Fe(CN)6 + КОН-------> (CH3)3C^QVo-
С(сн3)3 . , С(сн3)3
+ К4ре(СМ)6 + Н2О
Это — стабильные свободные радикалы. В отсутствие воздуха и реа-
гентов они способны существовать неопределенно долго. Мы встретимся
далее (стр. 224) с такого рода стабильными свободными радикалами,
у которых в a-положении к ароматическому ядру или, лучше, к двум
или трем ароматическим ядрам (при наличии углерода в а-положении)
находится атом с неспаренным электроном. Все они стабилизуются за
счет рассредоточения неспаренного электрона по всей ароматической си-
стеме, что выражается в символике резонанса так:
В символике английской школы выразить это явление труднее потому,
что кривая стрелка обозначает передвижение пары электронов, а в дан-
ном случае рассредоточивается лишь один электрон. Иногда это выра-
жают пунктирными кривыми стрелками:
ФЕНОлы
гл:
Ароксилы — темно-синие кристаллические вещества, раствош
в эфире и бензоле. Их парамагнитные свойства доказывают палю"116
в молекуле неспаренного электрона. 1Ие
Другой способ получения ароксилов состоит в элиминировании метал
лическпм серебром брома из продукта бромирования 2,4,6-гриалкип"
фенола (стр. Г17): ’ “ ь
R
О - +AgBr
R
2,4,6-Триалкилфенолы служат хорошими антиоксидантами и вообще
ингибиторами цепных реакций, так как «обезвреживают» активные сво-
бодные радикалы, ведущие цепь в цепных реакциях:
/R
R-^~^—ОН+А
\r
/R
—> R_A \_o «-РАН
\R
Сами они при этом превращаются в неактивные стабильные радикалы,
и реакционная цепь обрывается. Поэтому фенолы, способные давать арок-
силы, применяются как ингибиторы цепного окисления и связанного
с ним разрушения (старения) пластмасс, например, полипропилена.
В качестве примера практически применяющегося для этой цели фенола
приведем
ОН ОН
(СН’)зС\ A_CH,-Az С(СН’>з
II I с - Il J
y ।
СНз СНз
МНОГОАТОМНЫЕ ФЕНОЛЫ
Диоксибензолы
Изомерные диоксибензолы носят следующие названия: о-диоксибензол --
пирокатехин, .ч-изомер — резорцин и n-изомер гидрохинон. ?
хорошо растворимые в воде, твердые, лишенные запаха вещест
Свойства их представлены в табл. 66.
Пирокатехин известен как продукт декарооксилирования при на Р
вании протокатеховой кислоты, находимой в растениях.
+ СО2
МНОГОАТОМНЫЕ ФЕНОЛЫ
127
Его можно также получить гидролизом о-дихлорбензола (силикагель —
медная соль) перегретым паром.
Пирокатехин — сильный восстановитель, и, окисляясь гетеролити-
чески (например, ионом Ag+), он превращается в о-бензохинон:
Гомолитическое окисление кислородом воздуха протекает сложнее.
Как показывают исследования методом электронного парамагнитного резо-
нанса (ЭПР), сначала идет окисление до семихинона II, который легко
идентифицируется по ЭПР-спектру. Этот спектр потом сменяется спект-
ром свободного радикала VII, который окисляется в хинон VIII. Всю
картину можно представить следующим образом:
VIII
Регорцин (лг-оксибензол) получают в технике сплавлением со щелочью
л1-бенз олдисульфоната натрия:
>>O;(Na
2 NaOH
SO3Na
ОН
> + 2Na2SO3
ОН
128
ФЕНОЛЫ
Таблица 66. Многоатомные фенолы
Формула Название Температура плавления °C Температура кипения °C Плотность 4® Показатель преломления nD
—
С6Н4 (ОН)2 Пирокатехин, 1,2- или о-диоксибепзол 105 240; 245 1,371 1,615 (при 20° С)
С6Н4(ОН)2 Резорцин, 1,3- или л-диоксибензол ПО 276,5; 281 1,285 —
Свн4 (ОН)2 Гидрохинон, 1,4- или п-диоксибензол 170,5—173,1 286,2 1,358^°) —
СвЫз (ОН)з Пирогаллол, или' 1,2,3-триоксибензол 133,5 293 (разл.) 1,453 (rff) —
СвН3(ОН)3 Оксигидрохинон, или 1,2,4-триоксибензол 140,5 — — —
С6Нз(ОН)3 Флороглюцин, или 1,3,5-триоксибензол 219 (безводн.) Возг., разл. — —
Интересно, что о- и /г-бензолдисульфокислоты, а также о- и п-хлор-
бензолсульфокислоты при сплавлении со щелочью в жестких условиях
(выше 300° С) образуют тот же диоксибензол — резорцин. Возможно, что
сначала под действием щелочи происходит отщепление NaHSO3 с образо-
ванием производного дегидробензола (стр. 65), а затем присоединение
элементов воды пли щелочи (кинезамещение):
SOgNa
SOgNa
+ NaOH
-NaaSO3
SOgNa
-NaHSOa
ONa
ONa
Резорцин — устойчивее своих изомеров к окислению. Кислотные
свойства его выражены сильнее, чем у фенола. Уже водородом в момент
выделения (амальгама натрия и вода) он восстанавливается в дигидроре-
зорцин (циклогександион-1,3):
Н2С СН2
---> I I
Н2С ус=о
хсн2
Резорцин еще легче, чем фенол, воспринимает разноооразные зле'
фильные атаки, так как обе его гидроксильные группы осущест .
согласованную ориентацию. Поэтому резорцин легко галопдир)е
КЛАТРАТНЫЕ СОЕДИНЕНИЯ ГИДРОХИНОНА
129
сульфируется, нитруется, нитрозируется, вступает в реакции азосочета-
ния. Одно из его главных применений — синтез азокрасителей, в кото-
ром он служит азосоставляющей.
При исчерпывающем нитровании резорцина получается тринитроре-
зорцин, стифниновая кислота
ОН
O2N’ 1 n°2
Y он
NO 2
во многом напоминающая пикриновую кислоту. Для карбоксилирования
резорцина достаточно нагреть его в растворе бикарбоната натрия:
ONa
ОН
+ С О2
О=С—ONa
Получаемое соединение носит название резорциловой кислоты. Соответ-
ствующий резорциновый альдегид может быть получен по реакции Рей-
мера — Тимана, а также действием синильной кислоты и хлористого
водорода (стр. 139).
Гидрохинон получают восстановлением «-бензохинона (стр. 159):
О + 2Н
> НО^ОУ-ОН
Как и пирокатехин, гидрохинон — сильный восстановитель, при окис-
лении образующий «-бензохинон:
ОН
Пирокатехин и гидрохинон применяются как фотографические про-
явители, восстанавливающие бромистое серебро до металла. Гидрохинон
широко используется также как антиоксидант (стр. 164).
КЛАТРАТНЫЕ СОЕДИНЕНИЯ ГИДРОХИНОНА
Как и в случае фенола, молекулы гидрохинона ассоциированы за счет
водородных связей гидроксилов. Твердый гидрохинон (так называемая
^-модификация его) благодаря этим связям имеет пространственную
решетку (рис. 89), пронизанную пустотами длиной около 5А, в которые
9 Заказ 672
130
ФЕНОЛЫ
легко захватываются посторонние молекулы (SO2, H2S, СН3ОН и да;ке
благородные газы, например аргон). Образующиеся клатратные соедине-
ния гидрохинона (рис. 90) впервые получены и систематически изучены
Рис. 89. Образование каркаса гидрохинона
с помощью водородных связей.
Верх: каждый правильный шестиугольник оз-
начает шесть водородных связей между кислород-
ными атомами; шестиугольники различных уров-
ней изображены линиями разной толщины; ско-
шенные линии, представляющие О—О-оси моле-
кул гидрохинона, показывают способ связи, при
котором образуется бесконечное трехмерное кле-
точное соединение, каждая такая линия показана
снижающейся в направлении от наблюдателя.
Низ — перспективная проекция верхнего ри-
сунка: шестиугольники означают водородные свя-
зи; более длинные скошенные линии, связывающие
разные шестиугольники, представляют собой
О—О-оси молекул гидрохинона.
Рис. 90. Структура клатрата гидрохи-
нона. Окружность вверху выделяет
одну клетку.
Поуэллом. Образование клатрат-
ных соединений свойственно мно-
гим фенолам, в том числе и са-
мому фенолу, на примере которого
Б. А. Никитин открыл «молеку-
лярные соединения» с благород-
ными газами и сероводородом.
В отсутствие молекул «гостей» гидрохинон предпочтительно кристалли-
зуется без пустот, в более плотной а-модификации — плотность 1,35 а/см3;
плотность ^-модификации — 1,26 г/слг3.
«АНСА»-СОЕДИНЕНИЯ ГИДРОХИНОНА.
АТРОПЭНАНТИОМЕРИЯ. I
Алкилируя гидрохинон и гидрохинонкарбоновую кислоту серией дибро-
мидов Вг(СН2)„Вг, Люттрингхауз получил два ряда простых эфиров
типов I и II с мостиками у бензольного кольца (так называемые анса-
соединения):
I
II
(л =8,9 и 10)
ЛОЛПОКСИБЕНЗОЛЫ
131
Соединения типа II не имеют плоскости симметрии, и потому их зер-
кальное изображение несовместимо с оригиналом. Поэтому такие соедине-
ния могут быть разделены на антиподы обычными способами, например кри-
сталлизацией их солей с оптически активным алкалоидом (кп. I, стр. 390).
Этот случай оптической изомерии (именно энантиомерии) замечателен
тем, что в соединении отсутствует асимметрический атом; асимметрия
свойственна молекуле как целому вследствие невозможности (из-за про-
странственных препятствий) поворота бензольного кольца. Действи-
тельно, соединения II при п 8 обнаруживают полную устойчивость
левой и правой форм даже при химических превращениях. При п = 9
происходит (в результате поворота бензольного цикла) медленная рацеми-
зация, т. е. превращение левая форма правая форма, а при п = 10
рацемизация идет очень быстро.
Такой тип оптической изомерии называется атропэнантиоизомерией
(а — отрицание, тропо — вращаю). Мы еще встретимся с атропэпантио-
изомерией при рассмотрении бифенила (стр. 205).
Полиоксибензолы
Смежный триоксибензол называется пирогаллолом, так как получается
пиролизом (декарбоксилированием) галловой кислоты
НО
НО—(С)
но7
с—он
выделяемой из продуктов гидролиза дубильных веществ типа таннина
(стр. 186).
Пирогаллол в щелочных растворах легко окисляется даже кислородом
воздуха, поэтому такие растворы используются для поглощения кисло-
рода. В фотографии пирогаллол применяется как проявитель.
Симметрический триоксибензол — флороглюцин в виде его производ-
ных очень распространен в растительном мире. Многие из подобных про-
изводных получают синтетически через флороглюциновый альдегид,
в свою очередь синтезируемый из флороглюцина по Реймеру — Тиману,
или действием синильной кислоты и хлористого водорода (стр. 139).
Обычно флороглюцин получают гидролизом симметрического триами-
нобензола (его готовят восстановлением тринитробензола):
По свойствам флороглюцин похож на резорцин.
1,2,^-гГриоксибензол можно синтезировать, присоединяя к тг-бензохи-
нону уксусный ангидрид и гидролизуя образовавшийся ацетат (стр. 162).
9*
132
фЕНОЛЫ
Гексаоксибензол получают подкислением продукта соединения мета-г и
ческого калия и окиси углерода: 1ЛИ-
6 СО + 6К
6Н
ЭФИРЫ ФЕНОЛОВ
О синтезе сложных и простых эфиров фенолов было сказано на стр. Ц5.
Сложные эфиры гидролизуются обычным для этого типа веществ путем
в кислой и в щелочной среде. Простые эфиры фенолов, как всегда, трудно
гидролизуются, но все же легче, чем предельные алифатические эфиры.
Их гидролизуют в присутствии хлористого алюминия и хлористого водо-
рода. Они расщепляются также концентрированными бромистоводород-
ной и иодистоводородной кислотами с выделением галоидного алкила.
Из простейших и важных простых эфиров фенола назовем метиловый
(анизол) и этиловый (фенетол). Они находят применение в качестве раство-
рителей и главным образом в синтезе красителей и лекарственных веществ
после нитрования и последующего восстановления в о- и «-метоксианп-
лины (анизидины) и о- и ?г-этокспанилины (фенетидины). Полученные из
них гидразосоединения перегруппировываются соответственно в 2,2-
диметоксибензидин (дианизидин) и 2,2-диэтоксибензидин.
При нагревании с лыоисовыми кислотами алкоксибензолы перегруп-
пировываются в о- и n-алкилфенолы (Н. И. Курсанов):
ОСН3
ZnCb
----->
Очень интересно и легко (без участия кислоты Льюиса) протекает пере-
группировка аллилового эфира фенола (Клайзен). Аллильный радикал
перемещается только в орто-положение, если оно не занято, и в пара
положение, если заняты оба орто-положения. Посредством метки третьего
(крайнего) углеродного атома аллила путем введения в это положен
радиоактивного14 С доказано, что в первом случае аллил, перемещающи
в орто-положение, привязывается свопм третьим (меченым) углерод
атомом, очевидно, через циклическое переходное состояние:
ЭФИРЫ ФЕНОЛОВ
133
В случае клайзеновской пара-перегруппировки аллил оказывается свя-
занным первым углеродным атомом:
О—СН2—СН=СН2
ИзС^Л/СИз
(OJ
ОН
Это роднит клайзеповскую пара-перегруппировку с другими переме-
щениями группы из боковой цепи бензола в пара-положение: перегруп-
пировка фенилгидроксиламина в п-аминофенол, азоксибензола в н-окси-
азобензол и др. Разумеется, для того чтобы определить, где находится
14С в продукте реакции, нужно подвергнуть этот продукт деструкции так,
чтобы отделить крайний углеродный аллильный атом (например, озони-
рованием или иным окислением), и выяснить, какой из двух продуктов
деструкции радиоактивен.
Сложные эфиры фенолов при действии кислот Льюиса также претер-
певают перегруппировку с миграцией ацила в ядро, главным образом
в пара-положение, и образованием кетонофенолов (перегруппировка
Фриса):
Из простых эфиров диоксибензолов назовем монометиловып эфир
пирокатехина гваякол, встречающийся в дегте, особенно буковом,
и находящий фармацевтическое применение. Его получают из о-анизидина:
ОСНз ОСНз+ ОСНз
^\/NH2 + HCl+HNO2 —> J\/^n J /ОН +No + I.IC]
U । j । II
Вератрол (диметиловый эфир пирокатехина) синтезируют по реакции:
ONa ОСНз
Д/ON» А/0СПа
I II -|-2СвН58О2ОС11з --->• | || +2CeH.-,SOoONa
Гваякол состоит в родстве с важными душистыми веществами. Вводя
в гваякол тем пли иным способом альдегидную группу (в пара-поло-
жение к гидроксилу), получают ванилин. Такое формплпрование можно
131
ФЕНОЛЫ
осуществить действием
фосфора (реакция Вильсмейера):
ОН
метилформапилпда в присутствии хлорокиси
ОСНз
+c6h5-n-ch3
I /;0
ОН
k /ОСНз
—\ ||' -|-С6Н6ЫНСНз
н
О
Ванилин — пахучее начало ванили. Теперь его готовят проще —
окислением лигнина (стр. 137).
Пахучее начало гвоздики — эвгенол — представляет собой 4-аллил-
2-метоксифепол. Родство с ванилином устанавливается следующим путем:
аллильной перегруппировкой посредством кипячения со щелочью эвгенол
превращается в изоэвгенол (4-пропенил-2-метоксифенол), окисление кото-
рого (например, озоном) приводит к ванилину:
ОН ОН
Д /ОСНз ) /ОСНз
/Z У NaOH // у Оз; Н2О
СН2СН=СН2
СН-СНСНз
он
) /ОСНз
о
Аналогичны отношения сафрола (из сассафрасового эфирного масла)
и пиперонала, или гелиотропина, применяемого в парфюмерии и пахну-
щего гелиотропом:
О—СН2
о—сн2
о—сн2
сн2—сн=сн2
сафрол
У\/°
NaOH > | ||
сн=снсн3
изосафрол
Оз; н20
,0
I /О
пиперонал
о
ПРОСТЫЕ ЧИСТО АРОМАТИЧЕСКИЕ ЭФИРЫ
Простейший представитель этого класса — дифениловым эфир, называ-
емый также дифенилоксидом, — вещество с т. пл. 28° С, кипящее при
259° С, пахнущее листьями герани. Он применяется в парфюмерии,
а также как теплоноситель в обогревательных системах. Его можно полу
чить по Ульманну, нагревая до —200° С бромбензол и фенолят натрия
с медью:
Си
----->
Дифенилоксид образуется также как побочный продукт при гидролизе
Щелочью хлорбензола (в присутствии меди).
ТГИФЕИПЛОКСОНИЕВЫП CO.lH
135
ТРИФЕППЛОКСО1ПТЕВЫЕ СОЛИ
При разложении борфторида фенилдиазопия в ацетоновом растворе дифе-
нилового эфира с небольшим выходом получается борфторид трифепилок-
сопия в результате присоединения образующегося фенил-катиона (гетеро-
литически it распад фепилдиазония) к свободной паре электронов кисло-
рода (А. Н. Несмеянов, Т. 11. Толстая):
C(iH5N=N BF’.f + О— С6Н5 --►- C6H5:()':CttH5 BF\ + N2
с6н5
В отличие от других компонентов реакции эта оксониевая соль раство-
рима в воде и по отгонке ацетона может быть извлечена в водный слой,
затем осаждена в виде, например, малорастворимого иодида.
Соли трифенилоксония очень прочны термически и по отношению
к электрофильным реагентам, чем они отличаются от триалкилоксоние-
вых солей. Оксопиевый кислород, несущий положительный заряд, ведет
себя как сильно дезактивирующий (к электрофильному замещению) заме-
ститель, но направляет вступающий заместитель в пара-положение. Так,
при нитровании соли трифенилоксония (100° С, 60 ч, 100%-ная HNO3
+ 100%-ная H2SO4) образуется три-(?г-питрофенил)-оксониевая соль.
Трифепилоксопий — единственный из ароматических опиевых катио-
нов, нитрующийся в пара-положение; все остальные нитруются в мета-
положение. Причина, очевидно, та же, что и в хлорбензоле: наличие
свободной пары электронов у кислорода и благодаря этому + Г-эффект
при очень сильном —.г-эффекте положительно заряженного кислорода.
Трифепилоксоииевый катион гораздо более устойчив по отношению
к нуклеофилам, чем жирные оксонии, ио все же не очень прочен. Напри-
мер, при нагревании идут реакции:
(СвН.йзО Ш-7 I iNaOIJ -> CeH5OII + (CeH5)2O + NaBF4
(СбНгДзО BFj |-Nal\O2--> CeH5NO2+ (CeH5)2O + NaBEj
АРОМАТИЧЕСКИЕ СПИРТЫ
Простейший ароматический первичный спирт — бензиловый C(iH5CH.,OH —
может быть получен из бензальдегида (стр. 137), гидролизом хлористого
бензила
СбПьСПгС! |- П3О -> CelKCHoOII-)-НС1
или гриньяровой реакцией
C6H5MgBr-| СП2<) -—> Cell5-CH2OMgBr —СвЩСНоОН
Вторичные и третичные ароматические спирты получают реакцией
Гриньяра — взаимодействием фенилмагпийбромида с альдегидами и кето-
нами, а вторичные, кроме того, восстановлением кетонов.
Бензиловый спирт но функциональному поведению вполне сходен
с алифатическими первичными спиртами. Довольно существенной его
136
ФЕНОЛОСНИРТЫ И ИХ ПРОСТЫЕ ЭФИРЫ
чертой является легкая отдача им а-водородного атома с парой электронов
(гидрид-аниона); сам он при этом окисляется в бензальдегид:
свн5-с-о-:н +а
Н
CeH5-(i=O + AH + H
: I :
:Н;
где А+ — акцептор гидрид-аниона, например (СвН5)3С.
Кроме того, так же как от хлористого бензила отщепляется ион хлора,
так и из бензилового спирта легко удаляется кислотами гидроксил, оста-
вляя относительно устойчивый бензил-катион, претерпевающий дальней-
шие превращения.
Бензилацетат имеет свежий цветочный запах. Он входит в состав
эфирного масла жасмина и применяется в парфюмерии.
Р-Фенилэтанол, гидроксил которого более удален от фенильного ядра,
вполне подобен жирным спиртам. Готовят его из бензола и окиси этилена
по варианту реакции Фриделя — Крафтса:
С6Нв + Н2С СН2 -^-’+ С6Н5-СН2СН2ОН
Применяется в парфюмерии (запах его напоминает запах розы).
ФЕНОЛОСПИРТЫ И ИХ ПРОСТЫЕ ЭФИРЫ
Соединения этого класса важны, в частности, потому, что некоторые из них
находятся в ближайшем структурном родстве с лигнином, например:
сн=сн—СН2ОН
сн=сн-сн2он
он
'осн3
конисрериловый
спирт
синапиновый
спирт
Лигнин представляет собой инкрустирующее вещество древесины (30. о
от сухой массы) и связан в ней с целлюлозой и гемицеллюлозами. При
переработке древесины в целлюлозу сульфитным методом в макромоле-
кулы лигнина вступают сульфогруппы из сульфита и происходит разрыв
по кислородным связям. Лигнин переходит при этом в раствор, оставляя
целлюлозу. При «сульфатном» методе растворение лигнина достигается
действием щелочи за счет свободных фенольных гидроксилов лигнина-
При кислотном гидролизе древесины с целью получения глюкозы
лигнин остается нерастворенным. В качестве промышленных отхО^°_
получаются в огромных количествах гидролизный лигнин и лигносульфо
ФЕНОЛОСПИРТЫ II ИХ ПРОСТЫЕ ЭФИРЫ
137
новые кислоты. По существу это ценный материал, над более рациональ-
ным использованием которого следует работать.
Главный исследователь строения лигнина Фрейденберг воспроизвел
биосинтез лигнина, обрабатывая конифериловый спирт дегидрирующим
ферментом — дегидразой, выделенной из сока шампиньонов. О проходя-
щих при этом дегидрировании конденсационных процессах могут дать
представление получаемые побочные (или промежуточные) продукты:
СН3О
СН=СН—СН2ОН
СН=СН—С
СН3О
Меченный 14С конифериловый спирт, превращенный в глюкозид копи-
ферин, будучи введен в камбиальный сок сосны, действительно (как пока-
зывает радиоактивная метка) оказывается включенным в состав лигнина
сосны.
При окислении лигнина нитробензолом образуется ванилин, что и слу-
жит промышленным способом получения этого вещества.
СН3О
ванилин
АРОМАТИЧЕСКИЕ АЛЬДЕГИДЫ
Главное внимание здесь будет обращено на альдегиды, карбонильная
группа которых непосредственно связана с бензольным ядром, так как
именно они обладают некоторыми специфическими свойствами и синтези-
руются особыми методами. Типичным представителем их является бенз-
альдегид. Альдегиды, подобные фенилуксусному СвН5СН2СНО пли
138 АРОМАТИЧЕСКИЕ АЛЬДЕГИДЫ
----
имеющие еще более удаленную от бензол иного ядра альдегидную группу
например фепилгидрокоричпый Св1Т5СН2СП.,СНО, по способам синтеза
п реакциям вполне подобны жирным альдегидам и поэтому рассматри-
ваться не будут. Непредельные альдегиды, такие, как коричный
С6Н5СН = СНСНО (пахучее начало корицы), также существенно пе отли-
чаются от жирных альдегидов с сопряженной системой л-связей, типа
кротонового, и также могут быть получены конденсацией (на этот раз
двух разных альдегидов — бензойного и уксусного).
Бензальдегид, как уже сказано во введении, — это соединение, давно
известное как масло горького миндаля. Он получается при гидролизе
гликозида — амигдалина, содержащегося в горьком миндале (а также
в косточках вишен, абрикосов и т. д.). Гликозид распадается при гидро-
лизе на дисахарид — генциобиозу — и оксинитрил бензальдегида, легко
регенерирующий сам альдегид и синильную кислоту:
/Н Н
C6H5-C-C=N --> С8Н —Сч( +IICN
4 он о
Таким образом, бензальдегид стал известен химикам раньше бензола.
Синтез ароматических альдегидов
1. Методы, основанные на окислении метильной группы. Получение
бензальдегида промышленным путем сводилось ранее к гидролизу хлори-
стого бензилидена
СбН5-СНС12 c6II5-C<^° +2НС1
н
или к окислительному гидролизу хлористого бензила:
СвН5СН2С1
НЛМПО,,^ Cjll5_c Z0
н
Важным общим методом превращения группы —СН2С1 в альдегидную
служит метод Соммле, состоящий в кипячении хлористого бензила с вод-
ным раствором уротропина (см. кп. I, стр. 138).
N Н9СХС1^СН2 Н2С СН2СН2
С6Н5СН2С1 + | N I N > ЩССН, Ксснг iV CfiH5-CH2— N6x >N CH2 СГ CH2 > Cells-CH2—NH2-f-3CH2O4-3NH3 H
1+ .
С6Н5-СН2— NH2 + CH2O > С61]г,—CII2—N=CIl2 —СбН5—сн2—N=cl
СИНТЕЗ АЛЬДЕГИДОВ
139
н н
L I
СбН5-СН2— N=CH2 + Cell5—ci Io—NHo > C61I5-C1I2—N-CH3 + C6H5-CH=NH2
+ /Н
C6H5-CH = NH2+HoO > C6H5—c . +NHJ
'O
Возможно и прямое окисление толуола в бензальдегид (и вообще
метильных групп, связанных с ароматическим ядром, в альдегидные).
В лабораторных условиях это может быть осуществлено реакцией Этара
(действие хлористого хромила):
ЗСвН5—СН3 + 4СгОоС12 > ,ЗС6Н5-СНО + Н2О Р 4Сг(ОЛ)С12
В промышленности окисление метильных групп проводят хромовой
кислотой или перекисью марганца с серной кислотой при соблюдении
некоторых предосторожностей.
2. Не менее важны методы, заключающиеся во введении группы СНО
на место водорода ароматического ядра. Так, метод Гаттермана — Коха
заключается в действии на бензол (или вообще на ароматический угле-
водород) окисью углерода и хлористым водородом в присутствии А1С13
и СнС1. Окись углерода, по-видимому, образует с хлористым водородом
неустойчивый хлористый формил, который далее реагирует по реакции
Фриделя — Крафтса:
Н\
СО + НС1 —,с=о
Н\ А1С1
свнв+ ;с=о ——ceH5-c<f +нс1
сП н
Для введения альдегидной группы в фенолы и их эфиры используется
вариант реакции Гаттермана, в котором вместо окиси углерода приме-
няют синильную кислоту, а вместо хлористого алюминия — хлористый
цинк:
В орто-положение фенолов альдегидную группу можно вводить мето-
дом Реймера — Тимана. При этом на концентрированный щелочной рас-
твор фенола действуют хлороформом. По-видимому, дело сводится
140
АРОМАТИЧЕСКИЕ АЛЬДЕГИДЫ
к отщеплению от хлороформа хлористого водорода и образованию кар
бена: СС12, который и взаимодействует с фенолятом натрия:
ONa
2RaOH
------>
+ 2NaCl + Н2О
Так из фенола получается о-оксибензальдегид (салициловый альдегид).
На промежуточное образование карбена указывает побочное образо-
вание небольшого количества трополона:
ONa
он-
О ОН
трополон
Такое внедрение — типичная реакция карбенов (см. ч. II «Карбены»).
По методу Вильсмейера альдегидную группу вводят, действуя па аро-
матические соединения (достаточно нуклеофильно активные) формиль-
ными производными вторичных аминов, например N-метилформанилидом,
в присутствии хлорокиси фосфора:
+ С6Н5—N—С — Н
СН3
+ c6h5-n-ch3
н
Разумеется, альдегидную группу можно ввести в бензольное ядро
и посредством гриньяровой реакции:
+н-сб
XOR /О
(нахолоду). с н
-ROMgBr \Н
CeH5 —MgBf—
+H-C(OR)s .
-ROMgBr CeH5—С (ОВ)г
1
н
+ НЮ(Н+)^
-2ROH
С6Н5-С
\н
(А. Е. Чичпбабин)
СВОЙСТВА АЛЬДЕГИДОВ
141
3. Альдегиды можно также получить восстановлением функциональ-
ных производных карбоновых кислот, например восстановлением хлори-
стого бензоила по Зайцеву — Розенмунду:
c6H5-c<fO H,/pd--> с6н6-с^° +НС1
'Cl 41
Свойства ароматических альдегидов
Физические свойства ряда ароматических альдегидов, их формулы и наз-
вания приведены в табл. 67.
Бензальдегид и другие альдегиды, у которых группа —СНО связана
с ароматическим ядром, во многом похожи на жирные альдегиды. Так,
они восстанавливаются во вторичные спирты, окисляются в карбоновые
кислоты. На реакциях этого типа следует остановить внимание.
Окисление. Для ароматических альдегидов, как и для жирных,
существует два типа реакций окисления, в конечном результате приводя-
щих к образованию соответствующих кислот: гомолитическое цепное
и гетеролитическое.
а) Гомолитическое окисление. На воздухе налитый тонким слоем бенз-
альдегид через несколько часов превращается в бензойную кислоту.
Соли переходных металлов, например железа, марганца, ускоряют про-
цесс. Течение его таково:
О 1
,0 II
CglU—С<( -{-Инициатор —>• СвН5С--|-П +
Н
Например: ! Инициирование
О
z° II
C6H5-C<f + Fe3+ ---> c6U5-C«4-Fe2+4-IP
H
О О
И II
с6нй—с«+о2 —> С8н6—С-О-О.
О 0 0
II zO II II
C6H5-C-O-O.4-C6Il5-C^ -----> С6Н5-С-ООП+С6Н5-С-
\н
Звено
цепи
О
II
С6Н5-С-ООЩ- С6НЬ-
/О
2C6H5-Cf
\он
Реакция
вне цепи
б) Гетеролитическое окисление. Примером может служить гетероли-
тическое окисление перманганатом. Это катализируемая попом водорода
реакция, механизм которой исследован с применением меченного по
кислороду перманганата КМи1н()4 и меченного дейтерием бензальдегида
Таблица 67- Ароматические оксосоедпиеппя
Формула Название Температура плавления (и
А ль д е гиды
/° c6H6-cf хн /0 сн3с6н4-с^ +н Бензальдегид Толуиловый альдегид 0- м- п- —26
/О с6н6сн2-с+ н Фенил уксусный альдегид <-10
/° сйн5сн2си2-с+ \н /О Гидрокор ичный, или р-фенплпро- пионовый, альдегид +47
С6Н5—CH=CH-C<f 'Н /0 Коричный альдегид —7,5
ко2сйн4-сн+ +Н Нитробензальдегид о- м- п- 40 (а-); 37,9 (Р-) 58 106,5
Кетоны
С6Н5-С-СН3
II
О
CgHs—С—C2II5
II
О
С8Н5-С-С6Н6
Ацетофенон 20
Фенилэтилкетон, или пропиофенон 21
Бензофенон 49 (а-); 26 (Р+ 45
Температура кипения °C Дипольный момент р. в бензоле при 25° С D Температура плавления функциональных производных альдегидов
оксима фенилгидра- зона семикарба- зона
179,5 195,5 199 205 2,75 3,30 35 (а-син-изомер) 130 (Р-анши-изомер) 49 108—110 156 106 90 121 222 212 223—224 234
194 2,48 (при 20° С) 98,5 62—63 —
280 2,31 (при 20° С) 93—94,5 — 127
251 3,71 138,5 (а-); 64-65 (0-) 168 208
(разл.) (при 18° С)
156 4,30 154 (а-); 103(0-) 256
(при 15 JH3i рт. ст.)
164 3,28 85 (а-); 121(0-) — 246
(при 23 jhjh рт. ст.)
Возг. 2,4 182—184 (а-); 133(0-) 153—154 211
202,3 2,97 (при 18° С) 58 106 203
218 53—54 — 182
306 2,95 (при 13° С) 144 137 167
144 АРОМАТИЧЕСКИЕ альдегиды
/° о
СбН5С<^ . Оказалось, что скорость реакции для СвН5С<У и С«ШС^
D
весьма различна: дейтерированный бензальдегид окисляется в семь раз
медленнее! Это означает, что в определяющей скорость стадии реакции
участвуют связи С—D и С—Н. Поскольку полученная бензойная кислота
содержит 50% меченого 18О из перманганата, постольку кислород для
окисления бензальдегида доставляется не из воды, а из КМп18О4. На осно-
вании всех этих данных можно сделать вывод о следующем механизме
реакции гетеролитпческого окисления:
t ОН
CGH5-CX + Н+---► Свн5—С\
н н
+ /°н
сбн5 — с'
н
18 - 18
+ О Мп О3
он
> С6н5—С + НМп'о3
V VII <-Н2О VI
НМпОз + НМпО4 ---------> 2Н2МпО4
Механизм окисления приведен здесь не для противопоставления жирных и аромати-
ческих альдегидов — их поведение сходно в этих и дальнейших реакциях, а для того,
чтобы подчеркнуть, что механизмы реакций, одинаковых по конечному результату,
как, например, окисление воздухом и перманганатом, могут быть совершенно разными
и требуют изучения.
Фотолиз. При фотолизе бензальдегида образуется свободный
О
радикал бензоил С6Н5—С -, который устойчив при —200° С, а при 140 С
лишь медленно (минуты) присоединяет бром, превращаясь в бромистый
/°
бензоил С6Н5—С<( . Радикал бензоил — оранжево-красное вещество,
\Вг
дает сигнал ЭПР (см. ч. II «Свободные радикалы»), присоединяется по
олефиновым связям (У. Шмидт).
Ароматические альдегиды, так же как и жирные, присоединяют оисул
фит натрия, синильную кислоту, Гриньяров реактив (см. кн. I, стр- 1 Р
Ряд реакций замещения альдегидного кислорода проходит также
аналогично реакциям жирных альдегидов. Сюда относятся реакции с
разином, семикарбазидом, арилгидразинами (кн. I, стр. 140). Реакция с
роксиламином приводит к двум стереомерным оксимам бензальдег
различающимся не только по физическим константам, но и по реак ,
При действии уксусного ангидрида (и других в од о отнимающих сре
СВОЙСТВА АЛЬДЕГИДОВ
145
««mu-изомер отщепляет воду и образует бензонитрил, а cuu-изомер
ацетилируется по гидроксильной группе:
НС1;
(СН3СО)2О
:n—он
а-, или сип-
бензальдоксим
(Т.пл. 33ПС)
Na2 СО3
' "'1 с—н
:n—оссНо
II
но
р-, или чнти-
беизальдоксим
(т.пл. 130°С)
(СН3СО)2О
+ СН3СООН
+ 2СН3СООН
Си«-а«ти-изомерия, с которой мы уже встречались при азобензоле
<(стр. 79), вполне подобна г/ис-тра«с-изомерии соединений с углерод-
углеродной л-связью. Роль второго заместителя при азоте играет свобод-
ная пара электронов. 7\ра«с-элиминация (в данном случае отщепление Н
и ОН из транс-положения) является общим правилом (ср. ч. II «Реакции
элиминации»).
Реакции конденсации. Новой для ароматических альде-
гидов сравнительно с жирными является реакция конденсации с арома-
тическими же аминами, приводящая, к важному классу анилов (шиффовых
•основании):
/Н
С6Н5-С< -hH2N-CeH5 ---> CeH5-CH=N-C6H5-i-HoO
^0
(О реакции жирных альдегидов с анилинами см. раздел «Хинолин
и его производные», стр. 339.)
С аммиаком ароматические альдегиды также реагируют отлично от
жирных и дают не альдегидаммиаки, а гидробензамиды:
/°
ЗСбН5-С< +2NH3
C6m5-C=N—СН—N=C-C6II5 + 3H2O
I I I
н свн5 н
Одно из главных отличий ароматических альдегидов от жирных со-
стоит в том, что для них неизвестна реакция образования полимеров
вообще и, в частности, столь характерная в жирном ряду реакция образо-
вания тримеров, подобных паральдегиду.
Само собой разумеется, что для ароматических альдегидов невозможна
ни альдольная, ни кротоновая конденсация самих па себя, поскольку для
этих конденсаций необходимо наличие соседнего с альдегидной группой
метиленового звена. Зато бензальдегид и вообще ароматические а-альдегиды
10 Заказ 672
146
АРОМАТИЧЕСКИЕ АЛЬДЕГИДЫ
способны к иной — бензоиновой конденсации, протекающей под де^.
ствием цианистого калия:
Н
/н ।
С6П5-С/ +С-С6Н5
^0 II
о
н
свн5-с-с-с6н6
I II
но о
бензоин
Можно думать, что роль циан-иона состоит в первоначальном присое-
динении к альдегиду (по типу присоединения синильной кислоты) и оття-
гивании электронов связи от оставшегося водорода альдегидной группы
вплоть до его протонизации и сообщения ему способности перемещаться
в виде протона к карбонильному кислороду второй молекулы, после чего
происходит образование С—С-связи и выброс CN~:
Строение бензоина явствует из его окисления в 1,2-дикетон — бензил'.
С6Н5-С-СН-С6Н5 п
|| | С6Н5-С-С-СвН5 + Н2О
О ОН II ||
о о
При действии щелочи бензил претерпевает внутримолекулярную пере-
группировку в бензиловую кислоту:
с6н6-с—с-с6н5 + он
6 II II
о о
|С6н5^, I
----^С—С-С6н5
НО^
О" О'
/С6Н5
> но—с—с
II 1?с6н5
о о
—но—с-с^ + он
о он СбНз
Как уже упоминалось, бензальдегид не претерпевает под влиянием
Щелочи конденсаций сам на себя, но для него возможно иное проявление
действия щелочи: две молекулы бензальдегида диспропорционируются
в бензиловый спирт и бензойную кислоту (реакция Канниццаро):
2СвН5-С
/Н
%
+ NaOH --► С6Н5-С
/ONa
^0
+с6н6—сн2он
На механизм реакции Канниццаро проливает свет реакция ТиЩ ’
свойственная как жирным, так и ароматическим альдегидам и ПР°_ ов
Щая под действием алкоголятов алюминия, а также тетраалкоксити
СВОЙСТВА АЛЬДЕГИДОВ
147
(А. Н. Несмеянов, О. В. Ногина). По реакции Тищенко (кн. I, стр. 142)
две молекулы альдегида превращаются в молекулу сложного эфира соот-
ветствующих спирта и кислоты, в случае бензальдегида — в бензиловый
эфир бензойной кислоты. Можно думать, что реакция Канниццаро —
частный случай реакции Тищенко, отличающийся тем, что образующийся
и в этом случае сложный эфир гидролизуется щелочью в спирт и соль
кислоты. Смысл реакции состоит в гидридном перемещении, т. е. в пере-
ходе одного из водородных атомов со своей парой электронов к карбо-
нильному углероду другой молекулы альдегида:
I о lL J
с6н5—с—о-’ с—с6н5 —> с6н5—сн2—о—с—С6Н5
(он О "ОН о
Что касается альдольных и кротоновых конденсаций бензальдегида
с жирными предельными альдегидами, то эти процессы проходят нор-
мально и, например, с ацетальдегидом приводят к образованию сначала
§-окси-р-фенилпропионового альдегида, а затем коричного альдегида
(находится в корице и обусловливает ее запах):
/И /Н ,Н
сбн5с<С +СНзС< —> с6н5сн-сн2сА
^0 Ч) | ^0
он
свн5сн=снс
/н
%
-HjO^
Очень легко в присутствии разбавленных щелочей осуществляется
кротоновая конденсация бензальдегида с ацетоном (и другими аромати-
ческими кетонами). В зависимости от соотношения реагентов получается
бензальацетон или дибензальацетон:
СЙН5—СНО + СНз—С—СН3 —> СбН5-СН=СН—С-СНз + НоО
2СвН5-СНО4-СН3—С-СН8 > СйН5СН==СН-С-СН=СН-СвН5 + 2Н2О
Оба непредельных кетона — желтые кристаллические вещества.
Особенно важен тип конденсации Перкина (альдегид, ангидрид кислоты
и ее натриевая соль), приводящей к непредельным кислотам типа корич-
ной. Здесь уксуснокислый натрий играет роль основания и реакция
10*
148
АРОМАТИЧЕСКИЕ КЕТОНЫ
начинается как нуклеофильная атака карбаниона I ангидрида на карбо
нильный углерод: р
CH3-C<f
'.О + СН3 С- О' Na+ ДГ? CH2-C-O-C-CII3 + CH3-C-OH
СНз-Csk II || || ||
^0 О Na+ 0 0 О
С6Н5—С<^ + СН2—С—О—С—СНз > Сбн5—С—сн2—С—о—С—СПо
н Д II III ||
оо 0- Na+ 0 о
—> С6Н5-СН—СН2—С—о—С—СНз + СНз—С—О- Na+
Н20
СвН5-СН=СН-С-О-С-СНз
с6н5—сн=сн—с—он+СНз—С—он
Конечно, анион I мезомерен
СН2-С-0—С-СНз <-----> СН2=С—О—С—СНз
II II I II
0 0 О" о
I II
причем больший вес должен принадлежать структуре II. Так что по су-
ществу реакция этого аниона с альдегидом является реакцией с перенесе-
нием реакционного центра.
Реакции замещения бензальдегида в ядро. Альдегид-
ная группа относится к мета-ориентантам средней силы, дезактивирует
ароматическое ядро и направляет вступающий заместитель главным обра-
зом в мета-положение. При нитровании бензальдегида наряду с at-нитро-
бензальдегидом получается также значительное количество о-изомера.
АРОМАТИЧЕСКИЕ кетоны
связанньт1иеТОНОВ также Различают кетоны с карбонилом, непосредственно
Только пр ° аРОматическим ядром, и кетоны с удаленным карбонилом.
Обычнь)ВЫе имеют некоторую специфику свойств и путей синтеза,
углевопопотт названия кетонов строятся как названия ацилированных
бутппоДо.г °В--С пРи^авлением окончания он— ацетофенон, пропиофенон,
он, оензофенон (фен — старое лорановское название бензола).
СИНТЕЗ КЕТОНОВ
149
Синтез ароматических кетонов
1. Кетоны с карбонилом у ароматического ядра наиболее удобно синте-
зировать реакцией Фриделя — Крафтса:
СбНб+R-C-Cl CbH5-C-R4-HCI
CeHe+R—С-О-С—R СвЩ-C-RJ-R-C-OH
О
где R — ароматическим или алифатический радикал.
Хлористый алюминий в этой реакции приходится брать в эквивалент-
ном хлорангпдриду (или ангидриду) количестве, так как он полностью
связывается в комплекс и продолжает быть связанным с кетоном после
его образования. Для этого комплекса можно было бы предположить
структуру, аналогичную структуре известного комплекса Зееля
R—С—Г + ВЕ3
способного, хотя и слабо, ацилировать ароматическое ядро. Однако это
предположение не подтверждается, и в комплексах хлорангидридов
с хлористым алюминием связь осуществляется через кислород, как и в слу-
чае кетонов. Исследования УФ-спектра, имеющего, как и для кетонных
комплексов, сдвиги в более длинноволновую область, термохимические
исследования и данные о молекулярном весе позволили Н. Н. Лебедеву
предложить такую структуру обсуждаемого комплекса:
[А1С14]-
Естественно, что связь с алюминием усиливает положительный заряд
на карбонильном углероде и делает его более способным к электрофиль-
ной атаке.
Ароматическое ядро, пассивированное наличием мета-ориентанта, не
вступает в эту реакцию. Поэтому реакцией Фриделя — Крафтса не
удается ввести в бензольное ядро второй ацил. Таким путем нельзя про-
ацилировать также нитробензол, бепзолсульфокислоту и т. д.
2. В случае более нуклеофильного, чем бензол, ароматического ядра
(многоатомные фенолы, их эфиры) для синтеза кетонов применяют
150
АРОМАТИЧЕСКИЕ КЕТОНЫ
реакцию Геша — действие нитрила и хлористого водорода (вариант реак
ции Гаттермана):
^NH Н,о
CH3O~<Q) + R-C=N + HCI ---> сн;)оХПьс( —
о R
II
---> СН3О-\О/-С— R + NH3
3. Вполне применима также реакция гриньярова реактива с аромати-
ческими нитрилами:
/Вг
CeH5—C=N-f-RMgBr > C6II5-C=NMgBr > CeH5-C-R + Mg< + NH3
A o X°H
где R — ароматический или алифатический радикал.
4. Окисление кислородом воздуха этилбензола (и его гомологов),
инициированное в жидкой фазе солями марганца или кобальта, приводит
к образованию гидроперекиси и в результате ее разложения — к кетону
(цепная реакция):
С6Н5—СН2-С11з+О2 > С6Н5—СН-СНз > С6Н5-С-СПз + НгО
I I
ООН о
Свойства ароматических кетонов
Физические свойства ряда кетонов, их формулы и названия приведены
в табл. 67 (стр. 142).
Представителем жирноароматических кетенов может служить ацето-
фенон (т. пл. 20° С, т. кип. 202,3° С). Это жидкость с запахом цветов
черемухи, находящая применение в парфюмерии. Все сказанное о свой-
ствах ацетона (кн. 1, стр. 136 сл.) применимо и к ацетофенону. Он почти
в той же мере способен к реакциям присоединения по карбонильной двой-
ной связи и к реакциям замещения карбонильного кислорода. Как и все
кетоны, он не полимеризуется, но конденсируется. Продуктами кротоно-
вой конденсации ацетофенона являются дипнон (аналог окиси мезитила)
и трифенилбензол:
2С6Н5-С-СН3---►С6Н5-С=СН-С-С6Н5
II I II
О СНз о
с6н5
I
ЗСвН5-С-СНз---> I II
свн5//'^/хсвн5
Уже эти реакции демонстрируют обычную подвижность а-водородов
метила. Можно привести и другие подобные реакции.
СВОЙСТВА КЕТОНОВ
151
Конденсация с функциональными производными кислот в 1,3-дике-
тоны:
а) сложноэфирная конденсация:
R—С—OR -j-Na-j-СНз—С—CgHj >
R-C-CH2-C-CeH5 + R'dNa
II II
О о
б) конденсация с ангидридами кислот по Меервейну:
/О
СНЯ-С<
>о+сн3—с-свн5
СНз-|)
о
BFa
/ОН
СНз—с—СН2-С-СвН5 + СНз—с<С
II II ^0
о
Реакция Манниха. При действии на ацетофенон смеси формальдегида,
амина и соляной кислоты а-водород замещается на моноалкиламиноме-
тильную или диалкиламинометильную группу:
СвН5—С—СНз+ СН2=О + NH(CH3)2 + НС1 —>
I
о
--> СбН5-С-СН2-СН2-НН(СНз)2 CI-4-H2O
II
о
Промежуточным продуктом является образующееся при взаимодей-
ствии формальдегида с амином метилольное производное амина
/СНз
CH2O + HN(CH3)2 —> ho-ch2-n<
ХСН3
которое в кислой среде превращается в карбониевый катион, стабилизо-
ванный смещением свободной пары электронов азота:
СН3
НО—СН,—+ н +
СН3
Галоидирование ацетофенона по метильной группе:
СвНз—С—СН3-1-С12 —> СвН5—С—CHoCl-f-HCl
Хлорацетофенон, содержащий весьма подвижный галоид, очень сильно
раздражает слизистые оболочки. В прошлом — боевое слезоточивое ве-
щество. Он может быть получен также реакцией Фриделя — Крафтса
из бензола и С1СИ2С—С1, чем доказывается его строение.
II
О
Как ацетофенон, так и кетоны с удаленным положением карбонила
при действии серы и аммиака претерпевают замечательную реакцию
152 АРОМАТИЧЕСКИЕ КЕТОНЫ
Вильгеродта — превращение в кислоту с тем же числом углеродных
атомов:
Cell5-C-(CH2)«-CIl3 + S + NH3 — > С8Н5—(СН2)я+1—С—NH2
II 112 ||
о S
/ОН
* С8Н5 (СН2)л+1 — Сух -T-NH34-H2S
О
К этой реакции способны и жирные кетоны (Р. X. Френдлина).
Чисто ароматический кетон бензофенон заметно более пассивен, чем
ацетофенон, в реакциях присоединения по карбонилу (например, не при-
соединяет NaHSO3) и в реакциях замещения, которые проходят гораздо
медленнее.
Для чисто ароматических кетонов характерно расщепление концен-
трированной щелочью при высокой температуре:
СвЩ-С-СбНз-Н NaOH
II
О
--> С8Н5-С-ОNa 4-CeHe
О
Реакции электрофильного замещения направляются в этих кетонах
в основном в мета-положение к карбонилу.
Бензофенон переводится ближним ультрафиолетовым излучением в воз-
бужденное состояние и поэтому используется в фотохимических процес-
сах в качестве индуктора. Действие в качестве индуктора возбужденной
молекулы бензофенона на вторичный пропиловый спирт в отсутствие
кислорода и в его присутствии показано на следующих примерах (зве-
здочка обозначает фотохимически возбужденную молекулу):
с8н5-с-с8н5
11 у
—> ГСвН5-С-С6Н51*
о
(С6Н5)2С-С(С6Н5)2
I I
но он
>
2 СбН5-С-СбН5'|*4-СН3-С-СН3 --> 2С8Н5-С-С8Н5 + СН3-С-СН3
о
Н ОН
о
он
+02
00
2СвН6-С—С8Н5
СНз-С-СНз
н он
ООН
2С8Н5—с—С6Н5 -Г СНз—с—СНз
ОН
ОН
о
2С6115-С-Свн54- НО-ОН
о
ОКСИМЫ КЕТОНОВ; БЕКМАНОВСКЛЯ ПЕРЕГРУППИРОВКА
153
Некоторые производные бензофенона также возбуждаются светом,
отдавая поглощенную энергию в виде из лучения — флуоресцируют.
2,2'-Диокси-4,4'-диметоксибспзофеноп под названием увинол применяется
для защиты тканей и пластмасс от разрушающего действия света, так
как «принимает удар на себя» и затем отдает поглощенную энергию уже
в виде безвредного, более длинноволнового флуоресцентного излучения.
Бензофенон, так же как и ацетофенон, при освещении присоединяется
к олефинам, образуя четырехчленные гетероциклические простые эфиры:
СвН6ч , СвН.т'
\с=о+ С=С< -------> ZC—О
СвНь/ ' х свн5' ; ।
-с-с-
Оксикетоны типа ’ '
О
II ОН
где R ~ С18Н17 или С12Н25 под фирменным названием цианосорб при-
меняются для защиты пластмасс от разрушительного действия ультра-
фиолетовой, коротковолновой и видимой части спектра.
Своеобразная реакция присоединения натрия осуществляется при
действии металла на ароматические кетоны (Шленк). При этом обра-
зуются глубокоокрашенные парамагнитные (нечетный электрон!) металл-
кетилы с трехвалептным углеродом
(CeH5)2CO + Na > (CeII5)2C-O-Na
легко окисляющиеся в кетоны.
В жирном ряду существование металлкетилов для пространственно
затрудненных кетонов доказано А. Е. Фаворским и И. Н. Назаровым.
Оксимы кетонов и бекмановская перегруппировка
Несимметрично построенные кетоны R—С—-R', как ароматические, так
II
О
и жирные, образуют с гидроксиламином каждый два изомерных оксима,
различающихся физическими константами (в частности, температурой
плавления) и поведением в бекмановской перегруппировке (кн. I, стр. 490).
Так, о-метилбензофенон при действии NH2OH образует два изомерных
оксима с т. пл. 105 и 69° С.
Исследование этой изомерии привело к выводу, что это сии-анти-'азо-
мерия, подобная уже рассмотренной для азосоединенип (стр. 79)
о-СНзСвШ—С—СвН5 о-СИ.чСвП4—С—С6П5
II I'
:N—ОН 1IO-N:
еин-фепил-о-толилкетоксим ашни-фенил-о-толплкетоксим
154
АРОМАТИЧЕСКИЕ КЕТОНЫ
и аналогичная цис-транс-изомерии олефинов и их производных:
uuc-изомер
транс-из о м ер
Как было сказано ранее, в геометрической изомерии соединений азота
вместо приставки цис- применяют приставку син-, а вместо транс----
анти-.
В настоящее время син- или антпи-конфигурацию оксима можно уста-
новить по его дипольному моменту, если хотя бы один углеводородный
радикал кетона имеет дипольный момент, направленный известным нам
образом. Для этого требуется наличие в одном из радикалов заместителя,
такого, например, как галоид в пара-положении.
Чисто химическим путем конфигурация была установлена (Мейзенгей-
мер) для следующих оксимов:
НО—N:
син-
:N — ОН
анти-
син-2-Бром-5-нитроацетофеноноксим, содержащий подвижный благо-
даря наличию п-нитрогруппы атом брома, при действии щелочи замы-
кается в бензизоксазольный гетероцикл
но—N: о—n:
тогда как его «ниш-изомер не способен к такому замыканию.
Поскольку при циклизации не затрагиваются связи атомов, ответ
ственные за конфигурацию (^>C = N—), то замыкание в цикл происходило
без изменения конфигурации, и, значит, способный к замыканию изо
мер — снн-соединение, с близко расположенными бромом и гидроксилом^
Другой чисто химический путь прямо противоположен: он основ
на размыкании гетероцикла.
ОКСИМЫ КЕТОНОВ; БЕКМАН ОВСКАЯ ПЕРЕГРУППИРОВКА
155
Гетероцикл приведенного ниже строения подвергается озонированию,
затрагивающему только кратную связь С = С, но не C = N:
П Ш
R
o=c-c-R" , V
|| + R —С—ОН
НО—N
IV
Полученный в результате озонолиза сложный эфир оксима III гидро-
лизом превращается в монооксим IV дикетона R—С—С—R ". спн-Кон-
II II
О О
фигурация оксимной группы по отношению к радикалу R—С— следует
II
О
из способа его образования (тоже не затрагивающего связей группировки
>C=N—).
Оксимы при действии на них кислот, кислот Льюиса или РС15 пре-
вращаются в замещенные амиды кислот по схеме:
R С R н+
Г HO—C-R.
0—С—R' Нг0
N—ОН
R—С—R'
И Н+
НО—N
N—R
Г R-C-OH1
R'—N
NH-R
R С—О Н2о
R'—NH
О=С—R'
I -|-RNHa
ОН
R—С=О
I -J-R'NHj
ОН
Стереоизомерные оксимы при этом превращении, называемом по имени
автора (Бекманн, 1886 г.) бекмановской перегруппировкой, образуют струк-
турно изомерные замещенные амиды, строение которых легко устана-
вливается гидролизом в первичный амин и кислоту — разные для двух
стереоизомерных исходных оксимов.
Несколько слов о механизме перегруппировки, не разъясненной преды-
дущей схемой. При бекмановской перегруппировке сначала отщепляется
гидроксил оксимной группы в виде аниона, уводимого Н+-ионом, а к остав-
шемуся положительно заряженному азоту синхронно с отщеплением ОН-
мигрирует из анттш-п сложения по отношению к гидроксилу радикал
156 АРОМАТИЧЕСКИЕ КЕТ ОН Ы
(с парой электронов), оставляя карбониевый катион, захватывающий
гидроксил из среды:
R-C-R С— R' HO-C-R O=C-R'
i |l '—х —> || + ОН' —> II —► I
S-N—Ч>Н’Н* R—N R —N R — NH
,7 5 6 г
По правилу Эльтекова — Эрлепмепера промежуточное соединение в
с гидроксилом у углерода при двойной связи перегруппировывается
в амид г. То, что бекмановская перегруппировка совершается за счет
миграции антн-радикала, а не ближераспсложенного снн-радикала, дока-
зано Мейзенгепмером на примере приведенных выше оксимов с устано-
вленной им химическим путем конфигурацией:
R—С-С-ОН
II II
+ R—NH
хп„
N—ОН
Бекмановская перегруппировка известна и для жирных и алициклических оыя
мов. Она пмеет промышленное значение при получении капролактама и из него высок
молекулярного поликапроамида — капрона:
н+ t
нагревание
капролактам
ДИКЕТОНЫ
157
--► . . . — NH— (СН2)5—С—NH-(CH2)6—с—nh—(сн2)5—с—...
В некоторых случаях наряду с нормальной, описанно!! только что
бекмановскоп перегруппировкой происходит бекмановская перегруппи-
ровка второго пода. Например, при обработке стереоизомерных моноокси-
мов а-дикетона С6Н-;—С—С—С6Н5 толуолсульфохлоридом CH3CeH4SO,Cl
11 II
О О
в пиридине протекают следующие превращения с образованием бензой-
ной кислоты и, в случае одного стереоизомера оксима — бензонитрила,
а в случае другого — фенилкарбиламина (изонитрила):
С6Н5—С—С—Cglia
II II
НО—N О
с6н5-с—ОН ОН
II -----► С6н5—C=N-|-CeH5—С<\
СвН5—С—N Z
II
О
СбН6—с—с—С6Н5 но-с—с—свн5 он
II ------► I II -----------> CeH5-NsC:H-CeH5-C<x
О I О 'О
N—OH N—СвН5
Из приведенных схем видно, что бекмановская перегруппировка вто-
рого рода «ндги-кетоксима родственна уже упоминавшемуся расщеплению
.анти,-альдоксимов с образованием нитрилов (стр. 145):
С6Н5 С Н jcHeCObO^ c6H5~C=N-I-2CH3-С-ОН
HO—N II
О
ДИКЕТОНЫ
Простейший представитель ароматических и-дикетонов CSH5C—ССвНБ
II II
О О
носит исторически сложившееся название бензил (не путать с радикалом
СвН5СН2 —, также называемым бензилом). Этот 1,2-дикетон можно полу-
чить’.окислением бензоина (стр. 146):
СвНБ—СН—С-С6НБ
I II
ОН о
С6НБ—с—с—свнБ + н2о
Бензил образует три стереоизомерных диоксима
С6Н5—С----------С—С6Н5
II И
N\ ZN
он по
свнБ—с—с—с6н5
II II
/N N\
н 6 он
свн5-с—с-с6нБ
II II
/N yN
НО но
3-, или син-бепзилдиоксим
(т. пл. 206° С)
а-, или ангпи-бензил-
диоксим
(т. пл. 237° С)
7-, а.идви-, или син-анти-
бепзилдпоксим
(т. пл. 166° С)
158
АРОМАТИЧЕСКИЕ КЕТОНЫ
Наиболее значительное свойство бензила — его перегруппировка
в бензиловую кислоту, легко и количественно протекающая при Действии
щелочи на спиртовые растворы дикетона. Механизм бензиловой пере-
группировки представляют так:
С6Н5—С —С-С6Н5 + NaOH--> С6Н5 —С—С^С§Н^ >
О О Со О О-
Н Na+
С6Н5 о с6н5 о
> /С-С ----------^С—
С6н5х I ОН С6н5 I О' Na+
Перегруппировки типа бензиловой — довольно широко распростра-
ненное явление не только в ароматическом ряду. К ней способны также
алициклические 1,2-дикетоны, причем в этом случае перегруппировка
протекает с сокращением цикла на одно звено.
^-Дикетоны ароматического ряда получаются уже описанными мето-
дами конденсации (стр. 151). Как и их жирным аналогам, им свойственно
явление кето-енольной таутомерии
СвН6-С-СН2-С-СбН6 С6Н5-С=СН-С-СвН5
II II I II
0 0 он о
и способность образовывать хелатные соединения с катионами многих
металлов (кн. 1, стр. 416). Р-Дикетоны дают и натриевые производные,
которые алкилируются по углероду:
с6н6-с=сн-с-с6н5 — - С6Н5—С—СН—С—с6н5
I II II I II
О- Na+ О О R О
Непредельные кетоны с системой сопряженных связей С = С—С=0
образуются кротоновой конденсацией ароматических альдегидов с кето-
нами, например:
С6Н6-С=О4-СН3-С-СНз —► СвН6-СН=СН-С-СНз
I II II
НО о
бензальацетон
2С6Н5-С=О + СН3-С-СНз —> свн5-сн=сн-с-сн=сн-свн5.
I II II
но о
дибензальацетон
СвН6-С=О4-СНз—с-свн6 —> СвН5-СН=СН—с-с6н5
। о А
но о
бензальацетофенон
хиноны
159
Свойства их типичны для кетонов с сопряженными связями
С=С-С = 0 (кн. I, стр. 316).
ХИНОНЫ
При окислении пирокатехина и гидрохинона
•ственно п-бензохиноны:
^ОН
3 КЗГ + Н2Сг2С>7
Ч''Ч)Н
образуются о- и соответ-
+ Сг2О3 + 4 Н2О
n-Бензохинон открыт в 1837 г. А. А. Воскресенским.
Хиноны — окрашенные (и-хинон — желтое, о-хинон — красное) воз-
гоняющиеся при нагревании кристаллические вещества с резким запа-
хом. Они крайне активны химически и ведут себя как непредельные кетоны
с сопряженной системой связей С=С—С—0. Строение хинонов следует
из методов их синтеза и свойств.
Приведем методы синтеза и-хинонов.
1. n-Бензохинон образуется в ряде реакций окисления фенола. Поня-
тие о механизме этих реакций может дать хорошо изученное окисление
фенола солью Фреми• О—N(SO3K)2, которая представляет собой свобод-
ный радикал — одноэлектронный окислитель:
(KO3S)2N—О* + но^С?>
=О
(KO3S)2N—Оч
Н'
(KO3S)2NH
2. Получение и-бензохинона окислением анилина хромовой кислотой
проходит через стадию фенилгидроксиламина, который перегруппировы-
вается в п-аминофенол, окисляемый в монохинонимин. Хинонимин ги-
дролизуется в хинон:
3. Конденсацией в щелочной среде жирных 1,2-дикетонов получают
2,5-диалкилбензохиноны:
160
ХИНОНЫ
СНз СН3
С=о СНз с=сн
О=С + С=О -It, О-С С-0
CH1 °=f nc-cZ
СНз I
СН3
4. При взаимодействии пентакарбопилатжелеза Fe(CO)5 с диметил-
ацетиленом Реппе получил комплекс дурохинона с Fe(CO)3
О
II
н3с сн3 н3с zcx хсн3
С С Ге(СО), С С , v
III + III ---------> |L__[^=Fe(CO)3
С с с с
Н3С СНз Н3С V сн3
разложение которого кислотой дает сам дурохинон.
Из ацетилена и карбонила железа (через комплекс иного состава)
получается гидрохинон.
5. Действием царской водки (HNO3-|-HC1) на производные бензола,
в частности на анилин, получают тетрахлор-п-бензохинон (хлоранил)
О
С,\А/С1
II II
О
Реакции бензохинонов
Как уже было сказано (стр. 129), хиноны при восстановлении превра-
щаются в исходные диоксибензолы.
/г-Бензохинон при облучении димеризуется (Куксон):
О
О
о
Окисление n-бензохинона надсерной кислотой (в присутствии Ag )
приводит к образованию малеиновой кислоты:
НС— СООН
О — / \=О 35Оз(ООН)2'. Ag+ и
\=/ нс—соон
Эта реакция важна для доказательства строения бензохинона.
хиноны
161
ющи 3 реаКЦИЙ п‘бензохпн°на как непредельного кетона приведем следу-
С гидроксиламином и-бензохипон образует диоксим:
О N-OII
А /
li t|+2NH2OI-{ -> llA +2Н2О
II V"
О N—ОН
Монооксим «-бензохинона
рен с нитрозофенолом:
N—ОН N=O
таутоме-
Частичное превращение монооксима
в нитрозофенол наступает лишь в рас-
творах. На рис. 91 приведены кривые
поглощения «-нитрозофенола в различ-
ных растворителях. Интенсивность по-
глощения в области 720—735 ммк яв-
ляется мерой содержания в растворе
нитрозоформы, для которой характерен
максимум поглощения в этом диапазо-
Рпс. 91. Кривые поглощения га-пптро-
зофенола в разных растворителях:
1 — этилацетат; 2— ппридпп; 3 — диоксан;
4 — хлороформ.
не длин волн.
Неорганические кислоты (слабые и средней силы), органические кис-
лоты, спирты, первичные и вторичные амины реагируют с «-бензохиноном,
присоединяясь к нему, как к непредельному кетону, в 1,4-положение
(кн. I, стр. 318). В результате этого наступает превращение хиноидной
системы в бензоидную и образуется замещенный гидрохинон:
где НХ — кислота, спирт или амин; X = Cl, —N=N=N, ОССН3,
-S2Oi, -ОСН3, -NHC6H5 и др.
Этот замещенный гидрохинон может окислиться избытком хинона
в замещенный хинон
ОН
ОН
11 Заказ 67
162
хиноны
способный вновь присоединить молекулу НХ:
Сходным образом
<рид:
может быть присоединен к хинону и уксусный ангид-
0
II
|| || + (СН3-С)2О
II О
0=С-СНз
енолизация
о
О=С-СН3
I
О
А
у ।
Х//\Э-С-СН3
in «
В данном случае присоединение заканчивается на этой стадии. Таким
путем получают (после гидролиза) 1,2,4-триоксибензол:
О=С-СН3
I
О ОН
I I
н20(н^ /V +2СН.З-С-ОН
\^\)-С-СН8 \^0Н О
ОН А ОН
При освещении раствора хинона в альдегиде наступает (очевидно,
,гомолитическое) присоединение последнего к хинону (Чиамичиан):
О
Н
'Тпипг> ’ КРУГ веществ, присоединяющихся к хинону, значительно
ным ртггат В слУчае °бычных а, ^-непредельных кетонов. Дополнитель-
ная ПРИС°СДИН™ слУЖит здесь выделение энергии превраще-
ния хиноиднои формы в бензоидную.
серебра осмпп1иИ^т1Г^П'₽ОХИНОНа^в хпнон при проявлении освещенного бромистого
слоями биомпеопоН^1 Т3 спос,’бов цветной фотографии. Бумагу покрывают тремя
113 трех vtamJn Рянои эмульсии, из которых каждый сенсибилизирован к одному
слой введен пппаът™СПектра красному, желтому и сине-зеленому. В каждый
р итель-краситель, построенный по типу I (где К — остаток красителя
ХИНГИДРОН И СЕМИХИНОНЫ
163'
соответствующего цвета). На освещенных участках с «зародышами» металлического-
серебра проявитель-краситель окисляется в хинон II
ОН О
I II
ОН о
I II
количество которого соответствует количеству «проявленного» бромистого серебра и
почернению слоя в этом месте. После этого избыточный неиспользованный проявитель-
краситель вымывают щелочью, в которой он в отличие от хинона растворим. Остается
удалить закрывающее картину металлическое серебро, что и делается обработкой
окислителем в присутствии иона галоида и удалением образовавшегося галоидного
серебра тиосульфатом.
ХИНГИДРОН И СЕМИХИНОНЫ
При сливании растворов эквимолярных количеств п-бензохинона и гидро-
хинона выкристаллизовываются малорастворимые, почти черные кри-
сталлы хингидрона — молекулярного соединения этих двух веществ,
в котором молекулы связаны водородными связями и, кроме того, пере-
носом заряда от гидрохинона к хинону:
хкнгилрон
Если пометить, например, бензохинон, введя в него дейтерий, как
это сделали Миклухин и Грайгеров в лаборатории А. И. Бродского, то
оказывается, что две половины молекулы хингидрона не переходят, как
можно было бы ожидать, друг в друга.
Если же гидрохинон растворить в щелочи, т. е. удалить связующие
водородные атомы (протоны), то анион гидрохинона отдает один электрон-
хинону, и оба вещества превращаются в две одинаковые молекулы семи-
хинона:
Семихинон — это ион-радикал, имеющий бензоидную структуру, в кото-
ром мезомерно выравнено распределение электронов на обоих кислородных.
И*
ш
ХИНОНЫ
атомах, так что каждый кислород несет по половине электронного
заряда и по половине неснарешюго электрона, что символизируется фор
мулами:
Наличие неспаренпого электрона в семихинонах доказывается их пара-
магнитными свойствами. Таким образом, семихиноны — свободные ради-
калы со свободнорадикальными свойствами, рассредоточенными между
-обоими атомами кислорода. Вследствие такого рассредоточения семихи-
ноны довольно устойчивы и не проявляют обычной способности свободных
радикалов вырывать атом с неспаренным электроном из встретившейся
молекулы и таким образом инициировать цепную реакцию. Благодаря
способности превращаться в семихинон (под действием окислителей или
реакциями со свободными радикалами) гидрохинон является ингибито-
ром цепных реакций — вылавливает из реакционной среды свободные
радикалы, ведущие цепь:
анион
гидрохинона
семихинон
Поскольку семихинон (его анион) может заполучать один электрон,
превращаясь в анион гидрохинона, и терять один электрон, превращаясь
в хинон, семихиноны (и хингидрон) часто применяются в качестве «ре-
докс-систем», служащих посредниками в передаче электрона.
%
хинонимины
Подобно тому как при окислении гидрохинона получается п-бензохинон,.
окисление лг-фенилендиамина приводит к образованию п-бензохинонимина.
H2N
N Н2 —— > HN:
NH + Н2О
Гидролизуясь, хинонимин дает хинон.
хинонимины
165
Окислением смеси n-фенилендиамина с анилином получают простейший
индамин, аналогичной реакцией с фенолом — индофенол:
индамин
и н иол
Простые п замещенные индофенолы н индамины— спине красители. Их применяют
главным образом для синтеза более сложных красителей. В частности, индофенолы
разнообразного строения используют для получения так называемых сернистых краси-
телей. Для этого индофенолы обрабатывают полисульфидами натрия. Процессы, кото-
рые при этом разыгрываются, можно представить схемой:
д
Реакции а-*- б и в-+ г представляют собой обычные присоединения к хинонам
(стр. 1G1), реакции б-> в и г-> д это окисление в хиноидные формы. В последней
фазе идет циклизация в гетероцикл тиазин и сшивка фрагментов с дисульфидными
мостами. Вместо фенила в формуле а и последующих могут быть разнообразные арома-
тические ядра.
О сернистых красителях см. также в разделе «Гетероциклы», стр. 322.
Окисление смеси п-фенилендиамина с фенолом в пндофепол (см. выше реакцию 2)
может быть осуществлено, в частности, бромистым серебром. Этот процесс применяется
в другом способе цветной фотографии и цветного кино. В светочувствительный слой
вводят наряду с бромистым серебром вторую компоненту реакции — краситель
фенольного характера или с активной метиленовой группой. Лишь некоторые краси-
тели этого рода являются индофенолами. Однако по способу образования все эти
красители родственны между собой и потому упоминаются здесь вместе. Изображенные
ниже вещества I, Il u III служат такими компонентами для воспроизведения трех основ-
ХИНОНЫ
166
ных тонов- I — ДЛЯ спне-зелсного, II — для желтого и Ш — для пурпурно-красного.
От этих веществ требуется нерастворимость в воде и отсутствие способности диффун-
дировать в слое. Для этого и служат боковые цепи (С17Н35 пли С18Нз7). Подвергну-
тую экспозиции трехслойную пластинку, содержащую в каждом слое вещества I, щ
III и бромистое серебро, сенсибилизированное в данной,области спектра, проявляют
затем N' N'-замещенным п-фенилендиамином, который восстанавливает бромистое
серебро на освещенных при экспозиции участках и, окисляясь, сочетается в индофе-
нольпый (плп ему подобный) краситель по реакциям:
4-4Ag + 4HBr
4AgBr ,
0=С — NH — СО — С17Нзв
nh2+h..c .СООН
О=С— NH—
тт \COOH
О=С-< ^-NH-CO-C17H35
4-4Ag + 4HB₽
СООН
0=C-NH-^_^
\соон
Соль серебра и серебро затем удаляют,
R2N——N=C-----------С—С17Н354-
O=C N
4-4Ag+4HBr
о
как было описано ранее-
ТЕТЕРОФУНКЦИОНАЛЬНЫЕ АМИНЫ
167
ГЕТЕРОФУНКЦИОНАЛЬНЫЕ АМИНЫ
€ АМИНОГРУППОЙ В БОКОВОЙ ПЕГГИ
Мы не описывали способов получения и свойств простейших аминов
с аминогруппой в боковой цепи (и соответствующих четвертичных осно-
ваний), подобных бензиламину, дибензиламину, трибензиламину или
тетрабензиламмониевым солям, так как по способам получения и свой-
ствам они подобны своим алифатическим аналогам. Здесь мы приведем
несколько аминов с 0-положением аминогруппы в боковой цепи аромати-
ческого ядра, поскольку они имеют важное физиологическое или терапев-
тическое значение.
Бензедрин СвН5СН2—СН—СН3 — снимающее сон и усталость воз-
NHa
буждающее вещество; может быть получен из бензилметилкетона дей-
ствием формамида (реакция Лейкарта). Адреналин — гормон, выделяемый
корой надпочечников. Сужает кровеносные капилляры, ускоряет сердце-
биение, повышает кровяное давление, оказывает мобилизующее дейст-
вие: в момент опасности адреналин выделяется в кровь. Он служит
(наряду с норадреналином) медиатором нервов симпатической нервной
системы, играя здесь ту же роль, какую в парасимпатической нервной
системе (системе блуждающего нерва) играет ацетилхолин (кн. I, стр. 365).
Строение адреналина следует из превращения его сплавлением со щелочью
в протокатеховую кислоту и из данных функционального анализа,
согласно которым это вторичный амин и вторичный спирт, окисляющийся
в кетон — адреналон:
НО—сн—СН2—NHCH3
но/ ОН
НО-/ /—C-CH2-NHCH3
>=/ II
HOZ о
адреналин
НО—/ СН—CH2NH2
но/ он
норадреналин
адреналон
но—Ч—соон
но/
протокатеховая кислота
Адреналон можно синтезировать
из пирокатехина следующим путем:
С1СН2-С-С1+НО-<^
но/
О
CICH2—С-0-
перегруппировка
Фриса
НО—/ Ч-С-СН2С1
II
НО/ о
nh2ch,_
НО-/ V-C-CH2-NHCH3
>=/ II
НО/ о
168
гетерофункциональные амины
Таблица 68. Бензойная кислота и ее замещенные
Формула Название кислоты Температура плавления, °C Температура кипения, °с
свн5соон Бензойная 122,0 249,0
СНзСбЩСООН Толуиловая
о-, или 1,2- 104; 108 259,2
31-, пли 1,3- 109; 112 263
п-, или 1,4- 179,6 275
С1СвН4СООН Хлорбензойная
о-, или 1,2- 142 Возг.
м-, пли 1,3- 158 Возг.
и-, или 1,4- 243 Возг.
no2c6h4cooh Нитробензойная
о-, пли 1,2- 147,5 —
м-, или 1,3- 141,4 —
п-, или 1,4- 242,4 Возг.
nh2c6h4cooh Ампнобензойная
о-, или 1,2- (антраниловая) 145 Возг.
м-, или 1,3- 174; 179,5 Возг.
п-, или 1,4- 187 —
H0CeH4C00H Оксибензойная
о-, или 1,2- (салициловая) 159 76 (возг.)
м-, или 1,3- 201,3
п-, или 1,4- 213,0 76 (возг.)
ггтпгттт тановлением адреналона получают рацемический адреналин и рас-
ттопппШ0Т еГ° С ®иннои кислотой на антиподы. Природный адреналин —
ращающии, только он обладает сильным физиологическим действием.
ескалин алкалоид кактусов Anhalonium — вызывает отравление,
до ное опьянению — эйфорию, красочные галлюцинации. Он может
ыть получен присоединением синильной кислоты к альдегиду триметил-
алловои кислоты и каталитическим восстановлением полученного окси-
нитрила:
снзОх СН80^
СН3О-<^ СНзО—<^~4-CH-C=N
>=z Н \=z I
СНзСК СНзСИ он
сн3ох
--> СНзО-<^~Ч—сна-сн2—nh2
СН3(Х
мескалин
ароматические карбоновые кислоты
169
Плотность dl0 ка (при 25° C) Показатель преломления пд Дипольный момент в диоксане при 2 5° С D
1,2659 (dj®) 6,46-10-5 4,19 1,5397 (при 15° C) 1,78 (при 30° С)
1,062 (dp®) 1,350 • 10-5 3,91 1,512 (при 115° С) 1,70 (при 30° С в бензоле)
1,054 (при 112° С) 5,32 • 10-6 4,27 1,509 (при 111,6° С) 2,05 (при 30° С в бензоле)
— 4,33-10-5 4,36 — —
1,544 1,20-10-3 2,92 — 2,43 (при 30° С)
1,496 (dp) 1,51 • 10‘4 3,82 — 2,20 (при 30° С)
1,541 (dp) 1,04 • IO'4 3,98 — 2,00 (при 30° С)
1,575 (d|) 6,95- 10-3 (при 18° C) 2,16 — —
1,494 (dp 3,4 • IO’4 3,47 — 4,03
1,550 (dp) 3,93 • IO'4 3,41 — 4,02
1,07 • 10-7 4,97 — 1,51
1,511 1,67- 10-5 4,78 — 2,70
— 1,2-10-5 4,92 — 3,51
1,443 1,07-10'3 2,97 1,565 2,63
1,473 (df) 8,7 • IO-6 4,06 — 2,37
1,443 3,3 • 10-5 4,48 — 2,73
В растениях эфедра содержится ароматический аминоспирт — алка-
лоид эфедрин С6Н5СН (ОН) СН (NHCH3)CH3, применяемый как антиспаз-
матическое средство при бронхиальной астме. Два его асимметрических
углерода имеют эр^тпро-конфигурацию.
АРОМАТИЧЕСКИЕ КАРБОНОВЫЕ КИСЛОТЫ
Ароматические кислоты обычно носят тривиальные названия:
С6Н5СООН — бензойная кислота; СН3С6Н4СООН — толуиловые кислоты
(о-, м- и ц-); о-С6Н4(СООН)2 — фталевая кислота. Названия жирноаромати-
ческих (арилжирных) кислот, т. е. соединений с карбоксилом в боковой
цепи, производятся от названий соответствующих алифатических кислот,
например С6Н5СН2СООН — фенилуксусная, или же применяются три-
виальные названия (табл. 68).
Способы введения карбоксила в ароматическое ядро
Наибольшее значение имеют методы окисления ароматических углеводо-
родов. При энергичном окислении боковые цепи гомологов бензола «от-
горают» и образуются соответствующие ароматические кислоты.
170
АРОМАТИЧЕСКИЕ КАРБОНОВЫЕ КИСЛОТЫ
В качестве окислителя применяют хромовую кислоту, щелочной раств
перманганата. Так, из толуола получают бензойную кислоту, из ксил°-
лов — фталевую, изофталевую или терефталевую кислоты: °"
СН3
о=с—он
бензойная
кислота
сн3
о=с—он
о=с-он
терефталевая
кислота
Высшие гомологи бензола (с одним алкильным заместителем) также-
могут быть окислены до бензойной кислоты:
С6Н5С2Н5 --> СбНбСООН + СО2+2Н2О
Фталевую кислоту получают окислением нафталина кислородом
воздуха над пятиокисью ванадия при 450° С; при этом «отгорает» одно
из ароматических колец:
О
'V'c—он
Таким образом, углеводородная боковая цепь в ароматическом соеди-
нении окисляется по а-углеродному (по отношению к ароматическому
ядру) звену.
Специфическим для ароматического ряда является синтез карбоновых
кислот по Фриделю — Крафтсу; если в качестве ацилирующего агента
использовать фосген или хлоругольный эфир, то в ароматическое ядро
вступает карбоксильная группа:
Ar—Н + С1СОС1 Ar—COCI Ar—С-ОН
II
Аг—Н + С1С-ОС2Нб Аг-С-бС2Н5
II II
о о
Цианогруппу (а следовательно, потенциальный карбоксил)
ввести на место сульфогруппы сплавлением солей сульфокислот с Д
стым натрием или на место аминогруппы — через диазосоединен
CeH6SO3Na4-NaCN --> CeH5CN + Na2SO3
2C6H5N2 -SO4H + Cu2(CN)2 > 2CeH5CN + 2N2+Cu2SO4 + H2SO4
Эти методы также не имеют аналогии в алифатическом ряду.
БЕНЗОЙНАЯ КИСЛОТА И ЕЕ ФУНКЦИОНАЛЬНЫЕ ПРОИЗВОДНЫЕ
171
Магнийорганические соединения сохраняют свое значение и для син-
теза ароматических кислот:
ArMgBr + СОг > Аг—С—OMgBr Аг—С—OH-f-MgBrCl
II II
О О
Кислоты с карбоксилом в ароматическом ядре образуются также при
карбоксилировании натрий- и литийарилов, которые получаются при взаи-
модействии галоидного арила с металлом или металлированием аромати-
ческого соединения, например:
СвН§Т>г —2Li > СбН&Ы-р LiBr; CeHgLi-j-CC^ > СвНзС—OLi
II
О
CeHe-|-C^jHgNa -> CeHoNa+ С2Нв; CeHsNa-j-CC^ -> CeHjC—ONa
II
О
Что касается кислот с карбоксилом в боковой цепи, то они могут быть
синтезированы обычными методами, применяемыми в алифатическом ряду,
и в особых случаях — некоторыми специфическими методами (см. ниже).
БЕНЗОЙНАЯ КИСЛОТА И ЕЕ ФУНКЦИОНАЛЬНЫЕ
ПРОИЗВОДНЫЕ
Ароматические кислоты — кристаллические вещества, обычно несколько
растворимые в воде и хорошо растворимые в полярных органических
растворителях. Константы их диссоциации немного выше, чем у гомоло-
гов уксусной кислоты (но ниже, чем у муравьиной). Это связано с боль-
шим положительным индуктивным эффектом алкилов сравнительно
с фенилом. Ароматические кислоты обладают всеми общими свойствами
карбоновых кислот и в том, что касается поведения карбоксильной группы,
мало отличаются от карбоновых кислот алифатического ряда. Отличия,
когда они имеются, носят скорее количественный, чем качественный харак-
тер. Все известные функциональные производные получены и для бензой-
ной кислоты:
c6H5-c-o- +nh4 _в II О * NH, нон F СвНб-С-OR св11б—u—Oil II ШО(Н+) II >0 0 Na ОН vC6H5—C-ONa-|-C6H6- II О [2О * cell5—с—nh2 2 - C6H5-C=N 0 NH, NaNS- C^-C N3 0 —-СбНб-С-С!-2^- 'СбЩ-С-ОХ 11 A 0 \ 0 /2 C0115-C-OH II 0 -C-Cl > / Cello— C\ 0 II II 0 \ 0/2
172 АРОМАТИЧЕСКИЕ карбоновые КИСЛОТЫ
---
Бензойная кислота содержится в некоторых природных смолах
получается почти исключительно окислением толуола. Она примени Н°
в производстве красителей и некоторых лекарственных веществ ЭЛи^
бензойной кислоты используются как высококипящие растворите ти
Большое промышленное значение имеет хлористый бензоил — хлоп
ангидрид бензойной кислоты (ацил бензойной кислоты называется бен-
зоилом, а анион — бензоатом'). Хлористый бензоил получают неполным
гидролизом бензотрихлорида, который в свою очередь готовят хлориро-
ванием толуола на свету: "
г, пгт +ЗС12(спет) п +Н-0
СвЩ СПз -ЗНС1 "* ^6^5 CCI3 СвН6—С—Ci
II
О
Кроме того, хлористый бензоил можно получить хлорированием бенз-
альдегида (реакция, необычная для алифатического ряда):
С6Н5-С-II + С12 > СвН6-С-С1 Н-НС1
II II
о о
Хлористый бензоил употребляется для получения перекиси бензоила
и надбензойной кислоты (гидроперекиси бензоила); находит широкое при-
менение как ацилирующее средство. С его помощью бензоилируют спирты,
фенолы, амины, вводят бензоил по реакции Фриделя — Крафтса и др.
Хлористый бензоил несколько пассивнее, чем хлорангпдриды жирных
кислот, в частности значительно медленнее гидролизуется. Это обстоя-
тельство используется в реакции Шоттена — Бауманна, которая заклю-
чается в бензоилировании в водно-щелочной среде. В раствор, например,
фенола в разбавленной щелочи постепенно вводят хлористый бензоил
(скорость бензоилирования фенолята натрия значительно превышает
скорость гидролиза хлорангидрида):
Ar—С—Cl-pArONa > Ar—С—О—Аг
II II
О О
Бензоилирование фенолов и аминов проводят также в оезводных
растворах в присутствии пиридина или третичных аминов.
Высокая реакционная способность хлорангидридов кислот по отно
шению к нуклеофильным агентам (воде, аминам и др.) связана с РеЗК®
уменьшением электронной плотности на углероде карбонильной гРУ“аК
вследствие одновременного оттягивания электронов как кислородом,
и атомом хлора. „ отности
В случае хлористого бензоила это уменьшение электронной пл ид
на углероде компенсируется в некоторой мере подачей электро
бензольного кольца:
БЕНЗОЙНАЯ КИСЛОТА И ЕЕ ФУНКЦИОНАЛЬНЫЕ ПРОИЗВОДНЫЕ
17г
Бензольное кольцо может служить как акцептором, так и донором
электронов. Достаточно вспомнить, что ароматические амины — более
слабые основания, чем жирные, вследствие затягивания электронной
пары азота бензольным кольцом; ароматические хлорангидриды тоже
более пассивны, чем жирные, но уже вследствие смещения электронной
плотности из бензольного кольца. Следовательно, в ароматическом ряду
наблюдается частичное нивелирование, сглаживание свойств заместите-
лей; бензольное кольцо выступает при этом в качестве своеобразного
конденсатора электронов на молекулярном уровне. Чем сильнее выражены
электроноакцепторные свойства заместителя, чем больше его «требования»,
тем больше подача электронов бензольным кольцом. В случае хлорангид-
/0
ридов кислот такое требование со стороны группы —С4Х велико,
\С1
и положительным индуктивным эффектом алкилов оно удовлетворяется
в значительно меньшей степени, нежели сопряженной системой бензола.
В самих карбоновых кислотах ситуация другая. Карбоксильная
группа — относительно слабый акцептор электронов, ее требования могут
быть удовлетворены подачей электронов со стороны алкильных групп,
которые обладают большим положительным индуктивным эффектом,
нежели фенил. Поэтому гомологи уксусной кислоты, кроме муравьиной,
слабее бензойной.
Реакция хлористого бензоила с перекисью натрия приводит к перекиси
бензоила
2С6Нб—С—Cl-j-Na2O2 > С6Н5—С—0-0—C-C6H5 + 2NaCI
которая является распространенным инициатором многих радикальных
и цепных реакций, в особенности реакций полимеризации, имеющих
огромное промышленное значение.
Надбензойную кислоту (гидроперекись бензоила) получают бензоилиро-
ванием хлористым бензоилом перекиси водорода в щелочной среде
C6H5-C-Cl+H2O2+2NaOH ----> C6II5-C-0-0Na + NaCl + 2H20
II I
о о
или действием метилата натрия на перекись бензоила:
CeH6-C-O-O-C—CeH5 + NaOCH3 > СвГ1б-С-0-0Ха + CeII5-C-OCH3
h II II II
0 0 0 0
Надбензойная кислота более устойчива, чем гидроперекиси жирных
ацилов, и может быть получена в кристаллической форме (т. пл. 41е С).
Однако чаще всего ее употребляют в момент образования и не выделяют
в чистом виде. Надбензойная кислота применяется главным образом
для синтеза сс-окисей из непредельных соединений (реакция Н. А. Приле-
жаева):
RCH = CHR'-CfiIT5-C-0-011 -> RCll-CJlR' + CeH5-C-OU
II \ /х И
О 0 0
174 АРОМАТИЧЕСКИЕ КАРБОНОВЫЕ КИСЛОТЫ
Для этой цели она более удобна, чем жирные надкислоты.
Бензонитрил, который может быть получен из бензойной кислоты
нагреванием ее амида с фосфорным ангидридом или введением цианогруццЬ1
в ароматическое кольцо по одному из упомянутых ранее способов, обла-
дает свойствами, общими для всех нитрилов. Характерна конденсация под
действием металлического натрия:
3C6HS-C=N
бензонитрил
С6Н5
I
С
n' \т
II I
CeH5-C C-CeH6
\ //
N
киафенин
В табл. 69 приведены константы некоторых производных бензойной
кислоты.
Таблица 69. Некоторые производные бензойной кислоты
Формула Название Температура плавления, °C Температура кипения, °C Плотность dp 4
С6Н5СООН Бензойная кислота 122 249 1,2659
С6Н5СООСН3 Метнлбензоат —12,5 199,6 1,0886 (dl°)
(С6Н5СО)2 О Бензойный ангидрид 42 360 1,199
С6Н5СОС1 Хлористый бензоил —1 197 1,2187 (41)
C6H5CONH2 Бензамид 130 290 1,341 (<Ф
CeH5CN Бензонитрил -13 190,7 1,0102 (dp5)
ПОЛИКАРБОНОВЫЕ АРОМАТИЧЕСКИЕ КИСЛОТЫ
Дикарбоновые кислоты бензола представлены тремя возможными
изомерами (табл. 70):
II
/\Л'-ОН
^^с-он
о=с-он
фталевая?
кислота’
изофталевая
кислота
О=С-ОН
терефталевая
кислота
Наиболее важна фталевая кислота, которая, как уже ы силоЛа.
получается в промышленности окислением нафталина ил киСдота
В связи с орто-расположением функциональных групп Ф™ нацоминает
Дает ряд циклических производных (в этом отношении
Таблица 70. Бензолполикарбоновые кислоты
Формула Кислота Температура плавления °C Температура кипения рс Плотность с/20 4
о-С6Н4 (СООН)2 о-Фталевая 206—208 (разл.) 191 (разл.) 1,593
л«-СвН4 (СООН)2 ж-Фталевая, или изофталевая 330 Возг. —
и-С6Н4 (СООН)2 n-Фталевая, или терефталевая Возг. —300 (возг.) 1,510
С6Н3 (СООН)з Гемпмеллитовая, или 1,2,3-бензол- трикарбоновая 190 Разл. 1,546
С6Н3 (СООН)з Тримезиновая, или 1,3,5-бензол- трикарбоиовая 350 <300 (возг.) —
СвН3 (СООН)з Тримеллито- вая, или 1,2,4- бензолтрикарбо- новая 224—225 — —
С6Н2 (СООН)4 Пи ромеллитовая, или 1,2,4,5-бен- золтетракарбоно- вая 264 — —
СвН (СООН)6 Бепзолпентакар- бомовая 238 (безв.) — —
С6(СООП)6 Меллитовая, или бензолгексакар- боновая 286 Разл. —
Константы диссоциации (в воде при 20° С) Дипольный момент ц при 25° G D
Ki К1 К» к, к.
1,123- Ю’3 3,90 • IO’6 (при 30° С) — — __ — 2,30 (в диокса- не)
2,4 • 10"4 2,5 • 10-3 — — — — 2,37 (в диокса- не)
2,9 • 10~4 3,5 • 10’3 — — — — —
1,60 10-3 6,3 • 10’3 1,35 • 10-е — — — —
7,5 • IO’3 1,28-10-4 2,0 • IO"6 — — — —
3,0 • IO"3 1,45 • 10’4 6,3 • io-е — — — —
1,20 • IO’2 1,34-Ю-з 3,2 • 10-6 2,35 • IO"6 — — __
1,60-10-2 1,85 • IO'3 1,08- IO"4 5,6 • IO’6 3,5 • 10-’ — —
4,0 • IO’2 6,4 Ю-з 4,9- IO"4 1,65-10-6 1,28-10-6 1,10-10-7 —
176
АРОМАТИЧЕСКИЕ КАРБОНОВЫЕ КИСЛОТЫ
малеиновую или янтарную кислоты). Фталевый ангидрид образуется да»
при нагревании фталевой кислоты:
Он представляет собой бесцветное кристаллическое вещество с т. пл.
130—131° С. При нагревании со спиртами фталевый ангидрид присоеди-
няет молекулу спирта с образованием кислых (или неполных) эфиров
фталевой кислоты, которые могут быть превращены в полные эфиры обыч-
ной этерификацией (в присутствии минеральных кислот):
СгН5ОН(Н+)
О
II
С—ОС2Н5
С—ОС2Н5
II
Фталевый ангидрид легко вступает в реакцию Фриделя — Крафтса, напри-
мер с бензолом, с образованием о-бензоилбензойной кислоты, которая
в более жестких условиях может быть превращена в антрахинон:
А1С13
------->
В присутствии H2SOi или
при нагревании с фенолами:
ZnCl2 фталевый ангидрид конденсируется
H2SO4
-н,о
•фенолфталеин
поликарг.оновые кислоты
177
При действии аммиака на фталевый ангидрид могут получаться два
продукта: фталаминовая кислота и фталимид (имид фталевой кислоты).
Действие водного аммиака на холоду приводит к фталаминовой кислоте,
а фталимид образуется при пропускании газообразного аммиака над
нагретым фталевым ангидридом:
nh4oh
------>
NHn
----->-
Фталаминовая кислота получается также при неполном гидролизе
фталимида, в свою очередь образующегося при нагревании фталаминовой
кислоты. Из фталаминовой кислоты готовят технически важный продукт —
антраниловую кислоту (см. ниже). Фталимид находит применение в синте-
зах Габриэля для введения первичной аминогруппы на место галоида.
При этом исходят из фталимида калия, который реагирует с галоидными
алкилами с образованием замещенного фталимида; последний при гидро-
лизе дает соответствующий амин и фталевую кислоту. Особенно важен этот
метод для получения алифатических аминов, содержащих в цепи другие
заместители, например:
Фталимид калия получается при действии на фталимид сильных основа-
ний, например алкоголятов калия или спиртовых растворов щелочи.
Таким образом, фталимид обладает слабыми кислотными свойствами, что
связано с присутствием двух электроноакцепторных групп непосредственно
при атоме азота. Еще более сильными кислотными свойствами обладает,
12 Заказ 67
178 АРОМАТИЧЕСКИЕ КАРБОНОВЫЕ КИСЛотта
например, сахарин (или имид о-сульфобепзойпой кислоты), у котопог
к азоту привязан мощный акцептор электронов — группа SO2. v
сахарин
Фталевый ангидрид легко восстанавливается до фталида — лактона
2-оксиметплбензойной кислоты, который может быть получен также нагре-
ванием этой кислоты:
О
,СН2ОН
с—ОН
Этерификацией фталевого ангидрида многоатомными спиртами полу-
чают полиэфирные смолы; в качестве этерифицирующей компоненты часто
используется глицерин. Образующиеся при этом глифталевые смолы
служат основой лаков и пленок.
Хлорангидрид фталевой кислоты (фталилхлорид) способен реагировать
как нормально (в соответствии со структурой хлорангидрида), так и,
в некоторых случаях, в соответствии с изомерной ему (таутомерной?)
структурой:
Эфиры фталевой кислоты и бутилового и октилового спиртов приме
няются как высококипящие растворители и пластификаторы. Диметил ,
диэтил- и дибутилфталаты — средства, отпугивающие комаров и гну
(репелленты .
Калиевая соль фталевой кислоты при 400° С в присутствии фт
цинка или кадмия изомеризуется в соль терефталевой кислоты. •*-еР®^,ла
левую кислоту получают в больших количествах окислением п-кси^
Она применяется в производстве полиэтилентерефталата, из кот р
формуется синтетическое волокно терилен («лавсан»).
ПОЛИКАРБОНОВЫЕ КИСЛОТЫ
179
Практически полиэтилентерефталат получают в промышленном мас-
штабе путем переэтерификации этиленгликолем диметилтерефталата:
О ОСН3
С
+ носн2—сн2он----►
//X
О ОСНз
Другие поликарбоновые кислоты, приведенные в табл. 70, имеют
меньшее значение.
При конденсации малонового альдегида в момент его образования или
при окислении мезитилена образуется 1,3,5-бензолтрикарбоновая — три-
мезиновая кислота:
СН С<
О' / ХОН
о сн2
110/ н2с о
I + II
НС^ сн
0 сн2
-знго^
о
он
НзС ^ /СНз
I II
КМпО*
*----- I
I /О СНз
с\
хон
Бензолгексакарбоновая (меллитовая) кислота была получена из мине-
рала меллита, представляющего собой ее алюминиевую соль. Меллитовая
кислота образуется также в результате окисления графита азотной
кислотой.
Эта реакция показывает, что графит построен из полимерноконденси-
рованных ароматических колец:
СООН
ноос СООН
НООС соон
соон
меллитовая кислота
12*
180
АРОМАТИЧЕСКИЕ КАРБОНОВЫЕ
кислоты
ЗАМЕЩЕННЫЕ КАРБОНОВЫЕ КИСЛОТЫ
Карбоксильная группа — заместитель второго рода ^хотя и более слабый
О /О \
чем — С<^ и —С<\ I, поэтому нитрование, сульфирование и галои-
ХН ХСН3/
дирование бензойной кислоты приводит в основном к мета-замещенным
продуктам. При нитровании образуется также довольно много о-изомера.
Нитрованием смесью дымящей азотной и серной кислот может быть полу-
чена 3,5-дипитробеязойная кислота:
СООН
СООН
*
СООН
Хлорангидрид этой кислоты используется для идентификации спиртов
благодаря хорошим выходам образующихся эфиров, сравнительно высоким
температурам их плавления и, наконец, относительно большому молеку-
лярному весу динитробензойной кислоты.
п- и о-Нитробензойные кислоты получают нитрованием толуола
с последующим окислением метильной группы в карбоксильную. Этим
путем можно синтезировать и другие нефункциональные производные
бензойной кислоты с такими заместителями, как бром, хлор или сульфо-
группа.
Сахарин, т. е. имид о-сульфобензойной кислоты (сульфинпд бензойной кислоты),
синтезируют следующим образом. Толуол сульфируют хлорсульфоновой^ кислотой,
при этом получается смесь о- и n-толуолсульфохлоридов. Кристаллический п-пзомер
обычно вымораживается из раствора в жидком о-изомере. После этого о-пзомер еще
содержит значительную примесь n-изомера; от нее избавляются на следующей етэдии.
Обогащенный о-толуолсульфохлорид обрабатывают аммиаком и образующи
о-толуолсульфамид окисляют до о-сульфамида бензойной кислоты, который ле
циклизуется:
ЗАМЕЩЕННЫЕ КАРБОН ЕОВЬТ КИСЛОТЫ
181
Импдный водород сахарина обладает кислотными свойствами и замещается иа
атом металла при действии, оснований. Обычно применяют кристаллогидрат натриевой
соли имида о-сульфобензойпой кислоты, называемой кристаллозой.
кристаллоза
Это вещество в 400 раз «слэше» сахара, т. е. раствор сахарина 1/400 концентрации
имеет тот же сладкий вкус, что и раствор сахарозы при концентрации, равной единице.
Свойства бензойных кислот и их производных зависят от заместителей
в бензольном кольце (стр. 168). Так, электроноакцепторные группы, осо-
бенно в пара-положении, повышают константы диссоциации кислот и
реакционную способность хлорангидридов, а также скорость этерифика-
ции этих кислот. Обратное влияние оказывают электронодонорные группы.
Это связано с повышением или понижением электронной плотности реак-
ционного центра;
Однако присутствие двух заместителей в орто-положениях, независимо
от природы этих заместителей, пассивирует карбоксильную группу.
Так, мезитпленкарбоновая и о-динитробензойная кислоты плохо этери-
фицируются спиртами в обычных условиях, а эфиры этих кислот гидроли-
зуются с большим трудом. Это связано с блокированием реакционного
центра объемистыми заместителями, затрудняющими подход реагента.
Представления о такого рода пространственных затруднениях были
развиты В. Мейером в 80-х годах прошлого столетия. В последнее десяти-
летие они приобрели новый смысл. Вопрос о том, будут ли накопления
пространственных факторов затруднять реакцию, можно решить, проана-
лизировав пространственные затруднения в исходном и переходном состоя-
ниях. Если в переходном состоянии пространственные затруднения больше,
чем в исходном (при введении каких-либо объемистых групп вблизи реак-
ционного центра), реакция затрудняется, так как энергетический барьер
между исходным и переходным состояниями повышается. Поэтому для
оценки пространственных затруднений нужно знать механизм реакции.
Проанализируем, например, гидролиз эфиров о,о'-динитробензойной и
мезитиленкарбоновой кислот.
В щелочной среде гидролиз эфиров идет всегда по бимолекулярному
механизму. В переходном состоянии к реакционному центру присоединено
четыре группы, тогда как в исходном соединении их всего три.
.О
Аг—СГ + ОН
OR
о- -]
Х.О
АГ—С—OR
ОН
6- J
.О
Аг-С< + OR
ОН
182
ОКСИВЕНЗОЙНЫЕ КИСЛОТЫ (ФЕНОЛОКИСЛптЬ1-
Всякое дополнительное увеличение объема одного из заместителе"
резче повысит потенциальную энергию более пространственно затпудне И
ного переходного состояния, чем исходного соединения. Таким образом
щелочной гидролиз эфиров этих кислот сильно затрудняется. ’
Кислотный гидролиз сложных эфиров может идти и по мономолеку
лярному механизму:
/О н+ +
Аг-С<^ -----► [Ar-C=O] + HOR
\QR
hso I
t
/О
Для эфиров о, о'-динитробензойной кислоты мономолекулярный гидролиз
практически невозможен, поскольку нитрогруппы мешают образованию
положительно заряженного центра.
В соответствующем катионе мезитиленкарбоновой кислоты метильные
группы облегчают рассредоточение заряда. Такой катион оказывается
более устойчивым, и пространственные затруднения могут лишь ненамного
повысить его энергию; во всяком случае они больше скажутся на энергии
исходного эфира, поскольку в катионе к углероду присоединено всего два
остатка: кислород и ароматический радикал. Поэтому в сильнокислой
среде гидролиз эфира мезитиленкарбоновой кислоты проходит легко и
именно по мономолекулярному механизму *5^1:
ОКСИВЕНЗОЙНЫЕ КИСЛОТЫ (ФЕНОЛОКИСЛОТЫ)
Фенолокислоты могут быть получены введением карбоксилов в фенолы или
оксигруппы в бензойные кислоты.
Наиболее важную о-оксибензойную,или салициловую, кислоту получ
в промышленности по реакции Кольбе нагреванием (130 С) сухого Фе
лята натрия под давлением (5 ат) двуокиси углерода:
“О Na+
н
^/Дхс=о
ОКСИВЕПЗОЙН ЫЕ КИСЛОТЫ (ФЕНОЛОКИСЛОТЫ)
183
Если исходить из фенолята калия, получается не о-, а п-оксибензой-
на я кислота.
С многоатомными фенолами, например с резорцином, эта реакция
проходит легче. Так, резорциловая кислота образуется уже при нагре-
вании резорцина с раствором бикарбоната аммония:
I
ОН
Салициловую кислоту можно также получить окислением салицилового
альдегида или действием на фенолят натрия четыреххлористым углеро-
дом и щелочью (стр. 122).
Салициловая кислота значительно сильнее бензойной и своих изоме-
ров. Такой орто-эффект оксигруппы объясняется образованием водород-
ной связи между функциональными группами, что увеличивает 6+ заряд
на карбоксильном углероде:
Салициловая кислота образует два ряда производных (по обеим функци-
ональным группам). Хлористыми ацилами или ангидридами кислот она
ацилируется по оксигруппе (т. е. выступает в качестве фенола):
II
О—С—СН3
+ сну—с—ОН
II
о
аспирин
Хлорангидрид салициловой кислоты образует с фенолами или спир-
тами сложные эфиры другого типа (по карбоксилу), например:
|| • II
__С—ОН ос6н5
(Qf + РОС13 + С6н5он—> [of
^ОН ОН
салол
184
ОКСИ БЕНЗОЙНЫЕ КИСЛОТ
Ы (ФЕНОЛОКИСЛО ТЫ)
Аспирин употребляется в качестве жаропонижающего и анальгети
ского средства, салол является антисептиком, рекомендуемым при желуде?"
но-кишечных заболеваниях. Сама салициловая кислота — сильное дези4'
фицирующее средство; ее натриевая соль применяется как лекарство цп~
суставном ревматизме. Б больших масштабах салициловая кислота потпеб
ляется в производстве азокрасителей.
Гидрирование салициловой кислоты приводит к пимелиновой кислоте-
промежуточно образуется тетрагидросалициловая кислота, которая
гидролизуется по типу «кислотного расщепления» ацетоуксусного эфира-
тетрагидросалициловая кислота
--> НО—С—(СН2)5—С—ОН
пимелиновая кислота
Метиловый эфир салициловой кислоты встречается в виде гликозида
в различных растениях и применяется в медицине.
.ч-Оксибензойная кислота получается при щелочном плавлении
м-сульфобензойной кислоты
COONa
SO3Na
2NaOH
------>
COONa
+ Na2SO3 + H2O
и также употребляется в производстве красителей.
n-Оксибензойную кислоту синтезируют по Кольбе с применением
калиевой щелочи. Ее метиловый эфир (по оксигруппе), называемый ани
совой кислотой, получают окислением анетола (метилового эфира
п-пропенилфенола):
анисовая
анетол кислота
Они
Полиоксибензойные кислоты весьма Pacnp°^^^x во многих расте'
содержатся в виде гликозидов или других прои д' I и ее пр°и3'
ниях. Такими являются, например, IV кислоты-
водные: ванилиновая II, изованилиновая III и в р р
ОКСИВЕНЗОЙНЫЕ КИСЛОТЫ (ФЕНОЛОКИСЛОТЫ)
185
Из триоксибензойных кислот наибольшее значение имеет 3,4,5-триокси-
бензойная (галловая) кислота V, одна из наиболее распространенных
растительных кислот. Она содержится в чернильных орешках, листьях
чая, дубовой коре, корнях гранатового дерева и многих других растений.
Выделяют ее обычно из чернильных орешков. Галловая кислота приме-
нялась издавна для получения черных чернил и ряда красок. Висмутовая
основная соль галловой кислоты употребляется в качестве сильного анти-
септика под названием дерматол. Диметиловый эфир галловой кислоты —
сиреневая кислота VI также получается при расщеплении многих при-
родных соединений, в частности лигнина.
ОН
СООН
)
ОН
A/oc,,s
V1'
соон
п
ОСНз
соон
ОСНз
I ОСНз
соон
он
но
I
соон
он
СНзОк /I осп?
соон
VI
Амид, образованный 3,4,5-триметоксибензойной кислотой и гетероцик-
лическим амином — морфолином, называется андаксином и применяется
как транквилизирующий (успокаивающий) медикамент.
СН2—СН2
HN^ ^О
сн2-сн2
СНзО^
cH>°-<:ArN
СНзО/ О
СН2—СН2
сн2-сн2
морфолин
андаксин
Образованные двумя молекулами ароматических полиоксикислот
сложные эфиры, в которых одна выступает как кислота, а другая как
фенол, называются депсидами. Примерами могут служить леканоровая
кислота
ОН ОН
НО— Ч С-0— Д \-----------С—ОН
II II
СНз0 ОН 0
и приведенная на стр. 186 галлоилгалловая кислота.
Дубильные вещества, экстрагируемые из некоторых растительных тка-
ней, например, из коры дуба, представляют собой производные депсидов.
Дубильные вещества разделяются на два класса: гидролизуемые (именно
они и представляют собой сложные эфиры депсидов) и негидролизуемые.
186 Oh-СИГ,ЕНЗОЙНЫЕ КИСЛОТЫ (ФЕНОЛОКИСЛОТЫ)
Гидролизуемые~ дубильные вещества часто представляют собой прои.
водные галловой и л-галлоилгалловой («л-дигаллт вой») кислоты.
Синтез л-галлоилгалловой кислоты из галловой был осуществлен
следующей схеме: 0
О
О
НО
но
с—он + зс2н5о —с—С1
с2н-,о—с—о 0
C\H5O-C-O-/Q)>—C-QH
° /
с2н5о—с—о
лл - галлоилгалловая
(.и -дигалловая) кислота
Ганмины, извлекаемые из дубильных орешков, представляю
глюкозу, ацилированную остатками л-галлоилгалловои кисл ты. gff
При гидролизе таннинов образуется глюкоза, л-галлоил
кислота и небольшое количество галловой кислоты.
АРОМАТИЧЕСКИЕ АМИНОКИСЛОТЫ
187
Таннины, добываемые из разных видов дубильных орешков, несколько
различаются по числу ацилов и по соотношению между остатками галловой
и галлоилгалловой кислот. Даже таннины, выделяемые из одного и того же
растения, представляют собой смесь веществ.
н ОН
общая cpopMj'.ia таннииов
(D-остаток галловой
или лг-галлоилгалловой
кислоты, или Н)
Таннины применяются для протравки хлопчатобумажных тканей при их крашении
(для закрепления красителя), в медицине и в других областях.
Общей чертой всех дубильных веществ, в том числе и синтетических, является
большой молекулярный вес, ароматическая природа и кислотные свойства. Все это
позволяет им проявлять свое главное практическое свойство — связываясь с белками,
модифицировать их, например осаждать белки из раствора, как это делает таннин
с альбуминами, пли «дубить», превращая шкуру в кожу. Водный раствор таннина,
будучи нанесен на обожженное тело, связывает ядовитые белковые продукты распада
тканей и способствует заживлению тканей.
Связанный с таннином альбумин (танналъбин), попадая в пищеварительный тракт,
по мере переваривания альбумина, высвобождает таннин, который связывает бел-
ковые токсины болезнетворных бактерий.
АРОМАТИЧЕСКИЕ АМИНОКИСЛОТЫ
Аминобензойные кислоты могут быть получены восстановлением соответ-
ствующих нитробензойных кислот:
O2NC8H4COOH —> H2NC8H4COOH
В технике этим путем синтезируют п- и л<-аминобензойные кислоты.
о-Изомер (антраниловую кислоту) получают из фталаминовой кислоты
перегруппировкой Гофмана (кн. I, стр. 225):
,CONH2
+ NaOCl
COONa
+ СО2 + NaCl
Таким образом, сырьем для получения антраниловой кислоты служит
фталевый ангидрид, а в конечном счете — нафталин.
Диазотирование антраниловой кислоты приводит к о-диазобензолкар-
боновой кислоте, по существу являющейся внутренней солью. При
освещении она распадается с образованием дегидробензола:
,СООН
NH2
+ СОо + No
188
КАРВОНОВЫЕ КИСЛОТЫ С КАРБОКСИЛОМ 11 СОКОВОЙ ЦЕПи
Дегидробензол претерпевает далее ди- и тримеризацию (стр. Rex
Он может вступать и в другие, ранее описанные реакции присоединения
В частности, с бензолом осуществляется реакция присоединения, в котово“
дегидробензол выступает как диенофил, а бензол как диен: Р И
Антраниловая кислота является промежуточным продуктом при
синтезе индиго (стр. 307), а также ряда азокрасителей (в качестве диазосо-
ставляющей). Метиловый эфир антраниловой кислоты входит в состав
эфирного масла цветов жасмина (вместе с бензилацетатом и небольшим
количеством индола); он обладает запахом земляники и употребляется
в парфюмерии.
тг-Аминобензойная кислота применяется для синтеза ряда анестези-
рующих средств: анестезина (ее этиловый эфир), новокаина (диэтиламино-
этиловый эфир) и пантокаина.
О
/7Д\ II
H2N-(O)^C-O—сн2—сн2—n(c2h5)2
новокаин
О
сн3— (сн2)3— NH—<^ОУ-С-О—сн2—СН2—N(C2H5)2
пантокаин
n-Аминобензойная кислота — фактор роста многих микроорганизмов.
Действие сульфамидных препаратов, основанное на принципе подмена
метаболита бактерий, состоит именно в том, что эти препараты подменяют
необходимую для жизни бактерий /г-аминобензойную кислоту («вита-
мин Н»). Достаточная концентрация этих веществ в крови приводит
к гибели бактерий. п-Аминосалициловая кислота, получаемая из лг-ами
нофенола по реакции Кольбе, — важное антитуберкулезное лекарство
(ПАСК), действующее по тому же принципу.
КАРБОНОВЫЕ КИСЛОТЫ С КАРБОКСИЛОМ
В БОКОВОЙ ЦЕПИ
Простейший представитель этих соединений фенилуксусную кисл У
получают из хлористого бензила через ее нитрил
CeH5CH2Cl + KCN ——CeH5CH2CN СвН5 СН2 СООН
— K.G1
КАРБОНОВЫЕ КИСЛОТЫ С КАРБОКСИЛОМ В БОКОВОЙ ЦЕПИ
189
или из того же исходного вещества через реакцию Гриньяра. Следующий
гомолог — гидрокоричную кислоту приготовляют гидрированием корич-
ной кислоты:
СвП5—СН=СН—С—ОН СвТ15—СТТ2—СП2-С—OTI
Коричную кислоту синтезируют одним из следующих способов.
1. Промышленный способ — окисление бензальацетона (продукт
конденсации бензальдегида с ацетоном) хлорноватистой кислотой (гало-
формный распад):
ceH5-cno+cii3-с-сн3 св11Б-сп=С11-с-снз -NaOcl->
II II
О о
--► СвН6-СН=СН—С—СС1з CeH6-ClI=ClI-COONa4-CUCl3
2. Конденсация бензальдегида с уксусным ангидридом по реакции
Перкина (стр. 147):
CH.CONa
I
СвТ15-СНО4-(СНз—С)20 --5 * Свн5—С11=СВ—С—ONa + 2CH3COOH
3. Конденсация бензальдегида с малоновой кислотой в присутствии
оснований (Кневенагель):
Cell5-CIIO + CH2(COOR)2 mepg-> св115-СИ=С(СООН)2 —>
—> СвН5-С11=СН—СООН + СОг
Получаемая этими способами коричная кислота имеет тра/т-конфи-
гурацию. При УФ-облучении транс-изомер переходит в z/мс-изомер.
tfuc-Коричная кислота может существовать в виде кристаллических моди-
фикаций с разными температурами плавления.
н\ / н
\Q_QZ
СвН5/ хсоон
цис-коричная кислота
(т. пл. 57° С)
Нх /СООН
\г—с-
СвНь^ ХН
шрамс-коричная кислота
(т. пл. 133° С)
Такие цис-транс-изомеры, как коричные кислоты, имеющие водороды при двойной
связи, существенно различаются спектроскопически и легко могут быть распознаны.
На'рис. 92 приведен ЯМР (ПМР)-спектр этилового эфира коричной кислоты (отнесе-
ние линий приведено на спектре). Протоны этильной группы дают квадруплет б и трип-
лет а (кн. 1, стр. 596 сл.). Протоны фенила (бензола с одним заместителем), обычно
мало различающиеся по химическим сдвигам, дают уширенный инк или узкий мульти-
плет d. Сигналы же транс-протоиов при двойной связи (в и г) отличаются большой
константой сппн-спинового расщепления J, обычно почти в два раза (в гц) превышающей
величину J для цис-протонов.
190
КАРБОНОВЫЕ КИСЛОТЫ С КАРБОКСИЛОМ В БОКОВОЙ ЦЕПИ
цис- п трямс-Изомеры этого типа можно легко различить и с помощью ИК-спектп
или спектров комбинационного рассеяния. Так, для ИК-снектров транс-соединеви"
указанного типа характерна полоса внеплоскостного деформационного колебания поп
990—965 слГ1, отсутствующая в спектрах чистых i/uc-соединеппй. транс-Кротоповая
коричная п кумаровая кислоты и их эфиры также поглощают в области 980—974 слг^
Интенсивность полосы меняется для разных соединений от средней до высокой.
Рис. 92. ЯМР-Спектр
этилового эфира транс-
коричной кислоты (/=
= 15,8 гц.
Используя вместо бензальдегида замещенные альдегиды, можно
получить коричные кислоты с различными заместителями в ядре, например:
.О
СН=СН—С</
хон
CHsCONa
II
О
хсно
+ (СНз-С)2О
он
О
о-Оксикоричная кислота представлена двумя стереоизомерами: цис-
(кумариновая кислота) и транс- (о-кумаровая кислота). Первая известна
только в виде производных, так как свободная цис-кислота сразу же пре-
вращается в ее лактон — кумарин:
СН=СН
ОН
ОН
кумарин
Кумарин имеет запах сена; он — пахучее начало цветов донника.
Так же как и его оксипроизводные (эскулетин и умбеллиферон), кумари
широко распространен в растительном мире.
С=О
с=о
но/ Y о
он
эскулетин
умбеллиферон
Эскулетин и умбеллиферон могут быть синтезированы по ^е^вии
нагреванием соответствующего фенола с яблочной кислотой в р
КАРБОНОВЫЕ КИСЛОТЫ С КАРБОКСИЛОМ В БОКОВОЙ ЦЕПИ
191
серной кислоты. При этом яблочная кислота расщепляется до формилук-
сусной кислоты, которая и конденсируется с фенолом:
Из бензальдегида получают еще одну важную оксикислоту —
миндальную:
ceH6-ciio+ncN —> свп5-с^он НгО(н+)-> СвНБ—С^ОН
XCN \COOH
Как всякая сс-оксикислота, миндальная кислота образует два ряда
производных. Она легко отщепляет воду с образованием лактида:
О
О = С СН— Cgllj
2С6Н5—сн—СООН ---► | |
I С6Н5—НС с = о
он \ /
о
Миндальная кислота имеет один асимметрический атом углерода и
существует в виде двух оптически деятельных стереоизомеров: D (—)-
и L (4- )-миндальных кислот. Первая образуется при гидролизе глико-
зида горького миндаля — амигдалина, вторая может быть выделена из
гликозида, содержащегося в бузине. Рацемическая — параминдалъная
кислота, которая всегда образуется при синтезе, может быть разделена
на оптические антиподы с помощью цинхониновых солей. Миндальная
кислота издавна является излюбленным объектом для стереохимических
исследований (наряду с винной и молочной кислотами). Так, она послу-
жила одним из примеров, на котором было продемонстрировано вальденов-
ское обращение (см. кн. I, стр. 394).
Из аминокислот с функциональными группами в боковой цепи наиболь-
шее значение имеют фенилаланин и тирозин:
СН,—СИ—С—ОН
’ I II
nh2 о
фенилаланин
НО
сн2—сн—с—он
I II
nh2 о
тирозин
Обе эти аминокислоты входят в число 20 аминокислот, участвующих
в построении белков. Фенилаланин является одной из девяти незаменимых
аминокислот, т. е. он не может быть синтезирован организмом и поэтому
непременно должен присутствовать в составе белков пищи.
192 КАРБОНОВЫЕ КИСЛОТЫ С КАРБОКСИЛОМ В БОКОВОЙ ЦЕПИ
Тирозин был синтезирован конденсацией гиппуровой кислоты и п
оксибензальдегида с последующим гидрированием двойной связи и омыле-
нием продукта:
НОС6Н4—СНО-4-СН2—СООН ► НОС6Щ—СН=С-СООН „
I I —-
NH—СО—С6Нб NH—СО—СвН6
гиппуровая кислота
--> НОС6Н4—СН2—СН—СООН н2О(Н+) НОСвН4-СНа—СН-СООН
NH-СО-CeH6 NHa
тирозин
Природные L (—)-тирозин и L (—)-фенилаланин образуются при
гидролизе почти всех белков. Особенно много тирозина в составе казеина
и фиброина шелка. Важную физиологическую роль играют также некоторые
производные тирозина, в частности дииодтирозин и тироксин.
Тироксин — гормон, регулирующий обмен веществ; он продуцируется
щитовидной железой. Строение тироксина подтверждено синтезом по
следующей схеме (Харрингтон, 1926 г.):
1
он + 1—^Q)>-no2
I
как R-NOjI
+ Cu->(CNR SnCl.;H>0
R—NO2----->R—NH2------>R-N=N X"------------► '—>R-CHO
Hi
R-CHO + CHo-COOH------>R~CH=C—COOH --------> R—СН,-CH-COOH
I “ I I
NH- c-C6H5 nh-c-c6h5 nh2
II t II
о о
тироксин
Щитовидная железа вырабатывает определенное количество тироксина-
Недостаточное продуцирование тироксина в организме (гипотиреоз) ведет
к общей слабости, апатичности, а в тяжелых случаях — к кретинизму-
Гипертиреоз (чрезмерная деятельность щитовидной железы) приводи
к нарушению сердечной деятельности, повышенной нервозности, высоко У
кровяному давлению, пучеглазию (базедова болезнь). При гипотир6®
больные вынуждены постоянно принимать препараты, содержащие со
ветствующие гормоны; гипертиреоз понижается с помощью некотор
ПЕРЕДАЧА ВЛИЯНИЯ ЗАМЕСТИТЕЛЕЙ ЧЕРЕЗ КОЛЬЦО
193
лекарств, играющих роль антигормонов (например, метилтиоурацил),
а иногда требуется операционное вмешательство (удаляют часть щитовид-
ной железы).
Темные пигменты, содержащиеся в коже, волосяных покровах и рого-
вице глаз животных и человека, образуются при окислении в организме
другой аминокислоты — диоксифенилаланина (сокращенно ДОФА или
DOPA), которая в свою очередь образуется в результате окисления’из
тирозина. Диоксифенилаланин окисляется далее в хинон (ДОФА-хинон)
СНо —СН —С—ОН
I II
nh2 о
сн2—сн —с—он
nh2 о
конденсирующийся в краситель феноксазиновой структуры (стр. 375).
ПЕРЕДАЧА ВЛИЯНИЯ ЗАМЕСТИТЕЛЕЙ ЧЕРЕЗ БЕНЗОЛЬНОЕ
КОЛЬЦО. КОРРЕЛЯЦИОННЫЕ УРАВНЕНИЯ
Константы диссоциации карбоновых кислот зависят главным образом
от величины частичного положительного заряда 6+ на карбоксильном
атоме углерода. Связанные с бензольным ядром электронодонорные группы
уменьшают этот заряд (а следовательно, и константу диссоциации); электро-
ноакцепторные группы оказывают обратное действие. В табл. 68 (стр. 169)
приведены эти константы для ряда бензойных кислот. Как можно видеть,
сила бензойных кислот зависит и от электроноакцепторности заместителя,
и от положения его по отношению к карбоксилу.
Обычно влияние заместителя из пара-положения заметно больше, чем из
мета-положения, что связано с различием механизмов передачи влияния.
Из пара-положения передача влияния совершается по сопряженной системе
связей (мезомерный эффект), тогда как мета-заместители проявляют в основ-
ном индуктивный эффект. Особенно ярко это различие видно на примере
окси- или алкоксизаместителей. Метоксигруппа обладает небольшим
отрицательным индуктивным эффектом (—/); с другой стороны, она способ-
на подавать неспаренные электроны по сопряженной системе связей.
Индуктивный эффект проявляется в основном, когда метоксигруппа
находится в мета-положении, в котором нет сопряжения между замести-
телями и карбоксильной группой; поэтому введение метоксила в мета-
положение повышает константу диссоциации бензойной кислоты. В случае
и-метоксибензойной кислоты наблюдается уменьшение константы диссо-
циации, поскольку эффект сопряжения (мезомерный или резонансный)
превалирует над индуктивным.
Такая же конкуренция между индуктивным и мезомерным эффектом
хорошо известна в случае галоидных заместителей, однако при диссоциации
13 Заказ 672
194 ПЕРЕДАЧА ВЛИЯНИЯ ЗАМЕСТИТЕЛЕЙ ЧЕРЕЗ КОЛЬЦО
бензойных кислот преимущество остается на стороне индуктивного
эффекта, так как мезомерный эффект для галоидов очень мал.
Поэтому константы диссоциации падают в ряду:
•и-хлорбензойная 2> я-хлорбензойная 2> бензойная
При наличии нитрогруппы и подобных заместителей мезомерный и
индуктивный эффекты действуют в одном и том же направлении; введение
нитрогруппы, особенно в пара-положение, резко увеличивает константу
диссоциации.
Для орто-замещенных бензойных кислот наблюдаются аномалии
связанные с так называемым орто-эффектом (в отношении реакционной
способности кислот этот эффект рассмотрен на стр. 181). Орто-эффект
отражающийся на силе бензойных кислот, связан, в зависимости от природы
заместителя, с различными влияниями близкорасположенной группы. Это
может быть следствием образования водородной связи (в случае салици-
ловой кислоты), или пространственных затруднений (для о-алкилбензой-
ных кислот), или эффекта «поля» (т. е. непосредственного электростати-
ческого взаимодействия заместителей и карбоксила «через пространство»),
или, наконец, возможно наложение этих эффектов друг на друга.
На большинство реакций в боковой цепи ароматических соединений
заместители в кольце оказывают аналогичное полярное влияние. Поэтому
казалась естественной мысль, что в подобных системах константы скоро-
стей тех или иных реакций могут быть поставлены в прямую количествен-
ную зависимость от полярных влияний заместителя. На основании этих
соображений и обобщения большого количества экспериментальных данных
Гамметт в 1937 г. вывел уравнение, связывающее константы скоростей
реакций (а также константы равновесий) с природой заместителя в бен-
зольном кольце:
К,
1g -77— — ар или 1g К — 1g Ко = ао
Ао
где К — константа скорости или равновесия какой-либо реакции соедине-
ния, содержащего заместитель R в бензольном кольце; К$ — та же кон-
станта для незамещенного соединения. Например, если К — константа
скорости гидролиза хлористого /г-нитробензила в спирте при 20° С, то По-
такая же константа для хлористого бензила. Величина р в уравнении
Гамметта зависит от типа реакции, природы реакционного центра, удале-
ния его от бензольного кольца (в боковой цепи) и условий реакции
(растворитель, температура); о зависит только от свойств заместителя и
его положения в бензольном кольце *. Реакционная константа р для удоб-
ства (для начала отсчета) принимается за единицу в том случае, когда К
и Ко — константы диссоциации замещенной и незамещенной бензойной
кислоты (в воде при 25° С); тогда уравнение Гамметта принимает вид:
, к
18 &=’
Отсюда может быть определено значение другой константы уравнения
Гамметта — р.
* Поэтому различают значения а для мета- и пара-заместителей (а^» и ал_). Влия
ние орто-заместителей не подчиняется уравнению Гамметта.
передача влияния заместителей через кольни
195
Рис. 93. Графическое изображе-
ние уравнения Гамметта:
1 — для диссоциации бензойных ки-
слот в воде при 25° С (р = 1,00;
1g Ко = —4,203); 2 — для гидролиза
хлористых бензилов водой при 30° С
в 47%-ном этаноле (р = —2,20;
1g Ко = —5,61).
Зная значения а для ряда заместителей (в настоящее время эти значе-
ния можно найти в литературе, в частности в обзорных статьях) и константы
скоростей реакций нескольких соединений, можно рассчитать примерные
скорости реакций для соединений с любым
заместителем. Отложим по оси абсцисс
значения о, а по оси ординат величины
lg K/K.Q (рис. 93). Тогда графическое изо-
бражение уравнения Гамметта будет пред-
ставлять собой прямую, проходящую через
начало координат под углом а, причем
tg а=р.
Напротив, исходя из известных скоро-
стей реакций, можно вычислить о для ново-
го заместителя. Это одно из применений
уравнения Гамметта. Выведенные на его ос-
нове значения константы ст являются опре-
деленной количественной характеристикой
электроноакцепторности заместителя; о
может принимать положительные (в слу-
чае электроноакцепторных заместителей)
и отрицательные значения. Наибольшие
положительные значения о соответствуют
+
таким группам, как — NO2 и — N(CH3)3.
Для алкоксидов, так же как для окси-
группы, в зависимости от положения (мета-
или пара-) а принимает либо положитель-
ное, либо отрицательное значение. Алкильные группы обладают неболь-
шим отрицательным о. Приведем значения о для наиболее [характер-
ных заместителей в пара- и мета-положениях:
N (СН3)2........
ОН..............
СНз.............
С1..............
пара- мета- пара- мета-
—0,83 — СОСНз ..............+0,50 +0,376
—0,37 +0,12 CN .................+0,66 +0,56
—0,17 —0,069 NO2.................+0,78 +0,71
+0,227 +0,373 N (СН3)з..........+0.82 +0,88
При изучении реакций, в которых имеет большое значение сопряжение
заместителей с реакционным центром (т. е. Т-эффект, доходящий до реак-
ционного центра), уравнение Гамметта дает не всегда и не для всех замести-
телей хорошие результаты. Например, при реакциях электрофильного за-
мещения в ароматическом ядре резкое несоответствие наблюдается для'за-
местителей, подающих свободные электроны по сопряженной системе связей
(СН3О:, О-, :NH2 и др.). В таких случаях пользуются не значениями о,
а другими — <т+, выведенными Брауном и Окомото на основании соль-
волиза соединений
R
СН3
с—ci
СНз
13*
196
ПЕРЕДАЧА ВЛИЯНИЯ ЗАМЕСТИТЕЛЕЙ ЧЕРЕЗ Кг.-т,
----- ——---------—----------------
таким же образом, как это было сделано Гамметтом
диссоциации бензойных кислот.
Приведем для примера несколько
на основании
значений о+:
пара- мета-
СН3О..................—0,778 4-0,045
СНз...................—0,311 —0,066
F.....................—0,073 4-0,011
пара- мета-
.................. 4-0,114 4-0,399
CN......................+0.562 4-0,659
NO2 .................. 4-0,790 4-0,674
Уравнение Брауна — Окомото применяется обычно в тех случаях, когда
реакционный центр в переходном состоянии получает полный или значи-
тельный положительный заряд. Например, при электрофильном замеще-
нии атакующий агент создает такой заряд на углероде в ароматическом
кольце. В подобных реакциях особенно большое влияние оказывают
пара-заместители электронодонорного характера и тем более — облада-
ющие 4-Т-эффектом (ОН, OR, NH2). Для электронодонорных заместите-
лей значительно меньше о (но больше по абсолютной величине), а для
электроноакцепторных разница невелика. Для мета-заместителей <+ и о то-
же мало отличаются, поскольку 4-71-эффект в мета-положении не про-
является.
Сольволиз серии веществ Брауна и Окомото проходит, очевидно, через
диссоциацию (медленная стадия xS^l):
СН3
/=\ х + /снз
R—)>—С—С1 ч ± R—хх />—Сб 4- С1" • растворитель
\' \ \—z хСНз
СНз
Поэтому 4- Т-эффект заместителя R полностью достигает положительно
заряженного С+, например:
СНзО
СНз
=С\
ХСН3
Если на основании значений о (соответственно о+) можно судить
о природе заместителя и его влиянии на реакционную способность, то на
основании знака и величины реакционной константы р можно делать
иногда выводы о природе и механизме реакций. Положительные значе-
ния о означают, что с увеличением электроноакцепторности заместителя
(рост о) скорость реакции увеличивается. Если же о меньше 0, то, напротив,
повышение электроноакцепторности заместителя ведет к уменьшению
скорости реакции. Отсюда можно заключить, например, имеем ли мы дело
с электрофильным‘или с нуклеофильным присоединением (замещением),
каков механизм реакции — 5Л4 или SN2 и меняется ли он при изменении
условий реакции в тех или иных пределах. Так, для уже известного на
Sn2 механизма замещения галоида в галоидных алкилах константа
имеет положительные значения, поскольку увеличение электроноакд
торности заместителей ускоряет реакцию, например, при гидролизе
этому механизму галогенида
ПЕРЕДАЧА ВЛИЯНИЯ ЗАМЕСТИТЕЛЕЙ ЧЕРЕЗ КОЛЬЦО
197
Обратное действие электроноакцепторность заместителя оказывает при
механизме 6^1, следовательно, в таких случаях р<(0 (рис. 93).
Механизм реакции, протекающей в определенных условиях, может
меняться в какой-то точке при постепенном изменении величины о заме-
стителя. В таком случае графическое изображение уравнения Гамметта
представляет собой уже не одну, а две пересекающиеся прямые. Например,
подобное изменение механизма происходит при реакции между замещенны-
ми бензальдегидами и бутпламином при синтезе оснований Шиффа:
ОН
Н + (2)
С + NH2C4H9
I
Н
реакции при варьировании заместителя R
рис. 94, где по оси абсцисс отложены о+.
две прямые,
Рис. 94. Графическое изображение
уравнения Гамметта для синтеза
оснований Шиффа.
Данные изучения этой
изображены графически на
График представляет собой
пересекающиеся в начале координат, при-
чем, когда о+<(0, то р>0, и наоборот.
Следовательно, на этих двух участках
реакция идет по разным механизмам.
Левая прямая означает, что здесь элек-
тронодонорные заместители затрудняют
реакцию, и это естественно для стадии (1),
поскольку при возрастании электронной
плотности на группе С = 0 нуклеофиль-
ная атака карбонильного атома углерода
затрудняется. Следовательно, первая ста-
дия является наиболее медленной, опре-
деляющей общую скорость реакции, при
отрицательных значениях о+. Стадии же
(2) и (3) должны облегчаться с увеличением
электронодонорности заместителя. Заме-
ститель R со значениями о+(>0, напротив,
ускоряет присоединение амина и затруд-
няет протонизацию ОН (стадия 2) и отще-
пление воды (стадия 3). Поэтому в случае
электроноакцепторпых заместителей стадия
строй, и, таким образом, механизм меняется.
Уравнение Гамметта применимо к ряду бензола, а также
к некоторым другим ароматическим и гетероциклическим соединениям.
Недостаток этого уравнения в том, что некоторые типы реакций ему не
подчиняются и приходится вводить специальные значения о (например, о+).
(1) является наиболее бы-
198
ПЕРЕДАЧА ВЛИЯНИЯ ЗАМЕСТИТЕЛЕЙ ЧЕРЕЗ КОЛЬЦ
Это означает, что соотношение индуктивного и мезомерного влияв
заместителя изменяется в зависимости от типа реакции.
Тафт предположил, что изменение свободной энергии,
нальное 1g —можно приравнять сумме трех величин, одна
АО
пропорцио.
из которых
зависит только от индуктивного, другая — от мезомерного, а третья
от пространственного эффекта заместителя:
1g -^- = о*р* + М+ у
Если заместители проявляют только индуктивный эффект, то
lg =
А О
Это уравнение и известно под названием уравнения Тафта (или Гаммет-
та — Тафта). Конкретные значения о* были выведены на основании сле-
дующих соображений.
Ранее Инголдом было высказано соображение, что в случае «кислотного»
и «щелочного» гидролиза сложных эфиров пространственные влияния
одинаковы. Это следовало из механизмов гидролиза, катализируемого
щелочами и кислотами, и было подтверждено целым рядом наблюдений.
Ниже приведены эти механизмы:
9>
R—+ Н3О+
OR'
+ ОН
R—С
о-н
+ HOR'
.ОН
R—+ Н+
ХО
Г X .
R—С + "ОН R—С....OR
XOR' ХО—Н
+ HOR'
Таким образом, первое переходное состояние отличается от второго
только наличием двух протонов. Незначительные размеры протонов и их
удаленность от заместителя позволяют предположить, что оба переходных
состояния в равной мере чувствительны к пространственным изменениям
в радикале. Что касается «исходного состояния», то оно одинаково.
Тафт предложил выражать о* следующим уравнением:
где о* — мера полярного влияния заместителя; К и К() константы
скорости гидролиза замещенного и незамещенного эфира; индексы А и
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ С НЕПРЕДЕЛЬНОЙ БОКОВОЙ ЦЕПЬЮ
199
относятся к кислотному и, соответственно, щелочному гидролизу (в жир-
ном ряду за стандартную реакцию принимается гидролиз эфиров уксусной,
а не муравьиной кислоты). Коэффициент введен для удобства сравне-
ния о* с константой Гамметта сг. (В табл. 71 приведены значения о*).
Таблица 71. Константы о* для алифатического ряда
(мера полярности заместителей по Тафту)
R ст* к ’ 1 R ст*
СНз 0 fch2 +1,10 с6н5сн2 +0,215
С13С +2,65 С1СН2 + 1,05 с2н5 —0,10
f2ch +2,05 ВгСН2 + 1,00 Й-СЗН7 —0,115
СООСНз +2,00 1СН2 +0,85 изо-СэН? —0,125
(CH3)3NCH2 + 1,90 СбН3С=С + 1,35 1 1/.11КЛ0-С5Н9 —0,20
№ССН2 + 1,30 с6н5 н +0,60 +0,49 (СН3)3С —0,30
В этом уравнении о* — доля индуктивного влияния заместителя, без
пространственного и мезомерного влияния, поэтому уравнению подчиня-
ются только те реакционные ряды, в которых пространственный и мезомер-
ный факторы остаются постоянными (в том числе и алифатические).
Однако если, например, одна серия реакций подчиняется уравнению
lg-^- = o*P*
до
а другая уравнению
Ig ~ = о*Р*+Т
до
то ясно, что здесь у — доля пространственного и мезомерного эффекта.
Поэтому с помощью уравнения Тафта можно изучать не только общее
влияние заместителя, ио и отдельно — пространственное и индуктивное
или индуктивное и мезомерное.
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
С НЕПРЕДЕЛЬНОЙ БОКОВОЙ ЦЕПЬЮ
Простейший углеводород этого типа — стирол (винилбензол) приобрел
важное промышленное значение в производстве пластмасс и каучуков.
Стирол получают в настоящее время из этилбензола (его готовят конден-
сацией бензола с этиленом в присутствии А1С13) дегидрогенизацией над
катализатором, содержащим окись хрома (А. А. Баландин, Г. М. Мару-
кян):
С6Н5-С2Н5 Сг-*0’/А1г0 + Свн6-сн=сн2 + н2
Строение стирола может быть установлено как обратной реакцией
каталитического гидрирования (над Pt) в этилбензол, так и окислением
в бензойную кислоту и озонированием с последующим гидролизом
200 АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ С НЕПРЕДЕЛЬНОЙ БОКОВОЙ ЦЕПЬЮ
в бензальдегид. Строение его следует и из получения его декарбоксили
рованием коричной кислоты или дегидратацией метилфенилкарбинола'
СвН5-СН=СН—СООН --► СвНБ-СН=СН2 + СО2
СвН5—СН-СН3 —> С6Н5—СН=СН2 + Н2О
кип. 145° С; т. пл. —30,6° С.
активной олефиновой связи в боковой цепи
направление электрофильных атак на эту
ОН
Стирол — жидкость с т.
Естественно, что наличие
стирола определяет главное
цепь. Однако при нитровании стирола наряду с главным продуктом этой
реакции — со-нитростиролом образуются оба продукта нитрования
в ядро — о- и п-нитростиролы:
ch=ch-no2
СН=СН
2
что двойная связь, сопряженная с ароматическим
переходном состоянии при электрофильной атаке
Отсюда следует,
ядром, обладает (в
на ядро) электронодонорным характером:
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ С НЕПРЕДЕЛЬНОЙ БОКОВОЙ ЦЕПЬЮ
201
со-Нитростирол нитруется в орто- и пара-положения, но медленнее, чем
бензол. Таким образом, группа —CH = CHNO2 еще один пример редкого
сочетания пассивации с орто-пара-ориентацией.
Главное применение стирола — полимеризация его в полистирол ____
прозрачную твердую пластмассу с ценными электроизоляционными
свойствами. Полимеризация проводится под действием трехфтористого
бора (следы воды образуют сильную кислоту H+HOBF“) при низкой
температуре. Она протекает как ионный процесс, инициируемый Н+:
Об изотактическом полистироле см. кн. I, стр. 397.
При крекинге полистирол достаточно гладко деполимеризуется, т. е.
крекинг происходит по вновь образовавшимся в полистироле связям.
Большие количества стирола идут на сополимеризацию с бутадиеном
для производства сополимерного бутадиен-стирольного каучука (содер-
жащего до 30% стирола), отличающегося повышенной устойчивостью
к истиранию и потому особо ценного для изготовления автомобильных шин.
Стирол на воздухе постепенно густеет и затвердевает, самопроизвольно
полимеризуясь, так что его приходится хранить с добавлением инги-
битора.
Из диэтилбензола дегидрогенизацией над Сг2О3/А12О3 получают
дивинилбензол. В смеси со стиролом дивинил бензол полимеризуется
в «сшитый» пространственный (а не линейный) полимер, в котором допол-
нительные поперечные связи в макромолекуле образованы за счет второй
винильпой группы дивипилбензола. Такие пространственные полимеры
могут нести в бензольном ядре сульфогруппы. В этом случае они служат
ионообменниками, а именно катионообменными смолами:
---сн—сн2 —сн—сн2—сн—сн2
6 ф й
so3M so3M I
’ ’ ’ — СН — СМ2
+ лНХ
202 АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ С НЕПРЕДЕЛЬНОЙ БОКОВОЙ ЦЕПЬ©
Если сополимеры стирола и дивинилбензола пронитровать и восста
повить затем нитрогруппы в аминогруппы, получаются анионообменные
смолы:
Вся масса смолы представляет собою по существу одну макромолекулу
многоосновной кислоты (или основания), в силу своей огромности совершен-
но нерастворимую. При пропускании через смолу-ионообменник раствора
электролита происходит обмен катиона (или аниона), который в силу
нерастворимости смолы-кислоты или смолы-соли идет до конца. Этим
широко пользуются в современной технике и препаративной аналити-
ческой химии для выделения в чистом виде кислот, оснований и солей,
например для обессоливания воды. Стирольные сополимеры особенно
хороши для этой цели.
Фенилацетилен — простейший ароматический углеводород с тройной
связью в боковой цепи. Его получают обычным для ацетиленов путем —
присоединяя молекулу брома к олефину и отщепляя НВг действием
спиртовой щелочи:
СвН5-СН=СН2 + Вг2--> СвН5-СНВг-СН2Вг =2hbF* С8Н5-СееСН
Фенилацетилен (т. кип. 141,6° С) обладает обычными свойствами
монозамещенных ацетиленов. Промышленного применения не получил.
МНОГОЯДЕРНЫЕ
АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ
БЕНЗОЛЬНЫМИ ЯДРАМИ
Дифенил (или бифенил)
Наиболее просто этот углеводород можно получить по Бертло, пропуская
пары бензола через раскаленную докрасна трубку:
2С6Н6 —► СвН5-СвН6 + Н2
Строение его следует из синтеза по Вюрцу — Фиттигу (стр. 57):
2C6H5Br + 2Na -> СвНБ-С6НБ + 2NaBr
Он получается также в качестве побочного продукта при синтезе
гриньярова реактива из галогенбензола и магния по сходной реакции:
2СвН5Вг + Mg---► СвНБ-С6НБ + MgBr2
Дифенил и его разнообразные производные можно получить по реакции
Гомберга (стр. ПО), действуя солью фепилдиазония в присутствии ацетата
натрия на бензол и замещенные бецзолы:
X-С6Н4 г? N=N[-;-JO—С-СН3 + C6H5Y->
' 1 II
О
—>х—С6Н4—С6Н4—Y + N2 + сн3-С — ОН
о
Поскольку реакция проходит гомолитически, место вступления ради-
кала диазосоединепия в бензольное ядро мало зависит от характера ориен-
танта. Обычно получается смесь о-, м- и n-замещепных дифенилов с преоб-
ладанием n-из омера.
Дифенил с примесью терфенила С6НБ—С6Н4—СвНБ и кватерфенила
СБНБ—(СвН4)2—СвН5 — обычный побочный продукт при гомолитическом
разложении солей диазония (например, медью по Гаттермапу). Он полу-
чается за счет сдваивания промежуточно образующихся фенильных
свободных радикалов. Терфенил и кватерфепил образуются в результате
реакции Гомберга между дифенилом и солями диазопия.
ж
СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЯДРдад
Р. Я. Левиной и В. Р. Скварченко принадлежит вариант их же метода
синтеза ароматических углеводородов, приводящий к дифенилу и его
производным. Метод основан на диеновой конденсации 2-арилбутадиена
с малеиновым ангидридом:
СН2
с.н.-с^СНг сн-с<° «'‘-f )к-с<0
4 + <?н-с<°“> нсх/сн-с<0
\ш2 х° сн2
Рго» (ю0° G)
+ 2СО + Н2О
Из замещенных арилбутадиенов получаются при этом замещенные
дифенилы.
Наиболее важным производным дифенила является бензидин (п, п'-
диаминодифенил), получаемый в технике бензидиновой перегруппировкой
гидразобензола (стр. 80):
NH—NH—— * H2N—NH2
Аналогично получаются о-толидин и о-дианизидин:
H2N-^ ^-NH2
СН3о/ ^ОСНз
о-толидин
о-дианизидин
Строение их как производных дифенила может быть доказано диазоти-
рованием обеих аминогрупп и последующей элиминацией диазогрупп
путем замены их на водород (стр. 109). Эти реакции в случае бензидина
приводят к дифенилу, а в случае о-толидина и о-дианизидина — к соответ-
ствующим замещенным дифенилам. В бензидине пара-положение амино-
групп следует из того факта, что при введении электрофильного замести-
теля, например при галоидировании, бензидин (или его ацильное произ-
водное) дает единственный монозамещенный продукт. Бензидин и его
производные широко применяются в синтезе азокрасителей.
Дифенил — типичный ароматический углеводород, в котором оба
ядра более независимы друг от друга, чем можно было бы ожидать на осно-
вании его плоской структуры и уменьшенного сравнительно с нормой
расстояния между связанными простой связью углеродами двух бен-
зольных ядер (1,48 вместо 1,54 А). Это расстояние ясно выражает оттенок
двоесвязности, т. е. перекрывания л-облаков обоих ядер в смысле формул
205
АТРОПЭНАНТИОМЕРПЯ. II
При нитровании дифенила одна нитрогрунна вступает в основном
в пара-положение к атому углерода, несущему связь между ядрами,
а вторая — неожиданно тоже в пара-положение во второе ядро (ср'
с нитрованием со-нитростирола). Полученный п, п'-динитродифенил при
восстановлении дает бензидин.
Дифепил, в котором С—G-связь между фенилами осуществлена между
меченными 14С атомами, при действии на него А1С13 с примесью НА1С14
(в бензольном растворе) перераспределяет метку 14 С по всем о-, м- и п-угле-
родным атомам (Вайнберг и Вольф, 1963 г.), т. е. С—С-связь обоих фенилов
разрывается и вновь возникает уже между новыми углеродами. При этом,
однако, фенильные радикалы не выходят в объем и изменения соверша-
ются внутри одной и той же молекулы дифенила.
АТРОПЭНАНТ1ЮМЕРИЯ. II
Кеннер и Кристи (1922 г.) нашли, что 9, «/-замещенные дифеновые кислоты
могут быть разделены на оптические антиподы путем превращения их
в соли с оптически деятельным алка-
лоидом, разделения полученных диасте-
реомерных солей и выделения из них дей-
ствием кислоты свободных энантиоме-
ров замещенных дифеновых кислот. Легко
видеть, что ни дифенил,“ни дифеновые ки-
слоты не содержат (и не могут содержать)
асимметрических углеродных атомов,
так как и углероды ядер, и углероды
карбоксилов связаны всего с тремя (а не
с четырьмя) разными атомами или груп-
пами. Изомерия замещенных дифеновых
кислот — это оптическая изомерия без
асимметрического углеродного и вообще
какого бы то ни было асимметрического
атома. Она обязана ненлоскому строению
этих замещенных дифенилов, невозмож-
ности поворота вокруг оси С—С, связыва-
ющей два ядра. Повороту мешают задева-
ющие друг за друга группы, находящиеся
в орто-положении. В результате молекула
Рис. 95. УФ-Спектры дифенила
и его замещенных:
1 — дифенил; 2— 2,2'-диметилдифенил;
з — 3,3'-диметилдифенил; 4 — 4,1'-ди-
метилдифенил.
не имеет плоскости симметрии и ее зер-
кальное изображение не тождественно с предметом, как легко убедиться
на модели:
С такой стереоизомерией, связанной с невозможностью
поэтому называемой атропизомерией, мы уже встречались
поворота и
(стр/ 131).
206
СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ
ЯД₽ АМЦ
Особенно широко исследовал атропизомерию производных дифенил
Р. Адамс, выяснивший, какие орто-заместители мешают повороту и вызы
вают появление стереоизомерии. Ни сами дифеновые кислоты, ни их папа"
замещенные производные стереоизомеров не имеют.
Накопление заместителей в орто-положениях дифенила, затрудняющее
делающее невозможным сопряжение двух его колец вследствие вывода их из коиланагь
ностп, сразу же сказывается на УФ-спектре (см. кн. I, стр. 608) замещенных дифени-
лов. На рис. 95 приведены спектры дифенила и диметилдифенила с различным распо-
ложением метильных групп в кольцах. Особенно хорошо видно изменение второго
пика, который является, вероятно, комбинацией полос К и В. Высота этого пика
у мета- и пара-замещенных незначительно различается, интенсивность же полосы
орто-дизамещенного дифенила очень мала и напоминает уже 5-полосу бензола.
Дифенилен (или бифенилен)
Дифенилен, или дибензоциклобутадиен, имеющий строение
7А_А2 z^\=/\
J II II L <—> 1111
5 4
впервые получен Лотропом действием закиси меди на о, о'-дииоддифенил:
Позднее Виттиг нашел, что при полимеризации дегидробензола наряду
с тримером последнего — три-о-фениленом образуется и димер-дифени-
лен:
F
F
(3%)
(24%)
Дифенилен — твердое вещество (т. пл. 111
При окислении хромовой кислотой он образует ~
действии насыщенного водородом никеля Ренея дифенил (рвется оДИ®
С—С-связь). Дифенилен ацетилируется по Фриделю Крафтсу в полож
ние 2; сульфируется, образуя уже при комнатной температуре 2,6-дисул
фокислоту; нитруется с образованием 2-нитродифенилена, меркуриру6
и галоидируется также в положение 2. ы_
На ряде реакций сопряжение л-систем обоих бензольных ядер с
вается в большей степени, чем в дифениле. Так, при введении второ'
тильной группы по Фриделю — Крафтсу образуется только 2,Ь-диац
Дифенилен. Если бы влияние первого ацетила не передавалось Р
систему сопряжения, получился бы и 2,7-диацетилдифенилен.
°C) с запахом нафталина,
фталевый ангидрид, а при
1,2-ДИФЕНИЛЭТАН, 1,2-ДИФЕНИЛЭТИЛЕН, 1,2-ДИФЕНИЛАЦЕТИЛЕП
Представляет значительный интерес наличие в дифенилене циклобута-
диенового кольца, самого по себе неустойчивого.
1,2-Дифенилэтан, 1,2-дифенилэтплен,
1,2-дифенилацетилен
1,2-Дифенилэтан (т. пл. 52° С; т. кип. 285° С), обычно называемый
дибензилом, легко получается из хлористого бензила действием натрия
(реакция Вюрца) или взаимодействием хлористого бензила с бензилли-
тием или бензилмагнийгалогенидом. Разумеется, дибензил можно полу-
чить и из бензоина, а лучше всего реакцией Фриделя — Крафтса из
1,2-дихлорэтана и бензола.
1,2-Дифенилэтилен (стильбен) существует в цис- и транс-формах.
Устойчивая транс-форма имеет т. пл. 124° С, т. кип. 306° С, ^ас-изомер —
т. пл. +5° С. Обычные пути синтеза ведут к транс -изомеру; его можно
получить из фенилбензилкетона восстановлением в спирт и последующей
дегидратацией.
либензил
стильбен
^нс-Стильбен можно получить из транс-изомера индуцированным
облучением. Приводим здесь эту реакцию в качестве типичного примера
превращения с помощью индуцированного облучения. К транс-стиль-
бену добавляют бензофенон, который УФ-светом возбуждается в триплет-
ное состояние (н—>-л*-переход, кн. I, стр. 619). Далее идет обмен триплетным
состоянием с транс-стильбеном (Г —триплетное состояние, SQ—основное
состояние):
(С6Н5)2С=О —- (С8Н5)2С=О* (Т)
С6Н5\ /Н
(С8Н6)2С=О* (Т) + >с=с<
Hz с8н5
(So) —>
свн6\ /Н*
---> (С6Н5)2С=О (So) + /С-С( (Т)
HZ хс8н6
В триплетном состоянии mране -стильбена л-электроны двойной связи
приобретают параллельные спины, и эта связь становится ординарной.
Получая свободу вращения, стильбен перестает быть плоскостным. Из
Т -состояния возбужденная молекула стильбена возвращается как
в S д-состояние i^ac-стильбена (с несколько более высоким уровнем энергии,
как обычно для щгс-соединений), так и в ^-состояние транс-стильбена,
отдавая избыток энергии в виде тепла. В равновесную смесь цис-
208 СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЯДРАМИ
и транс-стильбенов превращается в тех же условиях и ^ис-стильбед
триплетное состояние которого энергетически разнится от его транс-
аналога (см. выше), и сначала превращается в последнее.
^ис-Стильбен может быть получен также неполным гидрированием
толана (см. ниже).
Из производных стильбена упомянем 4,4'-диоксистильбен
НО—СН =СН—он
названный за свое эстрогенное действие (стр. 674) стилбэстролом.
4,4'-Бис-(фенилуреидо)-2,2'-стильбендисульфокислота («бланкофор В»)
CeH6NHCNH—Ч-СН=СН-<^ 4-NHCNHC6H5
II Х=< >=Z II
О XSO3H HO3SZ о
применяется для подкраски белья. Она дает синеватую флуоресценцию
и играет роль активной подсиньки.
Дифенилацетшьен, или толан, получается обычными путями синтеза
ацетиленовых соединений, например:
С6П5-С-СН2-СвН5 C6H5-C-CH2-CeH5 2GlHa0Na^
О С1 С1
-->• СвН5—С=С—СбН54-2С2Н5ОН + 2NaCl
толан
При действии аммиаката цианистого никеля он тримеризуется в гекса-
фенилбензол. Над отравленным катализатором гидрогенизации толан
восстанавливается в г^ис-стильбен, над свежим — в дибензил.
Циклофаны
Краму удалось осуществить синтез разнообразных пара-циклофанов.
Так он назвал соединения типов
где тип минимально равны двум; А — одно или два алифатически
углеродных звена, несущих функциональные группы — ОН, =0 и т-
О методах синтеза циклофанов дают понятие следующие схемы:
(ВЫХОД 3% при
наименьшем -п)
209
ЦИКЛОФАНЫ
Ацилоиновая конденсация
1, стр. 571):
сложного эфира (кн.
т = 3
Наиболее интересной особенностью циклофанов является взаимодей-
ствие л-электронных систем расположенных друг над другом (сэндвиче-
образно) бензольных колец. Это взаимодействие можно проследить на
УФ-спектрах, которые сильнее всего отличаются (сдвиг максимума погло-
щения в длинноволновую область) от спектров открытых несэндвичеоб-
разных систем типа
н (с 1 b)o.-<6>- (с н2)„ -<б>- (С н2)у н
при п — 2, т = 2, т. е. при наибольшем сближении бензольных колец
(рис. 96). В таких циклофанах с искаженным УФ-спектром расстояние
между бензольными кольцами может сократиться до величины, меньшей,
чем между плоскостями бензольных ядер в кристалле (меньше 3,4 А).
Искажение спектра связано, по предположению Крама, с искажением
плоскостного строения бензольных колец и нарушением идеальной дело-
кализации л-электронной системы (т. е. со сдвигом в сторону непредельной
системы).
Очень интересны результаты ацетилирования циклофанов по Фриделю —
Крафтсу и их нитрования. 3,4- и 2,2-Пара-циклофаиы дали только моно-
замещенные продукты — кетон или, соответственно, нитросоединение.
Следовательно, в пара-циклофанах с искаженным (или сильно искаженным)
спектром трансаннулярное взаимодействие бензольных ядер таково, что
/ О \
замещение водорода на электроноакцепторную группу ^NO2 или —С\ J
делает второе ядро невосприимчивым к электронной атаке в резуль-
тате оттяжки из него л-электронов замещенным бензольным ядром.
Подобные отношения наблюдаются при образовании комплексов пара-
циклофанов с тетрацианэтиленом. Отношения ароматических ядер
14 Заказ 072
1 L____________I________|
?°° MO ~ 280
Л, пик
(I '
Рис. 96. Кривые поглощения циклофанов. С увеличением тип поглощение
приближается к поглощению их изологов с разомкнутым циклофановыц
кольцом.
ПОЛИФЕНИЛМЕТАНЫ
211
•с этим непредельным соединением являются примером отношения
л-основания с л-кислотой. л-Кислота забирает электрон от л-оспования,
образуя в предельном случае анион и превращая ароматическое ядро
в катион. Такой перенос электрона может быть и не полным, но оп связы-
вает оба компонента в молекулярное соединение «с переносом заряда»
{стр. 121). При этом поглощение света перемещается из ультрафиолетовой
в видимую область.
Для комплексов 2,2-пара-циклофанов с тетрацианэтиленом максимум
поглощения приходится на 5210 А, для комплексов 2,3-парациклофанов —
на 5110 А, в то время как комплекс модельного соединения с двумя бен-
зольными ядрами, связанными открытой предельной цепью углеродных
атомов, имеет максимум поглощения при 4460 А. Это различие объясняется
трансаннулярной «помощью» второго бензольного ядра, усиливающего
подачу циклофаном электрона:
Интересно протекает гидрирование пара-циклофанов. 6,6-Пара-цикл о-
фаны каталитически гидрируются до соответствующих циклогексановых
производных, причем каждое бензольное ядро гидрируется независимо
от другого. 2,2-Пара-циклофан восстанавливается литием в этиламине
(водород в момент выделения) и каталитически с одинаковым результатом—
присоединяется 4 моль водорода и получается диеновое соединение с не-
установленным положением двойных связей. Это соединение лишь с трудом
гидрируется на катализаторе до полностью циклогексанового производ-
ного.
Полифеннлметаны
.К ним относятся следующие углеводороды (начиная с дифенилметана):
Толуол СвНбСНз
Дифенилметан (СвН6)2СН2. • •
Трифенилметан (СеНБ)зСН . .
Тетрафенилметан (С6Н5)4С . .
Т. пл., °C
—95
+27
92
285
Т. кип., *С
262
359
Из них мы знакомы только с толуолом. Для синтеза дифенилметана
можно использовать два варианта реакции Фриделя — Крафтса:
2СвНв + СН2С12 СвН5-СН2-СвН6 + 2НС1
СвНв + СвНБСН2С1 —* СвН6-СН2-СвНб+ HG1
14*
212 СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ яДРДМИ
Трифенилметап проще всего синтезировать из хлороформа и бензола
той жешреакцией:
ЗС6Н6 + СНС1з А1СЬ > (С6НБ)зСН + ЗНС1
Для получения тетрафенилметана аналогичная реакция не пригодна
так как взаимодействие с четыреххлористым углеродом останавливается
на стадии образования трифенилхлорметана:
ЗС6Н6 + СС14 (СвНБ)3СС1 + ЗНС1
Четвертый фенил можно ввести, действуя на трифенилхлорметан
гриньяровым реактивом (выход плохой):
(СвН5)зСС1 + CeH5MgBr-► (С6НБ)4С + MgClBr
Некоторые уже рассмотренные нами соединения, такие, как бензо-
фенон С6Н5СОС6Н5, продукт его восстановления вторичный спирт бенз-
гидрол С6Н5—СН(ОН)—С6Н5 и так называемый кетон Михлера, получа-
емый действием фосгена на диметиланилин
СОС12 + 2С6НбЫ(СН3)2 -► (CH3)2N-CeH4-C-CeH4-N(CH3)2
О
являются производными дифенилметана.
При пропускании паров дифенилметана через накаленную докрасна
трубку образуется о, о'-дифениленметан — флуорен (т. пл. 115° С, т. кип.
294° С):
Пятичленное кольцо флуорена обнаруживает сходство с пятичленным
циклопентадиеном и инденом, которое проявляется прежде всего в легкой
протонизации водородных атомов метиленовой группы в реакциях
с высокоосновными реагентами, например с CH3MgI, и в реакциях конден-
сации с альдегидами:
Из производных полифенилметанов несравненно ^ее Л^дениЯг
и практически и теоретически являются трифенилме!
на которых следует остановиться подробнее.
ПОЛИФЕНИЛМЕТАНЫ
213
Кроме синтеза по Фриделю Крафтсу соединения трифенилметано-
вого ряда могут быть получены, во-первых, с помощью гриньярова реак-
тива:
CeHs—С—С6П5 + C6II5MgBr > (CelIs)3C—OMgBr
О
/ОТ!
—> (СвН5)зС-ОН + Mg<
•Вг
Cells—С—OC2IIs + 2CeH5MgBr > (СеНб)3С—OMgBr + QjIIsOMgBr
о lH>°
/ОН '
(СвН5)зС-ОН + Mg<
хВг
Во-вторых, многие соединения этого ряда получают конденсацией
ароматических оксосоединений с углеводородами в жестких условиях
(концентрированная серная кислота) или с ароматическими соединениями,
содержащими сильные орто-пара-ориентанты, в более мягких условиях:
СвН5 -С/° + 2СеГ1е (С6Н5)3 СН + Н2О
ХН
/О ZnCl« zCeHiN (СНз)2
C,H5-C<f + 2C6H5N(CH3)2—СбН5-СН< + Н2О
'll xCeH4N(CH3)2
(CH3)2N-CeH4-C-CeH4-N(CII3)2 + CeIT5-N (СН3)2 —-
О
Наконец находит
с бензотрихлоридом:
СС13
---> [(CH3)2N-CeH4]3C-OH
применение конденсация ароматических-аминов
2
С1
h2n-/OH-
2
Свойства соединений ряда трифенилметана. Наиболее замечательны
свойства центрального углеродного «метанового» атома производных
трифенилметана. В самом три фенил мета не водород, связанный с этим
углеродом, сравнительно легко (для углеводорода) протонизируется и при
действии таких сильных оснований, как KNH2 или NaCH3, замещается
на металл:
(СвН6)3СН + NaCH3-----> (CeH8)3CNa + СЩ
(СвН5)зСП + KNHo —> (С61Г5)гСК + Nils
214
СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЯДРАМИ
Трифенилметилнатрий можно получить также действием амальгамы
натрия на эфирный раствор трифеиилхлорметана. Трифенилметилнатрий_
ионно построенное соединение, его интенсивно вишнево-красный эфирный
раствор электропроводен.
Изучая оптически равновесия типа
RH + А’ R" + АН
(где RH — углеводород, аА~— анион сильного основания) можно оценить
кислотность подобного рода «кислых» углеводородов. Если сравнивать
даже не с кислородными кислотами, а с водой и спиртами, кислотность
эта чрезвычайно мала:
ка Ка
Трифенилметан . . . 10-зз Вода . . . . . 10-18
Дифенилметан . . . 10-35 СНзОН . . . . 10’17
Фенилацетилен . . . 10-21
Как ни малы эти величины, но на поведении ди- и трифенилметана они
ярко сказываются, прежде всего в приведенных выше реакциях металли-
рования.
Трифенилметилнатрий — исключительно реакционноспособное веще-
ство, тотчас разглагающееся водой или спиртом:
(CeH5)3CNa + Н2О —> (.СбН5)зСН + NaOH
С двуокисью углерода и оксосоединениями он реагирует подобно
гриньярову реактиву:
О
(CeH5)3CNa + С03-> (С6Н5)зС-С-ONa
Воздух окисляет трифенил натрий в перекись трифенилметила:
2 (СвНб)зСНа + 2О2 —> [(СвН&)зСО]2 + Na2O2
Еще гораздо легче протонизируется водород три-п-нитротрифенил-
метана. Для его нейтрализации достаточно действия щелочи в спирто-
вом растворе:
(02N—С6Н4)зСН + NaOH --> (О2Н-С6Щ)зСНа-|-Н2О
Три-п-нитротрифенилметилнатрий — также ионно построенное соед^
некие, но гораздо более устойчивое и менее реакционноспособное, я
трифенилметилнатрий. Спиртовой раствор этого интенсивно синего ве
ства устойчив на воздухе; вода медленно гидролизует его, обесцвечива ^.
Легко понять, что в этом нитросоединении заряд аниона не остается
ПОЛПФЕНИЛМЕТАНЫ
215
метановом углероде, а рассредоточивается по трем нитрогруппам (т. е. по
их кислородным атомам):
Отсюда и большая устойчивость этого аниона.
Что касается существования трифенил метил-аниона, хотя и очень
реакционноспособного, то оно возможно тоже благодаря рассредоточению
отрицательного заряда метанового С-атома (т. е. пары электронов) между
всеми о- и n-углеродными атомами бензольных ядер:
Таким образом, отрицательный заряд аниона не сосредоточен на цен-
тральном метановом атоме, но распределен между ним и девятью углерод-
ными атомами (всеми о-и n-атомами) бензольных циклов. Конечно, рассре-
доточение заряда по углеродам играет некоторую (но только подчиненную)
роль и в тринитротрифенилметил-анионе, где заряд в большей части сосре-
доточен уже на кислородных атомах нитрогрупп.
Сходно противоположная картина наблюдается у трифенилхлорметана.
В этом соединении хлор обладает довольно большой подвижностью и легко
гидролизуется водой:
(СвН5)зСС1 + Н2О -> (Св115)зСОН + 1IC1
Трифенилхлорметан применяется (в пиридиновом растворе) как трити-
лирующий (т. е. вводящий тритильную — трифенилметильную группу)
216 СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЯДРАМИ
.реагент. Так, тритилируют, например, первичные спиртовые группы
в сахарах (вторичные не тритилируются).
В растворе в жидком SO2 трифенилхлорметан электролитически диссо-
циирует (в воде и спиртах диссоциацию нельзя обнаружить из-за гидро-
лиза или алкоголиза) и образует трифенилметильный катион. Устойчивость
этоге карбониевого катиона достигается рассредоточением положитель-
ного заряда (подача электронов бензольными ядрами) между десятью
углеродными атомами:
Катион трифенилметила (например, раствор перхлората трифенил-
метила в уксусной кислоте) способен отнять гидрид-анион с парой электро-
нов от донора водорода, каким служит, например, бензиловый спирт:
(СвНб)зСС1О74-СбН5-С-ОН —> (С6Нб)зСН + свн5-с
+ НС104
Изящную реакцию такого гидридного перемещения осуществил
Бонтон [1959 г.], получивший окрашенный катион перинафтилия:
(с6н5)3с сю;
Можпо ли введением заместителей стабилизовать трифенилмет
катион подобно стабилизации аниона посредством введения нитрогрупп
Очевидно, что на этот раз нужно ввести в пара-положение электронод
норные группы типа —NH2, — N (СН3)2 и т. Д- Действительно, три-
ПОЛИФЕНИ.ЧМЕТАНЫ
217
(^-Диметил аминофенил )-хл орметан дает даже в водном растворе совер-
шенно устойчивый катион, в котором положительный заряд рассредоточен
по всем трем бензольным ядрам, а главным образом по атомам азота
диме тил аминогрупп:
Изображенный интенсивно окрашенный в фиолетовый цвет катион
(катион красителя кристаллического фиолетового) исключительно устой-
чив и не гидролизуется водой. Тольк сконцентрированная щелочь медленно
обесцвечивает его, превращая в бесцветный три-(п-диметиламинофенил)-
карбинол:
ОН
N(CH3)2
В катионе кристаллического фиолетового положительный заряд
рассредоточен в основном между тремя атомами азота, но также, в очень
малой степени, и между девятью о- и тг-углеродами бензольных ядер.'
Рассредоточение заряда катиона или аниона между двумя или более'
атомами, связанными системой л,л-сопряжепия, всегда имеет следствием^
уменьшение уровня энергии первого возбужденного состояния молекул!/!.'
218
СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЯДРАМИ
Поэтому такие окрашенные молекулы возбуждаются квантами меныпей
энергии, т. е. излучением с большей длиной волны сравнительно с Уф.
излучением, возбуждающим, например, бесцветный трифенилхлорметан.
Если трифенилхлорметап диссоциировал на ионы (например, в растворе
в S02), его катион возбуждается фиолетовыми квантами, поглощает их
и поэтому окрашен в дополнительный к фиолетовому желтый цвет. При
переходе к катиону кристаллического фиолетового, имеющему резко
пониженный энергетический уровень в возбужденном состоянии, длина
волны возбуждающих (и потому поглощенных) квантов резко перемеща-
ется в сторону желтой области спектра и возникает фиолетовая окраска.
Подобным образом дело обстоит со всеми трифенилметиловыми катионами
и анионами, и все они интенсивно окрашены.Однако практически только
соединения с более прочными катионами применяются в качестве кра-
сителей.
Главная группа трифенилметановых красителей это — аминопроиз-
водные трифенилхлорметана. Все они построены по одному из трех типов:
где R может быть водородом, алкилом, бензилом или фенилом. Для каждого
типа приведена только одна из резонансных структур, остальные легко
построит сам читатель. Красители типа I не имеют практического значения.
К типу II относятся важные красители: малахитовый зеленый (R=CH3),
бриллиантовый зеленый (R = C2H5), а также один из простейших по струк-
туре, но не применяемый практически фиолетовый Дебнера (R=H).
Обычные методы синтеза красителей этого типа проиллюстрируем на
примере фиолетового Дебнера и малахитового зеленого.
