/



























































































































Text
АКАДЕМИЯ НАУК УКРАИНСКОЙ ССР
ИНСТИТУТ ОБЩЕЙ И НЕОРГАНИЧЕСКОЙ ХИМИИ
Ю. Я. ФИАЛКОВ, В. Ф. ГРИЩЕНКО
ЭЛ ЕНТРОВЫ ДЕЛЕНИЕ
МЕТАЛЛОВ
ИЗ НЕВОДНЫХ
РАСТВОРОВ
КИЕВ НАУКОВА ДУМКА 1985
УДК 541.135.2+541.138:621.357
Электровыделение металлов из неводиых растворов t
Фиалков Ю. Я, Грищенко В. Ф —Киев: Наук. думка, 1985,—
240 с.
В монографии рассмотрены вопросы теории неводных элек-
тролитных растворов, имеющие значение при формировании
композиций для электроосаждения металлов. Представлен об-
зор по катодным процессам при электроосаждении металлов
различной природы из неводных сред. Отдельно рассматрива-
ются анодные процессы и вопросы коррозии металлов в невод-
ных средах. Заключительный раздел книги посвящен описа-
нию методов электровыделения металлов всех групп перио-
дической системы элементов из неводных растворов.
Для специалистов в области электрохимии, химии раство-
ров, гальваники и гидрометаллургии, а также для преподава-
телей и студентов соответствующих вузов.
Табл. 15. Ил. 24. Библиогр.: с. 203—238 (1277 назв.).
F
Ответственный редактор В. И. Шаповал
Рецензенты И. А. Шека, А. Л. Левинскас
Редакция химической литературы
1805000000-132
Ф------------------216-85
М221(04)-85
© Издательство «Наукова думка», 1985
ПРЕДИСЛОВИЕ
Из более чем восьмидесяти металлов, входящих в периодическую систему хи-
мических элементов Д. И. Менделеева, едва лишь три десятка могут быть вы-
делены электроосаждением из водных растворов. Стремление распространить
наиболее удобный и технологичный метод выделения и разделения элемен-
тов — электролиз — на все металлы закономерно привело исследователей и
технологов к неводным растворам.
Принципиальная возможность электровыделения металлов из неводных
растворов была показана более столетия назад. Однако прикладное значение
данный метод приобрел лишь в последнее время. Связано это с несколькими
обстоятельствами. Прежде всего лишь за последние два-три десятилетия теория
электролитных неводных растворов поднялась на качественно новый уровень, что
позволяет вплотную приблизиться к решению проблемы направленного подбо-
ра композиций для электроосаждения металлов; произошло существенное рас-
ширение круга неводных растворителей, доступных исследовательской и про-
мышленной практике, и наконец, химическая и электрохимическая технология
преодолела известный психологический барьер, связанный со своеобразием и
необычностью неводных растворов по сравнению с водными.
В настоящее время исследования по теории и практике электровыделения
металлов из неводных растворов развиваются чрезвычайно интенсивно.. Обра-
щение к неводным средам не только позволяет осуществлять низкотемператур-
ное электровыделение любого металла, но в ряде случаев приводит к образо-
ванию покрытий с такими эксплуатационными и декоративными характеристи-
ками, какие электролизом из водных растворов могут быть достигнуты с боль-
шим трудом, а то и вовсе недостижимы.
По-видимому, можно считать закономерным, что эта книга написана и вы-
ходит в свет именно в Киеве, где еще в начале века В. А. Плотниковым были
выполнены пионерские исследования по электроосаждению металлов из невод-
ных растворов, которые сразу привлекли внимание химиков прежде всего своей
несомненной оригинальностью, а также тем, что в этих работах была показа-
на принципиальная возможность электроосаждения из неводных растворов ще-
лочных металлов и алюминия — именно тех элементов, для которых пробле-
ма электроосаждения представляется наиболее актуальной.
Исследования по электроосаждению металлов из неводных растворов ин-
тенсивно велись школой В. А. Плотникова вплоть до 30-х годов, на исходе
которых В. А. Плотниковым совместно с И. А. Шекой и 3. А. Шекой была вы-
пущена монография «Электролитическое выделение металлов из неводных раст-
воров» — единственное до настоящего времени обобщение в мировой литерату-
ре, посвященное названной проблеме. Сегодня исследования теоретических и
прикладных аспектов неводной гальваники ведутся у нас в стране достаточно
широко. Помимо киевской электрохимической школы активное участие в разра-
ботке данной проблемы принимают научные силы Литвы (А. Л. Левинскас),
Ростова-на-Дону (В. П. Григорьев) и др.
Эта книга появилась как результат многолетнего сотрудничества кафедры
физической химии Киевского политехнического института и Института общей и
неорганической химии АН УССР — двух учреждений, в которых возникли и за-
тем развивались отечественные работы по электроосаждению металлов из не-
водных сред. Главы 1 и 3, а также раздел 2.1 написаны доктором химических
наук Ю. Я. Фиалковым; разделы 2.2—2.5 и глава 4 — кандидатом химических
наук В. Ф. Грищенко.
3
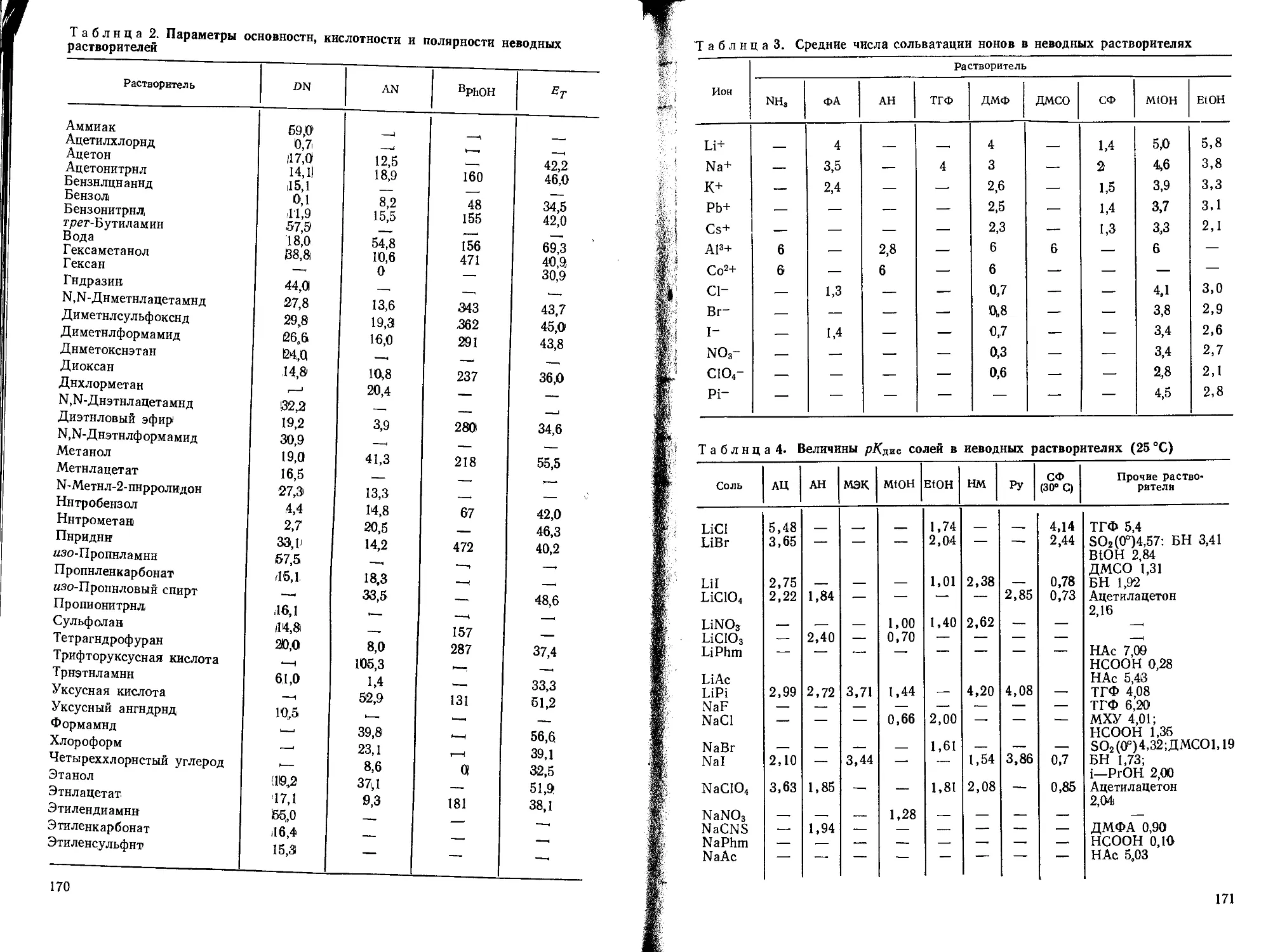
СПИСОК СОКРАЩЕНИЙ
АН — ацетон
АН — ацетонитрил
Б — бензол
БН — бензонитрил
ВЮН — бутанол
ГМФТА — гексаметиленфосфортриамид
Г — гексан
ДМА — диметиланилин
ДМАА — диметилацетамид
ДМСО — диметилсульфоксид
ДМФ — диметилформамид
MtOH — метанол
МАА — метилацетамид
МЭК — метилэтилкетон
НБ — нитробензол
НМ — нитрометан
Ру — пиридин
РгОН — пропанол
ПК — пропиленкарбонат
СФ — сульфолан
ТГФ — тетрагидрофуран
ТФУ — трифторуксусная кислота
ТХУ — трихлоруксуснаи кислота
НАс — уксусная кислота
ФА — формамид
EtOH — этанол
ЭГ — этиленгликоль
ЭДА— этилендиамин
ЭК — этиленкарбонат
Глава 1
НЕКОТОРЫЕ ПРОБЛЕМЫ ТЕОРИИ
ЭЛЕКТРОЛИТНЫХ НЕВОДНЫХ РАСТВОРОВ
1.1. ОБЩАЯ СХЕМА РАВНОВЕСИЙ В НЕВОДНЫХ
РАСТВОРАХ ЭЛЕКТРОЛИТОВ
УСЛОВИЯ ВОЗНИКНОВЕНИЯ ЭЛЕКТРОЛИТНОГО РАСТВОРА
1.1.1. Развитие представлений об основных типах
межмолекулярных процессов в растворах
История учения о растворах (примерно до 20-х годов нашего ве-
ка) детально изложена в работе Ю. И. Соловьева [468]. Здесь мы
кратко остановимся на некоторых • принципиальных достижениях
в этой области за последние десятилетия [533].
Изучение неводных растворов сыграло существенную роль в
разработке основных положений современной теории растворов.
Именно изучение неводных растворов позволило установить вза-
имосвязь физической и химической теорий растворов. При этом
было показано, что химическая теория справедливо отстаивала
тезис о химическом взаимодействии компонентов жидкой системы
как необходимом условии образования раствора вообще и элек-
тролитного раствора в особенности; физическая же теория раство-
ров бесспорно доказала плодотворность распространения положе-
ний молекулярно-кинетической теории и классической термодина-
мики на жидкие системы. В последние десятилетия в основном
завершен синтез этих двух генеральных направлений в теории
растворов.
Пионерами новой главы в учении о растворах стали отечест-
венные ученые И. А. Каблуков и В. А. Плотников. С именем
И. А. Каблукова связаны первые работы, в которых концентраци-
онный ход изотерм электропроводности объяснялся химическими
особенностями системы [183]. Основная заслуга В. А. Плотникова
и возглавляемой им школы заключается в привлечении и соедине-
нии наиболее передовых для того времени теорий электролитиче-
ской диссоциации, химической теории растворов и химии комп-
лексных соединений [453, 534].
Показав на ряде примеров, ставших классическими, что комп-
лексообразование существенно улучшает условия возникновения
электролитного раствора, В. А. Плотников, во-первых, применил
для изучения комплексообразования в растворах весь арсенал ме-
тодов теории электролитической диссоциации, а во-вторых, не
только не игнорировал подобно большинству представителей хи-
мической теории растворов «диссоциирующую силу» растворителя
(свойство, позже отождествленное с диэлектрической проницае-
5
мостью), но и достаточно подробно проанализировал случаи, ког-
да это свойство выступает на первый план в определении электро-
литных характеристик раствора, либо занимает подчиненное по
сравнению с чисто химическими взаимодействиями место.
Отметим роль выдающегося физикохимика Л. В. Писаржевско-
го в изучении электролитных неводных растворов [395]. Использо-
вав весьма остроумно прием составления гальванических цепей с
участием неводных растворителей, Л. В. Писаржевский определил
константы равновесия и термодинамические характеристики ряда
обменных реакций в неводных растворах.
Сближению физической и химической теорий растворов спо-
собствовало также интенсивное развитие теорий кислот и основа-
ний. Важную роль здесь сыграла протолитическая теория Брен-
стеда (библиографию см. в [614]), где вскрывается и обосновыва-
ется связь силы протонных (Н—) кислот с диэлектрической про-
ницаемостью растворителя.
Вопрос о влиянии физических (прежде всего, диэлектрической
проницаемости) и химических (энергии сольватационных процес-
сов) факторов на силу электролитов в растворах с классической
законченностью был решен Н. А. Измайловым [173]. Выдающийся
ученый создал основы общей теории электролитической диссоци-
ации, которая логично и естественно объясняет и учитывает физи-
ческие и химические свойства системы. Некоторые положения тео-
рии Н. А. Измайлова в той мере, как диктуется проблематикой
данногй книги, будут рассмотрены в разделе 1.2.
Весьма значительный вклад в развитие современной теории
электролитных растворов внесли результаты работ исследовате-
лей школ К. П. Мищенко [347] и Г. А. Крестова [228], в которых
получили плодотворное развитие термодинамика процессов раст-
ворения солей и ионной сольватации.
1.1.2. Гомомолекулярные (ассоциативно-
диссоциативные) процессы
В индивидуальном состоянии подавляющее большинство хи-
мических соединений ассоциировано. Рассмотрение причин, а так-
же физических и химических аспектов гомомолекулярной ассо-
циации, каким бы интересным оно ни представлялось, выходит за
рамки задач этой книги. Поэтому, адресовав читателя к соответ-
ствующей литературе [617, 616], рассмотрим равновесия, устанав-
ливающиеся в системе ассоциированный компонент (Аж) — индиф-
ферентный растворитель:
(S) (S) (S)
Ад. 2А*/2 .. • ^хА. (1—1)
Процесс (1—1), называемый гомомолекулярной диссоциацией,
может останавливаться на любой из стадий. В концентрирован-
ных растворах в основном сосуществует несколько форм различ-
ной степени ассоциации; в разбавленных растворах обычно уста-
навливается мономер-димерное равновесие.
6
Поскольку каждое индивидуальное химическое соединение под-
вергается автоионизации (частным случаем которой является авто-
протолиз), добавление растворителя к Аж приводит к сдвигу в ту
или иную сторону этого процесса:
(S) ,
А2^А+ + А“, (1-2, а)
<s> j.
(НА)2 НА+ + А-. (1—2, б)
. Так, процесс распада РегС16 на мономеры правильнее пред-
ставить схемой Fe2Cl6^±:FeC12++FeC14_ (подробнее о влиянии ра-
створителя на константу процесса (1—2, а) см. в разделе 1.5.3).
Таким образом, процесс гомомолекулярной диссоциации способ-
ствует образованию как нейтральных молекул (1—1), так и ионов
(1-2).
Природа химической связи в гомомолекулярных ассоциатах,
как и во всех иных химических соединениях, вступивших с раст-
ворителем в универсальную либо специфическую сольватацию,
обусловлена перекрыванием молекулярных орбиталей взаимодей-
ствующих молекул (химическая составляющая энергии химиче-
ской связи) и электростатическим взаимодействием. Поэтому энер-
гия химической связи в гомомолекулярных ассоциатах представ-
ляет собой сумму соответствующих составляющих:
Fa с = Fxhm Ч- Еэл.ст (1 3)
В первом и достаточно удовлетворительном приближении элек-
тростатическая составляющая в случае гомомолекулярных ассо-
циатов может рассматриваться как энергия диполь-дипольного
взаимодействия:
п2
£эл.ст = — AGd-d = a.d-d — [кДж/моль], (1—4)
rd-<F
где р. — дипольный момент мономерной молекулы (без учета вли-
яния растворителя на последний) ; rd-d — расстояние между моле-
кулами в димере; е — диэлектрическая проницаемость; ad-d — ко-
эффициент пропорциональности, связанный с выбором размерно-
стей и равный 120,7.
Растворители, не вступающие с растворителем в химическое
(специфическое) взаимодействие (далее такие растворители бу-
дем называть универсальными), влияют лишь на электростатиче-
скую составляющую общей энергии связи в ассоциате. Поскольку
свободная энергия процесса жестко связана с его константой рав-
новесия AG = —RT In А, между логарифмом константы димер-мо-
номерного равновесия и обратной диэлектрической проницаемо-
стью (1/е) в универсальных средах должна соблюдаться прямо
пропорциональная зависимость:
1п/Смон = п + 4. (1-5)
7
На рис. 1 эта зависимость иллюстрируется растворами уксус-
ной кислоты (равновесие (СН3СООН)2^2СН3СООН) в различ-
ных универсальных растворителях (к сожалению, привести пример
влияния растворителя на мономер-димерное равновесие соли за-
труднительно из-за граничащей с невозможностью трудностью
подбора универсального растворителя для соли).
Химическое взаимодействие (в литературе встречаются равно-
употребительные термины «специфическое взаимодействие», хотя,
Рис. 1. Зависимость логарифма
константы моиомеризации уксусной
кислоты от диэлектрической прони-
цаемости растворители:
1 — пар; 2 — Г; 3 — CCL*; 4 — сероугле-
род; 5 —ХВ; 6 НБ.
строго говоря, последнее включает
помимо химического и универсаль-
ное взаимодействие, «специфиче-
ская сольватация», «неуниверсаль-
ная сольватация» и т. п.) приводит
к тому, что распад полимерных мо-
лекул происходит даже в средах с
низкой диэлектрической проницае-
мостью:
5txASs, (1-6)
Аж -J- sxS -<?-
(S — молекула растворителя; схе-
ма (1—6) не учитывает молекуляр-
ное состояние растворителя и изме-
нение этого состояния при сольва-
тации).
Влияние химических свойств растворителя на молекулярное
состояние растворенного соединения можно проследить на боль-
шом числе примеров. Так, А1Вг3 димерен в бензоле, но мономерен
в гораздо более химически активном по отношению к нему и лишь
незначительно превышающем бензол по величине диэлектрической
проницаемости бромистом этиле [798] (о природе взаимодействия
компонентов и последней системы см. разделы 1.5.2 и 1.5.3). Отно-
шение равновесных концентраций мономера и димера в растворах
карбоновых кислот в каждом данном растворителе увеличивается
с повышением силы кислоты [558, 15]. Имеются все основания
ожидать аналогичного поведения апротонных (L)-кислот.
1.1.3. Гетеромолекулярные процессы (образование
продуктов присоединения)
Вследствие сольватации соединения А, В,..., растворенные в ра-
створителе S, находятся в виде сольватных комплексов ASs и BS<.
В тех случаях, когда А и В вступают в химическое взаимодейст-
вие, устанавливается равновесие:
mASs + nBSf 5±AmBnSr (ms -[-nt —r) S. (1—7)
Процесс (1—7) в общем случае сопряжен с десольватацией,
так как образование продукта присоединения происходит за счет
взаимного заполнения компонентами координационных вакансий.
8
т
Образование неионогенного соединения из неионогенных ком-
понентов является первой непременной стадией этого типа вза-
имодействий в растворе. Химия координационных соединений да-
ет многочисленные примеры подобного рода взаимодействий. Так,
хлорид алюминия в нитробензоле образует с пиридином эквимо-
лекулярный продукт присоединения АЮз-Ру.
Как и в случае гомомолекулярной ассоциации изменение сво-
бодной энергии системы в рассматриваемом случае гетеромолеку-
лярной ассоциации состоит из двух
составляющих (уравнение (1—3)).
Электростатическая составляющая
в данном случае
— АСэлект = RT In Кпр = ad-d ,
. rd~<fi
(1-8)
здесь обозначения такие же, как и
в уравнении (1—4).
Таким образом, в универсальных
Рис. 2. Зависимость логарифма
константы образования комплекса
12-Ру от обратной диэлектрической
проницаемости растворителя:
1 — ЦГ; 2 — СС1,; 3 — л-ксилол; 4 — Б;
S — м-ксилол.
средах в случае гетеромолекуляр-
ной ассоциации также должна со-
блюдаться прямолинейная зависи-
мость In К от 1/е. Примером, ил-
люстрирующим выполнение такой
зависимости, может быть электро-
нодонорно-акцепторное (ЭДА) взаимодействие в системе 12—Ру,
зависимость In К которого от 1/е приведена на рис. 2.
• я. Для исследования стехиометрии и термодинамики взаимодей-
ствия в системах А — В — (S) успешно используют методы физи-
ко-химического анализа [19, 530, 28].
1.1.4. Процесс ионизвции
(образование ионных пар)
Существует принципиальное различие между электролитами-
ионофорами типа K+CJ-, в которых ионы уже находятся в исход-;
НОМ растворяемом соединении, и электролитами-ионогенами, в!
которых ионная связь возникает в результате специфического вза-'
имодействия с растворителем. В последнем, наиболее распростра-
ненном типе электролитов переход продуктов присоединения не-
посредственно в разделенные ионы, т. е. процесс AB^K++L~, не-
возможен из-за весьма высокого энергетического барьера такого
процесса. Поэтому процессу электролитической диссоциации
практически всегда предшествует процесс ионизации_ДСИНОНИМЫ:
образование ионных пар, образование ионных ассоциатов):
АтВп5г^К^Ц-5г + (г-/)5. (1-9)
9
Примерами подобного рода процессов (без учета сольватации
участников равновесия) могут быть реакции: НАс-Ру^РуН+-Ас“,
А1С13-Ру^А1С12Ру+-Cl-
Энергию, необходимую для перестройки связей в продукте при-
соединения, система заимствует из энергии процессов специфиче-
ского взаимодействия ее компонентов А, В,...
Ионные пары в растворе могут быть контактными и сольватно-
разделенными (в последнем случае катион и анион разделены од-
igKL ной либо несколькими молекула-
Рис. 3. Зависимость логарифма кон-
станты ионизации в системе пикрино-
рая кислота — пиридин от обратной
диэлектрической проницаемости в рас-
творителях:
Г: 1— Б; 2- Б-ДХБ; 3- Б-ДХЭ; 4- три-
хлорбензол; 5—бромбензол; 6 — ХБ; 7 —
ДХВ; II—НАс в смеси с растворителями:
1 — В; 2 — трихлорбензол; 3 — бромбензол;
4-ХБ; 5 —ДХБ
ми растворителя). Спектрально
эти две разновидности ионных
пар часто различимы [181]. Вы-
сокий дипольный момент ионных
пар обусловливает, особенно в
растворителях с малыми диэлек-
трическими проницаемостями,
высокую степень полимеризации
ионных пар:
uKqp+Lp~ S; (№+ LP~SV)U +
+ u(l~v)S. (1 — Ю)
Качественно процесс (1—10) вы-
ражается в малой растворимости
ионных соединений в растворите-
лях с низкой диэлектрической
проницаемостью, а также в обра-
зовании осадков при взаимодей-
ствии компонентов системы в растворителе S. Количественно вли-
яние растворителя на равновесие процесса (1—10) изучено в ра-
боте [174].
Поскольку процесс (1—9) заключается в переходе комплекса,
взаимодействие в котором с указанной ранее степенью приближе-
ния описывается как диполь-дипольное, в комплекс, взаимодейст-
вие, в котором является ион-ионным, энергия процесса (1—9) мо-
жет быть представлена как энергия ион-дипольного взаимодей-
ствия:
— kGi-d = RT In Кион = [кДж/моль], (1—11)
ri~d6
где ze — заряд иона, а аг-_й=289.
Из уравнения (1—11) вытекает прямолинейность зависимости
логарифма константы равновесия процесса ионизации от обратной
диэлектрической проницаемости в универсальных растворителях
[543, 143]. Пример, соответствующий такому случаю, приведен на
рис. 3.
Не имея возможности более подробно останавливаться на ха-
рактеристике процесса ионизации, отметим, что в литературе, по-
10
т
священной изучению равновесий в растворах электролитов, неред-
ко встречаются ошибки, возникающие из-за неучета того, что ионы
в растворе могут находиться в различных формах: свободной, а
также в виде ионных ассоциатов различной химической природы
и различной степени полимеризации. В частности, этим обстоя-
тельством обусловлено нередко констатируемое различие констант
равновесия процесса электролитической диссоциации, определен-
ных различными методами: кондукто-, потенцио- и спектрофото-
метрическим.
1.Г.5. Электролитическая диссоциация
Заключительная из последних стадий процессов межмолекулярно-
го взаимодействия в растворах — процесс электролитической дис-
социации
pKq+Sa + qLp~S$ + Sz- а-₽ (1-12)
протекает постольку, поскольку реализуются перечисленные выше
предшествующие стадии; очевидное исключение представляют
лишь растворы ионофоров, в случае которых процесс электроли-
тической диссоциации состоит лишь из двух стадий — образования
сольватированного ионного ассоциата при растворении и распада
последнего на ионы.
Общая теория электролитической диссоциации достаточно об-
стоятельно разработана Н. А. Измайловым [173]. Некоторые по-
ложения этой теории кратко будут изложены в разделе 1.2. Здесь
же подчеркнем, что протекание процесса электролитической дис-
социации является следствием как специфической, так и универ-
сальной сольватации. При этом специфическая сольватация до-
ставляет системе энергию, необходимую для образования ионов,
а достаточно высокая диэлектрическая проницаемость обеспечи-
вает распад ионного ассоциата на ионы. Ослабление энергии элек-
тростатического взаимодействия в непрерывной среде с диэлектри-
ческой проницаемостью е происходит в соответствии с обычным
уравнением ион-ионного взаимодействия
AGz-z - RT In [кДж/моль], (1-13)
где az-z=1391.
Вопросы термодинамики процесса электролитической диссоци-
ации будут рассмотрены в параграфе 1.2.3.
1.1.6. Общая схема равновесий
в растворах электролитов.
Проблема индифферентности растворителя
Описанные выше процессы межмолекулярного взаимодействия в
растворах протекают последовательно и, следовательно, взаимо-
связаны. Это позволяет считать их отдельными стадиями общей
11
схемы равновесий в растворах, впервые предложенной Н. А. Из-
майловым [173] и затем детализированной в работе [547]. Общая
схема объединяет равновесия (1—1), (1—7), (1—9) и (1—12):
^мон
Аж + sxS xASs
Кмои
By + tyS yQSt
^пр ^иои
mASs + nBS£ AmBnSr АПГ^
(1-14)
кп
^pA’+Sa + <7Lp-S₽.
Последовательность равновесий (1—14) может обрываться на
любой из стадий. В растворителях с низкой диэлектрической про-
ницаемостью независимо от энергетики предшествующих стадий
процесс электролитической диссоциации характеризуется более
или менее малой величиной констант равновесия (Ад). Из общей
схемы равновесий (1—14) вытекает, что образование электролит-
ного раствора возможно лишь в случае протекания всех стадий,
предшествующих процессу электролитической диссоциацией.
Можно выявить общие черты, свойственные каждой из стадий,
объединяемых схемой (1—14). Отметим, что свободная энергия, а
следовательно, и константа равновесия каждого из процессов оп-
ределяются энергиями как специфической, так и унивеосальной
сольватации:
AG = — RT In А = f (Аспещ АуИИВ) = ф (Еспец> Пв). (1—15)
Будем далее называть среды, где £Спец<^Аунив, универсальны-
ми. Очевидно, что в универсальных средах будет соблюдаться пря-
молинейная зависимость логарифма константы соответствующего
процесса от 1/е, как это иллюстрировалось выше на ряде приме-
ров (см. рис. 1—3).
Прямолинейность In А от 1/е будет наблюдаться также в сре-
дах с постоянной энергией специфической сольватации — такие
среды будем называть условно универсальными. Выразительным
примером сопоставления влияния универсальных и условно уни-
версальных сред на константу равновесия процесса может слу-
жить процесс ионизации в системе пикриновая кислота — пиридин
[543]: НР14-Руч=ь ... 4=tHPy+Pi_. На рис. 3 приведена зависимость
lg Ai—1/е для данного процесса в универсальных и условно уни-
версальных средах. В последних специфическая сольватация сво-
дится к присоединению уксусной кислоты к пиридину и поэтому
величина £Спец всюду постоянна. В соответствии с этим в изоди-
электрических растворителях рК(=—lg Ai) в условно универсаль-
ной среде гораздо меньше, чем в универсальной среде, однако в
обоих случаях соблюдается прямолинейность зависимости
1g А,—1/е.
Среды, где Аспец^Аунивер, будем называть далее специфически-
ми. Введенная терминология дает основания для рассмотрения
вопроса об индифферентных (универсальных) и химически неин-
дифферентных (специфических) растворителях.
12
Отнесение данного растворителя к индифферентным либо к хи*
мически активным не может быть априорным — без учета химиче-
ских (а иногда и физических) характеристик растворенного ве-
щества. Энергетика даже электростатической составляющей раз-
личных типов межмолекулярного взаимодействия в растворах, оп-
ределяемая уравнениями (1—4), (1—8), (1—11) и (1—13), раз-
нится в весьма широких пределах.
Таблица 1. Величины свободных энергий ДО электростатических
взаимодействий в растворах, кДж/моль
Тип взаимодействия 8=170—60 8=60—10 8=10-2
Диполь-дипольное (d—d) {13} {0,2} 5
Иои-дипольное (i—d} {1,0} {1,5} 20
Ион-ионное (i—i\ 10 15 100
В табл. 1 приведены оце-
ночные величины изменения
свободной энергии для различ-
ных типов электростатическо-
го взаимодействия в средах с
различной диэлектрической
проницаемостью [532]. Из
данных таблицы следует, что
в общем случае переход от
d—d- к i—d-взаимодействию,
а от последнего к i—1-взаимо-
действию способствует увели-
чению Дб?эл.ст примерно на по-
Таблица 2. Термодинамические
характеристики образования
ЭДА-комплекса в системе 12—Ру
растворитель -дн, кДж/моль —AS, Дж/моль-К
Циклогексан 36,9. 78,1'
н-Гептан 34,0 71,8
ecu 29,3 53,4
Бензол 25,5 49,4
м-Ксилол 18,3 26,7
п-Кснлол 17,9 25,6
рядок.
В некоторых случаях величины AGan.CT соизмеримы с энергией
теплового движения молекул в жидкости [617, 616], иногда ниже
ее (в табл. 1 эти значения даны в скобках). Естественно, чем ниже
энергия соответствующего взаимодействия, тем сложнее подобрать
универсальный (индифферентный) растворитель. Так, в раствори-
телях с низкой е очень многие растворенные соединения в большей
или меньшей степени ассоциированы, в воде же гомомолекулярные
ассоциаты образуются весьма редко, а в N-метилацетамиде (е =
= 175) явление гомомолекулярной ассоциации не наблюдается во-
обще.
Выразительным примером «индифферентности» растворителя
могут служить термодинамические характеристики образования
ЭДА-комплекса в системе Ь-Ру, приведенные в табл. 2 [541]. Каж-
дый из практически изодиэлектрических растворителей, данные по
термодинамике взаимодействия в которых сведены в табл. 2, в
большинстве работ признается априорно «индифферентным». Меж-,
ду тем данные табл. 2 свидетельствуют о том, что специфическая
сольватация оказывает решающее влияние на термодинамические
13
теснение пиридином молекул
ки йода, тем, следовательно,
Рис. 4. Зависимость отрицательно-
го логарифма константы электро-
литической диссоциации (р/<дис)
триоктилметиламмонийметилсуль-
фата от обратной диэлектрической
проницаемости в растворителях:
1 — метилацетамид; 2— ПК; 3— ДМСО;
4 — ДМФ; 5 — НБ; 6 — Ру+ДМФ; 7 —
Ру+АН; 8 — Ру+ПК; S —Ру; 10 —
НБ+НАс; 11 — ПК+НАс; 12 — НАс.
характеристики взаимодействия йода с пиридином, которое, стро-
го говоря, является процессом пересольватации: I2-S+Py^I2-
•Py+S (из-за преимущественно донорной природы приведенных в
табл. 2 растворителей сольватация пиридина в этой схеме не учи-
тывается). Чем основнее растворитель, тем более затруднено вы-
растворителя из сольватной оболоч-
менее экзотермична реакциях образо-
вания продукта присоединения
1г-Ру. Естественное объяснение в
рамках данной модели нах-одит и
закономерное увеличение энтропии
процесса по мере основности рас-
творителя.
Чем более высокоэнергетичен
процесс химического взаимодейст-
вия в системе, тем легче подобрать
для нее универсальный раствори-
тель. Выразительным примером это-
го положения могут служить дан-
ные по константам электролитиче-
ской диссоциации электролита
СН3- (С8Н17)з1Ч+СНз5Оз- в разных
растворителях. Как видно из рис. 4,
несмотря на то что растворители
характеризуются самой различной
природой — от кислых (растворите-
ли на основе уксусной кислоты) до
сильно основных (растворители на
основе пиридина), экспериментальные данные укладываются на
одну прямую в координатах рКя—1/е, свидетельствуя о протека-
нии в этих системах лишь ион-ионных взаимодействий, значения
энергии которых, как отмечалось, не менее чем на ^порядок
превышают величины энергии иных видов электростатических
взаимодействий. Такое поведение электролита в данном слу-
чае обусловлено большим размером его ионов, резко умень-
шающим энергию специфической сольватации молекулами рас-
творителя. Итак, растворитель не может априорно, без учета энер-
гетики рассматриваемого процесса, считаться индифферентным по
отношению к протекающему в нем процессу.
1.2. ВЛИЯНИЕ РАСТВОРИТЕЛЯ НА СИЛУ ЭЛЕКТРОЛИТОВ
1.2.1. Единое уравнение электролитической
диссоциации по Н. А. Измайлову [173]
Представим общую схему равновесий в растворах (1—14) в сле-
дующем упрощенном виде Ч
1 Степень упрощения достаточно мала, так как в неводных растворах в
подавляющем большинстве случаев электролиты относятся к валентному типу
(1-1).
14
KA 4" pS KASp КСОЛЬВАСОЛЬВ Ксольв 4“ АСОЛЬВФ (1 16)
Анест
Если константа электролитической диссоциации определяется
методами, дающими возможность вычислить активность ионов, то
находимая таким образом константа является общей, т. е.
а , а
к+ а-
ту СОЛЬВ СОЛЬВ 1 /11 *7\
Лоб = ------, (1 — 1
иедис
где а — активность соответствующих форм; аиедис — суммарная актив-
ность форм КА, KASp и кХльв Асольв-
Рассмотрим свободную энергию в круговом процессе, объеди-
няющем стадии переноса электролита КА из растворителя S в
вакуум, диссоциации КА на ионы К+ и А~ и их сольватации при
переносе из вакуума в растворитель. Для этого случая Н. А. Из-
майлов вывел уравнение, связывающее величины Коб, Кдис(в> (кон-
станта диссоциации КА на ионы в вакууме — величина постоянная
для данного электролита) с работой переноса ионов из вакуума
в растворитель 2 ксольв и энергией £7Мол образования продукта
присоединения KASP:
Коб = Кдис(в) exp (^сольв~м<>^) . (1-18)
Детально уравнение (1—18) проанализировано в работе
Н. А. Измайлова [173], а также в [547].
Полное уравнение для Коб, относящееся к типу (1—15), связы-
вает эту величину с диэлектрической проницаемостью среды и ха-
рактеристиками ионов, образующимися при диссоциации:
In Коб — In Кдис(в) 4- In YKASp — In (1 4- Киест 4“ Кпр) 4-
(1-19)
। е2М) 2иои /, 1 ) , ^^сольв
+ rRT гиои RT ’
“к+ ’“а-
где у — коэффициент активности; Кпр =------------с°льв .
ак+ аА~
СОЛЬВ СОЛЬВ
Из уравнения (1—19) следует, что сила электролита КА в дан-
ном растворителе S зависит от энергии связи ионов в вакууме
UCp=RT In Кд(в) и энергии сольватации ионов и недиссоциирован-
ных молекул КА молекулами S. Чем меньше энергия средства
ионов в вакууме, чем больше энергия сольватации и диэлектри-
ческая проницаемость, тем выше константа электролитической
диссоциации.
В случае растворов ионофоров, которые в основном не образу-
ют прочных сольватов, уравнение (1—18) преобразуется
Коб = const exp [(2£/сольв — UKp)/RT], (1—20)
где St/сольв — сумма химических энергий сольватации; UKV —
энергия кристаллической решетки.
Уравнение (1—20) не пригодно не только для точного подсче-
та, но и для оценки константы диссоциации прежде всего из-за то-
го, что незначительная погрешность в разности (2£7СОльв—t/Kp)
приводит к существенной погрешности в расчете КОб- Поскольку
величины St/сольв и t/Kp рассчитываются со значительной погреш-
ностью, уравнение (1—20) позволяет только проследить общую
тенденцию в изменении величин Коб с изменением различных фак-
торов. Однако и в этом качестве уравнение (1—20) достаточно
полезно. Так, расчет разности (2£7СОЛьв—t/Kp) для водных раст-
воров галогенидов щелочных металлов (примерно 50 кДж/моль)
объясняет, почему эти ионогены в водном растворе весьма силь-
ные электролиты.
1.2.2. Дифференцирующее и нивелирующее
действие растворителя на силу электролитов
Анализ уравнения (1—19) четко определяет условия, при которых
будет соблюдаться прямолинейная зависимость рКя от 1/е: оче-
видно, что это будет наблюдаться в средах с пренебрежимо малой
по сравнению с иными членами уравнения (1—19) величиной
Рис. 5. Зависимость рКп электролита в одном растворителе Si от рКд в другом
растворителе S2:
а —общая схема; б — зависимость величин рКд в ацетоне от рКд в ацетонитриле:
/ — йодид калия; 2 — перхлорат цезия; 3 —роданид калия; 4 — пикрат калия; 5 —пикрат
натрия; 6 — пикрат лития.
2^сольв//?Т, т. е. в универсальных средах. Кроме того, прямоли-
нейной зависимость рКр от 1/е будет и в средах, где 2С/сольв =
= const (условно универсальные среды). С учетом этих условий,
а также в приближении, что ионные радиусы не зависят от е, бу-
дет выполняться условие
1пКд = А + 4’ (1-21)
которое иллюстрировалось выше (см. рис. 4).
16
Из уравнений (1—19) и (1—21) вытекает ряд значительных
следствий, среди которых важнейшее — требование прямолиней-
ности зависимости рА®1 — рК$‘, т. е. зависимости логарифма кон-
стант диссоциации ряда электролитов в одном растворителе Si
от логарифма констант диссоциации тех же электролитов в дру-
гом растворителе S2. Возможные типы такой зависимости приве-
дены на рис. 5, а. Если реализуется зависимость I, это означает,
что растворитель S2 обладает дифференцирующим действием на
силу растворенных в нем электролитов по сравнению с раствори-
телем Si; зависимость III свидетельствует о том, что растворитель
Si по сравнению с S2 дифференцирует силу электролитов; зависи-
мость II указывает на отсутствие дифференцирующего либо ни-
велирующего действия растворителей на силу электролитов.
В качестве примера на рис. 5,6 приведена-зависимость рКп
( = —1g Ад) ряда солей щелочных металлов в ацетоне от рАд тех
же солей в ацетонитриле. Видно, что АН обладает по сравнению с
АЦ дифференцирующим действием на силу солей: разности
ДрАд=1 в ацетоне соответствует ДрАд=1,5 в ацетонитриле.
Различные аспекты дифференцирующего либо нивелирующего
действия растворителей на силу электролитов достаточно подроб-
но рассмотрены в работе Н. А. Измайлова [173]. Учет этих аспек-
тов играет существенную роль при выборе композиций для элек-
троосаждения металлов из неводных растворов.
1.2.3. Термодинамика процесса электролитической
диссоциации
В большинстве случаев сведения о термодинамике процесса элек-
тролитической диссоциации, как и любого иного равновесного про-
цесса в растворах, получают из температурных зависимостей кон-
стант равновесия. Зависимость
Ад от температуры, рассматрива-
емая в широком температурном
интервале — от температуры
плавления растворителя до его
температуры кипения, экстре-
мальна (рис. 6). Однако при изу-
чении термодинамических харак-
теристик процесса электролити-
ческой диссоциации обычно огра-
ничиваются узким (50—75°)
температурным интервалом, в ко-
тором изменение Ад однозначно
с изменением температуры. Из
рис. 6 видно, что экспериментатор
констант равновесия процессов в рас-
творах.
в зависимости от того, выбирает
ли он интервал 7\—Т2 либо Т3—Т4, может получить произвольные
по абсолютной величине, а нередко и противоположные по знаку
величины термодинамических характеристик процесса. Это объяс-
няет имеющиеся в литературе неоднозначные результаты, а в не-
2 — 4-653
17
которых случаях взаимно исключающие выводы о термодинамике
процесса электролитической диссоциации, часть из которых к то-
му же противоречит физической картине данного процесса.
Если учесть, что по аналогии с выражением (1—3) энергия
процесса электролитической диссоциации, как и любое иное вза-
имодействие в растворе, состоит из химической и электростатиче-
ской составляющих:
Абд = А6ХИМ + Абэл.СТ, (1—22)
то уравнение (1—21) запишется в следующем виде:
In Кд = In Кхим + , (1-21а)
где Кмм и Кэл.ст — константы равновесия собственно химических
и электростатических взаимодействий в процессе электролитиче-
ской диссоциации.
Находимые из температурной зависимости Кд энтальпии и эн-
тропии процесса электролитической диссоциации, отражающие на-
ложение различных типов взаимодействий в растворах, будем да-
лее называть интегральными [565]. Итак, дифференцируя выраже-
ние (1—21—а) по 1/Т, получаем:
ДЯинтегр = AtfXHM + -1Г ДЯ, + К In Кэ (1-23)
Из выражения (1—23), учитывая, что Дб =—КТ’In К, и исполь-
зуя известную связь между гиббсовой энергией, энтальпией и эн-
тропией процесса, получаем выражение для энтропии процесса
электролитической диссоциации:
ASHHTerp = ASXHM + -А-[ДЗЭ + 1пКэ . (1-24)
Из уравнений (1—23, 1—24) видно, что термодинамические ха-
рактеристики процесса электролитической диссоциации (как, впро-
чем, и любого иного равновесного процесса в растворе) помимо
химической и электростатической составляющих определяются
также температурным изменением диэлектрической проницаемо-
сти. Для разделения влияния этих факторов авторы [567, 565J
предложили расчленить термодинамические характеристики рав-
новесного процесса на температурную и диэлектрическую состав-
ляющие:
АЯиитегр = ДЯг + АЯе, (1 -25)
АЗнитегр — ASt AS8. (1—25,а)
Температурные составляющие термодинамических характери-
стик процесса — это величины, которые определяют его течение в
среде с неизменной диэлектрической проницаемостью (т. е.
dine
д(1/Т) ~и>:
\НТ = АЯХИИ + , (1-26,а)
<5
18
ASy — ASXHM H-
(1-26,6)
Удобным приемом нахождения АЯт является использование
для расчета величин In К, взятых из изодиэлектрических разрезов
а—а', b—Ь'... (рис. 7, а). Величину ASr находят как разность
ДЯ_ —Дб
----=----. Соответственно диэлектрические составляющие — это
Рис. 7. Графический метод расчета температурных составляющих термодинамиче-
ских характеристик процесса электролитической диссоциации по изодиэлектриче-
ским разрезам (а—o', Ь—Ь’с—o'):
а — универсальные среды; б — специфические среды.
имодействия, которые определяются температурным изменением
диэлектрической проницаемости:
= (1-27,а)
ASe= А1п/<э^±. . (1-27,6)
Метод разделения, рассмотренный выше для случая прямоли-
нейной зависимости In к от 1/е, т. е. для случая выполнения урав-
нений (1—21) и (1—21, а), без труда может быть распространен
на непрямолинейные зависимости In К от 1/е. Для систем, харак-
теризующихся зависимостями In Д’—1/е последнего типа, темпе-
ратурные составляющие энтальпии и энтропии равновесного про-
цесса определяются расчетом, для которого берутся значения кон-
стант диссоциации при различных температурах и находимые из
изодиэлектрических разрезов а—ar, b—Ь',... (рис. 7,6).
Как показано [567, 565], величины АЯГ и ASt более адекватно
отражают физическую картину процесса электролитической дис-
социации, поэтому более информативны по сравнению с соответ-
2*
19
и
ствующими интегральными величинами. Приведем аналогичный
пример. В табл. 3 сопоставлены термодинамические характеристи-
ки электролитической диссоциации хлорида диспрозия в двойных
смешанных растворителях: вода — метанол и метанол — /г-пропа-
нол, в которых DyCl3 является 1—1-электролитом. Ход In Кд как
функции 1/е для двух температур представлен на рис. 7, а.
Сопоставление величин, приведенных в табл. 3, подтверждает
высказанный выше тезис о большей информативности величин
АЯу и АЗу. Действительно, в то
время как согласно значениям
А/7интегр процесс электролитиче-
ской диссоциации хлорида дис-
прозия во всех смешанных рас-
творителях экзотермичен, вели-
чины АНу показывают, что экзо-
термичным этот процесс в дей-
ствительности является только в
растворителях с диэлектрической
проницаемостью, достаточно вы-
сокой для того, чтобы обес-
печить распад ионной пары на
Рис. 8. Зависимость энтальпии от эн-
тропии процесса электролитической
диссоциации:
/ — температурные составляющие; 2— ин-
тегральные величины.
ионы.
Нетрудно убедиться, что величины АЗинтегр также не согласу-
ются с физической моделью процесса электролитической диссоци-
ации. Действительно, с понижением величины диэлектрической
проницаемости (т. е. при переходе от водно-метаноловых раство-
Таблица 3. Термодинамические характеристики электролитической
диссоциации хлорида диспрозия в смешанных растворителях вода—метанол
(M'tOH) и MtOH—н-пропанол (РгОН)
Состав растворителя, мол. % Е (298 °C) ДЯд, кДж-моль 1 Д5, Дж *моль 1ft 1
инте- гральная темпера- турная инте- гральная температурная
5он2о+а>мюн 47,1 —33,3 —18,7 —66 +41,6
26 НЮ+75 МЮН 38,9 —29,1 —12,6 —64 +20,1
мюн 32,7 —18,4 —11,5 —54 + 13,9
50 МЮН+Э0 РгОН 23,9 —14,9 —5,4 —51 —8,4
25 МЮН+ 75 РгОН 21,6 —16,0 +9,2 —27 —57,9!
РгОН 20,4 —7,0 + 36,7 —20 —154,8
рителей к пропаноловому) степень структурирования ионов, обра-
зовавшихся из электрически нейтральных ионных пар, должна по-
вышаться, следовательно, в том же направлении должна расти
экзоэнтропийность процесса. Это обстоятельство хорошо под-
тверждается изменением величин АЗу с составом растворителя и
в то же время противоречит изменению АЗинтегр.
Убедительной иллюстрацией преимуществ температурных со-
ставляющих величин термодинамических характеристик равновес-
ных процессов в растворах по сравнению с интегральными величи-
20
УК
нами может служить зависимость А/7— AS (рис. 8). Как извест-
но, в тех случаях, когда изменение внешних условий (в данном
случае состава растворителя) не приводит к изменению природы
процесса, эта зависимость должна быть прямолинейной. Этому
условию, как видно из рис. 8, соответствует зависимость АНГ —
АЗт, в то время как формальное рассмотрение зависимости
АЯинтегр — АЗинтегр должно было бы привести к заведомо невер-
ному выводу о том, что переход от водно-метаноловых раствори-
телей к пропаноловому вызывает изменение природы сольватации.
1.3. НЕКОТОРЫЕ ВОПРОСЫ ТЕОРИИ ЭЛЕКТРОПРОВОДНОСТИ
НЕВОДНЫХ РАСТВОРОВ
Электропроводность (удельная % и молекулярная, либо эквива-
лентная, %) относится к числу важнейших свойств электролитных
растворов. Большинство теоретических построений в области рас-
творов электролитов основывается на первичных данных по элек-
тропроводности. Теоретические вопросы электропроводности рас-
творов рассматриваются во многих работах [173, 530, 19, 547,
438, 638].
1.3.1. Общие положения
Электропроводность индивидуального химического соединения —
характеристика, обладающая известным своеобразием по сравне-
нию с большинством иных физических свойств. Точные значения
х определены лишь для жидких металлов, многих металлических
и гораздо меньшего числа ионных расплавов. Электропроводность
индивидуальных неорганических и органических жидкостей (воп-
рос, имеющий большое значение для проблематики данной книги,
поскольку он связан с подбором жидких композиций для электро-
осаждения металлов) существенно зависит от их чистоты. Кроме
того, величина % зависит от условий и способа измерений, в част-
ности от частоты тока [96] при измерениях на переменном токе.
Относительно последних в настоящее время нет установившейся
точки зрения. Поэтому нередки случаи, когда значения % индиви-
дуальных жидкостей по данным разных авторов (например, [79,
959, 136]) различны (до двух порядков).
Соответственно нет и не может быть четкой границы между
понятиями «проводящая» и «непроводящая» жидкость либо ра-
створ. Классификационные разграничения этих понятий предло-
жены в работе [530].
Электропроводность раствора в отличие от большинства ста-
ционарных либо транспортных свойств раствора — плотности, оп-
тического поглощения, вязкости и т. д. — возникает лишь в ре-
зультате необходимого сочетания ряда факторов: достаточно вы-
соких энергий специфической сольватации и диэлектрической про-
ницаемости (см. раздел 1.2). Это обстоятельство служит одной из
причин того, что количественная теория электропроводности,
21
объясняющая зависимость электропроводности от концентрации,
температуры, вязкости и диэлектрической проницаемости, с удов-
летворительной полнотой разработана только для весьма разбав-
ленных растворов электролитов и лишь в отдельных случаях по-
зволяет описывать растворы концентрации 10-2 м; в большинстве
случаев указанный верхний предел концентрации значительно ни-
же. Кроме того, абсолютная величина электропроводности сущест-
венно зависит от того, какой из механизмов переноса тока —
ионмиграционный либо ионотропный — превалирует в данном ра-
створе (о механизмах переноса тока и об оценке вклада различ-
ных механизмов в общий перенос тока в растворе см. пара-
граф 1.5.3).
Перечисленные обстоятельства обусловливают различный ме-
тодологический подход к теоретическому описанию свойств раз-
бавленных и концентрированных растворов. Поэтому здесь теоре-
тические вопросы электропроводности неводных растворов будут
рассмотрены раздельно для концентрированных растворов, в ча-
стности для двойных жидких систем, представляющих особый ин-
терес для практики электроосаждения металлов, и для разбавлен-
ных растворов электролитов.
1.3.2- Электропроводность двойных жидких
систем
К двойным жидким системам относятся системы, образованные
двумя жидкими при данной температуре компонентами [19, с. 374].
Системы, один из компонентов которых твердый при температуре
исследования либо эксплуатации раствора, но хорошо растворяет-
ся во втором — жидком и образует концентрированные растворы,
подчиняются большинству закономерностей, установленных для
двойных жидких систем. Таким образом, излагаемый в этом пара-
графе материал несколько шире его названия.
К системам рассматриваемого типа относятся растворы гало-
генидов многих металлов III—VI групп периодической системы,
т. е. достаточно широкий круг объектов.
Особенности кондуктометрии как метода физико-химического
анализа рассмотрены в работах [531, 19, 530, 547]. Существующая
классификация изотерм электропроводности [531] основана на
проводимости компонентов системы, а внутри каждого подразде-
ла — на геометрии изотерм.
Системы, образованные не проводящими ток компонентами.
Очевидно, что возникновение электропроводности в таких систе-
мах (точнее, электропроводности, на несколько порядков превы-
шающей низкую электропроводность компонентов) в соответствии
со схемой (1—14) однозначно свидетельствует о специфическом
взаимодействии компонентов. На рис. 9, а приведены основные
геометрические разновидности изотерм % данного класса систем.
Несмотря на различие геометрии, все изотермы генетически свя-
заны.
22
Появление минимума на изотермах х, наблюдающееся обычно
в системах с глубоким взаимодействием, обусловлено резким воз-
растанием вязкости т]. Поскольку максимум т] в таких системах в
основном приходится на состав образующегося в системе соеди-
нения [19, 530], минимум на изотерме х, в свою очередь, также мо-
жет свидетельствовать о стехиометрии взаимодействия. Повыше-
ние температуры способствует понижению вязкости, что ведет к
Рис. 9. Основные геометрические типы изотерм удельной электропроводности
двойных систем, образованных не проводящими ток компоиеитами:
«— геометрия изотерм; б — изотермы электропроводности системы треххлооистая сурьма —
м-амиловый спирт.
сглаживанию минимума на изотермах х, в конце концов минимум
исчезает, а изотерма х вырождается в кривую с одним максиму-
мом — наиболее часто встречающуюся форму изотерм электропро-
водности двойных систем.
Характерным примером влияния вязкости. (следовательно, и
температуры) на форму изотерм х может служить система
SbCU — н-амиловый спирт [535], ‘диаграмма электропроводности
которой приведена на рис. 9, б.
Роль вязкости как фактора, определяющего форму изотермы
х, проявляется также в том, что в соответствии с правилом
М. И. Усановича [507] в системах с взаимодействием на изотермах
коррегированной на вязкость удельной электропроводности о=хт,
сохраняется лишь один максимум (правило М. И. Усано-
вича уточнено в работе [530]). Системы этого типа представ-
ляют наибольший интерес для практики электроосаждения
металлов.
Системы, образованные компонентами, один из которых про-
водит ток в индивидуальном состоянии. Возможны четыре геомет-
23
рических типа изотерм данного класса (рис. 10, а). Если в систе-
мах предыдущего класса возникновение электропроводности, су-
щественно превышающей электропроводность компонентов, само
по себе свидетельствует о химическом взаимодействии, то установ-
ление факта взаимодействия по кондуктометрическим данным для
систем данного класса требует учета ряда обстоятельств.
Рассмотрим прежде всего системы, где второй компонент явля-
ется универсальным («индифферентным») растворителем по от-
ношению к первому — электролиту. Б таких системах максимум
Рис. 10. Удельная электропроводность двойных жидких систем с химически не-
взаимодействующими компонентами:
а — основные геометрические типы изотерм; б — системы, образованные ацндокомплексом
хлорного олова с Б (/); ХБ (2); ДХЭ (3).
на изотермах х появляется лишь в тех случаях, когда отношение
вязкости электролита к вязкости растворителя весьма велико (ко-
личественное рассмотрение этого вопроса см. в [530, 560]). При-
мером может служить система, диаграмма удельной электропро-
водности которой приведена надшс. 10,6.
Различие между системами с взаимодействием и без взаимо-
действия для рассматриваемого типа может быть установлено со-
поставлением экспериментальных изотерм % с изотермами, рассчи-
танными в случае отсутствия специфического взаимодействия по
уравнению [561]:
х = exp [in хат]а — — ’ (1—28)
L \ е ®А / J й
где обозначения с индексом А относятся к свойствам электролит-
ного компонента; обозначения без индексов — к свойствам раство-
ра; L — коэффициент пропорциональности, зависящий от соотно-
шения диэлектрических проницаем остей компонентов.
24
В работе [555] приведено более общее уравнение, связывающее
свойства электролитного компонента со свойствами растворителя:
In хц = const} + + у In С (1-29)
(С — концентрация электролитного компонента, моль-л-1), в ко-
тором величины постоянных определяются свойствами растворите-
ля, а также микрофизическими характеристиками молекул раст-
ворителя и растворенного элект-
ролита и рассчитываются по фор-
мулам, приведённым в работе
[555]. Уравнение (1—29) объяс-
Рис. 11. Изотермы логарифма исправ-
ленной на вязкость удельной электро-
проводности комплекса [НзО X
ХЗТБФ] +Т1СЦ+ в растворителях:
1 — л-ксилол; 2 — ХБ; 3 — ацетофенон; 4 —
НБ.
Рис. 12. Изотермы молекулярной элек-
тропроводности системы серная кисло-
та — диэтиловый эфир:
1 — расчет на концентрацию продукта при-
соединения; 2— расчет на аналитическую
концентрацию кислоты.
няет, почему для одного и того же электролита значения хц, от-
вечающие изодиэлектрическим растворам в различных универсаль-
ных растворителях, не совпадают.
Из уравнений (1—28), (1—29) вытекает важное следствие, со-
гласно которому в системах рассматриваемого типа зависимости
In иц—1/е прямолинейны (на том интервале концентраций, на ко-
тором сохраняется неизменным механизм электропроводности).
В качестве примера на рис. 11 приводится данная зависимость для
растворов жидкой соли [Н3О • ЗТБФ]+Т1СЦ~ в различных универ-
сальных растворителях [232].
Диаграммы молекулярная (эквивалентная) электропровод-
ность — состав практически не применяются для анализа концент-
рационного изменения электропроводности в двойных жидких си-
стемах. М. И. Усанович [509, с. 173] объяснил это тем, что истин-
ная концентрация электролитного компонента практически не
совпадает с аналитической концентрацией. Этим же объясняется
появление так называемых аномальных изотерм молекулярной
электропроводности. Действительно, в том случае, когда незави-
симым путем можно определить истинную концентрацию электро-
литного продукта, возникающего при взаимодействии компонентов
системы, аномальные кривые X превращаются в нормальные. На
рис. 12 сопоставляются изотермы %—состав для системы диэти-
ловый эфир — серная кислота, рассчитанные на определенную
25
спектроскопически истинную концентрацию электролитных про-
дуктов присоединения H2SO4-Et2O,\ H2SO4-2EtO и-2H2SO4-Et2O и
на аналитическую концентрацию серной кислоты [447]. Как видно
из рисунка, первому случаю, в отличие от второго, соответствует
нормальный ход изотермы X.
1.3.3. Зависимость электропроводности от концентрации
Недостаточность экспериментальных данных по зависимости элек-
тропроводности от концентрации и природы электролита концент-
рированных растворов ионофоров (солей) в неводных раствори-
телях [478, 323, 180, 662, 660, 661] вынуждает ограничиться лишь
качественными обобщениями, касающимися в основном солей ще-
лочных металлов либо алкил (арил) замещенных аммония.
В общем случае на кривой концентрационной зависимости
удельной электропроводности растворов солей в апротонных раст-
ворителях имеется максимум, локализующийся обычно в области
концентраций 6—7 мол. % (~1М). В растворителях с диэлектри-
ческой проницаемостью ниже 30 максимум % сдвигается в сторону
Рис. 13. Зависимость электропроводности от концентрации солей лития в ДМФ:
<1 — удельная электропроводность; б — электропроводность, исправленная на вязкость раст*
вора; 1 — перхлорат; 2 — иодид; 3 — бромид; 4 — пикрат; 5 — хлорид.
существенно более высоких концентраций соли (17—20 мол. %,
или 2,5 М), что, естественно, объясняется сдвигом равновесия в
таких растворителях в сторону ионных ассоциатов (ионных пар).
Исправление удельной электропроводности на вязкость приводит
к исчезновению максимума, зависимость от концентрации при-
мерно до 10—20 мол. % носит линейный характер.
26
В апротонных растворителях в ряду солей с одинаковым кати-
оном либо анионом величины % и хц возрастают с увеличением
кристаллографического радиуса противоиона.
По влиянию на электропроводность (исправленную электропровод-
ность) ноны можно расположить в следующие ряды: соли с общим
анионом Mt4N+ > Et4N+ > Cs+ > Rb+ > K+ > Pr4N+ > Na>Bt4N+>
2> Li+> Oct4N+, соли с общим катионом С1ОГ > Г’>ВЕГ~АзР(Г>
> CF3SO3~ > PFr > NOr > Br“> Pi> СГ > BPhp. На рис. 13 опи-
санные закономерности иллюстрируются растворами ряда солей
в ДМФ.
Аналогичные закономерности концентрационной зависимости
удельной электропроводности наблюдаются и в смешанных раст-
ворителях, например, ПК — диметоксиметан [1045], метанол —
ТГФ [58].
Исследованию зависимости молекулярной (эквивалентной)
электропроводности ‘посвящены многочисленные работы концент-
рации ([20, 173, 438, 547, 638, 768, 840]). Однако в большинстве
этих работ речь идет о весьма разбавленных растворах. Поэтому
данный вопрос, имеющий существенное значение для теории элек-
тролитных растворов, здесь будет затронут лишь постольку, по-
скольку электропроводность композиций, применяющихся для
электроосаждения металлов из неводных растворов и основанных
на растворах относительно высоких концентраций, в известной
степени коррелирует с электропроводностью разбавленных раст-
воров.
Теоретические вопросы концентрационной зависимости элект-
ропроводности неводных растворов менее изучены, чем для вод-
ных. Принципиальное отличие в данном случае заключается в
том, что природа носителей тока в неводных растворах в настоя-
щее время остается еще во многом предположительной. В то вре-
мя как большинство солей в водных растворах — сильные элект-
ролиты, в неводных растворах большая часть электролита при-
сутствует в незаряженной форме, а ионы могут существовать в
различных формах — одиночные ионы различной степени сольва-
тации и различные ионные ассоциаты: тройники и более высрко-
полимерные образования. По-видимому, именно поэтому теория
электропроводности развивалась преимущественно применительно
к водным растворам [638, 20].
Известны многочисленные попытки применения выводов теории
сильных электролитов к неводным растворам. В настоящее время
можно считать бесспорным, что верхняя граница концентрацион-
ного интервала, к которому приложимо уравнение Дебая — Онза-
гера, в случае неводных растворов примерно на порядок ниже, чем
в случае водных растворов. Интересно, что не только высокая ди-
электрическая проницаемость воды тому причиной. Растворы
электролитов в N-метилацетамиде, у которого диэлектрическая
проницаемость вдвое выше, чем у воды, уравнениями теории силь-
ных электролитов описываются не лучше, чем для большинства
иных неводных растворителей [559].
27
Основные направления исследований по обоснованию теорети-
ческой зависимости электропроводности от концентрации сводятся
к теоретическому расчету тангенса угла наклона (коэффициент В)
в эмпирическом уравнении Кольрауша
Х = Х0 — BVC, (1—30)
где X, Хо — соответственно электропроводность при малой конеч-
ной концентрации и при бесконечном разбавлении.
Уравнения теории сильных электролитов достаточно общеиз-
вестны [20]. Поскольку в неводных растворах согласие экспери-
мента с уравнениями этой теории чаще всего неудовлетворитель-
ное, предложено [638, 339, 756] сравнительно большое число урав-
нений типа
^ = /(^о, Кк, С, у, е, ц, а1( а2,...), (1—31)
связывающих %, и Хо с Кд, концентрацией электролита С, его ко-
эффициентами активности у, вязкостью г] и диэлектрической про-
ницаемостью е растворителя, а также содержащих различные эм-
пирические либо полуэмпирические параметры ai, а2,... Уравнения
типа (1—31) лежат в основе кондуктометрических методов опре-
деления констант электролитической диссоциации. Нельзя не за-
метить, что большое число предложенных в литературе уравне-
ний типа (1—31) свидетельствует скорее о неблагополучии в фи-
зической химии электролитных растворов. Приложения теории
сильных электролитов, связанные с учетом межионного взаимо-
действия в растворителях с относительно высокой концентрацией
(см., например, [639]), в случае неводных сред пока не достига-
ют желаемого согласия с экспериментом.
Простейшим из уравнений типа (1—31), ограниченность при-
менения которого обусловлена положениями классической теории
электролитической диссоциации, очевидно, является уравнение
Оствальда
= (1—32)
Для расчета констант диссоциации уравнение (1—32) часто при-
меняют в модификации Крауса — Брея:
Все уравнения, предлагаемые для описания концентрационной
зависимости молекулярной (эквивалентной) электропроводности
[339], относятся к типу
h = у, КД, (1-33)
где у — коэффициент активности. Анализ уравнений типа (1—33)
показывает, что точность определения Кд в случае больших зна-
чений этой величины (>10-1) весьма низка.
28
Кондуктометрическая методика определения величин Кц пре-
обладает над всеми иными методами: спектрофото-, потенцио-,
рефрактометрическим, крио- либо эбуллиоскопическим и т. п. Ос-
новная причина распространенности кондуктометрии в данном
случае обусловлена ее высокой чувствительностью, относительной
простотой выполнения измерений. К недостаткам кондуктометрии
следует отнести существенную зависимость определяемой величи-,
ны Кд от выбранной модели и соответствующего уравнения расче- '
та, а также необходимость тщательнейшей очистки исследуемого
электролита и растворителя.
1.3.4. Зависимость электропроводности от вязкости
и диэлектрической проницаемости [544]
Рассмотрим влияние указанных свойств растворителя на молеку-
лярную (эквивалентную) электропроводность в разбавленных ра-
створах электролитов (для концентрированных растворов данный
вопрос рассмотрен в параграфе 1.3.2).
На протяжении многих десятилетий влияние вязкости раство-
рителя на электропроводность описывается в литературе так на-
зываемым правилом Вальдена, согласно которому произведение
предельной эквивалентной электропроводности данного электро-
лита в различных растворителях есть величина постоянная:
Хот]о = const. (1—34)
Эмпирическое правило (1—34) получило впоследствии теоретиче-
скую трактовку, основывающуюся на гидродинамической теории,
которая рассматривает ион как жесткую сферу, движущуюся в
непрерывной изотропной среде. Радиус иона г в данном случае
определяется уравнением Стокса — Эйнштейна [438, с. 65]:
r — kT/6nD°, (1—35)
где k — константа Больцмана; т] — коэффициент вязкости; D° —
коэффициент диффузии. Коэффициент диффузии в бесконечно раз-
бавленном растворе связан с подвижностью уравнением Нернста
[438, 368]:
D°i = RTKoiIZiF^, (1—36)
где Zt — заряд иона; F — постоянная Фарадея. Объединение урав-
нений (1—35), (1—36) приводит к выражению
ЯЛодП = ZjF2IQnNQr0, (1—37)
из которого следует, что при постоянстве г вальденовское произ-
ведение действительно должно сохраняться постоянным для дан-
ного иона в любом растворителе. Однако экспериментальные ре-
зультаты по электропроводности неводных растворов показали,
что правило (1—34) не соблюдается. В табл. 4 приведены вели-
29
1
чины вальденовского произведения для некоторых ионов. Как вид-
но из данных таблицы, коррегированная подвижность не сохраня-
ется постоянной даже для весьма объемистых тетраалкиламмо-
нийных ионов.
Причина непостоянства величины Лот] обсуждалась в литерату-
ре и сводится в основном к постулированию факта об изменении
числа сольватации и эффективного
ионного радиуса при переходе от
одного растворителя к другому. Дей-
ствительно, произведение радиуса
сольватированного иона (см. пара-
граф 1.4.1) на макровязкость рас-
творителя и подвижность иона го-
раздо лучше сохраняет постоянство,
чем вальденовское произведение.
Предложено несколько эмпириче-
ских уравнений, связывающих ион-
ный радиус с диэлектрической про-
ницаемостью растворителя [839, 860,
1107].
Обработка экспериментально-
Рис. 14. Зависимость логарифма
предельной корригированной экви-
валентной электропроводности рас-
творов перхлората калия от обрат-
ной диэлектрической проницаемос-
ти растворителя:
/ — ПК; 2—ДМСО; 3— НМ, 4— НБ; 5—
ДМФ; 6 — АН; 7 — метаиол; 3 —ДМАА;
9- АЦ.
го материала показывает, что в.
универсальных либо условно универсальных средах вальденовское
произведение 'экспоненциально зависит от обратной диэлектричес-
кой проницаемости:
1пХоП = Д(Т) + -^-- (1- 38)
Таблица 4. Корригированная подвижность некоторых ионов в различных
растворителях при 298 К (Па-с-м2-См-кмоль—1 • 104)
Иои H,O MtOH EtOH ДМФ ДМСО ПК ФА АЦ АН РУ
Mt<N+ 0,40 0,37 0,31 0,37 0,41 0,29 0,33 0,41
Et<N+ 0,29 0,29 0,34 0,33 0,27 0,29 —
Pr«N+ 0,21 0,24 0,27 0,26 — 0,22 0,24 —
Bt4N+ 0,17 0,21 0,21 0,21' 0,24 0,24 0,22 0,20 0,21 0,23
H+ 3,1 0,79 0,65 0,!» 0,31 — 0,35 0,32 — 0,47
Li+ 0,34 0,22 0,18 0,19 0,22 0,18 0,28 0,27 0,24 0,22
Na+ 0,46 0,25 0,22 0,24 0,29 0,22 0,33 0,28 0,27 0,25
K+ 0,65 0,28 0,25 0,25 0,30 0,30 0,42 ' 0,24 0,29 0,30
Rb+ 0,69 0,!» 0,30 — — 0,30
ci- 0,68 0,28 0,24 0,45 0,46 0,51 0,56 0,35 0,31 —
Br- 0,70 0,311 0,26 0,42 0,47 0,48 0,56 0,39 0,35 0,49
J- 0,68 0,34 0,29 0,41' 0,46 0,47 0,54 0,38 0,35 0,46
ClOr 0,60 0,39 — 0,47 0,48 0,47 — 0,36 0,36 0,45
На эту зависимость впервые, по-видимому, указал А. М. Шко-
дин [624], однако уравнение (1—38) распространяется также на
корригированные ионные подвижности. Пример, иллюстрирующий
эту зависимость, приведен на рис. 14.
30
Применение принципа линейности свободных энергий ионной
миграции и вязкого течения приводит к выводу о том, что для од-
ного и того же электролита в ряду химически подобных раствори-
телей либо в условно-универсальных средах хорошо соблюдается
соотношение, учитывающее поправку величины корригированной
электропроводности на мольный объем растворителя:
%от]01/3 = const, (1—39)
где О — мольный объем в случае индивидуального растворителя
и псевдомольный объем [530, с. 41] в случае смешанного раство-
рителя. Уравнение (1—39) можно представить в более общей фор-
ме [544]:
ХоПВе(£-2/3) = const, (1-40)
где L — тангенс угла наклона прямолинейной в случае универ-
сальных либо условно универсальных сред зависимости свободной
энергии активации вязкого течения от свободной энергии актива-
ции ионной миграции.
В работах В. И. Ермакова и В. В. Щербакова [145, 144, 631]
установлена связь между удельной электропроводностью умеренно
концентрированных растворов, диэлектрической проницаемостью
растворителя и временем дипольной диэлектрической релаксации
е, согласно которой
х-^- = const. (1—41)
Так, для 0,1 М раствора нитрата кальция в воде, этаноле, пропа-
ноле и этиленгликоль величина хт/е постоянна в интервале тем-
ператур + Ю4--1-50 °C и составляет (1,85±0,1) • 10-12 с/Ом-см. Кро-
ме того, теми же авторами установлена связь между электропро-
водностью раствора, измеренной на конечной частоте х, и удель-
ной электропроводностью, экстраполированной на бесконечно
большую частоту х»:
х = Кхоо. (1—42)
Соотношение (1—42) позволяет связать электропроводность ра-
створов электролитов с электромагнитными свойствами раствори-
теля.
1.3.5. Зависимость электропроводности
от температуры, температурные коэффициенты
электропроводности.
Термодинамика активации процесса электропроводности
В большинстве случаев электропроводность электролитных раство-
ров — удельная и эквивалентная — экспоненциально зависит от
обратной температуры:
х = аи ехр (— Mfi/RT), (1—43)
X = ак exp (- Mfi/RT), (1-44)
31
где AGj* — свободные (гиббсовы) энергии активации удельной
(эквивалентной) электропроводности; а,- — соответствующие пред-
экспоненциальные множители. Заметим, что уравнение (1—41) в
неявной форме содержит зависимость % от температуры: зная тем-
пературную зависимость бит, можно прогнозировать изменение
удельной электропроводности от температуры.
Из выражения (1 — 28) можно вывести уравнение, связывающее
относительный температурный коэффициент удельной электропровод-
Х-р д 1п X
ности Вх =----с относительными температурными
G 2 О/
коэффициентами вязкости и диэлектрической проницаемости рЕ [562]:
+ (1-45)
из которого следует, что рх полностью определяется температур-
ными коэффициентами вязкости и диэлектрической проницаемо-
сти. Уравнение (1—45) позволяет прогнозировать возможность по-
явления отрицательного температурного коэффициента (т. е.
уменьшения электропроводности при повышении температуры) в
растворе данного состава. Очевидно, что знак рх определяется аб-
солютными величинами первого и второго слагаемых правой части
уравнения (1—45).
Относительный температурный коэффициент удельной электро-
проводности находит Щирокое применение в физико-химическом
анализе жидких систем [19, 530, 531, 562], поскольку диаграммы
этого свойства позволяют с достаточной точностью определять
стехиометрию продуктов присоединения.
Рассмотрение проблем термодинамики активации электропро-
водности следует начать с некоторых общих замечаний. Отметим
прежде всего, что уравнения (1—43), (1—44), позволяя из темпе-
ратурного хода электропроводности определять энтальпии акти-
вации электропроводности АНХ* и не дают возможности рас-
считывать энтропии активации, поскольку не известны абсолютные
величины предэкспоненциальных множителей в этих уравнениях и
их зависимость от температуры. Поэтому предложен ряд концеп-
ций, позволяющих теоретически рассчитывать величины ах%. Сре-
ди них наибольшее распространение получила теория переходно-
го состояния (ТПС), основы которой были сформулированы
Эйрингом [Ш]. За последние годы эта концепция была распро-
странена на транспортные процессы в растворах — вязкое тече-
ние, электропроводность, ионная миграция, диффузия [638]. Одна-
ко при этом часто не учитывали условный характер представле-
ний ТПС, вследствие чего полученные выводы не всегда оказыва-
ются физически обоснованными.
Общее уравнение константы скорости процесса в ТПС
kT I \ /1
ехр[----(1-46)
в принципе применимо для идеального газа [111, с. 389]. Переход
32
к жидкой фазе требует учета коэффициентов активности в раст-
воре, что в общем случае затруднительно, а при рассмотрении
скорости транспортных процессов, особенно электропроводности в
рамках данной теории, практически невыполнимо.
В рамках ТПС можно вывести уравнения для различных транс-
портных свойств, в частности для подвижности отдельного
иона [544]:
hi = (xz^) ©2/3^Г2/3 exp (- . (1-47)
Очевидно, что уравнение (1—47), позволяя из температурной
зависимости Хо,г- рассчитывать энтальпию ионной миграции АН^,
для расчета энтропии ионной миграции предполагает знание ве-
личины трансмиссионного коэффициента. В большинстве работ,
посвященных приложению ТПС к транспортным процессам, ве-
личина х явно либо неявно предполагается равной единице. Эта
точка зрения нашла отражение и в работе [638]. Здесь нелишним
будет вспомнить, что сами авторы ТПС полагали, что во многих
случаях х — весьма малая величина, и лишь предположительно
считали ее равной единице [111, с. 415]. В последнее время
М. И. Шахпаронов [618, с. 162] показал, что х не может быть боль-
ше 0,5.
Физическая модель процесса ионной миграции позволяет счи-
тать наиболее обоснованным значение х= 1/6 [716, 544].
С учетом этого обстоятельства в работе [159] предложено сле-
дующее уравнение для предельной эквивалентной электропровод-
ности:
или
zi f I е \2/3 / 1 / дхг \
- ТГ W «Р(- «Р (тН 48°)
В этих уравнениях величина предэкспоненциального множите-
ля может быть рассчитана (для чего, очевидно, требуется знать
легко находимую величину мольного объема индивидуального ли-
бо смешанного растворителя). С учетом числовых значений по-
стоянных величин уравнение (1—48) преобразуется в выражение,
позволяющее на основе экспериментальных данных рассчитывать
свободную энергию активации ионной миграции при любой тем-
пературе, следовательно, энтальпию и энтропию этого транспорт-
ного процесса:
= RT (8,6054 + 2/3 In Q - In Xo) Дж/моль (1—49)
(при выражении Q в м3- кмоль-1 и Хо— в м2-См-кмоль-1).
Отметим противоречие, лежащее в основе кондуктометрическо-
го определения констант электролитической диссоциации. Продиф-
ференцировав уравнение (1—32) по 1/7, получим с учетом (1—
3 - 4-653
33
1
44) выражение
+ с1-50)
связывающее энтальпии равновесного и активационного процессов.
В работе [145] показано, что Д/7Х — &НХ — &Не — разность
энтальпий активации диэлектрической релаксации и е, причем ДЯХ
раствора совпадает с таковой растворителя.
Рассмотрим влияние растворителя на термодинамические ха-
рактеристики активации ионной миграции [544]. Уравнение (1—44)
позволяет формально применить к ионной миграции соотношения
термодинамики равновесных процессов, положив a\=zeFl2l&h, где
I — среднее расстояние перескока иона [111].
В настоящее время термодинамические расчеты процессов вяз-
кого течения и электропроводности основаны на применении урав-
нения Эйринга с учетом величины мольного объема 0 раствори-
теля [158].
Согласно [544], величины ДС^, ДС^ определяются
— ДСх» = RT (In Xq — 2/3 In 0 4- tj),
Д6? = [In (т]0) 4-cj,
где 0 — мольный объем растворителя; С\ =—6,3019;
Из уравнений (1—51) и (1—52) следует, что
AG? —AG^
——— = In XqT] 4- 1/3 In 0 4- const
~ 3 (In %от] 4-1/3 In 0 4- const)
R ~ d(l/D
и
AlS^ ~ AlSn _ d [7* (In Ao r] 4- 1/3 In 0 + const)]
R “ dT
В общем случае, как отмечалось выше, Хо зависит от температу-
ры, а также от вязкости растворителя ц и диэлектрической про-
ницаемости е, т. е.
*0 = ДЛ п, 4 (1—54)
Последние два свойства в свою очередь зависят от температуры,
таким образом,
h = f[T, т] (7*), е(Т)]. (1-54,а)
Обычно в физической химии электролитных растворов рассмат-
ривают закономерности функции %р"Ц (In Хоц), поэтому представим
выражение (1—54) в виде
1пХот) = F[T, е(Т)],
или (1—54,6)
1га0= -1пт]4-ЛЛ е(Т)].
выражениями
(1-51)
(1-52)
С2=—0,8733.
(1-53)
(1-53,а)
(1—53,6)
34
В Продифференцировав (1—54,а) по МТ, получим
f d (1п %0) _ rf(lnn) , dF . dF de ,< rr.
4 d(l/T) ~ d(l/T) r d (l/T) de d(l/T) >
или
4- + Д^Го.е> (1—56)
где ДЯГ0, ДЯц — интегральные величины энтальпии активации ион-
ной миграции и вязкого течения, находимые из уравнений
“Г” = ± = Х° ИЛИ П)’ (1—57)
’ где &Нх0,т — температурная составляющая энтальпии активации ион-
ной миграции, свободная от влияния диэлектрической проницаемости;
— та часть интегральной энтальпии, которая связана с функ-
циональной зависимостью Х0 = /(е).
Температурная составляющая энтальпии активации ионной мигра-
ции, равная ЬН&т — — RdFId {МТ), рассчитывается по величине
тангенса угла наклона зависимости In fyyr]—1/7, по тем темпера-
Рис. 15. Графический метод расчета температурных составляющих термодинами-
ческих характеристик ионной миграции:
а — нахождение изодиэлектрическнх значений коррегированиой электропроводности; б — тем-
пературная зависимость изодиэлектрическнх значений коррегированиой электропроводности.
турным значениям In ЛоТ|, которые взяты из изодиэлектрического
сечения политермических зависимостей In Аю'п = /:’(е), т. е. при по-
стоянной е (рис. 15).
Следует отметить, что расчеты ДН/1 по выражению (1—44)
и по аналогичному экспоненциальному уравнению для вязкости, с
одной стороны, и расчет этих величин на основе уравнений (1—51)
и (1—52) —с другой, дают практически совпадающие результаты,
так как величина
[5 (In в)/д {МТ)] <с [3 (In К0)1д{МТ)] « [3(In n)/d (1/Т)].
3*
35
При рассмотрении закономерностей изменения АЯ^В в сме-
шанных растворителях на основе уравнения (1—55) в общем слу-
чае следует принимать во внимание изменения температурного
коэффициента е с составом смешанного растворителя, однако в
большинстве реальных систем величина де/др/Т) весьма мало
изменяется с составом растворите-
ля, и этим изменением можно пре-
небречь.
Универсальные растворители.
В данном случае под универсаль-
ными растворителями будем пони-
мать среды, для которых выполня-
ется соотношение (1—38). В этом
случае из выражений (1—55) и
(1—38) следует
ДЯ*
Ло,/
R
Рис. 16. Зависимость логарифма
коррегироваииой электропроводно-
сти от обратной диэлектрической
проницаемости тетраэтиламмоний-
пикрата в смешанном растворителе
вода — метанол.
да 1 дЬ
д(\/Т) ~Тд7Т/тТ ’
(1-58)
АЯМ . b d(l/e)
R
Выражение (1—53,а) с учетом уравнений (1—58) и
быть представлено в виде1
АД£-АЯи________ь d(l/e) да ..______________
R - ° d(l/T) д(1/Т) d(l/T) •
Специфические растворители. В общем случае диссоциации
электролита в неуниверсальных средах, как следует из уравнения
(I—55), величина АЯ^ — АЯ* определяется характером политер-
мических зависимостей F=f(l/e). Расчет квазитермодинамических
характеристик процессов ионной миграции и вязкого течения ряда
систем со специфической сольватацией показывает, что в этих
случаях зависимости In XpT] = f (1/е) нелинейны и описываются по-
линомом
d(l/T) ‘
(1-59)
(1-59)
может
дь
(1-60)
F — а(Т) + Ь/г 4- с/е 4-... 4- ае 4- ре2 4-..., (1—61)
от температуры зависит только свободный член а, а в
случаях он постоянен. Для примера на рис. 16 пред-
ставлена зависимость In Алл = f 1/е Для Et4NBr в смешанном раст-
ворителе MtOH—НгО.
В этом случае уравнение (1—60) преобразуется к виду
АЛ^-АЯ^ да
——^ = ^ + ф(П
’При учете изменении плотности с температурой в правой части уравнения
, 3(Ш0)
появится член 1/3 '•
в котором
некоторых
(1-62)
36
Из выражения (1—62) следует, что изотермическая величина
(ДЯ£-ДЯ*) не зависит от е и соответственно от состава смешан-
ного растворителя, т. е. зависимости ДЯ^0 и ДЯ^ как функции сос-
тава симбатны.
Таблица 5. Расчетные (по выражению (1—53)) и температурные
составляющие (по выражению (1—56)) энтальпии ионной миграции HSO3CH3(HA)
и CH3(C8H17)sN+ SO3CH^(R4NA) в н -алифатических спиртах (кДж/моль, 298 К)
Растворитель НА R.NA
А^интегр днт А^интегр днт
Метанол +9,5 —1,5 + 12,5 +3,7
Этанол + 10,3 —1,3 + 14,1 +3,9
Пропанол + 11,9 —1,2 +16,4 +4,2
Бутанол + 13,1 —1,1, +16,8 +4,3
Пентанол + 14,6 — 0,9 + 17,8 +4,7
Гексанол + 16,3 —0,9 +18,9 Н-4>9
Таким образом, если политермические зависимости In Хо ц =
= /(1/е) в растворителях симбатны, то ДЯх0— ДЯ^ = const, а в
общем случае миграция ионов полностью описывается и определяется
скоростью перескоков молекул растворителя в соседние положения
равновесия и температурно-диэлектрическим влиянием среды.
Величины ДЯ* т более информативны по сравнению с интег-
ральной характеристикой, включающей по крайней мере три эф-
фекта: температурные изменения подвижности ионов, вязкости и
диэлектрической проницаемости.
Иллюстрацией последнего могут служить данные табл. 5, где
сопоставляются интегральные энтальпии ионной миграции и их тем-
пературные составляющие для электролитов CH3SO3H и СН3(С8Н17)3-
•NSO3CH3 в «-алифатических спиртах. Обращает внимание, что ве-
личины ДЯдоТегр > 0 как для соли, так и для кислоты. Это противоре-
чит данным по механизму электропроводности в данных раство-
рах: в то время как в растворе кислоты перенос тока осуществля-
ется тоннельным эффектом (прототропный, эстафетный механиз-
мы), в растворах соли механизм переноса тока — естественно,
ионно-миграционный. Это различие в механизмах переноса тока
очень хорошо отражается величинами ДЯ+%,Г; в то время как для
растворов кислоты этот процесс экзотермичен, в растворах соли
он в соответствии с физической картиной переноса эндотермичен.
Величины ДЯ^х, иптегр. неоправданно велики и по абсолютной ве-
личине, так как значительно превышают энергию теплового дви-
жения молекул растворителя.
37
1.4. КИСЛОТНО-ОСНОВНОЕ ВЗАИМОДЕЙСТВИЕ МЕЖДУ
КОМПОНЕНТАМИ РАСТВОРА И ЕГО ВЛИЯНИЕ НА ИОННУЮ
МИГРАЦИЮ И СИЛУ ЭЛЕКТРОЛИТА
1.4.1. Кислотно-основные характеристики
электролита
Известно [20], что взаимодействие иона с окружающими нейтраль-
ными дипольными (ион-дипольное взаимодействие) либо заряжен-
ными молекулами (ион-ионное взаимодействие) в первую очередь
определяется двумя его феноменологическими характеристиками,
от которых зависит напряженность создаваемого ионом электро-
статического поля: зарядом и радиусом иона. Поэтому все попыт-
ки разработать систему классификаций либо теорий, позволяющих
прогнозировать поведение иона в конкретной среде, в явной либо
неявной форме учитывают названные характеристики.
Учет кислотно-основных взаимодействий в электролитной си-
стеме наиболее удобно проводить с помощью теории М. И. Усано-
вича [509]. Согласно этой теории каждый из катионов, включая
протон, является кислотой, а каждый анион, включая электрон —
основанием. Таким образом, в круг кислотно-основных равнове-
сий вводятся окислительно-восстановительные процессы.
Теорию кислот и оснований М. И. Усановича часто критикуют
[614, 173, 344]. Не останавливаясь на полемике вокруг этой теории,
отметим, что большинство критических тезисов игнорирует оче-
видное положение, согласно которому уменьшение числа класси-
фикационных признаков (а в теории Усановича они сведены к
минимуму и заключаются лишь в знаке отдаваемого либо прини-
маемого молекулой заряда) в любой теории ведет к адекватному
расширению ее юрисдикции и уменьшению степени конкретности
классифицируемого понятия. Впрочем, тезис о катионах-кислотах
и анионах-основаниях, столь удобный для прогнозирования пове-
дения иона в данной среде, по-видимому, никем не оспаривался.
Пользуясь теорией М. И. Усановича, можно утверждать, что в
ряду катионов одного химического типа и, следовательно, с оди-
наковым строением внешнего электронного слоя и одинаковым за-
рядом кислотность снижается по мере увеличения ионного ради-
уса (например, в рядах Li+>Na+>K+>Rb+>Cs+ и Са2+>
>Sr2+>Ba2+ и т. п.). В этом же направлении уменьшается и ос-
новность анионов (например, F~>Cl_>Br_>J_).