Нагревание аминов с бензотрихлоридом приводит непосредственно
к образованию катиона красителя. При конденсации аминов с бензаль-
дегидом образуется аминопроизводное трифенилметана — бесцветное
вещество, называемое лейкооснованием (от греч. leukos — белый), легко
окисляющееся уже воздухом или (как обычно делается) двуокисью
свинца в карбинол — основание красителя, при действии даже разбавлен-
ПОЛИФЕНИЛМЕТАНЫ
219
ной соляной (или иной) кислоты отдающее свой гидроксил и образующее
катион красителя:
фиолетовый Дебнера
лейкоосиоваиие
малахитового зеленого
малахитовый зеленый
(катион)
К красителям типа III относятся: парафуксин (R = H), фуксин
(то же, но в орто-положении к одной из аминогрупп имеется группа СН8),
кристаллический фиолетовый (R = CH3).
Первым представителем трифенилметановых красителей был фуксин
окислением мышьяковым ангидридом
примесь гомологов (о- и п-толуидинов):
(или розанилин), полученный
As2O5 анилина, содержащего
При окислении тем же окислителем или нитробензолом (Купье) смеси
анилина с ?г-толуидином был получен и низший гомолог фуксина —
парафуксин'. NH2
СГ
220
СОЕДИННЕИЯ С, ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЯДРдМи
Новый «фуксиновый процесс» (Гомолка) состоите нагревании анилина
с форма чином и окислителем. Предполагается такая последовательность
реакций:
СНоО
+ H..N—
Метилируя парафуксин иодистым метилом, получают краситель
метиловый фиолетовый — смесь метилированных по азоту три-н-амино-
трифенилхлорметанов с преобладанием пентаметильного производного,
но содержащую также и гексаметильное производное (кристаллический
фиолетовый).Метиловый фиолетовый (он очень похож по цвету на кристал-
лический фиолетовый) общеизвестен, так как из него делаются фиолетовые
чернила.
Кристаллический фиолетовый в чистом виде получается конденсацией
ди-(п-диметиламин)-бензофенона (кетона Михлера) с диметиланилином
и последующим подкислением:
С СГ
Ряд других находящих практическое применение трифенилметано-
вых красителей строится по тем же принципам, но содержит в одном
из фенильных остатков один или два атома хлора, одну или две сульфо-
группы. Сплавлением парафуксина с анилином и бензойной кислотой
(конденсирующий агент) получают краситель анилиновый синий
(<O>“NHX©>)aC Ci-
Как ясно из сказанного выше, аминогруппы трифенилметанового
красителя-катиона играют решающую роль и в устойчивости катиона
и в его вветопоглощении; такие группы, как мы знаем (стр. 108), назы
ваются ауксохромами. Оба эффекта являются следствием рассредоточен
ПОЛИФЕИИЛМЕТАНЫ
221
заряда с метанового углеродного атома на атомы азота, но эти эффекты
действуют непараллельно. При увеличении в трифенилметильном
катионе числа ауксохромов в ядре от одного до двух и далее до трех
устойчивость катиона растет, а скорость обесцвечивания гидроксильным
ионом падает. Это происходит вследствие снятия все большей части заряда
с метанового углерода и в связи с этим все большего возрастания энергии
активации взаимодействия соединения с гидроксильным ионом. Что
касается светопоглощенпя, то при увеличении числа ауксохромов от
одного до двух желтый или желто-оранжевый цвет (дополнительный
поглощаемый — фиолетовый) переходит в зеленый (поглощается пур-
пурно-красный), а при введении трех ауксохромов — в фиолетовый
(поглощается желтый или желто-оранжевый), т. е. батохромный эффект
максимален в случае двух, а не трех ауксохромов. Это происходит
потому, что уровень энергии первого возбужденного состояния наи-
более близок к невозбужденному, если заряд рассредоточен линейно,
.а не разветвленно, как в случае трех ауксохромных групп.
Ауксохромные группы действуют благодаря наличию у атомов азота
свободных пар электронов. Если эти электронные пары связать, присо-
единяя к ним ионы водорода действием кислоты (или превращением
в четвертичную аммониевую соль под действием СН31), то и химическое и
оптическое действие ауксохромов прекратится. Поэтому, приливая
к раствору кристаллического фиолетового концентрированную соляную
кислоту, можно наблюдать постепенный переход к зеленому цвету, в точ-
ности похожему на цвет малахитового зеленого (выключается один ауксо-
хром), затем к оранжево-желтому (выключаются два ауксохрома) и,
наконец, получается слабоокрашенный желтый раствор.
Строение парафуксина установлено Э. и О. Фишерами путем диазо-
тирования первичных аминогрупп его лейкооснования и элиминации
.{замены на водород) диазогрупп (стр. 109), в результате чего был получен
-трифенилметан:
(H2N-CeH4)3CH + ЗНС1 -> (С1-Н3Л-С6Н4)зСН 3HN0^
восстановление;
(Cl- N=Lc8H4j.3CH ------ЗСзНЮН (СвНб)зСН + 3N„ + зсНзСНО + ЗНС1
Диазотирование парафуксииа-катиона и замена диазогрупп на гидр-
оксилы кипячением с водой приводит к образованию аурина,-.
(Cl" N==N-CeH4)3G С1- + ЗН2О -> (НО-СвН4)зС СГ + 3N2 + ЗНС1
(-НС1
ОН
А
it 1
0=<2>U-<f voh
аурин
222 СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЯДРАМ11
— —
Из фуксина этим путем образуется розоловая кислота, а из фиолетового.
Дебнера — бензаурин:
ОН
розоловая кислота
бензаурин
Все три — красители трифенилметанового ряда с гидроксилами,
в качестве ауксохромов. Как видно из формул, окситрифенилметановые-
красители являются производными фуксона, который можно получить,,
отщепляя нагреванием воду от окситрифенилкарбинола:
НО~~^О/н~С(С6Н5)2
ОН
150°С
В щелочном растворе эти красители резко углубляют цвет, особенно
бензаурин (батохромный сдвиг области поглощения); желтый аурин со
щелочью становится красным, а бензаурин даже фиолетовым. Все три.
вещества и применяются как индикаторы:
или
X
Формулы изображают рассредоточение заряда в анионе бензаурина-
(Х = Н) и в аурипе (Х = ОН); в аурине, содержащем три ауксохрома,
рассредоточение разветвленное, углубление цвета меньше.
Фталеины, флуоресцеин, розамины и родамины
Фенол и фталевый ангидрид при нагревании с серной кислотой образуют
фенолфталеин. В химии это соединение известно как индикатор, в меди
цине, под названием пурген, как слабительное. При действии щелоч
ПОЛИФЕНИ ЛМЕТАНЫ
223
бесцветный фенолфталеин I образует красно-фиолетовый анион II с таким
же характером мезомерного распределения анионного заряда между двумя
бензольными ядрами, как в бензаурине. Избыток щелочи, как всегда,
•обесцвечивает трифенилметановый краситель, превращая его в карби-
нол III:
Известен ряд фталеинов — индикаторов.
При сплавлении фталевого ангидрида с резорцином и хлористым
щинком происходит подобная же конденсация, осложненная дополнитель-
ным выделением молекулы воды за счет двух фенольных гидроксилов
® замыканием гетероциклического кольца:
В щелочной среде с размыканием лактонного цикла образуется интен-
сивно флуоресцирующий желто-зеленым цветом анион флуоресцеин'.
Бромирование флуоресцеина приводит к образованию красителя
.эозина — тетрабромфлуоресцеина (все четыре орто-положения по отноше-
нию к гидроксилам замещены на бром).
224
СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЯДРАМ0
Конденсация фталевого ангидрида с л1-диал кил аминофенолами дает
имеющие практическое значение красители — родамины:
Лишенные карбоксила родамины называются розаминами. Их
получают конденсацией бензотрихлорида с ж-диалкиламинофенолами:
СВОБОДНЫЕ РАДИКАЛЫ РЯДА ТРИФЕНИЛМЕТИЛА
В 1900 г. Гомберг, стремясь получить реакцией Вюрца гексафенилэтан,
осуществил этот синтез, действуя на трифенилхлорметан, цинком, медью,
ртутью или серебром:
2 (СвН5)зСС1 +{Zn -> (СвН5)зС—С(СвН5)з -f- Z11CI2
Полученный углеводород обладает необычными свойствами. На возду*6
он окисляется в уже знакомую нам (стр. 214) перекись трифенилметила
(С8Н5)зС-С(С6Н5)з + О2
(С8Н5)3С-О-О-С(С6Н5)з
СВОБОДНЫЕ РАДИКАЛЫ РЯДА ТРИФЕНИЛМЕТИЛА
225-
с натрием дает трифенилметилнатрий
(СвН5)3С—С(СвН5)з + 2Na > 2(CeH5).1CNa
а с галоидами количественно образует трифенилгалоидметан:
(С8Н5)зС—С(СвН5)з + 12 > 2(С8Н5)3С1
Дальнейшие исследования Гомберга, А. Е. Чичибабина, Шленка,
Шмидлина с несомненностью доказали, что гексафенилэтан и вообще
подобные ему гексаарилэтаны в растворах (например, в эфире) гомоли-
тически диссоциируют на два свободных трпарилметильных радикала:
(СвН5)зС-С(СвН5)з 2(СоН5)3С.
За диссоциацией можно проследить по уменьшению молекулярного
веса гексафенилэтана, происходящему при разбавлении, что связано
с увеличением степени диссоциации при разбавлении (закон Оствальда).
Можно проследить за диссоциацией также и по изменению окраски
раствора, так как гексафенилэтан бесцветен, а трифенилметил — желтый.
Известны и триарилметилы, окрашенные достаточно интенсивно.
Для ряда гексаарилэтанов установлены следующие величины степени
гомолитической диссоциации в 0,1^7И растворе в бензоле при комнатной1
температуре (в %):
(СвН5)зС-С(СвН5)з................................
(п-СвНзСвЩ) (C8Hs)2C—СЦСвНб^СбНбСбЩ-га) . . . .
(и-С8Н5СвН4) (С8Нб)2С—C(CeHs) (С8Н5С8Н4-п)2 . . . .
(п-С8Н5С8Н4)зС—С(С8Н5С8Н4)3 .....................
(n-FC8H4) (C8H5)2C-C(C8H5)2(C8H4F-n) ............
(п-МОгС8Н4)зС—C(C8H4NO2-ra)3.....................
(о-СНзС8Н4)3С-С(С8Н4СНз-о)з .....................
То же, л1-изомер ................................
» п-изомер....................................
2-9
(по разным
источникам)
15
18
26
20
100
82
7
5,5
Наличие в растворах [а для (п-МО2С6Н4)3С • и в твердом состоянии]
свободных триарилметильных радикалов устанавливается по парамаг-
нитным свойствам этих частиц, имеющих один неспаренный электрон при
метановом углеродном атоме. Нескомпенсированный спин этого электрона
и создает магнитное поле вокруг каждого свободного радикала. Фактически-
именно триарильные свободные радикалы, а не гексаарилэтан реагируют
с кислородом, натрием и галоидами:
— - АгзС-О-О-САгз
2АгзС«------2ArsCNa
—* 2АгзШ
В то время как на диссоциацию этапа на два свободных метильных
радикала требуется 85 ккал! моль, для диссоциации гексафенилэтана
достаточно всего лишь 12 ккал/ моль. Вот почему этот процесс и совершается*-
15 Заказ 6/2
226 СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЯДрАМИ
так легко. Причина столь резкого понижения энергетического уровня
•трифенилметильного радикала сравнительно с метильным та, что неспарен
ный электрон в трифенилметиле не концентрируется на метильном угле-
родном атоме, а рассредоточивается по всем бензольным ядрам, точнее
говоря, по всем девяти их о- и n-углеродным атомам, приобретающим
-таким образом в некоторой степени свободнорадикальный характер-
О наличии доли свободнорадикальных свойств у n-углеродных атомов
трифеиилметила можно судить но тому, что гексафенилэтан при нагрева-
нии постепенно превращается в высокоплавкий (т. пл. 231° С) углеводо-
род, для которого А. Е. Чичибабин установил строение п-бензгидрилтетра-
фенилметана:
+ (С6Н5)3С------>
---> (СбНйЬс^ОХсн-^О/
Бензгидрилом называется остаток (С6Н6)2СН —.
Естественно, чем больше возможностей рассредоточения предоставля
неспаренному электрону примыкающие к метильному углеродво
атому арилы, тем выше константа гомолитической диссоциации ге
арилэтана. Поэтому гексабифенилэтан, (гексаксенилэтан) диссоцииру
сильнее, чем гексафенилэтан:
СВОБОДНЫЕ РАДИКАЛЫ РЯДА ТРИФЕНИЛМЕТИЛА
227
Очевидно, еще большее рассредоточение электрона от метильного-
углерода достигается оттягиванием электрона^посредством нитрогруппы:
или
Хотя основная причина гомолитической диссоциации полиарилэта-
нов — мезомерное рассредоточение свободного электрона в ароматических
ядрах, есть и второй фактор, способный усилить диссоциацию, а именно
пространственные препятствия рекомбинации радикала в димерную моле-
кулу. Это можно выразить и иначе: три громоздкие арильные группы
у каждого метильного углеродного атома испытывают большое взаимное
отталкивание (/-напряжение по Брауну) и отталкивание от трех арилов
у второго метильного углерода (/’-напряжение по Брауну). При диссоциа-
ции /’-напряжение исчезает полностью, а /-напряжение уменьшается
в результате перехода триарилметильного радикала в пропеллерообразную
форму. Из сопоставления значений степени диссоциации гексатолилэта-
нов (стр. 225) можно видеть, что орто-заместители резко повышают степень
диссоциации сравнительно с м- и «-заместителями. Этот эффект наблюдается
не только в гексаарилэтанах, но и в симметричных тетраарилэтанах.
Еще сильнее действует в том же направлении замещение в тетраарилэта-
нах оставшихся двух водородов на трет-6утильные радикалы.
Интересно отметить, что кремниевые, германиевые и оловянные аналоги гекса-
фенилэтана не диссоциируют с образованием свободных радикалов, что)лишь частично
может быть объяснено большей величиной атомов Si, Ge и Sn и, следовательно, мень-
шими пространственными препятствиями ассоциации.
15*
228
СОЕДИНЕНИЯ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЯДРдцщ
Таким образом, примыкающие к метаповому углеродному атом
фенильные и вообще арильные группы могут осуществить троякую
функцию: рассредоточить отрицательный заряд и сделать в той или иной
мере устойчивым анион Аг3С~; рассредоточить положительный заряд и
стабилизировать катион Аг3С+ ; наконец, за счет рассредоточения неспарен-
ного электрона превратить совершенно неустойчивый свободный радикал
метил в «Долгоживущий» устойчивый триарилметильный радикал.
Устойчивостью триарилметильного аниона объясняется «подвижность»
(легкая протонизируемость) метанового водорода триарилметанов, а устой-
чивостью триарильного аниона — подвижность хлора в триарилхлор-
метанах.
Следует отметить, что возможность рассредоточения заряда посредством
трех арильных ядер была бы еще больше, если бы триарильная группи-
ровка могла быть вполне плоской. На деле даже трифенилметильная группа
имеет пропеллерообразную форму. Если бы это было иначе, о-водородные
атомы не помещались бы и мешали бы друг другу. В этом можно убедиться
-если построить структуру трифенилметила, использовав приведенные
(кн. 1, стр. 358) длины межатомных расстояний, а для о-водородных атомов
проведя полуокружности с радиусом, равным ван-дер-ваальсовому радиусу
водорода.
Таким образом, перекрывание л-облаков электронов в трифенилмета-
новых производных несовершенно.
Особенно наглядно невыполнение пространственных требований
перекрытия электронов л-связей выражается в свойствах углеводорода
триптицена, синтез которого дан на стр. 247. Этот углеводород, казалось
-бы подобный трифенилметилу, совершенно не способен к протонизации
и вообще к каким-либо замещениям метинового водорода. Также «неподви-
•,жен» и хлор у метинового углерода в хлортриптицене. Неосуществим
триптинен
я «долгоживущий» триптицильный свободный радикал. Причина в том,
-что из-за жесткой структуры триптицена невозможно перекрывание
л-электронов любого из трех бензольных ядер ни с перпендикулярно
расположенной р-орбиталыо нечетного электрона метикового углерода
в триптицильном радикале, ни с р-орбиталью электронной пары в анИ0Н
триптицила, ни с вакантной р-орбиталыо катиона триптицена. Следстви
является невозможность рассредоточения заряда по бензольным яДРа^-
Есть и вторая причина «неподвижности» галоида в галоидтриптид
Если только что описанная причина исключает возможность оо
галоида по механизму то вторая объясняет неосуществимость
мена и по механизму SN2 (кн. I, стр. 82). Это — невозможность вальде^
некого обращения при обмене хлора в хлортриптицене по механизму
НАФТАЛИН II ЕГО ПРОИЗВОДНЫЕ
229
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
НАФТАЛИН И ЕГО ПРОИЗВОДНЫЕ
Нафталин содержится в каменноугольной смоле в количестве —5%
и выделяется (выкристаллизовывается) из ее средних (до 240° С) и тяжелых
(до 270° С) фракций. Получают его и из продуктов пиролиза нефти.
Нафталин — кристаллическое вещество с т. пл. 80° С, возгоняющееся,
летучее уже при комнатной температуре, перегоняющееся с водяным паром.
Запах его общеизвестен. Открыт нафталин Гарденом в 1819 г., т. е. раньше
бензола (1825 г.). Состав его установлен А. А. Воскресенским, а строение
как двух конденсированных бензольных ядер — Эрленмейером и Гребе,
использовавшими формулу бензола Кекуле.
8 1 а а
I U I ₽l II |P
4/\x*
5 4 а а
Доказательства в пользу этой формулы нафталина следующие. Нафта-
лин — явно ароматическое соединение, так как молекулярной формуле
С10Н8 должно отвечать при наличии одного цикла шесть двойных связей
(С10Н22—С10Н8= 14Н; 14/2 = 7; 7—1 = 6), а при наличии двух циклов — пять
двойных связей и т. д., но обычные реакции на двойные связи отсутствуют.
Вместе с тем при каталитическом присоединении водорода (над Ni),
протекающем в две фазы, сначала образуется тетралин — продукт
присоединения 4Н, затем декалин — продукт присоединения (в сумме)
10Н. Это указывает на наличие пяти ароматических («скрытых») двойных
«связей и, следовательно, двух циклов. Окисление нафталина кислородом
воздуха над V2O5 и серной кислотой с HgSO4 в качестве катализатора или
другими окислителями приводит к фталевому ангидриду, что доказывает
наличие одного бензольного цикла и орто-положение углеродных атомов
второго цикла. Нитрование нафталина дает а-нитронафталин, который при
окислении образует о-нитрофталевый ангидрид, а при восстановлении —
а-нафтиламин, превращающийся при окислении также во фталевый ангид-
рид — на этот раз отгорает ядро, содержащее аминогруппу и ею активи-
230
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Отсюда ясно, что молекула нафталина состоит из двух бензольнь
ядер, имеющих два общих о-углеродных атома.
В то время как в бензоле ароматическая шестерка л-электронов расппе
делена совершенно равномерно по отношению к шести углеродным атомам'
в нафталине выравнивание в такой мере невозможно, и он в результате
этого менее ароматичен и более непределен, чем бензол. Соответственно
меньше и понижение энергии нафталина сравнительно с рассчитанной
энергией непредельного углеводорода, отвечающего по структуре формуле-
Эрленмейера с фиксированными двойными связями (энергия резонанса)
Это понижение энергии для нафталина составляет 61 ккал/моль, т. е
заметно меньше удвоенной величины (36-2 = 72) для бензола.
Действительно, если использовать символику резонанса, структура
нафталина должна быть промежуточной между структурой Эрленмейера —
Гребе и структурами, в которых на одно из бензольных ядер приходятся
три л-связи, а на другое лишь две:
а б в
Меньшая симметрия каждого из бензольных ядер нафталина сказывается
и в чередовании расстояния С—С (по рентгеноструктурным данным):
Особенности структур бив, где одно из ядер бензольное, а другое
диеновое, находят отражение в особой активности а-углеродных атомов
к электрофильным атакам и к тем типам присоединений, к которым способны
диеновые углеводороды. Так, в отличие от бензола нафталин восстана-
вливается водородом в момент выделения, превращаясь в 1,4-дигиДрона-
фталин
I4-2H
и эта реакция экзотермична (4,5 ккал/моль). т
Бром присоединяется к нафталину в положение 1,4, причем проду
присоединения легко отщепляет НВг, образуя а-бромнафталия.
Н/\вг
(НАФТАЛИН И ЕГО ПРОИЗВОДНЫЕ
231
Для нафталина, так же как для бензола, стала известна (Куксон, Дане,
1962 г.) реакция присоединения сильных диенофилов в жестких условиях
ф~150 С):
CN
I
С
+ III
С
I
CN
Реакции электрофильного замещения (нитрование, сульфирование)
идут в нафталине в первую очередь в а-положения.
Подобно бензолу, но легче, нафталин способен захватывать электрон,
образуя радикал-анион. Эта реакция идет уже с натрием:
Na+
Изомерия производных нафталина. Уже монопроизводные нафталина
известны в виде двух рядов изомеров: а-замегценные и [3-замещенные.
Для двух одинаковых заместителей в соответствии с формулой Эрлен-
мейера — Гребе возможно 10 изомеров:
СНз СНз СНз СНз
8 СНо ।
’III! I 'I I I II I I II I
•yy vvy,,, vy y\^ HC/W
СНз СНз 3
1 2 3 4 5
6 7 8 9
iO
Для обозначения положения заместителей принята нумерация угле-
родных атомов (только несущих водороды, которые могут подвергнуться
замещению), указанная в формуле 1. Изображенный диметилнафталин
называется таким образом 1,2-диметплнафталин. Положения 1,2- или .,3-
232;КОНДЕНСПРОВАНН ЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ!
называются, как и для бензола, орто-положениями, но в случае нафталин
обозначение это неоднозначно; положение 1,3 — мета, 1,4 — пара, 18 —
пери, 2,6 — амфи. Приставки, придуманные для обозначения остальных
положений, не привились. Если в молекуле имеются два разных замести-
теля, число изомеров возрастает до 14. Все предвидения формулы нафта-
лина оправдываются на опыте.
Физические свойства нафталина и других конденсированных аромати-
ческих соединений приведены в табл. 72.
Таблица 72. Конденсированные ароматические соединения
Название Формула Цвет Энергия стабили- зации ккал) молъ Темпера- тура плавле- ния °C Темпера- тура кипения °C Плот- ность йГ
Нафталин Антрацен 1 II 1 А/\А АЛА Ч/\А\А Бесцвет- ный То же 71 104 80,2 217,0 218 354,5; 218,4 (при 40 рт. стп.) 1,145 1,25 (df)
Фенантрен <о-с> » 111 100 340,1 1,063 (при 100° с>
Флоурен Л—А X/X(iH2 » — 116 295 —
Пирен » — 150 >360 —
Хризен ^ол ^АХА » — 256 448 —
Нафтацен, или тетрацен 1 II 1 1 ) А/\А\А\А Оранжево- желтый 130 335 — —
Перилен АА Золотисто- желтый — 273—274 — •—
АА V\A
1НАФТАЛИН И ЕГО ПРОИЗВОДНЫЕ
233
•Синтез нафталина и его производных. При пропускании ацетилена над
.древесным углем при 400° С наряду с бензолом образуется нафталин
(Н. Д> Зелинский, Б. А. Казанский):
СН
сн
сн
нс%
МЛ1
СН
I + н2
ИСП
СН
Нри дегидроциклизации над платиной при 300° С гомологов бензола
< боковой цепью из четырех и более углеродов образуется нафталин и его
гомологи (Н. Д. Зелинский, Б. А. Казанский, А. Ф. Плата, С. И. Хромов):
СН2
/Y \сн2
I Pt(300° С)
/ /СНг
сн2
I + зн2
СНз
СНз
Различные варианты диенового
производным:
? СН2
о н
II сн2
синтеза
приводят
к нафталину и его
ОН
сн2
ОН
/СИ
//
сн2
\/ \ /сн
"|| сн2
О II
U
Л /сн
I сн2
он
-н2
ОН
Zn
о
| + ZuO
Наиболее давний путь, послуживший подтверждением
Эрленмейера — Гребе, — замыкание фенилвинилуксусной
•в а-нафтол:
формулы
кислоты
о
II
НОС
/г\ ХхСНг
Гомологи нафталина могут быть получены из нафталина алкилированием
или ацилированием по Фриделю - Крафтсу с последующим восстановле-
нием кетонов по Клемменсену в алкилнафталины. Ацетилирование по
Фриделю - Крафтсу с A1CJ3 в сероуглероде как растворителе идет
в большей степени в a-положение, а в нитробензоле - в 0-положение.
234
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ
СИСТЕМ^
Реакции электрофильного замещения в нафталине. 1. Уже сказано, ЧТ{>
в отсутствие катализатора галоиды (С12 и Вг2) присоединяются в 1 4
положения нафталина. В присутствии соли железа или других катализа'
торов галоидирования замещение идет преимущественно в а-положения
(при хлорировании — на 95% в а- и на 5% в Р~).
Галоидпроизводные нафталина обладают обычным малоподвижным
ароматическим галоидом и мало пригодны для реакций вторичного заме-
щения галоида на другие группы.
2. Нитрование нафталина проходит легко, и нитрогруппа вступает
в а-положенпе. Для введения еще одной нитрогруппы требуются более-
жесткие условия, причем вторая нитрогруппа вступает в другое ядро
нафталина тоже в a-положение и получается 1,8-динитронафталин наряду
с 1,5-изомером. Это указывает на некоторую автономию обоих ядер:
нитрогруппа пассивирует к электрофильным атакам в большей степени
то ядро, в которое она вступила.
Восстановлением а-нитронафталина Н. Н. Зинин получил а-нафтшь
амин.
p-Нитронафталин можно получить лишь обходным путем, например
окислением р-нафтиламина кислотой Каро или через (З-нафтилдиазошш по
Гаттермапу (NaNO24-Cu).
3. Сульфируется нафталин легко концентрированной серной кислотой,
причем ниже 100° С получается а-нафталинсульфокпслота, а выше 150° С—
Р-нафталинсульфокислота. Объясняется это тем, что благодаря высокой
нуклеофильной активности а-углеродных атомов нафталина а-сульфо-
группа при повышенной температуре в кислой среде легко отщепляется
в виде серной кислоты (электрофильная атака Н+) и замещается на водо-
род:
В подобной ацидолитической реакции может уцелеть только сульфо-
группа, попавшая в Р-положение.
Таким образом, оба изомера — а- и Р-нафталинсульфокислоты —
доступны и служат исходными веществами для синтезов в нафталиновом
ряду.
При избытке более концентрированной серной кислоты а-нафталинсуль-
фокислота сульфируется далее, образуя две изомерные дисульфокислоты—
1,5- и 1,6-; Р-нафталинсульфокислота также дает два изомера — 2,6- 2-
2,7-нафталиндисульфокислоты:
h2so«
------>•
SO3H
^\z W ОзН
I II I
SO3H
HO3s/
HO.S . z
I «. . + IL x J
SOjH
.НАФТАЛИН И ЕГО ПРОИЗВОДНЫЕ
235
Вторичное замещение в производных нафталина. Реакцией щелочного
плавления из а- и |3-нафталинсульфокислот в большом масштабе готовят
фенолы нафталина — а- и [3-нафтолы:
SO3Na ОН
! I
| К | +NaOH ------->1 || |+Na2SO3
^/WS°3Na ^\/W°H
I II I + NaOH --------> | II I + Na2SO3
Их применяют как вторые компоненты в азосочетании.
P-Нафтол служит единственным источником синтеза р-нафтиламина,
который получают действием на [3-нафтол NH4HSO3 (или, ранее, комплек-
-сом NH3-rZnCl2):
^\/\/0Н /7 , NH2
I II I + NH4HSO3----► I II I + H2SO3 + H2o
\/\^ \
Следующая схема дает понятие о моно- и дисульфокислотах, полу-
-чаемых сульфированием обоих нафтолов и нафтиламинов.
•Синтез нафтол сульфокислот
SO3H SO3H
H2SO* | I H2SO4+SO3_______________________________________t I H.SO.+SO.
105—120° С I I 70-80° С I 25—35°G
I 4. 4.
Р-кислота
SO3H
Г-кислота
SO3H
236
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ бИСТВЩд
Синтез пафтпламинсульфокислот
NH2
NH2
£>О3Н
SO3H
I znh2
45°—65° С
nh2
HO3S. .4 ,NH«
180—170° С
nh2
H2SO*+SO»
35—90° С
SO3H
so3h
YYY
ho3s/YY\^
SO3II
nh2
SO3H
В нафталиновом ядре, так же как и в бензольном, осуществляются
многочисленные перегруппировки. Мы уже рассмотрели перегруппировку
ct-нафталинсульфокислоты в |3-нафталинсульфокислоту под действием
серной кислоты. Это — интермолекулярная (т. е. межмолекулярная)
перегруппировка. По Н. Н. Ворожцову мл., при пропускании а- или
(3-хлорнафталина над окисью алюминия при высокой температуре и тот
и другой превращаются в равновесную смесь обоих изомеров, причем,
как показывает опыт с а-изомером, меченным 14С, перегруппировка проте-
кает по схеме
Cl
I 14
14 PI
т. e. интрамолекулярно (внутримолекулярно).
По Н. Н. Ворожцову ст. и В. В. Козлову, нагревание натриевой соли
1-аминонафталин-4-сульфокислоты приводит к ее изомеризации в соль
1-аминопафталин-2-сульфокислоты; 1-аминонафталин-2-сульфокислота (на-
триевая соль) перегруппировывается в 2-аминонафталин-6-сульфокислоту.
Подобные же перегруппировки происходят с солями нафтолсульфокислот
'k' А. Шилов и В. В. Козлов); 1-оксинафталин-4-сульфокислый натрия
при 1/0° С изомеризуется в 1-оксинафталин-2-сульфокислый натрия,
а 2-оксинафталин-1-сульфокислый натрий при более высокой темпера-
туре ( 200° С) превращается в 2-оксинафталин-6-сульфокислый натрия.
ttO.,s 'Х-'Х
нафталин и иго ПРОИЗВОДНЫЕ
237’
Бензидиновая перегруппировка нафтиламинов, подробно исследован-
ная В. О. Лукашевичем, проходит так же, как в бензольном ряду, но’
даже без минеральной кислоты:
Нафтолы и их производные, в основном моно- и дисульфокислоты,
нафтиламины и их сульфокислоты применяются главным образом для
синтеза разнообразных азокрасителей, примеры структур которых мы
приводим. Последовательность диазотирования и азосочетания понятна
из формул азокрасителей:
Моноазо красптели
оранжевый I
кислотный оранжевый
паракрасный
Н0\
HO3S—n=N-<^
О
прочный красный
НО /SO3H
Ч__N=N__Ч
\SO3H
оранжевый Р
НО\
HO3S-N = N—Ч
HO3S-/_J> '
Nso3h
яркий пунцовый
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИС
Дисазокрасители IIO3S— //~ HOsS\ \ //-^ / 4 SO3H НОХ N=N—<^~^>— N=N— НОз5-/_^ кроцеиновьш алый ЗБ /N=n-<^-n=n-<(2^ Ч ПзС/ HO3S-/_^ ^-so3H кроцеиновый яркий 9Б ^-N=N—n=n_> NH2 нафтиламин черный ОН нн2 _ К='\АА/ N=N-<_>N0‘ HO3SZ кислотный сине-черный
Прямые красители HzN~<C)>"N=N“C \vh2 nh2 он HOiS\A/vn=n' CH3| SO3I-I nh2oh >-O-N=N\J A/n=n-O 1 11 J НОз^^^^ОзН прямой черный 3 OH nh2 /' \ / \ AT— N ) zl ,SO3H 0/ \осн3 Y Y SO3H
прямой голубой светопрочный 23М
НАФТАЛИН И ЕГО ПРОИЗВОДНЫЕ
239-;
диаминогеновый синий
Нафтохиноны
При окислении хромовой кислотой 1,4-, 1,2- и 2 -диоксинафталнноь или
аминонафтолов получаются три нафтохинона;
л-яафтохппон
о-н.зфтохинон
амдОн-нафтохияои
По свойствам они представляют собой истинные хиноны и подобны
бензохинонам.
Производными тг-нафтохинона является ряд природных и синтетиче-
ских красителей. Из последних назовем лишь аналогичный хинизарину
(стр. 251, формула III) нафтазарин, получаемый действием серной кислоты-
и серы на 1,5-динитронафталин:
О ОН
Предполагается, что реакция идет через восстановление нитрогрупп
до NHOH. перегруппировку арилгидроксиламина в п-оксиариламин,
частичное (в одном ядре) окисление оксиариламина в хинонимин и гидро-
лиз иминной группы в оксогруппу и аминной в гидроксил.
Производными n-нафтохинона являются и витамины группы К: филло-
хинон (витамин Кт) и фарнохинон (витамин К2).
Витамин Кг содержится в зеленых частях растений, а витамин К2 —
в бактериях и рыбах.
Оба витамина К построены аналогично 2-метил-1,4-нафтохинону,
о чем можно судить по сходству спектров поглощения (во всех трех соеди-
нениях хромофорной является структура 1,4-нафтохинона).
:240
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Строение витаминов Кх и К2,
таково:
установленное Физером и Дойзи
О
zx 1 СНз
। К Y СНз I СНз \
^/ZX'{jZX'CH2CH=C-CH2-\CH2CH2CHCH2/3 н
о
витамин Kt (филлохинон,
или фитохинон)
О
zvJI СНз
Ак СИз / СНз \
Y'XCH2CH=C-CH2-\CH2CH =с-сн2/2н
о
витамин К, (фарнохинон)
Понятие о путях установления строения витаминов К мы даем на
^примере филлохинонал
О
СНз
СНз
СН2СН=С-СН2
СНз \
I
СН2СН2СНСН2/3Н
О
о
^\/\/СНз
хон
мягкое
окисление
энергичное
окисление
восстановитель
+(СНзСО)2О
СОСНз
О
/YYCH3 ^Нз f
^//\^ХСН2-СН=С-СН2\СН2СН2СНСН2/зН -2^
о
I
СОСНз
СОСНз
о
^yyGH3
Y\Асна_ч*
о u
I
СОСНз
/ СНз \
1
+ СН3ССН2 \СН2СН2СНСН2/«н
II
о
НАФТАЛИН И ЕГО ПРОИЗВОДНЫЕ
241
Образующийся кетон
С11 ;)СС 11 о (СIГ 2СН2с 11СII2)з 1 т
II I
О СНз
тот же, что и при озонировании спирта фитола.
Синтез филлохпнона из 2-метил-1,4-диоксинафталина и фитола был
осуществлен также Физером и Дойзи:
ОН
С1[, СНз / СПз \
I I
+ ПО-СН2—СН=С—С[Т2-ДсН2СН2С1-1СН2/зН -Si
ОН
,^\А/СПЗ , р„ X
। и । 9Нз f 9Нз
А хсн2-с 11=с- сн2 - \ с н2с н2снс н2 / з н —к-слс-~-
он
о
/\ Д/СНз
Y<Y а / А3
AYGH2~CI1==c~CIl2—^CH;:C[l2GHCIf2 311
о
Витамины Kj и К2 обладают кровеостанавливающим действием.
Это же действие свойственно синтетическому 2-метил-1,4-нафтохинону,
производство которого осуществляется по варианту метода Физера, раз-
работанному А. И. Королевым:
1(> Заказ 672
242
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Гидронафталины
Уже было сказано, что гидрирование нафталина амальгамой натрия в вод-
ном спирте дает главным образом 1,4-дигидронафталип (все же вместе
с 1,2-дигидропафталином).
При каталитическом гидрировании можно получить тетрагидронаф-
талин, или тетралин
H/Ni
I II I | || |
\/\/
который обладает ароматическим характером гомологов бензола. Это
жидкость с т. кип. 205—207° С. Подобно ксилолам, но еще легче, тетра-
лин окисляется воздухом уже при комнатной температуре в гидроперекись
тетралина (цепная реакция), при нагревании переходящую в кетон —
тетралон
Н ООН
Н2 О
который можно получить и прямым окислением тетралина хромовой
смесью.
Тетралин можно нитровать (и нитросоединения восстановить в ами-
ны), сульфировать и галоидировать. Всеми способами дегидрирова-
ния — каталитически (Pt, Сг2О3) и посредством Se и S тетралин может
быть превращен в нафталин. Тетралин используется как добавка к мотор-
ному топливу (в странах, бедных нефтью) и как растворитель.
При исчерпывающем гидрировании очень чистого нафталина водородом
над никелем (Сабатье, Сандеран) получают декагидронафталин, или дека-
лин, известный в виде двух стереоизомеров:
транс -декалин
(ткип 185 °C; теплота
сгорания 1497,1 ккал/моль)
и,не -декалип
(ткип. 195°С; теплота
сгорания 1499,9 ккал/моль)
Изомерные декалины были рассмотрены ранее в разделе о конденсиро
ванных алициклах (кн. I, стр. 566).
АЦЕНАФТЕН, ИНДЕН, ФЛУОРЕН
243
Гидрирование р-нафтола дает уже четыре стереоизомерных декалола,
одна пара которых имеет скелет рис-, а другая — транс-декалинов:
Окисление декалолов в кетоны — де калоны снова сводит стереоизо-
мерию к стереоизомерии скелета, и из двух первых декалолов образуется
один P-puc-декалон, а из двух вторых — его транс-изомер. Восстановле-
ние декалонов приводит соответственно к цис- и транс-декалинам. При
окислении puc-декалона образуются две кислоты — рис-2-карбоксицикло-
гексил-Р-пропионовая и рис-циклогексилен-1,2-диуксусная; окисление
транс-дскалона приводит к образованию транс-изомеров тех же кислот.
АЦЕНАФТЕН, ИНДЕН, ФЛУОРЕН
Эти углеводороды также содержатся в каменноугольной смоле.
Н2С-СН2
аценафтен
(т. пл. 95° С;
т. кип. 278° С)
инден
(т. кип. 182° С)
сн2
флуорен
(т. пл. 116° С;
т. кип. 295° С)
Аценафтен окисляется в аценафтенхинон (т. пл. 261° С) и далее в пери-
нафталиндикарбоновую (нафталевую) кислоту, чем и устанавливается
его строение:
16*
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Инден гидрируется в индан:
сн2 сн2
Инден при действии гриньярова реактива образует, подобно цикло-
пентадиену, магнийорганическое производное, которое можно исполь-
зовать в свою очередь как гриньяров реактив:
_________________________________________
| II II +CH3MgBr--->| II II Н-СЩ
\/"\/
СН2 CHMgBr
По активной СН2-группе инден может вступать в те же конденсации
с альдегидами и кедонами, что и циклопентадиен. При этом образуются
аналоги фульвенов:
II
R-C-R'
Флуорен окисляется в флуоренон:
Его метиленовая группа, хотя и в меньшей степени, чем в индене,
и особенно в циклопентадиене, обладает способностью к протонизации
водородов и вступает в реакции с гриньяровым реактивом и альдегидами.
Изучая оптическими методами состояние равновесия типа
RH-I-CH3O- -ГЗГ R- + CH3OH
оказалось возможным оценить кислотность индена и флуорена и срав-
нить ее с кислотностью дифенилметана и трифенилметана:
ка ка
Инден .........10-21 Дифенилметан . . 10-35
Флуорен .... 10-25 Трифенилметан . . 10-33
антрацен и его производные
Антрацен — высокоплавкий (т. пл. 217° С) углеводород с т. кип. 354с_С.
Он содержится в каменноугольной смоле в количестве от 0,25 до 0,5/о
и выкристаллизовывается вместе с более низкоплавким и более раствори-
мым своим изомером — фенантреном из самого высококипящего погона
каменноугольной смолы — антраценового, или зеленого, масла.
Строение антрацена установлено Армстронгом и Гинсбергом. Понятие
о его строении дают следующие факты. Антрацен гидрируется водородом
в момент выделения, присоединяя два атома водорода и образуя дигидро
АНТРАЦЕН II ЕГО ПРОИЗВОДНЫЕ
245
антрацен; при исчерпывающем каталитическом восстановлении (Ni) при-
соединяет 14 атомов водорода. Молекулярная формула антрацена С14Н10
отличается от формулы соответствующего предельного углеводорода
СцН30 на 20 Н, т. е. структура антрацена должна включать десять
л-связей, или один цикл и девять л-связей, два цикла и восемь л-связей,
три цикла и семь л-связей и т. д. Присоединение 14Н при гидрировании
подтверждает последнюю пз указанных возможностей. Характер антра-
цена ароматический. Окисление его приводит к антрахинону — дикетону,
сохраняющему число углеродных атомов антрацена и его циклическую
структуру, так как восстановлением антрахинона можно снова прийти
к антрацену. Строение антрахинона устанавливается синтезом из о-бен-
зоилбензойной кислоты, замыкающейся при действии пятиокиси фосфора:
3 0
Ci4Hj0 > C12HS(CO)2
6Н
Таким образом, для самого антрацена мы приходим к структуре кон-
денсированного ароматического трициклического углеводорода 2,3-бенз-
нафталина *. Такая структура, выражаемая совокупностью формул
находит подтверждение как в синтезах антрацена, так и в синтезах дигид-
роантрацена и антрахинона. Цифры в первой формуле — это принятая
нумерация атомов антрацена. Как всегда, нумеруются лишь углероды,
несущие водород. Ниже приведены диеновый синтез антрацена из бен-
зохинона и бутадиена
СН2 СЛГо СН2
•—> f II +—
\ /X /' / -H2S
сн2 сн2 сн2
гексагидроантрацен антрацен
* По этой номенклатуре приставка «бенз» обозначает наличие еще одного «прпкон-
Денсировапного» бензольного цикла, а цифры указывают места «прирастания» атомов
углерода, замыкающих новый «бепз»-цикл. Нафталин ио этой номенклатуре должен
был бы быть назван бепзбензолом.
246
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
и синтез дигидроантрацепа по Фриделю — Крафтсу:
С1Н2С
Дигидроантрацен легко дегидрируется (или окисляется) в антрацен.
По Р. Я. Левиной, можно получить антрацен и его производные,
нагревая о-бензилтетрагидробензойные кислоты с фосфорным ангидридом
и серой:
Синтез антрахинона может быть также осуществлен ацилированием
бензола по Фриделю — Крафтсу фталевым ангидридом в о-бензоилбен-
зойную кислоту и замыканием последней при действии Р2О5 в антрахинон:
антрахинон
Свойства антрацена. Рентгеноструктурный анализ кристаллов антрацена
Дает следующую картину его молекулы:
Уже из сказанного выше следует, что наиболее активными углерод-
ными атомами в антрацене (присоединение 2Н, окисление) являются
атомы 9 и 10, называемые также лгезо-углеродпымп атомами. Эти мезо
углеродные атомы отклоняются по поведению от ароматических оензоль
ных атомов еще больше, чем а-атомы нафталина, и приближаются к крап
антрацен и его производные
247
ним углеродам диеновых сопряженных систем, например бутадиена. Так,
антрацен легко присоединяет малеиновый ангидрид и другие «диенофилы»
(кн. 1,стр. 293) — реакция, используемая для количественного опре-
деления антрацена в смесях:
нс—С
I /°
НС — с
хо
Аддукт указанного строения легко (уже при несильном нагревании)
крекируется по вновь образованным о-связям, регенерируя исходные
продукты, чем пользуются для очистки антрацена.
Другой замечательный случай диенового синтеза с антраценом это
образование триптицена из антрацена и дегидробензола по Виттигу:
При освещении антрацен димеризуется в диантрацен, который в тем-
ноте самопроизвольно деполимеризуется в мономер:
Молекулярный кислород при освещении также присоединяется к антра-
цену в положения 9, 10:
+ О2
Av
---->
Перекись, получаемая аналогичным путем из 9, 10-дифенил антрацена,
При нагревании в вакууме до 180° С отдает молекулярный кислород, реге-
нерируя исходный углеводород (Дюфресс).
Подобно диеновым углеводородам, антрацен прямо присоединяет
щелочные металлы в положения 9 и 10:
Н Na
f II I I +2Na —> | || || I
II Na
248
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Связи натрия с углеродом имеют, как обычно, ионный характер.
С хлором и бромом сначала также идет присоединение к атомам 9
и 10 и лишь затем отщепление молекулы галогеноводорода:
11 Вг Вт
И Вг
Концентрированная азотная кислота окисляет антрацен в антрахинон,
но азотная кислота, растворенная в уксусной, нитрует его в положение 9.
Двуокись азота присоединяется в положения 9 и 10. Щелочь отщепляет
от продукта присоединения элементы азотистой кислоты, и образуется
тот же 9-нитроантрацен:
Концентрированная серная кислота сульфирует антрацен в «-положе-
ния (очевидно, в мезо-положении сульфогруппа слишком легко поддается
ацидолизу), а более разбавленная при нагревании — в [3-положения, так
что доступны четыре дисульфокислоты антрацена:
SO3H
I II I I
SO3H
SO3II SO3H
HO3S
I II I I
НОз5^/\^
\X\/WWSO3H
I II II
Несмотря па все эти возможности, антрацен используется в химии
красителей почти исключительно для окисления его в антрахинон, кото-
рый подвергают разным реакциям замещения. Не меньшее значение имеет
и синтез производных антрахинона из фталевого ангидрида и фенолов.
Антрахинон и его производные
Получение антрахинона окислением антрацена и синтезом из фталевого
ангидрида и бензола было уже рассмотрено.
Антрахинон — сравнительно высокоплавкое (т. пл. 86° С), нелетучее
(т. кип. 382° С), прочное вещество, вопреки своему названию мало по
АНТРАЦЕН II ЕГО ПРОИЗВОДНЫЕ
249
хожее на хиноны и совершенно лишенное химической активности непре-
дельного кетона. Его л-электроны, которые могли бы образовать угле-
род-углеродные л-связи, сопряженные с карбонильными л-связями,
заняты в ароматических секстетах. Точно так же присоединение по
л-связям (в данном случае по л-связям любого из двух крайних бензоль-
ных ядер) не только не дало бы выигрыша энергии, но привело бы к ее
затрате вследствие нарушения бензоидности этих крайних ядер. Таким
образом, антрахинон скорее похож на бензофенон и другие ароматиче-
ские кетоны. Он образует оксимы и другие азотистые производные; при
сплавлении со щелочью разлагается подобно бензофенону, что в данном
случае приводит к бензойной кислоте:
2 NaOH
Антрахинон восстанавливается в антрагидрохинон, и это одна из
немногих его «хинонных» реакций. Антрагидрохинон окисляется еще
легче, чем гидрохинон — прямо воздухом:
При этом образуется перекись водорода и таким путем получают кон-
центрированную перекись водорода.
Одной из важных промышленных операций является сульфирование
антрахинона. При этом первая сульфогруппа вступает в Р-положение
одного из ядер, а следующая — в a-положение второго ядра. При доба-
влении в реакционную массу соли двухвалентной ртути первая сульфо-
группа направляется в a-положение, а следующая в а-положепие другого
ядра (М. А. Ильинский). Все эти сульфопродукты применяются в синтезе
красителей.
При нагревании антрахинон-а-сульфокислоты (и соответственно антра-
хинон-р-сульфокислоты) со щелочью, например с Са(ОН)2, сульфогруппы
замещаются на гидроксилы и получаются п-окси- или, соответственно,
р-оксиантрахиноны. Нагревание сульфокислот с аммиаком и катализа-
тором - борной кислотой до —200° С приводит к образованию амино-
антрахинонов, используемых в синтезе так называемых индаптреновых
250 КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
красителей. Аналогичной реакцией с анилином получают фениламино-
антрахипоны.
антрахинон-
-р-сулырокислотг
N'H3; Н:!ВО3
антрахинон-
- а-сульфокислота
NH3; Н3ВО3
Наиболее давно известным и до сих пор не потерявшим значение антра-
хиноновым красителем является ализарин, который с незапамятных вре-
мен добывался из корня культивировавшегося в Европе растения марены
(краппа). В марене он содержится в виде гликозида (рубэритриновая
кислота), из которого в результате гидролиза получался сам али-
зарин.
Ализарин образует оранжево-красные кристаллы с т. пл. 289° С, воз-
гоняющиеся, растворимые в щелочи (раствор окрашен в фиолетовый
цвет).
Строение ализарина было установлено в 1868 г. Гребе и Либерманном,
и вслед за тем эти исследователи и Каро осуществили его синтез. В после-
дующие годы синтетический ализарин полностью вытеснил природный.
Факты, устанавливающие строение ализарина, таковы. В молекуле
его имеются две карбонильные и две гидроксильные группы. При пере-
гонке с цинковой пылью он превращается в антрацен. При окислении
образует фталевую кислоту, следовательно, оба гидроксила находятся
антрацен и его производные
251
в одном бензольном ядре. Для диоксиаптрахинона с гидроксилами в одном
ядре возможны следующие изомеры:
Формулы III и 1\ отвергаются на том основании, что синтетически
ализарин можно получить из фталевого ангидрида и пирокатехина:
О ОН
О
II
Из получаемых этим путем двух изомерных продуктов один тожде-
ствен с природным ализарином, а второй не обладает свойствами про-
травного красителя и не дает лаков с А1;!+ или Сг3+. Уже этот факт решает
выбор структуры ализарина в пользу формулы I. Такой выбор подтвер-
ждается и тем, что при электрофильных замещениях (галоидирование,
нитрование) из ализарина получается два изомерных производных, а одно-
временно образующийся из пирокатехина изомер II может давать лишь
один-единственный продукт замещения. Таким образом, ализарин имеет
строение I.
С трехвалентпыми металлами, например с алюминием, ализарин дает
внутрикомплексные окрашенные нерастворимые соединения, так назы-
ваемые «лаки», следующего строения:
О
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
На образовании лаков основан способ крашения ализарином.
Крашение ализарином всегда производится по «протраве». Для этого ткань сначала
пропитывают раствором соли сульфированного касторового масла, затем раствором
квасцов [алюминиевых для красного (кумачного) цвета, хромовых — для фиолетово-
черного]. При этом сульфокислота, закрепившаяся на ткани, превращается в нераство-
римую соль трехвалентного металла. При кипячении такой «протравленной» ткани
с ализарином последний образует с трехвалентным металлом окрашенный нераствори-
мый «лак».
Наиболее обычный путь синтеза ализарина состоит в одновременном
действии на антрахинон-[3-сульфокислоту щелочи и окислителя (проду-
вают воздух или добавляют в сплав хлорат натрия):
SOgNa
+ NaOH + О2
По всей вероятности дело начинается с нуклеофильной атаки ОН~на
наиболее сильно обедненное электронами a-положение сульфированного
ядра аптрахипоисульфокислоты (электроны оттянуты о-сульфогруппой
и о-карбонилом, а также, в меньшей степени, м-карбонил ом). Атака облег-
чается наличием окислителя, уводящего нуклеофильно замещаемый гид-
рид-анион. После внедрения гидроксила в a-положение происходит обыч-
ное, тоже нуклеофильное замещение SO3Na на ОН.
Синтезом из фталевого ангидрида (или замещенных фталевых ангид-
ридов) и фенолов получают множество других, уже чисто синтетических
антрахиноновых красителей. Таковы хинизарин (из гидрохинона), антра-
галлол (из пирогаллола), пурпурин'.
О ОН
хинизарин
ОН
пурпурин
Интересно, что антрахиноновые красители находят и в животном мире. Таковы
кермееовая кислота, до открытия Америки добывавшаяся из червецов (дубовой коше-
нили, пли кермеса) и впоследствии вытесненная карминовой кислотой, алюминиевый
лак которой называется кармином. Карминовую кислоту получают из насекомого, жи-
вущего на кактусах в Мексике.
Н3С О ОН °
с—сн3
но
он
ноос о он
кермесовая кислота
Н3С О ОН II
ДАД/ С-(СНОН)4-СН
Ж
НООС О ОН
3
карминовая
кислота
антрацен и ЕГО производные
253
- 3 0ДНЫМ '£етРагпДРоаптрацепа принадлежит антибиотик оливомииин,
к^пт7иТыеСИ0П сп°£обностыо угнетать биосинтез информационных нуклеи-
новых лот (ч. II, раздел «Нуклеотиды») и благодаря этому тормозить рост некоторых
3"ш’мЬ1т0ПуХ0ЛеЙ<< Выяснение полной пространственной формулы оливо-
мицин ( I. М. Шемякин, М. Н. Колосов) представляет типичный пример того, как
К0^1Тп₽пЛа^Н-,°е пР.пмененпе современных химических и физических методов иссле-
дован! я озволяет быстро устанавливать строение сложных молекул. Так, порядок
расположения моносахаридов в углеводных цепях оливомпцпна I определен путем
ступен атого гидролиза, который в конечном итоге привел к агликону — оливину II:
где Me-метил,
AC-auei ил
Атомы волорола на
концах связей не
проставлены
III
Выяснение структуры оливина только физическими методами представляет
значительную трудность, и поэтому он был подвергнут частичному окислению иодной
кислотой. В результате была получена оливиновая кислота III наряду с муравьиной
кислотой и ацетальдегидом, откуда вытекало строение окисленного участка боковой
цепи. С другой стороны, спектральное изучение производных олнвпновой кислоты,
в особенности их масс-спектров и спектров ЯМР, позволило сразу установить строение
всей остальной части молекулы агликона, а отсюда и самого оливомпцпна.
Так, в спектре ЯМР тетраацетата метилового эфира олнвпновой кислоты (рис. 97)
присутствуют шесть трехиротонных синглетов, которые, судя ио их химическим сдви-
гам, отвечают двум метоксильным и четырем ацетильным группам. В области 6—Ъ м. д.,
характерной для ароматических систем, имеется три однопротонных пика, причем
один из них представляет собой синглет (единственный протон в среднем кольце), а два
Других являются дублетами с малой константой сппп-синнового расщепления, что
Указывает на мета-расположение соответствующих атомов водорода, сигналы прото-
нов Н, и Н, расщеплены па дублеты вследствие их взаимодействия с одним и тем же
Н-атомом (это может быть показано при помощи двойного ядерного .
нанса, т. е. измерением ЯМР при наложении дополнительной p^in.ina.TOTH) Следо-
вательно, в молекуле содержится разветвленная группировка С112-СН(-СН) СН
и боковая цепь находится в положении 3 насыщенно! о ко. Ц.
254
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Аналогичное строение имеют родственные противоопухолевые анти-
биотики оливомицилы В и С, а также хромомицины.
Все они содержат в качестве агликона оливин или 7-метилоливин
и различаются только ацильными группами в углеводных остатках.
Рпс. 97. ЯМР-Спектр тетраацетата метилового эфира олпвиновой кислоты при частоте
генератора 60 Мгц в CDC13 с тетраметилсиланом в качестве внутреннего стандарта.
Индантреновые и полициклокетоновые красители
Из Р-аминоантрахпнона сплавлением его со щелочью и последующим окислением
получают прекрасный кубовый краситель индантрен синий I:
В присутствии SbCl5 конденсация протекает с образованием желтого красителя
флавантрена II:
О
И
АНТРАЦЕН II ЕГО ПРОИЗВОДНЫЕ
255
Наиоолее ценные кубовые красителя получают из бензантрона III, который синте-
зируют из антрахинона и глицерина, нагревая пх с серной кислотой до 120° С. При этом
глицерин образует акролеин, который и доставляет три углеродных атома, необходимых
для надстройки бенз-цикла бензантрона:
JbSO.^ сна=сн—с/°
- 2 И 2 О j j
сн2—сн—сн2
I I I
он он он
Понятие о структуре бензантрона дает синтез его действием хлористого алюминия
на а-бензоилнафталин и восстановление бензантрона в бензантрен:
А1С1;)
-Но
а-бензоилнафталин
бензантрон
бензантрен
При сплавлении бензантрона при 225° С с едким кали происходит
окислительная конденсация его в виолантрон — темно-синий кубовый
полициклокетоновый краситель IV, называемый также индантреном тем-
но-синим:
После предварительного хлорирования или бромирования бензантрона
и сплавления его со щелочью получается изомер виолантрона - сине-
фиолетовый кубовый краситель изовиолантрон
256
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
В этих красителях все циклы, кроме содержащих кетонную группу,
ароматические. Полициклокетоны в жестких условиях можно восстано-
вить в углеводороды — сплавление виолантрона с цинковой пылью дает
углеводород виолантрен — красные возгоняющиеся пластинки
(т. пл. 478° С). Восстановлением гидросульфитом натрия полициклокето-
новые красители переводят в растворимые в щелочи феноляты — «кубы»
типа V, в которых вываривают ткань, адсорбирующую «куб». На воздухе
«куб» снова окисляется в полициклокетон IV и окрашивает ткань несо-
крушимо прочной краской. Подобного рода крашение, в котором восста-
новленный в растворимый «куб» краситель окисляют воздухом на ткани,
и называется кубовым крашением.
Индантреновые и полициклокетоновые кубовые красители часто объ-
единяют под общим названием индантреновых. Они считаются одними из
наиболее ценных красителей, которые превосходят по качеству азокра-
сители и др.
ФЕНАНТРЕН
Фенантрен представляет собой ангулярный (т. е. «угловой») изомер линей-
ного антрацена и является простейшим ангулярным конденсированным
ароматическим углеводородом.
антрацен
сренантрен
Уже отмечалось, что он вместе с антраценом находится в антраценовом
масле — высшем погоне каменноугольного дегтя.
Фенантрен более легкоплавок (т. пл. 100° С; т. кип. 340 С) и л^чш
растворим, чем антрацен, благодаря чему может быть легко отделен о
последнего. Он имеет и большую энергию резонанса (99 lnp0Jn я
86 ккал/молъ для антрацена), другими словами, энергия его образов'
из С и И на 99 ккал) моль меньше, чем рассчитанная, с учетом тепл от
ФЕНАНТРЕН
257
зования связей С—Н, С—С и С = С, свойственных алифатическим и али-
циклическим соединениям.
Для фенантрена принята следующая нумерация атомов:
8=10 /=\
8==/—Х_1 z— <
' 11 т- д-
в~5 4~3
Строение фенантрена вытекает из следующих фактов. Фенантрен
каталитически гидрируется, присоединяя 14 водородных атомов и обра-
зуя пергидрофенантрен. Следовательно, он имеет 14 л-электронов и тем
самым три цикла. Он ароматичен. При окислении сначала образуется
фенантренхинон и далее дифеновая кислота, чем устанавливается наличие
двух бензольных ядер, связанных по дифенильному типу. Орто-положение
карбоксилов в дифеновой кислоте по отношению к связанным углеродным
атомам двух колец демонстрирует положение третьего бензольного цикла:
НоСГоО;
Стильбен при пропускании через раскаленные трубки образует фенан-
трен (синтез, аналогичный синтезу дифенила из бензола):
НС=СН
Синтез подтверждает структуру, выведенную на основании этих фактов.
Наличие в фенантрене нафталинной системы доказывается синтезом
Хеуорса:
Aids
----->
сн2
Zn;
HC1
сп2—с
। ;о
сн2—с/
\)
заказ 672
258
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
То же следует из изящного синтеза Р. Я. Левиной и В. Р. Скварченко
+ 2CO + H2O4-3H2S
Наиболее известен синтез фенантрена по Пшорру, начинающийся кон-
денсацией Перкина (стр. 147), а на последней стадии сводящийся к реак-
ции Гомберга (стр. 110):
N
С1-
Все три синтеза пригодны и для получения разнообразных производ-
ных фенантрена.
Фенантрен, так же как и антрацен, но в гораздо меньшей степени,
имеет активные 9, 10-положения. Гидрирование, окисление, электрофиль-
ные атаки направляются в первую очередь в эти положения. Так, при
нитровании (азотной кислотой в ледяной уксусной) главным продуктом
является 9-нитрофенантрен, а в меньших количествах образуются 2-
и 4-нитрофенантрены. При сульфировании при 60° С выход изомерных
сульфокислот распределяется так:
13%
18%
При повышении температуры сульфокислоты 9 и 10 не ооразуются.
Ацилирование по Фриделю — Крафтсу направляется в основном в поло-
жение 3. Бромирование осуществляется сначала как 9,10-присоединение
с последующим отщеплением НВт.
В отличие от антрацена фенантрен не получил существенного пр
нения в технике. Но производные, содержащие частично или полное
гидрированный скелет фенантрена, широко распространены в жив
ВЫСШИЕ КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
259
и растительном мире. Сюда относятся смоляные кислоты — абиетиновая
и левопимаровая, из которых состоит канифоль (стр. 657). Первая из них
при дегидрировании селеном, которое всегда сопровождается декарбо-
ксилированием, образует 1-метил-7-изопропилфенантрен (ретен):
Этот углеводород находят в ископаемых смолах.
Скелет фенантрена содержится во всех стероидах (стр. 658), в частности
в стероидных спиртах, желчных кислотах, гормонах пола, а также в алка-
лоидах группы морфина (стр. 694). При дегидрировании, сопровожда-
ющемся потерей части боковых алкилов, многие стероиды образуют кан-
церогенный метилхолантрен
Н2С—СН2
^\АА/СИз
I I II I
ВЫСШИЕ КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ
УГЛЕВОДОРОДЫ
Антрацен и фенантрен могут служить прообразами высших конденсиро-
ванных углеводородов. По типу антрацена построены линейно конденси-
рованные углеводороды с четырьмя, пятью, шестью и семью бензольными
циклами, так называемые ацены. Высшие ацены, по-видимому, неустой-
чивы и неизвестны, хотя описаны некоторые их производные, вплоть до
ундекацена.
По типу фенантрена построены ангулярные конденсированные угле-
водороды, более устойчивые, чем ацены, среди них замкнутые (такие, как
коронен), чрезвычайно устойчивые термически и химически. Все эти угле-
водороды легко образуют с тринитробензолом, пикриновой кислотой,
тетрацианэтиленом, тетранитрометаном и т. д. интенсивно окрашенные
комплексы с переносом заряда.
Ацены
Понятие о синтезе аценов могут дать следующие реакции. Исходя из фта-
левого ангидрида и тетралина, можно получить тетрацен:
17*
260
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИ ТЕМЫ
Аналогичная реакция нафталевого ангидрида с тетралином приводит к пент ацену,
который может быть синтезирован и по другому варианту:
О
О
пентацен
Ацены химически активны: окисляются кислородом воздуха, при-
соединяют диенофилы, гидрируются (в первую очередь по центральным
углеродам), при окислении образуют хиноны. Некоторые из этих реак-
ций показаны на примере тетрацена:
н2Сг2О7
<--------
ВЫСШИЕ КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
261
Ацены легко захватывают электроны щелочных металлов. С увеличе-
нием числа циклов в аценах растет интенсивность светопоглощения
и наблюдается резкий батохромный сдвиг максимума поглощения, так
что тетрацен — оранжевый, пентацен — глубоко фиолетово-синий, а гек-
сацен уже черно-зеленый.
Начальные члены ряда аценов имеют типичные «бензоидные» полосы
поглощения со следующими характеристиками (для сравнения приведен
бензол):
^макс., А емакс.
Бензол . . . 2 550 230
Нафталин .... . . . . 3 140 316
Антрацен .... . . . . 3 800 7 900
Тетрацен .... . . . . 4 800 11000
Пентацен .... . . . . 5 800 12 600
Чем больше шестичленных ароматических циклов линейно конденси-
ровано, тем глубже батохромный сдвиг светопоглощения, тем интенсив-
нее поглощение. Вместе с тем резко падает устойчивость соединения,
уменьшается его ароматичность, увеличивается непредельность и бли-
зость к полиеновому углеводороду с сопряженными связями. Это легко
понять, так как лишь единственный цикл ацена имеет секстет электронов.
Поскольку, однако, система любого ацена, как мы видели на примере
антрацена, симметрична, то можно считать, что два электрона секстета
подвижны и перемещаются из края в край системы, что, по Клару, выра-
жают такой схемой (стрелка обозначает перемещение пары электронов
секстета):
В ангулярных конденсированных углеводородах, начиная с фенан-
трена, больше полностью ароматических циклов и ближе перемещение
пар из секстетов.
Эти углеводороды поглощают свет в более длинноволновой области
спектра, более устойчивы и ароматичны
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Очень интересен 9,10,11,12-тетрафенилтетрацен, или рубрен, откры-
тый Дюфрессом при конденсации З-хлор-1,3,3-трифенилпропина при 100—-.
120° С в вакууме:
Растворы этого красного углеводорода на свету обесцвечиваются
кислородом, при этом образуется перекись
Н5С6 С6Н5
распадающаяся в вакууме при 140—150° С на молекулярный кислород
и рубрен. Кислород выделяется в активном (синглетном) состоянии,
в котором он способен присоединяться к сопряженным углерод-углерод-
ным л-связям по типу диенового синтеза:
/—\
+ О2 —> \о-о/
Частично гидрированные и замещенные тетрацены найдены в выделе-
ниях одноклеточных грибковых организмов и используются как анти-
биотики, объединяемые под названием тетрациклины. Важную группу
антибиотиков представляют собой тетрациклин и его производные:
7-хлортетрациклин (ауреомицин, или биомицин) и 5-окситетрациклин (тер-
рамицин). Они обладают высокой антибиотической активностью, широ-
ким спектром действия и применяются для лечения многих инфекцион-
ных заболеваний. Антибиотические свойства этих соединений обусло-
влены тем, что они подавляют в микробных клетках биосинтез белка
на рибосомах.
ВЫСШИЕ КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
263
Выяснением строения и синтезом антибиотиков этой группы начиная с 1950 г.
занимался ряд крупных химиков (Вудворд, Муксфелъд и др.), но только в 1967 г.
удалось осуществить полный синтез природного тетрациклина (М. М. Шемякин,
М. Н. Колосов). Этот двадцатпстадпйный синтез (см. схему) был спланирован таким
образом, что построение наиболее сложной части молекулы (кольцо А) и стереонапра-
вленное создание асимметрических центров требуемой конфигурации проводилось на
последних этапах, благодаря чему удалось на большинстве стадий избежать работы
со сложными и очень неустойчивыми веществами. Кроме того, в ходе синтеза оказалось
необходимым защищать или видоизменять имеющиеся в молекуле группировки с тем,
чтобы регенерировать их на более поздних этапах. Так, из двух кетогрупп в соединении
II более реакционноспособной является кетогруппа в положении 10; поэтому перед
проведением реакции Гриньяра ее избирательно восстанавливали во вторичную спир-
товую группу, которую позднее окисляли (вместе с гидроксилом в положении 4)
в карбонильную. Аналогичным образом оказалось целесообразным ароматизировать
уже готовое кольцо С в середине синтеза и лишь на заключительных стадиях снова
превратить его в циклогексанолоновое.
Схема синтеза тетрациклина
I. LiAim
2.С6Н5СН2Вг
3. CHsMg I
I. КОН
2. }ЬСг2О7
----------*
С6Н5СН2О о о
IV
1. 02NCH2COOC2Hs
2. HCI
---------------->
1. Zn + CH3COOH
2. СбЩ(СО)2Ь'СООС2Н9
3. CH3I + AgOH
2.
3.
кон
PCIs
о=с с=о
г N
ОН
С'ООС2Н5
C2H5OMgCHCONH2
осн3
CONH2
СООС2Н5
и
CHsSCH.Na
ИСТ + СНгСООН
СТ Г, 1
--------------у.
Vlir
C6H5CH2O
тетрациклин
264
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Ангулярные высшие ароматические конденсированные
углеводороды
В каменноугольном дегте содержатся в числе других следующие высоко
конденсированные ароматические углеводороды:
хризен (т. пл. 255° С;
бесцветен)
пирен (т. пл. 156° С; перилен (т. пл. 265° С; трифенилен
т. кип. 399° С; бес- золотисто-желтый) (т. пл. 199° С;
цветен) бесцветен)
Пирен впервые был синтезирован Вейценбеком следующим путем:
СНз СН2С1 CH2CN
\ clt; /г-; ксх , /=\ /==\ н»о; н+ ,
\=/ \=7 \=/
СНз CH2Cl CH2CN
О
сн2-соп
нос—сн2
Пирен окисляется в пиренхинон:
Шолль и Вейценбек получили перилен путем, доказывающим ег0
строение, из 1,8-дииоднафталина по Ульману. К тому же результату
ВЫСШИЕ КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
265
приводит (с плохим выходом) прямое действие хлористого алюминия
ла нафталин или а,а-динафтил:
С синтезом трифенилена мы уже
дегидробензол Виттига (стр. 66).
Синтез хризена можно осуществить
встречались — так тримерпзуется
по Робинсону и Фогелю:
Хризен может быть получен также по Пшорру (стр. 258):
Среди приведенной группы углеводородов золотисто-желтый пери-
леи самый активный, в частности, он легко присоединяет малеиновый анги-
дрид, образуя нестойкий аддукт. Если присоединение вести в присутствии
266
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
окислителя, такого, как СиО, или, лучше, тетрахлорхинона (хлоранил)
то аддукт ароматизуется и получается бензперилен:
Повторив тот же путь надстройки посредством малеинового ангидрида,
бензперилен превращают в коронен'.
Коронен — светло-желтое вещество (т. пл. 438—440° С), интенсивно
люминесцирующее под влиянием жестких излучений. Он применяется
для индикации и измерения интенсивности излучений. Интересно, что
Тодд нашел производные короненхинона в пигментах некоторых насе-
комых.
Коронен гидрируется; первый продукт его гидрирования — гексагид-
рокоронен является производным трифенплена. Коронен можно просуль-
фировать, пронитровать в моно-, ди-, три- и гексанитрокоронен, прогалои-
дировать; получен также аминокоронен.
Последним в группе высококонденсированных ароматических угле-
водородов мы рассмотрим гексагелицен (т. пл. 231—233 °C; растворы с голу-
бовато-красной флуоресценцией), интересный своей оптической актив-
ностью.
гексагелицен
ВЫСШИЕ КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
267
Синтез его осуществлен М. С. Ньюменом. Приводим сокращенную
и упрощенную схему синтеза (1,4-присоединение гриньярова реактива идет
по типу, аналогичному взаимодействию с п-непредельными кетонами):
Синтез гексагелицена
дегидроге-
низация
пал Ph
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Образуя из гексагелицена и оптически активной полинитроароматиче-
ской кислоты комплекс с переносом заряда, обычным путем разделяют
соединение на диастереомеры и из них выделяют в свободном виде два
антипода — левый и правый гексагелицены с необыкновенно высоким
молекулярным вращением [сс]Ь4 = 3640. Молекула гексагелицена (при
отсутствии асимметрических атомов углерода) несовместима со своим
зеркальным изображением, так как крайние циклы препятствуют рас-
положению молекулы в одной плоскости, и она принимает форму
спирали.
ГЕТЕРОЦИКЛ ИЧЕСКИЕ
СОЕДИНЕНИЯ
Гетероциклической называется любая циклическая молекула, содержа-
щая в цикле наряду с углеродными атомами по меньшей мере один неугле-
родный атом — гетероатом. Таким образом, уже изученные нами соеди-
нения — окись этилена, янтарный и фталевый ангидриды, бутиролактон,
гликолид, паральдегид, диоксан, фталимид, сукцинимид и т. д. относятся
по строгой систематике к гетероциклам, и в справочниках их следует
искать в отделе гетероциклических соединений. Но в то же время эти
вещества функционально так тесно связаны с родоначальными алифати-
ческими (или ароматическими) соединениями, что их удобнее было рас-
смотреть рядом с ними. Есть, однако, гетероциклические соединения,
включающие в свою электронную структуру ароматическую шестерку
электронов (и вообще 4~ 2 электрона, хотя иные системы, кроме
шестиэлектронных, изучены гораздо меньше). Им и будет посвящен
этот раздел книги. При этом мы параллельно рассмотрим и соответству-
ющие гидрированные системы, относящиеся к ароматическому гетеро-
циклу, как циклогексан и циклогексен к бензолу.
Такие отношения для пятичленного и шестичленного гетероциклов
можно проиллюстрировать следующими примерами:
НС—СН
II II
НС сн
\н
пиррол
НС—сн2
НС сн2
\н
дигидропиррол,
или пирролин
Н2С—сн2
I I
н2с сн2
NH
тстрагидропиррол.
пли пирролидин
СП
НС *СН
II I
нс СН
V
пиридин
сн2
НС сн
II II
нс сн
NH
1,4-дигидро-
пиридин
сн
НС^ ^СНг
I I
НС СН2
N
2<3-дигидро-
ппридпк
СН2
Н2С ^СНа
I I
Н2С сн2
\н
гексагидропирндин,
или пиперидин
272
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
В принципе гетероциклы могут включать любой гетероатом, если его
валентность не меньше двух. Особенно важны, однако, только гетеро-
циклы с О, S и N в качестве гетероатомов; рассмотрением их мы и огра-
ничимся.
Гетероциклы могут быть классифицированы как по признаку входя-
щих в них гетероатомов, так и по числу гетероатомов в цикле, например:
НС—СН
1(=)1
НС^СН
'сн
циклопента-
диенилий-анион
нс—СН
ini
нс^сн
NH
пиррол
нс—СН
HC^N
NH
пиразол
НС —N
in I
HC-^N
NH
триазол
НС—N
ini
N^N
NH
тетразол
N —N
О
NH
пентазол
Ряд начинается и кончается изоциклическими структурами, лишь
средние четыре формулы принадлежат гетероциклам.
Однако, может быть, наиболее важным признаком для классификации
гетероциклов служит число атомов в цикле. Практически дело ограни-
чится для нас изучением пяти- и шестичленных циклов. В некоторых слу-
чаях важны конденсированные гетероциклы. Такие конденсированные
системы, подобные конденсированным системам из двух бензольных ядер
в нафталине, могут быть построены из гетероцикла и бензольного ядра
или из двух гетероциклических ядер. Некоторые (немногие) системы
этого рода исключительно важны, и только они и будут освещены в изло-
жении.
Физические свойства ароматических гетероциклических систем, их
названия и принятая нумерация атомов приведены в табл. 73.
Ароматичность пятичленных гетероциклов с двумя л-связями объ-
ясняется тем, что одна свободная пара электронов О, S или N участвует
в ароматической делокализации и образовании ароматического секстета.
В символике резонанса для пятичленных и шестичленных циклов эта дело-
кализация л- (и р-)-электронов будет изображаться, например, следующим
образом.
пиррол
пиридин
ПЛрИМИДИН
Таблица 73- Гетероциклические соединения
Формула Название Энергия стабили- зации яяал/молъ Темпера- тура плавле- ния °C Температура кипения “С Плот- ность dl° к2ь5
О 1 4 ‘YV 1 4 3 5к /2 S 1 5/'| II’ 6\/\ /2 7 S «II Й2 NH 1 4 5/Y 3 J 1! IL / \ 7 NH 4|| ||3 5\/N2 NH 1 4,- N3 5\ /2 NH «1 N3 и 1 N2 \ / NH JI N2 * ' / NH 1 Фуран Кумарон Тиофен Бензтиофен, пли тионафтен Пиррол Индол Пиразол Имидазол 1,2,3-Триазол 1,2,4-Триазол 16 11 16 48 —18 —38,30 32 52,5 70 90 23 121 32 174 84,12 221 131 254 188 256 204 (при 7 40 лим рт. ст.) 260 0,9444 (при 15° С) 1,0776 (при 15°С) 1,0644 1,165 0,948 t 5,4-1()’15 3,0 • 10-12 1,1 • 10-7 2 • 10-72
18 Заказ 672
Продолжение табл. 7 g
Формула Название Энергия стабили- зации ккал/молъ Темпера- тура плавле- ния °C Температура кипения °C Плот- ность dl° — к?5
4N=N3 J NH2 1,2,3,4-Тетразол 155 Возг.
Ч z N 1 4|i N3 Оксазол 70
5 \ z2 0 II "3 II N2 Изоксазол
5 \ / 0 1 4(1 N3 Тиазол 116,8 1,198 3,3 • 10-12
s 1 4 A N 1 5 4 AX/;A J II I, Пиридин 21 —42 115,3 0,982 1,7 • 10-9
Хинолин 55 —19,5 237,7 1,095 (при 27° С) 6,46 • IO’6 8,7 • IO-8
7\x/\ 8 N 1 5 4 бА\/А 1 N2 Изохинолин +23 243 1,0986 6,3 • Ю-10 3,6 • Ю-10
7A\z 8 1 4 A N2 Пиридазин -8 208 1,107
6\// N 1 4 5/\ Пиримидин +22 124 2,0 • Ю-13
V2 1 при 2 0 ь)
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
275
Продолжение табл. 73
Формула Название Энергия стабили- зации ккал/молъ Темпера- тура плавле- ния °C Температура кипения °C Плот- ность к£5
4 N 5 II Г 6\ //2 N 1 1 О 2/ 7 Пиразин 53 118 1,031 4 • 10-и
Оксазин (при 61°С) (при 20° С)
3\ /5 NH 4 1 S Л NH 4 Тиазин 77
Такой способ изображения при всей своей громоздкости хорошо иллю-
стрирует делокализацию л-связей и р-пары гетероатома в пятичленах
и поясняет повышенную активность к электрофильному замещению
(повышенную нуклеофильность углеродов), ослабленную нуклеофильность
гетероатома, усиленную кислотность водорода при гетероатоме («участие
в резонансной структуре» аммониевых форм). Так же иллюстрируется
аналогия шестичленных гетероциклов с бензолом. Эта символика учиты-
вает возросшую электрофильность углеродов в шестичленах — пиридине
(или пиримидине), особенно в а- и у-положениях, и ослабление их нуклео-
фильности сравнительно с бензолом вследствие большей электроотрица-
тельности азота по сравнению с углеродом.
Образование ароматического секстета, вовлекающего свободную (непо-
деленную) пару электронов гетероатома, имеет следствием, с одной
стороны, сумму ароматических свойств гетероцикла, с другой стороны,
потерю нуклеофильных свойств гетероатомом. Так, азот пиррола лишен
основных свойств; азот, кислород и сера пиррола, фурана и тиофена
соответственно мало способны или вовсе не способны к комплексообразо-
ванию, алкилированию в ониевые катионы и к окислению по гетеро-
атому I например, к переходу ot>S к ^>S = O и у> S(y . Это, однако,
V ХМ
не значит, что системы, подобные тиофенсульфону, не могут существовать;
их просто приходится получать обходным путем.
Разумеется, так «обезличиваются» только те гетероатомы, электрон-
ные пары которых входят в ароматический секстет (или, более обще,
18*
276 ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
- ———
в группу 4п 4- 2), и это не относится ни ко второму гетероатому пяти-
членных циклов, ни даже к первому гетероатому шестичленных циклов.
«Двойные связи» гетероциклов имеют в разной степени ароматический
характер, иногда напоминая связи диеновых углеводородов, иногда —
бензола. Полностью или частично гидрированные циклы, такие, какдц-
или тетрагидропиридин, ди- и тетрагидрофуран и тиофен, гексагидро-
пиридин (пиперидин) по своим функциональным проявлениям похожи
на соответствующие алифатические соединения. Оставшиеся двойные связи
имеют обычную активность, гетероатом проявляет наличие свободной
пары электронов и нуклеофильно активен, как в жирных соединениях
соответствующих функций.
Наименования гетероциклов установились тривиальные и неунифици-
рованные ни по принципу построения, ни по окончаниям. Для замещен-
ных гетероциклов названия строятся так же, как для замещенных аро-
матических углеводородов. Положение заместителя обозначается цифрой
в соответствии с принятой для каждого гетероцикла нумерацией. Кроме
того, в простейших случаях положение заместителей метят как а,а',
или у (последнее для шестичленных циклов), обозначая углеродные
атомы, ближайшие к гетероатому, буквами а и а', следующие Р и Р'.
Существует еще один способ построения наименований гетероциклов,
по которому их рассматривают как системы, в которых СН-группа арома-
тического углеводорода (или более простого гетероцикла) замещена на
гетероатом. По этой системе название строится как название углеводорода
(или соответствующего более простого гетероцикла) с приставками окса,
аза, тиа, обозначающими замену СН на О, N, S, например:
пиридин, или
аза бензол
пиримидин, или тиазол, или
1,3-диазабензол, 3-азатиофен
или 3-азапири-
дии
Практически названия этого типа применяются в более сложных слу-
чаях — для наименования циклов, еще не получивших тривиальных
названий. Для гетероциклов, конденсированных с бензолом, можно
(тоже в более сложных случаях) применять приставку «бенз» (или бензо),
так же как для конденсированных ароматических соединений, по типу:
NH
карбазол, или
2,3,4,5-дибензпиррол
NH
индол, или
2,3-бензпиррол
Комиссией «Международного союза по чистой и прикладной химии»
была введена новая номенклатура гетероциклов. Названия строятся и
префикса, обозначающего гетероатом (N — аза, О — окса, S — тиац
* Для благозвучия перед гласной опускают последнюю гласную префиква.
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
277:
предваряемого, если имеется несколько гстероатомов, приставкой ди-,
три- и т. д.; затем следует корень, обозначающий членность цикла (трех-
членный — up, четырехчленный — ен, пятичленный — ол, шестичлен-
ный — ин); далее следует суффикс, разный для предельных и непредель-
ных соединений, как это принято и в обычной номенклатуре (только для
предельных азотистых гетероциклов приняты отличные суффиксы — ин
или идин).
Приводим для некоторых гетероциклов примеры корней -|- суффиксы
(число членов обозначено цифрой, в скобках дано обозначение для азо-
тистых гетероциклов):
Предельные
Непредельные
3 .
4 •
5 •
6 .
-иран (N-иридин)
-етан (N-етидин)
-слан (N-олидин)
-ан (N-пергидро . . . ин)
-ирен (N-ирпн)
-ет
-ол
-ин
Примеры пояснят сказанное:
Н2С------------СИ2 Н2С----СН2
NH
оксиран азиридин
диазирин
НС------СН
II II
НС сн
\п
азол
П2С—сн2
Н2С сн2
NH
аЗолидин
НС-----СН
II II
НС сн
оксол
н2с—сн3
I I
Н2С сн2
о
оксолан
нс—сн
II II
НС N
1,2-диазол
НС-----СН
II II
НС N
1,2-оксазол
НС--N
II II
нс сн
V
1,3-тиазол
СИ
нс %н
II I
нс сн
N
азин
сн2
н2с сн2
I I
н2с сн2
пергидроазин
// \
НС сн
I II
НС N
1,2-диазин
О
Н2С ^СНг
I I
Н2С СН2
о
1,4-диоксап
сн2
НаС7 \)
Н2С СН2
\)Z
1,3-диоксан
Как видно, в некоторых случаях новая номенклатура совпадает с при-
вычной, в других расходится. Для давно известных гетероциклов обычно
всегда пользуются тривиальными названиями (например, пиррол, пири-
дин и др.). Для трехчленных циклов получила распространение новая
номенклатура.
278
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
ЦИКЛЫ С ОДНИМ ГЕТЕРОАТОМОМ
Сюда относятся фуран, тиофен и пиррол, а также их гидрированные произ-
водные (на примере фурана
лов нумерация атомов):
4 з
НС---СН
показана общая для пятичленных гетероцик-
НС---СН
ИС---СП
нс—сн2
НС СН2
НС СН
НС СН
НС СН
о
дигидрофуран
О
1
Фуран
НС----СН2
НС СН2
S
дигидротиофен
тиофен
НС—сн2
НС СН2
Н2С—сн2
Н2С—сн2
NH
пиррол
нс=сн
Н2С СН2
NH
2,3- и 2,5-дигидропиррол,
или пирролин
Н2С—сн2
NH
Н2С СН2
тетрагидрофуран,
или фуранидпн
Н2С СН2
Н2С СН2 '
S
тетрагидротиофен,
или тиофан
NH
тетрагидропиррол,
или пирролидин
S
Рассмотрим общие способы синтеза ароматических пятичленных гетеро-
циклов с тем, чтобы методы, специфические для каждого из них, привести
при его описании. Затем рассмотрим общие свойства (в сравнении) и далее
перейдем к специфике каждого гетероцикла.
1. В одном из наиболее общих способов синтеза ароматических гетеро-
циклов с одним гетероатомом исходят из 1,4-диоксосоединений, на кото-
рые для получения фурана и его гомологов действуют водоотнимающими
веществами (Р2О5, СН3СОС1), для получения тиофена и его гомологов —
пятисернистым фосфором,
аммиаком:
для синтеза пиррола и его производных —
НС---СН
-Н8О
R—С С—R
Н2С---СН2
R—С С—R
НС—сн
II II ре
R—С C-R -^2
о
НС---СН
R-С С—R
о о
но он
енольная форма
S
НС—сн
NH,
R—С С—R
\ /
NH
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
279
2. Получение пятичленных гетероциклов из слизевой кислоты и дру-
гих дикарбоновых кислот —• продуктов окисления сахаров. Сухая пере-
гонка слизевой кислоты ведет к пирослизевой кислоте (так называется
а-фуранкарбоновая кислота), а пиролиз аммониевой соли слизевой кис-
лоты — обычный путь синтеза пиррола. В первом случае происходит
потеря одного карбоксила в виде СО2, во втором — полное декарбоксили-
рование:
НО—СН—СН—ОН
I I
но—сн снон
о=с с=о
I I
он он
NH,.
пиролиз
-|- СО2 4- ЗН2О
пиррослизевая
кислота
|| || + 2СО2+4Н2О
NH
пиррол
Для синтеза тиофена и его производных этого типа реакция не при-
меняется, но тиофен можно получить, действуя P2S3 на янтарную кислоту
(см. стр. 286).
3. Ю. К. Юрьев открыл реакцию взаимопревращения пятичленных
гетероциклов, ароматических и гидрированных, над дегидратирующим
катализатором (А12О3) при 400° С в токе H2S, NH3 или Н2О соответ-
ственно. Однако с хорошим выходом идут только превращения фурана
и фуранидина (тетрагидрофурана):
HgS
1 НгО^
И
____ NH3
4. Хьюсген обобщил ряд давно известных реакций (открытых Ганчем,
Димротом, Килико и др.) и нашел серию новых реакций «1,3-присоеди-
нения» к олефинам, ацетиленам, а также нитрилам, ведущих к замыканию
пятичленных гетероциклов. Примером присоединяющихся систем могут
служить жирные диазосоединения, азотистоводородная кислота и азиды,
окиси нитрилов, нитроны. Хьюсген доказывает, что эти реакции идут
как мономолекулярпое присоединение через пятичленное переходное
состояние, причем присоединяющиеся молекулы должны быть способны
к частичному сосредоточению (в резонансной структуре) положительного
и отрицательного зарядов на крайних атомах триады, например:
CH2=N=n СН2—N=N
диазомстан
СНз-N-N-N <-> СНз-N— N=N —> и т. д.
метилазид
280
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
По этой причине данный тип присоединения был назван диполярным
Присоединение по. кратной связи диполярофила (так по аналогии с диено-
филом называется присоединяющий диполярную триаду ацетилен, оле-
фин и т. д.) происходит по следующему типу:
R—С=:С-Щ
СНЧ—N—N=N
4 — +
R—С=С—R
I I
CH3--N^ N
Список способных к 1,3-присоединению потенциально диполярных
соединений, так же как типов диполярофилов, велик, а число возможных
комбинаций еще больше. Ниже приведены некоторые примеры:
R-CsC-R
R—С=С—R
N=N-CH2
:N CH2
R—С—С—R
II II
:N СН
R.R-пиразол
диазометан
R—СН=СН—R
N=N— СН2
R—СН—СН—R
R—С--СН—R
NH
R.R-пиразолин
R—С=С—R
N=N—N—R
R—С=С—R
I I
:N N-R
\ Z
N
замещенный
1,2.3-триазол
H-C=N
N=N—N—R
Н—C=N:
N—R
замещенный
тетразол
R-C=N-OH —R—C=N—О R—C=N—О
I окись нитрила
Cl
хлорангидрид
гидроксамовой
кислоты
Н—С=С—R
R—C=N—О
н—С=С—н
I I
R-С О
N
замещенный изоксазол
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
281
N=G—R N==C— R
_ --* 1 ।
R—C=N—0 R~G\ /°
замещенный
1,2,4-оксадиазол
R—CH=N—ОН
альдоксим
R I: °---* R—CH=N—0" ++ R—CH—N—0“
I , нитрон I ,
Н2С=СНН"
R—CH—N—0-
I
R
нитрон
H2C---CH—R*
I I
R—НС 0
^N—R'
замещенный
изоксазолидин
5. Ф. Я. Первеев и сотр. разработали (1948 г. и сл.) общий метод
синтеза пятичленных гетероциклов с одним гетероатомом, приведенный
здесь на примере синтеза тиофенов:
О
R—С^С—С——СН+ H2S ► RC=C—С(ОН)—CHS1I > || ГН'
.11 - I I Zz \ Z
R R" R' R" R S R"
Вместо сероводорода может быть применен селеноводород или аммиак
(в этом случае получаются пирролы),
6. Шульте разработал синтез пятичленных гетероциклов с одним
гетероатомом из диацетилена:
у [| h,S;-oc8h» HCssC—С=СН nh.-cuci, || j]
\ / 150° С \ /
S NH
Фуран
Синтез фурана и его производных. Общий метод синтеза фурана или его
производных состоит в дегидратации алифатических 1,4-диолов или
1,4-дикетонов.
1. При дегидратации пентоз образуется фурфурол:
НО—НС--СН—ОН ПС—сн
I I /О _зн,0-> II II /О
н2с СН-С-^ ’ ПС С—C<f
\ Z ЧН \ / Ml
2. При нагревании с водоотиимающими средствами у-дикетонов они
превращаются в гомологи фурана (см. стр. 278).
282
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
3. Тетрагидрофуран получают дегидратацией бутандиола-1,4:
ПОСНг-СНг-СПг—СН2ОН —>
н2с—сн2
I I
Н2С СНг
4. Фуран синтезируют декарбоксилированием пирослизевой кислоты-
О
ОН
ч / +со2
о
5. Замещенный а-аминофуран можно синтезировать по Т. II. Тем-
никовой:
Аг ~ОС2Н5“
СНС1 С=0
1 + 1 Na+
С=0 1 СН
1 Аг _C==N _
у ОС211б
Аг С<
I I 'О
СН—СН
I I
0 = С C=N
I
Аг
ZOC2H5
Ar С <
I I ^0
с-сн
II I
НО-С C=N
Аг
/О П
Ar—С---СН—С-^ Ar—С---С—C<f
II I ХОС2Н5 II II ХОС2Н5
Ar-C C=NH Аг—С С—NH2
''о'/
Циклизация с присоединением гидроксильной (или амино-) группы
к нитрильной — часто встречающийся метод синтеза фурановых производ-
ных. Он будет рассмотрен далее, на примере синтеза производных пурина
по Траубе.
Свойства фуранового цикла. Фуран обладает свойствами, промежуточ-
ными между свойствами ароматического соединения и обычного диена.
Способность к реакциям присоединения для фурана более характерна,
чем для других гетероциклов. Самым показательным в этом отношении
является вступление фурана в диеновый синтез с такими диенофилами,
как малеиновый ангидрид или эфир ацетилендикарбоновой кислоты:
CH=CII CH-C/Z
II Z0
сн=сн
/О
сн—сн—сн—с^
I > I /°
СН—СН—СН—
о
СН=СН
/° +
сн=сн
С-СООСНз
III
С—СООСНз
СН—СН—С—СООСИз
\° ||
СН—СН—С— СООСНз
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
283
Фуран значительно легче, чем тиофен, окисляется и осмоляется при
действии кислот. Его можно окислить до малеинового ангидрида:
НС---СИ о НС=СН
II II —> I I
ИС СП О = С с=о
Окисление фурана на воздухе сопровождается полимеризацией.
Осмоляющее действие минеральных кислот заключается в том, что
сначала происходит протонизация фурана по кислороду:
li +Н+ || ||
О: ОН
При этом пара электронов выключается из сопряжения с двойными
связями, ароматичность соединения нарушается и происходит полимери-
зация и осмоление диена.
Несмотря на свою малую стабильность, фуран способен к реакциям
электрофильного замещения (сульфирование, нитрование, ацилирование
по Фриделю — Крафтсу, галоидирование), хотя условия этих реакций
резко отличаются от условий их протекания в случае бензола. Так, фуран
удалось просульфировать пиридинсульфотриоксидом (А. П. Терентьев):
If fl+<f N.SO3—>|| [|
О О SO3
Фуран можно пронитровать ацетилнитратом в присутствии пиридина:
CH.3COONO2;
Непосредственное взаимодействие с бромом может привести к окисле-
нию и осмолению фурана. Для бромирования употребляют поэтому ком-
плексы пиридина или диоксана с бромом. Во всех случаях замещаются
в первую очередь а-водородные атомы.
Ацетильные заместители вводят в фуран или его производные по реак-
ции Фриделя — Крафтса, используя, однако, вместо хлористого алюми-
ния более мягкие катализаторы (Г. Л. Стадников, Я. Л. Гольдфарб):
ii i|+(CH8CO)2O -SnCL.^ jj j|
OZ O/'c-CH3
II
о
Производные фурана, содержащие электропоакцепторпые заместители,
такие, как СНО, СООН, NO2, более стабильны и не так легко осмо-
ляются. Поэтому удобнее нитровать или хлорировать соответствующее
284
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
производное, например пирослизевую кислоту, а
боксильную группу:
II II || ||ч
(/ ^СООИ NO2 oz boon
затем удалять кар-
0 NO г
/ \ / \ ~UU2 \ / \
Вг о СООН О В г
Наибольшее значение среди производных фурана имеет фурфурол —
альдегид, похожий по свойствам на ароматические альдегиды. Он всту-
пает в реакции, типичные для последних, например в реакции конденсации
типа бензоиновой, в реакцию Канниццаро:
NaOH ||
О СНО
О СН2ОН О COONa
С аммиаком фурфурол
амида):
образует гидрофурамид (аналог гидробенз-
О
СН
+ 2NH3
N
О СНО
О
СН
N
О СН
Образующийся при конденсации фурфурола под действием KCN кето-
спирт фуроин, подобно бензоину, легко окисляется до дикетона — фурила,
который при действии щелочи перегруппировывается в фуриловую кислоту
(бензиловая перегруппировка, стр. 158):
COOK
фуриловая кислота
фуроин фурил
Фурфурол легко вступает в различные конденсации, например с фено-
лами. При этом образуются смолы, близкие по свойствам к феноло-форм-
альдегидным (стр. 123). Фурфурол применяется также для получения
фурилового спирта (гидрирование), тетрагидрофурилового спирта и ДР-
Фуриловый спирт полимеризуется при действии минеральных кислот.
О СН2ОН
их
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
285
Эти смолы могут применяться в качестве покрытий и клеев.
Фуран гидрируется над никелевым или платиновым катализаторами
до тетрагидрофурана, который может быть получен также из бутан-
диола-1,4. Тетрагидрофуран (фуранидин, ТГФ) используется в некоторых
способах производства адипиновой кислоты и найлона. Это — ценный
растворитель. Например, оказалось, что в ТГФ можно синтезировать
магнпйорганические соединения с магнием при двойной связи (Норман);
как известно, реактив Гриньяра из галоидного винила нельзя синтезиро-
вать в диэтиловом эфире.
Тетрагпдрофуран лежит в основе пятичленных циклических форм саха-
ров, которые по этой причине и называются фуранозами (кн. I, стр. 444).
Его дипольный момент (р, = 1,777) значительно больше, чем у фурана
(р, =: 0,72?), что демонстрирует делокализацию р-электронов кислород-
ного атома фурана.
Конденсированные системы с фурановым циклом
Такие соединения содержатся в каменноугольной смоле, например:
кумарон дибензфуран
Строение кумарона явствует из следующего способа его получения:
Подобно фурану, он легко окисляется и осмоляется под действием
кислот. Галогены присоединяются к кумарону по двойной связи:
В отличие от фурана кумарон не вступает в диеновый синтез.
Дибензфуран по свойствам скорее следовало бы отнести к производ-
ным бифенила, из которых он легко получается:
/—ч /Г~ \ Cu(NaQH) Z=\ /=\ __________________ /==\_\
\=/ " 4 —HiO \ = /
НО Вт' V но он
Тиофен
Тиофен — спутник бензола при промышленном получении последнего из
каменного угля. Его открыл как примесь к бензолу и установил его стро-
ение В. Мейер. В техническом каменноугольном бензоле содержится около
0,5% тиофена. Температура кипения тиофена (84,1° С) близка к температуре
286
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
кипения бензола (80,4° С), и поэтому он нс может быть отделен обыч-
ной перегонкой. В промышленности тиофен получают из бутана или бути-
лена и серы при температурах порядка 700° С:
СН3СН2СН2СНз + 48 —>|| ||-f-3H2S
S
Тиофен и его гомологи получены из у-дикетонов и P2S5
R СН2—Н2С R R СН—НС R
\ / \/ — \ ^ Л/
с с с с
II II II
о о он но
а также нагреванием солей янтарной кислоты с P2S3, в этом случае игра-
ющим также роль восстановителя:
СН2—СН2
НООС соон
P.S3
Ацетилен реагирует с сероводородом при 400—450° С над окисью алю-
миния и дает тиофен (А. Е. Чичибабин):
СН СН
||] + in || ц + н2
СП СН \ /
H2s s
Производные тиофена образуются также при нагревании некоторых
олефинов с серой. Так, из стильбена получается тионессалъ — тетрафенил-
тиофен:
С6Н5— -----—С6Н5
2CeH5-CII=CII-CeH5+S —> II II
С6Н5//ЧудХ С6Н5
При действии сероводорода и хлористого водорода в спиртовом рас-
творе на 1-алкиноны-5 они замыкаются в производные тиофена (выход
до 70 %):
R
I р
Н— С—СН2—C=CII -H*S: HG1-> “1 [|
С R</\/4CH3
Многие производные и гомологи тиофена удобно получать из самого
тиофена, поскольку это достаточно прочное и в то же время реакционно-
способное соединение.
Химические свойства тиофена. Тиофен более, чем фуран и пиррол, напо-
минает бензол. л-Электроны тиофена не способны к реакциям присоеди-
нения и в отличие от фурана тиофен не реагирует с диенофилами.
Атом серы в тиофене инертен, не присоединяет йодистого метила-
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
287
Тиофен не окисляется перекисями и вообще какими-либо окисли-
телями ни в сульфоксид, ни в сульфон. Однако третичные оксониевые
соединения типа R3O BF7 алкилируют тиофен по сере с образованием
SR BF:
Сульфон тиофена все же существует и может быть получен 1,4-при-
соединением SO2 к бутадиену с последующим присоединением брома
и отщеплением щелочью бромистого водорода:
НС —СН
II II
НоС СН»
Вг Н Н Вг
+ SO 2
-2НВг
Тиофен вступает во многие реакции электрофильного замещения, и при-
том гораздо легче, чем бензол.
Хотя тиофен значительно устойчивее к действию кислот и окислите-
лей, чем фуран, однако он все же окисляется азотной кислотой с разруше-
нием молекулы. Поэтому тиофен нитруют ацетилнитратом:
|| H+CH3CONO2 —> II II 4-СНзСООН
\ / II \ /\
so s no2
В отличие от бензола тиофен сульфируется серной кислотой на холоду
II H + H2SO4 —>|| II
's SO3H
Эта реакция применяется для очистки каменноугольного бензола от
примеси тиофена.
Тиофен галоидируется бромом и хлором при низкой температуре без
катализатора. При этом могут образовываться как моно-, так и поли-
галоидтиофены:
288
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Ацилирование по Фриделю — Крафтсу гладко проходит в «-положе-
ние, если использовать в качестве катализатора ~ хлорное олово
(Г. Л. Стадников, Я. Л. Гольдфарб) или трехфтористый бор:
|| U4-G1COR-S££h^|| ||
С-R
Тиофен легко конденсируется с альдегидами и, в частности, вступает
в реакцию Манниха:
||^ ||+CH2O + HNR2—►II' 11^ +н2о
s'7 CH2-NR2
Бромтиофен образует магнийорганические соединения нормальным
образом, что может быть использовано в синтезе производных тиофена
например:
S MgBr
+ СН3-С-СвН5 >|| ||
|| \ /\ /СНз
О S С<
I Свн5
он
При восстановлении нитротиофена получается аминотиофен, который
так же, как и аминофуран, очень легко окисляется и не может быть про-
диазотирован. Однако аминотиофены, имеющие в ядре электроноакцеп-
торные заместители, диазотируются нормально.
Характерной для тиофена и фурана реакцией является меркурирова-
ние этих соединений ацетатом ртути уже на холоду, причем в зависимости
от условий могут получаться моно- и полимеркурированные соединения.
При обработке соляной кислотой ртутное производное регенерирует
тиофен:
II iu-Hg(ococH3)2—>11 II + снзсоон
^S7 s'7 HgOCOCH3
IKC1
if ii 11 ii+Hgci2
\/\gC] s
Эта реакция также применяется для отделения тиофена от бензола,
который меркурируется лишь при нагревании, и для определения содер-
жания тиофена в бензоле.
При действии на тиофен (и его производные) никеля Ренея сера уда-
ляется, а углеродный скелет гидрируется. Я. Л. Гольдфарб, основываясь
на легкости введения в тиофен электрофильных заместителей, предложил
использовать реакцию гидрирования для синтеза разнообразных органи-
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
289
ческих соединений, например алифатических кислот или аминов с длин-
ной предельной цепью:
li || + С1-С-(СН2)2—СООН SnC1< > li II H,/N1^
/\ / II /\ /\
С2Н5 S О С2Н5 s С—(СН2)2—СООН
II
о
---> C2H5-(CH2)7-COOH + H2O + H2S
ii Il+CH2O+NHR2'---> || ii R(CH2)5-NR'+II2S
r^s^ r^ s'ch2—nr;
Аналогичен по идее метод синтеза аминокислот (Я. Л. Гольдфарб,
Б. П. Фабричный, И. Ф. Шалавина):
ii 1 , rN у Пх й я
s' с/ S СН—CN S СН-СООН
^0 I I
nh2 nh2
По Зелинскому — Штреккеру получают а-тиенилаланин, который
при гидрировании дает соответствующую аминокислоту:
СН—СООН
I
nh2
—СНз-(СН2)з-СН-СООН
nh2
Исходя из тех или иных гомологов или замещенных тиофенового аль-
дегида, можно варьировать синтез.
Как мы видели, тиофен более ароматичен, чем фуран. Это проявляется
в большей устойчивости тиофенового цикла, в замаскированности харак-
терных свойств двойных связей, в большей энергии резонанса. В свою
очередь в фуране ярче проявляются свойства диена. Возможно, что такая
разница объясняется отчасти повышенной электроотрицательностью кис-
лорода по сравнению с серой и, как следствие, меньшей способностью
отдавать пару электронов для создания ароматического секстета элек-
тронов и разной степенью р- и s-характера свободных пар электронов
в О и S.
Тетрагидротиофен (тиофан)
При восстановлении тиофена (натрием в аммиаке и спирте) наряду с А
и Д3-дигидротиофенами получается также тетрагидротиофен (тиофан).
Никелевые и платиновые катализаторы для гидрирования по подходят,
так как они отравляются тиофеном и тиофаном. Тиофановое кольцо может
образоваться также в результате циклизации.
Тиофан и его гомологи содержатся наряду с алифатическими тпоэф
рами и меркаптанами в сернистых нефтях. В отличие от тиофена в ТИОФ^'
новом кольце электроны серы «свободны» — не участвуют в образована
ароматического секстета электронов. В связи с этим тиофан, как обычные
19 заказ 672
290
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
тпоэфпры, легко окисляется и может быть окислен до сульфоксида и суль-
фона; подпетыми алкилами тиофан алкилируется с образованием солей
сульфонил:
----j н-о, j------1
V SZ
II
О
СН31 j----1
S
СНз I’
КМпО4
------->
Важнейшими природными продуктами, в молекулу которых входит
тиофановое кольцо, являются биотины (стр. 315).
Бензтиофен и его производные
По свойствам бензтиофен напоминает нафталин. Важнейшее его произ-
водное — тиоиндоксил:
бензтиофен
Тиоиндоксил синтезируют подобно индоксилу (стр. 307):
/ч /СООН HNOz ; \/\nh2 /ч /СООН /, соон Na.S2O, • | II + I || С1СН2-СООН XN+ X- NsH
При окислении кислородом тиоиндоксил дает красный краситель —
тиоиндиго'.
О
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
291
Тиоиндоксил конденсируется с альдегидами и кетонами, образуя
тиоиндигоиды:
О
С
Clh+R-CHO —>• I
S
C=CHR
S
Некоторые тиоиндигоиды также находят применение в качестве краси-
телей.
Пиррол
Пиррол был обнаружен в костяном масле (продукт сухой перегонки костей)
и в небольшом количестве в каменноугольной смоле (Рунге, 1834 г.).
Строение его установлено А. Байером в 1870 г.
Пиррольный и гидрированный пиррольный циклы как структурная
единица входят в состав важных биогенных соединений — аминокислот
(пролин, оксипролин, триптофан), алкалоидов, гемоглобина, ряда кофер-
ментов окисления, хлорофилла и т. д.
Пиррол (или в ряде случаев его гомологи и производные) можно синте-
зировать следующими реакциями, которые дают понятие о его строении.
1. Действие аммиака на 1,4-диоксосоединения (Пиаль — Кнорр):
СН2—СН2 СН—СН НС—СН
II l| II +NH, II II
С С TZJ- НС СН —Чт-тч-* НС СН
/ \ /7 \ \ / \ /
Н 0 0 II НО ОН NH
Способ аналогичен синтезу фурана и тиофена и устанавливает родство
всех трех гетероциклов.
2. Обычный способ синтеза пиррола, родственный синтезу а-фуран-
карбоновой (пирослизевой) кислоты, состоит в пиролизе слизевокислого
аммония:
НО-СН---СН—ОН
I I
НО-СН СН—ОН НС—сн
I I ------► || || +2СО2 + 4П2О+Ш1з
о=с с=о нс сн
NHjb- О’- NI1J XxNH
19*
292
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Применяя вместо аммиака первичный амин, получают N-замещенные
пирролы (см. синтез никотина, стр. 704).
3. При перегонке сукцинимида с цинковой пылью образуется пиррол:
Н2С—СН2
о=с с=о
»2Zn ,
II II +2ZnO
^NH
4. Восстановление смеси кетона с изонитрозокетоном (кн. 1, стр. 153)
дает гомологи или замещенные пирролы (Кнорр):
R—СН2
I
R —G +
O=G—R"
СН
NOH
R' \’Н
Промежуточно образуется аминокетон.
5. В 1964 г. Хьюсген опубликовал новый метод синтеза производных
пиррола из азлактонов и, следовательно, из ацилированных по азоту
а-аминокислот (выход 60—90%);
/О ZC1
R-CH-C^ +R'-C4 —>
I \он
NH2
Z° z° /0
R— CH-Cf R-CH-C^ R-CH-C^
4)H । OH
* /О | ,OH Z°
NH—Cz’’ N=CZ N=C4
\R' zR' R'
азлактон, или
R, R'-оксазолон
,0
R—CH— R«_______R'"
I ,0 + R"*-C=C-R" —> Z\n,+CO2
N=C\O, Rz NH R
XK , .
6. Пропускание паров фурана с аммиаком над окисью алюминия при
400° С дает пиррол (Ю. К. Юрьев):
ir“jj+NH3 -Aho,^ j 1 + н2О
О NH
Свойства пиррола. При восстановлении пиррола водородом в момент
выделения (не в кислой среде) образуется \'л-пирролин, а при каталитиче-
ском гидрировании последнего или самого пиррола — пирролидин:
1Г II
XNH
НС=СН
I I
Н2С сн2
\н
H,/Pt
пиррол Д’-пирролин
п2с—сн2
I I
н2с сн2
NH
пирролидин
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
293
Пирролидин можно также получить сухой перегонкой солянокис-
лого путресцина:
СН2-СН2 Н2С----СН2
II II
сн2 сн2 —>• Н2с CH2+NII4C1+HC1
Cl- NH:; NH3 Cl- NH
Таким образом, пиррол по отношению к водороду в момент выделения
ведет себя как диен. Аналогично он реагирует и со свободным радикалом
трифенилметилом:
||\ || + 2(С6Н5)3С-----► \ |
NH (CeH5)3C NH С(СвН6)з
Других реакций диена, в отличие от фурана,, пиррол не обнаруживает,
и с малеиновым ангидридом он реагирует иначе, чем диены:
NH
СИ—с+
+ 11 4° —>
сн—с+
+о
ООО
у-СН-СНг
NH
Азот пиррола, как уже говорилось, полностью лишен основных свойств,
и его иминогруппа обладает очень слабыми кислотными свойствами,
такими, как фенол. При помощи ряда реакций можно получить N-ме-
таллические производные пиррола, широко применяемые в синтезах:
NaNH»
------>
II II +NII3
NNa
II
NH
1 CH.Mgl
и li+4- h*
nk
ll\ II+CH4
NMgl
При алкилировании и ацилировании этих металлических производ-
ных (в настоящее время предпочитают исходить из пиррилмагнийгалоге-
нидов) ниже 0° С образуются N-алкил- и соответственно N-ацилпирролы
СН,1
NMgl
N—СНз
СНаСОС! т---г
. ' /О
N-C-+
4 СПз
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
294
а при нагревании c-алкил- и а-ацилпиролы:
СИЛ
нагревание
NMgl
CfbCOCl
о
II
---II НСОСзН»
NMgl
Вместо хлорангидридов можно применять сложные эфиры. Из слож-
ного эфира муравьиной кислоты при низкой температуре получается
N-формилппррол, при высокой — пиррол-а-альдегид:
<°°с—>1. J
СИ О
нагревание;
Hz0
Vh СНО
При действии на пиррилмагнийгалогенид хлоругольным эфиром полу-
чается эфир а-пирролкарбоновой кислоты:
|| II +С1-С-ОС2Н5------->11 || +Mgicl
\ z II \ / \
N-Mgl О NH С-ОС2Н5
Гомологи пиррола можно также получать восстановлением оксосоедине-
ний пиррола (например, по Кижнеру — Вольфу).
N-Металлические производные пиррола по ряду реакций напоминают
феноляты щелочных металлов. Так, альдегиды можно синтезировать
и по реакции Реймера — Тиманна (стр. 139):
|| || +CHCl3+2NaOH > || || + 3NaCl-|-H2O
\ / \ /\ Z/O
NNa NH С<^
ХН
Хотя пиррол очень чувствителен к кислотам (полимеризуется), все же
альдегиды и кетоны пиррола можно получить, используя приложимую
к фенолам реакцию Геша:
l| ||+R-C=N + ncl —>|| || II +NH4CI
\ / \ /\ .R \ ZR
Nn NH (Х + NH ОС
ЧУН2 Cl- хо
где R = Н или углеводородный радикал.
Эти реакции демонстрируют особую (аналогичную орто-положениям
фенолов) доступность сс-положений пиррола для электрофильных атак.
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
295
Если оба a-положения замещены, большая часть этих и следующих реак-
ций проходит в ^-положение.
Пиррол может служить азосоставляющей в реакциях азосочетания:
II ||+Ar— N=N
NH
Ar-N = N'- ун +Н
Это еще одна черта, роднящая пиррол с фенолом.
Из главных реакций электрофильного ароматического замещения
упомянем бромирование и иодирование, приводящее к тетрабром- и тетра-
иодпирролу (антисептик иодол}, и хлорирование хлористым сульфурилом,
являющееся напболее простым путем к сс-хлорпирролу, а также сульфиро-
вание по А. П. Терентьеву пиридинсульфотриоксидом (метод сульфирова-
ния «ацидофобных» соединений):
|| || J-/ S+SO3---II + <^ /NH
\н - nh'sOs ~
Поскольку пиррол «ацидофобен», осуществить нитрование кислотой
нельзя, и а-нитропиррол получают, действуя на пиррол этилнитратом
п натрием.
Чрезвычайно важны реакции конденсации пиррола с формальдегидом
и муравьиной кислотой. В щелочной среде формальдегид оксиметилирует
пиррол, а в кислой среде реакция заходит дальше — 2 моль пиррола
сшиваются метиленовым мостом в a-положение с образованием дипиррил-
метана (пиррометин):
—FTji^
]j-)| + СН2О — NH СН2ОН
^NH “—ll\ Jl\ JI
NH CH2 NH
Реакция эта широко изучена и для гомологов пиррола.
Дипиррилметан окисляется хлорным железом, теряя два атома водо-
рода, и превращаясь в окрашенный пиррометен; к той же цели ведет
и прямая конденсация пиррола с муравьиной кислотой в кислой среде:
_____ /О
2 || ||+Н-C<J
\ Z ХО
NH
Пиррометены образуют прочные соли с кислотами, причем после при-
соединения протона пирролениновым циклом оба цикла приобретают оди-
наковую выровненную структуру и становятся неразличимы:
II II I 1 + И+ —► II II I I --> I I II II
\ /\ , \ /\ ^\+ > /\ /
NH СН N Nil СН NH Nil СН NH
296
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Пиррометены могут быть получены также конденсацией следующего
рода:
II II +11 II—>11 II I П + н2о
\ z° \ / \ /\+\
NH NH NH СН N
ХН
С гомологами пиррола (при условии свободного a-положения) все эти
реакции проходят лучше, чем с самим пирролом.
Порфин. Порфирины
При нагревании пиррол-а-альдегида с муравьиной кислотой образуется
с выходом около 1 % новая исключительной важности ароматическая гете-
роциклическая система, лежащая в основе хлорофилла и гема крови, —
порфин (Г. Фишер):
Эти высокоплавкие (разлагаются выше 360° С) темно-красные кри-
сталлы устойчивы к действию кислот. Два атома водорода, связанные
с азотами, могут быть замещены на металл при помощи MgO, FeCl3,
Сн(ОСОСН3)2. Г. Фишер разработал и другие методы синтеза порфинов
с несимметрично расположенными заместителями и тем открыл путь
синтеза гемина и хлорофилла. Среди них важен следующий метод:
ЙЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
297
Порфины — настоящие прочные ароматические соединения, способ-
ные сульфироваться, нитроваться, ацилироваться по Фриделю — Крафтсу
(если некоторые R = Н). Их л-электроны делокализованы, и на деле
молекула имеет симметричную структуру (двойные связи в данной фор-
муле написаны произвольно). Счет делокализованных электронов (11 двой-
ных связей —4 свободных пары азота; 22 + 8 = 30 электронов) показы-
вает, что система порфина удовлетворяет требованиям правила Хюккеля
для ароматических систем: 4п + 2 (7 X 4 + 2 = 30).
Порфины с углеродистыми заместителями в пиррольных и пирролени-
новых циклах называются порфиринами.
Гем. Красное вещество крови, переносящее с током крови кислород из
легких в каждую клетку тела, представляет собой белок глобин, связан-
ный с производным порфирина, содержащим в центре молекулы атом
двухвалентного железа (это гетероциклическое соединение называется
гемом). Связь глобина с гемом, по-видимому, координационная — азот
гистидина (одной из аминокислот белка глобина) связан с атомом железа
молекулы гема. Гем, координационно связанный с глобином, образует
гемоглобин, который координационно связывает О2 и с кровью разносит
его по всем клеткам организма.
Действием соляной кислоты гем отделяется от глобина. При этом
(на воздухе) железо гема окисляется в трехвалентное и образуется гемин,
по составу отличающийся от гема наличием аниона хлора. Гемин — ярко-
красные кристаллы. При деструкции иодистым водородом он образует
смесь гомологов пиррола (группа А) и 0-пиррилпропионовых кислот
(группа Б) (Ненцкий):
А СНз—----j—С2НЬ СНз — у-СгНбСНз- у С2НБ СНз-j--------р-С2Н5
^NH Nh\h3 НзС NH C1IS
CH2COOH
I
Б CH3—-------— CH2
II II
Nil
сщсоон
снз-д-г^2
НзС NH
CH2COOH
CH3
\'н\н3
CH2COOH
I
CH3- --CH2
Из*/ NH^blls
298
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Все эти вещества были идентифицированы и синтезированы Г. Фише-
ром с использованием метода Кнорра.
Таким образом, гемин С34Нз2К4О4РеС1 включает пиррольные ядра
с боковыми цепями. Анализ показывает, что таких пиррольных ядер
в гемине четыре.
При действии кислот в присутствии металлического железа гемин
отщепляет атом железа и превращается в протопорфирин. Функ-
циональным анализом в нем были обнаружены два карбоксила, очевидно,
те самые, которые фигурируют в группе Б продуктов разложения гемина
иодистым водородом. Кроме того, путем каталитического гидрирования
было установлено наличие двух активных двойных связей, каждая
из которых может быть прогидратирована во вторичноспиртовую
группу. Такой гликоль носит название гематопорфирин.
Гидрирование двух активных двойных связей протопорфирина приво-
дит к мезопорфирину. Окисление последнего дает продукты I и IIг
важные для установления строения всей группы веществ:
СНз—==—С2Н5
(Anh%
I
СН2СООН
CH3-j_-CH2
//\ А
О NH О
II
При окислении гемина образуется только продукт II.
Ясно, что пиррольные ядра в гемине были связаны по «-положениям.
Чем? Поскольку в продуктах деструкции иодистым водородом (как груп-
пы А, так и группы Б) остается или метил или водород, ясно, что эта
связь осуществлялась одним углеродным атомом. Так как вся система
гемина и протопорфирина по свойствам аналогична описанной ранее
ароматической системе порфина, естественно допустить, что связыва-
ющий пирролы углеродный атом выступает, как и в порфине, в виде
группы СН, соединенной двойной и ординарной связями. Обе непредель-
ные связи могли бы принадлежать винильным группам, при восстанов-
лении (каталитическом или иодистым водородом) превращающимся
в этильные.
Все эти соображения приводят для гемина к формуле III, для прото-
порфирина к формуле IV и для мезопорфирина к формуле V. Гему при-
надлежит соответственно структура VI.
I
ноос-сн2
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
299
ноос—сн2
Эти структуры были подтверждены синтезом (Г. Фишер):
ноос—сн2
НООС—СНо
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Хлорофилл. Зеленые части растений содержат растворимый в спирте
пигмент хлорофилл, при участии которого растения ассимилируют угле-
кислый газ из атмосферы, превращая его и воду в кислород и углеводы.
Как показал М. С. Цвет (изобретатель хроматографии, разработавший
ныне столь важный метод именно на этом примере), адсорбируя раствор
хлорофилла на твердом адсорбенте, можно разделить его на два индиви-
дуальных вещества — хлорофиллы а и Ъ, несколько различающиеся по
окраске. Функциональным анализом в хлорофилле а установлено нали-
чие метильной, а в хлорофилле b вместо нее — альдегидной группы.
В остальном они одинаковы. Оба хлорофилла — сложные эфиры. При их
переэтерификации, например этиловым спиртом, освобождается спирт
фитол (кн. 1, стр. 313), а сами они превращаются в этилхлорофиллиды.
Под действием минеральных кислот хлорофиллы отщепляют ком-
плексно связанный ион магния и одновременно гидролизуется сложно-
эфирная связь (опять-таки отщепляется фитол). В результате получаются
феофорбиды а и Ъ, исследование которых и дало главные данные о строе-
нии хлорофиллов. Действие иодистоводородной кислоты на хлорофиллы
(и, разумеется, на феофорбиды) приводит удивительным на первый взгляд
образом к той же смеси производных пиррола групп А и В (стр. 297),
что и аналогичная обработка гемина.
В результате описанных экспериментов и длинного ряда исследова-
ний, которые здесь нет возможности привести (ими мы обязаны Мархлев-
скому, Ненцкому, Вилыптеттеру и Г. Фишеру), для хлорофиллов были
установлены следующие структуры:
н3с сн=сн2 нс=<~>=сн НзС\А ? Д<снз 1 N—Мб-N _Н2<Г П N Усгн5 С20Н39ООС Н2С с—^)>=СН нС-\) ХСН3 с^° хосн3 хлорофилл а нзС НС=<~ НзС^Л N н2с/*у НООС-Н-.С с—< I >= н(г-% соон Н3С сн—сн.? У_/ нс=<^ J>=CH | N-MO-N . H2cy J yVh5 < н2с с-^_С=сн с20н39оос нс-ЧТсн, 1^0 Х)СН3 хлорофилл Ь сн=сн2 S=CH Н /ССН3 HN /1 у^с.^ ^СН сн3
феофорбид а
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
301
В формулах хлорофиллов С20Н39 — остаток фитола
СН3 СН,
—СН2—СН=С—СН2—(СН2—СН2—СН—СН2)3—Н
Полный синтез хлорофилла был осуществлен в 1960 г. Вудвордом.
Витамин В12. В 1948 г. американские (Фолькерс) и английские (Л. Смит)
исследователи выделили из печени теплокровных животных рубиновые
кристаллы витамина В12, или кобаламина — сложного порфириново-
кобальтового комплекса.
Этот витамин в дозах до 3 мкг в сутки излечивает смертельное заболе-
вание — пернициозную анемию (но не лейкемию) и в комплексе со специ-
фическим белком, очевидно как кофермент, участвует в работе кроветвор-
ных органов (костный мозг), а именно в продуцировании красных кровя-
ных шариков (эритроцитов).
Потребность человека в этом витамине ничтожна — 1 — 5 мкг в сутки,
но усвоение его через пищеварительный тракт происходит медленно
и неполно, и поэтому требуются в 100 раз большие дозировки. Дело в том,
что кобаламин может проникать через стенки кишечника только в сопро-
вождении так называемого внутреннего фактора — мукопротеина, выде-
ляемого слизистыми оболочками пищеварительного тракта, так что ави-
таминоз может развиться за счет недостатка этого фактора.
В настоящее время витамин В12 готовят в промышленном масштабе
микробиологически, культивируя актиномицеты, продуцирующие этот
витамин.
Строение кобаламина выяснено произведенными в Кембридже иссле-
дованиями Тодда
н н
О-Р-О-СН-СН3
о сн2
NH
СООН
I
сн2
СООН
и рентгено структурными изысканиями Дороти Ходжкин-Кроуфут.
_ „ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
3 1*4^ ____________________________________________________—----------
Индол (бензпиррол)
Строение индола следует из его синтеза путем замыкания в цикл о-амино-
<о-хлорстирола под действием алкоголята натрия:
+ C2H5ONa
NH
индол
Его можно
паров анилина
получить реакцией Чичибабина — пропусканием смеси
с ацетиленом через раскаленные трубки:
Эта реакция аналогична синтезу пиррола из ацетилена и аммиака.
Гомологи индола чаще всего синтезируют по реакции Э. Фишера,
нагревая гидразоны альдегидов или кетонов с хлористым цинком, или
по А. Е. Арбузову — нагреванием гидразонов с каталитическим количе-
ством хлористой меди. Для получения самого индола эти реакции мало
пригодны.
Смысл неожиданной реакции Э. Фишера заключается, по Р. Робин-
сону, в перегруппировке таутомерной формы фенилгидразона, аналогич-
ной бензидиновой:
Индол в небольшом количестве находится в каменноугольном дегте.
Он обладает приятным в малой концентрации и достаточно неприятным
в большой концентрации запахом. Находится он в эфирном масле жасмина
небелой акации. Применяется в парфюмерии.
Из производных индола важны: триптофан ({3-индолилаланин)
незаменимая (в пище) аминокислота, составная часть белка (стр. 726);
р-индолилуксусная (гетероауксин) и (3-индолилмасляная кислоты, при-
меняемые как стимуляторы роста и корнеобразования у растений; ин-
диго в прошлом природный, ныне синтетический краситель (стремление
синтезировать индиго и стимулировало последования ряда индола А. Байе-
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
303
Свойства индола. Индол, естественно, обладает многими свойствами
пиррола. Водород индола может быть замещен на металлы, причем обычно
пользуются реакцией с гриньяровым реактивом:
+ СН3МбВг
+ СН
NMgBr
4
С галоидным индолилмагнием можно осуществить те же реакции,
что и с галоидным пиррилмагнием, но отличием этих реакций является
вступление заместителей при повышенной температуре не в ос-, а в 13-
положение индольного ядра. Объясняется это тем, что в индоле одна
из электронных л-пар пиррольного ядра принадлежит также и бензолу
и связана в бензольном ядре жестче, чем в пиррольном. Таким образом,
в пирроле и в индоле лабильная пара электронов связи азота с магнием
вызывает разный эффект концентрации электронов:
Электрофильные замещения в самом индоле так же ограничены, как
и в пирроле (индол не выносит действия кислот), но те, что осуществляются,
ориентированы в [3-положение, причем в некоторых случаях описанный
механизм находит наглядное подтверждение.
Так, нитрозирование индола амилнитритом в щелочной среде проходит
по схеме:
NOH
Еще одним примером электрофильного замещения в [3-положение
является реакция Манниха:
ch2-n(ch3)2
NH
NH
грамин
2
304
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Образующийся по реакции Манниха алкалоид грамин служит исход-
ным веществом в синтезе незаменимой белковой аминокислоты — трипто-
фана:
j~CH2n(CH3)2 + Na(t_NHCOCH
^NH COOR
COOR
ch2-c-nhcoch,
COOR
H+;H2O
-CO2; -2 ROH
I
COOH
nh2
трин гофан
При гниении белков из триптофана образуются триптамин и р-метил-
индол {скатол). Скатол — вещество отвратительного запаха, обусло-
вливающее запах испражнений.
CH2CH2NH2
триптамин
СНз
скатол
В мексиканских грибах семейства Psilocybe находится производное
4-оксииндола — псилоцибин
НОЧ
>Р=О
НСГ |
б
I
----pCH2CH2N(CH3)2
/
NH
относящийся к числу так называемых психомиметических веществ. При
приеме внутрь —40 мг человек впадает в состояние, похожее на сон,
и у него возникают цветные галлюцинации, чем издавна пользовалось
древнее население Мексики при своих религиозных и ритуальных обрядах.
Для синтеза 4-оксизамещепных индолов удобен способ А. Н. Коста, в котором
исходят из 1,3-циклогександиона (дигидрорезорцина). Ниже приведены два варианта
этого способа:
0 0 0
ОН
ПЯТИ ЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
305
0 0 о
II II II
/\ + С-ОС2Н5 н+ /\____________
\/\ СН-С-ОС2Н5 \J\ J'-C-OCzHs
о ' 1| NH II
NO °
II
О
Изомером псилоцибина (по фенолу) является 5-окси-Ы-диметилтрип-
тамин, изолированный Виландом из ядовитых кожных выделений жаб.
Производным 5-оксииндола является также серотонин (5-окситрипта-
мин) — гормон, регулирующий кровяное давление и поток крови через
почки и связанный с нормальной деятельностью мозга. Нарушение его
концентрации в мозгу ведет к шизофрении.
CH2CH2N(CH3)2
\/\ /'
NH
CH2CH2NH2
5-окситриптамин (серотонин)
5-okcii-N .N-диметилтр иптамин
Производные индола с гидроксилом в положении 5 удобно синтезиро-
вать методом Неницеску, широко разработанным А. П. Терентьевым
и А. Н. Гриневым и заключающимся во взаимодействии п-бензохинона
с 1,3-дикетоном и аммиаком или амином:
+
NH3
II
CR"
СН2
С—R"
К ряду индола относятся и многие алкалоиды, которые рассматри-
ваются в специальном разделе II части книги.
индиго
В соке тропических растений вида Indigofera, возделывавшегося в Европе
растения вайда, содержится индикан, представляющий собой гликозид
индоксила. При вымачивании этих растений в воде индикан переходит
в водный раствор и гидролизуется до индоксила, который окисляется
кислородом воздуха в индиго, выпадающее и осадок в виде синих хлопьев.
Индиго с незапамятных времен применялось как краситель. Для окраски
тканей его восстанавливают в «белое индиго», растворимое в слабой
Щелочи, проваривают ткань и дают окислиться на воздухе в нераствори-
мое «синее индиго». Такой способ называется кубовым крашением, и индиго
относится к многочисленным кубовым красителям (стр. 254 сл.).
20 заказ 67 2
306
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
При сухой перегонке индиго получается анилин, а при окислении
азотной кислотой — изатин, который щелочью гидролизуется в о-амино-
фенилглиоксиловую (изатиновую) кислоту. Изатин является лактамом
этой кислоты, как явствует из легкого замыкания изатиновой кислоты
в изатин с потерей воды:
О О
V° 'н,° - fV хс=о
\/\ хон %н,о; +он-
NH2 NH
изатин
Отношение изатина к индолу и другим его оксопроизводным устана-
вливается следующими реакциями:
индол
ИНДОКСИЛ
(таутомерные формы)
изатин
Na; Hg;
оксиндол
Оксопроизводным индола (точнее индолина) свойственна таутомерия,
изображенная на приведенных выше схемах; изатину — лактим-лактам-
ная, индоксилу — кето-енольная. Индикан является гликозидом именно
енольной формы индоксила. Индоксил в щелочном растворе окисляется
кислородом воздуха в индиго, состав которого C16H10N2O2; Две молекулы
индоксила C8H7NO количественно образуют одну молекулу индиго, Т®РЯЯ
в виде воды четыре водорода. Одна молекула индиго окисляется HNO3
в две молекулы изатина C8H5NO2, утрачивая двойную связь и присоеди-
няя два атома кислорода. Отсюда, так же как из ряда Других соображении,
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
307
следует установленная А. Байером формула индиго, которую в отличие
от Байера мы напишем на основании новых данных в транс-форме:
HNO3 I | -2Н«О
изатин
изатин
индоксил
индоксил
После нескольких синтезов индиго, осуществленных А. Байером, были
разработаны два способа (Гейман), вошедших в промышленную практику
и позволивших производить индиго так дешево, что культивирование
индигоносных растений прекратилось.
NH2
О о
.11 II
С—ONa С-0 Na
4- сн2 --► fсн2
I \ z\ -/
Cl NH
NaNH2
(180—200° С)
О
NH
фенилглицин индоксил
(натриевая соль)
О
С—ONa
+ СН2-С^° ||z сн2-с^°
I 'ONa / xONa
Cl NH
фенплглицинкарбоновая
кислота (натриевая соль)
//\/ \ ,0
I II СН—С//
/ xONa
20*
308
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Оба способа приводят к индоксилу, который затем окисляют в индиго.
Чистое синтетическое индиго — темно-синий порошок, нераствори-
мый в обычных растворителях, немного растворимый в нитробензоле;
плавится при 390° С, образует красные пары. Синтетически получают
много иных индигоидных красителей (стр. 290). Древний пурпур — дра-
гоценная в древности фиолетовая краска, выделявшаяся из моллюсков,
по исследованию Фридлендера оказался 6,6'-диброминдиго.
Фталоцианины
Химики английского концерна ICI в 1928 г. сделали наблюдения, что
при изготовлении фтальимида из фталевого ангидрида и аммиака в мед-
ной посуде получается продукт, загрязненный синим веществом. Иссле-
дуя его, Линстед нашел, что это же вещество можно получить, нагревая
о-фталонитрил с медными солями, и установил строение нового краси-
теля — фталоцианина меди, нашедшего широкое применение (в печати,
окраске машин, синтетического волокна и в научных исследованиях).
Фталоцианин меди — ярко-синее вещество, возгоняющееся выше 550° С,
т. е. обладающее выдающейся термической прочностью. Уже эта проч-
ность и легкость образования указывают на ароматичность системы.
Действительно, фталоцианин меди имеет симметрическую структуру
тетрабензотетраазапорфина:
NC
,CN
NC
CN
CN
NC
Существуют фталоцианинные комплексы и других металлов, а также
фталоцианины с заместителями в ароматических ядрах, полученные син-
тезом из замещенных фталонитрилов или последующим электрофильным
замещением, например сульфированием. Медь из фталоцианинного ком-
плекса не удаляется ни кислотой, ни щелочью.
Карбазол
В каменноугольном дегте во фракции, называемой антраценовым маслом,
наряду с антраценом и фенантреном находится азотистый гетероцикл
дибензпиррол, или карбазол, который может быть отделен от нейтраль-
ных углеводородов благодаря присущим ему слабокислым свойствам,
папример способности образовывать металлические производные. Строе-
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
309
ние карбазола доказывается образованием его при нагревании соли о,о -
диаминодифенила:
CJ С1
NH
карбазол
NH4C1 + HCI
Карбазол может быть получен также пропусканием дифениламина
через раскаленные трубки (ср. получение бифенила из бензола):
NH nh
Это вещество больше похоже на дифениламин, чем на производные
пиррола, что и понятно, так как выигрыш энергии при ароматической
делокализации л-электронов по бензольным ядрам много выше, чем по
пиррольному ядру.
Карбазол находит ограниченное применение в производстве серни-
стых красителей и пластмасс на основе N-винилкарбазола. Последний
получают из ацетилена и карбазола под действием щелочи.
ПЯТИЧЛЕННЫЕ ЦИКЛЫ С НЕСКОЛЬКИМИ ОДИНАКОВЫМИ
ГЕТЕРОАТОМАМИ
Пиразол
Пиразол и его производные не найдены в природе. Сам пиразол
НС----сн
С 3
НС. /N
NH
а также его гомологи и производные могут быть получены следующими
реакциями.
1. Присоединением жирных диазосоединений к ацетиленам полу-
чаются пиразолы. Та же реакция в применении к олефинам приводит
к дигидропиразолам — пиразолинам (Пехмапн):
СН III сн2 сн2
II + N+ — нс—сн -> II II сн2 II II +N+ - Но с—сн -► 1 II
сн 11 НС N СН2 || Но с N
N" N-
NH NH
пиразол пиразолин
зю
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
2. При действии гидразина или замещенного гидразина на а,|3-непре-
дельные альдегиды или кетоны получаются пиразолины (образующиеся
сначала гидразоны претерпевают внутримолекулярное присоединение
к двойной связи). Пиразолины далее могут быть окислены в пиразолы.
Действие гидразинов на а,|3-ацетиленовые оксосоединения прямо дает
пиразолы (Клайзен):
/Н
СН—Cd
I +Хо
СН3-СН nh2
NH-R
СН-СН -1
II II
СПз-СН N
NH-R-
Н2С--СН
I II
СНз-НС N
\^-R
НС---СН
/Н
с-с4
II >
сн nh2
/
nh2
гС----CH-I
III II
СН N
- NH2 -
НС—сн
II II
НС N
\н
3. Самый старый способ синтеза, которым Кнорр и получил впервые
гомологи пиразола, состоит в действии гидразина (или монозамещенного
гидразина) на 1,3-диоксосоединения:
CH2-C-R' НС---С—R
I II 11 II
R—С О NH2—> R-C N
О NH2 NH
HC=C-R'
jzzd R-C NH
\ /
N
Изомерное образовавшемуся К,К'-замещенному пиразолу вещество
(формула справа), которое казалось бы должно было получиться при этом
синтезе, на деле тождественно с полученным по реакции замещенным
пиразолом. В пиразоле не только не локализованы двойные связи, как
и должно быть в ароматическом соединении, но и протон свободно пере-
мещается от одного азота к другому.
Как и пиррол, пиразол очень слабая кислота, дающая металлические
производные, например, с окисью серебра. Но в то же время наличие
второго атома азота (со свободной парой электронов, не занятой в арома-
тической шестерке) сообщает ему свойства слабого основания — с силь-
ными кислотами он образует соли.
Пиразол сульфируется олеумом, хлорируется и бромируется,
нитруется, меркурируется. Электрофильные замещения идут в положе-
ние 4. Он очень устойчив не только к действию кислот, но и к окислению.
Как и в гомологах бензола, действием перманганата можно окислить
в карбоксилы боковые цепи пиразола, не затрагивая гетероцикла. При
окислении N-фенилпиразола разрушается фенил и образуется пиразол.
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
311
Однако при действии сильных нуклеофилов пиразолы претерпевают
нитрилъное расщепление по связи N—N (А. П. Кост, Н. И. Грандберг):
НС—C-R"
ОН + II II
Nx C-R
N-R
НоО +
L.
Хлорпиразол обладает малоподвижным хлором; 4-аминопиразол, полу-
ченный восстановлением 4-нитропиразола, диазотируется.
Пиразолоны. При описании свойств ацетоуксусного эфира уже было
указано, что образование гидразона (например, фенилгидразона) сопро-
вождается отщеплением сложноэфирного этоксила и замыканием в цикл
метилпиразолона (или N-фенилметилпиразолона). Реакция эфиров (3-
оксокислот с гидразинами и служит простым, применяемым и в промыш-
ленности методом синтеза пиразолонов:
СНз-С—СН2
О С=О
H2N ‘ ОС2Н5
NH
С6Н5
СНз—с—СН2
II I
N С=0
NH ОС2НЙ
I
С6н5
—с2н5он
СНз—с—СН2
Г 41
||2 5'
N С=О
\1/
N
СНз-с—СН
1 с-он
с6н5
Енольная форма пиразолонов имеет пиразольную структуру и потому
ароматична.
Реакции электрофильного замещения в пиразолонах-5 проходят в поло-
жение 4. Йодистый метил атакует второй азот, образуя 1-фепил-2,3-диме-
тилпиразолон-5, в медицине называемый антипирином или фенизоном.
Электрофильные замещения для этого соединения проходят в то же четвер-
тое положение; как и фенилметилпиразолон, он питрозируется в нитрозо-
соединение, последнее восстанавливается в первичный амин. При метили-
ровании аминоантипирина образуется третичный амин — диметиламино-
312
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
антипирин (Кнорр) — широко распространенное анальгетическое средство,
известное под названиями пирамидона или амидопирина'.
СНз-С---СН2
II I
N С=О
С6н5
СНзТ
------>
СНз-С=СН
I I
СНз-N С=О
с6н5
СН3-С=С—N=O
I ।
CH3-N С=О
HNO» „ /
N
I
свн5
Zn; н+
--------*
антипирин
ch3-c=c-nh2
I I
СНз-N С=О
I
СбЩ
/СНз
CH3-C=C-N<
I I ХСН3
СНз—N С = О
снд \ /
* N'
I
СвН5
пирамидон
Еще более сильный анальгетик {анальгин, или новалъгин) получается,
если действовать на аминоантипирин бисульфитным соединением формаль-
дегида (ввести в первичную аминогруппу — СН2—SO3Na), а затем проме-
тилировать амин в третичный:
CH3-C=C-NH2
CH3-N С = О
CH3-C=C-NH-CH2SO3Na
СНз-N С = О
\'/
СвЩ
СНз—С=С—N—CH2SO3Na
--> СНз—N С=О
СвН5
анальгин
Сам 1-фенил-3-метилпиразолон-5 вступает в реакции азосочетания
с арилдиазониями:
СНз-С---СИ2
i | +C6H5NseN
N С=О
^N^
I
С8Н5
СНз-С---СН- N=N-С6Н5 + н+
'I I
N С=О
с6н5
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
—~~~----------------— —... _—_________________ о1 «5
Реакция эта используется в промышленности, так как образующиеся
азокрасители очень светостойки.
О
II
НО—С—С------СН—N=N—С6Н4—SO3H-Z1
N С=О
I
C6H4-SO3H-n
тартразин (желтый кислый азокраситель)
Имидазол
В противоположность изомерному пиразолу имидазол
НС---N
1£ 3
нс сн
\1/
NH
в виде своих производных (аминокислоты гистидина, гистамина, алка-
лоидов, пуриновых соединений) широко распространен в животном
и растительном мире и играет важную биологическую роль.
Сам имидазол был впервые получен Дебю действием аммиака на глиок-
саль. Лучший результат получается, если к глиоксалю добавить формаль-
дегид и затем аммиак. Гомологи и производные имидазола получаются
из других 1,2-диоксосоединений, альдегидов и аммиака:
НС=О NH3 НС---N
I 4* —зн5о^" II II
нс=о онсн 2 нс сн
NH3 \ /
NH
R—С=О NH.3 R-C---N
I -г II I1
R'—С=О OHC-R" 2 R'—С С—R"
NH3 \ '
Имидазол выдерживает действие хромовой кислоты, но при окислении
перманганатом превращается в амид щавелевой кислоты. Озон разрывает
олефиновую связь и приводит к диацильному производному амидина:
R'
R'
N---С^
J j
R NH R"
R'
II / 4 О n //'
О I —R„c</ +1I2O2
1 \/° \NH-C^°
V* Vi x \ D П
/ \ ТЭ tt П
R Nil
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
314
Из этих реакций следует строение имидазольного кольца. Восстанови-
тели на имидазол не действуют. Имидазол — довольно сильное основание
(сильнее пиридина), образующее с сильными кислотами твердые соли.
В то же время он обладает и выраженными кислотными свойствами за счет
своей N—Н-связи, образуя металлические производные, например,
с гриньяровыми реактивами:
N---СН N--СН
|| || +CH3MgBr----> || || +сн4
НС сн НС сн
'nH "''г/
I
MgBr
Эти производные могут быть использованы для получения N-алкиль-
ных (действие RHal) или 2-С-алкильных (то же, при нагревании) производ-
ных. Вследствие кислотных свойств N—Н-группы N-метилимидазол может
быть получен непосредственно действием диазометана на имидазол. Оба
азота имидазола и, следовательно, положения 4 и 5 равноценны. Водород
имидазола свободно перемещается от N1 к №. Таким образом, здесь
имеется случай прототропии с большой скоростью перемещения протона.
Имидазолы способны ко всем главным типам ароматических электро-
фильных замещений — галоидированию, сульфированию, нитрованию
(в положение 4 или, что то же самое, в положение 5).
Поражает необычайно высокая температура кипения имидазола
(250° С): изомерный пиразол кипит при 187° С, пиррол при 130° С. Это
было объяснено (В. Хюккель) ассоциацией через водородные связи
и подтверждено измерением расстояния N ... N двух молекул имидазола
(равного 3 А). Подобного рода водородные связи играют огромную роль
в нуклеиновых кислотах (см. ч. II, «Нуклеотиды и полинуклеотиды»).
Одним из важных природных производных имидазола является амино-
кислота белков — гистидин, представляющий собой Р-(4-имидазолпл)-ала-
нин, или, что то же самое, |3-(5-имидазолил)-аланин. Строение гистидина
ясно из синтеза:
N---СН
I II
НС С—CH2C1+Na+
nh
О=С—ОС2Н5
С—NH—СО—СНз
О = С—ОС2Н5
N---СН О=С-ОС2Н6
II II I
НС С—СН2—С—NH—СО—СН3
\ / I
NH О=С-ОС.,Н5
НгО(Н+) ,
N---GH
II U
НС С—СН2—СН—
NH NHj
О
I
С—ОН + 2С2Н5ОН
+СНзСООН+СОа
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
При ферментативном разложении гистидин, как и другие а-аминокис-
лоты, декарооксилируется, превращаясь в гистамин-.
N---СН
II II Р
нс с-сн2-сн-с-он
NH NH2
N---СН
I! II +СО2
нс с-сн2—сн2—nh2
• NH
Гистамин оказывает сильное физиологическое действие. Он уже в малых
концентрациях резко понижает кровяное давление и расширяет капил-
ляры, активизирует гладкую мускулатуру и, следовательно, усиливает
перистальтику. Действию гистамина приписывают некоторые аллерги-
ческие недомогания — крапивницу и т. д. Существует множество анти-
гистаминных препаратов, снимающих гистаминный эффект. Назовем один
из них — димедрол (С6Н5)2СН—О—СН2—СН3—N(CH3)2.
Биотины
Из конденсированных бигетероциклов следует назвать а- и ft-био-
тины, относящиеся к витаминам (витамин Н). [З-Биотин содержится в ма-
лых количествах в растениях, в печени животных, в молоке и в желтке
япца. Его отсутствие в пище нарушает обмен белков и жиров в организме;
замедляется рост, поражается дерматитом кожа.
Биотины—вещества, необходимые для жизнедеятельности не только
животных, но и низших одноклеточных организмов. Суточная потреб-
ность человека в биотинах равна 6—10 мкг.
P-Биотин входит в состав ферментных систем, например а-декарбо-
ксилаз, осуществляющих декарбоксилирование щавелевоуксусноп кислоты
в пировиноградную и щавелевоянтарной в а-кетоглутаровую, а также
обратную реакцию — введение карбоксила в кетопокислоты (см. ч. II,
«Ферменты»; кн. I, цикл Кребса, стр. 465).
а-Биотин был выделен Кеглем в 1936 г. из яичного желтка (1,1 мг
биотина из 250 кг сухого желтка); (3-биотин был выделен несколько позд-
нее Дю-Виньо из печени (и из молока).
Молекулы биотинов имеют в бициклах по три асимметрических атома
углерода. Для обоих биотинов установлена гщс-?ргс-/щс-конфигурация.
О
HN NH
I* *1
нс-бн
। *т
Н2СХ хСН-С11-СН(СН3)2
S СООН
а~ биотин
HN NH
I* *1
нс—сн
н2с СН-(СН.Д-СООН
^-биотин
Н
/_СО
{сХДснДсоон
S
цис-цис-аис-конфи-
гурация р-биотина
gjg ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Структура р-биотииа подтверждается следующими реакциями:
О НООС—(СН2)4—СООН
11 f
С КМпОл
HN NH I-I2N NH2
НС---СН нс---сн
II I 1
Н2С СН—(СН2)4-СООН Н2С СН—(СН2)<—СООН
Расщепление
I по Гофману
^V^CHah-СООН
Биотины получены синтезом.
Три азолы
Известны триазолы вицинальный (1, 2, 3-триазол) и симметричный (1,3,4-
триазол). Первый синтезируется взаимодействием азотистоводородной
кислоты с ацетиленом, а его производные — взаимодействием органи-
ческих азидов с ацетиленами:
НС N-
НС—N
НС N+
Hi |1
НС N
NH
R-C—N
Il II
R'—С N
N—R'
Реакции этого типа позднее были названы Хыосгеном 1,3-дпполярными
присоединениями (ср. стр. 279).
В 1,2,3-триазолах протон свободно перемещается по всем трем атомам
азота, л-электроны делокализованы, пара р-электронов одного из азото
занята в ароматическом секстете, оба положения СН эквпвалентн .
Соединение ароматично и весьма устойчиво. Имеет слабокислотные и о
временно слабоосновные свойства: дает соли как с Ag+, так п с сильных
кислотами. П11|ГГ,„
Вицинальный бензтриазол образуется в результате дпазотиро
о-фенилендиамина и представляет собой внутреннее диазоаминоео Д _
ние: реакция, однако, необратима в том смысле, что не происходит °
ного размыкания кислотой диазоаминосоединения в амин и соль диа ©
+ HNO2
НС1
+ 2 Н2О + НС1
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
317
1,3,4-Триазолы можно получить из симметричных диацилгидразинов
при действии аммиака или аминов:
NH—NI-I N--N
II 1 II
R—С C-R-j-R'NHj —► R— С С—R
|| || \т/
О О N
I ,
R
Триазольные циклы обоих типов выдерживают окисление, а боковые
цепи отгорают, превращаясь в карбоксильные группы.
Наиболее известным производным 1,3,4-триазола является нитрон,
уксуснокислая соль которого служит в аналитической химии для осажде-
ния нитрат-иона (нитрат нитрона в воде нерастворим). Нитрон получают
из трифениламиногуанидина и муравьиной кислоты:
C6H5-N---N
тт ,NHC6H5 II II -
Н-С^ +CeH5NH—N=C< -------------> НС C-N-CeTT.5 <----->
ОН \мнс6н5 \ ,/
N
I
с6н5
CeH5-N=N
I I
ч--> НС" C=N-CeH5
\
I
Свн5
Структура его биополярна (цвиттерион), а заряды, так же как
и л-электроны, рассредоточены.
Тетразол
Тетразол синтезирован присоединением азотистоводородной кислоты
к синильной кислоте, а его производные — аналогичной реакцией из
нитрилов (Ганч):
N"
II
НС N+ ПС—N
II + II —> II II
N N N N
Н NH
в тетразоле протон
перемещается по азотам. Тетразол — слабая
кислота, вполне устойчив к окислению, ароматичен, реакции электро-
фильного замещения проходят по СН-группе.
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
3f8
При действии на аминогуанидин азотистой кислоты образуется амино-
тетразол:
HN=C—NH—NH2 + HNO2 —> THN=C—NH—NH2I —> H2N—C—N
I I II II
NH2 L . +N=N J N N
NH
Это вещество диазотируется, образуя крайне взрывчатые даже в вод-
ном растворе тетразолдиазониевые соли:
H2N—С—N
II II
N N
NH
НМО-НГ) NesN— С—N
HNOz- НС1-> |1 || +2ЩО
Cl- N N
NH
Диазониевые соли вступают в реакции азосочетания.
Пентазол
Пентазол, конечно, уже не может быть отнесен к гетероциклическим соединениям.
Б последние годы были получены органические производные пентазола. Так, фенилпен-
тазол образуется при соединении азпд-иона с катионом фенилдиазония:
+ ZN=N
C6H5-N=N + N7 —> c8h5-n< I
XN=N
Это вещество устойчиво при температурах ниже —70° С (Хьюсген).
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С ДВУМЯ РАЗНЫМИ
ГЕТЕРО АТОМАМИ
Мы рассмотрим вкратце только важнейшие
НС—СН
II II
НС N
о7
изоксазол
НС—N
II II
НС сн
V
оксазол
гетероциклы этого рода
НС—N
II II
НС сн
V
тиазол
Изоксазол
Изоксазол можно получить, действуя гидроксиламином на пропаргиловый
альдегид:
/Н НС—СН
НС=С-С< +NH.2-OH —> ||* з[|
ХО нс N
^О
Это слабое основание, по запаху напоминающее пиридин. Он обладает
выраженными ароматическими свойствами; значительно менее склонен
подвергаться электрофильным атакам, чем фуран. Ярко выраженные
ароматические свойства изоксазола исследованы Н. К. Кочетковым. Им
синтезирован циклосерин — антибиотик ряда изоксазолидина (полностью
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
319
гидрированного изоксазола), представляющий собой 4-амино-З-изоксазо-
лидон
it2n—сн-с=о
Н2С NH
Х'о'
Оксазолы
циклосерин
Общим методом синтеза оксазолов служит конденсация а-бромкетонов
с амидами кислот. Конденсацией бромпировиноградной кислоты с форм-
амидом может быть получена оксазол-4-карбоновая кислота, а ее
декарбоксилированием — оксазол:
НО
;с-с—он nh
|| ||
СН + с—н
Вг ОН
НО
о
НС СН
НС—N
-со2
II!
СН
енол бромвино-
градной кислоты
оксазол
nh2
О=С-Н
Оксазол — жидкость пиридинового запаха, с т. кип. 69—70° С, обла-
дающая свойствами слабого основания.
Другой путь синтеза оксазолов заключается в действии пятихлори-
стого фосфора на ациламинокислоты:
R-C-NH—СН—R' N---С—R'
II I Д=± II II
О С R—С C-OC2II5
6Z OC2II5 НО ОН
N—С—R'
|| [| -t-POCb + HCl
R—С С—ОС2Н5
'"б''
Оксазолы менее других азотсодержащих циклов устойчивы к действию
кислот и к окислению. Они ароматичны, хотя с зтой стороны мало иссле-
дованы.
Тиазол
Тиазоловый цикл замыкается реакцией, аналогичной первой из описанных
реакций получения оксазолов. а-Галоидзамещенпые оксосоединения вво-
дят в реакцию с тиоамидами кислот. Таким путем из хлорацетальдегида
и тиоформамида получается сам тиазол
нс—он NH ПС—N
й + II
сн СН НС сн
тиазол
Н-С=О
I
С1Н2С
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
320
а из хлорацетальдегида и тиомочевины — 2-аминотиазол:
НС-ОН
II
СН
'cl
NH
+ с—nh2
SH
НС—N
II
НС c-nh2
Тиазол — жидкость с запахом и температурой кипения пиридина
и вообще так же близко напоминающая пиридин, как тиофен — бензол.
Тиазол очень устойчив к окислению, выдерживает действие перманганата
и горячей концентрированной азотной кислоты. Он нитруется в жестких
условиях в положение 5, следовательно, ему присуща совмещенная ориен-
тация — тиофена (a-положение к сере) и пиридина (0-положение к азоту),
но ядро сильно дезактивировано. В положение 5 проходит и сульфирова-
ние (200° С, олеум). Электрофильные замещения даже для метилтиазолов
(случай, который лучше исследован) протекают в жестких условиях
и с трудом; замещение идет в положение 5, если оно свободно, и в положе-
ние 4, если в положении 5 находится метил.
Тиазол настолько похож на пиридин, что при 150° С аминируется
амидом натрия в положение 4. Подобно пиридину, он представляет собой
умеренное по силе основание.
При действии галоидных алкилов образуются тиазолиниевые соли,
которые можно получить и замыканием Г\т-алкил-(пли арил-)тиоамидов
с а-хлороксосоединениями:
SH
N—R]
II
СН
НС----N-R С1-
II II
НС сн
Б7'
енол хлорацет-
альдегида
тиольная форма
К-тиоф ормамида
Из упомянутого выше
препарат сульфатиазол:
2-аминотиазола готовят важный сульфамидный
Большое значение имеют производные бензотиазола. Меркаптооепзо-
тиазол (называемый в технике каптаксом) применяется как ускоритель
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
32!
вулканизации каучука. Его готовят нагреванием анилина с серой и серо-
углеродом:
Подобно а-замещенным хинолинам, 2-метилбензотиазолиодалкилаты
используются для синтеза цианиновых красителей (в этом случае тио-
цианиновых), служащих сенсибилизаторами фотопленок к красным
и инфракрасным лучам. Простейший пример синтеза такого рода краси-
теля демонстрирует легкую протонизируемость водородов метильной
группы 2-метилбензотиазола, подобную подвижности водорода а- и у-ме-
тильных групп пиридина и хинолина:
HC(OR')3
Разумеется, левая и правая половины молекулы здесь совершенно-
симметричны, положительный заряд рассредоточен между обоими азотами,
а л-связи полиметиновой цепи делокализованы.
Основные исследования по цианиновым красителям в СССР принадле-
жат школе А. И. Киприанова и И. И. Левкоеву. Как видно из предыдущей
формулы, красители этого типа представляют собой сплошную систему
сопряжения л-электронов. Поэтому нарушение сопряжения, например,
выводом части молекулы из плоскости вследствие пространственных пре-
пятствий, неизбежно должно вызвать гипсохромный сдвиг поглощения
(стр. 108). Напротив, введение в сопряженную систему ауксохромной
группы, например N(CH3)2, вызовет батохромный сдвиг.
Таблица 74. Влияние заместителей на сдвиг максимума поглощения в цианиновом,
красителе
Красители R в/ R" ?макс.’ е-10-4
I н Н c2H5 5560 16,5
II СНз Н c2H6 5630 14,5
III С(СНз)з II СНз 5630 14.9
IV н N(CH3)2 C2H5 6080 8,2
V СНз N(CH.3)2 CH3 5750 10,1
VI С(СН3)з N(CH3)2 СНз 5700 14,1
VII Н NH(CH3) с2н5 5580 15,4»
21 Заказ 67 2
322
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
В табл. 74 приведены данные А. И. Киприапова о сдвиге области
поглощения при варьировании заместителей в тиоцианиновых красителях.
В красителе IV две диметиламиногруппы (R') играют роль ауксохромов,
дающих батохромный сдвиг сравнительно с красителем I. Однако, если
в орто-положение к этим диметиламиногруппам ввести метилы (В) и не-
сколько вывести тем самым диметиламиногруппы из плоскости сопряже-
ния (краситель V), батохромный сдвиг уменьшится. В красителе VI
о-трет-6утильные группы полностью выводят диметиламиногруппы из
плоскости, и батохромное влияние диметиламинных ауксохромов совер-
шенно исчезает (максимум поглощения у красителей III и VI почти сов-
падает).
Инвалидизация ауксохрома может быть достигнута, как всегда, не
только пространственными факторами, но и связыванием свободной пары
электронов ионом водорода (солеобразованием,как в случае красителя VII).
К производным бензотиазола относится также группа важных сернистых
красителей типа примулина, прочных к свету и стирке, имеющих цвета
от желтого до коричневого и черного. В процессе их получения в качестве
первой фазы протекает непосредственное «сульфидирование» ароматиче-
ского амина серой:
Полученный аминомеркаптан окисляется серой в смеси с п-толуидином
при повышенной температуре в производное бензотиазола:
СН3~<ОУ-НН2 + сн3-<ОУnh2 CH3~\Q) N
При еще более высокой температуре (>-100° С) процесс повторяется
с образовавшимся гетероциклическим амином, нарастают новые бензотиа-
зольные ядра и получаются структуры следующего типа:
Такие примулины используются как непосредственно, так и путем их
Диазотирования и азосочетания на ткани. (О сернистых красителях см.
®акже стр. 165.)
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
323.
Любопытно производное бензотиазола люциферин, окисление которого-
обусловливает свечение светлячков.
О
Н
с—ОН
D-люцифррпн
К тиазол иниевым солям относится также витамин Вх (аневрин), сведе-
ния о котором даны при рассмотрении пиримидина (стр. 351), поскольку
его молекула включает и пиримидиновый цикл.
Производными тиазолидина (т. е. тетрагидротиазола) является группа
антибиотиков пенициллинов, открытие которых в годы второй мировой,
войны ознаменовало вступление медицины в эпоху антибиотиков и победу
человека над бактериальными болезнями. Известно пять пенициллинов,
которые близки по антибиотическому действию и отличаются друг от
друга ацилирующим аминогруппу ацилом: С6Н5СН2СО—в бензилпени-
циллине, и-НОС6Н4СН2СО — в оксибензилпенициллине, n-C5HxlCO—
в амилпенициллине, СН3СН2СН = СНСН2СО — в пентенилпенициллине
и С6Н13СО— в гептилпенициллине. Эта группа антибиотиков была открыта
Флемингом и Флори, а строение установлено химиками Оксфордского
университета под руководством Р. Робинсона и рентгенографически уточ-
нено Дороти Ходжкин-Кроуфут. Самым трудным оказалось установление
наличия в пенициллине необычного лабильного и легко изомеризующегося
|3-лактамного цикла. На стр. 324 приведены реакции, доказывающие
структуру бензилпенициллина — наиболее обычного из этих антибио-
тиков.
В результате щелочного гидролиза раскрывается один из гетероциклов
(именно азотистый) бицикла бензилпенициллина, а меркуролиз сулемой
(атака по связи S—С) разрывает второй сульфидный цикл и приводит
к образованию двух осколков — пенальдовой кислоты и пеницилламина,
содержащего первичную аминогруппу, тиольную и карбоксильную группы
при изобутановом скелете. Пенальдовая кислота — это а-фенилацетил-
аминомалоновый полуальдегид. Малоновое расположение карбоксила
и альдегидной группы следует из легкого декарбоксилирования пеналь-
довой кислоты уже в растворе с превращением в фенилацетилглицин. То
обстоятельство, что пеницилламин и пенальдовая кислота представляют
собой действительно два фрагмента пенициллоиновой кислоты, образовав-
шиеся без глубоких нарушений структуры, доказывается ресинтезом
пенициллоиновой кислоты из этих половинок (пенальдовая кислота взята
в виде эфира).
Пенициллин, гидролизующийся в пенициллоиновую кислоту, по
составу отличается от последней тем, что в нем меньше на молекулу воды
и только один свободный карбоксил (вместо двух, имеющихся в кислоте).
Как замыкается при потере воды карбоксил и какой именно из двух?
Ответим сначала на вопрос, какой карбоксил? При действии на пени-
циллин амином происходит аминолиз с внедрением в молекулу (вместо
21*
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
.324
воды) амина и образованием амида кислоты за счет одного из карбоксилов.
Это тот карбоксил, который при нагревании пенициллоиновой кислоты
-теряется в виде СО2, причем образуется пениллоиновая кислота.
Доказательство структуры бензилпенициллина
О\
уС—СН—N—С—О
НО' | | |
(СН3)2С CH-CH-NHCCH2CeH5
\/ II
S о
бензилпенициллин
|он“
Ох /О
Z-C—СН—NH С"
HOZ | | | он
(СНз)оС СН-СП—NHCCH2CeH5
‘ \ / II
S о
r Hgcl
пенициллоиновая кислота
О\
^C-CH-NH
;HOZ | |
(CH3)2C CH—CH2—NHCCH2CeH5
\Z II
s о
(X /О
%C—CH—NH cZ
HOZ | | | XOH
(СНз)оС CH=C- NHCCH2C6H5
\ II
S—HgCl 0
пениллоиновая кислота
°\
yC—CH—NH2
oz I
(СНз)2С
S-HgCl
-J, XlgLJ 2
>0
c-Z
Ox |XOH
ZC—CH—NHCCH2CeH5
н/ II
0
пенальдовая кислота
H2S I
I l ~С0!
X:C-CH—NH2 ZC—CH2—NHCCH2C6H5
HOZ | Н/ ||
(CH3)2C-SH 0
пеницилл амин
jPeсинтез пенициллоиновой кислоты
0\ /О
ус— СН—NH2 cZ
НО' I Н\ | ЧОС2Н5 __w
(СНз)2С + >c-ch-nhcch2c6h5
\ oZ ||
SH о
0\ /О
Z-С—СН —NH C.Z
НО I I | чос2н5
(СНз)2С СН— CH-NHCCH2CeH5
.sz о
ПЯТ1Г1ЛЕННЫЕ ГЕТЕРОЦИКЛЫ
325
В пениллоиновой кислоте сохранился карбоксил, связанный с тиазо-
.лидиновым кольцом, что может быть доказано меркуролизом ее сулемой
(аналогично меркуролизу пенициллоиновой кислоты при этом образуется
пеницилламин). Значит, в молекуле пенициллина замкнут во второй цикл
карбоксил боковой цепи пенициллоиновой кислоты, так как иначе при
гидролизе не вводилась бы молекула воды, а происходил бы разрыв
молекулы пенициллина на две части.
Из двух наиболее вероятных структур бензилпенициллина
% О
%C-CH-N----С=О О\ ||
HOZ I »| *1 ^С—СН—Nil С-------О
(СНз)2С СН—CH-NIICCH2C6H5 и ПО III I
\gz (СНз)2С СН—СП—N=C—СН2С6Н5
s
I II
в пользу первой говорит отсутствие основных свойств у пенициллина.
Свободная группа NH в формуле II должна реагировать как основная
группа. Рентгеноструктурное исследование также подтвердило формулу I.
Мы не останавливаемся на сложной стереохимии пенициллина, имеющего
три асимметрических углерода. Она также выяснена химическими и рент-
геноструктурными исследованиями.
Синтез пенициллоиновой кислоты, как указывалось, прост. Синтез
исходных для нее пеницилламина и пенальдовой кислоты также не пред-
ставляет затруднений. Однако весьма многочисленные попытки замкнуть
неустойчивый [3-лактамный цикл и перейти от пенициллоиновой кислоты
к пенициллину окончились неудачей. Вряд ли в истории органической
химии можно назвать другой пример затраты таких огромных усилий
ряда крупных исследователей, не приведших к успеху. В 1948 г. Дю Виньо
удалось синтезировать пенициллин лишь с малым выходом по схеме:
С1- О
Ок + И
Нп>^П-КН-. СН’° ?-----------? <с.н.)Лч,
HIT | “Г II --------
(СН3)2С CH=C-N=C
\н СН2С6П5
О
Оч II
С—СН — NH С---О
--->НО | I I I
(СИз)2С СН=С—N=c
СН2СвН5
I пиридин;
1 пиридин-НС1
О
Ох II
^С—СН—N-----с О
110 I I I II
(СН3)2С CH-CH-NHC
CllaCglU
326
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
ГЕТЕРОЦИКЛЫ С ОДНИМ АТОМОМ АЗОТА
Пиридин
Рассмотрение шестичленных гетероциклов удобнее начать с простейшего
азотпстого гетероцикла пиридина, обладающего ярко выраженными арома-
тическими свойствами
СН
НСз 5СН
II I
НС2 6СН
\1^
N
пиридин
В небольшом количестве пиридин и его гомологи содержатся в каменно-
угольной смоле, при перегонке попадают в фракцию легкого масла, откуда
их легко извлечь водным раствором кислоты, выделить в свободном виде
щелочью и разогнать.
Пиридин имеет сильный неприятный запах. Он кипит при 115° С;
плавится при —38° С; смешивается с водой во всех отношениях; дипольный
момент 2,20 D; энергия резонанса 37 ккал!моль. Водный раствор его —
щелочной, окрашивает лакмус в синий цвет; константа основности Къ —
= 1,8-10“9. С более или менее сильными кислотами он образует соли.
Очень устойчив к окислению. Гомологи пиридина при окислении хромо-
вой смесью образуют (подобно гомологам бензола) соответствующие
пиридинкарбоновые кислоты.
Строение пиридина явствует из следующих фактов. При восстановле-
нии водородом в момент выделения (действуя натрием на спиртовой раствор
пиридина, А. Н. Вышнеградский) или над катализатором пиридин
превращается в пиперидин'.
СН2
Na; СДЦОН НгС СН2
Н2С СН2
\н
пиперидин
Строение пиперидина доказывается синтезом его путем нагревания
гидрохлорида пентаметилепдиамина
/СНг СН2
сн2 сн2 н2с сн2
| | + нагревание | | -j-NILiCl -f- НС1
СН2 СН3—NH3 Cl- * Н2С СН2
NH3 Cl- ''nh
ддаСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
327
а также деструкцией (исчерпывающим метилированием) по Гофману
СН2 сн2 СН2
н2с сн2 | | СПз! Н2С ^СНг AgOH Н/ СН2 нагревание - Н=О *
н2с сн2 '\ z 1 1 НоС сн2 н2с сн2
NH N(CH3)2 I- N(CH3)2OH -
СН2 сн2 СН2
СН2 СН V | || СН.Н сн2хсн * пгт СНг сн -AgOiI । у сн2 сн2 нагревание
сн2 сн2 1 II сн2 сн2
N(CH3)a N(CH3)3 Г 4N(CH3)3 ОН"
сн2
сн \н
---> II II + N(CH3)3-r Н20
СН2 CII2
сн2=сн-сн=сн-сн3
Гофмановская деструкция азотистых гетероциклов состоит в серии
последовательных метилирований подпетым метилом до стадии образова-
ния четвертичной аммониевой соли, замены аниона иода на гидроксил
и пиролиза аммониевого основания. При этом отщепляется вода и обра-
зуется непредельный амин, а на последней стадии — триметиламин
и диеновый или полиеновый углеводород, имеющий ту же последователь-
ность связей углеродных атомов, что и в исходном гетероцикле (последняя
фаза — превращение 1,4-пентадиена в 1,3-пентадиен вызвана большей
устойчивостью системы с сопряженными л-связями). Метод и приме-
няется для установления последовательности С—С-связей в гетероцикле.
В данном случае это — неразветвленная цепь из пяти углеродных атомов.
Подобным образом может быть установлено строение и гомологов пи-
ридина.
Переходя в пиперидин (гексагидропиридин), пиридин присоединяет
шесть водородных атомов, следовательно, он имеет три двойные связи.
Ароматические свойства пиридина доказывают бензоидность этих связей.
Все это приводит для пиридина к формуле Кернера I или ее современному
эквиваленту II. В символике резонанса строение пиридина выразится
рядом формул, подобных III:
+
Три правые формулы III выражают оттягивание л-электронов к азоту
(сравнительно с СН-группой в бензоле) и обеднение электронами (опять-
328
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
таки сравнительно с бензолом) остальных звеньев гетероцикла, что ярко
проявляется в описанных ниже свойствах пиридина.
Ясное подтверждение такой структуры дает и действие оснований
на продукты N-присоединения к пиридину галоидных алкилов или арилов
(Цинке) или веществ типа хлорангидридов, подобных хлорциану или эфиру
хлорсульфоновой кислоты. Азот пиридина располагает свободной парой
электронов и в связи с этим обладает основными свойствами. Поэтому он
легко присоединяет, как все амины, иодистый метил, а также упомянутые
галоидпропзводные.
Во всех подобных случаях получаются производные глутаконового
альдегида или его таутомерной енольной формы:
IR /Н Н ,Н
ЛС—СН=СП—СН3-С<\ Ас—сн=сн-сн=с<
Си Ч) СИ хон
При взаимодействии пиридина с эфиром хлорсульфоновой кислоты
и затем со щелочью образуется натриевый енолят глутаконового альде-
гида
. О
/Ч. и
Il 1+CI-S-0C2H5
o=s=o
ОС2Н5
сн
Zz Ч.
oNaOFT НС СН
----|| I /Н +NH2SO3Na + C2H5OH
НС С А
\ Ч
ONa
при взаимодействии с галоидным алкилом и вслед за этим — с анилином
получается енольная форма дианила глутаконового альдегида:
СН
/Ах /Ч, и НС СН 4.
К | + RX || I || | -f-C6H5NH3 X-
\ 7 \+И НС CH=NC6H5
N NX" \
| NH
R I
R
Такого рода размыкания пиридинового цикла применяются в синтезе
открытых систем с сопряженными двойными связями. Сами вещества типа
RNHCH=CH—СН=СН—CH=NC6H5
где R С6Н5 или замещенный фенил, обычно 2,4-(NO9)2C6H3, являются
интенсивно окрашенными /голи.не/пиноеымк красителями (см. также
синтез азулена по Циглеру, ч. II, стр. 513).
Направление реакции в сторону раскрытия цикла с образованием
производного глутаконового альдегида (направление а) или в сторону
отрыва цикла от RX (направление б) зависит от соотношения прочности
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
329
связей радикалов с азотом в полученной аммониевой (пиридиниевой)
соли *
r—nh—сн=сн—с=с—с—nhr’
I I I
Что касается радикалов R, то легкость отрыва их от азота и, следова-
тельно, преобладание направления б растет в ряду (М. И. Кабачник):
—CN > —SO2O- > —SO2OR > CeH3(NO2)2 > C6H2(NO2)3
C10H5(NO2)2
-COCeII5 > Вг
Усиление электронодонорных свойств заместителя R' в пиридиновом
ядре и перемещение его из [3- в у-положение уменьшает электрофильные
свойства а-углеродов и, значит, усиливает противодействие нуклеофиль-
ной атаке размыкающего амина. В итоге преобладает отщепление амином
RX — направление б. Чем более сильным основанием является атаку-
ющий амин, тем вероятнее направление а (А. Ф. Вомпе).
Синтез пиридинового цикла. Рамзай (1877 г.) получил пиридин изменен-
ным синтезом Бертло (стр. 18), пропуская смесь ацетилена с синильной
кислотой через раскаленную трубку:
СН
///
НС
НС
СН
СН
Пропуская через бензол азот, возбужденный электромагнитным полем
высокой частоты, Б. М. Михайлов констатировал образование пиридина.
Атом азота в этом случае заместил группу СН.
При всей наглядности этих синтезов они не имеют препаративного
значения и дают ничтожный выход продукта.
Для подтверждения строения пиридинового цикла важен синтез Ганча,
первая стадия которого состоит во взаимодействии ацетоуксусного эфира
с альдегидом и аммиаком (берут альдегпдаммиак) и приводит к этиловому
эфиру 2,4,6-триметил-1,4-дигидропиридин-3,5-дикарбоновой кислоты.
Образовавшийся эфир окисляют HNO2, удаляя два лишних водородных
атома, что приводит к ароматической системе пиридина. Затем гидроли-
зуют сложноэфпрные группы и декарбоксилируют (ароматические
* Примененный способ написания заместителя R' означает возможность нахож-
дения его при любом (2, 3 или 4) углеродном атоме.
330
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
карбоновые кислоты, как известно, легко теряют карбоксилы при нагре-
вании со щелочью). Так синтезируют 2,4,6-триметилпиридин — коллидин-.
О
II
с2н5о-с-сн2
СНз-С=(
СНз
о сн о
II / \ II
С2Н5О—С—С С—С—ОС:
II II
СНз-С С-СНз
NH
О
I!
но—с
СНз
СНз
СII о
II II
0 Н2С—С—ОС2Н
+ I
' О = С- СНз
nh8
о
СЛО J-
СНз-
-ЗН2О^
СНз
С о
/ II
-С С—С—ОС2Н5
II I н : н*°
С С—СНз
\
N
СНз
I
С о
/" II
С С—С-ОН
-С С-СНз ~с°2
/\ /\
С N СН з
Окисляя его метилы в карбоксилы и снова декарбоксилируя, можно,
прийти и к пиридину.
При пропускании смеси альдегида RCH2CHO с аммиаком над окисью
алюминия при 400° С получается смесь гомологов пиридина, образующаяся
по схеме, сходной со схемой синтеза Ганча (А. Е. Чичибабин, М. П. Опа-
рина, П. А. Мошкин):
I
сн2
с и
о
R—СН2 Н2С—R
I + I
нс сн
NH3
О
СН R
R—СН2 СН2
+ I
в-сн2 СН
\
сн о
+ ЗН2О + Н2
+ ЗН2О + Н2
II
О NH3
шестичленные гетероциклы
331
Гакпм образом, этот синтез дает смесь изомеров, подлежащую раз-
делению.
По схеме, сходной с предыдущими, протекает и синтез диалкилпири-
динмонокарбоновых кислот из Р-хлорвпнплкетонов, ацетоуксусного эфира
я аммиака (А. Н. Несмеянов, Н. К. Кочетков):
СНС1
о
II
СН2—С—ОС2Н5
С-СНз
(Z
NH3
При нагревании смеси нитрила и диенового углеводорода до 400° С
гладко образуются замещенные пиридины (Янц и Мак-Келлог):
СН
СН СНо _______<4
11 + " —* 8 А
сн= J~R
N
Пиперидин (получаемый синтезом из пентаметилендиамина) при
дегидрогенизации над катализатором (металлы VIII группы) превращается
в пиридин.
Пиридин как ароматический гетероцикл. Реакции электрофильного
замещения. Пиридин гораздо индифферентнее по отношению к электро-
фильным атакам, чем бензол и даже нитробензол. Лишь в жестких усло-
виях и с плохим выходом его удается пронитровать, просульфировать
и пробромировать в |3-положенпе. Схема показывает сходство с замеще-
нием в нитробензоле:
Второй заместитель ввести этими реакциями уже совсем не удается.
Таким образом, азот пиридина вследствие своей большей электроотрица-
тельности, чем группы СН, сильно обедняет электронами углеводородную
Цепь цикла. К этому добавляется и то, что в кислых средах (нитрование,
сульфирование) ппридип превращается в соль, азот становится аммоние-
вым — положительно заряженным и потому еще более электроноакцептор-
332
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
ным и пассивирующим ядро к электрофильным атакам. Ден Хертог раз-
работал метод преодоления этого ориентирующего действия азота, окис-
ляя пиридин перекисью водорода в N-окись пиридина:
Окиси третичных аминов, например (CH3)3N— О, обладают семиполяр-
ной связью N—О. В окиси пиридина, однако, в момент атаки электро-
фильного реагента в а- или у-положение возможна перестройка электрон-
ной системы по схемам, приведенным в трех формулах справа: кислород
примыкает двойной связью к азоту за счет перемещения одной л-пары
электронов к а- или у-углероду, благодаря чему становится возможным
а- или у-электрофильное замещение. Так, нитрование с выходом, близким
к количественному, проходит в у-положение. Кислород может быть затем
удален восстановителями. Путем восстановления у-нитропиридина легко
получить у-аминопиридин.
Замечательным свойством N-окиси пиридина является ее способность
принимать и нуклеофильные атаки, подобные описанным ниже для самого
пиридина. Это объясняется тем, что вне электрофильной атаки N-окись
пиридина близка к структуре а, и ее атом азота за счет своего положитель-
ного заряда еще более способен оттягивать электроны, чем азот в пиридине.
Это и проявляется при нуклеофильных атаках в а- и у-положения.
Нуклеофильные замещения в пиридине. А. Е. Чичибабин и О. А. Зейде
нашли, что при нагревании пиридина примерно до 130° С в растворе
ксилола с амидом натрия образуется с хорошим выходом а-аминопиридин.
С амидом калия получается также и у-изомер. Реакция представляет
собой типичную нуклеофильную атаку на ароматическое ядро:
И Na- NH2'
N
При значительно более высокой температуре (—400° С) подобная
реакция происходит и между пиридином и твердым едким кали:
Аналогично, но в мягких условиях, на пиридин действуют такие еще
более сильные нуклеофилы, как металлоорганические соединения щелоч-
ных металлов:
Н"
N'
Х'С6Н5
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
333'
Особенно важной оказалась реакция аминирования по Чичибабииу.
Множество а-замещенных производных пиридина было получено этим
путем. Среди них отметим сульфамидный препарат сульфидин, имевший
одно время большое применение в медицине;
Реакции боковых цепей гомологов (и производных) пиридина. Уже ука-
зывалось, что при окислении, например, хромовой смесью или перманга-
натом боковые цепи пиридина (подобно бензольным) отгорают, оставляя
карбоксилы. Особенно важно получение этим способом |3-пиридинкарбо-
новой (никотиновой) кислоты, которую готовят, окисляя алкалоиды
никотин или анабазин (см. ч. II, «Алкалоиды»). Подобным образом из
у-метилпиридина можно получить у-пиридинкарбоновую (изоникотин овую)
кислоту. О применении этих кислот см. ниже.
Окисление метилпиридинов воздухом над пятиокисыо ванадия при-
водит к соответствующим альдегидам.
Водороды а- и у-метильных групп метилпиридинов обладают повышен-
ной протонизируемостыо и похожи на водороды метильных групп о-
и n-нитротолуолов. Так, метилпиридины вступают в конденсацию крото-
нового типа с альдегидами, чем воспользовался Ладенбургв синтезе одного
из простейших алкалоидов — кониина (яд растения болиголова), представ-
ляющего собой /^-пропилпиперидин:
Н
|| Ч| +О=С-СНз
'у\сн.,
——II I С2Н6ОН; Na
Jtl2V \ //\ ~---------
хСН=СН-СНз
СНгСНгСНз
Конденсируя а-метилпиридин с формальдегидом и дегидратируй
образующийся (З-(а-пиридил)-этанол, получают а-винилпиридин, приме-
няемый для сополимеризации с бутадиеном при промышленном произ-
водстве специальных видов каучука.
Водород а-метилпиридииа при действии алкиллития легко замещается
на литий с образованием а-литийметилпиридина, который может быть
использован для обычных металлоорганических синтезов (папомним, что
В толуоле водород метильной группы замещается лишь при металлирова-
нии алкилнатрием, но не алкиллитием):
H-CjITaLi
N
+ С4Н10
CH2Li
334
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Протоппзируемость метилов еще более облегчается в галоидметилатах
гомологов пиридина, которые питрозируются, превращаясь в оксимы
соответствующих альдегидов (кн. 1, стр. 145):
Г И +HONO —> ( || —Г II
^/хсн3 ^/Xch2-no ^/Xch=n-oh
"1 X- IX- | X-
СНз СНз СНз
•Оксипиридины и аминопиридины
Окси- и аминопиридины (а- и у-) синтезируют путем описанного только что
прямого нуклеофильного замещения пиридина или посредством электро-
фильного замещения N-окиси пиридина. p-Производные получают через
Р-сульфо- и Р-нитропроизводные пиридина, которые синтезируют прямым
электрофильным замещением или гофмановской деструкцией (NaOCl)
амида никотиновой кислоты в Р-аминопиридин.
P-Окси- и Р-аминопроизводные пиридина резко отличаются от а- и
у-производных. Так, p-аминопроизводные более похожи на соответству-
ющие производные бензола. Например, в отличие от а- и у-аминопириди-
нов p-аминопиридин легко диазотируется; а- и у-оксипиридины тауто-
мерны соответствующим пиридонам:
Аналогичная таутомерия (аминопиридин — пиридонпмин) свойст-
венна и аминопиридинам (А. Е. Чичибабин). В частности, последним типом
таутомерии объясняется образование многочисленных циклических произ-
водных из а-аминопиридина и бифункциональных соединений, подобных
малоновому эфиру и др.:
, -+ | |
-г о = С С=О
С2Н5О ОС2Н5 \ Z
I I сн2
о=с с=о
Положение таутомерного равновесия в случае замещения аминопири-
Динов существенно зависит от природы радикала R. Например, в ряду
арил сульфаминопиридинов как показали М. И. Кабачник, Т. А. Мастрю-
кова, Ю. Н. Шейнкер и И. К. Кузнецова, в зависимости от характера ради-
кала R существенно изменяются кислотные свойства форм и меняется
положение равновесия. Так, в 80%-ном водном диоксане при R = ra-NO2
содержится 90% иминоформы II, а при R = ra-N(CH3)2 около 90.о
аминоформы I. Для этой системы осуществлено полное количественное
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
335
определение кислотности и положения таутомерного равновесия (спек-
троскопически и методом <т,р-анализа по Гамметту).
а-Алкокси- и легко доступный у-фен оксипир ид ин гладко обменивают
свою алкокси- (или фенокси-) группу с аминами, меркаптанами и т. п.,
превращаясь в соответствующие у-замещенные пиридины (А. Ф. Вомпе)^
SR
I
N
ОС6Н5 NHR
I I
физиологически активные вещества
с пиридиновым ядром
Ранее уже была упомянута 0-пиридинкарбоновая, или никотиновая,
кислота. Ее амид — никотинамид — не что иное, как витамин РР, или
ниацин. Отсутствие или недостаток его в пище (суточная потребность
человека — от 0,03 до 0,05 мг) вызывает заболевание — пеллагру, более
распространенное в местах, где питаются почти исключительно полиро-
ванным рисом. Дело в том, что никотинамид входит в состав некоторых
необходимых для жизнедеятельности ферментов, например кодегидраз I
и II, которые выполняют функции катализатора дегидрогенизации —
гидрогенизации в соответствии со схемой (значение R см. ч. II, раздел
«Ферменты»):
Н Н
/X /С-NH2
NH2
f II 4-н+
N
R
R
Диэтиламид никотиновой кислоты применяется как возбуждающее
сердечную деятельность средство под названием кордиамина. Производные
изоникотиновой кислоты — изоникотингидразид и фтивазид применяются-
как антитуберкулезные препараты.
CO-NH— NH2
СО—NH—N=CH—/ ^—011
I
изоникотингидразид
фтивазид
336
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Важны производные пиридина — алкалоиды никотин, его изомер
анабазин и никотирин (см. ч. II, «Алкалоиды»).
4-Амино-3,5,6-трихлорпиридин-2-карбоновая кислота является одним
из сильнейших гербицидов: 2 г этой кислоты способны уничтожить расти-
тельность на площади в 1 га.
Витамины группы В6, первоначально принятые за одно вещество —
пиридоксин, оказались смесью трех веществ — пиридоксаля I, пиридок-
сола II и пиридоксамина III (выделен из риса в 1939 г. Одаке):
Н—С=О
НОХ//^уСН2ОН
НзС/'//
I
СН2ОН
НО\/^/СН2ОН
Y V
Нзс/4'/
II
ch2nh2
НОу/^СНгОН
li \
H3CZ\Z
III
Эти витамины входят в структуру ферментов, регулирующих белковый
обмен и катализирующих, в частности, реакции переаминирования амино-
кислот и их декарбоксилирования.
Для примера рассмотрим установление строения пиридоксола CJ.1H11O3N,
выполненное Куном и др. Пиридоксол дает характерные реакции пириди-
нового цикла и имеет три активных водорода (по Церевитинову).
При бензоилировании по Шоттену — Бауману пиридоксол образует три-
бензоат, а с диазометаном дает только монометильное производное (простой
эфир фенола), т. е. два из трех подвижных водородов принадлежат
не фенольным, а спиртовым гидроксилам боковых цепей, не метили-
рующимся диазометаном. Цепи эти могут иметь только по одному угле-
роду, т. е. обе группировки должны быть метилольными (СН2ОН),
поскольку из восьми углеродов пять входят в пиридиновый цикл, а один
в метильную группу (наличие ее доказано окислением). Все эти соображе-
ния подтверждаются тем, что кислоты, образующиеся при окислении
пиридоксола, сохраняют число углеродных атомов исходного соединения.
Упомянутый выше монометиловый (по фенольному гидроксилу) эфир
можно последовательно окислить: в лактон V, дпкарбоновую кислоту VI
и в трикарбоновую кислоту VIII, причем метоксил сохраняется.
Образование лактона V доказывает, что спиртовые группы несут цепп,
занимающие соседние позиции. То же доказывает замыкание двухоснов-
ной кислоты VI, получившейся после окисления лактона в циклический
ангидрид VII. Наконец, при энергичном окислении в щелочной среде
исчезает метил и образуется трехосновная кислота VIII, при нагревании
декарбоксилирующаяся в двухосновную кислоту, способную давать
ангидрид IX. Кислота VIII дает характерную для а-пиридинкарбоновых
кислот окраску с хлорным железом, которую двухосновная кислота,
соответствующая ангидриду IX, не дает. Следовательно: а) метил, окис-
лившийся в карбоксил и затем удаленный, занимал a-положение, б) два
Других карбоксила, расположенных рядом, занимают не а-положеяия,
а находятся в положениях [3- и у-. Фенольная оксигруппа пиридоксола
занимает другое (3-положение, что было установлено на основании анало-
гии УФ-спектров пиридоксола и (3-оксипиридина. Окончательно вопрос
о расположении групп был решен синтезом кислоты VIII.
щестнчленные ГЕТЕРОЦИКЛЫ
337
СН2ОН
СН2ОН
СН2ОН
СНзО^/^/СНгОН
II I
НзС Z V
IV
КМпО4
Н2С—О
о=с—о
I Ао
СН,°х/^/
о он
I zOH
сн’°\/кХ„
КМпО4;
он-
V
I
о он
СНзО.
\
НзС7
I —Н2О
IX VIII
О=С---О
с=о
VII
Разработано множество методов синтеза витаминов группы В6, в част-
ности ппридоксола. Все они достаточно сложны. Приведем один из них:
СН2ОС2Н5
С=О
СН2
С
СП:? О
C=N
сн2
I
с
CH2OC2II5
HNO3;
(СНзСО)гО
H2N о
сн2ос2н5
СН2ОС2Н5
H2/Pd
СНгОСгНэ
СНгОН
НО
'll I
И3с/\^
М. М. Шемякин и А. Е. Браунштейн разработали теорию катализа
аминокислотных превращений пиридоксальсодержащими ферментами.
22 заказ 67 2
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Пиридоксаль, этерифицированный по спиртовому гидроксилу фосфорной
кислотой, ею же привязан к специфической белковой молекуле (апофер-
менту). С аминокислотой альдегидная функция образует шиффово осно-
вание — азометин (белковый апофермент обозначен буквой А):
ОН - А
СН2О—Р—ОН
| ЧОН
h2n-ch
I
R
ОН - А
ОН - А
I
СН2О—Р — ОН
с<^°
I ХОН
HN^ ^s=C—N=C
НзсГ О!?1 R
б
В таутомерной форме б аминокислотная часть молекулы становится
похожей на остаток а-кетокислоты типа, например, пировиноградной.
В пей имеется очень реакционноспособная группа —N = C(R)—СООН, спо-
собная обмениваться с другими кетокислотами и легко гидролизуемая
с отщеплением аминокислоты:
ОН - А
СН2О—Р—OIJ
J о с<?
HN^ \==с—N==C
Нз^ \ll Н R
О
ОН
ОН - А
f\OH сн2о-р-он 0 0
о=с 1 с/
। ,=1 0 Лон I хон
—2---> HNZ >=С—Х=С 4-О=С
>=< I ! I
НзС ОН Н RZ R
|+Н*°
он. А
I
С1120—Р—ОН
I II
_| о
С=0
>=/ ।
Н3С 0НН
с/
I хон
4- H2N-CI1
I
R'
ОН-А
СН2О—Р—ОН
J О
HN/— Ч,'=С—ОН
Н3С ОН11
/°
се
| 4 он
4- HN = C
R’
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
339
Таким образом, осуществляется превращение кетокис.тоты в амино-
кислоту. а также превращение одной аминокислоты в другую (переамини-
рование). Кроме того, через таутомерную форму б легко может осущест-
вляться и реакция декарбоксилирования а-кетокислот (а также а-амино-
кислот). например декарбоксилирование пировиноградной кислоты
в ацетальдегид
/Н
снз-с-соон —> cih—с.; + со2
I Ч)
о
и ряд других превращений, не затрагивающих функциональные группы
в радикале R аминокислоты, если они имеются.
Хинолин и его производные
Хинолин (2,3-бензопиридин) получают известным синтезом Скраупа,
состоящим в нагревании анилина с глицерином, серной кислотой и нитро-
бензолом в качестве окислителя. Глицерин с серной кислотой образует
акролеин, который и присоединяет анилин, а полученный [3-фениламино-
пропионовый альдегид (при участии окислителя, снимающего излишние
атомы водорода) замыкается в хинолин:
/°
СН2(ОН)-СН(ОН)-СН2(ОН) Тйьо* СН2=СП—
хинолин
Применение замещенных в ядре анилинов дает замещенные в бензоль-
ном цикле хинолины. Если глицерин заменить ацетальдегидом (или ег®
тримером — паральдегидом), то через промежуточную стадию присоеди-
нения анилина к образовавшемуся в результате конденсации кротоновому
альдегиду получается а-метилхпнолин — так называемый хиналъди н
(синтез Дебнера — Миллера):
сн—сн
В этом случае нитробензол не нужен, так как альдегид сам служит
окислителем (акцептором водорода).
Оба способа при использовании разных аминов и альдегидов дают
возможность синтезировать многочисленные разнообразные производные
хинолина, что находит применение в химико-фармацевтической и анили-
нокрасочной промышленности.
22*
340
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Доказательство структуры хинолина как 2,3-бензопиридина или, что
то же самое, 1-азанафталина, сводится к окислению его в хинолиновую
кислоту — аза-аналог фталевой кислоты:
НоСГоО;
^.С-ОН
Vх с-он
л II
Синтез из анилина также ясно доказывает а,[3-бензопиридиновое строе-
ние хинолина.
Хинолиновая кислота может быть декарбоксилирована в пиридин.
Орто-положение ее карбоксилов явствует из образования ангидрида,
аналогичного фталевому. Интересно, что пиридиновое ядро оказывается
при окислении более прочным, чем бензольное.
Хинолин — мало растворимая в воде жидкость с неприятным запахом,
кипящая при 338° С. Он еще более слабое основание, чем пиридин, однако
с сильными кислотами образует соли и с иодистым метилом —• иодметилат
хинолина:
+ СНа1
N
Электрофильные замещения (нитрование, сульфирование) проходят
в хинолине труднее, чем в бензоле, и по преимуществу в бензольное ядро,
в положения 5 и 8; бромирование в жестких условиях (180° С) идет в поло-
жение 3.
Сульфируя хинолин, получают 8-хинолинсульфокислоту, а из нее
щелочным плавлением 8-оксихинолин (оксин):
SO3H ОН
Это вещество нашло широкое применение в аналитической химии
вследствие своей выдающейся способности образовывать оксинаты, нера-
створимые внутрпкомплексные соли (хелаты), за счет участия свободной
пары электронов азота. Как пример приводим хелатный оксинат магния
с двумя молекулами комплексно связанной воды:
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
341
8-Оксихинолин и его сульфокислота находят применение как дезинфи-
цирующие средства (хина зол), 5-хлор-7-иод-8-оксихинолин (энтер осептол)
применяется для борьбы с кишечными заболеваниями. Он столь полно
связывает ионный кобальт (но не кобальт кобаламина — витамина В12),
что жизнь бактерий становится невозможной.
Нуклеофильные атаки направляются в пиридиновое ядро (в а- иу-поло-
ження). Так можно получить а-аминохинолин исс-оксихинолин (карбости-
рил), по свойствам, в частности по таутомерии, подобные своим пириди-
новым аналогам.
При восстановлении по А. Н. Вышнеградскому хинолин образует
тетр аги др охи н олин
СчН5ОН; Na
- ~
обладающий свойствами вторичного ароматического амина.
Среди природных производных хинолина следует назвать алкалоиды
хинин и цинхонин (см. ч. II, раздел «Алкалоиды»). Многие получаемые
синтезом производные хинолина применяются как лекарственные вещества
и красители.
Из лекарственных веществ отметим синтезируемый из изатина и ацето-
фенона по Фридлендеру атофан (цинхофен)
СООН
I
сн3
О=С-С6Н5
СООН
применяемый как противоартритное средство, примахин и плазмохин —
антималярийные средства, действующие на гаметы плазмодия малярии,
и анестетик — перкаин или совкаин.
342
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
о=с—nh(ch2)2n(c2h5)2
NH
CH3CH(CH2)3NH2
NH
СН3СН(СН2)зН(С2Н5)2
N О(СН2)3СН3
примахин
плазмохин
перкаин (совкаин)
Особенно важны цианиновые красители, применяемые для сенсибили-
зации фотографических эмульсий к красным и инфракрасным лучам, не
действующим на бромистое серебро без сенсибилизации. Простейший
краситель этого типа — этиловьщ красный получают конденсацией N-этил-
хинолона с иодэтилатом хинальдина (а-метилхинолина):
N-этилхи-
нолон
этиловый красный
N-Этилхинолон в свою очередь получают непосредственно в реакцион-
ной смеси, окисляя воздухом N-этил-а-окси-! ,2-дигидрохинолин — про-
дукт действия щелочи на иодэтилат хинолина:
Следует заметить, что в формуле этилового красного обе половины
'слева и справа от центральной группы СН) идентичны по расположению
атомов и разнятся лишь расположением электронов в системе сопряженных
д-связей. Аммониевый положительный заряд показан лишь на одном —
правом атоме азота; на деле, как всегда в подобных случаях, происходит
мезомерное выравнивание, что можно выразить в разных символиках так:
шестичленные гетероциклы
344
Красители, поглощающие свет в еще более длинноволновой части
спектра, имеют сходную структуру, отличающуюся заменой центральной
СН-группы на систему из п СН-групп, связанных сопряженной системой
л-связей:
В этой системе также происходит мезомерное выравнивание положи-
тельного заряда между обоими атомами азота по системе сопряженных
связей и, как следствие, сближение энергии первого возбужденного
состояния молекулы с основным состоянием.
Область поглощения таких красителей удается точно рассчитать на
основании простейшей модели «ящика» (ч. II, раздел «Небензоидные
ароматические системы», стр. 490).
Акридин
Акридин содержится в малом количестве в каменноугольном дегте (в зеле-
ном масле), где и открыт Гребе и Каро.
При окислении акридин образует акридиновую — 2.3-бензпиридин-
5,6-дикарбоновую кислоту:
акридин
СООН
СООН
Акридин плавится при 111° С; он имеет очень слабые основные
свойства.
При действии серной или фосфорной кислот на N-фенилантраниловую
кислоту замыкается цикл и образуется акридон-.
О 011
Действием РС15 или Р0С13 на акридон (на его таутомерную форму —
9-оксиакридин) получают 9-хлоракридин. Восстановлением 9-хлор-
о44 ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
—...........——------------------'-------“ - ----------------— —
акридина получается 9,10-дигидроакридин, который окисляется в акри-
дин:
Окси- и аминопроизводные акридина — красители. Некоторые из них
оказывают сильное бактерицидное действие. Таковы желтый риванол
(этакридин), в виде водных растворов применяемый для дезинфекции
полости рта и т. п., и акрихин (атебрин) — средство борьбы с шизон-
тами (бесполая стадия) малярийного плазмодия.
NH2
^\ZW\/OC2H5
I II I I
риванол (этакридин)
Синтез акрихина:
CH3CH(CH2)3N(C2H5)2
I
NH
х\А/х,-осн»
। I । ।
акрихин (атебрпн)
О
осн»
-СНз
Н2Сг2О7
С—ОН
С-ОН
/ /ОСНз
С1
nh2
росн
ОСНз
СНзСН(СН2)зК(СгН»)2
О
NH
CH3CH(CH2)3N(C2H5)2
NH
ОСНз
шестичленные гетероциклы
345
Нзохинолин
Изохинолин (3,ч-бензопирпдин) — изомер хинолина. При окислении пер-
манганатом он образует фталевую и 3,4-пиридиндикарбоновую (цинхо-
мероновую) кислоту, которая изомерна хинолиновом кислоте
6^-уКз КМпО4 ^/'оон HOOCX>i
в I ^^соон НООС/Х/
лзохинолин
чем доказывается наличие и относительное положение бенз- и пиридине-
вого ядер. Изохинолин (т. пл. 24° С, т. кип. 240° С) — несколько более
сильное основание, чем хинолин. Гидрированные в пиридиновом ядре
ди- и тетрагпдроизохинолины синтезируют из |3-аминоэтилбензола двумя
следующими путями:
где R=H, алкил или арил.
Осторожно окисляя или дегидрируя эти гидроизохинолины, получают
сам изохинолин.
Скелет изохинолина, обычно гидрированного, входит в структуру
ряда важных и сложно построенных алкалоидов — морфина, папаверина,
алкалоидов кураре и др. (см. ч. II, раздел «Алкалоиды»).
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С ДВУМЯ АТОМАМИ АЗОТА
(ДИ АЗИНЫ)
Возможны три структуры диазинов — 1,2-диазин, или пиридазин, 1,3-дпа-
зин, или пиримидин, и 1,4-диазин, или пиразин". 6/ з 6|| |3 N3 Ns JI 1, V N 1 1 пиридаэин пиримидин « N *1 II /2 N 1 пиразин
34б ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Первый из них —- не что иное, как азин малеинового диальдегида
и может быть получен соответственно из этого диальдегида и гид-
разина.
Пиридазин
Пиридазин (т. пл. 6,4° С, т. кип. 207° С) — очень слабое основание,
слабее пиридина, по все же сильнее пиримидина и пиразина. Рассчитана
энергия резонанса пиридазина, равная 22 ккал!моль. Электрофильным
замещениям пиридазин не поддается. Атомы хлора в хлорпиридазинах,
особенно в положениях 3 и 6, обладают такой же подвижностью, как
в п-нитрохлорбензоле. Диоксипиридазину (гидразиду малеиновой кислоты)
НО
б
более свойственны, однако, реакции в форме б. Так, метилирование диазо-
метаном дает О-метильное производное формы б.
Пиридазиновый цикл очень устойчив к окислению. Тетрагидропирид-
азины можно получать диеновым синтезом:
СН, О СН2 О
// ii II /\
СН N—С—OR НС N-C—OR „ n,w+, | N—СООН II NH
I II, __. I, I Н2О(Н+) ______,
’ ' I I I — *2ГО« '
СН N— С—OR НС N—С—OR N-COOII || NH
\ II \ II \/z \/
сн, О СН2 О
В присутствии щелочи и платины тетрагидропиридазины разлагаются
с выделением азота и образованием циклобутана (Р. Я. Левина,
Ю. С. Шабаров).
N Pt(OH-) ;
NH
СН2-СН2
I I + n2
СН2-СН,
Пиримидин
Очень важным гетероциклом является пиримидин. Пиримидиновые
структуры наряду с производными пурина (стр. 358) входят в состав
жизненно важных для любого организма нуклеиновых кислот, связанны-
с синтезом белка в клетках. Сам пиримидин (т. пл. 22° С; т. кип. 12 •
легко растворим в воде), удивительным образом не давая щелочной Ре*
ции, образует соли с сильными кислотами (с 1 экв). Его можно синте
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
347
ровать через барбитуровую кислоту (уреид малоновой кислоты, или мало-
нилмочевина), которая сама является важным производным пиримидина:
0 ?
II Г VII
С—OR H2N х
H2C<f + СО ------------>• Н2С С=О ДЛГ
С—OR H2N r._NH
он
С-ОН
С—N
I
ОН
барбитуровая
кислота
Действие на барбитуровую кислоту пятихлористого фосфора дает
2.4,6-трихлорпиримидин, при восстановлении которого иодистым водоро-
дом получается и сам пиримидин:
ОН
С
/ \
НС N
II I
НО-С С-ОН
РСЦ
С1
Cl/V^Cl
HI
пиримидин
Способность пиримидина к реакциям замещения довольно мало изу-
чена. Можно, однако, сказать, что в этом отношении он отличается от
пиридина, как пиридин от бензола. Так, к электрофильным замещениям
он способен еще меньше, чем пиридин, а если замещение происходит, то
заместитель вступает в положение 5 (^-положение по отношению к обоим
азотам). Действие амида натрия на 6-метилпиримидин приводит в резуль-
тате нуклеофильного замещения к 2-амино-6-метилпиримидину, хотя
метил и должен ослабить электрофильность пиримидинового ядра.
В метилпиримидинах метильные группы 2, 4 и 6 вступают в реакции кон-
денсации кротонового типа с альдегидами, как и метильные группы
а- и у-метилпиридинов. 2-,4- и 6-Хлорпиримидины обладают (подобно 2-,
4- и 6-хлорнитробензолам) реакционноспособным галоидом, который не
только замещается в результате нуклеофильных атак, но и вступает
в реакцию Фриделя — Крафтса с ароматическими углеводородами. Окси-
производные пиримидина обладают более сильными кислотными свой-
ствами, чем фенолы. Боковые алкильные группы гомологов пиримидина
можно окислить в карбоксилы, не затрагивая пиримидинового цикла,
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
очень устойчивого к окислению. Пиримидины, замещенные на ОН, NH2
и другие активирующие группы, нитруются, нитрозируются и азосоче-
таются в положение 5; заместители, естественно, находятся в положениях
2,4 или 6 и ориентируют в орто- и пара-положения по отношению к себе.
Аминогруппа в положении 5 способна диазотироваться, а аминогруппы
в положениях 2, 4 и б на холоду не затрагиваются азотистой кислотой,
а при легком подогревании замещаются на ОН.
Наиболее общий путь синтеза производных пиримидина состоит в кон-
денсации 1,3-диоксосоединений с веществами, имеющими при одном атоме
углерода две аминогруппы (или амино- и иминогруппу), такими, как
мочевина, гуанидин или амидины:
R
।
С
/ \
НС N
I I
R'—С С—R"
N
Мы уже видели на примере барбитуровой кислоты, что оксипроизвод-
ным пиримидина свойственна таутомерия. Для самой барбитуровой кис-
лоты это одновременно и кето-енольная и лактим-лактамная таутомерия
О ОН
II I
С С
Н2с NH НС \
I I k II |
О=с С=О НО—С С—он
\ z \
NH N
О 0-
II I
с с
НС^ NH НС XNH
II I — I
НО-С С=0 НО-С С-0-
NH XNH
в в'
причем главная структура, свойственная барбитуровой кислоте
в твердой форме, это структура в, т. е. енольно-лактамная, в которой,
однако, предполагают мезомерный сдвиг в сторону ароматической бетаин-
ной структуры в' (резонанс структур в и в')-
При конденсации мочевины с эфиром диэтплмалоновой кислоты полу-
чается диэтилбарбитуровая кислота (известное снотворное веронал, или
барбитал)
О
II
NH-С г „
о=с/ \с.
\ / хс2нй
NH—С
II
О
для которой
типа б, ни в
уже невозможно превращение ни в таутомерную структуру
структуру типа в—в', с чем связано резкое снижение кислот
П1ЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
349
ности (для барбитуровой кислоты К
Кя = 3,7 -IO-8).
Ю 4, для диэтилбарбитуровой
Кроме веронала (R — R' = С2Н5) известен целый ряд снотвор-
ных диалкилбарбитуровых кислот, например: нембутал [R = С ЛК; R' ==
== СН3СН2СН2СН(СН3)], амитал [R .-= С2Н5; R' = (СН3)2СНСН2СН21;
люминал, или фенобарбитал (R = С2Н5; R' = СвН5), эвипан ^R = СН3;
Н' = —/j, мединал (натриевая соль веронала) и др.—всего более
сотни препаратов.
Хорошим методом синтеза 2-окси- и 2-аминопиримидинов служит
циклизация р хлорвинилкетонов с мочевиной и гуанидином:
С=О
НС
II
НС
^С!
h2nx
;с=о
H2Nz
R
С
НС с=о
\н
R
I
С
нсГ Sj
4=2- II |
НС с-он
N
НСХ
II
НС
h2n,
+ 4C=NII------->
h2n
R
С
Z К
HC N
НС C=NH
\н
R
I
С
• Z \
HC N
ZZ2" II I
HC c—nh2
N
R
I
С = О
При гидролизе нуклеиновых кислот, находящихся в ядре клеток,
получаются три важных производных пиримидина:
урацил
тимин
350
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
цитозин
Их можно получить и синтетически, например, исходя из натриевого
енолята формилуксусного эфира и мочевины, тиомочевины пли их произ-
водных, например:
О
II
С-ОС3Н5
НС
II
НС
ONa
О
H2N\
+ /С=О
h2nz
О
он
ЦИТОНПН
В синтезе цитозина можно исходить и из |2,6-дихлорпиримидина
(в свою очередь получаемого из урацила и Р0С13), действуя на него после-
довательно аммиаком и затем водой:
С1
\
С1
П30ЦИТ''Г.Ьи
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
351
Главной таутомерной формой тимина, урацила и цитозина, по данным
преимущественно физических исследований, следует считать структуру б
(стр. 349), электронная система которой мезомерно смещена в сторону
структуры б', имеющей уже ароматический характер. Таким образом,
эти вещества сохраняют некоторую ароматичность, несмотря на то что
таутомер а присутствует только в растворах и притом в весьма незначи-
тельном количестве.
К пиримидиновым производным принадлежат лучшие из сульфамид-
ных препаратов — сулъфазин (другие его названия сульфадиазин, сульфа-
пиримидин), метилсульфазин (сульфамеразин) и сульфадимезин (суль-
фамезатин):
сульфазан
СНз
N Nil—S
-nij2
метнлсульфазин
суш фчдимезин
Витамин ЕЦ, или тиамин, тоже содержит пиримидиновый цикл
НОСН2-СЛ2—----
НзС^ У
тиамин
Отсутствие витамина Bi в пище, например при питании ободранным рисом
(он содержится в рисовых отрубях), вызывает известное на Востоке нерв-
ное заболевание бери-бери. Суточная потребность человека в витамине
В, — до 4 мг. Необходимость в нем связана с тем, что витамин (в виде
пирофосфата) входит в структуру фермента карбоксилазы, катализиру-
ющего некоторые стадии метаболизма сахаров в организме (отщепление
СО2 от пировиноградной кислоты) (см. кн. I, стр. 466).
Строение витамина В] было установлено Вильямсом и Греве. Основной
момент при установлении строения — расчленение тиамина действием
сульфита на тиазольный и пиримидиновый циклы:
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
352
СН2ОН
сна-
NH2
N—Н2С I
Na 2SO з
—NacT"”
СН2ОН
н3с S
НзС S
СНз
N
N
Восстановление пиримидинового осколка натрием в жидком аммиаке
привело к диметиламинопиримидину, строение которого доказано встреч-
ным синтезом:
В настоящее время в СССР витамин Вг синтезируют по варианту
метода Вильямса и Андерзага, разработанному Г. В. Челинцевым и
3. В. Беневоленской.
Синтез пиримидиновой части молекулы витамина В/.
CN CN
СН +С2Н5ОН > СН2
II I
СН2 с2н5осн2
о
н— с—Ос2н6;
CjHsONa
CN CN
---> С2Н5ОСН2— С -----------> с2н5осн2—с
II II
СН—ONa ' СН—ОСНз
енолят «-формил-Р-этоксипр опионитрила
C2H6OCH2
ОСНз
nh2
С-СНз
HN
NH
II
С2Н5ОСН2—с NH
II I
НС С-СНз
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
353
nh2
С2Н5ОСН2—С N
НС С—СНз
NH2
N СН3
Синтез тиазольной части молекулы:
н2с—сн
сь
Н2С---СНС1
Н2С С—СНз
I I /С1
Н2С с<
\ / СНз
о
О СНз
а-метилфуран
(сильван)
Н2С---CHC1
о
таутомер-
ная форма
тиоформ-
амида
НОСН2-СН2—п N4-2HCE
U II
Н3С S
Соединение обеих частей молекулы:
носн2—сн2
N
Н3С S
носн2—сн2
Превращение тиамина в
к веществу, тождественному
О О
С1Н2С
НзС S
витамин В
(соль)
NH2 • НС1
N
СНз
NH2-HC1
N СН3
эфир пирофосфорной кислоты приводит
коферменту кокарбоксилазе
II II
НО—Р—О—Р—О—СН2-СН2-7|—
I I II
О- он /\
НзС S
NH2
N СН3
Фермент карбоксилаза образуется соединением кофермента с соответ-
ствующим белком.
23 заказ 67 2
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
354
Пиразин
При самопроизвольной конденсации а-аминокетонов получаются диалкил-
дигидропиразины, при окислении воздухом или окисью ртути превраща-
ющиеся в дпалкилпиразины:
О H2N N Д
// \
СНз—С СН2 СНз-С СН2 о СН3—С СН
| 4- | —► I | —► I II + Н2о
Н»С С—СНз Н2с С—СНз НС С—СНз
“\ Z
NH2 О N N
Боковые группы гомологов пиразина при осторожном окислении могут
быть превращены в карбоксилы, а последние удалены декарбоксилирова-
нием. Так приходят к самому пиразину:
Пиразин — значительно более слабое основание, чем пиридин. В вод-
ном растворе он образует соли с 1 экв кислоты, в безводной среде — с 2 экв
кислоты: с 1 экв СН31 дает четвертичную аммониевую соль
N
У/
N
I
СНз
К электрофильным замещениям пиразин еще менее способен, чем
пиридин, и в настоящее время известна лишь реакция галоидирования.
Примером восприимчивости пиразина к нуклеофильным атакам может
служить аминирование действием амида натрия. Метилпиразин конден-
сируется с альдегидами, как а- и (3-метилпиридины:
+ Н2О
Пиразин гидрируется в пиперазин (тетрагидропиразин), который проще
получить из дибромэтана и аммиака (наряду с этилендиамином):
NH
^СНг
I I
н2с сн2
\н
Вг NH3 Вг
IbC^ СН2
I + I
н2с сн2
Вг NH3 Вг
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
355
С первичными аминами (вместо аммиака) получаются N-замещенные
пиперазины.
Пиперазин, в отличие от пиразина, — сильное основание. Дикето-
пиперазины, с которыми мы уже встречались (кн. I, стр. 493), получаются
нагреванием а-аминокислот:
Они могут быть восстановлены в пиперазины.
К пиразинам, точнее, к дибензопиразинам, называемым феназинами,
относятся и рассмотренные нами в разделе об антрацене красители типа
индантрена синего (стр. 254).
Проще индантрена синего по скелету феназиновые красители, получае-
мые, в частности, окислением смеси нитрозодиметиланилина с л^-фенилен-
диамином (или его гомологами). Реакция идет через стадию конденсации
в индамин:
N
феназин
NO
СНз
II I
/\^\
h2n nh2
(C1I3)2N
индамин
нейтральный красный
N-Фенилпрованные феназиновые красители называются сафранинами, простейший
из которых— феносафранин получается окислением смеси 1 моль п-фепплсндвамппа
и 2 моль анилина:
z\/NH2 А
। о + ii л
NHz
СвНь
О; НС1
-------►
HoN/
N
I Cl-
CelU
23*
.356
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Положительный аммониевый заряд, конечно, рассредоточен между всеми четырьмя
-атомами азота.
К этому же классу относится и первый (исторически) «анилиновый» краситель_
лювеин Перкина (1856 г,), полученный окислением нечистого анилина
CeH5NH2 +
NH2
-NII2
C0II5
N
о; HG1 ?
CeI-I5-NH/'j+Z\^NH2
I ci-
CeHa
<п потому содержащий наряду с веществом указанной структуры также примеси его го-
мологов. Получаемый окислением анилина бихроматом краситель анилиновый черный
предположительно имеет также феназиновую структуру:
СвЩ
NH
а:С1~
В настоящее время находят практическое применение лишь более сложно построен-
ные сафранины.
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С ТРЕМЯ АТОМАМИ АЗОТА
Из трех приведенных триазинов
N N
1,2,3-триазин,
или вицинальный
(смежный)
1.2,4-триазин,
или несимметри-
ческий
1,3,5-триазин,
или симметри-
ческий
мы рассмотрим вкратце только последний.
Сам симметрический 1,3,5-триазин можно получить трпмеризацией
•синильной кислоты в присутствии кислот (Грундманн и Крейцбергер):
X
С
/ \
N N
ЗХ— C=N > 'I | (Х--=Н)
Х-С С—X
\
N
Многочисленные производные этого триазина получаются полимери-
зацией (часто самопроизвольной) таких производных синильной кислоты,
как хлорциан (X — С1), эфиры циановой кислоты (Х-алкоксил), циан-
ЩЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
357
амид NH2 C^=N(X--NH2). Так, образующийся путем тримеризации
цианамида меламин применяется для изготовления ценных пластмасс,
которые получают из этого триамина и формалина, сшивая молекулы
меламина метиленовыми мостами по азотам аминогрупп, как это нам
знакомо по мочевино-формальдегидным смолам (кн. 1, стр. 372). Тример
хлорциана хлористый цианур также готовится в промышленном мас-
штабе и используется в производстве красителей и гербицидов.
NH2
С
N N
Н I
H2N—С с—nh2
\
N
меламин
С1
С
N N
II I
С1—С С—CL
\ Z
N
хлористый цианур
Гербициды ряда триазина, уничтожающие сорняки на полях,
отно-
сятся к трем типам соединений:
01
I
С
N N
RHN-C С—NHR
N
ОСНз
I
С
N N
II I
R'R"N—С С—NR'R*
SCH3
I
С
N N
II I
R"'HN— С С—NHR"'
Их коммерческие названия таковы:
Симазин .
Атразин .
Прометен
r=c2h5
r=c6h5
R'=CH(CH3)2,
R" = H
Симетон . . . R'==R”=C2Hs
Симетрии . • • R'"=C2H5
Прометрпп . . R"'=CH(CH3)2
Грундманн и Крейцбергер описали интересное превращение 1.3,5-три-
азина в 2-амино-1,3,4-триазол при действии аминогуанидина:
СН
N N
I II
НС СН
N
+ 3H2N-NH— c-nii2 ►
II
NH
N—N
3 II II + 3NH3
HC C-NII2
\fh
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
АЗОТИСТЫЕ БИГЕТЕРОЦИКЛЫ
Пурин и его производные
Бигетероцикл, представляющий собой конденсированную систему пири-
мидина и имидазола, называется пурином
в
СИ
Z \ 7
1N 5С— N
I И Ч
2НС 4С СНз
\н
3 9
Производные пурина имеют исключительное биологическое значение
прежде всего потому, что некоторые его окси- и аминопроизводные входят
наряду с пиримидиновыми основаниями в структуру нуклеиновых кислот
и имеют, таким образом, отношение к программированию синтеза белков
в организме и к явлениям наследственности. Сюда же относится ряд других
жизненно важных веществ, таких, как аденозинтрифосфат — переносчик
энергии в биохимических реакциях и фосфорилирующий агент. Алка-
лоиды кофеин и теобромин — также производные пурина.
Одним из ключевых соединений в синтезе пуриновых производных
служит мочевая, кислота, играющая у птиц и рептилий роль вещества,
выводящего из организма избыток азота (как мочевина у млекопитающих).
Соли мочевой кислоты {ураты} откладываются при неправильной работе
организма в суставах и в виде почечных камней.
Понятие о строении мочевой кислоты дает ее окисление. Перманганат
окисляет мочевую кислоту в аллантоин, разрушая пиримидиновый цикл;
при окислении азотной кислотой, наоборот, сохраняется пиримидиновый
цикл, а разрушается имидазольный и образуется аллоксан-.
ОН о
Z \ / \
N С—N HN С—NH
I II II I И I
IIO-C С С-ОН О=с с с=о
А / \ / \/\/
N NH NH NH
КМпО4
О=С—NH
H2N—С—NH—НС С=о
NH
О
мочевая кислота
аллантоин
С ОН
HN
С—NH
НЫОз ,
О=С
с=о
/
HN
I
о=с
NH| NH
ОН
аллоксан
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
359
Начиная с 1888 г. осуществлено несколько синтезов мочевой кислоты,
из которых мы приведем два: синтез Э. Фишера, одного из главных иссле-
дователей веществ группы пурина, и синтез Траубе. При синтезе по Фишеру
исходят из барбитуровой кислоты (стр. 347), нитрозируют ее по подвиж-
ной метиленовой группе, восстанавливают в амин (урамил) и, надстраивая
на аминогруппе действием изоциановой кислоты группировку мочевины,
получают псевдомочевую кислоту, которая при нагревании с соляной
кислотой замыкается в мочевую кислоту:
ООО
HN СН2 HNOi, HN СП—NO HN C=NOH н
О NH о О NH О О NH О
барбитуровая
кислота
HN CH-NII2 h_n=c=o hN Сп-------------NH HNZ X|j~NH
J\ zk zL C=O -h2o* I II I
6 NH (f N1A - (/W NH%
nu2 u
урамил псевломочевая мочевая кислота
кислота
Синтез Траубе:
О
О
О
RO—С
о
С
nh2
сн2
HN
СН2
oh- HN CHg HN02
HN CH—NO
О=С
C=N
О=С
C=N
NH2
О NH NH
О NH NH
nh2
О
ii
о
HN c-nh2
HN CH —NIC
О NH NH2
6 Nil NH
В этом синтезе первая стадия — ацилирование мочевины посредством
сложного эфира циапуксуспой кислоты, вторая — замыкание гетероцикла
путем внутримолекулярного присоединения группы Nil., к группе С N.
360
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Реакция переаминирования мочевины при нагревании ее с аминами
обычна: выделяется аммиак и образуются N-замещенные мочевины, в дан-
ном случае мочевая кислота.
О О
II
Hrf С—NH2 H2Nk HN II
I II + /с=°|-^йнГ I II I
o^nh\h2 H2N /nhnh%
мочевая кислота
Метод Траубе создает широкие возможности синтеза пуриновых осно-
ваний. Так, при ацилировании (или лучше сказать превращении в заме-
щенный амидин) 4,5-диаминоурацила действием сложных эфиров разных
кислот можно получить ксантин (действие муравьиного эфира) и всевоз-
можные его производные, замещенные в положении 8:
Il nh2
HN II
О NH NH2
О
II
с-н--->
Ref
О
II
o^nh'nh
ксантин
Напомним, что в имидазольном цикле водород свободно перемещается
между обоими азотами.
При действии пятихлористого фосфора на мочевую кислоту она, реаги-
руя согласно таутомерной (триоксипуриновой) структуре, образует
2,6,8-трихлорпурин:
рсц ?
С1
CI N NH С1
Три хлора в трихлорпурине проявляют разную подвижность по отно-
шению к действующему на молекулу нуклеофилу. Порядок их подви/К-
ности такой: 6 2 8. Поэтому при действии щелочи (100°С) или аммиака
замещается на ОН- или NH2-rpynny только хлор в положении 6. После
этого можно действием HI заменить на водород остальные два хлора
шестичленные гетероциклы
361
и получить два очень важных производных пурина — 6-оксипурин —
гипоксантин и 6-аминопурин — аденин'.
NH2 Cl
Cl V NHXci Cl V NН С1
a
Hd
nh2
z\ /
N NH
аденин
гипоксантин
Если же на стадии б подействовать более сильным нуклеофилом —
алкоголятом натрия или аммиаком в жестких условиях, то заместится
и хлор в положении 2, а после действия HI образуются два также весьма
важных производных пурина — ксантин и гуанин'.
ОН
Н2?Г n NH^ci
он
h/Ynh
гуанин
ксантин
Гуанин и аденин наряду с пиримидиновыми основаниями (стр. 349)
принимают участие в построении нуклеиновых кислот, обусловливающих
синтез специфических для данного организма видов белка и определяющих
•наследственные свойства организма.
Гуанин находится в рыбьей чешуе, от чего зависит ее блеск. Извлечен-
ный из чешуи растворителем, он применяется для получения искусствен-
ного жемчуга (выкристаллизовывается на внутренней стороне полых бус).
Нуклеотиды
Аденин, гипоксантин и гуанин играют огромную роль в обмене веществ
в организме. В соединении с моносахаридом (рибозой) и фосфорной кисло-
той они образуют так называемые нуклеотиды и их производные,
362
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
примерами которых могут служить адениловая кислота и аденозинтрифос-
сфат — переносчик фосфорной кислоты и энергии в организме.
адениловая кислота
Нуклеотиды, подобные адениловой кислоте, составляют звенья высоко-
молекулярных нуклеиновых кислот, в которых каждый из них связан
сложноэфирной связью остатка фосфорной кислоты с гидроксилом рибозы
следующего нуклеотида (см. ч. II, раздел «Нуклеотиды»),
Пуриновые алкалоиды
К пуриновым производным относятся алкалоиды какао (теобромин),
кофе и чая (кодбеин) и чая (теофиллин или эуфиллин). Они представляют
собой метилированные по азоту ксантины:
О
НзС-N С--N-СНз
I I! I
о=с С сн
'nZ
I
СНз
НзС— \-N
I II II
О=С С сн
СНз
теофиллин
кофеин
Н3С—N ХС--N-СНз
I II I
о=с с сн
/ \
NH N
теобромин
Кофеин и теофиллин содержатся в чайном листе (до 5% кофеина на
сухое вещество) и добываются из чайной пыли. Теобромин добывается
из бобов какао. Кофеин — важное средство, возбуждающее нервную
систему, стимулирующее работу сердца и учащающее пульс. Теобромин
и теофиллин — диуретики и расширяющие сосуды медикаменты, применя-
ющиеся при гипертонии. Представление о строении этих алкалоидов полу-
чают на основании их окисления. Кофеин при этом превращается в диме-
тилаллоксан и метилмочевину, теофиллин — в диметилаллоксан и моче-
вину, а теобромин — в метилаллоксан и метилмочевину. Кофеин можно
получить, метилируя ксантин, а теофиллин — синтезом Траубе (стр. 359),
исходя из симметричной диметилмочевины.
Наряду с извлечением кофеина из чайной пыли осуществляют и его
промышленный синтез, исходя из мочевой кислоты, получаемой из птичьего
шестичленные гетероциклы
363
помета. Действуя на мочевую кислоту уксусным ангидридом, замещают
гидроксил в имидазольном цикле на метил (т. е. заменяют остаток уголь-
ной кислоты на остаток уксусной), и полученный 8-метилксантин метили-
руют диметилсульфатом по трем азотам. Лишнюю метильную группу
элиминируют, хлорируя ее до трихлорметильной группы с последующим
гидролизом и декарбоксилированием:
О
IIn" Х||
о NH NH ОН
О
II
/nhVXch3
(СНзСОЦО
—СО,;
—СП,СООН
О
NH NH сн3
О
JI
(CH,),so< . НзС-ы |Р'N-СНз с1,
N/XcIl3
СНз
- О
НзС—N )| N СНз 2Н,0
| II I -ЗНС1
О N N СС13
СНз
НзС-N |1 N-СНз
СНз
-со,
О
НзС—N II ? СНз
О N N
СНз
кофеин
Кинетин
В 1955 г. Сноог выделил из растительных объектов кинетин — вещество,
резко ускоряющее рост растений за счет усиленного деления клеток.
Кинетин также оказался пуриновым производным:
O/XC1J2—NH
I
С
// \
N С---N
I II I1
НС с сн
XNZ NH
кинетин
364
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Птеридины
В отличие от давно известных пуринов (кофеин открыт в 1820 г., мочевая
кислота еще раньше) их аналоги птеридины стали известны лишь в 30-х
годах текущего столетия. В основе этого класса бигетероциклов лежит
птеридин — 1,3,5,8-тетраазанафталин, представляющий собой систему
конденсированных пиримидина и пиразина:
5
4 N
/>\/ \в
’Т
2 А. //ч
N А
1 8
птеридин
Птеридины найдены в природе; они являются красящими веществами
крыльев бабочек. Особое значение имеет наличие птеридиновой цикли-
ческой системы в структуре важных витаминов — фолевой кислоты и рибо-
флавина (витамин В2).
Примерами веществ, окрашивающих крылья бабочек, могут служить
белое красящее вещество крыльев капустницы — лейкоптерин и желтое —
ксантоптерин. Понятие о синтезе веществ этого класса можно получить
на примере синтеза лейкоптерина. Синтез начинают подобно синтезу
мочевой кислоты по Траубе, но вместо мочевины берут гуанидин:
О
о || о
с II
NH2 RO-С у \ /\
I | HN СН2 он- HN СН2 HN0.
С + СН2----> II —>|| —
I 6 C=N AviA тт
HN NH2 CsN у \ HN NH NH
HN NH2
О
II
HN CH—NO
--> I I
HN^NH^h
о OH
II I NH2
z\ ^\7
H HN CH-NH2 N
J\ /L /^/\
HN NH nh H2N n NH2
2,5,6-триамино-4-оксипирим идин
Полученный 2,5,6-триамино-4-оксипиримидин ацилируют сплавлением
с щавелевой кислотой:
ОН
H2n N NH2
ОН
| NH О
I II I
/\ z\
H2N n NH О
он
лейкоптерин
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
365-
Фолевая кислота
Одним из важных витаминов для многих организмов является фолевая
(птероилглута.чиновая) кислота, открытая в 1938 г. Штокштадтом
в экстракте печени. Фолевая кислота оказалась широко распространенной
в растительном мире, особенно в зеленых листьях. Она входит в состав
ферментов, обеспечивающих перенос одного углеродного атома в виде,
например, формильной группы. Строение фолевой кислоты подтверждено
синтезом (Чеше, Вейганд); освоено получение ее в промышленном масштабе.
Синтезируют ее исходя из 2.4,5-трпампно-6-оксипиримидина, действуя на
него продуктом присоединения брома к акролеину, а затем п-аминобензо-
плглутампновой кислотой. Таким образом приходят (хотя с небольшим-
выходом) к фолевой кислоте:
ОН
фолевая кислота
Рибофлавин
Наконец, к этому классу гетероциклов относится витамин В2 (рибофла-
вин), входящий в структуру окислительных ферментов организма человека
и животных. Впервые он был выделен Р. Куном из молочной сыворотки;
содержится также в яйцах. Синтез его был выполнен Р. Куном и Карце-
ром.
Витамин В 2 изготовляют в промышленном масштабе. Для этого-
из /)-глюкозы через /)-арабинозу и /)-арабоновую кислоту получают
D-рибоновую кислоту, восстанавливаемую через лактон в jD-рибозу.
£*-Рибозу конденсируют с о-ксилидином в шиффово основание, восстана-
вливаемое во вторичный амин. Азосочетанием вторичного амина с солью
фенилдиазония получают азосоединение, которое восстанавливают по
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
366_________________________ _______________ ______________________
азогруппе в амин, и этот амин конденсируют с аллоксаном в рибофлавин:
сн2он
НО—Н
но—Н
НО—Н
(Д'н
N СГ
сн2он
сн,он
НО-4-Н
ноД-н
сн2он
ноД-н
аллоксан
рибофлавин
Эт
СН О1Д1Д^аМ\И входит в виде своего фосфорного эфира (по первичной
передатчик- Ше П состав «желтого фермента» Варбурга it играет роль
атомам азот к?^°^°Да’ Два атома которого легко присоединяются к двум
а (один азот пиразинового, другой пиримидинового цикла/
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
367
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С АТОМОМ КИСЛОРОДА
Производные у-пирана
Из кислородных гетероциклов мы рассмотрим лишь производные у-пирана.
Так называется структура, наиболее известным производным которой
является у-пирон, а наиболее интересными дериватами — пирилиевые
соли, по строению аналогичные пиридиниевым.
О
'I
Cii if ii
ЧО О
7-пир ан
7-пирон
Самым доступным и изученным веществом этой группы является
у-диметилпирон III, который получается из дегидрацетовой кислоты II
в свою очередь синтезируемой сложноэфирной конденсацией двух тауто-
мерных форм ацетоуксусного эфира I, наступающей при нагревании его
паров: ООО
II II II
С С-СНз С О
СН ОС2Н5 сн2 НС сн—С-СПз НС1;
и + I ----------------------> I, । Нг0,
СН3-С С=О -2ЩН.0Н СНз-С С=О ---*
ОН С2Н5О о
I I II
о
II
с о
НС СН—С—СНз
II I
СНз-С С=0
но он
О О -
. II
СО СО
\ /' \ ;1
НС С—С-ОН НС С—С—он
I ,|| —> li II
СНз-С С—СНз -Н«О СНз-С С—СНз
но он ''"о7'
-со,
о
Н3С
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
368
Сама дегид рацетовая кислота (а-метил-0 -ацетилпирон) предста-
вляет собой производное а- и у-ппрона. При действии концентрированной
соляной кислоты опа превращается в а,а'-диметил-у-пирон.
Общий способ получения производных у-пирона состоит в действии
НС1 на 1,3,5-триоксосоединения:
О О
н2с \н2 нс1 нсГ ^сн
II Il II
R-G С—R’ R—С С—R'
\ Z \ /
0 0 но он
+НС1
-НзО^
Замечательной особенностью у-пирона и его гомологов являются его
основные свойства, более резко выраженные, чем у всех обычных кислород-
ных функций. Причиной этого служит то обстоятельство, что при пре-
вращении в пирилиевые соли, происходящем как под действием кислот,
так и йодистого метила, у-пирон ароматизируется, вовлекая одну из сво-
бодных электронных пар эфирного кислорода в ароматический секстет:
ОН
I
Z\ нс1
II I <—
/\+Z\
Н3С О СН3
О
II
А
Н3С 0 СНз
ОСНз
/\+^\
Н3С 0 СНз
С1- I-
+
Таким образом, хотя Н+ или СН3 атаковали кетонный кислород,
превратив его в гидроксил или алкоксил, роль основного кислорода
сыграл эфирный кислород, став оксониевым положительно заряженным
кислородом. Оксониевые соли этого типа, полученные и изученные Колли
и Тикле, известны наиболее давно (с 1899 г.). В отличие от продуктов
присоединения HG1 к спиртам, эфирам и оксосоединениям в этом случае
твердые характерные соли образуются также с анионами галоидов и силь-
ных кислородных кислот.
Строение таких солей, например продукта иодметилирования диметил-
пирона, доказывается его превращением под действием аммиака в 2.6-ди-
метил-4-метоксипиридин:
ОСН3 ОСНз
I \
||А f + н20 + NH4C1
/\+^\ zz\
Н3с о СНз Н3С N СНз
С1-
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
369
С гриньяровым реактивом у-пироны реагируют обычным образом по
карбонилу. Образовавшийся алкоголят при действии кислоты дает пири-
лиевую соль, уже лишенную в у-положении гидроксила (или алкоксида)
IMgO СНз
Л + CFIgMgl ---------->
НзС ° СНз
Л )
НзС О СНз
СНз
I
2НС1 /1
----* II I + Mg/ + н20
/\^\ С1
НзС О СНз
Cl-
Такая соль при действии аммиака также превращается в 2,4,6-триме-
тилпиридин.
Кислород пирилиевых солей можно заместить на СН-группу, превра-
тив соли в соответствующие производные бензола действием эквимолеку-
лярного количества реагента Виттига (Димрот, Вольф, Вахе):
R R
1 к
II-------------------| +СН2=Р(СбН5)з ---> f | + ОР(СвН5)з + НС1О4
/\+^\ /\ ^\
R ° R R сн R
C1OJ
Простой метод синтеза 2,4,6-триарилпирилиевых солей описан в 1964 г.
Р. Шмидтом. На примере трифенилпирилиевой соли реакция выражается
следующим уравнением:
С6Н5
I
/\
4СбН5—С=СН + 2С6Н5—С-С1 + SnCl4 ► 2 || |
II
О С6н8 ° Cells
SnClg
Бензпирилиевые и а-фенилбензпирилиевые (флавилиевые) соли можно
синтезировать по способу А. Н. Несмеянова, Н. К. Кочеткова и М. И. Ры-
бинской, исходя из фенолов и алкил- или арил-р-хлорвинилкетонов:
СН3—С—СН=СНС1
О
ГеС13 + НС1
ОН
+ Н2О
сн3
FeClf
феррихлорид
2-метил-5,6-бензпирнлия
С6Н5-С-СН=СНС1
о
феррихлорид
5,7-диоксифлавилия
24 Заказ 672
370
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Хромон, флавон и родственные им вещества
2,3-Бенз-у-пирон называется хромоном
О
& II 4
eY\/\3
\д
Y о
1
Этот скелет лежит в основе многих важных природных веществ. Сюда
относится группа витаминов Е (токоферолов), необходимых для размно-
жения животных. Впервые витамины этой группы были выделены из
масла ростков пшеницы. В настоящее время известно семь веществ, обла-
дающих действием витаминов Е, близких между собой по структуре
и частично изомерных друг другу. Приведем формулу одного из них —
0-токоферола:
СН3
Н0\А/\
I 11 1/Снз СН3 СНз СНз
Ч/\X \ I I
Y О ^СН2СН2СН2—СН—СН2СН2СН2—СН—СН2СН2СН2—СН—СНз
СНз
ос-Токоферол — гомолог p-токоферола, отличающийся наличием еще
одного метила в положении 7, а у-токоферол — изомер р-соединения,
несущий метильные группы в положениях 7 и 8. Длинный алифатический
скелет из 20 углеродов (включая замкнутые в дигидропирановый цикл,
связанный с бензольным ядром) тот же, что в изопреноидном спирте
фитоле (кн. I, стр. 313). Рацемический сс-токоферол был синтезирован Кар-
рером действием фитилбромида на 2,3,6-триметилгидрохинонв присутствии
ZnCl2. При этом первичный углеродный атом фитила (несущий бром)
связывался с ароматическим ядром, а орто-гидроксильная группа присое-
динялась по аллильной двойной связи фитильного остатка, замыкая
пирановый гетероцикл.
Производным хромона является келлин, содержащийся в семенах
растения Ammi Visnaga. Он применяется при стенокардии как сосудо*
расширяющее средство и как средство, действующее положительно на
сердечную мышцу. Строение келлина выяснено Шпетом.
Особенно распространены в растительном мире производные 2-фенил-
хромона, названного Костанецким (главным исследователем этой группы
веществ) флавоном
келлин
флавон
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
371
Природные производные флавона — это оксипроизводные, всегда
содержащие гидроксилы в положениях 5 и 7, иногда в положении 3 и
обычно в положениях 3' и 4'. Они окрашены в желтый цвет и представляют
собой обычные окрашивающие вещества желтых цветов. Примерами
могут служить лутеолин — красящее вещество почек тополя и резеды
и кверцетин — вещество, широко распространенное в желтых цветах.
НО О
лутеолин
В растениях они находятся в виде гликозидов. Гликозид кверцетина
с дисахаридом 6 [р-Л-рамнозпдо-7)-глюкозой] называется рутином. Это —
одно из веществ, относящихся к группе витаминов Р, при отсутствии
которых в пище капилляры кровеносных сосудов становятся проница-
емыми для крови, что ведет к «пурпуровой болезни» — множественным
точечным кровоизлияниям. Другие витамины этой группы близко род-
ственны по структуре.
Активностью витаминов Р обладают (А. Л. Курсанов) и находимые
в растительном мире (например, в чае) дубители — катехины, например
гидрированный цианидин (стр. 372). Гидрирование антоцианидинов (см.
ниже) устанавливает их родство с дубителями — катехинами.
Строение флавонов определил Костанецкий путем расщепления их
щелочью, как это показано на примере лутеолина:
кон; н2о
О
ОН II
I С—он
\ / /—X
I II + СНз—С—у—ОН
/\/\ II \=<
но он о он
Синтез лутеолина (и других флавонов) Костанецкий осуществил путем
конденсации эфира диметоксибензойной кислоты II с метиловым эфиром
24*
372
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
ацетилфлороглюцина I с последующим замыканием пиронового цикла
и деметилированием посредством HI:
ОСНз /О
I Cf
^\/ ^СНз
I II
/\/\
СНз О ОСНз
I
+ С2Н5О—С—Ч—ОСНз
II
0 ОСНз
II
Антоцианидины
При восстановлении оксифлавонов (например, магнием и кислотой)
можно получить антоцианидины. Так, кверцетин дает при этом циани-
дин — типичный представитель антоцианидинов. Гидрирование циани-
дина приводит к катехину:
ОН
| ОН
X к /—\ /-°н
но 0 . =\
он
сг
кверцетин
цианидин
Антоцианидины, как видно из этого примера, являются оксипроизвод-
ными а-фенилбензпирилиевой соли с оксониевым кислородом, входящим
в ароматический цикл. Это относительно прочные твердые соли, интен-
сивно окрашенные. В виде гликозидов — антоцианов они окрашивают
цветы, ягоды и овощи (антоциан — греч. краска цветов) *. Все анто-
* Овощи окрашены также каротиноидами: например, морковь — каротинами,
помидор — ликопином (стр. 656).
ШЕСТИ ЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
373
цианы — кислотно-основные индикаторы. В кислой среде их окраска
сдвигается в сторону красной, в щелочной — в сторону синей (или сине-
зеленой), в нейтральной она фиолетовая. Таким образом, цианидин
является причиной окраски и василька (щелочная среда сока) и красной
розы. Другие представители антоцианидинов — пеларгонидин (лежит
в основе окраски цветов пеларгонии), дельфинидин (окраска синих цветов
дельфиниума, а также красного винограда и вина). Первый содержит
в фенильном ядре на один гидроксил меньше, а второй на один гидроксил
больше, чем цианидин:
ОН
ОН
Cl-
пеларгонидин
ОН
Cl-
дел ьфинидии
На этих трех антоцианидинах построено великое множество оттенков
цветов сине-красной гаммы. Отдельные антоцианы — производные трех
приведенных антоцианидинов разнятся тем, что один или два гидроксила
в фенильном ядре метилированы, а гликозиды образованы разнообраз-
ными сахарами. Понятие о структуре антоцианидинов (Вильштеттер) дает
разложение щелочью. Например, пеларгонидин при этом разлагается,
образуя флороглюцин и п-оксибензойную кислоту.
Известны два классических синтеза антоцианидинов — Вилыптеттера и Р. Робин
сона, крупнейших исследователей этой группы веществ.
По Вилыптеттеру, из флороглюцинового альдегида получают 2,4-диметокси-6-
оксибензальдегид и конденсируют его по Перкину (стр. 147) с метоксиуксусной кисло-
той:
ОСНз/Н /ОСНз
^\/ % Н2С он-
I II +1 —>
С
СНзО ОН .z
На полученное производное кумарина (стр. 190) действуют гриньяровым реак-
тивом. Последняя стадия — деметилирование тетраметилового эфира посредством HI:
ОСНз
I ОСНз
f" Z~X
I I + BrMg—у у—OCH3---->
chZ^V^o
ОСНз
A/w0CHs
I I |>0MeBr
CHsO/^/^q/ \<^-^-OCH3
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
374
Cl" Cl-
пеларгонидин
Синтез Р. Робинсона очень типичен для замыкания пирилиевого цикла вообще
и необычайно изящен своей лаконичностью:
ОСНз
Jx/CHO СН2-ОСН3
I II +1 Z—X
сн ^С-<Э-0СН
СН3О о '—
СНз? сн
/Ч/ос«=
zU\ А\_>-°СНз
СНзО но о
НС1
ОСНз
—ОСНз + Н2О
Cl-
Далее следует деметилирование посредством HI.
Что касается путей генезиса всех описанных производных флороглю-
цина в растениях, то высказано предположение (Фодор) о таком пути
биосинтеза: сначала идет альдольное замыкание альдегидной формы
глюкозы в стереоизомерные инозиты, далее дегидратация во флороглюцин
и синтез антоциановых и флавоновых пигментов:
СНОН-СП2ОН СНОН
СНОН НОН!^ СНОН
СНОН * НОНС СНОН
\ /О \ /
СНОН—C<f СНОН
ХН
он
флороглюцин
инозит
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
375
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
С ДВУМЯ РАЗНЫМИ ГЕТЕРОАТОМАМИ
Цикл I называется оксазином, цикл II — тиазином. Упомянем также их
дибензопроизводные, называемые феноксазином III и фентиазином IV:
Некоторые производные феноксазина применяются как красители,
получаемые, в частности, нагреванием нитрозодиметил анилина с фено-
лами, например:
К фентиазиновым красителям относится метиленовый синий, получае-
мый из тг-аминодиметиланилина действием тиосульфата натрия и бихро-
мата натрия в присутствии хлористого цинка. Из тиосульфата выделяется
сера, которая и внедряется в цикл:
(Ch3)2n/
NH
(CJ13)2N
H2N\
T ii ZnCI,
I II -NH,
\N(CH3)2
NH
z\ / \ z\ окис-
2S у 'У >, ление|
—► I II I II HC1
(CH3)2N V XN(CH3)2
N
(C113)2N^\^44s//4^\n(C1I3)2
Cl"
Cl-
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
376
В 50-х годах нынешнего столетия фентиазиновые препараты нашли
совершенно новое применение в качестве нейролитиков при лечении шизо-
френии и маниакальных душевных заболеваний. Примером таких лекарств
могут служить хлорпромазин (аминазин) и стелазин.
СН2СН2СН2—N(CH3h
аминазин
GH2CH2CH2-N
стелазин
СН2—СН2
Z /NGH3
сн2-сн2
ЧАСТЬ
ВТО РАЯ
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
К середине прошлого века были открыты первые металлоорганические
соединения, которые были систематически исследованы Франкландом.
Эти летучие вещества, для которых можно было измерить плотность пара
и определить молекулярный вес, Франкланд использовал для установле-
ния понятия валентности. Металл связан в этих веществах химической
связью с углеродным атомом органического радикала, жирного или арома-
тического.
Между металлами и неметаллами (металлоидами) нет резкой границы.
Еще больше эта граница стирается в их органических производных,
так что черты сходства имеются, например, между простейшими угле-
родистыми производными: Bi, Sb, As и Р; Sn, Ge и Si; Al и В, что можно
видеть уже из сходства форм соединений элементов разной степени алки-
лирования (или арилирования). Например:
IV группа
V группа
RSiH3, B2SiH2, R3SiH, R4Si RSiCl3, R2SiCl2, R3SiCl RPH2, r-2ph, R3P RPCI2, R2PCI RPCI4, R2PC]3, R3PCI2, R4PC1. RsP
RGeH3, R2GeH2) R3GeH, R4Ge RGeCl3, R2GeCl2, R3GeCl
RAsH2, R2AsH, R3As RAsC12, R2AsC1 RAsC14, R2AsC13. R3AsC12. R4AsC1, R5As
RSnll;., R2SnH2, RaSnH, R4Sn RSnCl3, R2SnCl2, R3SnCl
RSbH2, R2SbH, R3Sb RSbCl2, R2SbCl RSbCl4, R2SbCI3, R3SbCI2, R4SbCl,; R6Sb
RPbH3, R2PbH2, R3PbH, R4Pb RPbCI3, R2PbCl2, R3PbCl
380
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Область металлоорганических соединений все расширялась и в сере-
дине XX века превратилась в элементоорганическую химию, т. е. химию
органических соединений всех элементов. По существу область элементо-
органических соединений — это область углеродистых соединений эле-
ментов, необычных для классической органической химии, тогда как
классическая органическая химия — область химии элементов «органо-
генов» (С, Н, О, N, S, С1, Вг, I и фосфора в форме эфиров фосфорной
кислоты). Фосфор, таким образом, стоит на грани (условной) обеих обла-
стей органической химии. Естественно, каждый элемент входит в состав
органической молекулы в форме какой-то (или каких-то) функциональной
группы. Это дополнит наши знания о функциональных группах — про-
изводных элементов органогенов.
Большая часть элементов периодической системы — металлы. Поэтому
больше половины соединений элементоорганической химии — металло-
органические соединения. Так называются соединения, имеющие прямую
связь (ковалентную или ионную) углерода с металлом. Однако не все
металлы, а только непереходные образуют прочные алкильные соедине-
ния (исключением являются Au и Pt).
Если попытаться синтезировать алкильное соединение переходного
металла, например кобальта, реакцией обмена
CH3MgI+CoCl2 —> CH3CoC14-MgICl,
то полученное алкильное производное кобальта распадется уже при темпе-
ратуре ниже 0° С:
СН3СоС1 —> СН3 • 4-СоС! •
2СН3. —> С2Нв
За малыми исключениями устойчивых чисто алкильных и чисто ариль-
ных соединений переходных металлов не существует. Однако для пере-
ходных металлов известны другие типы соединений с прямой связью
углерода с металлом. Эти вещества близки по природе к неорганическим
комплексным соединениям. Из них давно известны, например, комплекс-
ные цианиды K4Fe(CN)e. Сюда относятся также карбонилы металлов,
такие, как Fe(CO)5, Ni(CO)4, Cr(CO)e и др. Им родственны так называемые
л-комплексы олефинов с переходными металлами. Все эти вещества будут
рассмотрены позднее, во второй половине этого раздела.
АКТИВНЫЕ МЕТАЛЛООРГАНИЧЕСКИЕ (АМО)
СОЕДИНЕНИЯ I, II И III ГРУПП
Алкильные и арильные соединения щелочных и щелочноземельных метал-
лов, Be, Mg, Zn,CdnAl разлагаются кислотами, водой и спиртами с выде-
лением углеводорода, окисляются молекулярным кислородом, реагируют
с оксосоединениями и вступают во множество реакций сдругими кислород-
ными, сернистыми и азотистыми функциональными группами, при которых
разрывается связь металла с углеродом, отличаясь во всех этих реакциях
от алкильных и арильных соединений Hg, In, TI, Si, Ge, Sn, Pb, As,
Sb, Bi. Металлоорганические соединения щелочных металлов (кроме
литийорганических) построены ионно и их реакционным началом служит
АКТИВНЫЕ МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ I, II И III ГРУПП
381
алкил-(или арил-) анион. Остальные АМО-соединения хотя и имеют
ковалентную связь С—М, но она настолько сильно поляризована (С—М),
что реакции их протекают пусть не так энергично, как у ионных, но в общем
им подобны.
Так, органические соединения щелочных металлов (кроме RLi) реаги-
руют с галоидными алкилами и с галоидными арилами:
RM4-R'X —> RR'4-МХ
Уже магнийорганические соединения вступают в эту реакцию лишь
с галоидными аллилами, бензилами и другими типами соединений с под-
вижным галоидом (а-галоид галоидзамещенных кислот, эфиров и т. п.).
АМО-соединения реагируют с СО2 по известной реакции, образуя
кислоты, по алюминий- и цинкорганические соединения дают эту реак-
цию только в более жестких условиях. Нормальная реакция АМО-соеди-
нений с карбонильными соединениями протекает по известному типу:
\ \ /ом
RM+ >С=О -----> ;(У
/ / \R
Однако она сопровождается осложнениями (подробнее они будут разо-
браны при магнийорганических соединениях), которые особенно харак-
терны для наиболее активных соединений этого типа, т. е. для ионных
или более близких к ним АМО-соединений.
В случае оксосоединений общей структуры
О
II .
>СН—С—R"
реакция может итти в нескольких направлениях, и преобладание того
или иного направления зависит от характера радикала (его электроотри-
цательности), металла, от связи радикала и металла (более или менее
ионной), от пространственных препятствий, осуществляемых радикалами
R, В', R", R"': ОМ
г I R'"
--------------------------------4 СТТ-О'
(нормальная реак- /ии
ция)----------------------------------R_R
о ом
R'\ II II R \ I
z'CII С R -f- R М (восстановление) *
“ “ олефин
ОМ
ш R'\ I
(енолизация) ” п уС=С R -}-R 11
О
О
R\. II
+ /011—С—R*
R Z
(альдольная
конденсация)
382
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Пространственные препятствия подавляют нормальную реакцию I
и мало влияют на реакции II и III. Для осуществления реакции II (вос-
становление водородом за счет R'") необходимо наличие в радикале Rw
0-водорода, способного переместиться в виде гидрид-аниона к оксосоеди-
нению с тем, чтобы R'" превратился в олефин R'" (—Н). Для реакции Щ
необходимо наличие в оксосоединении а-водорода. Натрий- и калийорга-
нические соединения реагируют преимущественно по пути III, если
только в оксосоединении есть а-водород. Направление реакций литий-
и магнпйорганических соединений зависит от характера радикала, свя-
занного с металлом. С наименьшими осложнениями (т. е. по пути I) реаги-
руют метильные, бензильные, фенильные и ацетиленильные производные;
с наибольшими (по пути II и, в зависимости от типа оксосоединения, по
пути III) — соединения с алифатическими и алициклическими вторичными
радикалами. Побочные реакции (II), так же как и основная реакция I,
проходят, по-видимому, через шестичленный реакционный комплекс
(переходное состояние), в котором может участвовать дополнительная
молекула MgX2:
R"'
4Mg—Вг
/Вг
Mg-
Вг
R. ,RW
+ MgfBr2
R ХО-MgBr
ПЕРВАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
Металлоорганические соединения щелочных металлов. 1. Галоидные
алкилы и галоидные арилы реагируют со щелочными металлами в инерт-
ных растворителях (гексан, октан, для лития также бензол), образуя
алкильные или арильные производные щелочных металлов. Однако уже
Для натрия дело осложняется реакцией Вюрца, идущей через стадию (1)
и далее по реакции (2); для калия же реакцию редко удается задержать
на стадии (1).
RCI4-2M —> RM-f-MCl (1)
RMH-RC1 —► R—R-4-MC1 (2)
Полученные так соединения RNa можно использовать для многих
(хотя далеко не для всех) типов реакций, подобных реакциям реактивов
Гриньяра (П. П. Шорыгин, Мортон). Однако большее удобство работы
с литийорганическими соединениями, а также лучшая их растворимость
ПЕРВАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
383
создают такие преимущества, что из всех органических соединений щелоч
вещества, ни в чем нерастворимые. Бепзил-
Рис. 98. Модели тетрамера метиллития.
ных металлов только они находят широкое применение в синтезах, подоб-
ных гриньяровым. Кроме того, при синтезе литийорганических соедине-
ний меньше трудностей с выбором растворителя: их можно получать как
в парафиновых углеводородах, так и в бензоле, а во многих случаях
(метиллитий, фениллитий) даже предпочтительнее в эфире.
Алкил- и а рил натрий и соответствующие соединения калия твердые,
построенные ионно бесцветные
натрий и трифенилметилнат-
рий растворимы в эфире, ок-
рашены в вишнево-красный
цвет (цвет аниона), растворы
их электропроводны. Метил-
литий, этиллитий и арилли-
тий — твердые вещества, дру-
гие алкиллитии — жидкости.
Все они растворимы в индиф-
ферентных растворителях.
Бензиллитпй и трифенилме-
тпллитип подобны соответст-
вующим соединениям натрия.
Соединения калия, рубидия
и цезия изучены гораздо меньше. Алкиллитии в растворах ассоцииро-
ваны (степень их полимеризации от 2 до 6), метиллитий — тетрамер
(рис. 98), в котором связь осуществляется уже не парами электронов,
а посредством многоцентровых орбиталей. Углероды в (CH3Li)4 пятикоор-
динационны. Такого рода «мостиковые» метилы еще встретятся нам при
рассмотрении алюминийорганических соединений, а мостиковые водо-
роды — при рассмотрении гидридов бора. Все алкильные и арильные
соединения щелочных металлов самовоспламеняются на воздухе, бурно
разлагаются спиртами, водой и кислотами.
Кроме описанного выше синтеза из галоидпроизводных и металла
простейшие органические соединения щелочных металлов могут быть
получены следующими способами.
2. Действием металла на ртутноорганические соединения:
R2Hg4-2M —>2RM-f-Hg
3. Действием литийалкилов на арил- или алкилгалогениды:
RX-f-R'Li —> RLi-yR'X
4. Прямым металлированием (т. е. заменой водорода па металл) при
действии алкильных производных щелочных металлов на ароматические
углеводороды (реакция П. П. Шорыгина, для лития широко развитая
Гилманом) и вообще на вещества с подвижным водородом. Например:
CH3Na + CeHe —> CeII5Na + СН4
CH3Na + CeH5CH3 —> CeH5CH2Na + CH4
CeH5Na+C«H6CH3 —► CeH5CH2Na 4-СвНв
384
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
С4Нви+<^~^>—ОСНз > '\=/~ОСН3+С4Н10
Li
С4Н,Ы + |
сн2
Реакция металлирования протекает так, что металл вступает на место
более протонизируемого водорода. Таким образом, эта реакция дает
возможность построить шкалу «кислотности» углеводородов, которую
нельзя измерить обычными физико-химическими методами. Так, напри-
мер, из реакции Шорыгина ясно, что водороды метильной группы толуола
«кислее», чем водороды бензола.
Для наиболее «кислых» углеводородов, таких, как ацетилен, цикло-
пентадиен, инден и флуорен, этого рода реакции проходят особенно легко:
HC=CH + 2CeH5Na ---> NaC=CNa4-2CeHe
ГЛ + C4H0Li ------->- ®Li++ С4Н10
сн2
Металлирование этих углеводородов можно производить, даже не
применяя металлоорганического соединения, а действуя раствором щелоч-
ного металла в жидком аммиаке или амидом натрия
HC=CH + 2NaNH3 —-+ NaC=CNaNH3 + H2
HC=CH + NaNH2 —► NaC=CH + NH3
или (в случае флуорена и индена) едким кали (если отгонять воду).
5. Литий- и натрийорганические соединения вступают в следующую
реакцию обмена с бромарилами и ароматическими бромзамещенными
гетероциклами:
ArBr + RM > ArM + RBr
Этой реакцией весьма часто пользуются для синтеза литиевых и натри-
евых ароматических и гетероциклических производных с Li (или Na)
в определенном положении.
6. П. П. Шорыгин нашел, что при нагревании натрий расщепляет
ароматические простые эфиры по уравнению:
CeH5OCeH5 + 2Na —> CeH5Na + GeH5ONa
Особенно легко рвется связь в бензиловых эфирах. Такое расщепление
Циглер широко использовал для синтеза калийорганических соединений
бензильного типа:
СвН5С(СН3)2—ОСвН5+2К —> СвН5С(СН3)2К+4-СвН5ОК
Наконец, есть еще два важных пути образования металлоорганиче-
ских соединений щелочных металлов специфической структуры.
ПЕРИЛЛ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
385
7. Присоединение металла к углеводородам с сопряженными л-свя-
зями, в том числе к ароматическим. Оно начинается, по-видимому, всегда
с того, что углеводород принимает электрон щелочного металла (на низ-
шую из незанятых орбиталей) и образуется ионная пара [С„Н2 j|_M+.
Бензол присоединяет таким путем только калий:
СвНв + К—► [СвНвГК+
Нафталин дает аддукт уже с натрием
Na+
Далее может произойти новое присоединение металла.
Антрацен образует аддукт уже и с натрием и с литием, присоединяя
два атома металла:
н м
j+2M
Н М
Такие нерастворимые окрашенные соединения дают при гидролизе
дигидр оа р оматиче ские у гл ев од ор оды.
Основополагающие исследования по присоединению щелочных металлов
к непредельным и ароматическим соединениям были проведены Шленком.
Примеры поведения олефиновых связей, сопряженных с аромати-
ческими ядрами, при действии щелочных металлов:
CeH5CH=CHCeH5 + 2Na —> CeH5—СН-СН-СвН5
Na Na
2(CeH5)2C=CH2 + 2Na —> (Св116)2С-СНгСН2-С(СвН5)2
lia Na
Эти примеры иллюстрируют тот факт, что открытые л,л-сопряженные
системы присоединяют металл к тем углеродным атомам сопряженной
системы, которые несут ароматические заместители. Если лишь один атом
открытой непредельной цепи несет ароматические заместители, атом
металла присоединяется к нему и молекула димеризуется.
По сути дела речь идет о передаче атомами натрия электронов атомам
углерода и именно тем, которые легче рассредоточивают захваченные
электроны, например по ароматическим ядрам или по сопряженной
системе л-связей. Простые олефины не присоединяют натрий. В приведен-
ных ранее и последующих формулах продуктов присоединения щелочных
металлов к непредельным углеводородам связь щелочного металла с угле-
родом правильнее было бы изображать как электровалентную, например,
так:
CeH6-CH—СН—СвНБ
Na+ Na+
25 Заказ 672
386
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Чисто алифатические 1,3-диеновые системы при действии щелочных
металлов полимеризуются (на этом основан, например, синтез натрий-
бутадиенового каучука).
8. Присоединение металлоорганических соединении щелочных метал-
лов по сопряженным двойным связям. Алкильные соединения (часто
и арильные) щелочных металлов присоединяются к олефиновым двойным
связям, сопряженным с ароматическим ядром (1), к фульвенам (2) и дие-
нам (3):
(CeH5)2C=CH2+C4H9Li —► (С6Н5)2С—СН2С4Н9 (1)
Ы
ч /СНз
/=С -J-CeHsLi —►
Z ХСН3
/СН3
\7-C-CH3
' I хсвн5
Li
СНз СНз
I I /Н
СН2=С—CH=CH2-f-RLi > RCH2-C=C<
xCH2Li
(3)
II
В последнем случае металлоорганический продукт первичного присо-
единения далее способен присоединяться к следующей молекуле диена
и т. д., так что происходит полимеризация. При применении лптийорга-
нических соединений, например, для полимеризации изопрена процесс
идет стереоспецифично, и образуется tyizc-полиизопрен, идентичный
натуральному каучуку (кн. I, стр. 302):
СНз
СН3\ ХН |
,С=С< 4-СН2=С—сн=сп2 -->
R-CH2/ xCH2Li
СНз Н
С=С СН2 Н
r-ch2 \н2
СНз СН2—Li
III
СНз
Ш + СН2=С-СН=СН2 И т. д.
“ СНз Н
С=С СН2 Н
—СН2 ХчСН2ХС=С/
СН2-----Li
-1Л
Натрийизопреновый каучук не стереорегулярен. Кроме того, и он,
и натрийбутадиеновый каучук образуются как путем 1,4-, так и путем
1,2-присоединения, так что не только стереохимия, но и структура натрие-
вых каучуков имеет нерегулярный характер.
9. Отличительная черта литий- и натрийорганических соединений —
способность алкилироваться галоидными алкилами. Даже непрочные
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
387
аддукты двух атомов лития и натрия к ароматическим углеводородам
вступают в эту реакцию, образуя диалкилдигидроароматические углево-
дороды :
Гриньяров реактив удовлетворительно реагирует этим путем лишь
с бензилгалогенидами и галогенидами аллильного типа, в последнем
случае часто с аллильным перемещением двойной связи (Прево). В осталь-
ном реакции литийоргаппческих соединений и области их применения
в синтезе очень похожи па описываемые далее реакции магнийоргапиче-
ских соединений. То же можно сказать о патрпйоргаиических соединениях,
если говорить о типе реакций, а не о частных отклонениях. Однако натрий-
оргапические соединения имеют ряд неудобств. Они нерастворимы в индиф-
ферентных растворителях (образуют ионные пары); реагируют энергич-
нее, чем литий- и магнппоргапнческие соединения, и часто при их приме-
нении особенно быстро проходят такие реакции, как енолизация или
конденсация образовавшихся енолятов (стр. 395), т. е. преобладают
побочные, а не целевые реакции. Еще в большей степени это относится
к калийоргаиическим соединениям.
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
Магнийорганическпе соединения. Известны два типа магнийоргаииче-
ских соединений — R2Mg и RMgX (где X — галоид). Полные магпийоргани-
ческие соединения R2Mg можно получить через ртутнооргапические
(действием металлического магния) или из соединений типа RMgX. Эти
последние (гриньяровы реактивы) получают наиболее часто действием
галоидных алкилов и других галоидных углеводородов па металлический
магний в растворе диэтилового эфира, который существенно ускоряет
реакцию:
RX-f-Mg —> RMgX
Фактически комплексно связанный эфир входит в состав магиийорга-
нического соединения. Кроме того, по эбулиоскопическим определениям
А. П. Терентьева грипьяров реактив имеет удвоенный молекулярный вес.
При электролизе его эфирного раствора на катоде отлагается магний.
Все это дало основание Терентьеву предложить для реактива Гриньяра
такую структуру:
Mg2+
/С21Г5 2-
С1 о/
R : / ХС2Н6
>м<
Т>/ ' С2П6
С2Нб_
25*
388
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Эшби и Уокер (1967 г.) привели доводы в пользу того, что ассоциация
гриньярового реактива осуществляется галоидными мостами (так же, как
например, в хлорном железе Fe2Cle)
X
RMg/ MgR
Т ' /j\
(С2Щ)2О X О(С2Н5)2
Однако такая структура не объясняет факта электропроводности и элек-
тролиза эфирных растворов гриньярова реактива с переносом Mg2+ на
катод, если не учитывать установленного Шленком равновесия. Шленк
нашел, что в гриньяровом реактиве находятся в равновесии R2Mg, RMgX
nMgX2, связанные с эфиром. Добавлением диоксана (кн. 1, стр. 127) можно
осадить два последних соединения, причем в растворе остается только
R2Mg (равновесие устанавливается довольно медленно). Несмотря на
более сложную структуру гриньяровых реактивов, для описания всех
их реакций вполне удовлетворяет формула RMgX, которую обычно
и применяют. Вообще же обсуждение возможной структуры гриньярова
реактива нельзя считать завершенным.
В последнее время в качестве еще более благоприятной среды для
проведения гриньяровой реакции, чем этиловый эфир, предложен тетра-
гидрофуран, в котором магнийорганические соединения легко образуются
из таких галоидпроизводных, которые не реагируют в эфире. Например,
в тетрагидрофуране из СН2 = СНВг образуется CH2=CHMgBr (Норман).
Энергичнее всего реагируют с магнием алкилиодиды, но лучшие выходы
получаются с алкилбромидами и особенно с алкилхлоридами. Аромати-
ческие гриньяровы реактивы готовят обычно из арилиодидов или арил-
бромидов, так как арилхлориды вступают в реакцию только при темпера-
турах выше 100° С.
Галоидные аллилы, начиная с СН2 = СН—СН2С1, при взаимодействии
с магнием очень легко дают диаллил в результате реакции Вюрца. Все
же их удается ввести в гриньярову реакцию даже в эфире, если к магнию
медленно на холоду прикапывать галоидный аллил в эфирном растворе.
Из дигалоидалканов димагнийгалогениды образуются только из галоид-
производных с галоидами, удаленными друг от друга не менее чем на
четыре углеродных атома:
ClCH2CH2CH2CH2Cl + 2Mg —> ClMgCH2CH2CH2CH2MgCl
В ароматическом ряду м- и ге-дибромбензолы образуют CeH4(MgBr)2;
так же реагирует 1,4-дибромнафталин. Естественно, что галоидпроизвод-
ные жирного, алициклического и ароматического рядов, образующие
гриньяровы реактивы, не должны иметь реагирующих с реактивом замес-
тителей, таких, например, как —, \С=О, —С<^ ,—CN и Др-
\Н / XOR
В молекуле галоидпроизводного могут присутствовать простые эфир-
ные и третичные аминные группы, но не при том же углероде, с которым
соединен галоид, и не при соседнем с ним атоме углерода. Единственным
исключением являются сложные эфиры а-галоидкарбоновых кислот,
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
389
которые хотя и не дают изолируемых гриньяровых реактивов, но вступают
в реакцию с магнием в присутствии, например, карбонильных соединений
по реакции С. Н. Реформатского (ср. «Цинкорганические соединения»,
х;тр. 400):
R" о
Н\ I II
4 С=О+ Вг—СН—С—OR
R'Z
Mg;
затем HtO ,
ОН R’ О
R\ I I II
XC—CH—C—OR
R'/
Упомянем также о получивших большое значение перфторалкильных
гриньяровых реактивах. Йодиды общей структуры CnF2n+1I реагируют
с магнием на холоду в эфире, а лучше в тетрагидрофуране, образуя
C„F2n+1MgI. Труднее всего эта реакция осуществляется с CF3I (—72° С,
тетрагидропиран). Легко получаются CF2 = CFMgI и C6F5MgI.
Метод обмена галоида на металл при действии металлоорганического
соединения, важный в синтезе литийорганических соединений, в гринья-
ровой реакции значения не имеет. Зато очень важен метод замещения
активного водорода на MgX при действии гриньярова реактива. Следующие
реакции, с которыми мы уже встречались ранее, иллюстрируют получение
таким «косвенным» путем важных магнийорганических реагентов:
HC=CH + 2RMgX > XMgC=CMgX+2RH
реактив Иоцича
IIC=CH + RMgX ---> HC=CMgX-|-RH
(избыток
под
давлением)
Реактив Иоцича широко использовался в синтезах Ю. С. Залькиндом
и рядом других советских химиков.
-I- RMgX ----->- MgX + RH
CH2
циклопентадиен
Все магнийоргапические соединения, полученные замещением подвиж-
ного водорода при углероде, несколько менее активны, чем обычные
-алифатические и ароматические гриньяровы реактивы.
390
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Не менее важны магнийорганическпе производные пиррола, индола
и других азотистых пятичленных гетероциклов, имеющие связь N MgX;
|| || + RMgX — -> ||^ ^+RH
NH N MgX
\7 || 11 +RMgX - \/\ / NH -> 11+ RH 1 MgX
Они обычно реагируют с переносом реакционного центра по а-углероду
(пиррилмагнийгалогенид, стр. 369) или по ^-углероду (инденилмагний-
галогенид, стр. 244).
Подвижен а-водород также в фенилуксусном эфире или в фенилуксус-
ной кислоте и ее солях. Действуя гриньяровым реактивом на эти соедине-
ния, болгарский ученый Иванов получил и широко использовал в синтезе
своеобразные магнийорганическпе производные:
CeH5-CH2-C-OC2H5-|-CH3MgBr —> СбН5-СГ1-С-ОС2Н5 + СЩ
II I II
О BrMg О
С6Н5—СН2—с— OH + 2CH3MgBr —> CeH5—СН—С—OMgBr+ 2СН<
II I II
О BrMg О
Реакции гриньяровых реактивов. 1. Соединения
с «активным» водородом, типа Н—А, т. е. все кислоты, вода, спирты
и другие гидроксильные производные, аммиак, первичные и вторичные
амины и амиды и пр., реагируют с реактивом Гриньяра по схеме:
RMgCl + H-A —> R—H + A-MgCl
Например:
RMgCl+HOH —> RH-pHOMgCl
RMgCl + НОСНз —> RH + CH3OMgCl
RMgCl + RNH2 —> RH + RNHMgCl
Реакция положена в основу метода определения активного водорода
по количеству выделенного метана из CH3MgI (метод Церевитинова —
Чугаева, значительно усовершенствованный А. П. Терентьевым).
2. Галоиды, кислород, сера разрывают в гриньяровом реактиве
связь С—Mg:
RMgX Т Х2 —> RX-f-MgX2
RMgX+i/2O2 —> ROMgX ROH + MgZ°H
XX
RMgX+S —► RSMgX -5^ RSH+MgX2
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
391
3. Оксосоединения присоединяют гриньяров реактив (кн. I, стр. 101):
н.о ОН
RMgCl + CH2O > RCH2OMgCl RCH2OH-|-Mg<
Cl
/О R н о R- /Он
RMgCl-f-R'—CZ > /CHOMgCl >CHOH + MgZ
'll R' R'/ \q1
R R
R \ \ н.о \ /ОН
RMgCl-f- >C=O —> R' — COMgCl -U- R'—C0II4-Mg<
R'z Z Z Cl
R" R"
Сам Гриньяр приписывал аддуктам оксосоединений изображенные
здесь формулы, однако впоследствии (Гесс, 1921 г.) были приведены
доводы в пользу того, что эти аддукты имеют строение молекулярных
соединений типа
R\ /X
>СО-• -Mg(
R'/ XR
А. Н. Несмеянов и В. А. Сазонова доказали правильность точки зре-
ния Гриньяра, установив тождество веществ, полученных тремя разными
путями:
С2Н5 „ С2Н5 С2Нб
R'\ I I \ I П R\ I
>С=О -|-RMgX • О —> R — COMgX-O <— >C=O + R"MgX • О
R*/ I R"Z I R/ I
C2H5 C2H5 C2H6
1’П
R с2н5
\ I
R'—C—OH + CH3MgX • О
R" C2H5
4. Непредельные кетоны с сопряженной системой л-связей
С = С—С=О
присоединяют гриньяров реактив в 1,4-положение, что после гидролиза
приводит к образованию предельного кетона:
R' R'
I R\ I +н2о
RCH=CH—C=O + R"MgX —> >СН—CH=COMgX -----^~он*
R"Z -Mg/
хх
R'
R\ 1
—► >CH—CH2—c=o
R"/
392
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Обычно эта реакция идет наряду с 1,2-присоединением, дающим
непредельный третичный спирт:
R' R' R'
RCH=CH—C=O + R"MgX —► RCH = CH—COMgX +Н'°;он^ RCH = CH— C-OR
1 -Mg< I
R" R"
Как выяснил Колер, чем более объемист R", тем более он склонен
к 1,4-присоединению. То же относится и к радикалу R . Наиболее спосо-
бен к нормальному 1,2-присоединению CH3MgX. Объемистость радикала
R действует в обратном направлении. Из сказанного ясно, что альдегиды
(R' = Н) в большей степени, чем кетоны, способны к реакциям 1,2-при-
соединения.
Предполагается (Лутц и Ревели), что 1,4-присоединение идет через
шестицентровое переходное состояние:
Н
I
С
r-c^ch-r
cD-
Mg-x
Енолятное строение продукта 1,4-присоединения было доказано
Колером посредством озонирования.
В высшей степени интересны и неожиданны открытые Колером и иссле-
дованные также Кельшем и Фьюсоном 1,4-присоединения гриньяровых
реактивов к ароматическим кетонам с сопряженной системой С=С—С=0.
При этом радикал R гриньярова реактива внедряется в ароматическое
ядро:
С6Н5
(СвНа)2С=С—С—^> + CeH5MgBr --->
о
с6н5
—> (С6Н5)2С=С-С=/~\
BrMgO / \gH5
Свщ
I — \
(С6Н5)2С=С-С----< >
II /\
0 н свн5
|-2Н
СвЩ
(CeH5)2C=C—с—
II
о
С.нв
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
393
Очевидно, что такое направление реакции вызвано пространствен-
ными препятствиями присоединению но карбонильной двойной связи
и доступностью л-системы бензольного ядра, сопряженного с карбо-
нилом.
Бензантрон I реагирует с гриньяровым реактивом также в 1,4-поло-
жение:
I II I 11+RMgX
II /\
О Н R
III
IV
При этом образующийся промежуточный продукт III не удалось
выделить. Очевидно, отдавая водород, он ароматизуется в продукт IV.
В случае настолько пространственно загроможденных ароматических
кетонов, что орто-положение бензольного ядра недоступно, радикал
гриньярова реактива вступает в пара-положение, т. е. происходит 1,6-при-
соединение RMgX (Фьюсон):
Н3С СНз
„ /
Вг-/ -------С—CeHs + mpem-CJIgMgCl
/ \ 11
Н3С СНз°
При взаимодействии реактивов Гриньяра с оксосоединениями, осо-
бенно если радикал гриньярова реактива вторичный или третичный,
идут аномальные реакции, иногда преобладающие над нормальной.
394
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Аномальными являются реакции 1, 2, 3, 4:
1. Восстановление, приводящее после гидролиза к образованию
из альдегида первичного спирта, а из кетона не третичного, а вторич-
ного спирта:
R' R\
\C=O + RCH—СНз —> СН—OMgXH-R—СН=СН2
R"/ | R"z
MgX
Эта аномалия исследовалась многими учеными. Наиболее вероятное
объяснение дали Мошер и Лякомб. Оно сводится к тому, что реакция
идет через шестицентровое переходное состояние с гидридным перемеще-
нием (т. е. перемещением водорода с электронной парой ковалентной
связи) из [3-положения гриньярова реактива к карбонильному углероду:
R' Н
*- +
R OMg’X
ROM
СН.,
Доводом в пользу такой схемы служит действие оптически активного
гриньярова реактива /)-2-метилбутилмагния на пинаколин:
НдС „ СНд
(CH3)3c>,Q'4>c -*С2Н5
о) £сн2
Mg-
I
CI
(сн3)зсч ZCH3
>- * с
CIMgO н
7с2н5
+ с
II
сн2
При циклическом переходном состоянии двусторонняя атака концами Н
и MgX молекулы гриньярова реактива на плоскую молекулу кетона
совершается так, чтобы более громоздкие заместители — третичный
бутил и этил не оказались близко друг к другу по одну сторону цикла-
Поэтому гидридное перемещение (переход Н:) ведет к «передаче» (частич-
ной) оптической активности от гриньярова реактива полученному вто-
ричному спирту.
При этом после гидролиза получается правовращающий пинаколино-
вый спирт.
2. Дегидратация третичного спирта, полученного из кетона, с образо-
ванием непредельного углеводорода:
R'\
R„>C=o+RCH2MgX —>
R'\
>С—CH2R
R"' |
OMgX
R\ ZOH
>C=CHR + Mg<
R"/ \X
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
395
3. Енолизация при наличии пространственных затруднений, например,
если R = СвН2(СН3)3 (реакция идет только с кетонами):
R—С—CH2R' + R"MgX •—> R"1I + R—C=CHR'
II I
О OMgX
4. Связанная с енолизацией конденсация по альдольному или, чаще,
кротоновому типу:
R' R R' R
II II
RC=CH + C=CH2R' -> RC—СН—С—CH2R'
I II II I
OMgX О о OMgX
R' R
R' R
RC—СН—С—СН2Н'
II I
О OMgX
+н,0
/ОН
-Mg<
хх
/ОН
-Mg4
хх
I I
RC—СН—С—CH2R
II I
О он
R' R
RC—i=C—CH2R'
Возвращаемся к нормальным синтезам посредством гриньярова
реактива.
5. С реактивом Гриньяра аналогично кетонам реагируют нитрилы:
R\ тт Q
RMgGl+ R'C=N ----> yC = NMgCl ----
R'z -Mg/
XC)
R\ но R \
--->• ;C=NH > )C=O + NH3
R'z Rz/
Это наиболее важный метод синтеза кетонов.
6. При взаимодействии гриньярова реактива с хлорангидридами
кислот при охлаждении также получаются кетоны:
RMgCl-j-Cl—С—R' > R—С— R'-|-MgCl2
II II
О о
При избытке RMgCl в несколько более жестких условиях образуется
третичный спирт.
7. Сложные эфиры реагируют с гриньяровым реактивом с образова-
нием третичных спиртов:
R'
R'—C-OR" + 2RMgCl —> R—С—OMgCl + R''OMgCl
II /
О R
R' R'
\ и о \ / 011
R—С—OMgCl R—С—ОН 4-Mg/
/ , ХС1
R R
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Эту реакцию редко можно задержать на первой стадии: одна Молекула
гриньярова реактива с одной молекулой сложного эфира даст оксо-
соединение (по типу, аналогичному приведенному). Такая разновидность
реакции имеет значение только для эфиров муравьиной кислоты. При силь-
ном охлаждении они образуют с гриньяровым реактивом альдегиды:
OR' /Н
RMgCl 4-НС 4 —> RC< -j-R'OMgCl
XO XO
Реакции 5, 6, 7 начинаются с присоединения реактива Гриньяра
по кратной связи С = О или C=N (как и для альдегидов или кетонов).
В случае сложных эфиров вторая стадия состоит в обмене алкоксильного
остатка на радикал гриньярова реактива:
R"
R4 zOMgX /ОС2Н5
>С< 4- R"MgX —>• R-C-OMgX + Mgz
R'z XOC2H5 / ^X
R'
Возможно, что реакция на обеих стадиях проходит с,участием 2R"MgX
через шестицентровое переходное состояние (такой же механизм можно
предполагать и для реакций магнийорганических соединений с кетонами):
X Mg—х
R'O^H + „ -->
0
Mg-
X
R" r"
R~C xMg—X
XMgO i +yz —>
R4 A
, A ~ Mg—X
RO fc
Mg-
X
R\ A
R—C'v-y Mg—X
Mg-
A + R Mg-X
RO I
OMg-X
r"
r"—C — OMg-X + R^Mg-X
RZ +
R'OMg-X
В приведенных ниже примерах происходит прямой обмен алкоксила:
RMgX -j- (R'0)4C ---------------> RC(OR')3 -j- R'OMgCl
ортоугольный ортоэфир
эфнр кислоты R.COOH
RMgX 4- RC(OR')3 ->. R2C(OR')2 4~ R'OMgX
кеталь кетона R4CO
Н
RMgCl 4- HC(OR')3 -—► R—С—OR'R'OMgX
fclR'
ацеталь альдегида
Rch-о
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
397
Последняя реакция — главный метод синтеза альдегидов при помощи
магнийорганических соединений (А. Е. Чичибабин); ацеталь легко гидро-
лизуется в кислой среде в альдегид.
8. С окисью этилена гриньяров реактив образует первичный спирт
(происходит надстраивание цепи на два звена):
RMgX + СН2—СН2 > RCH2-CH2OMgX > RCH2-C1I2OH
V
С окисями типа R—СН—СН2 образуются вторичные спирты:
R'—СН—СН2 + RMgX > R'—СН—CH2R —* R'-CH-CH2R
\ / I I
О OMgX он
9. Чрезвычайно важной является реакция магнийорганических со-
единений с газообразной или твердой СО2, приводящая к синтезу кислот:
RMgCl + СО2 --> R— C-OMgCl — * R—С-ОН + MgCl2
10. С окисью азота гриньяровы реактивы реагируют по схеме:
NO „ о /NO .
RMgX +2NO —> R—N< R—N<
xOMgX \ОН
После гидролиза образуется алкил- или арилнитрозогидроксиламин.
11. Хлористый нитрозил дает с реактивом Гриньяра нитрозосоеди-
нения:
RMgCl + CINO -> R—NO + MgCl2
12. С хлорамином (Колеман), а лучше с О-эфиром гидроксиламина
(К. А. Кочешков и Н. И. Шевердина) образуются первичные амины:
RMgCl + C1NH2 —> RNH2 -j- MgCl2
RMgCl + NH2-OCH2CeH5 •—> RNH2-j- C6H5CH2OMgCl
Приведенные уравнения упрощены. На деле действуют избытком
гриньярова реактива, так как сначала идет замещение на MgX подвиж-
ного водорода аминогруппы.
13. Из шиффовых оснований и гриньярова реактива получают вто-
ричные амины (Буш):
/Аг гт q /Аг
ArN=CHAr + RMgCl > ArN— СН< -L-> ArN Н—СН<
| \r xR
MgCl
398
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
14. Алкил- или арилазиды с грипьяровым реактивом образуют трц-
азены (т. е. диазоаминосоединепия):
R н о
R—N = N = N-j-R'MgX > R—N==N—N< —*R-N=N-NH-R'
MgX
15. Нитрозосоединения превращаются в N-диалкил (диарил-)гидр-
оксил амины:
R но R\
R—N=O + R'MgX > OMgX ;N-OH
R'/ R'/
16. Обменной реакцией соли металла с гриньяровым реактивом полу-
чаются металлоорганические соединения (Пфейфер)
4RMgCl SnCU —> R4S11 -j- 4MgCl2
или
2RMgCl + SnCl4 -> R2SnCl2 + 2MgC]2
Таким путем наиболее просто получаются полные или смешанные
металлоорганические соединения всех непереходных металлов (и органи-
ческие,1 производные их металлоидных аналогов), за исключением металло-
органических соединений щелочных и щелочноземельных металлов.
Из алкильных и арильных соединений переходных металлов так могут
быть получены и изолированы производные Pt (IV), Au (III) и непрочные
соединения Си (I) и Ag.
Хараш?показал, что при добавлении малых количеств соли переход-
ного металла, например Со(П) (а также NiCl2, CuCl2 и др.), реакция
из гетеролитической становится гомолитической из-за первоначального
образования RCoCl, гомолизующегося на радикал и радикалообразный
галогенид переходного металла:
RCoCl -> R- + -CoCl
Реакцию с бензофеноном, которая в этих условиях дает только бенз-
пинакон, можно, например, изобразить так:
CeH5MgX + СоХ2 -—> С6Н5- + • СоХ + MgX2
2С6Н5СОС6Н5 + 2 -СоХ -> (СвН5)2С-С(СвН5)2 х*
। । «Ui 2
ХСоО ОСоХ
--> (С6Н5)2С-С(СбН5)2 — * (СвН5)2С-С(СбН5)
II II
XMgO OMgX но он
Разработано великое множество других типов реакций гриньярова
реактива с разнообразными азотистыми, сернистыми и т. п. функциональ-
ными группами, с комбинацией различных кислородных функциональных
групп — сотни различных реакций. Вероятно, можно без ошибки ска-
зать, что синтезы посредством магнийорганических соединений — самый
многообразный и широко применимый метод построения молекул в орга-
нической химии. Этот метод возник в 1900 г. в результате модификации
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
399
и широкого развития синтетического способа применения цинкоргани-
ческих соединении, найденного А. М. Бутлеровым (Казань) и разрабо-
танного его учениками А. М. Зайцевым, Е. Е. Вагнером, С. Н. Реформат-
ским и ДР-
Цинкорганические соединения. Действуя подпетыми алкилами на цин-
ковую стружку, получают RZnI, а при сухой перегонке этого типа солей
отгоняются жидкие, воспламеняющиеся на воздухе соединения R2Zn;
RI -|- Zn —> RZnI
2RZnI —> R2Zn + Znl2
Они, главным образом, и применялись в синтезе.
Реакции цинкорганических соединений, вполне аналогичные реакциям
магнийорганпческих соединений, дали возможность синтезировать пер-
вичные и вторичные спирты. Третичные спирты, существование которых
было предсказано Бутлеровым на основании теории строения, были им
открыты именно посредством синтеза с цинкорганическими соединениями:
R
СН3-С=О + ZnR2 --> СНз—С—OZnCl — -*
I I
Cl R
R
--> СНз-С-ОН + Zn(OII)Cl (A. M. Бутлеров)
R
R'
R\ \ н о
xC=0 + ZnR' ---> R-C-OZnR'
Rz Z
R
R\
--> R—C—Oil + Zn(OH)» + R'H (A. M. Зайцев)
R/
R \ t, n
R—C=0 + ZnR; ---> ДЗ-OZnR'
I r/|
H H
R\
--> R-^C—OH + Zn(OH)2 + R'H (E. E. Вагнер)
H\ R\ HO
)c=o + ZnR2 --> H—C-OZnR —
hz Н/
rx
--> H—>C—OH + Zn(OH)2 + RH (В. E. Тищенко)
Н/
Цинкорганические соединения, однако, более инертны, чем магний-
оргаиические, и не реагируют с углекислым газом в обычных условиях.
400
ЭЛЕ ME НТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
После открытия гриньяровой реакции неудобные в обращении диал-
килцинки почти не используются. Некоторую сферу применения сохра-
нили еще менее реакционноспособные соединения типа RZnX. Например,
действуя RZnX на хлорангидриды кислот, удается получить кетоны
с лучшим результатом, чем реакцией Гриньяра с теми же хлорангидри-
дамп, так как полученный кетон реагирует далее уже медленно. Кроме
того, с широким применением цинкорганических соединений, получаемых
по реакции Реформатского
R-сн— С— OR' + Zn > R—СН—С—OR
I II I II
Вг О BrZn О
мы уже встречались на примере синтеза ^-оксикислот (кн. 1, стр. 399).
Такого типа соединения {реактив Реформатского) в индивидуальном
виде выделил К. А. Кочешков.
К воде, кислотам, спиртам и аминам цинкорганические соединения
относятся так же, как и магнийорганические:
R2Zn + 2HA —> ZnA24-2RH
Кадмийорганические соединения по свойствам близки к цинкоргани-
ческим. При синтезе кетонов из хлорангидридов карбоновых кислот
часто добавляют хлористый кадмий к гриньярову реактиву для перевода
его в RCdCl, затем действуют хлорангидридом, что улучшает выход
кетона.
Металлоорганические соединения кальция, стронция и бария до сих пор
очень мало изучены. Они с трудом и в малом количестве получаются
при действии иодистых алкилов на металлическую стружку. Эти соедине-
ния еще более активны, чем магнийорганические.
Бериллийорганические соединения плохо получаются взаимодействием
металла с галоидными алкилами. Их лучше синтезировать через гринья-
ров реактив. Известны оба типа соединений — RBeX и R2Be. По свой-
ствам они ближе к алюминийорганическим соединениям, чем к магний-
органическим.
Ртутноорганические соединения. По способам получения и свойствам
ртутноорганические соединения резко отличаются от соединений осталь-
ных.Гэлементов II группы. Они ближе к металлоорганическим соединениям
Sn, Pb, Sb, Bi, тогда как соединения щелочноземельных металлов —
Be, Mg, Zn, Cd — ближе к металлоорганическим соединениям щелочных
металлов.
Ртутноорганические соединения бывают полные R2Hg и смешанные
RHgCl (ртутноорганические соли, где вместо G1 может быть любой анион).
Их можно получить общей реакцией синтеза металлоорганических соеди-
нений — действием гриньярова реактива на соответствующее количество
галогенида ртути:
RMgCl + HgCl2 --► RHgCl + MgCl2
2RMgCl + HgCl2 -► R2Hg + 2MgCla
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
401
Соединения RHgX превращаются в R2Hg реакцией симметризации,
вызываемой действием ряда веществ, способных связать соль ртути в комп-
лекс или иное неионпое соединение:
2RHgX + 4KCN ---> R2Hg + K2[Hg(CN)4p- + 2КХ
2RJIgX + 4KI --> H2Ug + K2[HgI4]2-+ 2KX
2RHgX + 2NH3----> Rillg + [Hg(NH3)212+ 2X‘
Обратиое превращение в RHgX совершается при нагревании раствора
(например, в спирте) эквивалентных количеств R2Hg и органической
или неорганической соли ртути:
R2Hg + HgX2 —> 2RHgX
Реакция практически идет до конца, быстрее, если R—арил или винил,
и медленнее, если R — алкил.
Известны и несимметрические полные ртутноорганические соединения
RHgR'. Их получают по Харашу действием гриньярова реактива на ртут-
ноорганическне соли:
RHgX + R'MgX ---> RHgR' + MgX2
Вместо гриньярова реактива можно брать литийорганическое соединение
или лучше (по А. Н. Несмеянову, К. А. Кочешкову и Р. X. Фрейдлиной)
действовать на RHgX в щелочном растворе R'B(OH)2, R'SnO(OH),
R'As(0H)2 и другими имеющими кислотную функцию металлоорганиче-
скими соединениями непереходных элементов. Например:
RHgX + R'B(OH)2+ ОН" ----> RHgR' + В(ОН)3+ X’
Несимметрические ртутноорганические соединения в общем неустой-
чивы и при стоянии, быстрее при нагревании, симметризуются:
2RHgR' —> R2Hg + R'Hg
Устойчивость их тем выше, чем больше разница электроотрицательно-
стей радикалов, так что наименее устойчив тип ArHgAr' с близкими ари-
лами, а наиболее устойчивы, не отличаясь от ртутнооргапических солей,
вещества RHgCN. Легкость симметризации RHgR' помогает попять
открытый О. А. Реутовым изумительный факт быстрого обмена атомами
ртути между молекулами диарилртути с обычной ртутью и с ее радиоак-
тивным изотопом:
Ат\ /Аг' Аг\ /Аг
4Hg-|-*Hg< ----> Mlg + *ilg<f
Аг/ 'Ar' Arz/ хАг
Подобного рода обмен может обеспечивать самопроизвольную симме-
тризацию несимметрических ртутнооргапических соединений RHgR'.
Отметим здесь же не менее поразительный факт, открытый также
О. А. Реутовым, — быстрый обмен металла диарилртути (а также дп-
хлорвинилртути) с металлической ртутью, что также было установлено
опытами с радиоактивной металлической ртутью.
26 заказ 672
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
402 ______________________________—------------- ------------------
В отличие от большинства других металлоорганических соединений,
для которых синтез при помощи гриньярова реактив.» - глав и, ртутно-
органические соединения можно синтезировать рядом других методов.
Приводим методы получения соединений жирного ряда.
1. Прямое замещение достаточно подвижного водорода на остаток HgX
при действии HgX2 па YCH3 или Y СН2 Z, где Л и — карбонилы,
карбоксилы или питрогруппа, а X ацетатный остаток.
О
/ О \ II
II /С-ОС2Н5
2 \СН3—С—О/2Hg-}~ Н2С
С-ОС2Н5
о о
II II
СНз-С-OHg. ,С-ОС2Н5
>С +2СН3СООН
СНз-С-OHg/ ЧС-ОС2Н5
3 /СН3-С— О\ Hg + СНз—с—ОП —> , СН3—С—Hg\ С—С—ОН < ЗСН3СООН
II II II II
\ О /2 О \ о /з о
Взаимодействие систем
X—СН2—С=СХ X—СН—С=О X—СН—C=N
т-е- аллилгалогенидов (Н. Н. Зинин), «-галоидоксосоединений и сложных
эфиров а-галоидкислот (О. А. Реутов) или их нитрилов (В. А. Абрамов)
с металлической ртутью, особенно при освещении:
X—СН2—С= • • • + Hg —> Xllg—С112—С=
Лучше реагируют иодиды, плохо — хлориды.
2. Присоединение ацетата ртути (или, более ограниченно, других
олеи) к непредельным соединениям в водном или спиртовом растворе
дает особый тип «квазикомплексных» соединений ртути:
НС=СН + HgCJ2—
в растворе Цх ,HgCl
—НЬ1... >С=С<
СК хп
в парах
cix с C\ngci
(Бигинелли)
(Р. X. Фрепдлина,
О. В. Ногина)
I,a. СН CH + 2HgX2+H2O -> СН-С^Ч-2НХ (М. Г. Кучеров)
XHgz 'О
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
403
но Н х zll
II. П2С=СП2 + 11g / ОССНзХ /С—С +CLI3COOH (Гофман, Заид)
II 1Г| | ХН
\ О /а ПО HgOCCH3
сн он /Н
III. П2С=СН2 4- 11g / ОСОСПз V ——------>• С-----С' (Шеллер,
II н ' | I ХП Шраут)
к О /а СН3О ]TgOCCH3
II
О
При присоединении соли ртути к несимметрично построенным олефи-
нам соблюдаются те же правила, что и при гетеролитическом присоедине-
нии галоидоводорода (правило Марковникова), так что из пропилена
получается
СПз-СП—CH2—Hg—О-С-СН3
I II
ОН о
Получаемые при присоединении солей ртути к ацетиленам (I и 1а)
и олефинам (реакции «оксимеркурирования» II и «алкоксимеркури-
рования» III) очень своеобразные по свойствам «квазикомплексные»
(А. Н. Несмеянов, Р. X. Фрейдлина) ртутноорганические соединения
отличаются той особенностью, что при действии на них реагентов,
атакующих ион ртути и связывающих ее в комплекс (I-, CN-, S2O32~
и Др.), уже на холоду протекает реакция элиминации — отщепляется
ртуть со своим анионом и из |3-положения к ней — С1, ОН или ОСН3
(для реакций I—III), и таким образом регенерируется олефин или аце-
тилен. Это напоминает поведение л-комплекспых соединений олефинов
и ацетилена; поэтому некоторое время продуктам присоединения прида-
вали строение комплексных соединений:
НС=СН • HgCl.
/ОН
Н2С=СН2 • Hg<
Ч)-С—СНз
о
Окончательная ясность в строение этих соединений внесена работами
Мидлтона, Р. Адамса и А. Н. Несмеянова, Р. X. Фрейдлиной, А. Е. Бо-
рисова. Последние авторы доказали существование tpc-тцянс-пзомерии
в продуктах присоединения солей ртути к ацетилену и его производным,
что несовместимо с формулами комплексных соединений. Кроме того,
они показали, что аддукты с олефинами превращаются по методу А. Н. Не-
смеянова, К. А. Кочешкова и Р. X. Фрейдлиной в несимметрические
полные ртутноорганические соединения
HOCH2-CH2ngCl + С611 ;,!’•(<) Н)2 — - IJ CCi 12 Cn2IlgCe115 + B(()n)3 + Cl-
26*
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
404 ------------------------------------------------------------
тогда как альтернативная структура с комплексно связанной ртутью
в условиях этой реакции дала бы дифенилртуть и этилен (см. ниже свой-
ства квазикомплексных соединений):
HOs>Hg*- СН2+2СвН5В(ОН)2 —(Св115)2^ + СН2=СН2 + 2В(ОН)з+С1-
СГ СНг
Еще раньше Марвел изолировал из продукта присоединения соли
ртути к L-ментиловому эфиру коричной кислоты два диастереоизомера,
что наиболее естественно объяснить появлением новых асимметрических
центров за счет присоединения соли ртути:
* , ~ , СНзОН
СвН5—СН=СН— С—О—Ментил+ Hg/О—С—СН3\ ----------*
II II
О \ О /2
о
* * || *
•—> С6II5—С Н—СН—С—О —Ментил + СН3С О 011
I I
СНзО HgOCCH3
II
о
О. А. Реутов и В. И. Соколов, применив метод количественного
расчета величины молекулярного вращения Брюстера (кн. 1, стр. 625),
установили на примере присоединения соли ртути к циклогексену, что
HgX и ОСН3 вступают в транс-положение:
HgO-C-CeH5 HgCl
f JI+ Hg/о-с-с6н5\ --зОН-> f Y 0 3L
\ о /2 4 ОСНз X'z ^ОСНз
Таким образом, аддукты соли ртути и олефинов (или ацетиленов)
являются продуктами присоединения с разрывом двойной связи. Приво-
дятся доводы (Уинстейн) о мимолетном образовании л-комплекса («мерку-
риниевый ион») в качестве нестойкой первой фазы реакции присоеди-
нения:
CH2=CII2 + IIg2+ —>
Hg2+
_СН2=СН2_
НОСН2—CH2HgCl + Н+
По данным Р. Я. Левиной, соли ртути j
цикл и присоединение идет аналогично присоединению
СН2
Н2с CII24-i-ig/O-C-Cn3\ +П2О —> HOCH2-CH2-CII2-HgO-C-ClI3 +
' II
\ о /2 о
раскрывают трехчленный
> к олефинам:
+ СН3СООН
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
405
Присоединение солей ртути к простым и сложным эфирам винилового
спирта и к кетенам протекает своеобразно и приводпт к ртутным произ-
водным альдегидов, кетонов и кислот (А. Н. Несмеянов, И. Ф. Луценко):
/О
CHi-CilOR + /СНз—С—О\ Ug —>- СПз-p—O-Tfg-CII2-C<f +
II ' II И
\ О /2 О
+ CH3COOH + ROH
/ /О \
2CH2=CHOR-MIgO ф Н2О —> Hg CII2—С-А + 2ROH
\ хН/2
СН2=С-СНз
) +/CH3-C-O\I!g► C1I3—С—О—Ug—СН2—С—СН3-|-2СН3СООП
О-С-СНз II II
II \ о /2 о о
о
СН2=С=О ф /СГГз-С—О\ Hg+ROII —> СН3-С-О—Hg—СН2—COR+CH3COOH
II II II
\ о/2 о о
(И. ср. Луценко)
3. В ароматическом ряду ртутноорганические соединения получаются
проще всего прямым меркурированием (Димрот), примером которого
может служить меркурирование бензола, протекающее при кипячении
бензола с ацетатом ртути или другими ее солями:
С6116фН8/О-С-СИ3\ —> СбПз—Hg-О-С-СНз+СНзСООН
\ О /2 О
Реакция представляет собой электрофильное замещение и, следова-
тельно, облегчается заместителями I рода, направляющими ртуть в орто-
пара-положение, и замедляется ориентантами II рода. Анилин, например,
меркурируется в водном растворе уже при комнатной температуре, при-
чем образуется смесь о- и н-ацетатмеркуранилипов. Легко меркури-
руются и фенолы. Многие меркурпрованные фенолы применяются как
антисептики, бактерициды и сельскохозяйственные фунгициды.
4. Второй метод синтеза, специфический для ароматического ряда,—
синтез ртутноорганических соединений через диазосоединения (А. Н. Не-
смеянов). Он состоит в действии порошка металлической меди на двойные
соли хлорида арилдиазония и сулемы:
+ р
Аг—HgClj + 2Cu —> ArllgC] фК2ф 2CuCI
2Ar—N==N IlgCJg +CCU+6NII3 —-> Ar2IIg + 2N2 + 6CuCI • NH3-|-Hg/
Этим методом можно получить (из соответствующего амина) любой
желаемый изомер ароматического ртутноорганического вещества, тогда
как прямое меркурирование по Димроту дает, обычно, смесь изомеров.
5. Действием на АгВг амальгамой натрия получают диарилртуть
(Дрехер, Отто):
2АгВг ф Na2Hg —> Ar2Hg-|-2NaBr
ЭЛЕМЕ НТО ОРГАНИЧЕСКИЕ СРЕДИНЕНИя
406
6. При действии сулемы на сульфиновые кислоты остаток SO2H заме-
няется на HgCl (Петерс):
ArSO2H + HgCl2 —> ArIIgCl + SO2 + НС1
7. Лабильные карбоксилы при действии соли ртути заменяются
на ртуть:
N02
NO2 \
z о- \
>_С< I Hg —•
ч о / у ,
NO2 /2 \ no2/2
CH3-C-CII-СООН-f-IIg/о—с—сн3\ —>
II I 11
OR \ О /2
- СНз-С-CI Г—11g—О—С-СПз + СО2 А СПзСООН
II ।
О R
<— ---- |lJg-f-2CO2 (Хараш)
О
(Хараш; А. II. Несмеянов и И. Ф. Луценко)
Ю. А. Ольдекопу, уксуснокислая ртуть
8. По Г. А. Разуваеву и
и ее гомологи при УФ-облучении декарбоксилируются по свободно-
радикальному механизму, образуя алкилмеркуркарбоксилат:
/СНзС-О\ Jig —> CII3Hg-O-CCH3 + CO2
II
О
2
Такая же реакция может быть инициирована перекисью.
9. Алкил- и арилборные кислоты реагируют с сулемой по схеме:
RB(OH)2+HgCl2 + H2O —> RHgCl-FB(OI])34-HCl
10. Все алкильные и арильные металлоорганические соединения
(кроме соединений пятивалентных мышьяка и сурьмы) при реакции
с солями ртути претерпевают обмен (А. Н. Несмеянов, К. А. Кочешков).
Например:
R4Sn + HgX2 •—> R3SnX4-RHgX
R3SnX-|-HgX2 —> R2SnX2+RHgX
R2SnX2 + HgX2 —> RSnX34-RHgX
RSnX3 + HgX2 > SnXi + RlIgX
2RSnX3 + IIgX2 + 20ir —> R2Hg + 2SnOf-- SX-4-3II2O
Проведение реакций 8 и 9 в щелочной среде приводит непосредственно
к R2Hg.
Свойства и реакции ртутнеорганических сое-
динен и и. В несимметрических RHgR' один из радикалов — более
электроотрицательный — легче подвергается атаке протоном. Система-
тически исследуя результат реакции
RHgR' + HCl —> RIIgCl+R'H
ИТОГА»! ГРУППА ПЕРИОДИЧЕСКОЙ системы элементов
407
Хараш построил ряд электроотрицательности радикалов:
—CN — ОСНз — Ч —<^ Ч—СП;,
ОСНз
Х —ci — // А, _
ci _ С1
-СНз -с2п5 —C3TI7 —сн2-<^
\ =г'
Из него следует, что кислоты отщепляют от RHgAr ароматические
радикалы легче, чем жирные радикалы, а ароматические радикалы,
содержащие в орто- и пара-положении заместители I рода, отщепляются
легче, чем незамещенный фенил. Таким образом, что касается аромати-
ческого ядра,связанного со ртутью, атака Н+ является обычной электро-
фильной атакой на ароматическое ядро, и именно на его углерод, связанный
с ртутью. С галоидоводородными кислотами реакция идет быстрее, чем
с другими сильными кислотами, что объясняется участием галоида в четы-
рехчленном переходном состоянии (стр. 593)-
RHgR г HC1
R-Hg-R
Й —Cl
--> RH + RHgCl
Радикал в RHgCl удерживается уже прочнее и для его отрыва в виде
RH необходимы более жесткие условия ацидолиза.
В реакции
RHgR' + *HgCl2 —> R'HgCl + R*HgCl
которая легко протекает при нагревании в растворе, ртуть сулемы (мече-
ной) забирает более отрицательный в ряду Хараша радикал R' (Н. А. Не-
смеянов и О. А. Реутов).
Реакции обмена геометрических изомеров [З-замещепных винильных
соединений ртути с сулемой, так же как п обратная реакция — симметри-
зации, идут с сохранением конфигурации (А. Н. Несмеянов, А. Е. Бори-
сов, 1948 г.):
,С к /Н\ Ск /Н
>С=С< irg+HgCl2 ------>2 >С=С<
А /2 Н/ \HgCl
? NH3 |
Обмены R2Hg с сулемой и реакции симметризации типа
2RHgX —> R2IIg
идут также с сохранением конфигурации (А. Н. Несмеянов, О. А. Реу-
тов, И. П. Белецкая). Разумеется, речь идет о случаях, когда асиммет-
рический радикал R связан с ртутью своим асимметрическим углеродом,
например:
CeHb-CH-COOR'
* Nil, I
2Ce[I.,-CIT-COOR' — i Jig
I I
KgCl свн5—ch-c.ooir
408
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Это были первые реакции типа 8^2, на которых было установлено
стереохимическое их течение, противоположное реакциям S1V2. При про-
ведении некоторых реакций обмена ртути в сильно сольватирующем
нуклеофильном растворителе (диметилсульфоксиде) оказалось, что они
текут по никогда ранее не констатированному механизму SE1, т. е.
с предварительной медленной ионизацией по связи С—Hg (О. А. Реутов,
И. П. Белецкая):
(-С-) Hg медлен11.?+ ^C: + HgC-
^Cr + HgClz быстро-> ^C-HgCl+CI-
Низшие диалкилртути — жидкие, перегоняющиеся без разложения,
очень ядовитые вещества со слабым неприятным запахом. Особенно
навязчив запах низших алкилмеркурхлоридов — твердых веществ с по-
движным галоидом, легко заменяемым на другие анионы. Диарилртути —
твердые вещества, более низкоплавкие, чем арплмеркурхлориды [так,
(C6H5)2Hg плавится при 125° С, C6H5HgCl при 272° С], и более, чем они,
растворимые в органических растворителях.
При действии на диалкилртуть концентрированных минеральных
кислот (но не слабых кислот или воды) от нее отщепляется одни из ради-
калов:
R2TTg + HCl —> RHgCl-pRlI
Диарилртуть еще легче, чем диалкилртуть, реагирует с НС1, галои-
дами и солями ртути.
Восстановление диалкилртути водородом в момент выделения
(NaHg+H2O; Zn + HCl в этаноле) или водородом при высокой темпера-
туре и давлении (Г. А. Разуваев) освобождает металлическую ртуть, и
выделяется углеводород:
R2llg + 2II —> 2RH4-Hg
Галоид отрывает один за другим оба радикала от диалкилртути:
R-Jlg + h —> RIIgl-J-RI
RHgI-pi2 --> Hgl2 + Rl
Никаких присоединений по карбонильной группе или обменов с галоид-
ными алкилами и каких-либо других реакций, подобных реакциям магний-
органических соединений, с обычными ртутноорганическими веществами
не происходит.
О. А. Реутов и И. П. Белецкая нашли, однако, что при «нуклеофиль-
ном содействии» в сильно ионизирующем апротонном растворителе,
например в диметилсульфоксиде, ртуть в диарплртути обменивается
на радикал, подобный триарилметилу. Нуклеофильное содействие —
одновременная атака на ртуть аниона, подобного иод-иону:
(CeH5)2Hg + ClC(CeII5)3 > CeII5C(CeH6)3 + C6H5IIgI+Cl-
I-
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
409
Единственные две реакции обмена, в которых ртутпоорганические
соединения широко использованы в синтезе металлоорганических соеди-
нений, эго — обмен с металлами и галогенидами металлов. В реакции
обмена вступают все непереходные металлы, значительно менее благород-
ные, чем ртуть, а именно: Li, Na, К и т. д.; Mg, Zn, Cd, Al, Ga, In.
Tl, Sn, Sb, Bi.
Например:
R-iHg-J-Zn—> R2Zn-|-llg
2R2Hg+ AsCl3 --> R2AsCl-J-2RHgCl
Ar2Ilg-|- AriС1-2-> Дг21С1-J-ArHgCI
ArJIg-J- IC13 -> ArlCl, + AriIgCl
В подобного рода реакции не вступают галоидные соли щелочных
и щелочноземельных металлов, но с большим пли меньшим трудом всту-
пают галогениды остальных непереходных элементов.
Обе реакции обмена исторически были важными методами, которыми
впервые были получены многие индивидуальные металлоорганические-
соединения непереходных элементов.
Изучая подобные реакции обмена металла в цис- и транс-изомерах
(3-хлорвинилмеркурхлорпда и бис- |3-хлорвипилмеркурхлорида, А. Н. Не-
смеянов и А. Е. Борисов установили правило: электрофильные и гомо-
литические замещения у олефинового атома углерода происходят с со-
хранением конфигурации (цис- или транс-).
Нагревание диарплртути с переходными металлами (Ag, Pd, Pt и др.)
приводит к образованию диарила (Г. А. Разуваев):
Pd
Ar2Hg —» Ar— Ar-f-Hg
Таким путем Виттиг получил о-дпфенилен:
Hg
Hg
Совершенно другими свойствами обладают квазикомплексные ртутно-
органические соединения. Как было ранее сказано, это аддукты солей
ртути с олефинами или ацетиленами, имеющие строение |3-окси-, |3-алкокси-,
р-галоид- и т. д. замещенных алкил- или алкенилртутноорганических
соединений. Аддукты олефинов реагируют подобно другим ртутпооргани-
ческим соединениям только при восстановлении амальгамой натрия, а ад-
дукты ацетилена — в реакциях с галоидами, металлами и галоидными
металлами.
Характернейшая реакция квазикомплексных соединений — ^-элими-
нация (стр. 601), наступающая при атаке как па P-заместитель, так и
на атом ртути и ведущая к выделению олефина (соответственно ацетилена)
и ртути в форме неорганического соединения:
НО—CIJ2CIJ2—IlgCl > ] Г—ОCH2=CH2-f-HgCl2
п- Cl- I,
HO-C1I2C112— HgCl + 31- -> Cll2=ClI2 + llgIj+OH-4-Cl-
Cl-CH=CII-JlgCl + 3I- -> IIC=CII4-HgIs4-2Cl-
ЭДЕМЕ НТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Аналогично идут реакции с CN , S2O32 и т. д.
Таким образом, реагенты, которые обычно симметризуют ртутноор-
ганические соли, разрушают квазикомплекспые соединения. Симмет-
ризовать их можно лишь действием аммиака:
2С1—СИ=СН—HgCl + 2NH3 > (ClCH=CH)2Hg + Hg(NH3)2Cl2
Формально эти реакции элиминации можно уподобить реакциям
1,4-присоединения к сопряженным системам С=С —С=С. Действительно,
например, расщепление этаиолмеркурхлорида соляной кислотой схемати-
чески изображается так:
НО—СН2—Cfl2—HgCl —> HOH4-CH2=CH24-HgCl2
Т ?
Н+ Cl-
Выделенные жирным шрифтом сг-связи играют роль сопряженных
л-связей диеновой системы (сг,сг-сопряжение, или гиперконъюгация).
Атака Н+ и С1~направлена на первый и четвертый атомы сопряженной
системы. Как и в системе С —С—С=С, в результате 1,4-присоединения
рвутся связи 1,2 и 3,4, а между углеродами 2 и 3 устанавливается
л-связь. Описанное выше присоединение солей ртути к простым или слож-
ным виниловым эфирам приводит (после замены аниона) к а-галоидмер-
куральдегидам и кетонам, которые тоже отличаются рядом особенностей
в своих реакциях. В отличие от алкильных и арильных соединений
ртуть в a-положении к оксогруппе «легко подвижна» и вступает в реакцию
обмена с органическими и неорганическими галоидпроизводными, причем
реакции протекают как без перенесения, так часто и с перенесением ре-
акционного центра.
Без перенесения реакционного центра:
/О /О
(C6H5)3CCl + ClHg-CH2-C< --> (С6Н5)3С -С112-с/ -1-llgCl,
хн \н
с перенесением реакционного центра (А. Н. Несмеянов, И. Ф. Лу-
ценко, Э. Г. Перевалова):
/О .0—С(С6114УО2-п)з
(«-NO2C6H4)3CCl + ClHg-Cfl2-C/ -> СН2=С( +HgCl2
41 н
/О ZO—С—С113
Cll3-C-Cl + Clllg-Cfl2-C4 ---> CII2=C li +Hgci2
II чЧ1 О
о
Реакции с перенесением реакционного центра протекают как электро-
фильные атаки по кислороду (а не по углероду) и дают производные
(в данном случае, например, ацетильное) таутомерной формы оксосоеди-
нения, хотя в отличие от самого оксосоединепия его ртутному производ-
ному не свойственна гтаутомерия, т. е. в системе
о
СШ£— СН2— с— о
н
ТРЕТЬЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
411
атака по кислороду облегчена одновременным «подводом электронов»
за счет легко поляризуемой связи Hg—С.
Реакцию такого типа формально можно рассматривать как 1/^при-
соединение по о,л-сопряжепной, т. е. гиперконъюгированной системе
связей (А. Н. Несмеянов):
/Н
сн2=с^
О-С-СНо
II 3
о
+ HgCI2
аналогично
R.
?:сн-сн
C-OMgCI
I
сн3
ТРЕТЬЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
Борорганические соединения. Неорганические и органические соедине-
ния бора изучаются в последнее время особенно интенсивно, и разно-
образие известных их типов быстро возрастает. Гидриды бора, как со-
держащие замещающие органические радикалы, так и чистые гидриды,
интересны своими «мостиковыми» водородными связями, например:
диборан
Н Нч Н
н3с> 'ЪГ н
метилдиборан
Водородные атомы этих водородных мостиков совершенно симме-
тричны по отношению к обоим атомам бора, и в этом отличие таких мости-
ков от обычных водородных связей, например, в уксусной кислоте
.О...Н-О\
СЩ-С^ ЩЗ-СИз
оно
где расстояния водородных атомов в димерном цикле от гидроксильного
кислорода «своей» и карбонильного кислорода «чужой» молекулы резко
отличны. В диборане четыре электрона «обслуживают» четыре атома
(2В и 2Н) и образуют четырехцентровые орбитали.
Известны еще гидриды бора состава В4Н10; В5119; В5Нн; В8Н12;
В8Н13 и, наконец, один из наиболее прочных — декаборан В10Н14.
Для некоторых из них известны алкильные и иные замещенные, получаемые
по Шлезингеру взаимодействием бороводородов с триалкилборамн-
412
ЭЛЕМЕНТООРГАНИЧЕСКПЕ СОЕДИНЕНИЯ
Интересно различное отношение диборана и декаборана к гриньярову
реактиву (Виберг):
B2II6 + RMgBr—> RB2H5 + IIMgBr
ВюНм + RMgBr > BioIInMgBr + RH
Действуя на B10H13MgBr галоидными алкилами, можно получить
и алкилдекабораны (Виберг):
Bi0Hi3MgBrH-RBr---> RB10II13 + MgBr2
Рис. 101. Строение декаборана В10Н14.
Строение гидридов бора (рис. 99—101) и их производных тем и инте-
ресно, что оно нарушает (как и ряд других структур элементоорганиче-
ских соединений) каноны классиче-
ской теории строения и позволяет
дальше совершенствовать наши
структурные представления.
Диборан и алкилдпбораны присо-
единяются (за счет В—Н-связей)
к олефинам и ацетиленам по следу-
ющему типу реакций (Херд, Броун):
B2He + 6RCH=CH2--->2(ВСН2-СН2-)зВ
B2(CH3).2lI4+4RCH=CH2-->
-----------------------------------------> 2(RCH2-CH2—)2ВСН3
Присоединение идет здесь всегда
как бы против правила Марковни-
кова. Это потому, что в гидридах бора
водород более отрицателен и отхо-
дит как гидрид-анион Н“, а бор более положителен, так что не формально,
а по существу правило Марковникова (кн. I, стр. 266) соблюдается.
Для синтеза борорганических соединений вместо гидридов бора,
самовозгорающихся па воздухе и очень ядовитых, можно действовать
на олефины ВС13 в присутствии металлического алюминия. Реакция,
видимо, идет через предварительное образование НВС12 и присоедине-
ние его к олефину:
RCn=CII2 + HBCl2 --> RC1I2—СН2ВС12
ТРЕТЬЯ ГРУППА ПЕРИОДИЧЕСКОЙ системы элементов
413
Широко использовано для синтеза борорганических соединений
взаимодействие трехгалоидпого бора (Краузе) или эфиров борной кислоты
(Е. Хотинский) с литийорганическими соединениями или гриньяровым
реактивом:
-Вг
BX3-|-3RMgBr --> R3B-|-3Mg^
ХХ
Таким же путем могут быть получены типы веществ R2B(OCH3),
RBG12 и R2BG1, которые при гидролизе дают алкил- и диалкилборные
кислоты RB(OH)2 и R2B(OH) еще более слабые, чем борная кислота, и до-
вольно легко отщепляющие алкилы при окислении кислородом воздуха и
при действии ряда металлгалогенидов (HgCl2, СпС12), но не при действии
кислот. Алкилборные кислоты образуют при высушивании или отнятии
воды химическим путем интересные тримерные циклические ангидриды
: BR
оХ О
RB BR
При действии па комплексную соль KBF4 гриньяровым реактивом
RMgBr в случае жирного радикала R получается R3B, а в случае арома-
тического ArMgBr реакция протекает по схеме (А. Н. Несмеянов,
В. А. Сазонова)
KBF4 +4ArMgBr > К+ "BAr4-|-2MgF2 + 2MgBr2
и получаются соли щелочных металлов с тетраарилбор-анионом. Эти
соли были впервые получены Виттигом посредством такой реакции:
BAr3-)-ArLi —> Li+ ~ВАг4
Здесь (Аг:)3В, имеющий вокруг бора секстет электронов, в стремлении
дополнить его до октета захватил радикал Аг: с парой электронов от
Li:Ar, оголив литий до катиона. В результате вокруг бора образуется
октет электронов и бор приобретает отрицательный заряд аниона. Такого
типа соли — превосходные реактивы на К, Rb и Cs, так как образуют
с этими элементами практически нерастворимые в воде соли.
Соединения типа R3B, исходя из той же тенденции, способны к сле-
дующим реакциям:
R:!BA:NR; ---> R3B:NR;
R3B+Na01I----> R3B:OJI+Na+
R3B-|-2Na. --> [R3B:Na]~Na+
В соединении R3B:NRg связь B:N должна рассматриваться как
семиполярная, ибо кроме ковалентной пары электронов атомы связывает
их полный (ионный) противоположный заряд, образовавшийся как
следствие одностороннего предоставления в совместное обладание пары
электронов одной нейтральной молекулой другой нейтральной молекуле.
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СРЕДИНЕ НИЯ
414
В анионе же В3В:ОН
вание, уже обычная
мального (полного)
на атоме бора (ОН-
и потерял, так как
связь В —О, несмотря на аналогичное ее образо-
ковалентная, поскольку кислород не несет фор-
заряда, противоположного заряду, возникшему
пришел с отрицательным зарядом, который он
односторонне предоставил бору пару электронов
в совместное обладание).
Соль В4В-М+ Виттиг назвал ат.-комплекс ом. Ее можно также назвать
обратным ониевым соединением, так как способ ее образования сходно
противоположен образованию оииевых, например аммониевых, катионов:
R3B-|-RNa --> ВдВ- Na+
R3N+ RC1 --> R4N+ Cl-
B 1962 г. Опак, Вильямс и Вейс открыли реакцию пентаборана с гомо-
логами ацетилена, протекающую по уравнению:
ПзС7
П3С
B4n6C.2RR'4-H3B:xN^
Н3С Z
На основании изучения спектров ЯМР подобным борорганическим
веществам приписана структура, изображенная на рис. 102. Так был
открыт первый из серии прочных ароматических карборанов с полиэдри-
ческими молекулами общей формулы B„C.2H/i+2. За ним последовало
открытие и остальных членов этой серии, изо-
браженных па рис. 103. Наиболее исследован-
ными оказались весьма интересные по свойст-
вам три прочных декакарборана — орто-(1.2-).
мета- (1,7-) и пара-(1,12-), изображенные на
рис. 104. Как и на рис. 103, каждый из атомов
бора и углерода в этих структурах несет по
одному атому водорода, не изображенному на
рисунке. Первый из декакарборанов (орто-
расположение ацетиленовых углеродов) был
открыт в 1963 г. практически одновременно
Л. II. Захаркиным, В. И. Станко и сотр.
(СССР), назвавшими его барен, и Фейном
(США), назвавшим его карборан-10. Он содер-
жит 10 атомов бора и два атома углерода,
т, расположенные по вершинам икосаэдра.
арен (о-карборап-10, или 1,2-карборан-10) получают нагреванием
^римерпо до 90 С комплексов декаборана B1OHUL., с ацетиленом, где
лиганд типа ацетонитрила или диэтил сульфида.’ Если нагревать его
иняпре11?е7СуТ”- Д0 465—500° С, он изомеризуется в .м-карборан-10 (или,
зуется л с о/Каро0рап'Ю)- При нагревании .м-карборана-10 до 615е С обра-
Все тпи • /011Ь1Мпых°Д°м тг-карборан-10 (или, что тоже, 1,12-карборан-Ю).
ниш „ С(’„едииеппя исключительно прочны термически, а также к окисле-
систвию кислот. Из всех трех лучше изучен барен.
ТРЕТЬЯ ГРУППА ПЕРИОДИЧЕСКОЙ системы элементов
IJo рентгеноструктурным данным расстояние С—С в бароне больше,
чем даже в предельных углеводородах. При действии алкиллития водороды
при бывших ацетиленовых углеродах замещаются па атомы лития
(Л. И. Захаркин, В. И. Станко). С этим литийорганическим соединением
можно осуществить обычные для литийорганических соединений реакции:
алкилирование посредством RBr, карбоксилирование при помощи СО.,,
с
1,5-В3Н3СгН2
2,4-В5Н5С2Н2
Рве. 103. Строение карборанов* общей формулы ВлС2Нл+2
присоединение альдегида, присоединение окиси этилена с образованием
спиртов, получение с солями металлов металлических производных
(О. Ю. Охлобыстин) и т. д. Замещение на натрий можно осуществить
действием карборана-10 па амид натрия в жидком аммиаке, замещение
на MgBr — действием C2H5MgBr (Хейинг, Эйджер, Кларки др., Л. И. За-
харкин, В. И. Станко и Др.).
Рис. 104. Строение декакарборанов* В10Н10С2Н2:
(1 — 1,2-карборан-1 и (т. пл. 287—288° С); б — 1,7-карборан-10 (т. пл. 266—269° С);
в — 1,12-карборан-10 (т. пл. 259—261° С).
При действии па барен спиртового раствора едкого кали происходит
отщепление одного атома бора и двух протонов, и образуется своеобразный
анион В9К2Щ2 (так называемый дикарболлид-анион) (рис. 105, а). Этот
анион, как установил Хоторн, может образовать сэндвичевые соединения
(стр. 481) с переходными металлами (рис. 105, бив). В барене можно
хлорированием на свету заместить один или несколько водородов при
боре па хлоры; такой хлоркарборан имеет малоподвижный галоид.
В общем, карбораны представляют собой явно ароматические системы
с низким энергетическим уровнем, обладающие электроноакцепторпым
характером, связанные воедино мпогоцептровыми электронными орби-
талями. Их структуру невозможно описать на основе классической
* Каждый В и II несут ио одному водороду, не изображенному на рисунке.
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
бутлеровской теории строения, используя привычные четырехвалентные
углероды и трехвалентные атомы бора, которые, кстати говоря, занимают
в карборане одинаково связанные симметричные положения. Карбораны —
одни из наиболее ярких типов соединений, выходящих за рамки клас-
сических структурных схем. Другими такими веществами являются
диборан и другие гидриды бора, а также описанные ниже димеры триметил-
алюминия и сэндвичеобразные комплексы переходных металлов.
Рис. 05. Строение дикарболлпд-анпона B9R2H27 и образуемых
им соединений с металлами:
а — анион; б — сэндвичеобразная структура комплекса с переходным
металлом; в — дикарболлид-циклопентадиенид металла М.
Интересным типом неорганического соединения бора, образующего
органические производные со связями R — N или R —В, является бор-
азол — «неорганический бензол»:
Органические производные боразола
реакциями:
можно получить следующими
3R3B+3NH3 —> 3R3B:NH3
BR
+/
HN NH
II I
R-B B-R
NH
ТРЕТЬЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
417
ВС1
+z z+
.= RN NR +
3BC13 + 9RNH2 —> II 1 +6RNJT3 С1‘
С1-В ВЫ
NR
Подобным образом можно синтезировать и шестичленные гетероциклы
с чередующимися атомами В и О или В и S, которые широко исследованы
Б. М. Михайловым.
Алюминийорганические соединения. За последнее время благодаря
открытию Циглером реакции прямого синтеза триалкильных соединений
алюминия из олефинов, водорода и мелкораздробленного металла
6СН2=СН2 + ЗЫ2 + 2А1 ------> 2(С2Н6)зА1
алюминийорганические соединения получают все большее промышлен-
ное значение, особенно при полимеризации олефинов (кн. 1, стр. 274).
Известны следующие типы алюминийорганических соединений:
RA1H2. r2aih, R3A1
RA1C12, R2A1C1, Aids
[R4A1]-Li+, [R3AlH]-Li+,i[R2AlH2]'Li+, [RA1H3]-Li+, [AlH4]’Li+
Алюминийорганические соединения с низшими алкилами (СН3, С2НБ)—
самовоспламеняющиеся на воздухе жидкости; солеобразпые соеди-
нения (нижняя строка) — твердые вещества.
Соединения RA1H2 и R2A1H получаются также реакцией Циглера
из чистого мелкодисперсного алюминия, водорода и олефина; RA1C12
и RgAlCl могут быть получены взаимодействием R3A1 с А1С13 или дей-
ствием RC1 на металлический алюминий в автоклаве.
Одним из резких противоречий с основами теории строения и четырех-
валентностью углерода, вообще строго соблюдаемыми для предельных
соединений, является димерность триметилалюминия, молекулярный
вес которого в парах соответствует молекулярной формуле (СН3)6А12.
Физическими методами установлено строение этого вещества, подобное
строению А12С16, с метильными мостами, соединяющими атомы алюминия:
с
(СНз)2А1 А1(СНз)2
X
Таким образом, в некоторых редких случаях углерод (метила), не рас-
ширяя своей электронной оболочки, т. е. не изменяя валентности в обыч-
ном смысле слова, связывает пять атомов. В этой формуле каждая валент-
ная связь уже не изображает более пары электронов, а лишь факт непо-
средственной связи атомов.
27 Заказ 672
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
418
Алюминийорганические соединения бурно разлагаются не только
кислотами, но и водой и спиртами:
A1R3+31I2O --> ЗКН+А1(ОН)з
Двуокись углерода реагирует с эфирным раствором триалкилалюми-
ниев при температуре 20—100° С (под давлением) с образованием R2A1OCR,
6
Второй алкил вступает в реакцию лишь при 180—200° С (Л. И. Захаркин,
В. В. Гавриленко).
Триалкилалюминий энергично реагирует и с оксосоединениями,
но способность восстанавливать последние, в зачаточной форме присущая
и магнийорганическим соединениям, выражена у алюминииорганическпх
соединений гораздо сильнее и делает их менее пригодными для «гринья-
ровых» синтезов. Однако они обладают замечательным свойством присое-
диняться к олефинам (Циглер), разделяя эту способность с боралкилами:
R3A1+3CH2=CH2 —> (RCH2CH2)3A1
Поскольку полученное новое алюминийорганическое соединение также
обладает способностью присоединяться к олефину, реакция не останавли-
вается на написанной первой стадии и, если имеется избыток олефина,
продолжается вплоть до образования высокомолекулярных соединений:
(НСН2СН2)зА1 + ЗСН2=СН2 —► (RCH2CH2CH2CH2)3A1 и т. д.
В итоге:
ЙзА1-гЗ«С112=СН2 —> [R(CII2CH2)n]3Al 3R(CH2-CH2)„H + А1(ОН)3
Через алюминийорганические соединения, действуя ими на соответ-
ствующие галоидные соли, можно синтезировать и металлоорганические
соединения тяжелых металлов. Алюминийорганические соединения окис-
ляются кислородом в алкоголяты.
Ароматические триарилалюминии удобно получать, нагревая диарил-
ртуть с металлическим алюминием:
3Ar2Hg+2Al —> 2Ar3Al + 3Hg
Металлоорганические соединения других металлов III группы. Скандий,
иттрий и редкоземельные элементы не образуют обычных металлооргани-
ческих алкильных и арильных соединений. Галлий, индий и таллий
дают все типы — RMX2, R2MX и R3M, исходя из МС13 и RLi или RMgX.
Таллипорганические соединения типов RT1X2 и R2T1X в некоторых
отношениях (по способам синтеза и инертности) подобны ртутнооргани-
ческим. Они устойчивы к воде и на воздухе, солеобразны, высокоплавки.
Ароматические таллийорганические соединения можно получить через
диазосоединеиия (А. Н. Несмеянов, Л. Г. Макарова) или прямым дей-
ствием маслянокислого таллия на ароматические соединения («таллиро-
H3HRe>Tir^ А' кочешков). В соединениях типа R3T1, полученных Гроллем
и RLi, один из радикалов отличается большой активностью.
ЧЕТВЕРТАЯ группа периодической системы элементов
419
Он отщепляется уже при действии слабых кислот или кислорода. Соеди-
нения RT1X2 склонны диспропорционироваться в И2Т1Х (наиболее
устойчивый тип) и Т1Х3.
ЧЕТВЕРТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
Кремнийоргапические соединения. Органические соединения кремния
впервые получены Ладепбургом и Фриделем и широко изучены Киплин-
гом. Однако промышленное значение они получили только после работ,
проведенных К. А. Андриановым.
Приведем известные типы мономерных кремнийорганических соедине-
ний и их названия:
RSiII3 RSiCl3 RSi(OCH3)3
алкилсилан алкилтрихлорсилан алкилтриметоксисилан
R2SiH2 R2SiCl2 R2Si(OCII3)2
диалкилсилан диалкилдихлорсилац диалкилдиметоксисилан
R3SiH RsSiCl R3Si(OCH3)
тр„алкилсилан триалкилхлоренлан триалкилметоксисплан
R4S1
тетраалкилеилап
Алкилхлорсилапы и арилхлорсиланы, а также тетраалкил- и тетра-
арилсиланы могут быть получены действием гриньярова реактива на че-
тыреххлористый кремний. Алкокси- и, в частности, метоксиалкилсила-
ны — действием гриньярова реактива на соответствующий эфир орто-
кремневой кислоты, например Si(OCH3)4, причем реакцию RC1 с магнием
можно вести прямо в среде ортокремневого эфира (К. А. Андрианов). Алкил-
силаны и арилспланы можно получить восстановлением алкилхлорсила-
нов или арилхлорсиланов, например, литийалюмипийгидридом.
Кроме того, по связи Si — Н силаны могут присоединяться к олефинам.
Таким образом, для синтеза алкильных производных кремния можно
использовать HSiCl3 или алкилсилан, содержащий группу Si—Н, и при-
соединять эти вещества к олефинам, папример:
HSiCl3+ СИ2=СН2 •—> С2Ф—SiCl3
Реакция эта может идти и как теломеризация (Р. X. Фрейдлина,
Е. Ц. Чуковская):
• »
НSiCI3-TИнициатор > »Я'С13
SiCl3 •J- С! 12= С! I i > Cl:iSiCII2—СП2«
Cl3SiClI2-CJl2’- СН2=С!12 -------> CI3SiCl]2Cll2Cri2ClJ2.
Cl3SiClI2CH2Cll2Ctl2 • -I- HS1CI3 -> Cl3SiCir2CH2Cn2CH3-!- • SiCl3
(продол-
жает
цепь)
Показательно, что диметилдихлорсилан (CH;i).2SiCl2 и диметнлди-
метоксисилап (CH3)2Si(OCH3)2 гидролизом могут быть превращены
27*
/90 ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
в высокомолекулярные продукты—полисилоксаны (связь Si О Si), обла-
дающие рядом ценных свойств:
СН3 СН3ч
n(CII3)2Si(OCH3)2 ПО—Si—I —О—Si—I—ОН
СН3V СН3/ п-1
Кроме линейных высокомолекулярных соединений диметоксисиланы
легко образуют устойчивые восьмичленные циклические тетрамеры и
менее стойкие шестичленные тримеры:
НзСч \/СНз Н3Сч \/снз
Si Si
О О О О
НгО П3С | 1 /СНз НзС | 1 /СНз
4(CH3)2Si(OCH3)2 > >Si Si; \si Si;
н3с/ 1 1 ХСН3 H3CZ \ ХСН3
О 0 О
'sC
НзС7 /ЧчСНз
Тримеры могут быть легко превращены в линейные высокомолекуляр-
ные соединения.
Полисилоксаны находят применение для получения термостойкой
электрической изоляции, каучуков, смазочных масел и т. Д.
Необходимый в качестве исходного материала диметилдихлорсилан
готовят действием хлористого метила на металлический кремний (в виде
сплава с медью, Рохов):
2CH3CI + Si -—> (CH3)2SiCl2
Получены и силоксановые смолы с другими, кроме метила, ра-
дикалами.
В свое время Киппинг направил усилия на получение кремниевых
аналогов соединений с углеродной цепью и действительно синтезировал
и изучил вещества типа
СНз
I
(CH3)3Si—Si-Si(CII3)3
СПз
(CII3)3Si—Si(CH3)3
с Из С Из
I I
(CH3)3Si—Si—Si—Si(CH3)3 и т. Д.
I I
СН3СН3
вплоть до нонасиланов, а также прочные циклические структуры типа
(CeHshSi—Si(CeH5)a
(CgHa^Si/ ^Si(CeHb)2
Si(CeIIs)2
с четырьмя, пятью и шестью
членами в цикле.
ЧЕТВЕРТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
4 21
Первые члены ряда можно получить реакцией Вюрца
2R3SiCl + 2Na —> R3Si—SiR3
или по методу Крауса — в жидком аммиаке:
R3SiCl + 2Li -> R3SiLi + LiCl
R3SiLi + R3SiCl -> R3Si—SiR3 + LiCl
Скелеты—Si — Si — Si— оказались неустойчивыми на воздухе и к дей-
ствию щелочей, и вещества такого типа не получили применения. Напро-
J. L I.
тив, скелеты типа —О—Si —О—Si—О — Si—О—, образующие силикаты
I I I
природные и искусственные (стекло), по почину К. А. Андрианова при-
обрели в виде своих органических производных важное значение для
практики.
В последнее время получены вещества типов
(CII3)3SiNa и (CH3)3SiMgCl
в известной мере подобные натрий- и магнийорганическим соединениям.
Из них синтезированы (Г. А. Разуваев) многие аналоги металлооргани-
ческих соединений со связью кремний — металл, например [(CH3)3Si]oHg,
[(CH3)3Si]4Sn.
Германипорганпческие соединения. Изучающиеся в последние годы
довольно интенсивно германийорганические соединения не получили
пока какого-либо применения. Начало изучению германийорганических
соединений положили Краус, Орндорф, Деннис.
Известны такие типы веществ с атомом германия (Х=галоид, 1/2О,
ОН и т. и.):
RGeII3 RGeX3
R2GeH2 R2GeX2 R2GeNa2
R3GelI R3GeX R3GeNa
R4Ge
а также полигерманы, например:
R3GeGeR3
R3GeGe(R)2Ge(R)2GeR3
R3GeGe(R)2Ge(R)2Ge(R)2GeR3
и смешанные Gc — Si- и Ge—Sn-соединения:
[R3Gc]3SiX, [R3Ge]4Si, R3GeSnRj
Связи Ge—С значительно прочнее к действию кислот и галоидов,
чем связи С—Sn, но уступают по прочности связям С—Si.
Основные пути синтеза германийорганических соединений — дей-
ствие гриньярова реактива и других металлоорганических соединений
на четыреххлористый германий и присоединение к олефинам органических
и неорганических производных германия, содержащих связь Ge—Н,
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
422
например HGeCl3. Как и для кремниевых аналогов, это присоединение
происходит формально «аптимарковниковски», например:
СПз—СН = СН2 + IlGeCl.i > СП3—СП 2-~CH2GeCl3
Поскольку гетеролиз связи Н—j Ge происходит так, что водород от-
ходит как гидрид-анион, по существу присоединение идет согласно пра-
вилу Марковпикова.
С. П. Колесников и О. М. Нефедов нашли, что л-системы нафталина
и анизола (но не бензола и толуола) также присоединяют HGeCl3. В слу-
чае анизола присоединение идет по всем трем связям и образуется три-
германиевое производное метоксициклогексапа CH3OC6H8(GeCl3)3.
И. Ф. Луценко и И. К). Бауков нашли, что реакция обмена
(CH3)3GeBr+ (C3H7)3SnCH2-C-Cll3 _(СзН.)38пв'Г
li
О
—> (CH3)3GeCH2C—СН3 + (CH3)3Ge-O-C=CH2
II I
О СНз
ведет к образованию таутомерной смеси кетонной и енольной форм
в следующих соотношениях:
(CH3)3GeCH2—С—СН3 (CH3)3Ge-O-C=CH2
II I
О CH3
(96%) (4%)
Это пока единственный пример металлотропной кето-енольной таутомерии.
Для аналогичного кремниевого производного кетонной формы те же
авторы констатировали только необратимую перегруппировку:
R3SiCH2-C—СНз —R3Si—О—С —СП2
о СНз
Металлоорганические соединения олова и свинца. Свинец- и олово-
органические соединения образуются при действии галоидных алкилов
(и арилов) на сплав этих металлов с натрием (Ладенбург, Левиг, Кагур)
,4С2Н5С1 + 4PbNa ---------------> (С2Н5)4РЬ Ц- 4NaCl 4- ЗРЬ
ИЛИ) действием гриньярова реактива на хлориды этих металлов (Пфейф-
4RMgCl 4- SnClj --------------------> R4Sn -j- 4MgCl2
4RMgCl + 2PbCl2 —> R4Pb %- Pb 4- 4MgCl2
Кроме R4Sn и R4Pb существует и весь ряд менее алкилированных
производных:
RSnCh R2SnCl2 R3SnCl R4Sn
RPbCl3 R2PbCl2 R3PbCl R4Pb
SnCl4 и соответственно Pb (OCOCH3)4 на правые члены
мо/кно получать левые (К. А. Кочешков). Действием хлористого
ЧЕТВЕРТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
водорода на R4M также можно отщеплять один радикал за другим, заме-
няя его на галоид. Из левых членов ряда действием гриньярова реактива
можно готовить правые, в том числе и смешанные. Полученные так соеди-
нения SnRR'R'R" Поп расщепил на антиподы.
Алкилируя иодистым алкилом станнит натрия, можно получить
(Р. Мейер) соль алкилстанноновой кислоты (продукт гидролиза RSnCl3):
и j v ONa
Rl !- Sn(ONa)2 --> RShxX + Nal
A.)
Как установил Краус, R3SnCI в жидком аммиаке при действии натрия
превращается в R3SnNa. В тех же условиях R2SnCl2 претерпевает пре-
вращения, выраженные схемой:
R2SnCl2 + 2Na ---> R2Sn + 2NaCl
R R
2R2Sn + 2Na --> NaSu—SnNa
R R
2Na
---> 2R2SnNa2
При разложении R3SnNa и R2SnNa2 в аммиачном растворе солью
аммония получают R3SnH и R2SnH2. Их можно получать, так же как
RSnH3, при восстановлении R3SnCl, R2SnCl2 и RSnCl3 посредством
RoAlH. Эти оловоорганические гидриды способны присоединяться (фор-
мально «антимарковниковски») по двойным и тройным связям (Вап-Керк).
Реакция присоединения инициируется свободными радикалами (например,
из азодинитрила изомаслянон кислоты) и может также протекать как
теломеризация. Из ацетиленов так получают олефиновые оловооргани-
ческие соединения
R\ II
R3S11II + R'feCR' --> )С=С/
RgSn^ R'
Из функционально замещенных олефинов, например из непредельных
кислот, образуются функционально замещенные оловоорганические сое-
динения алканов или циклоалканов:
СН2=С—С—ОСНз 4- (C2llo)2SnI[2 --> (C2IIo)2SnII-CII2—CII-C-OCH3
I I, I II
II3C О СПз О
Иные способы получения оловоорганических соединений жирного
ряда с функциональными заместителями разработал И. Ф. Луценко.