Весьма часто, основываясь на уравнении (1—37}, в качестве
меры сольватации иона, т. е. его способности вступать в ион-ди-
польное взаимодействие, используют величину обратного кристал-
лографического радиуса 7— (см., например, [399, с. 29]).
КР
Существует немало попыток количественно соотнести кислот-
ность либо основность различных по природе и заряду ионов [962].
В качестве примера приведем две шкалы. Первая из них предло-
жена Марксом и Драго [1035, 1036]. Пользуясь эмпирическими па-
раметрами этих авторов для кислоты А и основания В, можно со-
38
Таблица 6. Параметры кислотности и основности для некоторых
кислот-катнонов и основаниб-анионов [1035, 1036]
Ион 1 D 0 Ион D 0
Кислотные параметры
н+ 311,6 81,95 Mg2+ 253,7 17,75
Li+ 132,6 9,И Са2+ 2132 12,45
Na+ 112,6 5,86 Sr2+ 199,9 9,13
К+ 100,0 3,46 Ва2+ 190,6 7,50
Rb+ 95,6 2,65 М.п2+ 255,7 17,11
Cs+ 90,7 2,90 Fe2+ 270,8 22,49
Ag+ 168,0 18,87 Со2+ 273,1 20,47
T1+ 129,3 5,74 Ni2+ • 283,0 19,73
Mt<N+ 204,8 50,00 Cu2+ 293,0 21,63
Et4N+ 173,4 40,01 Zn2+ 281,4 22,44
n-Pr«N+ 164,0 41,33 Cd2+ 260,4 17,34
Ph4N+ 196,1 55,44 Sn2+ 294,5 6,87
J+ 196,5 25,73 Pb2+ 241,8 19,97
Be2+ 329,6 25,84 232,5 10,58
Основные параметры
F~ >-42,6 94,47 Se2- —122,9 9,77
Cb —16,0 63,57 он- —48,1 211,44
Br- —10,5 54,5 CN- —30,1 136,48
J- —3,6 47,06 ЙОГ —33,8 0,07
S2- —135,0 62,06 H~ —29,8 145,22
Таблица 7. Показатели жесткости 6 ионов по К. Б. Яцимирскому
Кислота Основание
Катион 6 Катион 6 Анион a Анион a
H+ 9 Ni2+ 02 он- 6,3 s2- —8,4
Zr*+ 4,9 Zn^ 0,2 F- 1,7 Cl~ —9,3
Cr3+ 3,8 Pb2+ о 3 NO3 0,9 Br- —13,7
Fe3+ 2,7 Cd2+ —1 Ac~ 0,8' J- —18,7
In3+ 1'2 CHsHg+ —2,8 SO42- 0t,5
Cu2+ 1,0 Ag+ —3,5 (H2O) 0,0
Fe2+ 0.6 Hg2+ —4,0 CN- —4,9
ставить относительную шкалу экзотермичности процессов нейтра-
лизации— присоединения катиона к аниону:
- ДЛГдв = [рА - Яв)2 + вАвв]1/2. (1-63)
Параметры Маркса и Драго для некоторых ионов приведены в
табл. 6.
Исходя из предпосылок теории жестких и мягких кислот и ос-
нований [547, 344], К. Б. Яцимирский [644] предложил шкалу по-
казателей кислотности некоторых ионов (табл. 7). Несколько
шкал жесткости (мягкости) катионов приведено в работе
[116, с. 366].
39
1.4.2. Кислотно-основные характеристики
растворителя
Вопросы классификации растворителей, применяющихся в прак-
тике электроосаждения металлов, будут подробно рассмотрены в
разделе 3.1.1. Здесь мы остановимся лишь на характеристиках
растворителей, имеющих значение для обсуждения вопросов, ко-
торым посвящен раздел 1.4.
Прежде всего отметим широко распространенный принцип де-
ления растворителей на протолитические и апротонные. В связи с
тем что не выработан единый подход к трактовке и содержанию
этих терминов, прокомментируем эту систему классификации.
К протолитическим относятся растворители, проявляющие по от-
ношению к растворенному веществу протонно-донорную либо про-
тонно-акцепторную функцию. К апротонным относят растворите-
ли, которые не могут принимать участие в процессах переноса
протона. Если отнесение растворителя к классу протолитических
в большинстве случаев может быть проведено априорно — на ос-
новании его химических свойств (так, к этому классу относятся
спирты, карбоновые кислоты, фенолы и т. д.), то отнесение раст-
ворителя к классу апротонных может быть проведено лишь с уче-
том особенностей системы растворенное вещество — растворитель
в целом. Так, бензол, который в системах с L-кислотами выступа-
ет как апротонный растворитель, будучи растворителем для очень
сильных Н-кислот (например, жидкого фтористого водорода) ли-
бо для очень сильных оснований (например, жидкого аммиака),
способен проявлять протонно-донорную либо протонно-акцептор-
ную функцию:
HF + СвН6 СвН6 • HF СвНГЕ~,
NH3 + CeH6 NH3 • ОД NH4+ • ОД~
В свою очередь, протолитические растворители в зависимости
от отношения к растворенному соединению подразделяются на
протогенные (кислые), протофильные и амфипротонные, т. е. ра-
створители, которые в зависимости от химической природы раст-
воренного вещества могут либо присоединять, либо отдавать про-
тон (помимо только что приведенного, впрочем, достаточно экстра-
вагантного примера с бензолом, к числу амфипротонных относятся
кетоны. Кроме того, каждый из перечисленных классов органиче-
ских соединений может в зависимости от природы партнера про-
являть либо протонно-донорную, либо протонно-акцепторную
функцию)1. К амфипротонным часто относят растворители, кото-
рые вступают с растворенным веществом лишь в универсальную
сольватацию. В табл. 8 приведены в рамках данной классифика-
1 Отметим, что, строго говоря, любое химическое соединение может и
должно быть амфотерным. На этом тезисе в известной мере основана хими-
ческая теория кислот и оснований [547].
40
ции примеры отнесения растворителей к каждой из указанных
групп.
Сложность проблемы количественного описания основности
(кислотности) растворителей и неоднозначность ее решения обус-
ловили появление большого числа работ в этом направлении, да-
же краткий обзор которых был бы вряд ли уместен в данной кни-
ге. Поэтому изложение этого вопроса ограничено здесь лишь при-
кладными аспектами и ни в коей мере не претендует на полноту.
С современным состоянием вопроса читатель ознакомится в рабо-
тах [926, 386, 101, 135, 435, 884, 326].
Таблица 8. Классификация растворителей по кислотно-основным свойствам
Растворители Протогенные (кислотные) Протофильные (основные) Амфи протонные
Протолитические Протонные H2SO4, НСООН, RCOOH, С6Н5ОН, CHCU НМ, НБ СН3СОС1, (СН3СО)2О NH3, n2h4, rnh2, ЭДА, ФА, МАА R3N, Ру, ГМФТА, ДМСО, ДМФА,- Диоксан, ТГФ, Et2O, RCOOR2 Н2О, ROH, rcor2 CCU, Г, ЦГ, Б
Наибольшее распространение получили спектральные и кало-
риметрические шкалы основности, базирующиеся соответственно
на сопоставлении спектральных характеристик какого-либо репер-
ного соединения в данном растворителе с растворителем, выбран-
ным в качестве стандарта (чаще всего w-гексаном), либо на теп-
ловых эффектах растворения определенного химического соеди-
нения.
Одной из наиболее важных характеристик является величина
основности В, введенная В. А. Пальмом [386] и представляющая
собой сдвиг полосы ОН фенола в ИК-спектрах этого реперного со-
единения в данном растворителе. Еще большее распространение
получила калориметрическая шкала В. Гутмана донорных чисел
(DN), представляющих собою энтальпию смешения пятихлори-
стой сурьмы с данным растворителем. По-видимому, нет основа-
ний предпочитать какую-либо разновидность этих параметров ос-
новности — спектральную либо калориметрическую. Установлено
[326], что шкала донорных чисел DN при условии учета парамет-
ра поляризуемости растворителя, вводимого функцией Лоренца —
Лорентца, хорошо коррелирует со спектральной шкалой основно-
сти В:
DN = 8,200 + 0,08765 — 35,734—^1 (г = 0,973, з = 3,32). (1—64)
В качестве параметров электронофильности (кислотности)
наибольшее распространение получили акцепторное число {AN),
представляющее химический сдвиг сигнала 31Р в ЯМР-спектрах
41
растворов оксида триэтилфосфина в данном растворителе [884,
962], а также параметр Ет, а именно энергия переноса заряда 1-
этил-4-метоксикарбонилпиридиниййодида в соответствующем раст-
ворителе.
В литературе предложены корреляционные уравнения, с помо-
щью которых учитываются неуклеофильность либо электрофиль-
ность растворителей. Так, в работе [995] выведено двухпараметро-
вое уравнение, согласно которому растворитель характеризуется
величиной
У = ад + + OzEj.. (1—65)
В. А. Пальм [386] предложил четырехпараметровое уравнение,
учитывающее полярные свойства растворителей (показатель пре-
ломления п и диэлектрическую проницаемость е):
„2_ 1 р _ 1
У = + а1 пг 0,2 2е + 1 "I" "I- аьЕт- (1—66)
Наконец, Р. Г. Макитра и Я. Н. Пириг [326] установили, что
величина у лучше всего описывается пятипараметровым уравне-
нием, учитывающим помимо ранее рассмотренных также пара-
метр когезии 62 — отношение мольной теплоты растворения к
мольному объему растворителя:
•7 2 — 1 В - - — 1
У — <2о + а1 га2_|_2 °2 2е+ 1 I- a^Z + "I- ^Ег'
(1-67)
Параметры основности и кислотности наиболее употребимых в
электрохимической практике растворителей приведены в табл. 2.
приложения.
1.4.3. Кислотность апротонных кислот
В большинстве случаев применяемые в электрохимической прак-
тике растворители относятся к электронно-донорным (основным),
по отношению к которым соли металлов проявляют свойства ап-
ротонных кислот. Соли металлов III—V групп периодической си-
стемы элементов классифицируются как кислоты в рамках элек-
тронной теории кислот и оснований. Что же касается солей вооб-
ще, то каждая из них считается апротонной кислотой в основном
растворителе, поскольку в таких системах преимущественно соль-
ватируется катион. Отсюда видно, что объекты, являющиеся кис-
лотами в рамках электронной теории (теории Льюиса), которые
мы далее будем обозначать как L-кислоты, являются частным слуг
чаем апротонных кислот, которые мы далее будем обозначать как
U-кислоты (т. е. кислоты по Усановичу).
Разработка меры кислотности неводных растворов относится к
числу весьма непростых проблем теории растворов. Здесь можно
выделить две основные задачи: сопоставление силы различных
42
кислот в одном растворителе; сопоставление силы одной кислоты
в различных растворителях. Первая из этих задач для Н- (водо-
родных, протонных) кислот полностью решена. В общем случае
эта задача сводится к определению равновесной концентрации
иона лиония, образующегося при присоединении протона к моле-
куле растворителя. Так, в случае воды кислотность раствора опре-
деляется активностью иона Н3О+, в случае жидкого аммиака —
активностью иона NH4+, в спиртовых растворах — активностью
ионов ROH2+ и т. д. Эти величины определяются по различным
экспериментальным методикам, и, поскольку, сопоставляя силу
различных Н-кислот в одном растворителе, приходится определять
концентрацию одного и того же катиона лиония, задача решается
сравнительно несложно с помощью экспериментальных либо ра-
счетных методик [173, 10, 101].
Заметим, что значения констант диссоциации, определенные
разными методами, различны, что обусловлено не только погреш-
ностями соответствующих методик, но и принципиальными их раз-
личиями. Так, кондуктометрия позволяет определять равновесную
концентрацию свободных (разумеется, сольватированных) ионов;
в то же время спектрофотометрия дает информацию о суммарной
концентрации ионов, находящихся как в свободном состоянии, так
и в виде ионных ассоциатов (см. схемы (1—14) и (1—16)).
Сопоставление силы различных U-кислот в одном растворите-
ле усложнено тем, что различные U-кислоты образуют в одном
растворителе S катионы различного состава. Так, кислотность ра-
створа трибромида алюминия в растворителе S определяется
активностью катиона [AlBr2S]+, а кислотность раствора триброми-
да сурьмы в том же растворителе — активностью катиона
]SbBr2S]+. (В этом примере растворитель S не вступает с галоге-
нидами в реакцию сольволиза; в противном случае задача еще
более осложнилась бы.)
В растворителях, автоионизация которых связана с перено-
сом не протона, а галогена, например Sb2Cl6:*i:SbCl2+4-SbCl4~,
2РОС1з5йьРОС12+4-РОС14- возможен в принципе такой же подход
к определению кислотности, как и в случае Н-кислот. Действи-
тельно, кислотность растворов двух соединений А1С13 и TiCl4 в
растворителе SbCh можно характеризовать активностью иона
’лиония SbCl4+, образующегося в результате реакций AlCl3-f-
+SbCl5^SbCl4++AlCl4- и TiCl4+2SbCl5=^2SbCl4++TiCl62- (не-
трудно заметить, что в приведенном примере растворенные в
SbCls соединения выступают по отношению к растворителю в ро-
ли основания). Однако подобные случаи весьма редки.
Более распространенным случаем можно считать тот, когда
растворенное в галогенотропном растворителе соединение, высту-
пая по отношению к растворителю в роли основания, будет повы-
шать концентрацию ионов лиония. Так, основность растворов LiCl
и NaCl в трихлориде сурьмы, в котором устанавливаются равно-
весия LiCl-|~SbCl3^Li++ SbCl4~ и NaCl+SbCl3^Na+-f-SbCl4_,
удобно характеризовать активностью ионов лиата SbCl4~
43
В соответствии со сказанным заключения об относительной си-
ле U (L)-кислот в одном растворителе выводят на основании раз-
личных теоретических либо экспериментальных предпосылок. Так,
можно полагать, что поскольку центральный атом в SbCl5 харак-
теризуется большим дефицитом электронов по сравнению с SbCl3,
то первое соединение будет более сильной L-кислотой по сравне-
нию с трихлоридом. (Заметим, впрочем, что и эти соображения
не являются априорными. Так, в работе [191] делается вывод о
том, что SbCl3— более сильная кислота, чем SbCl5).
Исходя из такого рода предпосылок, Д. Сэтчел и Р. Сэтчел
[483] предложили сопоставительные ряды для некоторых U-кислот:
а) ВХ3 > А1Х3 > FeX3 > GaX3 > SbX5 > InX3 > SnCl4 >
>AsX5>ZnX2>HgX2; 6) SnCl4>SnBr4>SnI4; в) ICl>IBr>
> I2> Br2> Cl2.
Закономерности, определяющие последовательность в измене-
нии апротонных кислот, которые представлены в этих рядах, до-
статочно очевидны. Отметим лишь полную логичность последнего
ряда, поскольку в нем уменьшается тенденция к отщеплению (и
соответственно — устойчивости) катиона; именно поэтому сила
кислот в ряду Х2 уменьшается от йода к хлору, так как молекула
йода гораздо легче отщепляет катион I+, чем молекула хлора —
весьма неустойчивый катион С1+.
В работе [358] индикаторным методом установлена следующая
последовательность силы кислот в эфирах карбоновых кислот:
SbCl5 > ТаС15 > NbCl5 > SnBr4 > SnCl4 > TiCl4 > GeCl4 >
> AsCl3 > SbBr3 > SbCl5.
В литературе имеются предложения считать мерой силы апро-
тонной кислоты тепловой эффект растворения соответствующей
кислоты в данном растворителе. Так, в работе [1112] калориметри-
чески найденные величины АЯ позволяют расположить кислоты
по силе в этилацетате в ряд: SbCl5«SO3>SnCl4>TiCl4>AlCl3:>
>AsCl3.
Не приходится говорить, что в рамках такого подхода нельзя
сопоставлять ионофорные и ионогенные U-кислоты, во всяком слу-
чае без учета энергии кристаллической решетки первых.
В качестве меры относительной силы кислоты в работе [672]
была принята скорость ее каталитического действия на процесс
деполимеризации паральдегида в эфирном растворе, в результате
чего установлена последовательность:
ZnCl2 > (НС1)» НВг > ВС13 > SnCl4 > FeCl3> А1С13 > РеВг^ПСЦ.
Очевидные расхождения в приведенных рядах не следует рас-
сматривать как противоречия. Причины этих расхождений могут
заключаться в различии механизмов взаимодействия разных апро-
тонных кислот с данным растворителем, в различии соотноситель-
ного влияния универсальной и специфической сольватаций на каж-
дую из кислот и т. д.
44
Решение аспекта кислотности неводных растворов апротонных
кислот — сопоставление силы одной кислоты в разных растворите-
лях — значительно труднее. В случае Н-кислот данная проблема
решается разработкой единой шкалы кислотности, предложенной
Н. А. Измайловым [173] и далее развитой В. В. Александровым
]10]. Существуют иные методы сопоставления кислотности, обзор
которых достаточно полно
приведен в монографиях
|[101, 10].
До сих пор отсутствует да-
же самый общий подход,
пользуясь которым можно бы-
ло бы сопоставить кислотность
двух растворов, чьи кислые
свойства определяются соот-
ветственно катионами
(MeXz_1S1]+ и [MeXz_iS2] +
Здесь можно назвать только
оценочные методы определе-
ния относительной силы U(L)-
кислот. В большинстве невод-
ных растворителей U-кислоты,
применяемые в композициях
для электроосаждения метал-
лов, являются слабыми элект-
Рис. 17. Избыточный мольный объем в
двойных жидких системах, образованных
трихлоридом сурьмы с «-алифатическими
спиртами:
1 — метанол; 2 — этанол; 3 — пропанол; 4 — бу-
танол; 5 — пентанол.
ролитами. Поэтому в соответствии со схемой (1—14) большая
часть U(L)-кислоты, подвергшейся специфической сольватации,
будет находиться в виде продукта присоединения либо в виде
ионного ассоциата, — относительный выход, а в ряде случаев и
константы образования которых определяются методами количе-
ственного физико-химического анализа [19, 530, 547]. Сопоставле-
ние глубины взаимодействия в системе U(L)-кислота — раствори-
тель может служить относительной мерой глубины взаимодейст-
вия, следовательно, силы данной кислоты по отношению к раство-
рителю.
Таблица 9. Свойства двойных жидких систем SbCl3 —н-алифатический спирт
(в точках экстремумов), 25 °C
Спирт Сжатие, % Отклонение от аддитивности (50 °C) х-10» (25 °C) ХЦ-102 (25 °C)
Плотность, г /см3 Мольный объем, см8-моль—1
С1 7,5 0,142 4,29 7,6 10,5
с2 5,5 0,102 3,78 7,2 9,7
Сз 4,3‘ 0,077 3,41 4,3 7J
с4 3,8 0,065 3,41 3,25 5,3
С5 8,0 0,054 3,03 2,6 4,9
45
В качестве примера на рис. 17 сопоставлены изотермы откло-
нения мольного объема от аддитивности двойных жидких систем
трихлорид сурьмы — н-алифатические спирты [535]. Данные, ха-
рактеризующие глубину взаимодействия в этих системах, приве-
дены в табл. 9. Как видно из данных таблицы, величины свойств
систем в точках экстремумов, т. е. степень химического взаимодей-
ствия и, следовательно, относительная кислотность растворов в
системах трихлорид сурьмы — н-алифатический спирт, закономер-
но уменьшаются с увеличением длины углеводородного радикала
спирта.
1.4.4. Кислотно-основное взаимодействие
и электропроводность неводных растворов 1
На антибатность кристаллографических радиусов и подвижность
ионов в водных растворах обращается внимание уже в элементар-
ных курсах физической химии. Действительно, подвижность кати-
онов лития, натрия, калия, рубидия и цезия (ряд, в котором кри-
сталлографический радиус возрастает) равна соответственно 38,6;
50,1; 73,5; 77,8; 77,2. Эта закономерность объясняется снижением
степени сольватации по мере роста Гкр и как следствие этого
уменьшением эффективного ионного радиуса. Отмеченную
закономерность можно считать общей. Сводится она к сле-
дующему:
1. С увеличением кристаллографического радиуса либо умень-
шением жесткости катионов степень их сольватации основными (в
частности, протофильными) растворителями уменьшается. Очевид-
но, что изменение подвижности катионов будет антибатно степени
сольватации.
2. В кислотных (протогенных) растворителях указанная зако-
номерность менее выражена, поскольку степень специфической
сольватации катионов кислотными растворителями существенно
меньше, чем основными. В некоторых случаях подвижность кати-
онов с ростом их кристаллографического радиуса в кислотных ра-
створителях снижается в соответствии с величиной Гкр.
3. Степень сольватации анионов в общем случае снижается с
увеличением их ионного радиуса, в связи с чем возрастает под-
вижность. Однако в сильноосновных растворителях, сольватация
анионов в которых существенно ослабляется,, эффективный ион-
ный радиус будет незначительно отличаться от кристаллографиче-
ского радиуса, т. е. с ростом последнего величина Хо будет умень-
шаться.
Закономерности изменения чисел сольватации ионов в зависи-
мости от относительной кислотности (основности) иона и раство-
рителя определяются высказанными положениями (табл. 3 при-
ложения) .
1 Данные по подвижности ионов в различных растворителях сведены в
табл. 5 приложения.
46
Действительно, в то время как в ряду Li+—Na+—К+—Rb+—Cs+,
кислотность в котором ослабевает, числа сольватации в основных
растворителях снижаются, в кислотном растворителе — серной
кислоте — при переходе от более кислого Li+ к менее кислому
Na+ число сольватации возрастает. Во многих случаях можно
проследить конкуренцию основности растворителя и анионов.
Рис. 18. Зависимость предельной эквивалентной электропроводности ионов от
кристаллографического радиуса:
а — катионы щелочных металлов: 1 — АН; 2 — ТГФ; 3 — метиловый спирт; 4 — вода; 5 —
ДМФ; 6 — МФА; 7 —ДМСО; в — ФМ; 9 — ПК; б — анионы галогенов: 1 — АН; 2 — вода; 3 —
НМ; 4 — ДМФ; 5 — метиловый спирт; б —ДМСО; 7 — НБ; 8 — ПК; 9 — я-пропанол; 10— СФ.
Закономерности изменения подвижности ионов, определяемые
их эффективным (стоксовым) радиусом, следовательно, числом
сольватации, также согласуются с рассмотренными положениями
о сольватации ионов.
На рис. 18 приводятся подвижности катионов Li+, Na+, К+ и
анионов Cl-, Br~, I- в ряде растворителей как функция кристал-
лографического радиуса ионов. Поскольку выбранные для иллю-
страции данной зависимости ионы однотипны, очевидно, что от-
носительная кислотность катионов и относительная основность
анионов снижаются с увеличением кристаллографического ради-
уса. Как видно из рис. 18, подвижность катионов с повышением
Гкр возрастет практически во всех растворителях, что неудивитель-
но, поскольку даже наиболее кислый из этих растворителей —
47
Рис. 19. Зависимость коррегированной
электропроводности от обратного кри-
сталлографического радиуса иона
(пунктир — кривая, отвечающая за-
кону Стокса).
муравьиная кислота — является электронодонором, следовательно,
вступает в специфическую сольватацию с катионами. В противо-
положность этому и в соответствии с высказанным выше в случае
анионов четко проявляется связь подвижности ионов с кислотно-
основными характеристиками электролитной системы: в сильноос-
< сульфолан, ДМФА, ДМСО (см.
табл. 2 приложения), подвиж-
ность анионов снижается с рос-
том гКр — в соответствии с фор-
мальным использованием уравне-
ния Стокса (1—35).
Более детально о соотноси-
тельном влиянии электронодо-
норных и электронофильных рас-
творителей на подвижность ио-
нов с изменением кристаллогра-
фического радиуса последних
можно судить из данных табл. 10,
в которой приведены величины
кристаллографических радиусов
ионов гКр, величины радиусов
сольватированных ионов rs (пос-
ледние взяты из работы [1137])
и подвижность ионов в двух рас-
творителях, относящихся к различным классам.
Данные табл. 10 подтверждают высказанные выше положения.
По величинам rs видно, что катионы в обоих растворителях соль-
ватированы достаточно сильно и что степень сольватации умень-
шается в ряду Li+ — Cs+. В отличие от катионов анионы весьма
слабо сольватированы в слабопротогенном растворителе — мета-
ноле, и совершенно не сольватированы в сильноосновном — ДМФ.
Таблица 10. Кристаллографические радиусы, радиусы сольватированных
ионов гХ10 нм, и ионные подвижности некоторых ионов в метаноле
и диметилформамиде
Ион гкри ст снаон ДМФ Ион гкри СТ CHjOH ДМФ
rs Xq rs rs %0 rs
Li+ 0,60 3,8 39,8 4,1 23,6 С1~ 1,81 — 52,4 1,9 55,1
Na+ 0,95 3,3 45,2 3,4 30,0 Вг“ 1,95 2,7 56,5 f ,9 53,6
к+ 1,33 2,9 52,4 3,3 31,7 J“ 2,16 2,4 62,7 2,0 52,3
Rb + Cs+ 1,48 1,69 — — 3,2 3,0 32,4 34,6 ClOf 2,4 2,1 71,0 2,0 52,4
В соответствии с этим подвижность катионов возрастает в ряду
Li+ — Cs+, подвижность же анионов с ростом снижается.
Дополним сказанное еще некоторыми фактами. Подвижность
ионов щелочных металлов в таком сильнокислотном растворителе,
48
I
W как жидкий цианистый водород, изменяется в полном соответствии
Ж с законом Стокса и кристаллографическими радиусами ионов.
$ Так, подвижность катиона Li+ (135,5) выше, чем подвижность ка-
, ’ тиона Na+ (132,4), что объясняется ослаблением (либо отсутстви-
ем) сольватации катионов-кислот кислотным растворителем.
Аналогичный ряд подвижностей, полностью соответствующий
кристаллографическим радиусам катионов, и обратный тому, ка-
кой имеет место в основных растворителях, наблюдается в абсо-
) лютной серной кислоте [363, с. 1.36].
Выразительным примером влияния сольватации на подвиж-
ность ионов может служить зависимость коррегированной электро-
проводности от обратной величины кристаллографического ради-
» уса иона (рис. 19). Из рис. 19 видно, что подвижности только
больших и поэтому несольватирующихся тетраалкиламмонийных
ионов укладываются на теоретическую кривую, построенную в со-
ответствии с уравнением Стокса (1—35). Начиная с иона
(CH3)4N+ и далее к катионам щелочных металлов эксперимен-
тальные точки сходят с теоретической кривой, причем в соответ-
ствии со сказанным выше отклонение тем больше, чем меньше
Гкр, т. е. чем выше степень сольватации.
1.4.5. Кислотно-основное взаимодействие
и сила электролитов в неводных растворах 1
Выше рассматривались общие вопросы соотносительного влия-
ния диэлектрической проницаемости и энергии сольватации на
константу электролитической диссоциации. Проблема направлен-
ного подбора электролитной композиции для процесса электровы-
деления металла требует более углубленно и с несколько иных
позиций рассмотреть влияние химических характеристик раство-
ра на силу электролита.
Как следует из изложенного, равновесие между ионной парой
(ионным ассоциатом) и свободными сольватированными ионами
(процесс (1 —12)) определяется величиной диэлектрической про-
ницаемости, которая, в свою очередь, диктует величину энергии
ион-ионного взаимодействия, зависящую прежде всего от эффек-
тивных радиусов катиона и аниона. Таким образом, при прочих
равных условиях прежде всего при постоянной диэлектрической
проницаемости, степень электролитической диссоциации будет
тем больше, чем больше сумма эффективных ионных радиусов
ионов электролита. Последние же, как было показано выше, оп-
ределяются природой и степенью сольватации, т. е. связаны с
процессами кислотно-основного взаимодействия ионов электроли-
та, с одной стороны, и молекул растворителя — с другой.
Можно утверждать, что без привлечения представлений о кис-
лотно-основном взаимодействии объяснение многих закономерно-
1 Данные по константам (рК) диссоциации электролитов в наиболее рас-
пространенных растворителях приведены в табл. 4 приложения.
4 - 4-653 49
стей изменения констант диссоциации с изменением природы элек-
тролита либо растворителя было бы осложнено. Приведем приме-
ры, иллюстрирующие это положение. Разобьем ряды изменения
величин Кр, (рКд) с изменением природы катиона (при одинаковом
анионе) либо с изменением аниона (при одинаковом катионе) на
две категории: нормальные и аномальные. К первым из них будем
относить ряды, в которых сила электролитов возрастает (следова-
тельно, снижается величина рКя) с ростом' суммы кристаллогра-
фических ионных радиусов, ко вторым — те, у которых наблюда-
ется обратное соотношение между силой электролитов и суммой
кристаллографических ионных радиусов. Очевидно, что к нормаль-
ным рядам будут относиться те системы электролит —раствори-
тель, где степень сольватации (и, следовательно, эффективный
ионный радиус) одного из ионов будет постоянной, а противо-
ионы будут несольватированы, либо степень сольватации послед-
них будет повышаться симбатно кристаллографическому радиусу.
В случае ионофоров с различными катионами-кислотами и од-
ним анионом появление нормальных рядов следует ожидать в кис-
лотных растворителях, практически не сольватирующих либо весь-
ма слабо сольватирующих катионы. Действительно, сила ацетатов
щелочных металлов в абсолютной уксусной кислоте повышается
в ряду Li+ — Na+—К+ (величины рКл равны соответственно 5,43;
5,03 и 4,50). Аналогичная картина наблюдается в случае броми-
дов щелочных металлов в жидком SO2: в том же ряду катионов
величины рКя равны соответственно 4,57; 4,32 и 3,85. Однако, по-
скольку в электрохимической практике гораздо чаще применяют-
ся электронодонорные (основные) растворители, соответственно
чаще встречаются аномальные ряды, в которых сольватация ка-
тиона-кислоты уменьшается с увеличением кристаллографическо-
го ионного радиуса.
Сила перхлоратов щелочных металлов в АН изменяется сле-
дующим образом (в скобках, как и всюду далее, приводятся ве-
личины рДд): Li+ (1,20), Na+ (1,23), К+ (1,43), Rb (1,53), Cs+
(1,56). Этот ряд свидетельствует о том, что эффективный ионный
радиус уменьшается, что совершенно естественно, поскольку в
этом же направлении снижается сила кислот-катионов. Интерес-
но, что в данном основном растворителе, где анионы сольватиру-
ются весьма слабо, ряд, составленный по анионам, является нор-
мальным. Так, для растворов солей тетраметиламмония величины
рКп изменяются следующим образом: Вг~ (1,77), J- (1,53), С1О4~
(1,32), BPh4" (1,12).
Аналогично к аномальным относятся ряды фторидов щелочных
металлов в ТГФ; Li+ (5,4), Na+ (6,20), Cs+ (7,86); йодидов ще-
лочных металлов в этаноле: Li+ (1,01), К+ (1,83), Cs+ (1,85)
(аналогичный ряд для хлоридов щелочных металлов в этаноле
см. в работе [116, с. 524]) и многие другие.
Для рядов, составленных по монотонному изменению аниона,
появление аномальных закономерностей следует ожидать в силь-
нокислотных растворителях с преимущественной сольватацией
50
1
f- аниона, но, к сожалению, соответствующие данные в литературе
отсутствуют.
Нередко можно обнаружить ряды ионофоров, для которых в
данном растворителе величины Лд изменяются немонотонно. Это
обстоятельство также находит логичное объяснение в рамках при-
нятой модели зависимости степени сольватации от кислотности
(основности) иона. Эффективный ионный радиус в первом и до-
статочном для данного рассмотрения приближении представляет
собой сумму кристаллографического ионного радиуса и диаметра
молекулы растворителя (в наиболее распространенном случае —
мономолекулярности сольватной оболочки): Гэф = Гкр+2гва. Умень-
шение степени сольватации при росте гКр может компенсироваться
последним, и тогда изменение величин Кд в ряду будет экстре-
мальным. В качестве примера можно привести растворы пикратов
щелочных металлов в пиридине: Li+ (4,08), Na+ (4,37), К+ (4,00),
а также галогениды калия в метаноле: С1~ (0,70), Вг~ (0,49), J-
(1,05).
У многозарядных катионов, являющихся сильнейшими кисло-
тами, сольватная оболочка особенно велика. Это обстоятельство
определяет известное своеобразие электролитных характеристик
солей таких металлов в растворах. Так, галогениды радкоземель-
ных элементов, катионы которых присоединяют в водных раство-
рах до 13, а в спиртовых до 6 молекул растворителя, при замеще-
нии сольватной оболочки на прочно связанные с центральным
ионом органическими лигандами, уменьшают константу диссоци-
ации (рКя хлорида европия в метаноле составляет 2,42, а рКд фе-
нантралинового комплекса европия в том же растворителе — 3,14
[123]). Это обусловлено тем, что эффективный радиус многозаряд-
ного иона, сольватированного растворителем, настолько велик, что
превышает радиус даже весьма объемного комплексного иона.
1.5. ПРИРОДА ЭЛЕКТРОЛИТОВ В НЕВОДНЫХ РАСТВОРАХ
1.5.1. Некоторые вопросы термодинамики
ионных растворов
В изучение термодинамики растворов электролитов наибольший
вклад внесли советские исследователи. Основополагающие иссле-
дования в этом направлении были выполнены школами Н. А. Из-
майлова [173], К. П. Мищенко и Г. М. Полторацкого [347],
А. Ф. Воробьева [166]. За последние годы проведены очень важ-
ные теоретические и экспериментальные работы школой Г. А. Кре-
стова [228].
По Г. А. Крестову [228], переход газообразного иона с элек-
тронной оболочкой инертного газа в раствор описывается схемой
III IV
: S, : +KSP, (1-68)
iii Liv
1 П\
Г(±> : S„ : Sp
I II/
4*
51
где индексы г и ж означают соответственно состояния в газе и в
растворе. Границы I—I и II—II оконтуривают первую сольватную
оболочку иона — ближнюю сольватацию. Далее следуют перифе-
рийные сольватные оболочки, и, наконец, за границей IV—IV
дальней сольватации молекулы растворителя уже не изменяют
своих термодинамических свойств под влиянием иона.
Собственно химической связью с центральным ионом соединены
молекулы растворителя Sn, находящиеся в первой сольватной обо-
лочке. Однако в сферу влияния этого иона входит по крайней ме-
ре еще два сольватных слоя. Схема (1—68), в частности, поясня-
ет причину неоднозначных литературных данных относительно чи-
сел сольватации (гидратации) ионов [173, с. 172]. Это обусловлено
тем, что различные методы определения чисел сольватации фик-
сируют разное число сольватных слоев, причем далеко не всегда
ясно, даже по модели, положенной в основу метода, какое именно.
Термодинамические характеристики ионов в растворах деталь-
но рассмотрены в работах [80, 173, 347, 228]. Большинство этих
характеристик относится к водным растворам. Необходимо отме-
тить, что достоверность экспериментальных термодинамических
свойств ионных неводных растворов часто оставляет желать луч-
шего.
Важной характеристикой, показывающей изменение свойств
данного электролита с изменением растворителя, являются термо-
динамические параметры процесса переноса одного моля этого
электролита из растворителя, принятого за стандартный (в боль-
шинстве случаев в качестве стандартного растворителя выбирают
воду), в неводный растворитель.
Изменение термодинамических характеристик процесса перено-
са сопряжено с разностью термодинамических свойств сольвата-
ции свободных ионов и недиссоциированных молекул электролита
в двух сравниваемых растворителях [173, с. 212]. В табл. 9 прило-
жения [116, с. 312] приведены величины энтальпии и энтропии про-
цесса переноса некоторых ионов из воды в неводный растворитель.
При определении термодинамических характеристик электро-
лита в растворе экспериментальные данные относятся, разумеется,
не к отдельным ионам, а к электролиту в целом (к стехиометриче-
ской смеси катионов и анионов). Поэтому проблема разделения
термодинамических параметров (отнесение суммарных термоди-
намических характеристик отдельно к катиону и аниону) решается
на основании модельного расчета. Поскольку разные авторы ис-
пользуют различные модели [173, 347, 228], значения свободных
энергий, энтальпий и энтропий определенных процессов для от-
дельных ионов, приведенные в разных работах, могут не совпа.-
дать. Следует учитывать, что степень достоверности получаемых
экспериментально суммарных термодинамических характеристик в
различных работах далеко не одинакова. Поэтому характер вы-
водов о сопоставлении термодинамических свойств отдельных
ионов в различных растворителях пока что только качественный.
Количественные обобщения должны делаться с разумной осторож-
52
ностью. Поэтому, как и в случае анализа ионной миграции, теоре-
тические обобщения на данном этапе развития физической химии
неэлектролитных растворов лучше относить не к отдельным ионам,
а к электролитам в целом (к стехиометрической смеси ионов).
Поскольку перенос электролита из одного растворителя в дру-
гой сопряжен с процессами пересольватации, термодинамики это-
го процесса определяется его сольватационными характеристика-
ми, общие закономерности которых были рассмотрены в разде-
ле 1.4.
Примером, иллюстрирующим зависимость процессов замены
гидратной оболочки на сольватную (из молекул неводного раство-
рителя) от донорности растворителя, может служить рис. 20, на
котором константа процесса .пересольватации Cl(H2O)--f-S->-
->C1(S)“+H2O представлена как функция параметра основности
Ет растворителя (см. параграф 1.4.2) [169].
Экзотермичность процессов переноса в основные растворители
возрастает с кислотностью катиона; эндотермичность переноса для
анионов в тех же растворителях по
мере повышения основности аниона
также возрастает.
Поскольку упорядоченность
сольватного комплекса изменяется
симбатно энергии связи в этом ком-
плексе, экзоэнтропийность процесса
переноса также возрастает с повы-
шением кислотности катиона и сни-
жается с увеличением основности
аниона. Можно констатировать из-
вестную корреляцию между основ-
ностью растворителя (например, его
донорным числом) и величиной эк-
зотермичности процесса переноса
катиона; естественно, что между
основностью аниона и величиной
экзотермичности его переноса на-
блюдается обратная зависимость.
Второй важнейшей группой тер-
галогенов как функция параметра
полярности Ет неводных раство-
рителей:
1 — 1-метил-2-пирролидон; 2 — ДМАА;
3 — ДМСО; 4 — ДМФ; 5 — СФ; 6 — АН;
Г — НМ; 8 — метанол; 9 — ФА; 10— вода.
модинамических характеристик
электролита в растворителе является свободная энергия, энтальпия
и энтропия процессов сольватации — величины, находимые пря-
мым калориметрическим либо потенциометрическим методом.
В табл. 10. приложения приводятся ионные составляющие энталь-
пии сольватации ряда ионов в некоторых неводных растворителях.
Термодинамические параметры сольватации, приведенные в табли-
цах Приложения, могут служить характеристикой интенсивности
взаимодействия в системе ионофор — растворитель, но ограничен-
ность данных по сольватации в неводных растворителях и упоми-
навшаяся несогласованность между различными шкалами деле-
ния суммарных характеристик на ионные составляющие не позво-
53
ляют судить о закономерностях изменения AGsoiv и A/Aoiv при пе-
реходе от одного растворителя к другому. Для каждого же дан-
ного растворителя практически во всех случаях наблюдается хо-
рошая корреляция между энтальпиями сольватации и величинами,
характеризующими кислотность (основность) ионов.
1.5.2. Комплексообразование в системах
U(L) -кислота — растворитель
Как следует из схем (1 —12) и (1 —14), образование электролит-
ного раствора определяется последовательностью стадий взаимо-
действия растворенного вещества с растворителем, одной из кото-
рых является стадия (1—7) гетеромолекулярной ассоциации (об-
разования продуктов присоединения). Поэтому возможность при-
менения определенной системы соль — растворитель в качестве ос-
новы композиции для электроосаждения металла в значительной
степени определяется стремлением компонентов этой системы к
комплексообразованию. Уделим особое внимание рассмотрению
апротонных растворителей, поскольку большинство достаточно
сильных L-кислот с протолитическими растворителями вступает в
реакции сольволиза, что существенно ухудшает условия либо да-
же делает невозможным электролитическое выделение металла.
L-кислоты, галогениды элементов III группы периодической си-
стемы— бора, алюминия, галлия и индия [623, 521)]—образуют
продукты присоединения 1 : 1 1 с галоидалкилами [710, 719, 645,
646], простыми [330, 400, 409, 289, 810, 1103, 870, 328, 797, 1057,
982, 502, 1262, 1013, 622] и сложными эфирами [1262, 1053, 1109,
1002, 1004], альдегидами [1098, 1141], кетонами [1098, 338, 337, 121],
галоидангидридами карбоновых кислот [690, 873, 730, 894, 385, 92],
N-алкилзамещенными амидами [1113, 1111, 1148, 859, 996, 432],
третичными аминами [797, 805, 1117], нитрилами [1194, 1195, 647,
1086], нитросоединениями [574, 965, 966, 964, 117, 98, 209, 403, 412,
431, 850, 880, 336, 334, 849], ДМСО [822, 1067, 762, 731],
ГМФТА [630].
Наряду с эквимолекулярными продуктами присоединения об-
разуются аддукты 1 : 2 хлорида алюминия с простыми эфирами
[797, 1057, 982], диоксаном [622], ТГФ [1267], этилацетатом [1002,
1004], АН [996], НБ [880, 336], Ру [1267]. Хлорид индия образует
аналогичные продукты присоединения с диоксаном [622], диэтило-
вым эфиром [1002, 1004], ацетоном [121], АН [1117], НМ [118], Ру
[1117]; соединения 1 : 2 GaCl3 дает с нитробензолом [574], а также
с Ру и пипередином [874]. Соединения 2 : 1 обнаружены для бро-
мида алюминия с диоксаном [622] и Ру [175].
Имеются указания на образование соединений более сложного
состава галогенидов металлов III группы с различными донорами:
бромид алюминия с Ру дает продукты 1 : 3 [1268] и 4 : 3 [401]. Хло-
1 Здесь и далее первая цифра — число молекул U(L)-кислоты, вторая —
число молекул лиганда в молекуле аддукта.
54
риды алюминия и галлия с АН и ДМСО образует продукты соста-
ва 2: 3 [1194, 647, 1067]. Наконец, имеются требующие подтверж-
дения данные, что хлорид алюминия с диэтиловым эфиром [502]
и ДМСО [1067, 762, 761] дает продукты присоединения 1 : 6.
В литературе приведены многочисленные сведения о продуктах
присоединения галогенидов металлов IV группы с различными до-
норными растворителями. Здесь будут названы важнейшие соеди-
нения, которые образуют тетрагалогениды германия, олова, тита-
на, циркония и гафния с наиболее распространенными раствори-
телями.
Таблица 14. Состав соединений тригалогенидов элементов V—-А подгруппы
органическими лигандами
Лиганд Состав соединений Литература Лиганд Состав соединений Литература
Простые эфиры Сложные эфиры Ру ДМФА Хлористый бензонл 1 : 1, 2 : 1 1:1, 1:2 1:1, 1:2, 1 : 3, 2 : 1 1 : 1 1 : 1) [508, 519, 486] [1100] [218, 345, 262] [996, 1215] [804] НБ ТБФ ДМСО 1:1, 1:2 1:1, 1:2 1:2, 1:3 [333, 49, 218] [1162] [1016, 1114]
Продукты присоединения 1 : 2 эти тетрагалогениды дают с ди-
оксаном [1075], сложными эфирами [1109, 266—268, 380, 379, 318,
319, 377, 306—308], альдегидами [1113, 1148, 996, 1083], нитрилами
[823, 1017, 1138], ДМСО [762, 761], ТБФ [374]. В ряде систем обра-
зуются продукты присоединения 1:1. Такие соединения известны
для тетрагалогенидов олова, титана, циркония с диоксаном [1075],
уксусноэтиловым эфиром [1109, 1002, 435, 1111, 1148, 859, 996, 432,
964, 380, 379, 318, 319, 377, 376] ацетилхлоридом [674, 770], бензо-
илхлоридом [465], пропионитрилом [1138], Ру [1008], ТБФ [374],
ДМСО [1003], кроме того, с последним лигандом SnCl4 дает про-
дукт 2 : 3 [168]. Данные по аддуктам Sn (IV) приведены в ра-
боте [362].
Пентагалогениды элементов V группы — фосфора, мышьяка,
сурьмы, ниобия и тантала — дают соединения состава 1:1с про-
стыми эфирами [769], со сложными эфирами [307, 369], альдегида-
ми и кетонами [818], бензоилхлоридом [465, 1108], ДМФ [1111, 996],
АН [1195, 647, 666, 986], ДМСО и дифенилсульфоксидом [1111,
1016]. Способностью к четко выраженному комплексообразованию
с донорными растворителями обладают оксигалогениды элементов
V группы, которые образуют соединения состава 1:1с ГМФТА
[365], 1 : 2 и 1 : 3 — с ДМСО [759, 1114].
Состав соединений тригалогенидов элементов V А подгруппы
(фосфора, мышьяка, сурьмы, висмута) с органическими донорны-
55
ми растворителями весьма разнообразен (табл. И). Данные по
стехиометрии аддуктов SbX5 приведены в работе [362]. Известны
соединения тетрахлорида ниобия с ДМФА, ДЭФА, ДММА и
ГМФТА состава 1 : 2 [940].
Молекулярные соединения галогенидов, оксигалогенидов и ок-
сидов элементов VI группы известны в меньшем разнообразии,
чем в случае L-кислот III—V групп периодической системы эле-
ментов.
Соединения эквимолекулярного состава образуются в системах
тетрахлорид теллура — сложные эфиры [1109, 388], кетоны и суль-
фоксиды [388]. Соединения состава 1 : 1 дает хлористый тионил с
простыми эфирами [493], ДМФ [327], ДМАА [493], АН [223] и Ру
[225]. СгС15 и СгО2С12 образуют комплексы состава 1:3с ДМСО
[762, 761, 1114].
Комплексные соединения галогенидов и межгалоидных соеди-
нений с различными органическими донорными растворителями
весьма разнообразны. Подробный обзор исследований систем
межгалоидное соединение — органический растворитель дан в ра-
Рис. 21. Свободная энергия об-
разования аддуктов ацетнлаце-
тоната ванадила с нейтральны-
ми и анионными лигандами как
функция донорного числа рас-
творителя:
Г —ПК; 2 — АЦ; 3 —ДМФ; 4 —
ДМАА; 5 — ДМСО; 6 — Ру; 7-
ГМФТА.
боте я. А. Фиалкова [570]; обзор ли-
тературы по молекулярным комплек-
сам 12 с различными донорными рас-
творителями — в [57].
Хлорное железо образует эквимо-
лекулярные продукты присоединения с
уксусноэтиловым эфиром [1109] и
бензоилхлоридом [894], с НБ [510] и
пиримидином [763] — комплексы со-
става 1:2, с Ру — комплексы 1 : 2 и
1:4 [510, 763], с пиразином — 2:5
[510]. Наконец, с ДМСО хлорное же-
лезо образует аддукты составов 1 : 2 и
1 :6 [762, 761].
Комплексообразование L-кислот
других металлов VIII группы в орга-
нических растворителях изучалось ме-
нее разнообразно, чем в случае гало-
генидов металлов III—V групп. Иссле-
дованию комплексов кобальта посвя-
щены работы [127, 1048].
Для молекулярных соединений U(L)-кислот справедливы все
закономерности относительно интенсивности взаимодействия меж-
ду компонентами аддукта и их кислотно-основными свойствами,
изложенные в разделе 1.4. В качестве примера на рис. 21 приве-
дена зависимость свободной энергии образования соответствующе-
го аддукта ванадил (IV) ацетилацетоната с различными нейтраль-
ными либо анионными лигандами от донорного числа растворите-
ля-лиганда [1048].
56
1.5.3. Механизмы электролитической диссоциации
в неводных растворах
Механизм электролитической диссоциации, т. е. природа ионов,
образующихся в системе электролит — растворитель и участвую-
щих в переносе тока, стал предметом исследований лишь в послед-
нее десятилетие. Причины несколько запоздалого обращения к
столь важной проблеме заключаются, по-видимому, в том, что
природа ионов, образующихся при электролитической диссоциации
в водных растворах, часто представлялась априорно очевидной, и
эта очевидность механически переносилась на неводные растворы.
Углубленное изучение схемы возникновения электролитного раст-
вора, в частности термодинамические исследования, показало, что
даже в водных растворах установление чисел гидратации, границ
ближней и дальней сольватации имеет решающее значение для
полного описания электролитного раствора. В неводных же сре-
дах, где, в отличие от большинства водных растворов, в системе
электролит — растворитель присутствует намного больше равно-
весных форм (см. схему (1—14)), определение природы и состава
ионов имеет первостепенное значение для понимания процессов,
происходящих в системе. Очевидно также и прикладное значение
проблемы природы ионов в неводных растворах: вряд ли процесс
электроосаждения металлов из неводных растворов можно эффек-
тивно осуществлять, если не известна эта важнейшая характери-
стика системы.
Для исследований использовали такие эффективные методы
изучений неводных растворов, как ЯМР-спектроскопию на ядрах
атомов, входящих в состав молекулы растворителя L-кислоты
[347, с. 108, 71], ИК-спектроскопию [454], рентгеновскую дифракто-
скопию, метод меченых атомов [536, 550, 548, 537, 38, 549, 553,
34, 440, 193, 231].
Схемы электролитической диссоциации, приводимые в литера-
туре, могут быть подразделены на четыре основных типа:
1. Ионизация L-к и с л о т ы. Все L-кислоты проявляют до-
статочно четкую способность к автоионизации :
2MeXn МеХ+_1 + Me X^i • (1 -69)
Сольватация (основным растворителем) смещает равновесие
диссоциации L-кислоты вправо:
Me Хп + aS ... [Me Хп-т (S)a}m+ + тХ~. (1 -69а)
В тех случаях, когда кислота находится в избытке, анион соль-
ватируется молекулой кислоты
2МеХп (или Me2X2n) + aS ... [MeX„_m (S)a]m+ + [MeX„+m]m-.
(1—696)
1 Здесь и далее схемы диссоциации приводятся для галогенидов металлов,
что оправдывается также тем, что в большинстве случаев в композициях для
электроосаждения металлов из неводных растворов применяются именно гало-
гениды металлов.
57
Отсутствие (либо весьма малая степень) сольватации аниона
в схеме (1—69, а) справедливо в случае наиболее часто применя-
ющихся в практике основных растворителей; если же процесс дис-
социации протекает по схеме (1—69, б), то отсутствие сольватации
аниона объясняется тем, что его центральный атом координацион-
но насыщен.
Рассматриваемая схема приводится во многих работах
М. И. Усановича и его школы [509]. Так, возникновение ионов в
растворах трихлорида сурьмы в абсолютной уксусной кислоте опи-
сывается уравнениями [514]:
SbCl3 + НАс = [SbCl2 (НАс)]+ + СГ,
2SbCl3 + НАс - [SbCl2 (НАс)]+ + SbCl?.
Аналогичный механизм диссоциации предлагается и для раст-
воров L-кислот в спиртах [516] и сложных эфирах [514]. С послед-
ней работой согласуется и точка зрения С. П. Мискиджьяна [343].
О. А. Осипов предложил схему (1—69, б) для определения при-
роды ионов в системе пентахлорид ниобия — сложные эфиры [480,
481]. В работах [621, 813, 276, 275, 222, 277] на основе данной схе-
мы объяснено возникновение ионов в растворах галогенидов алю-
миния в разнообразных органических растворителях. Аналогичные
схемы можно найти в литературе в приложении к галогенидам
многих иных металлов.
В работе [208] на основании изучения мессбауровских спектров
приводится следующая схема электролитической диссоциации
хлорного железа в нитробензоловых растворах: Fe2Cl6+PhNO24=b
^[Fe2Cl5PhNO2]++Cl-
Схема (1—69) предлагается также для растворов галогенидов
металлов в неорганических растворителях. Так, в системе
FeCls — РОС13 установлено образование катиона [FeCl2-POCls]+ и
аниона FeCLc [363, с. 204].
В работах Ю. А. Лысенко [305, 309—317] отстаивается следую-
щий вариант схемы (1—69 б):
2MeXn + (а + b) S [MeX„_m (S)Jm+ + [MeXm+„ (S)b]n“.
Например, для растворов GaBrs в сложных эфирах предлага-
ется схема
2GaBr3 + 7Э [GaBr234]+ [GaBr433]~.
Отметим малую вероятность реализации подобной схемы, так
как она предусматривает, что электронодонорный растворитель в
одинаковой степени сольватирует и электронодефицитный катион
и анион-основание, причем последний к тому же координационно
насыщен анионами галоида.
2 . Ионизация растворителя. Способность жидкости
подвергаться процессу автоионизации:
2S^S[t) +S£). (1-70)
58
является общим свойством жидкого состояния вещества. Концент-
рация ионов S+(l) (ионов лиония) и S~(2) (ионов лиата) в индиви-
дуальной жидкости связана с распределением при данной темпе-
ратуре молекул по энергиям, с макродиэлектрической проницае-
мостью жидкости и другими факторами.
В зависимости от природы растворителя процесс автоиониза-
ции передается одной из следующих схем:
2RH = RH^ + R“ (2С6Н6 = СеН^ + С6Н?), (1-71)
2ROH = ROH^ + RO“(2CeH5OH = С6Н5ОН^ + CeH5O~), (1-71,а)
2ROR' = [R2OR']++ R'O~ [2СН3СООС2Н5 = СН3СОО (С2Н5)| +
+ СН3СОО“], (1—71,6)
Х2 = Х+ + Х~ (12 = 1+ + Г), (1-71,в)
2MeXn = MeXt-i + MeX7+i (2SbCl3 = SbCl^ + SbCl?). (1—71,г)
Несмотря на то что существует ряд методов определения кон-
стант автоионизации, в основном эти величины оценены прибли-
зительно. В табл. 12 приводятся значения рКал некоторых раство-
рителей.
Таблица 12. Константы автоионизации некоторых индивидуальных
растворителей при 25 °C
Растворитель Р«ш Растворитель Р*ш Растворитель растворитель PKai
H2SO4 нсоон СНзСООН 3,1 6,2 12,6 Н2О СН3ОН с2н5он (СНз)2СО СН3СОС2Н5 14,0 16,9 19,5 25,9 25,6 NH3 NH2C2H4OH ДМФА C6H5NHz 32,7 5,1 21 27 дмсо АН ЭДА СН3СООС2Н5 33 28,5 15,3 29,8
Введение L-кислоты в растворитель обычно сдвигает вправо
равновесие процесса автоионизации (1—70):
MeXn + 2/nS ^... ^ [МеХп (S(2)m)f~ + mS& . (1-72)
Для часто встречающегося случая растворителей с протолити-
ческим равновесием процесс (1—72) выразится уравнением
МеХ„ + 2/nROH[MeXn(OR)mf~ + mROH2+. (1—72,а)
Мнения о справедливости последней схемы разделяют авторы
весьма большого числа работ. На основании разнообразных экс-
периментальных методик (обычный и радиометрический варианты
изучения чисел переноса ионов, ИК-спектроскопия и т. п.) эта
схема предлагается для растворов BF3, А1С13, SiCl4, SnCl4, TiCl4,
ZrCl4, SbF5, SbCU, MoCl5, FeCl3 в спиртах, фенолах, карбоновых
59
кислотах [514, 208, 516, 517, 511, 512, 518, 1110, 871, 872, 870J.
В работе [1100] по данным ИК-спектроскопии сделан вывод о том,
что в растворах AlCls, TiCl4, SbFs, SbCU в ароматических карбо-
новых кислотах образуются ионы [МеХ„ОН]~ и RCO+.
Сольволиз. Под сольоволизом в данном случае понимается
процесс, приводящий к образованию неэлектролитных продуктов.
С учетом обозначений, введенных в схеме (1—70), процесс можно
представить следующим образом:
МеХп 2mS ^LMeXn—m«$(2)OT /nS(i;X. (1—73)
Протекание сольволиза в системе не означает, что не образуются
электролитные соединения, но возникновение последних является
уже следствием последующих вторичных процессов:
S(1)X + S -> SS(t + X- (1-74)
(например, GeCl4+ROH-^-GeCl3(OR) 4-HCl->ROH+4-Cl_).
Сольволиз относится к числу наиболее распространенных про-
цессов в системах с участием L-кислот. Ниже приводится несколь-
ко примеров сольволиза галогенидов металлов различными ра-
створителями.
Из реакций данного типа после гидролиза наиболее обстоя-
тельно изучены процессы аммонолиза (в средах жидкого амми-
ака) [371, с. 98; 730]. Процесс аммонолиза может доходить до ста-
дий образования амидов (например, Hg(NH2)Cl), имидов
(Ge(NH)2) и, наконец, азидов (SbN, Te3N2). Примером сольво-
лиза в гидразине [372] может служить реакция
SeO2Cl2 + 4N2H4^ SeO2 (N2H3)2 + 2N2H^C1-.
Известно много реакций сольволиза в жидкой SO2 [363, с. 249,
371, с. 251], например, 2NaAc+2SO2^Na2SO3 + SOAc2.
По усложненной схеме часто идет сольволиз в абсолютной
серной кислоте [363, с. 158], например, Ph4Sn+ 14H2SO4 =
= H2Sn(HSO4)6+4PhSO3H+4H3O+4-4HSO4- В жидком фтори-
стом водороде сольволиз различных солей металлов чаще всего
приводит к образованию соответствующих фторидов [371, с. 229].
В расплавах 12 идет сольволитическая реакция, сопровожда-
ющаяся образованием солей I+, например, I2+KCN^ICN+KI
[363, с. 272]. Наиболее обстоятельно процессы сольволиза изучен
ны в спиртах и карбоновых кислотах.
Сольволиз тетрахлорида олова в «-алифатических спиртах
приводит к образованию продукта SnCl3OR-ROH [513]. В работе
[620] определены ступенчатые константы сольволиза в системе
GeCl4 — С2Н5ОН. В системе TiCl4 — С4НэОН установлено обра-
зование двух продуктов сольволиза — моно- и дибутоксизамещен-
ных [1073].
Сольволиз в системах трихлорид сурьмы — алифатические
спирты достаточно подробно описан в работах [535, 36, 536, 35,
537, 38], аналогичный процесс в системах, образованных пентахло-
60
ридом сурьмы с этанолом либо уксусной кислотой, изучен в рабо-
те [548]. Сольволизные процессы трихлорида железа в спиртах
рассмотрены в работе [549].
В феноле тетрахлориды олова и титана дают сольватирован-
ные молекулами растворителя продукты сольволиза различной
степени замещения: Sn(Ti) (OCeNs^Ch-x-CeHsOH [1027].
Сольволиз в карбоновых кислотах также был предметом мно-
гих исследований. Изучался сольволиз хлорного олова [550, 537,
698], трихлорида [537] и пентахлорида сурьмы [548], трихлорида
железа [537]. Подробное исследование продуктов сольволиза со-
лей алюминия, титана, циркония, тория, олова, сурьмы и теллура
в монохлоруксусной кислоте предпринято в работе [1028].
4. Образование карбкатионов. К реакциям этого ти-
па относятся процессы, протекающие по схеме
MeXn + RY^R++MeXnY~. (1-75)
Такая схема диссоциации предположена авторами работ [1261,
789] для объяснения возникновения электропроводности в гало-
идалкильных растворах А1С13; авторы работ [1054, 592] схемой
(1—75) объясняли природу взаимодействия А1С13 с галоидангид-
ридами карбоновых кислот.
Аналогичный механизм взаимодействия в системах трихлорид
сурьмы— алифатические спирты, предположенный одним из авто-
ров совместно с В. П. Басовым в работах [36, 536], по-видимому,
не верен, так как трактовка результатов эксперимента ПМР не
позволяет сделать однозначный выбор между обсуждаемой схе-
мой и схемами (1—73) и (1—74).
Схема (1—75) находит подтверждение в реакциях обмена, изу-
чавшихся с помощью радиоизотопов. Так, был обнаружен срав-
нительно быстрый обмен атомами хлора между СН3СОС1 и А1С13
1811], бромистым этилом и бромидом алюминия [60, 812, 1214].
В то же время нет обмена алкильными радикалами между
А1(С2Н5)3 и С2Н5ВГ [41, 42, 43, 44], так как карбкатион, вероятно,
образующийся в этой системе по реакции А1 (C2H5)34-C2H5Br5^ ...
... ^*С2Н5++[А1 (С2Н5)3Вг]~, не вступает в обмен ни с А1(С2Н5)3,
ни с анионом.
Обстоятельное сопоставление различных схем диссоциации га-
логенидов Sn (IV) и Sb (V) приведено в обзоре [362].
5. И о н-р адикальный механизм. В системах, образо-
ванных галогенидами двух элементов III— IV групп, в тех случаях,
когда в системе протекает взаимодействие, один из компонентов
(донор аниона) является кислотой, а второй (акцептор аниона) —
•основанием: MeiXn+Me2Xm4=t ... [Me1Xn-i]++ [Me2Xm+i]~ (оче-
видным условием протекания такого рода взаимодействия являет-
ся отсутствие растворителя, обычно нивелирующего силу L-кис-
лот) [362].
В системах указанного типа обнаружен помимо приведенного
«еще весьма своеобразный механизм взаимодействия, названный
61
ион-радикальным [94]:
Ме^ + Me2Xm^L... ^LMejX^• + М^Хй - (либо Ме^ + Ме2Хт-).
(1-76}
Возникновение радикалов в системах, образованных галогени-
дами алюминия либо галлия с полухлористой (либо полуброми-
стой) серой, было установлено с помощью ЭПР-спектроскопии,
причем было найдено, что максимум интенсивности сигнала ЭПР
приходится на состав раствора, отвечающий стехиометрии взаимо-
действия [362].
Существующие методы исследования природы взаимодействия
в системах L-кислоты — основный органический растворитель не
всегда позволяют выбрать какую-либо определенную схему диссо-
циации. Поэтому при выборе наиболее достоверной схемы необ-
ходимо нередко прибегать к анализу химических особенностей си-
стемы. При этом существенную роль играет природа основания, а
именно его способность принимать участие в кислотно-основном
взаимодействии только в молекулярной форме (апротонные осно-
вания) либо также или даже исключительно — в ионной форме
(протолитические основания).
Исследование различных систем, образованных Н- и L-кислота-
ми, с одной стороны, и апротонными либо протолитическими осно-
ваниями — с другой [34, 440, 193, 231], позволило выявить некото-
рые общие закономерности, имеющие значение для выбора наи-
более достоверной схемы взаимодействия.
При исследовании взаимодействия в системах, образованных
L-кислотой и апротонным основанием, необходимо учитывать, что
при автоионизации L-кислоты (схема (1—69)) образуется заря-
женная кислота [MeXn-i]+, являющаяся более сильной кислотой,
чем незаряженная МеХп. Поэтому катион [МеХп_]]+ в первую оче-
редь будет нейтрализоваться молекулой основания S, сдвигая рав-
новесие (1—69) вправо и приводя к реализации равновесия (1—
69, а), либо в растворах с достаточно высоким содержанием кис-
лоты—-к реализации равновесия (1—69,6) [37]. Справедливость
схемы (1—69, а), (1—69,6) была экспериментально доказана
изучением переноса ионов с помощью радиоизотопов 3Н, 14С, 36С1,
59Fe, II3Sn, I24Sb, I25Sb [38, 37, 553, 551, 194], ЯМР- и ИК-спектро-
скопией [71, 37].
Обоснованное заключение о природе ионов, образующихся в
системах апротонная кислота — протолитическое основание, может
быть сделано с учетом автоионизации компонентов системы — кис-
лотного (схема (1—69)) и основного (схема (1—70)). Очевидно,
что из всех ионных и молекулярных форм, входящих в упомянутые
схемы, наиболее сильной кислотой является ион лиония кислот-
ного компонента MeX+n_i, а наиболее сильным основанием — ион
лиата основного компонента S(2)“. Очевидно, что эти ионные формы
будут взаимодействовать в первую очередь, приводя к образова-
нию недиссоциирующего продукта сольволиза MeXn-iS(2). Возник-
новение же электропроводности в системе будет обусловлено вы-
62
свобождающимися в результате этого взаимодействия ионами ли-
ата кислоты Х~ (либо MeX_n+i) и лиония растворителя S+(ij.
Таким образом, наиболее общая схема взаимодействия в систе-
мах L-кислота — протолитическое основание должна передавать-
ся схемами — для области, богатой растворителем:
MeXn.+ 2S+... MeXn-jS^, + S& + Х“, (1—77)*
для области, богатой L-кислотой:
2MeXn + 2S ^ ... ^ MeXn-jS^ + S(t> + MeX7+i- (1—77,а)
Так, схема образования ионов в системах FeCl3 — спирты переда-
ется уравнением [537, 548]: FeCl3 + 2ROH^L...^tFeCl2OR+ ROH++
+ СГ либо 2FeCl3 + 2ROHq±... FeCl2OR + ROHJ~ + FeCl?. Спра-
ведливость этой схемы доказана экспериментами по электропере-
носу радиоизотопов: в то время как радиохлор мигрирует только
к аноду, радиоуглерод и тритий — только к катоду; в областях,
богатых L-кислотой, к аноду мигрирует также радиожелезо.
1.5.4. Механизмы переноса тока в неводных
растворах
В неводных растворах перенос тока может реализовываться за
счет каждого из трех основных механизмов переноса тока в раст-
ворах— ионмиграционного, ионотропного и электронного.
Ионотропный механизм переноса тока, известный для водных
растворов кислот [20, 638] (прототропный механизм, эстафетный
механизм), энергетически более выгоден, чем миграция гидрати-
рованного протона [55].
Исследования последних лет показали, что прототропный меха-
низм переноса тока в протолитических системах встречается го-
раздо чаще, чем это предполагали. Значительная доля тока( по
ряду признаков иногда достигающая 1) переносится по прото-
тропному механизму через жидкие индивидуальные сильные
Н-кислоты — серную [233, 450], селеновую [569, 568], ортофосфор-
ную [568, 546]. Соответственно по прототропному механизму пере-
носится ток в водных и неводных растворах этих кислот (в обла-
стях концентраций с преобладанием кислотного компонента) [237,
238, 239, 546, 569, 39]. Имеются достаточно веские аргументы в.
пользу того, что в некоторых случаях ток в карбоновых кислотах
как растворителях может также переноситься по прототропному
механизму [238]. После воды наиболее подробно прототропный ме-
ханизм переноса тока изучен для растворов в алифатических
спиртах [140, 757, 139].
С возможностью реализации прототропного (наряду с ионмиг-
рационным) механизма переноса тока всегда необходимо считать-
1 Для упрощения схемы (1—77, а) даны в простейшей стехиометрии, что,
впрочем, отвечает наиболее распространенным случаям взаимодействия в сис-
темах такого типа.
63
ся, когда идет речь о растворах L-кислот в протолитических раст-
ворителях, в которых в основном протекают процессы сольволиза,
приводящие к возникновению в растворе сольватированного про-
тона (см. схему (1—77)).
В отличие от водных в неводных растворах ионотропный меха-
низм переноса тока возможен и для более тяжелых, чем протон,
ионов.
Весьма высокая подвижность ионов С1_ в некоторых средах,
приближающаяся нередко к аномальной, дала основание предпо-
лагать в таких объектах наличие хлоротропного механизма прово-
димости. Именно так объяснялась высокая подвижность С1_ в ра-
створах на основе SbCl3 [363, с. 306; 527]. К аналогичному выводу
пришли авторы работы [886], установив аномально низкие числа
переноса ионов А1С14~ и SbCl4— в РОС13. На основании аналогич-
ных предпосылок был сделан вывод о бромотропном механизме
проводимости растворов РВг3 в Вг2 [119] и об йодотропном меха-
низме проводимости в растворах йодидов щелочных металлов в
жидком йоде [120].
Прямое экспериментальное подтверждение хлоротропного ме-
ханизма проводимости было получено в работе [38], где с помо-
щью радиоизотопной методики было установлено, что в растворах
SbCl3 в абсолютной уксусной кислоте, хлориде амила, этилацетате
и амидах уксусной кислоты ионы С1_ не мигрируют в электриче-
ском поле в областях концентраций SbCl3 0,8—0,9 мол. доли.
Для разработки оптимального режима электролиза немало-
важное значение имеет определение соотносительного вклада ион-
миграционного и ионотропного механизмов переноса тока.
В разбавленных и умеренно концентрированных растворах сво-
бодная энергия активации ионной миграции (AG?*) либо удель-
ной электропроводности (AGZ=*) практически совпадает со свобод-
ной энергией активации вязкого течения (AG^*) [638, с. 327].
Это дало основание [172] предложить отношение
“%
В
качестве критерия вклада прототропного механизма проводимо-
сти: при полностью ионмиграционном механизме это отношение
равно единице; при сосуществовании ионмиграционного и прото-
тропного механизмов переноса тока это отношение менее единицы,
причем, чем выше вклад прототропного механизма, тем в боль-
шей степени отношение энергий активации отклоняется от еди-
ницы.
Указанное отношение может служить критерием соотноситель-
ного вклада ионмиграционного и галогенотропного механизмов
переноса тока, что можно иллюстрировать рядом систем типа три-
Алорид сурьмы—апротонные основания [231], в которых наличие
галогенотропного механизма переноса тока было доказано пря-
мым экспериментом.
Описанный критерий, впрочем, не всегда строг. Так, в водных
растворах галогенидов щелочных металлов, где механизм прово-
64
димости полностью миграционный, отношение \Gr^/\G^ состав-
ляет приблизительно 0,9.
Для разбавленных водных растворов достаточно определенным
^0pr(-i
критерием может являться отношение —[638, с. 338], при-
Л°кс1
ближающееся к двум в растворах с доминирующим прототропным ме-
ханизмом переноса тока.
Для неводных растворов достаточно строгим критерием соотно-
сительного вклада обоих механизмов в общий перенос тока может
служить прямое определение чисел переноса, в частности по мето-
ду радиоактивной индикации [237]. Аналогичная методика оказы-
вается весьма пригодной для решения вопроса о соотносительном
вкладе ионмиграционного и галогенотропного механизмов [37, 38]:
при чисто ионмиграционном механизме сумма чисел переноса ка-
тиона и аниона /++/-=1. При сосуществовании обоих механизмов
эта сумма меньше единицы, причем, тем значительнее, чем выше
вклад ионотропного механизма переноса тока. Следует, впрочем,
заметить, что экспериментальное определение чисел переноса в
неводных растворах с удовлетворительной точностью — задача не-
простая.
Метод определения относительного вклада прототропного ме-
ханизма переноса тока в ХпрОт/х в общую проводимость раствора
х, основанный на прямом определении концентрации ионов в кон-
центрированных растворах [448, 447], предложен в работе [447].
Поскольку общая проводимость раствора х слагается из ионмиг-
рационной хион и прототропной хпрот:
* = Хион + Хпрот- (1-78)
как было установлено [447], коррегированная на вязкость величина
хнон(хион11) ПРЯМО пропорциональна концентрации ионов, т. е. хнонт)=
= йСион, ТО
хпрот = х — —, (1-79)
где значение а для каждой системы определяется прямым, напри-
мер ИК-спектроскопическим, экспериментом [448, 449].
Электронный механизм проводимости, встречающийся в раст-
ворах щелочных металлов в сильноосновных растворителях [20,
с. 135], где время жизни сольватированного электрона (сильней-
шее основание) достаточно велико, к предмету этой книги сколь-
нибудь прямого отношения не имеет. Поэтому ограничимся лишь
упоминанием об этом механизме переноса тока.
Глава 2
ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ В НЕВОДНЫХ
РАСТВОРАХ
Теория и практика электродных процессов в неводных растворах —
раздел электрохимии, развитие которого, начиная с основополага-
ющих работ В. А. Плотникова [453, 534] и Л. В. Писаржевского
[395], связано в основном с исследованиями советских физикохи-
миков. Существенный вклад в решение этой проблемы внесли
А. И. Бродский [61], В. А. Плесков [397], А. Н. Фрумкин [577],
Н. Е. Хомутов [589]. Важные теоретические исследования по вли-
янию растворителя на электродные потенциалы и ЭДС гальвани-
ческих элементов были проведены Н. А. Измайловым [173] и его
школой [Ю]. Большой вклад в теорию влияния растворителя на
характеристики электродных процессов внесли Дж. Батлер [723]
и Г. Штрелов [1220].
2.1. ЭЛЕКТРОДНОЕ РАВНОВЕСИЕ
2.1.1. Зависимость электродных потенциалов
и ЭДС от свойств растворителя
Н. А. Измайлов [173] вывел общее уравнение, связывающее свой-
ства металлов-электродов, а также энергетические характеристи-
ки взаимодействия ионов металлов с растворителем с величиной
ЭДС соответствующего гальванического элемента:
Е = [(AGcygj -|- АбИ0Н1 А0СОЛЬВ1) — (А0сУб2 -]- AGB0B2 -|- Д0СОЛЬВ2)] .
(2-1)
Здесь AGcy6 —• свободная энергия процесса атомизации 1 г-атома
металла; AGHOh — свободная энергия процесса ионизации 1 г-ато-
ма металла, отщепляющего z электронов; ДОсольв — свободная
энергия процесса сольватации 1 г-иона металла. Индексы 1 и 2
относятся соответственно к разным электродам гальванического
элемента.
Величины AGcy6 и ДОион для данного гальванического элемен-
та — величины постоянные и не связанные со свойствами раство-
66
рителя. Поэтому, считая, что активность ионов в растворе равна
единице (т. е. Е = Е°), приходим к выражению
Е° = const -|- (Аброльв! А6С0Льв2) > (2—2)
где
const = [(Абсуб, —АбсУб,) -Ь(АбИ0Н1 АбИОн2)] ^р~ • (2 3)
Для весьма важной в теории ЭДС в неводных средах цепи с пе-
реносом
(Pt) Н21 Н+ || Mez+1 Me (2—4)
уравнение (2—2) запишется как
Ео = const + (Абсольв.н+ - Абсольв.мг+) ± . (2-5)
Числитель второго слагаемого правой части уравнения (2—5)
представляет собой разность свободных энергий сольватации
(см. 1.5.1) протона и ионов металла-электрода, а постоянная в.
соответствии с (2—3) определяется как
COHstj = [(0,5АбднС.н2 + Абиои.н) (ДбСуб.Ме Ч" АбИ0Н.Ме)] ~~р 1 (2 6)
где Абдис.щ—свободная энергия процесса диссоциации молекулы
водорода на атомы, а Абиои.н—свободная энергия ионизации 1 г-
атома водорода.
Уравнение (2—5) широко применяется для определения раз-
ности свободных энергий сольватации ионов водорода и металла
(при условии отсутствия ограничений для использования во-
дородного электрода в данной неводной среде — см. параграф
2.2.1).
Цепь
Met | Mez+1| Mez+1 Ме2 (2__7)
в соответствии с уравнением (2—2) позволяет определить раз-
ность свободных энергий сольватации ионов двух металлов. Для
цепи без переноса, например,
(Pt)H2|H+(X")l|AgX, Ag (2-8)
уравнение (2—2) запишется как
£° = const, + (Аб„ „„„ н+ — Аб у_) 4г- (2—9)
* 1 X СОЛЬВ-Н СОЛЬВ-А ' р х f
Перечисление вариантов уравнения (2—2) применительно к
различным цепям можно было бы продолжить, однако читатель
без труда это сможет сделать сам. Важно подчеркнуть, что любое
из уравнений ЭДС будет состоять из двух слагаемых — постоян-
ного члена и члена, содержащего алгебраическую сумму свобод-
5*
67
ных энергий сольватации потенциалопределяющих ионов:
£° = const+--Gco;bB
2 г
(2-Ю)
Зависимость £° от свойств растворителя, выражаемая урав-
нением (2—10), может быть объяснена, если, представив процесс
сольватации как ион-дипольное взаимодействие, выразить свобод-
ную энергию этого процесса (1—11) в подходящей для данного
случая модификации:
^Gi- d= AGC0JlbB —
z2e2 /________1^
2fMez+ \ 8
niiez
rMez+.
No,
(2-H)
где rMez+— кристаллографический радиус иона металла; п. — чис-
ло сольватации. С учетом уравнения (2—11) уравнение (2—10)
может быть представлено в виде [173]:
Е» = const + » V±[| -Ik^^5 . (2-12)
2 rMe£ \ 8 /
Из уравнения (2—12) следует, что ЭДС данной цепи без пе-
реноса при изменении растворителя изменяется постольку, по-
скольку изменяются диэлектрическая проницаемость среды и сум-
марная энергия сольватации. Следующим очевидным следствием
из этого уравнения является заключение, согласно которому в
средах с постоянной энергией сольватации (условно универсаль-
ные среды) между величинами Е° и обратной диэлектрической
проницаемостью должна соблюдаться прямолинейная зависи-
мость— обстоятельство, обоснованное еще А. И. Бродским [61]
и многократно подтвержденное экспериментально. Примеры со-
блюдения этой зависимости приводятся в монографии [173]. Для
цепи с переносом, например (2—4) или (2—7), уравнение (2—10)
запишется в форме
£° = const + (1---Ц Ц-------') + — сольв,- А0СОЛЬВ2
(2-13)
Изменение ЭДС при переходе от воды (естественно выбранный
стандартный растворитель) к неводному растворителю S выразит-
ся уравнением
р0 р0 ____ Ze I \ 1 \ f 1 1 \ _1_ ^А^СОЛЬВ! ^Д^СОЛЬВ2
Ен,о - В, - (- - -Д - - —J + S ,
(2-14)
где бАбсольв — изменение свободной энергии сольватации при пе-
реходе от воды к растворителю S. Наконец, для цепей типа (2—
4) уравнение ЭДС (без учета диффузионного потенциала) за-
68
пишется как
£° = const - In Kasii+ + -у~ In as_ - ----1) X
__1_'
fMez+ )
r ' — AG - ।
сольвн"' сольвМе T
zF
(2-15)
где £asH+ — константа собственной кислотности кислоты НА, т. е.
константа равновесия процесса отщепления протона от НА в ва-
кууме и в отсутствие растворителя.
2.1.2. Ряды напряжений в неводных растворителях
Уравнение (2—15) позволяет, соблюдая условия эксперимента с
водородным электродом в неводных средах, определять в каждом
из растворителей стандартные электродные потенциалы е° метал-
лов, принимая за стандарт в воде е°н2/н+=0. В табл. 13 [1220,
547] приводятся значения стандартных электродных потенциалов
(ряды напряжений) металлов в некоторых растворителях.
Таблица 13. Стандартные электродные потенциалы в воде и неводных
растворителях, отнесенные к стандартному потенциалу водорода в воде
8н2(н2О)=018И]
Электрод 4
H2O NH3 CH3OH C2H5OH HCOOH
L1+/L1 —2,95 —3,24i —2,901 —2,79 —2,96
Na+/Na —2,71 —2,85 —2,58 —2,43 —2,90
К+/К —2,92 —2,98 —2,72, —2,60 —2,85
Rb+/Rb —2,93 —2,93 — —. —2,93
Cs+/Cs —2,93 —2,95 —2,95 —. —2,92
Са2+/Са —2,72 -2,64 1 1 —. —2,68
Zn2 +/Zn —0,76. —1,53 —0,54 —0,39 —0,73
Cd2+/Cd —0,40 —1,20 —0,23 —0,13 —0,23
H+/H2 0 —1,00 +0,20 +0,25 +0,52
Ag+/Ag +0,80 —0,18 +0,96 + 1,00 +0,69
I“/h +0,54 +0,45 4-0,56 +0,55
Br-/Br2 + 1,07 +0,83 + 1,04 + 1,03
I-/I2 + 1,34 + 1,03 + 1,32 + 1,30 + 1,51
Н. Е. Хомутовым [589] разработан метод определения стан-
дартных электродных потенциалов, предназначенный для цепей
типа электрод первого рода — водородный электрод:
Ме|МеАг(амег+> 1)|| НА(ан+ = 1) | Н2 (Pt)PH=laTM. (2_16)
69
ЭДС элемента (2—16) может быть представлена как
Е° = ЧГ0-Ь-А, (2—17)
где 4го — сумма свободных энергий образования 1 г-экв газооб-
разных ионов металла и сольватации этих ионов (выраженной в
вольтах); А — та же сумма для водородных ионов. Величина А
постоянна (при постоянной температуре) для каждого данного
растворителя. Например, для метанола А составляет — 4,839 В;
для этанола — 4,883 В; для ацетона — 4,865 В.
Уравнение (2—17) позволяет рассчитывать для каждого дан-
ного растворителя величины е° электродов, принимая в качестве
стандарта е°н2/н+=0.
Данные табл. 13 показывают, что растворитель может оказы-
вать существенное влияние как на абсолютную величину, так и на
знак е°. Так, медь в АН превышает по своей активности водород
и, следовательно, окисляется в ацетонитрильных растворах Н-кис-
лот. В сильноосновном жидком аммиаке потенциалы почти всех
щелочных металлов повышаются почти на 1 В.
Попытки разработать единый стандарт, по отношению к ко-
торому можно было бы сравнивать потенциалы электродов в раз-
личных растворителях, предпринимались неоднократно. Так, вы-
двигались предположения, что энергии сольватации достаточно
больших ионов (Cs+, Rb+, 1~) одинаковы в различных растворите-
лях, благодаря чему соответствующие электроды могут служить
точкой отсчета для построения единой шкалы е°. Однако, как сле-
дует из раздела 1.4, даже в случае цезия энергии сольватации не
остаются постоянными с изменением химической природы раство-
рителя.
Н. А. Измайлов [173] предложил исключить необходимость в
едином электроде сравнения, так как данные по энергиям соль-
ватации отдельных ионов (включая протон) в различных раство-
рителях позволяют вычислять изменение потенциала любого элек-
трода (для которого, разумеется, известны значения энергий соль-
ватации соответствующих ионов) при переходе из одного раство-
рителя в другой. При этом в качестве очевидного стандарта при-
нимается равным нулю потенциал водородного электрода в воде.
Подобные расчеты были проведены Н. А. Измайловым. Эти дан-
ные для некоторых электродов в жидком аммиаке, муравьиной
кислоте, а также в метаноле и этаноле в сравнении с водой
представлены в табл. 13.
Данные табл. 13 позволяют еще раз отметить определяющее
влияние сольватационных процессов на величину е°. Действитель-
но, сопоставив величины е° в жидком аммиаке и муравьиной кис-
лоте, видим, что в случае слабосольватированных ионов — Rb+ и
Cs+ — практически нет различий в величинах е° в каждом из рас-
творителей. Однако в случае протона, сольватированного в силь-
ноосновном аммиаке намного сильнее, чем в муравьиной кислоте,
различие в величинах стандартных потенциалов водородного
электрода составляет 1,5 В. Соответственно существенно различие
70
в величинах стандартных потенциалов в единой шкале для тех-
электродов, чьи ионы обладают ярко выраженной способностью
к комплексообразованию по отношению к одному из растворите-
лей. Так, для цинкового, кадмиевого и серебряного электродов
различия в величинах в жидком аммиаке и муравьиной кислоте
составляют соответственно 0,8; 0,97 и 0,87 В, в то время как, на-
пример, для кальциевого электрода — всего 0,04 В.
2.2 ВОЛЬТ-АМПЕРНЫЕ ИЗМЕРЕНИЯ В НЕВОДНЫХ РАСТВОРАХ
Вольт-амперометрия во всех ее многочисленных разновидностях —
основной метод при исследовании электродных процессов. Вольт-
амперные методы подробно описаны в литературе, часто приме-
нительно к неводным средам [322, 134, 129, 320, 659]. Кратко
остановимся лишь на тех методах, какие наиболее часто приме-
няются в неводных растворах, укажем некоторые особенности
электрохимических измерений, связанные с низкой электропровод-
ностью неводных сред.
2.2.1. Электроды сравнения
К обычным требованиям, предъявляемым к электроду сравнения
в водных растворах,— стабильности потенциала во времени, воз-
вращению к исходному состоянию после поляризации и подчине-
нию уравнению Нернста — при работе в неводных средах добав-
ляется еще несколько, среди которых важнейшее — отсутствие
взаимодействия (комплексообразования) с растворителем и ми-
нимальная растворимость (для электродов второго рода). Универ-
сального электрода сравнения, подобного насыщенному каломель-
ному электроду для водных сред, в неводных растворах нет [705].
Весьма часто в качестве электрода сравнения используется
ртутное дно — прием, позволяющий существенно уменьшить вли-
яние загрязнения, диффузионного потенциала и электросопротив-
ления. Более подробные сведения об условиях применения ртут-
ного дна приведены в работе [705, с. 25—26].
Для прецизионных измерений необходимо использовать трех-
электродную ячейку. Полный литературный обзор по электродам
сравнения в неводных средах в целом и в отдельных растворите-
лях составлен в работах [1116, 1224, 949, 807, 1275, 1153]. В ка-
честве электродов сравнения используются различные электроды
первого и второго рода как в исследуемом растворе, так и вынос-
ные. При исследованиях катодного выделения металлов наиболь-
шее распространение здесь получили электроды — водный насы-
щенный каломельный и серебряный (первого и второго рода) —
выносной, а также в исследуемой среде. Применение выносных
электродов особенно целесообразно при сопоставлении поведения
различных деполяризаторов в одном растворителе. Возникновение
скачка потенциала в месте соприкосновения водного и неводного
растворов препятствует количественному сопоставлению поведения
71
одного и того же деполяризатора в разных растворителях. Здесь
уместнее применение хлорсеребряного электрода, потенциал кото-
рого не зависит от состава среды, поскольку активность потенцио-.
определяющего иона будет постоянной, если поместить данный
электрод в насыщенный раствор соли с общим анионом (хлорид).
Однако измерения с хлорсеребряным электродом немногочисленны.
В целом применению выносных (водных) электродов сравне-
ния препятствуют наличие жидкостного потенциала на границе
вода — органический растворитель, часто неизвестной величины, и
диффузия воды из электрода сравнения в исследуемый раствор.
Жидкостные потенциалы для многих органических растворителей
определены [152, 949, 11, 1131, 726, 1093]. Что касается второго
фактора, то проблема в какой-то мере решается применением спе-
циально приготовленных диафрагм, замедляющих проникновение
через них воды и растворенных в ней солей. Часто в качестве
электродов сравнения используют электроды из исследуемого или
инертного металла [419, 749, 836, 346, 771, 711].
Из многочисленных систем металл — ион металла в качестве
электродов сравнения кроме Ag+/Ag успешно используются лишь
системы Hg22+/Hgo и Rb+/Rb. Впрочем, и эти электроды не уни-
версальны. Так, электрод Hg22+/Hg не может быть применен в АН,
ДМФ, ДМСО, так как в этих растворителях происходит диспро-
порционирование Hg22+ на Hg^ и Hg°. В. А. Плесков предложил
пару Rb/Rb+ в качестве электрода сравнения, считая, что вслед-
ствие большого кристаллографического радиуса сольватация иона
Rb+ должна быть минимальна в любом растворителе — предполо-
жение, оправдывающееся лишь с большой приблизительностью
(см. параграф 1.4.4).
Водородный электрод, столь хорошо зарекомендовавший себя
в водных растворах, для многих органических растворителей ока-
зывается непригодным, поскольку при потенциалах нормального
водородного электрода многие органические растворители гидри-
руются, особенно на платиновом электроде. Поэтому растворитель
загрязняется продуктами восстановления, и как следствие этого
потенциал электрода становится нестабильным. Впрочем, в неко-
торых важных для электроосаждения металлов растворителях —
ДМФ, ФМСО, ТГФ и ПК—водородный электрод ведет себя об-
ратимо [156]. Отметим, что активность сольватированных прото-
нов изменяется весьма существенно с изменением растворителя.
Нередки случаи, когда специфика исследований требует раз-
работки специальных электродов сравнения [836, 5, 163, 1085,
688]. В целом единый сравнительный электрод для неводных рас-
творов на основе органических растворителей не разработан. Наи-
большее число измерений в неводной вольтамперометрии проведе-
но с водным насыщенным каломельным электродом (нкэ).
72
2.2.2. Методы и приемы исследований
Исследования в основном проводятся в герметизированных ячей-
ках, заполненных сухим инертным газом, чаще всего азотом или
аргоном. В качестве вспомогательных электродов в классической
полярографии используют ртутное дно, при других вольт-ампер-
ных измерениях — инертный или исследуемый металл.
В качестве фоновых электролитов при электрохимических ис-
следованиях в неводных растворах наиболее часто применяют пер-
хлораты лития, натрия и тетраалкиламмония, йодиды и хлориды
лития и тетраалкиламмония [1116, 637, 153]. Однако и в присут-
ствии фоновых электролитов сопротивление неводных растворов
часто остается достаточно высоким, что вызывает искажение ре-
зультатов электрохимических измерений.
Для минимизации омического сопротивления ячейки, его рас-
чета либо исключения предложен ряд специальных методик ис-
следования [153, 707, 103, 1266] и приборов [162, 152, 975, 974,
366, 1181, 918].
Значительная часть работ, относящихся к катодному выделе-
нию металлов из неводных сред, сводится к полярографическим
исследованиям на ртутном капельном электроде. Наиболее полно
они представлены в библиографическом указателе по полярогра-
фии [50]. Поскольку ртуть в некоторых органических растворите-
лях окисляется при потенциалах, предшествующих потенциалам
восстановления ионов отдельных металлов (например, Ag+ в
ДМСО, ДМФ [796]), дальнейшим расширением границ поляро-
графических исследований явились вольт-амперные измерения на
твердых, преимущественно платиновых, электродах [796, 681, 766,
689, 588, 892, 1118, 814], гораздо реже — на электродах типа
Ме/Ме2+ [681, 479, 162, 609, 642]. Особого внимания заслуживает
применение вращающегося платинового электрода, который об-
ладает высокой чувствительностью, сочетающейся с иными пре-
имуществами твердых электродов (отсутствие колебаний силы
тока, обусловленных капанием на ртутном капельном электроде,
емкостного тока). На вращающихся платиновых электродах целе-
сообразно исследовать растворы деполяризаторов, в которых
вследствие низких коэффициентов диффузии весьма малы диффус
зионные токи, так как здесь предельный ток во много раз боль-
ше, чем на ртутном электроде. На таком электроде редко появля-
ются максимумы. Оптимальными условиями работы вращающего-
ся платинового электрода являются строго постоянные темпера-
тура и скорость вращения электрода, обеспечивающие постоянство
диффузионного тока и низкие концентрации деполяризатора, поз-
воляющие избежать изменения электродной поверхности из-за
осаждения металлов. Большое значение имеет форма электрода
[433]. При вольт-амперных измерениях на твердых электродах
довольно часто используют скорости изменения потенциала — го-
раздо большие, чем в классической полярографии на ртутном ка-
пельном электроде. Широкое распространение в последнее время
73
при исследовании неводных растворов получили циклическая
вольтамперометрия (осциллополярография) [925, 1136, 1046] и
метод хронопотенциометрии [74, 735, 1274], при этом применя-
ются как твердые, так и ртутный рабочие электроды.
В случае больших объемных сопротивлений, по существу, за-
висящих от поляризующего потенциала, эффективность методов их
компенсации сильно снижается. Одним из перспективных методов
изучения электрохимических систем, обладающих значительным
сопротивлением, является импульсная поляризация [134, 129, 23,
1033, 108]. Метод в отличие от поляризации постоянным током
дает возможность четко разграничить омическое сопротивление и
поляризационное сопротивление электрохимической реакции.
Принципиальная схема аппаратуры обычно представляет собой
вариант импульсной цепи, модернизированной для измерений в
неводных электролитах [23, 1033, 108].
В методе разностной осциллополярографии не требуется высо-
кой электропроводности раствора, что делает этот метод особенно
выгодным для исследования неводных сред [68].
Гораздо реже для определения механизма и кинетики катодно-
го выделения металлов из неводных растворов применяются дру-
гие методы: импедансометрия, позволяющая кроме кинетических
параметров (тока обмена, константы скорости, числа переноса)
определить также строение двойного электрического слоя [252,
793, 829]; электролиз при контролируемом потенциале, когда не-
обходимо изучить выделяющиеся на катоде продукты [1136, 1269,
750]; хроновольтамперометрия [150, 993]; различные виды про-
изводной полярографии [1179, 829, 1012]. Создана специальная
аппаратура для полярографических исследований в неводных рас-
творах при повышенных давлениях [1185].
Метод вольтамперометрии с медленным наложением потенциа-
лов на микроэлектрод, в частности классическая полярография,
способен дать довольно полную характеристику электродного про-
цесса и молекулярного состояния электролита. Из вольтамперо-
грамм (полярограмм) можно сделать заключение об обратимости
или необратимости электродных процессов, о наличии процессов
комплексообразования в электролитах, определить число электро-
нов, принимающих участие в электродном процессе, рассчитать
нормальные потенциалы, коэффициенты диффузии, кинетические
константы, а также коэффициенты активности.
В большинстве работ по приложению полярографии к изуче-
нию катодного выделения металлов из неводных сред исследова-
ния ограничиваются определением значений потенциалов полуволн
без учета величины контактного потенциала и коэффициента b
в известном уравнении
£ = £1/2 — & In-j-Ц-. (2—18)
ld 1
Полярографический потенциал полуволн £i/2, определяемый
как потенциал, при котором ток электродной реакции i равен по-
74
ловине предельного тока id, с некоторым приближением можно
считать стандартным электродным потенциалом. Для обратимых
процессов это приближение вполне достаточно, для необратимых—
весьма грубо.
Коэффициент b определяют из зависимости lg [i/id — i] — Е
(представляющей для обратимых процессов прямую линию) как
наклон, равный 2,"iRTInF, что при 25 °C составляет 59,1/пцВ. Кри-
терием обратимости может также служить разность между по-
тенциалами в точках, для которых ток реакции составляет 1/4 и
3/4 диффузионного тока, равная (RT/nF) 1g 9, т. е. 56,3/пр.В [320].
В качестве примеров исследований такого рода можно привести
работы [796, 681, 892, 814, 1009, 1266, 164, 751, 988, 1022, 382,
14, 1180].
Сопоставление потенциалов полуволн разных катионов в од-
ном растворителе и одного катиона в разных растворителях за-
труднено применением разных электродов сравнения. В некото-
рых работах использовались двухэлектродные ячейки [892, 751,
1180]. Для количественных определений наиболее серьезного вни-
мания заслуживают потенциалы полуволн, измеряемые по отно-
шению к водному насыщенному каломельному электроду; они как
уже отмечалось, наиболее многочисленны.
Вторую группу работ по данному методу составляют исследо-
вания, в которых наряду с определением Et/z и коэффициента b
рассчитаны коэффициенты диффузии ионов восстанавливающегося
на катоде металла, скорость протекающего электродного процесса,
количественные параметры комплексообразования ионов металлов
в электролите [892, 714, 855, 263, 919].
Изучение поляризации выделения металлов на твердых и жид-
ких катодах из неводных растворов дает ценную информацию для
выяснения специфики электроосаждения данных металлов с це-
лью получения их электролитическим путем. Поляризационные
кривые, снятые при различных скоростях поляризации в боль-
шинстве случаев в потенциостатическом режиме, позволяют опре-
делить характер электродного процесса (обратимый — необрати-
мый), его интенсивность (токи обмена, числа переноса катодного
и анодного процессов, константы скорости, энергию активации),
зависимость характера электродного процесса от концентрации
отдельных компонентов электролита, силы тока, поверхности элек-
трода [588, 479, 162, 419, 73, 186, 443, 640, 167, 16]. Метод поля-
ризационных кривых позволяет также изучать кинетику отдель-
ных стадий стадийных электродных процессов [643, 351].
Методы осциллополярографии и хронопотенциометрии [134,
320] служат для количественных определений кинетических пара-
метров катодного восстановления металлов из неводных сред [925,
735, 924, 994, 1183].
Наиболее важные экспериментальные параметры осциллопо-
лярографии— пиковый ток (ip) и потенциал пика (Ер или <рр).
Пиковый ток осциллограммы соответствует высоте волны поля-
рограммы и отличается от полярографического предельного тока
тем, что является функцией скорости развертки потенциала. Ве-
личина потенциала пика разнится от потенциала значения полу-
волны на постоянную величину и подобно последнему может ха-
рактеризовать потенциал выделения восстанавливающего иона
металла:
ЕР = Д1/2 - 1, IRT/nF. (2—19)
Иногда вместо потенциала пика пользуются потенциалом полупи-
ка (Ер/2). Ер и Ер/2 соотносятся очень просто:
Ер — Ер/2 = — l,857RT/anF, (2—20)
где а — коэффициент (число) переноса катодного процесса.
Пространственное расположение пиков катодного и анодного
токов позволяет судить об обратимости электродного процесса.
Количественная теория интерпретации кривых ток — напряже-
ние для обратимых и квазиобратимых электродных процессов да-
ется в работах [675, 778, 1043, 1042, 923]. Выведены временные
зависимости пикового тока (ip) для электродных процессов, конт-
ролируемых диффузией, переносом заряда, а также одновременно
обоими параметрами, позволяющие определить константы скоро-
сти катодного и анодного процессов, числа переноса, коэффициен-
ты диффузии разряжающихся ионов. Так, в случае катодного про-
цесса, контролируемого переносом заряда, зависимость величин
потенциалов пика и полупика от скорости изменения потенциалов
дается следующим выражением [923]:
(£р/2)2 - (£p/2)t = (Ер)2 - (Ер\ = (RT/anF) In (Vl/v2)1/2, (2-21)
где (Ep)i, (Ep/2)i, (Ep)2, (Ep/2)2 — значения потенциалов пика и полу-
пика, полученные при двух скоростях развертки Vi и v2. Выраже-
ние (2—21), как и (2—20), может быть использовано для опре-
деления коэффициента переноса а.
Для определения константы скорости процесса служит уравне-
ние тока у подножия катодной волны (i<10 % ip) [1154] :
i^o = nFAC0Ks exp [(— anF/RT) (E — Et)], (2—22)
где A — площадь поверхности электрода; Co — объемная концент-
рация ионов металла; k& — стандартная константа скорости элек-
тродного процесса; Е — потенциал катода по отношению к сравни-
тельному электроду; Ег — потенциал в начале развертки.
Хронопотенциометрия формально обратна вольтамперометрии
при заданном напряжении. Она заключается в наложении на ра-
бочий электрод контролирующего тока и измерении потенциала
во времени.
Основная экспериментальная величина, определяемая в хро-
нопотенциометрии, переходное время т — время, необходимое для
полного восстановления деполяризатора в приэлектродном слое
(т. е. время, за которое поверхностная концентрация реагирующе-
го вещества падает до нуля). В простейшем случае подачи реа-
76
тирующего вещества к электроду лишь через диффузию переход-
ное время определяется зависимостью
Ti/2 = ^/2nFCDx'2 .(2-23)
2l ’
где С—концентрация деполяризатора в объеме раствора; D —
коэффициент диффузии; i — плотность тока. Подобные зависимо-
сти получены и для более сложных случаев электродного процес-
са [134, 1154]. По виду зависимости силы тока от переходного
времени определяется характер диффузионного процесса. Эта за-
висимость позволяет также рассчитать коэффициенты диффузии
разряжающихся частиц. Зависимости потенциала от времени дают
возможность определить константы скорости электродных процес-
сов и число переноса катодного процесса а, которое характери-
зует долю потенциала, благоприятствующего данному про-
цессу.
Исследование катодных процессов электровыделения металлов
водной группы из органических сред направлено на решение сле-
дующих задач: выяснение возможности электроосаждения метал-
лов данной группы на активные поверхности, неустойчивые в вод-
ных растворах, и получение более качественных покрытий, чем в
воде; изучение особенностей электрохимического поведения метал-
лов, для чего неводные растворители, особенно апротонные, часто
представляют более широкие возможности, чем водные растворы;
определение возможностей аналитического определения металлов,
особенно их малых количеств, электрохимическим путем; изуче-
ние молекулярного состояния электролита.
Количественно механизм электровыделения металлов можно
охарактеризовать наиболее полно при помощи имеющихся в лите-
ратуре полярографических данных. В табл. 11 приложения сведе-
ны данные по потенциалам полуволн и предлогарифмическим ко-
эффициентам восстанавливающихся на ртутном капельном элект-
роде ионов металлов (анион — перхлорат) по отношению к водно-
му насыщенному каломельному электроду.
Величина £\/2 при данных условиях представляется наиболее
подходящей для характеристики механизма электродного процес-
са как возможно полно исключающая влияние побочных процес-
сов (в частности, комплексообразования) на электродный процесс.
В целом сходимость данных, полученных различными авторами в
неводных растворах, значительно хуже, чем в водных. Основные
причины этого заключаются в неодинаковой степени обезвожива-
ния и очистки растворителя, частичном разложении растворителя,
использовании различных фоновых электролитов. Кинетику про-
исходящих катодных процессов характеризуют данные табл. 7
приложения [134].
В некоторых работах для характеристики интенсивности про-
текания электродного процесса определялись ток обмена i0 и стан-
дартный ток обмена i0°. Эти величины взаимосвязаны со стандарт-
77
ной константой скорости следующими соотношениями:
to - nFks (С°ЬГ (2-24)
i°o = nFks, (2—25)
где п — число участвующих в реакции электронов; F — число Фа-
радея; Соа — начальная концентрация ионов металла в растворе;
Сь° — концентрация металла на электроде.
Величины токов обмена 1О и t0° приведены в табл. 8 прило-
жения наряду с коэффициентами диффузии D, характеризующими
скорость доставки восстанавливающихся частиц к электроду.
2.3. КАТОДНЫЕ ПРОЦЕССЫ ПРИ ЭЛЕКТРОВЫДЕЛЕНИИ
ЭЛЕМЕНТОВ ИЗ НЕВОДНЫХ РАСТВОРОВ
Как отмечалось, металлы по возможности их электролитического
выделения подразделяются на две группы — водную и неводную
[702]. Металлы группы могут быть электролитически выделены
в чистом виде из водных растворов. Металлы неводной группы из
водных растворов в принципе электролитически выделены быть
не могут. К неводной группе относятся также промежуточные в
рамках приведенной классификации металлы — вольфрам, молиб-
ден, германий, выделение которых из водных растворов в чистом
виде термодинамически разрешено, однако на практике электролиз
приводит лишь к осаждению сплавов.
Далее следует обзор данных по катодным процессам при
электровыделении металлов всех групп периодической системы
элементов Д. И. Менделеева.
2.3.1. Щелочные металлы
Катодное восстановление катионов щелочных металлов в невод-
ных органических растворителях представляет собой в основном
одноэлектронный обратимый процесс с образованием соответству-
ющего металла [681, 1153, 988, 1022, 963, 1096, 242, 708, 999, 887,
724, 819, 651, 1233, 987]. Исключение составляет ион лития, кото-
рый во многих растворителях восстанавливается необратимо бла-
годаря своей высокой способности сольватироваться. Об этом, в
частности, свидетельствуют значения полярографического коэффи-
циента Ь, приведенные в табл. 11 приложения (теоретическое зна-
чение для одноэлектронного обратимого процесса при комнатной
температуре 59 мВ). Образовавшийся на катоде щелочной металл
может вступать в различные вторичные реакции с растворите-
лем, следами воды, примесями [861, 414, 1184]. Щелочные метал-
лы могут проявлять свою активность даже в апротонных раство-
рителях. Так, по отношению к ДМСО не активен только литий.
При комнатной температуре натрий реагирует с ДМСО достаточ-
но быстро, а калий — бурно [722]. Катодное восстановление ионов
78
щелочных металлов иногда связано с образованием свободных
радикалов [946].
Область потенциалов восстановления ионов щелочных металлов
в большинстве органических растворителей шире, чем в воде, при
этом потенциалы отдельных ионов в большей мере удалены друг
от друга, чем в водных растворах, что можно использовать для
их разделения. И даже для растворителей, где первая закономер-
ность не сохраняется, вторая имеет силу. Так, по данным работы
[242], область восстановления щелочных металлов в ПК на
100 мВ уже, чем в воде, однако разность потенциалов полуволн
натрия и калия увеличивается по сравнению с водой.
Скорость реакций восстановления ионов щелочных металлов
закономерно увеличивается от Li+ к Cs+ в большинстве изученных
органических растворителей. Например, в ДМСО [925] восстанов-
ление иона Li+ происходит медленно (необратимо), восстановле-
ние иона Na+ быстрее (квазиобратимо), а К+, Rb+, Cs+ восстанав-
ливаются быстро и обратимо. Количественные данные немногочис-
ленны (табл. 7, 11 приложения) и относятся в основном к литию
и натрию. Для сравнительной характеристики водных и неводных
растворов следует отметить, что гетерогенная константа скорости
восстановления ионов щелочных металлов в воде находится в
пределах (от 2 до 9) • 10 3 см/с, а коэффициенты диффузии имеют
порядок 10~5 см/с.
Для отдельного некомплексообразующего неводного раствора
(деполяризатор добавлен в виде перхлората) параметры электро-
восстановления ионов щелочных металлов изменяются, когда из-
меняются природа растворителя, фоновый электролит или природа
электрода, причем данному влиянию в гораздо большей мере под-
вергаются кинетические параметры [681, 414, 1123, 891, 889, 1005,
48, 47, 709, 742, 744, 1056, 786, 260, 1244, 1175, 953, 1177]. Замена
жидкого электрода на твердый приводит к существенному сниже-
нию скорости электродного процесса и увеличению его необрати-
мости. При переходе от водных электролитов к электролитам на
основе органических растворителей замедляются скорости процес-
сов, протекающих на электродах металл/ион металла, хотя при
восстановлении щелочных металлов на чужеродном электроде до-
бавление к органическому растворителю воды часто вызывает уве-
личение гетерогенных констант разряда [1263, 1033].
Об изменении механизма процесса восстановления и его кине-
тических параметров для каждого отдельного иона при переходе
от одного растворителя к другому можно судить по данным
табл. 7, 11 приложения. Особенно сильное влияние на кинетиче-
ские параметры оказывает катион фонового электролита. При ис-
пользовании некомплексообразующего (перхлоратного) фонового
электролита во всех исследуемых случаях увеличение размеров
катиона фона способствует возрастанию константы скорости про-
цесса и коэффициента переноса [836, 657, 950, 952], т. е. облег-
чению процесса разряда. При этом часто изменяется механизм
процесса. Так, в ГМФТА в присутствии перхлората тетраэтилам-
79
мония с диффузионным контролем процесса полярографически
восстанавливаются лишь ионы рубидия и цезия, ион калия дает
кинетическую волну, а ионы натрия и лития не восстанавливаются
до восстановления фона. Замена ПТЭА на фон с катионом боль-
шего размера (LiClOi) приводит к тому, что уже все четыре иона
Cs+, Rb+, К+ и Na+ восстанавливаются обратимо при диффузион-
Iff'
-0,2
-w
Ka++e~Na(Hg)
АПОН
-0,6
ДМФ
-Т,4
ГИФА
-4,4 -4,6 -4,9
AG’fsoLVJ/eV
Рис. 22. Логарифм стандартной
константы скорости восстанов-
ном контроле, 1л+-ион дает кинетиче-
скую волну [950, 952].
М
CL
ления натрия на ртутном като-
де как функция свободной энер-
гии сольватапии иона Na+.
-0,9
-1,2-
•О 0,5
-0,1
0 ~0,2 0,4 AEvz/v
Рис. 23. Логарифм стандартной
константы скорости К, и кажу-
щийся коэффициент переноса а
в процессе электровосстановле-
иия Na+ на различных амаль-
гамных электродах в ДМФ как
функция сдвига потенциала по-
луволны.
Сильная зависимость электрохимического поведения ионов ще-
лочных металлов от размера катиона фона объясняется влиянием
катионов фона на свойства двойного слоя. Аналогичное влияние
на электродные процессы оказывает введение в раствор протонных
растворителей [1176]. Что же касается природы растворителя, то
с повышением его основности потенциалы восстановления ионов
£1/2 сдвигаются в отрицательную сторону. Стандартная константа
скорости восстановления на ртутном электроде при этом умень-
шается. На рис. 22 приведены данные по изменению стандартной
константы скорости восстановления натрия в зависимости от
стандартной свободной энергии сольватации некоторых органиче-
ских растворителей [657].
Закономерность в отношении скорости электродной реакции
нарушается при восстановлении катионов щелочных металлов на
электродах амальгамного типа в отдельном растворителе. Так, при
восстановлении Na+ из ДМФ (фон ПТЭА) на амальгамно-индие-
вых и индиево-галлиевых электродах во всех случаях, когда £1/2
становится более отрицательным, стандартная константа скорости
возрастает, а коэффициент переноса уменьшается [657]. При этом
существенном увеличении величина Ei/^JKs стремится к предель-
ному значению, а ап снижается до нуля (рис. 23).
80
Согласно терминологии Кришталика [229], такой электродный
процесс (а=0) может быть классифицирован как дезактиваци-
онный.
В литературе имеются также примеры установления связи
между другими характеристиками растворителя и параметрами
протекающего электродного процесса, например в работах [1188,
1187] рассматривается зависимость предельного тока полярогра-
фической волны щелочного металла от вязкости среды. В отдель-
ных случаях определяющими для восстановления щелочных ме-
таллов становятся процессы адсорбции и комплексообразования
[1263, 1126, 929].
Энергии активации электровосстановления ионов щелочных
металлов в некоторых органических раствори-Ллях составляют ве-
личины порядка 40 кДж/моль [746, 968, 329].
Учитывая важность теоретических и прикладных вопросов про-
цесса электровосстановления ионов щелочных металлов, представ-
ляющих одновременные процессы одноэлектронного переноса за-
ряда и одноатомного перемещения вещества, следует отметить
недостаточную изученность параметров этих электродных процес-
сов. Это в первую очередь относится к кинетическим параметрам,
а также к данным о форме разряжающихся частиц, о переходном
состоянии электролит — электрод.
2.3.2. Подгруппа меди
Из металлов подгруппы 16 периодической системы наиболее полно
изучено электрохимическое восстановление ионов меди. Данные
для золота практически отсутствуют.
Медь из всех переходных металлов, пожалуй, наиболее элект-
рохимически изученный металл в неводных средах [796, 681, 684,
766, 765, 689, 1118, 814, 74, 1009, 1153, 419, 1022, 714, 73, 351, 994,
722, 1052, 904, 906, 742, 745, 743, 712, 780, 1175, 1233, 837, 1134,
989, 680, 697, 718, 241, 885, 467, 1265, 913, 470, 247, 915, 1006, 68,
678, 428, 429, 430, 392, 391, 393, 394, 613, 841, 842, 720, 77, 78, 642,
353, 356, 354, 246]. Этому способствовала прежде всего стабилиза-
ция низших степеней окисления меди в органических растворите-
лях, что приводит при электрохимических измерениях к стадий-
ному протеканию процессов разряда—ионизации меди, как и иных
металлов со степенями окисления выше единицы [350, 304, 1078].
Закономерности протекания стадийных электродных процессов и
устойчивости ионов низших валентностей широко изучены на при-
мере переходных металлов, в первую очередь иона Си2+ [681, 684,
689, 1118, 643, 419, 351, 904, 917, 1226, 942, 899, 679, 1000, 802,.
984, 354, 1078, 827, 498, 937].
Для водной электрохимии меди характерна аномалия — обрат-
ный порядок потенциалов медных пар:
Си2+ + е. —---Си+ £° = — 0,089 В (нкэ-сравнения),
Си+ + е-------Си Е° = + 0,279 В (нкэ-сравнения)-
6 — 4-653
81
Особенность неожиданна, поскольку второй потенциал ионизации
меди выше первого, но благодаря ей в некомплексообразующих
водных растворах Си(II) на ртутных капельных электродах вос-
станавливается до металла в одну ступень, а ион Cu(I) нестаби-
лен, диспропорционируя до Си(II) и Си(0). Порядок может из-
мениться на нормальный, давая возможность Си2+ восстанавлива-
ться в две ступени в присутствии в водном растворе комплексооб-
разователей (лигандов), более прочно связывающих Си+, чем Си2*
[917, 1078, 179, 178].
В неводных растворителях для ионов меди характерно явление
селективной сольватации [745, 697, 1006, 1074], в результате чего
порядок потенциалов выделения ионов Си2+ и Си* чаще нормаль-
ный, чем обратный, и восстановление иона Си2+ происходит в две
ступени (см. табл. 11 приложения), а также появляется возмож-
ность приготовить устойчивые растворы Cu(I). Перемена порядка
потенциалов медных пар с обратного на нормальный по отноше-
нию к воде происходит в результате значительного увеличения
энергии сольватации Си+ или же уменьшения этой величины для
ионов Си2+. При этом энергия сольватации Си2+ очень чувстви-
тельна к изменениям основности растворителей; Так, в ряду рас-
творителей вода — метанол—этанол — 1-пропанол энергия соль-
ватации иона Си+ изменяется незначительно (потенциал пары
Cu(I)/Cu(0) почти постоянен), но уменьшение энергии сольвата-
ции Си2+-ионов достаточно большое, и уже в растворе 1-пропанола
ионы двухвалентной меди восстанавливаются в две ступени, а рас-
творы Си+ стабильны, поскольку протекает процесс диспропорци-
онирования. По данным некоторых авторов [814], двухступенчатое
восстановление меди характерно для всех спиртовых растворите-
лей, начиная с метанола. Однако одноступенчатость восстановле-
ния меди в неводных растворах может наблюдаться и при нор-
мальном порядке потенциалов медных пар в результате минималь-
ного различия потенциалов восстановления Си2* и Си+ в данных
растворителях. Такое явление наблюдается, например, для суль-
фокси-растворителей [899], хотя сближение потенциалов медных
пар в органических растворителях, даже при обратном порядке
их, в целом приводит к стабилизации одновалентной меди [642].
Особенно высока стабилизация ионов Си+ в нитрильных рас-
творителях. Энергия стабилизации Си+ в АН настолько высока,
что в ацетонитриле на медном электроде происходит самопроиз-
вольный процесс: Cu+Cu2+->2Cu+, в результате чего при элект-
роосаждении меди на медном электроде из растворов в АН в
электродном процессе практически участвуют только ионы Си+ [77].
Причиной нормального порядка потенциалов восстановления
ионов меди в неводных растворах, подобно водным, могут также
служить процессы комплексообразования [689, 1226, 915, 1029].
Характер катодных процессов восстановления ионов меди в
основном диффузионный: одноступенчатое двухэлектронное вос-
становление меди происходит чаще всего необратимо, для двухсту-
пенчатого восстановления более характерна обратимость или ква-
82
зиобратимость, из двух ступеней к необратимому протеканию бо-
лее склонна вторая ступень восстановления иона Си+. На меха-
низм восстановления большое влияние оказывают катионы фоно-
вых электролитов [743, 1175, 241, 720]. В случае восстановления
ионов меди на платиновом электроде процесс, как правило, не-
обратим.
Отдельную группу составляют работы, где исследование элект-
ровосстановления меди из комплексов ее с органическими, чаще
всего хелатными, лигандами проведено с целью использования
этого процесса в аналитических целях [68, 678, 428—430,
391—394].
Кинетика катодного процесса разряда меди исследовалась в
основном для стадии восстановления иона Си+ в отдельных рас-
творителях. Методом стационарных поляризационных кривых в
ледяной уксусной кислоте установлен первый порядок (по ионам
меди) катодной реакции восстановления Си+ [356]. Скорость ка-
тодного процесса реакции Cu+*Cu++e выражается уравнением
ih = kk [Cu+] exp (- aF<p//?T). (2-26)
Установлена величина тока обмена.
На основании изучения зависимостей приэлектродной концент-
рации меди от потенциала и плотности тока медного электрода
определены кинетические параметры обеих стадий разряда — ио-
низации меди в метаноле.
Потенциостатическим, гальванодинамическим и хронопотенцио-
метрическим методами изучена кинетика разряда — ионизации ме-
ди на твердых электродах в АН, ДМФ, ДМСО, MtOH [994, 679,
77, 78]. Судя по этим данным, скорость катодного процесса низ-
кая, в присутствии комплексообразующих добавок еще замедля-
ется [679], токи обмена падают с понижением диэлектрической
проницаемости, например в ряду Н2О—СН3ОН—СН3СООН [356].
Электровосстановление серебра в неводных растворах изучено
подробно [796, 766, 765, 814, 1022, 906, 742, 745, 743, 780, 1175,
1233, 1134, 989, 1226, 792, 17, 1121, 29]. Процесс является одно-
электронным, обратимым. В результате образования амальгамы
восстановление на ртутном электроде проходит особенно легко.
Однако во многих растворителях (например, ДМФА, ДМСО, АН
и др.) волна восстановления серебра лежит в области анодного
окисления ртути и поэтому может быть прослежена лишь на пла-
тиновом или другом инертном электроде. Восстановление на пла-
тине несколько затруднено, что приводит иногда к квазиобрати-
мому электродному процессу. Для ионов серебра прослеживается
корреляция между Е^2 и донорным числом растворителя. В целом
электрохимическое поведение ионов серебра аналогично электро-
химическому поведению ионов одновалентной меди, особенно в
нитрильных и спиртовых растворах, где наблюдается специфиче-
ское взаимодействие этих катионов с растворителем.
6*
83
2.3.3 Щелочноземельные металлы
Электрохимическое поведение бериллия в неводных средах изуче-
но весьма поверхностно. Наблюдается далеко идущее сходство с
электрохимическим поведением алюминия в органических раство-
рителях (см. параграф 2.3.5). По полярографическим данным для
обоих металлов характерно наложение на реакцию переноса заря-
да мешающего влияния адсорбции, и химических реакций. Катод-
ное восстановление идет одноступенчато до металла [742, 989, 848,
1039].
Катодное восстановление в органических растворителях ионов
щелочноземельных металлов до металлического состояния проис-
ходит, судя по полярографическим данным [988, 1022, 1052, 904,
743, 1233, 888, 837, 921], в основном необратимо в результате од-
ноступенчатого двухэлектронного процесса. Необратимость умень-
шается в ряду Mg2+—Ва2+ [925, 1005]. Обратимое восстановление
чаще всего наблюдается для иона бария (теоретическая величина
коэффициента b для двухэлектронного обратимого восстановления
при комнатной температуре составляет 29,5 мВ). Имеются отдель-
ные указания о двухступенчатом механизме разряда некоторых
ионов щелочноземельных металлов, например кальция [441] и
магния [837].
Как и для ионов щелочных металлов, с увеличением сольвати-
рующей способности растворителя потенциалы полуволн восста-
новления щелочноземельных металлов сдвигаются в отрицатель-
ную сторону. Имеются данные о корреляции между Е^2 ионов
щелочноземельных металлов и донорным числом, а также диэлек-
трической постоянной растворителя [1123, 164]. Прослеживается
качественная тенденция сдвига Е{/2 с увеличением донорного чис-
ла растворителя в отрицательную сторону.
В отдельных растворителях наблюдается большее по сравне-
нию с водой расстояние между потенциалами полуволн восста-
новления ионов щелочноземельных металлов [1153, 1197], что мо-
жет быть использовано для их аналитического определения и раз-
деления.
Ряды потенциалов полуволн восстановления щелочноземель-
ных металлов в различных растворителях часто не совпадают [892,
1134]. Процесс катодного восстановления для всех ионов протекает
медленно, значение параметров, характеризующих электродную
кинетику, в литературе почти не приводятся.
Восстановление ионов щелочноземельных металлов существен-
но зависит от катиона фона [950, 742, 780, 1175, 953, 888, 837].
Меняются не только количественные параметры электродных про-
цессов, но и механизм при замене одного фона на другой. Так,
восстановление ионов Са2+, Sr2+ и Ва2*' в диметилацетамидных
растворах в присутствии перхлората тетраэтиламмония или вовсе
не происходит, или же происходит с малым предельным током и
контролем предшествующим процессом. В присутствии перхлората
тетрабутиламмония восстановление происходит почти обратимо
84
[950, 952]. Аналогичная картина наблюдается и в ГМФТА [951].
Влияние объясняется, как и в случае щелочных металлов, разли-
чием в размерах катионов фона и соответственно разной абсорби-
руемостью на поверхности электрода.
2.3.4. Подгруппа цинка
Электровосстановление ионов цинка и кадмия в неводных раство-
рах изучено достаточно детально [681, 684, 766, 765, 892, 925, 74,
1009, 1266, 1153, 164, 988, 1022, 14, 714, 16, 924, 793, 150, 963, 722,
1052, 904, 1123, 891, 1005, 906, 745, 712, 780, 1233, 837, 1196, 1197,
739, 680, 718, 885, 913, 247, 1190, 997, 737, 1128, 1189, 1082, 1237,
591, 1206, 489, 654, 274, 504]. Разряд ионов Zn2+ и Cd2* в преоб-
ладающем большинстве случаев происходит обратимо и квази-
обратимо. По полярографическим данным процесс в обоих случаях
протекает в одну двухэлектронную ступень.
При исследовании кинетики разряда ионов кадмия и цинка
методами быстрой вольтамперометрии, фарадеевского импеданса
установлена стадийность их разряда, при этом замедленной ста-
дией является присоединение первого электрона [162, 146, 687,
828, 442, 487, 488]. Образующиеся ионы Zn+ и Cd+ сильно подвер-
жены диспропорционированию, и поэтому их фиксация возможна
лишь высокочувствительными быстродействующими методами.
Кроме электрохимических методов существование ионов кадмия
установлено в экспериментах по радиолизу [687]. Ионы цинка и
кадмия как промежуточная стадия стабилизируются в процессах
адсорбции или комплексообразования.
При изучении электрохимического поведения ионов цинка и
кадмия в органических растворителях уделено большое внимание
зависимости механизма и кинетики электродных процессов от при-
роды растворителя и анионного состава раствора. Механизм элек-
тродного процесса, в особенности параметры электродной кине-
тики, в целом определяются следующими факторами: взаимодей-
ствие восстанавливающегося иона с растворителем либо ионами
фона; взаимодействие между ионами фона и растворителем; поло-
жение равновесного потенциала на абсолютной шкале; состав и
диэлектрическая проницаемость раствора; распределение восста-
навливающихся ионов и ионов фона в двойном слое в начальном
и активном состояниях.
Полностью при измерениях очень трудно учесть все эти факто-
ры, возможно, поэтому приведенные в табл. 7 приложения зна-
чения для константы скорости системы Cd2+/Cd(Hg) в шести весь-
ма различных по физико-химическим свойствам растворителях из-
меняются весьма мало (0,454-0,01 см/с). Впрочем, основная за-
кономерность — понижение скорости катодного процесса в орга-
нических растворителях по сравнению с водой при прочих равных
условиях — четко проявляется как для цинка, так и для кадмия.
Подтверждают эту закономерность и величины токов обмена раз-
ряда-ионизации ионов цинка.
85
Изучение кинетики катодного процесса для кадмия и цинка в
водно-органических смесях показывает возрастание необратимости
реакции восстановления с увеличением содержания органического
растворителя, а также появление на зависимостях кинетических
параметров от состава смеси минимумов (ток обмена, константа
скорости) или максимумов (коэффициент переноса) [957, 146,
1070, 788, 687, 1208]. Существенное влияние на кинетику и меха-
низм процесса оказывает структура смешанного растворителя [53].
По полярографическим данным для кадмия и цинка установ-
лена почти прямолинейная зависимость между Ei/2 и донорным
числом растворителя [891]. Более поздние исследования [681, 684,
680] не подтвердили корреляцию между энергией сольватации
ионов переходных металлов в растворах и величиной напряжения
их катодного выделения (в частности, Е^).
Значительный объем исследований по электровосстановлению
кадмия и цинка проведен в комплексообразующих системах [443,
829, 150, 997, 737, 828, 442, 487, 488, 342, 67, 69, 62, 63, 504, 151,
64, 1165, 831]. Среди них особое место занимают работы по изу-
чению электровосстановления комплексов кадмия и цинка с ор-
ганическими лигандами, используемые в аналитических целях [997,
67, 69, 62, 63, 504, 68, 428, 429, 430, 391—394, 831].
Электроосаждение цинка и кадмия свидетельствует о большом
значении явлений адсорбции при электровосстановлении переход-
ных металлов [681, 684, 925, 73, 924, 82, 1175, 680, 470, 957, 1070,
131]. Адсорбции на электроде подвергаются частицы, которые
включают катион восстанавливающегося металла, молекулы раст-
ворителя, ионы фонового электролита, изменяя часто не только
параметры процесса, но и механизм. Сопоставление данных раз-
личных авторов позволяет утверждать [73], что в неводных рас-
творах соблюдается основное правило, характерное для адсорб-
ции на ртутном катоде в водных растворах: адсорбционная спо-
собность катионов возрастает по мере увеличения их радиусов, а
органические катионы проявляют бблыпую адсорбционную
способность, чем неорганические. Особенно велика роль ад-
сорбции молекул растворителя на твердых электродах [681, 684,
73, 680].
Блокируя поверхность инертного либо свежеосажденного ме-'
талла, органические молекулы сильно замедляют процесс восста-
новления иона металла. Как правило, скорость катодного процес-
са на твердом электроде ниже, чем при тех же условиях на ртут-
ном. Адсорбированные на электроде частицы в большой степени
определяют структуру двойного слоя, размеры слоя Гельмгольца,
непосредственно влияя на электродный процесс. Строение двойно-
го слоя в неводных растворах, а также влияние на него различных
параметров, в том числе и состава раствора, довольно подробно
описаны в работах [1116, 637, 1115, 170, 1105, 130].
Исследованию электрохимического поведения ртути посвящено
небольшое количество работ [796, 814, 780, 1134, 989, 720, 67, 838,
27, 815]. Восстановление на ртутном электроде, естественно, об-
86
ратимо, на платиновом — необратимо двухступенчато [796, 989],
Процессы электровосстановления из комплексообразующих рас-
творов [31] осложнены адсорбционными явлениями ]67, 838, 815].
2.3.5 Алюминий и металлы его подгруппы
Электровыделение алюминия — первый процесс, основанный на
применении неводных органических электролитов в промышленных
масштабах. Имеются многочисленные работы по низкотемператур-
ному электроосаждению алюминия. Однако большинство прове-
денных исследований направлено на выяснение условий получения
качественных алюминиевых покрытий, изучению электролитов алю-
минирования, в то время как работ, относящихся к изучению не-
посредственно электрохимического процесса выделения алюминия,
недостаточно, к тому же чаще всего они посвящены выяснению
молекулярного состояния разряжающихся частиц и носят полу-
количественный характер.
Основные электролиты для электроосаждения алюминия — это
различные композиции, основанные на эфирных либо ароматиче-
ских растворителях [164, 186, 702, 298, 295, 296, 299, 287, 24, 284,
282, 220, 475, 254].
Главными компонентами эфирных растворов является эфират
хлористого или бромистого алюминия А1Х3-Э или кислотный ион-
ный компллекс НЭ+-А1Х4- (X—С1 или Вг). Особенности элект-
родных процессов в эфирных электролитах в первую очередь оп-
ределяются электролитными свойствами и природой ионизациц
эфирата алюминия, что, естественно, связано с составом электро-
лита. В случае растворов диэтилового эфира, в среде которого га-
логениды алюминия наиболее стабильны, эфират хлорида алюми-
ния диссоциирует по схеме (1—696) [204, 12]:
2А1С13 (С2Н5)2 О А1С122 (С2Н2)2 О+ + AlClf.
Образующийся катион [А1С122(С2Н5)2О+] подвергается катодному
восстановлению. Мешающее сопутствующее восстановление катио-
на НЭ+ элиминируют повышением основности среды, применяя в
качестве оснований гидридную смесь, аммиак либо органические
основания: H3+-[-B5=tHB+-J-3.
При недостаточном сдвиге равновесия вправо необходимое по-
вышение основности среды для исключения побочного процесса
достигается проработкой электролита током. Во время такой про-
работки в диффузионном слое раствора у поверхности катода со-
держание ионов НЭ+ значительно понижается за счет электроли-
тического выделения водорода, в результате чего донорное воздей-
ствие основания быстро увеличивается.
Наиболее полное элиминирование процесса разряда катионов
водорода достигается в эфирно-гидридных электролитах.
Электровыделение алюминия из эфирно-гидридных электро-
литов часто сопровождается выделением (хотя и незначительным)
водорода, не имеющим связи с электродным процессом. Это побоч-
87
1
ное явление, вызываемое самопроизвольным разложением алано-
вых комплексов (например, ЫА1Н2С12) под каталитическим влия-
нием свежеобразующейся поверхности алюминиевого осадка.
При высоких плотностях тока (выше 0,005—0,010 А/см2) по-
бочные катодные процессы всегда остаются: слабое выделение
водорода за счет примесей катиона НЭ+, дополнительные элект-
роорганические процессы, например образование этилена и бу-
тана в результате электровосстановления примесей продуктов рас-
пада эфира [288].
Для исследования катодных процессов при выделении алюми-
ния из эфирных растворов применяли в основном метод поляри-
зационных кривых в гальваностатическом и потенциодинамиче-
ском режимах. Высокая молярность эфирных электролитов по
алюминию не позволяет обнаружить этими методами предельную
плотность тока при разряде эфирата алюминия. Разработана спе-
циальная импульсная методика обнаружения предельного тока по
катионам А1Х2-Э+ [297].
Потенциалы выделения алюминия из эфирных сред колеблются
в довольно широких пределах. Так, Евыя алюминия в эфирно-гид-
ридных электролитах и эфирном растворе хлорида алюминия раз-
личаются в среднем на 1—1,5 В. В области предельного тока на-
блюдаются значительные колебания потенциала выделения, что
свидетельствует о существовании диффузионных ограничений ка-
тодного процесса.
Кинетика восстановления катионов эфирата алюминия опреде-
ляется равновероятными замедленными реакциями А13++Зе->А1
или А12+-]-е->А1+. Механизм переходных стадий катодного процес-
са тождествен в электролитах различной основности. Порядок
плотностей токов обмена алюминиевого электрода различен в за-
висимости от метода определения, однако и минимальные значе-
ния его свидетельствуют о высокой интенсивности процесса и пре-
обладании диффузионной составляющей поляризации.
Электровыделение алюминия из ароматических растворителей
[186, 460, 626, 187, 254] в отношении механизма и кинетики элек-
тродных процессов изучалось недостаточно. Характер полученных
данных в основном качественный. Электровыделение алюминия из
сложных комплексов происходит (при устранении омической и
концентрационной составляющих поляризации) без заметного пе-
ренапряжения. Образование диссоциированных комплексов, кото-
рые определяют электропроводность раствора (по-видимому, они
же разряжаются на катоде), является замедленным процессом.
Роль диффузионных ограничений в кинетике процесса электровы-
деления алюминия уменьшается в ряду бензол > мезитилен > то-
луол > Л4-ксисол > этилбензол. Основное влияние на кинетику
процесса оказывают диссоциирующие и адсорбционные свойства
растворителя [254]. Катодное выделение алюминия из ароматиче-
ских электролитов сопровождается разрядом карбониевых ионов,
например, в случае этилбензолового электролита разрядом катио-
на С6Н5С2Н5Н+. В электролитах на основе четвертичных аммони-
88
евых солей обнаружено три катодные реакции: выделение водоро-
да при восстановлении карбониевых ионов, электроосаждение алю-
миния и образование органических соединений [137, 459, 650],
Большая группа работ, начатых школой В. А. Плотникова, ха-
рактеризует электродные процессы индивидуального электровыде-
ления алюминия и совместно со щелочными металлами из апро-
тонных растворителей с позиций возможности осаждения алюми-
ния и влияния на этот процесс состава электролита [414, 641].
В серии работ приведены результаты полярографического изу-
чения катодного восстановления алюминия [164, 1005, 1134, 989,
739, 846, 845, 777, 474, 981, 1023, 847] из различных органических
индивидуальных и смешанных растворителей, таких, как АН, ТГФ,
ДМФ. Процесс одноступенчатый, наблюдается заметное изменение
£1/2 с изменением природы растворителя. Необходимо отметить,
что в цитированных работах часто проводилось полярографирова-
ние растворов галогенидов алюминия, в которых основная карти-
на полярографического поведения в большей или меньшей степени
искажается процессами комплексообразования.
В некоторых работах [16, 904, 1128, 146, 1127, 45, 1223, 896]
изучено электрохимическое поведение индия в неводных раство-
рах. Установлено трехступенчатое восстановление его на ртутном
электроде до металла. Данные о замедленной стадии процесса
противоречивы [45, 146]. Возможно, замедленное присоединение
первого или третьего электрона связано с природой растворителя
и процессами комплексообразования. Предельные диффузионные
токи поляризационных кривых в основном меньше, чем в водных
растворах [16, 1128]. Высказано предположение, что в водно-
спиртовых растворах изменение потенциала полуволны связано с
перестройкой двойного слоя [16].
В значительном числе работ [766, 765, 1153, 1022, 963, 1052,
904, 891, 906, 743, 780, 1233, 1197, 1134, 739, 1226, 885, 1000, 984,
1190, 1128, 1189, 1082, 342, 67, 504, 987] уделено внимание элект-
ровосстановлению таллия из органических сред. По полярографи-
ческим данным процесс обратим, на ртутном электроде одноэлект-
ронное восстановление заканчивается образованием амальгамы. Не-
которая необратимость наблюдается лишь в отдельных раствори-
телях [1197]. Волны на поляризационных кривых имеют диффу-
зионный характер, в нескольких растворителях определены коэф-
фициенты диффузии ионов таллия. Прослеживается четкая зави-
симость fi/г от природы и концентрации фонового электролита
[1128], а также корреляция с донорным числом растворителя [891].
Ион таллия, характеризующийся малым эффективным зарядом, а
следовательно, небольшой склонностью к сольватации, как прави-
ло, показывает малое изменение в потенциалах восстановления
при переходе от одного растворителя к другому. Благодаря это-
му редокс-систему Т1(1)/Т1(0) можно использовать для некоторых
растворителей в качестве электрода сравнения [765, 766]. Элект-
родный процесс при восстановлении комплексов таллия с органи-
ческими лигандами осложнен адсорбцией [1082, 67, 69].
89
2.3.6. Лантаноиды и актиноиды
По полярографическим данным восстановление ионов РЗЭ быва-
ет двух типов. Ионы первой группы, которую составляет преобла-
дающее большинство лантаноидов, восстанавливаются непосред-
ственно до металлического состояния через трехэлектронную реак-
цию [890, 905, 1011, 877, 851]. Вторая группа характеризуется
двухступенчатым восстановлением: Ме3+-]-е->-Ме2+ и Ме2+-|-2е->-
->Ме, где Me34-— ионы самария, европия, иттербия [749, 1269, 750,
906, 989, 936, 890, 898, 1144, 1090, 265, 361]. В этаноловых рас-
творах Рг3* полярографически и спектроскопически обнаружено
трехступенчатое восстановление (Рг3+->Рг2+->-Рг+4-Рг) до метал-
лического состояния [747]. Наблюдаемое явление объясняется об-
разованием ионных ассоциатов Рг3+ и анионов фона (перхлораты,
хлориды, бромиды), которые восстанавливаются более легко, чем
сольватированный ион Рг3*-.
В остальных изученных растворителях Рг3+ восстанавливается
до металлического состояния непосредственно в одну ступень.
Судя по данным табл. 11 приложения, восстановление трехзаряд-
ных лантаноидов протекает преимущественно обратимо. Необра-
тимость катодного процесса наблюдается при ступенчатом восста-
новлении, причем вторая ступень (Ме2+->Ме) протекает более не-
обратимо, чем первая. Данные закономерности нарушаются чаще
всего при наличии процессов комплексообразования в растворах,
а также при восстановлении ионов
ских смесях [851, 1277].
Имеются указания на то, что
восстановления самария, европия,
лантаноидов в водно-органиче-
вторая двухэлектронная волна
иттербия также, как и един-
_________________4Ю,и)н2о
_________________
q______________________^дмсо
_________________
________________Ь7,0)АЦ
ц. ____________________
0___________________(П9)БН_
__I_________I_____________
0 ~0Д Е,В
Рис. 24. Потенциал £,/2 восста-
новления £u(iii) в различных
растворителях (в скобках — до-
норные числа растворителей).
ственная трехэлектронная волна ос-
тальных РЗЭ, обусловлена скорее вы-
делением водорода, чем образованием
амальгамы [745].
В работах, посвященных исследо-
ванию влияния растворителей на про-
цесс электровосстановления металлов,
часто встречается деление органиче-
ских растворителей на два класса:
растворители, в которых потенциалы
восстановления близки к значениям в
водных растворах, и растворители, в
которых потенциалы восстановления
значительно сдвинуты к менее отрица-
тельным значениям. У этих двух клас-
сов растворителей существенно разли-
чаются донорные числа. Такую классификацию можно убедитель-
но продемонстрировать на примере электровосстановления редко-
земельных элементов. На рис. 24 потенциалы полуволн для пары
Eu (III) |Eu (II) в различных растворителях сопоставлены с донор-
ными числами последних [667]. Прослеживается четкая зависи-
90
мость — с уменьшением донорного числа £1/2 сдвигается в более
положительную область.
По литературным данным, скорость восстановления большин-
ства катионов уменьшается при переходе от воды к органическим
растворителям. Судя по данным табл. 7 приложения для европия
и самария, эта закономерность полностью правомочна для ланта-
ноидов. Полученные значения констант скорости близки к соот-
ветствующим величинам в водных растворах или меньше их. Ано-
мальный порядок они имеют лишь в водно-органических раство-
рах, что можно объяснить специфической гидратацией [695, 1143].
Электровосстановление скандия и иттрия также происходит в
две ступени или же непосредственно до металлического состояния
в зависимости от природы растворителя [1178]. Электродные про-
цессы необратимы, с повышением pH раствора необратимость
уменьшается.
Для ионов тория (IV) характерно одно- или двухэлектронное
необратимое катодное восстановление [681, 684, 1180, 1039, 990,
878]. Согласно полярографическим данным восстановление тория
(IV), подобно цирконию и гафнию, часто сопровождается выделе-
нием водорода. Исследованы перхлоратные, нитратные и хлорид-
ные растворы тория. На полярограммах наблюдается от одной до
трех волн, природа которых в большинстве случаев не установле-
на. Кинетика катодного процесса рассчитана исходя из четырех-
электронного катодного процесса. При таком предположении ре-
зультаты исследований указывают на квазиобратимый электрод-
ный процесс. Значения коэффициентов диффузии диффундиру-
ющих ионов тория, рассчитанные из полярографических и потен-
циометрических данных в ДМСО, разнятся на два порядка.
Катодное восстановление тория из водных растворов сводится
практически к выделению водорода.
Электрохимические исследования восстановления остальных
актиноидов проведены в основном с целью установления низших
степеней окисления их в неводных средах. Сведения немногочис-
ленны [360, 359, 824] и касаются механизма процесса. Кинетиче-
ские данные отсутствуют. По-видимому, из-за сходства электрон-
ных структур низших степеней окисления актиноидов и щелочно-
земельных металлов полярографическое восстановление их
происходит одинаково. В частности, для трехвалентных ионов
трансурановых металлов электровосстановление осуществляется
двухступенчато через двухвалентное состояние или непосредствен-
но до металла. Потенциалы полуволн имеют близкие значения к
величинам для лантаноидов, электровосстановление происхо-
дит труднее, чем в случае РЗЭ. При ступенчатом восстановлении
двухзарядные катионы активно взаимодействуют со следами воды
в растворителе с образованием гидролизованной трехвалентной
формы.
Ступенчато до металла восстанавливаются на ртутном электро-
де и четырехзарядные ионы трансурановых элементов. Так, вос-
становление U4+, Np4+ и Рп4+ ионов в ДМСО и АН протекает в
91
две ступени. По полярографическим данным первая одноэлектрон-
ная волна отвечает процессу Ме^+е^-Ме^, вторая трехэлектрон-
ная— Ме3++ .Зе->Ме° (Hg). При условиях, соответствующих вос-
становлению сольватов строго определенного состава, наблюдает-
ся трехступенчатое восстановление [526].
В результате полярографического изучения восстановления
соединений U (IV) и U (V), по данным ряда авторов, обнаружена
только одна волна, относящаяся к электровосстановлению урана
[722, 1233, 820, 738], процесс двухэлектронный, используемый в
отдельных случаях для аналитического определения урана
[820, 738].
2.3.7. Подгруппа германия
Показана возможность электровосстановления галогенидов Ge (II)
и Ge (IV) до металлического состояния [1225, 482, 381, 147, 292,
971]. Особенно благоприятны для восстановления растворы в
гидроксилсодержащих органических соединениях, в частности в
многоатомных спиртах, содержащих группы ОН, находящиеся у
соседних атомов углерода (этиленгликоль, пропиленгликоль, гли-
церин и т. д.). Судя по кривым поляризации, можно предположить
ступенчатое восстановление Ge4+ через промежуточное образова-
ние Ge2+. Из комплексных частиц Ge (IV) восстанавливается по
полярографическим данным в четыре ступени. Кинетические па-
раметры процесса восстановления практически отсутствуют. Для
водно-органических систем наблюдается снижение степени необ-
ратимости катодного процесса с увеличением содержания органи-
ческой составляющей, в частности для смесей диолов с во-
дой [866].
Процессы электровосстановления олова из органических сред
изучены недостаточно, в основном за небольшим исключением
[1134], для галогенидных ионов и ионов с органическими лиган-
дами [1153, 164, 1243, 69, 343, 1047, 784]. По результатам элект-
ролиза и полярографических измерений [1243, 343, 261, 95] при
восстановлении тетрагалогенидов олова на катоде происходят ре-
акции: Sn (IV) +2e=Sn (II); Sn (IV)+4e = Sn(0); Sn(II)+2e =
= Sn(0). Течение той или иной реакции определяется фоновым
электролитом и плотностью тока. Процессы необратимы как на
инертном, так и на ртутном капельном электродах. На электроде
при этом выделяется смесь металлического олова и белого осадка
дихлорида олова. В случае галогенидов Sn (II) общий катодный
ток соответствует электровосстановлению Sn(II) до Sn(0).
Комплексы двухвалентного олова с органическими лигандами вос-
станавливаются на ртутном капельном электроде в адсорбиро-
ванном состоянии. Этот процесс применяется в аналитической хи-
мии [69, 64].
Электровосстановление свинца изучено в простых перхлорат-
ных и многих комплексных электролитах [766, 765, 1274, 1266,
1153, 164, 252, 963, 1052, 904, 1123, 906, 780, 1197, 1134, 739, 913,
92
247, .997, 342, 1206, 654, 69, 504, 1207, 932]. Восстановление Pb2+
в большинстве случаев протекает одноступенчато обратимо [892,
1128, 1082, 67, 1142]. Обычно полярографические измерения пока-
зывают линейную зависимость между предельным диффузионным
током и концентрацией ионов свинца в растворе. Механизм и
кинетика процесса электровосстановления во многом определяются
фоновым электролитом [925, 722, 837]. Скорость восстановления,
как правило, меньше, чем в водных растворах; в отдельных рас-
творителях определены константы скорости. Количественные по-
лярографические характеристики восстановления Pb (IV) не по-
лучены из-за сильного искажения его волны. Подобное искажение
основной волны предволной и максимумами в некоторых раствори-
телях наблюдается и для РЬ2+ [1022].
2.3.8. Подгруппа титана
Электровосстановление металлов подгруппы титана в неводных
растворах подобно процессам в водных растворах довольно часто
обрывается на степени окисления 3+ [992, 1204, 1102, 1039, 686].
В случае титана это относится к растворам его галогенидов в
более основных по сравнению с водой растворителях. Кислотно-
основная реакция между L-кислотой, например TiCl4, и раствори-
телем основного характера [892, 992] приводит к образованию
-стабильного продукта нейтрализации, который восстанавливается
.лишь до Ti (Ш). Результаты полярографических исследований
электродных процессов при восстановлении Ti (IV) в растворите-
.лях менее основного характера, чем вода, показывают, что воз-
можно восстановление до Ti (III), Ti (II) и Ti(O). В ДМСО по-
.лярографически наблюдалось четырехступенчатое восстановление
TiCl4 [922] с фиксированием всех промежуточных степеней окис-
ления титана. Потенциал полуволны электродной реакции
Ti (IV) -]-4e->Ti (О) близок к 2 В. Во всех исследованных случаях
электровосстановление титана происходит из комплексных час-
тиц, процесс необратим и носит диффузионный характер [855,
1233, 378, 1198, 931]. Определены коэффициенты диффузии диф-
фундирующих и разряжающихся комплексов титана (TiCU2-,
[TiC12Sn]2+), имеющие порядок (1—2) • 10-5 см2/с.
Изучение катодной поляризации платинового катода (анод ти-
тановый) при электролизе растворов четыреххлористого титана в
ДМСО и алифатических спиртах и их смесях с ДМСО и аромати-
ческими углеводородами, в простых и сложных эфирах, амидах,
НМ, АН, хлористых ацетиле и тиониле, ГМФТА, ПК показывает,
что из большинства этих растворителей титан не восстанавливает-
ся. Природа процесса восстановления титана из растворов TiCl4
в ДМСО и его смесях с этанолом не ясна. Возможно, что выделе-
ние титана на платиновом катоде в виде цветных пленок является
вторичным химическим процессом [294, 686].
Характер изменения поляризации титановых и кобальтовых
^электродов в глицериновом электролите при различных условиях
93
также свидетельствует о том, что лимитирующей стадией элект-
родного процесса являются диффузионные ограничения по доставке
ионов титана к поверхности катода [640]. В целом следует отме-
тить, что электровыделение металлического титана возможно
лишь из немногих органических растворителей и электродные про-
цессы, происходящие при этом, изучены недостаточно.
Характер восстановления ионов циркония и гафния по данным
полярографии очень похож [1179, 893, 1102, 1068, 773, 1039]. В ре-
зультате плохой растворимости в органических растворителях и
трудностей получения в безводном состоянии перхлоратов этих
металлов исследовались в основном растворы тетрахлоридов.
В зависимости от природы растворителя, фона и концентрации
восстанавливающихся частиц происходит ступенчатое восстанов-
ление или восстановление непосредственно до металла. Как и в
случае титана, нередки обрывы цепи восстановления. Часто хоро-
шо выраженные волны на полярограммах растворов тетрахлори-
дов циркония и гафния относятся к восстановлению водорода из
сольватного окружения ионов этих металлов. Этот процесс особен-
но характерен для спиртовых растворов [773] и смешанных вод-
но-органических растворов [138, 1039, 1068]. Как правило, элек-
тродные процессы носят диффузионный характер и, за небольшим
исключением, например обратимые ступени Zr(IV)->Zr(III) и
Hf(IV)(III) в ДМСО [1101] необратимы. Ступенчатость вос-
становления Me(V)->Me(III)->Me(II)->-Me(0) более характер-
на для циркония. Кинетика катодного восстановления этих метал-
лов не изучена.
2.3.9. Металлы пятой группы
Сведения об электрохимическом поведении металлов подгруппы
ванадия немногочисленны. Большинство исследований посвящено
выяснению возможности выделения их из органических раствори-
телей и носит качественный характер. Единственный вывод из этих
исследований — ступенчатое, часто одноэлектродное, восстановле-
ние до низших степеней окисления, на полярограммах иногда про-
является лишь одна ступень восстановления [421]. Полярографи-
рование растворов NbCls в некоторых органических растворителях
[893, 1030] приводит к полярограмме с одной волной. Возможно,
ее появление, как и в водных растворах [476], связано с образо-
ванием низшей степени окисления ниобия и каталитическим хи-
мическим процессом. Установлена зависимость высоты этих волн
от концентрации ниобия и возможность использования их для
аналитических целей. Определены параметры скорости реакции
V3++e^:V2+ в метилформамиде [687]. Как и следовало ожидать,
величина константы скорости значительно ниже, чем в воде. Изу-
чены также механизм и кинетические параметры восстановления
комплексных соединений ванадия с органическими лигандами
[1171, 1172].
94
По данным классической полярографии и осциллополярогра-
фии хорошо выраженные волны дают во многих органических рас-
творителях ионы трехвалентных сурьмы и висмута [892, 1153, 722,
1052, 904, 1123, 1066, 146, 1047, 785]. Процесс восстановления в
основном изучен на галогенидных солях. Наблюдалось как одно-
ступенчатое [1052, 1128, 785, 226], так и многоступенчатое [722,
146] восстановление до металла. Потенциалы выделения, как пра-
вило, более положительны, чем в водных растворах, что свиде-
тельствует о низкой энергии сольватации ионов в соединениях
Sb(III) и Bi(III) в органических средах. В случае двухступенча-
того разряда ионов соединения Sb(III) медленной ступенью слу-
жит первая ступень присоединения двух электронов [146]. Для
обоих металлов процесс электровосстановления имеет преимуще-
ственно диффузионный характер. В результате исследования элек-
трохимического поведения иона Bi(III) в спиртовых и водно-
спиртовых растворах отмечено нарушение пропорционально-
сти между концентрацией BiCl3 и величиной предельного тока
[1123].
Методом классической и импульсной полярографии изучено
электрохимическое поведение пятивалентной сурьмы в безводном
HF и его смесях с водой. В водном HF ион Sb^ электроактивен
в присутствии ионов С1~. Здесь наблюдается две волны: двухэлек-
тронная, соответствующая восстановлению Sb5^ до Sb3+, и трех-
электронная, соответствующая дальнейшему восстановлению Sb3+
до Sb(O) (£1/2=—0,460 В по отношению к нкэ). В безводном HF
обе волны исчезают вследствие образования полярографически
неактивного комплекса SbFe~ [785].
2.3.10. Металлы шестой группы
Катодное восстановление из неводных растворов металлов шестой
группы наиболее полно изучено для хрома. Хром (III) восстанав-
ливается в перхлоратных неводных растворах двухступенчато
[892, 925, 714, 1005, 742, 1233, 1134, 989, 718]. Первая ступень в
основном одноэлектронна, обратима; вторая — двухэлектронна, не-
обратима. Количественные полярографические данные по меха-
низму процесса электровосстановления немногочисленны. В рас-
творах в виде других солей, в том числе и комплексообразующих,
ступенчатость восстановления хрома сохраняется [1153, 722, 654,
1133, 331, 1001, 985]. Как и в случае многозарядных других d-
металлов, восстановление комплексов с органическими лигандами
осуществляется одноэлектронно [931, 1171, 1172, 1169, 1252, 985].
Установлена корреляция между значениями потенциалов полуволн
этих ступеней и электронным строением исследуемых комплексов.
Полярографическое поведение органических комплексов хрома в
неводных растворах используется в аналитических целях [498,
64]. Определены кинетические параметры процесса разряда иона
Сг3+ в некомплексообразующих [925] и комплексообразующих
[1171, 1172, 1169] электролитах.
95
Что касается вольфрама и молибдена, то исследование процес-
сов их электровосстановления, как и в случае металлов пятой
группы, сводится в основном к выяснению возможности их выде-
ления в металлическом виде из органических растворителей.
В этом плане более подробно изучен вольфрам. О механизме и
кинетике электродных процессов для обоих металлов известно
лишь, что восстановление происходит до низших степеней
окисления, а не до металла [1201, 1205, 1203, 1202, 1239, 264].
Так, в метаноловых растворах восстановление хлорида Mo(V)
протекает необратимо до Мо(Ш) [264], а оксокомплексы Mo(V)
в ДМФ-растворах претерпевают одноэлектронное восстановление
до соответствующих оксокомплексов Mo(IV), степень обратимости
которого существенно зависит от структуры комплекса и природы
лиганда [1239].
В отдельных случаях определены кинетические параметры
электровосстановления Mo(V) и W(V) [1171, 1172, 590, 787], в
основном для восстановления комплексов молибдена с органиче-
скими лигандами [1171, 1172,787].
Процесс электровосстановления четырехзарядных селена и тел-
лура изучался в уксуснокислых и спиртовых растворах [8, 9].
Методами поляризационных кривых и электролиза установлено,
что наряду с основной реакцией восстановления Те(IV) и Se(IV)
до металлического состояния происходит также частичное восста-
новление до двухзарядных ионов. Характер этих процессов не ис-
следован.
2.3.11. Подгруппа марганца
Марганец последний в ряду напряжений металл, который может
быть выделен электрохимически из водных растворов. Очевидно,
поэтому при изучении его электрохимического поведения в невод-
ных растворах [681, 684, 892, 1153, 714, 722, 1005, 780, 1233, 837,
1134, 680, 913, 853, 1066] много внимания уделено выяснению вли-
яния воды на электрохимические параметры [609, 164, 904, 891, 742,
989, 718, 885, 6], а также исследованию электровосстановления
марганца в смешанных водно-органических растворителях [853,
1256, 956].
При полярографических исследованиях в водных, неводных и
смешанных растворителях Мп(II) дает одну хорошо выраженную
волну, соответствующую двухэлектронному восстановлению. Прав-
да, в литературе имеются указания о замедленности стадии при-
соединения второго электрона при разряде иона Мп2+ и образова-
нии промежуточных соединений Мп(1) вблизи равновесного по-
тенциала [609, 6]. Катодный процесс в большинстве случаев ква-
зиобратим или обратим. Элемент необратимости более характерен
для водных растворов. Для неводных растворов необратимый про-
цесс электровосстановления марганца наблюдается в случае силь-
ного комплексообразования с растворителем, например, в раство-
рах триметилфосфата образование стабильных комплексов мар-
96
"f
ганца служит причиной необратимости процесса (Ь = 220 мВ)
[891]. Характер катодного процесса диффузионный, для большин-
ства растворов наблюдается прямолинейная зависимость между
предельным диффузионным током и концентрацией марганца в
растворе.
Во многих работах прослеживается корреляция потенциала по-
луволны и предельного тока с диэлектрической проницаемостью
и вязкостью раствора [1153, 891, 1005, 742, 989, 718, 885, 853, 1256,
956, 1190]. С повышением диэлектрической проницаемости Ец2
марганца сдвигается в сторону более отрицательных значений.
Отклонения наблюдаются лишь в случае наложения процессов;
комплексообразования. Для количественной интерпретации ре-
зультатов привлекается уравнение Борна (2—14).
Диффузионно контролируемый ток связан с коэффициентами
диффузии электроактивных частиц, и для многих растворителей
показано, что предельный ток находится в простом соотношении с
вязкостью. Связь устанавливается при помощи уравнения Сток-
са — Эйнштейна. Подчеркивается, что вязкость — не единствен-
ный фактор, влияющий на диффузионный коэффициент марганца..
Большое внимание уделено исследованию электровосстановле-
ния марганца в неводных растворах в присутствии комплексообра-
зующих добавок [891, 837, 956, 997, 737, 879, 1066], в том числе
комплексов Мп(III) [64, 272, 692, 920].
Скорость катодного процесса восстановления марганца, как
и других металлов в неводных растворах, низкая. Величины то-
ков обмена, полученные в перхлоратных растворах марганца в
ДМСО, на два порядка ниже, чем в воде [609]; на кинетику вы-
деления сильно влияет природа растворителя.
Для рения исследовано электровосстановление из комплексных
ионов [919]. Восстановление происходит многоступенчато, первые
ступени, как правило, одноэлектронны, в последующих могут при-
нимать участие и несколько электронов.
2.3.12. Металлы восьмой группы
Исследования процесса электровосстановления железа из невод-
ных растворов достаточно многочисленны [681, 684, 1136, 1153,
714, 1012, 722, 1005, 742, 780, 1233, 1134, 989, 718, 470, 997, 737,
879, 62, 63, 504, 1089, 897, 1173, 1260, 914], однако большинство
из них относится к комплексообразующим электролитам, и лишь
в некоторых работах уделено специальное внимание электрохими-
ческому поведению железа. По полярографическим данным в пер-
хлоратных растворах восстановление ионов Fe3+ и Fe^ происхо-
дит необратимо: первого — в две ступени, второго — одноступен-
чато. Первая ступень при восстановлении Fe3+ не всегда истинная,
поскольку восстановление Fe3+ часто наступает при потенциале,
более положительном, чем анодное растворение ртути [892].
Комплексные ионы двухвалентного и трехвалентного железа в
основном восстанавливаются многоступенчато, каждой ступени со-
7 - 4-653
97
ответствует одноэлектронное восстановление [777, 1066, 1238,
1097].
О кинетических параметрах процесса электровосстановления
ионов железа имеется лишь незначительная качественная инфор-
мация [1175, 653], количественные данные получены только для
комплексообразующих электролитов [1174].
Проведены обширные исследования по изучению электровос-
становления ионов никеля и кобальта из простых перхлоратных
и комплексообразующих неводных растворов [681, 684, 766, 765,
892, 1136, 153, 1009, 1153, 164, 1022, 382, 714, 963, 722, 1052, 904,
891, 1005, 906, 742, 743, 712, 780, 1175, 1233, 837, 1134, 989, 931,
718, 885, 1265, 913, 1190, 997, 737, 879, 654, 504, 852, 854, 1049].
По полярографическим данным восстановление в большинстве
случаев проходит необратимо. Степень необратимости выше в
случае никеля. Квазиобратимость для обоих металлов наблюдает-
ся лишь в нитрильных растворах. Определяющее значение при
электровосстановлении ионов Ni2+ и Со2+ имеет высокая комплек-
сообразующая способность этих металлов. При этом следует учи-
тывать образование комплексов с растворителем, фоном и комп-
лексообразующими добавками в случае присутствия последних.
В перхлоратных растворах, за редким исключением, восстанов-
ление происходит в одну ступень до металла. В комплексообра-
зующих электролитах наблюдается ступенчатое восстановление,
относящееся к комплексам различного состава [689, 1276, 916,
943—945, 938, 694, 66, 706, 928, 612]. Количество ступеней дости-
гает трех. Конечным продуктом восстановления может быть не
только металл, но и комплексы нулевой валентности.
Таблица 14. Кинетические параметры электровосстановления Ni2“*“ и Со2+
из водно-формамидных растворов
ФА, % 0,8 мМ Ni2+ в 0,1 М NaClO4 1 мМ СО2+ в 0,1 М NaClO4
“й Kf. см/с ап Кр см/с
0,0 0,737 2,5-ю-1» 0,90 2,5-10-*®
40,0 0,679 4,5-1О-1а 0,84 .5,6- 10-IS
60,0 0,650 4,0- ГО"11 0,80 4,5-10-*®
80,0 0,650 3>10—10 0,79 1,4-10-**
100,0 0,620 7,1 -10-10 0,84 .2,8- Н>-*5
Повышенной способностью к комплексообразованию, приводя-
щей к существованию прочных гидратов никеля и кобальта в вод-
ных растворах, объясняется и то, что прибавление неводного рас-
творителя (менее активного лиганда, чем ОН-) к водным раство-
рам этих металлов часто увеличивает обратимость их восстанов-
ления и константу скорости электродной реакции [766, 765], хотя
в целом при переходе от водных растворов к неводным растворам
наблюдается обратная закономерность. В табл. 14 приведены ве-
98
личины кажущихся констант скорости (К/0) и коэффициентов пе-
реноса (ап), демонстрирующие изменение скорости катодного про-
цесса при восстановлении Ni2+ и Со2+ из водно-формамидных сме-
сей [944, 945, 852, 854]. По данным таблицы в случае восстанов-
ления Ni2+ при переходе от воды к безводному формамиду наблю-
дается увеличение скорости на три порядка, в случае Со2^— на
один.
Процессу разряда кобальта и никеля часто, очевидно, предше-
ствует реакция десольватации или диссоциация комплексного иона
[925, 993, 955, 928]. Большое значение для электродного процесса
имеют обменные реакции между анионами фона и окружением
катионов Ni2+ и Со2+ [1276, 944, 945, 943, 938], а также адсорб-
ционные процессы на поверхности катодов с участием молекул
растворителя, фонового электролита и образующихся комплексов
исследуемых металлов [681, 684, 680, 470, 944, 945].
Особенно много внимания уделено изучению хелатов металлов
триады железа. Механизм их электровосстановления в неводных
растворах определяется в первую очередь природой центрального
атома. Так, полярографическое исследование восстановления ди-
тиокарбаминатов различных металлов на Hg-электроде в ДМФ
показало, что хелаты по своему электрохимическому поведению
делятся на две группы. Полярограммы, относящиеся к комплексам
Fe3+, Со3+, Ni2+, Сг3+, Мп3+, содержат п ступеней, соответствующих
последовательному переносу «-электронов. Продуктом конечной
необратимой стадии является металл на поверхности ртути. Хе-
латы металлов с заполненными d-оболочками (Zn2+, Cd2+, Sn^,
Hg2+, Pb2+ и т. д.) ведут себя иначе. Для комплексов данных ме-
таллов на полярограммах наблюдается одна волна, соответст-
вующая восстановлению центрального иона до металла, разряд
в большинстве случаев близок к обратимому. Работы по изуче-
нию электрохимического поведения хелатов переходных металлов
имеют практическое значение. Они позволяют решать вопросы
электрокатализа, гальваностегии, электросинтеза и электроанали-
тического определения металлов [68, 64, 65].
Электровосстановление платиновых металлов изучено лишь в
комплексообразующих неводных растворителях [1046, 1265, 1089,
1259, 808, 1106, 907, 1170—1172]. Основными объектами исследо-
вания служили комплексы осмия, родия, иридия с органическими
лигандами. Изучение их представляет как теоретический интерес
в плане выяснения основных закономерностей процессов комплек-
сообразования в неводных средах и стабилизации низших степе-
ней окисления элементов в апротонных растворителях, так и прак-
тический, поскольку некоторые из них люминесцентны и могут
служить объектами превращения световой энергии, в том числе
и солнечной, в химическую [1159, 1158, 1247].
Как правило, комплексы платиновых металлов восстанавлива-
ются многоступенчато (3—4 ступени). Обратимость процесса элек-
тровосстановления определяется природой лигандов и строением
комплекса, иногда температурой [755, 972, 648, 1258, 1160]. Пер-
7*
99
вые одна-две ступени одноэлектронны и обратимы, последующие—
необратимы, в процессах, соответствующих им, может принимать
участие несколько электронов [755, 648]. Аналогично при элект-
ровосстановлении ведут себя комплексы рения [919].
2.3.13. Электровосстановление металлов
из оксидов
В связи с широким использованием в последнее время оксидов
некоторых металлов в качестве электродного материала в химиче-
ских источниках тока представляет интерес рассмотреть отдельно
электровосстановление этих металлов из их оксидов в органиче-
ских растворителях. Литература по данному вопросу носит в ос-
новном патентный характер, однако отдельные работы посвящены
непосредственно выяснению механизма и кинетики катодных про-
цессов [492, 445, 446, 427, 426, 466, 425, 1163]. Большинство ра-
бот относится к исследованию оксидов V2O5, МоО3, Сг2О3, СгО3,
WO3, МпО2 [492, 445, 446, 427, 426, 466, 425]. Эти оксиды плохо
растворяются в органических растворителях.
Установлено, что в протонных диполярных растворителях (БЛ,
ДМФ, ДМА, ПК, ТГФ и др.) механизм восстановления оксидов,
по-видимому, аналогичен их механизму восстановления в водных
щелочных растворах и носит электронно-протонный характер. Со-
гласно этому механизму подвижной частицей, ответственной за
массоперенос в твердой фазе, является протон. Процесс восста-
новления оксида протекает через две основные стадии. Первая —
электрохимическая реакция перехода протона через межфазную
границу раствор — оксид, в результате которой поверхностный
слой оксида превращается в соединение нестехиометрического
состава. Вторая стадия, обеспечивающая восстановление более
глубоких слоев,— диффузия протона в глубь оксида с одновремен-
ным переходом электрона от одного иона металла к другому.
В стационарном состоянии вторая стадия является замедленной
и ее скорость определяется скоростью диффузии протонов в ре-
шетке оксида. В апротонных растворителях в роли подкислителя
выступает протон примесной воды или ион лития, который внед-
ряется в кристаллическую решетку оксида. Конечным продуктом
восстановления является оксидное соединение восстанавливаемого
металла низшей валентности. Так, в хлоридных растворах ДМА
процесс восстановления протекает с участием двух электронов,
конечным продуктом восстановления является смешанный оксид
состава xMoO3-z/MoO2-zLiO.
В хлоридном растворе ДМФ одноэлектронный катодный про-
цесс восстановления WO3 на границе оксид (электрод) — раствор
описывается уравнением 2WO3+Li++e->WO3-LiWO3. Конечный
продукт подобен литий-вольфрамовым бронзам [445, 446].
Процесс электровосстановления оксидов чаще всего необратим
и протекает стадийно. При участии ионов лития в процессе ско-
рость восстановления замедляется, поскольку механизм диффузии
100
т
ионов лития в оксиде отличен от диффузии протона и внедрение
его в кристаллическую решетку оксида происходит медленнее.
Например, коэффициент диффузии Li+ в пленках пентаоксида
• ванадия составляет величину 10~10—10-11 см2/с [426].
В целом процесс электровосстановления оксидных электродов
в органических растворителях изучен недостаточно. В общем слу-
чае механизм восстановления сложен, скорость процесса, очевид-
но, определяется скоростью диффузии в оксиде наиболее подвиж-
ной частицы электролита, влияние большинства факторов (напри-
мер, анионов) на течение процесса не установлено.
2.3.14. Неметаллы
Исследования по электровосстановлению неметаллов можно раз-
делить в основном на две группы: восстановление анионов и вос-
становление элементов в нейтральном состоянии. К электровос-
становлению анионов относится также электровосстановление
оксидов.
В случае восстановления анионов первостепенное значение при-
обретает природа органического растворителя. Большинство анио-
нов в протолитических растворителях стабилизируется водород-
ными связями. Ввиду отсутствия доноров протонов в апротонных
растворителях активность анионов повышается, и стабилизация их
часто происходит путем взаимодействия с катионами. Прочность
комплексов катионов с анионами в апротонных растворителях зна-
чительно выше, чем в протолитических, например в воде. Это
различие в молекулярном состоянии анионов в зависимости от
природы органической среды сказывается на протекании электрод-
ного процесса.
При восстановлении элементарных неметаллов основную роль
играет растворимость неметалла в органическом растворителе, ве-
личина которой изменяется в широких пределах. Наибольшее ко-
личество исследований приходится в обоих случаях на кислород,
серу и фосфор. Наименее изучены углерод (естественно, исключая
область органических соединений), кремний и азот. В табл. 8
приложения приведены электрохимические параметры, характери-
зующие процесс восстановления некоторых элементов из молеку-
лярного состояния.
Углерод, азот, кремний. Электровосстановление углевода про-
водили из СО2. Осуществление процесса затруднено [834]. В ра-
боте [1156], где исследование электровосстановления СО2 в ДМСО
проведено количественно, этот процесс рекомендован для исполь-
зования в аналитических целях. Возможны одноэлектронный и
двухэлектронный ступенчатый механизмы восстановления. В обоих
случаях конечным продуктов является сольватированный анион
НСО7. По данным работы [875], конечным продуктом восстанов-
ления СО2 в ДМФ и ДМСО служат оксалат и СО в отсутствие
воды, в ее присутствии образуются формиат и гликолят. В каче-
стве промежуточного продукта возможно образование анион-ра-
101
дикала С02~ [875, 834]. В растворителях (АН, ПК, ДМФ, ДМА,
ДМСО) установлена линейная зависимость между Е{/2 восстанов-
ления СО2 и донорным числом растворителя, с ростом которого
потенциалы полуволн сдвигаются в положительную сторону [834].
Проведено изучение катодного восстановления нитрат-иона
[722, 893, 1209, 185, 184, 903]. Установлено, что в безводных орга-
нических растворителях и их смесях с водой, концентрированных
по неводному компоненту, процессы электровосстановления NO3~-
иона сильно подавлены. Это выражается, в частности, в отсутст-
вие полярографических волн восстановления NO3~ на микроэлект-
родах из целого ряда металлов, а также в восстановлении NO3~
на электродах из многих металлов лишь до NO2_ [185, 184]. По-
следний факт свидетельствует об имеющихся затруднениях при
катодном восстановлении аниона NO2-. Глубину протекания про-
цесса электровосстановления аниона NO3~, а следовательно, и
NO2~ можно усилить путем изменения кислотности среды и вве-
дением в раствор многовалентных катионов металлов. Таким об-
разом удалось получить продукты восстановления NO2—NH4+ и
NH3OH+. Катодному восстановлению в неводных растворах под-
вержены и нейтральные соединения азота. Например, тетранитрид
серы (S4N4) в AH-растворе в присутствии доноров протонов элек-
тровосстановлением почти количественно превращается в
S4(NH)4 [903].
В нескольких работах наряду с другими элементами полярогра-
фически изучено электровосстановление кремния из его тетрага-
логенидов [722, 893]. Электровосстановление осуществляется в
две ступени, возможно, до элементарного состояния. Определен
потенциал полуволны первой ступени в ДМСО [893].
Фосфор. Изучено электровосстановление элементарного фосфо-
ра [182, 59, 496, 423] и его соединений [423, 656]. Элементарный
фосфор весьма реакционноспособен, он в равной мере способен
проявлять окислительные и восстановительные свойства, т. е.
должен вступать как в катодные, так и в анодные реакции. На
катоде желтый фосфор (Р4) в зависимости от материала катода,
растворителя и концентрации способен восстанавливаться до раз-
личных степеней окисления. В апротонных растворителях (АН,
ДМФ) на ртутном электроде при концентрациях Р4< 10~3 моль/л
происходит присоединение двух электронов с образованием двух-
зарядного бианиона Р42~, в концентрированных растворах фосфора
образуется однозарядный анион Р4-|-е^->-Р4_. Восстановление про-
текает через образование хемосорбированного комплекса P4Hgx
[59, 423]. Характер катодного процесса диффузионный. Анионы
Р4- и Р42- способны взаимодействовать с находящимися в при-
электродном слое органическими соединениями с образованием
фосфорорганических соединений [182, 59]. В протолитических рас-
творителях процесс восстановления протекает необратимо с при-
соединением трех электронов также через промежуточное образо-
вание поверхностного хемосорбированного соединения P4Hgx,
электрохимически восстанавливающегося до фосфористого водоро-
102
да [496, 423]: P4Hgx+12H++12e->4PH3+xHg. На свинцовых ка-
тодах возможно получение высших гидридов фосфора [423].
Возможно электровосстановление и анионов фосфора из орга-
нических растворителей. Так, в то время как в водных растворах
анион РО43- электрохимически не восстанавливается, в неводных
растворителях (сульфолане, 2-метоксиэтаноле, глицерине) проис-
ходит его восстановление до аниона РО3~. Суспендированный в
сульфолане пентаоксид фосфора также восстанавливается до фос-
фористой кислоты [656].
Восстановление как элементарного фосфора, так и его произ-
водных широко используется для электрохимического синтеза со-
единений фосфора.
Мышьяк. Показана возможность электровосстановления мы-
шьяка до элементарного аморфного состояния из растворов его
трихлорида в некоторых органических растворителях (ледяная
уксусная кислота, АЦ, МЭК) [1218, 1062].
Кислород. Электровосстановление кислорода в органических
растворителях изучено подробно методами полярографии, цикли-
ческой вольтамперометрии, электролиза при контролируемом по-
тенциале, хронопотенциометрии, нередко с привлечением неэлект-
рохимических измерений на различных катодах в присутствии раз-
нообразных фонов в многочисленных средах [925, 1022, 153, 963,
1052, 906, 1000, 984, 591, 991, 1034, 1122, 967, 868, 1146, 1248, 1018,
958, 663, 1019].
Особенно интересные результаты получены в апротонных раст-
ворителях. Восстановление кислорода в них протекает в две од-
ноэлектронные ступени, соответствующие им потенциалы полуволн
приведены в табл. 8 приложения. Первая ступень заключается
в переносе единственного электрона на молекулу кислорода с об-
разованием пероксидного радикал-аниона О2. Перенос осуществ-
ляется довольно быстро, обратимо или квазиобратимо в зависимо-
сти от природы растворителя его истинная константа скорости со-
ставляет 10-3 см/с [1183].
Вторая ступень определяется образованием пероксидного аниона
Ог-: I О2 + е^ОГ, II Ot + e-^O?T. В электродный процесс
включены химические реакции, в частности реакция диспропорциони-
рования пероксидного иона: ОГ + О О2 + Ог-.
В протолитических растворителях или апротонных, содержа-
щих доноры протонов, восстановление происходит в две двухэлек-
тронные необратимые ступени, как и в водных растворах. Первая
из них, как правило, определяющая скорость электродного про-
цесса, соответствует образованию пероксидного иона, вторая —
воды. Например, в формамидных растворах КС1 восстановление
растворенного молекулярного кислорода осуществляется в следу-
ющие две ступени: I — О2+2Ф+2е-»-Н2О2+2Ф_, II — Н2О2+2е->
->2ОН-(Н2О2+2Ф+2е->Н2О+2Ф_). Образующийся в первой ста-
дии восстановления кислорода во всех апротонных растворителях
радикал-анион О2~ очень нестабилен в присутствии доноров про-
103
ч
тонов и диспропорционирует до кислорода и пероксида водорода
02~-}-Н+->НО2, 2HOz->H2O2+O2. При этом донором протонов мо-
жет служить фоновый электролит [817].
Как в случае протолитических, так и апротонных растворите-
лей исследования первой стадии восстановления кислорода более
многочисленны, чем второй.
Сера. Развитие в последние годы электрохимии серы в невод-
ных растворах обусловлено, с одной стороны, практическим ис-
пользованием серы и ее диоксида в качестве перспективного ка-
тодного материала в источниках тока с электролитом на основе
апротонных растворителей; с другой стороны, некоторые серосо-
держащие анионы, например S2O82-, являются удобной моделью
для развития теории элементарного акта переноса заряда (вос-
становление S2O82- на катодной поверхности при отрицательных
зарядах исключает специфическую адсорбцию как реагирующей
частицы, так и продукта реакции). Соответственно этим направ-
лениям ведутся исследования электровосстановления элементар-
ной серы [681, 684, 1217, 155, 207, 1071, 444, 1246, 832, 833], ди-
оксида серы [693, 1037, 1026, 491, 628, 629, 717, 858] и ее анионов
[816, 132, 524, 523, 699, 131, 198, 195, 52, 133] в основном в апро-
тонных растворителях.
В протолитических растворителях конечным продуктом восста-
новления элементарной серы Ss на ртутном катоде является суль-
фидный анион [1217, 933, 155, 207, 1071]. Предполагается, что
акту электрохимического разряда предшествует быстрая химиче-
ская реакция элементарной серы с ртутью [155, 207]. Продуктами
восстановления сульфида ртути в зависимости от условий могут
быть сульфид (бисульфид)-ионы HgS-]-2e->Hg+S2~, HgS+2e-|-
+H+-s-Hg+HS~ или же сероводород HgS+2e+2H+-^-Hg+H2S.
В апротонных растворителях наряду с электровосстановлением
серы до S2- через промежуточное образование полисульфидов воз-
можно также одноэлектронное двухступенчатое восстановление с
образованием радикал-аниона S's, стабильного в ДМСО, ТГФ и
ДМФ в течение нескольких суток [681, 684, 487]. Образование
анион-радикала S s' наблюдалось на платиновом, графитовом, зо-
лотом электродах. Вторая ступень определяется образованием
двухзарядного аниона серы S 8 +e->S82-. Обе стадии процесса
необратимы, первая носит диффузионный характер, скорость вто-
рой определяется кинетикой или адсорбцией.