Виниловые эфиры (простые и сложные) оказались способными присоеди-
нять оловоорганические алкоксиды:
ОС4Н9
СН2=СН-ОС4119 4- R3SnOCH3 —> R3S11-Cll2-CIВ
4 ОСНз
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
424
Обменной реакцией ct-ртутпых производных оксосоединений с олово-
органическими галогенидами легко синтезировать соответствующие оло-
вянные производные оксосоединений:
О /О
CIHg—СН2—С-^ + R3Snd ---> R3Sn—СН2—С<( + HgCl2
\ц хн
Ароматические оловоорганические соединения с функциональными
заместителями можно получать по А. Н. Несмеянову и К. А. Кочешкову
действием хлористого олова на ароматические ртутноорганические со-
единения:
Ar2Hg + SnCl2 —> Ar2SnCl2 + Hg
При взаимодействии R3SnCl с R3SnNa образуются R3Sn—SnR3 (ин-
тересно, что в отличие от Аг3С—САг3 их оловянные аналоги не диссоции-
руют на свободные радикалы). Более длинные цепи атомов олова, всегда
непрочные, образуются при восстановлении R2SnCl2 менее энергичными
агентами, чем R2A1H. Из Ar2SnCl2 при этом получаются циклические
соединения:
Ar2Sn—SnAr2
I I
Ar2Sn SnAr2
SnAr2
SnAr2
Ar2Sn SnAr2
Ar2Sn SnAr2
SnAr2
Комбинируя натриевые производные оловоорганических соединений
с их хлоридами, можно осуществить синтез линейных цепей олова в 3—5
атомов:
R
I
2R3SnCl -f- Na2SnR2 —> R3Sn—Sn—SnR3
R
R R R
I I I
NaSn—Sn—SnNa + 2R3SnCl —-+
I I I
R R R ]
R R R R R
Ti I I I I I
R—Sn—Sn—Sn—Sn—Sn—R
I I I I I
R R R R R
Натриевые производные R2SnNa2 и R3SnNa могут быть использованы
для алкилирования по атому олова, например, для получения соединений
с функциональной группой в цепи:
R3SnNa + ClCH2COC2H5 —>' RsSnCH2COC2H51
II ’ II
О >0
/у Некоторые из оловоорганических соединений, в частности типа R3SnX
ст а™онл 0Казались очень сильно действующими фунгицидами и сред-
вами орьбы с грибковыми заболеваниями растений и животных. Али-
билизатКИе 0Л0В00РганичесКие соединения нашли применение как ста-
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
425
Для свинца также известны типы соединений R2Pb, RsPbPbR3,
RgPbNa (см. также стр. 379).
Тетраэтилсвинец в громадных масштабах применяется как анти-
детонатор в моторном топливе для двигателей внутреннего сгорания,
повышающий качество топлива (его действие открыто Мидгли в 1922 г.).
Недостатком «этилированных» бензинов является их сильная ядовитость.
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
Фосфорорганические соединения. Различают два типа органических
веществ, содержащих фосфор. Первый — это собственно фосфороргани-
ческие соединения, т. е. такие, в молекуле которых содержится фосфор,
непосредственно связанный с углеродом. Ко второму типу относятся
разнообразные производные неорганических кислот фосфора — эфиры,
тиоэфиры, амиды и т. д.; в этих соединениях фосфор связан не непосред-
ственно с углеродом, а через кислород, серу, азот. Соединения этого
последнего типа весьма распространены в природе. К ним относятся
некоторые важные ферменты и коферменты, переносчики энергии, такие,
как аденозиитрифосфат (стр. 362), и, наконец, нуклеиновые кислоты
клеточных ядер, рассмотренные в разделе «Нуклеотиды и полину-
клеотиды». Собственно фосфорорганические соединения в природе не
встречаются*; все они получены синтетическим путем.
Подобно аммиаку, фосфин РН3 является родоначальником ряда орга-
нических производных — фосфинов. В зависимости от того, сколько
атомов водорода фосфина замещено на органические остатки, различают
первичные, вторичные и третичные фосфины: RPH2, R2PH, R3P.
В отличие от аминов фосфины легко окисляются и вступают в разно-
образные реакции присоединения. Таким образом возникают ряды окисей
фосфинов, фосфинсульфидов, фосфиниминов:
Rx R4 /Н Rx R\ R\
>PZ /Р<х R^P=O R^P=S R4P=nR'
X) Rz X) Rz Rz Rz
Алкилированными производными фосфинов являются соли четвер-
тичных фосфониев R4P+X~, аналогичные солям четвертичных аммониев.
Из четвертичных солей фосфония, содержащих по соседству с фосфором
по меньшей мере одну СН3-группу, при действии очень сильных основа-
ний (CH3Li) получаются фосфинметилены, или метиленфосфораны.
называемые иногда илидами фосфора:
[(С6Н5)зР-СН3]С1- -> (CeH5)3P=CH2
-НС1
Для фосфорорганической химии характерны соответствующие фос-
финам галоидпроизводные типа RPC12h R2PC1. Их можно рассматривать
как производные треххлористого (и вообще, трехгалоидного) фосфора
РС13, в котором атомы хлора (галоида) замещены па органические
* Единственное исключение — Р-амипоэтилфосфоповая кислота, пли цилиатин
NH2CH2CH2PO(OH)2, найденная в некоторых микробах, морской анемоне, а также
в Rumen protozoan.
ЭЛЕМЕНТООРГАНИЧЕСКПЕ СОЕДИНЕНИЯ
426
радикалы. Впрочем, их можно также рассматривать и как фосфины, в кото-
рых атомы водорода замещены на хлор.
Существуют и более сложные производные фосфора, такие, как
R /R Сб115-Р-Р-СбН5
Х;Р—Р< I ! в др.
R R Сб115-Р-Р-С6Н3
Известны некоторые фосфорные аналоги нитросоединений, однако
они, в отличие от соединений азота, полимерны, например:
R
/О
R-P<
^0
ох О
%;
R' \ / R
О
В общем, эти соединения не характерны для фосфорорганической
химии. Зато их гидратированные формы, т. е. фосфорорганические
кислоты трех- и пятивалентного фосфора, представляют собой важный
класс фосфорорганических соединений. К нему относятся кислоты:
Z/W zOII
R-P^-OH R-Р.'
-п on
фосфонистые
соли и эфиры — фосфониты
R-P^OH
'он
фосфоповые
соли и эфиры —
фосфонаты
R\ .О R\
>р-^ ;р—он
Rz II R/
фосф пнистые
соли и эфиры — фосфинаты
Rx ,0
Rz ОН
фосфиновые
соли и эфиры —
фосфинаты
Для этих кислот известны разнообразные производные: ангидриды,
эфиры, амиды, тиопроизводные и т. п. Упомянутые выше алкил(арил)
дихлорфосфины ВРС12 можно рассматривать как хлорангидриды фос-
фоиистых кислот. Соответственно Для фосфоновых и фосфиновых кислот
характерны хлорангидриды:
zO R .О
R-P^Cl п
ХС1 Rz \С1
Наконец, замещением кислорода на два атома галоида (хлора) в окисях
третичных фосфинов можно получить соответствующие третичные дигало-
идфосфоры R3PC12. Это — уже производные пятихлористого фосфора,
от которого можно вывести все ступени замещения хлора на органиче-
ские радикалы:
RPC14 R0PCI3 R3PC12 R4PCI R5P
Все они известны и хорошо изучены, а некоторые из них играют важ-
ную роль полупродуктов фосфорорганического синтеза.
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ 427
Хотя фосфор является ближайшим соседом азота в пятой группе
периодической системы и многие типы органических соединений у них
формально одинаковы, тем не менее наблюдается резкое отличие в свой-
ствах и структуре их органических соединений. Это связано с различием
в строении электронных оболочек атомов азота и фосфора, поскольку
азот — элемент второго периода, а фосфор — третьего. Электронная
оболочка атома азота характеризуется заполнением слоя L, причем
на трех 2р-орбиталях находится по одному электрону. Поэтому для азота
характерны ковалентности 3 (гибридизация sp2) и 4 (гибридизация яр3).
Электронная оболочка атома фосфора содержит лишь частично запол-
ненный слон М
и кроме валентных состояний р3 (трехвалентный фосфор) и sp3 (фосфоние-
вый фосфор) фосфор способен находиться в валентном состоянии sp3d
(пятпковалентный фосфор) и даже sp3d2 (шестиковалептный фосфор,
например, в анионе PF"). Эта способность проявлять высшие ковалент-
ности очень характерна для фосфора. Благодаря ей для фосфора (и, как
мы увидим ниже, для мышьяка, сурьмы и висмута) возможны такие соеди-
нения, как, например, (СЛН5)5Р (симметрия связей фосфора — тригональная
бипирамида), которые невозможны для азота. Этим же объясняется пре-
обладание орто-форм кислородных кислот у фосфора и мета-форм у азота.
Так, для азота известна только одна азотная кислота HNO3, которая
относится к мета-ряду, подобно метафосфорной кислоте НРО3. Но для фос-
фора наиболее устойчива ортофосфорпая кислота Н3РО4, невозможная
для азота. Известны также производные полностью гидратированной
фосфорной кислоты, например (СЙН5О)5Р. Если для химии азота харак-
терны нитросоединения Ri\O2, то для химии фосфора — соответствующие
гидратированные формы: фосфоповые кислоты RP(O)(OH)2, их эфиры
RP(O)(OR)2 и даже ортоэфиры RP(OR),t.
В химии органических соединений азота наиболее устойчивыми фор-
мами являются амины, обладающие лишь очень слабыми восстановитель-
ными свойствами. Окиси третичных аминов R3N-+O и даже нитро-
соединения — вещества с выраженными окислительными свойствами
(окиси третичных аминов, например, выделяют иод из подкисленного
раствора йодистого калия). В химии фосфорорганических соединений
наиболее устойчивы производные фосфора в высших валентных состояниях;
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
428
фосфины — сильные восстановители (низшие фосфины самовоспла-
меняются на воздухе), а окиси третичных фосфинов, фосфоновые
и фосфиновые кислоты вовсе лишены окислительных свойств. С другой
стороны, есть некоторые функции азота, такие, как, например, азо-
соединения R—N = N—R, которых не существует в химии фосфора.
При попытках синтеза подобного рода веществ образуются только димеры
R—Р—Р—R
I I
R—Р—Р—R
Фундамент химии фосфорорганических соединений заложен работами
Михаэлиса и А. Е. Арбузова.
Фосфины. Первичные и вторичные фосфины, в отличие от аминов,
вещества трудно доступные. Они могут быть получены по одной из сле-
дующих реакций:
1) алкилированием фосфидов металлов
или
PH2Na+RI --> RPH2+NaI
RPHLi + R'Br —>• RR'PH + LiBr
2) восстановлением хлорфосфинов литийалюминийгидридом или три-
хлорсиланом (силикохлороформом) SiHCl3:
RPC12 RPII2
3) присоединением фосфина к непредельным соединениям по Харашу
(инициирование перекисями):
СПз
I
С8Н13СН=СН2 + РНз > С6Н13—сн—РН2
Третичные фосфины легко синтезировать действием реактивов Гринь-
яра или литийорганических соединений на треххлористый фосфор:
3RMgBr+PCl3 •—► R3P +3MgBrCl
Кроме того, они могут быть получены восстановлением окисей третич-
ных фосфинов трихлорсиланом:
(С6Н5)зРО (с6ц5)3р
В редких случаях третичные фосфины получают алкилированием
первичных или вторичных.
Первичные и вторичные фосфины при окислении в мягких условиях
превращаются в соответствующие фосфоновые и фосфиновые кислоты,
а третичные фосфины — в окиси:
о /ОН
RPH-2 —- R— PvcOH
Ч)
о R\ / О
r2ph —+ >р/
R/ \0Н
о R\
R3P —* R—Р=0
R-
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ 429
Впрочем, при очень мягком окислении можно получить и окиси пер-
Р11ЧНЫХ и вторичных фосфинов.
Низшие алифатические фосфины — очень легко окисляющиеся ве-
щества. Метилфосфин СН3РН2, диметилфосфин (СН3)2РН и триметил-
фосфин (СН3)3Р воспламеняются на воздухе.
фосфины являются лишь очень слабыми основаниями, и их соли
легко гидролизуются водой. Они легко образуют комплексные соединения
с солями тяжелых металлов, а также способны вытеснять СО из карбо-
нилов металлов:
Ni(CO)4+(C6TT5)3P —> (CGH5)3P---Ni(CO)3+CO
Некоторые комплексы трифенилфосфина с солями тяжелых металлов
являются катализаторами синтезов Реппе и аналогичных им реакций
(кн. 1, стр. 282).
При алкилировании третичных фосфинов образуются соли четвертич-
ных фосфониевых оснований, например:
(СН3)3Р + СП31 —> (СН3)4Р+ I-
(СвН5)зР + СПз1 —> (С6Н5)3РСН3 I-
Это соли сильных оснований, подобных аммониевым и сульфониевым.
Известны разнообразные соли фосфония, содержащие в углеводород-
ных радикалах другие функциональные группы, например оксигруппы.
Так тетраметилолфосфонийхлорид получается при конденсации фосфина
с формальдегидом в присутствии соляной кислоты:
РНз + 4СН2О + НС1 —> (ПОСН2)4Р + Cl-
Он применяется для пропитки тканей с последующей обработкой ве-
ществами, образующими с его гидроксильными группами полимеры.
Такая пропитка делает ткани негорючими.
Фосфониевая группа является сильным мета-ориентантом при заме-
щениях в бензольном кольце. Например, при нитровании триметил-
фенилфосфонийхлорида образуется количественно Л4-нитрозамещенное:
Р(СН3)з X- Р(СПз)з X-
1ч I
HNOHH.SOi)
V
no2
Четвертичные соли фосфония, содержащие хотя бы один а-водород,
при действии достаточно сильных оснований отщепляют молекулу НХ,
образуя фосфинметилены (Штаудингер, Виттиг):
(СвН5)3Р—CIIR2 ХЧ-СбНэЫ —> (C6H5)3P=CR2+C6ri6 + LiX
Распределение электронной плотности в фосфинметпленах среднее
между «иленовой» и «илидпой» формулами:
R3P=CR'R" <--> RaP-CR'R'
фосфорилеп фосфорилид
430
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Это весьма реакционноспособные соединения, особенно, если R'
и R" — алкил или водород; они быстро окисляются на воздухе и гидро-
лизуются. Значительно стабильнее фосфипметилены с электроноакцептор-
ными заместителями. Например:
R3P=CII-C—R <-> R;)P—СН —С—R
В соответствии с устойчивостью ацилфосфинометиленов их протони-
рованные формы, т. е. соответствующие соли фосфония, являются срав-
нительно сильными кислотами. Так, бромистый трифенилфенацилфосфо-
ний I — кислота, по силе близкая к уксусной, а бромистый трифенил-
дибепзоил метил фосфоний II — сильнее щавелевой (М. И. Кабачник,
Т. А. Мастрюкова):
(С6Н5)зРС112СОСвН5 Bi- (С811й)зРСН(СОСвН5)2 Вг-
I п
(РК'а 6,3 в 50%-пом спирте) (рк 2,6 в 50%-пом спирте)
Фосфинметилены — сильные нуклеофилы и, как таковые, реагируют
с галоидами, галоидными алкилами и ацилами и др. (Бестманн, Меркл):
R3P—CH(CH3)R' I’
+
| спя
R3?-CHR'-BF3 R3P—CHR' -(СНдС0^0^ R3P-CII(R')COCII3 ’ОСОСНз
R3P-CII(R')Br Вг- R3P=CR'—СОСН8
Стабильные фосфорилиды (с электроноакцепторными группами) гладко
присоединяют серный ангидрид и сулему (Н. А. Несмеянов, О. А. Ре-
утов):
R3P—CIIR' — Д- R3P-CH-SO3-
lngci2 R
Y
R3P-CH-UgCI Cl-
I
R'
Некоторые фосфорилиды реагируют с а-окисями образуя трехчленные
алициклы:
RsP-CIICOOC’Hs+R'HC—СП2 —> R3P-Cll—CIR—CIIR' ----->
V / I I
о СООС2На О-
—► R3P=O + R'IIC—СН-СООС2Н5
сн2
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ 431
Трехчленные циклы получаются и при взаимодействии илидов с со-
единениями, содержащими активированные двойные связи:
п сн—сн3
Р3Р=СН2 + СН3СН=СН-СООС2Н5 ----R3P-CH2^CH-COOC2H5 ------>-
СН— СН3
/ х
——>- R3P •+ Н2С—СНСООС2Нд
Наиболее важная реакция фосфинметиленов — реакция. Виттига —
взаимодействие фосфинметиленов с карбонильными соединениями (аль-
дегидами или кетонами). При этом образуются олефины и окись трифенил-
фосфина (стр. 437). Эта реакция позволяет вводить двойную связь одно-
временно с наращиванием углеродной цепи. Опа проходит в мягких
условиях с хорошими выходами, и положение двойной связи не вызывает
сомнений. В настоящее время реакция Виттига широко используется
в тонком органическом синтезе и особенно в синтезе природных соедине-
ний. Механизм этой реакции заключается в следующем:
(ceHs)3p=CRR'
O=<R„.
(csHs)3p=0 .
Первая стадия состоит в нуклеофильном присоединении реактива
Виттига (фосфинметилена) по двойной связи С = О. Далее, вероятно,
образуется промежуточное гетероциклическое соединение, которое необ-
ратимо распадается на окись фосфина и олефин.
Хлорфосфины. В большинстве своем хлорфосфины — жидко-
сти с резким запахом, дымящие на воздухе. Их получают несколькими
методами:
1) из ртутноорганических соединений и треххлористого фосфора
в довольно жестких условиях:
(C2II5)2IIg+PCl3 —> C2II5PCl2-|-C2I15HgCl
2) действием тетраалкилсвинца па треххлористый фосфор:
(C2II5)4Pb + PCl:, —> C2lIftPCl2+(C2Hr,):tPbCl
3) прямым алкилированием красного фосфора галоидными алкилами
в присутствии меди (Л. 3. Соборовскпй):
ЗСН3С1-Ь2Р —С113РС12 Ц-(СН3)2РС1
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
432
4) восстановлением комплексных соединении галоидного алкила, хло-
ристого алюминия и треххлористого фосфора.
[RPG131+[A1C14]- —> RPCl2 + AlCl3 + MgC]2
5) восстановлением соединений типа RPC14, например, метилдихлор-
фосфитом. RPC14+CII3OPC12 —> RPCI2+CH3CI+ РОС13
Ароматические хлорфосфины получаются по реакции Фриделя —
Крафтса из углеводородов и треххлорнстого фосфора под действием
хлористого алюминия (Михаэлис)
С6Нв + РС13 С6Н5РС12 + НС1
или прямым фосфорилированием бензола при высокой температуре
(Михаэлис, А. Е. Арбузов):
СвН6 + РС13 —> СвН5РС12 + НС1
Их можно синтезировать также из хлорбензола, фосфора и трех-
хлористого фосфора:
ЗС6Н6С1+2Р + РС13 —► ЗС8Н5РС12
Вторичные ароматические хлорфосфины получаются диспропорцио-
нированием первичных при каталитическом действии хлористого алюми-
ния (М. И. Кабачник, Т. Я. Медведь):
2С6Н5РС12 (С6Н5)2РС1 + РС13
Хлорфосфины, особенно ароматические, являются важными проме-
жуточными продуктами при синтезе фосфорорганических соединений
других типов. Например:
rp(or')2 ^_RJQ.ya
RPR' ^R'Kg.x
RP(NR’)2 R?NH
RPC12
RPOC12
RPSC12
RPCI4
CH2=CH-CH = CH2
Фосфонистые и фосфинистые
лоты образуются при осторожном гидролизе
фосфинов:
кислоты. Эти кис-
соответствующих хлор*
RPC12 + 2H2O —> RPO2H2 + 2HC1
Р2РС1 + Н2О > R2POH + HCI
ПЯТЛЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ 433
Они являются веществами таутомерными
/ОЛ R z0H Rx R H
R—p< ;р-он т=± >₽4
Х0Н I-K ^0 r/ r/ 'Q
фосфонпстая кислота фосфиипстая кислота
причем для кислот с алкильными или арильными группами у фосфора
равновесие в значительной степени смещено в сторону таутомеров с фор-
мально пятивалентным фосфором. Однако введение сильно электроотри-
цательных заместителей, таких, как CF3-rpynna, смещает равновесие
в противоположную сторону, и, например, бис-трифторметилфосфини-
СТая кислота соответствует гидроксильной форме.
F3CX
;р-он
F3CZ
Свободные фосфонистые и фосфинистые кислоты имеют меньшее зна-
чение. чем их эфиры и другие производные. Эфиры этих кислот — фос-
фониты и фосфиниты — получают реакцией хлорфосфинов с алкоголятами
или со спиртами в присутствии третичных аминов:
RPCI2 + 2NaOC2H5 —> RP(OC2H5)2 + 2NaCl
R2PC1 + C4II9OH + (C2H5)3N -> R2POC4H9 + (C2I-I5)3NII Cl-
Их получают также из хлорфосфитов (R'O).,PC1 или R'OPC12 по реак-
ции с магнийорганическими соединениями при низкой температуре
(М. И. Кабачник, Е. Н. Цветков).
Характерным свойством этих эфиров является их способность к пере-
группировке под действием галоидных алкилов (перегруппировка Арбу-
зова, стр. 434). Из фосфонитов при этом получаются эфиры диалкил-
фосфиновых кислот
R. .о
RP(OC21I5)2 + R'I -> РХ/ + С2Н51
R' 4ОС2Н5
а из фосфинитов — соответствующие окиси третичных фосфинов:
R2POC2H5 + RI —► R3P=O + C2H51
Эти эфиры легко присоединяют серу с образованием соответствующих
эфиров тиофосфоновых (и тиофосфиповых) кислот:
С2НБ\ z ОС2НБ
C2H5P(OC2ll5)2 + s -> />Р<
OC2U5
Фосфоновые и фосфиновые кислоты. Это, пожа-
луй, важнейшие классы фосфорорганических соединений как по много-
численности их представителей, так и по практической значимости.
Главные способы получения этих соединений следующие.
28 Заказ 672
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
434
1. Перегруппировка эфиров фосфористой кислоты (фосфитов)* или
алкилфосфонистых кислот (фосфопитов) под действием галоидных алкилов
(перегруппировка Арбузова):
гит C2II5, /ОС2Н5
Р(ОС2П5)з „
Естественно, что в случае эфиров фосфористой кислоты после пере-
группировки образуются эфиры алкилфосфотювых кислот, а из эфиров
алкилфосфонистых кислот — соответствующие эфиры диалкилфосфино-
вых кислот. При омылении эфиров легко выделяются сами кислоты.
2. Алкилирование натриевых солей диалкилфосфитов (реакция Ми-
хаэлиса и Беккера):
С2Н5. /ОС2Н5
(C2H50)oP0Na 4-С2Н51-> АР< -г Nal
са хос2н5
3. Алкилирование треххлористого фосфора галоидными алкилами
в присутствии хлористого алюминия:
RC1 + РС13 4- А1С13 [RPGl3]+[AICI4]"
Первоначально образующиеся комплексные соединения гидролизуются
водой и дают алкилфосфоновую кислоту:
[RPC13]+[A1C14]“ 4- ЗН2О -> RPO(OH)2 4- 4НС1 4- А1С13
4. Гидролиз хлорангидридов типа RPOC12 и R2POC1:
RPOCb 4- 2Н2О ---> RPO(OH)2 4- 2НС1
5. При получении ароматических фосфоновых кислот обычно исходят
из арилдихлорфосфинов:
СвН5РС12 + С12 —> СвНбРСП-^-* С6Н5РО(ОН)2 4- 4НС1
6. Специфической для синтеза ароматических фосфоновых кислот
является реакция борофторидов арилдиазониев с треххлористым фосфором
в присутствии меди (метод Фридмана и Дока):
IArN2]+[BF4[- 4- РС13 •—> [ArPCl3]+[BF4]’4-N2
[ArPCl3]+[BF4]- + ЗН2О -> АгРО(ОН)2 4- ЗНС1 4- HBF4
Существует много специфических методов синтеза фосфоновых и фос-
финовых кислот, содержащих в алкильном радикале разные функцио-
нальные группы.
Оксиалкилфосфоновые кислоты получают реакцией треххлористого
фосфора с альдегидами, обычно в среде ледяной уксусной кислоты. При
этом сначала образуется продукт присоединения сложного состава,
(пя,,.пЯ?<^П ФОСФПТЬ1 получают по Милобендзкомч' взаимодействием РС13 п спирта
(.даже первичного) в присутствии R3N.
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
435
который при гидролизе превращается в оксиалкилфосфоновую кислоту
/реакция Фоссека — Педжа — Конанта):
ОН О
zO Н2о I II , ОТТ
RCs + PCI3 ----->- Сложный продукт --> R—СП —Р<
ЧН ' "-он
Эфиры оксиалкилфосфоновых кислот легко получаются в результате
присоединения диалкилфосфитов к альдегидам и кетонам при каталити-
ческом действии алкоголятов (В. С. Абрамов):
ОН О
/О Нч /OR' | || /OR'
RC<f + ДрХ C2H5ONa^ R_CH_pZ
ХП 0х W \0R'
Из хлораля и диметилфосфита таким
распространенный инсектицид хлорофос:
СС1зС^° + Н> /ОСНЗ
ХН 0х х0СН3
образом синтезируют широко
ОН О
I II /ОСНз
СС13—СН—Р<
ХОСН3
а-Оксиалкилфосфиновые эфиры легко претерпевают при действии
щелочи перегруппировку в непредельные эфиры фосфорной кислоты
(Шрадер):
ОН О О
I II II
СС13—СН—P(OR)2 + NaOH •—* CCl2=CH-0-P(0R)2 + NaCI + H20
При нагревании альдегидов с треххлористым фосфором в запаянных
трубках или автоклавах до 200—250°С образуются хлорапгидриды
н-хлоралкплфосфоновых кислот (М. И. Кабачник, Е. С. Шепелева):
О
II /С1
СН2О + РС13 —> С1СН2Р<
ХС1
а-Аминоалкилфосфоновые кислоты получаются при конденсации
диалкилфосфитов, альдегидов (или кетонов) и аммиака или аминов
(М. И. Кабачник, Т. Я. Медведь; Филдс):
NH2 О
R\ Нч /OR R\ I || /OR'
>C=O + >P< + NH3 —> )C Px + 1I2O
R"'$5 ,0^ x0R R"x XOR'
Аминофосфоновые кислоты, как и аминокарбоновые, обладают спо-
собностью к образованию с солями тяжелых металлов внутрикомпле-
ксных соединений. Особенно сильными комплексообразователями явля-
ются полиаминополифосфоповые кислоты такого типа (М. И. Кабачник,
Н. М. Дятлова):
СН3 СНз
(НО)2ОР—С—NH—СН2—СН2—NH—С—Р0(0Н)2
I I
СНз СНз
фосфицин
28*
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
4 36
Фосфоновые кислоты обычно хорошо кристаллизуются^ они относятся
к сравнительно сильным кислотам (в водном растворе Ка О 10
При этерификации или алкилировании солеи в основном удается получать
лишь кислые эфиры этих кислот. Средние эфиры готовят через хлоран.
гидриды по схеме:
RPO(O!f)2 + 2РС15 -> RPOCU + 2POCI3 + 2UC1
R OR' +
RPOCI2 + 2R'OH + 2(C2H5)3N --> >гР- -г 2(С2Н5)зХН Cl-
0 'On
Производные фосфоновых и фосфиновых кислот находят применение
в качестве инсектицидов для борьбы с вредителями сельскохозяйствен-
ных культур. Кроме упомянутого выше хлорофоса отметим широко
используемый за рубежом для той же цели препарат ЕРЗ\ и его этильный
аналог — медицинский препарат армии, применяемый для лечения
глаукомы. OC2II5 OC2H5 j 1 ^-no2 C2H5-P-'S—no2 \=/ j] || x=z 0 0
EPN
армии
К производным метилфосфоновой кислоты относится боевое отрав-
ляющее вещество зарин
СН3х /ОСзНт-изо
Л
Это очень ядовитое вещество обладает приятным мятным запахом.
Если в фосфоновых или фосфиновых кислотах заместить кислород
на серу, то получаются тиофосфоновые и тиофосфиновые кислоты:
Rx /ОН
S^^OH
R ZS
Р^
Rz \OII
Производные тиофосфоновых кислот
серы к соответствующим соединениям
обычно получают присоединением
трехвалентного фосфора:
RPCI2 + S --->
Rx Cl
//Р<
Sz ^CI
Лучший способ получения тиофосфиповых кислот заключается в дей-
ствии магнийорганпческих соединений на диалкилфосфиты с последу-
ющим присоединением серы (М. И. Кабачник, Т. А. Маетрюкова):
R<\ /Н R\
пп/Р\п + 3R'MgX > >Р—OMgX + 2ROMgX + R'H
AlVJ ti /
^P-OMgX + S ---> R \p<^S 2^ R x.pz^S
R R'z xOMgX R'/ \он
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
437
Окиси третичных фосфинов. Эти вещества обычно
получают взаимодействием реактивов Гриньяра с хлорокисью фосфора:
,С1
РОС13 + 3RMgX —> R3P = O 4- 3Mg<
X
Их можно синтезировать также окислением третичных фосфинов
или арбузовской перегруппировкой эфиров диалкилфосфипистых кислот.
Недавно разработан интересный способ синтеза окисей третичных фосфи-
нов алкилированием иодистыми алкилами красного фосфора в присут-
ствии каталитических количеств иода (А. В. Кирсанов):
2Р + 212 —> Р214
6RI + Р214 —> 2R3PI2 + 3I2 —- r3po
Окиси третичных фосфинов — кристаллические вещества, трудно
окисляющиеся и трудно восстанавливающиеся. Они обладают слабыми
основными свойствами и легко дают комплексные соединения с солями
металлов. Эти окиси находят применение (например, окись триоктил-
фосфина) для экстракции солей урана и Других металлов из растворов.
Фосфорильная группа Р = О относится к числу электроноакцепторных
мета-ориентирующих. Так, при нитровании трифенилфосфиноксида обра-
зуется три-.и-нитротрифенил фосфиноксид:
/ Р—</ + HNO3 > 4 Р— Ч + Н2О
\=z || =/ || =/
о o2nz о \NO2
Несколько неожиданными оказались электроноакцепторные (—1- и
—М-эффекты) свойства группировок с трехвалентным фосфором. Фосфор
в этих группировках не/ет неподеленную пару электронов и, казалось бы,
подобно азоту аминогрупп, должен проявлять донорные свойства. Однако
дифенилфосфинобензойпая кислота оказалась сильнее бензойной, в то
время как дифепилайинобензойная — слабее:
(СвН5)2Р-СООН > Ч—СООН > (CeH6)2N-^ Ч_Соон
Это явление нашло объяснение в характере гибридизации неподелен-
ной пары фосфора и азота. В то время как у азота (гибридизация
близка к sp2) пеиоделеппая пара обладает выраженным р-характером,
У фосфора (гибридизация близка к р3) нелодслеиная пара находится
почти в чистом s-состоянии. Соответственно она не может взаимодей-
ствовать с л-электронами кольца (М. И. Кабачнпк, Е. II. Цветков,
Д- А. Бочвар).
Кроме фосфорорганических соединений с алкильными или арильными
группами у фосфора получены также соответствующие непредельные
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
438
соединения, производные этилена и его гомологов, ацетилена, диенов
и др., например: 0
II он /ОН
СРЬ=СН-р/ СН2=СН— СП-СП—Р
\<JH UH
винилфосфоновая кислота бутадиенилфосфоновая кислота
Винильная группа, соединенная с фосфором, легко присоединяет
в Р-поло?кение нуклеофильные реагенты.
! Фосфорорганические непредельные соединения спосооны полимери-
зоваться и сополимеризоваться с другими непредельными соединениями.
Полимеры, содержащие фосфоновые и аналогичные группы, негорючи
или трудно воспламеняются.
Известны фосфорорганические аналоги азотистых гетероциклических
систем пиррола и пиридина:
Р
Р
I
с6н5 с6н6
II
но эти соединения не обладают ароматическими свойствами, а являются
настоящими диенами (вступают в диеновый синтез и другие реакции).
Это связано с трудностью образования ароматического секстета с уча-
стием в соединении I Зр- и Зх-гибридизованных электронов фосфора (в об-
разовании ароматического секстета соединения II участвуют 3<7-элек-
троны).
Упомянем также устойчивые гетероциклические системы три- и тет-
рафосфонитрилов:
РЙ2 R2P=N-PR2
/ Ч 1 11
N Л N N
II I II I
R2₽ pr2 r2p— n=pr2
N
Молекулы трифосфонитрилов в отличие от триазинов неплоские.
Некоторые из них полимеризуются с размыканием цикла и образованием
каучукоподобных полимеров.
Стереохимия неуглеродных атомов тетраэд-
р и I е с к о и конфигурации. Катионы тетраалкилфосфония (ам-
еп₽НпИЯ’оарС0НИЯ)’ а также окиси третичных фосфинов построены подобно
“ ИЯМ углеР°Да< поскольку центральный атом связан с четырьмя
заместителями:
тэ
оптическпД16/^! °Т Симметрических солей фосфония следует ожидать
оптической стереоизомерии. Четвертичные соли аммония впервые были
jlflTAH ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
439
разделе:.и на оптические антиподы Попом в 1899 г.; четвертичные соли
фосфопия и фосфиноксиды, содержащие фосфор в цикле в качсствегею-
роатома, разделил па антиподы Мейзепхеймер в 20-х годах текущего сто-
летия. Обычные четвертичные соли фосфония удалось разделить па опти-
ческие антиподы Мак-Ивепу и Вандерверфу в 60-х годах нашего столе-
тия. Но наиболее интересное открытие было сделано Хорнером, который
в 1964 г. получил ряд оптически деятельных третичных фосфинов
R .
R' R"
Для этого он воспользовался установленным им же фактом, что чет-
вертичные соли фосфония, содержащие бензил при атоме фосфора, гладко
восстанавливаются электрохимически (катодное восстановление) до тре-
тичных фосфинов:
R\ + /К' В\
>Р< —> НЧР: + С6Н5СНз + НХ
В'/ \СН2СвНб RV
X-
Таким образом, в третичных фосфинах свободная пара электронов
фосфора удерживает конфигурацию и в этом смысле играет роль четвер-
того радикала. Это отличает третичные фосфины от третичных аминов.
Подобные стереохимические отношения были известны ранее для сульф-
оксидов, которые построены пирамидально, причем связь между кисло-
родом и серой семиполярная.
Вслед за оптически деятельными соединениями трех- и четырехкова-
лентного фосфора были выделены и соответствующие соединения мышьяка
и серы и изучена стереохимия некоторых их реакций.
•• •• ••
Р As S
rx/ r"
tx *x tx
Из соли фосфония, содержащей бензпльпую группу, можно получить
окись фосфина III либо с сохранением конфигурации, либо с обращением,
к зависимости от способа синтеза. При щелочном гидролизе происходит
инверсия:
R \ /
но + R'-^p—сн2сен5 —Н-^-1’-СН2—с6н5 —o=p-r' + CH3CGH;-
R" r" H-yOH R
I II 111
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
440
Промежуточное соединение II формально подобно переходному состо-
янию при нуклеофильном замещении у насыщенного атома углерода (^2).
Разница, однако, состоит в том, что фосфор, в отличие от углерода, спо-
собен расширять свою электронную оболочку до децета и образовывать
пятиковалентные соединения; поэтому энергия соединения II должна
лежать в минимуме па потенциальной кривой. Во второй стадии этой реак-
ции должна участвовать молекула воды, чтобы реализовать отрыв оензил-
аниона.
При действии фениллития на ту же соль I образуется реакционно-
способный илид IV, который реагирует с альдегидом по реакции Виттига
(стр. 431), давая олефин и окись фосфина Ша без изменения конфигура-
ции
R\ +
R —Р—СН2С6Н5
R"Z х_
I
СвНьЫ
(без инверсии)
Rx
RР= СН СвН5 + С6Н6 4- LiX
R"Z
IV
(без инвер-t (без инвер-1 + Н"'СНО
сип) | C.HSCH2X сии) Н'"СН=СНС«Н5
R Ог R
R'_\p. (без инверсии) q
R" ” R"Z “
Ша
так как при образовании илида не затрагивается асимметрический центр,
а реакция Виттига проходит через циклическое промежуточное соедине-
ние, которое не допускает инверсии.
При окислении третичного фосфина конфигурация также не меняется,
не меняется она и при алкилировании его галоидным алкилом, поскольку
алкильный радикал садится на свободную электронную пару, удержи-
вающую конфигурацию.
Органические соединения мышьяка. Действием гриньярова реактива
на треххлористый мышьяк можно получить триалкиларсин R3As, а при
иных соотношениях реагентов также алкилдихлорарсин RAsC12 и диал-
килхлорарсин R2AsC1. Сухой перегонкой As2O3 с уксуснокислым натрием
была получена окись какодила * (Бунзен):
О
II
4СН3—С—O\:a-:-AszO3 > (CH3)2As-O—As(ClI3)2 + Na2C03+2COa
Действии НС1 на окись какодила образуется диметилхлорарсин
\'-1 -П. з J 2 1.
Энергичное восстановление RAsC12 и R2AsC1 приводит к образованию
алкиларсипа RAsH2 и диалкиларсина R2AsH — очень ядовитых и легко
окисляющихся воздухом веществ.
Какодилом во времена теории радикалов был назван радикал (СН3)2Аз.
ПЯТАЯ ГРУППА периодической системы элементов
441
Действие иодистых алкилов на арсенит дает начало солям алкилар-
синовых (алкиларсоновых) кислот. Реакция идет с перенесением реак-
ционного центра:
СН3х /О
CH3I + Na3AsO3-> /AsK +NaI
NaCr xONa
арренал
Метиларсоновую кислоту можно восстановить, например, действием
S02 в метиларсинистую (или метилмышьяковистую) кислоту
ОН
снз—Аз/ . Метилирование ее соли приводит к соли диметилмышья-
\ОН
ковой кислоты:
ONa CH3s /О
CH3I-lCH3-As< ------> >As/ +NaI
ONa СИ/ ''-ONa
Галоидные алкилы присоединяются и к триалкиларсинам, образуя,
соли тетраалкиларсониев:
R3As/RI —> R4As+ I-
Соответствующие гидроокиси В4Аз+ОН~ — сильные основания; их
соли с сильными кислотами водой заметно не гидролизуются.
Так же как и алифатические мышьяковоорганические соединения,
известны и ароматические соединения следующих структур (Аг — арил,
X — галоид).
Соединения As (III):
ArAsH2, Ar2AsH, Ar3As; ArAsX2, Ar2AsX; ArAs(OH)2, Ar2AsOH
Аг—As—As—Аг Аг /Аг
| | (Ar As)n \As—Asz
Ar—As—As—Ar Аг Хдг
Соединения As(V):
ArAsX4, Ar2AsX3, Ar3AsX2, Ar4AsX, Ar5As;
ArAsO(OH)2, Ar2AsO(OH), Ar3AsO, Ar4AsOH~
В синтезе ароматических мышьяковоорганических соединений исходят
обычно из триариларсинов или из арилмышьяковых кислот.
Триариларсипы можно получать по Михаэлису своеобразной моди-
фикацией реакции Вгорца — действием натрия па смесь треххлористого
мышьяка и галоидного арила
ЗСвН5С16Na-j- AsCI3 ► (CgH^gAs/GNaCl
или проще, по Пфейфферу, действуя на треххлористый мышьяк феннл-
магпийгалогенидом:
3CeHbMgBr+ AsCI3 — -> (CeHehAs+ 3MgClBr
442
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Арилмышьяковые кислоты можно получить по методу Барта через диа-
зосоединения
+ CgHjx /ONa
C6II5—N=N Cl- + As(ONa)3—> AAs; +NaCl + N2
СИ xONa
пли прямым арсенировапием. В этом случае ароматическое ядро должно
быть очень активизировано к электрофильному замещению наличием
NH2, ОН и подобных заместителей. Приводим получение арсаниловой
кислоты по методу Бешама:
CeH6NIl2 + H3AsO4 -> n-NH2C6H4AsO(Ori)2-|-H2O
Через диазосоедипенпя можно получить и диарилмышьяковую кис-
лоту, если восстановить арилмышьяковую в арилмышьяковистую и затем
подействовать па нее солью диазония:
Аг ZOH ,ч ОН /О.\а
>As SO^HCD^ Ar_As Nao^ Аг-As/
OZ \OH XOH ONa
+ .ONa Ar /ONa
Ar'—N=N Cl~ + Ar—As -------> zAsW + N2 + NaCl
' ONa Ar' X)
Восстановлением диарилмышьяковой кислоты можно получить диарил-
мышьяковистую:
Аг .011 Агк
>As< SOHHCD^ \As_0[I
Ar'/ '0 Ar''
Арилмышьяковистые кислоты действием концентрированной НС1 пре-
вращаются в арил- или, соответственно, диарилхлорарсины
ArAs(OH)2 + 2HCl --> ArAsCl2 + 2H2O
Ar2AsOII Л-ИС1--> Ar2AsC14-H2O
которые в свою очередь при восстановлении цинковой пылью в кислой
среде дают арил- или диариларсины:
ArAsCl2 ArAsH2
Ar2AsCl —(Н Ar2AsH
Арил- и диариларсины не имеют сходства с аминами. Они лишены
основных свойств и являются сильными восстановителями.
При менее энергичном восстановлении ArAsCl, (соответственно Ar,AsCl)
в щелочной среде, например, фосфористой кислотой образуются арсено-
соединения и какодилы. Оба типа соединений можно также получить
взаимодействием ArAsH2 (соответственно Ar2AsH) с арил- и диарил-
хлорарсинами:
Аг—As—As—Ar
2ArAsII2 +2ArAsCl2 -► | | 4-2HC1
Ar—As—As—Ar
арсенобензол
Ar2AsII + Ar2AsCl -> Ar2As— AsAr24~HCl
фенилкакодил
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
443
Под действием галоида арсенобензол превращается в ArAsCl2 и далее
в ArAsCl4, а фенилкакодил в Ar2AsCl и затем в Ar2AsCl3.
К ArAsCl2 и At2AsC1 можно также прийти в результате диспропорцио-
нировання путем длительного нагревания AsCl3 с Ar3As:
Ar3As + AsCl3 > ArAsCl2 + Ar2AsCl
Следующими реакциями внутримолекулярного арсенирования полу-
чены гетероциклы с гетероатомом — мышьяком:
Zn(H+)
ASH
о, о'-бифениленарсин
(V
акридарсоновая
кислота
(2)
i°
(диспропорци-
онирование)
----l------->
ASC1
As Cl
+ ASClg
ди-о-фенилендихлордиарсин
(арсантренхлорид) три-о-фенилендиарсин
Арсониевые соли типа RR'R"R'"AsX- были разделены на оптические
антиподы. О расщеплении веществ типа RR'R"As уже было сказано.
Мышьяк расширяет электронную оболочку до децета легче, чем
фосфор. Пятиковалентные соединения типа Ar5As более характерны
Для мышьяка и еще более для сурьмы (Виттиг). Из арсониевых солей
типа R3As—CHR'R" X- действием оснований могут быть получены
«илиды» мышьяка — арсинметилены. В частности, синтезированы стабиль-
ные илиды типа I (Джексон) и типа II (Ник. А. Несмеянов, О. А. Реутов):
| / = AsR.3
R3As=CMCOR'
II
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
В илидах типа II радикал COR' — стабилизующая электроноакцеп-
ториая группа. Арсинметилены этого типа, подобно фосфипметиленам,
вступают с альдегидами в реакцию типа Виттига
R3As==CHCOR' + ArCHO -> ArCH = CHCOR' + R3AsO
п в некоторые другие реакции, характерные и для илидов фосфора:
+ /HgCl
R3As—СН< С1-
ХСОС6Н5
| Hgcl,
R3As=CH—СОС0Н5 — R3As-CHBr-C-C6H5
Вг- Д
|SOs
+ /SO3
R3As-CH<
ХСОС0Н5
3,3'-Диамнпо-4,4'-диоксиарсеиобензол сыграл историческую роль
в химиотерапии под названием сальварсана — первое эффективное химио-
терапевтическое средство против сифилиса (Эрлих). Ему приписывается
формула
НО—^>—As=As—ОН
h2n ^н2
Крафт доказал, что это вещество является полимером
НО-<^ Ч—As
= /
- H2N _х
Органические соединения сурьмы. Сурьма образует те же типы соединений,
что и мышьяк.
Соединения Sb(III):
RSbH2, R2SbH, R3Sb; RSbCl2, R2SbCl;
RSb=SbR; R2Sb—SbR.2
Соединения Sb(V):
RSbCl4, R2SbCl3, R3SbCl2, R4SbCl, R5Sb;
(только для
винильных и
ароматических
замещенных)
RSbO(OH)2, R2SbO(OIl), R3SbO, R4SbOH
Сурьма связана с углеродом менее прочно, чем мышьяк, и еще менее
прочно, чем фосфор. Однако эта связь еще не разрушается кислотами.
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ 445
Алифатические спшбины R3Sb проще всего получать по методу Пфейф-
фера действием гриньярова реактива на треххлористую сурьму; R3Sb
присоединяет галоид и образует R3SbX2, при пиролизе распадающийся
на R2SbX и RX. Взаимодействие R2SbX с галоидом приводит к R2SbX3,
при пиролизе которого получаются RSbX2 и RX.
Присоединение SbCl5 к ацетилену дает (ClCH = CH)3SbCl2 в виде смеси
стереоизомеров (цис- и транс-); разгонкой и восстановлением их
получают стереоизомеры (ClCH = CH)3Sb (А. Н. Несмеянов, А. Е. Бо-
рисов).
Ароматические соединения сурьмы типа Ar3Sb можно получать реак-
цией Гриньяра по Пфейфферу или более старым путем: — методом Михаэ-
лиса:
3ArCl + 6Na + SbCI3 > Ar3Sb + 6NaCl
Что касается соединений ArSbCl2 и Ar2SbCl, то их обычно получают
через диазосоединения по методу Шмидта (вполне аналогичному методу
Барта в ряду мышьяка)
+ /ОН Агх /ОН
ArN-X Cl’+NaO-Sb< --------> />Sb< + N2 + NaCl
XOH OZ XOH
+ /ONa Агч /О
ArN=N Cl- + Ar'—Sb/ ------> xSb</ +N2 + NaCl
'OH Ar'z OH
или по методу Несмеянова — Кочешкова, усовершенствованному А. Н. Не-
смеяновым, О. А. Реутовым и О. А. Птицыной (в зависимости от условий
проведения реакции можно получать один, второй или третий продукт
по выбору):
ArNsNfSbCRp-/ Zn ---> ArSbCb (или АггЭЬС!, или Ar3Sb)
Восстановлением (более или менее жестким) из ArSbCl2 получают
ArSbH2 или ArSb = SbAr. Последний, возможно, как и арсепобепзол,
представляет собой полимерное соединение. Из Ar2SbCl восстановлением
синтезируют Ar2SbH или Ar2Sb—SbAr2. Взаимодействием Ar'AsH2
с ArSbO можно получить смешанное арсеностибиносоединение с предпола-
гаемой структурой Ar'As=SbAr.
Тетраалкилстибониевые соли можно синтезировать, действуя подпе-
тыми алкилами па триалкилстибины:
(СНз)з8Ь + СН31 > (CH3)4SbI
Тетраарилстибониевые соли, в частности, можно получить по А. Н. Не-
смеянову и Л. Г. Макаровой разложением борофторида арилдиазония
в триарилстибине:
Аг*2 BFj-f-Ar3Sb •—> Аг4ьЬ
Описан ряд гетероциклов с сурьмой г качестве гетероатома.
Например:
,Sb
I
CeHB
Sb
С6Нб
446
ЭЛЕМЕНТООРГАНИЧЕСКИЕ
СОЕДИНЕНИЕ
Их получают действием двумагниевого гриньярова реактива
BrMg(CH2)„MgBr на фепилдихлорстибин (Грюттнер и Краузе^
о-Дифеиилилстибиндпхлорпд при нагревании отщепляет НС1 и .за-
мыкается в о,о'-дифенилхлорстибин (Морган).
SbCl-2
Нагреванием о-бензилфенилстибиновой кислоты с уксусным ангидри-
дом и каплей серной кислоты Морган получил стпибакридиновую кислоту
Ряд сурьмяноорганических соединений находит применение в качестве
химиотерапевтических средств для борьбы с тропическими заболеваниями,
вызванными трипанозомами и другими микроорганизмами (сонная бо-
лезнь, лейшманиоз, кала-азар и др.). Сюда относятся тг-ацетиламино-
фенилстибиновокислый натрий (стибенил, или стибазил'), Л4-хлор-тг-
ацетиламиностибиновокислый натрий (стибозан), стибаниловая кислота
и т. д.
Висмуторганические соединения. Известны следующие типы соединений
висмута:
RB1X2, R2B1X, R3Bi, R3B1X2, R4Bi+ Х~
Для ароматических соединений Виттигом получен тип Ar5Bi.
Алифатические висмуторганические соединения лучше всего получать,
действуя на трехгалоидный висмут гриньяровым реактивом (Ф. Гейн).
Полученный при избытке гриньярова реактива триалкилвисмут затем
подвергается действию галоида или BiCl3, причем образуются в зависи-
мости от соотношения реагентов R2BiCl или RBiCl,.
Так же можно получать и ароматические соединения. При действии
галоида можно изолировать R3BiX2, из которого при термическом раз-
ложении образуется R2BiX.
Ароматические висмуторганические соединения типа Ar3Bi можно
получить и через диазосоединения (Гилман; К. А. Кочешков, А. Н. Не-
смеянов)
3(ArN2Cl)2 • B;Cl3 + 2Bi -> 2Ar3Bi-j-6N2+3BiCl3
причем лучшие результаты дает метод А. Н. Несмеянова Т П Толстой
и Л. С. Исаевой: ’
3Ar№N BF*+2Bi — > Ar3Bi +3N2 + Bi(BF4)3
СЕДЬ
rxvjll СИСТЕМЫ ЭЛЕМЕНТОВ
447
ШЕСТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
Элементоорганические соединения селена и теллура. Эти вещества не па-
щли существенного применения и потому будут рассмотрены кратко.
Соединения селена впервые получены Велером. И селеновые и теллу-
ровые соединения изучались в основном Верноном и Дрыо.
Для селена наиболее важными типами соединений являются двух-
валентные производные — аналоги соответствующих сернистых соеди-
нений: RSeH, RSe — SeR, R2Se, RSeHal.
Соединения типа RSeH получаются, в частности, по реакции:
RMgX + Se --> RSeMgX —5 RSeH
Они легко окисляются в RSe—SeR.
Диалкилселениды R2Se получают алкилированием NaaSe. Соединения
типов RSeX и R2Se присоединяют галоид, переходя в RSeX3 и R2SeX2.
При действии RI на R2Se получается R3SeI — триалкилселеноний-
иодид—солеобразное, аналогичное сульфониевой соли вещество, в кото-
ром иод можно заменить на любой анион. R2Se окисляется К2Сг2О7
в R2SeO.
Ароматическое селенорганическое соединение C6H5Se—SeC6H5 можно
последовательно окислять в фенилселениновую и фенилселеноновую кислоты:
/О С6Н5ч /О
C6H5-Se^ />Se<f
ОН О' ХОН
фенилселениновая фенилселеноновая
кислота кислота
Известны гетероциклы с селеном в качестве гетероатома, например
аналог тиофена селенофен, широко изученный Б. А. Арбузовым и
Ю. К. Юрьевым. В ряду бензол — селенофен — тиофен — фуран по аро-
матическим свойствам ближе всего к бензолу селенофен.
Для теллура главными типами соединений являются: R„Te. R2TeX2,
RTeX3 и R3TeX.
Соединения с состоянием валентности теллура, аналогичным валент-
ности серы в серной кислоте, практически неизвестны.
СЕДЬМАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
Элементоорганические соединения галоидов достаточно подробно опи-
саны в разделах, посвященных галоидпроизводным жирного и аромати-
ческого ряда. Особого внимания заслуживают фторорганпческие
соединения.
Фторорганпческие соединения. Химия органических соединений фтора
начала бурно развиваться с конца 40-х годов этого столетия. Первона-
чальным стимулом явилась нужда в материалах, устойчивых к фториру-
ющему действию UF6, употребляющегося при разделении изотопов урана.
В дальнейшем оказалось, что фтороргапические полимеры, особенно
полностью фторированные, необычайно устойчивы также к окислению,
к действию различных агрессивных сред и некоторые из них превосходят
448
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
в этом отношении благородные металлы. В настоящее время применяются
пластмассы, каучуки, смазочные масла и покрытия на основе фторорга-
нических полимеров и теломеров. Некоторые фторорганические соедине-
ния употребляются в качестве хладоагентов (фреоны) и пропеллентов.
Полифторировапные соединения отличаются от обычных органических
соединений не только малой подвижностью фтора в органической молекуле,
но и особыми свойствами кратных связей и функциональных групп в этих
соединениях.
Номенклатура соединений с небольшим числом атомов фтора в моле-
куле подобна номенклатуре других галоидпроизводных. Полностью
фторированные углеводороды называются обычно перфторуглеводородами,
например CF3—CF=CF2— перфторпропилен, CF3—(CF2)3—CF3 —
перфторпептан; названия фторированных соединений, в которых имеется
один или несколько атомов водорода, хлора и др., строятся как названия
перфторсоединений, в которых определенные атомы фтора замещены
на атомы водорода, хлора и др. Например:
CF3-CF2-CFC1—СООН
а-хлорперфтормасляная кислота
CF3—CFH—CFH — CF2—CF3
2,3-дигидроперфторпентан
Методы получения,
ловой эффект фторирования
1. Фторирование углеводородов. Теп-
RH + F2 —> RF + HF
АН = — 104 ккал
значительно превосходит тепловой эффект разрыва С—С-связей. Поэтому
фторирование органических соединений фтором приводит к деструкции
органической молекулы (конечный продукт — четырехфтористый угле-
род). Для практического осуществления фторирования смесь реагирую-
щих газов разбавляют азотом п пропускают над медной сеткой или струж-
кой, покрытой кобальтом или никелем. Медная сетка служит для отвода
тепла; с углеводородами реагирует образующийся на ее поверхности
трехфтористый кобальт, а фтор регенерирует этот высший фторид:
2CoF3 + RH —> RF + 2C0F2 + HF
2C0F2+F2 —> 2CoF3
Для замещения водорода на фтор часто используют непосредственно
трехфтористый кобальт (можно применять также NiF3 или MnF4).
2. Электрохимическое фторирование (Саймонс). Метод применяется,
как правило, для получения полностью фторированных кислот, аминов,
простых эфиров и некоторых органических соединений серы. При этом
электролиз раствора органического соединения ведут в безводном фто-
ристом водороде (никелевый анод, напряжение 5—6 в, плотность тока
0,02 а!см2). Образующийся при электролизе фтор замещает атомв1
водорода:
IIF
(электролиз)
--------------* 2CF3COF
(СПзСО)2О
HF
(электролиз)
(CH3)3N
(CF3)3N
СЕДЬМАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ
44Я
Электрохимическое фторирование алифатических углеводородов за-
труднено из-за их плохой растворимости во фтористом водороде.
3. Замещение хлора на фтор. Хлор в органических соединениях
может быть замещен на фтор при помощи SbF3 (Сварте), а также AgFo
или HgF3. Подвижные атомы галоида (например, в а-хлорзамещенных
кислотах или в этиленхлоргидрине) замещаются при обменной реакции
с KF или NH4F в полярных растворителях. Для замещения хлора в хло-
ристых алкилах требуются значительно более жесткие условия: нагре-
вание с AgF2 или HgF3. Легче обменивается хлор в трихлорметильной
группе:
AikCCl3 + SbF3 —> AlkCCi2F + SbF2Ci
ArCCl3 + SbF3 —> ArCF3 + SbCl3
4. Замена гидроксила и карбонильного кислорода в спиртах, альде-
гидах, кетонах и кислотах с помощью четырехфтористой серы:
R2C=O —A R2CF2
RC<^° —A RCF3
"ОН
5. Присоединение фтористого водорода по кратным связям и к а-окисям
(эпоксидам):
>C=C<^+HF —> ^CII-CF^
Н2С—CH24-HF —> HOCH2-CH2F
\)
6. Получение перфторолефинов дегалоидированием галоидполифтор-
алканов:
C1CF—CF2 + Zn —> ClCF=CF2 + ZnCl2
Cl Cl
Это, в частности, промышленный способ синтеза трифторхлорэтилена.
7. Пиролиз тефлона (политетрафторэтилена) приводит к смеси трех
основных продуктов:
СЕзх
(CFo-CF2)ff 60°- 750°с-> CF2=CF2 + CF3-CF=CF2+ >C=CF2
“ CF3
Изменяя давление и температуру, можно сдвинуть процесс в сторону
образования одного из этих продуктов.
В промышленности так получают перфторпропплен. ,
Перфторэтилен готовят пиролизом хлордифторметана при 6ч0 С.
2HCC1F2 ► 2HC1 + CF2=CF2
Реакция, вероятно, идет через образование дпфторкарбена.
8. Ароматические соединения фтора, например фторбензол, получают
разложением борофторидов арилдиазониев (Ьальц и Шимапн, стр. 110).
29 Заказ G72
ЭЛЕМЕНТООРГАНИЧЕСКИЕ
450
Гексафторбепзол получен дефторированием перфторциклогексана прида.
греванпи над порошком железа:
CF2 F
+ \ F Д /F
F2C CF2 Fe (450° С) Д/
I I----------------J А,
f2c cf2 f \f
xcf2 f
По способу H. H. Ворожцова гексафторбепзол (наряду с хлорпента-
фторбепзолом) получается из гексахлорбензола по обменной реакции
с фтористым калием при нагревании под давлением:
С.С1« CsFsC1^c,Fg
Свойства фторзамещенных углеводородов. За-
мещение в органической молекуле одного из атомов водорода на фтор
ведет к небольшому повышению температуры кипения. Напротив, пер-
фторуглеводороды кипят ниже соответствующих углеводородов; исклю-
чение представляют только первые члены гомологического ряда.
Такое необычное явление говорит о малых межмолекулярных взаимо-
действиях в полифторнрованных углеводородах (табл. 75). Еще более
резкое понижение температур кипения происходит при переходе от соеди-
нений с функциональными группами к их перфторированным аналогам.
Так, ацетонитрил кипит при +78° С, а перфторацетонитрпл при —64° С.
Таблица 75- Температуры кипения фторированных углеводородов
(в скобках для сравнения приведены т. кип. углеводородов)
Формула Температура кипения °C Формула Температура кипения °C
Предельные
cf4 — 128 (—161,5) «-CeFi4 58 (68,7)
c2f6 -79 (-88,6) H-CvFie 82 (98,4)
«~Сз?8 —39 (—42,1) h-CsF18 —
;t-C4F4o + 1 (-0,5) H-CgF2o 125 (150,8)
и-СбРгг +30 (+36,1) 4-Ci0F22 238—240 (286.8)
Непредельные
cf2=cf2 —76 CF3—CF=CF2 —29
ch2=cf2 -82 CF3—CH=CHo —28,3
ch2=chf —72 CF3— CH=CHF — 16
chf=cf2 —51 CF3— CF2—CF=CFo + ‘
cf2=cfci —26,8 CF3—CF=CF—CF3 0
CFC1=CFC1 —22 (CF3)2C=CF2 +7
CF2=CFBr +2 CF3—(CF2)2—cf=cf2 +30
СЕДЬМАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ 451
Атомная рефракция фтора в фтороргапических соединениях непостоянна
и колеблется от 1,29 до 1,0ч6. Малое значение рефракции говорит о малой
поляризуемости связи С—F. Это, наряду с большой прочностью
С — F-связи (1U 1 — 114 ккал/моль против 78 ккал/моль для С—Cl-
связи), обусловливает особую неподвижность фтора в его органических
соединениях. Особенно инертны перфторалканы, которые ни в какие
реакции замещения не вступают. Другие атомы галогенов в полифтори-
рованиых соединениях (но не в полифторгалогенбензолах) тоже пас-
сивны, но в меньшей степени. Например, из соединений типа CF3—CF2I
получается магнийорганическое соединение CF3—CF2—Mgl (вспомним,
что из таких соединений, как СС13Вт илиСН2С1—СН2Вт, нельзя получить
магнийорганическое соединение; так, из СН2С1—СН2Вг вместо этого обра-
зуется этилен).
Фторсодержащие олефины способны присоединять сильные электро-
филы, но скорость такого присоединения меньше, чем для обычных олефи-
нов, и быстро падает по мере возрастания количества атомов фтора при двой-
ной связи или по соседству с ней. Фтор при двойной связи так же ориенти-
рует присоединение галоидоводорода, как и хлор (и по тем же причинам):
CH2=CF2 + HF---> СНз—CF3
Серный ангидрид присоединяется к перфторолефинам ^образованием
fi-сулътонов (аналоги (3-лактонов):
F\ ZF
• С_С z
F\ /F Fz I I ^F
;c=c; +so3 —> 0—s=o
F/ \F И
О
В безводной фтористоводородной кислоте азотная кислота диссоции-
рует с образованием нитроний-катиона. В этих условиях происходит
сопряженное присоединение нитроний-катиона и фтор-аниона по двойной
связи:
HF+2HNO3 —> HsCH + F' + NO^ + NOj
cf2=cf2+no++f-------> CF3-CF2NO2
Для перфторолефинов особенно характерны реакции нуклеофильного
присоединения, например спиртов и аминов, с образованием простых эфи-
ров и аминов:
CF2=CF2 + HOR > CHF2—CFZOR > CF2=CF—OR-J-1IF
CF2=CF2 + R2NII —> ciif2-cf2-nr2
Эти необычные для олефинов реакции осуществляются благодаря
крайнему обеднению электронами двойной связи из-за наличия атомов
Фтора. Естественно, что в следующем ряду возрастает склонность к нуклео-
фильному и уменьшается склонность к электрофильному присоединению:
CF2=CF—or cii2=cf2 cf2=cf2 cf3—cf=cf2 (CF3)2C=CF2
Оказалось что фтористый водород (а также фтористый нитрозил)
присоединяется к первым членам этого ряда по электрофильному, а
29*
459 ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
к последним членам ряда — по нуклеофильному механизму (И. Л. Кну-
нянц, Б. Л. Дяткин). Скорость присоединения фтористого водорода
падает в этом ряду и вновь возрастает для перфторизобутилена.
Электрофильное присоединение:
х—-х . в"
H+xCF^CF-OR ^-ДЛе-Н0> HCF2-CF-OR -CTp°> HCF2~CF2OR
Нуклеофильное присоединение:
CF3\ медленно CF3k _ быстоо
>C=CF2+F- >С->CF3 --->CH-CF3
CF3Z CF3Z CF3Z
Эта реакция катализируется солями фтора; значит, общую скорость
реакции определяет присоединение фтор-аниона к двойной связи. Так же
6 - 5 +
обстоит дело и с присоединением фтористого нитрозила F—NO: в присут-
ствии фторида калия он быстрее присоединяется к перфторпропилену
и перфторизобутилену.
Ориентация при нуклеофильном присоединении к перфторолефинам
объясняется следующим образом. Трифторметильные группы более элек-
троноакцепторны, чем атом фтора, поэтому в анионе I отрицательный
заряд более делокализован, чем в анионе II.
CF3t. _ F^ _ CF3
J>C—>CF3
CF3Z Fz I XCF3
F
i и
Таким образом, на первой стадии присоединения фтористого нитрозила
или фтористого водорода перфторизобутилен образует более энергети-
чески выгодный анион II (к которому присоединяется далее соответству-
ющий катион). По И. Л. Кнунянцу, это объясняется гиперконъюгацией
о-связей атомов фтора трифторметильных групп с л-связями или отри-
цательным зарядом (парой электронов):
Перфторолефины способны к реакциям радикального присоединения-
Так, они присоединяют хлор или бром только по свободнорадикальному
механизму при освещении. Таким же образом присоединяется и двуокись
азота:
CF2=CF2+2.NO2 O2N-CF2-CF2-ONO-LO2N-CF2-CF2-NO2
Радикальный характер имеет также полимеризация или сополимери-
зация перфторолефинов:
nCF2=CF2 —>----(— CF2— CF2--)„-
СЕДЬМАЯ ГРУППА ПЕРИОДИЧЕСКОЕ СИСТЕМЫ ЭЛЕМЕНТОВ
453
Перфторпропилен в отличие от перфторизобутилена способен к сопо-
лимеризации с обычными олефинами.
Из полимеров наибольшее значение имеет тефлон (политетрафтор-
этилен) — термопластик с температурой размягчения около 425° С.
Он совершенно инертен к действию кислот, щелочей и окислителей (кроме
фтора). Используется для производства химически стойких сосудов,
деталей механизмов, для нанесения антикоррозионных покрытий на метал-
лические поверхности. Тефлон обладает очень низким коэффициентом
трения и употребляется для тефлонирования или изготовления трущихся
поверхностей (поршней и пр.). Для производства пластмасс употребляется
также трифторхлорэтилен, синтезируемый по следующей схеме:
CCI3-CCI3 ——CCIF2-CFCI2 —•- CF2=CfC1
Трифторхлорэтиленовые теломеры используются в качестве химически
стойких смазочных масел. Химически стойкие Лторированные каучуки
получают сополимеризацией перфторпропилена с дпфторэтпленом и дру-
гими фторолефинами.
В химию полпфторароматических соединений большой вклад сделан
Н. Н. Ворожцовым мл.
Во фторбензоле фтор малоподвижен, подобно хлору в хлорбензоле.
Однако в п-нитро- и. особенно, в 2,4-дпнптрофторбензоле атом фтора
очень легко нуклеофильно замещается. Таким же образом в гексафтор-
бензоле два из шести атомов фтора способны к легкому нуклеофиль-
ному замещению. Гексафторбензол реагирует с аммиаком, аминами,
алкоголятами и др., обменивая в зависимости от условий один или два
атома фтора (в пара-положениях):
Очевидно, шесть атомов фтора настолько обедняют бензольное кольцо
электронами, что появляется возможность нуклеофильном атаки.
Своеобразным «нуклеофильным вариантом» реакции Фриделя ^Ра-
фтса является метод алкилирования перфторароматических соединении
фторолефинами в присутствии катализатора — фтористою калия.
F
+ CF3—CF=CF2
CF.4-CF-CF3
F I F
KF
F N f
F
F I F
CF3—CF N F
CFs
454 ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Первый акт реакции состоит в нуклеофильной атаке перфторолефина
фтор-аппоном. Образующийся при этом перфторалкил-анион нуклео-
фильно замещает один из атомов фтора в ароматическом кольце. Эта
реакция, так сказать, обратно аналогична реакции Фриделя — Крафтса
между ароматическими соединениями и олефинами в присутствии кислот
Льюиса. В классической реакции Фриделя — Крафтса роль катализатора
сводится к индуцированию положительного заряда на атакующей частице-
замещается водород, который уходит в виде протона. В приведенной
реакции, открытой Чамберсом, атакующая частица — анион, а замеща-
емый фтор уходит в виде аниона.
В отличие от перхлорвинильных групп перфторвинильные обменивают
атом фтора на органический радикал при действии литийарилов
(К. А. Кочешков, Т. В. Талалаева):
ArCF=CF2 + LiCeH4Li-ra ->• ArCF=CF— CeH4-CF=CFAr-n
Фторсодержащие соединения с функциональ-
ными группами. Фторированные соединения, содержащие функ-
циональные группы, получают либо электрохимическим фторированием
(стр. 448), замещением атомов хлора на фтор или же из перфторолефинов.
При окислении перфторолефинов могут получаться кислоты или кетоны:
CF3-CF2-CF=CF2 cf3-cf2-cooh
CF3\n Пг КМпи» CF3\
\C=CF2 ---------/0=0
CF3' CF3'
Последний способ наиболее удобен для получении перфторкетонов.
Окислением перфторолефинов в щелочной среде могут быть получены
а-окиси
CF3— OF— CF2
Нитрозо- и нитросоединения можно получить сопряженным при-
соединением соответствующих групп к двойным связям, о чем уже гово-
рилось. Присоединением спиртов и аминов к перфторолефинам получают
фторированные простые эфиры и амины.
Свойства фторированных кислот, кетонов, спиртов, аминов и Др-
тесно связаны с электроноакцепторным действием атомов фтора и поэтому
наиболее ярко выражены у перфторпроизводных, свойства которых мы,
главным образом, и рассмотрим. Отрицательный индуктивный эффект
фтора сказывается прежде всего на кислотности и основности функцио-
нальных групп. Так, перфтортриэтиламин не является основанием,
а фторированные спирты представляют собой слабые кислоты. Вот ряд
значений р/<а в водном растворе (И. Л. Кнунянц, Б. Л. Дяткин,
Н. П. Гамбарян, Е. М. Рохлин):
СРзСЩОН . . . 12,8 (CF3)2CHOH ... 9,3 (CF3)3COH . . .5,4
Свойства перфторкетонов также были исследованы И. Л. Кнунянцем
с сотр. Эти соединения обладают чрезвычайно реакционноспосоонои
СЕДЬМАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ ЭЛЕМЕНТОВ 455
карбонильной группой. Так, они способны присоединять воду или спирты,
образуя очень прочные гидраты или полуацетали:
CF3. /ОН CF3x /ОН
„ /С\ >С<
CF3 \ОН CF3Z \QR
Подобные аддукты не отщепляют воду или спирт даже при нагревании
в вакууме. Гидраты типа гидрата перфторацетопа являются слабыми
кислотами (порядка уксусной). Перфторкетоны дают устойчивые аддукты
с аммиаком и аминами
CF3s CF3X /ОН
'C=O + RNH2—> )с<
CF3Z CF3Z XNHR
которые даже перегоняются без дегидратации.
Имин гексафторацетона присоединяет аммиак, образуя геминальный
диамин — редкая комбинация атомов в органической химии:
CF3x Cf3x zNH2
>C=NH+NH3 —> C<
CFZ CF/ xnh2
Как можно видеть, для органической химии перфторированных соеди-
нений молекулы, содержащие при одном атоме углерода гидроксил
и аминогруппу, две аминогруппы, два гидроксила, гидроксил и фтор,—
нормальное явление. Устойчивость таких геминальных соединений
объясняется трудностью отрыва аниона (—OR, —ОН, —NH2) от положи-
тельно заряженного атома углерода.
Как уже говорилось, при присоединении спиртов и аминов к пер-
фторолефинам получаются простые эфиры и амины. В этих соединениях
геминальные ct-атомы фтора оказываются довольно подвижными и гидро-
лизуются с образованием производных фторкарбоповых кислот:
CF3-CF2-NR2 —° cf3-c~nr2
Фторсодержащие карбоновые кислоты отличаются высокими констан-
тами диссоциации:
ка-10~5
(при 20° С)
СНзСООН..................... 1-8
FCH2COOI-I................ 220
F2CHCOOH ................. 57 000
CF3COOH................... 59 000
CF3(CF2)3COOII ........... 68000
Трифторуксусная кислота по константе диссоциации приближается
к сильным минеральным кислотам. Эта кислота и ее ангидрид приме-
няются для защиты и ацилирования амино- и оксигрупп, поскольку
трифторацетильная защита легко снимается гидролизом. Смешанные
ангидриды трифторуксусной кислоты и других органических кислот
456
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
применяются как хорошие ацилирующие агенты, причем в этом случае
именно ацилы обычных органических кислот вступают на место водорода
в ацилируемые соединения. Трифторуксусная кислота используется
иногда как полярный протонный растворитель.
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ
Переходные металлы с незаполненными d-электронными уровнями не об-
разуют прочных алкильных и арильных производных (исключение со-
ставляет платина). В этом отношении особое место занимают металлы
2-й подгруппы I группы — медь, серебро, золото. Они завершают длинные
периоды, их d-уровни уже заполнены, но d-электропы еще способны
участвовать в химических превращениях. Образуемые ими металло-
органические соединения примыкают к соединениям переходных элемен-
тов. Медь и серебро способны образовывать крайне неустойчивые алифа-
тические производные RCu и RAg, которые получаются при действии
гриньярова реактива на Gul и AgCl при глубоком охлаждении, но уже
разлагаются при сравнительно низких температурах. Более устойчиво
полученное таким образом фенилсеребро G6H5Ag, распадающееся, однако,
уже при —18° С, и фенилмедь С6Н5Си, разлагающаяся при +80° С. Еще
прочнее металлоорганические соединения трехвалептного золота, из кото-
рых известны (Поп и Гибсон) типы [RAuCl2j, и [R2AuC1]2. Первый тип
можно синтезировать не только гриньяровой реакцией, но в аромати-
ческом ряду и прямым аурированием— действием АпС13на ароматические
соединения (Хараш):
АиС1з+С6Нб —>• CeH5AuGl2-LHGl
Известен ряд золотоорганических соединений жирного, аромати-
ческого и гетероциклического рядов. Эти вещества устойчивы не только
по отношению к воде, но и к кислотам и воздуху. Получены также
металлоорганические производные одновалентного золота, но обязательно
с координационно присоединенным вторым лигандом:
СНзАи • Р(С2Н5)3 (т. пл. 62° С); CeHsAu • Р(СбН5)3 (т. разл. 152° G) п ДР-
Одновалентное золото образует с олефинами л-комплексы АиС1-С,.Н2я
получаемые действием олефина (в избытке) на HAuCR в водном растворе.
При этом Au (III) восстанавливается в Ап(1), и комплекс выпадает в оса-
док. Описаны только соединения с циклоолефинами (Хюттель, 1965 г.)-
Как уже отмечалось, переходные металлы (за исключением платины
и в какой-то мере титана) не образуют устойчивых алкильных и даже
арильных соединений, если только не заполнить тем или иным путем их
свободные d-электронные уровни. В этом случае алкильные или арильные
соединения (обычно все же относительно малоустойчивые) получаются-
В качестве характерного примера приведем соединения хрома. Гало-
гениды трехвалептного хрома не дают алифатических металлоорганических
соединений, например, при действии гриньярова реактива, полученного
из галоидных алкилов или галоидного бензила. Но действие хло-
ристого бензила па водный раствор гидрата двухлористого хро^а
приводит к образованию необычного гидрата металлоорганическо
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ
457
катиона — бензилхрома, обязанного своим существованием заполнению
d-уровней хрома за счет комплексно связанных молекул воды:
C6Il5CH2Cl-J-2Cr(()JI2)6Cl2 -> [С6Н5СН2Сг(О112)5]С12 + Сг(ОН2)вС13 + Н2О
В качестве других примеров стабилизации алкильных производных
переходных металлов за счет заполнения их d-уровней комплексообразо-
ванием можно привести следующие (подробнее см. стр. 481):
(медленно разлагается
при 20°С, быстро при 100°С)
(т. разл. 80-100°С)
СН3Мп(СО)5
(т.пл. 95°С)
НС—СН
/О'
нс^сн
сн
'Ее-СНз
ОС СО
Р(С6Н5)3
СН3-Со(СО)3
(т. пл. 140°С)
НС—СН
/О\
нс<>сн
сн
СН3-\г-р(с6Н5)3
(т. разл. 78-82°С)
(т.пл. 115-118°С)
Ссн5>ря<р(с"из)3 (ceHs)5p cGH5 сн3>р Р(С2Н5)3 сн3 Р(с2н5)3 СН3 Р(С2Н5)3 , 3>Pt< 5/3 (с2н5)3р хсн3
(т. пл. 99-100°С) цг/е-изомер (т.пл. 81-82°С) тдаяс-изомер (т.пл. 76-79°С)
(двухвалентные Pd и Pt, квадратная координация)
Что касается металлоорганических соединений переходных металлов
с алкилами или арилами без лигандов, заполняющих d-уровни металла,
то еще в 1907 г. Поп и Пичи действием йодистого метилмагнпя на хлорную
платину получили (CH3)3PtI, а Гилман выделил CH3PtI3, (CH3)2PtI2
и (CH3)4Pt. Все эти соединения довольно устойчивы. Тетраметилплатина—
бесцветное, растворимое в бензоле вещество.
После десятилетий неудачных попыток неожиданностью явился син-
тез CH3TiCl3 и C2H5TiCl3 действием соответствующих алкилалюминиев
на TiCl4. Реакцию проводят при —80° С, однако и при комнатной темпе-
ратуре эти вещества некоторое время устойчивы; менее устойчив
(CH3)2TiCl2, a (CH3)4Ti устойчив лишь при температуре ниже —78° С.
Все они были получены в виде фиолетовых кристаллов, что, вероятно,
объясняется примесью трехвалентного титана.
Описаны также диметил- и дифепилмаргапец, которые были получены
при низкой температуре по реакции:
2RLi-(-MnI2 -> R2Mn-f-2LiI
Аналогично получен и неустойчивый СН3Мп1.
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
458 , —_________-------------------— __.
Дифенилмар ганец, как все ароматические соединения, более устойчив,
чем его алифатический аналог. Диметил марганец ни в чем нераствори-
мый взрывчатый порошок, самовоспламеняющийся на воздухе.
О саморазложении других алкильных и арильных соединений пере-
ходных металлов в реакционных растворах их солей и гриньярова реак-
тива см. стр. 398, опыты Хараша.
Если для непереходных металлов их алкильные и арильные соединения
являются по существу единственным типом истинных металлооргани-
ческих соединений, т. е. соединений с М — С-связью, то совершенно иначе
обстоит дело с переходными металлами. Главными типами металлоорга-
нических соединений переходных металлов являются следующие: а) кар-
бонилы металлов, например пентакарбонил железа; б) циклопентадиениль-
ные сэндвичеобразные производные — металлоцены (например, дицикло-
пентадиенилжелезо, или ферроцен, стр. 481 и приведенные на стр. 483
л-цпклопентадиенильные производные металлов); в) металлоарены, т. е.
комплексы переходного металла с ароматическими молекулами, например
дибензолхром (стр. 477); г) олефиновые (диеновые и т. д.) л-комплексы,
например давно известная соль Цейзе.
Fe(co)5
пентакарбо-
нил железа
дибензол-
хром
СН2 CI
II-->Pt Cl
СН2 Cl
дициклопента-
диенилжелезо
соль Цейзе
Между всеми этими типами существуют и комбинированные соедине-
ния — аренкарбонилы, металлоценкарбонилы, аренметаллоцены п т. Д.
Мы разберем вкратце металлоорганические соединения переходных
металлов в указанной последовательности, предпослав этому некоторые
соображения.
Уже из формул карбонилов металлов Fe(CO)5, Со2(СО)8, Ni(CO)4,
Сг(СО)6 и аренов (см. дибензолхром) ясно видно, что старые привычные
понятия валентности для этих соединений неприменимы. Это та же проб-
лема, которая встает перед химиком-неоргаником при изучении комп-
лексных соединений переходных металлов и которая уже в начале столе-
тия привела к необходимости введения понятия координационного
числа.
Хотя в сущности каждое отдельное соединение представляет собой
особый случай, можно, однако, подметить некоторые эмпирические
качественные правильности, которые не соблюдаются вполне строго,
но позволяют ориентироваться в области этих соединений без специальных
расчетов.
1. Правило эффективного атомного номера (Сиджвик). Лиганды
СО, С5Н5, С8Н8, олефины и т. п. присоединяются к переходному металлу,
дополняя его электронную оболочку до числа электронов ближайшего
инертного газа. Каждая молекула СО или олефин (а также амины,
фосфины, эфиры фосфористой кислоты и т. д.) поставляет металлу Два
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИИ ПЕРЕХОДНЫХ МЕТАЛЛОВ
459
электрона, каждый циклопентадиенил — пять, каждый бензольный
цикл — шесть. Приведем для ряда карбонилов металлов число электро-
нов, приходящееся на один атом металла (сумма собственных электронов
и электронов, поставляемых лигандом):
V2(CO)12
23+12=35
Сг(СО)в
24 + 12=36
Мп2(С0)10
25 + 10 = 35
Fe(CO)5
26+10 = 36
Со2(СО)8
27+8 = 35
Ni(CO)4
28 + 8 = 36
Как видно из подсчета, действительно эффективный атомный номер
в карбонилах (т. е. число электронов в оболочке металла) равен атомному
номеру ближайшего инертного газа — криптона (36). Для металлов
с нечетным атомным номером (V, Мп, Со) сумма электронов в расчете
на один атом металла составляет только 35, и эффективный атомный
номер, равный атомному номеру криптона, получается путем сдваивания
молекулы и обобщения атомами металла пары электронов. Это же может
быть достигнуто путем[захвата одного электрона от атома щелочного
металла или водорода в соединениях типа H2Fe(CO)4, NaMn(CO)5.
Такой же расчет справедлив и для наиболее устойчивых сэндвичей —
ферроцена, кобальтициний-катиона, дибензолхрома:
Fe-^ 26 + 10 = 36
Однако правило эффективного атомного номера определяет только
наиболее устойчивые системы. Существует множество веществ, у которых
число электронов больше, и особенно часто — меньше числа электронов
в ближайшем инертном газе. Таковы, например, катион, соответствующий
ферроцену, — феррицииий-катиои с 35 электронами, нейтральный ко-
бальтоцен с 37 электронами и т. д.
В соединениях, подобных карбонилам, дибензолхрому, состоящих
из атома металла и молекул, способных к самостоятельному существова-
нию (в наших примерах СО и CfiHe), металл формально считается нуль-
валентным. Условность такого определения очевидна. По Сиджвику,
валентность может быть определена как число электронов, предоставлен-
ное атомом партнеру (в полное и совместное обладание). Если считать,
чтов рассматриваемых соединениях только лиганд поставляет электроны,
а металл свои сохраняет, то нульвалентность металла соответствует
определению Сиджвика. На деле, однако, электроны металла, в част-
ности электроны d-уровней, участвуют в образовании связей (back-
donation — обратное предоставление электронов металлом).
2. В сэндвичеобразных цепах (и аренах), несмотря на делокализацию,
как бы соблюдается обычная формальная валентность металла, если
460
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
только допустить, что на присоединение цикла к металлу используется
одна единица валентности, как ясно из следующих сопоставлений:
железо (11)
титан (IV)
ванадий(V)
Как мы видели на примере борорганических и алюминийорганических
соединений, а также на примере всех только что приведенных типов
металлоорганических соединений переходных металлов, использование
языка метода валентных схем для описания их электронной структуры
встречается с большими трудностями. Даже чисто качественное описание
обычно заставляет обращаться к рассмотрению очень большого числа
резонансных структур. Так, Полинг для молекулы ферроцена, ограничи-
ваясь лишь ковалентными структурами, рассматривает 560 таких струк-
тур, сгруппированных в 11 классов, причем им вводились еще упроща-
ющие допущения о весе структур.
В методе валентных схем привлекается гибридизация орбиталей
металла, как это было описано (кн. I, стр. 251) для углерода в разных его
валентных состояниях: sp3, sp2, sp. В случае переходных металлов наряду
с s- и р-орбиталями гибридизации подвергаются также d-орбитали. Так,
например, наиболее обычный октаэдрический тип комплекса имеет
</2хр3-гибридизованные орбитали металла, направленные по осям окта-
эдра; квадратный тип комплекса, к которому, в частности, отно-
сятся многие соединения никеля, палладия и двухвалентной платины,
папример соль Цейзе [C2H4PtCl3l_K+, рассматривается как имеющий
йзр2-гибридизоваш1ые орбитали металла; тетраэдрические комплексы,
такие, как карбонил никеля, имеют хр3-гибридизованные орбитали ме-
талла. Гибридизация типа dsp3 положена в основу тригонально-бипира-
мидальных комплексов типа Fe(CO)5. Таким образом, число выравни-
ваемых гибридизацией орбиталей металла, входящего в соединение,
равно его координационному числу. Однако ряд аргументов, особенно
малая вероятность большого отрицательного заряда на металле в карбони-
лах илив анионе Fe(CN)4 при предположении об односвязности с металлом
каждой группы СО или CN и данные о сокращенном расстоянии М—G
и увеличенном С—О, заставляют предположить известную степень двое-
связностп в связях металла с углеродом групп СО и CN за счет предостав-
ления металлом своих электронов. Вследствие этого для ряда комплексов
приходится привлекать представление о резонансе с участием структур,
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ /£gj
содержащих двойные связи М = С. Например, для гексакарбонила хрома
и феррицианид-аниона даются следующие структуры:
°\\ ^0+
сХ
\з-/-
+О=С—Сг=С=О <--->
// \
о//С
% z C^N‘
-N=C=Fe-feN <----->
+ЧС (Z°
\з-/
О=С=Сг—С=О+
,cZ
+О^ ^0
-1\\ч
\с (X
N=C—Fe=C=N“
Z \
-Nz^C C\N
соединении пере-
Рис. 106. Образо-
вание октаэдриче-
ского комплекса.
Гораздо плодотворнее здесь метод молекулярных орбиталей (кн. 1,
стр. 241). С точки зрения молекулярно-орбитального описания электрон-
ной структуры рассматриваемых металлоорганических
ходных металлов связи лигандов с металлом осущест-
вляются в них общей системой электронов, распреде-
ление которых по молекулярным орбиталям и соот-
ветствующим уровням орбитальных энергий опреде-
ляется условием минимума полной энергии этой
системы электронов. Таким образом, в металлоорга-
нических соединениях этого типа содержатся дело-
кализованные связи металла с лигандами.
Рассмотрим молекулярно-орбитальный подход к опи-
санию электронной структуры наиболее распространен-
ных октаэдрических (рис. 106) комплексных соединений
переходных металлов. Пусть это будут металлы Сг, Мп,
Fe, Со с их атомными орбиталями 3d. 4s и 4р, которые предстоит
объединить в молекулярные орбитали с парами электронов, при-
соединяющихся к металлу лигандов. Это могут быть пеподеленные пары
электронов кислорода, серы, азота, фосфора, окиси углерода и т. д. или
л-пары олефинов, ароматических циклов и т.д. На рис. 107 представлены
атомные орбитали металла, используемые для образования октаэдриче-
ского комплекса и приближающиеся к ним орбитали электронов лигандов,
участвующие в образовании молекулярных орбиталей. Лишь Зс?с2-орби-
таль изображена без приближающихся орбиталей лиганда.
Разный тип перекрывания орбиталей металла и лиганда и разный
характер симметрии итоговой молекулярной орбитали обусловливают
ее так называемый о-, или л-, или 6-характер. Волновая функция ст-орби-
тали не меняет знака ври повороте на 180 вокруг оси симметрии. Вол-
новая функция л-орбитали при таком повороте меняет знак на проти-
воположный. Волновая функция 6-орбитали дважды меняет знак при по-
вороте иа 180°.
462
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Для металлоорганических комплексов важно, что не только металл
получает электроны лиганда на молекулярные орбитали комплекса
образованные с участием d-орбиталей, но что и металл дает свои (7-элек-
троны на новые молекулярные орбитали лиганда. Эти орбитали образу-
ются за счет перекрывания несвязывающих орбиталей лиганда и (7-ор-
биталей металла, как изображено, например, на рис. 106 для ком-
плекса металла с олефином. К такому принятию электронов от металла
(back-donation) способны такие лиганды, как олефины, арены, фосфины,
окись углерода, но не молекулы аммиака или воды, чем и объясняется
большая прочность карбонильных, фосфиновых и других комплексов
с обратной электроноотдачей.
На рис. 107 приведены изображения 4s-, р- и (7-орбиталей атомов (s-ор-
битали, как известно, имеют форму шара с центром в начале координат).
2 Z у В качестве представителя
Рис. 107. Атомные орбитали переходных метал-
лов, участвующие в образовании молекулярных
орбиталей октаэдрических комплексов. Зачер-
ненные контуры — области отрицательных зна-
чений орбитальных волновых функций.
трех р-орбиталей приведена
только р2-орбиталь; рх- и ру-
орбитали, имеющие точно та-
кую же форму, получаются
Y из нее поворотом на 90° в про-
странстве вокруг осей X и Y.
Точно так же из трех орбита-
лей dxy, dxz и dyz показана
лишь орбиталь dxy, обра-
зующая молекулярную орби-
таль л-характера. Остальные
орбитали — dxz и dyz также
получаются из нее вращением
в пространстве на 90°. Две
остающиеся (7-орбитали,
именно dX2-yz и (72г, приве-
дены внизу рисунка. Сложе-
ние атомной орбитали (т. е. ф-функции) металла с орбиталью лиганда
приводит к связывающей молекулярной орбитали. Вычитание атомной
орбитали металла из орбитали лиганда образует антисвязывающую моле-
кулярную орбиталь, заполняемую электронами в возбужденном состоя-
нии молекулы.
Молекулярные орбитали о-типа, как это обозначено на рис. 107 и
видно по характеру перекрывания (цилиндрическая симметрия), обра-
зуются за счет орбиталей dxt-y2, dzt, s, рх, ру, и р2.
Заметим, что в методе валентных схем эта группа орбиталей рассмат-
ривается как гибридизованная ((72хр3-гибридизация) в шесть одинаковых,
направленных по осям октаэдра орбиталей, включающих каждая пару
электронов связи.
Связи л-типа могут быть образованы как р- , так и dxy~, dxz-, dyz-aTOM~
ными орбиталями металла. Следующие «корреляционные диаграммы» пояс-
няют сказанное как для случая отсутствия л-связей, так и для случая нал и-
чия их. На рис. 108, относящемся к комплексным соединениям типа МХЙ,
жирные горизонтали обозначают энергетический уровень соответству-
ющей молекулярной орбитали (в средней части рисунков), слева и справа
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ
463
расположены энергетические уровни орбиталей металла и, соответственно,
лиганда. Черные кружки в средней части рисунков — всегда заполненные
МО, образованные комбинацией соответствующей атомной орбитали
металла и орбитали лиганда. Полузачериенпые кружки — орбитали,
которые могут быть, а могут и не быть заполнены электронами. Сюда
относятся столь характерные для переходных металлов с их переменной
валентностью d-электроны, обусловливающие и их характерные магнит-
ные и спектральные свойства.
Рис- 108,а отличается от 108,6 тем, что изображает неполярные
связи, обусловленные возрастающим положительным зарядом на атоме
Металл Лиганд
Металл лиганд
Рис. 108. Диаграмма энергетических уровней для комплексных соединений переход-
ных металлов типа МХ6:
а — неполярные связи; б — полярные связи; в — включена л-связь.
Черные кружки — орбитали, занятые электронными парами; светлые — антисвязывающие
вакантные орбитали; полузачерненные — орбитали, могущие быть заполненными парой или одним
электроном или остаться вакантными, в зависимости от того, о каком именно металле и о каком
ионном состоянии идет речь.
металла. Примером могут служить различные октаэдрические неоргани-
ческие комплексы переходных металлов. В неорганических комплексах,
и особенно в металлоорганических соединениях переходных металлов,
часто проявляется участие л-орбиталей лиганда. Этот особенно важный
случай в общей форме изображен на рис. 108,в.
Карбонилы металлов
Известны карбонилы металлов без мостиковых окисно-углеродных групп
между металлами (карбонилы «главной последовательности»); карбонилы,
содержащие обязательно два или более атомов металла, связанных мо-
стиками >С=О, предоставляющими по одному электрону связи каждому
из связываемых атомов металла; карбонилы, в которых три атома металла
связаны одной молекулой окиси углерода. Кроме того, во многих подоб-
ных «многоядерных» карбонилах, как и в простейших димерных карбо-
нилах, таких, как [Мп(СО)512, [Со(СО)4]2 л т. д., димеризация осуще-
ствляется за счет прямой связи между атомами металла.
Состав и физические свойства ряда карбонилов приведены в табл. 76.
Таблица 76. Карбонилы металлов
V VI VII V111
4 V(CO)e синий, возг. 45-60° С (10 мм рт. ст.) Уг(СО)12 оранже- B1JH Сг(СО)в бесцветный, твердый, neper, с паром [Мп(СО)5]2 з >лотн- стый, т. пл. 154—155° С (возг.) Fe(CO)5 желто-корич- Ni(CO)4 бесцв., невый, т. пл. т. нл. —20° С, —25е С, т. кип. (-103° С т. кип. +43° С Fe2(CO)9 золотистый, [Со(СО)4]2 оранжево- разл. 100“ С красный, т. пл. 51“ С [ Fe(CO)4]3 зеленый, [Со(СО)з]4 черный, разл. 140° С твердый, разл. 60° С
5 Мо(СО)в бесцвет- ный, возг., neper, с паром Ru(CO)5 бесцв., [Rh(CO)4)2 оранжевый, т. пл. —22° С разл. 76° С Ru2(CO)9 оранжевый, [АЬ(СОз)]х красный, ' возг. возг. [Ru(CO)4]3 зеленый, [RЬ4(СО)ц] разл. кристалл. -— 200 С
6 СО-г Пр имечание. Одной РУППЫ. W(C0)e бесцветный, возг., neper, с паром чертой подчеркнуты каре [Re(CO)5]2 бесцвет- ный, т. пл. 177° С (возг.) онилы со связью металл — Os(CO)a бесцветный, [1г(СО)4)-2 желто-зеле- т. нл.около ныи -15° С Os2(CO)9 желтый, т. пл. [1г(СО)3]ж желтый, 224° С (возг.) разл. 210° С металл, двумн чертами—имеющие, кроме того, и мостиковые
металлоорганические соединения переходных металлов
465
Строение карбонилов устанавливается рентгеноструктурным анализом.
Кроме того, указание на присутствие мостиковых окисно-углеродных
групп дает ИК-спектроскопия (мостиковые группы имеют частоту ниже
1900 см , а частота окиси углерода, связанной с одним атомом металла,
2000 см ). Отсутствие неспаренных электронов устанавливается
по отсутствию магнитного момента; о непосредственной связи металла
с металлом с^дят по величине расстояния М—М, измеренной рентгено-
графическим или электронографическим путем. Примеры строения карбо-
нилов приведены на рис. 109.
со
Рис. 109. Строение полиметаллических карбонилов:
а—марганца; б — железа; в — кобальта (октакарбонил и додекакарбонил).
Карбонилы никеля и железа легче всего получаются при прямом дей-
ствии окиси углерода на металл (быстрее на измельченный, при повышен-
ном давлении). Так, в 1890 г. Монд открыл карбонил никеля, что
способствовало широкому использованию в металлургии этого летучего
и легко разлагающегося с образованием чистого металла карбонила, к со-
жалению, очень ядовитого. П ентакарбонил железа также применяется для
получения чистейшего железа. Однако карбонилы других металлов так
просто не получаются. В большинстве случаев приходится действовать
окисью углерода под давлением на те или иные соли металла в момент их
восстановления, обычно в неводной среде. Жоб и Кассаль получили так
впервые гексакарбонилы хрома, молибдена и вольфрама, применив грппья-
ров реактив. А. Н. Несмеянов, К. А. Кочешков и К. Н. Анисимов с успе-
хом заменили Гриньяров реактив цинковой пылью. Открытый Бриммом
карбонил марганца лучше всего получать, применив в качестве восста-
новителя натрийкетил бензофенона (C8H5)2CONa (Клоссон).
«Многоядерные» карбонилы с мостиками окиси углерода получаются
из простых карбонилов в результате потери СО, обычно при действии света
2Fe(CO)5----* 17ег(СО)9-|-СО
или при нагревании
6Fe2(CO)B 2Fe3(CO)i2 + 6Fe(CO)5
30 Заказ 672
466 ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
' -—
Фундаментальные работы в области карбонилов принадлежат Хиберу
и его школе (Э. О. Фишер и др.).
Реакции обмена карбонилов. Все карбонилы металлов
с большей или меньшей легкостью обменивают молекулы окиси углерода
на иные лиганды. Такие обмены особенно легко проходят под действием
света.
1. Образование карбонилгидридов. При нагревании пентакарбонила
железа со щелочью происходит реакция:
Fe(CO)5+4NaOH —► Na2Fe(CO)4 + 2H2O + Na2CO3
Полученное соединение Na2Fe(CO)4 солеобразно, ионы натрия в нем
могут быть обменены на другие катионы. Так, например, было получено
ртутное производное HgFe(CO)4. При осторожном подкислении образуется
сначала кислая соль, а затем и кислота — карбонилгидрид H2Fe(CO),
(рХ\ = 4,4; р#2 = 14).
Подобные же карбонилгидриды можно получить восстановлением
димерных карбонилов. Так, [Мп(СО)5]2 восстанавливается водородом
при 200° С:
[Мп(СО)о]2 + Н2 > 2НМп(СО)5
Карбонилгидрид кобальта НСо(СО)4 играет важную роль в оксосин-
тезе по Реппе (кн. I, стр. 136). В этом синтезе олефин или ацетилен в при-
сутствии кобальтового катализатора, присоединяя Н2 и СО, образуют
альдегид. Фактическим переносчиком водорода и окиси углерода служит
промежуточно образующийся карбонилгидрид кобальта:
CH2=CH2-f-HCo(CO)4 —> СН3-СН2-Со(СО)4
СНз-СН2-Со(СО)4 + СО —> СНз-СН2-С-Со(СО)4 — *
II
О
—> СНз-СП2-С—Н+НСо(СО)4
II
о
2. Образование алкилкарбонилов металлов. Карбонилгидриды при-
соединяются по олефиновым и ацетиленовым л-связям и образуют алкил-
карбонилы металлов, для которых типично внедрение СО между металлом
и алкилом с превращением последнего в ацил. Алкил (и арил-) карбонилы
металлов можно получить из NaMn(CO)5 и его аналогов, действуя на них
либо СН31 либо его гомологами (Хибер, Клоссон), либо (C6H6)3S BF4
(А. Н. Несмеянов, Ю. А. Чаповский). Метильные производные получают
взаимодействием диазометана с карбонилгидридом. Например:
HMn(CO)54-CH2=N=N----> CH3Mn(CO)5+N2
Кроме того, пользуясь тем, что реакции алкилкарбонилов с СО оора-
тимы
RCo(CO)4 + CO RCOCo(CO)4
RMn(CO)5 + CO RCOMn(CO)5
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ '4Q7
' * -------------------------------------------—•— ---
можно ацилированием натриевых солей карбонилгидридов получать
ацилкарбонилметалл, который, при нагревании дает алкилкарбонил
металла (декарбонилирование).
В алкилкарбонилах металлов связь алкил — переходный металл хотя
и не отличается большой устойчивостью, все же существует, и некоторые
соединения способны выдержать температуру до ~ 100° С и выше.
Таково влияние заполнения вакантных d-уровней металла (в данном
случае за счет СО).
3. Образование металлкарбонилгалогенидов (обмен СО на галоид).
Такой тип соединении известен для металлов: Си, Au, Мп, Re, Fe, Ru,
Os, Co Rb, Ir, Pd, Pt. Эти вещества, в которых металл связан и с окисью
углерода и с галоидом, могут получаться двумя путями. Галогениды
меди и благородных металлов (если надо — в присутствии восстанови-
теля) поглощают окись углерода и образуют металлкарбонилгалогениды.
Таким путем получены: CICuCO, ClAuCO, I2Ru(CO)2. Платина при дей-
ствии окиси углерода и галоида образует
Ск /СО ОСХ /СС /С1
>Pt; П >Pt< /Pt<
Clz ZCO Clz ^CI7 co
Второй путь — действие на карбонилы металлов свободного галоида —
используется для получения металлкарбонилгалогенидов Мп, Fe и Со.
Реакция схематически изображается следующими уравнениями:
Fe(CO)5+I2 —> I2Fe(CO)4 + CO
[Мп(СО)5]г + Вг2 —> 2ВгМп(СО)5
[Со(СО)4]2 + С12 —► 2С1Со(СО)4
Эти галогениды обладают обычным обменоспособным, хотя и не ионным
галоидом. Так, действуя на них гриньяровым реактивом, можно получить
алкпл(или арил-)карбонил металла; с натрийкарбонилами металлов
получаются димерные карбонилы:
ClMn(CO)5 + NaMn(CO)5 —> [Mn(CO)5]2 + NaCl
Последняя реакция осуществима и с двумя разными карбонилами
металлов: ,
CIRe(CO)5 + NaMn(CO)5—> (СО)5Не-Мп(СО)5+NaCl
4. Обмен СО на лиганды со свободной (неподеленной) парой электронов.
Самый широко изученный класс реакций карбонилов металлов, а также
металлкарбонилгалогенидов - это реакции замещения одной, нескольких
или в редких случаях всех СО на другие лиганды: амины (главным обра-
зом, третичные), фосфины и РС13, арсины, стибины, циан ионы, изо
нитрилы и т. д.,что может быть показано на следующих примерах.
Ni(CO)4 + 2C5H5N ---> Ni(CO)8(NC6H6)a+2CO
пиридин
Cr(CO)e + 3C6H5N —> Cr(CO)3(NC5n5)3 + 3CO
30*
468
ЭЛЕ МЕНТООРГАНИЧЕСКИЕ СРЕДИНЕНИ Я
Сг(СО)в+2Р(С6Н5)з-------> Сг(СО)4[Р(СвН5)з12 + 2СО
С1Мп(СО)5 + 2С5Н5К -> С1Мп(СО)з (NC5H5)2+2CO
5. Обмен СО на непредельные углеводородные лиганды. Особенно
интересны для органика замещения окиси углерода на непредельные,
в частности диеновые и ароматические, углеводороды.
Олефины вытесняют окись углерода из карбонилов, если их л-связь
способна принимать нуклеофильные атаки или при наличии двух и более
сопряженных или несопряженных двойных связей, пространственно
расположенных удобным образом.
Типичным примером реакций первого рода служит действие на пен-
такарбонил железа тетрафторэтилена и акрилонитрила, приводящее
соответственно к
(F2C=CF2)2Fe(CO)3 и (CH2=CIICN)Fe(CO)4
Это явно указывает, что олефин играет не только роль донора за счет
л-электронов, но и служит акцептором для d-электронов металла.
Бутадиен образует с пентакарбонилом железа C4H6Fe(CO)<,.
Другие примеры структур продуктов, образующихся при вытеснении
СО посредством диенов, приведены в следующих схемах:
ос/сосо
ОС СО
Циклопенталпен вытесняет из Ni(CO)4 всю окись углерода, образуя
(C5H6)2Ni с нульвалентпым никелем. Это вещество, теряя водород,
легко превращается в никелецеп (C8H5),Ni (стр. 481).
Циклогептатриен замещает в пентакарбонпле железа три группы
СО. В гексакарбониле хрома он также замещает три карбонила (Уилкин-
сон), образуя комплекс, который, теряя гпдрид-анион, отбираемый,
например, катионом трифенилметила (Добен, Хоннен), превращается
в тропилийтрпкарбонплхром:
Сг
°с&со
Сг
оси > со
Это первый встретившийся нам представитель аренкарбонилов метал-
лов. Э. О. Фишер нашел, что аренкарбонплы металлов во многих случаях
могут быть получены вытеснением части СО из карбонилов при действии
ароматического углеводорода на карбонил металла в автоклаве; при этом
ароматическое ядро занимает место трех молекул СО:
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ
469
Сг(СО)6+С6Нв --> С6НвСг(СО)3 + ЗСО
Сг(СО)6 -f-
Сг
/| \
ОС СОСО
+ ЗСО
_ ^N-СНз I-
Мо(СО)6+^ /N-CH3-----> Mo +ЗСО
I' 1ос/сохсо
Роль арена (т. е. ароматического ядра) может играть и циклопента-
диенил-анион, обладающий ароматическими свойствами (см. кн. I, стр. 548
и кн. II, стр. 493). В некоторых случаях вытеснение окиси углерода
циклопентадиенилом совершается непосредственно при действии цикло-
пентадиена на карбонил металла и может привести к полному замещению
всей окиси углерода с образованием сэндвича—дициклопентадиенилме-
талла:
Выделяющийся из циклопентадиена водород идет на восстановление
избытка циклопентадиена.
В других случаях используют обратную реакцию. Так, циклопен-
тадиенилтрикарбонилмарганец получают действием окиси углерода на
дицикл опентадиенилмарганец:
+ зсо
Полимер
циклопентадиенила
ароматических циклопента-
Свойства этих в той или иной степени
Диенилкарбонилов металлов будут рассмотрены ниже в разделе <
зоидные ароматические системы». „„mmupn-
Существуют и циклопентадиенилкарбонилгалогениды, например.
[о>
ос — Ее—со
1
470
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Ацетилены, так же как и полиолефины, образуют комплексы с карбо-
нилами, которые легко претерпевают дальнейшие превращения с участием
окиси углерода пли путем циклической полимеризации. Так, по реакции
Реппе из Fe(CO)5 и ацетилена в водцо-спиртовохм растворе получаются
комплексы Fe-CjjP^OsH Fe-C8H4O4. Комплекс Fe-C11H7O5 разла-
гается кислотами с образованием хинона. В этом комплексе две молекулы
ацетилена связались с двумя молекулами СО:
2СО + 2СН=СН —> 0=<Z ^>=0
При образовании комплекса Fe-C8H4O4, который представляет собой
циклопентадиенонтрикарбонилжелезо
Fe
ОС СО
циклопентадиенон образуется, очевидно, из двух молекул ацетилена
и одной молекулы окиси углерода. Наконец, Оргел получил из ацетилена
и пентакарбонила железа комплекс, для которого была установлена
структура циклобутадиентрикарбонилжелеза:
ОС—Fe — СО
I
СО
Кроме этих комплексов в растворе находят этиловый эфир акриловой
кислоты (из ацетилена, СО и этанола, реакция Реппе, кн. I, стр. 283)
и гидрохинон.
Подобного типа реакции известны и для замещенных ацетиленов.
Наиболее легко реагируют карбонилы железа, кобальта и никеля.
В некоторых случаях наблюдается образование продуктов тримеризации
ацетиленов в замещенные бензолы (см. кн. 1, стр. 282).
Петтит получил циклобутадиентрикарбопилжелезо также взаимодей-
ствием дихлорциклобутена с нонакарбонилом железа:
С1 г,—77
у
СО со
При окислении солью церия (IV) происходит распад комплекса, и от-
рывающийся циклобутадиен димеризуется в трициклический непредель-
ный углеводород
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ
а также образует более сложные полимеры. Если в момент распада комп-
лекса подвести ацетилен RC=CR, то происходит его соединение с цикло-
бутадиеном и образуется производное «дыоаровского бензола»:
Циклобутадиентрикарбонилжелезо было получено еще одним необыч-
ным путем: рядом фотохимических превращений а-пирона с карбонилом
железа (Розенблюм и сотр., 1967 г.):
Fe(CO)4
pc_o+re(co)5-^fe_o^
ZC—о -СО \ zc—О
Fe(CO)4
Fe(CO)3 + СО
По данным Петтита, циклобутадиентрикарбонилжелезо обнаруживает,
наподобие ферроцена, способность к ряду электрофильных ароматпческих
замещений. Так, оно ацилируется по Фриделю — Крафтсу, меркури-
руется, дает альдегид по реакции Вильсмейера (с C6H5NH—СНО и РОС13),
вступает в реакцию Манниха (с СН2О и HNR2). Поскольку речь идет
не о моноциклпческом карбоцикле, правило Хюккеля 4п + 2 здесь,
очевидно, неприменимо.
Олефиновые л-комплексы переходных металлов
Наиболее давно известное координационное металлоорганическое со-
единение олефина и переходного металла — это соль Цейзе, получаемая
действием этилена на хлорплатинпт калия:
K+[PtC14]2-4-C2H4 —> К+ [СгЩРЮзГЧ-КС!
На примере этого классического вещества удобно дать понятие о типе
связи олефина с переходным металлом. Хотя еще Вернер подчеркивал
аналогию этой реакции с реакцией вытеснения хлора аммиаком во внеш-
нюю сферу
К + [PtCl4]2--(- NI-I3-> K+[NTl3PtCl3]--}-KCl
однако, аналогия поведения аммиака, присоединяющегося к металлу
своей свободной парой электронов, и олефина не полная.
Например, такие олефины, как CF2 — CF2 с «истощенной» л-связью,
связываются с металлом отнюдь не хуже, чем этилен. Таким образом,
связь олефин — металл должна иметь не только дативный, но и акцеп-
торный характер. Развивая представления, впервые высказанные Дьюа-
ром, Чатт и Дункансоп пришли к следующей концепции, схематически
472
ЭЛЕМЕ НТ ОО Р Г АН ИЧЕСКИЕ СОЕДИНЕНИЯ
изображенной на рис. 110. Олефин двояко связывается с платиной: связь
о-типа (заштрихованная на рисунке область) образуется за счет перекры-
вания л-орбитали олефина с 5d 6s 6р2-орбиталыо платины. Кроме того,
олефин и металл связываются дополнительно связью л-типа за счет
предоставления металлом электронов
Рис. 110. Схема перекрывания орбиталей
платины и этилена при образовании
л-комплекса типа соли Цензе.
Рис. 111. Схема строения аниона
л-комплекса платины и этилена (соли
Цензе).
5d бр-орбитали антпсвязывающим
п*-орбиталям олефина (зачерненные
на рисунке области перекрывания).
Строение аниона этого квадратного
комплекса двухвалентной платины
(Й8/?2-гибридизация на языке локали-
зованных пар) схематически изобра-
жено на рис. 111.
Итак, для образования этойдопол-
нительной (с точки зрения металла)
дативной связи л-типа требуются
электроны на cZ-орбиталях металла.
Вот почему именно элементы кон-
цов больших периодов (Ni, Pd, Pt,
Си, Ag, Au) с заполненными или почти
заполненными d-уровнями способны
образовать олефиновые л-комплексы.
Кроме комплексов типа соли
Цензе платина с олефинами образует
и нейтральные молекулы (а не анио-
ны) типа I
С1 л
I /с1'
С9Н4РЦ PtC2H4
г
С1
которые димерны за счет хлорных
мостов.
Хараш впервые получил аналогичные платиновым палладиевые оле-
финовые комплексы с Pd(II), осуществив вытеснение нитрильного лиганда
олефином:
(CeH5CN)2PdC12 + Олефин --->
’Олефин^
/PdCK
Олефин' J2
+ 2CeHaCN
Другим примером л-комплексов этого типа служат комплексы иона
серебра (рис. 112), констатированные Уинстейном и Лукасом при изуче-
нии закона распределения олефина между слоем углеводорода и водным
раствором азотнокислого серебра — при этом значительное количество
олефина переходит в водный раствор. Связи о-типа, образованные
s-орбиталыо Ag и л-орбиталыо олефина, изображены отдельно на
рис. 112, а, а дативная связь Ag+ на аитисвязывающие л*-орбитали оле-
фина на рис. 112, б. Как всегда, знаки плюс и минус обозначают не
знаки зарядов, а знаки областей функций.
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ
473
Полиолефины, особенно с сопряженными связями, еще легче, чем
моноолефины, образуют л-комплексы с переходными металлами. В этом
случае диен занимает два координационных места и реакция прямого
взаимодействия диена, например с K2PtCl4, разыгрывается так:
С1
2K2PtC]4 + Cn2=CII—СН=СП2 >• К£
С1 12-
ClPt—С4Нб— Ptci
Cl Cl
+ 2КС1
При дальнейшем воздействии бутадиена анион превращается в ней-
тральный зеленовато-бурый димер (А. Д. Гельман):
СК /С4Нв\ ХС1
xPtz Pt'
Clz С4Н6 z 4 Cl
При действии же бутадиена на димер I
(см. стр. 472) образуется изомер приведенной
выше димерной молекулы — темно-красное
вещество уже не с бутадиеновыми мостами,
а с хлорными. Подобные олефиновым димер-
ные комплексы, в которых диен занимает
два координационных места, образует с дие-
нами и Pd(II).
Ион серебра с полиенами образует уже
Рис. 112. Схема перекрывания
орбиталей иона серебра и оле-
фина в л-комплексе:
а и б — различного типа связи.
кристаллические л-комплексы, например, с циклопентадиеном — ком-
плекс I, с циклобутадиеном — комплекс II или, возможно, комплекс
полимерной структуры III:
а также комплексы с циклооктадиеном-1,5 и циклооктатетраеном.
Диены присоединяются к галогенидам одновалентной меди и дают
продукты присоединения диен — СиХ в соотношении 1 : 1 или 1 : 2.
Этим пользуются для отделения диенов от олефинов. Так, бутадиен обра-
зует с CuCl и СнВг комплексы состава 1 : 2, цпклооктадиен-1,5 — со-
става 1:1, циклооктатетраен с CuCl — состава 1 : 2, а с СнВг — состава
1 : 1.
По-видимому, лишь никель дает комплексы, в которых в качестве
лиганда фигурирует только полиолефин. Таков очень лабильный, окис-
ляющийся па воздухе циклододекатриеииикель C12H18Ni
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
темно-красное, кристаллизующееся в иглах вещество, которое получают
по Вильке — действуя на ацетилацетонат никеля транс-транс-транс-
циклододекатриеном в присутствии восстановителя (С2НЙ)2А1ОС2Н5.
Как можно видеть, в этом соединении никель формально нульвалентен.
Циклододекатриен добавляет к электронной свите никеля шесть л-электро-
нов, образуя в сумме 16-электронпую наружную оболочку. Это объясняет
почему данное вещество способно присоединить еще молекулу трифенил-
фосфина, дополнив оболочку свободной парой электронов до 18, т. е.
всего до 36 электронов — числа электронов криптона (правило эффектив-
ного атомного номера Сиджвика). Из этого удивительного вещества
другие полиены вытесняют циклододекатриен и образуют в свою очередь
комплексы с никелем, например, циклооктадиен-1,5 дает Ni(C8H12)2,
а циклооктатетраен — полимерный Ni(C8H8). Особенно интересен продукт
реакции с бутадиеном: образуется изомер циклододекатриенникеля,
тоже состава C12H18Ni, но с лигандом, представляющим собой открытую
цепь атомов. Это установлено гидрированием соединения, дающего я-доде-
кан (в случае циклододекатриена в результате гидрирования, естественно,
образуется циклододекан). Таким образом, три молекулы бутадиена ли-
нейно присоединились друг к другу, что можно изобразить схемой:
СН2=СН-СИ=СН2-|-СН2=СН-сн=сн2 + сн2=сн-сн=сн2 —>
—> СН2=СН—СН-СН2-С112-СН—сн-сн2—сн2-сн—сп=сн2 —>
—> CfI2=CH-CH-CH2-CH2-CH=CH-CH2-CH2-CH-CH=CH2
Такой бирадикал аллильного типа может предоставить для дополнения
числа электронов внешней сферы никеля до устойчивых 18 как раз 8
электронов (6 — от трех л-связей и 2 от двух радикалов аллильного
типа), так что в общем получается структура I, но так как электроны
делокализуются в пределах аллильной системы, лучше изображать ее
в виде II:
Ni
Здесь мы впервые сталкиваемся с так называемым л-аллильным лиган-
дом металлоорганических комплексов.
Если вспомнить, что цианид и аммиакат никеля в руках Реппе (см.
кн. 1, стр. 282) явились катализаторами полимеризации ацетилена в цикло-
октатетраен, циклодекапентаен и бензол, то становится ясным, что эти
реакции шли через комплексообразование ацетилена с никелем.
Хотя никель чрезвычайно активен в подобного рода полимеризациях,
он не единственный металл, вызывающий такие превращения. Так, «цпг-
леровские катализаторы» полимеризации (триэтилалюминий в смеси с
TiCl4 или TiCl3) также тримеризуют бутадиен в циклододекатриен. Это
обычный способ синтеза циклододекатриена, протекающий, по исследо-
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ 475
ванию Вильке, через такие стадии: а) образование бутадиенового л-комп-
лекса с переходным металлом; б) образование С—С-связей в ориентиро-
ванных вокруг атома металла трех молекулах бутадиена; в) вытеснение
образовавшегося так циклододекатриена бутадиеном.
Ранее мы уже встречались (стр. 19) с тримеризацией ацетилена
в бензол и замещенных ацетиленов в замещенные бензолы, протекающей
на разных комплексных соединениях переходных металлов. Мы еще
встретимся с такими тримеризациями ниже, в разделе «Металлоарены».
Напомним также, что линейная димеризация и линейная тримеризация
ацетилена в винилацетилен и дивинилацетилеи осуществляется, по Нью-
ленду (кн. I, стр. 281), в растворе хлористой меди в аммиаке и хлористом
аммонии. Таким образом, комплексные соединения переходных металлов
служат удивительными катализаторами необыкновенных синтезов, воз-
можности которых далеко не исчерпаны и которые интенсивно изучаются
в настоящее время.
Приняв во внимание дативно-акцепторный характер связей пере-
ходного металла с олефином, мы ближе подходим и к пониманию того,
что карбонилы металлов, заполняющие за счет СО-групп свою электрон-
ную оболочку (даже карбонилы металлов, удаленных от конца больших
периодов, таких, как Fe, Мп, Сг, V), образуют уже рассмотренные выше
олефин-карбонилы металлов, но не дают чисто олефиновых производных.
Следует кратко упомянуть о л-аллильных производных металлов,
с довольно сложным представителем которых мы встретились уже на при-
мере никелевого соединения II (стр. 474). Простейшие л-аллильные
производные переходных металлов образуются при действии аллилмагний-
галогенида или аллиллития на хлориды переходных металлов. Так,
из аллилмагнийхлорида и двухлористого никеля Вильке получил бис-л-
аллилникель I — летучее даже с парами эфира вещество. Бутадиен вытес-
няет из него'аллильные радикалы, спаивающиеся в диаллил, а сам через
комплекс II тримеризуется в циклододекатриен III:
Н2С
,/СН
Н2С Ni сн2 + ЗС4Н6
нсб
хсн2
I
Если же бутадиеном (2 моль) действуют в присутствии соединений
типа R3P, то через промежуточный л-аллильный комплекс IV образуется
циклооктадиен V.
476
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Приведем еще л-аллильные комплексы никеля с более сложными
циклическими аллильными системами (в скобках указаны температуры
выше которых эти комплексы неустойчивы):
(—40°С) (—40°С)
(ОТ 20 ДО 30°С)
Шире изучены л-аллильные комплексы (а также аллильные) с другими
лигандами. Следующие примеры дают понятие о них и о способах их
получения, например, получение л-аллилтрикарбонилиодида железа
(Мардох):
/ССН
H2CZ сН2
СН2=СН—CH2I + Fe2(CO)9-----> Fe-4 + Ге(со)5 + С0 ~
ОС (Iq СО
При восстановлении амальгамой натрия л-аллилтрикарбонилиодида
железа получается бис-л-аллилдикарбонил железа (А. Н. Несмеянов,
И. И. Крицкая):
^СН
н2с' ТН2
ОС---Fe---СО
Н2Сч ZCH2
хзн
Как никелевые, так и железные л-аллильпые комплексы могут быть
разложены (никелевые в присутствии СО) с образованием аллильных
оксосоединений — диаллилкетона и альдегида.
При попытке получить аллилпалладийхлорид СН2 = СН—CH2PdCl
(так называемый а-аллилпалладийхлорид) действием аллилмагнийхлорида
на хлористый палладий вместо ожидаемого вещества получается димерный
л-аллилпалладийхлорид, в котором аллильный радикал присоединен
к палладию л-связыо, притом так, что сверх двух л-электронов л-связи
в этом участвует и песпаренный, но рассредоточенный р-электрон аллиль-
ного радикала. Обе концевые метиленовые группы аллила оказываются
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ
477
совершенно одинаковыми (обнаружены лишь два рода сигналов ЯМР;
отношение площади ников 4:1), что соответствует такой структуре:
Cl Н2С
^сн2 xiz/ н2<^
Подобного типа л-аллильные соединения — распространенное явле-
ние среди переходных металлов, но наряду с аллилом эти вещества часто
содержат другие лиганды, обычно СО.
Аналогами л-аллильных комплексов являются л-бензильные. Впервые
такого рода комплекс был получен Кингом при нагревании о-бензил-
л-циклопентадиенилмолибдентрикарбонила:
©
Мо-СН2СсН5
ОС I со
со
Исходное соединение получило от лигандов на орбитали металла
54-2-34-1 =12 электронов, дополнившие электронную свиту молиб-
дена до соответствующей эффективному номеру атома ксенона (о-бензил
отдает один свой электрон). При нагревании бензил за счет одной из л-пар
бензола выталкивает одну молекулу окиси углерода и дает уже три
электрона— один радикальный и два л-бензольных —и становится л-бен-
зильным лигандом, в известной мере нарушив свою бензоидность.
Металлоарены
Поскольку бензол в какой-то мере обладает свойствами триена, не столь
удивительно, сколь неожиданно существование металлоорганических
л-комплексов переходных металлов с бензолом и другими ароматическими
соединениями. Мы уже видели, что ароматические соединения способны вы-
теснить три группы СО и заместить их на одно ароматическое ядро, оставив
лиганды СО связанными с нульвалентным металлом. Так получаются арен-
карбонилы металлов', сведения о некоторых из них приведены в табл. 77.
Однако бис-ареновые соединения сэндвичеобразного типа, в которых
два ароматических ядра связаны с атомом (или катионом) переходного
металла (табл. 78), получаются иначе.
Наиболее прост метод Э. О. Фишера. Бензольный раствор (или вообще
раствор в ароматическом соединении) или взвесь галоидной соли пере-
ходного металла нагревают с хлористым алюминием и алюминиевой пылью,
играющей роль восстановителя *. В результате получается бис-бензол-
металл (соответственно вообще бис-ареиметалл) с нульвалентным метал-
лом, который более или менее легко теряет электрон, окисляясь часто
уже на воздухе в бис-аренметалл-катион.
* Ср. с получением карбонилов металлов по способу Л. И. Несмеянова, К. А. Ко-
чешкова, К. Н. Аписпмова, стр. 465.
478
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Таблица 77- Аренкарбопилы металлов
Формула Температура плавления •с Цвет
СвНвСг(СО)з 161—163 Желтый
СюН8Сг(СО)з 150 (разл.) »
С1СвН5Сг(СО)з 102—103 »
NH2CeH5Cr(CO)3 173—175 (разл.) »
СвНвМо(СО)з 120—125 Зелено-желтый
CeHeW(CO)3 140—145 Желтый
1,3,5-СвНз(СН3)зМп(СО)з Желтый
Следует отметить, что первые бис-аренхромовые соединения были полу-
чены Гейном еще в 1919 г. действием на СгС13 гриньяровым реактивом
CeH6MgBr.
Однако впервые сэндвичеобразный тип структур был констатирован
лишь в 1952 г. на примере ферроцена (стр. 481). Гейн в 1919 г. принял
полученные им соединения за обычные металлоорганические, а именно
фенильные производные хрома:
Структуры Гейна Структуры, предложенные
(1919 г.) Онзагером (1954 г.)
МЕТАЛЛООРГА НИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ
479
Таблица 78. Бис-арены переходных металлов
Формула Температура плавления °C Цвет р'эфф.
(CeHehV 277—278 Красно-коричневый 1,73
(СвНб)гСг+ — Желтый 1,73
(CeHehCi-o 282—284 Темно-коричневый 0
(СвНв)гМе 115 (разл.) Зеленый 0
(C6H6)2W 110 (разл.) Желто-зеленый 0
[Св(СНз)в]гМп+ Розовый 0
(CeHe)2Re+ — Красно-коричневый 0
(CeHe)2Fe2+ — Желтый 0
[1,3,5-C6H3(CH3)3]2Ru+ — К расно-коричневый 0
[l,3,5-CeH3(CH3)3]2Os2+ — Желтый —
[ 1,3,5-СвН3(СН3)3]2Со3+ — Красно-коричневый —
{l,3,5-CeH3(CH3)3]2Rh3+ — Светло-коричневый —
{1,3,5-СвН3(СН3)3]21гЗ<- — Желто-коричневый —
Естественно, что по данным анализа эти структуры нельзя отличить:
структуры I Гейна и Онзагера изомерны, структуры III также, и лишь
структуры II разнятся между собой на один водород. Получение ди-
бензолхрома и серии его гомологов Фишером и их изучение решило
вопрос окончательно. Интересно отметить, что Цейссу, изучавшему сое-
динения Гейна и экспериментально установившему их тождество с бен-
золбифенилхромиодидом и бис-бифенилхромиодидом, удалось получить
и истинные фенильные производные хрома реакцией Гриньяра в тет-
рагидрофурановом (ТГФ) растворе. В этом случае три молекулы ТГФ
входят в комплекс в качестве лигандов и стабилизуют его.
ТГФ
ТГФ. । /ГГФ
Сгх
С6н5 7\ ^СбНб
CgHs
Вода разлагает это красное вещество, превращая его в зеленый ка-
тион Сг3+(ОН2)6, сулема также отщепляет фенилы, образуя CeH5HgCl.
Если вести реакцию в эфире, устойчивость эфпрата еще меньше (эфир
худший донор, чем ТГФ), и он самопроизвольно (а комплекс с ТГФ при на-
гревании) перегруппировывается в черное соединение, после гидролиза
которого образуется смесь бис-аренхромов:
нагревание) CeIIe СвНв
затем Н,0 Lf’r
(С6Н5)3Сг(ТГФ)8. - + £И5_.Свн5
Чрезвычайно интересно также открытое Цсйссом отношение три-
фенилхрома к ацетиленам. Ацетилены вытесняют и ТГФ и фенилы и, цик-
лизуясь в замещенные бензолы, образуют бис-аренхром:
480
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
ТГФ
ТГФ. | ^ТГФ
Сг + 3RC = CR
СвН5^ | ~^с6н5
Сб^5
При этом, например, из диметилацетилена получается бис-гекса-
метилбензолхром.
Таким образом, Цейсс открыл второй путь синтеза бис-аренхромов.
Интересно отметить, что фенилы принимают участие в реакции и, кон-
денсируясь с двумя молекулами диметилацетилена, дают некоторое коли-
чество 1, 2, 3, 4-тетраметплнафталина. Если вместо трифенилхрома взять
тримезитилхром в тетрагидрофуране, то наряду с бис-ареном I, за счет
присоединения к диметилацетилену мезитила, после гидролиза образуется
2, З-димезитилбутен-2 (II):
Соли никеля и кобальта в ТГФ с гриньяровым реактивом также обра-
зуют непрочные тетрагидрофуранаты диарилникеля и, соответственно,
кобальта. Более устойчивы димезитильные производные этих металлов.
Дифенилацетилен (Ph — фенил) реагирует над никелевым соединением так:
МЕТАЛЛООРГЛНИ^^^ ПЕРЕХОДНЫХ МЕТАЛЛОВ
481
ЭТОМ СЛучаО, Как п Bfiofimn
Дифенилкобальт—ТГФ, по-вмимом^аРеЫШТЛЬ Н° удаетСя из°лировать.
£ски в производные бензол^ нХп’я беН циклизовать каталити-
Однако ни для никеля, ни для 3™^° КОличества Метиленов,
В нескольких случаях получрн/ к Р НЫ Не выделены-
„довольно много аре^карбонилов ИР то -арены переходных металлов
Д „ р гетепоппкттттпгтгХ z не только с углеводородными аренами,
в л ТТЛ мп — Дрпи МП„ (напР11меР, с тиофеном) или с замещенными
°еНЗ 'а птта Л. лу^сУсн°и кислотой, анилином и его производными,
хлоро нз ь г др. днако наличие таких резко электроноакцепторных
групп, как, например, NO2, -C<f , CN и даже СООН. не дает возможности
ХН
бензольному ядру образовывать бис-арен или аренкарбонил. Напротив,
арены и аренкарбонилы, содержащие в ароматическом ядре алкильные
заместители, более устойчивы, чем бензольные.
Металлоцены
В 1951 г. молодой химик Посон в Шеффилде сделал неожиданное откры-
тие: желая получить дициклопентадиенил при действии хлорного железа
на гриньяров реактив из циклопентадиена
Г\/н
он выделил дициклопентадиенилжелезо, неожиданно обладающее исклю-
чительной прочностью — устойчивое до 400° С, перегоняющееся с водя-
ным паром, не разлагающееся ни кислотами, ни щелочами. (Алкильные
и арильные производные железа до сих пор не получены, и, конечно,
если они и существуют при очень низких температурах, то крайне
неустойчивы.) Вскоре рентгеноструктурными исследованиями Вудворд
и Уилкинсон установили необыкновенную сэндвичеобразную структуру
дициклопентадиенилжелеза. Затем стала известной первая реакция
ароматического замещения водорода в дициклопентадиенилжелезе — аци-
лирование по Фриделю — Крафтсу. Вудворд и Уилкинсон предложили
для новой ароматической системы название ферроцен.
Эти открытия положили начало повой области — химии сэндвиче-
образных цикл опентадиенил ьных металлоценов, изучению, осооенпо
на примере ферроцена, их ароматичности и имели решающее влияние
на зарождение и развитие также химии сэндвичеобразпых аренов.
Большинство переходных металлов образует сэндвичеобразные цены
с двумя циклопентадиепилами, а уран и редкоземельные элементы
с тремя циклопентадиенилами. На основании суммы физических и хими-
ческих свойств считают, что из трициклопентадиенилметаллов — трици-
кл опентадиенил редкоземельные металлы, а из дициклопентадиенплметал-
лов дициклопентадиепилмарганец и дициклопеитадпенилмагнпй имеют
ионные структуры хотя оба последних, так же как прочие дицикло-
пентадиенилметаллы, построены сэндвичеобразно. В остальных ценах
связь колец с металлом ферроценоподобиа. Понятие о ней может дать
31 3»к»» *72
482
.ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
следующая корреляционная диаграмма энергетических уровней (метод
молекулярных орбиталей), приведенная на рис. ИЗ.
Здесь уровни л-электронов ароматических шестерок циклопента-
диенил-анионов разбиты на три неравных уровня а, и е2 (см. стр. 14(
где описываются МО-уровни для бензола). Эти орбитали лиганда ком-
бинируются с атомными орбиталями металла. Наибольший вклад дает
комбинирование орбиталей циклов
Рис. ИЗ. Диаграмма энергетических
уровней ферроцена и аналогичных
соединений.
с близкими по энергии 3</л-орбита-
лями (т. е. 3dxy и т. д.) металла. В ре-
зультате — связь металла с цикл one н-
тадиенильными кольцами грубо может
быть описана как единственная, но име-
ющая ст-характер связь с каждым
кольцом. Как и на прежних диаграм-
мах, черные кружки — электроны, за-
нявшие данные орбитали, черно-белые
кружки — электроны, которые могут
быть, а могут и отсутствовать в том или
ином металлоцене или его катионе;
белые кружки — вакантные орбитали.
Так, в ионе Ti(C5H6)2+ все черно-белые
кружки вакантны, и их надо изобразить
белым. В ферроцене 2в'~ и 16'-орбитали
заняты, и кружки надо зачернить. Ион
Со(С5Н5)2 изоэлектронен Fe(C5H5)2.
Как раз этот кобальтициний-катион
и ферроцен (наиболее прочные среди
металлоценов) обладают и полным сидж-
виковым эффективным номером, т. е.
числом электронов ближайшего инерт-
ного газа —криптона, в целом 36, а во
внешней оболочке 18.
Приведем ряд известных металлоценов (в скобках указано число
неспаренных электронов, определяемое по наличию и величине парамаг-
нитности):
(C5H6)2Ti (0); (C5H5)2V (3); (С5Н5)2Сг (2); (C^I^Mn (5)
(С5Н5)2Со (1); (C5H5)2Ni (2); (C5II5)2Ru (0); (С5И6)20з (0)
Ведущие исследователи всего разнообразия металлоценовых соеди-
нений — Э. О. Фишер и Уилкинсон. Им принадлежит синтез и изучение
физических свойств и структур большинства металлоценов.
Все дициклопентадиенильные соединения элементов первого большого
периода имеют, каки ферроцен, т. пл. около 173° С и образуют изоморф-
ные с ним кристаллы. Особняком стоит ионно построенный (С5Н5)2^П’
Рутеноцен и осмоцен в общем химически очень похожи на ферроцен.
Многие элементы образуют дициклопентадиенилгалогениды, соединения
с иными апионоидпыми группами, а также гидриды. Их представителями
могут служить:
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ
483
IV V VI VII
4 (С5Н5)2Т1Х (С5Н5)2Т1Х2 (С5Н5)2УХз
5 (C5H5)2ZrCl2 (С5Н5)2ХЬХз (С5Н5)2МоХ2 (С5Н5)2МоН2
6 (С5Н5)2ТаХ3 (C5H5)2WH2 (CsH5)2ReH
(C5H5)3UX
Все они образуются при действии C5H5Na на соли соответствующих
металлов, иногда с последующим восстановлением.
Как уже сказано, ферроцен и особенно легко кобальтоцен окисляются,
теряя электрон и превращаясь в катионы феррициния и кобальтициния.
В отличие от исходных нейтральных соединений и подобно веществам
последнего перечня эти катионы не обладают какой-либо способностью
к реакциям замещения в циклопентадиенильных кольцах, например
к ароматическому электрофильному замещению.
Прочность связи циклопентадиенил —металл весьма различна в разных
соединениях: наибольшая она у ферроцена, рутеноцена и осмоцена,
а также у кобальтициний-катиона. По данным А. Н. Несмеянова и Э. Г. Пе-
реваловой, ферроцен гидрируется над никелем Ренея водородом лишь
при 300° С и — 300 атм, образуя циклопентан. А. Н. Несмеянов и
Н. А. Волькенау нашли, что одно кольцо ферроцена можно заменить
на бензол или другие ароматические системы кипячением с ними в при-
сутствии хлористого алюминия. С бензолом образуется катион
который можно выделить в виде солей с анионами BF4, (СвН5)4В~ или
PFg. Этот катион крайне устойчив к окислению и восстановлению. Он
является представителем немногочисленного пока класса метпаллоцено-
аренов, которые подробно рассматриваться не будут. Укажем лишь,
что этот тип соединений, получаемых действием ароматического угле-
водорода на циклопентадиенилкарбонилметаллы, изучен в основном
Э. О. Фишером. Наиболее удивительные превращения бензолциклопен-
тадиенилметаллов (Сг и Мп) — это реакции в условиях ацилирования
по Фриделю — Крафтсу, приводящие к превращению бензольного ли-
ганда в метилтропилиевый:
31*
484
ЭЛЕМЕНТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
А1С13;
затем гидролиз
(выход 10%)
В скобках указана формальная валентность металла в данном соеди-
нении.
В противоположность прочному ферроцену у некоторых титаноцено-
вых соединений, например (C5H6)2Ti(OC2H6)2, даже этанол при нагрева-
нии удаляет одно из колец в виде циклопентадиена, превращая вещество
в C6H6Ti(OC2H6)3, в котором в более жестких условиях можно заменить
и последний циклопентадиенил на этоксил, получив этплортотптанат
(C2H5O)4Ti (А. Н. Несмеянов, О. В. Ногина). Однако феррицпнпй-катион
уже легче, чем ферроцен, разрушается с частичным отщеплением цикло-
пентадиенильных ядер (например, щелочью), превращаясь в ферроцен,
с выделением металлического железа и органического полимера.
Во многих случаях известны цпклопентадиенильные полусэндвичи,
содержащие один л-связанный с металлом циклопентадиенил и другие
лиганды. С одним из таких веществ мы уже встретились на примере
C5H6Ti(OC2H5)3. Его хлорный аналог может быть получен реакцией:
(C5H5)2TiCl2 + TiCl.i > 2C5II5TiCl3
В сумме титан здесь как бы четырехвалентен. Вещества (C5H5)2Ti
и (C6H5)2TiCl получаются восстановлением соответствующих соединений
четырехвалентного титана (Г. А. Разуваев).
Другой пример моноциклопентадиенпльного полусэндвпча — про-
изводное двухвалентного палладия I, которое действием аллплмагнпй-
хлорида можно превратить в л-циклоиентадиенпл- л-аллилпалладий П-
В этом соединении л-аллильный лиганд удерживается прочнее, чем
л-циклопентадиенильный. Так, при действии сулемы снимается цикло-
пентадпенил (в виде C5H5HgCl) и остается довольно прочное соединение Ш
(А. Н. Несмеянов, С. П. Губин, А. 3. Рубежов):
।
Н
^СН2/С1\ СН2
Hcf Pd ^Pd^CH
СН2
in
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ 485
В большинстве случаев, однако, такие моноциклопентадиенильные
производные известны с лигандами типа окиси углерода. Их наиболее
просто получить, действуя циклопентадиеном на карбонил металла
(Э. О. Фишер). На примере никеля, где, однако, замещается вся окись
углерода, можно выделить промежуточную стадию — образование дицик-
лопентадиенникеля, который в более жестких условиях теряет Н2,
превращаясь в никелецен: ч .
Можно допустить подобное течение реакции и с другими карбонилами
металлов, но при этом вступает в реакцию вытеснения только один
циклопентадпен на один атом металла:
2Fe(CO)5 + ЗС5Н6
6СО + С5Н8
/к СО
[Со(СО)4]2 + ЗС5Н6----> 2 Со + 4 СО + С5Н8
со
2Сг(СО)6 + 2С5Н6
6СО + 2Н
Особенно интересен в высоко]! степени ароматичный циклопентадие-
нилтрикарбонилмарганец, который приходится получать обратной
Действием окиси углерода на дициклопентадиенилмарганец
(ионно построенный, как было сказано):
Mn2+ + ЗСО
НС —СИ
I ® /сн
нс—сн
полимер
486 ЭЛЕМЕПТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Известны и смешанные циклопентадиенилкарбонилгалогениды и гид-
риды переходных металлов. Мы ограничимся приведением их формул:
I—Мо—СО
осх Хсо
Н—Мо—СО
осх со
Аналогичные соединения дает вольфрам (Уилкинсон). Наряду с уди-
вительной способностью комплексов переходных металлов сшивать оле-
финовые молекулы между собой и с окисью углерода, может быть, наи-
больший интерес во всей органической химии переходных металлов
для химика-органика представляют необыкновенно ярко выраженные
ароматические свойства ферроцена, его рутениевого и осмиевого аналогов
и циклопентадиенилтрикарбонилмарганца. Эти чисто органические
реакции удобнее отнести к разделу «Небензоидные ароматические си-
стемы».
НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
В рассмотренных до сих пор изоциклических ароматических, в том числе
и конденсированных, системах в качестве ароматического всегда фигури-
ровало бензольное ядро.
Бензол содержит шесть обобщенных л-электронов. Он относится
к так называемым альтернантным ароматическим системам, в которых,
если мысленно закрепить электроны, л-связей по одному у каждого угле-
родного атома, их спины будут чередоваться. Альтернантные системы
отличаются тем, что их углеродные атомы, образующие ароматическую
систему, можно «рассчитать на первый, второй».
«Первые» и «вторые» атомы правильно чередуются. В неалътернантных
ароматических системах, с которыми мы вскоре встретимся и примером
которых является изомер нафталина — азулен
рассчитать атомы па «первый, второй» так, чтобы одинаковые номера
не стояли у соседних связанных друг с другом атомов, невозможно.
Столь ясно выраженное в бензоле альтернирование (чередование)
электрофильности или нуклеофильности углеродных атомов не свойственно
неальтерпаптным ароматическим системам.
Квантово-механический расчет по методу молекулярных орбиталей
(в приближении Хюккеля) приводит к выводу, что системами с резко
пониженной энергией (в основном состоянии молекулы) будут все непре-
дельные циклические соединения с сопряженными л-связями, если число
«-электронов в них равно 4п + 2, где п любое целое число (этот вывод
488
НЕБЕНЗОИДПЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
относится только к мопоциклическим системам). Таким образом, «арома-
тическая шестерка» электронов бензола (4 X 1 + 2 = G) является част-
ным, хотя и самым важным случаем. Поскольку метод молекулярных
орбиталей при рассмотрении молекулы подходит к ней так, как квантовая
механика подходит к рассмотрению атома, то устойчивую ароматическую
шестерку электронов и все группировки 4п + 2 можно сравнить с устой-
чивыми замкнутыми электронными октетами в атомах (и иоиах) элементов
верха (В—Ne) периодической системы. Таким образом, при п = 0. 1.2,
3, 4 устойчивые электронные группировки будут соответственно содер-
?кать 2, 6, 10, 14, 18 ^-электронов. Благодаря низкой энергпи системы,
содержащие это число электронов в л-связях, устойчивее других непре-
дельных систем. В них труднее нарушаются двойные связи; другими сло-
вами, эти системы обладают пониженной способностью к присоединению
(как это было уже показано па примере бензоидных ароматических систем),
и на первый план поэтому могут выступать реакции замещения.
Во всех ароматических системах имеется замкнутый цикл, и это отли-
чает их от открытых сопряженных полиметиповых систем:
X—СН=СН— СН=СН—.. . X
В изоциклических ароматических системах, альтернантных или не-
альтернантных, все атомы ароматического цикла находятся в состоянии
8р2-гибридизации (кн. I, стр. 257), и л-электроны свободно циркули-
руют по периферии цикла, как ток по бесконечно проводящему металлу.
Такое поведение электронов сообщает ароматическим системам особые
оптические свойства и свойства диамагнетизма.
ДИАМАГНИТНЫЕ СВОЙСТВА АРОМАТИЧЕСКИХ СИСТЕМ
При внесении системы атомов в магнитное поле каждый электрон получает
небольшой дополнительный момент количества движения и, как след-
ствие, дополнительный индуцированный магнитный момент, направлен-
ный противоположно индуцирующему полю и ослабляющий его. Все это
совершенно подобно поведению помещенного в магнитное поле соленоида
с текущим по нему током. Солепоид при этом становится перпендикулярно
к направлению поля и создает поле противоположного направления, про-
порциональное площади витка соленоида. Атомы, однако, закреплены
в молекулы. Поэтому получается некоторая усредненная картина, из
рассмотрения которой следует, что молекулярная диамагнитная воспри-
имчивость предельных систем является аддитивной величиной с вкладами,
вносимыми атомами каждого сорта. Системы с л-связями имеют повышен-
ную магнитную восприимчивость, так как л-связи вносят дополнитель-
ный вклад (инкремент).
Диамагнитная восприимчивость атомов (по Паскалю) %А-106:
—2,9 F . • • • . . . . —6,3
—6,0 С1 • . . . . —20,1
—5,55 Вг ... . . . . —30,6
-4,6 I . . . . . . . . —44,6
ДИАМАГНИТНЫЕ СВОЙСТВА
489
Инкременты связей % • 10°:
\с=с<^
—С=*С—
—N==N—
^C=N —
—C==N .
\с=о .
+5,5
+0,8
+ 1,85
+ 1,6
+0,8
+6,32
+4,1
+3,05
—0,98
+0,86
Слагая эти величины соответственно формуле органического соеди-
нения, находят рассчитанную па основании аддитивности величину моле-
кулярной диамагнитной восприимчивости. Экспериментальную величину
находят, измеряя ослабление магнитного поля при внесении в него иссле-
дуемого вещества при условии, что вещество это не содержит неспаренных
электронов, т. е. не обладает свободнорадикальными свойствами, и по-
этому спиновые магнитные моменты в нем попарно скомпенсированы
и не оказывают влияния. Практически измеряют силу выталкивания
образца вещества из неоднородного магнитного поля высокой напряжен-
ности, откуда находят объемную восприимчивость К и вычисляют молеку-
лярную восприимчивость из равенства:
КМ
где М — молекулярный вес, a d — плотность.
Для ароматических систем, т. е. плоских циклических систем из хр2-
гибридизованных углеродов, таким способом были обнаружены огромные
инкременты (экзальтации), пропорциональные числу бензольных ядер.
Таким образом, экзальтация диамагнитной восприимчивости служит
ясным признаком ароматичности (табл. 79).
Интересно приблизительно оцепить влияние замыкания в кольцо
сопряженной системы углерод-углеродных связей с открытой цепью на
энергию л-электронов молекулы. Следуя Хейльбропперу, это можно
сделать элементарными средствами, применив хорошо известное соотно-
шение Де-Бройля к движению электрона в одномерной потенциальной
яме с постоянным потенциалом и полностью отражающей стенкой. Тогда
mev
где h, _ постоянная Планка, а те и v — соответственно масса и скорость
электрона.
Условия квантования энергии для электрона в линейном потенциаль-
ном ящике и в кольце различны. Это понятно, так как при каждом
заданном значении энергии в линейном потенциальном ящике возможен
единственный вид движения («взад вперед» по ящику), тогда как
в кольце возможны два разных, но энергетически равноценных направле-
ния движения — «по часовой стрелке» и «против часовен стрелки». Иными
490
НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
словами, уровни энергии в линейном потенциальном ящике «невырождены»,
а в кольце, вообще говоря, дважды «вырождены» (как будет далее видно,
все уровни, кроме самого глубокого). Именно условия квантования тре-
буют, чтобы в линейном ящике длина L была кратна длине полуволны
Х/2, а в кольце — чтобы периметр L был ^кратным целой длине волны
(рис. 114).
Рис. 114. Модель свободного электрона для линей-
ных («ящик») и циклических («кольцо») л-элект-
ронных систем.
Следовательно, эти условия имеют вид:
линейный потенциальный ящик кольцо*
. (X/2)n’ = L (п' = 1, 2...) ln"=L (п' = 0, 1, 2. ..)
Теперь для кинетической энергии **
р mei>2 (тер)2 А2
кин,- 2 — 2те ~ 2теМ
Придавая длине волны значения, разрешенные условиями квантова-
ния, найдем:
линейный потенциальный ящик
= о h-Ti~n'2
(п'=1, 2.. .)
кольцо
р = „.2
Е“" 2meL%
(п"^0, 1, 2. . .)
* п"=(К), где, собственно, Хи есть истинное квантовое число К-0, ±1, *2, ....
позволяющее записать и вырождение. „„инимается постоянной п может
*♦ Потенциальная энергия в таких моделях р
быть положена равной 0.
ДИАМАГНИТНЫЕ СВОЙСТВА
491
Таблица 79. Экзальтация диамагнитной восприимчивости
как характеристика ароматичности
Соединения Э-эксп.” 10е *рассч.‘1 °' Разность Д Разность А
Число бензольных циклов
Бензол —55,6 —36.9 18,7 18,7
Нафталин —91,9 —-55,7 36,2 18,1
Антрацен — 129,4 —74,5 54,9 18,3
Фенантрен — 127,9 —74,5 53,4 17,8
Дифенил — 102,9 —68,0 34,9 17,5
Азулен -91 —55,7 35,3 —
Трополон —61 —45,8 15,2 —
Тропой —54 —41,2 12,8 —
Циклооктатетраен — 105,1 -79,3 25,8 —
Примечание. В системе CGS эти величины должны быть
помножены на 10”.
Заполнение электронами уровней энергии в линейном ящике и в кольце
(в основном состоянии систем) для случая бл-электронов (например,
для гексатриена и бензола) представлено на приведенном ранее рис. 114.
Три нижних уровня (п = 1, п = 2 и п = 3) заняты тремя парами л-элек-
тронов с противоположными спинами — случай, соответствующий шести
углеродным атомам, входящим в открытую сопряженную систему, т. е.,
например, гексатриену-1,3,5. Такова его грубая модель. Выше располо-
женные уровни — уровни возбужденной молекулы. Если же гексатриен
замкнуть в циклогексатриен — бензол, то возникнут новые условия кван-
тования — следствие непрерывности электронной волны. В периметре L
уже не полуволна, а волна должна уложиться целое число раз, включая 0.
т. е. лиг = L (пг = 0, 1, 2 и т. д.).
Подобно предыдущему, получаем значения кинетической энергии для
целочисленных пг (квантовые значения);
/г2„г2 Д2 2пг2
m 2meL2 8meL2
(m = 0, 1, 2 и т. д.)
Каждому значению Ет в линейной системе соответствует единственный
(линейный) вид движения электрона. В кольце же всем значениям Е,п
(кроме Ео) может соответствовать движение электрона по часовой стрелке
и против, что имеет следствием возможность нахождения в каждом таком
уровне четырех электронов (вырожденные уровни). Только уровню Е$
соответствует по-прежнему два электрона. Отсюда и следует правило
Хюккеля — ароматические, т. е. циклические сопряженные системы
должны иметь 4ге 2 электрона.
Замыкание кольца имеет своим следствием дополнительную стабили-
зацию л-электронной системы. В примере, представленном на рис. ,
492 НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
_ ------
эта дополнительная стабилизация равна 12 единицам, если за единицу
принять величину h2/8meL2. С точки зрения такой дополнительной стаби-
лизации процессы, нарушающие сопряжение в кольце (например, реак-
ции присоединения), термодинамически невыгодны, а переходные состоя-
ния, связанные с нарушениями сопряжения, имеют тенденцию к его вос-
становлению.
Принимая, что периметр кольца L пропорционален числу электронов
(4п + 2) в кольцевой л-электронной системе, т. е. что L = I (4п 4- 2) *,
можно найти величину энергии размыкания кольца в линейный потен-
циальный ящик длиной L:
f Л Т7Х
х&я )размыкания “ jfjm, £2
(» + 1)
(2п -|-1)
Как мы видим, энергия размыкания уменьшается с ростом п
и, следовательно, с увеличением размера кольца.
Таким образом, все ароматические системы можно классифицировать
по числу «ароматических» электронов.
АРОМАТИЧЕСКИЕ СИСТЕМЫ С ДВУМЯ л-ЭЛЕКТРОНАМИ
Единственной изоциклическоп ароматической системой этого ряда должен
быть гипотетический циклопропенилий-катион’.
СН
НС=СН
сн
НС—'СН
СН
или Х+)\
нс—СН
ч—>
Известен лишь трифенилциклопропенилий-катион, полученный
в 1958 г. Д. Н. Курсановым и М. Е. Вольпиным и почти одновременно
Дерингом. Это соединение отличается большой устойчивостью: ни при
действии брома, ни водорода над катализатором не обнаруживает обычных
реакций разрыва трехчленного цикла и присоединения по двойной связи.
АРОМАТИЧЕСКИЕ СИСТЕМЫ С ЧЕТЫРЬМЯ л -ЭЛЕКТРОНАМИ
По Хюккелю, циклобутадиен, имеющий две л-связи и, следовательно,
четыре л-электрона, не должен быть ароматичен. И действительно, цикло-
бутадиен в момент выделения из комплекса по реакции (Петтит, 1966 г.)
Fe
ocz 1 Хсо
Се4*
Fe
ОС7 1 >О + се
и
* 4~ 2)л-электрона
структурной формулы.
соответствуют (2м 4- 1) формальным двойным связям
СИСТЕМЫ С ШЕСТЬЮ ^-ЭЛЕКТРОНАМИ
493
обладает свойствами энергичного диена и одновременно диенофила
Будучи выделен, он тотчас димеризуется
а при реакции с ацетиленкарбоновым эфиром ведет себя как диен, обра-
зуя производные «дыоаровского бензола»:
Однако в виде комплекса I с трикарбонилом железа цикло-
бутадпен ведет себя как настоящее ароматическое соединение (это уже
не моноцпклическая система).
Петтпт и сотр., которым принадлежат и только что упомянутые работы,
осуществили ацилирование комплекса I по Фриделю — Крафтсу, дейтери-
рование его дейтероуксусной кислотой, дпметиламинометилпрованпе по
Манниху, хлорметилирование формальдегидом с соляной кислотой, фор-
милирование метилформанилидом и РОС13, меркурирование.
АРОМАТИЧЕСКИЕ СИСТЕМЫ С ШЕСТЬЮ л-ЭЛЕКТРОНАМИ
Кроме бензола и других бензоидных систем, шесть л-электронов содержат
ароматические гетероциклы:
пиррол
пиразол
фуран
тиофен
изоксазол
й----N
II
N
/
NH
триазол
тетразол
и др. Они были рассмотрены в разделе «Гетероциклы».
Из изоциклических небензоидных ароматических систем с шестью
л-электронами наиболее важны и просты по структуре циклопентадиенил-
анион и тропилий-катион.
494
НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Циклопеитадиенил-анион
Циклопентадиен (кн. 1, стр. 547), содержащийся в продуктах пиролиза
нефтяных фракций и в бензольной фракции каменноугольной смолы,
является типичным непредельным углеводородом с двумя сопряженными
л-связями. При действии натрия на циклопентадиен (в растворе в этило-
вом эфире или тетрагидрофуране) происходит реакция:
11^ II +Na
СН2
Na+ + i/2H2
Натрий передает свой электрон отрывающемуся от молекулы цикло-
пентадиена протону, превращая его в атомарный водород, частично рас-
ходуемый на восстановление второй молекулы циклопентадиена в цикло-
пентен. Натрий-катион оказывается электростатически связанным с анио-
ном циклопентадиенила, в котором четыре электрона л-связей и электрон-
ная пара углерода, оставшаяся после отрыва протона, образуют «обезли-
ченный» ароматический секстет. Пятичленный цикл образован теперь
пятью равноправными СН-группами, и циклопентадиенил-анион может
быть изображен так:
НС—СН
I ОЬ \
НС^У/СН
сн
Того же результата можно достигнуть, действуя на циклопентадиен
сильными основаниями (такими, как амид натрия) или гриньяровым реак-
тивом:
HC-CH
/ + CH3MgBr . --->- НС OjcH + MgBr + CH4
СН2 СН
Такая система беднее энергией, чем этого можно было ожидать от
циклопентадиенил-аниона с нормальными двойными связями и зарядом,
фиксированным на одном из пяти углеродных атомов. Ион водорода
в циклопентадиене так легко уходит из молекулы в приведенных выше
реакциях именно потому, что остается устойчивый, бедный энергией
анион, и процесс энергетически выгоден. Так же легко водород покидает
молекулу в виде протона и в ряде реакций конденсации (с альдегидами,
кетонами, кн. 1, стр. 146). В присутствии оснований циклопентадиен
по той же причине обменивает все свои шесть водородных атомов на дей-
терий при действии D2O (Д. Н. Курсанов и В. Н. Сеткина).
Хотя циклопентадиенил-анион, несомненно, ароматичен и предста-
вляет собой неальтернантно-ароматическую систему, продемонстрировать
это непосредственно обычным путем на трудности реакции присоединения
и способности к реакциям электрофильного замещения невозможно,
СИСТЕМЫ С ШЕСТЬЮ ^-ЭЛЕКТРОНАМИ
495
поскольку речь идет о заряженной молекуле — анионе. Любой электро-
фильный реагент атакует молекулу, имеющую избыточный электрон
(отрицательный заряд аниона), и присоединяется к одному (безразлично
к какому) из пяти углеродов, нарушая ароматичность системы и превра-
щая циклопентадиенил-анион в замещенный циклопентадиен:
Если, однако, на циклопентадиенилмагнийбромид подействовать хло-
ристым железом (можно взять и хлорное железо — оно восстановится
до хлористого), то образуется интереснейшая ароматическая система —
дициклопентадиенилжелезо, или ферроцен, построенная, как показал
рентгеноструктурный анализ, сэндвичеобразно (Посон, Вудворд, Уил-
кинсон):
2
г + FeCl2
Fe + MgBr2 + MgCl2
Циклопентадиенилы ферроцена, связанные с металлом л-связью
(стр. 482), несут не полный отрицательный заряд аниона, а лишь частич-
ный. По квантово-механическим расчетам методом молекулярных орбита-
лей (М. Е. Дяткина и Е. М. Шусторович) он составляет —0,35 электрон-
ного заряда на каждом ядре (на железе +0,7). Поэтому циклопентадие-
нилы ферроцена гораздо менее нуклеофильно активны, чем циклопента-
диенил-анион, и не способны присоединять электрофильные реагенты,
кроме как в жестких условиях с разрушением всей системы (например,
при действии брома, когда происходит отщепление циклопентадиенилов
в виде пентабромциклопентана). Ферроцен уже не обменивает, как цпкло-
пентадиеп, свои водороды па дейтерий в присутствии оснований: он всту-
пает в дейтерообмен лишь с сильными дейтерокпелотамп типа D2SO4,
подобно бензолу, но с гораздо больше)! скоростью (Д. Н. Курсаггсв,
В. Н> Сеткина).
НЕ БЕН 3 О ИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕ МЫ
496
Первой реакцией электрофильного замещения ферроцена было откры.
тое Вудвордом и Розенблюмом ацилирование ферроцена по Фриделю —
Крафтсу, приводящее к образованию моно- и дикетопов ферроцена
(здесь и далее ферроценовое кольцо для краткости изображено в виде
обычного пятиугольника):
13 D-f-“ + D-J-8
RCCI+A1C13+ Fe --> Fe_ q + Fe q
о \_l <J «-с-<21
О
Однако в этих условиях ацилируются и циклоалканы, и первое время
ароматичность ферроцена стояла под вопросом, тем более что обычные
реагенты электрофильного замещения в бензоле (азотная кислота, галоиды
и даже серная кислота) отнимают от ферроцена один электрон, превращая
его в феррициний-катпион, уже неспособный принимать электрофильные
атаки.
Дунитц и Оргел подробным рентгеноструктурным анализом ферроцена
установили, что длина его С—С-расстояний равна 1,41 А, что соответ-
ствует ароматическому соединению. Расстояние Fe—С, составляющее
2,03 А, равно сумме ковалентных радиусов железа и углерода (односвяз-
ных). Расстояние между плоскостями циклопентадиенилов составляет
3,41 А.
Вращение циклопентадиенилов вокруг оси, проходящей через атом
железа и центры циклопентадиенилов, не затруднено. Ряд производных
ферроцена исследовал рентгеноструктурным методом Ю. Т. Стручков.
А. Н. Несмеянову и его сотрудникам Э. Г. Переваловой, Р. В. Головне,
О. А. Несмеяновой, Н. С. Кочетковой, С. С. Чуранову принадлежат пер-
вые работы, твердо установившие химически ароматический характер
ферроцена. Им удалось (см. схему) промеркурпровать ферроцен (1),
проалкилировать его по Фриделю — Крафтсу (2), просульфировать
в условиях А. П. Терентьева серным ангидридом (3) (одновременно Уэйн-
майр осуществил сульфирование серной кислотой в уксусном ангидриде),
показать одновременно с Посоном, что ферроцен арилпруется (4) по реак-
ции Гомберга арилдиазониевыми солями (радикальная реакция, см.
стр. ПО) и, подобно «суперароматическим» соединениям, металлируется
бутиллитием (5) и фенилнатрием (5') (А. Н. Несмеянов, Э. Г. Перевалова,
Р. В. Головня и О. А. Несмеянова; Бенкезер). Посону удалось, применив
реакцию Вильсмейера, действием па ферроцен форманилпда (6) в присут;
ствии H2SO4 получить ферроценалъдегид, ведущий себя как ароматический
альдегид. С ферроценом проходит также реакция Манниха (7) — формаль-
дегид с диалкил аминами в результате электрофильной атаки диалкилами-
нометилирует ферроцен.
Аминоферроцен (ферроцепиламин), по А. Н. Несмеянову, Э. Г. Пе-
реваловой и Л. С. Шиловцевой, был получен реакцией К. А. Кочеш-
кова (8). Он оказался основанием, более сильным, чем анилин, и неспособ-
ным диазотироваться, но в остальном ведет себя подобно ароматическим
аминам: образует шиффовы основания с ароматическими альдегидами-
СИСТЕМЫ С ШЕСТЬЮ ^-ЭЛЕКТРОНАМИ
497
|_\-HgCl
Fe
(J)
|_>-HgCl
Fe_
CIHg-/ |
сне-снг; Aid»
(2)
|_>-c2h5
Fe
(СН3),СС1; А1С1»
или (CHi)tC=CHt; Aicia
(20
С(СН3)з +
Fe
(CH3)3C- .
_J>-C(CH3)3
Fe
(СНз)зС-
>- С(СН3)з
+ Fe
(СНз)зС-/ |“С(СНз>г
SOs
(3)
-SO3H
Fe
CeHsNs Ci-
te)
CaHtLi
(6)
CeHsNa
о
II
HCNHC«H,
(в)
• CH,O+N11R>
I CH2NR2
Fe
(7)
32 Заказ G72
ПЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Среди приведенных реакций — электрофильные замещения, типичные
для ароматических (точнее, суперароматических) ядер. Металлирование,
радикальные замещения, поведение амина и альдегида создают полную
аналогию если не с самим бензолом, то с суперароматическими соедине-
ниями типа тиофена, фурана и фенола.
Из ртутных производных ферроцена действием иода (9) были полу-
чены первые его галоидпроизводпые — иодферроцен и Диодферроцен
в которых иод не вступает в обмен с анионами без медного катали-
затора (О. А. Несмеянова):
\-HgCI
Fe
Cl
Металлоорганические соединения ферроцена использованы в многочи-
сленных синтезах (А. Н. Несмеянов, В. А. Сазонова, В. Н. Дрозд). В част-
ности, действием СО2 на литиевое производное получают]ферроценкарбо-
новую (10) и ферроцендикарбоновую кислоты, а действием В(ОС2Н5)3 —
ферроценборную (11) и ферроцендиборную кислоты. Из ферроценборных
кислот с галоидной медью (12) получены хлорферроцен и бромферроцен
и соответственно дигалоидные соединения. Галоид в этих производных
ведет себя как типично ароматический галоид и мало склонен к реакциям
обмена. Однако он обменивается с медными солями карбоновых кислот
(13) и с фтальимидом меди (14), образуя производные:
-О-С—СНз
СО
Гидролиз первого из них (15) дает ферроценол, а гидразинолиз второго
(16) — ферроцениламин [см. также реакцию (8)].
Ферроценол — втрое более слабая кислота, чем фенол, и вещество
очень неустойчивое.
Аналогичным путем были синтезированы 1,1'-диоксиферроцен и 1.1 "
ферроценилендиамин, а также [обменом с Cu2(CN)2] моно- и динитрилы
ферроценкарбоновых кислот.
СИСТЕМЫ С ШЕСТЬЮ ^-ЭЛЕКТРОНАМИ
499
Оказалось, что и прямое действие на ферроценилборную кислоту
медных (Си11) солей карбоновых кислот (17), а также медных N-производ-
ных, подобных фтальимиду меди (18), приводит к замене остатка борной
кислоты на анион. Даже с пиридином в присутствии карбоната меди (19)
образуется а-ферроценилпиридин, который с лучшим выходом, однако,
получается действием ферроцениллития на пиридин (20).
Иной путь получения ферроценилгетероциклов — действие гриньярова
реактива, производного соответствующего гетероцикла, на бромферроцен
32*
500
НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
в присутствии медной соли. При действии в этих условиях на бромферр0_
цен бромистого арилмагния (20а) получается арилферроцен — второй
независимый путь синтеза арилферроценов:
ArMgX+ Cu»+ .
(20а)
Действие меди на иодферроцен спаивает ферроценовые радикалы
в диферроценил, а из 1,1'-дииодферроцена и иодферроцена таким образом
получается терферроценил.
При взаимодействии бромферроцена (или ферроценилборной кислоты)
с азидом меди (21) получается ферроценилазид, а из него действием литий-
ферроцена (22) — диазоаминоферроцен, который ацидолизом (23) превра-
щается в смесь ферроцениламина и ферроценилдиазония, способного
к азосочетанию и образующего, например, с р-нафтолом (24) ферроценил-
1-азонафтол-2.
Необыкновенной реакцией в ряду ферроцена является прямое цианиро-
вание соответствующего катиона синильной кислотой, приводящее к ни-
трилу ферроценкарбоновой кислоты и к ее 1,1'-динитрилу. Цианированию
подвергаются и производные ферроцена, причем электроноакцепторные
заместители (см. ниже) направляют цианогруппу в другое ядро, а электро*
нодонорные — в то же (А. Н. Несмеянов, Э. Г. Перевалова, Л. П. Юрьева)-
Гомологи ферроцена получены прямым алкилированием по Фриделю —'
СИСТЕМЫ С ШЕСТЬЮ ^-ЭЛЕКТРОНАМИ
501
Крафтсу (Н. С. Кочеткова) восстановлением литпйалюмипийгидридом эфи-
ров ферроценкарбоповой кислоты (метильный гомолог, Э. Г. Перевалова)
и восстановлением ацилферроценов (Н. Л. Волькеиау).
Изучение реакций электрофильного замещения гомологов и других
производных ферроцена позволило придтп к выводу, что ферроцен пред-
ставляет собой единую ароматическую систему (а не два изолированных
циклопептадиенпльных ароматических кольца) и влияние заместителей
передается из одного кольца в другое примерно так, как в бензольном
кольце из мета-положения. Однако заместитель оказывает все же большее
влияние на то кольцо, в котором он находится, как это впервые выяснили
Н. А. Несмеянов и О. А. Реутов на целом ряде синтезированных ими
ферроценкарбоновых кислот, имеющих в другом циклопентадиепильном
кольце заместители типа
SO2NH2, SO2F, CN, R—С—, RO-C—, Aik
На примерах алкилирования и ацилирования ферроцена по’Фриделю —
Крафтсу можно видеть, что электроноакцепторная группа (например,
ацетил) пассивирует тот цпклопептадпеппл, в который опа вступила,
больше, чем другой, и следующий ацетил вступает в другой цикл. Этил
активирует тот циклопентадиенил, в который он вступил, больше, чем
другой, и второй этил замещает водород в том же цикле, причем образуется
смесь двух изомерных гомоапнулярпых диэтил ферроценов — 1,2-диэтил-
ферроцеп и 1,3-дпэтплферроцен с значительным преобладанием 1,3-изо-
мера. Такое замещение типично. Здесь сказываются свойства пеальтер-
нантной ароматической системы. Ориентация определяется общей усилен-
ной нуклеофильностью кольца за счет -(-/-эффекта этильной группы, без
альтернирования этой нуклеофильности у четных п нечетных атомов.
Преобладание 1,3-изомера над 1,2-изомером объясняется большими про-
странственными затруднениями при образовапип последнего. Неальтернан-
тность, видимо, является также и причиной отсутствия в ферроценовом
ядре столь типичных для бензольного ядра перегруппировок, таких, как
клайзеновская перегруппировка аллилового эфира фенола в о-аллил-
фенол (А. Н. Несмеянов, В. А. Сазонова):
СН
О—СНо
I
Fe СН2-=СН
НЕ БЕ НЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Из-за неальтернантности системы циклопентадиенила образование
реакционного комплекса с хиноидным расположением двойных связей,
как в бензоле, невозможно, и перегруппировка по осуществляется.
Не происходят и другие перегруппировки в ядро, характерные для
бензольного ядра. Так, полученные А. Н. Несмеяновым, Э. Г. Перевало-
вой п Т. В. Никитиной азоферроцены, будучи восстановлены в гидразо-
ферроцены, не перегруппировываются в соответствующие бензидины.
Чрезвычайно интересны результаты количественного изучения
С. П. Губиным и Э. Г. Переваловой (частью вместе с К. И. Грандберг)
влияния заместителей X в ферроценовом ядре на окислительно-восстано-
вительный потенциал обратимой реакции
ХС5Н4РеС5П5 XC5H4Fe+C5H5
+е
и на константы диссоциации замещенных карбоновых кислот ферроцена
XC5H4FeC5H4COOH. В первой реакции заместители уменьшают потен-
циал окисления, если это электронодонорные заместители, и направляют
электрофильное замещение в то же циклопентадиенильное ядро, где нахо-
дятся сами (X в этом случае — ориентанты первого рода). Если же это
электроноакцепторные заместители, то окислительно-восстановительный
потенциал повышается. Логарифмы этих потенциалов (ординаты) ложатся
на прямую, если по оси абсцисс отложить константы а“ара Тафта для ряда
заместителей X (уравнение Гамметта 1g К — ор, см. стр. 193). Эти кон-
станты выведены Тафтом на основании измерения констант диссоциации
ряда кислот типа
X—Ч—СН2—СОН
\=Z ||
о
т. е. таких, в которых влияние заместителя X, переданное бензольным
ядром, доходит до карбоксила только индуктивным эффектом через СН2-
группу, прерывающую сопряжение. Для констант диссоциации кислот
XC5H4FeC5H4COOH справедливы те же значения о’ара Для заместителей
X. Все это указывает, что влияние заместителя в одном циклопентадиениль-
ном ядре передается на окисляющийся атом железа или в другое ядро
индуктивным эффектом (/-эффектом), а не Т-эффектом, как из пара-поло-
жения в бензольном ядре.
Интересно, однако, отметить, что заместители X делятся по отношению
к ферроцену на электронодонорные и электроноакцепторные иначе, чем
по отношению к бензольному ядру. Так, лишь алкилы электронодонорны
по отношению к ферроцену (аминогруппа не измерялась); галоиды же,
ОН, ОСН3, —NH С—СН3 и даже —HgCl, не говоря уже о —С—ОН
О О
и О R, электроноакцепторны. Эти отношения выявляются и во взаимо-
0
действии бензольного ядра и ферроцена в фепилферроцене. При нитрова-
нии фенилферроцена образуется n-нитрофенилферроцеп, т. е. ферроцениль-
СИСТЕМЫ С ШЕСТЬЮ ^-ЭЛЕКТРОНАМИ
503
ный заместитель действует как электронодонорный ориентант первого
рода. Наоборот, в реакции диметиламинометилирования посредством
тетраметилдиаминометана (вариант реакции Манниха) был получен
1-фенил-1'-диметиламинометилферроцен, т. е. фенил как заместитель
второго рода направил реакцию электрофильного замещения в другое
циклопентадиенильное кольцо.
Е. О. Фишер синтезировал диинденилжелезо (бензферроцен), исходя
из галоидного инденил магния. Такое диинденилжелезо он прогидрировал
в ди-(тетрагидроинденил)-железо.
А. Н. Несмеянов, Н. А. Волькенау и В. Д. Вильчевская получили
последнее соединение, идентичное фишеровскому, следующей серией
реакций:
AICI. .
НООССН2СН2
|-\ I
с=°
Fe_
О=С-<" I
сн2сн2соон
Zn; нс! t
>—(СН2)3СООП
НД?2О, + Р2О5
Fe ------------>.