Поскольку сера интенсивно взаимодействует с ртутью с обра-
зованием сульфида, то, вероятно, во всех растворителях при ис-
пользовании ртутного катода конечным продуктом восстановления
будет анион S2-.
Электрохимическое поведение диоксида серы изучалось лишь в
апротонных растворителях. В таких средах SO2 подвергается
обратимому, квазиобратимому или необратимому одноэлектрон-
ному восстановлению с образованием радикал-аниона SO2~, кото-
104
рый затем в одних случаях димеризуется до аниона дитионата
S2O42- непосредственно, в других — через образование промежу-
точных комплексов (SO2)nSO2_ (n— 1, 2). Дитионат способен к
необратимой адсорбции на электроде, образуя слаборастворимую
пленку, пассивирующую электродную поверхность [858].
Подобно SO2 претерпевает в апротонных растворителях элек-
трохимическое восстановление хлористый тионил; двухступенчатый
процесс, сопровождающийся химическими реакциями, приводит к
образованию конечных продуктов SO2 и S [1253, 696]. Схемати-
чески механизм восстановления может быть записан следующим
образом: SOCl2+e-^SOCl+Cl-; SOCr+e-^SO+Cl'; SO-н
->1/2SO2+1/2S. Все стадии протекают с образованием сольватов
[1253]. До элементарной серы восстанавливается в апротонных
растворителях хлористая сера [835]. Процесс двухэлектронный.,
Электровосстановление персульфатного аниона в изученных
протонных и апротонных растворителях осуществляется согласно
схеме
S2O|~ + 2е 2SOl~.
Проведенные исследования в преобладающем большинстве отно-
сятся к изучению кинетики электровосстановления. В апротонных
растворителях скорость реакции восстановления замедлена по
сравнению с протонными. Так, в растворе перхлората натрия в
ДМСО истинная константа скорости восстановления S^Os2- на ртут-
ном электроде равна 5-10—3 см/с, в то время как при равных усло-
виях в водном растворе ее величина составляет 6,5-10—7 см/с.
Причиной может служить меньшая энергия сольватации анионов
в апротонных основных растворителях. Кроме того, скорость вос-
становления S2Os2~ в значительной мере определяется природой
катиона фонового электролита [816, 132, 524, 523, 699, 131, 198].
Изучен механизм катодного восстановления полисульфидных рас-
творов в апротонных растворителях [195, 52, 133].
Галогены. В органических средах исследовалось электровос-
становление как элементарных галогенов [862—864, 417, 947,
1024], так и их анионов [1047, 132, 772, 883, 882, 1166, 525]. Наи-
большее количество работ посвящено йоду и его производным
[1047, 132, 883, 882, 417, 1024, 864, 1166].
Восстановление анионов ХОГ, ХОГ, ХОГ(X = Cl,Вг, I), как пра-
вило, происходит ступенчато. Первые необратимые ступени про-
текают с участием нескольких электронов и образованием сво-
бодных галогенов. Вторая ступень одноэлектронна, продуктом ее
является галогенид-ион. На скорость протекающих электродных
процессов большое влияние оказывает природа фонового электро-
лита. В замедленной стадии переноса электрона принимают учас-
тие нейтральные молекулы растворителя.
Катодное восстановление элементарных йода и брома изучено
в апротонных растворителях. На инертных катодах процесс проис-
ходит в две двухэлектронные ступени. Первая ступень отвечает
105
I
образованию тригалогенид-иона: ЗХ2+2е->2Х3- вторая заканчи-
вается образованием галогенид-иона: 2Х3_+2е->ЗХ~ Скоростьоп-
ределяющей ступенью процесса часто является образование три-
галогенид-иона Х2 + Х_->Хз~. Обе ступени восстановления необра-
тимы как в случае йода, так и в случае брома в изученных рас-
творителях (АН, ДМФ, ДМСО).
Определены коэффициенты диффузии бромидных и йодидных час-
тиц Х2, X-, ХГ. Для отдельных растворителей найдены кинетические
параметры электродной реакции, энергии активации ступеней вос-
становления.
В заключение следует отметить, что в работе [522] сделана
попытка на основании обобщения кинетических закономерностей
процесса электровосстановления анионов, в том числе и анионов
металлов, создать теорию их разряда. Показано, что зависимость
скорости катодной реакции от потенциала электрода, концентра-
ции и заряда катионов фона, природы электрода и природы рас-
творителя может быть описана на основе феноменологической
теории замедленного разряда.
2.4. АНОДНЫЕ ПРОЦЕССЫ В СИСТЕМАХ МЕТАЛЛ —
НЕВОДНЫЙ РАСТВОРИТЕЛЬ
Катодному восстановлению металлов в органических средах в
•общем случае могут соответствовать анодный процесс ионизации
металла, разряда анионов (фона или специальных добавок), окис-
ление растворителя. В зависимости от материала анода, природы
восстанавливающегося на катоде металла, концентрации электро-
лита и условий эксперимента преобладать будет один из этих про-
цессов. Если анодным материалом служит металл, ионы которого
разряжаются катодно, анодное растворение металла происходит
более активно, чем в водных растворах. С анодным растворением
металлов неразрывно связан процесс коррозии.
Согласно современным представлениям [214, 128, 578, 494],
металлы в растворах электролитов растворяются преимущественно
по электрохимическому механизму. Подход к анодному растворе-
нию металлов и коррозии с единых позиций теории электрохими-
ческой кинетики, применение для изучения коррозии электрохими-
ческих методов исследования углубили и расширили теоретиче-
ские представления об этих процессах, и на их основе стали воз-
можны предварительные оценки коррозионной стойкости металлов
и сплавов в различных условиях, разработки принципов коррози-
онной защиты материалов. Однако коррозионная наука в послед-
ние три десятилетия развивалась в основном применительно к
водным растворам. Особенности процессов анодного растворе-
ния и коррозии металлов в органических электролитах изучены
недостаточно, хотя необходимость таких сведений в связи со все-
возрастающей ролью органических растворителей в качестве тех-
нологических средств очевидна.
106
Исследование анодных и коррозионных процессов проводилось
с помощью всего арсенала электрохимических методов, чаще все-
го поляризации в потенциостатическом, потенциодинамическом
и гальваностатическом режимах, в сочетании с чисто коррозион-
ными методами — весовым, рентгенографическим, спектральным и
т. д. [7].
Судя по экспериментальным данным, многие из теоретических
закономерностей в одинаковой степени правомочны для водных и
органических электролитов. Это представления о стадийности
процесса ионизации металла; о непосредственном участии в анод-
ном растворении металлов компонентов агрессивной среды — ани-
онов; о связи пассивации металлов с адсорбционными явлениями
и т. д. Однако в кинетике анодного растворения и коррозионного
разрушения металлов в водных и неводных средах имеются и
существенные различия. Как отмечалось, в целом металлы и
сплавы в органических растворителях подвергаются более актив-
ному растворению, многие из них теряют способность пассивиро-
ваться при анодной поляризации, резко снижается защитное дей-
ствие органических адсорбционных ингибиторов. До недавнего
времени вообще считалось необходимым условием пассивации ме-
таллов в органической среде некоторая критическая концентрация
воды, величина которой зависит от природы металла и состава
раствора или же растворенного молекулярного кислорода.
В связи с повышением агрессивности органических сред по
сравнению с водными возрастают трудности в подборе конструк-
ционных материалов для них и соответственно необходимость на-
копления данных о механизмах и кинетике сопряженных электро-
химических реакций коррозионного процесса. Имеющиеся в лите-
ратуре сведения в настоящее время несистематичны и ограничен-
ны. Это относится в первую очередь к металлам пятой, шестой и
седьмой групп.
2.4.1. Металлы первой группы
Анодное поведение и коррозионная устойчивость щелочных метал-
лов изучались в апротонных растворителях [479, 48, 47, 724, 1184,
1244, 968, 329, 202, 1232, 203, 253, 324, 1230, 961, 954, 677, 1055,
748, 1231, 826] в связи с их использованием в качестве электрод-
ного материала в источниках тока. Преобладающее большинство
работ относится к литиевому и литиево-амальгамному электродам.
Большое влияние на кинетику растворения литиевого электро-
да оказывают чистота электролита и атмосфера. В безводных ус-
ловиях такой электрод в исследованных растворителях обратим
[479, 1056, 1244], соответствующая ему электрохимическая реак-
ция одноэлектронна: Li^Li+4-е. Однако литиевые электроды, как
правило, показывают две ступени электрохимической активности.
Первая ступень характеризуется низкими токами обмена, на анод-
ной поляризационной кривой ей соответствует предельный ток.
Причина этого заключается в образовании на поверхности литие-
107
вого электрода плохо растворимых в электролите окисных, гидро-
окисных или солевых пленок в результате взаимодействия его со
следами воды, примесей, а иногда и компонентами электролита.
Часто пленки имеют полупроводниковый и полимерный характер
[418, 754]. Образующиеся пленки служат защитой литиевого элек-
трода от коррозии. Анодное растворение при высоких плотностях
тока (вторая ступень) является результатом частичного или пол-
ного разрушения пленки, которым объясняется увеличение стан-
дартной плотности тока обмена. Так, обычной «запассивирован-
ной» поверхности лития соответствуют токи обмена 0,80 [1056]
и 1,47 мА/см2 [908], на «свежем» срезе лития —10,2 [724] и
3,28 мА/см2 [908] для ПК и этиленкарбоната соответственно. На
жидких литиево-амальгамных электродах значения токов обмена
обычно выше, чем на твердых литиевых [47].
Установлена корреляция между величинами тока обмена и
природой растворителя. Для ряда растворителей (НМ, БЛ, ЭК,
ПК) проведенная корреляция показывает, что высокие значения
плотности тока могут быть получены в растворителе, имеющем
малую величину дипольного момента [260]. В этом случае худшим
растворителем является ПК, т. е. в нем литий меньше всего будет
подвержен коррозии. Эффективность анодного растворения лития
для данного растворителя существенно зависит от природы анио-
на. Так, в ПК она увеличивается в ряду Cl~<AlCLr<BF4_<;
ССЮл- [398]. Изучена также кинетика электроосаждения и рас-
творения на инертных электродах [383, 384]. По электрохимиче-
ским характеристикам в апротонных растворителях натрий ана-
логичен литию [48, 968, 329, 202, 1232, 373].
Исследования по анодному растворению меди в органических
растворителях проводились в двух направлениях. Часть работ
преследует цель установления стадийности процесса ионизации
меди и стабильности одновалентной меди в органических средах
[1118, 351, 904, 77, 78, 642, 1029, 356, 354, 355, 352, 1104], хотя в
некоторых из них затронуты и вопросы коррозионной устойчиво-
сти меди. В этих работах констатировано, как отмечалось при рас-
смотрении катодных процессов, для преобладающего большинства
исследованных органических растворителей двухстадийное проте-
кание процесса разряда — ионизации меди Cu^Cu+4-e; Cu+ч^
+±Cu2+-|-e. Определены количественные параметры кинетики этого
процесса. Кроме того, медь относится к тем металлам, при кон-
такте которых с растворами собственных устойчивых ионов обра-
зуются низковалентные ионы этого металла: Cu-f-Cu2+->-2Cu+, в
результате чего медь будет растворяться до тех пор, пока в объеме
раствора не установится равновесная концентрация ионов Си (I).
В больших объемах электролитов вследствие этой реакции наблю-
даются большие коррозионные потери, которые могут быть сни-
жены введением в раствор специальных ингибиторов [1092].
Вторая группа работ проведена непосредственно с целью уста-
новления коррозионной устойчивости меди как конструкционного
материала для органических сред, в основном это растворы элек-
108
w
тролитов в органических кислотах и спиртах [467, 247, 373, 520,
909, 910, 87, 90, 51, 32, 1095]. Во многих из этих работ наряду с
медью исследованы различные марки сталей и сплавов с целью
, замены корродирующей меди [520, 600, 606, 390]. Активное рас-
творение меди в ряде кислот, в частности монокарбоновых [32],
приводит к лимитирующей роли ступеней катодной реакции в
процессе растворения. Анодное поведение меди изучено в безвод-
ных и водных спиртовых растворах. При этом наблюдается инте-
ресная закономерность в некоторых спиртах (этанол, этиленгли-
коль) : вода по отношению к меди выступает в роли активного
агента, увеличивающего коррозию. Очевидно, процессы коррозии
меди и ее пассивации в водно-спиртовых растворах очень сложны,
и в них участвуют молекулы обоих компонентов раствора. В одних
случаях вода выступает в роли пассиватора, а спирт — активато-
ра, в других—наоборот, в зависимости от соотношения их кон-
центраций. Двойственная роль молекул воды отмечалась и в дру-
гих случаях [625]. Наряду с электрохимической для меди харак-
терна и химическая коррозия благодаря присутствию растворенно-
го кислорода [520, 87, 90].
Наряду с анодным растворением [1092] установлена возмож-
ность анодного окисления ионов серебра в неводных растворах
[713, 1186]. В жидком аммиаке получены соединения Ag (I) и
Ag (II); последние быстро разлагаются, окисляя растворитель.
В безводной уксусной кислоте в присутствии ацетата натрия на
платиновом аноде с очень низким выходом по току получен ди-
ацетат серебра.
Анодному окислению в органических растворителях подверже-
но и золото [1081, 1080, 869, 149]. В зависимости от природы при-
сутствующего в растворе лиганда окисление останавливается на
первой ступени, образуя Au (I), или идет дальше до образования
Au (III). В случае протекания процесса в две ступени лимитиру-
ющей является вторая, двухэлектронная. Предположено, что рас-
творение золота в жидком аммиаке в присутствии сольватирован-
ных электронов способствует образованию ионных пар типа
К+Аи- [1241].
2.4.2. Металлы второй группы
Анодному растворению металлов второй группы посвящено значи-
тельное количество работ [162, 443, 441, 487, 488, 373, 1230, 748,
909, 910, 473, 161, 500, 501, 280, 911, 682, 683, 76, 895, 607, 608,
1091], наиболее изучены цинковый и кадмиевый электроды.
Процессы растворения двухвалентных металлов могут проте-
кать по двум схемам: многостадийной (Ме->Ме+4-е, Ме+-^-Ме2++
-f-e) и одностадийной (Ме->Ме2++2е). Для двухстадийного про-
цесса, как отмечалось, установлены количественные закономерно-
сти не только для первой ступени ионизации, но и для сопряжен -
'ной реакции образования одновалентных ионов при контакте ме-
талла с раствором его двухвалентных ионов (Ме-->Ме+ + е, Ме2 +
109
+е-*-Ме+) [349]. Эти закономерности реализуются в преоблада-
ющем большинстве изученных случаев анодного растворения ме-
таллов второй группы, однако наблюдаются и одностадийные
процессы окисления металла до максимальной степени окисления
2 + . В работах уделено большое внимание выяснению влияния
природы растворителя и состава электролита на процесс иониза-
ции двухвалентных металлов
Растворение бериллия протекает в две одноэлектронные сту-
пени, причем некоторые из авторов [649] в безводных растворах
электрохимической считают только первую ступень: Ве-э-Ве++е
(электрохимическая ступень); Ве+-|-окислитель->-Ве2+ (химиче-
ская ступень, осуществляемая вдали от электрода).
Анодное окисление магния непосредственно до двухвалентного
состояния или же ступенчато через низшую промежуточную сте-
пень окисления часто зависит от состава электролита [1149, 1150,
1168]. Кальций, как правило, окисляется стадийно [441], замед-
ленная стадия — отщепление второго электрона. На анодных по-
ляризационных кривых обоих металлов наблюдаются предельные
токи, что свидетельствует об их пассивировании в определенной
области потенциалов [1230, 473]. В растворах ДМА и ДМФ пас-
сивация магния и кальция сопровождается появлением на их по-
верхности видимых пленок, не растворимых в электролите.
Механизм анодного растворения цинка преобладающе стадий-
ный [162, 443, 487, 488]. Кинетика процесса в значительной сте-
пени определяется природой присутствующих в растворе лиган-
дов. Так, образование прочных хлоридно-анионных комплексов
цинка в диметилацетамидных и диметилформамидных растворах
приводит к быстрому протеканию анодного процесса, но медлен-
ному катодного [162, 487]. Аналогичные явления происходят в
йодидных растворах ДМФ, хотя в отличие от хлоридных раство-
ров здесь наблюдаются более высокие токи обмена. При коррозии
в таких растворах торможение процесса будет катодным. Большое
внимание уделено поляризационным и коррозионным исследовани-
ям цинка в водно-органических растворах [162, 487, 634, 633].
Установлено сильное влияние состава водно-органических смесей
на механизм и особенно кинетику ионизации цинка.
Анодное растворение кадмия изучено на кадмиевом и кадмие-
во-амальгамном электродах. Процесс двухстадийный [1020, 76,
682, 683]:
Cd (С6+)адс Cd2+-растворитель.
В отсутствие специфически адсорбируемых анионов скорость образо-
вания (Сй)адс сравнима со скоростью образования Cd2+• растворитель.
В присутствии лигандов в основном замедленной является вторая
стадия образования Cd2+. Ионы кадмия значительно ускоряют
анодную реакцию. Поляризационные измерения в растворах на
основе АН и ДМФ на кадмиевом электроде в присутствии различ-
ных анионов: NO3~ BF4~, С1О4_, Br_, Cl- — показывают широкую
распространенность механизма анодного растворения металла в
ПО
органических растворителях с прямым участием анионов и молекул
растворителя [148, 76, 682, 683]. Предложены конкретные меха-
низмы для отдельных анионов. Попытка установить корреляцию
между анодным растворением кадмия и физико-химическими свой-
ствами органических растворителей осталась безуспешной. Изу-
чена коррозия полупроводниковых электродов типа CdTe [1091].
В зависимости от природы растворителя и состава ртуть окис-
ляется анодно до одно- или двухзарядных ионов [843, 865, 436].
2.4.3 Металлы третьей группы
Литературные сведения об анодном растворении и коррозионной
устойчивости металлов третьей группы относятся в основном к
подгруппе ША [108, 375, 186, 626, 220, 1127, 45, 1230, 677, 1240,
149, 1149, 1150, 911, 243, 1151, 114, 273, 673, 775, 978, 876, 973,
1094, 439, 124]. Единого мнения о механизме растворения алюми-
ния в органических растворителях не существует. Однако резуль-
таты большинства работ свидетельствуют о переходе алюминия
в раствор при анодной поляризации через промежуточные низко-
валентные частицы. Механизм и кинетика протекания процесса
во многом определяются не только составом электролита и состоя-
нием поверхности электрода, но и условиями эксперимента.
При изучении анодного поведения алюминия необходимо учи-
тывать наличие на его поверхности прочной оксидной пленки, ко-
торая способна в некоторых растворителях удерживаться даже
при длительной поляризации, а также омической и концентраци-
онной поляризации, после устранения которых в большинстве изу-
ченных растворителей растворение алюминия происходит без за-
метного перенапряжения.
Поведение алюминия изучено во многих растворителях, преж-
де всего в электролитах на основе эфира, из которых проводят
алюминирование [702, 298, 299, 204, 463, 286]. В таких электро-
литах анодный процесс сложный. Наряду с растворением алюми-
ния происходит газовыделение на аноде, способствующее его пас-
сивации. В эфирно-гидридных электролитах выделяется водород,
причем его выделение наблюдается даже при 100 %-ном выходе
алюминия. Высказано мнение, что газовыделение на аноде можно
объяснить самопроизвольным разложением гидридного аниона под
воздействием свежеобразующейся поверхности алюминия [298],
Еще более обильное газовыделение наблюдается на инертных ано-
дах, например платине, где разряд гидридного водорода является
основным процессом. В отсутствие гидридных анионов возможно
разложение эфира. Так, при разряде С2Н 5‘ происходит образо-
вание метана и этана [204]. Механизмы разряда анионов описа-
ны, например, для эфирата А1С13 в работе [299]. Анодное окис-
ление алюминия в электролитах ароматического ряда [186, 626]
происходит через образование сложных ионов, медленная иониза.-
ция которых, по-видимому, замедляет процесс.
111
В апротонных растворителях общую схему окисления алюми-
ниевого электрода можно представить следующим образом (вер-
тикальные строки соответствуют выходу частиц в раствор):
А1°=4>А1+=^>А13+
(а) (б)
| + а- | + ЗС1-
V V
А1С1 А1С13
В некоторых случаях в зависимости от условий эксперимента и
примененных методов исследования стадию а не удается зафик-
сировать [1044], хотя разряд многовалентных металлов представ-
ляет собой ряд последовательных одноэлектронных стадий [349,
113]. Вопрос о существовании в растворе нейтральных молекул
А1С12 в рамках используемых методик неразрешим. Положение
анодного потенциала зависит от природы присутствующих в рас-
творе анионов, природа катионов на него практически не влияет.
В ТГФ исследованы электроды из амальгамированного алюминия,
они ведут себя обратимо, воспроизводимо и могут быть исполь-
зованы в качестве электродов сравнения. Во многих случаях в
этих растворителях наступает анодное пассивирование алюминия,
часто с образованием видимых прочных оксидных пленок [161,
1228]. Характер процесса коррозии алюминия и сплавов на его
основе в апротонных растворителях электрохимический. Скорость
растворения алюминия, определяемая через силу тока растворения
i, в апротонных растворах, как и в протолитических, определяется
в основном характером влияния их координирующих и адсорб-
ционных свойств на состояние поверхности алюминиевого элект-
рода. В апротонных донорных растворителях, содержащих доста-
точное количество воды и активных анионов, монотонное снижение
lg i с ростом донорного числа позволяет предположить превали-
рующее влияние на i координирующей способности растворителя.
Так, высокая степень устойчивости алюминия (i->0) в хлорид-
ных растворах в ДМФ и ДМСО, содержащих воды выше 8 %,
объясняется, по-видимому, наряду с высокой адсорбируемостью
ДМФ и ДМСО возможным снижением по сравнению с водой по-
верхностной активности хлорид-ионов в результате увеличения
координирующей способности растворителя [243]. С другой сто-
роны, повышение скорости саморастворения алюминия в АН и
ПК в присутствии хлорид-ионов обусловлено, очевидно, более
высокой поверхностной активностью анионов в результате отсутст-
вия в этих растворителях специфической анионной сольватации.
Характер влияния роста концентрации хлорид-, а также бро-
мид-ионов на анодное поведение алюминия в водно-органических
растворителях противоположен при высоких содержаниях воды и
органического компонента. С одной стороны, с увеличением содер-
жания органического компонента активирующий эффект анодного
растворения С1_-ионов усиливается, с другой — по достижении
определенной для каждого органического растворителя концент-
112
рации в водно-органической смеси (АН — 76 %, ДМФ—63, аце-
тон — 45 % и выше) наступают торможение анодного растворения
алюминия и переход его поверхности в пассивное состояние [124].
Показано также, что в отдельных безводных растворах, на-
пример серной кислоты в ДМФ [655], кислород не в состоянии
образовывать оксидный слой на чистом алюминии. Анодное пове-
дение алюминия в таком растворе зависит от приложенного на-
пряжения, и при определенных условиях наблюдается эффект
анодного полирования алюминия.
Результаты исследования различных электролитов в жидком
аммиаке показали, что в отсутствие окисляющих частиц анодный
процесс на электроде представляет единственную реакцию А1 ->
АР+ + Зе. Если же в растворе присутствуют анионы, способные
восстанавливаться, например NO3, средняя степень окисления
алюминия ниже 3+, т. е. в растворе имеется смесь ионов А1+ и
А13+ [1050, 1051, 673].
Исследования по анодному поведению и коррозионной стойко-
сти алюминия в протогенных растворителях тесно взаимосвязаны.
Изучены растворы электролитов в органических кислотах и спир-
тах. В обоих типах растворов косвенно обнаружено стадийное
протекание растворения алюминия через образование А1+-ионов
[273, 479, 480], процесс осложнен выделением водорода [115,
775]. Потенциал анодно поляризованного алюминия в ряду без-
водных алифатических спиртов с добавками хлорид-ионов после
вычета омической составляющей линейно коррелируется с обрат-
ной диэлектрической проницаемостью растворителей.
Процесс коррозии во всех изученных протолитических рас-
творителях также электрохимический. В водно-спиртовых хлорид-
ных растворах величина скорости коррозии при постоянных кон-
центрациях воды и С1~-иона возрастает от метанола к бутанолу
[243]. Вода оказывает ускоряющее влияние на коррозию алюми-
ния в спиртах. Зависимость i от Сн2о описывается уравнением
прямой
i = а + &Сн2о- (2—27)
Последнюю закономерность, по-видимому, можно объяснить пре-
имущественным адсорбционным вытеснением молекул спирта на
поверхности электрода и сдвигом равновесия в сторону увеличе-
ния поверхностной концентрации С1_-ионов. В целом в таких рас-
творах пассивирование и активное растворение алюминия полно-
стью определяются конкурирующей адсорбцией молекул воды,
спиртов и хлор-ионов.
Коррозионная активность в ряду алифатических карбоновых
кислот муравьиная — стеариновая снижается с увеличением моле-
кулярной массы кислоты. Для первых трех водорастворимых кис-
лот ряда — муравьиной, уксусной, пропионовой — изучена также
коррозия в водных растворах. В них алюминий корродирует ин-
тенсивно, в концентрированных кислотах его стойкость повыша-
ется [206].
8 - 4-653
113
Анодное окисление галлия, индия и таллия во всех изученных
немногочисленных растворителях протекает стадийно [1127, 45,
713, 149, 1050, 1051, 775]. Во всех растворах обнаружено наличие
низшей степени окисления этих металлов Ме+ и высшей Ме3+.
В отдельных случаях установлен трехстадийный процесс, напри-
мер для индия в ацетамиде [149]. Скорость процесса растворения
контролируется отщеплением первого или последнего электрона.
Пассивирование электродов данных металлов обусловлено адсорб-
ционными явлениями на их поверхности. На специальных элект-
родах на основе индия получены анодные пленки в электролитах
сложного состава [469].
Оксидные пленки на иттрии и неодиме получены анодным окис-
лением в этиленгликолевых растворах [927, 867].
2.4.4. Металлы четвертой группы
Анодное поведение кремния прежде всего определяется природой
неводного раствора и типом проводимости кремния [373, 825, 13,
171, 72]. Результаты исследований в растворах R4NC1—НС1 — ме-
тилхлорсилана (R-алкильный радикал) и в растворах хлоридов
щелочных металлов в N-метилформамиде и диметилформамиде
позволяют предположить, что кремний переходит в раствор в че-
тырехвалентном состоянии и при растворении образуются Si—
Cl-связи [13].
В абсолютном этаноле в присутствии анионов Cl-, F-, SO42-
анодное растворение кремния осуществляется по радикальному
механизму через промежуточное образование радикалов соответ-
ствующих анионов [72]. Например, растворение кремния в форме
ферросилиция (ФС) можно объяснить химическим взаимодействи-
ем ФС с атомарным хлором, электрохимически генерируемым на
аноде, что приводит к образованию SiCl4, и далее процессом
SiCl4 + 4C2H5OH-^-Si(OC2H5)4 + 4HCl. На процессы пассивации
значительное влияние оказывает природа анионов электролита.
Большое внимание уделено получению анодированием диэлектри-
ческих пленок SiO2, которые образуются в присутствии кислород-
содержащих анионов.
Данные об анодном растворении германия немногочисленны
[373]. В анодном растворении олова в спиртовых растворах при-
нимают непосредственное участие галоидные анионы и молекулы
растворителя. Данных для установления механизма растворения
недостаточно.
Работы по анодному поведению свинца посвящены в основном
окислению Pb (II) до Pb (IV) [1186, 804, 575, 576, 2, 3]. Исполь-
зуются растворы в ледяной уксусной и пропионовой кислотах. На
инертных анодах и анодах из РЬО2 наблюдается близкое к 100 %-
ному окисление РЬА2 до РЬА4. Торможение реакции наступает в
результате экранирования поверхности электрода солями Pb (IV).
Во избежание этого необходимо подбирать состав электролита,
обеспечивающий растворение этой пленки. В присутствии следов
114
воды происходит гидролиз тетраацетата до РЬО2. Данный процесс
нежелателен, поскольку преимущество РЬА4 как сильного окисли-
теля перед РЬО2 заключается в его хорошей растворимости во
многих растворителях [575]. При использовании свинцового анода
окисление РЬ2+ до РЬ4+ не наблюдается, так как, по-видимому,
происходит растворение свинца [2, 3].
Относительно анодного поведения и коррозионной устойчиво-
сти в неводных средах титан является наиболее изученным ме-
таллом четвертой группы [600, 206, 495, 86, 601, 602, 1059, 1079,
1164]. Исследовались в основном спиртовые растворы и растворы
на основе органических кислот, в первую очередь уксусной, где
титан применяется как конструкционный материал.
В безводных уксуснокислых растворах титан не пассивируется.
Анодные поляризационные кривые в широком интервале потенци-
алов подчиняются уравнению Тафеля с наклоном, близким к
2,3 RT/F. Выход по току при анодном растворении титана близок
к 100 % при расчете на Ti (III) в пределах участка поляризаци-
онной кривой (г<10-3 А/см2) или Ti (IV) при более высоких по-
тенциалах и плотностях тока. При этом анализ полученных дан-
ных позволяет заключить, что в уксуснокислой среде при i^lX
X Ю~3 А/см2 титан растворяется в виде ионов [TiAcn]4-n и
[TiOAcn]2~n [598, 599, 603, 1041]. Анодное поведение сплавов на
основе титана, но содержащих молибден, существенно отличает-
ся от поведения титана. В безводных ускуснокислых растворах
такие сплавы, как и молибден, имеют область пассивного состоя-
ния с малой скоростью растворения [601, 602].
Работ, посвященных выяснению механизма растворения тита-
на в спиртовых растворах, недостаточно, например [1059]. Содер-
жание работ по поведению титана в спиртовых растворах сводит-
ся в основном к выяснению его коррозионной устойчивости и за-
висимости ее от состава электролита.
В безводных спиртовых растворах, как и в ацетатных, титан
не пассивируется. Для его пассивации в обоих типах растворов
необходимо присутствие воды или других соединений, содержащих
кислород с достаточно отрицательным эффективным зарядом. На-
пример, ацетон замедляет скорость коррозии титана в метаноло-
вых растворах хлороводорода, хотя и менее эффективно, чем вода
[86]. Механизм коррозии во всех исследованных растворах элект-
рохимический. Для HCl-спиртовых растворов наблюдается зако-
номерное уменьшение скорости коррозии с увеличением молеку-
лярной массы спирта [1079]. Ионы и молекулы галоидов служат
активаторами коррозии титана, непосредственно участвуя в про-
цессе. Так, при коррозии титана в растворах брома в метаноле
катодным процессом является ионизация брома, анодным — рас-
творение титана [495]. Вода необходима для пассивации титана
только в анодном процессе, способность титана к катодной пас-
сивации не зависит от наличия воды [495, 603, 86]. Титан — ме-
талл с механической пассивностью, в водных растворах он само-
пассивируется. Это его свойство сохраняется и в водно-спиртовых
8*
115
растворителях [227]. Наряду с общей коррозией для титана в
спиртовых и кислотных растворах характерны межкристаллитная
коррозия и процессы питтингообразования [495, 594, 1084, 341, 603,
1041]. Большинство сплавов на основе титана ведут себя анало-
гично титану. Процесс анодного растворения титана в эфирно-
гидридном электролите определяется концентрацией титановых
соединений в растворе [302].
В безводных апротонных растворителях титан также не пас-
сивируется. Например, в растворах НС1 в ДМФ титановый анод
при концентрациях воды менее 0,3 % находится в активном со-
стоянии, при концентрации более 0,6 % — в пассивном. В интер-
вале 0,6 °/о>Сн2о>0,3 % на титане наблюдается питтинги. В от-
сутствие тока с ростом содержания воды размеры питтингов и их
число уменьшаются [1164].
В связи с подбором конструкционных материалов для крем-
нийорганических производств исследовано электрохимическое и
коррозионное поведение циркония в безводных и водных раство-
рах спиртов [200, 367, 1079, 652, 25]. В безводных спиртах в при-
сутствии НС1 цирконий растворяется легче, чем в воде. Предпо-
лагается, что защитные свойства возникающих в данных услови-
ях хлоридных пассивирующих слоев хуже защитных свойств окис-
ных слоев, образующихся в водных растворах. Коррозия проте-
кает по электрохимическому механизму с водородной деполяриза-
цией, на поверхности циркония образуется черный порошок гидри-
да, в раствор переходят соединения типа Cl4_nZr(OR)n (в случае
спиртовых растворителей).
Введение в раствор воды резко замедляет коррозию циркония.
При концентрации воды выше 10 % цирконий не корродирует. Сле-
дует отметить, что в некоторых спиртах при содержании воды по-
рядка 10 % пассивное состояние циркония не нарушается даже
при анодной поляризации. Параметры анодного растворения цир-
кония в области химической поляризации находятся в линейной
зависимости от обратной величины диэлектрической проницаемо-
сти растворителя [25].
Влияние состава водно-спиртовых смесей на поведение цирко-
ния может быть объяснено изменением пассивирующей способно-
сти растворителя и активирующей ионов хлора при изменении со-
отношения компонентов раствора. В метаноловых растворах НС1
при поляризации в режиме предельного тока поверхность цирко-
ния электрополируется. Ток электрополировки уменьшается с уве-
личением концентрации НС1 [652]. В водно-органических смесях
на основе ДМФ и ДМСО в определенных областях их концентра-
ций также наблюдается торможение анодного растворения цирко-
ния и происходит смена активирующего действия ионов С1~ на
тормозящее [107].
116
2.4.5. Металлы пятой группы
Сведения об анодном поведении металлов пятой группы в орга-
, нических растворителях немногочисленны [149, 224, 734].
Анодное растворение сурьмы исследовано в растворах фторида
сурьмы (III) в ацетамиде. Процесс двухстадийный: Sb—e+*Sb+,
Sb+—2e=p±=Sb3+. Замедленной является вторая стадия отщепления
двух электронов от однозарядного иона сурьмы. Соединения
Sb (V) при электролизе не образуются [149].
Изучено анодное поведение ванадия в безводных АН и уксус-
ной кислоте в присутствии различных электролитов (LiCl, NaC104,
NaCl, CH3COONa, HC1, HC1O4, H3PO4). Установлено в зависимо-
сти от природы электролита и условий эксперимента существова-
ние одной или двух областей активного растворения ванадия [224].
Анодирование тантала в безводной муравьиной кислоте и дру-
гих органических растворах приводит к образованию двухслойной
пленки. Слой, прилегающий к металлу, по своим характеристикам
соответствует оксиду Ta2Og. Он растет по мере поляризации одно-
временно с внешним слоем, более растворимым, насыщенным мо-
бильными кислородными ионами [734]. Увеличение содержания
воды в электролите способствует возрастанию размеров внутренне-
го слоя пленки, т. е. пассированию тантала, и уменьшает скорость
растворения внешнего слоя.
2.4.6. Металлы шестой группы
Исследования по анодному окислению металлов шестой группы
относятся прежде всего к подгруппе хрома [600—602, 595—597,
364, 340, 217, 976, 983]. В целом для этих металлов при анодном
( растворении в неводных средах, по данным поляризационных из-
; мерений, характерно чередование активных областей растворения
J с областями пассивации и перепассивации.
Проведена сравнительная характеристика анодного растворе-
ния хрома, молибдена и вольфрама в метаноловых растворах
LiX(X = Cl, NO3-, С1О4-, ОСНз-) [976]. В зависимости от приро-
J ды фонового электролита и присутствия воды продуктами раство-
рения являются соединения этих металлов в степенях окисления
3+ или 6+. Особенно четко данная закономерность проявляется
в случае хрома. В безводных метаноловых растворах хлороводо-
I рода происходит активное растворение хрома с прямым участием
хлорид-ионов. Пассивация хрома наступает при введении в рас-
’ твор молекул воды [217]. Пассивируется хром добавками воды
и в других растворителях, причем добавки очень малые. Так, для
пассивации хрома в ледяной уксусной кислоте достаточно 0,01 %
воды [600]. В щелочном растворе этанола анодное растворение
молибдена осуществляется в результате последовательных квази-
; обратимых электрохимических реакций
Мо° =1£> Мо+С ~ > Мо2+с Мо3а+
117
1
с последующей десорбцией Мо3+ с поверхности металла в объем
раствора [983]. Кроме активной области растворения на поляри-
зационных кривых наблюдается область пассивации молибдена.
Перепассивация осуществляется по механизму точечной коррозии.
Молибден в безводных уксуснокислых растворах пассивирует-
ся, его растворение происходит лишь после достижения обратимо-
го потенциала образования оксида МоО3 и протекает с образова-
нием ионов высокой степени окисления (в ацетатных и хлоридных
растворах средняя степень окисления составляет 5,8+, в растворе
NaC104 — 4,85+). Специфическое влияние анионов на кинетику
анодного растворения и пассивацию отсутствует. Кислородные со-
единения молибдена образуются за счет кислорода молекул уксус-
ной кислоты. По полярографическим данным растворение протека-
ет в основном по реакциям
(1) Mo + 2п СН3СООН МоОп + п (СН3СО)2 О + 2п Н+ + 2пе,
(2) МоОл + 2т СН3СООН МоО„+т + т (СН3СО)2 О + 2тН+ + 2те.
Добавки воды облегчают растворение, уменьшая потенциал пере-
пассировации [494] и наклон анодной поляризационной кривой.
По-видимому, изменяется механизм процесса анодного растворе-
ния молибдена, вместо реакций (1) и (2) протекают термодинами-
ческие, более вероятные в присутствии воды реакции
Мо + п Н2О МоОп + 2п Н+ + 2пе,
МоОп + тН2О МоО„+т + 2тН+ + 2те.
Не зависящая от анионного состава кинетика анодного растворе-
ния описывается уравнением
ТИС1 = (0,96 ± 0.02) + F ‘Е ‘ (2 — 28)
Предполагается, что пассивное состояние поверхности молибдена
обеспечивается возможным образованием оксидов низших степе-
ней окисления
Mo + 2СН3СООН Z МоО + (СН3СО)2 О + 2Н+ + 2е.
Перепассивация молибдена начинается при потенциале, близком
к равновесному потенциалу системы Мо2О5/МоО3 [600—602].
Легирование сплавов молибденом обеспечивает их пассивацию
и, следовательно, высокую коррозионную стойкость в безводных
уксуснокислых растворах [595—597, 601, 602].
В безводном ДМФА молибден также имеет область пассивного
состояния и перепассивации. Здесь, как и в уксуснокислых рас-
творах, отсутствует специфическое влияние анионного состава на
кинетику анодного растворения и потенциал перепассивации. При
потенциалах, больших потенциала перепассивации, растворение
происходит с образованием молибдат-иона. Добавление воды к
диметилформамидным и спиртовым растворам уменьшает область
устойчивого пассивного состояния молибдена [595—597].
118
В исследованных неводных средах анодное поведение молибде-
на и вольфрама менее сходно, чем в водных [595—597]. В без-
водных уксуснокислых растворах две области пассивного состоя-
ния вольфрама наблюдаются только в случае присутствия в рас;
творе перхлората. В ацетатных и хлоридных растворах, содержа-
щих уксусный ангидрид, пассивность вольфрама сохраняется лишь
в первой области при малых плотностях тока. Далее следует рав-
номерное активное растворение до высоких плотностей тока со
степенью окисления 6+ с подчинением процесса тафелевской за-
висимости с угловым наклоном в «2,3 RT/F. Добавка 0,5 % воды
приводит к возникновению второй пассивной области, вероятно, в
результате взаимодействия растворимых кислородных соединений
вольфрама с образованием фазового окисла WO3:
WO2 + Н2О = WO3 + 2Н+ + 2е,
W2O5 + 2Н2О = WO3 + WO?" + 4Н+ + 2е.
Исследована кинетика образования анодных оксидных пленок
вольфрама в растворах неорганических и органических кислот и
солей в спиртах и ДМФ [364].
1.4.7. Металлы седьмой группы
Анодное поведение металлического марганца и его ионов иссле-
довано в апротонных и протолитических растворителях [6, 879,
-625, 216]. В ДМСО анодный процесс на марганце протекает ста-
дийно с участием молекул растворителя [6, 216]. Кинетическое
уравнение анодной реакции в кислых перхлоратных растворах в
ДМСО имеет вид
ia = Ка (Сц+Г1 ехР (°«5F<p/Z?T), (2-29)
Из уравнения видно, что скорость анодного растворения мар-
ганца в таких растворах при постоянном потенциале закономерно
снижается с ростом содержания кислоты. Наиболее вероятно, что
ингибирующее действие кислоты обусловлено протеканием хими-
ческой реакции, продуктом которой являются ионы водорода и
которая предшествует лимитирующей реакции ионизации марган-
ца. Высказано предположение [216], что такой химической реак-
цией является квазиравновесная адсорбция молекул растворите-
ля на поверхности марганца, ведущая к сдвигу вправо процесса
автопротолиза ДМСО:
Мп 4- 2 (СН3)2 SO Мп (CH3SOCH2)^c + (CH3SOCH3) Н+.
Ионизация металла протекает затем постадийно с участием хемо-
сорбированного комплекса:
Мп(СН35ОСН2)здС Мп (СН35ОСН2)адс + е,
Мп (СНзБОСН^адс Z Мп (CH3SOCH2)+ + е.
119
1
Хемосорбированные ионы (CH3SOCH2)_ облегчают процесс иони-
зации, координируясь с атомами марганца через кислород.
Характер влияния воды на скорость растворения марганца в
ДМСО зависит от потенциала. При небольших поляризациях с
увеличением содержания воды облегчается растворение марганца.
При высоких анодных потенциалах в богатых водой смесях повы-
шение концентрации воды приводит к уменьшению скорости рас-
творения, а затем появлению области пассивации. Влияние воды
на процесс ионизации марганца облегчается конкурирующей ад-
сорбцией молекул воды и ДМСО в присутствии ионов перхлора-
та [216].
Скорость коррозии марганца в разбавленных кислых перхло-
ратных растворах ДМСО лимитирует катодный процесс — диффу-
зию сольватированного протона [216].
Цикловольтометрически в ряде растворителей — дихлормета-
не, НМ, АН, ПК. т-пиролидине, ДМФ и ДМСО — установлено
существование трехзарядного марганца в виде триацетилацетона-
та и окисление его анодно до иона Мп(асас)3+ [879].
В растворах жидкого аммиака Мп2+ может быть также анодно
окислен до Мп (III), который в данной среде нестабилен и раз-
лагается, взаимодействуя с растворителем. Определены электро-
химические параметры для окислительно-восстановительной пары
Мп2+/Мп3+ [713].
2.4.8. Металлы восьмой группы
Анодное поведение и коррозионная устойчивость металлов группы
железа изучены достаточно детально [653, 1260, 600, 1240, 909,
910, 51, 713, 634, 633, 911, 76, 206, 601, 602, 340, 1199, 88, 1135,
505, 4, 779], причем большинство работ относится к железу (ста-
ли), которое широко применяется в качестве конструкционного ма-
териала для неводных сред [89, 396, 230, 632].
Анодное и коррозионное поведение железа исследовано в ос-
новном в спиртовых растворах, растворах органических кислот и
в апротонных растворителях. В большинстве случаев наряду с
безводными исследованы и водно-органические растворы. При
анодной поляризации железо переходит в раствор в двухвалент-
ном состоянии, в процессе ионизации принимают участие молеку-
лы растворителя и присутствующие в растворе анионы. Возмож-
ность прямого участия анионов в анодном растворении металлов
в органических средах намного больше, чем в водных растворах,
в случае железа эта возможность особенно проявляется. Для вод-
но-органических смесей отмечается участие в отдельных стадиях
растворения молекул воды и органического растворителя [653].
В механизме растворения большую роль играют адсорбционные
явления, примером его может служить последовательность стадий
при окислении железа в апротонных перхлоратных растворах [76]:
Fe + S Fe (S)n (S — растворитель)
Fe (S)n + С1ОГ Fe (С1О4)аДс + e + nS,
120
\ Fe (С104)адс 4- CIO4 Fe (С1О4)2адс 4*
к Fe (С1О4)2адс Fe (С1О4)2 + Ре2+сольв + 2С10Г.
Почти во всех исследованных растворителях на анодных поля-
ризационных кривых наблюдаются области активного растворения
и пассивации железа. Продолжительность и соотношение этих об-
ластей в шкале потенциалов определяются природой электролита,
в первую очередь его анионным составом [349, 977, 604, 605]. На
формирование его пассивной области большое влияние оказывает
присутствие воды. Природа пассивирующих пленок разнообразна
от адсорбционных молекул растворителя до фазовых оксидных и
солевых пленок [1199, 1227, 783]. Наряду с электрохимическим
механизмом при коррозии железа наблюдается и чисто химический
[632—635]. Уделено внимание теории подбора и практического
использования ингибиторов коррозии в неводных средах [632—
635, 125, 126, 230].
Анодное растворение кобальта в ДМФ, АН и ЭГ характеризу-
ется образованием на поверхности электрода фазовой оксидной
пленки и может быть использовано для электрохимического поли-
рования кобальта [499, 225, 368].
В уксусной кислоте и в растворах безводного аммиака, под-
кисленных введением NH4NO3, изучено электрохимическое поведе-
ние пар Со2/Со3. Установлены возможность окисления Со2+ до
Со3+ на инертных анодах и стабилизация последнего в этих рас-
творах [715, 225]. Ступенчатому окислению с образованием про-
межуточных метастабильных катион-радикалов в среде СН2С12 под-
вержены сложные комплексы кобальта с органическими лиганда-
ми [806].
В большом числе водно-органических и неводных растворов
изучено анодное поведение никеля (спирты, АЦ, АН, ФМ, ДМФ,
ДМСО, ПК, ТГФ, НАс) [600, 51, 125, 126, 4, 779, 106, 1129]. Во
всех изученных растворах при низких плотностях тока (почти во
всех случаях применялись кислые растворы) наблюдалось актив-
ное растворение никеля со 100 %-ным выходом при расчете на
Ni2+. Процесс необратимый, его протекание связано с участием
анионов, молекул растворителя и осложнено адсорбционными яв-
лениями [1200, 779]. При высоких плотностях тока (потенциалах)
в присутствии кислородсодержащих анионов (например, С1О4_) и
воды наступает пассивация электрода. В ДМСО скорость анодного
растворения никеля на несколько порядков ниже, чем в других
растворителях, в том числе и воде. Торможение анодной реакции,
вероятно, обусловлено хемосорбцией ДМСО [4, 1, 779]. Сделана
попытка корреляции анодного поведения никеля с физико-хими-
ческими свойствами протолитических и апротонных растворителей
[125, 126, 636]. В водно-органических смесях состав смешанного
растворителя влияет на поведение никелевого анода в определен-
ной области концентраций воды [636].
Установлено, что в солянокислых растворах на основе апро-
тонных растворителей (ДМФ, ДМСО, АЦ, ТГФ) в определенной
121
1
области концентраций органического растворителя происходит
смена активирующего действия хлорид-ионов на анодное растворе-
ние никеля на тормозящее [106]. Величина критической концент-
рации растворителя определяется его природой.
Изучено также анодное окисление ионов Ni^ на стационарных
инертных электродах в среде ДМФ, ДМСО, АН и АЦ, получены
соединения Ni®+ [505]. В подкисленных NH4NO3 растворах жидко-
го аммиака возможно образование соединений Ni (IV), которые
крайне неустойчивы и быстро разлагаются, окисляя растворитель.
На основании величины порядка реакции и тафелевского наклона
в последнем случае можно предположить, что никель окисляется
согласно схеме [713]:
Ni11 -Z.e>Nini^Nilv
окисление растворителя
Сведения о коррозионном поведении сталей на основе железа
и никеля немногочисленны, несмотря на важность таких исследо-
ваний как в практическом, так и в теоретическом плане [206, 598,
599, 601, 602, 595, 596, 597, 89, 105, 396, 230]. Работы посвящены
выбору конструкционных материалов для аппаратов электрохи-
мических производств в неводных средах. Поэтому большинство
исследований касается установления наличия пассивных участков
на поляризационных кривых и влияния на них различных факто-
ров (состав электролита, температура и т. д.). Обнаружены от-,
дельные закономерности. Так, в индивидуальных алифатических
кислотах коррозионная стойкость ряда сталей повышается с уве-
личением молекулярной массы кислоты [206].
Некоторые стали, как и индивидуальные металлы, приобрета-
ют способность пассивироваться при достижении определенной
критической концентрации воды [598, 599], при этом условии мо-
жет быть получена достаточная для пассивации поверхности сте-
пень покрытия пассивирующими кислородсодержащими частица-
ми. Если же органический растворитель в присутствии воды под-
вергается гидролизу с образованием кислоты, то такие смеси
агрессивны не только к углеродистым, но и к нержавеющим ста-
лям, особенно при повышенных температурах [593, 396]. Молиб-
ден увеличивает коррозионную стойкость сталей и сплавов [596,
597, 595, 601, 602].
Методами классической полярографии и циклической вольт-
амперометрии изучено анодное окисление органических производ-
ных двухвалентных рутения и осмия [1046, 1157—1159, 1247]. Во
всех случаях обнаружены одноэлектронные ступени окисления.
Платина в наибольшей мере отвечает требованиям, предъявля-
емым к анодному материалу в процессах электрохимического
окисления. В такой роли в преобладающем большинстве неводных
систем она ведет себя инертно [1046, 375, 373, 280, 463, 1200, 758].
Однако для платины характерно и анодное растворение в органи-
ческих растворителях, чаще всего при электроорганических синте-
зах [211, 213, 503]. Решающее влияние на процесс растворения
122
I
г
платины оказывает присутствие воды [211, 213]. В отдельных слу-
чаях исследована интенсивность растворения и других металлов
платиновой группы в зависимости от различных факторов [211].
2.5. АНОДНОЕ ОКИСЛЕНИЕ АНИОНОВ В НЕВОДНЫХ
РАСТВОРИТЕЛЯХ
Существенные различия в электрохимическом поведении анионов
в водных и неводных растворах в первую очередь касаются дипо-
лярных апротонных растворителей, поскольку анионы в них на-
много меньше сольватированы по сравнению с протолитическими
растворителями. Различия особенно значительны для малых лег-
кополяризуемых анионов. Имеющиеся в литературе сведения це-
лесообразно рассматривать отдельно для двух групп анионов: га-
логенидов (псевдогалогенидов) и кислородсодержащих анионов.
2.5.1. Галогениды и псевдогалогениды
Процессы анодного окисления галогенид-ионов в органических
растворителях более изучены, чем кислородсодержащих ионов.
Большинство работ относится к изучению анодного поведения йо-
дида [320, 722, 861, 745, 743, 989, 1260, 862, 863, 1024, 794, 1255,
1040, 1212, 1031, 33, 1095, 803].
Анодный разряд галогенидных ионов дает различные продукты
в зависимости от материала электрода, природы растворителя и
температуры. Так, на платиновом электроде во многих случаях
относительно легко достигается установление обратимых элект-
родов второго рода, поскольку растворение металла в пределах
потенциалов, соответствующих разряду галогенидных ионов, не-
значительно. В преобладающем большинстве случаев окисление
на инертном электроде происходит согласно схеме [861, 745, 743,
989, 1206, 862, 863, 1024, 33, 1132]: ЗХ-^Х<г+2е; 2Х3-^ЗХ2+2е.
Механизм реакции не зависит от природы растворителя, влияние
/ последнего сказывается лишь на величине кинетических парамет-
$ ров, главным образом токов обмена системы. Как правило, ско-
.£ рость определяющей ступенью является образование тригалогени-
v да. Для йодидов эта схема, пожалуй, единственна.
Для хлоридов и бромидов предлагается и другая схема окис-
I ления [1040, 1212, 1031]: 2Х~—2еч^Х2. .На ртутном капельном
электроде анионы X- на поляризационной кривой дают две анод-
ные волны, соответствующие последовательному образованию
HgX3~ и HgX2 [745, 743].
Молекулярный йод на платиновом электроде в органических
растворителях окисляется до катиона I+, который часто взаимо-
действует с растворителем [794, 1255, 1095]. Процесс протекает
* через промежуточное адсорбирование йода на электродной no-
у. верхности: 12^21адс,
7 21 аде — 2е^±21+.
123
Вольт-амперометрическим методом установлено, что при одновре-
менном присутствии в растворе йода и йодид-ионов в кислой среде
стабильны четыре частицы — 1~, 13_, 12 и I+, для которых рассчи-
таны константы устойчивости [1095].
Псевдогалогенидные анионы ведут себя подобно галогенидным,
в частности тиоцианатный анион [989, 1080, 1081, 1124, 1038, 731].
Электрохимическое окисление иона SCN- в различных апротонных
растворителях на платине осуществляется в области потенциалов»
где электрорастворение металла практически ничтожно. Анодный
разряд иона SCN~ в данных условиях при низких температурах,
как и в случае галогенидов, сводится к образованию окислитель-
но-восстановительной пары SCN~/(SCN)2Pt. Изучена электродная
кинетика системы SCN~/(SCN)2 в широком температурном ин-
тервале (—12-?- + 160оС) [1124, 1038]. Предлагается два меха-
низма образования родана:
(1) — SCN" + Pt Pt (SCN) 4- е,
2Pt (SCN) (SCN)2 + 2Pt;
(2) — SCN" + Pt ^Pt(SCN) + e,
Pt (SCN) + SCN" (SCN)2 + Pt + e.
Платина представлена в схемах как активная сторона электрод-
ной поверхности, a (SCN) — как адсорбированный радикал, обра-
зующийся во время начального переноса электрона. В первой схе-
ме медленная первая стадия, во второй — вторая. При высокой
концентрации иона SCN~, а также при повышенных температурах
(выше 60 °C) возможны реакции полимеризации окисленного про-
дукта (родана и радикала):
х (SCN)2^4(SCN)2x,
x(SCN)^(SCN)x.
В ацетонитрильных растворах полимеризация может протекать по
схеме (5СП)2-[-АН-^-полимер. Полимер образует пленку на элек-
троде, вызывая его пассивацию.
В ограниченной области потенциалов электродный процесс
3SCN~= (SCN)3-+2e протекает на золотом электроде. Для про-
цесса характерен смешанный диффузионно-кинетический контроль.
При повышении потенциала наступает растворение основного ме-
талла (золота) [1081, 1080].
Изучено также анодное окисление ионов смешанных галогенов
типа 1Вг2_, 1С12~, 1ВгС1~ [320]. Происходящие при этом электрод-
ные реакции определены полярографически, расшифрованы соот-
ветствующие анодные волны.
124
2.5.2. Кислородсодержащие анионы
Анодное окисление перхлорат-иона изучалось прежде всего сточ-
ки зрения его электрохимической устойчивости, поскольку соли
хлорной кислоты широко используются в качестве фоновых элект-
ролитов, причем ни в одном из обычных органических растворите-
лей С1О4- не восстанавливается [320]. Однако в некоторых апро-
тонных растворителях, в том числе и в АН, перхлорат-ион при
высоких анодных потенциалах способен окисляться [320, 1193].
Так, при электролизе растворов перхлората серебра в АН на пла-
тиновом аноде количественно образуется НС1О4. На катоде на-
ряду с осаждением серебра происходит выделение водорода. Для
образования хлорной кислоты предполагается радикальный меха-
низм по схеме [1193]: 2С1О4“—2е—>-С12О8; Cl2O3 + 2CH3CN->-
—>2HC1O4-|-2CH2CN. Электрохимическому окислению в диметил-
формамидных растворах подвержен также анион С1О2~, который
в конечном итоге превращается в хлорноватую кислоту и диоксид
хлора [1212].
Анодное окисление нитрат-иона в апротонных растворителях
(АН, ПК, НМ), согласно литературным данным [320, 1147, 752],
протекает по двум схемам:
I — NO3 —* NOj 4~ е,
2NO3-N2O6, N2O6-^N2O5 + l/2O2;
II - NO? -> NO2+ + 2е + 1 /2О2,
NOt + МОГ -*N2O5.
Основным продуктом по обеим схемам является пентаоксид азо-
та. Образующийся по схеме I ион-радикал NO3‘ очень активен,
относительно стабильный он только в присутствии перхлорат-
иона, в других случаях вступает в реакцию с присутствующими в
растворе катионами и анионами. Так, Ag(I) окисляется до Ag(II)
[1147]. Если в растворе отсутствуют реактивные катионы и анио-
ны, NO3‘ разлагает растворитель.
Для протолитических растворителей, в частности уксусной кис-
лоты, предложена третья схема окисления МО3--иона [752]:
III — КОГ + ПОз +е,
NO;-*NOt + 1/2О2 + е,
NO^ + NO3~ -*N2O5.
Скоростьопределяющей является первая ступень. Как и для двух
предыдущих схем, основным продуктом окисления нитрат-иона
является пентаоксид азота.
Изучена кинетика электрохимического окисления на платино-
вом электроде МО2_-иона в апротонных растворителях [320, 1264].
125
В процессе окисления, протекающем ступенчато, принимают уча-
стие адсорбированные промежуточные радикалы. В ДМСО-рас-
творах процесс одноэлектронный, протекающий с образованием
диоксида азота [1264]:
NO? + е = NO2 (2NO2 = N2O4).
Димеризованный диоксид азота образует с ДМСО продукты при-
соединения и окисления, способствующие дальнейшему образова-
нию NO2:
(CH3)2SO + N2O4 = (CH3)2SO-N2O4,
(CH3)2SO-N2O4 = (CH3)2SO2 + N2O3 (N2O3 = no + NO2).
В апротонных растворителях и их смесях с водой исследова-
но анодное поведение гидроксид-иона [989, 948]. Полярографиче-
ская анодная волна ОН_-иона в ацетонитрильных, диметилсуль-
фоксидных растворах проявляется при потенциалах, более отри-
цательных, чем в воде (например, в АН отрицательнее на 0,9 В),
что объясняется увеличением активности гидроксид-иона в этих
растворителях. Из высоты анодной волны рассчитан коэффициент
диффузии ОН_-иона в безводных растворах.
При изучении анодной поляризации в неводных растворах по-
лиэлектролитов установлено протекание разряда анионов и мак-
роанионов типа RCOO- [32, 210].
Глава 3
ХАРАКТЕРИСТИКИ ЭЛЕКТРОЛИТНЫХ
КОМПОЗИЦИЙ
ДЛЯ ЭЛЕКТРООСАЖДЕНИЯ МЕТАЛЛОВ
ИЗ НЕВОДНЫХ РАСТВОРОВ
3.1. ХАРАКТЕРИСТИКИ РАСТВОРИТЕЛЕЙ, ПРИМЕНЯЕМЫХ
В ПРАКТИКЕ ЭЛЕКТРООСАЖДЕНИЯ МЕТАЛЛОВ
3.1.1. Классификация растворителей
Очевидно, что одной рациональной системы классификации раст-
ворителей быть не может. В зависимости от количества и общнос-
ти признаков, лежащих в основе данной системы классификации,
может существовать громадное число таких систем. Поэтому мож-
но говорить лишь об общих системах классификации растворите-
лей, наиболее пригодных для практики.
а. Классификация растворителей, основанная
на физических свойствах*. Как следует из материала
предыдущих глав, одним из основных свойств растворителя, опре-
деляющим физико-химическую характеристику электролитной си-
стемы, является диэлектрическая проницаемость е. Терминология,
лежащая в основе градаций растворителей по величинам е, не во
всем определенна и последовательна. Чаще всего растворители с
высокими е называют «полярными» («высокополярными»), а раст-
ворители с низкими е—«неполярными» («низкополярными»), хотя
эта терминология, строго говоря, относится к дипольному моменту,
т. е. должна характеризовать не макро-, а микросвойство раство-
рителя. Для характеристики растворителей по дипольным момен-
там их молекул введены термины «дипольные» (ц=#0) и «апо-
лярные» (ц = 0).
К низкополярным относятся растворители, величина диэлектри-
ческой проницаемости которых находится в интервале 1,9—12, к
среднеполярным — растворители сев интервале 12—50, к высоко-
полярным — растворители с е>50 [547, с. 33; 435].
Несмотря на существующую связь между дипольным моментом
и диэлектрической проницаемостью индивидуальной жидкости
[547, с. 55], следует предостеречь от проведения параллелизма
между величиной 8 растворителя и дипольным моментом ц его мо-
лекул (такой параллелизм существует лишь в разбавленных рас-
творах), поскольку диполярные молекулы растворителей всегда
образуют ассоциаты большей или меньшей степени сложности, что
ведет к уменьшению е жидкости. Так, мономерные молекулы ряда
* Сведения о важнейших физических константах наиболее распространен-
ных в практике растворителей приведены в табл. 1 приложения.
127
растворителей в весьма широком интервале диэлектрических про-
ницаемостей от 24,3 (этанол) до 7,35 (ТГФ) характеризуются зна-
чением p.= l,7D [471].
Существенное влияние на полярность растворителя оказывают
его структурные особенности. Например, молекулы воды — весьма
высокополярного структурированного растворителя — имеют р,=
= 2-10~3 Па-с), средневязкие (т]=(2—10) • 10_3 Па-с) и высоко-
вязкие (r)> 10~2 Па-с).
Поскольку в общем случае электропроводность раствора тем
выше, чем выше е растворителя (см. параграф 1, 3, 4), при под-
боре композиций для электроосаждения металлов из растворов при
прочих равных условиях следует стремиться к растворителю с мак-
симально возможной е.
Вязкость г) растворителей изменяется в весьма широких пре-
делах от долей до тысяч паскаль-секунд. В соответствии с этим
по вязкости растворителя подразделяют на низковязкие (г) =
= 2-10—3 Па-с), средневязкие (т)= (2—10)-10~3 Па-с) и высоко-
вязкие ('Г)>10-г Па-с).
При подборе композиций, исходя из описанной выше (см. па-
раграфы и 1. 3. 4) связи между электропроводностью и вязкостью,
следует стремиться к растворителю с минимально возможной вяз-
костью.
Между термодинамическими параметрами активации вязкого
течения и структурными характеристиками жидкости существует
достаточно четкая связь [617, 616, 545]. Поэтому эти величины
позволяют судить о степени структурирования жидкости.
С очевидной ориентацией на температуру кипения воды раст-
ворители подразделяют на низкокипящие (температура кипения
ниже 100°C), среднекипящие (температура кипения 100—150°C)
и высококипящие (температура кипения выше 150 °C) [547, с. 34;
435, с. 29].
По летучести растворители предложено классифицировать по
относительной шкале, в которой за единицу принята летучесть ди-
этилового эфира при 20 °C и относительной влажности 65±5 %
[547, с. 29]. В соответствии с этим различают легколетучие (число
испарения ниже 10), среднелетучие (число испарения 10—35) и
труднолетучие (число испарения выше 35). Для удобства и без-
опасности работы (в тех случаях, когда растворители токсичны
либо огне- или взрывоопасны) при подборе электролитных компо-
зиций при прочих равных условиях следует выбирать раствори-
тель, наиболее высококипящий и труднолетучий.
б) . Классификация растворителей, основан-
ная на химических свойствах растворителей. Не-
редко растворители подразделяют в соответствии с рациональной
классификацией химических соединений. Очевидная прагматич-
ность такой системы в данном случае должна считаться достаточно
целесообразной, так как растворители одного химического типа
помимо аналогии в химическом поведении часто весьма подобны и
по физическим характеристикам (за исключением первых членов
128
гомологических рядов органических соединений—метанола, фор-
мальдегида, муравьиной кислоты и т. и.).
Классификация растворителей, основанная на их кислотно-ос-
новных свойствах, разбиралась в параграфе 1.4.2. Большинство
протолитических растворителей вступает в реакции сольватации с
растворенным веществом посредством Н-связи. В соответствии с
этим целесообразно классифицировать такие растворители по их
способности образовывать Н-связь [173, с. 250]. По данной клас-
сификации растворители подразделяются на пять групп: 1 — жид-
кости, способные к образованию трехмерной сетки Н-связи (вода,
серная кислота, гликоли); эти жидкости характеризуются высо-
кой диэлектрической проницаемостью и хорошей растворимостью
друг в друге; 2 — растворители с двухмерной сеткой Н-связей (фе-
нолы, большинство карбоновых кислот, одноатомные спирты); 3 —
жидкости, молекулы которых имеют в своем составе электронодо-
норные атомы, способные к образованию Н-связи с протоном рас-
творенного соединения (амины, эфиры, кетоны и др.); 4 — жидкос-
ти, молекулы которых содержат электронофильные атомы водоро-
да, способные вступать в Н-связь с электронодонорными атомами
молекул растворенного вещества (хлороформ, дихлорэтан и т. п.);
5 — растворители, не способные принимать участие в образовании
Н-связи (четыреххлористый углерод, гексан, пергалоидированные
углеводороды и т. п.).
Достаточно распространена система классификации, предложен-
ная Паркером. В основу ее положена способность растворителя
сольватировать ионы. Согласно Паркеру, растворители подразде-
ляются на три группы: 1 — аполярные апротонные растворители,
представляющие собой жидкости со сравнительно низкой диэлект-
рической проницаемостью (ниже 15), молекулы которых облада-
ют невысоким дипольным моментом (сероуглерод, четыреххлорис-
тый углерод, третичные амины и т. д.); 2 — диполярные апротон-
ные растворители, характеризующиеся сравнительно высокой ди-
электрической проницаемостью, молекулы которых имеют сравни-
тельно высокий дипольный момент; если в состав этих раствори-
телей входит водород, то он не может вступать в Н-связь (НБ,
НМ, АН, ПК, ДМ ФА, ДМСО и т. п.); 3 — протонные растворите-
ли, содержащие электронодефицитный водород, способный образо-
вывать Н-связь (спирты, карбоновые кислоты, фенолы и т. п.).
Сольватирующая способность растворителей по приведенной
классификации повышается от первой к третьей группе.
Иные классификации растворителей, основанные на химических
признаках, приведены в работах [547, 116, 435].
в) . Классификация растворителей, основанная
на сочетании физических и химических свойств
растворителей. Брёнстед предложил систему классифика-
ции, которая сочетает диэлектрическую проницаемость раствори-
телей и их протогенность (протофильность). В соответствии с эти-
ми признаками Брёнстед разделил растворители на восемь групп,
отличительные признаки которых сведены в табл. 15. Примерами
9 - 4-653 1 29
растворителей типов классификации по Брёнстеду могут служить:
I — вода; II — серная кислота, жидкий фтористый водород; III —
гидразин; IV — ПК, НМ, НБ, АН; V — одноатомные спирты, фе-
нолы; VI — жидкие галогеноводороды; VII—Ру; VIII—Г, Б. Клас-
сификация Брёнстеда позволяет прогнозировать проявление раст-
воренным веществом кислотно-основной функции, а также оцени-
вать нивелирующее либо дифференцирующее действие растворите-
ля на силу электролитов.
Таблица 15. Классификация растворителей по Брёнстеду
Свойство Тип растворителя
I II III IV V VI VII VIII
Диэлектрическая проницаемость Протогенность Протофильность + + + 1 ++ +1 + + + + + + —
Примечание. Знаки <-Ь» и «—»—в первой строке соответствуют высокой и низ-
кой е; в остальных строках эти знаки отвечают наличию либо отсутствию данного свойства.
В некоторых случаях может оказаться полезной классификация
растворителей, основанная на величинах констант автоионизации
(автопротолиза). В растворителях одного химического типа кис-
лотность увеличивается с повышением констант автоионизации
(данные по величинам pKai приведены в табл. 12). Предложены
также иные системы классификации растворителя, основанные на
сочетании физических и химических свойств [547, с. 38].
3.1.2. Смешанные растворители
Применение смешанных растворителей позволяет существенно рас-
ширить и разнообразить сферу применения неводных сред в теоре-
тико-экспериментальной и прикладной электрохимии и значительно
расширяет возможности направленного подбора электролитных
композиций.
Теоретические обобщения в области соотносительного влия-
ния физических и химических факторов на силу электролитов (см.
параграфы 1.2.2. и 1.4.5) позволяют осуществлять целенаправлен-
ный подбор растворителя, обеспечивающий максимально высокую
для данного электролита величину константы электролитической
диссоциации в неводных средах. При этом один из компонентов
смешанного растворителя может быть сольватирующим агентом,
доставляя системе энергию сольватации, необходимую для образо-
вания соответствующей ионной пары, второй из компонентов сме-
шанного растворителя определяет диэлектрическую проницаемость,
достаточно высокую для существенного распада ионной пары на
130
свободные сольватированные ионы. Так, в смешанном растворите-
ле, образованном, например, Ру и ПК, галогениды щелочных ме-
таллов будут диссоциировать намного лучше, чем в каждом из
w компонентов в отдельности, поскольку высокоосновный и, следова-
тельно, хорошо сольватирующий катионы Ру обладают сравнитель-
но низкой диэлектрической проницаемостью, а высокополярный ПК
весьма плохо сольватирует ионы электролита. Примеры направ-
ленного подбора состава смешанного растворителя с целью изме-
нить в нужном направлении силу электролитов можно найти в ра-
ботах [556, 564, 557, 542, 123].
Переход от индивидуальных к смешанным растворителям поз-
воляет существенно дифференцировать подвижность ионов. Так,
хлориды РЗЭ, характеризующиеся в воде практически одинако-
вой предельной эквивалентной электропроводностью, в смешанном
растворителе метанол — пропанол различаются по величине Хо поч-
ти вдвое [122, 611].
Используя установленные теоретические закономерности по
влиянию сольватации, диэлектрической проницаемости и вязкости
| на электропроводность, можно, направленно подбирая компонен-
, ты смешанного растворителя, добиваться максимальной для дан-
-' ного электролита электропроводности в неводных средах.
Очевидно, что сольватирующий компонент смешанного раство-
; рителя должен по возможности обладать наименьшим молекуляр-
* ным радиусом и минимальной вязкостью; второй же компонент
I растворителя достаточно высокую диэлектрическую проницаемость
। должен сочетать с возможно минимальной вязкостью.
« О направленном влиянии состава смешанного растворителя на
| электропроводность сообщается в работах [544, 158, 566, 449, 554].
? Количественная теория влияния состава и свойств смешанного ра-
створителя на эквивалентную электропроводность (на примере Н-
кислот) изложена в работе [70].
« Опираясь на сформулированные и обоснованные в работе [562]
условия появления в системе отрицательного температурного ко-
эффициента электропроводности, можно подбирать компоненты и
t. состав смешанного растворителя таким образом, чтобы достаточ-
но высокая электропроводность раствора достигалась при возмож-
/ но более низкой температуре (что весьма выгодно с точки зрения
эксплуатации и техники безопасности при работе с электролитной
композицией).
Поскольку растворимость ионофоров в неводных растворите-
лях, как и многие иные свойства неводных композиций, определя-
ется энергией сольватации и диэлектрической проницаемостью,
многое из того, что было сказано о влиянии смешанного раствори-
теля на силу электролитов, может быть перенесено и на раство-
• римость электролитов в неводных средах. Наконец, изменение со-
става смешанного растворителя оказывает существенное влияние
V. и на электродные процессы. Изменяя характер специфической соль-
\ ватации и диэлектрическую проницаемость, можно существенно
1. изменить величину стандартного электродного потенциала и рас-
4’.- - г
9* 131
положение металлов в ряду напряжений. Это обстоятельство поми-
мо очевидного его применения позволяет существенно повышать чи-
стоту выделяемого при электролизе металла.
В большинстве случаев компоненты смешанного растворителя,
применяемого в практике электроосаждения металлов, не должны
вступать друг с другом в высокоэнергетическое специфическое вза-
имодействие. В противном случае такое взаимодействие приводит
к образованию ионогенных продуктов, что существенно влияет на
режим электролиза, затрудняет, а то и вовсе делает невозможным
процесс электроосаждения металла. Поэтому чаще эти смешанные
растворители представляют собою системы с химически невзаимо-
действующими компонентами.
Физико-химические свойства многих таких систем приведены в
справочной литературе [1245]. При отсутствии этих сведений
свойства смеси двух химически невзаимодействующих жидкостей
можно с вполне удовлетворительной для практики точностью рас-
считать, исходя из свойств компонентов и их содержания в смеси
[19, 530, 547]. Приведем расчетные уравнения для трех важней-
ших макрофизических свойств растворителей — плотности (моль-
ного объема), вязкости и диэлектрической проницаемости.
Плотность смеси двух химически невзаимодействующих жид-
костей является объемно-аддитивным свойством [530, с. 58]:
d = d.V, + d2V2 = (dt - tQ Vj + dz, (3-1)
где d — соответственно плотности компонентов; Vi — их объемные
доли. Уравнение (3—1) адекватно уравнению мольно-долевой ад-
дитивности мольных объемов [547, с. 59]:
Vm -- + Ущгх2 — (Vm1 — Vm2) xt 4- Vm2- (3—2)
Здесь Vm. соответственно мольные объемы компонентов; xt — их
мольные доли.
Вязкость смеси с вполне удовлетворительной точностью описы-
вается уравнением степенной зависимости от мольно-долевого сос-
тава:
г) — r)[*r)^= = r)f ‘Ла — Х1) (3—3)
или
In г) = х41п л 1 + x2ln rl2 = ~ + In Яг- (3—За)
С несколько более высокой точностью вязкость смеси двух
жидкостей может быть рассчитана по аппроксимационным урав-
нениям, приведенным в работах [527—529; 530, с. 113, 19].
В первом приближении диэлектрическая проницаемость двой-
ной жидкой системы, образованной химически невзаимодействую-
щими компонентами, является объемно-аддитивной функцией со-
става [530, с. 91]
еад —- EjVJ + e2V2 = (sj — e2) Vi + e2. (3—4)
132
Более точный расчет е смеси двух жидкостей может быть про-
веден по уравнению [539]
е = (sj — е2) Vi + е2 + f (6S), (3 —5)
где зависящая от флюктуаций концентраций б8 системы функция
/(6S) для состава с объемной долей, равной 0,5, может быть рас-
считана по выражению
/(6S) = -0,043
(ei~ea)2
8аД
(3—5а)
Для систем, образованных ассоциированным компонентом с не-
ассоциированным, от величины 8, находимой расчетом по уравне-
нию (3—5), следует вычесть величину, постоянную для каждого
данного ассоциированного соединения, что составляет для равно-
объемной смеси двух жидкостей примерно единицу. Точные значе-
ния поправок для различных классов ассоциированных соединений
приведены в работах [538, 540].
3.1.3. Критерии выбора растворителей
для электролитных композиций
На основании теоретико-экспериментальных положений, приведен-
ных выше, можно утверждать, что индивидуальный либо смешан-
ный растворитель, предназначающийся для электролитной компо-
зиции, должен сочетать в оптимальном варианте следующие мак-
рофизические и химические характеристики: максимальную диэлек-
трическую проницаемость; минимальную вязкость; минимальную ле-
тучесть; максимальную донорность (основность).
Как отмечалось, задача оптимизации перечисленных свойств
рациональнее всего может быть решена составлением смешанных
растворителей. При этом надо учесть, что при достаточной кон-
центрации (>0,2—0,3 мольн. доли) донорного растворителя во
втором, индифферентном, основность смешанного растворителя
можно считать постоянной (поскольку остается постоянной энер-
гия сольватации),.
Необходимо учитывать также токсичность (сведения о токсич-
ности можно найти в руководстве [97]), стоимость, степень слож-
ности методов очистки, в частности осушки, гигроскопичность.
3.2. ХАРАКТЕРИСТИКИ ЭЛЕКТРОЛИТНЫХ КОМПОЗИЦИЙ
3.2.1. Растворимость соединений металлов
в неводных растворителях
Количественная теория растворимости относится к наименее раз-
работанным разделам общей теории растворов. До сих пор не най-
дена сколько-нибудь обоснованная связь растворимости с энерге-
тическими и химическими характеристиками электролита, а также
133
с химическими и физическими свойствами растворителя. С другой
стороны, хотя имеются многочисленные литературные данные по
растворимости разнообразных солей в различных неводных раст-
ворителях [320, 959], большинство из них не обладает необходи-
мой степенью достоверности. Поэтому даже нижеследующие ка-
чественные обобщения по закономерностям растворимости в не-
водных растворителях, основанные на литературных данных, ко-
торые приведены в табл. 6 приложения, в какой-то мере приблизи-
тельны.
Исходя из современных представлений о природе образования
электролитного раствора, растворимость должна определяться
энергией кристаллической решетки электролита, энергией специ-
фической сольватации в системе и диэлектрической проницаемос-
тью растворителя. Анализ данных, приведенных в табл. 8 прило-
жения, подтверждает это положение. Действительно, раствори-
мость галогенидов элементов III—IV А подгрупп периодической
системы, характеризующихся значительной долей ковалентности
связей и, следовательно, существенно меньшей энергией кристал-
лической решетки по сравнению с галогенидами элементов I и II
А—подгрупп, в каждом из растворителей значительно выше, чем
в случае указанных ионофоров. Уменьшение энергии кристалличе-
ской решетки с увеличением кристаллографического радиуса анио-
на практически во всех случаях (например, в рядах хлориды —
бромиды — йодиды) вызывает существенное повышение раствори-
мости. Однако при сопоставлении растворимости солей с одина-
ковым анионом в соответствии с представлениями о преимущест-
венной сольватации катионов в донорных растворителях (см. па-
раграф 1.4.5) рост кристаллографического радиуса катиона не
всегда ведет к адекватному росту растворимости. В данном слу-
чае рельефно отражается конкуренция между двумя процессами:
уменьшением энергии кристаллической решетки с ростом радиуса
катиона и уменьшением энергии специфической сольватации, иду-
щей в том же направлении. Действительно, растворимость хлори-
дов щелочных металлов в спиртах, АН и некоторых других раст-
ворителях в ряду литий—цезий сначала снижается, затем начина-
ет расти.
Увеличение кислотности катионов при переходе от щелочных к
щелочноземельным металлам в основном ведет к повышению раст-
воримости. Однако и в этом случае закономерности в изменении
растворимости в ряду магний — барий нарушаются конкуренцией
отмеченных выше противоположно направленных процессов.
Роль процессов специфической сольватации в определении раст-
воримости отчетливо проявляется при сопоставлении растворимос-
ти в сильноосновных и сильнокислых растворителях. Так, раст-
воримость галогенидов щелочных металлов в жидком аммиаке су-
щественно понижается с ростом кристаллографического радиуса
катиона и в гораздо меньшей степени зависит от аниона.
В то же время в уксуснокислых растворах наблюдается обрат-
ная картина.
134
г
' Термодинамика изменения растворимости при переходе от од-
ного растворителя к другому определяется разностями химических
энергий сольватации ионов и недиссоциированных молекул в этих
; двух растворителях.
Н. А. Измайлов [173, с. 217] вывел уравнение, связывающее
растворимость соли х в двух растворителях I и II с коэффициен-
тами активности соли у в этих растворителях, диэлектрической
проницаемостью растворителей и средним межионным расстояни-
ем г данной соли в растворе (в качестве стандартов для коэффи-
, циентов активности взят бесконечно разбавленный раствор в дан-
ном растворителе):
<з-б>
Yj Hl T \ Bj Bjj j
Уравнение (3—6) в неявном виде содержит допущение о неиз-
менности энергии сольватации в процессе растворения, следова-
) тельно, приложимо для универсальных либо условно универсаль-
г ных сред, для которых оно может быть преобразовано к виду
‘ In х = constj + со~~2 . (3—7)
(Примеры прямолинейной зависимости In х от 1/е, выполняющи-
еся в растворителях одной химической природы либо в двойных
смешанных растворителях с постоянной энергией сольватации, име-
ются в работах [173, с. 218; 547, с. 83].
Основной термодинамической характеристикой растворимости
является величина свободной энергии, пропорциональная логариф-
му произведения растворимости ПР,
AGp .-= — vflTln а± = — RT In ПР, (3—8)
| где v — число ионов, на которые распадается молекула соли; а± —
[средняя активность ^онов в насыщенном растворе данного электро-
лита. Для 1—1 электролитов в наиболее распространенных раство-
рителях, хотя и наблюдается тенденция к повышению растворимо-
сти (следовательно, к возрастанию экзоэнергетичности AGP) с
увеличением кристаллографического радиуса аниона, в некоторых
& случаях (соли цезия в метаноле, соли таллия в ПК, соли серебра)
наблюдаются отступления от этой тенденции. Термодинамические
Г характеристики растворимости находят из политермических дан-
I ных дифференцированием AGP по Т по известным уравнениям хи-
мической термодинамики.
Влияние энергии специфической сольватации и диэлектрической
проницаемости на величину растворимости наглядно выявляется
| при сопоставлении растворимости в ДМСО и ПК. Хотя е перво-
; го существенно ниже, чем второго, высокая донорность ДМСО обу-
( словливает намного большую растворимость в нем по сравне-
| иию с ПК.
135
№
3.2.2. Электрохимическая устойчивость
неводных электролитов
Одним из основных условий применения неводных растворов для
электрохимических исследований является электрохимическая ста-
бильность. Электрохимическая устойчивость электролита оп-
ределяется областью потенциалов, в пределах которой
не протекают электрохимические реакции с участием растворите-
ля. Понятие электрохимической устойчивости в последнее время
уточнено — ее определяют как область циклических вольт-ампер-
ных кривых с токами практически меньшими, чем менее
10 мкА-см-2 [676, 303]. Многие из органических растворителей
окисляются или восстанавливаются труднее, чем вода, что обус-
ловливает их стабильность в более широкой области потенциалов.
Наибольшая протяженность устойчивой области, достигающая
5,0—5,5 В, наблюдается в растворах перхлората лития в АН, изо-
пропаноле, ДМСО, ДМФА. В смесях органических растворителей
с водой протяженность области электрохимической стабильности
значительно сокращена, наибольшие изменения происходят при
малых концентрациях одного растворителя в другом, что связы-
вают с пересольватацией ионов [154].
Граничные величины потенциалов области электрохимической
устойчивости электролита определяются в основном тремя видами
электрохимических реакций: 1 — разряд катионов и анионов фо-
на; 2 — электрохимическое растворение анодного материала; 3 —
электрохимическое разложение самого растворителя.
От природы фонового электролита во многом зависит протя-
женность области электрохимической стабильности. Так, разряд
катионов фона во многих растворителях ограничивает область от-
рицательных потенциалов. При полярографических измерениях со-
ли .натрия позволяют работать до потенциалов — 1,95 В, соли ка-
лия— 2,0 В, соли лития — 2,1В (по отношению к насыщенному
водному каломельному электроду). Применяя тетраалкилзамещен-
ные соли аммония с длинными алкильными радикалами, можно
расширить предел измерений до —3,0 В [322]. Из обычно исполь-
зуемых фоновых электролитов для неводных сред в анодной облас-
ти наиболее устойчив перхлорат-ион. Близкие по природе фоновые
электролиты, как правило, обеспечивают электрохимическую ста-
бильность в практически одинаковом интервале потенциалов
[1254].
Поскольку большинство электрохимических измерений прово-
дится на инертных или ртутных электродах, анодное растворение
электрода в качестве причины предельного потенциала в положи-
тельной области стабильности электролита сводится в основном к
растворению ртути. Анодно ртуть растворяется легко. Использо-
вать ее в качестве анода можно в большинстве органических раст-
ворителей до +1,0 В (насыщенный каломельный электрод). Данные
о растворении ртутного капельного электрода в различных раст-
ворителях приведены в работе [320].
136
Что касается электрохимического разложения самих органичес-
ких растворителей, то восстановление их, как правило, происхо-
дит при потенциалах, более отрицательных, чем потенциалы, при
которых восстанавливаются неорганические ионы. С окислением
органического растворителя анодный предел области потенциалов
связан довольно тесно [320, 1072]. Сведения об анодном окисле-
нии всех классов органических растворителей содержатся в рабо-
тах [322, 320, 1116]. Продуктами электрохимических реакций ор-
ганических соединений являются обычно ионы или ион-радикалы,
которые затем реагируют с компонентами раствора. Так, ПК анод-
но окисляется с образованием пропилена и карбонат-иона [303].
Кроме природы и состава раствора, на область электрохимиче-
ской устойчивости сильно влияют природа и состояние поверхности
электрода. Например, нитрилы с низкими молекулярными масса-
ми, в первую очередь АН, с большим трудом окисляются и вос-
станавливаются электрохимически на обычно применяемых элект-
родах. В AH-растворах тетраалкиламмонийных солей обычно пред-
лагается, что катодные границы стабильности электролита опре-
деляются восстановлением катиона. Для ртутного электрода име-
ются прямые доказательства этого предположения — образование
амальгамы тетраалкиламмония. Однако на оловянном электроде
основной катодной реакцией является восстановление АН, приводя-
щее к разрыву связи S—N [821].
При использовании платинированного платинового электрода
анодная стабильность электролита КР1?6 — ПК намного ниже, чем
в тех же условиях в случае гладкого платинового электрода
[676]. На потенциодинамических кривых, снятых в растворе
LiC104 — ПК на электроде из никелевой черни, области стабиль-
ности электролита практически нет, в то время как на гладком
никеле она составляет 2,1 В [303].
Необходимо также отметить, что границы электрохимической
стабильности электролита могут определяться и присутствующими
в растворе примесями [676, 303, 920, 741]. Величины граничных
потенциалов, определяющих область электрохимической устойчи-
вости для наиболее употребительных неводных растворителей, при-
ведены в табл. 12 приложения. Растворители рассматриваются со-
гласно систематике, предложенной Ч. Манном [1116].
Глава 4
ПРАКТИКА ЭЛЕКТРОВЫДЕЛЕНИЯ МЕТАЛЛОВ
ИЗ НЕВОДНЫХ РАСТВОРОВ
4.1. МЕТАЛЛЫ ПЕРВОЙ ГРУППЫ
Первые литературные сведения по электролитическому выделению
щелочных металлов из неводных растворов относятся к концу
прошлого века. В. Латинский [1007] в 1895 г. из раствора хлори-
да лития в ацетоне на медной проволоке выделил металлический
литий в виде серой пленки. До середины настоящего столетия были
предприняты многочисленные попытки электроосаждения лития и
других щелочных металлов из неводных сред, в основном органи-
ческих. Однако характер этих работ эпизодический, в основном ка-
чественный и нередко малодоказательный. Катодные осадки час-
то представляют собой соединения щелочного металла и раствори-
теля. А основным доказательством присутствия щелочных метал-
лов во многих работах считается бурное взаимодействие продук-
тов электролиза с водой. Естественно, что такую же реакцию спо-
собны дать и металлоорганические соединения. В работах часто
не приводятся условия эксперимента, использование высоких на-
пряжений (100 В и выше) вызывает осмоление растворителя. Сов-
ременный термодинамический анализ возможности взаимодейст-
вия щелочных металлов со многими растворителями [203, 201]
показывает, что многие из них являются окислителями по отноше-
нию к щелочным металлам. В ранних работах часто использова-
лись растворители, заведомо активные по отношению к выделяе-
мому щелочному металлу. Таковы, например, работы по электро-
лизу спиртовых растворов щелочных металлов, где возможно
образование алкоголятов, а затем, в результате их электролиза,
эфиров.
И все же ценность этих работ неоспорима. В них впервые ус-
тановлена принципиальная возможность электроосаждения щелоч-
ных металлов из растворов, обнаружены отдельные закономерно-
сти по получению качественных осадков металлов. С этой точки
зрения в первую очередь следует отметить работы школы Плотни-
кова, где хорошо образованные осадки щелочных металлов (Li,
Na, К) получены из смешанных растворов хлоридов и бромидов
алюминия и галогенидов щелочного металла в нитросоединениях
[414, 411, 416]. Хотя нитросоединения в качестве растворителей
138
при элёктровосстановлении щелочных металлов мало пригодны из-
за частого образования взрывчатых катодных осадков, В. А. Плот-
ников и О. К. Кудра [410] разработали опытный электролизер для
электроосаждения из нитробензоловых растворов.
Результаты ранних работ по электроосаждению щелочных ме-
таллов из неводных сред критически рассмотрены и систематизи-
рованы в монографиях [414, 641, 702, 422].
Более поздние работы [588, 167, 702, 722, 641, 664, ИЗО, 81—
85, 1235, 420] по электроосаждению щелочных металлов из невод-
ных растворов представляют детальные исследования электродных
процессов, происходящих при выделении металла, влияние на них
параметров электролиза и различных добавок, зависимости каче-
ства осадка и выхода по току металла от условий электролиза.
Наибольшее количество работ посвящено исследованию лития, по-
скольку литий, во-первых, наименее электроотрицательный из
щелочных металлов, во-вторых, широко используется в каче-
стве электродного материала в разнообразных батареях [722,
664, 960].
Из всех классов растворителей для электроосаждения щелоч-
ных металлов наиболее перспективны апротонные растворители,
наиболее устойчивые по отношению к выделяемому металлу. Для
электроосаждения используют простые соли щелочных металлов
или в случае их недостаточной растворимости и степени диссоци-
ации двойные соли с галогенидами алюминия, фосфора, бора. Ка-
чество осадка и его выход в значительной мере определяются под-
бором оптимальной концентрации и состава ванны.
В качестве универсального растворителя, пригодного для вы-
деления всех щелочных металлов при комнатной температуре,
может быть рекомендован ПК и смешанные растворители на его
основе [702, 969, 670, 671].
Наилучшие результаты с высоким выходом по току получены при
осаждении щелочных металлов из их солей с анионами С ЮГ, РЕГ>
ВЕГ и частично А1С1Г’ концентрация соли порядка 1 моль на кило-
грамм электролитного раствора, плотность тока 1—10 мА/см2
[969]. При электролизе образуются мелкокристаллические осад-
ки щелочных металлов весьма высокой частоты.
Катодные осадки щелочных металлов и их амальгамы в раст-
воре ПК могут быть также с высоким выходом анодно растворены.
На основании этих данных на примере калия предложен способ
рафинирования щелочных металлов из амальгам, полученных в
водных хлоридно-ртутных ваннах. Качество осадка щелочного ме-
талла в пропиленкарбонатном электролите существенно зависит от
плотности тока. Мелкокристаллические серебристые гладкие осад-
ки можно получить лишь при низких плотностях тока. Для полу-
чения бездендритных осадков в случае осаждения лития в интенси-
фицированном процессе с использованием ванны на основе раст-
воров LiC104 в ПК предложено добавление в ванну солей метал-
лов, образующих с литием интерметаллиды [670, 671]. В качест-
139
1
ве подложки для осаждения лития из растворов ПК рекомендует-
ся титан.
Электроосаждение лития изучено в растворителях АН, ФА,
ДМАА, ДМФА, диэтилформамиде, ДМСО, ТГФ, ПК, АЦ и др. Де-
тально исследовано его выделение из растворов в ДМФ [588, 167,
81—85, 1235]. Выход по току определяется концентрацией соли ли-
тия в электролите и плотностью тока и всегда ниже 100 %. Так,
оптимальные характеристики электроосаждения лития из хлорид-
ных растворов в ДМФ с использованием никелевого катода и гра-
фитового анода следующие: концентрация ЫС1 — 4,25—42,5 г/л,
катодная плотность тока 0,25—0,75 А/дм2, выход по току 52—
78 % [81—85]. Электролиз 1М растворов L1C1 при силе тока
180 мА/см2 приводит к образованию на платиновом ка-
тоде кристаллических серебристых осадков лития с выходом
по току 85%. Добавка к электролиту 0,03 М СиС12 повышает вы-
ход до 84—90 % [167] и плотность тока приблизительно в 60 раз.
Это позволяет сделать заключение о лимитирующей роли стадии
кристаллизации при электроосаждении лития. Микроскопические
кристаллы электроосажденной меди, выполняя роль центров крис-
таллизации, существенно повышают скорость кристаллизации ли-
тия [167]. Другая трактовка пассивации электрода при электро-
осаждении лития и резкого смещения катодного потенциала в от-
рицательную сторону объясняется накоплением на электроде не-
растворимого малопроводящего продукта взаимодействия лития с
ДМФ [588, 861]. Определена скорость поверхностного электрохи-
мического взаимодействия (она составляет 5—6 А/см2). Вероятно,
пассивация литиевого электрода и низкий выход по току могут
быть следствием обеих причин, хотя объяснение на основании хи-
мического взаимодействия лития с растворителем более обобщаю-
щее и достоверно, поскольку максимальное значение фазовой по-
ляризации не превышает здесь 0,2 В.
Плотный мелкокристаллический осадок лития, содержащий
примеси в количестве ^0,01 %, может быть получен электролизом
на никелевом катоде растворов L1C1 в ДМФ концентрации 2,5—
42,5 г/л при плотности тока 1,0—2,5 мА/см2 [81—85].
Из растворов ЫС1 в ДМСО при электролизе осаждается литий
на вольфрамовую подложку с выходом 60—70 %. Такой же выход
по току наблюдается и при осаждении лйтия из растворов в ДМСО
LiBr на платину и никель. Относительно низкий выход по току
объясняется образованием сплавов Pt—Li, а также взаимодействи-
ем лития с растворителем или образованием ЫОН и Ы2О, пос-
кольку строгое обезвоживание не было проведено [722].
Сравнительное изучение электроосаждения лития из различных
растворителей в сочетании с его солями показало, что выход по
току лития зависит от используемого растворителя, а также от
природы аниона соли. В ДМА, ДМСО и ДМФ выход по току уве-
личивается в ряду LiCl<LiC104<LiN03 (концентрация электро-
лита 100 г/л, плотность тока порядка 2 А/дм2). Наиболее высокий
выход по току (выше 70 %) дают растворы нитрата лития в ди-
140
метилацетамиде. Для растворов LiCl выход по току увеличива-
ется в ряду: ДМСО<ДМФ<АН<ДМА, а с ЫС104 —в ряду
АН<ДМФ<ДМСО<ДМА [1235].
Эффект от прибавления воды к электролиту различный: в од-
них случаях (например, растворах LiNO3 в ДМФ) выход по току
повышается, в других—понижается [1235].
Электроосаждение лития на серебро и платину сопровождает-
ся сплавообразованием, сплавы не образуются при выделении ли-
тия на никель и нержавеющую сталь. Сплавообразование улучша-
ет сцепление литиевого осадка с основой, поэтому при изготовле-
нии литиевых электродов для батарей никелевую сетку часто се-
ребрят.
Электролизом растворов различных солей в ДМФ, Ру и АЦ
получены осадки калия и натрия при температурах ниже 20 °C, с
выходом, близким к 100 %. При комнатной температуре наблюда-
ется взаимодействие выделившегося металла с ДМФ и особенно
сильное с АЦ. Выход по току также существенно зависит от при-
роды соли щелочного металла: для хлоридов, бромидов и нитратов
выход выше, чем для перхлоратов, роданидов и йодидов, что объ-
ясняется различной способностью солей к образованию сольватов.
Электролиз проводился в течение 0,5 ч при плотности тока 0,5—
1 А/см2, катод — железный [ИЗО].
Электровыделение и рафинирование меди из водных раство-
ров изучено довольно детально. Но не всегда из обычных вод-
ных растворов можно получить электролитическую медь желаемо-
го качества. Кроме того, эксплуатационные характеристики мед-
ных ванн часто бывают неудовлетворительными. Так, лучшими
водными ваннами для получения медных гальванопокрытий до сих
пор считаются цианистые. В результате этого изучение процессов
электроосаждения меди из неводных растворов проводится в двух
направлениях: получение качественных высокочистых осадков ме-
ди и гальванопокрытий, в первую очередь, на активный металл.
Электролизу подвергаются растворы солей меди во многих, в
основном полярных растворителях [702, 414, 679, 243, 1161, 1234,
451, 1025, 1257, 405]. При этом более качественные осадки в боль-
шинстве случаев дают растворы солей одновалентной меди [243,
278, 190, 1025]. В целом электроосаждение происходит более труд-
но, чем в водных растворах, часто сопровождается как концент-
рационной, так и химической поляризацией.
Ровные блестящие и прочные осадки меди получены из бен-
золовых, толуоловых и ксилоловых растворов галогенидов одно-
валентной меди в присутствии хлорида алюминия [414, 190]. Од-
нако сцепляемость таких осадков с основой удовлетворительна
только в случае медного катода, электролиты во время электроли-
за меняют свой состав.
Из формамидных растворов CuCI в присутствии муравьиной
кислоты при плотности тока 3—32 мА/см2 медь осаж-
дается плотным мелкозернистым слоем с хорошим выходом
по току. Удовлетворительного качества осадки меди со 100 %-ным
141
выходом по току из формамидных и диметилформамидных ванн
получены электролизом трифторацетата меди [676]. В диметил-
формамидном растворе осаждение меди происходит только в при-
сутствии лимонной кислоты (без нее на катоде образуется Си2О).
Условия получения гладких мелкокристаллических осадков меди
следующие: состав — Си(СРзСОО)2— 500 г/л, лимонная кислота—
200 г/л; гк = 2,0—11,0 А/дм2; температура 60 °; ВТк = 75°/о.
Плотные мелкокристаллические осадки меди получены также из
других растворителей [702, 414]. Однако в большинстве случаев
медь осаждается в виде порошка или дендритов. По мнению ав-
торов [75], именно органические растворители являются благо-
приятной средой для получения переходных металлов в виде мел-
кодисперсных активных порошков.
В качестве перспективных растворителей для промышленного
электроосаждения и рафинирования меди рекомендуются смешан-
ные водно-органические растворители [258, 1025]. Выделение меди
из неорганических растворителей, в частности аммиака, представ-
ляет пока лишь теоретический интерес [702].
На основе ванн из различных органических растворителей про-
ведено соосаждение меди с цинком, свинцом, титаном и другими
металлами [1060, 257, 330, 56, 259]. Электроосаждение латуней
различного состава изучено в формамидных растворах Cui—
Znl2, а также в растворах ацетатов в смешанном растворителе —
уксусная кислота — пиридин [1060]. Состав и структура осадков
изменялись в зависимости от плотности тока и температуры. Ско-
ростное латунирование стальной проволоки проведено из водно-
органического электролита [370]. Оптимальные условия для по-
лучения латунного покрытия состава 70 % Си и 30 % Zn толщи-
ной 5—6 мк: электролит CuSO4-5H2O— 30, ZnSO4-7H2O—12,
глицерин —24, NaOH — 140, ДЦМ-2, NH4NO3 —5 г/л; 40—50 °C;
гЕ = 30—40 Д/дм2. В перхлоратном электролите на основе ТГФ по-
лучены светлые плотные осадки сплава меди со свинцом с содер-
жанием свинца до 90 % [1060]. Соосаждение меди с теллуром
проведено из деаэрированных растворов хлоридов этих металлов
в безводной уксусной кислоте, содержащей LiCl [330]. При этом
установлено соблюдение отдельных общих закономерностей элект-
роосаждения сплавов. Так, для соосаждения меди с цинком с по-
вышением температуры и понижением плотности тока характерно
обогащение сплава более благородным металлом (медью). Сов-
местное осаждение теллура и меди сопровождается деполяризаци-
ей при разряде обоих компонентов в сплав по сравнению с раз-
дельными процессами. Содержание титана в сплаве, полученном
электролизом биметаллического комплекса, который образован хе-
латами меди и четыреххлористым титаном, определяется не толь-
ко его концентрацией, но и структурой комплекса.
Благодаря высокому положительному потенциалу серебро лег-
ко и количественно выделяется из водных растворов. Поэтому и
для очистки серебра используют электрорафинирование. Из невод-
142
ных растворов гальванические покрытия серебра целесообразно
выделять на поверхности менее благородных металлов. В зависи-
мости от природы растворителя, плотности тока, состояния элек-
тролита серебро из неводных сред может быть выделено в виде
порошка, дендритов, гладких крупно- или мелкокристаллических
осадков [1192, 702, 414, 641, 1193, 278, 190, 408, 1229,
1272, 405].
Изменение структуры осадка серебра в зависимости от соста-
ва электролита и плотности тока исследовалось неоднократно, од-
нако лишь некоторые авторы пытались связать структуру осадка с
природой растворителя. Таким примером может служить работа
[452], где величины наиболее вероятного числа образующихся на
катоде кристаллических центров серебра (nz) поставлены в связь
с диэлектрической проницаемостью растворителя.
Закономерное увеличение nz с уменьшением е объясняется различ.
ной комплексообразующей способностью Ag'1’ по отношению к анио-
ну NOF в данных растворителях. Чем ниже е, тем большее количе-
ство комплексных ионов Ag(NO3)F содержит раствор, тем выше зна-
чение nz (из растворов комплексных соединений металлы выде-
ляются на катоде в более мелкокристаллической форме).
Плотные мелкокристаллические осадки серебра можно полу-
чить из растворов его солей в пиридине, амидах и аминах, про-
изводных бензола, этилгалогенидах [414, 190, 1058]. Так, плотный,
гладкий серебристый осадок серебра получается при электролизе
системы А1С13—AgCl — ксилол при плотности тока 0,6 А/дм2, вы-
ход по току 60—70 %. При этом хорошее плотное сцепление с ос-
новой наблюдается в случае медного катода и плохое — на желез-
ном катоде [190].
Подробно изучено электроосаждение серебра из растворов его
солей в жидком аммиаке. Электролизу подвергались нейтральные,
щелочные растворы и растворы подкисленные NH4NO3 [1192, 702,
414, 641, 1193]. Лучшие осадки серебра получены из последних.
На структуру осадков большое влияние оказывает температура
изучаемых растворов. Благоприятное влияние понижения темпе-
ратуры на получение мелкокристаллического гладкого осадка се-
ребра отмечается в случае водно-органических растворов [1229].
Электролиз растворов фторида серебра во фторсульфоновой
кислоте приводит к одновременному протеканию процессов восста-
новления и окисления ионов Ag+. В результате на катоде осажда-
ется металлическое серебро, а на аноде — черный осадок двухва-
лентного серебра, содержащий 40,4 % Ag [1272]. Легко получают-
ся из неводных растворов также сплавы серебра с различными
металлами [414].
Сведения об осаждении золота из неводных растворов немного-
численны, хотя и относятся к нескольким растворителям [414, 146,
1257, 1272, 1242]. Наилучшие результаты достигнуты при элект-
143
ролизе растворов солей золота в жидком аммиаке. В частности,
аммиачно-цианидные ванны дают результаты, аналогичные водно-
цианидным ваннам.
4.2. МЕТАЛЛЫ ВТОРОЙ ГРУППЫ
Экспериментальный материал по электровыделению щелочнозе-
мельных металлов из неводных растворов довольно обширный
[702, 414, 641, 753, 586, 46, 1125, 1213]. Анализ этих сведений пока-
зывает, что ни один из щелочноземельных металлов не может быть
электролитически осажден в простых условиях. Наиболее успешно
осуществлено электровыделение бериллия и магния.
Возможности получения металлических кальция, стронция, ба-
рия изучены преимущественно в спиртовых, ацетоновых, пириди-
новых, аммиачных растворах галогенидов этих металлов. В боль-
шинстве случаев в результате электролиза на катоде образуются
соединения щелочноземельных металлов (часто за счет взаимо-
действия первичного катодного продукта с растворителем) и лишь
в отдельных случаях — осадки металлического характера, содер-
жащие небольшие количества сильно загрязненных кальция, строн-
ция, бария.
Попытки получения металлического бериллия проводились во
многих растворителях различных классов. Чаще всего электролиз
органических растворов солей бериллия сопровождается катодным
газовыделением или образованием черных неметаллических осад-
ков. Первоначальные сведения об осаждении металлического бе-
риллия из жидкого аммиака [414] не подтвердились [1271, 1061].
Электровыделение бериллия наблюдается из очень ограниченного
числа растворителей. Так, гладкие черные металлические осадки
бериллия низкой чистоты получены электролизом диметилберил-
лия (iK = 0,15 А/дм2) и смеси Ве(СН3)2 и ВеС12 (гк=1,0—2,5 А/дм2)
в диэтиловом эфире [1061].
Вуд и Бреннер [1271] провели наиболее детальное изучение
электроосаждения бериллия из органических сред. Исследованы
растворы гидридов, алюмогидридов, борогидридов, галогенидов,
алкилов и арилов бериллия в различных неводных растворителях
(АЦ, Б, Ру, эфирах, аммиаке и др.). Наилучшие результаты полу-
чены в галогенидэфирных и алкилэфирных ваннах. Причем элект-
ролиз смешанных галогенидно-алкильных ванн дает серый, блес-
тящий когерентный чешуйчатый осадок бериллия, значительно
улучшенный по сравнению с осадками, полученными отдельно из
каждого бериллиевого электролита. Примером может служить
ванна: 3 моля Ве(СН3)2-]-(0,9—2,3) моля ВеС12 в диэтиловом эфи-
ре. Электролиз при напряжении 18—25 В и /1( = 0,1 А/дм2 дает на
катоде серый металлический хрупкий осадок, содержащий 93—
95 % Be. Из эфирного раствора борогидрида получен хрупкий ко-
герентный сплав, содержащий 70 % Be и 30 °/о В. Сплав бериллия
144
с алюминием образуется при электролизе смешанных эфирных
растворов Ве(А1Н4)2 и ВеС12. Осадок содержит 0—57 % Be. По
мере увеличения содержания ВеС12 в ванне цвет сплава изменя-
ется от белого через серый до черного, в этом же ряду возрастает
его хрупкость. Запатентованы способы электроосаждения берил-
лия и из простых солей в индивидуальных диполярных апротонных
растворителях (ДМФ, ДМСО, ДМА) [46].
Электролиз растворов простых солей магния в органических
растворителя (спиртах, пиридине, диметилформамиде, формами-
де, ацетонитриле, анилине и др.) чаще не приводит к осаждению
металлического магния [702, 414, 641]. Аналогичные результаты
[1140] наблюдаются в жидком аммиаке. Перспективнее для элек-
тровыделения магния оказались растворы его сложных солей. Это
в первую очередь относится к растворам реактива Гриньяра, пред-
ставляющего собой комплексное соединение алкильных и галоге-
нидных солей магния валового состава: RMgX (R—алкил, X—
С1, Вг, I).
С помощью радиоактивного магния показано, что в реактиве
Гриньяра магний входит в состав катиона [782]. При анодном раз-
ряде анионов образуются свободные алкил-радикалы. Возможны
следующие электродцые процессы:
на катоде:
2RMg+ + 2е -* Mg + R2Mg,
2MgX+ + 2e^Mg + MgX2.
на аноде:
R3Mg~ R’ 4- R2Mg + е,
R2MgX~->-R’ + RMgX 4-е,
RMgXr R’ 4- MgX2 4- e.
Для увеличения электропроводности раствора и улучшения ка-
чества осадка магния часто прибегают к различным добавкам.
Так, А. Бреннер [703] для получения светлого гладкого осадка
магния применил электролит, содержащий реактив Гриньяра (на-
пример, MgClC2H5, декаборан и хлорид магния в ТГФ). Хорошие
осадки магния со 100 %-ным выходом получаются при плотностях
тока 0,1-ь1,0 А/дм2. Осадок содержит менее 1 % В.
Результативным для получения сплавов магния оказался эфир-
ный раствор, содержащий магний в виде галогенида, алюмогид-
рида, борогидрида или реактивов Гриньяра. Наилучший осадок
(приблизительно 30 % Mg и 10 °/о В) получен из раствора бромис-
того магния и борогидрида лития в диэтиловом эфире. Сплав маг-
10 -4-653
145
ния с алюминием (7 % Mg+ 93 % Al) получен из диэтилового рас-
твора, содержащего MgBr2, А1Вгз и UAIH4. Плотные столбо- или
порошкообразные осадки сплавов циркония, титана и бериллия с
магнием получены также из гидридной или борогидридной ван-
ны [753].
Поиски оптимальных условий электровыделения из неводных
сред металлов подгруппы цинка направлены в первую очередь на
получение качественных гальванопокрытий [702, 414, 641, 1161,
75, 1257, 1060, 257, 330, 56, 1058, 370, 18, 934]. Электроосаждение
цинка изучалось преимущественно из растворов его простых солей
в органических растворителях — АЦ, ФА, ДМФА, АН, этаноле
[414, 1161, 278, 190, 75, 357]. В зависимости от природы раство-
рителя и условий электролиза цинк выделяется в виде мелкокри-
сталлического порошка или плотного покрытия, дендритов. Выход
по току в основном близок к 100%. Имеются данные также по
соосаждению цинка с другими металлами из неводных растворов,
в частности медью [1257, 1060, 370]. Так, латуни различного сос-
тава получены при электролизе Cui—Znl2 — формамидных раство-
ров. Содержание цинка в осадке увеличивалось с повышением плот-
ности тока при постоянной температуре. Причем в случае высоких
плотностей тока первые 1—2 мкм осадка обогащены цинком, внеш-
ние слои — медью [1060]. Равномерное соосаждение цинка и меди
происходит при низких плотностях тока.
Кадмий при электролизе неводных растворов его простых солей
осаждается преимущественно в крупнокристаллическом виде. Мел-
кокристаллические компактные осадки кадмия получаются при
введении в раствор комплексообразующих добавок. Так, кадмие-
вые покрытия хорошего качества получены из смешанных раство-
ров хлорида кадмия и четырехзамещенных иодидов тетрабутилам-
мония в ДМФ. В этих растворах кадмий присутствует преимуще-
ственно в виде ионов СсЩ2-. При перенапряжениях, превышающих
0,5 В, этот ион разряжается непосредственно с образованием мел-
кокристаллического осадка [1058]. Качественные осадки кадмия
из растворов Cdl2 в ДМФ можно получить также в присутствии
солей аммония. Оптимальные условия: электролит — раствор 2,34 н
Cdl2 и 0,3 н NH4CI в ДМФ, iK=0,5—1,0 А/дм2, температура —
(—50—[-100°C), выход по току приблизительно 100 % [18]. Мел-
кокристаллические компактные осадки кадмия на сталях марок
08КП и 20 получаются из уксуснокислых и метаноловых раство-
ров ацетата кадмия при введении в раствор триэтаноламина, да-
ющего комплексы с солями кадмия [219].
Практически отсутствуют сведения о применении поверхностно-
активных веществ для управления катодным процессом в невод-
ных средах. Поэтому особый интерес представляет работа [248], в
которой проведено электроосаждение кадмия из диметилформа-
мидных растворов в зависимости от наличия в растворе поверх-
ностно-активных веществ. В раствор кадмий вводили в виде солей
Cdl2, CdF2, Cd(C104)2 и Cd(CH3COO)2.
146
В качестве поверхностно-активных веществ изучена реакционная
серия анилов о-оксиальдегидов общей формулы
—ОН
к
! где К=Н, СН3, ОСН3, ОН, Cl, Br, NO2. Влияние поверхностно-актив-
а; ных веществ на процесс электроосаждения кадмия и эффектив-
W ность во многом определяется природой аниона. Хорошие осадки
Ж кадмия образуются в присутствии анионов, способствующих ад-
Ж сорбции растворителя на электроде, и поверхностно-активных ве-
SlmeCTB, выступающих в роли эффективных деполяризаторов реак-
Ж ции разряда ионов металла. Оптимальным оказался электролит
ж состава: ДМФ—1 л, СсПг— 0,2 моля, НС1О4—0,8 моля, га-ЬЮг —
S салицилальанилин — 0,02 моля. Катод — железный, анод — кад-
® миевый, iK=l А/дм2, t—18—25°С.
Ж Цинк и кадмий могут быть также выделены в металлическом
S состоянии из растворов хлоридов и бромидов в Ру [934]. Все три
Ж металла подгруппы цинка также осаждаются электролизом раство-
Ж ров их солей в жидком аммиаке ,1414]. Из электролитов на основе
Ж органических растворителей возможно и электроосаждение спла-^
№ вов цинка и кадмия с другими металлами, например, кадмия с
ж' танталом [259].
4.3. МЕТАЛЛЫ ТРЕТЬЕЙ ГРУППЫ
£? Сведения о получении бора в чистом виде из неводных растворов
& отсутствуют. Бор можно соосадить с легкими металлами, в част-
s. ности с магнием, бериллием [1271, 753] в эфирных растворах,
ж Сплавы, содержащие до 30 % бора, хрупки, плохо сцепляются с
я основой, часто порошкообразны.
ж Проблема низкотемпературного электролитического выделения
[ для алюминия особенно актуальна. Как известно, весь алюминий
ж практически получают сейчас электролизом из высокотемператур-
Пых солевых расплавов — метод, далеко небезукоризненный как с
В экономической, так и с технологической точки зрения. В частности.
при высокотемпературном электролизе алюминия почти исключе-
на возможность получения гальванических покрытий. Поэтому раз-
& работка методов электроосаждения алюминия из неводных раство-
'i ров является особенно важной задачей и, начиная с пионерских
В работ по выделению металлов из неводных сред, неизменно прив-
в лекала внимание исследователей. Электроосаждению алюминия из
неводных растворов посвящено множество работ [186, 702, 414,
298, 204, 12, 641, 302, 1271, 753, 1140, 586, 1125, 934, 721, 764, 665,
902, 1219, 205].
10*
147
Для электроосаждения алюминия в основном применяются эфи-
ры и ароматические углеводороды. Из предложенных до настоя-
щего времени электролитов алюминирования практическую цен-
ность представляют ванны на основе комплексных алюмооргани-
ческих соединений; четвертичных аммониевых солей; эфирногид-
ридные.
Эфирные электролиты наиболее эффективны и широко исполь-
зуемые. Приоритет исследований эфирных электролитов и элект-
ролитов на основе углеводородов ароматического ряда принадле-
жит В. А. Плотникову и его школе [414, 641, 399, 402, 407, 189,
406]. Экспериментальные данные по электроосаждению алюминия
из неводных сред и обобщения теоретического характера, по-
лученные школой В. А. Плотникова, являются основополагающи-
ми для всех последующих разработок неводных электролитов алю-
минирования. Позже большой вклад в совершенствование эфир-
ных ванн для электроосаждения алюминия внесла школа А. Брен-
нера [702, 764, 767]. На современном этапе исследование эфирных
электролитов проводится многими учеными различных стран, наи-
более широко и детально школой А. Л. Левинскаса [23, 298, 295,
296, 288, 297, 24, 282, 286, 302, 281, 283, 1211, 290, 289, 177, 176, 22,
301, 285, 104].
По природе и закономерностям процесса электроосаждения
алюминия эфирные электролиты делятся на несколько групп:
эфирноаминовые, эфирногидридные, эфиратные и эфирноаммиач-
ные. Последние две предложены и разработаны школой А. Л. Ле-
винскаса. В качестве эфирной среды в основном применяется ди-
этиловый эфир, поскольку он наиболее доступен и обеспечивает
наилучшие гальванотехнические показатели.
Эфирноаминовые электролиты представляют собой раствор сле-
дующих компонентов: галогенид алюминия (10—15%); алкила-
мин, содержащий до трех алкильных групп и до двенадцати ато-
мов углерода в молекуле (10—40%); алифатический простой
эфир, содержащий до шести атомов углерода (30—80 %). Готовят
электролиты добавлением эфирного раствора галогенида алюми-
ния к охлажденному разбавленному эфиром амину при перемеши-
вании [702, 282, 641, 1087, 1088, 1063]. Оптимальными компонен-
тами являются хлорид алюминия, n-бутиламин, этилгексиламин,
диэтиловый, этилбутиловый, дипропиловый эфиры, ТГФ. Практи-
чески электролит удовлетворительно работает во всем интервале
температур от комнатной до температуры кипения, рабочая тем-
пература в основном 20—30 °C. Необходима предварительная про-
работка током. В таких условиях чистый электролитический алю-
миний осаждается в широком диапазоне катодной плотности тока
(приблизительно до 0,140 А/см2) с выходом по току 50—80 %. Воз-
можно скоростное осаждение при катодной плотности тока 8—
9 А/дм2. Основным недостатком эфирноаминовых электролитов яв-
ляется их высокая чувствительность к чистоте компонентов. Толь-
148
ко применение свежесублимированного А1С1з и глубокой очистки
аминов и эфира дает возможность получить гомогенный раствор
самого электролита. Работающий электролит следует тщательно
герметизировать (герметизирующий газ-—сухие азот или аргон).
Растворяющиеся со 100 %-ным выходом алюминиевые аноды дол-
жны быть изготовлены из особо чистого металла. Ванна неустойчи-
ва во времени, требует частого обновления и корректировки.
Эфирногидридный электролит — основной неводный электролит
алюминирования промышленного масштаба. Исходный вариант
его был предложен и разработан А. Бреннером [702, 282, 764, 767]
под названием НБС (национальное бюро стандартов США). Сос-
тав эфирногидридного электролита следующий: хлорид алюминия
(1—4М), гидрид лития (0,5—1,0М) или смешанный литиевоалю-
миниевый гидрид (0,1—0,4М), абсолютированный диэтиловый
эфир. Ванну на основе электролита НБС обычно герметизируют
сухим азотом или аргоном, рабочая температура — комнатная.
Электроосаждение проведено на самые различные подложки от
активных металлов (уран) до инертных конструкционных матери-
алов (стали, латуни, медь, серебро), аноды — алюминиевые. В ин-
тервале плотностей тока до 0,1—0,15 А/см2 с 90—100 %-ным вы-
ходом катодно осаждается мелкокристаллический плотный элас-
тичный осадок алюминия, при этом могут быть получены гальвано-
пластические слои до 2—5 мм. Осадок алюминия содержит лишь
следы тяжелых металлов. Процесс электроосаждения включает
приемы, обеспечивающие выравнивание поверхности покрытия:
проточный, равномерно омывающий рабочий электрод электролит;
медленное вращение катода; непрерывное фильтрование электро-
лита и др. При тщательной герметизации, строгом соблюдении ус-
ловий электролиза и корректировки ванна может работать непре-
рывно в течение 18 месяцев. Основным недостатком ванны на ос-
нове НБС является высокая летучесть и легкая воспламеняемость.
Потребность в совершенствовании* эфирно-гидридной ванны
НБС привела к созданию новых модифицированных эфирногид-
ридных электролитов и режимов работы. Так, для улучшения вы-
равнивающего действия и скорости электрокристаллизации при-
меняют наложение на ванну импульсного тока [290, 22]. Предло-
жен «нелетучий» гидридный электролит с точкой воспламенения
85—90 °C (электролит ЛОВЕТ) [282]. По сравнению с электроли-
том НБС он содержит лишь незначительные количества свободно-
го эфира, однако обладает пониженной электропроводностью. Для
повышения электропроводности подобраны специальные ионизи-
рующие добавки. Наиболее эффективна из них хлорид 2-этокси-
этилтриметиламмония. Нелетучие гидридные электролиты, содер-
жащие в своем составе эту добавку, назвали ЭТМАК. Для сниже-
ния летучести и увеличения стабильности ванны предложен моди-
фицированный эфирногидридный электролит, в котором ДИЭТИЛО-
149
вый эфир заменен на ТГФ в смеси с ароматическими углеводоро-
дами [970, 941, 801]. Углеводородная добавка повышает электро-
проводность. Примером таких электролитов может служить раст-
вор А1С13 и ЫАШд в смеси Б и ТГФ [970, 941]. Превосходные
осадки алюминия с выходом 97—100 % были получены при моль-
ном соотношении А1С1з LiAlH4 = 3 : 1 даже при 10 А/дм2 (объем-
ная доля бензола не более 50 %).
Из эфирногидридных электролитов алюминирования получе-
ны сплавы алюминия с различными металлами (Be, Mg, In, Ti,
As, Sb) [302, 1271, 753, 301]. Механизм сокристаллизации алюми-
ния с легирующими компонентами определяется во многих случа-
ях долей последнего в формируемом осадке. Приемы получения
сплавов разнообразны. Покрытия повышенной микротвердости с
содержанием титана до 0,2 мае. % получены в простом эфирногид-
ридном электролите с использованием титанового анода [302]. Ча-
сто используются смешанные алюмогидридные ванны. Так, для по-
лучения сплавов алюминия с бериллием (до 57 % Be) применялся
раствор смеси Ве(А1Н4)2+ВеС12 в этиловом эфире [1271]. Сплав
алюминия с магнием (7 % Mg) получен из эфирного раствора, со-
держащего MgBr2, А1Вг3, ЫА1Н4. Твердость такого сплава ниже,
чем электролитического алюминия [753]. Для всех полученных из
гидридной ванны сплавов характерна общая черта — напряжен-
ность и хрупкость. Часто они формируются в виде порошка, поэто-
му процесс их электроосаждения имеет лишь теоретический ин-
терес.
Эфирный электролит алюминирования представляет собой рас-
твор исходной для синтеза всех эфирных электролитов системы га-
логенид алюминия — диэтиловый эфир [414, 282, 289, 177, 104].
Основным компонентом эфирных электролитов является кислот-
ный комплекс Виберга—Брауна НЭ+А1Х4_, где X—С1 или Вг. Сог-
ласно данным школы Левинскаса, восстановление алюминия из
этого комплекса с образованием белых плотных алюминиевых
осадков происходит при определенной основности среды, более
высокой, чем обеспечивает комплекс Виберга—Брауна.
Основность среды в эфирных электролитах обычно обеспечива-
ется введением депротонирующих компонентов — гидридов, орга-
нических оснований и др. [702, 24, 284, 970]. В эфирных распла-
вах основным компонентом, ответственным за электрокристаллиза-
цию алюминия, являются комплексные катионы эфирата А1Х2-
•эфир+. При соотношениях галогенид алюминия — диэтиловый
эфир, исключающих присутствие свободного эфира, уже в отсут-
ствие депротонизирующих компонентов только под действием то-
ка поддерживается необходимая степень основности (количествен-
но выражаемая быстрым понижением предельного тока водород-
ной волны кривой поляризации), обеспечивающая осаждение вы-
сококачественного кристаллического алюминия. Побочные про-
цессы при электролизе не возникают.
150
Эфиратные расплавы полностью термически устойчивы при тем-
пературах ниже 90 °C. Процесс электроосаждения алюминия осу-
ществляют при комнатных температурах. Так, при использовании
•системы А1Вг3—(С2Н5)2О в качестве эфиратного электролита алю-
минирования в интервале комнатных температур удобно приме-
нять расплавы состава 30—40 % эфира. Тщательная герметиза-
ция ванны на требуется, оптимальная плотность тока 1—2 А/дм2.
Электролиз ведется без перемешивания. Образуются плотные бе-
лые покрытия алюминия толщиной 12—15 мк, при реверсировании
тока — до 30 мк на различных катодных подложках. Выход по то-
ку 99—100 %. Ванны на основе эфирата бромида алюминия по
гальванотехническим качествам превосходят электролиты эфирата
хлорида алюминия [177]. Для легирования осадков алюминия в
эфиратных электролитах применяют растворимые аноды [289,
104]. Ванна корректируется добавками плавленого галогенида
алюминия.
Эфирноаммиачный электролит алюминирования принадлежит
к группе так называемых эфирнодативных электролитов, которые
содержат в составе, кроме эфирата галогенида алюминия, свобод-
ный эфир и L-оонование, необходимое для повышения основности
среды [702, 282, 176]. В качестве оснований могут служить али-
фатические либо гетероциклические амины [24, 284] или аммиак.
По гальванотехническим данным эфирноаммиачный электролит
превосходит другие электролиты данной группы. При взаимодей-
ствии с эфирным раствором галогенида алюминия аммиак не пол-
ностью связывает катион Н+-эфир. Поэтому условия восстановле-
ния катионов А1Х2_-Э+ в эфирноаммиачном электролите остаются
близкими к условиям, характерным для эфиратного раствора. При
комнатной температуре после проработки током на катоде из раз-
личных материалов (наиболее полно исследована медь) осажда-
ется белый плотный алюминиевый осадок с выходом по току,
близким к 100 %. Параллельные электроорганические реакции,
снижающие выход, начинаются лишь при плотности тока выше
0,01 А/см2. Максимальная толщина получаемых осадков 500 мк.
Разработаны условия получения осадков повышенной микротвер-
дости и износостойкости. Ванны герметизируются собственными
парами. Достоинством ванны служит корректировка электролита
в процессе длительной непрерывной работы растворением алюми-
ниевого анода.
Исследования электроосаждения алюминия из растворов в
ароматических углеводородах и их производных начаты школой
В. А. Плотникова [414, 402, 407] и продолжаются до настоящего
времени [641, 1015, 844, 472, 188, 727, 728, 1270, 1222, 1273].
Процесс выделения алюминия, а также вид и качество получаемо-
го осадка, в первую очередь, определяются природой растворите-
ля и молекулярным состоянием соединений алюминия в растворе.
Электроосаждение проводят из индивидуальных ароматических уг-
леводородов либо из их смесей. На течении катодного процесса и
151
качестве получаемого осадка алюминия сказывается не только за-
мена одного растворителя другим, но даже изомерная природа рас-
творителя. Так, при изучении электровыделения алюминия из о-, м-
и n-ксилольных растворов бромида алюминия установлено, что
оптимальные алюминиевые покрытия с наибольшим выходом по
току получаются из 40 % раствора А1Вг3 в jn-ксилоле при плотно-
сти тока 0,8—1 А/дм2 и 20—35 °C [461]. Электроосаждение алю-
миния производят из его простых галоидных солей [461, 998, 189],
чаще комплексных ионных солей типа МеА1Х4 и МеА12Х7 (Ме+ —
щелочной металл, NH4+; X—С1~, Вг~, I-), для образования кото-
рых в раствор добавляют соответствующие галогениды щелочных
металлов или аммония [1119, 1120, 1250, 1251, 1273, 402, 407].
Наиболее эффективны растворы комплексных алюмоорганических
соединений MeX-2Al(R)3 или 2A1X3-RX (Ме+ — щелочной металл;
X—С1-, Br-, I-, R — алкил) [1015, 844, 472, 1167, 1210, 1270].
Примерами могут служить электролиты на основе фторида натрия
и триэтилалюминия [472]. Подбор мольного соотношения NaF
с А1(С2Н5)3 и разбавления толуолом показали, что оптимальным
раствором для получения качественных алюминиевых покрытий яв-
ляется NaF-2Al (С2Н5)3, разбавленный толуолом в соотношении
1:1. Светлые компактные алюминиевые покрытия образуются при
небольших потенциалах катода, плотности тока приблизительно
0,8 А/дм2 и температуре 80—90°C. Выход по току 100 %. Для элек-
тровыделения и рафинирования алюминия могут быть использо-
ваны и индивидуальные жидкие комплексные алюмоорганические
соединения без разбавления ароматическим углеводородом [1015].
Этилбензоловые электролиты наиболее эффективны при ускорении
процесса осаждения алюминия по сравнению с бензолом, толуо-
лом и ксилолом [727, 728]. Алкилбензоловые электролиты исполь-
зуются также для получения сплавов алюминия с другими метал-
лами [472, 729]. Различие между потенциалами выделения алю-
миния и ряда металлов (Zn, Pb, Cd, Sn, Cu, Ag) в этих электроли-
тах в 10—30 раз меньше, чем в водных растворах, что благопри-
ятствует образованию сплавов [729].
В целом электролиты на основе ароматических растворителей
более просты в изготовлении и эксплуатации по сравнению с эфир-
ногидридными, наиболее широко используемыми промышленно-
стью. Характерной особенностью электролитов ароматического ря-
да является расслоение, наступающее в растворе при определен-
ном содержании соли [188, 189]. Верхний слой состоит в основ-
ном из растворителя, нижний из раствора соли в растворителе.
Электролиз ведут в нижнем слое, верхний при этом герметизиру-
ет нижний. Хотя тщательной герметизации ванны на основе элек-
тролитов бензолового ряда не требуется, допускаются лишь не-
большие примеси воды [1119, 1120, 1222]. Ванны стабильны, дол-
говечны, дают достаточно высокий выход алюминия по току. По-
крытия, полученные из этих электролитов, характеризуются высо-
кой чистотой, хорошими механическими и физико-химическими
свойствами.
152
Отдельную группу растворов с использованием ароматических
углеводородов составляют электролиты на основе четвертичных ам-
мониевых солей [1058, 137, 457, 458, 935, 459]. Готовят их в среде
тщательно осушенного инертного газа из обезвоженных реактивов.
Четвертичную аммониевую соль галогенида сплавляют с галоге-
нидом алюминия. Расплав разбавляют ароматическими углеводо-
родами до появления верхнего слоя ароматического углеводорода.
Наиболее исследован этиленпиридинбромидный электролит алю-
минирования в смеси с толуолом, ксилолом или бензолом. Получа-
ют его путем взаимодействия бромистого алюминия с бромистым
этилпиридинием, приводящего к образованию продукта А1гВг6-
•СгНбРуВг, способного диссоциировать по схеме [137]:
А12Вг6-С2Н5РуВг С2Н5Ру+ + А12Вг?.
Анион А12Вг7_ способен разряжаться на катоде с образованием
металлического алюминия. Оптимальное соотношение А1Вг3 и
СгНаРуВг—(1,2—9) : 1, разбавление ароматическим углеводоро-
дом до расслоения. Электролиз ведут при перемешивании элект-
ролита струей инертного газа. Оптимальный интервал катодных
плотностей тока 0,5—2,0 А/дм2. Светлые ровные компактные алю-
миниевые покрытия толщиной 30—40 мк получают при комнатной
температуре. Для получения более толстых осадков оптимальной
температурой является 35—40 °C. Катодный выход алюминия бли-
зок к 100 %. Весьма благоприятное влияние на катодный процесс
оказывает наложение переменного тока [137, 935]. Такая проце-
дура значительно повышает сцепление с подложкой, увеличивает
толщину без повышения хрупкости, уменьшает напряжение, при
этом наблюдается возрастание максимальной величины и эффек-
тивности постоянного тока. Сочетание наложения переменного тока
и перемешивания электролита дает возможность получить высоко-
качественные алюминиевые покрытия толщиной 150—200 мк. Как
и во всех остальных случаях электроосаждения алюминия, для
оптимального прохождения процесса получения качественного алю-
миниевого осадка необходимо 100 %-ное растворение алюминиево-
го анода. Для этилпиридинбромидного электролита это условие
выполняется при анодной плотности тока, не превышающей
3 А/дм2. При ia=3,0 А/дм2 выход по току быстро снижается и элек-
тролит выходит из строя. Оптимальные анодные плотности тока
0,5—1,5 А/дм2. Благоприятное влияние на толщину и качество
алюминиевых покрытий оказывают добавки некоторых органичес-
ких соединений. Из изученных наиболее эффективными оказались
диизоамиловый эфир, диметиланилин, этил-трет-бутиловый эфир и
триэтилалюминий. Основной недостаток ванн на основе четвертич-
ных аммониевых солей — необходимость тщательной гермети-
зации.
Результаты исследования электроосаждения алюминия из раз-
личных других растворителей (формамида, аммиака, ДМСО, АН
и т. п.) [702, 1140, 46, 462, 926] показали, что из некоторых раст-
153
ворителей алюминий вовсе не осаждается (аммиак), из других
могут быть выделены лишь его сплавы (формамидные растворы).
Предложены электролиты для получения композиционных по-
крытий алюминия [781, 1021, 455]. Наиболее обнадеживающие ре-
зультаты дали эфирные ванны, в частности эфирногидридные. В
качестве упрочняющего материала использованы углерод (в раз-
личных модификациях), А120з в высоко диспергированном состоя-
нии. Равномерное соосаждение с алюминием на матрице достига-
ется путем изменения количества диспергатора в ванне, катодной
плотности тока и температуры.
Остальные элементы главной подгруппы (галлий, индий, тал-
лий) могут быть электроосаждены из неводных сред как индиви-
дуально, так и в виде сплавов.
Наилучшие результаты по электроосаждению галлия достигну-
ты в глицериновых электролитах [641, 585, 580—583]. Изучено
катодное выделение галлия в чистом виде и совместно с отдель-
ными металлами (Cd, Си, Zn, Sb). Процесс сопровождается кон-
центрационной поляризацией и наиболее успешно протекает на ге-
терогенной поверхности (например, меди). Рекомендуемый темпе-
ратурный интервал — от комнатной до 60 °C. При соосаждении
увеличение плотности тока и уменьшение суммарной и относитель-
ной концентрации совместно разряжающихся ионов приводит к
снижению процентного содержания галлия в сплаве и ухудшению
качества последнего. Полученные электролитические сплавы пред-
ставляют собой твердые растворы с интерметаллическими соеди-
нениями типа GaCu4, Ga2Zn5, Ga2Cd5. Удовлетворительного качест-
ва осадки толщиной 10—15 мкм получаются при низкой катодной
плотности тока (до 10 мА/см2). Так, плотные, мелкозернистые, хо-
рошо сцепленные с основой светло-серые сплавы Ga—Cd, содер-
жащие 4—40 % Ga, получены при гк = 0,25-^5,0 мА/см2, температу-
ре 60 °C и перемешивании [585]. В глицериновых и этиленглико-
левых электролитах возможно также совместное осаждение гал-
лия и фосфора [580].
Электровыделение индия изучено в органических и водноорга-
нических растворах [702, 641, 146, 1127, 175, 301, 1076, 91]. В
большинстве случаев общей закономерностью для осуществления
процесса образования электролитических осадков индия и его
сплавов удовлетворительного качества является повышенная тем-
пература (до 60°C). В результате возможно получение жидких
сплавов, обладающих заданными свойствами. Так, глицериновая
ванна при температуре 140 °C была использована для получения
сплава индия с кадмием (75 % In) в виде отдельных шариков, на-
носимых на тонкую иглу и используемых в транзисторной технике
(температура плавления образующегося сплава 123 °C) [702]. В
общем случае преимущество жидкого сплава заключается в воз-
можности применения высокой плотности тока (до 50 А/см2).
Благодаря специфической способности индия поглощать тепло-
вые нейтроны особое внимание уделено его электроосаждению на
полупроводниковые материалы. Хорошо сцепленные с основой
154
осадки индия получены на кремнии и германии [146, Ю76]. Ин-
диевые покрытия толщиной 50—100 ммк в течение 2—3 с хорошо
формируются на кремниевой основе из электролита состава (г/л):
глицерин — 80, 1пС13—15, NH4CI — 8 с введением окислительной
добавки при температуре 160 °C. Напряжение между электродами
в проточном электролите 10—-15 В. Для получения тех же резуль-
татов при более низкой температуре (90 °C) необходимо повыше-
ние напряжения до 60 В. Сцепление с основой существенно зави-
сит от предварительной обработки кремниевой поверхности [1076].
Осажденный из ацетамидных растворов на германии p-типа ин-
дий образует омический контакт, на германии n-типа дает поверх-
ностно-барьерный переход. При определенных условиях можно по-
лучить своеобразный микросплавный р—«-переход [146].
Электроосаждение металлического таллия проведено йз отдель-
ных органических электролитов и аммиака [414, 641, 413, 424, 99,
100]. Для обоих растворителей характерно дендритообразование
на катоде. Вследствие крупнокристалличности таллиевые осадки
получаются загрязненными. Так, электролиз системы ТЦЗтг—-А1Втз
в бензоле и бромистом этилене приводит к образованию катодного
осадка, состоящего из металлического таллия и бромистого алю-
миния '[413, 424]. При расчете на одновалентный таллий выход по
току достигает высоких значений, например, в бензоловом раст-
воре при iK= 1,65-=-4,00 А/дм2, ВТК = 86 % [413].
Из водных растворов лантаноиды и актиноиды катодно либо
не осаждаются совсем, что используется для их отделения от при-
месей, либо осаждаются в форме преимущественно аморфных Гид-
роксидов [702, 414, 641, 387, 385]. Многочисленные попытки оса-
дить РЗЭ и актиноиды из неводных растворов в элементарном
виде также пока безуспешны. В лучшем случае катодные осадки
состоят приблизительно из 50 °/о металла и 50 % органических про-
дуктов. Утверждения о катодном выделении данных металлов в
отдельных работах недостаточно обоснованы. Например, в работе
[1077] предположение об электроосаждении металлического лан-
тана основывается лишь на факте взаимодействия термически об-
работанного катодного осадка с водой и кислотами. Дифракцион-
ные линии Х-лучей, соответствующие лантану, в этом осадке не
обнаружены. Необоснованы сведения также о выделении металли-
ческого урана [800]. Электролизом спиртовых растворов солей
РЗЭ с ртутным катодом удается получить амальгамы редких зе-
мель [702, 414, 464]. Максимальная концентрация РЗЭ в этих ама-
льгамах составляет 3 %, их разложением получают металл.
В целом при электролизе растворов солей лантаноидов и акти-
ноидов в неводных растворах на катоде образуются пленки окис-
ных соединений данных металлов [900, 223, 102, 587, 1145, 437, 901,
795, 881]. Такие пленки используют для получения циклотронных
мишеней, деталей радиационно-измерительных приборов и др.
Пленки должны быть осаждены на тонкие металлические (А1,
сталь, Ti, V, Та, Pt, Au) подложки и обладать высокой механичес-
кой и радиационной прочностью. Учитывая важность применения
155
/
данных пленок, хорошо разработаны методы их получения, в том
числе электролитический.
Изготовление слоев оксидов редкоземельных элементов, тория,
урана, протактиния, нептуния и транснептуниевых элементов элек-
троосаждением из неводных сред имеет неоспоримые преимущест-
ва по сравнению с водными растворами. Образующиеся на катоде
при электролизе в водной среде гидроксиды лантаноидов и актино-
идов аморфны. При дальнейшей термической обработке они обра-
зуют оксидные слои с большим количеством структурных дефек-
тов. При электролизе из органических растворов на катоде обра-
зуются кристаллические структуры, которые при прокаливании
легко переходят, теряя органическую составляющую, в кристалли-
ческие структуры оксидов РЗЭ и актиноидов. Кроме того, метод
электроосаждения из неводных растворов характеризует большая
скорость проведения процесса, полнота выделения металла, проч-
ность сцепления с подложкой слоев толщиной 1—5 мг/см2, равно-
мерность распределения покрытия на больших площадях. Наилуч-
шие результаты получены из спиртовых растворов нитратов и аце-
татов РЗЭ и актиноидов. Растворимость солей данных металлов в
органических растворителях низка, поэтому в основном применя-
ют насыщенные растворы. Из-за низкой проводимости растворов
и окисной пленки на электроде используются высокие напряжения
(порядка сотен вольт), плотности тока низкие. Большое значение
при подборе оптимальных условий осаждения имеют площадь
электродов, расстояние между ними, объем электролита, предва-
рительная обработка электродов. Катодный процесс сопровожда-
ется газовыделением, вызывающим образование неравномерной
пленки. Для уменьшения газовыделения добавляют специальные
добавки, в частности этиловый спирт [221]. Катодный продукт на-
ряду с металлом и кислородом содержит обычно азот, водород и
углерод. Результаты количественного анализа показывают загряз-
нение катодного осадка растворителем или продуктами его разло-
жения, но не образование соединений определенной стехиометрии
[1077]. При термической обработке катодного осадка происходит
уменьшение объема и перестройка кристаллической решетки, в ре-
зультате чего слои растрескиваются и осыпаются, и лишь в слу-
чае тонких слоев оказывается достаточно поверхностных молеку-
лярных сил сцепления для сохранения прочной связи с подложкой.
Для получения покрытий толщиной порядка 1—5 мг/см2 необхо-
димо многослойное нанесение продукта [1060].
Примером оптимальных условий может служить получение
циклотронных мишений из трансплутониевых и редкоземельных
элементов электроосаждением из изобутанола [221]: напряжение
600 В, концентрация по металлу — 300 мкг/мл, объем раствора
5—7 мл, площадь платинового анода — 0,25 мм2, расстояние анода
от катода — 35 мм, подложки — Al, Ti, Та, V, температура 20—
25 °C, время — 40 мин. Получены толстые (до 2 мг/см2) однослой-
ные покрытия, устойчивые при облучении ускоренными многоза-
рядными ионами.
156
Для получения окисных пленок лантаноидов и актиноидов ис-
пользуются также водно-органические растворы [900, 856, 857, 102,
387, 691, 385]. Присутствие воды обычно ухудшает качество
осадков.
Из фторидных растворов в этаноле, i-пропаноле и ацетоне элек-
троосаждение протактиния происходит анодно благодаря образо-
ванию анионом PaF72- с металлом анода комплексной соли, на-
пример А1г(Рар7)з [795]. Оксидные пленки лантаноидов и актино-
идов могут быть также соосаждены с некоторыми металлами (Sr,
Си, Ni, Со).
4.4. МЕТАЛЛЫ ЧЕТВЕРТОЙ ГРУППЫ
Электроосаждение из неводных сред металлов четвертой группы
представляет интерес прежде всего для германия и подгруппы ти-
тана, поскольку эти металлы электролитически из водных раство-
ров не осаждаются [484, 404]. Наилучшие результаты получены
в случае германия. Из спиртовых растворов (преимущественно
в двухатомных спиртах) галогенидов германия выделены тонкие
катодные пленки металлического германия [702, 641, 1225, 482,
381, 292, 650, 291, 293]. Наряду с осаждением германия на като-
де происходит выделение водорода, на последний процесс расхо-
дуется основная часть тока. Выход по току германия низкий (по-
рядка 1—3 %). Большое влияние на процесс электроосаждения
оказывает природа металлической подложки. При определенных
концентрациях галогенида германия, повышенных плотностях тока
и температурах возможно катодное образование диоксида герма-
ния [482, 196]. Пример оптимальных условий получения металли-
ческого германия: растворитель — этиленгликоль, концентрация
GeCl4 — 3—5 %, температура — комнатная, интервал плотности то-
ка 5—50 А/дм2. При этих условиях на подложках из меди, сереб-
ра, платины и алюминия осаждаются ровные, хорошо сцепленные
с подложкой, компактные германиевые покрытия светло-серого
цвета. В качестве анода использовали графит или германий, вы-
ход по току германия составляет 2 % [291, 293]. Возможно катод-
ное получение пленок германия и из других неводных сред, на-
пример из низкотемпературных расплавов ацетамида [147]. Из
растворов в ацетамиде с добавками хлорида аммония при темпе-
ратуре 90—130 °C двухвалентный германий восстанавливается, об-
разуя тонкослойные (1—2 мк) осадки, прочно сцепленные с под-
ложкой. Выход по току еще ниже, чем в спиртовых растворах
(приблизительно 0,1—0,5 %). Из-за выделяющегося водорода оса-
док германия при этом достаточно наводорожен.
Попытки получить электролитически из неводных растворов ме-
таллы подгруппы титана не столь успешны [702, 930, 1152, 294].
Электролиз проводили в многочисленных органических растворите-
лях различных классов: амидах, простых и сложных эфирах, али-
фатических спиртах, АН, ДМСО, НМ, ПК, ГМФА, хлористых аце-
тиле и тиониле, ледяной уксусной кислоте и др. Однако в боль-
157
шинстве случаев на катоде происходит образование низших сте-
пеней окисления данных металлов или же, в лучшем случае, спла-
вов [702, 641, 302, 1060, 753, 175, 301, 1152, 294]. Так, в работе
А. Бреннера [1152] с целью получения металлического титана ис-
следованы растворы TiCl3, TiCl4, TiBr4, TiF4, KyTiFg, бис-цикло-
пентадиенил титандибромида, а также их сочетаний с борогидри-
дами щелочных металлов, LiAlH4, галогенидами алюминия в 60
органических растворителях различных классов и нескольких не-
органических яеводных растворителях. И только из эфирногидрид-
ного электролита удалось получить металлические покрытия с со-
держанием титана до 6,5 %. Из подобных растворов выделены так-
же сплавы А1—Zr, содержащие до 45 % циркония.
Литературные данные по электроосаждению металлов подгруп-
пы титана из неводных сред, по выбору оптимальных растворите-
лей, степеней окисления металлов в растворе, необходимой степени
обезвоживания и т. д. весьма противоречивы. Получаемые резуль-
ты неоднозначны и часто недостаточно обоснованы [702, 641,
302, 1060, 753, 586, 46, 1125, 175, 301, 930, 1152, 484, 142, 294].
Лишь в отдельных работах получены обнадеживающие данные.
Так, совпадающие данные получены по электроосаждению титана
из растворов на основе ДМСО [46, 1182, 294]. Однако, по предпо-
ложению авторов работы [294], образование на катоде цветных
металлического вида пленок, содержащих до 20 % титана, явля-
ется вторичным химическим процессом в сульфоксидном электро-
лите. В целом сведений о получении чистых, незагрязненных ор-
ганикой катодных осадков титана, циркония и гафния в литерату-
ре нет. Удовлетворительного качества металлические осадки по-
лучены для данной подгруппы лишь в виде сплавов [302, 257, 175,
1152, 255, 256, 271]. Полученные сплавы с кадмием, медью, алю-
минием обладают повышенной микротвердостью и высокой корро-
зионной устойчивостью. Это, в первую очередь, относится к спла-
вам титана. В органических растворах соединения титана стаби-
лизируются, и на основе изучения взаимосвязи строения комплек-
сных соединений титана с их способностью к катодному разряду
возможно целенаправленное регулирование состава сплава и ско-
рости его осаждения.
Металлические осадки свинца и олова получены из многих ор-
ганических растворителей и аммиака [702, 414, 641, 261, 1161, 190,
1257, 257, 934, 1191, 251, 1249, 259]. В основном они не удовлетво-
ряют требованиям, предъявляемым к гальваническим покрытиям,
хотя осаждение преимущественно происходит количественно.
Лишь в отдельных случаях удалось получить плотные мелкокрис-
таллические хорошо сцепленные с основой осадки этих металлов.
Например, гладкие темно-серые осадки свинца получены из раст-
воров РЬ(МОз)г и РЬС12 в ДМФ при плотностях тока не выше
3 мА/см2 и температуре 40° [1249]. Хорошего качества осадки оло-
ва серебристо-белого цвета получены из электролита SnCl4-5H2O
в ЭГ при содержании олова 18 г/л и плотностях тока 0,2—
1,5 А/дм2 [482]. Но из этиленгликолевых растворов, содержащих
158
Sn2+, образуются губчатые осадки при плотностях тока зна-
чительно более низких — 0,2—0,3 А/дм2. В некоторых растворите-
лях, например, метаноле, SnCl4 в виде анионных комплексов пере-
носится к аноду [515].
Более высокого качества получены из неводных растворов спла-
вы свинца и олова [702, 1060, 257, 251, 579]. Так, качественный
сплав олова с никелем (65 °/о Sn) выделен из растворов хлоридов
данных металлов в этаноламине [1060]. Сплав свинца с медью
толщиной 16—19 ммк электроосажден в ТГФ. Сплав в зависимости
от условий электролиза содержит 78—93 % свинца [251]. В целом
неводные растворы для электроосаждения свинца и олова, а так-
же их сплавов, по-видимому, малоперспективны.
4.5. МЕТАЛЛЫ ПЯТОЙ ГРУППЫ
Металлические элементы подгруппы V-A — мышьяк, сурьма и вис-
мут — электролитически выделены из многих органических раст-
ворителей [702, 414, 641, 1272, 934, 406, 99, 100, 1191, 415, 234,
300]. Попытки электроосадить эти металлы из аммиачных рас-
творов из-за низкой растворимости в аммиаке их солей не дали
обнадеживающих результатов [702, 414].
Мышьяк электролитически получить из водных растворов труд-
но из-за его неметаллических свойств. Кроме того, его электровы-
деление из воды часто сопровождается образованием на катоде
мышьяковистого водорода, осадки, как правило, аморфны. Подбор
подходящих электролитов и оптимальных условий в неводных сре-
дах ведется в основном для получения гальванических покрытий
мышьяка [1062, 300]. Получены покрытия хорошего качества и
толщины при высоком катодном выходе в широком диапазоне ус-
ловий из растворов AsCh в АЦ, МЭК, диэтиловом эфире и др.
[416, 641, 1062, 497]. Так, тонкие (до 0,037 см) покрытия мышья-
ка высокой частоты (99,5 As) получены из растворов AsCh (20 —
40 %) при плотностях тока 0,02—0,03 А/см2 на железных и сталь-
ных подложках. Выход по току при расчете на трихлорид мышьяка
составляет 80 % [1062]. Такие покрытия могут быть использованы,
например, в качестве связующего звена между металлом и стек-
лянными эмалями. Толщина покрытия из водных растворов в ана-
логичных условиях достигает лишь 0,0012 см.
Сурьма в кристаллическом виде электролитически может быть
получена как из водных, так и из неводных сред. К сожалению,
нередко в зависимости от условий электролиза, в частности от кон-
центрации ионов сурьмы в растворе и температуры, на катоде
осаждается металл в смеси с солями Sb (III), что приводит к об-
разованию взрывоопасного осадка. Получение таких осадков ха-
рактерно для обеих сред. Осадки имеют губчатую форму и аморф-
ную структуру, после взрыва переходят в обычную кристалличес-
кую форму. Электроосаждением сурьму получают из многих не-
водных растворителей: спиртов, аминов, кислот, эфиров, Б, АЦ, НМ
[702, 414, 641, 146, 1218, 1272, 406, 99, 100, 1191, 415]. Использу-
159
ются обычно галогениды трехвалентной сурьмы в индивидуальном
виде [414, 1272, 234] или в виде двойных солей с галогенидами
щелочных металлов и алюминия [406, 99, 100, 1191]. Электролиз
растворов Sb (V) приводит частично к восстановлению Sb (V) до
Sb (0) и до Sb (Ш), например при электролизе SbFs в HSO3F
[1272]. Для обоих валентных состояний сурьмы электролиз в
большинстве случаев происходит количественно. При малых плот-
ностях тока и непродолжительном электролизе при комнатной тем-
пературе осадки сурьмы получаются ровные, хорошо сцепленные
с основой, с черным металлическим блеском. Повышение плотно-
сти тока, температуры и длительности электролиза способствует
образованию чешуйчатых губчатых порошкообразных осадков.
Электровыделение висмута из водных растворов, как и в слу-
чае мышьяка, затруднено, прежде всего склонностью катионов
трехвалентного висмута к гидролизу с образованием частиц ВЮ+
и ВЮН2+, присутствие которых в растворе не позволяет получить
катодные осадки висмута хорошего качества. Попытки осадить
висмут из спиртовых растворов, растворов в АЦ, ледяной уксус-
ной кислоте, Б, НБ тоже не привели к сколько-нибудь значитель-
ному успеху [702, 414, 641, 934]. В большинстве случаев выход по
току висмута низкий, осадок плохого качества, как и в воде, часто
образуются губчатые или порошкообразные осадки.
Все три металла подгруппы V-A способны в неводных средах
соосаждаться на катоде с другими металлами с образованием ср-
ответствующих сплавов. Например, сплавы алюминия с мышьяком
и сурьмой образуются в эфирногидридном электролите [301], спла-
вы сурьмы и галлия в глицериновом электролите [583]. Имеются
данные об осаждении фосфора с отдельными металлами [580].
Галогениды мышьяка и сурьмы, а также фосфора способны
с некоторыми соединениями образовывать настолько прочные
комплексы, входя в их анионную часть, что при электролизе их
перенос осуществляется в анодное пространство. Таковы комплекс-
ные соединения хлорида сурьмы с хлоридом йода [571], хлорида
фосфора с хлористым тетраметиламином и йодом [572, 573], хло-
рида мышьяка с нитрозилхлоридом [1014].
Относительно подгруппы ванадия в литературе имеются лишь
сведения о неудачных попытках выделить эти металлы электроли-
зом из неводных растворов [702, 641, 1182, 112]. Возможно толь-
ко их соосаждение с другими металлами. Так, плотные блестящие
никель-ниобиевые покрытия толщиной до- 1 мк с содержанием ни-
обия до 1 % получены из спиртовых растворов (EtOH, РгОН и
ВЮН). Состав электролита: NiCl3 —0,1—0,3 г/л; NbCl5— 40—
100 г/л; С2Н5ОН — 96—100 %. Выход по току 0,5—1 %, iK=
= 0,2 А/см2. Хрупкие покрытия можно получить с содержанием
ниобия до 10 %, выход такой же низкий [112]. В качестве легиру-
ющей добавки соосаждены также ванадий и тантал [1125, 271,
610]. В патентной литературе имеются сведения об электролити-
ческом выделении металлов подгруппы ванадия в чистом виде [46,
249, 669]. Металлы осаждаются в виде тонких пленок из сложных
160
органических электролитов. Пример состава электролита и ре-
жима работы для получения ниобиевой пленки: NbCls —10—
15 г/л, метанол — 300—500 мл/л, пропилацетатный эфир — 20—
30 мл/л, бензол — 500—600 мл, температура — 40—50 °C. Катод-
ная плотность тока 5—15 мА/см2, время электролиза—10 мин.
В качестве катода служила сталь, в качестве анода—ниобий. Тол-
щина пленки составляет 1 ммк [249].
4.6. МЕТАЛЛЫ ШЕСТОЙ ГРУППЫ
Из элементов шестой группы электроосаждением из неводных ра-
створов получены в элементарном состоянии селен и теллур, пред-
приняты многочисленные попытки по электровыделению металлов
и сплавов подгруппы хрома. Систематическое изучение электрохи-
мии селена и теллура, в частности электролиза их соединений в
органических растворителях, началось в 50—60-х годах нашего ве-
ка в связи с интенсивным использованием их в полупроводнико-
вой технике. Показана возможность электролитического получения
селена и теллура из растворов в спиртах, кислотах, смесях спир-
тов с бензолом и его производными.
Наилучшие результаты для селена получены в алифатических
спиртах, уксусной кислоте и этиленгликоле [89, 330, 26, 332]. Элек-
тролизу подвергают растворы SeCl4. Образующиеся на катоде
осадки селена в зависимости от условий электролиза, в первую
очередь, от температуры, имеют аморфную или кристаллическую
природу. Так, из электролитов, нагретых до 45—60 °C, выделя-
ется кристаллический селен, однако при этом электролит претер-
певает изменения и электролиз прекращается. Выход по току не
превышает 15 %, но в катодных осадках не обнаруживаются про-
межуточные продукты восстановления SeCl4. При добавлении в
электролит ароматических углеводородов выход по току возраста-
ет в два раза. Например, при прибавлении к раствору четырех-
хлористого селена в метаноле до 10 % ксилола выход по току до-
стигает 27,6 % [26].
Плотно пристающие к поверхности угольных и титановых ка-
тодов осадки металлического теллура получены из спиртовых, ук-
суснокислых, этиленгликолевых и водно-спиртовых (воды не более
2 %), кислых растворов ТеС14. Однако в составе всех электроли-
тических осадков теллура обнаруживается хлор, содержание кото-
рого зависит от условий электролиза и природы электролита. Наи-
большее количество хлора (до 7 %) находится в осадках, полу-
ченных из уксуснокислых растворов, наименьшее (до 0,04 %) —
из этиленгликолевых. Электроосаждение теллура из уксуснокис-
лых растворов может быть полезным для получения чистого тел-
лура с заданным содержанием хлора, что часто необходимо для
улучшения электрофизических свойств теллура, а также для нане-
сения слоев теллура с ТеОг любой толщины на поверхности гра-
фита и угля [8, 9]. Причиной появления хлора в осадках является
11 - 4-653
161
1
частичное неполное восстановление ТеС14, вследствие чего ТеС14
находится в катодном продукте в виде механической приме-
си. Пример оптимальных условий: 0,5—1,0М раствор ТеС14 в
уксусной кислоте, 1к = 5ч-10 мА/см2, температура 5—10°С. Об-
разуется плотный осадок теллура с низким выходом по току
[8, 9].
Теллур также способен в неводных растворах соосаждаться с
отдельными металлами, например с медью в деаэрированных ра-
створах ТеС14 и СнС12 в безводной уксусной кислоте, содержащей
LiCl [330]. Анодным растворением технического теллура в раст-
ворах соляной кислоты в уксусной кислоте можно приготовить
электролит, пригодный для электрорафинирования теллура [8, 9].
Катодные осадки теллура, полученные из указанного электроли-
та, по данным спектрального анализа имеют более высокую чисто-
ту по механическим примесям, чем исходный теллур.
Электролитическое выделение хрома из водных растворов тща-
тельно изучено и широко используется для получения разнообраз-
ных гальванопокрытий на других металлах. Преимущества его
электроосаждения из неводных сред заключается прежде всего
в увеличении скорости процесса (например, скорость осаждения
хрома в ДМФ в 6 раз выше, чем в стандартных водных электро-
литах), а также часто в улучшении кроющей и рассеивающей спо-
собности. Наилучшие результаты достигаются в случае растворов
солей трехвалентного хрома в диполярных апротонных раствори-
телях в смесях с водой. Количество воды варьируется в весьмц
широких пределах. Для улучшения качества осадка и увеличения
выхода по току в электролит добавляют различные буферирую-
щие и комплексообразующие вещества. Для получения хороших
осадков без трещин плотность тока должна быть 5—15 А/дм2 [641,
668, 669, 736].
Пример оптимальных условий: твердые блестящие осадки хро-
ма толщиной до 6 мк получены из водно-диметилформамидного
электролита (1:1) СгСЦз с добавками NH4C1, NaCl, Н3ВО3, анод
угольный, Zk=7-j-10 А/дм2, температура комнатная, напряжение
на клеммах 10—25 В, ВТК = 404-50 % [668]. Доброкачественные
осадки хрома могут быть получены также из электролита, содер-
жащего 100 г/л СгС13 и 20 мл/л воды в ацетоне при iK=14-
4-10 мА/см2 и 15 °C [732].
При добавлении в электролит солей некоторых металлов по-
следние соосаждаются о хромом, образуя сплавы [732, 733, 736,
658]. Так, при электролизе водно-диметилформамидного раствора
(соотношение Н2О : ДМФ изменяется от 8:20 до 40:60), содер-
жащего СгС13^0,8 М, FeCl2—0,1—1 М, NH4C1 —0,2 М, Н3ВО3 —
(0,1—1,0М) и диалкилфталат с 5—7 атомами углерода в алкиль-
ной группе, при гк=24-50 А/дм2, рН= 1,04-3,5 и температуре 20—
40 °C с воздушным перемешиванием образуются сплавы с содер-
жанием хрома от 18 до 57 % в зависимости от плотности тока и
концентрации железа в растворе. Выход по току колеблется от
24 до 46 %. Толщиной до 5 ммк осадки блестящие беспористые,
162
при большей толщине становятся матовыми и появляются микро-
сфероиды [658]. Получены также тройные сплавы, хрома, например,
с никелем и железом [733]. Состав и свойства сплавов можно ре-
гулировать при помощи буферирующих и комплексообразующих
добавок.
Имеются литературные указания о получении металлического
хрома из аммиачных растворов [702, 414].
Сведения об электровыделении из неводных сред остальных ме-
таллов подгруппы хрома противоречивы. Исследования по элек-
троосаждению этих металлов многочисленны, особенно в случае
вольфрама. Однако в основном получены отрицательные результа-
ты. По А. Бреннеру [700—702, 704], металлы «неводной» группы
по критерию невозможности их электроосаждения из водных раст-
воров подразделяются на два класса: металлы, которые не могут
быть осаждены из водных растворов, поскольку потенциалы их
выделения слишком отрицательны; металлы, имеющие потенциалы
электровыделения, теоретически достижимые в водных растворах,
но ионы которых не реагируют (не активны) на катоде. Металлы
первого класса (например, щелочные) довольно легко могут быть
осаждены из растворителей, содержащих ион водорода, менее
подвижный, чем у воды.
При электроосаждении металлов второго класса возникают
значительные трудности, поскольку не существует критерия для
предсказания типа ионов, способных реагировать катодно в дан-
ной среде. По-видимому, молибден и вольфрам принадлежит ко
второму классу металлов «неводной» группы. Для успешного ре-
шения проблемы их электроосаждения из неводных сред без опре-
деляющего критерия, очевидно, необходимо исследовать довольно
широкий круг органических растворителей и несколько десятков
соединений этих металлов.
Из изученных на сегодняшний день композиций лишь в случае
некоторых растворителей возможно получение молибдена и воль-
фрама [414, 586, 46, 279, 669, 725]. Так, в работе А. Левинскаса
[279] указывается, что из формамидных растворов высокой сте-
пени чистоты молибден и вольфрам могут быть электролитически
выделены из анионов ЭО42- на медных катодах. Для успешного
электроосаждения растворы должны быть выдержаны не менее
месяца, в таких растворах практически отсутствуют комплексные
соединения низших степеней окисления молибдена или вольфрама.
Если подвергнуть электролизу раствор, содержащий 1—5 г молиб-
дата натрия на 100 мл формамида, при токе 0,02—0,08 А/дм2, то
за 30—60 мин катод покрывается коричневым осадком сложной
смеси соединений молибдена и формамида (анод—графит). По
данным спектрального анализа основой его является молибден.
Добавка сульфат-ионов позволяет в некоторой мере разделить
процесс выделения тонкого металлического слоя молибдена и про-
цесс образования побочных продуктов. Электроосаждение молиб-
дена быстро прекращается, и при дальнейшем электролизе на ка-
тоде идет образование побочных продуктов. Изменение концентра-
11* 163
ции SO42~ и своевременное выключение тока позволяет получить
на катоде чистую поверхность молибдена (0,05—3,80 мг на 10 см2
поверхности) в течение 5—10 мин со средним выходом по току
10—12 %. Кроме SO42- процесс образования катодных продуктов
значительно тормозят и такие добавки, как ЭДТА, ацетамид, суль-
фониловая кислота, бензолсульфамид и особенно сильно — п-аце-
тамидбензойная кислота в присутствии SO42- или ацетамида. В
последнем случае время осаждения чистого молибдена может быть
доведено до 30—35 мин. По данным работы [725], молибден до
металлического состояния восстанавливается легче из пентагало-
генидов, но и здесь получены лишь тонкие пленки молибдена из
формамидных и гидразиновых растворов M0CI5, после чего про-
цесс обрывается. Возможно электроосаждение молибдена с от-
дельными металлами [725].
Тонкий серебристый слой металлического вольфрама может
быть получен из формамидного раствора вольфрама натрия с до-
бавкой ДМФ при плотности тока 0,035—0,080 А/дм2 в течение 10—
' 15 мин. После образования тонкого вольфрамового покрытия про-
цесс обрывается, как и в случае молибдена [279].
4.7. МЕТАЛЛЫ СЕДЬМОЙ ГРУППЫ
Сведения об электроосаждении из неводных сред металлов седь-
мой группы относятся в основном к марганцу. Марганец электро-
осажден из нескольких органических растворителей (ФА, ДМФ,
ДМСО, ДМАА, АН, ПК, АЦ, спирты) и их смесей с водой [641,
46, 669, 1139, 1069]. Наиболее детально процесс электроосажде-
ния марганца изучен в амидных растворах. Электролизу подверга-
ют в большинстве случаев растворы МпСЬ. В формамидных рас-
творах хлорид марганца по сравнению с водными растворами об-
ладает более высокой электропроводностью и выходами по току.
Так, используя ванну диафрагменного типа, где в качестве като-
лита применяется формамидный раствор МпС1г, а анолит — водный
раствор NH4C1 при следующих оптимальных условиях: концентра-
ция марганца в формамиде—10—15 г/л при pH 7,5; концентра-
ция NH4C1 — 90—100 г/л при pH 2,5; iK=50 мА/см2; температура
около 35 °C, анодом служит графит, катодом — сталь, получают
качественные осадки марганца чистотой 98 %, выход по току сос-
тавляет 76 % [1139]. Качественные прочные осадки марганца
осаждены также из насыщенных растворов его роданида в жид-
ком аммиаке при плотности тока 0,167 А/дм2 с выходом по току
97,8 % [414]. При этом использовался марганцевый анод, содер-
жащий 1,48 % железа. В катодном осадке примеси железа отсут-
ствовали.
Из водно-глицериновых растворов перрената калия с добавка-
ми сульфата аммония и серной кислоты получены качественные
покрытия рения. Режим электроосаждения рения из данного элек-
тролита рекомендуется для практического использования [165].
164
Ж 4.8. МЕТАЛЛЫ ВОСЬМОЙ ГРУППЫ
В' Изучение процессов электроосаждения металлов восьмой группы
Ж из неводных растворов ограничивается в основном триадой желе-
В. за. Никель, кобальт и железо электроосаждены катодно из орга-
В нических растворителей различных классов [414, 702, 75, 1234, 258,
В 46, 934, 387, 437, 732, 733, 669, 658, 579, 235, 236, 192, 109, 477,
В 240]. Процесс осаждения, как правило, сопровождается катод-
S ным торможением (особенно высоким в случае никеля и кобаль-
В та), вызываемым адсорбцией молекул органического растворите-
В ля на поверхности металла. Вследствие этого все три металла
ж склонны образовывать порошко- и дендритообразные осадки. Ka-
ff чество осадкй сильно зависит от состава электролита, плотности
В тока, температуры и других условий электролиза. Гладкие хороше-
го качества гальванические осадки чаще всего получаются при
В* низких плотностях тока, комнатной температуре из электролитов,
В содержащих различные добавки. Лучшие результаты получены в
В электролитах на основе диполярных апротонных растворителей
В [414, 1234, 244, 490]. Обычно качество осадка в одном и том же
В растворителе меняется в широких пределах в зависимости от сос-
В тавляющих его компонентов. Так, из ванны, представляющей со-
В бой раствор Ni(CF3COO)2 в ДМФ, при электролизе осаждается
В черный порошок никеля, при добавлении в ванну NH4C1, Н3ВО3,
В сульфамидной или хлоруксусной кислот образуется хорошо сцеп-
В ленный с основой светлый и гладкий осадок никеля при тех же na-
ff раметрах электролиза [1234]. В некоторых случаях электровыде-
В ление гладких блестящих металлических осадков и рыхлых высо-
В кодисперсных определяется соотношением между плотностью тока,
В концентрацией раствора и продолжительностью электролиза. Так,
В при электроосаждении кобальта из ацетоновых растворов галоге-
В нидов кобальта правомочна зависимость, установленная для вод-
В ных растворов c=aix°>5 (где с—концентрация соли кобальта, i —
В критическое значение плотности тока, выше которого наступает
и выделение металла в высокодисперсном состоянии, а — эмпириче-
В ская постоянная) [235, 236].
Е Наложение переменного тока закономерно способствует осаж-
Ц дению плотных гладких металлических покрытий при плотностях
К тока, значительно выше обычных (до 70—100 А/дм2) [250, 54].
К Высококачественные осадки железа из неводных растворов не
№ получены. В основном они плохо сцеплены с поверхностью подлож-
I ки [414, 490]. Однако количественно осадить железо из неводных
г сред можно. В частности, диметилформамидный раствор хлорного
| железа используют в электроаналитической химии для отделения
I его от иттрия. Электролиз раствора FeCl3 в ДМФ при 50 °C при-
I водит к 100 %-ному восстановлению железа на омедненном элек-
| троде Фишера [430].
Г Никель и кобальт при электроосаждении из органических рас-
| творителей способны образовывать качественные гальванопокры-
I тия. Примером может служить электролит, состоящий из 250 г
165
фторбората никеля в 1 л ледяной уксусной кислоты при катодной
плотности тока 1—30 мА/см2 и температуре 20 °, позволяющий по-
лучить блестящие эластичные осадки никеля, которые обладают
хорошим сцеплением с основой. Такие же результаты получены из
раствора 200 г фторбората никеля в ацетоне при 1К=1,5—
20 мА/см2 и температуре 15 °C [732]. Качественные осадки кобаль-
та получены из формамидных и ацетамидных ванн [414].
Все три металла в органических электролитах способны элек-
тролитически образовывать сплавы с другими металлами [1060,
733, 658, 579, 348]. Применяющиеся электролиты в большинстве
своем сложны по составу, основой их являются смешанные орга-
нические или водно-органические растворители. Однако сплавы,
полученные из этих растворов, обладают ценными физическими и
механическими свойствами. Таковы электролиты для получения
сплавов никеля и кобальта с оловом [579], железа с хромом
[658], тройного сплава Fe—Сг—Ni [733].
Использование органических электролитов представляет воз-
можности интенсификации процессов электроосаждения компози-
ционных покрытий и получения их с высоким содержанием частиц
второй фазы. Изучено формирование композиционных электрохи-
мических покрытий (КЭП) никеля и кобальта из неводных галь-
ванических ванн на основе апротонных (АН, кетоны, ДМФ, ДМСО,
ЭА, ПК, ТГФ, трибутилфосфат, Ру) и протолитических (алифати-
ческие спирты) растворителей. В качестве второй фазы применя-
лись в основном высокодисперсные порошки карбидов кремния и
титана, металлического молибдена. Установлено, что содержание
частиц второй фазы в никелевых и кобальтовых КЭП является
функцией природы металла, растворителя и факторов, определяю-
щих вероятность столкновения и время контакта частиц с- поверх-
ностью катода (степень дисперсности, концентрация в объеме
электролита, плотность тока, режим перемешивания и др.). Ско-
рость осаждения и состав образующихся КЭП из апротонных рас-
творителей определяется их способностью к координации в раст-
воре и на поверхности металла и имеет оптимальную величину в
растворителях средней донорной силы с пониженной адсорбцион-
ной способностью. В электролитах на основе спиртов скорость
формирования КЭП (iK) снижается в ряду СНзОН>С2Н5ОН>«-
СзН7ОН>н-С4Н9ОН>цзо-СзН7ОН, оптимальные условия — ма-
лая адсорбируемость и основность растворителя, небольшие разме-
ры его молекул и высокие сольватирующие свойства. Разработаны
практические рекомендации по технологии осаждения из органи-
ческих электролитов качественных КЭП кобальта и никеля
с высокими физико-механическими свойствами [244, 250, 54,
240].
Органические растворители используются также при бестоко-
вом осаждении металлов восьмой группы, в частности никеля [258,
109, 477]. Химическое осаждение никеля проведено из водно-спир-
товых и ацетамидных растворов путем восстановления ионов ни-
келя формиатами. Бестоковое осаждение металлов представляет
166
значительный интерес, поскольку позволяет получать покрытия по-
вышенной чистоты.
Никель, кобальт и железо дают хорошего качества гальвани-
ческие осадки при электролизе растворов их солей в жидком ам-
миаке. При этом необходимо использовать низкие плотности тока
(~0,1 А/дм2) и умеренно концентрированные растворы солей.
Наиболее высокие плотности тока (порядка 1 А/дм2) допустимы
при получении электролитического кобальта. Примером таких
ванн может служить электролиз раствора Ni(CNS)2-4NH3 (0,156г
соли в 4 см3 аммиака). Хорошо сцепленные с основой осадки ни-
келя, по цвету и прочности похожие на платиновые, получаются
при плотности тока 0,05 А/дм2 (напряжение—1,25 В) с выходом
по току 92,3 %. Газовыделение при этом не наблюдалось
[702, 414].
Из жидкого аммиака получены также электролитические осад-
ки палладия и платины [414]. Есть указания невозможности элек-
троосаждения из аммиачных растворов родия и осмия [702].
ПРИЛОЖЕНИЕ. Таблица 1. Свойства индивидуальных растворителей \ .
Растворитель ^шг °C ^кип- °с d-Ю-3 , кг/м’ (г/см*) Пр Вязкость ц-Ю’, Па-с Диэлектрическая проницаемость
н2о HF HNO3 NHa n2h< РОС13 SOC12 Анизол Анилин Ацетон Ацетонитрил Бензол Бензонитрил Бромбензол Бромистый этил Бутанол Бутиловый эфир Г ексаметнлфосфотриамид Гексан Гептан Глицерин М,М-Диметилацетамид Диметилсульфоксид N.N-Диметилформамид 1,4-Диоксан 1,1-Днхлорэтан 1,2-Дихлорэтан Диэтиленглнколь Диэтнлкарбонат •«-Ксилол N-Метилацетамнд Метанол N-Метилпирролидон N-Метнлформамид Метилцеллоз’ольв Метнлэтилкетон Нитробензол Нитрометан) Пиридин Пропиленкарбонат Пропанол «зо-Пропанол Сульфолан Тетрагидрофуран Толуол Трибутилфосфат Триэтнламин Уксусная кислота Уксуснобутиловый эфир Уксусноэтиловый эфир Формамид Фурфурол Хлорбензол Хлороформ Циклогексан Четыреххлорнстый углерод Этиленгликоль Этиленднамин Этиловый спирт 0,00 —89,37 —41,59 —77,7 2,0 1,13 —104,5 —37,5 —5,98 —95,35 —45,72 5,531 —12,9 —<30,82? —118,6 —89,53 —95,37 7,20 —95,34 —90,60 18,18 —20 18,4) —61 11,80 —96,6 —35,87 —10,45 —43,0 —47,87 29,5 —97,49 —16 —3,8 —85,1 —83,4 5,76 —28,5 —41,8 —49,2 —126,2 —89.5 28,86 —65 —94,99 —80 —1'14.7 16.63 —73,5 —83,97 2,55 —36,5 —45,58 —63,55 6,55 —22,99 —12,6 11,0 —114,5 100,00 19,51 83,901 —33,38 113,5 1081 75,6 153,7 184,40 56,24 81,60 80,10) 190,7 155,91 38,39 117,73 141,97 235 68,74 98,43 290,0 165,0 189 152,5 101,32 57,31 83,48 о 244,33 126,8 139,10 202,4 64,51 204 180 124,4 79,50 210,8 100,81 115,58 241,7 97,15 82,40 283 65,4 110,62 289 89,35 117,7 126,11 77,11 210,5 161,8 131,69 61,15 80,74 76,75 197,85 117,0 78,32 'V 0,99704 0,9546 1,457 0,6028 1,0040 1,648 1,638 0,9893 1,0175 0,7845 0,7767 0,87368 1,0088 1,4882 1,4505 0,8057 0,7646 1,0253(20 °С); 0,65482 0,67951- 1,2582 0,9366 1,096 0,9445 1,0269 1,1679 1,24554 1,1130 0,9693 0,8599 0,9503(30 °C)? 0,78658 1,0279 0,9961 0,9596 0,7995 1,1987 1,1312 0,9779 1,1995 0,7995 0,7810 1,2615(30 °C) 0,883 0,86228 0,9726 0,7235 1,04365 0,8764 0,8945 1,1292 1,1550 1,1009 1,4799 0,77387 1,5842 1,1'099 0,8934 0,78506 1,33250 1,1574 1,3904 1,325(16 °C) 1,4644(35 °C) 1,460 1,527 (10 °C) 1,5143 1,5832 1,3561 1,3416 1,497921 1,5265 1,5577 1,42201 1,3973 1,3968 1,457О| 1,37226 1,38512 1,4735 1,4360 1,4762 1,4269 1,42025 1,4145 1,4425 1,4461 1,3829 1,4946 1,4277(30 °C) 1,32663 1,4680 1,4300 1,4017 (20 °C) 1,3761 1,5506 1,3795 1,5067 1,4195 1,3835 1,3747 1,4817(30 °C) 1,4050(20 °C) 1,49414 1,4226(20 °C) 1,4010(20 °C)i 1,36995 1,3941 (20 °С)| 1,3698 1,4468 1,5162(20 °C) 1,5221 1,4433 1,42354 1,4576 1,4305 1,4540 1,35941 0,8903 0,256(0°) 0,749 0,135 0,905 1,15 1,008 3,77 0,302 0,341 0,6028 1,24 1,05 0,379 2,46 0,644 3,34 0,2923 0,3903 945 0,906 1,96 0,796 .1,253 0,505 0,785 ВО 0,748 0,581 3,885 (30 °C), 0,5428 1,666 1,65 30,84 0,407 1,823 0,627 0,8826 2,48 2,004 2,06 9,87(ЗО°С)| 2,21 0,5516 3,39 0,341? 1,13 0,688 0,426 3,30 1,49 0,756 0,540 0,898 0,901 16,79 1,54 1,092 78,35 60(19 °C) 50(14 °C)' 16,9 51,7 13,9(22 °C) 9,25 (20 °C) 4,33 6,99 20,70 36,02 2,297 25,2 5,40 9,20 17,7 3,06 29,6 1,883 1,927 42,5 38,5 46,6 36,71 2,21 10,46 10,16 29,4 2,82 (20 °C) 2,374 175,7 (30 °C) 32,63 32,0 182,4 15,9 18,4 34,82 38,6 12,3 65,1 20,1 18,3 44 (30 °C) 7,39 2,379 8,05 2,42 6,19 5,01 (20 °C) 6,02 109,5 41,7(20 °C) 5,62 4,72 2,02 2,23 37,67 12,9 24,30
168
169
Таблица 2. Параметры основности, кислотности и полярности неводных
растворителей
Растворитель DN AN BphOH E
Аммиак 69,0 —}
Ацетилхлорнд 0,7 »—t
Ацетон 117,0 12,5 42,2
Ацетонитрил 14,1) 18,9 160 46,0
Бензнлцнаннд ,15,1 — ,
Бензол, 0,1 8,2 48 34,5
Бензонитрил, ,11,9 15,5 155 42,0
трег-Бутиламин 57,9 —1 ' I —1
Вода 18,0 54,8 156 69,3
Гексаметанол 138,8 10,6 471 40,9,
Гексан —, 0 30,9
Гидразин 44,0 — —, •
М,М-Днметнлацетамнд 27,8 13,6 343 43,7
Диметнлсульфокснд 29,8 19,3 362 45,0
Диметнлформамид 26,6 16,0 291 43,8
Днметокснэтан 24Д —« —
Диоксан 14,8 10,8 237 36,0
Днхлорметан ,—’ 20,4 — —
М,М-Днэтнл ацетамид 32,2 — —
Диэтнловый эфир 19,2 3,9 280 34,6
N, N-Д нэтн лф орм а мид 30,9 —, — —
Метанол 19,0 41,3 218 55,5
Метнлацетат 16,5 — ,—.