НООС(СН2)з—/ |
В последнее время аналогичным путем действием фталевого ангидрида
получено производное которое легко окисляется в аналог антрахи-
нона — темно-фиолетовые кристаллы (А. Н. Несмеянов, Н. С. Кочеткова
и В. Д. Вильчевская):
МпО»
Интересны свойства атома углерода в а-положепии по отношению
к циклопентадиепильному ядру. Резкую стабилизацию а-карбониевого
НЕ БЕ Н ЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
504
катиона впервые отметил Ричардс, который объяснил ее непосредственной
мезомерной подачей электрона атомом железа карбкатиону
Э. Г. Перевалова и Ю. А. Устынюк проводили ферроценилметилирова-
ние четвертичными аммониевыми солями типа
|~СН2-N-СН2СвН5
F® НзС7 СПз
в реакции, например, с гидроксильным ионом, что позволило сравнить
способность к отрыву карбкатионов
~>-СН2
Fe и СН2СбНб
Как оказалось, гидроксил алкилируется всегда ферроценилметильным
ионом, а не бензильным; реакция идет как замещение типа S1. Значит,
ферроценилметил-катион значительно устойчивее, чем бензилкатион.
Совершенно иначе обстоит дело, по работам этих же авторов, со стаби-
лизацией ферроценилом карбаниона и радикала. Так, перегруппировка
Стивенса, которая идет с аммониевыми солями того же типа под действием
сильного основания, отрывающего протон от одной из СН2-групп, связан-
ных с аммонийным азотом, шла всегда в направлении, показывающем,
что фенил стабилизирует соседний анионный центр в большей степени,
чем ферроценил:
I CH2N—CH2CeH5 I \-CII2-N-CHCeII5 I 4;-CH2-CHCeIl5
I—/ Hl.i 1— ' /\ ___> I / I
H3C CH3 F-i НзС СПз ZF® N
НзС СНз
При восстановлении амальгамой натрия в воде отщепляется бензиль-
ный радикал в виде толуола и остается связанный с азотом ферро-
ценилметильный радикал.
В последнее время получен и триферроценилметилъный катион,
очевьустойчивый.
По исследованиям А. Н. Несмеянова, В. А. Сазоновой и В. И. Рома-
ненко, ферроценилметильные катионы неустойчивы по отношению к свету
и распадаются с образованием фульвенов, например:
1-х + /R
L>~G\.R ftv ,=ч ZR
Fe_ --------* /—+ Fe2++ H+-f- Полимер W
СИСТЕМЫ С ШЕСТЬЮ ^-ЭЛЕКТРОНАМИ
505
Оказалось, что все ферроценовые соединения, несущие на а-атоме
полный или частичный положительный заряд, подвержены фотолизу
с образованием фульвенов или илидов * (циклопентадиенилидов). Напри-
мер:
Реакции типа (3) представляют собой новый путь синтеза разнообраз-
ных илидов (см. ниже).
Ферроцен — не единственная ароматическая сэндвичеобразная система.
Исключительно прочен изоэлектронный^ферроцену кобалътициний-катион
Со*
который, однако, вследствие своего положительного заряда не способен
к реакциям электрофильного замещения.
Циклопентадиенильные производные рутения и осмия — рутеноцен
и осмоцен — подобны ферроцену, ио мепее суперароматичны, т. о. с не-
сколько меньшей легкостью, чем ферроцен, подвергаются электрофильным
замещениям.
Этот термин ввел Впттиг, который их впервые получил.
НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
506 ________________________'---------------------------------
На основании скорости дейтерирования в кислой среде Д. Н. Курси-
вов и В. Н. Сеткина установили следующий^ ряд ароматичности сэндвиче-
образных металлоорганических соединений:
Другие ароматические системы, в которых циклопентадиенил получает
отрицательный заряд, это так называемые илиды, примерами которых
могут служить структуры
+ /С6Н5
[@>-Р-С6Н5
хс6н5
В них отрицательный заряд пятичленного кольца внутримолекулярно
компенсируется положительным зарядом аммониевой или фосфониевой
части молекулы. Илиды обладают большим дипольным моментом; напри-
мер, дипольный момент приведенного только что трифенилфосфонийцикло-
пентадиенилилида равен 7D.
В качестве примеров ароматических реакций илидов можно привести
азосочетание с фенилдиазоний-катионом
-а +ХСбИ5 + /7^\ +/СбН5
О/—Р^С6.Н5 + С6Н5—N—N->С6Н5—N=N—(О)—Р—С6Н5 + н +
С6Н5 Ччс6н5
или осуществленное В. А. Сазоновой бромирование:
СИСТЕМЫ С ШЕСТЬЮ ^-ЭЛЕКТРОНАМИ
507
Еще одним примером илида может служить диазоциклопентадиен
со структурой
N=N
Наконец, отрицательно заряженный циклопентадиенил присутствует
в бициклической системе азулена (стр. 512).
Многие илиды легко захватывают протон кислот, превращаясь таким
образом в замещенные циклопентадиены:
+ Н+Х
При этом они резко меняют свойства, теряют ароматичность и изме-
няют светопоглощение, являясь индикаторами.
Тропилий-катион
В известном смысле антиподом циклопентадиенил-аниона является тропи-
лий-катион. Если к циклогептатриену присоединить молекулу брома
(1,6-присоединение) и затем щелочью отнять одну молекулу НВг, то
оставшийся атом брома одновременно сам покидает молекулу, уходя
в виде аниона и оставляя катион тропилия (Деринг):
---->
-НВг
Стремление образовать устойчивую ароматическую систему катиона
тропилия так велико, что, как независимо друг от друга нашли Д. Н. Кур-
санов с М. Е. Вольпиным и Даубен, циклогептатриен (тропилиден) легко
теряет гидрид-анион под действием акцептора последнего, каким может
служить, например, трифенилметил-катиоп или бензил-катион:
СН
с'-п СН2С6Н5 —> [ (+) > + сбн5сн3
L Н 1J
В то время как в циклопентадиенил-анионе шесть ароматических
электронов были обезличены между пятью углеродными атомами, в тропи-
лий-катионе ароматическая шестерка «размазана» уже между семью
углеродными атомами.
НЕ БЕ II3 ОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
508
Сопоставим еще раз три главные изоциклические ароматические
системы:
Все три системы — плоские. Это существенное условие ароматичности.
Циклопентадиенил-анион даже в форме сэндвичеобразных металличе-
ских производных с ослабленным отрицательным зарядом крайне воспри-
имчив к электрофильным атакам. Бензол — нейтральная молекула.
Тропилий-катион совершенно индифферентен к электрофильным атакам
и не подвергается обычным для бензола (и ферроцена) реакциям'замеще-
ния, но крайне восприимчив к нуклеофильным атакам. При этом, однако,
происходит не замещение, а присоединение с нарушением ароматичности
и превращением в систему циклогептатриена:
Н CN
Подобная же реакция протекает при действии активных металлоорга-
нических соединений (реакции с карбанионами). Так, действием гринья-
рова реактива тропилий-катион превращается в алкил- (или арил)цикло-
гептатриен:
Н R
+ RMgCl-----> j] + MgCl
В водных растворах тропилиевых солей устанавливается равновесие:
Н он
® ] + Н2О ч f I] + Н
Таким образом иоп тропилия создает в водном растворе кислую реак-
цию; он является лыоисовой кислотой, по силе равной уксусной кислоте.
Поэтому тропилиевые соли существуют только с анионами сильных
кислот. Анионы слабых кислот, т. е. анионы, склонные к образованию
ковалентных связей (поэтому и соответствующие им кислоты слабые),
реагируют подобно циан-аниону.
Интересны реакции превращения тропилий-катиона в производные
бензола. Так, при окислении соли тропилия хромовым ангидридом или
«СИСТЕМЫ С ШЕСТЬЮ ^-ЭЛЕКТРОНАМИ
509
перманганатом образуется бензальдегид. В свою очередь от бензола
ложно перейти к тропилий-катиону путем следующих реакций
+ СН
-и далее, как было указано раньше.
Среди производных тропилий-катиона особое место занимает тропон
(циклогептатриенон) — кетон, который может быть получен гидролизом
«бромтропилий-катиона:
О + НВг
Многие свойства этого кетона, как физические (высокий дипольный
момент, 4,17 D), так и химические (пассивность к действию брома), указы-
вают на то, что структура тропона сдвинута в сторону бетаина:
Если бы он имел полностью структуру этого бетаина, дипольный момент
был бы значительно больше.
Тропон может быть получен также путем расширения бензольного
цикла при действии диазометана на анизол. Первая фаза реакции состоит
•в превращении анизола в метоксигептатриен, который действием брома
превращают в тропон:
з “Ь CH9N2
^/ОСНз
С Но + No
+ Вг2
+ СН3Вг
Тропон — слабое основание, образующее с хлористым водородом
•соль:
510 НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
. - . .----------------------------------------------------------
Самым интересным веществом, имеющим в основе структуру тропона
является трополон. Его можно получить, дегидрогенизуя действием
бромсукципимида циклогептандион, который получают из суберона окис-
лением а-метиленов ого звена в карбонил посредством двуокиси селена:
Действие брома на 2-оксициклогептанон (дегидрогенизация бромом}
также приводит к устойчивой структуре трополона.
Трополон ароматичен. Он беднее энергией, чем требуется по аддитив-
ной схеме (на 29 ккал/моль). Дипольный момент трополона 3,59 D. Тропо-
лон — слабая кислота, располагающаяся по силе между уксусной кисло-
той и фенолом (рКг — 6,92). При диссоциации его образуется симметрично
построенный анион
Во многом трополон похож на фенолы. Как и фенолы, он дает окрашен-
ное соединение при действии хлорного железа, с ангидридами и хлорангид-
ридами (но не с кислотами) образует сложные эфиры. Простые эфиры
трополона, получаемые алкилированием диалкилсульфатами или диазо-
алканами, гидролизуются гораздо легче, чем простые эфиры фенолов,
и поэтому ближе к сложным эфирам кислот. Вместе с тем трополон —
основание, способное захватывать ион водорода и образовывать комплекс-
ные соединения, например, с ионом меди.
В отличие от тропонов трополоны легко вступают в реакции электро-
фильного замещения. Замещения протекают в положения 3, 5 и 7.
СИСТЕМЫ С ШЕСТЬЮ к-ЭЛЕКТРОНАМИ
511
Трополоны галоидируются, нитруются, нитроз ируются, образуют
с арилдиазоний-катионами арилазотрополоны, вступают в реакцию Ман-
ниха (аминометилирование), действием хлороформа в щелочной среде можно
ввести в трополон по реакции Реймера — Тимана альдегидную группу.
•Однако трополон не удается алкилировать и ацилировать по Фриделю —
Крафтсу. При действии нуклеофильных реагентов, например щелочи,
трополоны (особенно легко замещенные, имеющие электроноакцепторные
заместители) перегруппировываются в производные бензойной кислоты:
Эта реакция позволяет сравнительно легко выяснить положение за-
местителей в цикле и широко применяется для установления строения.
Как и многие устойчивые ароматические системы, трополон в виде
производных довольно широко распространен в растительном мире,
что стало известным лишь в 1940—1950 гг. Из природных веществ, вклю-
чающих структуру трополона, упомянем находящиеся в древесине кедра
терпеновые трополоны — а-, [3- и у-шуйяплицины'.
Строение их было установлено путем восстановления в соответству-
ющий изопропилциклогептандиол и окислением последнего в изопропил-
Пимелиновую кислоту с соответствующим положением изопропильной
. изопропилпимели-
а-туияплицин новая кислота
В плесенях разных видов были
найдены следующие производные
трополона:
стинитатовая
кислота
пуберуловая
кислота
В некоторых луковичных растениях семейства Lilaceae рода Colchicum
(крокус) содержится алкалоид колхицин, ставший особенно известным
НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
благодаря своей способности вызывать полиплоидию растений при обра-
ботке им зародышей семян. В 1945 г. Дьюар высказал предположение,
что в структуре{колхицина участвует трополонный цикл, а в дальнейшем
была установлена его структура:
ОСНд
АРОМАТИЧЕСКИЕ СИСТЕМЫ С ЧИСЛОМ л-ЭЛЕКТРОНОВ БОЛЕЕ ШЕСТИ
Азулен
Конденсированный бицикл из тропилиевого и циклопентадиенильного
колец — бицикло[5,3,0]декапентаен
за свой интенсивно синий цвет был назван азуленом. Дипольный момент
азулена равен всего 1,0 D. Это хотя и большая величина для углеводорода,
но гораздо меньшая, чем требует формула а. Поэтому следует допустить,
Что в описании строения азулена большее значение имеет совсем неполяр-
ная структура б. Однако азулен на 45 ккал/моль (энергия резонанса}
беднее энергией, чем соответствующий гипотетический углеводород
строения б с изолированными локализованными двойными связями.
Одним из первых путей синтеза азуленового бицикла явилась внутри-
молекулярная кротоновая конденсация дикетона — циклодекандиона-lA
приводящая к бициклическому кетону Д9П0)-бицикло [5, 3, 0]-деценону-4.
Его дегидрогенизация с одновременной дегидратацией приводит к азулену:
Другой путь состоит в действии на индан диазометана, разлагающегося
под влиянием ультрафиолетового света. Полученный циклопентаногепта-
диен дегидрируют над палладием (на угле) в азулен:
ch2n2
513
СИСТЕМЫ С ЧИСЛОМ ^-ЭЛЕКТРОНОВ БОЛЕЕ ШЕСТИ
При выборе надлежащих условий можно получить азулен дегидпоге-
нпзациеи над палладием, нанесенным на уголь, циклодекана Тыхопдо
50%) или циклодецена (выход до 70%). О^ыход до
В 1963 г. Димрот, Вольф и Вахе описали новый путь синтеза азуле-
нов действием избытка трифенилфосфинметилена (реагент Виттига)
на пприлиевые соли:
СН2=Р(С6Н5)3
+ 2 HR Г.,
4'
Выход достигает 30%. Изображенный в скобках промежуточный про-
дукт далее депротонпруется, раскрываются циклы с образованием кето-
нов; карбонильная группа в положении 5' конденсируется в а-положение-
4 разомкнувшегося в кетон верхнего цикла, а метиленовый углерод
виттиговского реагента, отщепляя трифенилфосфин, связывается с углеро-
дом С4, замыкая пятичлен и семичлен азулена. При недостатке реагента
Виттига реакция идет иначе: кислород пирилиевой соли замещается на СН
и образуется бензольный цикл.
Наконец, наиболее практичный путь — синтез азулена исходя из
пиридина (Циглер). Пиридин превращают в монометиланилиновое произ-
водное енольной формы глутаконового альдегида, которое за счет своей
второй альдегидной группы конденсируется с циклопентадиеном в соответ-
ствующий фульвен (кн. I, стр. 548). Этот фульвен при нагревании до 200 С
отщепляет метиланилин и с хорошим выходом образует азулен.
33 Заказ 672
514
НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Азулен обладает слабыми основными свойствами; с кислотами он обра-
зует соль азуления:
+ HCI <
н н
CI
При этом цвет его из синего переходит в желтый.
Азулен — изомер нафталина и при нагревании до 350° С и выше
количественно превращается в этот более устойчивый изомер.
Естественно, что азулен способен к электрофильным замещениям
в пятичленном кольце. Ацилирование его по Фриделю — Крафтсу дает
моно- и дикетоны:
R
Так же, хотя и с плохим выходом, проходит алкилирование азулена.
Вследствие того что азулен реагирует с ионом водорода, его нельзя нитро-
вать азотной кислотой, но при действии на азулен и алкилазулены тетра-
нитрометана в пиридиновом растворе осуществляется нитрование в поло-
жения 1 и 3 (в самом азулене эквивалентные); получен также 1,3-динитро-
. азулен. Сульфирование комплексным соединением серного ангидрида
с диоксаном или раствором серной кислоты в уксусном ангидриде приводит
к образованию 1,3-азулендисульфокислоты. Выделены также моносульфо-
кислоты алкилазуленов. В положения 1 и 3 осуществляется и галоидиро-
вание азулена галогенсукцинимидом.
Азулен легко подвергается реакции азосочетания при действии арил-
диазоний-катиона (на схеме показано, что азогруппа вступает в то же
положение 1 азулена, что и нитрогруппа при нитровании):
+ н
СИСТЕМЫ С ЧИСЛОМ ^-ЭЛЕКТРОНОВ БОЛЕЕ ШЕСТИ
515
Действие такого'нуклеофильного реагента, как метиллитий, напра-
вляется на положительно заряженное семичленное кольцо азулена.
Сначала, по-видимому, происходит присоединение метиллития по двойной
связилтак, что метил присоединяется в положение^, а литий в положение
7 или 5. После гидролиза образуется 8-метилдигидроазулен, который при
дегидрогенизации (хлоранилом) ^превращается в 8-метилазулен (или. что
то же, 4-метила.зулен).
Как выяснено сравнительно недавно, производные азулена довольно
широко распространены в растительном мире. В большинстве случаев
это так называемые азуленогены, относящиеся к сесквитерпенам (стр. 652)
и имеющие молекулярную формулу С1БН24 (или их кислородные дериваты).
Для превращения в гомологи азулена Эти вещества должны быть дегидро-
генизованы в С15Н18. Такая дегидрогенизация осуществляется нагрева-
нием с серой (200° С) или селеном (300э С), но выход составляет от 5 до
30%, так как при столь высоких температурах происходит изомеризация
в производные нафталина. Дегидрирование на палладии протекает еще
хуже. Лучший реагент дегидрогенизации в этом случае хлоранил (тетра-
хл op-n-бенз охинон).
Сесквитерпены азуленовой структуры были найдены во многих эфирных
маслах и самих растениях, например в полынях, в моркови, ветивере,
семенах аниса и фенхеля, в тысячелистнике, рыжиках.
Чтобы дать понятие о структурах этих азуленовых сесквитерпенов,
укажем, что главной составной частью эфирного масла древесины Guaia-
сит officinale является сесквитерпеновый спирт гвайол, который при нагре-
вании с кислотой сначала дегидратируется в гвайен, а затем действием
серы — в гвайазулен'.
Ветивазулен — изомер гвайазулена — получается дегидрированием
сесквитерпенового кетона Р-ветивона, содержащегося в ветиверном эфир-
ном масле. Грибы рыжики содержат лактаровиолин и лакпгаразулен
ветивазулен
лактаровиолин лактзразулен
33*
516
НЕБЕНЗОИДНЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
До сих пор не синтезирован углеводород пентален
Не поддается дегидрогенизации бицикло [0,3,3]октан, не привели
к успеху п другие многочисленные попытки синтеза пенталена. Этот
углеводород содержал бы 8, а не 6 и не 10 электронов, образующих л-
связи. Сама трудность его получения указывает Iна его нестабильность
и тем самым на неароматичность. Не обладает ароматическими свойствами
и циклооктатетраен, в котором имеется также лишь 8 л-электронов.
Однако циклооктатетраен способен захватить два электрона (например,
от металлического калия) и превратиться в результате этого в ароматиче-
ский дианион с 10 л-электронами (Д. Н. Курсанов):
О + 2К
Высший аналог циклооктатетраена циклопентадекаен
хотя и содержит 10 электронов л-связей, так же, как и циклооктатетраен,
обладает свойствами олефина. Такое несоответствие правилу Хюккеля
объясняют неплоской формой цикла, тогда как для непрерывного по циклу
перекрывания орбиталей л-электронов необходима (это условие обяза-
тельно, но недостаточно) плоская или достаточно близкая к плоской форма
цикла.
В циклододекагексаене С12Н12 число л-связей не соответствует правилу
Хюккеля. Кроме того, как легко видеть, приняв во внимание межатомные
расстояния и размер атомов водорода, в той конфигурации этого угле-
водорода, которая могла бы быть плоской (с чередующимися цис- и транс-
расположениями вокруг олефиновых связей)
атомы водорода в центре молекулы налагались бы друг на друга и не позво-
ляли бы молекуле принять плоскую форму.
Таким образом, ароматических свойств можно ожидать от циклополие-
нов С„Н„ с числом л-связей 7 (что соответствует 14 электронам), 9 (18
электронов), 11 (22 электрона) и т. д., могущих иметь плоскую конфигу-
рацию. Циклооктадеканонаен С18Н18 (один из аннуленов) получен
в 1959 г.
СИСТЕ МЫ С ЧИСЛОМ ^-ЭЛЕКТРОНОВ БОЛЕЕ ШЕСТИ
517
Аннулены
Так называются высшие циклические полиены со сплошь сопряженными
л-связями. Примером синтеза их может служить синтез циклооктадекано-
надиена, или 113}-аннулена (Зондхеймер), полученного следующей серией
реакций:
3 НС=С—СН2—СН2—С=СН
Сиг+(пиридин)
OR-
СН2—с=с — с=с— сн2
Первая стадия реакции проходит с выходом —6% в сторону цикличе-
ского полиина. Вторая стадия — это обычное перемещение л-связеп под
влиянием щелочного агента. Третья стадия — гидрирование тропных
связей над палладием, отравленным свинцом. Этот [18]-аннулен, как
и другие аннулены, имеет сплошную транс-конфигурацию, поэтому его
водороды резко делятся на два типа — экзоаннулярные и интрааннуляр-
ные, что ясно видно из спектра ЯМР (кн. I, стр. 596). Под влиянием пере-
менного магнитного поля (именно его вектора, перпендикулярного к плос-
кости ароматического цикла) л-электроны образуют круговой ток, вызы-
вающий со своей стороны относительно малый вектор магнитного поля,
противоположный приложенному полю и направленный из центра арома-
тической молекулы. Это экранирующий эффект, ослабляющий действие
внешнего поля на протоны, находящиеся в центре ароматической моле-
кулы, как в [18]-аннулене. Значит, необходимо приложить большее
внешнее поле, чтобы преодолеть этот экранирующий эффект и вызвать
ЯМР. Происходит «химический сдвиг» ЯМР внутрикольцевых (интра-
аннулярных) протонов в сторону сильных полей — даже более сильных,
чем для обычного стандарта — протонов тетраметилсилана (правее на
1,9 м. д.).
Напротив, внешнекольцевые (экзоаннулярные) протоны, как во всех
обычных ароматических системах, дают химический сдвиг ЯМР в сторону
518 НЕВЕНЗОИДПЫЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
--------------------------------—— — - -----------------------,
слабых полей (на 8,8 м. д.). Это объясняется тем, что снаружи аромати-
ческого кольца направление магнитных силовых линий, как всегда вокруг
соленоида, противоположно сравнительно с их направлением внутри
кольца-соленоида и таким образом вызывает дезэкранирующий эффект.
Порфирины
Широко распространены в природе и получены синтетически гетероцикли-
ческие ароматические системы с плоской формой молекулы и 18 л-электро-
нами — порфирины с их родоначалышм веществом порфином'.
порсрин
(пунктиром отмечены двоиные связи,
не участвующие в системе сопряжения)
Система порфирина лежит в оспове хлорофилла зеленого листа расте-
ний и красного вещества крови — гемоглобина. Она отличается высокой'
прочностью. (Химию порфиринов см. в разделе «Гетероциклы», стр. 296).
Сходное строение имеют фталоцианины, получаемые нагреванием нитрила
фталевой кислоты и его производных с солью меди (стр. 308).
И в порфиринах и во фталоцианинах водородные атомы при двух
атомах азота легко замещаются на разные металлы (магний, железо,
медь), причем атом металла симметрично связывается координационными
связями с остальными двумя азотами.
КАРБАНИОНЫ
Мы уже встречались с карбанионами как с промежуточными продуктами
в реакциях нуклеофильного замещения галоидов по ионному механизму
первого кинетического порядка (.S'jyl), а также нуклеофильного присоеди-
нения по л-связи олефинов. На протяжении курса рассматривались и дру-
гие реакции карбанионов. Примерами могут служить действие галоидных
алкилов на ионно построенные натрийалкилы или натрийарилы, алкили-
рование ацетиленида натрия, алкилирование натриймалонового эфира,
реакции конденсации по а-метиленовому звену кетонов в присутствии
оснований и многие другие важные реакции, приводящие к образованию
новых углерод-углеродных связей. Натриевые, калиевые и т. п. производ-
ные алканов, алкенов, алкинов, аренов являются ионными парами и со-
держат истинный карбанион в качестве активной в синтезе компоненты.
Литиевые, магниевые и другие металлоорганические соединения, о реак-
циях которых см. стр. 382 сл., часто имеют уже ковалентно связанный
-с углеродом металл, и, хотя их реакции во многом похожи на реакции
натрий и калийорганических соединений, говорить в этих случаях о реак-
циях истинных карбанионов нельзя. Натриймалоновый эфир и подобные
•ему соединения построены ионно, однако анионный заряд настолько
рассредоточен на кислородные атомы карбалкоксильных групп
•что о карбанионе можно говорить лишь условно.
Некоторое право па это дает лишь то обстоятельство, что любые элек-
трофильные атаки (например, при алкилировании галоидными алкилами)
проходят все-таки по углероду. Еще более условно (и обычно это не де-
лается) причисление к карбанионам аниона натрийацетоуксусного эфира
и подобных ему соединений, в которых анион несомненно имеет енолятную
структуру с большей частью заряда на кислороде. Однако и здесь, в силу
520
КАРБАНИОНЫ.
перенесения реакционного центра, алкилирование обычно проходит по-
углероду. Ароматический аналог енолята — фенолят натрия уже невоз-
можно отнести к веществам, включающим карбанион как структурную,
единицу, хотя по крайней мере некоторые реакции алкилирования, напри-
мер аллилпровапие, и здесь в определенных условиях проходят по угле-
роду. По углероду проходит и карбоксилирование двуокисью углерода
в реакции Кольбе.
Углеводородный остаток, входящий в молекулу, — метил, метилен
или метин — делается способным образовать карбанион прямым путем-
(отщепляя при действии оснований протон) под влиянием электроноакцеп-
торных групп, связанных с этим углеводородным остатком. По Арндту
и Эйстерту, электроноакцепторные группы располагаются по их способ-
ности к «ацидифпкацип» («протопизации» водорода, связанной с ними мети-
леновой группы) в такой последовательности:
—NO2 > — S—OR
II
О
Однако эти электроноакцепторные группы, принимая на себя бблыпую-
или значительную часть анионного заряда, тем самым вносят известную-
условность в самое понятие карбаниона.
В некоторых структурах карбанионов рассредоточение анионного-
заряда достигается и без участия названных электроноакцепторных групп;,
таким путем тоже создается устойчивость карбаниона даже в некоторых
протонных растворителях. Таковы анионы циклопентадиенпла, инденила
и флуоренила:
НС—СН
В циклопентадиенил-анионе лишний электрон, создающий анионный
заряд, входит в ароматическую шестерку (стр. 494) и принадлежит всей
системе ароматического пятичлена — одинаково каждому из пяти угле-
родных атомов.
Превращение углеводородного звена молекулы в карбанион совер-
шается путем отрыва протона под действием основания более сильного,
чем получаемый карбанион. Примеры:
CeHe + C2lI5Na+ —► СвЩНа+ + С2Нв (реакция Шорыгпна)
CHasCH + 2Na
NafcCNa 4- Н2
КАРБАНИОНЫ
521
ССН2 4- 2 Na+CeH5 --> fQ)> Na+ + C6H6
^COOR
XCOOR + ’°C2H5 ---№+[CH(COOR)2]- + C2H5OH
CH2—CH2
CH3-C-CH3 + HN\ ^CH2
£ CH2 —CH2
CH2=C—CH3 +
CH3NO2 + Na+ "OC2H5
Na + C2H5OH
В табл. 80 (по Краму) сопоставлены значения р/<а в округленных
числах, которые для слабых кислот получены косвенными методами
оценки.
Таблица 80. Шкала рЛГа Мак-Ивена — Стрейтвизера— Эпплквиста — Десен
.(шкала МСЭД)
Соединение Р*а Соединение РКа
Циклопентадиен 15 Бензол 37
Инден 18,5 Триптицен (а-положенпе) 38
Фенплацетплен 18,5 Циклопропан 39
•Флуорен 22,9 Метан 40
Ацетплеп 1 25 Этан 42
Трифенилметан 32,5 Циклобутан 43
Толуол (а-положение) 35 Неопентан 44
Пропен (а-положение) 35,5 Пропан (втор-положение) 44
Циклогептатрпен 36 Циклопентан 44
Этилен 2 36,5 Циклогексан 45
* По данным Вудинга и Хиггинсона (J. Chem. Soc., 1952, 774), приведенным в шкале МСЭД.
г С. Миллер и В. Ли [J. Am. Chem. Soc., 81, 6313 (1959)] для верхнего предела значений
фКа 1,2-дигалоидэтиленов дают величину 34—36.
Только для сравнительно сильных кислот начала таблицы возможно
прямое измерение констант Ка. Для более слабых помогает то, что между
логарифмом константы Ка равновесия
RH R: + Н+
и логарифмом константы скорости диссоциации в одномитомже раствори-
теле и при одной и той же температуре существует линейная зависимость.
КАРБАНИОНЫ
Зная эту зависимость, можно по данным измерения скорости иони-
зации рассчитать константу равновесия. Скорость ионизации RH можно
измерить по скорости бромирования (бромирование карбаниона R;
является электрофильной реакцией), по скорости изотопного обмена
D+
RH —> RD + Н+
или в некоторых случаях по скорости рацемизации R:, если анионный
заряд находится на асимметрическом углероде. На примере действия
алкоголят-аниона на стереоизомер
СН8
*1
СвН5-С-С-С2Н5
II I
о н
Инголд и Хыоз показали (1936—1938 гг.), что скорости всех трех реак-
ций — рацемизации, дейтерообмена и бромирования совпадают, так как
самой медленной стадией, определяющей итоговую скорость каждого
из процессов, является взаимодействие кетона с алкоголят-анионом (т. е.
вырывание протона из а-положения):
СНз
I
свн6—с—с—С2Н5
II I
о н
СНз
чИта- свн5-с-с-с2н6
о
СНз
с6н5—с==с—с2н5
0-
Этот процесс отличается от диссоциации обычных кислот только тем,
что он протекает очень медленно; ведь в диссоциации кислот тоже всегда
участвует основание в смысле Бренстедта — Льюиса, как, в частности,
напоминает такой тривиальный пример:
Н-С1 + Н20 —> Н30+ + Cl-
Кислоты, дающие при диссоциации карбанионы любого типа, обычно
относятся к числу псевдокислот (следуя Ганчу, так называют вещества,
анион которых структурно отличается от самой исходной псевдокислоты).
Примеры:
сн3—no2 -ь ОН- —> ch2=n< + Н20
ч0_
он-
СНз—С—СН2—С—ОС2Н5 —> СНз—с=сн—С—ОС2Н6 + Н20
О О
То обстоятельство, что по
мерном ионе
СН2=
СН2—N<^
\Q-
б
о- о
сути дела каждый раз речь вдет о мезо-
СНз—С=СН—С—OR > СНз—С—СНС-ОВ;
I II II И
б
а
КАРБАНИОНЫ
523
не меняет дела, поскольку форма а входит в описание мезомерного аниона
с таким большим весом, что формой б можно спокойно пренебречь.
Именно псевдокислоты тина —Н отличаются от обычных (кислород-
ных) кислот медленностью ионизации. В табл. 81 приведены скорости
прямой (к t) и обратной (/с2) реакции диссоциации
НА + Н2О А’ + Н3О+
kt
некоторых истинных и псевдокислот, а также их константы диссоциации
{приведенные в форме рХ), равные отношению /с1//с2:
pK=}gk2— lg fci
Интересно, что нитроэтан является кислотой, на много более сильной,
чем малоновый эфир, не потому, что он быстрее диссоциирует на ионы
(напротив, диссоциирует он медленнее), а потому, что для него очень
затруднена рекомбинация ионов (мала /с2, 1g /с2 = 1,2).
Таблица 81. Логарифмы констант диссоциации и скоростей ионизации
и рекомбинации ионов истинных кислот и псевдокислот
Формула РА’ bi Формула РК hi kt
Н2О 15,7 —4,6 11,1 СНзСОСНз 20 -9,3 10,7
Н3ВО3 9,1 1,0 10,1 СН2(СООС2Н5)2 13,3 —4,6 8,7
H2SO4 1,6 9,0 10,6 СН3СОСН2СОСН3 9,0 -1,8 7.2
СНзСООН 4,8 5,9 10,7 G2HgN О2 8,6 -7,4 1,2
СНзСОСООН 2,6 6,3 8,9 GH2(NO2)2 3,6 -0,1 3,5
Несмотря на то, что, как сказано, для большого числа кислот-^С—Н
•имеется приблизительная корреляция между Ка и скоростью отрыва
протона, число исключений столь велико, что это нельзя считать общим
правилом. Например, все нитросоединения совершенно не укладываются
в приводимую схему (В. М. Беликов, 1965 г.):
R2CH + ОН- Й2С + Н2О
no2 no2
Та же картина наблюдается и для обратной реакции протонирования
карбанионов. Отсутствие корреляции (рис. 115) связано с тем, что на
кислотность карбанионов и на скорость отрыва протона при их образова-
нии оказывают влияние как внутренние (строение), так и внешние (соль-
ватация) факторы. „ v _
Наиболее важным фактором строения является мезомерный эффект
заместителя, стабилизирующий карбанион. Чаще всего мезомерный
-и индуктивный эффекты заместителей действуют в одном направлении,
524
КАРБАНИОНЫ
хотя имеются исключения, например в случае фтора (И. Л. Кнунянц,
1964 г., Камлет, 1966 г.).
Внешние факторы (сольватация) играют очень большую и, возможно,
решающую роль. При переходе от воды к апротонным растворителям,
не способным стабилизовать карбанион за счет водородных связей, наблю-
даются следующие явления: константа диссоциации уменьшается на
10 порядков, а скорость диссоциации резко увеличивается, доходя до
величин, характерных для О—Н-кислот. По мнению Риччи (1964 г.),
последнее явление связано с тем, что при реакции в гидроксильных раство-
рителях перестраивается сольватная оболочка вокруг реагирующих моле-
кул, на что тратится основная часть энергии реакции. В апротонных рас-
творителях такой перестройки не требуется. Образование карбанионов,
ов
6,0-
щ-
2,0-
0-
—2,0 - 07
Off °9
~4,0~
2 OZ
e>°? г, 4
О ~е-°- 8 °?
°4 об о?J
j j -8,0j j 7T~^p/<c
a 6
Рис. 115. Константы скорости (в л • люль-1 • сек-1) отрыва протона поном ОН- (а) и
протонирования карбаниона водой (б) для ряда кислот RR'CHN02 и их р^»
(реакция RR'GH-f-OH" 7-^ RR'C"+H2O)
I I
no2 no2
1 ~ 2 —CjH.NO,; 3 — k-C3H,NO2; 4 — изо-С,Н,ХО,; 5 — н-С*Н,\'о.; 6— C.HSCH»NO«;
7 — C.HjOCOCHjNOj; 8 — CH,CH(NO,)l; 9 — CtHsCH(OH)CHtNO,.
в особенности из кислот типа RH, как уже было сказано, всегда требует
участия основания и протекает как медленная реакция
RH + В: R: + ВН
Раз это так, то очевидно, что любой (предшествующий или последу-
ющий) процесс, более быстрый, чем само образование карбаниона, уже
не поддается измерению в кинетических опытах и скорость всей совокуп-
ности реакций определяется этой медленно идущей стадией. Поскольку
берется огромный избыток В:, реакция идет по первому порядку в отно-
шении RH (псевдомономолекулярная), и любая реакция карбаниона
(бромирование, рацемизация, дейтероприсоединение и т. д.) также будет
реакцией первого порядка и будет обозначаться как 5£1 (т. е. замещение
электрофильное, I порядка).
КАРБАНИОНЫ
525-
Реакции SE1 противопоставляется реакция SE2, т. е. электрофильное
замещение, протекающее как реакция IT кинетического порядка, скорость
которой пропорциональна и концентрации RH, и концентрации действу-
ющего электрофильного реагента. Реакции SE2 протекают через переход-
ное состояние, в котором реагирующие молекулы связаны, например, так;
R\ /R
R'-C-Hg-C$-R'
R"/ \r-
/С1
+ Hg<
ХС1
“ RX Л "
{^/9—4s-c^R'
Hg—Cl R’
_ Cl
R\
--> 2R'^CHgCl
R"Z
В этих реакциях асимметрический углеродный атом — центр электро-
фильной атаки — сохраняет свою конфигурацию (О. А. Реутов).
Огромное влияние на скорость (и даже на тип) реакции оказывает
растворитель. О. А. Реутову и И. П, Белецкой удалось в реакции только
что приведенного типа, меняя растворитель (спирт на диметил сульфоксид),
заставить процесс проходить по типу вместо SE2, что явствует из
кинетики и из рацемизации, наступающей вследствие предварительной
ионизации ртутноорганического соединения.
Крам нашел, что отщепление протона от карбокислоты RH основа-
нием В: (например, алкоголят-анионом) в диметил сульфоксиде происходит
в 108раз быстрее, чем в метаноле. Это не может быть объяснено различием
диэлектрических проницаемостей двух растворителей, которое сравни-
тельно невелико. Крам объяснил это сольватацией основания спиртом
с образованием водородной связи В - • -Н—О—СН3, например:
СН3О- + СНзОН —> [СНз—О - Н-О-СНзГ
Сольватация резко ослабляет нуклеофильные свойства основания.
Особенно сильным основанием является тпреш-бутилат калия в диме-
тилсульфоксиде.
Выше был приведен пример рацемизации кетона с асимметрическим
а-углеродом при отщеплении основанием а-протона с превращением кетона
в соответствующий анион. Этот случай, однако, не может считаться типич-
ным, так как данный анион безусловно имеет структуру енолят-аниона,
и асимметрический тетраэдрический $р3-углерод становится углеродом
с олефиновой связью — плоской тригональной конфигурации.
Такая рацемизация неизбежна всегда, когда заряд аниона не только
рассредоточен, но, скорее, сосредоточен на примыкающей к реакцион-
ному центру группе с кислородом — акцептором электронного заряда
+ Z0" \
у:=о —N<C И т. д. .
V ХО /
По аналогии с отсутствием оптических изомеров у пирамидально-
построенных третичных аминов с тремя разными заместителями
R У zR"
N
R i, R"
R
526
КАРБАНИОНЫ
объясняемым быстрым вывертыванием пирамидальной конфигурации,
казалось бы, вполне вероятна невозможность существования по тем же
причинам оптической изомерии также и у истинных карбанионов:
R? R"
R С R"
R
На деле, однако, в реакциях карбанионов констатированы как случаи
частичного сохранения конфигурации, так и случаи частичного обращения
конфигурации (т. е. частичной рацемизации).
Крам исследовал скорость обмена D на Н (константа kJ и скорость
рацемизации — потери вращения плоскости поляризации (константа
Лр) при реакции стереоизомера 2-дейтеро-2-фенилбутана с о( нованием
в разных растворителях:
СН3 СН3
*1 *1
C2H5-C-D СгНь-С—Н
I I
свн5 С6Н5
Если дейтерообмен происходит с полным сохранением конфигурации,
должно соблюдаться отношение к0/кр — оо; при полной рацемизации
во время обмена kjko — 1; при полной инверсии конфигурации kjk^ ~
= 0,5.
Эксперимент дал следующие результаты:
Растворитель Основание Стереохимический результат
(СН3)3СОН (СН3)зСО-К+ 10 Сохранение конфи- гурации
(CH3)2SO+(CH3)3COH (СН3)зСО-К+ 1 Рацемизация
О(СН2СН2ОН)2 НОСН2СН2ОСН2СН2О- к+ 0,7 Обращение конфигу- рации
Ясно видно резкое влияние характера растворителя на стереохимиче-
ское течение процесса. Объяснение заключается в следующем. В трет-
бутиловом спирте как растворителе тпретп-бутилат калия хотя и ионизи-
рован, но не диссоциирует, т. е. образует неразошедшуюся ионную
пару, к тому же сольватированную растворителем. Эта сольватированная
ионная пара атакует стереоизомер 2-дейтеро-2-фенилбутана, отщепляя
дейтрон и поставляя на его место протон. Все это происходит с одной сто-
,роны карбаниона, и конфигурация при дейтерообмене сохраняется.
СНзч но—С(СН3)з
^2^ 5*^,0 D +
с6н5 к—бс(сн3)з
С2н5 СН3 о—С(СН3)3
14i +
С7 к
I "D. 1
сД '"-о-с(сн3)3
сн3
>С2Н5-~С~Н
с6н5
КАРБАНИОНЫ
527
В растворителе дпметилсульфокспде ионная пара трет-бутилата
калия полностью диссоциирована и атака совершается трсп-бутилат-
анионом, отрывающим дейтрон. Образовавшийся карбанион успевает
сделаться плоским и подвергается атаке введенного в растворитель н не-
большом количестве г??рс77г-бутилового спирта с обеих сторон плоскости.
Благодаря этому реакция дейтерообмепа проходит с рацемизацией:
сн< (CH3)2SO II 1_ II
С2Н5^С-Г) + б-С(СН3)з---------->-(СНз)зСОП +(CH3)2S—-с^—S(CH3)2
c6H5Z с2Н5 с6н5
(СНз)зСОН
ZH /СН3 СН3 Н
/С\ + ХС\
С2Н5 СбНз С2Н5 C6Hj.
В диэтиленглпколе ионная пара алкоголята хорошо диссоциирована
и атака на дейтерий, как и в предыдущем случае, совершается анионом
алкоголята. Но поскольку дпэтиленгликоль лучший донор протона, чем
тпре/тг-бутиловый спирт, синхронная атака «в спину» плоского карбаниона,
поставляющая протон, производится молекулой диэтилепгликоля. Резуль-
тат — обращение конфигурации (вальдеповское обращение):
ОСН2СН2ОСН2СН2ОН
НОСНоСНоОСНоСНоОН
сн3
> НОСН2СН2ОСН2СН2ОН—C"-D—-ОСН2СН2ОСН2СН2ОН —>
С2Н5 СсН5
Н СН3
\ /
--> НОСН2СН2ОСН2СН2О + ZC + DOCH2CH2OCH2CH2OH
С6Н5 С2Н5
Подобный же пример действия растворителя описан Крамом для рас-
щепления (реакции, аналогичной обратной альдольной конденсации)»
:528
КАРБАНИОНЫ
сн3 он
основанием спирта С2Н&—С-С—СН8 с образованием того же карбанио-
С6Н5СН3
«на (метилэтплфенил-аниона):
СН,С?к+ ?Нз ° «он |На
.^нДоД-сн,—► с,н5-с-к’ + С-СН,-----------►CjHs-C-H + ROK
CeHs сн3 Свн5 сн3 сен5
В этой реакции наблюдаются следующие стереохимические отношения:
растворитель
Основание
Стереохимический результат
(СНз)зСОН
(СНз)зСОН
(СНз)г8О+СНзОИ
>СН3ОН
(СН3)зСО-К+
(CH3)4NOH-
СН3О-К+
СН3О-К+
Сохранение конфигурации (90—99%)
Рацемизация (100%)
Рацемизация (100%)
Обращение конфигурации (40—60%)
В третп-бутиловом спирте, который не разделяет (не вызывает дис-
социации) ионных пар и сольватирует основание, по изложенным выше при-
чинам опять наблюдается сохранение конфигурации. Однако в той же
среде с гидроокисью тетраметиламмония — несольватируемым катио-
ном — происходит полная рацемизация. Такая же полная рацемизация
наблюдается в диметил сульфоксиде. Напротив, в метаноле — раствори-
теле с высокими диссоциирующими свойствами — атака совершается
метилат-анионом, а протон доставляется самим метиловым спиртом —
отличным донором протона. Результат — высокий процент вальденовского
обращения.
Все описанные выше реакции образования карбаниона как неустойчи-
вого промежуточного продукта реакции сводились к отрыву протона
(или, реже, катиона металла) от углеродного атома под действием основа-
ния или сольватирующего растворителя. Возможен и иной путь образо-
вания карбанионов — нуклеофильная атака по двойной углерод-углерод-
ной связи. Однако не всякая углерод-углеродная л-связь способна по-
двергаться нуклеофильным атакам. Для этого она должна быть или
сопряжена с электроноакцепторной группой, или углеродные атомы, не-
сущие л-электроны, должны быть непосредственно связаны с электроно-
акцепторными атомами или группами. Таким образом, нуклеофильным ата-
кам со стороны анионов НО-, RO-, R—С—О-, NH2 (и соответствующих
НА РГ> АНИОНЫ
529
производных), CN и ряда карбанионов подвержены системы следующих
типов;
I CF2 = CF2 (CN)2C = C(CN)2 CF3CH=CHt (CF3)2C = CH2 (CFj)2G = CF2
II CH2=CH—C—R СН2=СН—NO2 СН2=СН-C N СН2=СН—SO2R
О
HI R—С—CH=CH—C—R' R—C—CH=CH—NO2
n tl II
R—С—CH = CH—C N
R—C—CH = CH—SO.R
II
О
Реакция начинается с атаки анионом того атома углерода, партнер
которого, связанный с ним л-связью, способен лучше, чем атакуемый
С-атом, нести карбанионный заряд:
X—СН=СН—Y 4- Z: -> X—СН—СН—Y
I
Z
Полученный в результате атаки аниона карбанион должен быть, таким
образом, более устойчив (энергия его ниже), чем изомерный ему карб-
анион. Устойчивость карбаниона достигается большим рассредоточением
отрицательного заряда на прилегающие группы. Способность прилега-
ющих групп рассредоточить (т. е. частично принять на себя) заряд карб-
аниона уменьшается, по Арндту и Эйстерту, в следующем порядке:
—С=О > — С=О > —С==О > —C=eN-N02
III
Н R OR
В реакциях данного типа образование карбаниона — самая медленная
фаза реакции, определяющая ее кинетический ход. В общей форме, изо-
бражаемой приведенным выше уравнением, группа X лучше рассредото-
чивает электроны, чем группа Y (например, если Х=СН3—С—, а
I I
О
Y = CN). Если X и Y одинаковые группы, то атака идет с равной вероят-
ностью по тому или другому олефиновому углероду.
Затем наступает быстрая (на кинетике, следовательно, не отража-
ющаяся) фаза реакции — стабилизация аниона, осуществляющаяся одним
из двух путей. Анион может стабилизоваться посредством присоединения
протона из среды и завершения таким образом процесса, результатом
которого является присоединение молекулы АН по двойной связи (где
А- — начинающий реакцию анион). Часто встречающаяся альтернатив-
ная стабилизация осуществляется путем выброса из молекулы, связанной
с углеродом группы атомов в виде другого аниона и воссоздания этим
путем непредельной связи. Здесь итогом реакции является нуклеофиль-
ное замещение.
Примеры нуклеофильного присоединения:
СС13-СТТ=СН—NO2 -J- GII3O- —►
--► СС13—СП—СН—NO2 сн,0*--> cci3-ch-ch2-no2 -I- сн3о-
ОСН3 ОСНз
34 Заказ 672
530
КАРБАНИОНЫ
Н+
CCl3-CH=CH—NO2 Н- С!" > СС13—СП-СП—NO2 > cci3-cii-ch2-no2
Cl Cl
OR
I - ROH
CF3—C=CH— COOCH3 + RO’ > CF3—С—CH—C—OC1I3 >
I I II
СПз СНз О
OR
I
—> CF3—С—CII2—COCH3 + RO"
I II
CH3 О
SR
I RSH
C6II5C-CH=CH-COII + RS- -----> C6H5C-CH-CH-COH ----->
II II II II
0 0 0 0
SR
I
CeH5—С—CH2—CH—COH + RS-
Примеры нуклеофильного замещения, идущего через стадию нуклео-
фильной атаки по л-связям:
(^CN /CN
(NC)2C=C(CN)2 + СН3О" >(NC)26^C—CN ----->(NC)2C=C + CN
\>ch3 OCH3
RO2S. CN RO2S s'
+ CH3O"---> /С—c—so2r
NCX SO2R NC
ro2s. ,cn
/C=C. + rso2
NC OCH3
C6H5C— CH=CH— NOo + nh2c6h5—>C6H5C—CH — CHx +
II " II nh2c6h5
О о
>- с6н5с—ch=cii—NHC6H5 + H++ NOo
о
КАРВАНИОНЫ
531
CgHyC ОН — СН NOo 4~ Nз
О
о
> СсН5С-СН = СН — N3 + NO2
О
Особенно интересны с препаративной точки зрения реакции олефино-
вых связей, способных принимать нуклеофильные атаки, с карбанионами.
Сюда относятся конденсации Михаэля, например:
R—СН2—С—R' + В: >• R—СН—С—R' + B : Н
ГК-СИ—C-R' * R—СН=С— R'l
+ СН2=СН—X-->
_ Н:В
--> R —СП—СН2—СНХ > R—СН-СН2-СН2Х + В:
! I
O=CR' O = CR'
где X = —С—R; —NO2; —CN или другая группа, активирующая угле-
II
О
род-углеродную л-связь к нуклеофильным атакам, т. е. оттягивающая
электроны.
Частный случай реакций акрилонитрила с карбанионами, а также
с кислородными и Другими анионами называется реакцией цианэтилиро-
вания,. Реакция эта, открытая Брусоном, особенно широко изучена
А. П. Терентьевым и А. Н. Костом. Примеры цианэтилирования:
RO-;
CII2=CII—CN затем н--> rocit2ch2cn
C]I2=CH-CN rnhch2ch2cn
В: _
RCIR-CR'->• ГЙСН—CR' *---* RCH=CR'
II -II I
OOO"
ch2=chcn
_ В:н в:
--». R'C—CIT—C1I2CHCN > R'C— CHC1I2C112CN >
II I II I
OR OR
--> R'C—C—CH2CH2CN
CH.-CHCN , RrC_C/CH2CHCN B:H>
|| I x ch2ch2cn
0 R
ch2ch2cn
-—> R'C—C<
|| 1 XCH2CH2CN
О R
34*
532
КАРБАНИОНЫ
Реакция присоединения аниона к веществам типа RCO—СН=СН—X,
где Х = С1, CN, NO2 и т. д., часто сопровождается выбросом аниона X,
и, как сказано, превращается в нуклеофильное замещение. Это может,
очевидно, происходить в тех случаях, когда именно X является группой,
легко уходящей в виде аниона:
С6Н5С—СН=СН— X
II \
О А"
>с6н5с—сн—СН—X
О А
--->- С6Н5С—СН=СН—А + X
О
В этой реакции А- может быть анионом бензолсульфиновой кислоты,
которая образует сульфон *
О
C6H5-C-CH=CH-NO2 + CeH5-S<f -ТкгР
II ХО-
CeH5-C-CH=CH-S-C6H5
о
о о
или анионом азотистоводородной кислоты Ng. Реакции такого типа,
изученные подробно А. Н. Несмеяновым, Н. К. Кочетковым, М. И. Рыбин-
ской и Л. В. Рыбиным, называются кетовинилированием.
В иных случаях, когда гетеролиз связи С—X происходит более мед-
ленно (X = CN), можно выделить оба продукта (замещения и присое-
динения):
С6Н5-С—CH=CHCN .сн,°~
CeH5—С-СН—СН—CN
I
ОСНз
о
о
н+
С6Н5-с-сн=сносн3
о
о
Интересно,
двойственную
с анилином:
CcHs—С—СН2—CH-CN
ОСНз
что в случае p-нитровинилкетонов удалось обнаружить
ориентацию нуклеофильной атаки. Например, реакция
СвН5—С—СН=СН—NO2 + NH2CeH5 —> с6н5—с—сн—сн2—no2 +
II II I
о о nhc6h5
+ CeH5-C-CH=CHNHC6H5
II
_______________ о
* Анион сульфиновой кислоты, как и нитрит-анион, часто реагирует с переносом
реакционного центра с О на S (или, соответственно, на N):
R —> R —или О—N = O -< > O=N=O
хо~ ЧО
КАРБАНИОН Ы
533
Наконец, карбанионы (в большинстве случаев генерированные дей-
ствием оснований) реагируют с положительным углеродом карбонильной
группы альдегидов и кетонов. Это — хорошо известные реакции присо-
единения по карбонильной двойной связи аниона альдегида или кетона
(альдольная конденсация)
Г- z° /0-1 /О- /О
СН3—СХ -Г СН-2—С<^ > СН2=С/ ------> СН3-С-СН2—с<^
Чн _ \Ц \Н Хн н
или аниона уксусного ангидрида (конденсация Перкина):
/О
СвНб-СХ +
н
сн2—с
СНз-С
0- 0 0 0 0
/ II II II II
--> СвН5—С—СН2—С—О—С—СН3 > C6H5—С=СН—С—О'+СН3С—он
н н
В случае конденсации Перкина можно предположить в качестве первой
стадии реакции образование малого количества аниона уксусного ангид-
рида в результате обменной реакции с ацетатом натрия:
О
I
(СН3-С)20 + CTI3-C-O- Na+
II
О
сн2—с
СНз—с
О—
СН2=С
СНз— С/
\о
Na+ + CII3COOH
Во второй стадии происходит атака мезомерного аниона уксусного
ангидрида на карбонильный углерод и отщепление уксусной кислоты,
возможно, через циклическое переходное состояние:
О
-->С0Н5-СН=СН-С-О" + сн3-с-он
о
534
карбанионы
Нитрометан при действии оснований присоединяется к альдегидам;
R—С—II л- С1Г2
О
+ /О- + 0-
N' «—> CH2=N-
X) о ~j
Н
О"
Вообще подобные реакции карбанионов играют в синтетической химии
очень важную роль. К ним относятся такие хорошо известные читателю
реакции, как присоединение к оксосоединениям синильной кислоты,
аниона ацетилена НС:=С~ (А. Е. Фаворский, Реппе).
Уже упоминавшийся тетрацианэтплен способен полностью оторвать
электрон, например, от металлического калия или от иона иода, образуя
анион-радикал тетрацианэтилена:
(NC)2C=C(CN)2 + К---->К+ + (NC)2C=C(CN)2
2(NC)2C=C(CN)2 + 3KI------>21\++ 2 F(NC)2C=C(CN)2| + K+I3
Калиевая соль тетрацианэтилен-аниона — кристаллы бронзового цвета.
Сигнал ЭПР * этого аниона расщеплен на девять линий.
Тетрацианэтплен способен реагировать и с ароматическими соедине-
ниями, но с частичным переносом электрона от ароматического ядра
к тетрациапэтилену, так что обе молекулы связываются в окрашенные
парамагнитный комплекс. С дуролом (1,2,4,5-тетраметилбензолом) и с кон-
денсированными ароматическими углеводородами такие комплексы удается
получить в твердом индивидуальном состоянии, а не только в растворах.
Подобные же комплексы с частичным переносом электрона образуют
с ароматическими соединениями хиноны (см. хингидрон, стр. 163).
Тетрацианэтплен и хиноны относятся к числу веществ, обладающих
большим сродством к электрону и поэтому легко образующих анион-
радикалы или комплексы, получившие название комплексов с переносом
заряда (КПЗ). Однако в определенных условиях анион-радикалы обра-
зуются и из ароматических углеводородов. Так, бензол при более низкой
температуре восстанавливается металлическим калием в растворе дп-
метоксиэтапа с образованием анпон-радпкала. Диметокспэтан при этом
играет важную роль, сольватируя ион калия:
СН3
,.(У
/ \сн2
Celle + К. +СНз-О-СН2-СН2-О-СНз -> [С611в«Г4- К‘. '
0\сНз
* О спектрах ЭНР см. раздел «Свободные радикалы», стр. 538.
КАРБАНИОНЫ
535
В анион-радикале бензола лишний электрон принимается на первую
возбужденную орбиталь л*, охватывающую всю систему, так что плот-
ность его равномерно распределена по всем шести атомам углерода.
В соответствии с этим в спектре ЭПР наблюдается семь линий сверхтон-
кой структуры с отношением интенсивностей 1 : 6 : 15 : 20 : 15 : 6 : 1.
В аналогично образованном анион-радикале толуола плотность лишнего
электрона распределена иначе — на орто- и мета-углеродах кольца, что
приводит к пяти основным линиям спектра (В. В. Воеводский, С. П. Соло-
довников).
Нужно отметить, что к типу анион-радикалов относятся и металлке-
тилы, получаемые восстановлением калием или натрием ароматических
кетонов
СвН5-С-С6Н5 + К-
II
О
Г с6н5
С-С61Т51
I
6-
К +
и так называемые фосфилы — продукты восстановления окисей третичных
фосфинов, например
[(С6Н5)3Р-О-]К+
(М. И. Кабачник, В. В. Воеводский с сотр.).
Анион-радикалы могут быть получены и при восстановлении другими
методами, например электрохимически. В последнее время выяснилось,
что они играют важную роль во многих реакциях, начинающихся с пере-
носа электрона, например, в реакциях металлирования и особенно в био-
химических процессах восстановления (Л. А. Блюменфельд).
Анион-радикалы и комплексы с переносом заряда обычно глубоко
окрашены, так как неспаренный электрон в них занимает первую возбу-
жденную орбиталь, с которой возможны переходы на близлежащую следу-
ющую, что соответствует поглощению кванта видимого света.
В последнее время найдено, что некоторые непредельные сопряженные
системы способны присоединять два электрона с образованием двузаряд-
ного аниона — дианиона. Таков, например, изученный Д. Н. Кургано-
вым и др. дианион циклооктатетраена (стр. 516):
В таком диапиоие образуется ароматический децет электронов (4n Т 2),
и поэтому он устойчив.
Существуют и дианион-радикалы — частицы, образующиеся из арома-
тических карбанионов при присоединении к ним еще одного электрона.
Большую группу карбанионов составляют илиды, из которых
наибольшее значение для синтеза имеют фосфорилиды (реакция Виттига,
кн. I, стр. 264). Эти карбанионы представляют собой биполярные ионы,
536 ___________КАРБАНИО! 11,[
в которых положительный заряд сосредоточен па опиевом атоме, а отри-
цательный, в большей или меньшей мере, на углероде, например:
+ _ / В /Н
(СвП5)зР-С< ' (СяП5)зР = С/
R’ Н
Как и в других случаях, стабильность этих биполярных карбанионов
зависит от степени делокализации анионного заряда. Таким образом,
в ряду фосфорилидов (С„Н6)3Р—CHR стабильность повышается в такой
поел едов ател ьн ости:
/О .О
R = H<Celf6<Ce[I4NO3 <С^ <-С^
ХОВ \R
Фосфорилиды, несущие карбонильную группу, уже настолько ста-
бильны, что соответствующие им ацилфосфонии
+ /О “>’0
(С6Н5)зР-С112-С<;' Н+ + [(С6Н5)зР=СН-c<f < ►
+ _ /О + /0~ “I
<--> (С61[5)зР-С11-С^ <---> (С6Н5)зР-СН=С<
XR \R
являются кислотами, приближающимися по силе к карбоновым кислотам.
По данным М. И. Кабачника и Т. А. Мастрюковой, соли трифенилфен-
ацилфосфония [(CJI5)3PCH2COCeHjX~ являются кислотами с констан-
той диссоциации рАп — 6,5 в 50%-пом спирте *. При этом они нисколько
не енолизировапы. Ёнолизированпый примерно на 30% диацилфосфоний
г- О
I \С-СН3
II II
L 0 □
является уже сильной кислотой (р2£а — 2,59), гораздо более сильной, чем
соответствующий кетоенол бензоилацетона (рАа в воде равно 8,72).
* Для бензойной кислоты в 50%-ном спирте рАГа = 5,63.
СВОБОДНЫЕ РАДИКАЛЫ
В разных местах курса упоминались реакции, протекающие через стадию
образования свободных радикалов. Сюда относятся рассмотренные уже
реакции хлорирования и бромирования на свету (кн. I, стр. 72), сульфо-
хлорирования на свету (кн. I, стр. 73), окисления молекулярным кисло-
родом (кн. I, стр. 272 и кн. II, стр. 141), термического крекинга угле-
водородов (кн. I, стр. 71), распада некоторых азо- и диазосоединений
(стр. 108), радикальной полимеризации (кн. I, стр. 274) и теломери-
зации (кн. II, стр. 560).
Большая часть этих реакций протекает по цепному механизму
(кн. I, стр. 72).
Свободные радикалы, о которых идет речь, могут быть: а) неустойчи-
выми (термодинамически) частицами — осколками молекул с неспарен-
ным электроном, подобными СН3* и С6Н5* и их гомологам; б) атомами,
имеющими неспаренный электрон, например Н*, Cl*, Na*; в) наконец,
существуют термодинамически устойчивые радикалы, подобные триарил-
метилам (стр. 224) и семихинонам, в которых устойчивость достигается
рассредоточением неспаренного электрона по л-сопряженным (в данном
случае ароматическим) структурам. Устойчивым свободнорадикальным
частицам подобны также окислы азота (NO2, NO), так как они имеют
неспаренный электрон.
Нечетный электрон атома водорода — это s-электрон; в случае атома
хлора, а также любого С-радикала это р-электроп.
Устойчивый свободный радикал обычно не вступает в обменные
реакции с молекулой, а только соединяется с радикалом. Поэтому такие
радикалы не могут продолжать цепь, но могут ее обрывать (так как они
уводят свободные радикалы — продолжатели цепи) и служить, таким
образом, ингибиторами цепных реакций. Некоторые соединения переход-
ных металлов содержат нечетные электроны на d-уровнях. Это вполне
стабильные соединения, не обладающие реакционной способностью свобод-
ных радикалов, но иногда способные, оторвав электрон, породить свобод-
ный радикал и инициировать цепную реакцию.
Все эти частицы и молекулы с неспаренным электроном благодаря еп>
нескомпенсированному спину парамагнитны, что и можно установить.
538 СВОБОДНЫЕ РАДИКАЛЫ
при помощи магнитных весов (если речь идет об устойчивых частицах)
или путем измерения электронного парамагнитного резонанса (также
и для неустойчивых частиц в реакциях, если концентрация свободных
радикалов не слишком мала).
ЭЛЕКТРОННЫЙ ПАРАМАГНИТНЫЙ РЕЗОНАНС
Открытый Е. К. Завойским в Казани в 1945 г. метод электронного пара-
магнитного резонанса стал за короткое время мощным методом идентифи-
кации свободных радикалов и наблюдения за поведением нечетного элек-
трона в молекулах. В основе метода лежит следующее. В наложенном
постоянном магнитном поле Я неспаренный электрон должен ориентиро-
ваться в направлении поля или против него, причем обе ориентировки
разнятся энергией электрона. Эта разность энергии Е выражается фор-
мулой:
E = g$H
где g — постоянная величина, равная для свободного электрона 2,0023;
р — магнетон Бора, равный 0,927 • 10~ 20 эрг/гс.
Такое расщепление уровней энергии электрона в магнитном поле
делает возможным поглощение энергии Е электроном в нижнем энергети-
ческом состоянии с переводом его в верхнее состояние.
S Это совершается при облучении свободного радикала,
II находящегося в постоянном магнитном поле Я, коротко-
_______II,—— волновым излучением hv = Е = g$H. Именно при длине
I волны этого излучения, определяемой данным соотно-
I шением, наступает резонансное поглощение.
' Метод весьма чувствителен и позволяет обнару-
Рис. 116. Спектр жить концентрации свободного радикала до 10“12 молъ(л.
ЭПР свободного Кривая поглощения имеет форму с островыраженным
радикала. пиком (рис. 116). Однако такой простой вид кривой
получается только при отсутствии в радикале атомных
ядер, имеющих спин (и следовательно, магнитный момент) и способных
взаимодействовать с неспаренным электроном. Поскольку протон имеет
спин, то наиболее обычный случай — наличие такого взаимодействия, так
называемое спин-спиновое расщепление. При этом единственный пик превра-
щается в (п 1) пиков, где п — число одинаковых по положению водо-
родных атомов, взаимодействующих с неспаренным электроном. Напри-
мер, для метильного радикала п = 3, и на кривой поглощения появляются
четыре пика; для гидроксильного радикала НО-, очевидно, появляется
два пика; для иона семихинона
• О—о-
в котором электрон рассредоточен по обоим кислородным атомам и все
четыре водорода симметричны, т. е. п = 4, в спектре ЭПР имеется пять
пиков. Картина гораздо сложнее, если имеются «неодинаковые» водороды-
Однако как раз спин-спиновое расщепление позволяет хорошо идентифи-
цировать свободные радикалы по индивидуальной картине ЭПР, характер-
ной для каждого.
ОБРАЗОВАНИЕ СВОБОДНЫХ РАДИКАЛОВ
539
Другой, более старый классический способ обнаружения неспаренных
электронов и, в частности, свободных радикалов — спектроскопия.
Неспаренный электрон обнаруживает себя в триплетности соответству-
ющей линии спектра, так что «триплетное» и «свободнорадикальное»
состояние частицы стали синонимами. Используется также масс-спектро-
метрия. Поскольку свободнорадикальные реакции чаще всего протекают
по типу цепных реакций, используется измерение характерной кинетики
цепных реакций (см. стр. 562) с их S-образным развитием, включающим
начальный период индукции. О наличии свободных радикалов судят
также по признаку ингибирования цепных реакций добавкой «уловителей
радикалов» — ингибиторов.
Как мы видели на примере иона семихинона, из ЭПР-измерений совер-
шенно однозначно следует вывод об объективной реальности явления
делокализации (рассредоточения) свободного электрона по сложным
сопряженным системам. Аналогично спектр ЭПР трифенилметила одно-
значно указывает на то, что неспаренный электрон этого радикала взаимо-
действует со всеми 15 атомами водорода трех фенильных ядер.
ОБРАЗОВАНИЕ СВОБОДНЫХ РАДИКАЛОВ
Все свободнорадикальные реакции начинаются с акта образования свобод-
ного радикала, поэтому следует вкратце рассмотреть способы генерации
свободных радикалов.
Назовем четыре типа реакций образования свободных радикалов,
протекающих преимущественно в газообразной среде или в растворителе
с низкой диэлектрической проницаемостью и низкой сольватирующей
способностью. В растворителях же с высокой диэлектрической проница-
емостью и высокой сольватирующей способностью преобладают гетеролити-
ческие и ионные реакции.
Тип I. Гомолиз связей, например С—С, О—О, С—N=N, С—М
(М — металл), осуществляемый:
а) пиролизом — иногда при низких, например комнатных, температу-
рах, как для (С6Н5)3С—С(С6Н5)3, или при температурах < 100° С, как
для перекисей или азодинитрила изомасляной кислоты;
б) облучением (ультрафиолетовыми квантами или жестким излуче-
нием).
Энергии разрыва связей разных типов см. кп. I, стр. 344.
Тип II. Реакции, протекающие по принципу «радикал порождает
радикал», например:
RC14-Na« —> R«4-NaCl
СНз«+ СН3СН2СП3 CII4-рСНз—CIICIJ3
СГЩ-С!* ----------► СН3«4-НС1
R. 4-СН2=СН2—> rch2— СН2.
11 т. д.
Тип III. Реакции с участием редокс-систем, в которых нечет-
ность электрона достигается в результате передачи одного электрона
540 СВОБОДНЫЕ радикалы
молекулой с парными электронами окислителю (обычно катиону метал-
ла переменной валентности) или иногда обратной реакцией:
ROOH + C°3+ --* ВОО.-|-Со2+-|-Н+
ROOH + Co2+ --> ВО- + ОН_ + СоЗ*
R001I + Fe2+ -> R0* + 0H" + Fe3*
Тип IV. Электродные процессы, например типа реакции Кольбе
R —С—О'—е > R —С—О- > R.+СО2
II JI
о о
или электролиз гриньярова реактива:
RMgX-i-<? —> R* -j-Mg-J-X"
Реакции типа I. 1. Первой реакцией этого типа, в которой
было доказано образование свободных метильных и этильных радикалов,
был пиролиз паров тетраметил- и тетраэтилсвинца, осуществленный
в 1929 г. Панетом:
(С2Н5)4РЬ —> 4С2Н5- + РЬ
Существование метильных радикалов в быстром токе разреженного
водорода (2 мм рт. ст.) было доказано превращением металлов Hg, Pb,
Sn в их метильные производные. Можно было, зная скорость тока газа,
несущего радикалы, и расстояние, на котором прекращалось взаимодей-
ствие с металлом, вычислить время существования радикалов, оказав-
шееся порядка 0,01 сек. Постепенно они рекомбинировались
2СН3. —> СНз—СНз
и диспропорционировались:
2С2Н5« --> С2Н4 + С2Нв
Другие металлоорганические алкильные и арильные соединения ведут
себя так же.
Подобный гомолиз металлоорганических соединений можно осуще-
ствить и фотохимически, как показал Г. А. Разуваев, который явился
одним из первых исследователей свободнорадикальных органических
реакций в растворах еще в 30-х годах XX века.
2. Пиролиз алифатических углеводородов в жидкой и газообразной
фазах (крекинг) при температуре —500—700° С приводит к разрыву
С—С-связей, которые менее прочны по отношению к гомолизу
(84 ккал/молъ), чем С—Н-связи (100 ккал/молъ). Стоит оторваться метиль-
ному или этильному свободным радикалам, как они начинают цепную
реакцию: вырывают атом водорода из новых молекул алканов — предпо-
чтительно третичный водород, затем вторичный и лишь в последнюю оче-
редь первичный. Энергия активации отрыва свободным радикалом первич-
ного водорода на 2 ккал больше, чем вторичного, и на 4 ккал больше,
чем третичного, при одинаковом предэкспоненциальном факторе, что
соответствует отношению скорости реакции при 500° С
пере : втор : трет — 1 : 3,7 : 13,4
ОБРАЗОВАНИЕ СВОБОДНЫХ РАДИКАЛОВ
541
Образовавшийся новый алкильный радикал быстро распадается по
схеме, подобной следующей (мономолекулярная реакция):
СНз’+Сп112л+2 -> сщ+.слн2л+1
СПз—сн2 —СН2—СН2—СН—СНз > СНз—СН2—С11 = СН2-|-• СН2СН3
Не распадаются в условиях крекинга только метил и этил. Таким
образом, и тот, более длинный, чем этил, радикал, который остается после
отщепления метила или этила в первичном акте крекинга, тоже распа-
дается, и реакция продолжается. Кроме того, возможна изомеризация
радикала. Если радикал имеет цепь из шести и более углеродных атомов,
он может изомеризоваться, отрывая водородный атом от одного из соб-
ственных углеродов (обычно из пятого положения), благодаря чему
радикальный центр перемещается в энергетически наиболее выгодное
положение, после чего радикал распадается гомолитически. Эти отноше-
ния выяснены Райсом, построившим на их основе количественный расчет
выхода продуктов крекинга.
3. Пиролиз перекисей проходит при невысоких температурах (около
100° С). В результате пиролиза могут образоваться как жирные, так
и ароматические радикалы R«:
2R—С—О • --> R- +СО2
II
a) R—С—О—О-С—R — О
о о R- + R— С-О- +СО2
О
б) (СНз)зС-ООН --> (СНз)зСО. + -ОП
(СН3)зСО. + (СНз)зСООН > (СН3)зСОН + (СН3)3С-ОО•
2(СН3)зС-ОО- > 2(СНз)зСО.-|-О2
в) R0—С—О—О—С—OR > 2R—О—С—О- > 2RO-+CO2
II II II
0 0 О
пероксидикарбонат
4. Пиролиз нестойких азосоединепий, например азодинитрила изо-
масляной кислоты:
НзС СНз
N-C—CN СН3\ .
|| --->N2+2 ХС—CN
N-C-CN СНз '
НзС СНз
5. Распад ароматических диазосоединений в неполярных средах:
Ar—N—N—С1 —► Аг •-|-N2-| - CI •
6. Молекула дисульфидов H-S-S-R более пли менее легко
диссоциирует на два радикала RS-. Легкость диссоциации зависит от
542
СВОБОДНЫЕ РАДИКАЛЫ
способности R рассредоточить неспаренный электрон, так что наиболее бла-
гоприятны ароматические и гетероциклические остатки с системой гс-свя-
зей, доходящей до углерода, несущего S-. Таким соединением является
2,2-бензтпазолилдисульфид, уже при 100° С диссоциирующий по приведен-
ному ниже уравнению с константой равновесия 6-10 5:
г N
ГУ Vs-
Kv
N
С—S.
's^
2
Все реакции типа I могут быть использованы для инициирования
разнообразных цепных реакций, причем практически особенно применимы
реакции 3 и 4.
Реакции типа II. Образование свободных радикалов из других
радикалов нам уже встречалось. Такие реакции сопровождают, как вто-
ричные реакции дальнейшего превращения, любые, особенно высокотем-
пературные реакции первичного образования свободных радикалов.
Множество дальнейших примеров этого рода мы встретим при рассмотре-
нии реакций распада и присоединения свободных радикалов.
Многие молекулы, способные к термическому гомолизу, оказываются
способными к фотогомолизу (фотодиссоциация), но область поглощения
света у разных соединений различна. Так, перекиси поглощают свет
с длиной волны лишь ниже 3200 А и поэтому непригодны для фотолиза
в стекле. Азодинитрил изомасляной кислоты поглощает в области 3450—
4000 А и удобен для получения свободных радикалов фотолизом. Диметил-
ртуть и тетраэтилсвинец фотодиссоциируют под действием света с длиной
волны 2800 А. Легко подвергается гомолизу под действием света связь
С—Hal галоидных алкилов. Для гомолиза иодистых алкилов требуется
свет с длиной волны 3130 А, для бромидов и хлоридов — более коротко-
волновый.
Особенно хорошо изучена фотодиссоциация ацетона и других кетонов
(Нойес, Норриш), протекающая по типу
СНз—С-СН3 —>
II
О
СНз-+ СНз-С-
II
О
Реакции типа III. Что касается генерации радикалов в редокс-
системах, то примеры с участием ионов' Мп, Со и Fe разной степени
окисления уже были приведены. В таких системах ион металла, забирая
или отдавая электрон, превращает молекулу с четным числом электронов
в свободный радикал с неспаренным электроном или в сумму свободного
радикала с неспаренным электроном и иона (или молекулы) с четным
числом электронов. Однако существуют редокс-системы, выполняющие
ту же роль и без участия иона металла, например системы перекись —
амин:
СН3
ROOH + \n_YQ\ ----------->. RO* + ОН + *N—(О/
сн3
РЕАКЦИИ СВОБОДНЫХ РАДИКАЛОВ
543
Всякого рода редокс-системы применяются на практике как инициа-
торы цепных реакций, особенно для полимеризации в водной среде (поли-
меризация в эмульсии).
РЕАКЦИИ СВОБОДНЫХ РАДИКАЛОВ
Элементарные акты
I. Присоединяя электрон, свободный радикал превращается в анион. Сродство к электрону выражается следующими величинами (в газовой фазе): Сродство Сродство Радикал к электро- радикал к электро- ккал/молъ ккал/молъ
С1- .... 88,2 С6Н5О. . . . 27
F- . . . . 83,5 (СбН5)зС. . . 48
Вг- .... 81,6 СНз- • • . . 25
I. 74,6
НОО- . . . 70
НО- . . . 50
II. Отдавая электрон, свободный радикал образует катион. Уотерс
первым сделал вывод о том, что органические свободные радикалы и соот-
ветствующие им анионы и катионы составляют пары термодинамически
обратимых окислительно-восстановительных систем:
R. R* + e
e + R. R:“
Рочи и Мог привели доказательства того, что 2-циан-2-пропилрадикалы
СН3—С—СН3 восстанавливают соли двухвалентной меди в среде ацето-
CN
нитрила по механизму одноэлектронного переноса:
СЦ3—С—СПз + Си2+ ► СПз—С—СП3 + Си*
I I
CN CN
СПз-С-СНз
О
—+ СН3-С-СН3+Н* + П CN
+ С1Ь=С— СНз4-Н+
I
CN
Радикал, который при потере электрона дает сравнительно стабильный
катион, называют электроиодонорным. К таким радикалам относятся:
алкилы, а-арилалкилы, а-гидроксиалкилы, ацилы и а-аминоалкилы.
544
СВОБОДНЫЕ РАДИКАЛЫ
Радикал, который при приобретении электрона дает сравнительно
стабильный анион, называют электроноакцепторным. К таким радикалам
относятся: полигалоидалкилы, а-цианалкил, а-карбонильный и а-алкокси-
радикалы. Донорно-акцепторные свойства таких радикалов и соответ-
ствующих соединений, способных образовывать указанные радикалы,
проявляются в переходном состоянии реакции.
III. Реакция соединения двух свободных радикалов — одинаковых
или разных, называемая рекомбинацией, пе требует (или требует малой)
энергии активации и является обычной реакцией обрыва цепи в цепных
реакциях.
Примеры:
Cl- +С1. -> С12
R- + CI- —> RC1
СаНо* + Н- ----► С2Нв
обрывы цепи при хлори-
ровании
обрывы цепи при крекинге
СН3'4-С1-13. —> С2Нв
RO.-Ь -OR --> ROOR
О О .00
II II II 'I
R—C- + R-С—О- -> R—С—О—С—R
обрывы цепи в реакциях
окисления
IV. Распад радикалов, а) Алкильные радикалы старше С2Н5- неустой-
чивы в условиях пиролиза (т. е. при температурах около 500° С) и распа-
даются по типу:
RCnH2n----> R. + C„H2n
Судя по осуществляемым реакциям фотохимически инициируемого
гомолитического замещения водорода в алканах на хлор, —SO2C1 и т. д.
(стр. 546 сл.), радикалы •С„Н2л+1 с длинной цепью углеродных атомов при
невысоких температурах фигурируют как промежуточные продукты
реакции, не распадаясь.
б) Алкоксильные свободные радикалы способны распадаться с обра-
зованием оксосоединений и алкильных свободных радикалов:
(СНз)зС-О. —> (СН3)2С=О + СНЭ.
в) Ацильные радикалы способны распадаться (при более высокой
температуре) на алкильные свободные радикалы и СО:
R—С. —> R.-J-CO
г) Ацилатные радикалы очень неустойчивы; они декарбоксилируются,
образуя алкильные (или арильные) свободные радикалы:
О
R—С-О.
В.-ЬСОг
РЕАКЦИИ СВОБОДНЫХ РАДИКАЛОВ
д) Перекисные
распадаться так
хотя не исключен
радикалы типа II — О—0-, по-видимому, способны
2R—О—О. > 2R--0«4 02
и механизм
2R—0—0. —> R—0-0-R |-02
Если перекисный радикал имеет а-водород, то возможен другой про-
цесс распада:
2R2CII-О-О- —> R2C=0 + R2Cn0H4-02
V. Реакции внутримолекулярной перегруппировки свободных радика-
лов см. в разделе «Перегруппировки» (стр. 609).
VI. Диспропорционирование, т. е. превращение двух алкильных ра-
дикалов (не ниже этильных) в смесь алкана и алкена. Реакция эта, как
показывает опыт с меченым этилом CH3CD2., полученным фотолизом
CH8CD2GOCD2CH3, идет по механизму «голова к хвосту»:
CII3—CD2.~r II —С112—CD2.
CH3-CD2H + Cll2=CD2
Присутствие неспаренного электрона в радикале ослабляет окружа-
ющие свяли. Так, для -СН2СН2—Н энергия связи 40 ккал!молъ. в то
время как для СН3СН2—Н она составляет 98 ккал)моль.
VII. Реакция с кислородом является обычным звеном цепной реак-
ции окисления кислородом углеводородов, альдегидов, простых эфиров
и т. д.:
Радикал с нечетным электроном на углеродном атоме превращается
в радикал с нечетным электроном на кислороде.
VIII. Реакции с водород- или галоид содержащими молекулами —
важный частный случай реакций, текущих по принципу порождения
радикала радикалом. В этих реакциях свободный радикал вырывает
из молекул окружающей среды водородный или галоидный атом, образуя
новый свободный радикал. Такие реакции часто бывают также звеном
в цепной реакции, например в реакции окисления кислородом. Возможна
и изомеризация радикалов (стр. 636).
Примеры:
R„c_-o-o. I R-c-n —► R-c-o-o-H ; в—с-
и II if А
ооо о
О
ОН
о
R—с—0—0. ь СПз—СП - СНз — > R—С—О—О—H + CHs—с—СНз
ОН
о
СвП5- + С2Нг,ОН —► Се Не 4
стен»
I
он
35 Заказ 072
546
СВОБОДНЫЕ РАДИКАЛЫ
СвН5- + СН3-С-СН3 --> СвПв+СП3—С— СН2.
II II
О о
ССЛч- ЬСН3-СН-СН3 —> С11С1з + СНз-С-СНз
I I
он он
Свн5.+СС14 —> СвП5С1+СС13.
ci. 4-RH —> HC1 + R.
R.+SO2CI2---> RCI4-.SO2CI
Водородные атомы вырываются радикалами из алифатических соеди-
нений, но не из ядра ароматических. О поведении алифатических непре-
дельных соединений см. кн. I, стр. 266 сл., ароматических — кн. II,
стр. 550. Галоид вырывается углеводородными радикалами из полига-
лоидных алифатических соединений (в случае хлороформа предпочти-
тельно вырывается атом водорода).
Естественно, что радикалы с нечетным электроном на кислороде
вырывают водород, но не галоид.
IX. Реакции присоединения свободных радикалов к олефинам
R. + CH2=CH2 —> R—СН2—СН2«
порождают гомологичные свободные радикалы и фигурируют как первая
фаза реакций гомолитического присоединения, теломеризации и ради-
кальной полимеризации.
СВОБОДНОРАДИКАЛЬНЫЕ ЦЕПНЫЕ РЕАКЦИИ
Гомолитическое замещение
Галоидирование. 1. Гомолитическое галоидирование (фториро-
вание, хлорирование и бромирование) может совершаться элементарным
галоидом на свету по цепной реакции. Для примера в табл. 82 приведены
теплоты отдельных фаз цепной реакции фотохимического галоидирования
метана (Х2 — молекула галоида). Здесь —ДЯ, как всегда, энергия,
выделяемая при экзотермической реакции в газовой фазе.
Таблица 82. Теплоты отдельных фаз реакции галоидирования метана
(— \Н, ккал/моль)
F С1 Вг I
, hi а) Х2—>2Х. +37 +58 +46 +36
6) Х.+СН4->СНз-+НХ —33 —1 +15 +31
«) R. -|_Х2-> RX+X. —60 —23 —21 —17
Сумма б-\~ в -93 —24 —6 + 14
СВОБОДНОРАДИКАЛЬНЫЕ ЦЕПНЫЕ РЕАКЦИИ
Как видно, только атомы фтора и хлора вырывают из метана водород
экзотермично. Для брома, хотя процесс в итоге слабо экзотермичен,
но стадия превращения метана в метильный радикал эндотермична,
и реакция не идет. Лучше обстоит дело с фотохимическим бромированием
алканов — гомологов метана — и циклоалканов, однако реакционные
цепи и здесь очень коротки и квантовые выходы низки. Например, для
бромирования циклогексана квантовый выход при комнатной температуре
составляет '"'2, в то время как при хлорировании а при фторирова-
нии еще больше. Фотохимическое бромирование толуола в цепь из-за
большей стабильности бензильного радикала (и более низкой его энер-
гии — следствие рассредоточения электрона по сопряженной л-электрон-
ной системе) экзотермично уже на стадии отрыва водородного атома,
и реакция идет. Как показали Хараш и Броун, свободнорадикальное
хлорирование оптически активного 1-хлор-2-метилбутана
СНз СНз СН3
СНз-СН2-С-СН2С1 — -> СНз-СН2-С-СН2С1 -^СНз-СН2-С-СРЬС1 + СЬ
I • I
И С1
приводит к рацемическому 1,2-дихлор-2-метилбутану. Это общее правило —
свободные радикалы с радикальным центром на асимметрическом углероде
рацемизуются. Возможно, это происходит вследствие плоской структуры
свободного радикала, или же, будучи пирамидальным, он рацемизуется
в силу того же туннелъ-эффекта, в силу которого не существует оптиче-
ских антиподов у пирамидально построенных третичных аминов типа
Относительная реакционная способность различных водородных ато-
мов в реакции радикального хлорирования разных углеводородов может
быть проиллюстрирована следующим сопоставлением (100° С, газовая
фаза, равное время реакции):
СНз 1 СН2СНз 1 СII г—СН-С Из СН2С(СН3)з I СН2-С(СН3)2 1 1
1 н 1 Н । Н 1 и И 1 1 н н
100 ill 108 462 88 85 590
За сто здесь принята реакционная способность связи С—Н в ме-
тане. Цифры выражают выход данного хлорпроизводного, деленный
на число водородных атомов того же сорта, что и замещаемый (поправка
на статистический фактор). В соответствии с большей устойчивостью третич-
ных и вторичных свободных радикалов сравнительно с первичными заме-
щение у третичного углерода проходит скорее, чем у вторичного, и осо-
бенно, чем у первичного. Наиболее инертен метан.
При фотохимическом хлорировании в растворителях, образующих
л-комплекс с С1 •, разница в скоростях хлорирования в первичное,
35*
548
СВОБОДНЫЕ РАДИКАЛЫ
вторичное и третичное положения может быть резко увеличена. Так, в н-
пентане, применяемом в качестве растворителя, относительная реакционная
способность первичного и вторичного водорода по отношению к хлору
при 25° С составляет 1 : 3, в бензоле 1 : 5, а в сероуглероде 1 : 29.
Электронооттягивающие группы замедляют гомолитическое хлориро-
вание. Например, относительная реакционная способность а-, |3- и у-
звеньев в масляной кислоте и пропиопитриле выражается числами:
С—С—С—С—ОН C-C-C=N
1 3 0,24^ 1 0,5
2. Другой метод гомолитического хлорирования (Хараш) состоит
в действии на углеводороды хлористого сульфурила при инициировании
цепной реакции перекисью. Вся сумма реакций выражается схемой:
/СвН5-С-О\ —> 2СвН5-С-О---------> 2СвП5-+2СО2
II II
\ О / 2 о
RH + CeH5- > Cell5-IIH-R.
R.4-SO2CI2---> RCI4-.SO2CI
•SOCl2 SO2+CI.
RH+«SO2C1 ---> R.+ IICI + SO2
Rll + Cb --> R. + HC1 и т. д.
Гомолитическое бромирование осуществляют бромсукцинимидом. Го-
молитические цепные реакции с его участием идут только в средах с невы-
сокой диэлектрической проницаемостью или при действии твердого бром-
сукцинимида. Бромсукцинимид действует или с добавлением инициатора —
перекиси, или на свету, или при нагревании. Особенно большое значение
имеет бромирование в аллильное положение, не затрагивающее двойной
связи. Реакция развивается так: СНг—С^ СН2- I „ hi 1 N— Вг —-» сн2-схх СН2- Ч) /0 СН2—С" сн5 j /N.4-R—сн2-си=сн2—> | сн2—сн2 zO СН-г-С^ СН2-С^ 1 z N—ВгД-R —СИ —СН=С112 >1 4 СГ12—Схч гт.т с \0 z0 -СТ ^N._pBr. -ч Ч) '~С\ 4NH4-R—СИ—С1[ = СН2 Ч) 4N. + R-CH-C1I = CII2 И т. Д. 1 *0 Вг
СВОБОДНОРАДИКАЛЬНЫЕ ЦЕПНЫЕ РЕАКЦИИ
549
3. Гомолитическое бромирование и хлорирование могут быть осуще-
ствлены при действии галоида на серебряную соль карбоновой кислоты
(реакция Бородина — Хунсдиккера). При этом первоначально образу-
ющийся ацилат-радикал декарбоксилируется и на место карбоксила
встает атом галоида:
R—С—OAg + Br- > R—С—O.-J-AgBr
R—С—О. -----> R.-l-COz
R«-j-Br2 —> R—Вг-|-Вг« и т. д.
Особенность реакции та, что галоид может быть поставлен этим путем
у головы моста бициклов, куда его нельзя ввести ни при каком нуклео-
фильном замещении: замена любой группы X у головы моста на нуклео-
фил требует вальденовского обращения (кн I, стр. 394), которое в жест-
кой структуре бицикла невозможно у головы моста.
О=С—OAg Вг
Таким образом, это частный случай сохранения конфигурации свобод-
ным радикалом.
Сульфохлорирование. При действии на алкан сернистого
газа и хлора при освещении происходит цепная реакция, используемая
в практике для получения поверхностно-активных солей алкансульфо-
кислот (моющие средства):
С12 2С1.
RH4-C1- —► R- + HC1
r-+so2
О
II
R-S.
R-S. + C12 —> R-S-Cl + Cb
При этом нормальный алкан сульфохлорируется и в первичное,
и во вторичное положение (преимущественно во вторичное).
Ту же реакцию можно осуществить, действуя на алкан хлористым суль-
фурилом в присутствии пиридина, так как последний вызывает распад
SO2C12:
SO2C12 -> so2 + ci2
550
СВОБОДНЫЕ РАДИКАЛЫ
X л о р ф о р м и л и р о в а н и е. При облучении хлористый оксалил
и фосген действуют на предельные углеводороды по следующей цепной
реакции: С1 /С1
(С0С1)2 — > 2.схх --> С1.-|-СО+.С^
А) хо
RII + C1- -> R.-HIC1
/С1
R. + (COC1)2 —> R“c\0 + ,C0C1
•COCI---> C0-I-C1.
Введение сульфогруппы. Сульфогруппа может быть
введена действием О3 и SO2 на алканы и циклоалканы. Реакция идет
нод влиянием ультрафиолетовых лучей:
О
II
R.4-SO2+ 0-2 -> R—S—о-о.
II
о
О 0 0
II II SO. II
RH+R—S—0-0. > R. + R—S—0-0II —* R—S—он
II II II
О 0 0
Аналогична реакция с алканами треххлористого фосфора и кислорода
(Л. 3. Соборовский). Приводим лишь итоговое уравнение:
RH + PCI3-L1/2O2—> R-POCI2 + HCI
Гомолитическое замещение в ароматическом
ядре. Наиболее изученные реакции этого типа — арилирование бензола
и его производных посредством свободных арильных радикалов, получа-
емых разложением перекиси ароила или ароматического диазосоединения
(реакция Гомберга — Бахмана, 1924 г., стр. 110). При этом установлено
(Хей), что результат реакции один и тот же, каков бы ни был источник
арильного радикала. Реакция протекает по схемам:
Аг—С —О II L о _]2 Аг. + (Q)—X > Аг-<^>—X Ar—N=N—О—С—СН3 — О Аг. + <(О/~Х > Аг-^О^~~X 2Аг. + 2СО2 / Аг /Х \ ( + ^О)—х + <(О/~Аг) + [н«] —>- Аг. + No + «О—С—СНУ О /+ X + \О^~Аг j + [н«] \ Ar X /
СВОБОДНОРАДИКАЛЬНЫЕ ЦЕПНЫЕ РЕАКЦИИ
551
На деле замещаемый атом водорода, вероятно, не выделяется в свобод-
ном состоянии, а отщепляется вторым свободным радикалом от о-ком-
плекса (стр. 40), образованного первым свободным радикалом и аромати-
ческим соединением:
Аг» +
Аг
X + АгН
Интимный механизм этой стадии замещения не может быть выяснен
исследованием только кинетики и продуктов реакции, однако ясно, что
она не имеет характера цепной реакции и, следовательно, водородный
атом «не выходит в объем» в качестве свободного радикала.
В высшей степени интересно при этом ориентирующее действие заме-
стителя X в атакуемом бензольном ядре (табл. 83).
Таблица 83. Ориентирующий эффект заместителя X в бензольном ядре
при гомолитическом замещении арилом (радикальная атака)
Замести- тель X Содержание изоме- ров в продукте реакции, % Замести- тель X Содержание изо- меров в продукте реакции, % Замести- тель X Содержание изоме- ров в продукте реакции, %
О сС сз а О сС 03 а О н 03 сз а
о <5 с о к о 2 к
Атак у ю щ и й радикал * Свн». Атакующий радикал h-NO2C6H4«
no2 62 10 28 СН3 67 и 14 no2 58 15 27
F 54 31 15 С2Н5 53 28 19 Cl 60 24 16
Cl 50 32 18 С3Н7 31 42 27 СС1з 0 100 0
Вт 49 33 18 (СНз)зС 24 49 27
I 52 32 17 свн5 48,5 23 28,5
CN 60 10 30 CF3 29 41 30
СС1з 12 49 39
* Действие перекиси бензоила при 80° С.
Как показывают приведенные данные, ориентация замещения при
гомолитическом фенилировании гораздо менее избирательна, чем при элек-
трофильной атаке: все три изомерных Х-замещепных бифенила образуются
в сравнимых количествах. При этом разница в ориентирующем действии
групп NO2 и СН3 не слишком велика, значит, свободный фенильный
радикал не отличается ни выраженной электрофильной, ни нуклео-
фильной активностью. Положение может измениться, если придать ата-
кующему свободному фенильному радикалу большую электрофильность,
введя в него, например, нитрогруппу. В случае индуктивно оттягива-
ющей электроны группы СС13 картина резко меняется и СС13 ведет себя
как 100%-ный мета-ориентант. Однако, если заместитель в атакуемом
ароматическом соединении NOt или С1, картина не меняется. Более того,
552
СВОБОДНЫЕ РАДИКАЛЫ
если фенил диазоацетат разлагать в эквимолыюй смеси нитробензола
и толуола, то смеси нитродифепилов получается в четыре раза больше,
чем метилдпфенилов, т. е. нитробензол реагирует активнее, чем толуол.
Можно высказать предположение, что это зависит от большей возможности
нитрогруппы рассредоточить нечетный электрон в переходном состоянии
и понизить тем самым энергетический уровень переходного состояния
и о-комплекса при атаке в пара- или орто-положение к нитрогруппе.
Например, так:
С6Н5‘ +
или, выражая о-комплекс одной формулой (пунктирные стрелки — пере-
движения одного электрона)
И т.д.
Индуктивно действующие группы СН3, СС13 и т. д. не могут обеспечить
такого рассредоточения. Способность нитрогруппы в бензольном ядре
к рассредоточению нечетного электрона явствует из большей гомолитиче-
ской диссоциации гексанитрофенилэтана на свободные триарил метильные
радикалы сравнительно с гексафенилэтаном (стр. 225).
Реакции аутоокисления
Так называются цепные реакции окисления молекулярным кислородом.
Они развиваются в результате образования свободных радикалов за счет
каких-либо инициаторов. Подобными инициаторами могут быть искус-
СВОБОДНОРАДИКАЛЬНЫЕ ЦЕПНЫЕ РЕАКЦИИ
553
ственно введенные радикалы, ионы металлов переменной валентности
(Fe, Мп, Со и т. д.), а при повышенной температуре и сама молекула кисло-
рода, являющаяся бирадикалом, что следует из ее парамагнитных свойств
и триплетного спектра. В последнем случае реакция развивается так:
RH-f-Ог ► R «д-НО—О- (1)
।--> П0«-|-«0« (разветвление цепи)
R.4-O2 —► BOO- (2)
RH4-R00- ---> ROOH-~R« и т. д. (3)
R00-4-R- —> ROOR (обрыв цепи) (4)
Стадии 2—4 проходят и в случае образования радикала R« при дей-
ствии постороннего инициатора.
При более высоких температурах главными продуктами окисления
предельных углеводородов являются олефины, альдегиды и т. д., образо-
вание которых объясняется (Н. Н. Семенов) перегруппировкой радика-
лов R00« и последующим разложением их, например, по схеме:
СНз-СН-СНз
О—0«
перегруппировка
«СНг-СН—СН3---> СН2=СН-СНз4-НОО«
ООН
СНз—СН >
I
ООСНз
СНз—СНО + СНзО.
Примером аутоокисления является уже известная нам (стр. 141)
реакция окисления альдегидов. Она инициируется катионами переходных
металлов высшей валентности:
О
II
С6Н5—4-Fe3+ -------> с6Н5—C«4-Fe2+4-H+ (зарождение цепи)
ЧН
О О
С6Н5— С«4-О2 —> С6Н6-С—0-0«
о о о
I! /О Н II
С6Н5-С-О-О.4-СвН5-С^ •—> С6Н5-С-ООН4-С6Н5-С«
н
(продолжение
цепи)
О О 0 0
II II II II
C6H5—С—О—О«4-С6Н5—С« ----> С6п5—С—О—О—С—С6н5 (обрыв цепи)
О
II /О /О
С6Н6—С—ООН4-С6Н5—--------> 2С6Нб—С<4 (реакция вне цепи)*
Н ЧШ
* Последняя реакция происходит быстро и ускоряется бензойной кислотой, так
что в обычных условиях расходуется вся образующаяся пербензойная кислота.
Ее можно фиксировать, ведя реакцию в растворе уксусного ангидрида,
ацетилирующего пербензойную кислоту в более прочную смешанную перекись
О О
r, il II
С6Н5—С—О—О—С—СН3, которая и накапливается.
554
СВОБОДНЫЕ РАДИКАЛЫ
Относительные реакционные способности ряда замещенных бензальде-
гидов в этой реакции таковы (жирные альдегиды окисляются немного
быстрее, чем бензальдегид):
n-СНзО—СвН4СНО . . 3,7
n-СНз—С6Н4СНО . . . 2,29
Л4-СН3—С6Н4СНО ... 1,61
СвН5СНО...........1
п-С1—С6Н4СНО .... 0,54
n-N=C—СвН4СНО . . . 0.47
Таким образом, электронодонорные группы ускоряют реакцию окисле-
ния. В жирном ряду это соответствует большей устойчивости углеводород-
ных свободных радикалов, более нагруженных электр онодонорными
алкилами, т. е. последовательности
(СНз)зС. > (СН3)2СН. > СН3-СН2.
а следовательно, и более легкому отщеплению Н от С—Н-связи с образо-
ванием радикала в левой стороне ряда. Поэтому предельные углеводороды
с нормальной цепью при окислении кислородом образуют смесь вторичных
спиртов.
СН3(СН2)„СН2СНз+Соз+ —> СНз(СН2)пСНСНз + Со2+ + Н+ (зарождение цепи)
СН3(СН2)пСНСНз+О2-----> СН3(СН2)ЯСНСН3
О—О*
СНз(СН2)„СНСНз + СНз(СН2)пСН2С113 >
о—о.
--> СНз(СН2)лСНСНз + СНз(СН2)яСНСИз
ООН
(продолжение цепи;
1-е направление)
СН3(СП2)яСНСНз+Со2+ —> СНз(СН2)лСНСНз+О1Г + Соз+ 1 1 ООН о. СНз(СН2)яСНС Нз|- С Нз(С Н2)ЯС Н2С Нз > (продолжение цепи;
0- 2-е направление/
СНз(СН2)лСНС11зЧ-СНз(СН2)яСНСНз
I
ОН
СНз(СН2)яСНСН3
СНз(СН2)яСНСНз + СНз(СН2)яСНСНз —> О (обрыв цепи)
I I
О. СНз(СН2)яСНСНз
Если имеется разветвление скелета с третичным водородом, по нему
окисление идет еще быстрее.
СВОБОДНОРАДИКАЛЬНЫЕ ЦЕПНЫЕ РЕАКЦИИ
555
Особенно легко, как мы видели, образуются радикалы аллильного
(и бензильного) типа. Поэтому олефины аутоокисляются по схеме (И- —
инициатор):
RCH2CH=CH2 + H. > RCIICH=CII2-|-H—Н (зарождение цепи)
RCHCH=CII2+O2 —> RCIICH=CIIo
I
о—о.
RCHCH=CII2 + RCII2Cll=C}l2 -> RCHCH=CH2 +
i ।
0-0. OOH
4-rciicii=ch2
rchch=ch2
I
о
RCHCH==CH24-RCnCTI = CH2 --> 0
o—o. rchch=ch2
(продолжение цепи)
(обрыв цепи)
Толуол заметно не аутоокисляется при комнатных температурах, но
уже ксилолы при стоянии всегда образуют немного перекисей, что может
быть обнаружено по выделению иода из йодистого калия. Как уже упоми-
налось, изопропилбензол и тетралин могут быть полностью окислены
в гидроперекиси:
С6Н5СН(С1Тз)2-|-И. —> С6Н5С(СН3)2+ИН
С6Н6С(СН3)2 + О2---> С6Н5С(СН3)2
0—0
СвН5С(СНз)2 + С6115СН(С113)2 —>
о—о.
СвН5С(СНз)2 + СбН5С(СНз)2
ООН
Гидроперекись изопропилбензола используется при промышленном
синтезе фенола и ацетона, на которые она распадается при нагревании
С6Н5С(СН3)2 —> С6Н6ОН+(СНз)2С=О
ООН
Легко аутоокисляются простые эфиры, образуя гидроперекиси и пере-
киси:
R
R—о—с-оон
R
R R
R— О—С—0—0—С—О—R
I I
R R
556
СВОБОДНЫЕ РАДИКАЛЫ
Гидрохиноны в щелочном растворе окисляются кислородом воздуха,
превращая последний в перекись водорода:
ОН О
I Д
( 1+о2 —>ц и+иоон
I н
он о
Реакция представляет собой нецепной гомолитический процесс. Ана-
логичным процессом из антрагидрохинона получают в промышленном
масштабе перекись водорода. Антрагидрохинон получают каталитическим
восстановлением антрахинона водородом, так что итог процесса выра-
жается уравнением:
Н2 + О2 —> Н2О2
Радикальное присоединение по двойным связям
Это присоединение осуществляется по схеме
Х« + СН2=СН2 —> Х-СН2-СН2.
Х-СН2СН2. +XY —> X—СН2СН2—Y + X»
и является, следовательно, цепной реакцией.
Как видно из табл. 84, только для сероводорода, бромистого водорода,
хлорноватистой кислоты, хлора и брома оба звена цепной реакции экзо-
термичны. Для хлористого водорода второе звено было бы эндотермично.
Гомолитическое присоединение воды, иода, фтористого и подпетого водо-
рода к этилену невозможно.
Таблица 84. Теплота реакций радикального присоединения (ккал/моль)
X—Y х-+сн,=снг -> -> ХСНгСНг- XCH,CH»-+XY->- -> XCHzCHjY+X- X-Y X-CH,=CH, -> -> XCHjCHj- XCHaCHf+XY->- -r XCH.CHsY+X-
н—н -40 +6 НО—Cl —32 —17
HS-H —16 — 8 Cl—Cl — 19 — 19
С1-Н —19 + 5 Вг—Вт —5 —17
Вг—Н —5 — 11 I—I +9 -13
I—н +9 —27 H—OH — +37
Н—F — +37
Наиболее изучено открытое Харашем в 1932 г. радикальное присоеди-
нение бромистого водорода, инициируемое следами перекиси и ингибиру-
емое такими антиоксидантами, как гидрохинон. Первое наблюдение было
сделано на бромистом аллиле, который, тщательно очищенный от переки-
сей или в присутствии антиоксиданта, присоединял НВг «нормально»
гетеролитически по Марковникову
СП2=СН—СН2Вг-9НВг —♦ СНз—СНВг-СН2Вг
СВОБОДНОРАДИКАЛЬНЫЕ ЦЕПНЫЕ РЕАКЦИИ
557
а при добавке перекиси бензоила или аскаридола (стр. 643) — по
цепной свободнорадикальной реакции
СН2=СН —СН2Вг + Вг. -> ВгСН2—СН—СН2Вг
ВгСН2—СН—СН2Вг + НВг--► ВгСН2—СН2—СН2Вг+Вг. и т. д.
против правила Марковникова (так называемый перекисный эффект).
«Выбор» атомом брома места атаки олефинового углерода определяется,
очевидно, меньшим энергосодержанием * возникающего свободного ра-
дикала с вторичным свободнорадикальным углеродом ВтСН2СНСН2Вг
сравнительно с изомерным ему «СНгСНВгСНгВг.
Аналогично дело обстоит с пропиленом (и гомологами)
СН2=СН—СН3+Вг. —> ВгСН2—СН—СНз
ВгСН2— СН—СНз+НВг ► ВгСН2—СН2-СН3 + Вг«
тогда как гетеролитическое присоединение дает СН3СНВгСН3.
Интересны данные по радикальному присоединению НВт к 1-галоген-
циклогексену-1: оно происходит стереоспецифично с образованием цис-
дигалогенпроизводного, что необычно для свободнорадикальных процес-
сов. В данном случае это результат большей возможности атаки по акси-
альному, а не экваториальному направлению (кн. I, стр. 510, 526):
Н
Значит, второе звено цепной реакции происходит настолько быстро,
что данная конформация кресла не успевает превратиться в другой кон-
формер.
Хлористый водород, для присоединения которого общий энергетиче-
ский баланс благоприятен и лишь второе звено эндотермично (см. табл. 84),
в принципе может быть присоединен свободнорадикальным процессом.
Но его присоединение к этилену не инициируется перекисями ацилов,
а только ультрафиолетовым светом или разложением перекиси трет-
бутила при 140° С. Можно присоединить хлористый водород против пра-
вила Марковникова к щретп-бутил этилену, инициируя реакцию перекисью
бензоила. Образуется 24% первичного галогенида — 2,2-диметил-4-хлор-
бутана. Однако реакции присоединения к олефинам хлористого водорода
по свободнорадикальному пути идут короткими цепями и сопровождаются
побочными реакциями, например теломеризацией (стр. 560).
• О связи свободной энергии переходного состояния со скоростью реакции см. кн. I,
стр. 94.
558
СВОБОДНЫЕ РАДИКАЛЫ
В газовой фазе или в индифферентном растворителе при освещении
можно провести цепные реакции хлорирования и бромирования олефинов
и ацетилена
СЬ4 CII2=CIIR —> С1СН2 -CHR
CICH2—CHR-(-Cl2 —> CICH2-CIICIR-I Cl.
т. е. придти к тому же конечному результату, что и при обычном электро-
фильном присоединении галоида. Иод удается присоединить к олефинам
при освещении лишь при температуре около —55° С. Однако все эти
радикальные реакции идут с малым квантовым выходом (особенно с иодом)
и сильно ингибируются кислородом.
Бензол, как уже упоминалось (стр. 12), на свету хлорируется в смесь
стереоизомерных гексахлорциклогексанов. Реакция эта, как показывает
кинетика, протекает по цепному механизму с вероятной первой стадией
и далее:
Первую стадию можно зафиксировать, добавив в реакционную смесь
(раствор в СС14) малеиновый ангидрид. Реакция разыгрывается так:
В продуктах реакции присутствуют I и II.
СВОБОДНОРАДИКАЛЬНЫЕ ЦЕПНЫЕ РЕАКЦИИ
559
Меркаптаны под влиянием кислоты присоединяются к олефинам по
правилу Марковникова гетеролитически:
RCH=CH2 + H+ ->• RGH—СН3
RCH—СНз + R'SH > RCH—СНз+Н+
SR'
В присутствии инициаторов радикальных цепных реакций (И- —
инициатор) происходит антимарковниковское присоединение (Хараш):
R'SH-f-И» ----> R'S« 4-ИН (зарождение цепи)
R'S- + RCH=CH2---> RCHCH2SR' I (продолжение
RCHCH2SR'+R'SH-----> RCH2CH2SR' + R'S« J цепи)
R'S'-f-R'S* ---> R'—S—S—R' (обрыв цепи)
/О
Аналогично ведут себя тиоловые кислоты
SH
Хараш открыл, что в присутствии инициаторов — перекисей аци-
лов — олефины присоединяют СС14, СС13Вт. Позднее было найдено,
что многие другие полигалоидные алканы (CC13I, CF3I, CClaBr2, CF2Br2,
GF2BrCFClBr, СНС13, СНВг3, СН13, СВг4) и некоторые полигалоидные
производные, такие, как CC13CN, СС13—СОС1, также присоединяются
к непредельным соединениям в результате радикальных цепных реакций.
Представление о них могут дать примеры:
CCU+II- —> CCI3.-1-HCI
СС1з-+ch2=chr ----> СН2—CHR
СС13
CC13CH2CHR + CC14 ► CH2-CHR-f-CC]3«
cci3 Ai
При этом инициатор всегда отрывает атом галоида от полигалоид-
метильной группы. Иод отрывается легче, чем бром, бром легче, чем хлор.
От СНС13 легче отрывается водород.
Реакции эти, в особенности для этилена, сопровождаются полимери-
зацией, которая часто становится преобладающим процессом (см. стр. 560,
реакции теломеризации).
В присутствии перекисей ацилов (или при освещении) по двойным
0=С-связям могут присоединяться альдегиды с образованием кетонов.
560
СВОБОДНЫЕ РАДИКАЛЫ
Эта реакция также сопровождается теломеризацией, которая может стать
преобладающим процессом:
О О
II II
R—С-Н + И. —► R-C.-ИН
О О
II II
R—С*Н-СИ2=СИК' —> R—С—СН2—CHR
О О 0 0
II • II II „ , II
R—С—СГ12СГ1 R'+R—С—II —> R—С—CH2CH2R -f-R—С- и т. д.
Карбоновые кислоты с а-метиленовой группой в присутствии перекисей
или других инициаторов присоединяются к олефинам по реакции:
О О
II II
R—СП2-С—ОН + И. —> R-СН—С—ОНН-ИН
О О
. II II
R—СН-С— OH + CH2=CHR' —> R-CH-C—ОН
CII2-CH-R'
ООО
II II II
R-сн—С—ОН +R-CH2—С—ОН —> R—сн—с—он +
I . I
СН2—СН—R' СН2-СН2—R'
О
4-R-CH—С—ОН
Однако в этом взаимодействии преобладает реакция теломеризации.
РЕАКЦИИ ТЕЛОМЕРИЗАЦИИ (ОЛИГОМЕРИЗАЦИИ)
Кинетика реакций теломеризации будет рассмотрена на стр. 562.
Частные реакции теломеризации упоминались в кн. I на стр. 191.
Понятие о них дают следующие схемы:
I. ССЦ-рИ* -> СС1з.д-ИС1 (зарождение цепи)
СС13.+СН2=СН2--> СС13-СН2СН2.
СС13СН2СН2*+СН2=СН2--► ССКСНгСНгСНгСПг* (продолжение
СС13СНаСН2СН2СН2.+СН2=СН2—► Цепи)]
--► СС1зСН2СН2СН2СН2СН2СН2.
СС13СНгСН2СН2СН2СН2СН2. 4-CCI4 —*•
--> СС13СНгСН2СН2СН2СН2СН2С1+СС13« (передача цепи)
СС13.+СС13СН2СН2. —► СС13СН2СН2СС13 (обрыв цепи)
РЕАКЦИИ ТЕЛОМЕРИЗАЦИИ (ОЛИГОМЕРИЗАЦИИ)
561
II. СНС1з+И. —> СС13"+ИН
СС13- +СН2=СН2--► СС13СН2СН2.
СС1зСН2СН2« + СН2=СН2 —> СС1зСН2СН2СН2СН2.
СС13СН2СН2СН2СН2«+СНС13--> СС1зСН2СН2СН2СНз+СС1з«
III. СНз-С—ОН + И. > СН2—С—0Н4-ИН
II II
0 0
СН2—С—ОН 4-СН2=СН2 —> НО—С—СН2СН2СН2
II II
о о
НО—С—СН2СН2СН24- СН2=СН2 —> НО—С—СН2СН2СН2СН2СН2
ii л
о о
На этой стадии, как нашли Р. X. Фрейдлина и А. Б. Терентьев,
происходит изомеризация радикала
СН2-СН2
НО—С СНо ---------> НО—С—СН—СН2—СНо—СН2—СНз
II . / II
О СН2-СН2 О
в далее продолжает расти уже новый радикал
НО—С—СНСН2СН2СН2СНз+СН2=СН2 > НО—С—СН—СН2СНоСН2СНз
II II I
О О СНг-СНг-
НО-С-СНСН2СН2СН2СНз + СНз-С-ОН —> НО-С-СНСН2СН2СН2СНз +
II I II II I
О СН2СН2СН2СН2« о О СН2СН2СН2СНз
+ СН2—С—ОН
II
О
Разумеется, в каждой из этих реакций получается смесь теломер-
гомологов (олигомеров), построенных по общему типу, например:
СС13(СНаСН2)яС1, СС13(СНаСН2)яН, НОС—СНСН2СН2СН3
II I
О (СН2СН2)ЯН
Соотношение олигомеров с разным п зависит как от условий опыта
(давление, избыток олефина, температура), так и от соотношения кинети-
ческих констант, характерных для звеньев цепной реакции.
36 Заказ 872
562
СВОБОДНЫЕ РАДИКАЛЫ
КИНЕТИКА ЦЕПНЫХ РЕАКЦИЙ
В любом цепном свободнорадикальном процессе имеются следующие
стадии: инициирование (зарождение цепи), продолжение (развитие) цепи
и обрыв цепи. Так, например, в фотохимическом хлорировании алифати-
ческого углеводорода можно различить следующие элементарные стадии,
сумма которых представляет собой механизм реакции:
С!----* 2СЬ (инициирование цепи)
RH + C1. - I-IC1 + R’ ) ,
i (продолжение цепи)
R* -С12 !><.' J-С1« )
2R. —> Неактивные (об }
продукты 4 1
Инициирование. На этой стадии цепной реакции в системе,,
содержавшей только валентно насыщенные соединения, образуются сво-
бодные радикалы. Появление радикалов может быть следствием воздей-
ствия на исходное соединение или на специально добавленные вещества
физических факторов (температура, свет, рентгеновские лучи, у-л учи).
Свободные радикалы могут быть получены при помощи введения в систему
небольших количеств специальных добавок — инициаторов, способных
легко распадаться с образованием активных радикалов. К числу инициа-
торов относятся, например, перекиси алкилов (RO—OR), перекиси
ацилов (RC—О—О—CR), алифатические азосоединения (например,
О В
О О
RCH—N=N—CHR) (где X — CN, СООН, COOR'), металлоорганические
I I
X X
соединения (R4Pb) и др.
Продолжение кинетической цепи. На этих стадиях
цепной реакции происходит образование основных продуктов реакции
и расходование исходных веществ. Все элементарные реакции, составля-
ющие стадию продолжения цепи, идут с сохранением свободней валент-
ности. Атом или свободный радикал, расходуемый в первом акте продол-
жения цепи, должен образоваться в последнем акте этой стадии. Совокуп-
ность элементарных реакций продолжения цепи представляет звено цепи.
Многократное повторение этих звеньев и определяет характер реакции
как цепной. Длиной цепи называют число полных звеньев, приходящихся
в среднем на один свободный радикал, образовавшийся на стадии иници-
ирования и участвующий в зарождении цепи.
Большая реакционная способность радикалов, способных вступать
во взаимодействие с разнообразными органическими веществамп как по
типу реакций замещения (стр. 225), так и присоединения, например, ио
схеме
r.4-CH2=CHX
RCH2CHX
КИНЕТИКА ЦЕПНЫХ РЕАКЦИЙ
563
,-а также претерпевать изомеризацию
СС13-СН-СН2Х —> СС12—СНС1—СН2Х
или разложение
С8Н5СОО. —> СвН5- + СО2
обусловливают неисчерпаемое разнообразие вариантов продолжения цепи
в различных цепных процессах.
Для подавляющего числа цепных радикальных реакций органических
соединений механизм продолжения цепи характеризуется тем, что в любом
элементарном акте на один израсходованный свободный радикал (или атом)
образуется только один новый свободный радикал (или атом).
Существует, однако, специальный класс цепных реакций, включающих
элементарные акты, в которых на одну израсходованную частицу с нечет-
ным электроном появляются два или более радикалов. Примером может
служить окисление водорода кислородом, включающее стадию:
Н.4-О2 —> Н0- + .о»
Такие элементарные реакции называют разветвлением цепей. Горение,
взрыв протекают как разветвленные цепные реакции. Пониманием их
сущности и выяснением особенностей кинетики мы обязаны Н. Н. Семе-
нову и его школе.
В дальнейшем изложении мы будем рассматривать только неразветвлен-
•ные цепные реакции.
Обрыв цепи. На стадии обрыва цепи происходит гибель свобод-
ных радикалов, т. е. их превращение в стабильные молекулы, или обра-
зование малоактивных радикалов (подобных семихинонам), неспособных
продолжать цепь. Обрывом цепей объясняется тот факт, что длина цепей
в цепных реакциях не бесконечно велика.
В радикальных цепных реакциях, протекающих в жидкой фазе, обрыв
•цепи наступает обычно в результате взаимодействия двух радикалов
между собой (квадратичный обрыв). При этом происходит либо соединение
радикалов (рекомбинация)
СгНб’-рСгЩ* —> С4Н10
либо их диспропорционирование с образованием одной предельной и одной
непредельной молекулы:
СгНз’-рСгНб* —> СНг—СНгЧ-СНз—СНз
Малая концентрация радикалов в жидкой фазе делает обрыв
реакции как путем рекомбинации, так и путем диспропорционирования
сравнительно маловероятным. Это и определяет возможность многократ-
ного повторения взаимодействия радикала с окружающими молекулами
до того, как радикал погибнет в результате обрыва цепи.
Длина цепи реакции определяется соотношением скоростей процес-
сов продолжения и обрыва цепи.
Обрыв цепи в ряде случаев ведет к появлению побочных продуктов.
Соотношение выходов главных продуктов (получаемых на стадиях
36*
СВОБОДНЫЕ РАДИКАЛЫ
564
продолжения цепи) и побочных зависит от длины цепи. Длина цепи за-
висит от условий реакции и может даже меняться в течение реакции, поэ-
тому обычно радикальные цепные реакции нельзя описать одним стехи-
ометрическим уравнением. Только в случаях, когда цепные реакции
имеют большую длину цепи, их можно формально описать одним стехиомет-
рическим уравнением.
В любом цепном радикальном процессе могут быть экспериментально-
определены концентрации реагирующих соединений и скорость их расхо-
дования; концентрацию промежуточно образующихся свободных радика-
лов измерить трудно. Между тем в кинетические уравнения для любой
элементарной стадии входят концентрации радикалов. Так, например,
для реакции, звено цепи которой состоит всего из двух элементарных
актов
АВ + С. —* АС + В. (О
CD+B. —+ BD + O (2>
скорость расходования исходных веществ АВ и CD и образования продук-
тов реакции АС и BD выразится следующими уравнениями (3) и (4),
в которые входят концентрации радикалов:
(3)
d[CD] _ d[BD] .
-----dt--------^- = *2 [В.] [CD]
(4)
Для того чтобы исключить из уравнений концентрации радикалов,,
введено весьма плодотворное предположение о квазистационарном состоя-
нии системы.
В цепной реакции имеется начальная стадия, когда концентрация
радикалов растет от нулевой до «средней» — это нестационарная фаза
реакции. С увеличением концентрации радикалов возрастает скорость
их гибели. Когда скорости образования радикалов и их гибели становятся
близкими, наступает квазистационарная фаза реакции; в этой фазе кон-
центрацию радикалов можно считать постоянной.
К концу реакции при исчерпании источника образования новых
радикалов их концентрация быстро падает до нуля, и реакция опять при-
обретает нестационарный характер. Если длительность нестационарных
фаз реакции значительно меньше, чем длительность фазы с постоянной
концентрацией радикалов, то к такой реакции применим метод квазиста-
ционарного состояния.
Применяя метод квазистациоиарного состояния и некоторые други0'
упрощения, например вводя предположения, что обрыв цепи происходит
только путем соединения, или только путем диспропорционирования
радикалов, или что в реакциях обрыва расходуются радикалы преимуШе"
ственно одного типа (например, радикалы С> в схеме 3, радикалы В-
в схеме 4), удалось вывести ряд кинетических уравнений, хорошо совпади'
тощих с результатами опытов.
КИНЕТИКА ЦЕПНЫХ РЕАКЦИЙ
565
Наиболее глубоко разработаны методы исследования кинетики
радикальной цепной полимеризации виниловых мономеров строения:
СН2=СНХ и СН2=СХУ
Это объясняется важностью подобных процессов, а также тем, что мно-
гие такие реакции имеют большую длину кинетической цепи, текут прак-
тически без образования побочных продуктов, а в случае гомополимери-
зации реагирующая система состоит только из одного мономера.
Зависимость скорости полимеризации от концентрации мономера
часто бывает сложной. Так, например, для стирола найдено, что при ини-
циируемой полимеризации в массе до глубины превращения ~60%
реакция имеет I порядок относительно концентрации мономера, а в раз-
бавленных растворах та же реакция имеет II порядок. По более поздним
данным Майо, порядок реакции полимеризации стирола в растворе близок
к 5/2. В случае полимеризации винилацетата кажущийся порядок реакции
также зависит от интервала изучаемых концентраций мономера.
Механизм полимеризации может быть выражен следующей схемой,
в которой R* обозначает радикал, образовавшийся на стадии иницииро-
вания кинетической цепи, М — молекулу мономера, Rn — молекулу
полимера, содержащею п мономерных единиц, М* — радикал, образо-
вавшийся при отрыве какого-либо атома от молекулы мономера.
Инициирование:
Инициатор —-> 2R« (скорость образования R. = J)
R«+М —-> RM« (константа скорости кр)
Продолжение кинетической цепи:
RM«-f-nM--> Н(М)ЯМ« (константа скорости кр)
(рост радикала)
R(M)„M«-f-M -> RMn+i + M* (константа скорости kf)
(передача цепи)
Обрыв кинетической цепи:
2R(M)nM* —► R2n+2 (константа скорости kt)
2R(M)„M« -> 27?я+1 (константа скорости к})
Эта схема составлена с применением следующих упрощающих допуще-
ний. Предположено, что константа скорости роста радикалов (кр) не зави-
сит от длины цепи радикалов, а константы скорости передачи цепи (kj)
и обрыва цепи (kt и k’t) также не зависят от длины цепи радикалов и, кроме
того, постоянны при данной температуре. Предположено также, что обрыв
цепи квадратичный и длина цепи велика. Тогда с хорошим приближением
можно получить следующее уравнение для скорости расходования моно-
мера:
“-^I=2,,'₽[Rn JIMJ (5)
а
566
СВОБОДНЫЕ РАДИКАЛЫ
(6)
Для исключения из этого уравнения величины концентрации радика-
лов [R,; ] предполагается, что система находится в квазистационарном
состоянии, тогда уравнение (5) можно преобразовать в уравнение (6)
_1да=4„[М)( '
dt р 1 ' \. kt + kf /
в которое уже не входит значение концентрации радикалов.
Важным следствием уравнения (6) является пропорциональность
между скоростью полимеризации и корнем квадратным из скорости ини-
циирования. Это уравнение отражает характерные свойства цепных
радикальных реакций, а именно возможность увеличения скорости цепной
реакции путем увеличения скорости инициирования (например, с помощью
добавления небольших количеств инициаторов) и возможность уменьше-
ния скорости реакции путем увеличения скорости обрыва цепей, т. е.
увеличения kt -f- k't (например, с помощью добавления ингибиторов).
Вывод о пропорциональности скорости цепной реакции корню квадрат-
ному из скорости инициирования применим не только к реакциям поли-
меризации, а имеет общее значение для ценных реакций с большой дли-
ной цепи и квадратичным обрывом, удовлетворяющих принципу квази-
стационарного состояния.
Пользуясь уравнением (6), можно легко вывести и некоторые другие
соотношения, связывающие между собой константы скорости элементар-
ных стадий процесса. Так, например, для реакций полимеризации сред-
няя длина кинетической цепи х, по определению, может быть записана
выражением (7):
(7>
Введя в выражение (7) значение — " из УРавнения 161, получаем:
х = кр [М]-------—
(8)
Определение констант передачи цепи
в радикальной теломеризации
Как уже было сказано, под радикальной теломеризацией понимают цеп-
ную реакцию винилового мономера с каким-либо веществом — телогеном
(в приведенных выше примерах СС14, СН3СНО, СН3СООН), в результате
которой образуются низкомолекулярные продукты — теломеры. Их моле-
кулы построены присоединением молекул непредельного соединения
ДРУГ к другу, а концевые группы представляют собой фрагменты молекул
телогена. В общей форме эта реакция может быть выражена схемой:
A—B-f-nCH2=CXY —> А—(СН2—CHXY)„—В
где АВ — любое вещество, способное присоединяться к непредельному
соединению, а п = 1, 2, 3 и т. д.
Обозначив и в данном случае молекулу мономера через М, можно
описать механизм теломеризации схемой:
ОПРЕДЕЛЕНИЕ КОНСТАНТ ПЕРЕДАЧИ ЦЕПИ В РАДИКАЛЬНОЙ ТЕЛОМЕРИЗАЦИИ 567
Инициирование:
Инициатор —► 2R*
R» + A—В —> R—В + Д.
А« + М —> А—М.
Продолжение цепи:
£
А—Мл« + А—В —А—Мл—ВЦ-А* (передача цепи)
• k
А—Мя4-М —А — Мп+1 (рост радикала)
Обрывцепи:
2R • -> Неактивные продукты
где R« — любой радикал, образующийся в системе.
Общая кинетика таких реакций, очевидно, чрезвычайно сложна.
Ряд упрощающих допущений применим и при выводе кинетических
уравнений теломеризации, а именно метод квазистационарного состояния,
квадратичный обрыв цепи, большая длина кинетической цепи и т. д.
Однако некоторые упрощающие допущения, принятые при выводе кинети-
ческих уравнений полимеризации, здесь неприменимы, например большая
длина молекулярной цепи и независимость констант скоростей элементар-
ных реакций от длины цепи радикала. Кроме того, необходимо учитывать,
что в отличие от полимеризации скорость реакции передачи цепи влияет
на общую скорость теломеризации.
Весьма плодотворным в области теломеризации оказалось изучение
кинетики стадии продолжения цепи, состоящей из двух конкурирующих
реакций — передачи цепи и роста радикала. Расходование мономера
и телогена и образование основных продуктов реакции — теломеров —
происходят именно на этой стадии. При достаточной длине кинетической
цепи стадии инициирования и обрыва мало влияют на распределение
выходов теломеров. Поэтому отношение мольной доли теломера, содержа-
щего п мономерных единиц (А—Мп—В), к суммарной мольной доле всех
остальных теломеров, содержащих более чем п мономерных единиц
(00 \
2 А—Mz—В , определяется конкуренцией указанных двух элемента р-
п + 1 /
ных реакций. Отсюда получаем уравнение для расчета отношения кон-
станты скорости реакции передачи цепи к? к константе скорости реакции
роста кр (это отношение называют константой передачи цепи и обозн чают
dfA — Мл —В] kfn [А — М„«] [АВ] kf [АВ] [АВ] .
d[A —Mz— В] = А-рп [А —Мл«] [М] — кр * [М] п [М] 1
Мольный выход телогена с числом мономерных единиц п, а также сумма
мольных выходов всех телогенов с числом мономерных единиц больше п
могут быть легко определены экспериментально, так же как и концешра-
ции реагирующих веществ.
СВОБОДНЫЕ РАДИКАЛЫ
568
Предложен ряд других уравнений для определения констант передачи
цепи.
Для ряда систем, таких, как, например, теломеризации олефинов
четыреххлористым углеродом или хлороформом, найдено, что величина
Сп значительно меняется при переходе от п = 1 к п — 2 и п = 3; для
более высоких значений п величина С обычно становится постоянной.
В дальнейшем константы Сх, С2, С3, Сп мы будем называть частными
константами передачи цепи.
Величины Сп для данной реакции не должны зависеть от концентра-
ции исходных веществ; для разных систем величины Сп определяются
природой телогена и мономера.
Однажды определив значение частных констант в тщательно контроли-
руемой реакции, можно затем использовать эти значения для вычисления
выхода любого теломера при проведении реакции в любых конкретных
условиях. Для расчета используется выражение, в котором Fn обозначает
долю всех растущих цепей, определяющих выход теломера с числом моно-
мерных единиц и:
/-г
Константы передачи цепи могут служить достаточно простыми методом
оценки зависимости между структурой и реакционной способностью
мономера, телогена и образующихся теломерных радикалов.
Применение метода квазистационарного состояния и других упроща-
ющих предположений позволило вывести ряд формул (6), (8), (9) для ско-
рости расходования исходного соединения, средней длины кинетической
цепи, частных констант передачи цепи, в которые константы скорости
элементарных реакций кр, kf, kt всегда входят в виде отношений. Для
определения индивидуальных значений констант скоростей элементарных
реакций необходимы дополнительные данные, например измерение ста-
ционарных значений концентраций радикалов или изучение скорости
реакции при нестационарных условиях.
Особенно широко для определения индивидуальных констант скоро-
стей элементарных реакций цепного процесса используется метод враща-
ющегося сектора. Фотохимическую реакцию проводят в условиях переме-
жающегося освещения. Для периодического прерывания освещения при-
меняют вращающийся диск с вырезом в виде сектора. Измерение скоростей
реакции при непрерывном и перемежающемся освещении с известной дли-
тельностью темновых периодов позволяет определить длительность жизни
кинетической цепи.
Реакции теломеризации ценны для синтезов тем, что позволяют одним
приемом перейти от простейшего олефина и телогена к олигомерам (т. е.
теломерам) с цепью из нескольких углеродных атомов (от 2—3 до 20—30),
включающей одну или несколько функциональных групп. Недостатком
реакций теломеризации является невозможность получения единствен-
ного индивидуального теломера и необходимость разделять смесь, в кото-
рой каждый из теломеров составляет сравнительно небольшую долю.
определение констант передачи цепи в радикальной теломе Р ИЗА НИИ 569
Этот недостаток не имеет значения, если каждый из теломеров исполь-
зуется полноценно.
Реакции радикальной полимеризации ничем в сущности не отличаются
от теломеризации, кроме того что при получении высокомолекулярных
полимеров концевые группы, имеющие у теломера такое большое значение
в полимере незаметны из-за огромного количественного преобладания
тысячекратно повторяющегося фрагмента «мономера», связанного в еди-
ную цепь. Полимеризация, как и теломеризации, инициируется свободным
радикалом. Обычно это продукт разложения перекиси (R. или R—СО*
II
О
или R—О—О* п т. д.) или азодинитрила изомасляной кислоты
](CH3)2C(CN)], или диазоамипососдинспия (СвНБ или СвНБ—N=N«).
Этот свободный радикал остается связанным с началом скелета полимера.
Что касается конца молекулы полимера, то это может быть или водород,
вырванный из мономера, или какой-либо другой атом того же происхо-
ждения, или даже, как показал С. С. Медведев, это может быть свободно-
радикальный конец, иммобилизованный благодаря вязкости среды и лишь
медленно окисляющийся кислородом воздуха пли претерпевающий ту
или иную из реакций обрыва цепи, например рекомбинацию. Такие
«застрявшие» в полимере свободнорадикальные концы макромолекул
(а может быть, и перекиси, образовавшиеся из них и 02) ответственны
за то, что часто затравка полимера вызывает полимеризацию мономера.
КАРБЕНЫ
В широком ряду реакций, из которых некоторые приведены ниже, в ка-
честве неустойчивых промежуточных продуктов реакции, ясно, однако,
констатируемых, фигурируют частицы СН? и их всевозможные производ-
ные тина CHR, СН2, CRR', где R — углеводородные радикалы, их функ-
циональные замещенные, атомы галоида и т. д. Такие частицы с формально
двухвалентным углеродом названы карбенами (Деринг). Простейший
карбон — свободный метилен. Ни один карбен не получен в устойчивом
состоянии в виде вещества. Тем не менее по химическому поведению
и физическим свойствам карбены ясно идентифицируются как промежуточ-
ные продукты реакции.
Природа карбенов. Можно себе представить, что свободный метилен
(и карбены его производные) существует пли как бирадикал | f СН2
(sp-гпбрнднзацпя, триплетное оптическое состояние, спины обоих электро-
нов па пегпбрпдпзовапных орбиталях параллельны), пли он находится
в состоянии, при котором спины обоих свободных электронов спарены
($р2-гпбриднзацпя, синглетное состояние, fj,CH2). Решение этого вопроса
было длительным и трудным, так как концентрации карбена в газе или
растворе малы, а теоретический расчет в зависимости от принятия того
или другого приближения давал противоречивые результаты. В настоящее
время па основании оптических исследований Герцберга и определения
Мурреем спектра ЭПР основным состоянием, по крайней мере для угле-
водородных карбенов, можно считать бирадикальное триплетное. Синглет-
ное состояние, обладающее более высокой энергией, свойственно карбе-
нам, полученным фотолизом.
Способы образования карбенов. 1. Наиболее обычный способ генерации
метилена — фотолиз диазометана пли кетена (Норриш):
CHa=ft=N 2Д :Сц2 + n2
/.’V
СН2=С=О ----► :СИ2 + СО
Аналогичный жуть применим н для получения замещенных карбенов.
КАРБЕНЫ
571
Например, пз диазоуксусного эфира образуется соответствующий
карбен — карбэтоксиметилен (И. А. Дьяконов):
_ + ,СООС2НБ СООС2Н5
N=N=C< -->N24-:C<
ХП хп
2. Йодистый метилен реагиру» г з цинком, образуя I—СН2—Znl,
который распадается с выделением метилена:
I—CiI2-Zr.T--> :СН2 + Znl2
3. Распад ртутноорганнческих соединений типа С1--СН.,—HgCI
(О. А. Реутов), а также вообще соединений X—CRR'—HgX (Зейфэрт)
под влиянием агентов, связывающих HgX2, приводит к генерированию
карбена CRR' (R и R' могут быть радикалом или галоидом).
4. Галогенметаны при действии сильных оснований образуют карбены:
СН2С12 + C4H9Li -> :СНС1 + С4Ню + LiCl
CHCI3 + ОН' -> :СС12 + Н2О + С1-
5. Пиролиз CCl3COONa в инертном растворителе по В. Вагнеру дает
дихлоркарбен:
СС13—С—O"Na+ > :СС12 + NaCl + СО2
II
О
6. Получение синтина из окиси углерода и водорода над кобальтовым
и другими катализаторами (кн. I, стр. 67), вероятно, проходит через
стадию образования свободного метилена:
пСО -|- 2мН2 * ~ ** п:СН2 пН2О
1--> Н-СН2-(СН2)„_2-СН2-11
Интересный фотсраспад в метиловом спирте метоксиокисей указанного
строения с образованием и дальнейшим превращением карбена описала
Т. И. Темникова:
О О
CeHjs. / \ /CeHs ft* СвНБ\ / \ СвНй СвНвк , CgHfc
V—C<f —► )C. .C< ----> >C=O + :C<
CH3Cr XCeH5 CH3OZ xCeH5 СН3О/ xCeH5
I CH»OH;
j H,o
/CeHs
>C<
OHZ ХСвНб
Реакции карбенов. 1. Первой реакцией, явившейся по существу доказа-
тельством существования свободного метилена, было действие его в газо-
вой струе (в условиях, в которых Панет действовал свободным метилом
на свинцовое зеркало, см. стр. 540) на зеркало теллура или селена.
При этом образовывался полимер теллурового или селенового аналога
КАРБЕНЫ
572
формальдегида (CH2Se)„ и (СН2Те)„. На олово, свинец и ртуть сво-
бодный метилен не действовал.
2. Свободный метилен, генерируемый фотолизом диазометана,
присоединяется к окиси углерода, образуя с плохим выходом кетен
(Штаудингер, Купфер):
:СП2 + СО —> СН2=С=О
3. В последнее время доказано, что генерируемый тем же снособом
метилен присоединяется к азоту (азот был мечен изотопом) и с малым
выходом образует диазометан (меченный по азоту):
:СН2 + *N2 —► CH2=*N=N*
4. Характерны реакции присоединения карбенов по свободным р-парам
электронов фосфора, азота, кислорода, приводящие, по крайней мере
на первой стадии, к образованию илидов (см. стр. 536), которые в случае
триарилфосфоранов устойчивы, а в случае кислорода и азота подвергаются
дальнейшей перегруппировке:
(С8Н5)зР + :СН2 —► (СвН5)зР=СН2 (СвН5)3Р-СН2
СН3СН2Х СНзСН2х + _
^0: + ;СН2 —> >0- СН2 —► СН3СНа-О-СН2СН2—СН3
CH3CH2Z •’ СН3СН2/
1R;\ + _т R
R3N: + 1СН2 —> ' r^N-CH2 —► >N-CH2R
r/ RZ
5. Кароены присоединяются по двойной С=С-звязи, образуя цикло-
пропаны:
СН2
\c=C<f + :СН2 —>
СС12
+ :СС12 —> С12С—-СС12
Присоединяя карбэтоксиметилен к гомологам карбзтоксициклопро-
пена, полученным действием того же карбена на гомологи ацетилена
’ Дьяконов получил гомологи дикарбэтоксиби-
цикло11,1,0]бутана, например: 1
Н ^СООС2Н5 н СООС2НБ
/С\ Н
С3Н7_с=2с-СзН7 +N=N==C</ ---> с3Н,-сС> Г тт м
х соос2н6 3 7 СзНа + N«
С
с2н5оос
«КАРБЕНЫ
573
Карбены могут присоединяться и по л-связи к бензолу (Меервейн).
Образующийся норкарадиен изомеризуется далее в циклогептатриен
(об этой реакции см. также стр. 509), так что в результате в бензол как бы
внедряется метиленовое звено:
4- :сн2
норкарадиен циклогептатриен
Наряду с норкарадиеном образуется также немного толуола.
Подобным же образом идет превращение пиррола в |3-хлорпиридин
при действии :СС12
Присоединяя к бензолу хлоркарбен, Д. Н. Курсанов и М. Е. Вольпин
получили тропилиевый катион (см. раздел «Небензоидные ароматические
системы»):
+ :СНС!
Тройная связь также присоединяет карбены. Деринг путем присоеди-
нения метилена (из диазометана) к диметилацетилену синтезировал
.диметилциклопропен (сам циклопропен из ацетилена не образуется):
СН2
СНз—С=С—СНз + :СН2 > СНз—С=С—СНз
Присоединением фенилбромкарбена к дпфенилацетилену получен три-
‘фенилциклопропенилийкатион (Д. Н. Курсанов и М. Е. Вольпин):
В< /С6Н5 С6Н5
С Н
С6н5-с=с-с6н5 + :с< 6 5—>- с6н5-с=с-с6н5—> с6н5^^-с6н5
ВГ Вг-
6. Метилен, полученный фотолизом диазометана, реагирует с предель-
ными углеводородами, беспорядочно метилируя их, например:
н н 1 1 - Н " 1 н СНз 1 “ Н ' 1
1 1 Н—с—с— 1 —с— 1 1 —II + 3:СН2 > Н—С- 1 -С— 1 —с- 1 —н
1 1 н н 1 _н _ п Н3С 1 сна 1 _ н _ п
574
КАРБЕНЫ
Быстрее и избирательнее реагируют карбены, несущие электро-
ноакцепторные заместители. По скорости и избирательности реакции они
располагаются в следующий ряд (Деринг):
:СН2 < :СНСООС2НБ < :С(СООС2Н6)а
7. Карбены реагируют с хлоридами непереходных металлов. Таково,
например, превращение, описанное О. А. Реутовым:
HgCl2 + :СНС1 -► CI2CIIHgCl
8. Так же идет реакция с хлорангидридами кислот:
R—С—С1 + :СН2 ► R—С—СН2С1
9. Карбены способны внедряться по связи С—О (Меервейн). Такое
внедрение возможно и для связей С—О в цикле, например:
О О—сн2
НгС^ \н2 1 1 + :СН2 — Н2С сн2 Нг^ \ -> | сн2 н2с / О—СНг
10- Карбены более сложного строения способны к перегруппиров-
кам как типа, описанного в пунктах 4 и 9, так и приведенных в раз-
деле «Перегруппировки».
КАРБОНИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ)
В разных местах курса, начиная с раздела о нуклеофильном замещении
галоида в галоидных алкилах, мы встречались с реакциями катионов,
несущих положительный заряд на атоме углерода. Карбкатионы — зто
частицы разной энергии, и их энергия и реакционная способность всегда
связаны со степенью рассредоточения (делокализации) положительного
заряда. Делокализация может распространяться на другие углеродные
атомы или ароматические системы, связанные с карбониевым ионом, или
же на неуглеродные атомы. Стабильными (устойчивыми) карбониевыми
ионами будем условно называть ионы малой энергии. Влияние делокали-
зации заряда можно проиллюстрировать следующей серией катионов
«с постепенным возрастанием их стабильности (понижением энергии):
СН3
СН3х
СНз^С*
сн/
н-d' хс-н
i (+) ।
Н-С с-н
н
В то время как метильный катион не обнаруживается в растворах,
существование третичных карбкатионов типа щреттг-бутильного уже можно
КАРБОНИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ>
доказать в ряде случаев на основании кинетики реакций нуклеофильного
замещения (механизм *Sjyl), стереохимических данных (рацемизация при
нуклеофильном замещении), а иногда и по спектру ЯМР.
В третичных карбкатионах положительный заряд центрального атома
частично погашен подачей электронов со стороны алкильных групп за
счет индуктивного эффекта (4-Z). Таким путем осуществляется делокали-
зация заряда, его «деление» между центральным атомом углерода
и связанными с ним алкильными группами. Разумеется, большая доля
положительного заряда приходится на центральный атом углерода.
Трпфенилметильный катион, обладающий характерными частотами
поглощения в видимой области, может быть идентифицирован по спектрам
поглощения. Так, трифенилхлорметан в растворе в жидком SO2 диссо-
циирован на сольватированный карбкатион желтого цвета и анион. Дис-
социация трпфенилхлорметана происходит также в растворе в пятихлори-
стой сурьме (анион SbClg), в хлорном олове и в серной кислоте.
Гораздо большая устойчивость триарилкарбония обусловлена рассре-
доточением его центрального положительного заряда по трем ароматиче-
ским ядрам — по шести орто- и трем пара-углеродным атомам (в общей
сложности по десяти углеродным атомам). Больший заряд все же несет
центральный углерод, так как сопряжению (резонансу) и рассредоточению
мешает не вполне плоское в силу пространственных препятствий, а про-
пеллерообразное строение трифенилметильного катиона.
Идеальное рассредоточение заряда между семью углеродами плоского-
тропилий-катиона (см. раздел «Небензоидные ароматические системы»)
делает этот катион вполне устойчивым даже в водной и спиртовой среде,
хотя он способен реагировать с более сильными, чем вода и спирт, нуклео-
филами, присоединяя их к любому из семи своих углеродов:
Наконец, в катионе красителя кристаллического фиолетового главное
значение имеет рассредоточение заряда карбониевого центра уже не
столько между девятью орто- и пара-углеродными атомами фенилов,
сколько между тремя диметиламиногруппами, каждая из которых стано-
вится почти на х/3 аммониевой, и три эти группы принимают на себя
львиную долю катионного заряда.
Таким образом, катион трифенилметановых красителей лишь очень
условно может быть назван карбкатионом. Однако реакции рекомбинации
этого катиона с анионами очень слабых кислот, например с ОН", проходят
по почти беззарядному метильному углероду. Поэтому они идут медленно,
с значительной энергией активации и могут рассматриваться как реакции
с перенесением реакционного центра: вместо атаки по N+ совершается атака
по метильному углероду, что вполне понятно — азот не может присоеди-
нить пятый заместитель.
Поскольку в карбкатионах карбониевый углерод находится в состоя-
нии sp -гибридизации (той же, что и в олефинах), карбкатион должен иметь
плоскую тригональную форму. Здесь кроется дополнительный факторг
КАГБОИИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ)
снижающий энергию карбкатиона и способствующий его образованию,
когда молекула достаточно разветвлена и в ней имеются пространственные
затруднения. Роль этого фактора рассмотрена на стр. 227. Она сво-
дится к тому, что пространственное взаимодействие (отталкивание не свя-
занных друг с другом групп) в тетраэдрической молекуле больше, чем
в плоском катионе, поскольку при переходе к катиону валентные углы
увеличиваются от 109 до —120°. Наряду с электростатическими взаимо-
действиями это еще одна причина более легкого образования катиона
из, скажем, хлористого щре/п-бутила по сравнению с образованием катиона
из хлористого втор-пропила. Еще более существенную роль простран-
ственный фактор начинает играть в сильно разветвленных молекулах.
Как излагалось в кп. I, стр. 82, для соединений, способных в опре-
деленных условиях диссоциировать с образованием карбкатиона, реакции
замещения у насыщенного атома углерода могут идти по механизму 5^1.
Реакции по этому механизму двухстадийны и заключаются в медленном
обратимом образовании карбоний-катиона и быстрой реакции последнего
с нуклеофилом:
\ *i(i) \
—С—X —С+ + X-
/ /
Кинетику реакции определяет первая, медленная стадия. Поэтому
в предельном случае общая скорость реакции может подчиняться урав-
нению:
V = fcjtRX]
Этот, впрочем, довольно редкий случай выполняется, если только-
константа скорости к?> существенно превосходит константу скорости
реакции, обратной гетеролизу (/с2). Причина состоит в том, что в процессе
реакции накапливается постепенно ион X", тогда как концентрация
введенного реагента Y" все время падает, а следовательно, прямая реак-
ция (1) все более тормозится.
Более общее выражение, типичное для реакции 5^1, можно записать
в следующем виде *:
( гх] г1
v = A-1 [RX] jl-4-а [у] )
где а = /с2/А:3.
При малых значениях а это выражение упрощается и, как уже гово-
рилось, сводится к первой формуле.
Стереохимический результат реакции, идущей по механизму
это либо рацемизация, либо рацемизация, сопровождающаяся обращением
конфигурации (инверсией). Если катион, образующийся в рсз\льт
медленной стадии реакции, достаточно стабилен, то он успевает принят
плоскую конфигурацию до атаки нуклеофилом. В этом случае происходит
* Типичное выражение для скорости реакций
v = /C1[RX] [Y]
37 Заказ 672
57g КАРБОНИЕВЫВ ИОНЫ (КАРБКАТИОНЫ)
рацемизация, так как для плоского катиона направление атаки (сверху
или снизу его плоскости) безразлично. Если же катион атакуется прежде,
чем анион удалился на достаточное расстояние и, следовательно, прежде,
чем карбкатион принял идеальную плоскую форму, то направление атаки
уже не безразлично, и она будет осуществляться преимущественно со сто-
роны, противоположной уходящей группе. Тогда элементарных актов,
сопровождающихся обращением конфигурации, будет больше, чем актов
с ее сохранением. Таким образом, если полная рацемизация свидетель-
ствует о механизме и, следовательно, об образовании карбкатионов
в растворе, то полное обращение конфигурации говорит о механизме SN2.
Промежуточные случаи нельзя интерпретировать так строго.
Для реакций, текущих по механизму £#1, как, впрочем, и для боль-
шинства органических реакций, роль растворителя очень велика. Все
дело в том, что для гетеролиза связи С—X требуется большая энергия
и эту энергию доставляет растворитель. Для того чтобы реакция могла
пойти по механизму *9^1, растворитель должен обладать электрофильными
свойствами, т. е. должен быть способен связывать отделяющийся гало-
гьнид-ион. Этим требованиям удовлетворяют в первую очередь протонные
растворители (табл. 85), способные образовывать водородные связи или
связывать своим протоном уходящую группу. Таковы кислоты (особенно
более сильные), в меньшей степени вода, еще меньше — спирты.
Сольватация образующихся катионов также, по-видимому, имеет
значение, но этот вопрос менее ясен. Во всяком случае без сольватации
аниона нет реакций S^l. Так, в очень полярном, но нуклеофильном
Таблица 85- Диэлектрические проницаемости растворителей (при 20° С)
Протонные растворители Апротонные растворители
Растворитель Диэлектриче- ская проницаемость Растворитель Диэлектриче- ская проницаемость
СНзСООН СвН5ОН (СНз)зСОН NH3 С<>Н5ОН F3CCH2OH СН3ОН носн2сн2о.н нсоон Н2О 4 6—6,5 9—10 10—11 22 (при —33° С) 25 27 32—33 38 56 81 СНо—СН2 °\ Z0 сн..—сн2 С2НБОС2Н5 СН3ОСН2СН2ОСН3 сн2-сн2 )о сн2—сн2 СНзСОСНз CeH5NO2 CH3CN ch3no2 HCON(CH3)2 (CH3)2SO 0 P[ N (СН3)2]3* (гексаметапол) 2-2,5 4,5 6,5—7 7—7,5 20—21 35—36 36—37 36—37 37—38 45—49 30
КАРБОНИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ)
ЭТЭ'
и апротонном растворителе диметилсульфоксиде по механизму не-
йдут даже реакции трет-бутилгалогенидов.
Реакции типа легко осуществляются при действии солей серебра
в подходящих условиях. Ион серебра образует прочную связь с анионом
галоида и отрывает его от молекулы более энергично, чем растворитель;
кроме того, при этом анион уводится из сферы реакции, вследствие чего
замедляется (или предотвращается) реакция, обратная гетеролизу алкил-
галогепида.
Реакции, идущие по механизму SN2, напротив, затормаживаются
в протонных растворителях, так как, образуя водородные связи с реаген-
тами, эти растворители понижают их нуклеофильную активность. Так,
в протонных растворителях нуклеофильность галоид-анионог. понижается
от иод-пона ко фтор-иону:
I' > Вг" > Cl" > F*
Особенно низка активность фтор-иона, способного образовывать проч-
ные водородные связи. В апротонных растворителях разница между
этими нуклеофилами не гак заметна, и даже порядок в их ряду может
несколько меняться.
Хотя косвенными путями (кинетические измерения, стереохимическое
поведение в реакциях и пр.) доказано преходящее появление карбониевых
катионов во многих реакциях, снять спектры ряда алкильных катионов
только недавно удалось Олаху. Он наблюдал спектры ЯМР растворов,
фтористых алкилов в пятифтористой сурьме. По реакции
AlkF + SbF5---> Alk+ SbF6‘
образуется соль алкил-катиона, причем SbF6 — сильная кислота Льюиса
и акцептор фтор-аниона — находится в избытке. Для третл-бутильного
катиона спектр ЯМР показывает одиночный пик (как и для исходного
фторида), но сдвинутый в совершенно необычную для протонов метила
область слабых полей (4,35 м. д. по шкале б), тогда как сигнал протона
в (CH3)3CF проявляется при 1,30 м. д. На рис. 117 приведен спектр ка-
тиона (СН3)2ССН2СН3, снятый в тех же условиях. Справа при ~ 1,75 м. д.
находится триплет метила этильной группы (расщепление на про-
тонах СН2). Триплет при 3,9 м. д. — сигнал протонов двух эквивалент-
ных метильных групп; расщепление обусловлено взаимодействием про-
тонов этих групп с протонами СН2-группы через разделяющий их атом
углерода. Наконец, слева имеется малоинтенсивный мультиплет сигнал
метиленовой группы (расщепление на протонах метильных групп). Обра-
щает на себя внимание то, что сигнал метила, находящегося рядом с поло-
жительно заряженным углеродом, расположен почти при 4 м. д. случай,
не наблюдаемый для обычных органических соединений. Кроме того,
константа спин-спинов ого расщепления в сигнале протонов одиночных
метилов очень велика, несмотря на то что они отделены от метиленовой
группы атомом углерода.
Интересно, что в этих условиях не удалось наблюдать катионоа
типа R—СН2 (наименее стабильных катионов).
37*
КАРБОНИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ)
580
При действии Sb Fj на адамантилфторпд получен адамантил-катион.
На рис. 118 сопоставлены ЯМР-спектры исходного адамантилфторида
(кн. I, стр. 596) в СС]4 и адамантил-катиона.
При превращении C10HeF в С10Нд картина пиков ЯМР меняется
резко: пики под влиянием положительного заряда перемещаются в сто-
рону слабых полей и раздвигаются. В соответствии с тремя «сортами»
водородных атомов — а-метиленовых групп (6 атомов), [3-метинных групп
.(3 атома) и у-метиленовых групп — имеется три пика с отношением
Рис. 117. ЯМР-Спектр катиона
(СНзЯССНоСНз в растворе в SbF>.
Рис. 118. ЯМР-Спектр фтористого адамантила
в растворе в СС14 (сплошные линии) и обра-
зовавшегося из него при действии SbFg ада-
мантил-катиона (пунктирные линии).
площадей 3 : 6 : 6. В этом случае карбкатион, разумеется, не может при-
нять свойственную ему нормально плоскую конфигурацию. Так же об-
стоит дело со всеми карбкатионами бициклов, в которых положительный
заряд находится на углеродном атоме в «голове моста».
Нестойкие карбкатионы. При реакции олефинов (а также третичных
спиртов) с концентрированной серной кислотой возникает равновесие типа
+ R\
RCH=CH2 + H2SO4 4ХГ RCII—СНз HSOj- CH—О—SO3H
CH3Z
в котором карбкатион подвергается изомеризациям (см. ниже) и может
вступать в такие синтетические реакции, как реакция Коха с окисью
углерода (кн. I, стр. 168) или реакция алкилирования алканов.
(КАРБОНИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ)
581
Нестойкие алкильные катионы образуются также путем ионизации
тозилатов (n-толуолсульфокислых эфиров) третичных спиртов в среде,
например, ледяной уксусной кислоты:
Алкил-катионы можно получить путем окислительного декарбокси-
лирования карбоновых кислот, например, действуя на кислоты
тетраацетатом свинца:
2RCOO--f-Pb(CH3COO)4 > 2В++2СО2 + РЬ(СН3СОО)2 + 2СН3СОО-
Наконец, при действии азотистой кислоты на амины реакция проходит
перез стадии образования сначала диазония, а затем — после отщепления
молекулы азота — карбкатиона, чем и объясняются многочисленные
изомеризации, сопровождающие реакцию Демьянова (стр. 621), а также
возникновение двойной связи.
Обычное превращение алкил-(или циклоалкил-)катиона в олефин
путем выброса протона
ВСНСНз ---► R—СН—СНг + Н+
это то самое превращение, которое обратимо совершается в концентри-
рованной серной кислоте. Распад ароматического диазония в случае,
•если его катиону противостоит анион сильной кислоты, например BFj,
также совершается с промежуточным образованием арильного катиона:
CeH6N2BFi---> CeH' + N2 + BFf
Реакции арил-катионов. Арильный катион может быть фиксирован при-
соединением к свободной паре электронов любого аниона, а также
кислорода, например в (CgH5)2O, или галоида в хлор- или бромбензоле
С образованием соответственно катионов
(С6Н5)зО (С6Н5)2С1 (С6Н6)2Вг
Катион C6Hs может быть также обнаружен как электрофильный ре-
агент по вступлению в бензольное ядро в мета-положение к ориентантам
второго рода (А. Н. Несмеянов, Т. П. Толстая, Л. Г. Макарова,
Л. С. Исаева).
Реакции алкил-катиоиов. Уже было сказано, что одной из обычных реак-
ций алкил-катионов является выброс протона и превращение в олефин.
К этой реакции, разумеется, не способны метильный катион и катионы,
несущие положительный заряд на третичном углеродном атоме бицикла
(двойная связь не может образоваться у «головы моста» — пра-
вило Бредта).
Алкил-катионы реагируют с сильными анионами (т. е. с анионами
•слабых кислот), рекомбинируясь в молекулы
R+ + A- —> RA
582
КАРБОНИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ
и взаимодействуют с протонными растворителями, вытесняя протон,
например:
R*4- ЩО —► ROH + II*
По сути дела к последнему типу относится и алкилирование алканов •—
реакция, в которой выброс притона осуществляется труднее и требует
участия агента, способствующего этому отщеплению. Так, при действии
олефина в присутствии жидкого фтористого водорода на алканы реакция
протекает через стадию образования алкил-катиона (присоединение про-
тона олефином). Этот катион алкилирует третичный или вторичный угле-
род в алкане, от которого HF уводит протон в виде H2F.
Алкил-катионы способны присоединять окись углерода (реакция Коха).
Эта реакция осуществляется действием на раствор олефина в серной или
другой сильной кислоте непосредственно окисью углерода или (как это-
делал Кох вначале) муравьиной кислотой, разлагающейся по уравнению:
HCOOH + H2SO4—> CO + H2O.H2SO4
Вся реакция Коха протекает по схеме:
R-CH=CH2 + H* —► R—СН—СНз
<ь +со
R-СН-СНз—>• ГН—СН-СНз *
I
*с=о
ацилий-катион
В—СН—СНз'
I
с==о*
R-CI-1-CH3
—н+ ।
но—с=о
В результате присоединения к карбкатиону окиси углерода получаете®
ацилий-катион, способный существовать в концентрированной серной
кислоте или в виде солей с такими анионами, как BF4 и SbClg. Мы встре-
чались с ацилий-катионом при описании производных кислот (кн. I,
стр. 177) и при рассмотрении гидролиза сложных эфиров по механизму
(см. стр. 182).
Гидридные перемещения
Карбкатионы способны захватывать гидрид-анион, т. е. водород с парой
электронов связи. Такие реакции называются гидридными перемещениями.
Они могут быть межмолекулярными и внутримолекулярными.
В межмолекулярных гидридных перемещениях акцептором гидрид-
аниона является любой карбкатион, а донором — группировки
О I Р- п । ,o./R
Н—С—о—Н Н—С—о—R н— С—1<
1 1
. -
в частности Li N—СН — C6Hs
С6Н5
КАРБОНИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ) too
. ООО
Реакция разыгрывается на примере спирта как донора гидрид-аниона
? н
Аг+ 4- н—С-^о^-Н --->ArH + С=О + Н +
С6Н5 С6Н5
н
+ с=о + н+
С6Н5
н
(С6Н5)3С+ + СН3—с-^о^-н---->- (с6н5)3сн + сн3— с=о + н+
СН3 СН3
Аналогично другие доноры, отдавая Н“, превращаются следу-
ющим образом:
СН2—О. /Н СН2—0х+ СН2—бч сн2—о.
I --►Н"+| /С-R -> I ^C-R *—- I 'ЧС—R
СН2—СИ \R СН2—СИ сн2—о/ СН2—(У
R
I .. /К R\ + /R
Н-С—N< > Н~+ >C=N\
1 XR RZ XR
R
H
- I
Li+ N—CH—C6H5 ► Li+ + H- + CeH&—N=CH—C6H5
ieH5
Самый сильный из доноров гидридного аниона — последний. Он пере-
дает Н“ даже кетонам на их карбонильный углерод, а не только
аастоящим карбкатионам.
Д. Н. Курсанов и 3. Н. Парнес нашли, что углеводороды, имеющие
третичный углерод, в концентрированной дейтеросерной кислоте обмени-
вают весь свой водород на дейтерий. Первой стадией реакции является
окисление серной кислотой третичного атома углерода в относительно
устойчивый третичный карбкатион!
СН8 СНз
CH3-C-H + H2SO4---► CH3-C+ + HSOJ+H2
I I
CHs СНз
584
КАРБОНИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ)
Затем карбкатион обменивает постепенно все свои водороды на дей-
терий, вероятно, за счет выброса протона и присоединения дейтрона
к образовавшемуся олефину:
dig СНЭ СНз СН3 СНз
I | D+ I I +D+ |
СНО_С+ > ClI-z—C --------> DCH2—С+ > DCII=C -----------► D2CH—С+ п т. д.
I -н+ I | -и+ | |
СНз СПз СНз СНз СНз
В результате гидридного перемещения у того углеродного атома,
от которого мигрировал гидрид-анион, появляется положительный заряд.
Этот углерод берет теперь на себя роль карбкатиона. Данные табл. 86
Таблица 86. Энергии превращения R • -\-е ->R++2«“
п теплоты образования карбкатионов (е~ — электрон)
R+ Потенциал ионизации радикала Относитель- ная энергия превращения радикала ккал/м9лъ Теплоты образования иона ДН^ ккал/молъ
СН+ — — 262
СН3СН+ — — 224
CH3CH2CH2 8,69 29,2 216
CH3CHCH3 7,90 11,1 190
CH3CH2CH2CHt 8,64 28,1 207
СН3.
>СНСН£ 8,35 21,4 211
СН3/
сн3сн2снсн3 7,93 11,7 181
СНзх +
/ССНз 7,42 0 166
CH3Z
показывают, что энергетически наиболее устойчивыми являются третич-
ные карбкатионы, а наименее устойчивыми — первичные. Поэтому ги-
дридные перемещения совершаются в таком направлении, чтобы в равно-
весной смеси алкил-катионов преобладали третичные, затем вторичные
и меньше всего содержалось первичных. Переходы
пере- -> втор- --> трет-
экзотермичны.
Возможны также внутримолекулярные гидридные перемещения. Так,
П° 9‘ РеУтовУ и Т. Н. Шаткиной, при действии азотистой кислоты
на 1-пропиламин, меченный по углероду 1, образуется смесь про-
КАРБОНИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ)
585
панола-1-13С и пропанола-3-13С (конечно, наряду с изопропиловым
«спиртом):
HNO« + +HjO
СНз-СН2-13СНо—NH2 £ СНз—СН2—1зсн2 > СНз-СН2-13СН2ОН
л г -н+
I 1—3-гидридное
1 перемещение
+ +Н,О
СН2-СН2—13СНз ----> НОСН2—СН2—13СН3
-Н+
Такое 1—3-гидридное перемещение является, возможно, результатом
двух 1—2-гидридных перемещений. Другое внутримолекулярное 1—2-ги-
дридное перемещение наблюдал Олах. Он обнаружил, что, если нор-
борнилфторид растворить при 20° С в пятифтористой сурьме, спектр ЯМР
из сложного, изображенного на рис. 119 сплошной линией, превратится
в простой пик (пунктир), означающий, что в норборнил-катпоне все водо-
роды одинаковы. Это может быть понято только, если допустить быстрое
1—2-гидрпдное перемещение и одновременно быструю вагнеровскую
ретроппнаколпновую перегруппировку (стр. 613), усредняющие картину:
Последняя схема служит
уже нам перегруппировках
-будут подробнее рассмотрены
в разделе «Перегруппировки»
(стр. 609).
По Олаху, растворенный в
-«сверхкислоте» HSO3F + SF5
метан (под давлением, при
140° С) захватывает протон, об-
разуя метоний-катион СН*,
возникновение которого при
действии протона на метан
В. Л. Тальро.зе открыл ранее
посредством масс-спектрографа.
Метоний-катион в условиях
опыта нестоек и распадается
на карбкатион и молекулярный
водород:
также напоминанием о встречавшихся
карбкатионов, которые еще раз
СН£ —► СН^-рНа Рис. 119. ЯМР-Спектр порбориплфторида
1Г в растворе СС14 (сплошные линии) и образо-
Другими словами, протон вившегося из него при действии SbF5 норбор-
вырывает из метана гидрид- нил-катиона (пунктирная линия).
анион. Аналогично, по Олаху,
и другие предельные углеводороды способны захватывать протон
«сверхкислот» и далее претерпевать превращения, подобные описанным
выше.
586
КАРБОНИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ>
Оксосоединения в кислой среде принимают протон на карбонильный
кислород, а их углерод становится частично карбониевым с положитель-
ным зарядом, рассредоточенным между карбонильными углеродом и кис-
лородом:
СН;
с—о—н
с=о—н >
СН;
Поэтому альдегиды и кетоны в кислой среде ведут себя в некото-
рых реакциях как карбкатионы, например:
выброс
протона
ГСН;
_СН3'
СНзх +
>с—ОН
CH3Z
+ СНз\ ♦
с=о—н —-*
[СНз-
С-ОН +СНз—С-СНз
.гС—ОН (енолпзацпя)
CHZ
*н2о*
С—СН2—С-СНз —►
I -Н,О
О
О
о
о
СН3ч + II
/С—СН2— С—СНз—> /С=СН—С—СНз (кротоновая конденсация)
СНзХ -н+ CH3Z
Здесь нужно напомнить об устойчивых ацилий-катионах, образующихся
при действии кислот Льюиса или концентрированной H2SO4 на галоидан-
гидриды карбоновых кислот (или, соответственно, их эфиры) (Зеель):
R-CF + BF3 > [r—С=О + R-C=o]bF7
II
О
R—С—Cl+SbCl5 —> [R—С=О > R-C==6]sbCl;
Строение ацилиевых катионов характеризуется делокализацией поло-
жительного заряда между кислородом и углеродом.
Ацилиевые катионы являются важными промежуточными продуктами
кислотного гидролиза сложных эфиров (стр. 182) и других реакций
производных карбоновых кислот.
Карбкатионы иного типа образуются при присоединении протона
к ароматическим углеводородам. Хотя ароматические углеводороды
являются очень слабыми основаниями, тем не менее в определенных усло-
виях можно констатировать их кислотно-основное взаимодействие с про-
тонными кислотами. Так, при растворении бензола, толуола, нафталина
и всех многоядерных ароматических углеводородов с конденсированными
ядрами в жидком фтористом водороде устанавливается равновесие
ArH + HF ArH|4-F-
которое может быть существенно смещено вправо прибавлением, например,,
трехфтористого бора, связывающего анион фтора в прочный анион BF7-
КАРБОНИЕВЫЕ ИОНЫ (КАРБКАТИОНЫ)
587
По спектральным данным, при этом образуются о-комплексы, например
в которых карбонпсвый заряд рассредоточен в орто- и пара-положениях
кольца. В следующих формулах звездочкой отмечены возможные места
присоединения протона, т. е. места наибольшей основности!
Электропроводные окрашенные растворы, образующиеся при низкой
температуре в системе ароматический углеводород — хлористый водо-
род — хлористый алюминий, также содержат карбониевые соли галоид-
.алюмпниевых кислот. Так. при —80° С хлористый алюминий при про-
пускании НС1 растворяется в толуоле; при этом образуется зеленый
раствор, содержащий карбкатионы:
Н Н Н II н II
МЕХАНИЗМЫ ХИМИЧЕСКИХ РЕАКЦИЙ
Мы уже встречались с делением реакций на гомолитические и гетеро-
литические, и с классификацией последних на типы нуклеофильных
и электрофильных замещений (кн. I, стр. 24). Для каждого из двух типов
возможны кинетические подтипы реакций первого и второго порядка.
Используя в этой классификации Инголда его же символпку, мы получаем
четыре основных типа реакций гетеролитического замещения 8д-1, SE2,
SE1, Se2. В этих символах 5 обозначает реакцию замещения {Substitu-
tion), N и Е соответственно нуклеофильный и электрофильный характер,
а цифры указывают на кинетический порядок реакции.
К этим основным типам следует добавить еще типы S^i и SEi (от i —
intramolecular — внутримолекулярный), где весь символ обозначает
внутримолекулярное замещение — нуклеофильное или электрофильное.
По смыслу вещей это всегда реакция первого порядка, т. е. внутримоле-
кулярная гетер олитическая перегруппировка (см. раздел «Перегруп-
пировки»).
Возможны еще обменные реакции, проходящие по типу
AXH-BY
А-Х/
Y—В
--> AY + BX
а
когда одновременно обе части молекулы АХ атакуют обе половины моле-
кулы BY, т. е. обмен проходит через четырехцентровое (четырехчленное)
переходное состояние. Этот тип реакций замещения иногда обозначается
символом S р (от four — четыре). Очевидно, что это реакции второго кине-
тического порядка.
Реакции гетеролитического присоединения Ad * по кратной связи
могут быть классифицированы также по признаку нуклеофильной Adpi
или электрофильной AdE начальной атаки на кратную связь. Здесь также
возможна одновременная атака по типу
А=В+Х-Y—>
А—ВТ А—В
X-Vj X Y
* Ad. — addition — присоединение.
СТРУКТУРА ПЕРЕХОДНОГО СОСТОЯНИЯ
589'
проходящая через четырехцентровое переходное состояние и обознача-
емая AdF. По смыслу кинетический порядок в этом случае всегда второй.
Наконец, еще один тип реакций, о которых почти не было речи, это —
реакции элиминации, или отщепления, обозначаемые буквой Е и часто
А—В
обратные типу Ad. Поскольку, однако, отщепление X—Y от | |
X Y
с образованием А=В может осуществляться без воздействия реагента и про-
ходить мономолекулярно — это тип Ег. В случае необходимости
атаки на X или Y реагента (он может быть нуклеофилом или электро-
филом) реакция элиминации идет по второму порядку и обозначается
как Е2.
На деле приведенная классификация — только очень упрощенная
схема. В реакциях часто участвует третья, а иногда и четвертая частица,
и реакции формально третьего и четвертого, а также смешанных порядков
не редкость. Иногда эта третья частица — растворитель, иногда это тот же
реагент. Весьма часто участие третьей частицы проявляется по принципу
«тяни — толкай», например:
R.
Y** + R'-^C—X + HOR'"
R’
7
Y—С—X—HOR
Y-C
I
R
+ X-—HOR
Иногда это участие в циклическом переходном состоянии, примеры
чему можно найти в следующем разделе.
СТРУКТУРА ПЕРЕХОДНОГО СОСТОЯНИЯ
В разделе «Химическая реакция» (кн. I, стр. 85) уже рассматривалась
физико-химия переходного состояния (активированного комплекса). Изу-
чающему органическую химию должно быть ясно, насколько важно
знание параметров переходного состояния данной реакции для количе-
ственного предвидения ее течения. Между тем до настоящего времени
нет способов установления строения активированного комплекса, подоб-
ных уверенным химическим и физическим методам установления структур-
ной формулы обычных молекул. Это обусловлено тем, что концентрация
обязательно неустойчивого переходного состояния, находящегося в энер-
гетическом максимуме («горбе»), исчезающе мала по сравнению с осталь-
ной частью реакционной массы, и активированный комплекс не может
быть ни отделен от нее, ни индивидуализирован. Хотя можно высказать
надежду, что методы ЭПР, ЯМР и оптические методы, развиваясь, внесут
свой вклад в суждение о строении переходного состояния, в настоящее
время приходится выводить формулы переходного состояния косвенным
путем, основываясь на данных стереохимического течения реакции, ее
кинетики в разных растворителях и их смесях, на использовании изотоп-
ного эффекта, на энергетических и геометрических соображениях. На-
конец, полезен постулат Хэммонда.