Ь1-Метнл-2-пнрролидон 27,3 13,3 V
Нитробензол 4,4 14,8 67 42,0
Нитрометан 2,7 20,5 — 46,3
Пиридин 33, V 14,2 472 40,2
изо-Пропнламнн 67,5 —< *1 —-•
Пропнленкарбонат 45,1 18,3
изо-Пропнловый спирт —< 33,5 — 48,6
Пропионитрнл ,16,1
Сульфолан 114,8 — 157 —
Тетрагндрофуран 20,0 8,0 287 37,4
Трифторуксусная кислота •—1 105,3 *“—!
Трнэтнламнн 61,0 1,4 — 33,3
Уксусная кислота —4 52,9 131 51,2
Уксусный ангидрид 10,5 1 — 1
Формамид —1 39,8 *—i 56,6
Хлороформ . , 1 23,1 , 1 39,1
Четыреххлорнстый углерод 8,6 0 32,5
Этанол !Ш,2 37,1 51,9
Этнлацетат. 17,1 9,3 181 38,1
Этилендиамнн Б5„0 — .
Этиленкарбонат 46,4 — - —
Этиленсульфнт 15,3 — — —•
170
Таблица 3. Средние числа сольватации ионов в неводных растворителях
Ион Растворитель
NH, ФА АН ТГФ ДМФ ДМСО СФ мюн ЕЮН
Li+ — 4 — —, 4 1,4 5,0 5,8
Na+ — 3,5 — 4 3 — 2 4,6 3,8
К+ — 2,4 — — 2,6 — 1,5 3,9 3,3
Pb+ — — — — 2,5 — 1,4 3,7 3,1
Cs+ — — — — 2,3 — 1,3 3,3 2,1
А13+ 6 — 2,8 — 6 6 — 6 —
Со2+ 6 — 6 — 6 — — — —
ci- — 1,3 — 0,7 — — 4,1 3,0
Вг- — — — —' 0,8 — — 3,8 2,9
I- — 1,4 — — 0,7 — -— 3,4 2,6
NO3- — — — — 0,3 — •— 3,4 2,7
cio4- — — — — 0,6 — — 2,8 2,1
Pi- — — — — — — — 4,5 2,8
Таблица 4. Величины рКДИс солей в неводных растворителях (25°C)
Соль АЦ AH МЭК MtOH EtOH HM Py СФ (30° C) Прочие раство- рители
LiCl 5,48 — 1,74 4,14 ТГФ 5,4
LiBr 3,65 — — — 2,04 — — 2,44 SO2(0°)4,57: БН 3,41
Lil 2,75 1,01 2,38 0,78 BtOH 2,84 ДМСО 1,31 БН 1,92
LiC104 2,22 1,84 — — — — 2,85 0,73 Ацетилацетон
LiNO3 1,00 1,40 2,62 2,16
LiC103 — 2,40 — 0,70 — — — —
LiPhm НАс 7,09
LiAc LiPi 2,99 2,72 3,71 1,44 . 4,20 4,08 НСООН 0,28 НАс 5,43 ТГФ 4,08
NaF ТГФ 6,20
NaCl — — — 0,66 2,00 — —. МХУ 4,01;
NaBr — 1,61 — — НСООН 1,35 БО2(0р)4,32;ДМСО1,19
Nal 2,10 — 3,44 — — 1,54 3,86 0,7 БН 1,73;
NaClO4 3,63 1,85 — 1,81 2,08 0,85 i—РгОН 2,00 Ацетилацетон
NaNO3 1,28 2,04
NaCNS — 1,94 — — — — — — ДМФА 0,90
NaPhm НСООН 0,10
Na Ac — НАс 5,03
171
Соль АЦ ah МЭК MtOH EtOH HM Py СФ (30° C) Прочие растворители
NaPi 2,87 2,19 3,47 4,0 4,37 - НБ 4,50; МХУ 3,62
КС1 — — — 0,7 2,13 — — — НАс 3,78; НСООН 1,43 SO2(0°)4,13; ЭГА 1,19
КВг — .— —— 0,49 1,94 —. — — SO2(0p) 3,85; ДМСО 1,05
KI 2,10 1,19 2,72 1,05 1,83 1.44 3,68 0,80 SO2(0°) 3,52; БН 1,89
КС1О4 3,85 1,99 3,21 1,04 — — 3,36 0,90
KNO3 — — — 1,40 — — — — ДМФ А 1,07
KCNS 2,47 1,41 — 1,11 1,68 — — — НБ 1,11; ДМФ 2,46
KBPh4 .— 0,4 — — — — — — ТГФ 4,49
KPhm НСООН—0, 0,1
КАс ,— — — — — — —— —• НАс 4,50
KPi 2,46 1,82 3,04 1,32 — — 4,00 НБ 3,10
RbBPh4 — 1.8 — — "1 — — — ,—
RbC104 2,02 —— — — —. — —
RbNO3 — — —‘ 1,24 — — — —
Таблица 5. Предельные ионные подвижности (Хо) ионов в неводных
Ион Вода АЦ АН ГМФТА ДМАА дмсо ДМФА НСООН HCN (18° С) МАА (40’ С) МФА
Н+ 349,8 100 99 15,4 32,7 . 9,20
Li+ 38,6 72,8 69,3 5,3 21,3 11,2 26,0 14,5 135,5 6,69 15,0
Na+ 50,1 78,4 76,9 5,9 25,6 13,1 29,9 19,5 132,4 8,30 21,3
К+ 73,5 80,6 83,6 6,1 25,2 13,9 30,8 23,2 151,4 8,54 21,8
Pb+ 77,8 —— 85,6 5,9 27,0 — 32,4 — 153,2 9,И —.
Cs+ 77,2 —— 87,3 6,1 27,8 — 34,5 — 158,2 9,76 24,4
nh4+ 94,5 — 5,4 35,9 — 38,7 — —— 9,82 .—
Mt4N+ 44,9 97,7 94,5 .— 34,9 18,7 38,9 —— 12,17 .—
Et4N+ 32,6 89,0 84,8 9,4 32,7 17,2 35,6 19,2 —— 11,79 23,8
Pr4N+ 23,4 70 70,3 7,1 26,2 13,6 29,2 — — 9,29 —
BUN+ 19,5 67,3 61,4 6,1 22,8 11,8 25,4 13,0 — 7,90 17,1
Ag+ 61,9 ч— 86,0 5,6 27,2 16,6 35,2 — — 8,58 —
Am4N+ — 58,8 56,0 — —— 10,4 22,9 — 7,25
Mg2+ 9,88 —
Ca2+ — — —— —— — — — 10,4 ——.
Sr2+ 10,6
Ba2+ 10,3 ,—
ci- 76,4 105,2 98,4 21,9 45,9 24,4 — —, 210,0 11,41 20,8
Br- 78,1 115,9 100,7 18,0 43,2 24,7 55,1 — 211,4 12,66 21,9
I- 76,8 113,0 102,4 17,1 41,8 24,3 53,6 32,9 212,5 14,48 23,2
cio4- 67,4 115,3 103,7 15,2 42,8 24,6 52,3 — 23,4 16,66 21,1
NO3- — 120,1 106,4 20,0 46,3 27,0 52,4 25,1 201,4 14,36 —
Pi- 30,4 85,3 77,7 11,3 31,5 12,3 57,3 — 11,75 13,1
CNS- 123 113,4 20,1 48,8 29,2 31,5 __ 205,3 15,88
ВРЙ4- 19,7 — 58,3 6,0 — 11,1 59,8 ——
Ac- F~ 40,9 85,0 107 '— — 1— -— — — —
172
Продолжение табл. 4.
Соль АЦ AH мэк MtOH EtOH HM Py СФ (30° C) Прочие раство- рителя
CsCl 2,34 BtOH 3,32
CsI — — — 1,15 1,85 —— — — —
CsCIO4 2,35 1,56 — — — '— — 0,95 —
CsBPh4 — 1,28 — — — -— — ТГФ 5,72
NH4I — — — — — — 3,62 —i BtOH 2,71
nh4pi 2,96 — — — — —— 3,55 —♦ НБ 3,84
CuBF4 1,18 — — — — — 1 —
CuBFe — 1,15 — — — —— — —4 —
AgC104 — 1,67 — — 1,56 — 2,72 1 НБ 2,54
AgNOs — 1,86 — 1,87 2,66 1— 3,03 1 БН 3,30; ДМФ 1,42
AgPii — — 3,41 — — '— 2,51 НБ 4,60
4 MgBr2 — — — — 2,33 — — —t —
Mgl2 — — —— — 2,25 — ♦ —
T1BF4 1,15 — —- — -— J —
T1C1O4 — 1,50 — —. — *— — —4 —
MgCl2 — — — — 2,78 — — — —
р растворителях (25°C, См-м2кг-экв—МО-1)
МЭК НБ нм Ру ПК MtOH EtOH PrOH СФ (30°) ТГФ ФМА ЭГ
. 63 49,6 140,8 59,5 10,8 27,7
г 60,0 — 55 24,9 7,3 39,6 17,05 9,5 4,33 46,9 8,5 2,11
61,3 16,3 58 26,8 9,2 45,2 20,3 10,4 3,61 48,2 10,1 3,11
64,5 17,6 60 32,01 12,0 52,4 23,6 11,9 4,05 49,8 12,7 4,62
11.4 56,0 24,8 — 4,16 — 12,8 —
— — 12,0 60,8 26,6 — 4,27 — 13,5 —
18,4 64 46,8 57,9 19,3 — 4,97 — 15,6 —>
77,2 17,1 54,5 43,01 14,2 68,7 29,65 14,4 4,31 — 13,4 2,97
73,6 16,4 47,6 25,5 13,3 60,5 29,3 15,05 3,95 — н,о 2,20
13,3 39,1 10,5 46,1 23,0 12,2 3,23 — 8,12 1,74
56,3 11,9 34,1 24,0 9,4 38,9 19,7 10,7 2,80 84,8 6,83 1,51
52 11,3 50,1 19,2 —— — — — —
— — — — 8,2 — — — 2,50 — — —
— — — — 34,8 — — — — — —
— — — — —. — — — — — — —
— — . — — — —
78,4 22,2 62,5 20,2 52,4 21,9 10,5 9,3 17,1 5,07
84,5 21,6 62,9 51,3 19,3 56,5 23,9 12,2 8,92 17,2 4,98
86,5 20,4 62 49,И 18,8 62,8 27,0 13,9 7,22 16,6 4,61
85,5 20,9 64 47,61 18,8 71,0 — 16,4 9,69 — —-
22,6 64 52,6 20,4 61,1 25,65 —- 6,2 17,4 4,8
16,0 44 33,7 13,0 47,1 25,0 — 5,28 9,1 2,21
22,9 62,2 27,4 — 9,64 — 17,2 —
— 8,6 36,6 — — — 40,3 6,04 —
— — — 52,0 — 39,3 —' — 11,9 —
173
Таблица 6. Растворимость (моль. %) солей в некоторых растворителях при 25 °C
Соль Метиловый спирт Этиловый спирт АНетои Аммиак Гидразин
LiCl 24,3 21,2 1,38 0,56 1 160 s
LiBr 34,05 33,16 .11,67 — __
Lil 45,11' 46,34 18,88 —-
LiC104 35,44 39,66 42,70 —
LilNOg —1 — —- 9,57
NaF 0,33 0,11 0,14
NaCl 0,762 0,051 0,000335 1,28 80 6
NaBr 5,13 1,066 18,60 370 6
Nal 14,4 11,8 13,38 59,40 s 640 6
NaClO, 11,85 5,24 19,71 —
NaNOs 0,155 0,0193 .— —< 1000 6
KF 0,099 0,080 2,35-10-* —
KC1 0,23 0,1324 7,3-10-5 0,046 90е
KBr 0,60 0,185 0,011 1,89 13,927
KI 3,29 0,54 0,46 15,71 25,147
KCIO, 0,024 0,004 0,0065 — —
KNOa — — и—1 1,8 4,26 7
RbF — — 2,1-10-* .
RbCl 0,37 0,0297 1,12-10-4 0,041 1 506
RbBr 0,486 —1 1,7- Ю-3 2,24 1 1
Rbl 1,65 0,135 13,04 1 ,
RbClO, 0,0104 0,00224 0,02985 —
CsCl 0,57 •—1 —
CsBr 0,34 —
CsI 0,47 ,
CuCl2 6,93 15,02 1,26 !2 —
CuCl — —
CuBr —. — —
CuBr2 .— — —'
Cui —— — —1
Cu(NO3)3 — - 1— — 10=
AgCl 2,59-1O~6 2,03-10-6 0,527-10~6 0,098 —,
AgBr5 0,56-IO-5 0.013-io-5 0,534 —-
Agl5 1,519- 10-e 0,049-10-6 — — —1
AgC104 — — ' — —1
AgNOa 0,707 0,837 0,157 7,94 10 е
BeCl2 256,7 е 151,16 ,1 - — — —
BeBr2 —. —- — —
MgCl2 5,23 3,05 — —.
M.gBt'2 — - —
MgC164 6,93 4,71 10,04 — —
CaClz 8,31 9,67 7 6,18-10~3 — 160 е
CaBr? 8,67 — —. 7,67-10-4 1 —
Cal3 56,0 — 15,53 0,232 1 —
Ca(C104)2 24,14 24,25 13,05 — —
Ca(NO3)2 21,35 13,13 5,66 7,68 —.
SrCl2 6,38 17 1,086 17 — — 80е
SrBr2 13,57 10,68 0,1 5,51-104 1
Sr(ClO4)2 19,16 22,61 23,32 — —
Sr(NO3)2 t— 1,87-10-3 — 5,72 50е
BaCl2 0,34 8 — — — 3106
BaBr2 4,72 2 0,56 5,1-10-3 9,74-10~4 1;i —
Bal2 — 8,28 — 1,008-Ю-31 —
Ba (CIO4) 2 22,617 14,36 7 17,46 7 — —
Ba(NO3)2 0,0062 3,17-10~4 1,11- 1О-3 5,96 30 6
174
S $ Пиридин Ацето иитрнл Глицерин Уксусная кислота Нитрометан Нитро- бензол Диэтило- вый эфир
19,95 0,1356 19,452 —. — — ,—
— 4,00 — __
— 32,08 — — 25,2 s .—
л — — —1 44,20
29,90 1,75 . —
— 2,93-1О-з .
2,5-10-“ —. 0,076 4 — —-<
— 2,27-10“2 —. —( — —
—. 8,84 — 4,8 s — —
——5 —г — —< — —
f— -» — , 0,13 , —
» 1 2,41 • 10-3 — , > 1 '
— 1,88-IO-3 4,39 2 0,0237
—. 1,178-10-2 —, —
0,12 9 0,73 18,8 8 0,113 0,00012
—< — — —» — —
—• — ——• — —« —> —
— _ —
—« 1,74-10-3 1— r— —
— 1.66-10-2 r— —
Я —1 0,459 —-• — 0,5185 — —
——• 1—< —11 — — —
— 2,91 • 10-3 II 1— . —
—1 3,85-10-3 — —-» — —
— 0,22 —
0,204 0,48 12 0,007 4 —. —
—— 5,26 12 - - —
— 1,094 12 . —— —- —
— 4,30 I2 . — —
8в » 17,4 5 0,75 I2 — — —k — —
—. — __й —
0,85 — — 1— —-- —
,—, —> -1 — r— —
—— -- — -
9,16
Яр 14,76 21,35 . 0,0274 4
138,3 s к——I < — —
185,6 s — — —( .— — 2,21 6
fe — 1 — —— 1 __
5,4 5 133,5 s —k 1,1615
— 1—- —1 — 9,65-10-
1,17 — I 8,72 4 — — —
Illi- f—.
SSaafc,-. .''
1®, - 1 1 ! — 1 1 I — .4
— — — — —- —• 8,08-1'0-
— — — 3,27 —• — —
₽,. —‘ — — 5,5 —• — —
— — — — — — 1—
— __ __
№h ; 7,0 s — r— —
— —- 4,13 2 0,0077 4 —- 0,194 —
- —... I — —
82,2 s —1 — — — —
— 1 —— — —
— — — 4,98-10-4 ' — —
2
2
175
Соль Метиловый спирт Этиловый спирт Ацетон Аммиак Гидразин
ZnBr2 । 48,38
Znl2 — -1 м 5,341-10~9
Zn(NO3)2 — —- 2,54i . .
CdCl2 0,545 0,513
CdBr2 2,11 5,11 4,18 12 4,50'7
Cdl2 16,62 12,08 6,36 6ДЗ 7
HgCl2 7,за 7,75 23,17
HgBr2 5,91 4,77 7,91
Hgl2 0,28 0,22 0,3 6906
A1C1B —
AlBr3 — —
Alls — . —
YC13 12,602 —
Y(NO3)3 —
La(NO3)3 — < .
T1C1 9,22-10—1 - -н
CeCl3 — t 30 6
CeBra —
Ce(NO3)3 — —
PrCl3 — .
Pr(NO3)3 —
NdCl3 — 7,567 .
SmCla — 1
Er(NO3)3 — t—, , i
SiF, 13,06 21 20,2121 1,7521
SnCl2 —- 14,54 12
PbF2 — 60 е
PbCl2 — ,
PbBr2 .—
Pbl2 —
Pb(NO3)2 0,133 )S 5,577-10~a 52Ю 6
SbF3 1330 5 — 553 5
SbBr8 —
BiCig — — 3,20'2 320 8
TaCl8 — — — —
Примечания: > При О °C; 2 при 15 °C; 8 при 14,7 °C; 4 при 30 °C- 8 в г/л- 8 ппи 20 °C-
при 26,45 °C; 18 при 22 °C; 18 при 23,6 °C; 17 при 6 °C; 18 при 40,5 °C; 18 при 27,2 °C;’ 20 при 36 °C;
Таблица 7. Кинетические параметры электровосстановлении металлов из неводных
Ион Растворитель Рабочий электрод фоновый электролит Ks, См-с-1
Li+ АН у-БЛ ДМА ДМС ДМСО ДМФ Hg Li Hg Li Hg Li(Hg) Hg ПТБА LiC104 ПТЭА LiClO, ПТБА LiCl ПТЭА 1,5-10—1 2,9-10-5 пью-4
176
г Продолжение табл. 6
L пиридин Ацетонитрил Глицерин ^ислотГ Нитрометан бензол' в^й’спирт
] 44,0'2 _ _ _ _
126'2 __ Ю,358 _ _ —
—_ — _—>
‘0,395 — — — —
I 4,32 _ _ _ _ 0.0013 7
1 7,86 19 — — 0,68010 — 1,85 7
1 7,25 — 0,261 —
1 6,31 — — 0,0114 — — 0,158 19
2'8,83
I 0,8 — _ — — 47,2
1 0,158 — — — —
И' 2,562 — _ _ _ —
— 2,27 7
— 0,02 6
—
1 0,5031 — — — —
I о,449 _ _ _ _ —
— 0,0263 6
1 0,682 7 — — — — —
— 0,009 е
i 6,36 2 _ — _
1,93 2 — — — — —
— 16.26
I — — 5,08 2' 0,6421 _ —
1 - - - —-
__
! 0,1301s — — — —
; 0,126'4 _ _ __ _
i 3,4-10-2 ______ —
2,00 — — — — ——
1 — — — 59,68 — 0,565
Ч — — — 23,037 — 42,56
j —— —. —-1
' — — — — — 55,7®
-4-
, при 20 °C; • при 15,5 °C; 9 прн 10 °C; 10 при 24 °C; 11 при 28,03 °C- 12 при 18 °C; 13 при 25,3 °C:
4 " при 27-33 °C.
| растворов
К°, См-с-1 а 'о> мА. см—2 1° мА-см-2 В-10», см2, с—' Литература
а — 0,98 — — [657
1 — — 2>50 — — [4791
2,44 8891
| — 0,49 15,0 — — [1244
| 1.3-10-4 0,7 — — 1,8 925]
: — 0,75 — — — 746
: — 0,52 — — — [657
12-4-653
177
Растворитель Рабочий электрод Фоновый электролит Ks, См-с—I
ПТБА
Li(Hg) ТФБТБА 1,2-10“ 3
HM - LiCl .
— LiC104 —
—. LiAlCL —
ПК Hg ПТЭА 2,5-10“ 3
ПТБА —
Li LiC104 —
LiAlCl4 —
N;rf. ТГФ LijHg) LiC104 LiC104 3-10“5
АН Hg ПТЭА 5,7-10“'
ГМФА Hg ПТЭА 2,3-10“2
ДМА Hg ПТЭА —
ДМОЭ Na NaC104 —
дмсо Hg ПТЭА —
ДМФ Na(Hg) ТФБТБА 7,4-10“3
ПК Hg ПТБА —
Na NaC104 —
Na NaAlCl4
Na NaPF6 —
K+ ТГФ Na(Hg) NaC104 —
ТМФ Hg ПТЭА —
ДМА Hg ПТЭА —
ДМСО Hg ПТЭА —
ДМФ K(Hg) ТФБТБА 4,3-Ю~3
K(Hg) KI —
ПК Hg ПТБА —
Rb+ к KA1CU —
ТМФ Hg ПТЭА —
ДМА Hg ПТЭА —
Cs+ дмсо Hg ПТЭА —
ТМФ Hg ПТЭА —
ДМА Hg ПТЭА —
ДМСО Hg ПТЭА —
Cu+ ДМФ Hg ПТЭА 1,1 ч
Cu2+ Cs(Hg) ТФБТБА 9,4-Ю~3
АН Pt ПТЭА —
(2—>1) (l->m АН Pt ПТЭА
NH3 Hg — —
ДМФ Cu LiC104 —
СН3ОН Cu H2S04 —
сн3он Cu H2SO4 —
сн3он Cu NaC104—НС1О4 —
Be2+ СН3СООН Cu NaC104—HC1O4 —
ФА Hg NaC104 —
Mg2+ ЭС Hg ПТЭА —
ДМА Hg ПТЭА —
ДМА Hg ПТЭА —.
178 дмсо Hg ПТБА
Продолжение табл. 7
К°, См-с 1 а 10, мА. см ig, мА-см £>.10», см2с~1 Литература
0,82 —. —— 657]
—. ~0,75 — — — 786]
— 0,5 — — 506]
— 0,35 — — —. 260]
.— 2,00 — — 260]
~0 ,755 —. — 1,047 [1033]
1,3-10~4 0,7 17 1,78 2,4 [925] [746, 8 09]
— ~0,80 0,50 0,87 — [1056] [950,952]
— — 9,5—37 — — [ 486]
—. — — — — 657]
I» — —- — — 657]
— — —. — 4,54 889]
0,5 — — [329
1.4-10-2 — — . ,, — 2,4 [925]
-— ~0,75 — — — [786
>1 — —— 2,0 925'
— —- 11—21 — —— [486]
— — 28 — — [681, 684]
,— 0,002 — I 1 [681, 684]
— 0,43 4—20 [48,486]
— — — — 5,2 [891
——. — —. — 5,56 [889
>1 — —. — 2,8 925
— ~0,75 — — — [786
— —• 5 — — 506
^>1 — — — — 925
— — 4,58 — — [681, 684]
— — —• — 5,2 [891
— — —. — 6,42 [889]
^>1 — — —— 3,0 925]
— — —• — 4,9 [891]
— — — — 6,68 [889
^>1 — 1 — — 3,4 925
—• — — — 1 657]
— ~0,75 — — '— [786]
— 0,40 — — — [681, 684]
— 0,64 — — — [681, 684]
— — — 18 100С ]
— 1,47 — 78]
— —. 1,75 — — 6431
—. ,— 0,12 — 643
•— — ~0,07—0,7 — — 356]
— 0,66 — — — 356]
-— — —— — 1,10 714
— — — — 3,16 88b
— — — — 1,74 [837]
— — — — 1,78 [888
— 0,35 —- — 2,7 [925]
12*
179
И°н Растворитель Рабочий электрод Фоновый электролит Ks, См-с-1
Са2+ ДМА Са Lid
Hg ПТЭА —
ДМСО Hg ПТБА —
Sr2+ ДМА Hg ПТЭА
ДМСО Hg ПТЭА —
Ва2+ ДМА Hg ПТЭА —
ДМСО Hg ПТЭА 3-10“5
ТМФ Hg ПТЭА —-
Zn2+ Вода Hg NaC104 —-
LiC104 —
Ац Hg NaC104 —
ДМСО Hg КС1О4 —
ПТБА
ДМФ Hg LiC104 2,5-10—3
ДМФ Zn(Hg) LiC104 3,1 • 10-4
РгОН Hg LiC104 16
4 %С3Н7ОН+Н2О Hg LiC104 —
ТГФ Hg NaClO4 —
ТГФ+вода (1:1) Hg NaC104 —
ТМФ Hg ПТЭА —
ФА Hg NaC104 —
ЭС Hg ПТЭА —
Cd2+ Вода Hg NaClO4 4,5-10-'
Hg LiC104 9,2-Ю- 2
Вода+АН (1:1) Hg NaC104 1,45-10~2
АН Hg NaC104 1,9-10-
ДМФ Hg NaC104 1,5-10-'
Hg LiC104 2,8-10-2
ИМФ Hg NaC104 1,2-10-'
ДМФ+N—МФ Hg NaC104 8,9-10—2
(1:1)
НМ Hg ПТЭА —
ПК Hg NaC104 ~1-10~2
СН,ОН Hg NaC104 1,3-10- 2
СН3ОН+вода (1:1) Hg NaC104 2,8-10-2
ТМФ Hg ПТЭА —
ЭС Hg ПТЭА —
Al?+ ЭР Al(Hg) — —
Al —- —
ЭГЭ Al — —
ЭГЭ—ловет Al(Hg)
ЭГЭ—НБС Al(Hg) — —
Ti+ NH3 JHg —
НМ Hg ПТЭА —.
ТМФ Hg ПТЭА —
ЭС Hg ПТЭА —
М+ ДМА Hg ПТЭА
Се3+ ДМА Hg ПТЭА —
Рг 3+ ДМА Hg ПТЭА —
Nd3+ ДМА Hg ПТЭА —
180
П родолжение табл. 7
• К°. См. с-1 а i0, мА-см 2 I®, мА-см 2 D-10*. см2. с—1 Литература
0,08 0,0016 [441]
— — —- — 2,13 [888:
0,5 — — — [925]
— — — __ 2,42 [888]
— 0,5 — — 2,9 [925]
— — — 2,47 [888]
— 0,6 — 2,2 [925'
— — — — 1,8 [891]
— 0,30 8 [681, 957]
— — 18 — — 1070]
— <-1,5 — — 957]
5.6 10-® 0,27 — 2,7 925]
4,0-10-5 0,23 — — 1,9 925]
0,6б — — 3,5 793]
— 0,5з — — — [681, 684]
— 0,30 — — — [681, 684]
— 0,29 1 4,4 — [1070] [681, 957]
— 0,31 0,13 — — 957
— —. — 2,2 891]
— — 1,97 714
•— — — 2,7 [885]
0,15 __ [687]
— 0,20 — — — [489]
——- 687
0,15 — 687
0,24 — — 687]
0,24 — 489]
0,40 — —™ 687]
— 0,20 — — — [687]
— — — — 9,3 [1190
— —— —-- [687
0,27 — — — [687
0,14 — [687
— — — — 2,0 [891]
— — — — 2,7 1885
0,54 5—11 — 23]
— — 0,2—0,5 — — 282
— — 9—34 — — 24]
— — 0,43—1,6 — — 282]
— 0,51 28—34 —. .— 23]
— 0,51 28—34 — — [23]
34,7 [1000]
— — — 9,0 [1190]
3,5 [891]
— — — — 3,64 [885]
— — 2,5 [890
—_ — — — 2,96 [890
—_ — — — 2,44 [890
— '— — — 2,21 [890
181
Иои Растворитель Рабочий электрод Фоновый электролит Xs, Cm-c—1
Sm3+ ДМА Hg ПТЭА
ДМФ Hg ПТПА —
Eti3+ вода Hg NaC104 1,6-10-3
ДМА Hg ПТЭА —
(3—>2) ДМФ Hg ПТПА 1,6-10-4
NH4C1O4 7,1-Ю-3
NaC104 —
ДМФ4-вода (1:1) Hg NaClO4 —
NH4C1O4 3,3-ю-2
ФА Hg NaC104 ~4-10-4
Gd3+ ДМА Hg ПТЭА —
Tb3+ ДМА Hg ПТЭА —
Dy3+ ДМА Hg ПТЭА —
Ho3+ ДМА Hg ПТЭА —_
Er3+ ДМА Hg ПТЭА
Vb3+ ДМА Hg ПТЭА —
Pb2+ дмсо Hg КС1О4 —
ПТБА —
Th4+ ДМСО Hg LiCl 0,87
y3+ Вода Hg NH2'SO3H —
(3->2) N—МФ Hg nh2so3h —
Cr3+ дмсо Hg КС1О4
(3-2) ФА Hg NaC164 —
Mn2+ Вода Hg NaC104 5,57-Ю-4
АН Hg NaClO4 3,21-Ю-4
Диоксан—Н2О Hg NaC104 1 • Ю-3
ДМСО Mn NaC104 —
ДМФ Hg NaClO4 1,11-Ю-3
НМ Hg ПТЭА
ТМФ Hg ПТЭА —
ФА Hg NaC104 4,5-Ю-4
ЭС Hg ПТЭА —
Co2+ Вода Hg NaC104 —
АН Hg NaC104 —
ДМСО Hg KC1O4 —
ДМФ Hg NaC104
нм Hg ПТЭА —
ФА Hg NaC104 —
Ni2+ Вода Hg NaC104
ДМСО Hg KC1O4
ДМФ Hg NaC104 —
НМ Hg ПТЭА —
ФА Hg NaClO4 —
ЭС Hg ПТЭА —
182
Продолжение табл. 7
К®, См.с-1 а 1„, мА. см-2 .0 9 >0. мА»см 0.10е, СМ2. С 1 Литературе
2,7 [8901
1,8-Ю-4 1,1 — —- 3,3 [936
— —. — — 5,7 [263, 11441
— — — — 2,9 890
—. 0,74 — 2,5 936
— 0,65 " — 3,6 667
— — — — 1,3 1144]
1 • 10-3 0,60 — — — 1144]
— 0,65 — — 2,6 [667
—— — 863
» , । 2,53 890
— — —_ 2,49 890
— — —- 2,13 890
—. 1,97 890
—_ — — 2,30 890]
— — — — 2,64 [890
1,9-Ю-2 — — 1,4 [925]
3,0-Ю-3 — — — 1,5 [925]
— — — 13; 0,27 [735]
'3.36-10-3 0,60 — — — [687]
1,16 • 10—3 0,20 — •— — [687]
3,5-10-4 — —. 0,96 [925]
— — — — 1,84 [714
— 1,5 — — [853J
— 1,5 — — — [853
— — .— — — [1256]
—. — 7-10~7— — — [609
—4- IO-6
— 1,15 —. — — [853
? — — — । 9,2 [1190]
— — — — 2,0 [891]
S 1,2 — — 2,16 [714, 853]
i — — — — 3,16 [88Е >1
2,510—16 0,9 — — 13,6 [852, 854]
2,8 -10-6 0,66 — — 3,0 [852 , 854]
1.5-10-6 0,21 — — — [925]
5,0-10~13 0,70 — — 5,2 [852,854]
— — .— — 8,2 [1190 1
2,8-10~15 0,84 — — 2,92; 1,8 [714, 852]
2,25-10—3 0,737 — .— 2,75 [852, 854]
1,3-10~6— 0,33—0,25 — — — [925]
-6,7-10~7
1,4-10—9 0,56 — — 1,05 [852 , 854]
—— — 10,6 [1190]
7,1-10—10 0,62 0,66; 0,58 [714, 852]
— — — — 1,96 [885]
183
1
Таблица 8. Электрохимические параметры восстановления неметаллов в органически^
Элемент Фон Электрод
растворитель электрод сравнения
Pi АН ПТБА Hg Hg—дно
ДМФ ПТБА Hg Hg—дно
О, АН ПТБА Hg НКЭ
ПТЭА Hg НКЭ
ПТЭА Hg нкэ
Ац Вода ПТБА Hg, Pt нкэ
Hg нкэ
Hg нкэ
ДМСО ПТБА Pt, Hg нкэ
ПТБА Hg нкэ
ПТЭА Hg нкэ
ПТЭА Au, Hg, Pt нкэ
ПТЭА Hg нкэ
NaC104 Hg нкэ
ПТБА Hg нкэ
ПТЭА Au, Hg, Pt нкэ
ПТЭА Au нкэ
ПТЭА Hg нкэ
NaC104 Hg нкэ
NH4C1O4 Au, Hg, Pt нкэ
ДМФ ПТБА Hg, Pt нкэ
ПТЭА Hg нкэ
ПТЭА Hg нкэ
ПТЭА Hg нкэ
ПТЭА Hg нкэ
МА ПТЭА Hg нкэ
ПТЭА Hg нкэ
Метиленхлорид ПТБА Hg, Pt нкэ
Ру ПТБА Hg, Pt нкэ
ПК ПТЭА Hg нкэ
СФ ПТЭА Hg нкэ
ксю4+н2о Hg нкэ
СФ-рфеннл—СФ ПТЭА Hg нкэ
ФА КС1 Hg Hg—дно
S8 КС1 Hg Hg—Дно
ДМА LiCl СГЭ нкэ
ДМСО NaC104 Au нкэ
NaC104 Au нкэ
сн3он CH3COONa Hg нкэ
С2Н8ОН LiC104 Hg нкэ
Вг2 АН NaC104 Pt
Pt
Pt
АН NaC104 Pt
Pt
Pt
ДМФ NaNO3 Pt
Pt
* При восстановлении элементарной среды в ДМСО вместо Еу2 приведены осциллополярогра
184
юрнтелях (коэффициент диффузии относится к реагирующей на электроде частице)
t Электродная реакция —£1/2- В D. 10s, cm’/c Литература
1 4-е
71 Р4 D. Z 1,73 2,1 [59]
ж 4^Р?- 1,55 0,86 [59]
4-2е
О24~^~ч'О2 0,82 — [1122]
0,75 — [984]
1 О2—+е-+О22 1,4 — [984]
W О24-е—-О2— 0,88 — [1122]
О2+2е+2Н+->Н2О2 0,05 2,6 [984,1018]
Н2О24-2е—>2ОН~ 0,90 — [984]
О2Ч-6—>Од 0.77 — [1122]
0,73 — [1034]
0,72 — [984]
Ж 0,75 3,23 [11831
4g» 0,85 — [1019]
ж 0,72 — [991]
О2~+е->Оа2- 2,40 — [1034]
ж Ж- 2,02 1,08 [1183]
W' 2,05 0,73 [868]
1,13 [984]
1,2 — [1122]
О2+2е+2Н+->Н2О2 0,286 — [1183]
4, О2+е->О2“ 0,87 — [1122]
0,80 —. [817]
0,86 — [1019]
0,84 — [663]
О2 -f-e—Ю2^ 1,9 — [817]
О2-|-е—>О2 ’ 0,52 — [984]
О2' +е—>Оа 1,6 — [984]
0,79 — [1122]
0,89 — [1122]
0,77 0,805 [1019, 1018]
0,90 1,2 [1019]
О24-К+4-2е->КО2- 0,74 — [1019]
О2+н++2е->НОа- 0,58 — [1019]
Оа+2е+2Н+^Н2О2 0,14 1,4 [1146]
Н2О2+2е->2ОН~ 0,92 — [1146]
S+2e->S2" 0,36 — [444]
Sg-J-в—>Sg 0,62* 1,44 [1065]
Sg -J-e—>Sg2 1,29* 4,0 [1065]
S4-2e->S2- 0,58 — [933]
HgS+2e^Hg+S2- Br~ ~0,8 1,17 155] 947)
Br2 2,93 947]
Br3— — 1,97 947]
J— 1,68 10 24
I. 1,91 [1024
I3~ 1,37 [1024
I~ 0,80 [417
I2 — 0,60 [417
фВческие Ер,
185
Таблица 9. Энтальпия (кДж-моль-1) и энтропия (Дж-моль-1 К-1)
переноса ионов из воды в неводный растворитель
Ион H2O MtOH ФА ДМФ ДМСО ПК AH СФ
ДЯ
Li+ 0 —21,8 —5,4 —32,3 —26,4' 1,7
Na+ 0 —20,5 —16,3 —33,1 —27,6 —8,8 —13,0 —15,1
К+ 0 —18,4 —16,8 —39,4 —34,8 —21,0 —22,6 —25,1
Rb+ 0 —15,1 — 17,2 —37,7 —33,5 —23,5 —26,0 —26,7
Cs+ 0 —13,4 —17,2 —36,9, —32,3 —26,0 —24,7
Ag+ 0 —21,0 —22,6 —38,5 —54,9 —12,6 —52,8 —13,4
Mt4N+ 0 1,7 0,8 —16,3 —16,3 —19,3 —15,5
Et4N+ 0 9,2 7,5 —0,8 4,2 —0,8 —1,7
Bu4N+ 0 21,8 28,5 18,0 25,6 15,9 18,4
Ph4As+ 0 —1,7, —0,4 —19,7 —11,7 —15,1 —10,5 —10,5
ci- 0 8,4 3,4 21,4 18,8 28,1 19,7 26,0
Br- 0 3,8 —1,7 3,4 3,4 17,6 8,4 12,1
I- 0 —2,1 —7,5 —1'3,8 —13,4 —0,8 —7,1 —8,8
N3- 0 0,4 1,3 —2,5 16,3 8,8 16,5
cio4- 0 —2,5 —20,1 -25,1 —19,3 —15,5 —10,5
BPh4- 0 —1,7 —0,4 —19,7 —11,7 —15,1 —10,5 —
AS
Li+ —5,9 —28,1 4,2 —10,1 —11,7 —20,5 —
Na+ 4,2 -28,9 —6,7 —22,6 —43,0 —19,7 —26,0 12,1
K+ 13,4 —28,5 —10,5 —28,9 —22,6 —24,3 —30,6 21,0
Rb+ 17,2 —25,1 —11,7 —27,6 —22,6 —18,0 —32,7 18,0
Cs+ 18,0 —23,0 —9,6 —27,6 —19,7 —' н,з - 14,6
Ag+ 2,1 —28,5 —7,1 —21,4 —21,4 —26,4 —31,0 9,7
Et4N+ 5,4 8,4 —> 7,5 9,2 7,1
Ph4As+ —16,8 21,8 23,5 18,4 25,1 20,5 22,2 —25,0
ci- 8,4 —4,2 —10,5 —24,7 —119,7 —14,2 —22,6 26,8
Br- 12,6 —7,5 —13,0 —26,8 —22,2 —15,1 —19,3 27,8
I- 18,0 —8,8 —15,1 —32,7 —26,8 —20,1 —26,0 29,4
N3~ 18,9 —9,6 -— —33,1 —26,4 —13,4 —20,5 24,2
BPh4- — 16,8 21,8 23,5 18,4 25,1 20,5 22,2 —25,0
Таблица 10. Энтальпия сольватации ионов (—ДЯ, кДж г-иои-1) 298,15 К
Растворитель
Ион H2O NH. AH ГМФТА дмсо ДМФА MtOH МФА пк ФА ElOH эг
Li + 536 — — — 499 725 524 675 — 624 — —
Na+ 423 490 434 549 448 612 415 553 444 524 486 432
K+ 339 398 — 457 369 528 327 478 360 440 406 —
Rb+ 318 373 — 369 344 507 302 453 — 415 — —
Cs+ 281 335 — 339 310 473 368 406 — 381 — —
F 490 — — — — — 507 — — —- — —
Cl- 348 335 — — 398 168 373 226 — 260 — —
Br~ 318 306 — 352 377 159 344 205 — 226 264 —
I- 277 281 289 339 297 138 310 176 297 201 230 289
N0F 314 — — — — 168 339 — — — — —
Таблица 11. Потенциалы полуволн перхлоратов металлов на Hg-капельном
электроде относительно водного насыщенного каломельного электрода
в органических растворителях
Ион металла растворитель Фоновый электролит £1/2’ В bt мВ Литература
Li+ V. -> ♦ Na+ К+ Rb+ Вода АН БН ДМСО ДМФ ИБН Ру ПК ПН ФАН эк Вода АН Ац БН ДМА ДМСО ДМФ ИБН пдк Ру ПН ПК ТМФ ФАН эк Вода АН ГМФА ДМА ДМСО ДМФ ПК эк Вода ПТЭА ПТБА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТБА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА НТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТБА ПТЭА ПТЭА ПТЭА ПТЭА НТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА —2,332 —2,35 —2,345 —1,95 —1,97 —1,82 —2,578 —2,45 —2,385 —1,90 —2,17 —2,001 —2,071 —1,93 —1,83 —2,009 —2,113 —2,10 —2,100 —1,85 —1,85 —1,92 —1,73 —2,06 —2,075 —2,07 —2,07 —2,035 —1,77 —1,86 —1,89 —1,82 —1,916 —1,848 —2,07 —1,73 —1,861 —2,136 —2,13 —1,95 —2,049 —2,01 —2,08 —2,093 —2,11 —2,11 —2,005 —1,965 —2,020 —1,986 —2,131 —2,12 56 68 9Q 58 90 97 91 76 62 77 81 63 56 73 62 94 59 120 65 60 60 62 62 65 56 65 61 65 65 65 70 61 95 56 59 63 62 63 63 63 70 65 [1096 [708 242 988]' [1134 1005 1006 1052’ '1096' [743' [708' [1096' [242 [1005 [1005 [1096 [1096 [891' [242 [1134' [988 [745 [1006 [889 [1096 [1052 [893' [1096 [743' [891 [708 [1005 [842 [1096 [891 [1005' [1096 [1096 [89Г [11'34' [1022 [950, 952 [889 [1096' [893 [1062 [1096 [1096 [242 [1096 [1096] [891] 187
186
Продолжение табл. И
Продолжение табл. 11
Ион металла Растворитель Фоновый электролит £l/2- в b, мВ Литература
Rb+ Ся+ Сп+ Си2+ (2—0) (2-0) (2-1) (1-М>) (2-1) (1->0) (2-0) (2-1) (1^*01 АН Ац БН ГМФ ДМА ДМСО ДМФ ИБН пдк Ру ПК пн ТМФ ФАН ЭК Вода ГМФА ДМА ДМСО ДМФ Ру ПК ЭК АН 1-ВЮН ГАН ДВА ДИПА ДМА ДМФ ДЭА 3-МПН ПН 3-ХПН 3-ЭПН Вода АА АН Ац ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА LiClOa LiC104 ПТЭА LiC10« LiC104 LiC10« LiC104 LiC104 LiC104 ПТЭА LiC104 ПТЭА ПТЭА ПТЭА NaF LiC104 NaClO4 NaC104 LiC104 LiClO4 ПТЭА ПТЭА ПТЭА —1,978 —1,974 —1,876 —2,014 —1,99 —2,04 —2,075 —2,045 — 1,879 —1,97 —1,91 — 1,989 —1,937 —2,05 —1,89 —2,009 —2,108 —2,09 — 1,908 —1,96 —2,03 —2,047 —2,011 —1,89 —1,981 —2,011 -0,38 -0,33 —0,39 +0,31 —0,32 —0,30 + 0,07 +0,02 +0,05 0,00 +0,02 —0,36 —0,36 —0,40 -0,34 —0,36 —0,003 +0,03 —0,43 +0,22 + 1,0 —0,5 +0,2 —0,34 —0,30 +0,53 +0,31 62 53 70 60 59 60 62 50 50 63 53 59 120 75 65 59 59 61 58 63 60 77 59 54 59 60 52 96 100 94 65 94 57 58 58 59 56 37 52 60 Г742' . [742‘ [742 [1022 [950, 952] [889 [1096 [1096 [742 [891 [708 [1096 [742 : [891 [742 [1096] [10961 [708] [1022 [950,952 [889 [1096 (1096 [708 [13096’. [1096 [814' [742’ [814, 814 814' 814 697' '697' 697 697' '697 ,814 814’ 814 '814 '814 [1022] [947' [681,684 ! [917 '681,684’ 681,684 681,684 681,684' 681,684' [745' 1745]
Ион металла растворитель Фоновый электролит £l/2- b b, мВ Литература
(2—1) ЫС1О4 +0,52 63 [917]*
(1—2) LiC104 +0,33 65 917
(2—1) LiC104 +0,58 <— '913
(1-О) LiC104 +0,33 ,— [913]
(2—1) ПТЭА +0,55 — [913]
(Ь+Ю) ПТЭА +0,38 r— 913]
(2—0) БН ПТЭА —0,25 59 [681,684
(2—0) ГМФА ПТЭА —0,035 — [1022]
(2—0) ДВА LiC104 +0,10 120 [697]
(2-О) ДИПА LiC104 +0,07 110 [697]
(2—1) ДИППА LiC104 +0,17 89 697]
(1—0) LiClO4 —0,00 67 697
S (2—0) (C2H5)2SOa ПТЭА +0,60 62 899]
Л г (2—0) (C2H5)2SO4 ПТБА +0,63 52 [899]
(2—0) 2,4-Д МСЛ ПТЭА +0,52 99 899]
(2—0) ПТБА +0,56 100 899
(2—0) ДМСО ПТЭА —0,006 —— 722
(2—0) ПТЭА —0,06 — [1022
(2—0) ПТЭА —0,07 95 [899
(2—1) ПТЭА —0,03 66 [681,684
(1—0) ПТЭА —0,140 57 [681,684
(2—0) ПТБА —0,07 80 [899
Ж (2—1) NaClOa +0,004 59 [681,684
(1—О) NaClOa —0,120 57 [681, 684
(2—1) NaNO3 +0,04 59 [1052
(1—О) NaNO3 +0,04 57 [1052
(2—1) НТЭА —0,004 — [722
(1—0), НТЭА —0,12 — [722
(2—0) ДМФ ПТЭА 0,00 — [ / 8
$ ’ (2—1) ПТЭА +0,05 65 681,684
(2—0) ПТЭА —0,01 32 681,684
(2—1) NaClO4 —0,27 87 681,684
Ж- (1—0) NaClO4 0,00 37 681,684
(2—0) LiClOa +0,04 40 697
ИБН ПТЭА > +0,67 — 743
т (1—0), ПТЭА —0,25 — 743
42—>1) N-MA ПТЭА —0,10 — '984
(1—0) ПТЭА —0,37 — '984
(2—0) ММС ПТЭА +0,63 85 899
4 (2—0) ПТБА +0,64 85 '899
'(2—0) 3-МСЛ ПТЭА +0,50 93 899
(2—0) ПТБА +0,52 1'00 899
(2—1) НМ ПТЭА >+0,8 —— 917
(1—0), ПТЭА +0,22 68 '917
?- (2—1) Ру LiClOa >+0,1 — 917
(1—0), ЫС1О4 —0,64 93 917
(2—0) ПК LiClOa +0,18 55 [681, 684
(2—1) 1-РгОН LiClOa +0,39 59 917
(1—0) LiClOa +0,25 78 917
(2—0) СНзОН LiClOa +0,16 42 917
(2—1) LiClOa +0,21 — [681,684
(1—0) LiClOa +0,17 — [681,684
(2—0) С2Н5ОН LiClOa +0,28 64 917
(2—0) СФ ПТЭА +0,50 89 899
(2-0) ПТБА +0,46 1 91 [899
189
188
Продолжение табл. И
Ион металла Растворитель Фоновый электролит £1/2- В Ь, мВ Литература
(2+1) СНзСООН LiClOi +0,39 60 [765, 766]
(1+0) LiC10t +0,32 56 [765, 766]
(2+1) АУ LiC104 +0,51 , [917]
(1+0) LiC104 —0,16 82 [917'
(2+0) ФА NaC104 —0,12 77 1714]
(2+0) ЭС ПТЭА +0,41 , 885]
Ag+ Вода — +0,49 1—1 [765, 766]
— +0,557 — [1052
АКН ПТЭА +0,49 59 [1005]
LiC104 +0,39 80 4814]
АН ПТБА +0,42 94 [1134]
ПТЭА +0,42 60 [10051
NaC104 +0,32 58 [989]
ПТЭА + 0,37 50 [814]
Ац ПТЭА +0,68 —1 745]
БН ПТЭА +0,54 53 [1005]
БН LiC104 +0,34 59 [814]
ГМФА ПТЭА +0,386 — [1022]
ДМСО ПТЭА +0,30 — [1022]'
БН ПТЭА +0,49 [742].
з-мпн LiC104 +0,38 55 [814]
пн ПТЭА +0,45 62 [1005]
LiC104 +0,34 54 [814]
СНзСООН LiC104 +0,635 60 [765, 766]
ФАН ПТЭА +0,52 61; [1005]
з-хпн LiC104 +0,40' 58 [814]
Ве2+ Вода — . —1,80 — [989]
АН ПТЭА —1,6(0 ,— [989]
Mg2+ Вода — —2,20 , [745]
АН ПТЭА —1,84 г— [Гооб]
Ац ПТЭА —175 60 [745]
БН ПТЭА — 1Д2 82 [1005]
ДМА ПТЭА —2,30 130 4888]
ДМСО ПТЭА —2; 19 1 —« [963
НТЭА —2Д8 —, 893]
ИБН ПТЭА —1,68 — [743]
ПН ПТЭА —1,72 [1005'
ФАН ПТЭА —1,63 —> [1005]
Са2+ АН ПТЭА —1,82 45 [988]
ПТБА —1,85 [1134]
БН ПТЭА —1,73 58 [1005
ДМА ПТЭА —2,37 56 [888]
ПН ПТЭА —1,83 45 [1005]
ФАН ПТЭА —1,79 40 [1005]
Sr2+ АН ПТЭА —1,76 72 [988]
ПТБА —1,79 —_ [1134]
ВН ПТЭА —1,72 74 [1005'
ДМА ПТЭА —2,23 50 4888]
ДМСО NaNO3 —2,10 47 [1052'
ПН ПТЭА —1,78 31 [1005]
ФАН ПТЭА —1,73 —1 [1005'
Ва2+ Вода — —1,94 — [891]
АН ПТЭА —1,63 44 [1005]
Продолжение табл. 11
Иои металла Растворитель Фоновый электролит £1/2’ в 6, мВ Литература
Ва2+ ПТБА —1,63 [1134]
БН ПТЭА —1,58 32 [1005
ДМА ПТЭА —2,02 42 [888
ДМСО ПТЭА —2,08 — [891
ПТЭА —2,09 34 [Ю52
4 ДМФ ПТЭА —2,04 891
пдк ПТЭА —1,67 — 891
пн ПТЭА —1,63 37 [ 005
ТМФ ПТЭА —2,03 50 891
ФАН ПТЭА —1,58 33 [1005]
Zn2+ Вода — —1,02 — [891]
NaF —1,17 — [681, 684
АКН ПТЭА —0,56 30 [1005
АН ПТЭА —0,62 — [891
ПТБА —0,62 — [1134
NaC104 —0,70 35 [681, 684
NaClOj —0,66 38 [1005
АФ LiCIO4 —0,55 — 913
Ац ПТЭА —0,61 — 913
LiCiO4 —0,57 —— 913
БН ПТЭА —0,53 30 [681, 684
ДМСО ПТЭА —1,10 — [722
ПТЭА —1,09 — [891
NaNO3 —1,0 28 [1052
ДМФ ПТЭА —0,98 — [891
ДМФ NaNO3 —0,988 36 [681, 684
НВ нм ПТЭА —0,26 52 [891
пдк ПТЭА —0,54 — [891
явк, ПК ПТЭА —0,54 42 [681, 684
пн ПТЭА —0,59 35 [1005
ТМФ ПТЭА —0,82 32 [891
СН3СООН ПТЭА —1,085 40 [765, 766
LiClO4 —0,640 36 [765, 766
ФА NaC104 —1,07 43 [714
ФАН ПТЭА —0,51 34 [1005
ЭС ПТЭА —0,63 — [885]
Cd2+ Вода NaF —0,67 — [681, 684]
ПТЭА —0,593 — [1022
АН NaC104 —0,27 53 [1005
ПТЭА —0,26 — [891
ПТБА —0,27 39 [681, 684
АкН ПТЭА —0,19 28 [1005
№ АФ LiC;O4 —0,17 — 913
Ац ПТЭА —0,21 — 913
ПТЭА —0,18 — 745
ЫСЮ4 —0,20 — 913
БН ПТЭА —0,17 32 [1005
К ГМФА ПТЭА —0,531 — [1022
ДМФ ПТЭА —0,58 — [891
SB №С1О4 —0,60 22 [681, 684
ДМСО ПТЭА’ —0,70 — [891
мк. ПТБА —0,64 — [963
NaNO3 —0,60 31 [1052
К NaNO3 —0,70 31 [681, 684
190
191
Продолжение табл. 11
Продолжение табл. 11
Ион металла Растворитель Фоновый электролит В1/2- в Ь, мВ Литература
Cd2+ ксю4 —0,64 34 [681, 684 []
нм ПТЭА +0,03 35 [891
пдк — —0,11 — [891
ПК ПТЭА —0,11 42 [681, 6&
пн ПТЭА —0,25 33 [1005
СНзСООН ПТЭА —0,685 31 [765, 766
ЫС1О4 —0,295 31 [765, 766
ТМФ ПТЭА —0,49 53 [891
ФА NaC104 —0,12 77 [681, 684
ФАН ПТЭА —0,18 33 [1005
эд НТЭА —0,50 — [1197
НТЭА —0,54 — [1196
ЭС ПТЭА —0,21 — [885
Hg2+ ПТБА АН 0,42 94 [П34]
Al3+ Вода ПТЭА —1,75 — [989]
NH4C1 —1,70 — [164]
АН ПТЭА —1,42 — [9891
ПТБА —1,35 64 [П34]
БН ПТЭА —1,47 48 [1005]
ЫС1О4 —1,41 38 [10051
ДМФ ПТЭА —2,05 — 777]
СН3ОН NH4C1 —1,70 — 164]
С2Н5ОН nh4ci —1,73 — 164]
С3Н6(ОН)3 nh4ci — 1,54 — [164]
Ti+ Вода — —0,46 — [891
АН KNO3 ПТЭА —0,49 -0,27 [1226 [891
ПТБА —0,26 93 [1134
ГМФА ПТБА —0,246 — [1022
ДМСО ПТЭА —0,54 — [891
NaNO3 —0,46 52 [1052
ПТБА —0,50 — 963
ДМФ ПТЭА —0,45 — 891
НМ ПТЭА —0,21 — 891
ПТЭА — 63 [1190
пдк ПТЭА —0,24 — [8911
СНзСООН LiC104 —0,400 60 [765, 7661
ТМФ ПТЭА —0,39 61 [891]
ЭС ПТЭА —0,26 — [891]
La3+ Вода ПТЭА — 1,86 — [8901
ДМА ПТЭА -2,11 — 8901
ДМФ Bu4NI —2,01 55 [Ю111
Лантаноиды
Се3+ Вода ПТЭА —1,80 — 890
(3+0) ДМА ПТЭА —2,10 — 890
рг3+ Вода ПТЭА —1,84 — 890
(3+0) LiCl —1,80 — 745
АН ПТЭА —1,50 — 905
Ац ПТЭА —1,47 — 745
БН ПТЭА — 1,54 — 905
ДМА ПТЭА —2,06 — 8901
ДМФ Bu4NI —2,01 49 [ЮН]
192
Иои металла Растворитель Фоновый электролит в1/2> в Ь, мВ Литература
(3+2) C2H5OH ЫС1О4 —1,58 [747]
(2+0) C2H5OH ЫС1О4 —1,70 — [747]
Nd3+ (3+0) Вода AH Ац БН ДМА ПТЭА ИТЭА+ +H2SO4 ПТЭА ПТЭА ПТЭА — 1 ,84 — 1,82 —1,45 —1,57 —1,49 — 1,50 —2,07 [890] [745] [749] [905 745 [905 [890]
Sm3+ (3+2) (2+0) (3+2) (2+0) (3+2) (2+0) (3+2) (2+0) (3+2) (2+0) (3+2) <^0) : !£oi (3+2) ДМФ Вода АН Ац БН ДМА Bu4NI ПТЭА ПТЭА ИТЭА+ +H2SO4 ИТЭА+ +H2SO4 ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА —2,00 —1,80 — 1,96 — 1,80 —1,96 —1,02 — 1,69 —1,12 —1,66 —1,04 —1,55 —1,62 —1,13 —1,70 —1,00 — 1,58 —1,71 —2,08 47 58 67 60 [1011 [890] 890] [745] [745] [905] [905 265 265 749 749 989 745 745 [905 905' 890: 890
(3+2) (2+0) (3+2) (2+0) Eu3+ . (3+2) ; (2+o) ! (3+2) 1 (3+2) (3+2) (2+0) (3+2) (2+0) (3+2) (2+0) (3+2) (2+0) (3+2) (2+0) (3+2) (2+0) (3+2) 13-4-653 ДМФ Вода АН Ац Ац+Н2О (1:1) Bu4NI Bu4NI ПТПА ПТПА ПТЭА ПТЭА NH4C1 НСЮ4 ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА NaClO4 —1,81 —1,96 —1,842 —2,04 —0,77 —1,97 —0,67 —0,66 +0,15 —1,67 +0,19 —1,67 0,0 — 1,7 +0,10 —1,62 +0,17 — 1,75 -0,03 —1,79 —0,875 74 65 91 60 48 77 44 120 58 [1011 [1011 [936 [936[ 890] 890 745 [936] [989 989 905 [905 [1090 [1090 [749 749 361' 361! [745 745 [667 193
Продолжение табл. 11
Продолжение табл. 11
Ион металла Растворитель Фоновый электролит £1/2- в Ь, мВ Литературе Ион металла растворитель Фоновый электролит Е1/2, в Ь, мВ Литература
(3+2) (2+0) (3+2) (2+0) ПТЭА ПТЭА ПТЭА ПТЭА +0,18 — 1,68 —0,60 —2,10 60 905 905 890 [890 (3+2) (2+0) (3+2) (2+0) ДМА ДМФ ПТЭА ПТЭА Bu4NI Bu4NI —1,27 —2,53 —1,38 —2,05 59 70 [890] [890] [ЮН] [ЮН]
(3+2) ДМФ ПТПА —0,680 74 936 Актиноиды:
(2+0) ПТПА —2,05 96 936 Th4+
(3+2) (2+0) (3+2) Bu4NI Bu4NI NH4C1O4 —0,64 —1,96 —0,81 74 [1011 [1011 [667 (4+2) pu3+ АН АН ПТЭА ПТЭА —1,20 —2,2 — [1039] [360]
(3+2) ДМФ—Н2О (1:1) nh4cio4 —0,78 — [667j Am ’ (3+2) АН ПТЭА —1,65 — [360]
(3+0) ФА' NaC104 —0,73 62,5 [263 (2+0) ПТЭА —2,0 — [360]
Gd3+ Вода LiCl — 1,80 — [745 (3+2) ПТЭА —1,55 — 359]
(3+0) Ац ДМА ПТЭА ПТЭА — 1,49 —2,18 [745 [890 С f ' (3+2) АН ПТЭА — 1,00 — [824]
ДМФ Bu4NI —2,05 52 [1011 Ж (2+0) ПТЭА —1,43 — [824]
ть3+ Вода ПТЭА —1,85 — [745 Pb2+ Вода —0,38 [765, 766]
(3+0) Ац ПТЭА —1,38 — [745 АН ПТБА —0,04 [1134]
ДМА ПТЭА —2,20 — [890 Ж АФ ЫС1О4 —0,08 [913
ДМФ Bu4N1 — 42 [1011 Ац ПТЭА —0,12 [913]
Dy3+ Вода ПТЭА —1,85 — 745 1ЛС1О4 —0,12 [913'
(3+0) ПТЭА —1,80 — [890 ДМСО КС1О4 —0,53 [722
Ац ПТЭА — 1,44 — 745 КС1О4 —0,50 [963
ДМА ПТЭА —2,24 — 890 'Ж NaNO3 —0,40 32 [1052
ДМФ Bu4NI — 63 ПОП СН3СООН ЫС1О4 —0,226 30 [765, 766
Но3+ Вода ПТЭА —1,85 — 745 CH3COONH4 —0,570 30 [765', 766
(3+0) Ац ПТЭА ПТЭА 1,80 — 1,44 [890 745 В' Sn2+ АН ПТБА 0,42 94 [1134]
Ег3+ ДМА ДМФ Вода ПТЭА Bu4NI ПТЭА —2,25 — 1,85 43 [ 890 ОН 745 Ж/ Sb3+ Cr2+ ДМСО Вода NaNO3 ПТЭА —0,30 -1,47 39 [1052] [742]
(3+0) ПТЭА — 1,80 — 890 1' АКН ПТЭА — 1,05 — [1005]
Ац ПТЭА —1,43 — [745 АН ПТБА — 1,10 127 [П34]
ДМА ПТЭА —2,27 890 NaC104 —1,12 52 [1005]
ДМФ Bu4NI —2,2 24 [1011 NaClO^ —1,1 — 989]
Тш3+ Вода ПТЭА — 1,85 — [745 ьн ПТЭА —0,97 34 [ 005]
(3+0) NaCl — 1,86 — 905 в? дмсо ПТЭА —1,05 — [722]
АН ПТЭА — 1,50 — 905 пн ПТЭА —1,56 — [722]
Ац ПТЭА —1,44 745] ПТЭА —1,07 35 [1005]
БН ПТЭА —1,52 ___ 905 ФА NaClO4 — 1,56 119 [714]
Yb3+ ДМФ Bu4NI —2,2 32 [1011 ФАН ПТЭА —0,98 44 [1005]
(3+2) Вода ПТЭА — 1,45 — 890 ж Cr3+ Вода —0,91 [742] [742] 111341
(2+0) ПТЭА —2,03 — [890 “Г (3+2) АН ПТЭА 0,00
(2+0) ПТЭА —2,00 — 989] ПТБА -0,01
(3+2) NH4C1 — 1,17 — [745] NaC104 0,00 100 [989]
(3+2) АН ПТЭА —0,60 — [749 БН ПТЭА —0,40 742] 722] [891]
(2+0) ПТЭА —1,58 — [749 в ДМСО ПТЭА —0,82
(3+2) (2+0) ПТЭА ПТЭА —0,57 —1,69 62 60 989] 989] Mn2+ Вода — —1,48 —
(3—>2) ПТЭА —0,58 905 ж NH4C1 —1,54 — 164]
(2+0) ПТЭА —1,70 905 АКН NaC104 — 1,48(5 38 853]
(3+2) Ац ПТЭА —0,71 64 745 Ж ПТЭА — 1,05 34 [1005]
(2+0) ПТЭА — 1,71 — 745 АН ПТЭА — 1,08 — [891]
(3+2) БН ПТЭА —0,63 — 905 •Ж: ПТЭА — 1,12 —• [9131
(2+0) ПТЭА —1,64 — 905] I NaC104 —1,36 37 [853]
13*
195
194
Продолжение табл. 11
Продолжение табл. 11
Ион металла Растворитель Фоновый электролит В1/2, В Ъ, мВ Литература
Мп2+ NaC104 —1,08 — [742
NaC104 — 1,12 28 [1005
БН ПТЭА —0,98 32 [1005
LiC104 —1,11 48 [1005
ДМСО ПТЭА — 1,72 — [891
ПТБА — 1,68 — [963
NaC104 — 1,73 — [681, 684
ДМФ ПТЭА — 1,56 — [891
NaC104 — 1,476 43 [853
НМ ПТЭА —0,33 71 [891
пдк ПТЭА — 1,08 — [891
ПК ПТЭА —1,08 42 [681, 684
пн ПТЭА —1,10 36 [1005
СН3ОН NH4C1 —1,54 — [164
С2Н5ОН nh4ci — 1,58 — [164
ТМФ ПТЭА —1,85 20 [681, 684
ПТЭА — 1,85 220 [891
ФА NaC104 — 1,57 35 [714
NaClOa —1,516 41 [853
ФАН ПТЭА — 1,02 40 [1005
ЭС ПТЭА —1,19 — [885
Fe2+ АН ПТБА —1,04 — [1134]
NaC104 —1,00 55 1005]
БН ПТЭА —0,79 38 1005]
LiC104 -0,91 41 1005]
ДМСО КС1О4 —1,41 — [722]
пн ПТЭА —0,94 — [1005]
Fe3+ Вода — +0,49 — [765, 766]
(3->2) ДМСО КС1О4 —0,73 — [722]
СН3СООН LiClOa +0,210 72 [765, 766]
Со2+ Вода — —1,43 — [891]
— —1,20 — [765, 766
NaClOa —1,127 58 [852, 854
АКН ПТЭА —0,48 — [1005
АН ПТЭА —0,61 —— [891
ПТЭА —0,67 — [681, 684
ПТЭА —0,61 30 [943
ПТМА —0,62 31 [943
NaC104 —0,64 29 [943
NaC104 —0,545 100 [852, 854
NaClOa —0,65 37 [1005
LiClOa —0,63 30 [943
БН ПТЭА —0,49 — [1005
ГМФА ПТЭА <-0,7 — [681, 684
ДМСО ПТЭА —1,43 — [891
КС1О4 — 1,39 — [722
NaClOa — 1,40 — 681, 684
NaClOa — 1,32 — 681, 684
ДМФ ПТЭА —1,25 — 681, 684
ПТЭА —0,71 —— [891
NaClOa — 1,066 81 [852, 854
NaClOa — 116 [681, 684
ИБН ПТЭА —0,60 34 [743
НМ ПТЭА —0,26 75 [1190]
if ' ?. Ион металла Растворитель Фоновый электролит £1/2, В Ь, мВ Литература
Со2+ Ni2+ t i $ l л I jy i 1 ПДК ПК пн ТМФ СН3СООН ФА ФАН ЭС Вода АКН АН АФ Ац БН ГМФА ДМСО ДМФ ИБН НМ ПДК ПК ПН СН3ОН С2Н6ОН ТМФ СН2СООН ФА ЭС ПТЭА ПТЭА ПТЭА ПТЭА LidOa CH3COONH4 NaClOa NaClOa ПТЭА ПТЭА NaClOa ПТЭА ПТЭА ПТЭА ПТЭА NaClOa NaClOa LiClOa ПТЭА LiClOa ПТЭА ПТЭА ПТЭА као4 КСЮ4 NaNO3 ПТЭА NaClOa NaClOa NaClOa ПТЭА ПТЭА ПТЭА ПТЭА ПТЭА NH4C1 NH4C1 ПТЭА LiClOa CH3COONH4 NaClOa NaClOa NaClOa ПТЭА —0,73 —0,73 -0,64 -1,39 —0,825 -1,185 —1,110 — 1,20 —0,52 —0,67 — 1,01 —0,999 —1,10 —0,33 —0,29 —0,89 —0,45 —0,58 —0,33 —0,51 -0,57 —0,54 —0,29 —0,7 —1,07 — 1,08 —1,10 —1,08 —0,92 —0,856 —0,998 —0,33 —0,19 —0,48 —0,48 —0,36 —1,13 —1,14 —0,90 —0,645 -0,975 —0,949 —0,98 — 1,00 -0,60 64 33 215 61 54 67 72 37 75 85 120 34 80 31 59 69 100 79 105 130 132 121 127 86 82 83 79 [891] [681, 684 [1005 [891] [765, 766 [765, 766 [852, 854' [714 И1 [ООО [765, 7661 [852, 854] [891] [1005] [735 [681, 684 [681, 684 [681, 684 [1005 [913 [913] [913] [1005 [681, 684 [891] [722 [681, 684 [1052] [891 [852, 854' [681, 684 [681, 684 [743 [1190 [891 [681, 684 [1005 [164 [164 [891 [765, 766 [765, 766 [852, 854 [714 [681, 684 [891
F Примечание. j? Фоновые электролиты Электроды $ БТЭА — бромид тетраэтиламмоиня АПЭ — амальгамированный платино- $ ИТБА — нодид тетрабутнламмоння вый электрод ж ИТПА — ноднд тетрапропнламмоиня КРЭ — капельный ртутный электрод ® КТЭА — карбонат тетраэтнламмоння НКЭ — водный насыщенный кало- ша НТЭА — нитрат тетраэтнламмоння мельный электрод ПТАА — перхлорат тетраалкиламмония нор. КЭ — нормальный каломельный ПТБА •— перхлорат тетрабутнламмоння водный электрод £ ПТЭА —перхлорат тетраэтнламмоння СРЭ — стационарный ртутный элек- г» ПнТБА — пнкрат тетрабутнламмоння трод ТАА — тетраалкиламмоний СпЭ — электрод нз специальной гра- ХТМА — хлорид тетраметнламмония фитовой пасты Ц ПТПА — перхлорат тетрапропнламмо- СЭ — графитовый электрод В ния СГЭ — стеклографитовый электрод
196
197
Продолжение табл. 12
Таблица 12. Границы электрохимической устойчивости органических
растворителей
растворитель Фоновый электролит Рабочий электролт Электрод сравнения Область рабочих потенциалов Литература
Аммиак ЭДА ГМФА Морфолни Ру ДМФА ИТБА ИТБА NH4C1O4 nh4no3 LiC104 БТМА ИТБА ИТПА КТЭА НТЭА ПТЭА ПТЭА ПТЭА ПТ МА LiC104 LiC104 NaCIO4 NaC104 ПТАА ПТЭА ИТБА LiC104 LiC104 LiC104 LiCl LiCl LiCl LiCl A1CL KSCN KSCN NaJ NaBPh. ИТБА ИТБА ПИТБА ПТБА ПТЭА LiC104 LiC104 LiCl LiNO3 NaC104 крэ КРЭ Pt Pt Pt, сэ Pt Pt Pt Pt Pt Pt Pt КРЭ Pt Pt Pt СЭ Hg Pt Pt, СЭ КРЭ КРЭ КРЭ СЭ КРЭ КРЭ КРЭ КРЭ КРЭ КРЭ КРЭ КРЭ КРЭ Hg СЭ СЭ СЭ СЭ Pt Pt Pt Pt Hg СРЭ Pb|Pb(NO3)2 h2ih+ н3|н+ Zn—Hg|ZnCl2 нкэ нкэ нкэ нкэ нкэ нкэ нкэ нкэ нкэ AgAgNO3 Ag AgNO3 AgAgNO3 нкэ Ag|AgNO3 Ag|AgNO3 нкэ СРЭ СРЭ Ag|AgNO3, C5HbN СРЭ СРЭ СРЭ СРЭ Hg—дно СРЭ СРЭ СРЭ СРЭ НКЭ Ag|AgNO3, c5h5n AglAgNO3 c5h5n Ag|AgNO3, c5h6n Ag|AgNO3> c5h5n AglAgCl нкэ Ag|AgCl Ag|AgCl нкэ + 1,42 + 1,39 +5,0 —0,50 +0,75 +0,75 +0,85 +0,75 +0,85 —0,6 + 1,4 —1,00 —0,6 +0,1 + 1,2 +0,4 + 1,4 +1,9 +1,75 +1,15 + 1,8 +0,5 —LJ_LJ I l I I i I 1 i i i и i i i । i iii iii I i i । । । । । । । । । । । । । -2,3 —2,0 —1,1 —1,79 —2,25 —1,68 -2,12 —2,74 —1,80 —2,30 —2,60 —1,98 —3,6 —2,4 —2,8 —1,1 —3,0 —1,77 —1,65 -1,71 —1,59 —1,53 —1,47 —1,65 — 1,53 —1,18 —1,57 —2,2 -3,00 -2,80 -1,25 -2,5 [1116] [1116] [HO] [110] 1116 [1116 [1116 [1116 1116 1116 1116 1216 [1216 [1116 [Ш6] [H16] [14] [681, 684] [1116] [14] [H16] [И16] [1116] [1116] [1H6] [1116] [1116] 1116] [740] 11116 1116 1116 1116 [681, 684] [H16] [1H6] [1116] [1H6] [154] [676] [154] [154] [1И6]
Растворитель Фоновый электролит Рабочий электролит Электрод сравнения Область рабочих потенциалов Литература
ДМФА NaClO4 Hg AglAgCl, ДМФ +0,8 —1,5 i e [904]
NaC104 Pt НКЭ + 1,6 ‘1 j и [H16]
NaNO3 КРЭ Na—Hg|NaClO4 +2,48 +0,16 [1H6]
NaBFe Pt НКЭ +1,78 — [676] [1П6]
ИТБА Hg нкэ —0,4 —3,0
ПТБА КРЭ Ag|AgC104 —0,08 — —3,43 [837]
ПТБА Hg нкэ +0,5 — —3,0 [1116]
ПТБА Pt НКЭ +1,5 — —2,5 [1П6]
ПТЭА КРЭ Na—Hg|NaClO4 +2,42 — —0,78 [H16]
ПТЭА Hg нкэ +0,5 — —3,0 [1116]
ПТЭА Hg НКЭ +0,4 — —2,8 [681, 684]
ПТЭА Hg AglAgCl, ДМФ +0,8 — —2,4 [904] [1116]
ПТЭА Pt НКЭ + 1,6 —2,1
у МАА ПТЭА Hg НКЭ +0,35 — —2,75 [680]
к. ПТЭА Hg НКЭ +0,35 — —2,70 [984]
Тетраметил- NaNO3 КРЭ НКЭ +0,3 — —1,8 [1116] ‘
карбамид ПТБА КРЭ СРЭ +0,25 — —2,20 [1116]
W ФА NaC104 КРЭ нкэ — — —1,69 [1116]
КС1 КРЭ нкэ — ——• —1,69 [1116]
'V KNO3 КРЭ нкэ — — —1,60 [1116]
Муравьиная HCOONa КРЭ нкэ +0,2 — —0,8 [H16]
iT кислота HCOONa Pt СРЭ —1,4 — — [1116]
ч: НАс CH3COONa Pt нкэ +2,0 — — [1H6]
? Уксусный ангидрид LiC104 LiClO* Hg Pt Agl AgC104, LiC104 Agl AgCfo4, LiC104 +0,1 +2,1 — —2,0 —1,1 [1116] [1116]
LiC104 Ag Ag| AgCiO4, —0,1 — —1,1 [1116]
$ LiC104
1 LiC104 Au Ag| AgC104, + 1,4 — —1,0 [1H6]
LiC104+ Hg LiC104 Ag|AgC104, +0,1 — —0,5 [1116]
+HOAc LiC104 + Pt LiC104 AglAgClOj, + 1,4 — —0,7 [1П6]
к +HOAc LiC104+ Ag LiClO, Ag|AgC104, —0,1 —0,7 [1116]
+HOAc LiC104 —0,6 [1116]
LiClO,+ Au Ag|AgC104, + 1,4 —
W +HOAc LiC104 —2,0 [1116]
LiC104+ Hg Ag|AgC104, —0,3 —
4 +LiOAc LiC104+ Pt LiClO, Ag|AgClO4, +1,6 — —1,1 [1116]
+LiOAc LiC104+ Ag LiC104 Ag|AgC104, —0,2 — —1,1 [1116]
+LiOAc LiC104 —1,1 [1116]
LiC104+ Au Ag|AgC104, + 1,2 —
+LiOAc LiC104
i Акрилонит- рил ПТЭА КРЭ НКЭ — —1,5 [H16]
198
199
Продолжение табл. 12
Растворитель Фоновый электролит Рабочий электролн! Электрод сравнения Область рабочих потенциалов Литература
АН LiC104 Pt Ag|AgC104, +2,4 3,5 [1116]
LiCIO4
LidO4 Pt AglAgCl водн. +2,3 3,2 [154]
Lid Pt AglAgCl водн. + 1,15 3,15 [154]
LiNO3 Pt AglAgCl водн. +2,25 1,45 [154]
NaC104 КРЭ НКЭ +0,6 1,7 [1116]
NaC104 Pt нкэ + 1,8 1,5 [1Н6]
NaC104 Pt НКЭ +2,7 1,6 [680]
NaC104 Pt Ag|AgC104, +2,4 — —2,0 [1Н6]
LiC104
NaC104 Pt AglAgClHac , +2,7 - —1,2 [1Н6]
LiClfHac)
NadO4— —AgC104 Pt Ag|AgC104, NaC104 +2,0 — — [Н16]
NaBF6 Pt Ag|AgNO3 +4,0 — — [1116]
БТЭА КРЭ НКЭ —0,5 2,8 [1116]
ИТБА КРЭ НКЭ —0,6 2,8 [Н16]
ПТБА КРЭ Ag|AgC104 +0,28 3,23 [837]
ПТЭА КРЭ НКЭ +0,6 2,8 [1Н6]
ПТЭА СпЭ НКЭ + 1,1 — —0,7 [1032]
перхлораты КРЭ Ag +0,30 3,20 [1099]
перхлораты Pt Ag +2,00— —2,50 [1099]
ПТЭАА КРЭ Ag|Ag+ —0,2 2,25 [920]
БН ПТЭА КРЭ НКЭ +0,5 2,1 [681, 684]
Изобутиро- ПТЭА КРЭ НКЭ +0,67 2,82 [1116]
нитрил ПТЭА Pt НКЭ + 1,70 2,82 [1116]
дмсо LiC1O4 Pt AgAgCl + 1,55 3,40 [154]
Lid Hg НКЭ —0,21 2,17 [1Н6]
Lid Pt AgAgCl + 1,05 2,80 154]
LiNO3 Pt AgAgCl + 1,50 1,65 154]
LiPF6 Pt Li|Li+ +0,55 3,5 1254]
NaC104 Hg НКЭ +0,1 1,90 1116]
NaC104 Hg НКЭ +0,25 1,90 [681, 684]
NaC104 Hg НКЭ +0,25 1,9 [680]
NaC104 Pt нкэ —1,86— — [1116]
NaC104 Pt НКЭ —0,70 1,85 [1Н6]
(ПТЭА)
NaJ Pt НКЭ —1,81— — [1И6]
NaNO3 Hg НКЭ +0,31 1,85 [1116]
NaNO3 Hg нкэ +0,25 1,91 [1116
NaNO3 КРЭ Zn(Hg), ZnC104, + 1,33 0,83 [1052]
ДМСО
KC1O4 Hg нкэ +0,20— —1,84 [1116]
KC1O4 Hg нкэ +0,20 1,88 [1116]
ИТБА Hg нкэ —0,4 3,85 [1116]
НТЭА Hg нкэ +0,31 1,85 [1Н6]
НТЭА КРЭ нкэ —0,2 2,47 [893]
ПТБА Hg нкэ +0,30 3,00 [1116]
ПТБА Hg нкэ +0,37 2,85 [1116]
ПТЭА Hg нкэ +0,25 2,80 [1116]
ПТЭА Hg нкэ +0,28 2,77 [1116]
ПТЭА КРЭ Zn(Hg)|ZnC104, + 1,36 1,69 [1052]
дмсо 1
Продолжение табл. 12
растворитель Фоновый электролит рабочий электролит Электрод сравнения Область рабочих потенциалов Литература
Диметил- LiC104 КРЭ AglAgCl O4 +0,5 2,2 [H16]
сульфон ПТБА КРЭ Ag|AgC104 +0,5 3,0 [H16]
Диметил- LiC104 Pt НКЭ +2,4 [676]
сульфит
Сульфолан NaClO4 КРЭ AglAgCl + 1,2 1,3 [1116]
NaC104 Pt AglAgCl +3,3 1,3 [1116]
ПТЭА КРЭ AglAgCl + 1,2 2,3 [1116]
ПТЭА КРЭ Ag|AgC104 +0,04 3,5 [744]
ПТЭА Pt AglAgdO4 + 1,7 2,9 [744]
Этиленсуль- ПТЭА КРЭ нкэ +0,65 1,9 [885]
фит ПТЭА Hg НКЭ +0,57 2,3 [885]
ПТЭА Hg НКЭ +0,45 2,1 [891]
Метиловый LiC104 Pt Ag|AgClBQ£H + 1,30 1,25 [154]
спирт Lid Pt Ag|AgClBOm, + 1,00 1,15 [154]
LiCl КРЭ СРЭ 2,1 [H16]
LiNO3 Pt AglAgClB(^H + 1,25 1,20 [154]
NadO4 Pt Ag|Ag+ +0,81 — — [1221]
CH3ONa Pt Ag|Ag+ +0,87 [12?1]
БТЭА КРЭ СРЭ — 2,2 [H16]
Этиловый NaClO4 Pt AglAg+ +0,96 — — [1221]
спирт NaOC2H6 Pt Ag|Ag+ + 1,00 — — [1221]
LiC104 Pt Ag|AgClBOftH + 1,10 1,25 [154]
Lid Pt Ag|AgClBOflH + 1,65 1,30 [154]
LiNO3 Pt Ag|AgClBOftH + 1,60 1,25 [154]
Изопропило- LiC104 Pt Ag|AgC’BoW +2,35 3,25 [154]
вый спирт LiCl Pt Ag|AgdBOftB +0,95 3,20 [154]
LiNO3 Pt Ag|AgClBOftH + 1,9 1,4 [154]
1,2-Димето- ПТБА КРЭ НКЭ +0,65 2,95 1116]
ксиэтан ПТБА КРЭ Ag|AgC104 +0,9 3,6 1116]
ПТБА Pt AglAgNO3 + 1,0 — — 1116]
ПТБА АПЭ Ag|AgNO3 — 1,4 1116]
ТГФ LiC104 Hg Ag|AgC104 0 2,5 1116]
LiC104 Hg Ag|AgC104 —0,4 — —1,6 1116
LidO4 Pt Ag|AgC104 + 1,5 4,0 1116
LiC104 Pt AgjAgC104 + 1,8 3,6 1116
LiC104 Pt НКЭ +0,5 2,0 939]
LiC104 Ag Ag|AgC104 —0,5 3,6 1116
NaCICL Pt нкэ + 1,4 1116
ИТБА Hg Ag|AgC104 -1,2 3,6 1116
ИТБА Pt Ag]AgC104 -0,8 2,5 [1116]
нм LiC104 КРЭ Ag|Agd + 1,15 1,2 1116]
LiC104 Ag AglAgCl + 1,1 2,45 1116]
LiC104 Pt НКЭ +3,0 6761
LiC104 Pt НКЭ 2,6 1116]
LiC104 Pt AglAgCl — 2,4 1116]
Mg(C104)2 Pt НКЭ +2,2 2,6 1116]
Mg(C104)2 Pt AglAgCl — 2,2 1116]
KTMA Pt НКЭ +0,92 1,0 [1116]
200
201
1
Продолжение табл. 12
Растворитель Фоновый электролит Рабочий электролит Электрод сравнения Область рабочих потенциалов Литература
нм ПК ПТБА ПТЭА ПТЭА ПТЭА соль ТА А соль ТАА LiClO4 ЫС1О4 L1CIO4 LiClO, LiC104 LiCICL LiBF4 KPFe ПТЭА ПТЭА ПТЭА ПТЭА Pt КРЭ Hg СпЭ КРЭ Ag Pt Pt Pt Си Nj Ti Pt Pt Hg Hg Pt СпЭ AglAgCl НКЭ НКЭ НКЭ AglAgCl AglAgCl НКЭ НКЭ AgAgCl, ПК AgAgCl, ПК AgAgCl, ПК Ag AgCl, ПК AgAgCl, ПК НКЭ НКЭ НКЭ НКЭ НКЭ +0,55 +0,7 + 1,0 + 1,15 + 1,1 +2,2 +2,9 + 1,0 +0,2 +0,9 + 1,6 +0,75 +2,9 +0,5 +0,6 +1,7 +0,86 1 II II 1 1 II 1 1 1 II 1 II II | —1,0 —0,80 —1,3 -0,7 -0,85 -0,5 -3,2 —0,5 —3,0 —2,5 —1,0 —2,5 —2,9 —1,9 -0,6 [1116] [1190] [681, 6841 [1032] [1Н6] [1116] [681, 684] [676] [303 [303 [303 [303 [303 [676 [1Н6] [681, 684] [912] [Н16]
СПИСОК ЛИТЕРАТУРЫ
1. Абрашкина И. П., Агладзе Т. Р., Раскин Г. С.— Защита металлов, 1977,
13, № 6, с. 674—678.
2. Агасян П. К., Сираканян М. А.— Жури, аналит. химии, 1971, 26, №5,
с. 992—993.
3. Агасян П. К., Сираканян М. А„ Кристостурян Б. Г.— Арм. хим. жури.,
1978, 31, №2/3, с. 113—119.
4. Агладзе Т. Р., Абрашкина Н. П. Двойной слой и адсорбция на твердых
электродах, 5.— В кн.: Материалы Всесоюз. симпоз. 1978 г. Тарту: Изд-
во Тарт, ун-та, 1978, с. 9.
5. Агладзе Т. Р., Цурцумия Г. С., Абрашкина И. П.— Защита металлов,
1975, 11, №5, с. 609—612.
6. Агладзе Т. Р„ Цурцумия Г. С., Колотыркин Я. М.— Электрохимия. 1977,
13, №8, с. 1142—1146.
7. Акимов Г. В. Теория и методы исследования коррозии металлов.— М.:
Изд-во АН СССР, 1959,—252 с.
8. Алекперов А. И,— Электрохимия, 1968, 4, № 7, с. 847—850.
9. Алекперов А. И,-—Успехи химии, 1974, 43, № 4, с. 585—611.
10. Александров В. В. Кислотность неводных растворов.— Харьков : Вища
шк., 1981.—152 с.
11. Александров В. В., Безпалый Б. Н.— Электрохимия, 1978, 14, №1, с. 147—
149.
12. Алпатова Н. М., Дымова Т. Н., Кесслер Ю. М., Осипов О. Р.— Успехи
химии, 1968, 37, №2, с. 216—245.