"590
МЕХАНИЗМЫ ХИМИЧЕСКИХ РЕАКЦИЙ
Первый вопрос: сколько молекул того и другого реагента участвуют
в структуре переходного состояния — решается на основании порядка
реакции по каждому из реагентов *. Если реагируют вещества АВ и CD
и скорость реакции пропорциональна концентрации каждого, т. е. произ-
ведению [АГ.| LCD], то это — реакция второго порядка (первого по каж-
дому из реагентов), и в переходном состоянии участвует по одной молекуле
каждого вещества. Если скорость реакции пропорциональна [АВ] [CD]2 —
третий порядок, то в переходном состоянии участвует 1AD и 2CD и т. д.
(см. примечание).
Стереохимическое течение реакции, например сохранение конфигура-
ции вокруг атакуемого (нуклеофильно или электрофильно) тетраэдри-
ческого углеродного атома, или вальденовское обращение, позволяет
решить, в частности, вопрос, с какой стороны — со стороны замещаемой
группы или с противоположной — приближается атакующая частица.
Первый порядок реакции по одному из реагентов и нулевой по другому
свидетельствуют о предварительной диссоциации этого реагента, которая
обычно происходит медленно и потому определяет итоговую кинетику
реакции. В этом случае часто наступает рацемизация, конечно, если речь
идет о замещении у асимметрического тетраэдрического углерода. В пере-
ходном состоянии в этом случае участвует частица диссоциированной
молекулы (ион) и молекула второго реагента. Если ионизованное вещество
осталось с неразделенной ионной парой (в неполярном несольватирующем
R к
растворителе), то, например, катион в случае R'-^G" Na+ или анион в слу-
R"/
R\
чае R'-^C+Cr экранирует ион-партнер, и рацемизация (по крайней
R"Z
мере, полная) не наступает — оптическая активность сохраняется. Часто
рацемизация не наступает (или она не полна) в случае участия молекулы
растворителя (которое кинетически обычно не учитывается, так как при
огромном избытке растворителя его концентрация практически постоянна),
экранирующей ионный центр. Сменив растворитель или сильно разбавив
его инертным, можно, меняя концентрацию, выявить участие этого рас-
творителя в переходном состоянии.
Изотопный эффект используется главным образом в случае водорода
и дейтерия. Если в двух соединениях одинакового строения, различа-
ющихся лишь тем, что в одном из них один (или несколько) атом водорода
замещен дейтерием и именно этот атом (или атомы) вовлекается в реакцию
и участвует в структуре переходного состояния, наблюдается резкая
разница в скорости реакции водородного и дейтерированного соединения.
Это и называется изотопным эффектом. Не вовлекаемые в реакционный
комплекс атомы дейтерия существенно не влияют на скорость реакции
(пример см. кн. I, стр. 133, окисление спиртов).
Постулат Хэммонда заключается в следующем. Как уже сказано
кн. I, стр. 94), переходное состояние, структурно включающее в себя
Это утверждение строго только в применении к реакции, протекающей в одну
стадию, или к элементарной стадии сложной реакции.
СТРУКТУРА ПЕРЕХОДНОГО СОСТОЯНИЯ
591
две (или более) реагирующие частицы (молекулы, ионы, свободные ра-
дикалы), имеет максимум энергии и соответствует вершине горба на кри-
вой энергия системы (ордината) — координата реакции (абсцисса).
Координата реакции — это мера расстояний между атомами, основ-
ными участниками реакции.
Если реакция идет в несколько стадий, каждой из них соответствует
свое переходное состояние со своим максимумом. Чтобы поднять энергети-
ческий уровень реагирующих частиц до переходного состояния, нужно
________________________________________________________________Е.
затратить энергию активации Е (в уравнении Аррениуса k — Ае RT,
где к — константа скорости ре;
экспоненциальный множитель).
В том случае когда реагируют
два свободных радикала или два
иона, энергия активации обыч-
но мала, если не равна нулю (т. е.
равна немногим килокалориям на
моль). Если такая же неустойчи-
вая частица реагирует с молеку-
лой, энергия активации больше,
но обычно невелика. Если же ре-
агируют две молекулы и реакция
течет с измеримой при обычных
температурах скоростью, энергия
активации существенно выше и до-
стигает обычно 15—30 ккал/моль.
Эти случаи могут быть по Хэм-
монду изображены схематически
тремя кривыми (рис. 120).
Кривая А при переходе от ис-
ходных веществ до максимума
соответствует малой энергии подъема и затем глубокому энергетическому
спуску до энергии продуктов реакции. Здесь реагируют, следовательно,
энергетически богатые частицы, в реакции участвует, например, ион или
ионы. Это экзотермическая реакция. Кривая В имеет противополож-
ный характер: подъем высок — спуск мал, получаются богатые энергией
продукты. Реакция эндотермична. Кривая С соответствует довольно
большому подъему и довольно большому спуску, по она построена более
симметрично, чем предыдущие, и типична для реакции двух стабильных
молекул (реакция второго порядка).
Нужно сказать, что кривая А, в частности, изображающая реакцию
иона с молекулой, т. е. реакцию SN (или SE), по существу двугорба,
но первый — левый горб здесх. опущен — он соответствует переходному
состоянию диссоциации.
Постулат Хэммонда состоит в том, что переходное состояние по своей
структуре (т. е. по межатомным расстояниям и углам, словом, по гео-
метрии) в случае А близко к исходным частицам, а в случае В — к конеч-
ным продуктам. Этот оправдывающийся на деле постулат основывается
на том соображении, что раз энергия начального состояния мало
7 — температура,
Переходное
состояние
иаодные-
вещестВа
А — пред-
Переходное
состояние
Энергия
актиВаиии
Теплота _
реакиии лн
, конечные
вещества,
Д
Переходное
'-я стадия состояние
2-стадия
Конечные
Вещества
Энергия
активации
Рис. 120. Энергетическое состояние реа-
гирующих систем.
А — экзотермическая реакция; В — эндотерми-
ческая реакция; С — двухстадийная реакция.
В
с
МЕХАНИЗМЫ ХИМИЧЕСКИХ РЕАКЦИЙ
отличается от максимума, то и изменения по сравнению с начальным
состоянием невелики. Подобное соображение для случая В сближает пере-
ходное состояние с продуктами реакции.
Наконец, огромное значение имеют чисто геометрические соображе-
ния. Все межатомные расстояния в активированном комплексе остаются
обычными, за исключением длин вновь образующейся и разрыва-
ющейся связей, которые обе лишь немного превышают обычные ковалент-
ные расстояния. Валентные углы также меняются только у соединя-
ющихся и разрывающих связи атомов в соответствии с новым состоянием
гибридизации электронных орбиталей. Например, в случае реакции
электрофильного замещения водорода в бензоле
^атакуемый углерод в активированном комплексе из хр2-гибридизованного
(второе валентное состояние) переходит в хр3-гибрпдизованный (первое
валентное состояние — тетраэдрический углерод) и стремится изменить
валентные углы от 120 до 109°.
Подобно этому случаю, мы часто встречались с активированными
комплексами простейших структур и знаем, что, например нуклеофильное
замещение у тетраэдрического углерода SN2 проходит через переходное
-состояние, изображенное ниже, чем и объясняется вальденовское
обращение:
+ СГ
Поскольку в реакциях Se2, изученных гораздо меньше, обычно наблю-
дается сохранение конфигурации, то очевидно, что в переходном состо-
янии атакующая электрофильная частица подходит со стороны замеща-
емой группы. Например, в изученных О. А. Реутовым реакциях ртутно-
органических соединений предполагается поэтому четырехцентровое
(четырехчленное) циклическое переходное состояние:
С1
Hg-Cl н Н
С6н5—С—Hg—С—С6Н5 > 2С8Н5— С— HgCl
с С с
С2Н5О \) б^ОСгНь О^^ОСгНо
L L
СТРУКТУРА ПЕРЕХОДНОГО СОСТОЯНИЯ
593
Cells— HgCl
! I -------► CeHs*HgCl + HgCl2
»Hg—Cl
I
Cl
где *Hg — радиоактивный изотоп ртути.
Такое же четырехцентровое переходное состояние предполагает тот же
автор и Десси для разложения ртутноорганического соединения спирто-
вым раствором соляной кислоты:
R—Hg—С1
i ! --> R— H + HgCl2
Н—Cl
Действительно, скорость разложения ртутноорганических соединений
соляной кислотой много выше, чем кислородсодержащими кислотами
в тех же условиях, что и заставляет доцустить участие хлора в переходном
состоянии.
Уже на этих примерах простейших реакций мы столкнулись с необ-
ходимостью принять циклические переходные состояния, в данном случае
четырехцентровые. Циклические активированные комплексы — очень
распространенное явление в химии.
Для многочисленных реакций присоединения карбенов по кратной
связи (стр. 570) вероятно трехцентровое (трехчленное) переходное
состояние'.
:Сн2
сн.,=сн
сн2
нгс^'сн2
СН2
н2с—сн2
Электрофильная атака брома на олефиновую двойную связь проходит,
как было сказано (кн. 1, стр. 396), через трехчленный бромониевый
катион. Это доказывается тем, что в циклоолефинах 6poMj присое-
диняется в транс-положение:
<Q] + Вг2
Трехчленный бромониевый цикл атакуется бром-анионом и размы-
кается с вальденовским обращением, почему атомы брома и оказываются
в тпранс-положении друг к другу. Чтобы наблюдать подобное явление
в алифатическом ряду, надо выбрать такой олефин, в результате броми-
рования которого возник бы асимметрический атом. Таков случай бро-
мирования двух геометрических изомеров бутена-2:
38 Закав 672
МЯХАНИЗМЫ ХИМИЧЕСКИХ РЕАКЦИЙ
594
цис-бутен-2
+ В Го
рацемическая
форма
транс -бутен-2
Вг
Вг
ЧСН3
А<езс-форма
Образованию нестойкого трехчленного бромония, вероятно, ^пред-
шествует стадия образования л-комплекса олефина с молекулой орома,
который может превращаться в бромонин одним пз двух изоора-
женных ниже путей:
Как бы то ни было, при любом из этих путей реакция должна про-
ходить через трехцентровое переходное состояние, предшествующее ста-
дии циклического бромониевого катиона, и при размыкании последнего
бром-анионом дать эффект вальденовского обращения.
Многочисленные реакции изомеризации углеродного скелета типа пи-
наколиновых и ретропинаколиновых перегруппировок (см. стр. 610)
также идут через трехцентровое переходное состояние:
ZR.
R—С—
+
LH О"
R
R
R
>- R-C-C-R
К I
пинаколиновая перегруппировка
СТРУКТУРА ПЕРЕХОДНОГО СОСТОЯНИЯ
595
рстропинаколиновая перегруппировка
Отсутствие карбкатиона в подобных процессах доказано Д. Н. Кур-
сановым тем, что во время перегруппировки соединение не обменивает
водород на дейтерии из среды (D2SO4, D2O), как это свойственно карб-
катионам.
Четырехцентровое (четырехчленноё) переходное состояние фигурирует
в реакциях димеризации аллена и тетрафторэтилена, в которых обра-
зуются циклобутановые производные:
СН2=С=СН2 И2С—С=СН2
2СН2=С=СН2---> : ' ---> | |
сн2=с=сн2 сп2=с-сн2
f2c=cf2 f2c-cf2
2CF2=CF2 —> i i > I I
f2c=cf2 f2c-cf2
Весьма возможно, что присоединение литий- и магнийорганических
соединений к карбонильной группе также идет через четырехцентровое
переходное состояние.
Многочисленные реакции 1,3-диполярного присоединения к олефинам,
ацетиленам, нитрилам и т. д. (стр. 279), как доказывает Хьюсген, идут
через пятицентровое переходное состояние. Для того чтобы было ясно,
о чем идет речь, повторим несколько примеров циклизации этого типа:
|ch2=n=n —>- СН2—n=n] + СН=СН------->
_ XNH
Н2С XN Н2С N НС N
--> : ; > I I -----> II II
HC = CH-------------------HC = CH-HC—CH
пиразол
C6H5-N=N=N > C6H5—N—N=n] + R-C=N
c6h5-n n cgh5-n n
> | I
R-C^N r-c=n
дизамещенный
тетразол
38*
МЕХАНИЗМЫ ХИМИЧЕСКИХ РЕАКЦИЙ
596 _______________________________—-----
+
с6н5-с / ~
Н2С = СН—С\ос?1 !5
zNx
Н2С С ^ОС2Н5
3-фен iiJi-5-к арбэтокси-
изоксазолин
По Хьюстону, многочисленные подобные циклизации протекают би-
молекхлярно п стереоспецпфично. Так, циклы, получающиеся, например,
из "wuc-дифени л этилена, имеют фенилы в ^нс-положении; та же стерео-
специфичность свойственна и реакциям транс-олефинов. Мыслимы два
механизма реакции. На примере присоединения диазометана к олефину
они изобразятся так:
II
R
- zN\
Н2С''
Путь а предполагает непосредственное присоединение через пяти-
центровое переходное состояние, путь б — присоединение в две стадии,
сначала одним из концов диполя (в данном примере крайним азотом ди-
азометана), затем замыкание цикла. Оба процесса должны идти как би-
молекулярные, поскольку определяющим скорость (медленным) про-
цессом должны быть бимолекулярные реакции а и б. Против пути б гово-
рит практически одинаковая скорость присоединения в средах с резко
разнящейся диэлектрической проницаемостью. Высокая диэлектрическая
проницаемость среды будет способствовать стадии б, так как в результате
этой стадии образуется диполь с удаленными (+)-и (—)-зарядами, раз-
деленными предельным углеводородным звеном. Кроме того, высокая
диэлектрическая проницаемость среды должна способствовать ионизации,
уменьшая требуемую энергию. Путь а независим от свойств растворителя-
Второе соображение в пользу пути а — сравнительно низкие эптальпии
активации и высокие энтропии активации такого рода процессов, что
характерно для циклического переходного состояния, как это будет по-
казано далее.
„ подчерк11Ул Я- К- Сыркин, особенно широко распространены
переходные состояния с шестью центрами. Довольно очевидно, что к ним
СТРУКТУРА ПЕРЕХОДНОГО СОСТОЯНИЯ
597
относятся известные нам реакции Дильса — Альдера, так же как ди-
меризация диенов по Лебедеву:
НС^
I
нсч
Декарбоксилирование [3-кетонокислот, как показали Вестхеймер
и Джонс, идет через шестицептровое циклическое переходное состояние
и приводит к образованию енолов кетонов:
Н н
сх
СНз-С^С^О ___ ~^CHi
IK, -----> СО2 + сн3—
он
н I
/Сн3
СНз-С
о
Через шестичленное циклическое переходное состояние проходит
и клайзеновская перегруппировка аллилового эфира фенола в о-аллил-
фенол, для которой методом изотопного обмена доказано привязывание
аллильной группы к бензольному ядру у-углеродным атомом:
Реакция Кольбе и аномальная реакция хлористого бензилмагния
с формальдегидом явным образом идут через шестицентровое переходное
состояние (включая ионную пару):
Na+
МЕХАНИЗМЫ ХИМИЧЕСКИХ РЕАКЦИЙ
598
В этих случаях особенно наглядно циклическое перемещение электро-
нов, типичное вообще для циклических переходных состояний.
Я. К. Сыркин предполагает, что течение многих реакций кислотно-
щелочного катализа осуществляется также через шестичленный активи-
рованный комплекс. Сюда относится, например, дегидратация спиртов,
в которой течение реакции а может с успехом конкурировать с течением
реакции б через карбкатион:
/СН2— IK + а ^СН2 +
(СН3)гС 'ОН2 — -> (СИзНС + ОНз
ХО----IIZ 1U0
н
СПз б + . СНз н q
(СНз)2С —> (С11;!)2С —> (СПз)2С-СНг4-Н*
\ои
н+
Образование такого же рода циклов с участием воды, гидроксоний-
катиона или гидроксильных ионов можно предположить и для многих
других процессов кислотно-основного катализа.
Циклическое переходное состояние Я. К. Сыркин предлагает для
щелочного и кислотного гидролиза сложных эфиров и т. п.
R R'
0=С— О
-о^ ;н —>
н—o'
н
R R'
"О + II
Н20
R'°\ z°— Н\+/Н
>cz X ~ *
r/ Ro—if' хн
I
н
R'ox .он
/С'
R ХОН
R ОН
/ с~ °\
и далее R' —Qz Н
RH—О''
I
н
R\
/С==0
--> НО 4- н20
R'—он
для реакции Канниццаро (третий порядок;
первый по щелочи)
второй по бензальдегиду»
R
□ = С
“О
4- CH2-R
но
СТРУКТУРА ПЕРЕХОДНОГО СОСТОЯНИЯ
590
для альдольной конденсации в кислой среде
НзС. zO Н, /С н/ ' СН2—II' + ,н оС — \Ц ОН 1 * СНз-СН + ОНз 1
1 СН2—СН=О
сн=о
в щелочной среде
ZCH2—С—И а
1Г О * СН2=СН—ОП-^-ОН" (стадия енолизации)
'О'—Н-'
ОН
6 1
СПз—СН /° сиз-СН
'сп2=сп сн2—сн=о
сн2=-сн-он
. б, СПз—сн-он
или 1Г СН2—СП-0 —> | +О1Г
\q- —н/ СНг— СН=О
При подходящих скоростях стадий енолизации и собственно конденса-
ции в соответствующих вариантах по Сыркину могут получиться константы
в случае (а — б) третьего порядка (суммарно) и в случае (а — б') даже
четвертого порядка. В действительности щелочная альдолизация и дает
порядки между третьим и четвертым. Конечно, такие циклические акти-
вированные комплексы не более чем гипотеза.
Как видно из приведенных схем, в большинстве реакций, текущих
через циклические переходные состояния, происходит синхронное по циклу
передвижение электронных пар связи. Благодаря этому, естественно,
снижаются энергетические затраты, так как не требуется предваритель-
ного «взноса» энергии на разрыв связей одного или обоих реагентов для
превращения их в богатые энергией свободные радикалы или ионы. Уже
по одной этой причине энергия активации реакции, текущей через цикли-
ческое переходное состояние, ниже, чем той же реакции, идущей не через
циклический комплекс.
Напомним, что константа скорости к реакции, текущей через переход-
ное состояние, выражается формулой:
AS* АН*
к = е Н е ВТ
il
где Т — температура в °К; и, h и R — константы; Д5* и ДЯ* (приращения
энтропии и энтальпии переходного состояния сравнительно с исходным
состоянием). Приращение энтальпии в этом случае очень близко к энер-
гии активации. Таким образом, уменьшение Д/7* переходного состоя-
ния резко увеличивает скорость реакции. В этом и состоит влияние
циклического переходного состояния. По Сыркину, это влияние может
быть еще больше, если цикл активированного комплекса обладает квази-
ароматическим характером и вследствие этого еще более низким энерго-
содержанием. Однако цикл в силу жесткости построения обладает боль-
шей энтропией (и тем самым менып'й вероятностью), чем нециклическая
600
МЕХАНИЗМЫ ХИМИЧЕСКИХ РЕАКЦИЙ
система. Этот фактор действует в противоположном направлении и также
экспоненциально.
Такого рода энергетические соображения дополняются суждениями
о малой вероятности участия в реакции частиц, образование которых
требует большей затраты энергии, например несольватированных карб-
катионов или карбанионов, отщепление Н+ от Н3О+, и вообще предва-
рительного, ранее стадии образования активированного комплекса, раз-
рыва (гомолитического или гетеролитического) прочных связей с низким
знергетическим уровнем. В этом случае сразу возрастала бы Д//* акти-
вированного комплекса и скорость реакции уменьшалась бы до таких
величин, что реакция фактически бы не шла.
Легко видеть, что в синхронно протекающих циклических процессах,
подобных описанным выше, граница между нуклеофильными и электро-
фильными атаками становится условной, а понятие о реакционном центре
делается менее определенным, поскольку их появляется одновременно
несколько. Для иллюстрации этого положения стоит еще раз привести
в обобщенном виде схемы реакций, протекающих через циклическое
переходное состояние:
а—в
С~ D
л=в^
Е
Очевидно, что в циклическом активированном комплексе каждый
из атомов — его звеньев — является реакционным центром. О направле-
нии переходов электронных пар (кривые стрелки) мы судим главным обра-
зом на основании знания относительной электроотрицательности атомов.
В схеме 1а стрелка к D ставится на основании знания, что D более электро-
положителен, чем В, а А> С. Таким образом, В совершает в цикле нуклео-
фильную атаку на D, а С — на А. Поскольку энергетический эффект
реакции не зависит от пути перехода, то он будет таким же, если допу-
стить, что происходит не перемещение электронных пар, а гомолитическое
перемещение электронов. Например, для схемы 1а (пунктирные стрелки —
перемещение одного электрона): __R
A lx О
с —D
Схемы, подобные I и II, с гетеролитическим передвижением электронов
принимаются по аналогии с нециклическими, сходными по характеру
процессами. Например, реакция Кольбе и, очевидно, аналогичная ей
аномальная реакция хлористого бензилмагния с формальдегидом близки
к обычным реакциям электрофильного замещения в ароматическом ядре,
а значит, и перемещение электронов (их пар) аналогично во всех этих
реакциях.
РЕАКЦИИ ЭЛИМИНАЦИИ
В начале разделов об олефинах и ацетиленах мы рассмотрели ряд реак-
ций, ведущих к образованию углерод-углеродной л-связи. Это реакции
в известном смысле обратные реакциям присоединения по кратной
С—С-связи. Они носят название реакций ft-элиминации или Q-отщепления.
Среди многих реакций элиминации особенно большое значение имеют:
отщепление галоидоводорода от моногалоидпроизводных и гофмановская
деструкция — отщепление воды и третичного амина от гидроокисей чет-
вертичных аммониев. Касающееся первой из этих реакций правило
А. М. Зайцева (1875 г.) гласит: отщепление галоидоводорода и образо-
вание двойной связи происходит так, чтобы с непредельными углеродами
было связано максимально возможное число алкильных групп.
При гофмановской элиминации, напротив, предпочтительно отще-
пляется водород метильной группы, так что в большинстве случаев обра-
зуется группировка = СН2.
Кинетическими исследованиями Хьюза и Инголда было установлено,
что в зависимости от условии реакция Зайцева может идти двумя путями.
В сильно ионизирующих растворителях, например в спиртах (кн. I,
стр. 103), третичные и частично также вторичные галогениды способны
к медленной электролитической диссоциации. Карбкатион может затем
или соединиться с анионом или выкинуть протон и дать олефин. Две
реакции конкурируют между собой:
(СП3)2С1К /СН3 (СНзВСП. +
>С< —► >С— СПз —► (СН3)2С=С-СНз+Н+
(СН3)2С11/ ХС1 (СНз)2С11/ |
СН(СН3)а
I +C2HSOH
(СПз)2С11ч ..СПз
/С< 4-п +
(СПз)2СЦ/ хос.2н5
Отщепление галоидоводорода — реакция мономолекулярная. Ее ско-
рость, так же как и скорость сольволиза в целом, определяется скоростью
РЕАКЦИИ ЭЛИМИНАЦИИ
602
медленной стадии ионизации. Такая элиминация обозначается симво-
Если на то же галоидприизводиос действовать алкоголятом натрия,
реакция элиминации идет бимолекулярно и ооозначается Е 2. В этом
случае скорость реакции, следовательно, пропорциональна и концентра-
ции галоидпроизводного, и концентрации алкоксид-иона. Ориентация
остается прежней, подчиняющейся правилу Зайцева. ~ о „
Причина, лежащая в основе правильности, найденной Зайцевым,
та же, что в основе перемещений двойной связи с края в центр молекулы
при пропускании олефина над катализатором типа окиси алюминия:
термодинамически устойчивее те олефины, непредельные атомы которых
связаны с алкилами, а не с водородами. Здесь играет роль явление гипер-
конъюгации, или а,л-сопряжения (кн. I, стр. 425). Напомним, что в си-
стеме
Н Н
н—с—сп=сн—с—п
I I
II II
о-связи метильной группы сопряжены с л-связью, а это влечет за собой
небольшое понижение энергии.
Интересно, что бимолекулярное отщепление Е2 протекает, в отличие
от Ег, синхронно, т. е. атака алкоксид-иона на (В-водородный атом ф по от-
ношению к галоиду) и отрыв его в виде протона совершаются одновре-
менно с выбросом галоид-аниона:
СГ
Н\ . С1
н-^с—с~н —>
Н н
Нх /II
/С=СХ
II \Ц
с2н50-
С2Н5ОН
Это было доказано тем, что в среде, содержащей обменоспособный
дейтерий, например в C2H5OD, карбанион СН2—СН2С1, если бы
он существовал сколько-нибудь заметное время, должен" был бы всту-
пать в дейтерообмен и давать CDH—СН2С1. Из него выбрасы-
вался бы С1“ и в результате в^ качестве примеси получался бы дейте-
рированный этилен \с=С<^ . Между тем этилен получается чистый,
без примеси дейтерированного.
Гофмановское отщепление — также бимолекулярная реакция и от-
косится к типу Е2. По этому типу протекает деструкция не только гидро-
ЧеТВеРТИЧНЫХ аммОниев’ 110 и сульфониев и фосфониев. В случае
ег? га отщепления основание слабее (ОН" вместо OR"), и атака
Г ^ГМТСЯ на СаМЫЙ легК0 прозаизируемый водород. Например,
в случае аммониевого основания 11
N(CH3)3
R-CH2-CII—С1Г2-П
3 а р'
РЕАКЦИИ ЭЛИМИНАЦИИ
603
следует сравнивать кислотность водородов а, р, р'. Хотя легче всего
протонизируется а-водород, но его удаление никакого отношения к от-
щеплению группы N(CH3)3 иметь не может, так же как и вообще к де-
струкции молекулы: захват протона из среды тотчас компенсирует его
потерю и вернет молекуле первоначальное строение. Таким образом,
сравнивать надо водороды р и Р'. Если R — алкил, то его +/-эффект
делает Р-водород менее кислым, чем Р'-водород. Атака направляется
на последний, и в результате получается олефин RCH.2CH=CH2 («анти-
запцевский»). Если R проявляет — I- пли —У-эффект, например если
это галоид, нитрогруппа пли остаток бензола, то кислее будет Р-водород,
и олефин будет иметь структуру R—СН=СН—СН3.
Однако эта игра полярностей не единственный фактор, определяющий
направление элиминации. Второй важный фактор — пространственный.
Он важнее при гофмановской элиминации, так как группа NR3 всегда
объемистее, чем галоид. При синхронной элиминации молекула, под-
вергающаяся элиминации, всегда принимает в переходном состоянии
такую конформацию, что отщепляющийся протон и уходящий атом или
группа [С1 или N(CH3)3] находятся в транс-положении. Иногда это объ-
ясняют по аналогии с вальденовским обращением тем, что входящий за-
меститель (в данном случае углеродный атом, привязываемый л-связью)
должен присоединиться со стороны, противоположной отщепляемой
группе NR3 или С1~
Н
—с —с^-— ------> н + +CI-
С1
Однако такое объяснение несколько натянуто.
На примере RCH2C(CH3)2—X рассмотрим переходные состояния
и пространственные затруднения в элиминациях обоих типов (кроме
обычных формул приведены их ньюменовские проекции):
Зайцев ск ое отщепление Гофмановское отще-
п л е н и е
СН3О"
Н ^СН3
Р-О^-С—СН3
/ ХС1
п С1
ОН-
Н /CH2R
Н—с^-с —сн3
Н (_N(CH3)3
5
СН;)О-
а'
РЕАКЦИИ ЭЛИМИНАЦИИ
604
В зайцевском отщеплении, как видно на проекции а , группа 7?, если
она объемиста, мешает и СН3, и уходящей группе —• галоиду, еще боль-
шими были бы пространственные затруднения, если бы уходящей группой
была объемистая N(CH3)3. На проекции б' видно, что при иной конформа-
ции переходного состояния того же соединения пространственных помех
нет, и это способствует гофмановской ориентации отщепления, причем
в случае объемпстой группы N(CH3)3 пространственный фактор становится
категорическим.
Многочисленные элиминации диаммониевых солей, в частности не-
предельных, исследованы А. Т. Бабаян. Рассмотрим одну из этих реак-
ций, приводящую к пропенилацетилену:
СНз
+ I +
--- (CH3)3N—СН2—С=СН—СН2— N(CH3)3 —
ОН- он-
СНз СН3
I + он- + I он-
CH2=C-CII--=CH—N(CH3)3 j (CH3)3N—CII=C—CH=CII2 1
I OH- CH? 1 CH3
1 I /9
CHs (CH3)2N—CH=C—CH=CH2 ch3—ch=ch—c?
ch2=c-c=ch H
Требование предпочтительной транс-конфигурации обеих отщепля-
емых групп в случае элиминации от олефинов превращается в требование
о предпочтительной трапе-конфигурации отщепляемых групп, связанных
с непредельными атомами. Примером может служить исследованное
А. Н. Несмеяновым, А. Е. Борисовым и И. С. Савельевой отщепление
HgCl и С1 от транс- и г/нс-хлормеркурвинилхлоридов:
Н\/С1
с
II
с
ciiig\
и\/с1
С 31-
I) ----->
С
H^IgCl
II
с
I
II
°бе реакции протекают уже при комнатной температуре но тпанс-со-
Авторы обменяют ото л?“ииМи
Р странствепными условиями о,а-сопряжения связей Но- Г « Г—С1
» ^роке-соединении. Действительно, и другие рв“Х Йщенлёння от
реакции ЭЛИМИНАЦИИ
605
олефиновых углеродов проходят легче при тпранс-конфигурации отщепля-
емых групп.
Следующие два примера показывают, как /пранс-элиминация диктует
такую конформацию переходного состояния, что отщепление протекает
стереоспецифично и конфигурация получающегося олефина оказывается
определенной — либо цис-, либо транс-.
Пример 1.
нет
Н Аг '
Ar -С—С^-Н
в/ Вг
мезо-стереоизомер
НО'
н н
Аг — С—С^“Лг
вХ В г
только цчо-
D- или 6-стереоизомер
только транс -
Пример 2. НО"
трео-стереоизомер
только транс-
НО"
н
эршпро- стереоизомер
гольмо цис—
РЕАКЦИИ ЭЛИМИНАЦИИ
В примере 2 гофмановскому отщеплению подвергаются трео~
и эритро-днастереомеры вещества Ar(CH3)CHCHArN(CH3)3OH . Опять-
таки отщепление происходит в той конформации, в которой отщепля-
ющиеся группы Н и N(CH3)3 находятся в транс-положении, и отщепление
идет стереоспецифично: mpco-изомер образует только транс-олефпи,
а эритро--только гщс-изомер. Однако в последнем случае реакция идет
в десятки раз медленнее. Такое замедление объясняется пространствен-
ными помехами в переходном состоянии, создаваемыми двумя объемистыми
арильными группами друг другу и необходимостью преодолеть эти по-
мехи затратой большей энергии активации.
Интересно отметить, что пары соединений в примерах 1 и 2 отличаются
друг от друга противоположным зарядом одной из отщепляющихся групп
[Вг- и N(CH3)3]. Следовательно, требование траис-расположения эли-
минируемых групп нельзя свести к электростатическим причинам.
Из более редких реакций грш-элиминации назовем пиролиз сложных
эфиров карбоновых кислот и пиролиз эфиров ксантогеновых кислот (реак-
ция Л. А. Чугаева). Эти реакции проходят по схеме:
Н
I
R—С—Н
R-C-O-C=S
Н SCH3
R\/H
to С
;l +COS+CH3SH
RZ\
как
Обе элиминации протекают
вапии'^ яЧ "ST внУт₽им°лекулярно/ Отрицательная антр
’ Р' 94) 5'M3UE,»f‘T’ "ТО в переходном состоянии об
более жесткая структура, чем исходная, т. е.
предполагают такие циклические перехолные'
для обеих реакций:
реакции первого порядка, и, значит,
г энтропия акти-
—.....................образовалась
произошла циклизация,
состояния (стр. 593 ел )
реакции элиминации
607
Элиминации возможны, конечно, не только от углеродных атомов.
Примеры ^-транс-элиминаций мы уже встречали. Напомним элимина-
цию Н и ОН от анти- (т. е. транс-') альдоксимов и аналогичную бекма-
новскую перегруппировку II рода:
—С—Н R—С—Н R —
| +Ar-SO2Cl > || —> HI +Ar-SO2-O-H-
i-N Ar—SO2ON
R—C-R' R-C-R' R— C
II -гAr—SO2C1 > || ---> HI -J-Ar—SO2-OR'
НО—X Ar—SOoO—N N
До сих пор речь шла о |3-элпминации. Существуют, однако, и реакции
отрыва двух групп от одного углеродного атома, или а-элиминации.
Обычно они приводят к образованию карбенов (стр. 570) или продуктов
пх перегруппировки (стр. 625).
Все сказанное о протекании реакций элиминации в открытых системах
справедливо и для циклических структур, но особенности экваториального
и аксиального положения заместителей налагают здесь новые тре-
бования.
Очевидно, что обе элиминируемые группы в цикле должны находиться
в 1,2-положении. Если они располагаются в z^wc-положении, тогда не-
избежно одна из них аксиальна, а другая экваториальна, т. е. они не лежат
в одной плоскости, и реакция Е2 не происходит. Если же заместители
транс-расположены, то они или оба экваториальны, или оба аксиальны,
причем эти две конформации легко переходят друг в друга. Аксиально-
аксиальная конформация благоприятна для элиминации.
Приведем по Инголду и Хьюзу примеры дегидрохлорирования двух
стереоизомерных хлорпроизводных циклогексанового ряда — ментилхло-
рида и неоментилхлорида:
Ментилхлорид
608
РЕАКЦИИ ЭЛИМИНАЦИИ
неоменти.та.торид
Ментилхлорид может претерпеть отщепление НС1 только в единствен-
ной конформации, в которой Н и С1 аксиальны и лежат в одной плоскости
(эта конформация и изображена выше). Однако переходу в такую кон-
формацию препятствует аксиальное положение изопропильной группы —
такие объемистые группы предпочитают экваториальную конформацию,
поэтому реакция течет медленно. В неоментилхлориде дело обстоит по-
иному: благоприятная для отщепления конформация стабилизована
экваториальным положением изопропила, и реакция течет быстро, пре-
имущественно в сторону образования циклоолефина II.
ПЕРЕГРУППИРОВКИ
Самое название «реакции замещения» предполагает, что замещающая
группа вступает на место замещаемой. В огромном большинстве случаев
так именно и происходит. Структура меняется лишь у того атома (или ато-
мов), на который направлена атака реагента и у которого происходит
реакция. Это правило наименьших структурных изменений служит ком-
пасом при установлении строения химическим путем. Однако известны
и исключения из этого правила, и мы уже встречались с такими реакциями,
в которых заместитель вступает в молекулу не на место уходящей группы,
а к другому атому, особенно часто — к соседнему с тем, от которого оторва-
лась уходящая группа. Такие реакции — они называются перегруппиров-
ками — вызвали немало затруднений при установлении строения, и есте-
ственно, что их пытались объяснить протеканием реакции через ряд таких
стадий, в которых не нарушается принцип наименьших структурных
изменений. Эти попытки, однако, в общем оказались несостоятельными,
и было доказано, что по крайней мере многие перегруппировки совер-
шаются внутримолекулярно в одну стадию. Большинство известных
перегруппировок относится к гетеролитическим реакциям.
ПЕРЕГРУППИРОВКИ С ИЗМЕНЕНИЕМ УГЛЕРОДНОГОЧЖЕЛЕТА
Мы упоминали (кн. I, стр. НО и 558) о пинаколиновой перегруппировке
(Фпттиг, А. М. Бутлеров), происходящей при действии кислот, в част-
ности и лыоисовых, на пинаконы. В общей форме ее] можно выразить
так (R = углеводородные радикалы):
R. /R н
с—с. —
r'x I I R'"
но он
R. + /R
,С— С.
r'z I R'"
НО in
+н2о
Л /R"
---> R—С — С
1 4Rm
IV
Н
39 Заказ 672
610
ПЕРЕГРУППИРОВКИ
Расчленение на стадии здесь дано искусственно, для ясности. На деле
это синхронный процесс, в котором IV выражает переходное состо-
яние.
Подробно изучено, какие радикалы в этой перегруппировке легче
мигрируют (Тиффено, А. П. Орехов, Бахманн). В порядке легкости мигра-
ции радикалы располагаются в следующий ряд:
п-СП3ОС6Н4 > п-СН3СвН4 > С6Н5 > п-С1СвН4 > Алкилы > Н
Общий вывод таков: легче мигрирует радикал, в наибольшей степени
рассредоточивающий положительный заряд, появившийся в системе
в результате отрыва ОН~, или, другими словами, легче мигрирует ра-
дикал, наиболее способный подвергнуться электрофильной атаке.
То обстоятельство, что начальным моментом перегруппировки является
отрыв гидроксильного аниона и образование карбониевого катиона (в пере-
ходном состоянии тотчас рассредоточивающего свой заряд между С,
от которого оторван гидроксил, соседним С скелета и ключевым G мигри-
рующей R'-группы *) доказывается следующими аналогиями:
R \ zR' zR'
;С—С< +Ag+ > AgBr + R-C-C^-R'+П+
Rz/ I I R"' || \R'"
НО Вг О
(Здесь даны только исходный бромгидрин и конечный пинаколин;
механизм, очевидно, тот же.)
Еще одна аналогичная перегруппировка — «окислительное дезамини-
рование» Мак-Кензи:
VI
Движущей силой реакции является отщепление молекулярного азота,
а содействующим фактором — легкий отрыв протона от кислорода что
в итоге приводит к стабильной молекуле.
аром*атична?ЖН°’ ТйКЖе Внут₽и ГРУППЫ В', если она, например, непредельна или
ПЕРЕГРУППИРОВКИ G ИЗМЕНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
6И
Общий механизм реакции можно выразить одной схемой синхронного
превращения:
rf
нг
911
N
На примере окислительного дезаминирования впервые установлено
и стереохимическое течение перегруппировки пинаколинового типа.
Мигрирующий радикал R', конечно если с углеродом, несущим гидроксил,
связан асимметрический атом этого радикала, сохраняет свою (правую
или левую) конфигурацию. Группировка же вокруг углеродного атома,
к которому мигрирует R'. претерпевает вальденовское обращение (что
можно констатировать, конечно, только если исходный аминоспирт был
выделен в стереохимически индивидуальной форме). Все это безусловно
доказывает, что промежуточный карбкатион не появляется в свободном
состоянии: карбкатионы имеют плоскую конфигурацию, и когда они
фигурируют как промежуточные стадии реакции, т. е. когда реакции
разыгрываются по механизму iS'jvl, обычно происходит полная или
далеко идущая рацемизация.
Таким образом, стереохимическое течение пинаколиновых перегруп-
пировок такое же, как в реакциях S^2. Это как бы SN2-замещение групп
ОН или Вг (в окислительном дезаминировании — диазониевой группы
N=N) у несущего эти группы углеродного атома на радикал R' от
соседнего углерода.
В первых двух случаях выведению групп ОН и Вг благоприят-
+
ствует действие Н+ или ZnCl2 (льюисова кислота) и Ag, в третьем случае
перегруппировке содействует самопроизвольный распад непрочного али-
фатического диазония. Пинаколиновой перегруппировке содействует
также + Z-эффект гидроксила, благодаря которому реализуется превра-
щение спиртовой группы пинакона в кетонную группу пинаколина. Сле-
дует еще пояснить формулы IV и IX. Они изображают переходное состо-
яние, в котором R' еще не оторвался от материнского углерода, но уже
начал связываться с углеродом, принимающим мигрирующую группу.
Оба эти углерода и R' связаны трехцентровыми электронными орбиталями.
Если R' = Н, то речь идет о гидридном перемещении (стр. 635). Следует,
однако, напомнить, что, по данным Тиффено, Орехова и Бахманна
(стр. 610), водород мигрирует труднее, чем алкилы или арилы.
В отношении радикала R' процесс должен рассматриваться как элек-
трофильное замещение связанной с ним группы
39*
ПЕРЕГРУППИРОВКИ
612
Пинаколиновые перегруппировки многократно использовались
для изменепия структуры алициклов, расширения или сужения цикла.
Например:
Н. Д. Зелинский и
Н. И. Шуйкин осуществили следующий синтез
спиранового кетона:
Направление реакций в сторону образования производного цикло-
гексанона или циклопентильного кетона зависит, в частности, от харак-
тера R, но также и от стереохимии цикла.
Поскольку перегруппировка происходит с вальденовским обращением
у принимающего углерода, мигрирующий радикал должен поступать
со стороны, противоположной уходящей группе (НО, Вг, N=N в при-
веденных выше примерах). В случае фиксированного в цикле простран-
ственного расположения этих групп по отношению к остающемуся гидро-
ксилу, углеродному радикалу и СН2-группе цикла геометрический фактор
сказывается и иногда направляет реакцию по-разному. Пример (Куртин):
ПЕРЕГРУППИРОВКИ С ИЗМЕНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
613
Пинаколиновая перегруппировка проходит только для цис-амиво-
спирта (диазотированного азотистой кислотой): транс-аминоспирт сохра-
няет свой скелет.
Вторым основным типом перегруппировки с изменением углеродного
скелета является ретропинаколиновая перегруппировка. В 1901 г. Н. Д. Зе-
линский при дегидратации кислотой пинакол инов ого спирта вместо ожида-
емого 2,2-диметилбутена-З получил 2,3-диметилбутен-2:
СН3\ /СНз СН3\ /СНз
СНз-дС-СУ )С=С< +н20
СН3/ /\ СНзХ ХСН3
но н
Поскольку здесь, в противоположность пинаколиновой перегруп-
пировке, скелет из пинаколинового превращается в симметричный, как
у пинакона, перегруппировки этого типа называются ретропинаколино-
выми или иногда перегруппировками Вагнера — Меервейна, поскольку
первой разъясненной перегруппировкой этого типа была открытая
Е. Е. Вагнером в 1899 г. перегруппировка пинена в хлористый борнил
впоследствии (1910 г.) подробно изученная Меервейном и сопоставленная
им с простейшими примерами этого типа перегруппировок. На гетеро-
литический и, более того, ионный характер этих перегруппировок указы-
вает то, что они протекают гораздо быстрее в сольватирующих раство-
рителях с высокой диэлектрической проницаемостью. Как и для разных
типов пинаколиновых перегруппировок, покидающая молекулу группа
может быть не только гидроксилом, но и галоидом, азотом алифатического
диазония и сложноэфирной группой, например тозилатной. Примеры:
СН3\ Ag+ С1Ь + СН3\ +
СН3-7С-СП2ВГ —СНз-^С-СПг > >С—СН2-СН3
СН3/ СН3/ СНзХ
I II III
+HtO
-Н+ |
+
СПз
СПз
СНз\
>С-СН2-СНз
С Из7 I
(Уитмор)
ОН
IV
ПЕРЕГРУППИРОВКИ
614________________________
СНз\
СНз-С-СН-СПз
СНз/ I
Cl
VI
СПз /СНз
/С=С<
CIV чСПз
/СПз
с-с^-н
| ^СНз
С1
VIII
>С11-сп-*спз СНз)сн-сн-*сн3—>
z I Rz I
NH2 X К*
IX
СНЗ )СП-СП-*СИз СНз-СН-СН^ —-
в/ х*СНз
XI ХП
СН3\ /Н
—> )СН-С11-*С11з + СНз-СН-СН<
R/ | I \*СНз
он он
ХШ XIV
где *С —изотоп углерода 14С.
Нельзя считать доказанным, что во всех ретропинаколиновых перегруп-
пировках не фигурирует свободный карбкатион: стереохимическое течение
реакции известно не везде. Напротив, во многих случаях вполне вероятны
равновесия, подобные XI XII. Некоторым аргументом в пользу участия
свободного катиона может служить то, что в большинстве случаев резуль-
таты перегруппировки указывают на предпочтительное направление
реакции в сторону образования более устойчивого карбкатиона, т. е.
с более компенсированным положительным зарядом. Так, из катионов II
и III более устойчив последний, так как его положительный заряд по-
давляется -{-/-эффектом трех алкильных групп, тогда как в катионе II
-{-/-эффектом всего одной mpem-бутильной группы. Вообще по этой при-
чине карбкатионы с зарядом на третичном углероде устойчивее катионов
с зарядом на вторичном, а последние устойчивее карбкатионов типа — СН2.
Однако в случае В = С6Н5 формулы XI и XII предложено (Крам, 1949 г.)
заменить на XV — промежуточное соединение — фенониевый катион
СН3
с6н<
СН-СН-СНз—> сн3—сн—сн—*сн3
N
N
XV
X
ПЕРЕГРУППИРОВКИ С ИЗМЕНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
615
с равновероятным раскрытием цикла при действии на него воды и обра-
зованием соединений XIII и XIV.
Понятие о фенониевом катионе впервые ввел Крам с целью объяснить
стереоспецифическое поведение в ретропинаколиновой перегруппировке
двух диастереоизомеров З-фенил-2-бутилтозилата. При ацетолизе трео-сте-
реоизомер XVI полностью рацемизуется, а эритро-стереоизомер XVII
превращается в ацетат, сохраняющий в полной мере оптическую актив-
ность. Крам постулирует в качестве промежуточной формы в течение
перегруппировки структуры XVIII и XIX, включающие фенониевый
катион, привязанный симметрично к обоим углеродным атомам — старто-
вому и финишному:
СН3
Н----OTs
с6н5-|—н
СН3
XVI
трео-
СН3СООН
XVIII
сн3 сн3
Ас О----Н Н-------------ОАс
4*
н С6Н5 С6н5-----------н
сн3 СНз
антиподы трео-
(рацематД
СН3
Н----OTs
Н—С6Н3
СН3
вальд.
обр.
СНдСООН
-----Z-->
вальд. о-гр.
СН3
Н---ОАс
Н----С6Н5
XVII XIX оритро- (сохранение
конфигурации)
эритро-
где Ts= — S—СВН4СН3; Ас= —С—СН3
II 6 43 II
о о
Углеродный атом ароматического кольца, связанный первоначально
со стартовым атомом С, в трехчленной промежуточной форме стал тетра-
эдрическим. Значит, плоскость бензольного кольца стала перпендикуляр-
ной к плоскости чертежа. Положительный заряд финишного атома С
теперь рассредоточен в триаде углеродных атомов трехчлена, а следова-
тельно, и по всем атомам бензольного цикла, что, по Краму, снижает
энергетический уровень этой переходной формы и тем самым ускоряет
реакцию.
Разрыв под действием уксусной кислоты гипотетического трехчлен-
ного катиона в случае трео-изомера XVI, имеющего плоскость симметрии,
должен вести к рацемату, так как разрыв равновероятен слева и справа
(или, иначе, две симметричные молекулы при реакции не могут дать одной
оптически активной формы, но лишь рацемат). Напротив, в случае
ПЕРЕГРУППИРОВКИ
616
<?nizwpo-n зомера XVII ацетолиз несимметричной трехчленной структуры,
с какой бы стороны ни шла атака уксусной кислоты, приводит к исходной
конфигурации.
Следует обратить внимание па то, что и замыкание в трехчленный цикл
с удалением тозилат-апиопа, и разрыв трехчленного цикла с возвращением
фенонпевого катиона в бензоидную форму происходит с вальдеповским
обращением. В. Хюккель, оспаривающий трактовку Крама и самое суще-
ствование фенонпевого катиона и трехчленного цикла, указывает, что
стереоспецифичность рассматриваемой реакции можно объяснить просто
переменой местами фенильной группы с тозилатной (или с заменяющей ее
ацетатной) с двумя вальдеповскими обращениями:
СИ.»
II---О Л
—>
Свп6----II
СНз СНз
С6П5---II АО —|—И
= I
II--ОЛ II------СвН5
СПз
XVI
тпрео»
СНз СПз
XVIa
трео-аптпнод
СПз СПз СПз
XVII
tpumpo-
XVI la
вритро-
(сохрансинс
конфигурации)
Здесь ОА обозначает как уходящий тозилат-анион, так и вступающий
(в опытах Крама) ацетат-апиоп.
Само собой разумеется, что в условиях реакции ацетат XVIa может
перегруппировываться снова в ацетат XVI, так что рацемизацию обуслов-
ливают оба процесса XVI XVIa.
Возражения Хюккеля основательны, и широко распространенная
ныне концепция фенопиевого иона как промежуточной формы в пере-
группировках не может считаться бесспорно принятой в пауке.
Реакции, где мигрирует ароматическая группа, а не алкил или водород,
по мнению сторонников гипотезы фенопиевого катиона, протекают в сотни
и тысячи раз быстрое именно благодаря относительно низкому энерге-
тическому уровню фенопиевого катиона с его рассредоточенным по многим
атомам положительным карбониевым зарядом. Возражение Хюккеля
в этом случае состоит в том, что структуры типа XVIII и XIX по существу
являются «классическими» и никак не могут рассматриваться как переход-
ное состояние, а скорость реакции определяется только энергетическим
ПЕРЕГРУППИРОВКИ С ИЗМЕНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
617
уровнем переходного состояния (стр. 593). В самом деле, стабильные ка-
тионы подобного типа мы встречали, например, в видр.
СНУ СН.,
<о<СНз
7^\ снз
СН3 СН3
А1СЦ
при описании реакции Фриделя — Крафтса (стр. 37). Таким образом,
структуры XVIII и XIX отделены от начальных и конечных продуктов
реакции энергетическими барьерами, которые и определяют константу
скорости превращений исходного XVI в промежуточный продукт и послед-
него в продукт XVIa и соответственно XVII в промежуточный продукт
и далее — в XVIIa. Конечно, можно выдвинуть контрвозражение, что
если оба переходных состояния, отделяющие промежуточный продукт,
близки к нему по геометрии и структуре, то они и энергетически близки.
Тогда энергетические барьеры, отделяющие промежуточный продукт,
невысоки, а следовательно, энергетический уровень этого продукта все же
приблизительно определяет скорость реакции (постулат Хэммонда).
В 1967 г. Олах описал эксперимент, который, казалось бы, непосред-
ственно доказывает существование фенониевых ионов по крайней мере
в некоторых случаях. Оказалось, что п-(0-хлорэтил)-анизол, растворен-
ный в избытке SbF5 при —80° С, дает острый одиночный пик (синглет)
в спектре ЯМР в области метиленовых протонов. Это означает, что обе
метиленовые группы становятся эквивалентными после удаления хлор-
аниона, т. е. после образования карбониевого иона:
ОСН3 ОСН3
(О) + SbF5 ----> (a) SbF5Cl“
СН2СН2С1 сн2-сн2
Если бы метиленовые группы были неэквивалентны, то в спектре ЯМР
в этой области было бы два триплета, относящихся к разным метиленовым
группам. Однако на деле эти данные не исключают быстрой обратимой
изомеризации «неклассического» карбкатиона, т. е. равновесия типа:
ОСНз
сн2—СН2
н2с—сн2
ОСНз
При этом скорость взаимопревращений классических карбкатионов
должна быть настолько велика, чтобы ЯМР не мог уловить каждый карб-
катион как отдельную частицу.
618
ПЕРЕГРУППИРОВКИ
В некоторых случаях, например для перегруппировки Вагнера —
Меервейна, доказано, что ретроппнаколиновая перегруппировка про-
текает строго стереоспецпфичпо, с вальденовским обращением у при-
нимающего углеводорода и, кроме того, с резко повышенной скоростью
реакции. Все это делает несомненным и здесь «синхронный» механизм.
Этот факт огромного повышения скорости перегруппировки в случае подходящей
конфигурации (в данном случае эжзо-конфпгураццп) назван анхимерным (Уинстейн)
или, что то же самое, синартетическим содействием (Инголд).
В последние годы шла дискуссия между Уппстейном и Брауном о механизме пере-
группировок с участием катионов типа изоборнплыюго или норборнпльного. Как
установлено, скорость ацетолиза акзо-изомера норборпилтозплата в 1000 раз превосхо-
дит скорость ацетолиза эяЗо-пзомера и значительно больше скоростп той же реакции
для mpem-бутилтозилата. По Уппстейну, это объясняется тем, что пространственное
содействие со стороны вновь завязывающейся связп возможно только для экзо-изомера
(тогда завязывающаяся связь и уходящая группа будут располагаться на одной прямой,
как при обычном нуклеофильном замещении). Браун же считает, что образуются
обычные классические катионы, которые находятся в равновесии друг с другом:
**30' эндо-
ОАс
п
с 1жс^ро^отозде1п?ым1Х^кп^ПП»Р°В1^а плет Чере3 <(111'класспческ1ш» трехчленный ка_.
свободного капбгатпопл^Л^гпял1 лсР°яаУн положительным зарядом, без образования
Уинстейном он X , \е1'ласС|1Чес'<"И» катион (понятие о нем впервые введено
м) он описывает как резонанс структур, в общей форме выражаемый схемой
трехчленный катион
ПЕРЕГРУППИРОВКИ с изменением углеродного скелета
619
а на конкретном примере изоборнеола и камфена изображаемый так:
Однако эта трактовка вряд ли состоятельна (Хюккель), так как рассредоточение
электронов может происходить внутри определенной структуры с фиксированным поло-
жением атомных ядер. Между тем атомы углерода 1 и 3 в структурах А п В находятся
на расстоянии 2,5 А, а в структуре Б на расстоянии 1,5 А (длина ординарной связи).
Относительно малую скорость ацетолиза аядо-изомера Браун объясняет простран-
ственными затруднениями (со стороны атома водорода в положении 3), препятствующимп
•сольволизу, и уходу аяЗо-тозилатной группы.
Браун отрицает роль пространственного (синартетпческого) содействпя. Он основы-
вается на своих экспериментальных данных, показывающих, что введение в норборнил-
тозплат в положение 1 заместителей (СН3, С6НБ, п-СН3ОС6Н4), стабилизующих карб-
катион, увеличивает скорость ацетолиза и экзо- (гЗКз0) и аяЗо-изомера (v3Hg0). Далее,
поскольку пространственное содействие не должно зависеть от наличия заместителя,
•отношение скоростей vgHg0 : i-gK30 при переходе от незамещенного норборнилтозилата
к замещенному должно было бы заметно меняться (особенно если стабилизующее
карбкатион действие заместителя сравнимо с предполагаемым синартетическпм влия-
нием пли больше его). Однако в действительности это отношение остается постоянным
при переходе к производным, содержащим разные заместители, в том числе и упомя-
нутые выше.
Сопоставляя теоретические соображения Хюккеля и эксперименталь-
ные данные Брауна, можно сказать, что «неклассический» катион пра-
вильнее представлять не как самостоятельно существующую частицу
и не как сумму резонирующих «классических» катионов, а как переходное
состояние перегруппировки, которое находится не в минимуме, а в ма-
ксимуме потенциальной кривой (на вершине барьера, разделяющего два
переходящих друг в друга «классических» катиона I и II, стр. 618).
Разберем, например, перегруппировку изоборнеола в камфен и обрат-
ную перегруппировку под действием кислот:
В этом случае одновременно с отделением (при атаке Н+ на ОН) гидр-
оксила электронная пара, привязывающая группу СН2, выделенную на
схеме, начинает отрываться от четвертичного углерода и привязываться
«со спины» к углероду, теряющему гидроксил, содействуя отделению
гидроксила. Благодаря этому сипартетическому (или анхнмерному) содей-
ствию перегруппировка изоборнеола, а также изоборнилхлорида в камфен
совершается в тысячи раз быстрее, чем перегруппировка в тот же ьамфен
стереоизомерного изоборнеолу борнеола (или борнилхлорида).
ПЕРЕГРУППИРОВКИ
620
На последней стадии перегруппировки метильная группа теряет протон
и привязывавшая его пара электронов образует двойную связь с соседним
углеродом, ликвидируя тем самым положительный заряд карбкатиона.
Этот обычный процесс стабилизации карбкатиона путем превращения его
в олефин мы уже видели на примере превращения пинаколинового спирта
в симметрпческий тетраметилэтилен.
Кинетически был исследован ацетолиз n-толуолсульфонатов (тозила-
тов) пзоборнеола п борнеола и, для сравнения, ацетолиз циклогексил-п-то-
луолсульфоната. Скорость ацетолиза последнего принята за единицу.
Оказалось, что скорость ацетолиза изоборнилтозилата в 3,5-105 больше,
а скорость ацетолиза борнилтозилата только в 1,4 раза больше. Синарте-
тическое содействие наблюдалось, таким образом, только в случае изо-
борнплтозилата.
Как синартетический процесс разыгрывается и превращение аддукта
НС1 к пинену — «истинного пиненхлоргидрата» — в хлористый борнил
(см. раздел «Изопреноиды», стр. 639). При присоединении хлористого
водорода к камфену образуется очень лабильный камфенгидрохлорид,
который уже при растворении в сухом этаноле при 0° С превращается
в изоборнилхлорид со скоростью, в 6000 раз превышающей скорость алко-
голиза в тех же условиях трет-бутил хлорида. Опять налицо синартети-
ческое содействие отщеплению иона С1“ со стороны электронов связи
СН2-группы, мигрирующей к углероду карбкатиона через переходное
состояние — неклассический катион:
камсренгидро-
хлорид
изоборнилхлорид
медленно
борнилхлорид
Формула карбкатиона с фиксированным зарядом написана лишь для
ясности, в действительности такой карбкатион отдельно не существует,
а превращается в переходное состояние с неклассическим трехчленным
катионом, от которого ион хлора отделяется синхронно с миграцией
СН,-группы. При атаке ионом хлора переходное состояние быстро пре-
вращается в изоборнилхлорид, который лишь медленно стереоизомери-
зуется в борнил хлорид.
С. С. Наметкин открыл еще один тип ретропинаколиновой перегруп-
пировки в ряду терпенов, названный его именем. При перегруппировке
аметкина меняются местами углеводородный радикал из четвертичного
ложения и галоид (или гидроксил), находящиеся при двух соседних
ПЕРЕГРУППИРОВКИ С ИЗМЕНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
621
углеродах цикла. Перегруппировка Наметкина сопровождается пере-
группировкой Вагнера — Меервейна, и в целом процесс на примере
гомолога изоборнилхлорида — метилизоборнилхлорида — выглядит так
(А в данном случае метил):
перегруппировка
Вагнера—Меервейна
перегруппировка
Наметкина
---------------->
перегруппировка
Вагнера-Меервейна
зеркало
Если А не углеводородный радикал, а Н, перегруппировка Наметкина
приводит к превращению /^-изоборнилхлорида в А-изоборнилхлорид
(и обратно), т. е. к рацемизации. Если А — углеводородный радикал,
происходит изомеризация.
Изомеризации циклов при действии азотистой кислоты на алицикли-
ческие амины, открытые и систематически изученные Н. Я. Демьяновым
и именуемые перегруппировками Демьянова, также относятся к ретро-
пинаколиновым перегруппировкам. Основная схема этих перегруппировок
может быть выражена так:
СНа СН2
А. СН2 -2^-* СН2 \h-Ne=N
\сн2)/ \сн2)/
сн2----сн—сн2
Х(СН2)/
СН2-—с=сн2
Х(СН2)/
I —н+
i
сн
сн2 сн
\сн2)<
I Н2°
сн2---сн—снаон+н+
\сн2)/
ПЕРЕГРУППИРОВКИ
622
Б. СН»----СН—CThNHg HNO-^ cih---—СП—СН2 _N, *
^(СИг)^ Х(СН2)Л
—> С112---СН-СН2
Х(СН2)Х
СН2
сн2 ^сн
\сн2)/
сн
Х(СН2)/
С На
•----—------> СН2 ^СН—ОН+Н+
Х(СН2)/
Не все сужения и расширения цикла в этих перегруппировках про-
текают одинаково легко. Пятичленный цикл легко расширяется в шести-
членный, а семичленный — сужается в шестичленный, который, преиму-
щественно, остается неизменным. Что касается до взаимопревращений
трех- и четырехчленного циклов, то оно происходит, но, возможно, там
не такой простой механизм.
Наряду с продуктами перегруппировки образуются и спирты — про-
дукты нормальной реакции замещения группы NH2 на ОН-группу.
Как показал Н. И. Путохин, демьяновские перегруппировки при-
ложимы в гетероциклическом ряду, например:
---- HNO. / х'.
II || N2 + I-l2O + il 1 + Н+
\ /\ X
NH СН2—NH2 N
Н. Д. Зелинским и его школой найдены многочисленные перегруппи-
ровки алифатических и особенно алициклических углеводородов под
действием хлористого алюминия. Так, например, исходя из циклогексана
или из метилциклопентана можно прийти к равновесной смеси:
СНз СН3
Ряд таких реакций в жирном и алициклическом ряду нашел и Нени-
цеску с сотрудниками, трактовавший их как ретропинаколиновые пере-
группировки, протекающие ионноцепным путем. Хлористый алюминий
должен содержать небольшое количество влаги, с которой образуется
комплексная кислота:
Н2О + А1С13 ---> Н+
ГС1 С1"|-
А1
С1 ОН
ПЕРЕГРУППИРОВКИ С ИЗМЕНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА 623
Эта комплексная кислота действует на углеводород, отрывая (по пре-
имуществу из вторичного или третичного положения) гидрид-анион и вы-
деляя молекулярный водород. Образовавшийся карбкатион претерпевает
ретропинаколиновую перегруппировку в изомерный карбкатион, отры-
вающий от новой молекулы углеводорода гидрид-анион, превращая ее
в карбкатион и т. д., пока ионная цепь не оборвется выбросом протона
с образованием олефина:
СН3СН2СН2СН3 -}- Н+
ОН
СН3СН2СНСН3
ГС1 С1
А1
С1 ОН
+ Н2 зарожде-
ние цепи
СН3СН2СНСН3 ---> СН2—СН—СНз----> СНз—С-СНз
СНз СНз
ретропинаколиновая перегруппировка
СНз—С—СНз + СНзСН2СН2СНз---->СН3—СН—СН3 + СН3СН2СНСН3 продолжение
| | цепи
СНз СНз
и т. д.
СНз—С—СН3 > СНз—С=СН2 + Н+ обрыв цепи
СН3 СНз
Родственны ретропинаколиновым перегруппировкам и перегруппи-
ровки С. Н. Данилова и Э. Д. Венус-Даниловой — превращения альде-
гидов в кетоны под действием концентрированной серной кислоты:
R СН2—С—О + Н +
Н
>;r4ch2—с—он -
н
R
сн3—с=о + н+
Перегруппировка А. Е. Фаворского состоит в том, что а-хлорзамещенные
кетоны под действием щелочей превращаются в кислоты:
ОТТ“
R-CH2-C-CH2C1 ----- HO-C-CH2-CH2-R4-Cl-
Формально можно было бы рассматривать эту перегруппировку как
ретропинаколиновую, протекающую с миграцией группировки RCH2
на место хлора, но условия реакции совсем иные: нет атаки на CI ,
ПЕРЕГРУППИРОВКИ
624
действует щелочь, которая может уводить а-протон. Поэтому считают что
это другой тип перегруппировки, где движущим началом является обра-
зование карбаниона:
R-CH-C-CHoCl ---► R-СН — С— СН2С1
I II 'II
[н'7о о
он-;
:О:
СН2-С1 ---> R-СН— СН2 + CI
О
О
он-
н,о „О
R—СН—СН9---->-R—СН9 —СН9 —С + ОН-
С\ / ' ОН
НО О
Таким образом, перегруппировка Фаворского идет через образование
неустойчивого в щелочной среде производного циклопропанона. По отно-
шению к группе СН2С1 она является нуклеофильным замещением.
Важная для ароматического ряда (в алифатическом ряду она осуще-
ствляется редко) бензиловая перегруппировка проходит при действии
щелочи на а-дикетоны:
ОН“
Аг—с—С—Аг —
O<LI J t о^ Аг
ДС-С—Аг—> хс—С
НО Дэ НО I Аг
СИ "о
о,
Аг
о I Аг
он
О О
Кислота (С6Н5)2С(ОН)СООН называется бензиловой кислотой,
а дикетон СвН3—С—С—С6Н6 бензилом, откуда и название перегруппи-
О
ровки (сравните ее
пировки).
Промышленный
(стр. 114)
О
механизм с механизмом пинаколиновой перегруп-
метод получения фенола и ацетона по реакции
СНз
CeHj—С—
СНз
I н+
Н-рОо —► СвН5—С—ООН > С6Н5ОН + (СП3)2СО
СНз СНз
перегруппировки с промежуточным образованием карбенов
имеет в своей основе также перегруппировку углеродного скелета, выра-
жаемую схемой:
С6Н5
I
сн3—с—о—ОН
I
сн3
С'Но । МО СНч Н+ СН3
oc,.iis — »• >-освн5-------------► >=o + hoc,hs
ctV сн» in CH1
Сходное течение имеет перегруппировка Криге, примером которой может
служить превращение сложного эфира декалин-9-гидроперекиси в слож-
ный эфир 1-окси-1,6-эпоксициклодекана:
Перегруппировка проходит при нагревании в ионизирующем раство-
рителе и используется для раскрытия карбоциклов.
ПЕРЕГРУППИРОВКИ С КАРБЕНАМИ И КАРБЕНОИДАМИ
В КАЧЕСТВЕ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ
Перегруппировка Вольфа — Арндта. При каталитическом действии окиси
серебра а-диазокетоны перегруппировываются в кетены, и при дальней-
шем действии воды образуются кислоты с тем же числом углеродных
атомов, что и в исходных кетонах:
R—С—CH=N=N <--> R—С—СН—N=N ГТС—~С—СН + N2
li II ---II
0 0 о
________________________I
I
О=с=с/И °Чс-СИ2П
\Ц НО'
Существенное отличие от всех предыдущих типов перегруппировок
состоит в том, что миграция происходит не к карбкатиону, а к нейтраль-
ному углероду с неполной электронной оболочкой (с секстетом электро-
нов) — карбену.
40 Заказ 672
ПЕРЕГРУППИРОВКИ
626
Имеется серия аналогичных перегруппировок, где атомом с незапол-
ненной электронной оболочкой (тоже секстетом), принимающим мигриру-
ющую группу, является атом азота. ~
Перегруппировки с перемещением мигрирующей группы к азоту. На
предыдущую перегруппировку Вольфа — Арндта наиболее похожа пере-
группировка Курциуса. Она разыгрывается при нагревании азидов кислот
(кн. 1, стр. 184):
сн c_n=n=n:--<—>сн3—c-n—n=n:
11 о
о °
> СОо + ch3nh2
алкилизоцианат
алкилкарбамино-
вая кислота
Промежуточное неустойчивое соединение, образующееся сразу вслед
за отщеплением молекулы азота, содержит атом азота с секстетом электро-
нов, т. е. с незаполненной (вакантной) орбиталью. Такие атомы, как мы
видели выше, электрофильны наподобие катионов, хотя формально они
нейтральны. Они близки в этом отношении к атому углерода в карбенах
/В\ \
\С: и могут быть названы карбеноидными (Д. Н. Курсанов
\R/ J
и М. Е. Вольпин). Далее алкил в этом промежуточном соединении мигри-
рует к азоту вместе с парой электронов связи, и они заполняют вакантную
орбиталь азота. При этом на атоме азота не возникает отрицательного
заряда, поскольку одновременно свободная пара электронов азота ста-
новится л-парой связи с углеродом, и образуется молекула алкилизо-
цианата (эфира изоциановой кислоты):
Л’ r__rr--*N.-
сн3-с<4 --->Гсн3—> сн3—n=c=o
о - хо
Само удаление молекулы азота из азида может тоже происходить
одновременно или почти одновременно с начинающейся перегруппировкой,
так что «карбеноидная молекула» ни на момент не существует в свободном
состоянии.
Аналогично обстоит дело в перегруппировках Шмидта, Лоссена и Гоф-
мана, которые рассмотрены ниже более кратко. Продукт всех этих пере-
ПЕРЕГРУППИРОВКИ С ПРОМЕЖУТОЧНЫМ ОБРАЗОВАНИЕМ КАРБЕНОВ
627
группировок — алкилизоцианат, который в водной среде моментально
гидролизуется, причем образующаяся алкилкарбамиповая кислота не
существует в свободном состоянии (подобно угольной кислоте) и распа-
дается на СО2 и амин. Однако если разложение азидов проводить не в воде,
а в каком-либо спирте, то образуются продукты присоединения спирта
к алкилизоцианату, т. е. уретаны, которые вполне стабильны:
/О
СНз—N=C=O + C4H9OH > СНз—NH—Сч
ХОС4Н8
Перегруппировка Шмидта заключается в непосредственном превраще-
нии кислот в амины с цепью, содержащей на один углеродный атом меньше.
На раствор карбоновой кислоты в концентрированной серной кислоте
действуют азидом натрия. В таких условиях большинство карбоновых
кислот обратимо дает ацилониевые катионы, которые с азид-анионом обра-
зуют азиды кислот:
R—С—0Н4-Н+ R—С —R-C—N3 = R-C-N=N=N
и и II II
Далее реакция разыгрывается, как было только что описано при пере-
группировке Курциуса. Синтетически зта реакция имеет то преимущество
перед перегруппировками Курциуса или Гофмана, что минуются стадии
приготовления из кислоты ее азида или амида (разумеется, это выгодно,
лишь когда общие выходы существенно не различаются).
Знакомая нам уже перегруппировка Гофмана, исходящая из амидов
карбоновых кислот, проходит в водной среде и дает те же продукты, что
и реакция Шмидта или реакция Курциуса в водной среде. Действие ще-
лочного раствора брома приводит здесь прежде всего к неустойчивому
в этих условиях N-бромзамещенному амиду. Далее гидроксил щелочи
отрывает протон, а образующийся анион диссоциирует с отщеплением
бром-анпона. При этом получается то же промежуточное соединение, что
и в предыдущих перегруппировках. Возможно, что диссоциация с от-
щеплением бром-аниона происходит одновременно с перегруппировкой:
°Н у -/Вг I------‘“Ч t
О о
н2о
> O = C=N— R --> со2 + RNH2
По Уоллису, в перегруппировках Курциуса, Гофмана. Лоссена
и Вольфа мигрирующая группа сохраняет свою конфигурацию.
Перегруппировка Бекмана происходит при действии на оксимы кетонов
кислот Льюиса, PCI3, PCIfi, а также хлорангидридов сульфокислот
40*
ПЕРЕГРУППИРОВКИ
628
л концентрированных минеральных кислот типа серной. Эта перегруппи-
ровка приводит к амидам кислот следующим образом:
NHR
Если перегруппировка происходит под действием хлорангидридов
сульфокислот, вначале отщепляется хлористый водород и остаток кислоты
становится на место водорода гидроксила; такие соединения в ряде слу-
чаев можно легко выделить. Далее, в полярном растворителе, особенно
при нагревании, они диссоциируют (гетеролиз N—О-связи), так что у атома
азота остается незаполненная электронная оболочка. В эту лакуну и за-
тягивается электронная пара связи С—В, радикал мигрирует, а образу-
ющийся катион связывается с подходящим анионом:
R R' R R'
_। —у 4--0S0»R"
N N+
OSOoR"
I
R'
R’SO3H + C=O
NHR
Б
R’O2SO R'
c
-OSO2B"
Продукт перегруппировки типа А можно выделить. Он легко гидро-
лизуется до амида кислоты Б. Если реакция ведется в водной среде, то
к катиону присоединяется вода и сразу же получается амид. Перегруп-
пировка, как это строго доказано многими исследованиями (кн. I, стр. 490),
полностью стереоспецифична: мигрирует радикал, находящийся в анти-по-
ложении к уходящему гидроксилу. Ьекмановская перегруппировка ис-
пользована, в частности, для размыкания карбоцикла и получения капро-
лактама (в производстве капрона):
С=о +H2NOII
— I C_NOU -Bi- ( /~он — I )'°
N \-NH
перегруппировки с перемещением л-СВЯЗЕИ
Все рассмотренные перегруппировки носят название нуклеофильные
или секстетные перегруппировок. Первое название подчеркивает, что
миграция радикала осуществляется к обедненному электронами центру.
Для большинства этих перегруппировок было доказано, что мигрирует
в первую очередь и с большей скоростью более электроподопорный радикал
и арилы быстрее алкилов.
Перегруппировка Стивенса. Четвертичные аммониевые соли, имеющие
достаточно подвижный а-водородный атом (в группировках типа
—СН2С6Н5. —СН2С—В), при действии сильных оснований (NaOH,
II
0
LiC6H5) теряют этот водород в виде протона, причем образуется илид,
который и подвергается электрофильной перегруппировке
R
R\ + тш w R\ + R ч .. I
R-ЧУ—CHoCeH5 - R—N—CIICeII5 > ;N-CHC6H5
RZ RZ Rz
t. e. в этом случае мигрирующий радикал, оставляя пару электронов
связи на атоме азота, мигрирует к отрицательно заряженному углероду.
Перегруппировка Соммле происходит в том случае, если в предыдущей
схеме R — метилы. Ее течение таково:
сН2_Й(СН3)з CH2-N(CH3)2
1 NaNH2 А ” СН2
Oj Ч/ +NH3
+ Na+
ПЕРЕГРУППИРОВКИ С ПЕРЕМЕЩЕНИЕМ л-СВЯЗЕЙ
Аллильные перегруппировки. При нуклеофильных атаках на галоидный
аллил и его производные наряду с нормальным продуктом замещения
получаются продукты изомеризации: нуклеофильный реагент вступает
не на место замещаемого галоида, а к углероду 3, двойная же связь пере-
мещается. Для галоидного аллила, меченного по углероду (14С), это вы-
разится так:
*СН2=СН—СИ2-С1
__> ПО—СН2—СП=*СН2
+С1-
1—* сн2=сн—*СН2ОН
Поскольку галоидный аллил в средах с высокой диэлектрической про-
ницаемостью реагирует с нуклеофилами по механизму 5л-1, реакция
ПЕРЕГРУППИРОВКИ
630
должна идти с предварительной диссоциацией на ионы. Еще до широкого
применения в органической химии понятия мезомерии (примерно 30-е годы
нашего столетия) Прево и Кирманн для объяснения такого двойственного
реагирования выдвинули теорию синионии, т. е. в данном случае единой
структуры аллильного попа, что на современном языке выразится так:
СН2=^СН^СН2 или СН2=СН —СН2 > СН2 —СН = СН2
RCH==Cf4—СН2 или RCH = CH —СН2-< > RCH — СН = СН2
В этом случае отщепившийся в виде аниона хлор может рекомбиниро-
ваться с карбкатионом по любому из двух углеродов, несущих частичный
положительный заряд, с образованием
RGH=GH-GH2C1 или RCHC1-GH=CH2
Это — случай аллильной анионотропии, и явление по праву рассматри-
вается как анионотропная таутомерия'.
RCH=CH—СН2С1 RCH—СН=СНа
В написанной выше реакции с изотопом углерода структура карб-
катиона совершенно симметрична и заряд распределен равномерно между
крайними углеродами. Поэтому протекание реакции без перегруппировки
и с аллильной перегруппировкой равновероятно. Во втором случае это,
разумеется, не так: карбкатион с катионным центром у вторичного угле-
рода устойчивее, чем у первичного, и преобладать должно замещение
у вторичного углеродного атома.
Повышенная способность галоидного аллила (сравнительно с пре-
дельными галогенидами) к электролитической диссоциации и объяс-
няется устойчивостью его катиона — благодаря рассредоточению заряда
между двумя углеродами энергия такого карбкатиона снижается.
Однако аллильные перегруппировки происходят и при замещениях
по механизмам SN2 и SE2, т. е. без электролитической диссоциации.
Это типичный случай реакции с перенесением реакционного центра по
системе сопряженных связей, в данном случае о,л-сопряженных (кн. I,
стр. 311). Изображаем течение таких реакций, пользуясь символикой
английской школы.
Анионотропная аллильная перегруппировка (в среде с малой диэлек-
трической проницаемостью, механизм 8^2):
RO~ + СН2 = СН^СН2—С1---> RO—СГ12-СН=*СН2+С1-
ПЕРЕГРУППИРОВКИ С ПЕРЕМЕЩЕНИЕМ Л-СВЯЗЕЙ
631
Катионотропная аллильная перегруппировка:
Н /O-MgCl
> /С-^сн2—сн=сн2
н
Одним из важных простейших случаев анионотропной аллильной пере-
группировки может служить обратимая перегруппировка диметилвинил-
карбипола в изобутенилкарбинол (А. Е. Фаворский, А. Н. Лебедева):
ОН
СН3х | н+ СН3\
>с—сн=сн2 —-* >с=сн-СН2ОН
СНз/ *--- СН3/
По сути дела перегруппировкой этою же типа является открытое
И. н. назаровым превращение винилэтипилкарбинолов в дивинилкетоны
(под действием иона двухвалентной ртути):
„ |ОН' । ОН
R<L_J i Rx I
,/C—G = c—CH—CH2--->- /С=С=С—CH=CHo >
R R,X
r=CH—C— CH=CH2
R
Такой синтез дивинилкетонов из винилэтипилкарбинолов (в свою
R\
очередь получаемых действием на кетоны = О винилацетилена в
R/Z
присутствии КОН) позволил И. Н. Назарову широко применять дивп-
нилкетоны в синтезе циклических систем.
Простейшими примерами катионотропных аллильных перегруппировок
являются прототропные аллильные перегруппировки, например пере-
группировка у,у,у-трихлоркротоновой кислоты под действием пинка
и уксусной кислоты. Сначала, очевидно, в результате замещения хлора
на водород образуется дихлоркротоновая кислота, которая, как доказали
А. Н. Несмеянов, Р. X. Фрейдлица и Л. И. Захаркин, претерпевает пере-
группировку в дихлорвинилуксусную кислоту:
СС18\ /Н
\С=(Х -f-Zn-j- 11 + ►
нх хсоон
ГС12НСЧ /Н
\г—
ц/ ХЮОН
--► СС12=СН—СНа—С—ОН
632
ПЕРЕГРУППИРОВКИ
Дальнейшее восстановление дихлорвинилуксусной кислоты совер-
шается только под влиянием амальгамы натрия в спирте. При этом проис-
ходит обратная аллильная перегруппировка и получается сама кротоновая
кислота:
Na—Hg;
СС12= СН—СН2—С—O'Na* —гН5°Н > Гсн2=сн—СН2—с— O'Na*l —Н >
А п -
о о
—> СНо=СН—СН—с—о
>|--п II
Н I о
:он-1
> сн2—сн=сн-с—О" ►
н-он о
> СН3—СН=СН—с—О’
II
ОН о
Таким образом, на пути превращения трихлоркротоновой кислоты
в кротоновую восстановлением в две стадии лежат две прототропные
аллильные перегруппировки (одна в кислой, другая в щелочной среде).
Реакция протекает без изменения конфигурации только благодаря тому,
что и для исходной, и для конечной кислоты более устойчивы транс-мзо-
меры. В противном случае конфигурация не сохранялась бы, так как
на стадии дихлорвинилуксусной кислоты стереоизомерия исчезает. Между
тем Ауверс и вслед за ним другие многочисленные ученые ошибочно счи-
тали, что обнаруженное этим автором соответствие является доказатель-
ством транс-конфигурации кротоновой кислоты:
НООС /Н
\С=С-
ХСООН
гидролиз
С13СХ /Н
>с=с/
Н СООН
II3С Н
;zc=cz
Hz чюон
н
(неверная аргументация Ауверса)
Само по себе это верно, но никак не доказывается приведенными пре-
вращениями вследствие аллильной перегруппировки.
Приводим несколько примеров более сложных аллильных перегруп-
пировок. протекающих с перенесением реакционного центра по меха-
низму *5^2 (Р. X. Фрейдлина, Л. И. Захаркин и сотрА
н+ ’
!• Аг —СН —СН=СС12 + Н2О ----> Аг—СН—СН
I <1
осгн5 (ос2н5
н+
С1
•--> Аг —СН = СН—С—ОН + н+ + С2н5он
I
С1
633
ПЕРЕГРУППИРОВКИ G ПЕРЕМЕЩЕНИЕМ Л-СВЯЗЕЙ
CI
I /С1
Аг—CH=CII—С—ОН > Аг— СН=СН— СД - НС1
I ,
Cl I н2о
/ОН
Аг—СН=СН— Сх^о 4- НС1
II, При действии ряда нуклеофильных реагентов на изомеры
СН2=СН—СС1Н и СС12=СН—СН.,С1 получаются в обоих случаях про-
дукты замещения одного атома хлора на нуклеофил. Первый изомер
реагирует с аллильной перегруппировкой, второй — без нее:
CC12=CH-CH2-N(C2Tl5)2
| +(C2H.)2NH
I : 1
СН2=СН—СС13 СС12=СН-СНоС!
!________________________I
| +CHaONa
СС]2=СН—СНз-ОСНз
В первом случае атака нуклеофила идет, очевидно, на крайнюю
СН2-группу, например:
_Q
СН3О
сн2—сн—с—о
С1
---> СН3 —о —сн2—СН=СС]2 + Cl-
При гетерогенном катализе над основными катализаторами (напри-
мер, AloOs) или при действии оснований наблюдаются перемещения двой-
ных связей и протонов, протекающие по механизму прототропной ал-
лильной перегруппировки. Примеры.
I. Двойные связи, если их две или более, стремятся переместиться
в сопряженное положение, энергетически более устойчивое:
rch= сн-сн-сн=сн2 > rch=ch—сн—сн==сн2 —2 >
pr-i,
[он"; > Rch=ch—сн=сн—сн3 + он"
Если двойная связь находится в боковой цепи ароматического соеди-
нения, она также перемещается — в положение, сопряженное с арома-
тическим ядром.
ПЕРЕГРУППИРОВКИ
634
II. Двойные связи от края молекулы стремятся переместиться ближе
к центру;
r—сн—сн=сн2—► к—<дДсн=сн2 —- > RCH сн СНз + ОН
I -I
«ОН i
1 1
Не менее важным, чем перемещение кратных связей в перегруппиров-
ках этого типа, является результативное перемещение протона, превраща-
ющее метиленовую группу в метильную. Таким образом, смысл реакции
можно выразить схемой
независимо от того, переходит ли протон на новое место внутри молекулы,
или сначала он отрывается основанием, а на новое место доставляется
растворителем или сопряженной кислотой. Из последней схемы мы видим,
.что речь идет о прототропной аллильной перегруппировке.
Перемещение тройных связей. В кн. 1 на стр. 285 уже была описана
происходящая под действием сильных оснований ацетилен-аллен-диеновая
перегруппировка А. Е. Фаворского, выражаемая схемой
rch2-ch2-c=ch —> rch2-ch=c=ch2 —>• rch=ch-ch=ch2
и, очевидно, протекающая как прототропная (т. е. катионотропная) пере-
группировка — совершенно подобно описанным выше перемещениям
двойной связи.
Т. А. Фаворская нашла ацетилен-аллен-диеновые перегруппировки
галоидзамещенных гомологов ацетилена, протекающие под действием
аммиаката хлористой меди в хлористом аммонии и, по-видимому, пред-
ставляющие собой последовательность одной анионотропной (ацетилен ->
-► аллен) и одной прототропной (аллен -> диен) перегруппировок:
*- СН2 —С — СН = СНС1
СН;)
NH3
+ОН'
Ацилоиновые перегруппировки. А. Е. Фаворским открыто взаимопре-
вращение несимметрично построенных а-кетолов. Так метитбензоил-
карбинол Т иЙТеИ6М КИСЛ°ТЫ пеРегРУппировываетСЯ’ в фенилацетил-
кароинол. Т. И. Темникова, широко исследовавшая этого рода пере-
ГИДРИДНЫЕ ПЕРЕМЕЩЕНИЯ
635
группировки, предложила вероятное объяснение их течения с участием
соседней группы (ср. кн. I, стр. 394):
। >
н* + ♦ LlJ
с6н5-с—сн-сн3—> с6н5-с—сн—сн3 С6н5-с—с—сн3
II I I 'J -н / \ 7
о он н—о о—Н но о
с6н5—сн— с—сн3
I II 3
но о
Она показала, что в простых эфирах а-лактолов — модельных соеди-
нениях предполагаемых промежуточных продуктов перегруппировки —
действительно происходит перегруппировка окисного кольца в кето-
группу: _
I н
f IL н+ |
сбН5-С—С-СНз--------> С6Н5-С-С-СН3
/ \ / I II
сн;,о о СН3О о
ГИДРИДНЫЕ ПЕРЕМЕЩЕНИЯ
В сущности разобранные выше перемещения кратных связей в молекуле
можно рассматривать и как протонные перемещения: протон из аллиль-
ного положения перемещается к СН2-группе, превращая ее в СН3. Одно-
временно происходит превращение электронной пары связи С—Н в л-пару
и обратное превращение л-пары в пару, привязывающую протон (из рас-
творителя).
Довольно часто наблюдаются перемещения водородного атома вместе
с парой электронов связи (интрамолекулярные гидридные перемещения).
Они вполне подобны ретропинаколиновой перегруппировке, в которой
радикал перемещается также с парой электронов связи. Не следу,ет,
однако, забывать, что по легкости миграции радикалов (и атомов) в этой
перегруппировке (стр. 610) водород занимает последнее место. И все же1
иногда гидридное смещение преобладает. В качестве примера приведем
действие азотистой кислоты на н-пропиламин, меченный 14С по углероду,
несущему аминогруппу. О. А. Реутов установил, что реакция иде^
так:
CH3-CH2-*CH2-NH2 СН3-СН2—*CH2-N=N
СН2—СН2—*СН3 ч— СН3—СН2-*СН2 —> СНз—СН-*СНа
|нго |н,° |н,°
НОСН2-СН2—*СН3 СНз—СН2-*СН2ОН СНз-СН-*СНз
I
ОН
636
ПЕРЕГРУППИРОВКИ
Таким образом, гидроксил в полученном пропиловом спирте (первич-
ном и вторичном) оказывается у любого из трех углеродов.
Естественно, что гидридные перемещения осложняют и другие реакции,
промежуточной стадией которых является образование карбкатиона,
например перегруппировки Демьянова.
Пометив тритием (Т) молекулы алициклических аминов, О. А. Реутов
и Ю. Г. Бундель наблюдали следующие гидридные перемещения, сопро-
вождающие перегруппировки Демьянова:
В результате последующих гидридных сдвигов карбониевый центр
перемещается также в положения 3 и 4 циклогексанового кольца.
Гораздо шире изучены интермолекулярные гидридные перемещения,
в которых карбкатион вырывает гидрид-анион из другой (такой же или
иной) молекулы, превращая ее в карбкатион. Такие процессы могут про-
текать как цепные ионные реакции. В частности, подобного рода гидридные
перемещения широко изучены Д. Н. Кургановым, 3. Н. Парнес и сотр.
ГОМОЛИТИЧЕСКИЕ ПЕРЕГРУППИРОВКИ
Перегруппировки свободных радикалов (текущие, конечно, гомолити-
чески) стали известны между 1940—1950 гг. и изучены гораздо меньше,
чем гетеролитические.
Радикалы с неспаренным электроном у третичного углерода наиболее
устойчивы, с неспаренным электроном у первичного углерода — самые
неустойчивые. Поэтому при температуре 250° С первичные радикалы
изомеризуются во вторичные (или третичные), и эта реакция требует малой
энергии активации (~ 9 ккал). По-видимому, такая стабилизация нечет-
ного электрона у третичного (и вторичного) углерода происходит анало-
гично стабилизации карбкатиона, что может быть выражено на языке
резонанса так:
Н, + Ц+ /СНз Нх /СПз Нх .СП»
н—С-С-СНзч--+ И—С=С< ч---► 11+>С=С< ч-------> НАс=С< и т. д.
IE I „ ' \СПз HZ ХСН3 11+ \СН,
СН3 Н
стабилизация карбкатиона гпперконъюгацией
ГОМОЛИТИЧЕСКИЕ ПЕРЕГРУППИРОВКИ
637
Н\ Н. .н
Н—С—С—СТТ3 <---> Н /С—С—СНз <—•—* • • - <-> СНз—С=Ст-Н и т. д.
н/ I н' | | \н
СНз СНз СНз
стабилизация свободнорадикального центра
Еще позднее стали известны гомолитические перегруппировки, про-
текающие в жидкой фазе. Первой такой перегруппировкой, по-видимому,
была открытая Харашем в 1944 г. перегруппировка так называемого
неофилъного (2-фенил-2,2-диметилэтилового) радикала в третичный
радикал:
СНз СНз
С6Н5—С—СН2С1 _coci2 Cells—С—СН2 -----> C6Hs—С—СП2—СН3
СНз СНз СНз
Р. X. Фрейдлиной с сотрудниками (1950 —1965 гг.) были найдены
и систематически исследованы гомолитические перегруппировки чисто
алифатических непредельных галоидзамещенных соединений — это пере-
группировки аллильного типа. Перегруппировка всегда происходит
в сторону образования более устойчивого радикала, в данном случае
такого, у которого радикальный углерод более загружен атомами
галоида.
Бромистый водород в присутствии перекиси бензоила присоединяется
к 1,1,1-трихлорпропену с гомолитической аллильной изомеризацией:
О
С6Н5.--НВг --> С6Н6-|-Вг.
СС1з-СН=СН2+Вг. --> СС13-СН-СНгВг
СС]3—СН—СН2Вг > СС12—СНС1—СН2Вг (гомолитическая аллильная
перегруппировка радикала)
CCI2—СНС1—CH2Br-J-HBr > CHCI2-CHC1—CH2Br + Br. (возобновление цепи)
Другим примером служит изомеризация СС13—СВг—СП2
в СС12=СС1—СНоВг при освещении или при действии перекиси, т. е.
при инициировании гомолитической цепной реакции затравкой свобод-
ными радикалами.
Предполагается такая схема этой гомолитической изомеризации:
СС13—СВг=СН2 —* СС13-С=СН2 (Вг-
СС13—СВг=СН2 + Вг. > СС13—CBr-CH2Br
ПЕРЕГРУППИРОВКИ
638
Cl
ССЬ-СВг—CfbBr ► CCl»-CBr—СН2Вг (гомолитическая аллильная
Geis ьвг - перегруппировка радикала)
С1
СС1»—С—СН2Вг + СС1з—СВг=СН2 > СС12=СС1—СН2Вг + СС1з—СВг—СН2Вг
I (конечный
Вг продукт)
СС13—СВг—СН2Вг > (продолжение цепи)
Доказательством гомолитического цепного механизма реакции служит
инициирование его веществами, являющимися генераторами радикалов,
и прекращение ингибиторами цепных радикальных реакций, такими,
как гидрохинон.
Интересно, что гомолитическая аллильная перегруппировка
СС13—Вг=СН.> (I) приводит к совершенно иному продукту, чем анионо-
тропная перегруппировка (Л) того же вещества:
I. СС1з СВг—СИ2 * СС12=СС1—СН2Вг (цепная гомолитическая
перегруппировка)
II. СС13—СВг=СН2 i-»- СС12=СВг—СН2С1 (анионотропная перегруппи-
ровка; А. Н. Несмеянов,
Р. X. Фрейдлина, В. Н. Кост)
ИЗОПРЕНОИДЫ
ТЕРПЕНЫ И ТЕРПЕНОИДЫ
В растительном мире широко распространены вещества открытой и изо-
циклической структуры, которые формально могут быть рассматриваемы
как димеры (терпены), тримеры (сесквитерпены) или полимеры (поли-
терпены) изопрена и их кислородные производные (терпеноиды). Изучая
изопрен, мы уже встретились с терпеном — рацемическим лимоненом
(дипентеном) и политерпенами — каучуком и гуттаперчей. Монотерпены
С10Н ]6 обладают удвоенной молекулярной формулой изопрена и, следо-
вательно, в случае если они относятся к алифатическому ряду, должны
иметь три двойные связи, если они включают один алицикл — две двойные
связи, и, наконец, бициклические терпены должны иметь одну двойную
связь. Таким образом, по химической классификации уже монотерпены
относятся к разным классам углеводородов и выделены в одну группу
вопреки химической классификации, по фитохимическому признаку.
Впрочем, это находит свое оправдание в том, что существует много реак-
ций, связывающих взаимопревращениями алифатические, циклические
и бициклические терпены. Хотя некоторые терпены можно, как мы видели
на примере лимонена и каучука, получить из изопрена, в растениях
изопрен отсутствует, и путь биосинтеза терпенов и терпеноидов иной.
Из надрезов иа коре хвойных растений вытекает смола (или живица).
Живицу собирают в подсобных хозяйствах, главным образом из разных
пород сосны. При отгонке с паром отходит скипидар, или терпентинное
масло, а остается канифоль (см. ниже). Скипидар — смесь разных моно-
терпенов С10Н16, зависящая от пород сосны и местности. Он находит
применение в синтезе камфоры, непосредственно в медицине и в синтезе
таких медикаментов, как терпингидрат, некоторых душистых веществ,
например терпинеола. Скипидар применяется также как растворитель
и разбавитель для масляных красок.
При отгонке с паром лепестков цветов * или листьев некоторых растении
собирают верхний слой отгона — «эфирное масло» данного цветка.
* Используется также метод «апфлеража»: п замкнутую камеру между пластинами
со слоем жира илп вазелина помещают лепестки, затем извлекают растворителем насы-
тившие жпр эфпрпые масла.
640
ИЗОПРЕНОИДЫ
Оно в основном состоит из терпеноидов, т. е. кислородных производных
иерпенов. Эфирные масла применяются в парфюмерии как целое или из них
тзолируют отдельные компоненты, такие, как гераниол, цитронеллаль
и т. д.
Моно терпены и терпеноиды
Ациклические терпены и терпеноиды. Известны два главных алифати-
ческих терпена, содержащихся в эфирных маслах (например, хмеля) —
мирцен I и оцимен II:
СН3 СН2 СНз СПз
I II I I
СНз—С=СНСН2СП2—С—СН=СН2 СН2=С—СН2СН2СН=С—СН=СНч>
I II
При сильном нагревании бициклического терпена а-пинена Б. А. Ар-
бузов изомеризовал его в аллооцимен III
СПз СНз
I I
СНз-С=СН-СН=СН—С=СН—СНз
III
Строение каждого из этих углеводородов легко установить озо-
нолизом.
В эфирном масле герани (и ряде других эфирных масел) содержится
спирт гераниол
СН3 СПз
I I
СНз-С=СНСН2СН2-С=СН—СН2ОН
находящий применение в парфюмерии. Осторожным окислением (напри-
мер, по Оппенауэру) из него получают соответствующий альдегид (с ли-
монным запахом) — цитраль. Строение этого альдегида явствует из
разложения его при кипячении с К2СО;1 на 6-метилгептен-5-он-2 и ацет-
альдегид (обращение кротоновой конденсации):
СПз СНз И
I | | К2СО3;
СНз—С=СНСН2СН2—С=СН—С=О н-’° х
СПз СПз
I I /Н
--> СНз-С=СНСН2СН2-С=О + СНз-С<
Действуя на метилгептенон ацетиленом и КОН и подвергая тройную
связь частичному гидрированию, Ружичка получил рацемический ли-
налоол — изомер гераниола, находимый в лавандовом масле:
СНз СНз
СНз-С=С11СН2С112-С=ОЛ-1]С=СНН-КО11 ->
СНз СПз СПз СНз
I । 2Н I I
---> СНз-С=СНСН2СН2-С-С=СН ------------ СН3—С==СНСН2СН2—С—СН=СН>
I-------------------------------I
ОН Ан
ТЕРПЕНЫ И ТЕРПЕНОИДЫ
Сам линалоол и особенно его ацетаты и оксилиналоол широко при-
меняются в парфюмерии, так как пахнут ландышем. В цптронелловом
масле содержится цитронеллалъ, а в розовом масле и масле герани соот-
ветствующий спирт цитронеллол
СНз СН3
I 1
сн2=с—СН2СН2СН2—СН—СН2-С Н2О 11
также применяющийся в парфюмерии.
Алифатические терпеноиды легко циклизуются. Так, гераниол при
действии разбавленной серной кислоты, предварительно превращаясь
в свой zjize-изомер — нерол, гидратируется по двойным связям и замы-
кается в моноциклический терпеновый гликоль — терпин, в виде гидрата
применяемый как медикамент. В промышленности терпин получают
гидратацией серной же кислотой содержащегося в скипидаре терпена —
лимонена:
СН3
сн2он
I н
сн
I
с
СНз ^СНз
СНз
сн2он
сн
нерол
СНз
I—пгт
H2SO4 H2SO4
(НгО). | | ( (НгО)
С-ОН
СН^ ХСНз
геранвол
терпин лимонен
Кротоновая конденсация цитраля с ацетоном дает «псевдоионон».
Этот алифатический кетон при действии кислоты замыкается в изомерные
ему а- и (3-иононы:
Н3С СНз
С
н+
П(/ сн-сн=сп-с-сн3
. ! II II ---------
Н2С С-с Из о
сн2
НзС СНз
2<^уСН=СН-
х^ХСНз
а-ИОНОИ
псевдононон
С-СНз
НзС СПз
\/ .СН=СН-С-СИз
О
з
(З-ИОНОН
Иононы — вещества, пахнущие
вым деревом, а в разбавленном
в концентрированном виде кедро-
— пармской фиалкой и широко
41 Заказ 672
ИЗОПРЕНОИДЫ
642
Применя= з
а также корня ириса « , р 11 г
НзС
НзС\
х СН=СН-С-СНа
, V 11
• /7\ 0
\^\СНз
«-ирон
НзС?** ZCH3
НзС \</CII-CH-C-CHrf
I к ' о:
СНз
Р-ирон
СН—С—СНз
II
|О’
Т-ирон
Мопоциклические терпены. Уже упоминалось, что при нагревании изо-
прена в запаянной ампуле он превращается, димеризуясь, в дипентен
(С. В. Лебедев);
СНз СНз
н3с/^ н3с/^сн2
И обратно, при крекинге дипентена образуется изопрен. Дипентен
имеет один асимметрический атом углерода п по общему закону синтети-
чески получается рацемическим. В основных скипидарах и в эфирных
маслах находятся оптически деятельные (+)- и (—)-дипентены, которые
носят название лимоненов (пахнут сходно с лимонной цедрой). Каталити-
ческое гидрирование дипентена приводит к родоначальному углеводороду
группы моноциклических терпенов — 1-метил-4-изопропилциклогексану,
названному ментаномд
7
СНз
СН
Н2С6 2СН2
Н2С5 зсн2
сн
8СН
НзС СНз
9 10
ТЕРПЕНЫ и терпеноиды
643
Терпены ряда ментана должны, очевидно, иметь две двойные связи,
быть изомерны дипентену, называться, как и последний, ментадиенами.
В разных эфирных маслах находятся следующие ментадиены:
Терпинены и терпинолен не имеют асимметрического углерода. При-
родные фелландрены оптически деятельны. Их скелет установлен ката-
литическим гидрированием; как и их изомер лимонен, они образуют при
этом ментан. По типичным реакциям сопряженных л-связей оба феллан-
дрена и а-терпинен можно отличить от других изомеров. Для суждения
о расположении двойных связей, кроме методов озонирования и окисле-
ния, много дало исследование молекулярной рефракции: наличие семи-
циклической двойной связи, как в [3-терпинене или терпинолене, сказы-
вается на экзальтации молекулярной рефракции примерно на вели-
чину 0,5; наличие сопряженных л-связей, как в [3-фелландрене, дает
еще большую экзальтацию — до 1; сопряжение внутри цикла, как в а-тер-
пинене, не дает экзальтации. Терпинены получаются изомеризацией
лимонена при действии кислоты.
Терпены довольно легко окисляются кислородом воздуха по двум
типам, образуя перекиси или гидроперекиси: во-первых, в аллильное
положение
во-вторых, по сопряженным двойным связям. Так, а-терпинен дает пере-
кись — аскаридол. Окисление протекает как 1,4-присоединение О2 по
сопряженной системе л-связей:
Аскаридол — едва ли не единственная перекись, встречающаяся
в природе как вещество (в эфирном масле одного из видов полыни). Исполь-
зуется как противоглистное средство и как инициатор цепных реакций.
Именно по причине образования перекисей скипидары ускоряют вы-
сыхание масляной краски.
Среди кислородных производных ментана важное значение имеет
ментол — главная составная часть перечной мяты — кристаллы (т. пл.
41*
644
ИЗОПРЕНОИДЫ
43° С), пахнущие мятой и применяемые в производстве зубных порошков,
паст, в кулинарии и медицине. Строение ментола устанавливается полу-
чением его из 5-метил-2-изопропилфенола (тимола) гидрогенизацией:
H2/pt
Ментол имеет два асимметрических углеродных атома. Природный
ментол левовращающий. Конфигурация природного вещества такова,
что метил находится в цис-, а изопропил — в т/шнс-положении к гидро-
ксилу. Окисление ментола дает кетон — ментон. Его можно синтезиро-
вать внутренней сложноэфирной конденсацией эфира 1-изопропил-4-ме-
тилпимелиновой кислоты:
Н2С
I
--> Н9С
СНз
СН
Z \ /О
Н2С Clh-CZ
I „ „ ОС2Н5 NaoC.H.
I . ОС2115
Н2С С4
\ / хо
сн
сн
H3CZXCH3
с Из
41
/О
ртт_p/Z 1. гидролиз
, 1 тт 2. декарбоксилирование
| хОС2Нб З-кетонокислоты
с=о ----------------------------
с II
сн
НзС СПз
Следующая схема синтеза непредельного изолога ментона — пулегона
дает новый пример замыкания алифатического терпеноида в циклический
и размыкания цикла:
СНз
цитронеллаль
СНз
(СН»СО)2О ; j- \
^/ХоСОСНз
НзС СН2
СНз
I
Л
z/\он
Н3С СН2
изопулегол
600° С (над стекловолокном)
ТЕРПЕНЫ II ТЕРПЕНОИДЫ
645
пулегол
СНз
Н3С СНз
пулегон
Ранее упоминался гликоль — терпин, получаемый гидратацией в кис-
лой среде гераниола или лимонена. Приведем синтез терпина по Перкину:
на динатрийциануксусный эфир, который мы напишем упрощенно (он по-
строен, очевидно, ионно, с распределением заряда аниона в основном
между кислородом и азотом), действуют эфиром [3-иодпропионовой кис-
лоты, гидролизуют полученный нитрилоэфир, декарбоксилируют малоно-
вый карбоксил и замыкают (уксусный ангидрид) два карбоксила получен-
ной пентантрикарбоновой кислоты в циклогексанон-4-карбоновую
кислоту:
0 о 0 0
II II II
RO—С С—OR RO—С C-OR
\ /
Н2С -L- сн2 — -> н2с сн2
Н2С сн2 н2с сн2
I RO. /\
RO—С—С—C=N \с CsN
II I 0^
О Na
II II
НО-С С-ОН
Н2С
I
Н2С
сн2
но
о
о о
II II
НО-С с—он
НоС^ сн2
П2С сн2
^crf
с
но о
(СНзСО)аО
—со2; —н20
I I
Н2С сн2
с
но о
ИЗОПРЕНОИДЫ
646
Сложный эфир полученной циклогексанонкарбоновой кислоты под-
вергают действию избытка метилмагнийиодида.
О
CHS
НОЧ /СНз
RO—С=О
«»
н.о
СНз—С—OMgl
СНз—С—ОН
СНз
СНз
терпин
эфир циклогексанон-4-кар-
6оновой кислоты
При дегидратации терпина образуется два изомерных спирта —
терпинеола, находящих применение в парфюмерии (запах сирени):
СНз
НОХ .СНз
IIОх .СНз
-Н2О
СНз-С-ОН
СНз-С-ОН
СНз
а-терпинеол
НзС СН2
СНз
терцин
Бициклические терпены и терпеноиды. Как мы уже отмечали, бициклы
с молекулярной формулой С10Н1в должны включать одну двойную связь.
При гидрировании такие природные бициклические терпены образмют
родоначальные, изомерные
роды С10Н18. Их три:
друг другу
3 терпинеол
бициклические углеводо-
сн3
8
СН3
сн,
СН3
СНз
н3с-С7
н3с-с-сн3
каран
камфан
замкнувший изопропильную
пинан
Они представляют собой как бы ментан, ____________.___
группу в мостик бицикла в орто-, мета- или пара-положение?
/г \P4y “Па каРана- в шведском, немецком (Симонсен) и русском
а. Арбузов) скипидарах найдены изомерные карены:
сн3 сн3
а3 -карен
С-н3
д4-карен
з
г
5
в
ТЕРПЕНЫ II ТЕРПЕНОИДЫ
647
Группа пинана. Наиболее обычная составная часть скипидара
сосны — а-ппнен. В меньших количествах в нем содержится р-пинен,
пли «нопинен», который обычно получают из скипидара ели.
сг-пинен
пинен
Строение а-ппнена было установлено Е. Е. Вагнером на основании
деструкции окислением, последовательные стадии которой можно сокра-
щенно изобразить следующим образом:
а-пинен
Оч СНз
НО О \
V
н3с-сх
пиноновая
кислота
пиновая
кислота
При гидрировании а-пинена получается как пинан, так и открытый
С. С. Наметкиным его стереоизомер пинокамфан:
СН3
пинан
пинокамфан
P-Пинен в качестве первого продукта окисления дает кетон
О
Пинен способен к многим изомеризациям. Так, при 350е С он изомери-
зуется, как уже указано, в аллооцпмен (Б. А. Арбузов). При пропускании
648
ИЗОПРЕНОИДЫ
его примерно около 300° С над катализаторами, содержащими окиси алю-
миния и титана, он превращается в камфен (В. Е. Тищенко, Г. А. Руда-
ков), что является одним из известных путец полупения камфоры из ски-
пидара. Наконец, интереснейшая перегруппировка, открытая Е. Е. Ваг-
нером, происходит при пропускании хлористого водорода в пинен. Сна-
чала НС1 присоединяется по двойной связи в соответствии с правилом
Марковникова, далее хлор отщепляется как анион. Образовавшийся
катион подвергается ретропинаколиновой перегруппировке (стр. 621)
с изменением скелета пинана в скелет камфана, затем хлор-ион присоеди-
няется к новому карбониевому центру:
Таким образом, пинен через неустойчивый третичный хлорид II пре-
вращается в борнилхлорид V — твердое вещество, пахнущее камфорой
и являющееся промежуточным продуктом в синтезе камфена и камфоры
но старому способу.
Группа камфана. Камфан, как и его изомеры, в природе
не найден. Самым главным его производным является камфора — важное
средство, стимулирующее деятельность сердца. Очень большие количества
камфоры тратились на пластификацию нитрата и ацетата целлюлозы.
Камфора С]0Н16О добывается из листьев камфарного лавра отгонкой
с паром (Япония). В СССР она получается синтетически из пинена по спо-
собу В. Е. Тищенко и Г. А. Рудакова.
Строение камфоры установлено Бредтом. Доводы в пользу этой струк-
туры дало изучение продуктов окисления:
гф-СООН
\/СООН
ноос
СООН
СООН
камфора камфорная
кислота
камфороновая
кислота
СООН
Н3с—с—сн3
НООС—С—СООН
СНз
СООН
Н3с—С—сн3
I 3
нс—СООН
СНз
Исходя из камфорной кислоты, Бредт осуществил частичный синтез
камфоры. Восстанавливая ангидрид камфорной кислоты I, он получил
лактон — камфолид II (восстановление направляется на менее простран-
ственно затрудненный соседством метила карбонил). Под действием ци-
анистого калия лактон размыкался в нитрил III, который был прогидро-
ТЕРПЕНЫ II ТЕРПЕНОИДЫ
649
лизован в двухосновную гомона мфорную кислоту IV, замкнутую
в камфору VI путем сухой перегонки кальциевой соли V кислоты:
ZHch2cn
^х/СООК
Н+; Н-2О
гфсн.- соон
Ч/СООН
III IV
VI
+ СаСО3
Позднее Комппа синтезировал и саму камфорную кислоту, используя
сложноэфирную конденсацию щавелевого эфира с эфиром диметилглута-
ровой кислоты. Следует помнить, что получающийся при этом сложный
эфир Р-кетонокислоты способен замещать на натрий водород звена, за-
ключенного между карбонилом и карбоксилом. Натриевое производное
метилируют и получают дикетокамфорную кислоту, а затем и саму кам-
форную кислоту:
% /0 сн2-с х °\ /0 сн-са
С-OR I OR 4 р ""OR
1 + C-OR С(С1 Гз)2 1 zOR и -> 1 р С(СНз)2 NaOCgHs^ 1
0^ си2-схЧ А о^4 1 OR ^сн-сС ^0
/0 Ох /СН-С^ Vi- XoR > 1 С(СНз)2 1 /0R oz с-сс - ^0 Na+ Ох /СН-С^ СН-СООИ 4 р 1 OR тт с 1 | С(СН3)2 2| С(СН3)2 А ; .or А1 \с—С\ ХС—соон 1 % 1 СПз СНз
Камфора имеет два асимметрических атома углерода и должна была бы,
по общему правилу, иметь четыре стереоизомера, но так как два из них
должны были бы иметь мостик, замкнутый в транс-положении, что не-
возможно геометрически, то существуют лишь два стереоизомера, а именно
антиподы D- и А-камфора:
(форма ванны циклогексаноного кольца)
сн3
форма ванны
(замыкание в mpcmc-положение неосуществимо)
<ХС=О
ИЗОПРЕНОИДЫ
650
Природная камфора иа камфарного лавра — правовращающая^ Фрей-
денберг установил ее стереохимическое соответствие с Д>-глюкозои.
При восстановлении камфоры до вторичного спирта появляется новый
асимметрическим атом углерода и каждая D- и Z-камфора дает по два
спирта: D- и L-борнеолы и D- и L-изоборнеолы. Оба антипода борнеола
встречаются в природе и значительно более лабильны, чем борнеолы
в смысле ретронинаколиновой перегруппировки, к которой, однако, спо-
собны все четыре спирта.
Борнеолу принадлежит структура с эндо-положением гидроксила,
иаоборнеолу — с экзо-положением:
борнеол
или
изоборнеол
При действии на эти спирты агентов, уводящих гидроксил, например
кислот (реакция особенно легко совершается с изоборнеолом), остающийся
изоборнил-катион количественно перегруппировывается в камфен, вы-
брасывая протон (стр. 620) и образуя двойную связь:
Формулу камфена можно написать и так:
СН2
1 2
для чего отмеченные стрелками звенья цикла нужно
скостью бумаги и отогнуть, как показано на формуле
является терпеном с новым для нас бициклическим
назван^’к \ТеРПеЕ- КОТОРОМУ °РавУ Должно было бы принадлежать
неола пп мр ?еНаыназывается борниленом и может быть получен из бор-
неола по методу Чугаева, получившего его таким способом:
поднять над пло-
2.
ONa
метилксантогенат ,
борнеола оорнилен
+ COS + CH3SH
ТЕРПЕНЫ И ТЕРПЕНОИДЫ
651
Камфен I, полученный Е. Е. Вагнером и затем Меервейном, при действии
кислот (гидратация и отщепление ОН) претерпевает ретропинаколиновую
перегруппировку в изоборнеол V (камфеновая перегруппировка I рода
или перегруппировка Вагнера — Меервейна). рассматривавшуюся уже
в разделе «Перегруппировки» (стр. 620). Эта перегруппировка изображена
на первых двух строках схемы. Ответвлением III->VI изображена пере-
группировка Наметкина, или камфеновая перегруппировка ТТ рода.
Обе перегруппировки приводят в конечном счете к двум антиподам изо-
борнеола (V) и (VIII). Перегруппировки проходят через общий для них
карбониевый ион III.
В перегруппировке Вагнера — Меервейна заряд этого карбониевого
катиона компенсируется переброской связи с разрывом цикла и последу-
ющим присоединением ОН- к вновь возникающему карбониевому центру.
В перегруппировке Наметкина карбониевый катион III компенсирует
заряд переброской метила от изопропильного моста, в результате чего на
цептральном углероде этого моста возникает карбониевый катион, ком-
пенсируемый новой Вагнер — Меервейновской переброской связи
(VI—>-VII) с появлением карбониевого центра (VII), связывающего затем
ОН- из среды.
Обе перегруппировки идут параллельно, но с разной скоростью, в зави-
симости от условий. Поэтому изоборнеол получается оптически активный,
частично рацемизовапный. То, что эта частичная рацемизация в условиях
перегруппировки Вагнера — Меервейна объясняется перегруппировкой
II рода, доказано Наметкиным на 4-метилизоборнсоле. Для этого соеди-
нения, как легко понять, перегруппировки Наметкина и Вагнера —
Меервейна ведут к получению структурных изомеров (ср. стр. 621, раз-
дел «Перегруппировки»).
652
ИЗОПРЕНОИДЫ
Сесквитерпены и сесквитерпеноиды
Так называются углеводороды Ci5H24 — тримеры изопрена и их кисло-
родные дериваты. Так же как и терпены, эти вещества находятся в эфир-
ных маслах п бальзамах растений. Многие из них используются в парфю-
мерии. Мы ограничимся лишь несколькими примерами из огромного
количества изученных веществ такого рода.
Запах цветущей липы обусловлен сесквитерпеноидом — фарнезолом.
Дегидратацией этого спирта получается алифатический сесквитерпен —
фарнезен:
СНз СНз СНз
I I I
СН3-С=СНСН2СН2С=СНСН2СН2С=СН-СНгОН ——>
—На О
СНз СНз СНз
I I I
—> СН3-С=СНСН2СН2С=СНСН2СН=С-СН=СН2
Пахучее начало имбиря — цингиберен — является моноциклическим
сесквитерпеном
СНз СНз СН2
сн3-с=снсн2сн2-сн-нс/ ^СН
I II
НС С—СНз
СН
Примером бициклических сесквитерпенов может служить а-селинен.
Следующая схема демонстрирует структурное родство алифатических
сесквитерпеноидов с бициклическими и с азуленами (стр. 512), находи-
мыми также в эфирных маслах растений:
сн3 Н3С J ] н3с-У/у носн, (2н3 фарнезол СН3 Н3С СН3 СН3 иначе написанный фарнезол сн3 нзС ГТ) сн3 а-селинен <V^Y> СП сн3 н3с сн3 гвайззулен
ТЕРПЕНЫ И ТЕРПЕНОИДЫ
653
Дитерпены, тетраизопреноиды и каротиноиды
Среди тетраизопреноидов с открытой цепью атомов следует назвать фи-
тол — спирт в виде сложного эфира входящий в состав хлорофиллов
(кн. 1, стр. 313). Родство скелета фитола с фарнезолом доказывается
синтезом его из гексагидрофарнезилбромида (Ф. Г. Фишер):
/ СНз \ ONa О
I I II
Н \СН2СНСП2СН2/3Вг + СН3—С=С11 — С—ОС2Н5 >
/°
с/
/ СПз \ I ХОС2115
( ' СНз гидролиз;
> Н\СН2СНСН2СН2 'з—СН—С<( ^тонное расщепление
А)
/ с Из \
1 /с
---> Н\СН2СНСН2СН2/3-СН2-С<Ч
О
нс сн;
кон
СН3 \ СНз
СНСН2СН2 'з—сн2— с—с==сн
I
он
/ СН3 \ СНз
—> Н\СН2СНСН2СН2/з—сн2—с—сн=сн2
I
он
(СНзСоцо;
аллильная перегруппировка
/ СНз \ СНз
—> Н\СН2СНСН2СН2/з-СН2—С=СН—СН2О—С-СНз —
СНз / СНз \ СНз
1 1 I
---> СНзСНСН2СН2\СН2СНСН2СН2 >2—сн2—с=си— СН2ОН
фитол
Тетраизопреноидный скелет имеют витамины группы А. Отсутствие
в пище витамина Ах (аксерофтол, или ретинол) или соответствующего
провитамина — а-каротина (см. ниже) ведет к ксерофтальмии — высы-
ханию роговицы глаз и слепоте, похудению и общему плохому состоянию,
а недостаток — к куриной слепоте — ослаблению сумеречного зрения.
Последнее обстоятельство зависит от того, что витамин А! служит источ-
ником зрительного пурпура — родопсина, содержащегося в колбочках
и палочках сетчатки и обусловливающего зрение, в частности вечернее,
серое.
Родопсин представляет собою белок онсин, связанный с ретинином
(иначе — ретиналем) — альдегидом, соответствующим витамину Ах как
спирту. Цепь ретинина имеет сплошную щранг-конфигурацию. Поглощая
ИЗОПРЕНОИДЫ
654
квант света, ретинин, связанный в родопсин, изомеризуется в 11-цис-ре-
тинин
отделяющийся от опсина с освобождением его SH-группы, что и служит
начальным толчком нервного импульса зрения. Родопсин — хромопро-
теид, он окрашен интенсивнее, чем ретинин. Только в родопсине проис-
ходит описанная изомеризация ретинина. Белок играет здесь каталити-
ческую, ферментативную, роль.
Витамин Ах находится в молоке и сливочном масле, желтке яйца,
рыбьем жире; а-каротин (провитамин А1; свободно превращающийся
в организме в витамин Аг) — в моркови и зеленых листьях многих растений.
Витамин А2, имеющий меньшее значение, содержит на одну двойную
связь больше, чем витамин Ах. Окисляясь в организме, а-. (3- и у-каротины
образуют витамин Av Строение каротинов и их связь с витамином Ах
выяснены в основном исследованиями Вильштеттера, Каррера и Р. Куна.
_______________________А_____________________
Н,С\ /СН, си* 71* сн» н,С</<
/\^/СН=СНС-СНСН=СН—С-СНСН=СН-СН = С—СН = СНСН=С—СН=СН\5\
। к II
СНа Н»с/^/
«-каротин
(цикл р-ионона)
3-каротин
(цикл «-ионона)
Н3СХ ZCH3
А С
нс %н
II I
с сн2
Н3С \н2
т-ка>втин
ТЕРПЕНЫ И ТЕРПЕНОИДЫ
655
СНз СНз
Н3СХ /СН3 I I
/><^СН=СНС=СНСН=СН—С=СН—СН2ОН
витамин At
Р-Каротин в организме расщепляется на две молекулы вита-
мина Аг
Строение а-каротина установлено Цехмейстером, Каррером и Р. Ку-
ном в 1928 г. на основании окисления его в героновую I и изогероно-
вую II кислоты:
О
II/ОН
НзС\/Сс/°
НзС^/Г^
О СН2
\z \
но—с с
/СНз
\сн3
Самая окраска а-каротина (темно-красные кристаллы) указывает
на сплошную цепь сопряжения. Взаимоотношения с витамином Аг также
много говорят о строении каротинов.
Синтез витамина А1? выполненный многими путями и ныне осуще-
ствляемый в промышленности, полностью подтверждает принятое стро-
ение. В спнтезе его, так же как и в синтезе а-каротина, исходят
из (3-ионона. Приводим синтез витамина Аг по Ислеру:
СНз О
-Na-COa"*"
656
ИЗОПРЕНОИДЫ
Эта серия формул разъясняет идущую в одну стадию реакцию Дарзана,
посредством которой в данном случае наращивается (3-ионон в гомологич-
ный альдегид. Дальнейший синтез ясен из схемы:
СПз
СН3
затем
СНо-СН=С-СНО | Н2О
+BrMgC=C—С=СН—CH2OMgBr
СН3
СН3
, СН2—СН=С-СН—С=С-С=СН—СН2ОН H2/Pd(Pb‘+)
он
СНз СНз
\//СН2—СН=С-СН-СН=СН-С=СН—СН2ОН (пиридан0:
\А "н
СН3 СНз
,СН2—СН=С-СН-СН=СН-С=СН—СН-2—о-С-СН3пР°С!’н
/ хх [ И пиридин
I он О -н’°
с Из СНз
/СН=СН-С=СН-СН=СН-С=СН-СН2-О-С-СНз хт о. хтт.
' \ / и JtLfU» xl •
! Л о
СПз
СНз
I I
СН=СН—С=СН—СН=СН—С=СН—СН2ОН
витамин At
Изомерен каротинам алифатический углеводород ликопин — крася-
щее начало спелого помидора и многих других растительных объектов.
Его структура также включает длинную систему — именно 11 — сопря-
женных л-связей. Приведенная формула ликопина подчеркивает родство
его с каротинами
Н3С Cfl3 СНз СПз СНз
р I I I
/СН=СНС=СНСН=СНС=СНСН=СПСН=ССН=
н2с сн
тт L II
GH3
Н3С СНз
сн3 А
снсн=ссн=сысн ^сн
II I
с сн2
НзС СН2
ликопин
657
СМОЛЯНЫЕ КИСЛОТЫ
Интересен алифатический тритерпен — сквален, родственный тетра-
циклическим стероидам. Этот изопреноид, найденный в печени акулы
имеет приведенное ниже строение, доказанное осуществленным Каррером
синтезом исходя из фарнезола, превращенного заменой ОН на Вг в фар-
незилбромид и сдвоенного действием магния по реакции Вюрца:
СНз СН3 СН3
2СН3-С= СНСН2СН2—С=СНСН2СН2—С=СН—СН2Вг -*ме ,
- MgBr,
( СНз СНз СНз \
I I I
-->• \СН3—С=СНСН2СН2—с=снсн2сн2—с=сн—сн2/2
смоляные кислоты
Уже говорплось, что при перегонке смолы хвойных растений отгоняется
летучая часть — скипидар и остается канифоль — вещество, имеющее
характер карбоновой кислоты и употребляющееся как добавка к жирным
кислотам при мыловарении, так как ее натриевая соль тоже обладает
поверхностно-активными свойствами мыла. Канифоль находит и другие
многообразные применения. Ее соли с марганцем и другими металлами,
называемые резинатами, употребляются как сиккативы — добавки, ини-
циирующпе окисление, а следовательно, и полимеризацию льняного
масла при варке олифы (они растворимы в маслах). Канифоль применяют
при проклейке бумаги, она входит в состав замазок и клеев (например,
менделеевская замазка, защитные клеи от гусенпц). Состав канифоли
С19Н29СООН, она представляет собой смесь кислот — абиетиновой и лево-
и декстропимаровой. Приводим структуру двух первых кислот, которые
(особенно абиетиновая) являются главными составными частями канифоли:
абиетиновая кислота
левопичаровая кислота
Как можно видеть, эти кислоты представляют собою производные
гидрированного фенантрена и в этом отношении родственны стероидам
(см. ниже). Действительно, при ароматизации смоляных кислот действием
серы при высокой температуре (при этом происходит дегидрогенизация
в ароматическую систему замещенного фенантрена с одновременным
декарбоксилированием и потерей ангулярного метила в впдо метилмер-
каптана) получается так называемый ретен (2-изопроппл-8-метилфенан-
трен). Этот углеводород образуется из смол в течеппе геологических
периодов и найден как ископаемое. Вместе с тем абиетиновая кислота,
42 Заказ С72
ИЗОПРЕНОИДЫ
658 —--------------------—-----
как и другие смоляные кислоты (и
Это видно из следующего рассечения
стероиды), является изопреноидом,
ее формулы:
НООС СН3
СТЕРОИДЫ
В прямой связи с пзопренпдамп находятся стероиды. Все стероиды имеют
структурно общий циклический скелет. Они делятся на следующие ооль-
шпе группы: стерпны (или стеролы), желчные кислоты, стероидные гор-
моны, генины (в виде гликозидов встречающиеся в растениях и находящие
применение в качестве лечебных стимуляторов деятельности сердечной
мышцы), яды жаб, саиогенпны и стероидные алкалоиды, которые в отли-
чие от предыдущих групп содержат азот. Мы рассмотрим только три пер-
вые группы стероидов и дадим понятие о четвертой.
Общим для всех стероидов является тетрациклический скелет цпкло-
пентанофенантрена, углеродные атомы которого нумеруются нижеука-
занным образом
СНз
и который в большинстве случаев при углеродах 10 и 13 имеет «ангуляр-
ные» метильные группы, при углероде 17 — цепь пз нескольких углерод-
ных атомов. В желчных кислотах эта цепь оканчивается карбоксилом,
в генпнах она принимает форму кислородного гетероцикла, а в гормонах
она обычно отсутствует и заменена на гидроксил пли карбонильный кисло-
род. Кроме того, весьма часто в положении 3 находится гидроксил (или
карбонильный кислород).
Стероиды широко распространены в живой природе. Спирты — сте-
рины (стеролы), у которых X — ОН, В. — углеводородный остаток нахо-
дятся в животных и растительных тканях и играют важную, но не вполне
ясную роль. Вещество мозга содержит до 7% холестерина в расчете на
сухую массу. Он входит в состав животных жпров. Отложение холесте-
рина в кровеносных сосудах считают причиной заболевания атероскле-
розом. Вырабатываемый дрожжами эргостерин является провитамином Da
и превращается в последний при облучении солнечным светом, а находи-
ncJ11 /КИВОТНЫХ организмах 1-дегидрохолестерин является провитами-
м 3. /Келчные кислоты, вырабатываемые печенью, необходимы для
секпрНИЯ 'киров- Гормоны пола, вырабатываемые железами внутренней
реции самцов и самок (мужские — андрогенные и женские — эстро-
СТЕРОИДЫ
659
генные и лутоидные), регулируют нормальное развитие самцов и самок
и их специфические для каждого пола нормальные отправления.
Наиболее давно известны холестерин, выделенный впервые из желчных
камней, п желчные кислоты. Однако их строение было окончательно
установлено лишь в 30-х годах нашего столетия в результате длительных
исследований Виланда, Впндауса и ряда других ученых. В 1929 г. Буте-
нантом и др. был выделен первый женский гормон, а в 1933 г. — мужской
гормон. Вслед за этим началось быстрое и широкое развитие всей области
стероидов, которая теперь составляет обширную и своеобразную главу
органической химии.
Родство стероидов с изопреноидами устанавливается доказанной воз-
можностью синтеза их в организмах из типичного изопреноида — сква-
лена (стр. 657). Сопоставление структур сквалена и одного из стеринов —
ланостерина наглядно обнаруживает их родство (точки — метильные
группы).
сквален ланостерин
Кроме замыкания четырех циклов за счет исчезновения четырех двой-
ных связей, для превращения скелета сквалена в скелет этого стерина
необходимо перемещение метильной группы, что, как мы знаем из химии
терпенов, часто наблюдается.
На деле эти сложные изопреноиды, так же как и все изопреноиды,
образуются в растениях и животных не из изопрена, который и не найден
в живых объектах, а из уксусной кислоты, что надежно установлено
введением в растительные и животные организмы уксусной кислоты, ме-
ченной по метильной группе радиоактивным изотопом 14С. Этот изотоп
оказывается включенным в скелет изопреноида. Самый синтез изопреноида
(например, сквалена) из уксусной кислоты идет по схеме, напоминающей
сложноэфирную конденсацию, под влиянием катализатора — кофермента А
(стр. 777), который мы будем обозначать через RSH. С уксусной кислотой
он превращается в RS—СО—СН3, который реагирует в биохимических
процессах как ацетилирующий агент.
При этом через стадию ацетоуксусной кислоты I путем ее конденсации
как кетона с новой молекулой уксусной кислоты образуется кислота II,
восстанавливающаяся ферментативно в мевалоновую кислоту III, а по-
следняя с потерей СО2 и Н2О фосфорилируется в изопентенилиирофос-
фат IV, который и служит простейшим кирпичом в синтезе изопреноидов.
Изопентенилпирофосфат IV в присутствии Mg2+ и суммы ферментов
из дрожжей (дрожжевой сок) превращается в геранилпирофосфат V и фар-
незилпирофосфат VI. Эти вещества ферментативно превращаются в сква-
лен VII.
42*
ИЗОПРЕНОИДЫ
660
CH.COSR
СНз-с-SR + СНз-С-ОН ► СНз-С-СНг-С—он
» 11 л О
О о ° I °
СНз С**3
I П I Н*Р,О,
НО-С-СН2-С-СН2-С-ОН —* Н0-С-СН2-С-СН2-СН20Н _со2;-н,о*
о он о J ™
тт ш
О О
IV
2IV --->
СНз.
)С
СНз7
СН3 0 0
I II II
=СНСН2СН2—С=СН— СН2—О—Р—О—Р—он
I I
он он
СН3\ I
3IV —> >С=СНСН,—\СН2—С=СНСН2/2
СН3/
о о
II II
О—Р—о—Р—он
I I
он он
VI
/ СНз \ / СНз \
СН3\ I I /СНз
2VI—> >С=СНСН2-\СН2—С=СНСН2/2\СН2СН=С-СН2/2-СН2СН=С<
СН37 ХСНз
VII
Участие мевалоновой кислоты подтверждено введением в организм
меченой мевалоновой кислоты и получением в результате меченых изо-
преноидов.
Стерины (стероидные спирты)
Ниже приведены структуры и конфигурации нескольких важнейших
стеринов животного (холестерин, копростерин) и растительного (эрго-
стерин, стигмастерин) происхождения:
СТЕРОИДЫ
661
эргостерин стигмастерин
Пунктирные линии обозначают, что данный заместитель или Н рас-
положен под плоскостью рисунка (такие заместители обозначают как а)
при условии, что метил при углероде 10 расположен над плоскостью
чертежа. Сплошные линии валентностей обозначают, что заместитель
или Н расположен так же, как СН3 при углероде 10 — над плоскостью
чертежа (именуются |3). Отсюда ясно, что кольца А и В в копростерине
связаны в z/кс-положении, а кольца В и С, С и D во всех четырех остальных
приведенных стеринах в траке-положении (как в тракс-декалине, кн. I,
стр. 566). Вопрос о цис- или траке-связи колец А и В в остальных четырех
стеринах снимается из-за наличия двойной связи, связывающей угле-
родные атомы 5 и 6.
В организме холестерин синтезируется, как и другие изопреноиды,
из уксусной кислоты, что доказано введением в организм меченной по
метильной группе уксусной кислоты. Этот опыт позволяет также
сделать выбор между схемами образования скелета холестерина через
сквален по Робинсону и по Вудворду — Блоху в пользу последней.
При окислении в жестких условиях (по Куну — Роту) холестерина,
образовавшегося в организме из меченой уксусной кислоты *СН3СООН,
остаются только фрагменты из двух углеродных атомов (один из них —
метильная группа) в виде молекул уксусной кислоты. По схеме Робинсона
при переходе от сквалена к холестерину новые связи образуются так,
ИЗОПРЕНОИДЫ
662
что при окислении не может сохраниться ни одной связи между двумя
мечеными углеродами и должна образоваться только СН3 СООН.
По Вудворду — Блоху есть возможность сохранения связей С С
(у ангулярных групп), а следовательно, может образоваться и кислота
*СН *СООН, что и наблюдается на деле. В результате подробного иссле-
дования полученного так холестерина, меченного 14С, было установлено
приведенное на формуле распределение меченых (бывших метильных)
углеродов. Очевидно, что при этом метильные группы сквалена, нумеро-
ванные 1, 2, 4, теряются, а группа 3 мигрирует во^второе ангулярное
положение.
Холестерин явился ключевым соединением в исследовании многих
стероидов, строение которых было установлено, опираясь на знание их
родственных отношений к холестерину. Мы ограничимся поэтому более
подробной аргументацией структуры холестерина.
Холестерин — непредельный спирт, содержащий один гидроксил
и одну двойную связь, что легко устанавливается обычными методами.
При гидрировании над платиной холестерин поглощает 1 моль водорода
и образует дигидрохолестерин, (или холестанол С27Н48О), стероизомерный
копростерину (или копростанолу). Копростерин получается гидрирова-
нием холестерина в кишках животных и содержится в кале. Заменой
в дигидрохолестерине гидроксила на хлор (РС15) и элиминацией хлора
(Na -f- С5Н11ОН; сначала отщепление НС1 и образование непредельного
холестена, затем гидрирование последнего) можно превратить его в соот-
ветствующий углеводород — холестан С27Н48, стереоизомерный копро-
стану, образующемуся при этом вместе с холестаном. Подобным же обра-
зом его можно получить из копростерина. Поскольку и холестан и конро-
стан не имеют непредельных связей, а их молекулярная формула С,7Н5в
отличается от молекулярных формул изологичных им парафинов на восемь
водородных атомов, ясно, что холестан и копростан — алициклы, состо-
ящие каждый из четырех циклов. ’
Указание на то, какие это циклы, дает дегидрирование (ароматизация)
холестерина над селеном при 360° С. При этом крекируется боковая цепь
холестерина и отходят в виде метана ангулярные метильные группы.
В результате получается углеводород Дильса ~~ 3'-метил-1,2-цпклопентано
фенантрен:
„Л™”” ° Т°М’ ЧТОг,"’ СебЯ пРвДставляет боковая цепь, дает окисление
tSohT S ССЙ СН CHyS?CH Гевм УДаЛ0СЬ вылел“ть 2-да
ПзЬСН2СН2СН2СН(СНз) 2. Местоположение карбонила
У ает на то, к какому углеродному атому была присоединена боковая
стероиды
663
цепь. Если бы холестерин, будучи тетрациклическим однонепредельным
спиртом, имел только октановую боковую цепь, его молекулярная формула
была бы С25Н42О, а на деле она С27Н48О, т. е. богаче на 2СН2. Таким
образом, кроме боковой цепи (2-метилгептанона-6) холестерин содержит
еще две метильные боковые группы (или одну этильную). Положение
длинной боковой цепи при углероде 17 в холестерине и других стеринах
следует из описываемого ниже деструктивного окисления некоторых
из этих соединений в половые гормоны. При этом деструкции подвергается
длинная цепь и дело заканчивается образованием карбонильного кисло-
рода на месте прикрепления цепи. Это как раз место 17. Что же касается
двух СН3-групп, то, поскольку при ароматизации они не остаются свя-
занными с ароматическими ядрами, а отщепляются в виде метана, они
должны быть четвертичными — ангулярными — метилами. Только в этом
случае ароматизация не может быть осуществлена путем одной дегидро-
генизации, а требуется и отщепление боковых групп. Точное положение
боковых групп устанавливается лишь путем окислительной деструкции.
Деструкцией также решается вопрос о положении гидроксила и двойной
связи в холестерине. При этом находят и новое более строгое подтвержде-
ние характера всех четырех циклов холестерина и их стереохимического
цис- или ттгранс-характера связи друг с другом.
Окисление холестерина приводит, в частности, к бициклической три-
карбоновой кетонокислоте:
Поскольку окисление направляется в первую очередь по двойной
связи и по вторичной спиртовой группе (образуется кетон, который дает
дикарбоновую кислоту), ясно, что образование трикарбоновой кислоты
доказывает нахождение гидроксила и двойной связи холестерина в раз-
ных циклах.
Обстоятельство, что двойная связь находится в положении р — у
по отношению к гидроксилу, доказывается следующим образом. Осторож-
ным окислением двойной связи по Вагнеру перманганатом превращают
холестерин в триол II, последний окисляют далее в диоп-ол III, причем
одна спиртовая группа исходного триола (очевидно, третичная) не окис-
ляется. Дегидратируя ее и гидрируя полученный непредельный дикетон IV
в предельный дикетон V, убеждаются по типичным реакциям, что это
1,4-дикетон. Поскольку удаленный дегидратацией гидроксил был третич-
ным и, значит, занимал ангулярное положение, взаимоположенпе гидро-
ксила холестерина и его двойной связи определено.
ИЗОПРЕНОИДЫ
664
Однако нужно определить не только относительное положение гидро-
ксила и двойной связи (расстояние по цепи углеродных атомов), но и при-
вязать гидроксил к определенному углеродному атому и тем определить
положение двойной связи. В частности, этот вопрос решается на основании
окислительной деструкции дигидрохолестерина VI (холестанола) и холе-
стерина — процессы, еще раз подтверждающие строение цикла А как
циклогексанового кольца.
Полученный окислением дигидрохолестерина VI кетон VII далее окис-
ляется в дикарбоновую кислоту VIII. Разными путями можно доказать
наличие в этой кислоте четырех а-водородных атомов, т. е. наличие двух
метиленовых групп, к которым присоединены карбоксилы. Это делает
возможным наличие гидроксила в холестерине в положениях 2 или 3.
То обстоятельство, что кислота VIII при действии уксусного ангидрида
образует кетон (с потерей СО2 и Н2О), а не ангидрид двухосновной кис-
лоты, доказывает, что несущий гидроксил цикл А шестичленный, иначе,
оудь эта кислота производным глутаровой кислоты, а не адипиновой,
данная реакция привела бы к производному глутарового ангидрида,
не к кетону. Это общий метод, позволяющий отличать шестичленные
СТЕРОИДЫ
665
циклы от пятичленных (и низших). Что соединение IX действительно
пятичленный кетон, доказывается его окислением в производное глута-
ровой кислоты, замыкающееся уже не в кетон, а в ангидрид XI.
Холестерин I можно окислить посредством КВгО с разрывом цикла А,
не затрагивая двойной связи, причем образуется дикарбоновая кис-
лота XII, имеющая одно подвижное метиленовое звено (а-СН2). При
дальнейшем окислении SeO2 кислота XII превращается в кетонокис-
лоту XIII, которая окисляется с исчезновением двойной связи (следова-
тельно. с разрывом цикла В) в бис-а-кетонокислоту XIV, имеющую три
карбоксила. То, что обе кетонокислотвые группировки являются а-,
а не (3-кетонокислотными группировками, легко определяется характер-
ными реакциями и решает вопрос о положении гидроксила при углерод-
ном атоме 3 цикла А. Легко проследить, что, если бы гидроксил находился
при углероде 2, одна из кетонокислотных групп имела бы а-, а другая
р-кетонокислотный характер. При дальнейшем окислении вещества XIV
происходит отгорание двух карбоксилов (в СО2) и образуется трикарбо-
новая кислота XV, которая циклизуется с одновременным декарбокси-
лированием в кетонокислоту XVI. Эта последняя при окислении образует
трикарбоновую кислоту XVII, циклизующуюся также в кетоно-
кислоту XVIII:
XV
xvr
ИЗОПРЕНОИДЫ
666
Кетонокислота XVIII содержит уже карбонильную группу в пяти-
членном цикле, что можно установить окислением ее с разрывом этого
цикла в трикарбоновую кислоту, способную циклизоваться лишь в ан-
гидрид, а не в кетон. Отсюда следует, что кетонокислота XVI содержала
карбонил в шестичленном цикле, и, значит, в трикарбоновой кислоте
XV карбоксилы, участвующие в циклизации, отстояли друг от друга
на расстояние в семь углеродных атомов (один из семи углеродов уходит
при циклизации в виде СО2). Следовательно, формула XIV должна при-
надлежать а,а-дикетонокислоте и формула XIII — предыдущей стадии
окисления, сохранившей еще двойную связь холестерина. Таким образом
устанавливается шестичленное строение и кольца В холестерина.
Пятичленность цикла D подтверждается следующими реакциями.
Продукт гидрирования холестерина — копростерин XIX превращается
в соответствующий углеводород копростан путем замены в копростерине
группы ОН на С1 (действие РС15) и замены С1 на Н (Na в спирте). Копро-
стан XX при окислении образует 17-тестанон XXI, последний при даль-
нейшем окислении дает дикарбоновую этиобилиановую кислоту XXII,
из которой при циклизации получается ангидрид XXIII, а не кетон.
Следовательно, цикл D действительно являлся пятичленным.
ххп
XXIII
СТЕРОИДЫ
667
Изложенного достаточно, чтобы понять принципы установления струк-
туры стероидов путем окислительной деструкции. При этом выясняется
и положение метильных групп. Здесь, в частности, большие услуги ока-
зывает метод Барбье — Виланда (сокращение на одно звено СН2 цепи
карбоновой кислоты).
Понятие о методе дает следующая схема:
CH2-CH2-C<°R
О _|_
V'4/CH2-ch2-c<C6H5
1 6нСбН5---->-
2C6H5MgBr
С6Н
он
С6Н
Образующаяся кислота содержит карбоксил при четвертичном угле-
роде, что следует из невозможности дальнейшего сокращения числа
углеродных атомов по Барбье — Виланду.
Так выясняется местоположение четвертичного углерода (и тем
самым ангулярного метила).
Конфигурация стероидов
Тетрациклический скелет стероидов, не содержащих двойных связей,
может быть построен на основании цис- или транс-сочетания (как
в цис- и транс-декалинах) каждой пары циклов А и В, В и С, С и D.
Примером могут служить стереоизомерные углеводороды, соответству-
ющие холестерину — холестан и копростан (см. стр. 668).
В приведенных формулах стереоизомеров выделены водородные
атомы и метильные группы, относительно которых рассматриваются
Цис- и транс-положения.
В холестане в цикле А жирной чертой обозначена связь С—С, отно-
сительно которой циклы А и В находятся в транс-положении,
а в копростане — связь, относительно которой циклы А и В находятся
в цис-псложении.
ИЗОПРЕНОИДЫ
668
или
холестан (транс-, транс , транс-)
Вопрос о цис- или /пране-связи циклов А, В, G и D друг с другом
решается несколькими путями.
1. Посредством окислительной деструкции. Так, обстоятельство, что
при окислении холестанола VI (стр. 664) образуется кислота VIII, в ко-
торой обе группы НО—СО—СН2—находятся в транс-положении друг
к другу, доказывает транс-декалиновое расположение циклов А и В
в холестаноле. Очевидно, что подобная реакция с копростерипом должна
привести к г/нс-стереомеру кислоты VIII. Таким образом, вопрос сводится
к установлению цис- и транс - конфигурации замещенных циклогек-
санов.
2. Выяснение расположения атомов путем рентгеноструктурного ана-
лиза. Этот в то время весьма кропотливый путь был применен при исследо-
вании холестерина (Кроуфут и Карлайсл).
3. Установление стереохимического родства исследуемого стероида
с опорным веществом с известной стереохимией, например с холестери-
ном. Такое родство устанавливается по тождеству продуктов деструкции
в тех или иных превращениях.
4. Некоторое суждение можно получить на основании грубой аддитив-
ности молекулярного вращения плоскости поляризации.
Кроме стереохимии скелета стероиды характеризуются и стерео-
химией расположения заместителей. Так, гидроксил холестерина и копро-
стерина может быть в цис- или транс-положении по отношению к бли-
жайшему ангулярному метилу (при углероде 10). Заместитель, находя-
щийся в транс-положении по отношению к ближайшему ангулярному
метилу, обозначается буквой а, а заместитель в z/wc-положении — буквой 0-
В формулах принято ангулярный метил предполагать над плоскостью
формулы, т. е. соединять его сплошной чертой и так же поступать с 0-за-
местителями (цис-), а a-заместители (транс-) соединять с циклом пунк-
тиром. Соединения, стереоизомерные данному, по этому признаку (по
положению заместителя) принято обозначать приставкой эпи:
СТЕРОИДЫ
669
холестерин
холестанол
(дигидрохолестерин)
копростерин
(копростанол)
эпихолестерин
эпихолестанол эпикопростерин
Наиболее простой путь определения стереохимии оксигруппы (цис-
или (3~, транс- или а- по отношению к близлежащему ангулярному ме-
тилу) связан с конформацией молекулы стероида и с экваториальным
или аксиальным положением гидроксила. На приведенных ниже фор-
мулах замещающие группы в скелетах холестана — углеводорода, соот-
ветствующего дигидрохолестерину, и копростана — углеводорода, соот-
ветствующего копростерину, классифицированы на экваториальные <?
м аксиальные а:
холестан
Заместители с разной легкостью вступают в экваториальное и акси-
альное положения и покидают их. Это объясняется тем, что лежа-
щие близко к (средней) плоскости цикла экваториальные заместители
более удалены от соседних атомов и пространственные препятствия меньше
влияют на их вступление. Между тем, в отличие от жирного ряда, кон-
формация тетрациклического скелета закреплена и положение а- и с-за-
местителей постоянно.
На примере циклогексановых производных известен ряд реакции,
отличающих вступление аксиальных и экваториальных заместителей
га их элиминацию. Так, восстановление циклогексанонов натрием в спирте
ИЗОПРЕНОИДЫ
670
ведет к образованию экваториальной группы, а каталитическое гид-
рирование водородом — довольно часто к аксиальной. Дегидратация
кислотами легче происходит при аксиальном положении гидроксила
П т. д.
Поэтому кетон, образующийся при окислении дигидрохолестерина,—
холестанон-3 (I) при восстановлении его карбонильной группы натрием
в спирте дает дигидрохолестерин II с экваториальным расположением
группы ОН, которое, как видно на схеме, является ^uc-положением по
отношению к ближайшему ангулярному метилу. Таким образом устана-
вливается, что холестерин имеет конфигурацию холестанола-3-[3. По-
добным образом и продукт окисления копростерина — копростанон-3
(III) восстанавливается в спирт IV, причем опять-таки гидроксил занимает
экваториальное положение. Как видно из схемы, этот экваториальный
заместитель в данном случае находится уже в транс-положении по отно-
шению к ближайшему ангулярному метилу, т. е. конфигурация полу-
ченного спирта IV выражается термином копр оста цол-3-а:
° описа™ми стеринами следует назвать стерин соевых бобов —
стигшст^ин, конфигуративно тождественный холестерину, но отли-
иостиИИвСЯХР^УРОИ °£°В0Й цепи’ эргостерин — содержащийся, в"част-
тем что о/яипдат' ГрИ ростках-пшеницы п особенно интересный
провитамин “ пРОВитамином D2, а также 7-дегидрохолестерин -
стигмастерин
эргостерин (провитамин D2>
СТЕРОИДЫ
671
7-дегидрохолестерин (провитамин D3)
7-Дегидрохолестерин является животным стерином, содержится в коже
и нервной ткани животных и в особенно большом количестве — в мол-
люсках раковин. Его можно получить синтетически из эфира холесте-
рина, бромируя его бромсукцинимидом (гомолитическое бромирование __
бром вступает в аллильное положение) и отщепляя основанием НВт:
При облучении ультрафиолетовым светом эргостерин и 7-дегидро-
^олестерин (как таковые или в организме) превращаются в своп изомеры —
кальциферол (витамин D2) и холекалъциферол (витамин D3), обладающие
близкой активностью. Это самые активные из витаминов группы D. Ви-
тамины D — необходимая составная часть пищи человека (и животных),
регулирующая усвоение кальция и рост костей и зубов. Недополуче-
ние молодым организмом витаминов D ведет к заболеванию рахитом
с характерными для пего мягкостью и искривлением костей.
Другие витамины группы D отличаются только строением боковой
цепи, соответствующей у них разнообразным другим стерпнам, и обла-
дают значительно меньшей активностью. При облучении провитаминов
разрывается цикл В и взамен появляется третья двойная связь в сопря-
женном положении к двум другим, также сопряженным. На примере
эргостерина (провитамин D2), опуская промежуточные стадии, процесс
.можно изобразить следующим образом:
ИЗОПРЕНОИДЫ
672
Суточная доза витаминов D (D2 или D3) на севере для взрослого чело-
века 7—12 мкг в сутки, для ребенка 17—25 мкг, а при рахите значи-
тельно больше.
Наибольшее количество витаминов D находится в жире печени рыб,
особенно скумбрии. Они содержатся также в масле, молоке, яйцах (жел-
ток), грибах. В настоящее время витамины D выделяют из неомыляемой
части жира рыб или китов или получают облучением из провитаминов.
Желчные кислоты
В желчи животных и человека находятся желчные кислоты. Например,
в желчи человека — холевая I, дезоксихолевая II, литохолевая III и другие
кислоты. Это монокарбоновые оксикислоты с одинаковым скелетом,
разнящиеся по количеству гидроксилов.
Все они путем замены гидроксилов на водород (например, дегидра-
тацией с последующим гидрированием образовавшихся двойных связей)
превращаются в холановую кислоту V, в свою очередь полученную Ви-
ландом путем окисления копростана IV
Таким способом было установлено тождество циклическоготскелета
желчных кислот между собой и со скелетом копростана, т. е. идс-распо-
ложение циклов А и В и транс-расположение циклов В и С и С и D,
а равно и положение цепи при углероде 17 и строение ее.
СТЕРОИДЫ
67S
В этих кислотах только гидроксил при углероде 3 экваториален,
остальные гидроксилы аксиальны. Экваториальный гидроксил ацили-
руется гораздо легче, чем аксиальные, поэтому он может быть защищен
ацилированием. И наоборот, окисляется он наиболее трудно. Наиболее
легко окисляется аксиальный гидроксил в положении 7, поэтому при
осторожном окислении его можно окислить в карбонил, и затем по Киж-
неру — Вольфу восстановить в СН2-группу. Получается дезоксихолевая
кислота. При повторении такой операции то же произойдет с гидроксилом
в положении 12 и получится литохолевая кислота:
тезоксихолевая кислота
Желчные кислоты, важнейшая из которых холевая кислота, выраба-
тываются печенью. Они содержатся в желчи в виде ацилированных холе-
вой кислотой гликокола (кн. 1, стр. 485) — гликохолевая кислота и таурина-
H2NCH2CH2SO3H — таурохолевая кислота и имеют важное значение для
переваривания жиров, которые они эмульгируют. При этом играет роль
способность желчных кислот образовывать с жирными кислотами моле-
кулярные соединения. Генетически желчные кислоты связаны с холесте-
рином. Вводя в организм меченый холестерин, констатируют образование
меченых желчных кислот.
Стероидные гормоны
Половые гормоны. Понятие о железах внутренней секреции как об орга-
нах, вырабатывающих физиологически активные вещества — гормоны,
выделяемые в кровь и служащие для регулирования разнообразных
физиологических отправлений, связанных с обменом веществ, введено
в науку в середине прошлого столетия Клодом Бернаром. Давно несом-
ненный факт, что мужские и женские половые органы (гонады) играют
и роль желез внутренней секреции (у самцов — тестикулы, у самок —
яичники), лишь в 1930 г. удалось доказать непосредственно — выделе-
нием соответствующих гормонов, выяснением их специфического дей-
ствия и последующим установлением их структуры.
43 Заказ 672
ИЗОПРЕНОИДЫ
674
Все — и мужские и женские — половые гормоны оказались спиртами,
фенолами или кетонами (или оксикетонами) с циклопентанофенантрено-
?ым скелетом. Их поиски и нахождение связаны с разработкой биологи-
ческих тестов и с количественной оценкой содержания этих веществ по
их физиологическому эффекту. Мужские гормоны извлекаются из те-
стикул или из мочи самцов, где они содержатся в ничтожном количестве.
Они обладают способностью возвращать кастрированным в молодом
возрасте самцам утраченные вторичные половые признаки. Так, кастри-
рованные петухи имеют неразвитый гребень и общий вид курицы. Вспры-
скивание в их кровь андрогенно-активных (т. е. обладающих^своиствами
мужских гормонов) растворов приводит к быстрому росту гребня (хвоста,
шпор) и к развитию нормального петушиного облика. Установлены
количественные соотношения между быстротой роста гребня п содержа-
нием (и активностью) гормона. Подвергая обогащению вытяжки гормо-
нов путем экстрагирования источника гормона, убеждаются в повышении
концентрации экстракта путем подобных биологических проб. Таким
образом в 1931 г. Бутенант впервые выделил из 25 т мужской мочи
15 мг индивидуального кристаллического вещества, названного андро-
стероном. Позднее выяснилось, что концентрация андростерона в моче
достигает 1 мг!л (и ничтожный выход зависел от трудности и несовер-
шенства выделения). К 1935 г. было выяснено (Лякер, Давид), что
непосредственно из текстикул (быка, жеребца) можно экстрагировать
другой, в 10 раз более сильно действующий мужской гормон — тесто-
стерон. по отношению к которому андростерон является продуктом
восстановления.
Еще раньше, в 1929 г. Бутенант и Дойзи выделили первый женский
эстрогенный гормон — эстрон. Он содержится в большом количестве
в моче беременных женщин и самок, но также и в моче самцов, напри-
мер жеребцов, откуда и получается. Этому не следует удивляться, так
как организм освобождается таким путем от ненужного ему гормона.
Эстрон лишь один из серии выделенных в настоящее время родственных
по действию и структуре «эстрогенных» гормонов, вызывающих состояние
течки у животных и обеспечивающих первую фазу месячного цикла
у женщин. Еще более сильно действующим эстрогеном является эстра-
диол, выделенный из яичников животных. Индикатором действия этого
рода гормонов служит кастрированная самка мыши, у которой введение
некоторого минимального количества гормона («1 мышиная единица»)
вызывает признаки течки. Однако для организма женщин и самок харак-
терна еще и вторая группа гормонов, так называемого лутоидного дей-
ствия. Эти гормоны начинают вырабатываться «желтым телом», разви-
вающимся в яичнике после совершившегося в первой фазе месячного
цикла (женщина) — выделения яйца (овуляции). Примером таких гор-
монов служит лутеостерон. Назначение этого рода гормонов -— под-
готовка организма к беременности, в частности подготовка матки к вос-
приятию и развитию оплодотворенного яйца. Интересно, что эстрогенные
n°nnZH п ЭСтР°генн0 Действующие вещества) широко распространены
ПК НИ найдены в цветах и плодах многих растений. Эстрогены
-можно 21пСТИМуЛир1Ющее Действие на Р°ст и цветение растений. Воз-
’ навоз обязан частью своего полезного действия на полях
СТЕРОИДЫ
675-
всегда присутствующим в нем эстрогенам. В настоящее время эти гормоны
и некоторые их синтетические физиологически активные аналоги служат-
важными медицинскими средствами. Так, эстрон и тестостерон приме-
няются как тонизирующие и как лечебные препараты при недостаточной
продуктивности гонад, болезненной или возрастной. В некоторых слу-
чаях они эффективны против рака (предстательной и грудных желез
и ДР-)- Лутеостерон предупреждает выкидыши и обеспечивает нормаль-
ное течение беременности. В организме все эти гормоны вырабатываются
под воздействием «гонадотронных гормонов», выделяемых гипофизом
(нижний придаток мозга) и имеющих другую природу. Удаление гипо-
физа, так же как и удаление гонад, ведет к угасанию половой функции.
Приводим формулы наиболее важных половых гормонов:
андростерон
тестостерон
Эстрогенные (фолликулярные) гормоны
Лутоидные гормоны
Углеродный скелет всех этих веществ структурно один и тот же,
эстрогены отличаются отсутствием одной из ангулярных метильных
групп, а у прогестерона в положении 17 имеется ацетильная боковая
Цепь. Циклы В и С, С и D имеют, как всегда, /пранс-расположение.
43*
ИЗОПРЕНОИДЫ
<676----------------------------------------------------------------
Вопрос о цис- или транс-положении циклов А и В возникает только
в случае андростерона. Это — транс-связь, как показывает его получе-
ние (Ружичка, выход всего 0,5%) путем окисления хромовой смесью
ацетата эпидигидрохолестерина, в котором циклы А и В имеют транс-
декалиновую конфигурацию:
Эта же реакция доказывает тождество конфигурации гидроксилов
обоих веществ (а-гидроксил) и отличие ее от конфигурации у холесте-
рина (p-конфигурация, цис- по отношению к близлежащему метилу).
Гидроксилу в эпидигидрохолестерине принадлежит та же конфигурация,
что в стереоизомере андростерона — эпиандростероне.
Связь структур андростерона и тестостерона устанавливается следу-
ющим путем. Тестостерон IV и дегидроэпиандростерон II окисляются
.ацетоном в присутствии mpem-бутилата алюминия по Оппенауеру в один
и тот же дикетон III с сопряженной системой связей С=С—С=О. В свою
очередь дегидроэпиандростерон II получают постепенным окислением
холестерина I, двойная связь которого защищена присоединением брома,
отщепляемого затем цинком. Этот многостадийный переход символизи-
рован на схеме волнистой стрелкой:
Наблюдающееся при окислении II в
в сопряженное к карбонилу положение
точных реакциях.
III перемещение двойной связи
совершается во многих анало-
СТЕРОИДЫ
677
Дегидроэпиандростерон служит также источником для синтеза тесто-
стерона. оти отношения выражаются схематически так:
дегидроэпиандро-
стерон (ацилированный)
тестостерон
Синтез основан на том, что при любом восстановлении 17-кетогрулпа
восстанавливается в 17-р-оксисоединение с конфигурацией, соответству-
ющей тестостерону. Эфиры 17-оксигруппы труднее гидролизуются, чем
эфиры 3-оксигруппы. Как всегда, двойная связь из цикла В легко пере-
мещается в цикл А в сопряженное с С=О положение.
Все эстрогенные гормоны имеют фенольную группировку, а экви-
ленин, в частности, (3-нафтольную, что легко устанавливается обычными
реакциями ароматического ядра.
Восстановление кетонной группы эстрона дает два стереоизомерных
диола — эстрадиол, обладающий более активным гидроксилом с мень-
шими пространственными препятствиями (значит, |3-гидроксилом), и его
эпимер. Эстрадиол сначала был получен таким способом из эстрона и лишь
затем из яичников животных. Структура эстрона установлена окислительной
678
ИЗОПРЕНОИДЫ.
деструкцией пентанового цикла и сличением продуктов деструкции
(и дегидрогенизации) с синтетически полученными производными фе-
нантрена.
Строение прогестерона было установлено деструкцией и подтверждено
синтезом из дегидроэпиандростерона. В производстве этого важного-
для медицины гормона используется способ Бутенанта и Шмидт-Томэ,,
который приведен на следующей схеме:
° I?
Г Т___J (С11зСО)гО .f J HCN
io Г'
сн..
CN 1/ОН * /Д'У -Н,О> 1 1 J н сн..со-ХУХ II о снч - 1 с=о \ Т J н I 1 Еще проще синтез прогестерона из нанту: зашита ОН 1 1 —• и двойной связи 1 окислен не VC=C<CbH5 Cti“5 y-yAJ 1 . о Г roAAJ Г Т и о см /\-Ах CH3MgBr; 1 | затем H-.Q 1 Т Зн о сн3 сн3 с=о с=о '"у \ окисление по > 1 J j Оппенаузру^ , [ й стигмастерина по Слотта и Буте- А-°н II) С6Н5М£Вг(избыток); - ^.^11 [ затем Н2О СН3 сн3 с=о с=о । окисление по /''Х/Х Оппенауэру | хЛ : >- » 1 J 1 j Г । j
СТЕРОИДЫ
679
и полные синтезы всех глав-
БыЛ” “ЛЩеСТВлены <В"в°Р«. Джонсов) н полные синтезы всех глав-
"STnSZT"”’ которые мы ве приводим ввиду их многостадий-
ности. Приведем лишь сравнительно короткий полный синтез эстрона
ио Джонсону: ovipuiw
сравнительно короткий полный синтез эстрона
Гормоны коры надпочечников — кортикостероиды. В то время как «моз-
говое» вещество надпочечников выделяет в кровь гормон адреналин
(стр. 167), кора надпочечников синтезирует и выделяет в кровяное русло
серию стероидных гормонов. Без этих гормонов жизнь невозможна.
Оперативное удаление коры надпочечников ведет к смерти, недостаточная
ее работа (гипофункция коры надпочечников) вызывает Аддисонову бо-
лезнь. Всего из коры надпочечников извлечено до 40 индивидуальных
ИЗОПРЕНОИДЫ.
680
стероидов (Рейхштейн). Мы назовем только кортикостерон I, как ключе-
вую структуру, кортизон II и дезоксикортикостерон III:
СН2ОН СН2ОН СН2ОН
с=о с=о с=о
I п ш
Кортизон выполняет важную функцию в углеводном обмене, именно*
в синтезе гликогена и превращении белков в углеводы, участвует в вы-
ведении воды из организма; по-видимому, главная функция дезокси-
кортикостерона— поддержание нормальногосолевого баланса организма
(соотношение К+ : Na+). Практически для медицины особенно интере-
сен кортизон — важное средство для лечения ревматизма и артритов.
Некоторые чисто синтетические стероиды обладают тем же действием
и также применяются в медицине. Таков преднизон
СИ.,ОН
I -
С=о
Наиболее простую структуру из этой группы гормонов имеет дез-
оксикортикостерон III, который является 21-оксипрогестероном, что*
доказано заменой гидроксила боковой цепи на иод и восстановлением по-
следнего цинком:
О
цезоксикортикостерон
прогестерон
СТЕРОИДЫ
681
Все гормоны этой группы имеют группировку —С—СН2ОН, вследствие
II
О
чего отличаются рядом особенностей — образуют озазоны, дают специ-
фические реакции окисления. При окислении иодной кислотой группа
СН2ОН отгорает в виде формальдегида и карбоксил оказывается на
месте —С—СН2ОН.
II
О
Дезоксикортикостерон отличается от кортикостерона отсутствием од-
ной гидроксильной группы. Кортикостерон и кортизон (после изомери-
зации) дают при окислении одну и ту же непредельную дикетоно-
кислоту IV, откуда следует, что в обоих соединениях кислородный атом
(гидроксила в кортикостероне, карбонила в кортизоне) занимает одно
и то же место:
Установить положение необычного гидроксила в цикле С кортико-
стерона и карбонила в кортизоне было достаточно трудно, тем более, что
та карбонильная группа кортизона, о которой идет речь, инертна и не
дает обычных кетонных производных. Поскольку один карбонил занимает
положение 3, ацетил и гидроксил — положение 17, а двойная связь
начинается от четвертого углерода, для третьего карбонила остаются
положения 1; 2; 6; 8; 11; 12; 15 и 16. Однако кортизон не обладает до-
статочно характерными свойствами 1,2- и 1,3-дикетонов. Значит, поло-
жения 1; 2; 15 и 16 исключаются. Свойства карбопила в положениях 6;
7 и 12 известны по продуктам окисления желчных кислот, они отличны
от свойств кортизонного карбонила — карбонилы 6, 7 и 12 дают кетонные
реакции. Таким образом, для третьего карбонила кортизона, методом
исключения, остается положение 11.
ИЗОПРЕНОИДЫ
682
Синтез кортикостероидов представлял большие затруднения, так как
нужно было ввести гидроксил или карбонильный кислород в положение
11, где во всех доступных исходных стероидах нет функциональной группы.
С открытием того факта, что микроорганизмы способны производить
такого рода окисления и что, применяя окисление дрожжами, актино-
мицетами и разнообразными бактериями, можно вводить гидроксил
по желанию в положения 6; 7; 8; 9; 11; 14; 15; 16; 17 и 21, притом в а-
или p-конфигурации, стало возможным относительно просто получать
кортизон из прогестерона:
Генины и сапонины
В различных видах наперстянки (дигиталис) и в строфанте содержатся
гликозиды стероидных спиртов, которые давно уже употребляются в ме-
дицине в качестве средств, возбуждающих работу сердца. Это очень ядо-
витые вещества. Гидролиз приводит к отделению углеводной части мо-
лекулы от агликона (генина). Мы ограничимся приведением формул глав-
нейших получаемых так агликонов — дигитоксигенина I, гитоксигенина
11, строфантидина III:
СТЕРОИДЫ
683
Эти и другие генины получены Штоллем, Чеше и Рейхштейном. Осо-
бенностью их конфигурации, отличающей эти вещества от всех ранее
приведенных типов стероидов, является ^ис-соединение циклов С и D.
Циклы А и В также имеют цис- дека липовую конфигурацию и лишь
циклы В и С транс-св язавы.
В растениях довольно широко распространены сапонины (например,
в мыльнянке). Это ядовитые гликозиды с сильно выраженной поверх-
ностной активностью, вспенивающие водные растворы, как мыло. Их
агликоны называются сапогенинами и относятся к стероидам. Примером
может служить тигогенин:
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
Алкалоиды — группа веществ, так же как и терпены, выделяемая не
только на основании химического признака. Так называются азотистые
основания, извлекаемые из растений (сюда же иногда относят и некоторые
физиологически активные основания организма животных) и оказыва-
ющие сильное физиологическое действие на организм животных, обычно
на нервную систему. Основной характер и солеобразование позволили
уже в начале прошлого столетия индивидуализировать многие алкало-
иды (морфин — Сертюрнер, 1806 г.; хинин, колхицин — Пеллетье и
Каванту, 1820 г.; никотин — Боссельт и Рейман, 1828 г.; атропин —
Мейн, 1831 г.; теобромин — А. А. Воскресенский, 1842 г.).
Благодаря сильному физиологическому действию эта группа веществ
приобрела важное значение в качестве лекарств и многие алкалоиды
задолго до их индивидуализации применялись с этой целью в виде на-
стоек и экстрактов растений. Еще раньше соки алкалоидоносных расте-
ний нашли употребление в качестве ядов, например, содержащие кураре
и стрихнин — для нанесения на наконечники стрел.
Алкалоиды, наряду с физиологически активными гликозидами,
давно уже составляют существенную часть арсенала медицины и эта часть
пополняется до последнего времени новыми открытиями. Многие важ-
ные алкалоиды сложного строения выделены в индивидуальном состоянии
уже относительно давно, но строение их установлено только в текущем
столетии, иногда совсем недавно, а синтез наиболее сложных из них (хи-
нин, стрихнин, резерпин) осуществлен лишь в последние годы.
Алкалоидоносными растениями являются в основном растения несколь-
ких семейств: мотыльковых, бобовых, маковых, пасленовых, лютико-
вых, мареновых, сложноцветных. Обычно алкалоиды сосредоточены не-
во всех, а в каких-либо определенных тканях растения (никотин — в ли-
сте табака, хинин — в коре хинного дерева и т. д.). Часто один вид расте-
ния содержит несколько алкалоидов. Алкалоиды связаны в растениях
в соли, образованные или широко распространенными кислотами расти-
тельного мира — яблочной, винной, лимонной и т. д., или, в некоторых
случаях, какими-либо кислотами, специфичными для данного алкалоида.
ыделяют алкалоиды, обрабатывая алкалоидоносиую ткань щелочью*
АЛКАЛОИДЫ гетероциклического ряда
или аммиаком, затем извлекают свободный алкалоид из водного раствора
эфиром, хлороформом или другим органическим растворителем, после
чего снова переводят алкалоид в кислый водный раствор. Часто поль-
зуются также тем, что многие алкалоиды образуют малорастворимые
соли с пикриновой и фосфорновольфрамовой кислотами и осаждаются
таннином и анионом Hglg. Для разделения смеси алкалоидов применяют
экстракцию растворителями при разных pH или хроматографию.
В настоящее время известно немногим менее 1000 алкалоидов, причем
для значительной их части строение еще не установлено. Большинство
из них открыто в XX столетии. В исследовании алкалоидов выдающаяся
роль принадлежала Барджеру, Р. Робинсону, Виланду, Вилыптеттеру,
Шпету, Манске, Бартону, Карреру, Прелогу, Вудворду, А. П. Орехову
и его школе (Г. П. Меньшиков, Р. А. Коновалова, Н. Ф. Проскурина,
С. Ю. Юнусов, А. С. Садыков), А. Е. Чичибабину и его ученикам
(Н. А. Преображенский, М. Н. Щукина).
Алкалоиды классифицируют по их азотно-углеродному скелету. Не-
многие алкалоиды жирного ряда и ароматические, содержащие азот
в боковой цепи (эфедрин, колхицин), были уже рассмотрены (стр. 169,
512) и здесь не излагаются. Гетероциклические алкалоиды можно разде-
лить на рассматриваемые далее группы.
Алкалоиды с пятичленным гетероциклом
с одним атомом азота
Одним из простейших по строению алкалоидов является гигрин, выделен-
ний из листьев южноамериканского растения кока (Вёлер и Лоссен,
1862 г.), а позднее — из одного из видов вьюнка (Л. В. Лазурьев-
ский, 1939 г.). Это кетон, образующий при окислении N-метилпролин
(стр. 726), что доказывает его структурное родство с N-метилпирролиди-
ном:
Н2С---СН2 Н2С-СН«
I । II'
Н2С СНСНгССНз п Н2С GH-C-OH
\ / II \ / II
NO NO
I I
СН3 СН3
гигрин N-метилпролин
(гигринован кислота)
Н2С---СН2
I ' I
Н2С сн2
Y
СНз
N-метилпирролидин
Синтез его осуществлен по схеме:
CHaMgBT
-сн4
н.с—сн—сн,
|| II V
MgBr
II II
\^\ciI2CHCH3
OMgBr
н,о v
II II
\/\сн2снсн3
IN II ।
on
H,/Pt
I I CH,0 ,
X /^СНгСНСНз V^Ol^COH,
NH । N Ц
0H CH8 0
686
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
В последней стадии действием формальдегида одновременно мети-
лируется азот и окисляется вторичная спиртовая группа в карбонил
(реакция К. Гесса).
Родственным гигрину по строению является также содержащийся
в листьях кока (разновидность «куско») кускгигрин, синтез которого,
доказывающий строение, осуществлен Г. В. Лазурьевским:
Н2С—сн2 п2с 1 —сн2 н2с—сн2
Н2С СНСН2С—0- \ / II 1 „ „ пиролиз Ва2+ * X 1 1 1 сн—СН2ССН2—НС сн2 ч / II \ Z
N 0 NON
СНз 2 । । СНз СНз
Алкалоиды с системой двух конденсированных пятичленных циклов
с одним общим атомом азота
Большая группа алкалоидов, встречающаяся в некоторых сложноцвет-
ных (крестовник) и других растениях, имеет в основе бициклическую
структуру пирролизидина, а именно 1-метилпирролизидина, названного
гелиотриданом.
н?с—сн—сн2
I I I
Н2С N СН2
сн2\:н2
Н2с---СН---СН—СНз
I I I
Н2С N СН2
СН2 СН2
пирролизидин
гелиотридан
Самым важным с точки зрения лечебной практики является плати-
филлин — сложный эфир, при гидролизе распадающийся на двугпдр-
оксильный гетероциклический спирт платинецин и сенециониновую ки-
слоту. Платифиллин применяется как сосудорасширяющее и спазмо-
литическое средство (А. П. Орехов).
ОН
I
о=с—с—сн2—сн—с—с=о
СНСНз СНзСНз; Н+: Нг°->- НО~]------:---СН2ОН
1 N +
°—i----i-Г~СН2-0
I N I платинецин
платифиллин*
О он
II I
+ НО—С—С-СНо-СН—с-с—он
II I I II
СНСНз СНз СНзО
сенециоппновая кислота
* Не исключено, что сенециониновая кислота этерифицпрует платинецин в ином
и что в приведенной формуле платпфиллина ее остаток следует повернуть
Лй loll .
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
687
Исследование строения многочисленных алкалоидов этой группы'
выполнено школой А. П. Орехова — Г. П. Меньшиковым, С. Ю. Юну-
совым и др. Так, С. Ю. Юнусов установил структуру ряда алкалоидов
с пирролизидиновым ядром, из которых приведем следующие три:
N |
H2No/\/
норлолин
С*13 I N I
ЛОЛИН
О=с—СНз
лолинин
Г. П. Меньшиков же установил строение основного гетероцикличе-
ского ядра этих алкалоидов — гелиотридана путем гофмановской
деструкции:
AgOH I I I СНз
----* I /N+\ \
। \/
СН3 ОН-
СНз
-Н2О
СН3
N-СНз СН
сн2
СНа! у
AgOH
_сн-сн=сн2
/СНз
^СНз
СНз
СНз\ |
>Ы-СН2СН1СН2СН=С-СН=СН2
СН3/
Алкалоиды с пятичленным гетероциклом
с двумя атомами азота
Пилокарпин был изолирован (Арди, 1895 г.) из листьев Pilocarpus Jabo-
randi, произрастающего в Африке, и нашел широкое применение в борьбе
с глаукомой — глазной болезнью, связанной с повышением внутриглаз-
ного давления, причиной большей части случаев слепоты. Пилокарпин,
в противоположность атропину, суживает зрачок.
Строение пилокарпина как производного имидазола следует из пре-
вращения его при нагревании с известью в 1,5-диметилимидазол и 1-метил-
5-н-амилимидазол. С другой стороны, при окислении пилокарпина
перманганатом наряду с азотсодержащими продуктами распада
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
688
имидазольного цикла получены пилоповая кислота и ее гомолог — гомо-
пилоповая кислота:
С2Н6—СН—СП—СН2----с—N-СНз
СНз—С—N—СНз и-СбНц-C—N-СНз
о=с сн2
нс сн
Са(ОН).
НС СН
НС сн
О
пилокарпин
о=с сн2
N
1,5-диметилимидазол
/°
С2НВ-НС—СН— СН2—СУ
I I Х0Н 4-
1-метил-5-
н-амилимидазол
о=с сн2
О
пилоповая кислота
о
гомопилоповая кислота
-+-CH3NH-C-NH2 + NH3 + CO2
Гомопилоповая кислота при действии щелочи в
образует пентан-1,2,3-трикарбоновую кислоту
С2Н6СН---СН----СН2
жестких условиях
СООН СООН СООН
и, следовательно, является лактонокислотой. Строение
подтверждено синтезом (А. ~
/ОС2Н6
С2Н5—СН—СН2—Cd
о=с
пилоповои кислоты
Преображенский):
/ОС2Н5
С2Н6—СН—СН—С<
I I ^0
о=с нс=о
Е. Чичибабин и Н. А.
нсоос2н6;
C,H,ONa
С2Н5О
/ОС2Н6
С2Нв— сн-сн-с<
0=
сн2
С2НвО
/ОС2Н5
С2Н6—СН—сн-с<
нагревание
о=с сн2
N
О
О
о
н
ОН
С2Н56
О
этиловый эфир
пилоповой кислоты
Пилоповая кислота по Арндту — Вольфу (стр. 625) может быть превра-
щена в гомопилоповую, чем устанавливаются их взаимоотношения:
/С1
С2Н3—НС—СН—Cd + -
1 | CHt=N—N .
0=С СН2
о
„ тт ' zCHN2
С 2н5—НС— сн—с<<
II V)
О=с СН2
V
, Н,0 .он
(перегруппировка и нт г*._____г*и гп €''
Арндта—Вольфа) С.2Н5 HU ЬН СН2
* oU СН2
\>z
АЛКАЛОИДЫ гетероциклического ряда
689
Синтез пилокарпина осуществлен Н. А. Преображенским и сотр.,
исходя из хлорангидрида гомопилоповой кислоты:
С2Н6—НС— СН—СН2—С
О=с СН2
С1
О
ch.=n°n ,
ZCH=N=N
С2Н5—НС—СН—СН2—С<
--► II
О = С СН2
/СН2С1
С2Н5—НС—СН—СН2—СА
II ^0 __
0 = С СН2 (введение NHt по Габриэлю)
со
/ \^\
С2Н5— НС—СН—СН2—с—СНг—N | || н о
I I II “°
0=С СНг о СО
0^
С2Н5—НС—СН—СН2—С—CH2NH2
0=С СН2 О
СН«—N=C=S
Н3С
HN
c2h6-ch-=ch-ch2-c-ch2-nh-c
I I II II
0=С СН2 О S
н3с-
I------1 I
I ОН НI N
и—11
С2Н5—CH-CH—ch2-c=ch-n=c
I I
0=С СН2 SH
и
--► С2Н5-НС-СН—СН2—С—N-CH3
II II I
0=С СН2 НС С—SH
''o''
|hno,
С2Н6-ПС—СН-СНй-С—N-СНз
II II I
0=С СН2 НС СН
44
Заказ 672
690
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
В скобках приведены неизолируемые промежуточные стадии. Метил-
изотиоцианат присоединяется к аминогруппе, как в известной уже реак-
ции присоединения изоцианатов (см., например, синтез Траубе производ-
ных мочевой кислоты и других замещенных пуринов). Таутомерная форма
полученного производного тиомочевины циклизуется с отщеплением
воды
Алкалоиды с шестичленным гетероциклом
с одним атомом азоте
В основе целой группы алкалоидов лежит пиперидиновый цикл. Сам пи-
перидин также обнаружен в одном из растений семейства мареновых.
Кониин (главный алкалоид болиголова, сильный яд) представляет собой
а-пропилпиперидин, что явствует из его дегидрогенизации в а-пропил-
пиридин (конирин) и окисления последнего в а-пиридинкарбоновую ки-
слоту:
СН2
Н/)Н2 ~^А А 0
Н2С *СНСН2СН2СН3 " 112 Х/\СН2СН2СНз VXc/
а; х°н
кониин конирин в-пиридинкарбоновая
кислота
Ладенбург синтезировал
кониин следующим путем
Ох
II I + ^С-СНз------> II I
Н Х/^СН=СН-СНз
Na; С,Н»ОИ
х/нсн2сн2сн3
и расщепил полученный рацемический продукт на антиподы, кристал-
лизуя его соль с D-винной кислотой. Правовращающий изомер идентичен
с природным.
Вместе с кониином встречается ряд алкалоидов — его кислородных
производных: вторичные спирты конгидрин (гидроксил в боковой цепи)
и псевдоконгидрин (гидроксил в положении 5 пиперидинового ядра).
В коре гранатового дерева среди других алкалоидов содержится
пелътъерин — альдегид, который восстановлением по Кижнеру можно
превратить в кониин, чем и доказывается его строение.
А
XNHkCI-I2CH2CH=O
пельтьерин
NHg-NH;
CsHsONa ,
-N.
NHsch2ch2ch=n-nh2
NH СН2СН2СНз
кониин
АЛКАЛОИДЫ гетероциклического ряда
691
Пельтьерин применяется как антигельминтное средство.
Пиперин — алкалоид черного перца — гидролизуется в пиперидин
И пипериновую кислоту и может быть получен ацилированием пипери-
дина этой кислотой:
з
СН=СНСН=СНС—ОСН
II
О
CH=CHCH=CHC-N^3>
О
пиперин
Для пипериновой кислоты установлено приведенное выше строение
(при окислении она дает альдегид пиперонал, или гелиотропин).
Среди многих алкалоидов лобелии важнейшим является лобелин,
применяемый в медицине для возбуждения дыхательных центров мозга.
Это третичный амин и одновременно кетон и вторичный спирт; при оки-
слении он превращается в дикетон лобеланин, который при нагревании
с цинковой пылью образует столько ацетофенона, что ясно наличие двух
групп С6Н5ССН2—. Гофмановская деструкция лобеланина приводит
II
О
к 1,7-дибензоилгептадиену.
СНз1; затем NaOH
лобеланин
°\ /\
уС—НгСг n ХСН2-С<^
свн/ /\ \свн5
НзС СНз
он-
/°
'ЧС—СН=СНСН2СН2СН2СНСН0-С<
ceHZ I хсвн6
N(CH3)a
CHiI; затем NaOH
/°
Д>С-СН=СНСНйСНаСНаСНСНа-С< —
СвН6/ I ХСвН5 ~н,°
ОН- +N(CH3)3
л /0
%С-СН=СНСН2СН2СНаСН=СН-С< +N(CH3)s
CeHfi/ хс8н6
54*
алкалоиды гетероциклического ряда
692
За вычетом двух групп СвНБССН2- остается пять углеродных атомов,
I
О
которые и были замкнуты с одним атомом азота в пиперидиновыи цикл.
Синтез лобеланина был осуществлен следующим путем.
2С.Н.СН-О „
с6н6сн=сн/\^чсн==снсвн6
C.H.ONa , |
СвНБСНСН/\^ХСНСНСвНБ свнБс=с/\^с=сс6нБ
II II
ВгВг ВгВг
CHjC.HiSOgOCH.^
Л А
Vi-civ V xcH2-c<f
CeHfi/ хс«нБ
/О
сн2-с/
свнБ
СНз
H»/Pt ;
%
С8н/
-O3SC6H4CH3
лобеланин
Восстановление одной из кетогрупп лобеланина приводит к лобелину
Н2-СН-СвНБ
СН3
лобелии
ОН
В плодах арековой пальмы найдена группа родственных между собой
алкалоидов, из которых мы рассмотрим ареколин, и арекаидин, (арекаи-
дин кислота, ареколин — ее метиловый эфир). Ареколин применяется
в ветеринарии и в медицине. Оба они циклические третичные амины,
содержащие в цикле одну двойную связь. Гидрирование арекаидина
приводит к N-метилпиперидин-р-каобоновой кислоте. Синтез арекаидина
устанавливает положение двойной связи:
г-гг птг сгн,он; НС1
СН2=СН-СН=О ---------------- СН2С1-СН2—СН(ОС2НБ)2
(С2Н5О)2СН СН(ОС2НБ)2 (С2НБО)2СН СН(ОС2НБ)2
сн* сн2 сн2 сн2 нг
I I —> I I 2“
СНа ZCH2 СН2 СН2
Cl Cl
ch3nh2 сн«
алкалоиды гетероциклического ряда
693
“О=СН СН=О"
СН2 СН2
СН2 СН2
N
I
СНз
/СН = О
кротонизация ; | |' °
I
СНз
I
СНз
арекаидин
/\ /С/
СН,ОН . / у \0СНз
I
СНз
ареколин
В высохшем соке, выделенном из надрезов на головках мака (опий),
имеется алкалоид папаверин,, строение которого установил Гольдшмидт,
а синтез осуществил Пикте (1910 г.). Папаверин находит широкое приме-
нение в медицине как сосудорасширяющее средство (например, при гипер-
тонии).
Как показывает анализ, в молекуле папаверина содержится один
атом азота и четыре метоксильные группы. При деструкции папаверина
(сплавление со щелочью и окисление) получаются диметоксильные про-
изводные изохинолина и толуола или бензойной кислоты, откуда явст-
вует его строение:
СНз°\/\А.
I I N
СНзО/^/4^
сн2 —
СНзО
СНзО
СНзО/^
ОСНз
ОСНз
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
694 _________________________———— ------- '
Синтез Пикте:
О ‘ О -
м II
СНзОх /ч сн.сос); СН3О4 /ССНз СИзО^^/ССИгЯО
ьп3и\ z\ д1с1з \/ hno2 у >1 __
I II -----* 1 В * * 11 /\\ /
СНзО/^/ СНзО/^/ _CH3O/\Z
О 0
II II СНзО о
CII’°\^\/CCH=N0H н CH»°W\/CCH=™‘ сн.о->->_снЛ,
—► I II - I II. --------------
СНз0/^/ СНзС/^/
О
снз0\/^./“\
L И
СН3О/Х/
о=с
I
сн2
I
I II
СНзО7 у
ОСНз
-Н2о
СЫз0\^\/Ч..
I I. 7
СНзО/'^/ 'Y
СН2
I
I II
СНзО/^/
ОСНз
В соке недозрелых головок мака находятся близко родственные по
строению алкалоиды опия — морфин и кодеин. Оба они содержат тре-
тичный азот, несущий метильную группу и по три кислородных атома —
вторичноспиртовый (окисление в кетон), простоэфирный и в морфине —
фенольный, а в кодеине метилированный фенольный. Морфин метилиро-
ванием в щелочной среде превращается в кодеин. Таким образом, кодеин
зто простой метиловый зфир морфина как фенола. При перегонке с цинко-
вой пылью оба алкалоида образуют фенантрен. После длительной работы
многих химиков окончательную формулу этих алкалоидов установил
Р. Робинсон
морфин
кодеин
При гофмановской деструкции кодеин превращается в так называ-
емый а-метилморфиметин, который легко расщепляется соляной ки-
алкалоиды ГЕТЕРОЦИКЛИЧЕСКОГО ряда
695
слотой. При действии щелочи на а-метилморфиметин отщепляется та же
цепь, несущая азот, и образуется метиловый эфир морфенола:
соли кодеина
NaOH
Необычная легкость разрыва С—С-связи с отщеплением диметил-
аминоэтанола объясняется тем, что реакция сопровождается ароматиза-
цией фенантренового ядра и, следовательно, большим энергетическим
выигрышем.
Метилморфол и метилморфенол легко получаются синтезом Пшорра
(стр. 258) и таким образом устанавливается положение кислородного
моста и метоксила в кодеине (а значит, и кислородного моста и феноль-
ного гидроксила в морфине). Подобное же отщепление метиламиноэта-
нольной группировки и ароматизация фенантренового ядра происходит
и при длительном нагревании с уксусным ангидридом кетона кодеинона —
продукта окисления кодеина как вторичного спирта. В качестве фенан-
тренового производного при этом получается 3-метокси-4,6-диацетокси-
фенантрен, чем устанавливается нахождение третьего кислородного атома
кодеина (и морфина) в положении 6:
ch3coch2ch2nhch3
Легкость отрыва в этом случае группы HOCH2CH2NHCH3 и ранее —
группы HOCH2CH2N(CH3)2 с ароматизацией в фенантрен указывает на
то, что эта группировка была привязана к четвертичному циклическому
углероду и вероятность вхождения се в шестичлспный изохинолиновый
Цикл приводит к формуле Р. Робинсона для морфина. Эта структура
была установлена (1927 г.) почти через 120 лет после открытия морфина
(1806 г.) — первого изолированного алкалоида — и была подтверждена
синтезом его (Гатес, 1952) по схеме, приведенной на стр. 696.
Вг
алкалоиды гетероциклического ряда
697
Морфин имеет огромное значение как обезболивающий анальгетик,
хотя отрицательной стороной его применения является настолько силь-
ное привыкание, что организму становится трудно обходиться без мор-
фина (наркомания).
Диацетат морфина называется героином.
Алкалоиды с системой двух конденсированных
шестичленных циклов с общим атомом азота
Алкалоид семян некоторых видов лупина—лупиник—открыт Зивертом
в 1865 г. А. П. Орехов и Г. П. Меньшиков нашли его также в средне-
азиатском ежовнике безлистом. Это сильное третичное основание, содер-
жащее один атом азота, несущее первичноспиртовую функцию. Первично-
спиртовая группа может быть окислена в альдегидную и далее в карбок-
сильную. Декарбоксилирование этой лупининовой кислоты приводит
к норлупинину, который идентичен хпнолпзидину.
норлупинин
Гофмановское расщепление лупинина устанавливает узловое поло-
жение азота в бицпкле и положение первичноспиртовой группы, чем
доказывается строение лупинина. Каждый раз после метилирования
иодистым метилом, замены пона иода на ион ОН" и пиролиза с рас-
щеплением С—N-связи образующуюся С=С-связь тидрпруют и повто-
ряют гофмановское расщепление. Приходится повторить эту операцию
трижды, чтобы разорвать последнюю связь С—N и отщепить азот в виде
триметиламина. Это доказывает, что азот был циклически троесвязан,
т. е. лежал в узле бицикла:
698
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
ноианон-4
Алкалоиды с конденсированными пяти-
и шестичленными гетероциклами и одним атомом азота
Группа тропана. К этой группе алкалоидов, важных в терапевтиче-
ском отношении, относятся атропин (и гиосциамин) и кокаин. В их основе
лежит гетероцикл тропан
СН-СН2
HaC^I \
| NCHs СН2
Н2с | /
сн—сн2
представляющий собою комбинацию метилпирролидина и метилпипе-
ридина, имеющих общие группы а-СН и группу NCH3.
Атропин (алкалоид белладонны) и гиосциамин (алкалоид белены)
О
II
ОС—СИ—свн.
СНДЭН
являются стереоизомерами и гидролизуются с образованием одного и того
же гетероциклического спирта тропина и троповой кислоты (в случае
гиосциамина — оптически деятельной, в случае атропина — рацемиче-
ской). Троповая кислота имеет строение а-метилолфенилуксусноп, что
явствует из ее превращения при дегидратации в атроповую кислоту,
окисляющуюся в бензоилмуравьиную:
* лГ*
свн5-сн-с<
| хон
СН2ОН
троповая
кислота
-Н2О
СвН5-С-С^
II ХОН
сн2
атроповая
кислота
/О
CeH5-C-C<f
II хон
о
бензоилмуравьиная
кислота
алкалоиды гетероциклического ряда
699
Спирт тропин окисляется в кетон — тропинок, откуда следует,
что он вторичный спирт. Троппнон, наподобие ацетона, реагирует с двумя
молекулами бензальдегида, образуя дибензальтропиноп, откуда ясно
что он содержит группировку •—СН2—С—СН2—. Эта группировка сим-
11
О
метрична относительно остальной азотсодержащей части молекулы,
так как при окислении тропинона получается только одна (правило По-
пова!) двухосновная кислота — тропиновая кислота.
Гофмановская деструкция тропина доказывает наличие мостиковой
группы NCH3 в бигетероцикле, содержащем пзоциклический скелет
из семи углеродных атомов. Действительно, для удаления азота требуется
двукратное метилирование и двукратный пиролиз гидроокиси четвер-
тичного аммониевого основания, причем отщепляется триметиламин
и остается тропилиден — циклогептатриен (кн. 1, стр. 572). Все эти
факты приводят к следующей формуле тропина и схеме его превращений:
СН31; затем
A g’Ol4
тропинок
тропнновая кислота
он
2-й пиролиз
-Н,О *
-N(CH3)3
трогилнден
Р. Робинсон синтезировал тропин следующим изящным путем: ян-
тарный диальдегид в присутствии метиламина вступает с ацетоном в кон-
денсацию кротонового типа и по образовавшимся двойным связям присо-
единяется метиламин. Образуется тропинок, восстанавливаемый в тропин.
СН=О 3
700
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
Вместо ацетона можно взять ацетондикарбоновую кислоту. Тогда
получается тропинондикарбоновая кислота, которая, будучи кетокисло-
той, далее легко декарбоксилируется:
Этерификация
к атропину:
ОН
СООН
СООН
тропина рацемической троповой кислотой приводит
сн— с'
I хон
сн,он
ОС—СН—С6Н5
СН2ОН
Эфир тропина с вератровой кислотой
СН3О\
СН3О-<^ СОН
О
открытий в 1932 г. А. П. Ореховым и Р. А. Коноваловой, — алкалоид
конволамин, содержащийся в некоторых видах вьюнков. Конволамин,
в котором мОстиком служит He)>NCH3, a >NH, называется конволъвином
(алкалоид, открыт теми же авторами).
Атропин — важнейшее средство, снимающее спазмы. Обладает
мидриатическим действием (расширение зрачка) и широко применяется
в глазной практике.
Кокаин, открытый в 1860 г. Ниманом в листьях южноамериканского
растения Erythroxylon Соса
О
представляет собой сложный эфир оксикислоты ряда тропана, носящей
название экгонина, и может быть превращен гидролизом в экгонин, бен-
зойную кислоту и метиловый спирт. Последовательной этерификацией
экгонина хлористым бензоилом (по гидроксилу) и метанолом получают
кокаин, что устанавливает их отношения. В свою очередь экгонин оки-
сляется в кетокислоту, которая легко декарбоксилируется в водном
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
701
растворе подобно ₽-кетокислотам и превращается при этом в тропинон.
И обратно, натриевый енолят тропинона карбоксилируется действием
СО2 в кетокислоту:
экгонин
Таким образом выясняется строение экгонина и кокаина. Следо-
вательно, синтез тропинона ио Робинсону завершается и синтезом
кокаина (Вилыптеттер).
Кокаин был первым открытым обезболивающим средством и потому
приобрел огромное значение. Его недостатком является способность
вызывать наркоманию (кокаиноманию) вследствие привыкания нервной
системы к его приему. Этим объясняются многочисленные поиски лишен-
ных этого недостатка анестетиков, отталкивавшиеся первоначально
от структуры кокаина. Такими синтетическими и полусинтетическими
анестетиками являются, например, новокаин, перкаин, лупикаин и др.
О NH-CH2CII2NH(C2Il5)s
Cl-
H2N—С—OCH2CH2N(C2H5)2
\=/ у
О
новокаин перкаин
лупикаин
(М. M. Кацнельсон.
М. И. Кабачник, 1935)
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
702
Алкалоиды с двумя конденсированными
шестичленными гетероциклами с одним атомом азота
Псевдопельтьерин (мстил гранатанин) — алкалоид, открытый в 1878 г.
в коре гранатового дерева. Исследован Чамичаном, синтезирован Р. Ро-
бинсоном. Его строение аналогично тропинону, но в основе его лежит
не тропан, а гетероцикл из двух пиперидиновых циклов с общей системой
-CH-N-CH-.
СН3
Псевдопельтьерин окисляется в гранатовую кислоту с тем же числом
углеродных атомов (аналог тропиноновой кислоты):
СН2-СН—СН2
/ I \
Н2С NCH3 С=О
^СНг-СН-СНг
/°
СН2-СН-С<
о Z 1 0Н
Н2С NCH3
X I zO
сн2—СН—сн2—c<f
псевдопельтьерин
гранатовая кислота
Гофмановская деструкция гранатовой кислоты приводит к октадие-
новой кислоте, которая при гидрировании образует пробковую киелоту
(гександикарбоновую):
/О тт /р+ О\ /О
>С-CH=CHCH2CH2CH=CH—c<f - ы,/-\С-(СН2)6-с/
но/ \он но/ Хон
Гофмановская деструкция спирта — продукта восстановления псевдо-
пельтьерина, проходящая также в две стадии, но сопровождаемая дегидра-
тацией, приводит к циклооктатриену:
псевдопель-
тьерин
---->
СН3 1,
затем AgOH
------------->
I-н пиро-
лиз
-------->
— Н»О
СН31;
затем AgOH
------------>
2 й пиролиз
---------------->
N(CH3|3; — Н2О
цнклооктатриен
(кн. 1[™сЛУ573)И КСпо°л™гапСЯ ВильштеттеР для первого синтеза цпклооктатетраена
миду — трпметиламип f^«HH°My Цпкл0°ктатриену был присоединен бром, к бро-
аммониевого основания nXTeHeH Нг 1’ИДРОКСИЛ ” от гидроокиси четвертичного
то основания отщеплены по Гофману триметил амин и вода:
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
703
Вг2
Синтез псевдопельтьерина аналогичен синтезу тропинона (стр. 699),
но исходным является не янтарный, а глутаровый диальдегид:
.СН2-СНО сн-соон
Н2С\ + h2nch3 + с=о
СН2 —СНО /
сн2—СООН
----->
-2СОг
псевдопельтьерин
Алкалоиды с двумя изолированными гетероциклами
с двумя атомами азота
Среди 12 алкалоидов табака главным является никоти I. Это жидкость
(т. кип. 246° С при 730 мм рт. ст.) с запахом махорки, при ком-
натной температуре смешивающаяся с водой. Никотин сильный яд,
поражающий мозг и нервную систему, повышающий кровяное давле-
ние. Он применяется в сельскохозяйственной практике как инсектицид
и для этого извлекается из махорки.
Никотин включает два третичных атома азота и является силь-
ным основанием. При окислении (HNO3, КМпО4) он дает никотино-
вую ((З-пиридинкарбоновую) кислоту. При обработке никотина бромом
с последующим окислением продукта бромирования образуются: та же
|3-пиридинкарбоновая кислота, малоновая кислота и метиламин, что до-
казывает наличие в никотине следующих структурных групп
С-С-С
\n—СНз
Поскольку никотин двутретичпое основание, группа >> N—СН3
должна замыкать цикл. Группа из трех атомов углерода (малоновой
кислоты) должна была быть привязана к углеродному атому карбоксила
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
704
никотиновой кислоты, что дает группировку
с—с—С—с
Тогда становится вероятной структура никотина как (N-метил-а-
пирролидил)-|3-пиридина, так как из четырех углеродных атомов
цепи можно замкнуть посредством >>N—СН3 либо пятичленный пирро-
лидиновый цикл, либо низшие циклы, которые мало вероятны вследствие
их неустойчивости.
При окислении уксуснокислым серебром пирролидиновый цикл
никотина переходит в пиррольный и образуется никотирин, также
находящийся в табаке
никотин никотирин
А. Пикте следующим образом осуществил синтез никотина. Пиролизом
соли слизевой кислоты и p-аминопиридина был получен N-пиррил-
Р-пиридин (синтез аналогичен синтезу пиррола пиролизом слизевокис-
слого аммония):
СНОН—СНОН—COO" H3N
СНОН-СНОН-СОО- H3N
+ 4Н2О-2СО2
ное
Нагреванием N-пиррольное производное изомеризовалось в С-пирроль-
а-производное (стр. 293). Остальной путь синтеза ясен из схемы:
никотирин
никотин
алкалоиды гетероциклического ряда
705
Изомером никотина является анабазин, открытый в 1929 г. А. П. Оре-
ховым в среднеазиатском растении ежовнике безлистом. Анабазин сме-
шивается с водой. Он представляет собой сильное двухатомное основание;
один азот находится в виде вторичной аминогруппы. Окислением ана-
базин превращается в никотиновую кислоту, дегидрирование его дает
а,0-дипиридил. Все эти факты дали основание А. П. Орехову установить
для анабазина строение а-пиперидил-|3-пиридина:
анабазин
Синтез анабазина осуществлен Г. П. Меньшиковым по схеме:
+ IMg-(CH2)4-OCH3
NH 2ОН
НВг ?
/С-(СН2)4-ОСН3
NOH
N
//^ZCH-(CH2)4-OCH3
!1\ // nh2
N
Анабазин подобно никотину используется как инсектицид.
D-Тубокурарин — один из алкалоидов южноамериканских рас-
тений видов стрихнос, применявшийся индейцами в качестве стрельного
яда. Ему родственны по действию и структуре алкалоиды Л-курпн, про-
токуридин и др. Тубокурарин — самый сильный по действию из алкало-
идов кураре. Он обладает свойством задерживать проведение нервного
импульса, направленного к мышцам — в результате мышечное расслаб-
ление и при больших дозах — прекращение дыхания. Применяется
в хирургии для расслабления мышц. D-Тубокурарин (его строение уста-
новлено Кингом, Винтерштейнером и Дутгером) — двучетвертпчноам-
мониевая соль (хлорид), имеет молекулярную формулу С38 H44OeN2Cl2.
Оба атома хлора — ионные, из кислородных атомов два — фенольные,
два — метоксильные, связанные с ароматическими ядрами, и два —
простые эфирные. Гофмаповский распад может быть применен к тубо-
курарину непосредственно после замены иопов хлора на гидроксильные
ионы. По продуктам деструкции можно построить формулу тубокурарпна.
45 Заказ 672
706
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
Двукратная гофмановская деструкция и последующее окисление
«дезоснования» протекают, как показано ниже:
алкалоиды гетероциклического ряда
707
Алкалоиды с двумя конденсированными шестичленными
гетероциклами с общим атомом азота
и одним изолированным гетероциклом
Из коры хинного дерева в 1820 г. Пельтье и Кавенту выделили два ал-
калоида — хинин и цинхонин. Хинин приобрел огромное значение в
борьбе с плазмодиями малярии и как отправная структура в синтезе анти-
малярийных средств.
Хинин является метоксильным производным цинхонина. Действием
на хинин едкого кали в жестких условиях А. М. Бутлеров и А. Н. Вышне-
градский впервые получили хинолин (откуда и произошло его название).
Хинин — двухатомное основание. С иодистым метилом он дает ди-
иодметилат (по обоим азотам), с HI отщепляет СН31 (наличие метоксила),
с хлористым бензоилом образует монобензойный эфир (наличие гидр-
оксила). Хинин можно окислить в кетон (вторичность оксигруппы).
В молекуле хинина содержится одна активная двойная связь и окисление
перманганатом направляется на нее; при этом образуется карбоновая
кислота, содержащая на один углеродный атом меньше, чем хинин.
Следовательно, эта двойная связь принадлежит СН2 —СН-группе боковой
цепи. При более энергичном окислении и эта кислота и сам хинин образуют
идентичные продукты (кроме продуктов, происходящих из мерохинена,
см. далее). Это, во-первых, выделенная А. А. Воскресенским хининовая,
кислота, оказавшаяся 6-метоксихинолин-4-карбоновой кислотой (цин-
хонин дает при аналогичном окислении хинолин-4-карбоновую — цин-
хониновую кислоту). Кроме того, вторая структурная половина молекулы
хинина и цинхонина дает при окислении одинаковые осколки разной
степени деструкции: мерохинен, цинхолойпоновую кислоту и лойпоно-
вую кислоту, идентифицированные как производные пиперидина. Таким
образом, продукты окисления хинина имеют следующее строение:
О
II
с-он
хининовая
кислота
О
II
сн2-с—он
у^/СН=СН2
I 1
NH
мерохинен
О
СНг-С-ОП
ципхолойпоповая
кислота
О
II
с—он
NH
лойпоновая
кислота
45*
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
708
Во всех пиперидиновых продуктах окисления хинина и цинхонина
азот вторичен, тогда как в хинине нет вторичного азота.
Уже Пастер открыл, что хинин при нагревании с кислотами изомери-
зуется в кетон — хинотоксин (зто еще один алкалоид хинной коры).
Хинотоксин уже содержит вторичноаминный азот (ацилирование, нитро-
зирование no>NH); его можно обратно превратить в хинин (Рабе) путем
бромирования (NaOBr) по вторичноаминной группе в бромамин BrN<^
и последующего замыкания цикла при отщеплении НВг щелочью. Во-
дород в этом случае может оторваться только из a-положения к кетонной
группе. При этом получается кетон хининон — продукт окисления хи-
нина, который восстанавливают во вторичный спирт — хинин.
Все эти факты увязываются со следующими формулами хинина
и хинотоксина:
сн— сн=сн2
сн2
ХИНИНОН
Аналогичные превращения известны и
аким образом, оба эти а™—-------
Svr7oTttMKJIH4eCKHX ЯДеР ~ ----- - линдана.
ДУ собой углеродом с вторичноспиртовой группой.
из двТ/™» ~ Н0ВаЯ для нас гетероциклическая система,
иа двух конденсированных --
для цинхонина,
алкалоида представляют собой
- хинолина и хинуклидина.
сочетание
связанных
состоящая
по азоту и у-углероду пиперидиновых ядер.
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
709
Он был синтезирован (Рубцов) следующим путем:
СН2—СН2 —COAg СН2—СИ2Вг
1 II 1х
О вг2
С=О с=о
1 СбНз Св115
СН
/|\
NaOH Н2С СН.СН,
-Н,О* 1н_ 1Н2 +ЗД-1„-CONa
\|/ 0
N
хинуклидин
Рабе синтезировал дигидрохинин. Синтез самого хинина (с виниль-
ной группой) осуществлен в 1944 г. Вудвордом и Дерингом. Он состоял
из следующих стадий: 1) синтез рацемического гомомерохинена, содер-
жащего в боковой цепи лишнюю группу СН2 сравнительно с меро-
хиненом: 2) сложноэфирная конденсация хининовой кислоты с эфиром
гомомерохинена по а-метиленовому звену последнего; 3) омыление и де-
карбоксилирование (кетонное расщепление) образовавшегося эфира |3-ке-
токислоты и получение этим путем рацемического хинотоксина, расще-
пляемого на L- и 2)-хинотоксины; 4) превращение /^-изомера описанным
выше способом Рабе в хинин.
гидрирование в 2 стадии
с промежуточным ацили-
рованием по >NH
сн2 сн2
H2CZ ^сн сн2
I I I
С—N СН СН-ОН
\ // \ /
НзС о сн2 сн
СНз
окисление
(с выделением
цис-изомера)
СН2 СН2
Н2С сн сн2
С—N СН С=О
/ \ \ / \ /
НзС о сн2 СН
I
СНз
сн2 сн2
C62H6b°NaO: Ь^С7 \'Н XCH2-C^ Н
---5----+ I I \ос2н5 —i
С—N СН
НзС^Ь СН2 С—NOTI
СНз
с-сн2—сн2
/\ I
с2н5о о сн
Н2С CH-CH-NH2
-> III
Н2С СН2 СПз
I
с<
ХСНз
с-сн2-сн2
СгНбО7^ сн
CHlf;nn Н2С 'сн-СН-N(CH3)3
затем NaOH f । ।
Н2С СН2 СНз он-
\н
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
710
С—СН2—СН2
СН
С2Н50 О
СН
Н2с СН.СН—сн=сн2
Н2С
Н2С
CH-CH-N(CH3)3
I I TT -N(CH,)«;
CH2 CH3 OH -H,0
Н2С сн, сн2
С NH
NH
С2Н6О о
эфир гомомерохинена
Наиболее длинный ряд превращений понадобился для синтеза гомо-
мерохинена (он приведен здесь сокращенно). Сложноэфирная конденса-
ция эфира
лпрование
ХИНИНОВОЙ КИСЛОТЫ С
проводились по схеме:
эфиром гомомерохинена и декарбокси-
СНзО
О=С—OR
ch-\aa
Н2С
Н2С
СН
сн,сн-СН=СН2
сн, СН2
NH
RO О
СН
СН2 СН2 СН—СН=СН2 Н!о; н+
О=С—СН СН2 СН2
I/Or| /
у \С< NH
-со2
-нон
СН
СН2 СН2СН—СН=СН2
О=С-СН2 СН2 сн2
---> далее, как указано на стр. 708
NH
хинотоксин
Алкалоиды с тремя конденсированными гетероциклами
с двумя атомами азота
Мы разберем строение только двух алкалоидов этой группы — иохим-
оина и резерпина.
Иохил{би/1 — алкалоид коры африканского растения корианте
химбе — находится в этой коре вместе с рядом его изомеров.
N
N
С
N
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
711
Строение скелета иохимбина вытекает из следующих превращений:
гарман
+
тртрабирин
HNO3
Дегидрированием иохимбина селеном получают несколько веществ,
одно из которых — тетрабирин при окислении образует пиридин-2.4,5-
трикарбоновую, а другое — кетоиобирин — 2,3-диметилбензойную ки-
слоту. Первая кислота может образоваться только за счет сохранивше-
гося кольца D (кольцо С имеет всего две связи с углеродами и может дать
максимум пиридиндикарбоновую кислоту). 2,3-Диметилбензойная ки-
слота, очевидно, не может образоваться за счет циклов А, В, С, D и пред-
ставляет собой остаток цикла Е. Расположение карбоксилов в пиридин-
2,4,5-трикарбоновой кислоте, а также метила и карбоксила в 2,3-диме-
тилбензойной кислоте указывает места связи циклов D и Е и положение
карбоксила в кольце Е. На положение гидроксила в кольце Е указывает
разложение иохимбина перегонкой с цинковой пылью, в результате кото-
рой образуются в небольших количествах гарман и п-крезол.
Резерпин C33H40N2O9 — алкалоид индийского растения раувольфиа
серпентина. Начиная с 1950-х годов он применяется как одно из главных
гипотензивных (понижающих кровяное давление) средств. При щелочном
гидролизе он отщепляет метиловый спирт и 3,4,5-триметоксибензойную
кислоту, образуя резерпиновую кислоту C22H27O5N2. Последняя, будучи
прометилирована по карбоксилу диазометаном, этерифицируется 3,4,5-три-
метоксибензоилхлоридом в резерпин, чем и доказывается сохранение
♦структуры скелета резерпина в этих превращениях и роль метанола
712
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
и триметоксибензойной кислоты. Функциональным анализом в резерпине
обнаруживается пять метоксильных групп и одна карбметоксильная.
Таким образом, частичное строение резерпина может быть выражено
формулой I, а резерпиновой кислоты — формулой II
2ОСНэ
/ОСНз NaOH 2ОСН3 /ОН
C191l20l\2 ОСНз ” C19II20N2 • %
-о-с-< II ОСП3 ОН
О 4 ОСНз II
I
Ультрафиолетовый спектр поглощения резерпина, резерпиновой ки-
слоты, резерпинового спирта и смеси двух последних имеет форму, ха-
рактерную для производных индола, и близок к спектру 2,3-диметил-6-мет-
оксииндола (рис. 121). Инфракрасные спектры хлороформных растворов
резерпина (рис. 122) и эквимолекулярной смеси молекул, включающих
отдельные фрагменты его структуры, близки к полному совпадению.
Такая эквимолекулярная смесь составлена из 2,3-диметил-6-метоксиин-
дола (фрагмент, соответствующий кольцам А и В), N-этилпиперидина
(кольца Си D), метилового эфира циклогексанкарбоновой кислоты
(кольцо Е) и метилового эфира 3,4,5-триметоксибензойной кислоты. Сплош-
ная линия — спектр резерпина, линия с участками пунктира — спектр
указанной смеси (рис. 123). При сопоставлении со спектром синтезирован-
ной модельной пятициклической системы ABCDE близкой структуры
подтвердилась аналогия такой структуры с резерпиновой кислотой (и ио-
химбином). Одновременно при этом сравнении выяснилось и цнс-сочле-
нение колец D и Е в резерпиновой кислоте и в резерпине.
резерпин
резерпиновая кислота
модельная система
Сплавление резерпиновой кислоты со щелочью приводит (после мети-
лирования содержащей кислоту фракции сплава) к метиловому эфиру
3-метоксиизофталевой кислоты, что доказывает положение гидроксила
Рис. 121. УФ-Спектры (в метаноле):
1 — резерпина; 2 — эквимолярной смеси
резерпииового спирта и метилового эфира
3,4,5-триметоксибензойной кислоты; з —
метилового эфира 3,4,5-триметоксибензой-
ной кислоты; 4 — резерпииового спирта;
5 — 2,3-диметил-6-метоксииндола.
TlGc
-i-1-1-:_iiii;
3 5 6 7 8 9 Ю 11 12
yj, MK
Рис. 122. ИК-Спектры(в хлороформе):
I— 2,3-диметил-5,6-диметоксииидола; II —
2,3-диметил-6-метоксииндола; III — тетра-
гидроальстонилина; IV — резерпина.
Рис. 123. ИК-Спектры резерпина (сплошная линия) и эквимолярного раствора
2,3-диметил-6-метоксииидола, метилового эфира 3,4,5-трпметоксибензойпой
кислоты, метилового эфира циклогексанкарбоновой кислоты и N-этплпипери-
дина в хлороформе (сплошная линия с включениями точечного пунктира).
На верхней кривой видны калибровочные пики СО2 (4,224 мк) и Н2О (5,464
и 6,41 мк).
714
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
в резерпине (а-метоксил кольца Е при этом отщепился в виде СН3ОН
и образовал одну из двойных связей).
Синтез резерпина, осуществленный Вудвордом и сотр. в 1957 г.,
представляет собой очень сложную задачу, принимая во внимание стерео-
химию молекулы, отраженную в следующей формуле
резерпин
Синтез начинается с построения структуры и конфигурации циклов D
и Е, для чего по реакции Дильса — Альдера соединяют (^ис-присоедине-
ние) бензохинон с винилакриловой кислотой. Образующийся бицикл I
восстанавливается боргидридом натрия по менее пространственно за-
трудненной кетонной группе в соединение II, окисляемое далее надбен-
зойной кислотой в эпоксид III (кислородный мост присоединяется с внеш-
ней более открытой стороны пространственного угла, образуемого цик-
лами). При действии на III водоотнимающих веществ образуется лактон
IV за счет карбоксила и гидроксила, введенного при переходе от I к II.
Лактон IV можно переписать в виде перспективной формулы IVa,
тождественной IV:
IV
IVa
алкалоиды гетероциклического ряда
Восстановление лактона изопропиловым спиртом в присутствии изо-
пропилата алюминия приводит к превращению сохранившегося карбо-
нила во вторичноспиртовую группу, и одновременно происходит размы-
кание лактонного цикла и замыкание карбоксила на новый гидроксил.
Кроме того, разрывается эпоксидный цикл с образованием гликоля; один
из его гидроксилов замыкается с освобожденным гидроксилом в простой
эфирный мост, а другой отщепляется в виде воды с образованием в цикле
двойной связи. Все эти реакции, совершающиеся в одну фазу, в формулах
IVa — VII для ясности расчленены.
VII VIII
К VII по двойной С=С-связи, сопряженной с карбоксилом, присоеди-
няют метанол (VIII), а затем по второй двойной связи — бромноватистую
кислоту (IX). Вторичноспиртовую группу бромгидрина окисляют в же-
тонную и полученное вещество X обрабатывают цинком в кислой среДе-
При этом разрываются по С—О-связям оба гетероцикла лактонный
и простоэфирный (последний с одновременным отщеплением брома и об-
разованием на месте отщепившихся Вг и О двойной связи). Получается
716
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
вещество XI, формулу которого теперь проще писать в плоскости Xia.
Дальнейшие превращения ясны из формул Xia— XV:
Вещество XV послужит в дальнейшем для синтеза колец Е и D ре-
зерпина.
Теперь необходима заготовка остальной части скелета резерпина —
колец А и В. Для этого исходят из магниевого производного 6-метоксиин-
дола (XVI), на которое действуют нитрилом монохлоруксусной кислоты.
Как обычно в индоле, заместитель вступает в (3-положение и получается
нитрил 6-метокси-(3-индолилуксусной кислоты (XVII). Его восстанавли-
вают в амин 6-метокситриптамин XVIII, который взаимодействием
с альдегидом XV превращается в шиффово основание XIX. Шиффово осно-
вание восстанавливают в амин XX, замыкающийся с потерей СН3ОН
в циклический амид XXI (образование кольца D). Последнее кольцо С
замыкается при действии хлорокиси фосфора на XXI: вещество XXII
имеет уже все циклы: А, В, С, D, Е.
Действуя на XXII боргидридом натрия, восстанавливают аммоний-
mvTia30T В аМИННЫЙ’ а затем гидролизуют в XXIII обе сложноэфирные
РУ пировки. Освободившиеся карбоксил и гидроксил соединения XXIV
XXVI
XXVII
XXVIII
718
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
под действием дициклогексилкарбодиимида замыкаются в лактон XXV.
Последний нагреванием с разбавленной кислотой стереоизомеризуется
в отношении расположения вокруг углеродного атома, общего для цик-
лов С и D (стереоизомер XXVI). Затем лактонный цикл размыкается
подкисленным метанолом, и освобожденный гидроксил соединения
XXVII подвергается этерификации хлорангидридом триметилгалловой
кислоты.
Резерпин XXVIII, тождественный природному, синтезирован. Этот
синтез показан на схеме (стр. 717).
Стрихнин C21H22O2N2 и бруцин С23Н2eO4N2 — выделены в 1818 г.
Пельтье и Кавенту из «рвотных орешков» — семян Strychnos пих vomica,
произрастающего в Индонезии. Оба алкалоида, особенно стрихнин,
сильные яды (смертельная доза для человека 0,03 г), вызывающие судо-
роги. Это кристаллические вещества, растворимые в спирте, бензоле,
хлороформе, слабо — в воде и эфире, оптически активные. Стрих-
нин применяется в медицине как возбуждающее и тонизирующее
средство.
Бруцин—диметоксильное производное стрихнина — широко исполь-
зуется наряду со стрихнином в стереохимическом эксперименте для раз-
деления рацематов кислот. Окончательная формула стрихнина после
длительных исследований была установлена в 1945 г. Робинсоном, это
выяснило и формулу бруцина. Здесь могут быть лишь в общих чер-
тах приведены факты и соображения, на которых основываются эти
формулы.
При окислении стрихнина азотной кислотой образуется 5,7-динитро-
индол-2,3-дикарбоновая кислота, откуда явствует, что стрихнин (а значит,
и бруцин) содержат индольный цикл. Оба они содержат одну двойную
олефиновую связь (так как каталитически гидрируются в дигидроалка-
лоиды) и два атома азота. Атомы азота оба третичные, но лишь один из
них основной и способен присоединять иодистый метил; другой азот
лишен основных свойств, так как ацилирован (связан с СО-группой),
что следует из результатов гидролиза щелочью. При этом, очевидно
вследствие размыкания циклического лактамного кольца, получается
стрихниновая (или бруциновая — в случае бруцина) кислота в виде соли.
Кислота на Н2О богаче исходного основания и содержит вторичную,
уже основную, аминогруппу (ацилирование, действие HNO2).
Поскольку эти кислоты имеют два основных азота, ясно, что индоль-
ный цикл находится в них в гидрированном по гетероциклу состоянии
(2,3-дигид р оинд ол).
Обе кислоты легко замкнуть опять в алкалоиды. Рядом с лактамной
СО-группой, ацилирующей дигидроиндольный азот, находится СН2-груп-
па, поскольку и стрихнин и бруцин нитрозируются по углероду, образуя
изонитрозопроизводные, и конденсируются с бензальдегидом в бензилиде-
новые производные. Второй кислородный атом — простой эфирный
входит в цикл и на расстоянии двух атомов от него в цикле находится упо-
мянутая выше двойная олефиновая связь, которая первая разрывается
при окислении перманганатом, образуя с одной стороны разорванного
цикла карбоксил, а с другой — карбонил. После восстановления актив
ной СО-группы и нагревания отщепляется гликолевая кислота.
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
719
Окисление стрихнина, дальнейшую деструкцию молекулы и упомянутые
выше превращения можно изобразить схемой:
нагревание
-НОСН2-С*
(.Hl
д процессов деструкции дает некоторое понятие о строении и взаимо-
nBR3fR циклов I» И’ HI и VII. Превращение под действием щелочи
I U амино.)тил)-индол (триптамин) дает понятие о способе связи вто-
р го азота с индольным циклом. Серия окислении хромовой кислотой
(Двойная связь защищена), схема которых приведена на стр. 720,
дает основание для суждения о циклах IV, V и VI.
озможность дальнейшего упрощения молекулы с отщеплением
виднаЛСВ°и КИСЛОты и размыканием обоих пиперидиновых циклов оче-
Совсем недавно строение стрихнина было подтверждено его достаточно
головоломным синтезом (Вудворд). Особенно трудно в синтезе таких слож-
720
АЛКАЛОИДЫ ГЕТЕРОЦИКЛИЧЕСКОГО РЯДА
ных структур соблюдение стереохимических требований: стрихнин со-
держит шесть асимметрических атомов, в формуле помеченных звездоч-
ками.
Жирными точками отмечены (по Линстеду) те углеродные атомы,
которые несут водороды, располагающиеся над плоскостью.
БЕЛКИ (ПРОТЕИНЫ)
Каждая клетка животного, растительного и одноклеточного организма
содержит в качестве существеннейшей составной части белки, или про-
теины, строящие ферменты, органеллы и в целом протоплазму клетки.
Частицы простейших организмоподобных образований — вирусов — так-
же состоят из белковой оболочки и нуклеиновых кислот.
Белки являются субстратом всех известных нам форм жизни. Все
белки (кроме искусственно синтезированных в последние годы) созданы
жизнедеятельностью организмов, но не все они продолжают участвовать
в жизнедеятельности. Шерсть, рога, кожа, шелк, паутина —все это
частные примеры белков, называемых склеропротеинами. К белкам
относятся растворимые в воде или соляных растворах альбумины (яйца,
крови) и глобулины (крови), к белкам относятся миозин мышц, много-
численные белки растений, в частности их семян — глиадин пшеницы,
зеин кукурузы и т. д.
Белками являются многочисленные ферменты (энзимы) животных
и растительных организмов, такие, как ферменты пищеварения — пепсин,
химотрипсин, трипсин, различные пептидазы и другие протеолитические
ферменты, эстеразы, в частности холинэстераза (стр. 773) и т. д.
Некоторые важные гормоны (инсулин, андренокортикотропный гор-
мон, глюкагон и т. д.) также относятся к белкам. При этом, как теперь
установлено, строго индивидуальные белки, выполняющие идентичную
функцию, могут отличаться друг от друга в организмах разных видов.
Так, инсулин человека, свиньи и коровы в деталях различается по строе-
нию, хотя и сходен по функции и взаимосвязям.
Как видно из этого перечисления, белки представляют собой совер-
шенно разнородные по физическим свойствам вещества. В соответствии
с этими свойствами и с растворимостью была осуществлена их первичная
классификация: нерастворимые белки — склеропротеины (кератин волос,
шерсти, пера, коллаген кожи, сухожилий, фиброин шелка); растворимые
в воде белки альбумины (крови, яичного белка, молока); растворимые
в разбавлепньтх водных растворах солей, в кислотах и щелочах глобулины
(у-глобулин крови, белковые^фермеиты, гормоны и т. д.); близкие к ним
глутелины (белки семян злаков), растворимые в кислотах и щелочах,
46 Заказ 672
122
БЕЛКИ (ПРОТЕИНЫ)
но не в солевых растворах; находящиеся вместе с глутелинами в семе-
нах злаков глиадины (относящиеся к проламинам), отличающиеся рас-
творимостью в 70%-ном спирте; сильно основные гистоны и протамины,
растворимые в воде с щелочной реакцией (находятся в животных объек-
тах).
Протеидами называются белки, связанные с небелковой (простети-
ческой) группой, которая часто может быть отделена от белка гидролизом.
Это фосфопротеиды, содержащие связанную в виде эфира фосфорную
кислоту (пример: казеин молока); гликопротеиды, в которых простетиче-
ская группа углеводная; липопротеиды с фосфатидной (кн. 1, стр. 331)
простетпческой группой; хромопротеиды — с гетероциклической металл-
содержащей (порфиновой) простетической группой (пример: гемоглобин);
нуклеопротеиды — их простетической группой являются высокомолеку-
лярные нуклеиновые кислоты (стр. 743).
Как и полипептиды, белки амфотерны — в кислом растворе переме-
щаются к катоду, в щелочном — к аноду. Изоэлектрической точкой назы-
вается такая величина pH, характерная для каждого индивидуального
белка, при которой белок не перемещается ни к катоду, ни к аноду и, сле-
довательно, молекула его в целом электронейтральна.
Что же общего между собой имеют столь мало сходные по внешнему
виду и по физиологической функции вещества?
Уже в начале прошлого столетия было установлено, что все белки
близки не только по качественному, но и по количественному составу,
а именно содержат 50—52% С, 15—18% N, 6,8—7,7% Н, около 24% О,
0,5—2,0% S. В их состав часто входит малое количество фосфора в виде
Н3РО4 (протеиды), галоидов, металлов. Белки обладают свободными карб-
оксильными и (или) аминогруппами, в результате чего они амфотерны
(обычно с преобладанием кислотного или основного характера, в зависи-
мости от преобладания групп СООН или NH2 в молекуле). Поэтому белки
растворимы также в кислотах и щелочах. Постепенно был открыт ряд
цветных реакции на белки, исторически сыгравших большую роль. Не-
которые из этих реакций были общими для всех белков (или пептидов),
например биуретовая реакция (фиолетовое окрашивание с щелочным
раствором двухвалентной меди), нингидринная реакция (свойственная
аминокислотам и заключающаяся в возникновении фиолетовой окраски
при обработке смеси, содержащей а-аминокислоты, нингидрином — гидра-
том трикетогидриндена). Другие цветные реакции характерны для от-
дельных аминокислот, входящих в состав белка (например, ксантопротеи-
новая реакция — появление желтой окраски при нитровании азотной
кислотой тирозина и т. д.).
Уже к 1820 г. было установлено, что белки гидролизуются, и Браконно
выделил из гидролизата простейшую а-аминокпслоту — а-аминоуксус-
ную (за свой сладкий вкус и происхождение из желатина, т. е. из белко-
вого клея костей, она была названа гликоколом, а позднее — глицином).
Далее из гидролизатов белков были выделены и другие аминокислоты,
и вплоть до середины XX века продолжалось открытие более экзотиче-
ских аминокислот. В 90-х годах прошлого века Э. Фишер разработал
свой метод исследования аминокислотного состава гидролизатов белков.
Метод Фишера состоит в том, что смесь аминокислот, полученную в ре-
БЕЛКИ (ПРОТЕИНЫ)
723
зультате гидролиза с помощью концентрированной соляной кислоты
превращают этерификацией посредством этанола и НС1 в сложные эфиры
аминокислот, освобождают их от солеобразно связанной НС1 путем до-
бавления щелочи и разделяют эфиры фракционной перегонкой в вакууме.
Такой препаративный метод, несмотря на то что он далек от совершенства
(потеря от х/3 до х/2 всей массы аминокислот), был большим шагом вперед.
Вскоре было выяснено, что в состав подавляющего большинства исследо-
ванных белков входят L-a-аминокислоты из числа помещенных в табл. 87
и лишь редкие белки содержат какие-либо аминокислоты сверх этих.
В таблице выделены жирным шрифтом незаменимые в пище человека
и животных аминокислоты, потребление которых должно составлять
в сумме 21—31 г в сутки. Остальные аминокислоты организм способен
синтезировать сам, если ему доставляется с пищей источник азота (на-
пример, в виде глутаминовой кислоты). Эти аминокислоты требуются
в количестве 49—51 г в сутки, т. е. общее количество аминокислот соста-
вляет 70—80 г в сутки (по А. А. Покровскому).
Белки нацело гидролизуются в аминокислоты концентрированной
НС1 или 2%-ной НС1 при нагревании в автоклаве; протеиды при гидро-
лизе отделяют, кроме того, простетическую группу, которая во многих
случаях также способна гидролизоваться. Э. Фишер и одновременно с ним
Гофмейстер пришли к выводу об амидной (пептидной) связи аминокислот
в белках (кн. 1, стр. 501). Э. Фишер разработал синтетические методы
(1903 г.) построения полипептидов и синтезировал свой знаменитый окта-
декапептид, состоящий из 18 остатков аминокислот, связанных последо-
вательно амидными (пептидными) связями. Полипептиды, синтезированные
из Л-а-аминокислот, оказались подобными простейшим белкам и пеп-
тонам — осколкам белковых молекул, образующимся в результате пище-
варения в желудке. В частности, они обладали способностью гидролити-
чески расщепляться под влиянием соответствующих ферментов пищева-
рительного тракта.
Таким образом, теория строения белков как полипептидов, обосно-
ванная Э. Фишером, стала прочным фундаментом исследования белков.
Неясным оставалось, как при столь однообразном строении различных
белков объяснить их весьма разнообразные физические и биохимические
свойства. В 20-х годах XX века на примерах каучука, целлюлозы, крах-
мала были развиты представления о высокомолекулярных соединениях.
В то же время были разработаны методы определения молекулярного
веса высокомолекулярных соединений и, в частности, белков. Ранее
о минимальном молекулярном весе протеидов судили по содержанию
в них простетических групп (или каких-либо специфических атомов этих
групп, например атома железа в гемоглобине), исходя из предположения,
что одна простетическая группа содержится в одной молекуле протеида.
Молекулярные веса и таким путем получились огромные, например для
гемоглобина 68 000. Применение осмометрпческого метода определения
молекулярного веса (Серенсеп, 1917 г.) и особенно разработка ультрацен-
трифугального метода (Сведберг, 1926 г.) позволили систематически ис-
следовать молекулярные веса растворимых белков. Оказалось, что их
молекулярные веса располагаются в широком интервале величин от
10 000 и ниже для ряда ферментов и гормонов (6500 для инсулина) до
46*
Таблица 87. Важнейшие аминокислоты белка (жирным шрифтом выделены незаменимые аминокислоты)
Тривиальное название Сокращенное обозначение аминокислотного остатка Формула Темпе- ратура плавле- ния °C [«lz> в ледя- ной уксусной кислоте градусы Изоэлек- триче- ская точ ка PH Раство- римость в воде при 25° С г/100 г Суточная потреб- ность взрослого человека в амино- кислотах г
Гликокол (глицин) Gly 1I2N—CH2-COOH 292 — 5,97 25 3
Аланин Ala CH3CH—coon nh2 297 +33 6,00 16,6 3
Валин Vai (СНз)2СНСН-СООН 1 nii2 315 +62 5,96 8,85 4
Лейцин Leu (СНз)2СНСН2СН-СООН nh2 C2Il5\ 337 +22,5 5,98 2,2 4-6
Изолейцин He >CHCH—GOOH CH3Z 1 nh2 284 +49 6,02 4,12 3—4
Аспарагиновая кислота Asp HOOCCH2CH—GOOH N H2 270 +25,4 2,77 0,5 6
Аспарагин nh2 1 Asp (или Asn) 1I2N— 0—CH2CH-COOH II 1 0 nh2 236 +34,3 5,41 2,5
Глутаминовая кислота Glu НООС-(СН2)2СН—COOH 249 +31,8 3,22 0,84 16
1 1 nh2
Глутамин nh2 1 Glu (или Gin) H2NC-(CI12)2CH-COOH II 1 o nii2 185 +6,1 (в H2O) 5,65 4,2 —
Орнитин * О rn Н2Ы(СН2)зС11—-COOI1 i — — — — —
nh2
Лизин Lys h2n:cii2)4gh-cooh — +25,9 9,74 Хорошо 3-5
1 nh2 раств.
Аргинин A rg II2N-C-NH(GH2)3CH—COOH II 1 238 +29,4 10 76 15 6
NH NH2
Серин Ser IIOCH2CH—СООН 228 +15 5,68 5 3
nh2
Треонин Tre СНзСН-СН-СООН 253 -30 6,16 20,5 ?-3
1 1 OH NH2
Цистеин Cys HS-CH2CII—COOII 178 +13 5,02 X ,,<1шо 2-3
1 раств.
nh2
Цистин Cys- -S-S-Cys Г — S—CH2C1I—СОО1Г 260 -232 5,03 0,011 —
1 L nii2 J 2
Метионин Met CH3SCH2CH2CI-I-COOII 283 +20 5,74 3,5 2—4
1 nii2
Фенилаланин Phe CeH5CII2CH-COOH — —7,5 3,0 — 2-4
NH2 1
* Образуется в гидролизатах за счет гидролиза аргинина.
Продолжение табл. 87
Тривиальное наввание Сокращенное обозначение аминокислотного остатка Формула Темпе- ратура плавле- ния °C [Но в ледя- ной уксусной кислоте градусы Изоэлек- триче- ская точка pH Раство- римость в воде при 25° С г/100 г Суточная потреб- ность взрослого человека в амино- кислотах г
Тирозин Туг м-НОСвЩСПгСН—СООН 344 —10 5,66 — 3-4
nh2
Триптофан Try 7.-СН2-СН-СООН 1 II II 1 ^NH Nll> 382 -34 5,89 1,14 1
Пролин Pro н2с—сн2 299 -80 6,30 16.2 5
1 1 Н2с СНСООН
NH
Оксииролин Opr НО-СН-СНг 270 -77 5,83 36,1 —
1 1 Н2с СНСООН
NH
Гистидин His N —CII2CH-COOH II II 1 \ / nh2 NH 277 +7,5 7,6 4,3 2
БЕЛКИ (ПРОТЕИНЫ)
727
6 600 000 (гемоцианин улитки) и даже до 320 000 000 (белок вируса
гриппа). Если принять средний молекулярный вес аминокислотного
остатка, входящего в полипептидную цепь белка, равным 115, то ока-
жется, что число аминокислотных остатков в молекулах белков колеблется
от нескольких десятков до немногих миллионов. Таким образом, уже по
молекулярным весам белки представляют величайшее разнообразие. Про-
стейшие из них вряд ли могут быть отнесены к высокомолекулярным соеди-
нениям, между тем как некоторые представляются одними из высокомо-
лекулярных соединений с наиболее громоздкими молекулами. Существен-
нейшим отличием белков как высокомолекулярных соединений от таких
синтетических полимеров, как капрон, полистирол, и таких природных
высокомолекулярных соединений, как каучук, целлюлоза, крахмал,
является разнообразие элементарных звеньев («мономеров»), из которых
построены белки. Взамен одного «мономера» (например, остатка ы-ами-
нокапроновой кислоты или глюкозы, стирола, изопрена) в белки
входит более 20 разных аминокислотных остатков. Это было и вдох-
новляющим и обескураживающим обстоятельством. Если молекула
состоит всего из 20 разных аминокислотных остатков, для нее возможно
2 432 902 008 178 640 000 изомеров! А если задаться молекулой, состоя-
щей из 1000 аминокислотных остатков, то 20 аминокислот можно распо-
ложить в этой молекуле 2О1000 способами. Одна эта изомерия позволяет
не только каждому виду и индивидууму живого мира, но каждой клетке
иметь свои белки. Такое разнообразие возможностей должно было бы
обескураживать химиков. Вспомним, однако, что возможности изомерии,
например в химии предельных углеводородов, хотя и несравненно более
ограниченные, все же далеко превышают осуществимость синтеза всех
изомеров даже для первого десятка гомологов метана. Поскольку это
не смущает химика-исследователя, то и в случае белков дело не покажется
таким безнадежным. Ведь проблема синтеза всех возможных изомеров
и не стоит в науке. Актуальна проблема установления строения белков,
важнейших с той или иной (обычно физиологической) точки зрения, и син-
теза их, а это, как показывает состояние сегодняшней науки, хотя и труд-
ная, но посильная задача.
Методы индивидуализации белков
Нерастворимые белки приходится просто отмывать серией растворителей
от всех растворимых примесей, что, конечно, никак не дает гарантии их
индивидуальности. Растворимые (в воде, солевых растворах и т. д.)
белки, выделяемые из клеток после их механического разрушения, под-
вергают диализу, заключая в полупроницаемые (например, целлофановые)
мешочки и погружая в воду. Все электролиты и низкомолекулярные
вещества диффундируют через полупроницаемую пленку. Далее произ-
водится фракционное разделение смеси растворимых белков путем осажде-
ния постепенным добавлением этилового спирта или солей [например,
(NH4)2SO4] и хроматография. Более современный метод разделения
предусматривает применение ионообменппков (из сульфированного поли-
стирола) и молекулярных сит типа «сефадекс» (из декстранов, см. кн. 1,
стр. 482).
728
БЕЛКИ (ПРОТЕИНЫ)
В индивидуальности осаждаемых фракций убеждаются при помощи
хроматографии на бумаге или электрофореза. Многие белки после очистки
удается получить в кристаллическом состоянии, если медленно испарять
растворитель (например, упарить разбавленный водный раствор соли).
Этими методами современному исследователю удается получить белки
столь же чистые и индивидуальные, как и все другие органические соеди-
нения.
Методы ультрацентрпфугального фракционирования растворов белков
показывают, что хотя существуют и полидисперсные белки, представля-
ющие собой смеси полимергомологов различного молекулярного веса
(например, желатин — от 12 000 до 70 000), но в основном белки моно-
дисперсны п в отличие от целлюлозы, крахмала и синтетических полиме-
ров являются химическими индивидуумами, а не смесью полимергомоло-
гов (Сведберг).
Методы установления строения белков
Полипептидная цепь белков в общем виде имеет следующее строение:
NH2-CH-CO—NH—СН—СО—NH-CH-C—........—NH—СН—С—ОН
I I I II I II
R R R" О R" О
Первый вопрос, который возникает: каков суммарный аминокислотный
состав данного белка? Техника определения этого состава со времени
Э. Фишера ушла далеко вперед по точности (достигающей 99%) и по
скорости операции. После кислотного или щелочного гидролиза (кислот-
ный гидролиз разрушает триптофан и приходится комбинировать оба
способа расщепления) раствор смеси аминокислот при pH 2 пропускают
через ионообменник (сульфированный полистирол в виде натриевой соли),
который связывает все аминокислоты за счет их аммонийной функции,
и затем постепенно элюируют кислыми буферными растворами с pH,
увеличивающимися от 3,5 до 5,5 и затем до 11, т. е. все менее кислыми.
Аминокислоты вымываются в порядке уменьшающейся кислотности; под
конец, при буфере pH 11, идет вымывание дпаминокислот. В настоящее
время все операции разделения производятся на автоматических анализа-
торах, в которых смена буферных растворов совершается автоматическп,
а количество выделенных аминокислот определяется фотометрически
по интенсивности сине-фиолетовой окраски, возникающей при реакции
с нингидрином, который образует со всеми аминокислотами один и тот же
краситель в результате следующих реакций:
NH2
БЕЛКИ (ПРОТЕИНЫ)
729
Показания фотоэлемента градуируются по пятнам, образованным заве-
домо известными количествами аминокислот. Исключение составляют
пролин и оксипролин, которые дают в этих условиях желтое окрашивание.
Анализ смеси аминокислот автоматическим аминокислотным анализа-
тором Штейна и Мура отнимает всего несколько часов. Но такой анализ
ничего не говорит о последовательности связи аминокислотных остатков.
Для установления последовательности связи аминокислот разрабо-
тано несколько методов:
1. Во многих белках отдельные части молекулы оказались связанными
дисульфидными мостами за счет цистина
О=С—ОН О=С—ОН
NH—СН—С—NH—СН-СН2—S— S—СНа—СН—NH—С—СН—NH-------------
I II II I
R О OR'
Окислением серы до группы —SO3H удается разбить молекулу’ на
части по связи —S—S—. Того же можно достигнуть восстановлением
моста —S—S— до двух меркаптогрупп —SH и HS—, например, дейст-
вием HSCH2COOH, окисляющейся в (—S—СН2—СООН)2, или при
помощи меркаптоэтанола HSCH2CH2OH.
2. Действием на белок 2,4-динитрофторбензола — активного электро-
фильного реагента («подвижный фтор») — удается динитрофенилпровать
свободные аминогруппы белка. Это могут быть аминогруппы двух типов:
вторые аминогруппы диаминокислот, подобных лизину, или концевые
аминогруппы. Например:
H2NCHC—NHCHC-NHCHCOOH4-2F-/ ^>-NO2----►
I II I II I )=/
RO R' О (CH2)i N/2
NH2
--> O2N—IINCHC—NHCHC—NHCHCOOH
X=< I II I II I
NO2 Ro R' О (CIT2)4
NH— NO2
no2
730
БЕЛКИ (ПРОТЕИНЫ)
При осторожном гидролизе отщепляется написанная на схеме слева
2,4-динптрофенилированная аминокислота
O2N—Х—NH—СН—С—ОН
I II
\no2 R о
Отделяя эту дпнитрофениламинокпслоту и идентифицируя ее, напри-
мер, методом бумажной хроматографии, определяют одну из концевых
аминокислот молекулы белка, так называемую N-концевую (метод Сенд-
жера, 1949 г.). Метод имеет тот недостаток, что в условиях кислотного
гидролиза динитрофенильные производные глицина, цистина, пролина
и триптофана разрушаются.
Другой метод (Эдман. 1949 г.) состоит в обработке белка фенилизотио-
цпанатом, который со свободными аминогруппами образует производные
тпомочевины. При обработке такого производного разбавленной соляной
кислотой в нитрометане легко отщепляется тот остаток аминокислоты,
у которого фенилтпомочевпнная группировка находится в а-положении,
ибо. отщепляясь, он образует устойчивый пятичленнып гетероцикл —
фенилтпогидантоин:
(CH2)4NH2
H2NCHC-NHCIIC-NHCHC-NHCHCOOH4-2CeH5-X=C=S ------>
I II I II II I
RO R' О 0 R"
S
II
S (CH2)4NHCNHCeH5
II I HCl
--> C6H6NHCNHCHC-NHCHC—NHCHC--NHCHCOOH------>
I II I II II I
RO R' О 0 R*
S
II
(CH2)4NHCNHCeH5
--> C6H5—N—C=S + H2NCHC-NHCHC-NHCHCOOH
II I II II I
O=C NH R' О 0 R"
\h
I
R
фенилтиогиданТоин
Применяя этот метод, можно шаг за шагом отстригать по одному ами-
нокислотному остатку с N-конца. Так, например, френкель-Конрату уда-
лось определить последовательность первых восьми аминокислот в адрено-
кортикотропном гормоне.
3. Для отщепления аминокислотных остатков с G-конца (т. е. амино-
кислотных остатков со свободным карбоксилом) Акабори предложил
БЕЛКИ (ПРОТЕИНЫ)
731
использовать действие гидразина (при 100° С в течение нескольких часов).
Реакция может быть изображена так:
H2NCHC—NHCHC—NHCHCOOII + 2NHaNH2 >
I II I II I
RO R' О R"
—> II2NCHC— NHNHg-f-H2NCHC—NHNH2 + H2NCH-C-OII
I II I II I II
RO R O R" О
Из схемы видно, что вся цепь распадается на гидразиды аминокислот
кроме первой (с С-конца) аминокислоты, которая выделяется в свобод-
ном виде. После обработки полученной смеси подходящим альдегидом,
связывающим гидразиды в малорастворимые гидразоны, свободную амино-
кислоту выделяют хроматографически.
4. Проще всего для отщепления С-концевых аминокислот пользо-
ваться ферментативным гидролизом. Так, фермент карбоксипептидаза
(из поджелудочной железы) последовательно отрывает аминокислоты,
содержащие карбоксильную группу. Недостаток этого метода состоит
в том, что пролин и оксипролин карбоксипептидазой не отщепляются.
Конечно, цепь последовательных расщеплений гораздо проще при-
менить к полипептидам с небольшим числом аминокислотных остатков,
чем к белкам сложного строения. В случае сложных белков пользуются
тем, что гидролиз в разных условиях расщепляет их по разным связям
и, следовательно, дает разный набор более низкомолекулярных полипеп-
тидов. При гидролизе разбавленными кислотами разрываются прежде
всего связи двухосновных аминокислот, под действием концентрирован-
ных кислот рвутся в первую очередь связи оксиаминокислот, при щелочном
гидролизе разрываются связи треонина. Фермент пищеварения — трип-
син расщепляет пептидные связи лизина и аргинина с аминогруппами
других аминокислот, химотрипсин рвет связи ароматических аминоки-
слот, карбоксипептидаза, как уже было сказано, отщепляет С-концевые
аминокислоты со свободным карбоксилом, а аминопептидаза — Х-кон-
цевые аминокислоты со свободной а-МН2~группой. Следующая схема
иллюстрирует сказанное:
г-----------Трипсин------------.
;
Vai 4. Cys—Ciy—Giu—Arg-GIy—Phe-j-Leu—Tyr-j-Tre-Pro-Lys-Ser-j-AIa
1 * 1 1
I I 1 1
Аминопептидаза Химотрипсин Карбокси пептидаза
В результате гидролиза в разных условиях получается набор полипеп-
тидных осколков, которые разделяют хроматографическими методами,
разработанными Мартином и Синджем. Это методы бумажной хроматогра-
фии, распределительной хроматографии, а также электрофореза. После-
довательность связей аминокислот в каждом из полипептпдпых оскол-
ков может быть установлена приведенными выше методами, например
методом Эдмана. Сопоставляя эту последовательность в каждом осколке
732
БЕЛКИ (ПРОТЕИНЫ)
и принимая во внимание, что в некоторых осколках происходит наложе-
ние (они содержат последовательность аминокислот конца первого и на-
чала второго из пары осколков), можно установить полную последователь-
ность аминокислот в блоке. Для примера обозначим отдельные амино-
кислотные остатки буквами алфавита. Если в результате гидролиза иден-
тифицирована следующая серия полипептидных осколков
АВДБ
ДБГ
БГАЗ
ГА
ЗИ
то ясно, что последовательность аминокислот в исходном полипептиде
была АВДБГАЗИ.
5. В последнее время М. М. Шемякин, М. Н. Колосов и Н. С. Вульф-
сон, а также Ледерер разработали многообещающий масс-спектрометри-
ческий метод определения последовательности аминокислотных остатков
?2 I
—NH—СН—CO-f-NH—СН—CO—OY
^Па-^^Leu ——^-val—~~-Рго -^-та^-Рго — Val-^-ont-
Рис. 124. Масс-спектрограмма N-ацплпрованного метилового эфира нонапептида
(М. М. Шемякин, М. Н. Колосов, Н. С. Вульфсон).
в полипептидах, который был уже описан при рассмотрении метода масс-
спектрометрии в кн. 1 на стр. 591. Напоминаем, что полипептид, ацили-
рованный по свободной аминогруппе (N-конец, условно — левый конец)
и этерифицированный по свободному карбоксилу (правый конец),
в масс-спектрометре сначала теряет алкоксил (OY) и образует катион
с положительным зарядом на оставшемся — С=О-конце молекулы. Затем
идет последовательное отщепление справа налево аминокислотных остат-
ков, в результате чего получаются все более и более легкие фрагменты
(каждый раз на один аминокислотный остаток меньше, в порядке их
последовательности справа налево) — катионы с положительным заря-
дом на С "О-конце. Сопоставляя молекулярные веса этой серии остат-
БЕЛКИ (ПРОТЕИНЫ)
733
ков, можно сообразить, какие аминокислотные остатки каждый раз от-
щеплялись, и установить их последовательность. На рис. 124, который
это иллюстрирует, X—СО—обозначает стеароил, Y — метил.
Пример простого природного полипептида — антибиотика грамици-
дина С был приведен в кн. I, стр. 507. Ниже даются примеры достаточно
сложных белков, строение которых установлено ранее описанными мето-
дами за период с 1945 по 1965 г.
Гормоны:
S-----------------S
I nh2 nh2 I nii2
I III I
Cys-Tyr-Phe-Glu-Asp-Cys-Pro-Lys-Gly
вазопрессин свиньи
H2N-Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu-COOH
гипертензин лошади
NH2
His-Ser-Glu-Gly-Tre-Phe-Tre-Ser-Asp-Tyr-Ser-Lys-Tyr-Leu—।
I—Asp-Ser-Arg-Arg-Ala-Glu-Asp-Phe-Val-Glu-Try-Leu-Met-Asp-Tre
I I I
nh2 nh2 nh2
глюкагон
Ile-Gly NH2
I I
Vai NH2S--------------------S NH2 NH2 Asp
III I I II
Glu-Glu-Cys-Cys-Ala-Ser-Val-Cys-Ser-Leu-Tyr-Glu-Leu-Glu-Asp-Tyr-Cys
Glu-His-Leu-Cys-Gly-Ser-His-Leu-Val-Glu-Ala-Leu-Tyr-Leu-VarCys
I I
Asp—NH2 Gly
I I
Vai Glu
Phe Ala-Lys-Pro-Tre-Tyr-Phe-Phe-Gly-Arg
инсулин быка (гормон поджелудочной железы)
Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Tyr-Gly-Lys-Pro-Val-Gly-Lys-Lys-Arg-Arg-Pro-Val —
Lys-Val-Tyr-Pro-Asp-GIy-Ala-Glu-Asp-Glu-Leu-Ala-Glu-Ala-Phe-Pro-Leu-Glu-Phe
NH2
адренокортикотропный гормон
БЕЛКИ (ПРОТЕИНЫ)
734
Белки:
Ser-Tyr-Ser-Ile-Tre-Tre-Pro-Ser-Glu-Phe-Val-Phe-Leu-Ser-Ser-Ala-Try-Ala-Asp-Pro-Ile-
—Glu-Leu-Ile-Leu-Asp-Cys-Tre-Asp-Ala-Leu-Gly-Asp-Glu-Phe-Glu-Tre-Glu-Glu-Ala-
I I Illi!
NH2 NH2 NII2NH2 NH2 NH2NH2
l-Arg-Tre-Val-Glu-Val-Arg-Glu-Phe-Ser-Glu-Val-Try-Lys-Pro-Ser-Pro-Glu-Val-Tre-Val-
III I
nh2 nh2 nh2 nh2
-Arg-Phe-Pro-Asp-Ser-Asp-Phe-Lys-Val-Tyr-Arg-Tyr-Asp-Ala-Val-Leu-Asp-Pro-Leu-
NH2
-Vai-Tro-Ala-Leu-Leu-Gly-Ala-Phe-Asp-Tre-Arg-Asp-Arg-Ile-Ile-Glu-Val-Glu-Asp-
I III
nh2 nh2 nh2 nh2
—Glu-Ala-Asp-Pro-Tre-Tre-Ala-Glu-Tre-Leu-Asp-Ala-Tre-Arg-Arg-Val-Asp-Asp-Ala—
I I
nh2 nh2
—Tre-Val-Ala-Ile-Arg-Ser-Ala-Asp-Ile-Asp-Leu-Ile-Val-Glu-Leu-Ile-Arg-Gly-Tre-Glv-Ser—
I
NH2
—Tyr-Asp-Arg-Ser-Ser-Phe-Glu-Ser-Ser-Ser-Gly-Leu-Val-Try-Tre-Ser-Gly-Pro-Ala-Tre-COOH
NH2
белок вируса табачной мозаики
Синтез пептидов и белков
Принципы синтеза пептидов были изложены в кн. I в разделе «Амино-
кислоты». Синтез белков, строение которых в отношении последователь-
ности аминокислот известно, ничем кроме громоздкости не отличается
от синтеза пептидов.
Для защиты аминогруппы кроме уже описанных методов применяется
еще тозилирование, т. е. защита действием гг-толуолсульфохлорида. То-
зильная группа удаляется восстановлением металлическим натрием в жид-
ком аммиаке. Тем же путем удаляется и карбобензоксигруппа, которая,
что особенно важно, может быть удалена также каталитическим гидри-
рованием.
Меркаптогруппа может быть защищена бензилированием, а удаление
бензильной группы достигается действием натрия в жидком аммиаке.
Ацилирование пептида с защищенными прочими аминогруппами
осуществляется кроме методов, упомянутых в кн. I, также действием
смешанных ангидридов, подобных смешанному ангидриду данной амино-
БЕЛКИ (ПРОТЕИНЫ)
735
кислоты и изовалериановой. Не менее употребителен метод, состоящий
в действии одной аминокислоты (или полипептида с защищенными амино-
группами) на другую в присутствии эфира пирофосфористой кислоты
или уже упоминавшегося дициклогексилкарбодиимида. Некоторые при-
меры использования таких приемов читатель найдет ниже.
Приведем в качестве примера взятый из курса Каррера синтез гор-
мона окситоцина, осуществленный Дю Виньо. Этот гормон задней доли
гипофиза имеет сравнительно простую структуру
S---------------S
I \
H-Cys- Ту г-1 le- G1 u-As p-Cys- Р го - Leu- G ly-N Н 2
I I
nh2nh2
В дальнейшем остаток изовалериановой кислоты в смешанных ан
гидридах аминокислот и этой кислоты мы будем изображать для крат-
кости буквой V, а карбобензоксигруппу С6Н5СН2—ОСО— буквой Z;
тозильная группа, как обычно, обозначается Ts, так что формула сме-
шанного ангпдрида изовалериановой кислоты и карбобензоксилейцина,
имеющая вид
СНз~СН-С2Н5
I
C6H5CH2OC-NHCHC-O-CCH2CH(CH3)2
II II II
о о о
Z Leu V
изобразится так: Z — Leu — О — V.
Первая фаза синтеза состоит в ацилировании этилового эфира гли-
цина смешанным ангидридом изовалериановой кислоты и лейцина, за-
щищенного карбобензоксилированием. Таким образом, синтез начи-
нается с того конца молекулы окситоцина, который в приведенной выше
формуле написан справа:
Z-Leu-O-V + H-Gly-OC2H5 > Z-Leu-Gly-OC2H5
Z-Pro-O-V И./Pci
---> H-Leu-Gly-OC2Hs----------»- Z-Pro-Leu-Gly-OC2H5 "
Z-Cys-Pro-Leu-Gly-OC2H5 NaOH
. (ОМЫ"
I ленис)
Z Z S затем
/Cys-SS-Cys<^ | Ka+NH»^
---> H-Pro-Leu-Gly-OC2H5 S
Z-Cys-Pro-Leu-Gly-OColTn
---► H-Cys-Pro-Leu-Gly-OH C«H»CH,C1. H-Cvs-Pro-Leu-Gly-OCH2C6H5
I I
SH S-CH2CeIL
---► H-Cys-Pro-Leu-Gly-NH2
I
S-CH2CeI-I5
I
736
БЕЛКИ (ПРОТЕИНЫ)
CH2 NH, | CH2
zOH pC]5 Ts-Glu< - O=C/ ^СНг H-Asp-OH _ O=CZ ^СНг NH2
'OH Ts—N CH—COCI 1 1 1 Ts—N-—CHC—Asp—OH II 0
NH2 NH2 Na; NHs NH2 NH2 Ts-lie-ci NH2NH2
| | ---------* I I-----------------------* I |
--->. Ts-Glu-Asp-OH H-Glu-Asp-OH Ts-Ile-Glu-Asp-OH
II
nh2 nh2
I I
Ts-Ile-Glu-Asp-OH + H-Cys-Pro-Leu-Gly-NH2
I
S-CI-I2C6H5
II I
(C,HsO)2Py
(C.HaOhp/
O+(C2H8O)2POH
затем Na + NHs
nh2 nh2
---> H-Ile-Glu-Asp-Cys-Pro-Leu-Gly-NH2
S-CH2CeH6
NH2 NH2 (c2hsO)2pXo
H-Ile-Glu-Asp-Cys-Pro-Leu-Gly-NH2 + Z-Cys-Tyr-OH ^гН»0)гр/ ->
I I
S-CH2CeH5 S-CH2CeH5
nh2nh2
I I Na+NHs’
—> Z-Cys-Tyr-Ile-Glu-Asp-Cys-Pro-Leu-Gly-NH2 затем H2O + O2^
S-CH2C6H5 S-CH2C6H5
NH2NH2
I I
—► H-Cys-Tyr-Ile-Glu-Aps- Cys-Pro-Leu-G]y-N H2
I I
S------------------S
окситоцин
Более поздний синтез окситоцина Дю Виньо и Боданского более прост.
НВг;
Z-Leu-OCeH4NO24-H-Gly-OC2H5------> Z-Leu-Gly-OC2H5 —™аСООН^ Leu-Gly-OCjHs
nh2;
Z-Pro-OCeH4NO2+ Leu-Gly-OC2H6 —> Z-Pro-Leu-Gly-OC2H5 затем нвг+сн2соон^
-------------------------------> Pro-Leu-Gly-NH2
Z-Cys-OC6H4NO2-|-Pro-Leu-Gly-NH2 —> Z-Cys—Pro-Leu-Gly-NH2
I I
S-CH2CeH5 S-CH2CeH5
(затем НВг+СНзСООН)
БЕЛКИ (ПРОТЕИНЫ)
|^Н2 NHa
Z-Asp-OC6H4NO2 + Cys-Pro-Leu-Gly-NH2 > Z-Asp-Cys-Pro-Leu-Gly-NH2
S-CH2C6H5 S-CH2C6H5
(затем HBr-f-СПзСООН)
NHs NH2 NH2NH2
Z-Glu-OCeH4NO2 + Asp-Cys-Pro-Leu-Gly-NH2 —> Z-Glu-Asp-Cys-Pro-Leu-Gly-NH2
S-CH2C6H5 S-CH2C6H5
(затем HBr-f-CH3COOH)
NH2 nh2
Z-Ile-OCeH4NO2 Glu-Asp-Cys-Pro-Leu-Gly-NH2 --->
S-CH2C6H5
NH2NH2
---> Z-Ile-Glu-Asp-Cys-Pro-Leu-Gly-NH2 (затем HBr-4-CH3COOH)
I
S-CH2CeH5
nh2nh2
I I
Z-Tyr-OC6H4NO24- Ile-Glu-Asp-Cys-Pro-Leu-Gly-NH2 -->
I I
О-СН2СвН5 S-CH2C6H5
nh2nh2
Z-Tyr-Ile-Glu-Asp-Cys-Pro-Leu-Gly-NH2 (затем HBr-(-CH3COOH)
O-CH2CeH5 8“СН2СбНз
nh2nh2
Z-Cys-OCeH4N O2 + Tyr-Ile-Glu-Asp-Cys-Pro-Leu-Gly-N H2 —>
I I I
S-CH2CeH6 О-СН2СвН5 S-CH2C6Il5
nh2nh2 nh2
Z-Cys-Tyr-Ile-Glu-Asp-Cys-Pro-Leu-Gly (затем HBr-4-CH3COOH)
I I I
s O-CH2C6H5 s
CIIzCeHb CH2C6II5
4? Заказ 67 2
738
БЕЛКИ (ПРОТЕИНЫ)
NH2NH2 nh2
H-Cys-TjT-Ile-G'lu-Asp-Cys-Pro-Leu-Gly —Na’ N—*->
I I I
S О-СНгСвНь S
I I
СН2СвН5 СН2СвН5
NH2NH2 nh2
—> H-Cys-Tyr-Ile-Glu-Asp-Cys-Pro-Leu-Gly —*
SH SH
NH2NH2
---> II-Cys-Tyr-Ile-Glfu-Asp-Cys-Pro-Leu-Gly-NHo
I I
S------------------S
Белковые гормоны гипертензин, вазопрессин и адренокортикотропный
гормон, синтезированные подобным окситоцину образом, оказались по
леем свойствам, включая физиологическое действие, тождественными
А природными веществами.
В 1952 г. появилась первая публикация американского химика Мер-
-филда о новом способе синтеза полипептидов, который впоследствии
оказался чрезвычайно перспективным. В этом способе используется
ионообменник, на котором карбоксильным концом закрепляется одна
Аминокислота — первое звено будущего полипептида. Затем аминогруппа
первой аминокислоты ацилируется второй аминокислотой и т. д. В конце
концов полученный полипептид действием кислоты отделяют от ионооб-
менника:
R R
I I
ch2occhnhcchnhy
II II
о о
и т. д.
Таким путем удается достигнуть очень хорошего выхода целевого
продукта и сильно ускорить процесс.
Для идентификации белков важную роль играют иммунологические
реакции. Введенный в кровяное русло животного чужеродный белок
автоматически вызывает образование белкового же антитела, связыва-
ющего введенный белок. Антитело образуется из у-глобулиновой фрак-
ции белков крови. Иммунитет к новому заражению, вырабатываемый
в результате заболевания некоторыми болезнями или в результате при-
вивки, имеет в своей основе выработку антител к данным видам бакте-
риальных или вирусных белковых токсинов. Антитело дает и видимую
глазом реакцию с вызвавшим его появление белком (антигеном) — об-
разование осадка при смешении растворов. Пользуясь иммунологиче-
БЕЛКИ (ПРОТЕИНЫ)
739
сними реакциями, можно различать белки даже близкого строения.
Так, можно отличить, например, гемоглобин человека от гемоглобина
быка.
Следует иметь в виду, что антигенной активностью обладает не моле-
кула вводимого белка в целом, а определенные группировки, разные
в разных случаях. Такими группировками могут быть и искусственно
введенные в белок антигены. Например, диазотированная н-аминофенил-
арсоновая кислота вступает в реакцию азосочетания с тирозинным зве-
ном белка, и модифицированный таким образом белок (сыворотка крови
лошади), будучи введен кролику, вызывает образование специфического
антитела. Но это же антитело действенно и по отношению к обработанной
подобным образом сыворотке других животных. Таким образом, нельзя
переоценивать возможности иммунологической идентификации белков.
Мы не приводим гипотез механизма возникновения антител, поскольку
здесь ничего окончательного не имеется.
Вторичная и третичная структура белков
Сама по себе последовательность валентных связей пептида еще не объяс-
няет глубоких различий в физических свойствах разных белков. Рентге-
ноструктурное исследование кристаллических пептидов и нитеобразных
растянутых белков (шерсть, шелк) привело к следующим выводам.
1. В аминокислотных остатках пептидная связь —СО—NH— распола-
гается в одной плоскости, что может быть объяснено частичной двоесвя-
зностью атома азота и, следовательно, его частичной аммонийностью
(наличие частичного положительного заряда). Значит карбонильная
группа должна быть «раскрыта» более обычного с сосредоточением частич-
ного отрицательного заряда на кислороде. Это подтверждается умень-
шенным расстоянием С—N (1,35 А вместо 1,47 А) и увеличенной длиной
связи С=О (1,28 А вместо 1,24 А). Такая плоская структура относительно
жестка и закреплена так, что углеводородные остатки аминокислот на-
ходятся в игуане-расположении друг к другу:
R Н
I I
СН N
R Н
I I
СН +N
хс^ \н
I I
О- R
Вокруг осей С — С сохраняется обычно свободное вращение.
2. Существеннейшей чертой строения пептидов, определяющей
типы конформаций молекул, является образование водородных связей
N—Н • • • О—С, либо между двумя разными плоскорасположенными
цепями пептида ф-структура, рис. 125, а), либо между звеньями одной
и той же пептидной цепи, закрученной за счет свободного вращения вокруг
С—С-связей в спираль, навитую на цилиндр (а-спираль, рис. 125, б).
Более устойчивой и более распространенной в белках является а-спи-
раль. При растягивании, например, кератина волос или игл дикообраза
47*
БЕЛКИ (ПРОТЕИНЫ)
740 __________________________________________________________________
а-спираль переходит в ^-структуру. Независимо от величины и характера
остатков аминокислот (ароматических, жирных или гетероциклических)
длина одного аминокислотного остатка — NH—СН—С— в а-спирали
I О
R О
по поверхности цилиндра одна и та же (1,5 А), а шаг спирали (длина витка
по длине цилиндра) 5,4 А, так что на один виток приходится 3,6 амино-
кислотного остатка. Водородные связи в а-спирали образуются между
Рис. 125. Структура полппептидной цепи белка:
а — 3-конформация с межмолекулярными водородными связями; б — спиральная
а-конформация белка с внутримолекулярными водородными связями.
каждой первой и четвертой труппами -СО-NH- и идут по образующей
“"' По существу а- и («-структуры - ато конформаЦ=
пептидных цепей, называемые часто вторич синтетических
и белка. Открытие a-и ^-структур - заслуга Р°лЯНГпа-^Х ^пример,
полипептидах, построенных из остатков одной амяя переходов а-
на полплизине), можно показать ооратпмость вз „Полрйствпи пли
и ^-конформаций твердого вещества прп механпческ м _ полипеп-
измененпп влажности. При растворении в воде спнтетпческого шолпп^
тида (например, полиглутаминовой кислоты) или оелка тРмпепатур-
няется. Лишь прп повышении температуры, в довольно У3 ' х свЯЗей
ном интервале, происходит «плавление» — нарушение г одв г.-ю-
а-сиирали, образование новых водородных связей с водой P
БЕЛКИ (ПРОТЕИНЫ)
741
булярную структуру, сопровождающийся резким падением вязкости
раствора. Такое же нарушение а-спиралей происходит при растворении
их в водном растворе мочевины (например, в 8 М растворе) или в дихлор-
уксусной и трифторуксусной кислотах (вследствие образования межмо-
лекулярных водородных связей), тогда как диметилформамид не нарушает
ос-спирали. За процессом разрушения а-спиралей и обратным процессом
«спирализации» можно следить: 1) по изменению вязкости; 2) по ускоре-
нию и замедлению дейтерообмена; 3) по изменению вращения плоскости
поляризации; 4) по дисперсии оптического вращения. Каждый из этих
показателей допускает и количественную трактовку. Остановимся на
второй и третьей характеристиках.
Линдерштрем-Ланг предложил использовать для оценки количе-
ства внутримолекулярных водородных связей в белке скорость дейтеро-
обмена группы —С—N— при разных значениях pH. Простейшие пеп-
II I
О Н
тиды обменивают свой водород с неизмеримо большой скоростью. В ос-спи-
рали, где имеются водородные связи, в группах — N—Н • • • О=С<
дейтерообмен идет часами. Таким образом можно уценить степень спира-
ли за цип (в %).
Метод 3 основан на следующих соображениях. В принципе спираль
Z-аминокислотного полипептида может быть правой по ходу винта или
левой (зеркальное изображение). Из теории следует, что правая спираль,
независимо от наличия асимметрических атомов углерода в ее звеньях,
будет вращать плоскость поляризации вправо, и эта величина будет сум-
мироваться с вкладами, вносимыми асимметрическими атомами углерода
каждой Z-аминокислоты; поэтому при растворении в «щадящем» раствори-
теле наблюдается суммарная величина вращения, а в растворителе,
разрушающем а-спираль, остается только вклад асимметрических атомов.
В синтетических «полимерах» аминокислот до сих пор наблюдалась
только правая а-спираль; она же типична и для белков, но здесь встре-
чается, по-видимому, и левая спираль. Таким образом, меняя среду,
можно фибриллярные пептиды и белки (имеющие длинные макромолекулы
и образующие очень вязкие растворы) превращать в глобулярные —
с шарообразной формой молекул и с малой вязкостью раствора.
Некоторые белки при нагревании необратимо денатурируются —
«свертываются». Это процесс «плавления» а-спиралей с последующим хао-
тическим установлением межмолекулярных водородных связей и сшива-
нием макромолекул по новым направлениям. Денатурации менее под-
вержены белки со многими «вулканизирующими» —S—S-мостами, где
разрушение структуры а-спиралей иногда имеет обратимый характер,
так как дисульфидные мосты придают известную жесткость структуре.
Как показывают кропотливые и подробные рентгенографические ис-
следования сравнительно сложных белков, дело не ограничивается пер-
вичной и вторичной структурами, но существует еще и третичная струк-
тура, зависящая от складывания или закручивания цилиндрических
спиралей. Понятие о третичной структуре может дать выясненная рент-
генографически модель Кендрю молекулы миоглобина. Третичная струк-
тура иногда (например, в кератине или коллагене) представляет собою
742
БЕЛКИ (ПРОТЕИНЫ)
несколько а-спиралей, цилиндры которых свиты друг с другом в виде
кабеля. Иногда они имеют вид цилиндра а-спирали, многократно согну-
того или сложенного, как в миоглобине (рис. 126) или рибонуклеазе.
Третичная структура, как показано на стр. 774 при разборе строения
рибонуклеазы, также в основном обязана водородным связям, но при
этом не получается идеальной а-спирали. Кроме того, третичная структура
сильно зависит и от наличия дисульфидных цистиновых мостов, сшива-
ющих разные места а-спирали, сближенные
в силу изгиба цилиндра.
Характер связей простетических групп
с белком в протеидах далеко не всегда точно
установлен. Во многих случаях это легко
гидролизуемые связи. В фосфопротеидах,
таких, как казеин молока (1% Р), вителлин
яйца, фосфорная кислота этерифицирует
гидроксил остатка серина. Гликопротеиды
содержат наряду с сахарами и полисахари-
дами связанные с белком молекулы N-аце-
тилгексозамина. Это растворимые вещества,
не денатурирующиеся при нагревании. К
ним относятся альбумин яйца и альбумины
кровяной сыворотки, в частности, определя-
Рис. 126. Третичная структура ющие группы крови.
миоглобина. в гемоглобине, относящемся к хромо-
протеидам, в молекуле гема центральный
атом двухвалентного железа связан с четырьмя расположенными с ним
в одной плоскости атомами азота порфириновой части молекулы; над
этой плоскостью пятую вершину октаэдра образует молекула воды,
а под плоскостью шестую вершину — азот гистидинного остатка
белка — глобина. В оксигемоглобине молекула воды заменяется моле-
кулой кислорода (железо остается двухвалентным, кислород связан
координационно и обратимо диссоциирует). При отравлении окисью
углерода место воды или кислорода занимает прочно привязанная моле-
кула окиси углерода.
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
Синтез белка в организме
В ядре любой клетки и в цитоплазме содержатся полинуклеотиды —
высокомолекулярные нуклеиновые кислоты (от nucleus — ядро), связан-
ные с белками, по-видимому, солеобразной связью, а также водородными
связями в нуклеопротеиды. Во всяком случае связь нуклеиновых кислот
с белком легко разрывается, и тогда обе составные части могут быть от-
делены друг от друга.
Вирусы, которые не имеют клеточной структуры, являются с химиче-
ской точки зрения также нуклеопротеидами. Важная биологическая
роль нуклеиновых кислот в вирусах выясняется из того факта, что при
заражении вирусом (например, бактериофагом — вирусом бактерий) за-
ражаемая клетка получает от вируса только нуклеиновую кислоту,
а белковая часть (оболочка) вируса остается снаружи, в клетку не прони-
кает и отбрасывается. После заражения внутри клетки-хозяина за счет
нуклеотидных, аминокислотных и ферментных ресурсов этой клетки
вырастает множество частиц вируса (бактериофага). Эти новые частицы
состоят не только из многократно повторенных нуклеиновых кислот,
но имеют и белковые оболочки, тождественные с белком исходной зара-
жающей частицы вируса, хотя белок не проникал в зараженную клетку.
Отсюда ясно, что нуклеиновые кислоты принимают решающее участие
в биосинтезе белка, чему позднее мы приведем и другие доказательства.
Это поставило полинуклеотиды в центр интересов современного естество-
знания, тогда как отдельные нуклеотиды были известны еще со времен
Либиха.
Определение молекулярного веса нуклеиновых кислот (полинуклеоти-
дов) седиментационным методом на ультрацентрифуге дает величины от
200 000 и менее до нескольких миллионов. При полном гидролизе нуклеи-
новых кислот ядра клетки установлено наличие трех групп составных
частей этих так называемых дезоксирибонуклеиновых кислот (они обычно
обозначаются как ДНК). Это — фосфорная кислота, Р-2-дезоксирибоза
(т. е. /)-рибоза, у которой второй углеродный атом несет второй водород-
ный атом вместо гидроксила, кн. I, стр. 461) и смесь четырех гетероцик-
лов: двух пуриновых — аденина и гуанина и двух пиримидиновых
тимина и цитозина (см. стр. 349, 361). Полный гидролиз нуклеиновых
14А
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
кислот клеточной плазмы, так называемых рибонуклеиновых кислот (РНК)
также дает фосфорную кислоту, //-рибозу (вместо //-дезоксирибозы)
и смесь тех же аденина, гуанина, цитозина и, кроме того, урацила.
Тимин (метильный гомолог урацила) в них отсутствует.
//-Рибоза и ZZ-2-дезоксирибоза были выделены Левиным в 1929 г.
из соответствующих нуклеиновых кислот после их гидролиза (в случае
дезоксирибонуклеиновых кислот действием 0,01 н. НС1в течение 10 мин).
Строение и конфигурация //-рибозы установлены по тождеству ее озазона
с озазоном //-арабинозы, из чего следует эпимерия этих двух сахаров
(кн. I, стр. 448). Строение и конфигурация ZZ-2-дезоксирибозы доказаны
окислением метилированного сахара в 1,2-диметоксиглутаровую кислоту
известной конфигурации:
СН3О Н
При осторожном щелочном гидролизе рибонуклеиновых кислот или
гидролизе с помощью ферментов (например, рибонуклеазой для рибо-
нуклеиновых кислот, фосфодиэстеразой змеиного яда для дезоксирибо-
нуклеиновых кислот) можно расщепить высокомолекулярные полинуклео-
тиды на простые нуклеотиды. В молекуле простого нуклеотида тот или
иной из перечисленных выше гетероциклов связан с рибозой (в ДНК —
с дезоксирибозой) и фосфорной кислотой, этерифицирующей сахарную
часть нуклеотида. Это явствует из того, что среди продуктов гидролиза,
проведенного в соответствующих условиях, можно найти свободный
гетероцикл и изомерные фосфаты //-рибозы (соответственно //-2-дезоксири-
бозы). С другой стороны, гидролиз нуклеиновых кислот или изолирован-
ных нуклеотидов можно (ферментативно или действием аммиака) довести
и до соответствующих нуклеозидов, т. е. отщепить фосфорную кислоту,
оставив связанными гетероцикл и сахар. Таким образом, нуклеотиды —
«мономеры», поликонденсацией которых (с отщеплением воды) образуются
полинуклеотиды («полимеры»), — представляют собой фосфорные эфиры
нуклеозидов. Поскольку продукты гидролиза нуклеозидов — пиримиди-
новые и пуриновые гетероциклы (а также рибоза или дезоксирибоза),
идентифицируются сличением с известными образцами, остается устано-
вить место связи гетероцикла с сахаром, характер их циклизации, кон-
фигурацию гликозидного центра и, наконец, место фосфорилирования
сахарной части молекулы.
Трудная гидролизуемость связи гетероцикл — сахар свидетельствует
о том, что это не связь С—С (негидролизуемая) и не связь С—О (легко
гидролизуемая). Таким образом, это может быть только связь С N-
Со стороны сахара в этой связи участвует, как и следовало ожидать,
«альдегидный» атом углерода (С-1). Это доказывается, в частности, серией
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
реакций, осуществленных Тоддом на примере аденозина I, который при
окислении иодной кислотой образует диальдегид II, тождественный ди-
альдегиду, полученному при перйодатном окислении* омыленного N-глю-
козида V, синтезированного взаимодействием тетраацетилбромглюкозы III
с серебряной солью аденина IV:
Синтез V из тетраацетилбромглюкозы доказывает гликозидный ха
рактер связи глюкозы в соединении V, а тождество продуктов окисления I
и V устанавливает гликозидный характер связи в аденозине. Одновре-
менно, поскольку в производном V конфигурация первого атома глюкозы
Р-гликозидная, устанавливается также и р-гликозидная конфигурация
аденозина.
P-Конфигурация гликозидного центра подтверждается еще более
веско фактом легкого образования циклонуклеозидов. Внутримолекуляр-
ная циклизация 5'-тозил-2',3'-изопропилиденцитидина, 5'-тозил-3 -аце-
тилдезоксиаденозина и других аналогичных производных может про-
исходить только в том случае, если исходный гликозид является р-гли-
козидом (в этом случае СН2ОН-группа и гетероцикл находятся по одну
сторону кольца сахара).
* Об окислении ШО4 см. кн. I, стр. 457.
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
746
Рибоза нуклеотидов, как это доказано окислением их метильных
О-эфиров, приводящим только к диметоксиянтарной кислоте, имеет фура-
нозидную структуру (пятичленный цикл). Это подтверждается фактом
образования тритильных * эфиров рибонуклеотидов и дезоксирибонуклео-
тидов (трифенилхлорметан, как известно, алкилирует только первичный,
но не вторичный гидроксил спиртов).
Какой же атом азота пиримидиновых и пуриновых гетероциклов свя
зан с гликозидным углеродом? Что касается пиримидиновых нуклеотидов,
это не может быть азот аминогруппы цитидина, так как при действии
азотистой кислоты на цитидин VI его аминогруппа замещается на гидр
* Трифенилметильных.
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
747
оксил и он превращается в уридин VII без потери связи с сахаром. Эта
реакция доказывает также, что в уридине и цитидине связь с рибозой
осуществляется через один и тот же азот. Если уридин VII прометилиро-
вать иодистым метилом, то при последующем гидролизе с отщеплением
рибозы получится 3-метилурацил VIII (нумерация атомов в метилурациле,
принятая UPAC в 1968 г.):
Ясно, что связь с гликозидным углеродом осуществлялась через
первый атом азота.
Что касается пуриновых гетероциклов, то и их связь с сахаром также
не осуществляется через аминогруппу гуанина или аденина, так как
замена аминогруппы на гидроксил действием азотистой кислоты про-
исходит без нарушения связи с сахаром.
Пуриновые нуклеозиды типа инозина X, полученного из гуанозина IX
действием азотистой кислоты, при действии йодистого метила подвер-
гаются метилированию по обоим азотам шестичленного цикла, что ис-
ключает связь с ними гликозидного углерода.
748 н УК ЛЕ ОТ И,Ч 14 II ПОЛИНУКЛЕОТИДЫ
Выбор между двумя азотами пятичленного цикла был сделан, в част-
ности, на основании сравнения спектров поглощения аденозина, 9-метил-
аденина и 7-метпладенина, причем спектры двух первых оказались более
близкими. Кроме того, этот вопрос решен на основании синтеза.
Синтез нуклеотидов
В качестве примера синтеза пиримидиновых нуклеозидов приведем синтез
риботимидина по Фоксу (hg = -j- Hg):
ртутная
соль тимина
трибензоил рибофура-
нозилбромид'
Применяя вместо трибензоилрибофуранозилбромида дибензои тдез-
оксирибофуранозилбромид, получают соответствующие дезоксирибонук-
леозиды.
Аналогичный путь применим и для синтеза пуриновых нуклеозидов,
так как атом ртути замещает водород именно при девятом атоме азота.
Если пурин имеет свободную аминогруппу, как в гуанине или аденине,
ее защищают бензоилированием.
Для превращения нуклеозидов в нуклеотиды нужно проэтерифици-
ровать фосфорной кислотой, например, первичную спиртовую группу
сахара. Остальные гидроксильные группы должны быть защищены, что
может быть осуществлено разными способами, например ацетонпрованием
в кислом растворе двух гидроксилов рибозы, предварительно тритили-
рованной по первичноспиртовой группе:
(с6н5)3с-о
(С6Н5)3СС1>
но он
(СН3)2СО.
(С6Нз)3С-О
сн2.о N H2/pd
н
н
Н Н
° /°
ж
Н3С СНз
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
749
о
(С6Н5СН2О)2РО
(С6н5)3сн + НОСИЛО N (CeHsCHiO|.POC] SiL
—Кн н/ -------------------► Кн нД ---------~*
н\ / н н\,н
О\ /° °\ /°
н3с¥ сн3 н3с сн3
с6н5сн3 + (сн3)2со
Если фосфорилировать тритильное производное без защиты вторич-
ных спиртовых групп ацетонированием, остаток фосфорной кислоты
этерифицирует гидроксил при третьем углероде, и после снятия три-
тильной группы гидрогенолизом можно получить нуклеотид, изомерный
изображенному выше по положению остатка фосфорной кислоты. Поло-
жения 5' и 3' — это как раз те положения, которые остаток фосфорной
кислоты занимает в природных нуклеотидах, полученных гидролизом
полинуклеотидов.
ПОЛИНУКЛЕОТИДЫ
Полинуклеотиды, т. е. рибонуклеиновые кислоты (РНК) и дезоксирибо-
нуклеиновые кислоты (ДНК), представляют собой макромолекулярные
цепи, в которых, в соответствии с анализом, на 1 моль гетероцикла при-
ходится 1 моль сахара и 1 остаток фосфорной кислоты. По кривой тит-
рования ясно, что при каждом атоме фосфора имеется 1 гидроксил, т. е.
что полинуклеотиды представляют собой двузамещенные эфиры фосфор-
ной кислоты, сохранившей одну кислотную функцию. Все это позволяет
полностью установить тип первичной структуры РНК и ДНК. Однако
конкретная первичная структура каждой индивидуальной РНК и ДНК
определяется еще чередованием четырех гетероциклов — двух пуриновых
(аденин и гуанин) и двух пиримидиновых (урацил и цитозин — для РНК;
тимин и цитозин — для ДНК). Методы установления этого чередования
только разрабатываются. Метод, предложенный Корана, состоит в под-
боре специфических ферментов, один из которых (из змеиного яда) рас-
щепляет цепь по связи фосфорной кислоты с первичным гидроксилом
(С5<), а Другой (из селезенки) — по связи фосфорной кислоты с вторичной
гидроксильной группой (С3'):
750
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
(Г-остаток гетероцикла)
Таким образом, возникает возможность судить о характере концевых
групп и связанных с ними гетероциклов Г' и Г'".
В 1965 г. группе американских ученых во главе с Р. Холли удалось
установить полную последовательность нуклеотидов в одной из самых
низкомолекулярных растворимых РНК — транспортной РНК аланина.
Транспортные РНК служат для связывания аминокислот и доставки их
в рибосомы, где производится синтез белка сшиванием аминокислот
в последовательности, определяемой кодом ДНК ядра клетки, передава-
емым в рибосому матричной (информационной) РНК. Каждой аминоки-
слоте соответствует своя транспортная РНК. Мы считаем полезным вкрат-
це изложить работу по расшифровке транспортной РНК аланина в такой
мере, чтобы дать понятие о методе полного установления последователь-
ности нуклеотидов в нуклеиновой кислоте. При этом мы будем пользо-
ваться сокращенными обозначениями, примененными авторами этой
замечательной работы:
А Аденозин-3'-фосфат р или — Остаток фосфорной кислоты
G Гуанозин-З'-фосфат Р! 2',3'-циклический фосфат
I Инозин-3'-фосфат (например, 1р'—инозин-2',3'-
С и и* Цитидин-3' -фосфат Уридин-3'-фосфат Смесь U и DiH-U Ме- DiMe- циклический фосфат) Метил- Диметил-
Ф т Псевдоуридин-3 '-фо сфат Риботимидин-З'-фосфат DiH- -ОН Дпгидро- Свободный гидроксил
DE АЕ- Дпэтил аминоэтил-
Как видно из этого списка обозначений, даже самый низкомолеку-
лярный тип РНК содержит кроме четырех «классических» гетероциклов
также и ранее нами не упомянутые, более редкие, метилированные или
гидрированные их производные.
Последовательность нуклеотидов в транспортной РНК аланина, вы-
деленной из дрожжей и очищенной, устанавливалась путем ее гидролиза
двумя разными ферментами — рибонуклеазой поджелудочной железы
и рибонуклеазой TI такадиастазы. Гидролиз обоими ферментами прово-
дился до предела. Сумма полученных в каждом случае коротких полину-
клеотидов и мононуклеотидов разделялась хроматографически; эти поли-
нуклеотиды (по большей части ди- и три-, но не более окта-) идентифици-
НУКЛЕОТИДЫ II ПОЛИНУКЛЕОТИДЫ
751
ровались или дальнейшим гидролизом, или сличением с заведомыми
образцами по Rf (см. раздел о хроматографии, кн. I, стр. 41). Полу-
чены следующие результаты.
Гидролиз транспортной РНК аланина рибонуклеазой поджелудочной
железы:
С-ОН (терминальный фрагмент) 13С— Ф~ 6U- А—С— Ме-1—ф— DiMe-G—С— 2G-C— 3G-U- G—G—G—А—G—А—G—U* — Me-G—G—С— A—G—С— A—G—Dill-U— G—A—U — I—G—С— G—G—Т— G—G—DiH-U— G—G—А—С — pG—G—G—С—
Гидролиз транспортной РНК аланина рибонуклеазой TI такадиастаза:
9G pG—(начальный фрагмент) С—DiMe-Gp! U-Me-Gp! 4С—G— DiH-U—A—G — C—Me-I—ф—G— T—ф—C-G— A-C-U—C-G- U—С—C—A—C—С-ОН (терминальный
2А—G— фрагмент)
U—G— U*—C—U—C—C—G—
U-A-G— DiII-U-C—G— A—U—U—C—C—G — C—U-C—C-C—U—U—I —
Сравнение данных двух предшествующих таблиц позволяет вывести
заключение, что транспортная РНК аланина состоит из следующих
16 фрагментов:
pG—G—G—C— G-U-G- U—Me-G-G-C— C—DiMe-C— C—U—C—C—C—U—U—I—G—C— Me-I— ф—
G—C— G—G—G—A—G—A-G—U*-C-U-C-C-G—
G-U-A-G- Dill-U-C—G— G—Dill-U—A—G — C-G- G-T-ф—C-G— A—U—U—C—C—G— G—A—C—U—C—G — U-C-C-A—C-C—А-ОП
Фрагмент, содержащий свободный остаток фосфорной кислоты (обо-
значен буквой р), — это левый конец молекулы, фрагмент со свободным
гидроксилом — правый ее конец. Последовательность остальных 14 фраг-
ментов этими данными не определяется. Однако совпадение молекулярного
веса, найденного экспериментально и вычисленного на основании струк-
туры, включающей все 16 звеньев, подтверждает, что нет упущенных фраг-
ментов.
752
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
Далее следует гидролиз транспортной РНК аланина снова рибонукле-
азой TI, но в мягких условиях, что приводит к гораздо более длинным
фрагментам (а — к на схеме). Каждый из этих фрагментов гидролизуется,
как описано выше для исходной РНК, двумя ферментами, и по полученным
малым фрагментам устанавливается состав и последовательность нуклео-
тидов в больших фрагментах (от а до к). Поскольку принцип понятен,
подробности здесь опускаются.
Установив состав и последовательность нуклеотидов в больших фраг-
ментах, их можно расположить в определенной последовательности,
Строение транспортной
Me DiH DiH DiMe
I III
pG-G-G-C-G-U-G-U-G-G-C-G'C-G-U-A-G-U-C-G-G-U-A-G-C-G-C-G-C-U-C-C-C-U-U-l-G-
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 .30 31 32 33 34 35 36 37
Me
I
pG-G-G-C-G-U-G-U-G-G
123456789 10
a
DiMe
I
C-G-C-U-C-C-C-U-U-Ip.'
27 28 29 30 31 32 33 34 .35 36
b
DiH DiH DiMe
I I I
U-C-G-G-U-A-G-C-G-C-G-C-U-C-C-C-U-U-lp!
18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36
e
DiH DiH DiMe
I I I
U-A-G-U-C-G-G-U-A-G-C-G-C-G-C-U-C-C-C-U-U-lp!
15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36
A
Me DiH DiH DiMe
I III
pG-G-G-C-G-U-G-U-G-G-C-G-C-G-U-A-G-U-C-G-G-U-A-G-C-G-C-G-C-U-C-C-C-U-U-I-Gpf
t 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37
J
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
753
пользуясь тем, что они частично перекрываются последовательностью
нуклеотидов на своих концах. Так, например, последовательность нук-
леотидов в середине фрагмента i (Т — ф — С — G — А — U — U — С —)
доказывает, что фрагмент / с правым концом Т — ф — С — G — и фраг-
мент g с левым концом А — U — U — С — расположены непосредствен-
но (и без потерь) один за другим. Таким образом удается воссоздать всю
последовательность 77 нуклеотидов в транспортной РНК аланина, что
читатель без труда поймет из схемы.
рибонуклеиновой кислоты аланина
Me
C-i-4'-G-G-G-A-G-A-G-U:C-U-C-C-G-G-T-i|>-C-G-A-U-U-C-C-G-G-A-C-U-C-G-U-C-C-A-C-C-AOH
38 39 40 41 42 43 44 4а 4b 47 48 49 50 51 52 51 54 35 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 70 77
Me
C-I-l-G-G-G-A-G-A-G
38 39 40 41 42 43 44 45 46 47
С
A-C-U-C-G-U-C-C-A-C-C-AOH
66 67 68 69 70 71 72 73 74 75 76 77
а
Me
' I ....
C-l-'I'-G-G-G-A-G-A-G-U-C-U-C-C-G-G-T-t-C-G
38 39 40 41 42 43 44 45 46 47 48 49 50 51 53 53 54 53 aS 57 5S
f
A-U-U-C-C-G-G-A-C-U-C-G-U-C-C-A-C-C-AOH
j'J 60 61 62 63 64 65 66 67 68 69 70 71 73 73 74 75 76 77
9
Me
I
C-I-+-G-G-G-A-G-A-G-U-C-U-C-C-G-G-T-+-C-G-A-U-U-C-C-G
38 319 40 41 42 43 44 45 46 47 4S 49 50 51 52 53 54 55 56 57 53 59 60 61 63 63 64
i
Me
c-!-+-g-g-g-a-g-a-g-u-c-u-c-c-g-g-t-4-c-g-a-u-u-c-c-g-g-a-c-u-c-g-u-c-c-a-c-c-aqh
38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77
k
48 Заказ 672
7М
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
Б настоящее время идет интенсивное исследование транспортных РНК
других аминокислот и уже установлены структуры т-РНК серина, валина,
тирозина и фенилаланина. Конечно, трудности установления строения
РНК с высоким молекулярным весом будут несравнимо большими, хотя
принцип может остаться тем же.
Важное значение имел открытый Очоа и Гринберг-Манаго метод
синтеза полирибонуклеотидов из нуклеотида, превращенного в пирофос-
фат, путем действия на этот пирофосфат полинуклеотид-фосфорилазы —
фермента, выделенного из одного из видов азобактера
Так были синтезированы не встречающиеся в природе РНК монотон-
ного строения с одинаковыми гетероциклами — полиадениловая, поли-
уридиловая, полиинозиновая кислоты и т. д., послужившие простейшими
объектами для исследования вторичной структуры и свойств РНК.
Аналогичный синтез полидезоксирибонуклеотидов из дезоксинуклео-
зид-б^трифосфатов был осуществлен Корнбергом при помощи выделен-
ного им из бактерии Escherichia Coli фермента — дезоксирибонуклеотид-
полимеразы:
ОН ОН ОН
л-2
(Г-остаток гуанина)
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
755
Существенные отличия синтеза Корнберга от синтеза Очоа и Грин-
берг-Манаго заключаются (кроме субстрата и фермента) в следующем.
1. Синтез Очоа и Гринберг-Манаго может быть осуществлен с инди-
видуальным пирофосфатом нуклеозида или со смесью таких пирофосфатов.
Рис. 127. Модель участка мо-
лекулы ДНК (а) и схема струк-
туры ДНК в виде спиральной
лестницы (6), где ступени изо-
бражают пиримидиновые и пури-
новые основания, связанные
водородными связями (по Уот-
сону и Крику).
В первом случае возникает монотонно построенный полирибонуклеотид,
во втором — полинуклеотид со столь же правильным рибозофосфатным
скелетом, к которому присоединены гетероциклы в случайном порядке.
2. Для синтеза Корнберга требуется затравка готовой цепью ДНК
или ее обрывком и обязательно смесь трифосфатов всех четырех нуклео-
зидов (аденин, гуанин, тимин, цитозин). В этом синтезе поразительным
образом воссоздается именно та ДНК, которая служила затравкой,
с тем порядком чередования гетероциклов, который был в затравке. Фер-
мент катализирует синтез любой ДНК по образцу затравки. Еще быстрее
48*
756
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
действует затравка ДНК, если ее нагреть в воде до 100° G, т. е. выше тем-
пературы «плавления», при которой рвутся водородные связи, играющие
в ДНК и РНК столь же важную роль, как и в белках.
Переходим к вопросу о вторичной структуре ДНК и РНК.
Как для белков Полингом, так и для ДНК Криком и Уотсоном была
создана модель полимерной цепи, исходя из знания межатомных расстоя-
ний и валентных углов и из подтвержденного опытом предположения
об определяющем участии водородных связей в конформации молекулы.
На этот раз вторичная структура ДНК имеет вид спиральной лестницы
(рис. 127) из двух витков дезоксирибозофосфатной цепи с гетероциклами,
торчащими от этих двух цепей попарно навстречу друг другу и связанных
водородными связями (ступени лестницы). При этом масштаб позволяет
уложиться и связаться водородными связями в пару только пурину
с пиримидином, но не пиримидину с пиримидином (не достанут) и не пу-
рину с пурином (не поместятся). Возможность же образования водород-
ных связей существует (в этих рамках) у гуанина с цитозином и у аденина
с тимином. Таким образом, ступеньки спиральной^лестницьй ДНК будут
всегда только двух следующих типов:
аленин
цитозин
Крику и Уотсону (1953 г.) удалось рентгенограммой вытянутой нити
ДНК подтвердить основные выводы своей гипотезы. С тех пор она нашла
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
757
многочисленные подтверждения. Отсюда ясно, что в цепи ДНК число
молекул гуанина должно быть равно числу молекул цитозина, и то же
самое справедливо для другой комплементарной пары — аденина и ти-
мина. Таково простое объяснение правила Чаргаффа эмпирически уста-
новленного на основании анализа различных ДНК. Это правило конста-
тирует, что молекулярное отношение азотистых гетероциклов в ДНК
таково: 1 аденин к 1 тимину; 1 гуанин к 1 цитозину, а в РНК: 1 аденин
к 1 урацилу и 1 гуанин к 1 цитозину.
Синтез Корнберга на затравке или, как предпочитают говорить, на
матрице находит простое объяснение с точки зрения этой модели. Обо-
значим буквами участки макромолекулы с аденином через А, с гуанином
через G, с тимином через Тис цитозином через С, а водородные связи —
пунктиром из трех точек. Тогда написанные только что формулы примут
вид:
А-..Т
G-..C
Не включенные в макромолекулы мононуклеозиды, связанные с фос-
форной кислотой, будем обозначать теми же буквами. Тогда спиральная
лестница затравки (матрицы) ДНК изобразится какой-либо определенной
последовательностью букв, связанных пунктирами водородных связей
с соответствующими буквами, изображающими участки дополнительных
нуклеотидов.
Представим себе теперь, что водородные связи разорвались (сразу
или постепенно, во всей «лестнице» или в части ее), например, при нагре-
вании затравки в воде, что, как сказано, ускоряет процесс синтеза. Тогда
каждая из двух нитей будет забирать из среды и подбирать те нуклеозиды,
которые могут образовать с соответствующими звеньями водородные свя-
зи, а фермент будет их сшивать фосфорнокислотными связями, если нук-
леозиды подойдут по размерам. В противном случае рано пли поздно они
снова оборвутся и заменятся новым нуклеозидом и так до тех пор, пока
последний не сможет быть «пришит». Таким образом, неизбежно, если
хватит растворенных в среде нуклеозидов (в виде их трифосфатов), про-
изойдет редупликация — удвоение, причем обе полученные «лестницы»
будут точной копией матрицы:
А- • •т 1 А- ••Т Т-- 1 •А А-- •т т- • •А
Т- ••А Т- • •А 1 А-- •Т Т-- । • А 1 А- • •Т
С- ••G С- • •G G 1 С-. 1 •G G. ...с
1 1 — —1 + 1 -> 1 + 1 1
С- ••G с G-- 1 • С с-. 1 • G G- i • •С 1
т- 1 • •А т I 1 А 1 Т-- •А 1 А- • •т
G- 1 ••С 1 G- ••С С-. • G G-. •С С- • •G
(цепи разошлись,
и каждая наби-
рает из среды
комплементарные
нуклеотиды)
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
758
в опытах Корнберга достигалось увеличение количества дезоксири-
бонуклеиновой кислоты — затравки в 20 раз.
Тот же процесс, по современным представлениям, лежит в основе
целения клеток давно известным процессом, называемым в биологии
митозом или кариокинезом. Этот процесс состоит в том, что в ядре клетки
обособляются видимые под микроскопом шпилькообразные хромосомы,
состоящие из ДНК. Каждая расщепляется вдоль на две половины, они
расходятся в два противоположных конца клетки и каждая половина
дополняется до полной хромосомы. Затем клетка делится стенкой на две,
и попавшие в каждую новую клетку хромосомы снова образуют ядро.
Нетрудно видеть, что процесс редупликации хромосом имеет в своей ос-
нове только что описанный процесс редупликации нитей ДНК. Принимая
во внимание, что каждый организм начинает свою жизнь с одной клетки,
очевидно, что каждая клетка многоклеточного организма в результате
митотического деления получила полный набор хромосом и, значит, пол-
ный набор ДНК исходной клетки. Если последняя произошла от слияния
отцовской и материнской клеток (каждая из них имеет лишь половинный
набор хромосом), то в результате каждая клетка организма получила
половину хромосом (и ДНК) по отцовскому типу, а половину — по мате-
ринскому.
Данная выше картина редупликации ДНК в ходе митоза была под-
тверждена применением меченых атомов. Мы приведем только опыт с мет-
кой по азоту. Культура бактерий была выращена на среде, содержащей
только 15N, а затем была перенесена в среду с 14N, где и продолжала рост.
Время от времени отбирались пробы биомассы, извлекалась оттуда ДНК
и производилось центрифугирование ее макромолекул в растворе хлорис-
того цезия такой плотности, чтобы она была средней между плотностью
нитей ДНК с 14Х и 15N. Разделенные таким методом фракции ДНК, как
оказалось, не имеют макромолекул с хаотически перемешанными ато-
мами 14N и 15N, а четко делятся на три группы: ДНК, полностью постро-
енные на 16N; ДНК, содержащие только 14N, и ДНК с 50% 14N и 50%
16N. Таким образом, одна цепь двойной спирали ДНК действительно це-
лостно передается потомству, а вторая половина спирали набирается из
«подручного материала» нуклеозидфосфатов.
Другой опыт, иллюстрирующий роль ДНК, заключается в следующем.
Фаги, как уже упомянуто, это вирусы бактерий. Под электронным микро-
скопом можно наблюдать, как эти маленькие организмоподобные частицы,
прилипнув к стенке большой клетки — бактерии, прокалывают острым
выступом (точнее, растворяют с помощью выделяемого им фермента)
участок стенки бактерии и выпускают внутрь клетки содержимое своего
целкового мешка — дезоксирибонуклеиновую кислоту. Сам белковый
мешок легко отделяется от бактерии и в дальнейшем участия не прини-
мает. Был выращен бактериофаг (типа Т2), помеченный радиоактивным
фосфором по нуклеиновой кислоте и радиоактивной серой по белку (Херши
и Чейг). В результате радиохимического анализа было выяснено, что
в клетку бактерии проникает только радиоактивный фосфор (т. е. ДНК),
а радиоактивная сера практически вся остается в отпавших оболочках
фага. В дальнейшем идет обычное размножение фага в клетке бактерии,
распад последней и выпадение «взрослых» частиц фага в среду. Таким об-
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
759
разом, ясно, и это подтверждено множеством других аргументов и экспе-
риментов, что ДНК — первоначальная причина собственного размноже-
ния (повторной редупликации) и синтеза белка по типу, диктуемому
структурой ДНК. Иными словами, ДНК является носителем информации
будущей структуры белков, синтезируемых с каким-то ее участием.
Отвлекаясь от вирусов и фагов и возвращаясь к клеткам, напомним
что, как известно из цитологии, ДНК клетки сосредоточена в ядре (в хро-
мосомам), а синтез белка идет в частицах — рибосомах, расположенных
в цитоплазме, вне ядра, и содержащих только высокомолекулярную
рибонуклеиновую кислоту. Исследование этого вопроса привело к выводу,
что функции ДНК хромосом и РНК рибосом разделены так,что ДНК только
хранитель информации, а рибосомы — синтетическая фабрика любого
белка, строение которого задается ДНК. Как же передается эта информа-
ция от ДНК к рибосомам?
В настоящее время есть исчерпывающие доказательства существова-
ния в клетке относительно низкомолекулярных «информационных» рибо-
нуклеиновых кислот, синтезируемых на ДНК как на матрице, т. е. так,
что матричным A, G, Т и С отвечают соответственно U (урацил, а не тимин,
так как это уже не ДНК, а РНК), С, А и G. Эти и-РНК, берущие, так
сказать, отпечаток с ДНК, перемещаются в рибосомы и, закрепляясь
на рибосомальной РНК (р-РНК), передают туда информацию о типе
белков, которые нужно синтезировать рибосомам. В сущности, сборка
молекул белка идет именно на и-РНК, называемых поэтому также м-РНК—
матричными РНК (о коде информации речь пойдет позднее). Доказано
это было, в частности, так (Фолькин, Астрчан): бактерии заражали фагом,
а затем в среду вводили радиоактивный фосфат, меченный 32Р. Через
1—3 мин бактерии разрушались. Тогда отделяли всю РНК и в ней опреде-
ляли обычным путем суммарное количество каждого гетероциклического
основания (A, G, С, U) и, отдельно, соотношение меченых по фосфору
нуклеотидов с теми же гетероциклами. В то время как между составом
ДНК фага и РНК в целом отсутствовало какое-либо соответствие, между
составом ДНК фага и радиоактивных нуклеотидов из РНК было найдено
ожидаемое соответствие, т. е. следующее равенство:
д_1_т *Д I *ТТ
(звездочками обозначены нуклеотиды, меченные 82Р)
C-J-G *C-{-*G 14
(в ДНЮ (в РНК)
Следовательно, за короткий промежуток времени между введением
в среду 32Р и умерщвлением бактерий в них успел произойти синтез
только матричных (информационных) РНК, но не РНК рибосом, и только
и-РНК получили 32Р. Добавим, что в общем количестве РНК в клетке
на долю и-РНК приходится 1 %, па долю РНК рибосом 90%, а остальное
падает на долю низкомолекулярных растворимых рибонуклеиновых ки-
слот, роль которых будет освещена ниже. Поэтому на общий состав РНК
изменение состава и-РНК, составляющих лишь 1%, заметно не влияет.
После разрушения клеток (например, бактерий) рибосомы оказалось
возможным отцентрифугировать и, введи их в необходимую ферментную
среду, заставить вырабатывать белок. При этом можно было вместо и-Р
ввести чужую РНК, например бактериофага f-2 (этот фаг содержит только
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
РНК и белок). Эта чужая РНК функционировала в этом случае как
п-РНК, и рибосомы бактерии вырабатывали чужой белок — белок фаго-
вой оболочки (Ниренберг, Френкель-Конрат и др.). Не менее поразителен
опыт Ниренберга. К взвеси рибосом в растворе ферментов, в котором име-
ется набор всех 20 аминокислот, добавлялся в качестве и-РНК тот или иной
синтетический полинуклеотид, например полиуридпнфосфат. Рибосомы
вырабатывали в этом случае белок монотонного строения — именно
полифенилаланин. Заметим этот факт, фундаментальный для расшифровки
кода ДНК (стр. 761).
Какова же роль третьей группы РНК, составляющей до 10% общего
количества РНК клетки и не являющейся ни высокомолекулярной рибо-
сомальной РНК, ни матричной (информационной) РНК. Эта так назы-
ваемая транспортная РНК сравнительно низкомолекулярна (молекуляр-
ный вес порядка 25 000). Ее назначение — связывать свободные амино-
кислоты из раствора и в таком виде, снабженные кодовой меткой т-РНК,
нести их в рибосомы. По-видимому, смысл этого заключается, в част-
ности, в защите аминокислот от иных процессов метаболизма, например
от окисления и энергетического сгорания, на что организм все же тратит
большее или меньшее количество аминокислот — продуктов пищеваре-
ния белковой пищи. Главное, однако, — снабжение аминокислоты кодо-
вым ярлыком. Первой реакцией связывания аминокислоты является
взаимодействие ее карбоксила с аденозинтрифосфатом с образованием
фосфатной связи (для каждой из 20 аминокислот нужен свой специфи-
ческий фермент):
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
761
Далее аминокислотный остаток переходит в молекулу специфической
для данной аминокислоты растворимой т-РНК, причем аминокислота аци-
лирует один из двух вторичных гидрокислов рибозы концевого нуклео-
зида т-РНК. Это видно из того, что транспортная РНК, связавшая моле-
кулу аминокислоты, в отличие от свободной т-РНК не окисляется иодной
кислотой, как все цис-гликоли (кн. I, стр. 457), т. е. один из двух ее гидро-
ксилов ацилирован. На то, как затем осуществляется взаимодействие
рибосомальной, матричной и транспортной РНК со связанной амино-
кислотой, проливают свет опыты Липмана, согласно которым положение
аминокислоты в полипептидноп цепи определяется только взаимодейст-
вием кодона * (в и-РНК) и антикодона (в т-РНК).
Прямое доказательство этого положения таково.
Цистеиновая т-РНК предварительно была нагружена цистеином,
меченным 14С. Затем цистеин был мягко восстановлен (над никелем Ренея)
в аланин:
т-РНКс>—СН^О А . т-РНКСу— СН2 о А
н ] \ ы. нЧ К и
У он о он
O = C-CH-CH2SH О=С~СН~СН3
I I J
NH2 т^н2
Таким образом, цистеиновая т-РНК оказалась связанной уже с ала-
нином.
Затем эта т-РНК, связанная с аланином, была помещена в систему,
синтезирующую гемоглобин (система содержала и-РНК, специфичную
для гемоглобина). После анализа образовавшихся полипептидных цепей
гемоглобина было найдено, что цистеин в них замещен на аланин. Следо-
вательно, рибосомальная РНК в определении последовательности амино-
кислот в белке не участвовала.
Определение последовательности нуклеотидов в кодоне. Напомним, од-
нако, что порядок набора аминокислот в белке, синтезируемом в рибо-
соме, диктует ДНК хромосомы. Этот закодированный в ДНК порядок
передается в рибосому посредством матричной (информационной) РНК,
что твердо установлено. Очевидно, что код ДНК состоит в порядке чере-
дования гетероциклов в хромосоме, точнее, на каком-то ее участке, да-
ющем реплику на и-РНК. Поскольку гетероциклов в ДНК имеется четыре
сорта, а аминокислот 20, то один гетероцикл не может определять одну
аминокислоту. Не хватит и пары гетероциклов, так как 42 = 16. Но трех
гетероциклов (43 = 64) с избытком хватит. Таким образом, вероятно,
что это число именно 3, а не больше. Напомним опыты Ниренберга с поли-
* Кодон — участок, в данном случае участок и-РНК, состоящий пз трех нуклеози-
дов, кодирующих аминокислоту, которая войдет в состав белка. А нтикодон — три
других комплементарных нуклеозида, входящие в данном случае в состав т-нни
(см. далее).
НУКЛЕОТИДЫ и ПОЛИНУКЛЕОТИДЫ
уридпнфосфатом в качестве и-РНК. В этом случае рибосомы синтезиро-
вали полифепи.чалапин в качестве единственного белка. Отсюда можно
заключить, что кодон фенилаланина U—U—-U (три звена уридинфосфата
в молекуле п-РНК). Используя синтетические полинуклеотиды с моно-
тонным строением (один гетероцикл) и с хаотическим строением, но с раз-
ными соотношениями гетероциклов и применяя теорию вероятности,
v*'
Рис. 128. Схема синтеза белка в клетке:
ДНК — дезоксирибонуклеиновая кислота; и-РНК — информационная (матричная) рибонуклеино-
вая кислота, снимающая реплики UUU с кодона ААА ДНК; АТФ — аденозинтрифосфат, активиру-
ющий аминокислоты и связывающий их с РНК (т-РНК); левое кувшинообразное тело — одна из
рибосом, заполучившая и-РНК и подбирающая т-РНК (с несомыми аминокислотами) по их кодонам
в данном случае по ААА.
Порядок пяо т ИТЬ ТРОИКИ гетеРоциклов кодона для всех 20 аминокислот
берга и еотп 1 0Л05^ения тетероциклов в кодоне выяснен работами Нирен-
«выро-гпрнпт ’ опуг?ЛИКованнЬ1МИ в 1965 г. Во многих случаях код оказался
ляХя№ ЭТ° 2зналает’ ^о_одна и та же аминокислота опреде-
Как мстяст°Ц какоя либ° тР°йкой, а двумя и даже многими.
аминокисЛотаМОиИгвя?ИРеН 6РГ И r°TP‘ (1965 Т’РНК’ "^РУ^нные
на рибосомой ’ язываются с рибосомами только в том случае, если
полняетсиитР^аХ°ДИ™Я соответствующая и-РНК. Если роль и-РНК вы-
то с рибосомами а®скии полиРибонуклеотид (например, полиуридинфосфат),
язывается только та т-РНК, в структуре которой имеется
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
763
соответствующий антикодон. Так, для UUU это тройка А — А — А
присутствующая только в т-РНК фенилаланина. Схема всего синтеза
белка в клетке дана на рис. 128. Оказалось, что в этих опытах (связы-
вание т-РНК с рибосомами) полирибонуклеотид можно заменить тринукле-
отидом и таким образом точно определить последовательность нуклеоти-
дов в кодоне. Например, присоединение т-РНК лейцина к рибосомам
и включение лейцина в белок определяется кодоном UUG, причем если
те же три нуклеозида расположены в заменяющем и-РНК тринуклеотиде
в порядке GUU или UGU, то т-РНК лейцина.уже не связывается с рибо-
сомами.
Так была определена последовательность нуклеотидов (любая из
троек) в кодонах для всех аминокислот (табл. 88).
Таблица 88. Генетический код РНК
Аминокислота
Кодоны
Фенилаланин
Лейцин
Пролин
Серин
Изолейцин
Тирозин
Валин
Триптофан
Аланин
Арринин
Аспарагин
Цистеин
Глицин
Лизин
Треонин
Аспарагиновая кислота
Глутаминовая кислота
Гистидин
Глутамин
Метионин
UUU, UUC
UUA, UUG, CUU, CUC, CUA, CUG
CCU, ССС, CCA, CCG
UCU, ССС, UCA, UCG, AGU, AGC
AUU, AUC, AUA
UAU, UAC
GUU, GUC, CUA, GUG
UGG
GCU, GCC, GCA, GCG
CGU, CGC, CGA, CGG, AGA, AGG
AAU, ААС
UGU, UGC
GGU, GGC, GGA, GGG
AAA, AAG
ACU, ЛСС, АСА, ACG
GAU, GAC
GAA, GAG
CAU, CAG
САА, CAG
AUG
и-РНК доставляет в рибосому непрерывную цепь кодонов, связанных
в молекуле этой и-РНК. Транспортная РНК переносит в рибосому амино-
кислоты, снабженные антикодонами (комплементарными к кодону),
позволяющими аминокислотам закрепиться на и-РНК и начать «с юрку»
белковой молекулы. Нужно, однако, вскрыть еще одну существеннее
деталь этой сборки: знаки препинания в этой непрерывной цепи основа
ний. Очевидно, что результат «чтения» цепи триплетных кодов будет
разный, в зависимости от того, с какого места начать чтение этой ш пре
рывной строки. Выяснено, что, по-видимому, начало приходится bcci
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
на метиониновый код AUG и в начало белковой цени закладывается
метиошшовая молекула (а именно формилметионин), хотя бы в дальней-
шем ее пришлось отщепить. Дальнейшее «чтение» кода определено в виде
последующих троек. До каких пор? Оказывается до тех, пока не встре-
тится один из «бессмысленных» кодов, т. е. не соответствующих никакой
аминокислоте, а именно UAG или UAA (условно названные первая охрой,
вторая янтарем).
Интересно, что действие ряда антибиотиков сводится к помехе той
или иной стадии этого процесса. Тетрациклиновые антибиотики (стр. 263)
мешают т-РНК, несущей аминокислоту, закрепиться на комплементар-
ном кодоне и-РНК в рибосоме. Стрептомицин и актиномицин служат
помехой снятия реплики с ДНК и РНК. Пуромицин сам занимает место
концевого участка на т-РНК, обрывая таким образом синтез белка.
Левомицетин тормозит последнюю стадию синтеза белка — связывание
аминокислот в полипептидную цепь.
Синтез ДНК и РНК. Речь о синтезе ДНК и РНК придется вести несколько
издалека, начиная со связывания нуклеозида и нуклеотида в динуклеотид.
Трудность заключается в том, чтобы связать два нуклеотида фосфатным
мостом именно по третьему и пятому гидроксилам. Для этого надо за-
щитить гидроксилы, которые не должны вступать в связь, и, кроме того,
защитить аминогруппу (например, пиримидинового гетероцикла). Вто-
ричные гидроксилы защищают бензоилированием или ацетилированием,
первичные гидроксилы — тритилированием [действием (С6Н5)3СС1[, ами-
ногруппы — бензоилированием. Смыкание фосфатным мостом осущест-
вляется действием двукислотного фосфорного эфира нуклеозида (т. е.
нуклеотида) на защищенный описанным выше образом второй нуклеотид
в присутствии дегидратирующего агента—дициклогексилкарбодиимида.
На стр. 765 приведена схема синтеза динуклеотида, включающего в ка-
честве пиримидиновых гетероциклов цитозин и тимин.
Тот же синтез можно изобразить такой Сокращенной схемой:
CBz т
C6H„N=C=NC6H|1
BzC Т
l.OH
n , +
с т
Здесь (и далее):
С — цитозин;
Т — тимин;
G — гуанин;
А — аденин;
Тг — тритил (трифенилметил);
р — остаток фосфорной кислоты;
Bz — бензоил;
Ас — ацетил.
На схеме показаны лишь первичный гидроксил (пятый), вторичный
гидроксил (третий) и гетероциклическое основание. Остальные группи
ровки не показаны, так как они участия в синтезе не принимают.
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
765
NH-,
А’
NHCOC6H5
nhcoc6h5
N
СеН5СОС1
N
он
о
он
он’
он
сн.,осо
N
(СбН5)з
он
(С6Н5)3СС1
NHCOCeH5
О N
ОН
1 '-б _п
2.ОН
•в
NH,
А
О
N
3
Аналогично можно изобразить
О
О
BzA ABz
ОН
синтез тринуклеотидсг.
CBz
RO-Р—О
Р
-ОН
НО-Р-О —ОАс
С6И,1Х-"С=Хтсб»п
"О
"О
>
О
ABz ABz CBz
О
л
RO-P-O,
I
“О
ОН
— ОАс -----
-о-р-оф
“О
— ОН
766
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТ иды
Применяя этот принцип, Корана синтезировал тетрануклеотиды.
Из двух защищенных тетрануклеотидов был получен октануклеотид,
из него и тетрануклеотида — додекануклеотид и, наконец, из додекану-
клеотида и тетрануклеотида — гексадекануклеотид. Получены и другие
(м и гонуклеот иды.
Из приведенных схем ясно, что последовательность гетероциклических
оснований в таком синтетическом полинуклеотиде зависит от последова-
' тельности их во взятых для синтеза
Рис. 129.
фрагментарных полинуклеотидах,
т. е. от воли синтетика. Однако
такой многостадийный синтез не
только утомителен, но выход поли-
нуклеотида в конечном счете крайне
невелик, а цепь полинуклеотида,
включающую несколько десятков
нуклеотидных звеньев, так построить
вообще не удается.
Путь синтеза более длинных це-
пей ДНК основывается на примене-
нии ферментов для сшивания фос-
фатной связью закрепленных на
матрице сравнительно недлинных
полинуклеотидных цепей (олигонуклеотидов) в длинные цепи. Понятие
о действии ферментов в этом случае дают следующие иллюстрации.
На рис. 129 представлена спаренная двойная спираль ДНК, которая
ферментом экзонуклеазой частично разъединяется, и разъединенные концы
при действии фермента полимеразы достраиваются за счет комплементар-
ных нуклеотидов из среды, сшиваемых в цепь (заштрихованные участки
правой спирали) (Корнберг). В последние годы открыта также лигаза —
фермент, сшивающий фосфорнокислыми мостами два соседних, закреплен-
ных на матрице олигонуклеотида (рис. 130). Затрата энергии покрывается
3"~п—I—I—I—5'
А. Т G Т атф
Т А С А
3'---1 |—I------1---5'
А Т G Т
Т А С А
Рис. 130.
аденозинтрифосфатом (АТФ). В левой стороне рисунка изображен участок
сдвоенной спирали ДНК, цепи которой связаны между собой водород-
ными связями комплементарных гетероциклов А • • • Т и G • • б. Нпж^
няя цепь посредине разорвана, причем на конце одного из олигонуклеоти
дов имеется фосфатная группа, а на конце другого — гидроксил. 1 осл
действия фермента и АТФ два олигонуклеотида оказываются сшитым
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
767
Принцип комплементирования нуклеотидами матрицы ДНК и феимента-
тнвиого сшивания нуклеотидов на матрице фосфатными мостами позво-
ляет (Корана) построить цепь, комплементарную к исходной ДНК лаже
чисто синтетической. Схема на стр. 768 поясняет, как по плану Копана
можно синтезировать этим путем куски реальной ДНК и сшивать эти
куски в полную молекулу ДНК заданной структуры.
Строка а представляет собою последовательность нуклеотидов от 1
-п Т'"1г.а1т,"ИНа’ а стРока а' ~ последовательность нуклеотидов
21—эО тон же РНК.
Нуклеотид Нуклеотид
Раэры8
I I
______'пппнп ii । ii inn m 11 ii । и-
I
Разры8 п
г Лигаза
TTIIIIIIIITTTHIIIIIIIIIIIIIII
Рис. 131.
Строка б изображает полученный синтезом одноцепочечный фрагмент
ДНК, состоящий из звеньев, комплементарных к звеньям 1—20 исходной
т-РНК; строка б' — одноцепочечный
фрагмент синтетической ДНК из ну-
клеотидов, комплементарных к звеньям
21—40 строки а'. Нижние строки вив'
изображают также фрагменты синтети-
ческой ДНК, но в них только правые
концы из 10 звеньев комплементарны
к 10 нуклеотидам ДНК. изображаемых
строками б и б', и связаны с ними,
а левые концы (также из 10 звеньев)
свободны.
В присутствии смеси соответствующих мононуклеотидов свободные
концы синтетических ДНК строк б, б', в и в' могут дополняться компле-
ментирующими нуклеотидами, которые далее под действием полимеразы
свяжутся в цепь. Роль 10 средних нуклеотидов строк б, в и б', в' — это
роль кнопок, скрепляющих две цепи синтетического полинуклеотида.
Таким образом, в итоге, исходя из синтетических олигонуклеотидов по 20
нуклеотидов в каждом, получаются два отрезка ДНК-кодонов транспорт-
ной рибонуклеиновой кислоты, включающих: первый — 30 нуклеотидов
(от 1 до 30), второй также 30 (от 21 по 50)—нуклеотиды от 21 до 30 в стро-
ках в и б' являются комплементирующими. После их комплементаций
до полных двойных нитей и разъединения последних достроенные (что
не показано на рисунке) нити в и б' могут быть скреплены за счет этих
десяти комплементирующих нуклеотидов 21—30. Затем должно следо-
вать новое комплементирование свободных концов, т. е. 1—20 и 31 50.
Таким образом, синтез на матрице полинуклеотида с последовательностью
звеньев, комплементарных звеньям т-РНК аланина от 1 до 50, будет за-
вершен.
Видоизменение только что описанного плана синтеза отрезка цепи
ДНК, комплементарного к цепи 50 первых нуклеотидов т-РНК алапина,
иллюстрируется на рис. 131. Синтезируются четыре цепи нуклеотидов,
изображенные в верхней части рисунка горизонтальными линиями. Каж-
дая из четырех цепей содержит по 20 нуклеотидов. Цепи построены так.
что верхняя правая линия соответствует 20 нуклеотидам ДНК, компле-
ментарным к первым 20 нуклеотидам т-РНК аланина, верхняя левая
линия соответствует двадцати следующим. Две нижние горнзонтальньт
линии верхней части рисунка символизируют также отрезки Д г
20 нуклеотидов каждый, из них 20 + 10 нуклеотидов комплементарны
Схема синтеза фрагмента ДНК заданной структуры
DiH
(т-РНК) a U-C-U-C'C-G-G-T-<P-C-G-A-U-U-C-C-G-G-A-C-U-C-G-U-C-C-A-C-C-A
V ' 30 29 28 27 26 25 24 23 22 21 20 19 18 17 16 15 14 13 12 11 10 .9 8 7 6 5 4 3 2 I
(ДНК) s c-t-a-a-g-g-c-c-t-g-a-g-c-a-g-g-t-g-g-t
20 19 18 17 16 15 14 13 12 II 10 9 8 7 6 5 4 3 2 1
1111111111
(ДНК) в t-c-t-c-c-g-g-t-t-c-g-a-t-t-c-c-g-g-a-c
' ' 30 29 28 27 26 25 24 23 22 21 20 19 IS 17 16 15 14 13 12 11
DiMe Me DiH
III
G-C-U-C-C-C-U-U-I-G-C-I- + -G-G-G-A-G-A-G-U-C-U-C-C-G-G-T-I-C a' (r-PHK)
50 49 48 47 46 45 44 43 42 41 40 39 38 37 36 35 34 33 32 31 30 29 28 27 26 25 24 23 22 21
G-T-A-C-C-C-T-C-T-C-A-G-A-G-G-C-C-A-A-G !>’ (ДНК)
40 39 38 37 36 35 3 1 33 32 31 30 29 28 27 26 25 24 23 22 21
1111111111
G-C-T-C-C-C-T-T-A-G-C-A-T-G-G-G-A-G-A-G в' (ДНК)
50 49 48 47 46 45 44 43 42 41 40 39 38 37 36 35 34 33 32 31 '
НУКЛЕОТИДЫ И ПОЛИНУКЛЕОТИДЫ
769
к соответствующим 30 нуклеотидам двух верхних цепей. В целом получается
жестко связанная 30 парами комплементарных нуклеотидов система
четырех цепей ДНК со свободными концами по 10 нуклеотидов в верхней
и в нижней цепи. Имеются два разрыва фосфатных связей, отмеченные
стрелками. Действием фермента лигазы эти разрывы «зашиваются»,
и на их месте возникают фосфатные мосты. Таким образом, четыре перво-
начально взятые полинуклеотида превращаются в две цепи, в средней
части комплементарно связанные друг с другом. Осталось с помощью
полимеразы и набора свободных нуклеотидов комплементарно до-
строить оба свободных конца молекулы, и двойная спираль из 50 поли-
нуклеотидов готова. Одна из ее цепей комплементарна к 50 первым нуклео-
тидам т-РНК, другая — комплементарна к первой.
Следуя тому или другому из этих двух планов синтеза, можно по-
строить и ДНК из 77 полинуклеотидов, комплементарных к всей моле-
куле т-РНК аланина, а на ней осуществить и матричный синтез самой
т-РНК. Корана осуществил этот синтез в 1968 г.
Аминокислотный код и вся механика синтеза белков одна и та же для
всего живого — от бактерии до человека. Самое развитие организма
представляется так. В хромосомах той первой клетки, из которой разви-
вается организм, уже заложена в виде ДНК вся информация о тех белках,
которые предстоит синтезировать организму, и что особенно важно,
о тех белковых ферментах, которые организм будет синтезировать и кото-
рые определят всю биохимическую сторону его жизни. В клетке бактерии,
по грубой оценке, действует более 1000 различных ферментов, и это
основа ее жизнедеятельности. Структура каждого из них определяется
некоторым участком нити ДНК, который называется цистроном и пред-
ставляет собой ген, локализованный на нити хромосомы в виде определен-
ной последовательности нуклеотидов. Хромосомы с их цистронами пере-
даются при митотическом делении каждой клетке организма. При обра-
зовании половых клеток последние снабжаются половинным набором
хромосом (гаплоидные клетки); вторая половина дополняется при слиянии
отцовской и материнской клеток, и, таким образом, оплодотворенная клет-
ка — первая клетка будущего организма — получает наследственную
информацию с обеих сторон.
В последнее время появились экспериментальные основания считать
нуклеиновые кислоты ответственными за химию памяти от простейших
червей до высших животных.
49 Заказ 672
ФЕРМЕНТЫ
Катализ многочисленных биохимических реакций, протекающих в рас-
тительных и животных организмах и их клетках, так же как и в одно-
клеточных микроорганизмах, совершается ферментами (энзимами).
Ферменты представляют собой вещества или чисто белковой структуры,
или протеиды — белки, связанные с небелковой простетической группой.
Число уже известных ферментов очень велико. Считают, что одна клетка
бактерии использует до 1000 разных ферментов. Однако лишь для немно-
гих установлено строение. Примерами чисто белковых ферментов могут
служить протеолитические ферменты пищеварения, такие, как пепсин
и трипсин. Известны случаи, когда один и тот же белок несет в организме
и структурную и ферментативную функцию. Примером служит белок
мышц миозин, каталитически разлагающий аденозинтрифосфат — реак-
ция, в данном случае дающая энергию сокращения мышцы (В. А. Эн-
гельгардт, М. Н. Любимова).
Важная группа окислительно-восстановительных ферментов, как пра-
вило, содержит простатические группы, включающие азотистые гетеро-
циклы, которые и выполняют редокс-функции (окислительно-восстанови-
тельные). В случаях, когда именно небелковый компонент фермента
обусловливает специфичность ферментативной активности, простетиче-
ские группы называются коэнзимами или коферментами, а белковая
часть — апоэнзимом или апоферментом. Кофермент определяет специ-
фичность по типам реакций, апофермент — субстратную специфичность.
Апофермент и кофермент во многих случаях удается теми или иными
приемами отделить друг от друга, и часто можно осуществить обратную
реакцию, воссоздавая фермент. Один и тот же кофермент с разными апо-
ферментами образует серию ферментов с различным характером действия.
Кофермент часто тождествен одному из витаминов пли структурно
близок ему — является его производным. Витамины — группа веществ
обычно довольно сложной структуры, часто очень далеких химически
друг от друга и объединяемых только по биологическому признаку.
Это требуемые в микроколичествах, несовершенно необходимые состав-
ные части пищи, недостаток которых вызывает болезнь, а отсутствие
гибель организма. Используя витамины (но не только их), организм че-
ФЕРМЕНТЫ
771
ловека и животных и строит свои ферменты. Одно и то же вещество, слу-
жащее для организмов одного вида витамином, для других организмов
может и не быть витамином по двум причинам: либо организм второго
вида не пользуется данным ферментом, либо он может его синтезировать
сам. Так, аскорбиновая кислота — витамин для человека, но для крысы
она не является витамином, так как организм крысы создает ее из глю-
козы. Некоторые витамины имеют весьма универсальный характер и не-
обходимы всем организмам — от одноклеточных (дрожжей и бактерий)
до человека. Зеленые растения способны производить все свои вещества
из минеральных исходных веществ и СО2 и, следовательно, не нуждаются
в витаминах.
Для действия ряда белковых ферментов в организме необходимо при-
сутствие микроколичеств ионов некоторых металлов-активаторов (Mg,
Zn, Мо, Мп, Си), которые витаминами не считаются. В состав некоторых
коферментов и соответствующих витаминов входят металлы (железо, ко-
бальт). Очень существенна роль микроколичеств металлов (микроэлемен-
тов) для развития растений; по-видимому, эти микроэлементы исполь-
зуются для построения ферментов и служат как бы «витаминами растений».
Частная номенклатура ферментов строится по принципу субстрата,
на который они действуют, причем название фермента получает оконча-
ние аза, например протеаза — фермент, гидролизующий белок (протеин),
амилаза — фермент, гидролизующий крахмал (amylum), целлюлаза —
целлюлозу и т. д. Для групповых названий номенклатура часто строится
по принципу типа катализируемой реакции: эстеразы, трансфосфорилазы
и т. д. Такая номенклатура широко применялась в научной литературе
и с ней нужно быть знакомым, поэтому она и находит место в дальнейшем
изложении. В настоящее время создана международная систематическая
номенклатура ферментов — номенклатура К. Ф., в которой каждый
фермент получает шифр из четырех чисел, обозначающих класс, под-
класс, подподкласс и порядок данного фермента в подподклассе. Шесть
классов ферментов и примеры подклассов и номенклатурных шифров этой
системы приведены на стр. 772.
Ферменты являются катализаторами, но отличаются от обычных ката-
лизаторов. Так, им свойствены:
1. Гораздо большая специфичность по отношению к структуре ката-
лизируемого объекта и по отношению к реакции.
2. Полная стереохимическая специфичность.
3. Гораздо большая скорость протекания ферментативных реакций
по сравнению с теми же реакциями, катализируемыми обычными катали-
заторами.
4. Невысокие оптимумы температуры их действия (обычно активность
ферментов резко падает к 50° Сив интервале 50—80° С действие их пол-
ностью прекращается).
5. Денатурируемость — необратимая потеря каталитической актив-
ности при нагревании до 50—100° С. Существуют, однако, ферменты (трип-
син, рибонуклеаза), активность которых восстанавливается по охлажде-
нии даже после кипячения.
6. Существование оптимума кислотности среды для действия каждого
фермента.
49*
772
ФЕРМЕНТЫ
Подтверждение положения 1 мы увидим при рассмотрении протеоли-
тических ферментов. Положение 2 ясно из того, что фермент, активный
по отношению, например к Z-антиподу, никогда не действует на D-анти-
под, и обратно. Положение 3 можно иллюстрировать тем фактом, что
для гидролиза в единицу времени определенного количества сахарозы
в инвертный сахар необходима концентрация Н+ в 10 000 000 раз боль-
шая, чем фермента инвертазы.
Высокая скорость ферментативных реакций объясняется, с одной
стороны, как всегда при катализе, сильным снижением энергии актива-
ции реакции. Так, при гидролизе казеина кислотой энергия активации
20,6 ккал/молъ, а при гидролизе трипсином — только 12 ккал/молъ. Гид-
ролиз сахарозы кислотой требует энергии активации 25,5 ккал/молъ,
а’ферментативный (сахарозой) — лишь 12—13 ккал/молъ. С другой сто-
роны, в ферментативных реакциях не меньшую роль играет предэкспо-
ненциальный множитель уравнения Аррениуса, так как величина этого
множителя, как правило, на много порядков выше, чем в реакциях обыч-
ного типа. Есть доказательства того, что ферменты содержат центры
(«карманы»), фиксирующие субстрат на поверхности их молекул, и вторые
центры, осуществляющие реакцию. Фермент может быть активен в том
смысле, что он подтягивает активный центр к месту его действия, несколько
изменяя свою вторичную или третичную структуру.
КЛАССИФИКАЦИЯ ФЕРМЕНТОВ
В соответствии с типами реакций, катализируемых ферментами, их клас-
сифицируют в настоящее время следующим образом:
1. Оксидоредуктазы, осуществляющие реакции окисления и восста-
новления, в частности и дегидрирования (дегидрогеназы).
2. Трансферазы, осуществляющие перенос разнообразных остатков —
ацильных, фосфатных, гликозидных и т. д.
3. Гидролазы, осуществляющие гидролиз.
4. Лиазы, осуществляющие негидролитическое расщепление связей
С —С, С—О, С—S, С—N с образованием двойных связей и, в частности,
производящие декарбоксилирование, отщепление воды, сероводорода,
аммиака и т. д.; они осуществляют также реакции присоединения по двой-
ным связям.
5. Изомеразы осуществляют перегруппировки внутри молекулы, на-
пример фосфоглюкомутаза превращает глюкозо-1-фосфат в глюкозо-
6-фосфат.
6. Лигазы, или синтетазы, катализируют образование новых связей
С—С, С—О, С—S, С—N; реакция сопряжена с расщеплением пирофос-
фатной связи в аденозинтрифосфате, что и доставляет энергию синтеза.
Эти шесть классов делятся на подклассы (вторая цифра в шифре фер-
мента по систематической номенклатуре). Подклассы делятся на подпод-
классы (третья цифра в шифре). Наконец, цифрой же обозначается и по-
рядок данного конкретного фермента в подподклассе. Таким образом,
например, гидролизующая крахмал а-амилаза имеет шифр 3.2.1.1,
целлюлаза — 3.2.1.4, аминопептидаза — 3.4.1.2, пепсин — 3.4.4.1, трип-
син — 3.4.4.4 и т. д.
ФЕРМЕНТЫ
773
Из всех этих классов ферментов гидролазы (класс 3) обычно не имеют
простетических групп и являются чистыми белками. Поэтому рассмотре-
ние отдельных классов, которое мы осуществим лишь выборочно, удобно
начать с гидролаз.
Гидролазы
Гидролазы многочисленны. Примерами отдельных групп гидролаз могут
служить эстеразы с подгруппами липаз (гидролизующих [жиры), холин-
эстераз (гидролизующих ацетилхолин), фосфатаз (гидролизующих эфиры
фосфорной кислоты), гликозидаз (осуществляющих гидролиз гликозид-
ных связей), амидаз (гидролизующих амидную связь, например мочевины,
с отщеплением аммиака), протеаз (гидролизующих пептидные связи),
нуклеаз (гидролизующих нуклеиновые кислоты).
Здесь мы кратко рассмотрим лишь наиболее изученные протеазы,
которые делятся на две большие группы — экзопротеазы и эндопротеазы.
Экзопротеазы гидролизуют концевые аминокислоты белковой или поли-
пептидной цепи и в свою очередь делятся на карбоксипептидазы и амино-
пептидазы в зависимости от того, в какой конец белковой цепи — со сво-
бодной карбоксильной или аминогруппой — они отщепляют. Эндопро-
теазы расщепляют белковую молекулу где-либо посредине цепи.
Протеазы распространены в животном и растительном мире; сущест-
вуют клеточные протеазы, осуществляющие соответствующие реакции
внутри клеток. Особенно известен папаин, который выделяют из плодов
папайи. Но наиболее важны и наиболее изучены протеазы пищеваритель-
ного тракта животных и человека. Стенки желудка выделяют неактивный
белок (профермент) — пепсиноген. Под влиянием кислого желудочного
сока и готового находящегося в желудочном соке пепсина от пепсиногена
отщепляется полипептидная цепь, и он превращается в активный фермент
пепсин, имеющий молекулярный вес 35 000 и давно уже полученный
в кристаллическом виде. Пепсин при оптимальном pH 1,5—2,5 разры-
вает белки преимущественно по месту нахождения обеих ароматических
аминокислот (тирозин и фенилаланин) у их аминного конца. При этом
необходимо, чтобы аминокислота, соседняя с ароматической, имела такие
«зацепки» для пепсина, как остатки СООН или SH, и не имела свободной
NH2-rpynnbi. Этих условий оказывается, однако, достаточно для того,
чтобы в желудке произошел гидролиз макромолекул белков на сравни-
тельно небольшие пептидные цепи. Дальнейшее переваривание пищи
в двенадцатиперстной кишке и далее в тонких кишках происходит в усло-
виях уже щелочной среды. Двенадцатиперстную кишку снабжает фер-
ментами поджелудочная железа, которая выделяет проферменты — трип-
синоген, химотрипсиноген и профермент, соответствующий карбоксипеп-
тидазе. Эти проферменты (как и пепсиноген, см. выше) превращаются
в двенадцатиперстной кишке в ферменты—.трипсин, химотрипсин и карб-
оксипептидазу.
Химотрипсин в щелочной среде (pH 8) гидролизует в пептидах пре-
имущественно связи тех же ароматических кислот, что и пепсин, но
с другой стороны — со стороны карбоксила. Трипсин рвет пептидные
связи со стороны карбоксила у остатков лизина и аргинина. Карбокси-
ФЕРМЕНТЫ
пептидазы осуществляют гидролиз концевой аминокислоты, имеющей
свободный карбоксил. С аминного конца белка или пептида подобный
гидролиз осуществляют аминопептидазы. Они входят в состав фермент-
ного выделения стенок тонких кишок. Здесь пищеварение заканчивается
полным гидролизом до аминокислот в результате действия на дипептиды
фермента дипептидазы. Аминокислоты всасываются через стенки кишеч-
ника и поступают в кровь.
Мы уже отметили строгую специфичность соответствия действия каж-
дого фермента строению гидролизуемого участка белковой молекулы,
что устанавливается экспериментально испытанием гидролитического
действия фермента на простейшие пептиды известного строения (ди-,
трппептпды). Столь же строго специфично конфигуративное соответствие
ферментов: в частности, все упомянутые протеазы активны только по от-
ношению к пептидам, построенным из L-аминокислот. Интересно, что
специфичности в отношении вторичной и третичной структур гидроли-
зуемого белка, по-видимому, не существует, хотя наличие этих структур
обычно затрудняет расщепление белка протеазами. Сами ферменты,
денатурируясь, например при нагревании до 50—100° С, теряют актив-
ность, т. е. их активность тесно связана с их собственной структурой
высших порядков.
В настоящее время ведется интенсивная работа по выяснению струк-
туры ферментов пищеварения, и, по-видимому, в недалеком будущем
она будет установлена. Однако нельзя ожидать, что установление пер-
вичной структуры, и даже вторичной и третичной структур, сразу разре-
шит тайну каталитического действия ферментов. Так, строение одной
из фосфатаз — рибонуклеазы, гидролизующей рибонуклеиновые кислоты,
установлено Муром, Штейном и Анфинсеном. Приводим его для примера:
NH2
H2N-Lys-Glu-Tre-Ala-Ala-Ala-Lys-Phe-Glu-Arg-Glu-His-Met-Asp-Ser-Ser-Tre-Ser-Ala _
nh2 nh2nh2 nh2
Ala-Ser-Ser-Ser-Asp-Tyr-Cys-Asp-Glu-Met-Met-Lys-Ser-Arg-Asp-Leu-Tre-Lys-Asp-Arg-
NH2 nh2
2 | I 3
-Cys-Lys-Pro-Val-Asp-Tre-Phe-Val-His-Glu-Ser-Leu-Ala-Asp-Val-Glu-Ala-Val-Cys-Ser-
NH2 NH2 NH2 NHo nh2 nh3
I I 4 ! | | 4 I „ m
~Glu-Lys-Asp-Val-Ala-Cys-Lys-Asp-Gly-Tre-Asp-Glu-Cys-Tyr-Glu-Ser-Tyr-Ser-Tre—
NH2
i I 2
-Met-Ser-Ile-Tre-Asp-Cys-Arg-Glu-Ser-Tre-Gly-Ser-Lys-Tyr-Pro-Asp-Ala-Cys-Tyr-Lys
I NHa NH2 NH2
11 з 1
l-Tre-Tre-Asp-Ala-Glu-Lys-His-Ile-Ile-Val-Ala-Cys-Glu-Gly-Asp-Pro-Tyr-Val-Pro-Val
I- His-Phe-Asp-Ala-Ser-Val-COOH
ФЕРМЕНТЫ
775
В непрерывном полипептидной цепи рибонуклеазы попарно связаны
дисульфидными мостами остатки цистеина, обозначенные одинаковыми но-
мерами. Очевидно, что сама по себе последовательность аминокислот в моле-
куле рибонуклеазы еще совершенно ничего не говорит о ее каталитиче-
ском действии на связь фосфорной кислоты с рибозой в РНК. В высшей
степени интересны исследования рибонуклеазы, выполненные Анфинсе-
ном. Если подействовать [3-оксиэтилмеркаптаном на раствор рибонукле-
азы в водном ~50%-ном растворе мочевины (где а-спираль нарушена
полностью) и таким образом разорвать все S—S-мосты, то каталитические
свойства ферментов полностью исчезают. Однако если полученный белок,
содержащий SH-группы на месте S — S-мостов и освобожденный от моче-
вины, окислить воздухом, то все SH-группы попарно окисляются в S—S-мо-
сты, структура рибонуклеазы воссоздается, и вновь приобретается актив-
ность. Следовательно, S—S-мосты в данном белке (и это типично) возни-
кают на прежних местах и, несомненно, одновременно воссоздается не
только вторичная, но и третичная структура, свойственная рибонуклеазе.
Если же окисление производить в растворе мочевины, где обычные водо-
родные связи вторичной структуры нарушены, то сшивание происходит
в беспорядке или в ином порядке и активного фермента не получается.
Таким образом, как оказалось в этом случае, не дисульфидные мосты,
а водородные связи вторичной структуры предопределяют третичную
структуру, сближающую определенные цистеиновые SH-группы и созда-
ющие возможность окисления их в цистиновые мосты S—S. Вместе с тем
ясно, что за ферментативную активность ответственна совокупность пер-
вичной, вторичной и третичной структур.
Однако на примере ряда ферментов, и рибонуклеазы в частности, было
показано, что не вся молекула, а лишь некоторая ее часть (активный
участок) ответственна за каталитическую активность. Так, Ричардс,
используя фермент субтилизин, расщепил молекулу рибонуклеазы по
связи между звеньями 20 и 21 (пептидная связь Ala — Ser), и при этом
вторичная и третичная структуры удержали молекулу как целое. Сохра-
нились и ферментативные свойства. Но при хроматографии на кислом
ионообменнике короткий пептид из 20 аминокислотных остатков отде-
лился от остальной части. Обе части молекулы были лишены фермен-
тативной активности, однако при смешении их активность вновь возни-
кала. У отделенной большей части белковой молекулы еще сохранилась
способность связывать обычный для рибононуклеазы субстрат фермента-
тивной реакции, но не расщеплять его. При гидролизе рибонуклеазы
карбоксипептидазой и отщеплений с С-конца трех аминокислот валина,
серина и аланина активность рибонуклеазы не страдает. Прп гидролизе
пепсином разрывается четвертая связь с С-конца и отщепляется кроме
валина, серина и аланина еще и аспарагиновая кислота. Тогда остаток
рибонуклеазы полностью теряет активность. Подобным же образом уста
навливается существенность двух остатков His в положениях 12 и
Сказанное имеет целью дать понятие об исследовании структуры елка
как фермента.