13. Алпатова Н. М, Кесслер Ю. М., Горбанев А. И.— Электрохимия, 1965,
1, № 7, с. 844—850.
14. Алпатова И. М., Кесслер Ю. М., Кришталик Л. И. и др. Электрохимия
растворов в гексаметилфосфорамиде. М., 1975, с. 45—105 / ВИНИТИ. Сер.
Электрохимия; 1975. Т. 10.
15. Андреева А. А., Царевская М. Н.— Журн. физ. химии, 1973, 47, №10,
с. 2594—2598.
16. Андрусев М. М., Николаева-Федорович Н. В., Дамаскин Б. Б.— Электро-
химия, 1967, 3, № 10, с. 1247—1250.
17. Андрющенко Ф. К., Александров Ю. Л., Бекетов В. А.— Там же, 1976, 12,
№ 12, с. 1881—1884.
18. Андрющенко Ф. К., Бекетов В. А., Александров Ю. Л.— Bien. Харк. по-
л!техн. 1н-ту, 1972, вип. 4, №70, с. 32—35.
19. Аносов В. Я., Озерова М. И., Фиалков Ю. Я. Основы физико-химического
анализа.— М.: Наука, 1976.—504 с.
20. Антропов Л. И. Теоретическая электрохимия.— М.: Высш, шк., 1975.—
568 с.
21. Армалис С. Ю. Исследование электроосаждения алюминия из эфирно-
аминовых и эфириодативных электролитов: Автореф. дис. ... канд. хим.
наук.— Ростов н/Д, 1978.—24 с.
203
22. Армалис С. Ю.. Агафонов Г. В.. Левинскас А. Л.— В кн.: Теория и прак-
тика электроосаждения металлов и сплавов. Пенза: Приволжс. кн. изд-
во 1976. 111 с.
23. Армалис С. Ю., Левинскас А. Л.— Электрохимия, 1976, 12, № 11, с. 1697—
1701.
24. Армалис С. Ю., Левинскас А. Л.— Науч. тр. вузов ЛитССР. Сер. Химия
и хим. технология, 1973, 15, с. 227—232.
25. Атдаев О. Исследование анодного поведения циркония в спиртовых раст-
ворах хлористого водорода : Автореф. дис. ... канд. хим. наук.— М..,
1976.—24 с.
26. Бабаева М. А., Алекперов А. И.— В кн.: Исследования в области неор-
ганической и физической химии. Баку: Изд-во, АН АзССР 1971, с. 318—
322.
27. Бабенкова Л. Г., Балятинская Л. Н.— РЖ Химия, 1977, 24Б1433Деп.
28. Бабко А. К. Физико-химический анализ комплексных соединений в раст-
ворах.— Киев : Изд-во АН УССР, 1955.—205 с.
29. Байбарова Е. Л., Емельяненко Г. А., Мовчан В. В.— Укр. хим. журн.,
1978, 44, № 12, с. 1333—1335.
30. Байбарова Е. Я., Кайстре Л. Д., Емельяненко Г. А.— Там же, 1971, 37,
№ 4, с. 376—378.
31. Балятинская Л. Н.— Успехи химии, 1979, 48, №4, с. 772—791.
32. Барабанов В. П., Овсянников И. Н.— Электрохимия, 1969, 5, № 8,
с. 899—901.
33. Барбашева И. Е., Поваров Ю. М., Луковцев П. Д.— Там же, 1972, 8,
№9, с. 1275—1280.
34. Басов В. П. Физико-химический анализ двойных жидких систем, образо-
ванных треххлористой сурьмой со спиртами: Автореф. дис. ... канд. хим.
наук.— Киев, 1969,—20 с.
35. Басов В. П„ Бородин П. М., Кондратенков Г. П. и др.— Журн. общ. хи-
мии, 1971, 41, №6, с. 1181—1185.
36. Басов В. П., Бородин П. М., Кондратенков Г. П., Фиалков Ю. Я. Теорет.
и эксперим. химия, 1969, 5, № 4, с. 558—563.
37. Басов В. П., Карапетян Ю. А., Крысенка А. Д. и др.— Журн. общ. хи-
мии, 1975, 45, №7, с. 1428—1432.
38. Басов В. П„ Карапетян Ю. А., Крысенка А. Д. и др.— Докл. АН СССР,
1973, 211, №4, с. 851—852.
39. Басов В. П., Карапетян Ю. А., Крысенка А. Д., Фиалков Ю. Я.— Укр.
хим. журн., 1975, 41, №6, с. 583—585.
40. Басов В. П., Фиалков Ю. Я.— Журн. общ. химии, 1971, 41, №6, с. 1181—
1184.
41. Баталов А. П., Коршунов И. А.— Тр. по химии и хим. технологии, 1960,
3, с. 501—505.
42. Баталов А. П., Коршунов И. А,—Журн. общ. химии, 1961, 31, №7,
с. 1649—1652.
43. Баталов А. 77., Коршунов И. А.— Докл. АН СССР, 1961, 136, №1, с. 93—
95.
44. Баталов А. П„ Коршунов И. А.— Тр. по химии и хим. технологии, 1960,
3, с. 495—499.
45. Белецкая Т. Б., Стыркас А. Д„ Кочегаров В. М— Электрохимия, 1971,
7, № 11, с. 1628—1632.
46. Пат. 709886 (Бельгия). Procede pour le depot d’un revetement de metal
sur un article.— Опубл. 06.10.71.
47. Беляков E. А., Тихонов И. Я.—Электрохимия, 1978, 14, №10 с. 1543—
1548.
48. Беляков Е. А., Тихонов И. И., Ротинян А. Л,—Там же, с. 1549—1552.
49. Берштейн О. В.— Зап. Ин-та химии АН УССР, 1947, 9, с. 47—53.
50. Библиографический указатель литературы по полярографии.— Кишинев :
Штиинца, 1968—1980.— Вып. 1—28.
51. Богдашев Н. Н., Гонтмахер Н. М., Гарновский А. Д., Осипов О. А,—
В кн.: Ингибирование и пассирование металлов. Ростов н/Д, 1976, с. 111—
116, Изд-во Рост, ун-та.
204
52. Богомолова 3. В., Тихонов К. И., Ротинян А. Л.— РЖ Химия, 1980, 1,
Б1324Деп.
53. Боженко Л. Г. Влияние природы раствора и ПАВ на кинетику электро-
осаждения и контактного выделения некоторых металлов в водно-органи-
ческих средах: Автореф. дис. ... канд. хим. наук.— Ростов н/Д, 1980.—
27 с.
54. Боженко Л. Г., Кучеренко С. С., Бурлова А. С.— В кн.: 8-я Всесоюз. на-
уч.-техн. конф, по электрохим. технологии: Тез. докл. Казан, хим.-техн,
ин-та, Казань, 1977, с. 71.
55. Бокрис Дж., Конуэй Б. Современные аспекты электрохимии.— М.: Мир,
1967,—510 с.
56. Бондарь В. В., Гришина В. В.— Итоги науки и техники / ВИНИТИ. Сер.
Электрохимия; Т. 13. М., 1978, с. 155—187.
57. Боровиков А. Я. Влияние растворителя на термодинамические характе-
ристики электронодонорно-акцепторного взаимодействия (по — комплексы
йода) : Автореф. дис. ... канд. хим. наук.— Киев, 1976.—22 с.
58. Брагин Н. И., Авдеев В. П.— Исслед. в обл. хим. источников тока, 1977,
№5, с. 142—148.
59. Браго И. И., Томилов А. П.— Электрохимия, 1968, 4, №6, с. 697—699.
60. Брежнева Н. А., Рогинский С. Э., Шилинский А. И.— Журн. физ. химии,
1937, 10, № 3, с. 367.
61. Бродский А. И. Избранные труды : В 2-х т.— Киев : Наук, думка, 1974.—
Т. 1, с. 335. Т. 2, с. 519.
62. Будников Г. К-, Торопова В. Ф., Улахович Н. А., Витер И. П.— Журн.
аналит. химии, 1974, 29, №6, с. 1204—1209.
63. Будников Г. К., Торопова В. Ф., Улахович Н. А., Витер И. П.— Там же,
1975, 30, № 11, с. 2120—2124.
64. Будников Г. К-, Троепольская Т. В.— Успехи химии, 1979, 48, №5,
с. 829—853.
65. Будников Г. К-, Троепольская Т. В., Улахович Н. А. Электрохимия хе-
латов металлов в неводных средах.— М.: Наука, 1980.—192 с.
66. Будников Г. К., Шарапова Т. С., Стурис А. П., Торопова В. Ф.— Журн.
общ. химии, 1979, 49, № 5, с. 1113—1118.
67. Будников Г. К-, Улахович Н. А.— Журн. аналит. химии, 1973, 28, Ks3,
с. 448—453.
68. Будников Г. К-, Улахович Н. А.— Успехи химии, 1980, 49, №1, с. 147—
170.
69. Будников Г. К-, Улахович Н. А., Витер И. П., Колесникова Л. В.— Журн.
общ. химии, 1974, 44, № 4, с. 730—737.
70. Булейшвили М. И., Фиалков К). Я., Чумак В. Л.— Укр. хим. журн., 1984,
50, №2, с. 214—216.
71. Буслаев Ю. А.— В кн.: Термодинамика и строение растворов. Иваново,
Изд-во Иванов, хим.-техн, ин-та, 1977, с. 36—44.
72. Бухтиоров А. В., Томилов А. П., Будько В. П., Луканин М. Н.— Электро-
химия, 1979, 15, №3, с. 321—327.
73. Бяллозор С. Г.— Там же, 1970, 6, № 12, с. 1827—1834.
74. Бяллозор С. Г.— Там же, №8, с. 1190—1196.
75. Бяллозор С. Г.— Там же, 1972, 8, № 5, с. 755—757.
76. Бяллозор С. Г.— Там же, 1976, 12, №7, с. 1054—1060.
77. Бяллозор С. Г., Полэтэк Д.— Там же, 1979, 15, № 4, с. 472—476.
78. Бяллозор С. Г., Лисовска А.— Там же, с. 634—638.
79. Вайсбергер А., Проскауэр Э„ Риддик Дж., Тупс Э. Органические раство-
рители.— М.: Изд-во иностр, лит., 1958.—518 с.
80. Васильев В. П. Термодинамические свойства растворов электролитов.—
М.: Высш, шк., 1982.—320 с.
81. Ваулин Л. В., Кузнецов С. И.— Изв. вузов. Цв. металлургия, 1972, N°5,
с. 49—51.
82. Ваулин Л. В., Кузнецов С. И.— Там же, 1973, №5, с. 48—51.
83. Ваулин Л. В., Кузнецов С. И.— Там же, 1974, № 1, с. 31—33.
84. Ваулин Л. В., Кузнецов С. И.— Там же, 1978, № 1, с. 76—78.
85. Ваулин Л. В., Кузнецов С. И.— Там же, №2, с. 83—86.
205
86. Вигдорович В. И., Оше Е. К., Корнеева Т. В., Пчельников И. Т.— Защи-
та металлов, 1979, 15, № 1, с. 75—78.
87. Вигдорович В. И., Цыганкова Л. Е.— Там же, 1974, 10, №5, с. 549—
551.
88. Вигдорович В. И., Цыганкова Л. Е„ Осипова Н. В., Корнеева Т. В.—
Электрохимия, 1976, 12, № 12, с. 1791—1797.
89. Вигдорович В. И., Цыганкова Л. Е., Филиппова Н. В.— Защита металлов,
1974, 10, №4, с. 427—431.
90. Вигдорович В. И., Цыганкова Л. Е., Шарифулина И. И.— РЖ Химия,
1978, №10, Б1198Деп.
91. Владимирова В. Ф., Шабанова Т. М.— В кн.: Физико-химические мето-
ды анализа и контроля производства. Махачкала: Северокавказ. науч,
центр, высш. шк. 1976, вып. 2, с. 103—106.
92. Войтович Б. А., Звагольская Е. В.— Изв. АН СССР. Сер. Металлы, 1965,
с. 72—80.
93. Волков С. В., Яцимирский К. Б. Спектроскопия расплавленных солей.—
Киев : Наук, думка, 1978, с. 317.
94. Волков С. В., Трачевский В. В., Матящук Н. В.— Докл. АН СССР, 1977,
232, №6, с. 1336—1339.
95. Вольнов Ю. Н.— Журн. физ. химии, 1961, 35, № 1, с. 90—93.
96. Воробьев А. Ф., [Цербаков В. В., Ксенофонтова И. А.— Тр. МХТИ им.
Д. И. Менделеева, 1980, № 111, с. 21—34.
97. Вредные вещества в промышленности.— Л. : Химия, 1977.— Т. 1—3.
98. Галинкер В. С.— Журн. общ. химии, 1956, 26, №11, с. 1564—1566.
99. Галинкер В. С., Фикс А. Ф.— Там же, 1955, 25, №3, с. 463—468.
100. Галинкер В. С., Фикс А. Ф.— Изв. Киев, политехи, ин-та, 1960, 29,
с. 43—47.
101. Гаммет Л. П. Основы физической органической химии.— М.: Мир, 1971.—
715 с.
102. Гвоздев Б. А., Чубурков Ю. Т.— Радиохимия, 1963, 5, №6, с. 712—715.
103. Гейровский Я., Кута Л. Основы полярографии.— М.: Мир, 1965.—315 с.
104. Герасимович Ч. В. Электролитические свойства расплавов системы бро-
мистый алюминий — диэтиловый эфир : Автореф. дис.... канд. хим. наук.—
Вильнюс, 1971.—19 с.
105. Германова И. М., Григорьев В. П.— В кн.: Ингибирование и пассивиро-
вание металлов. Ростов н/Д : Изд-во Рост, ун-та, 1976, с. 48—53.
106. Германова И. М., Григорьев В. П., Иващенко О. А.— РЖ Химия, 1979,
17, Б1434Деп.
107. Германова И. М., Григорьев В. П., Иващенко О. А., Ребрин Е. И.— РЖ
Химия, 1979, 17, Б1436Деп.
108. Германова И. М., Кравченко В. М.— В кн.: Ингибирование и пассивиро-
вание металлов.— Ростов н/Д : Изд-во Рост, ун-та, 1976, с. 117—121.
109. Гериюв В. М., Суетин В. П.— В кн.: Химические и электрохимические ме-
тоды защиты металлов. Саратов : Приволж. кн. изд-во, 1977, с. 62—63.
НО. Гесслер Н. М„ Плесков В. А,—Журн. физ. химии, 1950, 24, №4, с. 445—
454.
111. Глесстон С., Лейдлер К-, Эйринг Г. Теория абсолютных скоростей реак-
ций.— М.: Изд-во иностр, лит., 1948.—585 с.
112. Годнева М. М.— Журн. неорган. химии, 1964, 9, №4, с. 996—1001.
113. Гонтмахер Н. М-, Григорьев В. П„ Абакумов Г. А. и др.— Докл. АН
СССР, 1976, 228, № 4, с. 846—848.
114. Гонтмахер И. М., Нечаева О. Н„ Григорьев В. П., Некрасов Л. Н,—Элек-
трохимия, 1977, 13, № 11, с. 1748—1751.
115. Гонтмахер Н. М„ Нечаева О. Н„ Кравченко В. М„ Бартенева О. И,—
В кн.: Ингибирование и пассивирование металлов. Ростов н/Д : Изд-во
Рост, ун-та, 1976, с. 122—128.
116. Гордон Дж. Органическая химия растворов электролитов.— М. : Мир,
1979,—712 с.
117. Горенбейн Е. Я.— Журн. неорган. химии, 1959, 4, №12, с. 1643—1646.
118. Горенбейн Е. Я-, Данилова В. Н.— Тр. Киев. вет. ин-та, 1957, 13, с. 301.
206
119. Горенбейн Е. Я., Горенбейн А. Е.— Журн. неорган. химии, 1967, 12, №7,
с. 1813—1820.
120. Горенбейн Е. Я., Кудра О. К., Горенбейн А. Е.— Журн. физ. химии, 1972,
46, № 4, с. 996—997.
121. Горенбейн Е. Я., Сухан В. В.— Журн. неорган. химии, 1963, 8, №3, с.
360—364.
122. Городыский А. В., Фиалков Ю. Я., Черный Д. Б.— Укр. хим. журн., 1982,
48, № 4, с. 433—435.
123. Городыский А. В., Фиалков Ю. Я-, Черный Д. Б.— Там же, 1981, 47, №8,
с. 879—881.
124. Григорьев В. П., Гонтмахер Н. М., Нечаева О. Н., Богдашев И. Н.—
Электрохимия, 1980, 16, №4, с. 489—494.
125. Григорьев В. П., Экилик Г. Н., Экилик В, В.— Защита металлов, 1979, 15,
№ 6, с. 667—672.
126. Григорьев В. П„ Экилик Г. И., Экилик В. В.— В кн.: Ингибирование и
пассивирование металлов. Ростов н/Д : Изд-во Рост, ун-та, 1976, с. 39—45.
127. Гутман В. Химия координационных соединений в неводных растворах.—
М.: Мир, 1976.—220 с.
128. Давыдов А. Д., Камкин А. Н.— Электрохимия, 1978, 14, №7, с. 979—992.
129. Дамаскин Б. Б. Принципы современных методов изучения электрохими-
ческих реакций.— М.: Изд-во МГУ, 1965.—180 с.
130. Дамаскин Ь. Б,, Иванова Р. В.— Успехи химии, 1979, 48, №10, с. 1747—
1772.
131. Джапаридзе Д. И„ Енукидзе Л. Г., Шавгунидзе В. В., Джапаридзе Ш. С.—
Электрохимия, 1980, 16, № 4, с. 472—476.
132. Джапаридзе Д. И., Шавгунидзе В. В„ Чачелишвили В. А.— Там же, 1974,
10, №9, с. 1414—1416.
133. Джусупов Е. А., Тихонов К. И.— Журн. прикл. химии, 1979, 52, №8, с.
1887—1889.
134. Длахей П. Новые приборы и методы в электрохимии.— М.: Изд-во
иностр, лит., 1957.—509 с.
135. Днепровский А. С., Темникова Т. И. Теоретические основы органической
химии.— Л.: Химия, 1979.—390 с.
136. Добош Д. Электрические константы.— М.: Мир, 1980.—365 с.
137. Добровольские П. Р. Исследование этилпиридинбромидного электролита
алюминия: Автореф. дис.... канд. хим. наук.— Вильнюс, 1972.—20 с.
138. Дорофеева В. Ф„ Рогатинская С. Л., Крешков А. П„ Жданов С. И.—
Журн. общ. химии, 1978, 48, №9, с. 1928—1932.
139. Дорофеева Н. Г., Ковальская В. П„ Фиалков Ю. Я., Чумак В. Л.— Укр.
хим. журн., 1982, 48, № И, с. 1147—1150.
140. Дорофеева Н. Г., Фиалков Ю. Я., Шляханова С. Н.— Там же, 1984, 50,
№3, с. 239—241.
141. Дорош А. К. Структура конденсированных систем.— Львов : Изд-во Льв.
ун-та, 1981.—175 с.
142. Драчевская Р. К., Алмазова Н. Г., Лукина А. Г.— В кн.: Научные труды
Казахского политехнического института. Алма-Ата: Каз. политехи, ин-т
1971, с. 548—550.
143. Дынер Л. Л.— Вест. Киев, политехи, ин-та. Сер. Хим. машиностроение и
технология, 1978, вып. 15, с. 64—65.
144. Ермаков В, И., Щербаков В. В.— Magy. Kern. Lapga, 1977, 31, №11, с.
560—570 (англ.).
145. Ермаков В. И., Щербаков В. В.— В кн.: Термодинамика и строение раст-
воров. Иваново : ИХТИ, 1979, с. 104—116.
146. Ерусалимчик И. Г., Ефимов Е. А.— В кн.: Электрохимические процессы
при электроосаждении и анодном растворении металлов. М.: Наука,
1969, с. 129—134.
147. Ерусалимчик И. Г., Ефимов Е. А.— Электрохимия, 1969, 5, №1, с. 15—19.
148. Ерусалимчик И. Г., Ефимов Е. А., Мотыле ва Н. В,—В кн.: Электрохи-
мические процессы при электроосаждении и анодном растворении метал-
лов. М.: Наука, 1969, 1, с. 155—161.
149. Ерусалимчик И. Г., Ефимов Е. А., Прилепина Т. И., Гериш Т. В.— Там
же, с. 162—169.
207
150. Ефимов А. Г., Тихонов К. И., Ламанский Н. Ю., Ротинян А. Л.— Элек-
трохимия, 1979, 15, №3, с. 365—369.
151. Ефимов А. Г, Тихонов К. И., Ротинян А. Л.— РЖ Химия, 1979,2Б1494Деп.
152. Жданов С. И. Полярография в органических средах.— В кн.: Успехи и
перспективы развития полярографического метода.— Кишинев : Штиинца,
1972 с. 73_____82.
153. Жданов С. И.— Успехи химии, 1973, 42, №9, с. 1698—1707.
154. Жданов С. И. Полярография: Пробл. и перспективы.— Рига: Зинатне,
1977,—412 с.
155. Жданов С. И., Киселев Б. А.— Collection, 1966, 31, №2, с. 788—805.
156. Жданов С. И., Поваров Ю. Л4.— №.; 1979, с. 46—63 (Итоги науки/
ВИНИТИ. Сер. Электрохимия ; Т. 9).
157. Житомирский А. И., Квитка А. А., Фиалков Ю. Я — Укр. хим. журн.,
1979, 45, №5, с. 412—415.
158. Житомирский А. И., Квитка А. А., Фиалков Ю. Я-, Эйчис В. Н.— Химия
и хим. технология, 1980, 23, №9, с. 1096—1100.
159. Житомирский А. Н., Стаднийчук П. М—Жури. физ. химии, 1974, 48,
№ 9, с. 2246—2249.
160. Жураев У., Халудханова Ш. 3., Юсупова Н„ Шереметьева В. Б.— Узб.
хим. журн., 1978, № 6, с. 21—24.
161. Заболоцкий В. И., Вольтер Д., Тихонов К- И.— Журн. прикл. химии, 1974,
47, № 1, с. 65—68.
162. Заболоцкий В. И., Тихонов К- И., Ротинян А. Л.— Электрохимия, 1973,
9, № 2, с. 222—227.
163. Заболоцкий В. И., Шишкина С. В., Тихонов К- И., Ротинян А. Л.— Там
же, №6, с. 799—801.
164. Занько А. М., Манусова Ф. Н.— Журн. общ. химии, 1940, 10, №13,
с. 1171—1176.
165. Затулина О. С., Ломакина Т. П., Дьякова А. П., Харин А. Н.— Изв.
Сев. кавк. науч, центра высш. шк. Сер. Естеств. науки, 1976, № 3, с. 54—
55.
166. Зеленкин В. At., Погодин В. П., Воробьев А- Ф. Теплопроводность фор-
мамидных растворов иодидов щелочных металлов при температурах 323,
348 и 373 °C.— В кн.: Термодинамические свойства растворов. М,: МХТИ,
1981, с. 118—121.
167. Зотов И. А., Хлыстова К- Б.— Электрохимия, 1966, 2, №12, с. 1444—1448.
168. Ибраева Т. Д., Невская Ю. А., Сумарокова Т. Н.— В кн.: Теория раст-
воров. Алма-Ата : Изд-во Казах, уи-та, 1971.—306 с.
169. Иванов И. М.— В кн.: Проблемы сольватации и комплексообразования.
Иваново : ИХТИ, 1980, с. 171—184.
170. Иванов В. Ф., Дамаскин Б. Б.— В кн.: Адсорбция и двойной электричес-
кий слой в электрохимии. №.: Наука, 1972, с. 15—24.
171. Изидинов С. О., Блохина А. П., Комыса Н. Г.— Журн. прикл. химии, 1974,
47, № 1, с. 158—162.
172. Измайлов А. В.— Журн. физ. химии, 1956, 30, №11, с. 2599—2601.
173. Измайлов Н. А. Электрохимия растворов.— №.: Химия, 1976.—575 с.
174, Измайлов И. А., Царевская М. И.— Укр. хим. журн., 1962, 28, №2, с. 101.
175. Ильясов Р. Ш., Нечаев А. В., Петрова Н. А. и др.— В кн.: 8-я Всесоюз.
науч.-техн. конф, по электрохим. технологии, Казань 13—16 сент. 1977 г.:
Тез. докл. Казань; КХТИ, 1977, с. 73.
176. Ингауните Б. И. Исследование эфирноаммиачного электролита алюмини-
рования : Автореф. дис. ... каид. хим. наук,— Вильнюс, 1974.—21 с.
177. Ингауните Б. И., Левинскас А. Л.— Науч. тр. вузов ЛитССР. Химия и
хим. технология, 1973, 15, с. 233—235.
178. Иозянос А. Л., Степанавичус А. А., Вишомирскис Р. Al.— Тр. АН
ЛитССР. Сер. Б, 1979, № 1, с. 9—17.
179. Иозянос А. Л., Степанавичус А. А., Вишомирскис Р. М.— Там же, №2,
с. 11—24.
180. Ионин М. В., Шерстнева Т. В., Рогозина At. Б. Рукопись деп. в ВИНИТИ,
30.08.77, №3563—77 Деп. Редкол. Изв. вузов. Сер. химия и хим. техноло-
гия.
208
181. Ионы и ионные пары в органических реакциях.— М.: Мир, 1975.—424 с.
182. Каабак Л. В., Кабачник М. И., Томилов А. П., Варшавский С. Л.— Журн.
общ. химии, 1966, 36, № 12, с. 2060—2063.
183. Каблуков И. А. Исторический обзор развития учения о неводных раст-
ворах : Сб„ посвящ. 35-летию науч, деятельности акад. Плотнико-
ва В. А.— Киев : Изд-во АН УССР, 1936.—288 с.
184. Кварацхели Р. К., Габриадзе Т. Ш.— Электрохимия, 1980, 16, №5,
с. 704—707.
185. Кварацхели Р. К-, Габриадзе Т. Ш.— In: 29 th. meet. Int. soc. electro-
chem., Budapest, 1978 : Extend. Abstr. SI. S. a., pt. 1, p. 628—638.
186. Казаков В. А., Титова В. И., Петрова И. В.— Электрохимия, 1976, 12,
№ 4, с. 576—579.
187. Казаков В. А., Титова В. Н., Петрова И. В.— In: 22 Int. Wiss. Kollog.
Techn. Hochson. Ilmenau, 1977. S. 1 : S. a., H. 3, s. 27—29.
188. Казаков В. А., Титова В. И., Смирнова С. А.— Защита металлов, 1979,
15, №2, с. 235—237.
189. Калюжная П. Ф.— Укр. хим. журн., 1952, 18, №6, с. 661—665.
190. Калюжная П. Ф., Зосимович Д. П.— Там же, 1955, 21, №1, с. 27—31.
191. Камысбаев Д. X. Автореф. дис. ... канд. хим. наук.— Алма-Ата, 1978.—
21 с.
192. Капустинский А. Ф., Солохин В. А.— Изв. вузов. Химия и хим. техноло-
гия, 1958, № 4, с. 3—7.
193. Карапетян Ю. А. Кислотно-основное взаимодействие и механизмы пере-
носа тока в двойных системах, образованных хлоридами Sn (IV), Sb (V),
Fe (III), Sb (III) с алифатическими спиртами и уксусной кислотой: Ав-
тореф. дис. ... канд. хим. наук.— Киев, 1971.—19 с.
194. Карапетян Ю. А., Крысенке А. Д., Фиалков Ю. Я.— Укр. хим. журн.,
1976, 42, №11, с. 1210—1213.
195. Каргин Ю. М., Латыпова В. 3., Устюгова И. А. и др.— Журн. общ. хи-
мии, 1979, 49, № 10, с. 2272—2275.
196. Катаев Г. А., Гудымович Е. Н., Панова Н. М.— В кн.: Реакционная спо-
собность веществ. Томск : Изд-во Том. ун-та 1977, с. 29—33.
197. Пат. 54—17300 (Япония). Органический электролит для осаждения А1.
198. Катиладзе Д. Д., Джапаридзе Дж. И. Двойной слой и адсорбция на
твердых электродах.— В кн.: Материалы Всесоюз. симпоз., Тарту, июль
1978 г. Тарту : Изд-во Тарт, ун-та, 1978, с. 88—89.
199. Кацнельсон I. Л.— Зап. 1н-ту х!мп АН УРСР, 1947, 9, Ks 1, с. 63—85.
200. Кащеева Г. П., Дубихина В. С., Гадасина Л. Ю.— Защита металлов,
1976, 12, №6, с. 681—683.
201. Кедринский И. А., Кузнецов Т. В., Морозов С. В. и др.— Электрохимия,
1976, 12, №9, с. 1458—1460.
202. Кедринский И. А., Кузнецова Т. В., Морозова О. Г., Потапова Г. П.—
В ки.: Химия и технология полимеров. Красноярск, 1975, 4, с. 133—136.
203. Кедринский И. А., Морозов С. В., Сухова Г. И., Соколов Л. А.— Электро-
химия, 1976, 12, №7, с. 1191.
204. Кесслер Ю. М., Алпатова Н. М., Осипов О. Р.— Успехи химии, 1964, 33,
№3, с. 261—295.
205. Кикец В. О.— Зап. 1н-ту х!мП АН УРСР, 1936, 3, №4, с. 489—507.
206. Кильчевская Т. Е.— Журн. прикл. химии, 1970, 43, №5, с. 1062—1068.
207. Киселев Б. А., Жданов С. И.— Изв. АН СССР. Сер. хим., 1965, №6,
с. 985—989.
208. Климов В., Усанович М., Сумарокова Т.— Изв. АН КазССР. Сер. хим.,
1957, №2, с. 14—17.
209. Клочко М. А.— Изв. АН СССР. Сер. хим., 1937, №5, с. 641—650.
210. Кобзева И. Т., Ковсман Е. П., Васильев Ю. Б. и др.—-Электрохимия,
1975, 11, №5, с. 718—724.
211. Ковсман Е. П., Тюрин Ю. М., Караваев Е. А.— Журн. прикл. химии,
1964, 37, № 1, с. 217—218.
212. Колесникова Т. И. Влияние природы растворителей и катиона фона иа
электроосаждение А1 из алкилбензольных электролитов: Автореф. дис. ...
... канд. хим. наук.— Ростов н/Д, 1981.—25 с.
14 — 4-653
209
213. Коломиец Б. С., Ковсман Е. П., Чемоданов А. Н. и др.— Защита метал-
лов, 1970, 6, №6, с. 678—681.
214. Колотыркин Я- М.— Успехи химии, 1962, 31, № 3, с. 322—335.
215. Колотыркин Я- At.— Журн. Всесоюз. хим. о-ва им. Д. И. Менделеева,
1971, 16, №6, с. 627—633.
216. Колотыркин Я- At., Агладзе Т. Р., Цурулия Г. С.— Электрохимия, 1975,
И, № 10, с. 1549—1555.
217. Колотыркин Я. К1., Коссый Г. Г.— Защита металлов, 1965, 1, №3, с. 272—
276.
218. Кондратенко В. П.— Журн. общ. химии, 1934, 4, №3, с. 244—246.
219. Коновалова Л. Л.— Тр. Естеств.-науч. ин-та/Перм. ун-т, 1975, 13, №3,
с. 66—75.
220. Коноплянцева Н. А.— Исслед. в обл. хим. источников тока, 1977, №5,
с. 162—170.
221. Коротким Ю. С.— Радиохимия, 1974, 16, №3, с. 377—382.
222. Коршак В. В., Лебедев Н. //.— Журн. общ. химии, 1948, 18, №12,
с. 1766—1768.
223. Косиенко Ю. Л., Овчаренко А. Н., Дмитровский В. Н.— Тр. Новочеркас.
политехи, ин-та, 1976, № 320, с. 31—33.
224. Костромин А. И., Абдулин И. Ф.— В кн.: 8-я Всесоюз. науч.-техн. конф,
по электрохим. технологии, Казань, 13—16 сент. 1977 г.: Тез. докл. Ка-
зань : КХТИ, 1977, с. 102.
225. Костромин А. Н., Абдулин И. Ф., Шалхметова Р. X.— РЖ Химия, 1978,
15Б1663Деп.
226. Костынюк В. П. Реакции в жидкой фазе.— Алма-Ата : Наука, 1979.—
118 с.
227. Коссый Г. Г., Соколова Л. А., Колотыркин -Я- М.— Защита металлов,
1974, 10, №6, с. 706—711.
228. Крестов Г. В. Термодинамика ионных процессов в растворе.— Л.: Химия,
1973._____302 с
229. Кришталик Л. И.— Успехи химии, 1965, 34, № 12, с. 1831—1845.
230. Кропачев В. С., Толстая М. А., Шиловская М. Е., Хворостухин Л. А.—
Защита металлов, 1980, 16, № 6, с. 721—724.
231. Крысенке А. Д. Кислотно-основное взаимодействие и механизм переноса
тока в системах, образованных Н и L — кислотами с амидами уксусной
кислоты: Автореф. дисс. ... канд. хим. наук.— Киев : 1981.—20 с.
232. Крысенке А. Д., Фиалков Ю. Я., Храненко С. П., Чучалин Л. К.— Изв.
Сиб. отд-ния АН СССР, 1971, 12, №5, с. 141—142.
233. Кудра О. К, Житомирский А. Н„ Фиалков Ю. Я.— Докл. АН СССР,
1963, 151, №2, с. 377—379.
234. Кудра О. К, Клейбс Г. С.— Зап. 1н-ту xiMii АН УРСР, 1941, 8, № 1,
с. 83—93.
235. Кудра О. К., Туров П. П.— Укр. хим. журн., 1950, 16, №2, с. 230—241.
236. Кудра О. К-, Туров П. П.— Журн. физ. химии, 1951, 25, №5, с. 513—522.
237. Кудра О. К, Фиалков Ю. Я-, Житомирский А. И.— Журн. неорган. химии,
1963, 8, Я» 7, с. 1737—1741.
238. Кудра О- К., Фиалков Ю. Я-, Житомирский А. Н.— Журн. неорган. хи-
мии, 1963, 8, №7, с. 1742—1747.
239. Кудра О. К., Фиалков Ю. Я., Житомирский А. Н.— Там же, 1964, 9, № 10,
с. 2454—2457.
240. Кузнецов В. В.— В кн.: Тез. основных докл. 1-й Укр. респ. конф, по кор-
розии и противокорроз. защите металлов. Львов, Изд-во Льв. ун-та, 1979,
с. 22—23.
241. Кузнецов В. В., Боженко Л. Г., Петрова И. В.— Изв. Сев. Кавк. иауч.
центра высш. шк. Сер. Естеств. наук, 1976, № 2, с. 47—49.
242. Кузнецов В. В., Василькевич Н. Т., Дамаскин Б. Б.— Электрохимия, 1969,
5, №8, с. 997—1000.
243. Кузнецов В. В., Гонтмахер Н. М., Боженко Л. Г., Нечаева О. Н.— В ки.:
Ингибирование и пассивирование металлов. Ростов н / Д : Изд-во Рост,
ун-та, 1976, с. 53—65.
244. Кузнецов В. В., Григорьев В. П., Атоян Л. К.— В кн.: 8-я Всесоюз. иауч.-
210
тех. конф, по электрохим. технологии. Казань, 13—16 сент. 1977 г.: Тез,
докл. Казань : КХТИ, 1977, с. 72—73.
245. Кузнецов В. В., Григорьев В. П„ Боженко Л. Г.—Электрохимия, 1980, 16,
№4, с. 495—501.
246. Кузнецов В. В., Григорьев В. П., Боженко Л. Г. и др.— В кн.: Двойной
слой и адсорбция на твердых электродах : Материалы Всесоюз. симпоз.
Тарту, июль 1978 г. Тарту : Изд-во Тарт, ун-та, 1978, с. 120—124.
247. Кузнецов В. В., Григорьев В. П., Боженко Л. Г., Шпанько С. П.— В кн.:
Ингибирование и пассивирование металлов. Ростов н / Д: Изд-во Рост, ун-
та, 1976, с. 157—163.
248. Кузнецов В. В., Григорьев В. П., Кучеренко С. С.— Защита металлов, 1978,
14, №4, с. 500—504.
249. Кузнецов В. В., Григорьев В. П„ Осипов О. А. и др. Способ электрохими-
ческого осаждения ниобия. А. с. 367178 (СССР).— Опубл, в Б. И., 1973,
№ 8. с. 77.
250. Кузнецов В. В., Григорьев В. П., Перерва В. В., Атоян Л. К-—В кн.: Ин-
гибирование и пассирование металлов. Ростов н / Д : Изд-во Рост, ун-та,
1976, с. 176—181.
251. А. с. 490869 (СССР). Электролит для осаждения свинец-медных сплавов/
В. В. Кузнецов, В. П. Григорьев, О. А. Осипов, С. П. Шпанько.— Открытие,
изобретение, 1975, №41, с. НО.
252. Кузнецов В . В., Григорьев В. П., Шпанько С. П., Боженко Л. Г.— Защи-
та металлов, 1975, 11, №5, с. 631—634.
253. Кузнецова Т. В., Жугаева В. И., Кедринский И. А., Райхельсон Л. Б.—
Электрохимия, 1976, 12, № 10, с. 1631—1634.
254. Кузнецов В. В., Казаков В. А., Григорьев В. А. и др.— Там же, 1980, 16,
№ 5, с. 646—650.
255. Кузнецов В. В., Коган В. А., Григорьев В. П. и др. А. с. 505755 (СССР).—
Опубл, в Б. И., 1976, № 9, с. 87.
256. Кузнецов В. В., Коган В. А., Григорьев В. П. и др. А. с. 506639 (СССР).—
Опубл, в Б. И., 1976, № 10, с. 73.
257. Кузнецов В. В., Коган В. А., Григорьев В. П. и др.— В кн.: Тез. докл. со-
вет.. Новая технология гальванических покрытий. Киров : Верхне-Волж. кн.
изд-во, 1974, с. 81—82.
258. Кузнецов В. В., Федорова О. В.— В кн.: Ингибирование и пассирование
металлов. Ростов н/Д : Изд-во Рост, ун-та, 1976, с. 181—185.
259. Кузнецов В. В., Шпанько С. П.— Изв. Севкавк. науч, центра высш. шк.
Сер. Естеств. науки, 1979, № 1, с. 36—43.
260. Кузнецова Т. В., Кедринский И. А., Иванов Е. Г., Потапова Г. П.— Элект-
рохимия, 1976, 12, №9, с. 1453—1454.
261. Кузьмина Е. Г., Вольнов Ю. Н.— Журн. физ. химии, 1954, 28, №2, с. 254—
286.
262. Куйдина Р. А., Сумарокова Т. Н.— Изв. АН КазССР. Сер. хим., 1972, №4,
с. 38.
263. Кулаковская С. И., Ефимов О. Н.— Журн. неорган. химии, 1979, 53, №4,
с. 1057—1060.
264. Кулаковская С. И., Ефимов О. Н.— Электрохимия, 1980, 16, №4, с. 593—
597.
265. Куляко Ю. М., Мясоедов Б. Ф., Скляренко И. С.—-Журн. неорган. химии,
1975, 20, №7, с. 1799—1805.
266. Курнаков Н. С., Воскресенская Н. В.— Изв. АН СССР. Сер. хим., 1936,
№ 3, с. 439—443; 1937, № 4, с. 797—803.
267. Курнаков Н. С., Перельмутер С. И., Капов Ф. П. Журн. Рус. физ.-хим.
о-ва, 1916, 48, №4, с. 1658—1671.
268. Курнаков И. С., Штернина Э. Б.— Изв. АН СССР. Сер. хим., 1936, №3,
с. 267—281.
269. Кутыгин Е. Н., Фомичев В. Т., Озеров А. М. Твердые износостойкие галь-
ванические покрытия,—М.: Химия, 1980, с. 184.
270. Кутыгин^ Е. Н., Фомичев В. Т., Озеров А. М. Теория и практика гальвано-
покрытий из коллоидных систем и нетоксичных электролитов.— Новочер-
касск, Изд-во Ростов н/Д ун-та, 1979.—НО с.
14*
211-
271. Кучеренко С. С. Влияние природы и строения раствора на процесс элект-
роосаждения некоторых металлов и сплавов в органических средах. Авто-
реф. дис. ... канд. хим. наук.— Ростов н/Д, 1980, 28 с.
272. Лаврухина А. К.., Юкина Л. В. Аналитическая химия марганца.— М.: Нау-
ка, 1974.-211 с.
273. Лазарев В. Ф., Суханова Л. С., Левин А. И.— Электрохимия, 1975, 11,
№ 5, с. 841—845.
274. Ламанский Л. Ю., Тихонов К. И.— Журн. неорган. химии, 1979, 24, №3,
с. 921_____926.
275. Лебедев И. Н.— Журн. фнз. химии, 1948, 22, № 11, с. 1505—1509,
276. Лебедев И. Н.— Журн. общ. химии, 1951, 21, №12, с. 1788—1791.
277. Лебедев Н. Н.— Успехи химии, 1952, 21, № 10, с. 1399—1422.
278. Левин А., Есин О.— Журн. общ. химии, 1937, 7, №10, с. 1478—1487.
279. Левинскас А. Л.— Электрохимия, 1965, 1, № 1, с. 115—117.
280. Левинскас А. Л.— Науч. тр. вузов ЛитССР. Химия и хим. технология,
1971, 13, с. 271—274.
281. Левинскас А. Л.— Там же, 1972, 14, с. 205—212.
282. Левинскас А. Л.— Успехи химии, 1975, 44, №7, с. 1236—1255.
283. Левинскас А. Л. Исследование процессов электроосаждения алюминия
из эфирных электролитов : Автореф. дис. ... д-ра хим. наук.— Вильнюс,
1976,—36 с.
284. Левинскас А. Л., Армолис С. Ю.— Науч. тр. вузов ЛитССР. Химия и хим.
технология, 1974, 16, с. 255—265.
285. Левинскас А. Л., Армолис С. Ю., Ингауните Б. И.— Электрохимия, 1978,
14, № 10, с. 1611—1614.
286. Левинскас А. Л., Герасимовичус Ч. В.— Науч. тр. вузов ЛитССР. Химия
и хим. технология, 1972, № 14, с. 213—219.
287. Левинскас А. Л., Герасимовичус Ч. В.— Там же, 1973, №15, с. 217—226.
288. Левинскас А. Л., Герасимовичус Ч. В., Армолис С. Ю.— Тр. АН ЛитССР.
Сер. Химия и хим. технология, 1971, 1, с. 95—100.
289. Левинскас А, Л., Герасимовичус Ч. В., Синюс Я. Ю.— Электрохимия,
1967, 3, №6, с. 781—784.
290. Левинскас А. Л., Симанавичус А. Ю.— Там же, 1966, 2, №2, с. 200—204.
291. Левинскас А. Л., Симанавичюс Л. Э.— Тр. АН ЛитССР. Сер. Б, 1971,
№ 2, с. 73—78.
292. Левинскас А. Л., Симанавичюс Л. Э.— Там же, 1972, №3, с. 117—124.
293. Левинскас А. Л., Симанавичюс Л. Э.— В кн.: Исследования в области
электроосаждения металлов. Вильнюс : ИХХТ, 1973, Т. 2, с. 147—152.
294. Левинскас А. Л., Симановичюс Л. Э., Акимов В. Я.— Тр. АН ЛитССР.
Сер. Б, 1977, 5, с. 37—46.
295. Левинскас А. Л., Синюс Я- Ю.— Там же, 1969, 4, с. 87—93.
296. Левинскас А. Л., Синюс Я. Ю.— Там же, 1970, 3, с. 93—106.
297. Левинскас А. Л., Синюс Я. ТО.— Электрохимия, 1972, 8, №7, с. 1053—
1055.
298. Левинскас А. Л., Синюс Я. Ю., Ингауните Б. И.— Там же, 1970, 6, № 10,
с. 1505—1508.
299. Левинскас А. Л., Синюс А. Ю., Ингауните Б. И., Сарапинес А. И.— Там
же, 1974, 10, №5, с. 712—717.
300. А. с. 533679 (СССР). Электролит для осаждения мышьяка/А. Л. Ле-
винскас, С. М. Шумаускайте.— Открытие, изобретение ... , 1976, № 40,
с. 78.
301. Левинскас А. Л., Шумаускайте С. М., Павидите 3. И.— Электрохимия,
1978, 14, № 12, с. 1885—1889.
302. Левинскас А. Л., Юстинович Я. Я-— В кн.: Электрохимические процессы
при электроосажденин и анодном растворении металлов. М.: Наука, 1969,
с. 52—56.
303. Липец Т. В., Старостина В. В., Спрыгина М. А.— Электрохимия, 1978,
14, № 10, с. 1560—1562.
304. Лосев В. В., Городецкий В. В.— Там же, 1967, 3, №9, с. 1061—1069.
305. Лысенко Ю. А.— Журн. общ. химии, 1967, 37, № 1, с. 11—13.
306. Лысенко Ю. А., Артемьева В. М., Ведмедская А. Н.— Там же, 1970, 40,
№ 6, с. 737—739.
212
307. Лысенко Ю. А., Ведмедская А. Н., Каплан Л. М.— Теорет. и эксперим.
химия, 1972, 8, №6, с. 681—685.
308. Лысенко Ю. А., Каплан Л. М„ Ведмедская А. Н., Артемова В. М.— Журн.
общ. химии, 1974, 41, № 10, с. 1132—1134.
309. Лысенко Ю. А., Краснянский М. Е.— Там же, 1968, 38, №12, с. 1913—
1915.
310. Лысенко Ю. А., Краснянский М. Е.— Теорет. и эксперим. химия, 1969,
5, №2, с. 115—118.
311. Лысенко Ю. А., Краснянский М. Е.— Журн. общ. химии, 1969, 39, №10,
с. 1676—1680.
312. Лысенко Ю. А., Краснянский М. Е.— Там же, №12, с. 1902—1904.
313. Лысенко Ю. А., Краснянский М. Е., Исачкина Л. Ф.— Там же, 1968, 38,
№ 7, с. 953—955.
314. Лысенко Ю. А., Медведева В. И.— Там же, 1969, 39, №10, с. 1205—1207.
315. Лысенко Ю. А., Медведева В. И.— Там же, 1970, 40, №7, с. 965—968.
316. Лысенко Ю. А., Медведева В. И., Краснянский М. Е.— Там же, №12,
с. 2002—2004.
317. Лысенко Ю. А., Медведева В. И., Курапатова А. А.— Изв. вузов. Сер.
Химия и хим. технология, 1970, № 12, с. 1257—1260.
318. Лысенко Ю. А., Осипов О. А.— Журн. общ. химии, 1954, 24, №1, с. 53—
56.
319. Лысенко Ю. А., Осипов О. А., Акопов Е. К. Журн. неорган. химии, 1956,
1, № 4, с. 536—539.
320. Манн Ч„ Барнес К. Электрохимические реакции в неводных системах.—
М.: Химия, 1974.—479 с.
321. Майрановский С. Г., Родионов Н. П., Тультяй В. П.— Завод, лаб., 1974,
40, №5, с. 518—519.
322. Майрановский С. Г., Страдынь Я. П., Безуглый В. Д. Полярография в
органической химии.— Л.: Химия, 1975.—350 с.
323. Макаренко Б. К, Поваров Ю. М., Середа П. Л.—Электрохимия, 1976,
12, №4, с. 518—521.
324. Макаренко Б. К., Поваров Ю. М., Середа П. А., Боброва Н. Ю.— Там
же, 1973, 9, №5, с. 705—707.
325. Макитра Р. Г., Макарук М. С., Дидыч М. Н. Деп. ВИНИТИ, 4281—72.
326. Макитра Р. Г., Пириг Я. Н.— Реакц. способность орган, соединений, 1980,
17, №2, с. 184—205.
327. Макитра Р. Г„ Середницкий Я. А., Никипанчук Н. В.— Укр. хим. журн.,
1971, 37, № 1, с. 46.
328. Макридин В. П., Есипов Г. 3.— Сб. науч. тр. Кузбас. политехи, ин-та,
1971, 36, с. 156—159.
329. Мамаев В. И., Тихонов К. И.— Электрохимия, 1975, 11, №8, с. 1086—
1089.
330. Мамедов М. Н., Алекперов А. И.— Азерб. хим. журн., 1977, №1, с. 119—
123.
331. Мат у лис Ю. Ю., Скоминас В. Ю„ Шяудинскас А. А.— Тр. АН ЛитССР.
Сер. Б, 1972, 3, с. 15—21.
332. Медяник В. Н„ Шула Г. В.—РЖ Химия, 1976, 18Б 1472К-
333. Меншуткин Б. Н.— Журн. Рус. физ.-хим. о-ва, 1906, 38, с. 1398—1415;
1907, 39, с. 102—110; 1911, 43, с. 425—440.
334. Меншуткин Б. Н.— Журн. Рус. физ.-хим. о-ва, 1909, 41, с. 1053—1070.
335. Меншуткин Б. Н.— Журн. Рус. физ.-хим. о-ва, 1910, 42, с. 1310—1322.
336. Меншуткин Б. Н.— Изв. Санкт-Петербург, политехи. ин-та, 1909, 11,
с. 261, 567.
337. Меншуткин Б. Н.— Там же, 1910, 13, № 1, с. 14—22.
338. Меншуткин Б. Н.— Журн. Рус. физ.-хим. о-ва, 1910, 42, с. 1298—1309.
339. Методы измерений в электрохимии.— М.: Мир, 1977.— Т. 2. 475 с.
340. Мещерякова И. Д„ Дубихина В. С., Кащеева Т. П., Рутковский М. Л,—
Защита металлов, 1968, 4, №3, с. 242—247.
341. Мещерякова И. Д., Кащеева Т. П., Рутковский М. Л.— Там же, 1970, 6,
№ 3, с. 286—289.
213
342. Мигель П. К-, Гринберг Н. X.— Журн. неорган. химии, 1961, 6, №3,
с 727______731.
343. Мискиджьян С. П.— Журн. общ. химии, 1953, 23, №11, с. 1947—1952.
344. Мискиджьян С. П., Гарновский А. Д. Введение в современную теорию
кислот и оснований.— Киев : Вища шк., 1979.—152 с.
345. Миркин В. А., Костынюк В. П.— В кн.: Теория растворов. Алма-Ата:
Изд-во Каз. ун-та, 1971.—309 с.
346. Миронов Б. Е„ Батяев И. М., Белькова Н. Л.— Электрохимия, 1970, 6,
№ 3, с. 354—356.
347. Мищенко К. П., Полторацкий Г. М. Термодинамика и строение водных и
неводных растворов электролитов.— Л.: Химия, 1976.—327 с.
348. Заявка на пат. №53—77843 (Япония). Электролит для осаждения спла-
ва олово—кобальт/А. Миясичэ.— Опубл. 10.07.78.
349. Молодое А. И.— Электрохимия, 1979, 13, № 11, с. 1625—1630.
350. Молодое А. И., Лосев В. В.— М., 1971, с. 65—113.— (Итоги науки/
ВИНИТИ. Сер. Электрохимия ; Т. 7).
351. Молодое А. И., Маркосьян Г. Н., Лосев В. В.— Электрохимия, 1971, 7,
№ 2, с. 263—267.
352. Молодое А. И., Янов Л. А.— Защита металлов, 1978, 14, №2, с. 194—
197.
353. Молодое А. И., Янов Л. А., Голодницкая Д. В.— Электрохимия, 1977, 13,
№ 2, с. 300—303.
354. Молодое А. И., Янов Л. А., Лосев В. В.— Там же, 1976, 12, №4, с. 513—
517.
355. Молодое А. И., Янов Л. А., Лосев В. В.— Защита металлов, 1976, 12,
№ 5, с. 578—581.
356. Молодое А. И., Янов Л. А., Лосев В. В. и др.— Электрохимия, 1979, 15,
№ 1, с. 122—124.
357. Муллаянов Р. X., Вахидов Р. С., Томилов А. П., Черных И. Н.— Там же,
1978, 14, №2, с. 334—338.
358. Мурыгина Н. Г., Дулова В. И.— Химия и хим. технология, 1968, 11, №5,
с. 511—515.
359. Мясоедов Б. Ф., Куляко Ю. М., Скляренко И. С.— Радиохимия, 1975, 17,
№ 3, с. 434—440.
360. Мясоедов Б. Ф., Мюзикас К.— Там же, 1970, 12, №6, с. 856—859.
361. Мясоедов Б. Ф., Скляренко И. С., Куляков Ю. М.— Жури, неорган. хи-
мии, 1972, 17, № 11, с. 2934—2937.
362. Невская Ю. А., Крючкова Е. И.— Тр. Ин-та хим. наук АН КазССР, 1983,
60, с. 31—87.
363. Неводные растворители / Под ред. Т. Баддингтона.— М.: Химия, 1971.—
374 с.
364. Ненов И. П., Иконописов С. М.~ Годишн. Висш. хим.-техиол. ин-т. Со-
фия, 1977(1978), 23, № 1, с. 129—136.
365. Норман А.— Успехи химии, 1970, 39, №7, с. 990—1004.
366. Нигматуллин Р. Ш., Вяселев М. Р. Полярография: Пробл. и перспекти-
вы.— Рига : Зинатне, 1977.—412 с.
367. Никитин К- Н., Атдаев О. В.— В кн.: 11-й Менделеев, съезд по общ. и
прикл. химии : Реф. докл. и сообщ. М.: Наука, 1975, № 3, с. 67.
368. Николаева Л. А., Басова С. К,—Защита металлов, 1980, 16, №4
с. 469—471.
369. Нурманова А. К., Усанович М. И., Сумарокова Т. Я.—Изв. АН КазССР.
Сер. хим., 1962, № 1, с. 32.
370. Озеров А. М., Филатов Н. Г., Литвиненко Н. П. и др.— Защита метал-
лов, 1969, 5, № 5, с. 583.
371. Одрит Л., Клейнберг Я- Неводные растворители.— М.: Изд-во иностр,
лит., 1955.—320 с.
372. @дРит Л., Огг Б. Химия гидразина.— М.: Изд-во иностр, лит., 1954.—
373. Осипов О. Р., Алпатова Н. М., Кесслер Ю. М.— Электрохимия, 1966 2,
№ 8, с. 984—988.
214
374. Осипов О. А., Гайворонский В. И., Швец А. А,—Журн. неорган. химии,
1963, 8, № 12, с. 2190—2194.
375. Осипов О. Р., Кесслер Ю. М.— Электрохимия, 1971, 7, №7, с. 923—927.
376. Осипов О. А., Клетеник Ю. В.— Журн. общ. химии, 1957, 27, №12,
с. 2921—2923; 1959, 29, №6, с. 1423—1425.
377. Осипов О. А., Кравцов Ю. Е.— Сб. ст. по общ. химии АН СССР, 1953,
1, с. 216—220.
378. Осипов О. А., Лысенко Ю. А.— Журн. неорган. химии, 1958, 3, №7,
с. 1605—1607.
379. Осипов О. А., Лысенко Ю. А., Акопов Е. К—Журн. общ. химии, 1955,
25, № 2, с. 249—252.
380. Осипов О. А., Сучков В. Л-—Там же, 1952, 22, №9, с. 1132—1135.
381. Остроумов В. В.— Жури, прикл. химии, 1974, 37, №7, с. 1483—1490.
382. Павлов В. Н., Бондарь В. В.— Успехи химии, 1973, 42, №6, с. 987—1008.
383. Павлович О. Ж., Войнович М. В.— Гласиик Хим. друшт. Београд, 1978,
43, №9, с. 591—602.
384. Павлович О. Ж., Войнович М. В.— Там же, № 10, с. 665—682.
385. Аналитическая химия урана и тория / Под ред. П. Н. Палея.— М.: Изд-
во иностр, лит., 1956.—365 с.
386. Пальм В. А. Основы количественной теории органических реакций.— Л.:
Химия, 1977.—359 с.
387. Палыиин Е. С., Мясоедов Б. Ф., Давыдов А. В. Аналитическая химия
протактиния.— М.: Наука, 1968.—210 с.
388. Пейсхалова М. Е., Гольдштейн И. П., Гурьянова Е. Н., Кочешков К- А.—
Докл. АН СССР, 1972, 203, с. 1316.
389. Перелыгин И. С.— В кн.: Термодинамика и строение растворов. Ивано-
во : ИХТИ, 1976, с. 135—148.
390. Персианцева В. П., Розенфельд И. Л., Зорина В. Е. и др.— Зашита ме-
таллов, 1979, 15, № 3, с. 309—319.
391. Пилипенко А. Т„ Шпак Э. А., Самчук А. И.— Журн. аналит. химии, 1975,
30, № 4, с. 717—720.
392. Пилипенко А. Т., Шпак Э. А., Самчук А. И.— Там же, №6, с. 1086—
1090.
393. Пилипенко А. Т., Шпак Э. А., Самчук А. И.— Завод, лаб., 1974, 40, № 3,
с. 250—252.
394. Пилипенко А. Т„ Шпак Э. А., Самчук А. И.— Укр. хим. журн., 1975, 41,
№ 2, с. 195—197.
395. Писаржевский Л. В. Избранные труды.— Киев : Изд-во АН УССР, 1936.—
700 с.
396. Писчик Л. М., Цинман А. И.— Защита металлов, 1979, 15, №2, с. 198—
200.
397. Плесков В. А.— Успехи химии, 1947, 16, №3, с. 254—278.
398. Плеханов В. Г., Кедринский И. А., Кузнецова Т. В. и др.— Электрохи-
мия, 1980, 16, №5, с. 677—678.
399. Плотников В. А.— В кн.: Работы по химии растворов и комплексных сое-
динений. Киев : Изд-во АН УССР, 1959, с. 3—71.
400. Плотников В. А.— Журн. Рус. физ.-хим. о-ва, 1907, 39, №1—2, с. 163—
190.
401. Плотников В. А., Балясный С. С.— Жури. общ. химии, 1931, 1, №6,
с. 823—830.
402. Плотников В. О., Бармашенко I. Б., Птман Е. Б — Зап. 1н-ту х!мп АН
УРСР, 1938, 5, № 1, с. 1—21.
403. Плотников В. О., Вайсберг Р. Г.— Там же, 1936, 3, с. 337—341.
404. Плотников В. А., Гитман Е. Б.— Жури, прикл.’ химии, 1946, 19, №8,
с. 826—832.
405. Плотников В. О., Горенбейн Е. Я.— Зап. 1и-ту xiMii АН УРСР, 1936, 3,
№ 1, с. 89—109.
406. Плотников В. О., Городисъкий В. /.—Там же, 1941, 8, №1, с. 1—22.
407. Плотников В. О., Д1брова А. Г,—Там же, 1940, 7, №3, с. 337—350.
408. Плотников В. О., Зосимович Д. П., Кириченко Е. I.— Там же, 1937, 4,
№ 1, с. 15—27.
215
409. Плотиков В. А., Каплан М. Л.— Изв. АН СССР. Сер. химия, 1948, №2,
с. 256—262.
410. Плотников В. О., Кудра О. К.— Зап. 1н-ту xiMi’i АН УРСР, 1937 , 4, №4,
с. 405—412.
411. Плотников В. А., Кудра О. К, Меженный Я- Ф.—Журн. общ. химии,
1936, 6, № 9, с. 1286—1290.
412. Плотников В. А., Подорван И. М.— Журн. общ. химии, 1953, 3, с. 787.
413. Плотников В. О., Спектор Б. В.— Зап. 1н-ту xiMii АН УРСР, 1940, 7,
№ 3, с. 429—437.
414. Плотников В., Шека И., Янкелевич 3. Електрол1тичне вилучення метал!в
з неводних розчишв.— К.: Вид-во АН УРСР, 1935.—110 с.
415. Плотников В. А., Якубсон С. И.— Журн. общ. химии, 1936, 6, №11,
с. 1694—1697.
416. Плотников В. О., Янкелевич 3. А.— Зап. 1н-ту xiMii’ АН УРСР, 1938, 5,
№ 1, с. 39—55.
417. Поваров Ю. М., Барабашева И. Е., Луковцев П. Д.— Электрохимия, 1967,
3, № 10, с. 1202—1206.
418. Поваров Ю. М., Бекетаева Л. А.— Там же, 1980, 16, № 8, с. 1252—
1256.
419. Поваров Ю. М., Ерошкина Л. В., Луковцев П. Д.— Там же, 1969, 5, №5,
с. 512—517.
420. Подорван И. М„ Пенкало И. И.— BicH. КиТв. ун-ту. Сер. ф1зики та xiMii,
1967, №7, с. 97—103.
421. Поздеева А. А., Вахрушева Н. А., Муллагашев И. Р. и др.— Журн. прикл.
химии, 1980, 53, № 1, с. 42—45.
422. Поспелов Д. П.— Там же, 1954, 27, №5, с. 552—557.
423. Преснов А. Е., Томилов А. П., Промоненков В. К, Фионин М. Я.— М..,
1971, с. 293—326.— (Итоги науки и техники / ВИНИТИ. Сер. электрохи-
мия ; Т. 6).
424. Приходченко В. Г.— Укр. хим. журн., 1953, 19, № 5, с. 479—483.
425. Птицын М. В., Алялина Т. В., Тихонов К. И.— Электрохимия, 1979, 15,
№ 12, с. 1782—1786.
426. Птицын М. В., Рахмилевич Я. Р., Тихонов К. И., Ротинян А. Л.— Элект-
рохимия, 1980, 16, №5, с. 740—742.
427. Птицын М. В., Тихонов К И., Ротинян А. А.— Там же, с. 737—740.
428. Пятницкий И. В., Ружанская Р. П.— Журн. аналит. химии, 1969, 24, №5,
с. 650—653.
429. Пятницкий И. В., Ружанская Р. П.— Там же, 1970, 25, №6, с. 1063—
1068.
430. Пятницкий И. В., Ружанская Р. П.— Там же, 1971, 26, №8, с. 1475—1478.
431. Рабинович В. Я-, Пономаренко А. Г.— Укр. хим. журн., 1953, 19, № 3,
с. 264—267.
432. Рабинович В. Я-, Пономаренко А. Г.— Журн. общ. химии, 1953, 23, №6,
с. 923—925.
433. Равдель Б. А., Ефимов А. Г., Карбасов Б. Г., Тихонов К- Я.— Электрохи-
мия, 1970, 16, №7, с. 1061—1064.
434. Равдель Б. А., Никольская Е. Ю., Тихонов К. И., Ротинян А. Л.— Там
же, 1980, 16, №7, с. 1057—1061.
435. Райхард X. Растворители в органической химии.— Л.: Хим-ия, 1973.—
107 с.
436. Рафиков Ф. М., Петросян В. А.— Изв. АН СССР. Сер. хим., 1980, с. 749—
754.
437. Реформатская И. А.— Радиохимия, 1971, 13, № 1, с. 89—94.
438. Робинсон Р-, Стокс Р. Растворы электролитов.— М.: Изд-во иностр, лит.,
1963.—646 с.
439. Розенбойм Г. Б., Осипов В. Н.— Защита металлов, 1980, 16, № 1, с. 63—
64.
440. Романенко Г. М. Механизмы кислотно-основного взаимодействия, элект-
ролитической диссоциации и переноса тока в системах апротонные кис-
лоты— апротонные органические основания: Автореф. дис. ... канд. хим.
наук.— Киев, 1974.—20 с.
216
441. Ротинян А. Л., Заболоцкий В. И., Тихонов К. И.— Электрохимия, 1973,
9, № 2, с. 225—227.
442. Ротинян А. Л., Заболоцкий В. И., Тихонов К. И.— Там же, №10,
с. 1511—1514.
443. Ротинян А. Л., Заболоцкий В. И., Тихонов К. И.— Там же, 1974, 10, №5,
с. 777—780.
444. Ротинян А. Л., Шишкина С. В., Тихонов К- И.— Там же, №4, с. 662—664.
445. Ротинян А. Л., Шишкина С. В., Тихонов К. И.— Там же, 1975, 11, №7,
с. 1094—1096.
446. Ротинян А. Л., Шишкина С. В., Тихонов К- И., Соколов Л. А.— Там же,
№ 10, с. 1493—1497.
447. Рудницкая А. А. Связь электролитических свойств с ионно-молекулярным
составом в двойных жидких системах серная (дейтеросерная) кислота —
эфиры : Автореф. дис. ... канд. хим. наук.— Киев, 1982.—20 с.
448. Рудницкая А. А., Майоров В. Д-, Либрович Н. Б., фиалков Ю. Я-— Изв.
АН СССР. Сер. хим., 1981, № 5, с. 960—966.
449. Рудницкая А. А., Майоров В. Д., Либрович Н. Б., Фиалков Ю. Я-— Там
же, № 11, с. 2478—2484.
450. Рудницкая А. А., Стаднийчук П. М., Фиалков Ю. Я-—Журн. физ. химии,
1981, 55, № 10, с. 2660—2662.
451. Савинцева С. А., Драговцева Н. А.— Электрохимия, 1976, 12, №12,
с. 1884—1888.
452. Саркисов Э. С.— Докл. АН СССР, 1936, 23, №7, с. 678—681.
453. Сборник, посвященный 35-летию научной деятельности акад. Плотнико-
ва В. А.— Киев : Изд-во АН УССР, 1936.—288 с.
454. Комплексообразование в неводных растворах / Под ред. Т. Н. Сумароко-
вой.— Алма-Ата: Наука, 1983.—211 с.
455. Сидельникова С. П-, Шерсткина В. Н., Ягубец А. Н.— Изв. АН МССР.
Сер. физ.-техн. и мат. наук, 1979, № 1, с. 69—74.
456. Симанавичюс Л. Э., Добровольские П. П.— Тр. АН ЛитССР. Сер. Б, 1972,
№ 3, с. 125—134.
457. Симанавичюс Л. Э-, Добровольские П. П.— Там же, №5, с. 99—ПО.
458. Симанавичюс Л. Э., Добровольские П. П.— В кн.: Исследования в облас-
ти электроосаждения металлов. Вильнюс, 1973, т. 2, с. 131—136.
459. Симанавичюс Л. Э., Добровольские П. П., Шаркис А. А.— В кн.: Тез.
докл. 5-го Всесоюз. совещ. по электрохимии. Янв. 1975, М.: ВИНИТИ,
1975, т. 2, с. 148—150.
460. Симанавичюс Л. Э., Карпавичюс А. П.— В кн.: Всесоюз. конф, по элект-
рохимии, 1969 : Тезисы докл. Тбилиси : Мицниереба, 1969, с. 589—590.
461. Симанавичюс Л. Э., Карпавичюс А. П.— Тр. АН ЛитССР. Сер. Б, 1971,
№ 1, с. 83—93.
462. Симанавичюс Л. Э., Левинскас А. Л., Карпавичюс А. П.— Электрохимия,
1966, 2, №2, с. 87—88.
463. Синюс А. Ю., Левинскас А. Л.— В кн.: Исследования в области электро-
осаждения металлов. Вильнюс : ИХХТ, 1973, т. 2, с. 144—147.
464. Скляренко С. И., Сахаров Б. А-—Журн. прикл. химии, 1940, 13, №6,
с. 841—845.
465. Славинская Р. А., Литвяк И. К-, Левченко А. В. и др.— Журн. общ. хи-
мии, 1969, 39, №3, с. 481—485.
466. Слайдинь Г. С., Залекснис Я- И., Багоцкий В. С.— Изв. АН ЛатССР.
Сер. хим., 1979, №2, с. 198—200.
467. Соколова И. Ю., Цыганов Г. А., Байбородов П. П.— Узб. хим. журн.,
1974, № 1, с. 44—46.
468. Соловьев Ю. И. История учения о растворах.— М.: Изд-во АН СССР,
1959,—581 с.
469. Сорокин И. Н„ Козлов В. И.— Изв. АН СССР. Сер. Неорган. материалы,
1979, 15, № 3, с. 537—538.
470. Сперанская Е. Ф., Чернова Р. Г.— В кн.: XI Менделеев, съезд по общ.
и прикл. химии. М.: Наука, 1975, № 3, с. 233—234.
471. Справочник по дипольным моментам/Под ред. О. А. Осипова.— М.:
Высш, шк., 1971.—414 с.
217
472. Стакенос А. П. Исследование электроосаждений алюминия из электро-
литов некоторых комплексов триэтилалюминия : Автореф. дис. ... канд.
хим. наук.— Вильнюс, 1972.—18 с.
473. Столяренко Л. И., Странков М. И., Ле махин А. Г.— Электрохимия, 1979,
15, № 3, с. 378—381.
474. Стрелец В- В., Бабкина О. Н.— РЖ Химия, 1978, 24 Б1682Деп.
475. Стрелец В. В., Бабкина О. И.— Журн. физ. химии, 1979, 53, №3, с. 790—
794.
476. Стромберг А. Г., Рейнус Л. М.— Там же, 1946, 20, №7, с. 693—705.
477. Суетин В. П., Ванага Н. Т.— Изв. АН ЛатвССР. Сер. хим., 1978, № 1,
с. 13—15.
478. Сурова М. Д., Жданов С. И.— Электрохимия, 1973, 9, № 3, с. 350—352.
479. Сурова М. Д., Жданов С. И.— Там же, 1974, 10, №6, с. 994—996.
480. Сурпина Л. В., Лещенко А. В., Осипов О. А.— Журн. общ. химии, 1970,
40, № 5, с. 975—979.
481. Сурпина Л. В., Лещенко А. В., Осипов О. А.— Там же, №9, с. 1915—
1918.
482. Сысоев А. Н., Гавырина Н. Н.— Журн. прикл. химии, 1961, 34, №9,
с. 2001—2009.
483. Сэтчел Д. Г., Сэтчел Р. С.— Успехи химии, 1973, 42, №6, с. 1009—1036.
484. Таубальд К., Швабе К.— Защита металлов, 1969, 5, №5, с. 579—580.
485. Терпугов Ф. И.— Журн. общ. химии, 1934, 4, №3, с. 235—239.
486. Тихонов К. И., Беляков Е. А.— Электрохимия, 1980, 16, №4, с. 600—602.
487. Тихонов К И., Заболоцкий В. И., Вольтер Д.— Там же, 1974, 10, №6,
с. 985—986.
488. Тихонов К И., Ламанский Л. Ю., Вольтер Д.— Там же, 1977, 13, №8,
с. 1207—1210.
489. Тихонов К. И., Птицын М. В-, Ламанский Л. Ю.— Там же, 1979, 15, №6,
с. 873—876.
490. Тихонов К. И., Черновьянц М. С., Багдасаров К. И., Феоктистова С. Г.—
Изв. вузов. Химия и хим. технология, 1978, 21, №8, с. 1121—1124.
491. Тихонова Л. С., Курчатова Н. А., Райхельсон Л. Б. и др.— Электрохимия,
1978, 14, № 10, с. 1563—1566.
492. Тихонова Л. С., Тихонов К- И.— Там же, № 4, с. 634.
493. Толопко Л. К-, Макитра Р. Г.— Укр. хим. журн., 1973, 39, №4, с. 395—
397.
494. Томашев Н. Д.— Успехи химии, 1955, 24, № 4, с. 453—470.
495. Томашев Н. Д., Альтовский Р. М., Владимиров В. Б.— В кн.: Коррозия
и защита конструкционных металлических материалов. М., 1961, с. 164—172.
496. Томилов А. П., Браго И. Н., Осадченко И. М.— Электрохимия, 1968, 4,
№ 10, с. 1153—1156.
497. Томилов А. П., Осадченко И. М., Хомутов Н. Е.—-Итоги науки и техники /
ВИНИТИ. Сер. Электрохимия, 1979, 14, с. 168—207.
498. Троепольская Г. В., Мунин Е. Н., Китаев Ю. Н.— Координац. химия,
1979, 5, №9, с. 1367—1371.
499. Турамов А. И., Мухамметгалиев Т. Г.— Защита металлов, 1971, 7, №6,
с. 741—744.
500. Турашев А. И., Губская В. П.— Там же, 1977, 13, №1, с. 121—123.
501. Турашев А. И., Губская В. П.— Там же, 1979, 15, №6, с. 724—726.
502. Турова Н. Я., Кедрова Н. С., Семененко К Н., Новоселова А. В.— Журн.
неорган. химии, 1964, 9, № 6, с. 905—909.
503. Тюрин Ю. М., Веселова М. В., Куратова В. А- и др.— Журн. прикл. хи-
мии, 1962, 35, №5, с. 1082—1092.
504. Улахович Н. А., Будников Г. К.— Жури. общ. химии, 1977, 47, №7,
с. 1588—1595.
505. Улахович Н. А., Будников Г. К, Дорогова Т. Н., Фомина Л. Г.— Там же,
1978, 48, №9, с. 2122—2127.
506. Улько И. Г., Блинникова В. Д.— Докл. Моск. с.-х. акад., 1978, №238,
с. 131—136.
507. Усанович М. И—Жури. общ. химии, 1940, 10, №8, с. 959—962.
508. Усанович М. И.— Z. Phys. Chem., 1926, 124, 427—430; 1929, 140, 429—
433 (нем).
218
509. Усанович М. И. Исследования в области теории растворов и теории кис-
лот и оснований.— Алма-Ата : Наука, 1970.—364 с.
510 Усанович М. И., Абланова Е. X — Изв. АН КазССР. Сер. хим., 1967, №6,
с. 85—88.
511. Усанович М. И., Абланова Е. X.— Там же, 1968, № 1, с. 26—30.
512. Усанович М. И., Абланова Е. X.— Там же, 1969, № 6, с. 23—27.
513. Усанович М. И., Абланова Е. X., Михайленко С. М — Там же, 1968, №5,
с. 33—37.
514. Усанович М. И., Дембицкий А. Д,—Жури. общ. химии, 1959, 29, с. 1771—
1781.
515 Усанович М. И., Климов В. И., Сумарокова Т. Н.— Докл. АН СССР,
1957, 113, №2, с. 364—365.
516 Усанович М. И., Нурманова А. К-, Сумарокова Т. Н.— Журн. общ. хи-
мии, 1961, 31, с. 3493—3498.
517. Усанович М. И., Нурманова А. К; Сумарокова Т. Н.— Изв. АН КазССР.
Сер. хим., 1962, № 1, с. 32—37.
518. Усанович М. И., Пичугина Е.~ Журн. общ. химии, 1956, 26, №8,
с. 2125—2127.
519. Усанович М. И., Розентретер Р.— Там же, 1932, 2, №7, с. 864—867.
520. Фархадов А. А., Асатрян В. Г., Абален Н. П. и др.— Защита металлов,
1968, 4, №3, с. 319—321.
521. Федоров П. И., Мохосоев М. В., Алексеев Ф. П. Химия галлия, индия и
таллия.— Новосибирск : Наука, 1977.—220 с.
522. Федорович Н. В.— Итоги науки и техники / ВИНИТИ. Сер. Электрохимия,
1978, 14, с. 5—56.
523. Федорович Н. В., Леви М. Д., Дамаскин Б. Б., Шлепаков А. В.— Докл.
АН СССР, 1975, 225, № 1, с. 148—151.
524. Федорович Н- В., Леви М. Д., Шлепаков А. В.— Электрохимия, 1977, 13,
№ 1, с. 89—92.
525. Федорович Н. В., Сарбаш Ф. С.— Докл. АН СССР, 1978, 243, № 5,
с. 1231—1234.
526. Федосеев А. Л4., Петрухин В. Ф.— Радиохимия, 1979, 21, №5, с. 650—
658.
527. Фиалков Ю. Я.— Журн. физ. химии, 1963, 37, №8, с. 1745—1749.
528. Фиалков Ю. Я.— Там же, № 10, с. 2149—2153.
529. Фиалков Ю. Я-— Там же, № 11, с. 2539—2542.
530. Фиалков Ю. Я. Двойные жидкие системы.— Киев: Техшка, 1969.—218 с,
531. Фиалков Ю. Я-—Вести. Харьк. ун-та, 1976, № 139, с. 9—15.
532. Фиалков Ю. Я.— В кн.: Проблемы сольватации и комплексообразования.
Иваново : ИХТИ, 1978, с. 138—146.
533. Фиалков Ю. Я-—История естествозиаиия и техники, 1984, №4.
534. Фиалков Ю. Я.— Журн. неорган. химии, 1984, 29, №2, с. 377—386.
535. Фиалков Ю. Я., Басов В. П.— Жури. общ. химии, 1968, 38, № 1, с. 7—12.
536. Фиалков Ю. Я., Басов В- П,— Укр. хим. жур., 1970, 36, № 6, с. 557—560.
537. Фиалков Ю. Я., Басов В. П., Карапетян Ю. А.— Журн. общ. химии, 1973,
43, №6, с. 1209—1212.
538. Фиалков Ю. Я., Боровиков Ю. Я.— Укр. хим. жури., 1966, 32, №6,
с. 590—594.
539. Фиалков Ю. Я-, Боровиков Ю. Я-— Журн. физ. химии, 1966, 40, № 2,
с. 371—376.
540. Фиалков Ю. Я., Боровиков Ю. Я-—Химия и хим. технология, 1967, 10,
№ 12, с. 1324—1326.
541. Фиалков Ю. Я., Боровиков Ю. Я-—Журн. физ. химии, 1976, 50, №4,
с. 1057. Деп. в ВИНИТИ № 3808—75.
542. Фиалков Ю. Я-, Дорофеева Н. Г.; Ковальская В. П.— Укр. хим. журн.,
1979, 45, №7, с. 668—670.
543. Фиалков Ю. Я., Дынер Л. Л.—Журн. общ. химии, 1978, 48, №2, с. 253—
256.
544. Фиалков Ю. Я-, Житомирский А. Н.— Изв. вузов. Сер. Химия и хим.
технология, 1983, 26, №9, с. 1068—1080.
545. Фиалков Ю. Я., Житомирский А. Н., Квитка А. А.— Журн. физ. химии,
1978, 52, №5, с. 520—521.
219
546. Фиалков Ю. Я., Житомирский А. Н., Кудра О. А.—Журн. неорган. хи-
мии, 1965, 10, № 4, с. 934—938.
547. Фиалков Ю. Я., Житомирский А. Н., Тарасенко Ю. А. Физическая химия
неводных растворов.— Л.: Химия, 1973.—375 с.
548. Фиалков Ю. Я., Карапетян Ю. А., Басов В. П.— Укр. хим. журн., 1973,
39, № 4, с. 320—322.
549. Фиалков Ю. Я., Карапетян Ю. А., Басов В. П.— Химия и хим. техноло-
гия, 1972, 15, №7, с. 1015—1017.
550. Фиалков Ю. Я., Карапетян Ю. А., Басов В. П.— Докл. АН СССР, 1971,
197, №5, с. 1113—1115.
551 Фиалков Ю. Я., Карапетян Ю. А.— Укр. хим. журн., 1971, 37, №4,
с. 398—401.
552. Фиалков Ю. Я., Карапетян Ю. А., Басов В. П.— Журн. неорган. химии,
1972, 17, №6, с. 1772—1773.
553. Фиалков Ю. Я., Карапетян Ю. А., Романенко Г. М.— Доп. АН УРСР.
Сер. Б, 1974, №2, с. 150—152.
554. Фиалков Ю. Я., Квитка А. А., Житомирский А. Н.— Там же, 1979. №2,
с. 129—131.
555. Фиалков Ю. Я-, Кулинич И. И., Чумак В. Л.— Электрохимия, 1982, 18,
№9, с. 1024—1027.
556. Фиалков Ю. Я., Лещинский Э. Л.— Химия и хим. технология, 1974, 17,
№ 12, с. 1762—1765.
557. Фиалков Ю. Я., Лещинский Э. Л.— Укр. хим. журн., 1976, 42, №4,
с. 428—430.
558. Фиалков Ю. Я., Пучковская Г. А., Ващинская В. В.— Журн. общ. химии,
1973, 43, №3, с. 482—486.
559. Фиалков Ю. Я., Супруненко А. А.— Укр. хим. журн., 1975, 41, №11,
с. 1214—1215.
560. Фиалков Ю. Я., Тарасенко Ю. А.— Теорет. и эксперим. химия, 1965, 1,
№ 4, с. 473—478.
561. Фиалков Ю. Я-, Тарасенко Ю. А.— Электрохимия, 1966, 2, №5, с. 610—
612.
562. Фиалков Ю. Я., Цендровская В. А.— Укр. хим. жури., 1968, 34, № 4,
с. 334—339.
563. Фиалков Ю. Я-, Чумак В. Л.— Журн. физ. химии, 1975, 49, №9, с. 2412—
2413.
564. Фиалков Ю. Я., Чумак В. Л.— Химия и хим. технология, 1976, 19, №2,
с. 265—268.
565. Фиалков Ю. Я-, Чумак В. Л.— Журн. физ. химии, 1979, 53, №4, с. 885—
887.
566. Фиалков Ю. Я., Чумак В. Л., Квитка А. А., Ковальская В. П.— Там же,
1980, 54, №7, с. 1829—1832.
567. Фиалков Ю. Я., Чумак В. Л., Ковальская В. П., Дорофеева Н. Г.— Укр.
хим. журн., 1978, 43, №7, с. 760—761.
568. Фиалков Ю. Я., Яковлева А. В.— Журн. неорган. химии, 1969, 14, №8,
с. 2174—2179.
569. Фиалков Ю. Я., Яковлева А. В.— Электрохимия, 1971, 7, № 4, с. 590—593.
570. Фиалков Я. А. Межгалоидные соединения.— Киев : Изд-во АН УССР,
1958,—358 с.
571. Фиалков Я. А., Абарбарчук И. Л.— Укр. хим. журн., 1955, 15, №1/2,
с. 116—135, с. 194—202.
572. Фиалков Я. А., Кузьменко А. А.— Журн. общ. химии, 1949, 19, № 5,
с. 812—825.
573. Фиалков Я. А., Кузьменко А. А., Костромина Н. А.— Укр. хим. журн.,
1955, 21, №2, с. 556—560.
574. Фиалков Я. А., Фокина 3. А.— Журн. неорган. химии, 1959, 4, с. 2611.
575. Фиошин М. Я., Гуськов В. А.—Докл. АН СССР, 1957, 112, №2, с. 303—
306.
576. Фиошин М. Я., Казакова Л. Я,—Журн. общ. химии, 1958, 28, №8,
с. 2005—2007.
577. Фрумкин А. Я.—Успехи химии, 1946, 15, №2, с. 385.
220
578. Фрумкин А. Н„ Багоцкий В. С., Иофа 3. А., Кабанов Б. Н. Кинетика
электродных процессов.— М.: Изд-во Моск, ун-та, 1952.—405 с.
579. Заявка на пат. №54—141130 (Япония). Электроосаждение сплавов оло-
во—никель или олово—кобальт (Фукуоко.).— Опубл. 8.12.78.
580. Хамудханова Ш. 3., Жураев А. У-, Шереметьева В. Б.— РЖ Химия,
1976, 4Л303. Деп.
581. Хамудханова Ш. 3., Шереметьева В. Б., Муратова С. Г.— В кн.: 5-е Все-
союз. совещ. по электрохимии. М.: ВИНИТИ, 1974, 2, с. 160—161.
582. Хамудханова Ш. 3., Шереметьева В. Б., Муратова С. Г.— В кн.: Химия
соединений редких и цветных металлов. Ташкент : Фан, 1977, с. 51—56.
583. Хамудханова Ш. 3., Рубничик Г. Ф., Муртазаев А. М.— Докл. АН УзССР,
1973, № 6, с. 47—48.
584. Хамудханова Ш. 3., Шереметьева В. Б., Муратова С. Г., Муртаза-
ев А. М.— Электрохимия, 1978, 14, №8, с. 1181—1185.
585. Хамудханова Ш. 3., Шереметьева В. Б., Муртазаев А. М., Цыга-
нов Г. А.— Докл. АН УзССР, 1969, № 11, с. 34—35.
586. Хаяси Тадао Дэнки казаку, денк1 кодаки, 1965, 33, № 7, с. 535—539.
587. Хлебников Г. И., Дергунов Е. П.— Атом, энергия, 1960, 9, №5, с. 406—
408.
588. Хлыстова К- Б., Коршунов В. Н.— Электрохимия, 1971, 7, №7,
с. 1021—1024.
589. Хомутов Н. Е.— Журн. физ. химии, 1968, 42, №12, с. 2229—2232.
590. Хоси И., Като И., Ямада А., Такака Н.— Rev. Polarogr., 1979, 25,
№ 1/6, 7.
591. Хоцяновский О. И.— Журн. неорган. химии, 1962, 7, №2, с. 390—393.
592. Хюккель В. Теоретические основы органической химии.— М.: ГХИ,
1935.— с. 582.
593. Цинман А. И., Катревич А. Н. Защита металлов, 1969, 5, №5, с. 517—523.
594. Цинман А. И., Ковсман Е. П., Кузуб В. С.— Укр. хим. журн., 1965, 31,
№ 9, с. 923—926.
595. Цинман А. И., Писчик Л. М.— Защита металлов, 1970, 6, №6, с. 684.
596. Цинман А. И., Писчик Л. М.— Электрохимия, 1975, 11, №3, с. 498—502.
597. Цинман А. И., Писчик Л. М.— Там же, 1979, 15, № 1, с. 87—90.
598. Цинман А. И., Писчик Л. М., Брусенцова В. М., Захаренкова Л. Е.—
Защита металлов, 1971, 7, №6, с. 700—704.
599. Цинман А. И., Писчик Л. М., Брусенцова В. М., Захаренкова Л. Е.—
Там же, 1972, 8, №5, с. 567—569.
600. Цинман А. И., Писчик Л. М„ Ванеева Р. А., Клочкова М. Р.— Там же,
1974, 10, №6, с. 677—683.
601. Цинман А. И., Писчик Л. М., Маковей Г. Л.— Электрохимия, 1974, 10,
№9, с. 1321—1327.
602. Цинман А. И., Писчик Л. М., Маковей Г. Л.—-Там же, 1975, 11, № 1,
с. 101—103.
603. Цинман А. И., Писчик Л. М., Маковей Г. Л.— Там же, №11, с. 1705—1709.
604. Цыганкова Л. Е., Вигдорович В. И.— Изв. вузов. Химия и хим. техноло-
гия, 1978, 21, №8, с. 1187—1191.
605. Цыганкова Л. Е., Вигдорович В. И., Данилина Т. С.— Там же, 1976, 19,
№ 10, с. 1557—1561.
606. Цыганкова Л. Е., Вигдорович В. И., Пчельников И. Т.— Защита метал-
лов, 1970, 1, №6, с. 648—652.
607. Цыганкова Л. Е., Корнеева Т. В., Вигдорович В. И., Оше Е. К-— Там
же, 1980, 16, №2, с. 150—154.
608. Цыганкова Л. Е., Черникова Л. А., Вигдорович В. И.— Изв. вузов. Хи-
мия и хим. технология, 1979, 22, № 2, с. 164—166.
609. Цурцулия Г. С., Агладзе Т. Р., Колотыркин Я. М.— Электрохимия, 1976,
12, № 10, с. 1582—1585.
610. А. с. 591532 (СССР). Способ алюминидирования ванадия/А. В. Чекав-
цев, Б. Н. Кабанов, И. Г. Киселева, Н. М. Матвеева.—Открытие, изобре-
тение ..., 1978, № 5, с. 90.
611. Черный Д. Б. Термодинамические характеристики электролитической дис-
социации и ионной миграции хлоридов редкоземельных элементов и их
221
комплексов в спиртовых и водно-спиртовых растворителях: Автореф-
дис. ... канд. хим. наук.— Киев, 1982.—20 с.
612. Шавгулидзе В. В., Гецадзе Э. А., Бекаури И. Т.— Сообщ. АН ГССР, 1979,
94, № 1, с. 101—104.
613. Шаповалов В. А., Солоутин В. И., Пашкевич К. И. и др.— Журн. общ.
химии, 1980, 50, № 1, с. 103—107.
614. Шатешитейн А. И. Теория кислот и оснований.— М.: Госхимиздат, 1948.—
332 с.
615. Шатешитейн. А. И. Изотопный обмен и замещение водорода в органи-
ческих соединениях.— М.: Изд-во АН СССР, 1960.—253 с.
616. Шахпаронов М. И. Введение в молекулярную теорию растворов.— М. :
Высш, шк., 1959.—258 с.
617. Шахпаронов М. И. Введение в современную теорию растворов.— М.:
Высш, шк., 1976.—310 с.
618. Шахпаронов М. И. Механизмы быстрых процессов в жидкостях.— М.:
Высш, шк., 1980.—352 с.
619. Швабе К., Зушке X. Д., Тиме М.— Защита металлов, 1974, 10, №5,
с. 491—507.
620. Шевченко Ф. Д., Кузина Л. А.— Укр. хим. журн., 1963, 29, №3, с. 351—
353.
621. Шека 3. А.— В кн.: Работы по химии растворов и комплексных соеди-
нений. Киев : Изд-во АН УССР, 1954, с. 113—128.
622. Шека И. А., Карлышева К. Ф.— Журн. общ. химии, 1951, 21, №6,
с. 833—836.
623. Шека И. А., Шека 3. А. Галогениды индия и их координационные соеди-
нения.— Киев : Наук, думка, 1981.—299 с.
624. Шкодин А. М.— Химия и хим. технология, 1961, 4, №6, с. 441—442.
625. Шмидт В.— Защита металлов, 1969, 5, № 6, с. 608—617.
626. Шоркис А. А., Симанавичюс Л. Э.— В кн/. Исследования в области
электроосаждения металлов. Вильнюс : ИХХТ, 1977, с. 97—101.
627. Шпак Э. А., Самчук А. И., Пилипенко А. Т.— Журн. аналит. химии, 1974,
29, №5, с. 938—941.
628. Шембель Е. М., Литвинова В. И., Максюта И. М., Ксенжек О. С.— Элект-
рохимия, 1979, 15, № 12, с. 1791—1795.
629. Шембель Е. М., Литвинова В. И., Максюта И. М. и др.— Там же, №11,
с. 1671—1677,
630. Щепкин Д. Н.— Теорет. и эксперим. химия, 1976, 2, №2, с. 276—279.
631. Щербаков В. В.— Труды Моск, хим.-технол. ин-та, 1982, вып. 121, с. 115—
127.
632. Экилик В. В., Григорьев В. П., Свирская С. Н., Маханько А. И.— Журн.
прикл. химии, 1980, 53, №6, с. 1303—1306.
633. Экилик В. В., Кудряшова Г. Б.— В кн.: Ингибирование и пассивирование
металлов. Ростов н / Д : Изд-во Рост, ун-та, 1976, с. 92—98.
634. Экилик В. В., Свирская С. Н.— Там же, с. 86—91.
635. Экилик В. В., Свирская С. Н., Андрейчиков Ю. Г., Трухан Г. Е.— Там
же, с. 98—103.
636. Экилик Г. И., Экилик В. В., Григорьев В. П.— Изв. вузов. Химия и хим.
технология, 1979, 22, № 6, с. 702—705.
Б37. Электрохимия металлов в неводных растворах.— М.: Мир, 1974.—440 с.
638. Эрдей-Груз Т. Явления переноса в водных растворах.— М.: Мир, 1976.—
596 с.
639. Юхновский И. Р., Головко М. Ф. Статистическая теория классических
равновесных систем.— Киев : Наук, думка, 1980.—372 с.
640. Яковлева И. Я-, Ханудханова Ш. 3., Гуревич Е. А.— В кн.: Химия соеди-
нений редких и цветных металлов. Ташкент : Фан, 1977, с. 47—51.
641. Якубсон С. И. Электролитные неводные растворы.— Киев : Наук, думка,
1967,—616 с.
642. Янов Л. А., Молодое А. И.— Электрохимия, 1975, 11, №7, с. 1112—1115.
643. Янов Л. А., Молодое А. И., Лосев В. В.— Там же, 1978, 14, №11.
с. 1663—1667.
644. Яиимиоский К. Б.— Журн. теорет. и эксперим. химии, 1970, 6, №4,
с. 462—468.
222
645. Adema E. H., Arnoldy H., Visser R., Sixma F. L. I.— Rec. trav. chim.,
1960, 79, N 10, p. 1111—1116.
646. Adema E. H„ Teunissen A. I. I. M., Tholen M. I. I.— Ibid., 1969, 85, N 3,
p. 377—381.
647. Ahmed I. J.— J. Magn. Reson., 1972, 7, N 2, p. 196—202.
648. Ahmed M., Mogel R. J.— J. Electroanal. Chem., 1980, 110, N 1/3, p. 335—
345.
649. Aida H., Epelboin I., Garreau M.— J. Electrochem. Soc., 1971, 118, N 2,
p. 243—248.
650. Aladjem A., Rafaeloff R.— Isr. Atom Energy Commis. (Repts), 1969,
N 1190, 173—174.
651. Antula Becker B. F.— J. Phys. Chem., 1975, 79, N 23, p. 2470—2473.
652. Aylward J. R., Whitener E. M.— J. Electrochem. Soc., 1962, 109, N 2,
p. 87—91.
653. Bacarella A. L. ,Sutton A. L.— Ibid., 1975, 122, N 1, p. 11—18.
654. Bachman G. B., Astle M. J.— J. Amer. Chem. Soc., 1942, 64, N 9, p. 2177—
2181.
655. Banas J., Schwabe K.— Oberflache Surface, 1979, 20, N 9, S. 200—204.
656. Bandler M., Shellenberg D.— Z. anorg. und allg. Chem., 1967, 356, N 3, 4,
S. 140—150.
657. Baranski A,, Fawcett W. R.— J. Electroanal. Chem., 1978, 94, N 3,
p. 237—240.
658. Пат. 1368747 (Англия). Electrodeposition of chromium/С. Barnes,
J. R. A., Christie, J. J. B. Ward.— Опубл. 02.10.74.
659. Barthel J.— Pure and Appl. Chem., 1979, 51, N 10, p. 2093—2124.
660. Barthel J.— Ber. Bunsengesellsch. phys. Chem., 1979, 83, N 6, S. 634—642.
661. Barthel J.— J. Solut. Chem., 1979, 9, N 3, p. 209—219.
662. Barthel J. et al.— Pure and Appl. Chem., 1979, 51, N 10, p. 2093—2124.
663. Bauer R., Beck J. P.— J. Electroanal. Chem., 1972, 40, N 2, p. 233—254.
664. Bauer R.— Chem. Ing. Techn., 1972, 44, N 4, p. 147—151, A123, A125.
665. Beach Л G., McGraw L. D., Faust C. L'.—Plating, 1968, 55, N 9, p. 936—
940.
666. Beattie I. R., Webster M.— J. Chem. Soc., 1963, N 1, p. 38—41.
667. Behr B., Borkowska Z., Elzanowska H.— J. Electroanal. Chem., 1979, 100,
N 1/2, p. 853—866.
668. Bharucha N. R., Ward J. J.— Australas. Eng., 1969, p. 30—33.
669. Пат. 441051 (Австралия). Electrodeposition of chromium and other me-
tal/ N. R. Bharucha.—Опубл. 02.10.73.
670. Пат. 3953302 (CHIA). Provention of dendrite plating of lithium / Bhaska-
ra M. L. Rao, C. R. Schlaikjer.— Опубл. 27.04.76.
671. Пат. 4091152 (США). Lithium—SO2 cell/M. L. Rao Bhaskara, C. R. Schlai-
kjer.— Опубл. 23.05.78.
672. Bell R. P., Skinner B. G.— J. Chem. Soc., 1952, Nil, p. 2955—2957.
673. Bennett W. E., Davidson A. W., Kleinberg J.—J. Amer. Chem. Soc., 1954,
74, N 3, p. 732—735.
674. Bertrand J.— Bull. Soc. chim. France, 1880, 33, p. 403.
675. Berzins T., Delahay P.— J. Amer. Chem. Soc., 1953, 75, N 3, p. 555—559.
676. Besenhard J. O., Eichinger G.— J. Electroanal. Chem., 1976, 68, N 1,
p. 1—18.
677. Besenhard J. O.—Ibid., 1978, 94, N 1, p. 77—81.
678. Bessette R. R., Olver J. W.— Ibid., 1968, 17, N 3 / 4, p. 327—334.
679. Biallozor S, G.— Electrochim. acta, 1972, 17, N 7, p. 1243—1249.
680. Biallozor S.— Wiadum. Chem., 1974, 28, N 12, s. 839—855.
681. Biallozor S.— Zesz. nauk. Politechn. grartsk., 1974, N215, s. 3—86.
682. Biallozor S.— Electrochim. acta, 1976, 21, N 11, p. 1089—1095.
683. Biallozor S.— Ibid., 1978, 23, N 12, p. 1309—1319.
684. Biallozor S.— Wiadum. Chem., 1979, 33, N 11, s. 727—748.
685. Biallozor S., Lisowska A.~ Zesz. nauk. Politechn. slaskiej., 1979, N631,
s. 221—231.
686. Biallozor S., Lisowska A.—Electrochim. acta, 1980, 25, N 9, p. 1209—1214.
223
687. Biegler T., Conzalez E. R., Persons R.—Collect. Czech. Chem. Communs.,
1971, 36, N 2, p. 414—425.
688. Bioadhead J., Elving P. J.— Anal. chim. acta, 1969, 48, N 2, p. 433—434.
689. Birch C. G., Iwamoto R. T.— Inorg. Chem., 1973, 12, N 1, p. 66—73.
690. Bocseken D.— Rec. trav. chim., 1901, 20, N 1, p. 102.
691. Bogenschutz A. F., Jostan J. L., Koch D. H. C. et al.— Metalloberflache,
1971, 25, NS, S. 267—276.
692. Bogges R. K-, Hughes I. W., Coleman W. M., Taylor L. T.— Inorg. chim.
acta, 1980, 38, N 2, p . 183—189.
693. Bonnaterre R., Cauquis A. G.— J. Electroanal.. Chem., 1971, 32, N 2,
p. 215—223.
694. Bontempelli G., Magno F., Corain B„ Schiavon G.— J. Electroanal. Chem.,
1979, 103, N 2, p. 243—250.
695. Borkowska Z., Elzanowska H.— In: 29th meet. Intern, soc. electrochem.
Budapest, 1978 : Extend, abstrs., pt 1, p. 577—580.
696. Bowaen W. L„ Dey A. N.— J. Electrochem. Soc., 1980, 127, N 7, p. 1419—
1426.
697. Boyer К W., Iwamoto R. T.— J. Electroanal. Chem., 1964, 7, N 6, p. 458—
464.
698. Bradley D. C., Caldmell E. V., Wardlaw W.— J. Chem. Soc., 1957, N 7,
p. 3039—3043.
699. Braun R. D.— J. Electroanal. Chem., 1977, 79, N 2, p. 381—390.
700. Brenner A.— J. Electrochem. Soc., 1956, 103, N 12, p. 652—656.
701. Brenner A.—Ibid., 1959, 106, N 2, 148—154.
702. Brenner A.— Adv. Electrochem. and Electrochem. Eng., 1967, 5, p. 205—
248.
703. Brenner A.— J. Electrochem. Soc., 1971, 118, N 1, p. 99—100.
704. Brenner A.—Ibid., 1975, 122, N 5, p. 156—166.
705. Brenet J.— J. Chim. phys. et phys.-chim biol., 1969, 66, N 6, p. 1057—1061.
706. Brinzoi V., Popa M. V.— Rev. chim. (Romin), 1979, 30, N 9, p. 883—889.
707. Britz D., Bauer H. H.— J. Electroanal. Chem., 1968, 18, N 1, p. 1—4.
708. Broadhead J., Elving P. J.— Anal. Chem., 1969, 41, N 13, p. 1814—1818.
709. Broadhead J., Elving P. J.— J. Electrochem. Soc., 1971, 118, N 1, p. 63—67.
710. Broun H. C„ Wallace W. L.— J. Amer. Chem. Soc, 1953, 75, N 12, p. 6279.
711. Brovo O., Iwamoto R . T.— J. Electroanal. Chem, 1969, 23, N 3, p. 419—
424.
712. Brown С. H., Al-Ureali R.— J. Amer. Chem. Soc., 1958, 80, N 9, p. 2113—
2115.
713. Brown O. R., Clarke S.— J. Electroanal. Chem, 1976, 70, N 1, p. 87—102,
349—356.
714. Brown G. H., Hsiung H.— J. Electrochem. Soc, 1960, 107, N 1, p. 57—58.
715. Brown O. R., Thornton S. A.— J. Chem. Soc. Faraday Trans, 1974, 1, N 1,
p. 14—26.
716. Brummer S. B., Hills G.— J. Trans. Faraday Soc, 1961, 57, N 10, p. 1816—
1822.
717. Bruno P., Caselli M., Trainl A.— J. Electroanal. Chem, 1980, 113, N1,
p. 99—111.
718. Bruss D. B., de Vries T.— J. Amer. Chem. Soc, 1956, 78, N 2, p. 733—736.
719. Burlage I. I., Garrett A. B.— J. Phys. Chem, 1952, 56, N5, p. 730.
720. Burnett I. N., Hiller L. K., Ir., Murray R. W.— J. Electrochem. Soc, 1970,
117, N 8, p. 1028—1032.
721. Buschow A. G„ Esola С. H.— Plating, 1968, 55, N 9, p. 931—935.
722. Butler I. N.— J. Electroanal. Chem, 1967, 14, N 1, p. 89—116.
723. Butler I. N.— Adv. Electrochem and Electrochem. Eng, 1970, 7, N. Y, p. 77.
724. Butler I. N„ Cogley D. K., Synnott I.—J. Phys. Chem, 1963, 73, Nil,
p. 4026—4027.
725. Campbell T. T.— J. Electrochem. Soc, 1959, 106, N 2, p. 119—123.
726. Cappellettl M., Longhi P., Mussini T — Gazz. chim. ital, 1976, 106, N 3/4,
p. 246—258.
727. Capuano G. A, Davenport W. G.— J. Electrochem. Soc, 1971, 118, N 10,
p. 1688—1695.
224
728. Capuano G. A., Davenport W. G.— Plating, 1973, 60, N 3, p. 251—255.
729. Capuano G. A., Ducasse R.— J. Appl. Electrochem., 1979, 9, N 1, p. 7—13.
730. Cassimatis P., Bonnin I. P., Theophanides T.— Can. J. Chem., 1970, 48,
N 11, p. 3860—3865.
731. Cauguis G., Pierre I-—Bull. Soc. Chim. France, 1972, N 6, p. 2244—2246.
732. Chisholm C. U.— Trans. Inst. Metal. Finish., 1969, 47, N 4, p. 134—140.
733. Chisholm C. U., Carnegie R. J. I.— Electrodepos. and Surface Treat., 1973,
1, N 5, p. 367—394.
734. Cheseldine D. M.— J. Electrochem. Soc., 1964, 111, N 10, p. 1128—1132.
735. Choudhury P., Khasgiwale K. A., Sundaresan M.— J. Electroanal. Chem.,
1972, 40, N 2, p. 444—447.
736. Пат. 1368749 (Англия). Electrodeposition of chromium / J. R. A. Christie,
J. J. B. Ward.— Опубл. 02.10.74.
737. Ciana A., Furlani C.— Electrochim. acta, 1965, 10, N 12, p. 1149—1160.
738. Cihalik J., Simek I., Riizicka J.— Collect. Czech, chem. Communs., 1958,
23, N 6, s. 1037—1047.
739. Cisak A., Elving P. J.— J. Electrochem. Soc., 1963, 110, N 2, p. 160—164.
740. Cisak A., Elving P. J.— Electrochim. acta, 1965, 10, N 9, p. 935—946.
741. Coetzee J. F., Cunningham G. P., Me Guire D. K., Radmanabhan G. R.—
Anal. Chem., 1962, 34, N 8, p. 1139—1143.
742. Coetzee J. F., Me Guire D. K., Hedrick J. L.— J. Phys. Chem., 1963, 67,
N 9, p. 1814—1820.
743. Coertce J. F., Hedrick J. L.— J. Phys. Chem., 1963, 67, N 2, p. 221—226.
744. Coetzee J. F., Simon J. M., Bertozzi R. J.— Anal. Chem., 1969, 41, N 6,
p. 766—772.
745. Coetzee J. F., Wei-San Siao.— Inorg. Chem., 1963, 2, N 1, p. 14—19.
746. Cogley D. R., Butler J. H.— L Phys. Chem., 1968, 72, N 12, p. 4568—4573.
747. Cohen S. H., Iwamoto T. R., Kleinbery J.— J. Phys. Chem., 1963, 67, N 7,
p. 1275—1278.
748. Coiola A., Guy H., Sohm J. C.— Electrochim. acta, 1970, 15, N 4, p. 555—
562, N 10, 1733—1746.
749. Cokal E. J., Wise E. N.— J. Electroanal. Chem., 1966, 11, N 6, p. 406—415.
750. Cokal E. L, Wise E. N.— Ibid., 12, N 2, p. 136—147.
751. Colichman E. L., Ludewig W. H.— Anal. Chem., 1953, 25, N 12, p. 1909—
1910.
752. Colombi M., Fiori G., Formaro L.— J. Electroanal. Chem., 1973, 44, Nl,
p. 22—24.
753. Connor J. H., Reid W. E., Wood G. B.— J. Electrochem. Soc., 1957, 104,
N 1, p. 38—41.
754. Contour J. P., Salesse A., Froment M. et al.—J. Microsc. et Spectrosc.
electron., 1979, 4, N 4, p. 483—491.
755. Contreras R., Heath G. A., Lindsay A. J., Stephenson T. A.— J. Organo-
metal. Chem., 1979, 179, N 4, p. 55—60.
756. Convington A.— Ann. Rep. Prog. Chem. A., 1977, 74, p. 5—21.
757. Conway B., Bockris L, Linton H.— J. Chem. Phys., 1956, 24, N 4, p. 834—
840.
758. Conway В. E., Hovak D. M.— In: 29th meet. Intern, soc. electrochem.,
Budapest, 1978 : Exfend, abstr. pt. 1. p. 556—570.
759. Copley В. B., Fairbrother F„ Grundy К. H., Thompson A.— J. Less—Com-
mon Metals, 1964, 6, N 5, p. 407—410.
760. Costa J. M., Cullere J. R., Franco C.— Electrochim. acta, 1977, 22, Nil,
p. 1325—1327.
761. Cotton F. A., Francis R., Horrocks W. D.— J. Phys. Chem., 1960, 64, N 8,
p. 1534—1538.
762. Cotton F. A., Francis R.— J. Amer. Chem. Soc., 1960, 82, N 6, p. 2986—2989.
763. Cotton S. A., Gibson I. F.— J. Chem. Soc., 1971, A, N 8, p. 1696—1698.
764. Couch D. E., Brenner A.— J. Electrochem. Soc., 1952, 99, N 2, p. 234—
244.
765. Coulter P. D. T., Iwamoto R. T.— J. Electroanal. Chem., 1967, 13, N 2,
p. 28—30.
766. Coulter P. D. T., Iwamoto R. T.— Ibid., N 1 / 2, p. 21—27.
15 - 4-653
225
767. Counor J. H., Brenner A.— J. Electrochem. Soc., 1956, N 12, p. 657—661.
768. Covington A. K., Pethybrige A. D.— Ann. Rep. Prog. Chem., A, 1977, 74,
p. 5—21.
769. Cowley A., Fairbrother F., Scott N.— J. Chem. Soc., 1958, N 10, p. 3133—
3136.
770. Cullinane J., Chard, Leyshon.— Ibid., 1952, N 12, p. 4106—4108.
771. Cuong N. H., Maiornikoff A., Hurwitz H. D.— J. Electroanal. Chem.,
1976, 72, N 1, p. 107—109.
772. Curti R., Locchi S.— Anal. Chem., 1957, 29, N 4, p. 534—537.
773. Czarnecka W., Perec M., Weglarczyk A.— Roczn. chem., 1962, 36, N 4,
s. 503—512.
774. Daenen T. E. /.— Nature, 1979, 280, N5721, p. 378—380.
775. Davidson A. W., Jirik F.— J. Amer. Chem. Soc., 1950, 72, N 12, p. 1700—
1705.
776. Davies A. G., Bughan E. C.— J. Chem., Soc., 1961, N 9, p. 1711—1715.
777. Dehn H., Gritzer G., Gutmann V.— Z. anal. Chem., 1969, 245, N 2,
s 172—176.
778. Delahay P.— J. Amer. Chem. Soc., 1953, 75, N 6, p. 1190—1196.
779. Delgado A. B., Posadas D., Arvia A. J.— Electrochim. acta, 1976, 21, N 6,
p . 385—393.
780. Desbarres J., Pichet P., Benoit R. L.— Ibid., 1968, 13, N 9, p. 1899—1904.
781. Deshpande P., Balachandra J.— Indian. J. Technol., 1974, 12, N 5,
p. 207—210.
782. Dessy R. E„ Haneller G. S.— J. Amer. Chem. Soc., 1958, 80, N21,
p. 5824—5826.
783. Derosa О. A. H., Macagno V. A., Giordano M. C.— Electrochim. acta, 1976,
21, N 4, p. 267—272.
784. Devand M., Le Moullec J.— Ibid., N 6, p. 395—400.
785. Devyuk J., Tremillon B., Menard H., Comarmand I.— Can. J. Chem., 1978,
56, N 5, p. 703—708.
786. Diggle J. W., Parker A. J., Owensly D. A.— Austral. J. Chem., 1975,
28, N2, p. 237—241.
787. Dix A. H., Schut F. H. M„ Schlebos P. P. J., Van der Linden J. I. M.—
J. Electroanal. Chem., 1980, 111, N 1, p. 71—79.
788. Dobkowska M.— Z. anal. Chem., 1962, 185, N 3, S. 222—226.
789. Dougherty D.— J. Amer. Soc., 1929, 51, N 3, p. 576—580.
790. Drago R. S., Hart D. M., Carlson R. L.— J. Amer. Chem. Soc., 1965, 87,
N 9, p. 1900.
791. Drago R. S., Meek D. W.—Ibid., 1961, 83, N 12, p. 4322—4327.
792. Dubois J. E., Lacaze P. C., Ficquelmont A. A. M.— C. r. Acad. Sci., 1966,
262, p. 181—184, 249—252.
793. Dunnett J. S., Gearey D.— Electrochim. acta, 1974, 19, N 12, p. 907—912.
794. Durand G., Tremillon B.— Anal. Chim., 1970, 49, N 1, p. 135—149.
795. Duschner H., Born H. J., Kim J. I.— Int. J. Appl. Radiat. and Isotopes,
1973, 24, N 8, p. 433—436.
796. Duschnek O., Gutmann V.— Z. anorg. und allgem. Chem., 1972, 394, N 4,
s. 243—253.
797. Dyke Van R. E., Grauford H. E.— J. Amer. Chem. Soc., 1950, 72, N 13,
p. 2829—2832.
798. Dyke Van R. E.— Ibid., N 8, p. 3619—3622.
799. Dyke Van R., Grauford H.— Ibid., 1951, 73, N 5, p. 2022—2025.
800. Eastman E. D., Fontana B. J.— Chem. Abstrs, 1959, 53, N 17, 15817 c.
801. Eckert J., Kolling H.— Wiss. Z. Techn. Univ. Dresden, 1975, 24, N 1, S. 19—
21.
802. Eichinger G., Fritz H. P.— Electrochim. acta, 1975, 20, N 10, p. 753—758.
803. Elamand M., Gilead! E.— J. Electrochem. Soc., 1980, 127, N 4, p. 815—820.
804. Elbs К—Z. Electrochem., 1896, N 3, S. 70—71.
805. Elew D. D., Watts H.— i. Chem. Soc., 1952, N 5, p. 1914—1921.
806. El Murr N„ Dusansoy У., Sheats J. E., Agnew M.— J. Chem. Soc. Dalton
Trans., 1979, N5, p. 901—905.
807. Elsemongy M. M.— J. Electroanal. Chem., 1978, 90, N 1, p. 49—96.
226
808 Endo A., Watanabe M., Hayashi S. et al.— Bull. Chem. Soc. Jap., 1978,
’ 54, N 3, p. 800—804.
809 Epelboin J., Froment M., Garrean M. et al.— J. Electrochem. Soc., 1980,
127, N 10, p. 2100—2104.
810. Evans G. G., Gibb Ir. T. R. P„ Kennedy I. K, Del Greco F. P.— J. Amer.
Chem. Soc., 1954, 76, N 12, p. 4861—4866.
811. Fairbrother F.— J. Chem. Soc., 1937, N 3, p. 503—506.
812. Fairbrother F.— Ibid., 1941, N 2, p. 293—299.
813. Fairbrother F., Scott N.— Ibid., 1955, N 2, p. 452—459.
814. Fama F. Ir., Iwamoto R. T.— J. Electroanal. Chem., 1964, 8, N 1, p. 55—64.
815. Fawcett W. R., Ikeda В. M., Sellan J. B.— Can. J. Chem., 1979, 57, N 17,
p. 2268—2277.
816. Fawcett W. R., Mackey M. D.— J. Electroanal. Chem., 1970, 27, N 2,
p. 219—232.
817. Feroci G., Roffia S.— Ibid., 1976, 71, N 2, p. 191—198.
818. Filippini F., Susz B.— Helv. chim. acta, 1971, 54, N 3, p. 835, N 4, p. 1175.
819. Findeis A. F., de Vries T.— Anal. Chem., 1956, 28, N 2, p. 209—211.
820. Fischer D. J., Thomason P. F.— Ibid., N 8, p. 1285—1288.
821. Fleischmann M., Mengoli G., Pletcher D.— J. Electroanal. Chem., 1973, 43,
N2, p. 308—310.
822. Forel M. T., Trauquille M., Fonassier M.— Spectrochim. acta, A, 1970, 26,
N 3, p. 1777—1780.
823. Fowles G. W. A., Walton R. A.— J. Chem. Soc., 1964, N 8, p. 2840.
824. Friedman H. A., Stonely J. R., Baybarz R. D.— J. Inorg. and Nucl, Chem.,
1972, 8, N 5, p. 433—441.
825. Fritzsche C. R.— J. Phys. Chem. Solids, 1969, 30, N 7, p. 1885—1895.
826. Froment M., Garreau M., Thevenin J., Warin D.— J. microsc. et spectrosc.
electron, 1979, 4, N2, p. 111—122.
827. Fronalus S., Johansson C. L.— J. Electroanal. Chem., 1980, 112, N 2,
p. 197—206.
828. Fronalus S., Johansson C. L., Palm B.— Ibid., 1978, 88, N 1, p. 1—13.
829. Fronalus S., Johansson C. L.— Ibid., 1977, 80, N 2, p. 283—294,
830. Froneus S., Palm B.— Acta chem. scand., A, 1978, 32, N 10, p. 909—918.
831. Fujinaga T., Hagaosa J.— Bull. Chem. Soc. Jap., 1980, 53, N 2, p. 416—420.
832. Fujinaga T„ Hajo M.— Rev. Polarogr., 1977, 23, N 1/6, p. 41.
833. Fujinaga T., Kuwamoto T., Okazaki S., Hajo M.— Bull. Chem. Soc. Jap.,
1980, 53, N 10, p. 2851—2855.
834. Fujinaga T., Okazani S., Hajo M.— Bull. Inst. Chem. Res. Kyoto Univ.,
1978, 56, N 4, p. 139—150.
835. Fujinaga T., Okazaki S., Hajo M.— J. Electroanal. Chem., 1980, 113 N 1,
p. 89—98.
836. Fujinaga T., Sakamoto J.— J. Electroanal. Chem., 1975, 67, N 2, p. 201—
204.
837. Fujinaga T., Sakamoto J.— Ibid., 1976, 73, N 2, p. 235—246.
838. Fujinaga T., Yamashita K.— Bull. Chem. Soc. Jap., 1964, 37, N 7, p 989—
994.
839. Fuoss R. M.— Proc. Nat. Acad. Sci., 1959, 45, N 6, p. 808—810.
840. Fuoss R. M.— J. Solut. Chem., 1978, 7, N 5, p. 771—782.
841. Furlani C., Sestili L., Ciana A., Garbassi F.— Electrochim. acta 1967 12
N 10, p. 1393—1408.
842. Furlani C., Sestili L., Ciana A., Garbassi F.— Ibid., 1970, 15, N 1 n 225—
236.
843. Gal J. У., Persin F.— Bull. Soc. Chim. France, 1979, pt 1 N 11/12
p. 1005—1010.
844. Gdlova M., Kladekova D.— Chem. prum., 1979, 29, N 2, s. 92—97.
845. Gdlova M., Lux L.— Chem. zv. 1975, 29, N 3, s. 279—289.
846. Gdlova M., Lux L — Ibid., 1976, 30, N 5, s. 599—610.
847. Galovd M., Lux L.— Zv. Ved. pr. VST Kosiciach, 1977, 2, s. 147—155.
848. Galovd M., Pantony D. A.—Taianta, 1971, 18, N 12, p. 1209—1216.
849. Gagnaux P„ lanjie P., Susz В. P — Helv. chim. acta, 1958, 41 N6,
p. 1322—1326.
15*
227
850. Gagnaux P., Susz В. P.— Ibid., 1961, 44, N 5, p. 1132—1140.
851. Gaur J. N., Bhargava S. R.— In: 29th meet Intern, soc. electrochem., Bu-
dapest, 1978; Extend, abstrs, pt 1, p. 694.
852. Gaur 1. N., Goswani H. R.— Electrochim. acta, 1966, 11, N 7, p. 939—943.
853. Gaur J. N„ Goswani H. R.— Ibid., 1967, 12, N 11, p. 1489—1494.
854. Gaur I. N„ Goswani H. R.— Ibid., 1970, 15, N 4, p. 519—524.
855. Gaur J. N., Zutshi R.— J. Electroanal. Chem., 1966, 11, N 5, p. 390—391.
856. Getoff N., Bildstein H., Proksch E.— Nucl. Instrum, and Meth., 1967, 46,
N 2, p. 305—308.
857. Getoff N., Bildstein H., Proksch E.— Ibid., 1969, 70, N3, p. 352—354.
858. Geronov J., Moshtev R. V., Buresheva B.— J. Electroanal. Chem., 1980,
108, N 3, p. 335—346.
859. Gerrard tt7., Lappert M. F„ Pyszora H., Wallis J. W.— J. Chem. Soc., 1960,
N 5, p. 2140—2144.
860. Gill D. S.—Electrochim. acta, 1979, 24, N 6, p. 701—703.
861. Giordano M. C„ Bazan J. C„ Arvia A. /.— Ibid., 1966, 11, N 7, p. 741—747.
862. Giordano M. C„ Bazan J. C., Arvia A. I.— Ibid., Nil, p. 1553—1578.
863. Giordano M. C„ Bazan J. C., Arvia A. J.— Ibid., 1967, 12, N 6 p. 723—
735.
864. Giordano M. C., Gopez B. A., Sereno R.— J. Electroanal. Chem., 1979, 101,
N 1, p. 39—49.
865. Given P. H., Peover M. E — Nature, 1958, 182, N 4644, p. 1226—1227.
866. Gael H. C., Dubey M. C., Mukhtar S.— Indian J. Chem., A, 1979, 18, N 2,
p. 176—177.
867. Goldstein R. M., Pettit J.— J. Electrochem. Soc., 1968, 115, N 9, p. 979—
980
868. Goolsby A. D., Sawyer D. T.— Anal. Chem., 1968, 40, N 1, p. 83—86.
869. Goolsby A. D., Sawyer D. T.— Ibid., N 13, p. 1978—1983.
870. Greenwood N. N., Martin R. L.— J. Chem. Soc., 1950, N 7, p. 3030—3035.
871. Greenwood N. N., Martin R. L.— Ibid., 1951, N 5, p. 1325—1328.
872. Greenwood N. N., Martin R. L.— Ibid., 1953, N 3, p. 757—760.
873. Greenwood N. N., Wade R.— Ibid., 1956, N 5, p. 1527—1530.
874. Greenwood N. H-, Wade R.— Ibid., 1958, N 5, p. 1663, 1671.
875. Gressin J. C., Michelet D., Hadjo L., Saveant J. M.— Nouv. J. Chim., 1979,
3, N 8/9, p. 545—555.
876. Grigorjev V. P., Gontmacher N. M., Hechneva O. N.— In: 29th meet. In-
tern. electrochem., Budapest, 1978 : Extend. Abstrs. Pt 2, p. 1008—1009.
877. Gritzner G., Gutmann V., Schmid R.— Electrochim. acta, 1968, 13, N 4,
p. 919—924
878. Gritzner G., Gutmann V., Michltnayr M.— Z. anal, chem., 1967, 224, N 2,
S. 245—251.
879. Gritzner G., Muraner H., Gutmann V.— J. Electroanal. Chem., 1979, 101,
N 2, p. 177—200.
880. Grossman R. E.— J. Org. Chem., 1957, 22, N 3, p. 581—585.
881. Grunner W-, Beutmann A.— Isotopenpraxis, 1978, 14, N 12, p. 421—425.
882. Gupta B. R., Jain D. S., Gaur J. H.— J. Electroanal. Chem., 1974, 52, N 1,
p. 147—150.
883. Gupta B. R., Jain D. S„ Gaur /. H.— Electrochim. acta, 1977, 22, N 4,
p. 479—484.
884. Gutmann V.— Chem. Technol., 1977, 7, N 4, p. 255—263.
885. Gutmann V., Duschek O.— Monatsch. Chem., 1969, 100, N 3, S. 1047—1056.
886. Gutmann V., Himml R.— Z. phys. Chem., 1955, 4, N 2, p. 157—164.
887. Gutmann V., Michltnayr M.— Z. anal. Chem., 1969, 245, N 2, p. 166—170.
888. Gutmann V., Michltnayr M., Peychai-Helling G.— J. Electroanal. Chem.,
1968, 17, N 1/2, p. 153—160.
889. Gutmann V., Michltnayr M., Peychal-Heiling G.— Anal. Chem., 1968, 40,
N 3, p. 619—623.
890. Gutmann V., Peychal-Heiling G.— Monatsh. Chem., 1969, 100, N 3, S. 813—
824.
891. Gutmann V-, Schmid R.— Ibid., N 6, S. 2113—2121.
892. Gutmann V., Schober G.— Angew. Chem., 1958, 70, N 1, p. 98—104.
228
893. Gutmann V., Schober G.— Z. anal. Chem., 1959, 171, N 2, S. 339—343.
894. Gutmann V., Utvary K.— Monatsh. Chem., 1959, 90, N 2, S. 751—761.
895. Habeed J. J., Tuck D. G., Zhandire S.— Can. J. Chem., 1979, 57, N 16,
p. 2196—2199.
896. Hagaosa J.— Taianta, 1979, 26, N 11, p. 987—990.
897. Haladjian J., Puggelli H., Biango P.— J. Chim. Phys, et phys.-chim. biol.,
1978, 75, N 2, p. 186—191.
898. Hall L. C., Flanigan D. A.— Anal. Chem., 1963, 35, N 3, p. 2108—2112.
899. Hanley J. L., Iwamoto R. T.— J. Electroanal. Chem., 1970, 24, N2/3,
p. 271—278.
900. Hansen P. У.— J. Inorg. and Nucl. Chem., 1961, 17, N3/4, p. 232—239.
901. Hardcastle J. E„ Levine N. E.— Radiochim. acta, 1972, 18, N 1, S. 59.
902. Harding W. B.— In: Modern Electroplating. New York : L. a., 1974, p. 63—
70.
903. Hajo M — Bull. Chem. Soc. Jap., 1980, 53, N 10, p. 2856—2860.
904. Headridge J. H., Ashraf M., Dodds H. L. H.— J. Electroanal. Chem., 1968,
16, N 1, p. 114—116.
905. Headridge I. B., Pletcher D.— Ibid., 1967, 15, N2/3, p. 312—314.
906. Headridge J. B., Pletchar D., Callingham M.— J. Chem. Soc. A, 1967, N 4,
p. 684—685.
907. Heath C. A., Martin R. G.— Austral. J. Chem., 1970, 23, N 9, p. 1721—1734.
908. Heerman L., Van Baelen J.— Bull. Soc. chim. Belg., 1972, 81, N 3,
p. 379—384.
909. Heitz V. E.— Ber. Bunsengesellsch. phys. Chem., 1969, 73, N 10, S. 1085—
1093.
910. Heitz E.— Werkst. und Korros, 1970, 21, N5, S. 360—367.
911. Heitz E.. Hunovic M., Maier K. H.~ Ibid., N 6, S. 457—462.
912. Helson R. F., Adans R. N.— J. Electroanal. Chem., 1967, 13, Nl/2,
p. 184—187.
913. Helson D. P., Ivory V — Text. J. Sci., 1977, 28, Nl/4, p. 223—229.
914. Helson I. V., Iwamoto R. T.— Anal. Chem., 1961, 33, N 12, p. 1795—1796.
915. Helson I. V., Iwamoto R. T.— Inorg. Chem., 1962, 1, N 1, p. 151—154.
916. Helson I. V., Iwamoto R. T.— J. Electroanal. Chem., 1963, 6, N 3, p. 234—
241.
917. Helson I. V., Larson R. C., Iwamoto R. T.— J. Inorg. and Nucl. Chem.,
1961, 22, N4, p. 279—284.
918. Hemes L.— J. Electroanal. Chem., 1964, 8, N 3, p. 166—170.
919. Hendriksma R. R., Van Leeuwen U. P.— Electrochim. acta, 1973, 18, N 1,
p. 39—44.
920. Heung L. L., Fujinaga T.— Bull. Inst. Chem. Res., Kyoto Univ., 1979, 57,
N 4, p. 285—296.
921. Heyman С. J.— J. Amer. Chem. Soc., 1949, 71, N 12, p. 3914—3916.
922. Неги H., Kaneko H., Ohaka A.—Rev. Polarogr., 1976, 22, N3/6, p. 115.
923. Hicolson R. S.— Anal. Chem., 1964, 36, N 3, p. 706—727.
924. Hills G. J., Peter L. M.— Electrochim. acta, 1974, 50, N 2, p. 187—194.
925. Hills G. J., Peter L. M.— J. Electroanal. Chem., 1974, 50, N 2, p. 175—185.
926. Hisamo T„ Terazowa T„ Mastsui //.— Chem. Lett., 1973, N 3, p. 219—222.
927. Hishikawa H„ Minami S. I.— Electrochim. acta, 1979, 24, N 3, p. 339—
343.
928. Hoa H. T. V., Magel R. J — J. Inorg. and Nucl. Chem., 1979, 41, N 3,
p. 351—356.
929. Hofmanova A., Koryta J., Brezina M. et al.— Inorg. chim. acta, 1979, 37,
N2, p 135—140.
930. Holt M. L.— J. Electrochem. Soc., 1951, 98, N 3, p. 33—35.
931. Hsiung H., Brown G. H.— J. Electrochem. Soc., 1963, 110, N 10, p. 1085—
1086.
932. Hubicki W., Dabkowska M.— Anal. Chem., 1961, 33, N 1, p. 90—92.
933. Humphrey R. E — Ibid., 1962, 34, N 1, p. 167—168.
934. Hurley F. H., Wier T. P.— J. Electrochem. Soc., 1951, 98, N 5, p. 203—206.
935. Hurley F. H., Wier T. P.— Ibid., p. 207—212.
936. Hush N. S., Dyke I. M.— J. Electroanal. Chem., 1974, 53, N 2, p. 253—260.
229
937. Ichimura A. J., Kitagawa T.— Bull. Chem. Soc. Jap., 1980, 53, N 9, p. 2528—
2530.
938. Ikeda S„ Itabashi E — Ibid., 1969, 42, N 7, p. 1901—1908.
939. Inocencio A. A.— Electrochim. acta, 1978, 23, N 10, p. 977—981.
940. lohnson D. P., Bereman R. D.— J. Inorg and Nucl. Chem., 1972, 34, N 9,
p. 2957.
941, Ishibashi N., loshio M.— Electrochim. acta, 1972, 17, N 8, p. 1348—1352.
942. Itabashi E.— J. Electroanal. Chem., 1972, 36, N 1, p. 179—188.
943. Itabashi E., Ikeda S.— Bull. Chem. Soc. Jap., 1968, 41, N 8, p. 1844—1854.
944. Itabashi E., Ikeda S.— J. Electroanal. Chem., 1970, 26, N 1, p. 97—112.
945. Itabashi E., Ikeda S.—Ibid., 1972, 36, N 1, p. 189—201.
946. Iwakura C., Hayashi T., Kikkawa S., Tamura H.— Electrochim. acta, 1972,
17, N 6, p. 1085—1093.
947. Iwasita T„ Giordano M. C.— Ibid., 1969, 14, N 10, p. 1045—1060.
948. Izutsu K, Adachi T., Fujinaga T.— Ibid., 1970, 15, N 1, p. 135—145.
949. Izutsu K, Nakamura T.t Kitano T.— Bull. Chem. Soc. Jap., 1978, 51, N 3,
p. 783—789.
950. Izutsu K, Sakura S., Fujinaga T.— Ibid., 1972, 45, N 2, p. 445—450.
951. Izutsu K-, Sakura S., Fujinaga T.— Bull. Chem. Soc. Jap., 1973, 46, N 2,
p. 493—495.
952. Izutsu K-, Sakura S., Fujinaga T.— Ibid., N 7, p. 2148—2151.
953. Izutsu K„ Sakura S., Kuroki K., Fujinaga T.— J. Electroanal. Chem., 1971,
32, N 2, p. 11—14.
954. Jackson C. W., Blomgren G. E.— J. Electrochem. Soc., 1969, 116, Nil,
p. 1483—1487.
955. Jha S. K, Srivastava S. N.— Bull. Chem. Soc. Jap., 1967, 40, N 4, p. 810.
956. Jain D. S., Gaur J. N.— Acta. Chim. Acad, sci., hung., 1964, 42, N 1,
p. 7—13.
957. Jalnicke V?., Schweitzer P. H.— Z. Phys. Chem. (BRD), 1967, 52, N1—4,
S. 104—122.
958. James H. J., Broman R. F.— J. Phys. Chem., 1971, 75, N 26, p. 4019—4024.
959. Janz G. J., Tomkins R. P. T. Nonaqueous electrolytes handbook: In 3 vol.—
New York : Acad, press, 1972.
960. Jasinski R.— J. Electroanal. Chem., 1970, 26, N 2/3, p. 189—194.
961. Jasinski R.— Collect. Czech. Chem. Communs, 1971, 36, N 3, S. 1079—
1089.
962. Jensen W. B.— Chem. Rev., 1978, 78, N 1, p. 1—22.
963. Jones J. L„ Fritsche H. A. Jr.— J. Electroanal. Chem., 1966, 12, N 4,
p. 334—340.
964. lines D. E. H., Wood J. L.— Spectrochim. acta, A, 1967, 23, N 2, p. 205—
210.
965. Jones D. E. H., Wood J. L.— J. Chem. Soc. (A), 1967, N 7, p. 1140—1144.
966. Jones D. E. H„ Wood J. L.— Ibid., 1971, N 20, p. 3132—3136.
967. lohnson E. L., Pool К. H., Hamm R. E.— Analyt. Chem., 1966 38, N 2,
p. 183—185.
968. Jorne I., Tobias C. W.— J. Electrochem. Soc., 1974, 121, N8, p. 994—1000.
969. Jorne J., Tobias C. W.— J. Appl. Electrochem., 1975, 5, N 4, p. 279—290.
970. Joshio M., Matsuyuki M., Iwasawa I., Hagamatsu M.— J. Metal. Finish
Soc. Jap., 1975, 26, N 9, p. 416—417.
971. Young L. K, Cobes M. E., Meux J. M. et al.— J. Electrochem. Soc., 1980,
127, N 7, p. 1525—1529.
972. Kahe J. L„ Hanck К W., Armond K. D.— J. Phys. Chem., 1979, 83, N 20,
p. 2611—2615.
973. Kasakow W. A., Titowa V. N„ Smirnowa S. A., Petrowa N. W.— In: 29th
meet. Intern, soc. electrochem., Budapest, 1978: Extend. Abstrs. pt 2,
p. 1010—1012.
974. Kelley M. T., Fischer D. J., Jones H. C.— Anal. Chem., 1960, 32, N 10,
p. 1262—1265.
975. Kelley M. T„ Jones H. C„ Fischer R. A—Ibid., 1959, 31, N9, p. 1475—1485.
976. Kelsey G. S — J. Electrochem. Soc., 1977, 124, N 6, p. 927—933.
977. Kiss L., Dongoc L., Varsanji M. L— Collection, 1971, 36, N2, p. 914—924.
230
978. Kiss L., Sziraki L., Varsanyi M. L.— Acta Chim. Acad, sci hung., 1978, 97,
N 4, p. 389—397.
979. Kiss L., Varsanyi M. L., Dudas E.— Ibid., 1973, 79, N 1, p. 73—80.
980. Kiss L., Varsanyi M. L., Dudas E.— Mag. kem. folyoirat, 1973, 79, N 5,
p. 216—219.
981. Kladenova D., Golova M., Lax L.— In: 29th meet. Intern, soc. electrochem.,
Budapest, 1978 : Extend. Abstrs. pt 1, p. 573—574.
982. Klages F., Meuresch H., Sleppich W.— Annalen. Chem., 1955, 522, N 1,
S. 61—72.
983. Klimmek M.— Z. Metallk., 1979, 70, N 4, S. 260—265.
984. Knecht L. A., Kolthoff I. M.— Inorg. Chem., 1962, 1, N 2, p. 195—203.
985. Koelle U„ Holzinger W., Muller J — Z. Naturforsch., 1979, 34, N 5, S. 759—
761.
986. Kolditz L„ Preiss H.— Z. anorg.. und allg. Chem., 1961, 310, N4/6,
S. 242—250.
987. Kolthoff I. M.— Pure and Appl. Chem., 1971, 25, N 2, p. 305—325.
988. Kolthoff I. M., Coetze I. F.— J. Amer. Chem. Soc., 1957, 79, N 4, p. 870—
874.
989. Kolthoff I. M„ Coetze I. F — Ibid., N 20, p. 1852—1858.
990. Kolthoff I. M., Ikeda S.— J. Phys. Chem., 1961, 65, N 6, p. 1020—1026.
991. Kolthoff I. M., Reddy T. B.~ J. Electrochem. Soc., 1961, 108, N9, p. 980—
985.
992. Kolthoff I. M., Thomas F. G — Ibid., 1964, 111, N9, p. 1065—1074.
993. Kowalski 7,—Rocz. Chem., 1977, 51, N 11, s. 2071—2077.
994. Kowalski T. A., Lingane P. I.— J. Electroanal. Chem., 1971, 31, N 1,
p. 1—8.
995. Krygowski T. M., Fawcett W. R.— J. Amer. Chem. Soc., 1975, 97, N 5,
p. 2143—2147.
996. Kuhn L., Heintyze I.— Can. J. Chem., 1965, 43, N 2, p. 375—380.
997. Kuta E. J.— Anal. Chem., 1960, 32, N 8, p. 1065—1068.
998. Lafortune R., Bourret J. T„ Capuano G. A.— Can. J. Chem., 1978, 56, N 18,
p. 2375—2380.
999. Laitinen H. A., Hyman С. J.— J. Amer. Chem. Soc., 1948, 70, N 6,
p. 2241—2244.
1000. Laitinen H. A., Shoemaker С. E.— Ibid., 1950, 72, N 10, p. 4975—4977.
1001. Landsberg R-, Janietz P., Prugel M.— Monatsh. Chem., 1979, 110, N 4,
S. 831—840.
1002. Lappert M. F.— J. Chem. Soc., 1961, N 3, p. 817; 1962, p. 542.
1003. Lappert M. F„ Smith I. K — Ibid., 1961, N 12, p. 3224.
1004. Lappert M. F., Smith I. K-—Ibid., 1965, N 11, p. 5826—5831.
1005. Larson R. C., Iwamoto R. T.— J. Amer. Chem. Soc., 1960, 82, N 12,
p. 3239—3244, N 14, 3526—3527.
1006. Larson R. C., Iwamoto R. T.— Inorg. Chem., 1962, 1, N 2, p. 316—323.
1007. Laszczynski V.— Z. Electrochem., 1895, 2, N 4, S. 55—57.
1008. Laubengayer A. W„ Smith W. C.— J. Amer. Chem. Soc., 1954, 76, N 23,
p. 5985.
1009. Leonard G. W., Ir„ Sellers D.— J. Electrochem. Soc., 1955, 102, N 2,
p. 95—97.
1010. Letav H. Ir„ Gropp A. H.— J. Phys. Chem., 1953, 57, N 12, p. 964—965.
1011. Leveque A., Rosset A. R.— Bull. Soc. chim. France, 1971, 8, Nil, p. 4205—
4212.
1012. Lever А. В. P„ Wilshire I. A—Inorg. Chem., 1978, 17, N5, 1145—1151.
1013. Lewis I., Miller I. R., Richards R. L.,Thompson S.— J. Chem. Soc., 1965,
N 11, p. 5850—5859.
1014. Lewis J., Sowerby D. B.— J. Chem. Soc., 1957, N 4, p. 1617—1622.
1015. Liegler K-, Lehmkuhl H.— Z. anorg. und allg. Chem., 1956, 283, S. 414—
424.
1016. Lindqvist L, Einarson A—Acta chem. scand., 1959, 13, N 2, p. 420—423.
1017. Lonis I. D. I., Clande D., Roger P.— Bull. Soc. chim. France, 1971, 8, N 8,
p. 2816—2822.
1018. Lormeau S., Calmar G.— Bull. Soc. Chim., 1971, 8, N 3, p. 714—719.
231
1019. Louis C., Benoit R. L.— Electrochim. acta, 1973, 18, N 1, p. 7—12.
1020. Lovrecek B„ Marincic H.— Ibid., 1966, 11, N 2, p. 237—249.
1021. Lozorgo L.— Prod. Eng., 1969, 40, N 14, p. 118—119.
1022. Luchrs D. G., Leddy D. G.— J. Electroanal. Chem., 1973, 41, N 1, p. 113—
118.
1023. Lux L„ Galova M.— In: 29th meet. Int. soc. electrochem. Budapest, 1978 :
Extend. Abstras. pt. 1, p. 575—576.
1024. Macagno V. A., Giordano M. C.— Electrochim. acta, 1969, 14, N 4, p. 335—
357.
1025. Mac Lead J. D., Muir D. M., Parker A. J., Singh P.— Austral. J. Chem.,
1977, 30, N 7, p. 1423—1437.
1026. Magno F., Mazzochin G. A., Bontempelli G.— J. Electroanal. Chem., 1974,
57, N 1, p. 89—96.
1027. Malhotra К. C., Mahajan R. K., Chandhky S. C.— Indian J. Chem., 1976,
N 6, p. 1017—1021.
1028. Malhotra К. C., Sud R. G.— J. Inorg. and Nucl. Chem., 1976, 38, N 3,
p. 393—396.
1029. Manahan S. E., Iwamoto R. T.— J. Electroanal. Chem., 1967, 13, N 4,
p. 411—417.
1030. Marchidan S.— Stud. cere, etari chim., 1973, 21, Nil, p. 1283—1305.
1031. Marchon J. C., Badoz-Lambling I.— Bull. Soc. chim. France, 1967, N 12,
p. 4660—4668.
1032. Marcoux L. S., Prater К. B., Prater B. G., Adams R. N.— Anal. Chem.,
1965, 37, N 6, p. 1446—1447.
1033. Marecek V., Honz J.— Collect. Czech. Chem. Communs, 1973, 38, N 12,
s. 3553—3560.
1034. Maricle D. L„ Hordson W. G.— Anal. Chem., 1965, 37, N 12, p. 1562—1565.
1035. Marks A. P„ Drago R. S.— J. Amer. Chem. Soc., 1975, 97, N8, p. 3324—
3328.
1036. Marks A. P., Drago R. S.— Inorg. Chem., 1976, 15, N 7, p. 1800—1806.
1037. Martin R. P., Sawyer D. T.—Ibid., 1972, 11, Nil, p. 2644—2647.
1038. Martinez C., Calandra A. J., Arvia A. J.— Electrochim. acta, 1972, 17, N 12,
p. 2153—2179.
1039. Mastragostino M.., Casalbore G., Valcher S.— J. Electroanal. Chem., 1973,
44, N 1, p. 37—45.
1040. Masek J.— Collect. Czech. Chem. Communs, 1959, 24, N 1, s. 159—169.
1041. Mathieu J. B., Landolt D.— J. Electrochem. Soc., 1978, 125, N 7, p. 1044—
1049.
1042. Matsuda H.— Elektrochemistry, 1957, 61, N 3, p. 489—506.
1043. Matsuda H., Ayabe I.— Z. Electrochem., 1955, 59, N 3, S. 494—503.
1044. Matsuda J., Onehi J.— i. Appl. Electrochem., 1974, 4, N 1, p. 53—56.
1045. Matsuda J., Satake U.— J. Electrochem. Soc., 1980, 127, N 4, p. 877—879.
1046. Matsumura-Inone T., Tominaga-Morimoto T.— J. Electroanal. Chem., 1978,
93, N2, p. 127—140.
1047. Matysik J.~ Z. anal. Chem., 1962, 189, N 2, S. 202—203.
1048. Mayer U.— Pure and Appl. Chem., 1975, 41, N 3, p. 291—326.
1049. Mayer U., Cotocova A., Gutmann V., Gerger W.— J. Electroanal. Chem.,
1979, 100, N 1 / 2, p. 875—883.
1050. McElroy A. D., Rleinberg I., Devidson A. W.— J. Amer. Chem. Soc., 1950,
72, N 11, p. 5178—5180.
1051. McElroy A. D., Rjleinberg L, Davison A. W.— Ibid., 1952, 74, N 3, p. 736—
739.
1052. McMosfers D. L., Dunlap R. B., Ruempel J. R. et al.— Anal. Chem., 1967,
39, N 1, p. 103—105.
1053. Meaume H. T„ Odiot S. /.— Mol. struct., 1972, 11, N 1, p. 147—151.
1054. Meerwein H.—Ann. Chem., 1927, 445, N 1, S. 227—237.
1055. Meibuhr S. G.— J. Electrochem. Soc., 1970, 117, N 1, p. 56—60.
1056. Meibuhr S. G.— Ibid., 1971, 118, N 8, p. 1320—1322.
1057. Menzel W.— Berichte, 1942, 75, N 8, S. 1055—1064.
1058. Menzies I. A.—Rev. Polytechn., 1973, N 1319, 1227, 1229, 1231.
1059. Menzies I. A., Averill A. F.— Electrochim. acta, 1968, 13, N 4, p. 807—824-
232
1060. Menzies I. A., Badelek P. S. C., Moreland P. J.— Ibid., N 3 p. 429—442.
1061. Menzies I. A., Hill D. L„ Owen L. W.— Nature, 1959, 183, N 4664, p 816—
817.
1062. Menzies I. A., Owen G. W.— Electrochim. acta, 1966, 11, N 2, p. 251—256.
1063. Menzies I. A., Salt D. B.— Trans. Inst. Metal Finish., 1965, 43, N 5,
p. 186—191.
1064. Menzies I. A., White B. S.— Electrochim. acta, 1969, 14, N 5, p. 413—425.
1065. Merritt W. V., Sawyer T.— Inorg. Chem., 1970, 9, N 2, p. 211—215.
1066. Messina A., Gritzner G.— J. Electroanal. Chem., 1979, 101, N 2, p. 201—
209.
1067. Meunier I., Forel M. T.— Can. J. Chem., 1972, 50, p. 1157.
1068. Michlmayr M., Gutmann V.— Electrochim. acta, 1968, 13, N 7, p. 1671—1680.
1069. Mldorikawa T., Tajima S„ Baba N.— РЖ Химия, 1977, 11, 382.
1070. Miles M. H., Gerischer H.— J. Electrochem. Soc., 1971, 118, N 6, p. 837—
841.
1071. Miles M. H., Harris W. S.— Ibid., 1970, 118, N 10, p. 1225—1229.
1072. Miller L. L.— Pure and Appl. Chem., 1979, 51, N 10, p. 2125—2129.
1073. Minami S., Ishino T.— J. Chem. Soc. Jap., 1958, 61, N 1, p. 66—69.
1074. Mine S., Libus W.— Roczn. Chem., 1955, 29, N 4, s. 1073—1080.
1075. Mishra B., Ramakrishna V.— Proc. Chem. Symp. Chandirn., 1969, N 1,
p. 135—139.
1076. Mitra R. W., Daw A. N.— Indian J. Technol., 1976, 14, N 6, p. 311—312.
1077. Moeller T., Cullen G. If.—J. Inorg. and Nucl. Chem., 1959, 10, Nl/2,
p. 148—153.
1078. Molodoo A. I., Markosyan G. N.> Losev V. V,— Electrochim. acta, 1972,
17, N 4, p. 701—721.
1079. Mori K-, Takamura A., Shimose T.— Corrosion, 1966, 22, N 2, p. 29—31.
1080 Martins M. E., Castellano C., Calandra A. J., Arvia A. J.— J. Electroanal.
Chem., 1977, 81, N 2, p. 291—300.
1081. Mortins M. E., Castellano C„ Calandra A. J., Arvia A. J — Ibid., 1978, 92,
N 1, p. 45—54.
1082. Mossaux J., Duyckarts G.— Ibid., 1975, 59, N 3, p. 311—322.
1083. Mowins W. G.— J. Inorg. and Nucl. Chem., 1972, 34, N 8, p. 3571—3583.
1084. Mozza F.— Werkst. und korros, 1969, 20, N 3, S. 199—205.
1085. Mukherjee L. M., Boden D. P.— Electrochim. acta, 1972, 17, N 5, p. 965—
972.
1086. Muller R.— Z. anorg. und allg. Chem., 1926, 156, N 1, S. 56—63.
1087. Murphy N. F., Doumas A. C.— Chem. Abstrs, 1957, 51, 102701.
1088. Murphy N. F., Doumas A. C.— Ibid., 1959, 53, 22191a.
1089. Murray R. W„ Hiller L. Л.—Anal. Chem., 1967, 39, Nil, p. 1221—1229.
1090. Myasoedov B. F., Kulyako Yu. M., Sklyarenko I. S.— J. Inorg. and Nucl.
Chem., 1975, 37, N 1, p. 113—117.
1091. Nadjo L.— J. plectroanal. Chem., 1980, 108, N 1, p. 29—47.
1092. Nakabayashi S., Fulishima A. F., Honda K.— Ibid., Ill, N2/3, p. 391—
395
1093. Nelson I. V., Iwamoto R. T.— Anal. Chem., 1963, 35, N 7, p. 867—871.
1094. Nigam R. K, Saini R. C., Kapoor R.— Indian J. Chem., A, 1979, 17, Nl,
p. 17—20.
1095. Nigretto J.-M., Jozefowiez M.— Electrochim. acta, 1974, 19, N 12, p. 809—
820.
1096. Niro M., Kisaburo U., Mosako W. et al.— Bull. Chem. Soc. Jap., 1974, 47,
N 4, p. 806—812.
1097. Ocura K-, Miyamoto K.— Electrochim. acta, 1978, 23, N 6, p. 509—512.
1098. Odette S., Rene R.— C. r. Acad, sci., 1970, 271, N 1, p. 208—212.
1099. Olson D. C., Vasilejkis J.—Inorg. Chem., 1971, 10, N 3, p. 463—470.
1100. Onlevey G., Susz B.— Helv. chim. acta, 1965, 48, N18, p. 1965 1973.
1101. Olwer J. W., Bessette R. R.— J. Inorg. and Nucl. Chem., 1968, 30, N 7,
p. 1791—1798.
1102. Olwer J. W., Ross J. W.— Ibid., 1963, 25, N 12, p. 1515—1519.
1103. Orgen P. I., Cannon I. P., Smith C. F.—J. Phys. Chem., 1971, 75, N 2,
p. 282—290.
233
1104. Pang J., Ritchie I. M., Gills D. E.— Electrochim. acta, 1975, 20, N12,
p. 923—928.
1105. Parsons R.— Ibid., 1976, 21, N 9, p. 681—686.
1106. Patterson G. S., Holm R. H.— Inorg. Chem., 1972, 11, N9, p. 2285—2289.
1107. Paul R.~ J. Chem. Phys., 1969, 50, N 3, p. 1491—1492.
1108. Paul R. C., Bains M. S., Singh D.— J. Indian Chem. Soc., 1958, 35,
p. 489—491.
1109. Paul R. C„ Malnotra К. C.— Z. anorg. und allg. Chem., 1963, 321, Nl,
S. 56—70.
1110. Paul R. C., Malhotra К. C.— Ibid., 325, N 3, S. 302—314.
1111. Paul R. C„ Rehatni S. K„ Chadha S. L.— Indian J. Chem., 1970, 8, N 5,
p. 829—831.
1112. Paul R. C„ Rehani S. K„ Pahl S. S.—Ibid., 1969, 7, N7, p. 712—714.
1113. Paul R. C., Singu S., Sreenothem B.— Ibid., 1964, 2, N 1, p. 97—100.
1114. Paul R. C„ Singh P„ Chadna S. L.— Ibid., 1969, 7, N4, p. 625—630; 1970,
8, N8, p. 1010—1014.
1115. Payne R.— J. Phys. Chem., 1969, 73, N 11, p. 3598—3608.
1116. Payne R.— Adv. Electrochem. and Electrochem. Eng., 1970, 7, N1,
p. 1—76.
1117. Pfeirffer P.— In: Organische MolekCluerbindungen. Stuttgart, 1922, S. 518.
1118. Pedraza J. F., Iwamoto R. T.— J. Electroanal. Chem., 1974, 52, N 3,
p. 480—482.
1119. Peled E., Gileadi E — Plating, 1975, 62, N 4, p. 342—347.
1120. Peled E., Gileadi E.— J. Electrochem. Soc., 1976, 123, N 1, p. 15—19-
1121. Peled E., Mitavski A., Reder A., Gileadi E.— J. Electroanal. Chem., 1977,
75, N 2, p. 677—695.
1122. Peover M. E., White B. S.— Electrochim. acta, 1966, 11, N 8, p. 1061—
1068.
1123. Peraccio E., Me’oche V.— J. Amer. Chem. Soc., 1938, 60, N 6, p. 1770—1775.
1124. Perciro R., Arvia A. J., Calandra A. J.— Electrochim. acta, 1972, 17, N 10,
p. 1723—1734.
1125. Peretti E. A.—Progr. Extr. Met, 1973, 1, N. Y„ p. 207—239.
1126. Petrii О .A., Khomchenko I. G.— J. Electroanal. Chem., 1980, 106, Nl/2,
p. 277—286.
1127. Pierey R„ Hampson N. A.— J. Appl. Electrochem., 1975, 5, N 1, p. 1—15.
1128. Pineold T. A., Sebba F.— J. Amer. Chem. Soc., 1956, 78, N20, p. 5193—
5196.
1129. Plontelli R„ Bertocci V., Sternhein G.— Z. Elektrochem., 1958, 62, N6/7,
S 772_________773.
1130. Pompe R.— Electrochim. acta, 1976, 21, N 12, p. 1111—1114.
1131. Pondinini S., Ardizzone S., Longhi P„ Mussini T.— J. Electroanal. Chem.,
1977. 89, N 1, p. 59—68.
1132. Ponillen P., Martre A. M., Martinet P.— Bull. Soc. Chim. France, 1979,
N9 / 10, pt. 1, p. 387—390.
1133. Ponillen P., Porrin M., Martinet P.— C. r. Acad. sci. C, 1977, 284, N 23,
p. 955—957.
1134. Popov A. J., Geske D. H.— J. Amer. Chem. Soc., 1957, 79, N 13, p. 2074—
2079.
1135. Posadas D., Arvia A. Podesta J. J.— Electrochim. acta, 1971, 16, N7,
p. 1025—1054.
1136. Prasad R., Scalfe D. B.— J. Electroanal. Chem., 1977, 84, N 2, p. 373—
386.
1137. Prue I. E., Sherrington P. J.— Trans. Faraday Soc., 1961, 57, N 10,
p. 1795—1808.
1138. Pushin N. A., Rlstic M., Parhomenko I., Ubovlc I.— Annalen der Chemie,
1942, 553, N 2, p. 278—290.
1139. Qazi M. A., Leja J.—J. Electrochem. Soc., 1971, 118, N 4, p. 548—551.
1140. Quiun R. K., Lagowskl I. /.— Inorg. Chem., 1979, 9, N 2, p. 414—415.
1141. Rabinovich M„ Grinvald A.—J. Amer. Chem. Soc., 1972, 94, N 14, p. 2724—
2726.
1142. Rabockai T — Can. J. Chem., 1979, 57, N 14, p. 1801—1803.
234
"4g?
1443. Rabockaj T., de S. Bulhoes L. O.— Electrochim. acta, 1980, 25, N 8,
p. 1041—1042.
1144. Rabockaj T., Jordan I.— Anal. Lett., 1974, 7, N 10, p. 674—658.
1145. Rajagopalan M., Ramaniah M. V.— Radiochim. acta, 1966, 5, N 4, S. 236—
237.
1146. Ratio F., Rampazzo L.— J. Electroanal. Chem., 1968, 16, N 1, p. 61—66.
1147. Rao R. R„ Milleken S. B., Robinson S. L„ Mann С. A.—Anal. Chem.,
1970, 42, N 9, p. 1076—1088.
1148. Raundall E. W„ Jodersilcox С. M., Zuckerman I.— Inorg. Chem., 1966, 5,
N 12, p. 2240—2243.
1149. Rausch M. D., McEwen W. E., Kleinberg J.— J. Amer. Chem. Soc., 1954,
76, N 14, p. 3622—3625.
1150. Rausch M. D., McEwen W. E., Kleinberg J.— Ibid., 1955, 77, N 8, p. 2093—
2096.
1151. Reger A., Peled E., Giliadi E.— J. Electrochem. Soc., 1976, 123, N 5,
p. 638—642.
1152. Reid W. E„ Bish J. M., Brenner A.—Ibid., 1957, 104, N1 p. 21—29.
1153. Reid D. S., Vincent C. A.— J. Electroanal. Chem., 1968, 18, N 4, p. 427—
466.
1154. Reinmuth W. H.— Anal. Chem., 1961, 33, N3, p. 323—325.
1155. Reita T., Hiroyuki K.— Electrochim. acta, 1980, 25, N 4, p. 391—397.
1156. Roberts I. L., Sawyer D. T.— J. Electroanal. Chem., 1965, 9, N I, p. 1—7.
1157. Roffia S„ Ciano M.— Ibid., 1977, 77, N 3, p. 349—359.
1158. Roffia S., Ciano M.— Ibid., 1978, 87, N 2, p. 267—274.
1159. Roffia S„ Ciano M.— Ibid., 1979, 100, N 1 / 2, p. 809—817.
1160. Roffia S., Raggi M. A —Ibid., 1980, 108, N 1, p. 69—76.
1161. Rohl er H.— Z. Elektrochem., 1910, 16, N 12, S. 419—436.
1162. Roland G., Gilbert B., Decerf I., Puychaerts G.— Spectrochim. acta, A,
1973, 29, N 6, p. 887.
1163. Rolison D. R., Kuo K-, Umana M. et al.— J. Electrochem. Soc., 1979, 126,
N 3, p. 407—414.
1164. Roques G„ Le Pen D., Petit J. A., Mathien J.—J. Chim. Phys, et Phys.—
Chim biol., 1979, 76, N 2, p. 173—182.
1165. Sachan N. P., Gupta С. M.— Indian J. Chem. A, 1979, 18, N 1, p. 82—83.
1166. Sadek H., Issa R. M., Abdel—Habey B. A.— Electrochim. acta, 1971, 16,
N 3, p. 401—408.
1167. Safranek W. H., Schickner W. C., Faust C. L.— J. Electrochem. Soc., 1952,
99, N 2, p. 53—59.
1168. Saito T., Ikeda H., Matsuda J., Tamura H.— J. Appl. Electrochem., 1976,
6, N 1, p. 85—88.
1169. Saji T., Aoyagui S.— Bull. Chem. Soc. Jap., 1974, 47, N 2, p. 389—392.
1170. Saji T., Aoyagui S.— J. Electroanal. Chem., 1975, 58, N 2, p. 401—410.
1171. Saji T„ Aoyagui S.—Ibid., 63, N 1, p. 31—37.
1172. Saji T., Aoyagui S.— Ibid., N 3, p. 405—419.
1173. Saji T., Fukaj T., Aoyagui S.— J. Electroanal. Chem., 1975, 66, N 1, p. 81—
84.
1174. Saji T., Jamado T., Aoyagni S.— Ibid., 61, N 2, p. 147—153.
1175. Sakura S.— Ibid., 1977, 80, N 2, p. 315—335.
1176. Sakura S„ Lee H. L., Fujunaga T.— Electrochim. acta., 1980, 25. N 10,
p. 1247—1250.
1177. Salomon M.— J. Electrochem. Soc., 1971, 118, N 10, p. 1609—16H.
1178. Sancho J., Aldaz A., Pujante A.— J. Electroanal. Chem., 1970, 25, N 3,
p. 505—511.
1179. Sancho J., Almagro J., Pujante A.— Ibid., 1966, 11, N 2, p. 122—127.
1180. Sancho J., Almagro J., Pujante A — Ibid., 1968, 16, N 1, p. 77—82.
1181. Santelices C„ Hawley M. D — Ibid.. 1977 84, N 2, p. 387—396.
1182. Santos E„ Dyment F.— Plating, 1973, 30, N 8, p. 821—822.
1183. Sawye' £>. T., Roberts J. L.— J. Electroanal. Chem., 1966, 12, N 2, p. 90—
101.
1184. Scarr R. E.— J. Electrochem. Soc., 1970, 117, N 3, p. 295—299.
235
1185. Schaap W. В., Conley R. F., Schmidt F. C.— Anal. Chem., 1961, 33, N 4,.
p. 498—500.
1186. Schall C., Melzer W.— Z. Elektrochem., 1922, 28, N21/22, S. 474—477.
1187. Schimizu 7.—РЖ Химия, 1980, 12Б1607.
1188. Schimizu T., Suzuki M.— Rev. Polarogr., 1979, 24, N 1/6, p. 47—50.
1189. Schneider H., Strehlow H.— J. Electroanal. Chem., 1966, 12, N5—6,
p. 530—534.
1190. Schmid R., Gutmann V.— Monatschefte, 1969, 100, N 5, S. 1662—1669.
1191. Schmidt H.— Z. anorg. und allg. Chem., 1952, 270, Nl/4, S. 188—200.
1192. Schmidt H., Meinert H.— Ibid., 1958, 295, S. 156—172.
1193. Schmidt H., Noack J.— Ibid., 296, S. 262—266.
1194. Schmulbach C. D.— J. Inorg. and Nucl. Chem., 1964, 26, N 5, p. 745—749.
1195. Schmulbach C. D., Ahmed I. J.— J. Chem. Soc., A, 1968, p. 3008—ЗОН.
1196. Schober G., Gutmann V.— Z. Elektrochem., 1959, 63, N 2, S. 274—277.
1197. Schober G., Gutmann V.— Z. anal. Chem., 1960, 173, N 1, S. 2—4.
1198. Schober G., Gutmann V., Hedbalek E.— Ibid., 1962, 186, N 1, S. 115—117.
1199. Schwabe K— Z. phys. Chem. (BRD), 1977, 108, N 1, S. 61—72.
1200. Schwabe K., Schmidt W.— Corros. sci., 1970, 10, N 3, p. 143—155.
1201. Seifert H. J„ Auel T.— Z. anorg. und allg. Chem., 1968, 360, N 1, S. 50—51.
1202. Seifert H. J., Petersen F., Wohrmann H.— J. Inorg. and Nucl. Chem., 1973,
35, N 8, p. 2735—2744.
1203. Seifert H. J., Sauerteig W.— Z. anorg. und allg. Chem., 1970, 376, N 3,
p. 245—253.
1204. Seifert H. J., Thum F. M.— Ibid., 372, N 1, p. 79—86.
1205. Seifert H. J., Wohrmann H — Inorg. Nucl. Chem. Lett., 1970, 6, N 3,
p. 295—298.
1206. Sellers D. E., Leonard G. IF.— Anal. Chem., 1961, 33, N 3, p. 334—336.
1207. Sellers D. E., Leonard I. W.— Ibid., 1962, 34, N 11, p. 1457—1460.
1208. Sharma S. S., Singh M.— Vishwakarma, 1978, 19, N7, p. 41—42.
1209. Shinozuka N., Maruma S., Hayano S.— РЖ Химия, 1979, 9Б1310.
1210. Silvestroni P., Ratio F.— Ann. chim. (Ital.), 1974, 64, Nil/12, p. 781—
790.
1211. Simanavicius L., Dobrovolskis P.— Technik, 1976, 31, N 5, S. 329—330.
1212. Sinicki C., Desports P., Breat M., Rosset R.— Bull. Soc. Chim. France,
1968, N 2, p. 829—832.
1213. Singh S., Balachandra 7.—Trans. SAEST, 1976, 11, N 4, p. 487—497.
1214. Sixma F., Hendriks H.— Rec. trav. chim., 1955, 75, p. 127—130.
1215. Smith T. D.~ J. Chem. Soc., A, 1966, N7, p. 841—845.
1216. Sock O., Lemoine P., Gross M.—- Electrochim. acta, 1980, 25, N 8, p. 1025—
1026.
1217. Stackelberg M. V., Hans W., Hauck G.— Z. Elektrochem., 1957, 61, N 4,
S. 473—480
1218. Stillwell C. W., Andrieth L. F.— J. Amer. Chem. Soc., 1932, 54, N 1,
p. 472—478.
1219. Пат. 2537285 (ФРГ). Vorrichtung und Verfahren zum galvanischen Absch-
liden von Aluminium / K. Stoger, K- Dotzer, J. Gekring.— Опубл. 11.08.77.
1220. Strehlow H. The chemistry of non-aqueons solvents.— New York, 1966,
vol. 1, p. 129, Pergamon-Press.
1221. Sundholm G.— J. Electroanal. Chem., 1971, 31, N 1, p. 265—267.
1222. Susie M. V., Mentus S. V.— Гласник Хем. друшт. Београд, 1978, 43, №7,
с. 411—418. Сербский.
1223. Suyan К. М., Shah S. К., Gupta С. М.— J. Indian. Chem. Soc., 1979, 56,
N 9, p. 923—925.
1224. Syhnot J. C., Butler J. //.— Anal, Chem., 1969, 41, N 13, p. 1890—1894.
1225. Szenely G.— J. Electrochem. Soc., 1951, 98, N 8, p. 318—324.
1226. Tachi T., Takahashi R.— Collect. Czech. Chem. Communs, 1960, 25, N 12,
s. 3111—3119.
1227. Tajlma S., Baba N.— Electrochim. acta, 1962, 7, Nl/6, p. 355—367.
1228. Tajama S., Baba N„ Mori T.— Electrochim. acta, 1964, 9, Nil, p. 1509—
1519.
1229. Tajima S„ Komatsu S., Baba N.— Ibid., 1974, 19, N 12, p. 921—928.
236
1230. Takada J., Miyake J.— Bull. Covt. Ind. Res. Inst. Osaka, 1973, 24, N 2,
p. 104—110.
1231. Takada J., Miyake J.— Ibid., 1979, 30, N 1, p. 27—31.
1232. Takada J., Miyake J.— РЖ Химия, 1979, 7Б1794.
1233. Takahashi R — Taianta, 1965, 12, N 12, p. 1211—1228.
1234. Takej T — Bull. Chem. Soc. Jap., 1974, 47, N 2, p. 249—264.
1235. Takej T.— J. Appl. Electrochem., 1979, 9, N 5, p. 587—594.
1236. Takej T.— Electrochim. acta, 1980, 25, N 10, p. 1231—1237.
1237. Tanaka N.— Ibid., 1976, 21, N 9, p. 701—710.
1238. Tanaka N., Sato J.— Ibid., 1968, 13, N 3, p. 335—346.
1239. Taylor R. D„ Streer J. P., Minelli M., Spence J. T.— Inorg. Chem., 1978,
17, N 11, p. 3207—3211.
1240. Teeple H. 0,—Corrosion, 1952, 8, N 1, p. 14—26.
1241. Teherani H. T., Peer W. J., Lagowski J. J.— J. Amer. Chem. Socu 1978,
100, N 24, p. 7768—7770.
1242. Teherani T. H., Peer W. J., Lagowski J. J., Bard A. J.— J. Electrochem.
Soc., 1978, 125, N 10, p. 1717—1718.
1243. Thomas F. G., Kolthoff I. M.— J. Electroanal. Chem., 1971, 31, N 2,
p. 423—439.
1244. Tiedemann W. H., Bennion D. N — Ibid., 1973, 120, N 12, p. 1624—1628.
1245. Timmermans J. The Phisico-chemical constants of binary systems in con-
centrated solutions.— New York; London: Intern, publ. ING, 1959.—
Vol. 1—4.
1246. Tlscher R. P., Ludwig F. A.— Adv. Electrochem. and Electrochem. Eng.,
1977, 10, p. 391—482.
1247. Torel-Tarvoryan H. E., Hemingway R. E., Bard A. J.— J. Amer. Chem.
Soc., 1973, 95, N 20, p. 6582—6589.
1248. Toni J. E. A.— J. Electrochem. Soc., 1969, 116, N 2, p. 212—217.
1249. Tsuru T., Kimura T., Kobayashi S., Inui T.— J. Metal. Finish. Soc. Jap.,
1976, 27, N 5, p. 230—234.
1250. Tsuru T., Shimokawa IF., Kabayashi S., Inui T.— Automation, 1975, 22,
N 6, p. 73—74.
1251. Tsuru T., Shimokawa IF., Kobayashi S., Inui T.— J. Metal. Finish. Soc.
Jap., 1977, 28, N 5, p. 272—276.
1252. Tucker В. V., Fitzgerald J. M., Hargis L. G., Rogers L. B.— J. Electro-
anal. Chem., 1967, 13, N 4, p. 400—410.
1253. Venkatasatty H. F.— J. Electrochem. Soc., 1980, 127, Nil, p. 2531—2533.
1254. Venkatasatty H. V., Saothoff D. L— Ibid., 1978, 125, N 12, p. 1974—1977.
1255. Verhoef J. C., Barendrecht E.— Electrochim. acta, 1978, 23, N5, p. 433—
438.
1256. Verma P. S., Chakraborty B., Jain D. S., Gaur J. N.— Ibid., 1976, 21, N 12,
p. 1183—1186.
1257. Пат. 3616280 (США). Nonaqueous electroplating solutions and processing/
A. E. Vernon.
1258. Vicek A., ViSek A. A.— Inorg. chim. acta, 1979, 34, N 1, p. 189—191.
1259. Vicek A., Vicek A. A.— J. Electroanal. Chem., 1979, 100, Nl/2, p. 867—
873.
1260. Voorhies J. D„ Schurdak E. J.— Anal. Chem., 1962, 34, N 8, p. 939—943.
1261. Walker J. IF.—J. Chem. Soc., 1904, 84, p. 1082—1098.
1262. Walker J. W., Spencer A.— J. Chem. Soc., 1904, 85, p. 1106—1111.
1263. Wanry R. K-, Wilson С. E.— J. Electrochem. Soc., India, 1978, 27, N 4,
p. 199—205.
1264. Wargon J. A., Awia A. J.— Electrochim acta, 1972, 17, N 4, p. 649—664.
1265. IFa/Z G. V., Vaughin I. IF.— J. Electrochem. Soc., 1963, 110, N 7, p. 723—
726.
1266. Wawzonek S., Runner M. E.— J. Electrochem. Soc., 1952, 99, Nil,
p. 457—459.
1267. Wick A. K.— Helv. chim. acta, 1968, 51, p. 85.
1268. Wilson J., Worrall I. J.— J. Chem. Soc., 1967, A, p. 392—395
.1269. Wise E. N., Cokal E. /.— Anal Chem., 1960, 32, p. 1417—1419.
237
1270. Wong J. J. Пат. США, кл. 204. 14N (с 25 D 3/44) N 645552, опубл.
18.01.1977.
1271. Wood G. В., Brenner A.— J. Electrochem. Soc., 1957, 104, N 1, p. 29—37.
1272. Woolf A. A.— J. Chem. Soc., 1955, N 2, p. 433—443.
1273. Ziegel S., Peled E., Gileadi E.— Electrochim. acta, 1978, 23, N 4, p. 363—
368.
1274. Zubaiz, Clande M. J.— Electrochim. acta, 1974, 19, N 9, p. 545—550.
1275. Zuman P.— Electrochim. acta, 1976, 21, N 9, p. 687—692.
1276. Zutshi K.— J. Electroanal. Chem., 1963, 6, N 3, p. 198—204.
1277. Zutshi K.. Gupta К. C.— Indian J. Chem., A, 1978, 16, N 5, p. 453—454.
ОГЛАВЛЕНИЕ
Предисловие.............................................................3
Список сокращений.......................................................4
Глава 1. Некоторые проблемы теории электролитных неводных растворов . 5
1.1. Общая схема равновесий в неводных растворах электролитов . . 5
Условия возникновения электролитного раствора ......................... 5
1.1.1. Развитие представлений об основных типах межмолекулярных про-
цессов в растворах .................................................... 5
1.1.2. Гомомолекулярные (ассоциативно-диссоциативные) процессы . . 6
1.1.3. Гетеромолекулярные процессы (образование продуктов присоедине-
ния) ...................................................................8
1.1.4. Процесс ионизации (образование ионных пар)......................9
1.1.5. Электролитическая диссоциация....................................11
1.1.6. Общая схема равновесий в растворах электролитов. Проблема ин-
дифферентности растворителя.............................................11
1.2. Влияние растворителя на силу электролитов..........................14
1.2.1. Единое уравнение электролитической диссоциации по Н. А. Измайлову 14
1.2.2. Дифференцирующее и нивелирующее действие растворителя на силу
электролитов .................................................... 16
1.2.3. Термодинамика процесса электролитической диссоциации ... 17
1.3. Некоторые вопросы теории электропроводности неводных растворов . 21
1.3.1. Общие положения.................................................21
1.3.2. Электропроводность двойных жидких систем........................22
1.3.3. Зависимость электропроводности от концентрации..................26
1.3.4. Зависимость электропроводности от вязкости и диэлектрической про-
ницаемости ............................................................29
1.3.5. Зависимость электропроводности от температуры, температурные
коэффициенты электропроводности. Термодинамика активации процес-
са электропроводности ................................................ 31
1.4. Кислотно-осиовное взаимодействие между компонентами раствора и его
влияние на ионную миграцию в силу электролита..........................38
1.4.1. Кислотно-основные характеристики электролита....................38
1.4.2. Кислотно-основные характеристики растворителя...................40
1.4.3. Кислотность апротонных кислот...................................42
1.4.4. Кислотно-основное взаимодействие и электропроводность неводных
растворов............................................................ 46
1.4.5. Кислотно-основное взаимодействие и сила электролитов в неводных
растворах..............................................................49
1.5. Природа электролитов в неводных растворах.........................51
1.5.1. Некоторые вопросы термодинамики ионных растворов .... 51
1.5.2. Комплексообразование в системах U(L)-кислота—растворитель . . 54
1.5.3. Механизмы электролитической диссоциации в неводных растворах . 57
1.5.4. Механизмы переноса тока в неводных растворах....................63
Глава 2. Электродные процессы в неводных растворах.....................66
2.1. Электродное равновесие............................................66
2.1.1. Зависимость электродных потенциалов и ЭДС от свойств растворителя 66
2.1.2. Ряды напряжений в неводных растворителях........................69
2.2. Вольт-амперные измерения в неводных растворах.....................71
2.2.1. Электроды сравнения.............................................71
2.2.2. Методы и приемы исследований....................................73
2.3. Катодные процессы при электровыделении элементов из неводных раст-
воров .................................................................78
2.3.1. Щелочные металлы...............................................78
2.3.2. Подгруппа меди..................................................°*
2.3.3. Щелочноземельные металлы......................................
2.3.4. Подгруппа цинка.................................................°6
2.3.5. Алюминий и металлы его подгруппы..............................
2.3.6. Лантаноиды н актиноиды....................................90
2.3.7. Подгруппа германия...............................................92
2.3.8. Подгруппа титана.................................................93
2.3.9. Металлы пятой группы.............................................94
2.3.10. Металлы шестой группы...........................................95
2.3.11. Подгруппа марганца..............................................96
2.3.12. Металлы восьмой группы..........................................97
2.3.13. Электровосстановление металлов из оксидов......................100
2.3.14. Неметаллы......................................................101
2.4. Анодные процессы в системах металл — неводный растворитель . .106
2.4.1. Металлы первой группы......................................107
2.4.2. Металлы второй группы......................................109
2.4.3. Металлы третьей группы.....................................111
2.4.4. Металлы четвертой группы...................................114
2.4.5. Металлы пятой группы.......................................117
2.4.6. Металлы шестой группы......................................117
2.4.7. Металлы седьмой группы.......................................119
2.4.8. Металлы восьмой группы.......................................120
2.5. Анодное окисление анионов в неводных растворителях .... 123
2.5.1. Галогениды и псевдогалогеннды................................123
2.5.2. Кислородсодержащие анноны....................................125
Глава 3. Характеристики электролитных композиций для электроосаждения
металлов из неводных растворов.......................................127
3.1. Характеристики растворителей, применяемых в практике электроосаж-
дения металлов......................................................127
3.1.1. Классификация растворителей..................................127
3.1.2. Смешанные растворители.......................................130
3.1.3. Критерии выбора растворителей для электролитных композиций . 133
3.2. Характеристики электролитных композиций........................133
3.2.1. Растворимость соединений металлов в неводных растворителях . 133
3.2.2. Электрохимическая устойчивость неводных электролитов . . . 136
Глава 4. Практика электровыделения металлов из неводных растворов 138
4.1. Металлы первой группы.........................................138
4.2. Металлы второй группы.........................................144
4.3. Металлы третьей группы........................................147
4.4. Металлы четвертой группы......................................157
4.5. Металлы пятой группы..........................................159
4.6. Металлы шестой группы.........................................161
4.7. Металлы седьмой группы........................................164
4.8. Металлы восьмой группы........................................165
Приложение..........................................................168
Список литературы....................................................203
Юрий Яковлевич Фиалков, Валентина Федоровна Грищенко
ЭЛЕКТРОВЫДЕЛЕНИЕ МЕТАЛЛОВ ИЗ НЕВОДНЫХ РАСТВОРОВ
Утверждено к печати ученым советом Института общей и неорганической химии АН УССР
Редактор Э. Е. Гриценко. Оформление художника А. Г. Самсонова. Художественный редак-
тор В. П. Кузь. Технический редактор Г. М. Ковалева. Корректоры Л. И. Ноур,
О. Е. Исарова, Е. А. Дубарь, Л. М. Тищенко.
Информ, бланк № 6933
Сдано в набор 07.09.84. Подп. в печ. 08.02.85. БФ 01532. Формат 60X90/16. Бум. тип. № 1.
Лит. гарн. Быс. печ. Усл. печ. л. 15,0. Усл. кр.-отт. 15,0. Уч.-изд. л. 20,08. Тираж
1000 экз. Заказ 4-653. Цена 3 р. 30 к.
Издательство «Наукова думка». 252601 Киев 4, ул. Репина, 3.
Киевская книжная типография научной книги. 252004 Киев 4, ул. Репина, 4.