В отличие от гидролаз — чисто белковых ферментов многие, н
далеко пе все другие типы ферментов содержат коферментные i руппы,
которые изучены, в то время как структура апофермента неизвестна.
776
ФЕРМЕНТЫ
Трансферазы
ферменты этой группы (шифр 2) делятся на следующие подгруппы:
2.1. Ферменты, переносящие одноуглеродпые остатки — метил, оксиметил
формил и т. д. ’
2.2. Трансферазы альдегидных и кетоиных групп.
2.3. Трансферазы ацильных групп.
2.4. Трансферазы гликозильных остатков.
2.5. Трансферазы алкилов.
2.6. Трансферазы амнпо-, амидипо- и оксиминогрупп.
2.7. Трансферазы фосфатных остатков.
2.8. Трансферазы групп, содержащих серу (в том числе переносящие кофермент
А, стр. 777).
Упомянем прежде всего о метилтрапсферазах (шифр 2.1.1), перенося-
щих метил (переносится, в частности, метильная группа молекулы метио-
нина). Примером может служить никотинамид-метилтрансфераза, которая
катализирует реакцию метилирования никотинамида по гетероцикли-
ческому азоту, перенося метил с метионина, связанного в S-аденозплме-
тионпн. Последний превращается при этом в S-адепозилгомоцистеин:
Полученный гомоцистеин снова метилируется, в частности за счет
метила бетаина, при посредстве фермента бетаин-а-гомоцистеин, метил
трансферазы:
ZO /Р фермент
HS-CH2CH2CH—+ (CH8)3N-CH2-C<^
| Уон 0
nh2
о
--> СНз-S—СН2СН2СН—с/ +(СН3)2Х-СИ2-С<^
| он °
nh2
ФЕРМЕНТЫ
777
Метилтрансферазы рассматриваются в настоящее время как одноком-
понентные ферменты-белки, действующие на два субстрата: на донор
(аденозилметионин) и на тот или иной акцептор метильной группы. Если
метилированный акцептор имеет ониевуго структуру (бетаины), то реак-
ция трансметилирования обратима. Таким образом, аденозилметионин
служит не коферментом, а метаболитом-переносчиком (косубстратом).
В отдельных случаях (биосинтез метионина, тимидина) в роли кофер-
ментов переноса СН3-групп выступают коферментные формы витамина
В12 или тетрагидрофолевые кислоты.
Ферменты подкласса 2.1.2 — переносчики одноуглеродных остатков
—СН2ОН и —СНО (но не —СООН) имеют в качестве кофермента
5,6,7,8-тетрагидрофолевую {тетрагидроптероилглутаминовую) кислоту
1 /NH /СН2—NH— CNH—СНСН2СН2СООН
N | Y Х=/ {> i00H
8
Группировка атомов 5, 6, 9 и 10 этой кислоты и является реакционным
центром в передаче оксиметильных и формильных групп по схеме, в ко-
торой анион X- служит акцептором этих групп:
5 10
—NH NH—
6СН—СН2
I 8
-СН2ОН
/О
сх
I ХН
—NH N—
СН—сн2
I
х-сн2он
Этого рода ферменты играют роль в синтезе пуриновых гетероциклов
и некоторых аминокислот.
Следующая группа трансфераз (шифр 2.3), которая должна быть здесь
упомянута, это ацилтрансферазы. Их кофермент называется коферментом
А и часто обозначается как КоА или HSKoA. Он включает структурные
единицы пантотеновой кислоты и 3'-фосфоаденозин-5'-пирофосфата и имеет
следующее строение:
Перенос ацила посредством HSKoA изобразится следующей схемой:
RCO-+AT<D + KoASH------> RC-SKoA-L АДФ-р НоРОт
II II
О о
(АТФ — аденозинтрифосфат, АДФ — аденозиндифосфат)
Роль аденозинтрифосфата — снабжение энергией для осуществления
процесса. RC—SKoA передает ацил на субстрат. Именно так совершается
И
о
биогенное превращение уксусной кислоты в изопреноиды через ацето-
уксусную кислоту и мевалоновую кислоту (стр. 660). Окисление жирных
кислот в организме происходит также через стадию образования RC—SKoA.
I
О
После этого под влиянием дегидрогенизующего фермента происходит
дегидрогенизация и в радикале R образуется в а, ^-положении двойная
связь (здесь мы, разумеется, выходим уже из круга трансфераз). Далее
следует гидратация, снова дегидрогенизация [3-оксисоединения в произ-
водное р-кетонокислоты и, наконец, отщепление СН3С—SKoA с образо-
0
ванием жирной кислоты, содержащей на два углеродных атома меньше,
чем было в исходной:
К'СНгСНгС—SKoA R'CH = CHC—SKoA R'CH-CH2C-SKoA -2Н
II "2Н || I И
О о ОН о
--> R'CCH2C—SKoA 2Д2 R'COIH-CHsC-SKoA
Далее процесс повторяется и т. д.
Синтез жирных кислот в организме идет обратным путем.
ферменты
779
Кофермент А осуществляет многочисленные реакции ацетилирования
пиптов оксикислот, аминов, цистеина и т. д., а также ацилирует
О второму углеродному атому карбоновые кислоты с образованием
R кетонокислот (в цикле Кребса, кн. I, стр. 465).
Кроме того, кофермент А вступает в конденсацию своим а-углеродом
с щавелевоуксусной кислотой, образуя лимонную кислоту (тоже одно
из звеньев цикла Кребса):
(X zO
/>ССН2СС/ +CH3C-SKoA + H2O
НО/ II ХОН ||
О О
ОН
ЧсСН2ССН2С^ +HSKOA
НО/ | 0 \он
\он
Из трансфераз подгруппы 2.7 назовем фосфат-трансферазы, перенося-
щие остаток фосфорной кислоты с аденозинтрифосфата (кн. I, стр. 462) на
субстрат, например на глюкозу (глюкокиназа) и фруктозу (фруктокиназа),
в процессе брожения.
К трансферазам же относятся трансаминазы, или аминотрансферазы
(А. Е. Браунштейн), например глутамат-трансаминаза, аланин-транс-
аминаза и т. д., обменивающие функции у соответствующих а-амино-
кислот и а-кетонокислот. Коферментом аминотрансфераз служит пирид-
оксалъ-5-фосфат, и, следовательно, именно трансферазы образуются
с участием витамина В6. Схема действия кофермента аминотрансфераз
выражается так:
/О
R-CH—C/z
| 4 ОН
NH2
+
? .ОН
но. I /СН2ОР<;
Y Y /ОН -НгО .
нзС/Х]/
/О
R—СН—С/
I /он
N
О
II/ОН
СН2ОР<
/он
+нго,
/ц,0
nh2
9Н2 II /ОН
Н°.Л/СН2°Р< .0
|| | /QH + r_с—
„ Г/\ Г II хон
H3cz N Q
Как правило, в живых клетках наиболее общим донором аминогрупп
служит глутаминовая кислота: всего активнее протекает обратимый
перенос аминогрупп между глутаминовой и щавелевоуксусной кислотами
(подробнее это было изложено на стр. 338).
780
ФЕРМЕНТЫ
Оксидоредуктазы
Эта группа ферментов (класс 1) весьма обширна. Все оксидоредуктазы
имеют коферменты. Наиболее обычные из пих это два никотинамидных
кофермента — никотинамидадениндинуклеотид (НАД) и никптнипу я
адениндинуклеотидфосфат (НАДФ), содержащие в качестве акцептора
водорода алкилированный по гетероциклическому азоту амид никотино-
вой кислоты (витамин РР);
R=H
НАД (или Кодегидраза {)
R-PO3H2 .. НАДФ (или Кодегидраза [[)
Имеются также флавиновые коферменты — флавинадениндинуклеотид
(ФАД) и флавинмононуклеотид (ФМН), в которых акцептором водорода
служит рибофлавин (витамин В2):
ФАД
о
CH2(CHOH)3CH2OP<qJJ
Н3С N^NyO
Л NH
Н3с
О
фмн %
Коферменты большой группы оксидоредуктаз эТ° Тк^а’^ы с такими
или близкие ему по строению железопорфирины; оксидореду
ФЕРМЕНТЫ
781
коферментами называются цитохромами. В цитохроме гем связан с белком
через атомы серы цистеина:
(в местах, помеченных звездочкой, группировка —S—СНа— для наглядности выде-
лена из символа Cys, т. е. знак Суз, обычно обозначающий остаток цистеина
—NH—СН—СО—, здесь соответствует только —NH—СН—СО—).
I 1
CHaSH
Окислительно-восстановительное действие цитохромов основано на
переходе центрального атома железа из двух- в трехвалентный и обратно.
Кроме цитохромов существует много других железосодержащих окси-
доредуктаз (каталазы, пероксидазы, ферредоксины, гидроксилазы), а также
оксидоредуктазы, содержащие медь (фенолазы, церулоплазмины, аминокси-
дазы и др.). Наконец, в ряде окислительно-восстановительных процес-
сов роль переносчика водорода играют липоевая кислота, глутатион
/О
O = CNHCH2C</
I /ОН
СН2 СН2
н,/ \h<cH!).cZ s У<сн*>«с\он
\s-s/ X°H ~2H HS SH
липоевая кислота
О\
/>cch2nhc=o
НО/ I
О\ I
'SCCH(CH2)2CNHCH
но/ I II I . ...
nh2 о ch2-s—S—сн2 о nh2
о о
°\ II II Z0
2 /CCH(CH2)2CNHCHCNIICH2C</
HOZ | I /он
nh2 ch2sh
глутатион
О +2Н
CHNHC(CH2)2CHC\’
782
ферменты
и производные /i-бензохинона и 1,4-нафтохинона — убихиноны и токо-
ферилхпноны.
Окисление в клетках совершается сложным путем. Происходит пере-
дача атомов водорода молекуле кислорода через ряд стадий при посред-
стве ряда ферментов. Понятие об этом может дать следующая схема:
I. НАД-Н2— фермент-]- Флавиновый НАД-фермент-|- Н2-Ф лавинов ый
(дегидрогеназа) фермепт фермент
II. 2Цитохром(Ее2+)Ч-1/202 -> 2Цитохром(Ее3+) + О2*
III. O2--f-2H+ -> Н2О
IV. 2Цитохром(Ге3+)+Н2-Флавииовый ---> 2Цитохром(Ге2+)+Флавиновый-|-2Н+
фермент фермент
Оксидоредуктазы (класс 1) делятся на подклассы (вторая цифра шифра)
по признаку окисляемой, т. е. отдающей водород или электроны, группы:
1.1. СНОН (донор водорода)
1.2. Альдегиды и кетоны
1.3. >СН—СН<
1.4. >CH-NH2
1.5. >C=NH
и н
1.6. НАД или НАДФ z
\н \Н
1.7. Другие соединения азота
1.8. Соединения серы
1.9. Гем
1.10. Бис-фенолы
1.11. Перекись водорода как акцептор
1.12. Н2
Внутри подкласса деление на подподклассы идет по признаку акцеп-
тора водорода (или электронов). Так, подкласс 1.1 (донор СНОН) де-
лится на подподклассы:
1.1.1. Акцептор НАД или НАДФ
1.1.2. Акцептор цптохром
1.1.3. Акцептор О2
Всего в списке известных в настоящее время оксидоредуктаз с приведен-
ными выше коферментами насчитывается около 250 ферментов. Мы, есте-
ственно, должны ограничиться лишь несколькими примерами, разъяс-
няющими принципы их действия.
Прежде всего рассмотрим группу коферментов НАД и НАДФ (более
старые их наименования — кодегидразы I и II). Эти коферменты распа-
даются при гидролизе на амид никотиновой кислоты, рибозу, фосфорную
кислоту и аденин-1\-рибозид. Окислительно-восстановительную функцию
несет .здесь остаток амида никотиновой кислоты:
б
ФЕРМЕНТЫ______________________________________________________
форма а дегидрирует субстрат, форма б — гидрирует. Строение и син-
тез никотинамида (витамина РР) см. на стр. 335. Кодегидразы участвуют
в реакциях обмена углеводов, жиров и белков.
Приведем наиболее характерные реакции, катализируемые фермен-
тами с коферментами НАД и НАДФ.
1. Дегидрирование фосфорного эфира глицеринового альдегида в ди-
фосфорный эфир глицериновой кислоты в процессе брожения сахаров:
/ „ОН „О О
С НС< С< с/
। | ХОРО3Н2 -2Н | \ОРОзН2 | ^ОН
СНОН +Н3РО4 -------> СНОН СНОН ^=> СНОН
I I н I I
СН2ОРО3Н2 СН2ОРО3Н2 СН2ОРО3Н2 СН2ОРО3Н2
2. Дегидрирование предельных жирных кислот в непредельные (участ-
вует НАДФ).
3. Гидрирование ацетальдегида в этанол при спиртовом брожении.
4. Гидрирование пировиноградной кислоты в молочную в мышцах
и при молочнокислом брожении.
5. Гидрирование иминоглутаровой кислоты в Д-глутамиповую как
стадия реакции восстановительного аминирования а-кетоглутаровой
кислоты:
°Чссн2сн2Сс/° °^ссн2сн2снс<^°
HOZ || ОН но/ | ХОН
NH NH2
Каждая из этих реакций требует своего специфического фермента.
Коферментами многих аэробных и некоторых анаэробных оксидоре-
дуктаз являются флавинадениндинуклеотид (ФАД) и, реже, флавинмоно-
нуклеотид (ФМН), в основе которых лежит молекула рибофлавина
(витамина В2). Забирая и отдавая водород, витаминная часть этих кофер-
ментов (полное строение см. стр. 780) следующим образом изменяет свою
структуру:
Витамин В 2 входит в молекулу известного желтого окислительного
фермента Варбурга (1932 г.), выделенного из дрожжей. Он выделен также
из молока, яйца и из микроорганизмов Eremothecium ashbyi, кото-
рые содержат до 2% рибофлавина и применяются для его нроиз-
784
ФЕРМЕНТЫ
Понятие о строении витамина В2 было получено путем деструкции
под действием света; оказалось, что в кислой среде он превращается
в люмихром, а в щелочной среде в — люмифлавин:
и 4-H2NCNH2
люмифлавин ||
О
Окончательно строение было доказано одновременно Р. Куном и Кар-
рером путем синтеза. Первой стадией синтеза Каррера было гидриро-
вание эквимолярной смеси ZJ-рибозы и 1-амино-2-карбэтоксиамино-4,5-ди-
метил бензола:
о
I II + ^>С-(СНОН)зСН2ОН
НзС/^/^ЫН-СОСаНб Н
II
О
H3CXZX/NHCH2(CHOH)3CH2OH
Y II
Н з N Н 2
Полученное
бофлавин:
соединение нагреванием с аллоксаном превращают в ри-
Н3С.
СН2(СНОН)3СН2ОН
| (X NH о
NH
NH ~гнйУ* Рибофлавин
h3c/xV\nh2
аллоксан
О
ФЕРМЕНТЫ
785
Лиазы
Эти ферменты (класс 4) в качестве первого подкласса включают фер-
менты, расщепляющие С—С-связп. К лиазам относятся прежде всего
многочисленные декарбоксилазы, отщепляющие карбоксил от карбоновых
кислот, в частности от окспкислот, оксокпслот, аминокислот. Затем
следуют альдегидлиазы, отщепляющие, например, от фруктозо-1-фосфата
глицериновый альдегид, который далее трансформируется в дпоксиацс-
тон (фермент альдолаза). Альдегидлиазы превращают также аминокис-
лоту треонин в глицин + ацетальдегид, отщепляют бензальдегид от его
оксинитрила и т. д.
Лиазы 2-го подкласса осуществляют разрыв С—0-связей и включают
ряд дегидратаз, дегидратирующих, например, яблочную кислоту в фу-
маровую; 3-п подкласс это С—N-лиазы, в основном отщепляющие аммиак
от аминокислот; 4-й подкласс С—S-лиазы; 5-й подкласс С—На1-лиазы
(единственный пока пример — ДДТ-гпдрохлоридлиаза насекомых).
Из коферментов лиаз приведем прежде всего кодекарбоксилазу —
пиридоксаль-Ъ-фосфат, имеющий в основе молекулу пиридоксаля (од-
ного из витаминов группы В6). Как было указано на стр. 338, пприд-
оксаль-5-фосфат является также коферментом трансаминаз, относящихся
к группе трансфераз и играющих важнейшую роль в белковом обмене
(реакция переампнпрования а-кетонокислот в а-аминокислоты).
Соотношение между взаимопревратпмыми в организме витаминами
группы В6 и пиридоксаль-5-фосфатом видно из приведенных формул:
Группа витампна Вв
СН2ОН
но\Л/СНгОН
i; I
НзС/V
пиридоксол
(адермин, пиридоксин)
CH2NH2
hox/^zcii2oh
11 )
Н3с/\/
пиридоксамип
Р0С13;
затем Н,0
сн2он о
пп I г. И /он
Н0\ А /СН2ОР<
[I Y он
НзС/V
МпОг
50 Заказ 672
НО
пиридоксаль
О
II /ОН
СН2ОР<
I CIbN.;
I затем окисление
Г
СНзО
Н3С
2-метил-З-метокси-
пиридиндикарбоновая
(метилмето кепцип-
хомероновая) кислота
П И ри докса ль-5-фосф ат
(ко декарбоксилаза)
ФЕРМЕНТЫ
786
Действительно, при действии хлорокиси фосфора на пиридоксол его
5-оксиметильная группа с наименьшими пространственными препятст-
виями этерифицируется, и после обработки водой образуется пиридок-
сол-5-фосфат, окисляемый затем в альдегид пиридоксаль-5-фосфат.
Структура кодекарбоксилазы (как и самого пиридоксаля) следует
из функционального анализа, показывающего наличие одной альдегидной
группы, одной первичноспиртовой (или ее фосфорного эфира), одного
фенольного гидроксила и одной метильной группы, а также из окисления
его после метилирования диазометаном в 2-метил-З-метоксипиридинди-
карбоновую кислоту — известную метилметоксицинхомероновую кислоту.
О синтезе витаминов группы Вв см. стр. 336.
В сочетании с различными апоферментами, строение которых далеко
не всегда известно, пиридоксальфосфат образует целую серию декарбок-
силаз, в частности декарбоксилаз аминокислот, превращающих их в соот-
ветствующие амины, причем для декарбоксилирования каждой амино-
кислоты требуется своя декарбоксилаза. В сочетании с другими апо-
ферментами пиридоксальфосфат образует трансаминазы, тоже специфи-
ческие для каждой аминокислоты.
К лиазам относится декарбоксилаза пировиноградной кислоты, ранее
обозначавшаяся как а-карбоксилаза. Ее коферментом является пирофос-
форный эфир тиамина (витамина В^, выделенного основателем учения
о витаминах Функом в 1912 г. из рисовых отрубей (экстракт из них лечит
авитаминоз — полиневрит «бери-бери», развивающийся при питании
исключительно ободранным рисом). О строении витамина Bj см.
стр. 351.
Декарбоксилиза, коферментом которой является тиаминпирофосфат,
катализирует отщепление двуокиси углерода от пировиноградной кислоты
в процессе деструкции глюкозы при спиртовом брожении и дыхании.
При недостатке тиамина в организме накапливается пировиноградная
кислота, что служит одной из причин поражения нервной системы и за-
болевания полиневритом.
Изомеразы
Изомеразы составляют 5-й класс ферментов и делятся на следующие под-
классы:
5.1. Рацемазы и эпимеразы, вызывающие стереоизомеризацию. Раце-
мазы аминокислот часто включают в качестве кофермента все тот же
пиридоксальфосфат.
5.2. Днс-пгранс-изомеразы.
5.3. Внутримолекулярные оксидоредуктазы, осуществляющие, на-
пример, взаимные превращения альдоз и кетоз.
5.4. Внутримолекулярные трансферазы, переносящие внутримолеку-
лярно ацильные, фосфорильные (фосфатные) остатки, метильную группу.
Коферментом некоторых из них служит витамин В12 (стр. 301).
о.о. Внутримолекулярные лиазы.
ферменты
787
Синтетазы
Синтетазы (лигазы), составляющие 6-й класс ферментов, делятся на об-
разующие С—О-связи (шифр 6.1), С—S-связи (6.2) и С—N-связи (6.3) и, на-
конец, С—С-связи (6.4).
Наиболее обычный тип реакций лигаз 1-го подкласса — связывание
аминокислот с рибонуклеиновыми кислотами при участии аденозинтри-
фосфата (АТФ), поставляющего энергию и превращающегося в аденозин-
дифосфат (АДФ).
Для 2-го подкласса лиаз типичны реакции ацетилирования кофермента
А (стр. 778) за счет ацетатного иона прп участии АТФ как источника
энергии, а также аналогичные реакции ацилирования.
Для 3-го подкласса характерны амидсинтетазы (например, превраща-
ющие аспарагиновую кпслоту в ее амид — аспарагин) и пептидсинтетазы
(например, синтезирующие глутатион).
Наконец, в качестве С—С-синтетаз (подкласс 6.4), характерны карб-
оксилазы, в которых простетической группой служит ft-биотин (стр. 315).
Они осуществляют введение карбоксила в пировиноградную кислоту
действием СО, при участии АТФ (образуется щавелевоуксусная кислота),
введение карбоксила в ацетпл-КоА (образуется малонил-КоА) и т. д.
Первая реакция имеет важнейшее значениев цикле Кребса (кн. I, стр. 465).
В биотпнилферментах р-биотин
HN—СН-СН(СН2)4СООН
o=cf S
HN-CH—СН2
ковалентно связан амидной связью с е-МН2-группой лизинового остатка
апофермента. При действии этих ферментов к уреидной группе биотина
присоединяется молекула СО, с образованием комплекса — N-карбокси-
биотинилфермента («активная» форма углекислоты). Реакция образования
комплекса сопряжена с дефосфорилированием АТФ до АДФ.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абиетиновая кислота 259, 657
Агликон (Генин) 682
Адамантип-катион 580
Адамантилфторид 580
Адениловая кислота 362
Аденин (6-Аминопурин) 361, 743 сл., 755 сл.,
765
N-рибозид 782
S-Аденозилг омоцистеин 776
S-Аденозилметионин 776, 777
Аденозин 745, 748
дифосфат (АДФ) 778, 787
трифосфат (АТФ) 760, 762, 766, 770,
772, 778, 779, 787
— функции в организме 362
Адермин (Пиридоксол) 336, 337, 785
Адипиновая кислота 285, 664
Адреналин 167, 168, 679
Адреналон 167, 168
Адренокортикотропные гормоны 721, 730,
734, 738
АДФ (Аденозиндифосфат) 778, 787
Азабензол см. Пиридин
1-Азанафталин см. Хинолин
З-Азапиридин см. Пиримидин
3-Азатиофен см. 1,3-Тиазол
Азатриптицен 91, 92
Азиды 316, 318, 398
Азимидобензол 95
Азин см. Пиридин
Азиридин 277
Азлактоиы (Оксазолоны) 292
Азобензол 74, 78 сл., 82, 145
Азокрасители 80, 89, 94, 96, 100, 104 сл.,
129, 184, 204, 236, 237 сл., 313
Азоксибензол 74, 75, 78, 79, 133
Азоксисоедпнения 79
Азол см. Пиррол
Азолидин СМ. Пирролидин
Азометин 338
Азосоединения 107, 537
пиролиз 541
Азосоставляющие 105, 129
Азосочетание 104, 318, 365, 500, 739
с пирролом 295
Азоферроцены 502
Азулен (Бицикло [0,3,5] декапентаен) 328,487,
507, 512 сл., 652
алкилпроизводные 514
диамагнитная восприимчивость 490
дисульфокислота 514
Азуления соль 514
Азуленогены 515
Акридарсоновая кислота 443
Акридин 343, 344
Акридиновая кислота (2,3-Бензпиридин-5,6-
дикарбоновая кислота) 343
Акридон 343,
Акрилонитрил 468, 531
Акрихин (Атебрин) 344
Акролеин 255, 339, 365
Аксерофтол (Витамин А,) 653 сл.
Активированный комплекс см. Переходное
состояние
Активные металлоорганические соединения
(АМО-соединения) 380 сл.
Актиномицин 765
Аланин 724, 763, 767, 775
т-РНК 750
Аланин-трансаминаза 779
Ализарин 2 50 сл.
Ализариновый лак 251, 252
Алкалоиды 120, 313, 333, 512
гетероциклические 684 сл.
с пятичленными гетероциклами 6S5 сл.,
698 СЛ., 704, 707 сл., 710 СЛ., 717, 719
р шестичленными гетероциклами 341,
345, 690 сл., 697 сл., 702 сл., 707 сл.,
710 сл., 717 сл.
стер скинье 658
ПРЕДМЕТНЫЙ указатель
789
длкилапилины 89 сл.
длкилдихлорарсин 440
длкилируюшие агенты 68
4ЛКПЛкарбаминовая кислота 623
длкил-катионы 581, 582
Алкильные радикалы 544
длкоксид-ион 602
Алкоксильные радикалы 54 4
Алкоксипиридин 335
Аллантоин 358
Аллен, димеризация 595
Аллиллитий 475
Аллилмагнийгалогениды 475
4-Аллил-2-мстоксифенол (Эвгенол) 134
Аллилпалладийхлорид, 476
Л-Аллилтрикарбонилиодид железа 4 76
дллилфеиолы 501, 597
Аллильные
л-комплексы 475, 47 7
перегруппировки 134, 629 сл., 637 ел., 653
— обратные 632
,-т-Аллил ьные производные металлов 475
Аллилы галоидные 388, 556
Аллоксан 358, 366, 784
Аллооцимен 640, 647, 648
Альбумин 187, 721, 742
Альбуцид 94
Альдегидаммиаки 14 5, 329
Альдегпдлпазы 785
Альдегиды
ароматические 137 сл.
ртутные производные 405
свойства 141 сл.
синтез 138 сл., 396, 397
Альдоксимы 281, 607
Альдолаза 785
Альдольная конденсация 147, 381, 395, 533, 599
Альтернантные ароматические системы 487
Алюминийорганические соединения 381, 400,
417, 418, 460
Амигдалин 191
Амидазы 773
Амидины 313, 348
Амидол 78
Амидопирин (Пирамидон, Диметиламииоапти-
пирии) 312
Амиды кислот 68, 628
а-Амилаза 771, 772
Амилнитрит 303
Амилпенициллпн 323
Аминазин (Хлорпромазин) 376
Амииоазобензол 95, 104
Аминоазосоедииенпя 104
а-Амипоалкилфосфоновые кислоты 435
Амииоаптипирии 311
Амшюантрахипопы 249, 254
п-Аминобенаоилглутамииовая кислота 365
о-Амипобензойиая (антраниловая) кислота 168,
169, 177, 187, 188
и-Аминобепзойная (1,3-аминобсизойпап) ки-
слота 168, 169
п-Аминобензойная (1,4-аминобензойная) ки-
слота 95, 168 сл., 187 сл.
диэтиламиноэтиловый эфир (Новокаин)
188, 701
этиловый-эфир (Анестезии) 188
о-Аминобензол сульф 'кислота (1,2-Амино-
бензолсульфокислота, Ортаниловая
кислота) 84, 85
л1-Аминобензолсульфокислота (Метапиловая
кислота) 84, 85, 94
n-Аминобензолсульфокислота см. Сульфани-
ловая кислота
Аминогруппа
защита 734
мезомерный эффект 85, 86
Аминогуанидии 318, 357
п-Аминодиметиланилин 96, 375
Аминодурол (2,3,5,6-Тетраметил-1 -аминобен-
зол) 86
4-Амино-З-изоксалидоп (Циклосерин) 318, 319
ы-Аминокапроповая кислота 727
1 -Амино-2-карбэтоксиамино-4,5-диметилбен-
зол 784
Аминокислоты 289, 291, 338, 339
ароматические 187 сл.
белка 723, 724, 728, 731
Аминокоронен 266
Аминоксидазы 781
Аминонафталинсульфокислоты 236
Аминонафтолы 237
Аминопептидазы 731, 773, 774
4-Аминопиразол 311
Аминопиридины 332, 334 сл., 704
2-АминОпиримидин 349
6-Аминопурин (Аденин) 361, 743 сл., 755 сл.,
765
п-Аминоеалициловая кислота (ПАСК) 188
Аминотетразол 318
2-Аминотиазол 320
Аминотиофен 288
Аминотрансферазы (Трансаминаза) 779, 785
2-Амино-1,3,4-триазол 357
4-Амино-3,5,6-трпхлорпиридпн-2-карбоновая
кислота 336
а-Амцноуксусная кислота 722
п-Аминофениларсоновая кислота 739
о-дминофенилглиоксиловая (изатиновая) ки-
слота 306
п-АминофснилсульфокиСлота см. Сульфани-
ловая кислота
Ампнофенолы 78 сл., ИЗ, 133
Аминоферроцен 4 96
Аминофосфоновыс кислоты 435
а-Ампнофуран 282, 288
а-Аминохинолин 341
о-Амино-ы-хлорстирол 302
р-Аминоэтилбеизол 34 5
Р-(Р-Амикоэтил)-индол (Триптамин) 304,
719
Р-Амипоэтилфосфонопап кислота (Цилиатин)
425
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
790
Амины
ароматические 82 сл.
— с аминогруппой в боковой цепи 167
бензоилирование 172
гетерофуньциональные 167 сл.
диазотирование 9 7
нитрозирование 90
Амитал 348
АМО-соединепие (Активные металлооргани-
ческие соединения) 380 сл.
Амд0и-И3омеры 15 7
Анабазин (а-Пиперидил-р-пиридпн) 333, 336,
705
Анальгин (Новальгин) 312
Ангидриды
бензойной кислоты 174
малеиновый 17, 21
фталевый 176
циклогексДдиен-3,5-дикарбоновой-1,2 ки-
слоты 17
Андаксин 185
Андростерон 674, 675, 676
Аневрин см. Витамин В,
Анестезин (n-Аминобензойная кислота, эти-
ловый эфир) 188
Анетол (n-Пропенилфенол, метиловый эфир)
184
Анизидиды 89
Анизидины (Метоксианилины) 83, 132, 133
Анизол (Фенол, метиловый эфир) 132, 422, 509
Анилиды 87 сл.
Анилин 79, 82, 84, 87, 93, 220
дипольный момент 85, 86
окисление 159, 165, 219
получение 60, 61, 74, 75, 79, 306
реакции 160, 302, 321, 339, 355, 356, 481
— замещение 86 сл.
— меркурирование 405
— сульфирование 94
Анилиновый синий 220
Анилиновый черный 356
Анилинсульфокислоты 84. 85, 87, 94 сл.
Анилы (Шиффовы основания) 77, 145, 338
Анионообменные смолы 202
Анионотропная
аллильная перегруппировка 630, 631, 637
таутомерия 630
Анион-радикалы 534, 535
Анисовая кислота (Метоксибензойная кислота)
184, 193
Аннулены 517 сл.
«Анса»-соединения 130
Антибиотики 95, 262 сл., 323
Антигены 739
Антидетонаторы 89, 425
Анти-изомеры 79, 145, 154, 157
Антикодон 761, 763
Антиметаболит 95
Антиоксиданты 126, 129, 556
Антипирин^ (1-Фенил-2,3-диметилпиразолон-5)
Антисвяэывающая молекулярная орбиталь
462
Антитела 738
Антифебрин (Ацетанилид, Фенилацетамид) 89
92
Антоцианидины 371, 372 сл.
Антоцианы 372, 373
Антрагаллол 252
Антрагидрохинон 249, 556
Антраниловая (о-аминобензойная) кислота
168, 169, 177, 187, 188
Антрахинон 245, 248, 503
получение 176, 247
реакции 249, 255, 556
Антрахинонсульфокислоты 249, 250, 252
Антрацен 23, 120, 232, 244 сл., 259, 308
аддукты с металлами 385
ароматичность 245, 490
спектр поглощения 261
Антрацендисульфокислоты 248
Антраценовое (зеленое) масло 23, 245, 256,
308, 309, 343
Анхимерное (синартетическое) содействие 618,
619, 620
Апоферменты (Апоэнзимы) 338, 770, 775, 786,
787
D-Арабиноза 365, 744
П-Арабоновая кислота 365
Аргинин 724, 731, 763
Арекаидин и ареколин 692, 693
Аренкарбонилы металлов 458, 468, 477 сл.
Аренметаллоцены (Металлоценоарены) 458,
483
Арилдиазонип 99
борфторид 112, 449
соли 97, 98, 100 сл., 496
Арилкарбонилметаллы 467
Арил-катионы 111, 581
Арилмышьяковая и арилмышьяковистая ки-
слоты 442
Арилферроцены 496, 497, 500
Арилхлорсиланы 442
Армин 436
Ароксилы 125, 126
Ароматизация 11, 20, 23
Ароматические спирты 135 сл.
Ароматические углеводороды
конденсированные 229 сл., 259 сл.,
264 сл.
ряда бензола 9 сл.
с изолированными бензольными ядрами
203 сл.
с непредельной боковой цепью 199 сл.
Ароматичность
бензола 13 сл.
гетероциклов 272, 275, 331
конденсированных систем 230, 245, 257,
261
небензоидных систем 487 сл.
Арренал 441
Арсаниловая кислота 442
п РЕДМЕТНЫЙ УКАЗАТЕЛЬ
791
дрсавтренхлорид (Ди-о-фенилендихлордиар-
син) 443
Арсенобензол 442, 443
дрсеносоединевия 44 2 сл.
Арсевостибивосоедивевия 44 5
Арсивметилены 443, 444
Арсиновые и арсоновые ароматические ки-
слоты 441 сл.
Аска ридол 5 57, 643
Аскорбиновая кислота 771
Аспарагин 724, 763, 787
Аспарагиновая кислота 724, 763, 775, 787
Аспирин 183, 184
Атебрин (Акрихин) 344
Ат-комплекс Виттига 414
Атофан (Цинхофен) 341
Атразнн 357
Атропин 684, 687, 698, 700
Атроповая кислота 69 8
Атропэнантиомерия 130, 131, 205 сл.
АТФ см. Аденозинтрифосфат
Ауксохромы 1 08. 220
Аценафтен 23, 243 сл.
Ацеиафтенхинон 243
Ацены 259 сл.
Ацетали 396
Ацетанилид (Фенилацетамид, Антифебрин)
89, 92
Ацетатмеркуранилины 405
n-Ацетиламинофенилстибиновая кислота, на-
триевая соль (Стабенил, Стибазил)
446
N-Ацетилгексозамин 742
Ацетилен
аниои 520, 521, 534
комплексы 403, 404, 470
реакции 286, 302, 316, 329, 384, 389,
414, 423, 466, 471
циклизация 18, 19, 233, 475, 480
Ацетилен-аллен-диеновая перегруппировка
634
Ацетилендикарбоновая кислота 282
Ацетилениды 21, 519
Ацетиленкарбоновый эфир 493
а, 3-Ацетиленовые оксосоединения 310
n-Ацетилфенетидин (Фенацетин) 89
Ацетилфлороглюцин 372
Ацетилхолин 167, 773
Ацетоуксусная кислота 660, 778
эфир 105, 106, 311, 329, 367
Ацетофенон 19, 148
реакции 150, 151, 153, 341, 691
Ацилатные радикалы 545
Ацилиевые катионы 582, 586
Ацилиевые кислоты 586
Ацилкарбонилметалл 467
Ацилоиновая конденсация 209
Ацилоиновые перегруппировки 634, 635
Ацилониевые катионы 627
Ацилтрансферазы 777
Ацжлферроцеиы 501
Ацильные радикалы 544
Ауксохромы 108, 220, 221, 222, 321, 322
Ауреомицин (7-Хлортетрациклин) 262
Аурин 221, 222
Аурирование 456
Аугоокисление 552 сл.
Баднонал 94
Бакелит 123
Бактериофаги 743, 758
Барбитал (Диэтилбарбнтуровая кислота) 348
Барбитуровая кислота (Малонилмочевина)
347, 348, 349, 359
Барен (о-Карборав-10; 1,2-Карборан-10; Дека.
карборан) 414, 415
Батохромный сдвиг 108, 261, 321, 322
Бекмановская перегруппировка 153, 155;
607, 628
второго рода 157
Белки (Протеины) 721 сл.
аминокислотный состав 191, 192, 724 сл.
методы установления строения 728 сл.
синтез 734 сл., 762
Бензальацетон 147, 158, 189
Бензальацетофенон 158
Бензальдегид 9, 61, 197, 509
конденсация альдольная 147
— бензоиновая 146
— кротоновая 147, 189
окисление 191
реакции 135, 147, 148, 200, 218, 598,
699, 718
хлорирование 172
Бензальдоксимы 145
Бензамид 174
Бензантрен 255
Бензантрон 255, 393
Бензаурин 222, 223
Бензвален 22
Бензгидрил-радикал 226
п-Бензгидрилтетрафенилметан 226
Бензгидрол 212
Бензедрин 167
Бензидин (п,п'-Диаминодифенил) 80, 81, 204,
205
Бензидиновая перегруппировка 80, 204, 237,
302
Бензизоксазол 154
Бензил (Дибензоил) 146, 157, 158, 624
Бензил
анион 440
галогениды 55, 61
— реакции 71, 135, 188, 207, 456
катион 504, 507
Бензиламнн 167
Бензилацетат 188
Бензилдиоксимы 157
Бензилиден, галогениды 52, 53, 55, 61
Бензилирование 60
Бензиллитий 207, 383
Беизилмагнийгалогениды 207, 597, 60#
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
792
Бензилметилкетон 167
Бензилнатрий 383
Бензиловая кислота 146, 158, 624
Бензиловая перегруппировка 158, 284,
624
Бензиловый спирт 135, 146, 216
Бензилпенициллин 323 сл.
o-Бензилтетрагидробензойная кислота 2 '6
о-Бензилфенилстибиновая кислота 446
Бензилхром 457
о-Бензил-л-циклопентадиенилмолибден-
трикарбонил 477
Бензимидазол(ы) 96
Бензин^см. Дегпдробензол
2,3-Бензнафталнн 245
Бензоил
гидроперекись (Надбензойпая кислота)
172. 173
перекись 172, 173. 557, 637
хлористый 61, 172 сл., 700
Бензоилацетон 536
о-Бензоилбензойная кислота 176, 245, 246
Бензоилирование 172, 336
Бензоилмуравьиная кислота 698
а-Бензоилнафталин 255
Бензоин 146, 157, 207, 284
Бензоиновая конденсация 146, 28л
Бензойная кислота 9, 168, 170, 249, 5оЗ, 700
ангидрид 174
образование 146, 170, 172, 199
производные 61, 147, 157, 1711сл., 174,
183, 511, 693
реакции 180 сл., 220
хлорангидрид (Бензоил хлористый) 61,
172 сл., 700
Бензол 9 сл., 15, 22, 30, 285
анион-радикал 34, 535
арилирование 550 сл.
ацилирование 37, 24.6
галоидирование 12, 51 сл., 558
дьюаровский (Бицикло [2,2,0] гекса-
Диен-2,5) 17, 18, 21, 471, 493
замещенные, галоидирование 44
клатратные соединения 35
комплексы 34, 477, 483
меркурирование 405
металлирование калием 385
молекулярные соединения 121
неорганический (Боразол) 16, 416
получение 18 сл., 233, 474
реакции 12, 13. 114, 151, 176, 188, 203',
283, 309, 331, 508, 509, 573, 586
— конденсации 199, 207, 212
— электрофильного замещения 31 сл.
резонанс структур 16, 17
сульфирование 66
условия квантования 489
фосфорилирование 432
Бензолбифенилхромиодид 478, 479
Бензолгеьсакарбоновая (меллитовая) ки-
слота 175, 179
о-Бензолдикарбоновая кислота см. Фталевая
кислота
п-Бензолдпкарбоновая кислота см. Терефта-
левая кислота
Бензолдисульфокислоты 67, 128
Бензолпентакарбоновая кислота 175
Бензолполикарбоновые кислоты 174 сл.
Бензолсульфиновая кислота 532
Бензолсульфокислота 31, 66 сл., 114, 149
Бензолтетракарбоновая (пиромеллитовая)
кислота 175
Бензолтрикарбоновые кислоты 20, 175, 179
Бензолциклопентадиенилметаллы 483
л-Бензольный лиганд 477, 483
Бензонитрил 145, 157, 174
2,3-Бензопиридин см. Хинолин
Бензотрифторид 55, 62
Бензотрихлорид 52, 53, 61, 62, 213
реакции 172, 218, 224
Бензофенон 148, 153, 207, 249, 398
натрийкетил 465
реакции 152, 212, 249
о-Бензохинон 127
п-Бензохиноп 131, 159, 238
реакции 129, 160 сл., 245, 305, 714
п-Бензохинонимин 164
п-Бензохинопмонооксим 122, 161
Бензперилен 266
Бензпинакон 398
Бензпирилиевыесоли 369
3,4-Бензопиридин см. Изохинолин
2,3-Бензопиридин-5,6-дикар боновая (акриди-
новая) кислота 343
2,3-Бенз-у-пирон (Хромой) 370 сл.
2,3-Бензпиррол см. Индол
Бензтиазол 320 сл.
2,2-Бензтиазолилдпсульфпд 542
Бензтиофен (Тионафтен) 273, 290 сл.
Бензтриазол 316
Беизферроцен (Диинденилжелезо) 503
Бензфуразан 77
Бериллийорганические соединения 400
Бетаин 509, 776
Бетаин-а-гомоцистеин-метилтрансфераза 776
Бигетероциклы 315, 358 сл.
Биомицин (7-Хлортетрациклин) 262
Биотинплферменты 787
Биотины 290, 315 сл., 787
Бирадикалы 474, 570
Бис-азокрасители 107, 238 сл.
Бис-л-аллплдикарбонил;келезо 476
Бис-л-аллилнпкель 475, 476
Бис-аренметалл-катион 477
Бис-аренникель 481
Бис-ареиовые соединения 477, 479
Бис-аренхромы 478, 479, 480
Бис-бензолметалл 477
Бис-бифеНилхромиодид 478, 479
Бис-гексаметилбензолхром 480
4,4 '-Б ис-оксифенил-2,2-пропан (Дифенилол-
пропан) 124
п РЕДМЕТНЫЙ указать ль
793
Бис-трифторметилфосфинистая кислота 433
4,4' -БиС-Ч фенилуреи до)-2,2' -стильбенднсул ьфо-
кислота (Бланкофор В) 208
Рдс-р-хлорвинилмеркурхлорид 409
БиУРетоваЯ РеакЧия 722
Бифенил см. Дифенил
Бифенилен (Дифенилен) 66, 206, 409
0 о'-Бифенпленарсин 443
Биникло [2,2,0] гексадиен-2,5 (Дьюаровский
бензол) 17, 18, 21, 471, 493
Бицикло [0,3,5] декапентаен см. Азулен
д9(Ю)_бицИКлО [5,3,0] деценон-4 512
Бидикло [0,3,3] октан 516
Бицикло [2,2] октатриен 12
Блаикофор В 208
Боразол 16, 416
Борнеол 620, 650
метилксантогспат 650
Борнилен 650
Борнилтозилат 620
Борнилхлорид 613, 620, 648
Бороводороды (Гидриды бора) 411 сл., 416
Борорганические соединения 411 сл., 460
Борфториды арилдиазониев 110 сл.. 449
Бриллиантовый зеленый 218
Бромарилы 384
Бромбензол 27, 52, 54, 56, 57, 111
реакции 64, 134
Бромирование 537, 558
алканов 547
гомолитическое 546 сл.
карбанионов 522, 524
переносчики брома 51
толуола 547
а-Бромнафталин 230
скн-2-Бром-5-нитроацетофеноноксим 1 54
Бромониевый цикл 593, 594
З-Бромпиридин 331
Бромпировиноградная кислота 319
Бромсукцинимид 510, 548, 671
2-Бромтиофен 287, 288
БроМтолуолы 5 5
Бромферроцен 499, 500
Бруцин 718, 720
Бруциновая кислота 718
Бутадиен 14, 17, 32
л-комплексы 473
полимеризация 474, 475
реакции 245, 247, 287, 468
Бутадиенидфосфоновая кислота 438
Бутандиол-1,4 282, 285
’претп-Бутила перекись 557
Бутиламин 197
’арещ-Бутилат-аниои 527
шрет-Бутилбензол 44
шрет-Бутил-катион 579
вгрет-Б утиловый спирт 527, 528
mpem-Бутилтолуол 71
’вретп-Бутилэтилеи 557
Бутиролактон 271
Бутирофеиои 148
Вазопрессин 733, 738
Валин 724, 754, 763, 775
Вальденовскос обращение 191, 228, 527 сл.
590, 592, 593, 594, 603
при перегруппировках 611, 612, 616, 618
Ванилин 133, 134, 137
Ванилиновая кислота 184
Вератровая кислота 184
производные 700
диметиловый эфир)
Вератрол (Пирокатехин,
133
Веронал (Диэтилбарбитуровая кислота) 348
Ветивазулен 515
Р-Ветивон 515
Винилакриловая кислота 714
Винилацетилен 475, 631
Винилбензол см. Стирол
N-Винилкарбазол 309
Виниловые эфиры в синтезе элементооргани-
ческих соединений 405, 423
а-ВинИлпиридин 333
Винилфосфоновая кислота 438
Винилэтинилкарбинол 631
Виопантрен 256
Виолантрон 255, 256
Висмуторганические соединения 446
Витамины 770, 771
Ai (Аксерофтол, Ретинол) 653 сл.
А2 654
Bi (Аневрин, Тиамин) 323, 351, 352, 786
В2 (Рибофлавин) 364, 365 сл., 780, 783 сл.
В« 336, 337 сл., 785
Bi2 (Кобаламин) 301, 341, 777
D2 (Кальциферол) 671
D3 (Холекальциферол) 671
Е (Токоферол) 370
Н (Биотины) 315, 787
Н, 95
Kt (Филлохинон, Фитохинон) 239, 240
К2 (Фарнохинон) 23 9, 240
Р (Рутин) 371
РР (Никотинамид, Ниацин) 334. 335,
776, 783
Вителлин 742
Водородные связи 194, 314, 41 1, 525
мостиковые 411
Вырожденные уровни электронов 4 90
Галловая (3,4,5-триоксибензойная) кислота
131, 185, 186
висмутовая основная соль (Дерматол) 185
диметиловый эфир (Сиреневая кислота)
185
м-Галлоилгалловая (м-дигалоилгалловая) кис-
лота 185, 186
Галоидирование 51, 546 сл., 548, 549
Галоидполифторалканы 449
Галоидпроизводные бензольного ряда 29,
51 сл.
Галоформный распад 181
Гаммексан (Гексахлорциклогексан, Гекса-
хлоран) 12, 558
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
791
Гарман 711
Гвайаэулен 515, 652
Гвайен 515
Гвайол 515
Гваякол (Пирокатехин, монометиловый эфир)
114, 133
Гексаарилэтаны 225, 227
Гексабпфепилэтан (Гексаксенилэтап) 226
Гексабромбепзол 54, 56
Гексагелицен 266, 267
Гексагидроантрацеп 245
Гексагидрокоронен 266
Гексагидрониридип см. Пиперидин
Гексагидрофарпезилбромид 653
Гексадекануклеотид 766
Гсксакарбонпл(ы)
вольфрама 465
молибдена 465
хрома 461, 465, 468
Гекса ксенилэтан (Гексабнфенилэтан) 226
Гексаметилбензол 31, 34, 37
Ге кса н птр офе нилэтан 552
Гсксаоксибепзол 132
Гексатолилзтаны 227
Гексафенилбензол 18, 19, 208
Гексафенилэтан 225 сл., 552
Гексафторацетон 455
Гексафторбепзол 54, 56, 450, 453
Гексахлоран (Гсксахлорциклогексан) 12, 558
Гексахлорбепзол 52, 54, 56, 450
Гексахлорциклогексан (Гексахлоран, Гам-
мексан) 12, 558
Гсксацеи 261
Гелиотридан (1-Метилпирролизидин) 686, 687
Гелиотропин (Пиперонол) 134, 691
Гем 297, 298, 780
Гематопорфирин 298
Гемимеллитовая (1,2,3-бензолтрикарбоновая)
кислота 175
Гемимеллитол (1,2,3-Триметилбензол рядо-
вой) 31
Гемин 296 сл., 300
Гемоглобин 291, 297, 722, 723, 739, 742, 518
Гении 682
Гсптилпенициллпн 323
Геранилпирофосфат 660
Гераниол 640, 641, 645
Гербициды 118, 336, 357
Германийоргаиические соединения 421, 422
Героин (Морфин, Диацетат) 695 сл.
Героновая кислота 655
Гетероаукснц (Индолплуксусная кислота) 302
Гетероциклическое соединение 269 сл.
ароматичность 272, 275, 331
бигетероцнклы азотистые 3 58
номенклатура 276 сл.
пиримидиновые 346, 744
пуриновые 358 сл., 744, 747
пятпчленные 278 сл., 309 сл., 318 сл.
свойства 2 73
шестичлеиные 326 сл., 356 сл.
Гибридизация атомных орбиталей 460, 472
в азоте и фосфоре 427, 437, 438
Гигрип 685, 686
Гигриновая кислота (N-Метилпролин) 685
Гидразобензол (Дифенилгидразин) 74, 80 сл.,
204
Гидразоферроцены 502
Гидрид-анион 49, 216, 382, 422, 468, 507
582 сл., 585, 623, 636
Гидридное перемещение 147, 394, 582 сл., 611
интрамолекулярное 63а, 636
Гидробепзамиды 145, 284
Гидрокоричпая кислота 189
Гидроксамовая кислота, хлорангидрид 280
Гидроксилазы 781
Гидроксоипй-катион 598
Гидролазы 772, 773 сл.. 775
Гидронафталины 242 сл.
Гидроперекиси 172, 173, 555, 625, 714
Гидрофурамид 284
Гидрохинон (1,4-Диоксибензол) 126, 128,
129 сл., 160, 163, 252
«анса»-соединения 130
клатратные соединения 129, 130
окисление 159, 162, 164, 556
Гиосциамин 698
Гиперконъюгация (о,о- и о,л-сопряжение
44, 45, 410, 411, 452, 602, 604
Гипертензин 733, 738
Гипоксантин (6-Оксипурпн) 361
Гиппуровая кислота 192
Гипсохромпый сдвиг 108, 321
Гистамин 313, 315
Гистидин 313 сл., 726, 763
Гистоны 722
Гитокспгенин 682
Г-Кислота 235
Глиадппы 721, 722
Гликозидазы 773
Гликокол (Глицин) 78, 724, 730, 735, 763,
785
Гликолевая кислота 118, 718, 720
Гликолид 271
Гликопротеины 722, 742
Глпкохолевая кислота 673
Глиоксаль И, 313
Глпфталевые смолы 178
Глицерин 178, 255, 339
Глицериновая кислота 783
Глицериновый альдегид 783, 785
Глицин (Гликокол) 78, 724, 730, 735, 763,
785
Глобин 297, 742
Глобулины 721
Глутаконовый альдегид 328, 513
Глутамат-трапсамппаза 779
Глутамин 725, 763
Глутаминовая кислота 724, 763, 779, 783
Глутаровая кислота, производные 664, >'65
Глутаровый диальдегид 703
Глутатион 781, 782, 787
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
795
Глутелины 721, 722
Глюкагон "21, 733
Глюкоза 186, 365, 650, 727, 771, 779, 786
фосфаты 772
Глюкокиназа 779
Гомокамфорная кислота 649
Гомолитические
перегруппировки 636 сл.
реакции 546 сл., 588
Гомолитический распад (Гомолиз) 109, 112,
539
Гомомерохинен 709, 710
Гомопилоповая кислота 688, 689
Гомоцистеин 776
Гормоны 658, 723, 733 сл.
андрогенные 658
гонадотронные 675
коры надпочечника (Кортикостероиды)
679, 682
лутоидные 659, 675
стероидные 673 сл.
эстрогенные (фо ликулярные^ 658, 675
Гофмановское расщепление 602, 603, 606, (17 .
691, 697, 699, 702, 705
Грамин 303, 304
Грамицидин 733
Гранатовая кислота 7 02
Графит 179
Гриньяров реактив 19. 35. 244 263, 373,
387 сл., 393 сл., 4(1 Г,4 45, 456 480,
508, 534, 540
аномальные реакции 394 сл.
взаимодействие с Сероводородами 412,
413
перфторалкильный 389
при получении аренхромов 478
— ароматических кетонов 150
— карбонилов хрома, молибдена, воль-
фрама 465
— мышьяковоорганических соединений
441
— стибинов 445, 446
реакции 189, 203, 212, 213, 285, 390 сл.,
401, 409 сл., 428, 436, 437, 440
— с имидазолом 314
— с индолом 303
— с пентафторбромбензолом 56
— с у-пироном 369
— с ферроценом 499 сл.
— с циклопентадиеном 481, 494
Гуанидин 348, 349, 364
Гуанин 361, 743 сл., 748, 755 сл., 765
Гуанозин 747
Гуттаперча 639
2,4-D (2,4-Дихлорфеноксиуксусная кислота)
118
ДДТ 38
Дегидратазы 785
Дегидрацетовая кислота (а-Метил-|3'-ацети.ч-
а-пирон) 367, 368
Дегидробензол (Бензин, Циклогексадиенин)
60, 65 сл., 83, 128, 187, 247
ди- и тримеризация 66, 188, 206, 265
Дегидрогеназы 772, 782
Дегпдрофторирование полифторциклогекса-
нов 56
7-Дегидрохолестерин (Провитамин Да) 658,
660, 670, 671
Дегидроциклизация 20, 233
Дегидроэпиандростерон 676, 677, 678
Деготь 133, 343
Дезоксикортикостерон 680, 681
.D-2-Дезоксирибоза 744
Дезоксирибонуклеиновые кислоты (ДНК) 743,
749, 756 сл.
матричный синтез 757 сл.
структура 756
Дезоксирибонуклеозиды 748
Дезоксирибонуклеотид-полимераза 759
Дезоксихолевая кислота 672 сл.
Дейтерообмен в карбанионах 522, 524 сл.
Дейтероуксусная кислота 493
2-Дейтеро-2-фенилбутан 526
Декагидронафталиин см. Декалин
Декаборан 411, 412, 414
Декакарборан (Барен) 414, 415
Декалин (Декагидронафталин) 229, 242
гидроперекись 625
стереоизомеры 242, 243, 661
Декалолы и декалоны 243
Декарбоксилазы 315, 785, 786
Декстропимаровая кислота 657
Дельфинидин 373
Депсиды 185
Дерматол 185
1,3-Диазабензол см. Пиримидин
Диазины 345 сл. (см. также Пиразин, Пири-
дазин, Пиримидин)
Диазиридин 277
Диазоаминобензол 104
Диазоаминосоединения (Триазены) 103, 398,
569
Диазоаминоферроцен 500
о-Диазобензолкарбоновая кислота 187
п-Диазобензолсульфойислота 102
Диазогидрат 100
а-Диазокетоны 625
1,2-Диазол см. Пиразол
Диазометан 509, 512
реакции 279, 280, 596
фотолиз 570, 572—573
Диазоний-катионы 99
соли 109, 110
Диазосоединсния 80, 97, 550
в синтезе металлоорганических соедине
НИЙ 405, 418, 442, 445, 446
реакции 103 сл., 108 сл., 170, 309, 537,
541
Диазосоставляющие 105
Диазотирование 95, 318
Диазоуксусный эфир 571
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
796
Диазоцианиды 101, 102
Диазоциклопентадиен 507
Диалкилбарбитуровые кислоты 348, 349
Диалкилпиридинмонокарбоновые кислоты 331
Диалкилртуть 408
Диалкилселениды 447
Диал килсиланы 419
Диалкилхлорарсин 440
Диаллил 18, 388, 47 5
Диаллилкетон 476
Диамагнитные свойства ароматических си-
стем 488 сл.
Диаминогеновый синий 239
3,3'-Диамино-4,4'-диоксиарсенобензол
(Сальварсан) 444
о,о'-Диаминодифенил 309
о,п-Диаминодифенил (Дифенилин) 81
п,л'-Диаминодифенил (Бензидин) 80, 81, 204,
205
4,5-Диаминоурацил 360
Диамины 95 сл.
Дианизидин(2,2-Диметоксибензидин) 132, 204
Дианионы и дианион-радикалы 535
Диантрацен 247
Диарилиодониевые соли 64
Диарилкобальт, тетрагидрофуранат 480
Диарилметан 37, 38
Диарилмышьяковая и диарилмышьяковистая
кислоты 44 2
Диарилникель, тетрагидрофуранат 480
Диарилртуть 405, 408
Диарилхлорарсины 442
Диастереомеры 606
Диацетилдифенилены 206
Диацилфосфоний 536
Дибензальацетон 147, 158
Дибензальтропинон 699
Дибензил (1,2-Дифенилэтан) 207, 208
Дибензиламин 167
Дибензоил (Бензил) 146, 157, 158, 624
1,7-Дибензоилгептадиен 691
Дибензолхром 458, 459, 479
Дибензопиразины (Феназины) 355
Дибензоциклобутадиен (Дифенилен, Бифени-
лен) 66, 206, 409
2,3,4,5-Дибензпиррол (Карбазол) 23, 276,
308 сл.
Дибензферроцен 244
Дибензфуран 285
Диборан 411, 412
Дибромбензолы 26, 54, 388
6,6'-Диброминдиго 308
1,4-Дибромнафталин 388
Дибутилфталат 178
Дивинилацетилен 475
Дивинилбензол 201, 202
Дивинилкетоны 631
м-Дигалловая (м-галлоилгалловая) кислота
185, 186
9,10-Дигидроакридин 344
Дигидроантрацен 245, 246
Дигидроароматические углеводороды 385, 387
Дигидроизохинолин 345
2,3-Дигидроиндол 718
Дигидронафталины 230, 242
2,3-Дигидроперфторпентан 448
Дигидропиразол (Пиразолин) 280, 309, 3iq
Дигидропиридин 271, 276
2,3-Дигидропиррол (Пирролин) 271, 278, 292
Дигидрорезорцин (Циклогександион-1,3) 128'
304 ’
Дигидротиофен 276, 278, 289
Дигидрофуран 276, 278
Дигидрохинин 709
Дигидрохолестерин (Холестанол) 662, 664, 670
Дигитоксигенин 682
Диеновый синтез 7 7, 696
антрацена 245, 247
бензперилена 266
пиридазинов 346
Диинденилжелезо (Бензферроцен) 503
о,о'-Дииоддифенил 206
1,8-Дииоднафталин 264, 265
Дииодтирозин 192
1,1'-Дииодферроцен 498, 500
Дикарболлиды 416
Дикарбонил-дитрифенилфосфинникель 19
Дикарбэтоксибицикло[1,1,0]бутан 572
Дикетокамфорная кислота 649
Дикетопиперазины 355
Димедрол 315
2,3-Димезитилбутен-2 480
Диметилаллоксан 362
Диметиламиноантипирин (Пирамидон, Амидо-
пирин) 312
л-Диметиламинонитроанилин 28
Диметиламинопиримидин 352
Диметиланилин 84, 85, 89, 90
реакции 212, 220
Диметилацетилен, реакции 160, 480, 573
Диметилбензолы см. Ксилолы
2,3-Диметилбензойная кислота 711
2,2-Диметилбутен-З и 2,3-диметилбутен-2 613
Диметилвинилкарбинол 631
Диметилглутаровая кислота, эфир 649
2,3-Диметил-5,6-диметоксииндол 713
Диметилдиметоксисилан 419
Диметилдифенилы 205
Диметилдихлорсилан 419
1,5-Диметилимидазол 687, 688
1,3-Диметил-4-иодбензол (4-Иод-л»-ксилол) 31
Диметилмарганец 457, 458
2,3-Диметил-6-метоксииндол 712, 713
2,6-Диметил-4-метоксипиридин 368
Диметилмочевина 362
Диметилмышьяковая кислота 441
1,2-Диметилнафталин 231
3,5-Диметил-4-нитроанилин 93
Диметилпироны 367, 368
Диметилртуть 542
Диметилсульфоксид 408, 525, 527 сл.
Диметилтерефталат 179
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
797
Диметилфосфин 429
Диметилфосфит 435
Диметилфталат 178
2,2-Диметил-4-хлорбутан 557
Диметилциклопропен 573
2,2-Диметоксибензидин (Дпанизидин) 132, 204
Диметоксибенэойная кислота, эфир 371
1,2-Диметоксиглутаровая кислота 744
2,4-Диметокси-6-оксибензальдегид 373
Диметоксисиланы 420
Диметоксиянтарная кислота 746
Динатрийциануксусный эфир 645
а,а-Динафтил 26 5
1,3-Динитроазулен 514
2,4-Динитроанилин 92, 93, 105
Динитробензойные кислоты 180 сл.
Динитробензолы 48, 49, 70, 72, 73, 83
реакции 95, 118
п,п'-Динитродифенил 205
о-Динитрозобензол 77
5,7-Динитроиндол-2,3-дикарбоновая кислота
718
Динитронафталины 234, 239
2,4-Динитротолуол 72, 73
Динитрофениламинокислота 730
Дпнитрофенолы 118, 119
2,4-Динитрофторбензол 60, 453, 729
Динитрохлорбензол 92, 118
Динуклеотиды, синтез 765
Диоксаны 271, 277, 388
Диоксиантрахинон 251
Диоксиацетон 785
Диоксибензолы 126, 160 (см, также Гидро-
хинон. Пирокатехин, Резорцин)
2,2'-Диокси-4,4'-диметокСибензофенон (Уви-
нол) 1 53
Диоксинафталины 239
Диоксипиридазин (Гидразид малеиновой
кислоты) 346
4,4'-Диоксистильбен 208
Диоксифенилаланин (ДОФА) 193
1,1'-Диоксиферроцен 498
5,7-Диоксифлавилий феррихлорид 369
Диоксосоединения в синтезе гетероциклов
291, 310
Дипентен см. Лимонен
Дипептидаза 774
и,₽-Дипиридил 705
Дипиррилметан (ПирроМетин) 295
Дипнон 150
Дипольные моменты
азулена 512
галоидпроизводных 27, 29, 58, 59
нитросоединений 29, 93
оксимов 154
трополона 510
1,3-Диполярное присоединение- 280, 316, 595
Диполярофил 280
Диспропорционирование 545, 563
Дисульфиды, диссоциация 541
2,6-Дисульфодифенилен 206
Дитерпены 653 сл.
Ди-(тетрагидроинденил)-железо 503
Дифенил (Бифенил) 23, 35, 57, 131, 490
образование 203 сл., 257
реакции 204 сл., 285, 309, 551
Дифениламин 84 сл., 90 сл., 309
Дифениламинобензойная кислота 437
9,10-Дифенилантрацен 247
Дифенилацетилен (Толан) 208
Дифенилбромоний 64, 65
борфторид 111
Дифенилгидразин (Гидразобензол) 74, 80 сл.,
204
Дифенилен (Бифенилен, Дибензоциклобута
диен) 66, 206, 409
Ди-о-фенилендихлордиарсин (Арсантренхло-
рид) 443
о,о'-Дифениленметан см. Флуорен
о-ДифенилилстибиндиХлорид 446
Дифенилип (о,п-Диаминодифенил) 81
Дифенилиодоний 62 сл.
борфторид 111
гидроокись 62, 63
Дифенилкобальт 481
Дифенплмарганец 457, 458
Дифенилметан 211, 212, 214, 244
кислотность 244
Дифениловый эфир (Днфенилоксид) 39, 111,
134
Дифенилолпропан (4,4-Бис-оксифенил-2,2-
пропан) 124
Дифенилртуть 62, 76, 404, 408
Дифенилсульфид 69, 70
Дифенилсульфоксид 69
Дифенилсульфон 66, 69
п-(М,М'-Дифенил)-фенилендиамин 91
Дифенилфосфинбензойная кислота 437
Дифенилхлороний 64, 65
борфторид 111
о,о'-Дифенилхлорстибин 446
1,2-Дифенилэтан (Дибензил) 207, 208
1,2-Дифенилэтилен см. Стильбен
Дифеновые кислоты 205, 257
Диферроценил 500
Дифторбензолы 54
3,5-Дифтор-2,6-дихлорбензол 59
Дифторкарбен 44 9
Дифторэтилен 453
Дихлорамины 68
Дпхлорбензолы 10, 27, 54, 127
Дихлорвинилуксусная кислота 631, 632
Дихлоркарбен 571
Дихлоркротоновая кислота 631
1,2-Дихлор-2-метилбутан 547
2,6-Дихлорпиримидин 350
2,4-Дихлорфеноксиуксусная кислота (Герби-
цид 2,4-D) 118
2,4-Дихлорфенол, эфир 118
Дихлорциклобутен, реакация с карбоннллми
железа 470
а,а'-Дихлорэтилбензол 52
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
798_________________________________
Дициклогексилкарбодиимид 718, 735
Дициклопентадиенил 481, 495
Дициклопентадиенплжелезо см. Ферроцен
Дициклопептадненилмарганец 469, 485
Дициклопентадиенилникель 485
Дициклопентадиеннльные соединения 481 сл.
металлов 469, 481 сл.
Диэлектрическая проницаемость 578, 596
Диэтилбарбптуровая кислота (Барбитал, Ве-
ронал) 348
Диэтилбензол 201
Днэтпленгликоль, донор протона 527
Дпэтилмалоновый эфир 34 8
Диэтилферроцены 501
2,2-Диэтоксибензидин 132
ДНК см. Дезоксирибонуклеиновые кислоты
Додекануклеотид 766
Донорно-акцепторные свойства радикалов
543, 544
ДОФА (Диоксифенилаланин) 193
ДОФА-хинон 193
Дубильные вещества 185, 371
Дурол (1,2,4,5-Тетраметилбснзол) 30, 93, 534
Дурохпнон 160
Дьюаровскпй бензол (Бицпкло [2,2,0] гскса-
диен-2,5) 17, 18, 21, 471, 493
Енолизация 387, 395
Желатин 728
Железо
л-аллнльныс комплексы 476
ди-(тетрагидроинденил) 503
карбонилы 458 сл., 465, 469, 470, 471
пентадиенил 458 сл.
сэндвичеобразные соединения 4 58 сл.,
481 сл.
циклобутадиенил 471, 492
циклопентадиепил см. Ферроцен
Железопорфирины 780
«Желтый фермент» Варбурга 366
Желчные кислоты 658, 672 сл.
Замещение
в аминогруппе 88
в бензольном ядре 38
внутримолекулярное 588
гомолитическое 550 сл.
нафталина 231
нуклеофильное см. Нуклеофильное заме-
щение
электрофильное см. Электрофильное за-
мещение
Зарин 436
Защитные группы в синтезе пептидов 734
Зеин 721
Зеленое масло см. Антраценовое масло
Золотоорганпческпе соединения 456
Изатин 305, 30G, 307, 341
Изатиновая (о-аминофенилглиоксиловая) ки-
слота 306
Изоборнеол 619, 620, 650
Изоборнил-катиоп 618, 650
Изоборнилтозилат 620
Изоборнилхлориды 620, 621
Изобутснилкарбинол 631
Изовалериановая кислота 735
Изованилиновая кислота 184
Изовиолантрон 255
Изогсроновая кислота 655
Изодиазогидрат 99, 100, 101
Изодурол (1,2,3,5-тетраметил-бензол) 30
Изоксазол (1,2-Оксазол) 274, 277, 280, 31
ароматичность 318, 493
производные 280, 319
Изоксазолидин 281, 319
Изолейцин 724, 763
Изомасляная кислота, азодинитрил 423, 539,
541, 542, 569
Изомеразы 772, 786 сл.
Изомерия
атропизомерпя 131
бензола дизамещенных 10
нитрозосоединений 75
оптическая (Энантиомерия) 131
син-анти- 79 сл., 100, 145, 1 53
трео-оритро- 616
цис-транс- 154, 403
Изопикотингидразид 335
Изонпкотиновая (?-пиридинкарбоновая) ки-
слота 333, 335
Изонитрозокетои 292
Изопентенилпирофосфат 660
Изопрен 639, 659, 727
ди- и полимеризация 386, 642, 652
Изопреноиды 639 сл., 657 сл., 778
Изопропилбензол (Кумол) 30, 37
гидроперекись 555
перекись 114
1-Изопропил-4-метилпимелиновая кислота 644
2-Изопропил-8-метилфенантрен (Ретен) 259,
657
Изопропилпимелиновая кислота 511
Изонроиилциклогсптандиол 511
Изэпулегол 644
Изосафроп 134
Изотопный эффект 590
Изофталевая (.u-фталевая) кислота 170, 174,
175
Изохпнолин (3,4-Бензпиридин) 274, 345 сл.
производные 345, 693
Изоцианаты 626, 627
Изоцианиды 102
Изоциановая кислота, эфир 626, 627
Изоцптозпн 350
Изоэвгенол (4-Пропенил-2-метокснфенол) 134
ИК-спектры резерпина н его моделей 712, 713
Илиды 505, 535, 536
мышьяка (Арсинметилены) 443, 444
фосфора 425, 429—431, 535, 536
Имидазол 273, 313, 314
производные 313, 687
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
799
р-(4-Имидазолил)-аланин (Гистидин) 313 сл.,
726, 763
Иминоглутаропая кислота 783
Инвертаза 772
Ингибиторы 126, 539, 638
Индамин 165, 355
Нидан 244, 512
Индантрон синий 254, 255, 355
Индаитрснон 254
Инден 212, 243, 244, 389, 521
металлирование 384
Инденил-анион 520
Индиго 82, 188, 302 сл., 308
белое 305
Индикан 305, 306
Индоксил 290, 305 сл.
Индол (2,3-Бензпиррол) 23, 188, 273, 276,
302 сл., 306
реакции 303, 390
Р-Индолилаланип (Триптофан) 291, 302, 304,
726, 728, 763
р-Индолилмасляная кислота 302
З-Индолилуксусная кислота (Гстсроауксин)
302
Ин долин 306
Индофенол 165
Индуктивный эффект (/-Эффект) 43 сл., 47,
49, 193, 194, 437, 552, 576, 614
в ароматических кислотах 193, 194, 171,
502
в галоидпроизводных 58
и карбанионах 523
в реакции электрофильного замещения
галоидбензолов 45
в реакциях элиминации 603
в трифенилоксоний-катионе 135
в ферроцене 501
влияние заместителей 199
нитрогруппы 93
соотношение I- и М-эффектов 198
фтора 4 54
Индуцированное облучение 207
Инициаторы 543, 552, 559
Инициирование (зарождение) цепи 562 сл.,
565, 567
скорость 566
Инкременты связей 489
Инозин 747
Инозит 374
Инсектициды 12, 435, 436, 705
Инсулин 721, 723, 733
Интраанпулярпые водороды 517
Иодбензол 53, 56 сл., 62, 111
реакции 63, 91
4-Иод-м-ксил ол (4 -ио д-1,3-диметил бензол)
31
Иодобензол 62, 63
Иодозобензол 62, 63
Иодол (Тетраиодпиррол) 295
Иодоииевые катионы 62 сл.
соли 64
Р-Иодпропиоповая кислота, эфир 645
Иодферроцеп 498, 500
п-Иодхлорбензол 63
а-Ионон 641
Р-Ионон 641 сл., 655 сл.
Йохимбин 710, 711
Ироны 642
Кадмийоргапические соединения 400
Казеин 192, 742
Какодил 442 сл.
окись 440
Калийорганические соединения 382, 384, 387,
519
Кальциферол (Витамин D2) 671
Каменноугольная смола 23, 82, 114, 228, 244,
291, 326, 494
Каменноугольный деготь 256, 264, 302, 308
Камфан 646, 648
Камфен 619, 620, 648 сл.
гидрохлорид 620
Камфеновые перегруппировки I и II
рода 651
Камфо лид 648
Камфора 649, 650
получение 648, 649
Камфорная кислота 648, 649
Камфороновая кислота 648
Канифоль 259, 639, 657
Капролактам 150, 628
Капрон 156, 628, 727
Каптакс (Меркаптобензотиазол) 320
Каран 646
Карбазол (2,3,4,5-Дибензпиррол) 23, 276,
308 сл.
Карбанионы 49, 519 сл.
биполярные 536
стереохимические соотношения 526 сл.,
Карбеноидные атомы углерода 626
Карбены 570 сл., 626, 607
Карбкатионы (карбониевые ионы) 32, 575 сл.<
595, 601
гидридные перемещения 582 сл., 636
«неклассические» 620
перегруппировки 609 сл.
стабилизация 620, 636, 637
Карбобензокси-защита 735 сл.
N-Карбоксибиотинилферменты 787
Карбоксилазы 351, 353, 786, 787
Карбоксипептидазы 731, 773, 775
Чнс-2-Карбоксициклогексил-р-пропионовая
кислота 243
Карбонилгидриды 466
Карбонилы металлов 380, 463 сл.
димерные 467
железа 458 сл., 465 сл.
кобальта 466, 470
марганца 465
миогоядерные 465 сл.
никеля 465, 468, 470, 474
рз акции 466, 467
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
800
Карбоновые кислоты 175, 180 сл., 397
ароматические 169 сл.
константы диссоциации 193
поликарбоновые 17 4 сл.
производные 405, 606, 628
Карбораны 414 сл.
Карбостирил (а-Оксихинолин) 341
Карбэтоксиметилен 571, 572
Карбэтоксицпклопропен 572
Карены 646
Кариокинез (Митоз) 758
Кармин 252
Карминовая кислота 252
а-Каротии (Провитамин 653 сл.
Каротиноиды 653 сл.
Каротины 372, 653, 654, 656
Каталазы 781
Катехины 371, 372
Катионотропная аллильная перегруппировка
631, 634
Каучуки 386, 420, 639, 727
бутадиен-винилпиридиновый 333
бутадиен-стирольный 199, 201
вулканизация 321
изопреновый 386
фторорганические 448, 453
Квадратичный обрыв 563
Квазиароматический активированный ком-
плекс 599
Квазикомплексные ртутноорганические со"
единения 403, 404, 409
Квазистационарное состояние системы 564,
566, 567
Кватерфеннл 203
Кверцетин 371, 372
Коллин 370
Кератин 721, 741
Кермесовая кислота 252
Кетены 405, 572
фотолиз 570
Кетовинилирование 532
а-Кетоглутаровая кислота 315, 783
Кетоиобирин 711
Кетон Михлера 212, 220
Кетонный мускус 71
Кетонокислоты
гидразоны 106
получение 315, 665 сл., 778
реакции 338 сл., 597, 700
Кетоны
ароматические 148 сл.
непредельные 310, 391 сл.
ртутные производные 405
Ферроцена 495
Киафеиин 174
Кинезамещение 61, 83, 128
Кинетика ценных реакций 562 сл
Кинетин 363
Кислотное расщепление 184
Кислотно-основной катализ
«Кислотность» углеводородов
598
384
Кислотный сине-черный 238
Клатратные соединения 35, 129, 130
Кобаламин (Витамин В1г) 301, 341, 777
Кобальтициний-катион 482, 483, 505
Кодегидразы 782
Кодеин 694, 695
Кодеинон 695
Кодекарбоксилаза (Пиродоксаль-5-фосфат)
779, 785, 786
Кодоны 761, 762, 763
Кокаин 698, 700, 701
Кокарбоксилаза 353
Коксовый газ 22
Коллаген 721, 741
Коллидин (2,4,6-Триметилпиридин) 328, 330
369
Колхицин 512, 684, 685
п-Комплексы 32, 33, 36, 42, 594
о-Комплексы 32, 33, 37, 42, 86, 552, 587
Комплекс Зееля 149
Комплексы переходных металлов 121, 404,
416, 471 сл.
Комплексы с переносом заряда (КПЗ, Анион-
радикалы) 259, 534 сл.
Конволамин 700
Конвольвин 700
Конгидрин 690
Конго (краситель) 107
Конденсации
альдольная 147, 395, 533, 599
ацилоиновая 209
бензоиновая 146, 284
кротоновая 19, 333, 347, 395, 512, 641. 699
Михаэля 531
Перкина 147, 148, 258, 533
сложноэфирная 367, 659
Конденсированные ароматические системы
229 сл.
Кониин (н-Пропилппперидин) 333, 690
Конирин (а-Пропилпиридин) 690
Константа скорости
диссоциации кислот и псевдокислот 521 сл.
передачи цепи при теломеризации 56" сл.
протонирования карбанионов 524
реакции 591
Конформация
аксиально-аксиальная 607
белков 739
переходного состояния 605, 606
«Координата реакции» 591
Координационное число 458
Копростан 662, 666 сл.' 672
Копростанон-3 и копростанол-3 670
Копростерин (Копростанол) 660 ся., 666,
668 сл.
Кордиамин (Никотиновая кислота, диэтил-
амид) 335
Коричная кислота 147, 189, 190
производные 189, 190, 404
реакции 189, 200
Коричный альдегид 147
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
801
Коронен 259, 266
Короненхинон 266
Корреляционные
диаграммы 462
уравнения 193 сл.
Кортизон 680 сл.
Кортикостероиды 679, 682
Кортикостерон 680, 681
Косубстрат 777
Кофеин 358. 362, 363, 364
Кофермент А 659, 777, 779, 787
структура 778
Коферменты (Коэнзимы) 291, 301, 353, 425,
770, 771, 779, 780, 785, 786
КПЗ (Комплексы с переносом заряда) 259,
534 сл.
Красителя (см. также Азокрасители) 105, 132,
172, 249, .302, 341, 344
антрахиноновые 248, 250, 252
индантреновые 249, 254
индигоидные 82, 188, 302 сл.. 308
кубовые 254, 255
полиметпновые 328
полициклокетоновые 254
сернистые 165, 309, 322
тиазиновые 96
тиоиндигоидные 290, 291
тиоцианиновые 322
триазиновые 357
трифенилметановые 89, 218 сл., 220, 223,
576
феназиновые (Сафранины) 193, 355, 356
фентиазиновые 375
цианиновые 321, 342
— мезомерное выравнивание 343
Крахмал 727, 771
Крашение кубовое 256
Крезолы 23, 60, ИЗ сл.. 124
Крекинг 537, 540 сл.
Кремнийорганические соединения 419 сл.
Кристаллический фиолетовый 217, 219 сл., 576
Кристаллоза (о-Сульфобензойная кислота,
имид, натриевая соль) 181
Кротоновая кислота 190, 632
Кротоновая конденсация 19, 333, 347, 395, 512,
641, 699
Кротоновый альдегид 339
Кроцеиновый алый ЗБ 238
Кроцеиновый нркий 9Б 238
Ксантин 360 сл.
Ксантогеновые кислоты 606
Ксантопротеиновая реакция 722
Ксантоптерин 364
Ксилидины 84, 365
n-Ксилол, анион-радикал 535
Ксилолы (Диметилбензолы) И, 12, 22, 23,
26, 30, 34, 105
аутоокисление 555
окисление 170, 174, 178
Кумарин 190
производные 373
51 Заказ 672
Кумариновая (цис-о-оксикоричная) кислота
190
о-Кумаровая (тпранс-о-оксикоричная) кислота
190
Кумарон 273, 285
Кумол см. Изопропилбензол
Кураре 345, 684
L-Курин 705
Кускгигрин 686
Лавсан 178
Лактаразулен 515
Лактаровио.тин 51 5
Ланостерин 659
Левомицетин 765
Левопимаровая кислота 259, 657
Лейкооснование 218
Лейкоптерин 364
Лейцин 724, 763
Леканоровая кислота 185
Лиазы 772, 785 сл.
Лигазы (Синтетазы) 766, 769, 772, 787
Лигнин 123, 134, 185
Лидит 120
Лизин 725, 729, 731, 763
Ликопин 372, 656
Лимонен (Дипентен) 639, 641, 642
гидратация 645
изомеризация 643
Лимонная кислота 684, 779
Линалоол 640, 641
Липазы 773
Липоевая кислота 781
Липопротеиды 722
а-Литийметилпиридин 333
Литийорганические соединения 380, 382 сл.,
389
ароматические (Литийарилы) 56, 171,5383,
440
реакции 386 сл., 401, 413, 415, 428, 595
Литохолевая кислота 672, 673
Лобеланин и лобелии 691, 692
Лойпоновая кислота 707
Лолинин и лолин 687
Лупикаин 701
Лупиннн и лупинпновая кислота 697
Лутеолин 371
Лутеостерон (Прогестерон) 674, 675, 678, 682.
Люминал (Фенобарбитал) 349
Люмифлавии 784
Люмихром 784
Л-Люциферин 323
Магнийорганические соединения (см. также
Гриньяров реактив и Металлоорга-
нические соединения) 285, 381 сл.
387 Сл., 399, 421, 457
реакции 171, 288, 390 сл., 39,3 сл., 401,
412 сл., 419 сл., 428, 433, 436 СЛ.,.
440 сл., 445 сл.. 451, 456
Магнитный момент 5 38
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
802
Малахитовый зеленый 218, 219
Малеиновая кислота, гидразид (Диоксипири-
дазин) 346
Малеиновый ангидрид 13, 1/, 21, 283
в диеновом синтезе 204, 265, 266 282
реакции 247, 293, 558
Малеиновый диальдегид, азин (Пиридазин)
274, 277, 345 СЛ.
Малонилмочевина (Барбитуровая.кислбта) 347,
348, 349, 359
Малоновая кислота 189, 703
ангидрид 179
полуальдсгид 19
Малоновый эфир 105, 106, 334, 523
С—Н-кислотность 49
Масло (а)
антраценовое (зеленое) 23, 245, 256, 30»,
309, 343
герани 640, 641
каменноугольной смолы 22, 23
касторовое 252
костяное 291
лавандовое 640
розовое 641
цитронелловое 641
Матричный синтез 759
Мевалоновая кислота 660, 778
Мединал 349
Мезитила окись 150
Мезитилен (1,3,5-Триметилбензол) 18 сл.,
31, 34, 179
молекулярные соединения 121
Мезитиленкарбоновая кислота 181, 182
Мезоксалевая кислота, арилгидразон 106
Мезомерия 28, 107, 522, 630
Мезомерный эффект (М-Эффект) 46, 437
аминогруппы 85, 86
в барбитуровой кислоте 348
в замещенных бензола, 58, 115, 193, 194
соотношение с /-эффектом 198
стабилизация карбаниона 523
Мезопорфирин 298
Мезотропия 28
Меламин 357
Меленит 120
Меллит 179
Меллитовая (бензолгсксакарбоновая) кислота
175, 179
Ментадиены 643
Ментан (1-Метил-4-изоиропилциклогексан)
642
Ментол 643, 644
Ментон 644
Ментилхлорид 607, 608
Меркаптаны (Тиоспирты, Тиолы) 67, 289, 559
меркаптобспзотиазол (Каптакс) 320
Меркаптоэтанол 729
Меркурпииевый ион 404
Меркурированне 402, 403 сл
Мерохпиеи 707
Мескалин 168, 169
Металлирование 3 5, 383, 384, 535
Металлкарбонилгалогениды 467
Металлкетилы 153, 535
Металлоарены 458, 477 сл.
Металлоорганические соединения
активные (АМО) 380 сл.
алюминия 381, 400, 417, 418, 460
бария и кальция 400
бериллия 400
бора 411 сл., 460
висмута 446
германия 421, 422
кадмия 400
калия 382, 384, 387, 519
лития см. Литийорганические соединения
магния см. Магнийорганические соеди-
нения
олова 422 сл.
переходных металлов 456 сл., 480
ртути см. Ртутноорганические соединения
свинца 422 сл., 431, 540, 542
стронция 400
сурьмы 444 сл.,
сэндвичеобразные 481 сл., 495 сл.
таллия 418
теллура 447
тяжелых металлов 418
цинка 389, 399, 400
щелочноземельных металлов 400
щелочных металлов 332, 382 сл., 421, 519
Металлоценкарбонилы 458
Металлоценоарены (Аренметаллоцены) 458,
483
Металлоцены 458 сл., 481 сл.
Метан
гомолоитическое галоидирование 546 сл.
ионизация 521
протинирование 585
Метаниловая кислота (м-Аминобензолсульфо-
кислота, .м-Анилинсульфокислота) 84,
85, 94
Метиладенины 748
Мстилазулен 515
Метилаллоксан 362
1-Метил-5-н-амилимидазол 687, 688
Метнланилин 84, 85, 89, 90, 513
Метнларсинистая и метиларсоновая кислоты
441
а-Метил-Р'-ацетил-а-пирон (Дегидрацетовая
кислота) 367, 368
Метилбензоилкарбинол 634
Метилбензол см. Толуол
2-Метилбензотиазол 321
2-Метилбензотиазолиодалкилаты 321
о-Метилбензофенон 153
2-Метил-5,6-бензпирилий феррихлорид 369
D-2-Метилбутилмагний 394
2-Метилгептанон-6 662, 663
6-Метшггептен-5-он-2 640
Метилглиоксаль 11
Метилгранатанин (Псевдопельтьерин) 702, 703
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
803
Мстилднборан 411
8-Мстилдигидразулеи 515
2-Лст>!Л-1,4-д11О1<синафталиц 241
Метилдифспил 552
Метилен 570, 571 сл.
Метиленовый синий 97, 375
Метиленфосфораны см. Илиды фосфора
4-Метилизоборнеол 651
Метилизоборнилхлорид G21
n-Метилизоиропнлбензол (п-Цимол) 31
1-Метил-7-изопропилфенацтрен (Ретеп) 259,
G57
5-Метил-2-пзопропилфенол (Тимол) 644
1-Метил-4-изонропилциклогексан (Ментан)
642
N-Метилимпдазол 314
р-Метилпндол (Скатол) 304
N-Метилирование 91, 92, 220, 311, 328, 707
исчерпывающее 327
8-Метилксантин 363
Метиллитий 383, 515
2-Метил-З-метоксипиридип дикарбон оная
(метилметоксицинхомерононвя) ки-
слота 786
Метилморфенол 695
а-Метилморфиметин 694, 695
Метилморфол 695
Метилмышьяковпстая (метиларсинистая) ки-
слота 441
Метилнафталины 23, 231, 233
2-Метил-1,4-нафтохинон 239, 241
Метиловый
оранжевый 107
фиолетовый 220
7-Ыетилоливии 254
а-Метплолфенилуксусная кислота 698
Метилпиперидин 698
Х-Метилниперидин-Р-карбоновая кислота 692
Метилпиразпн 354
Метилпиразолон 311
Метилпиридины 333, 354
6-Метнлпиримпдпн 347
(К'-Метил-а-пирролидил)-₽-11ирндин см. Нико-
тин
"М.етил пирролидин 685, 698
1-Метилпирролизидии (Гелиотридан) 686, 687
N-Метилпролин (Гигриновая кислота) 685
Метил-радикал 226, 540
Метилсульфазин (Сульфамеразин) 351
Метилтиоурацил 193
Метилтрансферазы 776, 777
Метилтропилиевый лиганд 483, 484
Метилфенилкарбинол 200
Метилфосфин 429
а-Метилфуран (Сильван) 353
а-МетИлхинолии (Хинальдин) 339, 342
Метилхолантрен 259
Метилциклопептан 21, 022
3'-Метил-1,2-циклопентанофеиантрен (Угле-
водород Дильса) 662
Метилэтилфенил-апион 528
51*
Метионин 725, 763, 776 сл.
Метод
Барбье — Виланда 667
Барта 442
Бсшама 442
валентных схем 460, 462
вращающегося сектора 568
квазистационарного состояния 567, 568
Кнорра 297
Крауса 421
молекулярных орбиталей 14, 461, 487, 488
Несмеянова 445, 446
Сенджера 730
Фпзера 241
Фишера 296
Френкель — Конрата 730
Фридмана — Дока 434
Церевитинова — Чугаева 390
Чугаева 650
Шмидта 445
Эдмана 730, 731
Метоксианилины (Аштзидины) 83, 132, 133
п-Мстокспбензойпая кислота 184, 193
Метоксигептатриен 509
3-Метоиси-4,6-диацетоксифенантрен 695
З-Метоксиизофталевая кислота 712
6-Метоксииндол 716, 717
6-Метокси-₽-индолилуксусная кислота, ни-
трил 716
6-Метокситриптамин 716, 717
о-Метоксифенилдиазоний 102
6-Метоксихинолин-4-карбоповая (хининовая)
кислота 707, 709, 710
1-Метокси-4-хлорбеизол 31
Метоксициклогексан, германиевое производ-
ное 422
Метол 78
Метоний-катион 585
Механизмы химических реакций 588 сл.
Миндальная кислота 191
Миоглобин 741
Миозин 721
Мирцен 640
Митоз (Кариокинез) 758
Мовеин Перкина 356
Молекулярные орбитали 14, 15, 461, 4s7
Молекулярные соединения с переносом за-
ряда 121
Молочная кислота 191, 783
Моиотерпены 640 ап.
Морфин 259, 345, 684, 694 сл.
диацетат (Героин) 697
синтез 69 5, 696
Морфолин 185
Мочевая кислота 359, 360, 364
реакции 362, 363
соли (Ураты) 358
Мочевина, реакции 348 сл., 360, 741, 773
Муконовая кислота 21
динитрил 96
Мукопротеин 301
804
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Мускус амбровый 71
Мышьяковоорганические соединения 440 сл.
НАД (Никотинамидадениндинуклеотид) 780,
782, 783
Надбензойная кислота (Бензоил, гидропере-
кись) 172, 173, 714
НАДФ (Никотпнамидаденнндинуклеотидфос-
фат) 780, 782, 783
Найлон 285
Натрийарилы 171
Натрийацетоуксусный эфир 519
Натрнйкарбонилы металлов 467
Натриймалоновый эфир 519
Натрийоргаиическис соединения 214, 225,
382 сл., 386, 387, 421, 519
Нафтазарин 239
Нафталевая (пери-нафталиндикарбоновая)
кислота 243
Нафталевый ангидрид 259
Нафталин 9, 22, 23, 120, 187, 229 сл., 233, 237
ароматичность 230
гидрирование 230, 242
молекулярные соединения 121
натриевый аддукт 385
окисление 170, 174, 229, 241
производные 422, 515
радикал-анпон 231
реакции 231, 385, 586
свойства 230, 232. 245, 261. 290, 491, 514
nepu-Нафталиндикарбоновая кислота (Нафта-
левая кислота) 243
Нафталинсульфокислоты 234, 235, 236
Нафтацен (Тетрацен) 232, 259 сл.
Нафтиламин черный 238
Нафтнламиносульфокислоты 235
Нафтиламины 229, 234, 235
реакции 234, 237
₽-Нафтилдиазоний 234
Ы-(Нафтил)-фениламин 91
Нафтолсульфокислоты 235
Нафтолы 233, 235, 500
Нафтохиноны 239, 240
Неальтсрнатные ароматические системы 487,
494
Небензоидные ароматические системы 487 сл.
Нейтральный красный 355
«Неклассический» катион 618, 619, 620
Нембутал 349
Неоментилхлорид 607, 608
Неофильный (2-фенил-2,2-диметилэтиловый)
радикал 637
Нерол 641
Нефть 20, 23, 52
пиролиз 228, 494
состав 289
Ниацин см. Никотинамид
Никелоцен 485
Никотин иИ-Метил-смтрролидил)-р-пиридин]
Ззз, 335, 684
синтез 292, 704
Никотинамид (Витамин РР, Ниацин) 334 335
776, 783
Никотинамидадениндинуклеотид (НАД) 780
782, 783
Никотинамидадениндинуклеотидфосфат
(НАДФ) 780, 782, 783
Никотинамид-метилтрансфераза 776
Никотиновая (Р-пиридинкарбоновая) кислота
333, 703, 705, 782
амид см. Никотинамид
диэтиламид (Кордиамин) 335
Никотирин 336, 704
Нингидрин (Трикетогидринден, гидрат) 72°
728
Нингидринная реакция 722
Нитрилы ароматические 150
Нитрильное расщепление пиразолов 311
Нитроаминодурол 93
Нитроанилины 28, 83, 84, 88, 92 сл., 118
9-Нитроантрацен 248
Нитроацетанилиды 87
Нитробензальдегиды 148
Нитробецзойные кислоты 168, 169, 180, 187
I- и М-эффект 194
Нитробензол 31, 70 сл., 149, 331, 552
восстановление 82, 83
л-комплекс 36
реакции 74 сл., 78, 79, 84, 111, 219, 339
.м-Нптробензолсульфокислота 94
Нптробромбензолы 72, 73
Нитрование ароматических соединений 71
п-Нитродиазобензол 102
31-Нитродифенил 111
2-Нитродифенилен 206
Нитрозобензол 74, 75
о,о'-Нитрозобифенил 76
Нитрозодиметпланилин 76, 96, 355
реакции 90, 373
Нитрозометиланилин 90
Нитрозосоединения 75, 90, 96, 161, 355, 373,
397
Нитрозофенолы 76, 90, 161
Нитроиодбензолы 72, 73
Нитрокоронены 266
Нитрометан, присоединение к альдегидам
534
Нитрон 281, 317
Нитронафталины 229, 234
Нитроний-катион 43
4-Нитропиразол 311
Нитропиридииы 331, 334
а-Нитропиррол 295
Нитросоединения ароматического ряда 29,
70 сл., 82, 149, 168, 200 сл., 331, 552
Нитростиролы 200, 201, 205
2-Нитротиофен 287, 288
Нитротолуолы 71 сл., 333
ауи-форма 72
Нитрофенантрены 258
Нитрофенилдиазоний 101, 102, 118
п-Нитрофенилферроцен 502
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
805
Нитрофеполы 28, 113, 118 сл.
таутомерия 122
п-Нптроформапнлпды 88
о-Иитрофталепый ангидрид 229
71-11 птрофторбепзол ВО, 453
Нитрохлорбсяэолы 50, 60, 72, 73, 346
о-Нитро-п-хлорфенплдназопий 102
Новальгин (Анальгии) 312
Новокаин (п-Аминобснзойнап кислота, диэтпл-
аминоэтиловый яфир) 188, 701
Нонакарбонил железа 4 70
Нонапептид 732
Ионасплапы 420
«Нопинен» (Р-П1И1СН) 647
Норборнпл-катпопы 585, 618
Норборяплтозилат 619
Норборнплфторид 585
Норкарадпен 573
Норлолин 687
Норлупннин 697
Нуклеазы 773
Нуклеиновые кислоты 346, 348, 358, 361, 721,
743 сл., 773
Нуклеозиды 744 сл.
Нуклеопротеиды 722, 743
Нуклеотиды 361, 362, 425, 743 сл., 748 сл.,
758, 766, 769
Нуклеофильное замещение 48 сл., 182, 196,
440, 468, 519, 529 сл., 576, 588, 592,
629
в галоидных арилах 57, 453. 575
в сульфопроизводных ароматического
ряда 252
в фенолах 148, 149
при перегруппировках 618, 624
Нуклеофильное присоединение 431, 4 51 сл.,
454, 508, 528 сл.
Нуклеофильные (сскстетпые) перегруппировки
629
Обрыв кинетической цепи 563, 565, 567,
623
Озазоны 681
Озонид бензола 11
а-Окиси 454
реакции 430, 449
Окиси
какодила 440
мезитила 150
пиридина 332, 334
триоктилфосфина 437
трифенилфосфипа 431
фосфинов 437, 438
этилена 271, 277, 397
Окислительное дезаминирование 610, 611
1,2,4-Окса диазол 281
Оксазин 275, 375
1,2- Оксазол см. Изооксазол
1,3- Оксазол (Оксазол) 274, 277, 318 сл.
ароматичность 493
Оксазол-4-карбоиовая кислота 319
Оксазолоны (Азлактопы) 292
п-Оксиазобспаол 133
Оксиазососдинения 79, 104
9-Оксиакридпп 343, 344
Оксиалкилфосфоиовыс кислоты 43. 4 35
Оке.пантрахипопы 249
71-Оксвбснзальдегпд 192
О ксибспзил пенициллин 323
о-Океибензойпая кислота см. Салициловая
кислота
Л1-Оксибен:юйпая (1,3-оксибепзойиап) кислота
168, 169, 184
п-Оксибспзойная (1,4-оксибепзойная) кислота
168, 169, 183, 184
метиловый эфир (Анисовая кислота)
184
Оксибензойные кислоты (Фенолокислоты) 122,
182 сл.
Оксигемоглобин 742
Оксигидрохинон (1,2,4-Триоксибензол) 128,
162
5-Окси-М,К-диметилтриптамин 305
Оксидоредуктаза 772, 780 сл.
Оксииндолы, производные 304, 305
транс-о-Оксикоричная (о-кумаровая) кислота
190
цис-о-Оксикоричпая (кумариновая) кислота
190
Оксилиналоол 641
2-Оксиметилбензойная кислота, лактон (Фта-
лид) 178
Оксимы 153 сл.
Оксин (8-Оксихинолин) 340, 341
Оксинафталинсульфокислоты, соли 236
Оксиидол 306
Оксипиридины 334 сл.
2-Оксиииримидин 349
21-Оксипрогестерон 680
Оксипролин 291, 726, 729, 731
6-Оксипурин (Гипоксантин) 361
Оксиран (Окись этилена) 271, 277, 397
5-Окситетрациклин (Террамицин) 262
Окситоцин 735, 736, 738
5-Окситрпптамин (Серотонин) 305
Окситрифенилкарбинол 222
Р-Окси-Р-фенилпропионовый альдегид 117
Оксифлавоны 372
8-Оксихинолип (Оксин) 340, 341
сульфокислота (Хипазол) 341
а-Оксихнполин (Карбостирил) 341
2-Оксицпклогептапоп 510
1-Окси-1,6-опоксициклодекан, эфир 625
Оксол, см. Фуран
Оксолан см. Тетрагидрофуран
Оксониевые соли 368
Оксосинтез но Реппе 466
О кеосоедииения
протопизация 586
реакции с АМО-соединениями 381
Окта дека пептид 723
Октануклеотид 766
806 _________________
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Олефины
комплексы с переходными металлами
471 сл.
л-орбитали 472
полимеризация 417
реакции присоединения И, 403, 404,
558, 559, 560
Оливин и оливиновая кислота 253, 254
Оливомицины 253, 254
Олигомеризация см. Теломеризация
Олигомеры (Теломеры) 453, 566, 568
Олигонуклеотиды 766, 767
Олифа 657
Оловоорганические соединения 422 сл.
Ониевое соединение «обратное» 414
Ониевые катионы 111
Опий 693
Орбитали атомные металлов 462
Орпентанты первого и второго рода 38
Ориентация замещения
галоидами 45 сл.
мета-ориентантами 47 сл.
нуклеофильного 48 сл.
орто-пара-ориентантами 38, 41, 43, 44, 116
электрофильного 31 сл.
Орнитин 725
Ортаниловая кислота см. о-Аминобензол-
сульфокислота
Ортоэфиры 396
Орто-эффект 194
Осмоцен 482, 483, 505
₽-Отщепление((3-Элиминация) 145, 409, 589,
601 сл.
Оцимен 640
Пантокаин 188
Пантотеновая кислота 777
Папаверин 345, 693
Парамагнетизм свободных радикалов 537
Параминдальная кислота 191
Парафуксин 219 сл.
Парациклофаны 211
ПАСК (n-Аминосалициловая кислота) 188
Пеларгонидин 373, 374
Пельтьерин 690, 691
Пенальдовая кислота (а-Фенилацетиламино-
малоновый полуальдегид) 323, 324,
325
Пениллоиновая кислота 324
Пеницилламин 324, 325
Пенициллины 95, 323 сл.
Пенициллоиповая кислота 323 сл.
Пентаборан 412, 414
Пентабромтолуол 55
Пентадиены, изомеризация 327
Пентазол 272, 318
Пентакарбонил железа 457, 465, 466, 468
комплекс с ацетиленом 470
Пентаметилбензол 30
Пентаметилендиамин 326
Пентантрикарбоновая кислота 645, 688
Пентафторанилин 56
Пентафторбромбензол 56
Пентафтортолуол 55
Пентахлортолуол 55
Пентацен 260, 261
Пентенилпенициллин 323
Пентозы, дегидратация 281
Пепсин 721, 770, 773
Пепсиноген 773
Пептидазы 721
Пептиды 734 сл.
Пептоны 723
Пербензойная кислота 553
Пергидроазин см. Пиперидин
Пергидрофенантрен 257
Переаминирование 339
Перегруппировки 609 сл.
алкоксибензолов 132
аллилового эфира фенола 132
аллильные 134, 629 сл., 633, 637, 638, 653
— обратная 632
Арбузова 433 сл., 437
ацетилен-аллен-диеновая 634
ацилоиновая 634, 635
бекмановская 153, 155, 607, 627, 628
— второго рода 157
бензидиновая 80, 204, 237, 302
бензиловая 158, 284, 624
борнеола 620
борнихлорида 620
Вагнера 648, 651
Вагнера—Меервейна 613, 618, 621
Данилова и Венус-Даниловой 623
Демьянова 621, 622, 636
изоборнеола в камфен 619, 620
изоборнильного (норборнильного) ка-
тионов 618 сл.
Валлаха 79
Вольфа — Арндта 625 сл., 688
гомолитические 636 сл.
Гофмана 187, 627
Клайзена 133, 501, 597
Криге 625
Курциуса 626, 627
Лоссена 627
камфеновые 651
карбеноидов и карбенов 572, 574, 625 сл.
Наметкина 621, 651
а-нафталинсульфокислоты в (3-кислоту 236
нуклеофильные (секстетные) 629 _
окислительного дезаминирования . Мак-
Кензи 610, 611
пинаколиновая 594, 609, 611, 612, 613,
радикалов 636 сл.
ретропинаколиновая 585, 594 сл., 613,
614, 618, 621—623, 635, 648, 650, 651
семидиновая 81
Соммле 629
с перемещением мигрирующей группы
к азоту 626
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
807
Перегруппировки
— — л-связей G29 сл.
Стивенса 504
Фаворского 623, 624, 634
фенплгпдроксиламина в п-аминофенол 133
Фриса 133, 167
Шмидта 627
Передача цепи 553 сл., 560 сл.
константа передачи 566
Перекиси органические 541, 569
ацилов 559
бензоила 170, 173, 557, 637
mpem-бутила 557
как инициаторы 428
кумола 114
Перекисный эффект 557
Переходное состояние (Активированный ком-
плекс) 33, 41, 589 сл., 617
конформация 605
при гидролизе сложных эфиров 198
при замещении в ароматическом ядре
552
при перегруппировках с изменением ске-
лета 610, 617, 619, 620
при электрофильном замещении 525
пространственные затруднения 181
пятицентровое 595, 596
трехцентровое 593, 594
циклическое 533, 593 сл., 599, 600, 606
четырехцентровое 593, 595
шестицентровое 382, 392, 394, 396, 596,
597, 598, 606
Перилен 232, 264
Перинафтилий-катион 216
Перкаин (Совкаин) 341, 701
Пероксидазы 781
Перфторалканы 451
Перфторалкил-анион 454
Перфторароматические соединения 56
Перфторацетон 455
Перфторацетонитрил 450
Перфторизобутилен 342, 453
Перфторкетоны 454, 455
Перфторолефины 449, 451
полимеризация 452
Перфторпентан 448
Перфторпропилен 448, 449, 452, 453
Перфтортриэтиламин 454
Перфторуглеводороды 448 сл.
Перфторциклогексан 450
Перхлорильные производные 65
Пиазтиол 96
Пикрамод (2,4,6-Тринитроанилин) 92, 93, 97
Пикриновая кислота (2,4,6-Тринитрофено;7)
118, 120, 129
комплексы с переносом заряда 120, 259
Пилокарпин 687, 688
полный синтез 689
Пилоповая кислота 688
Пимелиновая кислота 184
Пинаколин 394
Пинаколиновая перегруппировка 594, 609,
611, 612, 613, 624
стереохимия 611
Пинаколиновый спирт 620
Пинан 646, 647
Пинены 613, 640, 647, 648
Пинеихлоргидрат 620
Пиновая кислота 647
Пинокамфан 647
Пиноновая кислота 647
Пиперазин (Тетрагидропиразин) 354, 355
а-Пиперндил-р-пиридин (Анабазин) 333, 336,
705
Пиперидин 49, 271, 276 сл., 326, 327, 331
в алкалоидах 690, 691
Пиперин и пипериновая кислота 691
Пиперонал (Гелиотропин) 134, 691
Пиразин (1,4-Диазин) 275, 345, 346, 354, 364
Пиразол (1,2-Диазол) >272, 273, 277, 280,
309 сл., 493, 595
Пиразолин (Дигидропиразол) 280, 309, 310
Пиразолоны 311
Пирамидон 312
у-Пиран 367
Пирен 232, 264
Пиренхинон1264
Пиридазин (1,2-Диазин, Малеиновый диаль-
дегид, азин) 274, 277, 345 сл.
Пиридин (Азабензол) 22, 95, 271, 272, 277, 327
в природе 326
N-окись 332
производные 326, 329 сл., 333 сл.
— физиологическая активность 335
реакции 326 сл., 467, 498, 513, 54#
— замещения 331, 332, 334
свойства 274, .275, 320, 321, 326, 331,
347, 354, 493
синтез 329, 330, 340
3,4-Пиридиндикарбоновая (цинхомероновая)
кислота 345
а-Пиридинкарбоновая кислота 336, 690
Р-Пиридинкарбоновая (никотиновая) кислота
333, 703 сл.
у-Пиридинкарбоновая (изоникотиновая) ки-
слота 333, 335
Пиридинкарбоновые кислоты 326, 333, 337,
690
(З-Пиридинсульфокислота 331, 334
Пиридинсульфотриоксид 283, 295
Пиридинтрикарбоновые кислоты 711
(З-(а-Пиридил)-этанол 333
Пиридоксаль 336, 337
производные 338, 785
Пиридоксаль-5-фосфат (кодекарбоксилаза) 779,
785, 786
Пиридоксамин 336, 785
Пиридоксин 336
Пиридоксол (Адермин) 336, 337, 785
Пиридоксол-5-фосфат 786
Пиридонимцн 334
Пирилиевые соли 367, 368, Збв
808
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Пиримидин (1,3-Диазабензол, З-Азапиридин,
1,3-Диазшг) 95, 276, 345, 346 сл.
ароматичность 272, 274
производные 323, 348, 364, 746
Пировиноградная кислота 315, 338, 783,
786 сл.
реакции 339, 351, 787
Пирогаллол (1,2,3-Триоксибензол) 128, 131,
252
Пирокатехин (1,2-Диоксибензол) 126 сл.,
251
дпметиловый эфир (Вератрол) 133
монометиловый эфир (Гваякол) 114, 133
реакции 159, 167
Пиролиз 539 сл.
нефти 23
сложных эфиров 606
Пиромеллитовая (1,2,4,5-бензолтетракарбоно-
вая) кислота 175
а-Пирон 368, 471
у-Пирон 367—369
Пирослизевая (а-фуранкарбоновая) кислота
279, 291
реакции 282, 284
Пиррилмагиийгалогениды 293, 294, 390
N-Пиррил-Р-пиридин 704
Р-Пиррилпропионовые кислоты 297
Пиррол (Азол) 273, 277, 278, 291 сл., 302,
314, 704
ароматичность 272, 493
производные 294, 295, 297—300, 309, 390
реакции 292 сл., 295, 573
свойства 286, 293, 310
Пиррол-а-альдегид 294, 296
Пирролидин (Тетрагидропиррол, Азолидин)
271, 277, 278, 292, 293
Пирролизидин 686
Пирролин (2,5- или 2,3-Дигидропиррол) 271,
278, 292
а-Пирролкарбоновая кислота, эфир 2 94
Пиррометен 295, 296
Пиррометин (Дипиррплметан) 295
Плазмохин 341
Платинецин 686
Платифиллин 686
Полиаденпловая кислота 7 54
Полиаминополифоефоновые кислоты 435
Полигерманы 421
Полиглутаминовая кислота 740
Полидсзоксприбонуклотпды 7 54
цис-Полиизопрен 386
Полиинозиновая кислота 754
Поликарбоновые кислоты 174 сл.
Полимераза 766
Полимеризация 543, 565 сл.
1,3-диеновых систем 386
олефинов 417
перфторолефинов 452
радикальная 537, 569
этилена 559
Полимстиибензол 22
Полиметиновые красители 327
Полинуклеотид-фосфорилаза 754
Полинуклеотиды 743, 749 ст.
Полиоксибензойные кислоты 184 сл.
Полиоксибензолы 131
Полиолефины 470
^-комплексы с переходными металлами
472
Полипропилен 126
Полирибонуклеотиды 754
Полисилоксаны 420
Полистирол 727, 728
Политерпены 639
Политетрафторэтилен (Тефлон) 56, 449, 453
Полиуридиловая кислота 754
Полиуридинфосфат 762
Полифенилаланин 762
Полифенилметаны 211 сл.
Полифторбензолы 56
Полизтилентерефталат 178, 179
Полиэфирные смолы 178
Порфин 296 сл., 518
Порфирины 296 сл., 518
Постулат Хэммонда 589, 590, 591, 617
Правило
Брсдта 581
Зайцева 601, 602
Кернера 24, 41
Марковникова 36, 403, 412, 422, 556,
559, 648
наименьших структурных изменений 609
Попова 699
Сиджвика 458, 474
Хюккеля 16, 297, 491, 516
Эльтекова — Эрленмейера 156
Преднизон 680
Пренптол (1,2,3,4-Тетраметилбензол) 30
Примахин 341
Примулин 322
Пробковая кислота 702
Провитамин А, (а-Каротпн) 653 сл.
Провитамин D (Эргостерин) 658 сл., 670,
671
Провитамин D, (7-Дегидрохолестерин) 658,
660, 670, 671
Прогестерон (Лутеостерон) 674, 675, 682
получение 678
Пролин 291, 726, 729 СЛ., 763
Прометон 357
Прометрин 357
Проптальбпн 94
Пропанолы
гидридные перемещения 585, 636
индуцированное окисление 152
Пропаргиловый альдегид 318
Пропенплацетилеи 604
4-Пропенил-2-метоксифенол (Изоэвгенол) 134
п-Пропенилфенол, метиловый эфир (Анетол)
184
н-Пропиламин 584, 635
н-Пропилбензол 30
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
809
Пропилен
рКа 521
реакции присоединения 403, 557
а-Пропилпиперидин (Кониин) 333, 690
а-Пропилпиридин (Конирин) 690
Пропиофенон 148
Пространственный эффект заместителя 198
Протамины 722
Протеазы 771, 773
Протеиды 722
Протеин 771
Протеиназы 774
Протеины см. Белки
Протокатеховая кислота 126, 167, 184
Протокуридин 705
Протонирование карбанионов 523, 524
Протопорфирин 298
Прототропия 314
Прототропная аллильная перегруппировка
632—634
Проферменты 773
Прямой голубой светопрочный 23М, 238
Прямой черный 3 238
Псевдоионон 641
Псевдокислоты 99, 522 сл.
Псевдоконгидрин 690
Псевдокумол (1,2,4-Триметилбензол) 31
Псевдооснования 99
Псевдопельтьерин (Метилгранатанин) 702,
703
Псилоцибин 304, 305
Психомиметические вещества 304
Птеридин (1,3,5,8-Тетраазанафталин) 364 сл.
Птероипглутаминовая (фолевая) кислота 95,
364, 365
Пуберуловая кислота 511
Пулегол 645
Пулегон 644, 645
Пурген 222
Пурины 282, 313, 346, 358 сл., 364
в алкалоидах 358, 362 сл.
в нуклеиновых кислотах 743 сл.
синтез 360
Пуромицин 766
Пурпурин 252
Путресцин 298
Радикалы свободные 224 сп., 537 сл.
изомеризация 545, 561, 563
перегруппировка 553
перекисные 545
реакции 543 сл., 560 сл.
Радикальные реакции 173, 452, 543 сл.
Рацемазы 786
Рацемизация 547, 576 сл., 590, 611
карбанионов 522, 524 сл.
Реактив
Виттига 869, 431, 513
Гриньяра см. Гриньяров реактив
Иоцича 889
Реформатского 400
Реакции
азосочетания 104, 105, 312, 514
алкилирования 37
алкоксимсркурирования 403
аминирования 333
аурировання 456
аутоокисления 552 сл.
Бальца — Шимана 110
Барта 110
бензилирования 61
биуретовая 722
Бородина — Хунсдиккера 549
Вильгеродта 63, 152
Вильсмейера 134, 471, 496
Виттига 431, 440, 444, 535
Вюрца 57, 207, 224, 382, 388, 421, 441,
657
Вюрца — Фиттига 35, 203
Габриэля 689
Гаттермана 110, 150, 203
Геша 123, 150, 294
Гомберга 110, 111, 203, 258, 496
Гомберга — Бахмана 550
Гофмана (см. также Гофмановское рас-
щепление) 327, 602, 603, 606
Гриньяра см. Гриньяров реактив
Дарзана 656
декарбонилирования 467
Демьянова 581
Дильса — Альдера 597, 714
Зайцева 601, 603, 604
Зандмейера 110
Зинина 82
Иоцича 520
Ислера 655
Канниццаро 146, 284, 598
кетовинилирования 532
Кижнера — Вольфа 673, 696
Клемменсена 233
Кневенагеля 189
Кольбе 122, 182, 184, 188, 520, 540,
597, 600
Коха 580, 582
Курциуса 626, 627
Лейкарта 167
Манниха 151, 288, 303, 304, 471, 498,
503, 511
Меервейна 151
металлирования 35, 383, 384,535
Михаэлиса 441, 445
Михаэлиса — Бэккера 434
Несмеянова 110
нингидринпая 722
оксимеркурирования 403
Перкина 147. 189
Прилежаева 173
Пфейффера 441, 445
Реймера — Тимана 122, 129, 131, 294,
511
Реппе 429, 466, 470
Реформатского 389, 400
810
предметный указатель
Реакции
таллирования 418
теломеризации см. Теломеризации
Тищенко 146, 147
Ульмана 57
Фишера 302
Фоссеиа — Педжа — Конанта 435
Фриделя — Крафтса 35, 36, 123, 149,
151, 170, 172, 176, 206, 207, 211,
233, 246, 283, 288, 297, 347, 432,
453, 471, 481, 483, 495, 501, 514, 617
цианирования 500
цианэтилирования 531
Циглера 417
Чичибабипа 302, 333
Чугаева 606
Шимана 53
Шленка 153
Шмидта 627
Шорыгина 383, 384, 520
Шоттен-Бауманна 172, 336
элиминации 409, 589, 601 сл.
Редокс-системы 164, 539, 542, 543
Редупликация 757
Резерпин 684, 710, 711 сл., 718
Резерпиновая кислота 711, 712
Резерпиновый спирт 712, 713
Резинаты 657
Резит, резол, резитол 123, 124
Резорциловая кислота 129, 183
Резорциловый альдегид 129
Резорцин (1,3-Диоксибензол) 126, 127, 128
реакции 129, 183, 223
Рекомбинация радикалов 544
Репелленты 178J
Ретен (2-Изопропил-8-метилфенантрен) 259,
657
Ретиналь (Ретинин) 653, 654
Ретинол (Витамин А,) 653 сл.
Ретропинаколиновая перегруппировка 585,
594, 595, 613, 614, 618, 621 — 623,
635, 648 сл., 650, 651
Рибоза 361, 362, 365, 782, 789
Риботимидин 748
D-Рибоновая кислота 365
Рибонуклеазы 742, 750, 751, 771, 774, 775
Рибонуклеиновые кислоты (РНК) 744, 749,
751 сл., 756 сл., 760, 765, 787
информационная (матричная) 750, 762,
763
рибосом 759
транспортные 751 сл., 754, 761 сл., 767, 769
Рибосомы 7 50, 759
Рибофлавин (Витамин В2) 364, 365 сл., 780,
783 сл.
Риванол (Этакридин) 344
Р-кислота 235
РНК см. Рибонуклеиновые кислоты
Родамины 222 сл., 224
Родопсин 653, 654
Розамины 224
Розанилин (Фуксин) 219, 220, 222
Розоловая кислота 222
Ртутноорганические соединения 400 сл.
реакции 383, 387, 406 сл., 424, 431, 525
571, 592, 593
Рубрен (9,10,11,12-Тетрафенилтетрацен) 262
Рубэритриновая кислота 250
Рутсноцен 482, 505
Рутин (Витамин Р) 371
Салициловая (о-оксибензойная) кислота 168,
169, 182, 183, 184, 194
соли 122
Салициловый альдегид 122, 183
Салол 183, 184
Сальварсан 444
Сапогенины 658, 683
Сапонины 682, 683
Сассафрасовое масло 134
Сафранины 193, 355, 356
Сафрол 134
Сахарин (о-Сульфобензойная кислота, имид)
178, 180
Сахароза, инверсия 772
Светильный газ 9
Свинец, тетраацетат 18
Свинецорганические соединения 422 сл., 431
Свободнорадикальное (триплетное) состояние
539
Свободнорадикальные цепные реакции 546 сл.
Свободные радикалы 127, 159, 163, 164, 224 сл.,
537 сл.
ароксильные 125, 126
аутоокисленпе 552 сл.
перегруппировки 636 сл.
реакции 543 сл.
ЭПР 538
Секстетные (нуклеофильные) перегруппировки
629
Селепорганические соединения 447
Селенофен 447
а-Селинсн 652
Семидин 81
Семидиновая перегруппировка 81
Семихпноны 163, 164, 537 сл.
Сенецпониновая кислота 686, 687
Серин 725, 742, 754, 756
Сернистые красители 165, 309, 322
Серотонин (5-Окситриптамин) 305
Сесквитерпены 515, 639, 652 сл.
Сиккативы 657
Силаны замещенные 419
Силикохлороформ (Трихлорсилан) 428
Спльван (а-МетплфУран) 353
Симазин 357
Симетон 357
Симетрин 357
Син-анти-изомерия 79, 145, 15 4, 1 э7
Синартетическое (анхимерное) содействие 61 ,
619, 620
Синнония 630
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
811
Синтез
Вильштеттера 701
Габриэля 177
Дебнера — Миллера 339
ДНК Гринберг-Манаго 7 55
— Джонсона 679
— Корнберга 755 сл.
— Очоа 755
Пикте 694
Пшорра 695
Реппе 429
Скраупа 339
Синтетазы (Лигазы) 766, 769, 772, 787
Сиреневая кислота 185
Скатол (Р-Метилипдол) 304
Сквален 657, 659, 660, 661, 662
Скипидары 639, 642, 647, 657
Склеропротеины 721
Слизевая кислота 279, 291, 704
Смоляные кислоты 259, 657
Совкаин (Перкаин) 341, 701
Соль
Фремп 159
Цейзе 458, 460, 471, 472
Сольватация
карбанионов 524 сл.
карбкатионов 578 сл.
а,а- и а,л-Сопряжение (Гиперконъюгация)
44, 45, 410, 411, 452, 602, 604
Спектры
УФ 205
ЭПР 534, 535, 538, 539, 570
ЯМР 517, 579, 617
Спиновые магнитные моменты 489
Спин-спиновое расщепление 538
Спирты ароматические 135 сл.
Стелазин 376
Стереохимическое течение реакции 590
Стерины (Стеролы, Стероидные спирты) 658, 660
Стероидные гормоны 658 сл.
Стероиды 259, 658 сл.
конфигурация 667, 683
Стибазил 446
Стибакридиновая кислота 446
Стибенил 446
Стибины 445
Стибозан 446
Стигмастерин 660, 670
Стильбен (1,2-Дифенилэтилен) 207, 257, 286, 596
триплетное состояние 207
Стильбэстрол 208
Стипитатовая кислота 511
Стирол (Вппилбензол) 199, 200 сл., 727
полимеризация 201, 202, 565
Стифнипоная кислота (Трипитрорсзорцин) 129
Стрептомицин 764
Стрептоцид 94
Стрихнин 684, 718, 719
Стрихниновая кислота 718, 719
Стрихнинолоп и стрихиинолоноиая кислота
719 I
Строфантидин 682
Суберон 510
Субтилизин 775
Сукцинимид 271, 292
Сульгин 94
Р-Сультоны 451
Сульфагуанидин (Сульгин) 94
Сульфадиазин 351
Сульфадимезин 95, 351
Сульфазин 351
Сульфазол 95
Сульфамезатин 95, 351
Сульфамеразин (Метилсульфазин) 351
Сульфамидные препараты 95, 351
механизм действия 188
Сульфаниловая кислота 84, 85, 87, 94
амид 94
Сульфапиримидин 351
Сульфатиазол 320
Сульфиновые кислоты 69
Сульфидин 95, 333
Сульфиды ароматические 69
Л1-Сульфобензойная кислота 184
о-Сульфобензойная кислота, имид (Сахарин;
178, 180
Сульфокислоты
бензольного ряда 66 сл.
жирного ряда 549
нафталинового ряда 234 сл.
Сульфоксиды 290
Сульфониевые соли 290
Сульфоны 69, 532
Сульфохлорирование 537, 549
Сурьмяноорганические соединения 444 сл.
сэндвичеобразные соединения 416, 458 сл.,
481 СЛ.
Таллийорганические соединения 418
Таннальбин 187
Таннины 131, 186, 187
Тартразин 313
Таурохолевая кислота 673
Таутомерия
анионотропная 630
диазосоединений 100
изатина 306
имипо-енаминная 334
индоксила 306
кето-енольная 306, 348, 422
лактим-лактамная 306, 348
металлотронная 422
нитрозофеполов 76, 122
нитрофеполоп 119
триазенов 103
Таутомерный эффект (Электромерный эффект,
Г-эффскт) 4 3 сл., 49, 195, 196. 611
в галоидпроизводных бензола 59, 60
в ннтрозосоеДпиепиях 90
и реакциях элиминации 603
и трифеинлоксоний-катионе 135
питрогруплы 93
812______________________________________
ТГФ (Тетрагидрофуран) 276 сл.
Теллурорганические соединения 447
Телогены 566, 567, 568
Теломеризация (Олигомеризация) 419, 423,
537, 557, 560 сл., 568
радикальная 566 сл.
Теломеры (Олигомеры) 566, 568
фторированные 453
Теобромин 358, 362, 684
Теофиллин (Эуфиллин) 362
Терефталевая (n-фталевая, п-бензолдикарбо-
новая) кислота 10, 125, 170, 174,
175, 178
этиловый эфир 28
Терилен (Лавсан) 178
Терпеноиды 639 сл., 646
Терпены 639 сл., 684
Терпин 641, 645, 646
Терпинены 643
Терпинеол 646
Терпинолен 643
Террамицин (5-Окситетрациклин) 262
Терфенил 203
Терферроценил 500
17-Тестанон 666
Тестостерон 674 сл.
1,3,5,8-Тетраазанафталин (Птеридин) 364 сл.
Тетраалкилсвинец 431
Тетраалкоксититаны 146
Тетраарилбор-анион 413
Тетраарилстибониевые соли 44 5
Тетраарилэтаны 227
Тетраацетат свинца 17, 96, 581
Тетраацетилбромглюкоза 745
Тетрабензиламмония соли 167
Тетрабензотетраазапорфин 308
Тетрабприн 711
Тетрабромпиррол 295
Тетрабромфлуоресцеин (Эозин) 223
2,4.4,6-Тетрабромциклогексадиенон (Трибром-
фенолбром) 116, 117
Тетрагидроальстонилии 713
Тстрагидроантрацеп 253
Тетрагидроизохинолин 345
Тетрагидронафталин см. Тетралин
Тетрагидропиразин (Пиперазин) 354, 355
Тетрагидропиридазины 346
Тетрагидропиридин 276
Тстрагидропиррол (Азолидин, Пирролидин)
271, 277, 278, 292, 293
Тетрагидроптероилглутаминовая (5,6,7,8-те-
трагидрофолевая) кислота 777
Тетрагидросалициловая кислота 184
Тетрагидротиазол (Тиазолидин) 323
Тетрагидротиофен (Тиофан) 276, 278, 289 сл.
Тетрагидрофолевая кислота 777
Тетрагидрофурап (ТГФ, Фуранидип, Оксолан)
276 сл.
как лиганд 478 сл.
реакции 388, 389
Тетрагидрофуриловый спирт 284
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Тетрагидрохинолин 341
1,2,3,4-Тетразол 272, 274, 280, 317
Диазониевые соли 318
производные 595
Тетраиодпиррол (Иодол) 295
Тетракарбонил никеля 19
Тетралин (Тетрагидронафталин) 229, 242
гидроперекись 242, 555
реакции 259, 260
Тетралон 242
2,3,5,6-Тетраметил-1 -аминобензол (Аминоду-
рол) 86
1,2,3,4-Тетраметилбензол (Пренитол) 31
1,2,3,5-Тетраметилбензол (Изодурол) 31
1,2,4,5-Тетраметилбензол (Дурол) 31, 93, 534
Тетраметилдиаминометан 503
1,2,3,4-Тетраметилнафталин 480
Тетраметилплатина 457
Тетраметилсвинец 540
Тетраметилфосфопийхлорид 429
Тетраметилэтилен 620
Тетранитрометан 121, 259, 514
Тетрануклеотид 766
Тетрафенилметан 211, 212
9,10,11,12-Тетрафенилтетрацен (Рубрен) 262
Тетрафенилтиофен (Тионессаль) 286
Тетрафтор-Л1-фенилендиамин 56
Тетрафторэтилен 468, 595
Тетрахлор-п-бензохинон (Хлоранил) 160. 266
515
Тетрацен (Нафтацен) 232, 259 сл.
Тетрацианэтилен 121, 209, 534
л-кислота 211
комплексы с переносом заряда 259
Тетрациклины 262 сл.
Тетраэтилсвинец 425, 540, 542
Тетрил 89
Тефлон 449, 453
Тиазин 165, 275, 375
1,3-Тиазол (3-Азатиофен) 95, 276 сл., 318 сл.
493
Тиазолидин (Тетрагидротиазол) 323
Тиазолпниевые соли 320
Тиамин (Витамин БД 323, 351, 35-
производные 786
Тиаминпирофосфат 786
Тигогенин 683
а-Тиенилаланин 289
Тимидин 777
Тимин 349, 351, 743, 755 сл., 764
ртутная соль 748
Тимол (5-Метил-2-изопропилфенол) 6 и
Тиоиндиго и тиоипдигоиды 290, 291
Тиоиндоксил 290, 291
Тиомочевина 320, 350
Тиоиафтеп (Бензтиофен) 273, 290> сл.
Тионессаль (Тетрафенилтиофен) -8b
Тиофан (Тетрагидротиофен) 276, -
Тиофен 22, 35, 273, 278, 285’ сл.
ароматичность 287, 289, 493, *
выделение из бензола 288
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
813
Тиофен
гомологи 286
десульфуризация 288
получение 279, 281, 286, 291
производные 286, 289
реакции 283, 286 сл., 481
Тиофенальдегид 289
а-Тиофенсульфокислота 287
Тиофенсульфоксид 287
Тиофенсульфон 275
Тпоформамид 319, 320
Тиофосфиновые и тиофосфоновые кислоты 433,
436
Тирозин 191 СЛ., 726, 754, 773
реакции 193, 763
Тироксин 192
Титаноценовые соединения 484
Тозилат-анион 616
Тозилаты (п-Толуолсульфокислота, эфиры)
67, 581, 620
5' -Т озил-3' -а цетил дез о к сиа де но з ин 745
5' -Т озил-2', 3 '-из о пр опилиденцитидин 745
Токоферилхиноны 782
Токоферолы (Витамин Е) 370
Тол (2,4,6-Тринитротолуол, Тротил) 71 сл.,
120
Толан (Дифенилацетплен) 208
о-Толидин 204
Толуидиды 89
Толуидины 83, 84, 85, 322
реакции 219
Толуиловые кислоты 168, 169
Толуол 9. 20. 22, 23. 27, 30, 31, 44, 211
анион-радикал 535
аутоокисление 555
галоидирование 52, 172, 547
кислотность водородов 34, 384, 521
комплексы с BF, 34
нитрование 43, 44, 71
окисление 170, 172
реакции 53, 67, 552, 586, 693
фотохимическое бромирование 547
Толуолсульфамид 180
п-Толуолсульфокислота 67
эфиры (Тозилаты) 581, 620
Толуолсульфохлориды 157, 180 .734
Толуольный мускус 71
Трансаминазы 779, 785
Транспортная РНК 751 сл., 761 сл., 767, 769
Трансферазы 772, 776 сл., 787
Треонин 725, 731, 763, 785
Триазены (Диазоаминосоединения) 103, 398,
569
Триазины 3 56 сл.
Триазолы 272, 273, 280, 316
ароматичность 316
реакции 317
Триалкиларсин 440
Триалкилбор 411
Триалкилвисмут 446
Триалкилметоксисилан 419
Триалкилсилан 419
Триалкилфенолы 126
Триалкилхлорсилан 419
Триаминобензол симметрический 131
Триаминопиримидины 364, 365
Триариларснны 441
Триарилметан 37, 38
Триарилметил
анион 228
радикал 225, 227, 408, 537, 552
Триарилфосфораны 572
Триарилхлорметан 228
Трибензиламин 167
Трибензоплрибофуранозид 748
2,4,6-Трибромфенол 118
Трибромфенолбром (2,4,4,6-Тетрабромцикло-
гексадиенон) 116, 117
Трибромфторметан 56
Три-(п-диметиламинофенил)-кар бинол 217
Три-(п-диметиламинофенил)-хлорметан 217
2,4,6-Трииоданилин 87
Трикетогидринден, гидрат (Нингидрин) 722,
728
Тримезиновая (1,3,5-бензолтрикарбоновая)
кислота 20, 175, 179
Тримезитилхром 480
Тримеллитовая (1,2,4-бензолтрикарбоновая)
кислота 175
Триметила люминий 416, 417
Триметиламин 327, 702
1,2,3-Триметилбензол (Гемимеллитол) 31
1,2,4-Триметилбензол (Псевдокумол) 31
1,3,5-Триметилбензол (Мезитилен) 18 сл.,
31, 34, 179
Триметилгалловая кислота 718
Триметилгалловый альдегид 168
2,3,6-ТриМетнлгидрохинон 370
2,4,6-Триметил-1,4-дигидропиридин-3,5-ди-
карбоновая кислота 329
2,4,6-Триметилпиридин (Коллидин) 329, 330,
369
Триметилфенилфосфонийхлорид 429
Триметилфосфин 429
3,4,5-Триметоксибензоилхлорид 711
3,4,5-Триметоксибензойная кислота 711 сл.
2,4,6-Тринитроанизол 51
2,4,6-Тринитроанилин (Пнкрамид) 92, 93, 97
1,3,5-Тринитробензол 49, 70, 71, 120, 131
о-комплекс 50, 51
2,4,6-Тринитробепзол, молекулярные соеди-
нения 121
Тринитрорезорцин (Стифниновая кислота) 129
2,4,6-Трпнитротолуол (Тол, Тротил) 71 сл.,
120
Три-п-нитротрпфенилметан 214
Трииитротрифенплмстил-анион 215
Три-п-нитротрифенилметплнатрий 214
Три-.м-нитротрпфенплфосфииоксид 437
2,4.6 -Тринитрофе нилдиа з оний 105
Тринитрофенол см. Пикриновая кислота
2,4,6-Трийитрохлорбензол 92
814
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ.
Трияуклеотид 765
3,4,5-Триоксибеязойная кислота см. Галловая
кислота
1,2,3-Триоксибеязол (Пирогаллол) 128, 131,
252
1,2,4-Триоксибензол (Оксигидрохинон) 128,
162
1,3,5-Триоксибензол (Флороглюцин) 128, 131,
373, 374
Триоктилфосфина окись 437
Триплетное (свободнорадикальное) состоя-
ние 539
Трипсин 721, 731, 770 сл.
Трипсиноген 773
Триптамин 304, 719
Триптицея 228, 247, 521
Триптофан (Р-Индолипаланин) 291, 302, 304,
726, 728, 763
Трис-азокрасители 107
Тритерпены 657
Тритильная (трифенилметильная) группа 215,
765
Трифениламин 84, 85, 91
Трифениламиногуанипин 317
Трифенилбензол 18, 19, 150
Трифенилгалоидметан 225
Трифенилдибензоилметилфосфоний броми-
стый 430
Трифенилен 66, 206, 264, 265
Три-о-фенилендиарсин 443
Трифенилметан 211, 212, 221, 244
кислотность 214, 521
получение 213
Трифенилметановые красители 89, 218 сл.,
220, 223, 576
Трифенилметил
анион 215
катион 216, 218, 221, 468, 507, 576, 765
радикал (Тритил) 224 сл., 226
Трифенилметила перекись 214
Трифенилметиллитий 383
Трифенилметилнатрий 214, 225, 383
Трифенилметилперхлорат 216
Трпфенилметильная (тритильная) группа 215,
765
Трифенилоксоний 39, 47
катион 135
соли 135
Трифенилпирилиевая соль 369
Трифенилфенацилфосфониевые соли 430, 536
Трифенилфосфин 19, 429, 474
окись 431, 437
радикал 513
Трифенилфосфинметилен (Реактив Виттига)
369, 431, 513
Трифенилфосфит 116
Трифенилхлорметан 212, 214 сл., 218, 576
Трифенилхром 479, 480
Трифенилциклопропенилкатион 492, 573
Триферроценилметильный катион 504
Трифторацетильная защита 455
Трифторуксусная кислота 455, 456, 741
Трифторхлорэтилен 449, 453
Т,Т,У-Трихлоркротоновая кислота 631, 632
2,4,6-Трихлорпиримидин 347
1,1,1-Трихлорпропен 637
2,6,8-Трихлорпурин 360
Трихлорсилан (Силикохлороформ) 428
Трициклопентадиенилметаллы 481
Тропан 698 сл.
Тропилиден см. Циклогептатриен
Тропилийкарбонилхром 468
Тропилий-катион 493, 507 сл., 573, 576
Тропин 698 сл.
Тропиновая кислота 699
Тропинон 699, 701 сл.
Троповая кислота 698, 700
Трополон 491, 510, 511
Тропой (Циклогептатриенон) 491, 509, 510>
Тротил (2,4,6-Тринитротолуол) 71 сл., 126
Тубокурарин 705
Туйяплицины 511
Туннель-эффект 547
Убихиноны 782
Увинол (2,2'-Диокси-4,4'-диметоксибеязофе-
нон) 153
Углеводород Дильса (3'-Метил-1,2-циклопеи-
танофенантрен) 662
Углерод карбеноидный 626
нуклеофильность 60
электрофильность 50
Умбеллиферон 190
Ундекацен 259
Уравнение
Аррениуса 591
Брауна — Окомото 196
Гамметта 194 сл.
Тафта 198 сл.
Урамил 359
Ураты (Мочевая кислота, соли) 358
Урацил 349, 350, 744, 757, 759
таутомерия 351
Уросульфан 94
Фаги 758, 759
ФАД (Флавинадениндинуклеотид) 780, 783
Фарнезен 652
Фарнезилбромпд 657
Фарнезилпирофосфат 659, 660
Фарнезол 652, 657
Фарнохпнон (Витамин К) 239, 240
Фелландрены 643
Феназины (Дибензопиразины) 355
Фенантрен 23, 232, 245, 256 сл., 308,
694
производные 258, 657
Фенантренсульфокислоты 2 58
Фенантренхинон 257
Фенацетин (п-Ацетилфенетидин) _
Фенетидины (о-, п-Этоксианилины)
ПРЕДМЕТНЫ!! УКАЗАТЕЛЬ
815
‘Фенетол (Фенол, этиловый эфир) 132
•Фенизон 311
Фенилазид 103
Фенилаланин 191, 192, 725, 754, 763, 773
Фениламиноантрахиноны 250
43-Фениламинопропионовый альдегид 339
N-Фенилантраниловая кислота 343
Фенилацетамид (Ацетанилид) 89, 92
cz-Фенилацетиламиномалоновый полуальдегид
(Пенальдовая кислота) 323 сл.
Фенилацетилглицин 323
Фенилацетилен 202, 214, 521
Фенилацетилкарбинол 634, 635
Фенилбензилкетон 207
«z-Фенилбензопирилиевые (флавилиевые) соли
369, 372
Фенилбромкарбен 573
3 -Фенил-2-бутилтозилат 615
Фенилвинилуксусная кислота 233
Феяилгидразин 103
Фенилгидразоны 302
Фенилгидроксиламин 74, 75, 78, 80
перегруппировка 133
Фенилглицин 307
Фенилдиазоацетат 552
Фенилдиазоний 318, 506
соли 64, 102, 135, 203
1 -Фенил-1 '-диметиламинометилферроцен 503
1-Фенил-2,3-диметилпиразолон-5 (Антипирин,
Фенизон) 311
-2-Фенил-2,2-диметилэтиловый (неофильный)
радикал 637
Фенилдихлорстибин 446
Фенилендиамины 25, 83, 94 сл., 166
реакции 164 сл., 316, 355
о-Фенилендинитрен 96
Фенилизотиопианат 730
Фенилиодидхлорид 62, 63
Фенилкакодил 442, 443
Фенилкарбиламин 157
З-Фенил-5-карбэтоксиизоксазолин 596
Фенил-катион 135
Фениллитий 56, 383, 440
Фенилмедь 456
N-Фенилметилпнразолон 311
1-Фенил-3-метилпиразолон-5 312
Фенилнатрий 57, 496
Фениловый эфир фосфорной кислоты 116
Фенилпентазол 318
N-Фенилпиразол 310
Фенилселениновая и фенилселеноновая ки-
слоты 447
Фенилсульфаминовая кислота 87
Фенилтиогидантоин 730
Фенилтолилкетоксимы 153
Фенилуксусная кислота 169, 188, 481
производные 390
Фенилферроцен 502
Фенобарбитал 349
Феноксазин 375
Т-Феноксипиридин 335
Фенол(ы) 22, 23, 65, 68, 112 сл., 375
азосочетание 122
алкилирование 123
ацилирование 115
бензоилирование 172
конденсация с формальдегидом 123
— с фурфуролом 284
меркурирование 405
многоатомные 126 сл., 149
нптрозирование 121
окисление 159
получение 57, 69, 555, 624
реакции 100, 115 сл., 165, 222, 510
свойства 293, 497
сульфирование 118
эфиры 132 сл., 422, 509
— аллиловый 501, 597
Фенолазы 781
Феноло-альдегидные полимеры 123
Фенолоальдегиды 122
Фенолокислоты (Оксибензойные кислоты) 122,
182, 185
Фенолоспирты 122
Фенолсульфокислоты 118
Фенолфталеин 176, 222, 223
Фенониевый катион 614 сл.
Феносафранин 355
Фенотиазин 375
Феофорбиды 300
Ферменты 335 сл., 351, 425, 660, 721, 723,
770 сл.
классификация 772
Ферредоксины 781
Феррицианид-анион 461
Феррициний-катион 496
Ферроцен (Дицикл опентадиенилжелезо)
458 сл., 481 сл.
производные 104, 495, 498, 500, 502,
505
Ферроценальдегид 496
Ферроцендиборная кислота 498
Ферроцендикарбоновая кислота 498
Ферроценилазид 500
Ферроценил-1-азонафтол-2 500
Ферроцениламин 498, 500
Ферроценилборная кислота 498, 500
Ферроценилгетероциклы 499, 500
Ферроценилдиазоний 500
1,Г-Ферроценилидендиамин 498
Ферроценил-катион 504
Ферроцениллитий 498, 499, 500
Ферроценилметил-катион 504
а-Ферроценилппридин 498
Ферроценкарбоновые кислоты 498
константа диссоциации 502
Ферроценол 498
Ферроциний-катпон 459
Фиброин 192, 721
Физические методы установления строения
26 сл.
Филлохинон (Витамин Ki) 239, 240
816
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Фиолетовый
Дебнера 218, 219, 222
Лаута 96
Фитилбромид 370
Фитол 241, 301, 370, 653
Фитохинон (Витамин Ki) 240, 241
Флавантрен 254
Флавилиевые (а-фенилбензопирилиевые) соли
369, 372
Флавинадениндинуклеотид (ФАД) 780, 783
Флавинмононуклеотид (ФМН) 780, 783
Флавоны 370, 371, 372
Флороглюцин (1,3,5-Триоксибензол) 128, 131,
373, 374
Флороглюциновый альдегид 131, 373
Флуорен (о,о'-Дифениленметан) 23, 212, 232,
243 сл., 521
реакции 244, 384, 389
Флуоренил-анион 520
Флуоренон 244
Флуоресцеин 222, 223
ФМН (Флавинмононуклеотид) 780, 783
Фолевая (птероилглутаминовая) кислота 95,
364, 365
Фолликулин (Эстрон) 674, 675, 677, 679
Форманилид 496
N-формилпиррол 294
Формилуксусная кислота 191
Формилуксусный эфир 350
а-Формил-|3-этоксипропионитрил 352
Фосген 170, 212, 550
Фосфатазы 773
Фосфат-трансфераза 779
Фосфенистая кислота, производные 426
Фосфиды 535
Фосфинимины 425
Фосфинистая кислота, производные 432, 433
Фосфиниты 426, 433
Фосфинметилены (Метиленфосфораны, Имиды
фосфора) 425, 429, 430, 431
Фосфиновая кислота, производные 426, 428,
433, 434 сл.
Фосфинсульфиды 425
Фосфины 425, 426, 428 сл.
окиси 425, 428, 429, 437, 438, 439
Фосфиты 434 сл.
Фосфицин 435
3'-Фосфоаденозин-5'-пирофосфат 777
Фосфоглюкомутаза 772
Фосфодиэстераза 744
Фосфониевые четвертичные соли 425, 536
Фосфонистая кислота, производные 426, 432,
433
Фосфонитрилы 438
Фосфониты 426, 433 сл.
Фосфония соли 439
Фосфоновая кислота, производные 426, 428
433, 434 сл.
Фосфопротеиды 722, 742
Фосфорилен 429
Фосфорилиды 425. 429, 430, 431, 535, 536
Фосфорорганические соединения 425 сл
стереохимия 438 сл.
Фотогомолиз (Фотодиссоциация) 542
Фотохимические реакции 568, 570
бензофенона 152
галоидирование 547
метопсиокисей 571
ферроценовых соединений 505
Фреоны 448
Фруктоза 779
Фруктозо-1-фосфат 785
Фруктокиназа 779
Фталаминовая кислота 177, 187
Фталевая (о-бензолдикарбоновая) кислота 25,
26, 169, 170, 174 сл., 345
диэтиловый эфир (Диэтилфталат) 178
имид (Фталими1) 177, 271, 308
нитрил 518
реакции 174 сл., 250
хлорангидрид (Фталилхлорил) 178
•м-Фталевая (изофталевая кислота) 170, 174,
175
Фталевый ангидрид 176 сл., 271
получение 176, 206, 229
реакции 177, 178, 187, 222 сл., 246, 248,.
251 сл., 259, 308, 503
Фталеины 222 сл.
Фталил (2-Оксиметилбензойная кислота,
лактон) 178
Фталилхлорид (Фталевая кислота, хлор-
ангидрид) 178
Фталимид (Фталевая кислот, , имид) 177, 271 ,
308
калия 177
меди 498
о-Фталонитрил 308
Фталоцианины 308 сл., 518
Фтивазид 335
Фторбензол 54, 449, 453
о-Фторбромбензол 65
2,6-Фторбромтолуол 31
Фторкарбоновые кислоты 455, 456
Фторорганические полимеры 447, 4 48
Фторорганические соединения 447 сл.
Фтортолуолы 54
Фуксин (Розанилин) 219, 220, 222
Фуксой 222
Фульвен 21, 22, 504, 505
производные 244, 386, 513
Фумаровая кислота 785
Фуран (Оксол) 273, 277, 281 сл.
ароматичность 286, 489, 493, 497
ацетилирование 283
в диеновом синтезе 66, 282
гидрирование 285
меркурированпе 288
нитрование 283
окисление 282, 283
полимеризация 283
получение 281 сл., 291
производные 281 сл.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
817
Фуран (Оксол)
реакции 279, 282 сл., 292, 293
сульфиривание 283
Фуранидин (Оксолон, Тетрагидрофуран)
276 сл.
а-Фуранкарбоновая (пирослизевая) кислота
279, 282, 284, 291
Фуранозы 285
Фурил 284
Фуриловая кислота 284
Фуриловый спирт 284
Фуроин 284
Фурфурол 23, 281, 284
Хелатные соединения 158, 341
Химотрипсин 721, 773
Химотрипсиноген 773
Хпназол (8-Оксихинолин, сульфокислота)
341
Хинальдин (а-Метилхинолин) 339, 342
Хингидрон 163, 534
Хинизарин 238, 252
Хинин 341, 684, 707 сл.
Хининовая (6-метоксихинолин-4-карбоновая)
кислота 707, 709, 710
Хпнинон 708
Хиноксалины 96
Хинолизидин 697
Хинолин (2,3-Бензопиридин, 1-Азанафталин)
23, 274, 340, 341, 345, 707, 708
доказательство структуры 340
производные 340, 341
реакции 340, 341
Хпнолин-4-карбоновая (цинхониновая) ки-
слота 707
Хинолиновая кислота 340, 345
ангидрид 340
8-Хпнолинсульфокислота 340
Хинонимины 164
Хиноны 159 сл., 260, 470, 534
Хинотоксин 708 сл.
Хинуклидин 708, 709
9-Хлоракридин 343,344
а-Хлоралкилфосфоновые кислоты, хлор-
ангидриды 435
Хлораль 435
Хлорамины 68, 397
Хлоранил (Тетрахлор-п-бензохинон) 160, 266,
515
Хлоранилины 83 сл.
Хлорацетальдегид 319, 320
Л1-Х лор-п-ацетиламиностибипоная к исл ота,
Na-соль (Стибозап) 446
Хлорацетофенон 151
Хлорбензойные кислоты 168, 169
/-эффект 194
Хлорбензол 27, 30, 38, 50, 52, 54, 111
Дипольный момент 46
о-комплекс 59
нуклеофильное замещение 57
реакции 60, 64, 134, 135
52 Заказ 6 72
Хлорбензол
реакционная способность 56, 57 сл., 453,
481
скорость гидролиза 194
фосфорилирование 432
7-эффект 46, 59
Т-эффект 59
Хлорбензолсульфокислоты 128
Хлорбромбензолы 27
(З-Хлорвинилкетоны 349
З-Хлорвинилмеркурхлориды 409, 604
Хлордифторметан 449
5-Хлор-7-иодоксихинолин (Энтеросептол) 341
Хлорирование 537, 546 сл., 558
Хлоркарбен 573
Хлоркарборан 415
1-Хлор-2-метилбутан 547
Хлорметилирование 53
а-Хлорнафта.’пш 236
Хлорнитробензолы 31, 347
Хлорониевые катионы 64 сл., 111
Хлорофилл 291, 296, 300 сл., 518, 653
Хлорофос 435, 436
Хлорпентафторбензол 450
а-Хлорперфтормасляная кислота 448
Хлорпиразол 311
Хлорпиридазины 346
Р-Хлорпиридин 573
Хлорпиримидины 347
а-Хлорпиррол 295
Хлорпромазин (Аминазин) 376
7-Хлортетрациклин (Ауреомицин, Биомицин)
262
Хлортолуолы 27, 55, 60
Хлортриптииен 228
З-Хлор-1.3,3-трпфепилпропин 262
Хлоругольный эфир 170, 294
9-(о-Хлорфенил)-9,10-дпгидроакридин 91
Хлорфенолы ИЗ, 118
Хлорферроцен 498
Хлорформилирование 550
Хлорфосфины 428, 431 сл.
Хлорфосфиты 433
Хлорциан, полимеризация 356
п-($-Хлорэтил)-анизол 617
а-Хлорэтилбензол 52
Холановая кислота 672
Холевая кислота 672
Холекальциферол (Витамин Д») 671
Холестан 662, 667
Холестапол (Дипгдрохолестерин) 662, 664,
668 сл.
Холестанон-3 670
Холестерин 658, 660, 661. 662 сл., 666 сл.,
671, 673
конфигурация 676
окислительная деструкция 663, 66э
Холинэстераза 721, 773
Хризен 232, 264
Хромомицины 254
Хромой (2,3-Бенз-у-пироя) 370 сл.
818
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ»
Хромопротеиды 722, 742
Хроморганические соединения 456, 457
Хромосомы 759
цветность азокрасителей 107
Целлюлаза 771, 773
Целлюлоза 123, 727, 771
Цены сэндвпчеобразные 458 сл., 481 сл.
Цепные реакции 126, 543, 546 сл., 556
ингибиторы 164
инициаторы 173
кинетика 562 сл.
механизм 537
Церулоплазмины 781
Цианамид, полимеризация 357
Цианидин 372, 273
Цианиды комплексные 380
Цианиновые красители 321, 342, 343
Циановая кислота, эфиры 356
амид (Цианамид) 357
Цианосорб 153
2-Циан-2-пропилрадикалы 543
Цианур хлористый 357
Цианэтилирование 531
Цикл Кребса 779, 787
Цпклоалкил-катионы 581
Циклобутадиен 492, 493
димеризация 470
комплексы 473, 493
Циклобутадиентрикарбонилжелезо 470, 471
Циклобутан 346, 521
производные 595
Циклогексадиен 20
Циклогексадиендикарбоновая кислота 10
ангидрид 17
Циклогексадиенин см. Дегидробензол
Циклогексан 11, 13, 20, 271, 521
изомеризация 622
производные 211
Циклогександикарбоновая кислота 10
Циклогександион-1,3 (Дигидрорезорцин) 128,
304
Цнклогексанкарбоновая кислота 712, 713
Циклогексанон, производные 612
Циклогексанон-4-карбоновая кислота 645,
646
Циклогексатриен 13
Циклогексен 13, 20, 271, 404
чис-Циклогексилеи-1,2-диуксусная кислота
243
Циклогексил-п-толуолсульфонат 620
Ц иклог ептандион 510
Циклогептатриен (Тропилиден) 507, 508, 521,
649
получение 573
реакции 468
Циклогептатриенон (Тропой) 491, 509, 510
Циклодекан 513
Циклодекан диоп-1,6 512
Циклодекапентаен 474
Циклодецец 513
Циклододекаен 516
Циклододекан 474
Цинлододекатриен 474, 475
Циклододекатриенникель 473, 474
Циклооктадекапонадиен ([181-Аннулен) 516
Циклооктадеканонаеп 516
Циклооктадиен-1,5 474, 475
л-комплекс с металлами 473
Циклооктатетраен 16, 474, 491, 516
л-комплексы с металлами 473
Циклооктатетраен-дианион 535
Циклооктатриен 70?
Циклопентадекаен 516
Циклопентадиен 21, 105, 106, 212, 244, 52*
Гриньяров реактив 480
л-комплексы с металлами 469, 473
производные 468, 469, 484, 493 сл.
реакции 384, 389, 481, 485, 513
л-Циклопентадиенил-л-аллилпалладий 4 84
Циклопентадиенил-анион 272, 469, 483 сл._
494
ароматичность 493 сл., 508
свойства 482 сл., 507, 508, э20
Пиклопентадиенилиды 505 сл.
Циклопентадиенилкарбонилгалогениды 469
Циклопентадиенилкарбонилмарганец 485
Циклопентадиенилкарбонилметаллы 483 сл.^,
Циклопентадиенилмагнийбромпд 49 5
Циклопентадиенилтрикарбонилмарганец 469».
486
Циклопентадиенильные сэндвичеобразные про-
изводные 458 сл., 481 сл.
Циклопентадиенон 106, 470
Циклопентадиенонтрикарбонил-железо 476
Циклопентан 483, 521, 612
Циклопентаногептадиен 512
Циклопентанофенантрен 658
Циклополиены 516 сл.
Циклопропанон, производные 624
Циклопропаны 521, 572
Циклопропенилий-катион 492
Циклосерин (4-Амино-З-изоксалидон) 318, 31»
Циклофаны 208 сл., 211
Цилиатин (Р-Аминоэтилфосфоновая кислота>
425
n-Цимол (п-Метилизопропилбензол) 31
Цингиберен 652
Цинкорганические соединения 381, 389, 39»
Цинхолойпоновая кислота 707
Цинхомероновая (3,4-пиридиндикарбоновая)
кислота 345
Цинхонин 341, 707, 708
Цинхониновая (хинолин-4-карбоновая) ки-
слота 707
Цистеин 725, 763, 775, 779
производные 761, 775, 781
реакции 761, 779
Цистип 725, 729, 730
Цистрон ДНК 769
Цитозин 350, 351, 743, 755 СЛ.
производные 74 3, 765
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
819
Цитохромы 781, 782
Цитраль 640
кротоновая конденсация G41
Цитронеллаль 641, 644
цитронеллол 641
Четырехфтористый углерод 53, 448
Четыреххлористый углерод 53, 122, 568
Шимоза 120
Шиффовы основания (Анилы) 77, 145, 338
Шкала р7<а (Шкала МСЭД) 521
щавелевая кислота 364
производные 313, 649
Щавелевоуксусная кислота 315, 779, 787
Щавелевоянтарная кислота 315
Эвгенол (4-Аллил-2-метоксифенол) 134
Эвипан 349
Экгонин 700, 701
Экзальтация магнитной восприимчивости 490
сл.
Экзоаннулярные водороды 517
Экзонуклеазы 766
Экзопротеазы 773
Электромерный эффект см. Таутомерный
эффект
Электронный парамагнитный резонанс (ЭПР)
538, 539
Электроноакцепторные радикалы 544
Электронодонорные
заместители 197
радикалы 543
Электроотрицательность радикалов, ряд Ха-
раша 407
Электрофильное замещение 31, 36,116, 408 сл.,
442, 496, 510, 582, 588, 592, 600,
609, 611
в азулене 514
в ароматических аминах 86, 90
в бензофеноне 152
в имидазоле 314
в карбанионах 524, 525
•в нафталине 231, 234
н пиразоле 310
в пиридине 331
в пирроле 295
в резорцине 128
в стироле 200
в тетразоле 317
в тиазоле 320
в тиофене 287
в фенантрене 258
в ферроцене 495, 497, 503
в циклопентадиенильном анионе 494
в циклопентадиенильном кольце 483
Электрофильное замещение
мета-ориентация 47
образование л-комплексов 32
Электрофильные реакции
азосочетания 104, 122
меркурирования 405
присоединения 451
сульфирования 67
хлорметилирования 53
Элементоорганические соединения 379 сл.
Энантиомерия 131
Эндопротеазы 773
Энзимы см. Ферменты
Энтеросептол (5-Хлор-7-иоДоксихинолин) 341
Эозин (Тетрабромфлуоресцеин) 223
Эпиандростерон 676
Эпидигидрохолестерин, ацетат 676
Эпикопростерин 669
Эпимеразы 786
Эпихолестанол 669
Эпихолестерин 669
ЭПР (Электронный парамагнитный резонанс)
538, 539
Эргостерин (Провитамин D) 658 сл., 670, 671
Эскулетин 190
Эстеразы 721, 773
Эстрадиол 674, 675, 677
Эстрон (Фолликулин) 674, 675, 677, 679
Этакридин (Риванол) 344
Этанолмеркурхлорид 410
Этилбензол 21, 31, 37, 52, 199
дегидрогенизация 199
окисление 150
Этилен, окись (Оксиран) 271, 277, 397
Этиллитий 383
Этилнитрат 295
Этиловый красный 342
N-Этил-а-окси-! ,2-дигидрохинолин 342
Этилортотитанат 484
N-Этилпиперидин 713
N-Этилхинолин 342
Этилхлорофиллиды 300
Этиобилиановая кислота 666
Этоксианилины (Фенетидины) 83, 132, 133
Эуфиллин (Теофиллин) 362
Эфедрин 685
Эфиры ароматические 134
Эффект
Натана — Бэкера 45
поля 194
Яблочная кислота 190, 191, 684, 785
ЯМР-спсктры 254, 580, 585
Янтарная кислота 279, 286
ангидрид 271
Янтарный диальдегид 699
52*
СОДЕРЖАНИЕ КНИГИ 2
АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
Бензол и его производные ................................... 9
Бензол и его строение......................................... 9
Синтез бензола и его производных из алифатических и али-
циклических углеводородов ........................... 18
Превращения бензола в алифатические и алициклические
соединения .......................................... 21
Источники ароматических углеводородов................ 22
Изомерия замещенных бензолов......................... 23
Физические методы установления строения органических моле-
кул. III................................................. 26
Вопросы номенклатуры бензольных производных .... 29
Реакции электрофильного замещения в бензольных угле-
водородах ........................................... 31
Ориентация замещения ................................ 38
Причина явлений ориентации........................... 41
Гиперконъюгация и орто- и пара-ориентация ........... 44
Ориентирующий эффект галоидного заместителя .... 45
Мета-ориентация ..................................... 47
Ориентация нуклеофильного замещения.................. 48
Галоидпроизводные бензольного ряда........................... 51
Химические свойства галоидпроизводных ряда бензола . 56
Соединения с многовалентными галоидами................... 62
Дегидробензол (циклогексадиенин, «бензин»)................... 65
Сульфокислоты и другие ароматические сернистые соединения 66
Реакция сульфирования в ряду бензола................. 66
Функциональные производные сульфокислот ............. 67
Реакции сульфокислот с замещением сульфогруппы ... 69
Другие сернистые функции ароматического ряда .... 69
Нитросоединения ряда бензола................................. ^0
Функции, образуемые неполным восстановлением питросоединений 74
Нитрозобензол......................................
Фенилгидроксиламин................................... 1$
Азоксибензол ......................................
Азобензол............................................ 79
Ароматические амины .........................................
Основные свойства ароматических аминов..............
Реакции электрофильного замещения в бензольном ядре
аминов .............................................
Замещение в аминогруппе.............................
Ароматические вторичные и третичные амины...............
82
84
86
88
90
СОДЕРЖАНИЕ
821
Нитро анилины......................................... 92
Анилинсульфокислоты .................................. 94
Ароматические диамины.................................... 95
Диазосоединения.............................................. 97
Реакции диазосоединений без выделения азота........ ЮЗ
Реакции диазосоединений с выделением азота............ Ю8
Фенолы ..................................................... 112
Реакции гидроксила фенолов ........................... Ц5
Реакции ароматического ядра фенола.................... Иб
Молекулярные соединения с переносом заряда.............. 121
Ароксилы................................................ 125
Многоатомные фенолы . . 126
Диоксибензолы........................................ 126
Клатратные соединения гидрохинона....................... 129
Анса-соединения гидрохинона. Антропэнантиомерия. I . . . 130
Полиоксибензолы................................... 131
Эфиры фенолов .......................................... 132
Простые чистоароматические эфиры........................ 134
Трифенилоксониевые соли ................................ 135
Ароматические спирты ....................................... 135
Фенолоспирты и их простые эфиры............................. 136
Ароматические альдегиды ................................... 137
Синтез ароматических альдегидов..................... 138
Свойства ароматических альдегидов.................... 141
Ароматические кетоны ...................................... 148
Синтез ароматических кетонов........................ 149
Свойства ароматических кетонов....................... 150
Оксимы кетонов и бекмановская перегруппировка .... 153
Дикетоны................................................ 157
Хиноны...................................................... 159
Реакции бензохинонов ................................ 160
Хингидрон и семихинон ................... 163
Хинонимины.............................................. 164
етерофункциональные амины с аминогруппой в боковой цепи . - 167
Ароматические карбоновые кислоты........................... 169
Способы введения карбоксила в ароматическое ядро . . 169
Бензойная кислота и ее функциональные производные .... 171
Поликарбоновые ароматические кислоты.................... 174
Замещенные карбоновые кислоты........................... 180
Оксибензойные кислоты (фенолокислоты)....................... 182
Ароматические аминокислоты.................................. 187
Карбоновые кислоты с карбоксилом в боковой цепи ............ 188
Передача влияния заместителей через бензольное кольцо, кор-
реляционные уравнения................................ 19°
Ароматические углеводороды с непредельной боковой цепью 199
Многоядерные ароматические соединения
Соединения с изолированными бензольными ядрами...........
Дифенил (или бифенил) ...............................
А тропизомерия. II ......................................
Дифенилеп (или бифенилен) ...........................
1,2-Дифепилэтап. 1,2-дпфенилэтилен, 1,2-дифепилаце-
тилсн .............................................
203
203
205
206
207
822 СОДЕРЖАНИЕ
Циклофаны ....................................... . 208
Полифенилметаны ................................... 211
Фталеины, флуоресцеин, розампны и родамнны......... 222
Свободные радикалы ряда трифенил.метила............ 224
Конденсированные ароматические системы..................... 228
Нафталин и его производные............................. 228
Нафтохиноны......................................... 237
Гидронафталины ..................................... 241
Аценафтен, инден, флуорен.............................. 243
Антрацен и его производные ........................... 244
Антрахиноп п его производные........................ 248
Индантреповые и полицпклокетоновые красители .... 254
Фенантрен.............................................. 256
Высшпе конденсированные ароматические углеводороды . . 259
Ацены .............................................. 259
Ангулярные высшпе ароматические конденсированные
углеводороды........................................ 264
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Пятичленные гетероциклы .................................. 278
Циклы с одним гетероатомом............................... 278
Фуран ................................................ 281
Конденсированные системы с фурановым циклом .... 285
Тиофен ............................................... 285
Тетрагпдротиофен (тиофан)............................. 289
Бензтиофен и его производные.......................... 290
Пиррол ............................................... 291
Порфин. Порфирины..................................... 296
Индол (бензпиррол) ................................... 302
Индиго ............................................... 305
Фталоцианины.......................................... 308
Карбазол ............................................. 308
Пятичленные гетероциклы с несколькими одинаковыми гетеро-
атомами .......................................... 309
Пиразол ............................................ 309
Имидазол ............................................. 313
Биотины . ............................................ 315
Триазолы ............................................. 316
Тетразол ..... ....................................... 317
Пентазол........................................... 318
Пятичленные гетероциклы с двумя разными гетероатомами . . 318
Изоксазол............................................. 318
Оксазолы ............................................. 319
Тиазол................................................ 319
Шестичленные гетероциклы .................................
Гетероциклы с одним атомом азота......................
Пиридин ..........................................
Оксипиридины и аминопиридпны......................
Физиологически активные вещества с пиридиновым ядром
Хинолин и его производные.........................
Акридин ..........................................
Изохинолин - .....................................
Шестичленные гетероциклы с двумя атомами азота (диазины)
Пиридазин.............................................
Пиримидин . . ....................................
Пиразин ..........................................
Шестичленные гетероциклы с тремя атомами азота........
Азотистые бигетероциклы ..............................
326
326
326
334
335
339
343
345
345
346
346
354
356
358
СОДЕРЖАН ИЕ.82 3
Пурин и его производные ........................... 35g
Нуклеотиды......................................... 3g!
Пуриновые алкалоиды................................ 332
Кинетин............................................ 353
Птеридины........................................... 354
Фолевая кислота ................................... 305
Рибофлавин......................................... 305
Шестпчленные гетероциклы с атомом кислорода............. 337
Производные у-пирана .............................. 307
Хромоп, флавон п родственные им вещества .......... 37Q
Антоцианидины....................................... 372
Шестичленные гетероциклы с двумя разными гетероатомами 375
ЧАСТЬ ВТОРАЯ
Элементоорганические соединения ........................... 379
Активные металлоорганические соединения I, II и III групп 380
Первая группа периодической системы элементов.......... 332
Вторая группа периодической системы элементов.......... 337
Третья группа периодической системы элементов.......... 4ц
Четвертая группа периодической системы элементов....... 419
Пятая группа периодической системы элементов........... 425
Шестая группа периодической системы элементов.......... 447
Седьмая группа периодической системы элементов......... 447
Металлоорганические соединения переходных металлов . . . 450
Карбонилы металлов.................................. 463
Олефиновые л-комплексы переходных металлов.......... 471
Металлоарены........................................ 477
Металлоцены ........................................ 481
Небензоидные ароматические системы......................... 487
Диамагнитные свойства ароматических систем............. 488
Ароматические системы с двумя л-электронами............ 492
Ароматические системы с четырьмя л-электронами......... 492
Ароматические системы с шестью л-электронами........... 493
Циклопентадиенил-анион.............................. 494
Тропилий-катион..................................... 507
Ароматические системы с числом л-электронов более шести . . 512
Азулен.............................................. 512
Аннулены ........................................... 517
Порфирины........................................... 518
Карбанионы................................................. 519
Свободные радикалы ........................................ 537
Электронный парамагнитный резонанс..................... 538
Образование свободных радикалов ....................... 539
Реакции свободных радикалов............................ 543
Элементарные акты .................................. 543
Свободнорадикальные цеппые реакции..................... 546
Гомолитическое замещение............................ 546
Реакции аутоокислепия .............................. 552
Радикальное присоединение по двойным связям......... 556
Реакции теломеризации (олигомеризации)................. 560
Кинетика цепных реакций................................ 562
Определение констапт передачи цепи в радикальной теломе-
рпзацпп.......................................... 566
Карбены ................................................... 570
Карбониевые попы (карбкатионы)............................. 575
Гидридные перемещения .............................. 582
Механизмы химических реакций............................... 588
Структура переходного состояния ....................... 589
824
СОДЕРЖАНИЕ
Реакции элиминации .......................................
Перегруппировки...........................................
Перегруппировки с изменением углеродного скелета ....
Перегруппировки с карбенами и карбеноидами в качестве про-
межуточных продуктов .................................
Перегруппировки с перемещением л-связей...............
Гидридные перемещения ................................
Гомолитические перегруппировки .......................
Изопреноиды ..............................................
Терпены и терпеноиды..................................
Монотерпены и терпеноиды..........................
Сесквитерпены и сесквитерпеноиды..................
Дитерпены, тетраизопреноиды и каротиноиды.........
Смоляные кислоты .....................................
Стероиды .............................................
Стерины (стероидные спирты).......................
Конфигурация стероидов ...........................
Желчные кислоты ..................................
Стероидные гормоны................................
Генины и сапонины.................................
Алкалоиды гетероциклического ряда.........................
Алкалоиды с пятичленным гетероциклом с одним атомом
азота ............................................
Алкалоиды с системой двух конденсированных пятичлен-
ных циклов с одним общим атомом азота.............
Алкалоиды с пятичленным гетероциклом с двумя атомами
азота ............................................
Алкалоиды с шестичленным гетероциклом с одним атомом
азота ............................................
Алкалоиды с системой двух конденсированных шестичлен-
ных циклов с общим азотом.........................
Алкалоиды с конденсированными пяти- и шестичленными
гетероциклами и одним атомом азота................
Алкалоиды с двумя конденсированными шестичленными
гетероциклами с одним атомом азота................
Алкалоиды с двумя изолированными гетероциклами с двумя
атомами азота ....................................
Алкалоиды с двумя конденсированными шестичленными
гетероциклами с общим атомом азота и одним изолиро-
ванным гетероциклом...............................
Алкалоиды с тремя конденсированными гетероциклами
с двумя атомами азота.............................
Белки (протеины) .........................................
Методы индивидуализации белков ...................
Методы установления строения белков...............
Синтез пептидов и белков .........................
Вторичная и третичная структура белков ...........
Нуклеотиды и полинуклеотиды...............................
Синтез белка в организме .........................
Синтез нуклеотидов ...............................
Полинуклеотиды .......................................
Ферменты..................................................
Классификация ферментов ..............................
Гидролазы ........................................
Трансферазы ......................................
Оксидоредуктазы...................................
Лиазы.............................................
Изомеразы .......................................
Синтетазы........................................
Предметный указатель.....................................
601
609
609
625
629
635
636 ,
639
639
640
652
653
657
658
660
667
672
673
682
684
685
686
687
690
697
698
702
703
707
710
721
727
728
734
739
743
743
748
749
770
772
773
776
780
785
786
787
788
Читатель!
Просьба исправить на указанных страницах формулы,
поврежденные при печати
Стр.
71 2 св.
226 1 св.
284 2 сн.
287 1 сн.
291
292
295
337
375
3 сн.
3 св.
1 сн.
1 св.
4-R"—C=sC—R" ►
\ J\
NH СН2ОН
СН2ОС2Н5
H2N I CH2NH2
\/\/
)
Н3С N
ш
375 1 св.
3 св.
393 1 св.
395 1 св.
2 св.
405 1 св.
406 5 св.
579 1 сп.
624 1 св.
660
675
2 сп.
I св
^\Z\Z\
XMgO Н R
R-C-CH2R' +
R
I
+ С-СН2Н‘
II
о
-О CH.-C-0-Hs -сн, -с\ ..
II г
С)
CH3C-O\Hg
II
О /2
----► Alk+SbF;
П-СН=С-СН2С1
:О:
-С Н.ч
ХСН;1
тестостерон
П родолжение
Опечатки, указанные в этой таблице, исправлены в тексте
Фотографировал Семенюченко Владимир
chem_vova@mail.univ.kiev.ua; vova2002@mail.ru
I" Л.Н. НЕСМЕЯНОВ, Н.А.НЕСМЕЯНОВ
| НАЧАЛА
' ОРГАНИЧЕСКОЙ ХИМИИ
II